CN100582097C - 取代的苯并异噁唑磺酰胺广谱hiv蛋白酶抑制剂 - Google Patents
取代的苯并异噁唑磺酰胺广谱hiv蛋白酶抑制剂 Download PDFInfo
- Publication number
- CN100582097C CN100582097C CN03816458A CN03816458A CN100582097C CN 100582097 C CN100582097 C CN 100582097C CN 03816458 A CN03816458 A CN 03816458A CN 03816458 A CN03816458 A CN 03816458A CN 100582097 C CN100582097 C CN 100582097C
- Authority
- CN
- China
- Prior art keywords
- alkyl
- het
- amino
- group
- aryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/20—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Virology (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Molecular Biology (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
本发明涉及具有式(I)化合物及其N-氧化物、盐、立体异构形式、外消旋混合物、前药、酯和代谢物。它还涉及它们作为广谱HIV蛋白酶抑制剂的用途,它们的制备方法以及包含它们的药用组合物和诊断试剂盒。本发明还涉及本发明化合物与另一种抗逆转录病毒剂的组合,以及它们在试验中作为参考化合物或作为试剂的用途。
Description
本发明涉及取代的苯并异噁唑磺酰胺,它们作为天冬氨酸蛋白酶抑制剂,特别是作为广谱HIV蛋白酶抑制剂的用途,它们的制备方法以及包含它们的药用组合物和诊断试剂盒。本发明也涉及本发明的取代的苯并异噁唑磺酰胺与另一种抗逆转录病毒剂的组合,还涉及它们在试验中作为参考化合物或作为试剂的用途。
导致获得性免疫缺陷综合征(AIDS)的病毒有不同的名称,包括T-淋巴细胞病毒III(HTLV-III)或淋巴结病相关病毒(LAV)、AIDS-相关病毒(ARV)或人类免疫缺陷病毒(HIV)。迄今为止,已经鉴别出两种不同的类型,即HIV-1和HIV-2。以下将用HIV统称这些病毒。
在逆转录病毒的生命周期中的关键途径之一是多蛋白(polyprotein)前体经天冬氨酸蛋白酶的处理过程,例如在HIV病毒中,其gag-pol蛋白经过HIV蛋白酶的处理。感染病毒颗粒的装配需要前体多蛋白经天冬氨酸蛋白酶的正确处理,因此使得天冬氨酸蛋白酶成为抗病毒治疗的一个有吸引力的目标。特别是对于HIV的治疗,HIV蛋白酶是一个有吸引力的目标。
通常将HIV蛋白酶抑制剂(PIs)与其它抗HIV化合物如核苷逆转录酶抑制剂(NRTIs)、非核苷逆转录酶抑制剂(NNRTIs)、核苷酸逆转录酶抑制剂(NtRTIs)或其它蛋白酶抑制剂联合用药于AIDS患者。尽管事实上这些抗逆转录病毒剂非常有用,但它们有一个共同的限制,即HIV病毒中的靶向酶能够突变使得已知的药物药效降低,或甚至对这些突变的HIV病毒无效。或者换句话说,HIV病毒对现有的药物产生不断增长的耐药性。
逆转录病毒(特别是HIV病毒)对抑制剂的耐药性是治疗失败的主要原因。例如接受抗HIV联合疗法的半数患者对治疗没有完全反应,主要是因为病毒对所用的一种或多种药物有耐药性。而且,已显示有耐药性的病毒被带到新感染的个体后,可导致这些未曾用药(drug-naive)的患者的治疗选择严重受限。因此,在本领域中需要新的化合物用于逆转录病毒的治疗,尤其是用于AIDS的治疗。在本领域中特别急需不仅对野生型HIV病毒有活性,而且对日益增加的更常见的有耐药性的HIV病毒也有活性的化合物。
已知的抗逆转录病毒剂,通常以联合治疗方案给药,如上所述终将导致耐药性。这常迫使医生加大活性药物的血浆水平,以使得该抗逆转录病毒剂可再度有效地对抗突变的HIV病毒。其后果是极不愿有的药物负担的增加。增加血浆水平也导致增加处方治疗的不依从性(non-compliance)的风险。因此,不仅化合物对广泛的HIV突变显示活性是很重要的,在广泛的HIV突变株中对抗突变的HIV病毒的活性与对抗野生型HIV病毒的活性(也定义为折叠耐药性(foldresistance)或FR)之间存在很小的差异或无差异也是很重要的。如此,可以在较长时间内对患者维持相同的联合治疗方案,原因是突变的HIV病毒对于活性成分敏感的机会将增加。
寻找对于野生型和多种突变型有高度效力的化合物也很重要,因为如果治疗量可维持最小则可降低药物负担。一种降低此种药物负担的方法是发现具有高生物利用度,即具有良好的药代动力学和代谢特性的抗HIV化合物,使得每日剂量可最小化,因此取用的药物数量也可最小化。
良好的抗HIV化合物的另一个重要特征是抑制剂的血浆蛋白结合对其功效有很小的影响或甚至无影响。
因此,对于能够对抗广谱HIV病毒突变型的蛋白酶抑制剂有高度的医疗需求。其它重要的特征包括变异很小的折叠耐药性、良好的生物利用度,以及血浆蛋白结合对化合物功效影响很小或甚至无影响。
迄今为止,有数种蛋白酶抑制剂在市场上供应或正在研发中。一种特定的核心结构(描述如下)已经公布于多种参考文献中,例如WO95/06030、WO 96/22287、WO 96/28418、WO 96/28463、WO 96/28464、WO 96/28465和WO 97/18205。其中公开的化合物被称为逆转录病毒蛋白酶抑制剂。

WO 99/67254公开4-取代的-苯基磺酰胺可抑制对多种药物有耐药性的逆转录病毒蛋白酶。

发现本发明的取代的苯并异噁唑磺酰胺具有有利的药理学特性,它们不只是对抗野生型HIV病毒有活性,还对于已知的蛋白酶抑制剂显现耐药性的多种突变HIV病毒显示广谱对抗活性。
本发明涉及具有下式的取代的苯并异噁唑蛋白酶抑制剂

及其N-氧化物、盐、立体异构形式、外消旋混合物、药物前体、酯和代谢物,尤其是其N-氧化物、盐和立体异构形式,其中
R1及R8各自独立为氢、C1-6烷基、C2-6链烯基、芳基C1-6烷基、C3-7环烷基、C3-7环烷基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2、Het2C1-6烷基;
R1也可以是下式的基团

其中
R9、R10a及R10b各自独立为氢、C1-4烷氧基羰基、羧基、氨基羰基、一-或二(C1-4烷基)氨基羰基、C3-7环烷基、C2-6链烯基、C2-6炔基,或C1-4烷基,其任选被芳基、Het1、Het2、C3-7环烷基、C1-4烷氧基羰基、羧基、氨基羰基、一-或二(C1-4烷基)氨基羰基、氨基磺酰基、C1-4烷基S(O)t、羟基、氰基、卤素或任选被一-或二取代的氨基取代,其中所述氨基的取代基各自独立地选自C1-4烷基、芳基、芳基C1-4烷基、C3-7环烷基、C3-7环烷基C1-4烷基、Het1、Het2、Het1C1-4烷基及Het2C1-4烷基;其中R9、R10a与其连接的碳原子也可形成C3-7环烷基;
当L是-O-C1-6链烷二基-C(=O)-或-NR8-C1-6链烷二基-C(=O)-时,则R9也可为氧代基;
R11a是氢、C2-6链烯基、C2-6炔基、C3-7环烷基、芳基、任选被一-或二取代的氨基羰基、任选被一-或二取代的氨基C1-4烷基羰氧基、C1-4烷氧基羰基、芳氧基羰基、Het1氧基羰基、Het2氧基羰基、芳氧基羰基C1-4烷基、芳基C1-4烷氧基羰基、C1-4烷基羰基、C3-7环烷基羰基、C3-7环烷基C1-4烷氧基羰基、C3-7环烷基羰氧基、羧基C1-4烷基羰氧基、C1-4烷基羰氧基、芳基C1-4烷基羰氧基、芳基羰氧基、芳氧基羰基氧基、Het1羰基、Het1羰氧基、Het1C1-4烷氧基羰基、Het2羰氧基、Het2C1-4烷基羰氧基、Het2C1-4烷氧基羰氧基,或C1-4烷基,其任选被芳基、芳氧基、Het2、卤素或羟基取代;其中在氨基上的取代基各自独立地选自C1-4烷基、芳基、芳基C1-4烷基、C3- 7环烷基、C3-7环烷基C1-4烷基、Het1、Het2、Het1C1-4烷基及Het2C1-4烷基;
R11b是氢、C3-7环烷基、C2-6链烯基、C2-6炔基、芳基、Het1、Het2,或C1-4烷基,其任选被卤素、羟基、C1-4烷基S(=O)t、芳基、C3-7环烷基、Het1、Het2、任选被一-或二取代的氨基取代,
其中所述氨基的取代基各自独立地选自C1-4烷基、芳基、芳基C1-4烷基、C3-7环烷基、C3-7环烷基C1-4烷基、Het1、Het2、Het1C1-4烷基及Het2C1-4烷基;
其中R11b可通过磺酰基连接至分子的其余部分;
t各自独立地是0、1或2;
R2是氢或C1-6烷基;
L是-C(=O)-、-O-C(=O)-、-NR8-C(=O)-、-O-C1-6链烷二基-C(=O)-、-NR8-C1-6链烷二基-C(=O)-、-S(=O)2-、-O-S(=O)2-、-NR8-S(=O)2-,
其中C(=O)基团或S(=O)2基团连接至NR2部分;其中C1-6链烷二基任选被羟基、芳基、Het1和Het2取代;
R3是C1-6烷基、芳基、C3-7环烷基、C3-7环烷基C1-4烷基或芳基C1-4烷基;
R4是氢、C1-4烷氧基羰基、羧基、氨基羰基、一-或二(C1-4烷基)氨基羰基、C3-7环烷基、C2-6链烯基、C2-6炔基,或C1-6烷基,其任选被一或多个各自独立地选自以下的取代基所取代:芳基、Het1、Het2、C3-7环烷基、C1-4烷氧基羰基、羧基、氨基羰基、一-或二(C1-4烷基)氨基羰基、氨基磺酰基、C1-4烷基S(=O)t、羟基、氰基、卤素及任选被一-或二取代的氨基,其中所述氨基的取代基各自独立地选自C1-4烷基、芳基、芳基C1-4烷基、C3-7环烷基、C3-7环烷基-C1-4烷基、Het1、Het2、Het1C1-4烷基及Het2C1-4烷基;
R12是-NH2或-N(R5)(A-R6),其中
A是C1-6链烷二基、-C(=O)-、-C(=S)-、-S(=O)2-、C1-6链烷二基-C(=O)-、C1-6链烷二基-C(=S)-或C1-6链烷二基-S(=O)2-,其中A与取代的氨基的连接点为在含有所述C1-6链烷二基的A的这些定义中的C1-6链烷二基;
R5是氢、羟基、C1-6烷基、Het1C1-6烷基、Het2C1-6烷基、氨基C1-6烷基,其中所述氨基可以任选由C1-4烷基一-或二取代;
R6是氢、C1-6烷氧基、Het1、Het1氧基、Het2、Het2氧基、芳基、芳氧基、芳氧基C1-4烷基、C1-4烷氧基芳基、C1-4烷氧基Het1、C1-4烷氧基Het2、C1-4烷氧基羰基氨基、氨基C1-4烷基氨基、氨基或氨基C1-4烷氧基,并且如果A不是C1-6链烷二基时,
则R6也可以是C1-6烷基、Het1C1-4烷基、Het1氧基C1-4烷基、Het2C1-4烷基、Het2氧基C1-4烷基、芳基C1-4烷基、芳氧基C1-4烷基或氨基C1-4烷基;其中每个氨基可任选被C1-4烷基一取代或(可能时)被二取代;
-A-R6也可以是羟基C1-6烷基;
R5和-A-R6与其连接的氮原子一起也可形成Het1或Het2。
可用本领域普通技术人员已知的任何试剂,例如低级烷基卤化物、硫酸二烷基酯、长链卤化物及芳烷基卤化物,将本发明化合物中出现的碱性氮季铵化。
当术语“取代的”用于定义式(I)化合物时,是指在使用“取代的”表达中的指定原子上的一或多个氢被选自指定的基团取代,条件是不超过指定原子的正常价数,且取代产生化学稳定的化合物,即化合物足够稳定,能从反应混合物中分离成可用程度的纯度,且能配制成治疗药物。也可出现规定在指定的基团上的取代基数目的情形。例如,一-或二取代意指一个或两个取代基。
本文使用的作为基团或基团的一部份时的术语“卤基”或“卤素”通指氟、氯、溴或碘。
术语“C1-4烷基”作为基团或基团的一部份时,定义为含有1至4个碳原子的直链和支链饱和烃基,例如甲基、乙基、丙基、丁基及2-甲基-丙基等。
术语“C1-6烷基”作为基团或基团的一部份时,定义为含有1至6个碳原子的直链和支链饱和烃基,例如对C1-4烷基定义的基团及戊基、己基、2-甲基丁基、3-甲基戊基等。
术语“C1-6链烷二基”作为基团或基团的一部份时,定义为含有1至6个碳原子的二价直链和支链饱和烃基,例如亚甲基、乙-1,2-二基、丙-1,3-二基、丙-1,2-二基、丁-1,4-二基、戊-1,5-二基、己-1,6-二基、2-甲基丁-1,4-二基、3-甲基戊-1,5-二基等。
术语“C2-6链烯基”作为基团或基团的一部份时,定义为含有2至6个碳原子及含有至少一个双键的直链及支链烃基,例如乙烯基、丙烯基、丁烯基、戊烯基、己烯基等。
术语“C2-6炔基”作为基团或基团的一部份时,定义为含有2至6个碳原子及含有至少一个三键的直链及支链烃基,例如乙炔基、丙炔基、丁炔基、戊炔基、己炔基等。
术语“C3-7环烷基”作为基团或基团的一部份时,是指环丙基、环丁基、环戊基、环己基或环庚基。
术语“芳基”作为基团或基团的一部份时,是指包括苯基及萘基,两者都可任选被一或多个取代基取代,所述取代基独立地选自C1-6烷基、任选一-或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、任选一-或二取代的氨基、硝基、氰基、多卤代C1-6烷基、羟基C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、Het1、任选一-或二取代的氨基羰基、甲硫基、甲磺酰基及任选被一或多个取代基取代的苯基,所述取代基各自独立选自C1-6烷基、任选一-或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、任选一-或二取代的氨基、硝基、氰基、多卤代C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、Het1、任选一-或二取代的氨基羰基、甲硫基及甲磺酰基;其中在任何氨基官能团上任选的取代基独立地选自C1-6烷基、C1-6烷氧基-A-、Het1-A-、Het1C1-6烷基、Het1C1-6烷基-A-、Het1氧基-A-、Het1-氧基-C1-4烷基-A-、苯基-A-、苯氧基-A-、苯氧基C1-4烷基-A-、苯基C1-6烷基-A-、C1-6烷氧基羰基-氨基-A-、氨基-A-、氨基C1-6烷基及氨基C1-6烷基-A-,其中各氨基可任选被C1-4烷基单-或(可能时)二-取代。
术语“多卤代C1-6烷基”作为基团或基团的一部份时,定义为被一或多个卤原子,优选是氯或氟原子,更优选氟原子取代的C1-6烷基。优选的卤代C1-6烷基例如包括三氟甲基及二氟甲基。
术语“Het1”作为基团或基团的一部份时,定义为含有3至14个环成员优选含5至10个环成员且更优选含5至8个环成员的饱和或部份不饱和单环、二环或三环杂环,,它含一或多个杂原子环成员,杂原子各自独立地选自氮、氧或硫,且它在一或多个碳原子上任选被选自以下的取代基取代:C1-6烷基、任选一-或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、氧代基、任选一-或二取代的氨基、硝基、氰基、多卤代C1-6烷基、羟基C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、任选一-或二取代的氨基羰基、甲硫基、甲磺酰基、芳基及含3至14个环成员的饱和或部份不饱和单环、二环或三环杂环,所述杂环含一或多个杂原子环成员,杂原子各自独立选自氮、氧或硫,且其中在任何氨基官能团上的任选取代基独立选自C1-6烷基、C1-6烷氧基-A-、Het2-A-、Het2C1-6烷基、Het2C1-6烷基-A-、Het2氧基-A-、Het2氧基C1-4烷基-A-、芳基-A-、芳氧基-A-、芳氧基C1-4烷基-A-、芳基C1-6烷基-A-、C1-6烷氧基羰基氨基-A-、氨基-A-、氨基C1-6烷基及氨基C1-6烷基-A-,其中各氨基可任选被C1-4烷基单-或可能时二-取代。在“Het1”的定义中所列出的优选取代基为C1-6烷基、任选一或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、氧代基、任选一-或二取代的氨基、硝基、氰基、多卤代C1-6烷基、羟基C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、任选一-或二取代的氨基羰基、甲硫基、甲磺酰基、芳基及含3至14个环成员的饱和或部份不饱和单环、二环或三环杂环,所述杂环含一或多个各自独立选自氮、氧或硫的杂原子环成员,且其中在任何氨基官能团上任选的取代基独立地选自C1-6烷基、C1-6烷氧基-A-、苯基-A-、苯氧基-A-、苯氧基C1-4烷基-A-、苯基C1-6烷基-A-、C1-6烷氧基羰基-氨基-A-、氨基-A-、氨基C1-6烷基及氨基C1-6烷基-A-,其中各氨基可任选被C1-4烷基单-或可能时二-取代。
术语“Het2”作为基团或基团的一部份时,定义为含有3至14个环成员,优选含5至10个环成员且更优选含5至6个环成员的芳族单环、二环或三环杂环,它含一或多个杂原子环成员,杂原子各自独立选自氮、氧或硫,且它在一或多个碳原子上任选被选自以下的取代基取代:C1-6烷基、任选一-或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、任选一-或二取代的氨基、硝基、氰基、卤代C1- 6烷基、羟基C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、任选一-或二取代的氨基羰基、甲硫基、甲磺酰基、芳基、Het1及含3至14个环成员的芳族单环、二环或三环杂环;其中在任何氨基官能团上任选的取代基独立选自C1-6烷基、C1-6烷氧基-A-、Het1-A-、Het1C1-6烷基、Het1C1-6烷基-A-、Het1氧基-A-、Het1氧基C1-4烷基-A-、芳基-A-、芳氧基-A-、芳氧基C1-4烷基-A-、芳基C1-6烷基-A-、C1-6烷氧基羰基氨基-A-、氨基-A-、氨基C1-6烷基及氨基C1-6烷基-A-,其中各氨基可任选被C1-4烷基单-或可能时二-取代。在“Het2”的定义中所列出的优选取代基为C1-6烷基、任选一-或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、任选一-或二取代的氨基、硝基、氰基、多卤代C1-6烷基、羟基C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、任选一-或二取代的氨基羰基、甲硫基、甲磺酰基、芳基、Het1及含3至14个环成员的芳族单环、二环或三环杂环;其中在任何氨基官能团上任选的取代基独立选自C1-6烷基、C1-6烷氧基-A-、苯基-A-、苯氧基-A-、苯氧基C1-4烷基-A-、苯基C1-6烷基-A-、C1-6烷氧基羰基-氨基-A-、氨基-A-、氨基C1-6烷基及氨基C1-6烷基-A-,其中各氨基可任选被C1-4烷基单-或可能时二-取代。
本文使用的术语(=O)与其连接的碳原子形成羰基部分,术语(=O)与其连接的硫原子形成亚砜,术语(=O)2与其连接的硫原子形成磺酰基。
本文使用的术语(=S)与其连接的碳原子形成硫羰基。
本文前述中使用的术语“一或多个”涵盖全部可被取代基取代的H-原子(合适时),优选是一、二或三个取代。
当任何变量(例如卤基或C1-4烷基)在任何组成中出现一次以上时,各定义彼此独立。
在本文中使用的术语“药物前体”是指药学上可接受的衍生物,例如酯、酰胺及磷酸酯,所述衍生物在体内生物转化的产物是在式(I)化合物中定义的活性药物。Goodman及Gilman的文献全面介绍药物前体(The Pharmacological Basis of Therapeutics,第8版,McGraw-Hill,Int.Ed.1992,“Biotransformation of Drugs”,第13-15页),结合于本文供参考。通过修饰存在于化合物上的官能团制备本发明化合物的药物前体,修饰的方法使得当修饰物分解时,不论是常规操作还是在体内,都可形成母体化合物。药物前体包括其中羟基(例如在不对称碳原子上的羟基)或氨基与任何基团结合的本发明化合物,当将药物前体给予患者后,能分解而分别形成游离羟基或游离氨基。典型的药物前体实例在例如WO 99/33795、WO 99/33815、WO 99/33793及WO 99/33792中描述,全都专利内容通过引用结合到本文中。
药物前体的特征是有极佳的水溶性、增加的生物利用度且容易在体内代谢成活性抑制剂。
作为医疗用途的式(I)化合物的盐是其中抗衡离子为药学上或生理上可接受的那些盐。但是,含有药学上不可接受的抗衡离子的盐也可用在例如制备或纯化药学上可接受的式(I)化合物中。全部盐,不论是药学上可接受或不可接受的,都包括在本发明的范围内。
本发明化合物可以形成的药学上可接受或生理上可耐受的加成盐形式可以使用适当的酸方便地制备,例如无机酸,如氢卤酸,如氢氯酸或氢溴酸、硫酸、半硫酸、硝酸、磷酸等酸;或有机酸例如乙酸、天门冬氨酸、十二烷基硫酸、庚酸、己酸、烟酸、丙酸、羟基乙酸、乳酸、丙酮酸、草酸、丙二酸、琥珀酸、马来酸、富马酸、苹果酸、酒石酸、柠檬酸、甲磺酸、乙磺酸、苯磺酸、对-甲苯磺酸、环己烷氨基磺酸、水杨酸、对-氨基水杨酸、扑酸等酸。
反过来说,所述酸加成盐形式可通过用适当的碱处理而转化成游离碱形式。
含酸性质子的式(I)化合物通过用适当的有机及无机碱处理,也可转化成其无毒的金属或胺加成盐形式。适当的碱盐形式包括例如铵盐、碱金属及碱土金属盐例如锂、钠、钾、镁、钙盐等,与有机碱所成的盐例如苄星青霉素、N-甲基、-D-葡糖胺、海巴胺(hydrabamine)盐,及与氨基酸例如精氨酸、赖氨酸等所成的盐。
反过来说,所述碱加成盐形式可通过用适当的酸处理而转化成游离酸形式。
术语“盐”也包括本发明化合物可以形成的水合物及溶剂加成物形式。这种形式的实例是例如水合物、醇化物等。
本发明化合物的N-氧化物形式指包括其中一或数个氮原子被氧化成所谓的N-氧化物的式(I)化合物。
本发明化合物也可存在互变异构形式。这些形式虽然没有在上式中明确指出,但是也打算包括在本发明的范围内。
在本文中使用的术语本发明化合物的立体化学异构形式,定义为通过由相同顺序的键连接相同原子构成的、但是有无法互变的不同三维空间结构的本发明化合物可具有的全部可能的化合物。除非另外提到或指出,化合物的化学名称包括该化合物可有的全部可能的立体化学异构形式的混合物。该混合物可含该化合物的基本分子结构的全部的非对映异构体及/或对映异构体。本发明化合物的全部立体化学异构形式,不论是纯的形式或彼此的混合物,都包括在本发明的范围内。
本文提到的化合物及中间体的纯的立体异构形式定义为基本上不含该化合物或中间体的相同基本分子结构的其它对映异构体或非对映异构体形式的异构体。具体地说,术语“立体异构体纯的”是指具有立体异构过量至少80%(也就是最少90%的一种异构物及最多10%的其他可能的异构物)至高达立体异构过量100%(也就是100%的一种异构物及没有其他异构物)的化合物或中间体,更尤其是具有立体异构过量90%至高达100%,甚至更尤其是具有立体异构过量94%至高达100%,且最尤其是具有立体异构过量97%至高达100%的化合物或中间体。术语“对映异构体纯的”及“非对映异构体纯的”应以相同方式理解,但是分别指混合物的对映异构体过量及非对映异构体过量。
本发明化合物及中间体的纯的立体异构形式可通过应用本领域中已知的方法获得。例如,对映异构体可通过用光学活性酸或碱将其非对映异构体盐选择性结晶而彼此分离。其实例为酒石酸、二苯甲酰基酒石酸、二甲苯甲酰基酒石酸和樟脑磺酸。或者是,通过使用手性固定相的层析技术将对映异构体分离。所述纯的立体化学异构形式也可由对应的纯立体化学异构形式的适当的起始原料衍生制备,条件是反应是在立体特异性下进行。如果需要特异性立体异构体时,该化合物优选通过立体特异性的制备方法合成。这些方法将适宜使用对映异构体纯的起始原料。
式(I)的非对映异构体外消旋物可通过常规方法分离获得。可适宜使用的适当的物理分离方法是,例如选择性结晶法及层析法,例如柱层析法。
本领域的专业人员清楚了解式(I)化合物含有至少一个不对称中心,因此可存在不同的立体异构形式。在下图中,将该不对称中心用星号(*)表示。苯并异噁唑环的原子编号也在下面标明。

可存在于式(I)化合物中的各不对称中心的绝对构型可用立体化学描述符号R及S表示,此种R及S符号对应于在Pure Appl.Chem.1976,45,11-30中描述的规则。用星号(*)标示的碳原子优选具有R构型。
本发明也打算包括出现在本发明化合物中的原子的全部同位素。同位素包括具有相同原子数但是不同质量数的那些原子。例如(但不作限制),氢的同位素包括氚及氘。碳的同位素包括C-13及C-14。
当在下文中任何时候使用时,术语“式(I)化合物”或“本发明化合物”或类似的术语均包括通式(I)的化合物、其N-氧化物、盐、立体异构形式、外消旋混合物、药物前体、酯及代谢物,以及它们的季氮类系物。其一个重要的子集为式(I)的化合物,或其任何亚组、其N-氧化物、盐和立体异构形式。
一组特别的化合物是那些式(I)的化合物或其任何亚组的化合物,其中R1为芳基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2、Het2C1-6烷基;特别是R1为芳基、Het1、Het2、Het2C1-6烷基;更特别是R1为(i)含5至8个环成员的饱和单环或二环杂环,其中一个或两个环成员为氧原子,(ii)任选被一或多个独立选自以下的取代基取代的苯环:C1-6烷基、氨基C1-6烷基、一-或二(C1-6烷基)氨基-C1-6烷基、氨基、一-或二(C1-6烷基)氨基、多卤代C1-6烷基,(iii)具有5至6个环成员的芳族单环杂环,其含有一或两个各自独立选自氮、氧或硫的杂原子环成员,且其在一个或多个碳原子上任选被C1-6烷基、氨基C1-6烷基、一-或二(C1-6烷基)氨基-C1-6烷基、氨基、一-或二(C1-6烷基)氨基取代,(iv)通过C1-6烷基连接于变量L上的如在(iii)中定义的芳族单环杂环。
另一组特别的化合物是那些式(I)的化合物或其任何亚组的化合物,其中L与其连接的氮原子一起形成-O-C(=O)-NH-、-C(=O)-NH-、-O-C1-6链烷二基-C(=O)-NH-、-NR8-C1-6链烷二基-C(=O)-NH-;特别是-O-C(=O)-NH-、-C(=O)-NH-、-O-CH2-C(=O)-NH-、-NH-CH2-C(=O)-NH-。
另一组特别的化合物是那些式(I)的化合物或其任何亚组的化合物,其中R2是氢。
另一组特别的化合物是那些式(I)的化合物或其任何亚组的化合物,其中R3为芳基C1-4烷基,特别是芳基甲基;更特别是苯基甲基。
另一组特别的化合物是那些式(I)的化合物或其任何亚组的化合物,其中R4是C3-7环烷基、C2-6链烯基、C2-6炔基或任选被C3-7环烷基取代的C1-6烷基,特别是R4为C1-6烷基;更特别是R4为异丁基。
另一组特别的化合物是那些式(I)的化合物或其任何亚组的化合物,其中R12是-NH2或-N(R5)(A-R6),其中R5是氢或C1-6烷基,A是C1-6链烷二基和R6是氢或Het1,或R5和A-R6与它们连接的氮原子结合在一起形成Het1。
另一组特别的化合物是那些式(I)的化合物或其任何亚组的化合物,其中所述磺酰胺基团在下述的5-位连接于苯并异噁唑基团。

特别重要的是这样的式(I)化合物,其中在一个或多个上文直接提及的特殊的组中所限定的各变量的定义被组合,例如
(i)一组式(I)的化合物,其中R2是氢,R3为芳基C1-4烷基,R4是C3-7环烷基、C2-6链烯基、C2-6炔基或任选被C3-7环烷基取代的C1-6烷基;或
(ii)一组式(I)的化合物,其中R2是氢,R3为芳基C1-4烷基,R4是C3-7环烷基、C2-6链烯基、C2-6炔基或任选被C3-7环烷基取代的C1-6烷基,并且所述磺酰胺基团在5-位连接于苯并异噁唑基团;
(iii)一组式(I)的化合物,其中R1为芳基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2、Het2C1-6烷基,L与其连接的氮原子一起形成-O-C(=O)-NH-、-C(=O)-NH-、-O-C1-6链烷二基-C(=O)-NH-、-NR8-C1-6链烷二基-C(=O)-NH-;或
(iv)一组式(I)的化合物,其中R1为芳基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2、Het2C1-6烷基,L与其连接的氮原子一起形成-O-C(=O)-NH-、-C(=O)-NH-、-O-C1-6链烷二基-C(=O)-NH-、-NR8-C1-6链烷二基-C(=O)-NH-;且所述磺酰胺基团在5-位连接于苯并异噁唑基团;或
(v)一组式(I)的化合物,其中R12是-NH2或-N(R5)(A-R6),其中R5是氢或C1-6烷基,A是C1-6链烷二基和R6是氢或Het1,或R5和A-R6与它们连接的氮原子结合在一起形成Het1;或
(vi)一组式(I)的化合物,其中R12是-NH2或-N(R5)(A-R6),其中R5是氢或C1-6烷基,A是C1-6链烷二基和R6是氢或Het1,或R5和A-R6与它们连接的氮原子结合在一起形成Het1;且所述磺酰胺基团在5-位连接于苯并异噁唑基团;或
(vii)一组式(I)的化合物,其中R1为芳基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2、Het2C1-6烷基,L与其连接的氮原子一起形成-O-C(=O)-NH-、-C(=O)-NH-、-O-C1-6链烷二基-C(=O)-NH-、-NR8-C1-6链烷二基-C(=O)-NH-;R2是氢,R3为芳基C1-4烷基,R4是C3-7环烷基、C2-6链烯基、C2-6炔基或任选被C3-7环烷基取代的C1-6烷基;或
(viii)一组式(I)的化合物,其中R1为芳基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2、Het2C1-6烷基,L与其连接的氮原子一起形成-O-C(=O)-NH-、-C(=O)-NH-、-O-C1-6链烷二基-C(=O)-NH-、-NR8-C1-6链烷二基-C(=O)-NH-;R2是氢,R3为芳基C1-4烷基,R4是C3-7环烷基、C2-6链烯基、C2-6炔基或任选被C3-7环烷基取代的C1-6烷基;R12是-NH2或-N(R5)(A-R6),其中R5是氢或C1-6烷基,A是C1-6链烷二基和R6是氢或Het1,或R5和A-R6与它们连接的氮原子结合在一起形成Het1;且所述磺酰胺基团在5-位连接于苯并异噁唑基团;或
(ix)任何其它的可能组合。
重要的化合物为下列化合物、它们的N-氧化物、盐和立体异构形式:
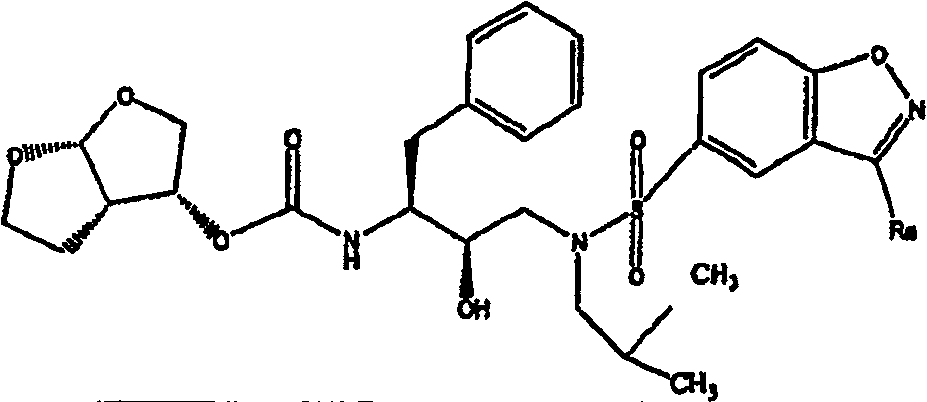
1 {3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-氨基甲酸六氢-呋喃并[2,3-b]呋喃-3-基酯;
2 3-氨基-N-{3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-2-甲基-苯甲酰胺;
3 N-{3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-2-(2,6-二甲基-苯氧基)-乙酰胺;
4 {3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-氨基甲酸四氢-呋喃-3-基酯;
5 5-甲基-异噁唑-4-甲酸{3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-酰胺;
6 {3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-氨基甲酸噻唑-5-基甲酯;
7 N-{3-[(3-氨基-苯并[d]异噁唑-6-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-2-(2,6-二甲基-苯基氨基)-乙酰胺;
8 {1-苄基-2-羟基-3-[异丁基-(3-甲基氨基-苯并[d]异噁唑-5-磺酰基)-氨基]-丙基}-氨基甲酸六氢-呋喃并[2,3-b]呋喃-3-基酯;
9 {1-苄基-3-[(3-二甲基氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-2-羟丙基}-氨基甲酸六氢-呋喃并[2,3-b]呋喃-3-基酯;
10 {1-苄基-2-羟基-3-[异丁基-(3-吡咯烷-1-基-苯并[d]异噁唑-5-磺酰基)-氨基]-丙基}-氨基甲酸六氢-呋喃并[2,3-b]呋喃-3-基酯;
11 (1-苄基-2-羟基-3-{异丁基-[3-(2-吡咯烷-1-基-乙基氨基)-苯并[d]异噁唑-5-磺酰基]-氨基}-丙基)-氨基甲酸六氢-呋喃并[2,3-b]呋喃-3-基酯。
一组特别的化合物是那些式(I)的化合物,其中
R1为氢、Het1、Het2、芳基、Het1C1-6烷基、Het2C1-6烷基、芳基C1-6烷基;更特别是R1为氢、具有5至8个环成员的单环或二环Het1,Het1含有一或多个各自独立选自氮、氧或硫的杂原子环成员,苯基、具有5至6个环成员的单环Het2,Het2含有一或多个各自独立选自氮、氧或硫的杂原子环成员,或Het2C1-6烷基,其中Het2为具有5至6个环成员的单环,其含有一或多个各自独立选自氮、氧或硫的杂原子环成员;
R2为氢;
L是-C(=O)-、-O-C(=O)-、-O-C1-6链烷二基-C(=O)-,更特别是L为-C(=O)-、-O-C(=O)-、-O-CH2-C(=O)-,其中在每种情况下,C(=O)基团连接于NR2部分;
R3为芳基C1-4烷基,特别是芳基甲基,更特别是苯基甲基;
R4是任选取代的C1-6烷基,特别是被芳基、Het1、Het2、C3-7环烷基或任选一-或二取代的氨基任选取代的C1-6烷基,其中所述取代基各自独立选自C1-4烷基、芳基、Het1和Het2;
R12是-NH2。
一组特别的化合物是那些式(I)的化合物,其中
R2为氢;
L是-C(=O)-、-O-C(=O)-、-O-CH2-C(=O)-,其中在每种情况下,C(=O)基团连接于NR2部分;
R3为苯基甲基;和
R4是C1-6烷基;和
R12是-NH2。
另一组特别的化合物是那些式(I)的化合物,其中
R2为氢;
L是-C(=O)-、-O-C(=O)-、-O-CH2-C(=O)-,其中在每种情况下,C(=O)基团连接于NR2部分;
R3为苯基甲基;
R4是C1-6烷基;
R5为氢;和
-A-R6是C1-6烷基。
另一组特别的化合物是那些式(I)的化合物,其中L是-O-C1-6链烷二基-C(=O)-。
一组特别的化合物是那些式(I)的化合物,其中R1-L是Het1-O-C(=O)-、Het2-C1-6链烷二基-O-C(=O)、芳基-O-C1-6链烷二基-C(=O)或芳基-C(=O)。
特别重要的是那些式(I)的化合物,其中R1是氢、C1-6烷基、C2-6链烯基、芳基C1-6烷基、C3-7环烷基、C3-7环烷基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2、Het2C1-6烷基,特别是R1是氢、C1-6烷基、C2-6链烯基、芳基C1-6烷基、C3-7环烷基、C3-7环烷基C1-6烷基、芳基、Het2、Het2C1-6烷基。
一组重要的化合物是那些式(I)的化合物,其中R1是氢、C1-6烷基、C2-6链烯基、芳基C1-6烷基、C3-7环烷基、C3-7环烷基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2、Het2C1-6烷基;其中Het1是具有5至6个环成员的饱和或部份不饱和单环杂环,它含一或多个选自氮、氧或硫的杂原子环成员,且它在一或多个碳原子上任选被取代。
一组优选的化合物是其中所述磺酰胺基团在5或6位(更优选在5-位)连接于苯并异噁唑基团的化合物。
一组合适的化合物是那些式(I)的化合物,其中R1为芳基或芳基C1-6烷基,特别是R1定义的芳基部分在一个或多个环成员上被进一步取代,其中每个取代基独立选自C1-4烷基、羟基、卤素、任选一-或二取代的氨基、任选一-或二取代的氨基C1-4烷基、硝基和氰基;优选所述取代基选自甲基、乙基、氯、碘、溴、羟基和氰基,特别是所述芳基部分含有6-12个环成员,更特别是在R1的定义中的芳基部分含有6个环成员;特别是,R1为含有至少一个取代基的苯基,L选自-C(=O)-、-O-C1-6链烷二基-C(=O)-,R12为-NH2。
一组合适的化合物是那些式(I)的化合物,其中R1为Het2和Het2C1-6烷基,其中在R1的定义中的Het2含一或多个各自独立选自氮、氧或硫的杂原子;特别是在R1的定义中的Het2在一个或多个环成员上被进一步取代,其中每个取代基独立选自C1-4烷基、羟基、卤素、任选一-或二取代的氨基和氰基;优选所述取代基选自甲基、乙基、氯、碘、溴、羟基、氨基和氰基。
另一组化合物是那些式(I)的化合物,其中R1为Het2或Het2C1-6烷基,L是-C(=O)-,-O-C(=O)-、-O-C1-6链烷二基-C(=O)-,R5和R6为氢;特别是在R1的定义中的Het2部分为含有5或6个环成员的单环,它含一或多个各自独立选自氮、氧或硫的杂原子环成员,更特别是Het2部分为含有5或6个环成员的单环,它含两个或更多个各自独立选自氮、氧或硫的杂原子环成员。
一组合适的化合物是那些式(I)的化合物,其中R1为Het1或Het1C1-6烷基,其中在R1的定义中的Het1含一或多个各自独立选自氮、氧或硫的杂原子;特别是在R1的定义中的Het2部分在一个或多个环成员上被进一步取代,其中每个取代基独立选自C1-4烷基、羟基、卤素、任选一-或二取代的氨基和氰基;优选所述取代基选自甲基、乙基、氯、碘、溴、羟基、氨基和氰基。
一组合适的化合物是那些式(I)的化合物,其中R1为Het1C1-6烷基或Het1,其中在R1的定义中的Het1为具有5或6个环成员的单环,其中Het1含一或多个各自独立选自氮、氧或硫的杂原子;特别是在R1的定义中的Het1部分在一个或多个碳原子上被进一步取代,其中每个取代基独立选自C1-4烷基、羟基、卤素、任选一-或二取代的氨基和氰基;优选所述取代基选自甲基、乙基、氯、碘、溴、羟基、氨基和氰基,优选R1为含有1个杂原子的5或6个环成员的单环,L为-O-C(=O)-,且R12为-NH2。
一组合适的化合物是那些式(I)的化合物,其中R1为Het1,其中所述Het1为含有7至10个环成员的双环,其中Het1含有一或多个各自独立选自氮、氧或硫的杂原子;特别是在R1的定义中的Het1部分在一个或多个碳原子上被进一步取代,其中每个取代基独立选自C1-4烷基、羟基、卤素、任选一-或二取代的氨基和氰基;优选所述取代基选自甲基、乙基、氯、碘、溴、羟基、氨基和氰基,特别是Het1部分含有2个或更多个选自氮、氧或硫的杂原子;在一个方面,R1为含有至少一个氧杂原子的二环Het1,L选自-C(C=O)-,且R12为-NH2。
一组合适的化合物是那些式(I)的化合物,其中R1为Het1,其中所述Het1为含有5至10个环成员的饱和双环,其中Het1含有一或多个各自独立选自氮、氧或硫的杂原子;特别是在R1的定义中的Het1部分在一个或多个碳原子上被进一步取代,其中每个取代基独立选自C1-4烷基、羟基、卤素、任选一-或二取代的氨基和氰基;优选所述取代基选自甲基、乙基、氯、碘、溴、羟基、氨基和氰基;特别是Het1含有5至8个环成员;特别是Het1部分含有6至8个环成员,其中Het1含有2个或更多个选自氮、氧或硫的杂原子。
一组重要的化合物是那些式(I)的化合物,其中R1为G或G-C1-6烷基,其中G选自噻唑基、咪唑基、噁唑基、噁二唑基、二噁唑基、吡唑基、吡嗪基、咪唑啉酮基、喹啉基、异喹啉基、吲哚基、哒嗪基、吡啶基、吡咯基、吡喃基、嘧啶基、呋喃基、三唑基、四唑基、苯并呋喃基、苯并噁唑基、异噁唑基、异噻唑基、噻二唑基、噻吩基、四氢呋喃并呋喃基、四氢吡喃并呋喃基、苯并噻吩基、咔唑基、咪唑酮基、噁唑酮基、中氮茚基、三嗪基、喹喔啉基、哌啶基、哌嗪基、吗啉基、硫代吗啉基、吡嗪基、噻吩基、四氢喹啉基、四氢异喹啉基、β-咔啉基、二氧六环基、二噻烷基、氧戊环基(oxolanyl)、二氧戊环基、四氢噻吩基、四氢吡喃基、四氢吡喃基;其中G任选被苯并稠合;其中G在一个或多个环成员上任选被进一步取代;优选G选自噻唑基、咪唑基、噁唑基、噁二唑基、吡唑基、吡啶基,任选在一个或多个环成员上被取代。
在Het1的定义中的具体的杂环为含一个或两个氧原子且其余的环原子为碳原子的5至8元饱和单环或二环杂环,更特别是四氢呋喃和六氢呋喃并[2,3-b]呋喃。
在Het2的定义中的具体的杂环为含一个或两个选自氮、氧或硫的杂原子的取代的或未取代的5元芳族单环杂环,更特别是取代的或未取代的噻唑、取代的或未取代的噁唑和取代的或未取代的异噁唑。在Het2的定义中的所述杂环上的合适的取代基为C1-4烷基和NH2,更优选为C1-4烷基。
一组合适的化合物为作为盐的那些式(I)化合物,其中所述盐选自三氟乙酸盐、富马酸盐、氯代乙酸盐和甲磺酸盐;重要的盐为三氟乙酸盐。
一组重要的化合物是具有折叠耐药性的那些式(I)化合物,所述折叠耐药性根据本文描述的方法测定,与选自10、71和84位的野生型序列(如M38432,K03455,gi 327742)比较,其对抗在HIV蛋白酶中具有至少一个突变体的HIV毒株的范围在0.01-100之间,特别是选自10、71和84中的至少二个突变体存在于HIV蛋白酶中;特别是所述化合物具有0.1-100范围内,更特别是0.1-50范围内,适合在0.2-35范围内的折叠耐药性。
一组重要的化合物是选自化合物编号1-10的那些化合物。式(I)化合物通常可使用描述于WO95/06030、WO 96/22287、WO 96/28418、WO 96/28463、WO 96/28464、WO 96/28465及WO 97/18205中的类似方法制备。
制备本发明化合物的具体反应步骤描述如下。在下述的制备中,反应产物可从介质中分离出来,并且如果必需的话,可根据本领域中一般已知的方法进一步纯化,例如萃取法、结晶法、研磨法及层析法。
流程A
根据美国专利5488162描述的方法制备化合物a-2。化合物a-4根据J.Heterocyclic Chem.,26,1293-1298(1989)中概述的方法制备。于室温(RT)下,向制备a-5,a-4(2.1g,0.015mol)中加入氯代磺酸(4.1ml,0.060mol)。在惰性气氛(如氮气)下,于60℃将所述反应混合物搅拌过夜。然后将该混合物倾入冰/水中。过滤出沉淀物,在一个Büchi-装置中用甲苯干燥(2.1g,得率60%)。
根据本领域已知的反应步骤,可以使中间体a-5或a-4(其中R12为氨基)进一步反应,制备其中R12为取代的氨基的类似中间体。
可以采用类似的方法制备式a-5的中间体,其中氯代磺酰基在4、6或7位。然而,优选在苯并异噁唑基的5位上的磺酰基的取代。
流程B

流程B是制备式b-4中间体的通用步骤,以下举例说明式(I)化合物(其中R2为氢,R3为苯基甲基,R4为异丁基,R12为氨基)的制备。本领域的技术人员将能应用类似的步骤,制备式b-4的其它中间体。
于室温下,将b-1(1.5g,0.0064mol)加入到作为碱的1ml三乙胺(ET3N)(0.0075mol)中,随后加入到在100ml有机溶剂如二氯甲烷中的b-2(2.0g,0.0060mol;见流程F)中。其它合适的有机溶剂包括乙酸乙酯、四氢呋喃。将该混合物搅拌3小时,用水洗涤。分离有机层,经硫酸镁干燥,蒸发溶剂,得到3.2g b-3。于室温下,将酸如三氟乙酸(4.6ml,0.060mol)加入到在50ml有机溶剂(如二氯甲烷)中的b-3(3.2g,0.0060mol)中。于室温下搅拌该混合物3小时,然后用水洗涤。分离有机层,经硫酸镁干燥并蒸发。残留物经硅胶纯化(洗脱液:二氯甲烷/甲醇96/4),得到1.2gb-4(总得率:48%)。
可以通过本领域技术人员已知的功能替换物或其功能衍生物替代用于流程B中的试剂和溶剂。也可以调整反应条件如搅拌时间、纯化和温度,以使反应条件达到最佳。
流程C1:

对于本发明的一组重要的化合物,R1-O-可以选自
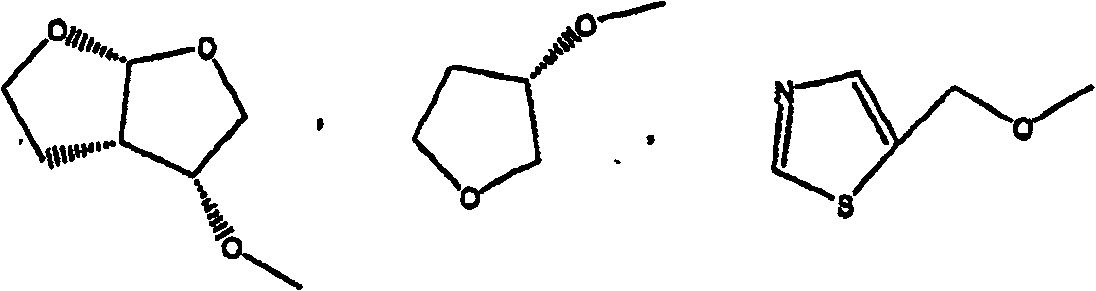
流程C-1是制备式c-3化合物的通用步骤。制备c-3的一个方法包括,在碱如三乙胺的存在下,在合适的溶剂如二氯甲烷中,使中间体c-2与式R1-L-(离去基团)c-1的中间体反应。在这个具体实例中,N-琥珀酰亚胺基用作离去基团,可以采用本领域技术人员已知的其它合适的离去基团。
本领域的技术人员将能应用类似的步骤制备式c-3的其它化合物。例如,可以通过本领域技术人员已知的功能替换物或其功能衍生物替代用于流程C1中的试剂和溶剂。也可以调整反应条件如搅拌时间、纯化和温度,以使反应条件达到最佳。
流程C2

流程C-2是制备式c-6化合物的通用步骤。制备c-6的一个方法包括,在1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)和1-羟基苯并三唑(HOBT)的存在下,在合适的溶剂如二氯甲烷中,使中间体c-5与式c-4的中间体反应。
本领域的技术人员将能应用与流程C1和C2中所述方法类似的步骤,制备式I的化合物,其中-L-R1具有不同于中间体c-1和c-4中的另外的含义。例如,流程G描述了制备中间体g-5的方法,然后可使中间体g-5进一步与式b-4中间体反应。
流程D:化合物1的合成

中间体d-1可如WO 01/25240中所述制备。
于室温下,在50ml二氯甲烷和0.18ml(1.30mmol)三乙胺中搅拌500mg d-2。然后加入d-1,于室温下搅拌该混合物过夜。用碳酸氢钠溶液洗涤该混合物,分离有机层,经硫酸镁干燥,过滤并蒸发溶剂。经硅胶纯化(二氯甲烷/甲醇98/2),得到300mg化合物1(得率45%)。
流程E:化合物6的合成

于室温下,在50ml二氯甲烷中搅拌176mg(1.38mmol)5-甲基异噁唑-4-甲酸(e-1);加入270mg(1.40mmol)EDC,于室温下搅拌该混合物1小时。使e-2溶于10ml二氯甲烷中,然后滴加到该混合物中,于室温下搅拌过夜,然后用水洗涤。分离有机层,经硫酸镁干燥,过滤并蒸发。经硅胶纯化(二氯甲烷/甲醇98/2),得到120mg化合物6(得率38%)。
流程F

中间体f-2,相应于流程B中的中间体b-2,可以通过将式H2N-R4的胺加入到在合适的溶剂如异丙醇中的中间体f-1中制备。
在流程F中,当f-1为对映体纯时,可以获得式f-2的对映体纯的化合物。如果f-1是立体异构体的混合物,则f-2应该也由立体异构体的混合物组成。
流程G
于0℃、氮气氛下,搅拌1g甲醇钠和10ml甲苯的混合物。滴加入1.9g氯代乙酸甲酯(g-1)和1.1g甲酸甲酯的混合物,同时维持温度在5-10℃之间。于0℃搅拌该混合物2小时。用水洗涤后,干燥有机层,减压蒸发得到2-氯-3-氧代-丙酸甲酯(g-2)。
将2.4g的g-2、20ml水和1.75g硫脲的混合物回流2小时。使该混合物冷却至室温,加入0.25g norit并过滤。加入2.5N氢氧化钠至滤液中,直至为中性pH。过滤得到1.23g(44%)2-氨基噻唑-5-甲酸甲酯(g-3)。将2.15g亚硝酸异戊酯及10ml二氧六环的混合物在80℃及氮气氛下搅拌。逐滴加入1.23g g-3在20ml二氧六环中的溶液。将混合物回流2小时。冷却至室温后,加入30ml乙酸乙酯。将混合物用盐水清洗并干燥,在减压下蒸发溶剂,将粗产物在硅胶上纯化,得到0.54g(48%)的噻唑-5-甲酸甲酯(g-4)。
将0.54g g-4及10ml四氢呋喃(THF)的混合物在0℃及氮气氛下搅拌。逐滴加入0.16g氢化铝锂及5ml乙醚的混合物。在0℃1小时后,加入水及20%氢氧化钠,并搅拌30分钟。将混合物通过decalite过滤,然后通过与甲苯共沸蒸馏将溶剂去除,得到0.3g(69%)的噻唑-5-基甲醇(g-5)。
流程H
流程H描述了制备乙酰胺取代的苯并异噁唑的具体方法。在碱如三乙胺的存在下,在溶剂如1,4-二氧六环中,可以使根据如上所述的类似方法制备的中间体h-1与氯代乙酰氯或功能类似物反应,以得到式h-2的酰胺。所述中间体h-2可以进一步与式NRaRb反应,其中Ra和Rb按在变量R12的氨基上的可能取代基的定义。
用于前述制备中的一些中间体和原料是已知的化合物,而其它的可根据本领域已知的制备所述或类似的化合物的方法制备。
本发明的化合物也可以根据流程J中所述的方法制备。
流程J

可使苯并异噁唑衍生物j-1与氯代磺酸反应,随后与亚硫酰氯反应,得到中间体j-2。所述中间体j-2可以与中间体j-3进一步反应,得到中间体j-4,其中PG指合适的保护基团如叔丁氧基羰基。所述反应可以在合适的溶剂(如2-甲基-四氢呋喃)中,任选在合适的碱(如三乙胺)的存在下进行。
然后,在合适的溶剂(如在乙醇中的2-甲基四氢呋喃)的存在下,可以使中间体j-4与合适的试剂如间-氯过氧苯甲酸(mCPBA)或一过氧邻苯二甲酸(phtalate)镁六水合物(MMPP)反应,由此得到中间体j-5和j-6。
中间体j-5和j-6可以进一步由式HN(R5)A-R6衍生,并经去保护反应后得到中间体j-7。然后,在碱如三乙胺的存在下,并任选在EDC或醇如叔丁醇的存在下,在合适的溶剂如二氯甲烷中,使中间体j-7与式R1-L-(离去基团)的中间体反应,由此获得为式(I)化合物的化合物j-8。
按照本领域已知的用于将三价氮转化为其N-氧化物形式的方法,也可以将式(I)化合物转化为相应的N-氧化物形式。进行该N-氧化反应通常是通过使式(I)的起始原料与适当的有机或无机过氧化物反应。合适的无机过氧化物包括例如过氧化氢、碱金属或碱土金属过氧化物,例如过氧化钠、过氧化钾;合适的有机过氧化物可包括过氧酸例如苯羰基过氧酸(benzenecarboperoxoic acid)或卤基取代的苯羰基过氧酸例如3-氯-苯羰基过氧酸、过氧烷酸例如过氧乙酸、烷基过氧化氢例如叔丁基过氧化氢。合适的溶剂为例如水、低级链烷醇例如乙醇等、烃例如甲苯、酮例如2-丁酮、卤代烃例如二氯甲烷及这些溶剂的混合物。
式(I)化合物或其任何亚组作为逆转录病毒的蛋白酶(如HIV蛋白酶)的抑制剂是有活性的。特别是,式(I)化合物或其亚组对抑制突变的HIV蛋白酶,更特别是抑制耐药性突变株HIV蛋白酶是有活性的。
HIV蛋白酶对药物的“敏感性”或者说是“耐药性”的标准可经市售获得的HIV蛋白酶抑制剂建立。如上所述,现有的可市售获得的HIV蛋白酶抑制剂对抗患者中的HIV病毒群体的活性会随时间而降低。其理由为,在具体的HIV蛋白酶抑制剂的存在的压力下,现有的HIV病毒群体(一般主要是野生型HIV蛋白酶)可突变为不同的突变株,这些突变株对相同的HIV蛋白酶抑制剂很不敏感。如果出现此种现象,人们一般会称之耐药突变株。如果这些突变株不仅对特定的HIV蛋白酶抑制剂产生抗性,而且对其它的可市售获得的多种HIV蛋白酶抑制剂也具有抗性,则人们会认为其为耐多重药物的HIV蛋白酶。表示突变株对一种具体的HIV蛋白酶抑制剂耐药的一种方式是测定所述HIV蛋白酶抑制剂对抗突变株HIV蛋白酶的EC50与所述HIV蛋白酶抑制剂对抗野生型HIV蛋白酶的EC50的比率。所述比率也称为折叠耐药性(DR)。
临床出现的许多突变株均具有抑制可市售获得的HIV蛋白酶抑制剂(如沙奎那韦、茚地那韦、利托那韦和奈非那韦)的100或100以上的折叠耐药性。HIV蛋白酶的临床有关的突变株可以以密码子位置10、71和/或84的突变为特征。此类临床有关的突变株HIV蛋白酶的实例列于下表2中。
本发明化合物或其任何亚组对至少一种临床有关的突变株HIV蛋白酶和对在几种情况下广谱的临床有关的突变株HIV蛋白酶的呈现出折叠耐药性的范围在0.01和100之间。一组特殊的式(I)化合物为对至少一种突变株HIV蛋白酶呈现的折叠耐药性的范围在0.1和100之间,适合在0.1和50之间,更适合在0.1和30之间的那些式(I)化合物。对至少一种突变株HIV蛋白酶呈现的折叠耐药性的范围在0.1和20之间的式(I)化合物具有特别的重要性,甚至更重要的是对至少一种突变株HIV蛋白酶呈现的折叠耐药性的范围在0.1和10之间的那些式(I)化合物。
由于它们的有利的药学性质,尤其是它们对抗广谱突变株蛋白酶,如突变株HIV蛋白酶的活性,本发明化合物可用于治疗HIV感染的个体及用于这些个体的预防。通常,本发明化合物可用于治疗感染病毒的温血动物,该病毒的存在通过蛋白酶的介导或依赖于蛋白酶。用本发明化合物可以预防或治疗的疾病,尤其是与HIV及其它病原性逆转录病毒相关的疾病,包括AIDS、AIDS相关复症(ARC)、进行性泛发性淋巴结病(PGL)以及逆转录病毒引起的慢性CNS疾病,例如HIV介导的痴呆及多发性硬化病。
因此,本发明化合物或任何其亚组可作为药物用于对抗上述疾病。所述作为药物的用途或治疗方法包括将有效量的本发明化合物系统给予感染逆病毒的哺乳动物(特别是感染HIV的哺乳动物)以对抗与HIV及其它致病的逆转录病毒(尤其是HIV-1)。因此,本发明化合物可用于制备用于治疗与HIV及其他致病的逆转录病毒相关的疾病的药物,尤其是用于治疗感染耐药的或其它突变HIV病毒的患者的药物。
在一个优选的实施方案中,本发明涉及式(I)化合物或任何其亚组在制备用于在哺乳动物中治疗或对抗对多种药物有耐药性的逆转录病毒感染相关的感染或疾病(尤其是HIV-1感染)的药物中的用途。因此,本发明也涉及一种治疗与对多种药物有耐药性的逆转录病毒感染相关疾病的方法,该方法包括将有效量的式(I)化合物或其亚组给予有需要的哺乳动物。
在另一个优选的实施方案中,本发明涉及式(I)化合物或任何其亚组在制备用于在感染逆转录病毒(特别是HIV-1逆转录病毒)的哺乳动物中抑制逆转录病毒蛋白酶(包括突变株蛋白酶、耐药性突变蛋白酶和对多种药物耐药的突变蛋白酶)的药物中的用途。
在另一个优选的实施方案中,本发明涉及式(I)化合物或其任何亚组在制备用于抑制逆转录病毒的复制,包括突变株逆转录病毒的复制,耐药的突变株逆转录病毒耐药株的复制和对多种药物耐药的突变株逆转录病毒的复制(特别是HIV-1复制)的药物中的用途。
也发现本发明化合物可用于在体外抑制含HIV的样本或预期暴露至HIV的样本,因此,本发明化合物可用于抑制存在于含有或怀疑含有或暴露至HIV的体液样本中的HIV。
抗逆转录病毒化合物与本发明化合物的组合也可作为药物使用。因此,本发明也涉及含(a)本发明化合物及(b)其它抗逆转录病毒化合物的产品,其作为联合制剂供同时、分开或依序使用以治疗逆转录病毒感染,尤其是治疗对多种药物有耐药性的逆转录病毒的感染。因此,为了对抗或治疗HIV感染,或与HIV感染相关的感染及疾病,例如获得性免疫缺陷综合征(AIDS)或AIDS相关复症(ARC),可将本发明化合物与下列药物组合以联合用药,例如结合抑制剂,例如葡聚糖硫酸盐、苏拉明、聚阴离子、可溶性CD4、PRO-542、BMS-806;融合抑制剂例如T20、T1249、5-螺旋化合物(5-helix)、D-肽ADS-J1;共同受体结合抑制剂,例如AMD3100、AMD-3465、AMD7049、AMD3451(Bicyclams)、TAK 779;SHC-C(SCH351125)、SHC-D、PRO-140RT抑制剂如膦甲酸(foscarnet)及药物前体;核苷RTIs,例如AZT、3TC、DDC、DDI、D4T、阿巴克(Abacavir)、FTC、DAPD、dOTC、DPC817;核苷酸RTIs,例如PMEA、PMPA(替诺福韦);NNRTIs,例如奈韦拉平、地拉韦啶、依法韦仑、8和9-Cl TIBO(替韦拉平)、洛韦胺、TMC-125、TMC-120、(dapivirine)、MKC-442、UC781、UC782、卡普韦林、DPC961、DPC963、DPC082、DPC083、calanolide A(红厚壳属植物提取物)、SJ-1366、TSAO、4”-去氨化TSAO、MV150、MV026048;RNAseH抑制剂,例如SP1093V、PD126338;TAT抑制剂,例如RO-5-3335、K12、K37;整合酶抑制剂,例如L708906、L731988;S-1360;蛋白酶抑制剂,例如氨普奈韦及前药GW908、利托那韦、奈非那韦、沙奎那韦、茚地那韦、洛匹那韦、帕利那韦、BMS186316、atazanavir、DPC681、DPC684、替拉那韦、AG1776、mozenavir、GS3333、KNI-413、KNI-272、L754394、L756425、LG-71350、PD161374、PD173606、PD 177298、PD178390、PD178392、PNU 140135、TMC114、山楂酸、U-140690;糖基化抑制剂,例如粟精胺、脱氧吉瑞霉素。
此组合在部份情形下可提供协同效应,因而可预防、基本上减轻或完全消除病毒感染度及其相关的症状。
本发明化合物也可与免疫调节剂(例如溴匹立明、抗人类α干扰素抗体、IL-2、甲硫氨酸脑啡肽、干扰素α及纳曲酮)、抗生素(例如依西酸喷他脒)、细胞因子(例如Th2)、细胞因子调节剂、趋化因子或其受体(例如CCR5)或激素(例如生长激素)联合用药,以减轻、对抗或消除HIV感染及其症状。这种在不同的制剂中的联合治疗可同时、顺序或彼此独立用药。或者,这种联合可作为单一制剂用药,其中活性成份是同时或分别从制剂中释出。
本发明化合物也可通过与代谢调节剂联合,然后一起将该药物给予患者进行给药。这些调节剂包括干扰细胞色素(例如细胞色素P450)代谢的调节剂。部份调节剂可抑制细胞色素P450。现已知数种同功酶存在于细胞色素P450中,其中一种是细胞色素P4503A4。利托那韦是一种通过细胞色素P450的代谢调节剂的实例。在不同的制剂中的这种联合治疗可同时、顺序或彼此独立用药。或者,这种联合疗法可作为单一制剂用药,其中活性成份是同时或分开从制剂中释出。这种调节剂可在与本发明化合物的相同或不同比例下用药。优选这种调节剂与本发明化合物的重量比(调节剂:本发明化合物)是1∶1或更低,更优选该比例是1∶3或更低,该比例合适是1∶10或更低,该比例是更适合是1∶30或更低。
因此,本发明化合物可以其本身作为药物或与其它药物的混合物或以药物制剂的形式用于动物,最好是哺乳动物中,特别是用于人类中。
因此,本发明涉及药物制剂,它含有有效剂量的至少一种式(I)化合物作为活性成份,加上常用的药学上无害的赋形剂及辅剂。所述药物制剂通常含0.1至90%(重量)的式(I)化合物。此药物制剂可用本领域的专业人员已知的方法制备。为此目的,将至少一种式(I)化合物以及一或多种固体或液体药用赋形剂和/或辅剂以及(如果需要)与其他药用活性化合物结合配制成合适的用药形式或剂型,它可随后作为药物用在人类用药或兽药中。
含根据本发明化合物的药物可经口服、胃肠道外(例如静脉内)、直肠、吸入或局部进行给药,较佳的用药由个别情形决定,例如所治疗疾病的具体情况。优选口服用药。
本领域的专业人员根据其基本专业知识可熟悉适用于所需药用制剂的辅剂。除了溶剂、凝胶形成剂、栓剂基质、片剂辅剂及其它活性化合物载体以外,也可使用抗氧化剂、分散剂、乳化剂、抗泡沫剂、矫味剂、防腐剂、增溶剂、达到储存效应的试剂、缓冲物质或着色剂。
对于口服用药形式,可将本发明化合物与适当的添加剂,例如赋形剂、稳定剂或惰性稀释剂混合,然后通过常用的方法制成合适的用药形式,例如片剂、包衣片剂、硬胶囊剂、水性、醇性或油性溶液剂。合适的惰性载体的实例是阿拉伯胶、氧化镁、碳酸镁、磷酸钾、乳糖、葡萄糖或淀粉尤其是玉米淀粉。在此情形下,可以以干燥及湿润颗粒形式进行制备。合适的油性赋形剂或溶剂是植物油或动物油,例如葵花油或鱼肝油。水性或醇性溶液的合适溶剂是水、乙醇、糖溶液或其混合物。聚乙二醇及聚丙二醇也可用作其它给药形式的辅剂。
对于皮下或静脉内用药,可将活性化合物,如果需要也可与常用物质例如助溶剂、乳化剂或其它辅剂一起,制成溶液剂、悬浮液或乳剂。也可将式(I)化合物冷冻干燥,并使用所得的冷冻干燥物制造注射或输注的制剂。合适的溶剂是例如水、生理盐水溶液或醇例如乙醇、丙醇、甘油,以及糖溶液例如葡萄糖或甘露醇溶液,或者是上述不同溶剂的混合物。
气溶胶或喷雾剂形式用药的合适药物制剂是例如式(I)化合物或其生理上可耐受的盐在药学上可接受的溶剂例如乙醇或水或这些溶剂的混合物中的溶液剂、悬浮液或乳剂。如果需要时,所述制剂中也可另外含其它药用辅剂,例如表面活性剂、乳化剂及稳定剂以及抛射剂。这种制剂通常含浓度从约0.1至50%,尤其是从约0.3至3%重量的活性化合物。
为了增加药用组合物中式(I)化合物的溶解度和/或稳定性,适于应用α-、β-或γ-环糊精或其衍生物。助溶剂(例如醇类)也可改进药用组合物中式(I)化合物的溶解度和/或稳定性。在制备水性组合物时,明显地更适于加入目标化合物的加成盐,原因是其水溶性增加。
适当的环糊精是α-、β-或γ-环糊精(CDs)或其醚及其混合的醚,其中环糊精无水葡萄糖单元的一或多个羟基被下列取代基取代:C1-6烷基,尤其是甲基、乙基或异丙基,例如无规甲基化的β-CD;羟基C1-6烷基,尤其是羟基乙基、羟基丙基或羟基丁基;羧基C1-6烷基,尤其是羧基甲基或羧基乙基;C1-6烷基-羰基,尤其是乙酰基;C1-6烷氧基羰基C1-6烷基或羧基C1-6烷氧基C1-6烷基,尤其是羧基甲氧基丙基或羧基乙氧基丙基;C1-6烷基羰基氧基C1-6烷基,尤其是2-乙酰氧基丙基。特别值得注意的作为络合剂和/或助溶剂的有β-CD、无规甲基化的β-CD、2,6-二甲基-β-CD、2-羟基乙基-β-CD、2-羟基乙基-γ-CD、2-羟基丙基-γ-CD及(2-羧基甲氧基)丙基-β-CD,且特别是2-羟基丙基-β-CD(2-HP-β-CD)。
术语混合的醚是指环糊精衍生物,其中至少两个环糊精羟基被不同的基团例如羟基丙基及羟基乙基醚化。
配制本发明化合物结合环糊精或其衍生物的一种重要方法已描述于EP-A-721,331中。虽然其中描述的制剂含有抗真菌活性成份,但它们同样也适于配制本发明的化合物。其中描述的制剂特别适于口服用药,其含作为活性成份的抗真菌剂、作为助溶剂的足量的环糊精或其衍生物、作为整个液体载体的水性酸性介质以及大幅简化组合物的制备的醇性助溶剂。所述制剂也可通过加入药学上可接受的甜味剂和/或矫味剂而使得更加可口。
增加药用组合物中本发明化合物的溶解度的其它常用方法描述于WO 94/05263、WO 98/42318、EP-A-499,299及WO 97/44014中,全都专利结合到本文中以供参考。
更确定地说,可将本发明化合物配制成药用组合物,其含有效治疗量的颗粒,该颗粒是由含(a)式(I)化合物及(b)一或多种药学上可接受的水溶性聚合物的固体悬浮液组成。
术语“固体分散体”是指含有至少两种成份的固体状态的系统(异于液体或气体状态),其中一种成份大概平均分散在另一种或多种成份中。当该成份的分散体是使此系统是化学及物理性一致或均匀分布或由热动力学定义的一个相组成时,此固态分散体被称为“固体溶液”。固体溶液是优选的物理系统,因为其中的成份通常容易被用药的有机体生物利用。
术语“固态分散体”也包括比固体溶液较不均匀分布的分散物。这种分散物不能化学或物理上均匀分布或含一个以上的相。
所述颗粒中的水溶性聚合物适合是当溶解于2%水溶液时,在20℃溶液中的表观粘度是1至100mPa.s的聚合物。
较佳的水溶性聚合物是羟基丙基甲基纤维素或HPMC,甲氧基取代程度从约0.8至约2.5且羟基丙基摩尔取代从约0.05至约3.0的HPMC通常具有水溶性。甲氧基取代程度是指纤维素分子每个无水葡萄糖单元存在的甲基醚基团的平均数。羟基丙基摩尔取代是指与纤维素分子各无水葡萄糖单元反应的环氧丙烷的平均摩尔数。
上文定义的颗粒可通过先制备各成份固体的分散物,随后任选粉碎或研磨该分散物制备。现存在可用于制备固态分散体的多种技术,包括熔化-挤压、喷雾-干燥及溶液-蒸发,优选熔化-挤压。
还可将本发明化合物方便地配制成为纳米颗粒的形式,其有足够量的表面改性剂吸附在其表面上以保持有效平均粒子大小低于1000纳米。现认为有用的表面改性剂包括那些能物理性吸附在抗逆转录病毒药物的表面但却没有化学性键接至抗逆转录病毒药物上的改性剂。
合适的表面改性剂可优选选自已知的有机及无机药用赋形剂。此类赋形剂包括多种聚合物、低分子量寡聚物、天然产物及表面活性剂。优选的表面改性剂包括非离子性及阴离子性表面活性剂。
配制本发明化合物的另一个重要方法涉及一种药用组合物,其中将本发明化合物掺混在亲水性聚合物中,然后将此混合物作为包衣薄膜包被在许多小珠粒上,因而得到有良好生物利用度的组合物,该组合物可方便地制备且适于制备供口服用药的药用剂型。
所述珠粒包括(a)中间圆形或球形的核心、(b)亲水性聚合物及抗逆转录病毒药物的包衣薄膜及(c)密封的包衣聚合物层。
在球珠中适于作为核心的物质有多种,条件是该物质是药学上可接受的且有合适的尺寸及硬度。这种物质的实例是聚合物、无机物质、有机物质及糖类及其衍生物。
用药途径取决于患者的疾病、辅助医疗的条件等。
本发明另一方面涉及一种药剂盒或容器,其中含有效量的可在用于测定潜在的药物抑制HIV蛋白酶、HIV生长或两者的能力的测试或试验中用作标准物或试剂的式(I)化合物。本发明的此方面可发现其在药物研究计划中的用途。
在产生耐药性的疾病(例如HIV)的临床管理中,本发明化合物可用于表型耐药性监视试验,例如已知的重组试验。特别有用的耐药性监视系统是称为AntivirogramTM的重组试验法。AntivirogramTM是一种高度自动化、高物料通过量、第二代的重组试验法,它可测量对本发明化合物的敏感性,尤其是病毒对本发明化合物的敏感性(Hertogs K,de Bethune MP,Miller V等,Antimicrob Agents Chemother,1998;42(2);269-276,结合于本文供参考)。
重要地,本发明化合物可含化学反应性部分,该部分能与局限部位形成共价键,使得该化合物增加了组织的保留时间及半衰期。术语“化学反应性基团”在本文中使用时是指可形成共价键的化学基团。反应性基团通常于水性环境中是稳定的,通常是羧基、磷酰基或常见的酰基,或者是酯或者是混酐,或酰亚氨酸酯(imidate),或马来酰亚氨酸酯(maleimidate),因而可与比如血液成份如白蛋白靶位上的官能团如氨基、羟基或硫醇基形成共价键。本发明化合物可连结至马来酰亚胺或其衍生物上而形成共轭体。
本发明化合物或其生理上可耐受的盐的用药剂量是由个体病例决定的,通常是根据个体病例的病情加以调整以得到最佳的效果。因此,其当然决定于用药频率及选用的化合物在治疗或预防各个病例时的功效与作用持续时间,以及感染和症状的性质和严重性、被治疗的人或动物的性别、年龄、体重、辅助治疗及个体反应,以及此医疗是否是急性或预防性的。通常,式(I)化合物在用药至体重约75千克的患者时,每日剂量是1毫克至1克,优选3毫克至0.5克。此给药剂量可以以单一剂量的形式或分成数个例如二、三或四个单独的剂量给予。
下列各表列出了按照以上反应流程之一制备的式(I)化合物。
表1a
根据上述方法制备本发明的化合物。如果未指明立体化学,本发明化合物为外消旋混合物。


表1b
根据上述的任一种合成方法的类似方法制备以下化合物。
| No. | R<sub>n</sub> |
| 12 | -NHCH<sub>3</sub> |
| 13 | -NH(CH<sub>3</sub>)<sub>2</sub> |
| 14 | 1-吡咯烷基 |
| 15 | -NH-CH<sub>2</sub>-CH<sub>2</sub>-(1-吡咯烷基) |
抗病毒分析:
在细胞试验中检测本发明化合物的抗病毒活性,此试验证明这些化合物对于野生型实验室HIV株(HIV-1株LAI)具有强力的抗HIV活性。此细胞试验根据下列步骤进行。
细胞试验的实验方法
在不同浓度抑制剂存在下,将HIV-或模拟-感染的MT4细胞培养5天。在培养期结束后,在无任何抑制剂存在的对照组培养液中,全部感染HIV的细胞都被复制的病毒杀死。通过测量MTT的浓度测量细胞存活率,黄色水溶性四唑鎓染料只有在活细胞的线粒体中转化成紫色非水溶性甲 。当所得的甲
。当所得的甲 晶体用异丙醇溶解后,在540纳米下监测溶液的吸收度。完成5天培养后在培养液中残留的活细胞数目直接对应于吸收值。对感染病毒的细胞监测化合物的抑制活性,并以EC50及EC90表示。这些值分别代表需要从病毒的细胞致病效应中保护50%及90%细胞所需的化合物量。在模拟感染的细胞中测量化合物的毒性,并以CC50表示,CC50代表需要抑制50%细胞生长所需的化合物浓度。选择性指数(SI)(CC50/EC50比率)表示抑制剂抗HIV活性的选择性。当以例如pEC50或pCC50值报导的结果时,该结果分别以EC50或CC50表示的结果的负对数表示。
晶体用异丙醇溶解后,在540纳米下监测溶液的吸收度。完成5天培养后在培养液中残留的活细胞数目直接对应于吸收值。对感染病毒的细胞监测化合物的抑制活性,并以EC50及EC90表示。这些值分别代表需要从病毒的细胞致病效应中保护50%及90%细胞所需的化合物量。在模拟感染的细胞中测量化合物的毒性,并以CC50表示,CC50代表需要抑制50%细胞生长所需的化合物浓度。选择性指数(SI)(CC50/EC50比率)表示抑制剂抗HIV活性的选择性。当以例如pEC50或pCC50值报导的结果时,该结果分别以EC50或CC50表示的结果的负对数表示。
测试化合物的SI范围是在400至28000之间。
抗病毒谱:
因为耐药性HIV株的出现日益增多,因此测试本发明化合物对隐藏数种突变的临床分离的HIV株(表2及3)的效应。这些突变与对蛋白酶抑制剂的抗性相关,并能导致病毒对目前市售药物例如沙奎那韦、利托那韦、奈非那韦、茚地那韦及氨普奈韦显现不同程度的表型交叉耐药性。
表2.所使用的HIV株(A至F)的蛋白酶基因中存在的突变表
结果:
测定定义为FR=EC50(突变株)/EC50(HIV-1株LAI)的折叠耐药性(FR),作为本发明化合物的广谱活性测定。表3显示以折叠耐药性表示的抗病毒测试的结果。从该表中可以看出,本发明化合物可以有效地抑制广泛范围的突变株:A栏:对突变A的FR值,B栏:对突变B的FR值,C栏:对突变C的FR值,D栏:对突变D的FR值,E栏:对突变E的FR值,F栏:对突变F的FR值。毒性(Tox)是以根据用模拟转染的细胞测试的pCC50值表示。WT栏显示对野生型HIV-LAI株的pEC50值。
表3.对抗A至F毒株的毒性测试与耐药性测试的结果(以FR表示)。ND表示没有测定。结果为计算的平均值。
| N° | A | B | C | D | E | F | G | Tox | WT |
| 1 | 0.63 | 0.78 | 0.49 | 0.35 | 0.30 | 0.85 | 6.9 | <4 | 8.1 |
| 2 | 7.1 | 2.4 | 1.7 | 1.5 | 1.4 | 27 | 85 | 4.3 | 7.52 |
| 3 | 1.8 | 1.9 | 2.2 | 2.3 | 2.4 | 11 | 54 | 4.35 | 7.62 |
| 4 | 49 | ND | ND | ND | 8.9 | ND | ND | <4 | 7.84 |
| 5 | 17 | 4.8 | 3.2 | 3.5 | 3.2 | 95 | 275 | <4.49 | 8.01 |
| 6 | 16 | 5.6 | 5.5 | 16 | 7.2 | 16 | 16 | ND | 6.2 |
| 7 | 6.3 | 4.6 | 4.1 | 15 | 5.6 | 44 | 257 | 4.12 | 8.03 |
| 11 | 13 | 11 | 10 | ND | ND | ND | ND | <4 | 6.64 |
肠吸收的Caco-2渗透性试验
根据Augustijns等(Augustijns et al.(1998),Int.J.of Pharm,166,45-54)描述的Caco-2测试法评估不同化合物的渗透性,其中使细胞传代数介于32及45之间的Caco-2细胞在24孔细胞培养板中生长21至25天。通过测量经表皮电阻(TEER)检查细胞单层的完整性。在pH7.4及100μM供体化合物浓度下进行此测试。
在不同pH水平下的水溶性
热力学条件下的模拟胃肠溶液中的平衡溶解度是化合物在胃及肠内不同部位溶解特性的良好测量指标。模拟的胃液(SGF)(没有胃蛋白酶)设定在pH1.5。模拟的肠液(SIF)(没有胆汁盐)设定在pH5、pH6.5、pH7及pH7.5。实验中使用96孔平底微量培养板,其中在各孔内加入1mg化合物(在甲醇中的储备溶液),然后蒸干。将化合物再度溶解在SGF及SIF中,并在37℃于水平摇动装置上培养过夜。过滤后,用UV-分光光度计测量化合物浓度。
在大鼠及狗中的口服利用率.
主要在雄性及雌性大鼠及次要在雄性及雌性狗中,采用一套标准动力实验评估所选择的化合物的口服利用率。将化合物配制成在DMSO、PEG400或环糊精40%(CD40%)水溶液中的20毫克/毫升溶液或悬浮液。对于在大鼠中的大部分实验,形成三个给药组:1/使用所述DMSO制剂在20毫克/千克的单一腹膜内剂量;2/使用所述PEG400制剂在20毫克/千克的单一口服剂量及3/使用所述环糊精制剂在20毫克/千克的单一口服剂量,在狗中只使用口服途径用药。给药后在规则的时间间隔下取血样,然后使用LC-MS生物分析法测定血清中药物浓度。将给药浓度归一化至10mg/kg后,以ng/g表示血清浓度。以下列出在30分钟和3小时时的血清浓度,因为这些数值反映了吸收程度(30’)和消除速率(180’)。大鼠的口服生物利用度采用DMSO和PEG制剂(参见上文)测定。给予10mg/kg的化合物1后的血清浓度在30分钟时分别为10.2ng/ml(DMSO)和22.2ng/ml(PEG)。给药30分钟和180分钟后,剂量10mg/kg (DMSO制剂)的腹膜内吸收分别为2076ng/ml和208ng/ml。
促升全身生物利用度
通过使用所描述类型的化合物(蛋白酶抑制剂),现已清楚通过减少在肝脏内的首过(first-pass)代谢作用及血浆中的代谢清除,抑制代谢分解历程可明显增加全身性的生物利用度。此‘促升’原理可临床应用至药物的药理作用中。也可通过同时给予可抑制Cyt-p450代谢酶的化合物,在大鼠及狗中探讨此作用机理。已知的阻断剂是例如利托那韦及酮康唑(ketoconazole)。
蛋白结合分析
现已知人类血清蛋白例如白蛋白(HSA)或α-1酸糖蛋白(AAG)可结合多种药物,从而可能导致这些化合物的作用降低。为了确定本发明化合物是否受这种结合的不利影响,在人血清存在下测量所述化合物的抗HIV活性,从而评估所述蛋白酶抑制剂与那些蛋白结合的影响。
以0.001-0.01CCID50的感染复数(MOI)(每个细胞50%细胞培养物感染剂量,CCID50)使MT4细胞感染HIV-1LAI。培养1小时后,清洗细胞,然后置于含有在10%FCS(胎牛血清)、10%FCS+1mg/mlAAG(α1-酸糖蛋白)、10%FCS+45mg/mlHSA(人类血清白蛋白)或50%人血清(HS)的存在下的序列稀释的化合物的96孔培养板中。培养5或6天后,通过测定细胞存活率或定量HIV复制水平而计算EC50(在细胞基质试验中的50%有效浓度)。使用上述方法测量细胞存活率。在10%FCS或10%FCS+1mg/mlAAG的存在下,向含序列稀释的化合物的96孔培养板中加入HIV(野生型或耐药性株)及MT4细胞使最终浓度分别为200-250CCID50/孔及30,000细胞/孔。培养5天后(37℃,5%CO2),通过四唑鎓比色性MTT(3-[4,5-二甲基噻唑-2-基]-2,5-二苯基四唑鎓溴化物)方法(Pauwels et al.J Virol.Methods1988,20,309-321)测定细胞的存活率。
制剂
将活性成分(即式(I)化合物)溶解在有机溶剂例如乙醇、甲醇或二氯甲烷中,优选是乙醇与二氯甲烷的混合物。将通常是5mPa.s的聚合物,例如聚乙烯基吡咯烷酮与乙酸乙烯酯的共聚物(PVP-VA)或羟基丙基甲基纤维素(HPMC)溶解在有机溶剂例如乙醇、甲醇或二氯甲烷中。适于将聚合物溶解在乙醇中。将聚合物及化合物溶液混合,随后喷雾干燥。化合物/聚合物的比率从1/1至1/6内选择。中间体范围是1/1.5及1/3。合适的比例是1/6。随后将喷雾干燥的粉末固态分散物填入供用药使用的胶囊内。填入每个胶囊内的药物范围介于50至100毫克之间,由使用的胶囊大小决定。
薄膜包衣片剂
制备片芯
将100克活性成份(即式(I)化合物)、570克乳糖及200克淀粉充分混合,随后用5克十二烷基硫酸钠及10克聚乙烯基吡咯烷酮在约200毫升水中的溶液湿润。将该湿的粉末混合物过筛、干燥,然后再度过筛。然后加入100克微晶纤维素及15克氢化植物油。将全部混合均匀,然后将其压制成片剂,得到10,000片各含10毫克活性成份的片剂。
包衣
向10克甲基纤维素的75毫升变性乙醇的溶液中加入5克乙基纤维素在150毫升二氯甲烷中的溶液。然后加入75毫升二氯甲烷及2.5毫升1,2,3-丙三醇。将10克聚乙二醇熔化,然后溶解在75毫升二氯甲烷中。将后者溶液添加至前者溶液中,然后加入2.5克十八烷酸镁、5克聚乙烯基吡咯烷酮及30毫升浓缩色素悬浮液,将整个混合液均匀化。在涂膜装置中用如此所得的混合物包覆片芯。
Claims (23)
1.一种具有下式的化合物

或其盐,其中
R1及R8各自独立为氢、C1-6烷基、C2-6链烯基、芳基C1-6烷基、C3-7环烷基、C3-7环烷基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2、Het2C1-6烷基;
R1如以上定义或是下式的基团

其中
R9、R10a及R10b各自独立为氢、C1-4烷氧基羰基、羧基、氨基羰基、一-或二(C1-4烷基)氨基羰基、C3-7环烷基、C2-6链烯基、C2-6炔基或C1-4烷基,其任选被芳基、Het1、Het2、C3-7环烷基、C1-4烷氧基羰基、羧基、氨基羰基、一-或二(C1-4烷基)氨基羰基、氨基磺酰基、C1-4烷基S(O)t、羟基、氰基、卤素或任选被一-或二取代的氨基取代,其中所述氨基的取代基各自独立地选自C1-4烷基、芳基、芳基C1-4烷基、C3-7环烷基、C3-7环烷基C1-4烷基、Het1、Het2、Het1C1-4烷基及Het2C1-4烷基;或其中R9、R10a与其连接的碳原子形成C3-7环烷基;当L是-O-C1-6链烷二基-C(=O)-或-NR8-C1-6链烷二基-C(=O)-时,则R9如上定义或为氧代基;
R11a是氢、C2-6链烯基、C2-6炔基、C3-7环烷基、芳基、任选被
一-或二取代的氨基羰基、任选被一-或二取代的氨基C1-4烷基羰氧基、C1-4烷氧基羰基、芳氧基羰基、Het1氧基羰基、Het2氧基羰基、芳氧基羰基C1-4烷基、芳基C1-4烷氧基羰基、C1-4烷基羰基、C3-7环烷基羰基、C3-7环烷基C1-4烷氧基羰基、C3-7环烷基羰氧基、羧基C1-4烷基羰氧基、C1-4烷基羰氧基、芳基C1-4烷基羰氧基、芳基羰氧基、芳氧基羰氧基、Het1羰基、Het1羰氧基、Het1C1-4烷氧基羰基、Het2羰氧基、Het2C1-4烷基羰氧基、Het2C1-4烷氧基羰基氧基或C1-4烷基,其任选被芳基、芳氧基、Het2、卤素或羟基取代;其中在氨基上的取代基各自独立地选自C1-4烷基、芳基、芳基C1-4烷基、C3-7环烷基、C3-7环烷基C1-4烷基、Het1、Het2、Het1C1-4烷基及Het2C1-4烷基;
R11b是氢、C3-7环烷基、C2-6链烯基、C2-6炔基、芳基、Het1、Het2,或C1-4烷基,其任选被卤素、羟基、C1-4烷基S(=O)t、芳基、C3-7环烷基、Het1、Het2、任选被一-或二取代的氨基取代,其中所述氨基的取代基各自独立地选自C1-4烷基、芳基、芳基C1-4烷基、C3-7环烷基、C3-7环烷基C1-4烷基、Het1、Het2、Het1C1-4烷基及Het2C1-4烷基;
或其中R11b通过磺酰基连接至分子的其余部分;
t各自独立是0、1或2;
R2是氢或C1-6烷基;
L是-C(=O)-、-O-C(=O)-、-NR8-C(=O)-、-O-C1-6链烷二基-C(=O)-、-NR8-C1-6链烷二基-C(=O)-、-S(=O)2-、-O-S(=O)2-、-NR8-S(=O)2-,
其中C(=O)基团或S(=O)2基团连接至NR2部分;其中C1-6链烷二
基部分任选被选自羟基、芳基、Het1及Het2的取代基取代;
R3是C1-6烷基、芳基、C3-7环烷基、C3-7环烷基C1-4烷基或芳基C1-4烷基;
R4是氢、C1-4烷氧基羰基、羧基、氨基羰基、一-或二(C1-4烷基)氨基羰基、C3-7环烷基、C2-6链烯基、C2-6炔基,或C1-6烷基,其任选被一或多个各自独立地选自以下的取代基所取代:芳基、Het1、Het2、C3-7环烷基、C1-4烷氧基羰基、羧基、氨基羰基、一-或二(C1-4烷基)氨基羰基、氨基磺酰基、C1-4烷基S(=O)t、羟基、氰基、卤素及任选被一-或二取代的氨基,其中所述氨基的取代基各自独立地选自C1-4烷基、芳基、芳基C1-4烷基、C3-7环烷基、C3-7环烷基-C1-4烷基、Het1、Het2、Het1C1-4烷基和Het2C1-4烷基;
R12是-NH2或-N(R5)(-AR6),其中
A是C1-6链烷二基、-C(=O)-、-C(=S)-、-S(=O)2-、C1-6链烷二基-C(=O)-、C1-6链烷二基-C(=S)-或C1-6链烷二基-S(=O)2-,其中A与取代的氨基官能团的连接点为在含有所述C1-6链烷二基的A的这些定义中的C1-6链烷二基;
R5是氢、羟基、C1-6烷基、Het1C1-6烷基、Het2C1-6烷基、氨基C1-6烷基,其中所述氨基任选由C1-4烷基一-或二取代;
R6是氢、C1-6烷氧基、Het1、Het1氧基、Het2、Het2氧基、芳基、芳氧基、芳氧基C1-4烷基、C1-4烷氧基芳基、C1-4烷氧基Het1、C1-4烷氧基Het2、C1-4烷氧基羰基氨基、氨基C1-4烷基氨基、氨基或氨基C1-4烷氧基,并且如果A不是C1-6链烷二基时,则R6如上定义或是C1-6烷基、Het1C1-4烷基、Het1氧基C1-4烷基、Het2C1-4烷基、Het2氧基C1-4烷基、芳基C1-4烷基、芳氧基C1-4烷基或氨基C1-4烷基;其中每个氨基任选被C1-4烷基一取代或被二取代;
或-A-R6是羟基C1-6烷基;
或R5和-A-R6与其连接的氮原子一起形成Het1或Het2,和
所述芳基作为基团或基团的一部份时,是指苯基或萘基,两者都任选被一或多个取代基取代,所述取代基独立地选自C1-6烷基、任选一-或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、任选一-或二取代的氨基、硝基、氰基、多卤代C1-6烷基、羟基C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、任选一-或二取代的氨基羰基、甲硫基、甲磺酰基及任选被一或多个取代基取代的苯基,该苯基的取代基各自独立选自C1-6烷基、任选一-或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、任选一-或二取代的氨基、硝基、氰基、多卤代C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、任选一-或二取代的氨基羰基、甲硫基及甲磺酰基;其中在任何氨基官能团上任选的取代基独立地选自C1-6烷基、C1-6烷氧基-A-、苯基-A-、苯氧基-A-、苯氧基C1-4烷基-A-、苯基C1-6烷基-A-、C1-6烷氧基羰基-氨基-A-、氨基-A-、氨基C1-6烷基及氨基C1-6烷基-A-,其中各氨基任选被C1-4烷基单-或二-取代;
Het1的定义为:含有3至14个环成员的饱和或部份不饱和单环、二环或三环杂环,它含一或多个杂原子环成员,杂原子各自独立地选自氮、氧或硫,且它在一或多个碳原子上任选被选自以下的取代基取代:C1-6烷基、任选一-或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、氧代基、任选一-或二取代的氨基、硝基、氰基、多卤代C1-6烷基、羟基C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、任选一-或二取代的氨基羰基、甲硫基、甲磺酰基、芳基及含3至14个环成员的饱和或部份不饱和单环、二环或三环杂环,所述杂环含一或多个杂原子环成员,杂原子各自独立选自氮、氧或硫,且其中在任何氨基官能团上的任选取代基独立选自C1-6烷基、C1-6烷氧基-A-、芳基-A-、芳氧基-A-、芳氧基C1-4烷基-A-、芳基C1-6烷基-A-、C1-6烷氧基羰基氨基-A-、氨基-A-、氨基C1-6烷基及氨基C1-6烷基-A-,其中各氨基任选被C1-4烷基单-或二-取代,以及
Het2的定义为:含有3至14个环成员的芳族单环、二环或三环杂环,它含一或多个杂原子环成员,杂原子各自独立选自氮、氧或硫,且它在一或多个碳原子上任选被选自以下的取代基取代:C1-6烷基、任选一-或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、任选一-或二取代的氨基、硝基、氰基、多卤代C1-6烷基、羟基C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、任选一-或二取代的氨基羰基、甲硫基、甲磺酰基、芳基、Het1及含3至14个环成员的芳族单环、二环或三环杂环;其中在任何氨基官能团上任选的取代基独立选自C1-6烷基、C1-6烷氧基-A-、Het1-A-、Het1C1-6烷基、Het1C1-6烷基-A-、Het1氧基-A-、Het1氧基C1-4烷基-A-、芳基-A-、芳氧基-A-、芳氧基C1-4烷基-A-、芳基C1-6烷基-A-、C1-6烷氧基羰基氨基-A-、氨基-A-、氨基C1-6烷基及氨基C1-6烷基-A-,其中各氨基任选被C1-4烷基单-或二-取代。
2.一种根据权利要求1的化合物,其中式(I)化合物具有以下结构
其中R1、R2、R3、R4、R12和L的定义同权利要求1。
3.一种根据权利要求1或2的化合物,其中R1为芳基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2或Het2C1-6烷基。
4.一种根据权利要求1或2的化合物,其中R1为芳基、Het1、Het2、Het2C1-6烷基。
5.一种根据权利要求1或2的化合物,其中R1为(i)含5至8个环成员的饱和单环或二环杂环,其中一个或两个环成员为氧原子,(ii)任选被一或多个独立选自以下的取代基取代的苯环:C1-6烷基、氨基C1-6烷基、一-或二(C1-6烷基)氨基C1-6烷基、氨基、一-或二(C1-6烷基)氨基、多卤代C1-6烷基,(iii)具有5至6个环成员的芳族单环杂环,其含有一或两个各自独立选自氮、氧或硫的杂原子环成员,且其在一个或多个碳原子上任选被C1-6烷基、氨基C1-6烷基、一-或二(C1-6烷基)氨基C1-6烷基、氨基、一-或二(C1-6烷基)氨基取代,(iv)通过C1-6烷基连接于变量L上的如在(iii)中定义的芳族单环杂环。
6.一种根据权利要求1或2的化合物,其中L与其连接的氮原子一起形成-O-C(=O)-NH-、-C(=O)-NH-、-O-C1-6链烷二基-C(=O)-NH-、-NR8-C1-6链烷二基-C(=O)-NH-。
7.一种根据权利要求1或2的化合物,其中L为-O-C1-6链烷二基-C(=O)-。
8.一种根据权利要求1或2的化合物,其中R2为氢。
9.一种根据权利要求1或2的化合物,其中R3为芳基C1-4烷基。
10.一种根据权利要求1或2的化合物,其中R4为C3-7环烷基、C2-6链烯基、C2-6炔基或C1-6烷基,其任选被C3-7环烷基取代。
11.一种根据权利要求1或2的化合物,其中在R12定义的-N(R5)(A-R6)中,R5是氢或C1-6烷基,A是C1-6链烷二基和R6是氢或Het1,或R5和A-R6与它们连接的氮原子结合在一起形成Het1。
12.一种根据权利要求1或2的化合物,其中R12为NH2。
13.一种根据权利要求1或2的化合物,其中R1为氢、C1-6烷基、C2-6链烯基、芳基C1-6烷基、C3-7环烷基、C3-7环烷基C1-6烷基、芳基、Het1、Het1C1-6烷基、Het2、Het2C1-6烷基;其中Het1是具有5或6个环成员的饱和或部分不饱和单环杂环,它含一或多个各自独立选自氮、氧或硫的杂原子环成员,且它在一或多个碳原子上任选被选自以下的取代基取代:C1-6烷基、任选一-或二取代的氨基C1-6烷基、C1-6烷氧基、卤素、羟基、氧代基、任选一-或二取代的氨基、硝基、氰基、多卤代C1-6烷基、羟基C1-6烷基、羧基、C1-6烷氧基羰基、C3-7环烷基、任选一-或二取代的氨基羰基、甲硫基、甲磺酰基、芳基及含3至14个环成员的饱和或部份不饱和单环、二环或三环杂环,所述杂环含一或多个杂原子环成员,杂原子各自独立选自氮、氧或硫,且其中在任何氨基官能团上的任选取代基独立选自C1-6烷基、C1-6烷氧基-A-、芳基-A-、芳氧基-A-、芳氧基C1-4烷基-A-、芳基C1-6烷基-A-、C1-6烷氧基羰基氨基-A-、氨基-A-、氨基C1-6烷基及氨基C1-6烷基-A-,其中各氨基任选被C1-4烷基单-或二-取代;
并且所述A的定义如同权利要求1。
14.一种根据权利要求1的化合物,其中所述化合物为:
1 {3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-氨基甲酸六氢-呋喃并[2,3-b]呋喃-3-基酯;
2 3-氨基-N-{3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-2-甲基-苯甲酰胺;
3 N-{3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-2-(2,6-二甲基-苯氧基)-乙酰胺;
4 {3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-氨基甲酸四氢-呋喃-3-基酯;
5 5-甲基-异噁唑-4-甲酸{3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-酰胺;
6 {3-[(3-氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-氨基甲酸噻唑-5-基甲酯;
7 N-{3-[(3-氨基-苯并[d]异噁唑-6-磺酰基)-异丁基-氨基]-1-苄基-2-羟基-丙基}-2-(2,6-二甲基-苯基氨基)-乙酰胺;
8 {1-苄基-2-羟基-3-[异丁基-(3-甲基氨基-苯并[d]异噁唑-5-磺酰基)-氨基]-丙基}-氨基甲酸六氢-呋喃并[2,3-b]呋喃-3-基酯;
9 {1-苄基-3-[(3-二甲基氨基-苯并[d]异噁唑-5-磺酰基)-异丁基-氨基]-2-羟丙基}-氨基甲酸六氢-呋喃并[2,3-b]呋喃-3-基酯;
10 {1-苄基-2-羟基-3-[异丁基-(3-吡咯烷-1-基-苯并[d]异噁唑-5-磺酰基)-氨基]-丙基}-氨基甲酸六氢-呋喃并[2,3-b]呋喃-3-基酯;
11 (1-苄基-2-羟基-3-{异丁基-[3-(2-吡咯烷-1-基-乙基氨基)-苯并[d]异噁唑-5-磺酰基]-氨基}-丙基)-氨基甲酸六氢-呋喃并[2,3-b]呋喃-3-基酯;
或它们的盐。
15.一种根据权利要求1或14的化合物,其中所述化合物是立体异构形式。
16.一种根据权利要求1的化合物的外消旋混合物。
17.一种药用组合物,它包含有效量的至少一种根据权利要求1至16中任一项的化合物及药学上可耐受的赋形剂。
18.一种组合药物,它含有(a)至少一种根据权利要求1至16中任一项的化合物和(b)另一种抗病毒化合物。
19.根据权利要求1至16中任一项的化合物在制备药物中的用途。
20.根据权利要求1至16中任一项的化合物在制备用于治疗与HIV和其它致病性逆转录病毒相关的疾病的药物中的用途。
21.根据权利要求20的用途,其中所述疾病与对多种药物耐药的逆转录病毒感染有关。
22.权利要求1至16中任一项的化合物在制备一种在感染逆转录病毒的哺乳动物中抑制所述逆转录病毒的蛋白酶的药物中的用途。
23.根据权利要求22的用途,其中所述逆转录病毒对多种药物有耐药性。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP02076957.6 | 2002-05-17 | ||
| EP02076957 | 2002-05-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1668605A CN1668605A (zh) | 2005-09-14 |
| CN100582097C true CN100582097C (zh) | 2010-01-20 |
Family
ID=29433159
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN03816458A Expired - Fee Related CN100582097C (zh) | 2002-05-17 | 2003-05-16 | 取代的苯并异噁唑磺酰胺广谱hiv蛋白酶抑制剂 |
Country Status (29)
| Country | Link |
|---|---|
| US (1) | US7462636B2 (zh) |
| EP (1) | EP1517899B1 (zh) |
| JP (2) | JP4674084B2 (zh) |
| KR (2) | KR20100130234A (zh) |
| CN (1) | CN100582097C (zh) |
| AP (1) | AP2004003190A0 (zh) |
| AR (1) | AR039887A1 (zh) |
| AT (1) | ATE371652T1 (zh) |
| AU (1) | AU2003238074B2 (zh) |
| BR (1) | BR0310089A (zh) |
| CA (1) | CA2485903C (zh) |
| CY (1) | CY1107040T1 (zh) |
| DE (1) | DE60315984T2 (zh) |
| DK (1) | DK1517899T3 (zh) |
| EA (1) | EA010908B1 (zh) |
| ES (1) | ES2292976T3 (zh) |
| HK (1) | HK1076099A1 (zh) |
| HR (1) | HRP20050607B1 (zh) |
| IL (1) | IL165043A0 (zh) |
| MX (1) | MXPA04011466A (zh) |
| MY (1) | MY135226A (zh) |
| NO (1) | NO330184B1 (zh) |
| NZ (1) | NZ536496A (zh) |
| PL (1) | PL373425A1 (zh) |
| PT (1) | PT1517899E (zh) |
| SI (1) | SI1517899T1 (zh) |
| TW (1) | TWI356700B (zh) |
| WO (1) | WO2003097616A1 (zh) |
| ZA (1) | ZA200410156B (zh) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EA010908B1 (ru) * | 2002-05-17 | 2008-12-30 | Тиботек Фармасьютикалз Лтд. | Ингибиторы вич-протеазы на основе замещённых бензизоксазолсульфонамидов широкого спектра действия |
| US7807845B2 (en) * | 2004-03-11 | 2010-10-05 | Sequoia Pharmaceuticals, Inc. | Resistance-repellent retroviral protease inhibitors |
| AU2005244121B2 (en) * | 2004-05-07 | 2012-01-19 | Sequoia Pharmaceuticals, Inc. | Resistance-repellent retroviral protease inhibitors |
| CA2595295C (en) | 2005-02-25 | 2014-07-08 | Tibotec Pharmaceuticals Ltd. | Protease inhibitor precursor synthesis |
| CN101702908A (zh) * | 2007-03-23 | 2010-05-05 | 麻萨诸塞州大学 | Hiv-1蛋白酶抑制剂 |
| ES2382618T3 (es) * | 2008-03-25 | 2012-06-11 | Formac Pharmaceuticals N.V. | Método de preparación para dispersiones sólidas |
| WO2011061590A1 (en) | 2009-11-17 | 2011-05-26 | Hetero Research Foundation | Novel carboxamide derivatives as hiv inhibitors |
| DE102010004957A1 (de) * | 2010-01-14 | 2011-07-21 | Universitätsklinikum Jena, 07743 | Biologisch wirksame Moleküle zur Beeinflussung von Virus-, Bakterien-, Parasiten-infizierten Zellen und/oder Tumorzellen und Verfahren zu deren Anwendung |
| EP2914587A1 (de) | 2012-10-31 | 2015-09-09 | Bayer CropScience AG | Neue heterocylische verbindungen als schädlingsbekämpfungsmittel |
| EP2964204B1 (en) * | 2013-03-07 | 2017-01-04 | Dow Global Technologies LLC | Novel esterified cellulose ethers of very low viscosity |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994005639A1 (en) * | 1992-09-08 | 1994-03-17 | Vertex Pharmaceuticals Incorporated | Sulfonamide inhibitors of hiv-aspartyl protease |
| US6143747A (en) * | 1995-01-20 | 2000-11-07 | G. D. Searle & Co. | Bis-sulfonamide hydroxyethylamino retroviral protease inhibitors |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5004751A (en) * | 1987-08-28 | 1991-04-02 | Mochida Pharmaceutical Co., Ltd. | Hydantoin derivatives |
| US5145684A (en) | 1991-01-25 | 1992-09-08 | Sterling Drug Inc. | Surface modified drug nanoparticles |
| EP0656887B1 (en) * | 1992-08-25 | 1998-10-28 | G.D. Searle & Co. | Hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| PH30929A (en) | 1992-09-03 | 1997-12-23 | Janssen Pharmaceutica Nv | Beads having a core coated with an antifungal and a polymer. |
| DK0715618T3 (da) | 1993-08-24 | 1999-08-23 | Searle & Co | Hydroxyethylaminosulfonamider til anvendelse som inhibitorer af retrovirale proteaser |
| JP3732508B2 (ja) | 1993-10-01 | 2006-01-05 | アストラ・アクチエボラーグ | 粉末状薬剤の製造方法 |
| US5756533A (en) * | 1995-03-10 | 1998-05-26 | G.D. Searle & Co. | Amino acid hydroxyethylamino sulfonamide retroviral protease inhibitors |
| US5705500A (en) | 1995-03-10 | 1998-01-06 | G.D. Searle & Co. | Sulfonylalkanoylamino hydroxyethylamino sulfonamide retroviral protease inhibitors |
| US6150556A (en) | 1995-03-10 | 2000-11-21 | G. D. Dearle & Co. | Bis-amino acid hydroxyethylamino sulfonamide retroviral protease inhibitors |
| PT815124E (pt) * | 1995-03-10 | 2003-04-30 | Searle & Co | Inibidores protease retroviral sulfonamida hidroxietilamino aminoacido heterociclocarbonilo |
| JP2000515488A (ja) * | 1995-11-15 | 2000-11-21 | ジー.ディー.サール アンド カンパニー | 置換スルホニルアルカノイルアミノヒドロキシエチルアミノスルホンアミド レトロウイルスプロテアーゼ阻害剤 |
| CN1165291C (zh) | 1996-05-20 | 2004-09-08 | 詹森药业有限公司 | 具有改进的生物利用度的抗真菌组合物 |
| SK284813B6 (sk) | 1997-03-26 | 2005-12-01 | Janssen Pharmaceutica N. V. | Peleta obsahujúca itrakonazol, spôsob jej prípravy a farmaceutická dávková forma s jej obsahom |
| US6436989B1 (en) | 1997-12-24 | 2002-08-20 | Vertex Pharmaceuticals, Incorporated | Prodrugs of aspartyl protease inhibitors |
| AU2012199A (en) | 1997-12-24 | 1999-07-19 | Vertex Pharmaceuticals Incorporated | Prodrugs of aspartyl protease inhibitors |
| WO1999033792A2 (en) | 1997-12-24 | 1999-07-08 | Vertex Pharmaceuticals Incorporated | Prodrugs os aspartyl protease inhibitors |
| JP2001527062A (ja) | 1997-12-24 | 2001-12-25 | バーテックス ファーマシューティカルズ インコーポレイテッド | アスパルチルプロテアーゼインヒビターのプロドラッグ |
| WO1999067254A2 (en) | 1998-06-23 | 1999-12-29 | The United States Of America Represented By The Secretary, Department Of Health And Human Services | Multi-drug resistant retroviral protease inhibitors and use thereof |
| ATE500823T1 (de) * | 1998-06-23 | 2011-03-15 | Us Of America Represented By The Secretary Dept Of Health And Human Services | Arzneimittel zur behandlung von hiv-infizierten säugetieren |
| ES2307533T3 (es) | 1999-10-06 | 2008-12-01 | Tibotec Pharmaceuticals Ltd. | Hexahidrofuro(2,3)furan-3-il-n-(3-((1,3-benzodioxol-5-ilsulfonil)(is obuti)amino)-1-bencil-2-hidroxipropil carbamato como inhibidor de la proteasa retroviral. |
| PL367084A1 (en) * | 2001-04-09 | 2005-02-21 | Tibotec Pharmaceuticals Ltd. | Broadspectrum 2-(substituted-amino)-benzoxazole sulfonamide hiv protease inhibitors |
| EA010908B1 (ru) * | 2002-05-17 | 2008-12-30 | Тиботек Фармасьютикалз Лтд. | Ингибиторы вич-протеазы на основе замещённых бензизоксазолсульфонамидов широкого спектра действия |
-
2003
- 2003-05-16 EA EA200401524A patent/EA010908B1/ru not_active IP Right Cessation
- 2003-05-16 AT AT03735707T patent/ATE371652T1/de active
- 2003-05-16 JP JP2004505349A patent/JP4674084B2/ja not_active Expired - Fee Related
- 2003-05-16 IL IL16504303A patent/IL165043A0/xx not_active IP Right Cessation
- 2003-05-16 PT PT03735707T patent/PT1517899E/pt unknown
- 2003-05-16 ES ES03735707T patent/ES2292976T3/es not_active Expired - Lifetime
- 2003-05-16 DK DK03735707T patent/DK1517899T3/da active
- 2003-05-16 NZ NZ536496A patent/NZ536496A/en not_active IP Right Cessation
- 2003-05-16 TW TW092113367A patent/TWI356700B/zh not_active IP Right Cessation
- 2003-05-16 DE DE60315984T patent/DE60315984T2/de not_active Expired - Lifetime
- 2003-05-16 AR ARP030101725A patent/AR039887A1/es active IP Right Grant
- 2003-05-16 US US10/514,539 patent/US7462636B2/en not_active Expired - Fee Related
- 2003-05-16 EP EP03735707A patent/EP1517899B1/en not_active Expired - Lifetime
- 2003-05-16 CA CA2485903A patent/CA2485903C/en not_active Expired - Fee Related
- 2003-05-16 AU AU2003238074A patent/AU2003238074B2/en not_active Ceased
- 2003-05-16 MX MXPA04011466A patent/MXPA04011466A/es active IP Right Grant
- 2003-05-16 BR BR0310089-8A patent/BR0310089A/pt not_active IP Right Cessation
- 2003-05-16 WO PCT/EP2003/050173 patent/WO2003097616A1/en active IP Right Grant
- 2003-05-16 KR KR1020107025714A patent/KR20100130234A/ko not_active Withdrawn
- 2003-05-16 AP AP2004003190A patent/AP2004003190A0/xx unknown
- 2003-05-16 MY MYPI20031819A patent/MY135226A/en unknown
- 2003-05-16 SI SI200331012T patent/SI1517899T1/sl unknown
- 2003-05-16 CN CN03816458A patent/CN100582097C/zh not_active Expired - Fee Related
- 2003-05-16 KR KR1020047018261A patent/KR101019648B1/ko not_active Expired - Fee Related
- 2003-05-16 PL PL03373425A patent/PL373425A1/xx unknown
-
2004
- 2004-12-14 NO NO20045444A patent/NO330184B1/no not_active IP Right Cessation
- 2004-12-15 ZA ZA2004/10156A patent/ZA200410156B/en unknown
-
2005
- 2005-06-29 HR HRP20050607AA patent/HRP20050607B1/hr not_active IP Right Cessation
- 2005-09-14 HK HK05108052A patent/HK1076099A1/xx not_active IP Right Cessation
-
2007
- 2007-11-29 CY CY20071101524T patent/CY1107040T1/el unknown
-
2010
- 2010-10-25 JP JP2010238833A patent/JP2011051999A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994005639A1 (en) * | 1992-09-08 | 1994-03-17 | Vertex Pharmaceuticals Incorporated | Sulfonamide inhibitors of hiv-aspartyl protease |
| US6143747A (en) * | 1995-01-20 | 2000-11-07 | G. D. Searle & Co. | Bis-sulfonamide hydroxyethylamino retroviral protease inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN100369904C (zh) | 2-(取代-氨基)-苯并噻唑磺酰胺hiv蛋白酶抑制剂 | |
| CN100549007C (zh) | 广谱2-氨基-苯并唑磺酰胺hiv蛋白酶抑制剂 | |
| JP2011051999A (ja) | 広域スペクトルの置換ベンズイソキサゾールスルホンアミドhivプロテアーゼ阻害剤 | |
| KR101419320B1 (ko) | 2-(치환된-아미노)-벤조티아졸 설폰아미드 hiv 프로테아제 저해제 | |
| CN100491360C (zh) | 广谱2-(取代的-氨基)-苯并唑磺酰胺hiv蛋白酶抑制剂 | |
| CN1688586B (zh) | 取代的羟吲哚磺酰胺类广谱hiv蛋白酶抑制剂 | |
| CN101316850A (zh) | 作为hiv蛋白酶抑制剂的取代的氨基苯基磺酰胺化合物 | |
| CN1671380B (zh) | 广谱2-氨基-苯并噻唑磺酰胺类hiv蛋白酶抑制剂 | |
| CN101935303B (zh) | 广谱取代的苯并咪唑磺酰胺hiv蛋白酶抑制剂 | |
| CN101445493A (zh) | 广谱2-氨基-苯并噻唑磺酰胺类hiv蛋白酶抑制剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20100120 Termination date: 20150516 |
|
| EXPY | Termination of patent right or utility model |