JP2016500130A - C6-C18-acylated derivative of hyaluronic acid and process for its preparation, nanomicelle composition based on it and process for its preparation, process for preparing stabilized nanomicelle composition and use thereof - Google Patents
C6-C18-acylated derivative of hyaluronic acid and process for its preparation, nanomicelle composition based on it and process for its preparation, process for preparing stabilized nanomicelle composition and use thereof Download PDFInfo
- Publication number
- JP2016500130A JP2016500130A JP2015543316A JP2015543316A JP2016500130A JP 2016500130 A JP2016500130 A JP 2016500130A JP 2015543316 A JP2015543316 A JP 2015543316A JP 2015543316 A JP2015543316 A JP 2015543316A JP 2016500130 A JP2016500130 A JP 2016500130A
- Authority
- JP
- Japan
- Prior art keywords
- acid
- hyaluronic acid
- preparation
- nanomicelle
- derivative
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 CC(*)(*)C(C(C1OC(C(C(C2O*)O)O*)OC2C(O*)=O)NC)OC(CO*)C1O Chemical compound CC(*)(*)C(C(C1OC(C(C(C2O*)O)O*)OC2C(O*)=O)NC)OC(CO*)C1O 0.000 description 1
Images












Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/61—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule the organic macromolecular compound being a polysaccharide or a derivative thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/12—Ketones
- A61K31/122—Ketones having the oxygen directly attached to a ring, e.g. quinones, vitamin K1, anthralin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
- A61K31/353—3,4-Dihydrobenzopyrans, e.g. chroman, catechin
- A61K31/355—Tocopherols, e.g. vitamin E
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/683—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols
- A61K31/685—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols one of the hydroxy compounds having nitrogen atoms, e.g. phosphatidylserine, lecithin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6905—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion
- A61K47/6907—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion the form being a microemulsion, nanoemulsion or micelle
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/02—Cosmetics or similar toiletry preparations characterised by special physical form
- A61K8/0291—Micelles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/72—Cosmetics or similar toiletry preparations characterised by the composition containing organic macromolecular compounds
- A61K8/73—Polysaccharides
- A61K8/735—Mucopolysaccharides, e.g. hyaluronic acid; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/107—Emulsions ; Emulsion preconcentrates; Micelles
- A61K9/1075—Microemulsions or submicron emulsions; Preconcentrates or solids thereof; Micelles, e.g. made of phospholipids or block copolymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61Q—SPECIFIC USE OF COSMETICS OR SIMILAR TOILETRY PREPARATIONS
- A61Q19/00—Preparations for care of the skin
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/006—Heteroglycans, i.e. polysaccharides having more than one sugar residue in the main chain in either alternating or less regular sequence; Gellans; Succinoglycans; Arabinogalactans; Tragacanth or gum tragacanth or traganth from Astragalus; Gum Karaya from Sterculia urens; Gum Ghatti from Anogeissus latifolia; Derivatives thereof
- C08B37/0063—Glycosaminoglycans or mucopolysaccharides, e.g. keratan sulfate; Derivatives thereof, e.g. fucoidan
- C08B37/0072—Hyaluronic acid, i.e. HA or hyaluronan; Derivatives thereof, e.g. crosslinked hyaluronic acid (hylan) or hyaluronates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/30—Sulfur-, selenium- or tellurium-containing compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2800/00—Properties of cosmetic compositions or active ingredients thereof or formulation aids used therein and process related aspects
- A61K2800/10—General cosmetic use
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2800/00—Properties of cosmetic compositions or active ingredients thereof or formulation aids used therein and process related aspects
- A61K2800/40—Chemical, physico-chemical or functional or structural properties of particular ingredients
- A61K2800/41—Particular ingredients further characterized by their size
- A61K2800/413—Nanosized, i.e. having sizes below 100 nm
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2800/00—Properties of cosmetic compositions or active ingredients thereof or formulation aids used therein and process related aspects
- A61K2800/40—Chemical, physico-chemical or functional or structural properties of particular ingredients
- A61K2800/56—Compounds, absorbed onto or entrapped into a solid carrier, e.g. encapsulated perfumes, inclusion compounds, sustained release forms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K36/00—Medicinal preparations of undetermined constitution containing material from algae, lichens, fungi or plants, or derivatives thereof, e.g. traditional herbal medicines
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/30—Sulfur-, selenium- or tellurium-containing compounds
- C08K2003/3045—Sulfates
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Polymers & Plastics (AREA)
- Birds (AREA)
- Dispersion Chemistry (AREA)
- Materials Engineering (AREA)
- Biochemistry (AREA)
- Inorganic Chemistry (AREA)
- Dermatology (AREA)
- Biophysics (AREA)
- Nanotechnology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Immunology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Natural Medicines & Medicinal Plants (AREA)
- Cosmetics (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Alternative & Traditional Medicine (AREA)
- Biotechnology (AREA)
- Botany (AREA)
Abstract
本発明は疎水化ヒアルロン酸(式I)の調製方法に関するものであり,更に生物学的に活性な疎水性物質の担体として作用する疎水化ヒアルロナンのナノミセルに生物学的活性物質をカプセル封入する方法に関する。ヒアルロナンの疎水化はヒアルロナンと長鎖カルボン酸とのエステル化反応を経て行われ,その長鎖カルボン酸は2,4,6‐トリクロロ安息香酸のハロゲン化物誘導体又は他の有機塩化物により活性化される。水性環境下では,水溶性であり疎水化された誘導体はナノミセルを形成することが可能であり,この内部では非極性物質が非共有的な物理的相互作用により結合することが可能である。ナノミセルのコアは疎水性アシル官能基により形成され,一方,ナノミセルのシェルはヒアルロン酸により形成される。ナノミセルへの物質のカプセル封入は溶媒交換法又は音波処理により行うことができる。ヒアルロン酸ナノミセルは,結合された物質が局所塗布部に浸透することを促進し,その結合された物質が個々の細胞に移行することを可能にする。疎水化ヒアルロナン誘導体から得られたナノミセルは化粧用途及び医療用途に有用である。The present invention relates to a process for the preparation of hydrophobized hyaluronic acid (formula I) and further a method for encapsulating a biologically active substance in nano micelles of hydrophobized hyaluronan which acts as a carrier for the biologically active hydrophobic substance. About. Hyaluronan is hydrophobized through esterification of hyaluronan with long-chain carboxylic acids, which are activated by halide derivatives of 2,4,6-trichlorobenzoic acid or other organic chlorides. The In an aqueous environment, water-soluble and hydrophobized derivatives can form nanomicelles, in which nonpolar substances can be bound by noncovalent physical interactions. Nanomicelle cores are formed by hydrophobic acyl functional groups, while nanomicelle shells are formed by hyaluronic acid. Encapsulation of substances in nanomicelles can be performed by solvent exchange or sonication. Hyaluronic acid nano micelles promote the penetration of the bound substance into the local application site and allow the bound substance to migrate to individual cells. Nano micelles obtained from hydrophobized hyaluronan derivatives are useful for cosmetic and medical applications.
Description
本発明は,生物学的に活性な疎水性物質を担持する疎水化ヒアルロン酸の調製方法及びその使用に関するものであり,ここでは生物学的活性物質を疎水化ヒアルロナンのナノミセルにカプセル封入している。ヒアルロナンの疎水化は長鎖カルボン酸によるヒアルロナンのエステル化反応を経て行われ,長鎖カルボン酸は2,4,6‐トリクロロ安息香酸のハロゲン化物誘導体又はR3‐CO‐Clの別の有機塩化物により活性化される。好適な水性環境では,水溶性疎水化誘導体はナノミセルを形成し,この中では非極性物質が非共有物理的相互作用により結合することが可能である。ナノミセルのコアは疎水性アシル官能基により形成され,一方,ナノミセルのシェルはヒアルロン酸により形成される。ナノミセルへの物質のカプセル封入は溶媒交換法又は音波処理により行うことが可能である。ヒアルロンナノミセルは局所塗布において結合した物質の浸透を支持し,その結合した物質が個々の細胞へと移行することを可能にする。本発明は更に,安定化ナノミセル組成物を調製する方法に関する。疎水化ヒアルロナン誘導体から得たナノミセルは化粧用途及び医薬用途に有用である。 The present invention relates to a method for the preparation of hydrophobized hyaluronic acid carrying a biologically active hydrophobic substance and the use thereof, wherein the biologically active substance is encapsulated in nano micelles of hydrophobized hyaluronan. . Hyaluronan is hydrophobized via esterification of hyaluronan with a long chain carboxylic acid, which is a halide derivative of 2,4,6-trichlorobenzoic acid or another organic chloride of R 3 -CO-Cl. It is activated by the object. In a suitable aqueous environment, the water-soluble hydrophobized derivatives form nanomicelles in which nonpolar substances can be bound by noncovalent physical interactions. Nanomicelle cores are formed by hydrophobic acyl functional groups, while nanomicelle shells are formed by hyaluronic acid. Encapsulation of substances in nanomicelles can be performed by solvent exchange or sonication. Hyaluronic nanomicelles support the penetration of bound substances in topical applications and allow the bound substances to migrate into individual cells. The invention further relates to a method of preparing a stabilized nanomicelle composition. Nano micelles obtained from hydrophobized hyaluronan derivatives are useful for cosmetic and pharmaceutical applications.
ヒアルロン酸は2つの繰り返し単位β‐(1,3)‐D‐グルクロン酸及びβ‐(1,4)‐N‐アセチル‐D‐グルコサミンから成る重要な多糖である。それは単離の方法及び初期の使用材料に依存して5×104〜5×106の範囲の高い分子量を特徴としている。ヒアルロン酸,特にヒアルロナンとして知られるそのナトリウム塩は,結合組織及び関節液の必須構成要素である。更にそれは水和,プロテオグリカンの組織化,細胞分化,増殖及び血管形成など多数の生物学的プロセスにおいて重要な役割を担う。強い親水性を有する多糖は全てのpHの範囲内で塩の形態で水溶性である。 Hyaluronic acid is an important polysaccharide consisting of two repeating units β- (1,3) -D-glucuronic acid and β- (1,4) -N-acetyl-D-glucosamine. It is characterized by a high molecular weight in the range of 5 × 10 4 to 5 × 10 6 depending on the method of isolation and the initial materials used. Hyaluronic acid, especially its sodium salt known as hyaluronan, is an essential component of connective tissue and joint fluid. Furthermore, it plays an important role in many biological processes such as hydration, organization of proteoglycans, cell differentiation, proliferation and angiogenesis. Polysaccharides with strong hydrophilicity are water soluble in salt form within the entire pH range.
ヒアルロン酸をベースとする担体系
天然形態で親水性であることから,ヒアルロン酸は疎水性物質の効果的な担体として作用することができない。この為,疎水性官能基をヒアルロン酸のポリマー鎖に結合しなければならない。このような疎水性官能基の量と長さが十分である場合,その官能基を含む自己凝集プロセスが開始し,その結果,ヒアルロナンの構造内で疎水性ドメインが形成され得る。その後,水不溶性物質の小型分子が非共有結合により,このようなドメインに結合することができる。文献では,得られた構造はポリマーナノミセルと呼ばれることが多く,ミセルのコアは疎水性であるので,小型の非極性分子が溶解され,一方,そのシェルは親水性であるのでそのポリマーミセルは水性環境で溶解することが可能となる。寸法(直径)が200nm未満のポリマーミセルは,ナノミセルと呼ばれる。
Hyaluronic acid-based carrier system Hyaluronic acid cannot act as an effective carrier for hydrophobic substances because it is hydrophilic in its natural form. For this reason, hydrophobic functional groups must be attached to the polymer chain of hyaluronic acid. If the amount and length of such a hydrophobic functional group is sufficient, a self-aggregation process involving that functional group is initiated, so that a hydrophobic domain can be formed within the structure of hyaluronan. Subsequently, small molecules of water-insoluble substances can bind to such domains by non-covalent bonds. In the literature, the resulting structure is often called polymer nanomicelle, and the micelle core is hydrophobic, so small non-polar molecules are dissolved, while the shell is hydrophilic, so the polymer micelle is aqueous. It becomes possible to dissolve in the environment. Polymer micelles with dimensions (diameter) of less than 200 nm are called nano micelles.
修飾されたヒアルロン酸をベースとするポリマーミセルにより形成された担体系は,ヒアルロナンとアルキルアミン(Liuら,2011年)及びギ酸との複合体で知られている。しかし,毒性が高く催奇形性であるホルムアミドの存在は,上記のヒアルロナン誘導体の調製には不可欠であると考えられている。酸化還元感受性ミセルを得る為,類似のポリマーミセル調製方法を採用した(Liら,2011年)。毒性の高い試薬が存在する為,このようなミセル系が生物学的用途に使用できないことは明らかである。 Support systems formed by polymer micelles based on modified hyaluronic acid are known for complexes of hyaluronan with alkylamines (Liu et al., 2011) and formic acid. However, the presence of formamide, which is highly toxic and teratogenic, is considered essential for the preparation of the above hyaluronan derivatives. In order to obtain redox-sensitive micelles, a similar polymer micelle preparation method was employed (Li et al., 2011). It is clear that such micellar systems cannot be used for biological applications due to the presence of highly toxic reagents.
D‐グルクロン酸のカルボキシル基により媒介された結合を経て,また可能性として低分子物質の取り込みを経て,ヒアルロン酸と他のポリマー(乳酸又はグリコール酸のポリマー)とが共役することは,特許U.S.7,767,806で特許請求されており,ここでは,著者らはポリマーの生体適合性を述べているが,試験により説明も証明もしていない。別のケースでは,低分子ヒアルロナン(9〜45kDa)は多様な鎖長の疎水性アミン及びカチオン性セグメントとして使用される正電荷スペルミンによって共有結合的に(グルクロン酸のカルボキシル基の位置で)修飾された(Shen,Li,Tu及びZhu,2009年)。後者の目的は,遺伝子の担体を調製することである。しかし,急性及び亜急性毒性により,上記目的の為のスペルミンの使用は制限されている(Til,Falke,Prinsen及びWillems,1997年)。直径が125〜555nmの形成されたポリマーミセルの臨界ミセル濃度は,0.04mg・mL-1以上と測定された。臨界ミセル濃度値が過剰に高くなり,よってミセル系が(例えば血流中で)最大に希釈されないということに加えて,上記寸法のミセルは,人体内における薬物の受動的分散の好適な候補薬にはならないと考える。なぜなら,ポリマーミセルのその寸法は,腫瘍部位又は静脈壁破裂による梗塞性病変を発症させるその能力の原因であるからである。このような場合は,直径20〜100nmのポリマーミセルが好ましい(Wang,Mongayt及びTorchilin,2005年)。 Conjugation of hyaluronic acid with other polymers (polymers of lactic acid or glycolic acid) via a bond mediated by the carboxyl group of D-glucuronic acid and possibly through the incorporation of low molecular weight substances is described in US Pat. , 767,806, where the authors describe the biocompatibility of the polymer, but have not been explained or proven by testing. In another case, the low molecular weight hyaluronan (9-45 kDa) is covalently modified (at the carboxyl group position of glucuronic acid) by various chain lengths of hydrophobic amines and positively charged spermines used as cationic segments. (Shen, Li, Tu and Zhu, 2009). The latter purpose is to prepare a carrier for the gene. However, acute and subacute toxicity limits the use of spermine for these purposes (Til, Falke, Prinsen and Willems, 1997). The critical micelle concentration of the formed polymer micelle having a diameter of 125 to 555 nm was measured to be 0.04 mg · mL −1 or more. In addition to the critical micelle concentration value becoming excessively high and thus the micelle system not being diluted to the maximum (eg in the bloodstream), micelles of the above dimensions are suitable candidates for passive dispersion of drugs in the human body. I don't think so. This is because the size of the polymer micelle is responsible for its ability to develop an infarcted lesion due to the tumor site or vein wall rupture. In such cases, polymer micelles with a diameter of 20-100 nm are preferred (Wang, Mongayt and Torchilin, 2005).
負電荷ヒアルロン酸と正電荷スチリルピリジニウムとの間の静電相互作用に基づいたポリマーヒアルロナンミセルの調製に,グルクロン酸のカルボキシル基の修飾を利用した(Tao,Xu,Chen,Bai及びLiu,2012年)。しかし,このような相互作用はニューロンから神経伝達物質を放出させる調節に関して重要な役割を担ってはいるが,スチリルピリジニウムと神経末端及びムスカリン性受容体との相互作用はこれまで十分に解明されてきていないことから,このようなポリマーミセルのインビボでの使用は限定的である(Mazzone,Mori,Burman,Palovich,Belmonte及びCanning,2006年)。 Modification of the carboxyl group of glucuronic acid was used to prepare polymer hyaluronan micelles based on electrostatic interaction between negatively charged hyaluronic acid and positively charged styrylpyridinium (Tao, Xu, Chen, Bai and Liu, 2012) ). However, such interactions play an important role in the regulation of neurotransmitter release from neurons, but the interaction of styrylpyridinium with nerve endings and muscarinic receptors has been well elucidated so far. The in vivo use of such polymeric micelles is limited (Mazzone, Mori, Burman, Palovich, Belmonte and Canning, 2006).
特許文献U.S.7,993,678及びWO2007/033677では,ヒアルロナンのアルキル/アリール‐コハク酸誘導体の調製方法を請求しており,このような誘導体は活性的非極性物質のカプセル封入にも利用可能である。後者の場合,修飾はヒアルロナンの第1級ヒドロキシル基を包含し,一方,カルボキシル基は未変化のままである。上記の修飾反応の短所は,環状無水物とヒアルロナンとの反応が起こるアルカリ性pH範囲(pH8.5〜9.0)である。実際,このようなアルカリ性pH値により無水物の加水分解が開始され,これにより修飾プロセスの効率が低下する。このことは工業規模で特に顕著である。上記の方法で調製し,置換度が44%であるヒアルロナンのアルキル/アリール‐コハク酸誘導体において,水性環境でミセルを形成(凝集)する能力は,個々の濃度が0.003〜0.004mg・mL-1超であるときに明らかになった。ポリマーミセルで観察された寸法は50〜200nmの範囲であった。しかし,このような誘導体の短所は,修飾アルキル/アリール‐コハク酸官能基中の付加的COO−基の存在に起因するヒアルロナンの全体的な負電荷の増加にある。分子の負電荷は細胞と個々の担体系との間の相互作用に対してかなり不利益な影響を与える可能性がある(Wang,Mongayt及びTorchilin,2005年)。上記の方法で調製した誘導体を注射に使用する際の制限因子の1つは,その低い溶解度にある(Eenschooten, Guillaumie, Kontogeorgis, Stenby及びSchwach‐Abdellaoui, 2010年)。このような誘導体の別の短所は,加圧滅菌などの加熱殺菌工程中におけるエステル結合の不安定性にある。特許文献U.S.7,993,678及びWO2007/033677では,安定的な乳液の形態などのヒアルロナンのアルキル/アリール‐コハク酸誘導体溶液中に非極性物質を直接溶解する方法が記載されているだけである。非極性物質をカプセル封入する直接的な方法の主な短所は,得られたポリマーミセルの結合能が低い所である(Kedar, Phutane, Shidhaye及びKadam,2010年)。上記特許は担体系の為の修飾ヒアルロナンの構造の利用を特許請求しているが,乳液系以外の所与のヒアルロナン構造に疎水性物質を結合できる可能性を有する更なる方法は提供していない。この理由で,適用性の現実的評価に必要である基本的な性質の一つとされている十分な結合能を有するポリマー担体系の提供が完全に欠落している。更に,細胞毒性及び細胞相互作用についての詳細は記載されておらず,よって,特許請求された構造が実際に疎水性物質の細胞内への活性的移行に適用可能であるか否かを結論付けることは不可能であり,この結論は医療への応用に不可欠である。 Patent documents US 7,993,678 and WO 2007/033677 claim a process for the preparation of alkyl / aryl-succinic acid derivatives of hyaluronan, which derivatives can also be used for the encapsulation of active non-polar substances. In the latter case, the modification includes the primary hydroxyl group of hyaluronan, while the carboxyl group remains unchanged. The disadvantage of the above modification reaction is the alkaline pH range (pH 8.5 to 9.0) where the reaction between the cyclic anhydride and hyaluronan occurs. In fact, such an alkaline pH value initiates the hydrolysis of the anhydride, which reduces the efficiency of the modification process. This is particularly noticeable on an industrial scale. In the alkyl / aryl-succinic acid derivative of hyaluronan prepared by the above method and having a substitution degree of 44%, the ability to form (aggregate) micelles in an aqueous environment has an individual concentration of 0.003 to 0.004 mg · Clarified when greater than mL -1 . The dimensions observed with polymer micelles ranged from 50 to 200 nm. However, disadvantages of such derivatives, modified alkyl / aryl - is the increase in overall negative charge of hyaluronan due to the presence of groups - in succinic acid functional groups additionally COO. The negative charge of the molecule can have a rather detrimental effect on the interaction between the cell and the individual carrier system (Wang, Mongayt and Torchilin, 2005). One of the limiting factors when using derivatives prepared by the above method for injection is its low solubility (Eenschooten, Guillaumie, Kontogeorgis, Stenby and Schwach-Abdellaoui, 2010). Another disadvantage of such derivatives is the instability of ester bonds during heat sterilization processes such as autoclaving. Patent documents US 7,993,678 and WO 2007/033677 only describe a method for directly dissolving a non-polar substance in an alkyl / aryl-succinic acid derivative solution of hyaluronan, such as a stable emulsion form. The main disadvantage of the direct method of encapsulating non-polar substances is the low binding capacity of the resulting polymer micelles (Kedar, Phutane, Shidhaye and Kadam, 2010). The above patent claims the use of a modified hyaluronan structure for a carrier system, but does not provide a further method with the potential to attach a hydrophobic substance to a given hyaluronan structure other than an emulsion system. . For this reason, there is a complete lack of providing a polymer support system with sufficient binding capacity, which is one of the fundamental properties required for realistic applicability evaluation. Furthermore, no details on cytotoxicity and cell interactions are given, so we conclude whether the claimed structure is actually applicable to the active transfer of hydrophobic substances into cells This is impossible and this conclusion is essential for medical applications.
別の刊行物(Smejkalova,Hermannova,Sulakova,Prusova,Kucerik及びVelebny,2012年)には,ヒアルロナンの疎水性ドメインが記載されており,これは共有結合によりヒアルロナンに結合したC6‐アシル鎖の凝集から生じるものである。しかし,この刊行物に記載された誘導体には残留溶媒が完全に無いとは言えない。また,上記刊行物にはポリマーミセルの形成も特徴付けも述べられていない。更に,上記刊行物に記載された対称性無水物は長アルキル鎖間の結合形成には利用できない。脂肪族鎖が長いカルボン酸は非常に高価である上に,1モルの最終試薬の調製中,少なくとも1モルの酸が失われる。その刊行物では,調製した誘導体の細胞毒性も述べられていない。 Another publication (Smejkalova, Hermannova, Sulakova, Prusova, Kucerik and Velebny, 2012) describes the hydrophobic domain of hyaluronan, from the aggregation of covalently linked C6-acyl chains to hyaluronan. It will occur. However, it cannot be said that the derivatives described in this publication are completely free of residual solvents. Also, the publication does not mention the formation or characterization of polymer micelles. Furthermore, the symmetric anhydrides described in the above publications cannot be used to form bonds between long alkyl chains. Carboxylic acids with long aliphatic chains are very expensive and at least one mole of acid is lost during the preparation of one mole of final reagent. The publication does not mention the cytotoxicity of the prepared derivatives.
同等の医薬組成物を含む多糖の酪酸エチルの調製は,特許EP0941253で特許請求されている。しかし,特許請求された調製方法は非常に低い置換度(最大3%)だけを可能にしている。非共有結合により調製された誘導体に結合した疎水性物質の量は不都合にもこのような低い置換度に影響される。非水性条件であること以外は特許WO2005/092929に従って,ヒアルロナンの酪酸エチルを更に調製した。従って,第4級アンモニウム塩へのヒアルロナンの変換はヒアルロナンの分解を伴う可能性がある。得られた置換度は0.1%未満であり,よってこのようなエステル誘導体は担体系の調製には適していない。ヒアルロナンと酪酸無水物及びレチノイン酸塩化物との同時エステル化を行ったところ,類似の結果が得られた(WO2004/056877)。 The preparation of the polysaccharide ethyl butyrate containing an equivalent pharmaceutical composition is claimed in patent EP0941253. However, the claimed preparation method allows only a very low degree of substitution (up to 3%). The amount of hydrophobic material bound to the derivative prepared by non-covalent bonding is adversely affected by such a low degree of substitution. Ethyl butyrate hyaluronan was further prepared according to patent WO2005 / 092929 except under non-aqueous conditions. Therefore, conversion of hyaluronan to quaternary ammonium salts may be accompanied by degradation of hyaluronan. The degree of substitution obtained is less than 0.1% and thus such ester derivatives are not suitable for the preparation of carrier systems. Similar results were obtained after simultaneous esterification of hyaluronan with butyric anhydride and retinoic acid chloride (WO2004 / 056877).
修飾ヒアルロナン(HA)‐[O(C=O)NH‐M]P(式中,Mはアルキル官能基C2−16から成る修飾単位を表し,pは3〜4の倍数を表す)及び薬学的活性分子をベースとするポリマーミセル組成物が特許U.S.2010/0316682及びEP1538166A1により特許請求されている。このような誘導体の主な短所は,免疫毒性及び催奇形の可能性を有する物質として知られ,ヒアルロナンの修飾を行う場合に使用されているジブチル錫ラウレートである。この物質は接着剤の製造に関連する修飾方法で使用されることが多く,急性毒性があることから欧州環境機構に収録されている(Boyer,1989年)。特許請求されたこの誘導体の別の短所は,ポリマー,例えばポリエチレングリコールと共役することであり,これは人体に対しては異質のものであり,静脈内投与又は局所投与に使用する場合,炎症反応を起こすか,或いは細胞毒性分解生成物を生じさせる。更に,ポリマーミセルを繰り返し塗布すると,ポリエチレングリコールが親水性セグメントを形成し,抗PEG性IgM抗体の形成により,血流からのこれらのミセルの排出が加速する(Gong,Chen,Zheng,Wang及びWang,2012年)。 Modified hyaluronan (HA)-[O (C = O) NH-M] P (wherein M represents a modified unit comprising an alkyl functional group C 2-16 and p represents a multiple of 3 to 4) and pharmaceutical Micelle compositions based on chemically active molecules are claimed by patents US2010 / 0316682 and EP1538166A1. The main disadvantage of such derivatives is dibutyltin laurate, which is known as a substance with immunotoxicity and teratogenic potential and is used in the modification of hyaluronan. This material is often used in modification methods related to the manufacture of adhesives and is recorded in the European Environmental Organization because of its acute toxicity (Boyer, 1989). Another disadvantage of the claimed derivative is that it is conjugated to a polymer, such as polyethylene glycol, which is heterogeneous to the human body, and when used for intravenous or topical administration, an inflammatory response Cause cytotoxic degradation products. In addition, when polymer micelles are repeatedly applied, polyethylene glycol forms hydrophilic segments and the formation of anti-PEGic IgM antibodies accelerates the elimination of these micelles from the bloodstream (Gong, Chen, Zheng, Wang and Wang ,year 2012).
乳酸‐グリコール酸共重合体(PLGA)により修飾されたヒアルロナンのミセルにパクリタキセルをうまく取り込んだ(Kim,Lee,Jang及びPark,2009年)。この取り込みは,ポリマーと個々の結合物質両方をDMSOに溶解させ,得られた溶液をH2Oに対して透析する透析方法を利用して行った。この場合,調製したポリマーミセルの結合能は4.5重量%であった。このような担体系の主要な短所はPLGAポリマーの存在であり,これは人体に対しては異質のものであり,完全な生分解系ではない。別の短所は最終生成物中にDMSOが残留することである。 Paclitaxel was successfully incorporated into hyaluronan micelles modified by lactic acid-glycolic acid copolymer (PLGA) (Kim, Lee, Jang and Park, 2009). This uptake was performed using a dialysis method in which both the polymer and the individual binding substances were dissolved in DMSO and the resulting solution was dialyzed against H 2 O. In this case, the binding capacity of the prepared polymer micelle was 4.5% by weight. The main disadvantage of such a carrier system is the presence of PLGA polymer, which is foreign to the human body and not a complete biodegradation system. Another disadvantage is that DMSO remains in the final product.
長鎖カルボン酸によるヒアルロナンの修飾
カルボン酸による多糖の修飾には,市販の所与の酸の無水物が必要である場合がほとんどである(WO1996/035720, WO2007/033677, (Smejkalova, Hermannova, Sulakova, Prusova, Kucerik及びVelebny, 2012年), EP0893451)。このような市販の無水物の主な短所は加水分解に対する感受性及び不純物が存在する可能性である。更に,数種の酸の無水物(例えばウンデカン‐カルボン酸)は市販されていない。入手可能な無水物の中には非常に高価なものもある(例えばオレイン酸,リノール酸又はリノレン酸の無水物)。従って,このような無水物の入手困難性,高価格及び不安定性により,修飾多糖の大規模調製が困難になる。
Modification of hyaluronan with long-chain carboxylic acids Modification of polysaccharides with carboxylic acids in most cases requires the anhydride of a given commercially available acid (WO1996 / 035720, WO2007 / 033677, (Smejkalova, Hermannova, Sulakova , Prusova, Kucerik and Velebny, 2012), EP0893451). The main disadvantage of such commercial anhydrides is the sensitivity to hydrolysis and the possible presence of impurities. In addition, several acid anhydrides (eg, undecane-carboxylic acid) are not commercially available. Some of the available anhydrides are very expensive (eg, oleic, linoleic or linolenic anhydrides). Therefore, the large scale preparation of modified polysaccharides becomes difficult due to the availability, high price and instability of such anhydrides.
酸無水物は,ヒアルロナンのエステル化に使用できる他の酸誘導体と置換可能である。特許WO2010/105582は,非水性条件でのクロロギ酸エチルによるカルボン酸の活性化法を特許請求しており,ここではO‐アシル‐O’‐アルキルカーボネートが形成され,その後これはヒアルロナンのエステル化に使用できる。このような活性化の短所は,毒性で爆発する可能性を有するガスが生成される点にある。クロロギ酸エチルによる同様の活性化法が特許U.S.3,720,662及びCZ20060605に開示されている。 The acid anhydride can be replaced with other acid derivatives that can be used for esterification of hyaluronan. Patent WO2010 / 105582 claims a method for the activation of carboxylic acids with ethyl chloroformate in non-aqueous conditions, where O-acyl-O'-alkyl carbonates are formed, which is then esterified with hyaluronan. Can be used for The disadvantage of such activation is that it produces toxic and potentially explosive gases. Similar activation methods with ethyl chloroformate are disclosed in patents U.S. 3,720,662 and CZ20060605.
別の公知の方法は,イミダゾールの存在下でのカルボン酸による多糖のエステル化に基づく(U.S.2012/0172587)。しかし,特許請求した調製方法は,高温下ではヒアルロナンは分解されてしまうことからヒアルロナンには応用できない高い反応温度(90〜200℃)を必要とする。 Another known method is based on the esterification of polysaccharides with carboxylic acids in the presence of imidazole (U.S. 2012/0172587). However, the claimed preparation method requires a high reaction temperature (90 to 200 ° C.) that cannot be applied to hyaluronan because hyaluronan is decomposed at high temperatures.
欧州特許EP0893451は超臨界抽出法を用いたカルボン酸の無水物による多糖のエステル化を特許請求している。このエステル化手技の短所は,高圧が必要であること及び設備費が高い点である。 European patent EP0893451 claims the esterification of polysaccharides with carboxylic anhydrides using a supercritical extraction method. The disadvantages of this esterification procedure are the high pressure required and the high equipment costs.
この理由から,原位置で利用できる方法としての長鎖カルボン酸活性化の代替法を見出すことは非常に重要である。可能な技術的解決策の1つは,無水物の形成を伴う2,4,6‐トリクロロ安息香酸の誘導体によるカルボン酸の活性化に基づく。温和な反応条件下,大環状物質を急速にエステル化する為,初めてDMAP触媒と組み合わせて2,4,6‐トリクロロ安息香酸の無水物を使用した(Inanaga,Hirata,Saeki,Katsuki及びYamaguchi,1979年)。しかし,発熱反応には個々の多糖の分解が伴う可能性があることから,このエステル化法は特にヒアルロン酸の多糖の修飾にはまだ応用していない。 For this reason, it is very important to find an alternative to long chain carboxylic acid activation as a method available in situ. One possible technical solution is based on the activation of carboxylic acids by derivatives of 2,4,6-trichlorobenzoic acid with the formation of anhydrides. For the first time, 2,4,6-trichlorobenzoic anhydride was used in combination with DMAP catalyst to rapidly esterify macrocycles under mild reaction conditions (Inanaga, Hirata, Saeki, Katsuki and Yamaguchi, 1979). Year). However, since the exothermic reaction may involve degradation of individual polysaccharides, this esterification method has not yet been applied to the modification of hyaluronic acid polysaccharides.
発明の概要
本発明の対象はヒアルロン酸の疎水化誘導体を合成すること,並びに水性環境で生物学的に活性な疎水性物質の為のポリマー担体の形態を取る前記誘導体を利用することであり,ここでは結合した物質の先天的性質及び生物学的活性は,未変化のままにしなければならない。
SUMMARY OF THE INVENTION The object of the present invention is to synthesize a hydrophobized derivative of hyaluronic acid and to utilize said derivative in the form of a polymer carrier for a biologically active hydrophobic substance in an aqueous environment, Here the innate nature and biological activity of the bound substance must remain unchanged.
長鎖脂肪族エステルによるヒアルロナン(C6‐C18)の疎水化は,2,4,6‐トリクロロ安息香酸の無水物の形成を伴う長鎖カルボン酸の直接的活性化に基づいている(スキーム1)。次の工程では,得られた無水物はヒアルロン酸と反応し,ヒアルロン酸のアシル化誘導体が形成される。前記活性化の主な利点は,脂肪族カルボン酸を直接的に利用できることにある。先行技術を構成する同様のヒアルロナン疎水化反応とは異なり,本発明の修飾には市販の無水物の使用は不要である。本発明の活性化の別の利点は,個々の反応が温和な条件下(室温〜50℃)で生じ,所要時間も短いところにある。更に,特許出願WO2010/105582で開示された方法とは異なり,本願の活性化には大量の脂肪酸も特別な無水条件も不要である。活性化試薬は安定的であり,また,クロロギ酸エチルとは異なり,反応で使用する際に毒性かつ爆発性のガスが生成されることもない。本発明の活性化試薬の別の利点は,当該試薬が副産物による反応を起こさないこと,また架橋反応を誘発しないことにある。 Hydrophobization of hyaluronan (C6-C18) with long chain aliphatic esters is based on the direct activation of long chain carboxylic acids with the formation of 2,4,6-trichlorobenzoic anhydride (Scheme 1) . In the next step, the resulting anhydride reacts with hyaluronic acid to form an acylated derivative of hyaluronic acid. The main advantage of the activation is that the aliphatic carboxylic acid can be used directly. Unlike similar hyaluronan hydrophobization reactions that constitute the prior art, the modification of the present invention does not require the use of commercially available anhydrides. Another advantage of the activation of the present invention is that the individual reactions occur under mild conditions (room temperature to 50 ° C.) and the required time is short. Furthermore, unlike the method disclosed in patent application WO2010 / 105582, activation of the present application does not require large amounts of fatty acids or special anhydrous conditions. The activation reagent is stable and, unlike ethyl chloroformate, does not produce toxic and explosive gases when used in the reaction. Another advantage of the activating reagent of the present invention is that the reagent does not cause a reaction by a by-product and does not induce a crosslinking reaction.
2,4,6‐トリクロロ安息香酸の誘導体によるカルボン酸の活性化は,得られた活性化生成物のヒアルロン酸エステル化へのその後の利用と共に下記スキーム1に表す。
更に本発明は,ナノミセル系を調製する為の疎水化ヒアルロン酸の使用,並びに皮膚,毛髪及び粘膜を治療する為のヒアルロン酸のナノミセル中で結合した物質の利用,更に可能性のある他の局所適用に関するものである。カプセル封入した生物学的活性物質は,ポリマー担体の疎水性官能基との非共有的な物理的相互作用により,ヒアルロナンのナノミセル中で結合する。上記の方法でカプセル封入すると,このような物質は表面皮膚層(表皮),毛髪の全体構造,粘膜上皮に効果的に浸透する。その利点の1つは,ナノミセル中で結合した活性物質の浸透深度が,直接に非極性溶媒に溶解された際の同物質の深度より大きい点にある。 Furthermore, the present invention relates to the use of hydrophobized hyaluronic acid to prepare nanomicelle systems and the use of substances bound in nanomicelles of hyaluronic acid to treat skin, hair and mucous membranes, as well as other possible topical applications. It is about application. The encapsulated biologically active substance is bound in hyaluronan nanomicelles by non-covalent physical interaction with the hydrophobic functional groups of the polymer carrier. When encapsulated in the manner described above, such substances effectively penetrate the surface skin layer (epidermis), the entire structure of the hair, and the mucosal epithelium. One advantage is that the depth of penetration of the active substance bound in the nanomicelle is greater than the depth of the substance when dissolved directly in a non-polar solvent.
類似のポリマー系(特許U.S.7,993,678及びWO2007/033677に開示されたポリマーなど)と比較して,ヒアルロナンのナノミセル抱合体は予想に反して3倍〜10倍低い臨界ミセル濃度を示すと同時に,塩分環境及び水性環境下で比較的高い安定性を示した。このような非常に低い臨界ミセル濃度の値はヒアルロナンミセルの静脈内投与が可能であるという点で特に有利である。先行技術を参照して述べた他の大半の公知のポリマーをベースとする類似の系とは異なり,凍結前に溶液へ凍結乾燥防止剤を添加することなく目的のカプセル封入を維持することができる為,ヒアルロナンミセルの高い安定性は多様な材料を凍結乾燥するという点で有利である可能性がある。
Compared to similar polymer systems (such as those disclosed in patents US7,993,678 and WO2007 / 033677), hyaluronan nanomicelle conjugates unexpectedly show a
特許請求されたヒアルロナンの疎水化誘導体の別の利点としては,その他の場合ではナノミセルの形成に必要であることが多く,反復投与されると抗体の形成を誘発する可能性がある合成ポリマー(PLGA,PEG等)及びコポリマーが不要であることが挙げられる(Gong,Chen,Wang&Wang)。 Another advantage of the hydrophobized derivative of the claimed hyaluronan is a synthetic polymer (PLGA), which is often necessary for nanomicelle formation in other cases and can induce antibody formation upon repeated administration. , PEG, etc.) and copolymers are unnecessary (Gong, Chen, Wang & Wang).
類似のポリマーミセル系とは異なり,ナノミセルの形成は全負電荷が増加した修飾ヒアルロナンをベースとしてはおらず,従って担体系と細胞との相互作用は,不都合にもこのような電荷の影響を受けない。 Unlike similar polymer micelle systems, the formation of nanomicelles is not based on modified hyaluronan with an increased total negative charge, and thus the interaction between the carrier system and the cells is unaffected by such charges. .
ナノミセル構造,特にヒアルロナンに結合した長めのアシル鎖を含む構造は,水性環境下でゲル相を形成し得るものもある。このようなゲル相は,個々の担体系の粘度増加を必要とする特定の用途において有利に使用し得る。 Some nanomicelle structures, particularly those containing longer acyl chains attached to hyaluronan, can form a gel phase in an aqueous environment. Such gel phases can be advantageously used in certain applications that require increased viscosity of the individual carrier system.
調製したナノミセル系の寸法は,大部分が20〜100nmであり,これは医療用途に最適な寸法であり,高浸透度及び保持効果(EPR効果)の有利性が得られる。先行技術を参照して説明したポリマーミセルにおいて,このような寸法は必ずしも得られるわけではない。 The size of the prepared nano micelle system is mostly 20 to 100 nm, which is the optimal size for medical use, and the advantages of high permeability and retention effect (EPR effect) are obtained. In the polymer micelles described with reference to the prior art, such dimensions are not always obtained.
ヒアルロナンミセルの別の利点は,結合した物質がナノミセルの疎水性ドメインから細胞へと移行することに関連している。 Another advantage of hyaluronan micelles is related to the transfer of bound material from the hydrophobic domain of nanomicelles to the cell.
この核心的なカプセル封入法は,水中に溶解したヒアルロナンと有機溶媒に溶解した生物学的活性物質との混合物の調製,次いでヒアルロナンの水和エンベロープのエネルギー崩壊,及びその後の溶液からの溶媒の除去に基づく。一般的なカプセル封入手技とは対照的に,上記の方法は有機溶媒へのポリマーの溶解度に基づいておらず,従って,特にヒアルロナンが細胞受容体により認識されることを可能にする重要なカルボキシル基の位置で,ヒアルロン酸の先天的性質が抑制されること,及び,ポリマー鎖が大幅に修飾されることを必要としない。有機溶媒は水より低い沸点を有するものが好ましい。ヒアルロナンナノミセルの結合能は,残余水相の完全な蒸発と,その後のナノミセルの再水和によって顕著に増加する。結合していない生物学的活性物質は濾過工程で除去し,得られたナノミセルは直接凍結乾燥し,その後の再水和の時間になるまで乾燥状態で保存することが可能である。 This core encapsulation method involves the preparation of a mixture of hyaluronan dissolved in water and a biologically active substance dissolved in an organic solvent, followed by the energy decay of the hyaluronan hydration envelope and subsequent removal of the solvent from the solution. based on. In contrast to common encapsulation procedures, the above method is not based on the solubility of the polymer in organic solvents, and thus, especially important carboxyl groups that allow hyaluronan to be recognized by cell receptors. In this position, it is not necessary that the innate properties of hyaluronic acid be suppressed and that the polymer chain be significantly modified. The organic solvent preferably has a lower boiling point than water. The binding capacity of hyaluronan nanomicelles is significantly increased by complete evaporation of the residual aqueous phase and subsequent rehydration of the nanomicelles. Unbound biologically active material can be removed by a filtration step, and the resulting nanomicelles can be directly lyophilized and stored dry until the time for subsequent rehydration.
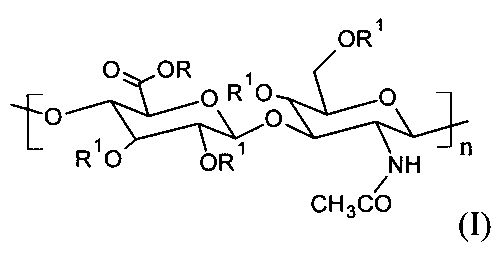
本発明は特に,一般式(I)に従ったヒアルロン酸のC6‐C18‐アシル化誘導体であって:
更に,本発明は,前記ヒアルロン酸の誘導体の調製方法に関し,水と水混和性非プロトン性溶媒との混合液中,塩基及び触媒の存在下で,ヒアルロン酸は,2,4,6‐トリクロロ安息香酸の塩化物又はR3‐CO‐Clの有機塩素化合物により活性化するC6‐C18‐カルボン酸と反応し,R3は,場合により複素芳香族又は芳香族官能基を含む脂肪族又は分岐C1‐C30アルキルである。例示的な複素芳香族官能基としては(例えば式(i)に従った)ピリジンとその誘導体が挙げられ,例示的な芳香族官能基としては(例えば式(ii)に従った)ベンゼンとそのハロゲン誘導体が挙げられる。
ヒアルロン酸は遊離酸形態,又はNa,K,Ca,Mg,Zn若しくはLi塩などの薬学的に許容可能な塩の形態を取り,分子量は好ましくは5×103〜1.6×106,より好ましくは15×103〜250×103,最も好ましくは15×103〜50×103である。本発明に従った調製方法は,ヒアルロン酸を水と水混和性の非プロトン性溶媒との混合液に溶解することにあり,ここではその溶媒は極性有機溶媒であり,水分含量は10〜99体積%,好ましくは50体積%である。水混和性の非プロトン性溶媒は,例えばジメチルスルホキシド(DMSO),テトラヒドロフラン(THF),アセトン,アセトニトリル又はイソプロパノール(IPA)であってもよい。反応混合液にはR’3N塩基が含まれ,R’は直鎖又は分岐CnHm炭化水素鎖であり,式中nは1〜4の整数であり,mは3〜9の整数であり,例えばトリエチルアミンの量はヒアルロン酸の二量体に対して0.01〜20当量,好ましくは6当量であり,触媒はジメチルアミノピリジンなどの置換ピリジンから成る群より選択され,その量はヒアルロン酸の二量体に対して0.01〜1当量,好ましくは0.05当量である。本発明に従った調製方法では,初めにC6‐C18‐カルボン酸の活性化を極性有機溶媒中,塩基及び2,4,6‐トリクロロ安息香酸又はその誘導体の存在下,或いは塩基及び有機塩化物の存在下で行い,その後,活性化C6‐C18‐カルボン酸を含む混合液をヒアルロン酸に添加し,これを水,有機溶媒,塩基及び触媒の混合液中に溶解し,その得られた反応の生成物は一般式(I)に従った誘導体である。前記C6‐C18‐カルボン酸はカプロン酸,エナント酸,カプリル酸,カプリン酸,パルミチン酸,ステアリン酸,オレイン酸,リノール酸及びリノレン酸から成る群より選択される。活性化C6‐C18‐カルボン酸の量はヒアルロン酸の二量体に対して0.01〜5当量,好ましくは0.5〜2当量である。C6‐C18‐カルボン酸の活性化は,20〜60℃,好ましくは25℃で,5〜120分間,好ましくは30分間行う。ヒアルロン酸と活性化C6‐C18‐カルボン酸との反応は20〜60℃,好ましくは25℃で,1〜24時間,好ましくは2〜3時間行う。その後,ヒアルロン酸のC6‐C18‐アシル化誘導体を反応混合液から分離し,洗浄し,乾燥し,凍結乾燥させることも可能である。誘導体はNaCl及びアルコールを使用して沈殿により反応混合液から分離させてもよい。その後,誘導体をアルコール,特にイソプロパノール又はエタノールで洗浄してもよい。 Hyaluronic acid takes the free acid form or the form of a pharmaceutically acceptable salt such as Na, K, Ca, Mg, Zn or Li salt, and the molecular weight is preferably 5 × 10 3 to 1.6 × 10 6 , more preferably 15 × 10 3 ~250 × 10 3 , and most preferably 15 × 10 3 ~50 × 10 3 . The preparation method according to the invention consists in dissolving hyaluronic acid in a mixture of water and a water-miscible aprotic solvent, where the solvent is a polar organic solvent and the water content is 10-99. % By volume, preferably 50% by volume. The water miscible aprotic solvent may be, for example, dimethyl sulfoxide (DMSO), tetrahydrofuran (THF), acetone, acetonitrile or isopropanol (IPA). The reaction mixture contains R ′ 3 N base, where R ′ is a linear or branched C n H m hydrocarbon chain, where n is an integer from 1 to 4 and m is an integer from 3 to 9. For example, the amount of triethylamine is 0.01 to 20 equivalents, preferably 6 equivalents, relative to the dimer of hyaluronic acid, and the catalyst is selected from the group consisting of substituted pyridines such as dimethylaminopyridine, It is 0.01-1 equivalent with respect to the dimer of hyaluronic acid, Preferably it is 0.05 equivalent. In the preparation process according to the invention, the activation of C 6 -C 18 -carboxylic acid is first carried out in a polar organic solvent in the presence of a base and 2,4,6-trichlorobenzoic acid or a derivative thereof, or In the presence of chloride, after which a mixture containing activated C 6 -C 18 -carboxylic acid is added to hyaluronic acid, which is dissolved in a mixture of water, organic solvent, base and catalyst, The product of the reaction obtained is a derivative according to general formula (I). The C 6 -C 18 -carboxylic acid is selected from the group consisting of caproic acid, enanthic acid, caprylic acid, capric acid, palmitic acid, stearic acid, oleic acid, linoleic acid and linolenic acid. The amount of activated C 6 -C 18 -carboxylic acid is 0.01-5 equivalents, preferably 0.5-2 equivalents, relative to the dimer of hyaluronic acid. Activation of the C 6 -C 18 -carboxylic acid is carried out at 20 to 60 ° C., preferably 25 ° C., for 5 to 120 minutes, preferably 30 minutes. The reaction between hyaluronic acid and activated C 6 -C 18 -carboxylic acid is carried out at 20-60 ° C., preferably 25 ° C., for 1-24 hours, preferably 2-3 hours. Thereafter, the C 6 -C 18 -acylated derivative of hyaluronic acid can be separated from the reaction mixture, washed, dried and lyophilized. The derivative may be separated from the reaction mixture by precipitation using NaCl and alcohol. The derivative may then be washed with alcohol, in particular isopropanol or ethanol.
更なる態様では,本発明は一般式(I)に従ったヒアルロン酸のC6‐C18‐アシル化誘導体をベースとするナノミセル組成物に関するものであり,この組成物はヒアルロン酸に結合したC6‐C18‐アシル基に形成された疎水性のコアとヒアルロン酸の親水性官能基に形成された親水性のシェルから成るナノミセルを含み,ここに1種以上の生物学的活性物質が各ナノミセル中で物理的に結合している。当該組成物は更に水を含み,また塩(例えば0.9%のNaCl)を含んでもよい。好ましい実施態様では,ナノミセル組成物はヒアルロン酸のC6‐C18‐アシル化誘導体の質量含有量に対して0.3〜50重量%の生物学的活性物質を含み,該生物学的活性物質は薬学的及び美容的活性物質,特にビタミン,医薬,細胞増殖抑制剤,植物エキス,植物複合体若しくは植物活性物質,鉱物或いは植物油,又はこれらの混合物から成る群より選択される。利用可能な生物学的活性物質の例としては,例えばトコフェロール,パクリタキセル,ホスファチジルコリン又はコエンザイムQ10が挙げられる。好ましい実施態様では,当該組成物はその臨界凝集濃度より高い濃度でヒアルロン酸のC6‐C18‐アシル化誘導体を含んでいる。当該組成物が水溶液の状態であるとき,ヒアルロン酸のC6‐C18‐アシル化誘導体の濃度は,0.0001mg・mL-1〜30mg・mL-1,好ましくは1〜20mg・mL-1の範囲である。別の好ましい実施態様では,生物学的活性物質は,ヒアルロン酸のC6‐C18‐アシル化誘導体の質量含有量に対して0.05〜40重量%,好ましくは1〜20重量%の量で含まれる鉱物又は植物油である。更に別の好ましい実施態様では,当該組成物は,液体で水不溶性の生物学的活性物質を含み,前記物質には,そこに溶解した状態の追加的生物学的活性物質が含まれている。液体で水不溶性のこのような生物学的活性物質は例えば鉱物又は植物油であってもよく,追加的生物学的活性物質は例えば薬学又は美容的活性物質,特にビタミン,医薬,細胞増殖抑制剤,植物エキス,植物複合体若しくは植物活性物質,又はこれらの混合物に属するものであってもよい。本発明に従ったナノミセル組成物は溶液,ナノエマルジョン,マイクロエマルジョン,コアセルベート又はゲルの形態を取ってもよい。
In a further aspect, the present invention relates to a nanomicelle composition based on a C 6 -C 18 -acylated derivative of hyaluronic acid according to general formula (I), which composition is C-linked to hyaluronic acid. A nanomicelle comprising a hydrophobic core formed on a 6- C 18 -acyl group and a hydrophilic shell formed on a hydrophilic functional group of hyaluronic acid, wherein one or more biologically active substances are each They are physically bonded in nanomicelles. The composition further includes water and may include a salt (eg, 0.9% NaCl). In a preferred embodiment, the nanomicelle composition comprises 0.3 to 50% by weight of biologically active substance relative to the mass content of the C 6 -C 18 -acylated derivative of hyaluronic acid, said biologically active substance Is selected from the group consisting of pharmaceutically and cosmetically active substances, in particular vitamins, medicaments, cytostatics, plant extracts, plant complexes or plant active substances, minerals or vegetable oils, or mixtures thereof. Examples of biologically active substances that can be used include tocopherol, paclitaxel, phosphatidylcholine or coenzyme Q10. In a preferred embodiment, the composition comprises a C 6 -C 18 -acylated derivative of hyaluronic acid at a concentration above its critical aggregation concentration. When the composition is in the form of an aqueous solution, the concentration of the C 6 -C 18 -acylated derivative of hyaluronic acid is 0.0001 mg ·
本発明は更に上記で定義したようなナノミセル組成物の調製方法に関連しており,ここでは,一般式(I)に従ったヒアルロン酸のC6‐C18‐アシル化誘導体を水に溶解し,生物学的活性物質を有機溶媒に溶解し,得られた溶液を混合し,その後,有機溶媒を除去する。有機溶媒はトリクロロメタンなどの揮発性塩化溶媒,又はエタノールやイソプロパノールなどのアルコールであってもよく,その除去は真空蒸発で行ってもよい。次に,水相を乾燥させ,そして再水和させ,得られたナノミセル構造物を濾過し,最後に凍結乾燥させる。或いは,有機溶媒は透析により除去してもよい。その後に,再度,得られたナノミセル構造を濾過し,最後に凍結乾燥させる。好ましい実施態様では,一般式(I)に従ったヒアルロン酸のC6‐C18‐アシル化誘導体を水に溶解し,次いで,液体で水不溶性の生物学的活性物質と混合し,そこで,得られた混合液を音波処理によりホモジナイズし,マイクロエマルジョン又はナノエマルジョンを形成する。別の好ましい実施態様では,一般式(I)に従ったヒアルロン酸のC6‐C18‐アシル化誘導体を水に溶解し,次いで,追加的生物学的活性物質が溶解されている液体で水不溶性の生物学的活性物質と混合し,そこで得られた混合液を音波処理によりホモジナイズし,マイクロエマルジョン又はナノエマルジョンを形成する。 The invention further relates to a process for the preparation of a nanomicelle composition as defined above, wherein a C 6 -C 18 -acylated derivative of hyaluronic acid according to general formula (I) is dissolved in water. The biologically active substance is dissolved in an organic solvent, the resulting solution is mixed, and then the organic solvent is removed. The organic solvent may be a volatile chlorinated solvent such as trichloromethane, or an alcohol such as ethanol or isopropanol, and the removal may be performed by vacuum evaporation. The aqueous phase is then dried and rehydrated and the resulting nanomicelle structure is filtered and finally lyophilized. Alternatively, the organic solvent may be removed by dialysis. Thereafter, the obtained nanomicelle structure is again filtered and finally freeze-dried. In a preferred embodiment, a C 6 -C 18 -acylated derivative of hyaluronic acid according to general formula (I) is dissolved in water and then mixed with a liquid, water-insoluble biologically active substance, where The resulting mixture is homogenized by sonication to form a microemulsion or nanoemulsion. In another preferred embodiment, a C 6 -C 18 -acylated derivative of hyaluronic acid according to general formula (I) is dissolved in water and then in a liquid in which the additional biologically active substance is dissolved. Mix with the insoluble biologically active material and homogenize the resulting mixture by sonication to form a microemulsion or nanoemulsion.
更に別の態様では,本発明は医療用途又は化粧用途,好ましくは局所適用でのナノミセル組成物の使用に関連している。 In yet another aspect, the present invention relates to the use of nanomicelle compositions for medical or cosmetic applications, preferably topical applications.
更に,安定化させたナノミセル組成物を調製してもよい。このような安定化ナノミセル組成物の調製方法は一般式(II)に従ったC6‐C18‐アシル化ヒアルロナンを調製することにある:
特に,安定化は以下の方法で行う:最初に,一般式(III)に従った誘導体を調製し:
DS=置換度=結合した置換基の100%*モル量/全多糖二量体のモル量
特段の規定がない限り,本明細書で使用する表現「当量」(eq)はヒアルロン酸の二量体に対するものを意味する。特段の規定がない限り,百分率は重量/重量(weight/weight basis)に基づいて計算されている。
大部分のヒアルロン酸の分子量はSEC‐MALLS法により測定した(ソース:コンティプロ ビオテック スポレチノスト エス ルチェニム オメゼニム(Contipro Biotech s.r.o),ドルニ ドブロウク,チェコ)。
DS = degree of substitution = 100% of bound substituents * molar amount / molar amount of total polysaccharide dimer Unless otherwise specified, the expression “equivalent” (eq) used herein is the dimer of hyaluronic acid. Means for the body. Unless otherwise specified, percentages are calculated on a weight / weight basis.
The molecular weight of most hyaluronic acids was determined by the SEC-MALLS method (source: Contipro Biotech srochenim omezenim, Dornidbroke, Czech).
実施例1 2,4,6‐トリクロロ安息香酸及びカプロン酸の混合アルデヒドによるヒアルロン酸のカプロニル(capronyl)(C6)誘導体の調製
1gのヒアルロン酸ナトリウム(2.5mmol,15kDa)を10mLの脱塩水に溶解した。その後,5mLのDMSOを徐々に添加した。次にTEA(1.05mL,3eq)及びDMAP(8.0mg,0.05eq)を溶液に添加した。同時にヘキサン酸(0.63mL,2eq)を5mLのDMSO及びTEA(1.05mL,3eq)に溶解し,その後,2,4,6‐トリクロロベンゾイルクロリド(1.6mL,4eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.25gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMSO及びDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS60%(NMRから測定)
1H NMR(D2O)アシルのシグナル:δ2.4ppm(m,2H,αCH2),δ1.6ppm(m,2H,βCH2),δ1.3ppm(m,4H,γ,δCH2),δ0.8(m,3H,CH3)
Example 1 Preparation of capronyl (C6) derivative of hyaluronic acid with mixed aldehyde of 2,4,6-trichlorobenzoic acid and caproic acid 1 g of sodium hyaluronate (2.5 mmol, 15 kDa) in 10 mL of demineralized water Dissolved. Thereafter, 5 mL of DMSO was gradually added. TEA (1.05 mL, 3 eq) and DMAP (8.0 mg, 0.05 eq) were then added to the solution. Simultaneously hexanoic acid (0.63 mL, 2 eq) is dissolved in 5 mL DMSO and TEA (1.05 mL, 3 eq), then 2,4,6-trichlorobenzoyl chloride (1.6 mL, 4 eq) is added to the solution did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.25 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with aqueous isopropanol (85% by volume) first to remove DMSO and DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
DS 60% (measured from NMR)
1 H NMR (D 2 O) acyl signal: δ 2.4 ppm (m, 2H, αCH 2 ), δ 1.6 ppm (m, 2H, βCH 2 ), δ 1.3 ppm (m, 4H, γ, δCH 2 ), δ0.8 (m, 3H, CH 3 )
実施例2 2,4,6‐トリクロロ安息香酸及びカプロン酸の混合アルデヒドによるヒアルロン酸のカプロニル(C6)誘導体の調製
1gのヒアルロン酸ナトリウム(2.5mmol,38kDa)を10mLの脱塩水に溶解した。その後,5mLのイソプロパノールを徐々に添加した。次にTEA(1.05mL,3eq)及びピリジン(0.4mL,2.0eq)を溶液に添加した。同時にヘキサン酸(0.32mL,1eq)を5mLのイソプロパノールに溶解し,その後,TEA(1.05mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.391mL,1eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.50gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からピリジンを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS15%(NMRから測定)
1H NMR(D2O)アシルのシグナル:δ2.4ppm(m,2H,αCH2),δ1.6ppm(m,2H,βCH2),δ1.3ppm(m,4H,γ,δCH2),δ0.8(m,3H,CH3)
Example 2 Preparation of Capronyl (C6) Derivative of Hyaluronic Acid with Mixed Aldehyde of 2,4,6-Trichlorobenzoic Acid and Caproic Acid 1 g of sodium hyaluronate (2.5 mmol, 38 kDa) was dissolved in 10 mL of demineralized water. Thereafter, 5 mL of isopropanol was gradually added. TEA (1.05 mL, 3 eq) and pyridine (0.4 mL, 2.0 eq) were then added to the solution. Simultaneously, hexanoic acid (0.32 mL, 1 eq) is dissolved in 5 mL isopropanol, then TEA (1.05 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.391 mL, 1 eq) are added to the solution did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.50 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with aqueous isopropanol (85% by volume) first to remove pyridine from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
DS15% (measured from NMR)
1 H NMR (D 2 O) acyl signal: δ 2.4 ppm (m, 2H, αCH 2 ), δ 1.6 ppm (m, 2H, βCH 2 ), δ 1.3 ppm (m, 4H, γ, δCH 2 ), δ0.8 (m, 3H, CH 3 )
実施例3 2,4,6‐トリクロロ安息香酸及びエナント酸の混合アルデヒドによるヒアルロン酸のエナンチル(C7)誘導体の調製
1gのヒアルロン酸ナトリウム(2.5mmol,15kDa)を10mLの脱塩水に溶解した。その後,5mLのアセトニトリルを徐々に添加した。次にTEA(0.70mL,2eq)及びDMAP(15.0mg,0.05eq)を溶液に添加した。同時にエナント酸(0.35mL,1eq)を5mLのアセトニトリルに溶解し,その後,TEA(0.70mL,2eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.39mL,1eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.75gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からアセトニトリル及びDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS12%(NMRから測定)
1H NMR(D2O)アシルのシグナル:δ2.4ppm(m,2H,αCH2),δ1.6ppm(m,2H,βCH2),δ1.3ppm(m,6H,γ,δ,ε(CH2)3),δ0.8(m,3H,CH3)
Example 3 Preparation of Enantyl (C7) Derivative of Hyaluronic Acid with Mixed Aldehyde of 2,4,6-Trichlorobenzoic Acid and Enanthic Acid 1 g of sodium hyaluronate (2.5 mmol, 15 kDa) was dissolved in 10 mL of demineralized water. Thereafter, 5 mL of acetonitrile was gradually added. TEA (0.70 mL, 2 eq) and DMAP (15.0 mg, 0.05 eq) were then added to the solution. At the same time, enanthic acid (0.35 mL, 1 eq) is dissolved in 5 mL of acetonitrile, and then TEA (0.70 mL, 2 eq) and 2,4,6-trichlorobenzoyl chloride (0.39 mL, 1 eq) are added to the solution. did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.75 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with aqueous isopropanol (85% by volume) first to remove acetonitrile and DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
DS12% (measured from NMR)
1 H NMR (D 2 O) acyl signal: δ 2.4 ppm (m, 2H, αCH 2 ), δ 1.6 ppm (m, 2H, βCH 2 ), δ 1.3 ppm (m, 6H, γ, δ, ε ( CH 2 ) 3 ), δ 0.8 (m, 3H, CH 3 )
実施例4 2,4,6‐トリクロロ安息香酸及びカプリル酸の混合アルデヒドによるヒアルロン酸のカプリリル(C8)誘導体の調製
1gのヒアルロン酸ナトリウム(2.5mmol,15kDa)を10mLの脱塩水に溶解した。その後,5mLのアセトニトリルを徐々に添加した。次にTEA(1.05mL,3eq)及びDMAP(8.0mg,0.05eq)を溶液に添加した。同時にオクタン酸(0.63g,4eq)を5mLのアセトニトリルに溶解し,その後,TEA(1.05mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.8mL,4eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.50gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からアセトニトリル及びDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS40%(NMRから測定)
1H NMR(D2O)アシルのシグナル:δ2.4ppm(m,2H,αCH2),δ1.6ppm(m,2H,βCH2),δ1.3ppm(m,8H,(CH2)4),δ0.8(m,3H,CH3)
Example 4 Preparation of Caprylyl (C8) Derivative of Hyaluronic Acid with Mixed Aldehyde of 2,4,6-Trichlorobenzoic Acid and Caprylic Acid 1 g of sodium hyaluronate (2.5 mmol, 15 kDa) was dissolved in 10 mL of demineralized water. Thereafter, 5 mL of acetonitrile was gradually added. TEA (1.05 mL, 3 eq) and DMAP (8.0 mg, 0.05 eq) were then added to the solution. At the same time, octanoic acid (0.63 g, 4 eq) is dissolved in 5 mL of acetonitrile, and then TEA (1.05 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.8 mL, 4 eq) are added to the solution. did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.50 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with aqueous isopropanol (85% by volume) first to remove acetonitrile and DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
1 H NMR (D 2 O) acyl signal: δ 2.4 ppm (m, 2H, αCH 2 ), δ 1.6 ppm (m, 2H, βCH 2 ), δ 1.3 ppm (m, 8H, (CH 2 ) 4 ) , Δ0.8 (m, 3H, CH 3 )
実施例5 2,4,6‐トリクロロ安息香酸及びカプリン酸の混合アルデヒドによるヒアルロン酸のカプリニル(caprinyl)(C10)誘導体の調製
1gのヒアルロン酸ナトリウム(2.5mmol,15kDa)を10mLの脱塩水に溶解した。その後,5mLのTHFを徐々に添加した。次にTEA(1.05mL,3eq)及びDMAP(8.0mg,0.025eq)を溶液に添加した。同時にデカン酸(0.8g,2eq)を5mLのTHFに溶解し,その後,TEA(1.05mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.8mL,2eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.25gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS15%(NMRから測定)
1H NMR(D2O)アシルのシグナル:δ2.4ppm(m,2H,αCH2),δ1.6ppm(m,2H,βCH2),δ1.3ppm(m,12H,γ,δ,ε,ζ,η,θCH2),δ0.8(m,3H,CH3)
Example 5 Preparation of caprinyl (C10) derivative of hyaluronic acid with mixed aldehydes of 2,4,6-trichlorobenzoic acid and capric acid 1 g of sodium hyaluronate (2.5 mmol, 15 kDa) in 10 mL of demineralized water Dissolved. Thereafter, 5 mL of THF was gradually added. TEA (1.05 mL, 3 eq) and DMAP (8.0 mg, 0.025 eq) were then added to the solution. At the same time decanoic acid (0.8 g, 2 eq) is dissolved in 5 mL of THF, then TEA (1.05 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.8 mL, 2 eq) are added to the solution did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.25 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
DS15% (measured from NMR)
1 H NMR (D 2 O) acyl signal: δ 2.4 ppm (m, 2H, αCH 2 ), δ 1.6 ppm (m, 2H, βCH 2 ), δ 1.3 ppm (m, 12H, γ, δ, ε, ζ, η, θCH 2 ), δ 0.8 (m, 3H, CH 3 )
実施例6 2,4,6‐トリクロロ安息香酸及びカプリン酸の混合アルデヒドによるヒアルロン酸のカプリニル(C10)誘導体の調製
1gのヒアルロン酸ナトリウム(2.5mmol,15kDa)を10mLの脱塩水に溶解した。その後,5mLのTHFを徐々に添加した。次にTEA(1.05mL,3eq)及びDMAP(8.0mg,0.025eq)を溶液に添加した。同時にデカン酸(0.8g,4eq)を5mLのTHFに溶解し,その後,TEA(1.05mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.8mL,4eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.25gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からTHF及びDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS40%(NMRから測定)
1H NMR(D2O)アシルのシグナル:δ2.4ppm(m,2H,αCH2),δ1.6ppm(m,2H,βCH2),δ1.3ppm(m,12H,γ,δ,ε,ζ,η,θCH2),δ0.8(m,3H,CH3)
Example 6 Preparation of Caprinyl (C10) Derivative of Hyaluronic Acid with Mixed Aldehyde of 2,4,6-Trichlorobenzoic Acid and Capric Acid 1 g of sodium hyaluronate (2.5 mmol, 15 kDa) was dissolved in 10 mL of demineralized water. Thereafter, 5 mL of THF was gradually added. TEA (1.05 mL, 3 eq) and DMAP (8.0 mg, 0.025 eq) were then added to the solution. At the same time, decanoic acid (0.8 g, 4 eq) is dissolved in 5 mL of THF, and then TEA (1.05 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.8 mL, 4 eq) are added to the solution. did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.25 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with aqueous isopropanol (85% by volume) first to remove THF and DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
1 H NMR (D 2 O) acyl signal: δ 2.4 ppm (m, 2H, αCH 2 ), δ 1.6 ppm (m, 2H, βCH 2 ), δ 1.3 ppm (m, 12H, γ, δ, ε, ζ, η, θCH 2 ), δ 0.8 (m, 3H, CH 3 )
実施例7 2,4,6‐トリクロロ安息香酸及びパルミチン酸の混合アルデヒドによるヒアルロン酸のパルミトイル(C16)誘導体の調製
0.5gのヒアルロン酸ナトリウム(1.25mmol,38kDa)を20mLの脱塩水に溶解した。その後,10mLのTHFを徐々に添加した。次にTEA(0.52mL,3eq)及びDMAP(8.0mg,0.05eq)を溶液に添加した。同時にパルミチン酸(0.16g,0.5eq)を10mLのTHFに溶解し,その後,TEA(0.52mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.098mL,0.5eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.25gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
1H NMR(D2O)アシルのシグナル:δ2.4ppm(m,2H,αCH2),δ1.6ppm(m,2H,βCH2),δ1.3ppm(m,24H,(CH2)12),δ0.8(m,3H,CH3).DS14%(NMRから測定)
Example 7 Preparation of palmitoyl (C16) derivative of hyaluronic acid with a mixed aldehyde of 2,4,6-trichlorobenzoic acid and palmitic acid 0.5 g sodium hyaluronate (1.25 mmol, 38 kDa) dissolved in 20 mL demineralized water did. Thereafter, 10 mL of THF was gradually added. TEA (0.52 mL, 3 eq) and DMAP (8.0 mg, 0.05 eq) were then added to the solution. At the same time, palmitic acid (0.16 g, 0.5 eq) is dissolved in 10 mL of THF, and then TEA (0.52 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.098 mL, 0.5 eq) are added. Added to the solution. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.25 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
1 H NMR (D 2 O) acyl signal: δ 2.4 ppm (m, 2H, αCH 2 ), δ 1.6 ppm (m, 2H, βCH 2 ), δ 1.3 ppm (m, 24H, (CH 2 ) 12 ) , Δ 0.8 (m, 3H, CH 3 ).
実施例8 2,4,6‐トリクロロ安息香酸及びステアリン酸の混合アルデヒドによるヒアルロン酸のステアリル(C18)誘導体の調製
0.5gのヒアルロン酸ナトリウム(1.25mmol,15kDa)を10mLの脱塩水に溶解した。その後,5mLのTHFを徐々に添加した。次にTEA(0.52mL,3eq)及びDMAP(8.0mg,0.05eq)を溶液に添加した。同時にステアリン酸(0.711g,2eq)を5mLのTHFに溶解し,その後,TEA(0.52mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.391mL,2eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた後,反応混合液を50℃で1時間加温した。その後,0.25gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS7%(NMRから測定)
1H NMR(D2O)アシルのシグナル:δ2.4ppm(m,2H,αCH2),δ1.6ppm(m,2H,βCH2),δ1.3ppm(m,28H,(CH2)14),δ0.8(m,3H,CH3)
Example 8 Preparation of stearyl (C18) derivative of hyaluronic acid with a mixed aldehyde of 2,4,6-trichlorobenzoic acid and stearic acid 0.5 g sodium hyaluronate (1.25 mmol, 15 kDa) dissolved in 10 mL demineralized water did. Thereafter, 5 mL of THF was gradually added. TEA (0.52 mL, 3 eq) and DMAP (8.0 mg, 0.05 eq) were then added to the solution. At the same time stearic acid (0.711 g, 2 eq) is dissolved in 5 mL of THF, then TEA (0.52 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.391 mL, 2 eq) are added to the solution did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. After reacting at room temperature for 3 hours, the reaction mixture was warmed at 50 ° C. for 1 hour. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.25 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
1 H NMR (D 2 O) acyl signal: δ 2.4 ppm (m, 2H, αCH 2 ), δ 1.6 ppm (m, 2H, βCH 2 ), δ 1.3 ppm (m, 28H, (CH 2 ) 14 ) , Δ0.8 (m, 3H, CH 3 )
実施例9 2,4,6‐トリクロロ安息香酸及びオレイン酸の混合アルデヒドによるヒアルロン酸のオレイル(C18:1)誘導体の調製
0.5gのヒアルロン酸ナトリウム(1.25mmol,15kDa)を10mLの脱塩水に溶解した。その後,5mLのTHFを徐々に添加した。次にTEA(0.52mL,3eq)及びDMAP(15.0mg,0.1eq)を溶液に添加した。同時にオレイン酸(0.18g,0.5eq)を5mLのTHFに溶解し,その後,TEA(0.52mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.098mL,0.5eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.25gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS10%(NMRから測定)
1H NMR(D2O):δ0.88(t,3H,‐CH2‐CH3),δ1.22‐1.35(m,20H,(‐CH2‐)10),
δ1.60(m,2H,‐CH2‐CH2‐CO‐),δ2.0ppm(m,4H,(CH2)2),δ2.41(t,2H,‐CH2‐CO‐),δ5.41(d,2H,CH=CH)
Example 9 Preparation of an oleyl (C18: 1) derivative of hyaluronic acid with a mixed aldehyde of 2,4,6-trichlorobenzoic acid and oleic acid 0.5 g of sodium hyaluronate (1.25 mmol, 15 kDa) in 10 mL of demineralized water Dissolved in. Thereafter, 5 mL of THF was gradually added. TEA (0.52 mL, 3 eq) and DMAP (15.0 mg, 0.1 eq) were then added to the solution. At the same time, oleic acid (0.18 g, 0.5 eq) is dissolved in 5 mL of THF followed by TEA (0.52 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.098 mL, 0.5 eq). Added to the solution. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.25 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
1 H NMR (D 2 O): δ 0.88 (t, 3H, —CH 2 —CH 3 ), δ 1.22-1.35 (m, 20H, (—CH 2 —) 10 ),
δ 1.60 (m, 2H, —CH 2 —CH 2 —CO—), δ 2.0 ppm (m, 4H, (CH 2 ) 2 ), δ 2.41 (t, 2H, —CH 2 —CO—), δ 5.41 (d, 2H, CH = CH)
実施例10 2,4,6‐トリクロロ安息香酸及びオレイン酸の混合アルデヒドによるヒアルロン酸のオレイル(C18:1)誘導体の調製
0.5gのヒアルロン酸ナトリウム(1.25mmol,130kDa)を5mLの脱塩水に溶解した。その後,3mLのイソプロパノールを徐々に添加した。次にTEA(0.52mL,3eq)及びDMAP(15.0mg,0.1eq)を溶液に添加した。同時にオレイン酸(0.4mL,1eq)を5mLのイソプロパノールに溶解し,その後,TEA(0.52mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.195mL,1eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.25gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS12%(NMRから測定)
1H NMR(D2O):δ0.88(t,3H,‐CH2‐CH3),δ1.22‐1.35(m,20H,(‐CH2‐)10),
δ1.60(m,2H,‐CH2‐CH2‐CO‐),δ2.0ppm(m,4H,(CH2)2),δ2.41(t,2H,‐CH2‐CO‐),δ5.41(d,2H,CH=CH)
Example 10 Preparation of an oleyl (C18: 1) derivative of hyaluronic acid with a mixed aldehyde of 2,4,6-trichlorobenzoic acid and oleic acid 0.5 g of sodium hyaluronate (1.25 mmol, 130 kDa) in 5 mL of demineralized water Dissolved in. Thereafter, 3 mL of isopropanol was gradually added. TEA (0.52 mL, 3 eq) and DMAP (15.0 mg, 0.1 eq) were then added to the solution. Simultaneously oleic acid (0.4 mL, 1 eq) is dissolved in 5 mL of isopropanol, then TEA (0.52 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.195 mL, 1 eq) are added to the solution did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.25 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
DS12% (measured from NMR)
1 H NMR (D 2 O): δ 0.88 (t, 3H, —CH 2 —CH 3 ), δ 1.22-1.35 (m, 20H, (—CH 2 —) 10 ),
δ 1.60 (m, 2H, —CH 2 —CH 2 —CO—), δ 2.0 ppm (m, 4H, (CH 2 ) 2 ), δ 2.41 (t, 2H, —CH 2 —CO—), δ 5.41 (d, 2H, CH = CH)
実施例11 塩化イソブチリル及びオレイン酸の混合アルデヒドによるヒアルロン酸のオレイル(C18:1)誘導体の調製
1.0gのヒアルロン酸ナトリウム(2.5mmol,15kDa)を10mLの脱塩水に溶解した。その後,5mLのTHFを徐々に添加した。次にTEA(1.05mL,3eq)及びDMAP(15.0mg,0.05eq)を溶液に添加した。同時にオレイン酸(0.787mL,1eq)を5mLのTHFに溶解し,その後,TEA(1.05mL,3eq)及び塩化イソブチリル(0.26mL,1eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.50gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS11%(NMRから測定)
1H NMR(D2O):δ0.88(t,3H,‐CH2‐CH3),δ1.22‐1.35(m,20H,(‐CH2‐)10),
δ1.60(m,2H,‐CH2‐CH2‐CO‐),δ2.0ppm(m,4H,(CH2)2),δ2.41(t,2H,‐CH2‐CO‐),δ5.41(d,2H,CH=CH)
Example 11 Preparation of an oleyl (C18: 1) derivative of hyaluronic acid with a mixed aldehyde of isobutyryl chloride and oleic acid 1.0 g of sodium hyaluronate (2.5 mmol, 15 kDa) was dissolved in 10 mL of demineralized water. Thereafter, 5 mL of THF was gradually added. TEA (1.05 mL, 3 eq) and DMAP (15.0 mg, 0.05 eq) were then added to the solution. At the same time, oleic acid (0.787 mL, 1 eq) was dissolved in 5 mL THF, and then TEA (1.05 mL, 3 eq) and isobutyryl chloride (0.26 mL, 1 eq) were added to the solution. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.50 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
1 H NMR (D 2 O): δ 0.88 (t, 3H, —CH 2 —CH 3 ), δ 1.22-1.35 (m, 20H, (—CH 2 —) 10 ),
δ 1.60 (m, 2H, —CH 2 —CH 2 —CO—), δ 2.0 ppm (m, 4H, (CH 2 ) 2 ), δ 2.41 (t, 2H, —CH 2 —CO—), δ 5.41 (d, 2H, CH = CH)
実施例12 2,4,6‐トリクロロ安息香酸及びオレイン酸の混合アルデヒドによるヒアルロン酸のオレイル(C18:1)誘導体の調製
0.5gのヒアルロン酸ナトリウム(1.25mmol,130kDa)を5mLの脱塩水に溶解した。その後,3mLのTHFを徐々に添加した。次にTEA(1.2mL,3eq)及びDMAP(15.0mg,0.1eq)を溶液に添加した。同時にオレイン酸(0.787g,2eq)を10mLのTHFに溶解し,その後,TEA(0.52mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.391mL,2eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.25gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS18%(NMRから測定)
1H NMR(D2O):δ0.88(t,3H,‐CH2‐CH3),δ1.22‐1.35(m,20H,(‐CH2‐)10),
δ1.60(m,2H,‐CH2‐CH2‐CO‐),δ2.0ppm(m,4H,(CH2)2),δ2.41(t,2H,‐CH2‐CO‐),δ5.41(d,2H,CH=CH)
Example 12 Preparation of an oleyl (C18: 1) derivative of hyaluronic acid with a mixed aldehyde of 2,4,6-trichlorobenzoic acid and oleic acid 0.5 g of sodium hyaluronate (1.25 mmol, 130 kDa) in 5 mL of demineralized water Dissolved in. Thereafter, 3 mL of THF was gradually added. TEA (1.2 mL, 3 eq) and DMAP (15.0 mg, 0.1 eq) were then added to the solution. At the same time oleic acid (0.787 g, 2 eq) is dissolved in 10 mL THF, then TEA (0.52 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.391 mL, 2 eq) are added to the solution. did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.25 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
DS18% (measured from NMR)
1 H NMR (D 2 O): δ 0.88 (t, 3H, —CH 2 —CH 3 ), δ 1.22-1.35 (m, 20H, (—CH 2 —) 10 ),
δ 1.60 (m, 2H, —CH 2 —CH 2 —CO—), δ 2.0 ppm (m, 4H, (CH 2 ) 2 ), δ 2.41 (t, 2H, —CH 2 —CO—), δ 5.41 (d, 2H, CH = CH)
実施例13 2,4,6‐トリクロロ安息香酸及びリノール酸の混合アルデヒドによるヒアルロン酸のリノレイル(C18:2)誘導体の調製
0.5gのヒアルロン酸ナトリウム(1.25mmol,15kDa)を10mLの脱塩水に溶解した。次にTEA(0.52mL,3eq)及びDMAP(8mg,0.05eq)を溶液に添加した。同時にリノール酸(0.77mL,2eq)を3mLのTHFに溶解し,その後,TEA(1.2mL,7eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.391mL,2eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.5gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS16%(NMRから測定)
1H NMR(D2O):δ0.88(t,3H,‐CH2‐CH3),δ1.22‐1.35(m,14H,(‐CH2‐)7),
δ1.63(m,2H,‐CH2‐CH2‐CO‐),δ2.0ppm(m,4H,(CH2)2),δ2.44(t,2H,‐CH2‐CO‐),δ2.83(m,2H,=CH‐CH2‐CH=),
δ5.45(m,4H,CH=CH)
Example 13 Preparation of linoleyl (C18: 2) derivative of hyaluronic acid with a mixed aldehyde of 2,4,6-trichlorobenzoic acid and linoleic acid 0.5 g of sodium hyaluronate (1.25 mmol, 15 kDa) in 10 mL of demineralized water Dissolved in. TEA (0.52 mL, 3 eq) and DMAP (8 mg, 0.05 eq) were then added to the solution. At the same time, linoleic acid (0.77 mL, 2 eq) is dissolved in 3 mL of THF, and then TEA (1.2 mL, 7 eq) and 2,4,6-trichlorobenzoyl chloride (0.391 mL, 2 eq) are added to the solution. did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.5 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
DS16% (measured from NMR)
1 H NMR (D 2 O): δ 0.88 (t, 3H, —CH 2 —CH 3 ), δ 1.22-1.35 (m, 14H, (—CH 2 —) 7 ),
δ 1.63 (m, 2H, —CH 2 —CH 2 —CO—), δ 2.0 ppm (m, 4H, (CH 2 ) 2 ), δ 2.44 (t, 2H, —CH 2 —CO—), δ2.83 (m, 2H, = CH -CH 2 -CH =),
δ 5.45 (m, 4H, CH = CH)
実施例14 2,4,6‐トリクロロ安息香酸及びリノール酸の混合アルデヒドによるヒアルロン酸のリノレイル(C18:2)誘導体の調製
0.5gのヒアルロン酸ナトリウム(1.25mmol,15kDa)を10mLの脱塩水に溶解した。次にTEA(0.52mL,3eq)及びDMAP(8mg,0.05eq)を溶液に添加した。同時にリノール酸(0.77mL,2eq)を3mLのTHFに溶解し,その後,TEA(1.2mL,7eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.391mL,2eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた後,反応混合液を50℃で1時間加温した。その後,0.75gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS20%(NMRから測定)
1H NMR(D2O):δ0.88(t,3H,‐CH2‐CH3),δ1.22‐1.35(m,14H,(‐CH2‐)7),
δ1.63(m,2H,‐CH2‐CH2‐CO‐),δ2.0ppm(m,4H,(CH2)2),δ2.44(t,2H,‐CH2‐CO‐),δ2.83(m,2H,=CH‐CH2‐CH=),
δ5.45(m,4H,CH=CH)
Example 14 Preparation of linoleyl (C18: 2) derivative of hyaluronic acid with a mixed aldehyde of 2,4,6-trichlorobenzoic acid and linoleic acid 0.5 g of sodium hyaluronate (1.25 mmol, 15 kDa) in 10 mL of demineralized water Dissolved in. TEA (0.52 mL, 3 eq) and DMAP (8 mg, 0.05 eq) were then added to the solution. At the same time, linoleic acid (0.77 mL, 2 eq) is dissolved in 3 mL of THF, and then TEA (1.2 mL, 7 eq) and 2,4,6-trichlorobenzoyl chloride (0.391 mL, 2 eq) are added to the solution. did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. After reacting at room temperature for 3 hours, the reaction mixture was warmed at 50 ° C. for 1 hour. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.75 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
1 H NMR (D 2 O): δ 0.88 (t, 3H, —CH 2 —CH 3 ), δ 1.22-1.35 (m, 14H, (—CH 2 —) 7 ),
δ 1.63 (m, 2H, —CH 2 —CH 2 —CO—), δ 2.0 ppm (m, 4H, (CH 2 ) 2 ), δ 2.44 (t, 2H, —CH 2 —CO—), δ2.83 (m, 2H, = CH -CH 2 -CH =),
δ 5.45 (m, 4H, CH = CH)
実施例15 2,4,6‐トリクロロ安息香酸及びリノレン酸の混合アルデヒドによるヒアルロン酸のリノレニル(C18:3)誘導体の調製
0.5gのヒアルロン酸ナトリウム(1.25mmol,15kDa)を10mLの脱塩水に溶解した。次にTEA(0.52mL,3eq)及びDMAP(8mg,0.05eq)を溶液に添加した。同時にリノレン酸(0.765mL,2.0eq)を5mLのTHFに溶解し,その後,TEA(0.52mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.391mL,2.0eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた。その後,0.75gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS15%(NMRから測定)
1H NMR(D2O):δ0.88(t,3H,‐CH2‐CH3),δ1.22‐1.35(m,8H,(‐CH2‐)4),
δ1.61(m,2H,‐CH2‐CH2‐CO‐),δ2.0ppm(m,4H,(CH2)2),δ2.43(t,2H,‐CH2‐CO‐),δ2.83(m,4H,=CH‐CH2‐CH=),
δ5.45(m,6H,CH=CH)
Example 15 Preparation of linolenyl (C18: 3) derivative of hyaluronic acid with a mixed aldehyde of 2,4,6-trichlorobenzoic acid and linolenic acid 0.5 g of sodium hyaluronate (1.25 mmol, 15 kDa) in 10 mL of demineralized water Dissolved in. TEA (0.52 mL, 3 eq) and DMAP (8 mg, 0.05 eq) were then added to the solution. At the same time, linolenic acid (0.765 mL, 2.0 eq) is dissolved in 5 mL of THF, and then TEA (0.52 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.391 mL, 2.0 eq) are added. Added to the solution. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. The reaction was allowed to proceed for 3 hours at room temperature. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.75 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
DS15% (measured from NMR)
1 H NMR (D 2 O): δ 0.88 (t, 3H, —CH 2 —CH 3 ), δ 1.22-1.35 (m, 8H, (—CH 2 —) 4 ),
δ 1.61 (m, 2H, —CH 2 —CH 2 —CO—), δ 2.0 ppm (m, 4H, (CH 2 ) 2 ), δ 2.43 (t, 2H, —CH 2 —CO—), δ 2.83 (m, 4H, = CH-CH 2 -CH =),
δ 5.45 (m, 6H, CH = CH)
実施例16 2,4,6‐トリクロロ安息香酸及びリノレン酸の混合アルデヒドによるヒアルロン酸のリノレニル(C18:3)誘導体の調製
0.5gのヒアルロン酸ナトリウム(1.25mmol,15kDa)を10mLの脱塩水に溶解した。次にTEA(0.52mL,3eq)及びDMAP(8mg,0.05eq)を溶液に添加した。同時にリノレン酸(0.382mL,1eq)を5mLのTHFに溶解し,その後,TEA(0.52mL,3eq)及び2,4,6‐トリクロロベンゾイルクロリド(0.195mL,1eq)をその溶液に添加した。酸の活性化の後,沈殿物を濾過し,HAの調製溶液に入れた。室温で3時間反応させた後,反応混合液を50℃で1時間加温した。その後,0.25gのNaClを添加した5mLの脱塩水で反応混合液を希釈した。その後の沈殿工程において,4倍の無水イソプロパノールを使用し,アシル化誘導体を反応混合液から単離した。デカンテーションを行った後,初めに誘導体からDMAPを除去する為にイソプロパノール水溶液(85体積%)で,次いで誘導体から水を除去する為に無水イソプロパノールで沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させ,次いで残留溶媒を除去する目的で凍結乾燥させた。
DS10%(NMRから測定)
1H NMR(D2O):δ0.88(t,3H,‐CH2‐CH3),δ1.22‐1.35(m,8H,(‐CH2‐)4),
δ1.61(m,2H,‐CH2‐CH2‐CO‐),δ2.0ppm(m,4H,(CH2)2),δ2.43(t,2H,‐CH2‐CO‐),δ2.83(m,4H,=CH‐CH2‐CH=),
δ5.45(m,6H,CH=CH)
Example 16 Preparation of linolenyl (C18: 3) derivative of hyaluronic acid with a mixed aldehyde of 2,4,6-trichlorobenzoic acid and linolenic acid 0.5 g of sodium hyaluronate (1.25 mmol, 15 kDa) in 10 mL of demineralized water Dissolved in. TEA (0.52 mL, 3 eq) and DMAP (8 mg, 0.05 eq) were then added to the solution. At the same time, linolenic acid (0.382 mL, 1 eq) is dissolved in 5 mL of THF, and then TEA (0.52 mL, 3 eq) and 2,4,6-trichlorobenzoyl chloride (0.195 mL, 1 eq) are added to the solution. did. After acid activation, the precipitate was filtered and placed in a prepared solution of HA. After reacting at room temperature for 3 hours, the reaction mixture was warmed at 50 ° C. for 1 hour. The reaction mixture was then diluted with 5 mL of demineralized water to which 0.25 g NaCl was added. In the subsequent precipitation step, 4 times anhydrous isopropanol was used and the acylated derivative was isolated from the reaction mixture. After decantation, the precipitate was washed repeatedly with an aqueous isopropanol solution (85% by volume) first to remove DMAP from the derivative and then with anhydrous isopropanol to remove water from the derivative. The precipitate was then dried at 40 ° C. for 48 hours and then lyophilized to remove residual solvent.
1 H NMR (D 2 O): δ 0.88 (t, 3H, —CH 2 —CH 3 ), δ 1.22-1.35 (m, 8H, (—CH 2 —) 4 ),
δ 1.61 (m, 2H, —CH 2 —CH 2 —CO—), δ 2.0 ppm (m, 4H, (CH 2 ) 2 ), δ 2.43 (t, 2H, —CH 2 —CO—), δ 2.83 (m, 4H, = CH-CH 2 -CH =),
δ 5.45 (m, 6H, CH = CH)
実施例17 ヒアルロン酸のカプロニル(C6)誘導体へのトコフェロール(ビタミンE)のカプセル封入
実施例1に従って調製したヒアルロナンのアシル化誘導体100mgを連続的に3時間撹拌しながら5mLの水に溶解した。得られた溶液に25〜40℃で連続的に撹拌しながらトコフェロール溶液(3mLのCHCl3中,10mg)を徐々に添加した後,更に3mLのCHCl3を徐々に添加した。次に,連続的蒸発工程で溶液からCHCl3を除去した。CHCl3の除去後,水相を完全に乾燥させ,水槽で再水和し,1μmのガラスフィルターで濾過した。濾過物を凍結乾燥した。
結合したトコフェロールの量(HPLC法により測定)は:2.3%(w/w)であった。
Example 17 Encapsulation of Tocopherol (Vitamin E) in Capronyl (C6) Derivative of
The amount of bound tocopherol (determined by HPLC method) was: 2.3% (w / w).
実施例18 ヒアルロン酸のカプロニル(C6)誘導体へのナイルレッドのカプセル封入
実施例1に従って調製したヒアルロナンのアシル化誘導体100mgを連続的に3時間撹拌しながら5mLの水に溶解した。得られた溶液に25〜40℃で連続的に撹拌しながらナイルレッド溶液(3mLのCHCl3中,10mg)を徐々に添加した後,更に3mLのCHCl3を徐々に添加した。次に,連続的蒸発工程で溶液からCHCl3を除去した。CHCl3の除去後,水相を完全に乾燥させ,水槽で再水和し,1μmのガラスフィルターで濾過した。濾過物を凍結乾燥した。
結合したナイルレッドの量(UV‐Vis法により測定)は:0.4%(w/w)であった。
Example 18 Encapsulation of Nile Red in Capronyl (C6) Derivative of
The amount of Nile Red bound (measured by UV-Vis method) was: 0.4% (w / w).
実施例19 ヒアルロン酸のカプロニル(C6)誘導体へのパクリタキセルのカプセル封入
実施例1に従って調製したヒアルロナンのアシル化誘導体100mgを連続的に3時間撹拌しながら5mLの水に溶解した。得られた溶液に25〜40℃で連続的に撹拌しながらパクリタキセル溶液(3mLのCHCl3中,10mg)を徐々に添加した後,更に3mLのCHCl3を徐々に添加した。次に,連続的蒸発工程で溶液からCHCl3を除去した。CHCl3の除去後,水相を完全に乾燥させ,水槽で再水和し,1μmのガラスフィルターで濾過した。濾過物を凍結乾燥した。
結合したパクリタキセルの量(HPLC法により測定):5%(w/w)
Example 19 Encapsulation of paclitaxel in capronyl (C6) derivative of
Bound paclitaxel amount (measured by HPLC method): 5% (w / w)
実施例20 ヒアルロン酸のカプロニル(C6)誘導体へのホスファチジルコリンのカプセル封入
実施例1に従って調製したヒアルロナンのアシル化誘導体100mgを連続的に3時間撹拌しながら5mLの水に溶解した。得られた溶液に連続的に撹拌しながらホスファチジルコリン溶液(5mLのEtOH中,10mg)を徐々に(滴下)添加した。連続的蒸発工程で溶液からEtOHを除去した。次に,残留水相を完全に乾燥させ,水槽で再水和し,1μmのガラスフィルターで濾過した。濾過物を凍結乾燥した。
結合したホスファチジルコリンの量(HPLC法により測定):3.0%(w/w)
Example 20 Encapsulation of phosphatidylcholine in capronyl (C6) derivative of
Amount of bound phosphatidylcholine (measured by HPLC method): 3.0% (w / w)
実施例21 ヒアルロン酸のパルミトイル(C16)誘導体へのコエンザイムQ10のカプセル封入
実施例7に従って調製したヒアルロナンのアシル化誘導体100mgを連続的に一晩撹拌しながら10mLの水に溶解した。得られた溶液に30〜40℃で連続的に撹拌しながらコエンザイムQ10溶液(5mLのCHCl3中,20mg)を徐々に添加した後,更に3mLのCHCl3を徐々に添加した。次に,連続的蒸発工程で溶液からCHCl3を除去した。CHCl3の除去後,水相を完全に乾燥させ,水槽で再水和し,1μmのガラスフィルターで濾過した。濾過物を凍結乾燥した。
結合したコエンザイムQ10の量(UV‐Vis法により測定)は:12%(w/w)であった。
生成物をNaClの0.9%溶液に溶解すると,溶解した生成物の濃度に依存してコアセルベート又はゲル状溶液が形成される。
Example 21 Encapsulation of Coenzyme Q10 in Palmitoyl (C16) Derivative of
The amount of coenzyme Q10 bound (measured by the UV-Vis method) was: 12% (w / w).
When the product is dissolved in a 0.9% solution of NaCl, a coacervate or gel solution is formed depending on the concentration of the dissolved product.
実施例22 ヒアルロン酸のステアリル(C18)誘導体へのトコフェロール(ビタミンE)のカプセル封入
実施例8に従って調製したヒアルロナンのアシル化誘導体100mgを連続的に一晩撹拌しながら10mLの水に溶解した。得られた溶液に25〜40℃で連続的に撹拌しながらトコフェロール(5mLのエタノール中,約50mg)を徐々に添加した。次に,連続的蒸発工程で溶液からエタノールを除去した。EtOHの除去後,水相を完全に乾燥させ,水槽で再水和し,1μmのガラスフィルターで濾過した。濾過物を凍結乾燥した。
結合したトコフェロールの量(UV‐Vis法により測定)は:30%(w/w)であった。
Example 22 Encapsulation of Tocopherol (Vitamin E) in Stearyl (C18) Derivative of
The amount of bound tocopherol (determined by the UV-Vis method) was 30% (w / w).
実施例23 ヒアルロン酸のオレイル(C18:1)誘導体へのトコフェロール(ビタミンE)のカプセル封入
実施例9に従って調製したヒアルロナンのアシル化誘導体100mgを連続的に一晩撹拌しながら10mLの水に溶解した。得られた溶液に25〜40℃で連続的に撹拌しながらトコフェロール(5mLのイソプロパノール中,約50mg)を徐々に添加した。次に,連続的蒸発工程で溶液からイソプロパノールを除去した。イソプロパノールの除去後,水相を完全に乾燥させ,水槽で再水和し,1μmのガラスフィルターで濾過した。濾過物を凍結乾燥した。
結合したトコフェロールの量(UV‐Vis法により測定)は:40%(w/w)であった。
Example 23 Encapsulation of tocopherol (vitamin E) in an oleyl (C18: 1) derivative of
The amount of bound tocopherol (determined by the UV-Vis method) was: 40% (w / w).
実施例24 ヒアルロン酸のパルミトイル(C16)誘導体へのコエンザイムQ10のカプセル封入
実施例7に従って調製したヒアルロナンのアシル化誘導体100mgを連続的に一晩撹拌しながら10mLの水に溶解した。得られた溶液に連続的に撹拌しながらコエンザイムQ10溶液(2mLのEtOH中,約30mg)を徐々に添加した。3時間撹拌した後,得られた混合液を30分間音波処理(100W)に供した。その後,混合液を蒸留水に対して集中的に透析し(2日間),1μmのガラスフィルターで濾過し,凍結乾燥した。
結合したコエンザイムQ10の量(UV‐Vis法により測定)は:4.6%(w/w)であった。
生成物をNaClの0.9%溶液に溶解すると,溶解した生成物の濃度に依存してコアセルベート又はゲル状溶液が形成される。
Example 24 Encapsulation of Coenzyme Q10 in Palmitoyl (C16) Derivative of
The amount of coenzyme Q10 bound (measured by the UV-Vis method) was: 4.6% (w / w).
When the product is dissolved in a 0.9% solution of NaCl, a coacervate or gel solution is formed depending on the concentration of the dissolved product.
実施例25 ヒアルロン酸のパルミトイル(C16)誘導体へのパクリタキセルのカプセル封入
実施例7に従って調製したヒアルロナンのアシル化誘導体100mgを連続的に一晩撹拌しながら10mLの水に溶解した。得られた溶液に連続的に撹拌しながらパクリタキセル溶液(2mLのEtOH中,約40mg)を徐々に添加した。3時間撹拌した後,得られた混合液を30分間音波処理(100W)に供した。その後,混合液を蒸留水に対して集中的に透析し(3.5kDaカットオフ),S4陶磁器フリットで濾過し,凍結乾燥した。
結合したパクリタキセルの量(HPLC法により測定):25%(w/w)
Example 25 Encapsulation of paclitaxel in palmitoyl (C16) derivative of
Bound paclitaxel amount (determined by HPLC method): 25% (w / w)
実施例26 ヒアルロン酸のオレイル(C18:1)誘導体へのホップエキスのカプセル封入
実施例9に従って調製したヒアルロナンのアシル化誘導体100mgを連続的に一晩撹拌しながら10mLの水に溶解した。得られた溶液に連続的に撹拌しながらホップエキスブレンド(5mLのイソプロパノール中,約50mg)を徐々に添加した。次に,連続的蒸発工程で溶液からイソプロパノールを除去した。イソプロパノールの除去後,水相を完全に乾燥させ,水槽で再水和し,1μmのガラスフィルターで濾過した。濾過物を凍結乾燥した。
結合したホップエキスの量(UV‐Vis法により測定)は:40%(w/w)であった。
Example 26 Encapsulation of hop extract in an oleyl (C18: 1) derivative of
The amount of bound hop extract (measured by UV-Vis method) was: 40% (w / w).
実施例27 ヒアルロン酸のアシル化誘導体の臨界ミセル(凝集)濃度の測定
(a)蛍光法
溶液濃度への蛍光強度の依存度から臨界ミセル(凝集)濃度を測定した(図1)。実施例18記載の手順に従って濃度範囲を0.00002〜1.5mg・mL-1に調整した,ナイルレッドが結合しているアシル化誘導体HAC6(DS=60%)及びHAC16(DS=14%)の水溶液の発光スペクトル(580〜700nm)を励起波長543nmで動作する蛍光測定装置RF‐5301(Shimadzu)において測定した。
以下の臨界ミセル(凝集)濃度が測定された:HAC6:0.001〜0.003mg・mL-1,HAC16:0.00006〜0.0002mg・mL-1(図1)。0.9%NaCl含有PBS溶液でも同様の測定値が得られた。
(b)静的光散乱法
溶液濃度への散乱光強度(I90)の依存度から臨界ミセル(凝集)濃度を測定した(図2)。実施例18記載の手順に従って濃度範囲を0.00002〜0.06mg・mL-1に調製した,ナイルレッドが結合しているアシル化誘導体HAC6(DS=60%)及びHAC16(DS=14%)の水溶液における散乱光強度を動作波長632nmの測光装置DAWN EOS(Wyatt Technology Corporation)において角度90°で測定した。
以下の臨界ミセル(凝集)濃度が測定された:HAC6:0.002〜0.004mg・mL-1,HAC16:0.00006〜0.0001mg・mL-1(図2)。0.9%NaCl含有PBS溶液でも同様の測定値が得られた。
Example 27 Measurement of critical micelle (aggregation) concentration of acylated derivative of hyaluronic acid (a) Fluorescence method The critical micelle (aggregation) concentration was measured from the dependence of fluorescence intensity on the solution concentration (FIG. 1). Nyl red bound acylated derivatives HAC6 (DS = 60%) and HAC16 (DS = 14%), the concentration range of which was adjusted to 0.00002-1.5 mg · mL −1 according to the procedure described in Example 18 The emission spectrum (580-700 nm) of the aqueous solution was measured with a fluorescence measuring apparatus RF-5301 (Shimadzu) operating at an excitation wavelength of 543 nm.
The following critical micelle (aggregation) concentrations were measured: HAC6: 0.001-0.003 mg · mL −1 , HAC16: 0.00006-0.0002 mg · mL −1 (FIG. 1). Similar measurement values were obtained with PBS solution containing 0.9% NaCl.
(B) Static Light Scattering Method The critical micelle (aggregation) concentration was measured from the dependence of the scattered light intensity (I 90 ) on the solution concentration (FIG. 2). Nyl red bound acylated derivatives HAC6 (DS = 60%) and HAC16 (DS = 14%) prepared in a concentration range of 0.00002 to 0.06 mg · mL −1 according to the procedure described in Example 18 The scattered light intensity in the aqueous solution was measured at an angle of 90 ° with a photometer DAWN EOS (Wyatt Technology Corporation) having an operating wavelength of 632 nm.
The following critical micelle (aggregation) concentrations were measured: HAC6: 0.002-0.004 mg · mL −1 , HAC16: 0.00006-0.0001 mg · mL −1 (FIG. 2). Similar measurement values were obtained with PBS solution containing 0.9% NaCl.
実施例28 ヒアルロナンナノミセルのゼータ電位の測定
He‐Neレーザー(633nm)を備えた装置Zetasizer Nano‐ZS(Malvern Instruments)でゼータ電位を測定した。カプセル封入した物質に関係なく,水溶液中のナノミセルが示したゼータ電位は5mg・mL-1では〜−50mVであり,10倍希釈した後では〜−60から−70mVであった。NaClの0.9%溶液中,ゼータ電位の範囲が減少した(−30から−23mV)。従って,ゼータ電位の絶対値は,水溶液中では調製ナノミセルの高い安定性を示し,塩溶液中では比較的高い安定性を示す。
Example 28 Measurement of zeta potential of hyaluronan nano micelles The zeta potential was measured with an apparatus Zetasizer Nano-ZS (Malvern Instruments) equipped with a He-Ne laser (633 nm). Regardless of the encapsulated material, the zeta potential exhibited by the nanomicelle in the aqueous solution was ˜-50 mV at 5 mg · mL −1 and ˜60 to −70 mV after 10-fold dilution. In 0.9% NaCl solution, the zeta potential range decreased (-30 to -23 mV). Therefore, the absolute value of the zeta potential shows a high stability of the prepared nanomicelle in an aqueous solution and a relatively high stability in a salt solution.
実施例29 ヒアルロナンナノミセルの形態学的分析
2kVのビーム加速電圧(すなわち微細ビームモード)で走査顕微鏡JEOL7401Fにおいて−135℃で顕微鏡分析を行った。上記の分析をする為に,2〜3μLの濃縮試料(約20mg/0.4mL)をAlプレートに滴下し,冷却チャンバAlta2500(Gatan)に充填した液体窒素に浸漬した。その後,それらを2分間かけてPt/Pd混合液で被覆した。
カプセル封入したビタミンEを含むヒアルロナンC6(実施例17)及びカプセル封入したパクリタキセルを含むヒアルロナンC16(実施例25)のナノミセルの寸法は範囲:20〜50nmであった(図3)。
Example 29 Morphological Analysis of Hyaluronan Nanomicelles Microscopic analysis was performed at −135 ° C. in a scanning microscope JEOL7401F with a beam acceleration voltage of 2 kV (ie fine beam mode). In order to perform the above analysis, 2-3 μL of a concentrated sample (about 20 mg / 0.4 mL) was dropped onto an Al plate and immersed in liquid nitrogen filled in a cooling chamber Alta2500 (Gatan). They were then coated with a Pt / Pd mixture for 2 minutes.
Nanomicelle dimensions of hyaluronan C6 with encapsulated vitamin E (Example 17) and hyaluronan C16 with encapsulated paclitaxel (Example 25) ranged from 20 to 50 nm (Figure 3).
実施例30 ヒアルロナンナノミセル中のアシル化鎖及び非極性物質の分布
カプセル封入したビタミンEを含む疎水化ヒアルロナンのナノミセルを,フリット入口分離チャネルを使用する流動場フローフラクショネーション(FlFFF)法により分離した。分析する目的で,(実施例23に記載の手順に従って表1に収載した誘導体から調製した)ビタミンEが結合した凍結乾燥アシル化ヒアルロナン10mgを1mLの移動相(50mMのNaNO3及び0.02%のNaN3)に溶解し,孔径1μmのガラスシリンジフィルターで濾過した。その後,100μlをFlFFF装置に注入した。
5分間隔での2mL/分〜0.1mL/分のクロスフロー勾配により分離を行った。検出器に供給される移動相の流速の設定値を1mL/分で一定に維持した。
実験温度で分離が行われた。光散乱検出器DAWN EOS,示差屈折率計Optilab rEX(両方ともWyatt Technology Corporation製)及び動作波長292nmのUV検出器(Shimadzu)により溶出液を観察した。
上記の方法を適用した場合,ナノミセルの内部に確実に取り込まれた結合物質及び疎水化ヒアルロナンの割合と,凝集構造の外部に存在するそれらの割合の両方を測定することが可能である(表1参照)。
表1.ヒアルロナン凝集体(ナノミセル)の内外でのアシル化鎖及びビタミンE(トコフェロール)の分布
Separations were performed with a cross-flow gradient of 2 mL / min to 0.1 mL / min at 5 min intervals. The set value of the flow rate of the mobile phase supplied to the detector was kept constant at 1 mL / min.
Separation took place at the experimental temperature. The eluate was observed with a light scattering detector DAWN EOS, a differential refractometer Optilab rEX (both manufactured by Wyatt Technology Corporation) and a UV detector (Shimadzu) with an operating wavelength of 292 nm.
When the above method is applied, it is possible to measure both the proportion of the binding substance and the hydrophobized hyaluronan that are reliably incorporated inside the nano micelles and the proportion of those existing outside the aggregated structure (Table 1). reference).
Table 1. Distribution of acylated chains and vitamin E (tocopherol) inside and outside hyaluronan aggregates (nano micelles)
実施例31 パクリタキセルをベースとする細胞増殖阻害剤を担持するナノミセルの細胞毒性
それぞれ実施例19及び25に記載の手順に従って調製したヒアルロナンC6及びC16のアシル化誘導体に結合したパクリタキセルを,培養液(10%のFBSを含む)に溶解し,最終濃度を100μg/mLにした。アシル化ヒアルロナンC6及びC16の誘導体に担持された濃度0.001,0.01,0.1,1.0,10.0及び100.0μg/mLのパクリタキセルを試験する為に,ヒト皮膚線維芽細胞(NHDF)の細胞,ヒト乳癌細胞株(MCF‐7)及びヒト結腸癌細胞株(HCT116)を使用し,細胞生存率の測定に基づいて試験した。アシル化ヒアルロナンC6及びC16の誘導体に担持されたパクリタキセルの効果を,そのヒアルロナン自体に対するパクリタキセルの効果と比較した(図4)。細胞生存率の測定は脱水素酵素の活性の検出に基づいており,この酵素は生細胞で活性的になって黄色い物質を紫色の溶液に変えるものである。540nmで検出される前記酵素の吸光度は生細胞の割合に比例している。
特にHAC16を使用した場合,担体の濃度が増加したとき,パクリタキセルの細胞増殖阻害効果がわずかに減少した(図4)。
アシル化誘導体自体は細胞増殖阻害効果を全く示さなかった。
Example 31 Cytotoxicity of Nanomicelles Carrying Paclitaxel-Based Cell Growth Inhibitors Paclitaxel conjugated to hyaluronan C6 and C16 acylated derivatives prepared according to the procedures described in Examples 19 and 25, respectively, was added to the culture medium (10 % Final FBS) to a final concentration of 100 μg / mL. To test paclitaxel at concentrations of 0.001, 0.01, 0.1, 1.0, 10.0 and 100.0 μg / mL supported on acylated hyaluronan C6 and C16 derivatives, Cells (NHDF) cells, human breast cancer cell line (MCF-7) and human colon cancer cell line (HCT116) were used and tested based on cell viability measurements. The effect of paclitaxel supported on acylated hyaluronan C6 and C16 derivatives was compared to the effect of paclitaxel on the hyaluronan itself (FIG. 4). Cell viability measurements are based on the detection of dehydrogenase activity, which becomes active in living cells and turns yellow substances into purple solutions. The absorbance of the enzyme detected at 540 nm is proportional to the proportion of living cells.
In particular, when HAC16 was used, the cell growth inhibitory effect of paclitaxel slightly decreased when the carrier concentration was increased (FIG. 4).
The acylated derivative itself did not show any cell growth inhibitory effect.
実施例32 細胞へのカプセル封入物質の移行
物質ドキソルビシン及び7‐アミノアクチノマイシンD(死細胞及び浸透化処理細胞にのみ浸透する物質である7‐AAD)を,実施例18に記載の手順に従って(ここではナイルレッドに7‐AADを代用した)ヒアルロナンC6のアシル化誘導体にカプセル封入した。溶液に物質を添加したときの物質の細胞への浸透について,その物質がそれ自体で存在する場合と,ヒアルロナンC6のアシル化誘導体中に存在する場合とで差異があるか否かを試験する為に,ヒト皮膚線維芽細胞(NHDF)の細胞,ヒト乳癌細胞株(MCF‐7)及びヒト結腸癌細胞株(HCT116)を使用した。試験は蛍光顕微鏡法により行った(倒立顕微鏡Nikon Eclipse Ti)。ドキソルビシンは5.0μg/mLの濃度で試験し(図5),7‐AADは15μg/mLの濃度で試験した(図6)。
担体から細胞への物質の移行は良好であった。
Example 32 Transfer of Encapsulated Substances to Cells The substances doxorubicin and 7-aminoactinomycin D (7-AAD, which is a substance that only penetrates dead and permeabilized cells), according to the procedure described in Example 18 ( This was encapsulated in an acylated derivative of hyaluronan C6 (where 7-AAD was substituted for Nile Red). To test whether there is a difference in the penetration of a substance into a cell when the substance is added to a solution, when the substance is present by itself and when it is present in an acylated derivative of hyaluronan C6 Human skin fibroblast (NHDF) cells, human breast cancer cell line (MCF-7) and human colon cancer cell line (HCT116) were used. The test was performed by fluorescence microscopy (inverted microscope Nikon Eclipse Ti). Doxorubicin was tested at a concentration of 5.0 μg / mL (FIG. 5) and 7-AAD was tested at a concentration of 15 μg / mL (FIG. 6).
The substance transfer from the carrier to the cells was good.
実施例33 ナノミセルから溶液へのカプセル封入オイルレッドの放出
実施例18に記載の手順に従って(ここではナイルレッドにオイルレッドOを代用した)HAC6にカプセル封入されたオイルレッド(オイルレッドO,溶媒レッド27)の溶媒への放出をインビトロで検討した。使用した標的溶液はPBS,及び1%のTWEEN80を添加したPBSであった。オイルレッドを結合したアシル化誘導体の水溶液(濃度1〜10mg・mL-1)をPBS,又は1%のTWEEN80を添加したPBSに溶解し,透析管(MWCO12〜14kDa,Spectrum Laboratories)に定量的に移し,温度37℃でPBS,又は1%のTWEEN80を添加したPBSに対して透析した。所定の時間間隔で,透析液を4mL採取し,新鮮な培地と交換した。オイルレッドの放出量はUV‐Vis法により測定した(図7)。
結合した物質の緩徐な放出は,PBS中で担体系の安定性が高いということの指標となる。
Example 33 Release of Encapsulated Oil Red from Nanomicelles into Solution Oil Red (Oil Red O, Solvent Red) encapsulated in HAC6 according to the procedure described in Example 18 (where Nile Red was replaced with Oil Red O) The release of 27) into the solvent was examined in vitro. The target solution used was PBS and PBS supplemented with 1% TWEEN80. An aqueous solution (
The slow release of the bound substance is an indication that the carrier system is highly stable in PBS.
実施例34 ナノミセルから溶液へのカプセル封入パクリタキセルの放出
それぞれ実施例19及び25に記載の手順に従ってHAC6及びHAC16にカプセル封入されたパクリタキセルの溶媒への放出を37℃の温度下,インビトロで検討した。総濃度0.2mgのパクリタキセルを含むアシル化誘導体水溶液をPBSに溶解し,透析管(MWCO12〜14kDa,Spectrum Laboratories)に定量的に移し,温度37℃で50mLのPBSに対して透析した。所定の時間間隔で,透析液を新鮮な培地と交換した。パクリタキセルの放出量は,それをクロロホルムへ抽出し,蒸発させ,次いでアセトニトリルに溶解した後にHPLC法により測定した(図8)。
HAC16で記録された放出はHAC6と比較して緩徐であった。結合した物質の緩徐な放出は,PBS中で担体系の安定性が高いということの指標となる。
Example 34 Release of Encapsulated Paclitaxel from Nanomicelles into Solution Release of paclitaxel encapsulated in HAC6 and HAC16 into solvent was studied in vitro at a temperature of 37 ° C. according to the procedures described in Examples 19 and 25, respectively. An acylated derivative aqueous solution containing paclitaxel having a total concentration of 0.2 mg was dissolved in PBS, quantitatively transferred to a dialysis tube (MWCO 12-14 kDa, Spectrum Laboratories), and dialyzed against 50 mL of PBS at a temperature of 37 ° C. The dialysate was replaced with fresh medium at predetermined time intervals. The amount of paclitaxel released was measured by the HPLC method after it was extracted into chloroform, evaporated and then dissolved in acetonitrile (FIG. 8).
The release recorded with HAC16 was slow compared to HAC6. The slow release of the bound substance is an indication that the carrier system is highly stable in PBS.
実施例35 ヒアルロン酸及びオレイン酸のオレイル誘導体(C18:1)からのナノエマルジョン及びマイクロエマルジョンの調製
実施例11に従って調製したヒアルロナンのアシル化誘導体80mgを連続的に一晩撹拌しながら4mLの水に溶解した。得られた溶液に,連続的に撹拌しながら8mgのオレイン酸を徐々に添加した。撹拌後,得られた混合液を2段階音波処理(Ultrasonic Processor,UPS 200S,200W出力)に供した。第1段階は連続的工程であり(50%の振幅),その後に,脈動的工程である第2段階が続き(0.8秒のパルス,70%の振幅),各段階は25分間継続された。音波処理工程では,過熱から保護する為に,処理中の混合液が入った容器は氷浴に浸漬した。
粒径(Zetasizerで測定):200〜300nm。寸法は混合液中の油相の量に依存していた。
Example 35 Preparation of nanoemulsions and microemulsions from oleyl derivatives of hyaluronic acid and oleic acid (C18: 1) 80 mg of acylated derivative of hyaluronan prepared according to Example 11 was stirred into 4 mL of water with continuous overnight stirring. Dissolved. To the resulting solution, 8 mg oleic acid was gradually added with continuous stirring. After stirring, the resulting mixture was subjected to two-stage sonication (Ultrasonic Processor, UPS 200S, 200 W output). The first stage is a continuous process (50% amplitude), followed by the second stage, which is a pulsating process (0.8 second pulse, 70% amplitude), each stage lasting 25 minutes. It was. In the sonication process, the container containing the mixed solution was immersed in an ice bath to protect it from overheating.
Particle size (measured with Zetasizer): 200-300 nm. The dimensions depended on the amount of oil phase in the mixture.
実施例36 コエンザイムQ10を溶解したヒアルロン酸及びオレイン酸のオレイル誘導体(C18:1)からのナノエマルジョン及びマイクロエマルジョンの調製
実施例11に従って調製したヒアルロナンのアシル化誘導体80mgを連続的に一晩撹拌しながら4mLの水に溶解した。得られた溶液に,連続的に撹拌しながらコエンザイムQ10(約0.5mg)を事前に溶解させた8mgのオレイン酸を徐々に添加した。撹拌後,得られた混合液を2段階音波処理(Ultrasonic Processor,UPS 200S,200W出力)に供した。第1段階は連続的工程であり(50%の振幅),その後に,脈動的工程である第2段階が続き(0.8秒のパルス,70%の振幅),各段階は25分間継続された。音波処理工程では,過熱から保護する為に,処理中の混合液が入った容器は氷浴に浸漬した。
粒径(Zetasizerで測定):200〜300nm。寸法は混合液中の油相の量に依存していた。
Example 36 Preparation of nanoemulsion and microemulsion from hyaluronic acid and oleyl derivative of oleic acid (C18: 1) in which coenzyme Q10 was dissolved 80 mg of acylated derivative of hyaluronan prepared according to example 11 was continuously stirred overnight. While dissolved in 4 mL of water. To the resulting solution, 8 mg of oleic acid in which Coenzyme Q10 (about 0.5 mg) was previously dissolved was continuously added with continuous stirring. After stirring, the resulting mixture was subjected to two-stage sonication (Ultrasonic Processor, UPS 200S, 200 W output). The first stage is a continuous process (50% amplitude), followed by the second stage, which is a pulsating process (0.8 second pulse, 70% amplitude), each stage lasting 25 minutes. It was. In the sonication process, the container containing the mixed solution was immersed in an ice bath to protect it from overheating.
Particle size (measured with Zetasizer): 200-300 nm. The dimensions depended on the amount of oil phase in the mixture.
実施例37 共有的架橋を介した安定化ナノミセルの調製
10gのヒアルロン酸ナトリウム(25mmol,38kDa)を200mLの脱塩水に溶解した。その後,TEA(6.97mL,HAの二量体に対して2eq)及びDMAP(153mg,0.05eq)を溶液に添加した。活性化:3.5gの3‐(2‐フリル)アクリル酸(25mmol)を50mLのテトラヒドロフラン及び19.2mLのTEA(2eq)に溶解した。次に,得られた溶液を氷浴中で冷却し,トリクロロベンゾイルクロリド(1.2mL)を添加した。15分間反応させた。その後,活性化した酸をヒアルロナン溶液に添加し,25℃で24時間反応させた。得られた反応混合液を水で希釈した。4倍の無水イソプロパノールを使用したその次の沈殿工程で,反応混合液からアクリル化誘導体を単離した。デカンテーションを行った後,イソプロパノール水溶液(85体積%)で沈殿物を繰り返し洗浄した。その後,沈殿物を40℃で48時間乾燥させた。得られたアシル化誘導体をアシル化し(実施例1参照),次いで水に溶解し,残留溶媒を除去する為に凍結乾燥させた。
実施例18に従って(ここではナイルレッドにオイルレッドを代用した),オイルレッド(Oil red O)及びアシル化誘導体から調製した担体系を溶解し,1%水溶液を形成した。得られた溶液中で,トリガーとしてペルオキシ二硫酸アンモニウム(10eq)を使用し,担体系を架橋した。
DS20%の光架橋用活性基(NMRから測定)
1H NMR(D2O):δ7.83,6.78(d,J=3.5),6.78,6.61(bs),7.83,7.59(Jtrans=16.01),6.39(Jtrans=15.85)
Example 37 Preparation of stabilized nanomicelles via covalent crosslinking 10 g of sodium hyaluronate (25 mmol, 38 kDa) was dissolved in 200 mL of demineralized water. TEA (6.97 mL, 2 eq for the dimer of HA) and DMAP (153 mg, 0.05 eq) were then added to the solution. Activation: 3.5 g 3- (2-furyl) acrylic acid (25 mmol) was dissolved in 50 mL tetrahydrofuran and 19.2 mL TEA (2 eq). The resulting solution was then cooled in an ice bath and trichlorobenzoyl chloride (1.2 mL) was added. The reaction was allowed for 15 minutes. Thereafter, the activated acid was added to the hyaluronan solution and reacted at 25 ° C. for 24 hours. The resulting reaction mixture was diluted with water. The acrylated derivative was isolated from the reaction mixture in the next precipitation step using 4X anhydrous isopropanol. After decantation, the precipitate was repeatedly washed with an isopropanol aqueous solution (85% by volume). The precipitate was then dried at 40 ° C. for 48 hours. The resulting acylated derivative was acylated (see Example 1), then dissolved in water and lyophilized to remove residual solvent.
According to Example 18 (here, oil red was substituted for Nile Red), a carrier system prepared from Oil Red (Oil red O) and an acylated derivative was dissolved to form a 1% aqueous solution. In the resulting solution, ammonium peroxydisulfate (10 eq) was used as a trigger to crosslink the support system.
DS20% active group for photocrosslinking (measured from NMR)
1 H NMR (D 2 O): δ 7.83, 6.78 (d, J = 3.5), 6.78, 6.61 (bs), 7.83, 7.59 (J trans = 16. 01), 6.39 (J trans = 15.85)
実施例38 ヒアルロナンナノミセルの局所塗布―皮膚,毛髪及び粘膜への浸透
ブタ皮膚,ウシ窒粘膜及びウシ頬粘膜はLetohrad,Kuncice243に拠点を置くMasoEko s.r.o.社により供与された。その取得直後に,カプセル封入したナイルレッドを含むアシル化ヒアルロナンC6及びC10(10mg・mL-1)溶液(実施例18に記載の手順に従って調製)の受動的作用に試料を供し,倒立顕微鏡Nikon Eclipse Ti(対物レンズPlan Fluor4xを備える)で蛍光値を測定することで試料への浸透を比較した。その後,37℃で:皮膚試料は20時間(図9),毛髪試料は15分間(図10),粘膜試料は4時間(図11及び図12)でインキュベーションを行った。
油性溶媒及び水性溶媒を用いて比較したところ,担体により皮膚,毛髪及び粘膜への浸透がより効率的になっていた。
Example 38 Topical application of hyaluronan nano micelles-penetration into skin, hair and mucosa Porcine skin, bovine nitrogen mucosa and bovine buccal mucosa were donated by MasoEko sro, based in Letohrad, Kuncice 243. Immediately after acquisition, the sample was subjected to the passive action of an acylated hyaluronan C6 and C10 (10 mg · mL −1 ) solution containing encapsulated Nile Red (prepared according to the procedure described in Example 18), and the inverted microscope Nikon Eclipse The penetration of the sample was compared by measuring the fluorescence value with Ti (with the objective lens Plan Fluor4x). Thereafter, incubation was performed at 37 ° C .: a skin sample for 20 hours (FIG. 9), a hair sample for 15 minutes (FIG. 10), and a mucosa sample for 4 hours (FIGS. 11 and 12).
When compared using an oily solvent and an aqueous solvent, the carrier was more efficient in penetrating the skin, hair and mucous membranes.
Claims (35)
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CZPV2012-842 | 2012-11-27 | ||
| CZ2012-842A CZ304654B6 (en) | 2012-11-27 | 2012-11-27 | C6-C18-acylated hyaluronate-based nanomicellar composition, process for preparing C6-C18-acylated hyaluronate, process for preparing nanomicellar composition and stabilized nanomicellar composition as well as use thereof |
| PCT/CZ2013/000156 WO2014082609A1 (en) | 2012-11-27 | 2013-11-26 | C6-c18-acylated derivative of hyaluronic acid, method of preparation thereof, nanomicellar composition on its basis, method of preparation thereof and method of preparation stabilized nanomicellar composition, and use thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2016500130A true JP2016500130A (en) | 2016-01-07 |
| JP2016500130A5 JP2016500130A5 (en) | 2018-02-08 |
Family
ID=50097520
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015543316A Pending JP2016500130A (en) | 2012-11-27 | 2013-11-26 | C6-C18-acylated derivative of hyaluronic acid and process for its preparation, nanomicelle composition based on it and process for its preparation, process for preparing stabilized nanomicelle composition and use thereof |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US9999678B2 (en) |
| EP (1) | EP2934592B1 (en) |
| JP (1) | JP2016500130A (en) |
| KR (1) | KR102191353B1 (en) |
| BR (1) | BR112015012191A8 (en) |
| CZ (1) | CZ304654B6 (en) |
| DK (1) | DK2934592T3 (en) |
| ES (1) | ES2683993T3 (en) |
| HU (1) | HUE038664T2 (en) |
| PL (1) | PL2934592T3 (en) |
| RU (1) | RU2640287C2 (en) |
| WO (1) | WO2014082609A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2021157665A1 (en) * | 2020-02-07 | 2021-08-12 | ||
| JPWO2021157677A1 (en) * | 2020-02-05 | 2021-08-12 |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CZ304654B6 (en) | 2012-11-27 | 2014-08-20 | Contipro Biotech S.R.O. | C6-C18-acylated hyaluronate-based nanomicellar composition, process for preparing C6-C18-acylated hyaluronate, process for preparing nanomicellar composition and stabilized nanomicellar composition as well as use thereof |
| CZ305153B6 (en) | 2014-03-11 | 2015-05-20 | Contipro Biotech S.R.O. | Conjugates of hyaluronic acid oligomer or a salt thereof, process for their preparation and use |
| CZ2014451A3 (en) | 2014-06-30 | 2016-01-13 | Contipro Pharma A.S. | Antitumor composition based on hyaluronic acid and inorganic nanoparticles, process of its preparation and use |
| CZ309295B6 (en) * | 2015-03-09 | 2022-08-10 | Contipro A.S. | Self-supporting, biodegradable film based on hydrophobized hyaluronic acid, method of its preparation and use |
| CN107708707B (en) * | 2015-05-29 | 2021-09-07 | 生化学工业株式会社 | Composition comprising glycosaminoglycan derivative and chemokine receptor activity modulating material |
| CZ306479B6 (en) | 2015-06-15 | 2017-02-08 | Contipro A.S. | A method of crosslinking polysaccharides by using photolabile protecting groups |
| CZ306662B6 (en) | 2015-06-26 | 2017-04-26 | Contipro A.S. | Sulphated polysaccharides derivatives, the method of their preparation, the method of their modification and the use |
| PL229276B1 (en) * | 2015-07-17 | 2018-06-29 | Univ Jagiellonski | Nanocapsule for transferring lipophile compound and method for producing it |
| CZ307511B6 (en) | 2015-12-23 | 2018-10-31 | Contipro A.S. | A fluorescent conjugate of hyaluronic acid or a salt thereof, a hydrophobized conjugate, a method of their preparation and use |
| CZ308106B6 (en) | 2016-06-27 | 2020-01-08 | Contipro A.S. | Unsaturated derivatives of polysaccharides, their preparation and their use |
| CZ2016826A3 (en) | 2016-12-22 | 2018-07-04 | Contipro A.S. | A medicinal agent with a carrier based on hyaluronan and/or its derivatives, a method of the manufacture and use |
| CZ307158B6 (en) * | 2016-12-23 | 2018-02-07 | Contipro A.S. | An ophthalmic preparation |
| US12324789B2 (en) * | 2017-08-28 | 2025-06-10 | Apirx Pharmaceutical Usa, Llc | Method to treat psoriasis |
| ES3040766T3 (en) | 2018-06-29 | 2025-11-04 | Merz Pharma Gmbh & Co Kgaa | Fatty acid-grafted hyaluronic acid, dermal filler formulations comprising same, process for preparation and use thereof |
| FR3105922B1 (en) | 2020-01-08 | 2022-12-02 | Le Rouge Francais | Cosmetic composition for the treatment of the lips in the form of a compact solid containing at least one probiotic agent and at least one compound chosen from hyaluronic acid and its derivatives. |
| KR102547183B1 (en) * | 2020-10-19 | 2023-06-26 | 주식회사 메피온 | Dermal filler and method for manufacturing the dermal filler |
| CN112159527B (en) * | 2020-10-29 | 2023-06-06 | 绍兴文理学院 | A kind of polyglycerol fatty acid ester HA-PG containing hyaluronic acid group and its synthesis method and its application in the preparation of pharmaceutical preparations |
| CN113476426B (en) * | 2021-06-29 | 2023-11-24 | 大连工业大学 | Preparation of a dual-targeted functional factor delivery system based on hyaluronic acid and its application in inflammatory bowel disease |
| CN114561827B (en) * | 2022-02-25 | 2023-04-21 | 华南理工大学 | Full biomass-based wax emulsion waterproof coating and preparation method and application thereof |
| CN116444695B (en) * | 2022-12-27 | 2024-09-06 | 山东丰金美业科技有限公司 | Preparation method of low-cost low-molecular-weight acetylated sodium hyaluronate |
| CN116459213A (en) * | 2023-02-20 | 2023-07-21 | 莫佳杰 | A kind of micelles with hydrogen peroxide responsiveness and its preparation method and application |
| FR3148720A1 (en) | 2023-05-18 | 2024-11-22 | Le Rouge Francais | Aqueous cosmetic composition containing a probiotic agent, a compound chosen from hyaluronic acid and its derivatives, a particular colorant and a vegetable oil. |
| FR3151215A1 (en) | 2023-07-23 | 2025-01-24 | Le Rouge Francais | Powdered anhydrous topical cosmetic composition comprising a probiotic agent, hyaluronic acid or one of its derivatives, a particular colorant and a particular mineral powder. |
| CN116874640B (en) * | 2023-07-24 | 2025-08-08 | 江南大学 | Long-residence sodium hyaluronate derivative for hair repair and preparation method thereof |
| WO2025029805A2 (en) * | 2023-08-03 | 2025-02-06 | Pmidg, Llc | Hemostatic compositions and methods |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0347801A (en) * | 1989-04-18 | 1991-02-28 | Agency Of Ind Science & Technol | Novel hyaluronic acid derivative and method for producing the same |
| JPH0625306A (en) * | 1992-04-21 | 1994-02-01 | Shiseido Co Ltd | Solvent-insoluble hyaluronic acid and its production |
| JP2004123785A (en) * | 2002-09-30 | 2004-04-22 | Chisso Corp | n-Alkanoylated hyaluronic acid or salt thereof and method for producing the same |
| JP2012520902A (en) * | 2009-03-17 | 2012-09-10 | コンティプロ ファーマ アクチオヴァ スポレチノスト | Method of modifying hyaluronic acid with (O-acyl-O'-alkyl carbonate-substituted pyridine) complexes |
Family Cites Families (153)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3720662A (en) | 1971-09-13 | 1973-03-13 | Nat Starch Chem Corp | Preparation of starch esters |
| US3728223A (en) | 1971-10-08 | 1973-04-17 | Amano Pharma Co Ltd | Production of hyaluronidase from a strain of streptomyces |
| GB1527592A (en) | 1974-08-05 | 1978-10-04 | Ici Ltd | Wound dressing |
| CH628088A5 (en) | 1975-09-17 | 1982-02-15 | Dresden Arzneimittel | Process for obtaining streptococcal metabolic products |
| US4205025A (en) | 1975-12-22 | 1980-05-27 | Champion International Corporation | Synthetic polymeric fibrids, fibrid products and process for their production |
| JPS6033474B2 (en) | 1978-05-11 | 1985-08-02 | 藤沢薬品工業株式会社 | Novel hyaluronidase BMP-8231 and its production method |
| US4716224A (en) | 1984-05-04 | 1987-12-29 | Seikagaku Kogyo Co. Ltd. | Crosslinked hyaluronic acid and its use |
| US4713448A (en) | 1985-03-12 | 1987-12-15 | Biomatrix, Inc. | Chemically modified hyaluronic acid preparation and method of recovery thereof from animal tissues |
| US4851521A (en) | 1985-07-08 | 1989-07-25 | Fidia, S.P.A. | Esters of hyaluronic acid |
| GB8519416D0 (en) | 1985-08-01 | 1985-09-04 | Unilever Plc | Oligosaccharides |
| JPS62104579A (en) | 1985-10-30 | 1987-05-15 | Kyowa Hakko Kogyo Co Ltd | Production of hyaluronidase |
| JPH0751064B2 (en) | 1986-08-13 | 1995-06-05 | 生化学工業株式会社 | Novel hyaluronidase SD-678 and method for producing the same |
| IT1219587B (en) | 1988-05-13 | 1990-05-18 | Fidia Farmaceutici | SELF-CROSS-LINKED CARBOXYLY POLYSACCHARIDES |
| JPH0214019A (en) | 1988-06-30 | 1990-01-18 | Tonen Corp | Fibrous shaped material and production thereof |
| US5522879A (en) | 1991-11-12 | 1996-06-04 | Ethicon, Inc. | Piezoelectric biomedical device |
| IT1254704B (en) | 1991-12-18 | 1995-10-09 | Mini Ricerca Scient Tecnolog | NON-WOVEN FABRIC ESSENTIALLY CONSTITUTED FROM DERIVATIVES OF HYALURONIC ACID |
| US5824335A (en) | 1991-12-18 | 1998-10-20 | Dorigatti; Franco | Non-woven fabric material comprising auto-crosslinked hyaluronic acid derivatives |
| JP2855307B2 (en) | 1992-02-05 | 1999-02-10 | 生化学工業株式会社 | Photoreactive glycosaminoglycans, cross-linked glycosaminoglycans and methods for producing them |
| FR2689131B1 (en) | 1992-03-30 | 1994-05-20 | Oreal | PROCESS FOR THE PREPARATION OF MONOESTERS MAJORITY IN THE 6 'POSITION OF D-MALTOSE AND THEIR USE IN THE COSMETIC, ORAL-DENTAL, PHARMACEUTICAL AND FOOD FIELDS. |
| IT1263316B (en) | 1993-02-12 | 1996-08-05 | Fidia Advanced Biopolymers Srl | MULTILAYER NON WOVEN FABRIC IN WHICH ONE OF THE LAYERS IS ESSENTIALS ESSENTIALS FROM HYALURONIC ACID ESTERS |
| NL9700003A (en) | 1993-09-28 | 1997-07-01 | House Foods Corp | Method of inoculating Fistulina hepatica |
| US5616568A (en) | 1993-11-30 | 1997-04-01 | The Research Foundation Of State University Of New York | Functionalized derivatives of hyaluronic acid |
| EP0701704B1 (en) | 1994-03-14 | 1999-12-15 | Seikagaku Corporation | Material to be worn on the eyeball |
| US5455349A (en) | 1994-05-13 | 1995-10-03 | Polaroid Corporation | Vinylbenzyl thymine monomers |
| ATE187079T1 (en) | 1994-09-27 | 1999-12-15 | Nycomed Imaging As | CONTRAST AGENTS |
| JP3308742B2 (en) | 1994-11-17 | 2002-07-29 | 生化学工業株式会社 | Photocrosslinkable hyaluronic acid derivative, crosslinked product thereof and methods for producing them |
| US6025444A (en) | 1994-11-17 | 2000-02-15 | Seikagaku Kogyo Kabushiki Kaisha (Seikagaku Corporation) | Cinnamic acid derivative |
| US5690961A (en) | 1994-12-22 | 1997-11-25 | Hercules Incorporated | Acidic polysaccharides crosslinked with polycarboxylic acids and their uses |
| AU708454B2 (en) | 1995-03-07 | 1999-08-05 | Novartis Ag | Photochemically cross-linked polysaccharide derivatives as supports for the chromatographic separation of enantiomers |
| IT1281877B1 (en) | 1995-05-10 | 1998-03-03 | Fidia Advanced Biopolymers Srl | Heavy metal salts of succinyl derivatives of hyaluronic acid and their use as potential therapeutic agents |
| IL123500A (en) | 1995-08-29 | 2003-06-24 | Fidia Advanced Biopolymers Srl | Composite biomaterials for preventing post-surgical adhesions of tissues |
| EP0763754B1 (en) | 1995-09-13 | 2003-01-08 | Seikagaku Kogyo Kabushiki Kaisha (Seikagaku Corporation) | Photocured crosslinked-hyaluronic acid contact lens |
| DE19604706A1 (en) | 1996-02-09 | 1997-08-14 | Merck Patent Gmbh | Crosslinking products of biopolymers containing amino groups |
| DE19616010C2 (en) | 1996-04-23 | 1998-07-09 | Seitz Filter Werke | Process and device for the production of fibrets (fibrids) from cellulose derivatives |
| US6632802B2 (en) | 1996-08-29 | 2003-10-14 | Fidia Advanced Biopolymers S.R.L. | Hyaluronic acid esters, threads and biomaterials containing them, and their use in surgery |
| IT1287698B1 (en) | 1996-08-29 | 1998-08-18 | Fidia Advanced Biopolymers Srl | SUTURE THREADS ESSENTIALLY MADE OF ESTERE DERIVATIVES OF HYALURONIC ACID |
| IT1286510B1 (en) | 1996-11-29 | 1998-07-15 | Cooperativa Centro Ricerche Po | BUTYRIC ESTERS WITH ANTI-PROLIFERATIVE ACTIVITY AND PHARMACEUTICAL COMPOSITIONS THAT CONTAIN THEM |
| JP4247846B2 (en) | 1997-07-03 | 2009-04-02 | デュピュイ・スパイン・インコーポレーテッド | Cross-linked polysaccharide drug carrier |
| US5977348A (en) | 1997-07-25 | 1999-11-02 | National Starch And Chemical Investment Holding Corporation | Polysaccharide modification in densified fluid |
| ATE302785T1 (en) | 1998-04-30 | 2005-09-15 | Maruha Corp | COMPOUNDS WHOSE STRUCTURE CONTAINS DERIVATIVES OF GLUCURONIC ACID AND GLUCOSAMINE, METHOD FOR THEIR PRODUCTION AND THEIR USE |
| CN1141322C (en) | 1998-05-07 | 2004-03-10 | 荷兰应用科学研究会(Tno) | Method for selective oxidation of primary alcohols |
| US6630457B1 (en) | 1998-09-18 | 2003-10-07 | Orthogene Llc | Functionalized derivatives of hyaluronic acid, formation of hydrogels in situ using same, and methods for making and using same |
| IT1302534B1 (en) | 1998-12-21 | 2000-09-05 | Fidia Advanced Biopolymers Srl | INJECTABLE, BIOCOMPATIBLE AND BIODEGRADABLE COMPOSITIONS INCLUDING AT LEAST A DERIVATIVE OF HYALURONIC ACID, CHONDROGENIC CELLS, FOR |
| JP2002533417A (en) | 1998-12-23 | 2002-10-08 | エスパルマ ゲゼルシャフト ミット ベシュレンクテル ハフツング | Hyaluronate lyase used to enhance penetration of topically applied drugs |
| DE19917614C2 (en) | 1999-04-19 | 2001-07-05 | Thueringisches Inst Textil | Process for the production of cellulosic moldings with high adsorption capacity |
| US6288043B1 (en) | 1999-06-18 | 2001-09-11 | Orquest, Inc. | Injectable hyaluronate-sulfated polysaccharide conjugates |
| US6592794B1 (en) | 1999-09-28 | 2003-07-15 | Organogenesis Inc. | Process of making bioengineered collagen fibrils |
| WO2001034657A1 (en) | 1999-11-08 | 2001-05-17 | Sca Hygiene Products Zeist B.V. | Process of oxidising primary alcohols |
| DE10003397A1 (en) | 2000-01-27 | 2001-08-09 | Hartmann Paul Ag | Polyelectrolyte solid system, process for producing the same and wound dressing |
| DE10009996B4 (en) * | 2000-03-02 | 2005-10-13 | Cognis Ip Management Gmbh | Solid granules with monodisperse particle size distribution, a process for their preparation and their use |
| IT1317358B1 (en) | 2000-08-31 | 2003-06-16 | Fidia Advanced Biopolymers Srl | CROSS-LINKATED DERIVATIVES OF HYALURONIC ACID. |
| IT1317359B1 (en) | 2000-08-31 | 2003-06-16 | Fidia Advanced Biopolymers Srl | PERCARBOXYLATE POLYSACCHARIDES, SUCH AS HYALURONIC ACID, PROCESS FOR THEIR PREPARATION AND USE IN THE PHARMACEUTICAL FIELD AND |
| US6498269B1 (en) | 2000-10-17 | 2002-12-24 | The University Of Connecticut | Method for the oxidation of aldehydes, hemiacetals and primary alcohols |
| AU2002219718A1 (en) | 2000-12-13 | 2002-06-24 | Sca Hygiene Products Zeist B.V. | Process for oxidising primary alcohols |
| EP1217008B1 (en) | 2000-12-19 | 2006-03-01 | Seikagaku Corporation | Photocurable hyaluronic acid derivative and process for producing the same, and photocured crosslinked hyaluronic acid derivative and medical material using the same |
| FR2819808B1 (en) | 2001-01-19 | 2003-04-18 | Simafex | STABILIZED COMPOSITIONS OF O-IODOXYBENZOIC ACID AND PROCESS FOR THEIR PREPARATION |
| CA2435491C (en) | 2001-01-31 | 2010-02-02 | Seikagaku Corporation | Crosslinked polysaccharide sponge |
| US6902548B1 (en) | 2001-03-19 | 2005-06-07 | Ed Schuler | Use of Streptomyces hyalurolyticus enzyme in ophthalmic treatments |
| US6673919B2 (en) | 2001-03-30 | 2004-01-06 | Chisso Cororation | Chemically modified hyaluronic acid or salts thereof, and a process for producing thereof |
| US6946284B2 (en) | 2001-11-16 | 2005-09-20 | University Of Massachusetts | Solubilizing cross-linked polymers with photolyase |
| US20060189516A1 (en) | 2002-02-19 | 2006-08-24 | Industrial Technology Research Institute | Method for producing cross-linked hyaluronic acid-protein bio-composites |
| ITPD20020064A1 (en) | 2002-03-12 | 2003-09-12 | Fidia Advanced Biopolymers Srl | FOREIGN DERIVATIVES OF HYALURONIC ACID FOR THE PREPARATION OF HYDROGELD FOR USE IN THE BIOMEDICAL, SANITARY AND SURGICAL FIELD AND AS A SYSTEM |
| JP3975267B2 (en) | 2002-06-03 | 2007-09-12 | 独立行政法人産業技術総合研究所 | Method for acylating polysaccharide substances |
| US20040101546A1 (en) | 2002-11-26 | 2004-05-27 | Gorman Anne Jessica | Hemostatic wound dressing containing aldehyde-modified polysaccharide and hemostatic agents |
| US7550136B2 (en) | 2002-12-20 | 2009-06-23 | University Of Massachusetts | Photo-reactive polymers and devices for use in hair treatments |
| ITMI20022745A1 (en) | 2002-12-23 | 2004-06-24 | Coimex Scrl United Companies | MIXED ESTERS OF HYALURONIC ACID FOR CYTOSTATIC AND MANUFACTURING ACTIVITIES AND PROCEDURE FOR THEIR PRODUCTION. |
| US6982298B2 (en) | 2003-01-10 | 2006-01-03 | The Cleveland Clinic Foundation | Hydroxyphenyl cross-linked macromolecular network and applications thereof |
| US7465766B2 (en) | 2004-01-08 | 2008-12-16 | The Cleveland Clinic Foundation | Hydroxyphenyl cross-linked macromolecular network and applications thereof |
| US20050126338A1 (en) * | 2003-02-24 | 2005-06-16 | Nanoproducts Corporation | Zinc comprising nanoparticles and related nanotechnology |
| FR2852012B1 (en) | 2003-03-04 | 2006-06-23 | Oreal | PROCESS FOR THE PREPARATION OF O-ACYLATED GLUCOSE DERIVATIVES |
| JP4813179B2 (en) | 2003-03-11 | 2011-11-09 | 生化学工業株式会社 | Photocrosslinked polysaccharide composition and method for producing the same |
| ES2226567B1 (en) * | 2003-06-20 | 2006-07-01 | Universidad De Santiago De Compostela | NANOPARTICULAS OF HIALURONIC ACID. |
| DE10331342B4 (en) | 2003-07-11 | 2009-03-12 | Thüringisches Institut für Textil- und Kunststoff-Forschung e.V. | Thermostable molding or spinning mass |
| WO2005014655A2 (en) | 2003-08-08 | 2005-02-17 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and a protein |
| WO2005023906A1 (en) | 2003-09-08 | 2005-03-17 | Chugai Seiyaku Kabushiki Kaisha | Hyaluronic acid modification product and drug carrier therefrom |
| WO2005025630A1 (en) | 2003-09-10 | 2005-03-24 | Cato T Laurencin | Polymeric nanofibers for tissue engineering and drug delivery |
| CA2538793C (en) | 2003-09-19 | 2011-01-11 | Colorado State University Research Foundation (Csurf) | Hyaluronan (ha) esterification via acylation technique for moldable devices |
| US20100330143A1 (en) | 2003-12-04 | 2010-12-30 | University Of Utah Research Foundation | Modified macromolecules and methods of making and using thereof |
| GB2408741B (en) * | 2003-12-04 | 2008-06-18 | Ind Tech Res Inst | Hyaluronic acid derivative with urethane linkage |
| US8313765B2 (en) | 2003-12-04 | 2012-11-20 | Industrial Technology Research Institute | Biodegradable hyaluronic acid derivative, biodegradable polymeric micelle composition and pharmaceutical or bioactive composition |
| ITMI20040605A1 (en) | 2004-03-29 | 2004-06-29 | Coimex S C R L United Companie | BUTYRICAL ESTERS OF HYALURONIC ACID WITH LOW DEGREE OF SUBSTITUTION PROCEDURE FOR THEIR PREPARATION AND USE |
| WO2006010066A2 (en) | 2004-07-09 | 2006-01-26 | The Cleveland Clinic Foundation | Hydroxyphenyl cross-linked macromolecular network and applications thereof |
| US7323425B2 (en) | 2004-08-27 | 2008-01-29 | Stony Brook Technology And Applied Research | Crosslinking of hyaluronan solutions and nanofiberous membranes made therefrom |
| PT1817347T (en) | 2004-11-24 | 2017-07-21 | Albumedix As | Method of cross-linking hyaluronic acid with divinylsulfone |
| US7214759B2 (en) | 2004-11-24 | 2007-05-08 | Advanced Cardiovascular Systems, Inc. | Biologically absorbable coatings for implantable devices based on polyesters and methods for fabricating the same |
| US7680038B1 (en) | 2005-04-25 | 2010-03-16 | Electronic Arts, Inc. | Dynamic bandwidth detection and response for online games |
| GB0513552D0 (en) | 2005-07-01 | 2005-08-10 | Bristol Myers Squibb Co | Bandage |
| RU2429018C2 (en) | 2005-07-06 | 2011-09-20 | Сейкагаку Корпорейшн | Gel produced of photo-cross-linked hyaluronic acid with administered drug |
| ITPD20050206A1 (en) | 2005-07-07 | 2007-01-08 | Fidia Advanced Biopolymers Srl | BIOMATERIALS IN THE FORM OF FIBER TO BE USED AS MEDICAL DEVICES IN THE TREATMENT OF WOUNDS AND THEIR PROCESSES OF PRODUCTION |
| ITMI20051415A1 (en) | 2005-07-22 | 2007-01-23 | Fidia Advanced Biopolymers Srl | BIOMATERIALS BASED ON SALT-MADE CORBOSSIMETHYLCELLULOSE WITH ZINC ASSOCIATED WITH IALURONIC ACID DERIVATIVES TO BE USED AS MEDICAL DEVICES WITH ANTIMICROBIAL AND ANTIFUNGAL ACTIVITY AND THEIR PRODUCTION PROCESS |
| CN101282985B (en) | 2005-09-21 | 2013-07-10 | 科德生物工程有限公司 | Cell surface coating with hyaluronic acid oligomer derivative |
| US7993678B2 (en) * | 2005-09-26 | 2011-08-09 | Novozymes Biopolymer A/S | Hyaluronic acid derivatives |
| US20070202084A1 (en) | 2005-12-14 | 2007-08-30 | Anika Therapeutics, Inc. | Bioabsorbable implant of hyaluronic acid derivative for treatment of osteochondral and chondral defects |
| US20070202570A1 (en) | 2006-02-24 | 2007-08-30 | Kikkoman Corporation | Enzyme composition, low molecular weight hyaluronan and process for preparing the same |
| EP1991588A1 (en) | 2006-02-28 | 2008-11-19 | Novozymes Biopolymer A/S | Derivatives of hyaluronic acids |
| US8071757B2 (en) * | 2006-03-02 | 2011-12-06 | Novozymes Biopharma Dk A/S | Aryl/alkyl vinyl sulfone hyaluronic acid derivatives |
| JP4892679B2 (en) | 2006-03-27 | 2012-03-07 | 国立大学法人弘前大学 | Hyaluronic acid fiber by gel spinning and method for producing the same |
| KR20070118730A (en) | 2006-06-13 | 2007-12-18 | 주식회사 코오롱 | Wound dressing with excellent moisturizing property and its manufacturing method |
| US20080124395A1 (en) | 2006-06-22 | 2008-05-29 | Weiliam Chen | Formulations and devices for treatment or prevention of neural ischemic damage |
| WO2008014787A1 (en) | 2006-08-04 | 2008-02-07 | Novozymes Biopolymer A/S | Branched hyaluronic acid and method of manufacture |
| US20080063617A1 (en) | 2006-09-07 | 2008-03-13 | Abrahams John M | Cosmetics formulations |
| ITMI20061726A1 (en) | 2006-09-11 | 2008-03-12 | Fidia Farmaceutici | CROSSLINKATI DERIVATIVES BASED ON HYALURONIC ACID RETICULATED VIA CLICK CHEMISTRY |
| CZ302856B6 (en) * | 2006-09-27 | 2011-12-14 | Cpn Spol. S R. O. | Process for preparing polysaccharide derivatives |
| CN101622017B (en) | 2006-12-22 | 2018-09-11 | 克罗马制药有限责任公司 | The purposes of polymer |
| KR20080062092A (en) | 2006-12-29 | 2008-07-03 | 주식회사 핸슨바이오텍 | Hyaluronic Acid Derivatives As Cellular Carriers And Methods For Making The Same |
| EP1942117A1 (en) | 2006-12-29 | 2008-07-09 | Sigea S.R.L. | Derivatives of acid polysaccharides |
| JP5329767B2 (en) | 2007-02-26 | 2013-10-30 | 帝人株式会社 | Aromatic copolyamide fiber production equipment |
| WO2008115799A1 (en) | 2007-03-21 | 2008-09-25 | William Marsh Rice University | Novel gene delivery vectors for human mesenchymal stem cells |
| CZ2007299A3 (en) | 2007-04-24 | 2009-02-04 | Cpn Spol. S R. O. | Preparation of nanofibers from polysaccharides and mixtures thereof with polyvinyl alcohol |
| JP5165281B2 (en) | 2007-06-01 | 2013-03-21 | 株式会社バイオベルデ | Two-reactor type water-containing medical gel-forming agent and hyaluronic acid gel obtained therefrom |
| US8288142B2 (en) | 2007-06-19 | 2012-10-16 | Uvarkina Tamara P | Hyaluronidase and method of use thereof |
| WO2009002869A2 (en) | 2007-06-22 | 2008-12-31 | Innovative Surface Technologies, Inc. | Nanofibers containing latent reactive groups |
| WO2009012372A1 (en) | 2007-07-18 | 2009-01-22 | Advantageous Systems, Llc | Methods and apparatuses for detecting analytes in biological fluid of an animal |
| FR2921675B1 (en) | 2007-09-28 | 2010-03-19 | Univ Claude Bernard Lyon | HYALURONIC ACID FILAMENT AND PROCESS FOR OBTAINING SAME |
| US7976825B2 (en) | 2007-12-06 | 2011-07-12 | Janos Borbely | Cancer cell diagnosis by targeting delivery of nanodevices |
| CA2713489A1 (en) | 2008-02-11 | 2009-08-20 | Basf Se | Producing porous structures from synthetic polymers |
| EP2254944B1 (en) | 2008-02-29 | 2018-12-19 | PVAC Medical Technologies Ltd. | Composition for the formation of gels |
| JP5563563B2 (en) | 2008-06-05 | 2014-07-30 | エージェンシー フォー サイエンス, テクノロジー アンド リサーチ | Method for forming hydrogels in the presence of peroxidase and low concentrations of hydrogen peroxide |
| JP2010014784A (en) | 2008-07-01 | 2010-01-21 | Fuji Xerox Co Ltd | Optical writing display apparatus, optical writing apparatus, and optical writing method |
| FR2934999B1 (en) | 2008-08-13 | 2011-07-29 | Adocia | POLYSACCHARIDES FUNCTIONALIZED BY TRYPTOPHAN DERIVATIVES |
| WO2010028025A1 (en) | 2008-09-02 | 2010-03-11 | Gurtner Geoffrey C | Threads of hyaluronic acid and/or derivatives thereof, methods of making thereof and uses thereof |
| CZ301555B6 (en) | 2008-11-06 | 2010-04-14 | Cpn S. R. O. | Process for preparing DTPA crosslinked derivatives of hyaluronic acid and modification thereof |
| ITRM20080636A1 (en) | 2008-11-28 | 2010-05-29 | Univ Palermo | PROCEDURE FOR THE PRODUCTION OF FUNCTIONAL DERIVATIVES OF HYALURONIC ACID AND RELATIVE HYDROGELS. |
| JP2010138276A (en) | 2008-12-11 | 2010-06-24 | Nipro Corp | Method for producing single yarn of hyaluronic acid |
| EP2398941B1 (en) | 2009-02-21 | 2016-07-13 | Sofradim Production | Crosslinked fibers and method of making same by extrusion |
| EP2398519A2 (en) | 2009-02-21 | 2011-12-28 | Sofradim Production | Compounds and medical devices activated with solvophobic linkers |
| AU2010215203B2 (en) | 2009-02-21 | 2015-07-16 | Covidien Lp | Medical devices with an activated coating |
| US8551378B2 (en) | 2009-03-24 | 2013-10-08 | North Carolina State University | Nanospinning of polymer fibers from sheared solutions |
| US20120219554A2 (en) | 2009-05-14 | 2012-08-30 | Fidia Farmaceutici S.P.A. | Extracellular yaluronidase from streptomyces koganeiensis |
| DE102009022805B4 (en) | 2009-05-27 | 2013-04-25 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Process for the preparation of polysaccharide esters or polysaccharide mixed esters |
| WO2010138074A1 (en) | 2009-05-29 | 2010-12-02 | Hilborn Joens | Hyaluronic acid based delivery systems |
| CA2764635C (en) * | 2009-06-09 | 2018-05-22 | Lux Biosciences, Inc. | Topical drug delivery systems for ophthalmic use |
| AU2010276574B2 (en) | 2009-07-30 | 2015-08-20 | Carbylan Therapeutics, Inc. | Modified hyaluronic acid polymer compositions and related methods |
| KR101103423B1 (en) | 2009-09-04 | 2012-01-06 | 아주대학교산학협력단 | Bio-injectable tissue adhesive hydrogels and their biomedical uses |
| WO2011059325A2 (en) | 2009-11-11 | 2011-05-19 | University Of Twente, Institute For Biomedical Technology And Technical Medicine (Mira) | Dextran-hyaluronic acid based hydrogels |
| EP3067069B1 (en) | 2009-11-11 | 2023-07-26 | Hy2Care B.V. | Hydrogels based on polymers of dextran tyramine and tyramine conjugates of natural polymers |
| US20110111012A1 (en) | 2009-11-12 | 2011-05-12 | Hemcon Medical Technologies, Inc. | Nanomaterial wound dressing assembly |
| CZ302504B6 (en) | 2009-12-11 | 2011-06-22 | Contipro C A.S. | Hyaluronic acid derivative oxidized selectively in position 6 of polysaccharide glucosamine portion to aldehyde and modification process thereof |
| CZ2009835A3 (en) | 2009-12-11 | 2011-06-22 | Contipro C A.S. | Process for preparing hyaluronic acid derivative oxidized in position 6 of saccharide glucosamine portion selectively to aldehyde and modification method thereof |
| US8197849B2 (en) | 2010-02-12 | 2012-06-12 | National Health Research Institutes | Cross-linked oxidated hyaluronic acid for use as a vitreous substitute |
| CN101897976A (en) * | 2010-07-16 | 2010-12-01 | 沈阳药科大学 | A drug solubilizing carrier and its preparation method and application |
| CZ20101001A3 (en) | 2010-12-31 | 2012-02-08 | Cpn S.R.O. | Hyaluronic acid fibers, process of their preparation and use |
| CZ304072B6 (en) | 2011-04-26 | 2013-09-25 | Contipro Biotech S.R.O. | Amphoteric material based on crosslinked hyaluronic acid, process for its preparation, materials containing active agents enclosed in hyaluronate network, process for their preparation and their use |
| CN102154738B (en) | 2011-05-10 | 2012-08-01 | 青岛大学 | Method for preparing red algae agar fiber |
| ITTO20110428A1 (en) | 2011-05-13 | 2012-11-14 | Rottapharm Spa | ESTERS OF HYALURONIC ACID, THEIR PREPARATION AND USE IN DERMATOLOGY |
| CN104024494B (en) | 2011-10-18 | 2017-11-10 | 海克私人有限公司 | Fiber caused by fiber-forming process and thus method |
| CZ2012282A3 (en) | 2012-04-25 | 2013-11-06 | Contipro Biotech S.R.O. | Crosslinked hyaluronate derivative, process of its preparation, hydrogel and microfibers based thereon |
| CZ304512B6 (en) | 2012-08-08 | 2014-06-11 | Contipro Biotech S.R.O. | Hyaluronic acid derivative, process for its preparation, modification process and use thereof |
| CZ2012844A3 (en) | 2012-11-27 | 2014-02-05 | Contipro Biotech S.R.O. | Photoreactive derivative of hyaluronic acid, process for its preparation, 3D crosslinked derivative of hyaluronic acid, process for its preparation and use |
| CZ304303B6 (en) | 2012-11-27 | 2014-02-19 | Contipro Biotech S.R.O. | Fibers based on hydrophobized hyaluronate, process for their preparation and use, fabric based thereon and use thereof |
| CZ304654B6 (en) | 2012-11-27 | 2014-08-20 | Contipro Biotech S.R.O. | C6-C18-acylated hyaluronate-based nanomicellar composition, process for preparing C6-C18-acylated hyaluronate, process for preparing nanomicellar composition and stabilized nanomicellar composition as well as use thereof |
| CN103505736A (en) | 2013-09-23 | 2014-01-15 | 天津大学 | Modified hyaluronic acid based macromolecule lipidosome and preparation method thereof |
| CZ305153B6 (en) | 2014-03-11 | 2015-05-20 | Contipro Biotech S.R.O. | Conjugates of hyaluronic acid oligomer or a salt thereof, process for their preparation and use |
-
2012
- 2012-11-27 CZ CZ2012-842A patent/CZ304654B6/en unknown
-
2013
- 2013-11-26 RU RU2015125076A patent/RU2640287C2/en active
- 2013-11-26 HU HUE13829034A patent/HUE038664T2/en unknown
- 2013-11-26 JP JP2015543316A patent/JP2016500130A/en active Pending
- 2013-11-26 US US14/647,626 patent/US9999678B2/en active Active
- 2013-11-26 PL PL13829034T patent/PL2934592T3/en unknown
- 2013-11-26 EP EP13829034.1A patent/EP2934592B1/en active Active
- 2013-11-26 DK DK13829034.1T patent/DK2934592T3/en active
- 2013-11-26 KR KR1020157016788A patent/KR102191353B1/en active Active
- 2013-11-26 WO PCT/CZ2013/000156 patent/WO2014082609A1/en not_active Ceased
- 2013-11-26 BR BR112015012191A patent/BR112015012191A8/en not_active Application Discontinuation
- 2013-11-26 ES ES13829034.1T patent/ES2683993T3/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0347801A (en) * | 1989-04-18 | 1991-02-28 | Agency Of Ind Science & Technol | Novel hyaluronic acid derivative and method for producing the same |
| JPH0625306A (en) * | 1992-04-21 | 1994-02-01 | Shiseido Co Ltd | Solvent-insoluble hyaluronic acid and its production |
| JP2004123785A (en) * | 2002-09-30 | 2004-04-22 | Chisso Corp | n-Alkanoylated hyaluronic acid or salt thereof and method for producing the same |
| JP2012520902A (en) * | 2009-03-17 | 2012-09-10 | コンティプロ ファーマ アクチオヴァ スポレチノスト | Method of modifying hyaluronic acid with (O-acyl-O'-alkyl carbonate-substituted pyridine) complexes |
Non-Patent Citations (1)
| Title |
|---|
| INANAGA, J. ET AL., BULLETIN OF THE CHEMICAL SOCIETY OF JAPAN, vol. 52, no. 7, JPN7017002018, 1979, pages 1989 - 1993, ISSN: 0003732525 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2021157677A1 (en) * | 2020-02-05 | 2021-08-12 | ||
| WO2021157677A1 (en) * | 2020-02-05 | 2021-08-12 | 旭化成株式会社 | Hyaluronic acid derivative composition, pharmaceutical composition and hyaluronic acid derivative-drug conjugate composition |
| JP7397103B2 (en) | 2020-02-05 | 2023-12-12 | 旭化成株式会社 | Hyaluronic acid derivative composition, pharmaceutical composition and hyaluronic acid derivative-drug conjugate composition |
| JPWO2021157665A1 (en) * | 2020-02-07 | 2021-08-12 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20150088848A (en) | 2015-08-03 |
| BR112015012191A8 (en) | 2018-01-23 |
| HUE038664T2 (en) | 2018-11-28 |
| EP2934592B1 (en) | 2018-05-16 |
| WO2014082609A1 (en) | 2014-06-05 |
| BR112015012191A2 (en) | 2017-07-11 |
| RU2640287C2 (en) | 2017-12-27 |
| DK2934592T3 (en) | 2018-07-02 |
| PL2934592T3 (en) | 2018-12-31 |
| US9999678B2 (en) | 2018-06-19 |
| CZ2012842A3 (en) | 2014-08-20 |
| EP2934592A1 (en) | 2015-10-28 |
| US20150320873A1 (en) | 2015-11-12 |
| KR102191353B1 (en) | 2020-12-16 |
| RU2015125076A (en) | 2017-01-13 |
| CZ304654B6 (en) | 2014-08-20 |
| ES2683993T3 (en) | 2018-10-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2016500130A (en) | C6-C18-acylated derivative of hyaluronic acid and process for its preparation, nanomicelle composition based on it and process for its preparation, process for preparing stabilized nanomicelle composition and use thereof | |
| Li et al. | Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel | |
| Lee et al. | Inflammation-responsive drug-conjugated dextran nanoparticles enhance anti-inflammatory drug efficacy | |
| Nosrati et al. | Folic acid conjugated bovine serum albumin: An efficient smart and tumor targeted biomacromolecule for inhibition folate receptor positive cancer cells | |
| Sun et al. | Temperature-sensitive gold nanoparticle-coated pluronic-PLL nanoparticles for drug delivery and chemo-photothermal therapy | |
| Li et al. | Redox-sensitive and intrinsically fluorescent photoclick hyaluronic acid nanogels for traceable and targeted delivery of cytochrome c to breast tumor in mice | |
| Jia et al. | Multi-functionalized hyaluronic acid nanogels crosslinked with carbon dots as dual receptor-mediated targeting tumor theranostics | |
| Wang et al. | Curcumin-loaded liposomes with the hepatic and lysosomal dual-targeted effects for therapy of hepatocellular carcinoma | |
| Song et al. | Construction of targeting-clickable and tumor-cleavable polyurethane nanomicelles for multifunctional intracellular drug delivery | |
| Deng et al. | Hydrophobic IR780 loaded sericin nanomicelles for phototherapy with enhanced antitumor efficiency | |
| Lee et al. | pH-Responsive hyaluronated liposomes for docetaxel delivery | |
| Toman et al. | Nanoparticles of alkylglyceryl-dextran-graft-poly (lactic acid) for drug delivery to the brain: Preparation and in vitro investigation | |
| Li et al. | Tailored Trojan horse nanocarriers for enhanced redox-responsive drug delivery | |
| Huang et al. | Amphiphilic prodrug-decorated graphene oxide as a multi-functional drug delivery system for efficient cancer therapy | |
| Hu et al. | Core cross-linked polyphosphoester micelles with folate-targeted and acid-cleavable features for pH-triggered drug delivery | |
| Liu et al. | Polymeric prodrug of bufalin for increasing solubility and stability: Synthesis and anticancer study in vitro and in vivo | |
| Lee et al. | pH-sensitive short worm-like micelles targeting tumors based on the extracellular pH | |
| CN104945538A (en) | Hyaluronic acid vitamin E derivative and preparation and application | |
| Liu et al. | A ROS-stimulus-responsive nanocarrier loading with guanidine-modified hydroxycamptothecin prodrug for enhanced anti-tumor efficacy | |
| Shirani et al. | Redox responsive polymeric micelles of gellan gum/abietic acid for targeted delivery of ribociclib | |
| CN103381273B (en) | Amycin prodrug and preparation method thereof and injectable compositions | |
| Liu et al. | pH-sensitive dextran-based micelles from copper-free click reaction for antitumor drug delivery | |
| ES2547326T3 (en) | Cellulose-based nanoparticles for drug delivery | |
| Mehata et al. | PLGA nanoplatform for the hypoxic tumor delivery: folate targeting, therapy, and ultrasound/photoacoustic imaging | |
| Cepraga et al. | Two-photon photosensitizer–polymer conjugates for combined cancer cell death induction and two-photon fluorescence imaging: structure/photodynamic therapy efficiency relationship |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20161124 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20170119 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170622 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20170907 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20171114 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20171221 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20180202 |