TWI767201B - 絲狀病毒科病毒感染之治療 - Google Patents
絲狀病毒科病毒感染之治療 Download PDFInfo
- Publication number
- TWI767201B TWI767201B TW109109475A TW109109475A TWI767201B TW I767201 B TWI767201 B TW I767201B TW 109109475 A TW109109475 A TW 109109475A TW 109109475 A TW109109475 A TW 109109475A TW I767201 B TWI767201 B TW I767201B
- Authority
- TW
- Taiwan
- Prior art keywords
- mmol
- compound
- group
- compounds
- alkyl
- Prior art date
Links
- 238000000034 method Methods 0.000 title abstract description 62
- 241000711950 Filoviridae Species 0.000 title abstract description 53
- 230000009385 viral infection Effects 0.000 title abstract description 12
- 150000001875 compounds Chemical class 0.000 claims abstract description 292
- 239000000203 mixture Substances 0.000 claims abstract description 190
- -1 riboside phosphates Chemical class 0.000 claims abstract description 136
- 241001115402 Ebolavirus Species 0.000 claims abstract description 44
- 238000002360 preparation method Methods 0.000 claims description 74
- 150000003839 salts Chemical class 0.000 claims description 55
- 239000003814 drug Substances 0.000 claims description 38
- 201000011001 Ebola Hemorrhagic Fever Diseases 0.000 claims description 24
- 229940124597 therapeutic agent Drugs 0.000 claims description 22
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 12
- 229950008454 favipiravir Drugs 0.000 claims description 11
- ZCGNOVWYSGBHAU-UHFFFAOYSA-N favipiravir Chemical compound NC(=O)C1=NC(F)=CNC1=O ZCGNOVWYSGBHAU-UHFFFAOYSA-N 0.000 claims description 9
- 239000003112 inhibitor Substances 0.000 claims description 8
- 239000003242 anti bacterial agent Substances 0.000 claims description 6
- 229940088710 antibiotic agent Drugs 0.000 claims description 5
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 claims description 4
- AMFDITJFBUXZQN-KUBHLMPHSA-N (2s,3s,4r,5r)-2-(4-amino-5h-pyrrolo[3,2-d]pyrimidin-7-yl)-5-(hydroxymethyl)pyrrolidine-3,4-diol Chemical compound C=1NC=2C(N)=NC=NC=2C=1[C@@H]1N[C@H](CO)[C@@H](O)[C@H]1O AMFDITJFBUXZQN-KUBHLMPHSA-N 0.000 claims description 3
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 claims description 3
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 claims description 3
- 229940121375 antifungal agent Drugs 0.000 claims description 3
- 239000003430 antimalarial agent Substances 0.000 claims description 3
- 229940124641 pain reliever Drugs 0.000 claims description 3
- 229960000329 ribavirin Drugs 0.000 claims description 3
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 claims description 3
- 229960005486 vaccine Drugs 0.000 claims description 3
- 229960001722 verapamil Drugs 0.000 claims description 3
- KSHJXDWYTZJUEI-UHFFFAOYSA-N 1-cyclopropyl-3-[[1-(4-hydroxybutyl)benzimidazol-2-yl]methyl]imidazo[4,5-c]pyridin-2-one Chemical compound N=1C2=CC=CC=C2N(CCCCO)C=1CN(C1=O)C2=CN=CC=C2N1C1CC1 KSHJXDWYTZJUEI-UHFFFAOYSA-N 0.000 claims description 2
- BLUJRJMLDHEMRX-UHFFFAOYSA-N 2-[[2-hydroxy-5-[(5-methyltetrazol-1-yl)iminomethyl]phenyl]-(4-hydroxyphenyl)methyl]-4-[(5-methyltetrazol-1-yl)iminomethyl]phenol Chemical compound Cc1nnnn1N=Cc1ccc(O)c(c1)C(c1ccc(O)cc1)c1cc(C=Nn2nnnc2C)ccc1O BLUJRJMLDHEMRX-UHFFFAOYSA-N 0.000 claims description 2
- ITPDYQOUSLNIHG-UHFFFAOYSA-N Amiodarone hydrochloride Chemical compound [Cl-].CCCCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(I)=C(OCC[NH+](CC)CC)C(I)=C1 ITPDYQOUSLNIHG-UHFFFAOYSA-N 0.000 claims description 2
- 229940124722 Ebola vaccine Drugs 0.000 claims description 2
- 101001124388 Homo sapiens NPC intracellular cholesterol transporter 1 Proteins 0.000 claims description 2
- 102100029565 NPC intracellular cholesterol transporter 1 Human genes 0.000 claims description 2
- 206010037660 Pyrexia Diseases 0.000 claims description 2
- WXJFKKQWPMNTIM-VWLOTQADSA-N [(2s)-1-(4-amino-2-oxopyrimidin-1-yl)-3-hydroxypropan-2-yl]oxymethyl-(3-hexadecoxypropoxy)phosphinic acid Chemical compound CCCCCCCCCCCCCCCCOCCCOP(O)(=O)CO[C@H](CO)CN1C=CC(N)=NC1=O WXJFKKQWPMNTIM-VWLOTQADSA-N 0.000 claims description 2
- 229960005260 amiodarone Drugs 0.000 claims description 2
- 230000003474 anti-emetic effect Effects 0.000 claims description 2
- 230000000843 anti-fungal effect Effects 0.000 claims description 2
- 239000003793 antidiarrheal agent Substances 0.000 claims description 2
- 229940125714 antidiarrheal agent Drugs 0.000 claims description 2
- 239000002111 antiemetic agent Substances 0.000 claims description 2
- 229940125683 antiemetic agent Drugs 0.000 claims description 2
- 229950005107 brincidofovir Drugs 0.000 claims description 2
- 239000011692 calcium ascorbate Substances 0.000 claims description 2
- ZQTNQVWKHCQYLQ-UHFFFAOYSA-N dronedarone Chemical compound C1=CC(OCCCN(CCCC)CCCC)=CC=C1C(=O)C1=C(CCCC)OC2=CC=C(NS(C)(=O)=O)C=C12 ZQTNQVWKHCQYLQ-UHFFFAOYSA-N 0.000 claims description 2
- 229960002084 dronedarone Drugs 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 235000020786 mineral supplement Nutrition 0.000 claims description 2
- 229940029985 mineral supplement Drugs 0.000 claims description 2
- 229960001521 motavizumab Drugs 0.000 claims description 2
- 229960000402 palivizumab Drugs 0.000 claims description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 2
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 claims description 2
- 235000011178 triphosphate Nutrition 0.000 claims description 2
- 239000001226 triphosphate Substances 0.000 claims description 2
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 claims description 2
- 241000701161 unidentified adenovirus Species 0.000 claims description 2
- 239000011782 vitamin Substances 0.000 claims description 2
- 229930003231 vitamin Natural products 0.000 claims description 2
- 229940088594 vitamin Drugs 0.000 claims description 2
- 150000003722 vitamin derivatives Chemical class 0.000 claims description 2
- 235000019195 vitamin supplement Nutrition 0.000 claims description 2
- MTPVBMVUENFFLL-HXUWFJFHSA-N 1-(2-fluorophenyl)-3-[(3s)-2-oxo-5-phenyl-1,3-dihydro-1,4-benzodiazepin-3-yl]urea Chemical compound FC1=CC=CC=C1NC(=O)N[C@@H]1C(=O)NC2=CC=CC=C2C(C=2C=CC=CC=2)=N1 MTPVBMVUENFFLL-HXUWFJFHSA-N 0.000 claims 1
- 208000000464 Henipavirus Infections Diseases 0.000 claims 1
- 206010064034 Nipah virus infection Diseases 0.000 claims 1
- 230000002757 inflammatory effect Effects 0.000 claims 1
- BRFMYUCUGXFMIO-UHFFFAOYSA-N phosphono dihydrogen phosphate phosphoric acid Chemical compound OP(O)(O)=O.OP(O)(=O)OP(O)(O)=O BRFMYUCUGXFMIO-UHFFFAOYSA-N 0.000 claims 1
- 238000011282 treatment Methods 0.000 abstract description 28
- 241001115401 Marburgvirus Species 0.000 abstract description 18
- 239000008194 pharmaceutical composition Substances 0.000 abstract description 12
- 229940002612 prodrug Drugs 0.000 abstract description 12
- 239000000651 prodrug Substances 0.000 abstract description 12
- 241000439488 Cuevavirus Species 0.000 abstract description 4
- 229910019142 PO4 Inorganic materials 0.000 abstract 1
- 235000021317 phosphate Nutrition 0.000 abstract 1
- 150000008223 ribosides Chemical class 0.000 abstract 1
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical group C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 131
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 95
- 239000000243 solution Substances 0.000 description 90
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 84
- 210000004027 cell Anatomy 0.000 description 80
- 238000006243 chemical reaction Methods 0.000 description 78
- 239000011541 reaction mixture Substances 0.000 description 77
- 125000004432 carbon atom Chemical group C* 0.000 description 75
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 74
- 238000005481 NMR spectroscopy Methods 0.000 description 72
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 69
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 67
- 125000000623 heterocyclic group Chemical group 0.000 description 62
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 58
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 56
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 54
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 52
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 51
- 125000000217 alkyl group Chemical group 0.000 description 50
- 229910052799 carbon Inorganic materials 0.000 description 49
- 239000000047 product Substances 0.000 description 46
- 239000004480 active ingredient Substances 0.000 description 45
- 208000007136 Filoviridae Infections Diseases 0.000 description 44
- 235000019439 ethyl acetate Nutrition 0.000 description 43
- 150000002148 esters Chemical class 0.000 description 42
- 125000003118 aryl group Chemical group 0.000 description 41
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 40
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 40
- 239000007787 solid Substances 0.000 description 38
- 230000002829 reductive effect Effects 0.000 description 37
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 35
- 238000009472 formulation Methods 0.000 description 35
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 35
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 34
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 34
- 150000001721 carbon Chemical group 0.000 description 32
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropyl acetate Chemical compound CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 32
- 238000003556 assay Methods 0.000 description 30
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 30
- 230000005764 inhibitory process Effects 0.000 description 30
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 30
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 29
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 28
- 208000015181 infectious disease Diseases 0.000 description 27
- 229910052760 oxygen Inorganic materials 0.000 description 27
- 229910052739 hydrogen Inorganic materials 0.000 description 26
- 239000012044 organic layer Substances 0.000 description 26
- 125000001424 substituent group Chemical group 0.000 description 26
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 25
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 24
- 241000700605 Viruses Species 0.000 description 24
- 239000002585 base Substances 0.000 description 24
- 238000010898 silica gel chromatography Methods 0.000 description 24
- 210000001519 tissue Anatomy 0.000 description 24
- 230000003612 virological effect Effects 0.000 description 24
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 23
- 238000010790 dilution Methods 0.000 description 22
- 239000012895 dilution Substances 0.000 description 22
- 229940011051 isopropyl acetate Drugs 0.000 description 22
- GWYFCOCPABKNJV-UHFFFAOYSA-M isovalerate Chemical compound CC(C)CC([O-])=O GWYFCOCPABKNJV-UHFFFAOYSA-M 0.000 description 22
- 239000002609 medium Substances 0.000 description 22
- 230000029812 viral genome replication Effects 0.000 description 22
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 21
- 229910052757 nitrogen Inorganic materials 0.000 description 21
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 20
- 239000011780 sodium chloride Substances 0.000 description 20
- 125000004648 C2-C8 alkenyl group Chemical group 0.000 description 19
- 239000002904 solvent Substances 0.000 description 19
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 18
- 239000007983 Tris buffer Substances 0.000 description 18
- 229940125904 compound 1 Drugs 0.000 description 18
- 239000010410 layer Substances 0.000 description 18
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 18
- 229920006395 saturated elastomer Polymers 0.000 description 18
- 125000004649 C2-C8 alkynyl group Chemical group 0.000 description 17
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 17
- 239000005090 green fluorescent protein Substances 0.000 description 17
- 125000006239 protecting group Chemical group 0.000 description 17
- 238000003756 stirring Methods 0.000 description 17
- 229910052717 sulfur Inorganic materials 0.000 description 17
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 16
- 125000000304 alkynyl group Chemical group 0.000 description 16
- 230000000694 effects Effects 0.000 description 16
- 238000011534 incubation Methods 0.000 description 16
- 239000012453 solvate Substances 0.000 description 16
- 238000012360 testing method Methods 0.000 description 16
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 15
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical group C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 15
- 241000282412 Homo Species 0.000 description 15
- 125000003342 alkenyl group Chemical group 0.000 description 15
- 125000004429 atom Chemical group 0.000 description 15
- 238000013207 serial dilution Methods 0.000 description 15
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 14
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 14
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 14
- 230000000840 anti-viral effect Effects 0.000 description 14
- CQRPUKWAZPZXTO-UHFFFAOYSA-M magnesium;2-methylpropane;chloride Chemical compound [Mg+2].[Cl-].C[C-](C)C CQRPUKWAZPZXTO-UHFFFAOYSA-M 0.000 description 14
- 235000019260 propionic acid Nutrition 0.000 description 14
- 239000002002 slurry Substances 0.000 description 14
- 235000017557 sodium bicarbonate Nutrition 0.000 description 14
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 14
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 13
- 239000006143 cell culture medium Substances 0.000 description 13
- 239000003153 chemical reaction reagent Substances 0.000 description 13
- 239000000460 chlorine Substances 0.000 description 13
- 238000004128 high performance liquid chromatography Methods 0.000 description 13
- 239000012074 organic phase Substances 0.000 description 13
- 230000001629 suppression Effects 0.000 description 13
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 12
- 239000002253 acid Substances 0.000 description 12
- 125000003710 aryl alkyl group Chemical group 0.000 description 12
- 125000005884 carbocyclylalkyl group Chemical group 0.000 description 12
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 12
- 125000001072 heteroaryl group Chemical group 0.000 description 12
- 230000002401 inhibitory effect Effects 0.000 description 12
- 239000000523 sample Substances 0.000 description 12
- 239000000126 substance Substances 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 12
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 11
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 11
- RZZIKHSWRWWPAN-UHFFFAOYSA-N oxolane-2-carbonitrile Chemical compound N#CC1CCCO1 RZZIKHSWRWWPAN-UHFFFAOYSA-N 0.000 description 11
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 11
- 125000003107 substituted aryl group Chemical group 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 10
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 10
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 10
- 239000013553 cell monolayer Substances 0.000 description 10
- 229960004106 citric acid Drugs 0.000 description 10
- 235000015165 citric acid Nutrition 0.000 description 10
- 229910052805 deuterium Inorganic materials 0.000 description 10
- 238000001727 in vivo Methods 0.000 description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- JNODDICFTDYODH-UHFFFAOYSA-N 2-hydroxytetrahydrofuran Chemical compound OC1CCCO1 JNODDICFTDYODH-UHFFFAOYSA-N 0.000 description 9
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 9
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 9
- 125000004452 carbocyclyl group Chemical group 0.000 description 9
- 235000019253 formic acid Nutrition 0.000 description 9
- 125000004404 heteroalkyl group Chemical group 0.000 description 9
- 125000005842 heteroatom Chemical group 0.000 description 9
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 9
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Chemical group C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 9
- 239000007937 lozenge Substances 0.000 description 9
- 239000000463 material Substances 0.000 description 9
- 238000002953 preparative HPLC Methods 0.000 description 9
- 241000894007 species Species 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 8
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 8
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 8
- 125000002947 alkylene group Chemical group 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 239000012267 brine Substances 0.000 description 8
- TXFOLHZMICYNRM-UHFFFAOYSA-N dichlorophosphoryloxybenzene Chemical compound ClP(Cl)(=O)OC1=CC=CC=C1 TXFOLHZMICYNRM-UHFFFAOYSA-N 0.000 description 8
- 201000010099 disease Diseases 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 239000003995 emulsifying agent Substances 0.000 description 8
- 239000000839 emulsion Substances 0.000 description 8
- 125000005843 halogen group Chemical group 0.000 description 8
- 239000004615 ingredient Substances 0.000 description 8
- 239000000543 intermediate Substances 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- IWCVDCOJSPWGRW-UHFFFAOYSA-M magnesium;benzene;chloride Chemical compound [Mg+2].[Cl-].C1=CC=[C-]C=C1 IWCVDCOJSPWGRW-UHFFFAOYSA-M 0.000 description 8
- 239000001301 oxygen Substances 0.000 description 8
- 229910052938 sodium sulfate Inorganic materials 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- 125000000547 substituted alkyl group Chemical group 0.000 description 8
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 7
- BTJIUGUIPKRLHP-UHFFFAOYSA-N 4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1 BTJIUGUIPKRLHP-UHFFFAOYSA-N 0.000 description 7
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 7
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical group C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 7
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 7
- 229960000583 acetic acid Drugs 0.000 description 7
- 229940024606 amino acid Drugs 0.000 description 7
- 235000001014 amino acid Nutrition 0.000 description 7
- 150000001413 amino acids Chemical class 0.000 description 7
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 7
- 238000004113 cell culture Methods 0.000 description 7
- 229910052801 chlorine Inorganic materials 0.000 description 7
- 239000000706 filtrate Substances 0.000 description 7
- 229910052736 halogen Inorganic materials 0.000 description 7
- 239000005457 ice water Substances 0.000 description 7
- 229910052740 iodine Inorganic materials 0.000 description 7
- 150000002596 lactones Chemical class 0.000 description 7
- 229910001629 magnesium chloride Inorganic materials 0.000 description 7
- 125000004433 nitrogen atom Chemical group N* 0.000 description 7
- 239000002777 nucleoside Substances 0.000 description 7
- 238000012216 screening Methods 0.000 description 7
- 238000013456 study Methods 0.000 description 7
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical group CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 7
- 125000006736 (C6-C20) aryl group Chemical group 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 6
- 102000003886 Glycoproteins Human genes 0.000 description 6
- 108090000288 Glycoproteins Proteins 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 241000439489 Lloviu cuevavirus Species 0.000 description 6
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical group CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 6
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical group C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 208000036142 Viral infection Diseases 0.000 description 6
- 125000002015 acyclic group Chemical group 0.000 description 6
- 125000003545 alkoxy group Chemical group 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 6
- 229910052794 bromium Inorganic materials 0.000 description 6
- 125000002837 carbocyclic group Chemical group 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 239000006071 cream Substances 0.000 description 6
- 125000000753 cycloalkyl group Chemical group 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 239000000796 flavoring agent Substances 0.000 description 6
- 229910052731 fluorine Inorganic materials 0.000 description 6
- 125000004449 heterocyclylalkenyl group Chemical group 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 230000002458 infectious effect Effects 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 230000002503 metabolic effect Effects 0.000 description 6
- 239000003607 modifier Substances 0.000 description 6
- MWUXSHHQAYIFBG-UHFFFAOYSA-N nitrogen oxide Inorganic materials O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 238000012856 packing Methods 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 125000005017 substituted alkenyl group Chemical group 0.000 description 6
- 125000004426 substituted alkynyl group Chemical group 0.000 description 6
- 239000000375 suspending agent Substances 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 5
- 240000007472 Leucaena leucocephala Species 0.000 description 5
- 208000030156 Marburg disease Diseases 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical group CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 5
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 241001115376 Sudan ebolavirus Species 0.000 description 5
- 235000011054 acetic acid Nutrition 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 229960003767 alanine Drugs 0.000 description 5
- 125000004450 alkenylene group Chemical group 0.000 description 5
- 125000004419 alkynylene group Chemical group 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 125000002619 bicyclic group Chemical group 0.000 description 5
- 239000012472 biological sample Substances 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 238000002648 combination therapy Methods 0.000 description 5
- 230000008878 coupling Effects 0.000 description 5
- 238000010168 coupling process Methods 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 230000000120 cytopathologic effect Effects 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- 239000002270 dispersing agent Substances 0.000 description 5
- 239000000284 extract Substances 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 150000004665 fatty acids Chemical class 0.000 description 5
- 125000000524 functional group Chemical group 0.000 description 5
- 239000008187 granular material Substances 0.000 description 5
- 150000002367 halogens Chemical class 0.000 description 5
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 5
- 239000002207 metabolite Substances 0.000 description 5
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 230000000704 physical effect Effects 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 239000003755 preservative agent Substances 0.000 description 5
- 230000002265 prevention Effects 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 238000004007 reversed phase HPLC Methods 0.000 description 5
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 5
- 229910000077 silane Inorganic materials 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- 239000003765 sweetening agent Substances 0.000 description 5
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 5
- 238000001195 ultra high performance liquid chromatography Methods 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 4
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 4
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 4
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 4
- RUKDVLFJSMVBLV-UHFFFAOYSA-N 5-iodo-1h-pyrazole Chemical compound IC1=CC=NN1 RUKDVLFJSMVBLV-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- 241000526636 Nipah henipavirus Species 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical group C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical group C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- 241001115394 Reston ebolavirus Species 0.000 description 4
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 4
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 241001115400 Zaire ebolavirus Species 0.000 description 4
- 125000003282 alkyl amino group Chemical group 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 239000007900 aqueous suspension Substances 0.000 description 4
- 125000005018 aryl alkenyl group Chemical group 0.000 description 4
- 125000005015 aryl alkynyl group Chemical group 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 238000012054 celltiter-glo Methods 0.000 description 4
- 239000007859 condensation product Substances 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 235000013355 food flavoring agent Nutrition 0.000 description 4
- 235000003599 food sweetener Nutrition 0.000 description 4
- 229940057995 liquid paraffin Drugs 0.000 description 4
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 4
- 235000019341 magnesium sulphate Nutrition 0.000 description 4
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 238000010899 nucleation Methods 0.000 description 4
- 150000003833 nucleoside derivatives Chemical class 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 229910052698 phosphorus Inorganic materials 0.000 description 4
- 235000015320 potassium carbonate Nutrition 0.000 description 4
- 125000006413 ring segment Chemical group 0.000 description 4
- 239000000741 silica gel Substances 0.000 description 4
- 229910002027 silica gel Inorganic materials 0.000 description 4
- 229910052710 silicon Inorganic materials 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 239000005720 sucrose Substances 0.000 description 4
- 239000011593 sulfur Substances 0.000 description 4
- 230000002195 synergetic effect Effects 0.000 description 4
- 125000001544 thienyl group Chemical group 0.000 description 4
- 238000004809 thin layer chromatography Methods 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 3
- HBZBAMXERPYTFS-SECBINFHSA-N (4S)-2-(6,7-dihydro-5H-pyrrolo[3,2-f][1,3]benzothiazol-2-yl)-4,5-dihydro-1,3-thiazole-4-carboxylic acid Chemical compound OC(=O)[C@H]1CSC(=N1)c1nc2cc3CCNc3cc2s1 HBZBAMXERPYTFS-SECBINFHSA-N 0.000 description 3
- HEWZVZIVELJPQZ-UHFFFAOYSA-N 2,2-dimethoxypropane Chemical compound COC(C)(C)OC HEWZVZIVELJPQZ-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- NPRYCHLHHVWLQZ-TURQNECASA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynylpurin-8-one Chemical compound NC1=NC=C2N(C(N(C2=N1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C NPRYCHLHHVWLQZ-TURQNECASA-N 0.000 description 3
- YJKXXWQTIJDTET-UHFFFAOYSA-N 2-ethylbutyl propanoate Chemical compound CCC(CC)COC(=O)CC YJKXXWQTIJDTET-UHFFFAOYSA-N 0.000 description 3
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 3
- 241000416162 Astragalus gummifer Species 0.000 description 3
- 241000884921 Bundibugyo ebolavirus Species 0.000 description 3
- NQSOMRSHXPZDCC-HHSZUNSFSA-N CCC(CC)COC(=O)[C@H](C)NP(=O)(Oc1ccccc1)Oc1c(F)c(F)c(F)c(F)c1F Chemical compound CCC(CC)COC(=O)[C@H](C)NP(=O)(Oc1ccccc1)Oc1c(F)c(F)c(F)c(F)c1F NQSOMRSHXPZDCC-HHSZUNSFSA-N 0.000 description 3
- 239000004215 Carbon black (E152) Substances 0.000 description 3
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 3
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical group C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 3
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 3
- 108060001084 Luciferase Proteins 0.000 description 3
- 239000005089 Luciferase Substances 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 235000019483 Peanut oil Nutrition 0.000 description 3
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical group C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 3
- 235000021355 Stearic acid Nutrition 0.000 description 3
- 241001115374 Tai Forest ebolavirus Species 0.000 description 3
- 229920001615 Tragacanth Polymers 0.000 description 3
- 108020000999 Viral RNA Proteins 0.000 description 3
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 3
- 150000001335 aliphatic alkanes Chemical class 0.000 description 3
- 150000001345 alkine derivatives Chemical class 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 235000006708 antioxidants Nutrition 0.000 description 3
- 229960005070 ascorbic acid Drugs 0.000 description 3
- 239000011668 ascorbic acid Substances 0.000 description 3
- 235000010323 ascorbic acid Nutrition 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- VYLVYHXQOHJDJL-UHFFFAOYSA-K cerium trichloride Chemical compound Cl[Ce](Cl)Cl VYLVYHXQOHJDJL-UHFFFAOYSA-K 0.000 description 3
- 229940126208 compound 22 Drugs 0.000 description 3
- 229940127204 compound 29 Drugs 0.000 description 3
- JNGZXGGOCLZBFB-IVCQMTBJSA-N compound E Chemical compound N([C@@H](C)C(=O)N[C@@H]1C(N(C)C2=CC=CC=C2C(C=2C=CC=CC=2)=N1)=O)C(=O)CC1=CC(F)=CC(F)=C1 JNGZXGGOCLZBFB-IVCQMTBJSA-N 0.000 description 3
- 238000013270 controlled release Methods 0.000 description 3
- 239000007822 coupling agent Substances 0.000 description 3
- 239000012351 deprotecting agent Substances 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 238000006073 displacement reaction Methods 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 230000036267 drug metabolism Effects 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- FPFQPLFYTKMCHN-PPHPATTJSA-N ethyl (2s)-2-amino-3-phenylpropanoate;hydron;chloride Chemical compound Cl.CCOC(=O)[C@@H](N)CC1=CC=CC=C1 FPFQPLFYTKMCHN-PPHPATTJSA-N 0.000 description 3
- YREPHXXOTHNLEV-UHFFFAOYSA-N ethyl 2-amino-2-methylpropanoate;hydrochloride Chemical compound Cl.CCOC(=O)C(C)(C)N YREPHXXOTHNLEV-UHFFFAOYSA-N 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- ZTQSADJAYQOCDD-UHFFFAOYSA-N ginsenoside-Rd2 Natural products C1CC(C2(CCC3C(C)(C)C(OC4C(C(O)C(O)C(CO)O4)O)CCC3(C)C2CC2O)C)(C)C2C1C(C)(CCC=C(C)C)OC(C(C(O)C1O)O)OC1COC1OCC(O)C(O)C1O ZTQSADJAYQOCDD-UHFFFAOYSA-N 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 229930195733 hydrocarbon Natural products 0.000 description 3
- 150000002430 hydrocarbons Chemical class 0.000 description 3
- 238000003384 imaging method Methods 0.000 description 3
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 3
- 125000000842 isoxazolyl group Chemical group 0.000 description 3
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 3
- DBTNVRCCIDISMV-UHFFFAOYSA-L lithium;magnesium;propane;dichloride Chemical compound [Li+].[Mg+2].[Cl-].[Cl-].C[CH-]C DBTNVRCCIDISMV-UHFFFAOYSA-L 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- 239000002480 mineral oil Substances 0.000 description 3
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 3
- 239000003471 mutagenic agent Substances 0.000 description 3
- BXWNKGSJHAJOGX-UHFFFAOYSA-N n-hexadecyl alcohol Natural products CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 3
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 239000000312 peanut oil Substances 0.000 description 3
- 239000011574 phosphorus Substances 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 230000002335 preservative effect Effects 0.000 description 3
- FNEIUFZZTHRBKT-UHFFFAOYSA-N propan-2-yl 2-amino-2-methylpropanoate;hydrochloride Chemical compound Cl.CC(C)OC(=O)C(C)(C)N FNEIUFZZTHRBKT-UHFFFAOYSA-N 0.000 description 3
- 238000011321 prophylaxis Methods 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- 230000009257 reactivity Effects 0.000 description 3
- 238000003753 real-time PCR Methods 0.000 description 3
- 238000010839 reverse transcription Methods 0.000 description 3
- 239000010703 silicon Substances 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 235000017550 sodium carbonate Nutrition 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 239000008117 stearic acid Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 235000010487 tragacanth Nutrition 0.000 description 3
- 239000000196 tragacanth Substances 0.000 description 3
- 229940116362 tragacanth Drugs 0.000 description 3
- WVLBCYQITXONBZ-UHFFFAOYSA-N trimethyl phosphate Chemical compound COP(=O)(OC)OC WVLBCYQITXONBZ-UHFFFAOYSA-N 0.000 description 3
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 3
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Substances C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 3
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 3
- 210000002700 urine Anatomy 0.000 description 3
- 210000002845 virion Anatomy 0.000 description 3
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 2
- DATPFTPVGIHCCM-BYPYZUCNSA-N (2s)-2-(ethylamino)propanoic acid Chemical compound CCN[C@@H](C)C(O)=O DATPFTPVGIHCCM-BYPYZUCNSA-N 0.000 description 2
- AYJORTVGNUTUMD-WCCKRBBISA-N (2s)-2-(ethylamino)propanoic acid;hydrochloride Chemical compound Cl.CCN[C@@H](C)C(O)=O AYJORTVGNUTUMD-WCCKRBBISA-N 0.000 description 2
- LDHBSABBBAUMCZ-UBFVSLLYSA-N (3r,4r,5r)-3,4-bis(phenylmethoxy)-5-(phenylmethoxymethyl)oxolan-2-one Chemical compound C([C@H]1OC([C@@H]([C@@H]1OCC=1C=CC=CC=1)OCC=1C=CC=CC=1)=O)OCC1=CC=CC=C1 LDHBSABBBAUMCZ-UBFVSLLYSA-N 0.000 description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 2
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 2
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 2
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Chemical compound C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 description 2
- XBNGYFFABRKICK-UHFFFAOYSA-N 2,3,4,5,6-pentafluorophenol Chemical compound OC1=C(F)C(F)=C(F)C(F)=C1F XBNGYFFABRKICK-UHFFFAOYSA-N 0.000 description 2
- BWZVCCNYKMEVEX-UHFFFAOYSA-N 2,4,6-Trimethylpyridine Chemical compound CC1=CC(C)=NC(C)=C1 BWZVCCNYKMEVEX-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- CYHSHMQZLJWIQQ-UHFFFAOYSA-N 2-(hydroxymethyl)oxolane-2-carbonitrile Chemical compound OCC1(CCCO1)C#N CYHSHMQZLJWIQQ-UHFFFAOYSA-N 0.000 description 2
- CSURKLFFMMBEFL-YDIHDLSKSA-N 2-ethylbutyl (2s)-2-[[(4-nitrophenoxy)-phenoxyphosphoryl]amino]propanoate Chemical compound C=1C=C([N+]([O-])=O)C=CC=1OP(=O)(N[C@@H](C)C(=O)OCC(CC)CC)OC1=CC=CC=C1 CSURKLFFMMBEFL-YDIHDLSKSA-N 0.000 description 2
- DEAGOVGXNPHPIJ-FJXQXJEOSA-N 2-ethylbutyl (2s)-2-aminopropanoate;hydrochloride Chemical compound Cl.CCC(CC)COC(=O)[C@H](C)N DEAGOVGXNPHPIJ-FJXQXJEOSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- 125000004398 2-methyl-2-butyl group Chemical group CC(C)(CC)* 0.000 description 2
- 125000004918 2-methyl-2-pentyl group Chemical group CC(C)(CCC)* 0.000 description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- 125000004917 3-methyl-2-butyl group Chemical group CC(C(C)*)C 0.000 description 2
- 125000004919 3-methyl-2-pentyl group Chemical group CC(C(C)*)CC 0.000 description 2
- 125000004921 3-methyl-3-pentyl group Chemical group CC(CC)(CC)* 0.000 description 2
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- 125000004920 4-methyl-2-pentyl group Chemical group CC(CC(C)*)C 0.000 description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 2
- SVXNJCYYMRMXNM-UHFFFAOYSA-N 5-amino-2h-1,2,4-triazin-3-one Chemical compound NC=1C=NNC(=O)N=1 SVXNJCYYMRMXNM-UHFFFAOYSA-N 0.000 description 2
- 125000004939 6-pyridyl group Chemical group N1=CC=CC=C1* 0.000 description 2
- 125000003341 7 membered heterocyclic group Chemical group 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical group C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 2
- 229930024421 Adenine Natural products 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- 229910015844 BCl3 Inorganic materials 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 229930186147 Cephalosporin Natural products 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 206010012735 Diarrhoea Diseases 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 239000005977 Ethylene Substances 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 206010061192 Haemorrhagic fever Diseases 0.000 description 2
- 208000032843 Hemorrhage Diseases 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- IJMWOMHMDSDKGK-UHFFFAOYSA-N Isopropyl propionate Chemical compound CCC(=O)OC(C)C IJMWOMHMDSDKGK-UHFFFAOYSA-N 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical class C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- 108010061100 Nucleoproteins Proteins 0.000 description 2
- 102000011931 Nucleoproteins Human genes 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- DLRVVLDZNNYCBX-UHFFFAOYSA-N Polydextrose Polymers OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(O)O1 DLRVVLDZNNYCBX-UHFFFAOYSA-N 0.000 description 2
- 206010036790 Productive cough Diseases 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical group O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 2
- 239000011149 active material Substances 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 229960000643 adenine Drugs 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 150000003973 alkyl amines Chemical class 0.000 description 2
- 125000004414 alkyl thio group Chemical group 0.000 description 2
- 125000001118 alkylidene group Chemical group 0.000 description 2
- 235000008206 alpha-amino acids Nutrition 0.000 description 2
- MTHSVFCYNBDYFN-UHFFFAOYSA-N anhydrous diethylene glycol Natural products OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical class C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 2
- 238000002832 anti-viral assay Methods 0.000 description 2
- 239000013011 aqueous formulation Substances 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 229960003121 arginine Drugs 0.000 description 2
- 229960005261 aspartic acid Drugs 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 239000012620 biological material Substances 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000000740 bleeding effect Effects 0.000 description 2
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 2
- 235000019437 butane-1,3-diol Nutrition 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 2
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- VAAUVRVFOQPIGI-SPQHTLEESA-N ceftriaxone Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NC(=O)C(=O)NN1C VAAUVRVFOQPIGI-SPQHTLEESA-N 0.000 description 2
- 229960004755 ceftriaxone Drugs 0.000 description 2
- 229940124587 cephalosporin Drugs 0.000 description 2
- 150000001780 cephalosporins Chemical class 0.000 description 2
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 2
- 229960000541 cetyl alcohol Drugs 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 238000011260 co-administration Methods 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 229940125961 compound 24 Drugs 0.000 description 2
- 229940125846 compound 25 Drugs 0.000 description 2
- 229940125877 compound 31 Drugs 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- WCGGWVOVFQNRRS-UHFFFAOYSA-N dichloroacetamide Chemical compound NC(=O)C(Cl)Cl WCGGWVOVFQNRRS-UHFFFAOYSA-N 0.000 description 2
- GGSUCNLOZRCGPQ-UHFFFAOYSA-N diethylaniline Chemical compound CCN(CC)C1=CC=CC=C1 GGSUCNLOZRCGPQ-UHFFFAOYSA-N 0.000 description 2
- 239000012897 dilution medium Substances 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical class C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 229940088679 drug related substance Drugs 0.000 description 2
- 241001493065 dsRNA viruses Species 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- GXESEMBEFHCNLW-UHFFFAOYSA-N ethyl 2-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound C(C)OC(C(C)(C)NC(=O)OC(C)(C)C)=O GXESEMBEFHCNLW-UHFFFAOYSA-N 0.000 description 2
- FKRCODPIKNYEAC-UHFFFAOYSA-N ethyl propionate Chemical compound CCOC(=O)CC FKRCODPIKNYEAC-UHFFFAOYSA-N 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 2
- 239000003925 fat Substances 0.000 description 2
- 235000019197 fats Nutrition 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 239000008098 formaldehyde solution Substances 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 229960002743 glutamine Drugs 0.000 description 2
- 235000004554 glutamine Nutrition 0.000 description 2
- 229940075507 glyceryl monostearate Drugs 0.000 description 2
- 229960002449 glycine Drugs 0.000 description 2
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 2
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical group C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 238000012744 immunostaining Methods 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 239000003701 inert diluent Substances 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 229940102223 injectable solution Drugs 0.000 description 2
- 229940102213 injectable suspension Drugs 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 230000000155 isotopic effect Effects 0.000 description 2
- 239000006101 laboratory sample Substances 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 229960003136 leucine Drugs 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 229960003646 lysine Drugs 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- TTWJBBZEZQICBI-UHFFFAOYSA-N metoclopramide Chemical compound CCN(CC)CCNC(=O)C1=CC(Cl)=C(N)C=C1OC TTWJBBZEZQICBI-UHFFFAOYSA-N 0.000 description 2
- 229960004503 metoclopramide Drugs 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 125000001620 monocyclic carbocycle group Chemical group 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 2
- 229940042880 natural phospholipid Drugs 0.000 description 2
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- 125000003835 nucleoside group Chemical group 0.000 description 2
- 235000014593 oils and fats Nutrition 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 239000003883 ointment base Substances 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- 230000035515 penetration Effects 0.000 description 2
- IZUPBVBPLAPZRR-UHFFFAOYSA-N pentachloro-phenol Natural products OC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl IZUPBVBPLAPZRR-UHFFFAOYSA-N 0.000 description 2
- 229960005190 phenylalanine Drugs 0.000 description 2
- 125000004437 phosphorous atom Chemical group 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 230000001766 physiological effect Effects 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 125000005412 pyrazyl group Chemical group 0.000 description 2
- 125000005495 pyridazyl group Chemical group 0.000 description 2
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 2
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 238000004445 quantitative analysis Methods 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 230000003362 replicative effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 210000003296 saliva Anatomy 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 229960001153 serine Drugs 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 238000002603 single-photon emission computed tomography Methods 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 210000003802 sputum Anatomy 0.000 description 2
- 208000024794 sputum Diseases 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- XOAAWQZATWQOTB-UHFFFAOYSA-N taurine Chemical compound NCCS(O)(=O)=O XOAAWQZATWQOTB-UHFFFAOYSA-N 0.000 description 2
- 210000001138 tear Anatomy 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- HLZKNKRTKFSKGZ-UHFFFAOYSA-N tetradecan-1-ol Chemical compound CCCCCCCCCCCCCCO HLZKNKRTKFSKGZ-UHFFFAOYSA-N 0.000 description 2
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 239000002562 thickening agent Substances 0.000 description 2
- 229960002898 threonine Drugs 0.000 description 2
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 2
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 2
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- AIFRHYZBTHREPW-UHFFFAOYSA-N β-carboline Chemical group N1=CC=C2C3=CC=CC=C3NC2=C1 AIFRHYZBTHREPW-UHFFFAOYSA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- FDKWRPBBCBCIGA-REOHCLBHSA-N (2r)-2-azaniumyl-3-$l^{1}-selanylpropanoate Chemical compound [Se]C[C@H](N)C(O)=O FDKWRPBBCBCIGA-REOHCLBHSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- XIQJZLBROXPFDC-ZETCQYMHSA-N (2s)-2-(2-ethylbutylamino)propanoic acid Chemical compound CCC(CC)CN[C@@H](C)C(O)=O XIQJZLBROXPFDC-ZETCQYMHSA-N 0.000 description 1
- NAQUAXSCBJPECG-NITSXXPLSA-N (3r,4r,5r)-3,4-bis(phenylmethoxy)-5-(phenylmethoxymethyl)oxolan-2-ol Chemical compound C([C@H]1OC([C@@H]([C@@H]1OCC=1C=CC=CC=1)OCC=1C=CC=CC=1)O)OCC1=CC=CC=C1 NAQUAXSCBJPECG-NITSXXPLSA-N 0.000 description 1
- 125000004642 (C1-C12) alkoxy group Chemical group 0.000 description 1
- 125000004641 (C1-C12) haloalkyl group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- QMMJWQMCMRUYTG-UHFFFAOYSA-N 1,2,4,5-tetrachloro-3-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=C(Cl)C(Cl)=CC(Cl)=C1Cl QMMJWQMCMRUYTG-UHFFFAOYSA-N 0.000 description 1
- AUHZEENZYGFFBQ-UHFFFAOYSA-N 1,3,5-Me3C6H3 Natural products CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 description 1
- SGUVLZREKBPKCE-UHFFFAOYSA-N 1,5-diazabicyclo[4.3.0]-non-5-ene Chemical compound C1CCN=C2CCCN21 SGUVLZREKBPKCE-UHFFFAOYSA-N 0.000 description 1
- YFVKHKCZBSGZPE-UHFFFAOYSA-N 1-(1,3-benzodioxol-5-yl)-2-(propylamino)propan-1-one Chemical compound CCCNC(C)C(=O)C1=CC=C2OCOC2=C1 YFVKHKCZBSGZPE-UHFFFAOYSA-N 0.000 description 1
- TUSDEZXZIZRFGC-UHFFFAOYSA-N 1-O-galloyl-3,6-(R)-HHDP-beta-D-glucose Natural products OC1C(O2)COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC1C(O)C2OC(=O)C1=CC(O)=C(O)C(O)=C1 TUSDEZXZIZRFGC-UHFFFAOYSA-N 0.000 description 1
- LOSXTWDYAWERDB-UHFFFAOYSA-N 1-[chloro(diphenyl)methyl]-2,3-dimethoxybenzene Chemical compound COC1=CC=CC(C(Cl)(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1OC LOSXTWDYAWERDB-UHFFFAOYSA-N 0.000 description 1
- VWCUMTCXBIRRSG-UHFFFAOYSA-N 1-[chloro(diphenyl)methyl]-2-methoxybenzene Chemical compound COC1=CC=CC=C1C(Cl)(C=1C=CC=CC=1)C1=CC=CC=C1 VWCUMTCXBIRRSG-UHFFFAOYSA-N 0.000 description 1
- JBWYRBLDOOOJEU-UHFFFAOYSA-N 1-[chloro-(4-methoxyphenyl)-phenylmethyl]-4-methoxybenzene Chemical compound C1=CC(OC)=CC=C1C(Cl)(C=1C=CC(OC)=CC=1)C1=CC=CC=C1 JBWYRBLDOOOJEU-UHFFFAOYSA-N 0.000 description 1
- MICMHFIQSAMEJG-UHFFFAOYSA-N 1-bromopyrrolidine-2,5-dione Chemical compound BrN1C(=O)CCC1=O.BrN1C(=O)CCC1=O MICMHFIQSAMEJG-UHFFFAOYSA-N 0.000 description 1
- VSTXCZGEEVFJES-UHFFFAOYSA-N 1-cycloundecyl-1,5-diazacycloundec-5-ene Chemical compound C1CCCCCC(CCCC1)N1CCCCCC=NCCC1 VSTXCZGEEVFJES-UHFFFAOYSA-N 0.000 description 1
- PZNPLUBHRSSFHT-RRHRGVEJSA-N 1-hexadecanoyl-2-octadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[C@@H](COP([O-])(=O)OCC[N+](C)(C)C)COC(=O)CCCCCCCCCCCCCCC PZNPLUBHRSSFHT-RRHRGVEJSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- HJNBCVWWPLRZLC-UHFFFAOYSA-N 1-methylpyrrolidin-2-one;oxolane Chemical compound C1CCOC1.CN1CCCC1=O HJNBCVWWPLRZLC-UHFFFAOYSA-N 0.000 description 1
- YUQCVPFHAFZFPV-UHFFFAOYSA-N 1-n,7-n-bis[3-(dimethylamino)propyl]-3,9-dimethylquinolino[8,7-h]quinoline-1,7-diamine;tetrahydrochloride Chemical compound Cl.Cl.Cl.Cl.C1=CC2=C(NCCCN(C)C)C=C(C)N=C2C(C=C2)=C1C1=C2C(NCCCN(C)C)=CC(C)=N1 YUQCVPFHAFZFPV-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- SPXOTSHWBDUUMT-UHFFFAOYSA-N 138-42-1 Chemical group OS(=O)(=O)C1=CC=C([N+]([O-])=O)C=C1 SPXOTSHWBDUUMT-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- LGEZTMRIZWCDLW-UHFFFAOYSA-N 14-methylpentadecyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCC(C)C LGEZTMRIZWCDLW-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical group C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- 125000005955 1H-indazolyl group Chemical group 0.000 description 1
- MGAXHFMCFLLMNG-UHFFFAOYSA-N 1h-pyrimidine-6-thione Chemical compound SC1=CC=NC=N1 MGAXHFMCFLLMNG-UHFFFAOYSA-N 0.000 description 1
- JHSXHKMJBWOMRU-UHFFFAOYSA-M 2,2-dimethylpropanethioate Chemical compound CC(C)(C)C([O-])=S JHSXHKMJBWOMRU-UHFFFAOYSA-M 0.000 description 1
- IHGAMSLGOJKVHJ-UHFFFAOYSA-N 2,3-bis(phenylmethoxy)-5-(phenylmethoxymethyl)oxolan-2-ol Chemical compound C(C1=CC=CC=C1)OC1C(OC(C1)COCC1=CC=CC=C1)(O)OCC1=CC=CC=C1 IHGAMSLGOJKVHJ-UHFFFAOYSA-N 0.000 description 1
- QZRKTYZKFOMKLV-UHFFFAOYSA-N 2,3-bis(phenylmethoxy)-5-(phenylmethoxymethyl)oxolane-2-carbonitrile Chemical compound C(C1=CC=CC=C1)OC1C(OC(C1)COCC1=CC=CC=C1)(C#N)OCC1=CC=CC=C1 QZRKTYZKFOMKLV-UHFFFAOYSA-N 0.000 description 1
- FFMBYMANYCDCMK-UHFFFAOYSA-N 2,5-dihydro-1h-imidazole Chemical group C1NCN=C1 FFMBYMANYCDCMK-UHFFFAOYSA-N 0.000 description 1
- LABFFVKLPSJCAN-UHFFFAOYSA-N 2-(propan-2-ylazaniumyl)propanoate Chemical compound CC(C)NC(C)C(O)=O LABFFVKLPSJCAN-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- WXHLLJAMBQLULT-UHFFFAOYSA-N 2-[[6-[4-(2-hydroxyethyl)piperazin-1-yl]-2-methylpyrimidin-4-yl]amino]-n-(2-methyl-6-sulfanylphenyl)-1,3-thiazole-5-carboxamide;hydrate Chemical compound O.C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1S WXHLLJAMBQLULT-UHFFFAOYSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 1
- NQSOMRSHXPZDCC-ZIROQRTJSA-N 2-ethylbutyl (2S)-2-[[(2,3,4,5,6-pentafluorophenoxy)-phenoxyphosphoryl]amino]propanoate Chemical compound FC1=C(O[P@@](=O)(OC2=CC=CC=C2)N[C@H](C(=O)OCC(CC)CC)C)C(=C(C(=C1F)F)F)F NQSOMRSHXPZDCC-ZIROQRTJSA-N 0.000 description 1
- SFAAOBGYWOUHLU-UHFFFAOYSA-N 2-ethylhexyl hexadecanoate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(CC)CCCC SFAAOBGYWOUHLU-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- VFPARGXUPUTTGL-UHFFFAOYSA-N 2-hydroxy-5-(hydroxymethyl)oxolane-2-carbonitrile Chemical compound OC1(OC(CC1)CO)C#N VFPARGXUPUTTGL-UHFFFAOYSA-N 0.000 description 1
- LXBGSDVWAMZHDD-UHFFFAOYSA-N 2-methyl-1h-imidazole Chemical compound CC1=NC=CN1 LXBGSDVWAMZHDD-UHFFFAOYSA-N 0.000 description 1
- 125000004922 2-methyl-3-pentyl group Chemical group CC(C)C(CC)* 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- OBOSXEWFRARQPU-UHFFFAOYSA-N 2-n,2-n-dimethylpyridine-2,5-diamine Chemical compound CN(C)C1=CC=C(N)C=N1 OBOSXEWFRARQPU-UHFFFAOYSA-N 0.000 description 1
- 125000003504 2-oxazolinyl group Chemical group O1C(=NCC1)* 0.000 description 1
- KRTGJZMJJVEKRX-UHFFFAOYSA-N 2-phenylethan-1-yl Chemical group [CH2]CC1=CC=CC=C1 KRTGJZMJJVEKRX-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 1
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical group C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- NAQUAXSCBJPECG-UHFFFAOYSA-N 3,4-bis(phenylmethoxy)-5-(phenylmethoxymethyl)oxolan-2-ol Chemical compound C=1C=CC=CC=1COC1C(OCC=2C=CC=CC=2)C(O)OC1COCC1=CC=CC=C1 NAQUAXSCBJPECG-UHFFFAOYSA-N 0.000 description 1
- DNNVRTZJRKIUFK-UHFFFAOYSA-N 3,4-dihydroquinoline Chemical compound C1=CC=C2N=CCCC2=C1 DNNVRTZJRKIUFK-UHFFFAOYSA-N 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-N 3-Hydroxy-2-naphthoate Chemical compound C1=CC=C2C=C(O)C(C(=O)O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-N 0.000 description 1
- CJYDQTAWSHWBIT-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-(2-hydroxy-2-methylpropyl)benzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NCC(C)(C)O)C=CC=1 CJYDQTAWSHWBIT-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- UOQHWNPVNXSDDO-UHFFFAOYSA-N 3-bromoimidazo[1,2-a]pyridine-6-carbonitrile Chemical compound C1=CC(C#N)=CN2C(Br)=CN=C21 UOQHWNPVNXSDDO-UHFFFAOYSA-N 0.000 description 1
- JVQIKJMSUIMUDI-UHFFFAOYSA-N 3-pyrroline Chemical group C1NCC=C1 JVQIKJMSUIMUDI-UHFFFAOYSA-N 0.000 description 1
- 238000004679 31P NMR spectroscopy Methods 0.000 description 1
- RTDAMORRDXWYPT-UHFFFAOYSA-N 4,5-dichloro-3,6-dioxocyclohexa-1,4-diene-1,2-dicarbonitrile Chemical compound ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O.ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O RTDAMORRDXWYPT-UHFFFAOYSA-N 0.000 description 1
- MCGBIXXDQFWVDW-UHFFFAOYSA-N 4,5-dihydro-1h-pyrazole Chemical group C1CC=NN1 MCGBIXXDQFWVDW-UHFFFAOYSA-N 0.000 description 1
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 1
- RJWBTWIBUIGANW-UHFFFAOYSA-M 4-chlorobenzenesulfonate Chemical group [O-]S(=O)(=O)C1=CC=C(Cl)C=C1 RJWBTWIBUIGANW-UHFFFAOYSA-M 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- USYVBQLOSZDDBE-UHFFFAOYSA-N 4-iodopyrimidine Chemical compound IC1=CC=NC=N1 USYVBQLOSZDDBE-UHFFFAOYSA-N 0.000 description 1
- 125000006181 4-methyl benzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])C([H])([H])* 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 5-Azacytidine Natural products O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- NPYPQKXJJZZSAX-UHFFFAOYSA-N 5-benzylpyrimidine Chemical class C=1N=CN=CC=1CC1=CC=CC=C1 NPYPQKXJJZZSAX-UHFFFAOYSA-N 0.000 description 1
- XHZWFUVEKDDQPF-UHFFFAOYSA-N 5-bromo-1h-pyrazole Chemical compound BrC1=CC=NN1 XHZWFUVEKDDQPF-UHFFFAOYSA-N 0.000 description 1
- LZGICTBSBIWVFA-UHFFFAOYSA-N 5-bromo-2-ethenylpyrimidine Chemical compound BrC1=CN=C(C=C)N=C1 LZGICTBSBIWVFA-UHFFFAOYSA-N 0.000 description 1
- HXXVIKZQIFTJOQ-UHFFFAOYSA-N 5-ethenylpyrimidine Chemical class C=CC1=CN=CN=C1 HXXVIKZQIFTJOQ-UHFFFAOYSA-N 0.000 description 1
- 125000006043 5-hexenyl group Chemical group 0.000 description 1
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 1
- NOYDQGFVFOQSAJ-UHFFFAOYSA-N 5-nitropyrimidine Chemical class [O-][N+](=O)C1=CN=CN=C1 NOYDQGFVFOQSAJ-UHFFFAOYSA-N 0.000 description 1
- 125000004938 5-pyridyl group Chemical group N1=CC=CC(=C1)* 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- ZKBQDFAWXLTYKS-UHFFFAOYSA-N 6-Chloro-1H-purine Chemical compound ClC1=NC=NC2=C1NC=N2 ZKBQDFAWXLTYKS-UHFFFAOYSA-N 0.000 description 1
- RDUORFDQRFHYBF-UHFFFAOYSA-N 6-methoxy-1-methyl-2,3,4,9-tetrahydro-1h-pyrido[3,4-b]indole Chemical compound CC1NCCC2=C1NC1=CC=C(OC)C=C12 RDUORFDQRFHYBF-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 230000035502 ADME Effects 0.000 description 1
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 1
- 208000004998 Abdominal Pain Diseases 0.000 description 1
- 206010000087 Abdominal pain upper Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical class N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 208000006820 Arthralgia Diseases 0.000 description 1
- 208000004429 Bacillary Dysentery Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 201000004569 Blindness Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical group [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 239000004358 Butane-1, 3-diol Substances 0.000 description 1
- 125000003860 C1-C20 alkoxy group Chemical group 0.000 description 1
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 1
- 125000003358 C2-C20 alkenyl group Chemical group 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 229910004664 Cerium(III) chloride Inorganic materials 0.000 description 1
- 206010008479 Chest Pain Diseases 0.000 description 1
- 244000060011 Cocos nucifera Species 0.000 description 1
- 235000013162 Cocos nucifera Nutrition 0.000 description 1
- 208000003322 Coinfection Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- MAMMVUWCKMOLSG-UHFFFAOYSA-N Cyclohexyl propionate Chemical compound CCC(=O)OC1CCCCC1 MAMMVUWCKMOLSG-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FDKWRPBBCBCIGA-UWTATZPHSA-N D-Selenocysteine Natural products [Se]C[C@@H](N)C(O)=O FDKWRPBBCBCIGA-UWTATZPHSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 239000004150 EU approved colour Substances 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 239000001263 FEMA 3042 Substances 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 241000282575 Gorilla Species 0.000 description 1
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 208000034507 Haematemesis Diseases 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 208000031361 Hiccup Diseases 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 239000002879 Lewis base Substances 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- RJUFJBKOKNCXHH-UHFFFAOYSA-N Methyl propionate Chemical compound CCC(=O)OC RJUFJBKOKNCXHH-UHFFFAOYSA-N 0.000 description 1
- 208000034486 Multi-organ failure Diseases 0.000 description 1
- 208000010718 Multiple Organ Failure Diseases 0.000 description 1
- 208000000112 Myalgia Diseases 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 240000007817 Olea europaea Species 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 206010068319 Oropharyngeal pain Diseases 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical group C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 241000282579 Pan Species 0.000 description 1
- 229920002230 Pectic acid Polymers 0.000 description 1
- LRBQNJMCXXYXIU-PPKXGCFTSA-N Penta-digallate-beta-D-glucose Natural products OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-PPKXGCFTSA-N 0.000 description 1
- 201000007100 Pharyngitis Diseases 0.000 description 1
- 229920001100 Polydextrose Polymers 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 108010020346 Polyglutamic Acid Proteins 0.000 description 1
- 241000282405 Pongo abelii Species 0.000 description 1
- 229910052777 Praseodymium Inorganic materials 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- 239000008156 Ringer's lactate solution Substances 0.000 description 1
- 206010040550 Shigella infections Diseases 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 108091027076 Spiegelmer Proteins 0.000 description 1
- 101710172711 Structural protein Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 108010017842 Telomerase Proteins 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 208000037386 Typhoid Diseases 0.000 description 1
- 101150010086 VP24 gene Proteins 0.000 description 1
- 101150026858 VP30 gene Proteins 0.000 description 1
- 101150077651 VP35 gene Proteins 0.000 description 1
- 101150036892 VP40 gene Proteins 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 208000028227 Viral hemorrhagic fever Diseases 0.000 description 1
- 229930003448 Vitamin K Natural products 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 125000005012 alkyl thioether group Chemical group 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001409 amidines Chemical class 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 1
- 229960000723 ampicillin Drugs 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 229960000981 artemether Drugs 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- CREXVNNSNOKDHW-UHFFFAOYSA-N azaniumylideneazanide Chemical group N[N] CREXVNNSNOKDHW-UHFFFAOYSA-N 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- 125000004931 azocinyl group Chemical group N1=C(C=CC=CC=C1)* 0.000 description 1
- 125000003943 azolyl group Chemical group 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004935 benzoxazolinyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 1
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 description 1
- 229960004853 betadex Drugs 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 239000004305 biphenyl Chemical class 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- SIOVKLKJSOKLIF-UHFFFAOYSA-N bis(trimethylsilyl)acetamide Chemical compound C[Si](C)(C)OC(C)=N[Si](C)(C)C SIOVKLKJSOKLIF-UHFFFAOYSA-N 0.000 description 1
- 239000007844 bleaching agent Substances 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 238000010504 bond cleavage reaction Methods 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- CDQSJQSWAWPGKG-UHFFFAOYSA-N butane-1,1-diol Chemical compound CCCC(O)O CDQSJQSWAWPGKG-UHFFFAOYSA-N 0.000 description 1
- SNCZNSNPXMPCGN-UHFFFAOYSA-N butanediamide Chemical compound NC(=O)CCC(N)=O SNCZNSNPXMPCGN-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- VTYYLEPIZMXCLO-UHFFFAOYSA-L calcium carbonate Substances [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- ZEWYCNBZMPELPF-UHFFFAOYSA-J calcium;potassium;sodium;2-hydroxypropanoic acid;sodium;tetrachloride Chemical compound [Na].[Na+].[Cl-].[Cl-].[Cl-].[Cl-].[K+].[Ca+2].CC(O)C(O)=O ZEWYCNBZMPELPF-UHFFFAOYSA-J 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 229960001668 cefuroxime Drugs 0.000 description 1
- JFPVXVDWJQMJEE-IZRZKJBUSA-N cefuroxime Chemical compound N([C@@H]1C(N2C(=C(COC(N)=O)CS[C@@H]21)C(O)=O)=O)C(=O)\C(=N/OC)C1=CC=CO1 JFPVXVDWJQMJEE-IZRZKJBUSA-N 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229940081733 cetearyl alcohol Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 230000009920 chelation Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 150000001805 chlorine compounds Chemical group 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 229940125833 compound 23 Drugs 0.000 description 1
- 229940125851 compound 27 Drugs 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 239000012045 crude solution Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- WTGNEPDJPOULKD-XKGYJNPMSA-N cyclobutyl (2S)-2-[[(4-nitrophenoxy)-phenoxyphosphoryl]amino]propanoate Chemical compound [N+](=O)([O-])C1=CC=C(OP(=O)(OC2=CC=CC=C2)N[C@H](C(=O)OC2CCC2)C)C=C1 WTGNEPDJPOULKD-XKGYJNPMSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000004850 cyclobutylmethyl group Chemical group C1(CCC1)C* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- MRKZAZMYXYSBDG-UHFFFAOYSA-N cyclopentyl propanoate Chemical compound CCC(=O)OC1CCCC1 MRKZAZMYXYSBDG-UHFFFAOYSA-N 0.000 description 1
- 125000004851 cyclopentylmethyl group Chemical group C1(CCCC1)C* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 229960002433 cysteine Drugs 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- SASYSVUEVMOWPL-NXVVXOECSA-N decyl oleate Chemical compound CCCCCCCCCCOC(=O)CCCCCCC\C=C/CCCCCCCC SASYSVUEVMOWPL-NXVVXOECSA-N 0.000 description 1
- 238000002716 delivery method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 239000012973 diazabicyclooctane Substances 0.000 description 1
- 230000001079 digestive effect Effects 0.000 description 1
- 210000002249 digestive system Anatomy 0.000 description 1
- SXYIRMFQILZOAM-HVNFFKDJSA-N dihydroartemisinin methyl ether Chemical compound C1C[C@H]2[C@H](C)CC[C@H]3[C@@H](C)[C@@H](OC)O[C@H]4[C@]32OO[C@@]1(C)O4 SXYIRMFQILZOAM-HVNFFKDJSA-N 0.000 description 1
- 125000004925 dihydropyridyl group Chemical group N1(CC=CC=C1)* 0.000 description 1
- JMRYOSQOYJBDOI-UHFFFAOYSA-N dilithium;di(propan-2-yl)azanide Chemical compound [Li+].CC(C)[N-]C(C)C.CC(C)N([Li])C(C)C JMRYOSQOYJBDOI-UHFFFAOYSA-N 0.000 description 1
- MCWXGJITAZMZEV-UHFFFAOYSA-N dimethoate Chemical compound CNC(=O)CSP(=S)(OC)OC MCWXGJITAZMZEV-UHFFFAOYSA-N 0.000 description 1
- BDUPRNVPXOHWIL-UHFFFAOYSA-N dimethyl sulfite Chemical compound COS(=O)OC BDUPRNVPXOHWIL-UHFFFAOYSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- UZZWBUYVTBPQIV-UHFFFAOYSA-N dme dimethoxyethane Chemical compound COCCOC.COCCOC UZZWBUYVTBPQIV-UHFFFAOYSA-N 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000008387 emulsifying waxe Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 238000007824 enzymatic assay Methods 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- FFPPNMDEWHTZHH-LJPZCQIFSA-N ethyl (2s)-2-[[(4-nitrophenoxy)-phenoxyphosphoryl]amino]-3-phenylpropanoate Chemical compound C([C@@H](C(=O)OCC)NP(=O)(OC=1C=CC=CC=1)OC=1C=CC(=CC=1)[N+]([O-])=O)C1=CC=CC=C1 FFPPNMDEWHTZHH-LJPZCQIFSA-N 0.000 description 1
- BQIVJVAZDJHDJF-LURJTMIESA-N ethyl (2s)-2-amino-3-methylbutanoate Chemical compound CCOC(=O)[C@@H](N)C(C)C BQIVJVAZDJHDJF-LURJTMIESA-N 0.000 description 1
- YDBJGWKZKPFTGO-STFFIMJZSA-N ethyl (2s)-3-methyl-2-[[(4-nitrophenoxy)-phenoxyphosphoryl]amino]butanoate Chemical compound C=1C=C([N+]([O-])=O)C=CC=1OP(=O)(N[C@H](C(=O)OCC)C(C)C)OC1=CC=CC=C1 YDBJGWKZKPFTGO-STFFIMJZSA-N 0.000 description 1
- WKJGDQUHBZZDKG-UHFFFAOYSA-N ethyl 2-methyl-2-[[(4-nitrophenoxy)-phenoxyphosphoryl]amino]propanoate Chemical compound CC(C(=O)OCC)(C)NP(=O)(OC1=CC=CC=C1)OC1=CC=C(C=C1)[N+](=O)[O-] WKJGDQUHBZZDKG-UHFFFAOYSA-N 0.000 description 1
- 239000004403 ethyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010228 ethyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229940043351 ethyl-p-hydroxybenzoate Drugs 0.000 description 1
- NUVBSKCKDOMJSU-UHFFFAOYSA-N ethylparaben Chemical compound CCOC(=O)C1=CC=C(O)C=C1 NUVBSKCKDOMJSU-UHFFFAOYSA-N 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 206010016256 fatigue Diseases 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- XRECTZIEBJDKEO-UHFFFAOYSA-N flucytosine Chemical compound NC1=NC(=O)NC=C1F XRECTZIEBJDKEO-UHFFFAOYSA-N 0.000 description 1
- 229960004413 flucytosine Drugs 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 150000002222 fluorine compounds Chemical class 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229960002598 fumaric acid Drugs 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- LRBQNJMCXXYXIU-QWKBTXIPSA-N gallotannic acid Chemical compound OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@H]2[C@@H]([C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-QWKBTXIPSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000004051 gastric juice Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- FVIZARNDLVOMSU-UHFFFAOYSA-N ginsenoside K Natural products C1CC(C2(CCC3C(C)(C)C(O)CCC3(C)C2CC2O)C)(C)C2C1C(C)(CCC=C(C)C)OC1OC(CO)C(O)C(O)C1O FVIZARNDLVOMSU-UHFFFAOYSA-N 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 229950006191 gluconic acid Drugs 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- UBHWBODXJBSFLH-UHFFFAOYSA-N hexadecan-1-ol;octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO.CCCCCCCCCCCCCCCCCCO UBHWBODXJBSFLH-UHFFFAOYSA-N 0.000 description 1
- 208000021760 high fever Diseases 0.000 description 1
- 229960002885 histidine Drugs 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- AFQIYTIJXGTIEY-UHFFFAOYSA-N hydrogen carbonate;triethylazanium Chemical compound OC(O)=O.CCN(CC)CC AFQIYTIJXGTIEY-UHFFFAOYSA-N 0.000 description 1
- 229910000040 hydrogen fluoride Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- ARRNBPCNZJXHRJ-UHFFFAOYSA-M hydron;tetrabutylazanium;phosphate Chemical compound OP(O)([O-])=O.CCCC[N+](CCCC)(CCCC)CCCC ARRNBPCNZJXHRJ-UHFFFAOYSA-M 0.000 description 1
- 239000008309 hydrophilic cream Substances 0.000 description 1
- 229920013821 hydroxy alkyl cellulose Polymers 0.000 description 1
- 150000002440 hydroxy compounds Chemical class 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 229940036998 hypertonic sodium chloride Drugs 0.000 description 1
- 239000000819 hypertonic solution Substances 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000005945 imidazopyridyl group Chemical group 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- QNLOWBMKUIXCOW-UHFFFAOYSA-N indol-2-one Chemical compound C1=CC=CC2=NC(=O)C=C21 QNLOWBMKUIXCOW-UHFFFAOYSA-N 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Chemical group CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Chemical group C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 125000004926 indolenyl group Chemical group 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- FGFUBBNNYLNVLJ-UHFFFAOYSA-N indolone Natural products C1=CC=C2C(=O)C=NC2=C1 FGFUBBNNYLNVLJ-UHFFFAOYSA-N 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 125000004936 isatinoyl group Chemical group N1(C(=O)C(=O)C2=CC=CC=C12)C(=O)* 0.000 description 1
- 229940045996 isethionic acid Drugs 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 229940078545 isocetyl stearate Drugs 0.000 description 1
- GWVMLCQWXVFZCN-UHFFFAOYSA-N isoindoline Chemical group C1=CC=C2CNCC2=C1 GWVMLCQWXVFZCN-UHFFFAOYSA-N 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- XUGNVMKQXJXZCD-UHFFFAOYSA-N isopropyl palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC(C)C XUGNVMKQXJXZCD-UHFFFAOYSA-N 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical group C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 230000005445 isotope effect Effects 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229940099563 lactobionic acid Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 150000007527 lewis bases Chemical class 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 229960004985 lumefantrine Drugs 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 210000004924 lung microvascular endothelial cell Anatomy 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 239000003120 macrolide antibiotic agent Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229940098895 maleic acid Drugs 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 229940099690 malic acid Drugs 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- BCVXHSPFUWZLGQ-UHFFFAOYSA-N mecn acetonitrile Chemical compound CC#N.CC#N BCVXHSPFUWZLGQ-UHFFFAOYSA-N 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 229960004452 methionine Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- GDOPTJXRTPNYNR-UHFFFAOYSA-N methyl-cyclopentane Natural products CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- XLSZMDLNRCVEIJ-UHFFFAOYSA-N methylimidazole Natural products CC1=CNC=N1 XLSZMDLNRCVEIJ-UHFFFAOYSA-N 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical compound CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 1
- 229960000282 metronidazole Drugs 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229940051866 mouthwash Drugs 0.000 description 1
- 208000029744 multiple organ dysfunction syndrome Diseases 0.000 description 1
- 208000013465 muscle pain Diseases 0.000 description 1
- 229940043348 myristyl alcohol Drugs 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- PEECTLLHENGOKU-UHFFFAOYSA-N n,n-dimethylpyridin-4-amine Chemical compound CN(C)C1=CC=NC=C1.CN(C)C1=CC=NC=C1 PEECTLLHENGOKU-UHFFFAOYSA-N 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical compound C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004923 naphthylmethyl group Chemical group C1(=CC=CC2=CC=CC=C12)C* 0.000 description 1
- 229920001206 natural gum Polymers 0.000 description 1
- ATINCSYRHURBSP-UHFFFAOYSA-K neodymium(iii) chloride Chemical compound Cl[Nd](Cl)Cl ATINCSYRHURBSP-UHFFFAOYSA-K 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 239000007764 o/w emulsion Substances 0.000 description 1
- 125000004930 octahydroisoquinolinyl group Chemical group C1(NCCC2CCCC=C12)* 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 229940006093 opthalmologic coloring agent diagnostic Drugs 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 229940116315 oxalic acid Drugs 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000005968 oxazolinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- YWWARDMVSMPOLR-UHFFFAOYSA-M oxolane;tetrabutylazanium;fluoride Chemical compound [F-].C1CCOC1.CCCC[N+](CCCC)(CCCC)CCCC YWWARDMVSMPOLR-UHFFFAOYSA-M 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229940098695 palmitic acid Drugs 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 125000004932 phenoxathinyl group Chemical group 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- WRMXOVHLRUVREB-UHFFFAOYSA-N phosphono phosphate;tributylazanium Chemical compound OP(O)(=O)OP([O-])([O-])=O.CCCC[NH+](CCCC)CCCC.CCCC[NH+](CCCC)CCCC WRMXOVHLRUVREB-UHFFFAOYSA-N 0.000 description 1
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- SHUZOJHMOBOZST-UHFFFAOYSA-N phylloquinone Natural products CC(C)CCCCC(C)CCC(C)CCCC(=CCC1=C(C)C(=O)c2ccccc2C1=O)C SHUZOJHMOBOZST-UHFFFAOYSA-N 0.000 description 1
- 230000006461 physiological response Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 230000000485 pigmenting effect Effects 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 239000001259 polydextrose Substances 0.000 description 1
- 235000013856 polydextrose Nutrition 0.000 description 1
- 229940035035 polydextrose Drugs 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000010318 polygalacturonic acid Substances 0.000 description 1
- 229920002643 polyglutamic acid Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 229940071643 prefilled syringe Drugs 0.000 description 1
- 229960002429 proline Drugs 0.000 description 1
- HQNCAWVCJWJLML-SKBWGGTPSA-N propan-2-yl (2S)-2-[[(4-nitrophenoxy)-phenoxyphosphoryl]amino]-3-phenylpropanoate Chemical compound [N+](=O)([O-])C1=CC=C(OP(=O)(OC2=CC=CC=C2)N[C@H](C(=O)OC(C)C)CC2=CC=CC=C2)C=C1 HQNCAWVCJWJLML-SKBWGGTPSA-N 0.000 description 1
- OVNGSPSAVXGYHI-UHFFFAOYSA-N propan-2-yl 2-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound C(C)(C)(C)OC(=O)NC(C(=O)OC(C)C)(C)C OVNGSPSAVXGYHI-UHFFFAOYSA-N 0.000 description 1
- MGMABNNGTAKDJF-UHFFFAOYSA-N propan-2-yl 2-methyl-2-[[(4-nitrophenoxy)-phenoxyphosphoryl]amino]propanoate Chemical compound C(C)(C)OC(C(C)(NP(=O)(OC1=CC=CC=C1)OC1=CC=C(C=C1)[N+](=O)[O-])C)=O MGMABNNGTAKDJF-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 239000013014 purified material Substances 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- DNXIASIHZYFFRO-UHFFFAOYSA-N pyrazoline Chemical group C1CN=NC1 DNXIASIHZYFFRO-UHFFFAOYSA-N 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- GRJJQCWNZGRKAU-UHFFFAOYSA-N pyridin-1-ium;fluoride Chemical compound F.C1=CC=NC=C1 GRJJQCWNZGRKAU-UHFFFAOYSA-N 0.000 description 1
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 1
- FVLAYJRLBLHIPV-UHFFFAOYSA-N pyrimidin-5-amine Chemical class NC1=CN=CN=C1 FVLAYJRLBLHIPV-UHFFFAOYSA-N 0.000 description 1
- 125000004943 pyrimidin-6-yl group Chemical group N1=CN=CC=C1* 0.000 description 1
- HBCQSNAFLVXVAY-UHFFFAOYSA-N pyrimidine-2-thiol Chemical compound SC1=NC=CC=N1 HBCQSNAFLVXVAY-UHFFFAOYSA-N 0.000 description 1
- XVIAPHVAGFEFFN-UHFFFAOYSA-N pyrimidine-5-carbonitrile Chemical compound N#CC1=CN=CN=C1 XVIAPHVAGFEFFN-UHFFFAOYSA-N 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 1
- 229930185107 quinolinone Natural products 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 150000003873 salicylate salts Chemical class 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229940055619 selenocysteine Drugs 0.000 description 1
- ZKZBPNGNEQAJSX-UHFFFAOYSA-N selenocysteine Natural products [SeH]CC(N)C(O)=O ZKZBPNGNEQAJSX-UHFFFAOYSA-N 0.000 description 1
- 235000016491 selenocysteine Nutrition 0.000 description 1
- 238000011452 sequencing regimen Methods 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 201000005113 shigellosis Diseases 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 239000008347 soybean phospholipid Substances 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 229940097346 sulfobutylether-beta-cyclodextrin Drugs 0.000 description 1
- 238000009120 supportive therapy Methods 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000015523 tannic acid Nutrition 0.000 description 1
- 229940033123 tannic acid Drugs 0.000 description 1
- 229920002258 tannic acid Polymers 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 229960001367 tartaric acid Drugs 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960003080 taurine Drugs 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- MLGCXEBRWGEOQX-UHFFFAOYSA-N tetradifon Chemical compound C1=CC(Cl)=CC=C1S(=O)(=O)C1=CC(Cl)=C(Cl)C=C1Cl MLGCXEBRWGEOQX-UHFFFAOYSA-N 0.000 description 1
- HWCKGOZZJDHMNC-UHFFFAOYSA-M tetraethylammonium bromide Chemical compound [Br-].CC[N+](CC)(CC)CC HWCKGOZZJDHMNC-UHFFFAOYSA-M 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000005942 tetrahydropyridyl group Chemical group 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 125000004927 thianaphthalenyl group Chemical group S1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005302 thiazolylmethyl group Chemical group [H]C1=C([H])N=C(S1)C([H])([H])* 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229930192474 thiophene Chemical group 0.000 description 1
- IBBLKSWSCDAPIF-UHFFFAOYSA-N thiopyran Chemical group S1C=CC=C=C1 IBBLKSWSCDAPIF-UHFFFAOYSA-N 0.000 description 1
- 229940113082 thymine Drugs 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000440 toxicity profile Toxicity 0.000 description 1
- 238000011277 treatment modality Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- NTJBWZHVSJNKAD-UHFFFAOYSA-N triethylazanium;fluoride Chemical compound [F-].CC[NH+](CC)CC NTJBWZHVSJNKAD-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical group [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 229960004799 tryptophan Drugs 0.000 description 1
- 201000008297 typhoid fever Diseases 0.000 description 1
- 229960004441 tyrosine Drugs 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- SCXZATZIVUFOFT-UHFFFAOYSA-N undec-5-ene Chemical compound [CH2]CCCC=CCCCCC SCXZATZIVUFOFT-UHFFFAOYSA-N 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 1
- 229940045145 uridine Drugs 0.000 description 1
- 229960004295 valine Drugs 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 235000014393 valine Nutrition 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000000277 virosome Substances 0.000 description 1
- 230000001018 virulence Effects 0.000 description 1
- 235000019168 vitamin K Nutrition 0.000 description 1
- 239000011712 vitamin K Substances 0.000 description 1
- 150000003721 vitamin K derivatives Chemical class 0.000 description 1
- 229940046010 vitamin k Drugs 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- NWONKYPBYAMBJT-UHFFFAOYSA-L zinc sulfate Chemical compound [Zn+2].[O-]S([O-])(=O)=O NWONKYPBYAMBJT-UHFFFAOYSA-L 0.000 description 1
- 229960001763 zinc sulfate Drugs 0.000 description 1
- 229910000368 zinc sulfate Inorganic materials 0.000 description 1
- 125000004933 β-carbolinyl group Chemical group C1(=NC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/18—Acyclic radicals, substituted by carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/683—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols
- A61K31/685—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols one of the hydroxy compounds having nitrogen atoms, e.g. phosphatidylserine, lecithin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/661—Phosphorus acids or esters thereof not having P—C bonds, e.g. fosfosal, dichlorvos, malathion or mevinphos
- A61K31/6615—Compounds having two or more esterified phosphorus acid groups, e.g. inositol triphosphate, phytic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/665—Phosphorus compounds having oxygen as a ring hetero atom, e.g. fosfomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/683—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/7056—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing five-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/22—Amides of acids of phosphorus
- C07F9/24—Esteramides
- C07F9/2404—Esteramides the ester moiety containing a substituent or a structure which is considered as characteristic
- C07F9/2429—Esteramides the ester moiety containing a substituent or a structure which is considered as characteristic of arylalkanols
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
- C07F9/65616—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings containing the ring system having three or more than three double bonds between ring members or between ring members and non-ring members, e.g. purine or analogs
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
- C07H1/02—Phosphorylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H11/00—Compounds containing saccharide radicals esterified by inorganic acids; Metal salts thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H13/00—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/02—Heterocyclic radicals containing only nitrogen as ring hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/14—Pyrrolo-pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/06—Heterocyclic radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H9/00—Compounds containing a hetero ring sharing at least two hetero atoms with a saccharide radical
- C07H9/02—Compounds containing a hetero ring sharing at least two hetero atoms with a saccharide radical the hetero ring containing only oxygen as ring hetero atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Molecular Biology (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本發明提供藉由投予式IV之核糖苷、核糖苷磷酸酯及其前藥來治療絲狀病毒科病毒感染之化合物、方法、及醫藥組成物:
Description
本發明大體關於用於治療絲狀病毒科病毒感染之方法和化合物,特別是治療伊波拉病毒(Ebola virus)、馬堡病毒(Marburg virus)和奎瓦病毒(Cueva virus)之方法和核苷。
絲狀病毒(例如,伊波拉病毒(EBOV)和馬堡病毒(MARV))是最致命及毀滅性的病毒之一。它們造成人類和非人靈長類動物(例如,猴子、大猩猩、和黑猩猩)之嚴重且往往是致命的病毒性出血熱。絲狀病毒特別值得關注的是其為可能的生物武器,因為它們具有噴霧劑傳播和武器化的潛力。
絲狀病毒感染之潛伏期範圍從2至21天。發病突然且以高燒、頭痛、關節和肌肉疼痛、喉嚨痛、乏力、腹瀉、嘔吐和胃痛為特徵。一些患者可看見皮疹、眼睛發紅、打嗝以及內外出血。在變為病毒感染一週內,大多數患者經驗胸部疼痛和和多重器官衰竭、進入休克,及死亡。一些患者死亡前亦經歷失明和廣泛性出血。
絲狀病毒科為RNA病毒之家族。已經確定絲狀病毒科家族之二成員:EBOV和MARV。已經確定二種關鍵致病型絲狀病毒科家族:伊波拉病毒和MARV。有一種MARV之確定變種和五種伊波拉病毒之確定變種:薩伊(Zaire)(即伊波拉病毒,EBOV)、蘇丹(Sudan)、大森林(Tai Forest)、班迪布交(Bundibugyo)、和雷斯頓(Reston)。絲狀病毒科的確切起源、地點和自然棲息地是未知的。但是,根據可用的證據和類似病毒的性質,可推測絲狀病毒科為人畜共通(即,動物源性)且通常保留在非洲大陸土生的動物宿主。
經過30年以上,在中非伊波拉病毒已經與在感染患者中產生嚴重疾病之出血熱的週期性爆發有關。暴發之死亡率範圍從伊波拉病之毒蘇丹種(SEBOV)的50%最高至伊波拉病毒之薩伊種(EBOV,ZEBOV)的90% (Sanchez等人,Filoviridae: Marburg and Ebola Viruses, in Fields Virology (eds. Knipe, D.M. & Howley, P.M.) 1409-1448 (Lippincott Williams & Wilkins, Philadelphia))。2007年底在烏干達由伊波拉病毒的明顯新物種引起之爆發導致約25%的致死率(Towner 等人., PLoS Pathog., 4:e1000212 (2008))。ZEBOV也使在非洲同一區的野生猩猩種群大批死亡(Walsh 等人., Nature, 422:611-614 (2003))。
絲狀病毒感染(包括伊波拉病毒(即EBOV))之預防和治療面臨許多挑戰。事實上,沒有可用於防止或管理EBOV感染之疫苗或暴露後治療模態。患者代替地接受支持療法,即,電解質和液體平衡、氧氣、血壓維持、及任何二次感染之治療。
因此,需要用於治療EBOV感染之組成物和方法。本發明解決了這些和其他需求。
發明概述
本發明提供一種治療需要該治療之人類的絲狀病毒科感染之方法,其包含投予治療有效量的式IV化合物: 或其醫藥上可接受的鹽、水合物或酯;
其中,
R7
係選自由下列組成之群組:
a) H、-C(=O)R11
、-C(=O)OR11
、-C(=O)NR11
R12
、-C(=O)SR11
、-S(O)R11
、-S(O)2
R11
、-S(O)(OR11
)、-S(O)2
(OR11
)、或-SO2
NR11
R12
;
b)
或其醫藥上可接受的鹽、水合物或酯;
其中,
R7
係選自由下列組成之群組:
a) H、-C(=O)R11
、-C(=O)OR11
、-C(=O)NR11
R12
、-C(=O)SR11
、-S(O)R11
、-S(O)2
R11
、-S(O)(OR11
)、-S(O)2
(OR11
)、或-SO2
NR11
R12
;
b) c) 選自下列之群組:
c) 選自下列之群組: 其中:
Rc
係選自下列之群組:苯基、1-萘基、2-萘基,
其中:
Rc
係選自下列之群組:苯基、1-萘基、2-萘基,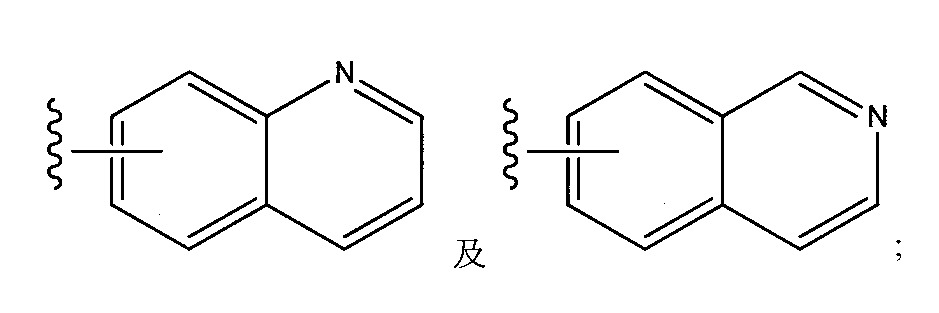 Rd
係選自下列之群組:H或CH3
;
Re1
和Re2
係各自獨立地選自下列之群組:H、(C1
-C6
)烷基或苯甲基;
Rf
係選自下列之群組:H、(C1
-C8
)烷基、苯甲基、(C3
-C6
)環烷基、和-CH2
-(C3
-C6
)環烷基;
Rg
係選自下列之群組:(C1
-C8
)烷基、-O-(C1
-C8
)烷基、苯甲基、-O-苯甲基、-CH2
-(C3
-C6
)環烷基、-O-CH2
-(C3
-C6
)環烷基、和CF3
;及
n’為選自下列群組之整數:1、2、3,及4;及
d) 下式之基團:
Rd
係選自下列之群組:H或CH3
;
Re1
和Re2
係各自獨立地選自下列之群組:H、(C1
-C6
)烷基或苯甲基;
Rf
係選自下列之群組:H、(C1
-C8
)烷基、苯甲基、(C3
-C6
)環烷基、和-CH2
-(C3
-C6
)環烷基;
Rg
係選自下列之群組:(C1
-C8
)烷基、-O-(C1
-C8
)烷基、苯甲基、-O-苯甲基、-CH2
-(C3
-C6
)環烷基、-O-CH2
-(C3
-C6
)環烷基、和CF3
;及
n’為選自下列群組之整數:1、2、3,及4;及
d) 下式之基團: 其中:
Q係選自下列之群組:O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;
Z1
和Z2
,當結合在一起時,為-Q1
(C(Ry)2
)3
Q1
-;
其中
各Q1
係獨立地選自下列之群組:O、S、或NR;及
各Ry
係獨立地選自下列之群組:H、F、Cl、Br、I、OH、R、-C(=Q2
)R、-C(=Q2
)OR、-C(=Q2
)N(R)2
、-N(R)2
、-+
N(R)3
、-SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、-OC(=Q1
)R、-OC(=Q2
)OR、-OC(=Q2
)(N(R)2
)、-SC(=Q2
)R、-SC(=Q2
)OR、-SC(=Q2
)(N(R)2
)、-N(R)C(=Q2
)R、-N(R)C(=Q2
)OR、-N(R)C(=Q2
)N(R)2
、-SO2
NR2
、-CN、-N3
、-NO2
、-OR、或Z3
;或當結合在一起時,在同一碳原子上之二個Ry
形成3-7個碳原子的碳環;
各Q2
獨立地為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;或
Z1
和Z2
係各自獨立地為式Ia之基團:
其中:
Q係選自下列之群組:O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;
Z1
和Z2
,當結合在一起時,為-Q1
(C(Ry)2
)3
Q1
-;
其中
各Q1
係獨立地選自下列之群組:O、S、或NR;及
各Ry
係獨立地選自下列之群組:H、F、Cl、Br、I、OH、R、-C(=Q2
)R、-C(=Q2
)OR、-C(=Q2
)N(R)2
、-N(R)2
、-+
N(R)3
、-SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、-OC(=Q1
)R、-OC(=Q2
)OR、-OC(=Q2
)(N(R)2
)、-SC(=Q2
)R、-SC(=Q2
)OR、-SC(=Q2
)(N(R)2
)、-N(R)C(=Q2
)R、-N(R)C(=Q2
)OR、-N(R)C(=Q2
)N(R)2
、-SO2
NR2
、-CN、-N3
、-NO2
、-OR、或Z3
;或當結合在一起時,在同一碳原子上之二個Ry
形成3-7個碳原子的碳環;
各Q2
獨立地為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;或
Z1
和Z2
係各自獨立地為式Ia之基團: 其中:
各Q3
係獨立地選自下列之群組:一鍵、O、CR2
、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、N-NR2
、S、S-S、S(O)、或S(O)2
;
M2為選自下列群組之整數:0、1或2;
各Rx
獨立地為Ry
或式:
其中:
各Q3
係獨立地選自下列之群組:一鍵、O、CR2
、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、N-NR2
、S、S-S、S(O)、或S(O)2
;
M2為選自下列群組之整數:0、1或2;
各Rx
獨立地為Ry
或式: 其中:
各M1a、M1c、和M1d為獨立地選自下列群組之整數:0或1;
M12c為選自下列群組之整數:0、1、2、3、4、5、6、7、8、9、10、11或12;
Z3
為Z4
或Z5
;
Z4
為R、-C(Q2
)Ry
、-C(Q2
)Z5
、-SO2
Ry
、或-SO2
Z5
;及
Z5
為碳環或雜環,其中Z5
獨立地經0至3個Ry
基團取代;
各R11
或R12
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C4
-C8
)碳環基烷基、(C6
-C20
)隨意地經取代之芳基、隨意地經取代之雜芳基、-C(=O)(C1
-C8
)烷基、-S(O)n
(C1
-C8
)烷基或(C6
-C20
)芳基(C1
-C8
)烷基;或R11
和R12
與它們所共同連接之氮原子一起形成3至7員雜環,其中該雜環之任何一個碳原子可隨意地經-O-、-S-或 -NRa
-置換;
各Ra
係獨立地選自下列之群組:H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C6
-C20
)芳基(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、-C(=O)R、-C(=O)OR、-C(=O)NR2
、 -C(=O)SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、或-SO2
NR2
;其中
各R係獨立地選自下列之群組:H、(C1
-C8
)烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基、(C2
-C8
)經取代之炔基、(C6
-C20
)芳基、(C6
-C20
)經取代之芳基、(C2
-C20
)雜環基、(C2
-C20
)經取代之雜環基、(C6
-C20
)芳基(C1
-C8
)烷基或經取代之(C6
-C20
)芳基(C1
-C8
)烷基;
各n為獨立地選自下列群組之整數:0、1、或2;及
其中各R11
或R12
之各(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基或(C6
-C20
)芳基(C1
-C8
)烷基係獨立地隨意地經一或多個選自下列群組之取代基取代:鹵基、羥基、CN、N3
、N(Ra
)2
或ORa
;及其中各該(C1
-C8
)烷基的非末端碳原子中之一或多者可隨意地經-O-、-S-或-NRa
-置換。
其中:
各M1a、M1c、和M1d為獨立地選自下列群組之整數:0或1;
M12c為選自下列群組之整數:0、1、2、3、4、5、6、7、8、9、10、11或12;
Z3
為Z4
或Z5
;
Z4
為R、-C(Q2
)Ry
、-C(Q2
)Z5
、-SO2
Ry
、或-SO2
Z5
;及
Z5
為碳環或雜環,其中Z5
獨立地經0至3個Ry
基團取代;
各R11
或R12
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C4
-C8
)碳環基烷基、(C6
-C20
)隨意地經取代之芳基、隨意地經取代之雜芳基、-C(=O)(C1
-C8
)烷基、-S(O)n
(C1
-C8
)烷基或(C6
-C20
)芳基(C1
-C8
)烷基;或R11
和R12
與它們所共同連接之氮原子一起形成3至7員雜環,其中該雜環之任何一個碳原子可隨意地經-O-、-S-或 -NRa
-置換;
各Ra
係獨立地選自下列之群組:H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C6
-C20
)芳基(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、-C(=O)R、-C(=O)OR、-C(=O)NR2
、 -C(=O)SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、或-SO2
NR2
;其中
各R係獨立地選自下列之群組:H、(C1
-C8
)烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基、(C2
-C8
)經取代之炔基、(C6
-C20
)芳基、(C6
-C20
)經取代之芳基、(C2
-C20
)雜環基、(C2
-C20
)經取代之雜環基、(C6
-C20
)芳基(C1
-C8
)烷基或經取代之(C6
-C20
)芳基(C1
-C8
)烷基;
各n為獨立地選自下列群組之整數:0、1、或2;及
其中各R11
或R12
之各(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基或(C6
-C20
)芳基(C1
-C8
)烷基係獨立地隨意地經一或多個選自下列群組之取代基取代:鹵基、羥基、CN、N3
、N(Ra
)2
或ORa
;及其中各該(C1
-C8
)烷基的非末端碳原子中之一或多者可隨意地經-O-、-S-或-NRa
-置換。
在另一實施態樣中,本發明提供一種化合物,其為

 或其醫藥上可接受的鹽、水合物、或其酯。
發明之詳細說明
I.定義
或其醫藥上可接受的鹽、水合物、或其酯。
發明之詳細說明
I.定義
除非另有說明,否則在此所使用的下列術語和片語係具有下列意義:
當本文使用商品名時,申請人的本意是獨立地包括該商品名產物以及該商品名產物的活性醫藥成分。
如使用於本文中者,“本發明化合物”或“式IV化合物”係指式IV化合物或其醫藥上可接受的鹽。同樣地,關於可單離的中間物,短語“式(編號)化合物”係指該式化合物或其醫藥上可接受的鹽。
“烷基”為含有直鏈、二級、三級或環狀碳原子的烴。例如,烷基可具有1至20個碳原子(即,C1
-C20
烷基)、1到8個碳原子(即,C1
-C8
烷基)、或1至6個碳原子(即,C1
-C6
烷基)。適當烷基的實例包括(但不限於)甲基(Me、-CH3
)、乙基(Et、-CH2
CH3
)、1-丙基(正Pr、正丙基、-CH2
CH2
CH3
)、2-丙基(異Pr、異丙基、-CH(CH3
)2
)、1-丁基(正Bu、正丁基、-CH2
CH2
CH2
CH3
)、2-甲基-1-丙基(異Bu、異丁基、-CH2
CH(CH3
)2
)、2-丁基(第二Bu、第二丁基、-CH(CH3
)CH2
CH3
)、2-甲基-2-丙基(第三-Bu、第三-丁基、-C(CH3
)3
)、1-戊基(正戊基、-CH2
CH2
CH2
CH2
CH3
)、2-戊基(-CH(CH3
)CH2
CH2
CH3
)、3-戊基(-CH(CH2
CH3
)2
)、2-甲基-2-丁基(-C(CH3
)2
CH2
CH3
)、3-甲基-2-丁基(-CH(CH3
)CH(CH3
)2
)、3-甲基-1-丁基(-CH2
CH2
CH(CH3
)2
)、2-甲基-1-丁基(-CH2
CH(CH3
)CH2
CH3
)、1-己基(-CH2
CH2
CH2
CH2
CH2
CH3
)、2-己基(-CH(CH3
)CH2
CH2
CH2
CH3
)、3-己基(-CH(CH2
CH3
)(CH2
CH2
CH3
))、2-甲基-2-戊基(-C(CH3
)2
CH2
CH2
CH3
)、3-甲基-2-戊基(-CH(CH3
)CH(CH3
)CH2
CH3
)、4-甲基-2-戊基(-CH(CH3
)CH2
CH(CH3
)2
)、3-甲基-3-戊基(-C(CH3
)(CH2
CH3
)2
)、2-甲基-3-戊基(-CH(CH2
CH3
)CH(CH3
)2
)、2,3-二甲基-2-丁基(-C(CH3
)2
CH(CH3
)2
)、3,3-二甲基-2-丁基(-CH(CH3
)C(CH3
)3
,及辛基(-(CH2
)7
CH3
)。
“烷氧基”意指具有式-O-烷基的基團,其中如上述所定義之烷基係經由一氧原子連接至母分子。烷氧基之烷基部份可具有1至20個碳原子(即,C1
-C20
烷氧基)、1至12個碳原子的(即,C1
-C12
烷氧基)、或1至6個碳原子(即,C1
-C6
烷氧基)。適當烷氧基的實例包括(但不限於)甲氧基(-O-CH3
或-OMe)、乙氧基(-OCH2
CH3
或-OEt)、第三丁氧基(-O-C(CH3
)3
或-OtBu)等等。
“鹵烷基”為如上述所定義之烷基,其中烷基之一或多個氫原子被鹵素原子置換。鹵烷基之烷基部份可具有1至20個碳原子(即,C1
-C20
鹵烷基)、1至12個碳原子 (即,C1
-C12
鹵烷基),或1至6個碳原子(即,C1
-C6
烷基)。適當鹵烷基的實例包括(但不限於)-CF3
、-CHF2
、-CFH2
、-CH2
CF3
,等等。
“烯基”為含有直鏈、二級、三級或環狀碳原子與至少一個不飽和位置(亦即,碳-碳sp2
雙鍵)的烴。例如,烯基可具有2至20個碳原子(即,C2
-C20
烯基)、2至8個碳原子(即,C2
-C8
烯基)、或2至6個碳原子(即,C2
-C6
烯基)。適當烯基的實例包括(但不限於)乙烯或乙烯基(-CH=CH2
)、烯丙基(-CH2
CH=CH2
)、環戊烯基(-C5
H7
),及5-己烯基(-CH2
CH2
CH2
CH2
CH=CH2
)。
“炔基”為含有直鏈、二級、三級或環狀碳原子與至少一個不飽和位置(亦即,碳-碳sp參鍵)的烴。例如,炔基可具有2至20個碳原子(即,C2
-C20
炔基)、2至8個碳原子(即,C2
-C8
炔),或2至6個碳原子(即,C2
-C6
炔基)。適當炔基的實例包括(但不限於)乙炔系(-C≡CH)、丙炔基(-CH2
C≡CH)、等等。
“伸烷基”係指具有從母烷之相同或兩個不同的碳原子移除兩個氫原子所衍生之兩個單價基中心的飽和支鏈或直鏈或環狀烴基團。例如,伸烷基可具有1至20個碳原子,1至10個碳原子,或1至6個碳原子。典型的伸烷基包括(但不限於)亞甲基(-CH2
-)、1,1-乙基 (-CH(CH3
)-)、1,2-乙基(-CH2
CH2
-)、1,1-丙基(-CH(CH2
CH3
)-)、1,2-丙基(-CH2
CH(CH3
)-)、1,3-丙基(-CH2
CH2
CH2
-)、1,4-丁基(-CH2
CH2
CH2
CH2
-),等等。
“伸烯基”係指具有從母烯之相同或兩個不同的碳原子移除兩個氫原子所衍生之兩個單價基中心的不飽和支鏈或直鏈或環狀烴基。例如,及伸烯基可具有1至20個碳原子、1至10個碳原子、或1至6個碳原子。典型的伸烯基包括(但不限於)1,2-乙烯(-CH=CH-)。
“伸炔基”係指具有從母炔之相同或兩個不同的碳原子移除兩個氫原子所衍生之兩個單價基中心的不飽和支鏈或直鏈或環狀烴基。例如,伸炔基可具有1至20個碳原子、1至10個碳原子,或1至6個碳原子。典型之伸炔基包括(但不限於)乙炔基(-C≡C-)、炔丙基(-CH2
C≡C-)及4-戊炔基(-CH2
CH2
CH2
C≡C-)。
“胺基”通常係指可被視為氨的衍生物之氮基團,其具有式-N(X)2
,其中各“X”獨立地為H、經取代或未經取代之烷基、經取代或未經取代之碳環基、經取代或未經取代之雜環基、等等。該氮之混成為約sp3
。胺基之非限制性類型包括-NH2
、-N(烷基)2
、-NH(烷基)、-N(碳環基)2
、-NH(碳環基)、-N(雜環基)2
、-NH(雜環基)、-N(芳基)2
、-NH(芳基)、-N(烷基)(芳基)、-N(烷基)(雜環基)、 -N(碳環基)(雜環基)、-N(芳基)(雜芳基)、-N(烷基)(雜芳基)、等等。術語“烷胺基”係指經至少一個烷基取代之胺基。胺基的非限制性實例包括-NH2
、-NH(CH3
)、-N(CH3
)2
、-NH(CH2
CH3
)、-N(CH2
CH3
)2
、-NH(苯基)、-N(苯基)2
、-NH(苯甲基)、-N(苯甲基)2
、等等。經取代之烷胺基通常係指如上述所定義之烷胺基,其中至少一個經取代之烷基(如上述所定義)係連接至胺基氮原子。經取代之烷胺基的非限制性實例包括-NH(伸烷基-C(O)-OH)、-NH(伸烷基-C(O)-O-烷基)、-N(伸烷基-C(O)-OH)2
、-N(伸烷基-C(O)-O-烷基)2
、等等。
“芳基”意指藉由從母芳族環系統之單一碳原子移除一個氫原子所衍生物之芳族烴基團。例如,芳基可具有6至20個碳原子、6至14個碳原子、或6至10個碳原子。典型芳基包括(但不限於)從苯(例如苯基)、經取代之苯、萘、蒽、聯苯、等等所衍生之基團。
“芳基烷基”係指其中鍵結於碳原子(通常為末端或sp3
碳原子)的氫原子中之一者係以芳基置換的非環烷基。典型的芳基烷基包括(但不限於)苯甲基、2-苯乙烷-1-基、萘甲基、2-萘乙烷-1-基、萘苯甲基、2-萘苯乙烷-1-基等等。該芳基烷基可包含7至20個碳原子,例如,該烷基部分為1至6個碳原子且該芳基部分為6至14個碳原子。
“芳基烯基”係指其中鍵結至碳原子(通常為末端或sp3
碳原子,但亦可為sp2
碳原子)的氫原子中之一者係經芳基置換的非環烯基。該芳基烯基之芳基部分可包括(例如)本文所揭示之芳基中的任一者,且該芳基烯基之烯基部分可包括(例如)本文所揭示之烯基的任一者。該芳基烯基可包含8至20個碳原子,例如,該烯基部分為2至6個碳原子且該芳基部分為6至14個碳原子。
“芳基炔基”係指其中鍵結至碳原子(通常為末端或sp3
碳原子,但亦可為sp碳原子)的氫原子中之一者係經芳基置換的非環炔基。該芳基炔基的芳基部分可包括(例如)本文所揭示之芳基中的任一者,且該芳基炔基的炔基部分可包括(例如)本文所揭示之炔基中的任一者。該芳基炔基可包含8至20個碳原子,例如,該炔基部分為2至6個碳原子和該芳基部分為6到14個碳原子。
關於烷基、伸烷基、芳基、芳基烷基、烷氧基、雜環基、雜芳基、碳環基等等之術語“經取代”,例如,“經取代之烷基、“經取代之伸烷基”、“經取代之芳基”、“經取代之芳基烷基”、“經取代之雜環基”,及“經取代之碳環基”分別意指其中一或多個氫原子各自獨立地經非氫取代基置換之烷基、伸烷基、芳基、芳基烷基、雜環基、碳環基。典型的取代基包括(但不限於)-X、-Rb
、-O-
、=O、-ORb
、-SRb
、-S-
、-NRb 2
、-N+
Rb 3
、=NRb
、-CX3
、 -CN、-OCN、-SCN、-N=C=O、-NCS、-NO、-NO2
、=N2
、-N3
、-NHC(=O)Rb
、-OC(=O)Rb
、-NHC(=O)NRb 2
、-S(=O)2
-、-S(=O)2
OH、-S(=O)2
Rb
、-OS(=O)2
ORb
、-S(=O)2
NRb 2
、-S(=O)Rb
、-OP(=O)(ORb
)2
、-P(=O)(ORb
)2
、-P(=O)(O-
)2
、-P(=O)(OH)2
、-P(O)(ORb
)(O-
)、-C(=O)Rb
、-C(=O)X、-C(S)Rb
、-C(O)ORb
、-C(O)O-
、-C(S)ORb
、-C(O)SRb
、-C(S)SRb
、-C(O)NRb 2
、-C(S)NRb 2
、-C(=NRb
)NRb 2
,其中各X獨立為鹵素:F、Cl、Br或I;且各Rb
獨立為H、烷基、芳基、芳基烷基、雜環或保護基、或前藥部分。伸烷基、伸烯基及伸炔基亦可經取代。除非另外指示,否則當術語“經取代”與諸如具有二或多個可取代部分的芳基烷基之基團併用時,該等取代基可連接至該芳基部分、該烷基部分或連接至該二者。
術語“前藥”如使用於本文中係指當投予至生物系統由於自發性化學反應、酵素催化的化學反應、光解反應、和/或新陳代謝化學反應而產生藥劑物質(即活性成分)的任何化合物。前藥因此為治療性活性化合物的共價修飾之類似物或潛在形式。
熟習該項技術者將理解:應選擇式IV化合物的取代基和其他部分,以提供足夠穩定的化合物而提供可調配成可接受之穩定醫藥組成物的醫藥上有用之化合物。預期具有該穩定性之式IV化合物落在本發明範圍內。
“雜烷基”係指其中1或多個碳原子已被雜原子如O、N、或S置換之烷基。例如,若其連接至母分子之烷基的碳原子經雜原子(例如,O、N、或S)置換,則所得雜烷基分別為烷氧基(例如,-OCH3
、等等)、胺(例如,-NHCH3
、-N(CH3
)2
、等等)、或烷硫基(例如,-SCH3
)。若未連接至母分子之烷基的非末端碳原子經雜原子(例如,O、N、或S)置換,則所得雜烷基分別為烷基醚(例如,-CH2
CH2
-O-CH3
、等等)、烷基胺(例如,-CH2
NHCH3
,-CH2
N(CH3
)2
、等等)、或烷硫基醚(例如,-CH2
-S-CH3
)。若烷基的末端碳原子經雜原子(例如,O、N、或S)置換,則所得雜烷基分別為羥基烷基(例如,-CH2
CH2
-OH)、胺烷基(例如,-CH2
NH2
)、或烷基硫醇基團(例如,-CH2
CH2
-SH)。雜烷基可具有(例如)1至20個碳原子、1至10個碳原子、或1至6個碳原子。C1
-C6
雜烷基意指具有1至6個碳原子之雜烷基。
“雜環”或“雜環基”如使用於本文中包括(例如且非限制地)Paquette, Leo A.;Principles of Modern Heterocyclic Chemistry (W.A. Benjamin,New York, 1968),特別是第1、3、4、6、7及9章;The Chemistry of Heterocyclic Compounds, A Series of Monographs” (John Wiley & Sons, New York, 1950至今),特別是第13、14、16、19及28冊;及 J. Am. Chem. Soc. (1960) 82:5566中所述之雜環。在本發明一特定實施態樣中,雜環包括如本文所定義之“碳環”,其中一或多個(例如,1、2、3或4個)碳原子已經雜原子(例如,O、N或S)置換。術語“雜環”或“雜環基”包括飽和環、部分不飽和環及芳族環(即,雜芳族環)。經取代之雜環基包括(例如)經本文所揭示之任一取代基(包括羰基)取代之雜環。經羰基取代之雜環基的非限制性實例為:
雜環的實例包括(例如且非限制地)吡啶基、二氫吡啶基、四氫吡啶基(哌啶基)、噻唑基、四氫硫苯基、硫氧化的四氫硫苯基、嘧啶基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、四唑基、苯並呋喃基、硫萘基(thianaphthalenyl)、吲哚基、吲哚烯基(indolenyl)、喹啉基、異喹啉基、苯並咪唑基、哌啶基、4-哌啶酮基、吡咯啶基、2-吡咯啶酮基、吡咯啉基、四氫呋喃基、四氫喹啉基、四氫異喹啉基、十氫喹啉基、八氫異喹啉基、氮㖕基(azocinyl)、三𠯤基、6H-1,2,5-噻二𠯤基、2H,6H-1,5,2-二噻𠯤基、噻吩基、噻嗯基、哌喃基、異苯並呋喃基、𠳭烯基、𠮿基(xanthenyl)、啡噻基(phenoxathinyl)、2H-吡咯基、異噻唑基、異㗁唑基、吡𠯤基、嗒𠯤基、吲 基、異吲哚基、3H-吲哚基、1H-吲唑基、嘌呤基、4H-喹𠯤基、呔𠯤基、㖠啶基、喹㗁啉基、喹唑啉基、㖕啉基、喋啶基、4aH-咔唑基、咔唑基、β-咔啉基、啡啶基、吖啶基、嘧啶基、啡啉基、啡𠯤基、啡噻𠯤基、呋吖基、啡㗁𠯤基、異克基、克基、咪唑啶基、咪唑啉基、吡唑啶基、吡唑啉基、哌𠯤基、吲哚啉基、異吲哚啉基、
基、異吲哚基、3H-吲哚基、1H-吲唑基、嘌呤基、4H-喹𠯤基、呔𠯤基、㖠啶基、喹㗁啉基、喹唑啉基、㖕啉基、喋啶基、4aH-咔唑基、咔唑基、β-咔啉基、啡啶基、吖啶基、嘧啶基、啡啉基、啡𠯤基、啡噻𠯤基、呋吖基、啡㗁𠯤基、異克基、克基、咪唑啶基、咪唑啉基、吡唑啶基、吡唑啉基、哌𠯤基、吲哚啉基、異吲哚啉基、 啶基、𠰌啉基、㗁唑啶基、苯並三唑基、苯並異㗁唑基、吲哚酮基、苯並㗁唑啉基、isatinoyl、和雙-四氫呋喃基:
啶基、𠰌啉基、㗁唑啶基、苯並三唑基、苯並異㗁唑基、吲哚酮基、苯並㗁唑啉基、isatinoyl、和雙-四氫呋喃基: 。
。
例如且非限制地,碳鍵結的雜環係鍵結在吡啶之2、3、4、5或6位置,嗒𠯤之3、4、5或6位置,嘧啶之2、4、5或6位置,吡𠯤之2、3、5或6位置,呋喃、四氫呋喃、噻喃、噻吩、吡咯或四氫吡咯之2、3、4或5位置,㗁唑、咪唑或噻唑之2、4或5位置,異㗁唑、吡唑或異噻唑之3、4或5位置,吖 之2或3位置,吖呾之2、3或4位置,喹啉之2、3、4、5、6、7或8位置或異喹啉之1、3、4、5、6、7或8位置。仍更典型地,碳鍵結的雜環包括2-吡啶基、3-吡啶基、4-吡啶基、5-吡啶基、6-吡啶基、3-嗒𠯤基、4-嗒𠯤基、5-嗒𠯤基、6-嗒𠯤基、2-嘧啶基、4-嘧啶基、5-嘧啶基、6-嘧啶基、2-吡𠯤基、3-吡𠯤基、5-吡𠯤基、6-吡𠯤基、2-噻唑基、4-噻唑基、或5-噻唑基。
之2或3位置,吖呾之2、3或4位置,喹啉之2、3、4、5、6、7或8位置或異喹啉之1、3、4、5、6、7或8位置。仍更典型地,碳鍵結的雜環包括2-吡啶基、3-吡啶基、4-吡啶基、5-吡啶基、6-吡啶基、3-嗒𠯤基、4-嗒𠯤基、5-嗒𠯤基、6-嗒𠯤基、2-嘧啶基、4-嘧啶基、5-嘧啶基、6-嘧啶基、2-吡𠯤基、3-吡𠯤基、5-吡𠯤基、6-吡𠯤基、2-噻唑基、4-噻唑基、或5-噻唑基。
例如且非限制地,氮鍵結的雜環係鍵結在吖、吖呾、吡咯、吡咯啶、2-吡咯啉、3-吡咯啉、咪唑、咪唑啶、2-咪唑啉、3-咪唑啉、吡唑、吡唑啉、2-吡唑啉、3-吡唑啉、哌啶、哌𠯤、吲哚、吲哚啉、1H-吲唑之1位置;異吲哚或異吲哚啉之2位置;佅啉之4位置,及咔唑或β-咔啉之9位置。又更典型地,氮鍵結的雜環包括1-吖基(1-aziridyl)、1-吖丁基(1-azetedyl)、1-吡咯基、1-咪唑基、1-吡唑基及1-哌啶基(1-piperidinyl)。
“雜環基烷基”係指其中鍵結至碳原子(通常終端或sp3
碳原子)的氫原子中之一者經雜環基置換之非環烷基(即,雜環基-伸烷基-部分)。典型的雜環基烷基包括但不限於:雜環基-CH2
-、2-(雜環基)乙烷-1-基、等等,其中“雜環基”部分包括上述所有雜環基,包括彼等Principles of Modern Heterocyclic Chemistry中所述者。熟習該項技術者亦將理解,雜環基可藉碳-碳鍵或碳-雜原子鍵連接至雜環基烷基的烷基部分,先決條件為所得基團為化學穩定的。雜環基烷基包含3至20個碳原子,例如,芳基烷基的烷基部分為1至6個碳原子及雜環基部分為2至14個碳原子。雜環基烷基的實例包括(例如且非限制地) 5員之含硫、氧、及/或氮的雜環(諸如噻唑基甲基、2-噻唑基乙烷-1-基、咪唑基甲基、㗁唑基甲基、噻二唑基甲基、等等)、6員的含硫、氧及/或氮的雜環(諸如,哌啶基甲基、六氫吡𠯤基甲基、𠰌啉基甲基、吡啶基甲基、嗒𠯤基(pyridizyl)甲基、嘧啶基甲基、吡𠯤基(pyrazinyl)甲基等等。
“雜環基烯基”係指其中鍵結至碳原子(通常為末端或sp3
碳原子,但亦為sp2
碳原子)的氫原子中之一者經雜環基置換之非環烯基(即,雜環基-伸烯基-部分)。雜環基烯基的雜環基部分包括本文中所述之任何雜環基,包括彼等Principles of Modern Heterocyclic Chemistry中所述者,且雜環基烯基的烯基部分包括本文中所揭示之任何烯基。熟習該項技術者亦將理解,雜環基可藉碳-碳鍵或碳-雜原子鍵連接至雜環基烯基的烯基部分,先決條件為所得基團為化學穩定的。雜環基烯基包含4至20個碳原子,例如,雜環基烯基的烯基部分為2至6個碳原子及雜環基部分為2至14個碳原子。
“雜環基炔基”係指其中鍵結至碳原子(通常為末端或sp3
碳原子,但亦為sp碳原子)的氫原子中之一者經雜環基置換之非環炔基(即,雜環基-伸炔基-部分)。雜環基炔基的雜環基部分包括本文中所述之任何雜環基,包括彼等Principles of Modern Heterocyclic Chemistry中所述者,且雜環基炔基的炔基部分包括本文中所揭示之任何炔基。熟習該項技術者亦將理解,雜環基可藉碳-碳鍵或碳-雜原子鍵連接至雜環基炔基的炔基部分,先決條件為所得基團為化學穩定的。雜環基炔基包含4至20個碳原子,例如,雜環基炔基的炔基部分為2至6個碳原子及雜環基部分為2至14個碳原子。
“雜芳基”係指於環中具有至少一個雜原子之芳族雜環基。芳族環中可包含之適當雜原子的非限制性實例包括氧、硫、及氮。雜芳基環之非限制性實例包括它們於“雜環基”定義中所列者全部,包括吡啶基、吡咯基、㗁唑基、吲哚基、異吲哚基、嘌呤基、呋喃基、噻吩基、苯並呋喃基、苯並硫苯基、咔唑基、咪唑基、噻唑基、異㗁唑基、吡唑基、異噻唑基、喹啉基、異喹啉基、嗒𠯤基(pyridazyl)、嘧啶基、吡𠯤基(pyrazyl)、等等。
“碳環”或“碳環基”係指飽和(即,環烷基)、部分不飽和(例如環烯基、環二烯基、等等)或芳族環,其具有3至7個碳原子作為單環、7至12個碳原子作為雙環,及至多達約20個碳原子作為多環。單環碳環具有3至7個環原子,又更典型為5或6個環原子。雙環碳環具有7至12個環原子,其係例如排列為雙環[4,5]、[5,5]、[5,6]或[6,6]系統,或9或10個環原子,其排列為雙環[5,6]或[6,6]系統或螺-稠合環。單環碳環之非限制性實例包括環丙基、環丁基、環戊基、1-環戊-1-烯基、1-環戊-2-烯基、1-環戊-3-烯基、環己基、1-環己-1-烯基、1-環己-2-烯基、1-環己-3-烯基、及苯基。雙環碳環之非限制性實例包括萘基、四氫萘及十氫萘。
“碳環基烷基”係指其中鍵結至碳原子的氫原子中之一者經如本文中所述之碳環基置換之非環烷基。碳環基烷基之典型但非限制性實例包括環丙基甲基、環丙基乙基、環丁基甲基、環戊基甲基和環己基甲基。
“芳基雜烷基”意指如本文所定義的雜烷基,其中氫原子(其可連結到碳原子或雜原子)已被如本文所定義的芳基置換。該芳基可鍵結至雜烷基的碳原子上,或鍵結至雜烷基的雜原子上,先決條件為所得芳基雜烷基提供化學穩定部分。例如,芳基雜烷基可具有通式-伸烷基-O-芳基、-伸烷基-O-伸烷基-芳基、-伸烷基-NH-芳基、-伸烷基-NH-伸烷基-芳基、-伸烷基-S-芳基、-伸烷基-S-伸烷基-芳基、等等。此外,在上述通式中的伸烷基部分中之任一者可進一步經本文中所定義或舉例說明的取代基中的任一者取代。
“雜芳基烷基”係指如本文所定義的烷基,其中氫原子已被如本文所定義的雜芳基置換。雜芳基烷基的非限制性實例包括-CH2
-吡啶基、-CH2
-吡咯基、-CH2
-㗁唑基、-CH2
-吲哚基、-CH2
-異吲哚基、-CH2
-嘌呤基、-CH2
-呋喃基、-CH2
-噻吩基、-CH2
-苯並呋喃基、-CH2
-苯並硫苯基、-CH2
-咔唑基、-CH2
-咪唑基、-CH2
-噻唑基、-CH2
-異㗁唑基、-CH2
-吡唑基、-CH2
-異噻唑基、-CH2
-喹啉基、-CH2
-異喹啉基、-CH2
-嗒𠯤基(pyridazyl)、-CH2
-嘧啶基、-CH2
-吡𠯤基(pyrazyl)、-CH(CH3
)-吡啶基、-CH(CH3
)-吡咯基、-CH(CH3
)-㗁唑基、-CH(CH3
)-吲哚基、-CH(CH3
)-異吲哚基、-CH(CH3
)-嘌呤基、-CH(CH3
)-呋喃基、-CH(CH3
)-噻吩基、-CH(CH3
)-苯並呋喃基、-CH(CH3
)-苯並硫苯基、-CH(CH3
)-咔唑基、-CH(CH3
)-咪唑基、-CH(CH3
)-噻唑基、-CH(CH3
)-異㗁唑基、-CH(CH3
)-吡唑基、-CH(CH3
)-異噻唑基、-CH(CH3
)-喹啉基、-CH(CH3
)-異喹啉基、-CH(CH3
)-嗒𠯤基(pyridazyl)、-CH(CH3
)-嘧啶基、-CH(CH3
)-吡𠯤基(pyrazyl)、等等。
關於式IV化合物之特定部分的術語“隨意地經取代”(例如,隨意地經取代之芳基)係指其中所有取代基為氫或其中該部分的氫之一或多者可經取代基置換之部分,諸如它們在“經取代”之定義下所列出者。
關於式IV化合物之特定部分的術語“隨意地經置換”(例如,該(C1
-C8
)烷基之碳原子可隨意地經-O-、 -S-、或-NRa
-置換)意指(C1
-C8
)烷基之亞甲基中之一或多者可經所指定基團(例如,-O-、-S-、或-NRa
-)置換0、1、2、或更多次。
關於烷基、烯基、炔基、伸烷基、伸烯基或伸炔基部份的術語“非末端碳原子”係指介於該部份的第一個碳原子和最後一個碳原子之間的部份中的碳原子。因此,例如且非限制地,烷基部份-CH2
(C*
)H2
(C*
)H2
CH3
或伸烷基部分-CH2
(C*
)H2
(C*
)H2
CH2
-中該C*原子應認為是非末端碳原子。
某些Q和Q1
替代品為氮氧化物諸如+
N(O)(R)或+
N(O)(OR)。這些氮氧化物,如此處所示地連接至碳原子,亦可分別以電荷分離基團表示,諸如 並且意欲與前述為了描述本發明的表示相等。
並且意欲與前述為了描述本發明的表示相等。
“連結子”或“連結”意指包含共價鍵或原子之鏈的化學部分。連結子包括烷氧基(例如聚伸乙氧基、PEG、聚亞甲氧基)和烷胺基(例如聚伸乙胺基、JeffamineTM
);二元酸酯或醯胺類(包括丁二酸酯、丁二醯胺、二乙二醇酯、丙二酸酯及己醯胺)之重複單元。
術語諸如“連結氧”、“連結氮”、“連結碳”、“連結硫”、或“連結磷”意指若可在一個部份中藉由使用一個以上的原子在兩個部份之間形成鍵,則在該等部份之間形成的鍵係經由所指定的原子。例如,連結氮的胺基酸將經由該胺基酸的氮原子而非該胺基酸的氧或碳原子進行鍵結。
在式IV化合物之一些實施態樣中,Z1
或Z2
之一或多者獨立為連結氮之天然α-胺基酸酯的基團。天然胺基酸的實例包括異白胺酸、白胺酸、離胺酸、甲硫胺酸、苯基丙胺酸、蘇胺酸、色胺酸、纈胺酸、丙胺酸、天冬醯胺酸、天冬胺酸、半胱胺酸、麩胺酸、麩醯胺、甘胺酸、脯胺酸、硒基半胱胺酸、絲胺酸、酪胺酸、精胺酸、組胺酸、鳥胺酸和牛磺酸。此等胺基酸類的酯類包含任何它們就取代基R所述者,特別是它們其中R為隨意地經取代之(C1
-C8
)烷基者。
術語“嘌呤”或“嘧啶”鹼包含(但不限於)、腺嘌呤、N6
-烷基嘌呤類、N6
-醯基嘌呤類(其中醯基為C(O)(烷基,芳基、烷基芳基、或芳基烷基)、N6
-苯甲基嘌呤、N6
-鹵嘌呤、N6
-乙烯基嘌呤、N6
炔屬嘌呤、N6
-醯基嘌呤、N6
-羥基烷基嘌呤、N6
-烯丙基胺基嘌呤、N6
-硫烯丙基嘌呤、N2
-烷基嘌呤類、N2
-烷基-6-硫嘌呤類、胸腺嘧碇、胞嘧啶、5-氟胞嘧啶、5-甲基胞嘧啶、6-氮雜嘧啶(包括6-氮雜胞嘧啶)、2-及/或4-巰基嘧啶、尿嘧啶、5-鹵尿嘧啶(包括5-氟尿嘧啶)、C5
-烷基嘧啶類、C5
-苯甲基嘧啶類、C5
-鹵嘧啶類、C5
-乙烯基嘧啶、C5
炔屬嘧啶、C5
-醯基嘧啶、C5
-羥基烷基嘌呤、C5
-醯胺基嘧啶、C5
-氰基嘧啶、C5
-5-碘嘧啶、C6
-碘-嘧啶、C5
-Br-乙烯基嘧啶、C6
-Br-乙烯基嘧啶、C5
-硝基嘧啶、C5
-胺基-嘧啶、N2
-烷基嘌呤類、N2
-烷基-6-硫嘌呤類、5-氮雜胞苷基、5-氮雜尿嘧啶基、三唑並吡啶基、咪唑並吡啶基、吡咯並嘧啶基,及吡唑並嘧啶基。嘌呤鹼類包括但不限於鳥嘌呤、腺嘌呤、次黃嘌呤、2,6-二胺基嘌呤,及6-氯嘌呤。嘌呤和嘧啶鹼係經由鹼之氮原子連結至核糖糖(ribose sugar)或其類似物。鹼上之功能性氧和氮基團如必要或需要時可經保護。適當保護基為熟習該項技術者熟知的,且包括三甲矽基、二甲基己矽基、第三-丁基二甲矽基,及第三-丁基二苯基矽基、三苯甲基、烷基,及醯基諸如乙醯基和丙醯基、甲烷磺醯基、及對甲苯磺醯基。
除非另有規定,否則式IV化合物之碳原子意欲具有四價。在一些其中碳原子沒有足夠數目之連結以產生四價的變數之化學結構表示中,提供四價所需之其餘碳取代基應應當假定為氫。例如, 具有與下式相同的意義
具有與下式相同的意義 。
。
“保護基”係指遮蔽或改變化合物之官能基的性質或化合物整體之性質的化合物之部分。保護基之化學子結構變化廣泛。保護基的一個功能係充當合成母藥物質中的中間物。化學保護基和用於保護/去保護之策略為該技術已知的。參見“Protective Groups in Organic Chemistry”,Theodora W. Greene (John Wiley & Sons, Inc., New York, 1991)。亦參見Protective Groups in Organic Chemistry, Peter G. M. Wuts和Theodora W. Greene,第四版,2006。保護基團通常用以遮蔽某些官能基團之反應性,以幫助所要化學反應之效率,例如以有次序及有計畫之方式製造及破壞化學鍵。化合物之官能基的保護除了改變經保護之官能基的反應性之外亦改變其他物理性質,諸如極性、親油性(疏水性)、及其他可藉由一般分析工具測量之性質。經化學保護之中間物本身可為生物活性或非活性。“羥基保護基”係指彼可用於保護羥基 (-OH)之保護基。
經保護之化合物在活體外及活體內亦可呈現出改變及(在某些情況下)最佳化的性質,諸如通過細胞膜與扺抗酵素降解或螯合性。在此作用中,具有所欲療效的經保護化合物可稱為前藥。保護基之另一功能為將母藥物轉化為前藥,藉此前藥在活體中轉化時釋出母藥物。因為活性前藥可比母藥被更有效地吸收,所以前藥在活體內可具有比母藥更大的效力。在活體外(在化學中間物的情況下),或活體內(在前藥的情況下)移除保護基。關於化學中間物,去保護後所得到的產物(例如,醇類)是否為生理上可接受的,並不特別重要,雖然一般而言,若該產物係藥理上無害的,則更理想。
術語“手性”係指具有鏡像夥伴的不重疊性之性質的分子,而“非手性”係指可與其鏡像夥伴重疊的分子。
術語“立體異構物”係指具相同化學組成但關於原子或基團在空間中的排列不同的化合物。
“非鏡像異構物”係指具二或多個手性中心且其分子彼此不為鏡像的立體異構物。非鏡像異構物具有不同物理性質,例如熔點、沸點、光譜性質、反應性和生物性質。例如,式IV化合物可具有手性磷原子,當R7
為, 且Z1
和Z2
為不同時。當Z1
或Z2
中之至少一者具有手性中心時,例如具有Z1
或Z2
為連結氮之手性天然α-胺基酸酯,則式IV化合物將因為分子中有兩個手性的中心而以非鏡像異構物存在。本發明涵蓋本文中所述之所有該等非鏡像異構物及它們的用途。非鏡像異構物之混合物可使用高解析分析程序諸如電泳、結晶及/或層析法分離。非鏡像異構物可具有不同的物理屬性,例如(但不限於)溶解度、化學穩定性和結晶性,也可具有不同的生物性質,諸如(但不限於)酵素穩定性、吸收及代謝穩定性。
且Z1
和Z2
為不同時。當Z1
或Z2
中之至少一者具有手性中心時,例如具有Z1
或Z2
為連結氮之手性天然α-胺基酸酯,則式IV化合物將因為分子中有兩個手性的中心而以非鏡像異構物存在。本發明涵蓋本文中所述之所有該等非鏡像異構物及它們的用途。非鏡像異構物之混合物可使用高解析分析程序諸如電泳、結晶及/或層析法分離。非鏡像異構物可具有不同的物理屬性,例如(但不限於)溶解度、化學穩定性和結晶性,也可具有不同的生物性質,諸如(但不限於)酵素穩定性、吸收及代謝穩定性。
“鏡像異構物”係指彼此為不重疊鏡像之化合物的二種立體異構體。
所使用之與數量有關的修飾詞“約”為包含所述之值且具有上下文所指明之意義(例如,包括與特定量之測量有關的誤差度)。
術語“治療(treating)”,如使用於本文中,除非另有指明,否則意指該術語所適用之病症或病況或此病症或病況的一或多個症狀的逆轉、緩和、抑制進展或預防。術語“治療(treatment)”,如使用於本文中,係指“治療 (treating)”的行為,如上述剛定義之治療(treating)。
術語“治療有效量”,如使用於本文中,為存在於本文中所述之組成物中的式IV化合物之量,該量為提供待治療之個體的呼吸道和肺之分泌物和組織內或血流中所要之藥物含量以當此組成物藉由所選取之投予途徑投予時,產生所預期之生理反應或所要生物作用所需之量。精確量將取決於數種因子,例如特定式IV化合物、組成物的特定活性、所使用之輸送裝置、組成物的物理特性、其所欲之用途、以及患者考量如疾病狀態的嚴重性、患者的合作性等等,且可由熟習該技術者根據本文中所提供之資訊而輕易地決定。
術語“生理鹽水”意指含有0.9%(w/v)NaCl之水溶液。
術語“高滲鹽水”意指含有大於0.9%(w/v)NaCl之水溶液。例如,3%高滲鹽水將含有3%(w/v)NaCl。
“形成反應混合物”係指使至少二種不同物質(species)接觸而使它們混合在一起且可反應的方法。然而,應當理解,所得反應產物可直接從所加試劑之間的反應或從可在反應混合物中產生的來自所加試劑之一或多者中間物製備。
“偶合劑”係指能夠偶合二種不同化合物的試劑。偶合劑可為催化或化學計量。例如,偶合劑可為鋰基偶合劑或鎂基偶合劑諸如格任亞(Grignard)試劑。示例性偶合劑包括(但不限於)n-BuLi、MgCl2
、iPrMgCl、tBuMgCl、PhMgCl或其組合。
“矽烷”係指具有式SiR4
之含矽基團,其中各R基團可為烷基、烯基、環烷基、苯基、或其他含矽基團。當矽烷係連結至另一化合物時,該矽烷稱為“矽基”且具有式-SiR3
。
“鹵-矽烷”係指具有至少一個鹵素基團連結至矽原子之矽烷。代表性鹵-矽烷類具有式鹵-SiR3
,其中各R基團可為烷基、烯基、環烷基、苯基、或其他含矽基團。特定鹵-矽烷類包括Cl-Si(CH3
)3
、和Cl-Si(CH3
)2
CH2
CH2
Si(CH3
)2
-Cl。
“非親核性鹼”係指電子予體、路易斯鹼,諸如氮鹼類包括三乙胺、二異丙基乙胺、N,N-二乙基苯胺、吡啶、2,6-二甲吡啶、2,4,6-三甲吡啶、4-二甲胺基吡啶,及啶。
“脫離基”係指在不均鍵裂解期間維持鍵結電子對之基團。例如,脫離基為在親核置換反應期間容易被置換。適當脫離基包括(但不限於)氯離子、溴離子、甲磺酸根離子、甲苯磺酸根離子、三氟甲磺酸根離子、4-硝基苯磺酸根離子、4-氯苯磺酸根離子、4-硝基苯氧基、五氟苯氧基、等等。熟習該項技術者將認知可使用於本發明中之其他脫離基。
“去保護劑”係指任何能夠移除保護基之試劑。去保護劑將取決於所使用之保護基的類型。代表性去保護劑在該項技術中為已知的且可發現於Protective Groups in Organic Chemistry, Peter G. M. Wuts and Theodora W. Greene,第四版,2006。
ii.本發明之化合物
現在將詳細地參考本發明之某些實施態樣,其實例係說明於所附的說明、結構和式中。雖然本發明將結合所列舉的實施態樣結合來描述,但應理解:它們並不意欲將本發明限制於它們的實施態樣。反之,本發明意欲涵蓋所有替代、修改和等價物,其可包括在本發明的範圍內。
所提供者為一種治療需要該治療之人類的絲狀病毒科感染之方法,其包含投予治療有效量的式I化合物: 或其醫藥上可接受的鹽、水合物或酯;
其中,
各R1
為H或鹵素;
各R2
、R3
、R4
或R5
獨立地為H、ORa
、N(Ra
)2
、N3
、CN、NO2
、S(O)n
Ra
、鹵素、(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基或(C2
-C8
)經取代之炔基;
其中
各Ra
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、芳基(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、-C(=O)R、-C(=O)OR、-C(=O)NR2
、-C(=O)SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、或-SO2
NR2
;
各R獨立地為H、(C1
-C8
)烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基、(C2
-C8
)經取代之炔基、C6
-C20
芳基、C6
-C20
經取代之芳基、C2
-C20
雜環基、C2
-C20
經取代之雜環基、芳基烷基或經取代之芳基烷基;
或相鄰碳原子上的任何二個R2
、R3
、R4
或R5
當一起為-O(CO)O-時或當與它們所連接之環碳原子一起形成雙鍵時;
R6
為ORa
、N(Ra
)2
、N3
、CN、NO2
、S(O)n
Ra
、-C(=O)R11
、-C(=O)OR11
、-C(=O)NR11
R12
、-C(=O)SR11
、-S(O)R11
、-S(O)2
R11
、-S(O)(OR11
)、-S(O)2
(OR11
)、-SO2
NR11
R12
、鹵素、(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基、(C2
-C8
)經取代之炔基、或芳基(C1
-C8
)烷基;
其中
各R11
或R12
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C4
-C8
)碳環基烷基、隨意地經取代之芳基、隨意地經取代之雜芳基、-C(=O)(C1
-C8
)烷基、-S(O)n
(C1
-C8
)烷基或芳基(C1
-C8
)烷基;或R11
和R12
與它們所共同連接之氮原子一起形成3至7員雜環,其中該雜環之任何一個碳原子可隨意地經-O-、-S-或-NRa
-置換;
各n獨立地為0、1、或2;
R7
係選自由下列組成之群組
a) H、-C(=O)R11
、-C(=O)OR11
、-C(=O)NR11
R12
、-C(=O)SR11
、-S(O)R11
、-S(O)2
R11
、-S(O)(OR11
)、-S(O)2
(OR11
)、或-SO2
NR11
R12
,
其中各R11
或R12
之各(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基或芳基(C1
-C8
)烷基係獨立地隨意地經一或多個鹵基、羥基、CN、N3
、N(Ra
)2
或ORa
取代;及其中各該(C1
-C8
)烷基的非末端碳原子中之一或多者可隨意地經-O-、-S-或-NRa
-置換,及
b)
或其醫藥上可接受的鹽、水合物或酯;
其中,
各R1
為H或鹵素;
各R2
、R3
、R4
或R5
獨立地為H、ORa
、N(Ra
)2
、N3
、CN、NO2
、S(O)n
Ra
、鹵素、(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基或(C2
-C8
)經取代之炔基;
其中
各Ra
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、芳基(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、-C(=O)R、-C(=O)OR、-C(=O)NR2
、-C(=O)SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、或-SO2
NR2
;
各R獨立地為H、(C1
-C8
)烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基、(C2
-C8
)經取代之炔基、C6
-C20
芳基、C6
-C20
經取代之芳基、C2
-C20
雜環基、C2
-C20
經取代之雜環基、芳基烷基或經取代之芳基烷基;
或相鄰碳原子上的任何二個R2
、R3
、R4
或R5
當一起為-O(CO)O-時或當與它們所連接之環碳原子一起形成雙鍵時;
R6
為ORa
、N(Ra
)2
、N3
、CN、NO2
、S(O)n
Ra
、-C(=O)R11
、-C(=O)OR11
、-C(=O)NR11
R12
、-C(=O)SR11
、-S(O)R11
、-S(O)2
R11
、-S(O)(OR11
)、-S(O)2
(OR11
)、-SO2
NR11
R12
、鹵素、(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基、(C2
-C8
)經取代之炔基、或芳基(C1
-C8
)烷基;
其中
各R11
或R12
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C4
-C8
)碳環基烷基、隨意地經取代之芳基、隨意地經取代之雜芳基、-C(=O)(C1
-C8
)烷基、-S(O)n
(C1
-C8
)烷基或芳基(C1
-C8
)烷基;或R11
和R12
與它們所共同連接之氮原子一起形成3至7員雜環,其中該雜環之任何一個碳原子可隨意地經-O-、-S-或-NRa
-置換;
各n獨立地為0、1、或2;
R7
係選自由下列組成之群組
a) H、-C(=O)R11
、-C(=O)OR11
、-C(=O)NR11
R12
、-C(=O)SR11
、-S(O)R11
、-S(O)2
R11
、-S(O)(OR11
)、-S(O)2
(OR11
)、或-SO2
NR11
R12
,
其中各R11
或R12
之各(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基或芳基(C1
-C8
)烷基係獨立地隨意地經一或多個鹵基、羥基、CN、N3
、N(Ra
)2
或ORa
取代;及其中各該(C1
-C8
)烷基的非末端碳原子中之一或多者可隨意地經-O-、-S-或-NRa
-置換,及
b) c) 選自下列之群組:
c) 選自下列之群組: 其中:
Rc
係選自苯基、1-萘基、2-萘基,
其中:
Rc
係選自苯基、1-萘基、2-萘基, Rd
為H或CH3
;
Re1
和Re2
各自獨立地為H、C1
-C6
烷基或苯甲基;
Rf
係選自H、C1
-C8
烷基、苯甲基、C3
-C6
環烷基、和-CH2
-C3
-C6
環烷基;
Rg
係選自C1
-C8
烷基、-O-C1
-C8
烷基、苯甲基、-O-苯甲基、-CH2
-C3
-C6
環烷基、-O-CH2
-C3
-C6
環烷基、和CF3
;及
n’係選自1、2、3、及4;及
d) 下式之基團:
Rd
為H或CH3
;
Re1
和Re2
各自獨立地為H、C1
-C6
烷基或苯甲基;
Rf
係選自H、C1
-C8
烷基、苯甲基、C3
-C6
環烷基、和-CH2
-C3
-C6
環烷基;
Rg
係選自C1
-C8
烷基、-O-C1
-C8
烷基、苯甲基、-O-苯甲基、-CH2
-C3
-C6
環烷基、-O-CH2
-C3
-C6
環烷基、和CF3
;及
n’係選自1、2、3、及4;及
d) 下式之基團: 其中
Q為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;
Z1
和Z2
,當結合在一起時,為-Q1
(C(Ry
)2
)3
Q1
-;
其中
各Q1
獨立地為O、S、或NR;及
各Ry
獨立地為H、F、Cl、Br、I、OH、R、-C(=Q2
)R、-C(=Q2
)OR、-C(=Q2
)N(R)2
、-N(R)2
、-+
N(R)3
、-SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、-OC(=Q1
)R、-OC(=Q2
)OR、-OC(=Q2
)(N(R)2
)、-SC(=Q2
)R、-SC(=Q2
)OR、-SC(=Q2
)(N(R)2
)、-N(R)C(=Q2
)R、-N(R)C(=Q2
)OR、-N(R)C(=Q2
)N(R)2
、-SO2
NR2
、-CN、-N3
、-NO2
、-OR、或Z3
;或當結合在一起時,在同一碳原子上之二個Ry
形成3-7個碳原子的碳環;
各Q2
獨立地為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;或
Z1
和Z2
係各自獨立地為式Ia之基團:
其中
Q為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;
Z1
和Z2
,當結合在一起時,為-Q1
(C(Ry
)2
)3
Q1
-;
其中
各Q1
獨立地為O、S、或NR;及
各Ry
獨立地為H、F、Cl、Br、I、OH、R、-C(=Q2
)R、-C(=Q2
)OR、-C(=Q2
)N(R)2
、-N(R)2
、-+
N(R)3
、-SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、-OC(=Q1
)R、-OC(=Q2
)OR、-OC(=Q2
)(N(R)2
)、-SC(=Q2
)R、-SC(=Q2
)OR、-SC(=Q2
)(N(R)2
)、-N(R)C(=Q2
)R、-N(R)C(=Q2
)OR、-N(R)C(=Q2
)N(R)2
、-SO2
NR2
、-CN、-N3
、-NO2
、-OR、或Z3
;或當結合在一起時,在同一碳原子上之二個Ry
形成3-7個碳原子的碳環;
各Q2
獨立地為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;或
Z1
和Z2
係各自獨立地為式Ia之基團: 其中:
各Q3
獨立地為一鍵、O、CR2
、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、N-NR2
、S、S-S、S(O)、或S(O)2
;
M2為0、1或2;
各Rx
獨立地為Ry
或式:
其中:
各Q3
獨立地為一鍵、O、CR2
、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、N-NR2
、S、S-S、S(O)、或S(O)2
;
M2為0、1或2;
各Rx
獨立地為Ry
或式: 其中:
各M1a、M1c、和M1d獨立地為0或1;
M12c為0、1、2、3、4、5、6、7、8、9、10、11或12;
Z3
為Z4
或Z5
;
Z4
為R、-C(Q2
)Ry
、-C(Q2
)Z5
、-SO2
Ry
、或-SO2
Z5
;及
Z5
為碳環或雜環,其中Z5
獨立地經0至3個Ry
基團取代;
各R8
為鹵素、NR11
R12
、N(R11
)OR11
、NR11
NR11
R12
、N3
、NO、NO2
、CHO、CN、-CH(=NR11
)、-CH=NNHR11
、-CH=N(OR11
)、-CH(OR11
)2
、-C(=O)NR11
R12
、-C(=S)NR11
R12
、-C(=O)OR11
、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C4
-C8
)碳環基烷基、隨意地經取代之芳基、隨意地經取代之雜芳基、-C(=O)(C1
-C8
)烷基、-S(O)n
(C1
-C8
)烷基、芳基(C1
-C8
)烷基、OR11
或SR11
;
各R9
或R10
獨立地為H、鹵素、NR11
R12
、N(R11
)OR11
、NR11
NR11
R12
、N3
、NO、NO2
、CHO、CN、-CH(=NR11
)、-CH=NHNR11
、-CH=N(OR11
)、-CH(OR11
)2
、-C(=O)NR11
R12
、-C(=S)NR11
R12
、-C(=O)OR11
、R11
、OR11
或SR11
;及
其中各R2
、R3
、R5
、或R6
之各(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基或芳基(C1
-C8
)烷基係獨立地隨意地經一或多個鹵基、羥基、CN、N3
、N(Ra
)2
或ORa
取代;及 其中各該(C1
-C8
)烷基的非末端碳原子之一或多者可隨意地經-O-、-S-或-NRa
-置換。
其中:
各M1a、M1c、和M1d獨立地為0或1;
M12c為0、1、2、3、4、5、6、7、8、9、10、11或12;
Z3
為Z4
或Z5
;
Z4
為R、-C(Q2
)Ry
、-C(Q2
)Z5
、-SO2
Ry
、或-SO2
Z5
;及
Z5
為碳環或雜環,其中Z5
獨立地經0至3個Ry
基團取代;
各R8
為鹵素、NR11
R12
、N(R11
)OR11
、NR11
NR11
R12
、N3
、NO、NO2
、CHO、CN、-CH(=NR11
)、-CH=NNHR11
、-CH=N(OR11
)、-CH(OR11
)2
、-C(=O)NR11
R12
、-C(=S)NR11
R12
、-C(=O)OR11
、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C4
-C8
)碳環基烷基、隨意地經取代之芳基、隨意地經取代之雜芳基、-C(=O)(C1
-C8
)烷基、-S(O)n
(C1
-C8
)烷基、芳基(C1
-C8
)烷基、OR11
或SR11
;
各R9
或R10
獨立地為H、鹵素、NR11
R12
、N(R11
)OR11
、NR11
NR11
R12
、N3
、NO、NO2
、CHO、CN、-CH(=NR11
)、-CH=NHNR11
、-CH=N(OR11
)、-CH(OR11
)2
、-C(=O)NR11
R12
、-C(=S)NR11
R12
、-C(=O)OR11
、R11
、OR11
或SR11
;及
其中各R2
、R3
、R5
、或R6
之各(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基或芳基(C1
-C8
)烷基係獨立地隨意地經一或多個鹵基、羥基、CN、N3
、N(Ra
)2
或ORa
取代;及 其中各該(C1
-C8
)烷基的非末端碳原子之一或多者可隨意地經-O-、-S-或-NRa
-置換。
在另一實施態樣中,所提供者為一種式IV之化合物: 或其醫藥上可接受的鹽、水合物或酯;
其中R7
係如上述就式I所定義者。
或其醫藥上可接受的鹽、水合物或酯;
其中R7
係如上述就式I所定義者。
所提供者為一種治療需要該治療之人類的絲狀病毒科感染之方法,其包含投予治療有效量的式IV化合物: 或其醫藥上可接受的鹽或酯;
其中:
R7
係選自下列之群組:
a) H、-C(=O)R11
、-C(=O)OR11
、-C(=O)NR11
R12
、-C(=O)SR11
、-S(O)R11
、-S(O)2
R11
、-S(O)(OR11
)、-S(O)2
(OR11
)、或-SO2
NR11
R12
,
其中
各R11
或R12
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C4
-C8
)碳環基烷基、隨意地經取代之芳基、隨意地經取代之雜芳基、-C(=O)(C1
-C8
)烷基、-S(O)n
(C1
-C8
)烷基或芳基(C1
-C8
)烷基;或R11
和R12
與它們所共同連接之氮原子一起形成3至7員雜環,其中該雜環之任何一個碳原子可隨意地經-O-、-S-或-NRa
-置換;
各Ra
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、芳基(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、-C(=O)R、-C(=O)OR、-C(=O)NR2
、-C(=O)SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、或-SO2
NR2
;
其中各R獨立地為H、(C1
-C8
)烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基、(C2
-C8
)經取代之炔基、C6
-C20
芳基、C6
-C20
經取代之芳基、C2
-C20
雜環基、C2
-C20
經取代之雜環基、芳基烷基或經取代之芳基烷基;及
其中各R11
或R12
之各(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基或芳基(C1
-C8
)烷基係獨立地隨意地經一或多個鹵基、羥基、CN、N3
、N(Ra
)2
或ORa
取代;及其中各該(C1
-C8
)烷基的非末端碳原子中之一或多者可隨意地經-O-、-S-或-NRa
-置換,及
b)
或其醫藥上可接受的鹽或酯;
其中:
R7
係選自下列之群組:
a) H、-C(=O)R11
、-C(=O)OR11
、-C(=O)NR11
R12
、-C(=O)SR11
、-S(O)R11
、-S(O)2
R11
、-S(O)(OR11
)、-S(O)2
(OR11
)、或-SO2
NR11
R12
,
其中
各R11
或R12
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C4
-C8
)碳環基烷基、隨意地經取代之芳基、隨意地經取代之雜芳基、-C(=O)(C1
-C8
)烷基、-S(O)n
(C1
-C8
)烷基或芳基(C1
-C8
)烷基;或R11
和R12
與它們所共同連接之氮原子一起形成3至7員雜環,其中該雜環之任何一個碳原子可隨意地經-O-、-S-或-NRa
-置換;
各Ra
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、芳基(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、-C(=O)R、-C(=O)OR、-C(=O)NR2
、-C(=O)SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、或-SO2
NR2
;
其中各R獨立地為H、(C1
-C8
)烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基、(C2
-C8
)經取代之炔基、C6
-C20
芳基、C6
-C20
經取代之芳基、C2
-C20
雜環基、C2
-C20
經取代之雜環基、芳基烷基或經取代之芳基烷基;及
其中各R11
或R12
之各(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基或芳基(C1
-C8
)烷基係獨立地隨意地經一或多個鹵基、羥基、CN、N3
、N(Ra
)2
或ORa
取代;及其中各該(C1
-C8
)烷基的非末端碳原子中之一或多者可隨意地經-O-、-S-或-NRa
-置換,及
b) c) 選自下列之群組:
c) 選自下列之群組: 其中:
Rc
係選自苯基、1-萘基、2-萘基,
其中:
Rc
係選自苯基、1-萘基、2-萘基, Rd
為H或CH3
;
Re
為H或C1
-C6
烷基;
Rf
係選自H、C1
-C8
烷基、苯甲基、C3
-C6
環烷基、和-CH2
-C3
-C6
環烷基;
Rg
係選自C1
-C8
烷基、-O-C1
-C8
烷基、苯甲基、-O-苯甲基、-CH2
-C3
-C6
環烷基、-O-CH2
-C3
-C6
環烷基、和CF3
;及
n’係選自1、2、3、及4;及
d) 下式之基團:
Rd
為H或CH3
;
Re
為H或C1
-C6
烷基;
Rf
係選自H、C1
-C8
烷基、苯甲基、C3
-C6
環烷基、和-CH2
-C3
-C6
環烷基;
Rg
係選自C1
-C8
烷基、-O-C1
-C8
烷基、苯甲基、-O-苯甲基、-CH2
-C3
-C6
環烷基、-O-CH2
-C3
-C6
環烷基、和CF3
;及
n’係選自1、2、3、及4;及
d) 下式之基團: 其中
Q為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;
Z1
和Z2
,當結合在一起時,為-Q1
(C(Ry
)2
)3
Q1
-;
其中
各Q1
獨立地為O、S、或NR;及
各Ry
獨立地為H、F、Cl、Br、I、OH、R、-C(=Q2
)R、-C(=Q2
)OR、-C(=Q2
)N(R)2
、-N(R)2
、-+
N(R)3
、-SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、-OC(=Q1
)R、-OC(=Q2
)OR、-OC(=Q2
)(N(R)2
)、-SC(=Q2
)R、-SC(=Q2
)OR、-SC(=Q2
)(N(R)2
)、-N(R)C(=Q2
)R、-N(R)C(=Q2
)OR、-N(R)C(=Q2
)N(R)2
、 -SO2
NR2
、-CN、-N3
、-NO2
、-OR、或Z3
;或當結合在一起時,在同一碳原子上之二個Ry
形成3-7個碳原子的碳環;
各Q2
獨立地為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;或
Z1
和Z2
係各自獨立地為式Ia之基團:
其中
Q為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;
Z1
和Z2
,當結合在一起時,為-Q1
(C(Ry
)2
)3
Q1
-;
其中
各Q1
獨立地為O、S、或NR;及
各Ry
獨立地為H、F、Cl、Br、I、OH、R、-C(=Q2
)R、-C(=Q2
)OR、-C(=Q2
)N(R)2
、-N(R)2
、-+
N(R)3
、-SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、-OC(=Q1
)R、-OC(=Q2
)OR、-OC(=Q2
)(N(R)2
)、-SC(=Q2
)R、-SC(=Q2
)OR、-SC(=Q2
)(N(R)2
)、-N(R)C(=Q2
)R、-N(R)C(=Q2
)OR、-N(R)C(=Q2
)N(R)2
、 -SO2
NR2
、-CN、-N3
、-NO2
、-OR、或Z3
;或當結合在一起時,在同一碳原子上之二個Ry
形成3-7個碳原子的碳環;
各Q2
獨立地為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;或
Z1
和Z2
係各自獨立地為式Ia之基團: 其中:
各Q3
獨立地為一鍵、O、CR2
、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、N-NR2
、S、S-S、S(O)、或S(O)2
;
M2為0、1或2;
各Rx
獨立地為Ry
或式:
其中:
各Q3
獨立地為一鍵、O、CR2
、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、N-NR2
、S、S-S、S(O)、或S(O)2
;
M2為0、1或2;
各Rx
獨立地為Ry
或式: 其中:
各M1a、M1c、和M1d獨立地為0或1;
M12c為0、1、2、3、4、5、6、7、8、9、10、11或12;
Z3
為Z4
或Z5
;
Z4
為R、-C(Q2
)Ry
、-C(Q2
)Z5
、-SO2
Ry
、或-SO2
Z5
;及
Z5
為碳環或雜環,其中Z5
獨立地經0至3個Ry
基團取代。
其中:
各M1a、M1c、和M1d獨立地為0或1;
M12c為0、1、2、3、4、5、6、7、8、9、10、11或12;
Z3
為Z4
或Z5
;
Z4
為R、-C(Q2
)Ry
、-C(Q2
)Z5
、-SO2
Ry
、或-SO2
Z5
;及
Z5
為碳環或雜環,其中Z5
獨立地經0至3個Ry
基團取代。
所提供者為一種治療需要該治療之人類的絲狀病毒科感染之方法,其包含投予治療有效量的式IV化合物: 或其醫藥上可接受的鹽、水合物或酯;
其中,
R7
係選自由下列組成之群組:
a) H、-C(=O)R11
、-C(=O)OR11
、-C(=O)NR11
R12
、-C(=O)SR11
、-S(O)R11
、-S(O)2
R11
、-S(O)(OR11
)、-S(O)2
(OR11
)、或-SO2
NR11
R12
;
b)
或其醫藥上可接受的鹽、水合物或酯;
其中,
R7
係選自由下列組成之群組:
a) H、-C(=O)R11
、-C(=O)OR11
、-C(=O)NR11
R12
、-C(=O)SR11
、-S(O)R11
、-S(O)2
R11
、-S(O)(OR11
)、-S(O)2
(OR11
)、或-SO2
NR11
R12
;
b) c) 選自下列之群組:
c) 選自下列之群組: 其中:
Rc
係選自下列之群組:苯基、1-萘基、2-萘基,
其中:
Rc
係選自下列之群組:苯基、1-萘基、2-萘基, Rd
係選自下列之群組:H或CH3
;
Re1
和Re2
係各自獨立地選自下列之群組:H、(C1
-C6
)烷基或苯甲基;
Rf
係選自下列之群組:H、(C1
-C8
)烷基、苯甲基、(C3
-C6
)環烷基、和-CH2
-(C3
-C6
)環烷基;
Rg
係選自下列之群組:(C1
-C8
)烷基、-O-(C1
-C8
)烷基、苯甲基、-O-苯甲基、-CH2
-(C3
-C6
)環烷基、-O-CH2
-(C3
-C6
)環烷基、和CF3
;及
n’為選自下列群組之整數:1、2、3、及4;及
d) 下式之基團:
Rd
係選自下列之群組:H或CH3
;
Re1
和Re2
係各自獨立地選自下列之群組:H、(C1
-C6
)烷基或苯甲基;
Rf
係選自下列之群組:H、(C1
-C8
)烷基、苯甲基、(C3
-C6
)環烷基、和-CH2
-(C3
-C6
)環烷基;
Rg
係選自下列之群組:(C1
-C8
)烷基、-O-(C1
-C8
)烷基、苯甲基、-O-苯甲基、-CH2
-(C3
-C6
)環烷基、-O-CH2
-(C3
-C6
)環烷基、和CF3
;及
n’為選自下列群組之整數:1、2、3、及4;及
d) 下式之基團: 其中:
Q係選自下列之群組:O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;
Z1
和Z2
,當結合在一起時,為-Q1
(C(Ry
)2
)3
Q1
-;
其中
各Q1
係獨立地選自下列之群組:O、S、或NR;及
各Ry
係獨立地選自下列之群組:H、F、Cl、Br、I、
OH、R、-C(=Q2
)R、-C(=Q2
)OR、-C(=Q2
)N(R)2
、-N(R)2
、-+
N(R)3
、-SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、-OC(=Q1
)R、-OC(=Q2
)OR、-OC(=Q2
)(N(R)2
)、-SC(=Q2
)R、-SC(=Q2
)OR、-SC(=Q2
)(N(R)2
)、-N(R)C(=Q2
)R、-N(R)C(=Q2
)OR、-N(R)C(=Q2
)N(R)2
、-SO2
NR2
、-CN、-N3
、-NO2
、-OR、或Z3
;或當結合在一起時,在同一碳原子上之二個Ry
形成3-7個碳原子的碳環;
各Q2
獨立地為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;或
Z1
和Z2
係各自獨立地為式Ia之基團:
其中:
Q係選自下列之群組:O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;
Z1
和Z2
,當結合在一起時,為-Q1
(C(Ry
)2
)3
Q1
-;
其中
各Q1
係獨立地選自下列之群組:O、S、或NR;及
各Ry
係獨立地選自下列之群組:H、F、Cl、Br、I、
OH、R、-C(=Q2
)R、-C(=Q2
)OR、-C(=Q2
)N(R)2
、-N(R)2
、-+
N(R)3
、-SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、-OC(=Q1
)R、-OC(=Q2
)OR、-OC(=Q2
)(N(R)2
)、-SC(=Q2
)R、-SC(=Q2
)OR、-SC(=Q2
)(N(R)2
)、-N(R)C(=Q2
)R、-N(R)C(=Q2
)OR、-N(R)C(=Q2
)N(R)2
、-SO2
NR2
、-CN、-N3
、-NO2
、-OR、或Z3
;或當結合在一起時,在同一碳原子上之二個Ry
形成3-7個碳原子的碳環;
各Q2
獨立地為O、S、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、或N-NR2
;或
Z1
和Z2
係各自獨立地為式Ia之基團: 其中:
各Q3
係獨立地選自下列之群組:一鍵、O、CR2
、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、N-NR2
、S、S-S、S(O)、或S(O)2
;
M2為選自下列群組之整數:0、1或2;
各Rx
獨立地為Ry
或式:
其中:
各Q3
係獨立地選自下列之群組:一鍵、O、CR2
、NR、+
N(O)(R)、N(OR)、+
N(O)(OR)、N-NR2
、S、S-S、S(O)、或S(O)2
;
M2為選自下列群組之整數:0、1或2;
各Rx
獨立地為Ry
或式: 其中:
各M1a、M1c、和M1d為獨立地選自下列群組之整數:0或1;
M12c為選自下列群組之整數:0、1、2、3、4、5、6、7、8、9、10、11或12;
Z3
為Z4
或Z5
;
Z4
為R、-C(Q2
)Ry
、-C(Q2
)Z5
、-SO2
Ry
、或-SO2
Z5
;及
Z5
為碳環或雜環,其中Z5
獨立地經0至3個Ry
基團取代;
各R11
或R12
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C4
-C8
)碳環基烷基、(C6
-C20
)隨意地經取代之芳基、隨意地經取代之雜芳基、-C(=O)(C1
-C8
)烷基、-S(O)n
(C1
-C8
)烷基或(C6
-C20
)芳基(C1
-C8
)烷基;或R11
和R12
與它們所共同連接之氮原子一起形成3至7員雜環,其中該雜環之任何一個碳原子可隨意地經-O-、-S-或 -NRa
-置換;
各Ra
係獨立地選自下列之群組:H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C6
-C20
)芳基(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、-C(=O)R、-C(=O)OR、-C(=O)NR2
、 -C(=O)SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、或-SO2
NR2
;其中
各R係獨立地選自下列之群組:H、(C1
-C8
)烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基、(C2
-C8
)經取代之炔基、(C6
-C20
)芳基、(C6
-C20
)經取代之芳基、(C2
-C20
)雜環基、(C2
-C20
)經取代之雜環基、(C6
-C20
)芳基(C1
-C8
)烷基或經取代之(C6
-C20
)芳基(C1
-C8
)烷基;
各n為獨立地選自下列群組之整數:0、1、或2;及
其中各R11
或R12
之各(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基或(C6
-C20
)芳基(C1
-C8
)烷基係獨立地為隨意地經選自下列群組的取代基中之一或多者取代:鹵基、羥基、CN、N3
、N(Ra
)2
或ORa
;及其中各該(C1
-C8
)烷基的非末端碳原子之一或多者可隨意地經-O-、-S-或-NRa
-置換。
其中:
各M1a、M1c、和M1d為獨立地選自下列群組之整數:0或1;
M12c為選自下列群組之整數:0、1、2、3、4、5、6、7、8、9、10、11或12;
Z3
為Z4
或Z5
;
Z4
為R、-C(Q2
)Ry
、-C(Q2
)Z5
、-SO2
Ry
、或-SO2
Z5
;及
Z5
為碳環或雜環,其中Z5
獨立地經0至3個Ry
基團取代;
各R11
或R12
獨立地為H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C4
-C8
)碳環基烷基、(C6
-C20
)隨意地經取代之芳基、隨意地經取代之雜芳基、-C(=O)(C1
-C8
)烷基、-S(O)n
(C1
-C8
)烷基或(C6
-C20
)芳基(C1
-C8
)烷基;或R11
和R12
與它們所共同連接之氮原子一起形成3至7員雜環,其中該雜環之任何一個碳原子可隨意地經-O-、-S-或 -NRa
-置換;
各Ra
係獨立地選自下列之群組:H、(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基、(C6
-C20
)芳基(C1
-C8
)烷基、(C4
-C8
)碳環基烷基、-C(=O)R、-C(=O)OR、-C(=O)NR2
、 -C(=O)SR、-S(O)R、-S(O)2
R、-S(O)(OR)、-S(O)2
(OR)、或-SO2
NR2
;其中
各R係獨立地選自下列之群組:H、(C1
-C8
)烷基、(C1
-C8
)經取代之烷基、(C2
-C8
)烯基、(C2
-C8
)經取代之烯基、(C2
-C8
)炔基、(C2
-C8
)經取代之炔基、(C6
-C20
)芳基、(C6
-C20
)經取代之芳基、(C2
-C20
)雜環基、(C2
-C20
)經取代之雜環基、(C6
-C20
)芳基(C1
-C8
)烷基或經取代之(C6
-C20
)芳基(C1
-C8
)烷基;
各n為獨立地選自下列群組之整數:0、1、或2;及
其中各R11
或R12
之各(C1
-C8
)烷基、(C2
-C8
)烯基、(C2
-C8
)炔基或(C6
-C20
)芳基(C1
-C8
)烷基係獨立地為隨意地經選自下列群組的取代基中之一或多者取代:鹵基、羥基、CN、N3
、N(Ra
)2
或ORa
;及其中各該(C1
-C8
)烷基的非末端碳原子之一或多者可隨意地經-O-、-S-或-NRa
-置換。
在式IV化合物之另一實施態樣中,R7
可為H。在式IV化合物之另一實施態樣中,R7
係選自下列之群組:如就式IV所定義之a)、b)、或c)。
在一些實施態樣中,Re1
和Re2
可各自獨立地為選自下列之群組:H、C1
-C6
烷基或苯甲基。在一些實施態樣中,Re1
可為H、C1
-C6
烷基或苯甲基,及Re2
可為H或C1
-C6
烷基。在一些實施態樣中,Re1
和Re2
可各自獨立地為H或C1
-C6
烷基。在一些實施態樣中,Re1
和Re2
可各自獨立地為H或苯甲基。在一些實施態樣中,Re1
可為H、甲基或苯甲基,及Re2
可為H或甲基。在一些實施態樣中,Re1
可為H或甲基,及Re2
可為H或甲基。在一些實施態樣中,Re1
可為甲基,及Re2
可為H或甲基。在一些實施態樣中,Re1
可為H或苯甲基,及Re2
可為H或甲基。
在式IV化合物之另一實施態樣中,R7
為 其中Rf
係選自下列之群組:H、C1
-C8
烷基、苯甲基、C3
-C6
環烷基、和-CH2
-C3
-C6
環烷基。在式IV化合物之另一實施態樣中,Rf
為C1
-C8
烷基。
其中Rf
係選自下列之群組:H、C1
-C8
烷基、苯甲基、C3
-C6
環烷基、和-CH2
-C3
-C6
環烷基。在式IV化合物之另一實施態樣中,Rf
為C1
-C8
烷基。
在式IV化合物之另一實施態樣中,R7
為 其中
Rf
係選自下列之群組:H、C1
-C8
烷基、苯甲基、C3
-C6
環烷基、和-CH2
-C3
-C6
環烷基;及
Rg
係選自下列之群組:C1
-C8
烷基、-O-C1
-C8
烷基、苯甲基、-O-苯甲基、-CH2
-C3
-C6
環烷基、-O-CH2
-C3
-C6
環烷基、和CF3
。
其中
Rf
係選自下列之群組:H、C1
-C8
烷基、苯甲基、C3
-C6
環烷基、和-CH2
-C3
-C6
環烷基;及
Rg
係選自下列之群組:C1
-C8
烷基、-O-C1
-C8
烷基、苯甲基、-O-苯甲基、-CH2
-C3
-C6
環烷基、-O-CH2
-C3
-C6
環烷基、和CF3
。
在式IV化合物之另一實施態樣中,R7
為 其中Rf
係選自下列之群組:H、C1
-C8
烷基、苯甲基、C3
-C6
環烷基、和-CH2
-C3
-C6
環烷基。在式IV化合物之另一實施態樣中,Rf
為C1
-C8
烷基。在式IV化合物之另一實施態樣中,Rf
為C1
-C6
烷基。
其中Rf
係選自下列之群組:H、C1
-C8
烷基、苯甲基、C3
-C6
環烷基、和-CH2
-C3
-C6
環烷基。在式IV化合物之另一實施態樣中,Rf
為C1
-C8
烷基。在式IV化合物之另一實施態樣中,Rf
為C1
-C6
烷基。
在式IV化合物之另一實施態樣中,R7
為: 其中Rg
係選自下列之群組:C1
-C8
烷基、-O-C1
-C8
烷基、苯甲基、-O-苯甲基、-CH2
-C3
-C6
環烷基、-O-CH2
-C3
-C6
環烷基、和CF3
。在式IV化合物之另一實施態樣中,Rf
為C1
-C8
烷基。在式IV化合物之另一實施態樣中,Rf
為C1
-C6
烷基。
其中Rg
係選自下列之群組:C1
-C8
烷基、-O-C1
-C8
烷基、苯甲基、-O-苯甲基、-CH2
-C3
-C6
環烷基、-O-CH2
-C3
-C6
環烷基、和CF3
。在式IV化合物之另一實施態樣中,Rf
為C1
-C8
烷基。在式IV化合物之另一實施態樣中,Rf
為C1
-C6
烷基。
在式IV化合物之另一實施態樣中,R7
係選自下列之群組: 。
。
在式IV化合物之另一實施態樣中,R7
為
在另一實施態樣中,所提供者為一種式IV化合物,其為: 或其醫藥上可接受的鹽或酯。
或其醫藥上可接受的鹽或酯。
在另一實施態樣中,所提供者為一種式IV化合物,其為:
 或其醫藥上可接受的鹽或酯。
或其醫藥上可接受的鹽或酯。
在另一實施態樣中,所提供者為一種式IV化合物,其為:
 或其醫藥上可接受的鹽、水合物、或其酯。
或其醫藥上可接受的鹽、水合物、或其酯。
在另一實施態樣中,所提供者為一種式IV化合物,其為

 或其醫藥上可接受的鹽或酯。
或其醫藥上可接受的鹽或酯。
在另一實施態樣中,本發明提供一種化合物,其為
 或其醫藥上可接受的鹽或酯。
或其醫藥上可接受的鹽或酯。
在另一實施態樣中,本發明提供一種化合物,其為

 或其醫藥上可接受的鹽、水合物、或其酯。
或其醫藥上可接受的鹽、水合物、或其酯。
在另一實施態樣中,本發明提供一種化合物,其為 或其醫藥上可接受的鹽或酯。
或其醫藥上可接受的鹽或酯。
在另一實施態樣中,本發明提供一種化合物,其為 或其醫藥上可接受的鹽或酯。
或其醫藥上可接受的鹽或酯。
在另一實施態樣中,本發明提供一種化合物,其為 或其醫藥上可接受的鹽或酯。
或其醫藥上可接受的鹽或酯。
本揭示之化合物的名稱係使用用於命名化合物之ACD/命名軟體(Advanced Chemistry Development, Inc., Toronto, Canada)提供。其他化合物或基團可使用一般名稱或系統或非系統名稱命名。以式IV之代表性化合物說明本揭示之化合物的命名及編號。 其命名為(2S)-2-((((2R,3S,4R,5R)-5-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基胺基)丙酸2-乙基丁酯。本發明的其他化合物包括:
其命名為(2S)-2-((((2R,3S,4R,5R)-5-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基胺基)丙酸2-乙基丁酯。本發明的其他化合物包括: 其命名為(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯,及
其命名為(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯,及 其命名為(S)-2-(((R)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯。
(S)-2-(((R)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯亦可顯示為
其命名為(S)-2-(((R)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯。
(S)-2-(((R)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯亦可顯示為 。
。
任何提及之本文所述之本發明化合物亦包括提及之其生理上可接受的鹽。本發明化合物之生理上可接受的鹽類之實例包括衍生自適當鹼(諸如鹼金屬或鹼土金屬(例如,Na+、Li+、K+、Ca+2
和Mg+2
)、銨和NR4 +
(其中R係定義於本文中))的鹽類。氮原子或胺基之生理上可接受的鹽類包括(a)與無機酸(例如,鹽酸、氫溴酸、硫酸、胺磺酸、磷酸、硝酸、等等)形成之酸加成鹽類;(b)與有機酸(諸如乙酸、草酸、酒石酸、琥珀酸、馬來酸、富馬酸、葡萄糖酸、檸檬酸、蘋果酸、抗壞血酸、苯甲酸、羥乙磺酸、乳糖酸、鞣酸、棕櫚酸、海藻酸、聚麩胺酸、萘磺酸、甲磺酸、對甲苯磺酸、苯磺酸、萘二磺酸、聚半乳糖醛酸、丙二酸、磺酸基水楊酸、羥乙酸、2-羥基-3-萘甲酸酯、雙羥萘酸酯、水楊酸、硬脂酸、酞酸、扁桃酸、乳酸、乙磺酸、離胺酸、精胺酸、麩胺酸、甘胺酸、絲胺酸、蘇胺酸、丙胺酸、異白胺酸、白胺酸等)形成之鹽類;及(c)由元素陰離子(例如氯、溴及碘)形成之鹽類。羥基化合物之生理上可接受的鹽類包括該化合物之陰離子與適當陽離子諸如Na+
和NR4 +
之組合。
式IV化合物和其醫藥上可接受的鹽類可以不同之多晶形物或假多晶形物存在。如使用於本文中,結晶多形現象意指結晶以不同結晶結構存在之能力。結晶多形現象可起因於晶體堆積之差異(堆積多形現象)或相同分子的不同構形異構物之間的堆積之差異(構形多形現象)。如使用於本文中,結晶假多形現象意指化合物之水合物或溶劑合物以不同結晶結構存在之能力。本發明之假多晶形物可由於晶體堆積之差異(堆積假多形現象)或由於相同分子的不同構形異構物之間的堆積之差異(構形假多形現象)存在。本發明包含式IV化合物及其藥學上可接受的鹽類之所有多晶形物和假多晶形物。
式IV化合物及其醫藥上可接受的鹽類亦可能以非晶形固體存在。如使用於本文中,非晶形固體為其中在固體內沒有原子的遠程有序的位置之固體。當晶體大小為二奈米或更小時,此定義亦適用。添加劑(包括溶劑)可用於產生本發明之非晶形形式。本發明包含式IV化合物及其醫藥上可接受的鹽類之所有非晶形形式。
關於治療用途,本發明化合物之活性成分的鹽類將為生理上可接受的,即,其可為衍生自生理上可接受的酸或鹼之鹽類。然而,非生理上可接受的酸或鹼的鹽類亦可發現(例如)在製備或純化生理上可接受的化合物上之用途。所有鹽類(不論是否衍生自生理上可接受的酸或鹼)皆在本發明範圍內。
最後,應理解的是:組成物在此包括於它們的非離子化以及兩性離子形式之本發明化合物、及呈水合物之與化學計量水的組合。
應指出的是:本發明包括在式IV範圍內的化合物及其醫藥上可接受的鹽類之所有鏡像異構物、非鏡像異構物、和外消旋混合物、互變異構物、多晶形物、假多晶形物。鏡像異構物和非鏡像異構物之所有混合物都在本發明的範圍內。
以式IV例示的本發明化合物可具有手性中心,例如手性碳或磷原子。本發明化合物因此包括所有的立體異構物(包括鏡像異構物、非鏡像異構物、和阻轉異構物(atropisomer))的外消旋混合物。此外,本發明化合物包括在任一或所有的不對稱手性原子經富集或解析的旋光異構物。易言之,從說明中顯而易見之手性中心係提供為手性異構物或外消旋混合物。外消旋和非鏡像異構物混合物二者、以及經分離或合成的實質上不含它們的鏡像異構物或非鏡像異構物夥伴之個別旋光異構物皆在本發明的範圍內。透過所熟知的技術,例如與光學添加劑(例如酸類或鹼類)形成的非鏡像異構物鹽類之分離,接著轉化回至該光學活性物質,而將該外消旋混合物分離成其個別的實質上光學純異構物。在大多數情形下,藉由立體特異性反應,而以所要起始材料的適當立體異構物開始來合成所要的光學異構物。
本文中所用之立體化學定義及常規通常遵循S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York;及 Eliel, E.和Wilen, S., Stereochemistry of Organic 化合物(1994) John Wiley & Sons, Inc., New York。許多有機化合物以光學活性形式存在,亦即,它們具有將平面偏極光的平面旋轉之能力。在描述光學活性化合物時,字首D及L或R及S係用以表示分子關於其手性中心之絕對構形。字首d及1、D及L、或(+)及(-)用以標示平面偏極光被化合物旋轉之符號,且S、(-)、或1意指化合物為左旋,而以R、(+)、或d為字首之化合物則為右旋。就給定的化學結構而言,這些立體異構體為相同的,除了彼此為鏡像之外。特定立體異構物亦稱為鏡像異構物,及該等異構體之混合物通常稱為鏡像異構物混合物。鏡像異構物混合物之50:50混合物稱為外消旋混合物或外消旋物,其可發生在化學反應或方法中沒有立體選擇或立體特異性之時。術語“外消旋混合物”及“外消旋物”意指無光學活性的二種鏡像異構物物質之等莫耳混合物。
在某些情況下,本發明之化合物亦可以互變異構物的形式存在。雖然,僅能繪出一個非定域共振結構,但所有該等形式皆預期在本發明之範圍內。例如,烯-胺互變異構物可存在於嘌呤、嘧啶、咪唑、胍、脒、以及四唑系統中且它們所有可能的互變異構形式皆在本發明的範圍內。
本文中給出之任何式或結構,包括式IV化合物,亦意欲表示化合物之未經標記的形式以及經同位素標記的形式。經同位素標記的化合物具有以本文中給出之式描述的結構,除了一或多個原子經具有所選原子質量或質量數之原子置換之外。可併入所揭示之化合物中的同位素之實例包括氫、碳、氮、氧、磷、氟及氯,諸如(但不限於)2
H (氘,D)、3
H(氚)、11
C、13
C、14
C、15
N、18
F、31
P、32
P、35
S、36
Cl和125
I。所揭示之各種經同位素標記的化合物例如為其中併有諸如3
H、13
C和14
C的放射性同位素之化合物。該等經同位素標記的化合物可用於代謝研究、反應動力學研究、偵測或成像技術(諸如正電子發射斷層攝影術(PET)或單光子發射電腦斷層攝影術(SPECT),包括藥物或受質組織分佈分析)中或用於患者之放射性治療中。
本揭示亦包括式IV化合物,其中連接於碳原子之1至n個氫經氘置換,其中n為分子中之氫的數目。該等化合物呈現抗代謝性增加且因此可用於增加任何式IV化合物當投予至哺乳動物時之半衰期。參見,例如Foster,“Deuterium Isotope Effects in Studies of Drug Metabolism”, Trends Pharmacol. Sci.5(12):524-527(1984)。該等化合物係藉由該項技術中所熟知的方法合成,例如藉由採用其中一或多個氫已被氘置換之起始材料合成。
本揭示之經氘標記或取代之治療化合物可具有關於分佈、代謝及排泄(ADME)之改良DMPK(藥物代謝及藥物動力學)性質。以較重之同位素(諸如氘)取代可提供由較大代謝穩定性所產生的某些治療優勢,例如活體內半衰期增加、劑量需求減少及/或治療指數改善。經18
F標記之化合物可使用於PET或SPECT研究。本揭示之經同位素標記的化合物及其前藥通常可藉由進行下述流程中或實例及製備中所揭示之步驟,藉由用可易於獲得之經同位素標記的試劑替代非經同位素標記的試劑而製得。據了解:在此情況下,氘被視為式(IV)化合物中之取代基。
該類較重同位素(特別是氘)之濃度可由同位素增濃因子定義。在本揭示之化合物中,未明確指定為特定同位素之任何原子皆意欲表示彼原子之任何穩定同位素。除非另有說明,否則當某一位置明確指定為 “H”或“氫”時,該位置應理解為具有在天然豐度同位素組成下之氫。因此,在本揭示之化合物中,明確指定為氘(D)之任何原子皆意欲表示氘。
每當文中所述之化合物經多於1個相同指定基團例如“R”或“R1
”取代,則其將被理解為該基團可為相同或不同,即,基團各自獨立地被選擇。波浪線表示連接至鄰接的次結構、基團、部分或原子之共價鍵的位置。
式IV化合物所包含的經選擇之取代基可呈現遞迴度(recursive degree)。在此情況下,“遞迴取代基(recursive substituent)”意指取代基可列舉其本身的另一實例。因為該等取代基之遞迴性質,所以理論上很多化合物可存在於任何所給定之實施態樣。例如,Rx
包含Ry
取代基。Ry
可為R。R可為Z3
。Z3
可為Z4
和Z4
可為R或包含包括Ry
之取代基。或者,Z3
可為Z5
,其包含包括Ry
之取代基。一般熟悉醫藥化學技術的人士均理解該取代基的總數合理地受限於所欲化合物的所要性質。該等性質包含(例如且非限制地)物理性質(諸如分子量、溶解度或log P)、應用性質(諸如對抗所欲標的之活性)、及實施性質(例如合成的容易性)。
例如且非限制地,在某些實施態樣中Z3
和Ry
為遞迴取代基。典型地,在一給定之實施態樣中,各遞迴取代基可獨立地出現20、19、18、17、16、15、14、13、12、11、10、9、8、7、6、5、4、3、2、1、或0次。更典型地,在一給定之實施態樣中,各遞迴取代基可獨立地出現12或更少次。甚至更典型地,在一給定之實施態樣中,各遞迴取代基可獨立地出現3或更少次。例如,在一給定之實施態樣中,Z3
將出現0至8次,Ry
將出現0至6次。甚至更典型地,在一給定之實施態樣中,Z3
將出現0至6次和Ry
將出現0至4次。
遞迴取代基為本發明之所欲態樣。熟習醫藥化學之技術者理解該等取代基的多樣性(versatility)。就遞迴取代基存在於本發明之實施態樣中來說,總數將如上所述決定。
本發明之化合物可藉由熟習該項技術者已知的方法製備。例如,本發明之化合物可根據美國專利第8,008,264號和美國申請案公開號US 2012/0027752中所述之方法製備。
A.本發明化合物之代謝產物
就該等產物相較於先前技術是新穎且非顯而易見的來說,文中所述之化合物的活體內代謝產物亦落在本發明範圍內。該等產物可由例如所投予之化合物的氧化、還原、水解、醯胺化、酯化等等而產生,主要是由於酵素過程。因此,本發明包括藉由一種包含以下之方法所產生之新穎且非顯而易見的化合物:使本發明化合物與哺乳動物接觸一段足以產生其代謝產物的時間。該等產物通常藉由製備本發明之輻射標記(例如,14
C或3
H)之化合物確認,以可檢測之劑量(例如大於約0.5 mg/kg)腸胃外投予至動物諸如大鼠、小鼠、天竺鼠、猴或人類,使經足以發生代謝之時間(通常約30秒至30小時),再由尿、血液或其它生物學樣品中分離其轉化產物。這些產物因為它們被標記而容易被分離出來(其他係藉由使用能結合殘存於代謝物之抗原決定基的抗體而分離出來)。代謝結構係以習用方式決定,例如,藉由MS或NMR分析。一般,代謝產物的分析是以與熟習該技術者熟知的習用藥物代謝研究相同的方式進行。只要轉化產物不是以其他方式在活體內發現,則它們可用於本發明化合物之治療劑量的診斷分析,即使它們本身不具抗絲狀病毒科活性。
用於測定化合物在替代胃腸分泌物中之穩定性的配方和方法是已知的。化合物在本文中被定義為在消化道是穩定的,其中小於約50莫耳%的經保護基團在替代腸道或胃液內於37℃培育1小時而去保護。僅因為化合物對胃腸道是穩定的,但不意謂其不會在體內水解。本發明之前藥在消化系統通常是穩定的,但實質上可在消化腔、肝或其他代謝器官,或一般在細胞內,被水解成母藥。
III.醫藥調合物
本發明化合物與將根據普通實務選擇的習用載劑和賦形劑調合。錠劑將包含賦形劑、滑動劑、填充劑、黏合劑等等。水性調合物係以無菌形式予以製備,且當欲以非口服投予輸送時一般將為等滲性。所有調合物將任意地包含賦形劑,諸如彼等“Handbook of Pharmaceutical Excipients”(1986)中所述者。賦形劑包括抗壞血酸和其他抗氧化劑、鉗合劑諸如EDTA、碳水化合物諸如聚葡萄糖、羥基烷基纖維素、羥烷基甲基纖維素、硬脂酸等等。調合物的pH範圍從約3至約11,但通常為約7至10。在一些實施態樣中,調合物的pH範圍從約2至約5,但通常為約3至約4。在一些實施態樣中,調合物的pH範圍從約2至約10,但通常為約3.5至約8.5。
雖然活性成分可能單獨投予,但其以醫藥調合物呈現更佳。本發明之供獸醫用和供人類用之調合物兩者皆包括至少一種如上所定義之活性成分,和一或多種為此之可接受的載劑,和任意地其他治療成分,特別是彼等如本文中所討論之額外治療成分。載劑就與調合物之其他成分相容之意義而言必須為“可接受的”且對其接受者為生理上無毒的。
調合物包括彼等適合於前述投予途徑者。調合物可以單位劑量形式方便地呈現,且可藉由製藥技術中已知的任何方法製備。技術和調合物通常見於Remington’s Pharmaceutical Sciences(Mack Publishing Co.,Easton,PA)。該等方法包括使活性成分與構成一或多種輔助成分的載劑結合之步驟。通常調合物藉由使活性成分與液體載劑或細碎載劑或兩者均勻地且密切地結合,且接著,若需要,將產物成形而製得。
適用於經口投予之本發明調合物可呈現為個別單位,諸如,膠囊、扁囊劑(cachet)或錠劑,各自含有既定之量的活性成分;粉劑或粒劑;在水性或非水性液體中的溶液或懸浮液;或水包油液態乳液或油包水液態乳液。該活性成分亦可以丸劑、糖劑或糊膏投予。
錠劑係藉由隨意與一或多種輔助成分一起壓縮或模製而製備。經壓縮的錠劑可藉由在適當的機器中壓縮自由流動形式(諸如,粉末或顆粒)的活性成分(其隨意與黏合劑、潤滑劑、惰性稀釋劑、防腐劑、表面活性劑或分散劑混合)而製得。模製錠劑可藉由在適當的機器中將以惰性液態稀釋劑潤濕的之活性成分粉末的混合物模製而製得。錠劑可隨意被塗覆或刻痕,及隨意地調合,以便提供活性成分從其慢慢或受控釋出。
關於眼或其他外部組織如嘴和皮膚之感染,調合物較佳以含有例如0.075至20% w/w(包括以0.1% w/w增量之介於0.1%與20%之範圍內的活性成分,諸如,例如0.6% w/w、0.7% w/w、等等),較佳為0.2至15% w/w,且最佳為0.5至10% w/w之量的活性成分之局部用軟膏或乳膏的形式施用。當調配成軟膏時,該活性成分可與石蠟或水溶混性軟膏基質一起使用。或者,活性成分可用水包油乳霜基質調配成乳霜。
如果需要的話,乳霜基質的水相可包括(例如)至少30% w/w的多元醇,即,具有二或更多個羥基之醇諸如丙二醇、丁烷1,3-二醇、甘露醇、山梨糖醇、甘油和聚乙二醇(包括PEG 400)及其混合物。局部調合物理想上可包括增強活性成分透過皮膚或其他受影響之區域之吸收或滲透的化合物。該等皮膚滲透增強劑的實例包括二甲基亞碸及相關類似物。
本發明乳液的油相可以已知方式由已知成分構。雖然該相可能僅包含乳化劑(或稱為乳化作用劑(emulgent)),但是希望包含至少一種乳化劑與脂或油或與脂和油二者的混合物。較佳地,包括親水性乳化劑連同充當穩定劑的親脂性乳化劑。較佳者也為包括油和脂二者。乳化劑可與或不與穩定劑一起組成所謂的乳化蠟,且該蠟可與油及脂一起組成所謂的乳化軟膏基質,形成乳霜調合物之油性分散相。
適合用於本發明調合物中之乳化作用劑及乳液穩定劑包括Tween®
60、Span®
80、鯨蠟基硬脂醇、苯甲醇、肉荳蔻醇、單硬脂酸甘油酯及月桂基硫酸鈉。其他適合用於本發明調合物中之乳化作用劑及乳液穩定劑包括Tween®
80。
適合用於調合物之油或脂肪的選擇係根據達成所要之化妝品性質。乳霜應較佳地為具有適當黏稠度以避免從管子或其它容器漏出之非油膩、不著色和可清洗的產品。可使用直鏈或支鏈的單-或二元烷酯類諸如二異己二酸酯、硬脂酸異鯨蠟酯、椰子脂肪酸的丙二醇二酯、肉豆蔻酸異丙酯、油酸癸酯、棕櫚酸異丙酯、硬脂酸丁酯、棕櫚酸2-乙基己酯或支鏈酯類的摻合物(稱為Crodamol CAP),後三者為較佳的酯類。這些酯類可依據所要求之性質而單獨使用或組合使用。或者,使用高熔點脂質如白色軟石蠟和/或液體石蠟或其他礦物油。
根據本發明之醫藥調合物包含根據本發明之組合物與一或多種醫藥上可接受的載劑或賦形劑和隨意其他治療劑。含有活性成分之醫藥調合物可為適合所欲之投予方法的任何形式。當用於口服使用時,可製備例如錠劑、喉錠、菱形錠、水性或油性懸浮液、可分散之粉末或顆粒、乳液、硬或軟膠囊、糖漿或酏劑。欲用於口服使用之組成物可根據用於製造醫藥組成物之技術已知之任何方法製備,且該等組成物可含有一或多種試劑,包括甜味劑、調味劑、著色劑和防腐劑,以提供可口製劑。可接受含有摻混適合製造錠劑之無毒醫藥上可接受的賦形劑之活性成分的錠劑。這些賦形劑可為例如惰性稀釋劑,諸如碳酸鈣或碳酸鈉、乳糖、磷酸鈣或磷酸鈉;粒化劑和崩散劑,諸如玉米澱粉、或海藻酸;黏合劑,例如澱粉、明膠或阿拉伯膠;和潤滑劑,諸如硬脂酸鎂、硬脂酸或滑石。錠劑可未經塗覆或可藉由已知技術(包括微膠囊化)塗覆,以延遲在胃腸道之崩散和吸附,而藉此提供較長時間的持續作用。例如,可使用時間延遲材料例如單硬脂酸甘油酯或二硬脂酸甘油酯,單獨或可與蠟一起。
用於口服使用之調合物亦可以硬明膠膠囊呈現,其中活性成分與惰性固態稀釋劑例如磷酸鈣或高嶺土混合,或以軟明膠膠囊呈現,其中活性成分與水或油性介質如花生油、液態石蠟或橄欖油混合。
本發明水性懸浮液包括摻混適合於製造水性懸浮液之賦形劑的活性材料。該等賦形劑包括懸浮劑諸如羧甲基纖維素鈉、甲基纖維素、羥丙基甲基纖維素、海藻酸鈉、聚乙烯基吡咯啶酮、黃蓍膠和阿拉伯膠,及分散劑或潤濕劑諸如天然磷脂質(例如,卵磷脂)、環氧烷與脂肪酸的縮合產物(例如,聚氧乙烯硬脂酸酯)、環氧乙烷與長鏈脂族醇的縮合產物(例如,十七氧乙烯鯨蠟醇)、環氧乙烷與衍生自脂肪酸和己糖醇酐之部分酯的縮合產物(例如,聚氧乙烯山梨糖醇單油酸酸酯)。懸浮劑的其他非限制性實例包括Captisol®(磺酸基丁基醚β-環糊精,SBE-β-CD)。水性懸浮液亦可包括一或多種防腐劑如對-羥基-苯甲酸乙酯或對-羥基-苯甲酸正丙酯,一或多種著色劑,一或多種調味劑和一或多種甜味劑如蔗糖或糖精。
油性懸浮液可藉由使活性成分懸浮於蔬菜油諸如花生油、橄欖油、芝麻油或椰子油,或於礦物油諸如液態石蠟而調配。口服懸浮液可包括增稠劑如蜂蠟、硬石蠟或鯨臘醇。可加入甜味劑諸如上述者和調味劑以提供可口的口服製劑。這些組成物可藉由添加抗氧化劑如抗壞血酸來防腐。
適合於藉由添加水製備水性懸浮液的本發明可分散粉末和顆粒提供摻混分散劑或潤濕劑、懸浮劑、和一或多種防腐劑之活性成分。合適之分散劑或濕潤劑及懸浮劑以它們上述所揭示者舉例說明。亦可存在額外的賦形劑,例如甜味劑、調味劑及著色劑。
本發明之醫藥組成物亦可為水包油乳液形式。油相可為蔬菜油(諸如橄欖油或花生油)、礦物油(例如液體石蠟)、或此等之混合物。適當乳化劑包括天然膠,諸如阿拉伯膠和黃蓍膠;天然磷脂,諸如大豆卵磷脂;衍生自脂肪酸和己醣醇酐的酯類或部分酯類,諸如去水山梨醇單油酸酯,和這些部分酯類與環氧乙烷的縮合產物,諸如聚氧乙烯去水山梨醇單油酸酯。乳液亦可包含甜味劑和調味劑。糖漿和酏劑可用甜味劑(諸如甘油、山梨醇或蔗糖)調配。該等調合物亦可包含緩和劑、防腐劑、調味劑或著色劑。
本發明醫藥組成物可為無菌可注射製劑之形式,諸如無菌可注射的水性或油質懸浮液。此懸浮液可根據已知技術使用上述之適當分散劑或潤濕劑和懸浮劑調配。無菌注射製劑亦可為在無毒腸胃外可接受的稀釋劑或溶劑中的無菌注射溶液或懸浮液,諸如在1,3-丁烷-二醇中的溶液,或製備為冷凍乾燥粉末形式。無菌注射製劑亦可為在腸胃外可接受的稀釋劑或溶劑中的無菌注射溶液或懸浮液。可使用的可接受的媒液和溶劑為水、林格氏(Ringer’s)溶液和等滲氯化鈉溶液。此外,無菌非揮發性油通常可用作為溶劑或懸浮介質。為此目的,可使用任何溫和的非揮發性油,包括合成單-或二甘油酯。此外,脂肪酸如油酸同樣地亦可用於製備注射劑。可使用的可接受的媒液和溶劑為水、林格氏溶液、等滲氯化鈉溶液、高滲氯化鈉溶液,及低滲氯化鈉溶液。
可與載劑材料組合以產生單一劑量形式的活性成分的量將視治療之宿主和投予之特定模式而變化。例如,欲口服投予至人類的時間-釋出調合物可包括約1至1000mg的活性材料與適當且方便量的載劑混合,其可從總組成物之約5改變至約95%(重量:重量)。可製備醫藥組成物以提供可輕易測量供投予之量。例如,欲用於靜脈輸注之水性溶液可包括從約3至500μg的活性成分/毫升溶液,以使得可發生速率約30mL/hr的適當體積輸注。
適合局部投予至眼的調合物亦包括眼藥水,其中活性成分溶於或懸浮於適當的載劑,特別是用於對活性成分之水性溶劑。活性成分較佳以0.5至20%,有利為0.5至10%,且特別是約1.5%重量/重量之濃度存在於該等調合物中。
適合於嘴內局部投予的調合物包括菱形錠,其包含在調味基質(通常為蔗糖和阿拉伯膠或黃蓍膠)中的活性成分;軟錠劑(pastille),其包含在惰性基質如明膠和甘油、或蔗糖和阿拉伯膠中的活性成分;及漱口水,其包含在適當液體載劑的活性成分。
用於直腸投予之調合物可以具有適當基質之栓劑呈現,該基質包括例如可可脂或水楊酸酯。
適合於肺內或鼻投予之調合物具有例如範圍在0.1至500微米如0.5、1、30、35等等之粒徑,其藉由通過鼻道之快速吸入而投予,或藉由通過嘴吸入而投予,以便到達肺泡囊。適當調合物包括活性成分的水性或油性溶液。適合於噴霧劑或乾燥粉末投予之調合物可根據習用方法製備,且可與其他治療劑諸如如下所述之過去用於治療或預防絲狀病毒科感染的化合物一起輸送。
適合於陰道投予的調合物可呈現為陰道栓劑、棉條、乳膏、凝膠、糊劑、泡沫或噴霧調合物,其除了活性成分之外,還含有諸如在該項技術已知為適當的載體。
適合於腸胃外投予的調合物包括水性及非水性無菌注射溶液,彼等可含有抗氧化劑、緩衝劑、抗菌劑以及使該調合物與預定接受者的血液呈等滲的溶質;以及水性及非水性的無菌懸浮液,其可包括懸浮劑及增稠劑。
該調合物可以單位劑量或多劑量容器(例如,密封的安瓿及小瓶)呈現且可儲存於冷凍乾燥條件下,而僅需在使用前立即添加無菌液態載體(例如,注射用水)。即時注射溶液及懸浮液可由前述種類的無菌粉末、粒劑及錠劑製備。較佳的單位劑量調合物為彼等含有每日劑量或單位每日亞劑量(如本文中所列舉者)或彼等之適當分量的活性成分者。
應理解,除了前文特別提及的成分之外,本發明之調合物可包括具有關於所討論的調合物之類型的技術中習用的其他試劑,例如,彼等適合於口服投予者可包括調味劑。
本發明進一步提供獸醫用組成物,其包括至少一種如上述定義之活性成分和為此之獸醫用載劑。
獸醫用載劑為用於投予組成物之目的的材料且可為固態、液態或氣態材料,其在獸醫技術中另為惰性或可接受的且與活性成分相容。這些獸醫用組成物可口服、腸胃外投予或藉由任何其他所要途徑投予。
本發明化合物係用於提供控制釋放之醫藥調合物,其含有作為活性成分之一或多種本發明化合物(“控制釋放調合物”),其中活性成分的釋出係經控制且經調整以允許較少頻率給藥或改善特定活性成分的藥物動力學或毒性狀況(toxicity profile)。
IV.投予途徑
一或多種本發明之化合物(在本文稱為活性成分)可藉由任何適合於所欲治療之病況的途徑來投予。適當的途徑包括:口服、經直腸、經鼻、經肺、局部(包括經頰及經舌下)、經陰道及腸胃外(包括皮下、肌內、靜脈內、經皮、鞘內及硬膜外)、等等。將可理解的是:較佳途徑理可隨著(例如)接受者的病況而改變。本發明化合物的優點之一在於它們係具有口服生物可用率且可經口給藥。
在本發明用於治療絲狀病毒科感染之方法中,本發明化合物可在任何時間投予至可能與罹患絲狀病毒科感染或已罹患絲狀病毒科感染的人接觸之人。在一些實施態樣中,本發明化合物可預防性投予至與罹患絲狀病毒科感染的人接觸之人。在一些實施態樣中,本發明化合物可投予至絲狀病毒科感染之測試為陽性但尚未顯示絲狀病毒科感染的症狀之人。在一些實施態樣中,本發明化合物可投予至絲狀病毒科感染的症狀開始時之人。
活性成分的有效劑量至少取決於待治療病況之性質、毒性、化合物是否正用於預防(較低劑量)或抵抗活性病毒感染、輸送方法、和醫藥調合物,且將由臨床醫師使用習用劑量遞增研究而決定。可預期在從約0.0001至約100 mg/kg體重/天;典型地,從約0.01至約10 mg/kg體重/天;更典型地,從約.01至約5 mg/kg體重/天;最典型地,從約.05至約0. 5mg/kg體重/天。例如,約70公斤體重的成人之每天候選劑量範圍將從1 mg至1000 mg,較佳在5 mg和500 mg之間,並可採取單一或多次劑量的形式。
本發明化合物用於治療絲狀病毒科感染之有效劑量可取決於該劑量係用於預防性或用於治療已罹患絲狀病毒科感染之人。此外,劑量可取決於罹患絲狀病毒科感染之人是否還沒有顯示症狀或已顯示絲狀病毒科感染之症狀。治療對絲狀病毒科感染之測試為陽性的人和已顯示絲狀病毒科感染之症狀的人相較於已接受預防性治療之人可能需要較大量。
預期投予本發明化合物之任何適當時間週期。例如,投予可經從1天至100天,包括2、3、4、5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、或90天。投予也可經從1週至15週,包括2、3、4、5、6、7、8、9、10、11、12、13、或14週。也考慮較長週期的投予。投予之時間可取決於化合物是否為預防性或治療罹患絲狀病毒科感染之人。例如,預防性投予可經定期接觸其他罹患絲狀病毒科感染之人的時間週期,及經與罹患絲狀病毒科感染之人最後接觸以後的適當時間週期。就已罹患絲狀病毒科感染之人而言,投予之週期可為經治療患者所需之時間長度和對絲狀病毒科感染之測試為陰性以後的適當時間週期以確保絲狀病毒科感染不復發。
V.組合治療
本發明組成物亦可與其他活性成分組合使用。關於絲狀病毒科病毒感染的治療,較佳地,其他活性治療劑具有抗絲狀病毒科病毒感染(特別是馬堡病毒(Marburg virus)、伊波拉病毒(Ebola virus)和奎瓦病毒(Cueva virus)感染)之活性。這些其他活性治療劑的非限制性實例為利巴韋林(ribavirin)、帕利珠單抗(palivizumab)、莫它珠單抗(motavizumab)、RSV-IGIV (RespiGam®
)、MEDI-557、A-60444、MDT-637、BMS-433771、胺碘酮(amiodarone)、決奈達隆(dronedarone)、維拉帕米(verapamil)、伊波拉(Ebola)恢復期血漿(ECP)、TKM-100201、BCX4430 ((2S,3S,4R,5R)-2-(4-胺基-5H-吡咯並[3,2-d]嘧啶-7-基)-5-(羥甲基)吡咯啶-3,4-二醇)、法匹拉韋(favipiravir)(亦稱為T-705或Avigan)、T-705 單磷酸鹽、T-705二磷酸鹽、T-705三磷酸鹽、FGI-106(1-N,7-N-雙[3-(二甲胺基)丙基]-3,9-二甲基喹啉並[[8,7-h]喹啉酮-1,7-二胺)、JK-05、TKM-伊波拉、ZMapp、rNAPc2、VRC-EBOADC076-00-VP、OS-2966、MVA-BN filo、伯利多韋(brincidofovir)、Vaxart 腺病毒載體5型伊波拉疫苗、Ad26-ZEBOV、FiloVax 疫苗、GOVX-E301、GOVX-E302、伊波拉病毒(Ebola virus)進入抑制劑(NPC1抑制劑)、和RVSV-EBOV、及其混合物。本發明之化合物和組成物亦可與胺基磷酸酯(phosphoramidate)佅啉基寡聚物(PMOs)組合使用,其為旨在藉由形成具有特定RNA序列之鹼對干擾轉譯過程之合成反義寡核苷酸類似物。PMOs的實例包括AVI-7287、AVI-7288、AVI-7537、AVI-7539、AVI-6002、及AVI-6003。本發明之化合物和組成物也意欲與提供具有絲狀病毒科感染的患者之一般護理(包括腸胃外的液體(包括葡萄糖生理鹽水和乳酸林格氏液(Ringer’s lactate))和營養)、抗生素(包括甲硝唑和頭孢菌素抗生素,諸如頭孢曲松(ceftriaxone)和頭孢呋辛(cefuroxime))及/或抗真菌預防性治療、發燒和止痛藥、止吐劑(諸如甲氧氯普胺(metoclopramide))及/或止瀉劑、維生素和礦物質補充劑(包括維生素K和硫酸鋅)、抗炎劑(諸如伊布洛芬(ibuprofen))、止痛藥、及用於在患者群體中其他常見疾病之藥物(諸如抗瘧劑(包括蒿甲醚(artemether)和青蒿琥酯(artesunate)-苯芴醇(lumefantrine)組合治療)、傷寒(包括喹啉酮抗生素,諸如塞普沙辛(ciprofloxacin)、巨環內酯抗生素(諸如亞藥索黴素(azithromycin))、頭孢菌素抗生素(諸如頭孢曲松)、或胺基青黴素類(諸如胺苄青黴素)、或志賀桿菌病(shigellosis)一起使用。
亦可能將任何本發明化合物與一或多種另外的活性治療劑組合於單一劑量形式以同時或依序投予至患者。組合治療可以同時或依序方案投予。當依序投予時,組合物可以二或更多次投予方式投予。
本發明化合物與一或多種其他活性治療劑的共投予一般意指同時或依序投予本發明化合物和一或多種其他活性治療劑,使得治療有效量的本發明化合物和一或多種其他活性治療劑同時存在於患者體內。
共投予包括在投予單位劑量的一或多種其他活性治療劑之前或之後,投予單位劑量的本發明化合物,例如,在投予一或多種其他活性治療劑後幾秒、幾分鐘或幾小時內,投予本發明化合物。例如,可先投予單位劑量的本發明化合物,接著在幾秒或幾分鐘內投予單位劑量的一或多種其他活性治療劑。或者,可先投予單位劑量的一或多種其他治療劑,接著在幾秒或幾分鐘內投予單位劑量的本發明化合物。在一些情況中,可能想要先投予單位劑量的本發明化合物,接著幾個小時後(例如,1-12小時)投予單位劑量的一或多種其他活性治療劑。在其他情況中,可能想要先投予單位劑量的一或多種其他活性治療劑,接著幾個小時後(例如,1-12小時)投予單位劑量的本發明化合物。
組合治療可提供“協同效果(synergy)”和“增效(synergistic)”,即,當一起使用的活性成分所達到的效果大於來自分開使用化合物所產生之效果的總和。當活性成分:(1)以組合調合物共調配和同時投予或輸送;(2)藉由以個別調合物方式交替或平行輸送;或(3)藉由一些其他療法,可能達到增效效果。當以交替治療輸送時,當化合物依序投予或輸送時,例如,以個別錠劑、丸或膠囊,或藉由在個別注射器內的不同注射液,可能達到增效效果。一般,在交替治療期間,依序地(即,連續地)投予有效劑量的各活性成分,然而組合治療時,一起投予有效劑量之二或更多種活性成分。增效抗病毒效果表示抗病毒效果大於組合物中個別化合物的所預測之純相加效果。
在又另一實施態樣中,本申請案提供抑制細胞中的絲狀病毒科聚合酶之方法,其包含:使感染絲狀病毒之細胞與有效量的式IV化合物、或其醫藥上可接受的鹽、溶劑合物、及/或酯接觸,藉此抑制絲狀病毒科聚合酶。
在又另一實施態樣中,本申請案提供抑制細胞中的絲狀病毒科聚合酶之方法,其包含:使感染絲狀病毒之細胞與有效量的式IV化合物、或其醫藥上可接受的鹽、溶劑合物、及/或酯,及至少一種另外的活性治療劑接觸,藉此抑制絲狀病毒科聚合酶。
在又另一實施態樣中,本申請案提供細胞中的抑制絲狀病毒科聚合酶之方法,其包含:使感染絲狀病毒之細胞與有效量的式IV化合物、或其醫藥上可接受的鹽、溶劑合物、及/或酯,及至少一種所選擇之另外的活性治療劑接觸。
在又另一實施態樣中,本申請案提供治療人類的絲狀病毒科病毒感染之方法,其包含:將治療有效量的式IV化合物、或其醫藥上可接受的鹽、溶劑合物、及/或酯投予至患者。
在又另一實施態樣中,本申請案提供治療人類的絲狀病毒科病毒感染之方法,其包含:將治療有效量的式IV化合物、或其醫藥上可接受的鹽、溶劑合物、及/或酯,及至少一種另外的活性治療劑投予至患者,藉此抑制絲狀病毒科聚合酶。
在又另一實施態樣中,本申請案提供人類的絲狀病毒科病毒感染之方法,其包含:將治療有效量的式IV化合物、或其醫藥上可接受的鹽、溶劑合物、及/或酯,及至少一種另外的活性治療劑投予至患者。
所提供者亦為一種套組,其包括式IV化合物、或其醫藥上可接受的鹽、醫藥上可接受的酯、立體異構物、立體異構物之混合物或互變異構物。在獨立實施態樣中提供個別套組,在此其包括選自式IV化合物,以及其各子群和實施態樣,包括個別化合物1、8、9、10、12、15、17、21、22、23、24、25、26、27、28、29、30、31,及32(化合物1-32)、或其醫藥上可接受的鹽、醫藥上可接受的酯、立體異構物、立體異構物之混合物或互變異構物。在一態樣中,套組包含式IV化合物、或其醫藥上可接受的鹽。各個本文中所述之個別套組可包含該化合物使用於治療需要該治療之個體(例如,人類)的疾病或病況之標籤及/或說明書。在一些實施態樣中,該疾病或病況為人類絲狀病毒科病毒感染,包括伊波拉病毒感染或馬堡病毒感染。在其他實施態樣中,各個獨立套組也可含有另外的藥劑與式IV化合物組合使用於治療需要該治療之個體(例如,人類)的疾病或病況之說明書。在此等實施態樣之某些中,該疾病或病況為人類絲狀病毒科病毒感染,包括伊波拉病毒感染或馬堡病毒感染。在本文中所述之各個套組中有另一實施態樣,其中該套組包含個別劑量單位之如本文所述之化合物、或其醫藥上可接受的鹽、外消旋物、鏡像異構物、非鏡像異構物、互變異構物、多晶形物、假多晶形物、非晶形式、水合物或溶劑合物。個別劑量單元的實例可包括丸、錠劑、膠囊、預充式注射器或注射器藥盒、IV袋、等等,各自包含治療有效量的所討論之化合物、或其醫藥上可接受的鹽、外消旋物、鏡像異構物、非鏡像異構物、互變異構物、多晶形物、假多晶形物、非晶形式、水合物或溶劑合物。在一些實施態樣中,該套組可含有單劑量單位及在其他中以多個劑量單位存在,諸如指定治療方案或週期所需劑量單位之數目。
亦提供者為製造之物件,其包括式IV化合物、或其醫藥上可接受的鹽、醫藥上可接受的酯、立體異構物、立體異構物之混合物或互變異構物;及容器。在一態樣中,製造之物件包含式IV化合物和個別化合物1、8、9、10、12、15、17、21、22、23、24、25、26、27、28、29、30、31、及32 (化合物1-32)、或其醫藥上可接受的鹽,及容器。在獨立實施態樣中,該製造之物件的容器可為小瓶、罐、安瓿、預裝注射器、泡罩包裝、罐頭、瓶、盒、或靜脈注射袋。
VI.抑制絲狀病毒科聚合酶之方法
本發明之另一態樣關於抑制絲狀病毒科聚合酶的活性之方法,其包含以本發明之化合物或組成物處理懷疑含有絲狀病毒科之樣品的步驟。
可使用本發明方法治療之絲狀病毒科為典型地感染靈長類動物之單股RNA病毒。絲狀病毒能夠在幾乎所有類型的細胞中繁殖。絲狀病毒抗原和病毒體主要發現於受感染個體的纖維母細胞和間質中。有三個已確定的絲狀病毒科:伊波拉病毒(Ebola virus) (EBOV;五種);馬堡病毒(Marburg virus)(MARV);及奎瓦病毒(Cueva virus),亦稱為Lloviu病毒(LLOV)。病毒體(病毒粒子)特徵形狀為長圓柱形絲狀粒子,其可為直線、彎曲、捲曲,或發現於“6”或“U”形構形。它們偶爾分支且粒子長度相差很大,但直徑(約80 nm)為一致。絲狀病毒基因組包含編碼4種病毒體結構蛋白質(VP30、VP35、核蛋白(NP)、及聚合酶蛋白質(L-pol))和3種膜相關蛋白(VP40、醣蛋白(GP)及VP24)之7種基因。
伊波拉病毒(Ebola virus) 屬包括5種已知的種:(1) 班迪布交(Bundibugyo)伊波拉病毒,亦稱為班迪布交病毒(BDBV,以前為BEBOV);(2) 雷斯頓(Reston)伊波拉病毒,亦稱為雷斯頓病毒或伊波拉-雷斯頓 (RESTV,以前為REBOV);(3) 蘇丹(Sudan)伊波拉病毒,亦稱為蘇丹病毒或伊波拉-蘇丹(SUDV,以前為SEBOV);(4) 大森林(Tai Forest)伊波拉病毒,亦稱為大森林病毒或伊波拉-大(TAFV,以前為CIEBOV);及(5) 薩伊(Zaire)伊波拉病毒,亦稱為伊波拉病毒或伊波拉-薩伊(EBOV,以前為ZEBOV)。
馬堡病毒屬包括馬堡馬堡病毒種,亦稱為馬堡病毒(MARV)或Ravn病毒(RAVV)。奎瓦病毒屬包括Lloviu奎瓦病毒種,亦稱為Lloviu病毒(LLOV)。
本發明之組成物可充當絲狀病毒科聚合酶之抑制劑,作為該抑制劑之中間物或具有如下所述之其他利用性。抑制劑將結合至絲狀病毒科聚合酶的表面上或空穴中之具有對絲狀病毒科聚合酶的獨特幾何之位置。結合絲狀病毒科聚合酶之組成物可具有不同程度的可逆性結合。它們實質上不可逆地結合之化合物為使用於本發明之方法的理想候選物。一旦標記,實質上不可逆地結合之組成物係用作為檢測絲狀病毒科聚合酶用的探針。因此,本發明關於檢測懷疑含有絲狀病毒科聚合酶之樣品中的絲狀病毒科聚合酶之方法,其包含以下步驟:以包含結合於標籤的本發明化合物之組成物處理懷疑含有絲狀病毒科聚合酶的樣品;並觀察樣品對標籤上的活性之作用。適當標籤在診斷領域為熟知的,包括穩定的自由基、螢光團、放射性同位素、酵素、化學發光基團和色素原。化合物在此以習用方式使用官能團如羥基、羧基、巰基或胺基標記。
在本發明的上下文中,懷疑含有絲狀病毒科聚合酶之樣品包括天然或人造材料,諸如活生物;組織或細胞培養物;生物樣品諸如生物材料樣品(血、血清、尿、腦脊髓液、淚液、痰、唾液、組織樣品、等等);實驗室樣品;食物、水或空氣樣品;生物製品樣品諸如細胞(特別是合成所需糖蛋白的重組細胞)之萃取物;等等。典型地,該樣品將被懷疑含有產生絲狀病毒科聚合酶之生物體(常為病原生物體諸如絲狀病毒病毒)。樣品可包含在包括水和有機溶劑\水混合物的任何介質中。樣品包括活生物體諸如人和人造材料諸如細胞培養物。
本發明之治療步驟包含將本發明之組成物加至樣品或其包含將組成物之前驅物加至樣品。添加步驟包含任何如上所述之投予方法。
如果需要的話,可藉由包括檢測絲狀病毒科聚合酶活性的直接和間接的方法之任何方法觀察施用組成物後的絲狀病毒科聚合酶之活性。確定絲狀病毒科聚合酶活性的定量、定性和半定量方法全被考慮。通常應用上述篩選方法之一,然而,任何其它方法諸如活生物體的生理性質的觀測也是適用的。
含有絲狀病毒科聚合酶之生物包括絲狀病毒科病毒。本發明化合物可用於治療或預防動物或人類的絲狀病毒科感染。
然而,在篩選能夠抑制人類絲狀病毒科病毒之化合物中,應牢記的是:酵素分析的結果可能與細胞培養分析無關。因此,以細胞為基礎之分析應為主要的篩選工具。
在另一實施態樣中,本申請案提供治療人類的絲狀病毒科病毒感染之方法,其包含:將治療有效量的式IV化合物、或其醫藥上可接受的鹽、溶劑合物、及/或其酯投予至患者。在一些實施態樣中,該絲狀病毒科感染係由絲狀病毒科病毒引起。在一些實施態樣中,該絲狀病毒科感染係由伊波拉病毒引起。在一些實施態樣中,該絲狀病毒科感染係由班迪布交伊波拉病毒、雷斯頓伊波拉病毒、蘇丹伊波拉病毒、大森林伊波拉病毒、或薩伊伊波拉病毒引起。在一些實施態樣中,該絲狀病毒科感染係由馬堡病毒感引起。在一些實施態樣中,該絲狀病毒科感染係由Lloviu病毒引起。在一些實施態樣中,抑制絲狀病毒科聚合酶。
本發明化合物可使用於治療已罹患 絲狀病毒科感染之人、或可預防性投予以減少或預防絲狀病毒科感染之機會。絲狀病毒科感染的特徵可為出血熱、吐血、腹瀉、胸骨後腹痛和衰竭。潛伏期為與罹患絲狀病毒科感染之人接觸後約21天。絲狀病毒科感染的結果通常為死亡。
亦提供作為獨立實施態樣者為選自本文各式之化合物,以及其各子群和實施態樣,包括選自式IV之群組的化合物、或本文中之實例的特定化合物中之一者,包括化合物1-32、或其醫藥上可接受的鹽、溶劑合物、及/或酯用於治療人類的絲狀病毒感染之方法的用途。亦提供作為各別實施態樣者為選自本文各式之化合物,以及其各子群和實施態樣,包括選自式IV之群組的化合物、或本文中之實例的特定化合物中之一者,包括化合物1-32、或其醫藥上可接受的鹽、溶劑合物、及/或酯用於治療人類的伊波拉病毒感染之方法的用途。亦提供作為獨立實施態樣者為選自本文各式之化合物,以及其各子群和實施態樣,包括選自式IV之群組的化合物、或本文中之實例的特定化合物中之一者,包括化合物1-32、或其醫藥上可接受的鹽、溶劑合物、及/或酯用於治療人類的馬堡病毒感染之方法的用途。在本文中各個實施態樣範圍內,其中該絲狀病毒科感染為伊波拉病毒,有使用它們之另外獨立實施態樣,其中該絲狀病毒科感染分別由班迪布交伊波拉病毒、雷斯頓伊波拉病毒、蘇丹伊波拉病毒、大森林伊波拉病毒、或薩伊波拉病毒引起。在一些實施態樣中,該絲狀病毒科感染係由馬堡病毒引起。在一些實施態樣中,該絲狀病毒科感染係由Lloviu病毒引起。
本發明也提供本文中各式之化合物,以及其各子群和實施態樣,包括選自式(IV)之群組的化合物、或本文中之實例的特定化合物中之一者,包括化合物1-32、或其醫藥上可接受的鹽、溶劑合物、及/或酯,用於任何如本文所定義之本發明方法的用途。
亦提供作為獨立實施態樣者為選自本文各式之化合物,以及其各子群和實施態樣,包括選自式IV之群組的化合物、或本文中之實例的特定化合物中之一者,包括化合物1-32、或其醫藥上可接受的鹽、溶劑合物、及/或酯用於製備供治療人類的絲狀病毒科感染用之藥劑的用途。亦提供作為獨立實施態樣者為選自本文各式之化合物,以及其各子群和實施態樣,包括選自式IV之群組的化合物、或本文中之實例的特定化合物中之一者,包括化合物1-32、或其醫藥上可接受的鹽、溶劑合物、及/或酯用於製備供治療人類伊波拉病毒感染用之藥劑的用途。亦提供作為獨立實施態樣者為選自本文各式之化合物,以及其各子群和實施態樣,包括選自式IV之群組的化合物、或本文中之實例的特定化合物中之一者,包括化合物1-32、或其醫藥上可接受的鹽、溶劑合物、及/或酯用於製備供治療人類的馬堡病毒感染用之藥劑的用途。
VII.絲狀病毒科聚合酶抑制劑之篩選。
藉由任何評估酶活性的習用技術篩選本發明之組成物的扺抗絲狀病毒科聚合酶之抑制活性。在本發明的情況下,典型地組成物首先篩選體外絲狀病毒科聚合酶之抑制,並接著篩選顯示抑制活性的組成物之活體內活性。組合物具有體外小於約5×10-6
M和較佳小於約1×10-7
M Ki (抑制常數)就體內使用而言為較佳。
已詳細描述有效的體外篩選且在此將不再闡述。然而,實施例描述適當的體外分析。
[相關申請案之交互參照]
本申請案根據35 U.S.C.119(e)主張2014年10月29日申請之美國臨時申請案第62/072,331號及2015年1月20日申請之美國臨時申請案第62/105,619號之權益。前述申請案的全部內容在此以引用方式併入本文中。
VIII.化合物的製備
實施例
某些縮寫和縮略語使用於描述實驗細節。雖然這些大多數將為熟習該項技術人員可理解的,但表1包含許多這些縮寫和縮略語的列表。
表1. 縮寫和縮略語之列表。
A.化合物的製備
實施例1. (2S)-2-(氯(苯氧基)磷醯基胺基)丙酸乙酯(氯化物A)
| 縮寫 | 意義 |
| Ac2 O | 乙酸酐 |
| AIBN | 2,2’-偶氮雙(2-甲基丙腈) |
| Bn | 苯甲基 |
| BnBr | 苯甲基溴 |
| BSA | 雙(三甲矽基)乙醯胺 |
| BzCl | 苯甲醯氯 |
| CDI | 羰基二咪唑 |
| DABCO | 1,4-二氮雜雙環[2.2.2]辛烷 |
| DBN | 1,5-二氮雜雙環[4.3.0]壬-5-烯 |
| DDQ | 2,3-二氯-5,6-二氰基-1,4-苯醌 |
| DBU | 1,5-二吖雙環[5.4.0]十一-5-烯 |
| DCA | 二氯乙醯胺 |
| DCC | 二環己碳二亞胺 |
| DCM | 二氯甲烷 |
| DMAP | 4-二甲胺基吡啶 |
| DME | 1,2-二甲氧基乙烷 |
| DMTCl | 二甲氧基三苯甲基氯 |
| DMSO | 二甲亞碸 |
| DMTr | 4, 4’-二甲氧基三苯甲基 |
| DMF | 二甲基甲醯胺 |
| EtOAc | 乙酸乙酯 |
| ESI | 電噴霧離子化 |
| EtOAc | 乙酸乙酯 |
| HMDS | 六甲基二矽氮烷 |
| HPLC | 高壓液相層析 |
| LDA | 二異丙基胺化鋰 |
| LRMS | 低解析質譜 |
| MCPBA | 間氯過苯甲酸 |
| MeCN | 乙腈 |
| MeOH | 甲醇 |
| MMTC | 單甲氧基三苯甲基氯 |
| m/z或m/e | 質荷比 |
| MH+ | 質量加1 |
| MH- | 質量減1 |
| MsOH | 甲磺酸 |
| MS或ms | 質譜 |
| MTBE | 第三丁基甲基醚 |
| NBS | N-溴琥珀醯亞胺 |
| Ph | 苯基 |
| rt或r.t. | 室溫 |
| TBAF | 四丁基氟化銨 |
| THF | 四氫呋喃 |
| TMSCl | 氯三甲基矽烷 |
| TMSBr | 溴三甲基矽烷 |
| TMSI | 碘三甲基矽烷 |
| TMSOTf | (三甲矽基)三氟甲磺酸酯 |
| TEA | 三乙胺 |
| TBA | 三丁胺 |
| TBAP | 焦磷酸三丁基銨 |
| TBSCl | 第三丁基二甲矽基氯 |
| TEAB | 三乙基碳酸氫銨 |
| TFA | 三氟乙酸 |
| TLC或tlc | 薄層層析法 |
| Tr | 三苯基甲基 |
| Tol | 4-甲基苯甲醯基 |
| Turbo格任亞 | 異丙基氯化鎂與氯化鋰之1:1混合物 |
| δ | 四甲基矽烷低場之ppm |
將乙基丙胺酸酯鹽酸鹽(1.69 g,11 mmol)溶解在無水CH2
Cl2
(10 mL)中並將混合物在N2
(g)下攪拌且冷卻至0℃。添加二氯磷酸苯酯(1.49 mL,10 mmol)接著經約10 min滴加Et3
N。然後將反應混合物慢慢升溫至RT並攪拌約12 h。添加無水Et2
O (50 mL)及將混合物攪拌約30 min。以過濾移除所形成之固體,並在減壓下濃縮濾液。將殘餘物進行用在己烷中之0-50% EtOAc溶析的矽膠層析法以提供中間物A。1
H NMR (300 MHz, CDCl3
) δ 7.397.27 (m, 5H), 4.27 (m, 3H), 1.52 (m, 3H), 1.32 (m, 3H)。31
P NMR (121.4 MHz, CDCl3
) δ 8.2, 7.8。
實施例2. (2S)-2-(氯(苯氧基)磷醯基胺基)丙酸2-乙基丁酯(氯化物B)
使用相同的程序製備2-乙基丁基丙胺酸氯胺基磷酸酯(phosphoramidate)B作為氯化物A,除了以2-乙基丁基丙胺酸酯取代乙基丙胺酸酯之外。該材料以粗製品使用於下一反應中。用甲醇或乙醇處理形成具有所要LCMS信號之置換產物。
實施例3. (2S)- 2-(氯(苯氧基)磷醯基胺基)丙酸異丙酯(氯化物C)
使用相同的程序製備異丙基丙胺酸氯胺基磷酸酯(phosphoramidate)C作為氯化物A,除了以異丙基丙胺酸酯取代乙基丙胺酸酯之外。該材料以粗製品使用於下一反應中。用甲醇或乙醇處理形成具有所要LCMS信號之置換產物。
實施例4. (2R, 3R, 4S, 5R)-2-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈 (化合物1)
(2R, 3R, 4S, 5R)-2-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈的製備係描述如下。
將市售內醚醇(10 g,23.8 mmol)在N2
(g)下溶解在無水DMSO (30 mL)中。添加Ac2
O (20 mL)且將所得反應混合物在RT下攪拌約48 h。將反應混合物倒入冰H2
O (500 mL)並將混合物攪拌20 min。將混合物用EtOAc (3×200 mL)萃取且然後將合併之有機萃取物用H2
O (3×200 mL)洗滌。將有機萃取物經無水MgSO4
乾燥,過濾並在減壓下濃縮。將殘餘物溶解在CH2
Cl2
中及進行用在己烷中之25%EtOAc溶析的矽膠層析法以提供內酯。1
H NMR (400 MHz, DMSO) δ 7.30-7.34 (m, 13H), 7.19-7.21 (m, 2H), 4.55-4.72 (m, 6H), 4.47 (s, 2H), 4.28 (d, J = 3.9 Hz,1H), 3.66 (m, 2H)。LCMS m/z 436.1 [M+H2
O], 435.2 [M+OH]- Tr = 2.82 min。HPLC Tr = 4.59 [2-98% ACN in H2)以 2 mL/min 流量經5 min。
將溴吡唑(根據WO2009/132135製備)(0.5 g,2.4 mmol)在N2
(g)下懸浮於無水THF(10 mL)中。攪拌懸浮液及添加TMSCl(0.67 mL,5.28 mmol)。將混合物在RT下攪拌20 min.且接著冷卻至約-78℃,之後慢慢添加nBuLi(6 mL,在己烷中之1.6 N,9.6 mmol)之溶液。將反應混合物在約-78℃下攪拌10 min.且接著經由注射器添加內酯(1 g,2.4 mmol)。當反應如用LCMS測定為完成時,添加AcOH 以淬滅反應。將混合物在減壓下濃縮並將殘餘物溶解在CH2
Cl2
和H2
O之混合物(100 mL,1:1)中。將有機層分離並用H2
O (50 mL)洗滌。然後將有機層經無水MgSO4
乾燥,過濾並在減壓下濃縮。將殘餘物進行用在己烷中之050% EtOAc溶析的矽膠層析法以提供產物,呈變旋異構物之1:1混合物。LCMS m/z 553 [M+H]。
將羥基核苷(1.1 g,2.0 mmol)溶解在無水CH2
Cl2
(40 mL)中及在N2
(g)下將溶液冷卻並攪拌至約-78℃。添加TMSCN(0.931 mL,7 mmol)並將混合物攪拌另10 min。將TMSOTf (1.63 mL,9.0 mmol)慢慢加至反應並將混合物攪拌1 h。接著用CH2
Cl2
(120 mL)稀釋反應混合物並添加NaHCO3
(120 mL)水溶液以淬滅反應。將反應混合物攪拌另10 min及分離有機層。將水層用CH2
Cl2
(150 mL)萃取且將合併之有機萃取物經無水MgSO4
乾燥,過濾並在減壓下濃縮。將殘餘物溶解在最小量的CH2
Cl2
中並進行用0-75% EtOAc和己烷之梯度溶析的矽膠層析法以提供三苯甲基氰基核苷,呈變旋異構物之混合物。1
H NMR (300 MHz, CD3
CN) δ 7.94 (s, 0.5H), 7.88 (s, 0.5H), 7.29-7.43 (m, 13H), 7.11-7.19 (m, 1H), 6.82-6.88 (m,1H), 6.706.76 (m, 1H), 6.41 (bs, 2H), 5.10 (d, J = 3.9 Hz, 0.5H), 4.96 (d, J = 5.1 Hz, 0.5H), 4.314.85 (m, 7H), 4.094.18 (m, 2H), 3.61-3.90 (m, 2H)。LCMS m/z 562 [M+H]。
將三苯甲基氰基核苷(70 mg,0.124 mmol)溶解在無水CH2
Cl2
(2 mL)中並在N2
(g)下冷卻至約-20℃。添加BCl3
之溶液(在CH2
Cl2
中之1N,0.506 mL,0.506 mmol)並將反應混合物在-78℃下攪拌1 h.。當反應藉由LC/MS為完成時,添加MeOH以淬滅反應。使反應混合物加溫至RT並在減壓下移除溶劑。將殘餘物進行C18逆相HPLC,用H2
O (0.1 % TFA)溶析5 min,接著0-70% MeCN在H2
O(0.1 % TFA)中之梯度經35 min,以溶析α-變旋異構物,及β-變旋異構物1。(α-變旋異構物)1
H NMR (300 MHz, D2
O) δ 7.96 (s, 1H), 7.20 (d, J = 4.8 Hz, 1H), 6.91 (d, J = 4.8 Hz, 1H), 4.97 (d, J = 4.4 Hz, 1H), 4.56-4.62 (m, 1H), 4.08-4.14 (m, 1H), 3.90 (dd, J = 12.9, 2.4 Hz, 1H), 3.70 (dd, J = 13.2, 4.5 Hz, 1H)。(β-變旋異構物)1
H NMR (400 MHz, DMSO) δ 7.91 (s, 1H), 7.80-8.00 (br s, 2H), 6.85-6.89 (m, 2H), 6.07 (d, J = 6.0 Hz, 1H), 5.17 (br s, 1H), 4.90 (br s, 1H), 4.63 (t, J = 3.9 Hz, 1H), 4.02-4.06 (m, 1H), 3.94 (br s, 1H), 3.48-3.64 (m, 2H)。LCMS m/z 292.2 [M+H], 290.0 [M-H]。Tr= 0.35 min。13C NMR (400 MHZ, DMSO), 156.0, 148.3, 124.3, 117.8, 117.0, 111.2, 101.3, 85.8, 79.0, 74.7, 70.5, 61.4。HPLC Tr = 1.32 min
使用LaCl3
-2LiCl之(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-醇的製備
製備7-碘吡咯並[2,1-f][1,2,4]三𠯤-4-胺(7.5 g,28.8 mmol,1.0 當量)在THF中(67 mL)之溶液。將溶液冷卻至約0℃,及添加TMSCl(3.3 mL,30.3 mmol,1.05 當量)。將反應混合物攪拌約30 min,及接著添加PhMgCl (在THF中之2 M;28 mL,56.8 mmol,1.97 當量),同時保持內溫低於5℃。將反應混合物在約0℃下攪拌約35 min,及接著冷卻至約-15℃。然後添加iPrMgCl (在THF中之2 M,14 mL,30.2 mmol,1.05 當量),同時保持內溫低於約-10℃。在約-15℃下約15分鐘之後,添加LaCl3
-2LiCl (在THF中之0.6 M,50 mL,14.4 mmol,0.5 當量),同時保持內溫低於約-15℃。將反應混合物在約-20℃下攪拌約25 min。
在獨立燒瓶中,製備(3R,4R,5R)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)二氫呋喃-2(3H)-酮 (10.0 g,23.9 mmol,0.83當量)在THF(45 mL)中之溶液。將溶液冷卻至約-20℃,及接著轉移至格任亞溶液,同時保持內溫低於約-15℃。將所得反應混合物在約-20℃下攪拌約30 min。
將反應用2 M HCl (53 mL)淬滅,並將混合物加熱至約15℃。添加iPrOAc (38 mL),及將有機相和水相分離。將下水層排出,及將上有機層依次用2.5 wt% NaHCO3
(53 mL)、2.5 wt% NaHCO3
(53 mL)、及10 wt% NaCl (53 mL)洗滌。
將有機相濃縮至約45 mL,及接著用iPrOAc(75 mL)稀釋。將溶液再濃縮至約45 mL,及接著用iPrOAc(23 mL)稀釋。將溶液濃縮至約45 mL,及接著經矽藻土墊過濾。將過濾溶液濃縮至約26 mL,及接著用MTBE (75 mL)稀釋。2h之後,慢慢添加庚烷(23 mL)並將漿料在約25℃下攪拌約2 h,及接著冷卻至約-5℃經約8 h。藉由過濾分離固體,及將濾餅用MTBE/庚烷(4:1, 23 mL)洗滌。將固體在真空烘箱中於不超過約35℃下乾燥以提供(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-醇。
使用CeCl3
之(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-醇的製備
將碘吡唑(5.02 g,19.3 mmol)溶解在THF(45 g)中並將溶液冷卻至約0℃同時攪拌。添加TMSCl(2.04 g,18.7 mmol),及約1 h之後,添加氯化苯鎂(在THF中之2.0 M,19.9 g,38.2 mmol)。將反應混合物冷卻至約 -20℃並慢慢添加氯化異丙鎂(在THF中之2.0 M,9.99 g,20.5 mmol)。約30 min之後,將反應混合物轉移至在約-20℃下的無水氯化鈰(4.75 g,19.3 mmol)在THF中(22 g)之混合物。約1.5 h之後,慢慢添加內酯(6.73 g,16.1 mmol)在THF中(22 g)之溶液,及將所得反應混合物攪拌約1 h。添加2 M HCl (41 g),將混合物加溫到約15℃,及添加乙酸異丙酯(35 g)。分離該等層並將有機層用2.5% NaHCO3
(2×40 g)、10% NaCl (1×35 g)洗滌且濃縮至約30 mL體積。進料乙酸異丙酯(44 g)並將溶液濃縮至約30 mL體積。進料乙酸異丙酯(43 g)並將溶液濃縮至約30 mL體積。將溶液過濾並將濾液濃縮至約18 mL體積。添加第三丁基甲基醚(37 g)接著產物種晶(10.7 mg)。約14 h之後,添加正庚烷(10.5 g)並將混合物冷卻至約-5℃和過濾。在約-5℃下將固體用第三丁基甲基醚(9 g)洗滌及在真空下於約34℃乾燥約15 h以提供產物。
使用CeCl3
和iPrMgCl-LiCl之(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-醇的製備
將碘吡唑(5.03 g,19.3 mmol)溶解在THF(45 g)中及將溶液冷卻至約0℃並在N2
(g)下攪拌。添加TMSCl(2.06 g,19.0 mmol),及約1 h之後添加氯化苯鎂(在THF中之2.0 M,20.23 g,38.8 mmol)。將反應混合物冷卻至約-20℃並慢慢添加氯化異丙鎂-氯化鋰錯合物(在THF中之2.0 M,15.37 g,21.0 mmol)。約1 h之後,在約-20℃下將反應混合物轉移至氯化鈰 (4.77 g,19.4 mmol)在THF(22 g)中之混合物。約1 h之後慢慢添加內酯(6.75 g,16.1 mmol)在THF(23 g)中之溶液,及將所得反應混合物攪拌約1.5 h。添加2 M HCl (40 g),將混合物加溫到約15℃及添加乙酸異丙酯(35 g)。分離該等層並將有機層用2.5% NaHCO3
(2×40 g)、10% NaCl (1×36 g)洗滌並濃縮至約30 mL體積。添加乙酸異丙酯(44 g)並將溶液濃縮至約30 mL體積。將溶液過濾並將濾液濃縮至約18 mL體積。添加第三丁基甲基醚(37 g)接著產物種晶(10.5 mg)。約14 h之後添加正庚烷(11 g)並將混合物冷卻至約 -5℃及過濾。將固體在約-5℃下用第三丁基甲基醚(9 g)洗滌並在真空下於約34℃乾燥約15 h以提供產物。
使用YCl3
之(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-醇的製備
將碘吡唑(4.99 g,19.2 mmol)溶解在THF(44 g)中並將溶液冷卻至約0℃同時攪拌。添加TMSCl(2.45 mL,19.4 mmol),及約30 min之後添加氯化苯鎂(在THF中之2.0 M,20.29 g,39.0 mmol)。將反應混合物冷卻至約-20℃並慢慢添加氯化異丙鎂(在THF中之2.0 M,9.85 g,20.1 mmol)。約30 min之後,將反應混合物轉入在約 -20℃下之無水氯化釔(3.76 g,19.3 mmol)和內酯(6.68 g,16.0 mml)在THF中(24 g)之混合物,約2.5 h之後添加2 M HCl (30 g),將混合物加溫到約15℃,及添加乙酸異丙酯(22 g)。分離該等層並將有機層用2.5% NaHCO3
(2×40 g)、10% NaCl (1×35 g)洗滌和濃縮至約30 mL體積。進料乙酸異丙酯(44 g)並將溶液濃縮至約30 mL體積。進料乙酸異丙酯(45 g)並將溶液濃縮至約30 mL體積。將溶液過濾並將濾液濃縮至約18 mL體積。添加第三丁基甲基醚(37 g)接著產物種晶(11.5 mg)。約1 h之後添加正庚烷(15 mL)並將混合物冷卻至約-5℃並攪拌約17 h。過濾漿料並將固體用預冷卻至約-5℃的第三丁基甲基醚(8 g)/正庚烷(2 g)混合物洗滌。將所得固體在真空下於約34℃乾燥約22 h以提供產物。
使用NdCl3
之(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-醇的製備
將碘吡唑(5.02 g,19.3 mmol)溶解在THF(38 g)中並將溶液在N2
(g)下冷卻至約0℃且攪拌。添加TMSCl(2.45 mL,19.4 mmol),及約1 h之後添加氯化苯鎂(在THF中之2.0 M,19.75 g,38.0 mmol)。將反應混合物冷卻至約-20℃並慢慢添加氯化異丙鎂(在THF中之2.0 M,9.40 g,19.2 mmol)。約1.5 h之後,將反應混合物轉入在約-20℃下的無水氯化釹(III)(4.03 g,16.1 mmol)和內酯(6.70 g,16.0 mml)在THF(22 g)中之混合物。約1.5 h之後將反應混合物加溫到-10℃,另外2 h之後,添加2M HCl(36 g)。將混合物加溫到約15℃並添加乙酸異丙酯(23 g)。分離該等層並將有機層用2.5% NaHCO3
(2×44 g)、10% NaCl (1×41 g)洗滌和濃縮至約30 mL體積。進料乙酸異丙酯(44 g)並將溶液濃縮至約30 mL體積。進料乙酸異丙酯(45 g)並將溶液濃縮至約30 mL體積。將溶液過濾並將濾液濃縮至約18 mL體積。添加第三丁基甲基醚(37 g)接著產物種晶(11.9 mg)。約1 h之後添加正庚烷(15 mL)並將混合物冷卻至約-5℃並攪拌約15 h。過濾漿料並將固體用預冷卻至約-5℃的第三丁基甲基醚(8 g)/正庚烷(11 g)混合物洗滌。將所得固體在真空下於約34℃乾燥約25 h以提供產物。
(2R,3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-甲腈的製備
將三氟乙酸(6.19克,54.3 mmols,3.0 當量),接著TMSOTf(24.1克,108.6 mmols,6.0 當量)和TMSCN (10.8克,108.6 mmols,6.0 當量)在DCM (50 mL)中之預冷卻(-30℃)之溶液進料至(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-醇(10.0克,18.1 mmols,1.0 當量)在DCM(100 mL)中之預冷卻溶液(-40℃)同時保持內溫低於約-25℃。將反應混合物在低於約-30℃下攪拌不少於10分鐘和淬滅於20 wt. % KOH之預冷卻(約-10℃)水溶液(120 mL)。將二相混合物加溫到周圍溫度。將有機層分離並用10 wt. % NaCl aq. (3×50 mL)洗滌。將有機相過濾,在真空下濃縮至約50 mL,用甲苯(200 mL)再稀釋並在真空下於約50℃濃縮至140 mL。將溶液在約55℃下用(2R,3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-甲腈種晶。在約55℃下攪拌約一小時並冷卻至約0℃經約6小時。固體藉由過濾分離及將濾餅用甲苯(30 mL)洗滌。將固體在真空下於約50℃乾燥。
經由流動化學量之(2R,3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-甲腈的製備
將(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-醇之溶液(23.0 g在 460.07 g的DCM中)、TMSOTf (55.81 g在138.07 g的DCM中)和TMSCN (25.03 g在 138.10 g 的DCM中)在約-40℃下依次泵進管式反應器。將反應混合物收集在保持於冰浴中且含有20% KOH水溶液(46.91 g KOH和210 g的水)之燒瓶中。分離該等層並將有機相用10% KOH水溶液(10 g KOH和90 mL的水)及用10%鹽水(2×100 g)依次洗滌。將有機相在真空下濃縮至約4體積,進料異丙醇 (162.89 g)並將混合物在真空下濃縮至約10體積。將內容物加溫到約60℃,接著調整至約0℃經約6.5 h並在約0℃下攪拌約15.5 h。過濾所得漿料,將固體用異丙醇(61.79 g)沖洗及接著在減壓下於約50℃乾燥過夜以提供產物。
(2R, 3R, 4S, 5R)-2-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈的製備
將三苯甲基氰基核苷(48.8 g,86.9 mmol,1.0 當量)溶解在無水CH2
Cl2
(244 mL)中並冷卻至約-20℃。滴加BCl3
之溶液(在CH2
Cl2
中之1M,295 mL,295 mmol,3.4 當量),將內溫保持低於約-15℃。添加之後,將反應混合物在約-20℃下攪拌1 h。滴加MeOH(340 ml),將內溫保持低於-15℃。將所得溶液蒸餾至約250 mL,接著再填充約250 ml MeOH。將所得溶液再蒸餾至約250 mL,接著再填充約250 ml MeOH,及最後蒸餾至約125 ml。添加水(125 ml),接著K2
CO3
溶液(在水中之20 wt%,125 ml)。檢查pH,及發現為~3。添加K2
CO3
溶液(在水中之20 wt%,50 ml),及發現pH為~8。將所得漿料攪拌過夜,接著過濾並用水(50 ml)和MeOH(50 ml)洗滌。濕產物餅於約40℃乾燥過夜。1
H NMR (300 MHz, D2
O) δ 7.96 (s, 1H), 7.20 (d, J = 4.8 Hz, 1H), 6.91 (d, J = 4.8 Hz, 1H), 4.97 (d, J = 4.4 Hz, 1H), 4.56-4.62 (m, 1H), 4.08-4.14 (m, 1H), 3.90 (dd, J = 12.9, 2.4 Hz, 1H), 3.70 (dd, J = 13.2, 4.5 Hz, 1H)。
實施例11. (2S)-2-((((2R,3S,4R,5R)-5-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)-磷醯基胺基)丙酸異丙酯(化合物8)
將核苷1(45mg, 0.15mmol)溶解在無水磷酸三甲酯(0.5 mL)中並在N2
(g)下於約0℃下攪拌溶液。將甲基咪唑(36 μL,0.45 mmol)加至溶液。將氯胺基磷酸酯(phosphoramidate) C (69 mg,0.225 mmol)溶解在無水THF(0.25 mL)中並加至核苷混合物。當藉由LCMS反應為完成時,將反應混合物用EtOAc稀釋並用飽和NaHCO3
水溶液、飽和NaCl洗滌,經無水Na2
SO4
乾燥,過濾並在減壓下濃縮。將殘餘物進行用在CH2
Cl2
中之0-5% MeOH溶析的矽膠層析法,接著製備級HPLC以產生產物。1
H NMR (300 MHz, CD3
OD) δ 7.95 (m, 1H), 7.31-6.97 (m, 7H), 4.94 (m, 1H), 4.78 (m, 1H), 4.43 (m, 3H), 4.20 (m, 1H), 3.80 (d, 1H), 1.30-1.18 (m, 9H)。31
P NMR (121.4 MHz, CD3
OD) δ 3.8。LCMS m/z 561.0 [M+H], 559.0 [M-H]。
實施例12. (2S)-2-((((2R,3S,4R,5R)-5-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基胺基)丙酸2-乙基丁酯(化合物9)
可藉由下述數種方法製備化合物9。
程序1
根據與化合物8的製備相同之方法從化合物1和氯化物B製備。1
H NMR (300 MHz, CD3
OD) δ 7.87 (m, 1H), 7.31-7.16 (m, 5H), 6.92-6.89 (m, 2H), 4.78 (m, 1H), 4.50-3.80 (m, 7H), 1.45-1.24 (m, 8H), 0.95-0.84 (m, 6H)。31
P NMR (121.4 MHz, CD3
OD) δ 3.7。LCMS m/z 603.1 [M+H], 601.0 [M-H]。
程序2
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯。將(2S)-2-(((4-硝基苯氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(1.08 g,2.4 mmol)溶解在無水DMF (9 mL)中並在氮氛圍下於RT攪拌。將(2R,3R,4S,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈(350 mg,1.2 mmol)以一整部分加至反應混合物。然後經約10分鐘將氯化三級丁鎂在THF中之溶液(1M,1.8 mL,1.8 mmol)滴加至反應。將反應攪拌約2 h,在此時將反應混合物用乙酸乙酯(50 mL)稀釋並用飽和碳酸氫鈉水溶液(3×15 mL)接著飽和氯化鈉水溶液(15 mL)洗滌。將有機層經無水硫酸鈉乾燥並在減壓下濃縮。用矽膠管柱層析法(在DCM中之0-10% MeOH)純化所得油以提供呈白色固體之(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(311 mg,43%,於磷之1:0.4非鏡像異構物的混合物)。1
H NMR (400 MHz, CD3
OD) δ 7.85 (m, 1H), 7.34 - 7.23 (m, 2H), 7.21 - 7.09 (m, 3H), 6.94 - 6.84 (m, 2H), 4.78 (d, J = 5.4 Hz, 1H), 4.46 - 4.33 (m, 2H), 4.33 - 4.24 (m, 1H), 4.18 (m, 1H), 4.05 - 3.80 (m, 3H), 1.52 - 1.39 (m, 1H), 1.38 - 1.20 (m, 7H), 0.85 (m, 6H)。31
P NMR (162 MHz, CD3
OD) δ 3.71, 3.65。LCMS m/z 603.1 [M+H], 600.9 [M-H]。HPLC (具有0.1%TFA改質劑之2-98% MeCN-H2
O梯度經8.5 min,1.5mL/min,管柱:Phenomenex Kinetex C18,2.6 um 100 Å,4.6×100 mm) tR
= 5.544 min,5.601 min
(S)和(R)非鏡像異構物的分離
將(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯溶解在乙腈中。將所得溶液裝載至Lux 纖維素-2手性管柱,在乙腈中平衡,及用等度乙腈/甲醇(95:5 vol/vol)溶析。第一溶析非鏡像異構物具有17.4 min之滯留時間,及第二溶析非鏡像異構物具有25.0 min之滯留時間。
第一溶析非鏡像異構物為(S)-2-(((R)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯: 1
HNMR (400 MHz, CD3
OD) δ 8.05 (s, 1H), 7.36 (d, J = 4.8 Hz, 1H), 7.29 (br t, J = 7.8 Hz, 2H), 7.19 - 7.13 (m, 3H), 7.11 (d, J = 4.8 Hz, 1H), 4.73 (d, J = 5.2 Hz, 1H), 4.48 - 4.38 (m, 2H), 4.37 - 4.28 (m, 1H), 4.17 (t, J = 5.6 Hz, 1H), 4.08 - 3.94 (m, 2H), 3.94 - 3.80 (m, 1H), 1.48 (sep, J = 12.0, 6.1 Hz, 1H), 1.34 (p, J = 7.3 Hz, 4H), 1.29 (d, J = 7.2 Hz, 3H), 0.87 (t, J = 7.4 Hz, 6H)。31
PNMR (162 MHz, CD3
OD) δ 3.71 (s)。HPLC (具有改質劑之2-98% MeCN-H2
O梯度0.1% TFA經 8.5 min,1.5mL/min,管柱:Phenomenex Kinetex C18,2.6 um 100 Å,4.6×100 mm) tR
= 5.585 min。
1
HNMR (400 MHz, CD3
OD) δ 8.05 (s, 1H), 7.36 (d, J = 4.8 Hz, 1H), 7.29 (br t, J = 7.8 Hz, 2H), 7.19 - 7.13 (m, 3H), 7.11 (d, J = 4.8 Hz, 1H), 4.73 (d, J = 5.2 Hz, 1H), 4.48 - 4.38 (m, 2H), 4.37 - 4.28 (m, 1H), 4.17 (t, J = 5.6 Hz, 1H), 4.08 - 3.94 (m, 2H), 3.94 - 3.80 (m, 1H), 1.48 (sep, J = 12.0, 6.1 Hz, 1H), 1.34 (p, J = 7.3 Hz, 4H), 1.29 (d, J = 7.2 Hz, 3H), 0.87 (t, J = 7.4 Hz, 6H)。31
PNMR (162 MHz, CD3
OD) δ 3.71 (s)。HPLC (具有改質劑之2-98% MeCN-H2
O梯度0.1% TFA經 8.5 min,1.5mL/min,管柱:Phenomenex Kinetex C18,2.6 um 100 Å,4.6×100 mm) tR
= 5.585 min。
第二溶析非鏡像異構物為(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯: 1
HNMR (400 MHz, CD3
OD) δ 8.08 (s, 1H), 7.36 - 7.28 (m, 3H), 7.23 - 7.14 (m, 3H), 7.08 (d, J = 4.8 Hz, 1H), 4.71 (d, J = 5.3 Hz, 1H), 4.45 - 4.34 (m, 2H), 4.32 - 4.24 (m, 1H), 4.14 (t, J = 5.8 Hz, 1H), 4.08 - 3.94 (m, 2H), 3.93 - 3.85 (m, 1H), 1.47 (sep, J = 6.2 Hz, 1H), 1.38 - 1.26 (m, 7H), 0.87 (t, J = 7.5 Hz, 6H)。31
PNMR (162 MHz, CD3
OD) δ 3.73 (s)。HPLC (具有0.1% TFA改質劑之2-98% MeCN-H2
O梯度經 8.5 min,1.5mL/min,管柱:Phenomenex Kinetex C18,2.6 um 100 Å,4.6×100 mm) tR
= 5.629 min。
實施例13. (2S)-2-((((2R,3S,4R,5R)-5-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基胺基)丙酸乙酯(化合物10)
1
HNMR (400 MHz, CD3
OD) δ 8.08 (s, 1H), 7.36 - 7.28 (m, 3H), 7.23 - 7.14 (m, 3H), 7.08 (d, J = 4.8 Hz, 1H), 4.71 (d, J = 5.3 Hz, 1H), 4.45 - 4.34 (m, 2H), 4.32 - 4.24 (m, 1H), 4.14 (t, J = 5.8 Hz, 1H), 4.08 - 3.94 (m, 2H), 3.93 - 3.85 (m, 1H), 1.47 (sep, J = 6.2 Hz, 1H), 1.38 - 1.26 (m, 7H), 0.87 (t, J = 7.5 Hz, 6H)。31
PNMR (162 MHz, CD3
OD) δ 3.73 (s)。HPLC (具有0.1% TFA改質劑之2-98% MeCN-H2
O梯度經 8.5 min,1.5mL/min,管柱:Phenomenex Kinetex C18,2.6 um 100 Å,4.6×100 mm) tR
= 5.629 min。
實施例13. (2S)-2-((((2R,3S,4R,5R)-5-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基胺基)丙酸乙酯(化合物10)
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸乙酯的製備係描述如下。
程序1. 經由氯化物A之製備
使用與製備化合物8相同的方法從化合物1和氯化物A製備。1
H NMR (300 MHz, CD3
OD) δ 7.95 (m, 1H), 7.32-6.97 (m, 7H), 4.78 (m, 1H), 4.43-4.08 (m, 6H), 3.83 (m, 1H), 1.31-1.18 (m, 6H)。31
P NMR (121.4 MHz, CD3
OD) δ 3.7。LCMS m/z 547.0 [M+H], 545.0 [M-H]。
程序2. 經由硝基-苯化合物L之製備
將化合物1 (50 mg,0.17 mmol)溶解在NMP-THF(1:1 mL))中並用冰浴冷卻。然後經約5 min添加tBuMgCl (0.257 mL,0.257 mmol)。使所得混合物加溫至RT並攪拌約30 min。接著添加化合物L (根據US20120009147製備,74.6 mg,0.189 mmol)在THF(2 mL)中之溶液。約30 min之後,藉由HPLC(在水中之乙腈10至80%)純化反應混合物以產生呈黃色固體之化合物29。用矽膠層析法(MeOH 0至20% DCM)進一步純化固體以提供化合物29。1
H NMR (400 MHz, CD3
OD) δ 7.76 (d, J = 6.0 Hz, 1H), 7.25 - 7.14 (m, 2H), 7.11 - 6.99 (m, 3H), 6.87 - 6.72 (m, 2H), 4.70 (d, J = 5.4 Hz, 1H), 4.39 - 4.24 (m, 2H), 4.20 (dddd, J = 9.7, 7.9, 5.1, 2.8 Hz, 1H), 4.10 (dt, J = 12.8, 5.5 Hz, 1H), 4.06 - 3.91 (m, 2H), 3.72 (ddq, J = 14.3, 9.3, 7.1 Hz, 1H), 1.17 (dd, J = 7.1, 1.0 Hz, 1H), 1.14 - 1.06 (m, 5H)。31
P NMR (162 MHz, CD3
OD) δ 3.73, 3.68。MS m/z = 547 (M+1)+
。
實施例15. (2S,2’S)-2,2’-((((2R,3S,4R,5R)-5-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)磷醯基)雙(脲二基)(azanediyl)二丙酸二乙酯(化合物12)
將核苷1(14.6 mg,0.05 mmol)溶解在無水磷酸三甲酯(0.5 mL)中並在N2
(g)下於RT攪拌。添加POCl3
(9.2 μL,0.1 mmol)並將混合物攪拌約60 min。添加丙胺酸乙酯鹽酸鹽(61 mg,0.4 mmol)及接著Et3
N (70 μL,0.5 mmol)。將所得混合物攪拌約15 min.及接著添加另外的Et3
N (70 μL,0.5 mmol)以產生9-10之溶液 pH。將混合物攪拌約2 h.及接著用EtOAc稀釋,用飽和NaHCO3
水溶液接著飽和NaCl水溶液洗滌。將有機層經無水Na2
SO4
乾燥並在減壓下濃縮。將殘餘物進行製備級 HPLC (C18
管柱)以產生產物12。1
H NMR (400 MHz, CD3
OD) δ 8.13 (s, 1H), 7.41 (d, J = 4.8 Hz, 1H), 7.18 (d, J = 4.8 Hz, 1H), 4.78 (d, J = 5.6 Hz, 1H), 4.36 (m, 1H), 4.25-4.08 (m, 7H), 3.83 (m, 2H), 1.33-1.23 (m, 12H)。31
P NMR (121.4 MHz, CD3
OD) δ 13.8。LCMS m/z 570.0 [M+H], 568.0 [M-H]。
實施例18. S,S’-2,2’-((((2R,3S,4R,5R)-5-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)磷醯基)雙(氧基)雙(乙烷-2,1-二基)雙(2,2-二甲基硫代丙酸酯(propanethioate))(化合物15)
將核苷1(0.028 g,0.096 mmol)溶解在磷酸三甲酯(1 mL)中。將反應在N2
(g)下攪拌及接著用1H-四唑(0.021 g,0.29 mmol)處理。將反應混合物冷卻至0℃並添加磷烷(Nucleoside Nucleotides, Nucleic acids; 14; 3-5; 1995; 763 - 766. Lefebvre, Isabelle; Pompon, Alain; Perigaud, Christian; Girardet, Jean-Luc; Gosselin, Gilles; 等人)(87 mg,0.192 mmol)。將反應攪拌2 h.及接著用30% 過氧化氫(0.120 mL)淬滅。將混合物在RT下攪拌30 min及接著用飽和硫代硫酸鈉水溶液(1 mL)處理。將混合物攪拌10 min.及接著在減壓下濃縮。將殘餘物進行製備級HPLC以分離標題產物15。1
H NMR (300 MHz, CD3
CN) δ 7.98 (s, 1H), 6.92 (d, 1H), 6.81 (d, 1H), 6.44 (bs, 2H), 4.82 (m, 2H), 4.47 (m, 1H), 4.24 (m, 2H), 4.00 (m, 4H), 3.80 (bs, 1H), 3.11 (m, 4H), 1.24 (s, 9H)。31
P NMR (121.4 MHz, CD3
CN) δ -1.85 (s)。LCMS m/z 661 [M+H]。
實施例20. 四氫三磷酸((2R, 3S, 4R, 5R)-5-(4-胺基吡咯並[1,2-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲酯(化合物17)
使用與化合物6的製備相似之程序從化合物1製備化合物17。將產物分離為鈉鹽。1
H NMR (400 MHz, D2
O) δ 7.76 (s, 1H), 6.88 (d, J = 4.8 Hz, 1H), 6.73 (d, J = 4.4 Hz, 1H), 4.86 (d, J = 5.2 Hz, 1H), 4.43 (m, 1H), 4.39 (m, 1H), 4.05 (m, 1H), 3.94 (m, 1H)。31
P NMR (121.4 MHz, D2
O) δ -5.4 (d, 1P), -10.8 (d, 1P), -21.1 (t, 1P)。LCMS m/z 530 [M-H], 531.9 [M+H] Tr = 0.22 min。HPLC離子交換Tr=9.95 min。
實施例24. (2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-3-苯基丙酸乙酯(21)
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-3-苯基丙酸乙酯的製備係描述如下。
(S)-2-胺基-3-苯基丙酸乙酯鹽酸鹽的製備。
將L-苯基丙胺酸(5 g,30 mmol)溶解在EtOH (30 mL)中。將TMSCl(6.915 mL,54 mmol)在RT下加至反應。將反應容器裝上回流冷凝器並將反應放入80℃浴中。將反應攪拌過夜。第二天將反應冷卻至RT,在減壓下濃縮並將所得殘餘物溶解在Et2
O中。將所得漿料過濾並將分離之固體用Et2
O進一步洗滌。將經洗滌之固體放置在高真空下以產生實施例(S)-2-胺基-3-苯基丙酸乙酯鹽酸鹽。1
H NMR (400 MHz, DMSO-d6
) δ 8.52 (s, 3H), 7.30 (m, 5H), 4.24 (ABX, JAX
= 7.8 Hz, JBX
= 6.2 Hz, 1H), 4.11 (m, 2H), 3.17, 3.05 (ABX, JAB
= -14 Hz, JBX
= 5.8 Hz, JAX
= 7.6 Hz, 2H), 1.09 (t, J =6.8 Hz, 3H)。
(2S)-2-(((4-硝基苯氧基)(苯氧基)磷醯基)胺基)-3-苯基丙酸乙酯(化合物D)的製備
將(S)-2-胺基-3-苯基丙酸乙酯鹽酸鹽(1.01 g,4.41 mmol)溶解在DCM(50 mL)中。將此溶液冷卻至約0℃及添加PhOP(O)Cl2
(0.656 mL,4.41 mmol),接著經5 min慢慢添加Et3
N(1.62 mL,11.5 mmol)。移除冷卻浴並使反應加溫至RT和攪拌80分鐘。添加對-NO2
PhOH(0.583 g,4.19 mmol),接著添加更多Et3
N (0.3 mL,2.1 mmol)。以LC/MS監控反應進展。一旦反應完成後,將其用Et2
O稀釋,並藉由過濾移除所得固體。將濾液濃縮並藉由矽膠管柱層析法(25 g乾負載筒,120 g 管柱;溶析液:100%己烷斜率變化至在己烷中之55% EtOAc)分離化合物D。1
H NMR (400 MHz, CD3
OD) δ 8.17 (m, 2H), 7.33 (m, 2H), 7.09-7.25 (m, 10H), 4.17 (m, 1H), 4.07 (m, 2H), 3.08 (m, 1H), 2.84 (m, 1H), 1.14 (m, 3H)。31
P NMR (162 MHz, DMSO-d6
) δ -1.479 (s), -1.719 (s)。MS m/z = 471.01 [M+1]。
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-3-苯基丙酸乙酯(化合物21)的製備
將化合物1 (0.030 g,0.103 mmol)溶解在DMF(1 mL)中及接著添加THF(0.5 mL)。以逐滴方式將t-BuMgCl (1M/THF,154.5 μL,0.154 μmol)加至反應並激烈攪拌。將所得白色漿料在RT下攪拌約30 min。將化合物D(0.058 g,0.124 mmol)在THF(1 mL)中之溶液在RT下以逐滴方式加至反應。以LC/MS監控反應進展。當反應進展至50%轉化時,將反應在冰浴中冷卻並用冰醋酸(70 μL)淬滅。將反應濃縮並藉由逆相HPLC從殘餘物分離出化合物21。1
H NMR (400 MHz, DMSO-d6
) δ 7.91 (d, J = 4 Hz, 1H), 7.90 (brs, 2H), 7.09-7.30 (m, 8H), 7.01, (t, J = 8.2 Hz, 2H), 6.89 (d, J = 4.4 Hz, 1H), 6.82 (t, J = 4.4 Hz, 1H), 6.27 (m, 1H), 6.14 (m, 1H), 5.34 (m, 1H), 4.62 (t, J = 5.6 Hz, 1H), 4.15 (m, 1H), 3.78-4.01 (m, 6H), 2.92 (m, 1H), 2.78 (m, 1H), 1.04 (m, 3H)。31
P NMR (162 MHz, DMSO-d6
) δ 3.69 (s), 3.34 (s)。MS m/z = 623.0 [M+H]。
實施例25. (2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-3-甲基丁酸乙酯(22)
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-3-甲基丁酸乙酯的製備係描述如下。
(2S)-3-甲基-2-(((4-硝基苯氧基)(苯氧基)磷醯基)胺基)丁酸乙酯(化合物E)的製備
將(S)-2-胺基-3-甲基丁酸乙酯(0.351 g,1.932 mmol)溶解在DCM(17 mL)中。將此溶液在冰浴中冷卻並添加PhOP(O)Cl2
(0.287 mL,1.932 mmol),接著經約5 min慢慢添加Et3
N(1.62 mL,11.4 mmol)。移除冷卻浴並使反應加溫至RT及攪拌經1h時段。添加對-NO2
PhOH(0.255 g,1.836 mmol),並以LC/MS監控反應進展。一旦反應完成後,將混合物用Et2
O稀釋,及以過濾移除所得固體。將濾液濃縮並藉由矽膠管柱層析法(12 g乾負載筒,80 g 管柱;溶析液:100%己烷斜率變化至在己烷中之55% EtOAc)分離化合物E。1
H NMR (400 MHz, DMSO-d6
) δ 8.30 (d, J = 9.2 Hz, 2H), 7.48 (t, J = 9.6 Hz, 2H), 7.40 (t, J = 7.8 Hz, 2H), 7.20-7.27 (m, 3H), 6.60 (quart, J = 11.6 Hz, 1H), 4.01 (m, 2H), 3.61 (m, 1H), 1.93 (m , 1H), 1.11 (m, 3H), 0.79 (m, 6H)。31
P NMR (162 MHz, DMSO-d6
) δ -0.342 (s), -0.578 (s)。MS m/z = 422.9 [M+H]。
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-3-甲基丁酸乙酯(化合物22)的製備
將化合物1 (0.040 g,0.137 mmol)溶解在NMP (1.5 mL)中及接著添加THF(0.25 mL)。將此溶液在冰浴中冷卻並以逐滴方式添加t-BuMgCl (1M/THF,425.7 μL,0.426 μmol)並激烈攪拌。移除冰浴並將所得白色漿料在RT下攪拌約15 min。將化合物E (0.081 g,0.192 mmol)在THF(0.5 mL)中之溶液在RT下以逐滴方式加至反應。以LC/MS監控反應進展。當反應進展至50%轉化時,將反應在冰浴中冷卻並用冰醋酸(70 μL)淬滅。將反應濃縮並藉由逆相HPLC從殘餘物半純化化合物22。藉由矽膠管柱層析法(12 g 乾負載筒,40 g 管柱;溶析液:100% EtOAc斜率變化至在EtOAc中之10% MeOH)進一步純化半純化之材料以產生化合物22。1
H NMR (400 MHz, DMSO-d6
) δ 7.91 (d, J = 1.6 Hz, 1H), 7.88 (brs, 2H), 7.32 (m, 2H), 7.15 (m, 3H), 6.90 (t, J = 4.2 Hz, 1H), 6.84 (d, J = 4.8 Hz, 1H), 6.26 (dd, J = 13.4, 6.2 Hz, 1H), 5.87 (quart. J = 11.2 Hz, 1H), 5.35 (m, 1H), 4.64 (m, 1H), 4.25 (m, 2H), 3.93-4.15 (m, 4H), 3.45 (m, 1H), 1.87 (m, 1H), 1.09-1.16 (m, 3H), 0.70-0.83 (m ,6H)。31
P NMR (162 MHz, DMSO-d6
) δ 4.59 (s), 4.47 (s)。MS m/z = 575.02 [M+H]。
實施例26. (S)-2-(((R)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸異丙酯(23)
(S)-2-(((R)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸異丙酯的製備係描述如下。
將化合物1 (60.0 mg,206 µmol)溶解在NMP(0.28 mL)中。在氬氛圍下於RT添加THF(0.2 mL)接著氯化第三丁鎂(在四氫呋喃中之1.0M溶液,0.309 mL)。20 min之後,添加化合物F(根據Cho, A. 等人 J. Med. Chem. 2014, 57, 1812-1825. 製備,81 mg,206 µmol)在THF(0.2 mL)中之溶液,及將所得混合物加溫到約50℃。3 h之後,使反應混合物冷卻至RT並以製備型HPLC (Phenominex Synergi 4u Hydro-RR 80Å 150×30 mm 管柱,5-100%乙腈/水梯度)直接炖化以提供化合物23。1
H NMR (400 MHz, CD3
OD) δ 7.86 (s, 1H), 7.34 - 7.26 (m, 2H), 7.21 - 7.12 (m, 3H), 6.91 (d, J = 4.6 Hz, 1H), 6.87 (d, J = 4.6 Hz, 1H), 4.92 (sept, J = 6.3 Hz, 1H), 4.80 (d, J = 5.4 Hz, 1H), 4.43 - 4.34 (m, 1H), 4.33 - 4.24 (m, 1H), 4.18 (t, J = 5.6 Hz, 1H), 3.82 (dq, J = 9.7, 7.1 Hz, 2H), 1.27 (dd, J = 7.1, 1.0 Hz, 3H), 1.18 (dd, J = 6.3, 4.8 Hz, 6H)。31
P NMR (162 MHz, CD3
OD) δ 3.72 (s)。LC/MS:tR
= 1.39 min,MS m/z = 561.11 [M+H];LC系統:Thermo Accela 1250 UHPLC;MS系統:Thermo LCQ Fleet;管柱:Kinetex 2.6µ XB-C18 100A,50×4.6 mm;溶劑:具有0.1%乙酸之ACN,具有0.1%乙酸之水;梯度:0 min-2.0 min 2-100% ACN,2.0 min-3.05 min 100% ACN,3.05 min-3.2 min 100%-2% ACN,3.2 min-3.5 min 2% ACN at 2µl/min。HPLC:tR
= 2.523 min;HPLC系統:Agilent 1100系列;管柱:Gemini 5µ C18 110A,50×4.6 mm;溶劑:具有0.1% TFA之ACN,具有0.1% TFA之水;梯度:0 min-5.0 min 2-98% ACN,5.0 min-6.0 min 98% ACN at 2 mL/min。
實施例27. (2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸環丁酯(24)
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸環丁酯的製備係描述如下。
(2S)-2-(((4-硝基苯氧基)(苯氧基)磷醯基)胺基)丙酸環丁酯(化合物G)的製備
將二氯磷酸苯酯(1.49 mL,10 mmol)溶解在10 mL的無水DCM中並在氮氛圍下在冰浴中攪拌。以一整部分添加L-丙胺酸異丁酯鹽酸鹽(0.9 g,5 mmol)。然後滴加三乙胺(765 µL,5.5 mmol)。將反應攪拌約1 h。滴加更多三乙胺(765 µL,5.5 mmol)並將反應攪拌約45 min。以一整部分添加對硝基酚(1.25g,9mmol)和並攪拌約30 min。添加三乙胺(765 µL,5.5 mmol)並將反應混合物攪拌約2 h。接著添加另外的對硝基酚(1.25g,9 mmol)和三乙胺(765 µL,5.5mmol),及將反應攪拌另一約2 h。將反應混合物在減壓下濃縮。將所得粗製物用EtOAc稀釋並用5%檸檬酸水溶液洗滌二次,接著用飽和氯化鈉水溶液洗滌。然後將有機層經無水硫酸鈉乾燥並在減壓下濃縮。將粗製殘餘物用矽膠管柱(在己烷中之0-20-50% EtOAc)純化以產生化合物G。1
H NMR (400 MHz, CD3
OD) δ 8.33 - 8.23 (m, 2H), 7.52 - 7.33 (m, 4H), 7.33 - 7.17 (m, 3H), 4.96 - 4.85 (m, 1H), 4.07 - 3.96 (m, 1H), 2.27 (m, 2H), 2.07 - 1.91 (m, 2H), 1.83 - 1.70 (m, 1H), 1.70 - 1.55 (m, 1H), 1.32 (m, 3H)。31
P NMR (162 MHz, CD3
OD) δ -1.36, -1.59。MS m/z = 420.9 [M+H]。
製備(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸環丁酯(化合物24)
將化合物1 (58 mg,0.2 mmol)與化合物G(101 mg,0.24 mmol)混合於2 mL的無水DMF中。以一整部分添加氯化鎂(42 mg,0.44 mmol)。將反應混合物加熱至約50℃。添加DIPEA(87 µL,0.5 mmol),及將反應在約50℃下攪拌約2 h。將反應混合物冷卻至室溫,用EtOAc稀釋並用5%檸檬酸水溶液接著飽和氯化鈉水溶液洗滌。然後將有機層經無水硫酸鈉乾燥並在減壓下濃縮。將粗製殘餘物用矽膠管柱(在DCM中之0-2-5% MeOH)純化以提供化合物24。1
H NMR (400 MHz, 甲醇-d4) δ 7.85 (m, 1H), 7.34 - 7.22 (m, 2H), 7.22 - 7.08 (m, 3H), 6.94 - 6.84 (m, 2H), 4.95 - 4.85 (m, 1H), 4.79 (m, 1H), 4.46 - 4.34 (m, 2H), 4.34 - 4.24 (m, 1H), 4.19 (m, 1H), 3.81 (m, 1H), 2.27 (m, 2H), 2.01 (m, 2H), 1.84 - 1.68 (m, 1H), 1.62 (m, 1H), 1.30 - 1.16 (m, 3H)。31
P NMR (162 MHz, cd3
od) δ 3.70, 3.65。MS m/z = 573.0 [M+H]。
實施例28. (2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-3-苯基丙酸異丙酯(25)
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-3-苯基丙酸異丙酯的製備係描述如下。
(2S)-2-(((4-硝基苯氧基)(苯氧基)磷醯基)胺基)-3-苯基丙酸異丙酯(化合物H)的製備
將二氯磷酸苯酯(718 µL,4.8 mmol)溶解在10 mL無水DCM中且在氮氛圍下在冰浴中攪拌。以一整個部分添加L-苯基丙胺酸異丙酯鹽酸鹽(1 g,4.1 mmol)。添加另一10 mL的無水DCM。滴加三乙胺(736 µL,5.3 mmol)並將反應混合物攪拌約30 min。然後滴加更多三乙胺(736 µL,5.3 mmol)並將反應混合物攪拌30 min。然後滴加另外的三乙胺(736 µL,5.3 mmol)並將反應混合物攪拌約15 min。然後添加對硝基酚(600 mg,4.32 mmol)。然後移除冰浴並使反應混合物加溫至室溫並攪拌約2 h。添加更多對硝基酚(50 mg)和三乙胺(736 µL,5.3 mmol)並將反應混合物攪拌約1 h。
然後將反應混合物在減壓下濃縮,及用EtOAc稀釋並用5%檸檬酸水溶液洗滌二次,接著用飽和氯化鈉水溶液洗滌。將有機層經無水硫酸鈉乾燥並在減壓下濃縮。將粗製物用矽膠管柱(在己烷中之0-15% EtOAc)純化以產生化合物H。1
H NMR (400 MHz, CDCl3
) δ 8.17 (m, 2H), 7.38 - 7.13 (m, 10H), 7.13 - 7.02 (m, 2H), 4.95 (m, 1H), 4.31 (m, 1H), 3.69 (m, 1H), 3.02 (dd, J = 6.1, 1.8 Hz, 2H), 1.21 - 1.08 (m, 6H)。31
P NMR (162 MHz, cdcl3) δ -2.96, -2.98。MS m/z = 485.0 [M+H]。
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-3-苯基丙酸異丙酯(化合物25)的製備
將化合物1 (58 mg,0.2 mmol)和化合物H(116 mg,0.24 mmol)混合及添加2 mL的無水DMF。在氮氛圍下於室溫攪拌反應混合物。經3分鐘滴加在THF(300 µL,0.3 mmol)中之1M tBuMgCl且然後將反應混合物攪拌約16 h。將反應混合物用EtOAc稀釋並用5%檸檬酸水溶液、飽和碳酸氫鈉水溶液及接著飽和氯化鈉水溶液洗滌。將有機層經無水硫酸鈉乾燥並在減壓下濃縮。將粗製殘餘物用矽膠管柱(在DCM中之0-5% MeOH)純化以產生化合物25。1
H NMR (400 MHz, CD3
OD) δ 7.84 (m, 1H), 7.27 - 7.08 (m, 8H), 7.08 - 6.97 (m, 2H), 6.88 (m, 2H), 4.91 - 4.84 (m, 1H), 4.74 (m, 1H), 4.26 (m, 1H), 4.19 - 4.04 (m, 2H), 4.04 - 3.91 (m, 2H), 2.97 (m, 1H), 2.82 (m, 1H), 1.14 (m, 3H), 1.06 (m, 3H)。31
P NMR (162 MHz, CD3
OD) δ 3.63, 3.25。MS m/z = 637.0 [M+H]。
實施例29. (S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸甲酯(26)
(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸甲酯的製備係描述如下。
將化合物1 (100 mg,0.34 mmol)溶解在THF(2 mL)中並用冰水浴冷卻。接著慢慢滴加1M t-BuMgCl (0.52 mL,0.77 mmol)。將所得混合物在室溫下攪拌約30 min。接著經5 min添加在THF(2 mL)中之化合物I (根據WO 2012142085製備,219 mg,0.52 mmol)並將所得混合物在室溫下攪拌約24 h。接著將反應混合物用EtOAc稀釋,在冰水浴下冷卻,用NaHCO3
(2 mL)水溶液洗滌,用鹽水洗滌,用硫酸鈉乾燥,並在真空中濃縮。將所得混合物藉由矽膠管柱層析法(在DCM中之MeOH 0至20%)和prep-HPLC(在水中之乙腈10至80%)純化以產生化合物26。1
H NMR (400 MHz, CD3
OD) δ 7.86 (s, 1H), 7.29 (dd, J = 8.6, 7.2 Hz, 2H), 7.21 - 7.09 (m, 3H), 6.94 - 6.81 (m, 2H), 4.79 (d, J = 5.4 Hz, 1H), 4.38 (ddq, J = 10.8, 5.3, 2.7 Hz, 2H), 4.33 - 4.23 (m, 1H), 4.18 (t, J = 5.5 Hz, 1H), 3.86 (dq, J = 9.9, 7.1 Hz, 1H), 3.62 (s, 3H), 1.27 (dd, J = 7.2, 1.1 Hz, 3H)。MS m/z = 533 (M+1)+
。
實施例30. (S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸新戊酯(27)
(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸新戊酯的製備係描述如下。
將化合物1 (100 mg,0.34 mmol)溶解在THF(2 mL)中並在冰水浴中冷卻。接著慢慢滴加1M t-BuMgCl (0.52 mL,0.77 mmol)。將所得混合物在室溫下攪拌約30 min。接著經約5 min添加化合物J(根據WO2012075140製備,248 mg,0.52 mmol)並將所得混合物在室溫下攪拌約24 h,用EtOAc稀釋,在冰水浴下冷卻,用NaHCO3
(2 mL)水溶液處理,用鹽水洗滌,用硫酸鈉乾燥,並在真空中濃縮。將所得混合物藉由矽膠管柱層析法(在DCM中之MeOH 0至20%)和prep-HPLC(在水中之乙腈10至80%)純化以產生化合物27。1
H NMR (400 MHz, CD3
OD) δ 7.86 (s, 1H), 7.36 - 7.24 (m, 2H), 7.23 - 7.10 (m, 3H), 6.96 - 6.85 (m, 2H), 4.78 (d, J = 5.4 Hz, 1H), 4.38 (tdd, J = 10.0, 4.9, 2.5 Hz, 2H), 4.32 - 4.24 (m, 1H), 4.17 (t, J = 5.6 Hz, 1H), 3.91 (dq, J = 9.8, 7.1 Hz, 1H), 3.81 (d, J = 10.5 Hz, 1H), 3.69 (d, J = 10.5 Hz, 1H), 1.31 (dd, J = 7.2, 1.1 Hz, 3H), 0.89 (s, 9H)。MS m/z = 589 (M+1)+
。
實施例31. (2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸環戊酯(28)
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸環戊酯的製備係描述如下。
將化合物1 (100 mg,0.34 mmol)溶解在THF(2 mL)中並在冰水浴中冷卻。接著慢慢滴加1M t-BuMgCl (0.52 mL,0.77 mmol)。將所得混合物在室溫下攪拌約30 min。接著經約5 min添加在THF(2 mL)之中化合物K(根據WO2012075140製備,247 mg,0.52 mmol)並將所得混合物在室溫下攪拌約24 h,用EtOAc稀釋,在冰水浴下冷卻,用NaHCO3
水溶液(2 mL)處理,用鹽水洗滌,用硫酸鈉乾燥,並在真空中濃縮。將所得混合物藉由矽膠管柱層析法(在DCM中之MeOH 0至20%)和prep-HPLC(在水中之乙腈10至80%)純化以產生實施例28。1
H NMR (400 MHz, CD3
OD) δ 7.85 (s, 1H), 7.33 - 7.22 (m, 2H), 7.14 (tdd, J = 7.6, 2.1, 1.1 Hz, 3H), 6.95 - 6.87 (m, 2H), 5.13 - 5.00 (m, 1H), 4.78 (d, J = 5.4 Hz, 1H), 4.48 - 4.35 (m, 2H), 4.30 (ddd, J = 10.6, 5.7, 3.6 Hz, 1H), 4.19 (t, J = 5.4 Hz, 1H), 3.78 (dq, J = 9.2, 7.1 Hz, 1H), 1.81 (dtd, J = 12.5, 5.9, 2.4 Hz, 2H), 1.74 - 1.49 (m, 6H), 1.21 (dd, J = 7.1, 1.2 Hz, 3H)。MS m/z = 587 (M+1)+
。
實施例32. (2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸環己酯(29)
經約5 min在約0℃下將二異丙基乙胺(0.075 mL,0.43 mmol)滴加至化合物1 (50 mg,0.343 mmol)、化合物M (根據US20130143835製備,93 mg,0.209 mmol)、和MgCl2
(24.5 mg,0.257 mmol)在DMF(1 mL)中之混合物。將所得混合物在約50℃下攪拌約1 h。接著將反應混合物用冰水浴冷卻,用1M檸檬酸(0.5 mL)處理,及直接藉由prep-HPLC(在水中之ACN 0至70%)純化以提供化合物29。1
H NMR (400 MHz, CD3
OD) δ 7.84 (s, 1H), 7.32 - 7.23 (m, 2H), 7.18 - 7.10 (m, 3H), 6.93 - 6.87 (m, 2H), 4.78 (d, J = 5.4 Hz, 1H), 4.67 (td, J = 8.7, 4.2 Hz, 1H), 4.48 - 4.35 (m, 2H), 4.30 (ddd, J = 10.8, 5.7, 3.7 Hz, 1H), 4.20 (t, J = 5.4 Hz, 1H), 3.88 - 3.71 (m, 1H), 1.83 - 1.63 (m, 4H), 1.58 - 1.46 (m, 1H), 1.46 - 1.24 (m, 5H), 1.24 (s, 3H)。31
P NMR (162 MHz, CD3
OD) δ 3.75。MS m/z = 601 (M+1)+
。
實施例33. 2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-2-甲基丙酸乙酯(30)
2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-2-甲基丙酸乙酯的製備係描述如下。
2-((第三丁氧羰基)胺基)-2-甲基丙酸乙酯的製備
將三苯膦(6.18 g,25.00 mmol)溶解在THF(30mL)中。接下來進料DIAD (4.92 mL,25.00 mmol)並在室溫下攪拌10 min。將2-((第三丁氧羰基)胺基)-2-甲基丙酸 (5.08 g,25.00 mmol)溶解在THF(20 mL)中並加至反應混合物,接著添加乙醇(2.19 mL,37.49 mmol)。使反應在室溫下攪拌約1 h。在減壓下移除溶劑並將粗製物溶解在1:1 Et2
O:己烷(120 mL)中。濾出固體氧化三苯膦並在減壓下移除溶劑。將粗製物溶解在最少CH2
Cl2
中和藉由矽膠層析法0-50% EtOAc/Hex純化以提供2-((第三丁氧羰基)胺基)-2-甲基丙酸乙酯。1
H NMR (400 MHz, 氯仿-d) δ 4.18 (q, J = 7.1 Hz, 2H), 1.49 (s, 6H), 1.43 (s, 9H), 1.27 (t, J = 7.1 Hz, 3H)。
2-胺基-2-甲基丙酸乙酯鹽酸鹽的製備
將2-((第三丁氧羰基)胺基)-2-甲基丙酸乙酯(2.71 g,11.72 mmol)溶解在CH2
Cl2
(25 mL)中並慢慢添加在二㗁烷(25 mmol)中之4N HCl並在室溫下攪拌。於1h時,藉由TLC測定反應為完成。在減壓下移除溶劑並將粗製物與乙醚一起蒸發兩次,接著放置在高真空下以提供2-胺基-2-甲基丙酸乙酯鹽酸鹽。1
H NMR (400 MHz, DMSO-d6
) δ 8.70 (s, 3H), 4.18 (q, J = 7.1 Hz, 2H), 1.46 (s, 6H), 1.21 (t, J = 7.1 Hz, 3H)。
2-甲基-2-(((4-硝基苯氧基)(苯氧基)磷醯基)胺基)丙酸乙酯(化合物N)的製備
將二氯磷酸苯酯(0.97mL,6.50mmol)和2-胺基-2-甲基丙酸乙酯鹽酸鹽(1.09 g,6.50 mmol)溶解在CH2
Cl2
(50 mL)中。將反應混合物冷卻至約0℃並慢慢添加TEA(1.75 mL,12.45 mmol)。移除冷卻浴和使反應混合物在室溫下攪拌。約2 h之後,以31
P NMR測定胺基酸之添加為完成。進料對硝基酚(0.860 g,6.17 mmol)接著添加TEA(0.87 g,7.69 mmol)。使反應在室溫下攪拌。約2 h之後,藉由LCMS測定反應為完成。將反應用Et2
O稀釋並濾出TEA•HCl鹽。將粗製物濃縮並藉由矽膠層析法(0-50% EtOAc/Hex)純化以提供化合物N。1
H NMR (400 MHz, DMSO-d6
) δ 8.37 - 8.21 (m, 2H), 7.55 - 7.44 (m, 2H), 7.43 - 7.33 (m, 2H), 7.30 - 7.09 (m, 3H), 6.57 (d, J = 10.1 Hz, 1H), 3.99 (q, J = 7.1 Hz, 2H), 1.39 (s, 6H), 1.08 (t, J = 7.1 Hz, 3H)。31
P NMR (162 MHz, DMSO-d6
) δ -2.87。LC/MS:tR
= 1.65 min,MS m/z = 408.97 [M+1];LC系統:Thermo Accela 1250 UHPLC;MS系統:Thermo LCQ Fleet;管柱:Kinetex 2.6µ XB-C18 100A,50×3.00 mm;溶劑:具有0.1%甲酸之乙腈,具有0.1%甲酸之水;梯度:0 min-2.4 min 2-100% ACN,2.4 min-2.80 min 100% ACN,2.8 min-2.85 min 100%-2% ACN,2.85 min-3.0 min 2% ACN於1.8mL/min。
2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-2-甲基丙酸乙酯(化合物30)的製備
將化合物1 (66 mg,0.23 mmol)溶解在NMP (2.0 mL)中。將混合物冷卻至約0℃並慢慢添加tBuMgCl (在THF中之1.0M,0.34 mL,0.34 mmol)。使反應在約0℃下攪拌約30 min,接著添加化合物N(139mg,0.34mmol)溶解在THF(1.0 mL)中之溶液。移除冷卻浴並將反應放進約50℃預熱油浴。約2 h之後,將反應冷卻至室溫並用乙酸和甲醇淬滅。將粗製物濃縮並藉由沒有改質劑之逆相HPLC純化以提供化合物30。1
H NMR (400 MHz, DMSO-d6
) δ 7.89 (m, 3H), 7.31 (q, J = 8.1 Hz, 2H), 7.22 - 7.05 (m, 3H), 6.87 (d, J = 4.5, 1H), 6.80 (d, J = 4.5 Hz, 1H), 6.27 (d, J = 11.7, 1H), 5.81 (d, J = 9.7, 1H), 5.35 (d, J = 5.6 Hz, 1H), 4.64 (dt, J = 9.0, 5.6 Hz, 1H), 4.24 (m, 2H), 4.11 (m, 1H), 4.04 - 3.90 (m, 3H), 1.39 - 1.23 (m, 6H), 1.10 (t, J = 7.1, 3H)。31
P NMR (162 MHz, DMSO-d6
) δ 2.45, 2.41。LC/MS:tR
= 1.03 min,MS m/z = 561.03 [M+1];LC系統:Thermo Accela 1250 UHPLC;MS系統:Thermo LCQ Fleet;管柱:Kinetex 2.6µ XB-C18 100A,50×3.00 mm;溶劑:具有0.1% 甲酸之乙腈,具有0.1%甲酸之水;梯度:0 min-2.4 min 2-100% ACN,2.4 min-2.80 min 100% ACN,2.8 min-2.85 min 100%-2% ACN,2.85 min-3.0 min 2% CAN於1.8mL/min。
實施例34. 2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-2-甲基丙酸異丙酯(31)
2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-2-甲基丙酸異丙酯的製備係描述如下。
2-((第三丁氧羰基)胺基)-2-甲基丙酸異丙酯的製備
將三苯膦(6.17 g,25.00 mmol)溶解在THF(30 mL)中。接下來進料DIAD(4.92 mL,25.00 mmol)並在室溫下攪拌約10 min。將2-((第三丁氧羰基)胺基)-2-甲基丙酸(5.07 g,25.00 mmol)溶解在THF(20mL)中並加至反應混合物,接著添加異丙醇(1.91 mL,25.00 mmol)。使反應在室溫下攪拌約1h。在減壓下移除溶劑並將粗製物溶解在1:1 Et2
O:己烷(120 mL)中。將固體氧化三苯膦濾出並在減壓下移除溶劑。將粗製物溶解在最少CH2
Cl2
中並藉由矽膠層析法(0-50% EtOAc/Hex)純化以提供2-((第三丁氧羰基)胺基)-2-甲基丙酸異丙酯。1
H NMR (400 MHz,氯仿-d) δ 5.03 (p, J = 6.2 Hz, 1H), 1.48 (s, 6H), 1.40 (d, J = 6.2 Hz, 9H), 1.24 (d, J = 6.3 Hz, 6H)。
2-胺基-2-甲基丙酸異丙酯鹽酸鹽的製備
將2-((第三丁氧羰基)胺基)-2-甲基丙酸異丙酯(4.09 g,16.67 mmol)溶解在CH2
Cl2
(50 mL)中並慢慢添加在二㗁烷(50 mmol)中之4N HCl及在室溫下攪拌。於約1 h時,藉由TLC測定反應為完成。在減壓下移除溶劑並將粗製物與乙醚一起蒸發兩次接著放置在高真空下以提供2-胺基-2-甲基丙酸異丙酯鹽酸鹽。1
H NMR (400 MHz, DMSO-d6
) δ 8.61 (s, 3H), 4.96 (p, J = 6.2 Hz, 1H), 1.44 (s, 6H), 1.22 (d, J = 6.2 Hz, 6H)。
2-甲基-2-(((4-硝基苯氧基)(苯氧基)磷醯基)胺基)丙酸異丙酯(化合物O)的製備
將二氯磷酸苯酯(0.83 mL,5.58 mmol)和2-胺基-2-甲基丙酸異丙酯鹽酸鹽(1.01 g,5.58 mmol)溶解在CH2
Cl2
(50 mL) 中。將反應混合物冷卻至0℃並慢慢添加TEA(1.61 mL,11.45 mmol)。移除冷卻浴並使反應混合物在室溫下攪拌。約2 h之後,藉由31
P NMR測定胺基酸之添加為完成。進料對硝基酚(0.74 g,5.30 mmol)接著添加TEA (0.81,5.84 mmol)。使反應在室溫下攪拌。約2 h之後,藉由LCMS測定反應為完成。用Et2
O稀釋反應和濾出TEA•HCl鹽。將粗製物濃縮並藉由矽膠層析法(0-50% EtOAc/Hex)純化以提供化合物O。1
H NMR (400 MHz, DMSO-d6
) δ 8.42 - 8.19 (m, 2H), 7.55 - 7.43 (m, 2H), 7.39 (dd, J = 8.6, 7.2 Hz, 2H), 7.30 - 7.12 (m, 3H), 6.53 (d, J = 10.1 Hz, 1H), 4.82 (hept, J = 6.3 Hz, 1H), 1.38 (s, 6H), 1.09 (d, J = 6.3, 6H)。31
P NMR (162 MHz, DMSO-d6
) δ -2.84。LC/MS:tR
= 1.73 min,MS m/z = 422.92 [M+1];LC系統:Thermo Accela 1250 UHPLC;MS系統:Thermo LCQ Fleet;管柱:Kinetex 2.6µ XB-C18 100A,50×3.00 mm;溶劑:具有0.1%甲酸之乙腈,具有0.1%甲酸之水;梯度:0 min-2.4 min 2-100% ACN,2.4 min-2.80 min 100% ACN,2.8 min-2.85 min 100%-2% ACN,2.85 min-3.0 min 2% ACN於1.8mL/min。
2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)-2-甲基丙酸異丙酯(化合物31)的製備
將化合物1 (66 mg,0.23 mmol)溶解在NMP (2.0 mL)中。將混合物冷卻至約0℃並慢慢添加tBuMgCl(在THF中之1.0M,0.57mL,0.57mmol)。使反應在約0℃下攪拌約30 min,接著添加化合物O(143 mg,0.34 mmol)溶解在THF(1.0 mL)中之溶液。移除冷卻浴並將反應放入約50℃預熱油浴。約2 h之後,將反應冷卻至室溫並用乙酸和甲醇淬滅。將粗製物濃縮且藉由無改質劑之改逆相HPLC純化以提供化合物31。1
H NMR (400 MHz, DMSO-d6
) δ 7.88 (m, 3H), 7.30 (td, J = 8.5, 7.0 Hz, 2H), 7.20 - 7.04 (m, 3H), 6.87 (d, J = 4.5, 1H), 6.80 (d, J = 4.5 Hz, 1H), 6.27 (d, 6.1 Hz, 1H), 5.75 (t, J = 9.1 Hz, 1H), 5.34 (d, J = 5.7 Hz, 1H), 4.81 (p, J = 6.3 Hz, 1H), 4.71 - 4.50 (m, 1H), 4.23 (m, 2H), 4.11 (m, 1H), 4.03 - 3.83 (m, 1H), 1.37 - 1.23 (m, 6H), 1.18 - 1.04 (m, 6H)。31
P NMR (162 MHz, DMSO) δ 2.47, 2.43。LC/MS:tR
= 1.08 min,MS m/z = 575.06 [M+1];LC系統:Thermo Accela 1250 UHPLC;MS系統:Thermo LCQ Fleet;管柱:Kinetex 2.6µ XB-C18 100A,50×3.00 mm;溶劑:具有0.1%甲酸之乙腈,具有0.1%甲酸之水;梯度:0 min-2.4 min 2-100% ACN,2.4 min-2.80 min 100% ACN,2.8 min-2.85 min 100%-2% ACN,2.85 min-3.0 min 2% ACN於1.8mL/min。
實施例35. (S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(32)
(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯的製備係描述如下。
(3R,4R,5R)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)二氫呋喃-2(3H)-酮的製備。
將(3R,4R,5R)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-醇(15.0 g)與MTBE (60.0 mL)、KBr (424.5 mg)、K2
HPO4
水溶液(2.5M,14.3 mL)及TEMPO(56 mg)組合。將此混合物冷卻至約1℃。分批慢慢進料漂白水溶液(7.9%wt.)直到經由澱粉/碘化物試驗指示起始材料完全消耗為止。分離該等層,及用MTBE萃取水層。將合併的有機相經MgSO4
乾燥並在減壓下濃縮以產生呈固體之產物。
製備(4-胺基-7-碘吡咯並[2,1-f][1,2,4]三𠯤)
將N-碘琥珀醯亞胺(17.01g;75.6 mmol)分批進料至4-胺基吡咯並[2,1-f][1,2,4]-三𠯤(10.03 g;74.8 mmol)在N,N-二甲基甲醯胺(70.27 g)中之冷溶液,同時將內容物保持在約0℃下。一旦反應完全完成(在約0℃下約3 h),將反應混合物轉入1 M 氫氧化鈉水溶液(11 g NaOH和276 mL 水),同時將內容物保持在約20-30℃下。將所得漿料在約22℃下攪拌1.5 h及接著過濾。用水(50 mL)沖洗固體並在真空下於約50℃乾燥以產生呈固體之4-胺基-7-碘吡咯並[2,1-f] [1,2,4]三𠯤。1
H NMR (400 MHz, DMSO-d6) δ 7.90 (s, 1H), 7.78 (br s, 2H), 6.98 (d, J = 4.4 Hz, 1H), 6.82 (d, J = 4.4 Hz, 1H)。13
C NMR (101 MHz, DMSO-d6) δ 155.7, 149.1, 118.8, 118.1, 104.4, 71.9。MS m/z = 260.97 [M+H]。
經由(4-胺基-7-碘吡咯並[2,1-f] [1,2,4]三𠯤)製備(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙(苯甲氧基)-5-((苯甲氧基)甲基)四氫呋喃-2-醇
在氮氛圍下將碘鹼2 (81 g)和THF(1.6 L)進料至反應器。將所得溶液冷卻至約5℃,及進料TMSCl(68 g)。然後慢慢進料PhMgCl (345mL,在THF中之1.8 M)同時將內溫保持在約≤ 5℃下。將反應混合物在約0℃下攪拌30 min,及接著冷卻至約-15℃。慢慢進料iPrMgCl-LiCl (311 mL,在THF中之1.1 M),同時保持內溫低於約-12℃。在約-15℃下攪拌約10分鐘之後,將反應混合物冷卻至約-20℃,及進料內酯1 (130 g)在THF(400 mL)中之溶液。接著將反應混合物在約-20℃下攪拌約1 h並用AcOH (57 mL)淬滅。將反應混合物加溫到約0℃並用NaHCO3
水溶液(5 wt%,1300 mL)調整至pH 7-8。接著將反應混合物用EtOAc(1300 mL)稀釋,及分離有機層和水層。將有機層用1N HCl (1300 mL)、NaHCO3
水溶液
(5 wt%,1300 mL)和鹽水(1300 mL)洗滌,及接著經無水Na2
SO4
乾燥並濃縮至乾燥。藉由使用由MeOH和EtOAc之混合物組成的梯度之矽膠管柱層析法的純化提供產物。
((2S)-2-(((全氟苯氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯)(Sp和Rp的混合物)製備:
將L-丙胺酸2-乙基丁酯鹽酸鹽(5.0 g,23.84 mmol)與二氯甲烷(40 mL)組合,冷卻至約-78℃,及添加二氯磷酸苯酯(3.65 mL,23.84 mmol)。在約-78℃下經約60 min添加三乙胺(6.6 mL,47.68 mmol)並將所得混合物在周圍溫度下攪拌3h。將反應混合物冷卻至約0℃並添加五氟酚(4.4 g,23.84 mmol)。經約60 min添加三乙胺(3.3 mL,23.84 mmol)。將混合物在周圍溫度下攪拌約3h 並在減壓下濃縮。將殘餘物溶解在EtOAc中,用碳酸鈉水溶液洗滌幾次,並在減壓下濃縮。藉由使用EtOAc和己烷之梯度(0至30%)的矽膠管柱層析法純化殘餘物。在減壓下濃縮含產物的部分以產生呈固體之(2S)-2-(((全氟苯氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯。1
H NMR (400 MHz, 氯仿-d) δ 7.41 - 7.32 (m, 4H), 7.30 - 7.17 (m, 6H), 4.24 - 4.16 (m, 1H), 4.13 - 4.03 (m, 4H), 4.01 - 3.89 (m, 1H), 1.59 - 1.42 (m, 8H), 1.40 - 1.31 (m, 8H), 0.88 (t, J = 7.5 Hz, 12H)。31
P NMR (162 MHz, 氯仿-d) δ -1.52。19
F NMR (377 MHz, 氯仿-d) δ -153.63, -153.93 (m), -160.05 (td, J = 21.9, 3.6 Hz), -162.65 (qd, J = 22.4, 20.5, 4.5 Hz)。MS m/z = 496 [M+H]。
((2S)-2-(((全氟苯氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯)製備: 將L-丙胺酸-2-乙基丁酯鹽酸鹽(40.10 g,0.191 mmol)溶解在二氯甲烷(533 g)中及將溶液在N2
(g)下冷卻並攪拌至約-15℃。添加二氯磷酸苯酯(40.32 g,0.191 mol),接著慢慢添加三乙胺(41.58 g,0.411 mmol)並將反應混合物在約-15℃下攪拌約1.5 h。添加五氟酚(35.14 g,0.191 mol),接著三乙胺(19.23 g,0.190 mol)並將反應混合物攪拌約2 h。將反應混合物加溫到約0℃和添加0.5 M HCl (279.19 g)。將混合物加溫到約22℃並將有機層分離並用5% KHCO3
水溶液(281 g),接著水(281 g)洗滌。將一等份的有機層(453.10 g之604.30 g 溶液)濃縮至約120 mL體積,添加乙酸異丙酯(157 g)並將溶液濃縮至乾燥。將殘餘物溶解在乙酸異丙酯(158 g)中。將所得溶液濃縮至約120 mL體積及將溫度調整至約45℃,添加正庚烷(165 g)並將混合物冷卻至22℃經約1 h。添加正庚烷(167 g)並將混合物冷卻至約0℃。添加三乙胺(2.90 g,0.0287 mol)並將混合物在0℃下攪拌約17 h。將混合物過濾,用正庚烷(145 g)沖洗固體並在真空下於約40℃乾燥固體經約15 h以提供((S)-(五氟苯氧基)(苯氧基)磷醯基)-L-丙胺酸2-乙基丁酯。
((S)-(4-硝基苯氧基)(苯氧基)磷醯基)-L-丙胺酸2-乙基丁酯製備:
將L-丙胺酸-2-乙基丁酯鹽酸鹽(40.10 g,0.191 mmol)溶解在二氯甲烷(533 g)中及將溶液在N2
(g)下冷卻並攪拌至約-15℃。添加二氯磷酸苯酯(40.32 g,0.191 mol),接著慢慢添加三乙胺(41.58 g,0.411 mmol)並將反應混合物在約-15℃下攪拌約1.5 h。添加五氟酚(35.14 g,0.191 mol),接著三乙胺(19.23 g,0.190 mol)並將反應混合物攪拌約2 h。將反應混合物加溫到約0℃和添加0.5 M HCl (279.19 g)。將混合物加溫到約22℃並將有機層分離並用5% KHCO3
水溶液(281 g),接著水(281 g)洗滌。將一等份的有機層(453.10 g之604.30 g 溶液)濃縮至約120 mL體積,添加乙酸異丙酯(157 g)並將溶液濃縮至乾燥。將殘餘物溶解在乙酸異丙酯(158 g)中。將所得溶液濃縮至約120 mL體積及將溫度調整至約45℃,添加正庚烷(165 g)並將混合物冷卻至22℃經約1 h。添加正庚烷(167 g)並將混合物冷卻至約0℃。添加三乙胺(2.90 g,0.0287 mol)並將混合物在0℃下攪拌約17 h。將混合物過濾,用正庚烷(145 g)沖洗固體並在真空下於約40℃乾燥固體經約15 h以提供((S)-(五氟苯氧基)(苯氧基)磷醯基)-L-丙胺酸2-乙基丁酯。
((S)-(4-硝基苯氧基)(苯氧基)磷醯基)-L-丙胺酸2-乙基丁酯製備: 將L-丙胺酸-2-乙基丁酯鹽酸鹽(20.08 g,95.8 mmol)和乙酸異丙酯(174 g)之漿料冷卻並攪拌至約-20℃。添加二氯磷酸苯酯(20.37 g,96.5 mmol),接著慢慢添加三乙胺(20.97 g,207.2 mmol)並將混合物在約-20℃下攪拌約1 h。添加4-硝基酚(13.23 g,95.1 mmol),接著慢慢添加三乙胺(10.01 g,98.8 mmol)並將反應混合物攪拌約1.5 h。將反應混合物加溫到約0℃及添加0.5 M HCl (140 g)。將有機層分離並用5% Na2
CO3
(2×100 g)和10% NaCl (2×100 g)洗滌。接著將有機層濃縮至約80 mL體積及添加乙酸異丙酯(4 g),接著正庚烷(110 g)。添加產物種晶(0.100 g)接著第二部分的正庚烷(110 g)並將混合物冷卻至約0℃。添加1,8-二氮雜雙環十一-7-烯(1.49 g,9.79 mmol)並將混合物在約0℃下攪拌約21 h。將所得固體過濾及先用正庚烷(61 g)洗滌並接著用H2
O (2×100 g)洗滌。將固體與H2
O (200 g)一起攪拌約1.5 h,過濾,並用H2
O (3×100 g),接著正庚烷(61 g)沖洗。將所得固體在真空下於約40℃乾燥約19 h 以提供((S)-(4-硝基苯氧基)(苯氧基)磷醯基)-L-丙胺酸2-乙基丁酯。
標題化合物(Sp和Rp的混合物)的製備:
將L-丙胺酸-2-乙基丁酯鹽酸鹽(20.08 g,95.8 mmol)和乙酸異丙酯(174 g)之漿料冷卻並攪拌至約-20℃。添加二氯磷酸苯酯(20.37 g,96.5 mmol),接著慢慢添加三乙胺(20.97 g,207.2 mmol)並將混合物在約-20℃下攪拌約1 h。添加4-硝基酚(13.23 g,95.1 mmol),接著慢慢添加三乙胺(10.01 g,98.8 mmol)並將反應混合物攪拌約1.5 h。將反應混合物加溫到約0℃及添加0.5 M HCl (140 g)。將有機層分離並用5% Na2
CO3
(2×100 g)和10% NaCl (2×100 g)洗滌。接著將有機層濃縮至約80 mL體積及添加乙酸異丙酯(4 g),接著正庚烷(110 g)。添加產物種晶(0.100 g)接著第二部分的正庚烷(110 g)並將混合物冷卻至約0℃。添加1,8-二氮雜雙環十一-7-烯(1.49 g,9.79 mmol)並將混合物在約0℃下攪拌約21 h。將所得固體過濾及先用正庚烷(61 g)洗滌並接著用H2
O (2×100 g)洗滌。將固體與H2
O (200 g)一起攪拌約1.5 h,過濾,並用H2
O (3×100 g),接著正庚烷(61 g)沖洗。將所得固體在真空下於約40℃乾燥約19 h 以提供((S)-(4-硝基苯氧基)(苯氧基)磷醯基)-L-丙胺酸2-乙基丁酯。
標題化合物(Sp和Rp的混合物)的製備:
在周圍溫度下組合核苷(29 mg,0.1 mmol)和膦醯胺(60 mg,0.12 mmol)及N,N-二甲基甲醯胺(2 mL)。慢慢添加氯化第三丁鎂(在THF中之1M,0.15 mL)。約1h之後,將反應用乙酸乙酯稀釋,用檸檬酸水溶液(5%wt.)、飽和NaHCO3
水溶液和飽和鹽水溶液洗滌。將有機相經Na2
SO4
乾燥並在減壓下濃縮。藉由使用甲醇和CH2
Cl2
(0至5%)之梯度的矽膠管柱層析法純化殘餘物。在減壓下濃縮含產物的部分以提供產物。
(3aR,4R,6R,6aR)-4-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-6-(羥甲基)-2,2-二甲基四氫呋喃並[3,4-d][1,3]二氧呃-4-甲腈的製備:
在周圍溫度下將硫酸(18M,1.44 mL)加至(2R,3R,4S,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈(5.8g,0.02 mol)、2,2-二甲氧基丙烷(11.59 mL,0.09 mol)和丙酮(145 mL)之混合物。將混合物加溫到約45℃。約30 min之後,將混合物冷卻至周圍溫度並添加碳酸氫鈉(5.8 g)和水(5.8 mL)。15 min之後,將混合物在減壓下濃縮。殘餘物溶解在乙酸乙酯(150 mL)和水(50 mL)中。用乙酸乙酯(2×50 mL)萃取水層。將合併的有機相經硫酸鈉乾燥並在減壓下濃縮以產生粗製(2R,3R,4S,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈。1
H NMR (400 MHz, CD3
OD) δ 7.84 (s, 1H), 6.93 (d, J = 4.6 Hz, 1H), 6.89 (d, J = 4.6 Hz, 1H), 5.40 (d, J = 6.7 Hz, 1H), 5.00 (dd, J = 6.7, 3.3 Hz, 1H), 4.48 - 4.40 (m, 1H), 3.81 - 3.72 (m, 2H), 1.71 (s, 3H), 1.40 (s, 3H)。MS m/z = 332.23 [M+1]。
(3aR,4R,6R,6aR)-4-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-6-(羥甲基)-2,2-二甲基四氫呋喃並[3,4-d][1,3]二氧呃-4-甲腈 TsOH鹽的製備:
在周圍溫度下將對甲苯磺酸(3.59 g,1.1 當量)加至(2R,3R,4S,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈(5.0 g,17.2 mmol,1.0 當量)、2,2-二甲氧基丙烷(10.5 mL,86 mmol,5.0 當量)和丙酮(25 mL)之混合物。在周圍溫度下混合物攪拌。約30 min之後,經約一小時添加乙酸異丙酯(25 mL)。將所得漿料過濾並用2:1 庚烷:乙酸異丙酯(25 ml)沖洗。將產物在真空下於約40℃乾燥。
(3aR,4R,6R,6aR)-4-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-6-(羥甲基)-2,2-二甲基四氫呋喃並[3,4-d][1,3]二氧呃-4-甲腈的製備:
在周圍溫度下將對甲苯磺酸(3.59 g,1.1 當量)加至(2R,3R,4S,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈(5 g,17.2 mmol,1.0 當量)、2,2-二甲氧基丙烷(10.5 mL,86 mmol,5.0 當量)和丙酮(25 mL)之混合物。在周圍溫度下混合物攪拌。30 min之後,經一小時添加乙酸異丙酯(25 mL)。將所得漿料過濾並用2:1 庚烷:乙酸異丙酯(25 ml)沖洗。將產物在真空下於約40℃乾燥。將分離之固體加至反應器和添加5% K2
CO3
溶液(50 ml)和乙酸乙酯(50 mL)。分離該等層,及用乙酸乙酯(25 ml)洗滌水層。將合併有機層用(25 ml)水洗滌,接著濃縮至約25 ml。將反應器再填充乙酸異丙酯(25 ml)並濃縮至約25 ml。將反應器再次填充乙酸異丙酯(25 ml)並濃縮至25 ml。將所得溶液種晶,產生稠漿料。經一小時對此添加庚烷(25 ml)。將所得漿料過濾並用2:1 庚烷:乙酸異丙酯(25 ml)沖洗。將產物在真空下於40℃乾燥。() (2R,3R,4S,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈。1
H NMR (400 MHz, CD3
OD) δ 7.84 (s, 1H), 6.93 (d, J = 4.6 Hz, 1H), 6.89 (d, J = 4.6 Hz, 1H), 5.40 (d, J = 6.7 Hz, 1H), 5.00 (dd, J = 6.7, 3.3 Hz, 1H), 4.48 - 4.40 (m, 1H), 3.81 - 3.72 (m, 2H), 1.71 (s, 3H), 1.40 (s, 3H)。MS m/z = 332.23 [M+1]。
(2S)-2-(((((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯的製備:
在周圍溫度下將乙腈(100 mL)與(2S)-2-(((4-硝基苯氧基)(苯氧基)磷醯基)-胺基)丙酸2-乙基丁酯(9.6 g,21.31 mmol)、反應物醇(6.6 g,0.02 mol)、氯化鎂(1.9 g,19.91 mmol)組合。將混合物攪拌約15 min並添加N,N-二異丙基乙胺(8.67 mL,49.78 mmol)。約4h之後,將反應用乙酸乙酯(100 mL)稀釋,冷卻至約0℃且與檸檬酸水溶液 (5%wt.,100 mL)組合。將有機相用檸檬酸水溶液(5%wt.,100 mL)和飽和水氯化銨溶液(40 mL)、碳酸鉀水溶液(10%wt.,2×100 mL),及飽和鹽水溶液(100 mL)洗滌。將有機相用硫酸鈉乾燥並在減壓下濃縮以提供粗製產物。1
H NMR (400 MHz, CD3
OD) δ 7.86 (s, 1H), 7.31 - 7.22 (m, 2H), 7.17 - 7.09 (m, 3H), 6.93 - 6.84 (m, 2H), 5.34 (d, J = 6.7 Hz, 1H), 4.98 (dd, J = 6.6, 3.5 Hz, 1H), 4.59 - 4.50 (m, 1H), 4.36 - 4.22 (m, 2H), 4.02 (dd, J = 10.9, 5.7 Hz, 1H), 3.91 (dd, J = 10.9, 5.7 Hz, 1H), 3.83 (dq, J = 9.7, 7.1 Hz, 1H), 1.70 (s, 3H), 1.50 - 1.41 (m, 1H), 1.39 (s, 3H), 1.36 - 1.21 (m, 7H), 0.86 (t, J = 7.4 Hz, 6H)。MS m/z = 643.21 [M+1]。
(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(化合物32)的製備
將粗製丙酮化合物(12.85 g)與四氫呋喃(50 mL)組合並在減壓下濃縮。將殘餘物溶解在四氫呋喃(100 mL)中,冷卻至約0℃並慢慢添加濃HCl (20 mL)。使混合物加溫至周圍溫度。藉由HPLC分析指示起始丙酮化合物的消耗之後,添加水(100 mL)接著飽和碳酸氫鈉水溶液(200 mL)。將混合物用乙酸乙酯(100 mL)萃取,將有機相用飽和鹽水溶液(50 mL)洗滌,經硫酸鈉乾燥並在減壓下濃縮。將殘餘物藉由使用甲醇和乙酸乙酯(0至20%)之梯度的矽膠管柱層析法純化。將含產物的部分在減壓下濃縮以提供產物。
(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(化合物32)的製備 將80%甲酸水溶液(1.5 mL)加至含有(S)- 2-(((S)-(((3aR,4R,6R,6aR)-6-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-6-氰基-2,2-二甲基四氫呋喃並[3,4-d][1,3]二氧呃-4-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(30 mg,0.05 mmol)之小瓶。在約20℃下18 h之後用HPLC和LC-MS確認完全轉化。MS (m/z) = 603(M+1)+
。
經由直接偶合之(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(化合物32)的製備
將80%甲酸水溶液(1.5 mL)加至含有(S)- 2-(((S)-(((3aR,4R,6R,6aR)-6-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-6-氰基-2,2-二甲基四氫呋喃並[3,4-d][1,3]二氧呃-4-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(30 mg,0.05 mmol)之小瓶。在約20℃下18 h之後用HPLC和LC-MS確認完全轉化。MS (m/z) = 603(M+1)+
。
經由直接偶合之(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(化合物32)的製備
將N,N-二甲基乙醯胺(10 mL)進料至(2R,3R,4S,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈(0.5 g,2mmol)、(S)-2-(((S)-(4-硝基苯氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(0.9 g,2 mmol)和MgCl2
(0.2 g,2 mmol)之混合物。將所得混合物加溫到約30℃並恆定攪拌。然後慢慢添加N,N-二異丙基乙胺(0.7 mL,4 mmol),及將反應混合物攪拌約6 h。將水(10 mL)進料H2
O,接著2-MeTHF(10 mL),及將有機相和水相分離。然後將水層用2-MeTHF(10 mL)回萃取。將有機層合併,及用10 wt%檸檬酸溶液(10 mL)、接著10 wt% K2
CO3
溶液(10 mL),及H2
O(10 mL) 洗滌。在分離該等層之前添加少量的鹽水以將乳液溶解在水洗液中。將有機層蒸發至乾燥以提供0.65 g泡沫。添加iPrOAc (2.6 mL),及將混合物加溫到約40℃以達成溶解。將溶液冷卻至約20℃,及將混合物攪拌約3天。藉由過濾分離固體,及將濾餅用少量的iPrOAc洗滌。乾燥固體 以提供(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯。
將N,N-二甲基乙醯胺(4 mL)進料至(2R,3R,4S,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-二羥基-5-(羥甲基)四氫呋喃-2-甲腈(0.2 g,0.7 mmol)、(S)-2-(((S)-(全氟苯氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(0.3 g,0.7 mmol)和MgCl2
(0.1 g,1 mmol)之混合物。將所得混合物加溫到約30℃並恆定攪拌。然後慢慢添加N,N-二異丙基乙胺(0.3 mL,2 mmol),及將反應混合物攪拌5 h。經由UPLC分析確認轉化至產物。
(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-(((第三丁基二甲矽基)氧基)甲基)四氫呋喃-2-醇的製備
製備7-碘吡咯並[2,1-f][1,2,4]三𠯤-4-胺(13.9 g,53.5 mmol)在THF中(280 mL)之溶液。將溶液冷卻至約0℃,及添加TMSCl(13.6 mL,107 mmol)。將反應混合物攪拌約20 min,及接著添加PhMgCl (在THF中之2 M;53.5 mL,56.8 mmol) 同時保持內溫低於約5℃。將反應混合物在約0℃下攪拌約30 min,及接著冷卻至約 -20℃。然後添加iPrMgCl-LiCl (在THF中之1.3 M,43.1 mL,56 mmol)同時保持內溫低於約-15℃。將反應混合物在約-20℃下攪拌約30 min。
在一個單獨燒瓶中,製備(3R,4R,5R)-3,4-雙((第三丁基二甲矽基)氧基)-5-(((第三丁基二甲矽基)氧基)甲基)二氫呋喃-2(3H)-酮(25.0 g,50.9 mmol,0.83 當量)在LaCl3
-2LiCl(在THF中之0.6 M,85 mL,50.9 mmol)之溶液。然後將溶液轉移至格任亞溶液同時保持內溫低於 -20℃。將所得反應混合物在約-20℃下攪拌約4 h。
將反應用1 M HCl (140 mL)淬滅,及將混合物加溫至周圍溫度。添加EtOAc (140 mL),及將有機相和水相分離。用EtOAc(200 mL)萃取水層。合併之 EtOAc層依次用飽和NaHCO3
水溶液(2×200 mL)、水(200 mL)及鹽水(200 mL)萃取。將有機層濃縮,及然後藉由矽膠層析法(30% EtOAc/己烷)純化以提供(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-(((第三丁基二甲矽基)氧基)甲基)四氫呋喃-2-醇。1
H NMR (300 MHz, CDCl3
) δ 8.15 - 7.88 (m, 1H), 7.51 (d, J = 4.8 Hz, 0.5H), 7.02 - 6.92 (m, 0.5H), 6.65 - 6.57 (m, 1H), 5.66 - 5.24 (m, 3H), 4.49 - 3.50 (m, 4H), 0.97 - 0.78 (26H), 0.65 (s, 1.5H), 0.19 - 0.00 (m, 15.5H), -0.22 (s, 1H), -0.55 (s, 1H)。MS m/z = 626 (M+H)。
(2R,3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-(羥甲基)四氫呋喃-2-甲腈的製備
將(3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-(((第三丁基二甲矽基)氧基)甲基)四氫呋喃-2-醇(1.50 g,2.40 mmol)在CH2
Cl2
(15 mL)中之溶液冷卻至約-40℃。添加三氟乙酸(0.555 mL,7.20 mmol),保持溫度低於-20℃。在一單獨燒瓶中,將三氟甲磺酸三甲矽基酯(2.60 mL,14.4 mmol)在約15℃下加至5 ml的CH2
Cl2
(5 mL),接著氰化三甲矽基(1.92 mL,14.4 mmol),及將溶液冷卻至約-30℃。將冷卻溶液加至 (3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-(((第三丁基二甲矽基)氧基)甲基)四氫呋喃-2-醇之溶液同時保持溫度低於-25℃。將反應混合物在約-30℃下攪拌15 min。將反應用三乙胺(3.34 mL,24.0 mmol)淬滅並將混合物加溫到約0℃。加水(50 mL),同時保持溫度低於約20℃。當添加完成時,將混合物在室溫下攪拌15 min。分離該等層並將有機層依次用KOH(20 mL)、水(20 mL)、及鹽水(20 mL)洗滌。將有機層經Na2
SO4
乾燥,濃縮,及然後藉由矽膠層析法(30% EtOAc / 己烷)純化以提供產物,呈非鏡像異構物之3.8:1混合物)。將混合物進一步藉由prep-HPLC(在水中之ACN 0至95%)純化以提供產物,呈單一非鏡像異構物。1
H NMR (400 MHz, DMSO-d6) δ 8.14-7.92 (m, 2H), 7.89 (s, 1H), 6.95 (d, J = 4.8 Hz, 1H), 6.88 (d, J = 4.4 Hz, 1H),5.27 (d, J = 4.6 Hz, 1H), 5.10 (dd, J = 7.7, 4.6 Hz, 1H), 4.31 (dd, J = 4.7, 1.4 Hz, 1H), 4.12 (ddd, J = 5.9, 4.1, 1.4 Hz, 1H), 3.80 - 3.69 (m, 1H), 3.56 (td, J = 7.8, 3.9 Hz, 1H), 0.93 (s, 9H), 0.75 (s, 9H), 0.11 (s, 3H), 0.09 (s, 3H), -0.15 (s, 3H), -0.62 (s, 3H)。MS m/z = 520 (M+H)。
(S)-2-(((S)-(((2R,3R,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-氰基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯的製備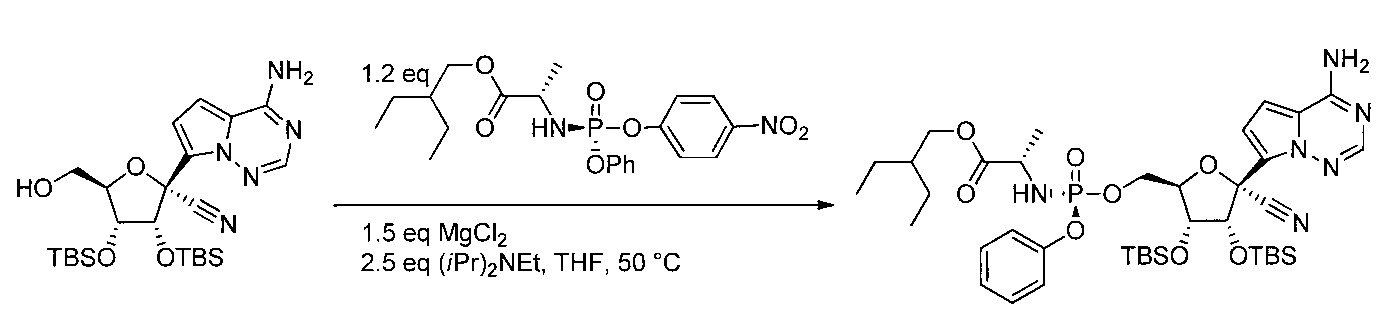 將THF(0.3 mL)進料至(2R,3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-(羥甲基)四氫呋喃-2-甲腈(16 mg,0.03 mmol)、(S)-2-(((S)-(4-硝基苯氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(17 mg,0.04 mmol)和MgCl2
(4 mg,0.05 mmol)之混合物。將所得混合物加溫到約50℃並恆定攪拌。然後添加N,N-二異丙基乙胺(0.013 mL,0.08 mmol),及將反應混合物攪拌21 h。經由UPLC和LC-MS分析確認轉化至產物。MS m/z = 831 (M+H)。
將THF(0.3 mL)進料至(2R,3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-(羥甲基)四氫呋喃-2-甲腈(16 mg,0.03 mmol)、(S)-2-(((S)-(4-硝基苯氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(17 mg,0.04 mmol)和MgCl2
(4 mg,0.05 mmol)之混合物。將所得混合物加溫到約50℃並恆定攪拌。然後添加N,N-二異丙基乙胺(0.013 mL,0.08 mmol),及將反應混合物攪拌21 h。經由UPLC和LC-MS分析確認轉化至產物。MS m/z = 831 (M+H)。 將(2R,3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-(羥甲基)四氫呋喃-2-甲腈(16 mg,0.03 mmol)在THF中(0.3 mL)之溶液冷卻至-10℃。滴加tBuMgCl (0.07 mL,0.07 mmol),接著(S)-2-(((S)-(全氟苯氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(22 mg,0.04 mmol)在THF(0.15 mL)中之溶液。將反應混合物加溫到5℃,及攪拌16 h。將反應用MeOH淬滅、濃縮,及然後藉由矽膠層析法(EtOAc /己烷)純化以提供產物。1H NMR (400 MHz, CDCl3
) δ 7.97 (s, 1H), 7.38 - 7.29 (m, 2H), 7.25 - 7.21 (m, 2H), 7.21 - 7.13 (m, 1H), 7.11 (d, J = 4.6 Hz, 1H), 6.65 (d, J = 4.6 Hz, 1H), 5.88 (br s, 2H), 5.35 (d, J = 4.4 Hz, 1H), 4.49 - 4.41 (m, 1H), 4.41 - 4.35 (m, 1H), 4.32 - 4.26 (m, 1H), 4.24 (dd, J = 4.5, 1.7 Hz, 1H), 4.10 - 3.99 (m, 2H), 3.96 (dd, J = 10.9, 5.7 Hz, 1H), 3.80 - 3.72 (m, 1H), 1.48 (h, J = 6.2 Hz, 1H), 1.39 - 1.28 (m, 7H), 0.96 (s, 9H), 0.85 (t, J = 7.5 Hz, 6H), 0.80 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H), -0.13 (s, 3H), -0.56 (s, 3H)。31P NMR (162 MHz, CDCl3) δ 2.74 (s)。MS m/z = 831 (M+H)。
(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯的製備
將(2R,3R,4R,5R)-2-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-(羥甲基)四氫呋喃-2-甲腈(16 mg,0.03 mmol)在THF中(0.3 mL)之溶液冷卻至-10℃。滴加tBuMgCl (0.07 mL,0.07 mmol),接著(S)-2-(((S)-(全氟苯氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯(22 mg,0.04 mmol)在THF(0.15 mL)中之溶液。將反應混合物加溫到5℃,及攪拌16 h。將反應用MeOH淬滅、濃縮,及然後藉由矽膠層析法(EtOAc /己烷)純化以提供產物。1H NMR (400 MHz, CDCl3
) δ 7.97 (s, 1H), 7.38 - 7.29 (m, 2H), 7.25 - 7.21 (m, 2H), 7.21 - 7.13 (m, 1H), 7.11 (d, J = 4.6 Hz, 1H), 6.65 (d, J = 4.6 Hz, 1H), 5.88 (br s, 2H), 5.35 (d, J = 4.4 Hz, 1H), 4.49 - 4.41 (m, 1H), 4.41 - 4.35 (m, 1H), 4.32 - 4.26 (m, 1H), 4.24 (dd, J = 4.5, 1.7 Hz, 1H), 4.10 - 3.99 (m, 2H), 3.96 (dd, J = 10.9, 5.7 Hz, 1H), 3.80 - 3.72 (m, 1H), 1.48 (h, J = 6.2 Hz, 1H), 1.39 - 1.28 (m, 7H), 0.96 (s, 9H), 0.85 (t, J = 7.5 Hz, 6H), 0.80 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H), -0.13 (s, 3H), -0.56 (s, 3H)。31P NMR (162 MHz, CDCl3) δ 2.74 (s)。MS m/z = 831 (M+H)。
(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯的製備 將(S)-2-(((S)-(((2R,3R,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-氰基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯之粗製溶液冷卻至約0℃並慢慢添加濃HCl(0.05 mL,0.62 mmol)。將反應混合物在約20℃下攪拌約72小時。經由UPLC和LC-MS分析確認轉化至產物。MS m/z = 603 (M+H)。
將(S)-2-(((S)-(((2R,3R,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-氰基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯之粗製溶液冷卻至約0℃並慢慢添加濃HCl(0.05 mL,0.62 mmol)。將反應混合物在約20℃下攪拌約72小時。經由UPLC和LC-MS分析確認轉化至產物。MS m/z = 603 (M+H)。 (S)-2-(((S)-(((2R,3R,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-氰基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯在氟化物或酸中之溶液可去保護至(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯之溶液。代表性氟化物包括但不限於TBAF、KF、氫氟化吡啶、氫氟化三乙銨、氟化氫、氫氯酸、甲苯磺酸、或任何其他適當氟化物來源。代表性酸包括但不限於它們在Greene, T. W.;Wuts, P. G. M. Protective Groups In Organic Synthesis, 第四版, John Wiley & Sons:New York, 2006中所發現者。
B.抗病毒活性
(S)-2-(((S)-(((2R,3R,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-3,4-雙((第三丁基二甲矽基)氧基)-5-氰基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯在氟化物或酸中之溶液可去保護至(S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-胺基吡咯並[2,1-f][1,2,4]三𠯤-7-基)-5-氰基-3,4-二羥基四氫呋喃-2-基)甲氧基)(苯氧基)磷醯基)胺基)丙酸2-乙基丁酯之溶液。代表性氟化物包括但不限於TBAF、KF、氫氟化吡啶、氫氟化三乙銨、氟化氫、氫氯酸、甲苯磺酸、或任何其他適當氟化物來源。代表性酸包括但不限於它們在Greene, T. W.;Wuts, P. G. M. Protective Groups In Organic Synthesis, 第四版, John Wiley & Sons:New York, 2006中所發現者。
B.抗病毒活性
本發明之另一態樣關於抑制病毒感染之方法,其包含以本發明組成物治療懷疑需要該抑制之樣品或個體的步驟。
在本發明的上下文中,懷疑含有病毒之樣品包括天然或人造材料,諸如活生物;組織或細胞培養物;生物樣品諸如生物材料樣品(血、血清、尿、腦脊髓液、淚液、痰、唾液、組織樣品、等等);實驗室樣品;食物、水或空氣樣品;生物製品樣品諸如細胞(特別是合成所需糖蛋白的重組細胞)之提取物;等等。典型地,該樣品將被懷疑含有誘發病毒感染之生物體(常為病原生物體諸如腫瘤病毒)。樣品可包含在包括水和有機溶劑\水混合物的任何介質中。樣品包括活生物體諸如人和人造材料諸如細胞培養物。
如果需要的話,可藉由包括檢測該活性的直接和間接之方法的任何方法觀察施用組成物後的本發明化合物之抗病毒活性。確定該活性的定量、定性和半定量方法全被考慮。通常應用上述篩選方法之一,然而,任何其它方法諸如活生物體的生理性質的觀測也是適用的。
本發明化合物之抗病毒活性可使用已知的標準篩選方案測量。例如,化合物之抗病毒活性可使用下列一般方案測量。 實施例36. 伊波拉病毒(Ebola virus)抗病毒活性和細胞毒性分析
實施例36. 伊波拉病毒(Ebola virus)抗病毒活性和細胞毒性分析
使用表現螢光素酶或綠色螢光蛋白質(GFP)之完全複製報導病毒(Uebelhoer, L.S., 2014. AVR;Hoenen, T., 2013. AVR)測定化合物1和化合物9對抗伊波拉病毒(EBOV)、馬堡病毒(MARV)(表2)及Nipah病毒(NiV)(表3)的抗病毒活性。使用表現螢光素酶或綠色螢光蛋白質(GFP)之完全複製報導病毒(Uebelhoer, L.S., 2014. AVR;Hoenen, T., 2013. AVR)測量化合物1和化合物9對抗伊波拉病毒(EBOV)、馬堡病毒(MARV)之其他抗病毒活性(表2-a)。所有研究都是在疾病管制與預防中心(Centers for Disease Control and Prevention (CDC))以生物安全等級-4防護(containment)(BSL-4)進行。伊波拉病毒抗病毒分析在以端粒酶催化蛋白質(HMVEC-TERT) 永生化之原代人微血管內皮細胞和Huh-7細胞(Shao, R., 2004, BBRC)中進行。Nipah病毒抗病毒活性係在HMVEC-TERT和海拉細胞中測量。
在96孔盤中進行抗病毒分析。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種細胞單層之盤。將該等盤轉移至BSL-4防護及將病毒原料(virus stock)之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養3至4天。培養後,在Envision盤式讀取機中以GFP報導病毒之直接螢光或後續添加用於螢光素酶報導病毒的螢光素酶受質之後測量病毒複製。關於病毒產率試驗,從經感染的細胞移除培養基,將一部分用於以反轉錄定量聚合酶鏈反應(RT-qPCR)定量病毒RNA。將剩餘培養基連續稀釋,並藉由使用稀釋培養基來感染新鮮的細胞單層來測定感染病毒的量以使用Cell TiterGlo試劑(Promega, Madison, WI)確定導致50%細胞病變效應(TCID50)的組織培養感染劑量。關於病毒細胞病變效應(CPE)分析,使用CellTiter Glo試劑測定感染細胞的生存力。
計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例37. EBOV-GFP HMVEC-TERT細胞
將HMVEC-TERT細胞接種於96孔盤。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種HMVEC-TERT單層之96孔盤。將該等盤轉移至BSL-4防護及將EBOV-GFP病毒原料之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養3至4天。培養後,在Envision盤式讀取機中以直接螢光測量病毒複製以測量來自報導病毒之GFP表現。計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例38. EBOV-GFP Huh-7細胞
將Huh-7細胞接種於96孔盤。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種Huh-7單層之96孔盤。將該等盤轉移至BSL-4防護及將EBOV-GFP病毒原料之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養3至4天。培養後,在Envision盤式讀取機中以直接螢光測量病毒複製以測量來自報導病毒之GFP表現。計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例39. EBOV-Luc Huh-7細胞
將Huh-7細胞接種於96孔盤。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種細胞單層之盤。將該等盤轉移至BSL-4防護及將EBOV-Luc病毒原料之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養3至4天。培養後,在Envision盤式讀取機中後續添加螢光素酶受質之後測量病毒複製。計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例40. MARV-GFP Huh-7細胞
將Huh-7細胞接種於96孔盤。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種Huh-7單層之96孔盤。將該等盤轉移至BSL-4防護及將MARV-GFP病毒原料之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養3至4天。培養後,在Envision盤式讀取機中以直接螢光測量病毒複製以測量來自報導病毒之GFP表現。計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例41. Ebola Huh-7 (RNA)
將Huh-7細胞接種於96孔盤。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種Huh-7細朐單層之盤。將該等盤轉移至BSL-4防護及將EBOV病毒原料之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養3至4天。培養後,從被感染的細胞移除培養基,且將一部分用於以反轉錄定量聚合酶鏈反應(RT-qPCR)定量病毒RNA。計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例42. 伊波拉Huh-7 (產率)
將Huh-7細胞接種於96孔盤。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種Huh-7細胞單層之盤。將該等盤轉移至BSL-4防護及將EBOV病毒原料之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養3至4天。培養後,從被感染的細胞移除培養基及稀釋於10-倍連續稀釋液。藉由使用稀釋培養基來感染新鮮的細胞單層來測定感染病毒的量以使用Cell TiterGlo試劑(Promega, Madison, WI)確定導致50%細胞病變效應(TCID50)的組織培養感染劑量。計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例43. 伊波拉海拉細胞
測定所選化合物對抗伊波拉病毒(EBOV)薩伊株的抗病毒活性,其係在美國陸軍傳染病醫學研究所(US Army Medical Research Institute for Infections Disease)(USAMRIID)以生物安全等級-4防護(防護)(BSL-4)進行。將海拉細胞以5000個細胞/孔接種於384孔盤。測定各化合物的抗病毒活性一式四份。將八至十個濃度的化合物在感染之前2h使用HP300數字分配器以3倍連續稀釋增量直接加至細胞培養物。將盤轉移至BSL-4防護和適當稀釋之病毒原料(預先以滴定測定且製備於細胞培養基中)加至含有細胞和連續稀釋化合物之試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養2天。培養之後,將細胞固定在甲醛溶液中及藉由疫染色和使用Perkin Elmer Opera共聚焦顯微鏡儀器免高通量成像之後,定量伊波拉醣蛋白含量來測定病毒複製。計算每個測試濃度相對於0%和100%抑制對照組之抑制百分比且藉由非線性回歸測定每個化合物的EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例44. 伊波拉巨噬細胞培養
測定所選化合物對抗伊波拉病毒(EBOV)薩伊株的抗病毒活性,其係在美國陸軍傳染病醫學研究所(USAMRIID)以生物安全等級-4防護(防護)(BSL-4)進行。從新鮮人類PBMC分離巨噬細胞培養物並在5ng/ml GM-CSF和50uM B-巰基乙醇的存在下分化。每2天更換培養基,且7天後將附著於組織培養盤之細胞用在1X PBS中之0.5M EDTA移除,藉由以200 xg離心10分鐘濃縮並以40,000個細胞/孔鋪在384孔分析盤。測定各化合物的抗病毒活性一式四份。將八至十個濃度的化合物在感染之前2h使用HP300數字分配器以3倍連續稀釋增量直接加至細胞培養物。將盤轉移至BSL-4防護和適當稀釋之病毒原料(預先以滴定測定且製備於細胞培養基中)加至含有細胞和連續稀釋化合物之試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養2天。培養之後,將細胞固定在甲醛溶液中及藉由免疫染色和使用Perkin Elmer Opera共聚焦顯微鏡儀器高通量成像之後,定量伊波拉醣蛋白含量測定病毒複製。計算每個測試濃度相對於0%和100%抑制對照組之抑制百分比且藉由非線性回歸測定每個化合物的EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例45. Nipah-GFP HMVEC-TERT細胞
將HMVEC-TERT細胞接種於96孔盤。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種HMVEC-TERT單層之96孔盤。將該等盤轉移至BSL-4防護及將NiV-GFP病毒原料之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養3至4天。培養後,在Envision盤式讀取機中以直接螢光測量病毒複製以測量來自報導病毒之GFP表現。計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例46. NiV-Luc HMVEC-TERT
將HMVEC-TERT細胞接種於96孔盤。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種細胞單層之盤。將該等盤轉移至BSL-4防護及將Niv-Luc病毒原料之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養3至4天。培養後,在Envision盤式讀取機中後續添加螢光素酶受質之後測量病毒複製。計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例47. NiV Hela (產率)
將海拉細胞接種於96孔盤。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種海拉細胞單層之盤。將該等盤轉移至BSL-4防護及將Niv病毒原料之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養4天。培養後,從被感染的細胞移除培養基及稀釋於10-倍連續稀釋液。藉由使用稀釋培養基感染新鮮的細胞單層來測定感染病毒的量以使用Cell TiterGlo試劑(Promega, Madison, WI)確定導致50%細胞病變效應(TCID50)的組織培養感染劑量。計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。
實施例48. Niv Hela (RNA)
將Huh-7細胞接種於96孔盤。將八至十個濃度的化合物以3-倍連續稀釋增量稀釋於培養基中及將100 uL/孔之各稀釋液一式三份轉移至含有預接種海拉細胞單層之盤。將該等盤轉移至BSL-4防護及將Niv病毒原料之適當稀釋液(預先以滴定測定且製備於細胞培養基中)加至包含細胞和連續稀釋之化合物的試驗盤。各盤包括三個經感染而未經治療之細胞的孔和三個未經感染之細胞的孔,分別充當0%和100%病毒抑制對照組。感染之後,將試驗盤在組織培養箱中培養3至4天。培養後,從被感染的細胞移除培養基,且將一部分用於以反轉錄定量聚合酶鏈反應(RT-qPCR)定量病毒RNA。計算各測試濃度相對於0%和100%抑制對照組的百分比抑制及以非線性回歸確定各化合物之EC50
值作為抑制50%病毒複製之化合物的有效濃度。


本文中所引用的所有公開、專利及專利文獻皆以引用方式併入本文中,如同以引用方式個別併入。
本發明已參照各種具體及較佳實施態樣及技術說明。然而,熟習該項技術者應理解:在保留本發明精神及範圍下可進行許多變化及改良。

Claims (7)
- 一種治療有效量之下式化合物或其醫藥上可接受之鹽之用途,

- 如請求項1之用途,其中該藥劑包含醫藥上可接受之載劑或賦形劑。
- 如請求項1之用途,其中該藥劑進一步包含至少一種其它治療劑,或係用於與至少一種其它治療劑組合使用。
- 如請求項3之用途,其中該至少一種其它治療劑係選自由下列所組成之群:抗生素、抗真菌藥、發燒和止痛藥、止吐劑、止瀉劑、維生素和礦物質補充劑、抗炎劑及抗瘧劑;或其混合物。
- 如請求項4之用途,其中該至少一種其它治療劑為抗瘧劑。
- 如請求項3之用途,其中該至少一種其它治療劑係選自由下列所組成之群:利巴韋林(ribavirin)、帕利珠單抗(palivizumab)、莫它珠單抗(motavizumab)、RSV-IGIV、MEDI-557、A-60444、MDT-637、BMS-433771、胺碘酮(amiodarone)、決奈達隆(dronedarone)、維拉帕米 (verapamil)、伊波拉(Ebola)恢復期血漿(ECP)、TKM-100201、(2S,3S,4R,5R)-2-(4-胺基-5H-吡咯並[3,2-d]嘧啶-7-基)-5-(羥甲基)吡咯啶-3,4-二醇、法匹拉韋(favipiravir)(亦稱為T-705)、T-705單磷酸鹽、T-705二磷酸鹽、T-705三磷酸、1-N,7-N-雙[3-(二甲胺基)丙基]-3,9-二甲基喹啉並[[8,7-h]喹啉酮-1,7-二胺、JK-05、TKM-伊波拉、ZMapp、rNAPc2、VRC-EBOADC076-00-VP、OS-2966、MVA-BN filo、伯利多韋(brincidofovir)、Vaxart腺病毒載體5型伊波拉疫苗、Ad26-ZEBOV、FiloVax疫苗、GOVX-E301、GOVX-E302、伊波拉病毒(Ebola virus)進入抑制劑(NPC1抑制劑)及rVSV-EBOV;或其混合物。
- 如請求項6之用途,其中該至少一種其它治療劑為利巴韋林。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462072331P | 2014-10-29 | 2014-10-29 | |
| US62/072,331 | 2014-10-29 | ||
| US201562105619P | 2015-01-20 | 2015-01-20 | |
| US62/105,619 | 2015-01-20 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW202039526A TW202039526A (zh) | 2020-11-01 |
| TWI767201B true TWI767201B (zh) | 2022-06-11 |
Family
ID=54557474
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW109109475A TWI767201B (zh) | 2014-10-29 | 2015-10-27 | 絲狀病毒科病毒感染之治療 |
| TW109120726A TWI740546B (zh) | 2014-10-29 | 2015-10-27 | 製備核糖苷的方法 |
| TW104135248A TWI698444B (zh) | 2014-10-29 | 2015-10-27 | 製備核糖苷的方法 |
| TW104135247A TWI687432B (zh) | 2014-10-29 | 2015-10-27 | 絲狀病毒科病毒感染之治療 |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW109120726A TWI740546B (zh) | 2014-10-29 | 2015-10-27 | 製備核糖苷的方法 |
| TW104135248A TWI698444B (zh) | 2014-10-29 | 2015-10-27 | 製備核糖苷的方法 |
| TW104135247A TWI687432B (zh) | 2014-10-29 | 2015-10-27 | 絲狀病毒科病毒感染之治療 |
Country Status (46)
Families Citing this family (113)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10023600B2 (en) | 2009-09-21 | 2018-07-17 | Gilead Sciences, Inc. | Processes and intermediates for the preparation of 1′-substituted carba-nucleoside analogs |
| ME01924B (me) | 2010-07-22 | 2015-05-20 | Gilead Sciences Inc | Metode i jedinjenja za lečenje infekcija izazvanih Paramyxoviridae virusom |
| TWI767201B (zh) | 2014-10-29 | 2022-06-11 | 美商基利科學股份有限公司 | 絲狀病毒科病毒感染之治療 |
| CN115887465A (zh) | 2015-09-16 | 2023-04-04 | 吉利德科学公司 | 治疗沙粒病毒科和冠状病毒科病毒感染的方法 |
| CA3040540A1 (en) * | 2016-11-10 | 2018-05-17 | Oyagen, Inc. | Methods of treating and inhibiting ebola virus infection |
| KR20190085976A (ko) * | 2016-11-18 | 2019-07-19 | 뉴로바이브 파마슈티컬 에이비 | 미토콘드리아 프로톤 이오노포어의 간 프로드러그 |
| CN116036112A (zh) * | 2017-03-14 | 2023-05-02 | 吉利德科学公司 | 治疗猫冠状病毒感染的方法 |
| JP6923112B2 (ja) * | 2017-03-31 | 2021-08-18 | 国立大学法人北海道大学 | フィロウイルスの細胞侵入阻害活性を有するビアリールスルフォンアミド誘導体 |
| CN110636884B (zh) | 2017-05-01 | 2022-10-04 | 吉利德科学公司 | 新结晶形式 |
| WO2019014247A1 (en) | 2017-07-11 | 2019-01-17 | Gilead Sciences, Inc. | COMPOSITIONS COMPRISING POLYMERASE RNA INHIBITOR AND CYCLODEXTRIN FOR THE TREATMENT OF VIRAL INFECTIONS |
| AU2018332540B2 (en) | 2017-09-18 | 2023-10-05 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| CN109535200A (zh) * | 2017-09-21 | 2019-03-29 | 杭州和正医药有限公司 | 一种核苷类似物的磷酰胺酯前药、药物组合物及其应用 |
| CN109748944B (zh) * | 2017-11-03 | 2021-12-10 | 中国科学院上海药物研究所 | 5’-脱氧-5’-异丙基取代氨基核苷类化合物、其制备方法和用途 |
| CN109748943A (zh) * | 2017-11-03 | 2019-05-14 | 中国科学院上海药物研究所 | 2’-c-甲基取代核苷类化合物及其制备与用途 |
| CN110330540A (zh) * | 2019-08-08 | 2019-10-15 | 木天(济南)生物科技有限公司 | 核苷盐及其制备方法 |
| CN110613726B (zh) * | 2019-09-25 | 2020-10-16 | 广州六顺生物科技股份有限公司 | 核苷化合物的应用 |
| WO2021095091A1 (ja) * | 2019-11-11 | 2021-05-20 | 藤本化学製品株式会社 | アミノアリール誘導体及びその中間体、並びにそれらの製造方法 |
| CN110776512A (zh) * | 2019-11-28 | 2020-02-11 | 成都傲飞生物化学品有限责任公司 | 一种核苷类似物的制备方法 |
| CN118662520A (zh) | 2020-01-27 | 2024-09-20 | 吉利德科学公司 | 用于治疗SARS CoV-2感染的方法 |
| WO2021158248A1 (en) | 2020-02-04 | 2021-08-12 | Oyagen, Inc. | Method for treating coronavirus infections |
| CN113214263B (zh) * | 2020-02-06 | 2022-09-30 | 北京桦冠医药科技有限公司 | 瑞德西韦关键中间体的一种合成方法 |
| US20210244705A1 (en) * | 2020-02-07 | 2021-08-12 | Centre For Digestive Diseases | Therapeutic compositions, products of manufacture and methods for ameliorating or preventing coronavirus infection |
| CN111205327B (zh) * | 2020-02-17 | 2022-05-31 | 南京法恩化学有限公司 | 一种瑞德西韦的制备方法 |
| CA3171648A1 (en) | 2020-02-18 | 2021-08-26 | Gilead Sciences, Inc. | Antiviral compounds |
| TWI794742B (zh) | 2020-02-18 | 2023-03-01 | 美商基利科學股份有限公司 | 抗病毒化合物 |
| TWI775313B (zh) | 2020-02-18 | 2022-08-21 | 美商基利科學股份有限公司 | 抗病毒化合物 |
| TWI791193B (zh) * | 2020-02-18 | 2023-02-01 | 美商基利科學股份有限公司 | 抗病毒化合物 |
| CN113292565B (zh) * | 2020-02-24 | 2023-01-31 | 浙江森科建设有限公司 | 核苷类化合物及其制备方法和应用 |
| CN111187269A (zh) * | 2020-02-27 | 2020-05-22 | 江苏阿尔法药业有限公司 | 一种瑞德西韦中间体的合成方法 |
| CN111171078B (zh) * | 2020-02-27 | 2022-04-22 | 江苏阿尔法药业股份有限公司 | 一种瑞德西韦的合成方法 |
| CN111205294B (zh) * | 2020-02-27 | 2021-10-01 | 江苏阿尔法药业股份有限公司 | 一种瑞德西韦中间体的制备方法 |
| CN111233930B (zh) * | 2020-03-04 | 2022-05-13 | 江苏阿尔法药业股份有限公司 | 一种瑞德西韦的制备方法 |
| WO2021175296A1 (zh) * | 2020-03-04 | 2021-09-10 | 中国科学院上海药物研究所 | 瑞德西韦的中间体及其制备方法 |
| CN111269248A (zh) * | 2020-03-05 | 2020-06-12 | 江苏福瑞康泰药业有限公司 | 一种核苷氨基磷酸酯类药物母液回收的新方法 |
| CN111269263A (zh) * | 2020-03-09 | 2020-06-12 | 上海龙翔生物医药开发有限公司 | 一种瑞德西韦侧链中间体及其制备方法 |
| CN111233870A (zh) * | 2020-03-11 | 2020-06-05 | 中国科学技术大学 | 用于快速制备瑞德西韦药物中间体的方法 |
| CN113387954B (zh) * | 2020-03-11 | 2024-03-19 | 上海特化医药科技有限公司 | 一种瑞德西韦中间体的制备方法 |
| CN111233869B (zh) * | 2020-03-12 | 2022-09-16 | 杭州新博思生物医药有限公司 | 用于制备瑞德西韦关键中间体的新化合物及其制备方法 |
| CN115298181B (zh) * | 2020-03-12 | 2024-08-16 | 吉利德科学公司 | 制备1’-氰基核苷的方法 |
| WO2021194861A1 (en) | 2020-03-23 | 2021-09-30 | Genentech, Inc. | Biomarkers for predicting response to il-6 antagonist in covid-19 pneumonia |
| US20230174656A1 (en) | 2020-03-23 | 2023-06-08 | Genentech, Inc. | Tocilizumab and remdesivir combination therapy for covid-19 pneumonia |
| CN111233931B (zh) * | 2020-03-26 | 2022-03-22 | 宿迁盛基医药科技有限公司 | 一种瑞德西韦的合成方法 |
| CN111398464B (zh) * | 2020-04-02 | 2022-03-08 | 广州隽沐生物科技股份有限公司 | 2-乙基丁基((全氟苯氧基)(苯氧基)磷酰基)-l-丙氨酸酯的检测方法 |
| CN111423443A (zh) * | 2020-04-03 | 2020-07-17 | 广州科锐特生物科技有限公司 | 一种4-氨基-7-碘吡咯并[2,1-f][1,2,4]三嗪的制备方法 |
| CN113493480A (zh) * | 2020-04-03 | 2021-10-12 | 南京正大天晴制药有限公司 | 一种瑞德西韦异构体的制备及其分析方法 |
| CA3172483A1 (en) * | 2020-04-06 | 2021-10-14 | Scott Ellis | Inhalation formulations of 1'-cyano substituted carbanucleoside analogs |
| US20230158054A1 (en) * | 2020-04-07 | 2023-05-25 | Biocryst Pharmaceuticals, Inc. | Methods, compositions, and dosing regimens for treatment of sars-cov-2 infections |
| CN113811309B (zh) * | 2020-04-13 | 2023-05-05 | 山东华铂凯盛生物科技有限公司 | 用于治疗病毒感染的聚合物制剂及制备方法和用途 |
| CN111393494A (zh) * | 2020-04-17 | 2020-07-10 | 广东帕派恩生物科技有限公司 | 基于核苷酸结构的化合物、制备方法、用途 |
| CN112778310A (zh) * | 2020-04-20 | 2021-05-11 | 中国科学院上海药物研究所 | 核苷类似物或含有核苷类似物的组合制剂在抗病毒中的应用 |
| CN111471070B (zh) * | 2020-04-26 | 2022-05-13 | 江苏阿尔法药业股份有限公司 | 瑞德西韦的合成方法 |
| CN111454270B (zh) * | 2020-04-27 | 2024-05-14 | 南通伟顺生物科技有限公司 | 一种含六元环的核苷类化合物及其制备方法 |
| CN111440176B (zh) * | 2020-04-28 | 2022-04-26 | 江苏大学 | 金属配合物促进的瑞德西韦中间体的合成方法 |
| US10988503B1 (en) * | 2020-05-06 | 2021-04-27 | Nantong Weishun Biotechnology Co., Ltd | Six-membered ring-containing nucleoside compound and preparation method thereof |
| CN113637041B (zh) * | 2020-05-11 | 2024-02-27 | 上海科胜药物研发有限公司 | 一种核糖核苷的制备方法 |
| RU2740660C1 (ru) * | 2020-05-20 | 2021-01-19 | Общество С Ограниченной Ответственностью "Промомед Рус" | Противовирусная композиция |
| US20230277524A1 (en) | 2020-05-22 | 2023-09-07 | Trailhead Biosystems Inc. | Combination therapy for treatment of viral infections |
| CN111961057A (zh) * | 2020-05-26 | 2020-11-20 | 李小冬 | 一种α构型核苷及其在治疗猫冠状病毒感染的应用 |
| IN202011022634A (zh) | 2020-05-29 | 2020-10-09 | Jubilant Generics Limited | |
| EP3915548A1 (en) | 2020-05-29 | 2021-12-01 | Jubilant Generics Limited | Transmucosal pharmaceutical compositions of antiviral drugs |
| TW202203941A (zh) | 2020-05-29 | 2022-02-01 | 美商基利科學股份有限公司 | 瑞德西韋之治療方法 |
| CN113754694B (zh) * | 2020-06-03 | 2022-10-25 | 上海交通大学 | 一种不对称催化无保护基核苷合成瑞德西韦的方法 |
| CN113754693B (zh) * | 2020-06-03 | 2022-09-09 | 上海交通大学 | 一种磷手性核苷衍生物的不对称催化合成方法及催化剂 |
| CN113754692B (zh) * | 2020-06-03 | 2022-06-10 | 上海交通大学 | 瑞德西韦中间体(s,s)-氨基磷酸酯的不对称催化合成方法 |
| US20230241225A1 (en) | 2020-06-15 | 2023-08-03 | Metro International Biotech, Llc | Anti-viral compounds and methods of use |
| WO2021257569A1 (en) * | 2020-06-15 | 2021-12-23 | Metro International Biotech, Llc | Anti-viral compounds and methods of use |
| CN111620903A (zh) * | 2020-06-17 | 2020-09-04 | 安徽贝克联合制药有限公司 | C-核苷类似物以及用于合成瑞德西韦合成的含腈c-核苷类化合物的制备方法及其应用 |
| US20210393663A1 (en) * | 2020-06-23 | 2021-12-23 | Cai Gu Huang | Pharmaceutical formulation containing active metabolites of remdesivir or its analog for inhalation |
| PE20230618A1 (es) | 2020-06-24 | 2023-04-14 | Gilead Sciences Inc | Analogos de nucleosido de 1'-ciano y usos de los mismos |
| CN111732618A (zh) * | 2020-07-03 | 2020-10-02 | 镇江巨杰新材料技术研发中心(有限合伙) | 瑞德西韦关键片段的合成方法 |
| CN111961079A (zh) * | 2020-07-17 | 2020-11-20 | 南京正济医药研究有限公司 | 一种瑞德西韦有关物质及其制备方法和应用 |
| EP4185300A4 (en) * | 2020-07-24 | 2024-09-25 | The Regents of the University of California | ANTIVIRAL PRODRUGS, PHARMACEUTICAL FORMULATIONS AND METHODS |
| EP4192839A1 (en) | 2020-08-06 | 2023-06-14 | Richter Gedeon Nyrt. | Remdesivir intermediates |
| CN113004330A (zh) * | 2020-08-22 | 2021-06-22 | 齐鲁制药有限公司 | 一种高纯度瑞德西韦的制备方法 |
| CN116323630A (zh) | 2020-08-24 | 2023-06-23 | 吉利德科学公司 | 磷脂化合物及其用途 |
| CN116568688A (zh) * | 2020-08-27 | 2023-08-08 | 吉利德科学公司 | 用于治疗病毒感染的化合物和方法 |
| CA3190702A1 (en) | 2020-08-27 | 2022-03-03 | Elaine Bunyan | Compounds and methods for treatment of viral infections |
| CN114105989A (zh) * | 2020-08-31 | 2022-03-01 | 中国科学院大连化学物理研究所 | 一种碘代吡咯并三嗪胺类化合物的制备方法和应用 |
| CN116685351A (zh) | 2020-09-17 | 2023-09-01 | 基因泰克公司 | Empacta的结果:一项用于评估托珠单抗在患有covid-19肺炎的住院患者中的功效和安全性的随机、双盲、安慰剂对照、多中心研究 |
| CN112321642A (zh) * | 2020-09-28 | 2021-02-05 | 南京正济医药研究有限公司 | 一种瑞德西韦有关物质及其制备方法和用途 |
| WO2022074630A1 (en) * | 2020-10-09 | 2022-04-14 | Auxilla Pharmaceuticals And Research Llp | Liquid oral suspension of favipiravir |
| CN112194661B (zh) * | 2020-10-22 | 2021-06-08 | 威海同丰海洋生物科技有限公司 | 一种4-氨基-7-碘吡咯并[2,l-f][l,2,4]三嗪的制备方法 |
| CN114507256B (zh) * | 2020-11-16 | 2025-02-28 | 上海医药集团股份有限公司 | 一种瑞德西韦工艺手性异构体、其制备方法及其应用 |
| RU2756921C1 (ru) * | 2020-11-20 | 2021-10-07 | Общество С Ограниченной Ответственностью "Технология Лекарств" | Способ получения ремдесивира и фосфорамидаты |
| JP2023554592A (ja) | 2020-11-23 | 2023-12-28 | ジェネンテック, インコーポレイテッド | Sars-cov-2と宿主細胞の表面との相互作用を調節するための方法 |
| CN112358513A (zh) * | 2020-12-02 | 2021-02-12 | 上海朴颐化学科技有限公司 | 一种利用连续流反应器制备瑞德西韦中间体的方法 |
| CN114621229B (zh) * | 2020-12-11 | 2024-07-02 | 嘉兴金派特生物科技有限公司 | 治疗或预防猫传染性腹膜炎的化合物或组合物 |
| CN114685558A (zh) * | 2020-12-28 | 2022-07-01 | 尚科生物医药(上海)有限公司 | 一种瑞德西韦中间体的制备方法 |
| CN113754665B (zh) * | 2020-12-30 | 2022-08-19 | 南方科技大学 | 一种核苷类化合物的制备方法 |
| CN113735862B (zh) * | 2020-12-30 | 2024-02-02 | 南方科技大学 | 一种治疗病毒感染的核苷类化合物及其用途 |
| US11998562B2 (en) | 2021-01-25 | 2024-06-04 | University Of South Florida | Ophthalmological formulations for the prevention of a coronavirus infection |
| WO2022166581A1 (zh) * | 2021-02-07 | 2022-08-11 | 石家庄迪斯凯威医药科技有限公司 | 一种核苷酸衍生物及其药物组合物和用途 |
| WO2022174779A1 (zh) * | 2021-02-19 | 2022-08-25 | 南京赛弗斯医药科技有限公司 | 一种具有抗肿瘤活性的核苷酸衍生物及其药物组合物和用途 |
| CN112979736B (zh) * | 2021-03-04 | 2022-03-25 | 南京欧信医药技术有限公司 | 一种瑞德西韦的制备方法 |
| US11697666B2 (en) | 2021-04-16 | 2023-07-11 | Gilead Sciences, Inc. | Methods of preparing carbanucleosides using amides |
| JP2024513986A (ja) | 2021-04-16 | 2024-03-27 | ギリアード サイエンシーズ, インコーポレイテッド | アミドを使用してカルバヌクレオシドを調製する方法 |
| US20230000873A1 (en) | 2021-05-26 | 2023-01-05 | Gilead Sciences, Inc. | Phospholipid formulations of 1'-cyano substituted carba-nucleoside analogs |
| CN113683640B (zh) * | 2021-08-10 | 2025-01-14 | 浙江华海药业股份有限公司 | 一种丁基瑞德西韦的制备方法 |
| CA3228162A1 (en) | 2021-08-18 | 2023-02-23 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| AU2022331233A1 (en) | 2021-08-20 | 2024-02-29 | Shionogi & Co., Ltd. | Nucleoside derivatives and prodrugs thereof having viral growth inhibitory action |
| CN113999237B (zh) * | 2021-10-29 | 2023-08-08 | 润佳(苏州)医药科技有限公司 | 一种核苷类前药及其用途 |
| KR102675967B1 (ko) | 2021-11-02 | 2024-06-17 | 충남대학교산학협력단 | 렘데시비르를 함유하는 나노지질담체, 이의 제조방법 및 이를 포함하는 약학적 조성물 |
| CN114409655A (zh) * | 2022-01-26 | 2022-04-29 | 郑州大学 | 3′,4′-不饱和核糖c-核苷类似物及其制备方法 |
| AU2023227794A1 (en) | 2022-03-02 | 2024-10-17 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| CN115028672A (zh) * | 2022-06-14 | 2022-09-09 | 南京正济医药研究有限公司 | 瑞德西韦中间体晶体及其制备方法 |
| US11655255B2 (en) | 2022-10-17 | 2023-05-23 | Sph No.1 Biochemical & Pharmaceutical Co., Ltd. | Method for catalytic asymmetric synthesis of phosphorus-stereogenic (P-stereogenic) nucleoside derivative and catalyst used therein |
| TW202421167A (zh) | 2022-11-18 | 2024-06-01 | 美商基利科學股份有限公司 | 用於治療痘病毒感染之方法 |
| CN115737675B (zh) * | 2022-11-28 | 2024-03-08 | 安徽农业大学 | 氧化型硒代硫酸钠的制备及其在治疗冠状病毒感染中的应用 |
| CN115947759A (zh) * | 2022-12-13 | 2023-04-11 | 安徽大学 | 一种治疗新冠药物瑞德西韦的制备方法 |
| CN116332996A (zh) * | 2023-05-04 | 2023-06-27 | 南京颐媛生物医学研究院有限公司 | 抗冠状病毒化合物及其制备方法和应用 |
| HUP2300153A1 (hu) | 2023-05-09 | 2024-11-28 | Rotachrom Tech Zrt | Eljárás remdesivir foszfor sztereoizomereinek elválasztására akirális folyadék-folyadék kromatográfiával |
| CN116554176A (zh) * | 2023-05-11 | 2023-08-08 | 南京正济医药研究有限公司 | 一种核苷类化合物的制备方法 |
| CN117126199B (zh) * | 2023-05-17 | 2024-12-06 | 江西师范大学 | 一种氟代瑞德西韦合成方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012012776A1 (en) * | 2010-07-22 | 2012-01-26 | Gilead Sciences, Inc. | Methods and compounds for treating paramyxoviridae virus infections |
Family Cites Families (154)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816570A (en) | 1982-11-30 | 1989-03-28 | The Board Of Regents Of The University Of Texas System | Biologically reversible phosphate and phosphonate protective groups |
| US4968788A (en) | 1986-04-04 | 1990-11-06 | Board Of Regents, The University Of Texas System | Biologically reversible phosphate and phosphonate protective gruops |
| DE69115694T2 (de) | 1990-06-13 | 1996-10-17 | Arnold Newton Mass. Glazier | Phosphorylierte prodrugs |
| EP0481214B1 (en) | 1990-09-14 | 1998-06-24 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Prodrugs of phosphonates |
| JPH1017629A (ja) | 1996-07-04 | 1998-01-20 | Kayaku Akzo Kk | ハードコート用樹脂組成物及びその硬化方法 |
| US6887707B2 (en) | 1996-10-28 | 2005-05-03 | University Of Washington | Induction of viral mutation by incorporation of miscoding ribonucleoside analogs into viral RNA |
| KR20010041533A (ko) | 1998-03-03 | 2001-05-25 | 한센 핀 베네드, 안네 제헤르, 웨이콥 마리안느 | (2e)-5-아미노-5-메틸헥스-2-엔오산n-메틸-n-((1r)-1-(n-메틸-n-((1r)-1-(메틸카르바모일)-2-페닐에틸)카르바모일)-2-(2-나프틸)에틸)아미드의신규한 염 형태 |
| US6312662B1 (en) | 1998-03-06 | 2001-11-06 | Metabasis Therapeutics, Inc. | Prodrugs phosphorus-containing compounds |
| US6475985B1 (en) | 1998-03-27 | 2002-11-05 | Regents Of The University Of Minnesota | Nucleosides with antiviral and anticancer activity |
| AU760688B2 (en) | 1998-10-16 | 2003-05-22 | Merck Sharp & Dohme Limited | Pyrazolo-triazine derivatives as ligands for GABA receptors |
| DE19912636A1 (de) | 1999-03-20 | 2000-09-21 | Aventis Cropscience Gmbh | Bicyclische Heterocyclen, Verfahren zu ihrer Herstellung und ihre Verwendung als Herbizide und pharmazeutische Mittel |
| IL145496A0 (en) * | 1999-03-24 | 2002-06-30 | Exiqon As | Improved synthesis of [2.2.1] bicyclo nucleosides |
| MXPA01012444A (es) * | 1999-06-03 | 2002-07-30 | Abbott Lab | Sintesis de oligonucleotido con acidos de lewis como activadores. |
| AUPQ105499A0 (en) | 1999-06-18 | 1999-07-08 | Biota Scientific Management Pty Ltd | Antiviral agents |
| WO2001019375A1 (en) | 1999-09-15 | 2001-03-22 | Biocryst Pharmaceuticals, Inc. | Inhibiting t-cell proliferation |
| CA2389745C (en) | 1999-11-04 | 2010-03-23 | Shire Biochem Inc. | Method for the treatment or prevention of flaviviridae viral infection using nucleoside analogues |
| JP2003523978A (ja) | 2000-02-18 | 2003-08-12 | シャイアー・バイオケム・インコーポレイテッド | ヌクレオシドアナログを用いるflavivirus感染の処置もしくは予防するための方法 |
| MY164523A (en) | 2000-05-23 | 2017-12-29 | Univ Degli Studi Cagliari | Methods and compositions for treating hepatitis c virus |
| CZ301182B6 (cs) | 2000-05-26 | 2009-12-02 | Idenix (Cayman) Limited | Použití nukleosidových derivátu pro výrobu farmaceutických prostredku pro lécení infekcí vyvolaných flaviviry a pestiviry |
| IL153658A0 (en) | 2000-07-21 | 2003-07-06 | Gilead Sciences Inc | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same |
| US20030008841A1 (en) | 2000-08-30 | 2003-01-09 | Rene Devos | Anti-HCV nucleoside derivatives |
| AU2874902A (en) | 2000-10-18 | 2002-04-29 | Pharmasset Ltd | Modified nucleosides for treatment of viral infections and abnormal cellular proliferation |
| AUPR213700A0 (en) | 2000-12-18 | 2001-01-25 | Biota Scientific Management Pty Ltd | Antiviral agents |
| CZ20032005A3 (en) | 2001-01-22 | 2004-04-14 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of RNA-dependent RNA viral polymerase |
| DE10145223A1 (de) | 2001-09-13 | 2003-04-03 | Basf Ag | Verfahren zur Herstellung von meso-Zeaxanthin |
| WO2003026675A1 (en) | 2001-09-28 | 2003-04-03 | Idenix (Cayman) Limited | Methods and compositions for treating flaviviruses and pestiviruses using 4'-modified nucleoside |
| AT410792B (de) | 2001-12-28 | 2003-07-25 | Dsm Fine Chem Austria Gmbh | Verfahren zur herstellung von geschützten, enantiomeren-angereicherten cyanhydrinen durch in-situ-derivatisierung |
| AU2003217863B9 (en) | 2002-02-28 | 2009-10-29 | Biota Scientific Management Pty Ltd | Nucleotide mimics and their prodrugs |
| EP1485396A2 (en) | 2002-02-28 | 2004-12-15 | Biota, Inc. | Nucleoside 5'-monophosphate mimics and their prodrugs |
| US20040138170A1 (en) | 2002-03-06 | 2004-07-15 | Montgomery John A. | Nucleosides, preparation thereof and use as inhibitors of rna viral polymerases |
| GB0210124D0 (en) | 2002-05-02 | 2002-06-12 | Merck Sharp & Dohme | Therapeutic agents |
| GB0210127D0 (en) | 2002-05-02 | 2002-06-12 | Merck Sharp & Dohme | Therapeutic agents |
| US20040063658A1 (en) | 2002-05-06 | 2004-04-01 | Roberts Christopher Don | Nucleoside derivatives for treating hepatitis C virus infection |
| WO2003100009A2 (en) | 2002-05-23 | 2003-12-04 | Biocryst Pharmaceuticals, Inc. | Enhancing the efficacy of reverse transcriptase and dna polymerase inhibitors (nucleoside analogs) using pnp inhibitors and/or 2'-deoxyguanosine and/or prodrug thereof |
| RU2005118421A (ru) | 2002-11-15 | 2006-01-20 | Айденикс (Кайман) Лимитед (Ky) | 2'-разветвленные нуклеозиды и мутация flaviviridae |
| EP1628685B1 (en) | 2003-04-25 | 2010-12-08 | Gilead Sciences, Inc. | Antiviral phosphonate analogs |
| CA2529949C (en) | 2003-06-26 | 2013-08-06 | Biotron Limited | Antiviral compounds and methods |
| GB0317009D0 (en) | 2003-07-21 | 2003-08-27 | Univ Cardiff | Chemical compounds |
| WO2005009418A2 (en) | 2003-07-25 | 2005-02-03 | Idenix (Cayman) Limited | Purine nucleoside analogues for treating diseases caused by flaviviridae including hepatitis c |
| CN1863813B (zh) | 2003-08-27 | 2011-03-30 | 生物区科学管理控股有限公司 | 作为治疗剂的三环核苷或核苷酸 |
| JP2005185235A (ja) * | 2003-12-26 | 2005-07-14 | Univ Of Tokyo | ウイルス感染に対する感受性診断のための方法およびキット。 |
| JP2005187428A (ja) * | 2003-12-26 | 2005-07-14 | Univ Of Tokyo | 抗mgl抗体によるフィロウイルス治療薬 |
| CN103435581B (zh) * | 2004-03-16 | 2015-08-19 | 贝林格尔.英格海姆国际有限公司 | 吡喃葡萄糖基取代的苯基衍生物、含该化合物的药物、其用途及其制造方法 |
| JP5055564B2 (ja) | 2004-06-15 | 2012-10-24 | メルク・シャープ・エンド・ドーム・コーポレイション | Rna依存性rnaウイルスポリメラーゼの阻害剤としてのc−プリンヌクレオシド類似体 |
| US7560434B2 (en) | 2004-06-22 | 2009-07-14 | Biocryst Pharmaceuticals, Inc. | AZA nucleosides, preparation thereof and use as inhibitors of RNA viral polymerases |
| EP1809301B1 (en) | 2004-09-14 | 2019-11-06 | Gilead Pharmasset LLC | 2-fluoro-2-alkyl-substituted d-ribonolactone intermediates |
| CN101043893A (zh) | 2004-10-21 | 2007-09-26 | 默克公司 | 治疗RNA-依赖性RNA病毒感染的氟化吡咯并[2,3-d]嘧啶核苷 |
| JP2008517912A (ja) | 2004-10-21 | 2008-05-29 | メルク エンド カムパニー インコーポレーテッド | RNA依存性RNAウイルス感染治療用フッ素化ピロロ[2,3−d]ピリミジンヌクレオシド |
| CN101166750A (zh) | 2004-10-29 | 2008-04-23 | 拜奥克里斯特制药公司 | 治疗用呋喃并嘧啶类化合物和噻吩并嘧啶类化合物 |
| JP2008524162A (ja) | 2004-12-16 | 2008-07-10 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | グルコピラノシル置換ベンゼン誘導体、該化合物を含む薬物、その使用及びその製造方法 |
| WO2006094347A1 (en) | 2005-03-08 | 2006-09-14 | Biota Scientific Management Pty Ltd. | Bicyclic nucleosides and nucleotides as therapeutic agents |
| JP5107228B2 (ja) | 2005-03-29 | 2012-12-26 | バイオクライスト ファーマシューティカルズ, インコーポレイテッド | C型肝炎治療 |
| AR056327A1 (es) | 2005-04-25 | 2007-10-03 | Genelabs Tech Inc | Compuestos de nucleosidos para el tratamiento de infecciones virales |
| WO2006121820A1 (en) | 2005-05-05 | 2006-11-16 | Valeant Research & Development | Phosphoramidate prodrugs for treatment of viral infection |
| WO2007027248A2 (en) | 2005-05-16 | 2007-03-08 | Valeant Research & Development | 3', 5' - cyclic nucleoside analogues for treatment of hcv |
| AU2006261593B2 (en) | 2005-06-24 | 2013-01-24 | Biotron Limited | Antiviral compounds and methods |
| WO2007038860A2 (en) | 2005-10-03 | 2007-04-12 | University Health Network | Odcase inhibitors as anti-virals and antibiotics |
| WO2007056170A2 (en) | 2005-11-02 | 2007-05-18 | Bayer Healthcare Ag | Pyrrolo[2,1-f] [1,2,4] triazin-4-ylamines igf-1r kinase inhibitors for the treatment of cancer and other hyperproliferative diseases |
| PL1954264T3 (pl) | 2005-12-01 | 2010-02-26 | Basilea Pharmaceutica Ag | Sposób wytwarzania epoksybutanolowych związków pośrednich |
| US8143393B2 (en) | 2005-12-02 | 2012-03-27 | Bayer Healthcare Llc | Substituted 4-amino-pyrrolotriazine derivatives useful for treating hyper-proliferative disorders and diseases associated with angiogenesis |
| PE20070855A1 (es) | 2005-12-02 | 2007-10-14 | Bayer Pharmaceuticals Corp | Derivados de 4-amino-pirrolotriazina sustituida como inhibidores de quinasas |
| EP1957510A1 (en) | 2005-12-09 | 2008-08-20 | F.Hoffmann-La Roche Ag | Antiviral nucleosides |
| EP1971331A2 (en) | 2005-12-09 | 2008-09-24 | Basilea Pharmaceutica AG | 4-oxo-(iso)tretinoin for the topical treatment of severe dermatological disorders |
| WO2007097991A2 (en) | 2006-02-16 | 2007-08-30 | Pharmasset, Inc. | Methods and kits for dosing of antiviral agents |
| DE102006015378A1 (de) | 2006-04-03 | 2007-10-04 | Ludwig-Maximilians-Universität München | Verfahren zur Synthese von Organoelementverbindungen |
| AR061024A1 (es) | 2006-05-22 | 2008-07-30 | Novartis Ag | Maleato de 5-amino-3- (2'3'-di-o-acetil-beta-d-ribofuranosil)-3h-tiazolo[4,5-d] pirimidin-2-ona. |
| WO2008005542A2 (en) | 2006-07-07 | 2008-01-10 | Gilead Sciences, Inc., | Antiviral phosphinate compounds |
| US20080161324A1 (en) | 2006-09-14 | 2008-07-03 | Johansen Lisa M | Compositions and methods for treatment of viral diseases |
| CL2007003187A1 (es) * | 2006-11-06 | 2008-03-07 | Boehringer Ingelheim Int | Compuestos derivados de bencil-benzonitrilo sustituido con glucopiranosilo y sus sales; procedimiento de preparacion; compuestos intermediarios; proceso para preparar los compuestos intermediarios; composicion farmaceutica; y uso en el tratamiento de |
| EP2120565B1 (en) | 2006-12-20 | 2012-11-28 | Merck Sharp & Dohme Corp. | Nucleoside cyclic phosphoramidates for the treatment of rna-dependent rna viral infection |
| US7951789B2 (en) | 2006-12-28 | 2011-05-31 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| US8071568B2 (en) | 2007-01-05 | 2011-12-06 | Merck Sharp & Dohme Corp. | Nucleoside aryl phosphoramidates for the treatment of RNA-dependent RNA viral infection |
| SG177974A1 (en) | 2007-01-12 | 2012-02-28 | Biocryst Pharm Inc | Antiviral nucleoside analogs |
| CN101730699A (zh) | 2007-03-21 | 2010-06-09 | 百时美施贵宝公司 | 可用于治疗增殖性、变应性、自身免疫性和炎症性疾病的稠合杂环化合物 |
| US7964580B2 (en) | 2007-03-30 | 2011-06-21 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| DK2155758T3 (da) | 2007-05-10 | 2012-11-05 | Biocryst Pharm Inc | Tetrahydrofuro[3,4-d]dioxolanforbindelser til anvendelse i behandlingen af virusinfektioner og cancer |
| CN100532388C (zh) | 2007-07-16 | 2009-08-26 | 郑州大学 | 2’-氟-4’-取代-核苷类似物、其制备方法及应用 |
| NZ583652A (en) | 2007-08-03 | 2012-06-29 | Biotron Ltd | Hepatitis c antiviral compositions and methods |
| KR101502533B1 (ko) | 2007-11-22 | 2015-03-13 | 에스케이케미칼주식회사 | 우수한 안정성을 갖는 택산 유도체 함유 주사제용동결건조 조성물 및 이의 제조방법 |
| TW200942243A (en) | 2008-03-05 | 2009-10-16 | Biocryst Pharm Inc | Antiviral therapeutic agents |
| US8227431B2 (en) | 2008-03-17 | 2012-07-24 | Hetero Drugs Limited | Nucleoside derivatives |
| US7863291B2 (en) | 2008-04-23 | 2011-01-04 | Bristol-Myers Squibb Company | Quinuclidine compounds as alpha-7 nicotinic acetylcholine receptor ligands |
| DK2937350T3 (en) | 2008-04-23 | 2018-04-23 | Gilead Sciences Inc | 1'-SUBSTITUTED CARBA NUCLEOSIDE ANALYSIS FOR ANTIVIRAL TREATMENT |
| WO2010036407A2 (en) | 2008-05-15 | 2010-04-01 | Biocryst Pharmaceuticals, Inc. | Antiviral nucleoside analogs |
| WO2010002877A2 (en) | 2008-07-03 | 2010-01-07 | Biota Scientific Management | Bycyclic nucleosides and nucleotides as therapeutic agents |
| US8012942B2 (en) | 2009-02-10 | 2011-09-06 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| AR075584A1 (es) | 2009-02-27 | 2011-04-20 | Intermune Inc | COMPOSICIONES TERAPEUTICAS QUE COMPRENDEN beta-D-2'-DESOXI-2'-FLUORO-2'-C-METILCITIDINA Y UN DERIVADO DE ACIDO ISOINDOL CARBOXILICO Y SUS USOS. COMPUESTO. |
| AU2010226466A1 (en) | 2009-03-20 | 2011-10-20 | Alios Biopharma, Inc. | Substituted nucleoside and nucleotide analogs |
| CA2755950C (en) | 2009-03-24 | 2017-04-25 | Biocryst Pharmaceuticals, Inc. | Useful pharmaceutical salts of 7-[(3r,4r)-3-hydroxy-4-hydroxymethyl-pyrrolidin-1-ylmethyl]-3,5-dihydro-pyrrolo[3,2-d]pyrimidin-4-one |
| TWI598358B (zh) | 2009-05-20 | 2017-09-11 | 基利法瑪席特有限責任公司 | 核苷磷醯胺 |
| CN102471327A (zh) | 2009-07-21 | 2012-05-23 | 吉里德科学公司 | 黄病毒科病毒的抑制剂 |
| US7973013B2 (en) | 2009-09-21 | 2011-07-05 | Gilead Sciences, Inc. | 2'-fluoro substituted carba-nucleoside analogs for antiviral treatment |
| US10023600B2 (en) | 2009-09-21 | 2018-07-17 | Gilead Sciences, Inc. | Processes and intermediates for the preparation of 1′-substituted carba-nucleoside analogs |
| BR122021012877B8 (pt) | 2009-09-21 | 2021-12-14 | Gilead Sciences Inc | Compostos análogos de 2'-flúor substituído carbanucleosídeo, uso dos mesmos e composição farmacêutica que os compreende |
| US8455451B2 (en) | 2009-09-21 | 2013-06-04 | Gilead Sciences, Inc. | 2'-fluoro substituted carba-nucleoside analogs for antiviral treatment |
| SG181757A1 (en) | 2009-12-28 | 2012-07-30 | Dev Center Biotechnology | Novel pyrimidine compounds as mtor and pi3k inhibitors |
| JP5872539B2 (ja) | 2010-03-31 | 2016-03-01 | ギリアド ファーマセット エルエルシー | プリンヌクレオシドホスホルアミダート |
| KR101715981B1 (ko) | 2010-03-31 | 2017-03-13 | 길리애드 파마셋 엘엘씨 | 뉴클레오사이드 포스포르아미데이트 |
| TW201201815A (en) * | 2010-05-28 | 2012-01-16 | Gilead Sciences Inc | 1'-substituted-carba-nucleoside prodrugs for antiviral treatment |
| US9090642B2 (en) * | 2010-07-19 | 2015-07-28 | Gilead Sciences, Inc. | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs |
| TW201305185A (zh) | 2010-09-13 | 2013-02-01 | Gilead Sciences Inc | 用於抗病毒治療之2’-氟取代之碳-核苷類似物 |
| MX2013003179A (es) | 2010-09-20 | 2013-04-24 | Gilead Sciences Inc | Analogos de carba-nucleosidos sustituidos con 2 '-fluoro para tratamiento antiviral. |
| AP3584A (en) | 2010-09-22 | 2016-02-09 | Alios Biopharma Inc | Substituted nucleotide analogs |
| HUE036387T2 (hu) | 2010-10-15 | 2018-07-30 | Biocryst Pharm Inc | Pirrolopirimidin származékok vírusfertõzések kezelésében való felhasználásra |
| PT3042910T (pt) | 2010-11-30 | 2019-04-16 | Gilead Pharmasset Llc | 2'-espiro-nucleósidos para utilização na terapia da hepatite c |
| NZ613370A (en) | 2010-12-20 | 2015-05-29 | Gilead Sciences Inc | Combinations for treating hcv |
| JP2014515023A (ja) | 2011-04-13 | 2014-06-26 | メルク・シャープ・アンド・ドーム・コーポレーション | 2’−置換ヌクレオシド誘導体およびウイルス疾患の処置のためのその使用方法 |
| BR112013026219A2 (pt) | 2011-04-13 | 2016-07-26 | Gilead Sciences Inc | análogos de n-nucleosídeo 1'-substituída pirimidina para tratamento antiviral |
| US20150050305A1 (en) | 2011-05-13 | 2015-02-19 | Zoetis Llc | Hendra and nipah virus g glycoprotein immunogenic compositions |
| WO2013039861A2 (en) * | 2011-09-12 | 2013-03-21 | modeRNA Therapeutics | Engineered nucleic acids and methods of use thereof |
| NZ623396A (en) | 2011-09-16 | 2016-07-29 | Gilead Pharmasset Llc | Methods for treating hcv |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| US20130143835A1 (en) * | 2011-12-05 | 2013-06-06 | Medivir Ab | HCV Polymerase Inhibitors |
| WO2013084165A1 (en) | 2011-12-05 | 2013-06-13 | Medivir Ab | Hcv polymerase inhibitors |
| CA2860234A1 (en) * | 2011-12-22 | 2013-06-27 | Alios Biopharma, Inc. | Substituted phosphorothioate nucleotide analogs |
| PE20150132A1 (es) | 2012-05-22 | 2015-02-14 | Idenix Pharmaceuticals Inc | Compuestos d-aminoacidos para enfermedad hepatica |
| US9556216B2 (en) | 2012-08-31 | 2017-01-31 | Novartis Ag | 2′-Ethynyl nucleoside derivatives for treatment of viral infections |
| KR20150054795A (ko) | 2012-09-10 | 2015-05-20 | 에프. 호프만-라 로슈 아게 | B형 간염 바이러스 감염의 치료 및 예방을 위한 6-아미노산 헤테로아릴다이하이드로피리미딘 |
| WO2014042433A2 (en) * | 2012-09-14 | 2014-03-20 | Kainos Medicine, Inc. | Compounds and compositions for modulating adenosine a3 receptor activity |
| US20150291596A1 (en) | 2012-11-16 | 2015-10-15 | Biocryst Pharmaceuticals, Inc. | Antiviral azasugar-containing nucleosides |
| RU2015123641A (ru) | 2012-11-19 | 2017-01-10 | Мерк Шарп И Доум Корп. | 2-алкинилзамещенные производные нуклеозидов, предназначенные для лечения вирусных заболеваний |
| LT2935303T (lt) * | 2012-12-21 | 2021-03-25 | Janssen Biopharma, Inc. | 4'-fluor-nukleozidai, 4'-fluor-nukleotidai ir jų analogai, skirti hcv gydymui |
| WO2014116755A1 (en) | 2013-01-22 | 2014-07-31 | Massachusetts Institute Of Technology | Uses of dihydro bases |
| US10034893B2 (en) | 2013-02-01 | 2018-07-31 | Enanta Pharmaceuticals, Inc. | 5, 6-D2 uridine nucleoside/tide derivatives |
| AU2014250762A1 (en) | 2013-04-12 | 2015-10-29 | Achillion Pharmaceuticals, Inc. | Highly active nucleoside derivative for the treatment of HCV |
| US9542154B2 (en) | 2013-06-25 | 2017-01-10 | Intel Corporation | Fused multiply add operations using bit masks |
| UA117375C2 (uk) * | 2013-09-04 | 2018-07-25 | Медівір Аб | Інгібітори полімерази hcv |
| CA2923090C (en) | 2013-09-11 | 2018-08-21 | Poxel | Methods and pharmaceutical compositions for the treatment of hepatitis b virus infection |
| UA119050C2 (uk) | 2013-11-11 | 2019-04-25 | Ґілеад Саєнсиз, Інк. | ПІРОЛО[1.2-f][1.2.4]ТРИАЗИНИ, ЯКІ ВИКОРИСТОВУЮТЬСЯ ДЛЯ ЛІКУВАННЯ РЕСПІРАТОРНО-СИНЦИТІАЛЬНИХ ВІРУСНИХ ІНФЕКЦІЙ |
| CN105899508B (zh) | 2014-01-30 | 2017-07-04 | 豪夫迈·罗氏有限公司 | 用于治疗和预防乙型肝炎病毒感染的新型二氢喹嗪酮类化合物 |
| SI3114128T1 (sl) | 2014-03-07 | 2019-04-30 | F. Hoffmann-La Roche Ag | Novi 6-spojeni heteroarildihidropirimidini za zdravljenje in preprečevanje okužbe z virusom hepatitisa B |
| WO2015173164A1 (en) | 2014-05-13 | 2015-11-19 | F. Hoffmann-La Roche Ag | Novel dihydroquinolizinones for the treatment and prophylaxis of hepatitis b virus infection |
| US9504701B2 (en) | 2014-06-02 | 2016-11-29 | The Board Of Regents Of The University Of Texas System | Methods for treating viral infections using hydrogen sulfide donors |
| US9616076B2 (en) * | 2014-06-02 | 2017-04-11 | The Board Of Regents Of The University Of Texas Systems | Methods for treating viral infections using hydrogen sulfide donors |
| WO2016012470A1 (en) | 2014-07-25 | 2016-01-28 | F. Hoffmann-La Roche Ag | New amorphous and crystalline forms of (3s)-4-[[(4r)-4-(2-chloro-4-fluoro-phenyl)-5-methoxycarbonyl-2-thiazol-2-yl-1, 4-dihydropyrimidin-6-yl]methyl]morpholine-3-carboxylic acid |
| BR112017002970B1 (pt) | 2014-08-14 | 2023-04-11 | F. Hoffmann-La Roche Ag | Novas piridazonas e triazinonas para o tratamento e profilaxia da infecção pelo vírus da hepatite b |
| TWI767201B (zh) | 2014-10-29 | 2022-06-11 | 美商基利科學股份有限公司 | 絲狀病毒科病毒感染之治療 |
| US9637485B2 (en) | 2014-11-03 | 2017-05-02 | Hoffmann-La Roche Inc. | 6,7-dihydrobenzo[a]quinolizin-2-one derivatives for the treatment and prophylaxis of hepatitis B virus infection |
| US9676793B2 (en) | 2014-12-23 | 2017-06-13 | Hoffmann-Laroche Inc. | Co-crystals of 5-amino-2-oxothiazolo[4,5-d]pyrimidin-3(2H)-yl-5-hydroxymethyl tetrahydrofuran-3-yl acetate and methods for preparing and using the same |
| AR103222A1 (es) | 2014-12-23 | 2017-04-26 | Hoffmann La Roche | Procedimiento para la preparación de análogos de 4-fenil-5-alcoxicarbonil-2-tiazol-2-il-1,4-dihidropirimidina |
| CN107108610B (zh) | 2014-12-30 | 2019-06-04 | 豪夫迈·罗氏有限公司 | 用于治疗和预防肝炎b病毒感染的新的四氢吡啶并嘧啶和四氢吡啶并吡啶化合物 |
| JP2018504891A (ja) | 2014-12-31 | 2018-02-22 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | リアルタイムPCRによる細胞溶解物からのHBV cccDNA定量化のための新規ハイスループット法 |
| MA41338B1 (fr) | 2015-01-16 | 2019-07-31 | Hoffmann La Roche | Composés de pyrazine pour le traitement de maladies infectieuses |
| EP3250685A1 (en) | 2015-01-27 | 2017-12-06 | F. Hoffmann-La Roche AG | Recombinant hbv cccdna, the method to generate thereof and the use thereof |
| CN107207505B (zh) | 2015-02-11 | 2018-12-14 | 豪夫迈·罗氏有限公司 | 治疗和预防乙型肝炎病毒感染的 2-氧代-6,7-二氢苯并[a]喹嗪-3-甲酸衍生物 |
| CN115887465A (zh) | 2015-09-16 | 2023-04-04 | 吉利德科学公司 | 治疗沙粒病毒科和冠状病毒科病毒感染的方法 |
| WO2017184668A1 (en) | 2016-04-20 | 2017-10-26 | Gilead Sciences, Inc. | Methods for treating flaviviridae virus infections |
| AU2018218179B2 (en) | 2017-02-08 | 2023-05-04 | Biotron Limited | Methods of treating influenza |
| CN116036112A (zh) | 2017-03-14 | 2023-05-02 | 吉利德科学公司 | 治疗猫冠状病毒感染的方法 |
| CN110636884B (zh) | 2017-05-01 | 2022-10-04 | 吉利德科学公司 | 新结晶形式 |
| WO2019014247A1 (en) | 2017-07-11 | 2019-01-17 | Gilead Sciences, Inc. | COMPOSITIONS COMPRISING POLYMERASE RNA INHIBITOR AND CYCLODEXTRIN FOR THE TREATMENT OF VIRAL INFECTIONS |
| CN111265532A (zh) | 2020-01-21 | 2020-06-12 | 中国人民解放军军事科学院军事医学研究院 | 取代氨基丙酸酯类化合物在治疗2019-nCoV感染中的应用 |
| CN118662520A (zh) | 2020-01-27 | 2024-09-20 | 吉利德科学公司 | 用于治疗SARS CoV-2感染的方法 |
| CN115298181B (zh) * | 2020-03-12 | 2024-08-16 | 吉利德科学公司 | 制备1’-氰基核苷的方法 |
-
2015
- 2015-10-27 TW TW109109475A patent/TWI767201B/zh active
- 2015-10-27 TW TW109120726A patent/TWI740546B/zh active
- 2015-10-27 TW TW104135248A patent/TWI698444B/zh active
- 2015-10-27 TW TW104135247A patent/TWI687432B/zh active
- 2015-10-28 BR BR102015027413A patent/BR102015027413A2/pt not_active Application Discontinuation
- 2015-10-29 SM SM20180381T patent/SMT201800381T1/it unknown
- 2015-10-29 WO PCT/US2015/057932 patent/WO2016069825A1/en active Application Filing
- 2015-10-29 US US14/926,062 patent/US9724360B2/en active Active
- 2015-10-29 SG SG11201702904RA patent/SG11201702904RA/en unknown
- 2015-10-29 EP EP20152978.1A patent/EP3695844A1/en active Pending
- 2015-10-29 EP EP15797205.0A patent/EP3212174B1/en active Active
- 2015-10-29 JP JP2017520934A patent/JP6487547B2/ja active Active
- 2015-10-29 TR TR2018/09518T patent/TR201809518T4/tr unknown
- 2015-10-29 SI SI201531150T patent/SI3366295T1/sl unknown
- 2015-10-29 CN CN202410806375.0A patent/CN118812607A/zh active Pending
- 2015-10-29 EP EP18167340.1A patent/EP3366295B1/en active Active
- 2015-10-29 CA CA2963832A patent/CA2963832C/en active Active
- 2015-10-29 HU HUE15797205A patent/HUE039231T2/hu unknown
- 2015-10-29 CR CR20170483A patent/CR20170483A/es unknown
- 2015-10-29 NZ NZ730803A patent/NZ730803A/en unknown
- 2015-10-29 MD MDA20170046A patent/MD20170046A2/ro not_active Application Discontinuation
- 2015-10-29 LT LTEP15797205.0T patent/LT3212174T/lt unknown
- 2015-10-29 RS RS20180822A patent/RS57425B1/sr unknown
- 2015-10-29 MX MX2017005250A patent/MX2017005250A/es unknown
- 2015-10-29 EA EA201790597A patent/EA032239B1/ru unknown
- 2015-10-29 KR KR1020177014110A patent/KR102337664B1/ko active Active
- 2015-10-29 SI SI201530258T patent/SI3212174T1/en unknown
- 2015-10-29 PL PL18167340T patent/PL3366295T3/pl unknown
- 2015-10-29 HU HUE18167340A patent/HUE049192T2/hu unknown
- 2015-10-29 DK DK15797205.0T patent/DK3212174T3/en active
- 2015-10-29 KR KR1020217039965A patent/KR102453808B1/ko active Active
- 2015-10-29 CN CN202111041161.1A patent/CN113620992B/zh active Active
- 2015-10-29 PL PL15801008T patent/PL3212175T3/pl unknown
- 2015-10-29 UY UY0001036376A patent/UY36376A/es unknown
- 2015-10-29 SG SG10202008772UA patent/SG10202008772UA/en unknown
- 2015-10-29 MA MA040872A patent/MA40872A/fr unknown
- 2015-10-29 CA CA3184285A patent/CA3184285A1/en active Pending
- 2015-10-29 SI SI201531764T patent/SI3212175T1/sl unknown
- 2015-10-29 CN CN202111480947.3A patent/CN114191438A/zh active Pending
- 2015-10-29 ES ES15797205.0T patent/ES2674806T3/es active Active
- 2015-10-29 CN CN201580059611.6A patent/CN107073005B/zh active Active
- 2015-10-29 PT PT157972050T patent/PT3212174T/pt unknown
- 2015-10-29 NZ NZ730809A patent/NZ730809A/en unknown
- 2015-10-29 ES ES15801008T patent/ES2904298T3/es active Active
- 2015-10-29 UA UAA201703584A patent/UA121485C2/uk unknown
- 2015-10-29 EA EA201790630A patent/EA201790630A1/ru unknown
- 2015-10-29 CN CN202110878274.0A patent/CN113549120A/zh active Pending
- 2015-10-29 JP JP2017520938A patent/JP6220484B1/ja active Active
- 2015-10-29 DK DK18167340.1T patent/DK3366295T3/da active
- 2015-10-29 ES ES18167340T patent/ES2785034T3/es active Active
- 2015-10-29 MA MA052506A patent/MA52506A/fr unknown
- 2015-10-29 CA CA2963907A patent/CA2963907C/en active Active
- 2015-10-29 PL PL15797205T patent/PL3212174T3/pl unknown
- 2015-10-29 KR KR1020177014043A patent/KR102694960B1/ko active Active
- 2015-10-29 PE PE2017000745A patent/PE20171439A1/es unknown
- 2015-10-29 PT PT158010082T patent/PT3212175T/pt unknown
- 2015-10-29 AR ARP150103506A patent/AR102468A1/es unknown
- 2015-10-29 BR BR112017007636A patent/BR112017007636A2/pt not_active Application Discontinuation
- 2015-10-29 WO PCT/US2015/057933 patent/WO2016069826A1/en active Application Filing
- 2015-10-29 AU AU2015339223A patent/AU2015339223B2/en active Active
- 2015-10-29 AU AU2015339222A patent/AU2015339222B2/en active Active
- 2015-10-29 MA MA040867A patent/MA40867A/fr unknown
- 2015-10-29 KR KR1020247026762A patent/KR20240125990A/ko active Pending
- 2015-10-29 CN CN201580059613.5A patent/CN107074902A/zh active Pending
- 2015-10-29 EP EP21209687.9A patent/EP4036099B1/en active Active
- 2015-10-29 PT PT181673401T patent/PT3366295T/pt unknown
- 2015-10-29 ME MEP-2018-183A patent/ME03070B/me unknown
- 2015-10-29 NZ NZ745328A patent/NZ745328A/en unknown
- 2015-10-29 MY MYPI2017701244A patent/MY195823A/en unknown
- 2015-10-29 AR ARP150103505A patent/AR102467A1/es not_active Application Discontinuation
- 2015-10-29 PE PE2017002319A patent/PE20180202A1/es unknown
- 2015-10-29 CU CUP2017000056A patent/CU20170056A7/es unknown
- 2015-10-29 SG SG11201702903TA patent/SG11201702903TA/en unknown
- 2015-10-29 EP EP15801008.2A patent/EP3212175B1/en active Active
- 2015-10-29 US US14/926,063 patent/US11344565B2/en active Active
- 2015-10-29 CR CR20170165A patent/CR20170165A/es unknown
- 2015-10-29 WO PCT/US2015/057934 patent/WO2016069827A1/en active Application Filing
- 2015-10-29 PH PH1/2017/500631A patent/PH12017500631B1/en unknown
- 2015-10-29 LT LTEP18167340.1T patent/LT3366295T/lt unknown
- 2015-10-29 KR KR1020227034888A patent/KR20220140656A/ko not_active Withdrawn
- 2015-10-29 MX MX2017005252A patent/MX2017005252A/es unknown
- 2015-10-29 KR KR1020177014042A patent/KR101822348B1/ko active IP Right Grant
- 2015-10-29 CU CUP2017000145A patent/CU20170145A7/xx unknown
-
2016
- 2016-08-24 US US15/246,240 patent/US9949994B2/en active Active
-
2017
- 2017-04-04 IL IL251560A patent/IL251560A0/en unknown
- 2017-04-12 IL IL251707A patent/IL251707B/en active IP Right Grant
- 2017-04-21 MX MX2020012560A patent/MX2020012560A/es unknown
- 2017-04-24 SV SV2017005424A patent/SV2017005424A/es unknown
- 2017-04-24 DO DO2017000103A patent/DOP2017000103A/es unknown
- 2017-04-24 CO CONC2017/0003960A patent/CO2017003960A2/es unknown
- 2017-04-24 EC ECIEPI201725261A patent/ECSP17025261A/es unknown
- 2017-04-26 CL CL2017001040A patent/CL2017001040A1/es unknown
- 2017-04-27 SA SA517381419A patent/SA517381419B1/ar unknown
- 2017-06-06 JP JP2017111470A patent/JP6757294B2/ja active Active
- 2017-10-24 CL CL2017002693A patent/CL2017002693A1/es unknown
- 2017-10-30 EC ECIEPI201772474A patent/ECSP17072474A/es unknown
- 2017-11-01 UY UY0001037464A patent/UY37464A/es not_active Application Discontinuation
- 2017-11-03 SV SV2017005561A patent/SV2017005561A/es unknown
-
2018
- 2018-01-19 ZA ZA201800414A patent/ZA201800414B/en unknown
- 2018-02-22 US US15/902,690 patent/US10251898B2/en active Active
- 2018-07-06 JP JP2018129006A patent/JP6671424B2/ja active Active
- 2018-07-17 HR HRP20181130TT patent/HRP20181130T1/hr unknown
- 2018-08-10 CY CY20181100844T patent/CY1120893T1/el unknown
- 2018-10-23 AU AU2018253483A patent/AU2018253483B2/en active Active
-
2019
- 2019-01-07 JP JP2019000664A patent/JP2019048901A/ja not_active Withdrawn
- 2019-01-23 HK HK19101159.4A patent/HK1258795A1/zh unknown
- 2019-02-12 US US16/274,049 patent/US10695357B2/en active Active
- 2019-02-22 AU AU2019201232A patent/AU2019201232B2/en active Active
- 2019-11-22 US US16/692,966 patent/US20200197422A1/en not_active Abandoned
-
2020
- 2020-02-14 JP JP2020023613A patent/JP2020090536A/ja not_active Withdrawn
- 2020-03-06 JP JP2020038878A patent/JP7158428B2/ja active Active
- 2020-04-14 CY CY20201100351T patent/CY1122946T1/el unknown
- 2020-05-22 US US16/881,419 patent/US11266666B2/en active Active
- 2020-06-12 AU AU2020203892A patent/AU2020203892A1/en not_active Abandoned
- 2020-07-08 PH PH12020551055A patent/PH12020551055A1/en unknown
-
2021
- 2021-03-09 AU AU2021201474A patent/AU2021201474B2/en active Active
-
2022
- 2022-02-07 US US17/665,724 patent/US20220354873A1/en active Pending
- 2022-02-10 JP JP2022019651A patent/JP2022065066A/ja not_active Withdrawn
- 2022-02-18 JP JP2022024119A patent/JP2022068297A/ja active Pending
- 2022-12-09 AU AU2022283772A patent/AU2022283772A1/en not_active Abandoned
-
2023
- 2023-05-02 AU AU2023202679A patent/AU2023202679A1/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012012776A1 (en) * | 2010-07-22 | 2012-01-26 | Gilead Sciences, Inc. | Methods and compounds for treating paramyxoviridae virus infections |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI767201B (zh) | 絲狀病毒科病毒感染之治療 | |
| AU2020233714B2 (en) | Methods for treating Arenaviridae and Coronaviridae virus infections | |
| OA20831A (en) | Methods for treating filoviridae virus infections. |