BR112012008267B1 - Derivados hidroxila, ceto e glucuronida de 3-(4-(7h-pirrolo[2,3-d]pirimidin-4-il)-1h-pirazol-1- il)-3-ciclopentilpropanonitrila - Google Patents
Derivados hidroxila, ceto e glucuronida de 3-(4-(7h-pirrolo[2,3-d]pirimidin-4-il)-1h-pirazol-1- il)-3-ciclopentilpropanonitrila Download PDFInfo
- Publication number
- BR112012008267B1 BR112012008267B1 BR112012008267-1A BR112012008267A BR112012008267B1 BR 112012008267 B1 BR112012008267 B1 BR 112012008267B1 BR 112012008267 A BR112012008267 A BR 112012008267A BR 112012008267 B1 BR112012008267 B1 BR 112012008267B1
- Authority
- BR
- Brazil
- Prior art keywords
- pyrazol
- pyrimidin
- pyrrolo
- propanenitrile
- hydroxy
- Prior art date
Links
- 229930182480 glucuronide Natural products 0.000 title abstract description 17
- 150000008134 glucuronides Chemical class 0.000 title abstract description 17
- 150000001875 compounds Chemical class 0.000 claims description 206
- 239000000203 mixture Substances 0.000 claims description 98
- 150000003839 salts Chemical class 0.000 claims description 57
- 239000002253 acid Substances 0.000 claims description 24
- 229910014033 C-OH Inorganic materials 0.000 claims description 20
- 229910014570 C—OH Inorganic materials 0.000 claims description 20
- 229910052799 carbon Inorganic materials 0.000 claims description 18
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 11
- 229920006395 saturated elastomer Polymers 0.000 claims description 6
- YIOHYHBNMPEYIR-UHFFFAOYSA-N 3-(2-hydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound OC1CCCC1C(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 YIOHYHBNMPEYIR-UHFFFAOYSA-N 0.000 claims description 4
- HJNFDLVNGCFTBD-UHFFFAOYSA-N 3-(1-hydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(CC#N)C1(O)CCCC1 HJNFDLVNGCFTBD-UHFFFAOYSA-N 0.000 claims description 3
- DBJFGIPGWLPPQS-UHFFFAOYSA-N 3-(3-oxocyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1C(=O)CCC1C(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 DBJFGIPGWLPPQS-UHFFFAOYSA-N 0.000 claims description 3
- PEDGDCYQFZPRCX-UHFFFAOYSA-N 3-(3-hydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1C(O)CCC1C(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 PEDGDCYQFZPRCX-UHFFFAOYSA-N 0.000 claims description 2
- YEURRWPOSAWASX-UHFFFAOYSA-N 3-cyclopentyl-2-hydroxy-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(C(C#N)O)C1CCCC1 YEURRWPOSAWASX-UHFFFAOYSA-N 0.000 claims description 2
- HHNARICGCSVDAS-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(2-oxo-1,3-dihydropyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C=CNC2=NC(O)=NC=1C(=C1)C=NN1C(CC#N)C1CCCC1 HHNARICGCSVDAS-UHFFFAOYSA-N 0.000 claims description 2
- OHDYFGLFUXOHSG-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(5,6-dihydroxy-6,7-dihydro-5h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C(O)C(O)NC2=NC=NC=1C(=C1)C=NN1C(CC#N)C1CCCC1 OHDYFGLFUXOHSG-UHFFFAOYSA-N 0.000 claims description 2
- WBDZJBCZCBJJQQ-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(5-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C(O)=CNC2=NC=NC=1C(=C1)C=NN1C(CC#N)C1CCCC1 WBDZJBCZCBJJQQ-UHFFFAOYSA-N 0.000 claims description 2
- ZVOWMVHSOALPOW-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(6-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound N1=CN=C2NC(O)=CC2=C1C(=C1)C=NN1C(CC#N)C1CCCC1 ZVOWMVHSOALPOW-UHFFFAOYSA-N 0.000 claims description 2
- OBYLXIZDTJLUTM-UHFFFAOYSA-N 3-cyclopentyl-3-[5-oxo-4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-2-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C(O)=NN1C(CC#N)C1CCCC1 OBYLXIZDTJLUTM-UHFFFAOYSA-N 0.000 claims description 2
- CUJVLJISMAZUKA-UHFFFAOYSA-N 3-(1,2-dihydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound OC1CCCC1(O)C(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 CUJVLJISMAZUKA-UHFFFAOYSA-N 0.000 claims 2
- MJUANLVGHZGODT-UHFFFAOYSA-N 3-(3-hydroxycyclopentyl)-3-[4-(2-oxo-1,3-dihydropyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1C(O)CCC1C(CC#N)N1N=CC(C=2C=3C=CNC=3N=C(O)N=2)=C1 MJUANLVGHZGODT-UHFFFAOYSA-N 0.000 claims 2
- CUPRBWRGYKRDQU-UHFFFAOYSA-N 3-(3-hydroxycyclopentyl)-3-[4-(5-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1C(O)CCC1C(CC#N)N1N=CC(C=2C=3C(O)=CNC=3N=CN=2)=C1 CUPRBWRGYKRDQU-UHFFFAOYSA-N 0.000 claims 2
- IBESKZNPEIGRCH-UHFFFAOYSA-N 3-(3-hydroxycyclopentyl)-3-[4-(6-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1C(O)CCC1C(CC#N)N1N=CC(C=2C=3C=C(O)NC=3N=CN=2)=C1 IBESKZNPEIGRCH-UHFFFAOYSA-N 0.000 claims 2
- PKDZVKTUOKFVRS-UHFFFAOYSA-N 3-(3-hydroxycyclopentyl)-3-[5-oxo-4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-2-yl]propanenitrile Chemical compound C1C(O)CCC1C(CC#N)N1N=C(O)C(C=2C=3C=CNC=3N=CN=2)=C1 PKDZVKTUOKFVRS-UHFFFAOYSA-N 0.000 claims 2
- ZNJSVMDHWRMHNO-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(2,6-dioxo-5,7-dihydro-3h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound N=1C(O)=NC=2NC(=O)CC=2C=1C(=C1)C=NN1C(CC#N)C1CCCC1 ZNJSVMDHWRMHNO-UHFFFAOYSA-N 0.000 claims 2
- 125000004432 carbon atom Chemical group C* 0.000 claims 2
- KWRPLELDSVYAGK-UHFFFAOYSA-N 2-cyclopentyl-2-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]acetyl cyanide Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(C(C#N)=O)C1CCCC1 KWRPLELDSVYAGK-UHFFFAOYSA-N 0.000 claims 1
- HSAAROHAJQCOER-UHFFFAOYSA-N 2-hydroxy-3-(1-hydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(C(C#N)O)C1(O)CCCC1 HSAAROHAJQCOER-UHFFFAOYSA-N 0.000 claims 1
- XQRDNUYNKUQMEW-UHFFFAOYSA-N 2-hydroxy-3-(2-hydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(C(C#N)O)C1CCCC1O XQRDNUYNKUQMEW-UHFFFAOYSA-N 0.000 claims 1
- VDXDCRBASIIWBO-UHFFFAOYSA-N 2-hydroxy-3-(3-hydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(C(C#N)O)C1CCC(O)C1 VDXDCRBASIIWBO-UHFFFAOYSA-N 0.000 claims 1
- VZFAXXRRGWEKPV-UHFFFAOYSA-N 3-(1,3-dihydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1C(O)CCC1(O)C(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 VZFAXXRRGWEKPV-UHFFFAOYSA-N 0.000 claims 1
- NYCZEXXAROJVCL-UHFFFAOYSA-N 3-(1-hydroxycyclopentyl)-3-[4-(2-oxo-1,3-dihydropyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C=CNC2=NC(O)=NC=1C(=C1)C=NN1C(CC#N)C1(O)CCCC1 NYCZEXXAROJVCL-UHFFFAOYSA-N 0.000 claims 1
- WBYLIJXCVJBOBV-UHFFFAOYSA-N 3-(1-hydroxycyclopentyl)-3-[4-(5-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C(O)=CNC2=NC=NC=1C(=C1)C=NN1C(CC#N)C1(O)CCCC1 WBYLIJXCVJBOBV-UHFFFAOYSA-N 0.000 claims 1
- PAZKSWLWHYICJN-UHFFFAOYSA-N 3-(1-hydroxycyclopentyl)-3-[4-(6-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound N1=CN=C2NC(O)=CC2=C1C(=C1)C=NN1C(CC#N)C1(O)CCCC1 PAZKSWLWHYICJN-UHFFFAOYSA-N 0.000 claims 1
- LCAPFWOTNIJZHU-UHFFFAOYSA-N 3-(1-hydroxycyclopentyl)-3-[5-oxo-4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-2-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C(O)=NN1C(CC#N)C1(O)CCCC1 LCAPFWOTNIJZHU-UHFFFAOYSA-N 0.000 claims 1
- KLWMFSZCVQLXFB-UHFFFAOYSA-N 3-(2,3-dihydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound OC1C(O)CCC1C(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 KLWMFSZCVQLXFB-UHFFFAOYSA-N 0.000 claims 1
- QSABKWFPAMQNSN-UHFFFAOYSA-N 3-(2,4-dihydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1C(O)CC(O)C1C(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 QSABKWFPAMQNSN-UHFFFAOYSA-N 0.000 claims 1
- JFPNABLFFDROSS-UHFFFAOYSA-N 3-(2,5-dihydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound OC1CCC(O)C1C(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 JFPNABLFFDROSS-UHFFFAOYSA-N 0.000 claims 1
- QFRLIMHBRORCAB-UHFFFAOYSA-N 3-(2-hydroxycyclopentyl)-3-[4-(2-oxo-1,3-dihydropyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound OC1CCCC1C(CC#N)N1N=CC(C=2C=3C=CNC=3N=C(O)N=2)=C1 QFRLIMHBRORCAB-UHFFFAOYSA-N 0.000 claims 1
- ZEDDBYPJWZSBLK-UHFFFAOYSA-N 3-(2-hydroxycyclopentyl)-3-[4-(5-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound OC1CCCC1C(CC#N)N1N=CC(C=2C=3C(O)=CNC=3N=CN=2)=C1 ZEDDBYPJWZSBLK-UHFFFAOYSA-N 0.000 claims 1
- XPCPJOPRVQKFFV-UHFFFAOYSA-N 3-(2-hydroxycyclopentyl)-3-[4-(6-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound OC1CCCC1C(CC#N)N1N=CC(C=2C=3C=C(O)NC=3N=CN=2)=C1 XPCPJOPRVQKFFV-UHFFFAOYSA-N 0.000 claims 1
- BBWXMQMJQVNODH-UHFFFAOYSA-N 3-(2-hydroxycyclopentyl)-3-[5-oxo-4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-2-yl]propanenitrile Chemical compound OC1CCCC1C(CC#N)N1N=C(O)C(C=2C=3C=CNC=3N=CN=2)=C1 BBWXMQMJQVNODH-UHFFFAOYSA-N 0.000 claims 1
- JNFPBLRRNITRTG-UHFFFAOYSA-N 3-(2-oxocyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound O=C1CCCC1C(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 JNFPBLRRNITRTG-UHFFFAOYSA-N 0.000 claims 1
- GVMURUAGAAZVBP-UHFFFAOYSA-N 3-(3,4-dihydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1C(O)C(O)CC1C(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 GVMURUAGAAZVBP-UHFFFAOYSA-N 0.000 claims 1
- FRACHNQNEVQPGS-UHFFFAOYSA-N 3-(3-hydroxycyclopentyl)-3-[4-(6-oxo-5,7-dihydropyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1C(O)CCC1C(CC#N)N1N=CC(C=2C=3CC(=O)NC=3N=CN=2)=C1 FRACHNQNEVQPGS-UHFFFAOYSA-N 0.000 claims 1
- DDFWDYZCHQPHAE-UHFFFAOYSA-N 3-cyclopentyl-2,3-dihydroxy-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(O)(C(C#N)O)C1CCCC1 DDFWDYZCHQPHAE-UHFFFAOYSA-N 0.000 claims 1
- HRKBQSHTNDXSCI-UHFFFAOYSA-N 3-cyclopentyl-2-hydroxy-3-[4-(2-oxo-1,3-dihydropyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=C(O)N=2)C=NN1C(C(C#N)O)C1CCCC1 HRKBQSHTNDXSCI-UHFFFAOYSA-N 0.000 claims 1
- FECAFTOUEXMDSC-UHFFFAOYSA-N 3-cyclopentyl-2-hydroxy-3-[4-(5-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C(O)=CNC=3N=CN=2)C=NN1C(C(C#N)O)C1CCCC1 FECAFTOUEXMDSC-UHFFFAOYSA-N 0.000 claims 1
- YAIJKKHKCXGYNI-UHFFFAOYSA-N 3-cyclopentyl-2-hydroxy-3-[4-(6-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C=C(O)NC=3N=CN=2)C=NN1C(C(C#N)O)C1CCCC1 YAIJKKHKCXGYNI-UHFFFAOYSA-N 0.000 claims 1
- OASDHOZTXCBNRU-UHFFFAOYSA-N 3-cyclopentyl-2-hydroxy-3-[5-oxo-4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-2-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C(O)=NN1C(C(C#N)O)C1CCCC1 OASDHOZTXCBNRU-UHFFFAOYSA-N 0.000 claims 1
- XFDRMMOLOFYVMI-UHFFFAOYSA-N 3-cyclopentyl-3-[3-hydroxy-5-oxo-4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-2-yl]propanenitrile Chemical compound OC1=C(C=2C=3C=CNC=3N=CN=2)C(O)=NN1C(CC#N)C1CCCC1 XFDRMMOLOFYVMI-UHFFFAOYSA-N 0.000 claims 1
- ZMVNSFISJOYCJR-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(5,6-dihydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C(O)=C(O)NC2=NC=NC=1C(=C1)C=NN1C(CC#N)C1CCCC1 ZMVNSFISJOYCJR-UHFFFAOYSA-N 0.000 claims 1
- UZPQJRRLNUDGSY-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(5-hydroxy-2-oxo-1,3-dihydropyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C(O)=CNC2=NC(O)=NC=1C(=C1)C=NN1C(CC#N)C1CCCC1 UZPQJRRLNUDGSY-UHFFFAOYSA-N 0.000 claims 1
- PVDINXKUTVCCMS-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(5-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)-5-oxo-1h-pyrazol-2-yl]propanenitrile Chemical compound C1=C(C=2C=3C(O)=CNC=3N=CN=2)C(O)=NN1C(CC#N)C1CCCC1 PVDINXKUTVCCMS-UHFFFAOYSA-N 0.000 claims 1
- PQZUSBNVPBEKQZ-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(6-hydroxy-2-oxo-3,7-dihydropyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound N1=C(O)N=C2NC(O)=CC2=C1C(=C1)C=NN1C(CC#N)C1CCCC1 PQZUSBNVPBEKQZ-UHFFFAOYSA-N 0.000 claims 1
- LMLJKZCZAXRHLK-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(6-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)-5-oxo-1h-pyrazol-2-yl]propanenitrile Chemical compound N1=CN=C2NC(O)=CC2=C1C(C(=N1)O)=CN1C(CC#N)C1CCCC1 LMLJKZCZAXRHLK-UHFFFAOYSA-N 0.000 claims 1
- ORLQCDBUTPCEPE-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(6-oxo-5,7-dihydropyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound N1C(=O)CC2=C1N=CN=C2C(=C1)C=NN1C(CC#N)C1CCCC1 ORLQCDBUTPCEPE-UHFFFAOYSA-N 0.000 claims 1
- MUVUVZBRRQFXCV-UHFFFAOYSA-N 3-cyclopentyl-3-[5-oxo-4-(2-oxo-1,3-dihydropyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-2-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=C(O)N=2)C(O)=NN1C(CC#N)C1CCCC1 MUVUVZBRRQFXCV-UHFFFAOYSA-N 0.000 claims 1
- FKTKNQCDPJWNAZ-UHFFFAOYSA-N 3-cyclopentyl-3-hydroxy-3-[4-(2-oxo-1,3-dihydropyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C=CNC2=NC(O)=NC=1C(=C1)C=NN1C(O)(CC#N)C1CCCC1 FKTKNQCDPJWNAZ-UHFFFAOYSA-N 0.000 claims 1
- IUWKAJWSVUKBBV-UHFFFAOYSA-N 3-cyclopentyl-3-hydroxy-3-[4-(5-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C(O)=CNC2=NC=NC=1C(=C1)C=NN1C(O)(CC#N)C1CCCC1 IUWKAJWSVUKBBV-UHFFFAOYSA-N 0.000 claims 1
- ARTAHQADYHBTBW-UHFFFAOYSA-N 3-cyclopentyl-3-hydroxy-3-[4-(6-hydroxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound N1=CN=C2NC(O)=CC2=C1C(=C1)C=NN1C(O)(CC#N)C1CCCC1 ARTAHQADYHBTBW-UHFFFAOYSA-N 0.000 claims 1
- SFINNIXZZVCBTK-UHFFFAOYSA-N 3-cyclopentyl-3-hydroxy-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(CC#N)(O)C1CCCC1 SFINNIXZZVCBTK-UHFFFAOYSA-N 0.000 claims 1
- KGDUFMKGHJYMDM-UHFFFAOYSA-N 3-cyclopentyl-3-hydroxy-3-[5-oxo-4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-2-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C(O)=NN1C(O)(CC#N)C1CCCC1 KGDUFMKGHJYMDM-UHFFFAOYSA-N 0.000 claims 1
- RNKCJRPQRDOIHW-UHFFFAOYSA-N 3-hydroxy-3-(1-hydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(CC#N)(O)C1(O)CCCC1 RNKCJRPQRDOIHW-UHFFFAOYSA-N 0.000 claims 1
- YYGVSNQKCFEHQT-UHFFFAOYSA-N 3-hydroxy-3-(2-hydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound OC1CCCC1C(O)(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 YYGVSNQKCFEHQT-UHFFFAOYSA-N 0.000 claims 1
- NBBXMFWBMBFZIO-UHFFFAOYSA-N 3-hydroxy-3-(3-hydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C1C(O)CCC1C(O)(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 NBBXMFWBMBFZIO-UHFFFAOYSA-N 0.000 claims 1
- PZTNVVDBMDOYRG-UHFFFAOYSA-N C1C(O)CCC1C(CC#N)N1C(O)=C(C=2C=3C=CNC=3N=CN=2)C=N1 Chemical compound C1C(O)CCC1C(CC#N)N1C(O)=C(C=2C=3C=CNC=3N=CN=2)C=N1 PZTNVVDBMDOYRG-UHFFFAOYSA-N 0.000 claims 1
- QKGMTHXRVADEIM-UHFFFAOYSA-N C=12C(O)=CNC2=NC=NC=1C(=C1O)C=NN1C(CC#N)C1CCCC1 Chemical compound C=12C(O)=CNC2=NC=NC=1C(=C1O)C=NN1C(CC#N)C1CCCC1 QKGMTHXRVADEIM-UHFFFAOYSA-N 0.000 claims 1
- VUCQZVZGLYQJMK-UHFFFAOYSA-N N1=CC(C=2C=3C=CNC=3N=CN=2)=C(O)N1C(C(C#N)O)C1CCCC1 Chemical compound N1=CC(C=2C=3C=CNC=3N=CN=2)=C(O)N1C(C(C#N)O)C1CCCC1 VUCQZVZGLYQJMK-UHFFFAOYSA-N 0.000 claims 1
- ILNYHPQDRGZJGF-UHFFFAOYSA-N N1=CN=C2NC(O)=CC2=C1C(=C1O)C=NN1C(CC#N)C1CCCC1 Chemical compound N1=CN=C2NC(O)=CC2=C1C(=C1O)C=NN1C(CC#N)C1CCCC1 ILNYHPQDRGZJGF-UHFFFAOYSA-N 0.000 claims 1
- ZTLPUQBOJLYNJG-UHFFFAOYSA-N OC1=C(C=2C=3C=CNC=3N=C(O)N=2)C=NN1C(CC#N)C1CCCC1 Chemical compound OC1=C(C=2C=3C=CNC=3N=C(O)N=2)C=NN1C(CC#N)C1CCCC1 ZTLPUQBOJLYNJG-UHFFFAOYSA-N 0.000 claims 1
- XQVDGVJOZVNQPS-UHFFFAOYSA-N OC1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(CC#N)C1(O)CCCC1 Chemical compound OC1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(CC#N)C1(O)CCCC1 XQVDGVJOZVNQPS-UHFFFAOYSA-N 0.000 claims 1
- ZHZLDCAFBCJTDF-UHFFFAOYSA-N OC1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(CC#N)C1CCCC1 Chemical compound OC1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(CC#N)C1CCCC1 ZHZLDCAFBCJTDF-UHFFFAOYSA-N 0.000 claims 1
- FSSPWQTVBUETAX-UHFFFAOYSA-N OC1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(O)(CC#N)C1CCCC1 Chemical compound OC1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(O)(CC#N)C1CCCC1 FSSPWQTVBUETAX-UHFFFAOYSA-N 0.000 claims 1
- FIYRMIRCBMGBID-UHFFFAOYSA-N OC1CCCC1C(CC#N)N1C(O)=C(C=2C=3C=CNC=3N=CN=2)C=N1 Chemical compound OC1CCCC1C(CC#N)N1C(O)=C(C=2C=3C=CNC=3N=CN=2)C=N1 FIYRMIRCBMGBID-UHFFFAOYSA-N 0.000 claims 1
- 125000002887 hydroxy group Chemical class [H]O* 0.000 abstract description 14
- 229930194542 Keto Chemical class 0.000 abstract description 4
- 125000000468 ketone group Chemical class 0.000 abstract description 4
- PPUBMRYNGIUSBF-UHFFFAOYSA-N 3-cyclopentylpropanenitrile Chemical compound N#CCCC1CCCC1 PPUBMRYNGIUSBF-UHFFFAOYSA-N 0.000 abstract 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 72
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 66
- 239000002207 metabolite Substances 0.000 description 62
- 108010024121 Janus Kinases Proteins 0.000 description 59
- 102000015617 Janus Kinases Human genes 0.000 description 59
- 201000010099 disease Diseases 0.000 description 55
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 54
- 150000002500 ions Chemical class 0.000 description 46
- 238000000034 method Methods 0.000 description 42
- 235000002639 sodium chloride Nutrition 0.000 description 41
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 37
- 229910001868 water Inorganic materials 0.000 description 34
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 33
- 239000000047 product Substances 0.000 description 31
- 239000012071 phase Substances 0.000 description 30
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 28
- 210000002700 urine Anatomy 0.000 description 28
- 241000282414 Homo sapiens Species 0.000 description 26
- 238000004128 high performance liquid chromatography Methods 0.000 description 26
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 26
- 206010028980 Neoplasm Diseases 0.000 description 24
- 238000006243 chemical reaction Methods 0.000 description 24
- 239000000243 solution Substances 0.000 description 24
- 239000003814 drug Substances 0.000 description 23
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 22
- -1 organic acid salts Chemical class 0.000 description 20
- 201000011510 cancer Diseases 0.000 description 19
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 19
- 238000005160 1H NMR spectroscopy Methods 0.000 description 18
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 18
- 239000012634 fragment Substances 0.000 description 18
- 239000004480 active ingredient Substances 0.000 description 17
- 208000035475 disorder Diseases 0.000 description 17
- 239000002904 solvent Substances 0.000 description 17
- 238000004458 analytical method Methods 0.000 description 15
- 230000000694 effects Effects 0.000 description 15
- 229940124597 therapeutic agent Drugs 0.000 description 15
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 14
- 229940122245 Janus kinase inhibitor Drugs 0.000 description 14
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 14
- 102100025387 Tyrosine-protein kinase JAK3 Human genes 0.000 description 14
- 238000010828 elution Methods 0.000 description 14
- 235000019253 formic acid Nutrition 0.000 description 14
- 208000003556 Dry Eye Syndromes Diseases 0.000 description 13
- 239000002245 particle Substances 0.000 description 13
- 239000000546 pharmaceutical excipient Substances 0.000 description 13
- 229920000642 polymer Polymers 0.000 description 13
- 238000011282 treatment Methods 0.000 description 13
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 12
- 208000027866 inflammatory disease Diseases 0.000 description 12
- 239000000523 sample Substances 0.000 description 12
- 238000012360 testing method Methods 0.000 description 12
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 11
- 206010013774 Dry eye Diseases 0.000 description 11
- VQGISNOMGHCEPX-UHFFFAOYSA-N propanenitrile Chemical compound C[CH]C#N VQGISNOMGHCEPX-UHFFFAOYSA-N 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- 239000002552 dosage form Substances 0.000 description 10
- 238000013467 fragmentation Methods 0.000 description 10
- 238000006062 fragmentation reaction Methods 0.000 description 10
- 238000004949 mass spectrometry Methods 0.000 description 10
- 239000011159 matrix material Substances 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 9
- JQZPQJMJBSXUTQ-UHFFFAOYSA-N C=1C=NNC=1.N1C=NC=C2N=CC=C21 Chemical compound C=1C=NNC=1.N1C=NC=C2N=CC=C21 JQZPQJMJBSXUTQ-UHFFFAOYSA-N 0.000 description 9
- 206010035226 Plasma cell myeloma Diseases 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 9
- 239000003480 eluent Substances 0.000 description 9
- 239000000284 extract Substances 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 239000008194 pharmaceutical composition Substances 0.000 description 9
- 239000006228 supernatant Substances 0.000 description 9
- 208000034578 Multiple myelomas Diseases 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 8
- 239000002674 ointment Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 7
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 7
- 238000003556 assay Methods 0.000 description 7
- 150000001793 charged compounds Chemical class 0.000 description 7
- 230000001419 dependent effect Effects 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 238000000605 extraction Methods 0.000 description 7
- 229910052938 sodium sulfate Inorganic materials 0.000 description 7
- 235000011152 sodium sulphate Nutrition 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 210000001519 tissue Anatomy 0.000 description 7
- 239000003643 water by type Substances 0.000 description 7
- RAXXELZNTBOGNW-UHFFFAOYSA-N 1H-imidazole Chemical compound C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- 201000011152 Pemphigus Diseases 0.000 description 6
- 108091000080 Phosphotransferase Proteins 0.000 description 6
- 102000001253 Protein Kinase Human genes 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 150000001336 alkenes Chemical class 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 150000001721 carbon Chemical group 0.000 description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 6
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 6
- 239000010408 film Substances 0.000 description 6
- 239000003112 inhibitor Substances 0.000 description 6
- 238000002955 isolation Methods 0.000 description 6
- 230000014759 maintenance of location Effects 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- 201000005962 mycosis fungoides Diseases 0.000 description 6
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 6
- 201000001976 pemphigus vulgaris Diseases 0.000 description 6
- 102000020233 phosphotransferase Human genes 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 108060006633 protein kinase Proteins 0.000 description 6
- 210000002345 respiratory system Anatomy 0.000 description 6
- 238000000926 separation method Methods 0.000 description 6
- 208000017520 skin disease Diseases 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 238000004885 tandem mass spectrometry Methods 0.000 description 6
- 208000023275 Autoimmune disease Diseases 0.000 description 5
- 241000282472 Canis lupus familiaris Species 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 201000004681 Psoriasis Diseases 0.000 description 5
- 150000001299 aldehydes Chemical class 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 5
- 229960003957 dexamethasone Drugs 0.000 description 5
- 210000003608 fece Anatomy 0.000 description 5
- 239000012530 fluid Substances 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 239000004005 microsphere Substances 0.000 description 5
- 239000013580 millipore water Substances 0.000 description 5
- 239000008177 pharmaceutical agent Substances 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 230000000717 retained effect Effects 0.000 description 5
- 210000003491 skin Anatomy 0.000 description 5
- 239000008247 solid mixture Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 230000000699 topical effect Effects 0.000 description 5
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 4
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 4
- KDOPAZIWBAHVJB-UHFFFAOYSA-N 5h-pyrrolo[3,2-d]pyrimidine Chemical compound C1=NC=C2NC=CC2=N1 KDOPAZIWBAHVJB-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 4
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- 208000032027 Essential Thrombocythemia Diseases 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- 241000701085 Human alphaherpesvirus 3 Species 0.000 description 4
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 4
- 241000701806 Human papillomavirus Species 0.000 description 4
- 206010048643 Hypereosinophilic syndrome Diseases 0.000 description 4
- 206010025323 Lymphomas Diseases 0.000 description 4
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 4
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 4
- 206010034277 Pemphigoid Diseases 0.000 description 4
- 206010039705 Scleritis Diseases 0.000 description 4
- 201000008736 Systemic mastocytosis Diseases 0.000 description 4
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 description 4
- 206010046851 Uveitis Diseases 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 235000011114 ammonium hydroxide Nutrition 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 229920002988 biodegradable polymer Polymers 0.000 description 4
- 239000004621 biodegradable polymer Substances 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 208000000594 bullous pemphigoid Diseases 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 238000005119 centrifugation Methods 0.000 description 4
- 229940125877 compound 31 Drugs 0.000 description 4
- 208000010247 contact dermatitis Diseases 0.000 description 4
- 229920001577 copolymer Polymers 0.000 description 4
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- FESLGQSAIKKHKS-NWDGAFQWSA-N ethyl (1s,2r)-2-[tert-butyl(dimethyl)silyl]oxycyclopentane-1-carboxylate Chemical compound CCOC(=O)[C@H]1CCC[C@H]1O[Si](C)(C)C(C)(C)C FESLGQSAIKKHKS-NWDGAFQWSA-N 0.000 description 4
- 230000002550 fecal effect Effects 0.000 description 4
- 238000003818 flash chromatography Methods 0.000 description 4
- 230000014509 gene expression Effects 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 238000005805 hydroxylation reaction Methods 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 239000003456 ion exchange resin Substances 0.000 description 4
- 229920003303 ion-exchange polymer Polymers 0.000 description 4
- 230000000302 ischemic effect Effects 0.000 description 4
- 229940043355 kinase inhibitor Drugs 0.000 description 4
- 238000001819 mass spectrum Methods 0.000 description 4
- 239000002105 nanoparticle Substances 0.000 description 4
- 230000007170 pathology Effects 0.000 description 4
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 4
- 239000006187 pill Substances 0.000 description 4
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 125000003226 pyrazolyl group Chemical group 0.000 description 4
- 150000004944 pyrrolopyrimidines Chemical class 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 230000035945 sensitivity Effects 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- 230000001954 sterilising effect Effects 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 3
- TWCMVXMQHSVIOJ-UHFFFAOYSA-N Aglycone of yadanzioside D Natural products COC(=O)C12OCC34C(CC5C(=CC(O)C(O)C5(C)C3C(O)C1O)C)OC(=O)C(OC(=O)C)C24 TWCMVXMQHSVIOJ-UHFFFAOYSA-N 0.000 description 3
- PLMKQQMDOMTZGG-UHFFFAOYSA-N Astrantiagenin E-methylester Natural products CC12CCC(O)C(C)(CO)C1CCC1(C)C2CC=C2C3CC(C)(C)CCC3(C(=O)OC)CCC21C PLMKQQMDOMTZGG-UHFFFAOYSA-N 0.000 description 3
- 206010010741 Conjunctivitis Diseases 0.000 description 3
- 206010012438 Dermatitis atopic Diseases 0.000 description 3
- 206010012442 Dermatitis contact Diseases 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 102100037813 Focal adhesion kinase 1 Human genes 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- 102000042838 JAK family Human genes 0.000 description 3
- 108091082332 JAK family Proteins 0.000 description 3
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 3
- 208000014767 Myeloproliferative disease Diseases 0.000 description 3
- 208000009525 Myocarditis Diseases 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 206010060862 Prostate cancer Diseases 0.000 description 3
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 229920002125 Sokalan® Polymers 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 206010052779 Transplant rejections Diseases 0.000 description 3
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 3
- 239000012491 analyte Substances 0.000 description 3
- 239000007900 aqueous suspension Substances 0.000 description 3
- 201000008937 atopic dermatitis Diseases 0.000 description 3
- 210000000941 bile Anatomy 0.000 description 3
- 230000036983 biotransformation Effects 0.000 description 3
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 3
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 3
- 229940125936 compound 42 Drugs 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 238000000502 dialysis Methods 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 238000003821 enantio-separation Methods 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 210000000744 eyelid Anatomy 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- PFOARMALXZGCHY-UHFFFAOYSA-N homoegonol Natural products C1=C(OC)C(OC)=CC=C1C1=CC2=CC(CCCO)=CC(OC)=C2O1 PFOARMALXZGCHY-UHFFFAOYSA-N 0.000 description 3
- 230000033444 hydroxylation Effects 0.000 description 3
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 3
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 201000004614 iritis Diseases 0.000 description 3
- 238000005567 liquid scintillation counting Methods 0.000 description 3
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 3
- 229960001924 melphalan Drugs 0.000 description 3
- 239000006199 nebulizer Substances 0.000 description 3
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 229940124531 pharmaceutical excipient Drugs 0.000 description 3
- 210000002381 plasma Anatomy 0.000 description 3
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 3
- 239000004584 polyacrylic acid Substances 0.000 description 3
- 229920001610 polycaprolactone Polymers 0.000 description 3
- 239000004632 polycaprolactone Substances 0.000 description 3
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 3
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 3
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 125000000168 pyrrolyl group Chemical group 0.000 description 3
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 238000001228 spectrum Methods 0.000 description 3
- 238000004659 sterilization and disinfection Methods 0.000 description 3
- 150000003431 steroids Chemical class 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 238000010189 synthetic method Methods 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 231100000607 toxicokinetics Toxicity 0.000 description 3
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- HVMCMDMMBWWWCI-QWRGUYRKSA-N (1r,2s)-2-[tert-butyl(dimethyl)silyl]oxycyclopentane-1-carbaldehyde Chemical compound CC(C)(C)[Si](C)(C)O[C@H]1CCC[C@H]1C=O HVMCMDMMBWWWCI-QWRGUYRKSA-N 0.000 description 2
- HVMCMDMMBWWWCI-GHMZBOCLSA-N (1s,2r)-2-[tert-butyl(dimethyl)silyl]oxycyclopentane-1-carbaldehyde Chemical compound CC(C)(C)[Si](C)(C)O[C@@H]1CCC[C@@H]1C=O HVMCMDMMBWWWCI-GHMZBOCLSA-N 0.000 description 2
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 2
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 2
- YIOHYHBNMPEYIR-QEJZJMRPSA-N (3s)-3-[(1s,2s)-2-hydroxycyclopentyl]-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound O[C@H]1CCC[C@H]1[C@H](CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 YIOHYHBNMPEYIR-QEJZJMRPSA-N 0.000 description 2
- NCLRZUXUGYGOGG-JKOXKOMZSA-N (z)-3-[(1r,2r)-2-[tert-butyl(dimethyl)silyl]oxycyclopentyl]prop-2-enenitrile Chemical compound CC(C)(C)[Si](C)(C)O[C@@H]1CCC[C@@H]1\C=C/C#N NCLRZUXUGYGOGG-JKOXKOMZSA-N 0.000 description 2
- NCLRZUXUGYGOGG-JWRBQHRDSA-N (z)-3-[(1s,2s)-2-[tert-butyl(dimethyl)silyl]oxycyclopentyl]prop-2-enenitrile Chemical compound CC(C)(C)[Si](C)(C)O[C@H]1CCC[C@H]1\C=C/C#N NCLRZUXUGYGOGG-JWRBQHRDSA-N 0.000 description 2
- RHCXSJZUSMLFES-UHFFFAOYSA-N 1-trimethylsilyloxycyclopentane-1-carbaldehyde Chemical compound C[Si](C)(C)OC1(C=O)CCCC1 RHCXSJZUSMLFES-UHFFFAOYSA-N 0.000 description 2
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 2
- UIUQLPYCKLGRKP-UHFFFAOYSA-N 2-cyclopentylpropanenitrile Chemical compound N#CC(C)C1CCCC1 UIUQLPYCKLGRKP-UHFFFAOYSA-N 0.000 description 2
- AWVWKPGJWARLMT-UHFFFAOYSA-N 3-(1-hydroxycyclopentyl)-3-[4-[7-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl]propanenitrile Chemical compound N1=CN=C2N(COCC[Si](C)(C)C)C=CC2=C1C(=C1)C=NN1C(CC#N)C1(O)CCCC1 AWVWKPGJWARLMT-UHFFFAOYSA-N 0.000 description 2
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 2
- 208000006386 Bone Resorption Diseases 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- 206010006895 Cachexia Diseases 0.000 description 2
- 208000005024 Castleman disease Diseases 0.000 description 2
- 208000011231 Crohn disease Diseases 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 108010075944 Erythropoietin Receptors Proteins 0.000 description 2
- 102100036509 Erythropoietin receptor Human genes 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 2
- 208000010201 Exanthema Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 102000053187 Glucuronidase Human genes 0.000 description 2
- 108010060309 Glucuronidase Proteins 0.000 description 2
- 208000005176 Hepatitis C Diseases 0.000 description 2
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 2
- 241000598436 Human T-cell lymphotropic virus Species 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 208000003456 Juvenile Arthritis Diseases 0.000 description 2
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 2
- 208000007766 Kaposi sarcoma Diseases 0.000 description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 206010028561 Myeloid metaplasia Diseases 0.000 description 2
- 201000002481 Myositis Diseases 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- 239000004696 Poly ether ether ketone Substances 0.000 description 2
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 2
- 206010038389 Renal cancer Diseases 0.000 description 2
- 206010070834 Sensitisation Diseases 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- 208000021386 Sjogren Syndrome Diseases 0.000 description 2
- 208000000453 Skin Neoplasms Diseases 0.000 description 2
- 206010040880 Skin irritation Diseases 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 206010051379 Systemic Inflammatory Response Syndrome Diseases 0.000 description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 description 2
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 208000017733 acquired polycythemia vera Diseases 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 230000000172 allergic effect Effects 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 208000022531 anorexia Diseases 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 208000010668 atopic eczema Diseases 0.000 description 2
- 230000001363 autoimmune Effects 0.000 description 2
- JUPQTSLXMOCDHR-UHFFFAOYSA-N benzene-1,4-diol;bis(4-fluorophenyl)methanone Chemical compound OC1=CC=C(O)C=C1.C1=CC(F)=CC=C1C(=O)C1=CC=C(F)C=C1 JUPQTSLXMOCDHR-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 230000024279 bone resorption Effects 0.000 description 2
- 229960001467 bortezomib Drugs 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 230000000973 chemotherapeutic effect Effects 0.000 description 2
- 238000004296 chiral HPLC Methods 0.000 description 2
- 201000010902 chronic myelomonocytic leukemia Diseases 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 239000003246 corticosteroid Substances 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 206010061428 decreased appetite Diseases 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 239000012055 enteric layer Substances 0.000 description 2
- 201000005884 exanthem Diseases 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 230000002489 hematologic effect Effects 0.000 description 2
- 208000002672 hepatitis B Diseases 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 229960002411 imatinib Drugs 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 230000001506 immunosuppresive effect Effects 0.000 description 2
- 229960003444 immunosuppressant agent Drugs 0.000 description 2
- 239000003018 immunosuppressive agent Substances 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 150000002497 iodine compounds Chemical class 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 201000010982 kidney cancer Diseases 0.000 description 2
- 208000017169 kidney disease Diseases 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 208000014018 liver neoplasm Diseases 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- 206010025135 lupus erythematosus Diseases 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 238000003801 milling Methods 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 206010028417 myasthenia gravis Diseases 0.000 description 2
- 206010028537 myelofibrosis Diseases 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 238000006772 olefination reaction Methods 0.000 description 2
- 108091008819 oncoproteins Proteins 0.000 description 2
- 102000027450 oncoproteins Human genes 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 208000037244 polycythemia vera Diseases 0.000 description 2
- 229920002530 polyetherether ketone Polymers 0.000 description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 2
- 229960004618 prednisone Drugs 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 239000012264 purified product Substances 0.000 description 2
- 206010037844 rash Diseases 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 201000000306 sarcoidosis Diseases 0.000 description 2
- 238000007423 screening assay Methods 0.000 description 2
- 230000008313 sensitization Effects 0.000 description 2
- 201000000849 skin cancer Diseases 0.000 description 2
- 230000036556 skin irritation Effects 0.000 description 2
- 231100000475 skin irritation Toxicity 0.000 description 2
- 231100000046 skin rash Toxicity 0.000 description 2
- 229960001922 sodium perborate Drugs 0.000 description 2
- YKLJGMBLPUQQOI-UHFFFAOYSA-M sodium;oxidooxy(oxo)borane Chemical compound [Na+].[O-]OB=O YKLJGMBLPUQQOI-UHFFFAOYSA-M 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 2
- 239000002562 thickening agent Substances 0.000 description 2
- 201000002510 thyroid cancer Diseases 0.000 description 2
- 230000017423 tissue regeneration Effects 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- PVFOMCVHYWHZJE-UHFFFAOYSA-N trichloroacetyl chloride Chemical compound ClC(=O)C(Cl)(Cl)Cl PVFOMCVHYWHZJE-UHFFFAOYSA-N 0.000 description 2
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 229910052722 tritium Inorganic materials 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 239000003039 volatile agent Substances 0.000 description 2
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 1
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 1
- CAQAIGDORNMTCH-LLVKDONJSA-N (3r)-3-cyclopentyl-3-[4-(2,5-dioxo-3,6-dihydro-1h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanamide Chemical compound C1([C@@H](CC(=O)N)N2N=CC(=C2)C=2C=3C(=O)CNC=3N=C(O)N=2)CCCC1 CAQAIGDORNMTCH-LLVKDONJSA-N 0.000 description 1
- LJZLJWDLFHJEOC-OAHLLOKOSA-N (3r)-3-cyclopentyl-3-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazol-1-yl]propanenitrile Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN([C@H](CC#N)C2CCCC2)N=C1 LJZLJWDLFHJEOC-OAHLLOKOSA-N 0.000 description 1
- YIOHYHBNMPEYIR-VHDGCEQUSA-N (3s)-3-[(1r,2r)-2-hydroxycyclopentyl]-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound O[C@@H]1CCC[C@@H]1[C@H](CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 YIOHYHBNMPEYIR-VHDGCEQUSA-N 0.000 description 1
- ZYZCALPXKGUGJI-DDVDASKDSA-M (e,3r,5s)-7-[3-(4-fluorophenyl)-2-phenyl-5-propan-2-ylimidazol-4-yl]-3,5-dihydroxyhept-6-enoate Chemical compound C=1C=C(F)C=CC=1N1C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C(C(C)C)N=C1C1=CC=CC=C1 ZYZCALPXKGUGJI-DDVDASKDSA-M 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- SXDXUPKVDBZOPR-UHFFFAOYSA-N 1-hydroxypyrrolidin-2-ol Chemical compound OC1CCCN1O SXDXUPKVDBZOPR-UHFFFAOYSA-N 0.000 description 1
- WJTZXPFJXASXAH-UHFFFAOYSA-N 1-trimethylsilyloxycyclopentane-1-carbonitrile Chemical compound C[Si](C)(C)OC1(C#N)CCCC1 WJTZXPFJXASXAH-UHFFFAOYSA-N 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- 125000003821 2-(trimethylsilyl)ethoxymethyl group Chemical group [H]C([H])([H])[Si](C([H])([H])[H])(C([H])([H])[H])C([H])([H])C(OC([H])([H])[*])([H])[H] 0.000 description 1
- KWMBADTWRIGGGG-UHFFFAOYSA-N 2-diethoxyphosphorylacetonitrile Chemical compound CCOP(=O)(CC#N)OCC KWMBADTWRIGGGG-UHFFFAOYSA-N 0.000 description 1
- AUVALWUPUHHNQV-UHFFFAOYSA-N 2-hydroxy-3-propylbenzoic acid Chemical class CCCC1=CC=CC(C(O)=O)=C1O AUVALWUPUHHNQV-UHFFFAOYSA-N 0.000 description 1
- LOIXPDIJYVEECO-UHFFFAOYSA-N 3-(1-hydroxycyclopentyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(CC#N)C1(O)CCCC1 LOIXPDIJYVEECO-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- CAQAIGDORNMTCH-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(2,5-dioxo-3,6-dihydro-1h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanamide Chemical compound C1=C(C=2C=3C(=O)CNC=3N=C(O)N=2)C=NN1C(CC(=O)N)C1CCCC1 CAQAIGDORNMTCH-UHFFFAOYSA-N 0.000 description 1
- CQXXNXSZRDUNCD-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(2-methoxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C=CNC2=NC(OC)=NC=1C(=C1)C=NN1C(CC#N)C1CCCC1 CQXXNXSZRDUNCD-UHFFFAOYSA-N 0.000 description 1
- LMFHEMPDLJSPGW-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(5-iodo-2-methoxy-7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound C=12C(I)=CNC2=NC(OC)=NC=1C(=C1)C=NN1C(CC#N)C1CCCC1 LMFHEMPDLJSPGW-UHFFFAOYSA-N 0.000 description 1
- HFNKQEVNSGCOJV-UHFFFAOYSA-N 3-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1-pyrazolyl]propanenitrile Chemical compound C1=C(C=2C=3C=CNC=3N=CN=2)C=NN1C(CC#N)C1CCCC1 HFNKQEVNSGCOJV-UHFFFAOYSA-N 0.000 description 1
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- DBCAQXHNJOFNGC-UHFFFAOYSA-N 4-bromo-1,1,1-trifluorobutane Chemical compound FC(F)(F)CCCBr DBCAQXHNJOFNGC-UHFFFAOYSA-N 0.000 description 1
- SWQQJGKVGZQZDT-UHFFFAOYSA-N 4-chloro-2-methoxy-7h-pyrrolo[2,3-d]pyrimidine Chemical compound COC1=NC(Cl)=C2C=CNC2=N1 SWQQJGKVGZQZDT-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 235000006491 Acacia senegal Nutrition 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 206010027654 Allergic conditions Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- GHXZTYHSJHQHIJ-UHFFFAOYSA-N Chlorhexidine Chemical compound C=1C=C(Cl)C=CC=1NC(N)=NC(N)=NCCCCCCN=C(N)N=C(N)NC1=CC=C(Cl)C=C1 GHXZTYHSJHQHIJ-UHFFFAOYSA-N 0.000 description 1
- 208000002691 Choroiditis Diseases 0.000 description 1
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 244000303965 Cyamopsis psoralioides Species 0.000 description 1
- 229920001651 Cyanoacrylate Polymers 0.000 description 1
- 206010011715 Cyclitis Diseases 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 102100038497 Cytokine receptor-like factor 2 Human genes 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 1
- 239000012623 DNA damaging agent Substances 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- 206010015084 Episcleritis Diseases 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 229920003134 Eudragit® polymer Polymers 0.000 description 1
- 229940124783 FAK inhibitor Drugs 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 108010091824 Focal Adhesion Kinase 1 Proteins 0.000 description 1
- 208000004262 Food Hypersensitivity Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 206010018341 Gliosis Diseases 0.000 description 1
- 201000005569 Gout Diseases 0.000 description 1
- 208000009329 Graft vs Host Disease Diseases 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000010496 Heart Arrest Diseases 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 101000956427 Homo sapiens Cytokine receptor-like factor 2 Proteins 0.000 description 1
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 1
- LELOWRISYMNNSU-UHFFFAOYSA-N Hydrocyanic acid Natural products N#C LELOWRISYMNNSU-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- 101150009057 JAK2 gene Proteins 0.000 description 1
- 101150069380 JAK3 gene Proteins 0.000 description 1
- 108010000837 Janus Kinase 1 Proteins 0.000 description 1
- 108010019437 Janus Kinase 2 Proteins 0.000 description 1
- 108010019421 Janus Kinase 3 Proteins 0.000 description 1
- 229940123241 Janus kinase 3 inhibitor Drugs 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- MWCLLHOVUTZFKS-UHFFFAOYSA-N Methyl cyanoacrylate Chemical compound COC(=O)C(=C)C#N MWCLLHOVUTZFKS-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 101100346764 Mus musculus Mtln gene Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 208000021642 Muscular disease Diseases 0.000 description 1
- 201000007224 Myeloproliferative neoplasm Diseases 0.000 description 1
- 201000009623 Myopathy Diseases 0.000 description 1
- GCTFALKRTYVYFB-UHFFFAOYSA-N N1=CNC2=CC=NC2=C1Cl Chemical compound N1=CNC2=CC=NC2=C1Cl GCTFALKRTYVYFB-UHFFFAOYSA-N 0.000 description 1
- 238000012565 NMR experiment Methods 0.000 description 1
- 206010029113 Neovascularisation Diseases 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- JQGPBDSMTHZOMO-UHFFFAOYSA-N OBO.C=1C=NNC=1 Chemical compound OBO.C=1C=NNC=1 JQGPBDSMTHZOMO-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 206010051482 Prostatomegaly Diseases 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 229940079156 Proteasome inhibitor Drugs 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 206010038910 Retinitis Diseases 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 208000009359 Sezary Syndrome Diseases 0.000 description 1
- 208000021388 Sezary disease Diseases 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 238000006859 Swern oxidation reaction Methods 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 1
- 206010047571 Visual impairment Diseases 0.000 description 1
- 229910021536 Zeolite Inorganic materials 0.000 description 1
- VHJDXVGRVZTYQX-UHFFFAOYSA-N [4-[1-(2-cyano-1-cyclopentylethyl)pyrazol-4-yl]-2-methoxy-7h-pyrrolo[2,3-d]pyrimidin-5-yl] acetate Chemical compound C=12C(OC(C)=O)=CNC2=NC(OC)=NC=1C(=C1)C=NN1C(CC#N)C1CCCC1 VHJDXVGRVZTYQX-UHFFFAOYSA-N 0.000 description 1
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 229940081735 acetylcellulose Drugs 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 208000002029 allergic contact dermatitis Diseases 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 230000019552 anatomical structure morphogenesis Effects 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 208000037875 astrocytosis Diseases 0.000 description 1
- 230000007341 astrogliosis Effects 0.000 description 1
- 208000010928 autoimmune thyroid disease Diseases 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 238000010504 bond cleavage reaction Methods 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 229910021538 borax Inorganic materials 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- 229960003340 calcium silicate Drugs 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000001733 carboxylic acid esters Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 229960001777 castor oil Drugs 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 108020001778 catalytic domains Proteins 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229960003260 chlorhexidine Drugs 0.000 description 1
- 238000011210 chromatographic step Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000002485 combustion reaction Methods 0.000 description 1
- 239000002361 compost Substances 0.000 description 1
- 229940125878 compound 36 Drugs 0.000 description 1
- 229940125807 compound 37 Drugs 0.000 description 1
- 229940127573 compound 38 Drugs 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 230000001143 conditioned effect Effects 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- NKKMVIVFRUYPLQ-NSCUHMNNSA-N crotononitrile Chemical class C\C=C\C#N NKKMVIVFRUYPLQ-NSCUHMNNSA-N 0.000 description 1
- RALDHUZFXJKFQB-UHFFFAOYSA-N cyclopentene-1-carbaldehyde Chemical compound O=CC1=CCCC1 RALDHUZFXJKFQB-UHFFFAOYSA-N 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- YCVGBTRTVKMLSH-UHFFFAOYSA-L disodium;1,3,4-thiadiazole-2,5-dithiolate Chemical compound [Na+].[Na+].[S-]C1=NN=C([S-])S1 YCVGBTRTVKMLSH-UHFFFAOYSA-L 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 210000003717 douglas' pouch Anatomy 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 230000007071 enzymatic hydrolysis Effects 0.000 description 1
- 238000006047 enzymatic hydrolysis reaction Methods 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 1
- 238000003810 ethyl acetate extraction Methods 0.000 description 1
- STVZJERGLQHEKB-UHFFFAOYSA-N ethylene glycol dimethacrylate Substances CC(=C)C(=O)OCCOC(=O)C(C)=C STVZJERGLQHEKB-UHFFFAOYSA-N 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 230000003090 exacerbative effect Effects 0.000 description 1
- 206010016256 fatigue Diseases 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012054 flavored emulsion Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000020375 flavoured syrup Nutrition 0.000 description 1
- 235000020932 food allergy Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 229940014259 gelatin Drugs 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 208000024908 graft versus host disease Diseases 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 238000001794 hormone therapy Methods 0.000 description 1
- 102000049918 human JAK1 Human genes 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 230000001861 immunosuppressant effect Effects 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229940030980 inova Drugs 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 238000005040 ion trap Methods 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 238000000021 kinase assay Methods 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 239000011344 liquid material Substances 0.000 description 1
- 210000001853 liver microsome Anatomy 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 238000007884 metabolite profiling Methods 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 210000001589 microsome Anatomy 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 238000003032 molecular docking Methods 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- OVHHHVAVHBHXAK-UHFFFAOYSA-N n,n-diethylprop-2-enamide Chemical compound CCN(CC)C(=O)C=C OVHHHVAVHBHXAK-UHFFFAOYSA-N 0.000 description 1
- 230000010352 nasal breathing Effects 0.000 description 1
- 230000010807 negative regulation of binding Effects 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 125000002524 organometallic group Chemical group 0.000 description 1
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 1
- 239000012285 osmium tetroxide Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229920000771 poly (alkylcyanoacrylate) Polymers 0.000 description 1
- 229920000962 poly(amidoamine) Polymers 0.000 description 1
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920002338 polyhydroxyethylmethacrylate Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 229920001299 polypropylene fumarate Polymers 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 239000003207 proteasome inhibitor Substances 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- 150000003217 pyrazoles Chemical class 0.000 description 1
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 230000025053 regulation of cell proliferation Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000009711 regulatory function Effects 0.000 description 1
- 230000002105 relative biological effectiveness Effects 0.000 description 1
- 230000010410 reperfusion Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 206010039083 rhinitis Diseases 0.000 description 1
- 102200084388 rs121918345 Human genes 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 210000003786 sclera Anatomy 0.000 description 1
- 239000012056 semi-solid material Substances 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 201000009890 sinusitis Diseases 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 1
- 229960002218 sodium chlorite Drugs 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000011121 sodium hydroxide Nutrition 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 235000011008 sodium phosphates Nutrition 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- 235000010339 sodium tetraborate Nutrition 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000002594 sorbent Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000012058 sterile packaged powder Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- AVMLPTWVYQXRSV-UHFFFAOYSA-N trimethyl-[2-[[4-(1h-pyrazol-4-yl)pyrrolo[2,3-d]pyrimidin-7-yl]methoxy]ethyl]silane Chemical compound N1=CN=C2N(COCC[Si](C)(C)C)C=CC2=C1C=1C=NNC=1 AVMLPTWVYQXRSV-UHFFFAOYSA-N 0.000 description 1
- JSPLKZUTYZBBKA-UHFFFAOYSA-N trioxidane Chemical class OOO JSPLKZUTYZBBKA-UHFFFAOYSA-N 0.000 description 1
- BSVBQGMMJUBVOD-UHFFFAOYSA-N trisodium borate Chemical compound [Na+].[Na+].[Na+].[O-]B([O-])[O-] BSVBQGMMJUBVOD-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 230000005740 tumor formation Effects 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 238000009827 uniform distribution Methods 0.000 description 1
- 229940099039 velcade Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 239000004034 viscosity adjusting agent Substances 0.000 description 1
- 230000004304 visual acuity Effects 0.000 description 1
- 238000003260 vortexing Methods 0.000 description 1
- 238000001238 wet grinding Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
- PAPBSGBWRJIAAV-UHFFFAOYSA-N ε-Caprolactone Chemical compound O=C1CCCCCO1 PAPBSGBWRJIAAV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/02—Heterocyclic radicals containing only nitrogen as ring hetero atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/26—Acyclic or carbocyclic radicals, substituted by hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Diabetes (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Endocrinology (AREA)
- Physical Education & Sports Medicine (AREA)
- Neurosurgery (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Dermatology (AREA)
- Pain & Pain Management (AREA)
- Cardiology (AREA)
- Hospice & Palliative Care (AREA)
- Heart & Thoracic Surgery (AREA)
- Psychiatry (AREA)
- Reproductive Health (AREA)
- Urology & Nephrology (AREA)
- Transplantation (AREA)
Abstract
DERIVADOS HIDROXIL, CETO E GLICURONIDA DE 3-(4(7H-PIRROLO[2,3-D]PIRIMIDIN-4-IL)-1H-PIRAZOL-1-IL)-3-CICLO-PENTILPROPANONITRILA. A presente invenção fornece derivados hidroxil, ceto e glicuronida de 3-(4-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)3-ciclopentilpropanonitrila.
Description
Este pedido reivindica prioridade do Pedido Provisório US 61/250.387, depositado em 9 de outubro de 2009, e Pedido Provisório US 61/316.218, depositado em 22 de março de 2010. As revelações destes documentos são incorporadas aqui como referência em suas totalidades.
A presente invenção fornece derivados derivados hidroxila, ceto, e glicuronidade 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol~1 -il)-3-ciclo- pentilpropanonitrila.
As proteínas quinase (PKs) são um grupo de enzimas que regulam diversos e importantes processos biológicos incluindo crescimento celular, sobrevivência e diferenciação, formação de órgão e morfogênese, neovascularização, reparo de tecido e regeneração, entre outros. Proteínas quinase exercem suas funções fisiológicas através da catalisação da fos- forilação de proteínas (ou substratos) e assim moduiam as atividades ceiu- lares dos substratos em vários contextos biológicos. Além das funções nos tecidos/órgãos normais, muitas proteínas quinase também desempenham papeis mais especializados em uma série de doenças humanas incluindo câncer. Um subconjunto de proteínas quinase (também chamado de proteínas quinase oncogênicas), quando desreguladas, pode levar a formação e crescimento de tumor, e ainda contribuir para a manutenção e progressão do tumor (Blume-Jensen P et al, Nature 2001, 411(6835):355-365). Até aqui, as proteínas quinase oncogênicas representam um dos maiores e mais atrativos grupos de proteínas alvos para intervenção de câncer e desenvolvimento de drogas.
A família Janus Quinase (JAK) desempenha um papel na regulação dependente de citocina da proliferação de células envolvidas em resposta imune. Atualmente, há quatro membros conhecidos da família JAK: JAK1 (também conhecida como Janus quinase-1), JAK2 (também conhecida como Janus quinase-2), JAK3 (também conhecida como Janus quinase, leucócito; JAKL; L-JAK e Janus quinase-3) e TYK2 (também conhecida como proteína tirosina quinase 2). As proteínas JAK variam em tamanho de 120 a 140 kDa e compreendem sete domínios de homologia JAK conservada (JH); um destes é um domínio funcional de quinase catalítica, e outro é um domínio pseudoquinase potencialmente servindo a uma função regulató- ria e/ou servindo como um sítio de ancoragem para STATs (Scott, Godshall et al. 2002, supra).
A transdução de sinal de bloqueio no nível de JAK quinase mantém a promessa para o desenvolvimento de tratamentos para cânceres humanos. A inibição de JAK é ainda imaginada como tendo benefícios terapêuticos em pacientes que sofrem de distúrbios imunes de pele como psorí- ase e sensibilidade de pele. Assim, inibidores de Janus quinase ou quinases relacionadas são amplamente procurados e vários publicações reportam classes efetivas de compostos. Por exemplo, certos inibidores de JAK, in- cluindo(R)-3-(4-(7H-pirrolo[2,3-d]pirimidÍn-4-il)-1 H-pirazol-1 -il)-3-ciclopentil- propanonitrila mostrados abaixo, são reportados em US 11/637.545 (US 2007/0135461), depositado em 12 de dezembro de 2006; US 12/138.082 (US 2009/0181959), depositado em 16 de julho de 2009; e certos metabolites de Composto I são reportados em US 12/137.883 (US 2008/0312258), depositado em 12 de junho de 2008, cada um dos quais está incorporado aqui como referência em sua totalidade.
Assim, agentes novos ou melhorados que inibem quinases, tal como Janus quinase, são continuamente necessários para o desenvolvimen- to de novos e mais efetivos medicamentos para tratar o câncer e outras doenças. Os metabolites, composições e métodos descritos aqui são direcionados para estas necessidades e outras finalidades.
A presente invenção fornece, entre outros, derivados hidroxila, ceto, e glicuronida de um composto de Fórmula I: ou um sal farmacêutica mente aceitável do mesmo.
ou um sal farmacêutica mente aceitável do mesmo.
Assim, em um aspecto, é fornecido aqui um composto de Fór ou um sal farmaceuticamente aceitável do mesmo, em que: grupos n C-H são cada qual independentemente substituídos por C-OH; ou, um grupo CH2 é independentemente substituído por C=O; ou, um grupo C-H é substituído por:
ou um sal farmaceuticamente aceitável do mesmo, em que: grupos n C-H são cada qual independentemente substituídos por C-OH; ou, um grupo CH2 é independentemente substituído por C=O; ou, um grupo C-H é substituído por:  dois grupos C-H são cada qual independentemente substituídos por C-OH e um grupo C-H é substituído por:
dois grupos C-H são cada qual independentemente substituídos por C-OH e um grupo C-H é substituído por:  n é 1, 2, 3, ou 4; contanto que o composto não seja selecionado de: 3-(4-(7H-pirrolo[2,3-d]pírimidin-4-íl)-1 H-pirazol-1-il)-3-(3-hidroxici- 5 clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidÍn-4-il)-1 H-pirazol-1-il)-3-(2-hidroxíci- clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-3-(3-oxociclo- pentil)propanonitrila; 10 e sais farmaceuticamente aceitáveis do mesmo.
n é 1, 2, 3, ou 4; contanto que o composto não seja selecionado de: 3-(4-(7H-pirrolo[2,3-d]pírimidin-4-íl)-1 H-pirazol-1-il)-3-(3-hidroxici- 5 clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidÍn-4-il)-1 H-pirazol-1-il)-3-(2-hidroxíci- clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-3-(3-oxociclo- pentil)propanonitrila; 10 e sais farmaceuticamente aceitáveis do mesmo.
Em uma modalidade de Fórmula I, o átomo de carbono alfa para o grupo ciano não é substituído por um C-OH ou Grupo C=O; e o átomo de carbono beta para o grupo ciano não é substituído 15 por um grupo C-OH.
Em outra modalidade do composto, grupos n C-H são cada qual independentemente substituídos por C-OH. Em outra modalidade, n é 1. Ainda em outra modalidade, n é 2. Ainda em outra modalidade, n é 3. Em outra modalidade, n é 4.
Em outra modalidade de Fórmula I, um grupo CH2 é independen temente substituído por C=O. Ainda em outra modalidade, um grupo C-H é substituído por
Em outra modalidade, um grupo C-H saturado é substituído por CO2H OJTOH Ainda em outra modalidade, dois grupos C-H são cada qual independentemente substituídos por C-OH e um grupo C-H são substituídoa por:
Ainda em outra modalidade, dois grupos C-H são cada qual independentemente substituídos por C-OH e um grupo C-H são substituídoa por: 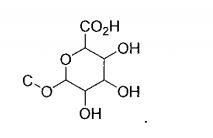
A presente invenção ainda fornece composições compreenden- dos compostos descritos aqui, ou um sal farmaceuticamente aceitável do mesmo, e pelo menos um carreador farmaceuticamente aceitável.
A presente invenção ainda fornece métodos para modular uma atividade de JAK compreendendo contatar JAK com certos compostos descritos aqui, ou sal farmaceuticamente aceitável do mesmo. A presente invenção ainda fornece métodos de tratar uma doença em um paciente, compreendendo administrar ao paciente uma quantidade terapeuticamente efetiva de certos compostos descritos aqui, ou sal farmaceuticamente aceitável do mesmo. Em uma modalidade particular, a doença é associada com atividade de JAK. Tais doenças incluem, por exemplo, rejeição de enxerto heterólogo ou doença enxerto versus hospedeiro. A doença pode ainda ser uma doença autoimune, incluindo, entre outras, um distúrbio de pele, escle- rose múltipla, artrite reumatoide, artrite juvenila, diabetes tipo I, lúpus, doença intestinal inflamatória, Doença de Crohn, miastenia gravis, nefropatias de imunoglobulina, miocardite, ou doença da tireoide autoimune. A doença autoimune pode ainda ser distúrbio de pele bolhoso, por exemplo, pênfigo vulgar (PV) ou penfigoide bolhoso (BP). O distúrbio de pele pode ser dermatite atópica, psoríase, sensibilidade na pele, irritação na pele, rash cutâneo, dermatite de contato ou sensibilidade de contato alérgico.
Em outra modalidade, a doença é uma doença virai. Exemplos de doenças virais que podem ser tratadas pelos compostos descritos aqui incluem vírus de Epstein Barr (EBV), Hepatite B, Hepatite C, HIV, HTLV 1, vírus da Varicela-Zoster (VZV) ou Papiloma vírus humano (HPV).
Em outra modalidade, a doença é câncer, por exemplo, um tumor sólido. O câncer a ser tratado pode ser câncer de próstata, câncer renal, câncer hepático, câncer de mama, câncer pulmonar, câncer de tireoide, Sarcoma de Kaposi, doença de Castleman ou câncer pancreático. Em uma modalidade particular, o câncer é câncer de próstata. O câncer pode ser hematológico. O câncer pode ser ainda um linfoma, leucemia, ou mieloma múltiplo. Em outra modalidade, o câncer é um câncer de pele, por exemplo, linfoma de célula T cutâneo ou linfoma de célula B cutâneo. Em outra modalidade, o câncer é mieloma múltiplo. A doença a ser tratada pode ser uma fadiga resultante de ou associada com câncer, ou anorexia ou caquexia resultante de ou associada com câncer.
Em outra modalidade, a doença a ser tratada é um distúrbio mieloproliferativo, por exemplo, policitemia vera (PV), trombocitemia essencial (ET), metaplasia mieloide com mielofibrose (MMM), leucemia mieloide crônica (CML), leucemia mielomonocítica crônica (CMML), síndrome hipere- osinofílica (HES), ou doença de mastócito sistêmica (SMCD).
Em outra modalidade, a doença a ser tratada é uma doença inflamatória. A doença inflamatória pode ser uma doença inflamatória dos olhos, por exemplo, irite, uveíte, esclerite ou conjuntivite. Uma doença inflamatória pode ser uma doença inflamatória do trato respiratório, por exemplo, o trato respiratório superior ou o trato respiratório inferior. A doença inflamatória pode ser uma miopatia ou miopatia inflamatória ou miocardite.
Em outra modalidade, a doença é reperfusão isquêmica ou relacionada a um evento isquêmico.
A presente invenção fornece, entre outros, derivados hidroxila, ceto, e glicuronida de 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-ÍI)~1 H-pirazol-1 -il)-3- ciclopentilpropanonitrila. Em algumas modalidades, o composto é um metabolite do composto I. Em algumas modalidades, o composto é um metabolite ativo que pode modular a atividade de uma ou mais JAKs e pode ser útila, por exemplo, no tratamento de doenças associadas com expressão ou atividade de JAK. Em algumas modalidades, o nível de um composto metabolite descrito aqui é medido e perfilado em para auxiliar o médico no ajuste dos níveis de dosagem do composto de Fórmula I.
Assim, a presente invenção fornece um composto de Fórmula I: z-CN ou um sal farmaceuticamente aceitável do mesmo, em que: n grupos C-H são cada qual independentemente substituído por C-OH; ou, um grupo CH2 é independentemente substituído por C=O; ou, um grupo C-H é substituído por:
ou um sal farmaceuticamente aceitável do mesmo, em que: n grupos C-H são cada qual independentemente substituído por C-OH; ou, um grupo CH2 é independentemente substituído por C=O; ou, um grupo C-H é substituído por:  dois grupos C-H são cada qual independentemente substituído por C-OH e um grupo C-H é substituído por:
dois grupos C-H são cada qual independentemente substituído por C-OH e um grupo C-H é substituído por:  n é 1, 2, 3, ou 4; contanto que o composto não seja selecionado de: 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-3-(3-hidroxici- clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-3-(2-hidroxici- clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 “il)-3-(3-oxociclo- pentil)propanonitrila; e sais farmaceuticamente aceitáveis do mesmo.
n é 1, 2, 3, ou 4; contanto que o composto não seja selecionado de: 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-3-(3-hidroxici- clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-3-(2-hidroxici- clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 “il)-3-(3-oxociclo- pentil)propanonitrila; e sais farmaceuticamente aceitáveis do mesmo.
Em algumas modalidades, n grupos C-H são cada qual independentemente substituídos por C-OH. Em algumas modalidades, n é 1. Em al- gumas modalidades, n é 2. Em algumas modalidades, n é 3. Em algumas modalidades, n é 4.
Em algumas modalidades, um grupo CH2 é independentemente substituído por C=O. Em algumas modalidades, um grupo C-H é substituído por CO2H Em algumas modalidades, um grupo C-H saturado é substituído por
Em algumas modalidades, um grupo C-H saturado é substituído por 
Em algumas modalidades, dois grupos C-H são cada qual índe- pendentemente substituído por C-OH e um grupo C-H é substituído por:
Em algumas modalidades: o átomo de carbono alfa para o grupo ciano não é substituído CO2H oArOH C\T^yX)H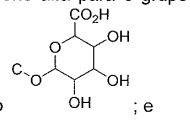 por um C-OH, C=O, ou grupo OH ; e o átomo de carbono beta para o grupo ciano não é substituído
por um C-OH, C=O, ou grupo OH ; e o átomo de carbono beta para o grupo ciano não é substituído  por um C-OH ou grupo 0H
por um C-OH ou grupo 0H
Em algumas modalidades: o átomo de carbono alfa para o grupo ciano não é substituído por um C-OH ou Grupo C=O; e o átomo de carbono beta para o grupo ciano não é substituído por um grupo C-OH.
Em outra modalidade, a presente invenção fornece um composto, caracterizado pelo fato de que tem a Fórmula II: ou um sal farmaceuticamente aceitável do mesmo, em que: 1, 2, 3, ou 4 de carbonos a, b, c, d, e, f, g, h, i, j, k ou m são cada um independentemente substituídod por OH; ou, ”10 um dos carbonos h, i, j, k, ou m é índependentemente substituído por =0; ou, um dos carbonos a, b, c, d, e, f, g, h, i, j, k ou m é substituído por:
ou um sal farmaceuticamente aceitável do mesmo, em que: 1, 2, 3, ou 4 de carbonos a, b, c, d, e, f, g, h, i, j, k ou m são cada um independentemente substituídod por OH; ou, ”10 um dos carbonos h, i, j, k, ou m é índependentemente substituído por =0; ou, um dos carbonos a, b, c, d, e, f, g, h, i, j, k ou m é substituído por:  15 dois dos carbonos a, b, c, d, e, f, g, h, i, j, k ou m são cada um independentemente substituídos por OH e:
15 dois dos carbonos a, b, c, d, e, f, g, h, i, j, k ou m são cada um independentemente substituídos por OH e:  contanto que o composto não seja selecionado de: 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-3-(3-hidroxici- 20 clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-3-(2-hid roxici- clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-3-(3-oxociclo- pentil)propanonitrila; e sais farmaceuticamente aceitáveis dos mesmos.
contanto que o composto não seja selecionado de: 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-3-(3-hidroxici- 20 clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-3-(2-hid roxici- clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-3-(3-oxociclo- pentil)propanonitrila; e sais farmaceuticamente aceitáveis dos mesmos.
Em algumas modalidades de Fórmula II, 1, 2, 3, ou 4 de carbonos a, b, c, d, e, f, g, h, i, j, k ou m são cada um independentemente substituído por OH. Em algumas modalidades, um dos carbonos a, b, c, d, e, f, g, h, i, j, k ou m é substituído por OH. Em algumas modalidades, 2 de carbonos a, b, c, d, e, f, g, h, i, j, k ou m são cada um independentemente substituído por OH. Em algumas modalidades, 3 de carbonos a, b, c, d, e, f, g, h, i, j, k ou m são cada um independentemente substituído por OH. Em algumas modalidades, 4 de carbonos a, b, c, d, e, f, g, h, i, j, k ou m são cada um independentemente substituído por OH. Em algumas modalidades, um dos carbonos h, i, j, k, ou m é substituído por =0.
Em algumas modalidades, um dos carbonos a, b, c, d, e, f, g, h, i, j, k ou m é substituído por: CO2H °H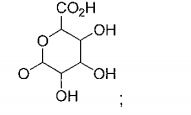 Em algumas modalidades, um dos carbonos f, g, h, i, j, k ou m é substituído por:
Em algumas modalidades, um dos carbonos f, g, h, i, j, k ou m é substituído por: 
Em algumas modalidades, dois dos carbonos a, b, c, d, e, f, g, h, i, j, k ou m são cada um independentemente substituídos por OH e um dos carbonos a, b, c, d, e, f, g, h, i, j, k ou m é substituído por: CO2H
Em algumas modalidades: o carbono m não é substituído por um grupo OH, =0, ou grupo o carbono f não é substituído por um grupo OH ou grupo CO2H
o carbono f não é substituído por um grupo OH ou grupo CO2H 
Em algumas modalidades: o carbono m não é substituído por um grupo OH ou =0; e o carbono f não é substituído por um grupo OH.
Cada uma das modalidades acima mencionadas assume que as regras para a valência apropriada são cumpridas. Em algumas modalidades, o composto é selecionado de:
Ácido 6 (3 (1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazoí-1-il)-2- cianoetil)ciclopentilóxí)-3,4,5-tri-hidroxitetra-hidr-2H-piran-2-carboxílico; 3-(4-(7 H-pirrolo[2,3-d] p i ri m id i n-4-i I)-1 H-pirazoM -il)-3-(1,2-di-h i- droxic!c!openf:!)propanonitrila; e 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-iI)-1 H-pirazol-1 -il)-3-(1 -hidroxici- clopentil)propanonitrila; ou um sal farmaceuticamente aceitável dos anteriores.
Em algumas modalidades, o composto é selecionado de: 3-(4-(7 H-pirrolo[2,3-d]pirimid in-4-íl)-1 H-pirazoM -il)-3-( 1 -hid roxici- clopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazoM-il)-3-ciclopentil- 3-hidroxipropanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimídin-4-il)-1 H-pirazol-1-il)-3-ciclopentií- 2-hidroxipropanonitrila; 3-ciclopentil-3-(5-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H- pirazoM-il)propanonitrila; 3-ciclopentil-3-(3-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(5-hidróxí-7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(6-hid róxi-7H-pirrolo[2,3-d]pirimid in-4-il)-1 H- pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(2-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H- pirazol-1-il)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazoM -il)-3-( 1,2-di-hi- droxiciclopentil)propanonitrila; 3-ciclopentil-3-(4-(5,6-di-hidróxi-6,7-di-hidro-5H-pirroio[2,3-d]pi- rimidin-4-il)-1 H-pirazol-1-il)propanonitπla; e
Ácido 6-(4-(1 -(2-ciano-1 -ciclopentiletil)-1 H-pirazol-4-il)-7H-pirro- lo[2,3-d]pirimidiri-7-il)-3,4,5-tri-hidroxitetra-hidr-2H-piran-2-carboxi[ico; ou um sal farmaceuticamente aceitável dos anteriores.
Em algumas modalidades, os compostos descritos aqui podem incluir os compostos mostrados nos quadros abaixo 1 a 7, e enantiômeros, diastereoisômeros, e racematos dos mesmos.


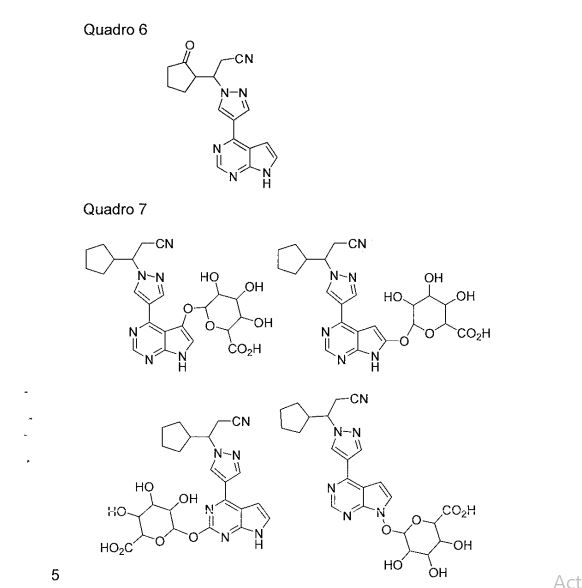
Certos metabolites são indicados na Tabela 1 abaixo. As estru turas se destinam a incluir todos os possíveis estereoisômeros.
 * Os números em parênteses referem-se aos números dos compostos na Tabela 2 (infra)
* Os números em parênteses referem-se aos números dos compostos na Tabela 2 (infra)
Os compostos descritos aqui são metabólitos de (R)-3-(4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-3-ciclopentilpropanonÍtrila. Os me- 5 tabólitos da invenção foram isolados de amostras de urina de humanos, ratos ou cães coletadas a partir de estudos de farmacocinética e toxicociné- tica do inibidor JAK (R)-3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-3- ciclopentilpropanonitrila (Composto I). Certos metabólitos podem ser inibidores de JAK, e têm propriedades vantajosas relacionadas às frações livres significativamente maiores e estabilidade metabólica maior em microssomas humanos comparada com o Composto I. Os presentes metabolites podem desejavelmente ter uma meia-vida de eliminação maior em humanos do que o Composto I.
Em algumas modalidades, os metabolites da invenção são subs tancialmente isolados. Por “substancialmente isolado” entende-se que o composto é pelo menos parcialmente ou substancialmente separado do ambiente no qual este é formado ou detectado. A separação parcial pode incluir, por exemplo, uma composição enriquecida no composto da invenção.
A separação substancial pode incluir composições contendo pelo menos cerca de 50%, pelo menos cerca de 60%, pelo menos cerca de 70%, pelo menos cerca de 80%, pelo menos cerca de 90%, pelo menos cerca de 95%, pelo menos cerca de 97%, ou pelo menos cerca de 99% em peso do metabolite.
A presente invenção ainda inclui sais farmaceuticamente acei táveis dos compostos descritos aqui. Conforme usado aqui, “sais farmaceuti-camente aceitáveis” referem-se aos derivados dos compostos revelados em que o composto parente é modificado pela conversão de uma fração de ácido ou base existente em sua forma de sal. Exemplos de sais farmaceutica- 20 mente aceitáveis incluem, entre outros, sais de ácidos minerais ou orgânicos de resíduos básicos como aminas; sais alcalinos ou orgânicos de resíduos ácidos como ácidos carboxílicos; e semelhantes. Os sais farmaceuticamente aceitáveis da presente invenção incluem os sais não tóxicos convencionais do composto parente formado, por exemplo, de ácidos não tóxicos orgânicos ou inorgânicos. Os sais farmaceuticamente aceitáveis da presente invenção podem ser sintetizados a partir do composto parente que contém uma fração básica ou ácida por métodos químicos convencionais. Geralmente, ditos sais pode ser preparados pela reação de formas livres de ácidos ou bases destes compostos com uma quantidade estequiométrica da base ou ácido apropria- do em água ou em um solvente orgânico, ou em uma mistura dos dois; geralmente, meio não aquoso como éter, acetato de etila, etanol, isopropanol, ou acetonitrila são preferenciais. Listas de saís apropriados são encontrados em Remington’s Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, p. 1418 e Journal of Pharmaceutical Science, 66, 2 (1977), cada um dos quais está incorporado aqui como referência em sua totalidade.
A frase “farmaceuticamente aceitável” é empregada aqui para referir-se a estes compostos, materiais, composições, e/ou formas de dosagem que estão, dentro do escopo do julgamento clínico, apropriado para uso em contato com os tecidos de humanos e animais sem toxicidade excessiva, irritação, resposta alérgica, ou outros problemas ou complicações, proporcional com uma razoável relação benefício/risco.
Os compostos descritos aqui são assimétricos (por exemplo, contendo um ou mais estereocentros). Todos estereoisômeros, como enan- tiômeros e diastereoisômeros, são pretendidos, salvo se de outra forma indicado. Os métodos sobre como preparar formas opticamente ativas a partir de materiais de partida opticamente ativos são conhecidos na técnica, como por resolução de misturas racêmicas ou por síntese estereoseletiva.
Os compostos descritos aqui ainda incluem todos os isótopos de átomos que ocorrem nos metabólitos. Os isótopos incluem aqueles átomos contendo o mesmo número atômico, mas diferentes números de massa. Por exemplo, isótopos de hidrogênio incluem trítio e deutério. Os compostos podem ainda incluir solvatos e hidratos dos compostos ou formas de sal.
O termo “composto,” conforme usado aqui pretende incluir todos os estereoisômeros, isômeros geométricos, tautômeros, e isótopos das estruturas representadas.
Os compostos descritos aqui, incluindo sais dos mesmos, podem ser preparados usando técnicas de síntese orgânica e podem ser sintetizados de acordo com qualquer número de possíveis vias sintéticas.
As reações para preparação dos compostos descritos aqui podem ser conduzidas em solventes apropriados que podem ser prontamente selecionados por um especialista na técnica de síntese orgânica. Solventes apropriados podem ser substancialmente não reativos com os materiais de partida (reagentes), os intermediários ou produtos em temperaturas nas quais as reações são conduzidas, por exemplo, temperaturas que podem variar desde a temperatura de congelamento do solvente até a temperatura de ebulição do solvente. Certa reação pode ser conduzida em um solvente ou 5 uma mistura de mais do que um solvente. Dependendo da etapa da reação particular, solventes apropriados para uma etapa particular da reação podem ser selecionados pelos especialistas na técnica.
A preparação dos compostos descritos aqui pode envolver a proteção e desproteção de vários grupos químicos. A necessidade para pro- 10 teção e desproteção, e a seleção de grupos de proteção apropriados, pode ser prontamente determinada por um especialista na técnica. A química de grupos de proteção pode ser encontrada, por exemplo, em T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd. Ed., Wiley & Sons, Inc., New York (1999), que está aqui incorporada como referência em 15 sua totalidade.
As reações podem ser monitoradas de acordo com qualquer método apropriado conhecido na técnica. Por exemplo, a formação de produto pode ser monitorada por meios espectroscópicos, como espectroscopia de ressonância magnética nuclear (por exempio, 1H ou 13C), espectroscopia de 20 infravermelho, espectrofotometria (por exemplo, UV visível), ou espectrome- tria de massa, ou por cromatografia como cromatografia líquida de alta performance (HPLC) ou cromatografia de camada fina.
Os compostos descritos aqui podem ser preparados de acordo com várias vias preparatórias conhecidas na literatura. Por exemplo, os com- 25 postos descritos aqui podem ser feitos por processes análogos aos descritos em US 11/637.545 (US 2007/0135461), depositado em 12 de dezembro de 2006; US 12/138.082 (US 2009/0181959), depositado em 16 de julho de 2009; e US 12/137.883 (US 2008/0312258), depositado em 12 de junho de 2008, cada um dos quais está incorporado aqui como referência em sua 30 totalidade. Exemplos de métodos sintéticos para preparação dos compostos descritos aqui são fornecidos nos Esquemas abaixo.
Conforme mostrado no Esquema 1, a síntese do composto 32 começa com 4-cloro pirrolopírimidina S1. O acoplamento de Suzuki de S1 com o pirazol boronato S2 em condições básicas na presença de catalisador de Pd(0) pode fornecer o composto tricíclico S3 que é subsequentemente convertido seletivamente ao composto de iodo S4 usando N-iodosuccini- mida. O composto de iodo S4 é transformado no acetato correspondente S5. O tratamento de S5 com brometo de hidrogênio 4M em ácido acético fornece S6 com a oxidação desejada no anel pirrol junto com a desproteção do grupo metóxi ao hidroxil livre. O grupo ciano que foi hidrolisado a amida sob estas condições é reinstalado em 32 pelo tratamento de S6 com cloreto de tricloroacetil e trietilamina para efetuar a desidratação.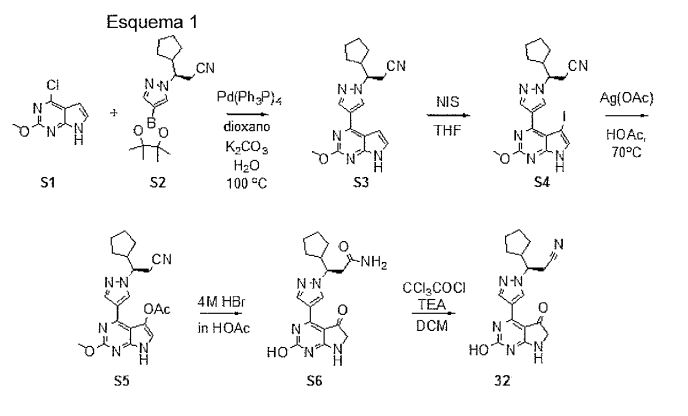
O composto mono-hidroxilado 42 (metabólito 8) foi relatado em US 12/137.883, depositado em 12 de junho de 2008. Ainda, compostos contendo um grupo hidroxil no anel ciclopentila podem ainda ser sintetizados como no Esquema 2. Por exemplo, o racêmico comercialmente disponível S14 no qual o álcool é protegido seguid por redução de éster carboxílico ao aldeído seguido por uma olefinação do aldeído para fornecer S17. A adição do conjugado de S9 a S17 em condições básicas fornece S18 do qual os grupos de proteção no álcool e anel pirrol são removidos sequencialmente para fornecer uma mistura de diastereoisômeros que são então separados usando várias colunas quirais de HPLC para fornecer os quatro diastereoisô- meros de S19. Em algumas modalidades, a presente invenção fornece um processo para produzir o composto 42, ou um sal farmaceuticamente aceitá vel do mesmo, compreendendo uma ou mais das etapas no Esquema 2.
Outro exemplo de composto mono-hidroxilado 36 é obtido pela sequência mostrada no Esquema 3. O cianohidrin protegido S31 pode ser reduzido com DIBAL ao aldeído correspondente seguido por olefinação com 10 o fosfonato como S7 para fornecer o crotontirila S33. A adição do conjugado de S9 a S33 em condições básicas fornece S34 do qual o grupo de proteção no anel pirrol é removido para fornecer uma mistura de diastereoisômeros que pode, então, ser separada usando várias colunas quirais de HPLC para fornecer os estereoisômeros individuais de 36.
Os metabolites di-hidroxilados podem ser obtidos de modo se-melhante como mostrado no Esquema 4: ciclopenteno carboxaldeído S6A 5 pode ser tratado diretamente com o ileto S7 para gerar o derivado crotonitrila S8. A nitrila S8, então, pode ser reagida com o pirazol S9 na presença de uma base como DBU para gerar S10 como uma mistura de diastereoisô- meros, que pode ser di-hidroxiiado com tetróxido de ósmio para fornecer os cis-álcoois CÍS-S12A, após a remoção do grupo SEM. Os estereoisômeros 10 individuais desta mistura (cis-S12A) podem ser separados por cromatografia quiral para gerar os álcoois enantiomericamente puros. O trans-S12A pode ser obtido por primeiro epoxidação da oiefina com m-CPBA seguido por abertura do epóxido em condições ácidas. Os estereoisômeros individuais desta mistura (trans-S12A) podem ser separados por cromatografia quiral 15 para gerar os álcoois enantiomericamente puros. As mesmas vias sintéticas podem ser adaptadas para obter os isoméricos S12B e S12C substituindo o aldeído inicial S6A com S6B e S6C.
Uma combinação dos métodos descritos acima junto com aqueles revelados no Pedido de Patente US 11/637.545, depositado em 12 de dezembro de 2006; e no Pedido de Patente US 12/137.883, depositado em 12 de junho de 2008, que são incorporados aqui em suas totalidades, pode ser utilizada para obter os compostos tri-hidróxi da invenção. Por exemplo, compostos hidróxi e di-hidróxi descritos aqui podem ser oxidados sob condições de oxidação de Swern descritas no Pedido de Patente US 12/137.883, depositado em 12 de junho de 2008. As glicuronidas de metabólitos contendo grupos hidroxil podem ser sintetizadas de acordo com o procedimento de Suzuki et al. (Bioorg. Med. Chem. Lett. (1999), 9(5), 659-662 que está incorporado aqui em sua totalidade).
Certos compostos descritos aqui podem modular a atividade de uma ou mais Janus quinase (JAKs). O termo “modular” pretende referir-se a uma capacidade de aumentar ou diminuir a atividade de um ou mais membros da família JAK de quinases. Assim, certos compostos descritos aqui podem ser usados nos métodos de modulação de uma JAK ao contatar a JAK com qualquer um ou mais dos compostos ou composições desceritos aqui. Em algumas modalidades, certos compostos podem atuar como inibidores de uma ou mais JAKs. Em algumas modalidades, certos compostos descritos aqui podem atuar para estimular a atividade de uma ou mais JAKs. Em outras modalidades, certos compostos podem ser usados para modular a atividade de uma JAK em um indivíduo em necessidade de modulação do receptor por administração de uma quantidade modulante de um composto da invenção. JAKs as quais os compostos ligam e/ou modulam podem incluir qualquer membro da família JAK. Em algumas modalidades, a JAK é JAK1, JAK2, JAK3 ou TYK2. Em algumas modalidades, a JAK é JAK1 ou JAK2. Em algumas modalidades, a JAK é JAK2. Em algumas modalidades, a JAK é JAK3.
Em algumas modalidades, compostos ativos podem ser seletivos. Por “seletivo” entende-se que o composto se liga a ou inibe uma JAK com maior afinidade ou potência, respectivamente, comparada a pelo menos outra JAK (por exemplo, inibidores seletivos de JAK1 ou JAK2 sobre JAK3 e/ou TYK2). Em algumas modalidades, seletivo significa inibição seletiva de JAK2 (por exemplo, over JAK1, JAK3 e TYK2). Sem querer se ligar a qualquer teoria, porque os inibidores de JAK3 podem levar a efeitos imunossu- pressores, um composto que é seletivo para JAK2 sobre JAK3 e que é útil no tratamento de câncer (como mieloma múltiplo, por exemplo) pode oferecer a vantagem adicional de conter menores efeitos colaterais imunossu- pressores. A seletividade pode ser pelo menos cerca de 5 vezes, pelo menos cerca de 10 vezes, pelo menos cerca de 20 vezes, pelo menos cerca de 50 vezes, pelo menos cerca de 100 vezes, pelo menos cerca de 200 vezes, pelo menos cerca de 500 vezes ou pelo menos cerca de 1000 vezes. A seletividade pode ser medida por métodos de rotina na técnica. Em algumas modalidades, a seletividade pode ser testada no Km de cada enzima. Em algumas modalidades, a seletividade para JAK2 sobre JAK3 pode ser determinada pela concentração celular de ATP.
Outro aspecto da presente invenção pertence aos métodos de tratar uma doença ou distúrbio associado a JAK em um indivíduo (por exemplo, paciente) ao adminsitrar ao indivíduo em necessidade de dito tratamento uma quantidade terapeuticamente efetiva ou dose de certos compostos descritos aqui ou uma composição farmacêutica dos mesmos. Uma doença associada a JAK pode incluir qualquer doença, distúrbio ou condição que é diretamente ou indiretamente ligada à expressão ou atividade de JAK, incluir superexpressão e/ou níveis de atividade anormais. Uma doença associada a JAK pode ainda incluir qualquer doença, distúrbio ou condição que pode ser prevenida, amenizada, ou curada por modulação de atividade de JAK.
Exemplos de doenças associadas a JAK incluem doenças que envolvem o sistema imune incluindo, por exemplo, rejeição de transplante de órgão (por exemplo, rejeição de enxerto homólogo e doença enxerto-hospedeiro).
Outros exemplos de doenças associadas a JAK incluem doenças autoimune como esclerose múltipla, artrite reumatoide, artrite juvenila, diabetes tipo I, lúpus, psoríase, doença intestinal inflamatória, colite ulce- rativa, Doença de Crohn, miastenia gravis, nefropatias de imunoglobulina, doença da tireoide autoimunes, e semelhantes. Em algumas modalidades, a doença autoimune é um distúrbio autoimune de pele bolhoso como pênfigo vulgar (PV) ou penfigoide bolhoso (BP).
Outros exemplos de doenças associadas com JAK incluem condições alérgicas como asma, alergias alimentares, dermatite atópica e rinite. Outros exemplos de doenças associadas a JAK incluem doenças virais como vírus de Epstein Barr (EBV), Hepatite B, Hepatite C, HIV, HTLV 1, vírus de Varicela-Zoster (VZV) e Papiloma vi rs humano (HPV).
Outros exemplos de doenças ou condições associadas a JAK in- cluem distúrbios de pele como psoríase (por exemplo, psoríase vulgaris), dermatite atópica, rash cutâneo, irritação na pele, sensibilidade na pele (por exemplo, dermatite de contato, dermatite alérgica de contato, ou sensibilida-de de contato alérgico). Por exemplo, certas substâncias incluindo alguns medicamentos quando topicamente aplicados pode causar sensibilidade na pele. Em algumas modalidades, co-administração ou administração sequen-cial de pelo menos um inibidor de JAK junto com o agente que causa sen-sibilização não desejada pode ser útil no tratamento de dita sensibilização indesejada ou dermatite. Em algumas modalidades, o distúrbio de pele é tra-tado por administração tópica de pelo menos um inibidor de JAK.
Em outras modalidades, a doença associada a JAK é câncer incluindo aqueles caracterizados por tumores sólidos (por exemplo, câncer de próstata, câncer renal, câncer hepático, câncer pancreático, câncer gástrico, câncer de mama, câncer pulmonar, cânceres de cabeça e pescoço, câncer de tireoide, glioblastoma, Sarcoma de Kaposi, doença de Castleman, melanoma etc.), cânceres hematológicos (por exemplo, linfoma, leucemia como leucemia linfoblástica aguda, leucemia mieloide aguda (AML) ou mieloma múltiplo), e câncer de pele como linfoma de célula T cutâneo (CTCL) e linfoma de célula 6 cutâneo. Exemplos de iinfoma de céiuta T cutâneos inciuem síndrome de Sezary e micose fungoide.
Doenças associadas a JAK podem ainda incluir aquelas caracterizadas por expressão de: JAK2 mutantes como aquelas contendo pelo menos uma mutação no domínio pseudo-quinase (por exemplo, JAK2V617F); JAK2 mutantes contendo pelo menos uma mutação foram do domínio pseudo-quinase; JAK1 mutantes; JAK3 mutantes; mutantes de receptor de eritropoietina (EPOR); ou expressão desregulada de CRLF2.
Doenças associadas a JAK podem ainda incluir distúrbios mielo- proliferativos (MPDs) como policitemia vera (PV), trombocitemia essencial (ET), metaplasia mieloide com mielofibrose (MMM), leucemia mieloide crônica (CML), leucemia mielomonocítica crônica (CMML), síndrome hipereosi- nofílica (HES), doença de mastócito sistêmica (SMCD), e semelhantes.
Outras doenças associadas com JAK incluem inflamação e doenças inflamatórias. Doenças inflamatórias incluem sarcoidose, doenças inflamatórias dos olhos (por exemplo, olho seco, irite, uveíte, esclerite, con- juntivite, ou doença relacionada), doenças inflamatórias do trato respiratório (por exemplo, trato respiratório superior incluindo nariz e sinos como rinite ou sinusite ou trato respiratório inferior incluindo bronquite, doença pulmonar obstrutiva crônica, e semelhantes), miopatia inflamatória como miocardite, e outras doenças inflamatórias. Outras doenças inflamatórias tratáveis por inibidores de JAK incluem síndrome de resposta inflamatória sistêmica (SIRS) e choque séptico.
Conforme usado aqui, “distúrbio de olho seco” pretende incluir os estados de doença resumidos em um relatório oficial recente do Dry Eye Workshop (DEWS), que definiu olho seco como “uma doença multifatorial das lágrimas e superfície ocular que resulta em sintomas de desconforto, distúrbio visual, e instabilidade de filme da lágrima com dano potencial à superfície ocular. É acompanhado por osmolaridade aumentada do filme de lágrima e inflamação da superfície ocular.” Lemp, “The Definition and Classi-fication of Dry Eye Disease: Report of the Definition and Classification Subcommittee of the International Dry Eye Workshop”, The Ocular Surface, 5(2), 75-92 Abrii de 2007, que está aqui incorporada como referência em sua totalidade. Em algumas modalidades, o distúrbio do olho seco é selecionado olho seco deficiente de lágrima aquosa (ADDE) ou distúrbio de olho seco evaporativo, ou combinações apropriadas dos mesmos. Em algumas modalidades, o distúrbio de olho seco é síndrome de Sjogren de olho seco (SSDE). Em algumas modalidades, o distúrbio de olho seco é uma síndrome de não Sjogren de olho seco (NSSDE).
Em outro aspecto, a presente invenção fornece um método de tratar conjuntivite, uveíte (incluindo uveíte crônica), corioidite, retinite, ciclite, esclierite, epiesclerite, ou irite; tratar inflamação ou dor relacionada ao transplante de córnea, LASIK (queratomileuse in situ assistida com laser), quera- tectomia fotorefrativa, ou LASEK (queratmileuse sub-epitelial assistida com laser); inibir perda de acuidade visual relacionada ao transplante de córnea, LASIK, queratectomia fotorefrativa, ou LASEK; ou inibir rejeição de trans- plante em um paciente em necessidade do mesmo, compreendendo administrar ao paciente uma quantidade terapeuticamente efetiva do composto da invenção, ou um sal farmaceuticamente aceitável do mesmo.
Inibidores de JAK podem ainda ser usados para tratar danos de reperfusão isquêmica ou uma doença ou condição relacionada a um evento isquêmico inflamatório como infarto ou parada cardíaca. Inibidores de JAK podem ainda ser usados para tratara anorexia, caquexia, ou fadiga como que resultante de ou associada com câncer. Inibidores de JAK podem ainda ser usados para tratar restenose, esclerodermite, ou fibrose. Inibidores de JAK podem ainda ser usados para tratar condições associadas com hipóxia ou astrogliose como, por exemplo, retinopatia diabética, câncer, ou neurode- generação. Vide, por exemplo, Dudley, A.C. et al. Biochem. J. 2005, 390(Pt 2):427-36 e Sriram, K. et al. J. Biol. Chem. 2004, 279(19): 19936-47. Epub 2 de março de 2004.
Inibidores de JAK podem ainda ser usados para tratar gota e tamanho de próstata aumentado devido a, por exemplo, hipertrofia benigna da próstata ou hiperplasia benigna da próstata.
Outras doenças associadas a JAK incluem doenças de reabsorção óssea como osteoporose, osteoartrite. A reabsorção óssea pode ainda estar associada com outras condições como desequilíbrio hormonal e/ou terapia hormonal, doença autoimune (por exemplo sarcoidose óssea), ou câncer (por exemplo mieloma). A redução de reabsorção óssea devido a inibidores de JAK pode ser cerca de 10%, cerca de 20%, cerca de 30%, cerca de 40%, cerca de 50%, cerca de 60%, cerca de 70%, cerca de 80%, ou cerca de 90%.
Conforme usado aqui, o termo “contatar” refere-se a colocar em contato as frações indicadas em um sistema in vitro ou um sistema in vivo. Por exemplo, “contatar” uma JAK com um composto inclui a administração de um composto da presente invenção a um indivíduo ou paciente, como um humano, contendo uma JAK, bem como, por exemplo, introduzir um composto em uma amostra contendo uma preparação celular ou purificada contendo a JAK.
Conforme usado aqui, o termo “indivíduo” ou “paciente,” usados de modo intercambiável, refere-se a qualquer anima), incluindo mamíferos, preferencialmente camundongos, ratos, outros roedores, coelhos, cães, gatos, suínos, gado, ovelha, cavalos, ou primatas e mais preferencialmente hu- 5 manos.
Conforme usado aqui, a frase “quantidade terapeuticamente efetiva” refere-se a uma quantidade de composto ativo ou agente farmacêutico que induz resposta biológica ou medicinal que está procurada em um tecido, sistema, animal, indivíduo ou humano por um pesquisador, veterinário, mé~ 10 dico ou outro profissional de saúde.
Conforme usado aqui, o termo “tratar” ou “tratamento” refere-se a um ou mais de (1) prevenir a doença; por exemplo, prevenir uma doença, condição ou distúrbio em um indivíduo que pode estar predisposto à doença, condição ou distúrbio, mas não experimentou ainda ou apresenta a patologia 15 ou sintomatologia da doença; (2) inibir a doença; por exemplo, inibir a doença, condição ou distúrbio em um indivíduo que está experimentando ou apresentando a patologia ou sintomatologia da doença, condição ou distúrbio; e (3) amenizar a doença; por exemplo, amenizar a doença, condição ou distúrbio em um indivíduo que está experimentando ou apresentando a patologia 20 ou sintomatologia da doença, condição ou distúrbio (ou seja, reverter a pato-logia e/ou sintomatologia) como diminuição da gravidade da doença.
Os níveis de metabolites em um paciente após a administração do composto I em um indivíduo podem ser medidos e perfilados. Esse perfil de metabolite pode então ser usado para ajustar os regimes de dosagens 25 (por exemplo, para administração do composto I) naquele indivíduo. Por exemplo, depuração mais rápida do composto I como mostrado pelos níveis de vários metabolites após certo período de tempo pode significar que as dosagens iniciais poderiam ser ajustadas para cima.
Um ou mais agentes farmacêuticos adicionais como, por exem plo, quimioterapêuticos, agentes anti-inflamatórios, esteroides, imunossu- pressores, bem como Bcr-Abl, Flt-3, RAF e inibidores de FAK quinase como, por exemplo, aqueles descritos em WO 2006/056399, ou outros agentes podem ser usados em combinação com certos compostos descritos aqui para tratamento de doenças distúrbios ou condições associados a JAK. O um ou mais agentes farmacêuticos adicionais podem ser administrados a um paciente simultaneamente ou sequencialmente.
Exemplos de quimioterápicos incluem inibidores de proteossoma (por exemplo, bortezomibe), talidomida, revlimide, e agentes que danificam o DNA como melfalan, doxorrubicina, ciclofosfamida, vincristina, etoposideo, carmustina, e semelhantes.
Exemplos de esteroides incluem corticosteroides como dexame- tasona ou prednisona. Exemplos de inibidores de Bcr-AbI incluem os compostos, e sais farmaceuticamente aceitáveis do mesmo, dos gêneros e espécies descritos na Patente US 5.521.184, WO 04/005281, e US 60/578.491.
Exemplos apropriados de inibidores de Flt-3 incluem compostos e seus sais farmaceuticamente aceitáveis, conforme revelado em WO 03/037347, WO 03/099771, e WO 04/046120.
Exemplos apropriados de inibidores de RAF incluem compostos, e seus sais farmaceuticamente aceitáveis, conforme revelado em 'WO 00/09495 e WO 05/028444.
Exemplos apropriados de inibidores de FAK incluem compostos, e seus sais farmaceuticamente aceitáveis, conforme revelado em WO 04/080980, WO 04/056786, WO 03/024967, WO 01/064655, WO 00/053595, eWO 01/014402.
Em algumas modalidades, um ou mais compostos descritos aqui podem ser usados em combinação com um ou mais outros inibidores de quinase incluindo imatinibe, particularmente para tratar pacientes resistentes a imatinibe ou outros inibidores de quinase.
Em algumas modalidades, um ou mais compostos podem ser usados em combinação com um quimioterápico no tratamento de câncer, como mieloma múltiplo, e pode melhorar a resposta ao tratamento comparada com a resposta de agente quimioterápico isolado, sem exacerbação de seus efeitos tóxicos. Exemplos de agentes farmacêuticos adicionais usados no tratamento de mieloma múltiplo, por exemplo, podem incluir, entre outros, melfalan, melfalan mais prednisona [MP], doxorrubicina, dexametasona, e Velcade (bortezomibe). Outros agentes adicionais usados no tratamento de mieloma múltiplo incluem inibidores de Bcr-Abl, Flt-3, RAF e FAK quinase. Efeitos aditivos ou sinergísticos são resultados desejáveis de combinação de um composto da presente invenção com um agente adicional. Além disso, a resistência de células de mieloma múltiplo aos agentes como dexametasona podem ser reversíveis no tratamento com um composto da presente invenção. Os gentes podem ser combinados com o inibidor de JAK em uma forma de dosagem única ou contínua, ou os agentes podem ser administrados simultaneamente ou sequencialmente como formas de dosagem separadas.
Em algumas modalidades, um corticosteroide como dexametasona é administrado a um paciente em combinação com pelo menos um inibidor de JAK onde a dexametasona é administrada intermitentemente em oposição a continuamente.
Em algumas outras modalidades, combinações de um ou mais compostos com outros agentes terapêuticos podem ser administradas a um pacientes antes, durante, e/ou após um transplante de medula óssea ou transplante de célula tronco.
Quando empregados como medicamentos, os compostos descritos aqui podem ser administrados na forma de composições farmacêuticas. Estas composições podem ser preparadas em um modo conhecido na técnica farmacêutica, e podem ser administrados por uma variedade de vias, dependendo se o tratamento local ou sistêmico é desejado e da área a ser tratada. Administração pode ser tópica (incluindo transdérmica, epidérmica, oftálmica e em membranas mucosas incluindo liberação intranasal, vaginal e retal), pulmonar (por exemplo, por inalação ou insuflação de pós ou aerossóis, incluindo por nebulizador; intratraqueal ou intranasal), oral ou parenteral. Em algumas modalidades, a composição é apropriada para administração oral. A administração parenteral inclui injeção ou infusão intravenosa, intraarterial, subcutânea, intraperitoneal, intramuscular; ou administração in-tracranial, por exemplo, intratecal ou intraventricular. Administração parenteral pode ser na forma de urn bolus único, ou pode ser, por exemplo, por uma bomba de perfusão contínua. Composições farmacêuticas e formulações para administração tópica podem incluir adesivos transdérmicos, pomadas, loções, cremes, géis, gotas, supositórios, sprays, líquidos e pós. Carreadores farmacêuticos convencionais, aquosos, pós ou bases oleosas, espessantes e semelhantes podem ser necessários ou desejáveis. Preservativos revestidos, luvas e semelhantes podem ser ainda úteis.
Esta invenção ainda inclui composições farmacêuticas que contêm, como o ingrediente ativo, um ou mais dos compostos descritos aqui em combinação com um ou mais carreadores farmaceuticamente aceitáveis (ex- cipientes). Ao produzir as composições da invenção, o ingrediente ativo é tipicamente misturado com um excipiente, diluído por um excipiente ou englobado dentro de dito carreador na forma de, por exemplo, uma cápsula, um sachê, papel, ou outro recipiente. Quando o excipiente serve como um diluente, este pode ser um material sólido, semissólido ou líquido, que atua como um veículo, carreador ou meio para o ingrediente ativo. Assim, as composições podem estar na forma de comprimidos, píiuias, pós, pastilhas, sanchês, caches, elixires, suspensões, emulsões, soluções, xaropes, aerossóis (como um sólido ou em um meio líquido), pomadas contendo, por exemplo, até 10% em peso do composto ativo, cápsulas gelatinosas moles e duras, supositórios, soluções injetáveis estéreis, e pós embalados estéreis.
Na preparação de uma formulação, o composto ativo pode ser moído para fomecer o tamanho de partícula apropriado antes da combinação com os outros ingredientes. Se o composto ativo é substancialmente insolúvel, este pode ser moído para um tamanho de partícula de menos do que 200 mesh. Se o composto ativo é substancialmente solúvel em água, o tamanho de partícula pode ser ajustado por moagem para fornecer uma distribuição substancialmente uniforme na formulação, por exemplo, cerca de malha 40.
O ingrediente ativo pode se moído usando procedimentos conhecidos de moagem como moagem úmida para obter um tamanho de partícula apropriada para formação de comprimido e para outros tipos de formulação. Preparações finamente divididas (nanoparticulados) do ingrediente ativo podem ser preparados por processos conhecidos na técnica, por exemplo, vide Pedido de Patente Internacional WO 2002/000196.
Alguns exemplos de excipientes apropriados incluem lactose, dextrose, sacarose, sorbitol, manitol, amidos, goma acácia, fosfato de cálcio, alginatos, tragacante, gelatina, silciato de cálcio, celulose microcristalina, po- livinilpirrolidona, celulose, água, xarope, e metil celulose. A formulação pode adicionalmente incluir: agentes lubrificantes como talco, estearato de magnésio, e óleo mineral; agentes molhantes; agentes emulsificantes e de suspensão; agentes preservativos como metil- e propilhidróxi-benzoatos; agentes adoçantes; e agentes flavorizantes. As composições da invenção podem ser formuladas de modo a fornecer liberação rápida, sustentada ou prolongada do ingrediente ativo após administração ao paciente ao empregar procedimentos conhecidos na técnica.
As composições podem ser formuladas em uma forma de dosagem unitária, cada dosagem contendo de cerca de 5 a cerca de 1000 mg (1 g), mais usuaimente cerca de 100 a cerca de 500 mg, do ingrediente ativo. O termo "formas de dosagem unitária" referem-se a unidades fisicamente discretas apropriadas como dosagens unitárias para sujeitos humanos e outros mamíferos, cada unidade contendo uma quantidade predeterminada de material ativo calculado para produzir o efeito terapêutico desejado, em associação com um excipiente farmacêutico apropriado.
Em algumas modalidades, as composições da invenção contêm de cerca de 5 mg a cerca de 50 mg do ingrediente ativo. Um especialista na técnica irá apreciar que isto realiza compostos ou composições contendo cerca de 5 mg a cerca de 10 mg, cerca de 10 mg a cerca de 15 mg, cerca de 15 mg a cerca de 20 mg, cerca de 20 mg a cerca de 25 mg, cerca de 25 mg a cerca de 30 mg, cerca de 30 mg a cerca de 35 mg, cerca de 35 mg a cerca de 40 mg, cerca de 40 mg a cerca de 45 mg, ou cerca de 45 mg a cerca de 50 mg do ingrediente ativo.
Em algumas modalidades, as composições da invenção contêm de cerca de 50 mg a cerca de 500 mg do ingrediente ativo. Um especialista na técnica apreciará que isto produz compostos ou composições contendo cerca de 50 mg a cerca de 100 mg, cerca de 100 mg a cerca de 150 mg, 5 cerca de 150 mg a cerca de 200 mg, cerca de 200 mg a cerca de 250 mg, cerca de 250 mg a cerca de 300 mg, cerca de 350 mg a cerca de 400 mg, ou cerca de 450 mg a cerca de 500 mg do ingrediente ativo.
Em algumas modalidades, as composições da invenção contêm de cerca de 500 mg a cerca de 1.000 mg do ingrediente ativo. Um espe- 10 cialista na técnica apreciará que isto produz compostos ou composições contendo cerca de 500 mg a cerca de 550 mg, cerca de 550 mg a cerca de 600 mg, cerca de 600 mg a cerca de 650 mg, cerca de 650 mg a cerca de 700 mg, cerca de 700 mg a cerca de 750 mg, cerca de 750 mg a cerca de 800 mg, cerca de 800 mg a cerca de 850 mg, cerca de 850 mg a cerca de 15 900 mg, cerca de 900 mg a cerca de 950 mg, ou cerca de 950 mg a cerca de 1.000 mg do ingrediente ativo.
O composto ativo pode ser efetivo sobre uma ampla faixa de do-sagens e é geralmente administrado em uma quantidade farmaceuticamente efetiva. Deve ser entendido, no entanto, que a quantidade do composto reai- 20 mente administrada irá geralmente ser determinada por um médico, de acordo com circunstâncias relevantes, incluindo a condição a ser tratada, a via de administração escolhida, o composto real administrado, a idade, peso e resposta do paciente individual, a gravidade dos sintomas do paciente, e semelhantes. Para preparar composições sólidas, tal como comprimidos, o ingrediente ativo principal é misturado com um excipiente farmacêutico para formar uma composição de pré-formulação contendo uma mistura homogênea de um composto da presente invenção. Quando referindo a estas composições de pré-formulação como homogêneas, o ingrediente ativo é tipica- 30 mente disperso uniformemente por toda a composição de modo que a composição seja prontamente subdividida em formas de dosagem unitárias igualmente efetivas como comprimidos, pílulas e cápsulas. Esta pré-formula- ção sólida é então subdividida em formas de dosagem unitárias do tipo descrito acima contendo de, por exemplo, cerca de 0,1 a cerca de 1000 mg do ingrediente ativo da presente invenção.
Os comprimidos ou pílulas da presente invenção podem ser revestidos ou de outra forma manipulados para fornecer uma forma de dosagem que obtém a vantagem de ação prolongada. Por exemplo, o comprimido ou pílula podem compreender um componente de dosagem interna e um de dosagem externa, o último estando na forma de um envelope sobre o anterior. Os dois componentes podem ser separados por uma camada entérica que serve para resistir a desintegração no estômago e permite que o componente do interior passe intacto no duodeno ou seja atrasado na liberação. Uma variedade de materiais pode ser usada para camadas entéricas ou revestimentos, como materiais incluindo um número de ácidos poli- méricos e misturas de ácidos poliméricos com ditos materiais como goma laca, álcool cetílico, e acetato de celulose.
As formas líquidas em que os compostos e composições podem ser incorporados para administração por via oral ou por injeção incluem soluções aquosas, xaropes apropriadamente flavorizados, suspensões aquosas ou oleosas, e emulsões fíavorizadas com óleos comestíveis como óleo de semente de algodão, óleo de gergelim, óleo de coco, ou óleo de amendoim, bem como elixires e veículos farmacêuticos semelhantes.
Composições para inalação ou insuflação incluem soluções e suspensões em solventes farmaceuticamente aceitáveis, aquosos ou orgânicos, ou misturas dos mesmos, e pós. As composições líquidas ou sólidas podem conter excipientes farmaceuticamente aceitáveis conforme descrito acima. Em algumas modalidades, as composições são administradas pela via oral ou respiratória nasal para efeito local ou sistêmico. As composições podem ser nebulizadas pelo uso de gases inertes. Soluções nebulizadas podem ser aspiradas diretamente a partir do dispositivo de nebulização ou o dispositivo de nebulização pode ser conectado a máscaras faciais, ou máquina de respiração de pressão positiva intermitente. Composições em solução, suspensão ou pó podem ser administradas por via oral ou nasal a partir de dispositivos que liberam a formulação de modo apropriado.
A quantidade do composto ou composição administrada a um paciente irá variar dependendo do que está sendo administrado, a finalidade da administração, tal como profilaxia ou terapia, o estado do paciente, o mo- 5 do de administração, e semelhantes. Em aplicações terapêuticas, as composições podem ser administradas a um paciente que já sobre da doença em uma quantidade suficiente para curar ou pelo menos parcialmente reprimir os sintomas da doença e suas complicações. Doses eficazes dependerão da condição da doença a ser tratada bem como o julgamento do médico 10 dependendo dos fatores como a gravidade da doença, a idade, peso e condição geral do paciente, e semelhantes.
As composições administradas a um paciente podem estar na forma de composições farmacêuticas descritas acima. Estas composições podem ser esterilizadas por técnicas convencionais de esterilização, ou po- 15 dem passar por filtração estéril. Soluções aquosas podem ser empacotadas para uso como tal, ou liofilizadas, a preparação liofilizada sendo combinada com um carreador estéril antes da administração. O pH das preparações de composto tipicamente irão estar entre 3 e 11, mais preferencialmente de 5 a 9 e mais preferencialmente de 7 a 8. Deve ser entendido que o uso de 20 alguns dos excipientes anteriores, carreadores ou estabilizadores irá resultar na formação de sais farmacêuticos.
As dosagens terapêuticas dos compostos podem variar de acordo com, por exemplo, o uso particular para o qual o tratamento é feito, o modo de administração do composto, a saúde e condição do paciente, e o jul- 25 gamento do médico. A proporção ou concentração de um composto em uma composição farmacêutica pode variar dependendo do número de fatores incluindo dosagem, características químicas (por exemplo, hidrofobicidade), e a via de administração. Por exemplo, os compostos podem ser fornecidos em uma solução de tampão fisiológico aquoso contendo cerca de 0,1 a 30 cerca de 10% p/v do composto para administração parenteral. Algumas faixas de doses típicas são de cerca de 1 μg/kg a cerca de 1 g/kg de peso corporal por dia. Em algumas modalidades, a faixa de dose é de cerca de 0.01 mg/kg a cerca de 100 mg/kg de peso corporal por dia. A dosagem é possivelmente dependente de ditas variáveis como o tipo e extensão da progressão da doença ou distúrbio, o estado geral de saúde do paciente particular, a eficácia biológica relativa do composto selecionado, formulação do excipien- te, e sua via de administração. Doses efetivas podem ser extrapoladas de curvas de dose-resposta derivadas de sistemas de teste de modelo in vitro ou animal.
As composições da invenção podem ainda incluir um ou mais agentes farmacêuticos adicionais como um quimioterápico, esteroide, composto anti-inflamatório, ou imunossupressor, exemplos dos quais estão listados acima.
Em algumas modalidades, o composto, ou sal farmaceuticamente aceitável do mesmo, é administrado como uma composição oftálmica. Assim, em algumas modalidades, os métodos compreendem administração do composto, ou sal farmaceuticamente aceitável do mesmo, e um carreador oftalmicamente aceitável. Em algumas modalidades, a composição oftálmica é uma composição líquida, composição semissóüda, inserto, filme, micropar- tículas ou nanopartículas.
Em algumas modalidades, a composição oftálmica é uma com-posição líquida. Em algumas modalidades, a composição oftálmica é uma composição semissólida. Em algumas modalidades, a composição oftálmica é uma composição tópica. As composições tópicas incluem, entre outras, composições líquidas e semissólidas. Em algumas modalidades, a composição oftálmica é uma composição tópica. Em algumas modalidades, a composição tópica compreende solução aquosa, uma suspensão aquosa, uma pomada ou um gel. Em algumas modalidades, a composição oftálmica é topicamente aplicada na frente doolho, sob a pálpebra superior, na pálpebra inferior e no cul-de-sac. Em algumas modalidades, a composição oftálmica é esterilizada. A esterilização pode ser conseguida por técnicas conhecidas como filtração esterilizante da solução ou por aquecimento da solução na ampola pronta para uso. A composição oftálmica da invenção pode ainda conter excipientes farmacêuticos apropriados para a preparação de formula ções oftálmicas. Exemplos de ditos excipientes são agentes de preservação, agentes tamponantes, agentes quelantes, agentes antioxidantes e sais para regulação da pressão osmótica.
Conforme usado aqui, o termo “carreador aceitável oftalmica- mente” refere-se a qualquer material que pode conter e liberar o composto, ou sal farmaceuticamente aceitável do mesmo, e que é compatível com o olho. Em algumas modalidades, o carreador oftalmicamente aceitável é água ou uma solução ou suspensão aquosa, mas também indica óleos como aqueles usados para proparar pomadas e matrizes de polímero como usadas em insertos oculares. Em algumas modalidades, a composição pode ser uma suspensão aquosa compreendendo o composto, ou sal farmaceuticamente aceitável do mesmo. Composições oftálmicas líquidas, incluindo pomadas e suspensões, podem ter uma viscosidade que é apropriada para a via de administração selecionada. Em algumas modalidades, a composição oftálmica tem uma viscosidade na faixa de cerca de 1.000 a cerca de 30.000 centipoise.
Em algumas modalidades, as composições oftálmicas podem ainda compreender um ou mais de surfactantes, adjuvantes, tampões, antioxidantes, ajustadores de tonicidade, preservativos (por exemplo, EDTA, BAK (cloreto de benzalcônio), clorito de sódio, perborato de sódio, poliqua- terium-1), espessantes ou modificadores de viscosidade (por exemplo, car- boximetil celulose, hidroximetil celulose, polivinil álcool, polietileno glícol, gli- col 400, propileno glicol, hidroximetil celulose, hidroxpropil-guar, ácido hialu- rônico, e hidroxipropil celulose) e semelhantes. Aditivos na formulação podem incluir, entre outros, cloreto de sódio, bicarbonato de sódio, ácido sórbi- co, meti! parabeno, propil parabeno, clorexidina, óleo de rícino, e perborato de sódio.
Composições oftálmicas aquosas (soluções ou suspensões) geralmente não contêm constituintes fisiologicamente ou oftalmicamente danosos. Em algumas modalidades, água purificada ou dionizada é usada na composição. O pH pode ser ajustado pela adição de qualquer ácido, base ou tampão fisiologicamente e oftalmicamente aceitável ajustador de pH para dentro da faixa de cerca de 5,0 a 8,5. Exemplos oftalmicamente apropriados de ácidos include acético, bórico, cítrico, láctico, fosfórico, clorídrico, e semelhantes, e exemplos de bases incluem hidróxido de sódio, fosfato de sódio, borato de sódio, citrato de sódio, acetato de sódio, lactato de sódio, trometa- mina, trishidroximetilamino-metano, e semelhantes. Sais e tampões incluem citrato/dextrose, bicarbonato de sódio, cloreto de amónio e misturas dos ácidos e bases acima mencionados.
Em algumas modalidades, os métodos que envolvem a formação ou suprimento de um depot do agente terapêutico em contato com a superfície externa do olho. Um depot refere-se a uma fonte de agente terapêutico que não é rapifamente removido por lágrimas ou outros mecanismos de eliminação do olho. Isto permite concentrações altas continuadas e sustentadas de agente terapêutico para estar presente no fluido na superfície externa do olho por uma única aplicação. Sem querer se ligar a qualquer teoria, acredita-se que a absorção e penetração podem depender da con- centraca da droga dissolvida e da duração do contato do tecido externo com o fluindo contendo a droga. Como a droga é removida por depuração do fluido ocular e/ou absorção no tecido do olho, mais droga é fornecida, por exemplo, dissolvida, no fluido ocular reabastecido a partir do depot. Assim, o uso de um depot pode mais facilmente carregar o tecido ocular para agentes terapêuticos mais insolúveis. Em algumas modalidades, o depot pode permanecer por até oito horas ou mais. Em algumas modalidades, as formas de depot oftálmico incluem, entre outras, suspensões poliméricas aquosas, pomadas, e insertos sólidos.
Em algumas modalidades, a composição oftálmica é uma pomada ou gel. Em alguma modalidade, a composição oftálmica é um veículo de liberação a base de óleo. Em algumas modalidades, a composição compreende uma base de petróleo ou lanolina a qual é adicionado o ingrediente ativo, geralmente como 0,1 a 2%, e excipientes. Bases comuns podem incluir, entre outras, óleo mineral, petrolato e combinações dos mesmos. Em algumas modalidades, a pomada é aplicada como uma fita sobre a pálpebra inferior.
Em alguma modalidade, a composição oftálmica é um inserto oftálmico. Em algumas modalidades, o inserto oftálmico é inerte biologicamente, macio, bio-degradável, viscoelástico, estável à esterilização após exposição a agentes terapêuticos, resistente a infeções de bactérias do ar, 5 biodegradável, biocompatível, e/ou viscoelástico. Em algumas modalidades, o inserto compreende uma matriz oftalmicamente aceitável, por exemplo, uma matriz de polímero. A matriz é tipicamente um polímero e o agente terapêutico é geralmente disperso neste ou ligado à matriz de polímero. Em algumas modalidades, o agente terapêutico pode ser lentamente liberado da 10 matriz por dissolução ou hidrólise da ligação covalente. Em algumas modalidades, o polímero é biodegradável (solúvel) e a taxa de dissolução do mesmo pode controlar a taxa de liberação do agente terapêutico disperso neste. Em outra forma, a matriz de polímero é um polímero biodegradável que quebra como por hidrólise para liberar o agente terapêutico ligado a este ou • 15 disperso neste. Em outras modalidades, a matriz e agente terapêutico podem ser circundados com um revestimento polimérico adicional para ainda controlada a liberação. Em algumas modalidades, o inserto compreende um polímero biodegradável como policaprolactona (PCL), um copolímero de eti- leno/acetato de vinila (EVA), poliaíquil cianoacrilato, poliuretano, um náilon, 20 ou poli (dl-lactide-co-glicolide) (PLGA), ou um copolímero de qualquer um destes. Em algumas modalidades, o agente terapêutico é disperso em um material de matriz ou disperso entre a composição de monômero usada para produzir o material de matriz antes da polimerização. Em algumas modalidades, a quantidade de agente terapêutico é de cerca de 0,1 a cerca de 25 50%, ou de cerca de 2 a cerca de 20%. Em outras modalidades, a matriz de polímero biodegradável é usada de modo que o inserto gasto não precise ser removido. Conforme o polímero biodegradável é degradado ou dissolvido, o agente terapêutico é liberado.
Em outras modalidades, o inserto oftálmico compreende um polí- 30 mero, incluindo, entre outros, aqueles descritos em Wagh, et al., “Polimers used in ocular dosage form and drug delivery systems”, Asian J. Pharm., páginas 12-17 (Jan. 2008), que está aqui incorporada como referência em sua totalidade. Em algumas modalidades, o inserto compreende um polímero selecionado de polivinilpirrolidona (PVP), um polímero ou copolímero de acrilato ou metacrilato (por exemplo, Eudragit® família de polímeros de Rohm ou Degussa), hidroximetil celulose, ácido poliacrílíco, poli(amidoami- na) dendrimeros, poli(dimetil siloxano), óxido de poíietileno, poli(lactide-co- glicolide), poli(2-hidroxietilmetacrilato), poli(vinil álcool), ou poli(propiieno fu- marato). Em algumas modalidades, o inserto compreende Gelfoam® R. Em algumas modalidades, o inserto é um conjugado de ácido poliacrílíco de 450 kDa-cisteína.
Em algumas modalidades, a composição oftálmica é um filme oftálmico. Polímeros apropriados para ditos filmes incluem, entre outros, aqueles descritos em Wagh, et al. (ibid), Em algumas modalidades, o filme é uma lente de contato mole, como aquelas feitas de copolímeros de N,N- dietilacrilamida e ácido metacrílico de ligação cruzada com etileneglícol di- metacrilato.
Em algumas modalidades, a composição oftálmica compreende microesferas ou nanopartículas. Em alguma modalidade, as microesferas compreendem gelatian. Em algumas modalidades, as microesferas são injetadas no segmento posterior do olho, no espaço coroidal, na esclera, intravi- teralmente ou sub-retinalmente. Em algumas modalidades, as microesferas ou nanopartículas compreendem um polímero incluindo, entre outros, aqueles descritos em Wagh, et al. (ibid), que está aqui incorporada como referência em sua totalidade. Em algumas modalidades, o polímero é quitosana, um ácido policarboxílico como ácido poliacrílíco, partículas de albumina, ésteres de ácido hialurônico, ácido poliitaconico, poli(butil)cianoacrilato, polica- prolactona, poli(isobutil)caprolactona, poli(ácido láctico -co-ácido glícolico), or poli(ácido láctico). Em algumas modalidades, as microesferas ou nanopartículas compreendem partículas de lipídio sólidas.
Em algumas modalidades, a composição oftálmica compreende uma resina de troca iônica. Em algumas modalidades, a resina de troca iô- nica é um zeolito inorgânico ou resina orgânica sintética. Em algumas modalidades, a resina de troca iônica inclui, entre outras, aquelas desceritas em
Wagh, et al. (ibid), que está aqui incorporada como referência em sua totalidade. Em algumas modalidades, a resina de troca iônica é um ácido poliacrílico parcialmente neutralizado.
Em algumas modalidades, a composição oftálmica é uma suspensão polimérica aquosa. Em algumas modalidades, o agente terapêutico ou um agente de suspensão polimérico é suspendido em um meio aquoso. Em algumas modalidades, as suspensões poliméricas aquosas podem ser formuladas de modo que retenham a mesma ou substancialmente a mesma viscosidade no olho de que tinham antes da administração do olho. Em algumas modalidades, podem ser formulados de modo que tenha gelificação aumentada no contato com fluido lacrimoso.
Outro aspecto da presente invenção refere-se a versão marcada de certos compostos descritos aqui (radio-marcados, marcados com fluorescência, etc.) que poderia ser úteis não somente em técnias de imagens mas também em ensaios, in vitro e in vivo, para localização e quantificação de JAK emamostras de tecidos, incluindo humanos, e para identificar os ligan- tes de JAK por inibição de ligação de um composto marcado. Assim, a presente invenção inclui ensaios de JAK que contêm ditos compostos marcados.
A presente invenção ainda inclui compostos marcados isotopi- camente. Um composto “isotopicamente” ou “radiorrotulado” é um composto descrito aqui onde um ou mais átomos são substituídos por um átomo contendo uma massa atômica ou número de massa diferente da massa atômica ou número de massa tipicamente encontrado na natureza (ou seja, de ocorrência natural). Radionuclídeos apropriados que podem ser incorporados nos compostos descritos aqui incluem, entre outros, 2H (também escrito como D para deuterio), 3H (também escrito como T para trítio), 11C, 13C, 14C, 13N, 16N, 15O, 17O, 18O, 18F, 35S, 36CI, 82Br, 75Br, 76Br, 77Br, 123l, 124l, 125l e 1311. O radionuclídeo que está incorporado nos compostos radiomarca- dos irá depender da aplicação específica do composto radimarcado. Por exemplo, para marcação in vitro de meta I oprotease e ensaios de competi ção, os compostos que incorporam 3H, 14C, 82Br, 125l, 131l, 35S ou irão geralmente ser mais úteis. Para aplicações de radio-imagem 11C, 18F, 125l, 123l, 124l, 131l, 75Br, 76Br ou 77Br irão geralmente ser mais úteis.
Deve ser entendido que um composto “radiomarcado” ou “composto marcado” é um composto que tem incorporado pelo menos um radio- nuclídeo. Em algumas modalidades o radionuclídeo é selecionado do grupo que consiste de 3H, 14C, 1251,35S e 82Br.
A presente invenção pode ainda incluir métodos sintéticos para incorporar radio-isótopos nos compostos descritos aqui. Métodos sintéticos para incorporar radio-isótopos em compostos orgânicos são bem conhecidos na técnica, e uma pessoa especialista na técnica irá prontamente reconhecer os métodos aplicáveis para os compostos descritos aqui.
Um composto marcado da invenção pode ser usado em um ensaio de triagem para identificar /avaliar os compostos. Por exemplo, um composto identificado ou recém identificado (ou seja, composto teste) que é marcado para ser avaliado para sua capacidade de se ligar a JAK por monitoramento de sua variação de concentração quando contatando com JAK, através de rastreamento da marcação. Por exemplo, um composto teste (marcado) pode ser avaliado para sua capacidade de reduzir a ligação a outro composto que é conhecido por se ligar a uma JAK (ou seja, composto padrão). Assim, a capacidade de um composto teste em competir com o composto padrão para ligação a JAK diretamente se correlaciona com sua afinidade de ligação. De forma recíproca, em alguns outros ensaios de triagem, o composto padrão é marcado e os compostos de teste são não marcados. Assim, a concentração do composto padrão é monitorada para avaliar a competição entre o composto padrão e o composto teste, e a afinidade de ligação relativa do composto teste é assim, apurada. KITS
A presente invenção ainda inclui kits farmacêuticos úteis, por exemplo, no tratamento ou prevenção de doenças ou distúrbios associados a JAK, como câncer, que inclui um ou mais recipientes contendo uma composição farmacêutica compreendendo uma quantidade terapeuticamente efetiva de um composto ativo. Ditos kits podem ainda incluir, se desejado, um ou mais de vários componentes convencionais de kits farmacêuticos, como, por exemplo, recipientes com um ou mais carreadores farmaceuticamente aceitáveis, recipientes adicionais, etc., como será prontamente aparente aos especialistas na técnica. Instruções, como bulas ou rótulos, indicando as quantidades dos componentes a ser administradas, instruções para administração, e/ou instruções para mistura dos componentes, podem ainda ser incluídos no kit
A invenção irá agora ser descrita em maiores detalhes por meio de exemplos específicos. Os seguintes exemplos são oferecidos para fins ilustrativos e não têm a intenção de limitar a invenção de qualquer modo. Os especialistas na técnica irão prontamente reconhecer uma variedade de parâmetros não críticos que podem ser alterados ou modificados para gerar essencialmente os mesmos resultados.
Exemplo 1: (R)-3-ciclopentil-3-[4-(2-hidróxi-5-oxo-6,7-di-hidro-5H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -íljpropanonitrila
Etapa 1. (R)-3-ciclopentil-3-[4-(2-metóxi- 7H-pÍrrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1 -Íljpropanonitrila 4-Cloro-2-metóxi-7H-pirrolo[2,3-d]pirimidina (0,4 g, 2.18 mmol, Toronto Research Chemicals) e (R)-3-ciclopentil-3-[4-(4,4,5,5-tetrametil- 1,3,2-dioxaborolan-2-il)-1 H-pirazol-1-iljpropanonitrila (0,824 g, 2,61 mmol, preparado conforme descrito em Org. Lett., 2009, 11(9), 1999-2002.) foram dissolvidos em 1,4-dioxano (4 mL) e carbonato de potássio (0,903 g, 6,54 mmol) em água (2 mL) foi adicionado. A mistura foi desgaseificada e te- trakis(trifenilfosfino)paládio(0) (0,126 g, 0,109 mmol) foi adicionado. A mistura de reação foi aquecida a 100°C por 16 h. A mistura de reação foi particionada entre água e acetato de etila. A camada aquosa foi extraída com acetato de etila três vezes. Os extratos combinados foram secos em sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida, eluin- do com um gradiente de 0-10% MeOH em cloreto de metileno foi usada para purificar o produto (670 mg, 91%). 1H RMN (300 MHz, CD3OD): Ô 8,59 (s, 1H), 8,35 (s, 1H), 7,24 (d, 1H), 6,81 (d, 1H), 4,47 (dt, 1H), 4,04 (s, 3H), 3,21 (dd, 1H), 3,10 (dd, 1H), 2,62-2,44 (m, 1H), 2,02-1,86 (m, 1H), 1,81-1,20 (m, 7H); LCMS (M+H)+: 337,0.
4-Cloro-2-metóxi-7H-pirrolo[2,3-d]pirimidina (0,4 g, 2.18 mmol, Toronto Research Chemicals) e (R)-3-ciclopentil-3-[4-(4,4,5,5-tetrametil- 1,3,2-dioxaborolan-2-il)-1 H-pirazol-1-iljpropanonitrila (0,824 g, 2,61 mmol, preparado conforme descrito em Org. Lett., 2009, 11(9), 1999-2002.) foram dissolvidos em 1,4-dioxano (4 mL) e carbonato de potássio (0,903 g, 6,54 mmol) em água (2 mL) foi adicionado. A mistura foi desgaseificada e te- trakis(trifenilfosfino)paládio(0) (0,126 g, 0,109 mmol) foi adicionado. A mistura de reação foi aquecida a 100°C por 16 h. A mistura de reação foi particionada entre água e acetato de etila. A camada aquosa foi extraída com acetato de etila três vezes. Os extratos combinados foram secos em sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida, eluin- do com um gradiente de 0-10% MeOH em cloreto de metileno foi usada para purificar o produto (670 mg, 91%). 1H RMN (300 MHz, CD3OD): Ô 8,59 (s, 1H), 8,35 (s, 1H), 7,24 (d, 1H), 6,81 (d, 1H), 4,47 (dt, 1H), 4,04 (s, 3H), 3,21 (dd, 1H), 3,10 (dd, 1H), 2,62-2,44 (m, 1H), 2,02-1,86 (m, 1H), 1,81-1,20 (m, 7H); LCMS (M+H)+: 337,0.
Etapa 2. (R)~3-ciclopentil~3-[4-(5dodo-2-metóxi~7H-pirrolo[2,3-d]pirímídíri-4- il) -1 H-pirazol-1 -il]propanonitrila A uma solução de 3-ciclopentil-3-[4-(2-metóxi-7H-pirrolo[2,3-d]pi- rimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (0,532 g, 1,58 mmol) em tetra-hidro- furano (20 mL) foi adicionado N-iodosuccinimida (0,36 g, 1,6 mmol). A reação foi agitada por 30 min e o solvente foi removido à vácuo. O resísudo foi purificado por cromatografia de coluna rápida, eluindo com um gradiente de 0 a 65% de acetato de etila em hexanos para obter um sólido amarelo (250 mg, 34%). 1H RMN (300 MHz, CDCI3): δ 10,31 (br s, 1H), 8,27 (s, 1H), 8,23 (s, 1H), 7,29 (s, 1H), 4,34-4,21 (m, 1H), 4,06 (s, 3H), 3,14 (dd, 1H), 3,03-2,90 (m, 1H), 2,66-2,49 (m, 1H), 2,02-1,17 (m, 8H); LCMS (M+H)+: 463,0.
A uma solução de 3-ciclopentil-3-[4-(2-metóxi-7H-pirrolo[2,3-d]pi- rimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (0,532 g, 1,58 mmol) em tetra-hidro- furano (20 mL) foi adicionado N-iodosuccinimida (0,36 g, 1,6 mmol). A reação foi agitada por 30 min e o solvente foi removido à vácuo. O resísudo foi purificado por cromatografia de coluna rápida, eluindo com um gradiente de 0 a 65% de acetato de etila em hexanos para obter um sólido amarelo (250 mg, 34%). 1H RMN (300 MHz, CDCI3): δ 10,31 (br s, 1H), 8,27 (s, 1H), 8,23 (s, 1H), 7,29 (s, 1H), 4,34-4,21 (m, 1H), 4,06 (s, 3H), 3,14 (dd, 1H), 3,03-2,90 (m, 1H), 2,66-2,49 (m, 1H), 2,02-1,17 (m, 8H); LCMS (M+H)+: 463,0.
Etapa 3. acetato de 4-[1-(2-ciano-1-ciclopentiletil)-1H-pirazol~4-il]-2-metóxi- 7H-pirrolo[2,3~d]pirimidin-5~ila Uma solução de 3-ciclopentil-3-[4-(5-iodo-2-metóxi-7H-pirrolo[2,- 3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila (0.25 g, 0,54 mmol) em ácido acético (3 mL) foi tratada com acetato de parta (0,27 g, 1,6 mmol) e aquecida a 70°C por 16 h. A mistura foi filtrada, enxaguada com MeCN, água foi adicionada ao filtrado e esta mistura foi agitada por 20 min. Cloreto de sódio sólido foi adicionado à esta solução. O produto foi obtido por extração desta mistura aquosa com três partes acetato de etila. Os extratos combinados foram secos em sulfato de sódio, decantados e concentrados. Uma parte do protudo foi usada na etapa de hidrólise (Etapa 4) sem outra purificação. 1H RMN (300 MHz, CDCI3): δ 9,87 (br s, 1H), 8,39 (s, 1H), 8,37 (s, 1H), 7,33 (s, 1H), 4,22 (dt, ÍH), 4,06 (s, 3H), 3,14 (dd, 1H), 2,94 (dd, 1H), 2,64-2,47 (m, 1H), 2,36 (s, 3H), 2,03-1,86 (m, 1H), 1,79-1,12 (m, 7H); LCMS (M+H)+: 395,1.
Uma solução de 3-ciclopentil-3-[4-(5-iodo-2-metóxi-7H-pirrolo[2,- 3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila (0.25 g, 0,54 mmol) em ácido acético (3 mL) foi tratada com acetato de parta (0,27 g, 1,6 mmol) e aquecida a 70°C por 16 h. A mistura foi filtrada, enxaguada com MeCN, água foi adicionada ao filtrado e esta mistura foi agitada por 20 min. Cloreto de sódio sólido foi adicionado à esta solução. O produto foi obtido por extração desta mistura aquosa com três partes acetato de etila. Os extratos combinados foram secos em sulfato de sódio, decantados e concentrados. Uma parte do protudo foi usada na etapa de hidrólise (Etapa 4) sem outra purificação. 1H RMN (300 MHz, CDCI3): δ 9,87 (br s, 1H), 8,39 (s, 1H), 8,37 (s, 1H), 7,33 (s, 1H), 4,22 (dt, ÍH), 4,06 (s, 3H), 3,14 (dd, 1H), 2,94 (dd, 1H), 2,64-2,47 (m, 1H), 2,36 (s, 3H), 2,03-1,86 (m, 1H), 1,79-1,12 (m, 7H); LCMS (M+H)+: 395,1.
Etapa 4. (R)-3-ciclopentil-3-[4-(2-hidróxi-5-oxo-6,7-di-hidro~5H-pirrolo[2,3- d]pírímidin-4-il)-1 H-pirazol-1 -il]propanamida 4 M de HBr em ácido acético (2 mL, 8 mmol) foi adicionado a acetato de 4-[1 -(2-ciano-1 -ciclopentiletiI)-1 H-pirazol-4-il]-2-metóxi-7H-pirro- lo[2,3-d]pirimidin-5-ila (0,050 g, 0,13 mmol) e a reação foi agitada por 1 h. Os voláteis foram removidos a vácuo. O resíduo foi reconstituído e HPLC-MS preparativa (eluindo com um gradiente de MeCN/H2O contendo 0,15% NH4OH) foi usado para obter produto purificado (12 mg, 26%). 1H RMN (500 MHz, de-DMSO): ô 9,20 (s, 1H), 8,69 (s, 1H), 7,34 (s, 1H), 6,75 (s, 1H), 4,49 (dt, 1H), 3,89 (s, 2H), 2,80 (dd, 1H), 2,64 (dd, 1H), 2,36-2,26 (m, 1H), 1,83- 1,74 (m, 1H), 1,63-1,36 (m, 4H), 1,32-1,20 (m, 2H), 1,15-1,05 (m, 1H); LCMS (M+H)+: 357,0.
4 M de HBr em ácido acético (2 mL, 8 mmol) foi adicionado a acetato de 4-[1 -(2-ciano-1 -ciclopentiletiI)-1 H-pirazol-4-il]-2-metóxi-7H-pirro- lo[2,3-d]pirimidin-5-ila (0,050 g, 0,13 mmol) e a reação foi agitada por 1 h. Os voláteis foram removidos a vácuo. O resíduo foi reconstituído e HPLC-MS preparativa (eluindo com um gradiente de MeCN/H2O contendo 0,15% NH4OH) foi usado para obter produto purificado (12 mg, 26%). 1H RMN (500 MHz, de-DMSO): ô 9,20 (s, 1H), 8,69 (s, 1H), 7,34 (s, 1H), 6,75 (s, 1H), 4,49 (dt, 1H), 3,89 (s, 2H), 2,80 (dd, 1H), 2,64 (dd, 1H), 2,36-2,26 (m, 1H), 1,83- 1,74 (m, 1H), 1,63-1,36 (m, 4H), 1,32-1,20 (m, 2H), 1,15-1,05 (m, 1H); LCMS (M+H)+: 357,0.
Etapa 5. (R)-3-ciclopentil-3-[4-(2-hidróxi-5~oxo-6; 7-di-hidro-5H-pÍrrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitriía
A uma solução de 3-ciclopentil-3-[4-(2-hidróxi-5-oxo-6,7-di-hidro- 5H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanamida (0,006 g, 0,02 mmol) em cloreto de metileno (0,5 mL) contendo trietilamina (20 TL, 0,2 mmol) foi adicionado cloreto de tricloroacetil (20 TL, 0,2 mmol). Quando a reação estava concluída, HPLC-MS preparativa (MeCN/H2O contendo 0,15% de NH4OH) foi usada para obter produto purificado (3 mg, 52%). 1H RMN (400 MHz, d6-DMSO): δ 11,39 (br s, 1H), 9,37 (s, 1H), 8,77 (s, 1H), 8,63 (s, 1H), 4,68-4,59 (m, 1H), 3,94 (s, 2H), 3,19-3,15 (m, 2H), 2,42-2,30 (m, 1H), 1,86-1,75 (m, 1H), 1,69-1,20 (m, 6H), 1,18-1,05 (m, 1H); LCMS (M+Hf: 339,1.
Exemplo 2: (3R)- e (3S)-3-[(1R,2R)-2-hidroxiciclopentil]-3-[4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila e (3R)- e (3S)-3- [(1S,2S)-2-hidroxiciclopentil]-3-[4-(7H-pirroIo[2,3-d]pirimidín-4-íl)-1H- pirazol-1-il]propanonitrila
Etapa 1. 2-(terc-butildimetilsi!ióxi)ciclopentanocarboxilato de (1S,2R)-etila e 2-(terc-butildimetilsilióxi)ciclopentanocarboxilato de (1S,2R)-etila A uma solução de cloreto de terc-butildimetilsilila (0,524 g, 3,48 mmol) e 1H-lmidazol (0,473 g, 6,95 mmol) em N,N-dimetilformamida (15 mL) foi adicionado etil cis-2-hidróxi-1~ciclopentanocarboxilato (racêmica, Acros) (0,50 g, 0,0032 mol). A reação foi agitada por 16 h. Mais imidazol (0,40 g, 5,8 mmol) e cloreto de terc-butildimetilsilil (0,50 g, 3,3 mmol) foram adicionados em porções e a reação agitada por mais 24 h. O produto foi extraído com hexano. Os extratos foram lavados com água, secos em sulfato de só-dio, filtrados e evaporados para obter hidroxiester protegido por TBS racêmi- co (0,9 g) que foram usados sem outra purificação. 1H RMN (400 MHz, CDCI3): δ 4,46 (ddd, 1H), 4,19 (dq, 1H), 4,01 (dq, 1H), 2,72 (dt, 1H), 2,22- 2,11 (m, 1H), 1,96-1,49 (m, 5H), 1,26 (t, 3H), 0,84 (s, 9H), 0,03 (s, 3H), 0,01 (s, 3H).
A uma solução de cloreto de terc-butildimetilsilila (0,524 g, 3,48 mmol) e 1H-lmidazol (0,473 g, 6,95 mmol) em N,N-dimetilformamida (15 mL) foi adicionado etil cis-2-hidróxi-1~ciclopentanocarboxilato (racêmica, Acros) (0,50 g, 0,0032 mol). A reação foi agitada por 16 h. Mais imidazol (0,40 g, 5,8 mmol) e cloreto de terc-butildimetilsilil (0,50 g, 3,3 mmol) foram adicionados em porções e a reação agitada por mais 24 h. O produto foi extraído com hexano. Os extratos foram lavados com água, secos em sulfato de só-dio, filtrados e evaporados para obter hidroxiester protegido por TBS racêmi- co (0,9 g) que foram usados sem outra purificação. 1H RMN (400 MHz, CDCI3): δ 4,46 (ddd, 1H), 4,19 (dq, 1H), 4,01 (dq, 1H), 2,72 (dt, 1H), 2,22- 2,11 (m, 1H), 1,96-1,49 (m, 5H), 1,26 (t, 3H), 0,84 (s, 9H), 0,03 (s, 3H), 0,01 (s, 3H).
Etapa 2. (1S,2R)~2-(terc-butildimetilsililóxi)ciclopentanocarbaldeído e (1R, 2S)-2- (terc-butildime tiisHilóxi) ciclope n tanocarbaideído A uma solução de 2-(terc-butildimeti!silióxi)ciclopentanocarboxi- lato de (1S,2R)-etila e 2-(terc-butildimetilsilióxi)ciclopentanocarboxilato de (1S,2R)-etila da Etapa 1 (0,86 g, 3,2 mmol) em hexanos (40 mL) a -78°C foi adicionado sob gotejamento uma solução de 1,0 M of diisobutilalumínio hi- dreto em tolueno (3,5 mL, 3,5 mmol). A mistura de reação foi agitada por 1 h a -78°C e foi extinta esta temperatura pela adição sob gotejamento de metanol (2 mL). O resfriamento foi descontinuado e a mistura foi deixada alcançar a temperatura ambiente. Uma solução aquosa de sal de Rochelle foi adicionada. A mistura bifásica foi agitada vigorosamente por 2 h e as camadas resultantes foram separadas. A camada aquosa foi extraída uma vez mais com hexanos, em seguida com três partes de acetato de etila. Os extratos combinados foram lavados com salmoura, secos em sulfato de sódio, decantados e concentrados para obter o produto aldeído racêmico, usado sem outra purificação (0,7 g, 97%). 1H RMN (400 MHz, CDCI3): δ 9,74 (d, 1H), 4,62 (ddd, 1H), 2,68-2,61 (m, 1H), 2,22-2,11 (m, 1H), 1,95-1,83 (m, 1H), 1,80-1,57 (m, 4H), 0,85 (s, 9H), 0,05 (s, 3H), 0,04 (s, 3H).
A uma solução de 2-(terc-butildimeti!silióxi)ciclopentanocarboxi- lato de (1S,2R)-etila e 2-(terc-butildimetilsilióxi)ciclopentanocarboxilato de (1S,2R)-etila da Etapa 1 (0,86 g, 3,2 mmol) em hexanos (40 mL) a -78°C foi adicionado sob gotejamento uma solução de 1,0 M of diisobutilalumínio hi- dreto em tolueno (3,5 mL, 3,5 mmol). A mistura de reação foi agitada por 1 h a -78°C e foi extinta esta temperatura pela adição sob gotejamento de metanol (2 mL). O resfriamento foi descontinuado e a mistura foi deixada alcançar a temperatura ambiente. Uma solução aquosa de sal de Rochelle foi adicionada. A mistura bifásica foi agitada vigorosamente por 2 h e as camadas resultantes foram separadas. A camada aquosa foi extraída uma vez mais com hexanos, em seguida com três partes de acetato de etila. Os extratos combinados foram lavados com salmoura, secos em sulfato de sódio, decantados e concentrados para obter o produto aldeído racêmico, usado sem outra purificação (0,7 g, 97%). 1H RMN (400 MHz, CDCI3): δ 9,74 (d, 1H), 4,62 (ddd, 1H), 2,68-2,61 (m, 1H), 2,22-2,11 (m, 1H), 1,95-1,83 (m, 1H), 1,80-1,57 (m, 4H), 0,85 (s, 9H), 0,05 (s, 3H), 0,04 (s, 3H).
Etapa 3. (E)- e (Z)-3-((1 R,2R)-2~(terc-butildimetilsililóxi}ciclopentil)acrilonitrila e (E)~ e (Z)-3-((1 S,2S)-2-(terc-butildimetilsililóxi}ciclopentil)acrilonitrila A uma solução de (1S,2R)-2-(terc-butildimetilsililóxi)ciclopen- tanocarbaldeído e (1 R,2S)-2-(terc-butildimetilsililóxi)ciclopentanocarbaldeído (0,36 g, 1,6 mmol, da Etapa 2) em tolueno (9 mL) foi adicionado (trifenilfos- foranilideno)acetonitrila (0,475 g, 1,58 mmol) e a reação foi aquecida a 80°C for 2 h. A reação foi resfriada até temperatura ambiente e água foi adicionada. O produto foi extraído com três partes de éter de etila. Os extratos foram lavados com salmoura, secos em sulfato de sódio, decantados e concentrados para fornecer a mistura racêmica de isômeros E-e Z- olefina que foi usada sem outra purificação. 1H RMN (400 MHz, CDCI3): δ 6,82 (dd, 1H, trans olefin), 6,63 (dd, 1H, cis olefin), 5,307 (dd, 1H, trans olefina), 5,305 (dd, 1H, cis olefina), 4,24 (ddd, 1H), 4,20 (ddd, 1H), 2,96-2,86 (m, 1H), 2,53-2,43 (m, 1H), 1,95-1,56 (m, 12H), 0,87 (s, 9H), 0,86 (s, 9H), 0,04-0,01 (singletos, juntos 12H).
A uma solução de (1S,2R)-2-(terc-butildimetilsililóxi)ciclopen- tanocarbaldeído e (1 R,2S)-2-(terc-butildimetilsililóxi)ciclopentanocarbaldeído (0,36 g, 1,6 mmol, da Etapa 2) em tolueno (9 mL) foi adicionado (trifenilfos- foranilideno)acetonitrila (0,475 g, 1,58 mmol) e a reação foi aquecida a 80°C for 2 h. A reação foi resfriada até temperatura ambiente e água foi adicionada. O produto foi extraído com três partes de éter de etila. Os extratos foram lavados com salmoura, secos em sulfato de sódio, decantados e concentrados para fornecer a mistura racêmica de isômeros E-e Z- olefina que foi usada sem outra purificação. 1H RMN (400 MHz, CDCI3): δ 6,82 (dd, 1H, trans olefin), 6,63 (dd, 1H, cis olefin), 5,307 (dd, 1H, trans olefina), 5,305 (dd, 1H, cis olefina), 4,24 (ddd, 1H), 4,20 (ddd, 1H), 2,96-2,86 (m, 1H), 2,53-2,43 (m, 1H), 1,95-1,56 (m, 12H), 0,87 (s, 9H), 0,86 (s, 9H), 0,04-0,01 (singletos, juntos 12H).
Etapa 4. (3R)- e (3S)-3-[(1R,2R)-2-hidroxiciclopentn]-3-[4-(7H-pirrolo[2>3-d]pi- rimidin-4-il)-1 H-pirazol-1 -il]propanonitríla e (3R)- e (3S)-3-[(1S, 2S)-2-hidroxi- ciclopentil]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)~1 H-pirazol-1 -il]propanonitrila
A uma solução de (E)- e (Z)-3-((1 R,2R)-2-(terc-butildimetilsililóxi) ciclopentil)acrilonitrila e (E)- e (Z)-3-((1 S,2S)-2-(terc-butildimetilsililóxi) ciclo- pentil) acrilonitrila (0,40 g, 1,6 mmol, produto bruto da Etapa 3) em aceto- nitrila (20 mL) foi adicionado 4-(1 H-pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidina (0,50 g, 1,6 mmol) e 1,8-diazabiciclo[5,4,0]undec- 7-eno (DBU, 0,24 mL, 1,6 mmol). A reação foi agitada por 2 h em temperatura ambiente, e mais DBU (0,24 mL, 1,6 mmol) foi adicionado. A reação foi 5 agitada por 3 dias e foi concentrada. Cromatografia de coluna rápida (eluin- do com um gradiente de 10-40% acetato de etila/hexanos) foi usada para purificar o produto que foi então tratado com 20% TFA em DCM por 3 h, evaporado, e tratado com excesso de etilenodiamina em solução de metanol durante a noite. Quando a remoção do grupo de proteção SEM estava con- 10 cluída, qualquer grupo de proteção restante TBS foi removido por agitação com EtOH/H2O/c.HCI por 3 horas (10:4:3 proporção em volume). Os produtos completamente desprotegidos foram purificados em HPLC-MS preparativa (0,15% NH4OH em um gradiente de MeCN/H2O). Todas as frações de M+H=323 foram agrupadas e evaporadas (aproximadamente 80 mg). Os 15 produtos foram submetidos a uma série de purificações em cromatografia quiral como a seguir: Chiral Technologies Chiralcel OJ-H (3 x 25 cm, 5 Tm) eluindo com 20% EtOH/80% Hexanos em uma vazão de 25 mL/min para obter Pico 1 (19 mg), o pico menor seguinte não coletado; Pico 2 (60 mg), Pico 3 (6 mg). Pico 2 foi uma mistura que foi então separada usando Chirai
Technologies Chiralpak IA (2 x 25 cm, 5 Tm) eluindo com um gradiente de 70% EtOH/30% Hexanos em uma vazão de 8 mL/min em três componentes. Estes foram rotulados Pico 2-1 (corrida 2, pico 1, 32 mg) que foi uma mistura de produtos, Pico 2-2 (6,5 mg) e Pico 2-3 (13,7 mg). Pico 2-1 foi ainda separado em três componentes usando Chiral Technologies Chiralpak IA (2 25 x 25 cm, 5 Tm) eluindo com um gradiente de 25% EtOH/75% Hexanos em uma vazão de 12 mL/min. Os produtos isolados foram rotulados Pico 2-1-1 (10,5 mg); 2-1-2 (13 mg); e 2-1-3 (2,3 mg). Pico 1: 1H RMN (500 MHz, CD3OD): δ 8,65 (s, 1H), 8,62 (s, 1H), 8,37 (s, 1H), 7,49 (d, 1H), 6,95 (d, 1H), 4,71 (ddd, 1H), 4,28 (br t, 1H), 3,26 30 (dd, 1H), 3,21 (dd, 1H), 2,53-2,45 (m, 1H), 1,98-1,73 (m, 3H), 1,62-1,47 (m, 2H), 1,38-1,29 (m, 1H); LCMS (M+H)+: 323. Pico 2-1-1: 1H RMN (300 MHz, CD3OD): δ 8,64 (s, 1H), 8,59 (s, 1H), 8,38 (s, 1H), 7,49 (d, 1H), 6,94 (d, 1H), 4,90-4,78 (m, 1H), 3,64 (br t, 1H), 3,21 (dd, 1H), 3,07 (dd, 1H), 2,55-2,40 (m, 1H), 2,01-1,58 (m, 6H); LCMS (M+H)+: 323. Pico 2-1-2: 1H RMN (500 MHz, CD3OD): Ô 8,65 (s, 1H), 8,62 (s, 1H), 8,37 (s, 1H), 7,49 (d, 1H), 6,95 (d, 1H), 4,71 (ddd, 1H), 4,28 (br t, 1H), 3,26 (dd, 1H), 3,21 (dd, 1H), 2,53-2,45 (m, 1H), 1,98-1,73 (m, 3H), 1,62-1,48 (m, 2H), 1,38-1,26 (m, 1H); LCMS (M+H)+: 323. Pico 2-3: 1H RMN (300 MHz, CD3OD): Ô 8,64 (s, 1H), 8,59 (s, 1H), 8,38 (s, 1H), 7,48 (d, 1H), 6,93 (d, 1H), 4,90-4,78 (m, 1H), 3,64 (br t, 1H), 3,21 (dd, 1H), 3,07 (dd, 1H), 2,55-2,40 (m, 1H), 2,01-1,58 (m, 6H); LCMS (M+H)+: 323.
Exemplo 3: Sal racêmico de trifluoracetato de 3-(1-Hidroxiciclopentil)-3- [4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila
Etapa 1. 1-[(trimetilsilil)óxi]ciclopentanocarbaldeído A reação foi conduzida em um procedimento semelhante ao descrito em Tetrahedron, 50(9), 2821-30; 1994: A uma solução de 1-[(trime- tilsilil)óxi]ciclopentanocarbonitrila (2,25 g, 12,3 mmol, preparado conforme descrito em Organometallics, 3(11), 1660-5; 1984) em tolueno (18 mL) a - 45°C foi adicionado sob gotejamento 1,0 M de diisobutilalumínio hidreto em hexano (17,2 mL, 17,2 mmol). A solução foi então deixada aquecer a 0°C e agitada por 1 h nesta temperatura. A mistura de reação foi vertida em uma mistura de éter dietila (25 mL) e cloreto de amónio (25 mL, saturado). À mistura resultante, a 15°C, foi adicionado uma solução diluída de ácudo sulfúrico (preparado por diluição 1,53 mL de H2SO4 concentrado com 50 mL de água). A solução foi então agitada em temperatura de 5°C durante a noite. A mistura foi extraída com três partes de éter dietila, os extratos combinados foram lavados com salmoura, secos em sulfato de sódio, decanta- 5 dos e concentrados para obter product (0,84 g, 36%), que foi usado sem outra purificação na Etapa 2. 1H RMN (300 MHz, CDCI3): δ 9,47 (s, 1H), 1,88-1,46 (m, 8H), 0,00 (s, 9H).
A reação foi conduzida em um procedimento semelhante ao descrito em Tetrahedron, 50(9), 2821-30; 1994: A uma solução de 1-[(trime- tilsilil)óxi]ciclopentanocarbonitrila (2,25 g, 12,3 mmol, preparado conforme descrito em Organometallics, 3(11), 1660-5; 1984) em tolueno (18 mL) a - 45°C foi adicionado sob gotejamento 1,0 M de diisobutilalumínio hidreto em hexano (17,2 mL, 17,2 mmol). A solução foi então deixada aquecer a 0°C e agitada por 1 h nesta temperatura. A mistura de reação foi vertida em uma mistura de éter dietila (25 mL) e cloreto de amónio (25 mL, saturado). À mistura resultante, a 15°C, foi adicionado uma solução diluída de ácudo sulfúrico (preparado por diluição 1,53 mL de H2SO4 concentrado com 50 mL de água). A solução foi então agitada em temperatura de 5°C durante a noite. A mistura foi extraída com três partes de éter dietila, os extratos combinados foram lavados com salmoura, secos em sulfato de sódio, decanta- 5 dos e concentrados para obter product (0,84 g, 36%), que foi usado sem outra purificação na Etapa 2. 1H RMN (300 MHz, CDCI3): δ 9,47 (s, 1H), 1,88-1,46 (m, 8H), 0,00 (s, 9H).
Etapa 2. (2E)~ e (2Z)~3-{1-[(trimetilsilil)óxi]ciclopentil}acrilonitrila Cianometilfosfonato de dietila (0,912 mL, 5,64 mmol) foi adicio nado sob gotejamento a uma suspensão de hidreto de sódio (0,198 g, 4,96 mmol) em tetra-hidrfurano (10 mL) a 0°C. Após a adição, a reação foi aquecida até temperatura ambiente e agitada por 45 minutos. A mistura foi novamente resfriada a 0°C e 1-[(trimetilsilil)óxi]ciclopentanocarbaldeído (0,84 g, 15 4,5 mmol) em tetra-hidrfurano (20 mL) foi introduzido. A reação was deixada aquecer até temperatura ambiente e agitada por 2 horas. Agua e acetato de etila foram adicionados na reação e as camadas separadas. A camada aquosa foi extraída com duas oturas partes de acetato de etila. Os extratos orgânicos combinados foram lavados com salmoura, secos em sulfato de 20 sódio, decantados e concentrados para obter produto como uma mistura de isômeros de olefina, usados sem outra purificação na Etapa 3. 1H RMN (300 MHz, CDCh): δ 6,80 (d, 1H, trans/produto principal), 6,53 (d, 1H, cis/produto menor), 5,52 (d, 1H, trans), 5,28 (d, 1H, cis), 2,06-0,76 (m, 16H total para ambos isômeros), 0,18 (s, 9H, produto menor), 0,13 (s, 9H, produto pnn- 25 cipal).
Cianometilfosfonato de dietila (0,912 mL, 5,64 mmol) foi adicio nado sob gotejamento a uma suspensão de hidreto de sódio (0,198 g, 4,96 mmol) em tetra-hidrfurano (10 mL) a 0°C. Após a adição, a reação foi aquecida até temperatura ambiente e agitada por 45 minutos. A mistura foi novamente resfriada a 0°C e 1-[(trimetilsilil)óxi]ciclopentanocarbaldeído (0,84 g, 15 4,5 mmol) em tetra-hidrfurano (20 mL) foi introduzido. A reação was deixada aquecer até temperatura ambiente e agitada por 2 horas. Agua e acetato de etila foram adicionados na reação e as camadas separadas. A camada aquosa foi extraída com duas oturas partes de acetato de etila. Os extratos orgânicos combinados foram lavados com salmoura, secos em sulfato de 20 sódio, decantados e concentrados para obter produto como uma mistura de isômeros de olefina, usados sem outra purificação na Etapa 3. 1H RMN (300 MHz, CDCh): δ 6,80 (d, 1H, trans/produto principal), 6,53 (d, 1H, cis/produto menor), 5,52 (d, 1H, trans), 5,28 (d, 1H, cis), 2,06-0,76 (m, 16H total para ambos isômeros), 0,18 (s, 9H, produto menor), 0,13 (s, 9H, produto pnn- 25 cipal).
Etapa 3. Racêmico - 3-(1-hidroxiciclopentil)-3-(4-(7-((2-(trimetilsilil)etóxi)me- til)-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)propanonitrila A uma suspensão de (2E)- e (2Z)-3-{1-[(trimetilsilil)óxi]ciclopen- tiljacrilonitrila (0,94 g, 4,5 mmol) e 4 (1 H-pirazol-4-íI)~7-{[2-(trímetilsilíl)etóxi]- metil}-7H-pirro)o[2,3-d]pirimidina (1,4 g, 4,5 mmol) em acetonitríla (20 mL) foi adicionado 1,8-diazabiciclo[5,4,0]undec-7-eno (0,67 mL, 4,5 mmol). A reação foi deixada agitar em temperatura ambiente por 6 dias. A acetonitríla foi evaporada. O produto desejado não protegido álcool foi isolado da misturade álcool não protegido e álcool protegido com TMS usando cromatografia de coluna rápida, eluindo com um gradiente de 0 a 80% de acetato de etila em hexanos (890 mg, 44%). 1H RMN (300 MHz, CDCI3): δ 8,87 (s, 1H), 8,56 (br s, 1H), 8,35 (s, 1H), 7,46 (d, 1H), 6,84 (d, 1H), 5,69 (s, 2H), 4,40 (dd, 1H), 4,03 (s, 1H), 3,55 (dd, 2H), 3,46 (dd, 1H), 2,97 (dd, 1H), 2,04-1,27 (m, 8H), 0,93 (dd, 2H), -0,05 (s, 9H); LCMS (M+H)+: 453,1.
A uma suspensão de (2E)- e (2Z)-3-{1-[(trimetilsilil)óxi]ciclopen- tiljacrilonitrila (0,94 g, 4,5 mmol) e 4 (1 H-pirazol-4-íI)~7-{[2-(trímetilsilíl)etóxi]- metil}-7H-pirro)o[2,3-d]pirimidina (1,4 g, 4,5 mmol) em acetonitríla (20 mL) foi adicionado 1,8-diazabiciclo[5,4,0]undec-7-eno (0,67 mL, 4,5 mmol). A reação foi deixada agitar em temperatura ambiente por 6 dias. A acetonitríla foi evaporada. O produto desejado não protegido álcool foi isolado da misturade álcool não protegido e álcool protegido com TMS usando cromatografia de coluna rápida, eluindo com um gradiente de 0 a 80% de acetato de etila em hexanos (890 mg, 44%). 1H RMN (300 MHz, CDCI3): δ 8,87 (s, 1H), 8,56 (br s, 1H), 8,35 (s, 1H), 7,46 (d, 1H), 6,84 (d, 1H), 5,69 (s, 2H), 4,40 (dd, 1H), 4,03 (s, 1H), 3,55 (dd, 2H), 3,46 (dd, 1H), 2,97 (dd, 1H), 2,04-1,27 (m, 8H), 0,93 (dd, 2H), -0,05 (s, 9H); LCMS (M+H)+: 453,1.
Etapa 4. Sal Racêmico 3-(1-hidroxiciclopentil)-3-[4-(7H-pirrolo[2,3-d]pirimidin~ H-pirazol-1 -il]propanonitrila trifluoracetato Uma solução de racêmico 3-(1 -hidroxiciclopentil)-3-[4-(7-{[2-(tri- metilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanoni- trila (0,100 g, 0,221 mmol) em cloreto de metileno (4 mL) e ácido trifluoracé- tico (1 mL) foi agitada por 2 horas e os solventes foram evaporados. O resíduo foi agitado com etilenodiamina (0,1 mL, 2 mmol) em metanol (4,5 mL) por 1 hora e evaporado. O produto bruto foi reconstituído em metanol e purificado por duas etapas suecessivas de cromatografia usando HPLC-MS preparativa (eluindo com um gradiente de acetonitrila/H2O contendo 0,15% NH4OH para a primeira corrida, seguido por um gradiente de acetonitrila/H2O contendo 0,1% TFA para a segunda corrida) para obter os produtos racêmi- cos como o sal trifluoracetato. 1H RMN (400 MHz, d6-dmso): δ 12,72 (br s, 1H), 8,94 (s, 1H), 8,86 (s, 1H), 8,49 (s, 1H), 7,82 (s, 1H), 7,21 (s, 1H), 4,80 (dd, 1H), 3,51 (dd, 1H), 3,22 (dd, 1H), 1,79-1,42 (m, 7H), 1,25-1,15 (m, 1H); LCMS (M+H)+: 323,1.
Os metabolites 1-39 foram isolados de plasma, urina ou fezes de humano, rato, ou cão após administração do composto I em relação com estudos de farmacocinética e toxicocinética. A identidade dos metabolites foi determinada após isolamento de metabolite usando métodos HPLC. O método usado e os tempos de retenção correspondentes dos metabolites são apresentados na Tabela 2. Análise tandem MS/MS e experimentos MSn de ordem superior foram conduzidos como requerido para elucidar a informação estrutural.
Composto I demonstra um íon molecular protonado em m/z 307, e um espectro de íon do produto mostrando sinais em m/z 266, 186, e 159. Oíon de fragmento em m/z 266 é consistente com a perda de fração carbonitrila. Oíon de fragmento diagnóstico em m/z 186 é indicatido da perda de fração intacta de ciclopentilpropanonitrila. Oíon de fragmento em m/z 159 indica a perda de ciclopentilpropanonitrila em conjunto com divagem através do anel pirazol do composto i.
Compostos de metabolite 1, 2, 27, e 29 foram observados prin-cipalmente na urina de sujeitos humanos, com níveis de traços dos compostos 27 e 29 observados no plasma. Espectrometroa de massa de varredura completa demonstrou um íon molecular protonado em m/z 339, consistente com bis-hidroxilação do composto I. Fragmentação do íon do produto destes íons m/z 339 gera virtualmente espectros idênticos para estes metabolites, com íons observados em m/z 321, 186, 159 e 154. O íon em m/z 321 é consistente com a perda de água e sugere que pelo menos uma hidroxilação pode ser no anel ciclopentil. Os íons em m/z 186 e m/z 159 são consistentes com um intacto pirazole-pirrolopirimidina. O íon em m/z 154 é consistente com uma perda neutra da fração inteira não modificada de pirazole/pirrolopirimidina. Fragmentos de íons menores em m/z 298 e m/z 280 sugere a perda de elementos de acetonitrila, com e sem a perda fácil de água, ainda restringindo a localização da hidroxilação à fração ciclopentila. Isto é ainda suportado pelo íon em m/z 237, que é consistente com a perda de ciclopentila modificado, deixando o resto da molécula não modificada.
Composto 31 foi encontrado em urina de sujeitos humanos. Espectrometria de massa de varredura total demonstrou um íon molecular protonado em m/z 339, consistente com a adição de 32 amu para Composto I. Fragmentação do íon do produto de m/z 339 íon gera fragmentos de íons em m/z 311, 218, e 191. O principal fragmento em m/z 218 que é consistente com a adição de 32 amu a fração pirazole-pirrolopirimidina. Oíon de fragmento em m/z 191 é consistente com a perda de CHN da fração pirazol do bis putativo hidroxilado pirazole-pirrolopirimidina, restingindo a localização da modificação ao pirrolopirimidina. Oíon de fragmento em m/z 311 possivelmente origina de clivagem inicial da ligação amida, seguida por perda de CO de pirrolidinona. A designação estrutural do composto 31 usando espectrometria de massa isolada leva a um resultado ambíguo, portanto Composto 31 foi isolado de urina humana e analisado por 1H e 13C RMN. A estrutura de 31 é identificada como um metabólíto amida-álcool da fração saturada pirrolopirimidina do composto I. O espectro de próton RMN de 31 tem um singleto em Õ 3,56 com uma intensidade de 2H. Este síngieto tem uma correlação de longo alcance a um carbono em δ 177,9, consistente com uma amida. Além disso, uma intensificação nuclear Overhauser (nOe) ocorre entre H3 e H9.
Composto 32 foi encontrado em plasma e urina de sujeitos humanos. Espectrometria de massa de varredura total demonstrou um íon molecular protonado em m/z 339, consistente com a adição de 32 amu ao Composto I. Fragmentação do íon do produto de m/z 339 íon gera fragmentos de íons em m/z 218, e 191. O principal fragmento em m/z 218 que é consistente com a adição de 32 amu a fração pirazol-pirrolopirimidina. Oíon de fragmento em m/z 191 é consistente com a perdad e CHN da fração pirazol do putativo bis hidroxilado pirazole-pirrolopirimidina, restingindo a localização das modificações ao pirrolopirimidina. Designação estrutural do composto 32 usando espectrometria de massa isolada levou a um resultado ambíguo, portanto, o Composto 32 foi isolado de urina humana e analisado por 1H e 13C RMN. A estrutura do composto 32 é identificada como um ceto-álcool metabólito da fração saturada de pirrolopirimidina do composto I. O espectro de RMN de próton de 32 tem um singleto em δ 3,82 que tem uma intensidade de 2H e mostra uma longa correlação ao carbono em δ 191,5, consis- 5 tente com uma cetona. Não houve intensificação nuclear Overhauser (nOe) observada de H2 para qualquer outro próton.
Composto 40 foi observado em plasma e urina de sujeitos humanos. Espectrometria de massa de varredura total demonstrou um íon molecular protonado em m/z 341, consistente com a adição de 34 amu ao 10 Composto I. Fragmentação do íon do produto de m/z 341 íon gera fragmentos de ions em m/z 323, 220, 202, e 175. Oíon de fragmento em m/z 323 se origina da perda de água do pirrolidina diol. Fragmentos de ions em m/z 220 e 202 são consistentes com uma duplamente hidroxilada sautrada pir-rolopirimidina, com e sem a perda de água fácil observada. O íon observado 15 em m/z 175 é consistente com perda de CHN da fração pirazol de duplamente hidroxilada e saturada pirazol pirrolopirimidina após a perda fácil de água. Composto 40 é identificado como 3-ciclopentil-3-(4-(5,6-di-hidróxi-6,7- di-hidro-5H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)propanonitrila.
Conjugados de meiabóiítos do composto i simplesmente hidroxi- 20 lados contendo um m/z observado de 499 foram observados em urina de humano, com dois dos sete conjugados ainda observados em quantidades traços em plasma humano. A fragmentação inicial do íon do produto geral espectros virtualmente idênticos para seis destes metabólitos, com um íon observado em m/z 323, consistente com a perda do conjugado glicuronida. A 25 fragmentação do íon do produto (MS3) destes m/z 323 de fragmentos de íons novamente demonstrou espectros de massa virtualmente idênticos, com fragmentos de íons em m/z 305, 186, e 159. Os íons de fragmento encontrados em m/z 305 são consistentes com a perda de água (18 amu), sugerindo hidroxilação e subsequente colisão induzida por fragmentação em uma 30 parte saturada da molécula, restringindo o sítio de modificação par a fração ciclopentila. Os íons de fragmento em m/z 186 e m/z 159 são consistentes com uma não modificada pirazol-pirrolopirimidina. Fragmentos adicionais menores de íons foram muito fracos para designar com qualquer certeza. A identidade destes metabólitos putativos foi confirmada por hidrólise dos conjugados isolados de glicuronida com β-Glicuronidase para revelar a aglicona, que foi então submetida a análise por HPLC-MS onde os tempos de retecao e espectros de massa das agliconas liberadas foram coicindentes com aqueles de padrões de metabolite unicamente hidroxilado. As agliconas destes metabólitos conjugados de glicuronida correspondem a 2-hidroxil metabólitos (Metabolite 9, Tabela 1) e 3-hidroxil metabólitos (Metabolite 8, Tabela 1). A aglicona do sétimo metabolite conjugado de glicuronida, composto 42, cor-responde a um unicamente hidroxilado metabolite de fração pirazol-pirrolopi- rimidina do composto I, que não foi independentemente observada em plasma ou urina de sujeitos humanos. Espectrometria de massa de varredura total demonstrou um íon molecular protonado em m/z 323, consistente com a adição de 16 amu ao Composto I. A fragmentação de íon de produto de m/z 323 íon rende um fragmento primário em m/z 202 que é consistente com a adição de 16 amu a fração pirazole-pirrolopirimidina. Oíon de fragmento em m/z 175 é consistente com a perda de CHN da fração pirazol do hidroxilado putativo pirazole-pirrolopirimidina, restingindo a localização da modificação a pirrolopirimidina. A estrutura de composto metabolite 42 é identificada como um conjugado glicuronida de uma fração unicamente hidroxilada de pirrolopirimidina do composto I.
Composto metabolite 41 foi encontrado em urina de sujeitos humanos. Espectrometria de massa de varredura total indicou um íon molecular protonado em m/z 583, consistente com conjugação de glicuronida de não modificado Composto I. Fragmentação de íon de produto de m/z 583 íon rende um fragmento primário em m/z 307 que é consistente com a perda de uma glicuronida intacta. Fragmentos menores de íons em m/z 186 e m/z 159 são consistentes com aqueles observados para o Composto I. MS3 fragmentação de fragmento m/z 307 íon ainda revelou fragmentos de íons em m/z 186 e m/z 159. Composto 41 é identificado como um conjugado N-ligado de glicuronida do composto I.



MÉTODO A1: Biotransformação de 14C-Composto I em Amostras de Plasma, Urina e Fezes Humanas, de Cão e de Camundongo
Amostras de plasma de todos os indivíduos foram agrupadas por assunto e /ou por ponto de tempo. As amostras de plasma reunidas foram preparadas para análise por HPLC-radiométrica como se segue: Alíquotas das amostras de plasma reunidas foram inicialmente extraídas com aproximadamente dois equivalentes (P/V) volumes de 1% de ácido fórmico em acetonitrila de grau HPLC, seguida por vortex vigoroso, e centrifugação para remover as proteínas piasmáticas desnaturadas. O sobrenadante foi reservado e passado através de cartuchos pré-condicionados Waters C-18 de extração em fase sólida, e o filtrado retido. O cartucho de SPE foi retido. O restante do pellet de plasma foi então extraído três vezes com 1 % de ácido fórmico em acetonitrila grau HPLC/água (90/10), com centrifugação após cada extração. Todos os sobrenadantes foram reservados. Cada extração de sobrenadante em ácido fórmico/acetonitrila/água foi usada para eluir o cartucho SPE. O cartucho SPE foi então rinsado com 1% de ácido fórmico em metanol. Todos os eluentes foram retidos, combinados, e evaporados sob uma corrente de nitrogênio a 30°C. O restante desta extração foi reconstituído em aproximadamente 0,6 ml de 0,1% de ácido fórmico em água, agitada em vórtex e centrifugado para remover partículas. O sobrenadante foi então analisado por HPLC-Fluxo de Radiometria e HPLC-MS. Urina:
As alíquotas de quantidades aproximadamente iguais de urina a partir de cada um dos sujeitos foram reunidas para representar intervalos de tempo apropriados pós-dose. Antes de análise HPLC-radiométrica, cada amostra de urina reunida foi centrifugada a 4.000 rpm durante 10 min para remover as partículas, então, analisada diretamente. Fezes:
Alíquotas de homogenatos fecais de cada sujeito foram reunidas para representar intervalos de tempo apropriados pós-dose. As amostras fecais agrupadas de homogenatos foram preparados para análise por HPLC- radiométrica como se segue:
Alíquotas de cerca de 5 gramas de homogenato fecal foram inicialmente extraídas com aproximadamente dois equivalentes (W/V) volumes de água Millipore, seguido por centrifugação para remover os sólidos. O sobrenadante foi reservado e passado através de cartuchos pré-condiciona- dos Waters C-18 de extração em fase sólida. O cartucho de SPE foi retido. O restante do pellet fecal foi então extraído três vezes com 1% de ácido fórmico em acetonitríla grau HPLC/água (90/10), com centrifugação após cada extração. Todos os sobrenadantes foram reservados. Cada extração de sobrenadante em ácido fórmico/aceionitrüa/água foi usada para eiuir o cartucho SPE. Todos os eluentes foram retidos, combinados, e evaporados sob uma corrente de nitrogênio a 30°C. O restante desta extração foi reconstituído em aproximadamente 2,0 ml de 0,1% de ácido fórmico em água, agitada em vórtex e centrifugado para remover partículas. O sobrenadante foi, então, analisado por HPLC-Radiometria de Fluxo e HPLC-MS.
Método Analítico HPLC-Radiometria de Fluxo: Todas as amostras foram analisadas por HPLC-Radiometria de Fluxo ou HPLC-MS utilizando sistemas analíticos que eram tão idênticas quanto possível. O sistema HPLC-Radiometria de Fluxo consistiu em uma bomba Agilent HP1100 series Quaternary, Autoamostrador, e detector UV interfaceado com um detector de cintilação de fluxo Raytest Ramona-90 com uma célula de cintilação líquida de 500 μL. Separação do composto I e os seus metabólitos foi conseguida com uma coluna C-18 Waters Symmetry de
HPLC, 4,6 x 250 mm, 5 μm partícula tamanho da coluna de HPLC. Fase móvel “A” consistiu de 0,1% ácido fórmico em água grau HPLC, com fase móvel “B” consistindo de metanol /acetonitrila grau HPLC (1:1). A eluição dos analitos foi conseguida através da utilização de um gradiente linear crescente de fase móvel “B”. Dois métodos de eluição de gradientes diferentes foram empregados em vários momentos, conforme necessário. Condições cromatográficas para estes dois métodos são descritos a seguir nas Tabelas 3e4.
O sistema de HPLC-Radiometria de Fluxo consistiu em uma bomba Agilent HP1100 series Quaternary pump, e Autoamostrador Leap Technologies CTC-PAL interfaceado com um Applied Biosystems API 4000 Qtrap operado em modo de detecção positiva de íon. Varredura total (MS) e dados dependentes de varreduras de produtos de íons (MS2) foram empregados na caracterização de metabólitos não composto I. Transições MRM selecionadas também foram utilizadas para procurar e identificar metabólitos previamente identificados e caracterizados. Separação do composto I e os seus metabólitos foi conseguida com uma coluna C-18 Waters Symmetry de HPLC, 4,6 x 250 mm, 5 μm partícula tamanho da coluna de HPLC. Fase móvel “A” consistiu de 0,1% ácido fórmico em água grau HPLC, com fase móvel “B” consistindo de metanol /acetonitrila grau HPLC (1:1). A eluição dos analitos foi conseguida através da utilização de um gradiente linear crescente de fase móvel “B”. Dois métodos de eluição de gradientes diferentes foram empregados em vários momentos, conforme necessário. Condições cromatográficas para estes dois métodos são descritos a seguir nas Tabelas 3 e 4.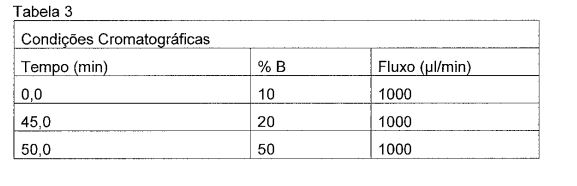

MÉTODO A2: Biotransformação do composto I: Isolamento de Metabólitos Isolamento de metabólitos da urina humana: 5 Extração em fase sólida, utilizando 20cc Waters cartuchos de HLB SPE (1 grama de sorvente) foram usados para concentrar metabólitos do composto I a partir de amostras de urina agrupadas. Os cartuchos foram condicionados primeiro com 100% de metanol grau HPLC, em seguida, com água Millipore. O cartucho foi, então, imediatamente carregado com até 10 10 ml de urina não processada bruta. O cartucho foi sequencialmente eluída uti- lizando várias concentrações de grau HPLC metanol in água (Tabela 5)

Alíquotas da urina inicial, o último volume de carga, e cada volu me de lavagem/eluição foram analisados por LC-MS para a presença do composto I e metabólitos de interesse. Não houve uma quantidade significativa de composto I, ou seus metabólitos, em qualquer dos volumes de carga, 5 ou lavagens/eluições até metanol a 25% em água Millipore. Nenhuma quantidade significativa de droga ou metabólitos foi encontrada nas alíquotas de eluição de 100% metanol. Volumes de eluição de 50% e 75% metanol em eluições foram reduzidos em volume usando o evaporador centrífugo Gene- vac e reconstituídos para um volume final de ~ 2 mL antes de serem injeta- 10 dos em LC-MS para coleta de frações dos metabólitos de interesse.
As amostras foram analisadas por LC-MS utilizando um Finnigan LCQ Deca XP Plus interfaceado com uma Shimadzu Binary HPLC stack consistindo de um amostrador Sil-HTC e o sistema controlador, e duas 15 bombas Sil 10ADVp de alta pressão LC. O espectrômetro de massa foi operado em modo positivo de detecção de íon, usando varredura dependente de dados para gerar dados MS e MS2 dependentes de dados. Um detector de UV Shimadzu, Sil SPD10 AVp foi utilizado no comprimento de onda de detecção de 254 nm para monitorar o eluente de HPLC em concerto com o 20 espectrômetro de massa. Fase móvel “A” consistiu em formato de amónio 5 mM que teve pH ajustado para pH 3,2 com ácido fórmico (aproximadamente 0,1% emvolume). A fase móvel “B” consistiu de 90% acetonitrila /10% metanol. A coluna de HPLC utilizado foi um Zobax XDB C-18, 3,0 mm x 150 mm, 5,0 25 Tm de tamanho de partícula. A eluição do analito foi através da utilização de um gradiente linear crescente de fase móvel “B”. As condições cromatográficas para a análise de escala analítica são descritos na Tabela 6 abaixo.

As amostras foram analisadas por LC-MS como acima com as seguintes alterações: A coluna de HPLC utilizada foi uma Phenomenex Polar RP 10 5 mm x 150 mm 5 Tm de tamanho de partícula.
O fracionamento inicial foi conduzido em acetato de amónio neutro como a fase “A”, com uma segunda etapa de limpeza de picos coletados individuais conduzidos usando 0,025% de ácido fórmico em água como a fase “A”. A fase móvel “B” consistiu de 90% acetonitríla /10% metanol.
A eluição inicial do analito e da coleta de fração automatizada foi através da utilização de um gradiente linear crescente de fase móvel “B”. Condições cromatográficas para a análise semi preparativa inicial estão descritos na Tabela 7 abaixo, mas % B foi otimizada conforme necessário para cada metabólito individual na segunda etapa de limpeza. de fracionamento
de fracionamento
O fluxo de eluente das análises semi-prep foram divididos usando um PEEK tee e tubulação, com ~ 1,65 ml/min do fluxo de eluente a ser utilizada para a coleta da fração, e o restante indo para o espectrômetro de massa. A composição de fluxo eluente foi monitorada por experimentos de MS e MS-MS selecionado. As frações contendo o analito de interesse foram recolhidas automaticamente, utilizando a válvula de desvio em concerto com um coletor de frações.
MÉTODO A3: Biotransformação do Composto I em amostras de urina e plasma humano e de rato
As amostras de plasma e urina dos animais e sujeitos humanos que tinham sido oralmente doseadas com Composto I foram obtidos após dose única e/ou estudos de doses múltiplas. Amostras de plasma residual para cada sujeito foram reunidas por agrupamento médio ponderado por tempo. As alíquotas das amostras de plasma agrupadas (150 μl_) foram precipitadas com dois volumes de acetonitrila, misturados em vortex e em seguida centrifugadas. Os sobrenadantes foram removidos e usados para a anáiise por LC-MS. Para humano, as amostras de urina de cada indivíduo no estudo foram agrupadas para representar 0-12 horas no Dia 1 (estudo de dose única) e Dia 10 (estudo de dose múltipla) do estudo clínico. As amostras de urina foram centrifugadas a 15.000 xg ~ durante 10 minutos antes da análise e injetadas diretamente.
As amostras foram analisadas usando ionização por eletrospray LC-MS com uma Thermo Finnigan LCQ Deca-XP Plus Ion-Trap Espectrômetro de Massa (Thermo Fisher Scientific, Waltham MA, EUA), operado em modo de ionização positivo. A varredura dependente dos dados foi usada para gerar MS inicial para os dados MS2. Experimentos de ordem superior MSn foram conduzidos como requerido para elucidar informação estrutural. O espectrômetro de massa foi acoplado a uma Shimadzu Sil HT-C combinada com autoamostrador /controlador combinado com um sistema de bomba de gradiente binário Shimadzu LC-10A (Shimadzu Scientific Instruments, Columbia, MD, EUA). As condições de gradiente são descritas na Tabela 8. A separação do composto I e seus metabólitos foi conseguida usando uma coluna Zorbax XDB C-18 HPLC (3,0 x 150 mm, 3,5 μm) (Agilent, Santa Clara, CA EUA) com uma vazão de fase móvel de 300 pL/min. A fase móvel “A” consistiu de 5 mM de formato de amónio em água Millipore que teve pH ajustado a pH 3,2 com ácido fórmico. A fase móvel “B” consistiu de 90% de acetonitríla /10% de metanol. Tabela 8 Esquema de Gradiente de Eluição de HPLC
Amostras de urina, fezes e bile de ratos administrados com Composto I foram reunidas. Aproximadamente 30% de cada amostra foi reunida a partir de cada ponto de tempo tal que 90% da dose excretada de um rato estava contida em uma única amostra. Amostras de cada rato foram analisadas separadamente. As amostras de fezes foram extraídas utilizando acetonitríla e recuperação de extração determinada por contagem de cintilação líquida (LSC) do extrato e combustão e LSC do pellet não extraído. Quando disponível, padrões de metabólito foram utilizados para confirmar estruturas. Picos representando <5% da dose administrada não foram relatados, a menos que indicado de outra maneira.
Amostras de urina, fezes e/ou bile foram analisadas por HPLC e espectroscopia de massa para determinar o peso molecular de quaisquer metabólitos marcados radioativamente e obter informação estrutural destes metabólitos.
As amostras de urina do Composto I de estudos de farmacoci- nética e toxicocinética foram reunidas a partir de animais individuais da mes- 5 ma espécie, ou seja, cão ou de rato. As amostras de urina foram extraídas com acetato de etila em uma proporção em volume de 1 para 1 três vezes. As frações de extração em acetato de etila foram reunidas e os voláteis foram removidos usando um rotavapor. O resíduo resultante foi dissolvido em acetonitrila:água (1:1 v/v) e a solução foi submetida a purificação HPLC 10 preparativa. Isolamento de cada metabolito foi conseguido usando as três condições de HPLC abaixo em ordem sequencial.
O extrato concentrado foi purificado usando uma fase móvel que consiste de 0,1% TFA em água (Solvente A) e 0,1% TFA em acetonitrila (Solvente B), em uma coluna Zobax SB C18 (19x150mm, 5 Tm) usando um 15 gradiente de 5% a 60% de solvente B em 20 minutos, e uma vazão de 15 mL/min. 1. As frações recolhidas a partir da primeira purificação por HPLC foram então purificadas usando uma fase móvel consistindo de 0,1% de TFA em água (solvente A) e metanoi (Solvente B), em uma coluna Zobax 20 SB C18 (9,6 x150mm, 5 Tm), utilizando um gradiente de 5% a 95% do solvente B durante 25 minutos, e uma vazão de 4 mL/min. 2. Cada fração coletada a partir da segunda purificação com [M H]+ = 321 e 323 foi submetida a separação quiral usando uma fase móvel consistindo de etanol 15% e 85% hexanos ou etanol 30% e 70% hexanos, 25 em uma coluna quiral (Chiralcel OD-H, 20x250mm, 5 Tm) com uma vazão de 15 mL/min.
Os tempos de retenção para 35, 37 e 38 foram obtidos a partir da análise de HPLC usando coluna Zorbax SB C18 (4,6x150 mm, 80Â, 3,5 30 Tm) em uma temperatura de coluna de 40°C usando uma fase móvel que consiste de 0,05% TFA em água (Solvente A) e 0,05% TFA em Acetonitrila (Solvente B) com uma vazão de 1 mL/min usando um esquema de gradiente de eluição mostrado na Tabela 9. Uma detecção por UV em linha foi realizada a 220 nm. Tabela 9 Esquema de Gradiente de eluição
Os tempos de retenção para 40, 41 e 42 foram ontidos a partir de análise de HPLC usando uma coluna Waters Atlantis® T-3 HPLC (4,6 x 150 mm, 3,5μm) (Waters Corporation, Milford, MA, EUA) com uma vazão de fase móvel de 400 pL/min. A fase móvel “A” consistiu de 5 mM de formato de amónio em água Millipore que teve pH ajustado para pH 3,2 com ácido fórmico. A fase móvel “B” consistiu de 100% metanol. A condição de fase móvel inicial foi 90% de fase móvel “A” / 10% de fase móvel “B” com uma etapa de gradiente a 27% de fase móvel “B” em um minuto. O gradiente inicial, então, progrediu de modo linear a 52% de fase móvel “B” em 57 minutos, seguido por um segundo gradiente linear para 95% de fase móvel “B” em 10 minutos. Um período de lavagem da coluna de cinco minutos a 95% de fase móvel “B” ocorreu e foi seguido por um retorno para as condições inicias e um reequilíbrio da coluna de 8 minutos antes da próxima injeção analítica. 100% do eluente da coluna foi encaminhado através de um detector de UV Shimadzu comprimento de onda fixo, SPD-10AV (Shimadzu Scientific Instruments, Columbia, MD, EUA) monitorando X.254nm. Ao sair do detector de UV, o fluxo de eluente foi dividido usando um PEEK tee para permitir aproximadamente 100 μl de eluente para ser introduzido através de ionização por eletrospray para o espectrômetro de massa. A tensão da fonte de eletrospray foi fixada em 4,5 kV, com uma Temperatura capilar de 325°C, bainha e gases de varrimento foram fixados em 50 e 30 (unidades arbitrárias), respectivamente. Conjuntos MS/MS de dados iniciais dependentes in- cluíram uma largura de isolamento de 2,0 amu e uma configuração de energia de colisão de 42. Todas as configurações de outros instrumentos e potencialidades foram otimizados para atingir máximo do sinal-ruído para Composto I.
Metabolites isolados glicuronida conjugadas do Composto I foram incubadas com β-glucuronidase com um processo de reação até conclusão em todos os casos, obtendo-se a aglicona associada, a qual foi analisada utilizando o método de LC-MS mesmo como descrito acima. Os tempos de retenção e de massa espectros das agliconas libertadas foram comparados com os tempos de retenção e espectros de massa dos isolados padrões hidroxilados metabolites do Composto I para atribuir a identidade do conjugado.
As amostras de metabólito de peso molecular de 323, foram pre-sumidas como sendo hidroxil contendo metabolites e foram dissolvidos in 200 μL de CD3OD, obtido a partir de Isotec. Amostras de peso molecular 321 foram presumidas por conter uma fração cetona e foram dissolvidas em 200 μL de CDCI3, obtido a partir de Isotec. As amostras que eram de um peso molecular diferente do descrito acima foram dissolvidas in 200 μL de DMSO- d6, também obtido a partir de Isotec. Após dissolução, cada amostra foi colocada num tubo de RMN 3 mm, obtidas a partir de Wilmad Glass. As amostras foram preparadas imediatamente antes da análise.
As amostras foram analisadas usando um espectrômetro Varian INOVA RMN de RMN a 500 MHz para 1H e 125 MHz para 13C. Uma sonda de gradiente de detecção inversa de tripla ressonância de 3mm foi usada. A amostra foi mantida a 30°C durante o tempo que toda a aquisição de dados ocorreu. A sonda foi bloqueada e enchida para cada amostra. Outros experimentos de RMN foram realizados para obter os dados necessários para determinar a estrutura.
Composto 31 1H(500 MHz, DMSO-D6): δ 8,58 (s, 1H), 8,23 (s, 1H), 4,47 (td, 1H), 3,56 (s, 2H), 3,18 (dd, 1H), 3,13 (dd, 1H), 2,35 (m, 1H), 1,79 (m, 2H), 1,59-1,39 (m, 4H), 1,29 (m, 2H).
Composto 32 1H(500 MHz, DMSO-D6): δ 9,24 (s, 1H), 8,66 (s, 1H), 4,56 (td, 1H), 3,82 (s, 2H), 3,17 (dd, 1H), 3,11 (dd, 1H), 2,35 (m, 1H), 1,79-1,12 (m, 4H), 1,61-1,45 (m, 4H).
Composto 35 1H(500 MHz, DMSO-D6): δ 12,25 (s, 1H), 8,82 (s, 1H), 8,73 (s, 1H), 8,37 (s, 1H), 7,64 (m, 1H), 7,08 (m, 1H), 4,93 (dd, J=11,6, 3,3, 1H), 3,76 (d, J=4,8, 1H), 3,52 (dd, J=17,2, 11,7, 1H), 3,08 (dd, J=17,4, 3,6), 2,04 (m, 1H), 1,76 (m,1H), 1,63 (m, 1H), 1,54 (m, 2H), 0,95 (m, 1H).
Composto 37 1H(500 MHz, CDCI3): δ 8,71 (s, 1H), 8,17 (s, 1H), 8,00 (s, 1H), 4,22 (td, J=9,6, 3,7, 1H), 3,71 (s, 2H), 3,09 (dd, J=17,0, 9,1, 1H), 2,91 (dd, J=16,8, 3,7, 1H), 2,55 (m, 1H), 1,94 (m, 1H), 1,80-1,45 (m, 5H), 1,27 (m, 1H), 1,19 (m, 1H).
Composto 38 1H(500 MHz, DMSO-D6): δ 11,35 (s, 1H), 8,60 (s, 1H), 8,54 (s, 1H), 8,09 (s, 1H), 4,55 (td, J=9,8, 3,8, 1H), 4,10 (m, 1H), 3,79 (s, 2H), 3,19 (dd, J=16,9, 9,5, 1H), 3,13 (dd, J=17,5, 4,5, 1H), 2,39 (m, 1H), 2,01 (m, 1H), 1,58 (m, 1H), 1,48-1,07 (m, 4H).
Os dados de atividade para os Metabólitos 1-39, junto com a tração livre e dados de depuração intrínsecos, podem ser comparados com aqueles para o composto parente, Composto I. Ensaios da atividade de JAK, ensaios de fração livre, e ensaios de depuração intrínseco são descritos abaixo. Os pontos de dados podem ser obtidos para estereoisômeros individuais de metabólitos 1-39. Metabólitos podem ser potentes inibidores de JAK1, JAK2, e JAK3, como Composto I.
Os compostos descritos aqui podem ser testados para atividade inibitória de alvos de JAK de acordo com o seguinte ensaio in vitro descrito em Park et al., Analytical Biochemistry 1999, 269, 94-104. Os domínios catalíticos de JAK1 humana (a 837-1142), JAK2 (a e JAK3 (a 781-1124) com tag Hís N-terminal podem ser expressos usando baculovirus em células de inseto e purificados. A atividade catalítica de JAK1, JAK2 ou JAK3 pode ser ensaiada por medição da fosforílação de um peptídeo biotinilado. O peptídeo fosforilado pode ser detectado por uma fluorescência resolvida de tempo homogêneo (HTRF). lC50s dos compostos podem ser medidas para cada quinase nas reações que contêm a enzima, ATP e 500 nM de peptídeo em tampão Tris 50 mM (pH 7,8) com NaCI 100 mM, DTT 5 mM e 0,1 mg/mL (0,01%) BSA. A concentração de ATP nas reações pode ser de 90 μM para Jak1, 30 μM para Jak2 e 3 μM para Jak3. As reações podem ser conduzidas em temperatura ambiente por 1 hr e em seguida interrompidas com 20 μL EDTA 45 mM, SA-APC 300 nM, Eu-Py20 6 nM em tampão do ensaio (Perkin Elmer, Boston, MA). Ligação ao anticorpo marcado com európio pode ser, por 40 minutos e sinal HTRF pode ser medido com um leitor de placas de Fusão (Perkin Elmer, Boston, MA). Compostos com uma IC50 de 10 μM ou menos para qualquer um dos alvos JAK acima mencionados pode ser considerado ativo.
A proteína de ligação de um composto de teste pode ser determinada por diálise de equilíbrio utilizando um sistema Dianorm de Harvard Apparatus (Holliston, MA). A diálise pode ser realizada a 37°C durante 2 horas em soro humano. Os metabólitos podem ser incubados a 3 μM, e Composto I em 3 e 10 μM. As concentrações do composto no soro e tampão pós- diálise podem ser determinadas por análise LC/MS/MS. A fração livre é definida como a razão entre o tampão versus concentração de soro. ENSAIO DE DEPURAÇÃO INTRÍNSECA
Depuração intrínseca pode ser determinada por incubação de 1 μM do composto teste em humano misturado suavemente como microsso- mas hepáticos (0,5 mg/mL proteína) a 37°C na presença de 1 mM NADPH. O desaparecimento do composto teste pode ser monitorado por LC/MS a 0, 5, 10, 20 e 30 min. A inclinação do declínio na concentração do composto pode ser usada para calcular a depuração intrínseca humana empregando métodos reportados na literatura.
Várias modificações da invenção, além daquelas descritas aqui, ficarão aparentes aos versados na técnica a partir da descrição anterior. Tais modificações são ainda pretendidas para estar dentro do escopo das reivindicações anexadas. Cada referência citada no presente pedido está incorporada aqui como referência em sua totalidade.
Claims (18)
1. Composto, caracterizado pelo fato de que apresenta a Fórmula I: ou um sal farmaceuticamente aceitável do mesmo, em que: grupos n C-H são cada qual independentemente substituídos por C-OH; ou um grupo CH2 é independentemente substituído por C=O; ou um grupo C-H é substituído por:
ou um sal farmaceuticamente aceitável do mesmo, em que: grupos n C-H são cada qual independentemente substituídos por C-OH; ou um grupo CH2 é independentemente substituído por C=O; ou um grupo C-H é substituído por: 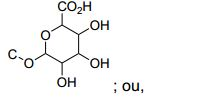 dois grupos C-H são cada qual independentemente substituídos por C-OH e um grupo C-H é substituído por:
dois grupos C-H são cada qual independentemente substituídos por C-OH e um grupo C-H é substituído por:  n é 1, 2, 3 ou 4; desde que o composto não seja selecionado de: 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- oxociclopentil)propanonitrila; e sais farmaceuticamente aceitáveis do mesmo.
n é 1, 2, 3 ou 4; desde que o composto não seja selecionado de: 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- oxociclopentil)propanonitrila; e sais farmaceuticamente aceitáveis do mesmo.
2. Composto, de acordo com a reivindicação 1, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que: o átomo de carbono alfa para o grupo ciano não é substituído por um C-OH ou grupo C=O; e o átomo de carbono beta para o grupo ciano não é substituído por um grupo C-OH.
3. Composto, de acordo com a reivindicação 1 ou 2, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que os grupos n C-H são cada qual independentemente substituídos por C-OH.
4. Composto, de acordo com a reivindicação 1 ou 2, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que n é 1.
5. Composto, de acordo com a reivindicação 1 ou 2, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que n é 2.
6. Composto, de acordo com a reivindicação 1 ou 2, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que n é 3.
7. Composto, de acordo com a reivindicação 1 ou 2, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que n é 4.
8. Composto, de acordo com a reivindicação 1 ou 2, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que um grupo CH2 é independentemente substituído por um C=O.
9. Composto, de acordo com a reivindicação 1 ou 2, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que um grupo C-H é substituído por
10. Composto, de acordo com a reivindicação 1 ou 2, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que um grupo C-H saturado é substiituído por
11. Composto, de acordo com a reivindicação 1 ou 2, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que dois grupos C-H são cada qual independentemente substituídos por C-OH e um grupo C-H é substituído por:
12. Composto, de acordo com a reivindicação 1, caracterizado pelo fato de que é selecionado de: Ácido 6-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2- cianoetil)ciclopentilóxi)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1,2-di- hidroxiciclopentil)propanonitrila; e 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1- hidroxiciclopentil)propanonitrila; ou um sal farmaceuticamente aceitável dos mesmos.
13. Composto, de acordo com a reivindicação 1, selecionado de: 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-ciclopentil- 3-hidroxipropanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-ciclopentil- 2-hidroxipropanonitrila; 3-ciclopentil-3-(5-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il)propanonitrila; 3-ciclopentil-3-(3-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(5-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(6-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(2-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1,2-di- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1,3-di- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2-hidróxi-3-(1- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-hidróxi-3-(1- hidroxiciclopentil)propanonitrila; 3-(3-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1- hidroxiciclopentil)propanonitrila; 3-(4-(2-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1- hidroxiciclopentil)propanonitrila; 3-(4-(5-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1- hidroxiciclopentil)propanonitrila; 3-(4-(6-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1- hidroxiciclopentil)propanonitrila; 3-(5-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2-hidróxi-3-(2- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2,3-di- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2,4-di- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2,5-di- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-hidróxi-3-(2- hidroxiciclopentil)propanonitrila; 3-(3-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2- hidroxiciclopentil)propanonitrila; 3-(4-(2-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2- hidroxiciclopentil)propanonitrila; 3-(4-(5-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2- hidroxiciclopentil)propanonitrila; 3-(4-(6-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2- hidroxiciclopentil)propanonitrila; 3-(5-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2-hidróxi-3-(3- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3,4-di- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-hidróxi-3-(3- hidroxiciclopentil)propanonitrila; 3-(5-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(3-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(4-(2-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(4-(5-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(4-(6-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(3-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(4-(2-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(4-(5-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(4-(6-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(3- hidroxiciclopentil)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-ciclopentil- 2,3-di-hidroxipropanonitrila; 3-ciclopentil-3-hidróxi-3-(3-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-hidróxi-3-(4-(2-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-hidróxi-3-(4-(5-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-hidróxi-3-(4-(6-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-hidróxi-3-(5-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-2-hidróxi-3-(3-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-2-hidróxi-3-(4-(2-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-2-hidróxi-3-(4-(5-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-2-hidróxi-3-(4-(6-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-2-hidróxi-3-(5-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(3,5-di-hidróxi-4-(7H-pirrolo[2,3-d]pirimidin-4-il)- 1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(3-hidróxi-4-(2-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(3-hidróxi-4-(5-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(3-hidróxi-4-(6-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(2,5-di-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)- 1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(2,6-di-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)- 1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(5,6-di-hidróxi-7H-pirrolo[2,3-d]pirimidin-4-il)- 1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(5-hidróxi-4-(2-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(5-hidróxi-4-(5-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(5-hidróxi-4-(6-hidróxi-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 2-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2- ciclopentilacetil cianida; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(2- oxociclopentil)propanonitrila; ácido 6-(1-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2- cianoetil)ciclopentiletóxi)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; ácido 6-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2- ciano-1-ciclopentiletóxi)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; ácido 6-(2-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2- cianoetil)ciclopentilóxi)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; ácido 6-(2-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-1- ciano-2-ciclopentiletóxi)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; e ácido 6-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2- cianoetil)ciclopentilóxi)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; ou um sal farmaceuticamente aceitável dos mesmos.
14. Composto, caracterizado pelo fato de que é selecionado de: 3-ciclopentil-3-(4-(2,6-dioxo-3,5,6,7-tetra-hidr-2H-pirrolo[2,3- d]pirimidin-4-il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(2-hidróxi-6-oxo-6,7-di-hidro-5H-pirrolo[2,3- d]pirimidin-4-il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(6-oxo-6,7-di-hidro-5H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il)propanonitrila; 3-(3-hidroxiciclopentil)-3-(4-(6-oxo-6,7-di-hidro-5H-pirrolo[2,3- d]pirimidin-4-il)-1H-pirazol-1-il)propanonitrila; 3-ciclopentil-3-(4-(5,6-di-hidróxi-6,7-di-hidro-5H-pirrolo[2,3- d]pirimidin-4-il)-1H-pirazol-1-il)propanonitrila; e Ácido 6-(4-(1-(2-ciano-1-ciclopentiletil)-1H-pirazol-4-il)-7H- pirrolo[2,3-d]pirimidin-7-il)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; ou um sal farmaceuticamente aceitável dos mesmos.
15. Composto, caracterizado pelo fato de que é selecionado de: Ácido 6-(4-(1-(2-ciano-1-ciclopentiletil)-1H-pirazol-4-il)-7H- pirrolo[2,3-d]pirimidin-5-ilóxi)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; Ácido 6-(4-(1-(2-ciano-1-ciclopentiletil)-1H-pirazol-4-il)-7H- pirrolo[2,3-d]pirimidin-6-ilóxi)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; Ácido 6-(4-(1-(2-ciano-1-ciclopentiletil)-1H-pirazol-4-il)-7H- pirrolo[2,3-d]pirimidin-2-ilóxi)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; e Ácido 6-(4-(1-(2-ciano-1-ciclopentiletil)-1H-pirazol-4-il)-7H- pirrolo[2,3-d]pirimidin-7-ilóxi)-3,4,5-triidroxitetra-hidro-2H-piran-2-carboxílico; ou um sal farmaceuticamente aceitável dos mesmos.
16. Composto, de acordo com qualquer uma das reivindicações 1 a 15, ou sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que é isolado.
17. Composição, caracterizada pelo fato de que compreende um composto como definido em qualquer uma das reivindicações 1 a 15, ou um sal farmaceuticamente aceitável do mesmo, e um veículo farmaceuticamente aceitável.
18. Composição, de acordo com a reivindicação 17, caracterizado pelo fato de que é para administração oral.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US25038709P | 2009-10-09 | 2009-10-09 | |
| US61/250,387 | 2009-10-09 | ||
| US31621810P | 2010-03-22 | 2010-03-22 | |
| US61/316,218 | 2010-03-22 | ||
| PCT/US2010/052011 WO2011044481A1 (en) | 2009-10-09 | 2010-10-08 | Hydroxyl, keto, and glucuronide derivatives of 3-(4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| BR112012008267A2 BR112012008267A2 (pt) | 2021-11-03 |
| BR112012008267B1 true BR112012008267B1 (pt) | 2022-10-04 |
Family
ID=43037845
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BR112012008267-1A BR112012008267B1 (pt) | 2009-10-09 | 2010-10-08 | Derivados hidroxila, ceto e glucuronida de 3-(4-(7h-pirrolo[2,3-d]pirimidin-4-il)-1h-pirazol-1- il)-3-ciclopentilpropanonitrila |
Country Status (13)
| Country | Link |
|---|---|
| US (3) | US8486902B2 (pt) |
| EP (1) | EP2486041B1 (pt) |
| JP (3) | JP5946768B2 (pt) |
| KR (3) | KR20180126619A (pt) |
| CN (2) | CN105541847B (pt) |
| BR (1) | BR112012008267B1 (pt) |
| CA (1) | CA2777114C (pt) |
| EA (1) | EA021478B1 (pt) |
| ES (1) | ES2435491T3 (pt) |
| MX (1) | MX2012004180A (pt) |
| PL (1) | PL2486041T3 (pt) |
| PT (1) | PT2486041E (pt) |
| WO (1) | WO2011044481A1 (pt) |
Families Citing this family (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2343298B9 (en) | 2005-12-13 | 2020-05-06 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| RS53245B2 (sr) | 2007-06-13 | 2022-10-31 | Incyte Holdings Corp | Soli inhibitora janus kinaze (r)-3-(4-(7h-pirolo(2,3-d) pirimidin-4-il)-1h-pirazol-1-il)-3-ciklopentilpropan-nitrila |
| CL2008001709A1 (es) | 2007-06-13 | 2008-11-03 | Incyte Corp | Compuestos derivados de pirrolo [2,3-b]pirimidina, moduladores de quinasas jak; composicion farmaceutica; y uso en el tratamiento de enfermedades tales como cancer, psoriasis, artritis reumatoide, entre otras. |
| CL2009001884A1 (es) * | 2008-10-02 | 2010-05-14 | Incyte Holdings Corp | Uso de 3-ciclopentil-3-[4-(7h-pirrolo[2,3-d]pirimidin-4-il)-1h-pirazol-1-il)propanonitrilo, inhibidor de janus quinasa, y uso de una composición que lo comprende para el tratamiento del ojo seco. |
| JOP20190230A1 (ar) | 2009-01-15 | 2017-06-16 | Incyte Corp | طرق لاصلاح مثبطات انزيم jak و المركبات الوسيطة المتعلقة به |
| HRP20192203T1 (hr) | 2009-05-22 | 2020-03-06 | Incyte Holdings Corporation | 3-[4-(7h-pirolo[2,3-d]pirimidin-4-il)-1h-pirazol-1-il]oktan- ili heptan-nitril kao jak inhibitori |
| WO2010135650A1 (en) * | 2009-05-22 | 2010-11-25 | Incyte Corporation | N-(HETERO)ARYL-PYRROLIDINE DERIVATIVES OF PYRAZOL-4-YL-PYRROLO[2,3-d]PYRIMIDINES AND PYRROL-3-YL-PYRROLO[2,3-d]PYRIMIDINES AS JANUS KINASE INHIBITORS |
| TW201113285A (en) | 2009-09-01 | 2011-04-16 | Incyte Corp | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| BR112012008267B1 (pt) * | 2009-10-09 | 2022-10-04 | Incyte Holdings Corporation | Derivados hidroxila, ceto e glucuronida de 3-(4-(7h-pirrolo[2,3-d]pirimidin-4-il)-1h-pirazol-1- il)-3-ciclopentilpropanonitrila |
| JP5858434B2 (ja) * | 2010-02-18 | 2016-02-10 | インサイト・ホールディングス・コーポレイションIncyte Holdings Corporation | Janusキナーゼ阻害薬としてのシクロブタンおよびメチルシクロブタン誘導体 |
| SI3354652T1 (sl) | 2010-03-10 | 2020-08-31 | Incyte Holdings Corporation | Derivati piperidin-4-il azetidina kot inhibitorji JAK1 |
| MY161078A (en) | 2010-05-21 | 2017-04-14 | Incyte Holdings Corp | Topical formulation for a jak inhibitor |
| JP5917545B2 (ja) | 2010-11-19 | 2016-05-18 | インサイト・ホールディングス・コーポレイションIncyte Holdings Corporation | Jak阻害剤としてのシクロブチル置換ピロロピリジンおよびピロロピリミジン誘導体 |
| WO2012068440A1 (en) | 2010-11-19 | 2012-05-24 | Incyte Corporation | Heterocyclic-substituted pyrrolopyridines and pyrrolopyrimidines as jak inhibitors |
| JP5936628B2 (ja) | 2011-02-18 | 2016-06-22 | ノバルティス・ファルマ・アクチェンゲゼルシャフトNovartis Pharma AG | mTOR/JAK阻害剤併用療法 |
| PE20140832A1 (es) | 2011-06-20 | 2014-07-14 | Incyte Corp | Derivados de azetidinil fenil, piridil o pirazinil carboxamida como inhibidores de jak |
| EP2741747A1 (en) | 2011-08-10 | 2014-06-18 | Novartis Pharma AG | JAK P13K/mTOR COMBINATION THERAPY |
| TW201313721A (zh) | 2011-08-18 | 2013-04-01 | Incyte Corp | 作為jak抑制劑之環己基氮雜環丁烷衍生物 |
| UA111854C2 (uk) | 2011-09-07 | 2016-06-24 | Інсайт Холдінгс Корпорейшн | Способи і проміжні сполуки для отримання інгібіторів jak |
| WO2013082476A1 (en) | 2011-11-30 | 2013-06-06 | Emory University | Antiviral jak inhibitors useful in treating or preventing retroviral and other viral infections |
| US9193733B2 (en) | 2012-05-18 | 2015-11-24 | Incyte Holdings Corporation | Piperidinylcyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| CA2879603A1 (en) | 2012-07-27 | 2014-01-30 | Ratiopharm Gmbh | Oral dosage forms for modified release comprising ruxolitinib |
| CA2888816A1 (en) | 2012-11-01 | 2014-05-08 | Incyte Corporation | Tricyclic fused thiophene derivatives as jak inhibitors |
| TWI702057B (zh) | 2012-11-15 | 2020-08-21 | 美商英塞特控股公司 | 盧梭利替尼之緩釋性劑型 |
| WO2014118388A1 (en) | 2013-02-04 | 2014-08-07 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for assaying jak2 activity in red blood cells and uses thereof |
| ES2900492T3 (es) | 2013-03-06 | 2022-03-17 | Incyte Holdings Corp | Procesos y productos intermedios para elaborar un inhibidor de JAK |
| PL3786162T3 (pl) | 2013-05-17 | 2024-04-08 | Incyte Holdings Corporation | Pochodne bipirazolu jako inhibitory jak |
| RS60469B1 (sr) | 2013-08-07 | 2020-07-31 | Incyte Corp | Dozni oblici sa produženim oslobađanjem za jak1 inhibitor |
| MA39987A (fr) | 2014-04-30 | 2017-03-08 | Incyte Corp | Procédés de préparation d'un inhibiteur de jak1 et nouvelles formes associées |
| US9498467B2 (en) | 2014-05-30 | 2016-11-22 | Incyte Corporation | Treatment of chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML) by inhibitors of JAK1 |
| JP6149810B2 (ja) * | 2014-06-13 | 2017-06-21 | 株式会社島津製作所 | 代謝物解析システム及び代謝物解析方法 |
| PL3179991T3 (pl) | 2014-08-11 | 2022-02-14 | Acerta Pharma B.V. | Kombinacje terapeutyczne inhibitora btk i inhibitora bcl-2 |
| US20170239351A1 (en) | 2014-08-11 | 2017-08-24 | Acerta Pharma B.V. | Therapeutic Combinations of a BTK Inhibitor, a PI3K Inhibitor, a JAK-2 Inhibitor, a PD-1 Inhibitor, and/or a PD-L1 Inhibitor |
| TW201618773A (zh) | 2014-08-11 | 2016-06-01 | 艾森塔製藥公司 | Btk抑制劑、pi3k抑制劑、jak-2抑制劑、及/或cdk4/6抑制劑的治療組合物 |
| CN105777754B (zh) * | 2014-12-16 | 2019-07-26 | 北京赛林泰医药技术有限公司 | 吡咯并嘧啶化合物 |
| US10435428B2 (en) | 2015-11-24 | 2019-10-08 | Theravance Biopharma R&D Ip, Llc | Prodrugs of a JAK inhibitor compound for treatment of gastrointestinal inflammatory disease |
| CN107513067A (zh) * | 2016-06-16 | 2017-12-26 | 北京赛林泰医药技术有限公司 | 含有取代环戊基的吡咯并嘧啶化合物 |
| CN107759623B (zh) * | 2016-08-23 | 2020-08-14 | 苏州旺山旺水生物医药有限公司 | Jak抑制剂的中间体及其制备方法 |
| CN110418796A (zh) | 2017-03-08 | 2019-11-05 | 施万生物制药研发Ip有限责任公司 | 托法替尼(tofacitinib)的葡萄糖苷酸前药 |
| WO2018167283A1 (en) | 2017-03-17 | 2018-09-20 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the diagnosis and treatment of pancreatic ductal adenocarcinoma associated neural remodeling |
| WO2018189335A1 (en) | 2017-04-13 | 2018-10-18 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the diagnosis and treatment of pancreatic ductal adenocarcinoma |
| CN108794480A (zh) * | 2017-04-28 | 2018-11-13 | 天津药物研究院有限公司 | 吡咯并嘧啶类化合物、其制备方法和用途 |
| WO2018217699A1 (en) | 2017-05-23 | 2018-11-29 | Theravance Biopharma R&D Ip, Llc | Thiocarbamate prodrugs of tofacitinib |
| JP2020520955A (ja) | 2017-05-23 | 2020-07-16 | セラヴァンス バイオファーマ アール&ディー アイピー, エルエルシー | ヤヌスキナーゼ阻害剤のグルクロニドプロドラッグ |
| TW201924683A (zh) | 2017-12-08 | 2019-07-01 | 美商英塞特公司 | 用於治療骨髓增生性贅瘤的低劑量組合療法 |
| PT3746429T (pt) | 2018-01-30 | 2022-06-20 | Incyte Corp | Processos para a preparação de (1-(3-fluoro-2-(trifluorometil)isonicotinoíl)piperidin-4-ona) |
| BR122023022189A2 (pt) | 2018-02-16 | 2024-02-20 | Incyte Corporation | Usos de inibidores da via de jak1 para o tratamento de distúrbios relacionados a citocinas |
| EP4424328A3 (en) | 2018-03-30 | 2024-12-04 | Incyte Corporation | Treatment of hidradenitis suppurativa using jak inhibitors |
| JP7565798B2 (ja) | 2018-03-30 | 2024-10-11 | インサイト・コーポレイション | 炎症性皮膚疾患のバイオマーカー |
| CA3097025A1 (en) | 2018-04-13 | 2019-10-17 | Incyte Corporation | Biomarkers for graft-versus-host disease |
| US12109193B2 (en) | 2018-07-31 | 2024-10-08 | Loxo Oncology Inc. | Spray-dried dispersions, formulations, and polymorphs of (s)-5-amino-3-(4-((5-fluoro-2-methoxybenzamido)methyl)phenyl)-1-(1,1,1-trifluoropropan-2-yl)-1H-pyrazole-4-carboxamide |
| CN119751336A (zh) * | 2018-09-06 | 2025-04-04 | 艾尼纳制药公司 | 可用于治疗自身免疫性病症和炎性病症的化合物 |
| EP3873433A1 (en) | 2018-10-31 | 2021-09-08 | Incyte Corporation | Combination therapy for treatment of hematological diseases |
| WO2020092015A1 (en) | 2018-11-02 | 2020-05-07 | University Of Rochester | Therapeutic mitigation of epithelial infection |
| JP2022515198A (ja) | 2018-12-19 | 2022-02-17 | アレイ バイオファーマ インコーポレイテッド | FGFRチロシンキナーゼの阻害剤としての置換ピラゾロ[1,5-a]ピリジン化合物 |
| US12351571B2 (en) | 2018-12-19 | 2025-07-08 | Array Biopharma Inc. | Substituted quinoxaline compounds as inhibitors of FGFR tyrosine kinases |
| WO2021062163A1 (en) | 2019-09-27 | 2021-04-01 | Disc Medicine, Inc. | Methods for treating myelofibrosis and related conditions |
| CN110538183B (zh) * | 2019-10-09 | 2021-05-04 | 吉林大学 | 一种预防和治疗小儿湿疹的组合物及其制备方法 |
| JP2022551649A (ja) | 2019-10-10 | 2022-12-12 | インサイト・コーポレイション | 移植片対宿主病のバイオマーカー |
| US12360120B2 (en) | 2019-10-10 | 2025-07-15 | Incyte Corporation | Biomarkers for graft-versus-host disease |
| MX2022006176A (es) | 2019-11-22 | 2022-08-17 | Incyte Corp | Terapia combinada que comprende un inhibidor de alk2 y un inhibidor de jak2. |
| WO2021231798A1 (en) | 2020-05-13 | 2021-11-18 | Disc Medicine, Inc. | Anti-hemojuvelin (hjv) antibodies for treating myelofibrosis |
| HRP20241560T1 (hr) | 2020-06-02 | 2025-01-17 | Incyte Corporation | Postupci za proizvodnju inhibitora jak1 |
| US11833155B2 (en) | 2020-06-03 | 2023-12-05 | Incyte Corporation | Combination therapy for treatment of myeloproliferative neoplasms |
| US11905292B2 (en) | 2020-08-18 | 2024-02-20 | Incyte Corporation | Process and intermediates for preparing a JAK inhibitor |
| AR123268A1 (es) | 2020-08-18 | 2022-11-16 | Incyte Corp | Proceso e intermediarios para preparar un inhibidor de jak1 |
| IL303238A (en) | 2020-12-08 | 2023-07-01 | Incyte Corp | Jak1 pathway inhibitors for the treatment of vitiligo |
| TW202308610A (zh) | 2021-05-03 | 2023-03-01 | 美商英塞特公司 | 用於治療結節性癢疹之jak1途徑抑制劑 |
| CN118317946A (zh) | 2021-07-12 | 2024-07-09 | 因赛特公司 | 用于制备巴瑞替尼的方法和中间体 |
Family Cites Families (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3036390A1 (de) | 1980-09-26 | 1982-05-13 | Troponwerke GmbH & Co KG, 5000 Köln | Neue pyrrolo-pyrimidine, verfahren zu ihrer herstellung und ihre verwendung bei der herstellung von biologischen wirkstoffen |
| US5521184A (en) | 1992-04-03 | 1996-05-28 | Ciba-Geigy Corporation | Pyrimidine derivatives and processes for the preparation thereof |
| AU6478396A (en) | 1995-07-05 | 1997-02-05 | E.I. Du Pont De Nemours And Company | Fungicidal pyrimidinones |
| TR199800012T1 (xx) | 1995-07-06 | 1998-04-21 | Novartis Ag | Piroloprimidinler ve preparasyon i�in tatbikler. |
| US6232320B1 (en) | 1998-06-04 | 2001-05-15 | Abbott Laboratories | Cell adhesion-inhibiting antiinflammatory compounds |
| JP2002517396A (ja) | 1998-06-04 | 2002-06-18 | アボット・ラボラトリーズ | 細胞接着阻害抗炎症性化合物 |
| PA8474101A1 (es) | 1998-06-19 | 2000-09-29 | Pfizer Prod Inc | Compuestos de pirrolo [2,3-d] pirimidina |
| HRP20000885B1 (hr) | 1998-06-19 | 2007-03-31 | Pfizer Products Inc. | PIROLO[2,3-d]PIRIMIDINSKI SPOJEVI |
| ES2342240T3 (es) | 1998-08-11 | 2010-07-02 | Novartis Ag | Derivados de isoquinolina con actividad que inhibe la angiogenia. |
| US6133031A (en) | 1999-08-19 | 2000-10-17 | Isis Pharmaceuticals Inc. | Antisense inhibition of focal adhesion kinase expression |
| GB9905075D0 (en) | 1999-03-06 | 1999-04-28 | Zeneca Ltd | Chemical compounds |
| JP4078074B2 (ja) | 1999-12-10 | 2008-04-23 | ファイザー・プロダクツ・インク | ピロロ[2,3−d]ピリミジン化合物 |
| CN1615873A (zh) | 1999-12-24 | 2005-05-18 | 阿文蒂斯药物有限公司 | 氮杂吲哚类化合物 |
| GB0004890D0 (en) | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| US6335342B1 (en) | 2000-06-19 | 2002-01-01 | Pharmacia & Upjohn S.P.A. | Azaindole derivatives, process for their preparation, and their use as antitumor agents |
| AU2001274598A1 (en) | 2000-06-23 | 2002-01-02 | Mitsubishi Pharma Corporation | Antitumor effect potentiators |
| CA2412560C (en) | 2000-06-26 | 2008-12-30 | Pfizer Products Inc. | Pyrrolo[2,3-d]pyrimidine compounds as immunosuppressive agents |
| KR100786927B1 (ko) | 2000-06-28 | 2007-12-17 | 스미스클라인비이참피이엘시이 | 습식 분쇄방법 |
| GB0100622D0 (en) | 2001-01-10 | 2001-02-21 | Vernalis Res Ltd | Chemical compounds V111 |
| EP1363702A4 (en) | 2001-01-30 | 2007-08-22 | Cytopia Pty Ltd | PROCESS FOR INHIBITING KINASES |
| US7301023B2 (en) | 2001-05-31 | 2007-11-27 | Pfizer Inc. | Chiral salt resolution |
| WO2003011285A1 (en) | 2001-08-01 | 2003-02-13 | Merck & Co., Inc. | BENZIMIDAZO[4,5-f]ISOQUINOLINONE DERIVATIVES |
| HUP0401982A3 (en) | 2001-09-19 | 2012-09-28 | Aventis Pharma Sa | Indolizine derivates, process for their preparation and pharmaceutical compositions containing the compounds |
| HU230798B1 (hu) | 2001-10-30 | 2018-06-28 | Novartis Ag | Staurosporin-származékok mint az FLT3 receptor tirozin-kináz aktivitás inhibitorai |
| GT200200234A (es) | 2001-12-06 | 2003-06-27 | Compuestos cristalinos novedosos | |
| TW200403058A (en) | 2002-04-19 | 2004-03-01 | Bristol Myers Squibb Co | Heterocyclo inhibitors of potassium channel function |
| PE20040522A1 (es) | 2002-05-29 | 2004-09-28 | Novartis Ag | Derivados de diarilurea dependientes de la cinasa de proteina |
| GB0215676D0 (en) | 2002-07-05 | 2002-08-14 | Novartis Ag | Organic compounds |
| EP1562938B1 (en) | 2002-11-04 | 2007-08-29 | Vertex Pharmaceuticals Incorporated | Heteroaryl-pyrimidine derivatives as jak inhibitors |
| TWI335913B (en) | 2002-11-15 | 2011-01-11 | Vertex Pharma | Diaminotriazoles useful as inhibitors of protein kinases |
| BR0316487A (pt) | 2002-11-26 | 2005-10-11 | Pfizer Prod Inc | Método todo de tratamento da rejeição de transplantes |
| UA80767C2 (en) | 2002-12-20 | 2007-10-25 | Pfizer Prod Inc | Pyrimidine derivatives for the treatment of abnormal cell growth |
| AU2004212421B2 (en) | 2003-02-07 | 2009-08-20 | Vertex Pharmaceuticals Incorporated | Heteroaryl substituted pyrolls useful as inhibitors of protein kinases |
| GB0305929D0 (en) | 2003-03-14 | 2003-04-23 | Novartis Ag | Organic compounds |
| SE0301373D0 (sv) | 2003-05-09 | 2003-05-09 | Astrazeneca Ab | Novel compounds |
| SE0301372D0 (sv) | 2003-05-09 | 2003-05-09 | Astrazeneca Ab | Novel compounds |
| US20050043346A1 (en) | 2003-08-08 | 2005-02-24 | Pharmacia Italia S.P.A. | Pyridylpyrrole derivatives active as kinase inhibitors |
| AR045944A1 (es) | 2003-09-24 | 2005-11-16 | Novartis Ag | Derivados de isoquinolina 1.4-disustituidas |
| JP2007512316A (ja) | 2003-11-25 | 2007-05-17 | ファイザー・プロダクツ・インク | アテローム性動脈硬化症の治療方法 |
| BRPI0417803A (pt) | 2003-12-17 | 2007-04-10 | Pfizer Prod Inc | método de tratamento de rejeição de transplantes |
| US20050153989A1 (en) | 2004-01-13 | 2005-07-14 | Ambit Biosciences Corporation | Pyrrolopyrimidine derivatives and analogs and their use in the treatment and prevention of diseases |
| KR101298951B1 (ko) | 2004-03-30 | 2013-09-30 | 버텍스 파마슈티칼스 인코포레이티드 | Jak 및 기타 단백질 키나제의 억제제로서 유용한아자인돌 |
| US7558717B2 (en) | 2004-04-28 | 2009-07-07 | Vertex Pharmaceuticals Incorporated | Crystal structure of human JAK3 kinase domain complex and binding pockets thereof |
| WO2005105814A1 (en) | 2004-04-28 | 2005-11-10 | Incyte Corporation | Tetracyclic inhibitors of janus kinases |
| WO2005105146A1 (en) | 2004-05-03 | 2005-11-10 | Novartis Ag | Combinations comprising a s1p receptor agonist and a jak3 kinase inhibitor |
| WO2005110410A2 (en) | 2004-05-14 | 2005-11-24 | Abbott Laboratories | Kinase inhibitors as therapeutic agents |
| PE20060426A1 (es) | 2004-06-02 | 2006-06-28 | Schering Corp | DERIVADOS DE ACIDO TARTARICO COMO INHIBIDORES DE MMPs, ADAMs, TACE Y TNF-alfa |
| WO2006013114A1 (en) | 2004-08-06 | 2006-02-09 | Develogen Aktiengesellschaft | Use of a timp-2 secreted protein product for preventing and treating pancreatic diseases and/or obesity and/or metabolic syndrome |
| UY29177A1 (es) | 2004-10-25 | 2006-05-31 | Astex Therapeutics Ltd | Derivados sustituidos de purina, purinona y deazapurina, composiciones que los contienen métodos para su preparación y sus usos |
| MY179032A (en) | 2004-10-25 | 2020-10-26 | Cancer Research Tech Ltd | Ortho-condensed pyridine and pyrimidine derivatives (e.g.purines) as protein kinase inhibitors |
| MX2007006204A (es) | 2004-11-24 | 2007-06-20 | Novartis Ag | Combinaciones que comprenden inhibidores de jak y cuando menos uno de entre inhibidores de bcr-abl, flt-3, fak o raf cinasa. |
| AR054416A1 (es) | 2004-12-22 | 2007-06-27 | Incyte Corp | Pirrolo [2,3-b]piridin-4-il-aminas y pirrolo [2,3-b]pirimidin-4-il-aminas como inhibidores de las quinasas janus. composiciones farmaceuticas. |
| AP2672A (en) * | 2005-01-31 | 2013-05-23 | Mylan Lab Inc | Glucuronidate nebivoloi metabolites |
| DE602006004844D1 (de) * | 2005-02-03 | 2009-03-05 | Vertex Pharma | Pyrrolopyrimidine verwendbar als protein kinase inhibitoren |
| US7019626B1 (en) * | 2005-03-03 | 2006-03-28 | Omnitek Engineering, Inc. | Multi-fuel engine conversion system and method |
| WO2006127587A1 (en) | 2005-05-20 | 2006-11-30 | Vertex Pharmaceuticals Incorporated | Pyrrolopyridines useful as inhibitors of protein kinase |
| US20070049591A1 (en) | 2005-08-25 | 2007-03-01 | Kalypsys, Inc. | Inhibitors of MAPK/Erk Kinase |
| EP1926735A1 (en) | 2005-09-22 | 2008-06-04 | Incyte Corporation | Tetracyclic inhibitors of janus kinases |
| ZA200802685B (en) | 2005-09-30 | 2009-10-28 | Vertex Pharma | Deazapurines useful as inhibitors of janus kinases |
| EP2343298B9 (en) | 2005-12-13 | 2020-05-06 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| EP1968568A4 (en) | 2005-12-22 | 2011-04-13 | Glaxosmithkline Llc | HEMMER OF NUTS ACTIVITY |
| TWI423976B (zh) | 2006-01-17 | 2014-01-21 | Vertex Pharma | 適合作為傑納斯激酶(janus kinase)抑制劑之氮雜吲哚 |
| SG170828A1 (en) | 2006-04-05 | 2011-05-30 | Vertex Pharmaceuticals Inc Us | Deazapurines useful as inhibitors of janus kinases |
| JP5492565B2 (ja) | 2006-12-22 | 2014-05-14 | インサイト・コーポレイション | Janusキナーゼ阻害剤としての置換複素環 |
| RS53245B2 (sr) * | 2007-06-13 | 2022-10-31 | Incyte Holdings Corp | Soli inhibitora janus kinaze (r)-3-(4-(7h-pirolo(2,3-d) pirimidin-4-il)-1h-pirazol-1-il)-3-ciklopentilpropan-nitrila |
| CL2008001709A1 (es) | 2007-06-13 | 2008-11-03 | Incyte Corp | Compuestos derivados de pirrolo [2,3-b]pirimidina, moduladores de quinasas jak; composicion farmaceutica; y uso en el tratamiento de enfermedades tales como cancer, psoriasis, artritis reumatoide, entre otras. |
| MY165582A (en) | 2008-03-11 | 2018-04-05 | Incyte Holdings Corp | Azetidine and cyclobutane derivatives as jak inhibitors |
| CL2009001884A1 (es) * | 2008-10-02 | 2010-05-14 | Incyte Holdings Corp | Uso de 3-ciclopentil-3-[4-(7h-pirrolo[2,3-d]pirimidin-4-il)-1h-pirazol-1-il)propanonitrilo, inhibidor de janus quinasa, y uso de una composición que lo comprende para el tratamiento del ojo seco. |
| JOP20190230A1 (ar) | 2009-01-15 | 2017-06-16 | Incyte Corp | طرق لاصلاح مثبطات انزيم jak و المركبات الوسيطة المتعلقة به |
| WO2010135650A1 (en) | 2009-05-22 | 2010-11-25 | Incyte Corporation | N-(HETERO)ARYL-PYRROLIDINE DERIVATIVES OF PYRAZOL-4-YL-PYRROLO[2,3-d]PYRIMIDINES AND PYRROL-3-YL-PYRROLO[2,3-d]PYRIMIDINES AS JANUS KINASE INHIBITORS |
| TW201113285A (en) | 2009-09-01 | 2011-04-16 | Incyte Corp | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| BR112012008267B1 (pt) | 2009-10-09 | 2022-10-04 | Incyte Holdings Corporation | Derivados hidroxila, ceto e glucuronida de 3-(4-(7h-pirrolo[2,3-d]pirimidin-4-il)-1h-pirazol-1- il)-3-ciclopentilpropanonitrila |
| JP5858434B2 (ja) | 2010-02-18 | 2016-02-10 | インサイト・ホールディングス・コーポレイションIncyte Holdings Corporation | Janusキナーゼ阻害薬としてのシクロブタンおよびメチルシクロブタン誘導体 |
-
2010
- 2010-10-08 BR BR112012008267-1A patent/BR112012008267B1/pt active IP Right Grant
- 2010-10-08 EP EP10765557.3A patent/EP2486041B1/en active Active
- 2010-10-08 PT PT107655573T patent/PT2486041E/pt unknown
- 2010-10-08 PL PL10765557T patent/PL2486041T3/pl unknown
- 2010-10-08 KR KR1020187033300A patent/KR20180126619A/ko not_active Ceased
- 2010-10-08 KR KR1020127009995A patent/KR101805936B1/ko active Active
- 2010-10-08 CN CN201510984778.5A patent/CN105541847B/zh active Active
- 2010-10-08 US US12/901,001 patent/US8486902B2/en active Active
- 2010-10-08 WO PCT/US2010/052011 patent/WO2011044481A1/en not_active Ceased
- 2010-10-08 KR KR1020177023122A patent/KR101921850B1/ko active Active
- 2010-10-08 MX MX2012004180A patent/MX2012004180A/es active IP Right Grant
- 2010-10-08 JP JP2012533354A patent/JP5946768B2/ja active Active
- 2010-10-08 EA EA201200565A patent/EA021478B1/ru not_active IP Right Cessation
- 2010-10-08 CN CN201080049430.2A patent/CN102596960B/zh active Active
- 2010-10-08 CA CA2777114A patent/CA2777114C/en active Active
- 2010-10-08 ES ES10765557T patent/ES2435491T3/es active Active
-
2013
- 2013-06-13 US US13/917,124 patent/US8748401B2/en active Active
-
2014
- 2014-04-28 US US14/263,476 patent/US9512161B2/en active Active
-
2015
- 2015-12-17 JP JP2015246515A patent/JP6251232B2/ja active Active
-
2017
- 2017-09-08 JP JP2017172989A patent/JP2017226695A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BR112012008267B1 (pt) | Derivados hidroxila, ceto e glucuronida de 3-(4-(7h-pirrolo[2,3-d]pirimidin-4-il)-1h-pirazol-1- il)-3-ciclopentilpropanonitrila | |
| CA2689158C (en) | Metabolites of the janus kinase inhibitor (r)-3-(4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-1-yl)-3-cyclopentylpropanenitrile | |
| CA2689663C (en) | Salts of the janus kinase inhibitor (r)-3-(4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-1-yl)-3-cyclopentylpropanenitrile | |
| AU2010303245B2 (en) | Hydroxyl, keto, and glucuronide derivatives of 3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile | |
| HK1174899B (en) | Hydroxyl, keto, and glucuronide derivatives of 3-(4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h-pyrazol-1-yl)-3-cyclopentylpropanenitrile | |
| HK1141020B (en) | Metabolites of the janus kinase inhibitor (r)-3-(4-(7h-pyrrolo[2,3-d] pyrimidin-4-yl)-lh-pyrazol-l-yl)-3-cyclopentylpropanenitrile |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B25G | Requested change of headquarter approved |
Owner name: INCYTE CORPORATION (US) |
|
| B25A | Requested transfer of rights approved |
Owner name: INCYTE HOLDINGS CORPORATION (US) |
|
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 08/10/2010, OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF, QUE DETERMINA A ALTERACAO DO PRAZO DE CONCESSAO. |