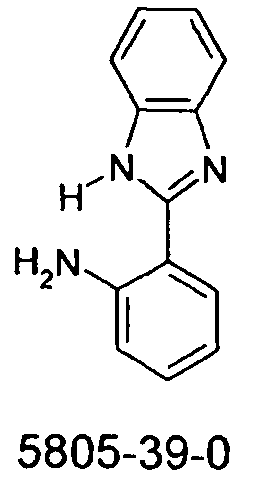
-
Die vorliegende Erfindung betrifft Metallkomplexe, welche sich für den Einsatz als Emitter in organischen Elektrolumineszenzvorrichtungen eignen.
-
Der Aufbau organischer Elektrolumineszenzvorrichtungen (OLEDs), in denen organische Halbleiter als funktionelle Materialien eingesetzt werden, ist beispielsweise in
US 4539507 ,
US 5151629 ,
EP 0676461 und
WO 98/27136 beschrieben. Dabei werden als emittierende Materialien zunehmend metallorganische Komplexe eingesetzt, die Phosphoreszenz statt Fluoreszenz zeigen (M. A. Baldo et al., Appl. Phys. Lett. 1999, 75, 4-6). Aus quantenmechanischen Gründen ist unter Verwendung metallorganischer Verbindungen als Phosphoreszenzemitter eine bis zu vierfache Energie- und Leistungseffizienz möglich. Generell gibt es bei OLEDs, die Triplettemission zeigen, immer noch Verbesserungsbedarf, insbesondere im Hinblick auf Effizienz, Betriebsspannung und Lebensdauer. Dies gilt insbesondere für OLEDs, welche im kürzerwelligen Bereich, also grün und insbesondere blau, emittieren.
-
Gemäß dem Stand der Technik werden in phosphoreszierenden OLEDs als Triplettemitter insbesondere Iridiumkomplexe eingesetzt. Eine Verbesserung dieser OLEDs konnte dadurch erzielt werden, dass Metallkomplexe mit polypodalem Liganden bzw. Kryptate eingesetzt wurden, wodurch die Komplexe eine höhere thermische Stabilität aufweisen, was zu einer höheren Lebensdauer der OLEDs führt (
WO 2004/081017 ,
WO 2005/113563 ,
WO 2006/008069 ). Für blaue Emission, insbesondere für gesättigte tiefblaue Emission, sind diese Komplexe ebenso wie die nicht überbrückten Komplexe jedoch weniger geeignet.
-
Aus dem Stand der Technik sind weiterhin Iridiumkomplexe bekannt, welche als Liganden Imidazophenanthridin-Derivate bzw. Diimidazochinazolin-Derivate enthalten (
WO 2007/095118 ). Diese Komplexe können bei Anwendung in organischen Elektrolumineszenzvorrichtungen, je nach genauer Struktur des Liganden, zu blauer Phosphoreszenz führen. Auch hier sind noch weitere Verbesserungen hinsichtlich Effizienz, Betriebsspannung und Lebensdauer, wünschenswert. Weiterhin besteht hier auch noch Verbesserungsbedarf in Bezug auf die Farbkoordinaten, um tiefblaue Emission erzielen zu können.
-
Aus
WO 2010/086089 sind Metallkomplexe bekannt, welche als Liganden Imidazo-isochinolin-Derivate enthalten. Mit derartigen Komplexen wurden bereits gute Fortschritte in der Entwicklung blauer Triplettemitter erzielt. Jedoch sind auch hier noch weitere Verbesserungen hinsichtlich Effizienz, Betriebsspannung und Lebensdauer wünschenswert. Insbesondere besteht hier auch noch Verbesserungsbedarf in Bezug auf die Farbkoordinaten, um tiefblaue Emission erzielen zu können, sowie in Bezug auf die Ausbeute, mit der sich die Komplexe synthetisieren lassen.
-
Aufgabe der vorliegenden Erfindung ist daher die Bereitstellung neuer Metallkomplexe, welche sich als Emitter für die Verwendung in OLEDs eignen. Insbesondere ist die Aufgabe, Emitter bereitzustellen, welche sich für blau phosphoreszierende OLEDs eignen, und welche dabei verbesserte Eigenschaften in Bezug auf Effizienz, Betriebsspannung, Lebensdauer und/oder Farbkoordinaten zeigen und/oder welche sich mit verbesserter Ausbeute herstellen lassen.
-
Überraschend wurde gefunden, dass bestimmte, unten näher beschriebene Metallchelatkomplexe diese Aufgabe lösen und zu Verbesserungen der organischen Elektrolumineszenzvorrichtung führen. Weiterhin sind diese Metallkomplexe in hoher Ausbeute zugänglich. Diese Metallkomplexe und organische Elektrolumineszenzvorrichtungen, welche diese Komplexe enthalten, sind daher der Gegenstand der vorliegenden Erfindung.
-
Gegenstand der Erfindung ist somit eine Verbindung gemäß Formel (1),
M(L)n(L')m Formel (1) wobei die Verbindung der allgemeinen Formel (1) eine Teilstruktur M(L)
n gemäß einer der Formeln (7) bis (38) enthält:
wobei für die verwendeten Symbole und Indizes gilt:
- M
- ist Iridium oder Platin;
- R
- ist bei jedem Auftreten gleich oder verschieden H, D, F, CN, eine geradkettige Alkylgruppe mit 1 bis 10 C-Atomen oder eine verzweigte oder cyclische Alkylgruppe mit 3 bis 10 C-Atomen, wobei ein oder mehrere H-Atome durch D oder F ersetzt sein können, oder ein aromatisches oder heteroaromatisches Ringsystem mit 5 bis 30 aromatischen Ringatomen, das jeweils durch einen oder mehrere Reste R2 substituiert sein kann; dabei können zwei benachbarte Reste R oder R mit R1 auch miteinander ein mono- oder polycyclisches, aliphatisches Ringsystem bilden;
- R1
- ist bei jedem Auftreten gleich oder verschieden CF3, OCF3, eine verzweigte oder cyclische Alkyl- oder Alkoxygruppe mit 3 bis 20 C-Atomen, wobei ein oder mehrere H-Atome durch D oder F ersetzt sein können, oder Si(R2)3, wobei R2 ungleich H oder D ist, oder ein aromatisches oder heteroaromatisches Ringsystem mit 5 bis 24 aromatischen Ringatomen, das jeweils durch einen oder mehrere Reste R2 substituiert sein kann, oder eine Aralkyl- oder Heteroaralkylgruppe mit 5 bis 40 aromatischen Ringatomen, die durch einen oder mehrere Reste R2 substituiert sein kann;
- R2
- ist bei jedem Auftreten gleich oder verschieden H, D, F, CN, eine geradkettige Alkylgruppe mit 1 bis 20 C-Atomen oder eine verzweigte oder cyclische Alkylgruppe mit 3 bis 20 C-Atomen, wobei ein oder mehrere H-Atome durch D oder F ersetzt sein können, oder ein aromatisches oder heteroaromatisches Ringsystem mit 5 bis 60 aromatischen Ringatomen, das jeweils durch einen oder mehrere Reste R3 substituiert sein kann; dabei können zwei oder mehrere benachbarte Reste R3 miteinander ein mono- oder polycyclisches, aliphatisches Ringsystem bilden;
- R3
- ist bei jedem Auftreten gleich oder verschieden H, D, F oder ein aliphatischer und/oder aromatischer Kohlenwasserstoffrest mit 1 bis 20 C-Atomen;
- L'
- ist gleich oder verschieden bei jedem Auftreten ein beliebiger Coligand;
- n
- ist 1, 2 oder 3;
- m
- ist 0, 1, 2, 3 oder 4;
dabei können auch mehrere Liganden L miteinander oder L mit L' über eine Einfachbindung oder eine beliebige Brücke V verknüpft sein und so ein tridentates, tetradentates, pentadentates oder hexadentates Ligandensystem aufspannen;
dabei kann auch ein Substituent R oder R1 zusätzlich an das Metall koordinieren;
mit der Maßgabe, dass R1 in den Formeln (7) und (8) für eine verzweigte oder cyclische Alkylgruppe mit 4 bis 20 C-Atomen, wobei ein oder mehrere H-Atome durch D oder F ersetzt sein können, oder für eine Bi- oder Oligoaryl- bzw. -heteroarylgruppe mit 10 bis 24 aromatischen Ringatomen oder für eine Aryl- oder Heteroarylgruppe, welche in mindestens einer ortho-Position zur Verknüpfung mit dem Liganden mit einem Rest R2 ungleich H oder D substituiert ist, steht.
-
Dabei ist es erfindungswesentlich, dass das Ligandengrundgerüst mindestens ein Stickstoffatom enthält und dass eine Gruppe, die diesem Stickstoffatom benachbart ist, für eine Gruppe CR1 steht, also für ein Kohlenstoffatom, welches mit einer der oben definierten Gruppen R1 substituiert ist.
-
Dabei werden in den Komplexen der Formel (1) die Indizes n und m so gewählt, dass die Koordinationszahl am Metall M insgesamt, je nach Metall, der für dieses Metall üblichen Koordinationszahl entspricht. Dies ist für Übergangsmetalle je nach Metall üblicherweise die Koordinationszahl 4, 5 oder 6. Es ist generell bekannt, dass Metallkoordinationsverbindungen abhängig vom Metall und von der Oxidationsstufe des Metalls unterschiedliche Koordinationszahlen aufweisen, also eine unterschiedliche Anzahl von Liganden binden. Da die bevorzugten Koordinationszahlen von Metallen bzw. Metallionen in verschiedenen Oxidationsstufen zum allgemeinen Fachwissen des Fachmanns auf dem Gebiet der metallorganischen Chemie bzw. der Koordinationschemie gehören, ist es für den Fachmann ein Leichtes, je nach Metall und dessen Oxidationsstufe und je nach genauer Struktur des Liganden L eine geeignete Anzahl Liganden zu verwenden und somit die Indizes n und m geeignet zu wählen.
-
Eine Arylgruppe im Sinne dieser Erfindung enthält 6 bis 40 C-Atome; eine Heteroarylgruppe im Sinne dieser Erfindung enthält 2 bis 40 C-Atome und mindestens ein Heteroatom, mit der Maßgabe, dass die Summe aus C-Atomen und Heteroatomen mindestens 5 ergibt. Die Heteroatome sind bevorzugt ausgewählt aus N, O und/oder S. Dabei wird unter einer Arylgruppe bzw. Heteroarylgruppe entweder ein einfacher aromatischer Cyclus, also Benzol, bzw. ein einfacher heteroaromatischer Cyclus, beispielsweise Pyridin, Pyrimidin, Thiophen, etc., oder eine kondensierte Aryl- oder Heteroarylgruppe, beispielsweise Naphthalin, Anthracen, Phenanthren, Chinolin, Isochinolin, etc., verstanden.
-
Ein aromatisches Ringsystem im Sinne dieser Erfindung enthält 6 bis 60 C-Atome im Ringsystem. Ein heteroaromatisches Ringsystem im Sinne dieser Erfindung enthält 1 bis 60 C-Atome und mindestens ein Heteroatom im Ringsystem, mit der Maßgabe, dass die Summe aus C-Atomen und Heteroatomen mindestens 5 ergibt. Die Heteroatome sind bevorzugt ausgewählt aus N, O und/oder S. Unter einem aromatischen oder heteroaromatischen Ringsystem im Sinne dieser Erfindung soll ein System verstanden werden, das nicht notwendigerweise nur Aryl- oder Heteroarylgruppen enthält, sondern in dem auch mehrere Aryl- oder Heteroarylgruppen durch eine nicht-aromatische Einheit (bevorzugt weniger als 10 % der von H verschiedenen Atome), wie z. B. ein C-, N- oder O-Atom oder eine Carbonylgruppe, unterbrochen sein können. So sollen beispielsweise auch Systeme wie 9,9'-Spirobifluoren, 9,9-Diarylfluoren, Triarylamin, Diarylether, Stilben, etc. als aromatische Ringsysteme im Sinne dieser Erfindung verstanden werden, und ebenso Systeme, in denen zwei oder mehrere Arylgruppen beispielsweise durch eine lineare oder cyclische Alkylgruppe oder durch eine Silylgruppe unterbrochen sind. Weiterhin sollen Systeme, in denen zwei oder mehrere Aryl- oder Heteroarylgruppen direkt aneinander gebunden sind, wie z. B. Biphenyl oder Terphenyl, ebenfalls als aromatisches bzw. heteroaromatisches Ringsystem verstanden werden.
-
Unter einer cyclischen Alkyl-, Alkoxy- oder Thioalkoxygruppe im Sinne dieser Erfindung wird eine monocyclische, eine bicyclische oder eine polycyclische Gruppe verstanden.
-
Im Rahmen der vorliegenden Erfindung werden unter einer C1- bis C40-Alkylgruppe, in der auch einzelne H-Atome oder CH2-Gruppen durch die oben genannten Gruppen substituiert sein können, beispielsweise die Reste Methyl, Ethyl, n-Propyl, i-Propyl, Cyclopropyl, n-Butyl, i-Butyl, s-Butyl, t-Butyl, Cyclobutyl, 2-Methylbutyl, n-Pentyl, s-Pentyl, tert-Pentyl, 2-Pentyl, neo-Pentyl, Cyclopentyl, n-Hexyl, s-Hexyl, tert-Hexyl, 2-Hexyl, 3-Hexyl, neo-Hexyl, Cyclohexyl, 1-Methylcyclopentyl, 2-Methylpentyl, n-Heptyl, 2-Heptyl, 3-Heptyl, 4-Heptyl, Cycloheptyl, 1-Methylcyclohexyl, n-Octyl, 2-Ethylhexyl, Cyclooctyl, 1-Bicyclo[2,2,2]octyl, 2-Bicyclo[2,2,2]-octyl, 2-(2,6-Dimethyl)octyl, 3-(3,7-Dimethyl)octyl, Adamantyl, Trifluormethyl, Pentafluorethyl oder 2,2,2-Trifluorethyl verstanden. Unter einer Alkenylgruppe werden beispielsweise Ethenyl, Propenyl, Butenyl, Pentenyl, Cyclopentenyl, Hexenyl, Cyclohexenyl, Heptenyl, Cycloheptenyl, Octenyl, Cyclooctenyl oder Cyclooctadienyl verstanden. Unter einer Alkinylgruppe werden beispielsweise Ethinyl, Propinyl, Butinyl, Pentinyl, Hexinyl, Heptinyl oder Octinyl verstanden. Unter einer C1- bis C40-Alkoxygruppe werden beispielsweise Methoxy, Trifluormethoxy, Ethoxy, n-Propoxy, i-Propoxy, n-Butoxy, i-Butoxy, s-Butoxy, t-Butoxy oder 2-Methylbutoxy verstanden.
-
Unter einem aromatischen oder heteroaromatischen Ringsystem mit 5 - 60 aromatischen Ringatomen, welches noch jeweils mit den oben genannten Resten R substituiert sein kann und welches über beliebige Positionen am Aromaten bzw. Heteroaromaten verknüpft sein kann, werden beispielsweise Gruppen verstanden, die abgeleitet sind von Benzol, Naphthalin, Anthracen, Benzanthracen, Phenanthren, Benzophenanthren, Pyren, Chrysen, Perylen, Fluoranthen, Benzfluoranthen, Naphthacen, Pentacen, Benzpyren, Biphenyl, Biphenylen, Terphenyl, Terphenylen, Fluoren, Spirobifluoren, Dihydrophenanthren, Dihydropyren, Tetrahydropyren, cis- oder trans-Indenofluoren, cis- oder trans-Monobenzoindenofluoren, cis- oder trans-Dibenzoindenofluoren, Truxen, Isotruxen, Spirotruxen, Spiroisotruxen, Furan, Benzofuran, Isobenzofuran, Dibenzofuran, Thiophen, Benzothiophen, Isobenzothiophen, Dibenzothiophen, Pyrrol, Indol, Isoindol, Carbazol, Indolocarbazol, Indenocarbazol, Pyridin, Chinolin, Isochinolin, Acridin, Phenanthridin, Benzo-5,6-chinolin, Benzo-6,7-chinolin, Benzo-7,8-chinolin, Phenothiazin, Phenoxazin, Pyrazol, Indazol, Imidazol, Benzimidazol, Naphthimidazol, Phenanthrimidazol, Pyridimidazol, Pyrazinimidazol, Chinoxalinimidazol, Oxazol, Benzoxazol, Naphthoxazol, Anthroxazol, Phenanthroxazol, Isoxazol, 1,2-Thiazol, 1,3-Thiazol, Benzothiazol, Pyridazin, Benzopyridazin, Pyrimidin, Benzpyrimidin, Chinoxalin, 1,5-Diazaanthracen, 2,7-Diazapyren, 2,3-Diazapyren, 1,6-Diazapyren, 1,8-Diazapyren, 4,5-Diazapyren, 4,5,9,10-Tetraazaperylen, Pyrazin, Phenazin, Phenoxazin, Phenothiazin, Fluorubin, Naphthyridin, Azacarbazol, Benzocarbolin, Phenanthrolin, 1,2,3-Triazol, 1,2,4-Triazol, Benzotriazol, 1,2,3-Oxadiazol, 1,2,4-Oxadiazol, 1,2,5-Oxadiazol, 1,3,4-Oxadiazol, 1,2,3-Thiadiazol, 1,2,4-Thiadiazol, 1,2,5-Thiadiazol, 1,3,4-Thiadiazol, 1,3,5-Triazin, 1,2,4-Triazin, 1,2,3-Triazin, Tetrazol, 1,2,4,5-Tetrazin, 1,2,3,4-Tetrazin, 1,2,3,5-Tetrazin, Purin, Pteridin, Indolizin und Benzothiadiazol.
-
Bevorzugt sind Verbindungen gemäß Formel (1), dadurch gekennzeichnet, dass diese nicht geladen, d. h. elektrisch neutral, sind. Dies wird auf einfache Weise dadurch erreicht, dass die Ladung der Liganden L und L' so gewählt werden, dass sie die Ladung des komplexierten Metallatoms M kompensieren.
-
Bevorzugt sind weiterhin Verbindungen gemäß Formel (1), dadurch gekennzeichnet, dass die Summe der Valenzelektronen um das Metallatom in vierfach koordinierten Komplexen 16 und in fünffach koordinierten Komplexen 16 oder 18 und in sechsfach koordinierten Komplexen 18 beträgt. Diese Bevorzugung ist durch die besondere Stabilität dieser Metallkomplexe begründet.
-
Bevorzugt sind Verbindungen gemäß Formel (1), in denen M für Ir(III) und Pt(II) steht.
-
In einer bevorzugten Ausführungsform der Erfindung ist M Pt(II), und der Index n steht für 1 oder 2. Wenn der Index n = 1 ist, sind noch ein bidentater oder zwei monodentate Liganden L', bevorzugt ein bidentater Ligand L', an das Metall M koordiniert. Wenn der Index n = 2 ist, ist der Index m = 0.
-
In einer weiteren bevorzugten Ausführungsform der Erfindung ist M Ir(III), und der Index n steht für 1, 2 oder 3, bevorzugt für 2 oder 3. Wenn der Index n = 1 ist, sind noch vier monodentate oder zwei bidentate oder ein bidentater und zwei monodentate oder ein tridentater und ein monodentater oder ein tetradentater Ligand L', bevorzugt zwei bidentate Liganden L', an das Metall koordiniert. Wenn der Index n = 2 ist, sind noch ein bidentater oder zwei monodentate Liganden L', bevorzugt ein bidentater Ligand L', an das Metall koordiniert. Wenn der Index n = 3 ist, ist der Index m = 0.
-
In einer bevorzugten Ausführungsform der Erfindung stehen in den Formeln (26), (29), (34) und (38) nicht beide Gruppen R1 für eine tertiäre Alkylgruppe.
-
Wie oben definiert, ist benachbart zu mindestens einem Stickstoffatom im Ligadnen eine Gruppe R1 als Substituent gebunden. Dabei ist R1, wie oben definiert, eine Gruppe, ausgewählt aus CF3, OCF3, verzweigten oder cyclischen Alkyl- oder Alkoxygruppen mit mindestens 3 C-Atomen, dreifach substituierten Silylgruppen, aromatischen bzw. heteroaromatischen Ringsystemen oder Aralkyl- bzw. Heteroaralkylgruppen. Es handelt sich bei diesen Gruppen um sterisch anspruchsvolle Gruppen.
-
Wenn R
1 für eine Alkylgruppe steht, dann weist diese Alkylgruppe bevorzugt 4 bis 10 C-Atome auf. Bevorzugt handelt es sich weiterhin um eine sekundäre oder tertiäre Alkylgruppe, bei der das sekundäre oder tertiäre C-Atom entweder direkt an den Liganden gebunden ist oder über eine CH
2-Gruppe an den Liganden gebunden ist. Besonders bevorzugt ist diese Alkylgruppe ausgewählt aus den Strukturen der folgenden Formeln (R
1-1) bis (R
1-33), wobei jeweils auch die Anknüpfung dieser Gruppen an den Liganden mit eingezeichnet ist:
wobei Lig die Anknüpfung der Alkylgruppe an den Liganden kennzeichnet.
-
Wenn R
1 für eine Alkoxygruppe steht, dann weist diese Alkoxygruppe bevorzugt 3 bis 10 C-Atome auf. Bevorzugt ist diese Alkoxygruppe ausgewählt aus den Strukturen der folgenden Formeln (R
1-34) bis (R
1-47), wobei jeweils auch die Anknüpfung dieser Gruppen an den Liganden mit eingezeichnet ist:
wobei Lig die Anknüpfung der Alkylgruppe an den Liganden kennzeichnet.
-
Wenn R
1 für eine Aralkylgruppe steht, dann ist diese Aralkylgruppe bevorzugt ausgewählt aus den Strukturen der folgenden Formeln (R
1-56) bis (R
1-69), wobei jeweils auch die Anknüpfung dieser Gruppen an den Liganden eingezeichnet ist:
wobei Lig die Anknüpfung der Aralkylgruppe an den Liganden kennzeichnet und die Phenylgruppen jeweils durch einen oder mehrere Reste R
2 substituiert sein können.
-
Die Alkyl-, Alkoxy- und Aralkylgruppen können, je nach genauer Struktur, auch ein oder mehrere Stereozentren aufweisen. Da es sich bei der Grundstruktur des Komplexes auch um eine chirale Struktur handeln kann, ist die Bildung von Diastereomeren möglich, insbesondere auch, wenn mehrere solcher Alkyl-, Alkoxy- und Aralkylgruppen mit Stereozentren vorliegen. Die erfindungsgemäßen Komplexe umfassen dann sowohl die Mischungen der verschiedenen Diastereomere bzw. die entsprechenden Racemate wie auch die einzelnen isolierten Diastereomere bzw. Enantiomere.
-
Wenn R
1 für ein aromatisches bzw. heteroaromatisches Ringsystem steht, dann weist dieses aromatische bzw. heteroaromatische Ringsystem 5 bis 24 aromatische Ringatome auf. Weiterhin enthält dieses aromatische bzw. heteroaromatische Ringsystem bevorzugt keine Aryl- bzw. Heteroarylgruppen, in denen mehr als zwei aromatische Sechsringe direkt aneinander kondensiert sind. Besonders bevorzugt enthält das aromatische bzw. heteroaromatische Ringsystem überhaupt keine kondensierten Aryl- bzw. Heteroarylgruppen, und ganz besonders bevorzugt enthält es nur Phenylgruppen. Dabei ist das aromatische Ringsystem bevorzugt ausgewählt aus den Strukturen der folgenden Formeln (R
1-70) bis (R
1-84), wobei jeweils auch die Anknüpfung dieser Gruppen an den Liganden eingezeichnet ist:
wobei Lig die Anknüpfung des aromatischen oder heteroaromatischen Ringsystems an den Liganden kennzeichnet und die Phenylgruppen jeweils durch einen oder mehrere Reste R
2 substituiert sein können.
-
Weiterhin ist das heteroaromatische Ringsystem bevorzugt ausgewählt aus den Strukturen der folgenden Formeln (R
1-85) bis (R
1-112), wobei jeweils auch die Anknüpfung dieser Gruppen an den Liganden eingezeichnet ist:
wobei Lig die Anknüpfung des aromatischen oder heteroaromatischen Ringsystems an den Liganden kennzeichnet und die aromatischen und heteroaromatischen Gruppen jeweils durch einen oder mehrere Reste R
2 substituiert sein können.
-
Wenn in der Teilstruktur der Formel (2) außer den Resten R1 noch weitere Reste R gebunden sind, so sind diese Reste R bei jedem Auftreten gleich oder verschieden bevorzugt ausgewählt aus der Gruppe bestehend aus H, D, F, einer geradkettigen Alkylgruppe mit 1 bis 6 C-Atomen oder einer verzweigten oder cyclischen Alkylgruppe mit 3 bis 10 C-Atomen, wobei ein oder mehrere H-Atome durch D oder F ersetzt sein können, oder einem aromatischen oder heteroaromatischen Ringsystem mit 5 bis 24 aromatischen Ringatomen, das jeweils durch einen oder mehrere Reste R2 substituiert sein kann; dabei können zwei benachbarte Reste R oder R mit R1 auch miteinander ein mono- oder polycyclisches, aliphatisches Ringsystem bilden.
-
Wenn in der Teilstruktur der Formel (2) des erfindungsgemäßen Komplexes noch Substituenten R gebunden sind, welche einen +M- oder einen -M-Effekt aufweisen, so sind diese bevorzugt in den folgenden Positionen gebunden:
- Wenn es sich um einen Substituenten R handelt, welcher einen +M-Effekt aufweist, so ist dieser bevorzugt an dem Cyclus, der über den Kohlenstoff an das Metall bindet, in meta-Position zum Metall gebunden.
-
Wenn es sich um einen Substituenten R handelt, welcher einen -M-Effekt aufweist, so ist dieser bevorzugt an dem Cyclus, der über den Kohlenstoff an das Metall bindet, in para-Position zum Metall gebunden.
-
Welche Substituenten einen +M-Effekt und welche Substituenten einen -M-Effekt aufweisen, ist dem Fachmann der organischen Chemie bekannt. Ein Beispiel für einen Substituenten, der einen +M-Effekt aufweist, ist F. Ein Beispiel für einen Substituenten, der einen -M-Effekt aufweist, ist CN.
-
Die bevorzugten Positionen, an denen Substituenten mit +M-Effekt und -M-Effekt gebunden sind, sind im Folgenden schematisch dargestellt:
wobei die Teilliganden enthaltend X ausgewählt sind aus den Strukturen der Formeln (7) bis (38), wie oben definiert.
-
Weiterhin ist es möglich, dass der Substituent R, der in der ortho-Position zur Metallkoordination gebunden ist, eine Gruppe darstellt, die ebenfalls an das Metall M koordiniert bzw. bindet. Bevorzugte koordinierende Gruppen R sind Aryl- bzw. Heteroarylgruppen, beispielsweise Phenyl oder Pyridyl.
-
Hier sind beispielsweise die Teilstrukturen ML der folgenden Formeln (39) bis (44) zugänglich:
wobei die Teilliganden enthaltend X ausgewählt sind aus den Strukturen der Formeln (7) bis (38), wie oben definiert, und wobei X in der zusätzlichen koordinierenden Aryl- bzw. Heteroarylgruppe für CR oder N steht, X
1 gleich oder verschieden bei jedem Auftreten für C oder N steht und W gleich oder verschieden bei jedem Auftreten für S, O oder NR
2 steht.
-
Die Formeln (39) bis (44) zeigen nur exemplarisch, wie der Substituent R zusätzlich an das Metall koordinieren kann. Ganz analog sind ohne weiteres erfinderisches Zutun auch andere an das Metall koordinierende Gruppen R zugänglich, beispielsweise auch Carbene.
-
Wie oben beschrieben, kann auch statt einem der Reste R eine verbrückende Einheit V vorhanden sein, die diesen Liganden L mit einem oder mehreren weiteren Liganden L bzw. L' verknüpft. In einer bevorzugten Ausführungsform der Erfindung ist statt einem der Reste R, insbesondere statt der Reste R, die in ortho- oder meta-Position zum koordinierenden Atom stehen, eine verbrückende Einheit V vorhanden, so dass die Liganden dreizähnigen oder mehrzähnigen oder polypodalen Charakter aufweisen. Es können auch zwei solcher verbrückenden Einheiten V vorhanden sein. Dies führt zur Bildung makrocyclischer Liganden bzw. zur Bildung von Kryptaten.
-
Bevorzugte Strukturen mit mehrzähnigen Liganden bzw. mit polydentaten Liganden sind die Metallkomplexe der folgenden Formeln (45) bis (50),
wobei die Teilliganden enthaltend X ausgewählt sind aus den Strukturen der Formeln (7) bis (38), wie oben definiert, wobei V bevorzugt eine Einfachbindung oder eine verbrückende Einheit darstellt, enthaltend 1 bis 80 Atome aus der dritten, vierten, fünften und/oder sechsten Hauptgruppe (Gruppe 13, 14, 15 oder 16 gemäß IUPAC) oder einen 3- bis 6-gliedrigen Homo- oder Heterocyclus, die die Teilliganden L miteinander oder L mit L' miteinander kovalent verbindet. Dabei kann die verbrückende Einheit V auch unsymmetrisch aufgebaut sein, d. h. die Verknüpfung von V zu L bzw. L' muss nicht identisch sein. Die verbrückende Einheit V kann neutral, einfach, zweifach oder dreifach negativ oder einfach, zweifach oder dreifach positiv geladen sein. Bevorzugt ist V neutral oder einfach negativ oder einfach positiv geladen, besonders bevorzugt neutral. Dabei wird die Ladung von V bevorzugt so gewählt, dass insgesamt ein neutraler Komplex entsteht. Dabei gelten für die Liganden die oben für die Teilstruktur MLn genannten Bevorzugungen und n ist bevorzugt mindestens 2.
-
Es ist auch möglich, dass zwei Liganden L
1 außer über eine Einfachbindung auch zusätzlich über eine weitere bivalente Brücke miteinander verknüpft sind. Dadurch entstehen Strukturen der folgenden Formel (45a):
wobei die Teilliganden enthaltend X ausgewählt sind aus den Strukturen der Formeln (7) bis (38), wie oben definiert und V
1 für CR
2, NR, O oder S steht.
-
Die genaue Struktur und chemische Zusammensetzung der Gruppe V hat keinen wesentlichen Einfluss auf die elektronischen Eigenschaften des Komplexes, da die Aufgabe dieser Gruppe im Wesentlichen darin liegt, durch die Verbrückung von L miteinander bzw. mit L' die chemische und thermische Stabilität der Komplexe zu erhöhen.
-
Wenn V eine trivalente Gruppe ist, also drei Liganden L miteinander bzw. zwei Liganden L mit L' oder einen Liganden L mit zwei Liganden L' verbrückt, ist V bevorzugt gleich oder verschieden bei jedem Auftreten gewählt aus der Gruppe bestehend aus B, B(R
2)
-, B(C(R
2)
2)
3, (R
2)B(C(R
2)
2)
3 -, B(O)
3, (R
2)B(O)
3 -, B(C(R
2)
2C(R
2)
2)
3, (R
2)B(C(R
2)
2C(R
2)
2)
3 -, B(C(R
2)
2O)
3, (R
2)B(C(R
2)
2O)
3 -, B(OC(R
2)
2)
3, (R
2)B(OC(R
2)
2)
3 -, C(R
2), CO
-, CN(R
2)
2, (R
2)C(C(R
2)
2)
3, (R
2)C(O)
3, (R
2)C(C(R
2)
2C(R
2)
2)
3, (R
2)C(C(R
2)
2O)
3, (R
2)C(OC(R
2)
2)
3, (R
2)C(Si(R
2)
2)
3, (R
2)C(Si(R
2)
2C(R
2)
2)
3, (R
2)C(C(R
2)
2Si(R
2)
2)
3, (R
2)C(Si(R
2)
2Si(R
2)
2)
3, Si(R
2), (R
2)Si(C(R
2)
2)
3, (R
2)Si(O)
3, (R
2)Si(C(R
2)
2C(R
2)
2)
3, (R
2)Si(OC(R
2)
2)
3, (R
2)Si(C(R
2)
2O)
3, (R
2)Si(Si(R
2)
2)
3, (R
2)Si(Si(R
2)
2C(R
2)
2)
3, (R
2)Si(C(R
2)
2Si(R
2)
2)
3, (R
2)Si(Si(R
2)
2Si(R
2)
2)
3, N, NO, N(R
2)
+, N(C(R
2)
2)
3, (R
2)N(C(R
2)
2)
3 +, N(C=O)
3, N(C(R
2)
2C(R
2)
2)
3, (R
2)N(C(R
2)
2C(R
2)
2)
+, P, P(R
2)
+, PO, PS, PSe, PTe, P(O)
3, PO(O)
3, P(OC(R
2)
2)
3, PO(OC(R
2)
2)
3, P(C(R
2)
2)
3, P(R
2)(C(R
2)
2)
3 +, PO(C(R
2)
2)
3, P(C(R
2)2C(R
2)
2)
3, P(R
2) (C(R
2)
2C(R
2)
2)
3 +, PO(C(R
2)
2C(R
2)
2)
3, S
+, S(C(R
2)
2)
3 +, S(C(R
2)
2C(R
2)
2)
3 +, oder eine Einheit gemäß Formel (51), (52), (53) oder (54),
wobei die gestrichelten Bindungen jeweils die Bindung zu den Teilliganden L bzw. L' andeuten und Z gleich oder verschieden bei jedem Auftreten ausgewählt ist aus der Gruppe bestehend aus einer Einfachbindung, O, S, S(=O), S(=O)
2, NR
2, PR
2, P(=O)R
2, P(=NR
2), C(R
2)
2, C(=O), C(=NR
2), C(=C(R
2)
2), Si(R
2)
2 oder BR
2. Die weiteren verwendeten Symbole haben die oben genannten Bedeutungen.
-
Wenn V für eine Gruppe CR2 steht, so können die beiden Reste R auch miteinander verknüpft sein, so dass auch Strukturen wie zum Beispiele 9,9-Fluoren geeignete Gruppen V sind.
-
Wenn V eine bivalente Gruppe ist, also zwei Liganden L miteinander bzw. einen Liganden L mit L' verbrückt, ist V bevorzugt gleich oder verschieden bei jedem Auftreten gewählt aus der Gruppe bestehend aus BR
2, B(R
2)
2 -, C(R
2)
2, C(=O), Si(R
2)
2, NR
2, PR
2, P(R
2)
2 +, P(=O)(R
2), P(=S)(R
2), AsR
2, As(=O)(R
2), As(=S)(R
2), O, S, Se, oder eine Einheit gemäß Formel (55) bis (64),
wobei die gestrichelten Bindungen jeweils die Bindung zu den Teilliganden L bzw. L' andeuten, Y bei jedem Auftreten gleich oder verschieden für C(R
2)
2, N(R
2), O oder S steht und die weiteren verwendeten Symbole jeweils die oben aufgeführten Bedeutungen haben.
-
Im Folgenden werden bevorzugte Liganden L' beschrieben, wie sie in Formel (1) vorkommen. Entsprechend können auch die Ligandengruppen L' gewählt sein, wenn diese über eine verbrückende Einheit V an L gebunden sind, wie in Formeln (45) bis (50) angedeutet.
-
Die Liganden L' sind bevorzugt neutrale, monoanionische, dianionische oder trianionische Liganden, besonders bevorzugt neutrale oder monoanionische Liganden. Sie können monodentat, bidentat, tridentat oder tetradentat sein und sind bevorzugt bidentat, weisen also bevorzugt zwei Koordinationsstellen auf. Wie oben beschrieben, können die Liganden L' auch über eine verbrückende Gruppe V an L gebunden sein.
-
Bevorzugte neutrale, monodentate Liganden L' sind ausgewählt aus der Gruppe bestehend aus Kohlenmonoxid, Stickstoffmonoxid, Alkylcyaniden, wie z. B. Acetonitril, Arylcyaniden, wie z. B. Benzonitril, Alkylisocyaniden, wie z. B. Methylisonitril, Arylisocyaniden, wie z. B. Benzoisonitril, Aminen, wie z. B. Trimethylamin, Triethylamin, Morpholin, Phosphinen, insbesondere Halogenphosphine, Trialkylphosphine, Triarylphosphine oder Alkylarylphosphine, wie z. B. Trifluorphosphin, Trimethylphosphin, Tricyclohexylphosphin, Tri-tert-butylphosphin, Triphenylphosphin, Tris(pentafluorphenyl)phosphin, Dimethylphenylphosphin, Methyldiphenylphosphin, Bis(tert-butyl)phenylphosphin, Phosphiten, wie z. B. Trimethylphosphit, Triethylphosphit, Arsinen, wie z. B. Trifluorarsin, Trimethylarsin, Tricyclohexylarsin, Tri-tert-butylarsin, Triphenylarsin, Tris(pentafluorphenyl)arsin, Stibinen, wie z. B. Trifluorstibin, Trimethylstibin, Tricyclohexylstibin, Tri-tert-butylstibin, Triphenylstibin, Tris(pentafluorphenyl)stibin, stickstoffhaltigen Heterocyclen, wie z. B. Pyridin, Pyridazin, Pyrazin, Pyrimidin, Triazin, und Carbenen, insbesondere Arduengo-Carbenen.
-
Bevorzugte monoanionische, monodentate Liganden L' sind ausgewählt aus Hydrid, Deuterid, den Halogeniden F-, Cl-, Br- und I-, Alkylacetyliden, wie z. B. Methyl-C≡C-, tert-Butyl-C≡C-, Arylacetyliden, wie z. B. Phenyl-C≡C-, Cyanid, Cyanat, Isocyanat, Thiocyanat, Isothiocyanat, aliphatischen oder aromatischen Alkoholaten, wie z. B. Methanolat, Ethanolat, Propanolat, iso-Propanolat, tert-Butylat, Phenolat, aliphatischen oder aromatischen Thioalkoholaten, wie z. B. Methanthiolat, Ethanthiolat, Propanthiolat, iso-Propanthiolat, tert-Thiobutylat, Thiophenolat, Amiden, wie z. B. Dimethylamid, Diethylamid, Di-iso-propylamid, Morpholid, Carboxylaten, wie z. B. Acetat, Trifluoracetat, Propionat, Benzoat, Arylgruppen, wie z. B. Phenyl, Naphthyl, und anionischen, stickstoffhaltigen Heterocyclen, wie Pyrrolid, Imidazolid, Pyrazolid. Dabei sind die Alkylgruppen in diesen Gruppen bevorzugt C1-C20-Alkylgruppen, besonders bevorzugt C1-C10-Alkylgruppen, ganz besonders bevorzugt C1-C4-Alkylgruppen. Unter einer Arylgruppe werden auch Heteroarylgruppen verstanden. Diese Gruppen sind wie oben definiert.
-
Bevorzugte di- bzw. trianionische Liganden sind O2-, S2-, Carbide, welche zu einer Koordination der Form R-C≡M führen, und Nitrene, welche zu einer Koordination der Form R-N=M führen, wobei R allgemein für einen Substituenten steht, oder N3-.
-
Bevorzugte neutrale oder mono- oder dianionische, bidentate oder höherdentate Liganden L' sind ausgewählt aus Diaminen, wie z. B. Ethylendiamin, N,N,N',N'-Tetramethylethylendiamin, Propylendiamin, N,N,N',N'-Tetramethylpropylendiamin, cis- oder trans-Diaminocyclohexan, cis- oder trans-N,N,N',N'-Tetramethyldiaminocyclohexan, Iminen, wie z. B. 2-[1-(Phenylimino)ethyl]pyridin, 2-[1-(2-Methylphenylimino)ethyl]pyridin, 2-[1-(2,6-Di-iso-propylphenylimino)ethyl]pyridin, 2-[1-(Methylimino)ethyl]pyridin, 2-[1-(ethylimino)ethyl]pyridin, 2-[1-(iso-Propylimino)ethyl]pyridin, 2-[1-(Tert-Butylimino)ethyl]pyridin, Diiminen, wie z. B. 1,2-Bis(methylimino)ethan, 1,2-Bis(ethylimino)ethan, 1,2-Bis(iso-propylimino)ethan, 1,2-Bis(tert-butylimino)ethan, 2,3-Bis(methylimino)butan, 2,3-Bis(ethylimino)butan, 2,3-Bis(iso-propylimino)butan, 2,3-Bis(tert-butylimino)butan, 1,2-Bis(phenylimino)ethan, 1,2-Bis(2-methylphenylimino)ethan, 1,2-Bis(2,6-di-iso-propylphenylimino)ethan, 1,2-Bis(2,6-di-tert-butylphenylimino)ethan, 2,3-Bis(phenylimino)butan, 2,3-Bis(2-methylphenylimino)butan, 2,3-Bis(2,6-di-iso-propylphenylimino)butan, 2,3-Bis(2,6-di-tert-butylphenylimino)butan, Heterocyclen enthaltend zwei Stickstoffatome, wie z. B. 2,2'-Bipyridin, o-Phenanthrolin, Diphosphinen, wie z. B. Bis(diphenylphosphino)methan, Bis(diphenylphosphino)ethan, Bis(diphenylphosphino)propan, Bis(diphenylphosphino)butan, Bis(dimethylphosphino)methan, Bis(dimethylphosphino)ethan, Bis(dimethylphosphino)propan, Bis(diethylphosphino)methan, Bis(diethylphosphino)ethan, Bis(diethylphosphino)propan, Bis(di-tert-butylphosphino)methan, Bis(di-tert-butylphosphino)ethan, Bis(tert-butylphosphino)propan, 1,3-Diketonaten abgeleitet von 1,3-Diketonen, wie z. B. Acetylaceton, Benzoylaceton, 1,5-Diphenylacetylaceton, Dibenzoylmethan, Bis(1,1,1-trifluoracetyl)methan, 3-Ketonaten abgeleitet von 3-Ketoestern, wie z. B. Acetessigsäureethylester, Carboxylate, abgeleitet von Aminocarbonsäuren, wie z. B. Pyridin-2-carbonsäure, Chinolin-2-carbonsäure, Glycin, N,N-Dimethylglycin, Alanin, N,N-Dimethylaminoalanin, Salicyliminaten abgeleitet von Salicyliminen, wie z. B. Methylsalicylimin, Ethylsalicylimin, Phenylsalicylimin, Dialkoholaten abgeleitet von Dialkoholen, wie z. B. Ethylenglykol, 1,3-Propylenglykol und Dithiolaten abgeleitet von Dithiolen, wie z. B. 1,2-Ethylendithiol, 1,3-Propylendithiol.
-
Bevorzugte tridentate Liganden sind Borate stickstoffhaltiger Heterocyclen, wie z. B. Tetrakis(1-imidazolyl)borat und Tetrakis(1-pyrazolyl)borat.
-
Bevorzugt sind weiterhin bidentate monoanionische, neutrale oder dianionische Liganden L', insbesondere monoanionische Liganden, welche mit dem Metall einen cyclometallierten Fünfring oder Sechsring mit mindestens einer Metall-Kohlenstoff-Bindung aufweisen, insbesondere einen cyclometallierten Fünfring. Dies sind insbesondere Liganden, wie sie allgemein im Gebiet der phosphoreszierenden Metallkomplexe für organische Elektrolumineszenzvorrichtungen verwendet werden, also Liganden vom Typ Phenylpyridin, Naphthylpyridin, Phenylchinolin, Phenylisochinolin, etc., welche jeweils durch einen oder mehrere Reste R substituiert sein können. Dem Fachmann auf dem Gebiet der phosphoreszierenden Elektrolumineszenzvorrichtungen ist eine Vielzahl derartiger Liganden bekannt, und er kann ohne erfinderisches Zutun weitere derartige Liganden als Ligand L' für Verbindungen gemäß Formel (1) auswählen. Generell eignet sich dafür besonders die Kombination aus zwei Gruppen, wie sie durch die folgenden Formeln (65) bis (92) dargestellt sind, wobei eine Gruppe bevorzugt über ein neutrales Stickstoffatom oder ein Carbenkohlenstoffatom bindet und die andere Gruppe bevorzugt über ein negativ geladenes Kohlenstoffatom oder ein negativ geladenes Stickstoffatom bindet. Der Ligand L' kann dann aus den Gruppen der Formeln (65) bis (92) gebildet werden, indem diese Gruppen jeweils an der durch # gekennzeichneten Position aneinander binden. Die Position, an der die Gruppen an das Metall koordinieren, sind durch * gekennzeichnet. Diese Gruppen können auch über eine oder zwei verbrückende Einheiten V an den Liganden L gebunden sein.
-
Dabei steht X bei jedem Auftreten gleich oder verschieden für CR oder N, und R hat dieselbe Bedeutung wie oben beschrieben. Bevorzugt stehen maximal drei Symbole X in jeder Gruppe für N, besonders bevorzugt stehen maximal zwei Symbole X in jeder Gruppe für N, ganz besonders bevorzugt steht maximal ein Symbol X in jeder Gruppe für N. Insbesondere bevorzugt stehen alle Symbole X für CR.
-
Weiterhin können die Formeln (76) bis (80) statt des Schwefels auch Sauerstoff enthalten.
-
Ebenfalls bevorzugte Liganden L' sind η5-Cyclopentadienyl, η5-Pentamethylcyclopentadienyl, η6-Benzol oder η7-Cycloheptatrienyl, welche jeweils durch einen oder mehrere Reste R substituiert sein können.
-
Ebenfalls bevorzugte Liganden L' sind 1,3,5-cis,cis-Cyclohexanderivate, insbesondere der Formel (93), 1,1,1-Tri(methylen)methanderivate, insbesondere der Formel (94) und 1,1,1-trisubstituierte Methane, insbesondere der Formel (95) und (96),
wobei in den Formeln jeweils die Koordination an das Metall M dargestellt ist, R die oben genannte Bedeutung hat und A, gleich oder verschieden bei jedem Auftreten, für O
-, S
-, COO
-, PR
2 oder NR
2 steht.
-
Bevorzugte Reste R in den oben aufgeführten Strukturen sind bei jedem Auftreten gleich oder verschieden ausgewählt aus der Gruppe bestehend aus H, D, F, CN, einer geradkettigen Alkylgruppe mit 1 bis 5 C-Atomen, insbesondere Methyl, oder einer verzweigten oder cyclischen Alkylgruppe mit 3 bis 5 C-Atomen, insbesondere iso-Propyl oder tert-Butyl, wobei ein oder mehrere H-Atome durch D oder F ersetzt sein können, oder einem aromatischen oder heteroaromatischen Ringsystem mit 5 bis 12 aromatischen Ringatomen, das jeweils durch einen oder mehrere Reste R2 substituiert sein kann; dabei können zwei oder mehrere Reste R auch miteinander ein mono- oder polycyclisches, aliphatisches, aromatisches und/oder benzoannelliertes Ringsystem bilden.
-
Die erfindungsgemäßen Komplexe können facial bzw. pseudofacial sein, oder sie können meridional bzw. pseudomeridional sein.
-
Die oben genannten bevorzugten Ausführungsformen sind beliebig miteinander kombinierbar. In einer besonders bevorzugten Ausführungsform der Erfindung gelten die oben genannten bevorzugten Ausführungsformen gleichzeitig.
-
Die erfindungsgemäßen Metallkomplexe sind prinzipiell durch verschiedene Verfahren darstellbar. Es haben sich jedoch die im Folgenden beschriebenen Verfahren als besonders geeignet herausgestellt.
-
Daher ist ein weiterer Gegenstand der vorliegenden Erfindung ein Verfahren zur Herstellung der Metallkomplex-Verbindungen gemäß Formel (1) durch Umsetzung der entsprechenden freien Liganden mit Metallalkoholaten der Formel (97), mit Metallketoketonaten der Formel (98), mit Metallhalogeniden der Formel (99) oder mit dimeren Metallkomplexen der Formel (100),
wobei die Symbole M, m, n und R die oben angegebenen Bedeutungen haben und Hal = F, Cl, Br oder I ist.
-
Es können ebenfalls Metallverbindungen, insbesondere Iridiumverbindungen, die sowohl Alkoholat- und/oder Halogenid- und/oder Hydroxy- wie auch Ketoketonatreste tragen, verwendet werden. Diese Verbindungen können auch geladen sein. Entsprechende Iridiumverbindungen, die als Edukte besonders geeignet sind, sind in
WO 2004/085449 offenbart. Besonders geeignet sind [IrCl
2(acac)
2]
-, beispielsweise Na[IrCl
2(acac)
2], Metallkomplexe mit Acetylacetonat-Derivaten als Ligand, beispielsweise Ir(acac)
3 oder Tris(2,2,6,6-Tetramethylheptan-3,5-dionato)iridium, und IrCl
3·xH
2O, wobei x üblicherweise für eine Zahl zwischen 2 und 4 steht.
-
Geeignete Platin-Edukte sind beispielsweise PtCl2, K2[PtCl4], PtCl2(DMSO)2, Pt(Me)2(DMSO)2 oder PtCl2(Benzonitril)2.
-
Die Synthese der Komplexe wird bevorzugt durchgeführt wie in
WO 2002/060910 ,
WO 2004/085449 und
WO 2007/065523 beschrieben. Heteroleptische Komplexe können beispielsweise auch gemäß
WO 2005/042548 synthetisiert werden. Dabei kann die Synthese beispielsweise auch thermisch, photochemisch und/oder durch Mikrowellenstrahlung aktiviert werden. In einer bevorzugten Ausführungsform der Erfindung wird die Reaktion ohne die Verwendung eines zusätzlichen Lösemittels in der Schmelze durchgeführt. Dabei bedeutet „Schmelze“, dass der Ligand geschmolzen vorliegt und die Metall-Vorstufe in dieser Schmelze gelöst oder suspendiert ist.
-
Durch diese Verfahren, gegebenenfalls gefolgt von Aufreinigung, wie z. B. Umkristallisation oder Sublimation, lassen sich die erfindungsgemäßen Verbindungen gemäß Formel (1) in hoher Reinheit, bevorzugt mehr als 99 % (bestimmt mittels 1H-NMR und/oder HPLC) erhalten.
-
Die erfindungsgemäßen Verbindungen können auch durch geeignete Substitution, beispielsweise durch längere Alkylgruppen (ca. 4 bis 20 C-Atome), insbesondere verzweigte Alkylgruppen, oder gegebenenfalls substituierte Arylgruppen, beispielsweise Xylyl-, Mesityl- oder verzweigte Terphenyl- oder Quaterphenylgruppen, löslich gemacht werden. Solche Verbindungen sind dann in gängigen organischen Lösemitteln, wie beispielsweise Toluol oder Xylol bei Raumtemperatur in ausreichender Konzentration löslich, um die Komplexe aus Lösung verarbeiten zu können. Diese löslichen Verbindungen eignen sich besonders gut für die Verarbeitung aus Lösung, beispielsweise durch Druckverfahren.
-
Die oben beschriebenen Komplexe gemäß Formel (1) bzw. die oben aufgeführten bevorzugten Ausführungsformen können in der elektronischen Vorrichtung als aktive Komponente verwendet werden. Unter einer elektronischen Vorrichtung wird eine Vorrichtung verstanden, welche Anode, Kathode und mindestens eine Schicht enthält, wobei diese Schicht mindestens eine organische bzw. metallorganische Verbindung enthält. Die erfindungsgemäße elektronische Vorrichtung enthält also Anode, Kathode und mindestens eine Schicht, welche mindestens eine Verbindung der oben aufgeführten Formel (1) enthält. Dabei sind bevorzugte elektronische Vorrichtungen ausgewählt aus der Gruppe bestehend aus organischen Elektrolumineszenzvorrichtungen (OLEDs, PLEDs), organischen integrierten Schaltungen (O-ICs), organischen Feld-Effekt-Transistoren (O-FETs), organischen Dünnfilmtransistoren (O-TFTs), organischen lichtemittierenden Transistoren (O-LETs), organischen Solarzellen (O-SCs), organischen optischen Detektoren, organischen Photorezeptoren, organischen Feld-Quench-Devices (O-FQDs), lichtemittierenden elektrochemischen Zellen (LECs) oder organischen Laserdioden (O-Laser), enthaltend in mindestens einer Schicht mindestens eine Verbindung gemäß der oben aufgeführten Formel (1). Besonders bevorzugt sind organische Elektrolumineszenzvorrichtungen. Aktive Komponenten sind generell die organischen oder anorganischen Materialien, welche zwischen Anode und Kathode eingebracht sind, beispielsweise Ladungsinjektions-, Ladungstransport- oder Ladungsblockiermaterialien, insbesondere aber Emissionsmaterialien und Matrixmaterialien. Die erfindungsgemäßen Verbindungen zeigen besonders gute Eigenschaften als Emissionsmaterial in organischen Elektrolumineszenzvorrichtungen. Eine bevorzugte Ausführungsform der Erfindung sind daher organische Elektrolumineszenzvorrichtungen.
-
Die organische Elektrolumineszenzvorrichtung enthält Kathode, Anode und mindestens eine emittierende Schicht. Außer diesen Schichten kann sie noch weitere Schichten enthalten, beispielsweise jeweils eine oder mehrere Lochinjektionsschichten, Lochtransportschichten, Lochblockierschichten, Elektronentransportschichten, Elektroneninjektionsschichten, Exzitonenblockierschichten, Elektronenblockierschichten, Ladungserzeugungsschichten und/oder organische oder anorganische p/n-Übergänge. Ebenso können zwischen zwei emittierende Schichten Interlayers eingebracht sein, welche beispielsweise eine Excitonen-blockierende Funktion aufweisen und/oder die Ladungsbalance in der Elektrolumineszenzvorrichtung steuern. Es sei aber darauf hingewiesen, dass nicht notwendigerweise jede dieser Schichten vorhanden sein muss.
-
Dabei kann die organische Elektrolumineszenzvorrichtung eine emittierende Schicht enthalten, oder sie kann mehrere emittierende Schichten enthalten. Wenn mehrere Emissionsschichten vorhanden sind, weisen diese bevorzugt insgesamt mehrere Emissionsmaxima zwischen 380 nm und 750 nm auf, so dass insgesamt weiße Emission resultiert, d. h. in den emittierenden Schichten werden verschiedene emittierende Verbindungen verwendet, die fluoreszieren oder phosphoreszieren können. Insbesondere bevorzugt sind Dreischichtsysteme, wobei die drei Schichten blaue, grüne und orange oder rote Emission zeigen (für den prinzipiellen Aufbau siehe z. B.
WO 2005/011013 ) bzw. Systeme, welche mehr als drei emittierende Schichten aufweisen. Es kann sich auch um ein Hybrid-System handeln, wobei eine oder mehrere Schichten fluoreszieren und eine oder mehrere andere Schichten phosphoreszieren.
-
In einer bevorzugten Ausführungsform der Erfindung enthält die organische Elektrolumineszenzvorrichtung die Verbindung gemäß Formel (1) bzw. die oben aufgeführten bevorzugten Ausführungsformen als emittierende Verbindung in einer oder mehreren emittierenden Schichten.
-
Wenn die Verbindung gemäß Formel (1) als emittierende Verbindung in einer emittierenden Schicht eingesetzt wird, wird sie bevorzugt in Kombination mit einem oder mehreren Matrixmaterialien eingesetzt. Die Mischung aus der Verbindung gemäß Formel (1) und dem Matrixmaterial enthält zwischen 0.1 und 99 Vol.-%, vorzugsweise zwischen 1 und 90 Vol.-%, besonders bevorzugt zwischen 3 und 40 Vol.-%, insbesondere zwischen 5 und 15 Vol.-% der Verbindung gemäß Formel (1) bezogen auf die Gesamtmischung aus Emitter und Matrixmaterial. Entsprechend enthält die Mischung zwischen 99.9 und 1 Vol.-%, vorzugsweise zwischen 99 und 10 Vol.-%, besonders bevorzugt zwischen 97 und 60 Vol.-%, insbesondere zwischen 95 und 85 Vol.-% des Matrixmaterials bezogen auf die Gesamtmischung aus Emitter und Matrixmaterial.
-
Als Matrixmaterial können generell alle Materialien eingesetzt werden, die gemäß dem Stand der Technik hierfür bekannt sind. Bevorzugt ist das Triplett-Niveau des Matrixmaterials höher als das Triplett-Niveau des Emitters.
-
Geeignete Matrixmaterialien für die erfindungsgemäßen Verbindungen sind Ketone, Phosphinoxide, Sulfoxide und Sulfone, z. B. gemäß
WO 2004/013080 ,
WO 2004/093207 ,
WO 2006/005627 oder
WO 2010/006680 , Triarylamine, Carbazolderivate, z. B. CBP (N,N-Biscarbazolylbiphenyl), m-CBP oder die in
WO 2005/039246 ,
US 2005/0069729 ,
JP 2004/288381 ,
EP 1205527 ,
WO 2008/086851 oder
US 2009/0134784 offenbarten Carbazolderivate, Indolocarbazolderivate, z. B. gemäß
WO 2007/063754 oder
WO 2008/056746 , Indenocarbazolderivate, z. B. gemäß
WO 2010/136109 oder
WO 2011/000455 , Azacarbazole, z. B. gemäß
EP 1617710 ,
EP 1617711 ,
EP 1731584 ,
JP 2005/347160 , bipolare Matrixmaterialien, z. B. gemäß
WO 2007/137725 , Silane, z. B. gemäß
WO 2005/111172 , Azaborole oder Boronester, z. B. gemäß
WO 2006/117052 , Diazasilolderivate, z. B. gemäß
WO 2010/054729 , Diazaphospholderivate, z. B. gemäß
WO 2010/054730 , Triazinderivate, z. B. gemäß
WO 2010/015306 ,
WO 2007/063754 oder
WO 2008/056746 , Zinkkomplexe, z. B. gemäß
EP 652273 oder
WO 2009/062578 , Dibenzofuranderivate, z. B. gemäß
WO 2009/148015 , oder verbrückte Carbazolderivate, z. B. gemäß
US 2009/0136779 ,
WO 2010/050778 oder den nicht offen gelegten Anmeldungen
DE 102009048791.3 und
DE 102010005697.9 .
-
Es kann auch bevorzugt sein, mehrere verschiedene Matrixmaterialien als Mischung einzusetzen, insbesondere mindestens ein elektronenleitendes Matrixmaterial und mindestens ein lochleitendes Matrixmaterial. Eine bevorzugte Kombination ist beispielsweise die Verwendung eines aromatischen Ketons, eines Triazin-Derivats oder eines Phosphinoxid-Derivats mit einem Triarylamin-Derivat oder einem Carbazol-Derivat als gemischte Matrix für den erfindungsgemäßen Metallkomplex. Ebenso bevorzugt ist die Verwendung einer Mischung aus einem ladungstransportierenden Matrixmaterial und einem elektrisch inerten Matrixmaterial, welches nicht bzw. nicht in wesentlichem Maße am Ladungstransport beteiligt ist, wie z. B. in
WO 2010/108579 beschrieben.
-
Weiterhin bevorzugt ist es, eine Mischung aus zwei oder mehr Triplett-Emittern zusammen mit einer Matrix einzusetzen. Dabei dient der Triplett-Emitter mit dem kürzerwelligen Emissionsspektrum als Co-Matrix für den Triplett-Emitter mit dem längerwelligen Emissionsspektrum. So können beispielsweise die erfindungsgemäßen Komplexe gemäß Formel (1) als Co-Matrix für längerwellig emittierende Triplettemitter, beispielsweise für grün oder rot emittierende Triplettemitter, eingesetzt werden.
-
Die erfindungsgemäßen Verbindungen lassen sich auch in anderen Funktionen in der elektronischen Vorrichtung einsetzen, beispielsweise als Lochtransportmaterial in einer Lochinjektions- oder -transportschicht, als Ladungserzeugungsmaterial oder als Elektronenblockiermaterial. Ebenso lassen sich die erfindungsgemäßen Komplexe als Matrixmaterial für andere phosphoreszierende Metallkomplexe in einer emittierenden Schicht einsetzen.
-
Als Kathode sind Metalle mit geringer Austrittsarbeit, Metalllegierungen oder mehrlagige Strukturen aus verschiedenen Metallen bevorzugt, wie beispielsweise Erdalkalimetalle, Alkalimetalle, Hauptgruppenmetalle oder Lanthanoide (z. B. Ca, Ba, Mg, Al, In, Mg, Yb, Sm, etc.). Weiterhin eignen sich Legierungen aus einem Alkali- oder Erdalkalimetall und Silber, beispielsweise eine Legierung aus Magnesium und Silber. Bei mehrlagigen Strukturen können auch zusätzlich zu den genannten Metallen weitere Metalle verwendet werden, die eine relativ hohe Austrittsarbeit aufweisen, wie z. B. Ag, wobei dann in der Regel Kombinationen der Metalle, wie beispielsweise Mg/Ag, Ca/Ag oder Ba/Ag verwendet werden. Es kann auch bevorzugt sein, zwischen einer metallischen Kathode und dem organischen Halbleiter eine dünne Zwischenschicht eines Materials mit einer hohen Dielektrizitätskonstante einzubringen. Hierfür kommen beispielsweise Alkalimetall- oder Erdalkalimetallfluoride, aber auch die entsprechenden Oxide oder Carbonate in Frage (z. B. LiF, Li2O, BaF2, MgO, NaF, CsF, Cs2CO3, etc.). Ebenso kommen hierfür organische Alkalimetallkomplexe in Frage, z. B. Liq (Lithiumchinolinat). Die Schichtdicke dieser Schicht beträgt bevorzugt zwischen 0.5 und 5 nm.
-
Als Anode sind Materialien mit hoher Austrittsarbeit bevorzugt. Bevorzugt weist die Anode eine Austrittsarbeit größer 4.5 eV vs. Vakuum auf. Hierfür sind einerseits Metalle mit hohem Redoxpotential geeignet, wie beispielsweise Ag, Pt oder Au. Es können andererseits auch Metall/Metalloxid-Elektroden (z. B. Al/Ni/NiOx, Al/PtOx) bevorzugt sein. Für einige Anwendungen muss mindestens eine der Elektroden transparent oder teiltransparent sein, um entweder die Bestrahlung des organischen Materials (O-SC) oder die Auskopplung von Licht (OLED/PLED, O-LASER) zu ermöglichen. Bevorzugte Anodenmaterialien sind hier leitfähige gemischte Metalloxide. Besonders bevorzugt sind Indium-Zinn-Oxid (ITO) oder Indium-Zink-Oxid (IZO). Bevorzugt sind weiterhin leitfähige, dotierte organische Materialien, insbesondere leitfähige dotierte Polymere, z. B. PEDOT, PANI oder Derivate dieser Polymere.
-
In den weiteren Schichten können generell alle Materialien verwendet werden, wie sie gemäß dem Stand der Technik für die Schichten verwendet werden, und der Fachmann kann ohne erfinderisches Zutun jedes dieser Materialien in einer elektronischen Vorrichtung mit den erfindungsgemäßen Materialien kombinieren.
-
Die Vorrichtung wird entsprechend (je nach Anwendung) strukturiert, kontaktiert und schließlich hermetisch versiegelt, da sich die Lebensdauer derartiger Vorrichtungen bei Anwesenheit von Wasser und/oder Luft drastisch verkürzt.
-
Weiterhin bevorzugt ist eine organische Elektrolumineszenzvorrichtung, dadurch gekennzeichnet, dass eine oder mehrere Schichten mit einem Sublimationsverfahren beschichtet werden. Dabei werden die Materialien in Vakuum-Sublimationsanlagen bei einem Anfangsdruck von üblicherweise kleiner 10-5 mbar, bevorzugt kleiner 10-6 mbar aufgedampft. Es ist auch möglich, dass der Anfangsdruck noch geringer oder noch höher ist, beispielsweise kleiner 10-7 mbar.
-
Bevorzugt ist ebenfalls eine organische Elektrolumineszenzvorrichtung, dadurch gekennzeichnet, dass eine oder mehrere Schichten mit dem OVPD (Organic Vapour Phase Deposition) Verfahren oder mit Hilfe einer Trägergassublimation beschichtet werden. Dabei werden die Materialien bei einem Druck zwischen 10-5 mbar und 1 bar aufgebracht. Ein Spezialfall dieses Verfahrens ist das OVJP (Organic Vapour Jet Printing) Verfahren, bei dem die Materialien direkt durch eine Düse aufgebracht und so strukturiert werden (z. B. M. S. Arnold et al., Appl. Phys. Lett. 2008, 92, 053301).
-
Weiterhin bevorzugt ist eine organische Elektrolumineszenzvorrichtung, dadurch gekennzeichnet, dass eine oder mehrere Schichten aus Lösung, wie z. B. durch Spincoating, oder mit einem beliebigen Druckverfahren, wie z. B. Siebdruck, Flexodruck, Offsetdruck oder Nozzle-Printing, besonders bevorzugt aber LITI (Light Induced Thermal Imaging, Thermotransferdruck) oder Ink-Jet Druck (Tintenstrahldruck), hergestellt werden. Hierfür sind lösliche Verbindungen nötig, welche beispielsweise durch geeignete Substitution erhalten werden.
-
Die organische Elektrolumineszenzvorrichtung kann auch als Hybrid-system hergestellt werden, indem eine oder mehrere Schichten aus Lösung aufgebracht werden und eine oder mehrere andere Schichten aufgedampft werden. So ist es beispielsweise möglich, eine emittierende Schicht enthaltend eine Verbindung gemäß Formel (1) und ein Matrixmaterial aus Lösung aufzubringen und darauf eine Lochblockierschicht und/oder eine Elektronentransportschicht im Vakuum aufzudampfen.
-
Diese Verfahren sind dem Fachmann generell bekannt und können von ihm ohne Probleme auf organische Elektrolumineszenzvorrichtungen enthaltend Verbindungen gemäß Formel (1) bzw. die oben aufgeführten bevorzugten Ausführungsformen angewandt werden.
-
Die erfindungsgemäßen elektronischen Vorrichtungen, insbesondere organische Elektrolumineszenzvorrichtungen, zeichnen sich durch folgende überraschende Vorteile gegenüber dem Stand der Technik aus:
- 1. Organische Elektrolumineszenzvorrichtungen enthaltend Verbindungen gemäß Formel (1) als emittierende Materialien weisen eine sehr gute Lebensdauer auf.
- 2. Organische Elektrolumineszenzvorrichtungen enthaltend Verbindungen gemäß Formel (1) als emittierende Materialien weisen eine hervorragende Effizienz auf.
- 3. Mit den erfindungsgemäßen Metallkomplexen sind organische Elektrolumineszenzvorrichtungen zugänglich, welche im blauen Farbbereich phosphoreszieren. Insbesondere blaue Phosphoreszenz ist gemäß dem Stand der Technik nur sehr schwierig mit guten Effizienzen und Lebensdauern zu verwirklichen.
- 4. Die erfindungsgemäßen Metallkomplexe sind synthetisch gut und in hoher Ausbeute zugänglich.
-
Diese oben genannten Vorteile gehen nicht mit einer Verschlechterung der weiteren elektronischen Eigenschaften einher.
-
Die Erfindung wird durch die nachfolgenden Beispiele näher erläutert, ohne sie dadurch einschränken zu wollen. Der Fachmann kann aus den Schilderungen ohne erfinderisches Zutun weitere erfindungsgemäße elektronische Vorrichtungen herstellen und somit die Erfindung im gesamten beanspruchten Bereich ausführen.
-
Beispiele:
-
Die nachfolgenden Synthesen werden, sofern nicht anders angegeben, unter einer Schutzgasatmosphäre in getrockneten Lösungsmitteln durchgeführt. Die Metallkomplexe werden zusätzlich unter Ausschluss von Licht gehandhabt. Die Lösungsmittel und Reagenzien können z.B. von Sigma-ALDRICH bzw. ABCR bezogen werden.
-
A: Synthese von Synthonen S:
-
1) 3,5-Bisphenyl-benzoesäurechlorid, S1
-
-
Darstellung nach Organikum, VEB Deutscher Verlag der Wissenschaften, Berlin, 5. Auflage, 1965, Seite 409 durch Kochen von 3,5-Bisphenylbenzoesäure [99710-75-5] mit 2 Äquivalenten Thionylchlorid unter Zusatz von 2 Tropfen DMF. Ausbeute: Quantitativ. Reinheit: > 98 % nach 1H-NMR.
-
2) 2-(2-Amino-phenyl)-benzimidazol-Derivate:
-
Darstellung von 2-(2-Amino-4-methyl-phenyl)benzimidazol, S2
-
Darstellung analog zu B. Saha et al., Synth. Commun. 2007, 37, 19, 3455. Eine Lösung von 13.5 g (100 mmol) 2-Nitro-4-methylbenzaldehyd [20357-22-6] und 11.9 g (110 mmol) 1,2-Diaminobenzol in einem Gemisch aus 150 ml DMF und 5 ml Wasser wird unter Rühren und Kühlung auf 20 °C portionsweise so mit 33.8 g (55 mmol) Oxone [70693-62-8] versetzt, dass die Temperatur nicht über 35 °C ansteigt. Anschließend wird die Reaktionsmischung bei Raumtemperatur bis zum vollständigen Umsatz des Aldehyds gerührt (ca. 4 h). Man rührt die Reaktionsmischung in eine Lösung von 40 g Kaliumcarbonat in 2000 ml Wasser ein, rührt 15 min. nach, extrahiert mit je drei 300 ml Portionen Dichlormethan, wäscht die organische Phase zweimal mit 300 ml Wasser, einmal mit 500 ml ges. Kochsalzlösung und trocknet über Natriumsulfat. Die Dichlormethan-Lösung wird über Kieselgel filtriert und das Dichlormethan wird im Vakuum entfernt. Der gelbe Rückstand wird in 500 ml Methanol aufgenommen, unter Rühren mit Stickstoff inertisiert, mit 3 g 10 % Pd/C versetzt und bei Raumtemperatur im Autoklaven bei 2 bar Wasserstoffdruck hydriert. Nach beendeter Wasserstoffaufnahme wird der Katalysator über ein Celite-Bett abfiltriert und das Methanol wird im Vakuum entfernt. Ausbeute: 18.5 g (83 mmol), 83 %. Reinheit: > 95 % nach 1H-NMR.
-
Analog werden folgende Derivate dargestellt:
| Bsp. | Aldehyd | 1,2-Diaminobenzol | Produkt | Ausbeute |
| S2 | | | | 83 % |
| S3 | | | | 76 % |
| S4 | | | | 67 % |
| S5 | | | | 47 % |
| S6 | | | | 88 % |
| S7 | | | | 77 % |
| S8 | | | | 67 % |
-
3) 2-(2-Brom-phenyl)-benzimidazol-Derivate:
-
3.1) Variante A, Oxone als Oxidationsmittel
-
Darstellung analog zu B. Saha et al., Synth. Commun. 2007, 37, 3455. Eine Lösung von 100 mmol des Aldehyds und 110 mmol des 1,2-Diaminobenzols in einem Gemisch aus 150 ml DMF und 5 ml Wasser wird in einem Kaltwasserbad (ca. 1000 ml bei ca. 10 °C) platziert und dann unter Rühren portionsweise so mit 33.8 g (55 mmol) Oxone [70693-62-8] versetzt, dass die Temperatur nicht über 35 °C ansteigt. Nach Abklingen der exothermen Reaktion rührt man bei Raumtemperatur bis zum vollständigen Umsatz des Aldehyds (1 - 6 h). Man rührt die Reaktionsmischung in eine Lösung von 40 g Kaliumcarbonat in 2000 ml Wasser ein und rührt 15 min. nach. Ausgefallene Feststoffe werden abgesaugt, dreimal mit je 100 ml Wasser gewaschen und dann trockengesaugt. Öle werden mit je drei 300 ml Portionen Dichlormethan extrahiert, die organische Phase wird zweimal mit je 300 ml Wasser und einmal mit 500 ml ges. Kochsalzlösung gewaschen, und über Natriumsulfat getrocknet. Die Dichlormethan-Lösung wird über eine kurze Kieselgel-Säule filtriert, das Dichlormethan wird im Vakuum entfernt und der Rückstand aus Ethylacetat / Ether oder Ethanol / Wasser umkristallisiert.
-
Darstellung von 2-(2-Brom-4-fluor-phenyl)-D4-benzimidazol, S9
-
Eine auf 10 °C gekühlte Lösung von 20.3 g (100 mmol) 2-Brom-4-fluorbenzaldehyd [59142-68-6] und 11.9 g (110 mmol) 1,2-Diaminobenzol-D4 [291765-93-0] in einem Gemisch aus 150 ml DMF und 5 ml Wasser wird unter Rühren portionsweise so mit 33.8 g (55 mmol) Oxone [70693-62-8] versetzt, dass die Temperatur 35 °C nicht übersteigt. Nach Abklingen der exothermen Reaktion rührt man bei Raumtemperatur bis zum vollständigen Umsatz des Aldehyds nach (ca. 2 h). Man rührt die Reaktionsmischung in eine Lösung von 40 g Kaliumcarbonat in 2000 ml Wasser ein, rührt 15 min. nach, saugt den braunen Feststoff ab, wäscht diesen dreimal mit je 100 ml Wasser und trocknet diesen im Vakuum. Man nimmt den Feststoff in 200 ml Dichlormethan auf, filtriert über eine kurze Kieselgel-Säule, entfernt das Dichlormethan im Vakuum und kristallisiert den Rückstand aus Ethylacetat / Ether um. Ausbeute: 12.2 g (42 mmol), 42 %. Reinheit: > 95 % nach 1H-NMR.
-
3.2) Variante B, Nitrobenzol als Oxidationsmittel
-
Darstellung analog zu D. Jerchel et al., Ann. Chem. 1952, 575, 162. Eine Lösung von 100 mmol des Aldehyds und 110 mmol des 1,2-Diaminobenzols in 100 ml Ethanol wird in einer Apparatur, bestehend aus einem 500 ml Kolben mit Wasserabscheider und Rückflusskühler, platziert und 30 min. bei Raumtemperatur gerührt. Anschließend setzt man 40 ml Nitrobenzol zu und erhitzt die Reaktionsmischung auf schwachen Rückfluss (Ölbadtemperatur ca. 220 °C), wobei man das Ethanol und gebildetes Wasser abdestilliert. Nach 45 min. unter schwachem Rückfluss lässt man erkalten, versetzt mit 40 ml Diethylether, rührt 30 min. nach, saugt vom Feststoff ab und wäscht diesen einmal mit 50 ml Diethylether. Man nimmt den Feststoff in 200 ml Dichlormethan auf, filtriert über eine kurze Kieselgel-Säule, entfernt das Dichlormethan im Vakuum und kristallisiert den Rückstand aus Ethylacetat / Ether oder Ethanol / Wasser um.
-
Darstellung von 2-(2-Brom-4-fluor-phenyl)-D4-benzimidazol, S9
-
Eine Lösung von 20.3 g (100 mmol) 2-Brom-4-fluorbenzaldehyd [59142-68-6] und 11.9 g (110 mmol) 1,2-Diaminobenzol in 100 ml Ethanol wird in einer Apparatur bestehend aus einem 500 ml Kolben mit Wasserabscheider und Rückflusskühler platziert und 30 min. bei Raumtemperatur gerührt. Anschließend setzt man 40 ml Nitrobenzol zu, und erhitzt die Reaktionsmischung auf schwachen Rückfluss (Ölbadtemperatur ca. 220 °C), wobei man das Ethanol und gebildetes Wasser abdestilliert. Nach 45 min. unter schwachem Rückfluss lässt man erkalten, versetzt mit 40 ml Diethylether, rührt 30 min. nach, saugt vom Feststoff ab und wäscht diesen einmal mit 50 ml Diethylether. Man nimmt den Feststoff in 200 ml Dichlormethan auf, filtriert über eine kurze Kieselgel-Säule, entfernt das Dichlormethan im Vakuum und kristallisiert den Rückstand aus Ethylacetat / Ether um. Ausbeute: 19.7 g (68 mmol), 68 %. Reinheit: > 95 % nach 1H-NMR.
-
Analog werden folgende Derivate dargestellt:
| Bsp | Variante | Aldehyd | 1,2-Diaminobenzol | Produkt | Ausbeute |
| S9 | A | | | | 42 % |
| S9 | B | | | | 68 % |
| S10 | A | | | | 40 % |
| S11 | A | | | | 31 % |
| S12 | A | | | | 42 % |
| S13 | A | | | | 38 % |
| S14 | B | | | | 63 % |
| S15 | A | | | | 37 % |
| S16 | A | | | | 47 % |
| S16 | B | | | | 61 % |
| S17 | A | | | | 56 % |
-
4) 2-(2-Amino-phenyl)-benzimidazol-Derivate:
-
Darstellung von 2-(2-Amino-4-fluor-phenyl)-D4-benzimidazol, S18
-
Darstellung analog zu N. Xiua et al, Angew. Chem. Int. Ed. 2009, 48, 337. Ein Autoklav wird mit 29.5 g (100 mmol) 2-(2-Brom-4-fluor-phenyl)-D4-benzimidazol (S9), 65.2 g (200 mmol) Cäsiumcarbonat, 200 ml DMF, 30 ml konz. Ammoniak-Lösung, 1.3 g (5 mmol) Kupfer(II)acetylacetonat und 2.1 ml (20 mmol) Acetylaceton beschickt und verschlossen. Die Reaktionsmischung wird 24 h bei 90 °C gerührt. Nach Erkalten wird die Reaktionsmischung im Vakuum eingeengt, der Rückstand wird mit 500 ml Wasser versetzt und fünfmal mit 200 ml Dichlormethan extrahiert. Die vereinigten org. Phasen werden dreimal mit je 200 ml Wasser und einmal mit 300 ml ges. Kochsalzlösung gewaschen und über Natriumsulfat getrocknet. Nach Entfernen des Lösungsmittels wird der Rückstand mit Diethylether ausgerührt. Ausbeute: 16.9 g (73 mmol), 73 %. Reinheit: > 95 % nach 1H-NMR.
-
Analog werden folgende Derivate dargestellt:
| Bsp. | 2-(2-Bromphenyl)-benzimidazol | Produkt | Ausbeute |
| S18 | | | 73 % |
| S19 | | | 65 % |
| S20 | | | 58 % |
| S21 | | | 51 % |
| S22 | | | 55 % |
| S23 | | | 67 % |
| S24 | | | 61 % |
| S25 | | | 70 % |
| S26 | | | 24 % |
-
5) 2-tert-Butyl-3-brom-4-iod-pyridin, S27:
-
-
5.1) 2-tert-Butyl-3-brom-6-trimethylsilyl-pyridin:
-
Durchführung analog zu
WO 2007/073303 : Ein Gemisch aus 128.8 g (560 mmol) 3-Brom-6-trimethylsilylpyridin [291312-74-8], 286.0 g (2.8 mol) Pivalinsäure [75-98-9] und 400 ml Wasser wird mit 12.4 g (73 mmol) Silbernitrat versetzt und 30 min. bei Raumtemperatur gerührt. Man versetzt die Reaktionsmischung tropfenweise während 15 min. mit 1000 ml 10 Gew.-% iger Schwefelsäure, erwärmt die Mischung auf 70 °C und versetzt tropfenweise während 30 min. mit einer Lösung von 168.5 g (730 mmol) Ammoniumperoxodisulfat in 300 ml Wasser. Nach beendeter Kohlendioxidentwicklung rührt man noch 3 h bei 70 °C nach, lässt die Reaktionsmischung erkalten, versetzt mit 500 ml Ethylacetat, trennt die wässrige Phase ab, extrahiert diese erneut mit 500 ml Ethylacetat, vereinigt die organischen Phasen, wäscht diese zehn mal mit je 300 ml ges. Natriumhydrogencarbonat-Lösung, wäscht abschließend einmal mit 500 ml ges. Kochsalzlösung, trocknet über Magnesiumsulfat, entfernt das Lösungsmittel im Vakuum und trocknet das so erhaltene Öl bei 60 °C im Ölpumpenvakuum. Ausbeute: 59.4 g (207 mmol), 37 %. Reinheit: > 95 % nach
1H-NMR.
-
5.2) 2-tert-Butyl-3-brom-4-iod-6-trimethylsilyl-pyridin:
-
Durchführung analog zu P. N. W. Baxter, Chem. Eur. J. 2003, 9, 2531: Eine auf -78 °C gekühlte, gut gerührte Mischung aus 14.0 ml (100 mmol) Di-iso-propylamin und 500 ml THF wird tropfenweise während 15 min. mit 65.6 ml (105 mmol) n-BuLi, 1.6 M in Hexan versetzt. Die Reaktionsmischung wird 45 min. bei -78 °C gerührt und dann auf -90 °C abgekühlt. Eine auf -90 °C vorgekühlte Lösung von 30.1 g (105 mmol) 2-tert-Butyl-3-brom-6-trimethylsilyl-pyridin in 100 ml THF wird so zugetropft, dass die Temperatur -80 °C nicht übersteigt. Nach 1 h Nachrühren bei -90 °C erwärmt man die Reaktionsmischung auf -75 °C, rührt weitere 15 min. bei -75 °C nach und kühlt diese dann erneut auf - 90 °C ab. Dann versetzt man tropfenweise so mit einer Lösung von 30.5 g (120 mmol) lod in 80 ml THF, dass die Temperatur -75 °C nicht übersteigt, rührt die Reaktionsmischung weitere 4 h bei -75 °C nach und lässt diese auf Raumtemperatur erwärmen. Nach Zugabe von 10 ml Wasser und Entfernen des THFs im Vakuum setzt man 1000 ml tert-Butyl-methyl-ether zu und versetzt die Mischung tropfenweise mit 100 ml gesättigter Natriumsulfit-Lösung, um überschüssiges Jod zu reduzieren. Man trennt die org. Phase ab, wäscht diese dreimal mit je 300 ml Wasser, trocknet über Natriumsulfat, entfernt das Lösungsmittel im Vakuum und kristallisiert den Rückstand einmal aus Cyclohexan um. Ausbeute: 14.8 g (36 mmol), 34 %. Reinheit: > 95 % nach
1H-NMR.
-
5.3) 2-tert-Butyl-3-brom-4-iod-pyridin:
-
-
Eine Lösung von 14.8 g (36 mmol) 2-tert-Butyl-3-brom-4-iod-6-trimethylsilyl-pyridin und 14.0 g (40 mmol) Tetrabutylammoniumfluorid-Trihydrat in 150 ml THF wird 5 min. unter Rückfluss erhitzt. Nach Erkalten und Entfernen des THF im Vakuum wird der Rückstand in 200 ml Dichlormethan aufgenommen, die org. Phase wird fünfmal mit je 100 ml Wasser gewaschen und über Natriumsulfat getrocknet. Nach Entfernen des Dichlormethans im Vakuum wird der Rückstand aus Ethanol umkristallisiert. Ausbeute: 9.2 g (27 mmol), 75 %. Reinheit: > 95 % nach 1H-NMR.
-
6) 2-Chlor-3-cyano-6-tert-butyl-pyridin, S28:
-
Durchführung analog 5.1, wobei anstelle von 128.8 g (560 mmol) 3-Brom-6-trimethylsilylpyridin 77.6 g (560 mmol) 2-Chlor-3-cyano-pyridin [6602-54-6] eingesetzt wird. Ausbeute: 92.5 g (475 mmol), 85 %. Reinheit: > 95 % nach
1H-NMR.
-
7) 2-(2-Chlor-6-tert-butyl-pyridin-3-yl)-benzimidazol-Derivate Darstellung von 2-(2-Chlor-6-tert-butyl-pyridin-3-yl)-benzimidazol, S29
-
Ein im Mörser homogenisiertes Gemisch aus 19.5 g (100 mmol) 2-Chlor-3-cyano-6-tert-butyl-pyridin (S28), 36.2 g (200 mmol) o-Phenylendiamin und 38.0 g (200 mmol) p-Toluolsulfonsäure-monohydrat wird 3 h auf 220 °C (Ölbadtemperatur) erhitzt. Nach Erkalten wird der schwarze, glasige Sinterkuchen unter gutem Rühren in einem Gemisch aus 100 ml Ethanol und 100 ml 1N Salzsäure aufgenommen. Nach 30 min. Rühren saugt man den grau-grünen Feststoff ab, wäscht diesen dreimal mit je 50 ml Wasser und trocknet im Vakuum. Man nimmt den Feststoff in 200 ml Ethylacetat auf, chromatographiert an Kieselgel (Ethylacetat-Heptan 1:1, Rf ca. 0.7), um 2-tert-Butyl-6,11-dihydro-benzo[b]pyrido[2,3-e][1,4]diazepin-5-on und braune Nebenprodukte zu entfernen. Das so erhaltene Rohprodukt wird in Ethylacetat in der Siedehitze gelöst und tropfenweise mit der vierfachen Menge an Cyclohexan versetzt. Nach 18 h rühren bei Raumtemperatur saugt man von den gebildeten Kristallen ab, wäscht diese mit n-Heptan und trocknet im Vakuum. Ausbeute: 7.5 g (26 mmol), 26 %. Reinheit: > 95 % nach 1H-NMR.
-
Analog werden folgende Derivate dargestellt:
| Bsp. | Cyanopyridin | 1,2-Diaminobenzol | Produkt | Ausbeute |
| S29 | | | | 26 % |
| S30 | | | | 22 % |
| S31 | | | | 31 % |
-
8) 3-Cyano-4-chlor-6-tert-butyl-pyridin, S32:
-
Durchführung analog 5.1, wobei anstelle von 128.8 g (560 mmol) 3-Brom-6-trimethylsilylpyridin 77.6 g (560 mmol) 3-Cyano-4-chlor-pyridin [89284-61-7] eingesetzt wird. Ausbeute: 60.9 g (313 mmol), 56 %. Reinheit: > 95 % nach
1H-NMR.
-
9) 2-(4-Chor-6-tert-butyl-pyridin-3-yl)benzimidazol-Derivate:
-
Darstellung von 2-(4-Chor-6-tert-butyl-pyridin-3-yl)benzimidazol, S33
-
Ein im Mörser homogenisiertes Gemisch aus 19.5 g (100 mmol) 2-tert-butyl-3-chlor-4-cyano-pyridin (S32), 36.2 g (200 mmol) o-Phenylendiamin und 38.0 g (200 mmol) p-Toluolsulfonsäure-monohydrat wird 3 h auf 220 °C (Ölbadtemperatur) erhitzt. Nach Erkalten wird der schwarze, glasige Sinterkuchen unter gutem Rühren in einem Gemisch aus 100 ml Ethanol und 100 ml 1N Salzsäure aufgenommen. Nach 30 min. Rühren saugt man den grau-grünen Feststoff ab, wäscht diesen dreimal mit je 50 ml Wasser und trocknet im Vakuum. Man nimmt den Feststoff in 200 ml Ethylacetat auf, chromatographiert an Kieselgel (Ethylacetat-Heptan 1:1, Rf ca. 0.7), um 3-tert-Butyl-5,10-dihydro-benzo[b]pyrido[4,3-e][1,4]diazepin-11-on und braune Nebenprodukte zu entfernen. Das so erhaltene Rohprodukt wird in Ethylacetat in der Siedehitze gelöst und tropfenweise mit der vierfachen Menge an Cyclohexan versetzt. Nach 18 h rühren bei Raumtemperatur saugt man von den gebildeten Kristallen ab, wäscht diese mit n-Heptan und trocknet im Vakuum. Ausbeute: 7.5 g (26 mmol), 26 %. Reinheit: > 95 % nach 1H-NMR.
-
Analog werden folgende Derivate dargestellt:
| Bsp. | Cyanopyridin | 1,2-Diaminobenzol | Produkt | Ausbeute |
| S33 | | | | 36 % |
| S34 | | | | 40 % |
| S35 | | | | 33 % |
-
10) 2-tert-Butyl-5-chlor-1,6-naphthyridin, S36
-
-
Ein Gemisch aus 15.7 g (100 mmol) 2-Chlor-3-formyl-4-amino-pyridin [338452-92-9], 37.6 ml (300 mmol) tert-Butyl-methyl-keton, 1.0 ml (10 mmol) Piperidin und 100 ml Ethanol wird 60 h unter Rückfluss erhitzt. Nach Erkalten verdünnt man mit 500 ml Dichlormethan, wäscht die Reaktionsmischung fünf mal mit je 500 ml Wasser, trocknet über Magnesiumsulfat und entfernt das Lösungsmittel im Vakuum. Der Rückstand wird aus Ethanol umkristallisiert. Ausbeute: 8.2 g (37 mmol), 37 %. Reinheit: > 95 % nach 1H-NMR.
-
11) 8-Phenyl-5-chlor-1,6-naphthyridin, S37
-
-
Eine Suspension von 22.2 g (100 mmol) 8-Phenyl-1,6-naphthyridin-5(6H)-on [173773-04-1] in 100 ml Toluol wird bei Raumtemperatur tropfenweise mit 12.1 ml (130 mmol) Phosphorylchlorid versetzt. Nach Zugabe von 5 Tropfen N,N-Dimethylanilin wird die Reaktionsmischung 5 h unter Rückfluss erhitzt. Nach Erkalten verdünnt man mit 300 ml Toluol, gießt die Reaktionsmischung auf 1000 g Eis und stellt durch Zugabe von 5 N NaOH alkalisch (pH ca. 9). Man trennt die organische Phase ab, wäscht diese einmal mit 300 ml ges. Kochsalzlösung, trocknet über Magnesiumsulfat und entfernt das Toluol im Vakuum. Ausbeute: 22.9 g (95 mmol), 95 %. Reinheit: > 95 % nach 1H-NMR.
-
12) 5-Amino-1,6-naphthyridin-Derivate:
-
Darstellung von 2-tert-Butyl-5-amino-1,6-naphthyridin, S38
-
Ein Gemisch aus 22.1 g (100 mmol) 2-tert-Butyl-5-chlor-1,6-naphthyridin (S36), 32.1 g (600 mmol) Ammoniumchlorid in 100 ml Sulfolan wird 20 h bei 200 °C gerührt. Die erkaltete Mischung wird mit 300 ml Wasser versetzt und 2 h bei Raumtemperatur gerührt. Der Feststoff wird abgesaugt, zweimal mit je 50 ml Wasser gewaschen und anschließend in einem Gemisch aus 50 ml Methanol und 150 ml konz. Ammoniak-Lösung suspendiert. Die Suspension wird 20 h bei Raumtemperatur gerührt. Der Feststoff wird abfiltriert, dreimal mit je 50 ml Wasser gewaschen, im Vakuum getrocknet und einer Sublimation (p ca. 1 × 10-2 mbar, T = 150 °C) unterzogen. Ausbeute: 16.7 g (83 mmol), 83 %. Reinheit: > 97 % nach 1H-NMR.
-
Analog werden folgende Derivate dargestellt:
| Bsp. | 5-Chlor-1,6-naphthyridin | Produkt | Ausbeute |
| S38 | | | 83 % |
| S39 | | | 71 % |
-
13) 3-Brom-4-amino-6-tert-butyl-pyridin, S40
-
-
Durchführung analog zu V. Canibano et al., Synthesis 2001, 14, 2175. Eine gut gerührte, lichtgeschützte Lösung von 15.0 g (100 mmol) 2-tert-Butyl-4-amino-pyridin [39919-69-2] in 500 ml Acetonitril wird bei 40 °C portionsweise mit 18.7 g (105 mmol) N-Bromsuccinimid versetzt und weitere 30 h gerührt. Man entfernt das Lösungsmittel im Vakuum, nimmt den Rückstand in 500 ml Dichlormethan auf, wäscht fünfmal mit je 500 ml Wasser und einmal mit 300 ml ges. Kochsalzlösung, trocknet die organische Phase über Natriumsulfat und entfernt dann das Lösungsmittel im Vakuum. Das Rohprodukt wird aus Cyclohexan umkristallisiert. Ausbeute: 17.9 g (78 mmol), 78 %. Reinheit: > 95 % nach 1H-NMR.
-
14) 1-Amino-6-tert-butyl-2,7-naphthyridin, S41
-
-
Durchführung analog zu A. Zhang et al., J. Combi. Chem. 2007, 9, 6, 916: Ein Gemisch aus 17.4 g (100 mmol) 3-Cyano-4-methyl-6-tert-butyl-pyridin [942938-45-6], 14.0 ml (105 mmol) N,N-Dimethylformamind-dimethylacetal [4637-24-5] und 150 ml DMF wird 16 h unter Rückfluss erhitzt. Dann wird das DMF im Vakuum bei 70 °C entfernt. Der ölige Rückstand wird mit 46.3 g (600 mmol) wasserfreiem Ammoniumacetat versetzt, homogenisiert, im Ölbad (Temperatur ca. 135 °C) zum Schmelzen erhitzt und 3 h gerührt. Nach Erkalten nimmt man die Schmelze in einem Gemisch aus 200 ml Wasser und 100 ml Ethanol auf, stellt durch Zugabe von konz. Ammoniaklösung alkalisch (pH ca. 9) und extrahiert dreimal mit je 300 ml Dichlormethan. Die vereinigten org. Phasen werden zweimal mit je 300 ml Wasser gewaschen und über Natriumsulfat getrocknet. Nach Einengen der org. Phase im Vakuum wird der verbleibende Rückstand einer Sublimation (p ca. 1 × 10-2 mbar, T = 150 °C) unterzogen. Ausbeute: 13.7 g (68 mmol), 68 %. Reinheit: > 95 % nach 1H-NMR.
-
15) 2-N-Pivaloylamido-3-cyano-6-tert-butyl-pyridin, S42
-
-
Ein Gemisch aus 19.5 g (100 mmol) 2-Chlor-3-cyano-6-tert-butyl-pyridin (S28), 14.2 g (140 mmol) Pivalinsäureamid [754-10-9], 48.9 g (150 mmol) Cäsiumcarbonat, 1.7 g (3 mmol) 9,9-Dimethyl-4,5-bis(diphenylphosphino)-xanthen und 630 mg (2.8 mmol) Palladium(II)acetat in 400 ml Dioxan wird 12 h bei 100 °C gerührt. Nach Erkalten wird das Lösungsmittel im Vakuum entfernt, der Rückstand wird in 1000 ml Ethylacetat aufgenommen, die organische Phase wird dreimal mit je 300 ml Wasser und einmal mit 300 ml gesättigter Kochsalzlösung gewaschen und über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels verbleibt ein brauner Feststoff. Ausbeute: 24.9 g (96 mmol), 96 %. Reinheit: > 95 % nach 1H-NMR.
-
16) 2-(2-Amino-6-tert-butyl-pyridin-3-yl)benzimidazol-Derivate Darstellung von 2-(2-Amino-6-tert-butyl-pyridin-3-yl)benzimidazot, S43
-
Ein im Mörser homogenisiertes Gemisch aus 25.9 g (100 mmol) 2-N-Pivaloylamido-3-cyano-6-tert-butyl-pyridin (S42) und 90.5 g (500 mmol) o-Phenylendiamin-dihydrochlorid [615-28-1] wird in ein auf 240 °C vorgeheiztes Ölbad eingestellt und 3.5 h bei dieser Temperatur belassen. Nach Erkalten wird die tiefblaue Schmelze in einem Gemisch aus 150 ml Ethanol und 300 ml Wasser in der Wärme gelöst und dann tropfenweise unter gutem Rühren mit einer Lösung von 40 g Natriumcarbonat in 200 ml Wasser versetzt (Achtung: Schäumen, Kohlendioxidentwicklung). Nach beendeter Zugabe rührt man noch 30 min. nach, saugt dann vom grauen Feststoff ab, wäscht diesen dreimal mit je 100 ml Wasser und trocknet im Vakuum. Ausbeute: 38.0 g, 86 % eines 1:1 Gemischs aus dem Produkt und 2-tert-Butyl-benzimidazol, das ohne weitere Reinigung umgesetzt wird.
-
Analog werden folgende Derivate dargestellt:
| Bsp. | Cyanopyridin | 1,2-Diaminobenzol | Produkt | Ausbeute |
| S43 | | | | 86 % |
| S44 | | | | 69 % |
| S45 | | | | 63 % |
-
17) 3-Cyano-4-N-pivaloylamido-6-tert-butyl-pyridin, S46
-
-
Ein Gemisch aus 19.5 g (100 mmol) 3-Cyano-4-chlor-6-tert-butyl-pyridin, (S32), 14.2 g (140 mmol) Pivalinsäureamid [754-10-9], 48.9 g (150 mmol) Cäsiumcarbonat, 1.7 g (3 mmol) 9,9-Dimethyl-4,5-bis(diphenylphosphino)-xanthen und 630 mg (2.8 mmol) Palladium(II)acetat in 400 ml Dioxan wird 12 h bei 100 °C gerührt. Nach Erkalten wird das Lösungsmittel im Vakuum entfernt, der Rückstand wird in 1000 ml Ethylacetat aufgenommen, die org. Phase wird dreimal mit je 300 ml Wasser und einmal mit 300 ml ges. Kochsalzlösung gewaschen und über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels verbleibt ein brauner Feststoff. Ausbeute: 24.4 g (94 mmol), 94 %. Reinheit: > 95 % nach 1H-NMR.
-
18) 2-(4-Amino-6-tert-butyl-pyridin-3-yl)benzimidazol, S47
-
-
Ein im Mörser homogenisiertes Gemisch aus 25.9 g (100 mmol) 3-Cyano-4-N-pivaloylamido-6-tert-butyl-pyridin (S46), 90.5 g (500 mmol) o-Phenylendiamin-dihydrochlorid [615-28-1] wird in ein auf 240 °C vorgeheiztes Ölbad eingestellt und 3.5 h bei dieser Temperatur belassen. Nach Erkalten wird die tiefblaue Schmelze in einem Gemisch aus 150 ml Ethanol und 300 ml Wasser in der Wärme gelöst und dann tropfenweise unter gutem Rühren mit einer Lösung von 40 g Natriumcarbonat in 200 ml Wasser versetzt (Achtung: Schäumen, Kohlendioxidentwicklung). Nach beendeter Zugabe rührt man noch 30 min. nach, saugt dann vom grauen FS ab, wäscht diesen dreimal mit je 100 ml Wasser und trocknet im Vakuum. Ausbeute: 33.5 g, 76 % eines 1:1 Gemischs aus dem Produkt und 2-tert-Butyl-benzimidazol, das ohne weitere Reinigung umgesetzt wird.
-
19) 2-(2-tert-Butyl-pyrimidin-5-yl)benzimidazol, S48
-
-
Eine Lösung von 16.4 g (100 mmol) 2-tert-Butyl-pyrimidin-5-carboxaldehyd [104461-06-5] und 11.9 g (110 mmol) 1,2-Diaminobenzol in einem Gemisch aus 100 ml DMF und 3 ml Wasser wird bei 10 °C unter Rühren portionsweise mit 16.9 g (55 mmol) Oxone [70693-62-8] versetzt und anschließend bei Raumtemperatur bis zum vollständigen Umsatz des Aldehyds gerührt (ca. 2 h). Man rührt die Reaktionsmischung in eine Lösung von 40 g Kaliumcarbonat-Lösung in 2000 ml Wasser ein, rührt 15 min. nach, saugt vom gebildeten Feststoff ab, wäscht diesen dreimal mit je 100 ml Wasser und trocknet im Vakuum. Man löst den Feststoff in ca. 50 ml heißem Ethylacetat und fügt beim Abkühlen Diethylether bis zur leichten Trübung zu, rührt 12 h nach, saugt von den Kristallen ab, wäscht diese einmal mit 50 ml Diethylether und trocknet im Vakuum. Ausbeute: 16.6 g (66 mmol), 66 %. Reinheit: > 95 % nach 1H-NMR.
-
20) 4-Chlor-8-tert-Butyl-chinazolin, S49
-
-
20.1) 8-tert-Butyl-4(3H)chinazolinon
-
-
Durchführung analog zu M. Berg et al., Chem. Med. Chem. 2009, 4, 2, 249: Ein Gemisch aus 19.3 g (100 mmol) 2-Amino-3-tert-butyl-benzoesäure [917874-35-2], 31.4 g (300 mmol) Formamidin-acetat [3473-63-0] und 4.4 ml (110 mmol) Formamid wird 12 h bei 160 °C gerührt. Nach Erkalten auf 60 °C versetzt man tropfenweise mit einem Gemisch aus 200 ml Ethanol und 200 ml 2 N Natronlauge, filtriert über eine mit Seesand belegte P4-Fritte ab, um polymeres Material zu entfernen, und stellt dann durch Zugabe von 2 N Salzsäure neutral. Nach 12 h Rühren saugt man von den gebildeten Kristallen ab und wäscht diese dreimal mit je 100 ml Wasser. Ausbeute: 13.9 g (68 mmol), 68 %. Reinheit: > 95 % nach 1H-NMR.
-
20.2) 4-Chlor-8-tert-butyl-chinazolin, S49
-
Eine Suspension von 10.2 g (50 mmol) 8-tert-Butyl-4(3H)chinazolinon in 100 ml Toluol wird bei Raumtemperatur tropfenweise mit 18.6 ml (200 mmol) Phosphorylchlorid versetzt. Nach Zugabe von 5 Tropfen N,N-Dimethylanilin wird die Reaktionsmischung 5 h unter Rückfluss erhitzt. Nach Erkalten verdünnt man mit 300 ml Toluol, gießt die Reaktionsmischung auf 1000 g Eis und stellt durch Zugabe von konz. Ammoniak-Lösung alkalisch (pH ca. 9). Man trennt die organische Phase ab, wäscht diese einmal mit 300 ml ges. Kochsalzlösung, trocknet über Magnesiumsulfat und entfernt das Toluol im Vakuum. Der Rückstand wird an Kieselgel chromatographiert (n-Heptan : Ethylacetat 3 : 1 v : v). Ausbeute: 9.3 g (42 mmol), 84 %. Reinheit: > 95 % nach 1H-NMR.
-
21) 3,4-Diamino-6-tert-butyl-pyridin-di-hydrochlorid, S50
-
Durchführung analog zu 4) S18 wobei anstatt 29.5 g (100 mmol) 2-(2-Brom-4-fluor-phenyl)-D4-benzimidazol (S9) 22.9 g (100 mmol) 3-Brom-4-amino-6-tert-butyl-pyridin (S40) verwendet wird. Das Rohprodukt wird durch Lösen in 100 ml Ethanol und Einleiten von gasförmiger Salzsäure in das Dihydrochlorid überführt. Die so erhaltenen Kristalle werden abgesaugt und im Vakuum getrocknet. Ausbeute: 13.3 g (56 mmol), 56 %. Reinheit: > 95 % nach
1H-NMR.
-
22) 2-(2-Amino-6-tert-butyl-pyridin-3-yl)-6-tert-butyl-3H-imidazo-[4,5-c]pyridin, S51
-
Durchführung analog zu 16) S43, wobei anstatt 90.5 g (500 mmol) o-Phenylendiamin-dihydrochlorid 71.4 g (300 mmol) 3,4-Diamino-6-tert-butyl-pyridin-di-hydrochlorid (S50) eingesetzt wird. Nach Erkalten wird die tiefblaue Schmelze in einem Gemisch aus 150 ml Ethanol und 300 ml Wasser in der Wärme gelöst und dann tropfenweise unter gutem Rühren mit einer Lösung von 25 g Natriumcarbonat in 100 ml Wasser versetzt (Achtung: Schäumen, Kohlendioxidentwicklung). Nach beendeter Zugabe rührt man noch 30 min. nach, saugt dann vom braunen Feststoff ab, wäscht diesen dreimal mit je 100 ml Wasser und trocknet im Vakuum. Ausbeute: 43.8 g, 79 % eines 1:1 Gemischs aus dem Produkt und 2,6-Ditert-butyl-3H-imidazo-[4,5-c]pyridin, das ohne weitere Reinigung umgesetzt wird.
-
23) 2-(2-tert-butyl-pyrimidin-5-yl)-6-tert-butyl-3H-imidazo[4,5-c]pyridin, S52
-
Darstellung analog zu 16), S43 wobei anstatt 25.9 g (100 mmol) 2-N-Pivaloylamido-3-cyano-6-tert-butyl-pyridin (S42) und 90.5 g (500 mmol) o-Phenylendiamin-dihydrochlorid [615-28-1] 16.1 g (100 mmol) 2-tert-Butyl-5-cyano-pyrimidin [126230-72-6] und 71.4 g (300 mmol) 3,4-Diamino-6-tert-butyl-pyridin-di-hydrochlorid (S50) eingesetzt wird. Nach Erkalten wird die violette Schmelze in einem Gemisch aus 150 ml Ethanol und 300 ml Wasser in der Wärme gelöst und dann tropfenweise unter gutem Rühren mit einer Lösung von 25 g Natriumcarbonat in 100 ml Wasser versetzt (Achtung: Schäumen, Kohlendioxidentwicklung). Nach beendeter Zugabe rührt man noch 30 min. nach, saugt dann vom braunen Feststoff ab, wäscht diesen dreimal mit je 100 ml Wasser und trocknet im Vakuum. Ausbeute: 23.8 g (77 mmol), 77 %. Reinheit: > 95 % nach
1H-NMR.
-
24) 2-(4-Amino-6-tert-butyl-pyridin-3-yl)-6-tert-butyl-3H-imidazo-[4,5-c]pyridin, S53
-
Durchführung analog zu 18) S47, wobei anstatt 90.5 g (500 mmol) o-Phenylendiamin-dihydrochlorid 71.4 g (300 mmol) 3,4-Diamino-6-tert-butyl-pyridin-di-hydrochlorid (S50) eingesetzt wird. Nach Erkalten wird die tiefblaue Schmelze in einem Gemisch aus 150 ml Ethanol und 300 ml Wasser in der Wärme gelöst und dann tropfenweise unter gutem Rühren mit einer Lösung von 25 g Natriumcarbonat in 100 ml Wasser versetzt (Achtung: Schäumen, Kohlendioxidentwicklung). Nach beendeter Zugabe rührt man noch 30 min. nach, saugt dann vom braunen Feststoff ab, wäscht diesen dreimal mit je 100 ml Wasser und trocknet im Vakuum. Ausbeute: 40.5 g, 74 % eines 1:1 Gemischs aus dem Produkt und 2,6-Ditert-butyl-3H-imidazo-[4,5-c]pyridin, das ohne weitere Reinigung umgesetzt wird.
-
25) 2-tert-Butyl-4-(N-pivaloyl-amido)-pyrimidin-5-carbonsäuremethylester, S54
-
Ein Gemisch aus 22.9 g (100 mmol) 2-tert-Butyl-4-chlor-pyrimidin-5-carbonsäuremethylester [897375-22-3], 14.2 g (140 mmol) Pivalinsäureamid [754-10-9], 48.9 g (150 mmol) Cäsiumcarbonat, 1.7 g (3 mmol) 9,9-Dimethyl-4,5-bis(diphenylphosphino)-xanthen und 630 mg (2.8 mmol) Palladium(II)acetat in 400 ml Dioxan wird 12 h bei 100 °C gerührt. Nach Erkalten wird das Lösungsmittel im Vakuum entfernt, der Rückstand wird in 1000 ml Ethylacetat aufgenommen, die org. Phase wird dreimal mit je 200 ml Wasser und einmal mit 300 ml ges. Kochsalzlösung gewaschen und über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels verbleibt ein brauner Feststoff. Ausbeute: 28.2 g (96 mmol), 96 %. Reinheit: > 95 % nach
1H-NMR.
-
26) 2-(2-tert-Butyl-4-amino-pyrimidin-5-yl)benzimidazol, S55
-
Darstellung analog zu 16) S43, wobei anstatt 25.9 g (100 mmol) 2-N-Pivaloylamido-3-cyano-6-tert-butyl-pyridin (S42) 29.3 g (100 mmol) 2-tert-Butyl-4-(N-pivaloyl-amido)-pyrimidin-5-carbonsäure-methylester (S54) eingesetzt wird. Während des Erhitzens der Reaktionsmischung gebildetes Methanol und Wasser werden durch einen schwachen Argonstrom abgetrieben. Nach Erkalten wird die schwarze Schmelze in einem Gemisch aus 150 ml Ethanol und 300 ml Wasser in der Wärme gelöst und dann tropfenweise unter gutem Rühren mit einer Lösung von 40 g Natriumcarbonat in 2700 ml Wasser versetzt (Achtung: Schäumen, Kohlendioxidentwicklung). Nach beendeter Zugabe rührt man noch 30 min. nach, saugt dann vom braunen Feststoff ab, wäscht diesen dreimal mit je 100 ml Wasser und trocknet im Vakuum. Ausbeute: 28.3 g, 64 % eines 1:1 Gemischs aus dem Produkt und 2-tert-Butyl-benzimidazol, das ohne weitere Reinigung umgesetzt wird.
-
27) 2-(2-tert-Butyl-4-amino-pyrimidin-5-yl)-6-tert-butyl-3H-imidazo-[4,5-c]pyridin, S56
-
-
Darstellung analog zu 16) S43, wobei anstatt 25.9 g (100 mmol) 2-N-Pivaloylamido-3-cyano-6-tert-butyl-pyridin (S42) 29.3 g (100 mmol) 2-tert-Butyl-4-(N-pivaloyl-amido)-pyrimidin-5-carbonsäure-methylester (S54) und anstatt 90.5 g (500 mmol) o-Phenylendiamin-dihydrochlorid 71.4 g (300 mmol) 3,4-Diamino-6-tert-butyl-pyridin-di-hydrochlorid (S50) eingesetzt werden. Während des Erhitzens der Reaktionsmischung gebildetes Methanol und Wasser werden durch einen schwachen Argonstrom abgetrieben. Nach Erkalten wird die schwarze Schmelze in einem Gemisch aus 150 ml Ethanol und 300 ml Wasser in der Wärme gelöst und tropfenweise unter gutem Rühren mit einer Lösung von 25 g Natriumcarbonat in 100 ml Wasser versetzt (Achtung: Schäumen, Kohlendioxidentwicklung). Nach beendeter Zugabe rührt man noch 30 min. nach, saugt dann vom braunen Feststoff ab, wäscht diesen dreimal mit je 100 ml Wasser und trocknet im Vakuum. Ausbeute: 38.9 g, 70 % eines 1:1 Gemischs aus dem Produkt und 2,6-Di-tert-butyl-3H-imidazo-[4,5-c]pyridin, das ohne weitere Reinigung umgesetzt wird.
-
28) 2-(N-Alkylamido)-benzaldehyd-Derivate
-
Darstellung von 2-(N-Pivaloylamido)-4-fluor-benzaldehyd, S57:
-
Ein Gemisch aus 20.3 g (100 mmol) 2-Brom-4-fluor-benzaldehyd [59142-68-6], 14.2 g (140 mmol) Pivalinsäureamid [754-10-9], 81.5 g (250 mmol) Cäsiumcarbonat, 1.7 g (3 mmol) 9,9-Dimethyl-4,5-bis(diphenylphosphino)-xanthen und 630 mg (2.8 mmol) Palladium(II)acetat in 400 ml Dioxan wird 4 h bei 100 °C gerührt. Nach Erkalten wird das Lösungsmittel im Vakuum entfernt, der Rückstand wird in 1000 ml Ethylacetat aufgenommen, die organische Phase wird dreimal mit je 300 ml Wasser und einmal mit 300 ml ges. Kochsalzlösung gewaschen und über eine kurze Kieselgel-Säule filtriert. Die nach Abziehen des Lösungsmittels im Vakuum erhaltenen Feststoffe werden weiter umgesetzt. Ausbeute: 20.8 g (93 mmol), 95 %. Reinheit: > 95 % nach 1H-NMR.
-
Analog werden folgende Derivate dargestellt:
| Bsp. | 2-Brom- 4-fluor benzaldehyd | Amid | Produkt | Ausbeute |
| S57 | | | | 93 % |
| S58 | | | | 95 % |
-
29) 2-(2-(N-Alkylamido)-phenyl)-benzimidazol-Derivate Darstellung von 2-(2-(N-Pivaloylamido)-4-fluor-phenyl)-benzimidazol, S59:
-
Eine Lösung von 22.3 g (100 mmol) 2-(N-Pivaloylamido)-4-fluor-benzaldehyd (S57) und 11.9 g (110 mmol) o-Phenylendiamin [95-54-5] in 50 ml Ethanol wird in einem 500 ml Rundkolben mit Wasserabscheider platziert und 30 min. bei 50 °C gerührt. Dann gibt man 50 ml Nitrobenzol zu und steigert die Temperatur bis zum schwachen Rückfluss des Nitrobenzols, wobei man beim Hochheizen das Ethanol und gebildetes Wasser abdestilliert. Nach 2 h unter schwachem Rückfluss lässt man auf 50 °C erkalten, gibt 40 ml Methanol zu, lässt dann unter Rühren ganz erkalten, rührt 2 h bei Raumtemperatur nach, saugt dann von den gebildeten Kristallen ab, wäscht diese zweimal mit je 20 ml Methanol und trocknet im Vakuum. Ausbeute: 28.6 g (92 mmol), 92 %. Reinheit: > 95 % nach 1H-NMR.
-
Analog werden folgende Derivate dargestellt:
| Bsp. | 2-(2-(N-Alkylamido)-phenyl)-benzimidazolDerivate | Amin | Produkt | Ausbeute |
| S59 | | | | 92 % |
| S60 | | | | 79 % |
-
30) 6-Chlor-benzo[4,5]imidazo[1,2-c]chinazolin, S61:
-
Ein Gemisch aus 23.5 g (100 mmol) Benzimidazo[1,2-c]quinazolin-6(5H)-on [16367-99-0], 22.9 g (110 mmol) Phosphorpentachlorid und 250 ml Phosphorylchlorid wird 24 h unter Rückfluss erhitzt. Man entfernt das überschüssige Phosphorylchlorid im Vakuum, versetzt den Rückstand mit 1000 ml Dichlormethan, hydrolysiert durch Zugabe von 1000 g Eis, stellt durch Zugabe von 10 Gew.-% iger Natronlauge schwach alkalisch, trennt die organische Phase ab, trocknet diese über Natriumsulfat und entfernt dann das Dichlormethan im Vakuum. Ausbeute: 24.1 g (94 mmol), 94 %. Reinheit: > 95 % nach
1H-NMR.
-
31) 6-tert-Butyl-2-(3,3-dimethyl-but-1-ynyl)-nicotinonitril, S62
-
Eine Lösung von 194.7 g (1 mol) 2-Chlor-3-cyano-6-tert-butyl-pyridin, S28 in einem Gemisch aus 1800 ml DMF und 1000 ml Triethylamin wird konsekutiv mit 15.7 g (60 mmol) Triphenylphosphin, 6.7 g (30 mmol) Palladium(II)acetat, 5.7 g (30 mmol) Kupfer(I)iodid und 155.6 g (1.9 mmol) tert-Butylacetylen versetzt und 4 h bei 65 °C gerührt. Nach Erkalten wird vom ausgefallenen Triethylammonium-hydrochlorid abgesaugt, dieses wird mit 300 ml DMF nachgewaschen. Das Filtrat wird im Vakuum von den Lösungsmitteln befreit. Der ölige Rückstand wird in 1000 ml Ethylacetat aufgenommen, die Lösung wird fünfmal mit je 500 ml Wasser und einmal mit 500 ml gesättigter Kochsalzlösung gewaschen, und die organische Phase wird über Magnesiumsulfat getrocknet. Nach Entfernen des Ethylacetats im Vakuum wird der schwarze ölige Rückstand einer Kugelrohrdestillation unterzogen (p ca. 10
-2 mbar, T = 120 - 140 °C). Ausbeute: 190.6 g (793 mmol), 79 %. Reinheit: > 97 % nach
1H-NMR.
-
Analog werden folgende Derivate dargestellt:
| Bsp. | Pyridin | Alkin | Produkt | Ausbeute |
| S63 | | Das Produkt wird ohne Kugelrohrdestillation weiter umgesetzt. | | 46 % |
| S64 | | Das Alkin wird in die Reaktionsmischung eingeleitet. | | 77 % |
| S65 | | Das Alkin wird in die Reaktionsmischung eingeleitet. | | 74 % |
| S66 | | | | 86 % |
| S67 | | | | 81 % |
| S68 | | | | 79 % |
-
32) 6-tert-Butyl-2-(3,3-dimethyl-but-1-ynyl)-pyridin-3-carboxaldehyd, S69
-
Eine auf -78 °C gekühlte Lösung von 72.1 g (300 mmol) 6-tert-Butyl-2-(3,3-dimethyl-but-1-ynyl)-nicotinonitril, S62 in 1500 ml Dichlormethan wird tropfenweise so mit 310 ml (315 mmol) Di-iso-butylaluminiumhydrid, 1 M in Toluol versetzt, dass die Temperatur -65 °C nicht übersteigt. Nach beendeter Zugabe wird weitere 2 h bei -78 °C gerührt, dann lässt man die Reaktionsmischung langsam auf Raumtemperatur erwärmen und rührt 12 h nach. Nach erneutem Abkühlen auf -10 °C setzt man 300 ml THF und dann unter gutem Rühren 400 ml 2 N Schwefelsäure (exotherm!) zu und rührt 12 h bei Raumtemperatur nach. Nach erneuten Abkühlen auf -10 °C gibt man eine Lösung von 70 g NaOH in 300 ml Wasser zu, trennt die wässrige Phase ab, wäscht die organische Phase dreimal mit je 1000 ml Wasser, einmal mit 500 ml gesättigter Kochsalzlösung, trocknet über Magnesiumsulfat und entfernt das Lösungsmittel im Vakuum. Ausbeute: 69.6 g (286 mmol), 95 %. Reinheit: > 95 % nach
1H-NMR.
-
Analog werden folgende Derivate dargestellt:
| Bsp. | Nitril | Produkt Pyridin-3-carboxaldehyde | Ausbeute |
| S70 | | | 87 % |
| S71 | | | 55 % |
| S72 | | | 58 % |
| S73 | | | 93 % |
| S74 | | | 96 % |
| S75 | | | 57 % |
-
B. Synthese der Liganden L:
-
1) Benzimidazo[1,2-c]chinazoJin-Systeme
-
-
1.1) Aus 2-(2-Amino-phenyl)-benzimidazol-Derivaten Allgemeine Ligandensynthese Variante A:
-
Eine gut gerührte Mischung aus 100 mmol des 2-(2-Amino-phenyl)-benzimidazol-Derivats, 350 mmol des Carbonsäurechlorids und 300 mmol der Carbonsäure wird 24 bis 100 h unter Rückfluss bei Carbonsäurechloriden, die unter 150 °C sieden, bzw. auf 150 °C bis 180 °C bei Carbonsäurechloriden, die über 150 °C sieden, erhitzt, bis das 2-(2-Amino-phenyl)-benzimidazol-Derivat umgesetzt ist. Nach Abkühlen wird die Reaktionsmischung in Ethanol oder Ethylacetat (50 - 200 ml) aufgenommen. Die Reaktionsmischung wird unter gutem Rühren in ein Gemisch aus 500 g Eis und 500 ml wässrigem konz. Ammoniak eingerührt. Fällt das Produkt als Feststoff an, wird dieser abgesaugt, mit Wasser gewaschen und trocken gesaugt. Fällt das Produkt als Öl an, wird dieses mit drei Portionen zu je 300 ml Ethylacetat extrahiert. Die organische Phase wird abgetrennt, mit 500 ml Wasser gewaschen und im Vakuum eingeengt. Das Rohprodukt wird in Ethylacetat oder Dichlormethan aufgenommen, über eine kurze Säule aus Alox, basisch, Aktivitätsstufe 1 oder Kieselgel filtriert, um braune Verunreinigungen zu entfernen. Nach zweimaliger Umkristallisation (Methanol, Ethanol, Aceton, Dioxan, etc.) des so erhaltenen Produktes wird dieses, durch Kugelrohrdestillation oder Sublimation (p ca. 1 × 10-5 mbar, T ca. 150 - 230 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Allgemeine Ligandensynthese Variante B:
-
Analoge Durchführung zu Variante A, jedoch werden anstatt der Carbonsäure 50 mmol Wasser zugesetzt.
-
Allgemeine Ligandensynthese Variante C:
-
Analoge Durchführung zu Variante A, jedoch wird keine Carbonsäure zugesetzt.
-
Darstellung von L13:
-
Ein Gemisch aus 20.9 g (100 mmol) 2-(2-Aminophenyl)-benzimidazol, 42.2 g (350 mmol) Pivalinsäurechlorid und 30.6 g (300 mmol) Pivalinsäure wird 50 h unter Rückfluss erhitzt. Man lässt die Reaktionsmischung auf ca. 60 °C erkalten, gibt 100 ml Ethanol zu, rührt die so erhaltene Mischung in ein Gemisch aus 500 g Eis und 500 ml konz. Ammoniak ein, rührt 15 min. nach, saugt dann vom ausgefallenen Feststoff ab, wäscht diesen zweimal mit je 100 ml Wasser und saugt diesen trocken. Man nimmt das Rohprodukt in 200 ml Dichlormethan auf, filtriert dieses über eine kurze Kieselgel-Säule, wäscht mit 200 ml Dichlormethan nach, entfernt das Dichlormethan im Vakuum, kristallisiert den Rückstand zweimal aus ca. 600 ml Ethanol um und sublimiert das Produkt abschließend zweimal im Vakuum (p = 1 × 10-5 mbar, T = 160 °C). Ausbeute: 21.2 g (77 mmol), 77 %. Reinheit: > 99.5 % nach 1H-NMR.
-
Darstellung von L23:
-
Ein Gemisch aus 20.9 g (100 mmol) 2-(2-Aminophenyl)-benzimidazol, 47.1 g (350 mmol) 3,3-Dimethylbuttersäurechlorid und 34.8 g (300 mmol) 3,3-Dimethylbuttersäure wird 20 h unter Rückfluss erhitzt. Man lässt die Reaktionsmischung auf ca. 60 °C erkalten, gibt 100 ml Ethanol zu, rührt die so erhaltene Mischung in ein Gemisch aus 500 g Eis und 500 ml konz. Ammoniak ein, rührt 15 min. nach, saugt dann vom ausgefallenen Feststoff ab, wäscht diesen dreimal mit je 100 ml Wasser und saugt diesen trocken. Man nimmt das Rohprodukt in 200 ml Dichlorrmethan auf, filtriert dieses über eine kurze Kieselgel-Säule , wäscht die Kieselgel-Säule mit 200 ml Dichlormethan nach, entfernt das Dichlormethan im Vakuum, kristallisiert den Rückstand zweimal aus ca. 400 ml Ethanol um und sublimiert das Produkt abschließend zweimal im Vakuum (p = 1 × 10-5 mbar, T = 170 °C). Ausbeute: 25.2 g (87 mmol), 87 %. Reinheit: > 99.5 % nach 1H-NMR.
-
Darstellung von L42:
-
Ein Gemisch aus 20.9 g (100 mmol) 2-(2-Aminophenyl)-benzimidazol, 63.9 g (350 mmol) 2,4,6-Trimethylbenzoesäurechlorid und 0.9 ml Wasser wird 24 h unter Rückfluss erhitzt. Man lässt die Reaktionsmischung auf ca. 40 °C erkalten, gibt 50 ml Ethylacetat zu, rührt die so erhaltene Mischung in ein Gemisch aus 500 g Eis und 500 ml konz. Ammoniak ein, rührt 15 min. nach, extrahiert die wässrige Phase dreimal mit je 300 ml Ethylacetat, trocknet die vereinigten organischen Phasen über Natriumsulfat, filtriert die organische Phase über eine kurze Kieselgel-Säule, wäscht die Kieselgel-Säule mit 200 ml Ethylacetat nach, entfernt das Ethylacetat im Vakuum, kristallisiert den Rückstand einmal aus ca. 300 ml Methanol und einmal aus ca. 100 ml Aceton um und sublimiert das Produkt abschließend zweimal im Vakuum (p = 1 × 10-5 mbar, T = 170 °C). Ausbeute: 21.9 g (65 mmol), 65 %. Reinheit: > 99.5 % nach 1H-NMR.
-
1.2) Aus 2-(2-(N-Alkylamido)-phenyl)-benzimidazol-Derivaten Allgemeine Ligandensynthese Variante D:
-
Eine gut gerührte Mischung aus 100 mmol des 2-(2-(N-Alkylamido)-phenyl)-benzimidazol-Derivats und 350 mmol des Carbonsäurechlorids wird 24 bis 100 h unter Rückfluss - bei Carbonsäurechloriden, die unter 150 °C sieden - bzw. auf 150 °C bis 180 °C - bei Carbonsäurechloriden, die über 150 °C sieden - erhitzt, bis das 2-(2-(N-Alkylamido)-phenyl)-benzimidazol-Derivat umgesetzt ist. Falls die Reaktionsmischung zu breiig ist, wird mit einem inerten, im Siedepunkt an das verwendete Carbonsäurechlorid angepassten Lösungsmittel, z. B. Dioxan oder Diethylenglykoldimethylether, verdünnt. Nach Abkühlen wird die Reaktionsmischung in Dioxan (50 - 200 ml) aufgenommen. Die Reaktionsmischung wird unter gutem Rühren in ein Gemisch aus 500 g Eis und 500 ml wässrigem konz. Ammoniak eingerührt. Fällt das Produkt als Feststoff an, wird dieser abgesaugt, mit Wasser gewaschen und trockengesaugt. Fällt das Produkt als Öl an, wird dieses mit drei Portionen zu je 300 ml Ethylacetat extrahiert. Die organische Phase wird abgetrennt, mit Wasser gewaschen und im Vakuum eingeengt. Das Rohprodukt wird in Ethylacetat oder Dichlormethan aufgenommen, über eine kurze Säule aus Alox, basisch, Aktivitätsstufe 1 oder Kieselgel filtriert, um braune Verunreinigungen zu entfernen. Nach zweimaliger Umkristallisation des so erhaltenen Produktes (Methanol, Ethanol, Aceton, Dioxan, etc.) wird dieses durch Kugelrohrdestillation oder Sublimation (p ca. 1 × 10-5 mbar, T ca. 150 - 230 °C) von Leichtsiedern und braunen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
1.3) Aus 6-Chlor-benzo[4,Slimidazo[1,2-c]chinazolin-Derivaten Allgemeine Ligandensynthese Variante E:
-
Ein Gemisch aus 25.4 g (100 mmol) 6-Chlor-benzo[4,5]imidazo[1,2-c]chinazolin, S61 und 200 mmol des Natrium-Alkoholats wird in 200 ml des entsprechenden Alkohol so lang unter Rückfluss erhitzt, bis das 6-Chlorbenzo[4,5]imidazo[1,2-c]chinazolin umgesetzt ist (2-12 h). Man destilliert den Alkohol bzw. das Lösungsmittel ab, nimmt den Feststoff in 300 ml Wasser auf und rührt diesen aus. Nach Absaugen wird der Feststoff zweimal mit 100 ml Wasser und einmal mit 30 ml kaltem Methanol gewaschen und dann im Vakuum getrocknet. Nach zweimaliger Umkristallisation des so erhaltenen Produktes (Ethanol, Aceton, Dioxan, etc.) wird dieses durch Kugelrohrdestillation oder Sublimation (p ca. 1 × 10-5 mbar, T ca. 150 - 230 °C) von Leichtsiedern befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Analog werden foiende Derivate daraestellt:
| Bsp. | Variante | Carbonylkomponenten X = CI [CAS] OH [CAS] | 2-(2-Aminophenyl)-benzimidazol-Derivat bzw. 6-Chlorbenzo[4,5]imidazo[1,2-c]chinazolin | Produkt | Ausbeute |
| L1 | A | | | | 71 % |
| L2 | A | | | | 70 % |
| L3 | B | | | | 74 % |
| L4 | B | | | | 61 % |
| L5 | B | | | | 68 % |
| L6 | A | | | | 41 % |
| L7 | C | | | | 70 % |
| L8 | C | | | | 71 % |
| L9 | B | | | | 65 % |
| L10 | B | | | | 40 % |
| L11 | B | | | | 66 % |
| L12 | B | | | | 60 % |
| L13 | A | | | | 78 % |
| L14 | B | | | | 73 % |
| L15 | B | | | | 73 % |
| L16 | B | | | | 80 % |
| L17 | B | | | | 55 % |
| L18 | B | | | | 51 % |
| L19 | B | | | | 57 % |
| L20 | C | | | | 49 % |
| L21 | B | | | | 65 % |
| L22 | B | | | | 67 % |
| L23 | A | | | | 69 % |
| L24 | A | | | | 66 % |
| L25 | B | | | | 74 % |
| L26 | B | | | | 36 % |
| L27 | B | | | | 58 % |
| L28 | B | | | | 61 % |
| L29 | B | | | | 67 % |
| L30 | B | | | | 65 % |
| L31 | B | | | | 70 % |
| L32 | B | | | | 62 % |
| L33 | C | | | | 55 % |
| L34 | B | | | | 60 % |
| L35 | C | | | | 78 % |
| L36 | C | | | | 45 % |
| L37 | C | | | | 52 % |
| L38 | C | | | | 66 % |
| L39 | C | | | | 28 % |
| L40 | B | | | | 70 % |
| L41 | B | | | | 61 % |
| L42 | B | | | | 48 % |
| L43 | B | | | | 51 % |
| L44 | B | | | | 50 % |
| L45 | B | | | | 38 % |
| L46 | B | | | | 42 % |
| L47 | B | | | | 54 % |
| L48 | B | | | | 30 % |
| L49 | B | | | | 56 % |
| L50 | B | | | | 63 % |
| L51 | B | | | | 66 % |
| L52 | C | | | | 72 % |
| L53 | B | | | | 65 % |
| L54 | B | | | | 58 % |
| L55 | B | | | | 40 % |
| L56 | B | | | | 45 % |
| L57 | C | | | | 50 % |
| L58 | C | | | | 56 & |
| L59 | C | | | | 61 % |
| L60 | C | | | | 57 % |
| L61 | C | | | | 60 % |
| L62 | C | | | | 45 % |
| L63 | B | | | | 55 % |
| L64 | B | | | | 56 % |
| L65 | B | | | | 59 % |
| L66 | B | | | | 66 % |
| L67 | A | | | | 70 % |
| L68 | A | | | | 76 % |
| L69 | C | | | | 68 % |
| L70 | B | | | | 57 % |
| L71 | B | | | | 58 % |
| L72 | C | | | | 73 % |
| L73 | A | | | | 65 % |
| L74 | B | | | | 65 % |
| L75 | C | | | | 44 % |
| L76 | A | | | | 52 % |
| L77 | B | | | | 49 % |
| L78 | A | | | | 51 % |
| L79 | B | | | | 54 % |
| L80 | C | | | | 63 % |
| L81 | A | | | | 74 % |
| L82 | A | | | | 72 % |
| L83 | A | | | | 74 % |
| L84 | C | | | | 56 % |
| L85 | C | | | | 75 % |
| L86 | A | | | | 65 % |
| L87 | C | | | | 61 % |
| L88 | C | | | | 65 % |
| L89 | B | | | | 67 % |
| L90 | B | | | | 35 % |
| L91 | A | | | | 74 % |
| L92 | C | | | | 58 % |
| L93 | C | | | | 72 % |
| L94 | C | | | | 59 % |
| L95 | B | | | | 70 % |
| L96 | A | | | | 68 % |
| L97 | A | | | | 69 % |
| L98 | A | | | | 46 % |
| L99 | A | | | | 55 % |
| L100 | A | | | | 68 % |
| L101 | A | | | | 68 % |
| L102 | A | | | | 71 % |
| L103 | C | | | | 76 % |
| L104 | B | | | | 70 % |
| L91 | D | | | | 74 % |
| L93 | D | | | | 76 % |
| L159 | E | | | | 64 % |
| L 160 | E | | | | 67 % |
| L161 | E | | | | 42 % |
-
2) 2,6a,11-Triaza-benzo[a]fluoren-Systeme
-
-
2.1) Aus 2-(4-Chlor-pyridin-3-yl)-benzimidazolen und terminalen Alkinen
-
Ein Gemisch aus 14.3 g (50 mmol) des 2-(4-Chlor-6-tert-butyl-pyridin-3-yl)-benzimidazol-Derivats, 55 mmol des terminalen Alkins, 191 mg (1 mmol) Kupfer(I)iodid, 112 mg (0.5 mmol) Palladium(II)acetat, 315 mg (1.2 mmol) Triphenylphosphin, 100 ml Triethylamin und 100 ml Dioxan wird 16 h bei 120 °C im Autoklaven gerührt. Nach Erkalten wird das Lösungsmittel im Vakuum entfernt, der Rückstand wird in 300 ml Ethylacetat aufgenommen, die org. Phase wird dreimal mit je 100 ml Wasser und einmal mit 100 ml ges. Kochsalzlösung gewaschen, über Natriumsulfat getrocket und über eine kurze Kieselgel-Säule filtriert. Nach Entfernen des Lösungsmittels im Vakuum wird der Rückstand zweimal aus Methanol umkristallisiert. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 180 - 230 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | Imidazol-Derivat | Alkin | Produkt | Ausbeute |
| L105 | | | | 46 % |
| L106 | | | | 51 % |
| L107 | | | | 33 % |
| L108 | | | | 47 % |
-
3) 4,6a,11-Triaza-benzo[a]fluoren-Systeme
-
-
3.1) Aus 2-(2-Chlor-pyridin-3-yl)-benzimidazolen und terminalen Alkinen
-
Ein Gemisch aus 14.3 g (50 mmol) des 2(2-Chlor-6-tert-butyl-pyridin-3-yl)-benzimidazol-Derivats, 55 mmol des terminalen Alkins, 191 mg (1 mmol) Kupfer(I)iodid, 112 mg (0.5 mmol) Palladium(II)acetat, 315 mg (1.2 mmol) Triphenylphosphin, 100 ml Triethylamin und 100 ml Dioxan wird 16 h bei 120 °C im Autoklaven gerührt. Nach Erkalten wird das Lösungsmittel im Vakuum entfernt, der Rückstand wird in 300 ml Ethylacetat aufgenommen, die organische Phase wird dreimal mit je 100 ml Wasser und einmal mit 100 ml gesättigter Kochsalzlösung gewaschen, über Natriumsulfat getrocknet und über eine kurze Kieselgel-Säule filtriert. Nach Entfernen des Lösungsmittels im Vakuum wird der Rückstand zweimal aus Methanol umkristallisiert. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 180 - 230 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | Imidazol-Derivat | Alkin | Produkt | Ausbeute |
| L109 | | | | 47 % |
| L110 | | | | 31 % |
| L111 | | | | 40 % |
| L112 | | | | 38 % |
-
3.2) Aus 2-(2-Chlor-pyridin-3-yl)-benzimidazolen und internen Alkinen
-
Ein Gemisch aus 14.3 g (50 mmol) des 2(2-Chlor-6-tert-butyl-pyridin-3-yl)-benzimidazol-Derivats, 55 mmol des internen Alkins, 225 mg (1 mmol) Palladium(II)acetat, 1.1 g (4 mmol) Triphenylphosphin, 14 g (100 mmol) Kaliumcarbonat und 200 ml Xylol wird 16 h bei unter Rückfluss erhitzt. Nach Erkalten wird über ein Kieselgel-Bett abfiltriert, mit 500 ml THF nachgewaschen und das Filtrat zur Trockene eingeengt. Der Rückstand wird in 50 ml Essigsäureethylester in der Siedehitze gelöst und langsam mit 200 ml n-Heptan versetzt. Nach Erkalten wird von auskristallisiertem Feststoff abgesaugt, dieser wird zweimal mit je 50 ml n-Heptan gewaschen und im Vakuum getrocknet. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 200 - 250 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | Imidazol-Derivat | Alkin | Produkt | Ausbeute |
| L113 | | | | 40 % |
| L114 | | | | 31 % |
-
3.3) Aus Pyridin-3-carboxaldehyden und 1,2-Diaminobenzolen
-
Eine Lösung von 500 mmol Pyridin-3-carboxaldehyd und 550 mmol 1,2-Diaminobenzol in 1000 ml Nitrobenzol wird in einer Apparatur bestehend aus einem 2000 ml Einhalskolben mit Hahnstück und aufgesetzter Destillationsbrücke platziert und 2 h bei 200 °C gerührt, wobei das gebildete Wasser abdestilliert. Dann steigert man die Temperatur auf ca. 215 °C und destilliert das Nitrobenzol im Argonstrom ab. Gegen Ende der Destillation legt man ein Vakuum von ca. 100 mbar an, um letzte Reste von Nitrobenzol zu entfernen, dann lässt man die Reaktionsmischung erkalten. Fällt das Rohprodukt glasartig an, wird das Glas mechanisch zerkleinert, Öle versetzt man direkt mit 200 - 400 ml Methanol und erhitzt die Mischung unter Rückfluss, wobei sich das Glas bzw. das Öl löst und das Produkt auskristallisiert. Die so erhaltenen Rohprodukte weisen schon eine hohe Reinheit auf (1H-NMR typischerweise 97 - 99 %ig).
-
Gegebenenfalls werden sie erneut umkristallisiert und dann durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 200 - 250 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | Pyridin-3carboxaldehyd | 1,2-Diaminobenzol | Produkt | Ausbeute |
| L109 | | | | 64 % |
| L163 | | | | 66 % |
| L111 | | | | 58 % |
| L165 | | | | 55 % |
| L166 | | | | 55 % |
| L167 | | | | 63 % |
| L168 | | | | 59 % |
| L169 | | | | 67 % |
| L170 | | | | 46 % |
| L171 | | | | 30 % |
| L172 | | | | 23 % |
| L173 | | | | 21 % |
| L174 | | | | 33 % |
| L175 | | chromatographisch isoliert aus der Mutterlauge von L174 | | 14 % |
| L176 | | | | 71 % |
| L177 | | | | 68 % |
| L178 | | | | 59 % |
| L179 | | | | 60 % |
| L180 | | | | 47 % |
| L181 | | | | 65 % |
| L182 | | | | 70 % |
| L183 | | | | 62 % |
| L184 | | | | 53 % |
| L185 | | | | 39 % |
| L186 | | | | 38 % |
| L187 | | | | 27 % |
| L188 | | | | 30 % |
| L189 | | | | 34 % |
| L190 | | | | 39 % |
| L191 | | | | 62 % |
| L192 | | | | 62 % |
| L193 | | | | 57 % |
| L194 | | | | 27 % |
| L195 | | | | 49 % |
| L196 | | | | 28 % |
| L197 | | | | 66 % |
| L198 | | | | 34 % |
| L199 | | | | 69% |
| L200 | | | | 64 % |
| L201 | | | | 60 % |
| L202 | | | | 37 % |
| L203 | | | | 63 % |
| L204 | | | | 67 % |
| L205 | | | | 35 % |
| L206 | | | | 57 % |
| L207 | | | | 51 % |
| L208 | | | | 47 % |
| L209 | | | | 25 % |
| L210 | | | | 22 % |
| L211 | | | | 47 % |
| L212 | | | | 41 % |
| L213 | | | | 45 % |
| L214 | | | | 43 % |
| L215 | | | | 39 % |
-
4) 6a,8,11-Triaza-benzo[a]fluoren-Systeme
-
-
4.1) Aus 1-Amino-iso-chinolin-Derivaten
-
Durchführung analog zu K. T. J. Loones, et al. Tetrahedron 2007, 63, 3818: Eine gut gerührte Mischung von 100 mmol des 1-Amino-isochinolin-Derivats, 37.4 g (110 mmol) 2-tert-Butyl-3-brom-4-iod-pyridin (S27) bzw. 27.9 g (110 mmol) 2-Trifluormethyl-4-iod-5-chlor-pyridin, 35.0 g (250 mmol) Kaliumcarbonat, 200 g Glaskugeln (3 mm Durchmesser), 2.6 g (10 mmol) Triphenylphosphin und 450 mg (2 mmol) Palladium(II)-acetat in 500 ml o-Xylol wird unter Rückfluss erhitzt, bis das 1-Amino-isochinolin-Derivat verbraucht ist (typischerweise 3-30 h). Nach Erkalten wird über ein Kieselgel-Bett abfiltriert, mit 1000 ml THF nachgewaschen und das Filtrat zur Trockene eingeengt. Der Rückstand wird in 100 ml Essigsäureethylester in der Siedehitze gelöst und langsam mit 250 ml n-Heptan versetzt. Nach Erkalten wird vom auskristallisierten Feststoff abgesaugt, dieser wird zweimal mit je 50 ml n-Heptan gewaschen und im Vakuum getrocknet. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 180 - 230 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 1-Aminoisochinolin-Derivat | PyridinDerivat | Produkt | Ausbeute |
| L115 | | | | 61 % |
| L116 | | | | 28 % |
| L117 | | | | 23 % |
| L118 | | | | 34 % |
| L119 | | | | 60 % |
| L120 | | | | 55 % |
| L121 | | | | 58 % |
| L122 | | | | 66 % |
| L123 | | | | 56 % |
-
4.2) Aus 1-Chlor-isochinolin-Derivaten
-
Eine gut gerührte Mischung von 100 mmol des 1-Chlor-iso-chinolin-Derivats, 29.8 g (130 mmol) 3-Brom-4-amino-6-tert-butyl-pyridin (S40), 35.0 g (250 mmol) Kaliumcarbonat, 200 g Glaskugeln (3 mm Durchmesser), 2.6 g (10 mmol) Triphenylphosphin und 450 mg (2 mmol) Palladium(II)acetat in 500 ml o-Xylol wird unter Rückfluss erhitzt, bis das 1-Chlor-isochinolin-Derivat verbraucht ist (typischerweise 3-30 h). Nach Erkalten wird über ein Kieselgel-Bett abfiltriert, mit 1000 ml THF nachgewaschen und das Filtrat zur Trockene eingeengt. Der Rückstand wird in 75 ml Essigsäureethylester in der Siedehitze gelöst und langsam mit 250 ml n-Heptan versetzt. Nach Erkalten wird vom auskristallisierten Feststoff abgesaugt, dieser wird zweimal mit je 50 ml n-Heptan gewaschen und im Vakuum getrocknet. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 150 - 230 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 1-Chlor-isochinolin-Derivat | Pyridin | Produkt | Ausbeute |
| L124 | | | | 73 % |
| L125 | | | | 67 % |
| L126 | | | | 73 % |
| L127 | | | | 58 % |
| L128 | | | | 61 % |
| L129 | | | | 65 % |
| L130 | | | | 46 % |
| L131 | | | | 50 % |
-
5) 4,6a,8,11-Tetraaza-benzo[a]fluoren-Systeme
-
-
5.1) Aus 5-Chlor-1,6-naphthyridin-Derivaten
-
Eine gut gerührte Mischung von 100 mmol des 5-Chlor-1,6-naphthyridin - Derivats, 29.8 g (130 mmol) 2-tert-Butyl-4-amino-5-brom-pyridin (S40), 35.0 g (250 mmol) Kaliumcarbonat, 200 g Glaskugeln (3 mm Durchmesser), 2.6 g (10 mmol) Triphenylphosphin und 450 mg (2 mmol) Palladium(II)acetat in 500 ml o-Xylol wird unter Rückfluss erhitzt, bis das 5-Chlor-1,6-naphthyridin-Derivat verbraucht ist (typischerweise 3-30 h). Nach Erkalten wird über ein Kieselgel-Bett abfiltriert, mit 1000 ml THF nachgewaschen und das Filtrat zur Trockene eingeengt. Der Rückstand wird in 75 ml Essigsäureethylester in der Siedehitze gelöst und langsam mit 250 ml n-Heptan versetzt. Nach Erkalten wird vom auskristallisierten Feststoff abgesaugt, dieser wird zweimal mit je 50 ml n-Heptan gewaschen und im Vakuum getrocknet. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 180 - 230 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 5-Chlor-1,6-Naphthyridin-Derivaten | Pyridin | Produkt | Ausbeute |
| L132 | | | | 73 % |
| L133 | | | | 67 % |
-
5.2) Aus 5-Amino-1,6-naphthyridin-Derivaten
-
Eine gut gerührte Mischung von 100 mmol des 5-Amino-1,6-Naphthyridin-Derivats, 37.4 g (110 mmol) 2-tert-Butyl-3-brom-4-iod-pyridin (S27) bzw. 27.9 g (110 mmol) 2-Trifluormethyl-4-iod-5-chlor-pyridin [823221-95-0], 35.0 g (250 mmol) Kaliumcarbonat, 200 g Glaskugeln (3 mm Durchmesser), 2.6 g (10 mmol) Triphenylphosphin und 450 mg (2 mmol) Palladium(II)acetat in 500 ml o-Xylol wird unter Rückfluss erhitzt, bis das 5-Amino-1,6-naphthyridin-Derivat verbraucht ist (typischerweise 3-30 h). Nach Erkalten wird über ein Kieselgel-Bett abfiltriert, mit 1000 ml THF nachgewaschen und das Filtrat zur Trockene eingeengt. Der Rückstand wird in 75 ml Essigsäureethylester in der Siedehitze gelöst und langsam mit 250 ml n-Heptan versetzt. Nach Erkalten wird vom auskristallisierten FS abgesaugt, dieser wird zweimal mit je 50 ml n-Heptan gewaschen und im Vakuum getrocknet. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 180 - 230 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 5-Amino-1,6-naphthyridin-Derivat | PyridinDerivat | Produkt | Ausbeute |
| L134 | | | | 67 % |
| L135 | | | | 50 % |
| L136 | | | | 47 % |
-
6) 2,6a,8,11-Tetraaza-benzo[a]fluoren-Systeme
-
-
Eine gut gerührte Mischung von 20.1 g (100 mmol) 1-Amino-6-tert-butyl-2,7-naphthyridin S41, 37.4 g (110 mmol) 2-tert-Butyl-3-brom-4-iod-pyridin (S27) bzw. 27.9 g (110 mmol) 2-Trifluormethyl-4-iod-5-chlor-pyridin, 35.0 g (250 mmol) Kaliumcarbonat, 200 g Glaskugeln (3 mm Durchmesser), 2.6 g (10 mmol) Triphenylphosphin und 450 mg (2 mmol) Palladium(II)acetat in 500 ml o-Xylolwird unter Rückfluss erhitzt, bis das 1-Amino-6-tert-butyl-2,7-naphthyridin verbraucht ist (typischerweise 3-30 h). Nach Erkalten wird über ein Kieselgel-Bett abfiltriert, mit 1000 ml THF nachgewaschen und das Filtrat zur Trockene eingeengt. Der Rückstand wird in 75 ml Essigsäureethylester in der Siedehitze gelöst und langsam mit 250 ml n-Heptan versetzt. Nach Erkalten wird vom auskristallisierten Feststoff abgesaugt, dieser wird zweimal mit je 50 ml n-Heptan gewaschen und im Vakuum getrocknet. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 170 - 200 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 1-Amino-6-tert-butyl-2,7-naphthyridin | PyridinDerivat | Produkt | Ausbeute |
| L137 | | | | 56 % |
| L138 | | | | 48 % |
-
7) 4,5,6a,11-Tetraaza-benzo[alf1uoren-Systeme
-
-
Ein Gemisch aus 100 mmol des 2-(2-Amino-pyridin-3-yl)-benzimidazol-Derivats (eingesetzt als äquimolares Gemisch mit dem entsprechenden 2-tert-Butyl-benzimidazol-Derivat wie aus Synthese 16) erhalten) und 1 mol des entsprechenden Carbonsäurechlorids wird 8 bis 40 h unter Rückfluss bei Carbonsäurechloriden, die unter 150 °C sieden, bzw. auf 150 °C bis 180 °C bei Carbonsäurechloriden, die über 150 °C sieden, erhitzt, bis das 2-(2-Amino-pyridin-3-yl)-benzimidazol-Derivat umgesetzt ist. Man lässt die Reaktionsmischung auf 80 °C abkühlen, verdünnt gegebenenfalls mit 100 ml Dioxan, rührt dann in ein Gemisch aus 500 ml konz. Ammoniak-Lösung und 500 g Eis ein und rührt 3 h nach. Anschließend saugt man vom Feststoff ab, wäscht diesen zweimal mit je 100 ml Wasser und trocknet im Vakuum. Man nimmt den Feststoff in 1000 ml Ethylacetat auf, filtriert über eine kurze Kieselgel-Säule, wäscht diese mit 500 ml Ethylacetat nach, entfernt das Ethylacetat im Vakuum und kristallisiert den braunen Rückstand aus Methanol um. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 170 - 200 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 %ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 2-(2-Aminopyridin-3-yl)-benzimidazol-Derivat | Carbonsäure chlorid | Produkt | Ausbeute |
| L139 | | | | 38 % |
| L140 | | | | 35 % |
| L141 | | | | 41 % |
| L142 | | | | 27 % |
-
8) 2,5,6a,11-Tetraaza-benzo[a]fluoren-Systeme
-
-
Ein Gemisch aus 26.6 g (100 mmol) 2-(4-Amino-6-tert-butyl-pyridin-3-yl)-benzimidazol (eingesetzt als äquimolares Gemisch mit 2-tert-Butyl-benzimidazol wie aus Synthese 18) erhalten) und 1 mol des entsprechenden Carbonsäurechlorids wird 8 bis 40 h unter Rückfluss bei Carbonsäurechloriden, die unter 150 °C sieden, bzw. auf 150 °C bis 180 °C, bei Carbonsäurechloriden, die über 150 °C sieden, erhitzt, bis das 2-(4-Amino-pyridin-3-yl)-benzimidazol-Derivat umgesetzt ist. Man lässt die Reaktionsmischung auf 80 °C abkühlen, verdünnt gegebenenfalls mit 100 ml Dioxan, rührt dann in ein Gemisch aus 500 ml konz. Ammoniak-Lösung und 500 g Eis ein und rührt 3 h nach. Anschließend saugt man vom Feststoff ab, wäscht diesen zweimal mit je 100 ml Wasser und trocknet im Vakuum. Man nimmt den Feststoff in 1000 ml Ethylacetat auf, filtriert über eine kurze Kieselgel-Säule, wäscht diese mit 500 ml Ethylacetat nach, entfernt das Ethylacetat im Vakuum und kristallisiert den braunen Rückstand aus Methanol um. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 180 - 220 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 2-(4-Aminopyridin-3-yl)-benzimidazol-Derivat | Carbonsäure chlorid | Produkt | Ausbeute |
| L143 | | | | 40 % |
| L144 | | | | 39 % |
-
9) 2,4,6a,11-Tetraaza-benzo[alfIuoren-Systeme
-
-
Darstellung analog zu N. Umeda et al., Angew. Chem Int. Ed. 2008, 47, 4019: In einem Druckschlenkrohr wird eine Lösung von 25.2 g (100 mmol) 2-(2-tert-Butyl-pyrimidin-5-yl)-benzimidazol (S48) und 110 mmol des Alkins in 400 ml DMF vorgelegt, mit 1.5 g (4 mmol) Tetraphenylcyclopentadien, 547 mg (1 mmol) Pentamethylcyclopentadienyl-rhodiumchlorid-dimer und 21.0 (105 mmol) Kupfer(II)acetat-monohydrat versetzt und 18 h bei 100 °C verschlossen gerührt. Nach Erkalten wird das DMF im Vakuum entfernt, der Rückstand wird in 1000 ml THF aufgenommen und über eine kurze Kieselgel-Säule filtriert. Nach Entfernen des THF im Vakuum wird der ölige Rückstand in heißem Methanol (ca. 75 ml) aufgenommen. Nach Erkalten saugt man von den gebildeten Kristallen ab und kristallisiert diese erneut aus Methanol unter Zusatz von wenig Ethylacetat um. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10
-5 mbar, T ca. 180 - 220 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach
1H-NMR typischerweise > 99.5 % ig.
| Bsp. | 2-(2-tert-Butylpyrimidin-5-yl)-benzimidazol | Alkin | Produkt | Ausbeute |
| L145 | | | | 31 % |
| L146 | | | | 26 % |
-
10) 5,6a,8,11 -Tetraaza-benzo[a]fluoren-Systeme
-
-
Eine gut gerührte Mischung von 100 mmol des 4-Chlor-chinazolin-Derivats, 29.8 g (130 mmol) 3-Brom-4-amino-6-tert-butyl-pyridin (S40), 35.0 g (250 mmol) Kaliumcarbonat, 200 g Glaskugeln (3 mm Durchmesser), 2.6 g (10 mmol) Triphenylphosphin und 450 mg (2 mmol) Palladium(II)acetat in 500 ml o-Xylol wird unter Rückfluss erhitzt, bis das 4-Chlor-chinazolin-Derivat verbraucht ist (typischerweise 16 h). Nach Erkalten wird über ein Kieselgel-Bett abfiltriert, mit 1000 ml THF nachgewaschen und das Filtrat zur Trockene eingeengt. Der Rückstand wird in 75 ml Essigsäureethylester in der Siedehitze gelöst und langsam mit 250 ml n-Heptan versetzt. Nach Erkalten wird vom auskristallisierten Feststoff abgesaugt, dieser wird zweimal mit je 50 ml n-Heptan gewaschen und im Vakuum getrocknet und anschließend an Kieselgel (Heptan : Ethylacetat, 3:1 vv) gesäult. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 150 - 230 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 4-Chlor-chinazolin-Derivat | Pyridin | Produkt | Ausbeute |
| L147 | | | | 33 % |
| L148 | | | | 41 % |
-
11) 4,5,6a,8,11-Pentaaza-benzo[a]fluoren
-
-
Darstellung analog zu 7), wobei anstatt 100 mmol des 2-(2-Amino-pyridin-3-yl)-benzimidazol-Derivats 32,3 g (100 mmol) 2-(2-Amino-6-tert-butylpyridin-3-yl)-6-tert-butyl-3H-imidazo-[4,5-c]pyridin (S51) (eingesetzt als äquimolares Gemisch mit 2,6-Di-tert-butyl-3H-imidazo-[4,5-c]pyridin wie aus Synthese 22) erhalten) eingesetzt werden. Nach Kristallisation aus Methanol werden die Rohprodukte an Kieselgel (Heptan:Ethylacetat, 3:1 vv) gesäult. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 200 - 240 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 2-(2-Aminopyridin-3-yl)-3H-imidazo-[4,5-c]pyridinDerivat | Carbonsäure chlorid | Produkt | Ausbeute |
| L149 | | | | 21 % |
| L150 | | | | 24 % |
-
12) 2,4,6a,8,11-Pentaaza-benzo[a]fluoren
-
-
Darstellung analog 9), wobei anstatt 25.2 g (100 mmol) 2-(2-tert-Butylpyrimidin-5-yl)-benzimidazol (S48) 30.9 g (100 mmol) 2-(2-tert-butylpyrimidin-5-yl)-6-tert-butyl-3H-imidazo[4,5-c]pyridin (S52) eingesetzt werden. Nach Kristallisation aus Methanol werden die Rohprodukte an Kieselgel (Heptan:Ethylacetat, 3:1 vv) gesäult. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 200 - 240 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 2-(Pyrimidin-5-yl)-3H-imidazo-[4,5-c]pyridinDerivat | Alkin | Produkt | Ausbeute |
| L151 | | | | 19 % |
| L152 | | | | 16 % |
-
13) 2,5,63,8,11 -Pentaaza-benzo[a]fluoren
-
-
Darstellung analog zu 7), wobei anstatt 100 mmol des 2-(2-Amino-pyridin-3-yl)-benzimidazol-Derivats 32.3 g (100 mmol) 2-(4-Amino-6-tert-butylpyridin-3-yl)-6-tert-butyl-3H-imidazo-[4,5-c]pyridin (S53) (eingesetzt als äquimolares Gemisch mit 2,6-Di-tert-butyl-3H-imidazo-[4,5-c]pyridin wie aus Synthese 24) erhalten) eingesetzt werden. Nach Kristallisation aus Methanol werden die Rohprodukte an Kieselgel (Heptan:Ethylacetat, 3:1 vv) gesäult. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 200 - 240 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 2-(4-Aminopyridin-3-yl)-3H-imidazo-[4,5-c]pyridinDerivat | Carbonsäurechlorid | Produkt | Ausbeute |
| L153 | | | | 25 % |
| L154 | | | | 27 % |
-
14)) 2,4,5,6a,11-Pentaaza-benzo[a]fluoren
-
-
Darstellung analog zu 7), wobei anstatt 100 mmol des 2-(2-Amino-pyridin-3-yl)-benzimidazol-Derivats 26.7 g (100 mmol) 2-(2-tert-Butyl-4-aminopyrimidin-5-yl)benzimidazol (S55) (eingesetzt als äquimolares Gemisch mit 2-tert-Butyl-benzimidazol wie aus Synthese 26) erhalten) eingesetzt werden. Nach Kristallisation aus Methanol werden die Rohprodukte an Kieselgel (Heptan:Ethylacetat, 3:1 vv) gesäult. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 200 - 240 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 2-(4-aminopyrimidin-5-yl)-benzimidazol-Derivat | Carbonsäurechlorid | Produkt | Ausbeute |
| L155 | | | | 20 % |
| L156 | | | | 25 % |
-
15) 2,4,5,6a,8,11-Hexaaza-benzo[a]fluoren
-
-
Darstellung analog zu 7), wobei anstatt 100 mmol des 2-(2-Amino-pyridin-3-yl)-benzimidazol-Derivats 32.4 g (100 mmol) 2-(2-tert-Butyl-4-aminopyrimidin-5-yl)-6-tert-butyl-3H-imidazo-[4,5-c]pyridin (S56) (eingesetzt als äquimolares Gemisch mit 2,6-Di-tert-butyl-3H-imidazo-[4,5-c]pyridin wie aus Synthese 27) erhalten) eingesetzt werden. Nach Kristallisation aus Methanol werden die Rohprodukte an Kieselgel (Heptan:Ethylacetat, 3:1 vv) gesäult. Die so erhaltenen Feststoffe werden durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 200 - 240 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Reinheit nach 1H-NMR typischerweise > 99.5 % ig.
-
Es werden folgende Derivate dargestellt:
| Bsp. | 2-(4-Aminopyrimidin-5-yl)-3H-imidazo-[4,5-c]pyridinDerivat | Carbonsäurechlorid | Produkt | Ausbeute |
| L157 | | | | 27 % |
| L158 | | | | 19 % |
-
16) Tetradentate Liganden
-
-
A) 9,9'-Dibrom-3,6,8,3',6',8'-hexa-tert-butyl-[10,10]bi[4,6a,11-triazabenzo[a]fluorenyl], S76
-
-
Eine auf 100 °C erwärmte Lösung von 77.3 g (100 mmol) 3,6,8,3',6',8'-Hexa-tert-butyl-[10,10]bi[4,6a,11-triazabenzo[a]fluorenyl], L213 in 300 ml DMF wird portionsweise mit 39.2 g (220 mmol) NBS versetzt und anschließend 6 h nachgerührt. Man engt die Reaktionsmischung im Vakuum auf ca. 100 ml ein, versetzt tropfenweise mit 200 ml Methanol, rührt 2 h nach, saugt dann von den ausgefallenen Kristallen ab und wäscht diese abschließend zweimal mit je 50 ml Methanol. Ausbeute: 67.0 g (72 mmol), 72 %. Reinheit: 97 % nach 1H-NMR.
-
B) L216, X = S
-
Ein auf -78 °C gekühlte Lösung von 23.3 g (25 mmol) 9,9'-Dibrom-3,6,8,3',6',8'-hexa-tert-butyl-[10,10]bi[4,6a,11-triazabenzo[a]fluorenyl] in 1000 THF wird unter Rühren tropfenweise mit 22.0 ml (55 mmol) n-Butyllithium (2.5 M in Hexan) versetzt und anschließend 1 h nachgerührt. Dann tropft man ein Gemisch aus 2.8 ml (35 mmol) Dischwefeldichlorid und 50 ml THF zu. Nach langsamem Erwärmen auf Raumtemperatur entfernt man das THF im Vakuum, rührt den Rückstand einmal mit 200 ml heißem Methanol aus und kristallisiert dann zweimal aus DMF um. Der Feststoff wird durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 340 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Ausbeute: 8.4 g (10.5 mmol), 42 %. Reinheit: 99 % nach 1H-NMR.
-
C) L217, X = CH2
-
Ein auf -78 °C gekühlte Lösung von 23.3 g (25 mmol) 9,9'-Dibrom-3,6,8,3',6',8'-hexa-tert-butyl-[10,10]bi[4,6a,11-triazabenzo[a]fluorenyl] in 1000 THF wird unter Rühren tropfenweise mit 22.0 ml (55 mmol) n-Butyllithium (2.5 M in Hexan) versetzt und anschließend 1 h nachgerührt. Dann tropft man ein Gemisch aus 2.7ml (35 mmol) Chlorameisensäuremethylester und 50 ml THF zu. Nach langsamem Erwärmen auf Raumtemperatur entfernt man das THF im Vakuum. Man nimmt den Feststoff in 100 ml Diethylenglykol auf, setzt 4 ml Hydrazin-Hydrat zu und erhitzt langsam am Wasserabscheider auf 190 °C. Nach 16 h lässt man auf Raumtemperatur erkalten, verdünnt mit 50 ml Methanol, saugt von den ausgefallenen Kristallen ab, wäscht diese dreimal mit je 30 ml Methanol und kristallisiert zweimal aus DMF um. Der Feststoff wird durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 340 °C) von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Ausbeute: 7.3 g (9.3 mmol), 39 %. Reinheit: 99 % nach 1H-NMR.
-
17 Hexadentate Liganden
-
-
A) 10-Brom-3,6-di-tert-butyl-8,9-dimethyl-4,6a,11-triazabenzo[a]fluoren, S77
-
-
Eine auf 100 °C erwärmte Lösung von 36.0 g (100 mmol) 3,6-Di-tert-butyl-8,9-dimethyl-4,6a,11-triaza-benzo[a]fluoren, L111 in 300 ml DMF wird portionsweise mit 19.6 g (110 mmol) NBS versetzt und anschließend 6 h nachgerührt. Man engt die Reaktionsmischung im Vakuum auf ca. 150 ml ein, rührt 2 h nach, saugt von den ausgefallenen Kristallen ab und wäscht diese abschließend zweimal mit je 50 ml Methanol. Ausbeute: 33.8 g (77 mmol), 77 %. Reinheit: 97 % nach 1H-NMR.
-
B) 10-Hydroxy-3,6-di-tert-butyl-8,9-dimethyl-4,6a,11-triazabenzo[a]fluoren, S78
-
-
Eine auf -78 °C gekühlte Lösung von 13.2 g (30 mmol) 10-Brom-3,6-ditert-butyl-8,9-dimethyl-4,6a,11-triaza-benzo[a]fluoren in 300 ml THF wird unter gutem Rühren tropfenweise mit 13.2 ml (33 mmol) n-Butyllithium (2.5 M in Hexan) versetzt und 30 min. nachgerührt. Zu dieser Lösung gibt man 4.7 ml (42 mmol) Trimethylborat auf ein Mal zu, rührt 1 h nach und lässt dann auf Raumtemperatur erwärmen. Man entfernt das Lösungsmittel im Vakuum, nimmt den Rückstand in 1000 ml Ethylacetat auf, kühlt die Lösung auf 5 °C ab, versetzt unter gutem Rühren mit 19 ml wässriger H2O2-Lösung (30 Gew.-%) und tropft dann eine Lösung von 825 mg NaOH in 20 ml Wasser zu. Nach 3 h Rühren gibt man 300 ml gesättigte Ammoniumchloridlösung zu, trennt die organische Phase ab, wäscht diese zweimal mit je 200 ml Wasser, trocknet über Magnesiumsulfat und engt die org. Phase dann im Vakuum auf ein Volumen von ca. 50 ml ein. Man versetzt den Kristallbrei mit 200 ml Methanol, saugt ab, wäscht die Kristalle einmal mit 50 ml Methanol und Trocknet im Vakuum.
Ausbeute: 8.1 g (22 mmol), 72 %. Reinheit: 95 % nach 1H-NMR.
-
C) L218
-
Eine Suspension von 5.63 g (15 mmol) 10-Hydroxy-3,6-di-tert-butyl-8,9-dimethyl-4,6a,11-triaza-benzo[a]fluoren in einem Gemisch aus 100 ml Toluol und 50 ml Methanol wird mit 0.5 ml einer 1N Natriummethanolat-Lösung in Methanol versetzt und 1h bei 50 °C gerührt. Dann gibt man 681 mg Trimethoxymethylsilan zu, rührt 2 h nach und destilliert dann das Methanol langsam ab, steigert dann die Temperatur, bis auch das Toluol komplett abdestilliert ist. Gegen Ende legt man ein Vakuum an, um letzte Reste an Toluol zu entfernen. Der so erhaltene farblose Schaum wird ohne Reinigung weiter umgesetzt. Ausbeute: 5.82 g (5 mmol), quantitativ. Reinheit: 90 % nach 1H-NMR.
-
18) Makrocyclische tetradentate Liganden, L219
-
-
A) 6-tert-Butyl-2-chloro-5-methyl-nicotinonitril, S79
-
-
Durchführung analog 5.1, wobei anstelle von 128.8 g (560 mmol) 3-Brom-6-trimethylsilylpyridin 85.4 g (560 mmol) 2-Chlor-3-cyano-5-methyl-pyridin [66909-34-0] eingesetzt wird. Ausbeute: 78.4 g (376 mmol), 67 %. Reinheit: > 95 % nach 1H-NMR.
-
B) 6-tert-Butyl-2-(3,3-dimethyl-but-1-ynyl)-5-methyl-nicotinonitril, S80
-
-
Durchführung analog 31, wobei anstelle von 194.7 g (1 mol) 2-Chlor-3-cyano-6-tert-butyl-pyridin, S28 62.6 g (300 mmol) 6-tert-Butyl-2-chloro-5-methyl-nicotinonitril, S79 eingesetzt und die restlichen Reagenzien entsprechend molar skaliert werden. Ausbeute: 68.9 g (271 mmol), 90 %. Reinheit: > 95 % nach 1H-NMR.
-
c) 6-tert-Butyl-2-(3,3-dimethyl-but-1 -ynyl)-5-methyl-pyridin-3-carboxaldehyd, S81
-
-
Durchführung analog 32, wobei anstelle von 72.1 g (300 mmol) 6-tert-Butyl-2-(3,3-dimethyl-but-1-ynyl)-nicotinonitril, S62 63.6 g (250 mmol) 6-tert-Butyl-2-(3,3-dimethyl-but-1-ynyl)-5-methyl-nicotinonitril, S80 eingesetzt und die restlichen Reagenzien entsprechend molar skaliert werden. Ausbeute: 57.7 g (224 mmol), 90 %. Reinheit: > 95 % nach 1H-NMR.
-
D) 3,6,8,3',6',8'-Hexa-tert-butyl-2,2'-dimethyl-[10,10]bi[4,6a,11-triazabenzo[a]fluorenyl], S82
-
-
Durchführung analog 3.3, L213. Ausbeute 38 %.
-
E) L219
-
Eine auf -5 °C gekühlte Suspension von 40.0 g (50 mmol) 3,6,8,3',6',8'-Hexa-tert-butyl-2,2'-dimethyl-[10,10]bi[4,6a, 11-triazabenzo[a]fluorenyl], S82 in 1000 ml Diethylether wird mit 200 g Glasperlen (5 mm Durchmesser) versetzt. Unter gutem Rühren tropft man langsam 40 ml (100 mmol) n-Butyllithium (2.5 M in n-Hexan) zu, rührt 30 min. nach und gibt dann auf ein Mal 10.3 ml (120 mmol) 1,2-Dibromethan zu und lässt unter Rühren auf Raumtemperatur erwärmen. Man gibt 50 ml Ethanol zu, dekantiert von den Glasperlen ab, wäscht die org. Phase einmal mit 200 ml Wasser und engt im Vakuum auf etwa 200 ml ein. Nach Zugabe von 100 ml Methanol saugt man vom Feststoff ab, wäscht diesen zweimal mit 200 ml Methanol und trocknet im Vakuum. Man kristallisiert den Feststoff dreimal aus DMF um, durch Sublimation (p ca. 1 × 10-5 mbar, T ca. 350 °C) wird er von Leichtsiedern und nicht flüchtigen Nebenkomponenten befreit. Ausbeute: 20.8 g (26 mmol), 52 %. Reinheit: 99 % nach 1H-NMR.
-
C. Synthese der Metallkomplexe
-
1) Homoleptische tris-faciale Iridium-Komplexe:
-
Variante A: Tris-acetylacetonato-iridium(III) als Iridium-Edukt
-
Ein Gemisch aus 10 mmol Tris-acetylacetonato-iridium(III) [15635-87-7] und 60 mmol des Liganden L wird unter Vakuum (10-5 mbar) in eine 50 ml Glasampulle abgeschmolzen. Die Ampulle wird für die angegebene Zeit bei der angegebenen Temperatur getempert, wobei das aufgeschmolzene Gemisch mit Hilfe eines Magnetrührers gerührt wird. Nach Erkalten (ACHTUNG: die Ampullen stehen meist unter Druck!) wird die Ampulle geöffnet, der Sinterkuchen wird mit 100 g Glaskugeln (3 mm Durchmessser) in 100 ml des angegebenen Suspensionsmittels 3 h gerührt und dabei mechanisch aufgeschlossen. Man dekantiert die feine Suspension von den Glaskugeln ab, saugt den Feststoff ab und trocknet diesen im Vakuum. Der trockene Feststoff wird in einem Heißextraktor auf einem 10 cm hohen Alox-Bett (Alox, basisch Aktivitätsstufe 1) platziert und dann mit dem angegebenen Extraktionsmittel (Vorlagemenge ca. 500 ml) extrahiert. Nach beendeter Extraktion wird das Extraktionsmittel im Vakuum auf ca. 100 ml eingeengt. Metallkomplexe, die im Extraktionsmittel eine zu gute Löslichkeit aufweisen, werden durch Zutropfen von 200 ml Methanol zur Kristallisation gebracht. Der Feststoff der so erhaltenen Suspensionen wird abgesaugt, einmal mit ca. 50 ml Methanol gewaschen und getrocknet. Nach Trocknen wird die Reinheit des Metall-Komplexes mittels NMR und / oder HPLC bestimmt. Liegt die Reinheit unter 99.5 % wird der Heißextraktionsschritt wiederholt, ist eine Reinheit von 99.5 - 99.9 % erreicht, wird der Metallkomplex getempert oder sublimiert. Das Tempern erfolgt im Hochvakuum (p ca. 10-6 mbar) im Temperaturbereich von 200 - 300 °C. Die Sublimation erfolgt im Hochvakuum (p ca. 10-6 mbar) im Temperaturbereich von ca. 320 bis ca. 400 °C, wobei die Sublimation bevorzugt in Form einer fraktionierten Sublimation durchgeführt wird. Bei Liganden der Punktgruppe C1 fallen die abgeleiteten Metallkomplexe als Diastereomerenmischung an.
-
Variante B: Tris-(2,2,6,6-tetramethyl-3,5-heptandionato)iridium als Iridium-Edukt
-
Durchführung analog zu Variante A, wobei anstelle von 10 mmol Trisacetylacetonato-iridium(III) [15635-87-7] 10 mmol Tris-(2,2,6,6-tetramethyl-3,5-heptandionato)iridium eingesetzt werden.
| Bsp. | Ligand L | Ir-Komplex | Variante Reaktionstemp./ Reaktionszeit Suspensionsmittel Extraktionsmittel | Ausbeute |
| Ir(L1)3 | L1 | Ir(L1)3 | A 270 °C / 100 h DCM THF | 46 % |
| Ir(L2)3 | L2 | Ir(L2)3 | wie Bsp. Ir(L1)3. | 51 % |
| Ir(L3)3 | L3 | Ir(L3)3 | wie Bsp. Ir(L13) | 38 % |
| Ir(L4)3 | L4 | Ir(L4)3 | wie Bsp. Ir(L1)3 | 37 % |
| Ir(L5)3 | L5 | Ir(L5)3 | A
270 °C 1100 h
DCM / EtOH, 2:1
THF | 46 % |
| Ir(L6)3 | L6 | Ir(L6)3 | wie Bsp. Ir(L5)3 | 18 % |
| Ir(L7)3 | L7 | Ir(L7)3 | wie Bsp. Ir(L1)3 | 31 % |
| Ir(L8)3 | L8 | Ir(L8)3 | wie Bsp. Ir(L1)3 | 47 % |
| Ir(L9)3 | L9 | Ir(L9)3 | wie Bsp. Ir(L1)3 | 30 % |
| Ir(L10)3 | L10 | Ir(L10)3 | wie Bsp. Ir(L1)3 | 35 % |
| Ir(L11)3 | L11 | Ir(L11)3 | wie Bsp. Ir(L1)3 | 36 % |
| Ir(L12)3 | L12 | Ir(L12)3 | wie Bsp. Ir(L1)3 | 39 % |
| Ir(L 13)3 | L13 | Ir(L13)3 | wie Bsp. Ir(L1)3 | 42 % |
| Ir(L13)3 | L13 | Ir(L13)3 | B
270 °C / 120 h
DCM
THF | 52 % |
| Ir(L14)3 | L14 | Ir(L14)3 | wie Bsp. Ir(L5)3 | 41 % |
| Ir(L15)3 | L15 | Ir(L15)3 | wie Bsp. Ir(L5)3 | 41 % |
| Ir(L16)3 | L16 | Ir(L16)3 | wie Bsp. Ir(L5)3 | 40 % |
| Ir(L17)3 | L17 | Ir(L 17)3 | wie Bsp. Ir(L5)3 | 35 % |
| Ir(L18)3 | L18 | Ir(L18)3 | wie Bsp. Ir(L5)3 | 22 % |
| Ir(L19)3 | L19 | Ir(L19)3 | wie Bsp. Ir(L5)3 | 33 % |
| Ir(L20)3 | L20 | Ir(L20)3 | wie Bsp. Ir(L5)3 | 27 % |
| Ir(L21)3 | L21 | Ir(L21)3 | wie Bsp. Ir(L5)3 | 36 % |
| Ir(L22)3 | L22 | Ir(L22)3 | wie Bsp. Ir(L5)3 | 41 % |
| Ir(L23)3 | L23 | Ir(L23)3 | wie Bsp. Ir(L5)3 | 50% |
| Ir(L24)3 | L24 | Ir(L24)3 | wie Bsp. Ir(L5)3 | 30 % |
| Ir(L25)3 | L25 | Ir(L25)3 | wie Bsp. Ir(L5)3 | 32 % |
| Ir(L26)3 | L26 | Ir(L26)3 | wie Bsp. Ir(L5)3 | 35 % |
| Ir(L27)3 | L27 | Ir(L27)3 | wie Bsp. Ir(L5)3 | 38 % |
| Ir(L28)3 | L28 | Ir(L28)3 | wie Bsp. Ir(L5)3 | 48 % |
| Ir(L29)3 | L29 | Ir(L29)3 | wie Bsp. Ir(L5)3 | 20 % |
| Ir(L30)3 | L30 | Ir(L30)3 | wie Bsp. I.r(L5)3 | 26 % |
| Ir(L31)3 | L31 | Ir(L31)3 | wie Bsp. Ir(L5)3 | 34 % |
| Ir(L32)3 | L32 | Ir(L32)3 | wie Bsp. Ir(L5)3 | 37 % |
| Ir(L33)3 | L33 | lr(L33)3 | wie Bsp. Ir(L5)3 | 17 % |
| Ir(L34)3 | L34 | Ir(L34)3 | wie Bsp. Ir(L5)3 | 41 % |
| Ir(L35)3 | L35 | Ir(L35)3 | wie Bsp. Ir(L5)3 | 43 % |
| Ir(L36)3 | L36 | Ir(L36)3 | wie Bsp. Ir(L1)3 | 35 % |
| Ir(L37)3 | L37 | Ir(L37)3 | wie Bsp. Ir(L1)3 | 30 % |
| Ir(L38)3 | L38 | Ir(L38)3 | wie Bsp. Ir(L1)3 | 32 % |
| Ir(L39)3 | L39 | Ir(L39)3 | wie Bsp. Ir(L5)3 | 9 % |
| Ir(L40)3 | L40 | Ir(L40)3 | wie Bsp. Ir(L1)3 | 27 % |
| Ir(L41)3 | L41 | Ir(L41)3 | wie Bsp. Ir(L1)3 | 36 % |
| Ir(L42)3 | L42 | Ir(L42)3 | wie Bsp. tr(L1)3 | 35 % |
| Ir(L43)3 | L43 | Ir(L43)3 | wie Bsp. Ir(L5)3 | 38 % |
| Ir(L44)3 | L44 | Ir(L44)3 | wie Bsp. Ir(L5)3 | 40 % |
| Ir(L45)3 | L45 | Ir(L45)3 | wie Bsp. Ir(L5)3 | 45 % |
| Ir(L46)3 | L46 | Ir(L46)3 | wie Bsp. Ir(L5)3 | 40 % |
| Ir(L47)3 | L47 | Ir(L47)3 | wie Bsp. Ir(L5)3 | 42 % |
| Ir(L48)3 | L48 | Ir(L48)3 | wie Bsp. Ir(L5)3 | 38 % |
| Ir(L49)3 | L49 | Ir(L49)3 | wie Bsp. Ir(L5)3 | 45 % |
| Ir(L50)3 | L50 | Ir(L50)3 | wie Bsp. Ir(L5)3 | 44 % |
| Ir(L51)3 | L51 | Ir(L51)3 | wie Bsp. Ir(L5)3 | 28 % |
| Ir(L52)3 | L52 | Ir(L52)3 | wie Bsp. Ir(L5)3 | 43 % |
| Ir(L53)3 | L53 | Ir(L53)3 | wie Bsp. Ir(L5)3 | 32 % |
| Ir(L54)3 | L54 | Ir(L54)s | wie Bsp. Ir(L5)3 | 34 % |
| Ir(L55)3 | L55 | Ir(L55)3 | wie Bsp. Ir(L5)3 | 25 % |
| Ir(L56)3 | L56 | Ir(L56)3 | wie Bsp. IrL53 | 20 % |
| Ir(L57)3 | L57 | Ir(L57)3 | wie Bsp. Ir(L5)3 | 39 % |
| Ir(L58)3 | L58 | Ir(L58)3 | wie Bsp. Ir(L5)3 | 37 % |
| Ir(L59)3 | L59 | Ir(L59)3 | wie Bsp. Ir(L5)3 | 34 % |
| Ir(L60)3 | L60 | Ir(L60)3 | wie Bsp. Ir(L5)3 | 45 % |
| Ir(L61)3 | L61 | Ir(L61)3 | wie Bsp. Ir(L5)3 | 48 % |
| Ir(L62)3 | L62 | Ir(L62)3 | wie Bsp. Ir(L5)3 | 40 % |
| Ir(L63)3 | L63 | Ir(L63)3 | wie Bsp. Ir(L5)3 | 39 % |
| Ir(L64)3 | L64 | Ir(L64)3 | wie Bsp. Ir(L5)3 | 36 % |
| Ir(L65)3 | L65 | Ir(L65)3 | wie Bsp. Ir(L5)3 | 40 % |
| Ir(L66)3 | L66 | Ir(L66)3 | wie Bsp. Ir(L5)3 | 41 % |
| Ir(L67)3 | L67 | Ir(L67)3 | wie Bsp. Ir(L1)3 | 44 % |
| Ir(L67)3 | L67 | Ir(L67)3 | B
270 °C /110 h
DCM
THF | 53 % |
| Ir(L68)3 | L68 | Ir(L68)3 | wie Bsp. Ir(L1)3 | 42 % |
| Ir(L69)3 | L69 | Ir(L69)3 | wie Bsp. Ir(L1)3 | 43 % |
| Ir(L70)3 | L70 | Ir(L70)3 | wie Bsp. Ir(L5)3 | 35 % |
| Ir(L71)3 | L71 | Ir(L71)3 | wie Bsp. Ir(L5)3 | 36 % |
| Ir(L72)3 | L72 | Ir(L72)3 | wie Bsp. Ir(L5)3 | 32 % |
| Ir(L73)3 | L73 | L Ir(L73)3 | wie Bsp. Ir(L1)3 | 17 % |
| Ir(L74)3 | L74 | Ir(L74)3 | wie Bsp. Ir(L5)3 | 15 % |
| Ir(L75)3 | L75 | Ir(L75)3 | wie Bsp. Ir(L5)3 | 11 % |
| Ir(L76)3 | L76 | Ir(L76)3 | wie Bsp. Ir(L5)3 | 16 % |
| Ir(L77)3 | L77 | Ir(L77)3 | wie Bsp. Ir(L5)3 | 19 % |
| Ir(L78)3 | L78 | Ir(L78)3 | wie Bsp. Ir(L5)3 | 47 % |
| Ir(L79)3 | L79 | Ir(L79)3 | wie Bsp. Ir(L5)3 | 41 % |
| Ir(L80)3 | L80 | Ir(L80)3 | wie Bsp. Ir(L5)3 | 46 % |
| Ir(L81)3 | L81 | Ir(L81)3 | wie Bsp. Ir(L5)3 | 36 % |
| Ir(L82)3 | L82 | Ir(L82)3 | wie Bsp. Ir(L5)3 | 47 % |
| Ir(L83)3 | L83 | Ir(L83)3 | wie Bsp. Ir(L5)3 | 45 % |
| Ir(L84)3 | L84 | Ir(L84)3 | wie Bsp. Ir(L5)3 | 50 % |
| Ir(L85)3 | L85 | Ir(L85)3 | wie Bsp. Ir(L5)3 | 49 % |
| Ir(L86)3 | L86 | Ir(L86)3 | wie Bsp. Ir(L5)3 | 44 % |
| Ir(L87)3 | L87 | Ir(L87)3 | wie Bsp. Ir(L5)3. | 45 % |
| Ir(L88)3 | L88 | Ir(L88)3 | wie Bsp. Ir(L5)3 | 39 % |
| Ir(L89)3 | L89 | Ir(L89)3 | wie Bsp. Ir(L5)3 | 32 % |
| Ir(L90)3 | L90 | Ir(L90)3 | wie Bsp. Ir(L5)3 | 22 % |
| Ir(L91)3 | L91 | Ir(L91)3 | wie Bsp. Ir(L5)3 | 50 % |
| Ir(L92)3 | L92 | Ir(L92)3 | wie Bsp. Ir(L5)3 | 56 % |
| Ir(L93)3 | L93 | Ir(L93)3 | wie Bsp. Ir(L5)3 | 45 % |
| Ir(L94)3 | L94 | Ir(L94)3 | wie Bsp. Ir(L5)3 | 46 % |
| Ir(L95)3 | L95 | Ir(L95)3 | wie Bsp. Ir(L5)3 | 48 % |
| Ir(L96)3 | L96 | Ir(L96)3 | wie Bsp. Ir(L5)3 | 36 % |
| Ir(L97)3 | L97 | Ir(L97)3 | wie Bsp. Ir(L5)3 | 47 % |
| Ir(L98)3 | L98 | Ir(L98)3 | wie Bsp. Ir(L5)3 | 28 % |
| Ir(L99)3 | L99 | Ir(L99)3 | wie Bsp. Ir(L5)3 | 31 % |
| Ir(L100)3 | L100 | Ir(L100)3 | wie Bsp. Ir(L5)3 | 39 % |
| Ir(L101)3 | L101 | Ir(L101)3 | wie Bsp. Ir(L5)3 | 41 % |
| Ir(L102)3 | L102 | Ir(L102)3 | wie Bsp. Ir(L5)3 | 35 % |
| Ir(L103)3 | L103 | Ir(L103)3 | wie Bsp. Ir(L5)3 | 30 % |
| Ir(L104)3 | L104 | Ir(L104)3 | wie Bsp. Ir(L5)3 | 49 % |
| Ir(L105)3 | L105 | Ir(L105)3 | A
290 °C/100 h
DCM / EtOH, 2:1
THF | 23 % |
| Ir(L106)3 | L106 | Ir(L106)3 | wie Bsp. Ir(L105)3 | 19 % |
| Ir(L107)3 | L107 | Ir(L107)3 | wie Bsp. Ir(L105)3 | 17 % |
| Ir(L108)3 | L108 | Ir(L108)3 | A
300 °C / 100 h
DCM / EtOH, 2:1
THF | 9 % |
| Ir(L109)3 | L109 | Ir(L109)3 | wie Bsp. Ir(L105)3 | 17 % |
| Ir(L110)3 | L110 | Ir(L110)3 | wie Bsp. Ir(L105)3 | 15 % |
| Ir(L111)3 | L111 | Ir(L111)3 | wie Bsp. Ir(L105)3 | 19 % |
| Ir(L112)3 | L112 | Ir(L112)3 | wie Bsp. Ir(L108)3 | 8 % |
| Ir(L113)3 | L113 | Ir(L113)3 | wie Bsp. Ir(L105)3 | 23 % |
| Ir(L114)3 | L114 | Ir(L114)3 | A
300 °C / 100 h
Toluol / EtOH, 1:1
DCM | 24 % |
| Ir(L115)3 | L115 | Ir(L115)3 | A
280 °C 1130 h
DCM / EtOH, 2:1
THF | 34 % |
| Ir(L116)3 | L116 | Ir(L116)3 | wie Bsp. Ir(L115)3 | 35 % |
| Ir(L117)3 | L117 | Ir(L117)3 | wie Bsp. Ir(L115)3 | 41 % |
| Ir(L118)3 | L118 | Ir(L118)3 | wie Bsp. Ir(L115)3 | 30 % |
| Ir(L119)3 | L119 | Ir(L119)3 | wie Bsp. Ir(L115)3 | 32 % |
| Ir(L120)3 | L120 | Ir(L120)3 | wie Bsp. Ir(L115)3 | 25 % |
| Ir(L121)3 | L121 | Ir(L121)3 | wie Bsp. Ir(L115)3 | 27 % |
| Ir(L122)3 | L122 | Ir(L122)3 | wie Bsp. Ir(L115)3 | 23 % |
| Ir(L123)3 | L123 | Ir(L123)3 | wie Bsp. Ir(L115)3 | 9 % |
| Ir(L124)3 | L124 | Ir(L124)3 | wie Bsp. Ir(L115)3 | 20 % |
| Ir(L125)3 | L125 | Ir(L125)3 | wie Bsp. Ir(L115)3 | 19 % |
| Ir(L126)3 | L126 | Ir(L126)3 | wie Bsp. Ir(L115)3 | 22 % |
| Ir(L127)3 | L127 | Ir(L127)3 | wie Bsp. Ir(L115)3 | 20 % |
| Ir(L128)3 | L128 | Ir(L128)3 | wie Bsp. Ir(L115)3 | 16 % |
| Ir(L129)3 | L129 | Ir(L129)3 | wie Bsp. Ir(L115)3 | 24 % |
| Ir(L130)3 | L130 | Ir(L130)3 | wie Bsp. Ir(L114)3 | 21 % |
| Ir(L131)3 | L131 | Ir(L131)3 | wie Bsp. Ir(L115)3 | 10 % |
| Ir(L132)3 | L132 | Ir(L132)3 | A
315 °C /130 h
DCM / EtOH, 2:1
THF | 17 % |
| Ir(L133)3 | L133 | Ir(L133)3 | wie Bsp. Ir(L132)3 | 17 % |
| Ir(L134)3 | L134 | Ir(L134)3 | wie Bsp. Ir(L132)3 | 24 % |
| Ir(L135)3 | L135 | Ir(L135)3 | wie Bsp. Ir(L132)3 | 11 % |
| Ir(L136)3 | L136 | Ir(L136)3 | wie Bsp. Ir(L132)3 | 22 % |
| Ir(L137)3 | L137 | Ir(L137)3 | wie Bsp. Ir(L132)3 | 9 % |
| Ir(L138)3 | L138 | Ir(L138)3 | wie Bsp. Ir(L132)3 | 5 % |
| Ir(L139)3 | L139 | Ir(L139)3 | wie Bsp. Ir(L132)3 | 10 % |
| Ir(L140)3 | L140 | Ir(L140)3 | wie Bsp. Ir(L132)3 | 12 % |
| Ir(L141)3 | L141 | Ir(L141)3 | wie Bsp. Ir(L132)3 | 9 % |
| Ir(L142)3 | L142 | Ir(L142)3 | wie Bsp. Ir(L132)3 | 7 % |
| Ir(L143)3 | L143 | Ir(L143)3 | A
320 °C / 120 h
DCM / EtOH, 2:1
THF | 9 % |
| Ir(L144)3 | L144 | Ir(L144)3 | wie Bsp. Ir(L143)3 | 11 % |
| Ir(L145)3 | L145 | Ir(L145)3 | A
320 °C / 130 h
Toluol
THF | 11 % |
| Ir(L146)3 | L146 | Ir(L146)3 | wie Bsp. Ir(L143)3 | 13 % |
| Ir(L147)3 | L147 | Ir(L147)3 | wie Bsp. Ir(L115)3 | 17 % |
| Ir(L148)3 | L148 | Ir(L148)3 | wie Bsp. Ir(L115)3 | 20 % |
| Ir(L149)3 | L149 | Ir(L149)3 | A
315 °C/ 130 h
DCM
THF | 7 % |
| Ir(L150)3 | L150 | Ir(L150)3 | wie Bsp. Ir(L149)3 | 9 % |
| Ir(L151)3 | L151 | Ir(L151)3 | wie Bsp. Ir(L145)3 | 5 % |
| Ir(L152)3 | L152 | Ir(L152)3 | wie Bsp. Ir(L145)3 | 6 % |
| Ir(L153)3 | L153 | Ir(L153)3 | wie Bsp. Ir(L149)3 | 5 % |
| Ir(L154)3 | L154 | Ir(L154)3 | wie Bsp. Ir(L149)3 | 8 % |
| Ir(L155)3 | L155 | Ir(L155)3 | wie Bsp. Ir(L143)3 | 4 % |
| Ir(L156)3 | L156 | Ir(L156)3 | wie Bsp. Ir(L143)3 | 6 % |
| Ir(L157)3 | L157 | Ir(L157)3 | A
325 °C / 80 h
DCM
THF | 7 % |
| Ir(L158)3 | L158 | Ir(L158)3 | wie Bsp. Ir(L157)3 | 6 % |
| Ir(L159)3 | L159 | Ir(L159)3 | A
265 °C / 80 h
DCM
THF | 37 % |
| Ir(L160)3 | L160 | Ir(L160)3 | wie Bsp. Ir(L159)3 | 43 % |
| Ir(L161)3 | L161 | Ir(L161)3 | wie Bsp. Ir(L159)3 | 30 % |
| Ir(L162)3 | L162 | Ir(L162)3 | B
305 °C / 130 h
Aceton
Toluol | 46 % |
| Ir(L163)3 | L163 | Ir(L163)3 | wie Bsp. Ir(L162)3 | 44 % |
| Ir(L164)3 | L164 | Ir(L164)3 | B
305 °C / 130 h
Aceton
Cyclohexan | 41 % |
| Ir(L165)3 | L165 | Ir(L165)3 | wie Bsp. Ir(L164)3 | 43 % |
| Ir(L166)3 | L166 | Ir(L166)3 | wie Bsp. Ir(L164)3 | 43 % |
| Ir(L167)3 | L167 | Ir(L167)3 | wie Bsp. Ir(L164)3 | 32 % |
| Ir(L168)3 | L168 | Ir(L168)3 | wie Bsp. Ir(L162) | 39 % |
| Ir(L169)3 | L169 | Ir(L169)3 | wie Bsp. Ir(L162) | 40 % |
| Ir(L170)3 | L170 | Ir(L170)3 | wie Bsp. Ir(L162) | 40 % |
| Ir(L171)3 | L171 | Ir(L171)3 | wie Bsp. Ir(L162) | 19 % |
| Ir(L172)3 | L172 | Ir(L172)3 | wie Bsp. Ir(L162) | 31 % |
| Ir(L173)3 | L173 | Ir(L173)3 | wie Bsp. Ir(L162) | 36 % |
| Ir(L174)3 | L174 | Ir(L174)3 | wie Bsp. Ir(L162) | 35 % |
| Ir(L175)3 | L175 | Ir(L175)3 | wie Bsp. Ir(L162) | 33 % |
| Ir(L176)3 | L176 | Ir(L1763 | wie Bsp. Ir(L162) | 45 % |
| Ir(L177)3 | L177 | Ir(L177)3 | wie Bsp. Ir(L162) | 44 % |
| Ir(L178)3 | L178 | Ir(L178)3 | wie Bsp. Ir(L162) | 41 % |
| Ir(L179)3 | L179 | Ir(L179)3 | wie Bsp. Ir(L162) | 42 % |
| Ir(L180)3 | L180 | Ir(L180)3 | wie Bsp. Ir(L162) | 29 % |
| Ir(L181)3 | L181 | Ir(L181)3 | wie Bsp. Ir(L162) | 36 % |
| Ir(L182)3 | L182 | Ir(L182)3 | wie Bsp. Ir(L162) | 44 % |
| Ir(L183)3 | L183 | Ir(L183)3 | wie Bsp. Ir(L162) | 38 % |
| Ir(L184)3 | L184 | Ir(L184)3 | wie Bsp. Ir(L162) | 37 % |
| Ir(L185)3 | L185 | Ir(L185)3 | wie Bsp. Ir(L162) | 26 % |
| Ir(L186)3 | L186 | Ir(L186)3 | wie Bsp. Ir(L162) | 40 % |
| Ir(L187)3 | L187 | Ir(L187)3 | wie Bsp. Ir(L162) | 35 % |
| Ir(L188)3 | L188 | Ir(L188)3 | wie Bsp. Ir(L162) | 36 % |
| Ir(L189)3 | L189 | Ir(L189)3 | wie Bsp. Ir(L162) | 39 % |
| Ir(L190)3 | L190 | Ir(L190)3 | wie Bsp. Ir(L162) | 30 % |
| Ir(L191)3 | L191 | Ir(L191)3 | wie Bsp. Ir(L162) | 39 % |
| Ir(L192)3 | L192 | Ir(L192)3 | wie Bsp. Ir(L164)3 | 41 % |
| Ir(L193)3 | L192 | Ir(L193)3 | wie Bsp. Ir(L162) | 36 % |
| Ir(L194)3 | L194 | Ir(L194)3 | wie Bsp. Ir(L162) | 28 % |
| Ir(L195)3 | L192’5 | Ir(L195)3 | wie Bsp. Ir(L162) | 37 % |
| Ir(L196)3 | L196 | Ir(L196)3 | wie Bsp. Ir(L162) | 22 % |
| Ir(L197)3 | L197 | Ir(L197)3 | wie Bsp. Ir(L162) | 34 % |
| Ir(L198)3 | L198 | Ir(L198)3 | wie Bsp. Ir(L162) | 36 % |
| Ir(L199)3 | L199 | Ir(L199)3 | wie Bsp. Ir(L162) | 35 % |
| Ir(L200)3 | L200 | Ir(L200)3 | wie Bsp. Ir(L164) | 27 % |
| Ir(L201)3 | L201 | Ir(L201)3 | wie Bsp. Ir(L162) | 40 % |
| Ir(L202)3 | L202 | Ir(L202)3 | wie Bsp. Ir(L162) | 30 % |
| Ir(L203)3 | L203 | Ir(L203)3 | wie Bsp. Ir(L162) | 35 % |
| Ir(L204)3 | L204 | Ir(L204)3 | wie Bsp. Ir(L162) | 39 % |
| Ir(L205)3 | L205 | Ir(L205)3 | wie Bsp. Ir(L162) | 25 % |
| Ir(L206)3 | L206 | Ir(L206)3 | wie Bsp. Ir(L164)3 | 21 % |
| Ir(L207)3 | L207 | Ir(L207)3 | wie Bsp. Ir(L164) | 43 % |
| Ir(L208)3 | L208 | Ir(L208)3 | wie Bsp. Ir(L162) | 26 % |
| Ir(L209)3 | L209 | Ir(L209)3 | wie Bsp. Ir(L162) | 30 % |
| Ir(L210)3 | L210 | Ir(L210)3 | wie Bsp. Ir(L162) | 29 % |
| Ir(L218) | L218 | Tris-(2,2,6,6-tetramethyl-3,5-heptandionato)iridium: L218
1:1
Zusatz von 3 ml Tridecan | B
305 °C / 130 h
Aceton
Toluol | 12 % |
-
2) Heteroleptische Iridium-Komplexe:
-
Variante A:
-
Schritt 1:
-
Ein Gemisch aus 10 mmol Natrium-bis-acetylacetonato-dichloro-iridat(III) [770720-50-8] und 24 mmol des Liganden L wird unter Vakuum (10-3 mbar) in eine 50 ml Glasampulle abgeschmolzen. Die Ampulle wird für die angegebene Zeit bei der angegebenen Temperatur getempert, wobei das aufgeschmolzene Gemisch mit Hilfe eines Magnetrührers gerührt wird. Nach Erkalten - ACHTUNG: die Ampullen stehen meist unter Druck! - wird die Ampulle geöffnet, der Sinterkuchen wird mit 100 g Glaskugeln (3 mm Durchmessser) in 100 ml des angegebenen Suspensionsmittels 3 h gerührt und dabei mechanisch aufgeschlossen. Man dekantiert die feine Suspension von den Glaskugeln ab, saugt den Feststoff ab und trocknet diesen im Vakuum.
-
Schritt 2:
-
Das so erhaltene rohe Chloro-Dimer der Formel [Ir(L)
2Cl]
2 wird in einem Gemisch aus 75 ml 2-Ethoxyethanol und 25 ml Wasser suspendiert, mit 13 mmol des Co-Liganden CL bzw. der Co-Liganden-Verbindung CL und 15 mmol Natriumcarbonat versetzt. Nach 20 h unter Rückfluss gibt man weitere 75 ml Wasser tropfenweise zu, saugt nach Erkalten vom Feststoff ab, wäscht diesen dreimal mit je 50 ml Wasser und dreimal mit je 50 ml Methanol und trocknet diesen im Vakuum. Der trockene Feststoff wird in einem Heißextraktor auf einem 10 cm hohen Alox-Bett (Alox, basisch Aktivitätsstufe 1) platziert und dann mit dem angegebenen Extraktionsmittel (Vorlagemenge ca. 500 ml) extrahiert. Nach beendeter Extraktion wird das Extraktionsmittel im Vakuum auf ca. 100 ml eingeengt. Metallkomplexe, die im Extraktionsmittel eine zu gute Löslichkeit aufweisen, werden durch Zutropfen von 200 ml Methanol zur Kristallisation gebracht. Der Feststoff der so erhaltenen Suspensionen wird abgesaugt, einmal mit ca. 50 ml Methanol gewaschen und getrocknet. Nach Trocknen wird die Reinheit des Metall-Komplexes mittels NMR und / oder HPLC bestimmt. Liegt die Reinheit unter 99.5 % wird der Heißextraktionsschritt wiederholt, ist eine Reinheit von 99.5 - 99.9 % erreicht, wird der Metallkomplex getempert oder sublimiert. Das Tempern erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von 200 - 300 °C. Die Sublimation erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von ca. 300 bis ca. 390 °C, wobei die Sublimation bevorzugt in Form einer fraktionierten Sublimation durchgeführt wird.
| Bsp. | Ligand L | Co-Ligand CL | Ir-Komplex Schritt 1: Reaktionstemp. Reaktionszeit Suspensionsmittel Schritt 2: Extraktionsmittel | Ausbeute |
| Ir(L1)2(CL1) | L1 | | 260 °C / 60 h / DCM THF | 68 % |
| Ir(L13)2(CL1) | L13 | CL1 | wie Bsp. Ir(L1)2(CL1) | 65 % |
| Ir(L23)2(CL1) | L23 | CL1 | wie Bsp. Ir(L1)2(CL1) | 56 % |
| Ir(L41)2(CL2) | L41 | | wie Bsp. Ir(L1)2(CL1) | 64 % |
| Ir(L91)2(CL2) | L91 | CL2 | wie Bsp. Ir(L1)2(CL1) | 60 % |
| Ir(L91)2(CL3) | L91 | | wie Bsp. Ir(L1)2(CL1) | 58 % |
| Ir(L91)2(CL4) | L91 | | wie Bsp. Ir(L1)2(CL1) | 47 % |
| Ir(L91)2(CL5) | L91 | | wie Bsp. Ir(L1)2(CL1) | 50 % |
| Ir(L92)2(CL6) | L92 | | wie Bsp. Ir(L1)2(CL1) | 56 % |
| Ir(L111)2(CL3) | L111 | CL3 | 275 °C / 70 h / DCM THF | 35 % |
| Ir(L120)Z(CL3) | L120 | CL3 | 270 °C / 70 h / Toluol THF | 52 % |
| Ir(L141)2(CL1) | L141 | CL1 | 300 °C / 80 h / DCM THF | 23 % |
| Ir(L13)2(CL17) | L13 | | wie Bsp. Ir(L1)2(CL1) | 55 % |
-
Variante B:
-
Schritt 1:
-
Siehe Variante A, Schritt 1.
-
Schritt 2:
-
Das rohe Chloro-Dimer der Formel [Ir(L)
2Cl]
2 wird nach
WO 2007/065523 , Beispiel 5 in Gegenwart von 80 mmol des Co-Liganden CL und 75 mmol N,N-Dimethylglycin in 1000 ml eines Dioxan-Wasser-Gemischs (1:1, vv) weiter umgesetzt. Der so erhaltene Feststoff wird in einem Heißextraktor auf einem 10 cm hohen Alox-Bett (Alox, basisch Aktivitätsstufe 1) platziert und dann mit dem angegebenen Extraktionsmittel (Vorlagemenge ca. 500 ml) extrahiert. Nach beendeter Extraktion wird das Extraktionsmittel im Vakuum auf ca. 100 ml eingeengt. Metallkomplexe, die im Extraktionsmittel eine zu gute Löslichkeit aufweisen, werden durch Zutropfen von 200 ml Methanol zur Kristallisation gebracht. Der Feststoff der so erhaltenen Suspensionen wird abgesaugt, einmal mit ca. 50 ml Methanol gewaschen und getrocknet. Nach Trocknen wird die Reinheit des Metall-Komplexes mittels NMR und / oder HPLC bestimmt. Liegt die Reinheit unter 99.5 % wird der Heißextraktionsschritt wiederholt, ist eine Reinheit von 99.5 - 99.9 % erreicht, wird der Metallkomplex getempert oder sublimiert. Das Tempern erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von 200 - 300 °C. Die Sublimation erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von ca. 300 bis ca. 390 °C, wobei die Sublimation bevorzugt in Form einer fraktionierten Sublimation durchgeführt wird.
| Bsp. | Ligand L | Co-Ligand CL | Ir-Komplex Schritt 1: Reaktionstemp. / Reaktionszeit / Suspensionsmittel Schritt 2: Extraktionsmittel | Ausbeute |
| Ir(L1)2(CL7) | L1 | | 260 °C / 60 h / DCM THF | 61 % |
| Ir(L13)2(CL7) | L13 | CL7 | wie Bsp. Ir(L1)2(CL7) | 55 % |
| lr(L23)2(CL7) | L23 | CL7 | wie Bsp. Ir(L1)2(CL7) | 43 % |
| Ir(L91)2(CL7) | L91 | CL7 | wie Bsp. Ir(L1)2(CL7) | 48 % |
| Ir(L91)2(CL8) | L91 | | wie Bsp. Ir(L1)2(CL7) | 39 % |
| Ir(L92)2(CL8) | L92 | CL8 | wie Bsp. Ir(L1)2(CL7) | 42 % |
| Ir(L111)2(CL9) | L111 | | 280 °C / 50 h / DCM THF | 45 % |
| Ir(L120)2(CL7) | L120 | CL7 | 270 °C / 50 h / DCM THF | 37 % |
| Ir(L147)2(CL9) | L147 | CL9 | 275 °C / 80 h / DCM THF | 29 % |
-
Variante C:
-
Schritt 1:
-
Siehe Variante A, Schritt 1.
-
Schritt 2:
-
Das so erhaltene rohe Chloro-Dimer der Formel [Ir(L)
2Cl]
2 wird in 100 ml THF suspendiert, die Suspension wird mit 40 mmol des Co-Liganden CL, 20 mmol Silber(l)trifluoracetat und 80 mmol Kaliumcarbonat versetzt und 24 h unter Rückfluss erhitzt. Nach Erkalten wird das THF im Vakuum entfernt. Der Rückstand wird in 200 ml eines Gemischs aus Ethanol und konz. Ammoniak-Lösung (1:1, vv) aufgenommen. Die Suspension wird 1 h bei Raumtemperatur gerührt, der Feststoff wird abgesaugt, zweimal mit je 50 ml eines Gemischs aus Ethanol und konz. Ammoniak-Lösung (1:1, vv) und zweimal mit je 50 ml Ethanol gewaschen und dann im Vakuum getrocknet. Der so erhaltene Feststoff wird in einem Heißextraktor auf einem 10 cm hohen Alox-Bett (Alox, basisch Aktivitätsstufe 1) platziert und dann mit dem angegebenen Extraktionsmittel (Vorlagemenge ca. 500 ml) extrahiert. Nach beendeter Extraktion wird das Extraktionsmittel im Vakuum auf ca. 100 ml eingeengt. Metallkomplexe, die im Extraktionsmittel eine zu gute Löslichkeit aufweisen, werden durch Zutropfen von 200 ml Methanol zur Kristallisation gebracht. Der Feststoff der so erhaltenen Suspensionen wird abgesaugt, einmal mit ca. 50 ml Methanol gewaschen und getrocknet. Nach Trocknen wird die Reinheit des Metall-Komplexes mittels NMR und / oder HPLC bestimmt. Liegt die Reinheit unter 99.5 % wird der Heißextraktionsschritt wiederholt, ist eine Reinheit von 99.5 - 99.9 % erreicht, wird der Metallkomplex getempert oder sublimiert. Das Tempern erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von 200 - 300 °C. Die Sublimation erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von ca. 300 bis ca. 390 °C, wobei die Sublimation bevorzugt in Form einer fraktionierten Sublimation durchgeführt wird.
| Bsp. | Ligand L | Co-Ligand CL | Ir-Komplex Schritt 1: Reaktionstemp. / Reaktionszeit / Suspensionsmittel Schritt 2: Extra ktionsmittel | Ausbeute |
| Ir(L9)2(CL7) | L9 | CL7 | 260 °C / 70 h / DCM THF | 40 % |
| Ir(L22)2(CL7) | L22 | CL7 | wie Bsp. Ir(L9)2(CL7) | 46 % |
| Ir(L52)2(CL10) | L52 | | wie Bsp. Ir(L9)2(CL7) | 34 % |
| Ir(L91)2(CL11) | L91 | | wie Bsp. Ir(L9)2(CL7) | 47 % |
| Ir(L91)2(CL8) | L91 | CL8 | wie Bsp. Ir(L9),(CL7) | 45 % |
| Ir(L92)2(CL12) | L92 | | wie Bsp. Ir(L9)2(CL7) | 34 % |
| Ir(L111)2(CL9) | L111 | CL9 | wie Bsp. Ir(L111)2(CL9) | 36 % |
| Ir(L120)2(CL7) | L120 | CL7 | 270 °C / 40 h / DCM THF | 33 % |
| Ir(L124)2(CL13) | L124 | | 270 °C / 75 h DCM / THF | 24 % |
-
Variante D:
-
Schritt 1:
-
Siehe Variante A, Schritt 1.
-
Schritt 2:
-
Das so erhaltene rohe Chloro-Dimer der Formel [Ir(L)
2Cl]
2 wird in 1000 ml Dichlormethan und 150 ml Ethanol suspendiert, die Suspension wird mit 40 mmol Silber(I)trifluormethansulfonat versetzt und 24 h bei Raumtemperatur gerührt. Man saugt vom ausgefallen Feststoff (AgCI) über eine kurzes Celite-Bett ab und engt das Filtrat im Vakuum zur Trockene ein. Der so erhaltene Feststoff wird in 100 ml Ethanol aufgenommen, mit 30 mmol des Co-Liganden CL versetzt und dann 30 h unter Rückfluss erhitzt. Nach Erkalten saugt man vom Feststoff ab, wäscht diesen zweimal mit je 50 ml Ethanol und trocknet im Vakuum. Der so erhaltene Feststoff wird in einem Heißextraktor auf einem 10 cm hohen Alox-Bett (Alox, basisch Aktivitätsstufe 1) platziert und dann mit dem angegebenen Extraktionsmittel (Vorlagemenge ca. 500 ml) extrahiert. Nach beendeter Extraktion wird das Extraktionsmittel im Vakuum auf ca. 100 ml eingeengt. Metallkomplexe, die im Extraktionsmittel eine zu gute Löslichkeit aufweisen, werden durch Zutropfen von 200 ml Methanol zur Kristallisation gebracht. Der Feststoff der so erhaltenen Suspensionen wird abgesaugt, einmal mit ca. 50 ml Methanol gewaschen und getrocknet. Nach Trocknen wird die Reinheit des Metall-Komplexes mittels NMR und / oder HPLC bestimmt. Liegt die Reinheit unter 99.5 % wird der Heißextraktionsschritt wiederholt, ist eine Reinheit von 99.5 - 99.9 % erreicht, wird der Metallkomplex getempert oder sublimiert. Das Tempern erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von 200 - 300 °C. Die Sublimation erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von ca. 300 bis ca. 390 °C, wobei die Sublimation bevorzugt in Form einer fraktionierten Sublimation durchgeführt wird. Bei ionischen Metallkomplexen wird im Heißextraktionsschritt Alox durch Celite ersetzt.
| Bsp. | Ligand L | Co-Ligand CL | Ir-Komplex Schritt 1: Reaktionstemp. / Reaktionszeit / Suspensionsmittel Schritt 2: Extraktionsmittel | Ausbeute |
| Ir(L91)2(CL14) | L91 | | wie Bsp. Ir(L9)2(CL7) | 56 % |
| Ir(L91)2(CL15) | L91 | CL15 | wie Bsp. Ir(L9)2(CL7) | 38 % |
| Ir(L111)2(CL16) | L111 | | wie Bsp. Ir(L111)2(CL9) | 44 % |
| [Ir(L91)2 (CL18)]OTf | L91 | | wie Bsp. Ir(L9)2(CL7) | 27 % |
| [Ir(L91)2 (CL19)]OTf | L91 | | wie Bsp. Ir(L9)2(CL7) | 49 % |
| [Bu4N][Ir(L91)2 (CL20)] | L91 | | wie Bsp. Ir(L9)2(CL7) Zusatz von 30 mmol Bu4NCl und 30 mml Na2CO3 in Schritt 2 | 23 % |
| [Bu4N][Ir(L91)2 (CL19)] | L91 | | wie Bsp. Ir(L9)2(CL7) Zusatz von 30 mmol Bu4NCl in Schritt 2 | 37 % |
-
Variante E:
-
Ein Gemisch aus 10 mmol des Ir-Komplexes Ir(L)
2(CL) und 30 mmol des Liganden L wird unter Vakuum (10
-5 mbar) in eine 50 ml Glasampulle abgeschmolzen. Die Ampulle wird für die angegebene Zeit bei der angegebenen Temperatur getempert, wobei das aufgeschmolzene Gemisch mit Hilfe eines Magnetrührers gerührt wird. Nach Erkalten (ACHTUNG: die Ampullen stehen meist unter Druck!) wird die Ampulle geöffnet, der Sinterkuchen wird mit 100 g Glaskugeln (3 mm Durchmessser) in 100 ml des angegebenen Suspensionsmittels 3 h gerührt und dabei mechanisch aufgeschlossen. Man dekantiert die feine Suspension von den Glaskugeln ab, saugt den Feststoff ab und trocknet diesen im Vakuum. Der trockene Feststoff wird in einem Heißextraktor auf einem 10 cm hohen Alox-Bett (Alox, basisch Aktivitätsstufe 1) platziert und dann mit dem angegebenen Extraktionsmittel (Vorlagemenge ca. 500 ml) extrahiert. Nach beendeter Extraktion wird das Extraktionsmittel im Vakuum auf ca. 100 ml eingeengt. Metallkomplexe, die im Extraktionsmedium eine zu gute Löslichkeit aufweisen, werden durch Zutropfen von 200 ml Methanol zur Kristallisation gebracht. Der Feststoff der so erhaltenen Suspensionen wird abgesaugt, einmal mit ca. 50 ml Methanol gewaschen und getrocknet. Nach Trocknen wird die Reinheit des Metall-Komplexes mittels NMR und / oder HPLC bestimmt. Liegt die Reinheit unter 99.5 % wird der Heißextraktionsschritt wiederholt, ist eine Reinheit von 99.5 - 99.9 % erreicht, wird der Metallkomplex getempert oder sublimiert. Das Tempern erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von 200 - 300 °C. Die Sublimation erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von ca. 340 bis ca. 400 °C, wobei die Sublimation bevorzugt in Form einer fraktionierten Sublimation durchgeführt wird.
| Bsp. | Ir-Komplex Ir(L) 2 (CL) | Ligand L | Ir-Komplex Schritt 1: Reaktionstemp. / Reaktionszeit / Suspensionsmittel Schritt 2: Extraktionsmittel | Ausbeute |
| Ir(L13)2(L23) | Ir(L13)2(CL1) | L23 | wie Bsp. Ir(L1)3 | 35 % |
| Ir(L23)2(L13) | Ir(L23)2(CL1 ) | L13 | wie Bsp. Ir(L1)3 | 28 % |
| Ir(L41)2(L13) | Ir(L41)2(CL2) | L13 | wie Bsp. Ir(L1)3 | 34 % |
| Ir(L91)2(L111) | Ir(L91)2(CL2) | L111 | wie Bsp. Ir(L105)3 | 26 % |
-
Variante F:
-
Schritt 1:
-
Ein Gemisch aus 10 mmol lridium(III)chlorid-Hydrat, 21 mmol des Liganden L, 60 ml 2-Ethoyxethanol und 30 ml Wasser wird 160 h unter Rückfluss erhitzt. Nach Erkalten saugt man vom Feststoff ab, wäscht diesen einmal mit 20 ml eines Gemischs aus Ethanol und Wasser (1:1, vv) und dreimal mit je 10 ml Ethanol und trocknet im Vakuum.
-
Schritt 2:
-
Das so erhaltene rohe Chloro-Dimer der Formel [Ir(L)2Cl]2 kann als Edukt in Variante A, B, C und D Schritt 2 eingesetzt werden.
-
3) Heteroleptische Platin-Komplexe:
-
Ein Gemisch aus 10 mmol Platin(II)chlorid, 12 mmol des Liganden L, 1 mmol Tetra-n-butyl-ammoniumchlorid in 30 ml Dichlormethan wird 12 h unter Rückfluss erhitzt. Nach tropfenweiser Zugabe von 100 ml Methanol wird vom feinen Feststoff abgesaugt, dieser wird zweimal mit 25 ml Methanol gewaschen und im Vakuum getrocknet. Das so erhaltene rohe Chloro-Dimer der Formel [Pt(L)Cl]
2 wird in einem Gemisch aus 60 ml 2-Ethoxyethanol und 20 ml Wasser suspendiert und mit 12 mmol des Co-Liganden CL bzw. der Co-Liganden-Verbindung CL und 12 mmol Natriumcarbonat versetzt. Nach 20 h unter Rückfluss gibt man weitere 100 ml Wasser tropfenweise zu, saugt nach Erkalten vom Feststoff ab, wäscht diesen dreimal mit je 50 ml Wasser und dreimal mit je 50 ml Methanol und trocknet diesen im Vakuum. Der so erhaltene Feststoff wird in einem Heißextraktor auf einem 10 cm hohen Celite-Bett (Alox, basisch Aktivitätsstufe 1) platziert und dann mit dem angegebenen Extraktionsmittel (Vorlagemenge ca. 500 ml) extrahiert. Nach beendeter Extraktion wird das Extraktionsmittel im Vakuum auf ca. 100 ml eingeengt. Metallkomplexe, die im Extraktionmittel eine zu gute Löslichkeit aufweisen, werden durch Zutropfen von 200 ml Methanol zur Kristallisation gebracht. Der Feststoff der so erhaltenen Suspensionen wird abgesaugt, einmal mit ca. 50 ml Methanol gewaschen und getrocknet. Nach Trocknen wird die Reinheit des Metall-Komplexes mittels NMR und / oder HPLC bestimmt. Liegt die Reinheit unter 99.5 % wird der Heißextraktionsschritt wiederholt, ist eine Reinheit von 99.5 - 99.9 % erreicht, wird der Metallkomplex getempert oder sublimiert. Das Tempern erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von 200 - 300 °C. Die Sublimation erfolgt im Hochvakuum (p ca. 10
-6 mbar) im Temperaturbereich von ca. 300 bis ca. 390 °C, wobei die Sublimation bevorzugt in Form einer fraktionierten Sublimation durchgeführt wird.
| Bsp. | Ligand L | Co-Ligand CL | Pt-Komplex | Ausbeute |
| Pt(L13)(CL1) | L13 | CL1 | | 20 % |
| Pt(L91)(CL17) | L91 | CL17 | | 23 % |
-
4) Platin-Komplexe tetradentater Liganden:
-
Variante A:
-
Ein Gemisch aus 10 mmol Kalium-tetrachloroplatinat, 10 mmol des Liganden L, 50 mmol Lithiumacetat, wasserfrei in 100 ml Eisessig wird 60 h unter Rückfluss erhitzt. Nach tropfenweiser Zugabe von 100 ml Methanol und 100 ml Wasser zur erkalteten Reaktionsmischung wird vom Feststoff abgesaugt, dieser wird fünfmal mit je 25 ml Methanol gewaschen und im Vakuum getrocknet. Der so erhaltene Feststoff wird in einem Heißextraktor auf einem 3 cm hohen Celite-Bett (Alox, basisch Aktivitätsstufe 1) platziert und dann mit dem angegebenen Extraktionsmittel (Vorlagemenge ca. 300 ml) extrahiert. Nach beendeter Extraktion wird das Extraktionsmittel im Vakuum auf ca. 100 ml eingeengt. Metallkomplexe, die im Extraktionsmittel eine zu gute Löslichkeit aufweisen, werden durch Zutropfen von 200 ml Methanol zur Kristallisation gebracht. Der Feststoff der so erhaltenen Suspensionen wird abgesaugt, einmal mit ca. 50 ml Methanol gewaschen und getrocknet. Nach Trocknen wird die Reinheit des Metall-Komplexes mittels NMR und / oder HPLC bestimmt. Liegt die Reinheit unter 99.5 % wird der Heißextraktionsschritt wiederholt; ist eine Reinheit von 99.5 - 99.9 % erreicht, wird der Pt-Komplex sublimiert. Die Sublimation erfolgt im Hochvakuum (p ca. 10-6 mbar) im Temperaturbereich von ca. 350 bis ca. 390 °C, wobei die Sublimation bevorzugt in Form einer fraktionierten Sublimation durchgeführt wird.
-
Variante B:
-
Ein Gemisch aus 10 mmol Bis(benzonitril)-dichloro-platin(II) und 10 mmol des Liganden L in 50 ml Benzonitril wird 24 h unter Rückfluss erhitzt. Nach tropfenweiser Zugabe von 100 ml Methanol zur erkalteten Reaktionsmischung wird vom Feststoff abgesaugt, dieser wird fünfmal mit je 25 ml Methanol gewaschen und im Vakuum getrocknet. Restliche Aufarbeitung wie bei Variante A beschrieben.
| Bsp. | Ligand L | Pt-Komplex | Variante Extraktionsmittel | Ausbeute |
| Pt(L211) | L211 | Pt(L211) | A p-Xylol | 43 % |
| Pt(L212) | L212 | Pt(L212) | wie Bsp. Pt(L211) | 39 % |
| Pt(L213) | L213 | Pt(L213) | A Toluol | 28 % |
| Pt(L214) | L214 | Pt(L214) | wie Bsp. Pt(L211) | 33 % |
| Pt(L215) | L215 | Pt(L215) | wie Bsp. Pt(L213) | 34 % |
| Pt(L216) | L216 | Pt(L216) | B p-Xylol | 20 % |
| Pt(L217) | L217 | Pt(L217) | wie Bsp. Pt(L216) | 31 % |
| Pt(L219) | L219 | Pt(L219) | wie Bsp. Pt(L216) | 18 % |
-
Beispiel: Herstellung der OLEDs
-
Die Herstellung von erfindungsgemäßen OLEDs sowie OLEDs nach dem Stand der Technik erfolgt nach einem allgemeinen Verfahren gemäß
WO 2004/058911 , das auf die hier beschriebenen Gegebenheiten (Schichtdickenvariation, verwendete Materialien) angepasst wird.
-
In den folgenden Beispielen 1 bis 146 (siehe Tabellen 1 und 2) werden die Ergebnisse verschiedener OLEDs vorgestellt. Glasplättchen, die mit strukturiertem ITO (Indium Zinn Oxid) der Dicke 150 nm beschichtet sind, werden zur verbesserten Prozessierung mit 20 nm PEDOT beschichtet (Poly(3,4-ethylendioxy-2,5-thiophen), aus Wasser aufgeschleudert; bezogen von H. C. Starck, Goslar, Deutschland). Diese beschichteten Glasplättchen bilden die Substrate, auf welche die OLEDs aufgebracht werden. Die OLEDs haben prinzipiell folgenden Schichtaufbau: Substrat / optionale Lochinjektionsschicht (HIL) / Lochtransportschicht (HTL) / Elektronenblockerschicht (EBL) / Emissionsschicht (EML) / optionale Lochblockierschicht (HBL) / Elektronentransportschicht (ETL) / optionale Elektroneninjektionsschicht (EIL) und abschließend eine Kathode. Die Kathode wird durch eine 100 nm dicke Aluminiumschicht gebildet
-
Zunächst werden vakuumprozessierte OLEDs beschrieben. Hierfür werden alle Materialien in einer Vakuumkammer thermisch aufgedampft. Dabei besteht die Emissionsschicht immer aus mindestens einem Matrixmaterial (Hostmaterial, Wirtsmaterial) und einem emittierenden Dotierstoff (Dotand, Emitter), der dem Matrixmaterial bzw. den Matrixmaterialien durch Coverdampfung in einem bestimmten Volumenanteil beigemischt wird. Eine Angabe wie M3:M2:Ir(L1)3 (55%:35%:10%) bedeutet hierbei, dass das Material M3 in einem Volumenanteil von 55%, M2 in einem Anteil von 35% und Ir(L1)3 in einem Anteil von 10% in der Schicht vorliegt. Analog kann auch die Elektronentransportschicht aus einer Mischung zweier Materialien bestehen. Der genaue Aufbau der OLEDs ist Tabelle 1 zu entnehmen. Die zur Herstellung der OLEDs verwendeten Materialien sind in Tabelle 3 gezeigt.
-
Die OLEDs werden standardmäßig charakterisiert. Hierfür werden die Elektrolumineszenzspektren, die Stromeffizienz (gemessen in cd/A) und die Spannung (gemessen bei 1000 cd/m2 in V) bestimmt aus Strom-Spannungs-Helligkeits-Kennlinien (IUL-Kennlinien). Für ausgewählte Versuche wird die Lebensdauer bestimmt. Als Lebensdauer wird die Zeit definiert, nach der die Leuchtdichte von einer bestimmten Startleuchtdichte aus auf einen gewissen Anteil abgesunken ist. Die Angabe LD50 bedeutet, dass es sich bei der genannten Lebensdauer um die Zeit handelt, bei der die Leuchtdichte auf 50% der Startleuchtdichte abgefallen ist, also von z.B. 4000 cd/m2 auf 2000 cd/m2. Je nach Emissionsfarbe wurden unterschiedliche Starthelligkeiten gewählt. Die Werte für die Lebensdauer können mit Hilfe dem Fachmann bekannten Umrechnungsformeln auf eine Angabe für andere Startleuchtdichten umgerechnet werden. Hierbei ist die Lebensdauer für eine Startleuchtdichte von 1000 cd/m2 eine übliche Angabe.
-
Verwendung von erfindungsgemäßen Verbindungen als Emittermaterialien in phosphoreszierenden OLEDs
-
Die erfindungsgemäßen Verbindungen lassen sich unter anderem als phosphoreszierende Emittermaterialien in der Emissionsschicht in OLEDs einsetzen. Hierbei kommen die Metallkomplexe mit den Zentralatomen Ir und Pt zum Einsatz. Als Vergleich gemäß dem Stand der Technik wird die Verbindungen lr(ref)
3 verwendet. Die Ergebnisse der OLEDs sind in Tabelle 2 zusammengefasst. Bei den OLEDs zeigt sich hier, dass die erfindungsgemäßen Materialien zu effizienten blau und grün emittierenden OLEDs führen. Tabelle 1: Aufbau der OLEDs
| Bsp. | HTL1 Dicke | HTL2 Dicke | EBL Dicke | EML Dicke | HBL Dicke | ETL Dicke | EIL Dicke |
| 1 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L1)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 2 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L2)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 3 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L3)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 4 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L6)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 5 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L7)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 6 | HIM 20 nm | HTM 5 nm | EBM1 15 nm | M2:M3:Ir(L9)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 7 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L10)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 8 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L13)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 9 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | EBM2:M3:Ir(L13)3 (60%:30%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 10 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M1:Ir(L13)3 (90%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 11 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L13)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 12 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L14)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 13 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L15)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 14 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L18)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 15 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L21)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 16 | HIM 20 nm | HTM 5 nm | EBM1 15 nm | M2:M3:Ir(L22)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 17 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L23)3 (80%:10%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 18 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L23)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 19 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L24)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 20 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L25)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 21 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L26)3 (85%:10%:5%) 40 nm | - | ETM1 30 nm | LiQ 2nm |
| 22 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L27)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 23 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L28)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 24 | HIM 20 nm | HTM 5 nm | EBM1 15 nm | M4:M3:Ir(L31)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 25 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M3:Ir(L35)3 (90%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 26 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M3:Ir(L36)3 (90%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 27 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M3:Ir(L37)3 (90%:10%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 28 | HIM 20 nm | HTM 5nm | EBM1 15 nm | M4:M3:Ir(L39)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 29 | HIM 20 nm | HTM 5 nm | EBM1 15 nm | M4:M3:Ir(L40)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 30 | HIM 20 nm | HTM 5 nm | EBM1 15 nm | M4:M3:Ir(L41)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 31 | HIM 20 nm | HTM 5 nm | EBM1 15 nm | M4:M3:Ir(L42)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 32 | HIM 20 nm | HTM 5 nm | EBM1 15 nm | M4:M3:Ir(L43)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 33 | HIM 20 nm | HTM 5 nm | EBM1 15 nm | M4:M3:Ir(L44)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 34 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L59)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 35 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L60)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 36 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L61)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 37 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L62)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 38 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L63)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 39 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L64)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 40 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L67)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 41 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(C68)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 42 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L69)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 43 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L71)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 44 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L73)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 45 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L74)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 46 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M2:M3:Ir(L75)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 47 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L78)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 48 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L79)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 49 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L80)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 50 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L81)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 51 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M2:M3:Ir(L82)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 52 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L83)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 53 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L85)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 54 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2:M3:Ir(L86)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 55 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L87)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 56 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L88)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 57 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L91)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 58 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L92)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 59 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L93)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 60 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L95)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 61 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L96)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 62 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L97)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 63 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L100)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 64 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L101)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 65 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L103)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 66 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L104)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 67 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L105)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 68 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4:M3:Ir(L106)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 69 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4.M3:Ir(L107)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 70 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4:M3:Ir(L108)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 71 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4:M3:Ir(L109)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 72 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L110)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 73 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4:M3:Ir(L111)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 74 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L112)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 75 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L115)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 76 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L116)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 77 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L117)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 78 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L118)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 79 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L119)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 80 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L120)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 81 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L121)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 82 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L122)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 83 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L123)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 84 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L124)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 85 | HIM 20 nm | HTM 20 nm | EBM2 15 nm | M4:M3:Ir(L125)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 86 | HIM 20 nm | HTM 20 nm | EBM2 15 nm | M4:M3:Ir(L126)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 87 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L128)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 88 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L129)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 89 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L131)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 90 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L132)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 91 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4M3:Ir(L134)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 92 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L135)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 93 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L137)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 94 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4:M3:Ir(L138)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 95 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4:M3:Ir(L139)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 96 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4:M3:Ir(L140)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 97 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L141)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 98 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L142)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 99 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L143)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 100 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L144)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 101 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L146)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 102 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L147)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 103 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L148)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 104 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L149)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 105 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L150)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 106 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L152)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 107 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L153)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 108 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L154)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 109 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4:M3:Ir(L155)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 110 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L156)3 (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 111 | HIM 20 nm | HTM 5 nm | EBM1 15 nm | M2:M3:Ir(L1)2(CL1) (88%:12%) 30 nm | --- | ETM1 30 nm | LiQ 2nm |
| 112 | HIM 20 nm | HTM 5 nm | EBM1 15 nm | M2:M3:Ir(L13)2(CL1) (88%:12%) 30 nm | --- | ETM1 30 nm | LiQ 2nm |
| 113 | HIM 20 nm | HTM 5nm | EBM1 15 nm | M2:M3:Ir(L23)2(CL1) (88%:12%) 30 nm | --- | ETM1 30 nm | LiQ 2nm |
| 114 | HIM 20 nm | HTM 5nm | EBM1 15 nm | M2:M3:Ir(L41)2(CL2) (88%:12%) 30 nm | --- | ETM1 30 nm | LiQ 2nm |
| 115 | HIM 20 nm | HTM 5nm | EBM1 15 nm | M2:M3:Ir(L91)2(CL2) (88%:12%) 30 nm | --- | ETM1 30 nm | LiQ 2nm |
| 116 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L91)2(CL3) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 117 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4:M3:Ir(L91)2(CL4) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 118 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L91)2(CL5) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 119 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3:Ir(L92)2(CL6) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 120 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M4:M3:Ir(L111)2(CL3) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 121 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L120)2(CL3) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 122 | HIM 20 nm | HIM 5 nm | EBM1 15 nm | M4:M3: Ir(L141)2(CL1) (88%:12%) 30 nm | --- | ETM1 30 nm | LiQ 2nm |
| 123 | HIM 20 nm | HTM 5 nm | EBM2' 15 nm | M4:M3: Ir(L13)2(CL17) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 124 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L1)2(CLT) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 125 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L13)2(CL7) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 126 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L23)2(CL7) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 127 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L91)2(CL7) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 128 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L91)2(CL8) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 129 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L92)2(CL8) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 130 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L111)2(CL9) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 131 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L120)2(CL7) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 132 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L147)2(CL9) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 133 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L9)2(CL7) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 134 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L22)2(CL7) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 135 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L91)2(CL11) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 136 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4: M3: Ir(L92)2(CL12) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 137 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L124)2(CL13) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 138 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L91)2(CL14) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 139 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L91)2(CL15) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 140 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L111)2(CL16) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 141 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L13)2(L23) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 142 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L23)2(L13) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 143 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L41)2(L13) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 144 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Ir(L91)2(L111) (85%:10%:5%) 40 nm | --- | ETM1 30 nm | LiQ 2nm |
| 145 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Pt(L13)2(CL1) (88%:12%) 30 nm | --- | ETM1 30 nm | LiQ 2nm |
| 146 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M4:M3: Pt(L91)2(CL17) (85%:10%:5%) 30 nm | --- | ETM1 30 nm | LiQ 2nm |
| Vgl. | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M2: Ir(ref)
(85%:15%) 40 nm | M3 10 nm | Alq3 20 nm | LiF |
| 203 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L163)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 204 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L165)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 205 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L167)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 206 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L168)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 207 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L169)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 208 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:1r(L170)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 209 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L171)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 210 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L172)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 211 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L173)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 212 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L174)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 213 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:)Ir(L175)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 214 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:)Ir(L176)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 215 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L177)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 216 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L178)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 217 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L179)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 218 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2.Ir(L181)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 219 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L183)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 220 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L184)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 221 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L185)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 222 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L186)3 (10%:85%:5%) 40 nm | M5 10nm | ETM1 30 nm | LiQ 2nm |
| 223 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | MS:M2:Ir(L187)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 224 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L188)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 225 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L189)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 226 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L190)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 227 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L191)9 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 228 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M5:M2:Ir(L192)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 229 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L196)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 230 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L198)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 231 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L199)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 232 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L203)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 233 | HIM 20 nm | HTM 5nm | EBM2 15 nm | M5:M2:Ir(L207)3 (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 234 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Pt(L211) (45%:45%:10%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 235 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Pt(L212) (40%:55%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 236 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Pt(L213) (40%:55%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 237 | HIM 20 nm | HIM 5 nm | EBM2 15 nm | M5:M2:Pt(L214) (50%:45%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 238 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Pt(L215) (40%:50%:10%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 239 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Pt(L216) (40%:50%:10%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 240 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Pt(L217) (40%:50%:10%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 241 | HIM 20 nm | HTM 5 nm | EBM2 15 nm | M5:M2:Ir(L218) (10%:85%:5%) 40 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
| 242 | HIM 20 nm | HTM 5 nm | EBM2 10 nm | M5:M2:Pt(L219) (40%:55%:5%) 30 nm | M5 10 nm | ETM1 30 nm | LiQ 2nm |
Tabelle 2: Verwendung von erfindungsgemäßen Verbindungen als Emitter in phosphoreszenten OLEDs
| Bsp. | Effizienz (cd/A) bei 1000 cd/m2 | Spannung (V) 1000 cd/m2 | CIE x/y bei 1000 cd/m1 | LD50 (h) bei 1000 cd/m2 |
| 1 | 13.2 | 6.9 | 0.15/0.28 | 750 |
| 2 | 12.5 | 5.5 | 0.15/0.29 | 260 |
| 3 | 8.7 | 8.5 | 0.16/0.31 | 40 |
| 4 | 15.4 | 5.8 | 0.15/0.30 | 560 |
| 5 | 11.0 | 6.7 | 0.14/0.29 | 430 |
| 6 | 7.9 | 6.5 | 0.15/0.31 | --- |
| 7 | 6.5 | 8.2 | 0.16/0.31 | --- |
| 8 | 26.8 | 7.2 | 0.15/0.30 | 1100 |
| 9 | 14.7 | 5.6 | 0.15/0.28 | 330 |
| 10 | 22.4 | 6.4 | 0.16/033 | --- |
| 11 | 21.1 | 5.5 | 0.15/0.30 | 580 |
| 12 | 29.5 | 6.7 | 0.14/0.29 | 900 |
| 13 | 17.6 | 7.2 | 0.16/0.30 | 650 |
| 14 | 23.3 | 5.9 | 0.16/0.29 | 530 |
| 15 | 11.3 | 5.4 | 0.17/0.29 | --- |
| 16 | 9.7 | 8.6 | 0.16/0.29 | --- |
| 17 | 8.3 | 7.0 | 0.15/0.29 | 580 |
| 18 | 14.0 | 8.2 | 0.15/0.29 | 570 |
| 19 | 9.5 | 5.2 | 0.16/0.29 | 470 |
| 20 | 8.3 | 5.9 | 0.15/0.28 | --- |
| 21 | 10.4 | 6.1 | 0.17/0.33 | --- |
| 22 | 9.7 | 6.4 | 0.16/0.29 | --- |
| 23 | 16.1 | 6.4 | 0.15/0.28 | --- |
| 24 | 12.0 | 7.3 | 0.17/0.32 | --- |
| 25 | 48.1 | 4.0 | 0.36/0.60 | 29000 |
| 26 | 59.7 | 3.5 | 0.36/0.61 | 45000 |
| 27 | 35.2 | 3.8 | 0.17/0.35 | 2000 |
| 28 | 11.2 | 6.3 | 0.16/0.29 | 600 |
| 29 | 22.4 | 5.4 | 0.15/0.33 | 930 |
| 30 | 17.4 | 5.3 | 0.15/0.29 | 990 |
| 31 | 24.3 | 5.5 | 0.15/0.28 | 1000 |
| 32 | 26.3 | 6.3 | 0.15/0.29 | 640 |
| 33 | 12.4 | 5.9 | 0.15/0.30 | --- |
| 34 | 30.9 | 5.7 | 0.14/0.28 | --- |
| 35 | 11.3 | 6.1 | 0.15/0.29 | --- |
| 36 | 9.8 | 7.3 | 0.15/0.29 | --- |
| 37 | 6.5 | 7.1 | 0.15/0.30 | --- |
| 38 | 24.4 | 5.5 | 0.15/0.28 | 1100 |
| 39 | 12.7 | 6.0 | 0.17/0.34 | 450 |
| 40 | 30.9 | 7.0 | 0.15/0.29 | 850 |
| 41 | 14.1 | 8.1 | 0.16/0.30 | 430 |
| 42 | 19.5 | 6.5 | 0.14/0.27 | 520 |
| 43 | 9.9 | 5.7 | 0.15/0.30 | --- |
| 44 | 7.3 | 6.8 | 0.17/0.35 | 470 |
| 45 | 22.5 | 5.2 | 0.18/0.37 | 1500 |
| 46 | 6.4 | 7.6 | 0.18/0.38 | 1450 |
| 47 | 28.3 | 6.2 | 0.17/0.35 | 960 |
| 48 | 14.1 | 7.8 | 0.14/0.26 | 870 |
| 49 | 13.9 | 7.1 | 0.14/0.24 | 540 |
| 50 | 15.8 | 6.1 | 0.15/0.29 | --- |
| 51 | 20.0 | 5.4 | 0.15/0.28 | 1050 |
| 52 | 21.2 | 5.4 | 0.15/0.33 | 780 |
| 53 | 17.9 | 5.6 | 0.15/0.28 | --- |
| 54 | 16.3 | 5.7 | 0.14/0.26 | 560 |
| 55 | 6.3 | 6.8 | 0.17/0.38 | --- |
| 56 | 21.7 | 6.2 | 0.17/0.35 | 350 |
| 57 | 15.7 | 5.3 | 0.15/0.23 | 550 |
| 58 | 16.9 | 5.6 | 0.15/0.21 | 560 |
| 59 | 21.9 | 5.4 | 0.15/0.20 | 680 |
| 60 | 16.3 | 8.1 | 0.15/0.28 | 720 |
| 61 | 25.7 | 6.1 | 0.15/0.26 | 910 |
| 62 | 19.9 | 5.9 | 0.15/0.28 | --- |
| 63 | 15.7 | 6.1 | 0.15/0.24 | 620 |
| 64 | 13.4 | 5.7 | 0.15/0.25 | 710 |
| 65 | 19.8 | 5.6 | 0.15/0.26 | 450 |
| 66 | 17.7 | 8.5 | 0.14/0.23 | 680 |
| 67 | 25.7 | 4.8 | 0.16/0.35 | 1300 |
| 68 | 12.7 | 6.1 | 0.17/0.39 | 2100 |
| 69 | 19.6 | 5.2 | 0.17/0.35 | 2600 |
| 70 | 235 | 5.3 | 0.16/0.33 | 2300 |
| 71 | 16.5 | 4.8 | 0.15/0.23 | 1400 |
| 72 | 16.4 | 4.9 | 0.15/0.24 | 1500 |
| 73 | 18.3 | 4.7 | 0.15/0.22 | --- |
| 74 | 14.8 | 5.5 | 0.15/0.25 | --- |
| 75 | 29.6 | 4.8 | 0.15/0.29 | 970 |
| 76 | 7.6 | 6.8 | 0.16/0.31 | 150 |
| 77 | 12.5 | 6.7 | 0.15/0.30 | 540 |
| 78 | 24.6 | 6.3 | 0.14/0.26 | 360 |
| 79 | 16.7 | 5.2 | 0.15/0.29 | 720 |
| 80 | 16.5 | 6.3 | 0.15/0.23 | 250 |
| 81 | 13.1 | 5.2 | 0.15/0.28 | 760 |
| 82 | 14.9 | 5.5 | 0.15/0.22 | 110 |
| 83 | 9.0 | 5.8 | 0.15/0.25 | 730 |
| 84 | 6.5 | 7.8 | 0.16/0.29 | --- |
| 85 | 8.8 | 7.3 | 0.16/0.30 | --- |
| 86 | 8.6 | 7.4 | 0.16/0.31 | 810 |
| 87 | 11.1 | 7.4 | 0.15/0.24 | 170 |
| 88 | 16.4 | 7.3 | 0.15/0.32 | 870 |
| 89 | 13.1 | 6.4 | 0.15/0.25 | 210 |
| 90 | 18.4 | 5.8 | 0.16/0.28 | 590 |
| 91 | 23.1 | 6.2 | 0.15/0.30 | --- |
| 92 | 12.3 | 7.6 | 0.15/0.28 | --- |
| 93 | 6.4 | 8.0 | 0.16/0.33 | --- |
| 94 | 9.7 | 7.1 | 0.16/0.31 | 180 |
| 95 | 13.4 | 6.5 | 0.15/0.20 | 220 |
| 96 | 12.9 | 5.8 | 0.15/0.24 | 250 |
| 97 | 10.7 | 6.1 | 0.15/0.23 | 270 |
| 98 | 16.4 | 5.7 | 0.15/0.21 | 160 |
| 99 | 6.3 | 5.2 | 0.15/0.25 | --- |
| 100 | 9.6 | 5.6 | 0.15/0.24 | --- |
| 101 | 6.1 | 6.5 | 0.15/0.14 | --- |
| 102 | 26.8 | 5.7 | 0.15/0.29 | 330 |
| 103 | 18.3 | 5.6 | 0.15/0.28 | 370 |
| 104 | 5.5 | 6.0 | 0.15/0.20 | --- |
| 105 | 7.8 | 6.3 | 0.15/0.28 | --- |
| 106 | 11.0 | 7.5 | 0.15/0.19 | 190 |
| 107 | 6.5 | 7.4 | 0.15/0.22 | --- |
| 108 | 7.3 | 7.6 | 0.15/0.21 | --- |
| 109 | 6.2 | 8.1 | 0.15/0.18 | --- |
| 110 | 4.5 | 8.3 | 0.15/0.22 | --- |
| 111 | 241 | 6.3 | 0.15/0.29 | 670 |
| 112 | 22.7 | 5.9 | 0.15/0.30 | 1100 |
| 113 | 18.9 | 5.5 | 0.16/0.31 | 690 |
| 114 | 26.4 | 6.1 | 0.15/0.28 | 1200 |
| 115 | 23.1 | 5.7 | 0.16/0.32 | 270 |
| 116 | 17.2 | 5.1 | 0.15/0.26 | 180 |
| 117 | 14.3 | 5.7 | 0.15/0.28 | 240 |
| 118 | 16.8 | 6.7 | 0.15/0.29 | 170 |
| 119 | 11.2 | 6.2 | 0.15/0.31 | 210 |
| 120 | 160 | 5.1 | 0.15/0.29 | 350 |
| 121 | 14.3 | 5.9 | 0.15/0.25 | 430 |
| 122 | 15.8 | 6.1 | 0.15/0.26 | 2200 |
| 123 | 16.8 | 7.1 | 0.15/0.28 | 480 |
| 124 | 8.3 | 7.0 | 0.15/0.28 | 330 |
| 125 | 11.1 | 7.7 | 0.16/0.30 | 610 |
| 126 | 24.0 | 7.2 | 0.15/0.29 | 510 |
| 127 | 16.5 | 5.6 | 0.15/0.28 | 170 |
| 128 | 32.0 | 5.5 | 0.15/0.23 | 450 |
| 129 | 14.7 | 5.6 | 0.15/0.28 | --- |
| 130 | 19.7 | 6.4 | 0.16/0.33 | --- |
| 131 | 6.6 | 7.1 | 0.15/0.27 | --- |
| 132 | 10.4 | 7.0 | 0.15/0.29 | 320 |
| 133 | 25.6 | 6.1 | 0.15/0.26 | 470 |
| 134 | 6.5 | 7.2 | 0.15/0.27 | 360 |
| 135 | 22.7 | 4.5 | 0.21/0.43 | --- |
| 136 | 27.8 | 4.2 | 0.19/0.42 | --- |
| 137 | 9.6 | 8.7 | 0.16/0.30 | --- |
| 138 | 13.5 | 5.0 | 0.15/0.28 | --- |
| 139 | 19.0 | 6.5 | 0.14/0.25 | --- |
| 140 | 17.7 | 6.5 | 0.15/0.28 | --- |
| 141 | 13.4 | 6.2 | 0.15/0.32 | 1200 |
| 142 | 17.6 | 5.3 | 0.15/0.30 | 990 |
| 143 | 8.8 | 7.0 | 0.16/0.34 | 870 |
| 144 | 6.4 | 6.9 | 0.17/0.35 | 890 |
| 145 | 28.0 | 5.0 | 0.16/0.31 | 360 |
| 146 | 16.0 | 6.3 | 0.15/0.28 | 780 |
| Vgl. | 6.7 | 9.6 | 0.18/0.30 | 300 |
| 203 | 15.8 | 4.8 | 0.15/0.23 | 950 |
| 204 | 9.8 | 5.3 | 0.15/0.22 | --- |
| 205 | 10.2 | 5.4 | 0.15/0.22 | --- |
| 206 | 13.3 | 4.8 | 0.15/0.22 | --- |
| 207 | 14.7 | 4.6 | 0.15/0.26 | --- |
| 208 | 7.3 | 6.2 | 0.15/0.21 | --- |
| 209 | 14.0 | 5.0 | 0.16/0.22 | --- |
| 210 | 21.5 | 4.8 | 0.17/0.26 | 1200 |
| 211 | 17.8 | 5.0 | 0.16/0.25 | --- |
| 212 | 16.4 | 4.9 | 0.15/0.24 | --- |
| 213 | 18.8 | 5.2 | 0.15/0.24 | --- |
| 214 | 17.8 | 4.6 | 0.15/0.23 | --- |
| 215 | 169 | 4.6 | 0.15/0.23 | --- |
| 216 | 19.3 | 4.7 | 0.15/0.23 | --- |
| 217 | 19.7 | 4.7 | 0.1510.23 | --- |
| 218 | 17.5 | 5.0 | 0.15/0.24 | --- |
| 219 | 19.7 | 4.6 | 0.15/0.26 | --- |
| 220 | 12.0 | 5.9 | 0.15/0.21 | --- |
| 221 | 16.5 | 5.1 | 0.16/0.22 | --- |
| 222 | 15.0 | 5.1 | 0.16/0.22 | --- |
| 223 | 19.6 | 4.7 | 0.17/0.26 | --- |
| 224 | 19.0 | 4.8 | 0.1610.25 | --- |
| 225 | 19.5 | 4.8 | 0.15/0.24 | --- |
| 226 | 17.6 | 4.7 | 0.15/0.24 | --- |
| 227 | 15.9 | 4.7 | 0.15/0.23 | --- |
| 228 | 16.0 | 4.9 | 0.15/0.23 | --- |
| 229 | 14.5 | 5.0 | 0.16/0.22 | --- |
| 230 | 10.1 | 5.6 | 0.17/0.26 | --- |
| 231 | 17.8 | 4.8 | 0.15/0.23 | --- |
| 232 | 17.5 | 4.8 | 0.15/0.23 | --- |
| 233 | 17.3 | 4.9 | 0.15/0.23 | --- |
| 234 | 68.0 | 4.3 | 0.30/0.65 | 8000 |
| 235 | 70.0 | 4.2 | 0.29/0.68 | 11000 |
| 236 | 65.3 | 4.2 | 0.30/0.65 | 9500 |
| 237 | 67.4 | 4.3 | 0.30/0.67 | 13000 |
| 238 | 69.0 | 4.3 | 0.30/0.66 | 12000 |
| 239 | 71.2 | 4.2 | 0.25/0.60 | 16000 |
| 240 | 64.6 | 4.4 | 0.27/0.62 | 15000 |
| 241 | 15.6 | 6.5 | 0.16/0.31 | --- |
| 242 | 64.3 | 4.4 | 0.26/0.64 | 20000 |
Tabelle 3: Strukturformeln der verwendeten Materialien
| | |
| HIM | HTM |
| | |
| EBM1 | EBM2 |
| | |
| M1 | M2 |
| | |
| M3 | M4 |
| | |
| M5 | M6 |
| | |
| Ir(ref) | ETM1 |
| | |
| LiQ | Alq3 |
-
Erfindungsgemäße Materialien können auch aus Lösung verwendet werden und führen dort zu wesentlich einfacheren OLEDs gegenüber vakuumprozessierten OLEDs mit dennoch guten Eigenschaften. Die Herstellung solcher Bauteile lehnt sich an die Herstellung polymerer Leuchtdioden (PLEDs) an, die in der Literatur bereits vielfach beschrieben ist (z. B. in der
WO 2004/037887 ). Der Aufbau setzt sich aus Substrat / ITO / PEDOT (80 nm) / Interlayer / Emissionsschicht (80 nm) / Kathode zusammen. Die verwendete Interlayer dient der Lochinjektion; in diesem Fall wird HIL-012 von Merck verwendet. Im vorliegenden Fall werden die erfindungsgemäßen Emitter für die Emissionsschicht neben der Matrices in Toluol gelöst. Der typische Feststoffgehalt solcher Lösungen liegt zwischen 16 und 25 g/L, wenn, wie hier, die für eine Device typische Schichtdicke von 80 nm mittels Spincoating erzielt werden soll. Die Emissionsschicht wird in einer Inertgasatmosphäre, im vorliegenden Fall Argon, aufgeschleudert und 10 min bei 120 °C ausgeheizt. Zuletzt wird eine Kathode aus Barium und Aluminium im Vakuum aufgedampft. Zwischen EML und Kathode können auch die in den vorgenannten Beispielen verwendeten Schichten HBL und ETL per Bedampfung aufgebracht werden, auch kann die Interlayer durch eine oder mehrere Schichten ersetzt werden, die lediglich die Bedingung erfüllen müssen, durch den nachgelagerten Prozessierungsschritt der EML-Abscheidung aus Lösung nicht wieder abgelöst zu werden. Die lösungsprozessierten Devices werden standardmäßig in den Matrices PS (Polystyrol):M6:M1: Ir(LX)
3 (26%:14%:42%:20%) charakterisiert, die genannten OLED-Beispiele sind noch nicht optimiert. Tabelle 4 fasst die erhaltenen Daten zusammen. Bei den prozessierten OLEDs zeigt sich hier, dass die erfindungsgemäßen Materialien zu effizienten blau emittierenden OLEDs führen. Tabelle 4: Ergebnisse mit aus Lösung prozessierten Materialien
| Bsp. | EML mit Emitter 80 nm | Spannung [V] bei 100 cd/m2 | Max. Eff. [cd/A] | CIE (x,y) |
| 147 | Ir(L4)3 | 8.3 | 9.6 | 0.15/0.32 |
| 148 | Ir(L5)3 | 8.6 | 6.6 | 0.16/0.32 |
| 149 | Ir(L8)3 | 7.9 | 10.4 | 0.16/0.33 |
| 150 | Ir(L11)3 | 7.7 | 8.3 | 0.16/0.35 |
| 151 | Ir(L12)3 | 8.6 | 19.1 | 0.16/0.36 |
| 152 | Ir(L16)3 | 7.6 | 14.7 | 0.16/0.35 |
| 153 | Ir(L17)3 | 6.5 | 6.4 | 0.16/0.33 |
| 154 | Ir(L19)3 | 7.4 | 4.8 | 0.17/0.35 |
| 155 | Ir(L29)3 | 8.6 | 9.1 | 0.16/0.35 |
| 156 | Ir(L29)3 | 9.7 | 12.6 | 0.15/0.32 |
| 157 | Ir(L30)3 | 8.0 | 7.6 | 0.15/0.30 |
| 158 | Ir(L32)3 | 7.8 | 9.4 | 0.16/0.33 |
| 159 | Ir(L33)3 | 7.9 | 13.4 | 0.16/0.33 |
| 160 | Ir(L34)3 | 7.9 | 12.8 | 0.1610.32 |
| 161 | Ir(L38)3 | 7.3 | 28.0 | 0.17/0.40 |
| 162 | Ir(L45)3 | 8.1 | 20.3 | 0.16/0.30 |
| 163 | Ir(L46)3 | 8.4 | 6.7 | 0.17/0.35 |
| 164 | Ir(L47)3 | 8.7 | 25.7 | 0.17/0.36 |
| 165 | Ir(L48)3 | 8.0 | 19.8 | 0.16/0.34 |
| 166 | Ir(L49)3 | 7.9 | 8.4 | 0.15/0.34 |
| 167 | Ir(L50)3 | 8.3 | 17.2 | 0.16/0.33 |
| 168 | Ir(L51)3 | 9.5 | 11.2 | 0.15/0.32 |
| 169 | Ir(L52)3 | 10.1 | 16.3 | 0.16/0.33 |
| 170 | Ir(L53)3 | 7.4 | 10.5 | 0.15/0.30 |
| 171 | Ir(L54)3 | 8.0 | 12.7 | 0.16/0.31 |
| 172 | Ir(L55)3 | 7.8 | 21.3 | 0.16/.35 |
| 173 | Ir(L56)3 | 8.1 | 9.0 | 0.16/0.33 |
| 174 | Ir(L57)3 | 7.7 | 27.5 | 0.17/0.38 |
| 175 | Ir(L58)3 | 8.1 | 31.2 | 0.17/0.37 |
| 176 | Ir(L65)3 | 9.1 | 6.5 | 0.17/0.34 |
| 177 | Ir(L66)3 | 7.7 | 5.2 | 0.15/0.30 |
| 178 | Ir(L70)3 | 8.5 | 10.0 | 0.15/0.31 |
| 179 | Ir(L72)3 | 8.5 | 9.9 | 0.15/0.29 |
| 180 | Ir(L76)3 | 8.9 | 19.9 | 0.16/0.28 |
| 181 | Ir(L77)3 | 8.5 | 8.6 | 0.16/0.27 |
| 182 | Ir(L84)3 | 7.9 | 22.5 | 0.18/0.37 |
| 183 | Ir(L89)3 | 9.1 | 4.5 | 0.16/0.33 |
| 184 | Ir(L90)3 | 8.2 | 18.4 | 0.16/0.31 |
| 185 | Ir(L94)3 | 7.8 | 12.1 | 0.15/0.27 |
| 186 | Ir(L98)3 | 9.6 | 8.5 | 0.15/0.26 |
| 187 | Ir(L99)3 | 10.5 | 20.1 | 0.16/0.31 |
| 188 | Ir(L102)3 | 7.6 | 11.9 | 0.15/0.29 |
| 189 | Ir(L113)3 | 9.2 | 14.1 | 0.15/0.30 |
| 190 | Ir(L114)3 | 8.7 | 14.0 | 0.15/0.26 |
| 191 | Ir(L127)3 | 7.8 | 4.5 | 0.16/0.31 |
| 192 | Ir(L130)3 | 8.5 | 7.6 | 0.16/0.35 |
| 193 | Ir(L133)3 | 9.9 | 21.1 | 0.16/0.32 |
| 194 | Ir(Ll 36)3 | 8.6 | 9.0 | 0.16/0.33 |
| 195 | Ir(L145)3 | 7.9 | 5.5 | 0.15/0.30 |
| 196 | Ir(L151)3 | 7.5 | 8.1 | 0.15/0.25 |
| 197 | Ir(L157)3 | 9.4 | 17.1 | 0.15/0.18 |
| 198 | Ir(L158)3 | 7.9 | 6.9 | 0.16/0.19 |
| 199 | Ir(L159)3 | 8.4 | 21.0 | 0.15/0.23 |
| 200 | Ir(L160)3 | 7.6 | 18.6 | 0.16/0.24 |
| 201 | Ir(L161)3 | 7.1 | 20.1 | 0.15/0.24 |
| 202 | Ir(L52)2(CL10) | 32.1 | 18.0 | 0.16/0.38 |
-
Beispiel 243: Weiß emittierende OLEDs
-
Gemäß den allgemeinen Verfahren wird eine weiß emittierende OLED mit folgendem Schichtaufbau hergestellt: Tabelle 5: Aufbau der weißen OLEDs
| Bsp. | HTL1 Dicke | HTL2 Dicke | EML Rot Dicke | EML Blau Dicke | EML Grün Dicke | HBL Dicke | ETL Dicke |
| 243 | HIM 250 nm | HTM 10 nm | EBM2:Ir-R (97%:3%) 9 nm | M2:M5:Ir(L13)3 (45%:50%:5%) 8 nm | M5:Ir-G (90%:10%) 7 nm | M5 10 nm | ETM1:LiQ (50%:50%) 30 nm |
-
Strukturformeln der verwendeten Materialien
Tabelle 5: Deviceergebnisse
| Bsp. | Effizienz (cd/A) bei 1000 cd/m2 | Spannung (V) 1000 cd/m2 | CIE x/y bei 1000 cd/m2 CRI | LD50 (h) bei 1000 cd/m2 |
| 243 | 33.0 | 6.2 | 0.45/0.44 80 | 1500 |