WO2010036407A2 - Antiviral nucleoside analogs - Google Patents
Antiviral nucleoside analogs Download PDFInfo
- Publication number
- WO2010036407A2 WO2010036407A2 PCT/US2009/044214 US2009044214W WO2010036407A2 WO 2010036407 A2 WO2010036407 A2 WO 2010036407A2 US 2009044214 W US2009044214 W US 2009044214W WO 2010036407 A2 WO2010036407 A2 WO 2010036407A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- substituted
- compound
- aryl
- heteroaryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC1C(*)(**)C(*)(*)*(*)C1 Chemical compound CC1C(*)(**)C(*)(*)*(*)C1 0.000 description 4
- YGNXFCPNJCLCLM-WCFLWFBJSA-N C[C@@H]1[C@H](C(N)=O)O[C@H](COCc2ccccc2)C1 Chemical compound C[C@@H]1[C@H](C(N)=O)O[C@H](COCc2ccccc2)C1 YGNXFCPNJCLCLM-WCFLWFBJSA-N 0.000 description 1
- COHHGYUFUQLCNO-SVCMUZATSA-N C[C@@]1([C@H]([C@H](C2C(OC)=O)OC=C2C(OC)=O)O[C@H](COCc2ccccc2)[C@H]1OCc1ccccc1)OCc1ccccc1 Chemical compound C[C@@]1([C@H]([C@H](C2C(OC)=O)OC=C2C(OC)=O)O[C@H](COCc2ccccc2)[C@H]1OCc1ccccc1)OCc1ccccc1 COHHGYUFUQLCNO-SVCMUZATSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- Viral diseases are a major cause of death and economic loss in the world.
- the Flaviviridae family of viruses includes the three genera flaviviruses (including dengue, West Nile, and yellow fever viruses), hepacivirus (including hepatitus C virus (HCV)), and pesti viruses (including bovine viral diarrhea virus, BVDV).
- the disease states and conditions caused by members of this family include yellow fever, dengue, Japanese encephalitis, St. Louis encephalitis, Hepatitis B and C, West Nile disease, and AIDS.
- human immunodeficiency virus (HIV), hepatitis B virus (HBV) and hepatitis C virus (HCV) infections are responsible for the largest number of viral related deaths worldwide.
- Ribavirin (l- ⁇ -D-ribofuranosyl-l-l,2,4-triazole-3-carboxamide) is a synthetic, non-interferon-inducing, broad spectrum antiviral nucleoside. Ribavirin is structurally similar to guanosine, and has in vitro activity against several DNA and RNA viruses including Flaviviridae (Davis. Gastroenterology 118:S1O4-S114, 2000). Ribavirin reduces serum amino transferase levels to normal in 40% of patients, but it does not lower serum levels of HCV-RNA (Davis. Gastroenterology 118:S104-S114, 2000). Thus, ribavirin alone is not effective in reducing viral RNA levels. Additionally, ribavirin has significant toxicity and is known to induce anemia.
- NS 5 B apoenzyme The X-ray crystal structure of NS 5 B apoenzyme has been determined and three recent publications describe the unusual shape of the molecule. This unique shape for a polymerase, resembling a flat sphere, is attributed to extensive interactions between the fingers and thumb subdomains in such a way that the active site is completely encircled, forming a cavity 15 A across and 20 A deep. Modeling studies showed that the NS5B apoenzyme can accommodate the template-primer without large movement of the subdomains, suggesting that the structure is preserved during the polymerization reaction.
- the RdRp polypeptides from various members of the Flaviviridae family and other viral families have been shown to be conserved (J. A. Bruenn, Nucleic Acids Research, Vol. 19, No. 2 p. 217, 1991).
- compositions comprising a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof, and a pharmaceutically acceptable carrier.
- the composition can optionally comprise one or more additional anti-viral or anti-cancer agents.
- Certain embodiments of the present invention provide a method for inhibiting a viral RNA or DNA polymerase comprising contacting the polymerase (in vitro or in vivo) with an effective inhibitory amount of a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof.
- Certain embodiments of the present invention provide a method for treating cancer in an animal comprising administering to the animal an effective amount of a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof.
- Certain embodiments of the present invention provide a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof, for use in medical therapy (e.g., for use in treating a viral infection or for use in treating cancer).
- Certain embodiments of the present invention provide the use of a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof, to prepare a medicament useful for treating a viral infection in an animal (e.g., a human). Certain embodiments of the present invention provide the use of a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof, to prepare a medicament useful for treating cancer in an animal (e.g., a human).
- Certain embodiments of the present invention provide a compound of the invention for use in therapy.
- Certain embodiments of the present invention provide a compound of the invention for treating cancer. Certain embodiments of the present invention provide novel synthetic intermediates and synthetic methods that are disclosed herein as being useful for preparing compounds of Formula I to III, or a salt or prodrug thereof. Some compounds of Formula I to III may be useful as synthetic intermediates for preparing other compounds of Formula I to III. Certain embodiments of the present invention provide a compound as described herein for use in medical treatment or diagnosis.
- Certain embodiments of the present invention provide the use of a compound as described herein to prepare a medicament useful for treating a viral infection in an animal. Certain embodiments of the invention provide the use of a compound as described herein to prepare a medicament useful for treating cancer in an animal.
- pharmaceutically acceptable salt refers to a compound of the present disclosure derived from pharmaceutically acceptable bases, inorganic or organic acids.
- suitable acids include, but are not limited to, hydrochloric, hydrobromic, sulfuric, nitric, perchloric, fumaric, maleic, phosphoric, glycollic, lactic, salicyclic, succinic, toluene-p-sulfonic, tartaric, acetic, citric, methanesulfonic, formic, benzoic, malonic, naphthalene-2- sulfonic, trifluoroacetic and benzenesulfonic acids.
- alkyl refers to alkyl groups having from 1 to 6 carbon atoms. This term is exemplified by groups such as methyl, ethyl, n- propyl, iso-propyl, n-butyl, t-butyl, n-pentyl and the like. In a specific embodiment, the alkyl groups have from 1-4 carbon atoms and are referred to as lower alkyl.
- substituted alkyl refers to an alkyl group having from 1 to 3 substituents, said substituents being selected from the group consisting of alkoxy, alkoxyalkyl, tri(C 1 -C 4 alkyl)silyl, substituted alkoxy, acyl, substituted acyl, acylamino, acyloxy, oxyacyl, amino, substituted amino, aminoacyl, aryl, substituted aryl, aryloxy, substituted aryloxy, cyano, halogen, hydroxyl, nitro, N 3 , carboxyl, carboxyl esters, thiol, thioalkyl, substituted thioalkyl, thioaryl, substituted thioaryl, thioheteroaryl, substituted thioheteroaryl, thiocycloalkyl, substituted thiocycloalkyl, substituted thiocycloallcyl, thioheterocyclic, substituted thi
- substituted alkenyl refers to alkenyl groups having from 1 to 3 substituents, said substituents being selected from those describe above for a substituted alkyl.
- substituted alkynyl refers to alkynyl groups having from 1 to 3 substituents, said substituents being selected those describe above for a substituted alkyl.
- acyl refers to the groups alkyl-C(O) -, alkenyl- C(O) -, alkynyl-C(O) -, cycloalkyl-C(O) -, aryl-C(O) -, heteroaryl-C(O) -, and heterocyclic-C(O).
- substituted acyl refers to the groups substituted alkyl-C(O)-, substituted alkenyl-C(O) -, substituted alkynyl-C(O) -, substituted cycloalkyl-C(O) -, substituted aryl-C (O)-, substituted heteroaryl-C(O), and substituted heterocyclic-C(O) -.
- acylamino refers to the group-C(O)NZ!Z2 where each Z 1 and Z 2 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl, and the substituents described above in the definition of substituted alkyl.
- oxyacyl refers to the groups alkyl-OC(O) -, substituted alkyl-OC(O)-, alkenyl-OC(O)-, substituted alkenyl-OC(O) -, alkynyl- OC(O) -, substituted alkynyl-OC(O)-, aryl-OC(O)-, substituted aryl-OC(O)-, cycloalkyl-OC(O)-, substituted cycloalkyl-OC(O)-, heteroaryl-OC(O) -, substituted heteroaryl-OC(O) -, heterocyclic- OC(O)-, and substituted heterocyclic-OC(O)-.
- amino refers to the group -NH 2 .
- substituted amino refers to the group-N Z 1 Z 2 where Z 1 and Z 2 are as described above in the definition of acylamino, provided that Z 1 and Z 2 are both not hydrogen.
- aryl refers to a monovalent aromatic cyclic group of from 6 to 14 carbon atoms having a single ring (e.g., phenyl) or multiple condensed rings (e.g., naphthyl or anthryl) which condensed rings may or may not be aromatic.
- exemplary aryls include, but are not limited to, phenyl and naphthyl.
- substituted aryl refers to aryl groups which are substituted with from 1 to 3 substituents selected from alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl, and those substituents described above in the definition of substituted alkyl.
- aryloxy refers to the group aryl-O- that includes, by way of example but not limitation, phenoxy, naphthoxy, and the like.
- substituted aryloxy refers to substituted aryl- O-groups.
- carboxyl refers to -COOH or salts thereof.
- carboxyl esters refers to the groups-C(O)O- alkyl, -C (O)O-substituted alkyl, -C(O)O-aryl, and-C(O)O-substituted aryl.
- cycloalkyl refers to a saturated or unsaturated cyclic hydrocarbon ring systems, such as those containing 1 to 3 rings and 3 to 7 carbons per ring.
- exemplary groups include but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and adamantyl.
- cycloalkoxy refers to -O-cycloalkyl groups.
- substituted cycloalkoxy refers to-O-substituted cycloalkyl groups.
- heteroaryl refers to an aromatic group of from 1 to 10 carbon atoms and 1 to 4 heteroatoms selected from the group consisting of oxygen, nitrogen, sulfur in the ring.
- the sulfur and nitrogen heteroatoms atoms may also be present in their oxidized forms.
- Such heteroaryl groups can have a single ring (e.g., pyridyl or furyl) or multiple condensed rings (e.g., indolizinyl or benzothienyl) wherein the condensed rings may or may not be aromatic and/or contain a heteroatom.
- Exemplary heteroaryl groups include, but are not limited to, heteroaryls include pyridyl, pyrrolyl, thienyl, indolyl, thiophenyl, and furyl.
- substituted heteroaryl refers to heteroaryl groups that are substituted with from 1 to 3 substituents selected from the same group of substituents defined for substituted aryl.
- heteroaryloxy refers to the group -O- heteroaryl.
- heterocycle or “heterocyclic” or “heterocycloalkyl” refers to a saturated or unsaturated group (but not heteroaryl) having a single ring or multiple condensed rings, from 1 to 10 carbon atoms and from 1 to 4 hetero atoms selected from the group consisting of nitrogen, oxygen, sulfur, within the ring wherein, in fused ring systems, one or more the rings can be cycloalkyl, aryl or heteroaryl provided that the point of attachment is through the heterocyclic ring.
- the sulfur and nitrogen heteroatoms atoms may also be present in their oxidized forms.
- substituted heterocycle or “substituted heterocyclic” or “substituted heterocycloalkyl” refers to heterocycle groups that are substituted with from 1 to 3 of the same substituents as defined for substituted aryl.
- heterocycles and heteroaryls include, but are not limited to, azetidine, pyrrole, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, phenanthroline, isothiazole, phenazine, isoxazole, phenoxazine, phenothiazine, imidazolidine, imidazoline, piperidine, piperazine, indoline, phthalimide, 1,2, 3,4-tetrahydroisoquinoline, 4,5, 6,
- heterocyclyloxy refers to the group -O- heterocyclic.
- substituted heterocyclyloxy refers to the group-O-substituted heterocyclic.
- phosphate refers to the groups- OP(O)(OH) 2 (monophosphate or phospho), -OP(O)(OH)OP(O)(OH) 2 (diphosphate or diphospho) and -OP(O)(OH)OP(O)(OH)OP(O)(OH) 2 (triphosphate or triphospho) or salts thereof including partial salts thereof. It is understood that the initial oxygen of the mono-, di-, and triphosphate may include the oxygen atom of a sugar.
- phosphate esters refers to the mono-, di-and tri-phosphate groups described above wherein one or more of the hydroxyl groups is replaced by an alkoxy group.
- phosphonate refers to the groups -0P(0)(Z4)(0H) or-OP(O)
- each Z 4 is independently selected from hydrogen, alkyl, substituted alkyl, carboxylic acid, and carboxyl ester. It is understood that the initial oxygen of the phosphonate may include the oxygen of a sugar.
- thiol refers to the group -SH.
- thioalkyl or "alkylthioether” or “thioalkoxy” refers to the group- S-alkyl.
- substituted thioalkyl or “substituted alkylthioether” or “substituted thioalkoxy” refers to the group -S-substituted alkyl.
- thiocycloalkyl refers to the group -S- cycloalkyl.
- substituted thiocycloalkyl refers to the group - S-substituted cycloalkyl.
- thioaryl refers to the group -S-aryl.
- thioheterocyclic refers to the group -S- heterocyclic.
- amino acid sidechain refers to the Z 7 substituent of ⁇ -amino acids of the formula Z 6 NHCH(Z 7 )COOH where Z 7 is selected from the group consisting of hydrogen, alkyl, substituted alkyl and aryl and Z 6 is hydrogen or together with Z 7 and the nitrogen and carbon atoms bound thereto respectively form a heterocyclic ring.
- the ⁇ -amino acid sidechain is the sidechain one of the twenty naturally occurring L amino acids.
- R 1 is H, NR 3 R b , Cl, F, 0R a , SR 3 , NHCOR 3 , NHSO 2 R 3 , NHCONHR 3 , CN, alkyl, aryl, 0NR 3 R b , or NR 3 C(0)0R b ;
- R is a nucleoside sugar group
- R 3 , R b , R c , and R d are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, SO 2 -alkyl, amino, substituted amino, substituted cycloalkyl, heteroaryl, substituted heteroaryl , and NO; or R 3 and R b together with the nitrogen to which they are attached form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; or R b and R 0 together with the nitrogen to which they are attached form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; or R 0 and R d together with the nitrogen to which they are attached form a heterocycloalkyl, substitute
- X is N, Y is N and Z is CH; or X is C, Y is CR 3 and Z is O;
- R 1 is H, NR a R b , Cl, F, OR 3 , SR 3 , NHCOR 3 , NHSO 2 R 3 , NHCONHR 3 , CN, alkyl, aryl, 0NR a R b , or NR 3 C(0)0R b ;
- R 2 is a nucleoside sugar group;
- R a , R b , R c , and R d are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, S ⁇ 2 -alkyl, amino, substituted amino, and NO; or R a and R b together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidin
- R c , and R d are independently selected from the group consisting of H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heteroaryl, and substituted heteroaryl; or R 0 , and R d together with N atom to which they are attached may form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; or a pharmaceutically acceptable salt or prodrug thereof.
- Compounds of the invention include compounds of Formula I and II:
- R 1 is H, NR 3 R b , Cl, F, OR a , SR 3 , NHC0R a , NHSO 2 R 3 , NHCONHR 3 , CN, alkyl, aryl, 0NR a R b , or NR a C(0)0R b ;
- R 2 is a nucleoside sugar group
- the invention provides a compound of Formula I, II, or III as described above, wherein R is OR 3 , Cl, SR a , NR 3 R b , or NR 3 NRbRc; or a pharmaceutically acceptable salt or prodrug thereof.
- the invention provides a compound of Formula I, II, or III as described above, wherein R is NR 3 R b ;
- R 8 is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, SO 2 -alkyl and NO;
- R b is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, SO 2 -alkyl and NO; or R a and R b together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azet
- the invention provides a compound of Formula I, II, or III as described above, wherein R is NR 3 NR b R c , alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, (CH 2 ) n -CH(NHR a )CO 2 R b , Cl, F, Br, I, CN, COOR a , CONR a R b ,
- the invention provides a compound of Formula I, II, or III as described above, wherein R 1 is H or NR a
- the invention provides a compound of Formula I, II, or III as described above, wherein R 2 is a nucleoside sugar group of Group A, B, C, D, E, or F described herein below; or a pharmaceutically acceptable salt or prodrug thereof.
- the invention provides a compound of Formula I, II, or III as described above, wherein R 2 is ribose, 2-methylribose, 2- deoxyribose; 2-deoxy-2-fluororibose; arabinose; 2-deoxy-2-fluoroarabinose; 2,3- dideoxyribose; 2,3-dideoxy-2-fluoroarabinose; 2,3-dideoxy-3-fluororibose; 2,3- dideoxy-2,3-didehydroribose; 2,3-dideoxy-3-azidoribose; 2,3-dideoxy-3- thiaribose; or 2,3-dideoxy-3-oxaribose; or a pharmaceutically acceptable salt or prodrug thereof.
- R 2 is ribose, 2-methylribose, 2- deoxyribose; 2-deoxy-2-fluororibose; arabinose; 2-deoxy-2-fluoroarabinose; 2,3- dide
- the invention provides a compound of Formula I, II, or III as described above, wherein R 2 is thioribose, 2-deoxythioribose; 2- deoxy-2-fluorothioribose; thioarabinose; 2-deoxy-2-fluorothioarabinose; 2,3- dideoxythioribose; 2,3-dideoxy-2-fluorothioarabinose; 2,3-dideoxy-3- fluorothioribose; 2,3-dideoxy-2,3-didehydrothioribose; or 2,3-dideoxy-3- azidothioribose; or a pharmaceutically acceptable salt or prodrug thereof.
- the invention provides a compound of Formula I, II, or III as described above, wherein R 2 is 4-hydroxymethylcyclopent-2-ene; 2,3-dihydroxy-4-hydroxyrnethylcyclopent-4-ene; 3-hydroxy-4- hydroxymethylcyclopentane; 2-hydroxy-4-hydroxymethylcyclopentene; 2- fluoro-3-hydroxy-4-hydroxymethylcyclopentane; 2,3-dihydroxy-4- hydroxymethyl-5-methylenecyclopentane; 4-hydroxymethylcyclopentane, 2,3- dihydroxy-4-hydroxymethylcyclopentane; or 2,3 -dihydroxymethylcyclobutane; or a pharmaceutically acceptable salt or prodrug thereof.
- the invention provides a compound of Formula I, II, or III as described above, wherein R 2 is 4-hydroxymethylpyrrolidine; 2,3- dihydroxy-4-hydroxymethylpyrrolidine; 2/3-hydroxy-4- hydroxymethylpyrrolidine; 2-fluoro-3 -hydroxy-4-hydroxymethylpyrrolidine; or 3-fluoro-2-hydroxy-4-hydroxymethyl-pyrrolidine; or a pharmaceutically acceptable salt or prodrug thereof.
- R 2 is 4-hydroxymethylpyrrolidine; 2,3- dihydroxy-4-hydroxymethylpyrrolidine; 2/3-hydroxy-4- hydroxymethylpyrrolidine; 2-fluoro-3 -hydroxy-4-hydroxymethylpyrrolidine; or 3-fluoro-2-hydroxy-4-hydroxymethyl-pyrrolidine; or a pharmaceutically acceptable salt or prodrug thereof.
- the invention provides a compound of Formula I,
- R a , R b , R c , and Rd are independently selected from the group consisting of H, alkyl, and substituted alkyl; or R a and R b together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; or R b and R 0 together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring.
- the invention provides a compound of Formula II, which is a compound of the following formula (IV):
- the compound of the invention is not a compound of the following formula:
- B is selected from:
- W is O, S, or NH
- X is N, Y is N and Z is CH; or X is C, Y is CR 23 and Z is O;
- R 3 , R 4 , R 5 , and R 6 are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, SO 2 -alkyl and NO; or R 3 and R 4 together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; or R 4 and R 5 together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; and
- R c is selected from the group consisting of hydrogen, alkyl, alkenyl, and alkynyl wherein each alkyl, alkenyl, and alkynyl is optionally substituted with one or more substituents independently selected from halo, amino, hydroxy, and alkoxy; or a pharmaceutically acceptable salt thereof.
- prodrug refers to a compound that can be metabolized in vivo to provide a compound of Formula I to III.
- one or more pendent hydroxyl groups from a mono-, di-, or triphosphate functionality in a compound of Formula I to III can be converted to an alkoxy, substituted alkoxy, aryloxy, or substituted aryloxy group.
- the term prodrug includes a compound wherein one or more hydroxy groups on a nucleoside sugar group (e.g., a 2', 3', or 5' hydroxy group) have been converted to a group that can be metabolized in vivo to provide a compound of Formula I to III.
- the invention provides a compound wherein one or more hydroxy groups on a nucleoside sugar group (e.g., a 2', 3', or 5' hydroxy group) have been converted to an acyloxy, acylamino or R-O group, wherein R is a carboxy-linked amino acid.
- the term prodrug includes a compound wherein one or more pendent hydroxyl groups from a mono-, di-, or tri-phosphate functionality in a compound of Formula I to III is converted to a group R 2 -N-; wherein each R 2 is a residue of an amino acid.
- administering includes administration of a compound of Formula I to III, as well as administration of a prodrug which converts to a compound of Formula I to III or a salt thereof in vivo.
- R 15 is H, alkyl, substituted alkyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, heteroaryl, substituted heteroaryl, heterocyclic, substituted heterocyclic, and an amino acid;
- R 16 is H, optionally substituted monocyclic aryl, or optionally substituted monocyclic heteroaryl; and R 17 is H, halogen, CN, -CO-R 20 , -CON(R 2 O 2 , - CO 2 R 20 , -SO 2 R 20 , -SO 2 N(R 21 ) 2 , -OR 2 I, -SR 21 , -R 21 , -N(R 21 ) 2 , -0-COR 20 , -O- CO 2 R 20 , -SCOR 20 , -S-CO 2 R 20 , -NHCOR 21 , -NHCO 2 R 21 , -(CH 2 ) P -OR 22 , or - (CH 2 ) P -SR 22 ; or R 16 and R 17 are connected via an additional 3-5 atoms to form a cyclic group, optionally containing one heteroatom, that is fused to an aryl group at the beta and gamma position to the O attached
- R 18 and R 19 are each independently H, alkyl, aryl, heterocycloalkyl, aralkyl, optionally substituted monocyclic aryl or optionally substituted monocyclic heteroaryl; or R 18 and R 19 are connected via an additional 2-5 atoms to form a cyclic group, optionally containing 0-2 heteroatoms; or R 17 and R 18 are connected via an additional 3-5 atoms to form a cyclic group, optionally containing one heteroatom and R 19 is H, alkyl, aryl, heterocycloalkyl, aralkyl, optionally substituted monocyclic aryl or optionally substituted monocyclic heteroaryl; and R 20 is alkyl, aryl, heterocycloalkyl, or arylalkyl;
- R 21 is H, alkyl, aryl, heterocycloalkyl, or arylalkyl;
- R 22 is H or lower acyl; n is an integer from 2-5; m is an integer from 10-20; and p is an integer from 2-3.
- Prodrug forms of carboxyl -bearing compounds include esters (-CO 2 R m ) where the R m group corresponds to any alcohol whose release in the body through enzymatic or hydrolytic processes would be at pharmaceutically acceptable levels.
- Another prodrug derived from a carboxylic acid form of the disclosure may be a quaternary salt type of structure described by Bodor et al. , J. Med. Chem. 1980, 23, 469.
- nucleoside sugar group as used herein includes cyclic and acyclic groups that can be included as the sugar portion of a nucleoside analog of Formula I to III. Many examples of such groups are known in the field of nucleoside chemistry (See for example Antiviral Drugs by John S. Driscoll (2002) published by Ashgate Publishing Ltd.).
- nucleoside sugar group includes substituted and unsubstituted tetrahydrofuranyl and dihydrofuranyl compounds including those set forth in group (A) below, substituted and unsubstituted tetrahydrothiophenyl and dihydrothiophenyl compounds including those set forth in group (B) below, substituted and unsubstituted alkyl compounds including those set forth in group (C) below, substituted and unsubstituted cycloalkyl and cycloalkenyl compounds including those set forth in group (D) below, substituted and unsubstituted dihydropyrrolidinyl and tetrahydropyrrolidinyl compounds including those set forth in group (E) below, and substituted and unsubstituted dioxolane, substituted and unsubstituted thioxolane, and substituted and unsubstituted dithiolane compounds including those set forth in group (F) below.
- group A substituted and unsub
- substituted tetrahydro and dihydrofuranyl compounds include those compounds represented by the general structures:
- substituted tetrahydrothiophenyl and dihydrothiophenyl compounds include those compounds represented by the general structures:
- substituted alkyl compounds include those compounds represented by:
- substituted cycloalkyl and cycloalkenyl compounds include those compounds represented by the general structures:
- substituted dihydropyrrolidinyl and tetrahydropyrrolidinyl compounds include those compounds represented by the general structures:
- substituted dioxolane, substituted thioxolane and substituted dithiolane compounds include those compounds represented by the general structures:
- R 10 and R 11 are each independently H, alkyl, aryl, substituted aryl, acyloxyalkyl, or (CH 2 ) nl -O-(CH 2 ) m CH 3 ; nl is 2-5; m is 10-20;
- Ri 2 is an N-linked amino acid residue (e.g., -NH-CH(CH 3 )C0 2 alkyl or - NH-CH(isopropyl)-CO 2 alkyl);
- R 13 is H, CH 3 , C 2 H 5 , CH 2 F, CH 2 OH, CH 2 CH 2 F, CH 2 CH 2 OH, CH 2 N 3 , CH 2 CH 2 N 3 , CH 2 NH 2 , or CH 2 CH 2 NH 2 ; and R 14 is H.
- Processes for preparing compounds of Formula I-III, or a pharmaceutically acceptable salts or prodrugs thereof, as well as processes for preparing intermediate compounds that can be used to prepare compounds of Formula I-III or pharmaceutically acceptable salts or prodrugs thereof are provided as further embodiments of the invention.
- the invention provides a method for preparing a pharmaceutically acceptable salt of compound of Formula I-III comprising converting a corresponding compound of Formula I-III to the salt.
- the invention provides a method for preparing a prodrug of a compound of Formula I-III comprising converting a corresponding compound of Formula I-III to the prodrug.
- the invention provides a method for preparing a compound of Formula I-III comprising deprotecting a corresponding compound of Formula I-III that comprises one or more protecting groups to provide the compound of Formula I-III.
- a representative compound of formula III (and formula II), compound lie, can be prepared as follows. Synthetic scheme for the preparation of (2R,3R,4R,5R)-2-(4-aminofuro[3,4- d]pyrimidin-7-yI)-5-(hydroxymethyl)-3-methyltetrahydrofuran-3,4-diol J. Chem. Soc. Perkin Trans. 1 ; 1982; 903-908
- the invention also provides synthetic intermediates that are useful for preparing compounds of Formula I to III or a salt or prodrug thereof.
- the invention provides novel synthetic intermediates such as those described in the Schemes and Examples herein. Isomers and Physical Forms
- the present disclosure provides compounds of the general Formula I to III as detailed above which are inhibitors of DNA and/or RNA viral polymerases and anticancer agents.
- Various forms of DNA and RNA viral polymerases are inhibited by the compounds disclosed, such as but not limited to viral RdRps.
- the compounds of the present disclosure therefore have utility in treating and/or preventing viral infections in a host and in treatment and/or preventing a variety of disease states and/or conditions caused by or related to such viral infections.
- the compounds are useful in the above mentioned treating and/or preventing by inhibiting a viral RNA and DNA polymerases.
- the present disclosure provides for a compound of the general Formula I to III and a pharmaceutical composition comprising a pharmaceutically effective amount of at least one compound of general Formula I toIII as described herein.
- Such compounds and/or pharmaceutical compositions may be used in the manufacture of a medicament for treating and/or preventing a disease or condition in which it is desirable to inhibit a viral RNA and DNA polymerases.
- Such pharmaceutical compositions may also comprise a pharmaceutically acceptable carrier and other ingredients known in the art, or may comprise solely a compound of the general Formula I to III.
- the pharmaceutically acceptable carriers described herein, including, but not limited to, vehicles, adjuvants, excipients, or diluents, are well-known to those who are skilled in the art.
- the pharmaceutically acceptable carrier is chemically inert to the active compounds and has no detrimental side effects or toxicity under the conditions of use.
- the pharmaceutically acceptable carriers can include polymers and polymer matrices.
- the compounds described in the instant disclosure can be administered by any conventional method available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in combination with additional therapeutic agents.
- the total amount is between about 0.1 mg/kg and about 100 mg/kg of body weight per day; in an alternate embodiment between about 1.1 mg/kg and about 50 mg/kg of body weight per day; in yet another alternate embodiment between 0.1 mg/kg and about 30 mg/kg of body weight per day.
- the above described amounts may be administered as a series of smaller doses over a period of time if desired.
- the pharmaceutically effective amount can be calculated based on the weight of the parent compound to be delivered. If the salt or prodrug exhibits activity in itself, the pharmaceutically effective amount can be estimated as above using the weight of the salt or prodrug, or by other means known to those skilled in the art.
- the dosage of active ingredient may be given other than daily if desired.
- the active ingredient may be administered to achieve peak plasma concentrations of the active ingredient of from about 0.2 to 70 ⁇ M, or from about 1.0 to 10 ⁇ M.
- the active ingredient can be administered orally in solid dosage forms, such as capsules, tablets, and powders, or in liquid dosage forms, such as elixirs, syrups and suspensions. It can also be administered parenterally, in sterile liquid dosage forms.
- the active ingredient can also be administered intranasally (nose drops) or by inhalation via the pulmonary system, such as by propellant based metered dose inhalers or dry powders inhalation devices.
- Other dosage forms are potentially possible such as administration transdermally, via patch mechanisms or ointment.
- Formulations suitable for oral administration can include (a) liquid solutions, such as a pharmaceutically effective amount of the compound dissolved in diluents, such as water, saline, or orange juice; (b) capsules, sachets, tablets, lozenges, and troches, each containing a predetermined pharmaceutically effective amount of the active ingredient, as solids or granules; (c) powders; (d) suspensions in an appropriate liquid; and (e) suitable emulsions.
- Liquid formulations may include diluents, such as water and alcohols, for example, ethanol, benzyl alcohol, propylene glycol, glycerin, and the polyethylene alcohols, either with or without the addition of a pharmaceutically acceptable surfactant, suspending agent, or emulsifying agent.
- diluents such as water and alcohols, for example, ethanol, benzyl alcohol, propylene glycol, glycerin, and the polyethylene alcohols, either with or without the addition of a pharmaceutically acceptable surfactant, suspending agent, or emulsifying agent.
- Capsule forms can be of the ordinary hard- or soft-shelled gelatin type containing, for example, surfactants, lubricants, and inert fillers, such as lactose, sucrose, calcium phosphate, and corn starch.
- Tablet forms can include one or more of the following: lactose, sucrose, mannitol, corn starch, potato starch, alginic acid, microcrystalline cellulose, acacia, gelatin, guar gum, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, calcium stearate, zinc stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, disintegrating agents, moistening agents, preservatives, flavoring agents, and pharmacologically compatible carriers.
- Lozenge forms can comprise the active ingredient in a flavor, usually sucrose and acacia or tragacanth, as well as pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin, or sucrose and acadia, emulsions, and gels containing, in addition to the active ingredient, such carriers as are known in the art.
- a flavor usually sucrose and acacia or tragacanth
- pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin, or sucrose and acadia, emulsions, and gels containing, in addition to the active ingredient, such carriers as are known in the art.
- Formulations suitable for parenteral administration include aqueous and non-aqueous, isotonic sterile injection solutions, which can contain antioxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the patient, and aqueous and non-aqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives.
- the compound can be administered in a physiologically acceptable diluent in a pharmaceutically acceptable carrier, such as a sterile liquid or mixture of liquids, including water, saline, aqueous dextrose and related sugar solutions, an alcohol, such as ethanol, isopropanol, or hexadecyl alcohol, glycols, such as propylene glycol or polyethylene glycol such as poly(ethyleneglycol) 400, glycerol ketals, such as 2,2-dimethyl-l,3-dioxolane-4- methanol, ethers, an oil, a fatty acid, a fatty acid ester or glyceride, or an acetylated fatty acid glyceride with or without the addition of a pharmaceutically acceptable surfactant, such as a soap or a detergent, suspending agent, such as pectin, carbomers, methylcellulose, hydroxypropylmethylcellulose, or carboxymethylcellulose, or emulsifying agents and
- Oils which can be used in parenteral formulations, include petroleum, animal, vegetable, or synthetic oils. Specific examples of oils include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral. Suitable fatty acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters.
- Suitable soaps for use in parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts
- suitable detergents include (a) cationic detergents such as, for example, dimethyldialkylammonium halides, and alkylpyridinium halides, (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyl, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine oxides, fatty acid alkanolamides, and polyoxyethylene polypropylene copolymers, (d) amphoteric detergents such as, for example, alkyl .beta.- aminopropionates, and 2-alkylimidazoline quaternary ammonium salts, and (e) mixtures thereof.
- the parenteral formulations
- compositions may contain one or more nonionic surfactants having a hydrophile-lipophile balance (HLB) of from about 12 to about 17.
- HLB hydrophile-lipophile balance
- Suitable surfactants include polyethylene sorbitan fatty acid esters, such as sorbitan monooleate and the high molecular weight adducts of ethylene oxide with a hydrophobic base, formed by the condensation of propylene oxide with propylene glycol.
- HLB hydrophile-lipophile balance
- Suitable surfactants include polyethylene sorbitan fatty acid esters, such as sorbitan monooleate and the high molecular weight adducts of ethylene oxide with a hydrophobic base, formed by the condensation of propylene oxide with propylene glycol.
- Pharmaceutically acceptable excipients are also well-known to those who are skilled in the art.
- excipient will be determined in part by the particular compound, as well as by the particular method used to administer the composition. Accordingly, there is a wide variety of suitable formulations of the pharmaceutical composition of the present invention. The following methods and excipients are merely exemplary and are in no way limiting.
- the pharmaceutically acceptable excipients preferably do not interfere with the action of the active ingredients and do not cause adverse side-effects.
- Suitable carriers and excipients include solvents such as water, alcohol, and propylene glycol, solid absorbants and diluents, surface active agents, suspending agent, tableting binders, lubricants, flavors, and coloring agents.
- the compounds of the present invention can be made into aerosol formulations to be administered via inhalation.
- aerosol formulations can be placed into pressurized acceptable propellants, such as dichlorodifluoromethane, propane, and nitrogen.
- Such aerosol formulations may be administered by metered dose inhalers. They also may be formulated as pharmaceuticals for non-pressured preparations, such as in a nebulizer or an atomizer.
- the formulations can be presented in unit-dose or multi-dose sealed containers, such as ampules and vials, and can be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid excipient, for example, water, for injections, immediately prior to use.
- sterile liquid excipient for example, water
- Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets.
- the requirements for effective pharmaceutically acceptable carriers for injectable compositions are well known to those of ordinary skill in the art. See Pharmaceutics and Pharmacy Practice, J.B. Lippincott Co., Philadelphia, Pa., Banker and Chalmers, Eds., 238-250 (1982) and ASHP Handbook on Injectable Drugs, Toissel, 4th ed., 622-630 (1986).
- Formulations suitable for topical administration include pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin, or sucrose and acacia, as well as creams, emulsions, and gels containing, in addition to the active ingredient, such carriers as are known in the art.
- transdermal patches can be prepared using methods known in the art.
- formulations suitable for rectal administration may be presented as suppositories by mixing with a variety of bases such as emulsifying bases or water-soluble bases.
- Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams, or spray formulas containing, in addition to the active ingredient, such carriers as are known in the art to be appropriate.
- Suitable methods of administering a compound of the present invention to an patient are available, and, although more than one route can be used to administer a particular compound, a particular route can provide a more immediate and more effective reaction than another route.
- a mixture of active ingredient in a digestible oil such as soybean oil, cottonseed oil or olive oil is prepared and injected by means of a positive displacement pump into molten gelatin to form soft gelatin capsules containing 100 mg of the active ingredient.
- the capsules are washed and dried.
- the active ingredient can be dissolved in a mixture of polyethylene glycol, glycerin and sorbitol to prepare a water miscible medicine mix.
- a large number of tablets are prepared by conventional procedures so that the dosage unit is 100 mg of active ingredient, 0.2 mg of colloidal silicon dioxide, 5 mg of magnesium stearate, 275 mg of microcrystalline cellulose, 11 mg of starch, and 98.8 mg of lactose.
- Appropriate aqueous and non-aqueous coatings may be applied to increase palatability, improve elegance and stability or delay absorption.
- Immediate release tablets/capsules are solid oral dosage forms made by conventional and novel processes. These units are taken orally without water for immediate dissolution and delivery of the medication.
- the active ingredient is mixed in a liquid containing ingredient such as sugar, gelatin, pectin and sweeteners. These liquids are solidified into solid tablets or caplets by freeze drying and solid state extraction techniques.
- the drug compounds may be compressed with viscoelastic and thermoelastic sugars and polymers or effervescent components to produce porous matrices intended for immediate release, without the need of water.
- the compounds of the present invention can be administered in the form of nose drops, or metered dose and a nasal or buccal inhaler.
- the drug is delivered from a nasal solution as a fine mist or from a powder as an aerosol.
- teachings of the present disclosure provide for the use of such pharmaceutical compositions and medicaments in a method of treating a viral infection or treating a disease state and/or condition caused by or related to such viral infection.
- the treatment is the result of the inhibition of a viral RNA or DNA polymerase, such as but not limited to a RdRp. Such treatment or inhibition need not be complete to be useful.
- the method of treatment comprises the steps of: (i) identifying a patient in need of such treatment; (ii) providing such pharmaceutical composition containing at least one compound of the invention; and (iii) administering such pharmaceutical composition in a therapeutically effective amount to treat the viral infection in a patient in need of such treatment or to inhibit the activity of a viral RNA or DNA polymerase in a patient in need of such treatment.
- the teachings of the present disclosure provide for, among other things, the use of such pharmaceutical compositions and medicaments in a method of preventing or suppressing a viral infection or preventing or suppressing a disease state and/or condition caused by or related to such viral infection.
- the prevention or suppression is the result of the inhibition of a viral RNA or DNA polymerase, such as but not limited to a RdRp.
- a viral RNA or DNA polymerase such as but not limited to a RdRp.
- the method of preventing or suppressing can optionally comprises the steps of: (i) identifying a patient in need of such prevention; (ii) providing such pharmaceutical composition containing at least one compound of the general Formula I to III; and (iii) administering such pharmaceutical composition in a therapeutically effective amount to prevent or suppress the viral infection in a patient in need of such treatment or to inhibit the activity of a viral RNA and DNA polymerase in a patient in need of such treatment.
- the methods of the treating and preventing a viral infection or a disease state and/or condition caused by or related to said viral infection may further comprise administering a therapeutically effective amount of a compound of the present invention in combination with a therapeutically effective amount of another anti-viral agent which , in particular, may be active against HCV.
- Cancers include, but are not limited to, leukemias and lymphomas such as acute lymphocytic leukemia, acute nonlymphocytic leukemias, chronic lymphocytic leukemia, chrome myelogenous leukemia, Hodgkin's Disease, non- Hodgkin's lymphomas, and multiple myeloma, childhood solid tumors such as brain tumors, neuroblastoma, retinoblastoma, Wilms Tumor, bone tumors, and soft-tissue sarcomas, common solid tumors of adults such as lung cancer, colon and rectum cancer, breast cancer, prostate cancer, urinary cancers, uterine cancers, oral cancers, pancreatic cancer, melanoma and other skin cancers, stomach cancer, ovarian cancer, brain tumors, liver cancer, laryngeal cancer, thyroid cancer, esophageal cancer, and
- the methods of the treating and preventing cancer may also comprises further administering of a chemotherapeutic agent in combination with any of the compounds or pharmaceutical compositions of the present disclosure.
- a chemotherapeutic agent can be employed for this purpose.
- the chemotherapeutic agent is typically selected from the group consisting of alkylating agents, antimetabolites, natural products, hormonal agents, and miscellaneous agents.
- alkylating chemotherapeutic agents include carmustine, chlorambucil, cisplatin, lomustine, cyclophosphamide, melphalan, mechlorethamine, procarbazine, thiotepa, uracil mustard, triethylenemelamine, busulfan, pipobroman, streptozocin, ifosfamide, dacarbazine, carboplatin, and hexamethylmelamine.
- chemotherapeutic agents that are antimetabolites include cytosine arabinoside, fluorouracil, gemcitabine, hydroxyurea, mercaptopurine, methotrexate, azaserine, thioguanine, floxuridine, fludarabine, cladribine and L- asparaginase.
- chemotherapeutic agents that are natural products include actinomycin D, bleomycin, camptothecins, daunomycin, doxorubicin, etoposide, mitomycin C, TAXOLTM (paclitaxel), taxotere, teniposide, vincristine, vinorelbine, mithramycin, idarubicin, MITHRACINTM. (plicamycin), and deoxycoformycin.
- hormonal chemotherapeutic agent includes tamoxifen.
- miscellaneous chemotherapeutic agents include mitotane, mitoxantrone, vinblastine, and levamisole.
- prodrugs of the invention can be been tested in vitro or in vivo to confirm that the prodrug is converted, e.g., by hydrolysis, to provide an active compound.
- the ability of a compound to inhibit viral polymerases can be evaluated using known assays.
- the ability of a compound to inhibit HCV NS5B polymerase can be evaluated using the following assay.
- HCV NS5B Polymerase Assay Antiviral activity of the test compounds can be assessed (Okuse et al ,
- Compounds can be added to dividing cultures once daily for three days. Media can be changed with each addition of compound. Cultures generally start the assay at 30-50% confluence and reach confluence during the last day of treatment. Intracellular HCV RNA levels and cytotoxicity can be assessed 24 hours after the last dose of compound.
- Triplicate cultures for HCV RNA levels (on 48-well and 96-well plates) and cytotoxicity (on 96-well plates) can be used.
- Intracellular HCV RNA levels can be measured using a conventional blot hybridization method in which HCV RNA levels are normalized to the levels of B-actin RNA in each individual culture (Okuse et al, Antivir. Res. 2005, 65, 23- 34). Cytotoxicity can be measured using a neutral red dye uptake assay (Korba and Gerin, Antivir. Res. 1992, 19, 55). HCV RNA levels in the treated cultures can be expressed as a percentage of the mean levels of RNA detected in untreated cultures.
- Compound Synthesis Compounds of Formula I-III can be prepared using synthetic intermediates and synthetic procedures that are known, or they can be prepared using the synthetic intermediates and synthetic procedures described in the Schemes and Examples herein.
- Example 1 The invention will now be illustrated by the following non-limiting Example.
- Example 1 The invention will now be illustrated by the following non-limiting Example.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
The invention provides compounds as described herein, as well as pharmaceutical compositions comprising the compounds, and synthetic methods and intermediates that are useful for preparing the compounds. The compounds are useful as anti-viral agents and/or as anti-cancer agents.
Description
ANTIVIRAL NUCLEOSIDE ANALOGS
Cross-reference to Related Application (s)
This patent application claims the benefit of priority of the following application, which application is herein incorporated by reference: U.S. application serial No. 61/053,488, filed May 15, 2008.
Background
Viral diseases are a major cause of death and economic loss in the world. The Flaviviridae family of viruses includes the three genera flaviviruses (including dengue, West Nile, and yellow fever viruses), hepacivirus (including hepatitus C virus (HCV)), and pesti viruses (including bovine viral diarrhea virus, BVDV). The disease states and conditions caused by members of this family include yellow fever, dengue, Japanese encephalitis, St. Louis encephalitis, Hepatitis B and C, West Nile disease, and AIDS. Currently, human immunodeficiency virus (HIV), hepatitis B virus (HBV) and hepatitis C virus (HCV) infections are responsible for the largest number of viral related deaths worldwide. Although there are some drugs useful for treating HIV, there are only a few drugs useful for treating HBV, and no drugs that are broadly useful for treating HCV. Ribavirin (l-β-D-ribofuranosyl-l-l,2,4-triazole-3-carboxamide) is a synthetic, non-interferon-inducing, broad spectrum antiviral nucleoside. Ribavirin is structurally similar to guanosine, and has in vitro activity against several DNA and RNA viruses including Flaviviridae (Davis. Gastroenterology 118:S1O4-S114, 2000). Ribavirin reduces serum amino transferase levels to normal in 40% of patients, but it does not lower serum levels of HCV-RNA (Davis. Gastroenterology 118:S104-S114, 2000). Thus, ribavirin alone is not effective in reducing viral RNA levels. Additionally, ribavirin has significant toxicity and is known to induce anemia.
Interferons (IFNs) are compounds which have been commercially available for the treatment of chronic hepatitis for nearly a decade. IFNs are glycoproteins produced by immune cells in response to viral infection. IFNs inhibit viral replication of many viruses, including HCV. When used as the sole treatment for hepatitis C infection, IFN suppresses serum HCV-RNA to
undetectable levels. Additionally, IFN normalizes serum amino transferase levels. Unfortunately, the effects of IFN are temporary and a sustained response occurs in only 8%-9% of patients chronically infected with HCV (Davis. Gastroenterology 118:S104-Sl 14, 2000). HCV is a positive stranded ss RNA virus with a well characterized RNA- dependent RNA polymerase (RdRp) and a well characterized disease progression. HCV has infected an estimated 170 million people worldwide, leading to a major health crisis as a result of the disease. Indeed, during the next few years the number of deaths from HCV-related liver disease and hepatocellular carcinoma may overtake those caused by AIDS. Egypt is the hardest hit country in the world, with 23% of the population estimated to be carrying the virus; whereas, in the USA the prevalence of chronic infections has recently been determined to be around 1.87% (2.7 million persons). HCV infections become chronic in about 50% of cases. Of these, about 20% develop liver cirrhosis that can lead to liver failure, including hepatocellular carcinoma.
The NS5B region of HCV encodes a 65 KDa RdRp thought to be responsible for viral genome replication. RdRps function as the catalytic subunit of the viral replicase required for the replication of all positive-strand viruses. The NS5B protein has been well characterized, shown to possess the conserved GDD motif of RdRps and in vitro assay systems have been reported. Cellular localization studies revealed that NS5B is membrane-associated in the endoplasmic reticulum like NS5A, suggesting that those two proteins may remain associated with one another after proteolytic processing. Additional evidence suggests that NS3, NS4A and NS5B interact with each other to form a complex that functions as part of the replication machinery of HCV.
The X-ray crystal structure of NS 5 B apoenzyme has been determined and three recent publications describe the unusual shape of the molecule. This unique shape for a polymerase, resembling a flat sphere, is attributed to extensive interactions between the fingers and thumb subdomains in such a way that the active site is completely encircled, forming a cavity 15 A across and 20 A deep. Modeling studies showed that the NS5B apoenzyme can accommodate the template-primer without large movement of the subdomains, suggesting that the structure is preserved during the polymerization reaction. The RdRp polypeptides from various members of the Flaviviridae family and other viral
families have been shown to be conserved (J. A. Bruenn, Nucleic Acids Research, Vol. 19, No. 2 p. 217, 1991).
Currently, there are no safe and effective therapeutic agents on the market that target HCV polymerase. There is currently a need for therapeutic agents and therapeutic methods that are useful for treating viral infections, such as HCV, HIV, and HBV.
In addition, there is also a current need for therapeutic agents and therapeutic methods that are useful for treating cancer. Even though significant advances have occurred in the treatment of cancer, it still remains a major health concern. It has been reported that cancer is the cause of death of up to one of every four Americans. Notwithstanding the advances in treatments for cancer and other diseases there is still a need for novel drugs that are effective to treat cancer.
Summary of Certain Embodiments
Certain embodiments of the present invention provide compounds that are inhibitors of viral RNA and DNA polymerases (e.g., polymerases from hepatitis B, hepatitis C, human immunodeficiency virus, Polio, Coxsackie A and B, Rhino, Echo, small pox, Ebola, and West Nile virus) and that are useful for treating HCV, as well as other viral infections (e.g., flaviviral infections), and cancer. Accordingly, certain embodiments of the present invention provide a novel compound of Formula I to III as described here below, or a pharmaceutically acceptable salt or prodrug thereof.
Certain embodiments of the present invention provide a pharmaceutical composition comprising a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof, and a pharmaceutically acceptable carrier. The composition can optionally comprise one or more additional anti-viral or anti-cancer agents.
Certain embodiments of the present invention provide a method for treating a viral infection in an animal comprising administering to the animal an effective amount of a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof.
Certain embodiments of the present invention provide a method for inhibiting a viral RNA or DNA polymerase comprising contacting the polymerase (in vitro or in vivo) with an effective inhibitory amount of a
compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof.
Certain embodiments of the present invention provide a method for treating cancer in an animal comprising administering to the animal an effective amount of a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof.
Certain embodiments of the present invention provide a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof, for use in medical therapy (e.g., for use in treating a viral infection or for use in treating cancer).
Certain embodiments of the present invention provide the use of a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof, to prepare a medicament useful for treating a viral infection in an animal (e.g., a human). Certain embodiments of the present invention provide the use of a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof, to prepare a medicament useful for treating cancer in an animal (e.g., a human).
Certain embodiments of the present invention provide a compound of the invention for use in therapy.
Certain embodiments of the present invention provide a compound of the invention for treating a viral infection.
Certain embodiments of the present invention provide a compound of the invention for treating cancer. Certain embodiments of the present invention provide novel synthetic intermediates and synthetic methods that are disclosed herein as being useful for preparing compounds of Formula I to III, or a salt or prodrug thereof. Some compounds of Formula I to III may be useful as synthetic intermediates for preparing other compounds of Formula I to III. Certain embodiments of the present invention provide a compound as described herein for use in medical treatment or diagnosis.
Certain embodiments of the present invention provide the use of a compound as described herein to prepare a medicament useful for treating a viral infection in an animal.
Certain embodiments of the invention provide the use of a compound as described herein to prepare a medicament useful for treating cancer in an animal.
Certain embodiments of the invention provide a compound as described herein for use in therapy. Certain embodiments of the invention provide the use of a compound as described herein for treating a viral infection in an animal.
Certain embodiments of the invention provide the use of a compound as described herein for treating cancer in an animal.
Detailed Description Definitions
The term "pharmaceutically acceptable salt" as used herein refers to a compound of the present disclosure derived from pharmaceutically acceptable bases, inorganic or organic acids. Examples of suitable acids include, but are not limited to, hydrochloric, hydrobromic, sulfuric, nitric, perchloric, fumaric, maleic, phosphoric, glycollic, lactic, salicyclic, succinic, toluene-p-sulfonic, tartaric, acetic, citric, methanesulfonic, formic, benzoic, malonic, naphthalene-2- sulfonic, trifluoroacetic and benzenesulfonic acids. Salts derived from appropriate bases include, but are not limited to, alkali such as sodium and ammonia. Te terms "treat", "treating" and "treatment" as used herein include administering a compound prior to the onset of clinical symptoms of a disease state/condition so as to prevent any symptom, as well as administering a compound after the onset of clinical symptoms of a disease state/condition so as to reduce or eliminate any symptom, aspect or characteristic of the disease state/condition. Such treating need not be absolute to be useful.
The term "animal" as used herein refers to any animal (e.g., an animal in need of treatment), including mammals, such as, but not limited to, mice, rats, other rodents, rabbits, dogs, cats, swine, cattle, sheep, horses, and primates. In one specific embodiment of the invention the animal is a male or female human. The term "therapeutically effective amount", in reference to treating a disease state/condition, refers to an amount of a compound either alone or as contained in a pharmaceutical composition that is capable of having any detectable, positive effect on any symptom, aspect, or characteristics of a disease
state/condition when administered as a single dose or in multiple doses. Such effect need not be absolute to be beneficial.
The term "alkyl" as used herein refers to alkyl groups having from 1 to 6 carbon atoms. This term is exemplified by groups such as methyl, ethyl, n- propyl, iso-propyl, n-butyl, t-butyl, n-pentyl and the like. In a specific embodiment, the alkyl groups have from 1-4 carbon atoms and are referred to as lower alkyl.
The term "substituted alkyl" as used herein refers to an alkyl group having from 1 to 3 substituents, said substituents being selected from the group consisting of alkoxy, alkoxyalkyl, tri(C1-C4alkyl)silyl, substituted alkoxy, acyl, substituted acyl, acylamino, acyloxy, oxyacyl, amino, substituted amino, aminoacyl, aryl, substituted aryl, aryloxy, substituted aryloxy, cyano, halogen, hydroxyl, nitro, N3, carboxyl, carboxyl esters, thiol, thioalkyl, substituted thioalkyl, thioaryl, substituted thioaryl, thioheteroaryl, substituted thioheteroaryl, thiocycloalkyl, substituted thiocycloallcyl, thioheterocyclic, substituted thioheterocycliccycloalkyl, substituted cycloalkyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic. In one specific embodiment of the invention, the term "substituted alkyl" refers to an alkyl group substituted with 1 to 3 substituents, said substituents being selected from the group consisting of alkoxy, alkoxyalkyl, tri(CrC4alkyl)silyl, acyl, acylamino, acyloxy, oxyacyl, amino, aminoacyl, aryl, aryloxy, cyano, halogen, hydroxyl, nitro, N3, carboxyl, carboxyl esters, thiol, thioalkyl, thioaryl, thioheteroaryl, thiocycloalkyl, thioheterocyclic, cycloalkyl, heteroaryl, and heterocyclic. The terms "alkenyl" or "alkene" as used herein refers to an alkenyl group having from 2 to 10 carbon atoms and having at least 1 site of alkenyl unsaturation. Such groups are exemplified by vinyl(ethen-l-yl), allyl, but-3-en- 1-yl, and the like.
The term "substituted alkenyl" as used herein refers to alkenyl groups having from 1 to 3 substituents, said substituents being selected from those describe above for a substituted alkyl.
The term "alkynyl" or "alkyne" as used herein refers to an alkynyl group having from 2-10 carbon atoms and having at least 1 site of alkynyl unsaturation. Such groups are exemplified by, but not limited to, ethyn-1-yl, propyn-1-yl,
propyn-2-yl, l-methylprop-2-yn-l-yl, butyn-1-yl, butyn-2-yl, butyn-3-yl, and the like.
The term "substituted alkynyl" as used herein refers to alkynyl groups having from 1 to 3 substituents, said substituents being selected those describe above for a substituted alkyl.
The term "alkoxy" refers to the group alkyl-O-.
The term "substituted alkoxy" as used herein refers to the group substituted alkyl-O-.
The term "acyl" as used herein refers to the groups alkyl-C(O) -, alkenyl- C(O) -, alkynyl-C(O) -, cycloalkyl-C(O) -, aryl-C(O) -, heteroaryl-C(O) -, and heterocyclic-C(O).
The term "substituted acyl" as used herein refers to the groups substituted alkyl-C(O)-, substituted alkenyl-C(O) -, substituted alkynyl-C(O) -, substituted cycloalkyl-C(O) -, substituted aryl-C (O)-, substituted heteroaryl-C(O), and substituted heterocyclic-C(O) -.
The term "acylamino" as used herein refers to the group-C(O)NZ!Z2 where each Z1 and Z2 are independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl, and the substituents described above in the definition of substituted alkyl.
The term "acyloxy" as used herein refers to the groups alkyl-C(O)O-, substituted alkyl- C(O)O-, alkenyl-C(O)O-, substituted alkenyl-C(O)O-, alkynyl- C(O)O-, substituted alkynyl-C(O)O-, aryl-C(O)O-, substituted aryl-C(O)O-, cycloalkyl-C(O)O-, substituted cycloalkyl-C(O)O-, heteroaryl-C(O)O-, substituted heteroaryl-C(O)O-, heterocyclic-C(O)O-, and substituted heterocyclic-C(O)O-.
The term "oxyacyl" as used herein refers to the groups alkyl-OC(O) -, substituted alkyl-OC(O)-, alkenyl-OC(O)-, substituted alkenyl-OC(O) -, alkynyl- OC(O) -, substituted alkynyl-OC(O)-, aryl-OC(O)-, substituted aryl-OC(O)-, cycloalkyl-OC(O)-, substituted cycloalkyl-OC(O)-, heteroaryl-OC(O) -, substituted heteroaryl-OC(O) -, heterocyclic- OC(O)-, and substituted heterocyclic-OC(O)-.
The term "amino" as used herein refers to the group -NH2.
The term "substituted amino" as used herein refers to the group-N Z1Z2 where Z1 and Z2 are as described above in the definition of acylamino, provided that Z1 and Z2 are both not hydrogen.
The term "aminoacyl" as used herein refers to the groups -NZ3C(O)alkyl, -NZ3C(O)substituted alkyl, -NZ3C(O)cycloalkyl, -NZ3C(O)substituted cycloalkyl, -NZ3C(O)alkenyl, -NZ3C(O)substituted alkenyl, -NZ3C(O)alkynyl, - NZ3C(O)substituted alkynyl, -NZ3C(O)aryl, -NZ3C(O)substituted aryl, - NZ3C(O)heteroaryl, - NZ3C(O)substituted heteroaryl, -NZ3C(O)heterocyclic, and -NZ3C(O)substituted heterocyclic, where Z3 is hydrogen or alkyl. The term "aryl" as used herein refers to a monovalent aromatic cyclic group of from 6 to 14 carbon atoms having a single ring (e.g., phenyl) or multiple condensed rings (e.g., naphthyl or anthryl) which condensed rings may or may not be aromatic. Exemplary aryls include, but are not limited to, phenyl and naphthyl. The term "substituted aryl" as used herein refers to aryl groups which are substituted with from 1 to 3 substituents selected from alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl, and those substituents described above in the definition of substituted alkyl.
The term "aryloxy" as used herein refers to the group aryl-O- that includes, by way of example but not limitation, phenoxy, naphthoxy, and the like.
The term "substituted aryloxy" as used herein refers to substituted aryl- O-groups.
The term "carboxyl" as used herein refers to -COOH or salts thereof. The term "carboxyl esters" as used herein refers to the groups-C(O)O- alkyl, -C (O)O-substituted alkyl, -C(O)O-aryl, and-C(O)O-substituted aryl.
The term "cycloalkyl" as used herein refers to a saturated or unsaturated cyclic hydrocarbon ring systems, such as those containing 1 to 3 rings and 3 to 7 carbons per ring. Exemplary groups include but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and adamantyl.
The term "substituted cycloalkyl" as used herein refers to a cycloalkyl having from 1 to 5 substituents selected from the group consisting of oxo (=0), thioxo (=S), alkyl, substituted alkyl, and those substituents described in the definition of substituted alkyl.
The term "cycloalkoxy" as used herein refers to -O-cycloalkyl groups.
The term "substituted cycloalkoxy" as used herein refers to-O-substituted cycloalkyl groups.
The term "formyl" as used herein refers to HC(O)-. The term "halogen" as used herein refers to fluoro, chloro, bromo and iodo.
The term "heteroaryl" as used herein refers to an aromatic group of from 1 to 10 carbon atoms and 1 to 4 heteroatoms selected from the group consisting of oxygen, nitrogen, sulfur in the ring. The sulfur and nitrogen heteroatoms atoms may also be present in their oxidized forms. Such heteroaryl groups can have a single ring (e.g., pyridyl or furyl) or multiple condensed rings (e.g., indolizinyl or benzothienyl) wherein the condensed rings may or may not be aromatic and/or contain a heteroatom. Exemplary heteroaryl groups include, but are not limited to, heteroaryls include pyridyl, pyrrolyl, thienyl, indolyl, thiophenyl, and furyl.
The term "substituted heteroaryl" as used herein refers to heteroaryl groups that are substituted with from 1 to 3 substituents selected from the same group of substituents defined for substituted aryl.
The term "heteroaryloxy" as used herein refers to the group -O- heteroaryl.
The term "substituted heteroaryloxy" as used herein refers to the group - O-substituted heteroaryl.
The term "heterocycle" or "heterocyclic" or "heterocycloalkyl" refers to a saturated or unsaturated group (but not heteroaryl) having a single ring or multiple condensed rings, from 1 to 10 carbon atoms and from 1 to 4 hetero atoms selected from the group consisting of nitrogen, oxygen, sulfur, within the ring wherein, in fused ring systems, one or more the rings can be cycloalkyl, aryl or heteroaryl provided that the point of attachment is through the heterocyclic ring. The sulfur and nitrogen heteroatoms atoms may also be present in their oxidized forms.
The term "substituted heterocycle" or "substituted heterocyclic" or "substituted heterocycloalkyl" refers to heterocycle groups that are substituted with from 1 to 3 of the same substituents as defined for substituted aryl.
Examples of heterocycles and heteroaryls include, but are not limited to, azetidine, pyrrole, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, phenanthroline, isothiazole, phenazine, isoxazole, phenoxazine, phenothiazine, imidazolidine, imidazoline, piperidine, piperazine, indoline, phthalimide, 1,2, 3,4-tetrahydroisoquinoline, 4,5, 6,7-tetrahydrobenzo [b] thiophene, thiazole, thiazolidine, thiophene, benzo [b] thiophene, morpholinyl, thiomorpholinyl (also referred to as thiamorpholinyl), piperidinyl, pyrrolidine, tetrahydrofuranyl, and the like.
The term "heterocyclyloxy" as used herein refers to the group -O- heterocyclic.
The term "substituted heterocyclyloxy" as used herein refers to the group-O-substituted heterocyclic.
The term "phosphate" as used herein refers to the groups- OP(O)(OH)2 (monophosphate or phospho), -OP(O)(OH)OP(O)(OH)2 (diphosphate or diphospho) and -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 (triphosphate or triphospho) or salts thereof including partial salts thereof. It is understood that the initial oxygen of the mono-, di-, and triphosphate may include the oxygen atom of a sugar.
The term "phosphate esters" as used herein refers to the mono-, di-and tri-phosphate groups described above wherein one or more of the hydroxyl groups is replaced by an alkoxy group. The term "phosphonate" refers to the groups -0P(0)(Z4)(0H) or-OP(O)
(Z4)(OZ4) or salts thereof including partial salts thereof, wherein each Z4 is independently selected from hydrogen, alkyl, substituted alkyl, carboxylic acid, and carboxyl ester. It is understood that the initial oxygen of the phosphonate may include the oxygen of a sugar. The term "thiol" as used herein refers to the group -SH.
The term "thioalkyl" or "alkylthioether" or "thioalkoxy" refers to the group- S-alkyl.
The term "substituted thioalkyl" or "substituted alkylthioether" or "substituted thioalkoxy" refers to the group -S-substituted alkyl.
The term "thiocycloalkyl" as used herein refers to the group -S- cycloalkyl.
The term "substituted thiocycloalkyl" as used herein refers to the group - S-substituted cycloalkyl. The term "thioaryl" as used herein refers to the group -S-aryl.
The term "substituted thioaryl" as used herein refers to the group-S- substituted aryl.
The term "thioheteroaryl" as used herein refers to the group -S- heteroaryl. The term "substituted thioheteroaryl" as used herein refers to the group -
S-substituted heteroaryl.
The term "thioheterocyclic" as used herein refers to the group -S- heterocyclic.
The term "substituted thioheterocyclic as used herein refers to the group - S-substituted heterocyclic.
The term "amino acid sidechain" refers to the Z7 substituent of α-amino acids of the formula Z6NHCH(Z7)COOH where Z7 is selected from the group consisting of hydrogen, alkyl, substituted alkyl and aryl and Z6 is hydrogen or together with Z7 and the nitrogen and carbon atoms bound thereto respectively form a heterocyclic ring. In one embodiment, the α-amino acid sidechain is the sidechain one of the twenty naturally occurring L amino acids.
Sugars described herein may either be in D or L configuration. Compounds of Formula III
In one embodiment the invention provides a compound of Formula III:

III wherein:
X is N, Y is N and Z is CH; or X is C, Y is CR3 and Z is O; R is ORa, SR3, NRaRb, NR3NRbR0, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, (CH2)n-
CH(NHR3)CO2Rb, (CH2)n-S-alkyl,,(CH2)n-S-aryl, Cl, F, Br, I, CN, COORa, CONR3Rb, NHC(=NRa)NHRb, NR3ORb, NR3NO, NHCONHR3, NRaN=NRb, NRaN=CHRb, NR3C(O)NRbR0, NR3C(S)NRbRc, NR3C(0)0Rb, CH=N-OR3, NRaC(=NH)NRbRc, NR3C(O)NRbNR0Rd, 0-C(O)R3, OC(O)-OR3, ONH- C(0)0-alkyl, 0NHC(0)0-aryl, 0NR3Rb, SNR3Rb, S-0NR3Rb, CHO, C(=S)N RaRb, nitro, C (=NR3)0Rb, or SO2NR3Rb; and R3 is H, CN, NO2, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, CH=CF2, CH(=N R3)ORb, CHO, CH=CH-OCH3, NHCONH2, NHCSNH2, CONR3Rb, CSNR3Rb, CO2R3, alkoxy, NH2, alkylamino, dialkylamino, halogen, (l,3-oxazol-2-yl), (l,3-oxazol-5-yl), (l,3-thiazol-2-yl), (imidazol-2-yl), (2- oxo[l, 3]dithiol-4-yl), (foran-2-yl), (2H[l,2,3]triazol-4-yl), C(=NH)NH2, C(=NH)NH0H, C(=N0H)NH2, acyl, substituted acyl, OR3, C(=NRa)Rb, CH=NNR3Rb, CH=NOR3, CH(OR3)2, B(ORa)2, C≡C-C(=O)NRaRb, CH=NNR3Rb, CH=NOR3, CH(ORa)2, B(ORa)2, C≡C-C(=O)NRaRb, (CH2)n-S- alkyl, (CH2)n-S-aryl, (CH2)n-S(O)-alkyl, (CH2)n-S(O)-aryl, (CH2)n-S(O2)-alkyl, (CH2)n-S(O2)-aryl, (CH2)n-SO2NR3Rb, or (CH2)n-OR3; or R and R3 together with atoms to which they are attached may form a cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; n is 0-5;
R1 is H, NR3Rb, Cl, F, 0Ra, SR3, NHCOR3, NHSO2R3, NHCONHR3, CN, alkyl, aryl, 0NR3Rb, or NR3C(0)0Rb;
R is a nucleoside sugar group; and
R3, Rb, Rc, and Rd are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, SO2-alkyl, amino, substituted amino, substituted cycloalkyl, heteroaryl, substituted heteroaryl , and NO; or R3 and Rb together with the nitrogen to which they are attached form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; or Rb and R0 together with the nitrogen to which they are attached form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; or R0 and Rd together with the nitrogen to which they are attached form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl;
or a pharmaceutically acceptable salt or prodrug thereof. In one embodiment the invention provides a compound of Formula III:

III wherein:
X is N, Y is N and Z is CH; or X is C, Y is CR3 and Z is O; R is ORa, SR3, NRaRb, NRaNRbRc, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, (CH2)n- CH(NHRa)CO2Rb, (CH2)n-S-alkyl, (CH2)n-S-aryl, Cl, F, Br, I, CN, COORa, CONR3Rb, NHC(=NRa)NHRb, NRaORb, NR3NO, NHCONHR3, NRaN=NRb, NRaN=CHRb, NRaC(O)NRbRc, NR3C(S)NRbRc, NR3C(O)OR1,, CH=N-OR3, NRaC(=NH)NRbRc, NR3C(O)NRbNRcRd, 0-C(O)R3, OC(O)-OR3, ONH- C(O)O-alkyl, ONHC(O)O-aryl, ONR3R5, SNR3Rb, S-ONRaRb, CHO, C(=S)N R3Rb, nitro, CH(=NR3)0Rb, or SO2NR3Rb; and R3 is H, CN, NO2, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, CH=CF2, CH(=N R3)OR5, CHO, CH=CH-OCH3, NHCONH2, NHCSNH2, CONR3Rb, CSNR3Rb, CO2R3, alkoxy, NH2, alkylamino, dialkylamino, halogen, (l,3-oxazol-2-yl), (l,3-oxazol-5-yl), (l,3-thiazol-2-yl), (imidazol-2-yl), (2- oxo[l, 3]dithiol-4-yl), (furan-2-yl), (2H[l,2,3]triazol-4-yl), C(=NH)NH2, C(=NH)NH0H, C(=N0H)NH2, acyl, substituted acyl, OR3, C(=NRa)Rb,
CH=NNRaRb, CH=NOR3, CH(0Ra)2, B(OR3)2, C≡C-C(=0)NR3Rb, (CH2)n-S- alkyl, (CH2)n-S-aryl, (CH2)n-S(O)-alkyl, (CH2)n-S(O)-aryl, (CH2)n-S(O2)-alkyl, (CH2)n-S(O2)-aryl, (CH2)n-SO2NRaRb, or (CH2)n-0Ra; or R and R3 together with atoms to which they are attached may form a cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; n is 0-5;
R1 is H, NRaRb, Cl, F, OR3, SR3, NHCOR3, NHSO2R3, NHCONHR3, CN, alkyl, aryl, 0NRaRb, or NR3C(0)0Rb; R2 is a nucleoside sugar group;
Ra, Rb, Rc, and Rd are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, Sθ2-alkyl, amino, substituted amino, and NO; or Raand Rb together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; or Rb and R0 together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; and
Rc, and Rd are independently selected from the group consisting of H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heteroaryl, and substituted heteroaryl; or R0, and Rd together with N atom to which they are attached may form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; or a pharmaceutically acceptable salt or prodrug thereof. Compounds of Formula I and II
Compounds of the invention include compounds of Formula I and II:

I II.
In certain embodiments:
R is OR3, SRa, NR3Rb, NRaNRbRc, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, (CH2)n- CH(NHRa)CO2Rb, (CH2)n-S-alkyl, (CH2)n-S-aryl, Cl, F, Br, I, CN, COOR3, CONR3Rb, NHC(=NR3)NHRb, NRaORb, NR3NO, NHCONHR3, NRaN=NRb, NRaN=CHRb, NR3C(O)NRbR0, NR3C(S)NRbRc, NRaC(O)ORb, CH=N-OR3, NRaC(=NH)NRbRc, NRaC(O)NRbNRcRd, 0-C(O)R3, OC(O)-OR3, ONH- C(O)O-alkyl, ONHC(O)O-aryl, 0NR3Rb, SNRaRb, S-0NRaRb, CHO, C(=S)N RaRb, nitro, CH(NR3)ORb, or SO2NR3Rb; and R3 is H, CN, NO2, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, CH=CF2, CH(=NRa)0Rb, CHO, CH=CH-OCH3, NHCONH2, NHCSNH2, CONR3Rb, CSNRaRb, CO2R3, alkoxy, NH2, alkylamino, dialkylamino, halogen,
(l,3-oxazol-2-yl), (l,3-oxazol-5-yl), (l,3-thiazol-2-yl), (imidazol-2-yl), (2- oxo[l, 3]dithiol-4-yl), (furan-2-yl), (2H[l,2,3]triazol-4-yl), C(=NH)NH2, C(=NH)NHOH, C(=NOH)NH2, acyl, substituted acyl, ORa, C(=NRa)Rb, CH=NNRaRb, CH=NORa, CH(ORa)2, B(ORa)2, C≡C-C(=O)NRaRb, (CH2VS- alkyl, (CH2)n-S-aryl, (CH2)n-S(O)-alkyl, (CH2)n-S(O)-aryl, (CH2)n-S(O2)-alkyl, (CH2)n-S(O2)-aryl, (CH2)n-SO2NRaRb, or (CH2)n-0Ra; or R and R3 together with atoms to which they are attached may form a cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; n is 0-5;
R1 is H, NR3Rb, Cl, F, ORa, SR3, NHC0Ra, NHSO2R3, NHCONHR3, CN, alkyl, aryl, 0NRaRb, or NRaC(0)0Rb;
R2 is a nucleoside sugar group;
Ra, Rb, Rc, and Ra are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, SO2-alkyl, amino, substituted amino, and NO; or R3 and Rb together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; or Rb and R0 together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; and
Rc, and Ra are independently selected from the group consisting of H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heteroaryl, and substituted heteroaryl; or R0, and Rd together with N atom to which they are attached may form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; or a pharmaceutically acceptable salt or prodrug thereof.
In one embodiment the invention provides a compound of Formula I, II, or III as described above, wherein R is OR3, Cl, SRa, NR3Rb, or NR3NRbRc; or a pharmaceutically acceptable salt or prodrug thereof.
In one embodiment the invention provides a compound of Formula I, II, or III as described above, wherein R is NR3Rb; R8 is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl,
substituted acyl, SO2-alkyl and NO; and Rb is selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, SO2-alkyl and NO; or Ra and Rb together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring.
In one embodiment the invention provides a compound of Formula I, II, or III as described above, wherein R is NR3NRbRc, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, (CH2)n-CH(NHRa)CO2Rb, Cl, F, Br, I, CN, COORa, CONRaRb,
NHC(=NRa)NHRb, NRaORb, NRaNO, NHCONHR3, NRaN=NRb, NRaN=CHRb, NRaC(O)NRbR0, NRaC(S)NRbRc, NRaC(O)ORb, CH=N-OR3, NRaC(=NH)NRbRc, NR3C(O)NRbNR0R11, 0-C(O)R3, OC(O)-OR3, ONH- C(O)O-alkyl, ONHC(O)O-aryl, 0NRaRb, SNR3Rb, S-ONR3Rb, or SO2NR3Rb. In one embodiment the invention provides a compound of Formula I, II, or III as described above, wherein R1 is H or NRaRb; or a pharmaceutically acceptable salt or prodrug thereof.
In one embodiment the invention provides a compound of Formula I, II, or III as described above, wherein R2 is a nucleoside sugar group of Group A, B, C, D, E, or F described herein below; or a pharmaceutically acceptable salt or prodrug thereof.
In another embodiment the invention provides a compound of Formula I, II, or III as described above, wherein R2 is ribose, 2-methylribose, 2- deoxyribose; 2-deoxy-2-fluororibose; arabinose; 2-deoxy-2-fluoroarabinose; 2,3- dideoxyribose; 2,3-dideoxy-2-fluoroarabinose; 2,3-dideoxy-3-fluororibose; 2,3- dideoxy-2,3-didehydroribose; 2,3-dideoxy-3-azidoribose; 2,3-dideoxy-3- thiaribose; or 2,3-dideoxy-3-oxaribose; or a pharmaceutically acceptable salt or prodrug thereof.
In another embodiment the invention provides a compound of Formula I, II, or III as described above, wherein R2 is thioribose, 2-deoxythioribose; 2- deoxy-2-fluorothioribose; thioarabinose; 2-deoxy-2-fluorothioarabinose; 2,3- dideoxythioribose; 2,3-dideoxy-2-fluorothioarabinose; 2,3-dideoxy-3- fluorothioribose; 2,3-dideoxy-2,3-didehydrothioribose; or 2,3-dideoxy-3- azidothioribose; or a pharmaceutically acceptable salt or prodrug thereof.
In another embodiment the invention provides a compound of Formula I, II, or III as described above, wherein R2 is 4-hydroxymethylcyclopent-2-ene; 2,3-dihydroxy-4-hydroxyrnethylcyclopent-4-ene; 3-hydroxy-4- hydroxymethylcyclopentane; 2-hydroxy-4-hydroxymethylcyclopentene; 2- fluoro-3-hydroxy-4-hydroxymethylcyclopentane; 2,3-dihydroxy-4- hydroxymethyl-5-methylenecyclopentane; 4-hydroxymethylcyclopentane, 2,3- dihydroxy-4-hydroxymethylcyclopentane; or 2,3 -dihydroxymethylcyclobutane; or a pharmaceutically acceptable salt or prodrug thereof.
In another embodiment the invention provides a compound of Formula I, II, or III as described above, wherein R2 is 4-hydroxymethylpyrrolidine; 2,3- dihydroxy-4-hydroxymethylpyrrolidine; 2/3-hydroxy-4- hydroxymethylpyrrolidine; 2-fluoro-3 -hydroxy-4-hydroxymethylpyrrolidine; or 3-fluoro-2-hydroxy-4-hydroxymethyl-pyrrolidine; or a pharmaceutically acceptable salt or prodrug thereof. In another embodiment the invention provides a compound of Formula I,
II, or III as described above, wherein Ra, Rb, Rc, and Rd are independently selected from the group consisting of H, alkyl, and substituted alkyl; or Raand Rb together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; or Rb and R0 together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring.
In another embodiment the invention provides a compound of Formula I, II, or III which is a compound of the following formula (11a or lib).

11a lib or a pharmaceutically acceptable salt or prodrug thereof.
In another embodiment the invention provides a compound of Formula II, which is a compound of the following formula (IV):

IV or a pharmaceutically acceptable salt or prodrug thereof; wherein X is H or alkyl.
In certain embodiments the invention, the compound of the invention is not a compound of the following formula:

B is selected from:
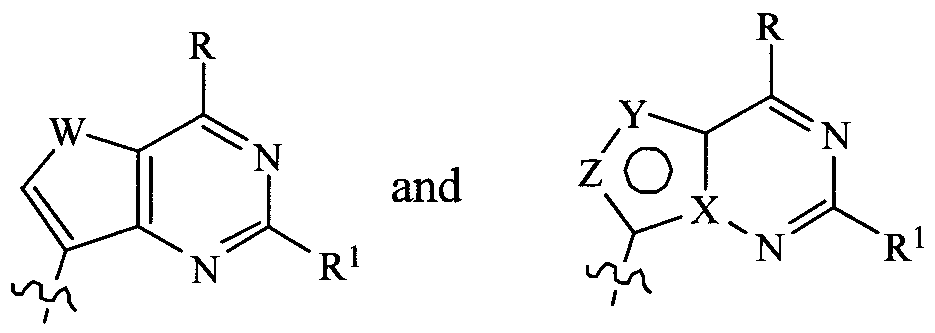
W is O, S, or NH;
X is N, Y is N and Z is CH; or X is C, Y is CR23 and Z is O;
R is OR3, SR3, NR3R4, NR3NR4R5, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, (CH2),,- CH(NHR3)CO2R4, (CH2)n-S-alkyl, (CH2)n-S-aryl, Cl, F, Br, I, CN, COOR3, CONR3R4, MKX=NR3)NHR4, NR3OR4, NR3NO, NHCONHR3, NR3N=NR4, NR3N=CHR4, NR3C(O)NR4R5, NR3C(S)NR4R5, NR3C(O)OR4, CH=N-OR3, NR3Q=NH)NR4R5, NR3C(O)NR4NR5R6, 0-C(O)R3, OC(O)-OR3, ONH- C(O)O-alkyl, ONHC(O)O-aryl, ONR3R4, SNR3R4, S-ONR3R4, or SO2NR3R4; n is 0-5;
R1 is H, NR3R4, Cl, F, OR3, SR3, NHCOR3, NHSO2R3, NHCONHR3, CN, alkyl, aryl, ONR3R4, Or NR3C(O)OR4;
R3, R4, R5, and R6 are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, SO2-alkyl and NO; or R3 and R4 together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; or R4 and R5 together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; and
Rc is selected from the group consisting of hydrogen, alkyl, alkenyl, and alkynyl wherein each alkyl, alkenyl, and alkynyl is optionally substituted with one or more substituents independently selected from halo, amino, hydroxy, and alkoxy; or a pharmaceutically acceptable salt thereof.
Prodrugs
The term "prodrug" as used herein refers to a compound that can be metabolized in vivo to provide a compound of Formula I to III. Thus prodrugs include compounds that can be prepared by modifying one or more functional groups in a compound of Formula I to III to provide a corresponding compound that can be metabolized in vivo to provide a compound of Formula I to III. Such modifications are known in the art. For example, one or more hydroxy groups or amine groups in a compound of Formula I to III can be acylated with alkyl- C(=O)-groups or with residues from amino acids to provide a prodrug. Alternatively, one or more pendent hydroxyl groups from a mono-, di-, or triphosphate functionality in a compound of Formula I to III can be converted to an alkoxy, substituted alkoxy, aryloxy, or substituted aryloxy group.
In one embodiment, the term prodrug includes a compound wherein one or more hydroxy groups on a nucleoside sugar group (e.g., a 2', 3', or 5' hydroxy group) have been converted to a group that can be metabolized in vivo to provide a compound of Formula I to III. For example, the invention provides a compound wherein one or more hydroxy groups on a nucleoside sugar group (e.g., a 2', 3', or 5' hydroxy group) have been converted to an acyloxy, acylamino or R-O group, wherein R is a carboxy-linked amino acid.
In one embodiment, the term prodrug includes a compound wherein one or more pendent hydroxyl groups from a mono-, di-, or tri-phosphate functionality in a compound of Formula I to III is converted to a group Ry-O-; wherein each Ry is independently a 1-20 carbon branched or unbranched, saturated or unsaturated chain, wherein one or more (e.g., 1, 2, 3, or 4) of the carbon atoms is optionally replaced with -O- or -S- and wherein one or more of the carbon atoms is optionally substituted with oxo (=0) or thioxo (=S) (See Lefebvre et al, J. Med. Chem. 1995, 38, 3941-50).
In another embodiment, the term prodrug includes a compound wherein one or more pendent hydroxyl groups from a mono-, di-, or tri-phosphate functionality in a compound of Formula I to III is converted to a group R2-N-; wherein each R2 is a residue of an amino acid. Thus, in the methods of treatment of the present invention, the term "administering" includes administration of a compound of Formula I to III, as well as administration of a prodrug which converts to a compound of Formula I to III or a salt thereof in vivo.
Conventional procedures for the selection and preparation of prodrug derivatives are described, for example, in "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985; and in International Patent Application Publication Number WO 2005/084192. A variety of prodrugs are also described in International Patent Application Number PCT US2004/013063 , which was published as International Publication Number WO 2004/096286 and PCT US2006/030549, which was published as International Publication Number WO 2007/021610.
In another embodiment the prodrug comprises one of more groups of formula:
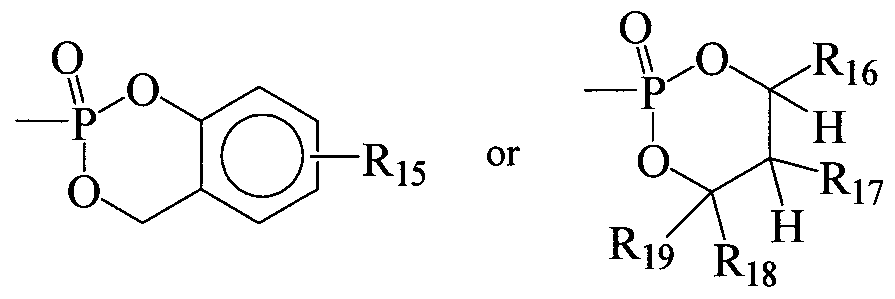
R15 is H, alkyl, substituted alkyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, heteroaryl, substituted heteroaryl, heterocyclic, substituted heterocyclic, and an amino acid;
R16 is H, optionally substituted monocyclic aryl, or optionally substituted monocyclic heteroaryl; and R17 is H, halogen, CN, -CO-R20, -CON(R2O2, -
CO2R20, -SO2R20, -SO2N(R21)2, -OR2I, -SR21, -R21, -N(R21)2, -0-COR20, -O- CO2R20, -SCOR20, -S-CO2R20, -NHCOR21, -NHCO2R21, -(CH2)P-OR22, or - (CH2)P-SR22; or R16 and R17 are connected via an additional 3-5 atoms to form a cyclic group, optionally containing one heteroatom, that is fused to an aryl group at the beta and gamma position to the O attached to the phosphorus; or R17 and R18 are connected as described below;
R18 and R19 are each independently H, alkyl, aryl, heterocycloalkyl, aralkyl, optionally substituted monocyclic aryl or optionally substituted monocyclic heteroaryl; or R18 and R19 are connected via an additional 2-5 atoms to form a cyclic group, optionally containing 0-2 heteroatoms; or R17 and R18 are connected via an additional 3-5 atoms to form a cyclic group, optionally containing one heteroatom and R19 is H, alkyl, aryl, heterocycloalkyl, aralkyl, optionally substituted monocyclic aryl or optionally substituted monocyclic heteroaryl; and R20 is alkyl, aryl, heterocycloalkyl, or arylalkyl;
R21 is H, alkyl, aryl, heterocycloalkyl, or arylalkyl;
R22 is H or lower acyl; n is an integer from 2-5; m is an integer from 10-20; and p is an integer from 2-3.
Prodrug forms of a compound bearing various nitrogen functions (amino, hydroxyamino, amide, etc.) may include the following types of derivatives where each Rp group individually may be hydrogen, substituted or unsubstituted alkyl, aryl, alkenyl, alkynyl, heterocycle, alkylaryl, aralkyl, aralkenyl, aralkynyl, cycloalkyl or cycloalkenyl groups as defined earlier.
(a) Carboxamides, represented as -NHC(O)Rp
(b) Carbamates, represented as -NHC(O)ORp
(c) (Acyloxy)alkyl Carbamates, represented as NHC(O)OROC(O)Rp
(d) Enamines, represented as -NHCR(=CHCO2RP) or -NHCR(=CHCONRpRp)
(e) Schiff Bases, represented as -N=CRPRP
(f) Mannich Bases (from carboximide compounds), represented as RCONHCH2NRpRp
Preparations of such prodrug derivatives are discussed in various literature sources (examples are: Alexander et al, J. Med. Chem. 1988, 31, 318; Aligas- Martin et al, PCT WO0041531, p.30).
Prodrug forms of carboxyl -bearing compounds include esters (-CO2Rm) where the Rm group corresponds to any alcohol whose release in the body through enzymatic or hydrolytic processes would be at pharmaceutically acceptable levels. Another prodrug derived from a carboxylic acid form of the disclosure may be a quaternary salt type of structure described by Bodor et al. , J. Med. Chem. 1980, 23, 469.

R
Nucleoside Sugar Groups
The term "nucleoside sugar group" as used herein includes cyclic and acyclic groups that can be included as the sugar portion of a nucleoside analog of Formula I to III. Many examples of such groups are known in the field of nucleoside chemistry (See for example Antiviral Drugs by John S. Driscoll (2002) published by Ashgate Publishing Ltd.).
The term nucleoside sugar group includes substituted and unsubstituted tetrahydrofuranyl and dihydrofuranyl compounds including those set forth in group (A) below, substituted and unsubstituted tetrahydrothiophenyl and dihydrothiophenyl compounds including those set forth in group (B) below, substituted and unsubstituted alkyl compounds including those set forth in group (C) below, substituted and unsubstituted cycloalkyl and cycloalkenyl compounds including those set forth in group (D) below, substituted and unsubstituted dihydropyrrolidinyl and tetrahydropyrrolidinyl compounds including those set forth in group (E) below, and substituted and unsubstituted dioxolane, substituted and unsubstituted thioxolane, and substituted and unsubstituted dithiolane compounds including those set forth in group (F) below.
Group A
Examples of substituted tetrahydro and dihydrofuranyl compounds include those compounds represented by the general structures:

Specific examples include, but are not limited to, the following compounds:

(d) (1) (d) (1)
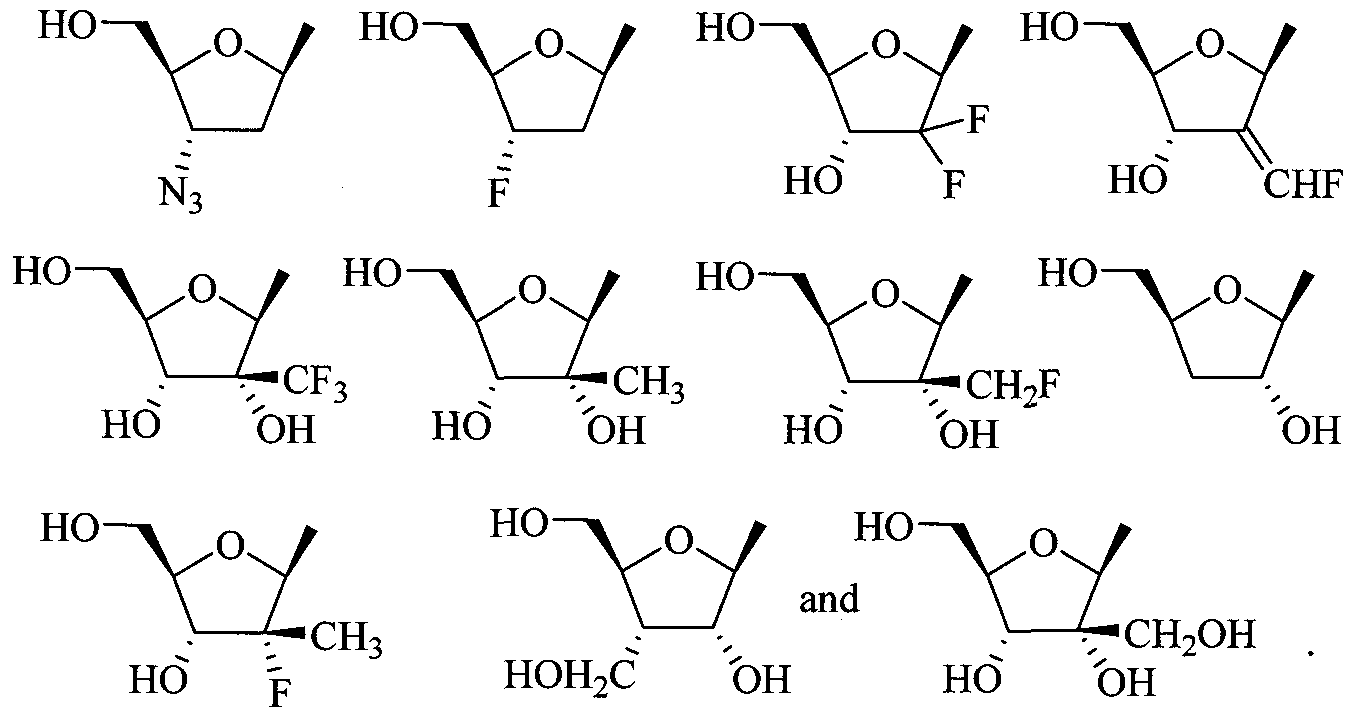
Group B
Examples of substituted tetrahydrothiophenyl and dihydrothiophenyl compounds include those compounds represented by the general structures:

Specific examples include, but are not limited to, the following compounds:

Group C
Examples of substituted alkyl compounds include those compounds represented by:
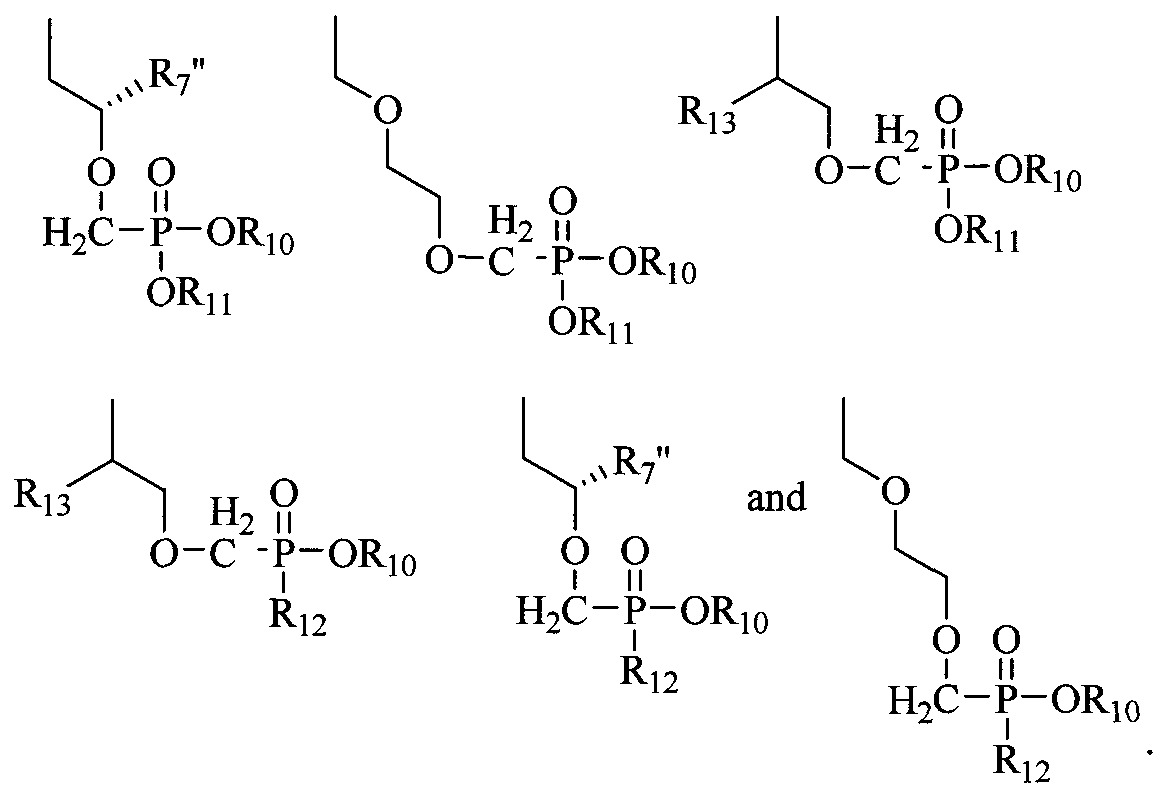

Group D Examples of substituted cycloalkyl and cycloalkenyl compounds include those compounds represented by the general structures:

Specific examples include, but are not limited to, the following compounds:
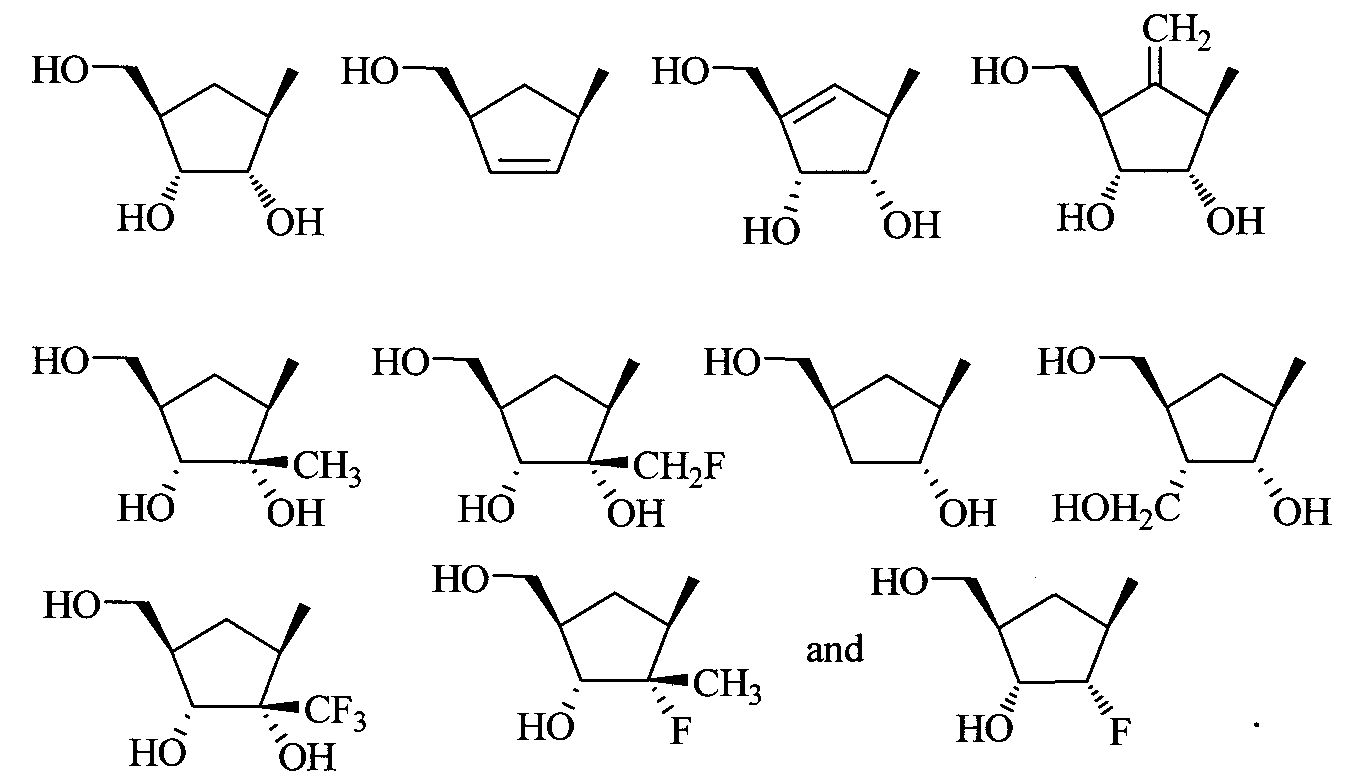
Group E
Examples of substituted dihydropyrrolidinyl and tetrahydropyrrolidinyl compounds include those compounds represented by the general structures:
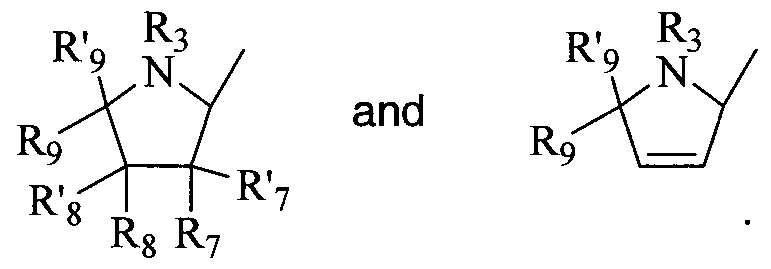

Group F
Examples of substituted dioxolane, substituted thioxolane and substituted dithiolane compounds include those compounds represented by the general structures:
R .q9 π n / / R IVq9 ς /

Specific examples include, but are not limited to, the following compounds:
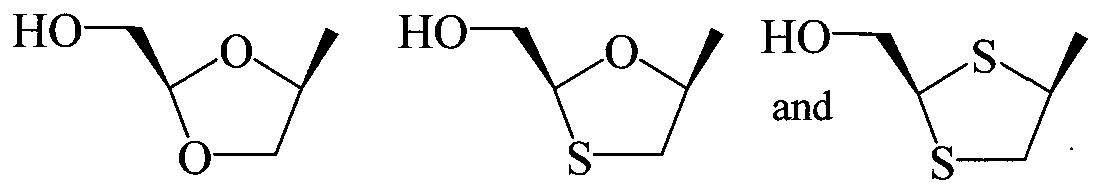
For the structures in Groups A-F, the following definitions apply: R7 is H, OR14, N3, NH2, or F; and R'7 is H, F, OH, O-alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R7 and R7 together may be =CH2, =CHF; wherein both R7 and R7 are not OH; and when one of R7 and R7 is NH2, the other is not OH; and when one of R7 and R'7 is N3, the other is not OH; R7" is alkyl or substituted alkyl. R8 is H, OR14, N3, NH2, or F; and R'8 is H, F, OH, O alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R8 and R8 together may be =CH2, =CHF; wherein both R8 and R'8 are not OH; and when one of R8 and R8 is NH2, the other is not OH; and when one of R8 and R'8 is N3, the other is not OH;
or R7 and R8 together can form
O O
R100
wherein: RJOO is C1-12 alkyl C3-8 cycloalkyl, aryl or heteroaryl; wherein any C1-12 alkyl and C3-8 cycloalkyl OfR100 is unsubstituted or is substituted with 1-3 substituents selected from halogen, hydroxy, carboxy, and C1-4 alkoxy; and wherein any aryl or heteroaryl of R1Oo is unsubstituted or is substituted with 1-5 substituents independently selected from R1Oi; each R101 is independently halo, C1-4 alkyl, C1-4 alkoxy, C1-4 alkylthio, C1-
4 alkylsulfoyl, cyano, nitro, amino, phenyl, carboxy, trifluoromethyl, trifluoromethoxy, C1-4 alkylamino, di(C1-4 alkyl) amino, C1-4 alkanoyl, C1-4 alkanoyloxy, or C1-4 alkyloxycarbonyl; R9 is H, CH3, C2H5, or N3; R9 is CH2OR14, CH2F, CH2SH, CHFOH, CF2OH,
— o-C -P-OR10 ;

R10 and R11 are each independently H, alkyl, aryl, substituted aryl, acyloxyalkyl, or (CH2)nl-O-(CH2)mCH3; nl is 2-5; m is 10-20;
Ri2 is an N-linked amino acid residue (e.g., -NH-CH(CH3)C02alkyl or - NH-CH(isopropyl)-CO2alkyl);
R13 is H, CH3, C2H5, CH2F, CH2OH, CH2CH2F, CH2CH2OH, CH2N3, CH2CH2N3, CH2NH2, or CH2CH2NH2; and R14 is H.
In one embodiment, for the structures in Groups A-F, R7 is H, OR14, N3, NH2, or F; and R'7 is H, F, OH, O-alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R7 and R7 together may be =CH2, =CHF; wherein both R7 and R7 are not OH; and when one of R7 and R7 is NH2, the other is not OH; and when one of R7 and R7 is N3, the other is not OH; R7" is alkyl or substituted alkyl; and R8 is H, OR14, N3, NH2, or F; and
R'8 is H, F, OH, O alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R8 and R'8 together may be =CH2, =CHF; wherein both R8 and R'8 are not OH; and when one of R8 and R8 is NH2, the other is not OH; and when one of R8 and R'8 is N3, the other is not OH; In one embodiment, for a compound of Formula I to III, R14 is replaced to form a pharmaceutically acceptable prodrug, for example, a prodrug selected from the group consisting of: acyl, oxyacyl, phosphonate, phosphate, phosphate esters, phosphonamidate, phosphorodiamidate, phosphoramidate mono ester, cyclic phosphoramidate, cyclic phosphorodiamidate, phosphoramidate diester, C(O) CHR15NH2,

R15 is H, alkyl, substituted alkyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, heteroaryl, substituted heteroaryl, heterocyclic, substituted heterocyclic, or an amino acid; R16 is H, optionally substituted monocyclic aryl, or optionally substituted monocyclic heteroaryl; and R17 is H, halogen, CN, -CO-R20, -CON(R21)2, - CO2R20, -SO2R20, -SO2N(R21)2, -OR21, -SR21, -R21, -N(R21)2, -0-COR20, -O- CO2R20, -SCOR20, -S-CO2R20, -NHCOR21, -NHCO2R21, -(CH2)P-OR22, or - (CH2)P-SR22; or R16 and R17 are connected via an additional 3-5 atoms to form a cyclic group, optionally containing one heteroatom, that is fused to an aryl group at the beta and gamma position to the O attached to the phosphorus; or R17 and R18 are connected as described below;
Ri8 and R19 are each independently H, alkyl, aryl, heterocycloalkyl, aralkyl, optionally substituted monocyclic aryl or optionally substituted monocyclic heteroaryl; or R18 and R19 are connected via an additional 2-5 atoms to form a cyclic group, optionally containing 0-2 heteroatoms; or R17 and R18 are connected via an additional 3-5 atoms to form a cyclic group, optionally containing one heteroatom and R19 is H, alkyl, aryl, heterocycloalkyl, aralkyl, optionally substituted monocyclic aryl or optionally substituted monocyclic heteroaryl; and
R20 is alkyl, aryl, heterocycloalkyl, or arylalkyl; R21 is H, alkyl, aryl, heterocycloalkyl, or arylalkyl; R22 is H or lower acyl; n is an integer from 2-5; m is an integer from 10-20; and p is an integer from 2-3.
Synthetic Processes
Processes for preparing compounds of Formula I-III, or a pharmaceutically acceptable salts or prodrugs thereof, as well as processes for preparing intermediate compounds that can be used to prepare compounds of Formula I-III or pharmaceutically acceptable salts or prodrugs thereof are provided as further embodiments of the invention. For example in one embodiment the invention provides a method for preparing a pharmaceutically acceptable salt of compound of Formula I-III comprising converting a corresponding compound of Formula I-III to the salt.
In another embodiment the invention provides a method for preparing a prodrug of a compound of Formula I-III comprising converting a corresponding compound of Formula I-III to the prodrug. In another embodiment the invention provides a method for preparing a compound of Formula I-III comprising deprotecting a corresponding compound of Formula I-III that comprises one or more protecting groups to provide the compound of Formula I-III.
A representative compound of formula III (and formula I), compound lib, can be prepared as follows.
Synthetic schemes for the preparation of (3R,4R)-2-(4-aminoimidazo[l,2- f][l,2,4]triazin-7-yl)-5-(hydroxymethyI)-3-methyltetrahydrofuran-3,4-diol

A representative compound of formula III (and formula II), compound lie, can be prepared as follows.
Synthetic scheme for the preparation of (2R,3R,4R,5R)-2-(4-aminofuro[3,4- d]pyrimidin-7-yI)-5-(hydroxymethyl)-3-methyltetrahydrofuran-3,4-diol
 J. Chem. Soc. Perkin Trans. 1 ; 1982; 903-908
J. Chem. Soc. Perkin Trans. 1 ; 1982; 903-908


BnO OBn Synthesis, 2004, 1359-1362
46


1 , NaOH
1, NaOH 2,DPPA 2,EDCI/H0Bt NEt3, Toluene NH4CI, DIPEA t-BuOH
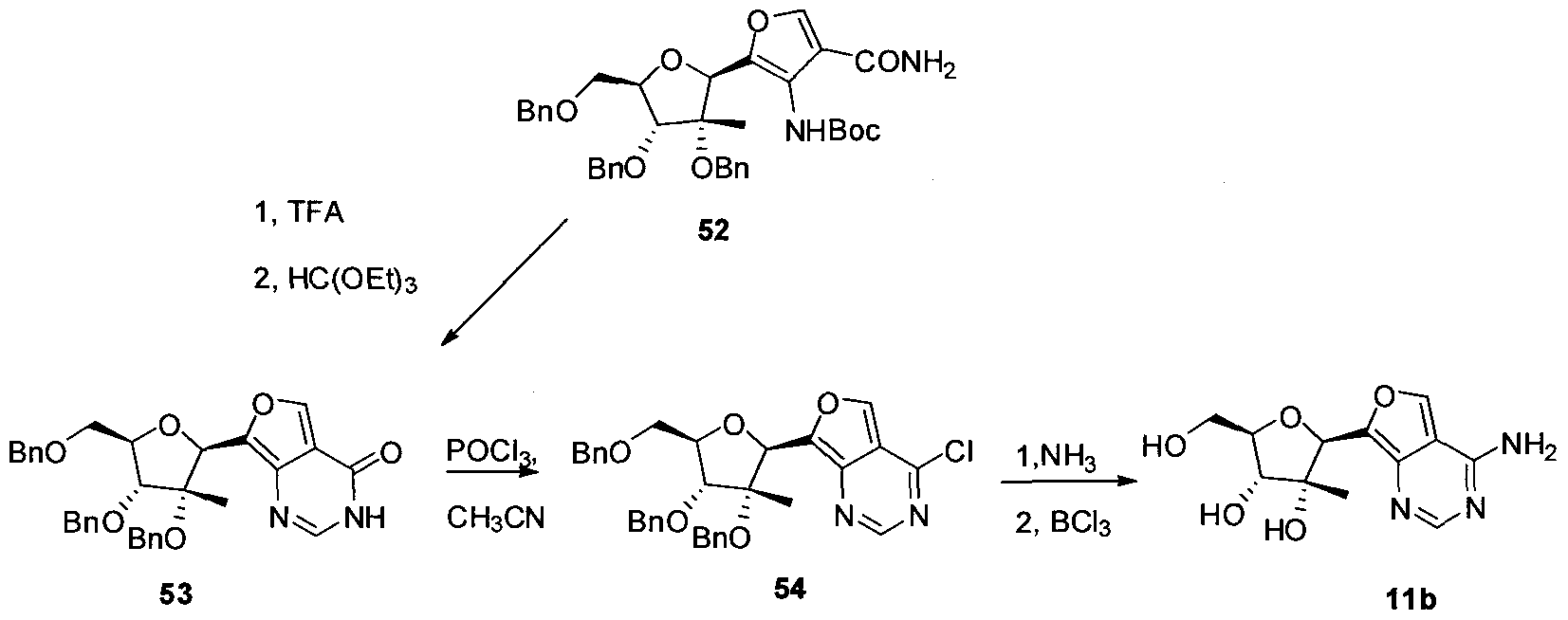
Synthetic Intermediates
The invention also provides synthetic intermediates that are useful for preparing compounds of Formula I to III or a salt or prodrug thereof. For example, the invention provides novel synthetic intermediates such as those described in the Schemes and Examples herein. Isomers and Physical Forms
It will be appreciated by those skilled in the art that compounds of the invention having a chiral center may exist in and be isolated in optically active and racemic forms. Some compounds may exhibit polymorphism. It is to be understood that the present invention encompasses any racemic, optically-active, polymorphic, tautomeric, or stereoisomeric form, or mixtures thereof, of a compound of the invention (e.g., a compound of Formula I to III, which possess the useful properties described herein, it being well known in the art how to prepare optically active forms (for example, by resolution of the racemic form by recrystallization techniques, by synthesis from optically-active starting materials, by chiral synthesis, or by chromatographic separation using a chiral stationary
phase) and how to determine anti-viral or anti-cancer activity using the standard tests described herein, or using other similar tests which are well known in the art. Although the invention includes all isomeric forms of the compounds described herein, one embodiment of the invention provides compounds having the absolute stereochemistry depicted in the Examples herein below. Pharmaceutical Compositions, Modes of Administration and Methods of Treatment
The present disclosure provides compounds of the general Formula I to III as detailed above which are inhibitors of DNA and/or RNA viral polymerases and anticancer agents. Various forms of DNA and RNA viral polymerases are inhibited by the compounds disclosed, such as but not limited to viral RdRps. The compounds of the present disclosure therefore have utility in treating and/or preventing viral infections in a host and in treatment and/or preventing a variety of disease states and/or conditions caused by or related to such viral infections. In one embodiment, the compounds are useful in the above mentioned treating and/or preventing by inhibiting a viral RNA and DNA polymerases. Such viral agents include, but are not limited to, hepatitis B, hepatitis C, human immunodeficiency virus, Polio, Coxsackie A and B, Rhino, Echo, small pox, Ebola, and West Nile virus. In a particular embodiment, the causative agent of the viral infection is a flavivirus.
The present disclosure provides for a compound of the general Formula I to III and a pharmaceutical composition comprising a pharmaceutically effective amount of at least one compound of general Formula I toIII as described herein. Such compounds and/or pharmaceutical compositions may be used in the manufacture of a medicament for treating and/or preventing a disease or condition in which it is desirable to inhibit a viral RNA and DNA polymerases. Such pharmaceutical compositions may also comprise a pharmaceutically acceptable carrier and other ingredients known in the art, or may comprise solely a compound of the general Formula I to III. The pharmaceutically acceptable carriers described herein, including, but not limited to, vehicles, adjuvants, excipients, or diluents, are well-known to those who are skilled in the art. Typically, the pharmaceutically acceptable carrier is chemically inert to the active compounds and has no detrimental side
effects or toxicity under the conditions of use. The pharmaceutically acceptable carriers can include polymers and polymer matrices.
The compounds described in the instant disclosure can be administered by any conventional method available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in combination with additional therapeutic agents.
The compounds described are administered in a pharmaceutically effective amount. The pharmaceutically effective amount of the compound and the dosage of the pharmaceutical composition administered will, of course, vary depending upon known factors, such as the pharmacodynamic characteristics of the particular agent and its mode and route of administration; the age, health and weight of the recipient; the severity and stage of the disease state or condition; the kind of concurrent treatment; the frequency of treatment; and the effect desired. A daily dosage of active ingredient can be expected to be about 0.001 to
1000 milligrams (mg) per kilogram (kg) of body weight per day. In one embodiment, the total amount is between about 0.1 mg/kg and about 100 mg/kg of body weight per day; in an alternate embodiment between about 1.1 mg/kg and about 50 mg/kg of body weight per day; in yet another alternate embodiment between 0.1 mg/kg and about 30 mg/kg of body weight per day. The above described amounts may be administered as a series of smaller doses over a period of time if desired. The pharmaceutically effective amount can be calculated based on the weight of the parent compound to be delivered. If the salt or prodrug exhibits activity in itself, the pharmaceutically effective amount can be estimated as above using the weight of the salt or prodrug, or by other means known to those skilled in the art. The dosage of active ingredient may be given other than daily if desired.
The total amount of the compound administered will also be determined by the route, timing and frequency of administration as well as the existence, nature, and extent of any adverse side effects that might accompany the administration of the compound and the desired physiological effect. It will be appreciated by one skilled in the art that various conditions or disease states, in particular chronic conditions or disease states, may require prolonged treatment involving multiple administrations.
Dosage forms of the pharmaceutical compositions described herein (forms of the pharmaceutical compositions suitable for administration) contain from about 0.1 mg to about 3000 mg of active ingredient (i.e., the compounds disclosed) per unit. In these pharmaceutical compositions, the active ingredient will ordinarily be present in an amount of about 0.5-95% weight based on the total weight of the composition. Multiple dosage forms may be administered as part of a single treatment. The active ingredient may be administered to achieve peak plasma concentrations of the active ingredient of from about 0.2 to 70 μM, or from about 1.0 to 10 μM. The active ingredient can be administered orally in solid dosage forms, such as capsules, tablets, and powders, or in liquid dosage forms, such as elixirs, syrups and suspensions. It can also be administered parenterally, in sterile liquid dosage forms. The active ingredient can also be administered intranasally (nose drops) or by inhalation via the pulmonary system, such as by propellant based metered dose inhalers or dry powders inhalation devices. Other dosage forms are potentially possible such as administration transdermally, via patch mechanisms or ointment.
Formulations suitable for oral administration can include (a) liquid solutions, such as a pharmaceutically effective amount of the compound dissolved in diluents, such as water, saline, or orange juice; (b) capsules, sachets, tablets, lozenges, and troches, each containing a predetermined pharmaceutically effective amount of the active ingredient, as solids or granules; (c) powders; (d) suspensions in an appropriate liquid; and (e) suitable emulsions. Liquid formulations may include diluents, such as water and alcohols, for example, ethanol, benzyl alcohol, propylene glycol, glycerin, and the polyethylene alcohols, either with or without the addition of a pharmaceutically acceptable surfactant, suspending agent, or emulsifying agent. Capsule forms can be of the ordinary hard- or soft-shelled gelatin type containing, for example, surfactants, lubricants, and inert fillers, such as lactose, sucrose, calcium phosphate, and corn starch. Tablet forms can include one or more of the following: lactose, sucrose, mannitol, corn starch, potato starch, alginic acid, microcrystalline cellulose, acacia, gelatin, guar gum, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, calcium stearate, zinc stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, disintegrating agents,
moistening agents, preservatives, flavoring agents, and pharmacologically compatible carriers. Lozenge forms can comprise the active ingredient in a flavor, usually sucrose and acacia or tragacanth, as well as pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin, or sucrose and acadia, emulsions, and gels containing, in addition to the active ingredient, such carriers as are known in the art.
Formulations suitable for parenteral administration include aqueous and non-aqueous, isotonic sterile injection solutions, which can contain antioxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the patient, and aqueous and non-aqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives. The compound can be administered in a physiologically acceptable diluent in a pharmaceutically acceptable carrier, such as a sterile liquid or mixture of liquids, including water, saline, aqueous dextrose and related sugar solutions, an alcohol, such as ethanol, isopropanol, or hexadecyl alcohol, glycols, such as propylene glycol or polyethylene glycol such as poly(ethyleneglycol) 400, glycerol ketals, such as 2,2-dimethyl-l,3-dioxolane-4- methanol, ethers, an oil, a fatty acid, a fatty acid ester or glyceride, or an acetylated fatty acid glyceride with or without the addition of a pharmaceutically acceptable surfactant, such as a soap or a detergent, suspending agent, such as pectin, carbomers, methylcellulose, hydroxypropylmethylcellulose, or carboxymethylcellulose, or emulsifying agents and other pharmaceutical adjuvants.
Oils, which can be used in parenteral formulations, include petroleum, animal, vegetable, or synthetic oils. Specific examples of oils include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral. Suitable fatty acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters. Suitable soaps for use in parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts, and suitable detergents include (a) cationic detergents such as, for example, dimethyldialkylammonium halides, and alkylpyridinium halides, (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyl, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine
oxides, fatty acid alkanolamides, and polyoxyethylene polypropylene copolymers, (d) amphoteric detergents such as, for example, alkyl .beta.- aminopropionates, and 2-alkylimidazoline quaternary ammonium salts, and (e) mixtures thereof. The parenteral formulations typically contain from about 0.5% to about
25% by weight of the active ingredient in solution. Suitable preservatives and buffers can be used in such formulations. In order to minimize or eliminate irritation at the site of injection, such compositions may contain one or more nonionic surfactants having a hydrophile-lipophile balance (HLB) of from about 12 to about 17. The quantity of surfactant in such formulations ranges from about 5% to about 15% by weight. Suitable surfactants include polyethylene sorbitan fatty acid esters, such as sorbitan monooleate and the high molecular weight adducts of ethylene oxide with a hydrophobic base, formed by the condensation of propylene oxide with propylene glycol. Pharmaceutically acceptable excipients are also well-known to those who are skilled in the art. The choice of excipient will be determined in part by the particular compound, as well as by the particular method used to administer the composition. Accordingly, there is a wide variety of suitable formulations of the pharmaceutical composition of the present invention. The following methods and excipients are merely exemplary and are in no way limiting. The pharmaceutically acceptable excipients preferably do not interfere with the action of the active ingredients and do not cause adverse side-effects. Suitable carriers and excipients include solvents such as water, alcohol, and propylene glycol, solid absorbants and diluents, surface active agents, suspending agent, tableting binders, lubricants, flavors, and coloring agents.
The compounds of the present invention, alone or in combination with other suitable components, can be made into aerosol formulations to be administered via inhalation. These aerosol formulations can be placed into pressurized acceptable propellants, such as dichlorodifluoromethane, propane, and nitrogen. Such aerosol formulations may be administered by metered dose inhalers. They also may be formulated as pharmaceuticals for non-pressured preparations, such as in a nebulizer or an atomizer.
The formulations can be presented in unit-dose or multi-dose sealed containers, such as ampules and vials, and can be stored in a freeze-dried
(lyophilized) condition requiring only the addition of the sterile liquid excipient, for example, water, for injections, immediately prior to use. Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets. The requirements for effective pharmaceutically acceptable carriers for injectable compositions are well known to those of ordinary skill in the art. See Pharmaceutics and Pharmacy Practice, J.B. Lippincott Co., Philadelphia, Pa., Banker and Chalmers, Eds., 238-250 (1982) and ASHP Handbook on Injectable Drugs, Toissel, 4th ed., 622-630 (1986).
Formulations suitable for topical administration include pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin, or sucrose and acacia, as well as creams, emulsions, and gels containing, in addition to the active ingredient, such carriers as are known in the art. Furthermore, transdermal patches can be prepared using methods known in the art. Additionally, formulations suitable for rectal administration may be presented as suppositories by mixing with a variety of bases such as emulsifying bases or water-soluble bases. Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams, or spray formulas containing, in addition to the active ingredient, such carriers as are known in the art to be appropriate.
One skilled in the art will appreciate that suitable methods of administering a compound of the present invention to an patient are available, and, although more than one route can be used to administer a particular compound, a particular route can provide a more immediate and more effective reaction than another route.
Useful embodiments of pharmaceutical dosage forms for administration of the compounds according to the present invention can be illustrated as follows.
A large number of hard-shell capsules are prepared by filling standard two-piece hard gelatine capsules each with 100 mg of powdered active ingredient, 150 mg of lactose, 50 mg of cellulose and 6 mg of magnesium stearate.
A mixture of active ingredient in a digestible oil such as soybean oil, cottonseed oil or olive oil is prepared and injected by means of a positive
displacement pump into molten gelatin to form soft gelatin capsules containing 100 mg of the active ingredient. The capsules are washed and dried. The active ingredient can be dissolved in a mixture of polyethylene glycol, glycerin and sorbitol to prepare a water miscible medicine mix. A large number of tablets are prepared by conventional procedures so that the dosage unit is 100 mg of active ingredient, 0.2 mg of colloidal silicon dioxide, 5 mg of magnesium stearate, 275 mg of microcrystalline cellulose, 11 mg of starch, and 98.8 mg of lactose. Appropriate aqueous and non-aqueous coatings may be applied to increase palatability, improve elegance and stability or delay absorption.
Immediate release tablets/capsules are solid oral dosage forms made by conventional and novel processes. These units are taken orally without water for immediate dissolution and delivery of the medication. The active ingredient is mixed in a liquid containing ingredient such as sugar, gelatin, pectin and sweeteners. These liquids are solidified into solid tablets or caplets by freeze drying and solid state extraction techniques. The drug compounds may be compressed with viscoelastic and thermoelastic sugars and polymers or effervescent components to produce porous matrices intended for immediate release, without the need of water. Moreover, the compounds of the present invention can be administered in the form of nose drops, or metered dose and a nasal or buccal inhaler. The drug is delivered from a nasal solution as a fine mist or from a powder as an aerosol. In one embodiment, the teachings of the present disclosure provide for the use of such pharmaceutical compositions and medicaments in a method of treating a viral infection or treating a disease state and/or condition caused by or related to such viral infection. In one embodiment, the treatment is the result of the inhibition of a viral RNA or DNA polymerase, such as but not limited to a RdRp. Such treatment or inhibition need not be complete to be useful. The method of treatment comprises the steps of: (i) identifying a patient in need of such treatment; (ii) providing such pharmaceutical composition containing at least one compound of the invention; and (iii) administering such pharmaceutical composition in a therapeutically effective amount to treat the viral infection in a patient in need of such treatment or to inhibit the activity of a viral RNA or DNA polymerase in a patient in need of such treatment.
The teachings of the present disclosure provide for, among other things, the use of such pharmaceutical compositions and medicaments in a method of preventing or suppressing a viral infection or preventing or suppressing a disease state and/or condition caused by or related to such viral infection. In one embodiment, the prevention or suppression is the result of the inhibition of a viral RNA or DNA polymerase, such as but not limited to a RdRp. Such prevention, suppression or inhibition need not be complete to be useful. The method of preventing or suppressing can optionally comprises the steps of: (i) identifying a patient in need of such prevention; (ii) providing such pharmaceutical composition containing at least one compound of the general Formula I to III; and (iii) administering such pharmaceutical composition in a therapeutically effective amount to prevent or suppress the viral infection in a patient in need of such treatment or to inhibit the activity of a viral RNA and DNA polymerase in a patient in need of such treatment. The methods of the treating and preventing a viral infection or a disease state and/or condition caused by or related to said viral infection may further comprise administering a therapeutically effective amount of a compound of the present invention in combination with a therapeutically effective amount of another anti-viral agent which , in particular, may be active against HCV. Agents active against HCV include, but are not limited to, ribavirin, levovirin, viramidine, thymosin alpha- 1, an inhibitor of HCV NS3 serine protease, an inhibitor of inosine monophosphatedehydrognease, interferon-α, pegylated interferon-α (peginterferon-α), a combination of interferon-α and ribavirin, a combination of peginterferon-α and ribavirin, a combination of interferon-α and levovirin, and a combination of peginterferon-α and levovirin. Interferon-α includes, but is not limited to, recombinant interferon-α2a, interferon-α2b, a consensus interferon, and a purified interferon-α product.
The compounds and pharmaceutical compositions of the present disclosure can be administered to patients to prevent and/or treat a number of cancers. Cancers include, but are not limited to, leukemias and lymphomas such as acute lymphocytic leukemia, acute nonlymphocytic leukemias, chronic lymphocytic leukemia, chrome myelogenous leukemia, Hodgkin's Disease, non- Hodgkin's lymphomas, and multiple myeloma, childhood solid tumors such as brain tumors, neuroblastoma, retinoblastoma, Wilms Tumor, bone tumors, and
soft-tissue sarcomas, common solid tumors of adults such as lung cancer, colon and rectum cancer, breast cancer, prostate cancer, urinary cancers, uterine cancers, oral cancers, pancreatic cancer, melanoma and other skin cancers, stomach cancer, ovarian cancer, brain tumors, liver cancer, laryngeal cancer, thyroid cancer, esophageal cancer, and testicular cancer. The cancer may be related to a viral infection or an activity of a viral DNA or RNA polymerase.
The methods of the treating and preventing cancer may also comprises further administering of a chemotherapeutic agent in combination with any of the compounds or pharmaceutical compositions of the present disclosure. Any suitable chemotherapeutic agent can be employed for this purpose. The chemotherapeutic agent is typically selected from the group consisting of alkylating agents, antimetabolites, natural products, hormonal agents, and miscellaneous agents.
Examples of alkylating chemotherapeutic agents include carmustine, chlorambucil, cisplatin, lomustine, cyclophosphamide, melphalan, mechlorethamine, procarbazine, thiotepa, uracil mustard, triethylenemelamine, busulfan, pipobroman, streptozocin, ifosfamide, dacarbazine, carboplatin, and hexamethylmelamine.
Examples of chemotherapeutic agents that are antimetabolites include cytosine arabinoside, fluorouracil, gemcitabine, hydroxyurea, mercaptopurine, methotrexate, azaserine, thioguanine, floxuridine, fludarabine, cladribine and L- asparaginase.
Examples of chemotherapeutic agents that are natural products include actinomycin D, bleomycin, camptothecins, daunomycin, doxorubicin, etoposide, mitomycin C, TAXOL™ (paclitaxel), taxotere, teniposide, vincristine, vinorelbine, mithramycin, idarubicin, MITHRACIN™. (plicamycin), and deoxycoformycin.
An example of a hormonal chemotherapeutic agent includes tamoxifen. Examples of the aforesaid miscellaneous chemotherapeutic agents include mitotane, mitoxantrone, vinblastine, and levamisole.
Representative prodrugs of the invention can be been tested in vitro or in vivo to confirm that the prodrug is converted, e.g., by hydrolysis, to provide an active compound.
The ability of a compound to inhibit viral polymerases can be evaluated using known assays. The ability of a compound to inhibit HCV NS5B polymerase can be evaluated using the following assay. HCV NS5B Polymerase Assay Antiviral activity of the test compounds can be assessed (Okuse et al ,
Antiviral Res. 2005, 65, 23-34) in the stably HCV RNA-replicating cell line, AVA5, derived by transfection of the human hepatoblastoma cell line, Huh7 (Blight et al, Sci. 2000, 290, 1972). Compounds can be added to dividing cultures once daily for three days. Media can be changed with each addition of compound. Cultures generally start the assay at 30-50% confluence and reach confluence during the last day of treatment. Intracellular HCV RNA levels and cytotoxicity can be assessed 24 hours after the last dose of compound.
Triplicate cultures for HCV RNA levels (on 48-well and 96-well plates) and cytotoxicity (on 96-well plates) can be used. A total of six untreated control cultures, and triplicate cultures treated with α-interferon and ribavirin can serve as positive antiviral and toxicity controls.
Intracellular HCV RNA levels can be measured using a conventional blot hybridization method in which HCV RNA levels are normalized to the levels of B-actin RNA in each individual culture (Okuse et al, Antivir. Res. 2005, 65, 23- 34). Cytotoxicity can be measured using a neutral red dye uptake assay (Korba and Gerin, Antivir. Res. 1992, 19, 55). HCV RNA levels in the treated cultures can be expressed as a percentage of the mean levels of RNA detected in untreated cultures. Compound Synthesis Compounds of Formula I-III can be prepared using synthetic intermediates and synthetic procedures that are known, or they can be prepared using the synthetic intermediates and synthetic procedures described in the Schemes and Examples herein.
The invention will now be illustrated by the following non-limiting Example.
Example 1
The following illustrate representative pharmaceutical dosage forms, containing a compound of Formula I to III, or a pharmaceutically acceptable salt or prodrug thereof ('Compound X'), for therapeutic or prophylactic use in humans.
Ci) Tablet 1 mg/tablet
Compound X= 100.0
Lactose 77.5
Povidone 15.0
Croscarmellose sodium 12.0
Microcrystalline cellulose 92.5
Magnesium stearate 3.0
300.0
(iϊ) Tablet 2 mg/tablet
Compound X= 20.0
Microcrystalline cellulose 410.0
Starch 50.0
Sodium starch glycolate 15.0
Magnesium stearate
500.0
(iii) Capsule mg/capsule
Compound X= 10.0
Colloidal silicon dioxide 1.5
Lactose 465.5
Pregelatinized starch 120.0
Magnesium stearate 3.0
600.0
(iv) Injection 1 Cl mR/ml) mg/ml
Compound X= (free acid form) 1.0
Dibasic sodium phosphate 12.0
Monobasic sodium phosphate 0.7
Sodium chloride 4.5
1.0 N Sodium hydroxide solution
(pH adjustment to 7.0-7.5) q.s.
Water for injection q.s. ad 1 mL
Cv) Injection 2 (10 mε/ml) mg/ml
Compound X= (free acid form) 10.0
Monobasic sodium phosphate 0.3
Dibasic sodium phosphate 1.1
Polyethylene glycol 400 200.0
01 N Sodium hydroxide solution
(pH adjustment to 7.0-7.5) q.s.
Water for injection q.s. ad 1 niL
(vi) Aerosol mR/can
Compound X= 20.0
Oleic acid 10.0
Trichloromonofluoromethane 5,000.0
Dichlorodifluoromethane 10,000.0
Dichlorotetrafluoroethane 5,000.0
The above formulations may be obtained by conventional procedures well known in the pharmaceutical art.
All publications, patents, and patent documents are incorporated by reference herein, as though individually incorporated by reference. The invention has been described with reference to various specific and preferred embodiments and techniques. However, it should be understood that many variations and modifications may be made while remaining within the spirit and scope of the invention.
Claims
1. A compound of Formula III :

III wherein:
X is N, Y is N and Z is CH; or X is C, Y is CR3 and Z is O;
R is OR3, SR3, NR3Rb, NR3NRbR0, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, (CH2)n- CH(NHR3)CO2Rb, (CH2)n-S-alkyl, (CH2)n-S-aryl, Cl, F, Br, I, CN, COORa, CONRaRb, NHC(=NRa)NHRb, NRa0Rb, NR3NO, NHCONHR3, NRaN=NRb, NRaN=CHRb, NR3C(O)NRbRc, NR3C(S)NRbRc, NR3C(O)ORb, CH=N-OR3, NR3Q=NH)NRbRc, NRaC(O)NRbNRcRd, 0-C(O)R3, OC(O)-OR3, ONH- C(O)O-alkyl, ONHC(O)O-aryl, 0NR3Rb, SNR3Rb, S-ONRaRb, CHO, C(=S)N R3Rb, nitro, C (=NR3)0Rb, or SO2NR3Rb; and R3 is H, CN, NO2, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, CH=CF2, CH(=N R3)ORb, CHO, CH=CH-OCH3, NHCONH2, NHCSNH2, CONR3Rb, CSNR3Rb, CO2R3, alkoxy, NH2, alkylamino, dialkylamino, halogen, (l,3-oxazol-2-yl), (l,3-oxazol-5-yl), (l,3-thiazol-2-yl), (imidazol-2-yl), (2- oxo[l, 3]dithiol-4-yl), (furan-2-yl), (2H[l,2,3]triazol-4-yl), C(=NH)NH2, C(=NH)NH0H, C(=N0H)NH2, acyl, substituted acyl, OR3, C(=NRa)Rb, CH=NNR3Rb, CH=NOR3, CH(OR3)2, B(OR3)2, C≡C-C(=O)NRaRb, CH=NNR3Rb, CH=NOR3, CH(0Ra)2, B(ORa)2, C≡C-C(=0)NRaRb, (CH2)n-S- alkyl, (CH2)n-S-aryl, (CH2)n-S(O)-alkyl, (CH2)n-S(O)-aryl, (CH2)n-S(O2)-alkyl, (CH2)n-S(O2)-aryl, (CH2)H-SO2NR3Rb, or (CH2)n-0Ra; or R and R3 together with atoms to which they are attached may form a cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; n is 0-5; R1 is H, NR3Rb, Cl, F, ORa, SRa, NHCOR3, NHSO2R3, NHCONHRa, CN, alkyl, aryl, ONRaRb, or NR3C(0)0Rb;
R2 is a nucleoside sugar group; and
Ra, Rb, Rc, and Rj are independently selected from the group consisting of H, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, heterocyclic, aryl, substituted aryl, acyl, substituted acyl, SO2-alkyl, amino, substituted amino, substituted cycloalkyl, heteroaryl, substituted heteroaryl , and NO; or R3 and Rb together with the nitrogen to which they are attached form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; or Rb and Rς together with the nitrogen to which they are attached form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; or R0 and Rd together with the nitrogen to which they are attached form a heterocycloalkyl, substituted heterocycloalkyl, heteroaryl, or substituted heteroaryl; or a pharmaceutically acceptable salt or prodrug thereof.
2. The compound of claim 1 wherein the compound of formula III is a compound of Formula I or II:

II.
3. The compound of claim 2 wherein the compound of formula III is a compound of Formula I:

I.
4. The compound of claim 2 wherein the compound of formula III is a compound of Formula II:

5. The compound of any one of claims 1-4 wherein R is ORa, Cl, SRa, NRaRb5 Or NRaNRbR0.
6. The compound of any one of claims 1-4 wherein R is hydroxy, chloro, methoxy, mercapto, amino, methylamino, isopropylamino, propylamino, ethylamino, dimethylamino, cyclopropylamino, 2-aminoethylamino, l-(2- hydroxyethyl)hydrazino, hydrazino, 1-methylhydrazino, azetidino, or pyrrolidino.
7. The compound of any one of claims 1 -6 wherein R1 is H or NRaRb.
8. The compound of any one of claims 1-7 wherein R2 is
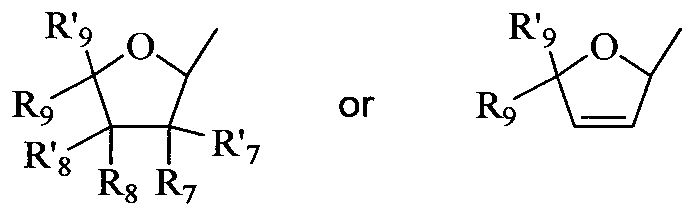
wherein:
R7 is H, OR14, N3, NH2, or F; and R7 is H, F, OH, O-alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R7 and R7 together may be =CH2, =CHF; wherein both R7 and R'7 are not OH; and when one of R7 and R'7 is NH2, the other is not OH; and when one of R7 and R'7 is N3, the other is not OH;
R8 is H, OR14, N3, NH2, or F; and R'8 is H, F, OH, O alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R8 and R8 together may be =CH2, =CHF; wherein both R8 and R8 are not OH; and when one of R8 and R'8 is NH2, the other is not OH; and when one of R8 and R'8 is N3, the other is not OH; or R7 and R8 together can form
wherein: R1Oo is C1-12 alkyl C3-8 cycloalkyl, aryl or heteroaryl; wherein any C1-12 alkyl and C3-8 cycloalkyl of R1O0 is unsubstituted or is substituted with 1-3 substituents selected from halogen, hydroxy, carboxy, and C1-4 alkoxy; and wherein any aryl or heteroaryl OfR100 is unsubstituted or is substituted with 1-5 substituents independently selected from R101; each R101 is independently halo, C1-4 alkyl, C1-4 alkoxy, C1-4 alkylthio, C1- 4 alkylsulfoyl, cyano, nitro, amino, phenyl, carboxy, trifluoromethyl, trifluoromethoxy, C1-4 alkylamino, di(C1-4 alkyl) amino, C1-4 alkanoyl, C1-4 alkanoyloxy, or C1-4 alkyloxycarbonyl;
R9 is H, CH35 C2H5, or N3;
R'9 is CH2OR14, CH2F, CH2SH, CHFOH, CF2OH,
— 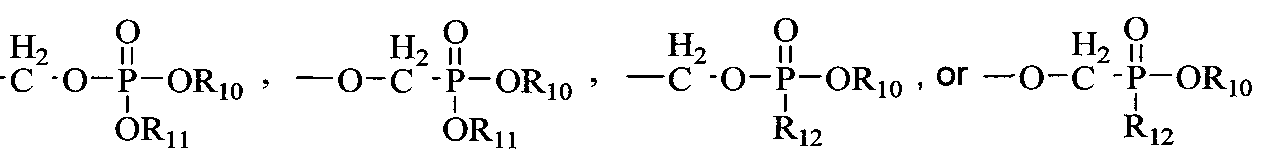
R1O and R11 are each independently H, alkyl, aryl, substituted aryl, acyloxyalkyl, or (CH2)nl-O-(CH2)mCH3;
R12 is an N-linked amino acid residue (e.g., -NH-CH(CH3)Cθ2alkyl or NH-CH(isopropyl)-CO2alkyl); and
Ri4 is H; nl is 2-5; and m is 10-20.
9. The compound of any one of claims 1-7 wherein R2 is selected from:

(d) (1) (d) (1)
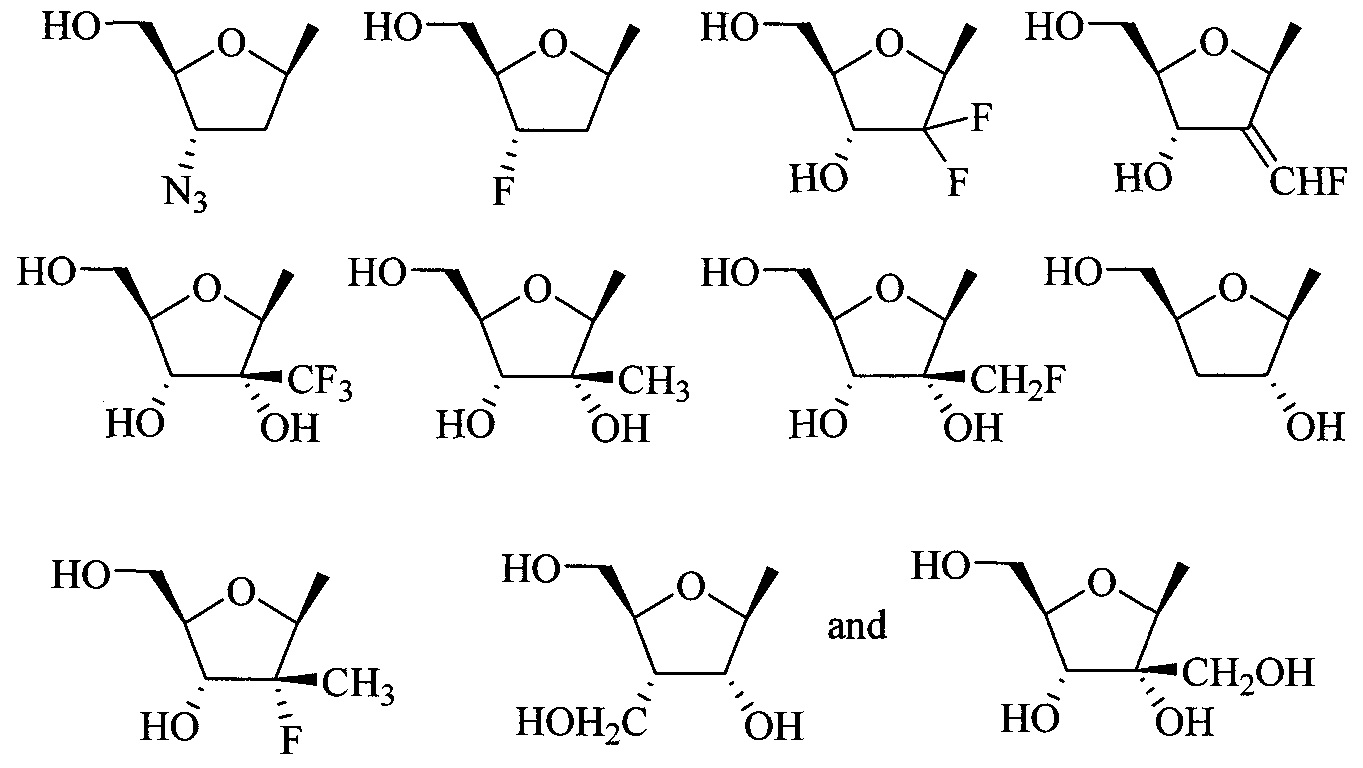
10. The compound of any one of claims 1-7 wherein R2 is:

wherein:
R7 is H, OR14, N3, NH2, or F; and R7 is H, F, OH, O-alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R7 and R'7 together may be =CH2, =CHF; wherein both R7 and R7 are not OH; and when one of R7 and R7 is NH2, the other is not OH; and when one of R7 and R7 is N3, the other is not OH;
R8 is H, OR14, N3, NH2, or F; and R'8 is H, F, OH, O alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R8 and R8 together may be =CH2, =CHF; wherein both R8 and R8 are not OH; and when one of Rg and R8 is NH2, the other is not OH; and when one of R8 and R'8 is N3, the other is not OH; or R7 and R8 together can form
wherein: R100 is C1-12 alkyl C3-8 cycloalkyl, aryl or heteroaryl; wherein any C1-12 alkyl and C3-8 cycloalkyl OfR1O0 is unsubstituted or is substituted with 1-3 substituents selected from halogen, hydroxy, carboxy, and C1-4 alkoxy; and wherein any aryl or heteroaryl OfR100 is unsubstituted or is substituted with 1-5 substituents independently selected from Ri01; each Rιoi is independently halo, C1-4 alkyl, C1-4 alkoxy, C1-4 alkylthio, C1- 4 alkylsulfoyl, cyano, nitro, amino, phenyl, carboxy, trifluoromethyl, trifluoromethoxy, Ci-4 alkylamino, di(C1-4 alkyl) amino, C1-4 alkanoyl, C1-4 alkanoyloxy, or C1-4 alkyloxycarbonyl;
R9 is H, CH3, C2H5, or N3;
R'9 is CH2OR14, CH2F, CH2SH, CHFOH, CF2OH,
— o-C -P-OR10 ; 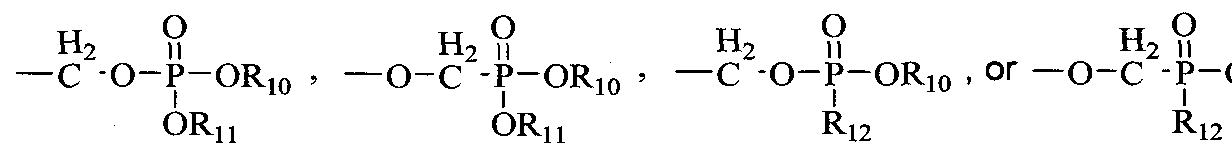
RIO and R11 are each independently H, alkyl, aryl, substituted aryl, acyloxyalkyl, or (CH2)nl-O-(CH2)mCH3;
R12 is an N-linked amino acid residue (e.g., -NH-CH(CH3)C02alkyl or - NH-CH(isopropyl)-CO2alkyl);
R14 is H; nl is 2-5; and m is 10-20.
11. The compound of any one of claims 1 -7 wherein R is selected from:

12. The compound of any one of claims 1-7 wherein R2 is:
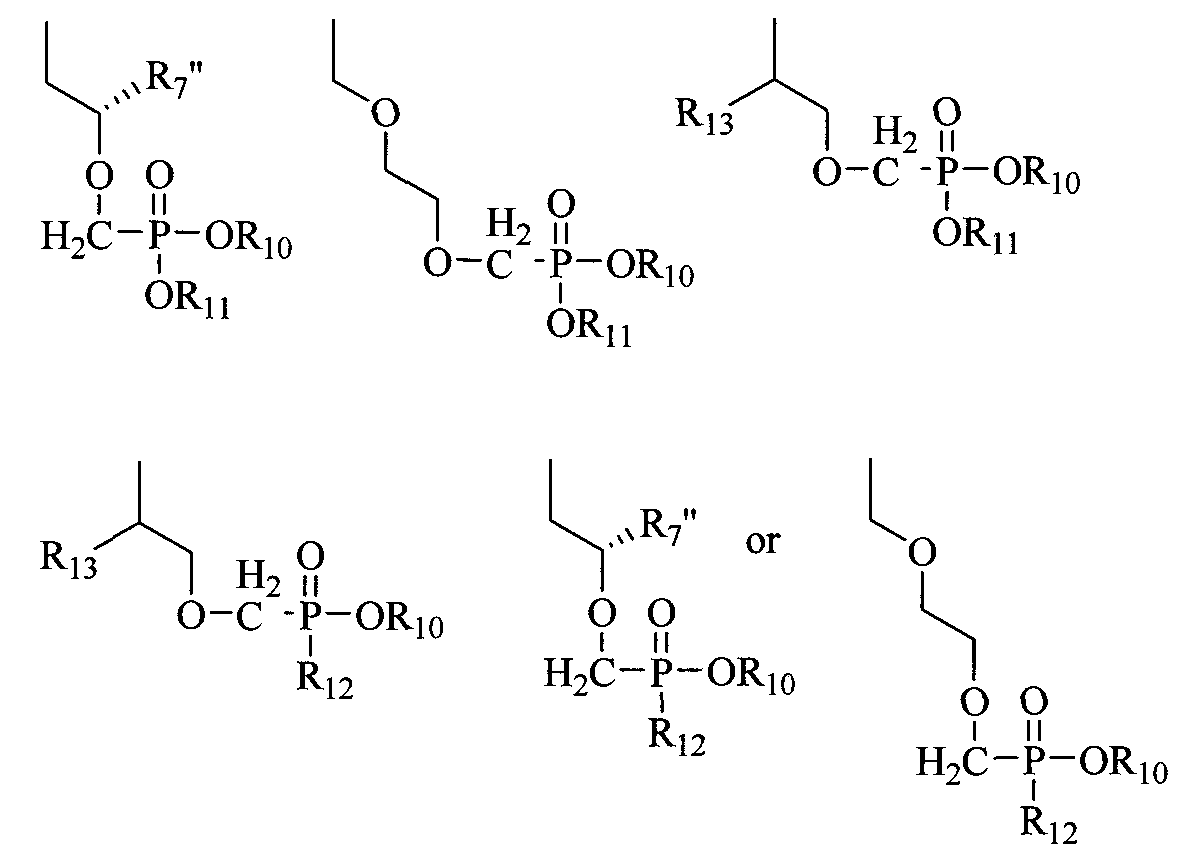
R7" is alkyl or substituted alkyl;
R10 and R11 are each independently H, alkyl, aryl, substituted aryl, acyloxyalkyl, or (CH2)nl-O-(CH2)mCH3; nl is 2-5; and m is 10-20;
R12 is an N-linked amino acid residue (e.g., -NH-CH(CH3)C02alkyl or NH-CH(isopropyl)-CO2alkyl); and
R13 is H, CH3, C2H5, CH2F, CH2OH, CH2CH2F, CH2CH2OH, CH2N3, CH2CH2N3, CH2NH2, or CH2CH2NH2.
13. The compound of any one of claims 1 -7 wherein R2 is:

14. The compound of any one of claims 1-7 wherein R is:
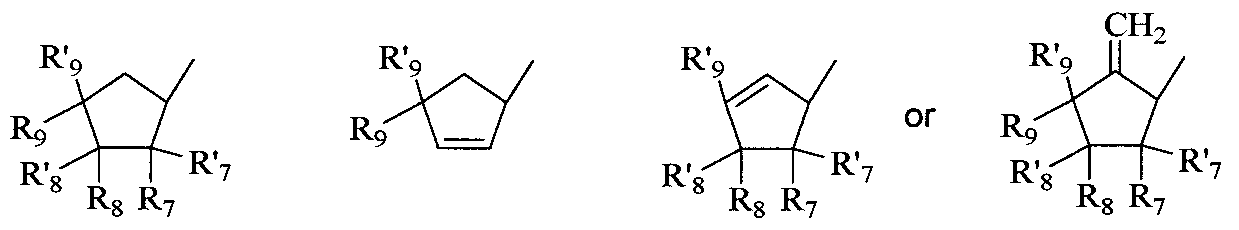
wherein:
R7 is H, OR14, N3, NH2, or F; and R'7 is H, F, OH, O-alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R7 and R'7 together may be =CH2, =CHF; wherein both R7 and R'7 are not OH; and when one of R7 and R'7 is NH2, the other is not OH; and when one of R7 and R7 is N3, the other is not OH;
R8 is H, OR14, N3, NH2, or F; and R'8 is H, F, OH, 0 alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R8 and R8 together may be =CH2, =CHF; wherein both R8 and R8 are not OH; and when one of R8 and R8 is NH2, the other is not OH; and when one of R8 and R's is N3, the other is not OH; or R7 and R8 together can form
R9 is H, CH3, C2H5, orN3;
R9 is CH2OR14, CH2F, CH2SH, CHFOH, CF2OH,
— , 
R10 and R11 are each independently H, alkyl, aryl, substituted aryl, acyloxyalkyl, or (CH2)nl-O-(CH2)mCH3;
R12 is an N-linked amino acid residue {e.g., -NH-CH(CH3)C02alkyl or - NH-CH(isopropyl)-CO2alkyl); and
R14 is H; nl is 2-5; and m is 10-20.
15. The compound of any one of claims 1-7 wherein R2 is selected from:

16. The compound of any one of claims 1 -7 wherein R2 is:
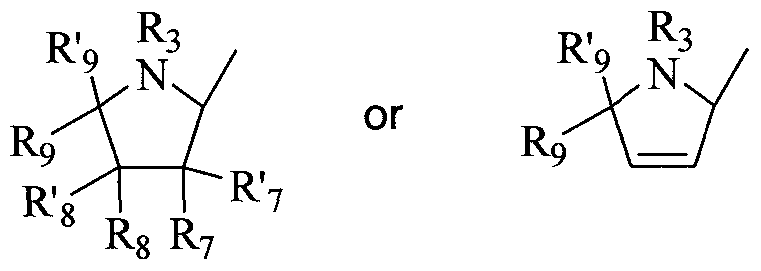
wherein:
R7 is H, ORi4, N3, NH2, or F; and R'7 is H, F, OH, O-alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R7 and R'7 together may be =CH2, =CHF; wherein both R7 and R'7 are not OH; and when one of R7 and R'7 is NH2, the other is not OH; and when one of R7 and R'7 is N3, the other is not OH;
R8 is H, ORi4, N3, NH2, or F; and R'8 is H, F, OH, O alkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, or substituted alkynyl; or R8 and R8 together may be =CH2, =CHF; wherein both R8 and R'8 are not OH; and when one of R8 and R8 is NH2, the other is not OH; and when one of R8 and R'8 is N3, the other is not OH; or R7 and R8 together can form
O. ,O
Y R M, 00
wherein: R100 is C1-12 alkyl C3-8 cycloalkyl, aryl or heteroaryl; wherein any C1-12 alkyl and C3-8 cycloalkyl of R100 is unsubstituted or is substituted with 1-3 substituents selected from halogen, hydroxy, carboxy, and C1-4 alkoxy; and wherein any aryl or heteroaryl OfRi00 is unsubstituted or is substituted with 1-5 substituents independently selected from Ri01; each RjO1 is independently halo, C1-4 alkyl, C1-4 alkoxy, C1-4 alkylthio, C1- 4 alkylsulfoyl, cyano, nitro, amino, phenyl, carboxy, trifluoromethyl, trifluoromethoxy, C1-4 alkylamino, di(C1-4 alkyl) amino, C1-4 alkanoyl, Ci-4 alkanoyloxy, or Ci-4 alkyloxycarbonyl; R9 is H, CH3, C2H5, or N3;
R'9 is CH2OR14, CH2F, CH2SH, CHFOH, CF2OH,
— O~C P-OR10 I 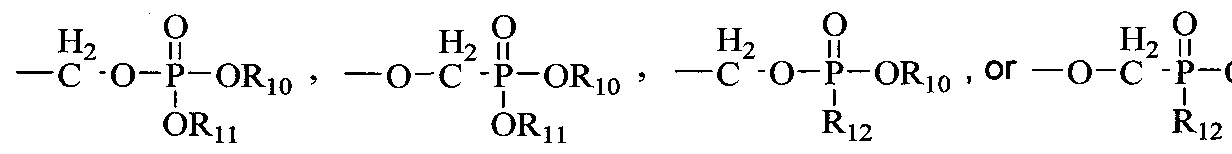
R10 and R11 are each independently H, alkyl, aryl, substituted aryl, acyloxyalkyl, or (CH2)ni-O-(CH2)mCH3;
R12 is an N-linked amino acid residue (e.g., -NH-CH(CH3)C02alkyl or - NH-CH(isopropyl)-CO2alkyl); and
R14 is H; nl is 2-5; and m is 10-20.
17. The compound of any one of claims 1-7 wherein R2 is:
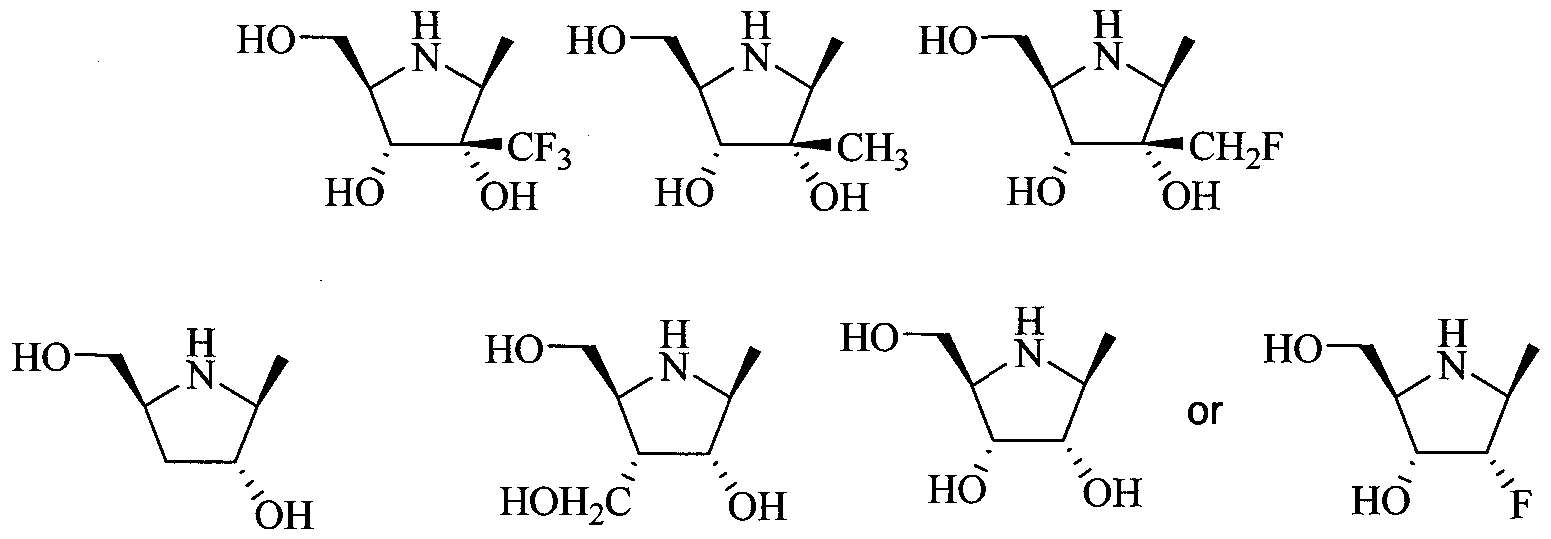
18. The compound of any one of claims 1 -7 wherein R2 is:

R9 is H, CH3, C2H5, or N3;
R'9 is CH2OR14, CH2F, CH2SH, CHFOH, CF2OH,
— O-C -P-OR10 ; 
R1O and R11 are each independently H, alkyl, aryl, substituted aryl, acyloxyalkyl, or (CH2)nl-O-(CH2)mCH3; R12 is an N-linked amino acid residue (e.g., -NH-CH(CH3)C02alkyl or - NH-CH(isopropyl)-CO2alkyl); R14 is H; nl is 2-5; and m is 10-20.
19. The compound of any one of claims 1-7 wherein R is:

20. The compound of any one of claims 1-7 wherein R is ribose, 2- methylribose, 2-deoxyribose; 2-deoxy-2-fluororibose; arabinose; 2-deoxy-2- fluoroarabinose; 2,3-dideoxyribose; 2,3-dideoxy-2-fluoroarabinose; 2,3-dideoxy- 3-fluororibose; 2,3-dideoxy-2,3-didehydroribose; 2,3-dideoxy-3-azidoribose; 2,3-dideoxy-3-thiaribose; or 2,3-dideoxy-3-oxaribose; or a pharmaceutically acceptable salt or prodrug thereof.
21. The compound of any one of claims 1-7 wherein R2 is thioribose, 2- deoxythioribose; 2-deoxy-2-fluorothioribose; thioarabinose; 2-deoxy-2- fluorothioarabinose; 2,3 -dideoxythioribose; 2,3 -dideoxy-2-fluorothioarabinose; 2,3-dideoxy-3-fluorothioribose; 2,3-dideoxy-2,3-didehydrothioribose; or 2,3- dideoxy-3-azidothioribose; or a pharmaceutically acceptable salt or prodrug thereof.
22. The compound of any one of claims 1-7 wherein R2 is 4-hydroxymethyl- cyclopent-2-ene; 2,3-dihydroxy-4-hydroxymethylcyclopent-4-ene; 3-hydroxy-4- hydroxymethylcyclopentane; 2-hydroxy-4-hydroxymethylcyclopentene; 2- fluoro-3 -hydroxy-4-hydroxymethylcyclopentane; 2,3 -dihydroxy-4- hydroxymethyl-5-methylenecyclopentane; 4-hydroxymethylcyclopentane, 2,3- dihydroxy-4-hydroxymethylcyclopentane; or 2,3 -dihydroxymethylcyclobutane; or a pharmaceutically acceptable salt or prodrug thereof.
23. The compound of any one of claims 1-7 wherein R2 is 4-hydroxymethyl- pyrrolidine; 2,3-dihydroxy-4-hydroxymethylpyrrolidine; 2/3-hydroxy-4- hydroxymethylpyrrolidine; 2-fluoro-3-hydroxy-4-hydroxymethylpyrrolidine; or 3-fluoro-2-hydroxy-4-hydroxymethyl-pyrrolidine; or a pharmaceutically acceptable salt or prodrug thereof.
24. The compound of any one of claims 1-23 wherein Ra, Rb, R0, and Ra are independently selected from the group consisting of H, alkyl, and substituted alkyl; or Ra and Rb together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring; or Rb and R0 together with the nitrogen to which they are attached form a pyrrolidino, piperidino, piperazino, azetidino, morpholino, or thiomorpholino ring.
25. The compound of claim 1 which is a compound of the following formula 11a or lib:

11a lib. or a pharmaceutically acceptable salt or prodrug thereof.
26. The compound of claim 1 which is a compound of the following formula:

or a pharmaceutically acceptable salt or prodrug thereof; wherein X is H or alkyl.
27. The compound of any one of claims 1 -26 which is a prodrug.
28. The compound of any one of claims 1-26 which comprises one or more mono-, di-, or tri-phosphate groups.
29. The compound of any one of claims 1-26 which comprises one or more mono-phosphate groups.
30. The compound of claim 28 or 29 which is a prodrug.
31. The compound of claim 28 wherein one or more pendent hydroxyl groups from the mono-, di-, or tri-phosphate group has been converted to an alkoxy, substituted alkoxy, aryloxy, or substituted aryloxy group.
32. The compound of claim 28 wherein one or more pendent hydroxyl groups from the mono-, di-, or tri-phosphate group has been converted to a group RyO-; wherein each Ry is independently a 1-20 carbon branched or unbranched, saturated or unsaturated chain, wherein one or more of the carbon atoms is optionally replaced with -O- or -S- and wherein one or more of the carbon atoms is optionally substituted with oxo (=0) or thioxo (=S).
33. The compound of claim 28 wherein one or more pendent hydroxyl groups from the mono-, di-, or tri-phosphate group has been converted to a group R2-N-; wherein each R2 is a residue of an amino acid.
34. The compound of claim 33 wherein the amino acid is a natural amino acid.
35. The compound of claim 30 which comprises one or more groups of formula:

R15 is H, alkyl, substituted alkyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, heteroaryl, substituted heteroaryl, heterocyclic, substituted heterocyclic, or an amino acid;
R16 is H, optionally substituted monocyclic aryl, or optionally substituted monocyclic heteroaryl; and R17 is H, halogen, CN, -CO-R20, -CON(R2O2, - CO2R20, -SO2R20, -SO2N(R21)2, -OR21, -SR21, -R21, -N(R2O2, -0-COR20, -O- CO2R20, -SCOR20, -S-CO2R20, -NHCOR21, -NHCO2R21, -(CH2)P-OR22, or - (CH2)P-SR22; or R16 and R17 are connected via an additional 3-5 atoms to form a cyclic group, optionally containing one heteroatom, that is fused to an aryl group at the beta and gamma position to the O attached to the phosphorus; or R17 and R18 are connected as described below;
R18 and R19 are each independently H, alkyl, aryl, heterocycloalkyl, aralkyl, optionally substituted monocyclic aryl or optionally substituted monocyclic heteroaryl; or R18 and R19 are connected via an additional 2-5 atoms to form a cyclic group, optionally containing 0-2 heteroatoms; or R17 and R18 are connected via an additional 3-5 atoms to form a cyclic group, optionally containing one heteroatom and R19 is H, alkyl, aryl, heterocycloalkyl, aralkyl, optionally substituted monocyclic aryl or optionally substituted monocyclic heteroaryl;
R20 is alkyl, aryl, heterocycloalkyl, or arylalkyl;
R21 is H, alkyl, aryl, heterocycloalkyl, or arylalkyl;
R22 is H or lower acyl; n is an integer from 2-5; m is an integer from 10-20; and p is an integer from 2-3.
36. A pharmaceutical composition comprising a compound as described in any one of claims 1-35 and a pharmaceutically acceptable carrier.
37. The composition of claim 36 which further comprises one or more additional anti- viral agents.
38. The composition of claim 37 wherein the one or more anti-viral agents are selected from ribavirin, levovirin, viramidine, thymosin alpha- 1, an inhibitor of a serine proteases, an inhibitor of inosine monophosphatedehydrognease, interferon-α, and pegylated interferon-α (peginterferon-α).
39. The composition of any one of claims 36-38 which further comprises one or more additional HCV polymerase inhibitors.
40. The composition of any one of claims 36-39 which further comprises one or more protease inhibitors.
41. The composition of any one of claims 36-40 which further comprises ribavirin.
42. The composition of any one of claims 36-41 which further comprises interferon-α or pegylated interferon-α (peginterferon-α).
43. The composition of claim 36 which further comprises one or more anticancer agents.
44. The composition of claim 43 wherein the one or more anti-cancer agents are selected from alkylating agents, antimetabolites, natural products, and hormonal agents.
45. A method for treating a viral infection in an animal comprising administering to the animal an effective amount of a compound as described in any one of claims 1-35, or a composition as described in any one of claims 36- 44.
46. The method of claim 45 wherein the viral infection is selected from the group consisting of: hepatitis B, hepatitis C, human immunodeficiency virus, Polio, Coxsackie A and B, Rhino, Echo, small pox, Ebola, and West Nile virus.
47. The method of claim 45 wherein the viral infection is HCV.
48. The method of any one of claims 45-47 which further comprises administering to the animal one or more additional HCV polymerase inhibitors.
49. The method of any one of claims 45-48 which further comprises administering to the animal, one or more protease inhibitors.
50. The method of any one of claims 45-49 which further comprises administering ribavirin to the animal.
51. The method of any one of claims 45-50 which further comprises administering interferon-α or pegylated interferon-α (peginterferon-α) to the animal.
52. A method for treating cancer in an animal comprising administering to the animal an effective amount of a compound as described in any one of claims 1-35, or a composition as described in any one of claims 36 and 43-44.
53. The method of claim 52 wherein one or more additional anti-cancer compounds are administered.
54. The method of any one of claims 45-53 wherein the animal is a human.
55. A method for inhibiting a viral RNA or DNA polymerase comprising contacting the polymerase in vitro or in vivo with an effective inhibitory amount of a compound as described in any one of claims 1-35.
56. The method of claim 55 wherein the viral polymerase is an RdRp.
57. A compound as described in any one of claims 1-35 for use in medical therapy.
58. A compound according to any one of claims 1-35 for use in therapy.
59. The use of a compound according to any one of claims 1-35 for treating a viral infection.
60. The use of a compound according to any one of claims 1-35 for treating cancer.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US5348808P | 2008-05-15 | 2008-05-15 | |
| US61/053,488 | 2008-05-15 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2010036407A2 true WO2010036407A2 (en) | 2010-04-01 |
| WO2010036407A3 WO2010036407A3 (en) | 2010-05-20 |
Family
ID=41716509
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/044214 Ceased WO2010036407A2 (en) | 2008-05-15 | 2009-05-15 | Antiviral nucleoside analogs |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2010036407A2 (en) |
Cited By (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010093608A1 (en) * | 2009-02-10 | 2010-08-19 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| US8008264B2 (en) | 2008-04-23 | 2011-08-30 | Gilead Sciences, Inc. | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| WO2012075140A1 (en) | 2010-11-30 | 2012-06-07 | Pharmasset, Inc. | Compounds |
| JP2013538230A (en) * | 2010-09-20 | 2013-10-10 | ギリアード サイエンシーズ, インコーポレイテッド | 2'-Fluoro substituted carbanucleoside analogues for antiviral therapy |
| US8716282B2 (en) | 2009-10-30 | 2014-05-06 | Janssen Pharmaceutica Nv | Imidazo[1,2-b]pyridazine derivatives and their use as PDE10 inhibitors |
| CN104086553A (en) * | 2014-06-13 | 2014-10-08 | 南京药石药物研发有限公司 | Method for preparing 7-bromoimidazo[2,1-f][1,2,4]triazin-4-amine |
| US8859543B2 (en) | 2010-03-09 | 2014-10-14 | Janssen Pharmaceutica Nv | Imidazo[1,2-a]pyrazine derivatives and their use for the prevention or treatment of neurological, psychiatric and metabolic disorders and diseases |
| US8877731B2 (en) | 2010-09-22 | 2014-11-04 | Alios Biopharma, Inc. | Azido nucleosides and nucleotide analogs |
| JP2015509983A (en) * | 2012-03-13 | 2015-04-02 | ギリアード サイエンシーズ, インコーポレイテッド | 2'-substituted carbnucleoside analogues for antiviral treatment |
| US9073960B2 (en) | 2011-12-22 | 2015-07-07 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9090642B2 (en) | 2010-07-19 | 2015-07-28 | Gilead Sciences, Inc. | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs |
| US9441007B2 (en) | 2012-03-21 | 2016-09-13 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9550784B2 (en) | 2012-07-09 | 2017-01-24 | Beerse Pharmaceutica NV | Inhibitors of phosphodiesterase 10 enzyme |
| US9669035B2 (en) | 2012-06-26 | 2017-06-06 | Janssen Pharmaceutica Nv | Combinations comprising PDE 2 inhibitors such as 1-aryl-4-methyl-[1,2,4]triazolo-[4,3-A]]quinoxaline compounds and PDE 10 inhibitors for use in the treatment of neurological of metabolic disorders |
| US9724360B2 (en) | 2014-10-29 | 2017-08-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US9777035B2 (en) | 2014-03-28 | 2017-10-03 | Merck Sharp & Dohme Corp. | 4′-substituted nucleoside reverse transcriptase inhibitors |
| US9862743B2 (en) | 2013-10-11 | 2018-01-09 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10023600B2 (en) | 2009-09-21 | 2018-07-17 | Gilead Sciences, Inc. | Processes and intermediates for the preparation of 1′-substituted carba-nucleoside analogs |
| US10065958B2 (en) | 2010-07-22 | 2018-09-04 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US10251904B2 (en) | 2015-09-16 | 2019-04-09 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US10604523B2 (en) | 2011-06-27 | 2020-03-31 | Janssen Pharmaceutica Nv | 1-aryl-4-methyl-[1,2,4]triazolo[4,3-a]quinoxaline derivatives |
| US10675296B2 (en) | 2017-07-11 | 2020-06-09 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US10682368B2 (en) | 2017-03-14 | 2020-06-16 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| WO2020121123A2 (en) | 2018-12-12 | 2020-06-18 | Janssen Biopharma, Inc. | Cyclopentyl nucleoside analogs as anti-virals |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10836787B2 (en) | 2017-05-01 | 2020-11-17 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5- (4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy) phosphoryl)amino)propanoate |
| US10953029B2 (en) | 2015-09-23 | 2021-03-23 | Merck Sharp & Dohme Corp. | 4′-Substituted nucleoside reverse transcriptase inhibitors and preparations thereof |
| US11040975B2 (en) | 2017-12-08 | 2021-06-22 | Merck Sharp & Dohme Corp. | Carbocyclic nucleoside reverse transcriptase inhibitors |
| US11491169B2 (en) | 2020-05-29 | 2022-11-08 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| US11660307B2 (en) | 2020-01-27 | 2023-05-30 | Gilead Sciences, Inc. | Methods for treating SARS CoV-2 infections |
| US11697666B2 (en) | 2021-04-16 | 2023-07-11 | Gilead Sciences, Inc. | Methods of preparing carbanucleosides using amides |
| US11701372B2 (en) | 2020-04-06 | 2023-07-18 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carba-nucleoside analogs |
| US11767337B2 (en) | 2020-02-18 | 2023-09-26 | Gilead Sciences, Inc. | Antiviral compounds |
| US11780844B2 (en) | 2022-03-02 | 2023-10-10 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11814406B2 (en) | 2020-08-27 | 2023-11-14 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11939347B2 (en) | 2020-06-24 | 2024-03-26 | Gilead Sciences, Inc. | 1′-cyano nucleoside analogs and uses thereof |
| US12030903B2 (en) | 2020-02-18 | 2024-07-09 | Gilead Sciences, Inc. | Antiviral compounds |
| US12054507B2 (en) | 2020-02-18 | 2024-08-06 | Gilead Sciences, Inc. | Antiviral compounds |
| US12116380B2 (en) | 2021-08-18 | 2024-10-15 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| US12357577B1 (en) | 2024-02-02 | 2025-07-15 | Gilead Sciences, Inc. | Pharmaceutical formulations and uses thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000041531A2 (en) | 1999-01-13 | 2000-07-20 | Genentech, Inc. | Serine protease inhibitors |
| WO2004096286A2 (en) | 2003-04-25 | 2004-11-11 | Gilead Sciences, Inc. | Antiviral phosphonate analogs |
| WO2005084192A2 (en) | 2004-02-13 | 2005-09-15 | Merck & Co., Inc. | Novel 2’-c-methyl nucleoside derivatives |
| WO2007021610A2 (en) | 2005-08-09 | 2007-02-22 | Merck & Co., Inc. | Ribonucleoside cyclic acetal derivatives for the treatment of rna-dependent rna viral infection |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19912636A1 (en) * | 1999-03-20 | 2000-09-21 | Aventis Cropscience Gmbh | Bicyclic heterocycles, processes for their preparation and their use as herbicides and pharmaceutical agents |
| US20070078136A1 (en) * | 2005-09-22 | 2007-04-05 | Bristol-Myers Squibb Company | Fused heterocyclic compounds useful as kinase modulators |
| CN101730699A (en) * | 2007-03-21 | 2010-06-09 | 百时美施贵宝公司 | Can be used for treating the condensed heterocyclic compouds of proliferative, allergy, autoimmunity and diseases associated with inflammation |
| ES2393038T3 (en) * | 2007-05-10 | 2012-12-18 | Biocryst Pharmaceuticals, Inc. | Tretrahydrofuro [3,4-D] dioxolane compounds for use in the treatment of viral infections and cancer |
-
2009
- 2009-05-15 WO PCT/US2009/044214 patent/WO2010036407A2/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000041531A2 (en) | 1999-01-13 | 2000-07-20 | Genentech, Inc. | Serine protease inhibitors |
| WO2004096286A2 (en) | 2003-04-25 | 2004-11-11 | Gilead Sciences, Inc. | Antiviral phosphonate analogs |
| WO2005084192A2 (en) | 2004-02-13 | 2005-09-15 | Merck & Co., Inc. | Novel 2’-c-methyl nucleoside derivatives |
| WO2007021610A2 (en) | 2005-08-09 | 2007-02-22 | Merck & Co., Inc. | Ribonucleoside cyclic acetal derivatives for the treatment of rna-dependent rna viral infection |
Non-Patent Citations (14)
| Title |
|---|
| "ASHP Handbook on Injectable Drugs", 1986, TOISSEL, pages: 622 - 630 |
| "Design of Prodrugs", 1985, ELSEVIER |
| "Pharmaceutics and Pharmacy Practice", 1982, J.B. LIPPINCOTT CO., pages: 238 - 250 |
| ALEXANDER ET AL., J. MED. CHEM., vol. 31, 1988, pages 318 |
| BLIGHT ET AL., SCI., vol. 290, 2000, pages 1972 |
| BODOR ET AL., J. MED. CHEM., vol. 23, 1980, pages 469 |
| DAVIS, GASTROENTEROLOGY, vol. 118, 2000, pages S 104 - S 114 |
| DAVIS, GASTROENTEROLOGY, vol. 118, 2000, pages S104 - S114 |
| J.A. BRUENN, NUCLEIC ACIDS RESEARCH, vol. 19, no. 2, 1991, pages 217 |
| JOHN S. DRISCOLL: "Antiviral Drugs", 2002, ASHGATE PUBLISHING LTD. |
| KORBA; GERIN, ANTIVIR. RES., vol. 19, 1992, pages 55 |
| LEFEBVRE ET AL., J. MED. CHEM., vol. 38, 1995, pages 3941 - 50 |
| OKUSE ET AL., ANTIVIR. RES., vol. 65, 2005, pages 23 - 34 |
| OKUSE, ANTIVIRAL RES., vol. 65, 2005, pages 23 - 34 |
Cited By (91)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8318682B2 (en) | 2008-04-23 | 2012-11-27 | Gilead Sciences, Inc. | 1′substituted carba-nucleoside analogs for antiviral treatment |
| US8008264B2 (en) | 2008-04-23 | 2011-08-30 | Gilead Sciences, Inc. | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| US8853171B2 (en) | 2008-04-23 | 2014-10-07 | Gilead Sciences, Inc. | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| US8012941B2 (en) * | 2008-04-23 | 2011-09-06 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| USRE46762E1 (en) | 2008-04-23 | 2018-03-27 | Gilead Sciences, Inc | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| AP2922A (en) * | 2009-02-10 | 2014-05-31 | Gilead Sciences Inc | Carbra-nucleoside analogs for antiviral treatment |
| AU2017202413B2 (en) * | 2009-02-10 | 2018-09-13 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| AU2010213873B2 (en) * | 2009-02-10 | 2015-07-09 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| WO2010093608A1 (en) * | 2009-02-10 | 2010-08-19 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| JP2014185183A (en) * | 2009-02-10 | 2014-10-02 | Gilead Sciences Inc | Carba-nucleoside analogs for antiviral treatment |
| US8012942B2 (en) | 2009-02-10 | 2011-09-06 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| AU2015238785B2 (en) * | 2009-02-10 | 2017-02-16 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| EA025085B1 (en) * | 2009-02-10 | 2016-11-30 | Джилид Сайэнс, Инк. | Carba-nucleoside analogs for antiviral treatment |
| US10988498B2 (en) | 2009-09-21 | 2021-04-27 | Gilead Sciences, Inc. | Processes and intermediates for the preparation of 1′-substituted carba-nucleoside analogs |
| US10023600B2 (en) | 2009-09-21 | 2018-07-17 | Gilead Sciences, Inc. | Processes and intermediates for the preparation of 1′-substituted carba-nucleoside analogs |
| US8716282B2 (en) | 2009-10-30 | 2014-05-06 | Janssen Pharmaceutica Nv | Imidazo[1,2-b]pyridazine derivatives and their use as PDE10 inhibitors |
| US8859543B2 (en) | 2010-03-09 | 2014-10-14 | Janssen Pharmaceutica Nv | Imidazo[1,2-a]pyrazine derivatives and their use for the prevention or treatment of neurological, psychiatric and metabolic disorders and diseases |
| US9090642B2 (en) | 2010-07-19 | 2015-07-28 | Gilead Sciences, Inc. | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs |
| US9487544B2 (en) | 2010-07-19 | 2016-11-08 | Gilead Sciences, Inc. | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs |
| US10696679B2 (en) | 2010-07-22 | 2020-06-30 | Gilead Sciences, Inc. | Methods and compounds for treating paramyxoviridae virus infections |
| US10065958B2 (en) | 2010-07-22 | 2018-09-04 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US11492353B2 (en) | 2010-07-22 | 2022-11-08 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US12509466B2 (en) | 2010-07-22 | 2025-12-30 | Gilead Sciences, Inc. | Methods and compounds for treating paramyxoviridae virus infections |
| JP2016074732A (en) * | 2010-09-20 | 2016-05-12 | ギリアード サイエンシーズ, インコーポレイテッド | 2'-fluoro substituted carba-nucleoside analogs for antiviral treatment |
| JP2013538230A (en) * | 2010-09-20 | 2013-10-10 | ギリアード サイエンシーズ, インコーポレイテッド | 2'-Fluoro substituted carbanucleoside analogues for antiviral therapy |
| JP2017119726A (en) * | 2010-09-20 | 2017-07-06 | ギリアード サイエンシーズ, インコーポレイテッド | 2'-fluoro substituted carba-nucleoside analogs for antiviral treatment |
| US9346848B2 (en) | 2010-09-22 | 2016-05-24 | Alios Biopharma, Inc. | Azido nucleosides and nucleotide analogs |
| US8877731B2 (en) | 2010-09-22 | 2014-11-04 | Alios Biopharma, Inc. | Azido nucleosides and nucleotide analogs |
| EP3042910A2 (en) | 2010-11-30 | 2016-07-13 | Gilead Pharmasset LLC | 2'-spiro-nucleosides for use in the therapy of hepatitis c |
| WO2012075140A1 (en) | 2010-11-30 | 2012-06-07 | Pharmasset, Inc. | Compounds |
| US10604523B2 (en) | 2011-06-27 | 2020-03-31 | Janssen Pharmaceutica Nv | 1-aryl-4-methyl-[1,2,4]triazolo[4,3-a]quinoxaline derivatives |
| US10464965B2 (en) | 2011-12-22 | 2019-11-05 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US11021509B2 (en) | 2011-12-22 | 2021-06-01 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9073960B2 (en) | 2011-12-22 | 2015-07-07 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| JP2015509983A (en) * | 2012-03-13 | 2015-04-02 | ギリアード サイエンシーズ, インコーポレイテッド | 2'-substituted carbnucleoside analogues for antiviral treatment |
| JP2017206528A (en) * | 2012-03-13 | 2017-11-24 | ギリアード サイエンシーズ, インコーポレイテッド | 2'-substituted carbnucleoside analogues for antiviral treatment |
| US11787832B2 (en) | 2012-03-13 | 2023-10-17 | Gilead Sciences, Inc. | 2′-substituted carba-nucleoside analogs for antiviral treatment |
| US10941177B2 (en) | 2012-03-13 | 2021-03-09 | Gilead Sciences, Inc. | 2′-substituted carba-nucleoside analogs for antiviral treatment |
| US9441007B2 (en) | 2012-03-21 | 2016-09-13 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10485815B2 (en) | 2012-03-21 | 2019-11-26 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9669035B2 (en) | 2012-06-26 | 2017-06-06 | Janssen Pharmaceutica Nv | Combinations comprising PDE 2 inhibitors such as 1-aryl-4-methyl-[1,2,4]triazolo-[4,3-A]]quinoxaline compounds and PDE 10 inhibitors for use in the treatment of neurological of metabolic disorders |
| US9550784B2 (en) | 2012-07-09 | 2017-01-24 | Beerse Pharmaceutica NV | Inhibitors of phosphodiesterase 10 enzyme |
| US9862743B2 (en) | 2013-10-11 | 2018-01-09 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10370401B2 (en) | 2013-10-11 | 2019-08-06 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9777035B2 (en) | 2014-03-28 | 2017-10-03 | Merck Sharp & Dohme Corp. | 4′-substituted nucleoside reverse transcriptase inhibitors |
| CN104086553A (en) * | 2014-06-13 | 2014-10-08 | 南京药石药物研发有限公司 | Method for preparing 7-bromoimidazo[2,1-f][1,2,4]triazin-4-amine |
| US10695357B2 (en) | 2014-10-29 | 2020-06-30 | Gilead Sciences, Inc. | Methods for treating filoviridae virus infections |
| US10251898B2 (en) | 2014-10-29 | 2019-04-09 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US11266666B2 (en) | 2014-10-29 | 2022-03-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US9949994B2 (en) | 2014-10-29 | 2018-04-24 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US11344565B2 (en) | 2014-10-29 | 2022-05-31 | Gilead Sciences, Inc. | Methods for the preparation of ribosides |
| US9724360B2 (en) | 2014-10-29 | 2017-08-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US10251904B2 (en) | 2015-09-16 | 2019-04-09 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US11382926B2 (en) | 2015-09-16 | 2022-07-12 | Gilead Sciences, Inc. | Methods for treating Arenaviridae and Coronaviridae virus infections |
| US11007208B2 (en) | 2015-09-16 | 2021-05-18 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US10695361B2 (en) | 2015-09-16 | 2020-06-30 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US10953029B2 (en) | 2015-09-23 | 2021-03-23 | Merck Sharp & Dohme Corp. | 4′-Substituted nucleoside reverse transcriptase inhibitors and preparations thereof |
| US11260070B2 (en) | 2017-03-14 | 2022-03-01 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| US10682368B2 (en) | 2017-03-14 | 2020-06-16 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| US12030906B2 (en) | 2017-05-01 | 2024-07-09 | Gilead Sciences, Inc. | Crystalline forms of (s)-2-ethylbutyl 2-(((s)-(((2r,3s,4r,5r)-5-(4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy) (phenoxy) phosphoryl)amino)propanoate |
| US11597742B2 (en) | 2017-05-01 | 2023-03-07 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy) (phenoxy) phosphoryl)amino)propanoate |
| US10836787B2 (en) | 2017-05-01 | 2020-11-17 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5- (4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy) phosphoryl)amino)propanoate |
| US10675296B2 (en) | 2017-07-11 | 2020-06-09 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US11266681B2 (en) | 2017-07-11 | 2022-03-08 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US11975017B2 (en) | 2017-07-11 | 2024-05-07 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US11040975B2 (en) | 2017-12-08 | 2021-06-22 | Merck Sharp & Dohme Corp. | Carbocyclic nucleoside reverse transcriptase inhibitors |
| WO2020121123A2 (en) | 2018-12-12 | 2020-06-18 | Janssen Biopharma, Inc. | Cyclopentyl nucleoside analogs as anti-virals |
| US11660307B2 (en) | 2020-01-27 | 2023-05-30 | Gilead Sciences, Inc. | Methods for treating SARS CoV-2 infections |
| US11767337B2 (en) | 2020-02-18 | 2023-09-26 | Gilead Sciences, Inc. | Antiviral compounds |
| US12264173B2 (en) | 2020-02-18 | 2025-04-01 | Gilead Sciences, Inc. | Antiviral compounds |
| US12054507B2 (en) | 2020-02-18 | 2024-08-06 | Gilead Sciences, Inc. | Antiviral compounds |
| US12030903B2 (en) | 2020-02-18 | 2024-07-09 | Gilead Sciences, Inc. | Antiviral compounds |
| US12012431B2 (en) | 2020-03-12 | 2024-06-18 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| US11701372B2 (en) | 2020-04-06 | 2023-07-18 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carba-nucleoside analogs |
| US11903953B2 (en) | 2020-05-29 | 2024-02-20 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11491169B2 (en) | 2020-05-29 | 2022-11-08 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11975012B2 (en) | 2020-05-29 | 2024-05-07 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11939347B2 (en) | 2020-06-24 | 2024-03-26 | Gilead Sciences, Inc. | 1′-cyano nucleoside analogs and uses thereof |
| US12404289B2 (en) | 2020-06-24 | 2025-09-02 | Gilead Sciences, Inc. | 1′-cyano nucleoside analogs and uses thereof |
| US11926645B2 (en) | 2020-08-27 | 2024-03-12 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11814406B2 (en) | 2020-08-27 | 2023-11-14 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US12297226B2 (en) | 2020-08-27 | 2025-05-13 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11697666B2 (en) | 2021-04-16 | 2023-07-11 | Gilead Sciences, Inc. | Methods of preparing carbanucleosides using amides |
| US12116380B2 (en) | 2021-08-18 | 2024-10-15 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| US11845755B2 (en) | 2022-03-02 | 2023-12-19 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US12180217B2 (en) | 2022-03-02 | 2024-12-31 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11780844B2 (en) | 2022-03-02 | 2023-10-10 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US12448383B2 (en) | 2022-03-02 | 2025-10-21 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US12357577B1 (en) | 2024-02-02 | 2025-07-15 | Gilead Sciences, Inc. | Pharmaceutical formulations and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010036407A3 (en) | 2010-05-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2010036407A2 (en) | Antiviral nucleoside analogs | |
| EP2155758B1 (en) | Tetrahydrofuro[3,4-d]dioxolane compounds for use in the treatment of viral infections and cancer | |
| US8440813B2 (en) | Antiviral nucleoside analogs | |
| US7994139B2 (en) | Antiviral therapeutic agents | |
| JP5107228B2 (en) | Hepatitis C treatment | |
| US8133870B2 (en) | Therapeutic furopyrimidines and thienopyrimidines | |
| HK1139656B (en) | Tetrahydrofuro [3,4-d] dioxolane compounds for use in the treatment of viral infections and cancer | |
| AU2012201345A1 (en) | Hepatitis C therapies | |
| AU2012268918A1 (en) | Antiviral nucleoside analogs |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09799772 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09799772 Country of ref document: EP Kind code of ref document: A2 |