CN1060479C - 咪唑衍生物的制备方法 - Google Patents
咪唑衍生物的制备方法 Download PDFInfo
- Publication number
- CN1060479C CN1060479C CN96121796A CN96121796A CN1060479C CN 1060479 C CN1060479 C CN 1060479C CN 96121796 A CN96121796 A CN 96121796A CN 96121796 A CN96121796 A CN 96121796A CN 1060479 C CN1060479 C CN 1060479C
- Authority
- CN
- China
- Prior art keywords
- indol
- formula
- group
- methyl
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Public Health (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Epoxy Resins (AREA)
- Paints Or Removers (AREA)
- Lubricants (AREA)
- Adhesives Or Adhesive Processes (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Photoreceptors In Electrophotography (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Dental Preparations (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Steroid Compounds (AREA)
Abstract
本发明涉及咪唑衍生物的制备方法。该方法详述于说明书中。该咪唑衍生物是5-HT1状受体的选择性激动剂,由于已确认它们是这些受体的选择性激动剂,因而可用于临床治疗,特别是对偏头痛和有关疾病的治疗。
Description
本专利申请是CN92101189.X的分案申请。原申请的申请日为1992年2月1日;原申请的发明发称是“咪唑、三唑和四唑衍生物”。
本发明涉及—类作用于5—羟色胺(5—HT)受体的取代的咪唑、三唑和四唑衍生物,它们是称之为“5—HT1状”受体的选择性激动剂。因为已确定它们是这些受体的选择性激动剂,因而它们可用于临床治疗。
具有选择性血管收缩活性的5—HT1状受体激动剂最近已被认为可用于治疗偏头痛(见例如A.Doenicke等,The Lancet,1988,Vol.1,1390—11)。因此,作为选择性5—HT1状受体激动剂的本发明化合物可特别地用于偏头痛和有关的疾病的冶疗,例如偏头神经痛、慢性阵发性偏头痛、与脉管障碍有关的头痛、紧张性头痛和小儿偏头痛。
EP—A—0313397描述了一类被五元杂脂族环取代的色胺衍生物,阐明它对“5—HT1状”受体特殊类型是有特效的并因此可成为需此活性的临床治疗有效的治疗剂,特别是偏头痛。但是,EP—A—0313397既没有公开也没有指示本发明所涉及的咪唑、三唑和四唑衍生物。
本发明提供式Ⅰ化合物或其盐或药物前体。其中虚线环表示两个五元环中任何位置上非相邻的双键;
V、W、X、Y和Z中的两个、三个或四个表示氮、余下的表 示碳,若当V、W、Y和Z中的两个表示氮,余下的表示碳时,则所述氮原子在五元环的非相邻位置上;
示碳,若当V、W、Y和Z中的两个表示氮,余下的表示碳时,则所述氮原子在五元环的非相邻位置上;
A1表示氢、烃、杂环基、卤素、氰基、三氟甲基、—ORX、—SRX、—NRXRY、—NRXCORY、—NRXCO2RY、—NRXSO2RY或—NRZCTNRXRY;
当V、W、X、Y和Z中的四个表示氮,其他的表示碳,A2表示非成键电子对;或当V、W、X、Y和Z中的两个或三个表示氮,余下的表示碳时,A2表示氢、烃、杂环基、卤素、氰基、三氟甲基、—ORX、—SRX、—NRXRY、—NRXCORY、—NRXCO2RY、—NRXSO2RY或—NRZCTNRXRY;
E表示一个键或含1至4个碳原子的直或支链亚烷基链;
F表示下式基团
U表示氮或C—R2;
B表示氧、硫或N—R3;
R1表示—CH2·CHR4·NR6R7或下式基团 或
或
 其中虚线表示任意的化学键;
其中虚线表示任意的化学键;
R2、R3、R4、R5、R6和R7各自独立地表示氢或C1—6烷基;
RX和RY各自独立地表示氢、烃或杂环基,或RX和RY一起表示C2—6亚烷基;
RZ表示氢、烃或杂环基;
T表示氧、硫或基团=N·G;和
G表示烃、杂环基或吸电子基团。
本发明还涉及上述式Ⅰ化合物,其中V、W、Y和Z中的三个或四个表示氮,余下的表示碳;
当V、W、X、Y和Z中的四个表示氮,其它的表示碳时,A2表示非成键电子对;或当V、W、X、Y和Z中的三个表示氮,余下的表示碳时,A2表示氢、烃、杂环基、卤素、氰基、三氟甲基、—ORX、—SRX、—NRXRY、—NRXCORY、—NRXCO2RY、—NRXSO2RY或—NRZCTNRXRY;和
A1、E、F、RX、RY、RZ和T如上述定义。
式Ⅰ化合物的盐将以无毒的药学上可接受的盐用于药物中。而其它的盐可用于制备根据本发明的化合物或它们的无毒的药学上可接受的盐。本发明化合物合适的药学上可接受的盐包括酸加成盐,其可以是,例如,通过将本发明化合物的溶液与药学上可接受的无毒酸溶液混合形成的盐,如盐酸、富马酸、马来酸、琥珀酸、乙酸、苯甲酸、草酸、柠檬酸、酒石酸、碳酸或磷酸。此外,当本发明化合物带有酸性部分时,其合适的药学上可接受的盐可以包括碱金属盐,例如钠或钾盐;碱土金属盐,例如钙或镁盐;和与合适的有机配位体形成的盐,例如季铵盐。
此处所用的术语“烃”包括含至多18个碳原子,较合适的是至多15个碳原子和合适的是至多12个碳原子的直链、支链和环状基团。合适的烃基包括C1—6烷基、C2—6链烯基、C2—6链炔基、C3—7环烷基、C3—7环烷基(C1—6)烷基、芳基和芳基(C1—6)烷基。
此处所用的术语“杂环基”包括含至多18个碳原子和至少一个较优选地选自氧、氮和硫的杂原子的环状基团。杂环基较合适地是含至多15个碳原子和合适地是含至多12个碳原子,并且较优选地是通过碳键合。合适的杂环基的例子包括C3—7杂环烷基、C3—7杂环烷基(C1—6)烷基、杂芳基和杂芳基(C1—6)烷基。
合适的烷基包括含1至6个碳原子的直链和支链的烷基。典型的实例包括甲基和乙基,和直链或支链的丙基和丁基。特殊的烷基是甲基、乙基和叔丁基。
合适的链烯基包括含2至6个碳原子的直链和支链的链烯基。典型的实例包括乙烯基和烯丙基。
合适的链炔基包括含2至6个碳原子的直链和支链的链炔基。典型的实例包括乙炔基和丙炔基。
合适的环烷基包括含3至7个碳原子的基团。特殊的环烷基是环丙基和环己基。
特殊的芳基是苯基。
特殊的芳基(C1—6)烷基包括苄基、苯乙基和苯丙基。
合适的杂环烷基包括氮杂环丁烷基、吡咯烷基、哌啶基、哌嗪基和吗啉基。
合适的杂芳基包括吡啶基、喹啉基、异喹啉基、哒嗪基、嘧啶基、吡嗪基、吡喃基、呋喃基、苯并呋喃基、二苯并呋喃基、噻吩基、苯并噻吩基、咪唑基、噁二唑基和噻二唑基。
特殊的杂芳基(C1—6)烷基包括吡啶甲基和吡嗪甲基。
烃和杂环基可以依次任意地被选自下列的一或多个基团取代:C1—6烷基、金刚烷基、苯基、卤素C1—6卤代烷基、C1—6氨基烷基、三氟甲基、羟基、C1—6烷氧基、芳氧基、酮基、C1—3亚烷基二氧基、硝基、氰基、羧基、C2—6烷氧羰基、C2—6烷氧羰基(C1—6)烷基、C2—6烷羰氧基、芳羰氧基、C2—6烷羰基、芳羰基、C1—6烷硫基、C1—6烷烷基亚磺酰基、C1—6烷磺酰基、芳磺酰基、NRVRW、—NRVCORW、—NRVCO2RW、—NRVSO2RW、—CH2NRVSO2RW、—NHCONRVRW、—CONRVRW、—SO2NRVRW和—CH2SO2NRVRW,其中RV和RW各自独立地表示氢、C1—6烷基、芳基或芳基(C1—6)烷基,或RV和RW一起表示C2—6亚烷基。
当RX和RY,或RV和RW一起表示C2—6亚烷基时,此基团可以是1,2—亚乙基、1,2—亚丙基、1,2—亚丁基、1,5—亚戊基或1,6—亚己基,较优选的是1,2—亚丁基或1,5—亚戊基。
当基团G表示吸电子基团时,此基团较合适地是氰基、硝基、—CORX、—CO2RX或—SO2RX,其中RX如上定义。
此处所用的术语“卤素”包括氟、氯、溴和碘,特别是氟。
在本发明的范围内包括上述式Ⅰ化合物的药物前体。一般地,此种药物前体作为式Ⅰ化合物的功能衍生物,它可在体内容易地转变成所需要的式Ⅰ化合物。合适的药物前体衍生物的选择和制备的传统方法已有描述,例如,“Design of Prodrugs”编辑H.Bundgaard,Elsevier,1985。
当本发明化合物中至少一个不对称中心时,则它们可以以对映体存在。当本发明化合物中具有两个或更多不对称中心时,则它们可以以非对映异构体存在。显然,所有这种异构体和其混合物都包含在本
发明的范围内。
式Ⅰ的咪唑、三唑或四唑环可以以各种不同的正则形式存在。它们较合适地是下列式ⅠA至ⅠT表示的形式: 其中A1、A2、E和F如上定义。较优选的式Ⅰ的咪唑、三唑和四唑环包括上述式ⅠA、ⅠC、ⅠG、ⅠH、ⅠL、ⅠM、ⅠN、ⅠP和ⅠQ表示的环,特别是ⅠH。
其中A1、A2、E和F如上定义。较优选的式Ⅰ的咪唑、三唑和四唑环包括上述式ⅠA、ⅠC、ⅠG、ⅠH、ⅠL、ⅠM、ⅠN、ⅠP和ⅠQ表示的环,特别是ⅠH。
亚烷基链E可以是例如亚甲基、1,2—亚乙基、1—甲基—1,2—亚乙基、1,2—亚丙基或2—甲基—1,2—亚丙基。或者,基团E可以表示一个单键,则式Ⅰ中的基团F直接与五元杂芳环相连。
基团F较合适地是式FA的吲哚、苯并呋喃或苯并噻吩部分,或式FB的吲唑部分:其中B、R1、R2和R3如上定义。较优选地是,基团F表示结构FC的吲哚部分: 其中R1、R2和R3如上定义,特别是其中的R2和R3两者都是氢。
其中R1、R2和R3如上定义,特别是其中的R2和R3两者都是氢。
当V、W、X、Y和Z中的四个表示氮,其它的表示碳时,即式Ⅰ环为四唑环时,则基团A2为非成键电子对。否则,A1和A2各自独立地表示氢、烃、杂环基、卤素、氰基、三氟甲基、—ORX、—SRX、—NRXRY、—NRXCORY、—NRXCO2RY、—NRXSO2RY或—NRZCTNRXRY。
与基团A1/和/或A2等同的较合适的基团包括C1—6烷基、C6—7环烷基、芳基、芳基(C1—6)烷基、C3—7杂环烷基、杂芳基或杂芳基(C1—6)烷基,其中的任一基团可被任意取代;和氢、卤素、氰基、三氟甲基、C1—6烷氧基、C1—6烷硫基或—NRXRY,其中RX和RY如上定义。基团A1和/或A2上较合适的任意取代基的实例包括三氟甲基、C1—6烷氧基、C2—6烷氧羰基、C2—6烷羰基、C1—6烷基磺酰基、芳磺酰基、氨基、一或二(C1—6)烷基氨基、C2—6烷羰基氨基、芳羰基氨基、C2—6烷氧羰基氨基、C1—6烷基磺酰氨基、芳基磺酰氨基、C1—6烷基磺酰氨基甲基、氨羰基氨基、一或二(C1—6)烷氨基羰氨基、一或二芳氨基羰氨基、吡咯烷基羰氨基、氨基羰基、一或二(C1—6)烷氨基羰基、C1—6烷氨基磺酰基、氨基磺酰甲基和一或二(C1—6)烷氨基磺酰甲基。
A1和/或A2等同的基团特别是包括氢、甲基、甲氧甲基、氨甲基、二甲氨基甲基、乙酰氨甲基、苯甲酰氨甲基、叔丁氧羰氨基甲基、甲磺酰氨基甲基、苯磺酰氨基甲基、氨羰基甲基、乙基、氨乙基、乙酰氨乙基、苯甲酰氨乙基、甲氧羰基氨基乙基、乙氧羰基氨基乙基、叔丁氧羰基氨基乙基、甲磺酰氨基乙基、氨羰基氨基乙基、甲氨基羰基氨基乙基、叔丁基氨基羰基氨基乙基、苯基氨基羰氨基乙基、吡咯烷基羰基氨基乙基、环丙基、苯基、甲磺酰氨基苯基、氨羰基苯基、甲氨基羰基苯基、甲磺酰氨基甲苯基、氨基磺酰基甲苯基、甲氨磺酰基甲苯基、二甲氨基磺酰基甲苯基、苄基、三氟甲基苄基、甲氧基苄基、乙酰氨基苄基、甲磺酰氨基苄基、氨基羰基氨基苄基、氨羰基苄基、甲氨基羰基苄基、甲磺酰基苄基、甲氨基磺酰基苄基、吡啶甲基、甲氧基吡啶甲基、氨基、甲氨基、苄氨基、二甲氨基、叔丁氧羰基氨基乙氨基和甲磺酰氨基乙氨基。
A1和/或A2优选的等同基团包括氢、甲基、乙基、苄基和氨基。
R1典型的等同基团包括氨乙基、N—甲基氨乙基、N,N—二甲基氨乙基、4—哌啶基、1—甲基—4—哌啶基、3—吡咯烷基和1—甲基—3—吡咯烷基。
R2至R7较优选的等同基团是氢和甲基。
根据本发明的特殊的次级化合物及其盐和药物前体是如式ⅡA所示的化合物: 其中
其中
X1表示氮或A12—C;
n是零、1、2或3;
B1表示氧、硫或N—R13;
A11和A12各自独立地表示C1—6烷基、C2—6链烯基、C2—6链炔基、C3—7环烷基、芳基、芳基(C1—6)烷基、C3—7杂环烷基、杂芳基或杂芳基(C1—6)烷基,其中的任一基团均可被任意取代;或氢、卤素、氰基、三氟甲基、C1—6烷氧基、C1—6烷硫基或—NRXRY;
R12、R13、R14、R16和R17各自独立地表示氢或C1—6烷基;和
RX和RY各自独立地表示氢、烃或杂环基,或RX和RY一起表示C2—6亚烷基。
A11和A12上任意的取代基的实例较合适地包括三氟甲基、C1—6烷氧基、C2—6烷氧羰基、C2—6烷羰基、C1—6烷基磺酰基、芳基磺酰基、氨基、一或二(C1—6)烷氨基、C2—6烷羰基氨基、芳羰基氨基、C2—6烷氧基氨基、C1—6烷基磺酰氨基、芳磺酰氨基、C1—6烷基磺酰氨基甲基、氨羰基氨基、一或二(C1—6)烷氨基羰基氨基、一或二芳氨基羰基氨基、吡咯烷基羰基氨基、氨基羰基、一或二(C1—6)烷氨基羰基、C1—6烷氨基磺酰基、氨基磺酰基甲基和一或二(C1—6)烷氨基磺酰甲基。
式ⅡA中A11和A12等同基团特别是包括氢、甲基、乙基、苄基和氨基。当X1表示A12—C时,基团A11较优选地是氢或甲基。
R12、R13和R14较优选地是各自表示氢。式ⅡA中R16和R17的较优选的等同基团包括氢和甲基。
根据本发明的另一次级化合物及其盐和药物前体是式ⅡB所示的化合物: 其中
其中
Y1表示氮或A22—C;
n是零、1、2或3;
B2表示氧、硫或N—R23;
A21和A22各自独立地表示C1—6烷基、C2—6链烯基、C2—6链炔基、C3—7环烷基、芳基、芳基(C1—6)烷基、C3—7杂环烷基、杂芳基或杂芳基(C1—6)烷基、其中的任一基团均可被任意取代;或氢、卤素、氰基、三氟甲基、C1—6烷氧基、C1—6烷硫基或—NRXYY;
R22、R23、R24、R26和R27各自独立地表示氢或C1—6烷;和
RX和RY各自独立地表示氢、烃或杂环基,或RX和RY一起表示C2—6亚烷基。
基团A21和A22上的任意的取代基的寮例与上述式ⅡA中指示的基团A11和A12的取代基相一致。式ⅡB中A21和A22的特殊等同基团包括氢、甲基、乙基和苄基。
R22、R23和R24较优选地是各自表示氢。式ⅡB中R26和R27较优选的等同基团包括氢和甲基。
根据本发明的另一次级化合物及其盐和药物前体是如式ⅡC所示的化合物:其中
Y2表示氮或A32—C;
Z1表示氮或CH;
n是零、1、2或3;
B3表示氧、硫或N—R23;
A31和A32各自独立地表示C1—6烷基、C2—6链烯基、C2—6链炔基、C3—7环烷基、芳基、芳基(C1—6)烷基、C3—7杂环烷基、杂芳基或杂芳基(C1—6)烷基,其中的任一基团均可被任意取代;或氢、卤素、氰基、三氟甲基、C1—6烷氧基、C1—6烷硫基或—NRXRY;
R31表示—CH2·CHR34·NR36R37或下式基团
R32、R33、R34、R35、R36和R37各自独立地表示氢或C1—6烷基;和
RX和RY各自独立地表示氢、烃或杂环基,或RX和RY一起表示C2—6烷亚烷基。
A31和A32基上任意的取代基的实例与上述式ⅡA中指明的A11和A12上的取代基相一致。式ⅡC中A31和A32特殊的等同基团包括氢、甲基和氨基。
R32、R33和R34较优选地是各自表示氢。R35、R36和R37较优选的等同基团包括氢和甲基。
根据本发明的另一次级化合物及其盐和药物前体是式ⅡD所示化合物: 其中
其中
W1表示氮或C—A42;
n是零、1、2或3;
B4表示氧、硫或N—R43;
A41和A42各自独立地表示C1—6烷基、C2—6链烯基、C2—6链炔基、C3—7环烷基、芳基、芳基(C1—6)烷基、C3—7杂环烷基、杂芳基或杂芳基(C1—6)烷基、其中的任一基团均可被任意取代;或氢、卤素、氰基、三氟甲基、C1—6烷氧基、C1—6烷硫基或—NRXRY;
R41表示—CH2·CHR44·NR46R47或下式基团
R42、R43、R44、R45、R46和R47各种独立地表示氢或C1—6烷基;和
RX和RY各自独立地表示氢、烃或杂环基,或RX和RY一起表示C2—6亚烷基。
基团A41和A42上任意的取代基的实例与上述式ⅡA中指明的基团A11和A12上的取代基相一致。式ⅡD中A41和A42特殊的等同基团包括氢和甲基。
较优选地是,R42、R43和R44各自表示氢。R45、R46和R47较优选的等同基团包括氢和甲基。
本发明范围内的具体化舍物及其盐和药物前体包括:
2—[5—(2—苄基四唑—5—基甲基)—1H—吲哚—3—基]乙胺;
2—[5—(1—苄基四唑—5—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(1—甲基四唑—5—基甲基)—1H—引哚—3—基]乙胺;
N,N—二甲基—2—[5—(2—甲基四唑—5—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(四唑—2—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(四唑—1—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(1—甲基—1,2,4—三唑—5—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(1—甲基—1,2,4—三唑—3—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(1,2,3—三唑—1—基甲基)—1H—吲哚—3—基]乙胺;
3—(2—氨乙基)—5—(1—甲基四唑—5—基)苯并[b]噻吩;
3—(2—氨乙基)—5—(2—甲基四唑—5—基)苯并[b]噻吩;
3—[2—(N,N—二甲氨基)乙基]—5—(2—甲基四唑—5—基)苯并[b]噻吩;
N,N—二甲基—2—[5—(2—甲基咪唑—1—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(咪唑—1—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(2—乙基四唑—5—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(1—乙基四唑—5—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(1,2,4—三唑—1—基)—1H—吲哚—3—基]乙胺;
1—甲基—4—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]哌啶;
1—甲基—4—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]哌啶;
4—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]哌啶;
4—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]哌啶;
3—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]吡咯烷;
1—甲基—3—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]吡咯烷;
4—[5—(咪唑—1—基)—1H—吲哚—3—基]哌啶;
4—[5—(1,2,3—三唑—1—基)—1H—吲哚—3—基]哌啶;
1—甲基—4—[5—(咪唑—1—基)—1H—吲哚—3—基]哌啶;
1—甲基—4—[5—(1,2,3—三唑—1—基)—1H—吲哚—3—基]哌啶;
1—甲基—3—[5—(1,2,3—三唑—1—基)—1H—吲哚—3—基]吡咯烷;
1—甲基—3—[5—(2—甲基咪唑—1—基甲基)—1H—吲哚—3—基)吡咯烷;
1—甲基—3—[5—(咪唑—1—基)—1H—吲哚—3—基]吡咯烷;
1—甲基—3—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]吡咯烷;
1—甲基—3—[5—(咪唑—1—基甲基)—1H—吲哚—3—基]吡咯烷;
N,N—二甲基—2—[5—(2—氨基咪唑—1—基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(2—氨基咪唑—1—基甲基)—1H—吲哚—3—基]乙胺;
N—甲基—2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺。
本发明还提供含—或多种本发明化合物及有关的药物上可接受的载体的药物组合物。这些组合物较优选地是对口服、非肠道、鼻内、舌下或直肠给药,或通过吸入法或吹入法给药的单位剂型、如片剂。丸剂、胶囊剂、粉剂、颗粒剂、灭菌注射液或悬浮液、定量气雾剂或液体喷雾剂、滴剂、安瓿、自动注射器或栓。对于制备如片剂的固体组合物,可将主要的活性成分与药物载体,例如常规的片剂成分如玉米淀粉、乳糖、蔗糖、山梨醇、滑石、硬脂酸、硬脂酸镁、磷酸钙或树胶,和其它药物稀释剂,例如水,相混合以形成含有本发明化合物或其药学上可接受的无毒盐的均匀混合物的剂型确定前的固体组合物,这里所指的均匀状的剂型确定前的组合物,其含义是活性成分完全均匀地分散于组合物中,以至于组合物可以很容易地细分成等效的单位剂型如片剂、丸剂和胶囊剂,然后再将剂型确定前的固体组合物细分成含有0.1至大约500mg本发明活性成分的上述类型的单位剂型。新颖的组合物的片剂或丸剂可以被包衣或者另外混合以获得一种具有长效优点的剂型。例如,片剂或丸剂可以由内剂量和外剂量成分构成,后者是以包裹在前者上的形式存在。此两个成分可以通过肠溶衣层分开,它可以起到耐胃中分解并允许内成分完整地达十二指肠或延迟释放的作用。许多材料都可用作这种肠溶衣层或包衣,此种材料包括很多聚酸和聚酸与如虫胶、十六烷醇和乙酸纤维素这类材料的混合物。
本发明新颖的口服或通过注射药的液体剂型是由包括水溶液、合适的调味糖浆、水或油的悬浮液和调味乳液与食用油如棉花仔油、芝麻油、椰子油或花生油以及酏剂和类似的药物载体混合而成。对于水悬浮液合适的分散剂或悬浮剂包括合成的和天然的树胶如西黄蓍胶、阿拉伯胶、藻酸酯、葡聚糖、羧甲基纤维素钠、甲基纤维素、聚乙烯吡咯烷酮或明胶。
在偏头痛的治疗中,合适的剂量水平是每天约0.01至250mg/kg,较优选地是每天约0.05至100mg/kg,以及特别是每天约0.05至5mg/kg。对于偏头痛患者施用化合物每天可1至4次。
本发明的1,2,4—三唑化合物或其盐可以通过式Ra—CO2H的羧酸活性衍生物与或式Ⅲ或式Ⅳ化合物反应的方法制备:其中根据上述式Ⅰ的定义,Ra、Rb、Rc中的一个为式A1基团,另一个为式A2基团,第三个为式—E—F基团。
酸Ra—CO2H的合适的活性衍生物包括酯,例如C1—4烷基酯;硫酯,例如吡啶基硫酯;酸酐,例如(Ra—CO)2O;酰卤,例如酰氯;原酸酯;及伯、仲和叔酰胺。
酸Ra—CO2H的较优选的活性衍生物是下式Ⅴ的亚氨醚衍生物:其中RC1—4烷基。
式Ⅲ试剂可以于反应混合物中就地产生。例如反应可以通过用烷基肼,如甲基肼,随后用合适的羧酸如甲酸处理上述式Ⅴ化合物实现。
反应通常任意在溶剂中,如四氢呋喃、二甲基甲酰胺或低纸链烷醇如乙醇、丙醇或异丙醇,于大约20℃至100℃下通过将试剂一起加热约1至6小时进行。
当Ra为式—E—F基团和基团F为如上述定义的结构FC的吲哚部分时,式HO2O—E—F的羧酸活性衍生物可以通过将式Ⅵ化合物: 其中Q表示活性羧酸部分和E如上定义;与式Ⅶ化舍物或其羰基被保护的形式反应:
其中Q表示活性羧酸部分和E如上定义;与式Ⅶ化舍物或其羰基被保护的形式反应: 其中R2如上定义和R11与上述定义的基团R1相同或者表示式—CH2·CHR4D1基团,其中R4如上定义和D1表示容易被置换的基团;随即,如果需要,用常规的方法通过N—烷基化作用引入基团R3制得。
其中R2如上定义和R11与上述定义的基团R1相同或者表示式—CH2·CHR4D1基团,其中R4如上定义和D1表示容易被置换的基团;随即,如果需要,用常规的方法通过N—烷基化作用引入基团R3制得。
式Ⅶ化合物合适的羰基保护形式包括二甲基乙缩醛或缩酮衍生物。
式Ⅶ化合物中容易被置换的基团D1较合适地表示卤素原子,优选地是氯。当式Ⅶ化合物中基团R11为式—CH2·CHR4D1基团时,取代基D1在普通反应条件下就地被置换以获得其中R1表示式—CH2·CHR4·NH2基团的式Ⅰ最终产物。如果需要,末端氨基随后可用本技术领域已知的方法进一步处理,以给出其中R1表示所需式—CH2·HR4·NR6R7基团的式Ⅰ化合物。
化合物Ⅵ和Ⅶ的反应可以一步进行(费歇尔吲哚合成法)或通过低温下初始非环化步骤给出式Ⅷ化合物:其中Q、E、R2和R11如上定义;随即用合适的试剂如多磷酸酯成环给式Q—E—F化合物。
式Ⅵ的肼可以由相应的式Ⅸ苯胺通过重氮化作用,随即通过还原作用制得:其中Q和E如上定义。重氮化作用典型地是用亚硝酸钠/浓HCl进行,并用例如氯化锡(Ⅱ)/浓HCl或亚硫酸钠/浓HCl将获得的重氮基产物就地还原。
式Ⅸ的苯胺可以通过相应的式Ⅹ硝基化合物还原制得: 其中Q和E如上定义;典型地是通过催化氢化或用氯化锡(Ⅱ)还原。
其中Q和E如上定义;典型地是通过催化氢化或用氯化锡(Ⅱ)还原。
当式Ⅹ的硝基化合物商业不上能得到时,它们可以通过本领域技术人员公知的常规方法合成。
当Ra为式—E—F基团和基团F为上述定义的结构FB吲唑残基时,式HO2C—E—F的羧酸活性衍生物可以通过式Ⅺ化合物的环合作用制得,其中Q、E和R1如上定义,D2表示容易被置换的基团;随即,如果需要,用常规方法通过N—烷基化作用引入基团R3。
化合物Ⅺ的环化作用通常是合适的有机溶剂中,在升高的温度下,例如在间二甲苯和2,6—二甲基吡啶的混合物中于140℃左右温度下完成。
式Ⅺ化合物中容易被置换的基团D2较合适地表示C1—4烷酰氧基,较优选地是乙酰氧基。当所需式Ⅺ化合物中D2表示乙酰氧基时,此化合物通常可通过用羟胺盐酸化物处理式Ⅻ的羰基化合物或其被保护的衍生物制得,其中R1、E和Q如上定义;有利地是在吡啶中于溶剂的回流温度下处理,随即用乙酸酐,有利地是在催化量的4—二甲氨基吡啶存在下,在二氯甲烷中室温下通过乙酰化制得。
式Ⅻ中间体的N—甲酰基保护的衍生物,通常可以通过式ⅩⅢ吲哚衍生物的臭氧分解, 其中R1、E和Q如上定义;随即通过还原处理,有利地是用二甲亚砜处理制得。
其中R1、E和Q如上定义;随即通过还原处理,有利地是用二甲亚砜处理制得。
式ⅩⅢ吲哚衍生物可通过与随后实施例中描述的相似的方法,或通过本领域公知的方法制得。
另一方法是,根据本发明的三唑化合物可以通过包括使式ⅩⅣ化合物:
其中A1、E和F如上定义,Hal表示卤素和V3、W3、X3、Y3和Z3中的两个,其中一个与基团Hal相连,表示碳,余下的表示氮;与能够提供阴离子—A2的试剂(其中A2如前定义)反应的方法制得。
能够提供阴离子—A2的试剂包括格氏试剂A2MgHal(其中Hal=卤素);有机铜酸盐试剂如LiA2 2Cu;有机锂试剂A2Li;或通过相邻活性基团如酯或能烯醇化的酮官能团稳定阴离子的化合物。这种情况下,此步完成后相邻的酯或酮官脂团可以被保留,也可以被除去。例如,酯残基可以被水解和脱去羧基。
根据本发明的1,2,3—三唑化合物可以通过包括式R3—C≡C—Rb的炔与式Rc—N3的叠氮化物环合加成的方法制得,其中R3、Rb和Rc如上定义。
环合加成反应通常可以在合适的溶剂如四氢呋喃中,较理想地是通过在反应釜中加热8小时来完成。
根据本发明的四唑化合物可以通过包括式N≡C—Rd的腈化物与式Re—N3的叠氮化物环合加成的方法制得,其中Rd和Re中的一个表示如上述定义的式A1基团,而另一个是如上述定义的式—E—F基团。
环合加成反应通常是在升高的温度下,例如在150℃左右的温度下,在合适的溶剂如N—甲基吡咯烷—2—酮中,有利地是在三乙胺盐酸化物存在下,通过一起加热反应物进行。由环合加成反应获得的产物通常是分别与上述定义的结构IL和IM相一致的四唑环位置1和2上被A1基团取代的异构体混合物。这些异构体通常可以用常规方法例如色谱法分离。
在另一个方法中,本发明四唑化合物可以通过包括在碱如三惭胺存在下,将式Re—L化合物与式ⅩⅤ四唑衍生物反应的方法制得:其中Rd和Re中的一个表示如上定义的式A1基团,另一个是上述定义的式—E—F基团以及L表示合适的离去基团。
离去基团L较合适地表示卤素,例如溴或碘,或磺酸酯衍生物如甲苯磺酸酯或甲磺酸酯。
反应通常是在合适的有机溶剂中,例如乙腈,于室温下进行。
式ⅩⅤ四唑衍生物可以通过式N≡C—Rd腈化物与叠氮化钠,较有利地是在上述腈化物N≡C—Rd与叠氮化物Re—N3的反应条件下环合加成;随即用无机酸如盐酸酸化制得。
在另一方法中,根据本发明的其中基团F是上述定义的结构FC的吲哚残基的化合物可以通过包括将式ⅩⅥ化合物: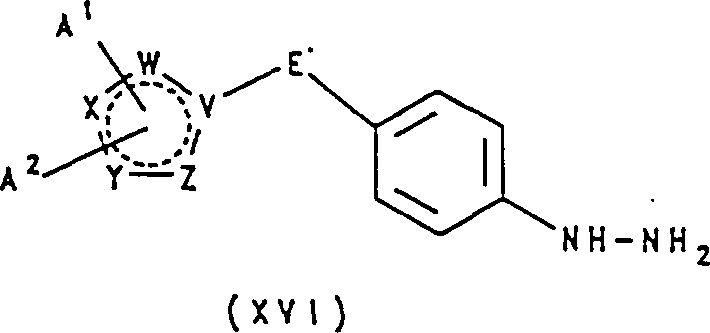 其中V、W、X、Y、Z、A1、A2和E如上定义;与上述定义的式Ⅶ化合物,或其羰基被保护的形式如二甲基乙缩醛或缩酮反应制得;随即,如果需要,用常规方法通过N—烷基化作用引入基团R3。
其中V、W、X、Y、Z、A1、A2和E如上定义;与上述定义的式Ⅶ化合物,或其羰基被保护的形式如二甲基乙缩醛或缩酮反应制得;随即,如果需要,用常规方法通过N—烷基化作用引入基团R3。
如化合物Ⅵ和Ⅶ的反应,化合物ⅩⅥ和Ⅶ的反应可以一步进行(费歇尔吲哚合成法)或通过低温下初始非环化步骤给出式ⅩⅦ化合物: 其中V、W、X、Y、Z、A1、A2、E、R2和R11如上定义;随即用合适的试剂如多磷酸酯成环进行。
其中V、W、X、Y、Z、A1、A2、E、R2和R11如上定义;随即用合适的试剂如多磷酸酯成环进行。
式ⅩⅦ的肼可以由相应的式ⅩⅧ的苯胺化合物: 其中V、W、X、Y、Z、A1、A2和E如上定义;用上述式Ⅸ化合物中描述的相似的方法所得。
其中V、W、X、Y、Z、A1、A2和E如上定义;用上述式Ⅸ化合物中描述的相似的方法所得。
式ⅩⅧ的苯胺可以由相应的式ⅪⅩ的硝基化合物:其中V、W、Y、Z、A1、A2和E如上定义;用上述式Ⅹ化合物中描述的相似的方法制得。
式ⅩⅨ的硝基化合物可以通过本领域技术人员显而易见的方法制得。例如,当Ⅴ表示氮原子时,相应的式ⅩⅨ化合物可以通过式ⅩⅩ化合物的阴离子与式ⅩⅪ化合物反应制得:其中W、X、Y、Z、A1、A2和E如上定义,和D3表示容易被置换的基团。
当化合物ⅩⅩ是三唑或四唑衍生物时,其阴离子可以通过在碱如三乙胺中进行反应生成。当化合物ⅩⅩ是咪唑衍生物时,若用N,N—二甲基甲酰胺作溶剂在氢化钠中进行反应,则很容易生成其阴离子。当式ⅩⅩ化合物的盐在商业上可以得到时,例如1,2,4—三唑的钠盐,较有利地是,利用在N,N—二甲基甲酰胺溶液中的化合物的盐代替式ⅩⅩ化合物本身,在此情况下在反应混合物中无需再加入另外的碱。
式ⅩⅪ化合物中容易被置换的基团D3合适地是卤素原子,较优选地是溴;除非当基团D3是与芳环直接相连时,即当E表示单键时,此时D3较优选地是氟。
当上述式ⅩⅪ的硝基化合物在商业上不能得到时,它们可以通过下面实施例中描述的相似的方法,或通过本领域公知的方法制得。
对于1,2,4—三唑衍生物一种可选择的方法是,式ⅩⅨ的硝基化合物可以由上述基团Q有合适的限定定义的式Ⅹ化合物,用例如与上述涉及式Ⅲ和Ⅳ化合物的相似的方法制得。例如,当式Ⅹ化合物中WQ表示活性羧酸酯基时,式ⅩⅨ化合物可以通过与式A2—C(=NNHA1)NH2或A2—C(=NNH2)NHA1的反应制得。
还有另一种方法是,本发明其中基团F是上述定义的结构FB的吲唑残基的化合物,可以通过包括式ⅩⅫ化合物成环反应制得: 其中V、W、X、Y、Z、A1、A2、E、R1和D2如上定义;如果需要,随即用常规方法通过N—烷基化作用引入基团R3。
其中V、W、X、Y、Z、A1、A2、E、R1和D2如上定义;如果需要,随即用常规方法通过N—烷基化作用引入基团R3。
如式Ⅺ化合物的成环反应,式ⅩⅫ化合物的成环反应较便利地是在合适的有机溶剂中,于升高的温度下,例如在间二甲苯和2、6—二甲基吡啶混合物中,于140℃左右的温度下完成。
式ⅩⅫ化合物例如可以由相应的式ⅩⅩⅢ化合物或其被保护的衍生物: V、W、X、Y、Z、A1、A2、E和R1如上定义;其依次由相应的式ⅩⅩⅣ化合物制得:
V、W、X、Y、Z、A1、A2、E和R1如上定义;其依次由相应的式ⅩⅩⅣ化合物制得: 其中V、W、X、Y、Z、A1、A2、E和R1如上定义;用涉及上述式Ⅻ和ⅩⅢ化合物的相似的方法制得。例如,当式ⅩⅢ化合物中Q表示活性羧酸酯基时,式ⅩⅩⅣ的1,2,4—三唑衍生物可以通过与式A2—C(=NNHA1)NH2或A2—C(=NNH2)NHA1的反应制得。
其中V、W、X、Y、Z、A1、A2、E和R1如上定义;用涉及上述式Ⅻ和ⅩⅢ化合物的相似的方法制得。例如,当式ⅩⅢ化合物中Q表示活性羧酸酯基时,式ⅩⅩⅣ的1,2,4—三唑衍生物可以通过与式A2—C(=NNHA1)NH2或A2—C(=NNH2)NHA1的反应制得。
还有另一个方法是,本发明其中基团F是苯并呋喃或苯并噻吩残基的化合物,可以通过包括式ⅩⅩⅤ化合物成环反应: 其中V、W、X、Y、Z、A1、A2、E和R2如上定义,B3表示氧或硫,和R21与上述定义的基团R1相同或表示下述的前体基团,如果需要,随即用常规方法将基团R21转变成所需的基团R1。
其中V、W、X、Y、Z、A1、A2、E和R2如上定义,B3表示氧或硫,和R21与上述定义的基团R1相同或表示下述的前体基团,如果需要,随即用常规方法将基团R21转变成所需的基团R1。
成环反应通常通过用多磷酸或多磷酸酯,有利地是在升高的温度下进行。
式ⅩⅩⅤ化合物可以通过式ⅩⅩⅥ化舍物与式ⅩⅩⅦ化舍物反应所得: 其中V、W、X、Y、Z、A1、A2、E、Ba、R2和R21如上定义,和Hal表示卤素。
其中V、W、X、Y、Z、A1、A2、E、Ba、R2和R21如上定义,和Hal表示卤素。
反应较方便地是在碱如氢氧化钠的存在下进行。
式ⅩⅩⅥ的羟基和巯基衍生物可以通过各种本领域技术人员显而易见的方法制得,其中的一个方法是,将上述定义的式ⅩⅩ化合物的阴离子与式ⅩⅩⅧ化合物反应: 其中D3、E和B3如上定义;以获得式ⅩⅩⅥ的中间体,其中Ⅴ是氮。
其中D3、E和B3如上定义;以获得式ⅩⅩⅥ的中间体,其中Ⅴ是氮。
当式ⅩⅩⅦ和ⅩⅩⅧ化合物在商业上不能得到时,它们可以通过本领域公知的常规方法制得。
当然,当适合时可以由上述任一种方法最初获得的任一种式Ⅰ化合物,用本领域公知的方法依次制得另一种式Ⅰ化合物。的确当合适时,其中Rd是式—E—F基团的上述式ⅩⅤ倾倒中物就是其中A1是氢和A2表示非成键电子对的式Ⅰ化合物本身。特别是,通过常规方法如烷基化作用,例如用烷基碘处理(如碘甲烷)典型地是在碱性条件下(如二甲基甲酰胺中的氢化钠或乙腈中的三乙胺)可以将最初获得的其中R3是氢的式Ⅰ化合物转变成其中R3表示C1—6烷基、C2—6链烯基或C2—6链炔基的式Ⅰ化合物。相似地,例如通过常规的N—烷基化或N—芳基化的方法,例如在还原剂氰氢硼化钠存在下用合适的醛处理,可以将最初获得的其中R1表示式—CH2·CHR4·NH2基团的式Ⅰ化合物转变成其中R1表示式—CH2·CHR4·NR6R7基团的式沦合物,其中R6和R7除氢外如上述定义。
当根据本发明化合物的上述制备方法给出的是立体异构体混合物时,此异构体可以用常规方法如制备色谱法分离。
新化合物可以以外消旋体的形式制得,或单一的对映体可以或者通过对映体定向合成法或者通过拆分制得。例如,因常规方法,如与光学活性的酸如(—)—二—对—甲苯酰—d—酒石酸和/或(+)—二—对—甲苯酰—1—酒石酸成盐以形成非对映异构体对,随即通过分级结晶和游离碱的再生过程,将新颖的化合物拆分成其对映体组分。还可以通过形成非对映异构体的酯或酰胺,随即通过色谱分离和除去手性助剂拆分新化合物。
在上述任一合成步骤中,有必要和/或需要注意保护任何分子中的敏感的或活性基团,此可通过常规保护基团的方法实现,如在ProtectiveGroups in Organic Chemistry,编辑J.F.W.McOmie,Plenum出版,1973;和T.W.Greene,Protective Groups in Organic Synthesis,John Wiley &Sons,1981中描述的方法。保护基可以用本领域已知的方法在随后的步骤中方便地除去。
另外,所需产物中的某种官能团可以以前体基团贯穿于反应过程中,并且在整个合成过程最后一步时由这些前体基团再生。例如在所需式Ⅰ化合物中R1表示式—(CH2)2NH2基团时,此基团可以通过用例如甲硼烷/四氢呋喃还原作用由氰基前体—CH2CN产生。氰基前体可以依次以甲基—CH3贯穿于反应过程中,其可以通过在有光源存在下,用N—溴代琥珀酰亚妥和过氧化苯甲酰处理,随即通过将获得的溴中间体在二甲亚砜中与氰化钠反应转变成—CH2CN。
下列实施例说明了本发明化合物的制备方法。
试验化合物与5—HT1状受体结合的能力可用J.Neurosci.,1987,7,894中描述的方法在由猪尾制得的膜上测定。用2nM5—羟色胺肌酸酐硫酸盐,5—[1,2—3H(N)]作放射配体测定结合情况。在检测中分别用氰基心得乐(Cyanopindolol)(10nM)和mesulergine(100nM)大致找到5—HT1A和5—HT1C结合位置。在每种情况下,需要置换50%特异性结合(IC50)的下列实施例化合物的浓度低于1μM。
作为5—HT1状受体激动剂的试验化合物的活性可以用Arch.pharm.,1990,342,111中描述的方法,根据它们介导新西兰白兔隐静脉收缩的能力测定。由5—HT(1μm)应答百分率对激动剂浓度作图,计算—log10EC50(pEC50)的值作为激动剂的效力。在此检测中每种情况下发现,下列实施例化合物所具有的pEC50不小于5.0。
实施例1
2—[5—(2—苄基四唑—5—基甲基)—1H—吲哚—3—基]乙胺,草酸盐1.4—肼基苄基氰,盐酸化物
将NaNO2(80g,1.16mol)怕溶液滴加到冷却(—10℃)了的,搅拌着的4—氨基苄基氰(153.5g,1.16mol)的浓HCl(1500ml)悬浮液中,其滴加速度应不使温度升至—10℃以上,将该混合物于—10℃下搅拌0.25小时,然后于真空下将其迅速过滤到加料漏头中。在约0.25小时期间,将上述溶液分份迅速加入到SnCl2·2H2O(1.05kg,4.46mol)在浓Hcl(800ml)搅拌着的混合物中,并使温度维持在—5℃以下,将混合物温热至室温并搅拌0.25小时,然后于真空下滤出沙状的有色沉淀,用乙醚洗涤(5×500ml)。所得固体于真空烘箱(80℃)用P2O5干燥16小时,得标题化合物(213g,100%)。m.p.181—183℃;1H NMR(360MHz,D2O)δ3.90(2H,s,CH2);7.06(2H,d,J=8.7Hz,Ar—H);7.40(2H,d,J=8.7Hz,Ar—H)。
2.2—(5—氰甲基—1H—吲哚—3—基)乙胺,盐酸化物
将4—氯丁醛的二甲基缩醛(37.07g,0.24mol)加入到搅拌着的4—肼基苄基氰盐酸化物(47.0g,0.26mol)在EtOH/H2O(5∶1,21)中的溶液中,并回流4.5小时。于真空下把反应混合物蒸发至干,加入MeOH(150ml),再将混合物于0℃下放置10小时。于真空下过滤所产生的淡黄色沉淀,用Et2O/MeOH(5∶1,2 × 100ml)洗并干燥。该产品不须进一步提纯可用于下步反应(24.1g,40%)。m.p.239—241℃;Rf 0.4 inCH2Cl2/EtOH/NH3(40∶8∶1);1H NMR(360MHz,D2O)3.18(2H,t,J=7.1Hz,CH2);3.36(2H,t,J=7.1Hz,CH2);4.02(2H,s,CH2);7.22(1H,dd,J=1.5和8.4Hz,Ar—H);7.36(1H,s,Ar—H);7.56(1H,d,J=8,4Hz,Ar—H);7.66(1H,s,Ar—H)。3.2—(5—四唑—5—基甲基—1H—吲哚—3—基)乙胺
于140℃下将2—(5—氰甲基—1H—吲哚—3—基)乙基胺盐酸化物(2.5g,10.6mmol),三乙胺盐酸化物(2.2g,16.0mmol)和叠氮化钠(2.1g,32.3mmol)的1—甲基吡咯烷—2—酮(30ml)溶液加热8小时。加5N盐酸(3ml)真空下蒸馏除掉溶剂,残留物在硅胶上层析,用EtOH/Et2O/H2O/NH3(20∶30∶8∶1)洗脱得标题的四唑化合物(1.76g,69%)。δ(360MHz,CD3OD)3.06(2H,t,J=7.2Hz,CH2);3.19(2H,t,J=7.2Hz,CH2);4.29(2H,s,CH2);7.07(1 H,d,J=8.4Hz,Ar—H);7.13(1H,s,Ar—H);7.29(1H,d,J=8.4Hz,Ar—H);7.44(1H,s,Ar—H)。4.N—叔丁氧羰基—2—(5—四唑—5—基甲基—1H—吲哚—3—基)乙胺
往搅拌着的2—(5—四唑—5—基甲基—1H—吲哚—3—基)乙胺(1.76g,7.27mmol)的无水CH2Cl2(40ml)悬液中加入三乙胺(1.5g,14.9mmol)和(BOC)2O(1.9g,7.3mmol),并将混合物搅拌16小时。真空下除掉溶剂,残留物在硅胶上层析,用EtOH/Et2O/H2O/NH3(20∶60∶8∶1)洗脱,得标题产品(1.6g,64%)。δ(360MHz,CD3OD)1.41(9H,s,3 of CH3);2.87(2H,t,J=7.4Hz,CH2);3.30(2H,t,J=7.4Hz,CH2);4.32(2H,s,CH2);6.99(1H,d,J=8.3Hz,Ar—H);7.04(1H,s,Ar—H);7.26(1H,d,J=8.3Hz,Ar—H);7.49(1H,s,Ar—H)。5.N—叔丁氧羰基—2—[5—(2—苄基四唑—5—基甲基)—1H—吲哚—3—基]乙胺和N—叔丁氧羰基—2—[5—(1—苄基四唑—5—基甲基)—1H—吲哚—3—基]乙胺
把苄基溴(0.31g,1.8mmol)加入到步骤4制得的四唑(0.62g,1.8mmol)和三乙胺(0.37g,3.6mmol)的无水乙腈(20ml)溶液中。将混合物于室温下搅2小时。于70℃下加热1小时,再于室温下搅16小时。真空下除去溶剂,残留物用硅胶层析,用CH2Cl2/MeOH(97∶3)洗脱,得到分开的两种苄基四唑。经鉴定极性较小的异构体是2—苄基四唑(0.17g,22.4%),δ(360MHz,CDCl3)1.43(9H,s,3 of CH3);2.90(2H,t,J=6.8Hz,CH2);3.41(2H,brt,CH2);4.32(2H,s,CH2);5.70(2H,s,CH2Ph);7.00(1H,s,Ar—H);7.15(1H,d,J=8.4H2,Ar—H);7.28(1H,d,J=8,4Hz,Ar—H);7.34(5H,s,Ar—H);7.50(1H,s,Ar—H);7.96(1H,brs,NH)。极性较高的组份是1—苄基四唑(0.2g,26.4%)。δ(360MHz,CDCl3)1.43(9H,s,3 of CH3);2.88(2H,t,J=7.0Hz,CH2);3.40(1H,br,t,CH2);4.26(2H,s,CH2);5.29(2H,s,CH2—Ph);6.92(1H,d,J=8.4Hz,Ar—H);7.01—7.05(3H,m,Ar—H);7.27—7.30(5H,m,Ar—H);8.08(1H,br s,NH)。6.2—[5—2—苄基四唑—5—基甲基)—1H—吲哚—3—基]乙胺草酸盐
把三氟乙酸(1.5ml)加入到从步骤5分离出的极性较小的组分(0.17g,0.4mmol)的CH2Cl2(5ml)溶液中并于室温下搅拌1小时。真空下除去溶剂,残留物用硅胶层析,用CH2Cl2/EtOH/NH3(40∶8∶1)洗脱得标题的四唑。再制成草酸盐(65mg),m.p.169—171℃;(测定值:C,59.23;H,5.07;N,19.60。C19H20N6·1.05(C2H2O4)计算值C,59.36;H,5.22;N,19.68%);δ(360MHz,D2O)3.09(2H,t,J=6.9Hz,CH2);3.29(2H,t,J=6.9Hz,CH2);4.30(2H,s,CH2);5.77(2H,s,CH2);7.11(1H,dd,J=1.6和8.4Hz,Ar—H);7.28(1H,s,Ar—H);7.32—7.34和7.39—7.41(5H,m,Ar—H);7.43(1H,d,J=8.41Hz,Ar—H);7.51(1H,s,Ar—H)。
实施例22—[5—(1—苄基四唑—5—基甲基)—1H—吲哚—3—基]乙胺,盐酸化物,半水合物
使用实施例1步骤6所述方法,从实施例1步骤5分离出的极性较高组分制备。制成其盐酸化物半水合盐。m.p.210—213℃;(测定值:C,60.39;H,5.88;N,22.14。C19H20N6·HCl·0.5H2O计算值C,60.39;H,5.87;N,22.24%);δ(250MHz,D2O)3.02(2H,t,J=6.8Hz,CH2);3.19(2H,t,J=6.8Hz,CH2);4.44(2H,s,CH2);5.60(2H,s,CH2);6.95—7.02(3H,m,Ar—H);7.16—7.25(4H,m,Ar—H);7.28(1H,s,Ar—H);7.40(1H,d,J=8.4Hz,Ar—H)。
实施例3N,N—二甲基—2—[5—(2—甲基四唑—5—基甲基)—1H—吲哚—3—基]乙胺,草酸盐1.N—叔丁氧羰基—2—[5—(2—甲基四唑—5—基甲基)1H—吲哚—3—基]乙胺和N—叔丁氧羰基—2—[5—(1—甲基四唑—5—基甲基)1H—吲哚—3—基]乙胺
把甲基碘化物(0.44g,3.1mmol)加入到搅拌着的实施例1步骤4制备的四唑(0.95g,2.78mmol)和三乙胺(0.56g,5.5mmol)的无水乙腈(15ml)溶液中,10小时后,再加1当量的甲基碘并搅拌16小时。真空除掉溶剂。残留物用硅胶层析,用CH2Cl2/MeOH(97∶3)洗脱,得标题的1—和2—甲基四唑的混合物(0.6g,61%)。δ(360MHz,CDCl3);1.43(9H,m,ofCH3);2.89—2.92(2H,m,CH2);3.38—3.48(2H,m,CH2);3.83(2H,s,CH2);4.28和4.40(共3H,s,CH3);6.98和7.17(共1H,d,J=8.4Hz,Ar—H);7.02和7.06(共1H,s,Ar—H);7.30和7.31(共1H,d,J=8.4Hz,Ar—H);7.43和7.54(共1H,s,Ar—H);8.00和8.10(共1H,br s,NH)。2.2—[5—(2—甲基四唑—5—基甲基)—1H—吲哚—3—基]乙胺和2—[5—(1—甲基四唑—5—基甲基)—1H—吲哚—3—基]乙胺
用实施例1步骤6所述方法,从上述甲基四唑起始制备。粗产品用硅胶层析,用CH2Cl2/EtOH/NH3(40∶8∶1)洗脱;得到两个分开的组分。经鉴定极性较小的组分是2—甲基四唑(0.1g,24%)。δ(360MHz,CDCl3)1.38(9H,s,3 of CH3);2.88(2H,t,J=6.6Hz,CH2);3.00(2H,t,J=6.6Hz,CH2);4.28(3H,s,CH3);4.33(2H,s,CH2);7.00(1H,d,J=8.4Hz,Ar—H);7.06(1H,d,J=2.1Hz,Ar—H);7.17(1H,d,J=8.4Hz,Ar—H);7.56(1H,s,Ar—H);8.04(1H,br s,NH)。
极性较高的组分(0.13g,31%)经鉴定是1—甲基四唑。δ(360MHz,CDCl3)1.38(9H,s,3 ofCH3);2.86(2H,t,J=6.6Hz,CH2);3.00(2H,t,J=6.6Hz,CH2);3.82(3H,s,CH3);4.40(2H,s,CH2);6.98(1H,dd,J=1.6和8.3Hz,Ar—H);7.06(1H,d,J=1.6Hz,Ar—H);7.31(1H,d,J=8.3Hz,Ar—H);7.41(1H,s,Ar—H);8.18(1H,br s,NH)。3.N,N—二甲基—2—[5—(2—甲基四唑—5—基甲基)—1H—吲哚—3—基)乙胺,草酸盐
将甲醛(80mg 30%的溶液)的甲醇(15ml)溶液加入到搅拌着的2—[5—(2—甲基四唑—5—基甲基)—1H—吲哚—3—基]乙胺(0.1g,0.4mmol),NaCNBH3(60mg)和冰醋酸(0.12g)的甲醇(15ml)溶液中,搅拌2小时,用K2CO3溶液碱化,再于真空下除去甲醇。用乙酸乙酯萃取,再除去溶剂,如此得到的粗产品用硅胶层析,用CH2Cl2/EtOH/Et2O/NH3(40∶8∶1)洗脱,得到所需的N,N—二甲基色铵(96mg,87%)再制成草酸盐.m.p.185—187℃(MeOH/Et2O):(测定值:C,54.42;H,5.74;N,22.53。C15H20N6·C2H2O4计算值C,54.54;H,5.92;N,22.45%);δ(360MHz,D2O)2.91(6H,s,2 of CH3);3.21(2H,t,J=7.4Hz,CH2);3.47(2H,t,J=7.4Hz,CH2);4.30(3H,s,CH3);4.34(2H,s,CH2);7.17(1H,dd,J=1.5和8.4Hz,Ar—H);7.33(1H,s,Ar—H);7.48(1H,d,J=8.4Hz,Ar—H);7.59(1H,s,Ar—H)。
实施例4N,N—二甲基—2—[5—(1—甲基四唑—5—基甲基)—1H—吲哚—3—基]乙胺,草酸盐
采用实施例3步骤3所述方法,从2—[5—(1—甲基四唑—5—基甲基)—1H—引哚—3—基]乙胺(0.125g,0.49mmol)起始制备。将所得到的游离碱(0.11g,80%)转化成草酸盐,再用MeOH/Et2O重结晶。m.p.176—177℃;(测定值:C,54.21;H,5.84;N,22.36。C15H20N6·C2H2O4计算值C,54.54;H,5.92;N,22.45%);δ(360MHz,D2O);2.91(6H,s,2 of CH3);3.21(2H,t,J=7.4Hz,CH2);3.40(2H,t,J=7.4Hz,CH2);4.00(3H,s,CH3);4.43(2H,s,CH2);7.13(1H,dd,J=1.5和8.4Hz,Ar—H);7.35(1H,s,Ar—H);7.50(1H,d,J=8.4Hz,Ar—H);7.54(1H,s,Ar—H)。
实施例5N,N—二甲基—2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺,草酸盐半水合物1.1—(4—硝基苯基)甲基—1,2,4—三唑
将4—硝基苄基溴(21.6g,0.1mol)加入到快速搅拌着的1,2,4—三唑钠盐(9.1g,0.1mol)的无水DMF(100ml)悬浮液中,再把混合物于室温下搅拌16小时。加乙酸乙酯(400ml),再加水(250ml)使分层,用水(3×250ml)洗有机相,用MgSO4干燥,蒸发。残留物用硅胶层析,用乙酸乙酯洗脱,得标题产品(10.6g,52%)。m.p.98—100℃。δ(360MHz,CDCl3)5.47(2H,s,CH2)7.40(2H,d,J=9Hz,Ar—H);8.02(1H,s,Ar—H);8.18(1H,s,Ar—H);8.32(2H,d,J=9Hz,Ar—H)。2.1—(4—氨基苯基)甲基—1,2,4—三唑,盐酸化物
1—(4—硝基苯基)甲基—1,2,4—三唑(10.0g,49mmol)的乙醇(50ml)溶液,乙酸乙酯(50ml),5NHCl(10ml)和水(10ml)在40p.s.i.下用10%Pd/C(1.0g)在帕尔加压器(Parr apparatus)中氢化,直到压力增至188p.s.i.(约10分钟),用hyflo硅藻土过滤,除去催化剂,并于真空下除去溶剂。残留物同乙醇(X2)共沸,得标题的胺的盐酸化物(10.6g,100%)。δ(360MHz,D2O)5.53(2H,s,CH2),7.37—7.48(4H,m,Ar—H),8.12(1H,s,Ar—H);8.66(1H,s,Ar—H)。3.1—(4—肼基苯基)甲基—1,2,4—三唑
将亚硝酸钠(3.28g,48mmol)的水(20ml)溶液加到上述胺的盐酸化物(10.0g,48mmol)的浓HCl(40ml)溶液中,其加入速度应使反应温度不超过—10℃。加完后将溶液于0℃下搅0.25小时,然后滴加到快速搅拌着的SnCl2·2H2O(40g)的浓HCl(40ml)溶液中。将溶液温热至室温,用20%NaOH水溶液碱化。用乙酸乙酯萃取(3×250ml),将合并在一起的萃取液干燥(MgSO4),用hyflo硅藻土过滤。将溶液蒸发至干,得所需的肼(5.0g,56%)。m.p.109—112℃,δ(360MHz,D6—SMSO)3.93(2H,br,s,NH2),5.20(2H,s,CH2),6.73(2H,d,J=8Hz,Ar—H),7.08(2H,d,J=Hz,Ar—H),7.92(1H,s,Ar—H),8.57(1H,s,Ar—H)。4.2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺
将4—氯丁醛的二甲基缩醛(3.22g,21.1mmol)加入到搅拌着的上述肼(5.0g,26.4mmol)的乙醇/水(5∶1,180ml)和5NHCl(4.5ml)的溶液中,并将该溶液回流4小时。真空下除去溶剂,残留物用硅胶层析,用CH2Cl2/EtOH/NH3(30∶8∶1)洗脱,得所需的色胺(2.4g,38%),δ(360MHz,CDCl3)2.90(2H,t,J=7Hz,CH2)2.99(2H,t,J=7Hz,CH2),5.43(2H,s,CH2),7.10(1H,s,Ar—H),7.11(1H,d,J=8Hz,Ar—H),7.39(1H,d,J=8Hz,Ar—H),7.57(1H,s,Ar—H),7.94(1H,s,Ar—H),8.08(1H,s,Ar—H)。5.N,N—二甲基—2—[5—(1、2、4—三唑—1—基—甲基)—1H—吲哚—3—基]乙胺草酸盐半水合物
将甲醛(37% W/W溶液,0.19g)的甲醇(10ml)溶液加入到前述色胺(0.36g,1.5mmol),NaCNBH3(0.225g,3.6mmol)和冰醋酸(0.45g)于甲醇(10ml)中的混合物中。将混合物于室温下搅拌2小时,然后加入饱和K2CO3(50ml)并蒸发掉甲醇。残留物用乙酸乙酯萃取(3×100ml),合并在一起的萃取液用食盐水(100ml)洗涤,干燥(K2CO3)并蒸发。粗产品用硅胶层析,用CH2Cl2/EtOH/NH3(20∶8∶1)洗脱,得标题化合物的游离碱(0.21g,52%),再制备成其草酸盐半水合物。m.p.165—167℃(MeOH/Et2O);(测定值:C,55.53;H,6.04;N,18.59。C15H19N5·C2H2O4·0.55H2O计算值C,55.29;H,6.03;N,18.96%);m/e 269(M+);δ(360MHz,D2O)2.91(6H,s,Nme2),3.22(2H,t,J=7Hz,CH2),3.47(2H,t,J=7Hz,CH2),5.52(2H,s,CH2),7.21(1H,dd,J=1.6)和(8.4Hz,Ar—H),7.36(1H,s,Ar—H),7.52(1H,d,J=8.4Hz,Ar—H),7.65(1H,s,Ar—H),8.06(1H,s,Ar—H),8.56(1H,s,Ar—H)。
实施例6N,N—二甲基—2—[5—(1,2,3,4—四唑—2—基甲基)—1H—吲哚—3—基]乙胺草酸盐1.1—(4—硝基苯基)甲基—1,2,3,4—四唑和2—(4—硝基苯基)甲基—1,2,3,4—四唑
把4—硝基苄基溴(15.42g,71.3mmol)加到搅拌着的1H—四唑(5.0g,71.3mol)和三乙胺(7.9g,78.0mmol)的乙腈(100ml)溶液中,于室温搅拌混合物16小时,真空除掉溶剂,残余物用硅胶层析,用二氯甲烷洗脱,得两种异构体。极性较小的是2—烷基化产物(2.47g,17%)。δ(360MHz,CDCl3)5.92(2H,s,CH2),7.35(2H,d,J=8.7Hz,Ar—H),8.25(2H,d,J=8.7Hz,Ar—H),8.56(1H,s,Ar—H)。极性较高的主要异构体是1—烷基化产物(11g,75%)。δ(360MHz,CDCl3)5.73(2H,s,CH2),7.46(2H,d,J=8.7Hz,Ar—H),8.27(2H,d,J=8.7Hz,Ar—H),8.64(1H,s,Ar—H)。2.2—(4—氨基苯基)甲基—1,2,3,4—四唑,盐酸化物
2—(4—硝基苯基)甲基—1,2,3,4—四唑(2.47g,12.1mmol)如实施例5步骤2所述方法加氢。所得产品是其盐酸化物(2.55g,100%)。δ(250MHz,D2O)5.86(2H,s,CH2),7.40(2H,d,J=8.7Hz,Ar—H),7.36(2H,d,J=8.7Hz,Ar—H),8.74(1H,s,Ar—H)。3.N,N—二甲基—2—[5—(1,2,3,4—四唑—2—基甲基)—1H—吲哚—3—基]乙胺,草酸盐
前述的胺用实施例5步骤3—5所述一般方法转化成标题化合物。制成草酸盐并用MeOH/Et2O重结晶。m.p.198—199℃;(测定值:C,53.38;H,5.55;N,22.63。C14H18N6·C2H2O4·0.2(EtOH)计算值C,53.30;H,5.78;N,22.74%);δ(360MHz,D2O)2.91(6H,s,Nme2),3.23(2H,t,J=7.4Hz,CH2),3.48(2H,t,J=7.4Hz,CH2),6.01(2H,s,CH2),7.30(1H,dd,J=1.6和8.4Hz,Ar—H),7.37(1H,s,Ar—H),7.53(1H,d,J=8.4Hz,Ar—H),7.76(1H,s,Ar—H),8.74(1H,s,Ar—H)。
实施例7N,N—二甲基—2—[5—(1,2,3,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺,琥珀酸盐1—(4—硝基苯基)甲基—1,2,3,4—四唑用实施例5所述方法转化成标题化合物。制成琥珀酸盐。m.p.55—56℃(异丙醇);(测定值C,57.08;H,6.14;N,23.34。C14H18N6·0.75(C4H6O4)计算值C,56.89;H,6.32;N,23.42%);δ(360MHz,D2O)2.93(6H,s,Nme2),3.23(2H,t,J=7.5Hz,CH2),3.48(2H,t,J=7.5Hz,CH2),5.81(2H,s,CH2),7.28(1H,dd,J=1.7和8.4Hz,Ar—H),7.39(1H,s,Ar—H),7.56(1H,d,J=8.4Hz,Ar—H),7.75(1H,s,Ar—H),9.20(1H,s,Ar—H)。
实施例8N,N—二甲基—2—[5—(1—甲基—1,2,4—三唑—5—基甲基)—1H—吲哚—3—基]乙胺,二草酸盐1.3—[2—(二甲氨基)乙基]—1H—吲哚—5—甲基碳亚胺乙酯,盐酸化物
N,N—二甲基—2—(5—氰甲基—1H—吲哚—3—基)乙胺(5g,22.01mmol)的乙醇溶液用HCl气体饱和并将溶液于室温下搅拌16小时。真空下除去溶剂,得标题的产品(6g,92%)。δ(360MHz,D6—DMSO)1.29(3H,t,J=7.0Hz,CH2);2.83(6H,s,Nme2),3.13(2H,t,J=7.5Hz,CH2),3.31(2H,m,CH2),4.04(2H,s,CH2),4.42(2H,q,J=7.0Hz,CH2),7.08(1H,dd,J=1.5和8.4Hz,Ar—H),7.27(1H,s,Ar—H),7.37(1H,d,J=8.4Hz,Ar—H),7.48(1H,br s,NH),7.71(1H,s,Ar—H)。2.N,N—二甲基—2—[5—(1—甲基—1,2,4—三唑—5—基甲基)—1H—吲哚—3—基]乙胺,二草酸盐将前述亚胺酯(3g,10.15mmol),甲基肼(0.8ml)和三乙胺(3.54ml)在乙醇(30ml)中的混合物于室温下搅拌3小时,真空下除去溶剂并把所得产品溶在甲醇(98%,3.3ml)中,将溶液于室温下搅拌0.5小时,再回流2小时,溶液冷至室温后倾入K2CO3(75ml)水溶液中,用乙酸惭酯萃取(4×200ml),将合并在一起的萃取液干燥(MgSO4),蒸发·残留物用硅胶层析,用CH2Cl2/EtOH/NH3(40∶8∶1)洗脱,得两个组分。经鉴定极性较小的异构体是标题的1—甲基—1,2,4—三唑(360mg)。制成双草酸盐,m.p.135—137℃;(测定值:C,50.91;H,5.38;N,13.86。C16H21N5·0.25(乙醇)计算值:C,50.70;H,5.47;N,14.08%);δ(360MHz,D2O)2.91(6H,s,NMeCH2);3.23(2H,t,J=7.3Hz,CH2),3.48(2H,t,J=7.3Hz,CH2),3.95(3H,s,Me),4.48(2H,s,CH2),7.13(1H,dd,J=1.5和8.4Hz,Ar—H),7.37(1H,s,Ar—H),7.53(1H,d,J=8.4Hz,Ar—H),7.57(1H,s,Ar—H),8.32(1H,s,Ar—H)。
实施例9N,N—二甲基—2—[5—(1—甲基—1,2,4—三唑—3—基甲基)—1H—吲哚—3—基]乙胺,三盐酸化物
实施例8步骤2得到的极性较高的异构体被鉴定是标题的三唑(180mg)。制成三盐酸化物,m.p.<40℃(吸湿);测定值:C,49.80,H,6.56;N,16.69。C16H21N5·3HCl·0.35(Et2O)计算值:C,49.91;H,6.62;N,16.73%);δ(360MHz,D2O)2.91(6H,s,Nme2);3.23(2H,t,J=7.4Hz,CH2),3.49(2H,t,J=7.4Hz,CH2),3.95(3H,s,Me),4.27(2H,s,CH2),7.17(1H,dd,J=1.5和8.5Hz,Ar—H),7.34(1H,s,Ar—H),7.50(1H,d,J=8.5Hz,Ar—H),7.60(1H,s,Ar—H),8.88(1H,s,Ar—H)。
实施例10N,N—二甲基—2—[5—(1,2,3—三唑—1—基甲基)—1H—吲哚—3—基]乙胺,草酸盐1.1—(4—硝基苯基)甲基—1,2,3—三唑
将4—硝基苄基溴化物(25.4g,0.12mol)加到—1,2,3—三唑(8.12g,0.12mol)和三乙胺(11.88g,0.12mol)的无水乙腈溶液中,将混合物回流1小时,冷至室温并沉淀。滤出NEt3·HBr,真空下除去溶剂。残留物用硅胶层析,用CH2Cl2(100)—CH2Cl2/MeOH(95.5)洗脱,得到2个产品,极性较高的产品是标题的1—异构体(13g,54%),m.p.114—116℃δ(250MHz,CDCl3)5.72(2H,s,CH2);7.38(2H,d,J=9Hz,Ar—H),7.64(1H,s,Ar—H),7.78(1H,s,Ar—H),8.18(2H,d,J=9Hz,Ar—H)。极性较小的较少的异构体是2—烷基化产品(2.25g,9%),m.p.112—113℃δ(250MHz,CDCl3)5.72(2H,s,CH2);7.40(2H,d,J=9Hz,Ar—H),7.66(2H,s,Ar—H),8.18(2H,d,J=9Hz,Ar—H)。2.N,N—二甲基—2—[5—(1,2,3—三唑—1—基甲基)—1H—吲哚—3—基]乙胺,草酸盐
采用实施例5所述的一般方法,将1—(4—硝基苯基)甲基—1,2,3—三唑转化成标题的吲哚。制成草酸盐。m.p.210—212℃(测定值:C,55.88,H,5.75;N,18.69。C16H21N5·1.1(C2H2O4)0.15H2O计算值:C,55.67;H,5.84;N,18.87%);δ(360MHz,D2O)2.90(6H,s,NMe2);3.22(2H,t,J=7.4Hz,CH2),3.46(2H,t,J=7.4Hz,CH2),5.72(2H,s,CH2),7.24(1H,dd,J=1.6和8.4Hz,Ar—H),7.36(1H,s,Ar—H),7.52(1H,d,J=8.4Hz,Ar—H),7.66(1H,s,Ar—H),7.79(1H,s,Ar—H),8.00(1H,d,J=1Hz,Ar—H)。
实施例113—(2—氨乙基)—5—(2—甲基—四唑—5—基)苯并[b]噻吩,草酸盐步骤14—溴苯硫基丙酮
往搅拌着的4—溴代苯硫酚(5.09g,26.9mmol)于NaOH(1.08g,26.9mmol)和水(32ml)中的溶液中加入氯丙酮(2.17ml,27.3mmol),将混合物于氮气氛中搅拌45分钟,然后用乙醚萃取,用水洗涤,干燥(Na2SO4),真空蒸发,得6.89g(100%)标题化合物,为白色固。δ(CDCl3)2.27(3H,s),3.65(2H,s),7.20(2H,d,J=8.5Hz),7.41(2H,d,J=8.5Hz)。步骤25—溴—3—甲基苯并[b]噻吩
在1小时期间,往温和回流着的多磷酸(4.47g)和氯苯(100ml)的混合物中分次加入4—溴苯硫基丙酮(2.24g,0.14mmol),将混合物于回流下加热8天。冷后将有机相倾出,残留物用水(约100ml)分解,用CH2Cl2(2×75ml)萃取,干燥(MgSO4),和倾泻出的有机相合并,真空蒸发得2.096g棕色油状物,在Kugelrohr仪器中蒸馏得1.83g(88%)标题化合物,为浅黄色液体。bp100—110℃/0.35毫巴。δ(CDCl3)2.41(3H,s),7.10(1H,s),7.43(1H,dd,J=8.5和1.9Hz),7.69(1H,d,J=9.5Hz),7.64(1H,d,J=1.9Hz)。步骤35—氰基—3—甲基苯并[b]噻吩
往氰化铜(Ⅰ)(0.569g,6.35mmol)中加入于N—甲基吡咯烷酮(10ml)中的5—溴—3—甲基苯并[b]噻吩(1.179g,5.19mmol),于180—190℃将该混合物搅拌17小时。然后将其在乙醚(75ml)和氨水(75ml)之间分配,分出醚层,用更多的氨水洗涤(2×50ml),干燥(Na2SO4),真空蒸发,得0.81g灰白的固体。用闪式硅胶层析,用10%乙酸乙酯/石油醚洗脱,得0.76g(85%)标题化合物,是白色固体。δ(CDCl3)2.47(3H,s),7.23(1H,s),7.55(1H,dd,J = 8.3和1.5Hz),7.93(1H,d,J=8.4Hz),8.03(1H,d,J=1.4Hz)。步骤43—甲基—5—(四唑—5—基)—苯并[b]噻吩
于氮气氛下,往5—氰基—3—甲基苯并[b]噻吩(0.194g,1.12mmol)的N—甲基吡咯烷酮(5ml)溶液中加三乙胺盐酸化物(0.231g,1.68mmol),随后加叠氮化钠(0.234g,3.59mmol),混合物用乙醚(4×50ml)萃取。合并在一起的醚萃取液用MgSO4干燥,真空蒸发得0.78g白色固体,用闪式硅胶层析,用CH2Cl2/MeOH/NH3(aq)(40∶8∶1—30∶8∶1)洗脱,得0.246g(100%)标题产品,为白色固体。δ(DMSO)2.46(3H,s),7.41(1H,s),7.98(1H,d,J=8.4Hz),8.03(1H,dd,J=8.4和1.5Hz),8.36(1H,d,J=0.9Hz).m/z(CI—,NH3)215(M—H)—,160。步骤53—甲基—5—(2—甲基四唑—5—基)—苯并[b]噻吩和3—甲基—5—(1—甲基四唑—5—基)苯并[b]噻吩
往3—甲基—5—(四唑—5—基)苯并[b]噻吩(0.241g,1.12mmol)于乙腈(5ml)的混合物中加三乙胺(0.28ml,2.01mmol),然后加碘甲烷(0.486ml,7.81mmol),随后再加DMF(3ml),直到得到一清澈的溶液,将该溶液于氮气氛下搅拌过夜,再于真空下蒸发,在水(50ml)和乙醚(25ml)之间分配残留物。分出水层,用更多的乙醚(2×25ml)萃取,将合并在一起的醚层干燥(MgSO4),真空蒸发得0.241g黄色固体,在闪式硅胶上层析,用25—40%乙酸乙酯/石油醚洗脱,得0.168g(65%)标题化合物的2—异构体(是白色固体)和0.063g(24%)标题化合物的1—异构体(也是白色固体)。2—异构体:δ(CDCl3)2.52(3H,s)4.42(3H,s),7.14(1H,s),7.94(1H,d,J=8.4Hz),8.10(1H,dd,J=8.4和1.5Hz),8.51(1H,s).m/z(Cl+,NH3)231(M+H)+1—异构体δ(CDCl3)2.50(3H,s),4.22(3H,s),4.22(3H,s),7.23(1H,s),7.64(1H,dd,J=8.3和1.5Hz),8.03(1H,d,J=8.4Hz),8.12(1H,d,J=1.6Hz).m/z(Cl+,NH3)231(M+H)+,202,172。步骤63—氰甲基—5—(2—甲基四唑—5—基)苯并[b]噻吩
在两盏台灯(2×60W)照射下,往回流着的3—甲基—5—(2—甲基四唑—5—基)苯并[b]噻吩(0.162g,0.703mmol)和过氧化苯甲酰(10.6mg)于四氯化碳(10ml)中的混合物中分成小份加入N—溴代琥珀酸亚胺(0.126g,0.707mmol),加完后把混合物于回流情况下再加热90分钟,过滤,真空下蒸发滤液得油/固混合物。用闪式硅胶层析,用二氯甲烷洗脱得0.161g粗3—溴甲基—5—(2—甲基四唑—5—基)苯并[b]噻吩,是无色油状物。
将于DMSO(0.3ml)中的粗3—溴甲基—5—(2—甲基—四唑—5—基)苯并[b]噻吩(0.145g)加到于DMSO(0.2ml)中的氰化钠(29.9mg,061mmol)混合物中,于100℃搅拌混合物2小时。冷后,将其倾入水(10ml)中,滤出棕色固体,用水洗,于真空枪中干燥,残留物为73.5mg,滤液用二氯甲烷萃取(3×30ml),干燥合并在一起的萃取液(Na2SO4),真空蒸发得44.7mg残留物。与前述残留物合并,用闪式硅胶层析,用20—50%乙酸乙酯/石油醚洗脱,得61.5mg(38%)标题化合物,为白色固体。δ(CDCl3)3.99(2H,s),4.43(3H,s),7.59(1H,s),8.00(1H,d,J=8.5Hz),8.19(1H,dd,J=8.5和1.5Hz),8.47(1H,s)。步骤73—(2—氨乙基)—5—(2—甲基—四唑—5—基)苯并[b]噻吩,草酸盐
在氮气氛下,往3—氰甲基—5—(2—甲基—四唑—5—基)苯并[b]噻吩(0.434g,1.70mmol)的THF(16ml)溶液中滴加于THF(5.10ml,5.10mmol)中的1.0M甲硼烷—四氢呋喃配合物,于回流下加热混合物6小时。在冰浴中冷后将混合物用2NHCl(22ml)停止反应,然后再加热回流1小时。真空下除去THF,用50%的氢氧化钠溶液(4ml)碱化残留物,再用二氨甲烷萃取(3×75ml)。干燥合并在一起的萃取液(K2CO3),真空蒸发得0.45g残留物。用闪式硅胶层析,用CH2Cl2/MeOH/NH3(aq)(60∶8∶1)洗脱,得0.383(87%)标题化合物,是白色固体。用于甲醇/乙醚中的草酸制备草酸盐,得标题产品的草酸盐,是白色固体。m.p.204—209℃测定值:C,47.75;H,4.28;N,19.28%。计算值:C12H13N5S·1.1C2H2O4:C,47.59;H,4.28;N,19.54%。δ(DMSO)3.17—3.21(4H,m),4.46(3H,s),7.72(1H,s),8.06(1H,dd,J=8.4和1.4Hz),8.52(1H,s)m/z(Cl+,NH3)260(M+H)+,230。
实施例123—(2—氨乙基)—5—(1—甲基四唑—5—基)苯并[b]噻吩,草酸盐步骤13—氰甲基—5—(1—甲基四唑—5—基)苯并[b]噻吩
按照实施例11步骤6的方法,使0.666g(2.89mmol)3—甲基—5—(1—甲基四唑—5—基)苯并[b]噻吩和0.515g(2.89mmol)N—溴代琥珀酰亚胺和38.1mg过氧化苯甲酰在30ml四氯化碳中反应。真空蒸发反应混合物,用闪式硅胶层析,用0—3%甲醇/二氯甲烷洗脱,得0.532g粗的3—溴—5—(1—甲基四唑—5—基)苯并[b]噻吩。
于100℃使粗的3—溴—5—(1—甲基四唑—5—基)苯并[b]噻吩(0.504g)和97.7mg(1.99mmol)氰化钠在1.5ml DMSO中反应2小时冷后,把反应混合物倾入水(25ml)中,用二氯甲烷(6×50ml)萃取。干燥合并在一起的萃取液(Na2SO4),真空蒸发得0.37g残留物。用闪式硅胶层析,用30—60%乙酸乙酯/石油醚洗脱,得28.0mg(4%)标题化合物。δ(CDCl3)4.00(2H,s),4.23(3H,s),7.73(1H,dd),8.08(1H,d),8.15(1H,d)。步骤23—(2—氨乙基)—5—(1—甲基四唑—5—基)苯并[b]噻吩,草酸盐
按照实施例11,步骤7的方法,使2ml THF中的26.1mg(0.102mmol)3—氰乙基—5—(1—甲基四唑—5—基)苯并[b]噻吩和THF中的0.36ml(0.36mmol)的1.0M甲硼烷—四氢呋喃配合物反应。用闪式硅胶层析,用CH2Cl2/MeOH/NH3(aq)(60∶8∶1)洗脱,得17.7mg(67%)标题化合物,是无色油状物。用甲醇/乙醚中的草酸制成草酸盐,得到标题产品的草酸盐,为白色固体。m.p.206—212℃。测定值:C,47.55;H,4.05;N,19.65%。计算值:C12H13N5S·1.1C2H2O4: C,47.59;H,4.28;N,19.54%。δ(D2O)3.32—3.35(2H,m),3.40—3.44(2H,m),4.22(3H,s),7.64(1H,s),7.73(1H,d,J = 8.4Hz),8.19(1H,s),8.22(1H,d,8.5Hz)。
实施例133—[2—(N,N—二甲基氨基)乙基]—5—(2—甲基四唑—5—基)苯并[b]噻吩,草酸盐在约5分钟期间内,在冰浴冷却下,往3—(2—氨乙基)—5—(2—甲基四唑—5—基)苯并[b]噻吩(0.372g,1.43mmol)和氰基氢硼化钠(0.135g,2.15mmol)于甲醇(3ml)和乙酸(0.247ml,4.30mmol)中的混合物中滴加在甲醇(3ml)中的38%W/V甲醛溶液(0.453ml,5.74mmol),将混合物于室温下搅3小时。此后加饱和酸钾溶液(30ml),用乙酸乙酯萃取混合(3×5ml)。真空蒸发合并的萃取液,得0.53g残留物,用闪式硅胶层析,用20—30%甲醇/二氯甲烷洗脱,得0.335g(81%)标题产品,是无色油状物。用甲醇/乙醚中的草酸制备其草酸盐,得标题化合物的草酸盐,是白色固体。m.p.214—218℃,测定值:C,50.58;H,4.80;N,18.28%。计算值C14H17N5S.C2H2O4:C2H2O4:C,50.92;H,5.07;N,18.56%。δ(DMSO)2.84(6H,s),3.30—3.42(4H,m),4.46(3H,s),7.69(1H,s),8.06(1H,dd,J=8.4和1.4Hz),8.20(1H,d,J=8.4Hz),8.56(1H,s).m/z(Cl+,NH3)288(M+H)+。
实施例14N,N—二甲基—2—[5—(2—甲基咪唑—1—基甲基)—1H—吲哚—3—基]乙胺,三草酸盐1.1—(4—硝基苯基)甲基—2—甲基咪唑
把氢化钠(2.45g,61.0mmol,油中的60%分散体)加到2—甲基咪唑(5.0g,60.9mmol)的DMF(100ml)溶液中。将混合物在室温下搅拌0.25小时,然后加4—硝基苄基溴(13.2g,61.0mmol),并于110℃下加热2小时,随后于室温下搅拌16小时。加水(200ml)和乙酸乙酯(500ml),分离水层,用乙酸乙酯萃取(2×500ml)。合并的萃取液用水洗(3×250ml),干燥(MgSO4),蒸发。粗产品用硅胶层析,用CH2Cl2/MeOH(4%)洗脱,得标题的产品(1.58g,10.5%)。δ(360MHz,CDCl3)2.34(3H,s,Me);5.16(2H,s,CH2);6.67(1H,d,J=1.3Hz,Ar—H);7.03(1H,d,J=1.3Hz,Ar—H);7.19(2H,d,J=9.5Hz,Ar—H);8.22(2H,d,J=9.5Hz,Ar—H)。2.N,N—二甲基—2—[5—(2—甲基咪唑—1—基甲基)—1H—吲哚—3—基)乙胺,三草酸盐
采用实施例5所述一般方法,从上述4—硝基苄基咪唑起始制备。制成三草酸盐。m.p.160—163℃(MeOH/Et2O);(测定值:C,50.57;H,5.25;N,10.60。C17H22N4·2.8(C2H2O4)计算值:C,50.79;H,5.21;N,10.48%);m/e 282(M+);δ(360MHz,D2O)2.65(3H,s,Me);2.92(6H,s,Nme2);3.25(2H,t,J=7.3Hz,CH2);3.50(2H,t,J=7.3Hz,CH2);5.42(2H,s,CH2);7.18(1H,d,J=8.4Hz,Ar—H);7.31—7.40(2H,m,Ar—H);7.40(1H,s,Ar—H);7.56(1H,d,J=8.4Hz,Ar—H);7.66(1H,s,Ar—H)。
实施例15N,N—二甲基—2—[5—咪唑—1—基甲基—1H—吲哚—3—基]乙胺,二草酸盐
采用实施例5所述方法,从咪唑和4—硝基苄基溴起始制备,制成二草酸盐。165—166℃(MeOH/Et2O);(测定值:C,53.30;H,5.34;N,12.18。C16H20N4·2.05(C2H2O4)计算值:C,53.30;H,5.36;N,12.37%);δ(360MHz,D2O)2.92(6H,s,Nme2);3.24(2H,t,J=7.7Hz,CH2);3.48(2H,t,J=7.7Hz,CH2);5.50(2H,s,CH2);7.27(1H,dd,J=1.5和8.4Hz,Ar—H);7.37(1H,s,Ar—H);7.45(1H,s,Ar—H);7.49(1H,s,Ar—H);7.56(1H,d,J=8.4Hz,Ar—H);7.75(1H,s,Ar—H);8.78(1H,s,Ar—H)。
实施例16N,N—二甲基—2—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]乙胺倍半草酸盐1.1—(4—硝基苯基)—2—甲基咪唑
把氢化钠(4.87g,122.0mmol,在油中60%的分散体)加入到2—甲基咪唑(10g,122.0mmol)的DMF(100ml)溶液中,于室温搅拌0.25小时。往反应混合物中加1—氟—4—硝基苯(17.18g,122.0mmol),于室温下搅16小时,加水(150ml)和乙酸乙酯(250ml)分出水层,用乙酸乙酯萃取(3×150ml),用水洗合并的萃取液(3×150ml),干燥(Na2SO4),蒸发得所需产品(11.5g,47%)。δ(360MHz,CDCl3)2.24(3H,s,Me);7.06(1H,d,J=1.5Hz,Ar—H);7.10(1H,d,J=1.5Hz,Ar—H);7.50(2H,d,J=9.5z,Ar—H);8.38(2H,d,J=9.5Hz,Ar—H)。2.N,N—二甲基—2—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]乙胺倍半草酸盐
采用实施例5所述方法,从前述4—硝基苯基咪唑起始制备,再制成倍半草酸盐。m.p.185—186℃(iPA/MeOH);(测定值:C,56.17;H,5.99;N,13.46。C16H20N4·1.55(C2H2O4)·0.1EtOH计算值:C,56.19;H,5.79;N,13.58%);δ(360MHz,D2O)2.55(3H,s,Me);2.93(6H,s,Nme2);3.26(2H,t,J=7.4Hz,CH2);3.51(2H,t,J=7.4Hz,CH2);7.30(1H,dd,J=2.0和8.7Hz,Ar—H);7.48(1H,d,J=2.1Hz,Ar—H);7.51—7.53(2H,m,Ar—H);7.70(1H,d,J=8.7Hz,Ar—H);7.79(1H,d,J=2.0Hz,Ar—H)。
实施例17N,N—二甲基—2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺,琥珀酸盐,方法B
把1—(4—肼基苯基)甲基—1,2,4—三唑的双盐酸化物(2g,7.6mmol,实施例5,步骤3)和4—N,N—二甲基氨基丁醛的二甲基缩醛(1.8g,11.2mmol)在4%硫酸水溶液(70ml)中加热回流2小时。反应混合物冷至室温后,加乙酸乙酯(200ml),水相用K2CO3碱化。分出水相,再用乙酸乙酯萃取(2×150ml)。干燥后并在有机相(Na2SO4)并蒸发,残留物用硅胶层析,用CH2Cl2/EtOH/NH3(30∶8∶1)洗脱,得标题的三唑(610mg,30%),将琥珀酸(0.27g,2.3mmol)的甲醇(3ml)溶液加到上述三唑(0.61g,2.3mmol)的甲醇(5ml)溶液中以制备琥珀酸盐。真空下除去溶剂,所得产物用异丙醇重结晶,m.p.118—120℃;(测定值:C,58.76;H,6.27;N,17.79。C15H19N3·C4H6O4计算值:C,58.90;H,6.50;N,18.08%)。
实施例18N,N—二甲基—2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺,苯甲酸盐
往其游离碱的乙醇/乙醚(1∶4)溶液中加苯甲酸的乙醚溶液可制得N,N—二甲基—2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺的苯甲酸盐。沉出的盐再用乙醇重结晶。m.p.178—180℃;(测定值:C,67.28;H,6.55;N,17.66。C15H19N3·C6H5CO2H计算值:C,67.50;H,6.44;N,17.89%);1HNMR(360MHz,D2O)δ 2.92(6H,s,Nme2);3.22(2H,t,J=7.3Hz,CH2);3.46(2H,t,J=7.3Hz,CH2);5.52(2H,s,CH2);7.22(1H,dd,J=1.6和8.4Hz,Ar—H);7.36(1H,s,Ar—H);7.44—7.58(4H,m,Ar—H);7.65(1H,s,Ar—H);7.87—7.91(2H,m,Ar—H);8.06(1H,s,Ar—H);8.54(1H,s,Ar—H)。
实施例19N,N—二甲基—2—[5—(2—乙基四唑—5—基甲基)—1H—吲哚—3—基]乙胺,草酸盐
采用实施例3所述方法,用乙基碘来制备。再制成草酸盐。m.p.140—142℃:(测定值:C,55.71;H,6.26;N,21.35。C16H22N6·C2H2O4计算值:C,55.66;H,6.23;N,21.64%);1HNMR(360MHz,D2O)δ1.54(3H,t,J=7.4Hz,CH3);2.91(6H,s,Nme2);3.21(2H,t,J=7.4Hz,CH2);3.47(2H,t,J=7.4Hz,CH2);4.34(2H,s,CH2);4.64(2H,q,J=7.4Hz,CH2CH3);7.17(1H,dd,J=1.5和8.4Hz,Ar—H);7.33(1H,s,Ar—H);7.48(1H,d,J=8.4Hz,Ar—H);7.59(1H,s,Ar—H)。
实施例20N,N—二甲基—2—[5—(1—乙基四唑—5—基甲基)—1H—吲哚—3—基]乙胺,草酸盐
采用实施例4所述方法,用乙基碘制备。再制成草酸盐,m.p.179℃,(Me/OH/Et2O);(测定值:C,55.59;H,6.23;N,21.49。C16H22N6.C2H2O4计算值C,55.66;H,6.23;N,21.64%);1HNMR(360MHz,D2O)δ1.32(3H,t,J=7.4Hz,CH3);2.90(6H,s,Nme2);3.21(2H,t,J=7.4Hz,CH2);3.46(2H,t,J=7.4Hz,CH2);4.38(2H,q,J=7.4Hz,CH2);4.47(2H,s,CH2);7.14(1H,dd,J=1.5和8.4Hz,Ar—H);7.35(1H,s,Ar—H);7.50(1H,d,J=8.4Hz,Ar—H);7.53(1H,s,Ar—H)。
实施例21N,N—二甲基—2—[5—(1,2,4—三唑—1—基)—1H—吲哚—3—基]乙胺,二草酸盐
依照实施例16所述,由1,2,4—三唑钠衍生物和1—氟—4—硝基苯制备。制成二草酸盐。m.p.210℃(MeOH/Et2O);(测定值:C,50.11;H,4.78;N,16.35。C14H17N5·1.9(C2H2O4)计算值C,50.14;H,4.92;N,16.43%);1HNMR(360MHz,D2O)δ 2.92(6H,s,Nme2);3.25(2H,t,J=7.4Hz,CH2);3.50(2H,t,J=7.4Hz,CH2);7.44(1H,s,Ar—H);7.47(1H,dd,J=2.0和8.7Hz,Ar—H);7.63(1H,d,J=8.7Hz,Ar—H);7.88(1H,d,J=2.0Hz,Ar—H);8.36(1H,s,Ar—H);9.05(1H,s,Ar—H)。
实施例224—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]—N—甲基哌啶,二草酸盐倍半水合物
N—甲基—4—(甲酰基甲基)哌啶(0.25g,1.8mmol)和4—(2—甲基咪唑基)苯肼盐酸化物(0.48g,2.1mmol)的4%H2SO4(25ml)溶液于回流下加热16小时。将混合物冷至室温,用K2CO3溶液碱化,再用CH2Cl2(3×75ml)萃取。干燥合并的萃取液(Na2SO4),蒸发,残留物硅胶层析提纯,用CH2Cl2/EtOH/NH3(60∶8∶1)洗脱,得标题化合物(0.12g),再制备二草酸盐倍半水合物。m.p.65—70℃(吸湿);(测定值:C,52.97;H,5.51;N,11.07。C16H22N4·2(C2H2O4)·1.5H2O计算值:C,52.69;H,5.83;N,11.17%);1HNMR(360MHz,D2O)δ1.96—2.08(2H,m,CH2);2.31—2.40(2H,m,CH2);2.56(3H,s,CH3);2.95(3H,s,CH3);3.20—3.27(3H,m,CH和CH2);3.64—3.68(2H,m,CH2);7.28(1H,dd,J=2和8.7Hz,Ar—H);7.53(1H,d,J=2Hz,Ar—H);7.69(1H,d,J=8.7Hz,Ar—H);7.81(1H,d,J=2Hz,Ar—H)。
实施例234—[5—(1,2,4—三咪—1—基甲基)—1H—吲哚—3—基]—N—甲基哌啶,草酸盐
N—甲基—4—(甲酰基甲基)哌啶(0.1g,0.71mmol)和4—(1,2,4—三唑基甲基)苯肼双盐酸化物(0.185g,0.71mmol)的4%H2SO4溶液于回流下加热2小时。将混合物冷至室温,用K2CO3溶液碱化,再用CH2Cl2(2×100ml)萃取。粗产品用硅胶层析,用CH2Cl2/EtOH/NH3洗脱(40∶8∶1),得标题化合物(60mg),制成草酸盐。m.p.219—220℃,(测定值:C,58.61;H,6.03;N,17.94。C17H21N5·1.02(C2H2O4)计算值:C,58.96;H,6.38;N,17.56%);1HNMR(360MHz,D2O)δ 1.88—2.02(2H,m,CH2);2.20—2.34(2H,m,CH2);2.92(3H,s,CH3);3.10—3.24(3H,m,CH和CH2);3.60—3.64(2H,m,CH2);5.51(2H,s,CH2);7.21(1H,dd,J=1.5和8.4Hz,Ar—H);7.26(1H,s,Ar—H);7.51(1H,d,J=8.4Hz,Ar—H);7.69(1H,s,Ar—H);8.05(1H,s,Ar—H);8.55(1H,s,Ar—H)。
实施例241H—4—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]哌啶,二草酸盐二水合物1.4—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]—N—苄基哌啶
用实施例22所述方法,由N—苄基—4—(甲酰基甲基)哌啶制备。1HNMR(360MHz,CDCl3)δ1.8—1.94(2H,m,CH2);1.98—2.06(2H,m,CH2);2.14—2.24(2H,m,CH2);2.33(3H,s,CH3);2.76—2.85(1H,m,CH);3.02—3.08(2H,m,CH2);3.60(2H,s,CH2);7.03—7.10(4H,m,Ar—H);7.26—7.38(5H,m,Ar—H);7.41(1H,d,J=8.5Hz,Ar—H);7.52(1H,d,J=1.8Hz,Ar—H);8.30(1H,br s,NH)。2.1H—4—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]哌啶,二草酸盐二水合物
往甲酸铵(O.32g,5.07mmol)和4—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]—N—苄基哌啶(O.4g,1.08mmol)的甲醇(40ml)溶液中加Pd/C(10%,0.4g),将混合物于60℃搅3小时。通过硅藻土过滤除掉催化剂,真空去除溶剂。残留物放入水(30ml)中,用NH3溶液碱化,用乙酸乙酯(3×100ml)萃取。干燥合并的萃取液(Na2SO4),蒸发。残留物用硅胶层析,用CH2Cl2/MeOH/NH3(30∶8∶1)洗脱,得所需的哌啶(0.2g),制备双草酸盐二水合物。m.p.80℃(吸湿);(测定值:C,50.53;H,5.54;N,10.87。C17H20N4·2(C2H2O4)·2.2H2O计算值:C,50.43;H,5.72;N,11.20%);1HNMR(360MHz,D2O)δ 1.91—2.03(2H,m,CH2);2.30—2.34(2H,m,CH2);2.55(3H,s,CH3);3.19—3.36(3H,m,CH和CH2);3.55—3.62(2H,m,CH2);7.28(1H,dd,J=1.2和8.6Hz,Ar—H);7.44(1H,s,Ar—H);7.47(1H,d,J=2.0Hz,Ar—H);7.52(1H,d,J=2.OHz,Ar—H);7.69(1H,d,J=8.6Hz,Ar—H);7.82(1H,d,J=1.2Hz,Ar—H)。
实施例251H—4—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]哌啶,草酸盐
用实施例23和24所述方法,由N—苄基—4—(甲酰基甲基)哌啶和4—(1,2,4—三唑基甲基)苯肼双盐酸化物制备。制成草酸盐,m.p.272℃;(测定值:C,58.27;H,5.56;N,18.79。C16H19N5·C2H2O4计算值:C,58.21;H,5.70;N,18.86%);1HNMR(360MHz,D2O)δ1.86—1.98(2H,m,CH2);2.24—2.28(2H,m,CH2);3.15—3.36(3H,m,CH和CH2);3.52—3.56(2H,m,CH2);5.51(2H,s,CH2);7.21(1H,dd,J=1.6和8.5Hz,Ar—H);7.27(1H,s,Ar—H);7.52(1H,d,J=8.5Hz,Ar—H);7.70(1H,d,J=1.6Hz,Ar—H);8.09(1H,s,Ar—H);8.60(1H,s,Ar—H)。
实施例261H—3—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]吡咯烷,二草酸盐1.3—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]—N—苄基吡咯烷
依实施例22所述,用N—苄基—3—(甲酰基甲基)吡咯烷和4—(2—甲基咪唑基)苯肼盐酸化物来制备。1HNMR(360MHz,CDCl3)δ1.98—2.06(1H,m,CH2中的CH);2.34(3H,s,CH3);2.34—2.44(2H,m,2个CH2中的CH);2.71(1H,t,J=7.4Hz,CH2中的CH);2.80(1H,t,J=6.9Hz,CH2中的CH);3.05(1H,t,J=8.7Hz,CH2中的CH)3.61—3.73(1H,m,CH);3.72(2H,Abq,J=13Hz,CH2);6.95—7.14(4H,m,Ar—H);7.22—7.41(5H,m,Ar—H);7.40(1H,d,J=8.5Hz,Ar—H);7.66(1H,s,Ar—H);8.30(1H,br sNH)。2.1H—3—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]吡咯烷,二草酸盐
依实施例24所述方法,由前述N—苄基吡咯烷起始制备。再制成双草酸盐。m.p.210—213℃(甲醇/乙醚);(测定值:C,53.93;H,5.22;N,12.50。C16H18N4·2(C2H2O4)计算值C,53.81;H,4.97;N,12.55%);1HNMR(360MHz,D2O)δ 2.91—2.30(1H,m,CH of CH2);2.55(3H,s,CH3);2.55—2.60(1H,m,CH of CH2);3.35—3.64(3H,m,CH和CH2);3.80—3.90(2H,m,CH2);7.30(1H,dd,J=2和8.6Hz,Ar—H);7.47(1H,d,2Hz,Ar—H);7.50(1H,s,Ar—H);7.53(1H,d,J=2Hz,Ar—H);7.70(1H,d,J=8.6Hz,Ar—H);7.80(1H,d,J=2Hz,Ar—H)。
实施例27N—甲基—3—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]吡咯烷,二草酸盐
往经冷却(0℃)的,搅拌着的1H—3—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]吡咯烷(0.12g,O.45mmol),乙酸(O.136g,2.3mmol)和NaCNBH3(7.1mg,1.1mmol)于甲醇(15ml)中的混合物中滴加甲醛(89mg,38%W/W水溶液,1.1mmol)的甲醇(10ml)溶液。将混合物于0℃搅0.1小时,然后温热至室温,继续搅拌1.5小时。加饱和K2CO3溶液(10ml),真空下除掉溶剂。残留物用乙酸乙酯萃取(3×100ml),干燥合并的萃取液(Na2SO4),蒸发。粗产品用硅胶层析,用CH2Cl2/MeOH/NH3(60∶8∶1)洗脱,得标题产品(O.1g),制成双草酸盐,m.p.191—194℃,(MeOH/Et2O);(测定值:C,54.39;H,5.30;N,11.87。C17H20N4·2(C2H2O4)·0.2H2O计算值:C,54.36;H,5.30;N,12.07%);1H NMR(360MHz,D2O)δ 2.26—2.45(1H,m,CH of CH2);2.55(3H,s,Me);2.62—2.75(1H,m,CH2中的CH);3.02和3.03(共3H,s,Me);3.23—3.45(2H,m,CH2);3.60—3.68,3.77—4.1和4.12—4.15(共3H,各为m,CH和CH2);7.30(1H,d,J=8.9Hz,Ar—H);7.48(1H,d,J=2.2Hz,Ar—H);7.52(1H,s,Ar—H);7.53(1H,d,J=2.2Hz,Ar—H);7.70(1H,d,J=8.9Hz,Ar—H);7.78(1H,s,Ar—H)。
实施例281H—4—[5—咪唑—1—基—1H—吲哚—3—基]哌啶,二草酸盐
使用实施例22和24所述方法,从N—苄基—4—(甲酰基甲基)哌啶和4—(咪唑基)苯肼盐酸化物制备。制成二草酸盐。m.p.155—157℃;(测定值:C,54.32;H,5.50;N,11.66。C16H18N4·2(C2H2O4).0.3(Et2O)计算值C,54.33;H,5.38;N,11.96%);1H NMR(360MHz,D2O)δ 1.90—2.04(2H,m,CH2);2.32(2H,br d,J=13Hz,CH2);3.20—3.32(3H,m,CH和CH2);3.55—3.60(2H,m,CH2);7.41—7.44(2H,m,Ar—H);7.64(1H,s,Ar—H);7.68(1H,d,J=8.7Hz,Ar—H);7.85(1H,s,Ar—H);7.92(1H,d,J=2Hz,Ar—H);9.06(1H,s,Ar—H)。
实施例291H—4—[5—(1,2,3—三唑—1—基)—1H—吲哚—3—基]哌啶,半草酸盐
使用实施例22和24所述方法,由N—苄基—4—(甲酰基甲基)哌啶和4—(1,2,3—三唑基)苯肼盐酸化物制备。制成半草酸盐。m.p.278℃(MeOH/Et2O);(测定值:C,61.84;H,6.10;N,22.21。C15H17N5.0.5(C2H2O4)计算值:C,61.53;H,5.81;N,22.42%);1H NMR δ(360MHz,D6—DMSO)δ 1.66—1.82(2H,m,CH2);1.98—2.06(2H,m,CH2);2.83—2.89(2H,m,CH2);2.98—3.08(1H,m,CH);3.21(2H,br d,J=12.5Hz,CH2);7.28(1H,s,Ar—H);7.51—7.56(2H,m,Ar—H);7.93(1H,s,Ar—H);8.05(1H,s,Ar—H);8.73(1H,s,Ar—H)。
实施例30N—甲基—4—[5—咪唑—1—基—1H—吲哚—3—基]哌啶,倍半草酸盐
依实施例22所述,由N—甲基—4—(甲酰基甲基)哌啶和4—(咪唑基)苯肼盐酸化物制备。制成倍半草酸盐。m.p.217℃,(测定值:C,57.41;H,5.83;N,13.30。C17H20N4·1.5(C2H2O4)·O.14(CH3OH)计算值:C,57.61;H,5.66;N,13.34%);1H NMR(360MHz,D2O)δ 1.94—2.06(2H,m,CH2);2.34—2.38(2H,m,CH2);2.94(3H,s,CH3);3.20—3.27(3H,m,CH和CH2);3.63—3.67(2H,m,CH2);7.40—7.43(2H,m,Ar—H);7.64(1H,s,Ar—H);7.68(1 H,d,J=8.7Hz,Ar—H);7.84(1H,s,Ar—H);7.90(1H,d,J=1.3Hz,Ar—H);9.07(1H,s,Ar—H)。
实施例31N—甲基—4—[5—(1,2,3—三唑—1—基)—1H—吲哚—3—基]哌啶,半草酸盐
依实施例22所述,由N—甲基—4—(甲酰基甲基)哌啶和4—(1,2,3—三唑基)苯肼盐酸化物制备。制成半草酸盐。m.p.215—254℃(MeOH/Et2O);(测定值:C,62.21;H,6.49;N,21.21·C16H19N5·0.5(C2H2O4).0.1H2O计算值:C,62.22;H,6.20;N,21.34%);1H NMR(360MHz,D2O)δ1.69—2.01(2H,m,CH2);2.25—2.31(2H,m,CH2);2.94(3H,s,CH3);3.04—3.20(3H,mm CH和CH2);3.61—3.65(2H,m,CH2);7.32(1H,s,Ar—H);7.44(1H,dd,J=1.9和8.7Hz,Ar—H);7.58(1H,d,J=8.7Hz,Ar—H);7.86(1H,d,J=1.8Hz,Ar—H);7.94(1H,s,Ar—H);8.29(1H,s,Ar—H)。
实施例32N—甲基—3—[5—(1,2,3—三唑—1—基)—1H—吲哚—3—基]吡咯烷,草酸盐
依实施例26和27所述,由N—苄基—3—(甲酰基甲基)吡咯烷和4—(1,2,3—三唑基)苯肼盐酸化物制备。制成草酸盐。m.p.154—156℃(MeOH/Et2O);(测定值:C,57.06;H,5.39;N,19.43。C15H17N5·C2H2O4计算值:C,57.14;H,5.36;N,19.60%);1H NMR(360MHz,D2O)δ 2.23—2.38(1H,m,CH of CH2);2.55—2.69(1H,m,CH of CH2);3.01(3H,s,Me);3.13—3.42和3.55—3.60(共2H,各为M,CH2);3.70—4.09(3H,m,CH和CH2);7.39(1H,d,J=8.7Hz,Ar—H);7.42—7.46(1H,m,Ar—H);7.58(1H,d,J=8.7Hz,Ar—H);7.62(1H,s,Ar—H);7.93(1H,s,Ar—H);8.30(1H,s,Ar—H)。
实施例33N—甲基—3—[5—(2—甲基咪唑—1—基甲基)—1H—吲哚—3—基]吡咯烷,二草酸盐
依实施例26和27所述,由N—苄基—3—(甲酰基甲基)吡咯烷和4—(2—(甲基)咪唑—1—基甲基)苯肼盐酸化物制备。制成二草酸盐。m.p.152—153℃;(测定值:C,55.41;H,5.51;N,11.61。C18H22N4·2(C2H2O4)计算值:C,55.69;H,5.52;N,11.81%);1H NMR(360MHz,D2O)δ 2.22—2.46(1H,m,CH of CH2);2.58—2.76(1H,m,CH of CH2);2.65(3H,s,Me);3.02和3.03(共3H,s,Me);3.21—3.44,3.60—3.67,3.75—3.95和4.09—4.14(共5H,各为m,CH和2 of CH2);5.42(2H,s,CH2);7.17—7.19(1H,m,Ar—H);7.32(2H,s,Ar—H);7.39(1H,d,J=8.4Hz,Ar—H);7.56(1H,d,J=8.4Hz,Ar—H);7.67(1H,s,Ar—H)。
实施例34N—甲基—3—[5—咪唑—1—基—1H—吲哚—3—基]吡咯烷,二草酸盐
用实施例26和27所述方法,由N—苄基—3—(甲酰基甲基)吡咯烷和4—(咪唑基)苯肼盐酸化物制备。制成二草酸盐。m.p.173—175℃(MeOH/Et2O);(测定值:C,53.94;H,5.07;N,12.51。C16H18N4·2(C2H2O4)计算值:C,53.81;H,4.97;N,12.55%);1H NMR(360MHz,D2O)δ 2.26—2.45和2.60—2.78(各为1H,各为m,CH2),3.02和3.30(共3H,各为S,Me),3.23—3.45,3.61—3.66,3.78—3.95和4.11—4.16(共5H,各为m,2个CH2和CH),7.42和7.45(共1H,每s,Ar—H),7.49(1H,d,J=9.2Hz,Ar—H),7.65(1H,s,Ar—H),7.69(1H,d,J=9.2Hz,Ar—H),7.86—7.89(2H,m,Ar—H),9.09(1H,s,Ar—H)。
实施例35N—甲基—3—[5—(1,2,4—三唑—1—基)—1H—吲哚—3—基]吡咯烷,倍半草酸盐,半水合物
依实施例26和27所述,由N—苄基—3—(甲酰基甲基)吡咯烷和4—(1,2,4—三唑基甲基)苯肼二盐酸化物制备。制得倍半草酸盐半水合物。m.p.59—61℃(异丙醇/Et2O);(测定值:C,55.10;H,5.79;N,16.99。C16H19N5·1.3(C2H2O4)·0.4H2O计算值:C,55.08;H,5.57;N,17.27%);1H NMR(360MHz,D2O)δ 2.20—2.42和2.54—2.72(每个1H,每个m,CH2),3.00和3.02(共3H,每个s,Me),3.16—3.42,3.56—3.62,3.72—3.76,3.82—3.94和3.98—4.10(共5H,各为m,2 of CH2和CH),5.52(2H,s,CH2),7.22和7.24(共1H,每个s,Ar—H),7.34(1H,d,J=8.6Hz,Ar—H),7.52(1H,d,J=8.6Hz,Ar—H),7.66(1H,s,Ar—H),8.06(1H,s,Ar—H),8.58(1H,s,Ar—H)。
实施例36N—甲基—3—[5—咪唑—1—基甲基—1H—吲哚—3—基]吡咯烷,草酸盐,半水合物
依实施例26和27所述,由N—苄基—3—(甲酰基甲基)吡咯烷和4—(咪唑—1—基甲基)苯肼盐酸化物制备。制成草酸盐半水合物。m.p.101—104℃(异丙醇/Et2O);(测定值:C,59.51;H,6.35;N,14.54。C17H20N4·C2H2O4·0.6H2O.0.1(iPrOH)计算值:C,59.86;H,6.25;N,14.47%);1H NMR(360MHz,D2O)δ 2.26—2.42(1H,m,CH of CH2),2.60—2.74(1H,m,CH2中的CH),3.03(3H,s,Me),3.16—4.12(5H,br m,2 of CH2和CH),5.45(3H,s,Me),7.27(1H,dd,J=1.6和8.5Hz,Ar—H),7.31(1H,s,Ar—H),7.38—7.40(2H,m,Ar—H),7.58(1H,d,J=8.5Hz,Ar—H),7.70(1H,s,Ar—H),8.39(1H,s,Ar—H)。
实施例37N,N—二甲基—2—[5—(2—氨基咪唑—1—基)—1H—吲哚—3—基]乙胺,二草酸盐
依实施例16所述,由2—氨基咪唑和4—氟硝基苯制备。为防止在重氮化条件下,氨基咪唑和亚硝酸钠反应,在加氢和形成肼之前,应使用Ac2O/AcOH将氨基以乙酰氨基的形式保护起来。4—[2—(甲基羰基氨基)咪唑—1—基]苯肼和N,N—二甲基氨基丁醛的二甲基缩醛之间进行费歇尔反应(Fischer)得到标题产品。制成二草酸盐。m.p.199—200℃(MeOH/Et2O):(测定值:C,50.35;H,5.06;N,15.05。C15H19N5·2.1(C2H2O4)计算值:C,50.31;H,5.10;N,15.28%);1H NMR(360MHz,D2O)δ 2.91(6H,s,N(Me)2),3.27(2H,t,J=7.4Hz,CH2),3.50(2H,t,J=7.4Hz,CH2),6.97(2H,s,Ar—H),7.29(1H,dd,J=1.8和8.7Hz,Ar—H),7.48(1H,s,Ar—H),7.67(1H,d,J=8.7Hz,Ar—H),7.78(1H,d,J=1.8Hz,Ar—H)。
实施例38N,N—二甲基—2—[5—(2—氨基咪唑—1—基甲基)—1H—吲哚—3—基]乙胺,倍半草酸盐1.4—氰基苯肼,盐酸化物
往冷却的(—15℃)搅拌着的4—氨基苄基(50g,423mmol)于浓盐酸(550ml)中的悬浮体中滴加亚硝酸钠(31.5g,457mmol)的水(200ml)溶液,其滴加速度应维持温度低于—10℃。加完后,迅速过滤反应混合物以除去固体。将滤液分次加到冷却的(—20℃)搅拌着的氯化锡(Ⅱ)二水合物(477g,2.1mol)的浓盐酸(370ml)溶液中,其加入速度应维持温度低于—10℃。于—10°—0℃再维持15分钟后,过滤收集白色沉淀,用乙醚洗(4×250ml),干燥后得56g(78%)标题化合物。m.p.235—237℃(乙醇—水1∶1);1H NMR(250MHz,DMSO—d6)δ 10.50(3H,br s,—N+H3),9.10(1H,br s,—NH),7.71(2H,d,J=8.8Hz,Ar—H)7.03(2H,d,J=8.8Hz,Ar—H);m/z(Cl)132(M+—1)。2.2—[5—氰基—1H—吲哚—3—基]乙胺,盐酸化物
往搅拌着的4—氰基苯肼(50g)于乙醇和水(5∶1,21)的混合物中的悬本中加4—氯丁醛的二甲基醛(45g),将所得混合物回流18小时。真空下除掉溶剂,残留物与甲苯共沸,得—棕色固体,用甲醇(150ml)将该粗产品重结晶,得23g(35%)标题化合物,是黄色固体。m.p.270—274℃;1H NMR(250MHz,DMSO—d6)δ 11.60(1H,br s吲哚N—H),8.17(1H,d,J=1.1Hz,Ar—H),7.97(3H,br s,—N+H3),7.54(1H,d,J=8.5Hz,Ar—H),7.46(1H,s Ar—H),7.44(1H,dd,J=8.5和1.1Hz,Ar—H),3.05(4H,br s,—CH2CH2N—);m/z(Cl)184(M+—1)。3.N—叔丁氧羰基—2—[5—氰基—1H—吲哚—3—基]乙胺
使用实施例1(步骤4)所述条件,标题化合物由前述的色胺以58%的产率被制备。为白色固体。m.p.123—134℃(乙烷—乙酸乙酯);1HNMR(250MHz,CDCl3)δ 8.42(1H,br s,吲哚N—H3),7.93(1H,s,Ar—H),7.41(2H,s,Ar—H)7.12(1H,d,J=2.2Hz,Ar—H);4.71(1H,br s,—NH—),3.44(2H,q,J=6.9Hz,—CH2NH—),2.94(2H,t,J=6.9Hz,Ar—CH2—),1.45(9H,s,t—Bu);m/z(CI)286(M++1)。4.N—叔丁氧羰基—2—[5—氨甲基—1H—吲哚—3—基]乙胺
前述步骤产品(11.3g)于绝对乙醇(750ml)和氯仿(22ml)混合物中的溶液在50psi下用氧化铂(Ⅳ)(1g)氢化28小时。过滤除掉催化剂,真空下除去溶剂。残留物经闪式层析(硅胶,二氯甲烷—甲醇—氨(90∶10∶1)得9.5g(82%)标题化合物,为白色固体。m.p.147—149℃,1H NMR(360MHz,CDCl3)δ 8.04(1H,br s,引哚N—H),7.52(1H,s,Ar—H),7.33(1H,d,J=8.4Hz,Ar—H),7.16(1H,d,J=8.4Hz,Ar—H),7.03(1H,s,Ar—H),4.61(1H,br s,—NHBOC),3.96(2H,s,Ar—CH2NH2),3.45(2H,br q,—CH2NHBOC),2.95(2H,t,J=6.8Hz,Ar—CH2),1.43(9H,s,t—Bu);m/z(CI)288(M+—1)。5.N—叔丁氧羰基—2—[5—二甲基氨甲基]—1H—吲哚—3—基]乙胺
使用实施例3(步骤3)所述条件,从前述步骤得到的产品制备标题化合物,产率71%,无色粘稠油状物。1H NMR(250MHz,CDCl3)δ 8.07(1H,br s,吲哚N—H),7.50(1H,s,Ar—H),7.31(1H,d,J=8.3Hz,Ar—H)7.16(1H,d,J=8.3Hz,Ar—H);7.02(1 H,s,Ar—H),4.61(1H,br s,—NH—),3.54(2H,s,Ar—CH2N—),3.45(2H,q,J=6.2Hz,—CH2NH—),2.94(2H,t,J=6.2Hz,Ar—CH2—),2.27(6H,s,—Nme2),1.43(9H,s,t—Bu)。6.N—叔丁氧羰基—2—[5—三甲基铵甲基—1H—吲哚—3—基]乙胺,碘化物
步骤5的产品(2.9g)于无水乙醚(170ml)和碘甲烷(36ml)的混合物中的溶液于室温下在暗处静置16小时。过滤收集白色固体,用乙醚洗,于50℃下在真空中用五氧化二磷干燥,得到4.2g(100%)标题化合物。m.p.199—202℃(分解);1H NMR(360MHz,DMSO—d6)δ 11.09(1H,br s,吲哚N—H),7.69(1H,s,Ar—H),7.44(1H,d,J=8.3Hz,Ar—H),7.26(1H,s,Ar—H),7.19(1H,d,J=8.3Hz,Ar—H),6.89(1H,br t,—NH—),4.57(2H,s,Ar—CH2N—),3.23(2H,q,J=7.6Hz,—CH2NH—),3.01(9H,s,—N+Me3),2.83(2H,t,J=7.6Hz,Ar—CH2—),1.37(9H,s,t—Bu);m/z(FAB)332。(测定值:C,49.30;H,6.55;N,8.79。C19H30IN3O2计算值:C,49.68;H,6.58;N,9.15%)。7.N—叔丁氧羰基—2—[5—(2—硝基咪唑—1—基甲基)—1H—吲哚—3—基]乙胺
于室温下将氢化钠(0.6g,油中60%的分散体)加到搅拌着的2—硝基咪唑(1.61g,14.2mmol)的DMF(65ml)溶液中,0.5小时后加入前述的甲碘化物(3.26g,7.1mmol)的DMF(40ml)溶液并将混合物回流2小时,然后于室温搅拌18小时。水洗后将粗产品进行闪式层析,得标题化合物(2.6g);1H NMR(360MHz,CDCl3)δ1.43(9H,s,t—Bu),2.94(2H,t,J=7.0Hz,CH2),3.40—3.48(2H,m,CH2)5.69(2H,s,CH2),7.01(1H,s,Ar—H),7.09(1H,d,J=8.4Hz,Ar—H),7.10(2H,s,Ar—H),7.37(1H,d,J=8.4Hz,Ar—H),7.54(1H,s,Ar—H),8.12(1H,s,吲哚—NH)。8.2—[5—(2—硝基咪唑—1—基甲基)—1H—吲哚—3—基]乙胺
前述咪唑(2.6g,6.7mmol)在90%HCO2H(150ml)中的溶液于室温搅拌1.25小时加入MeOH停止反应,真空下降掉溶剂。粗产品用硅胶经闪式层析法提纯,用CH2Cl2/EtOH/NH3(30∶8∶1)洗脱,所得产品(0.73g)为黄色油状物。1H NMR(360MHz,d4—MeOH)δ 2.87—2.94(4H,m,2个CH2),5.71(2H,s,CH2),7.05(1H,d,J=8.4Hz,Ar—H),7.11(1H,s,Ar—H),7.12(1H,s,Ar—H),7.35(1H,d,J=8.4Hz,Ar—H),7.39(1H,s,Ar—H),7.55(1H,s,Ar—H)。9.N,N—二甲基—2—[5—(2—硝基咪唑—1—基甲基)—1H—吲哚—3—基]乙胺
使用实施例3(步骤3)所述条件,从前述的色胺制备。1H NMR(250MHz,CDCl3)δ 2.33(6H,s N(Me)2),2.62(2H,t,J=7.4Hz,CH2),2.92(2H,t,J=7.4Hz,CH2),5.68(2H,s,CH2),7.00(1H,d,J=1.0Hz,Ar—H),7.07(1H,dd,J=1.0和8.2Hz,Ar—H),7.09(1H,d,J=2.4Hz,Ar—H),7.10(1H,d,J=2.4Hz,Ar—H),7.35(1H,d,J=8.2Hz,Ar—H),7.53(1H,s,Ar—H),8.19(1H,br s,吲哚—NH)。10.N,N—二甲基—2—[5—(2—氨基咪唑—1—基甲基)—1H—吲哚—3—基]乙胺,倍半草酸盐
标题化合物采用实施例5(步骤2)所述条件从上述步骤9产品制备。制成倍半草酸盐。m.p.211—212℃(MeOH/Et2O);(测定值:C,54.46;H,6.08;N,16.53。C16H21N5·1.5(C2H2O4)·0.06(MeOH)计算值:C,54.46;H,5.81;N,16.66%);1H NMR(360MHz,D2O)δ 2.91(6H,s,N(Me)2),3.25(2H,t,J=7.4Hz,CH2),3.49(2H,t,J=7.4Hz,CH2),5.16(2H,s,CH2),6.77(1H,d,J=2.3Hz,Ar—H),6.83(1H,d,J=2.3Hz,Ar—H),7.19(1H,dd,J=1.5和8.5Hz,Ar—H),7.39(1H,s,Ar—H),7.56(1H,d,J=8.5Hz,Ar—H),7.61(1H,s,Ar—H)。
实施例39N—甲基—2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺,草酸盐1.N—苄基—2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺
往2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺(1.5g,6.2mmol)的EtOH(30ml)溶液中加新蒸出的苯甲醛(0.66g,6.2mmol),将该溶液室温下搅拌21小时,10分钟内室温下分次加入NaBH4(0.24g,6.3mmol),将所得混合物搅0.5小时,然后真空除掉溶剂。将所得残留物放入水(10ml)中,用1NHCl(15ml)酸化。再将混合物用2NNaOH碱化。用EtOAc(4×50ml)萃取。合并有机相,用食盐水(30ml)洗,干燥,浓缩。残留物在硅胶上层析,用CH2Cl2/MeOH(85∶15)洗脱,得标题产品(1.38g,67%),1H NMR(360Mhz,CDCl3)δ 2.94(4H,s,2 of CH2),3.80(2H,s,CH2),5.38(2H,s,CH2),7.04(1H,d,J=2Hz,Ar—H),708(1H,dd,J=1.5和8.4Hz,Ar—H),7.18—7.30(5H,m,Ar—H),7.32(1H,d,J=8.4Hz,Ar—H),7.54(1H,s,Ar—H),7.94(1H,d,J=2Hz,Ar—H),8.17(1H,br s,吲哚—NH)。2.N—苄基—N—甲基—2—[5—(1,2,4—三唑—1—基甲基)1H—吲哚—3—基]乙胺
往搅拌着的前述胺(1.14g,3.4mmol)的无水DMF(45ml)溶液中加K2CO3(0.89g,6.4mmol)和硫酸二甲酯(0.46g,3.7mmol)。将混合物于室温搅3.5小时后加水(90ml),用EtOAc(2×100ml)萃取。用食盐水(40ml)洗合并的有机溶液,干燥,浓缩。残留物用硅胶层析,用CH2Cl2/MeOH(90∶10)洗脱,得所需产品(0.69g)。1H NMR(360MHz,CDCl3)δ 2.34(3H,s,CH3),2.70—2.76(2H,m,CH2),2.94—3.00(2H,m,CH2),3.60(2H,s,CH2),5.38(2H,s,CH2),7.04(1H,d,J=2Hz,Ar—H),7.08(1H,dd,J=2和8.4hz,Ar—H),7.20—7.36(6H,m,Ar—H),7.44(1H,s,Ar—H),7.94(1H,s,Ar—H),7.96(1H,s,Ar—H),8.18(1H,br s,吲哚—NH)。3.N—甲基—2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺,草酸盐
前述苄胺(0.69g,2.0mmol)于乙醇(100ml)和2NHCl(2ml)中的溶液在30psi下用10%Pd/C(0.6g)氢化4小时,用hyflo硅藻土,过滤除去催化剂,真空下除掉溶剂,残留物用硅胶层析,用CH2Cl2/EtOH/NH3(40∶8∶1)洗脱,得标题的N—甲基胺(0.34g,68%),制成草酸盐并用异丙醇重结晶。m.p.149—150℃;(测定值:C,55.42;H,5.72;N,19.55。C14H17N5·C2H2O4.0.15(iPA)计算值C,55.72;H,5.75;N,19.76%);1H NMR(360MHz,D2O)δ 2.44(3H,s,CH3),2.87—2.98(4H,m,2 of CH2),5.41(2H,s,CH2),7.05(1H,s,Ar—H),7.09(1H,dd,J=1.6和8.4Hz,Ar—H),7.31(1H,d,J=8.4Hz,Ar—H),7.57(1H,s,Ar—H),7.96(1H,s,Ar—H),7.99(1H,s,Ar—H)。
实施例40
片剂的制备
分别含1.0,2.0,25.0,26.0,50.0和100.0mg下述化合物的片剂制法如下:N,N—二甲基—2—[5—(2—甲基四唑—5—基甲基)—1H—吲哚—3—基]乙胺,草酸盐;N,N—二甲基—2—[5—(1,2,4—三唑—1—基甲基)—1H—吲哚—3—基]乙胺,苯甲酸盐;N,N—二甲基—2—[5—(1,2,3,4—四唑—1—基甲基)—1H—吲哚—3—基]乙胺,琥珀酸盐;N—甲基—4—[5—咪唑—1—基—1H—吲哚—3—基]哌啶,倍半草酸盐;N—甲基—3—[5—(1,2,3—三唑—1—基)—1H—吲哚—3—基]吡咯烷,草酸盐。
含1—25mg活性化合物剂量的片剂
量(mg)活性化合物 1.0 2.0 25.0微晶纤维素 49.25 48.75 37.25改性食用玉米淀粉 49.25 48.75 37.25硬脂酸镁 0.50 0.50 0.50
含26—100mg活性化合物剂量的片剂
量(mg)活性化合物 26.0 50.0 100.0微晶纤维素 52.0 100.0 200.0改性食用玉米淀粉 2.21 4.25 8.5硬脂酸镁 0.39 0.75 1.5
把所有活性化合物,纤维素和部分玉米淀粉混合,并制成含10%玉米淀粉浆的颗粒,将所得颗粒过筛,干燥,再和其余玉米淀粉及硬脂酸镁掺合。将所得颗粒压成每片含1.0mg,2.0mg,25.0mg,26.0mg,50.0mg和100mg活性化合物的片剂。
Claims (3)
1.式Ⅰ化舍物或其盐或药物前体的制备方法: 其中虚线环表示五元环中任何位置上非相邻的双键;
其中虚线环表示五元环中任何位置上非相邻的双键;
V、W、X、Y和Z中的二个表示氮,余下的表示碳,所述氮原子在五元环的非相邻位置上;
A1表示氢、甲基、乙基、苄基或氨基;
A2表示氢、甲基、乙基、苄基或氨基;
E表示一个键或含1至4个碳原子的直或支链亚烷基链;
F表示下式基团;
U表示C—R2;
B表示氧、硫或N—R3;
R1表示—CH2·CHR4·NR6R7或下式基团其中虚线表示任选的化学键;
R2、R3、R4、R5、R6和R7各自独立地表示氢或C1—6烷基;
该方法包括:
(G)将式ⅩⅥ化合物: 其中V、W、X、Y、Z、A1、A2和E如上定义;与式Ⅶ化合物或其羰基被保护的式Ⅶ化合物反应:
其中V、W、X、Y、Z、A1、A2和E如上定义;与式Ⅶ化合物或其羰基被保护的式Ⅶ化合物反应: 其中R2如上定义且R11与上述定义的基团R1相一致或者表示式—CH2·CHR4D1基团,其中R4如上定义和D1表示容易被置换的基团;如果需要,随即用常规方法通式N—烷基化引入基团R3;或
其中R2如上定义且R11与上述定义的基团R1相一致或者表示式—CH2·CHR4D1基团,其中R4如上定义和D1表示容易被置换的基团;如果需要,随即用常规方法通式N—烷基化引入基团R3;或
(J)使式ⅩⅩⅤ化合物成环:其中V、W、X、Y、Z、A1、A2、E和R2如上定义,Ba表示氧或硫,和R21与上述定义的基团R1相一致或表示前体基团;如果需要,随即将基团R21转变成所需的基团R1;和
(K)随后,如果合适,可用常规方法将起始获得的式Ⅰ化合物转变成另一种式Ⅰ化合物。
2.根据权利要求1的方法,其中制得的化合物是式ⅡC表示的化合物的其盐和药物前体: 其中
其中
Y2表示A32—C;
Z1表示CH;
n是零、1、2或3;
B3表示氧、硫或N—R33;
A31和A32各自独立地表示氢、甲基或氨基;
R31表示—CH2·CHR34NR36R37或下式基团: 或
或
R32、R33、R34、R35、R36和R37各自独立地表示氢或C1—6烷基。
3.根据权利要求1的方法,其中制备的是选自下列的化合物和其盐和药物前体:
N,N—二甲基—2—[5—(2—甲基咪唑—1—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(咪唑—1—基甲基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]乙胺;
1—甲基—4—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]哌啶;
4—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]哌啶;
3—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]吡咯烷;
1—甲基—3—[5—(2—甲基咪唑—1—基)—1H—吲哚—3—基]吡咯烷;
4—[5—(咪唑—1—基)—1H—吲哚—3—基]哌啶;
1—甲基—4—[5—(咪唑—1—基)—1H—吲哚—3—基]哌啶;
1—甲基—3—[5—(2—甲基咪唑—1—基甲基)—1H—吲哚—3—基]吡咯烷;
1—甲基—3—[5—(咪唑—1—基)—1H—吲哚—3—基]吡咯烷;
1—甲基—3—[5—(咪唑—1—基甲基)—1H—吲哚—3—基]吡咯烷;
N,N—二甲基—2—[5—(2—氨基咪唑—1—基)—1H—吲哚—3—基]乙胺;
N,N—二甲基—2—[5—(2—氨基咪唑—1—基甲基)—1H—吲哚—3—基]乙胺。
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9102222.8 | 1991-02-01 | ||
| GB919102222A GB9102222D0 (en) | 1991-02-01 | 1991-02-01 | Therapeutic agents |
| GB9106917.9 | 1991-04-03 | ||
| GB919106917A GB9106917D0 (en) | 1991-04-03 | 1991-04-03 | Therapeutic agents |
| GB9113415.5 | 1991-06-21 | ||
| GB919113415A GB9113415D0 (en) | 1991-06-21 | 1991-06-21 | Therapeutic agents |
| GB9122451.9 | 1991-10-23 | ||
| GB919122451A GB9122451D0 (en) | 1991-10-23 | 1991-10-23 | Therapeutic agents |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN92101189A Division CN1038681C (zh) | 1991-02-01 | 1992-02-01 | 三唑衍生物的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1161967A CN1161967A (zh) | 1997-10-15 |
| CN1060479C true CN1060479C (zh) | 2001-01-10 |
Family
ID=27450619
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN92101189A Expired - Lifetime CN1038681C (zh) | 1991-02-01 | 1992-02-01 | 三唑衍生物的制备方法 |
| CN96121797A Expired - Lifetime CN1128617C (zh) | 1991-02-01 | 1996-11-25 | 含三唑衍生物的药物组合物的制备方法 |
| CN96121796A Expired - Lifetime CN1060479C (zh) | 1991-02-01 | 1996-11-25 | 咪唑衍生物的制备方法 |
| CN96117268A Expired - Lifetime CN1061654C (zh) | 1991-02-01 | 1996-11-25 | 四唑衍生物的制备方法 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN92101189A Expired - Lifetime CN1038681C (zh) | 1991-02-01 | 1992-02-01 | 三唑衍生物的制备方法 |
| CN96121797A Expired - Lifetime CN1128617C (zh) | 1991-02-01 | 1996-11-25 | 含三唑衍生物的药物组合物的制备方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN96117268A Expired - Lifetime CN1061654C (zh) | 1991-02-01 | 1996-11-25 | 四唑衍生物的制备方法 |
Country Status (30)
| Country | Link |
|---|---|
| US (4) | US5298520A (zh) |
| EP (2) | EP0497512B1 (zh) |
| JP (1) | JP2500280B2 (zh) |
| KR (1) | KR100212111B1 (zh) |
| CN (4) | CN1038681C (zh) |
| AT (1) | ATE158582T1 (zh) |
| BG (1) | BG62024B2 (zh) |
| CA (2) | CA2060139C (zh) |
| CY (1) | CY2095B1 (zh) |
| CZ (1) | CZ283018B6 (zh) |
| DE (2) | DE69222329T2 (zh) |
| DK (1) | DK0497512T3 (zh) |
| ES (2) | ES2162188T3 (zh) |
| FI (1) | FI107154B (zh) |
| GR (2) | GR3025026T3 (zh) |
| HR (1) | HRP930777B1 (zh) |
| HU (2) | HU222350B1 (zh) |
| IE (1) | IE920342A1 (zh) |
| IL (1) | IL100756A (zh) |
| LU (1) | LU90338I2 (zh) |
| LV (1) | LV12090B (zh) |
| MX (1) | MX9200405A (zh) |
| NL (1) | NL980019I1 (zh) |
| NO (2) | NO180447C (zh) |
| NZ (1) | NZ241394A (zh) |
| SA (1) | SA94140534B1 (zh) |
| SG (1) | SG50409A1 (zh) |
| SI (1) | SI9210101B (zh) |
| SK (1) | SK278998B6 (zh) |
| YU (1) | YU48236B (zh) |
Families Citing this family (123)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL114690A (en) * | 1990-06-07 | 1997-02-18 | Wellcome Found | Antimigraine oxazolidinone substituted indole derivatives |
| US5545644A (en) * | 1990-10-15 | 1996-08-13 | Pfizer Inc. | Indole derivatives |
| US5607951A (en) * | 1990-10-15 | 1997-03-04 | Pfizer Inc | Indole derivatives |
| US5578612A (en) * | 1990-10-15 | 1996-11-26 | Pfizer Inc. | Indole derivatives |
| US5559129A (en) * | 1990-10-15 | 1996-09-24 | Pfizer Inc | Indole derivatives |
| US5559246A (en) * | 1990-10-15 | 1996-09-24 | Pfizer Inc. | Indole derivatives |
| AU2363492A (en) * | 1991-08-03 | 1993-03-02 | Smithkline Beecham Plc | 5-ht4 receptor antagonists |
| GB9201038D0 (en) * | 1992-01-16 | 1992-03-11 | Glaxo Group Ltd | Chemical compounds |
| TW288010B (zh) * | 1992-03-05 | 1996-10-11 | Pfizer | |
| EP0630374B1 (en) * | 1992-03-13 | 2001-09-05 | MERCK SHARP & DOHME LTD. | Imidazole, triazole and tetrazole derivatives |
| GB9207396D0 (en) * | 1992-04-03 | 1992-05-13 | Merck Sharp & Dohme | Therapeutic agents |
| CA2133413C (en) | 1992-04-07 | 2001-04-10 | John Eugene Macor | Indole derivatives as 5-ht1 antagonists |
| US6380233B1 (en) | 1992-04-07 | 2002-04-30 | Pfizer Inc | Indole derivatives as 5-HT1 agonists |
| US5498626A (en) * | 1992-04-10 | 1996-03-12 | Pfizer Inc. | Acylaminoindole derivatives as 5-ht1 agonists |
| GB9207930D0 (en) * | 1992-04-10 | 1992-05-27 | Pfizer Ltd | Indoles |
| GB9208463D0 (en) * | 1992-04-16 | 1992-06-03 | Merck Sharp & Dohme | Therapeutic agents |
| GB9210400D0 (en) * | 1992-05-15 | 1992-07-01 | Merck Sharp & Dohme | Therapeutic agents |
| GB9211277D0 (en) | 1992-05-28 | 1992-07-15 | Glaxo Group Inc | Pharmaceutical compositions |
| AU4337293A (en) * | 1992-06-05 | 1994-01-04 | Merck Sharp & Dohme Limited | The sulphate salt of a substituted triazole, pharmaceutical compositions thereof, and their use in therapy |
| GB9215526D0 (en) * | 1992-07-22 | 1992-09-02 | Merck Sharp & Dohme | Chemical process |
| JPH07509452A (ja) * | 1992-07-24 | 1995-10-19 | メルク シヤープ エンド ドーム リミテツド | イミダゾール,トリアゾール及びテトラゾール誘導体 |
| IL106445A (en) * | 1992-07-30 | 1998-01-04 | Merck Sharp & Dohme | History of 1,2,4-Trans-Triazole 4-Transform, Preparation and Pharmaceutical Preparations Containing Them |
| TW251284B (zh) * | 1992-11-02 | 1995-07-11 | Pfizer | |
| GB9226537D0 (en) * | 1992-12-21 | 1993-02-17 | Smithkline Beecham Plc | Compounds |
| FR2701026B1 (fr) * | 1993-02-02 | 1995-03-31 | Adir | Nouveaux dérivés de l'indole, de l'indazole et du benzisoxazole, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent. |
| US6060473A (en) * | 1993-04-01 | 2000-05-09 | Ucb S.A. - Dtb | 7-azabicyclo[2.2.1]-heptane and -heptene derivatives as cholinergic receptor ligands |
| US5817679A (en) | 1993-04-01 | 1998-10-06 | University Of Virginia | 7-Azabicyclo 2.2.1!-heptane and -heptene derivatives as cholinergic receptor ligands |
| CZ59996A3 (en) * | 1993-08-31 | 1996-06-12 | Pfizer | 5-arylindole derivatives per se and for treating diseases, intermediates for their preparation and pharmaceutical compositions based thereon |
| CA2171440A1 (en) | 1993-09-10 | 1995-03-16 | Changgeng Qian | Epibatidine and derivatives thereof as cholinergic receptor agonists and antagonists |
| GB9402016D0 (en) * | 1994-02-02 | 1994-03-30 | Merck Sharp & Dohme | Therapeutic agents |
| GB9402011D0 (en) * | 1994-02-02 | 1994-03-30 | Merck Sharp & Dohme | Therapeutic agents |
| US6117889A (en) * | 1994-04-01 | 2000-09-12 | University Of Virginia | 7-Azabicyclo-[2.2.1]-heptane and -heptene derivatives as analgesics and anti-inflammatory agents |
| US5552402A (en) * | 1994-05-19 | 1996-09-03 | Merck, Sharp & Dohme Ltd. | Five-membered heteroaromatic compounds as 5-HT receptor agonists |
| US5807857A (en) * | 1994-05-19 | 1998-09-15 | Merck Sharp & Dohme Ltd. | Piperazine, piperidine and tetrahydropyridine derivative of indol-3-alkyl as 5-HT1D-α agonists |
| GB9410031D0 (en) * | 1994-05-19 | 1994-07-06 | Merck Sharp & Dohme | Therapeutic agents |
| US5567824A (en) * | 1994-05-24 | 1996-10-22 | Merck & Co., Inc. | Palladium catalyzed ring closure of triazolyltryptamine |
| WO1996003400A1 (en) * | 1994-07-26 | 1996-02-08 | Pfizer Inc. | 4-indole derivatives as serotonin agonists and antagonists |
| CA2195107A1 (en) * | 1994-08-02 | 1996-02-15 | Richard Alexander Jelley | Azetidine, pyrrolidine and piperidine derivatives |
| GB9417310D0 (en) | 1994-08-27 | 1994-10-19 | Pfizer Ltd | Therapeutic agents |
| US5521196A (en) * | 1994-10-05 | 1996-05-28 | Eli Lilly And Company | 5-HT1F agonists for the treatment of migraine |
| GB9423460D0 (en) * | 1994-11-21 | 1995-01-11 | Merck Sharp & Dohme | Therapeutic agents |
| US5521197A (en) * | 1994-12-01 | 1996-05-28 | Eli Lilly And Company | 3-<1-alkylenearyl>-4-<1,2,3,6-tetrahydropyridinyl>-and 3-<1-alkylenearyl>-4-piperidinyl-1h-indoles: new 5-HT1F agonists |
| GB9501865D0 (en) * | 1995-01-31 | 1995-03-22 | Merck Sharp & Dohme | Therapeutic agents |
| US5484763A (en) * | 1995-02-10 | 1996-01-16 | American Cyanamid Company | Substituted benzisoxazole and benzisothiazole herbicidal agents |
| NZ305166A (en) * | 1995-03-20 | 1998-12-23 | Lilly Co Eli | 5-substituted-3-(1,2,3,6-tetrahydropyridin-4-yl)- and 3-(piperidin-4-yl)-1h-indoles; preparation and medicaments |
| US5602168A (en) * | 1995-07-10 | 1997-02-11 | Neurogen Corporation | 1-(N'-(arylalkylaminoalkyl)) aminoisoindoles; dopamine receptor subtype specific ligands |
| US6245768B1 (en) | 1995-06-05 | 2001-06-12 | Neurogen Corporation | 1-(N′-(arylalkylaminoalkyl)) aminoisoindoles; a new class of dopamine receptor subtype specific ligands |
| US5656632A (en) * | 1995-06-05 | 1997-08-12 | Neurogen Corporation | 1-(N'-(arylalkylaminoalkyl)) aminoisoindoles; dopamine receptor subtype specific ligands |
| ATE207485T1 (de) * | 1995-07-11 | 2001-11-15 | Merck & Co Inc | Ein triazolylmethyl-indol-ethylamin-bisulfat-salz |
| ZA965837B (en) * | 1995-07-11 | 1997-01-31 | Merck & Co Inc | A triazolylmethyl-indole ethylamine bisulfate salt |
| CA2234166A1 (en) * | 1995-10-10 | 1997-04-17 | Patric James Hahn | N-¬2-substituted-3-(2-aminoethyl)-1h-indol-5-yl|-amides: new 5-ht1f agonists |
| WO1997016446A1 (en) * | 1995-11-02 | 1997-05-09 | Merck Sharp & Dohme Limited | Bicyclic heteroaryl-alkylene-(homo)piperazinones and thione analogues thereof, their preparation and their use as selective agonists of 5-ht1-like receptors |
| GB9523583D0 (en) * | 1995-11-17 | 1996-01-17 | Merck Sharp & Dohme | Therapeutic agents |
| GB9610978D0 (en) * | 1996-05-24 | 1996-07-31 | Merck Sharp & Dohme | Therapeutic agents |
| US5877329A (en) * | 1996-08-13 | 1999-03-02 | Merck & Co., Inc. | Palladium catalyzed indolization |
| US5811551A (en) * | 1996-08-13 | 1998-09-22 | Merck & Co., Inc. | Palladium catalyzed indolization |
| US5808064A (en) * | 1996-08-13 | 1998-09-15 | Merck & Co., Inc. | Palladium catalyzed indolization |
| DE59710447D1 (de) | 1996-08-19 | 2003-08-21 | Volkswagen Ag | FREMDGEZÜNDETE BRENNKRAFTMASCHINE MIT EINEM NOx-ADSORBER |
| WO1998007504A1 (de) | 1996-08-19 | 1998-02-26 | Volkswagen Aktiengesellschaft | NOx-ABSORBER |
| GB9620777D0 (en) * | 1996-10-07 | 1996-11-20 | Merck Sharp & Dohme | Therapeutic use |
| US5998438A (en) * | 1996-11-26 | 1999-12-07 | Allelix Biopharmaceuticals, Inc. | 5-cyclo indole compounds |
| US7189753B1 (en) | 1997-11-06 | 2007-03-13 | Cady Roger K | Preemptive prophylaxis of migraine |
| US6066092A (en) * | 1997-11-06 | 2000-05-23 | Cady; Roger K. | Preemptive prophylaxis of migraine device and method |
| ZA991893B (en) * | 1998-03-09 | 1999-09-27 | Lundbeck & Co As H | 5-Heteroaryl substituted indoles. |
| ES2234324T3 (es) * | 1998-11-02 | 2005-06-16 | MERCK & CO., INC. | Combinaciones de un agonista 5ht1b/1d y un inhibidor selectivo de cox-2 para el tratamiento de la migraña. |
| US5994352A (en) * | 1998-11-13 | 1999-11-30 | Pfizer Inc. | 5-arylindole derivatives |
| CO5190664A1 (es) * | 1999-06-30 | 2002-08-29 | Pfizer Prod Inc | Terapia de combinacion para el tratamiento de migrana administracion de un receptor 5ht, cafeina y un inhibidor de ciclooxigenasa-2 |
| WO2003079972A2 (en) | 2002-02-22 | 2003-10-02 | New River Parmaceuticals Inc. | Active agent delivery systems and methods for protecting and administering active agents |
| CA2427098A1 (en) * | 2000-11-29 | 2002-06-06 | John Mehnert Schaus | 1-(2-m-methanesulfonamidophenylethyl)-4-(m-trifluoromethylphenyl)piperazine and pharmaceutically acceptable salts and solvates thereof and their use in the treatment of incontinence |
| US20030096379A1 (en) * | 2001-03-28 | 2003-05-22 | Kilgore James L. | Method for producing tryptamine derivatives |
| DE10214228A1 (de) * | 2002-03-22 | 2003-10-02 | Bdd Group Holding Ag Zug | Deuterierte substitutierte Indole sowie diese Verbindungen enthaltende Arzneimittel |
| WO2004000845A1 (en) | 2002-06-21 | 2003-12-31 | Suven Life Sciences Limited | Arylalkyl indoles having sertonin receptor affinity useful as therapeutic agents, process for their preparation and pharmaceutical compositions containing them |
| JP4473120B2 (ja) | 2002-06-21 | 2010-06-02 | スベン ライフ サイエンシズ リミティド | 治療薬剤として有用なセロトニン受容体親和性を有するアリールスルホニル・インドール、その製造方法、並びにそれらを含む医薬組成物 |
| ES2204303B2 (es) * | 2002-08-07 | 2004-12-16 | Laboratorios Vita, S.A. | Procedimiento para la obtencion de un compuesto farmaceuticamente activo. |
| EA009193B1 (ru) | 2002-11-28 | 2007-12-28 | Сувен Лайф Сайенсиз Лимитед | N-арилсульфонил-3-аминоалкоксииндолы |
| KR100818508B1 (ko) | 2002-11-28 | 2008-03-31 | 수벤 라이프 사이언시스 리미티드 | 세로토닌 수용체 친화력을 가지는 엔-아릴술포닐-3-치환인돌, 그 제조방법 및 그를 포함하는 약제학적 조성물 |
| DE60312226T2 (de) | 2002-12-18 | 2007-11-15 | Suven Life Sciences Ltd. | Tetrazyklische 3-substituierte indole mit serotoninrezeptoraffinität |
| EP2666772B1 (en) | 2002-12-20 | 2017-03-01 | Basf Se | Synthesis of amines and intermediates for the synthesis thereof |
| KR100572325B1 (ko) * | 2003-12-17 | 2006-04-19 | 삼성전자주식회사 | 이온 주입 장치 및 이를 이용한 이온 주입 방법 |
| CA2552663A1 (en) * | 2004-01-09 | 2005-07-28 | Ratiopharm Gmbh | Crystalline forms of rizatriptan benzoate |
| DK1751104T3 (da) * | 2004-01-28 | 2010-06-07 | Ratiopharm Gmbh | Syntesemetoder og mellemprodukter til fremstilling af rizatriptan |
| WO2006053116A2 (en) * | 2004-11-10 | 2006-05-18 | Dr. Reddy's Laboratories Ltd. | Rizatriptan process |
| MX2007005940A (es) * | 2004-11-18 | 2007-06-19 | Synta Pharmaceuticals Corp | Compuestos de triazol que modulan la actividad hsp90. |
| WO2006082598A2 (en) * | 2005-02-01 | 2006-08-10 | Natco Pharma Limited | Novel crystalline forms of rizatriptan benzoate |
| WO2006137083A1 (en) * | 2005-06-20 | 2006-12-28 | Natco Pharma Limited | Improved process for the preparation of rizatriptan benzoate |
| ES2270717B1 (es) * | 2005-08-08 | 2008-03-01 | Ragactives, S.L. | Procedimiento para la obtencion de derivados de triptamina. |
| CA2618586A1 (en) * | 2005-08-11 | 2007-02-15 | Merck Frosst Canada Ltd. | Novel substituted 1,2,3-triazolylmethyl-benzothiophene or -indole and their use as leukotriene biosynthesis inhibitors |
| WO2007094819A2 (en) * | 2005-08-18 | 2007-08-23 | Synta Pharmaceuticals Corp. | Triazole compounds that modulate hsp90 activity |
| EP1951713A1 (en) * | 2005-11-14 | 2008-08-06 | Matrix Laboratories Ltd | Process for the large scale production of rizatriptan benzoate |
| US20110313171A1 (en) | 2006-01-19 | 2011-12-22 | Chandra Purna Ray | Conversion of aromatic diazonium salt to aryl hydrazine |
| WO2007139960A2 (en) * | 2006-05-25 | 2007-12-06 | Synta Pharmaceuticals Corp. | Compounds that modulate hsp90 activity and methods for identifying same |
| RS53446B (en) | 2006-05-25 | 2014-12-31 | Synta Pharmaceuticals Corp. | TRIAZOLE UNITS MODULATING HSP90 ACTIVITY |
| US8318790B2 (en) * | 2006-05-25 | 2012-11-27 | Synta Pharmaceuticals Corp. | Triazole compounds that modulate HSP90 activity |
| CA2653332A1 (en) * | 2006-05-25 | 2007-12-06 | Synta Pharmaceuticals Corp. | Triazole compounds that modulate hsp90 activity |
| ES2524556T3 (es) | 2006-10-09 | 2014-12-10 | Charleston Laboratories, Inc. | Composiciones farmacéuticas |
| WO2008149152A1 (en) * | 2007-06-04 | 2008-12-11 | Generics [Uk] Limited | Novel process |
| WO2009049030A1 (en) * | 2007-10-09 | 2009-04-16 | Triton Biopharma, Llc | Method to use compositions having antidepressant anxiolytic and other neurological activity and compositions of matter |
| JP5714910B2 (ja) | 2008-01-09 | 2015-05-07 | チャールストン ラボラトリーズ,インコーポレイテッド | 薬学的組成物 |
| US20090294028A1 (en) * | 2008-06-03 | 2009-12-03 | Nanochip, Inc. | Process for fabricating high density storage device with high-temperature media |
| WO2011006012A1 (en) | 2009-07-08 | 2011-01-13 | Charleston Laboratories Inc. | Pharmaceutical compositions |
| US20110082450A1 (en) * | 2009-10-02 | 2011-04-07 | Cardiofocus, Inc. | Cardiac ablation system with inflatable member having multiple inflation settings |
| HUE030794T2 (en) * | 2010-02-08 | 2017-06-28 | Medivation Technologies Inc | Synthesis Processes of Dihydro-Pyrido-Phthalazinone Derivatives |
| EP2560640A1 (en) | 2010-04-19 | 2013-02-27 | Synta Pharmaceuticals Corp. | Cancer therapy using a combination of a hsp90 inhibitory compounds and a egfr inhibitor |
| JP2014534228A (ja) | 2011-11-02 | 2014-12-18 | シンタ ファーマシューティカルズ コーポレーション | 白金含有剤とhsp90阻害剤の組合せ療法 |
| EP2773345A1 (en) | 2011-11-02 | 2014-09-10 | Synta Pharmaceuticals Corp. | Cancer therapy using a combination of hsp90 inhibitors with topoisomerase i inhibitors |
| EP2780010A1 (en) | 2011-11-14 | 2014-09-24 | Synta Pharmaceuticals Corp. | Combination therapy of hsp90 inhibitors with braf inhibitors |
| WO2013120045A1 (en) | 2012-02-10 | 2013-08-15 | University Of Utah Research Foundation | Substituted 1h-indazol-1-ol analogs as inhibitors of beta catenin/tcf protein-protein interactions |
| CN103739594A (zh) * | 2012-10-17 | 2014-04-23 | 南京大学 | 一类含吡嗪环和三氮唑结构的席夫碱类衍生物及其制法 |
| WO2016187308A1 (en) | 2015-05-20 | 2016-11-24 | Amgen Inc. | Triazole agonists of the apj receptor |
| US20170119738A1 (en) * | 2015-10-28 | 2017-05-04 | Dr. Reddy's Laboratories, Ltd. | Pharmaceutical compositions for rizatriptan |
| EP3407869A1 (en) | 2016-01-27 | 2018-12-05 | Instar Technologies A.S. | Oromucosal nanofiber carriers for therapeutic treatment |
| WO2017152130A1 (en) | 2016-03-04 | 2017-09-08 | Charleston Laboratories, Inc. | Pharmaceutical compositions |
| WO2017189893A1 (en) | 2016-04-27 | 2017-11-02 | University Of Puerto Rico | 1,5-disubstituted 1,2,3-triazoles are inhibitors of rac/cdc42 gtpases |
| US9988369B2 (en) | 2016-05-03 | 2018-06-05 | Amgen Inc. | Heterocyclic triazole compounds as agonists of the APJ receptor |
| EP3541805B1 (en) | 2016-11-16 | 2020-10-14 | Amgen Inc. | Heteroaryl-substituted triazoles as apj receptor agonists |
| EP3541792B1 (en) | 2016-11-16 | 2020-12-23 | Amgen Inc. | Triazole furan compounds as agonists of the apj receptor |
| US10689367B2 (en) | 2016-11-16 | 2020-06-23 | Amgen Inc. | Triazole pyridyl compounds as agonists of the APJ receptor |
| US11191762B2 (en) | 2016-11-16 | 2021-12-07 | Amgen Inc. | Alkyl substituted triazole compounds as agonists of the APJ Receptor |
| US11020395B2 (en) | 2016-11-16 | 2021-06-01 | Amgen Inc. | Cycloalkyl substituted triazole compounds as agonists of the APJ receptor |
| EP3541810B1 (en) | 2016-11-16 | 2020-12-23 | Amgen Inc. | Triazole phenyl compounds as agonists of the apj receptor |
| WO2019089335A1 (en) | 2017-11-03 | 2019-05-09 | Amgen Inc. | Fused triazole agonists of the apj receptor |
| WO2019213006A1 (en) | 2018-05-01 | 2019-11-07 | Amgen Inc. | Substituted pyrimidinones as agonists of the apj receptor |
| EP3766483A1 (en) | 2019-07-19 | 2021-01-20 | BioPharma Synergies, S. L. | Orodispersible powder composition comprising a triptan |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0200322A1 (en) * | 1985-04-10 | 1986-11-05 | H. Lundbeck A/S | Heterocyclic compounds |
| EP0313397A1 (en) * | 1987-10-23 | 1989-04-26 | The Wellcome Foundation Limited | Therapeutic heterocyclic compounds |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3475437A (en) * | 1964-06-05 | 1969-10-28 | Boehringer Sohn Ingelheim | 3-tertiary amino lower alkylpseudoindoles |
| GB1237008A (en) * | 1968-12-18 | 1971-06-30 | Pfizer Ltd | Novel indoline derivatives |
| US3686213A (en) * | 1970-08-28 | 1972-08-22 | American Cyanamid Co | Substituted aminoethyl indoles |
| DE2520816C3 (de) * | 1975-05-09 | 1979-02-15 | Bayer Ag, 5090 Leverkusen | Methinfarbstoffe |
| ZA795239B (en) * | 1978-10-12 | 1980-11-26 | Glaxo Group Ltd | Heterocyclic compounds |
| IE51491B1 (en) * | 1980-08-12 | 1987-01-07 | Glaxo Group Ltd | Heterocyclic compounds |
| ZW19381A1 (en) * | 1980-08-12 | 1983-03-09 | Glaxo Group Ltd | Heterocyclic compounds |
| GB2083463B (en) * | 1980-08-12 | 1984-05-10 | Glaxo Group Ltd | Heterocyclic compounds |
| BE889931A (fr) * | 1980-08-12 | 1982-02-11 | Glaxo Group Ltd | Derives indoliques, leur preparation et leurs applications en tant que medicaments |
| US4453001A (en) * | 1982-02-22 | 1984-06-05 | Sandoz, Inc. | Isoxazolyl indolamines as intermediates |
| JPS58150576A (ja) * | 1982-03-03 | 1983-09-07 | Sumitomo Chem Co Ltd | 新規な1,2,4−オキサジアゾ−ル誘導体 |
| US4692531A (en) * | 1984-06-22 | 1987-09-08 | Bristol-Myers Company | Substituted 3,4-diamino-1,2,5-thiadiazoles having histamine H2 -receptor antagonist activity |
| GB8419575D0 (en) * | 1984-08-01 | 1984-09-05 | Glaxo Group Ltd | Chemical compounds |
| DE3430284A1 (de) * | 1984-08-17 | 1986-02-27 | Troponwerke GmbH & Co KG, 5000 Köln | Neue tryptamin-derivate, ein verfahren zu ihrer herstellung und ihre verwendung |
| US4663526A (en) * | 1984-12-26 | 1987-05-05 | Emil Kamieniecki | Nondestructive readout of a latent electrostatic image formed on an insulating material |
| DE3531658A1 (de) * | 1985-09-05 | 1987-03-12 | Boehringer Mannheim Gmbh | Heterocyclisch substituierte indole, zwischenprodukte, verfahren zu ihrer herstellung und arzneimittel |
| EP0225726B1 (en) * | 1985-11-08 | 1991-04-17 | Glaxo Group Limited | Indole derivatives |
| GB8527619D0 (en) * | 1985-11-08 | 1985-12-11 | Glaxo Group Ltd | Chemical compounds |
| US4881967A (en) * | 1986-12-10 | 1989-11-21 | E. I. Du Pont De Nemours And Company | Heterocyclic 2,3-dihydrobenzofuran herbicides |
| US5225431A (en) * | 1987-10-23 | 1993-07-06 | Burroughs Wellcome Co. | Therapeutic substituted indole compounds and compositions thereof |
| NZ227841A (en) * | 1988-02-12 | 1991-08-27 | Merck Sharp & Dohme | Heterocyclic compounds with at least two non-condensed five membered rings and pharmaceutical compositions |
| IL114690A (en) * | 1990-06-07 | 1997-02-18 | Wellcome Found | Antimigraine oxazolidinone substituted indole derivatives |
| AU2363492A (en) * | 1991-08-03 | 1993-03-02 | Smithkline Beecham Plc | 5-ht4 receptor antagonists |
| KR930011238A (ko) * | 1991-11-12 | 1993-06-24 | 오리 노리오 | 스태틱 알에이엠(ram)의 메모리셀 및 그 메모리셀어레이 |
| US5436246A (en) * | 1992-09-17 | 1995-07-25 | Merrell Dow Pharmaceuticals Inc. | Serotonin receptor agents |
-
1992
- 1992-01-23 CZ CS92198A patent/CZ283018B6/cs not_active IP Right Cessation
- 1992-01-23 SK SK198-92A patent/SK278998B6/sk not_active IP Right Cessation
- 1992-01-24 ES ES97200467T patent/ES2162188T3/es not_active Expired - Lifetime
- 1992-01-24 ES ES92300617T patent/ES2106822T3/es not_active Expired - Lifetime
- 1992-01-24 SG SG1996000418A patent/SG50409A1/en unknown
- 1992-01-24 EP EP92300617A patent/EP0497512B1/en not_active Expired - Lifetime
- 1992-01-24 EP EP97200467A patent/EP0778275B1/en not_active Expired - Lifetime
- 1992-01-24 DK DK92300617.5T patent/DK0497512T3/da active
- 1992-01-24 DE DE69222329T patent/DE69222329T2/de not_active Expired - Lifetime
- 1992-01-24 DE DE1999175007 patent/DE19975007I2/de active Active
- 1992-01-24 NZ NZ241394A patent/NZ241394A/en not_active IP Right Cessation
- 1992-01-24 IL IL10075692A patent/IL100756A/en not_active IP Right Cessation
- 1992-01-24 AT AT92300617T patent/ATE158582T1/de active
- 1992-01-28 CA CA002060139A patent/CA2060139C/en not_active Expired - Lifetime
- 1992-01-28 US US07/827,187 patent/US5298520A/en not_active Expired - Lifetime
- 1992-01-28 CA CA002238140A patent/CA2238140C/en not_active Expired - Lifetime
- 1992-01-30 MX MX9200405A patent/MX9200405A/es unknown
- 1992-01-30 SI SI9210101A patent/SI9210101B/sl unknown
- 1992-01-30 YU YU10192A patent/YU48236B/sh unknown
- 1992-01-31 JP JP4059849A patent/JP2500280B2/ja not_active Expired - Lifetime
- 1992-01-31 IE IE034292A patent/IE920342A1/en active Protection Beyond IP Right Term
- 1992-01-31 NO NO920424A patent/NO180447C/no not_active IP Right Cessation
- 1992-01-31 FI FI920442A patent/FI107154B/fi active Protection Beyond IP Right Term
- 1992-01-31 HU HU9200299A patent/HU222350B1/hu active IP Right Grant
- 1992-02-01 KR KR1019920001592A patent/KR100212111B1/ko not_active Expired - Lifetime
- 1992-02-01 CN CN92101189A patent/CN1038681C/zh not_active Expired - Lifetime
-
1993
- 1993-04-02 HR HR930777A patent/HRP930777B1/xx not_active IP Right Cessation
- 1993-07-14 BG BG097943A patent/BG62024B2/bg unknown
- 1993-11-22 US US08/156,140 patent/US5451588A/en not_active Expired - Lifetime
-
1994
- 1994-01-30 SA SA94140534A patent/SA94140534B1/ar unknown
-
1995
- 1995-05-10 US US08/438,621 patent/US5602162A/en not_active Expired - Lifetime
- 1995-05-25 US US08/452,185 patent/US5602163A/en not_active Expired - Lifetime
- 1995-06-28 HU HU95P/P00500P patent/HU211516A9/hu unknown
-
1996
- 1996-11-25 CN CN96121797A patent/CN1128617C/zh not_active Expired - Lifetime
- 1996-11-25 CN CN96121796A patent/CN1060479C/zh not_active Expired - Lifetime
- 1996-11-25 CN CN96117268A patent/CN1061654C/zh not_active Expired - Lifetime
-
1997
- 1997-10-15 GR GR970402671T patent/GR3025026T3/el unknown
-
1998
- 1998-03-17 LV LVP-98-54A patent/LV12090B/en unknown
- 1998-06-08 NL NL980019C patent/NL980019I1/nl unknown
- 1998-07-17 CY CY9800017A patent/CY2095B1/xx unknown
-
1999
- 1999-01-13 LU LU90338C patent/LU90338I2/fr unknown
- 1999-04-09 NO NO1999005C patent/NO1999005I1/no unknown
-
2001
- 2001-10-11 GR GR20010401586T patent/GR3036864T3/el unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0200322A1 (en) * | 1985-04-10 | 1986-11-05 | H. Lundbeck A/S | Heterocyclic compounds |
| EP0313397A1 (en) * | 1987-10-23 | 1989-04-26 | The Wellcome Foundation Limited | Therapeutic heterocyclic compounds |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1060479C (zh) | 咪唑衍生物的制备方法 | |
| CN1036456C (zh) | 氨基取代的吡唑 | |
| CN1072669C (zh) | 苯并咪唑化合物及其用作gabaa受体复合物调节器的用途 | |
| CN1260225C (zh) | 四氢吡啶基或哌啶基杂环衍生物 | |
| TWI421073B (zh) | 作為活化激酶抑制劑之經取代吲唑衍生物 | |
| CN1720225B (zh) | 具有5-羟色胺受体亲和性的n-芳基磺酰-3-取代吲哚及其制备方法和含有其的药物组合物 | |
| CN1191239C (zh) | 用于治疗病毒性疾病的吡唑衍生物 | |
| CN1198741A (zh) | 具有α2受体亲合活性的咪唑衍生物 | |
| CN1349503A (zh) | 肾素抑制剂 | |
| CN1331688A (zh) | 氨基吡啶衍生物 | |
| CN1816529A (zh) | 用作激酶抑制剂的芳基取代的吡唑-酰胺化合物 | |
| CN1234031A (zh) | 吲唑衍生物及其作为磷酸二酯酶(pde)iv型和肿瘤坏死因子(tnf)产生的抑制剂的应用 | |
| CN1149051A (zh) | 用作α1-肾上腺素能受体拮抗剂的嘧啶二酮、嘧啶三酮、三嗪二酮、四氢喹唑啉二酮衍生物 | |
| CN1378547A (zh) | 用作磷酸二酯酶抑制剂的5-(2-取代的-5-杂环基磺酰基吡啶-3-基)-二氢吡唑并[4,3-d]嘧啶-7-酮类化合物 | |
| CN1281852A (zh) | 三唑及咪唑衍生物 | |
| CN1118598A (zh) | 吡咯并吡啶衍生物 | |
| CN1600783A (zh) | 咪唑并吡啶衍生物 | |
| CN1058779A (zh) | 喹诺酮衍生物和含有该化合物的药物组合物 | |
| CN1072219C (zh) | 二氮杂䓬酮、其生产和用途 | |
| CN1249052C (zh) | 作为hiv逆转录酶抑制剂的吡唑衍生物 | |
| CN1319094A (zh) | 咪唑基衍生物 | |
| CN1136038A (zh) | 吲哚酰基胍衍生物 | |
| CN1230432C (zh) | 取代的苯基-哌嗪衍生物及其制备和用途 | |
| CN87103504A (zh) | 杂环羧酰胺 | |
| CN1023602C (zh) | 制备抑制精神的3-哌嗪基苯并唑类衍生物的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CX01 | Expiry of patent term |
Expiration termination date: 20120201 Granted publication date: 20010110 |