US20150119319A1 - Macrocyclic hepatitis c serine protease inhibitors - Google Patents
Macrocyclic hepatitis c serine protease inhibitors Download PDFInfo
- Publication number
- US20150119319A1 US20150119319A1 US14/588,805 US201514588805A US2015119319A1 US 20150119319 A1 US20150119319 A1 US 20150119319A1 US 201514588805 A US201514588805 A US 201514588805A US 2015119319 A1 US2015119319 A1 US 2015119319A1
- Authority
- US
- United States
- Prior art keywords
- optionally substituted
- alkyl
- independently
- alkenyl
- alkynyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 208000006454 hepatitis Diseases 0.000 title description 2
- 231100000283 hepatitis Toxicity 0.000 title description 2
- 239000003001 serine protease inhibitor Substances 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 175
- 238000000034 method Methods 0.000 claims abstract description 62
- 239000000651 prodrug Substances 0.000 claims abstract description 38
- 229940002612 prodrug Drugs 0.000 claims abstract description 38
- 150000003839 salts Chemical class 0.000 claims abstract description 35
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 27
- 150000002148 esters Chemical class 0.000 claims abstract description 25
- 239000000546 pharmaceutical excipient Substances 0.000 claims abstract description 12
- 239000003937 drug carrier Substances 0.000 claims abstract description 8
- -1 substituted Chemical class 0.000 claims description 207
- 125000000623 heterocyclic group Chemical group 0.000 claims description 113
- 125000001072 heteroaryl group Chemical group 0.000 claims description 78
- 125000003107 substituted aryl group Chemical group 0.000 claims description 56
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 55
- 229910052736 halogen Inorganic materials 0.000 claims description 54
- 150000002367 halogens Chemical class 0.000 claims description 52
- 125000001424 substituent group Chemical group 0.000 claims description 46
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 45
- 125000002837 carbocyclic group Chemical group 0.000 claims description 45
- 229910052760 oxygen Inorganic materials 0.000 claims description 45
- 229910052717 sulfur Inorganic materials 0.000 claims description 44
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 42
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 40
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 40
- 125000005842 heteroatom Chemical group 0.000 claims description 40
- 229910052739 hydrogen Inorganic materials 0.000 claims description 38
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 36
- 239000001257 hydrogen Substances 0.000 claims description 34
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 29
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 claims description 25
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 claims description 25
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 24
- 125000005017 substituted alkenyl group Chemical group 0.000 claims description 24
- 125000004426 substituted alkynyl group Chemical group 0.000 claims description 24
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 claims description 21
- 229910052757 nitrogen Inorganic materials 0.000 claims description 21
- 229920006395 saturated elastomer Polymers 0.000 claims description 21
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 20
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 claims description 20
- 125000003118 aryl group Chemical group 0.000 claims description 19
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 18
- 125000000217 alkyl group Chemical group 0.000 claims description 15
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 15
- 125000005843 halogen group Chemical group 0.000 claims description 14
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 12
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 12
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 11
- 125000001188 haloalkyl group Chemical group 0.000 claims description 11
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 11
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 11
- 125000004076 pyridyl group Chemical group 0.000 claims description 11
- 229910052701 rubidium Inorganic materials 0.000 claims description 11
- 125000001544 thienyl group Chemical group 0.000 claims description 11
- 125000004432 carbon atom Chemical group C* 0.000 claims description 10
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 10
- 239000012453 solvate Substances 0.000 claims description 10
- 125000002541 furyl group Chemical group 0.000 claims description 9
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 9
- 125000004043 oxo group Chemical group O=* 0.000 claims description 9
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 9
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 9
- 229910052705 radium Inorganic materials 0.000 claims description 9
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 9
- 208000036142 Viral infection Diseases 0.000 claims description 8
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 8
- 239000011203 carbon fibre reinforced carbon Substances 0.000 claims description 8
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 8
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 8
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 8
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 8
- 125000004306 triazinyl group Chemical group 0.000 claims description 8
- 230000009385 viral infection Effects 0.000 claims description 8
- JJWKPURADFRFRB-UHFFFAOYSA-N carbonyl sulfide Chemical compound O=C=S JJWKPURADFRFRB-UHFFFAOYSA-N 0.000 claims description 7
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 7
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 7
- 125000004648 C2-C8 alkenyl group Chemical group 0.000 claims description 6
- 125000003342 alkenyl group Chemical group 0.000 claims description 6
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 6
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 6
- 125000000160 oxazolidinyl group Chemical group 0.000 claims description 6
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 claims description 6
- 125000004450 alkenylene group Chemical group 0.000 claims description 5
- 125000002947 alkylene group Chemical group 0.000 claims description 5
- 125000000304 alkynyl group Chemical group 0.000 claims description 5
- 125000004419 alkynylene group Chemical group 0.000 claims description 5
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 claims description 5
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims description 5
- 150000004677 hydrates Chemical class 0.000 claims description 5
- 125000000335 thiazolyl group Chemical group 0.000 claims description 5
- 125000002632 imidazolidinyl group Chemical group 0.000 claims description 4
- 125000002636 imidazolinyl group Chemical group 0.000 claims description 4
- 125000002883 imidazolyl group Chemical group 0.000 claims description 4
- 125000001786 isothiazolyl group Chemical group 0.000 claims description 4
- 125000002757 morpholinyl group Chemical group 0.000 claims description 4
- 125000002971 oxazolyl group Chemical group 0.000 claims description 4
- 125000004193 piperazinyl group Chemical group 0.000 claims description 4
- 125000003386 piperidinyl group Chemical group 0.000 claims description 4
- 125000003072 pyrazolidinyl group Chemical group 0.000 claims description 4
- 125000002755 pyrazolinyl group Chemical group 0.000 claims description 4
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 4
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 4
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 4
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 claims description 3
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 claims description 3
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 claims description 3
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 claims description 3
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 claims description 3
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 claims description 3
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 claims description 3
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 claims description 3
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 claims description 3
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 claims description 3
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 claims description 3
- 125000001422 pyrrolinyl group Chemical group 0.000 claims description 3
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 claims description 3
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 claims description 2
- 125000004511 1,2,3-thiadiazolyl group Chemical group 0.000 claims description 2
- 125000001399 1,2,3-triazolyl group Chemical group N1N=NC(=C1)* 0.000 claims description 2
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 claims description 2
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 claims description 2
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 claims description 2
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 claims description 2
- GAMYYCRTACQSBR-UHFFFAOYSA-N 4-azabenzimidazole Chemical compound C1=CC=C2NC=NC2=N1 GAMYYCRTACQSBR-UHFFFAOYSA-N 0.000 claims description 2
- 125000002471 4H-quinolizinyl group Chemical group C=1(C=CCN2C=CC=CC12)* 0.000 claims description 2
- BBAWTPDTGRXPDG-UHFFFAOYSA-N [1,3]thiazolo[4,5-b]pyridine Chemical compound C1=CC=C2SC=NC2=N1 BBAWTPDTGRXPDG-UHFFFAOYSA-N 0.000 claims description 2
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 claims description 2
- 125000004931 azocinyl group Chemical group N1=C(C=CC=CC=C1)* 0.000 claims description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 claims description 2
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 claims description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 claims description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000004623 carbolinyl group Chemical group 0.000 claims description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 2
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 claims description 2
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 claims description 2
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 claims description 2
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 claims description 2
- 125000003838 furazanyl group Chemical group 0.000 claims description 2
- 125000004926 indolenyl group Chemical group 0.000 claims description 2
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 claims description 2
- 125000001041 indolyl group Chemical group 0.000 claims description 2
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 claims description 2
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 claims description 2
- 125000005438 isoindazolyl group Chemical group 0.000 claims description 2
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 claims description 2
- 125000004930 octahydroisoquinolinyl group Chemical group C1(NCCC2CCCC=C12)* 0.000 claims description 2
- 125000001715 oxadiazolyl group Chemical group 0.000 claims description 2
- QNNHQVPFZIFNFK-UHFFFAOYSA-N oxazolo[4,5-b]pyridine Chemical compound C1=CC=C2OC=NC2=N1 QNNHQVPFZIFNFK-UHFFFAOYSA-N 0.000 claims description 2
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 claims description 2
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 claims description 2
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 claims description 2
- 125000005954 phenoxathiinyl group Chemical group 0.000 claims description 2
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 claims description 2
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 claims description 2
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 claims description 2
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 claims description 2
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 claims description 2
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 claims description 2
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 claims description 2
- 125000004627 thianthrenyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3SC12)* 0.000 claims description 2
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 claims description 2
- 125000004517 1,2,5-thiadiazolyl group Chemical group 0.000 claims 1
- 125000005512 benztetrazolyl group Chemical group 0.000 claims 1
- 239000012038 nucleophile Substances 0.000 claims 1
- 208000005176 Hepatitis C Diseases 0.000 abstract description 15
- 208000010710 hepatitis C virus infection Diseases 0.000 abstract description 6
- 150000002678 macrocyclic compounds Chemical class 0.000 abstract description 6
- 238000002560 therapeutic procedure Methods 0.000 abstract description 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 198
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 144
- 239000000203 mixture Substances 0.000 description 82
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 78
- 239000000243 solution Substances 0.000 description 78
- 239000011541 reaction mixture Substances 0.000 description 66
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 55
- 210000004027 cell Anatomy 0.000 description 52
- 239000007787 solid Substances 0.000 description 49
- 230000002829 reductive effect Effects 0.000 description 45
- 125000004008 6 membered carbocyclic group Chemical group 0.000 description 43
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 40
- 239000000741 silica gel Substances 0.000 description 39
- 229910002027 silica gel Inorganic materials 0.000 description 39
- 238000003556 assay Methods 0.000 description 38
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 36
- 239000000047 product Substances 0.000 description 33
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 32
- 241000711549 Hepacivirus C Species 0.000 description 31
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 31
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 31
- 238000001952 enzyme assay Methods 0.000 description 30
- 238000003818 flash chromatography Methods 0.000 description 28
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 27
- 239000012044 organic layer Substances 0.000 description 27
- 238000002360 preparation method Methods 0.000 description 24
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 20
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 18
- 0 *CN([3*])[C@H]1CC*Cc(C)(C)c(C)(C)[C@@H]2C[C@@]2(C(C)=O)N([3*])C(=O)[C@@H]2C[C@@H](CC)CN2C1=O.C Chemical compound *CN([3*])[C@H]1CC*Cc(C)(C)c(C)(C)[C@@H]2C[C@@]2(C(C)=O)N([3*])C(=O)[C@@H]2C[C@@H](CC)CN2C1=O.C 0.000 description 17
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 17
- 239000003814 drug Substances 0.000 description 17
- 239000002904 solvent Substances 0.000 description 17
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 description 16
- 125000006643 (C2-C6) haloalkenyl group Chemical group 0.000 description 16
- 230000000694 effects Effects 0.000 description 16
- 239000003112 inhibitor Substances 0.000 description 16
- 239000003921 oil Substances 0.000 description 16
- 235000019198 oils Nutrition 0.000 description 16
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 15
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 15
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 15
- 125000006239 protecting group Chemical group 0.000 description 15
- 238000011282 treatment Methods 0.000 description 15
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 13
- 239000003795 chemical substances by application Substances 0.000 description 13
- 239000000463 material Substances 0.000 description 13
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 description 12
- 229940079593 drug Drugs 0.000 description 12
- 230000002401 inhibitory effect Effects 0.000 description 12
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical group N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 description 12
- 229960000311 ritonavir Drugs 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 11
- 229910052740 iodine Inorganic materials 0.000 description 11
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 125000000753 cycloalkyl group Chemical group 0.000 description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000012472 biological sample Substances 0.000 description 9
- 150000003857 carboxamides Chemical class 0.000 description 9
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 9
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 8
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 8
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 235000019441 ethanol Nutrition 0.000 description 8
- 239000012091 fetal bovine serum Substances 0.000 description 8
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 7
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 7
- 230000008901 benefit Effects 0.000 description 7
- 229910052731 fluorine Inorganic materials 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 230000008569 process Effects 0.000 description 7
- 108090000623 proteins and genes Proteins 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 238000004809 thin layer chromatography Methods 0.000 description 7
- BNMPIJWVMVNSRD-UHFFFAOYSA-N 5-methyl-1,2-oxazole-3-carboxylic acid Chemical compound CC1=CC(C(O)=O)=NO1 BNMPIJWVMVNSRD-UHFFFAOYSA-N 0.000 description 6
- 208000003311 Cytochrome P-450 Enzyme Inhibitors Diseases 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 108020004684 Internal Ribosome Entry Sites Proteins 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 241000700605 Viruses Species 0.000 description 6
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 6
- WMSPXQIQBQAWLL-UHFFFAOYSA-N cyclopropanesulfonamide Chemical compound NS(=O)(=O)C1CC1 WMSPXQIQBQAWLL-UHFFFAOYSA-N 0.000 description 6
- 239000002609 medium Substances 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 230000010076 replication Effects 0.000 description 6
- 238000004007 reversed phase HPLC Methods 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 230000001052 transient effect Effects 0.000 description 6
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 5
- 125000004649 C2-C8 alkynyl group Chemical group 0.000 description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 5
- 108060001084 Luciferase Proteins 0.000 description 5
- 239000005089 Luciferase Substances 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 101800001014 Non-structural protein 5A Proteins 0.000 description 5
- WXIONIWNXBAHRU-UHFFFAOYSA-N [dimethylamino(triazolo[4,5-b]pyridin-3-yloxy)methylidene]-dimethylazanium Chemical compound C1=CN=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 WXIONIWNXBAHRU-UHFFFAOYSA-N 0.000 description 5
- 239000003443 antiviral agent Substances 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 125000004452 carbocyclyl group Chemical group 0.000 description 5
- 239000013058 crude material Substances 0.000 description 5
- 238000010790 dilution Methods 0.000 description 5
- 239000012895 dilution Substances 0.000 description 5
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 5
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 5
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 5
- BDEDJVAZBRIIOL-DGYZZDTHSA-N n-((2r,6s,13as,14ar,16as)-2-(3-(benzo [d]thiazol-2-yl)quinoxalin-2- yloxy)-14a-(cyclopropylsulfonylcarbamoy1)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-yl)-5-methylisoxazole-3-carboxamide Chemical compound O1C(C)=CC(C(=O)N[C@@H]2C(N3C[C@@H](C[C@H]3C(=O)N[C@@]3(C[C@H]3CC(F)(F)CCCCC2)C(=O)NS(=O)(=O)C2CC2)OC=2C(=NC3=CC=CC=C3N=2)C=2SC3=CC=CC=C3N=2)=O)=N1 BDEDJVAZBRIIOL-DGYZZDTHSA-N 0.000 description 5
- 238000012746 preparative thin layer chromatography Methods 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 125000006413 ring segment Chemical group 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- LKUOXWZPVMDETG-ASUJFGITSA-N tert-butyl (2r,6s,13as,14ar,16as)-2-(3-(benzo[d]thiazol-2-yl)quinoxalin-2-yloxy)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate Chemical compound O=C([C@@]12C[C@H]1CC(F)(F)CCCCC[C@@H](C(N1C[C@@H](C[C@H]1C(=O)N2)OC=1C(=NC2=CC=CC=C2N=1)C=1SC2=CC=CC=C2N=1)=O)NC(=O)OC(C)(C)C)NS(=O)(=O)C1CC1 LKUOXWZPVMDETG-ASUJFGITSA-N 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- YQUCBFIQSJVCOR-JOCHJYFZSA-N (7r)-14-cyclohexyl-7-{[2-(dimethylamino)ethyl](methyl)amino}-7,8-dihydro-6h-indolo[1,2-e][1,5]benzoxazocine-11-carboxylic acid Chemical compound C([C@@H](CN1C2=CC(=CC=C22)C(O)=O)N(C)CCN(C)C)OC3=CC=CC=C3C1=C2C1CCCCC1 YQUCBFIQSJVCOR-JOCHJYFZSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- QUGADGXAROHCKR-UHFFFAOYSA-N 4-methyl-1,2-oxazole-3-carboxamide Chemical compound CC1=CON=C1C(N)=O QUGADGXAROHCKR-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- 108091026890 Coding region Proteins 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 102000006992 Interferon-alpha Human genes 0.000 description 4
- 108010047761 Interferon-alpha Proteins 0.000 description 4
- 102000014150 Interferons Human genes 0.000 description 4
- 108010050904 Interferons Proteins 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 4
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 4
- 239000003701 inert diluent Substances 0.000 description 4
- 229940079322 interferon Drugs 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 230000035772 mutation Effects 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- DEKOYVOWOVJMPM-RLHIPHHXSA-N setrobuvir Chemical compound N1([C@H]2[C@@H]3CC[C@@H](C3)[C@H]2C(O)=C(C1=O)C=1NC2=CC=C(C=C2S(=O)(=O)N=1)NS(=O)(=O)C)CC1=CC=C(F)C=C1 DEKOYVOWOVJMPM-RLHIPHHXSA-N 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- KGKNRORBBYKDQE-FDSIQNTBSA-N tert-butyl (2r,6s ,13as,14ar,16as)-14a-(cyclopropylsulfonylcarbamoyl)-2-(3-ethylquinoxalin-2-yloxy)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate Chemical compound O=C([C@@]12C[C@H]1CC(F)(F)CCCCC[C@@H](C(=O)N1[C@H](C(N2)=O)C[C@H](C1)OC1=NC2=CC=CC=C2N=C1CC)NC(=O)OC(C)(C)C)NS(=O)(=O)C1CC1 KGKNRORBBYKDQE-FDSIQNTBSA-N 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- YAWYPRFHANAKRU-WZJOZTAQSA-N (2r,6s,13as,14ar,16as)-2-(3-(benzo[d]thiazol-2-yl)quinoxalin-2-yloxy)-6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid Chemical compound O=C1[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(O)=O)NC(=O)[C@@H]2C[C@@H](OC=3C(=NC4=CC=CC=C4N=3)C=3SC4=CC=CC=C4N=3)CN21 YAWYPRFHANAKRU-WZJOZTAQSA-N 0.000 description 3
- DFWDEDXXKQLXJS-FDSIQNTBSA-N (2r,6s,13as,14ar,16as)-6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadeca hydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid Chemical compound O=C1[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(O)=O)NC(=O)[C@@H]2C[C@@H](OC=3C4=CC=CC=C4C4=CC=CC=C4N=3)CN21 DFWDEDXXKQLXJS-FDSIQNTBSA-N 0.000 description 3
- ZYWJDCMCFJSJNT-QCVATHDWSA-N (2r,6s,13as,14ar,16as)-6-amino-2-(3-(benzo [d]thiazol-2-yl)quinoxalin-2-yloxy)-n-(cyclopropylsulfonyl)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecine-14a-carboxamide Chemical compound O=C([C@@]12C[C@H]1CC(F)(F)CCCCC[C@@H](C(N1C[C@@H](C[C@H]1C(=O)N2)OC=1C(=NC2=CC=CC=C2N=1)C=1SC2=CC=CC=C2N=1)=O)N)NS(=O)(=O)C1CC1 ZYWJDCMCFJSJNT-QCVATHDWSA-N 0.000 description 3
- VCSOMDNTHXRNGP-YDAXWHBXSA-N (2r,6s,13as,14ar,16as)-6-amino-n-(cyclopropylsulfonyl)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide Chemical compound O=C([C@@]12C[C@H]1CC(F)(F)CCCCC[C@@H](C(N1C[C@@H](C[C@H]1C(=O)N2)OC=1C2=CC=CC=C2C2=CC=CC=C2N=1)=O)N)NS(=O)(=O)C1CC1 VCSOMDNTHXRNGP-YDAXWHBXSA-N 0.000 description 3
- XCIHXPUUVKKIMP-SLFWVYJPSA-N (2r,6s,13as,14ar,16as)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound C1C(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@H](O)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]21 XCIHXPUUVKKIMP-SLFWVYJPSA-N 0.000 description 3
- PGHKWYUPJRBILL-JRHDMDNSSA-N (2r,6s,13as,14ar,16as,z)-ethyl 6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound N([C@H]1CCCCC\C=C/[C@@H]2C[C@]2(NC(=O)[C@@H]2C[C@H](CN2C1=O)OC=1C2=CC=CC=C2C2=CC=CC=C2N=1)C(=O)OCC)C(=O)C=1C=C(C)ON=1 PGHKWYUPJRBILL-JRHDMDNSSA-N 0.000 description 3
- VGIHXCUJTHADQJ-DYPBWMGKSA-N (2r,6s,13as,14ar,16as,z)-ethyl 6-(tert-butoxycarbonylamino)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound C/1=C/CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@H](OC=3C4=CC=CC=C4C4=CC=CC=C4N=3)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]2\1 VGIHXCUJTHADQJ-DYPBWMGKSA-N 0.000 description 3
- BQAKBZPMKFYYKM-YZNFRCNNSA-N (2r,6s,13as,14ar,16as,z)-ethyl 6-amino-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound C/1=C/CCCCC[C@H](N)C(=O)N2C[C@H](OC=3C4=CC=CC=C4C4=CC=CC=C4N=3)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]2\1 BQAKBZPMKFYYKM-YZNFRCNNSA-N 0.000 description 3
- RVMGRWWHHHMBLB-UHFFFAOYSA-N 3-(1,3-benzothiazol-2-yl)-1h-quinoxalin-2-one Chemical compound C1=CC=C2SC(C3=NC4=CC=CC=C4N=C3O)=NC2=C1 RVMGRWWHHHMBLB-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- XRWNGRFEIJXNRX-UULNMNGPSA-N CC(C)(C)/C=C\C(C)(C)C.CC(C)(C)CCC(C)(C)C Chemical compound CC(C)(C)/C=C\C(C)(C)C.CC(C)(C)CCC(C)(C)C XRWNGRFEIJXNRX-UULNMNGPSA-N 0.000 description 3
- 239000004215 Carbon black (E152) Substances 0.000 description 3
- 229940124683 HCV polymerase inhibitor Drugs 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 108060004795 Methyltransferase Proteins 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 3
- 101800001838 Serine protease/helicase NS3 Proteins 0.000 description 3
- 108091027544 Subgenomic mRNA Proteins 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 230000003044 adaptive effect Effects 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 230000000840 anti-viral effect Effects 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- ZLPZSKQDLBNGEJ-KZULUSFZSA-N ethyl (1r,2s)-1-[bis[(2-methylpropan-2-yl)oxycarbonyl]amino]-2-(2-hydroxyethyl)cyclopropane-1-carboxylate Chemical compound CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)[C@]1(C(=O)OCC)C[C@H]1CCO ZLPZSKQDLBNGEJ-KZULUSFZSA-N 0.000 description 3
- VAHLBBPNTKTZDN-IVZQSRNASA-N ethyl (1r,2s)-1-[bis[(2-methylpropan-2-yl)oxycarbonyl]amino]-2-(2-oxohex-5-enyl)cyclopropane-1-carboxylate Chemical compound CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)[C@]1(C(=O)OCC)C[C@H]1CC(=O)CCC=C VAHLBBPNTKTZDN-IVZQSRNASA-N 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 229930195733 hydrocarbon Natural products 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 239000008297 liquid dosage form Substances 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 125000001624 naphthyl group Chemical group 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 108090000765 processed proteins & peptides Chemical group 0.000 description 3
- FFRYUAVNPBUEIC-UHFFFAOYSA-N quinoxalin-2-ol Chemical compound C1=CC=CC2=NC(O)=CN=C21 FFRYUAVNPBUEIC-UHFFFAOYSA-N 0.000 description 3
- 229960000329 ribavirin Drugs 0.000 description 3
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 3
- CGRKYEALWSRNJS-UHFFFAOYSA-N sodium;2-methylbutan-2-olate Chemical compound [Na+].CCC(C)(C)[O-] CGRKYEALWSRNJS-UHFFFAOYSA-N 0.000 description 3
- 238000000935 solvent evaporation Methods 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 238000001890 transfection Methods 0.000 description 3
- UFXIRMVZNARBDL-UHFFFAOYSA-N trifluoro(morpholin-4-yl)-$l^{4}-sulfane Chemical compound FS(F)(F)N1CCOCC1 UFXIRMVZNARBDL-UHFFFAOYSA-N 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- JUJOMMPRVKISQV-HOZLVPSRSA-N (2r,6s,13as,14ar,16as)-12,12-difluoro-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid Chemical compound O1C(C)=CC(C(=O)N[C@@H]2C(N3C[C@@H](C[C@H]3C(=O)N[C@@]3(C[C@H]3CC(F)(F)CCCCC2)C(O)=O)OC=2C3=CC=CC=C3C3=CC=CC=C3N=2)=O)=N1 JUJOMMPRVKISQV-HOZLVPSRSA-N 0.000 description 2
- XGOMEBNQKFVGCR-MDBVHUGHSA-N (2r,6s,13as,14ar,16as)-6-(tert-butoxycarbonylamino)-2-(3-ethylquinoxalin-2-yloxy)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid Chemical compound C1N2C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(O)=O)NC(=O)[C@@H]2C[C@H]1OC1=NC2=CC=CC=C2N=C1CC XGOMEBNQKFVGCR-MDBVHUGHSA-N 0.000 description 2
- VKINFPAVKOXPLC-CTBDIBJJSA-N (2r,6s,13as,14ar,16as)-6-amino-n-(cyclopropylsulfonyl)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocyclo propa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide Chemical compound O=C([C@@]12C[C@H]1CC(F)(F)CCCCC[C@@H](C(N1C[C@@H](C[C@H]1C(=O)N2)OC=1N=C2C=CC=CC2=NC=1)=O)N)NS(=O)(=O)C1CC1 VKINFPAVKOXPLC-CTBDIBJJSA-N 0.000 description 2
- CFMCQKGWFUEUJM-DNQHFQCQSA-N (2r,6s,13as,14ar,16as)-ethyl 6-(tert-butoxycarbonylamino)-12-hydroxy-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound C1C(O)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@H](OC=3C4=CC=CC=C4C4=CC=CC=C4N=3)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]21 CFMCQKGWFUEUJM-DNQHFQCQSA-N 0.000 description 2
- ONYDVYNMYRYXSS-VFLDZSNKSA-N (2r,6s,13as,14ar,16as)-n-(cyclopropylsulfonyl)-6-(1,5-dimethyl-1h-pyrazole-3-carboxamido)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecine-14a-carboxamide Chemical compound CN1C(C)=CC(C(=O)N[C@@H]2C(N3C[C@@H](C[C@H]3C(=O)N[C@@]3(C[C@H]3CC(F)(F)CCCCC2)C(=O)NS(=O)(=O)C2CC2)OC=2C3=CC=CC=C3C3=CC=CC=C3N=2)=O)=N1 ONYDVYNMYRYXSS-VFLDZSNKSA-N 0.000 description 2
- XQMYXERTJPRWOT-ZZDLXCAUSA-N (2r,6s,13as,14ar,16as)-tert-butyl 12,12-difluoro-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound O1C(C)=CC(C(=O)N[C@@H]2C(N3C[C@@H](C[C@H]3C(=O)N[C@@]3(C[C@H]3CC(F)(F)CCCCC2)C(=O)OC(C)(C)C)OC=2C3=CC=CC=C3C3=CC=CC=C3N=2)=O)=N1 XQMYXERTJPRWOT-ZZDLXCAUSA-N 0.000 description 2
- GSGXVXJXISXRSQ-DXNKKRDQSA-N (2r,6s,13as,14ar,16as,e)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxo-1,2,3,5,6,7,10,11,12,13,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound C1C(F)(F)CC\C=C\C[C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@H](O)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]21 GSGXVXJXISXRSQ-DXNKKRDQSA-N 0.000 description 2
- WMWUBTVHBDRTGR-DNIQWYRDSA-N (2r,6s,13as,14ar,16as,z)-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid Chemical compound O1C(C)=CC(C(=O)N[C@@H]2C(N3C[C@@H](C[C@H]3C(=O)N[C@@]3(C[C@H]3\C=C/CCCCC2)C(O)=O)OC=2C3=CC=CC=C3C3=CC=CC=C3N=2)=O)=N1 WMWUBTVHBDRTGR-DNIQWYRDSA-N 0.000 description 2
- UENCTQHLZDQVRU-VWYCJHECSA-N (2s,4r)-4-hydroxy-1-[(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]pent-4-enoyl]pyrrolidine-2-carboxylic acid Chemical compound CC(C)(C)OC(=O)N[C@@H](CC=C)C(=O)N1C[C@H](O)C[C@H]1C(O)=O UENCTQHLZDQVRU-VWYCJHECSA-N 0.000 description 2
- ICUMOWKLBPFMCR-BMCYYIBGSA-N (2s,6s,12s,13as,14ar,16as)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonylamino)-12-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound O([C@@H]1CN2C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC[C@H](O)C[C@@H]3C[C@]3(NC(=O)[C@@H]2C1)C(=O)OCC)C(=O)C1=CC=CC=C1 ICUMOWKLBPFMCR-BMCYYIBGSA-N 0.000 description 2
- QZXRTRIXDZZYEW-BONHKBQFSA-N (2s,6s,13as,14ar,16as)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound O([C@@H]1CN2C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]3C[C@]3(NC(=O)[C@@H]2C1)C(=O)OCC)C(=O)C1=CC=CC=C1 QZXRTRIXDZZYEW-BONHKBQFSA-N 0.000 description 2
- DBINXKABOKVYDJ-YUQYVICNSA-N (2s,6s,13as,14ar,16as,z)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonylamino)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound O([C@@H]1CN2C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC\C=C/[C@@H]3C[C@]3(NC(=O)[C@@H]2C1)C(=O)OCC)C(=O)C1=CC=CC=C1 DBINXKABOKVYDJ-YUQYVICNSA-N 0.000 description 2
- LKYNGTHMKCTTQC-UHFFFAOYSA-N 1,2-oxazole-3-carboxamide Chemical compound NC(=O)C=1C=CON=1 LKYNGTHMKCTTQC-UHFFFAOYSA-N 0.000 description 2
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 2
- AWNXKZVIZARMME-UHFFFAOYSA-N 1-[[5-[2-[(2-chloropyridin-4-yl)amino]pyrimidin-4-yl]-4-(cyclopropylmethyl)pyrimidin-2-yl]amino]-2-methylpropan-2-ol Chemical compound N=1C(NCC(C)(O)C)=NC=C(C=2N=C(NC=3C=C(Cl)N=CC=3)N=CC=2)C=1CC1CC1 AWNXKZVIZARMME-UHFFFAOYSA-N 0.000 description 2
- IANQTJSKSUMEQM-UHFFFAOYSA-N 1-benzofuran Chemical compound C1=CC=C2OC=CC2=C1 IANQTJSKSUMEQM-UHFFFAOYSA-N 0.000 description 2
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 2
- NHWZRIQVTHVGSL-UHFFFAOYSA-N 2-(1-benzofuran-2-yl)-3-chloroquinoxaline Chemical compound C1=CC=C2OC(C3=NC4=CC=CC=C4N=C3Cl)=CC2=C1 NHWZRIQVTHVGSL-UHFFFAOYSA-N 0.000 description 2
- MTHNWHRBVIADBZ-UHFFFAOYSA-N 2-chloro-3-ethylquinoxaline Chemical compound C1=CC=C2N=C(Cl)C(CC)=NC2=C1 MTHNWHRBVIADBZ-UHFFFAOYSA-N 0.000 description 2
- MIVILCNCIWEHAD-UHFFFAOYSA-N 3-(1-benzofuran-2-yl)-1h-quinoxalin-2-one Chemical compound C1=CC=C2OC(C3=NC4=CC=CC=C4N=C3O)=CC2=C1 MIVILCNCIWEHAD-UHFFFAOYSA-N 0.000 description 2
- XVXWUBIIHFDOJO-KQYNXXCUSA-N 4-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-3-oxopyrazine-2-carboxamide Chemical compound O=C1C(C(=O)N)=NC=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 XVXWUBIIHFDOJO-KQYNXXCUSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- XBEQSQDCBSKCHJ-UHFFFAOYSA-N 5-[[6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl]methyl]-2-(2-fluorophenyl)imidazo[4,5-c]pyridine Chemical compound FC1=CC=CC=C1C1=NC2=CN(CC=3N=NC(=CC=3)C=3C(=CC(=CC=3)C(F)(F)F)C(F)(F)F)C=CC2=N1 XBEQSQDCBSKCHJ-UHFFFAOYSA-N 0.000 description 2
- UPPWMBQIDFTBEQ-UHFFFAOYSA-N 6-(3,4-dimethoxyphenyl)-n-[4-(1,2,4-triazol-1-yl)phenyl]quinazolin-4-amine Chemical compound C1=C(OC)C(OC)=CC=C1C1=CC=C(N=CN=C2NC=3C=CC(=CC=3)N3N=CN=C3)C2=C1 UPPWMBQIDFTBEQ-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- OLROWHGDTNFZBH-XEMWPYQTSA-N Alisporivir Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)N(CC)C(=O)[C@@H](C)N(C)C1=O OLROWHGDTNFZBH-XEMWPYQTSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- JMVRQMYDBRVRSG-UHFFFAOYSA-N CC(C)C1=CC=CC=C1.CC(C)C1=CC=NC=C1.CC(C)C1=CN=CC=C1.CC(C)C1=CNC=C1.CC(C)C1=COC=C1.CC(C)C1=CSC=C1.CC(C)C1=NC=CC=C1.CC(C)C1=NC=CN=C1.CC(C)C1=NC=NC=C1.CC(C)C1=NC=NC=N1.CC1=CC(C(C)C)=NN1C.CC1=CC(C(C)C)=NO1 Chemical compound CC(C)C1=CC=CC=C1.CC(C)C1=CC=NC=C1.CC(C)C1=CN=CC=C1.CC(C)C1=CNC=C1.CC(C)C1=COC=C1.CC(C)C1=CSC=C1.CC(C)C1=NC=CC=C1.CC(C)C1=NC=CN=C1.CC(C)C1=NC=NC=C1.CC(C)C1=NC=NC=N1.CC1=CC(C(C)C)=NN1C.CC1=CC(C(C)C)=NO1 JMVRQMYDBRVRSG-UHFFFAOYSA-N 0.000 description 2
- BUVUEXFQHCLAPI-WTNQCIMSSA-N CC.CC.CC(C)N1CC2=CC=CC=C2C1.CC1=[Y]C2=CC=CC=C2N=C1C(C)C Chemical compound CC.CC.CC(C)N1CC2=CC=CC=C2C1.CC1=[Y]C2=CC=CC=C2N=C1C(C)C BUVUEXFQHCLAPI-WTNQCIMSSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 108090000331 Firefly luciferases Proteins 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- 102000003996 Interferon-beta Human genes 0.000 description 2
- 108090000467 Interferon-beta Proteins 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- 101800001554 RNA-directed RNA polymerase Proteins 0.000 description 2
- MHFMTUBUVQZIRE-WINRQGAFSA-N Sovaprevir Chemical compound C([C@H](C(=O)N1[C@@H](C[C@H](C1)OC=1C2=CC=C(C=C2N=C(C=1)C=1C=CC=CC=1)OC)C(=O)N[C@]1([C@@H](C1)C=C)C(=O)NS(=O)(=O)C1CC1)C(C)(C)C)C(=O)N1CCCCC1 MHFMTUBUVQZIRE-WINRQGAFSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- YQNQNVDNTFHQSW-UHFFFAOYSA-N acetic acid [2-[[(5-nitro-2-thiazolyl)amino]-oxomethyl]phenyl] ester Chemical compound CC(=O)OC1=CC=CC=C1C(=O)NC1=NC=C([N+]([O-])=O)S1 YQNQNVDNTFHQSW-UHFFFAOYSA-N 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- 108010058359 alisporivir Proteins 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 125000005907 alkyl ester group Chemical group 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 2
- 229960003805 amantadine Drugs 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000000844 anti-bacterial effect Effects 0.000 description 2
- 230000000843 anti-fungal effect Effects 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 229960002118 asunaprevir Drugs 0.000 description 2
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- ZTTKEBYSXUCBSE-QDFUAKMASA-N beclabuvir Chemical compound C1([C@@H]2C[C@@]2(CN2C3=CC(=CC=C33)C(=O)NS(=O)(=O)N(C)C)C(=O)N4[C@@H]5CC[C@H]4CN(C)C5)=CC(OC)=CC=C1C2=C3C1CCCCC1 ZTTKEBYSXUCBSE-QDFUAKMASA-N 0.000 description 2
- 229950010541 beclabuvir Drugs 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- UCMIRNVEIXFBKS-UHFFFAOYSA-N beta-alanine Chemical compound NCCC(O)=O UCMIRNVEIXFBKS-UHFFFAOYSA-N 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 229960000517 boceprevir Drugs 0.000 description 2
- LHHCSNFAOIFYRV-DOVBMPENSA-N boceprevir Chemical compound O=C([C@@H]1[C@@H]2[C@@H](C2(C)C)CN1C(=O)[C@@H](NC(=O)NC(C)(C)C)C(C)(C)C)NC(C(=O)C(N)=O)CC1CCC1 LHHCSNFAOIFYRV-DOVBMPENSA-N 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 235000019437 butane-1,3-diol Nutrition 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- YMGUBTXCNDTFJI-UHFFFAOYSA-M cyclopropanecarboxylate Chemical compound [O-]C(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-M 0.000 description 2
- FKRSSPOQAMALKA-CUPIEXAXSA-N daclatasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C1=NC(C=2C=CC(=CC=2)C=2C=CC(=CC=2)C=2N=C(NC=2)[C@H]2N(CCC2)C(=O)[C@@H](NC(=O)OC)C(C)C)=CN1 FKRSSPOQAMALKA-CUPIEXAXSA-N 0.000 description 2
- 229960005449 daclatasvir Drugs 0.000 description 2
- ZVTDLPBHTSMEJZ-JSZLBQEHSA-N danoprevir Chemical compound O=C([C@@]12C[C@H]1\C=C/CCCCC[C@@H](C(N1C[C@@H](C[C@H]1C(=O)N2)OC(=O)N1CC2=C(F)C=CC=C2C1)=O)NC(=O)OC(C)(C)C)NS(=O)(=O)C1CC1 ZVTDLPBHTSMEJZ-JSZLBQEHSA-N 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 2
- 229940043264 dodecyl sulfate Drugs 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- ZNVHRWXBOBYFMS-QQJVIXOFSA-N ethyl (1S,4R,6S,14S,18R)-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,8,15-trioxo-18-phenanthridin-6-yloxy-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxylate Chemical compound C1C(=O)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@H](OC=3C4=CC=CC=C4C4=CC=CC=C4N=3)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]21 ZNVHRWXBOBYFMS-QQJVIXOFSA-N 0.000 description 2
- OBDZWMRIICYVQP-IVZQSRNASA-N ethyl (1r,2s)-1-[bis[(2-methylpropan-2-yl)oxycarbonyl]amino]-2-(2,2-difluorohex-5-enyl)cyclopropane-1-carboxylate Chemical compound CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)[C@]1(C(=O)OCC)C[C@H]1CC(F)(F)CCC=C OBDZWMRIICYVQP-IVZQSRNASA-N 0.000 description 2
- WWSXFIPIYSQEKJ-XIESDHNKSA-N ethyl (1r,2s)-1-[bis[(2-methylpropan-2-yl)oxycarbonyl]amino]-2-(2-hydroxyhex-5-enyl)cyclopropane-1-carboxylate Chemical compound CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)[C@]1(C(=O)OCC)C[C@H]1CC(O)CCC=C WWSXFIPIYSQEKJ-XIESDHNKSA-N 0.000 description 2
- INGYLSXYLFSQFW-KZULUSFZSA-N ethyl (1r,2s)-1-[bis[(2-methylpropan-2-yl)oxycarbonyl]amino]-2-ethenylcyclopropane-1-carboxylate Chemical compound CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)[C@]1(C(=O)OCC)C[C@H]1C=C INGYLSXYLFSQFW-KZULUSFZSA-N 0.000 description 2
- CMUHFHCPBNFFOS-BXKDBHETSA-N ethyl (1r,2s)-1-amino-2-(2,2-difluorohex-5-enyl)cyclopropane-1-carboxylate Chemical compound CCOC(=O)[C@@]1(N)C[C@H]1CC(F)(F)CCC=C CMUHFHCPBNFFOS-BXKDBHETSA-N 0.000 description 2
- XWOIZBDKZDPDQY-UHFFFAOYSA-N ethyl 2-(1,3-benzothiazol-2-yl)-2-oxoacetate Chemical compound C1=CC=C2SC(C(=O)C(=O)OCC)=NC2=C1 XWOIZBDKZDPDQY-UHFFFAOYSA-N 0.000 description 2
- ZEYOYRUFYAFJHE-UHFFFAOYSA-N ethyl 2-(1-benzofuran-2-yl)-2-oxoacetate Chemical compound C1=CC=C2OC(C(=O)C(=O)OCC)=CC2=C1 ZEYOYRUFYAFJHE-UHFFFAOYSA-N 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- SLVAPEZTBDBAPI-GDLZYMKVSA-N filibuvir Chemical compound CCC1=NC(CC)=CC(CC[C@]2(OC(=O)C(CC3=NN4C(C)=CC(C)=NC4=N3)=C(O)C2)C2CCCC2)=C1 SLVAPEZTBDBAPI-GDLZYMKVSA-N 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 230000007794 irritation Effects 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 2
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 238000003670 luciferase enzyme activity assay Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- BHAZIAUOPHGQFL-WOPDTQHZSA-N methyl (2s,4r)-4-hydroxy-1-[(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]pent-4-enoyl]pyrrolidine-2-carboxylate Chemical compound COC(=O)[C@@H]1C[C@@H](O)CN1C(=O)[C@H](CC=C)NC(=O)OC(C)(C)C BHAZIAUOPHGQFL-WOPDTQHZSA-N 0.000 description 2
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 2
- 239000002808 molecular sieve Substances 0.000 description 2
- JWLSJBFATXHRSL-RYCLYDRISA-N n-((2r,6s ,13as,14ar,16as)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecin-6-yl)-5-methylisoxazole-3-carboxamide Chemical compound O1C(C)=CC(C(=O)N[C@@H]2C(N3C[C@@H](C[C@H]3C(=O)N[C@@]3(C[C@H]3CC(F)(F)CCCCC2)C(=O)NS(=O)(=O)C2CC2)OC=2C3=CC=CC=C3C3=CC=CC=C3N=2)=O)=N1 JWLSJBFATXHRSL-RYCLYDRISA-N 0.000 description 2
- QOWJRGRGPLNCFM-LWAHBMJCSA-N n-((2r,6s,13as,14ar,16as)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-yl)-5-methylisoxazole-3-carboxamide Chemical compound O1C(C)=CC(C(=O)N[C@@H]2C(N3C[C@@H](C[C@H]3C(=O)N[C@@]3(C[C@H]3CC(F)(F)CCCCC2)C(=O)NS(=O)(=O)C2CC2)OC=2N=C3C=CC=CC3=NC=2)=O)=N1 QOWJRGRGPLNCFM-LWAHBMJCSA-N 0.000 description 2
- 229960002480 nitazoxanide Drugs 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 239000001301 oxygen Chemical group 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- YNPNZTXNASCQKK-UHFFFAOYSA-N phenanthrene Chemical compound C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 2
- RZFVLEJOHSLEFR-UHFFFAOYSA-N phenanthridone Chemical compound C1=CC=C2C(O)=NC3=CC=CC=C3C2=C1 RZFVLEJOHSLEFR-UHFFFAOYSA-N 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- RYXIBQLRUHDYEE-UHFFFAOYSA-M potassium;5-(cyclohexen-1-yl)-3-[(4-methoxycyclohexyl)-(4-methylcyclohexanecarbonyl)amino]thiophene-2-carboxylate Chemical compound [K+].C1CC(OC)CCC1N(C1=C(SC(=C1)C=1CCCCC=1)C([O-])=O)C(=O)C1CCC(C)CC1 RYXIBQLRUHDYEE-UHFFFAOYSA-M 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- TTZHDVOVKQGIBA-IAAJYNJHSA-N propan-2-yl (2s)-2-[[[(2r,3r,4r,5r)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate Chemical compound N1([C@@H]2O[C@@H]([C@H]([C@]2(F)C)O)COP(=O)(N[C@@H](C)C(=O)OC(C)C)OC=2C=CC=CC=2)C=CC(=O)NC1=O TTZHDVOVKQGIBA-IAAJYNJHSA-N 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000013207 serial dilution Methods 0.000 description 2
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- 229960002091 simeprevir Drugs 0.000 description 2
- JTZZSQYMACOLNN-VDWJNHBNSA-N simeprevir Chemical compound O=C([C@@]12C[C@H]1\C=C/CCCCN(C)C(=O)[C@H]1[C@H](C(N2)=O)C[C@H](C1)OC=1C2=CC=C(C(=C2N=C(C=1)C=1SC=C(N=1)C(C)C)C)OC)NS(=O)(=O)C1CC1 JTZZSQYMACOLNN-VDWJNHBNSA-N 0.000 description 2
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 2
- 239000012418 sodium perborate tetrahydrate Substances 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- 235000019345 sodium thiosulphate Nutrition 0.000 description 2
- SSERCMQZZYTNBY-UHFFFAOYSA-M sodium;3-[(4-hydroxycyclohexyl)-(4-methylcyclohexanecarbonyl)amino]-5-phenylthiophene-2-carboxylate Chemical compound [Na+].C1CC(C)CCC1C(=O)N(C1=C(SC(=C1)C=1C=CC=CC=1)C([O-])=O)C1CCC(O)CC1 SSERCMQZZYTNBY-UHFFFAOYSA-M 0.000 description 2
- IBDSNZLUHYKHQP-UHFFFAOYSA-N sodium;3-oxidodioxaborirane;tetrahydrate Chemical compound O.O.O.O.[Na+].[O-]B1OO1 IBDSNZLUHYKHQP-UHFFFAOYSA-N 0.000 description 2
- 239000007962 solid dispersion Substances 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 125000003003 spiro group Chemical group 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 239000011593 sulfur Chemical group 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- NHKZSTHOYNWEEZ-AFCXAGJDSA-N taribavirin Chemical compound N1=C(C(=N)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NHKZSTHOYNWEEZ-AFCXAGJDSA-N 0.000 description 2
- 229950006081 taribavirin Drugs 0.000 description 2
- 229960002935 telaprevir Drugs 0.000 description 2
- BBAWEDCPNXPBQM-GDEBMMAJSA-N telaprevir Chemical compound N([C@H](C(=O)N[C@H](C(=O)N1C[C@@H]2CCC[C@@H]2[C@H]1C(=O)N[C@@H](CCC)C(=O)C(=O)NC1CC1)C(C)(C)C)C1CCCCC1)C(=O)C1=CN=CC=N1 BBAWEDCPNXPBQM-GDEBMMAJSA-N 0.000 description 2
- 108010017101 telaprevir Proteins 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- YOQSDZIWODERTO-RUZMOVHESA-N tert-butyl (2r,6s,13as,14ar,16as)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate Chemical compound O=C([C@@]12C[C@H]1CC(F)(F)CCCCC[C@@H](C(N1C[C@@H](C[C@H]1C(=O)N2)OC=1C2=CC=CC=C2C2=CC=CC=C2N=1)=O)NC(=O)OC(C)(C)C)NS(=O)(=O)C1CC1 YOQSDZIWODERTO-RUZMOVHESA-N 0.000 description 2
- JDENMLANLIKEPC-LSRHAZJQSA-N tert-butyl (2r,6s,13as,14ar,16as)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate Chemical compound O=C([C@@]12C[C@H]1CC(F)(F)CCCCC[C@@H](C(N1C[C@@H](C[C@H]1C(=O)N2)OC=1N=C2C=CC=CC2=NC=1)=O)NC(=O)OC(C)(C)C)NS(=O)(=O)C1CC1 JDENMLANLIKEPC-LSRHAZJQSA-N 0.000 description 2
- 125000001984 thiazolidinyl group Chemical group 0.000 description 2
- 230000036964 tight binding Effects 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- 238000003146 transient transfection Methods 0.000 description 2
- 238000011269 treatment regimen Methods 0.000 description 2
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 2
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- UDGCXALFXWTQSC-LUXWINNMSA-N (1S,4R,6S,14S,18R)-14-amino-N-cyclopropylsulfonyl-18-(3-ethylquinoxalin-2-yl)oxy-8,8-difluoro-2,15-dioxo-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxamide Chemical compound O=C([C@@]12C[C@H]1CC(F)(F)CCCCC[C@H](N)C(=O)N1[C@H](C(N2)=O)C[C@H](C1)OC1=NC2=CC=CC=C2N=C1CC)NS(=O)(=O)C1CC1 UDGCXALFXWTQSC-LUXWINNMSA-N 0.000 description 1
- MXLBNAXBCVNIIA-FKFVTETFSA-N (1S,4R,6S,14S,18R)-14-amino-N-cyclopropylsulfonyl-8,18-difluoro-2,15-dioxo-18-quinoxalin-2-yloxy-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxamide Chemical compound N[C@H]1CCCCCC(F)C[C@@H]2C[C@]2(NC(=O)[C@@H]2C[C@@](F)(CN2C1=O)Oc1cnc2ccccc2n1)C(=O)NS(=O)(=O)C1CC1 MXLBNAXBCVNIIA-FKFVTETFSA-N 0.000 description 1
- WRZJXXQSWXEWDB-MVEVPWQMSA-N (1S,4R,6S,14S,18R)-14-amino-N-cyclopropylsulfonyl-8,8-difluoro-18-(3-methylquinoxalin-2-yl)oxy-2,15-dioxo-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxamide Chemical compound O=C([C@@]12C[C@H]1CC(F)(F)CCCCC[C@H](N)C(=O)N1[C@H](C(N2)=O)C[C@H](C1)OC1=NC2=CC=CC=C2N=C1C)NS(=O)(=O)C1CC1 WRZJXXQSWXEWDB-MVEVPWQMSA-N 0.000 description 1
- FCSVTSKOKYSUAB-ZGEPUCGGSA-N (1S,4R,6S,14S,18R)-14-amino-N-cyclopropylsulfonyl-8,8-difluoro-2,15-dioxo-18-(3-phenylquinoxalin-2-yl)oxy-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxamide Chemical compound O([C@@H]1C[C@H]2C(=O)N[C@@]3(C[C@H]3CC(F)(F)CCCCC[C@@H](C(N2C1)=O)N)C(=O)NS(=O)(=O)C1CC1)C1=NC2=CC=CC=C2N=C1C1=CC=CC=C1 FCSVTSKOKYSUAB-ZGEPUCGGSA-N 0.000 description 1
- UAVXUHAONFCAAI-DFSUGDSSSA-N (1S,4R,6S,14S,18R)-18-[3-(1-benzofuran-2-yl)quinoxalin-2-yl]oxy-8,8-difluoro-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxylic acid Chemical compound O=C1[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(O)=O)NC(=O)[C@@H]2C[C@@H](OC=3C(=NC4=CC=CC=C4N=3)C=3OC4=CC=CC=C4C=3)CN21 UAVXUHAONFCAAI-DFSUGDSSSA-N 0.000 description 1
- AUXPYFJIPNFHMT-ODGWHSJUSA-N (1S,4R,6S,14S,18R)-8,18-difluoro-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-18-quinoxalin-2-yloxy-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxylic acid Chemical compound CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)C[C@@H]2C[C@]2(NC(=O)[C@@H]2C[C@@](F)(CN2C1=O)Oc1cnc2ccccc2n1)C(O)=O AUXPYFJIPNFHMT-ODGWHSJUSA-N 0.000 description 1
- ODZWBUIPABMGBV-LDQZHNCJSA-N (1S,4R,6S,14S,18R)-8,8-difluoro-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-18-(3-phenylquinoxalin-2-yl)oxy-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxylic acid Chemical compound O([C@@H]1C[C@H]2C(=O)N[C@@]3(C[C@H]3CC(F)(F)CCCCC[C@@H](C(N2C1)=O)NC(=O)OC(C)(C)C)C(O)=O)C1=NC2=CC=CC=C2N=C1C1=CC=CC=C1 ODZWBUIPABMGBV-LDQZHNCJSA-N 0.000 description 1
- KROLYRCAOQSRNR-FDSIQNTBSA-N (1S,4R,6S,14S,18R)-N-cyclopropylsulfonyl-14-(2,2-dimethylpropanoylamino)-18-(3-ethylquinoxalin-2-yl)oxy-8,8-difluoro-2,15-dioxo-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxamide Chemical compound O=C([C@@]12C[C@H]1CC(F)(F)CCCCC[C@@H](C(=O)N1[C@H](C(N2)=O)C[C@H](C1)OC1=NC2=CC=CC=C2N=C1CC)NC(=O)C(C)(C)C)NS(=O)(=O)C1CC1 KROLYRCAOQSRNR-FDSIQNTBSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- LRQIULSUXYLAQU-BSYKTYROSA-N (2r,6s,13as,14ar,16as)-6-(tert-butoxycarbonylamino)-12,12-difluoro-2-(3-methylquinoxalin-2-yloxy)-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid Chemical compound C1N2C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(O)=O)NC(=O)[C@@H]2C[C@H]1OC1=NC2=CC=CC=C2N=C1C LRQIULSUXYLAQU-BSYKTYROSA-N 0.000 description 1
- KKIPIHFNBCGHOL-MKELMXMHSA-N (2r,6s,13as,14ar,16as)-6-(tert-butoxycarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid Chemical compound O=C1[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(O)=O)NC(=O)[C@@H]2C[C@@H](O)CN21 KKIPIHFNBCGHOL-MKELMXMHSA-N 0.000 description 1
- YRFFWHSNNWHNPF-IISFBIFZSA-N (2r,6s,13as,14ar,16as)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclo pentadecine-14a-carboxylate Chemical compound C1C(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@H](OC=3C4=CC=CC=C4C4=CC=CC=C4N=3)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]21 YRFFWHSNNWHNPF-IISFBIFZSA-N 0.000 description 1
- HFKDYAZXVDIILC-WIFIPGKVSA-N (2r,6s,13as,14ar,16as)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound C1C(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@H](OC=3N=C4C=CC=CC4=NC=3)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]21 HFKDYAZXVDIILC-WIFIPGKVSA-N 0.000 description 1
- IAFHOFINSVHYNN-DUOQQSOHSA-N (2r,6s,13as,14ar,16as)-tert-butyl 12-hydroxy-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclo propa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound O1C(C)=CC(C(=O)N[C@@H]2C(N3C[C@@H](C[C@H]3C(=O)N[C@@]3(C[C@H]3CC(O)CCCCC2)C(=O)OC(C)(C)C)OC=2C3=CC=CC=C3C3=CC=CC=C3N=2)=O)=N1 IAFHOFINSVHYNN-DUOQQSOHSA-N 0.000 description 1
- OKFINMMHJOXSSN-PBGAFUIBSA-N (2r,6s,13as,14ar,16as)-tert-butyl 6-(5-methylisoxazole-3-carboxamido)-5,12,16-trioxo-2-(phenanthridin-6-yloxy)o ctadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound O1C(C)=CC(C(=O)N[C@@H]2C(N3C[C@@H](C[C@H]3C(=O)N[C@@]3(C[C@H]3CC(=O)CCCCC2)C(=O)OC(C)(C)C)OC=2C3=CC=CC=C3C3=CC=CC=C3N=2)=O)=N1 OKFINMMHJOXSSN-PBGAFUIBSA-N 0.000 description 1
- APOAOKPRKWJTMI-IWEPMSHRSA-N (2r,6s,13as,14ar,16as,z)-tert-butyl 6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound O1C(C)=CC(C(=O)N[C@@H]2C(N3C[C@@H](C[C@H]3C(=O)N[C@@]3(C[C@H]3\C=C/CCCCC2)C(=O)OC(C)(C)C)OC=2C3=CC=CC=C3C3=CC=CC=C3N=2)=O)=N1 APOAOKPRKWJTMI-IWEPMSHRSA-N 0.000 description 1
- INGSRYLDPRZHDF-DBFOEXOYSA-N (2s ,6s ,13as,14ar,16as,z)-ethyl 2-(4-bromophenylsulfonyloxy)-6-(tert-butoxycarbonylamino)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound O([C@@H]1CN2C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC\C=C/[C@@H]3C[C@]3(NC(=O)[C@@H]2C1)C(=O)OCC)S(=O)(=O)C1=CC=C(Br)C=C1 INGSRYLDPRZHDF-DBFOEXOYSA-N 0.000 description 1
- BUPDPLXLAKNJMI-ZETCQYMHSA-N (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]pent-4-enoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CC=C BUPDPLXLAKNJMI-ZETCQYMHSA-N 0.000 description 1
- KJQVROMUJPFBGA-OUJMPZBYSA-N (2s,6s,13as,14ar,16as)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonylamino)-5,12,16-trioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound O([C@@H]1CN2C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(=O)C[C@@H]3C[C@]3(NC(=O)[C@@H]2C1)C(=O)OCC)C(=O)C1=CC=CC=C1 KJQVROMUJPFBGA-OUJMPZBYSA-N 0.000 description 1
- XCIHXPUUVKKIMP-QTJBUOKZSA-N (2s,6s,13as,14ar,16as)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo [1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound C1C(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@@H](O)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]21 XCIHXPUUVKKIMP-QTJBUOKZSA-N 0.000 description 1
- LNACKEHWUDDQJJ-ZAGOMNEGSA-N (2s,6s,13as,14ar,16as,z)-ethyl 6-(tert-butoxycarbonylamino)-2-hydroxy-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate Chemical compound C/1=C/CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@@H](O)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]2\1 LNACKEHWUDDQJJ-ZAGOMNEGSA-N 0.000 description 1
- 125000006727 (C1-C6) alkenyl group Chemical group 0.000 description 1
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- 108010030583 (melle-4)cyclosporin Proteins 0.000 description 1
- UKAUYVFTDYCKQA-UHFFFAOYSA-N -2-Amino-4-hydroxybutanoic acid Natural products OC(=O)C(N)CCO UKAUYVFTDYCKQA-UHFFFAOYSA-N 0.000 description 1
- 125000006033 1,1-dimethyl-2-propenyl group Chemical group 0.000 description 1
- 125000004529 1,2,3-triazinyl group Chemical group N1=NN=C(C=C1)* 0.000 description 1
- UXYRXGFUANQKTA-UHFFFAOYSA-N 1,2-oxazole-3-carboxylic acid Chemical compound OC(=O)C=1C=CON=1 UXYRXGFUANQKTA-UHFFFAOYSA-N 0.000 description 1
- 125000004520 1,3,4-thiadiazolyl group Chemical group 0.000 description 1
- MNXBVXVIRAIAEG-UHFFFAOYSA-N 1,3-benzothiazol-2-yl(trimethyl)silane Chemical compound C1=CC=C2SC([Si](C)(C)C)=NC2=C1 MNXBVXVIRAIAEG-UHFFFAOYSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical group C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- LUQVIGCZOGBWKB-UHFFFAOYSA-N 1,4-diazacyclopentadeca-2,4,6,8,10,12,14-heptaen-6-ylcarbamic acid Chemical compound C1=CC=CC=C(C=NC=CNC=CC=C1)NC(=O)O LUQVIGCZOGBWKB-UHFFFAOYSA-N 0.000 description 1
- PXRXGHUTKHXUGF-UHFFFAOYSA-N 1,5-dimethylpyrazole-3-carboxylic acid Chemical compound CC1=CC(C(O)=O)=NN1C PXRXGHUTKHXUGF-UHFFFAOYSA-N 0.000 description 1
- AZUYLZMQTIKGSC-UHFFFAOYSA-N 1-[6-[4-(5-chloro-6-methyl-1H-indazol-4-yl)-5-methyl-3-(1-methylindazol-5-yl)pyrazol-1-yl]-2-azaspiro[3.3]heptan-2-yl]prop-2-en-1-one Chemical compound ClC=1C(=C2C=NNC2=CC=1C)C=1C(=NN(C=1C)C1CC2(CN(C2)C(C=C)=O)C1)C=1C=C2C=NN(C2=CC=1)C AZUYLZMQTIKGSC-UHFFFAOYSA-N 0.000 description 1
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 1
- YBFIKNNFQIBIQZ-UHFFFAOYSA-N 1-methyl-pyrazole-3-carboxylic acid Chemical compound CN1C=CC(C(O)=O)=N1 YBFIKNNFQIBIQZ-UHFFFAOYSA-N 0.000 description 1
- ATJVVVCODTXRAE-UHFFFAOYSA-N 1-methylcyclopropane-1-sulfonamide Chemical compound NS(=O)(=O)C1(C)CC1 ATJVVVCODTXRAE-UHFFFAOYSA-N 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- 125000005955 1H-indazolyl group Chemical group 0.000 description 1
- 125000000453 2,2,2-trichloroethyl group Chemical group [H]C([H])(*)C(Cl)(Cl)Cl 0.000 description 1
- NOGFHTGYPKWWRX-UHFFFAOYSA-N 2,2,6,6-tetramethyloxan-4-one Chemical compound CC1(C)CC(=O)CC(C)(C)O1 NOGFHTGYPKWWRX-UHFFFAOYSA-N 0.000 description 1
- DFNMITDSNZEASO-UHFFFAOYSA-N 2-(3-chloroquinoxalin-2-yl)-1,3-benzothiazole Chemical compound C1=CC=C2SC(C3=NC4=CC=CC=C4N=C3Cl)=NC2=C1 DFNMITDSNZEASO-UHFFFAOYSA-N 0.000 description 1
- RVHOBHMAPRVOLO-UHFFFAOYSA-N 2-ethylbutanedioic acid Chemical class CCC(C(O)=O)CC(O)=O RVHOBHMAPRVOLO-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- WYKHFQKONWMWQM-UHFFFAOYSA-N 2-sulfanylidene-1h-pyridine-3-carboxylic acid Chemical compound OC(=O)C1=CC=CN=C1S WYKHFQKONWMWQM-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- BRMWTNUJHUMWMS-UHFFFAOYSA-N 3-Methylhistidine Natural products CN1C=NC(CC(N)C(O)=O)=C1 BRMWTNUJHUMWMS-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- BMIMNRPAEPIYDN-UHFFFAOYSA-N 3-methyl-2-quinoxalinol Chemical compound C1=CC=C2N=C(O)C(C)=NC2=C1 BMIMNRPAEPIYDN-UHFFFAOYSA-N 0.000 description 1
- 125000006032 3-methyl-3-butenyl group Chemical group 0.000 description 1
- ZBBQSGVRBQKLLE-UHFFFAOYSA-N 3-phenyl-1H-quinoxalin-2-one Chemical compound OC1=NC2=CC=CC=C2N=C1C1=CC=CC=C1 ZBBQSGVRBQKLLE-UHFFFAOYSA-N 0.000 description 1
- WCKQPPQRFNHPRJ-UHFFFAOYSA-N 4-[[4-(dimethylamino)phenyl]diazenyl]benzoic acid Chemical compound C1=CC(N(C)C)=CC=C1N=NC1=CC=C(C(O)=O)C=C1 WCKQPPQRFNHPRJ-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-M 4-bromobenzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-M 0.000 description 1
- OTLNPYWUJOZPPA-UHFFFAOYSA-M 4-nitrobenzoate Chemical compound [O-]C(=O)C1=CC=C([N+]([O-])=O)C=C1 OTLNPYWUJOZPPA-UHFFFAOYSA-M 0.000 description 1
- SJQRQOKXQKVJGJ-UHFFFAOYSA-N 5-(2-aminoethylamino)naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(NCCN)=CC=CC2=C1S(O)(=O)=O SJQRQOKXQKVJGJ-UHFFFAOYSA-N 0.000 description 1
- RBYJWCRKFLGNDB-UHFFFAOYSA-N 5-methylpyrazine-2-carboxylic acid Chemical compound CC1=CN=C(C(O)=O)C=N1 RBYJWCRKFLGNDB-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 235000003276 Apios tuberosa Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000010744 Arachis villosulicarpa Nutrition 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- LWZOMEUAXCSUJF-GUGXNSJQSA-N B.C1CCOC1.C=C[C@@H]1C[C@]1(CC(=O)OC(C)(C)C)C(=O)OCC.CCOC(=O)[C@@]1(N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)C[C@H]1CCO Chemical compound B.C1CCOC1.C=C[C@@H]1C[C@]1(CC(=O)OC(C)(C)C)C(=O)OCC.CCOC(=O)[C@@]1(N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)C[C@H]1CCO LWZOMEUAXCSUJF-GUGXNSJQSA-N 0.000 description 1
- ZYEKDIBXSGREPV-GRZUGJMDSA-N B.C1CCOC1.CC1=CC(C(=O)N[C@H]2CCCCC/C=C\[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCCC(O)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.OO.[Na+].[OH-] Chemical compound B.C1CCOC1.CC1=CC(C(=O)N[C@H]2CCCCC/C=C\[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCCC(O)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.OO.[Na+].[OH-] ZYEKDIBXSGREPV-GRZUGJMDSA-N 0.000 description 1
- YTQIVNNVHGHINU-MZPIIDKYSA-N B.C1CCOC1.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(O)C[C@@H]1C2.OO.[Na+].[OH-] Chemical compound B.C1CCOC1.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(O)C[C@@H]1C2.OO.[Na+].[OH-] YTQIVNNVHGHINU-MZPIIDKYSA-N 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- MEKDPHXPVMKCON-UHFFFAOYSA-N C.CC Chemical compound C.CC MEKDPHXPVMKCON-UHFFFAOYSA-N 0.000 description 1
- KFLXTWAUJUYXKE-UHFFFAOYSA-N C1=CC2=C(C=C1)OC=C2.CCOC(=O)C(=O)C1=CC2=C(C=CC=C2)O1.CCOC(=O)C(=O)OCC.[Li]CCCC Chemical compound C1=CC2=C(C=C1)OC=C2.CCOC(=O)C(=O)C1=CC2=C(C=CC=C2)O1.CCOC(=O)C(=O)OCC.[Li]CCCC KFLXTWAUJUYXKE-UHFFFAOYSA-N 0.000 description 1
- FVJPZEXUYPBZBW-OMLZERRLSA-N C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(C)OC(=O)/N=N/C(=O)OC(C)C.CC1=C(O)N=C2C=CC=CC2=N1.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](O)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2 Chemical compound C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(C)OC(=O)/N=N/C(=O)OC(C)C.CC1=C(O)N=C2C=CC=CC2=N1.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](O)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2 FVJPZEXUYPBZBW-OMLZERRLSA-N 0.000 description 1
- FCGXQWKJKIXVMY-IUWQYWSCSA-N C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(C)OC(=O)/N=N/C(=O)OC(C)C.CCOC(=O)[C@@]12C[C@H]1CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.OC1=CN=C2C=CC=CC2=N1 Chemical compound C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(C)OC(=O)/N=N/C(=O)OC(C)C.CCOC(=O)[C@@]12C[C@H]1CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.OC1=CN=C2C=CC=CC2=N1 FCGXQWKJKIXVMY-IUWQYWSCSA-N 0.000 description 1
- UIKZLHMPKJQPNP-FZRSONGNSA-N C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(C)OC(=O)/N=N/C(=O)OC(C)C.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](O)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.OC1=C(C2=CC=CC=C2)N=C2C=CC=CC2=N1 Chemical compound C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(C)OC(=O)/N=N/C(=O)OC(C)C.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](O)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.OC1=C(C2=CC=CC=C2)N=C2C=CC=CC2=N1 UIKZLHMPKJQPNP-FZRSONGNSA-N 0.000 description 1
- TVOOROHMWHOSPF-ONAYEHSQSA-N C1=CC=NC=C1.CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4)CN3C2=O)=NO1.CC1=CC(C(=O)O)=NO1.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[PH](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O Chemical compound C1=CC=NC=C1.CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4)CN3C2=O)=NO1.CC1=CC(C(=O)O)=NO1.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[PH](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O TVOOROHMWHOSPF-ONAYEHSQSA-N 0.000 description 1
- BGCHCLISBVVLOR-UIILLBIHSA-N C1=CC=NC=C1.CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C)CN3C2=O)=NO1.CC1=CC(C(=O)O)=NO1.CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[PH](F)(F)(F)(F)F Chemical compound C1=CC=NC=C1.CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C)CN3C2=O)=NO1.CC1=CC(C(=O)O)=NO1.CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[PH](F)(F)(F)(F)F BGCHCLISBVVLOR-UIILLBIHSA-N 0.000 description 1
- BMXNNRXAOBCCBI-RFRJPPHRSA-N C1=CC=NC=C1.CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C4=CC=CC=C4)CN3C2=O)=NO1.CC1=CC(C(=O)O)=NO1.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[P-](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O Chemical compound C1=CC=NC=C1.CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C4=CC=CC=C4)CN3C2=O)=NO1.CC1=CC(C(=O)O)=NO1.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[P-](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O BMXNNRXAOBCCBI-RFRJPPHRSA-N 0.000 description 1
- CPLBWEZILLCYGR-AJJSHMDZSA-N C1CCC2=NCCCN2CC1.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 Chemical compound C1CCC2=NCCCN2CC1.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 CPLBWEZILLCYGR-AJJSHMDZSA-N 0.000 description 1
- CCOZSAVOMBODDX-LTESZVTASA-N C1CCC2=NCCCN2CC1.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 Chemical compound C1CCC2=NCCCN2CC1.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 CCOZSAVOMBODDX-LTESZVTASA-N 0.000 description 1
- CHIJTQZXBXBFFO-BWIAWXRBSA-N C1CCC2=NCCCN2CC1.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 Chemical compound C1CCC2=NCCCN2CC1.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 CHIJTQZXBXBFFO-BWIAWXRBSA-N 0.000 description 1
- QHJWILVLHGPPMB-YVHKYISWSA-N C1CCC2=NCCCN2CC1.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 Chemical compound C1CCC2=NCCCN2CC1.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 QHJWILVLHGPPMB-YVHKYISWSA-N 0.000 description 1
- XKBZMUFWPMBQPB-VAKDIUPASA-N C1CCC2=NCCCN2CC1.CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O.CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 Chemical compound C1CCC2=NCCCN2CC1.CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O.CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 XKBZMUFWPMBQPB-VAKDIUPASA-N 0.000 description 1
- WNGMCCZPSCJRDT-CDMWPUBCSA-N C1CCC2=NCCCN2CC1.CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)O)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 Chemical compound C1CCC2=NCCCN2CC1.CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)O)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 WNGMCCZPSCJRDT-CDMWPUBCSA-N 0.000 description 1
- GFZOKIIBKDBLIW-RHPBKZKASA-N C1CCC2=NCCCN2CC1.CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1.CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)O)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 Chemical compound C1CCC2=NCCCN2CC1.CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1.CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)O)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1.NS(=O)(=O)C1CC1.O=C(N1C=CN=C1)N1C=CN=C1 GFZOKIIBKDBLIW-RHPBKZKASA-N 0.000 description 1
- RILFFTXULJGFTO-VCQXAOINSA-N C=CCCC(=O)C[C@@H]1C[C@@]1(C(=O)OCC)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C.C=CCCC(O)C[C@@H]1C[C@@]1(C(=O)OCC)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C Chemical compound C=CCCC(=O)C[C@@H]1C[C@@]1(C(=O)OCC)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C.C=CCCC(O)C[C@@H]1C[C@@]1(C(=O)OCC)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C RILFFTXULJGFTO-VCQXAOINSA-N 0.000 description 1
- IRHJBHKRZDXTCJ-YFKYFRGLSA-N C=CCCC(F)(F)C[C@@H]1C[C@@]1(C(=O)OCC)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C.C=CCCC(F)(F)C[C@@H]1C[C@]1(N)C(=O)OCC.N=N/N=N/Cl Chemical compound C=CCCC(F)(F)C[C@@H]1C[C@@]1(C(=O)OCC)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C.C=CCCC(F)(F)C[C@@H]1C[C@]1(N)C(=O)OCC.N=N/N=N/Cl IRHJBHKRZDXTCJ-YFKYFRGLSA-N 0.000 description 1
- TWYVUGGXDDLZFX-HFJVRAEPSA-N C=CCCC(F)(F)C[C@@H]1C[C@]1(CC(=O)[C@@H]1C[C@@H](O)CN1C(=O)[C@H](CC=C)NC(=O)OC(C)(C)C)C(=O)OCC.C=CCCC(F)(F)C[C@@H]1C[C@]1(N)C(=O)OCC.C=CC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](O)C[C@H]1C(=O)O.Cl Chemical compound C=CCCC(F)(F)C[C@@H]1C[C@]1(CC(=O)[C@@H]1C[C@@H](O)CN1C(=O)[C@H](CC=C)NC(=O)OC(C)(C)C)C(=O)OCC.C=CCCC(F)(F)C[C@@H]1C[C@]1(N)C(=O)OCC.C=CC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](O)C[C@H]1C(=O)O.Cl TWYVUGGXDDLZFX-HFJVRAEPSA-N 0.000 description 1
- WWNOWQDMMXDMLM-RVYCNEAISA-N C=CCCC(F)(F)C[C@@H]1C[C@]1(CC(=O)[C@@H]1C[C@@H](O)CN1C(=O)[C@H](CC=C)NC(=O)OC(C)(C)C)C(=O)OCC.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](O)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CC=CCCC(F)(F)C[C@@H]1C2 Chemical compound C=CCCC(F)(F)C[C@@H]1C[C@]1(CC(=O)[C@@H]1C[C@@H](O)CN1C(=O)[C@H](CC=C)NC(=O)OC(C)(C)C)C(=O)OCC.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](O)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CC=CCCC(F)(F)C[C@@H]1C2 WWNOWQDMMXDMLM-RVYCNEAISA-N 0.000 description 1
- SYZIWRRNKXPFPC-UVYRFMFCSA-M C=CCCC(O)C[C@@H]1C[C@@]1(C(=O)OCC)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C.C=CCC[Mg]Br.CCOC(=O)[C@@]1(N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)C[C@H]1CCO Chemical compound C=CCCC(O)C[C@@H]1C[C@@]1(C(=O)OCC)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C.C=CCC[Mg]Br.CCOC(=O)[C@@]1(N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)C[C@H]1CCO SYZIWRRNKXPFPC-UVYRFMFCSA-M 0.000 description 1
- PQFZQXLRYCUWIX-DDFUFULDSA-N C=CC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](O)C[C@H]1C(=O)O.C=CC[C@H](NC(=O)OC(C)(C)C)C(=O)O.COC(=O)[C@@H]1C[C@@H](O)CN1 Chemical compound C=CC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](O)C[C@H]1C(=O)O.C=CC[C@H](NC(=O)OC(C)(C)C)C(=O)O.COC(=O)[C@@H]1C[C@@H](O)CN1 PQFZQXLRYCUWIX-DDFUFULDSA-N 0.000 description 1
- IDMRIUXWRGUTRB-UHFFFAOYSA-N CC(C)(C)C1=CN=CC=N1.CC(C)C(C(C)C)N1CCCNC1=O.CC(C)C1=C(F)C=CC=C1.CC(C)C1=C(F)C=NC=C1.CC(C)C1=CC(C(C)C)=NO1.CC(C)C1=CC(F)=CC=C1.CC(C)C1=CC=C(F)C=C1.CC(C)C1=CC=C(F)C=C1.CC(C)C1=CC=NN1C.CC(C)C1=CSC=N1.CC(C)C1=NC=CS1.CC(C)C1=NC=NC=C1.CC(C)C1=NN(C)C=C1.CC(C)C1=NOC(C2CC2)=C1.CC(C)C1=NOC=C1.CC1=C(C(C)C)SC=C1.CC1=C(C(C)C)SC=N1.CC1=C(C)C(C(C)C)=NO1.CC1=CC(C(C)C)=NN1.CC1=CC(C(C)C)=NN1C.CC1=CC(C(C)C)=NO1.CC1=CC=C(C(C)C)O1.CC1=CC=C(C(C)C)S1.CC1=CN=C(C(C)C)S1.CC1=NC=C(C(C)C)N=C1.CC1=NC=C(C(C)C)N=C1.CC1=NN(C)C(C(C)C)=C1.CC1=NNC(C(C)C)=C1.CC1=NOC(C(C)C)=C1.CCC1=CC(C(C)C)=NO1 Chemical compound CC(C)(C)C1=CN=CC=N1.CC(C)C(C(C)C)N1CCCNC1=O.CC(C)C1=C(F)C=CC=C1.CC(C)C1=C(F)C=NC=C1.CC(C)C1=CC(C(C)C)=NO1.CC(C)C1=CC(F)=CC=C1.CC(C)C1=CC=C(F)C=C1.CC(C)C1=CC=C(F)C=C1.CC(C)C1=CC=NN1C.CC(C)C1=CSC=N1.CC(C)C1=NC=CS1.CC(C)C1=NC=NC=C1.CC(C)C1=NN(C)C=C1.CC(C)C1=NOC(C2CC2)=C1.CC(C)C1=NOC=C1.CC1=C(C(C)C)SC=C1.CC1=C(C(C)C)SC=N1.CC1=C(C)C(C(C)C)=NO1.CC1=CC(C(C)C)=NN1.CC1=CC(C(C)C)=NN1C.CC1=CC(C(C)C)=NO1.CC1=CC=C(C(C)C)O1.CC1=CC=C(C(C)C)S1.CC1=CN=C(C(C)C)S1.CC1=NC=C(C(C)C)N=C1.CC1=NC=C(C(C)C)N=C1.CC1=NN(C)C(C(C)C)=C1.CC1=NNC(C(C)C)=C1.CC1=NOC(C(C)C)=C1.CCC1=CC(C(C)C)=NO1 IDMRIUXWRGUTRB-UHFFFAOYSA-N 0.000 description 1
- RASMJAMBSSPFDT-WBNCSAJTSA-N CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O.CCN(C(C)C)C(C)C.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](O)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.CN1C=CN=C1.O=C(OC(=O)C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O.CCN(C(C)C)C(C)C.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](O)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.CN1C=CN=C1.O=C(OC(=O)C1=CC=CC=C1)C1=CC=CC=C1 RASMJAMBSSPFDT-WBNCSAJTSA-N 0.000 description 1
- HSCVOFCKIFMUJK-XSASELIFSA-N CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O.Cl.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O.Cl.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O HSCVOFCKIFMUJK-XSASELIFSA-N 0.000 description 1
- YSXLLWSNRBBAEJ-MHWYJLRCSA-N CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O YSXLLWSNRBBAEJ-MHWYJLRCSA-N 0.000 description 1
- KHBIZESTKRYKRD-NQDGPSJFSA-N CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O.Cl.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O.Cl.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O KHBIZESTKRYKRD-NQDGPSJFSA-N 0.000 description 1
- IGXVHJVCQHHUEW-ZGALVPQSSA-N CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O.Cl.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O.Cl.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O IGXVHJVCQHHUEW-ZGALVPQSSA-N 0.000 description 1
- HWMIBEBGTFBNPD-QUNQYOGUSA-N CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O HWMIBEBGTFBNPD-QUNQYOGUSA-N 0.000 description 1
- WYYDGBPBQYDFTJ-DPIFWTARSA-N CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O.Cl.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O.Cl.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O WYYDGBPBQYDFTJ-DPIFWTARSA-N 0.000 description 1
- XMIXMZHXZHHVJS-SPTSXRSISA-N CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](O)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O.CCC(C)(C)O.ClC1=NC2=C(C=CC=C2)N=C1/C1=C/C2=C(C=CC=C2)O1.[Na+] Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](O)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O.CCC(C)(C)O.ClC1=NC2=C(C=CC=C2)N=C1/C1=C/C2=C(C=CC=C2)O1.[Na+] XMIXMZHXZHHVJS-SPTSXRSISA-N 0.000 description 1
- IBINPFVMFUQVKT-CJKKZHRPSA-N CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](O)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O.CCC(C)(C)O.ClC1=NC2=C(C=CC=C2)N=C1/C1=N/C2=C(C=CC=C2)S1.[Na+] Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](O)CN2C1=O.CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O.CCC(C)(C)O.ClC1=NC2=C(C=CC=C2)N=C1/C1=N/C2=C(C=CC=C2)S1.[Na+] IBINPFVMFUQVKT-CJKKZHRPSA-N 0.000 description 1
- VIWGFUMGOXOFAK-UYPLZCGLSA-M CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[Li]O Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3)CN2C1=O.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[Li]O VIWGFUMGOXOFAK-UYPLZCGLSA-M 0.000 description 1
- IZIGTOKCFJXUBZ-SBFWTJIMSA-M CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[Li]O Chemical compound CC(C)(C)OC(=O)C[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3C3=CC=CC=C3)CN2C1=O.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[Li]O IZIGTOKCFJXUBZ-SBFWTJIMSA-M 0.000 description 1
- WLFSWFVXNFEIBH-IWQUPONHSA-N CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O Chemical compound CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O WLFSWFVXNFEIBH-IWQUPONHSA-N 0.000 description 1
- OIYZTCIGPVILHE-VPHFUGEYSA-N CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O.Cl.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O Chemical compound CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O.Cl.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O OIYZTCIGPVILHE-VPHFUGEYSA-N 0.000 description 1
- SEOQMDIUSZTGPB-KYIOFCNXSA-M CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](O)CN2C1=O.CCC(C)(C)[Na]O.CCC1=C(Cl)N=C2/C=C\C=C/C2=N1.CCC1=C(O[C@@H]2C[C@H]3C(=O)N[C@]4(C(=O)O)C[C@H]4CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C2)N=C2C=CC=CC2=N1 Chemical compound CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](O)CN2C1=O.CCC(C)(C)[Na]O.CCC1=C(Cl)N=C2/C=C\C=C/C2=N1.CCC1=C(O[C@@H]2C[C@H]3C(=O)N[C@]4(C(=O)O)C[C@H]4CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C2)N=C2C=CC=CC2=N1 SEOQMDIUSZTGPB-KYIOFCNXSA-M 0.000 description 1
- KHSHYESZTJWXNC-MOGMHANRSA-M CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](O)CN2C1=O.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](O)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[Li]O Chemical compound CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](O)CN2C1=O.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](O)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[Li]O KHSHYESZTJWXNC-MOGMHANRSA-M 0.000 description 1
- IKIBTZPPYGSCAI-FDIQEMGLSA-N CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[Li+].[OH-] Chemical compound CC(C)(C)OC(=O)N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)O)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[Li+].[OH-] IKIBTZPPYGSCAI-FDIQEMGLSA-N 0.000 description 1
- DGGGVIXZPQZMBI-XDKSRMPOSA-N CC(C)(C)OC(N[C@@H](CCCCCC([C@H](C1)C1(C1)[C@]1(C(NS(C1CC1)(=O)=O)=O)NC([C@H](C1)N2C[C@@H]1Oc1nc3ccccc3nc1)=O)(F)F)C2=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CCCCCC([C@H](C1)C1(C1)[C@]1(C(NS(C1CC1)(=O)=O)=O)NC([C@H](C1)N2C[C@@H]1Oc1nc3ccccc3nc1)=O)(F)F)C2=O)=O DGGGVIXZPQZMBI-XDKSRMPOSA-N 0.000 description 1
- ABPZYIVXBSZSTP-VDXOJYAPSA-N CC(C)/C=C\C(C)(C)C.CC(C)CCC(C)(C)C Chemical compound CC(C)/C=C\C(C)(C)C.CC(C)CCC(C)(C)C ABPZYIVXBSZSTP-VDXOJYAPSA-N 0.000 description 1
- COWZGDLCGNPALY-UHFFFAOYSA-N CC(C)C1=CC2=CC=CC=C2O1.CC(C)C1=CC=CC=C1.CC(C)C1=NC2=CC=CC=C2S1 Chemical compound CC(C)C1=CC2=CC=CC=C2O1.CC(C)C1=CC=CC=C1.CC(C)C1=NC2=CC=CC=C2S1 COWZGDLCGNPALY-UHFFFAOYSA-N 0.000 description 1
- WTJKJHFRNATZOM-UHFFFAOYSA-N CC(N1CC1)=[IH] Chemical compound CC(N1CC1)=[IH] WTJKJHFRNATZOM-UHFFFAOYSA-N 0.000 description 1
- QUFBMYLKDQPJBV-GRYOFQPXSA-N CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](O/C4=N/C5C=CC=CC5C5=C4C=CC=C5)CN3C2=O)=NO1 Chemical compound CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](O/C4=N/C5C=CC=CC5C5=C4C=CC=C5)CN3C2=O)=NO1 QUFBMYLKDQPJBV-GRYOFQPXSA-N 0.000 description 1
- QUNKZKZNGUZQTJ-OCAVINHWSA-N CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4/C4=C/C5=C(C=CC=C5)O4)CN3C2=O)=NO1.CC1=CC(C(=O)O)=NO1.CCN(C(C)C)C(C)C.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[P-](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O Chemical compound CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4/C4=C/C5=C(C=CC=C5)O4)CN3C2=O)=NO1.CC1=CC(C(=O)O)=NO1.CCN(C(C)C)C(C)C.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[P-](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O QUNKZKZNGUZQTJ-OCAVINHWSA-N 0.000 description 1
- XXMVHLFSEVGZOT-LISXCZPOSA-N CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C)CN3C2=O)=NO1 Chemical compound CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C)CN3C2=O)=NO1 XXMVHLFSEVGZOT-LISXCZPOSA-N 0.000 description 1
- SETBJPVFTDVLQY-ONZCCOAMSA-N CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C4=CC5=C(C=CC=C5)O4)CN3C2=O)=NO1 Chemical compound CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C4=CC5=C(C=CC=C5)O4)CN3C2=O)=NO1 SETBJPVFTDVLQY-ONZCCOAMSA-N 0.000 description 1
- DXKGOOJAYKNBLA-WMCXDDCWSA-N CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C4=CC=CC=C4)CN3C2=O)=NO1 Chemical compound CC1=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C4=CC=CC=C4)CN3C2=O)=NO1 DXKGOOJAYKNBLA-WMCXDDCWSA-N 0.000 description 1
- WKVLOZUJSFWUAS-HBLZMGMFSA-N CC1=CC(C(=O)N[C@H]2CCCCC/C=C\[C@@H]3C[C@@]3(C(=O)O)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCC/C=C\[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CN(C)C(OC(C)(C)C)OC(C)(C)C Chemical compound CC1=CC(C(=O)N[C@H]2CCCCC/C=C\[C@@H]3C[C@@]3(C(=O)O)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCC/C=C\[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CN(C)C(OC(C)(C)C)OC(C)(C)C WKVLOZUJSFWUAS-HBLZMGMFSA-N 0.000 description 1
- AIFZFAUXONPKQS-WCSBRSRUSA-N CC1=CC(C(=O)N[C@H]2CCCCC/C=C\[C@@H]3C[C@@]3(C(=O)O)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)C3=NOC(C)=C3)CCCCC/C=C\[C@@H]1C2.[Li+].[OH-] Chemical compound CC1=CC(C(=O)N[C@H]2CCCCC/C=C\[C@@H]3C[C@@]3(C(=O)O)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)C3=NOC(C)=C3)CCCCC/C=C\[C@@H]1C2.[Li+].[OH-] AIFZFAUXONPKQS-WCSBRSRUSA-N 0.000 description 1
- SRNOQGDGCLRGMH-CMOBXNIXSA-N CC1=CC(C(=O)N[C@H]2CCCCCC(=O)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.FS(F)(F)N1CCOCC1 Chemical compound CC1=CC(C(=O)N[C@H]2CCCCCC(=O)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.FS(F)(F)N1CCOCC1 SRNOQGDGCLRGMH-CMOBXNIXSA-N 0.000 description 1
- TUHQXVJNJWYYEK-WUYKZKIWSA-N CC1=CC(C(=O)N[C@H]2CCCCCC(=O)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCCC(O)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CCN(CC)CC.CS(C)=O.O=C(Cl)C(=O)Cl Chemical compound CC1=CC(C(=O)N[C@H]2CCCCCC(=O)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCCC(O)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CCN(CC)CC.CS(C)=O.O=C(Cl)C(=O)Cl TUHQXVJNJWYYEK-WUYKZKIWSA-N 0.000 description 1
- UBTMMNDYVUDHNS-OIPONERXSA-N CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)C3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4/C4=N/C5=C(C=CC=C5)S4)CN3C2=O)=NO1.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O Chemical compound CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)C3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4/C4=N/C5=C(C=CC=C5)S4)CN3C2=O)=NO1.Cl.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=N/C4=C(C=CC=C4)S3)CN2C1=O UBTMMNDYVUDHNS-OIPONERXSA-N 0.000 description 1
- DJKFARKHEDCCIU-NFRYGHHESA-N CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NN1C Chemical compound CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NN1C DJKFARKHEDCCIU-NFRYGHHESA-N 0.000 description 1
- DTVHLAHBOUDABB-FHNRMANUSA-N CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NN1C.CC1=CC(C(=O)O)=NN1C.CCN(C(C)C)C(C)C.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[P-](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O Chemical compound CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NN1C.CC1=CC(C(=O)O)=NN1C.CCN(C(C)C)C(C)C.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[P-](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O DTVHLAHBOUDABB-FHNRMANUSA-N 0.000 description 1
- QIKMMRCUOLLFAS-PIQFXFIDSA-N CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)O)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.Cl Chemical compound CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)O)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.CC1=CC(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)OC(C)(C)C)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)=NO1.Cl QIKMMRCUOLLFAS-PIQFXFIDSA-N 0.000 description 1
- YKYQCPXNWCJTKI-MCCZRPCWSA-N CC1=CC(C(=O)O)=NO1.CCN(C(C)C)C(C)C.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](N)CCCCC/C=C\[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)C3=NOC(C)=C3)CCCCC/C=C\[C@@H]1C2.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[P-](F)(F)(F)(F)F Chemical compound CC1=CC(C(=O)O)=NO1.CCN(C(C)C)C(C)C.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](N)CCCCC/C=C\[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)C3=NOC(C)=C3)CCCCC/C=C\[C@@H]1C2.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[P-](F)(F)(F)(F)F YKYQCPXNWCJTKI-MCCZRPCWSA-N 0.000 description 1
- DJCKWSQZHGJZJK-VTVLMTLPSA-N CC1=CC(CC[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4)CN3C2=O)=NO1 Chemical compound CC1=CC(CC[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4)CN3C2=O)=NO1 DJCKWSQZHGJZJK-VTVLMTLPSA-N 0.000 description 1
- ATKJQRROPAJVAB-NFRYGHHESA-N CC1=CN=C(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)C=N1 Chemical compound CC1=CN=C(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)C=N1 ATKJQRROPAJVAB-NFRYGHHESA-N 0.000 description 1
- UTMLCVJQYUAJTJ-FHNRMANUSA-N CC1=CN=C(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)C=N1.CC1=CN=C(C(=O)O)C=N1.CCN(C(C)C)C(C)C.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[P-](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O Chemical compound CC1=CN=C(C(=O)N[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C2=O)C=N1.CC1=CN=C(C(=O)O)C=N1.CCN(C(C)C)C(C)C.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.F[P-](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=CC=CC=C4C4=C3C=CC=C4)CN2C1=O UTMLCVJQYUAJTJ-FHNRMANUSA-N 0.000 description 1
- WUMBMXVXRBIXSX-XZFGXIHVSA-N CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1 Chemical compound CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1 WUMBMXVXRBIXSX-XZFGXIHVSA-N 0.000 description 1
- VAJRRRJJHLYORI-HPPVEERWSA-N CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1.CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.Cl.Cl Chemical compound CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1.CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.Cl.Cl VAJRRRJJHLYORI-HPPVEERWSA-N 0.000 description 1
- AKPJCMBTIQQZPP-WRRKBBOPSA-M CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)O)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[Li]O Chemical compound CC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)O)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4C)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[Li]O AKPJCMBTIQQZPP-WRRKBBOPSA-M 0.000 description 1
- AHYJYLSXXQYKCE-PUCYAHNDSA-N CCC(CC(C)=O)C(=O)O[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(N)=O.P.P Chemical compound CCC(CC(C)=O)C(=O)O[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(N)=O.P.P AHYJYLSXXQYKCE-PUCYAHNDSA-N 0.000 description 1
- IBDJTGUHIKXJGN-HXGLBWRBSA-N CCC1=C(O[C@@H]2C[C@H]3C(=O)N[C@]4(C(=O)NS(=O)(=O)C5(C)CC5)C[C@H]4CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C2)/N=C2/C=CC=C/C2=N\1.CCC1=C(O[C@@H]2C[C@H]3C(=O)N[C@]4(C(=O)O)C[C@H]4CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C2)/N=C2/C=CC=C/C2=N\1 Chemical compound CCC1=C(O[C@@H]2C[C@H]3C(=O)N[C@]4(C(=O)NS(=O)(=O)C5(C)CC5)C[C@H]4CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C2)/N=C2/C=CC=C/C2=N\1.CCC1=C(O[C@@H]2C[C@H]3C(=O)N[C@]4(C(=O)O)C[C@H]4CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C2)/N=C2/C=CC=C/C2=N\1 IBDJTGUHIKXJGN-HXGLBWRBSA-N 0.000 description 1
- ATWZSFABWSFIDW-PIWNTJIRSA-N CCC1=C(O[C@@H]2C[C@H]3C(=O)N[C@]4(C(=O)NS(=O)(=O)C5CC5)C[C@H]4CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C2)/N=C2/C=CC=C/C2=N\1.CCC1=C(O[C@@H]2C[C@H]3C(=O)N[C@]4(C(=O)O)C[C@H]4CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C2)/N=C2/C=CC=C/C2=N\1 Chemical compound CCC1=C(O[C@@H]2C[C@H]3C(=O)N[C@]4(C(=O)NS(=O)(=O)C5CC5)C[C@H]4CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C2)/N=C2/C=CC=C/C2=N\1.CCC1=C(O[C@@H]2C[C@H]3C(=O)N[C@]4(C(=O)O)C[C@H]4CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C2)/N=C2/C=CC=C/C2=N\1 ATWZSFABWSFIDW-PIWNTJIRSA-N 0.000 description 1
- GYNSXTKNUFVFFB-ASOXUZBCSA-N CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4(C)CC4)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1.CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4(C)CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.Cl.Cl Chemical compound CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4(C)CC4)C[C@H]3CC(F)(F)CCCCC[C@H](CC(=O)OC(C)(C)C)C(=O)N2C1.CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4(C)CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.Cl.Cl GYNSXTKNUFVFFB-ASOXUZBCSA-N 0.000 description 1
- GYZIBDRVASYGNY-OYIXWIFZSA-N CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4(C)CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4(C)CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C3=NOC(C)=C3)C(=O)N2C1.Cl Chemical compound CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4(C)CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4(C)CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C3=NOC(C)=C3)C(=O)N2C1.Cl GYZIBDRVASYGNY-OYIXWIFZSA-N 0.000 description 1
- VDBJJNBPKMCFHS-KJANWCMYSA-N CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4(C)CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C3=NOC(C)=C3)C(=O)N2C1 Chemical compound CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4(C)CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C3=NOC(C)=C3)C(=O)N2C1 VDBJJNBPKMCFHS-KJANWCMYSA-N 0.000 description 1
- MOYIDUYDRRMZFO-RRRDKLQJSA-N CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C3=NOC(C)=C3)C(=O)N2C1.Cl Chemical compound CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C3=NOC(C)=C3)C(=O)N2C1.Cl MOYIDUYDRRMZFO-RRRDKLQJSA-N 0.000 description 1
- WWOQIOOECSYJLP-VFMPNEQRSA-N CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1.Cl.Cl Chemical compound CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](N)C(=O)N2C1.CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1.Cl.Cl WWOQIOOECSYJLP-VFMPNEQRSA-N 0.000 description 1
- UATLPZZAELSDQU-JUXUBQHBSA-N CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C(C)(C)C)C(=O)N2C1 Chemical compound CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C(C)(C)C)C(=O)N2C1 UATLPZZAELSDQU-JUXUBQHBSA-N 0.000 description 1
- YVHMUSIWJVEBNU-JGMLGMLCSA-N CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C3=NOC(C)=C3)C(=O)N2C1 Chemical compound CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C3=NOC(C)=C3)C(=O)N2C1 YVHMUSIWJVEBNU-JGMLGMLCSA-N 0.000 description 1
- GHFYQYKKRVVDDF-NVJQFURWSA-N CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C3=NOC=C3)C(=O)N2C1 Chemical compound CCC1=NC2=C(C=CC=C2)N=C1O[C@@H]1C[C@H]2C(=O)C[C@]3(C(=O)NS(=O)(=O)C4CC4)C[C@H]3CC(F)(F)CCCCC[C@H](NC(=O)C3=NOC=C3)C(=O)N2C1 GHFYQYKKRVVDDF-NVJQFURWSA-N 0.000 description 1
- IFNITRYFMQKSNG-OCAVINHWSA-N CCN(C(C)C)C(C)C.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.CN1C=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4/C4=C/C5=C(C=CC=C5)O4)CN3C2=O)=N1.CN1C=CC(C(=O)O)=N1.F[P-](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O Chemical compound CCN(C(C)C)C(C)C.CN(C)C(ON1N=NC2=C1N=CC=C2)=[N+](C)C.CN1C=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4/C4=C/C5=C(C=CC=C5)O4)CN3C2=O)=N1.CN1C=CC(C(=O)O)=N1.F[P-](F)(F)(F)(F)F.N[C@H]1CCCCCC(F)(F)C[C@@H]2C[C@@]2(C(=O)NS(=O)(=O)C2CC2)CC(=O)[C@@H]2C[C@@H](OC3=NC4=C(C=CC=C4)N=C3/C3=C/C4=C(C=CC=C4)O3)CN2C1=O IFNITRYFMQKSNG-OCAVINHWSA-N 0.000 description 1
- KDFCBPWRKYKRQQ-BQHZEIDVSA-N CCN(CC)CC.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(=O)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(O)C[C@@H]1C2.CS(C)=O.O=C(Cl)C(=O)Cl Chemical compound CCN(CC)CC.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(=O)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(O)C[C@@H]1C2.CS(C)=O.O=C(Cl)C(=O)Cl KDFCBPWRKYKRQQ-BQHZEIDVSA-N 0.000 description 1
- ZUVJVDTYZJIIBY-ZFNKPWALSA-N CCN(CC)CC.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(=O)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCC[C@H](O)C[C@@H]1C2.CS(C)=O.O=C(Cl)C(=O)Cl Chemical compound CCN(CC)CC.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(=O)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCC[C@H](O)C[C@@H]1C2.CS(C)=O.O=C(Cl)C(=O)Cl ZUVJVDTYZJIIBY-ZFNKPWALSA-N 0.000 description 1
- FXJUHBJDKMWTIS-UHFFFAOYSA-N CCOC(=O)C(=O)C1=CC2=C(/C=C\C=C/2)O1.NC1=C(N)C=CC=C1.OC1=C(/C2=C/C3=C(C=CC=C3)O2)N=C2C=CC=CC2=N1 Chemical compound CCOC(=O)C(=O)C1=CC2=C(/C=C\C=C/2)O1.NC1=C(N)C=CC=C1.OC1=C(/C2=C/C3=C(C=CC=C3)O2)N=C2C=CC=CC2=N1 FXJUHBJDKMWTIS-UHFFFAOYSA-N 0.000 description 1
- HBZONQISMCPHPO-UHFFFAOYSA-N CCOC(=O)C(=O)C1=NC2=C(/C=C\C=C/2)S1.NC1=C(N)C=CC=C1.OC1=C(/C2=N/C3=C(C=CC=C3)S2)N=C2C=CC=CC2=N1 Chemical compound CCOC(=O)C(=O)C1=NC2=C(/C=C\C=C/2)S1.NC1=C(N)C=CC=C1.OC1=C(/C2=N/C3=C(C=CC=C3)S2)N=C2C=CC=CC2=N1 HBZONQISMCPHPO-UHFFFAOYSA-N 0.000 description 1
- RQXGAUHIPOMFJY-UHFFFAOYSA-N CCOC(=O)C(=O)C1=NC2=C(C=CC=C2)S1.CCOC(=O)C(=O)Cl.C[Si](C)(C)C1=NC2=C(C=CC=C2)S1 Chemical compound CCOC(=O)C(=O)C1=NC2=C(C=CC=C2)S1.CCOC(=O)C(=O)Cl.C[Si](C)(C)C1=NC2=C(C=CC=C2)S1 RQXGAUHIPOMFJY-UHFFFAOYSA-N 0.000 description 1
- OCMYBXRTJVZBGM-HBBHLGOCSA-N CCOC(=O)[C@]1(C[C@H]1CC(F)(F)CCC=C)NC(=O)[C@@H]1C[C@@H](O)CN1C(=O)[C@H](CC=C)NC(=O)OC(C)(C)C Chemical compound CCOC(=O)[C@]1(C[C@H]1CC(F)(F)CCC=C)NC(=O)[C@@H]1C[C@@H](O)CN1C(=O)[C@H](CC=C)NC(=O)OC(C)(C)C OCMYBXRTJVZBGM-HBBHLGOCSA-N 0.000 description 1
- MDTHQMSTVDFRNE-RGPGPGMBSA-N CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](O)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CC=CCCC(F)(F)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](O)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[HH] Chemical compound CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](O)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CC=CCCC(F)(F)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](O)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.[HH] MDTHQMSTVDFRNE-RGPGPGMBSA-N 0.000 description 1
- NAVXXLKAGPAONS-WCXMYOPHSA-N CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](N)CCCCC/C=C\[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.Cl.Cl Chemical compound CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](N)CCCCC/C=C\[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.Cl.Cl NAVXXLKAGPAONS-WCXMYOPHSA-N 0.000 description 1
- DXBZCQHBZSPQJV-YUNVNBHVSA-M CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OS(=O)(=O)C4=CC=C(Br)C=C4)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.O=C([O-])O.O=C1NC2=C(C=CC=C2)C2=C1/C=C\C=C/2.[Cs+].[Cs+] Chemical compound CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OS(=O)(=O)C4=CC=C(Br)C=C4)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCC/C=C\[C@@H]1C2.O=C([O-])O.O=C1NC2=C(C=CC=C2)C2=C1/C=C\C=C/2.[Cs+].[Cs+] DXBZCQHBZSPQJV-YUNVNBHVSA-M 0.000 description 1
- LWAUNLLCOFRFPC-OMEQPHOJSA-N CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(=O)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.FS(F)(F)N1CCOCC1 Chemical compound CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(=O)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@@H](OC4=NC5=CC=CC=C5C5=C4C=CC=C5)CN3C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.FS(F)(F)N1CCOCC1 LWAUNLLCOFRFPC-OMEQPHOJSA-N 0.000 description 1
- MPTCHINYWLMVHA-VVAGCLHISA-N CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](O)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2 Chemical compound CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](O)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2 MPTCHINYWLMVHA-VVAGCLHISA-N 0.000 description 1
- ULSAUGHTSXZEAT-IIPHRGETSA-N CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(=O)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.FS(F)(F)N1CCOCC1 Chemical compound CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(=O)C[C@@H]1C2.CCOC(=O)[C@]12CC(=O)[C@@H]3C[C@H](OC(=O)C4=CC=CC=C4)CN3C(=O)[C@@H](CC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]1C2.FS(F)(F)N1CCOCC1 ULSAUGHTSXZEAT-IIPHRGETSA-N 0.000 description 1
- 102100027221 CD81 antigen Human genes 0.000 description 1
- SPJMBLCFFHEMOW-UHFFFAOYSA-N CN(C)C=O.ClC1=C(C2=CC3=C(C=CC=C3)O2)N=C2C=CC=CC2=N1.O=S(Cl)Cl.OC1=C(C2=CC3=C(C=CC=C3)O2)N=C2C=CC=CC2=N1 Chemical compound CN(C)C=O.ClC1=C(C2=CC3=C(C=CC=C3)O2)N=C2C=CC=CC2=N1.O=S(Cl)Cl.OC1=C(C2=CC3=C(C=CC=C3)O2)N=C2C=CC=CC2=N1 SPJMBLCFFHEMOW-UHFFFAOYSA-N 0.000 description 1
- LYTUOTYYQNOTDI-UHFFFAOYSA-N CN(C)C=O.ClC1=C(C2=NC3=C(C=CC=C3)S2)N=C2C=CC=CC2=N1.O=S(Cl)Cl.OC1=C(C2=NC3=C(C=CC=C3)S2)N=C2C=CC=CC2=N1 Chemical compound CN(C)C=O.ClC1=C(C2=NC3=C(C=CC=C3)S2)N=C2C=CC=CC2=N1.O=S(Cl)Cl.OC1=C(C2=NC3=C(C=CC=C3)S2)N=C2C=CC=CC2=N1 LYTUOTYYQNOTDI-UHFFFAOYSA-N 0.000 description 1
- XFMUUPHDMDPFBW-ONZCCOAMSA-N CN1C=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4/C4=C/C5=C(C=CC=C5)O4)CN3C2=O)=N1 Chemical compound CN1C=CC(C(=O)C[C@H]2CCCCCC(F)(F)C[C@@H]3C[C@@]3(C(=O)NS(=O)(=O)C3CC3)CC(=O)[C@@H]3C[C@@H](OC4=NC5=C(C=CC=C5)N=C4/C4=C/C5=C(C=CC=C5)O4)CN3C2=O)=N1 XFMUUPHDMDPFBW-ONZCCOAMSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- MTENYGSJPBMBDS-VFLDZSNKSA-N Cc(nc1)cnc1C(N[C@@H](CCCCCC(C[C@H](C1)[C@]1(C(NS(C1CC1)(=O)=O)=O)NC([C@H](C1)N2C[C@@H]1Oc1c(cccc3)c3c(cccc3)c3n1)=O)(F)F)C2=O)=O Chemical compound Cc(nc1)cnc1C(N[C@@H](CCCCCC(C[C@H](C1)[C@]1(C(NS(C1CC1)(=O)=O)=O)NC([C@H](C1)N2C[C@@H]1Oc1c(cccc3)c3c(cccc3)c3n1)=O)(F)F)C2=O)=O MTENYGSJPBMBDS-VFLDZSNKSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 102000001493 Cyclophilins Human genes 0.000 description 1
- 108010068682 Cyclophilins Proteins 0.000 description 1
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- CYCGRDQQIOGCKX-UHFFFAOYSA-N Dehydro-luciferin Natural products OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 1
- 241000710198 Foot-and-mouth disease virus Species 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 229940122604 HCV protease inhibitor Drugs 0.000 description 1
- 229940121759 Helicase inhibitor Drugs 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 101000914479 Homo sapiens CD81 antigen Proteins 0.000 description 1
- 101000600434 Homo sapiens Putative uncharacterized protein encoded by MIR7-3HG Proteins 0.000 description 1
- LCWXJXMHJVIJFK-UHFFFAOYSA-N Hydroxylysine Natural products NCC(O)CC(N)CC(O)=O LCWXJXMHJVIJFK-UHFFFAOYSA-N 0.000 description 1
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 108010025815 Kanamycin Kinase Proteins 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- QAQJMLQRFWZOBN-LAUBAEHRSA-N L-ascorbyl-6-palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](O)[C@H]1OC(=O)C(O)=C1O QAQJMLQRFWZOBN-LAUBAEHRSA-N 0.000 description 1
- 239000011786 L-ascorbyl-6-palmitate Substances 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 1
- FFFHZYDWPBMWHY-VKHMYHEASA-N L-homocysteine Chemical compound OC(=O)[C@@H](N)CCS FFFHZYDWPBMWHY-VKHMYHEASA-N 0.000 description 1
- UKAUYVFTDYCKQA-VKHMYHEASA-N L-homoserine Chemical compound OC(=O)[C@@H](N)CCO UKAUYVFTDYCKQA-VKHMYHEASA-N 0.000 description 1
- UCUNFLYVYCGDHP-BYPYZUCNSA-N L-methionine sulfone Chemical compound CS(=O)(=O)CC[C@H](N)C(O)=O UCUNFLYVYCGDHP-BYPYZUCNSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 102000008109 Mixed Function Oxygenases Human genes 0.000 description 1
- 108010074633 Mixed Function Oxygenases Proteins 0.000 description 1
- JDHILDINMRGULE-LURJTMIESA-N N(pros)-methyl-L-histidine Chemical compound CN1C=NC=C1C[C@H](N)C(O)=O JDHILDINMRGULE-LURJTMIESA-N 0.000 description 1
- FFDGPVCHZBVARC-UHFFFAOYSA-N N,N-dimethylglycine Chemical class CN(C)CC(O)=O FFDGPVCHZBVARC-UHFFFAOYSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- OBHBKFCOGMYRMU-IQDCAMADSA-N N[C@@H](CCCCCC([C@H](C1)C1(C1)[C@]1(C(NS(C1CC1)(=O)=O)=O)NC([C@H](C1)N2C[C@@H]1Oc1cnc(cccc3)c3n1)=O)(F)F)C2=O Chemical compound N[C@@H](CCCCCC([C@H](C1)C1(C1)[C@]1(C(NS(C1CC1)(=O)=O)=O)NC([C@H](C1)N2C[C@@H]1Oc1cnc(cccc3)c3n1)=O)(F)F)C2=O OBHBKFCOGMYRMU-IQDCAMADSA-N 0.000 description 1
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 101800001020 Non-structural protein 4A Proteins 0.000 description 1
- 240000007817 Olea europaea Species 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 208000037581 Persistent Infection Diseases 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical group OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 description 1
- 102100037401 Putative uncharacterized protein encoded by MIR7-3HG Human genes 0.000 description 1
- 238000002123 RNA extraction Methods 0.000 description 1
- 238000011529 RT qPCR Methods 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- 229910006074 SO2NH2 Inorganic materials 0.000 description 1
- 229910007161 Si(CH3)3 Inorganic materials 0.000 description 1
- 241000862969 Stella Species 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 150000007933 aliphatic carboxylic acids Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000010385 ascorbyl palmitate Nutrition 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- XYOVOXDWRFGKEX-UHFFFAOYSA-N azepine Chemical compound N1C=CC=CC=C1 XYOVOXDWRFGKEX-UHFFFAOYSA-N 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 235000012216 bentonite Nutrition 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000004622 benzoxazinyl group Chemical group O1NC(=CC2=C1C=CC=C2)* 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 229940000635 beta-alanine Drugs 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 238000010256 biochemical assay Methods 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 150000004648 butanoic acid derivatives Chemical class 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 229960002173 citrulline Drugs 0.000 description 1
- 235000013477 citrulline Nutrition 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 238000007398 colorimetric assay Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000058 cyclopentadienyl group Chemical group C1(=CC=CC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 125000005508 decahydronaphthalenyl group Chemical group 0.000 description 1
- 125000004855 decalinyl group Chemical group C1(CCCC2CCCCC12)* 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- YSMODUONRAFBET-UHFFFAOYSA-N delta-DL-hydroxylysine Natural products NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 125000002576 diazepinyl group Chemical group N1N=C(C=CC=C1)* 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- WYACBZDAHNBPPB-UHFFFAOYSA-N diethyl oxalate Chemical compound CCOC(=O)C(=O)OCC WYACBZDAHNBPPB-UHFFFAOYSA-N 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- NLEBIOOXCVAHBD-QKMCSOCLSA-N dodecyl beta-D-maltoside Chemical compound O[C@@H]1[C@@H](O)[C@H](OCCCCCCCCCCCC)O[C@H](CO)[C@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 NLEBIOOXCVAHBD-QKMCSOCLSA-N 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 229940047652 ear drops Drugs 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- ZSWFCLXCOIISFI-UHFFFAOYSA-N endo-cyclopentadiene Natural products C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- YSMODUONRAFBET-UHNVWZDZSA-N erythro-5-hydroxy-L-lysine Chemical compound NC[C@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-UHNVWZDZSA-N 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- NNRLZTHBTVXDSL-WYUVZMMLSA-N ethyl (1R,2S)-1-amino-2-(2,2-difluorohex-5-enyl)cyclopropane-1-carboxylate hydrochloride Chemical compound Cl.CCOC(=O)[C@@]1(N)C[C@H]1CC(F)(F)CCC=C NNRLZTHBTVXDSL-WYUVZMMLSA-N 0.000 description 1
- AHXXLGSGZRARGS-DVHILSRVSA-N ethyl (1S,4R,6R,7R,14S,18R)-7-hydroxy-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-18-phenanthridin-6-yloxy-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxylate Chemical compound O[C@@H]1CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@H](OC=3C4=CC=CC=C4C4=CC=CC=C4N=3)C[C@H]2C(=O)N[C@]2(C(=O)OCC)C[C@H]21 AHXXLGSGZRARGS-DVHILSRVSA-N 0.000 description 1
- VDSJKGLWVSAEGH-ISGWBCADSA-N ethyl (1S,4R,6S,14S,18R)-8,8-difluoro-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-18-(3-phenylquinoxalin-2-yl)oxy-3,16-diazatricyclo[14.3.0.04,6]nonadecane-4-carboxylate Chemical compound O([C@H]1CN2C(=O)[C@@H](NC(=O)OC(C)(C)C)CCCCCC(F)(F)C[C@@H]3C[C@]3(NC(=O)[C@@H]2C1)C(=O)OCC)C1=NC2=CC=CC=C2N=C1C1=CC=CC=C1 VDSJKGLWVSAEGH-ISGWBCADSA-N 0.000 description 1
- UBIGRFSDNDAYMW-KZULUSFZSA-N ethyl (1r,2s)-1-[bis[(2-methylpropan-2-yl)oxycarbonyl]amino]-2-(2-oxoethyl)cyclopropane-1-carboxylate Chemical compound CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)[C@]1(C(=O)OCC)C[C@H]1CC=O UBIGRFSDNDAYMW-KZULUSFZSA-N 0.000 description 1
- MUWAMLYKLZSGPE-NOZJJQNGSA-N ethyl (1r,2s)-2-ethenyl-1-[(2-methylpropan-2-yl)oxycarbonylamino]cyclopropane-1-carboxylate Chemical compound CC(C)(C)OC(=O)N[C@]1(C(=O)OCC)C[C@H]1C=C MUWAMLYKLZSGPE-NOZJJQNGSA-N 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000001125 extrusion Methods 0.000 description 1
- 239000003885 eye ointment Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 125000001183 hydrocarbyl group Chemical group 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- QJHBJHUKURJDLG-UHFFFAOYSA-N hydroxy-L-lysine Natural products NCCCCC(NO)C(O)=O QJHBJHUKURJDLG-UHFFFAOYSA-N 0.000 description 1
- 229960002591 hydroxyproline Drugs 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- 125000005969 isothiazolinyl group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- JAUWOQLHLFMTON-UHFFFAOYSA-M magnesium;but-1-ene;bromide Chemical compound [Mg+2].[Br-].[CH2-]CC=C JAUWOQLHLFMTON-UHFFFAOYSA-M 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- ZORHSASAYVIBLY-UHNVWZDZSA-N methyl (2s,4r)-4-hydroxypyrrolidine-2-carboxylate Chemical compound COC(=O)[C@@H]1C[C@@H](O)CN1 ZORHSASAYVIBLY-UHNVWZDZSA-N 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- TYKALBWEPNHUGZ-UHFFFAOYSA-N methylsulfonylcyclopropane Chemical compound CS(=O)(=O)C1CC1 TYKALBWEPNHUGZ-UHFFFAOYSA-N 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- DBNQIOANXZVWIP-UHFFFAOYSA-N n,n-dimethyl-1,1-bis[(2-methylpropan-2-yl)oxy]methanamine Chemical compound CC(C)(C)OC(N(C)C)OC(C)(C)C DBNQIOANXZVWIP-UHFFFAOYSA-N 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- RPJPZDVUUKWPGT-FOIHOXPVSA-N nim811 Chemical compound CC[C@H](C)[C@@H]1N(C)C(=O)CN(C)C(=O)[C@H](CC)NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC1=O RPJPZDVUUKWPGT-FOIHOXPVSA-N 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 125000004365 octenyl group Chemical group C(=CCCCCCC)* 0.000 description 1
- 125000005069 octynyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C#C* 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 229940041678 oral spray Drugs 0.000 description 1
- 239000000668 oral spray Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 125000003585 oxepinyl group Chemical group 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 239000008180 pharmaceutical surfactant Substances 0.000 description 1
- 125000001828 phenalenyl group Chemical group C1(C=CC2=CC=CC3=CC=CC1=C23)* 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 125000005541 phosphonamide group Chemical group 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920001184 polypeptide Chemical group 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229950008882 polysorbate Drugs 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 229960004063 propylene glycol Drugs 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- FGHMGRXAHIXTBM-TWFJNEQDSA-N s-[2-[[(2r,3r,4r,5r)-5-(2-amino-6-oxo-3h-purin-9-yl)-3,4-dihydroxy-4-methyloxolan-2-yl]methoxy-(benzylamino)phosphoryl]oxyethyl] 3-hydroxy-2,2-dimethylpropanethioate Chemical compound C([C@@H]1[C@H]([C@@](C)(O)[C@H](N2C3=C(C(NC(N)=N3)=O)N=C2)O1)O)OP(=O)(OCCSC(=O)C(C)(CO)C)NCC1=CC=CC=C1 FGHMGRXAHIXTBM-TWFJNEQDSA-N 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- ABZLKHKQJHEPAX-UHFFFAOYSA-N tetramethylrhodamine Chemical compound C=12C=CC(N(C)C)=CC2=[O+]C2=CC(N(C)C)=CC=C2C=1C1=CC=CC=C1C([O-])=O ABZLKHKQJHEPAX-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000003777 thiepinyl group Chemical group 0.000 description 1
- 125000005032 thiofuranyl group Chemical group S1C(=CC=C1)* 0.000 description 1
- 125000000464 thioxo group Chemical group S=* 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000006168 tricyclic group Chemical group 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical class CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 235000019871 vegetable fat Nutrition 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 235000014692 zinc oxide Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0827—Tripeptides containing heteroatoms different from O, S, or N
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- the present invention relates to novel fluorinated macrocycles having activity against the hepatitis C virus (HCV) and useful in the treatment of HCV infections. More particularly, the invention relates to macrocyclic compounds, compositions containing such compounds and methods for using the same, as well as processes for making such compounds.
- HCV hepatitis C virus
- HCV is the principal cause of non-A, non-B hepatitis and is an increasingly severe public health problem both in the developed and developing world. It is estimated that the virus infects over 200 million people worldwide, surpassing the number of individuals infected with the human immunodeficiency virus (HIV) by nearly five fold. HCV infected patients, due to the high percentage of individuals inflicted with chronic infections, are at an elevated risk of developing cirrhosis of the liver, subsequent hepatocellular carcinoma and terminal liver disease. HCV is the most prevalent cause of hepatocellular cancer and cause of patients requiring liver transplantations in the western world.
- HIV human immunodeficiency virus
- anti-HCV therapeutics There are considerable barriers to the development of anti-HCV therapeutics, which include, but are not limited to, the persistence of the virus, the genetic diversity of the virus during replication in the host, the high incident rate of the virus developing drug-resistant mutants, and the lack of reproducible infectious culture systems and small-animal models for HCV replication and pathogenesis. In a majority of cases, given the mild course of the infection and the complex biology of the liver, careful consideration must be given to antiviral drugs, which often have significant side effects.
- the present invention relates to novel macrocyclic compounds and methods of treating a hepatitis C infection in a subject in need of such therapy with said macrocyclic compounds.
- the compounds of the present invention interfere with the life cycle of the hepatitis C virus and are useful as antiviral agents.
- the present invention further relates to pharmaceutical compositions comprising the compounds of the present invention, or pharmaceutically acceptable salts, esters or prodrugs thereof, in combination with a pharmaceutically acceptable carrier or excipient.
- the invention provides a compound of formula I:
- X is an optionally substituted carbocyclic, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl;
- J is —C(O)—, —O—C(O)—, —C(O)O—, —N(R 3 )—C(O)—, —C(S)—, —C( ⁇ NR 4 )—, —S(O)—, or —S(O 2 )— or absent;
- A is selected from the group consisting of the following:
- E is -G-R 5 ;
- each of R a , R b , R c , and R d are each independently H or halo, wherein at least two of R a , R b , R c , and R d are halo; and R b and R d are absent if is a double bond
- each R 3 and R 4 is independently selected at each occurrence from the following: H, optionally substituted alkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; and optionally substituted carbocyclic;
- L is absent or a C 2 -C 5 saturated or unsaturated chain, optionally containing one to three heteroatoms independently selected from O, N and S(O) q , wherein L is optionally substituted with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl and C 2 -C 6 alkynyl, wherein each C 1 -C 6 alkyl, C 2 -C 6 alkenyl and C 2 -C 6 alkynyl, group is optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, formyl and cyano;
- j independently 0, 1, 2, 3, or 4;
- k independently 0, 1, 2, or 3;
- each m is independently 0, 1, or 2;
- q is independently 0, 1, or 2.
- the present invention also features the compounds of Formula I′, and pharmaceutically acceptable salts, esters, hydrates, solvates, polyrnorphs, or prodrugs thereof,
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of any of the formulae herein (e.g., Formula I or I′), or a pharmaceutically acceptable salt, ester, or prodrug thereof, in combination with an excipient.
- the invention provides a method of treating a viral infection in a subject, comprising administering to the subject a therapeutically effective amount of a compound of any of the formulae herein (e.g., Formula I or I′, or a pharmaceutically acceptable salt, ester or prodrug thereof; or a pharmaceutical composition comprising the same.
- a compound of any of the formulae herein e.g., Formula I or I′, or a pharmaceutically acceptable salt, ester or prodrug thereof; or a pharmaceutical composition comprising the same.
- the invention provides a compound of formula I:
- X is an optionally substituted carbocyclic, optionally substituted heterocycyl, optionally substituted aryl, or optionally substituted heteroaryl;
- J is —C(O)—, —O—C(O)—, —C(O)O—, —N(R 3 )—C(O)—, —C(S)—, —C( ⁇ NR 4 )—, N(R 3 )—, —S(O)—, or —S(O 2 )— or absent;
- A is selected from the group consisting of the following:
- E is -G-R 5 ;
- each of R a , R b , R c , and R d are each independently H or halo, wherein at least two of R a , R b , R c , and R d are halo; and R b and R d are absent if is a double bond
- each R 3 and R 4 is independently selected at each occurrence from the following: H, optionally substituted alkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; and optionally substituted carbocyclic;
- L is absent or a C 2 -C 5 saturated or unsaturated chain, optionally containing one to three heteroatoms independently selected from O, N and S(O) q , wherein L is optionally substituted with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl and C 2 -C 6 alkynyl, wherein each C 1 -C 6 alkyl, C 2 -C 6 alkenyl and C 2 -C 6 alkynyl, group is optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, formyl and cyano;
- j independently 0, 1, 2, 3, or 4;
- k independently 0, 1, 2, or 3;
- each m is independently 0, 1, or 2;
- q is independently 0, 1, or 2.
- the present invention also features the compounds of Formula I′, and pharmaceutically acceptable salts, esters, hydrates, solvates, polymorphs, or prodrugs thereof,
- Z is O or OC(O); and ring X is cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, adamantyl, cyclooctyl, [3.3.0]bicyclooctanyl, [4.3.0]bicyclononanyl, [4.4.0]bicyclodecanyl, [2.2.2]bicyclooctanyl, fluorenyl, phenyl, naphthyl, indanyl, tetrahydronaphthyl, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetraz benzisoxazolyl, benzisothiazolyl, benzimidazol
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl.
- ring X is
- Y is N or —C(R′′)—
- R′ is H, alkyl, alkenyl, alkynyl, carbocyclic, heterocyclyl, aryl, or heteroaryl, each of which may be optionally substituted;
- each n is independently 0, 1, 2, 3, or 4;
- each R 3 and R 4 are independently selected at each occurrence from the following: optionally substituted alkyl., optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen; and
- the invention provides a compound of formula I-A:
- J is —C(O)—, —O—C(O)—, N(R 3 )—C(O)—, —C(S)—, —C( ⁇ NR 4 )—, —N(R 3 )—, —S(O)—, or —S(O 2 )— or absent;
- A is optionally substituted alkyl, optionally substituted heteroaryl, optionally substituted aryl, optionally substituted heterocyclic, or optionally substituted carbocyclic;
- Y is N or —C(R′′)—
- each R 1 is independently selected from H, halogen, hydroxy, amino, —CN, —CF 3 , —N 3 , —NO 2 , —OR 4 , —SR 4 ., —SOR 4 , —SO 2 R 4 , —N(R 3 )S(O) 2 —R 4 , —N(R 3 )(SO 2 )NR 3 R 4 , —NR 3 R 4 , —C(O)—OR 4 , —C(O)R 4 , —C(O)NR 3 R 4 , —N(R 3 )C(O)R 4 ; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl, or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N
- each p is independently 0, 1, or 2;
- R 5 is FE, optionally substituted alkyl, optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl; each of R 2a , R 2b , R a , and R 2d are each independently H or F, wherein at least two of R 2a , R 2b , R 2c , and R 2d are F;
- each R 3 and R 4 are independently selected at each occurrence from the following: optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen;
- L is absent or is selected from optionally substituted alkylene, optionally substituted alkenylene or optionally substituted alkynylene, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N;
- j independently 0, 1, 2, 3, or 4;
- k independently 0, 1, 2, or 3;
- n is independently 0, 1, 2, 3, or 4.
- Y is —C(R′′)—; and R′ and R′′, taken together with the atoms to which each is attached, form a ring.
- the ring formed by R′ and R′′ is an optionally substituted an optionally substituted heteroaryl, an optionally substituted carbocyclic or an optionally substituted heterocyclic.
- Y is N and R′ is H, optionally substituted alkyl, optionally substituted aryl, or optionally substituted heteroaryl.
- G is —N(R 3 )S(O) p —; —N(R 3 )C(O)—, —OS(O) p —, or —C(O)S(O) p —; and p is 2.
- R 5 is optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl. In certain embodiments, R 5 is carbocyclic.
- R 2a and R 2h are F. In other embodiments, R 2c and R 2d are F. In still other embodiments, R 2a and R 2c are F.
- J is —C(O)—, —O—C(O)—, —S(O)—, or —S(O 2 )—.
- A is an optionally substituted alkyl, optionally substituted heteroaryl, or optionally substituted aryl. In certain embodiments, A is heteroaryl or alkyl.
- the invention provides a compound of formula I-A, or a pharmaceutically acceptable salt, ester or prodrug thereof,
- R′ and R′′ taken together with the atoms to which each is attached, form a phenyl which is optionally substituted with one or more R 1 ;
- J is —C(O)—, —O—C(O)—, N(R 3 )—C(O)—, —S(O)—, or —S(O 2 )—;
- A is optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted heteroaryl, optionally substituted aryl, optionally substituted heterocyclic, or optionally substituted carbocyclic;
- each R 1 is independently selected from H, halogen, hydroxy, amino, —CN, —CF 3 , —N 3 , —NO 2 , —OR 4 , —SR 4 , —NR 3 R 4 , optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; optionally substituted haloalkyl, or optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl;
- G is —N(R 3 )S(O) p —;
- p 0, 1, or 2;
- each of R 2a , R 2b , R 2c and R 2d are each independently H or F, wherein at least two of R 2a , R 2b , R 2c , and R 2d are F;
- R 5 is optionally substituted carbocyclic or heterocyclic
- each R 3 and R 4 is independently selected at each occurrence from the following: optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen; and
- n 0, 1, 2, 3, or 4.
- J is —C(O)—, or —O—C(O)—.
- A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl, phenyl, pyridyl, pyrimidyl, pyrazinyl, triazinyl, furanyl, thiophenyl, pyrrolyl, isoxazolyl, or pyrazolyl; each of which may be optionally substituted.
- G is —N(R 3 )S(O) p — and R 5 is cyclopropyl, cyclobutyl, cyclopentyl, cyciohexyl, or cycloheptyl, each of which is optionally substituted.
- R 2a and R 2b are F. In another embodiment, R 2a and R 2c are F. In another embodiment, R 2c and R 2d are F.
- A-J- is A-C(O)— or A-O—C(O)—;
- A is C r C 8 alkyl, C 2 -C 8 alkenyl or C 2 -C 8 alkynyl, each of which is optionally substituted with one or more R H , or
- A is 5- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more R G ;
- -G-R 5 is —N(R 3 )S(O) 2 —R 5 ; and
- R 5 is 3- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more R G .
- Each R H is independently selected at each occurrence from independently selected at each occurrence from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, or cyano; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R H is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkeny
- Each R 1 is independently selected at each occurrence from hydrogen or R G .
- Each R G is independently selected at each occurrence from halo, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C 3 -C 6 carbocycle or 3- to 6-membered heterocycle, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkyn
- Each R 3 is independently selected at each occurrence from hydrogen; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R 3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl., C 2 -C 6 alkynyl, C 1 -C 6 haloalkyl, C 2 -C 6 haloal
- A-J- is A-C(O)— or A-O—C(O)—;
- A is 5- to 6-membered aryl or heteroaryl which is optionally substituted with one or more R G ;
- -G-R 5 is —N(R 3 )S(O) 2 —R 5 (preferably —N(H)S(O) 2 —R 5 ); and
- R 5 is 3- to 6-membered cycloalkyl (preferably cyclopropyl) which is optionally substituted with one or more R G .
- Each R 1 is independently selected at each occurrence from hydrogen or R G .
- R G is as defined above.
- Each R 3 is independently selected at each occurrence from hydrogen; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R 3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 haloalkyl, C 2 -C 6 haloalken
- the invention provides a compound of formula I-A, or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein:
- Y is —C(R′′)—
- R′ and R′′ taken together with the atoms to which each is attached, form a phenyl which is optionally substituted with one or more R 1 ;
- J is —C(O)— or —O—C(O)—;
- A is optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted heteroaryl, optionally substituted aryl, optionally substituted heterocyclic, or optionally substituted carbocyclic;
- each R 1 is independently selected from H, halogen, hydroxy, amino, —CN, —CF 3 , —N 3 , —NO 2 , —OR 4 , —SR 4 , —NR 3 R 4 , optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; optionally substituted haloalkyl, or optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl;
- G is —N(R 3 )S(O) p —;
- p 0, 1., or 2;
- R 5 is optionally substituted carbocyclic or heterocyclic
- R 2a and R 2b are F, and R 2 and R 2d are H;
- each R 3 and R 4 is independently selected at each occurrence from the following: optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen; and
- n 0, I, 2, 3, or 4.
- A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl,
- R 5 is optionally substituted cyclopropyl.
- n 0.
- A-J- is A-C(O)— or A-O—C(O)—;
- A is C 1 -C 8 alkyl, C 2 -C 8 alkenyl or C 2 -C 8 alkynyl, each of which is optionally substituted with one or more R H , or
- A is 5- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more R G ;
- -G-R 5 is —N(R 3 )S(O) 2 —R 5 ; and
- R 5 is 3- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more R G .
- Each R H is independently selected at each occurrence from independently selected at each occurrence from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, or cyano; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R H is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkeny
- Each R 1 is independently selected at each occurrence from hydrogen or R G .
- Each R G is independently selected at each occurrence from halo, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C 3 -C 6 carbocycle or 3- to 6-membered heterocycle, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkyn
- Each R 3 is independently selected at each occurrence from hydrogen; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-meinbered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R 3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 haloalkyl, C 2 -C 6 haloal
- A-J- is A-C(O)— or A-O—C(O)—;
- A is 5- to 6-membered aryl or heteroaryl which is optionally substituted with one or more R G ;
- -G-R 5 is —N(R 3 )S(O) 2 —R 5 (preferably —N(H)S(O) 2 —R 5 ); and
- R 5 is 3- to 6-membered cycloalkyl (preferably cyclopropyl) which is optionally substituted with one or more R G .
- Each R 1 is independently selected at each occurrence from hydrogen or R G .
- R G is as defined above.
- Each R 3 is independently selected at each occurrence from hydrogen; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R 3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 haloalkyl, C 2 -C 6 haloalken
- the invention provides a compound of formula II:
- J is —C(O)—, —O—C(O)—, —N(R 3 )—C(O)—, —S(O)—, or —S(O 2 )—;
- A is optionally substituted alkyl, or optionally substituted heteroaryl, optionally substituted aryl, optionally substituted heterocyclic, or optionally substituted carbocyclic;
- R′ is H, optionally substituted aryl; optionally substituted heteroaryl; or optionally substituted alkyl;
- G is —N(R 3 )S(O) p —;
- p 0, 1, or 2;
- each of R 2a , R 2b , R 2c , and R 2d are each independently H or F, wherein at least two of R 2a , R 2b , R 2c , and R 2d are F;
- R 5 is optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl;
- each R 3 and R 4 is independently selected at each occurrence from the following: optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen.
- R′ is H, optionally substituted alkyl; optionally substituted aryl; or optionally substituted heteroaryl.
- J is —C(O)—, or —O—C(O),
- A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl, phenyl, pyridyl, pyrimidyl, pyrazinyl, triazinyl, furanyl, thiophenyl, pyrrolyl, isoxazolyl, or pyrazolyl; each of which may be optionally substituted.
- G is —N(R 3 )S(O) p — and R 5 is cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, each of which is optionally substituted.
- R 2a and R 2b are F. In another embodiment, R 2a and R 2c , are F. In another embodiment, R 2c and R 2d are F.
- A-J- is A-C(O)— or A-O—C(O)—;
- A is C 1 -C 8 alkyl, C 2 -C 8 alkenyl or C 2 -C 8 alkynyl, each of which is optionally substituted with one or more R H , or
- A is 5- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more R G ;
- -G-R 5 is —N(R 3 )S(O) 2 —R 5 ; and
- R 5 is 3- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more R G .
- Each R H is independently selected at each occurrence from independently selected at each occurrence from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, or cyano; or C 1 -C 6 alkyl, C 1 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R FI is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alken
- Each R 1 is independently selected at each occurrence from hydrogen or R G .
- Each R G is independently selected at each occurrence from halo, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C 3 -C 6 carbocycle or 3- to 6-membered heterocycle, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkyn
- Each R 3 is independently selected at each occurrence from hydrogen; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R 3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 haloalkyl, C 2 -C 6 haloalkenyl or C 2 -C 6
- A-J- is A-C(O)— or A-O—C(O)—;
- A is 5- to 6-membered aryl or heteroaryl which is optionally substituted with one or more R G ;
- -G-R 5 is —N(R 3 )S(O) 2 —R 5 (preferably —N(H)S(O) 2 —R 5 ); and
- R 5 is 3- to 6-membered cycloalkyl (preferably cyclopropyl) which is optionally substituted with one or more R G .
- Each R 1 is independently selected at each occurrence from hydrogen or R G .
- R G is as defined above.
- Each R 3 is independently selected at each occurrence from hydrogen; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R 3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 haloalkyl, C 2 -C 6 haloalken
- the invention provides a compound of formula II-a:
- J is —C(O)— or —O—C(O)—;
- A is optionally substituted alkyl, or optionally substituted heteroaryl, optionally substituted aryl, optionally substituted heterocyclic, or optionally substituted carbocyclic;
- R′ is H, optionally substituted alkyl; optionally substituted aryl; or optionally substituted heteroaryl;
- G is —N(R 3 )S(O) p —;
- p 0, 1, or 2;
- R 5 is optionally substituted carbocyclic
- each R 3 is optionally substituted alkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen.
- R′ is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, phenyl, pyridyl, pyrimidyl, pyrazinyl, triazinyl, furanyl, thiophenyl, pyrrolyl, isoxazolyl, or pyrazolyl; each of which may be optionally substituted.
- R′ is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl,
- A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl, phenyl, pyridyl, pyrimidyl, pyrazinyl, triazinyl, furanyl, thiophenyl, pyrrolyl, isoxazolyl, or pyrazolyl; each of which may be optionally substituted.
- A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl,
- (1 is —N(R 3 )S(O) p — and R 5 is cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, each of which is optionally substituted.
- R 5 is optionally substituted cyclopropyl.
- A-J- is A-C(O)— or A-O—C(O)—;
- A is C 1 -C 8 alkyl, C 2 -C 8 alkenyl or C 2 -C 8 alkynyl, each of which is optionally substituted with one or more R H , or
- A is 5- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more R G ;
- -G-R 5 is —N(R 3 )S(O) 2 —R 5 ; and
- R 5 is 3- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more R G .
- Each R H is independently selected at each occurrence from independently selected at each occurrence from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, or cyano; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R H is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2
- Each R 1 is independently selected at each occurrence from hydrogen or R G .
- Each R G is independently selected at each occurrence from halo, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C 3 -C 6 carbocycle or 3- to 6-membered heterocycle, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkyn
- Each R 3 is independently selected at each occurrence from hydrogen; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R 3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 haloalkyl, C 2 -C 6 haloalken
- A is 5- to 6-membered aryl or heteroaryl which is optionally substituted with one or more R G ;
- -G-R 5 is —N(R 3 )S(O) 2 —R 5 (preferably —N(H)S(O) 2 —R 5 ); and
- R 5 is 3- to 6-membered cycloalkyl (preferably cyclopropyl) which is optionally substituted with one or more R G .
- Each R 1 is independently selected at each occurrence from hydrogen or R G .
- R G is as defined above.
- Each R 3 is independently selected at each occurrence from hydrogen; or C 1 -C 6 alkyl, C 2 -C 6 alkenyl or C 2 -C 6 alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R 3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 haloalkyl, C 2 -C 6 haloalken
- the invention provides a compound of formula IV:
- Z is OC(O), C(O), C(O)O, or C(O)NR x ;
- R x is H or alkyl
- k 0, 1, or 2;
- each R 1 is independently selected from the group consisting of:
- n 0, 1, 2, 3, or 4;
- R 6 is selected from aryl; substituted aryl; heteroaryl; substituted heteroaryl; heterocyclyl, substituted heterocyclyl; —C 1 -C 5 alkyl, substituted —C 1 -C 8 alkyl, —C 3 -C 12 carbocyclic, and substituted —C 3 -C 12 carbocyclic;
- G is -E-R 5 ;
- each of R a , R b , R c , and R d are each independently H or F, wherein at least two of R a , R b , R c , and R d are F;
- R 3 is H, optionally substituted alkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; or optionally substituted carbocyclic.
- each R 1 is substituted with one, two, three or four independent R E , wherein each R E is independently selected from alkyl, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, halogen, hydroxy, amino, —CN, —CF 3 , —N 3 , —NO 2 , —OR F , —SR, —SOR F , —SO 2 R F , —N(R F )S(O 2 )—R F , —N(R F ) S(O 2 )NR F R F , —NR F R F , —C(O)OR F , —C(O)R F , —C(O)NR F R F , or —N(R F )C(O)R F ; wherein each R F is independently hydrogen, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, or C 2 -C 6
- E is G-R 5 ; and G is —O—, —S—, —N(R 3 )—, —N(R 3 )S(O) p —, —N(R 3 )C(O)—, —N(R 3 ) C(O)S(O p )—, —OS(O p )—, —C(O)S(O p )—, or —C(O)N(R 3 )S(O p )—; and each p is independently 0, 1, or 1
- E is —N(R 3 )S(O) p —.
- R 5 is H; optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl.
- R 5 is cyclopropyl optionally substituted with C 1 -C 6 alkyl, C 1 -C 6 alkoxyC 1 -C 6 alkyl, —C(O)OH, —C(O)NH 2 , or —C(O)O—C 1 -C 6 alkyl.
- R a , R b , R c , and R d are F.
- R a and R b are F.
- R c and R d are F.
- R a and R c are F.
- three of R a , R b , R c , and R d are F.
- R a , R b , and R c are F. In one embodiment of each of the above-described aspects of the invention, R a , R c , and R d are F. In another embodiment of each of the above-described aspects of the invention, all of R a , R b , R c , and R d are F.
- R a and R c are F.
- J is —(C ⁇ O)—; and A is optionally substituted heteroaryl.
- J is —(C ⁇ O)—; and A is optionally substituted aryl.
- J is —(C ⁇ O)—; and A is optionally substituted alkyl.
- J is —(C ⁇ O)—;
- A is optionally substituted alkyl;
- Z is O,
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of R a , R b , R c , and R d are each independently F.
- J is —(C ⁇ O)—;
- A is optionally substituted aryl;
- Z is O,
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of R a , R b , R c , and R d are each independently F.
- J is —(C ⁇ O)—;
- A is optionally substituted heteroaryl;
- Z is O,
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of R a , R b , R c , and R d are each independently F.
- J is —(C ⁇ O)—;
- A is optionally substituted alkyl;
- Z is O,
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of R a , R b , R c , and R d are each independently F.
- J is —(C ⁇ O)O—;
- A is optionally substituted aryl;
- Z is O,
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of R a , R b , R c , and R d are each independently F.
- J is —(C ⁇ O)O—;
- A is optionally substituted heteroaryl;
- Z is O,
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of R a , R b , R c , and R d are each independently F.
- J is —(C ⁇ O)O— or —(C ⁇ O)—;
- A is optionally substituted alkyl;
- Z is O or (C ⁇ O),
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and
- R a and R b are F, and R c and R d are H.
- J is —(C ⁇ O)O— or —(C ⁇ O)—;
- A is optionally substituted alkyl;
- Z is O or (C ⁇ O),
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and
- R a and R d are F, and R c and R d are H.
- J is —(C ⁇ O)O— or —(C ⁇ O)—;
- A is optionally substituted alkyl;
- Z is O or (C ⁇ O),
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and
- R a and R b are H, and R c , and R d are F.
- J is —(C ⁇ O)O— or —(C ⁇ O)—;
- A is optionally substituted aryl;
- Z is O or (C ⁇ O),
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and
- R a and R b are F, and R c and R d are H.
- J is —(C ⁇ O)O— or —(C ⁇ O)—;
- A is optionally substituted aryl;
- Z is O or (C ⁇ O),
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and
- R a and R c are F, and R b and R d are H.
- J is —(C ⁇ O)O— or —(C ⁇ O)—;
- A is optionally substituted aryl;
- Z is O or (C ⁇ O),
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and
- R a and R b are H, and R c and R d are F.
- J is —(C ⁇ O)O— or —(C ⁇ O)—;
- A is optionally substituted heteroaryl;
- Z is O or (C ⁇ O),
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and
- R a and R b are F, and R c and R d are H.
- J is —(C ⁇ O)O— or —(C ⁇ O)—;
- A is optionally substituted heteroaryl;
- Z is O or (C ⁇ O),
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and
- R a and R c are F, and R b and R d are H.
- J is —(C ⁇ O)O— or —(C ⁇ O)—;
- A is optionally substituted heteroaryl;
- Z is O or (C ⁇ O),
- ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and
- R a and R b are H, and R c and R d are F.
- E is —NHS(O) 2 R 5 ;
- R 5 is independently cyclopropyl optionally substituted with C 1 -C 6 alkyl;
- J is —(C ⁇ O)—;
- A is optionally substituted alkyl, Z is O;
- ring X is phenanthridinyl or quinoxalinyl; and any two of R a , R b , R c , and R d are each independently F.
- E is —NHS(O) 2 R 5 ;
- R 5 is independently cyclopropyl optionally substituted with C 1 -C 6 alkyl;
- J is —(C ⁇ O)—;
- A is optionally substituted aryl, Z is O;
- ring X is phenanthridinyl or quinoxalinyl; and any two of R a , R b , R c , and R d are each independently F.
- E is —NHS(O) 2 —R 5 ;
- R 5 is independently cyclopropyl optionally substituted with C 1 -C 6 alkyl;
- J is —(C ⁇ O)—;
- A is optionally substituted heteroaryl, Z is O;
- ring X is phenanthridinyl or quinoxalinyl; and any two of R a , R b , R c , and R d are each independently F.
- E is —NHS(O) 2 —R 5 ;
- R 5 is independently cyclopropyl optionally substituted with C 1 -C 6 alkyl;
- J is —(C ⁇ O)O—;
- A is optionally substituted alkyl, Z is O;
- ring X is phenanthridinyl or quinoxalinyl; and any two of R a , R b , R c , and R d are each independently F.
- E is —NHS(O) 2 —R 5 ;
- R 5 is independently cyclopropyl optionally substituted with C 1 -C 6 alkyl;
- J is —(C ⁇ O)O—;
- A is optionally substituted aryl, Z is O;
- ring X is phenanthridinyl or quinoxalinyl; and any two of R a , R b , R c , and R d are each independently F.
- E is —NHS(O) 2 —R 5 ;
- R 5 is independently cyclopropyl optionally substituted with C 1 -C 6 alkyl;
- J is —(C ⁇ O)O—;
- A is optionally substituted heteroaryl, Z is O;
- ring X is phenanthridinyl or quinoxalinyl; and any two of R a , R b , R c , and R d are each independently F.
- A is an optionally substituted nitrogen-containing heteroaryl.
- A is optionally substituted isoxazolyl, optionally substituted thiazolyl, optionally substituted pyrazolyl, optionally substituted pyrazinyl, optionally substituted pyridinyl, or optionally substituted pyrimidinyl.
- A is alkyl-substituted isoxazolyl.
- J can be, for example, —(C ⁇ O)— or —(C ⁇ O)O—; and A is selected from the following groups:
- Representative compounds include, but are not limited to, the following compounds:
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of the invention (e.g., a compound of formula I, or any formulae as described herein), or a pharmaceutically acceptable salt, ester, or prodrug thereof, in combination with a pharmaceutically acceptable carrier or excipient.
- a compound of the invention e.g., a compound of formula I, or any formulae as described herein
- a pharmaceutically acceptable salt, ester, or prodrug thereof in combination with a pharmaceutically acceptable carrier or excipient.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound described herein, or an embodiment or example described herein, or a pharmaceutically acceptable salt, ester, hydrate, solvate, polymorph, or prodrug thereof, in combination with a pharmaceutically acceptable carrier or excipient.
- the pharmaceutical compositions of the present invention may further contain one or more other anti-HCV agents.
- anti-HCV agents include, but are not limited to, ⁇ -interferon; ⁇ -interferon; pegylated interferon- ⁇ ; pegylated interferon-lambda; ribavirin; viramidine; R-5158; nitazoxanide; amantadine; Debio-025, NEM-811; HCV polymerase inhibitors such as R7128, R1626, R4048, T-1106, PSI-7851, PF-00868554, ANA-598, IDX184, IDX102, IDX375, GS-9190, VCH-759, VCH-916, MK-3281, BCX-4678, MK-3281, VBY708, ANA598, GL59728 or GL60667; BMS-790052; BMS-791325; BMS-650032; HCV entry, helicase or
- compositions of the present invention may further contain another HCV protease inhibitor, such as telaprevir, boceprevir, ITMN-191, B1-201335, TMC-435, MK-7009, VBY-376, VX-500, VX-813, PHX-B, ACH-1625, IDX136, or IDX316.
- another HCV protease inhibitor such as telaprevir, boceprevir, ITMN-191, B1-201335, TMC-435, MK-7009, VBY-376, VX-500, VX-813, PHX-B, ACH-1625, IDX136, or IDX316.
- the invention provides a pharmaceutical composition further comprising pegylated interferon, another anti-viral, anti-bacterial, anti-fungal or anti-cancer agent, or an immune modulator, and/or further comprising a cytochrome P450 monooxygenase inhibitor or a pharmaceutically acceptable salt thereof.
- the cytochrome P450 monooxygenase inhibitor is ritonavir.
- the invention provides for the use of a compound of the invention to manufacture an agent for preventing or treating viral infection.
- the invention provides for the use of a compound of the invention to manufacture an agent for preventing or treating hepatitis C viral infection.
- the present invention also contemplates the use of a solvate (e.g., hydrate) of a compound of the invention to manufacture pharmaceutical compositions for preventing or treating hepatitis C infection.
- solvate refers to the physical association of a compound of the invention with one or more solvent molecule, whether organic or inorganic. This physical association often includes hydrogen bonding. In certain instances, the solvate is capable of isolation, for example, when one or more solvate molecules are incorporated in the crystal lattice of the crystalline solid.
- the compounds or pharmaceutical compositions of the invention are administered with ritonavir, either simultaneously or sequentially.
- a compound or a pharmaceutical composition of the invention is administered in the same composition as ritonavir.
- a compound or a pharmaceutical composition thereof of the invention is administered in a different composition than ritonavir.
- compositions of the present invention may further comprise inhibitor(s) of other targets in the HCV life cycle, including, but not limited to, helicase, polyrnerase, metalloprotease, CD81, NS5A, cyclophilin, and internal ribosome entry site (IRES).
- inhibitor(s) of other targets in the HCV life cycle including, but not limited to, helicase, polyrnerase, metalloprotease, CD81, NS5A, cyclophilin, and internal ribosome entry site (IRES).
- the invention provides a method of treating a viral infection in a subject, comprising administering to the subject a therapeutically effective amount of a compound of the invention (e.g., a compound of formula I or I′ described herein), or a pharmaceutically acceptable salt, ester or prodrug thereof, or a pharmaceutical composition comprising the same.
- a compound of the invention e.g., a compound of formula I or I′ described herein
- a pharmaceutically acceptable salt, ester or prodrug thereof e.g., a pharmaceutically acceptable salt, ester or prodrug thereof, or a pharmaceutical composition comprising the same.
- the present invention includes methods of treating hepatitis C infections in a subject in need of such treatment by administering to said subject an anti-HCV virally effective amount or an inhibitory amount of the compounds or pharmaceutical compositions of the present invention.
- the present invention includes methods of treating hepatitis C infections in a subject in need of such treatment by administering to said subject a compound or a pharmaceutical composition of the present invention.
- the methods can further include administration of an additional therapeutic agent, including another antiviral agent or an anti-HCV agent as described hereinabove.
- the additional agent can be co-administered (such as concurrently administered or sequentially administered) with a compound (a pharmaceutically acceptable salt, ester or prodrug thereof) or a pharmaceutical composition of the present invention.
- the additional agent(s) and a compound (or a pharmaceutically acceptable salt, ester or prodrug thereof) of the present invention can be formulated in the same composition, or in different compositions but co-administered concurrently or sequentially.
- the methods herein can further include the step of identifying that the subject is in need of treatment for hepatitis C infection. The identification can be by subjective (e.g., health care provider determination) or objective (e.g., diagnostic test) means.
- the invention provides a method as described above, further comprising administering an additional anti-hepatitis C virus agent.
- anti-hepatitis C virus agents include, but are not limited to, ⁇ -interferon; ⁇ -interferon; pegylated interferon- ⁇ ; pegylated interferon-lambda; ribavirin; viramidine; R-5158; nitazoxanide; amantadine; Debio-025, NIM-811; HCV polymerase inhibitors such as R7128, R1626, R4048, T-1106, PSI-7851, PF-00868554, ANA-598, IDXI84, IDX102, IDX375, GS-9190, VCH-759, VCH-916, MK-3281, BCX-4678, MK-3281, VBY708, ANA598, GL59728 or GL60667; BMS-790052; BMS-791325; BMS-6500
- a compound or a pharmaceutical composition of the present invention is co-administered with, or used in combination with, pegylated interferon (e.g., pegylated interferon alpha-2a or 2b) and ribavirin.
- pegylated interferon e.g., pegylated interferon alpha-2a or 2b
- Ritonavir or another cytochrome P450 monooxygenase inhibitor can also be used to enhance the pharmacokinetics of the compound of the present invention.
- the patient being treated is preferably infected with HCV genotype-1 (e.g., genotype 1a or 1b), Patients infected with other HCV genotypes, such as genotypes 2, 3, 4, 5 or 6, can also be treated with a compound or a pharmaceutical composition of the present invention.
- the invention provides a method as described above, further comprising administering another 1-ICV protease inhibitor, an HCV polymerase inhibitor, an HCV helicase inhibitor, or an internal ribosome entry site (IRES) inhibitor, such as telaprevir, boceprevir, ITMN-191, 131-201335, TMC-435, MK-7009, VBY-376, VX-500, VX-813, PHX-B, ACH-1625, IDX136, IDX316, pegylated interferon, another anti-viral, anti-bacterial, anti-fungal or anti-cancer agent, or an immune modulator, and/or further comprising a cytochrome P450 monooxygenase inhibitor or a pharmaceutically acceptable salt thereof.
- the cytochrome P450 monooxygenase inhibitor is ritonavir.
- An additional embodiment of the present invention includes methods of treating biological samples by contacting the biological samples with the compounds of the present invention.
- Yet another aspect of the present invention is a process of making any of the compounds delineated herein employing any of the synthetic means delineated herein.
- C x -C y The number of carbon atoms in a hydrocarbyl substituent can be indicated by the prefix “C x -C y ,” where x is the minimum and y is the maximum number of carbon atoms in the substituent.
- a C, chain means a hydrocarbyl chain containing x carbon atoms.
- halo indicates that the substituent to which the prefix is attached is substituted with one or more independently selected halogen radicals.
- C 1 -C 8 haloalkyl means a C 1 -C 8 alkyl substituent wherein at least one hydrogen radical is replaced with a halogen radical.
- a linking element in a depicted structure is “absent”, then the left element in the depicted structure is directly linked to the right element in the depicted structure.
- a chemical structure is depicted as X-(L) n -Y wherein L is absent or n is 0, then the chemical structure is X—Y.
- alkyl refers to a saturated, straight- or branched-chain hydrocarbon radical preferably containing one to eight carbon atoms (C 1 -C 8 alkyl).
- alkyl radicals include, but are not limited to, methyl, ethyl, propyl, isopropyl, n-butyl, tert-butyl, neopentyl, n-hexyl, heptyl, octyl radicals and the like.
- alkenyl denotes a straight- or branched-chain hydrocarbon radical containing one or more double bonds and preferably containing two to eight carbon atoms (C 2 -C 8 alkenyl).
- Alkenyl groups include, but are not limited to, for example, ethenyl, propenyl, butenyl, 1-methyl-2-buten-1-yl, heptenyl, octenyl and the like.
- alkynyl denotes a straight- or branched-chain hydrocarbon radical containing one or more triple bonds and preferably containing two to eight carbon atoms (C 2 -C 8 alkynyl).
- Representative alkynyl groups include, but are not limited to, for example, ethynyl, 1-propynyl, 1-butyny-1, heptynyl, octynyl and the like.
- cycloalkyl denotes a monovalent group derived from a monocyclic or polycyclic saturated carbocyclic ring compound and preferably containing 3 to 14 carbon ring atoms (C 3 -C 14 cycloalkyl).
- Examples of cycloalkyl include, but not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyciohexyl, bicyclo[2.2.1]heptyl, and bicyclo[2.2.2]octyl and the like.
- Carbocycle or “carbocyclic” or “carbocyclyl” refer to a saturated (e.g., “cycloalkyl”), partially saturated (e.g., “cycloalkenyl” or “cycloalkynyl”) or completely unsaturated (e.g., “aryl”) ring system containing zero heteroatom ring atom and preferably containing 3 to 14 carbon ring atoms (C 3 -C 14 carbocyclyl, such as C 3 -C 14 cycloalkyl).
- a carbocyclyl may be, without limitation, a single ring, or two or more fused or covalently inked rings, or bridged or Spiro rings.
- a substituted carbocyclyl may have either cis or trans geometry.
- Representative examples of carbocyclyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclopentenyl, cyclopentadienyl, cyclohexadienyl, adamantyl, decahydro-naphthalenyl, octahydro-indenyl, cyclohexenyl, phenyl, naphthyl, fluorenyl, indanyl, 1,2,3,4-tetrahydro-naphthyl, indenyl, isoindenyl, bicyclodecanyl, anthracenyl, phenanthrene, benzonaphthenyl (also known as “phenalenyl”), decalinyl,
- aryl refers to an aromatic carbocyclyl containing from 6 to 14 carbon ring atoms.
- Non-limiting examples of aryls include phenyl, naphthalenyl, anthracenyl, and indenyl and the like.
- An aryl group can be connected to the parent molecular moiety through any substitutable carbon atom of the group.
- heteroaryl means an aromatic heterocyclyl typically containing from 5 to 14 ring atoms.
- five-membered heteroaryls include imidazolyl; furanyl; thiophenyl (or thienyl or thiofuranyl); pyrazolyl; oxazolyl; isoxazolyl; thiazolyl; 1,2,3-, 1,2,4-, 1,2,5-, and 1,3,4-oxadiazolyl; and isothiazolyl.
- Non-limiting examples of six-membered heteroaryls include pyridinyl; pyrazinyl; pyrimidinyl; pyridazinyl; and 1,3,5-, 1,2,4-, and 1,2,3-triazinyl.
- Non-limiting examples of 6/5-membered fused ring heteroaryls include benzothiofiiranyl, isobenzothiofuranyl, benzisoxazolyl, benzoxazolyl, purinyl, and anthranilyl.
- Non-limiting examples of 6/6-membered fused ring heteroaryls include quinolinyl; isoquinolinyl; and benzoxazinyl (including ciimolinyl and quinazolinyl).
- heterocycloalkyl refers to a non-aromatic 3-, 4-, 5-, 6- or 7-membered ring or a bi- or tri-cyclic group fused system, where at least one of the ring atoms is a heteroatom, and where (i) each 5-membered ring has 0 to 1 double bonds and each 6-membered ring has 0 to 2 double bonds, (ii) the nitrogen and sulfur heteroatoms may optionally be oxidized, (iii) the nitrogen heteroatom may optionally be quaternized, and (iv) any of the above rings may be fused to a benzene ring.
- heterocycloalkyl groups include, but are not limited to, [1,3]dioxolane, pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, piperidinyl, piperazinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, and tetrahydrofuryl and the like.
- heterocyclic or “heterocycle” or “heterocyclyl” refer to a saturated (e.g., “heterocycloalkyl”), partially unsaturated (e.g., “heterocycloalkenyl” or “heterocycloalkynyl”) or completely unsaturated (e.g., “heteroaryl”) ring system preferably containing 3 to 14 ring atoms, where at least one of the ring atoms is a heteroatom (i.e., nitrogen, oxygen or sulfur), with the remaining ring atoms being independently selected from the group consisting of carbon, nitrogen, oxygen and sulfur.
- heteroatom i.e., nitrogen, oxygen or sulfur
- a heterocyclic or heterocycle or heterocyclyl may be, without limitation, a single ring, or two or more fused or covalently linked rings, or bridged or Spiro rings.
- a heterocyclyl group can be linked to the parent molecular moiety via any substitutable carbon or nitrogen atom in the group, provided that a stable molecule results.
- a heterocyclyl may be, without limitation, a single ring.
- Non-limiting examples of single-ring heterocyclyls include furanyl, dihydrofuranyl, pyrrolyl, isopyrrolyl, pyrrolinyl, pyrrolidinyl, imidazolyl, isoimidazolyl, imidazolinyl, imidazolidinyl, pyrazolyl, pyrazolinyl, pyrazolidinyl, triazolyl, tetrazolyl, dithiolyl, oxathiolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiazoliriyl, isothiazolinyl, thiazolidinyl, isothiazolidinyl, thiodiazolyl, oxathiazolyl, oxadiazoly, pyranyl, dihydropyranyl, pyrridinyl, piperidinyl, pyridazinyl,
- a heterocyclyl may also include, without limitation, two or more rings fused together, such as, for example, naphthyridinyl, thiazolpyrimidinyl, thienopyrimidinyl, pyrimidopyrimidinyl, or pyridopyrimidinyl.
- a heterocyclyl may comprise one or more sulfur atoms as ring members; and in some cases, the sulfur atom(s) is oxidized to SO or SO 2 .
- the nitrogen heteroatom(s) in a heterocyclyl may or may not be quaternized, and may or may not be oxidized to N-oxide. In addition, the nitrogen heteroatom(s) may or may not be N-protected.
- optionally substituted refers to groups that are substituted or unsubstituted by independent replacement of one, two, or three or more of the hydrogen atoms thereon with typical substituents including, but not limited to:
- —NH 2 protected amino, —NH alkyl, —NH alkenyl, —NH alkynyl, —NH cycloalkyl, —NH— aryl, —NH-heteroaryl, —NH-heterocyclic, -dialkylamino, -diarylamino, -diheteroarylamino,
- —O— alkyl —O— alkenyl. —O— alkynyl, —O— cycloalkyl, —O-aryl, —O-heteroaryl, —O— heterocyclic,
- -alkyl -alkenyl, -alkynyl, -aryl, -arylalkyl, -heteroaryl, -heteroarylalkyl, -heterocycloalkyl, -cycloalkyl, -carbocyclic, -heterocyclic, polyalkoxyalkyl, polyalkoxy, methoxymethoxy, -methoxyethoxy, —SH, —S— alkyl, —S— alkenyl, —S— alkynyl, —S— carbocyclyl, —S-aryl, —S-heteroaryl, —S-heterocyclyl, or methylthiomethyl.
- aryls, heteroaryls, carbocycles, heterocycles, alkyls, and the like can be further substituted.
- halo and “halogen,” as used herein, refer to an atom selected from fluorine, chlorine, bromine and iodine.
- Any divalent group having two points of connection to two other groups may be aligned in either direction, e.g., the term “—C(O)O-” is interpreted to include both —C(O)O— and —OC(O)—.
- subject refers to a mammal.
- a subject therefore refers to, for example, dogs, cats, horses, cows, pigs, guinea pigs, and the like.
- the subject is a human.
- the subject may be either a patient or a healthy human.
- LG refers to any group that leaves in the course of a chemical reaction involving the group and includes but is not limited to halogen, brosylate, mesylate, tosylate, triflate, p-nitrobenzoate, phosphonate groups, for example.
- protected hydroxy refers to a hydroxy group protected with a hydroxy protecting group, as defined above, including benzoyl, acetyl, trirnethylsilyl, triethylsilyl, methoxyrnethyl groups, for example.
- hydroxy protecting group refers to a labile chemical moiety which is known in the art to protect a hydroxy group against undesired reactions during synthetic procedures. After said synthetic procedure(s) the hydroxy protecting group as described herein may be selectively removed. Hydroxy protecting groups as known in the are described generally in T. H. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999).
- hydroxy protecting groups include benzyloxycarbonyl, 4-nitrobenzyloxycarbonyl, 4-bromobenzyloxycarbonyl, 4-methoxybenzyloxy-carbonyl, methoxycarbonyl, tert-butoxycarbonyl, isopropoxycarbonyl, diphenylmethoxycarbonyl, 2,2,2-trichloroethoxycarbonyl, 2-(trimethylsilypethoxycarbonyl, 2-furfuryloxycarbonyl, allyloxycarbonyl, acetyl, formyl, chloroacetyl, trifluoroacetyl, methoxyacetyl, phenoxyacetyl, benzoyl, methyl, t-butyl, 2,2,2-trichloroethyl, 2-trimethylsilyl ethyl, 1,1-dimethyl-2-propenyl, 3-methyl-3-butenyl, allyl, benzyl, para-methoxybenzyldipheny
- Preferred hydroxy protecting groups for the present invention are acetyl (Ac or —C(O)CH 3 ), benzoyl (Bz or —C(O)C 6 H 5 ), and trimethylsilyl (TMS or —Si(CH 3 ) 3 ).
- amino protecting group refers to a labile chemical moiety which is known in the art to protect an amino group against undesired reactions during synthetic procedures. After said synthetic procedure(s) the amino protecting group as described herein may be selectively removed.
- Amino protecting groups as known in the are described generally in T. H. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999). Examples of amino protecting groups include, but are not limited to, t-butoxycarbonyl, 9-fluorenylmethoxycarbonyl, benzyloxycarbonyl, and the like.
- protected amino refers to an amino group protected with an amino protecting group as defined above.
- alkylamino refers to a group having the structure —N(R a R b ), where R a and R b are independent 1-1 or alkyl.
- the term “pharmaceutically acceptable salt” refers to those salts of the compounds formed by the process of the present invention which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio.
- Pharmaceutically acceptable salts are well known in the art. For example, S. M. Berge, et al. describes pharmaceutically acceptable salts in detail in J. Pharmaceutical Sciences, 66: 1-19 (1977). The salts can be prepared in situ during the final isolation and purification of the compounds of the invention, or separately by reacting the tree base function with a suitable organic acid.
- salts include, but are not limited to, nontoxic acid addition salts, or salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange.
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid
- organic acids such as acetic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange.
- salts include, but are not limited to, adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanestilfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate
- alkali or alkaline earth metal salts include sodium, lithium, potassium, calcium, or magnesium salts, and the like.
- Further pharmaceutically acceptable salts include, when appropriate, nontoxic ammonium, quaternary ammonium, and amine cations formed using counterions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, alkyl having from 1 to 6 carbon atoms, sulfonate and aryl sulfonate.
- ester refers to esters of the compounds formed by the process of the present invention which hydrolyze in vivo and include those that break down readily in the human body to leave the parent compound or a salt thereof.
- Suitable ester groups include, for example, those derived from pharmaceutically acceptable aliphatic carboxylic acids, particularly alkanoic, alkenoic, cycloalkanoic and alkanedioic acids, in which each alkyl or alkenyl moiety advantageously has not more than 6 carbon atoms.
- esters include, but are not limited to, formates, acetates, propionates, butyrates, acrylates and ethylsuccinates.
- prodrugs refers to those prodrugs of the compounds formed by the process of the present invention which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals with undue toxicity, irritation, allergic response, and the like, commensurate with a reasonable benefit/risk ratio, and effective for their intended use, as well as the zwitterionic forms, where possible, of the compounds of the present invention.
- Prodrug as used herein means a compound which is convertible in vivo by metabolic means (e.g. by hydrolysis) to afford any compound delineated by the formulae of the instant invention.
- prodrugs are known in the art, for example, as discussed in Bundgaard, (ed.), Design of Prodrugs, Elsevier (1985); Widder, et al. (ed.), Methods in Enzymology, vol. 4, Academic Press (1985); Krogsgaard-Larsen, et al., (ed). “Design and Application of Prodrugs, Textbook of Drug Design and Development, Chapter 5, 113-191 (1991); Bundgaard, et al., Journal of Drug Deliver Reviews, 8:1-38(1992); Bundgaard, J. of Pharmaceutical Sciences, 77:285 et seq.
- This invention also encompasses pharmaceutical compositions containing, and methods of treating viral infections through administering, pharmaceutically acceptable prodrugs of compounds of the invention.
- compounds of the invention having free amino, amido, hydroxy or carboxylic groups can be converted into prodrugs.
- Prodrugs include compounds wherein an amino acid residue, or a polypeptide chain of two or more (e.g., two, three or four) amino acid residues is covalently joined through an amide or ester bond to a free amino, hydroxy or carboxylic acid group of compounds of the invention.
- the amino acid residues include but are not limited to the 20 naturally occurring amino acids commonly designated by three letter symbols and also includes 4-hydroxyproline, hydroxylysine, demosine, isodemosine, 3-methylhistidine, norvalin, beta-alanine, gamma-aminobutyric acid, citrulline, homocysteine, homoserine, ornithine and methionine sulfone. Additional types of prodrugs are also encompassed. For instance, free carboxyl groups can be derivatized as amides or alkyl esters.
- Free hydroxy groups may be derivatized using groups including but not limited to hemisuccinates, phosphate esters, dimethylaminoacetates, and phosphoryloxymethyloxy carbonyls, as outlined in Advanced Drug Delivery Reviews, 1996, 19, 1 15.
- Carbamate prodrugs of hydroxy and amino groups are also included, as are carbonate prodrugs, sulfonate esters and sulfate esters of hydroxy groups.
- stable refers to compounds which possess stability sufficient to allow manufacture and which maintains the integrity of the compound for a sufficient period of time to be useful for the purposes detailed herein (e.g., therapeutic or prophylactic administration to a subject).
- compositions of the present invention comprise a therapeutically effective amount of a compound of the present invention formulated together with one or more pharmaceutically acceptable carriers.
- pharmaceutically acceptable carrier means a non-toxic, inert solid, semi-solid or liquid filler, diluent, encapsulating material or formulation auxiliary of any type.
- the pharmaceutical compositions of this invention can be administered to humans and other animals orally, rectally, parenterally, intracisternally, intravaginally, intraperitoneally, topically (as by powders, ointments, or drops), buccally, or as an oral or nasal spray.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups, and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art such as, for example, water, alcohol or other solvents, solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, polysorbate, dimethylformamide, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), mono- or di-glycerides, glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof.
- inert diluents commonly used in the art such as
- the oral compositions can also include adjuvants such as wetting agents, emulsifying and suspending agents, antioxidants, sweetening, flavoring, and perfuming agents.
- adjuvants such as wetting agents, emulsifying and suspending agents, antioxidants, sweetening, flavoring, and perfuming agents.
- the liquid dosage form can also be encapsulated in a gelatin capsule, wherein a compound of the present invention can be dissolved in a pharmaceutically acceptable canter containing, for example, one or more soltibilizating agents (e.g., polysorbate 80 and mono and diglycerides), and other suitable excipients (e.g., an antioxidants such as ascorbyl palmitate, or a sweetening or flavoring agent).
- soltibilizating agents e.g., polysorbate 80 and mono and diglycerides
- other suitable excipients e.g., an antioxidants such as ascorbyl palmitate, or a sweetening or flavoring agent
- sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution, suspension or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution, U.S.P. and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil can be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid are used in the preparation of injectables.
- compositions for rectal or vaginal administration are preferably suppositories which can be prepared by mixing the compounds of this invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
- the active compounds can also be in micro-encapsulated form with one or more excipients as noted above
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings, release controlling coatings and other coatings well known in the pharmaceutical formulating art.
- the active compound may be admixed with at least one inert diluent such as sucrose, lactose or starch.
- Such dosage forms may also comprise, as is normal practice, additional substances other than inert diluents, e.g., tableting lubricants and other tableting aids such a magnesium stearate and microcrystalline cellulose.
- the dosage forms may also comprise buffering agents.
- a compound of the invention is formulated in a solid dispersion, where the compound can be molecularly dispersed in a matrix which comprises a pharmaceutically acceptable, hydrophilic polymer.
- the matrix may also contain a pharmaceutically acceptable surfactant.
- Suitable solid dispersion technology for formulating a compound of the invention includes, but is not limited to, melt extrusion, spray drying, or solvent evaporization.
- Dosage forms for topical or transdermal administration of a compound of this invention include ointments, pastes, creams, lotions, gels, powders, solutions, sprays, inhalants or patches.
- the active component is admixed under sterile conditions with a pharmaceutically acceptable carrier and any needed preservatives or buffers as may be required.
- Ophthalmic formulation, ear drops, eye ointments, powders and solutions are also contemplated as being within the scope of this invention.
- the ointments, pastes, creams and gels may contain, in addition to an active compound of this invention, excipients such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- excipients such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- Powders and sprays can contain, in addition to the compounds of this invention, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances.
- Sprays can additionally contain customary propellants such as chlorafluorohydrocarbons.
- Transdermal patches have the added advantage of providing controlled delivery of a compound to the body.
- dosage forms can be made by dissolving or dispensing the compound in the proper medium.
- Absorption enhancers can also be used to increase the flux of the compound across the skin.
- the rate can be controlled by either providing a rate controlling membrane or by dispersing the compound in a polymer matrix or gel.
- viral infections are treated or prevented in a subject, such as a human or another animal, by administering to the subject a therapeutically effective amount of a compound of the invention (or a pharmaceutically acceptable salt, ester or prodrug thereof), in such amounts and for such time as is necessary to achieve the desired result.
- a therapeutically effective amount of a compound of the invention means a sufficient amount of the compound so as to decrease the viral load in a subject and/or decrease the subject's HCV symptoms.
- a therapeutically effective amount of a compound of this invention will be at a reasonable benefit/risk ratio applicable to any medical treatment.
- An inhibitory amount or dose of the compounds of the present invention may range from about 0.1 mg/Kg to about 500 mg/Kg, alternatively from about 1 to about 50 mg/Kg. Inhibitory amounts or doses will also vary depending on route of administration, as well as the possibility of co-usage with other agents.
- viral infections are treated or prevented in a subject such as a human or lower mammal by administering to the subject an anti-hepatitis C virally effective amount or an inhibitory amount of a compound of the present invention, in such amounts and for such time as is necessary to achieve the desired result.
- An additional method of the present invention is the treatment of biological samples with an inhibitory amount of a compound of composition of the present invention in such amounts and for such time as is necessary to achieve the desired result.
- anti-hepatitis C virally effective amount of a compound of the invention, as used herein, means a sufficient amount of the compound so as to decrease the viral load in a biological sample or in a subject.
- an anti-hepatitis C virally effective amount of a compound of this invention will be at a reasonable benefit/risk ratio applicable to any medical treatment.
- inhibitory amount of a compound of the present invention means a sufficient amount to decrease the hepatitis C viral load in a biological sample or a subject. It is understood that when said inhibitory amount of a compound of the present invention is administered to a subject it will be at a reasonable benefit/risk ratio applicable to any medical treatment as determined by a physician.
- biological sample(s),” as used herein means a substance of biological origin, which may be intended for administration to a subject. Examples of biological samples include, but are not limited to, blood and components thereof such as plasma, platelets, subpopulations of blood cells and the like; organs such as kidney, liver, heart, lung, and the like; sperm and ova; bone marrow and components thereof; or stem cells.
- another embodiment of the present invention is a method of treating a biological sample by contacting said biological sample with an inhibitory amount of a compound or pharmaceutical composition of the present invention.
- a maintenance dose of a compound, composition or combination of this invention may be administered, if necessary. Subsequently, the dosage or frequency of administration, or both, may be reduced, as a function of the symptoms, to a level at which the improved condition is retained when the symptoms have been alleviated to the desired level, treatment should cease.
- the subject may, however, require intermittent treatment on a long-term basis upon any recurrence of disease symptoms.
- the total daily usage of the compounds and compositions of the present invention will be decided by the attending physician within the scope of sound medical judgrient.
- the specific inhibitory dose for any particular patient will depend upon a variety of factors including the disorder being treated and the severity of the disorder; the activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed; and like factors well known in the medical arts.
- the total daily inhibitory dose of the compounds of this invention administered to a subject in single or in divided doses can be in amounts, for example, from 0.01 to 50 mg/kg body weight or more usually from 0.1 to 25 mg/kg body w eight.
- Single dose compositions may contain such amounts or submultiples thereof to make up the daily dose.
- treatment regimens according to the present invention comprise administration to a patient in need of such treatment from about 10 mg to about 1000 mg of the compound(s) of this invention per day in single or multiple doses.
- the treatment regimen comprises administration to a patient in need of such treatment from about 25 mg to about 6000 mg of a compound(s) of this invention per day in single or multiple doses, either with or without a cytochrome P450 monooxygenase inhibitor such as ritonavir.
- the suitable daily dose for the co-administered cytochrorne P450 monooxygenase inhibitor can range, without limitation, from 10 to 200 mg.
- a compound(s) of the present invention, or a combination of a compound(s) of the invention and ritonavir is administered once daily or twice daily to achieve the desired daily dose amount.
- a compound of the present invention when used without ritonavir, can be administered to a patient twice a day with a total daily dose of 4000, 4200, 4400, 4600, 4800 or 5000 mg.
- a compound of the present invention when used in combination with ritonavir, can be administered to a patient once or twice a day with a total daily dose of 200, 400, 600 or 800 mg, where the amount of ritonavir can be 25, 50 or 100 mg per administration.
- the compounds described herein contain one or more asymmetric centers and thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)— or (S)—, or as (D)- or (L)- for amino acids.
- the present invention is meant to include all such possible isomers, as well as their racemic and optically pure forms.
- Optical isomers may be prepared from their respective optically active precursors by the procedures described above, or by resolving the racernic mixtures. The resolution can be carried out in the presence of a resolving agent, by chromatography or by repeated crystallization or by some combination of these techniques which are known to those skilled in the art.
- the synthesized compounds can be separated from a reaction mixture and further purified by a method such as column chromatography, high pressure liquid chromatography, or recrystallization.
- a method such as column chromatography, high pressure liquid chromatography, or recrystallization.
- further methods of synthesizing the compounds of the formulae herein will be evident to those of ordinary skill in the art.
- the various synthetic steps may be performed in an alternate sequence or order to give the desired compounds.
- the solvents, temperatures, reaction durations, etc. delineated herein are for purposes of illustration only and one of ordinary skill in the art will recognize that variation of the reaction conditions can produce the desired bridged macrocyclic products of the present invention.
- Synthetic chemistry transformations and protecting group methodologies useful in synthesizing the compounds described herein are known in the art and include, for example, those such as described in R. Larock, Comprehensive Organic Transformations , VCR Publishers (1989); T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis , 2d. Ed., John Wiley and Sons (1991); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis , John Wiley and Sons (1994); and L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis , John Wiley and Sons (1995), and subsequent editions thereof
- the compounds of this invention may be modified by appending various functionalities via any synthetic means delineated herein to enhance selective biological properties.
- modifications are known in the art and include those which increase biological penetration into a given biological system (e.g., blood, lymphatic system, central nervous system), increase oral availability, increase solubility to allow administration by injection, alter metabolism and alter rate of excretion.
- the chemical structures herein contain certain —NH—, —NH 2 (amino) and OH (hydroxyl) groups where the corresponding hydrogen atom(s) may not explicitly appear; however they are to be read as —NH—, —NH 2 or —OH as the case may be.
- N-ethyl-N-isopropylpropan-2-amine (2.83 ml, 16.2 mmol) and 1-methylimidazole (0.097 ml, 1.2 mmol) followed by benzoic anhydride (1.83 g, 8.10 mmol).
- the solution was stirred at room temperature for 16 h, DMAP (9.3 mg, 0.076 mmol) was added and the reaction mixture stirred for 2.5 h.
- the reaction mixture was diluted with ethyl acetate (150 ml) and washed with HCl (1 N, 50 ml ⁇ 2), water and brine.
- the mixture was stirred at 0° C. for 20 min and then at room temperature for 20 min.
- the reaction mixture was quenched by adding water (70 ml) dropwise at 0° C. followed by the addition of sodium perborate tetrahydrate (2.92 g, 19.0 mmol).
- the mixture was stirred at room temperature overnight.
- the reaction mixture was diluted with ethyl acetate (300 ml) and washed with water (50 ml ⁇ 2) and brine (50 ml).
- the combined aqueous layer was back-extracted with ethyl acetate (75 ml ⁇ 2).
- the organic layers were dried (anhydrous sodium sulfate) and concentrated to give a white foamy solid (3.2 g).
- reaction mixture was stirred at room temperature overnight, diluted with ethyl acetate and cooled to 0° C.
- the reaction mixture was neutralized with HCl (4 N in dioxane, 25 ⁇ l; diluted with ethyl acetate), stirred at room temperature and dried over anhydrous sodium sulfate.
- reaction mixture was stirred at room temperature overnight, diluted with ethyl acetate, cooled to 0° C. and neutralized with HCl (4 N in dioxane, 22 ⁇ l; diluted with ethyl acetate).
- Example 2 provided an IC 50 of >1.0 nM in a 1a enzyme assay; an IC 50 of >1.0 nM in a 1b enzyme assay; a HLM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of >5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of >5.0 nM in a replicon cell line assay in a 1b con1 background.
- Example 3a was prepared according to the procedure utilized for the preparation of Example if, replacing 2-quinoxalinol with 3-methyl-2-quinoxalinol.
- Example 3b was prepared according to the procedure utilized for the preparation of Example 1 g, replacing the product of Example 1 f with the product of Example 3a.
- Example 3 provided an IC 50 of between 0.1 and 0.5 nM in a 1a enzyme assay; and an IC 50 of between 0.1 and 0.5 nM in a 1b enzyme assay.
- Example 4a was prepared according to the procedure utilized for the preparation of Example 2a, replacing the product of Example 1 with the product of Example 3.
- Example 4 provided an IC 50 of between 0.1 and 0.5 nM in a 1a enzyme assay; an IC 50 of between 0.5 and 1.0 nM in a 1b enzyme assay; a FILM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of >5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1 b-con1 background.
- Example 5a was prepared according to the procedure utilized for the preparation of Example if, replacing 2-quinoxalinol with 3-phenyl-2-quinoxalinol.
- Example 5b was prepared according to the procedure utilized for the preparation of Example 1 g, replacing the product of Example if with the product of Example 5a.
- Example 5 provided an IC 50 of between 0.5 and 1.0 MV in a 1a enzyme assay; and an IC 50 of between 0.5 and 1.0 nM in a 1b enzyme assay.
- Example 6a was prepared according to the procedure utilized for the preparation of Example 2a, replacing the product of Example 1 with the product of Example 5.
- Example 6 provided an IC 50 of ⁇ 0.1 nM in a 1a enzyme assay; an IC 50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a HLM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
- reaction mixture was diluted with ethyl acetate and water, and the resulting organic layer was washed with saturated aqueous sodium chloride solution, separated, dried over anhydrous magnesium sulfate, filtered, and the solvent evaporated under reduced pressure.
- the residue was purified via flash chromatography on silica gel (10-20% acetone/hex).
- reaction mixture was washed with 1 N aqueous sodium bicarbonate solution followed by 1 N HCl and organic layer was separated, dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (5% methanol/dichloromethane).
- Example 7 provided an IC 50 of between 0.5 and 1.0 nM in a 1a enzyme assay; an IC 50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a HLM stability value of >100 ⁇ l/min/mg; an EC 50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
- Example 8 provided an IC 50 of between 0.1 to 0.5 nM in a 1a enzyme assay; an IC 50 of between 0.1 to 0.5 nM in a 1b enzyme assay; a HLM stability value of >100 ⁇ l/min/mg; an EC 50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
- Example 9a was prepared according to the procedure utilized for the preparation of Example 2a, replacing the product of Example 1 with the product of Example 7.
- Example 9 provided an IC 50 of between 0.5 and 1.0 nM in a 1a enzyme assay; an IC 50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a FILM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
- Example 10a was prepared according to the procedure utilized for the preparation of Example 2a, replacing the product of Example 1 with the product of Example 8.
- Example 10 provided a HLM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
- Example 11 provided an IC 50 of between 0.1 and 0.5 nM in a 1a enzyme assay; an IC 50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a FILM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of >5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1b-con1 background.
- Example 12 provided an IC 50 of between 0.5 and 1.0 nM in a 1a enzyme assay; an IC 50 of between 0.5 and 1.0 nM in a 1b enzyme assay; a HLM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of >5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of >5.0 nM in a replicon cell line assay in a 1b-con1 background.
- Example 14 provided an IC of between 0.1 and 0.5 nM in a 1a enzyme assay; an IC 50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a HLM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
- Example 16 provided an IC 50 of between 0.1 and 0.5 nM in a 1a enzyme assay; an IC 50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a HUM stability value of between 50 and 100 ⁇ l/min/mg; an EC 50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of ⁇ 1.0 nM in a replicon cell line assay in a 1b-con1 background.
- Example 17 provided an IC 50 of between 0.1 and 0.5 nM in a 1a enzyme assay; an IC 50 of ⁇ 0.1 nM in a 1b enzyme assay; a FILM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of ⁇ 1.0 nM in a replicon cell line assay in a 1b-con1 background.
- the mixture was stirred at 50° C. for 2 h, the solvent was evaporated under reduced pressure, and the residue partitioned between water and ethyl acetate. The aqueous layer was separated, diluted with ethyl acetate, and washed with 1N HCl.
- the reaction mixture was stirred for 20 min at ⁇ 60° C., and triethylamine (0.123 ml, 0.882 mmol) was added dropwise.
- the mixture was stirred at ⁇ 60° C. for 5 min, and allowed to warm to room temperature with stirring for 10 min.
- the reaction mixture was quenched with water, stirred for 15 min, and diluted with dichloromethane.
- the organic layer was separated, dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure to give a foamy material.
- Example 18 provided a HLM stability value of between 50 and 100 ⁇ l/min/mg; an EC 50 of ⁇ 1.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of ⁇ 1.0 nM in a replicon cell line assay in a 1b-con1 background.
- the mixture was cooled to 0° C. and sodium hydroxide (11.5 ml, 28.6 mmol) was added dropwise followed by hydrogen peroxide (4.0 ml, 43 mmol), and the reaction mixture stirred for 1.5 h at 0° C.
- the mixture was diluted with ethyl acetate and water, and the organic layer was washed with saturated aqueous sodium chloride solution.
- the reaction mixture was stirred for 20 min ⁇ 60° C., and triethylamine (1.716 ml, 12.31 mmol) was added. The mixture was stirred for 10 min at ⁇ 60° C., and then stirred for another 20 min at room temperature. The reaction mixture was quenched with water and stirred for 15 mm, and diluted with dichloromethane. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure to give a foamy material. The crude product was purified by flash chromatography on silica gel (5% methanolldichloromethane).
- reaction mixture was dissolved in 2 mL dichloromethane and added dropwise into a cooled solution of saturated aqueous sodium carbonate solution, stirred for 10 min, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure.
- Example 19 provided an IC 50 of ⁇ 0.1 nM in a 1a enzyme assay; an IC 50 of ⁇ 0.1 0 nM in a 1b enzyme assay; a HLM stability value of >100 ⁇ l/min/mg; an EC 50 of ⁇ 1.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of ⁇ 1.0 nM in a replicon cell line assay in a 1b-con1 background.
- Example 20a (2R,6S,13aS,14aR,16aS)-6-amino-N-(cyclopropylsulfonyl)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopenta decine-14a-carboxamide
- Example 20 provided a HLM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1 b-con1 background.
- Example 21 provided a FILM stability value of ⁇ 50 ⁇ l/min/mg; an EC 50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC 50 of ⁇ 1.0 nM in a replicon cell line assay in a 1b-con1 background.
- the substrate is labeled with a fluor and a fluorescence quencher. Cleavage results in release of the quencher and an increase in fluorescence.
- NS3 protease is incubated with a dilution series of inhibitor in 150 mM NaCl, 10% Glycerol, 5 mM. DTT, with or without 0.01% dodecyl maltoside for either 30 minutes or 300 minutes. Substrate is added at a concentration of 5 uM to initiate the reaction, and fluorescence is measured at 2 minute intervals for 30 minutes. Enzyme concentrations range from 10 to 100 nM in the absence of detergent, or 10-fold lower in the presence of detergent.
- Substrate peptides are labeled with either EDANS and DABCYL (excitation 355 nm, emission 485 nm) or TAMRA and QSY (excitation 544 nm, emission 590 nm).
- EDANS and DABCYL excitation 355 nm, emission 485 nm
- TAMRA and QSY excitation 544 nm, emission 590 nm.
- 3-fold serial dilutions starting with initial concentrations of 100 ⁇ M, 200 ⁇ M, or 2 mM are used.
- K i values approaching or lower than the enzyme concentration
- K i values are calculated using the tight binding assay format, according to the following equation:
- V A ⁇ [( K+I ⁇ E ) 2 +4 KE ]) 1/2 ⁇ ( K+I ⁇ E ) ⁇ ,
- genotype 1a Two subgenomic replicon cell lines can be used for compound characterization in cell culture: one derived from genotype 1a and one derived from genotype 1 b. Both replicon constructs are bicistronic subgenomic replicons essentially similar to those described by Bartenschlager and coworkers (Lohmann et al., Science (1999) 285(5424):110-113).
- the genotype 1a replicon construct contains the NS3-NS5B coding region derived from the H77 strain of HCV (1a-H77) (Blight et al. J Virol (2003) 77(5):3181-3190).
- the first cistron of the construct consists of the first 36 nucleotides of the HCV1a-H77 core gene fused to a firefly luciferase reporter and a neomycin phosphotransferase (Neo) selectable marker.
- the luciferase and Neo coding regions are separated by the FMDV 2a protease.
- the second cistron contains the NS3-NS5B coding region derived from 1a-H77 with the addition of adaptive mutations E1202G in NS3, K1691R in NS4A, and K2040R and S2204I in NS5A.
- the 1b-Con-1 replicon construct is identical to the 1a-H77 replicon, except that the 5′ and 3′ NTRs and the NS3-NS5B coding region can be derived from the 1b-Con-1 strain (Blight et al., Science (2000) 290(5498):1972-1974), and the adaptive mutations are E1202G and T12801 in NS3 and S2204I in NS5A.
- Replicon cell lines can be maintained in Dulbecco's modified Eagles medium (DMEM) containing 100 IU/ml penicillin, 100 mg/ml streptomycin (Invitrogen), 200 mg/ml G418 (Invitrogen) and 10% (v/v) fetal bovine serum (FBS).
- DMEM Dulbecco's modified Eagles medium
- FBS fetal bovine serum
- Replicon-containing cells can be seeded into 96 well plates at a density of 5000 cells per well in 100 ⁇ l DMEM containing 5% FBS.
- the compound can be initially diluted in dimethyl sulfoxide (DMSO) to generate a 200 ⁇ stock of the inhibitor in a series of 8 half-log dilutions.
- DMSO dimethyl sulfoxide
- the dilution series can then be diluted 100-fold in the medium containing 5% FBS.
- One hundred microliters of medium with the inhibitor can be added to each well of the overnight cell culture plate already containing 100 ⁇ l of DMEM with 5% FBS.
- the medium from the overnight cell culture plates can be replaced with 200 ⁇ l DMEM containing 40% human plasma (innovative Research) plus 5% FBS as well as compound.
- the cells can be grown for 4 days in tissue culture incubators.
- the inhibitory effects of compounds against the replicons can be determined by measuring either the level of luciferase or HCV RNA.
- the luciferase assay can be conducted using a Luciferase Assay System kit (Promega) following the manufacturer's instructions. Briefly, the cell culture medium is removed and wells are washed with 200 ⁇ l of phosphate-buffered saline. To each well Passive Lysis buffer (Promega, WI) is added and the plates are incubated for 30 min with rocking to lyse the cells. Luciferin solution (50 ⁇ l, Promega) is added, and luciferase activity is measured with a Victor II luminometer (Perkin-Elmer).
- Luciferin solution 50 ⁇ l, Promega
- RNA extractions can be performed using the CellsDirect kit (Invitrogen), and the HCV RNA copy number can be measured using the SuperScript III Platinum One-Step qRT-PCR system (Invitrogen) and primers specific to the HCV 5′ nontranslated region.
- Cytotoxicity can be determined by the 3-[4,5-dimethythiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay as follows. Replicon cells is plated in 96-well plates (4000 cells per well), the next day compound dilutions are added as in the activity assay, and the cells are grown in the presence of the inhibitors for 4 days.
- MTT 3-[4,5-dimethythiazol-2-yl]-2,5-diphenyltetrazolium bromide
- the MTT solution is diluted in DMEM containing 5% FBS and 60 ⁇ l of the solution is added to the cells. After 4 hrs, the cells are solubilized by the addition of 30 ⁇ l SDS (20% in 0.02 N HCl). The plates are incubated overnight and the optical density can be measured at 570 nm. To determine compounds' EC 50 and TD 50 , luciferase, RNA inhibition and MTT data can be analyzed using the GraphPad Prism 4 software (equation: sigmoidal dose-response variable slope).
- Mutations detected in resistance selection studies can be introduced into wild type transient replicon constructs based on genotypes 1a-H77 and 1b-N. Both replicons are bicistronic sub-genomic constructs containing a firefly luciferase reporter similar to those described above, but they do not contain a Neo selectable marker and are therefore only suitable for transient replication assays.
- the 1a-H77 replicon for transient assays further differs from the replicon in the stable cell line in that it contains NS2 through NS5B in the second cistron.
- the 1b-N strain replicon contains NS3 through NS5B in the second cistron, with adaptive mutations E1202G in NS3 and S2204I in NS5A.
- Mutagenesis can be performed using the Stratagene QuikChange XL 11 site-directed mutagenesis kit. Mutants' sequences can be confirmed, plasmids can be linearized with Xba I restriction enzyme and used as template for in vitro transcription reactions to make mutant replicon RNA for transient transfections. In vitro transcription can be performed with the T7 Megascript kit (Ambion).
- Transient replicon transfections can be performed essentially as described by Mo et al. ( Antimierob Agents Chemother (2005) 49(10):4305-4314) with slight modifications. Fifteen micrograms of template RNA can be used to electroporate 3 ⁇ 10 6 cells in a 200 ⁇ l volume in a 0.2 cm cuvette. The cells used for transient transfections can be Huh7 cells obtained by curing replicon-containing cells with IFN (Mo et al., supra). Electroporation can be done with a Gene Pulser II (Bio-Rad, CA) at 480V and 25 ⁇ F, using two manual pulses.
- Gene Pulser II Bio-Rad, CA
- Transfected cells can be diluted to 7.5 ⁇ 10 cells/ml and plated in 96 well plates at 7.5 ⁇ 10 3 cells per well in DMEM with 5% FBS and 100 IU/ml penicillin, 100 mg/ml streptomycin (Invitrogen). Four hours post-transfection, one plate is harvested for luciferase measurement; this plate may provide a measure of the amount of input RNA that can be translated, and thus of transfection efficiency. To the remaining plates, test compound serial dilutions in DMSO can be added (0.5% DMSO final concentration), and plates are incubated for 4 days.
- Exemplary compounds of the present invention were tested for their anti-HCV activities. Many of the compounds tested showed unexpected anti-HCV activities, including excellent activities in biochemical assays against HCV proteases representing various HCV genotypes, superior activities in standard HCV replicon assays including activity against 1a-H77 and 1b-con1 HCV strains in the absence or presence of 40% human plasma, and/or excellent activities in transient replicon assays against drug-resistant mutants in a number of different HCV genetic backgrounds.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
The present invention relates to novel fluorinated macrocyclic compounds and methods of treating a hepatitis C infection in a subject in need of such therapy with said macrocyclic compounds. The present invention further relates to pharmaceutical compositions comprising the compounds of the present invention, or pharmaceutically acceptable salts, esters, or prodrugs thereof, in combination with a pharmaceutically acceptable carrier or excipient.
Description
- This application claims the benefit of U.S. Provisional application 61/428,446, filed on Dec. 30, 2010. The entire contents of the above application are incorporated herein by reference.
- Inventions described in this application were made by or on behalf of Abbott Laboratories and Enanta Pharmaceuticals, Inc. whom are parties to a joint research agreement, that was in effect on or before the date such inventions were made and such inventions were made as a result of activities undertaken within the scope of the joint research agreement.
- The present invention relates to novel fluorinated macrocycles having activity against the hepatitis C virus (HCV) and useful in the treatment of HCV infections. More particularly, the invention relates to macrocyclic compounds, compositions containing such compounds and methods for using the same, as well as processes for making such compounds.
- HCV is the principal cause of non-A, non-B hepatitis and is an increasingly severe public health problem both in the developed and developing world. It is estimated that the virus infects over 200 million people worldwide, surpassing the number of individuals infected with the human immunodeficiency virus (HIV) by nearly five fold. HCV infected patients, due to the high percentage of individuals inflicted with chronic infections, are at an elevated risk of developing cirrhosis of the liver, subsequent hepatocellular carcinoma and terminal liver disease. HCV is the most prevalent cause of hepatocellular cancer and cause of patients requiring liver transplantations in the western world.
- There are considerable barriers to the development of anti-HCV therapeutics, which include, but are not limited to, the persistence of the virus, the genetic diversity of the virus during replication in the host, the high incident rate of the virus developing drug-resistant mutants, and the lack of reproducible infectious culture systems and small-animal models for HCV replication and pathogenesis. In a majority of cases, given the mild course of the infection and the complex biology of the liver, careful consideration must be given to antiviral drugs, which often have significant side effects.
- The present invention relates to novel macrocyclic compounds and methods of treating a hepatitis C infection in a subject in need of such therapy with said macrocyclic compounds. The compounds of the present invention interfere with the life cycle of the hepatitis C virus and are useful as antiviral agents. The present invention further relates to pharmaceutical compositions comprising the compounds of the present invention, or pharmaceutically acceptable salts, esters or prodrugs thereof, in combination with a pharmaceutically acceptable carrier or excipient.
- In one aspect, the invention provides a compound of formula I:
-

- or pharmaceutically acceptable salts, esters, hydrates, solvates, polymorphs, or prodrugs thereof,
- wherein:
- X is an optionally substituted carbocyclic, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl;
-
- Z is O, S(O)m, NRx, OC(O), C(O), C(O)O, NRxC(O), or C(O)NRx;
- each Rx is independently H or alkyl;
- J is —C(O)—, —O—C(O)—, —C(O)O—, —N(R3)—C(O)—, —C(S)—, —C(═NR4)—, —S(O)—, or —S(O2)— or absent;
- A is selected from the group consisting of the following:
-
- (i) H, aryl; substituted aryl; heteroaryl; substituted heteroaryl;
- (ii) heterocyclyl or substituted heterocyclyl; and
- (iii) —C1-C8 alkyl, —C2-C8 alkenyl or —C2-C8 alkynyl, each containing 0, 1, 2, or 3 heteroatoms independently selected from O, S, and N; substituted —C1-C8 alkyl, substituted —C2-C8 alkenyl or substituted —C2-C8 alkynyl, each containing 0, 1, 2, or 3 heteroatoms independently selected from O, S and N; —C3-C12 carbocyclic, substituted —C3-C12 carbocyclic; —C3-C12 cycloalkenyl, or substituted —C3-C12 cycloalkenyl;
- E is -G-R5;
-
- wherein G is absent; optionally substituted alkylene, optionally substituted alkenylene, optionally substituted alkynylene, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N;
- or —O—, —S—, —N(R3)—, —N(R3)S(Op)—, —N(R3)C(O)—, —N(R3) C(O)S(Op)—, —OS(Op)—, —C(O)S(Op)—, or —C(O)N(R3)S(Op)—;
- each p is independently 0, 1, or 2;
- R5 is H; optionally substituted alkyl, optionally substituted alkenyl, or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl;
- denotes a carbon-carbon single or double bond (i.e.,

-

- means
-

- each of Ra, Rb, Rc, and Rd are each independently H or halo, wherein at least two of Ra, Rb, Rc, and Rd are halo; and Rb and Rd are absent ifis a double bond
- each R3 and R4 is independently selected at each occurrence from the following: H, optionally substituted alkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; and optionally substituted carbocyclic;
- L is absent or a C2-C5 saturated or unsaturated chain, optionally containing one to three heteroatoms independently selected from O, N and S(O)q, wherein L is optionally substituted with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl and C2-C6alkynyl, wherein each C1-C6alkyl, C2-C6alkenyl and C2-C6alkynyl, group is optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, formyl and cyano;
- j=independently 0, 1, 2, 3, or 4;
- k=independently 0, 1, 2, or 3;
- each m is independently 0, 1, or 2; and
- q is independently 0, 1, or 2.
- The present invention also features the compounds of Formula I′, and pharmaceutically acceptable salts, esters, hydrates, solvates, polyrnorphs, or prodrugs thereof,
-
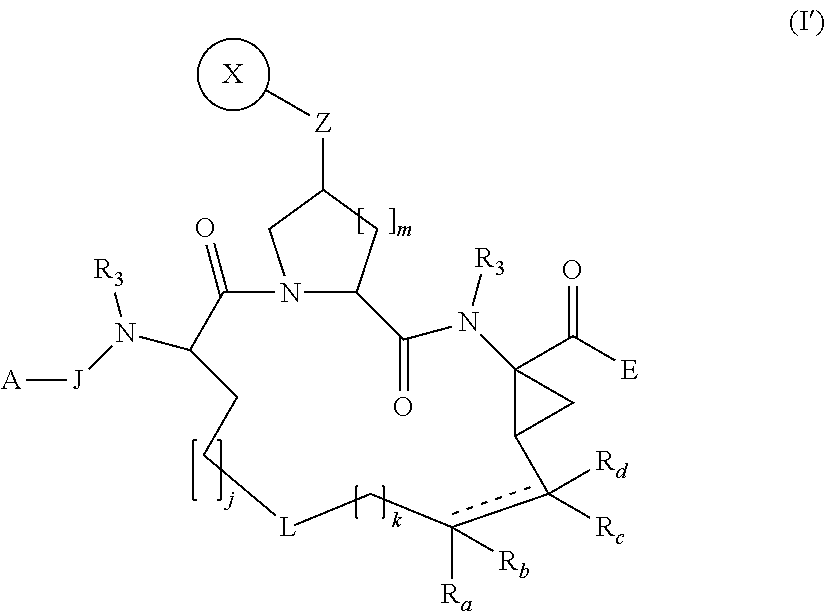
- wherein Z, X, A, L, R3, J, E, j, k, Ra, Rb, Rc, and Rd, are as defined in Formula I.
- In another aspect, the invention provides a pharmaceutical composition comprising a compound of any of the formulae herein (e.g., Formula I or I′), or a pharmaceutically acceptable salt, ester, or prodrug thereof, in combination with an excipient.
- In one aspect, the invention provides a method of treating a viral infection in a subject, comprising administering to the subject a therapeutically effective amount of a compound of any of the formulae herein (e.g., Formula I or I′, or a pharmaceutically acceptable salt, ester or prodrug thereof; or a pharmaceutical composition comprising the same.
- In one aspect, the invention provides a compound of formula I:
-
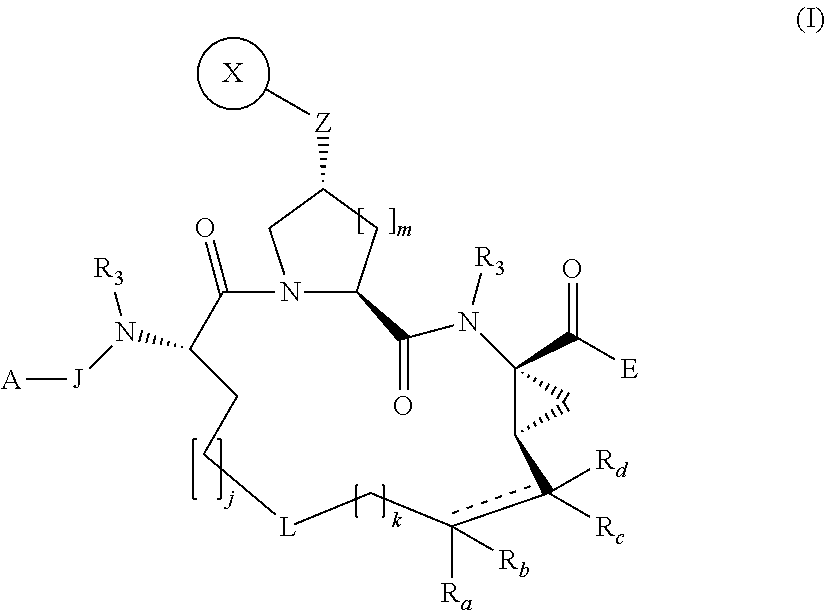
- or pharmaceutically acceptable salts, esters, hydrates, solvates, polymorphs, or prodrugs thereof,
- wherein:
- X is an optionally substituted carbocyclic, optionally substituted heterocycyl, optionally substituted aryl, or optionally substituted heteroaryl;
-
- Z is O, S(O)m, NRA, OC(O), C(O), C(O)O, NRxC(O), or C(O)NRx;
- each Rx is independently H or alkyl;
- J is —C(O)—, —O—C(O)—, —C(O)O—, —N(R3)—C(O)—, —C(S)—, —C(═NR4)—, N(R3)—, —S(O)—, or —S(O2)— or absent;
- A is selected from the group consisting of the following:
-
- (i) H, aryl; substituted aryl; heteroaryl; substituted heteroaryl;
- (ii) heterocyclyl or substituted heterocyclyl; and
- (iii) —C1-C8 alkyl, —C2-C8 alkenyl or —C2-C8 alkynyl, each containing 0, 1, 2, or 3 heteroatoms independently selected from O, S, and N; substituted —C1-C8 alkyl, substituted —C2-C8 alkenyl or substituted —C2-C8 alkynyl, each containing 0, 1, 2, or 3 heteroatoms independently selected from O, S and N; —C3-C12 carbocyclic, substituted —C3-C12 carbocyclic; —C3-C12 cycloalkenyl, or substituted —C1-C12 cycloalkenyl;
- E is -G-R5;
-
- wherein G is absent; optionally substituted alkylene, optionally substituted alkenylene, optionally substituted alkynylene, each containing 0, 1, or 3 heteroatoms selected from O, S, or N;
- or —O—, —S—, —N(R3)—, —N(R3)S(Op)—, —N(R3)C(O)—, —N(R3) C(O)S(O)—, —OS(Op)—, —C(O)S(Op)—, or —C(O)N(R3)S(Op)—;
- each p is independently 0, 1, or 2;
- R5 is U; optionally substituted alkyl, optionally substituted alkenyl, or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl;
- denotes a carbon-carbon single or double bond (i.e.,
-

- means
-

- each of Ra, Rb, Rc, and Rd are each independently H or halo, wherein at least two of Ra, Rb, Rc, and Rd are halo; and Rb and Rd are absent ifis a double bond
- each R3 and R4 is independently selected at each occurrence from the following: H, optionally substituted alkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; and optionally substituted carbocyclic;
- L is absent or a C2-C5 saturated or unsaturated chain, optionally containing one to three heteroatoms independently selected from O, N and S(O)q, wherein L is optionally substituted with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl and C2-C6alkynyl, wherein each C1-C6alkyl, C2-C6alkenyl and C2-C6alkynyl, group is optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, formyl and cyano;
- j=independently 0, 1, 2, 3, or 4;
- k=independently 0, 1, 2, or 3;
- each m is independently 0, 1, or 2; and
- q is independently 0, 1, or 2.
- The present invention also features the compounds of Formula I′, and pharmaceutically acceptable salts, esters, hydrates, solvates, polymorphs, or prodrugs thereof,
-
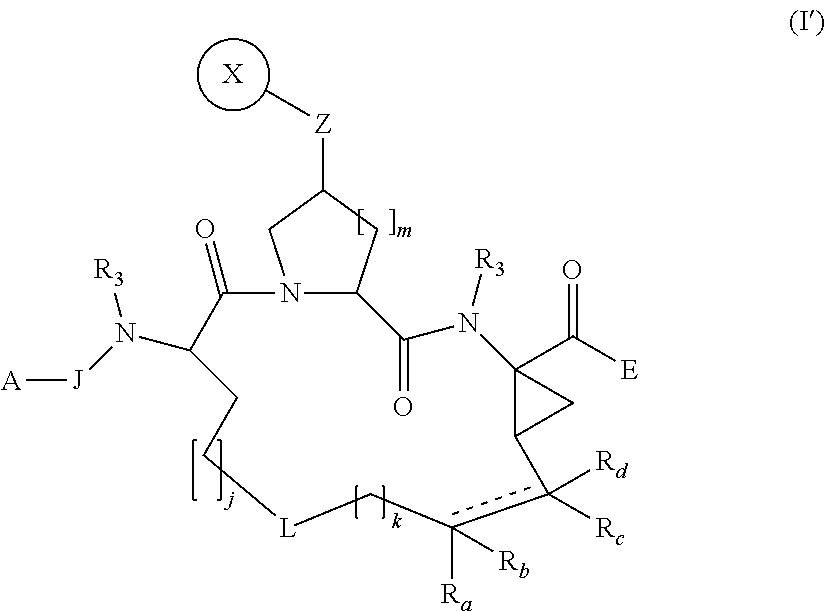
- wherein Z, X, A, L, R3, J, E, j, k, in, Ra, Rb, Rc, and Rd, are as defined in Formula I.
- In certain embodiments, Z is O or OC(O); and ring X is cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, adamantyl, cyclooctyl, [3.3.0]bicyclooctanyl, [4.3.0]bicyclononanyl, [4.4.0]bicyclodecanyl, [2.2.2]bicyclooctanyl, fluorenyl, phenyl, naphthyl, indanyl, tetrahydronaphthyl, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetraz benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, NH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H,6H-1,5,2-dithiazinyl, dihydrofuro [2,3-h]tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, 1H-indazolyl, indolenyl, indolinyl., indolizinyl, indolyl, isoindolinyl, isoindolenyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isoxazolyl, morpholinyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-oxadiazolyl; 1,2,5oxadiazolyl, 1,3,4-oxadiazolyl, oxazolidinyl, oxazolyl, oxazolidinyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyridyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, pyrrolyl, quinazolinyl, quinolinyl, 4H-quinolizinyl, quinoxalinyl, quinuclidinyl, tetrahydrofuranyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, 6H-1,2,5-thiadiazinyl, 1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,3,4thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl, 1,2,3-triazolyl, 1,2,4-triazolyl, 1,3,4-triazolyl, or xanthenyl; each of which may be optionally substituted.
- In a further embodiment, ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl.
- In a further embodiment, ring X is
-
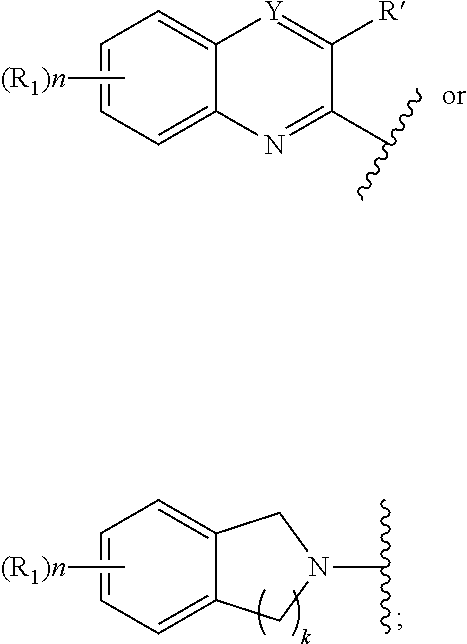
- wherein
- Y is N or —C(R″)—;
- wherein if Y is —C(R″)—, then R′ and R″ taken together with the carbon atoms to which they are attached form a ring, which is optionally substituted;
- R′ is H, alkyl, alkenyl, alkynyl, carbocyclic, heterocyclyl, aryl, or heteroaryl, each of which may be optionally substituted;
- each R1 is independently selected from halogen, hydroxy, amino, —CN, —CF3, —NO2, —OR4, —SR4, —SOR4, —SO2R4, —N(R3)S(O)2—R4, —N(R3)(SO2)NR3R4, —NR3R4, —C(O)—OR1, —C(O)R4, —C(O)NR3R4, —N(R3)C(O)R4; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl, or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N;
- each n is independently 0, 1, 2, 3, or 4;
- each R3 and R4 are independently selected at each occurrence from the following: optionally substituted alkyl., optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen; and
-
- k is 0, 1, or 2.
- In another aspect, the invention provides a compound of formula I-A:
-
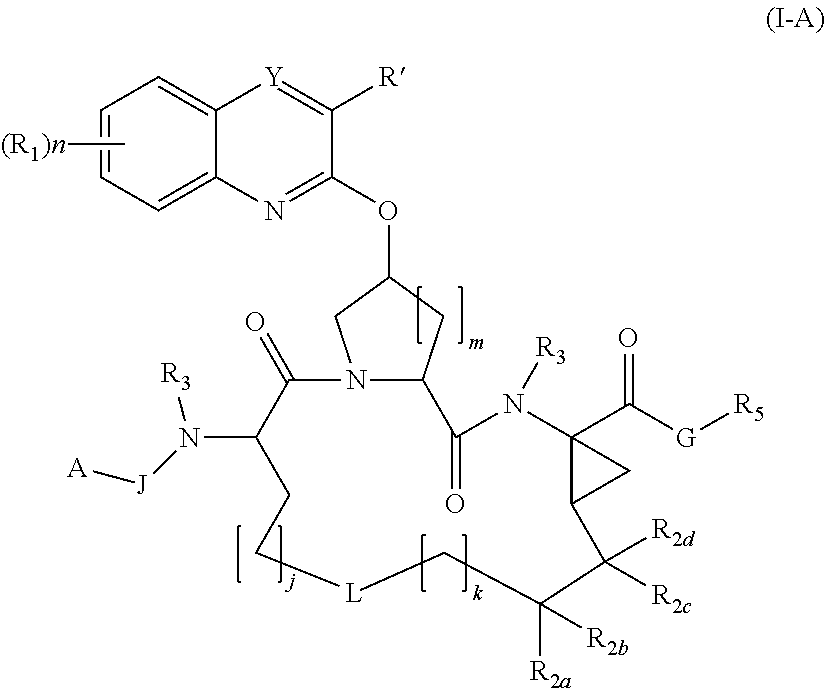
- or a pharmaceutically acceptable salt, ester or prodrug thereof,
- wherein:
- J is —C(O)—, —O—C(O)—, N(R3)—C(O)—, —C(S)—, —C(═NR4)—, —N(R3)—, —S(O)—, or —S(O2)— or absent;
- A is optionally substituted alkyl, optionally substituted heteroaryl, optionally substituted aryl, optionally substituted heterocyclic, or optionally substituted carbocyclic;
- Y is N or —C(R″)—;
- R′ is H, optionally substituted alkyl, optionally substituted aryl, optionally substituted heteroaryl; optionally substituted carbocyclic, or optionally substituted heterocyclic; or
- R′ and R″, together with the atoms to which each is attached, can form a ring;
- each R1 is independently selected from H, halogen, hydroxy, amino, —CN, —CF3, —N3, —NO2, —OR4, —SR4., —SOR4, —SO2R4, —N(R3)S(O)2—R4, —N(R3)(SO2)NR3R4, —NR3R4, —C(O)—OR4, —C(O)R4, —C(O)NR3R4, —N(R3)C(O)R4; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl, or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N;
- G is —N(R3)S(O)p—; —N(R3)C(O)—, —N(R3)C(O)S(O), —OS(O)p—, —C(O)S(O)p, or —C(O)N(R3)S(O)p—;
- each p is independently 0, 1, or 2;
- R5 is FE, optionally substituted alkyl, optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl; each of R2a, R2b, Ra, and R2d are each independently H or F, wherein at least two of R2a, R2b, R2c, and R2d are F;
- each R3 and R4 are independently selected at each occurrence from the following: optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen;
- L is absent or is selected from optionally substituted alkylene, optionally substituted alkenylene or optionally substituted alkynylene, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N;
- j=independently 0, 1, 2, 3, or 4;
- k=independently 0, 1, 2, or 3;
- m=independently 0, 1, or 2; and
- n is independently 0, 1, 2, 3, or 4.
- In certain embodiments of this aspect, Y is —C(R″)—; and R′ and R″, taken together with the atoms to which each is attached, form a ring.
- In a further embodiment, the ring formed by R′ and R″ is an optionally substituted an optionally substituted heteroaryl, an optionally substituted carbocyclic or an optionally substituted heterocyclic.
- In other embodiments, Y is N and R′ is H, optionally substituted alkyl, optionally substituted aryl, or optionally substituted heteroaryl.
- In certain embodiments, G is —N(R3)S(O)p—; —N(R3)C(O)—, —OS(O)p—, or —C(O)S(O)p—; and p is 2.
- In a further embodiment, R5 is optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl. In certain embodiments, R5 is carbocyclic.
- In various embodiments, R2a and R2h are F. In other embodiments, R2c and R2d are F. In still other embodiments, R2a and R2c are F.
- In another embodiments, J is —C(O)—, —O—C(O)—, —S(O)—, or —S(O2)—.
- In other embodiments, A is an optionally substituted alkyl, optionally substituted heteroaryl, or optionally substituted aryl. In certain embodiments, A is heteroaryl or alkyl.
- In yet another aspect, the invention provides a compound of formula I-A, or a pharmaceutically acceptable salt, ester or prodrug thereof,
- wherein: Y is —C(R″),
- R′ and R″, taken together with the atoms to which each is attached, form a phenyl which is optionally substituted with one or more R1;
- m is 1, j is 2, k is 2, L is absent;
- J is —C(O)—, —O—C(O)—, N(R3)—C(O)—, —S(O)—, or —S(O2)—;
- A is optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted heteroaryl, optionally substituted aryl, optionally substituted heterocyclic, or optionally substituted carbocyclic;
- each R1 is independently selected from H, halogen, hydroxy, amino, —CN, —CF3, —N3, —NO2, —OR4, —SR4, —NR3R4, optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; optionally substituted haloalkyl, or optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl;
- G is —N(R3)S(O)p—;
- p is 0, 1, or 2;
- each of R2a, R2b, R2c and R2d are each independently H or F, wherein at least two of R2a, R2b, R2c, and R2d are F;
- R5 is optionally substituted carbocyclic or heterocyclic;
- each R3 and R4 is independently selected at each occurrence from the following: optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen; and
- n is 0, 1, 2, 3, or 4.
- In one embodiment of this aspect. J is —C(O)—, or —O—C(O)—.
- In another embodiment of this aspect, A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl, phenyl, pyridyl, pyrimidyl, pyrazinyl, triazinyl, furanyl, thiophenyl, pyrrolyl, isoxazolyl, or pyrazolyl; each of which may be optionally substituted.
- In certain embodiments of this aspect, G is —N(R3)S(O)p— and R5 is cyclopropyl, cyclobutyl, cyclopentyl, cyciohexyl, or cycloheptyl, each of which is optionally substituted.
- In one embodiment of aspect, R2a and R2b are F. In another embodiment, R2a and R2c are F. In another embodiment, R2c and R2d are F.
- In still another embodiment of this aspect, A-J- is A-C(O)— or A-O—C(O)—; A is Cr C8alkyl, C2-C8alkenyl or C2-C8alkynyl, each of which is optionally substituted with one or more RH, or A is 5- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more RG; -G-R5 is —N(R3)S(O)2—R5; and R5 is 3- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more RG. Each RH is independently selected at each occurrence from independently selected at each occurrence from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, or cyano; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in RH is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Each R1 is independently selected at each occurrence from hydrogen or RG. Each RG is independently selected at each occurrence from halo, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C3-C6carbocycle or 3- to 6-membered heterocycle, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Each R3 is independently selected at each occurrence from hydrogen; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl., C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Preferably, R3 is hydrogen.
- In still another embodiment of this aspect, A-J- is A-C(O)— or A-O—C(O)—; A is 5- to 6-membered aryl or heteroaryl which is optionally substituted with one or more RG; -G-R5 is —N(R3)S(O)2—R5 (preferably —N(H)S(O)2—R5); and R5 is 3- to 6-membered cycloalkyl (preferably cyclopropyl) which is optionally substituted with one or more RG. Each R1 is independently selected at each occurrence from hydrogen or RG. RG is as defined above. Each R3 is independently selected at each occurrence from hydrogen; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Preferably, R3 is hydrogen.
- In still another aspect, the invention provides a compound of formula I-A, or a pharmaceutically acceptable salt, ester or prodrug thereof, wherein:
- Y is —C(R″)—;
- R′ and R″, taken together with the atoms to which each is attached, form a phenyl which is optionally substituted with one or more R1;
-
- m is 1, j is 2, k is 2, L is absent;
- J is —C(O)— or —O—C(O)—;
- A is optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted heteroaryl, optionally substituted aryl, optionally substituted heterocyclic, or optionally substituted carbocyclic;
- each R1 is independently selected from H, halogen, hydroxy, amino, —CN, —CF3, —N3, —NO2, —OR4, —SR4, —NR3R4, optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; optionally substituted haloalkyl, or optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl;
- G is —N(R3)S(O)p—;
- p is 0, 1., or 2;
- R5 is optionally substituted carbocyclic or heterocyclic;
- R2a and R2b are F, and R2 and R2d are H;
- each R3 and R4 is independently selected at each occurrence from the following: optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen; and
- n is 0, I, 2, 3, or 4.
- In certain embodiments, A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl,
-
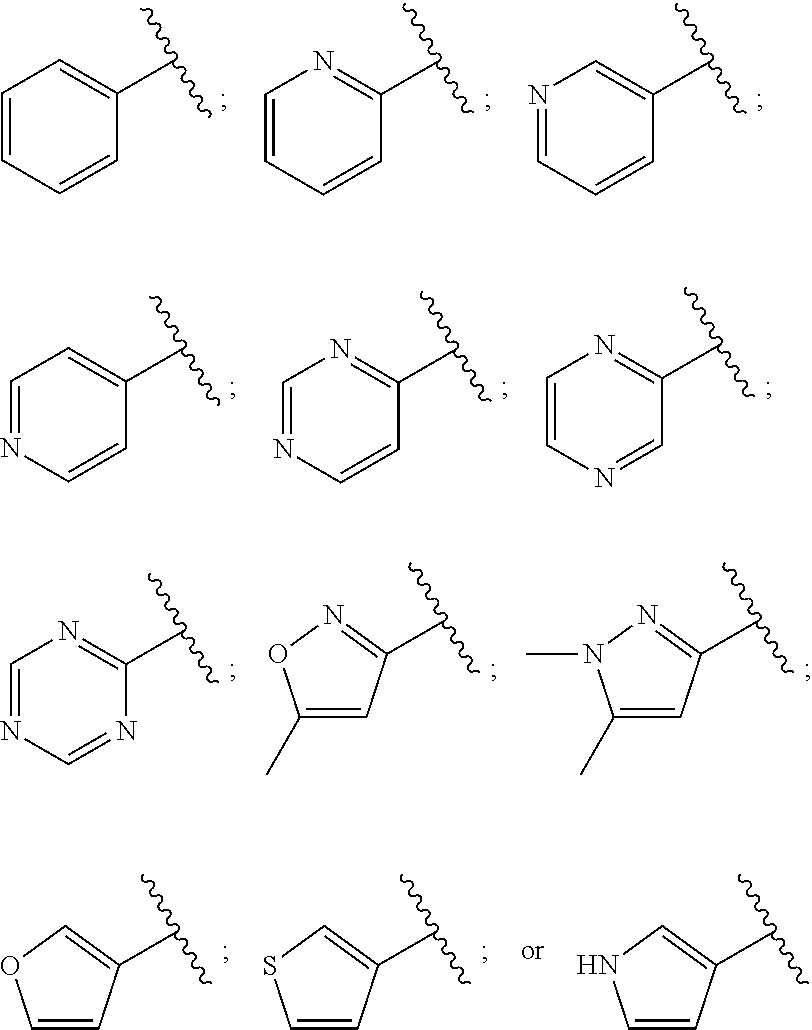
- In another embodiment, R5 is optionally substituted cyclopropyl.
- In various embodiments, n is 0.
- In still another embodiment of this aspect, A-J- is A-C(O)— or A-O—C(O)—; A is C1-C8alkyl, C2-C8alkenyl or C2-C8alkynyl, each of which is optionally substituted with one or more RH, or A is 5- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more RG; -G-R5 is —N(R3)S(O)2—R5; and R5 is 3- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more RG. Each RH is independently selected at each occurrence from independently selected at each occurrence from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, or cyano; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in RH is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Each R1 is independently selected at each occurrence from hydrogen or RG. Each RG is independently selected at each occurrence from halo, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C3-C6carbocycle or 3- to 6-membered heterocycle, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Each R3 is independently selected at each occurrence from hydrogen; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-meinbered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Preferably, R3 is hydrogen.
- In still another embodiment of this aspect, A-J- is A-C(O)— or A-O—C(O)—; A is 5- to 6-membered aryl or heteroaryl which is optionally substituted with one or more RG; -G-R5 is —N(R3)S(O)2—R5 (preferably —N(H)S(O)2—R5); and R5 is 3- to 6-membered cycloalkyl (preferably cyclopropyl) which is optionally substituted with one or more RG. Each R1 is independently selected at each occurrence from hydrogen or RG. RG is as defined above. Each R3 is independently selected at each occurrence from hydrogen; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Preferably, R3 is hydrogen.
- In a further aspect, the invention provides a compound of formula II:
-
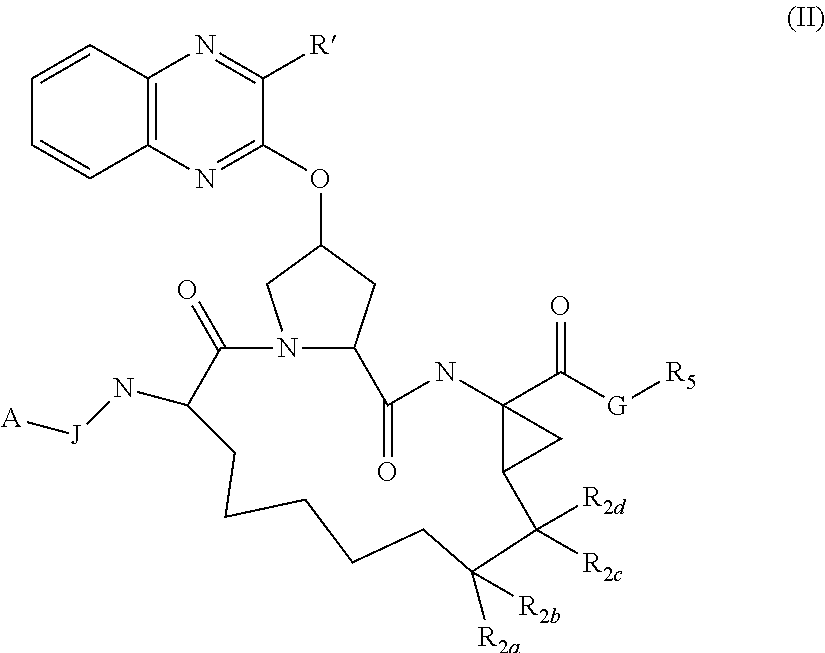
- or a pharmaceutically acceptable salt, ester or prodrug thereof,
- wherein:
- J is —C(O)—, —O—C(O)—, —N(R3)—C(O)—, —S(O)—, or —S(O2)—;
- A is optionally substituted alkyl, or optionally substituted heteroaryl, optionally substituted aryl, optionally substituted heterocyclic, or optionally substituted carbocyclic;
- R′ is H, optionally substituted aryl; optionally substituted heteroaryl; or optionally substituted alkyl;
- G is —N(R3)S(O)p—;
- p is 0, 1, or 2;
- each of R2a, R2b, R2c, and R2d are each independently H or F, wherein at least two of R2a, R2b, R2c, and R2d are F;
- R5 is optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl; and
- each R3 and R4 is independently selected at each occurrence from the following: optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen.
- In certain embodiments, R′ is H, optionally substituted alkyl; optionally substituted aryl; or optionally substituted heteroaryl.
- In another embodiment, J is —C(O)—, or —O—C(O),
- In other embodiments, A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl, phenyl, pyridyl, pyrimidyl, pyrazinyl, triazinyl, furanyl, thiophenyl, pyrrolyl, isoxazolyl, or pyrazolyl; each of which may be optionally substituted.
- In various embodiments, G is —N(R3)S(O)p— and R5 is cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, each of which is optionally substituted.
- In another embodiment, R2a and R2b are F. In another embodiment, R2a and R2c, are F. In another embodiment, R2c and R2d are F.
- In still another embodiment of this aspect, A-J- is A-C(O)— or A-O—C(O)—; A is C1-C8alkyl, C2-C8alkenyl or C2-C8alkynyl, each of which is optionally substituted with one or more RH, or A is 5- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more RG; -G-R5 is —N(R3)S(O)2—R5; and R5 is 3- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more RG. Each RH is independently selected at each occurrence from independently selected at each occurrence from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, or cyano; or C1-C6alkyl, C1-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in RFI is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Each R1 is independently selected at each occurrence from hydrogen or RG. Each RG is independently selected at each occurrence from halo, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C3-C6carbocycle or 3- to 6-membered heterocycle, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Each R3 is independently selected at each occurrence from hydrogen; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Preferably, R3 is hydrogen.
- In still another embodiment of this aspect, A-J- is A-C(O)— or A-O—C(O)—; A is 5- to 6-membered aryl or heteroaryl which is optionally substituted with one or more RG; -G-R5 is —N(R3)S(O)2—R5 (preferably —N(H)S(O)2—R5); and R5 is 3- to 6-membered cycloalkyl (preferably cyclopropyl) which is optionally substituted with one or more RG. Each R1 is independently selected at each occurrence from hydrogen or RG. RG is as defined above. Each R3 is independently selected at each occurrence from hydrogen; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Preferably, R3 is hydrogen.
- In another aspect, the invention provides a compound of formula II-a:
-

- or a pharmaceutically acceptable salt, ester or prodrug thereof;
- wherein:
- J is —C(O)— or —O—C(O)—;
- A is optionally substituted alkyl, or optionally substituted heteroaryl, optionally substituted aryl, optionally substituted heterocyclic, or optionally substituted carbocyclic;
- R′ is H, optionally substituted alkyl; optionally substituted aryl; or optionally substituted heteroaryl;
- G is —N(R3)S(O)p—;
- p is 0, 1, or 2;
- R5 is optionally substituted carbocyclic; and
- each R3 is optionally substituted alkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen.
- In one embodiment, R′ is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, phenyl, pyridyl, pyrimidyl, pyrazinyl, triazinyl, furanyl, thiophenyl, pyrrolyl, isoxazolyl, or pyrazolyl; each of which may be optionally substituted.
- In another embodiment, R′ is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl,
-
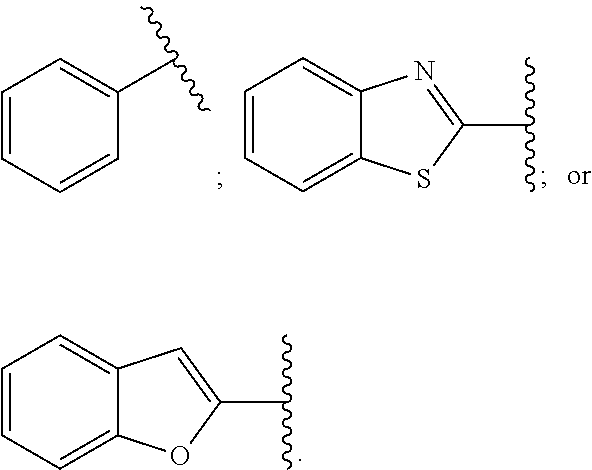
- In another embodiment, A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl, phenyl, pyridyl, pyrimidyl, pyrazinyl, triazinyl, furanyl, thiophenyl, pyrrolyl, isoxazolyl, or pyrazolyl; each of which may be optionally substituted.
- In a further embodiment, A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl,
-
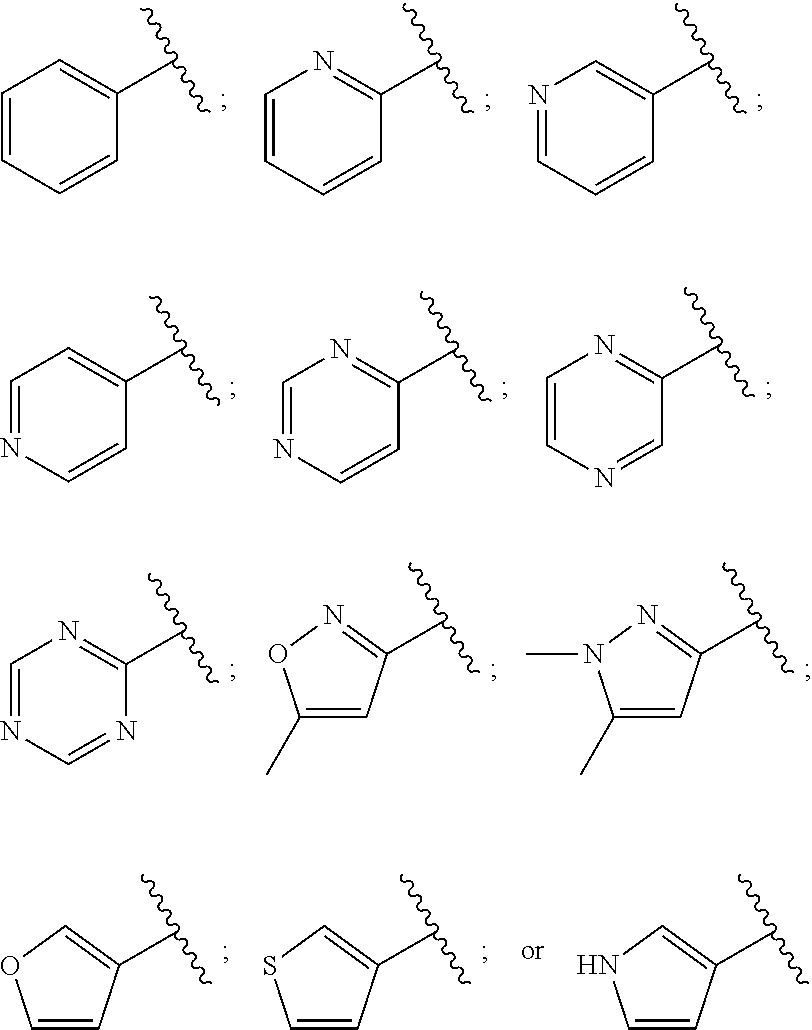
- In other embodiments, (1 is —N(R3)S(O)p— and R5 is cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, each of which is optionally substituted.
- In a further embodiment, R5 is optionally substituted cyclopropyl.
- In still another embodiment of this aspect, A-J- is A-C(O)— or A-O—C(O)—; A is C1-C8alkyl, C2-C8alkenyl or C2-C8alkynyl, each of which is optionally substituted with one or more RH, or A is 5- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more RG; -G-R5 is —N(R3)S(O)2—R5; and R5 is 3- to 6-membered carbocyclic or heterocyclic which is optionally substituted with one or more RG. Each RH is independently selected at each occurrence from independently selected at each occurrence from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, or cyano; or C1-C6alkyl, C2-C6alkenyl or C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in RH is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Each R1 is independently selected at each occurrence from hydrogen or RG. Each RG is independently selected at each occurrence from halo, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano; or C3-C6carbocycle or 3- to 6-membered heterocycle, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Each R3 is independently selected at each occurrence from hydrogen; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Preferably, R3 is hydrogen.
- In still another embodiment of this aspect, is A-C(O)— or A-O—C(O)—; A is 5- to 6-membered aryl or heteroaryl which is optionally substituted with one or more RG; -G-R5 is —N(R3)S(O)2—R5 (preferably —N(H)S(O)2—R5); and R5 is 3- to 6-membered cycloalkyl (preferably cyclopropyl) which is optionally substituted with one or more RG. Each R1 is independently selected at each occurrence from hydrogen or RG. RG is as defined above. Each R3 is independently selected at each occurrence from hydrogen; or C1-C6alkyl, C2-C6alkenyl or C2-C6alkynyl, each of which is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano or 3- to 6-membered carbocycle or heterocycle; or 3- to 6-membered carbocycle or heterocycle; wherein each 3- to 6-membered carbocycle or heterocycle in R3 is independently optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, nitro, oxo, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl, C2-C6alkynyl, C1-C6haloalkyl, C2-C6haloalkenyl or C2-C6haloalkynyl. Preferably, R3 is hydrogen.
- In yet another aspect, the invention provides a compound of formula IV:
-

- wherein,
- Z is OC(O), C(O), C(O)O, or C(O)NRx;
- Rx is H or alkyl;
- k is 0, 1, or 2;
- each R1 is independently selected from the group consisting of:
-
- (i) H, aryl; substituted aryl; heteroaryl; substituted heteroaryl;
- (ii) heterocyclyl or substituted heterocyclyl; and
- (iii) —C1-C8 alkyl, —C2-C8 alkenyl or —C2-C8 alkynyl, each containing 0, 1, or 3 heteroatoms independently selected from O, S, and N; substituted —C1-C8 alkyl, substituted —C2-C8 alkenyl or substituted —C2-C8 alkynyl, each containing 0, 1, 2, or 3 heteroatoms independently selected from O, S and N; —C3-C12 carbocyclic, substituted —C3-C12 carbocyclic; —C3-C12 cycloalkenyl, or substituted —C3-C12 cycloalkenyl;
- n is 0, 1, 2, 3, or 4;
- R6 is selected from aryl; substituted aryl; heteroaryl; substituted heteroaryl; heterocyclyl, substituted heterocyclyl; —C1-C5 alkyl, substituted —C1-C8 alkyl, —C3-C12 carbocyclic, and substituted —C3-C12 carbocyclic;
- G is -E-R5;
-
- wherein E is —N(R3)S(O)p—, —N(R3)C(O)—, or —N(R3)C(O)S(O)p—;
- each p is independently 0, 1, or 2;
- R5 is optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl;
- each of Ra, Rb, Rc, and Rd are each independently H or F, wherein at least two of Ra, Rb, Rc, and Rd are F; and
- R3 is H, optionally substituted alkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; or optionally substituted carbocyclic.
- In another embodiment, the invention provides a compound wherein each R1 is substituted with one, two, three or four independent RE, wherein each RE is independently selected from alkyl, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, halogen, hydroxy, amino, —CN, —CF3, —N3, —NO2, —ORF, —SR, —SORF, —SO2RF, —N(RF)S(O2)—RF, —N(RF) S(O2)NRFRF, —NRFRF, —C(O)ORF, —C(O)RF, —C(O)NRFRF, or —N(RF)C(O)RF; wherein each RF is independently hydrogen, C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl.
- In certain embodiments. E is G-R5; and G is —O—, —S—, —N(R3)—, —N(R3)S(O)p—, —N(R3)C(O)—, —N(R3) C(O)S(Op)—, —OS(Op)—, —C(O)S(Op)—, or —C(O)N(R3)S(Op)—; and each p is independently 0, 1, or 1 In a further embodiment, E is —N(R3)S(O)p—.
- In various embodiments, R5 is H; optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl. In a further embodiment, R5 is cyclopropyl optionally substituted with C1-C6alkyl, C1-C6alkoxyC1-C6alkyl, —C(O)OH, —C(O)NH2, or —C(O)O—C1-C6alkyl.
- In certain embodiments of formulae I and I′,denotes a carbon-carbon single bond. In a further embodiment of each of the above-described aspects of the invention, at least two of Ra, Rb, Rc, and Rd are F. In one embodiment, Ra and Rb are F. In one embodiment of each of the above-described aspects of the invention, Rc and Rd are F. In one embodiment of each of the above-described aspects of the invention, Ra and Rc are F. In another embodiment of each of the above-described aspects of the invention, three of Ra, Rb, Rc, and Rd are F. In one embodiment of each of the above-described aspects of the invention, Ra, Rb, and Rc are F. In one embodiment of each of the above-described aspects of the invention, Ra, Rc, and Rd are F. In another embodiment of each of the above-described aspects of the invention, all of Ra, Rb, Rc, and Rd are F.

- In certain embodiments for formulae I and I′,denotes a carbon-carbon double bond. In certain embodiments of each of the above-described aspects of the invention, Ra and Rc are F.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)—; and A is optionally substituted heteroaryl.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)—; and A is optionally substituted aryl.
- In another embodiment of each of the above-described aspects of the invention J is —(C═O)—; and A is optionally substituted alkyl.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)—; A is optionally substituted alkyl; Z is O, ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)—; A is optionally substituted aryl; Z is O, ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention. J is —(C═O)—; A is optionally substituted heteroaryl; Z is O, ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)—; A is optionally substituted alkyl; Z is O, ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O—; A is optionally substituted aryl; Z is O, ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O—; A is optionally substituted heteroaryl; Z is O, ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O— or —(C═O)—; A is optionally substituted alkyl; Z is O or (C═O), ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and Ra and Rb are F, and Rc and Rd are H.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O— or —(C═O)—; A is optionally substituted alkyl; Z is O or (C═O), ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and Ra and Rd are F, and Rc and Rd are H.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O— or —(C═O)—; A is optionally substituted alkyl; Z is O or (C═O), ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and Ra and Rb are H, and Rc, and Rd are F.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O— or —(C═O)—; A is optionally substituted aryl; Z is O or (C═O), ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and Ra and Rb are F, and Rc and Rd are H.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O— or —(C═O)—; A is optionally substituted aryl; Z is O or (C═O), ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and Ra and Rc are F, and Rb and Rd are H.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O— or —(C═O)—; A is optionally substituted aryl; Z is O or (C═O), ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and Ra and Rb, are H, and Rc and Rd are F.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O— or —(C═O)—; A is optionally substituted heteroaryl; Z is O or (C═O), ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and Ra and Rb are F, and Rc and Rd are H.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O— or —(C═O)—; A is optionally substituted heteroaryl; Z is O or (C═O), ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and Ra and Rc are F, and Rb and Rd are H.
- In another embodiment of each of the above-described aspects of the invention, J is —(C═O)O— or —(C═O)—; A is optionally substituted heteroaryl; Z is O or (C═O), ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl; and Ra and Rb are H, and Rc and Rd are F.
- In another embodiment of each of the above-described aspects of the invention, E is —NHS(O)2R5; R5 is independently cyclopropyl optionally substituted with C1-C6alkyl; J is —(C═O)—; A is optionally substituted alkyl, Z is O; ring X is phenanthridinyl or quinoxalinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, E is —NHS(O)2R5; R5 is independently cyclopropyl optionally substituted with C1-C6alkyl; J is —(C═O)—; A is optionally substituted aryl, Z is O; ring X is phenanthridinyl or quinoxalinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, E is —NHS(O)2—R5; R5 is independently cyclopropyl optionally substituted with C1-C6alkyl; J is —(C═O)—; A is optionally substituted heteroaryl, Z is O; ring X is phenanthridinyl or quinoxalinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, E is —NHS(O)2—R5; R5 is independently cyclopropyl optionally substituted with C1-C6alkyl; J is —(C═O)O—; A is optionally substituted alkyl, Z is O; ring X is phenanthridinyl or quinoxalinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, E is —NHS(O)2—R5; R5 is independently cyclopropyl optionally substituted with C1-C6alkyl; J is —(C═O)O—; A is optionally substituted aryl, Z is O; ring X is phenanthridinyl or quinoxalinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, E is —NHS(O)2—R5; R5 is independently cyclopropyl optionally substituted with C1-C6alkyl; J is —(C═O)O—; A is optionally substituted heteroaryl, Z is O; ring X is phenanthridinyl or quinoxalinyl; and any two of Ra, Rb, Rc, and Rd are each independently F.
- In another embodiment of each of the above-described aspects of the invention, A is an optionally substituted nitrogen-containing heteroaryl.
- In another embodiment of each of the above-described aspects of the invention, A is optionally substituted isoxazolyl, optionally substituted thiazolyl, optionally substituted pyrazolyl, optionally substituted pyrazinyl, optionally substituted pyridinyl, or optionally substituted pyrimidinyl.
- In another embodiment of each of the above-described aspects of the invention, A is alkyl-substituted isoxazolyl.
- In another embodiment of each of the above-described aspects of the invention, J can be, for example, —(C═O)— or —(C═O)O—; and A is selected from the following groups:
-
- Representative compounds include, but are not limited to, the following compounds:
-
N-((2R,6S,13aS,14aR,16aS)-14a- tert-butyl (2R,6S,13aS,14aR,16aS)-14a- (cyclopropyl sulfonylcarbamoyl)-12,12- (cyclopropylsulfonylcarbamoyl)-12,12- difluoro-5,16-dioxo-2-(phenanthridin-6- difluoro-5,16-dioxo-2-(phenanthridin-6- yloxy)octadecahydro yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a] cyclopropa[e]pyrrolo[1,2-a][1,4]diaza [1,4]diazacyclopentactecin-6-ylcarbamate cyclopentadecin-6-yl)-5-methylisoxazole-3- carboxamide (2R,6S,13aS,14aR,16aS)-N-(cyclopropyl (2R,6S,13aS,14aR,16aS)-N-(cyclopropyl sulfonyl)-12,12-difluoro-6-(5- sulfonyl)-6-(1,5-dimethyl-1H-pyrazole-3- methylpyrazine-2-carboxamido)-5,16-dioxo- carboxamido)-12,12-difluoro-5,16-dioxo-2- 2-(phenanthridin-6- (phenanthridin-6-yloxy)octadecahydrocyclo yloxy)octadecahydrocyclopropa[e]pyrrolo[1, propa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine- 2-a][1,4]diazacyclopentadecine-14a- 14a-carboxamide carboxamide tert-butyl (2R,6S,13aS,14aR,16aS)-14a- tert-butyl (2R,6S,13aS,14aR,16aS)-14a- (cyclopropylsulfonylcarbamoyl)-12,12- (cyclopropylsulfonylcarbamoyl)-12,12- difluoro-5,16-dioxo-2-(quinoxalin-2- difluoro-5,16-dioxo-2-(3-phenylquinoxalin- yloxy)octadecahydrocyclopropa[e]pyrrolo[1, 2-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a] 2-a][1,4]diazacyclopentadecin-6-ylcarbamate [1,4]diazacyclopentadecin-6-ylcarbamate tert-butyl (2R,6S,13aS,14aR,16aS)-14a- N-((2R,6S,13aS,14aR,16aS)-14a- (cyclopropylsulfonylcarbamoyl)-12,12- (cyclopropylsulfonylcarbamoyl)-12,12- difluoro-2-(3-methylquinoxalin-2-yloxy)- difluoro-5,16-dioxo-2-(quinoxalin-2- 5,16-dioxooctadecahydrocyclopropa[e]pyrrolo yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a] [1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate [1,4]diazacyclopentadecin-6-yl)-5- methylisoxazole-3-carboxamide N-((2R,6S,13aS,14aR,16aS)-14a- N-((2R,6S,13aS,14aR,16aS)-14a- (cyclopropylsulfonylcarbamoyl)-12,12- (cyclopropylsulfonylcarbamoyl)-12,12- difluoro-2-(3-methylquinoxalin-2-yloxy)- difluoro-5,16-dioxo-2-(3-phenylquinoxalin-2- 5,16-dioxooctadecahydrocyclopropa[e]pyrrolo yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a] [1,2-a][1,4]diazacyclopentadecin-6-yl)-5- [1,4]diazacyclopentadecin-6-yl)-5 methylisoxazole-3-carboxamide methylisoxazole-3-carboxamide (2R,6S,13aS,14aR,16aS)-6-(tert- N-((2R,6S,13aS,14aR,16aS)-2-(3- butoxycarbonylamino)-14a- (benzo[d]thiazol-2-yl)quinoxalin-2-yloxy)- (cyclopropylsulfonylcarbamoyl)-12,12- 14a-(cyclopropylsulfonylcarbamoyl)-12,12- difluoro-5,16-dioxooetadecahydrocyclo difluoro-5,16- propa[e]pyrrolo[1,2-a][1,4]diazacyclo dioxooctadecahydrocyclopropa[e] pentadecin-2-yl 4-fluoroisoindoline-2- pyrrolo[1,2-a][1,4]diazacyclopentadecin-6- carboxylate yl)-5-methylisoxazole-3-carboxamide (2R,6S,13aS,14aR,16aS)-2-(3-(benzofuran- N-((2R,6S,13aS,14aR,16aS)-2-(3- 2-yl)quinoxalin-2-yloxy)-N- (benzofuran-2-yl)quinoxalin-2-yloxy)-14a- (cyclopropylsulfonyl)-12,12-difluoro-6-(1- cyclopropylsulfonylcarbamoyl)-12,12- methyl-1H-pyrazole-3-carboxamido)-5,16- difluoro-5,16-dioxooctadecahydrocyclo dioxooctadecahydro propa[e]pyrrolo[1,2-a][1,4]diazacyclo cyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclo pentadecin-6-yl)-5-methylisoxazole-3- pentadecine-14a-carboxamide carboxamide tert-butyl (2R,6S,13aS,14aR,16aS)-14a- N-((2R,6S,13aS,14aR,16aS)-14a- (cyclopropylsulfonylcarbamoyl)-2-(3- (cyclopropylsulfonylcarbamoyl)-2-(3- ethylquinoxalin-2-yloxy)-12,12-difluoro-5,16- ethylquinoxalin-2-yloxy)-12,12-difluoro-5,16- dioxooetadecahydrocyclopropa[e]pyrrol[1,2-a] dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a] [1,4]diazacyclopentadecin-6-ylcarbamate [1,4]diazacyclopentadecin-6-yl)-5- methylisoxazole-3-carboxamide tert-butyl (2R,6S,13aS,14aR,16aS)-2-(3- N-((2R,6S,13aS,14aR,16aS)-2-(3- ethylquinoxalin-2-yloxy)-12,12-difluoro-14a-(1- ethylquinoxalin-2-yloxy)-12,12-difluoro-14a-(1- methylcyclopropylsulfonylcarbamoyl)-5,16- methylcyclopropylsulfonylcarbamoyl)-5,16- dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a] dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a] [1,4]diazacyclopentadecin-6-ylcarbamate [1,4]diazacyclopentadecin-6-yl)-5- methylisoxazole-3-carboxamide N-((2R,6S,13aS,14aR,16aS)-14a-(cyclo (2R,6S,13aS,14aR,16aS)-N- propylsulfonylcarbamoyl)-2-(3- (cyclopropylsulfonyl)-2-(3-ethylquinoxalin- ethylquinoxalin-2-yloxy)-12,12-difluoro- 2-yloxy)-12,12-difluoro-5,16-dioxo-6- 5,16-dioxo pivalamidooctadecahydrocyclopropa[e]pyrrolo octadecahydrocyclopropa[e]pyrrolo[1,2- [1,2-a][1,4]diazacyclopentadecine-14a- a][1,4]diazacyclopentadecin-6-yl)isoxazole- carboxamide 3-carboxamide tert-butyl (2R,6S,13aS,14aR,16aS)-2-(3- tert-butyl (2R,6S,13aS,14aR,16aS)-2-(3- benzo[d]thiazol-2-yl)quinoxalin-2-yloxy)- (benzofuran-2-yl)quinoxalin-2-yloxy)-14a- 14a-(cyclopropylsulfonylcarbamoyl)-12,12- (cyclopropylsulfonylcarbamoyl)-12,12- difluoro-5,16- difluoro-5,16- dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a] dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a] [1,4]diazacyclopentadecin-6-ylcarbamate [1,4]diazacyclopentadecin-6-ylcarbamate - Other compounds contemplated by the invention include the following:
-
(2R,6S,13aS,14aR,16aS)-12,12-difluoro-2- (2R,6S,13aS,14aR,16aS)-N- (3-fluorophenanthridin-6-yloxy)-6-(1- (cyclopropylsulfonyl)-12,12-difluoro-2- methyl-1H-pyrazole-3-carboxamido)-N-(1- (3-fluorophenanthridin-6-yloxy)-6-(1- methylcyclopropylsulfonyl)-5,16- methyl-1H-pyrazole-3-carboxamido)- dioxooctadecahydrocyclopropa[e]pyrrolo[1, 5,16-dioxooctadecahydrocyclopropa[e]pyrrolo 2-a][1,4]diazacyclopentadecine-14a- [1,2-a][1,4]diazacyclopentadecine-14a- carboxamide carboxamide (2R,6S,13aS,14aR,16aS)-12,12-difluoro-2- (2R,6S,13aS,14aR,16aS)-N- (9-fluorophenanthridin-6-yloxy)-6-(1- (cyclopropylsulfonyl)-12,12-difluoro-2- methyl-1H-pyrazole-3-carboxamido)-N-(1- (9-fluorophenanthridin-6-yloxy)-6-(1- methylcyclopropylsulfonyl)-5,16- methyl-1H-pyrazole-3-carboxamido)- dioxooetadecahydroxycyclopropa[e]pyrrolo[1, 5,16-dioxooctadecahydrocyclopropa[e]pyrrolo 2-a][1,4]diazacyclopentadecine-14a- [1,2-a][1,4]diazacyclopentadecine-14a- carboxamide carboxamide N-((2R,6S,13aS,14aR,16aS)-12,12-difluoro- N-((2R,6S,13aS,14aR,16aS)-14a- 2-(3-fluorophenanthridin-6-yloxy)-14a-(1- (cyclopropylsulfonylcarbamoyl)-12,12- methylcyclopropylsulfonylcarbamoyl)-5,16- difluoro-2-(3-fluorophenanthridin-6- dioxooctadecahydrocyclopropa[e]pyrrolo[1, yloxy)-5,16- 2-a][1,4]diazacyclopentadecin-6-yl)-5- dioxooctadecahydrocyclopropa[e]pyrrolo methylisoxazole-3-carboxamide [1,2-a][1,4]diazacyclopentadecin-6-yl)-5- methylisoxazole-3-carboxamide N-((2R,6S,13aS,14aR,16aS)-12,12-difluoro- N-((2R,6S,13aS,14aR,16aS)-14a- 2-(9-fluorophenanthridin-6-yloxy)-14a-(1- (cyclopropylsulfonylcarbamoyl)-12,12- methylcyclopropylsulfonylcarbamoyl)-5,16- difluoro-2-(9-fluorophenanthridin-6- dioxooctadecahydrocyclopropa[e]pyrrolo[1, yloxy)-5,16- 2-a][1,4]diazacyclopentadecin-6-yl)-5- dioxooctadecahydrocyclopropa[e]pyrrolo methylisoxazole-3-carboxamide [1,2-a][1,4]diazacyclopentadecin-6-yl)-5- methylisoxazole-3-carboxamide (2R,6S,13aS,14aR,16aS)-12,12-difluoro-6- (2R,6S,13aS,14aR,16aS)-N- (1-methyl-1H-pyrazole-3-carboxamido)-N- (cyclopropylsulfonyl)-12,12-difluoro-6- (1-methylcyclopropylsulfonyl))-5,16-dioxo- (1-methyl-1H-pyrazole-3-carboxamido)- 2-(3-trifluoromethoxy)phenanthridin-6- 5,16-dioxo-2-(3- yloxy)octadecahydrocyclopropa[e]pyrrolo[1, (trifluoromethoxy)phenanthridin-6- 2-a][1,4]diazacyclopentadecine-14a- yloxy)octadecahydrocyclopropa[e]pyrrolo carboxamide [1,2-a][1,4]diazacyclopentadecine-14a- carboxamide (2R,6S,13aS,14aR,16aS)-12,12-difluoro-6- (2R,6S,13aS,14aR,16aS)-N- (1-methyl-1H-pyrazole-3-carboxamido)-N- (cyclopropylsulfonyl)-12,12-difluoro-6- (1-methylcyclopropylsulfonyl)-5,16-dioxo- (1-methyl-1H-pyrazole-3-carboxamido)- 2-(3-(trifluoromethyl)phenanthridin-6- 5,16-dioxo-2-(3- yloxy)octadecahydrocyclopropa[e]pyrrolo[1, (trifluoromethyl)phenanthridin-6- 2-a][1,4]diazacyclopentadecine-14a- yloxy)octadecahydrocyclopropa[e]pyrrolo carboxamide [1,2-a][1,4]diazacyclopentadecine-14a- carboxamide N-((2R,6S,13aS,14aR,16aS)-12,12-difluoro- N-((2R,6S,13aS,14aR,16aS)-14a- 14a-(1- (cyclopropylsulfonylcarbamoyl)-12,12- methylcyclopropylsulfonylcarbamoyl)-5,16- difluoro-5,16-dioxo-2-(3- dioxo-243-(trifluoromethoxy)phenanthridin- (trifluoromethoxy)phenanthridin-6- 6- yloxy)octadecahydrocyclopropa[e]pyrrolo yloxy)octadecahydrocyclopropa[e]pyrrolo[1, [1,2-a][1,4]diazacyclopentadecin-6-yl)-5- 2-a][1,4]diazacyclopentadecin-6-yl)-5- methylisoxazole-3-carboxamide methylisoxazole-3-carboxamide N-((2R,6S,13aS,14aR,16aS)-12,12-difluoro- N-((2R,6S,13aS,14aR,16aS)-14a- 14a-(1- (cyclopropylsulfonylcarbamoyl)-12,12- methylcyclopropylsulfonylcarbamoyl)-5,16- difluoro-5,16-dioxo-2(3 dioxo-243-(trifluoromethyl)phenanthridin- (trifluoromethyl)phenanthridin-6- 6- yloxy)octadecahydrocyclopropa[e]pyrrolo yloxy)octadecahydrocyclopropa[e]pyrrolo[1, [1,2-a][1,4]diazacyclopentadecin-6-yl)-5- 2-a][1,4]diazacyclopentadecin-6-yl)-5- methylisoxazole-3-carboxamide methylisoxazole-3-carboxamide (2R,6S,13aS,14aR,16aS)-12,12-difluoro-2- (2R,6S,13aS,14aR,16aS)-N- (3-methoxyphenanthridin-6-yloxy)-6-(1- (cyclopropylsulfonyl)-12,12-difluoro-2- methyl-1H-pyrazole-3-carboxamido)-N-(1 - (3-methoxyphenanthridin-6-yloxy)-6-(1- methylcyclopropylsulfonyl)-5,16- methyl-1H-pyrazole-3-carboxamido)- dioxooctadecahydrocyclopropa[e]pyrrolo[1, 5,16-dioxooctadecahydrocyclopropa[e]pyrrolo 2-a][1,4]diazacyclopentadecine-14a- [1,2-a][1,4]diazacyclopentadecine-14a- carboxamide carboxamide (2R,6S,13aS,14aR,16aS)-2-(3,9- (2R,6S,13aS,14aR,16aS)-N- difluorophenanthridin-6-yloxy)-12,12- (cyclopropylsulfonyl)-2-(3,9- difluoro-6-(1-methyl-1H-pyrazole-3- difluorophenanthridin-6-yloxy)-12,12- carboxamido)-N-(1- difluoro-6-(1-methyl-1H-pyrazole-3- methylcyclopropylsulfonyl)-5,16- carboxamido)-5,16- dioxooetadecahydrocyclopropa[e]pyrrolo[1, dioxooctadecahydrocyclopropa[e]pyrrolo 2-a][1,4]diazacyclopentadecine-14a- [1,2-a][1,4]diazacyclopentadecine-14a carboxamide carboxamide N-((2R,6S,13aS,14aR,16aS)-12,12-difluoro- N-((2R,6S,13aS,14aR,16aS)-14a- 2-(3-methoxyphenanthridin-6-yloxy)-14a- (cyclopropylsulfonylcarbamoyl)-12,12- (1-methylcyclopropylsulfonylcarbamoyl)- difluoro-2-(3-methoxyphenanthridin-6- 5,16- yloxy)-5,16- dioxooctadecahydrocyclopropa[e]pyrrolo[1, dioxoodadecahydrocyclopropa[e]pyrrolo 2-a][1,4]diazacyclopentadecin-6-yl)-5- [1,2-a][1,4]diazacyclopentadecine-14a- methylisoxazole-3-carboxamide methylisoxazole-3-carboxamide N-((2R,6S,13aS,14aR,16aS)-2-(3,9- N-((2R,6S,13aS,14aR,16aS)-14a- difluorophenanthridin-6-yloxy)-12,12- (cyclopropylsulfonylcarbamoyl)-2-(3,9- difluoro-14a-(1- difluorophenanthridin-6-yloxy)-12,12- methylcyclopropylsulfonylcarbamoyl)-5,16- difluoro-5,16- dioxooctadecahydrocyclopropa[e]pyrrolo[1, dioxooctadecahydrocyclopropa[e]pyrrolo 2-a][1,4]diazacyclopentadecin-6-yl)-5- [1,2-a][1,4]diazacyclopentadecin-6-yl)-5- methylisoxazole-3-carboxamide methylisoxazole-3-carboxamide - It is understood that the embodiments of the invention discussed below with respect to the preferred variable selections can be taken alone or in combination with one or more of the other embodiments, or preferred variable selections, of the invention, as if each combination were explicitly listed herein.
- In other aspects, the invention provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of the invention (e.g., a compound of formula I, or any formulae as described herein), or a pharmaceutically acceptable salt, ester, or prodrug thereof, in combination with a pharmaceutically acceptable carrier or excipient.
- In another aspect, the invention provides a pharmaceutical composition comprising a therapeutically effective amount of a compound described herein, or an embodiment or example described herein, or a pharmaceutically acceptable salt, ester, hydrate, solvate, polymorph, or prodrug thereof, in combination with a pharmaceutically acceptable carrier or excipient.
- According to another embodiment, the pharmaceutical compositions of the present invention may further contain one or more other anti-HCV agents. Examples of anti-HCV agents include, but are not limited to, α-interferon; β-interferon; pegylated interferon-α; pegylated interferon-lambda; ribavirin; viramidine; R-5158; nitazoxanide; amantadine; Debio-025, NEM-811; HCV polymerase inhibitors such as R7128, R1626, R4048, T-1106, PSI-7851, PF-00868554, ANA-598, IDX184, IDX102, IDX375, GS-9190, VCH-759, VCH-916, MK-3281, BCX-4678, MK-3281, VBY708, ANA598, GL59728 or GL60667; BMS-790052; BMS-791325; BMS-650032; HCV entry, helicase or internal ribosome entry site inhibitors; HCV NS5A inhibitors; or other HCV replication inhibitors such as GS-9132, ACH-1095, AP-H005, A-831, A-689, AZD2836. For further details see S. Tan, A. Pause, Y. Shi, N. Sonenberg, Hepatitis C Therapeutics: Current Status and Emerging Strategies, Nature Rev. Drug Discov., 1, 867-881 (2002); WO 00/59929 (2000); WO 99/07733 (1999); WO 00/09543 (2000); WO 99/50230 (1999); U.S. Pat. No. 5,861,297 (1999); and US2002/0037998 (2002).
- According to an additional embodiment, the pharmaceutical compositions of the present invention may further contain another HCV protease inhibitor, such as telaprevir, boceprevir, ITMN-191, B1-201335, TMC-435, MK-7009, VBY-376, VX-500, VX-813, PHX-B, ACH-1625, IDX136, or IDX316.
- In other embodiments, the invention provides a pharmaceutical composition further comprising pegylated interferon, another anti-viral, anti-bacterial, anti-fungal or anti-cancer agent, or an immune modulator, and/or further comprising a cytochrome P450 monooxygenase inhibitor or a pharmaceutically acceptable salt thereof. In certain embodiments, the cytochrome P450 monooxygenase inhibitor is ritonavir.
- In another aspect, the invention provides for the use of a compound of the invention to manufacture an agent for preventing or treating viral infection. In another aspect, the invention provides for the use of a compound of the invention to manufacture an agent for preventing or treating hepatitis C viral infection. The present invention also contemplates the use of a solvate (e.g., hydrate) of a compound of the invention to manufacture pharmaceutical compositions for preventing or treating hepatitis C infection. As used herein, “solvate” refers to the physical association of a compound of the invention with one or more solvent molecule, whether organic or inorganic. This physical association often includes hydrogen bonding. In certain instances, the solvate is capable of isolation, for example, when one or more solvate molecules are incorporated in the crystal lattice of the crystalline solid.
- In another embodiment, the compounds or pharmaceutical compositions of the invention are administered with ritonavir, either simultaneously or sequentially. In certain embodiments, a compound or a pharmaceutical composition of the invention is administered in the same composition as ritonavir. In another embodiment, a compound or a pharmaceutical composition thereof of the invention is administered in a different composition than ritonavir.
- According to yet another embodiment, the pharmaceutical compositions of the present invention may further comprise inhibitor(s) of other targets in the HCV life cycle, including, but not limited to, helicase, polyrnerase, metalloprotease, CD81, NS5A, cyclophilin, and internal ribosome entry site (IRES).
- In one aspect, the invention provides a method of treating a viral infection in a subject, comprising administering to the subject a therapeutically effective amount of a compound of the invention (e.g., a compound of formula I or I′ described herein), or a pharmaceutically acceptable salt, ester or prodrug thereof, or a pharmaceutical composition comprising the same.
- According to a further embodiment, the present invention includes methods of treating hepatitis C infections in a subject in need of such treatment by administering to said subject an anti-HCV virally effective amount or an inhibitory amount of the compounds or pharmaceutical compositions of the present invention.
- According to another embodiment, the present invention includes methods of treating hepatitis C infections in a subject in need of such treatment by administering to said subject a compound or a pharmaceutical composition of the present invention. The methods can further include administration of an additional therapeutic agent, including another antiviral agent or an anti-HCV agent as described hereinabove. The additional agent can be co-administered (such as concurrently administered or sequentially administered) with a compound (a pharmaceutically acceptable salt, ester or prodrug thereof) or a pharmaceutical composition of the present invention. The additional agent(s) and a compound (or a pharmaceutically acceptable salt, ester or prodrug thereof) of the present invention can be formulated in the same composition, or in different compositions but co-administered concurrently or sequentially. The methods herein can further include the step of identifying that the subject is in need of treatment for hepatitis C infection. The identification can be by subjective (e.g., health care provider determination) or objective (e.g., diagnostic test) means.
- In one aspect, the invention provides a method of inhibiting the replication of hepatitis C virus, the method comprising contacting a hepatitis C virus with an effective amount of a compound or pharmaceutical composition of the invention.
- In another embodiment, the invention provides a method as described above, further comprising administering an additional anti-hepatitis C virus agent. Examples of anti-hepatitis C virus agents include, but are not limited to, α-interferon; β-interferon; pegylated interferon-α; pegylated interferon-lambda; ribavirin; viramidine; R-5158; nitazoxanide; amantadine; Debio-025, NIM-811; HCV polymerase inhibitors such as R7128, R1626, R4048, T-1106, PSI-7851, PF-00868554, ANA-598, IDXI84, IDX102, IDX375, GS-9190, VCH-759, VCH-916, MK-3281, BCX-4678, MK-3281, VBY708, ANA598, GL59728 or GL60667; BMS-790052; BMS-791325; BMS-650032; HCV entry, helicase or internal ribosome entry site inhibitors; or other HCV replication inhibitors such as GS-9132, ACH-1095, AP-H005, A-831, A-689, AZD2836. For further details see S. Tan, A. Pause, Y. Shi, N. Sonenberg, Hepatitis C Therapeutics: Current Status and Emerging Strategies, Nature Rev. Drug Discov., 1, 867-881 (2002); WO 00/59929 (2000); WO 99/07733 (1999); WO 00/09543 (2000); WO 99/50230 (1999); U.S. Pat. No. 5,861,297 (1999); and US2002/0037998 (2002). Preferably, a compound or a pharmaceutical composition of the present invention is co-administered with, or used in combination with, pegylated interferon (e.g., pegylated interferon alpha-2a or 2b) and ribavirin. Ritonavir or another cytochrome P450 monooxygenase inhibitor can also be used to enhance the pharmacokinetics of the compound of the present invention. The patient being treated is preferably infected with HCV genotype-1 (e.g., genotype 1a or 1b), Patients infected with other HCV genotypes, such as genotypes 2, 3, 4, 5 or 6, can also be treated with a compound or a pharmaceutical composition of the present invention.
- In another embodiment, the invention provides a method as described above, further comprising administering another 1-ICV protease inhibitor, an HCV polymerase inhibitor, an HCV helicase inhibitor, or an internal ribosome entry site (IRES) inhibitor, such as telaprevir, boceprevir, ITMN-191, 131-201335, TMC-435, MK-7009, VBY-376, VX-500, VX-813, PHX-B, ACH-1625, IDX136, IDX316, pegylated interferon, another anti-viral, anti-bacterial, anti-fungal or anti-cancer agent, or an immune modulator, and/or further comprising a cytochrome P450 monooxygenase inhibitor or a pharmaceutically acceptable salt thereof. In certain embodiments, the cytochrome P450 monooxygenase inhibitor is ritonavir.
- An additional embodiment of the present invention includes methods of treating biological samples by contacting the biological samples with the compounds of the present invention.
- Yet another aspect of the present invention is a process of making any of the compounds delineated herein employing any of the synthetic means delineated herein.
- Listed below are definitions of various terms used to describe this invention. These definitions apply to the terms as they are used throughout this specification and claims, unless otherwise limited in specific instances, either individually or as part of a larger group. The number of carbon atoms in a hydrocarbyl substituent can be indicated by the prefix “Cx-Cy,” where x is the minimum and y is the maximum number of carbon atoms in the substituent. Likewise, a C, chain means a hydrocarbyl chain containing x carbon atoms.
- The prefix “halo” indicates that the substituent to which the prefix is attached is substituted with one or more independently selected halogen radicals. For example, “C1-C8haloalkyl” means a C1-C8alkyl substituent wherein at least one hydrogen radical is replaced with a halogen radical.
- If a linking element in a depicted structure is “absent”, then the left element in the depicted structure is directly linked to the right element in the depicted structure. For example, if a chemical structure is depicted as X-(L)n-Y wherein L is absent or n is 0, then the chemical structure is X—Y.
- The term “alkyl” as used herein, refers to a saturated, straight- or branched-chain hydrocarbon radical preferably containing one to eight carbon atoms (C1-C8 alkyl). Examples of alkyl radicals include, but are not limited to, methyl, ethyl, propyl, isopropyl, n-butyl, tert-butyl, neopentyl, n-hexyl, heptyl, octyl radicals and the like.
- The term “alkenyl” as used herein, denotes a straight- or branched-chain hydrocarbon radical containing one or more double bonds and preferably containing two to eight carbon atoms (C2-C8 alkenyl). Alkenyl groups include, but are not limited to, for example, ethenyl, propenyl, butenyl, 1-methyl-2-buten-1-yl, heptenyl, octenyl and the like.
- The term “alkynyl” as used herein, denotes a straight- or branched-chain hydrocarbon radical containing one or more triple bonds and preferably containing two to eight carbon atoms (C2-C8 alkynyl). Representative alkynyl groups include, but are not limited to, for example, ethynyl, 1-propynyl, 1-butyny-1, heptynyl, octynyl and the like.
- The term “cycloalkyl” denotes a monovalent group derived from a monocyclic or polycyclic saturated carbocyclic ring compound and preferably containing 3 to 14 carbon ring atoms (C3-C14 cycloalkyl). Examples of cycloalkyl include, but not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyciohexyl, bicyclo[2.2.1]heptyl, and bicyclo[2.2.2]octyl and the like.
- The terms “carbocycle” or “carbocyclic” or “carbocyclyl” refer to a saturated (e.g., “cycloalkyl”), partially saturated (e.g., “cycloalkenyl” or “cycloalkynyl”) or completely unsaturated (e.g., “aryl”) ring system containing zero heteroatom ring atom and preferably containing 3 to 14 carbon ring atoms (C3-C14carbocyclyl, such as C3-C14cycloalkyl). A carbocyclyl may be, without limitation, a single ring, or two or more fused or covalently inked rings, or bridged or Spiro rings. A substituted carbocyclyl may have either cis or trans geometry. Representative examples of carbocyclyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclopentenyl, cyclopentadienyl, cyclohexadienyl, adamantyl, decahydro-naphthalenyl, octahydro-indenyl, cyclohexenyl, phenyl, naphthyl, fluorenyl, indanyl, 1,2,3,4-tetrahydro-naphthyl, indenyl, isoindenyl, bicyclodecanyl, anthracenyl, phenanthrene, benzonaphthenyl (also known as “phenalenyl”), decalinyl, and norpinanyl and the like. A carbocyclyl group can be attached to the parent molecular moiety through any substitutable carbon atom of the group.
- The term “aryl” refers to an aromatic carbocyclyl containing from 6 to 14 carbon ring atoms. Non-limiting examples of aryls include phenyl, naphthalenyl, anthracenyl, and indenyl and the like. An aryl group can be connected to the parent molecular moiety through any substitutable carbon atom of the group.
- The term “heteroaryl” means an aromatic heterocyclyl typically containing from 5 to 14 ring atoms. Non-limiting examples of five-membered heteroaryls include imidazolyl; furanyl; thiophenyl (or thienyl or thiofuranyl); pyrazolyl; oxazolyl; isoxazolyl; thiazolyl; 1,2,3-, 1,2,4-, 1,2,5-, and 1,3,4-oxadiazolyl; and isothiazolyl. Non-limiting examples of six-membered heteroaryls include pyridinyl; pyrazinyl; pyrimidinyl; pyridazinyl; and 1,3,5-, 1,2,4-, and 1,2,3-triazinyl. Non-limiting examples of 6/5-membered fused ring heteroaryls include benzothiofiiranyl, isobenzothiofuranyl, benzisoxazolyl, benzoxazolyl, purinyl, and anthranilyl. Non-limiting examples of 6/6-membered fused ring heteroaryls include quinolinyl; isoquinolinyl; and benzoxazinyl (including ciimolinyl and quinazolinyl).
- The term “heterocycloalkyl” refers to a non-aromatic 3-, 4-, 5-, 6- or 7-membered ring or a bi- or tri-cyclic group fused system, where at least one of the ring atoms is a heteroatom, and where (i) each 5-membered ring has 0 to 1 double bonds and each 6-membered ring has 0 to 2 double bonds, (ii) the nitrogen and sulfur heteroatoms may optionally be oxidized, (iii) the nitrogen heteroatom may optionally be quaternized, and (iv) any of the above rings may be fused to a benzene ring. Representative heterocycloalkyl groups include, but are not limited to, [1,3]dioxolane, pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, piperidinyl, piperazinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, and tetrahydrofuryl and the like.
- The terms “heterocyclic” or “heterocycle” or “heterocyclyl” refer to a saturated (e.g., “heterocycloalkyl”), partially unsaturated (e.g., “heterocycloalkenyl” or “heterocycloalkynyl”) or completely unsaturated (e.g., “heteroaryl”) ring system preferably containing 3 to 14 ring atoms, where at least one of the ring atoms is a heteroatom (i.e., nitrogen, oxygen or sulfur), with the remaining ring atoms being independently selected from the group consisting of carbon, nitrogen, oxygen and sulfur. A heterocyclic or heterocycle or heterocyclyl may be, without limitation, a single ring, or two or more fused or covalently linked rings, or bridged or Spiro rings. A heterocyclyl group can be linked to the parent molecular moiety via any substitutable carbon or nitrogen atom in the group, provided that a stable molecule results. A heterocyclyl may be, without limitation, a single ring. Non-limiting examples of single-ring heterocyclyls include furanyl, dihydrofuranyl, pyrrolyl, isopyrrolyl, pyrrolinyl, pyrrolidinyl, imidazolyl, isoimidazolyl, imidazolinyl, imidazolidinyl, pyrazolyl, pyrazolinyl, pyrazolidinyl, triazolyl, tetrazolyl, dithiolyl, oxathiolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiazoliriyl, isothiazolinyl, thiazolidinyl, isothiazolidinyl, thiodiazolyl, oxathiazolyl, oxadiazoly, pyranyl, dihydropyranyl, pyrridinyl, piperidinyl, pyridazinyl, pyrimidinyl, pyrazinyl, piperazinyl, triazinyl, isoxazinyl, oxazolidinyl, isoxazolidinyl, oxathiazinyl, oxadiazinyl, morpholinyl, azepinyl, oxepinyl, thiepinyl, or diazepinyl. A heterocyclyl may also include, without limitation, two or more rings fused together, such as, for example, naphthyridinyl, thiazolpyrimidinyl, thienopyrimidinyl, pyrimidopyrimidinyl, or pyridopyrimidinyl. A heterocyclyl may comprise one or more sulfur atoms as ring members; and in some cases, the sulfur atom(s) is oxidized to SO or SO2. The nitrogen heteroatom(s) in a heterocyclyl may or may not be quaternized, and may or may not be oxidized to N-oxide. In addition, the nitrogen heteroatom(s) may or may not be N-protected.
- The terms “optionally substituted”, “optionally substituted alkyl,” “optionally substituted “optionally substituted alkenyl,” “optionally substituted alkynyl”, “optionally substituted carbocyclic,” “optionally substituted aryl”, “optionally substituted heteroaryl,” “optionally substituted heterocyclic,” and any other optionally substituted group as used herein, refer to groups that are substituted or unsubstituted by independent replacement of one, two, or three or more of the hydrogen atoms thereon with typical substituents including, but not limited to:
- —F, —Cl, —Br, —I,
- —OH, protected hydroxy, alkoxy, oxo, thiooxo,
- —NO2, —CN, CF3, N3,
- —NH2, protected amino, —NH alkyl, —NH alkenyl, —NH alkynyl, —NH cycloalkyl, —NH— aryl, —NH-heteroaryl, —NH-heterocyclic, -dialkylamino, -diarylamino, -diheteroarylamino,
- —O— alkyl, —O— alkenyl. —O— alkynyl, —O— cycloalkyl, —O-aryl, —O-heteroaryl, —O— heterocyclic,
- —C(O)— alkyl, —C(O)— alkenyl, —C(O)— alkynyl, —C(O)— carbocyclyl, —C(O)-aryl, —C(O)— heteroaryl, —C(O)-heterocyclyl,
- —CONH2, —CONH— alkyl, —CONH— alkenyl, —CONH— alkynyl, —CONH-carbocyclyl, —CONH-aryl, —CONH-heteroaryl, —CONH-heterocyclyl,
- —OCO2— alkyl, —OCO2— alkenyl, —OCO2— alkynyl, —OCO2— carbocyclyl, —OCO2-aryl, —OCO2-heteroaryl, —OCO2-heterocyclyl, —OCONH2, —OCONH— alkyl, —OCONH— alkenyl, OCONH— alkynyl, —OCONH— carbocyclyl, —OCONEE- aryl, —OCONH— heteroaryl, —OCONH— heterocyclyl,
- —NHC(O)— alkyl, —NHC(O)— alkenyl, —NHC(O)— alkynyl, —NHC(O)— carbocyclyl, —NHC(O)-aryl, —NHC(O)-heteroaryl, —NHC(O)-heterocyclyl, —NHCO2— alkyl, —NHCO2— alkenyl, —NHCO2— alkynyl, —NHCO2— carbocyclyl, —NHCO2— aryl, —NHCO2— heteroaryl, —NHCO2-heterocyclyl, —NHC(O)NH2, —NHC(O)NH— alkyl, —NHC(O)NH— alkenyl, —NHC(O)NH— alkenyl, —NHC(O)NH— carbocyclyl, —NHC(O)NH-aryl, —NHC(O)NH-heteroaryl, —NHC(O)NH— heterocyclyl, NHC(S)NH2, —NHC(S)NH— alkyl, —NHC(S)NH— alkenyl, —NHC(S)NH— alkynyl, —NHC(S)NH— carbocyclyl, —NHC(S)NH-aryl, —NHC(S)NH-heteroaryl, —NHC(S)NH-heterocyclyl, —NHC(NH)NH2, —NHC(NH)NH— alkyl, —NHC(NH)NH— -alkenyl, —NHC(NH)NH— alkenyl, —NHC(NH)NH— carbocyclyl, —NHC(NE)NH-aryl, —NHC(NH)NH-heteroaryl, —NHC(NH)NH-heterocyclyl, —NHC(NH)— alkyl, —NHC(NH)— alkenyl, —NHC(NH)— alkenyl, —NHC(NH)-carbocyclyl, —NHC(NH)-aryl, —NHC(NH)-heteroaryl, —NHC(NH)-heterocyclyl,
- —C(NH)NH— alkyl, —C(NH)NH— alkenyl, —C(NH)NH— alkynyl, —C(NH)NH— carbocyclyl, C(NH)NH-aryl, —C(NH)NH-heteroaryl, —C(NH)NH-heterocyclyl,
- —S(O)— alkyl, —S(O)— alkenyl, —S(O)— alkynyl, —S(O)— carbocyclyl, —S(O)-aryl, —S(O)— heteroaryl, —S(O)-heterocyclyl —SO2NH2, —SO2NH— alkyl, —SO2NH— alkenyl, —SO2NH— alkynyl, —SO2NH— carbocyclyl, —SO2NH— aryl, —SO2NH— heteroaryl, —SO2NH— heterocyclyl,
- —NHSO2— alkyl, —NHSO2— alkenyl, NIESO2— alkynyl, —NHSO2— carbocyclyl, —NHSO2-aryl, —NHSO2-heteroaryl, —NHSO2-heterocyclyl,
- —CH2NH2, —CH2SO2CH3,
- -alkyl, -alkenyl, -alkynyl, -aryl, -arylalkyl, -heteroaryl, -heteroarylalkyl, -heterocycloalkyl, -cycloalkyl, -carbocyclic, -heterocyclic, polyalkoxyalkyl, polyalkoxy, methoxymethoxy, -methoxyethoxy, —SH, —S— alkyl, —S— alkenyl, —S— alkynyl, —S— carbocyclyl, —S-aryl, —S-heteroaryl, —S-heterocyclyl, or methylthiomethyl.
- It is understood that the aryls, heteroaryls, carbocycles, heterocycles, alkyls, and the like can be further substituted.
- The terms “halo” and “halogen,” as used herein, refer to an atom selected from fluorine, chlorine, bromine and iodine.
- Any divalent group having two points of connection to two other groups may be aligned in either direction, e.g., the term “—C(O)O-” is interpreted to include both —C(O)O— and —OC(O)—.
- The term “subject” as used herein refers to a mammal. A subject therefore refers to, for example, dogs, cats, horses, cows, pigs, guinea pigs, and the like. Preferably the subject is a human. When the subject is a human, the subject may be either a patient or a healthy human.
- The term “leaving group,” or “LG”, as used herein, refers to any group that leaves in the course of a chemical reaction involving the group and includes but is not limited to halogen, brosylate, mesylate, tosylate, triflate, p-nitrobenzoate, phosphonate groups, for example.
- The term “protected hydroxy,” as used herein, refers to a hydroxy group protected with a hydroxy protecting group, as defined above, including benzoyl, acetyl, trirnethylsilyl, triethylsilyl, methoxyrnethyl groups, for example.
- The term “hydroxy protecting group,” as used herein, refers to a labile chemical moiety which is known in the art to protect a hydroxy group against undesired reactions during synthetic procedures. After said synthetic procedure(s) the hydroxy protecting group as described herein may be selectively removed. Hydroxy protecting groups as known in the are described generally in T. H. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999). Examples of hydroxy protecting groups include benzyloxycarbonyl, 4-nitrobenzyloxycarbonyl, 4-bromobenzyloxycarbonyl, 4-methoxybenzyloxy-carbonyl, methoxycarbonyl, tert-butoxycarbonyl, isopropoxycarbonyl, diphenylmethoxycarbonyl, 2,2,2-trichloroethoxycarbonyl, 2-(trimethylsilypethoxycarbonyl, 2-furfuryloxycarbonyl, allyloxycarbonyl, acetyl, formyl, chloroacetyl, trifluoroacetyl, methoxyacetyl, phenoxyacetyl, benzoyl, methyl, t-butyl, 2,2,2-trichloroethyl, 2-trimethylsilyl ethyl, 1,1-dimethyl-2-propenyl, 3-methyl-3-butenyl, allyl, benzyl, para-methoxybenzyldiphenylmethyl, triphenylmethyl(trityl), tetrahydrofuryl, methoxymethyl, methylthiomethyl, benzyloxymethyl, 2,2,2-triehloroethoxymethyl, 2-(trimethylsilypethoxymethyl, methanesulfonyl, para-toluenesulfonyl, tri methyl silyl, triethylsilyl, triisopropylsilyl, and the like. Preferred hydroxy protecting groups for the present invention are acetyl (Ac or —C(O)CH3), benzoyl (Bz or —C(O)C6H5), and trimethylsilyl (TMS or —Si(CH3)3).
- The term “amino protecting group,” as used herein, refers to a labile chemical moiety which is known in the art to protect an amino group against undesired reactions during synthetic procedures. After said synthetic procedure(s) the amino protecting group as described herein may be selectively removed. Amino protecting groups as known in the are described generally in T. H. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999). Examples of amino protecting groups include, but are not limited to, t-butoxycarbonyl, 9-fluorenylmethoxycarbonyl, benzyloxycarbonyl, and the like.
- The term “protected amino,” as used herein, refers to an amino group protected with an amino protecting group as defined above.
- The term “alkylamino” refers to a group having the structure —N(RaRb), where Ra and Rb are independent 1-1 or alkyl.
- As used herein, the term “pharmaceutically acceptable salt” refers to those salts of the compounds formed by the process of the present invention which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio. Pharmaceutically acceptable salts are well known in the art. For example, S. M. Berge, et al. describes pharmaceutically acceptable salts in detail in J. Pharmaceutical Sciences, 66: 1-19 (1977). The salts can be prepared in situ during the final isolation and purification of the compounds of the invention, or separately by reacting the tree base function with a suitable organic acid. Examples of pharmaceutically acceptable salts include, but are not limited to, nontoxic acid addition salts, or salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange. Other pharmaceutically acceptable salts include, but are not limited to, adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanestilfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-plienylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, valerate salts, and the like. Representative alkali or alkaline earth metal salts include sodium, lithium, potassium, calcium, or magnesium salts, and the like. Further pharmaceutically acceptable salts include, when appropriate, nontoxic ammonium, quaternary ammonium, and amine cations formed using counterions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, alkyl having from 1 to 6 carbon atoms, sulfonate and aryl sulfonate.
- As used herein, the term “pharmaceutically acceptable ester” refers to esters of the compounds formed by the process of the present invention which hydrolyze in vivo and include those that break down readily in the human body to leave the parent compound or a salt thereof. Suitable ester groups include, for example, those derived from pharmaceutically acceptable aliphatic carboxylic acids, particularly alkanoic, alkenoic, cycloalkanoic and alkanedioic acids, in which each alkyl or alkenyl moiety advantageously has not more than 6 carbon atoms. Examples of particular esters include, but are not limited to, formates, acetates, propionates, butyrates, acrylates and ethylsuccinates.
- The term “pharmaceutically acceptable prodrugs” as used herein refers to those prodrugs of the compounds formed by the process of the present invention which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals with undue toxicity, irritation, allergic response, and the like, commensurate with a reasonable benefit/risk ratio, and effective for their intended use, as well as the zwitterionic forms, where possible, of the compounds of the present invention. “Prodrug”, as used herein means a compound which is convertible in vivo by metabolic means (e.g. by hydrolysis) to afford any compound delineated by the formulae of the instant invention. Various forms of prodrugs are known in the art, for example, as discussed in Bundgaard, (ed.), Design of Prodrugs, Elsevier (1985); Widder, et al. (ed.), Methods in Enzymology, vol. 4, Academic Press (1985); Krogsgaard-Larsen, et al., (ed). “Design and Application of Prodrugs, Textbook of Drug Design and Development, Chapter 5, 113-191 (1991); Bundgaard, et al., Journal of Drug Deliver Reviews, 8:1-38(1992); Bundgaard, J. of Pharmaceutical Sciences, 77:285 et seq. (1988); Iliguchi and Stella (eds.) Prodrugs as Novel Drug Delivery Systems, American Chemical Society (1975); and Bernard Testa & Joachim Mayer, “Hydrolysis In Drug And Prodrug Metabolism: Chemistry, Biochemistry And Enzymology,” john Wiley and Sons, Ltd. (2002).
- This invention also encompasses pharmaceutical compositions containing, and methods of treating viral infections through administering, pharmaceutically acceptable prodrugs of compounds of the invention. For example, compounds of the invention having free amino, amido, hydroxy or carboxylic groups can be converted into prodrugs. Prodrugs include compounds wherein an amino acid residue, or a polypeptide chain of two or more (e.g., two, three or four) amino acid residues is covalently joined through an amide or ester bond to a free amino, hydroxy or carboxylic acid group of compounds of the invention. The amino acid residues include but are not limited to the 20 naturally occurring amino acids commonly designated by three letter symbols and also includes 4-hydroxyproline, hydroxylysine, demosine, isodemosine, 3-methylhistidine, norvalin, beta-alanine, gamma-aminobutyric acid, citrulline, homocysteine, homoserine, ornithine and methionine sulfone. Additional types of prodrugs are also encompassed. For instance, free carboxyl groups can be derivatized as amides or alkyl esters. Free hydroxy groups may be derivatized using groups including but not limited to hemisuccinates, phosphate esters, dimethylaminoacetates, and phosphoryloxymethyloxy carbonyls, as outlined in Advanced Drug Delivery Reviews, 1996, 19, 1 15. Carbamate prodrugs of hydroxy and amino groups are also included, as are carbonate prodrugs, sulfonate esters and sulfate esters of hydroxy groups. Derivatization of hydroxy groups as (acyloxy)methyl and (acyloxy)ethyl ethers wherein the acyl group may be an alkyl ester, optionally substituted with groups including but not limited to ether, amine and carboxylic acid functionalities, or where the acyl group is an amino acid ester as described above, are also encompassed. Prodrugs of this type are described in J. Med. Chem, 1996, 39, 10. Free amines can also be derivatized as amides, sulfonamides or phosphonamides. All of these prodrug moieties may incorporate groups including but not limited to ether, amine and carboxylic acid functionalities
- Combinations of substituents and variables envisioned by this invention are only those that result in the formation of stable compounds. The term “stable”, as used herein, refers to compounds which possess stability sufficient to allow manufacture and which maintains the integrity of the compound for a sufficient period of time to be useful for the purposes detailed herein (e.g., therapeutic or prophylactic administration to a subject).
- The pharmaceutical compositions of the present invention comprise a therapeutically effective amount of a compound of the present invention formulated together with one or more pharmaceutically acceptable carriers. As used herein, the term “pharmaceutically acceptable carrier” means a non-toxic, inert solid, semi-solid or liquid filler, diluent, encapsulating material or formulation auxiliary of any type. The pharmaceutical compositions of this invention can be administered to humans and other animals orally, rectally, parenterally, intracisternally, intravaginally, intraperitoneally, topically (as by powders, ointments, or drops), buccally, or as an oral or nasal spray.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups, and elixirs. In addition to the active compounds, the liquid dosage forms may contain inert diluents commonly used in the art such as, for example, water, alcohol or other solvents, solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, polysorbate, dimethylformamide, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), mono- or di-glycerides, glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof. Besides inert diluents, the oral compositions can also include adjuvants such as wetting agents, emulsifying and suspending agents, antioxidants, sweetening, flavoring, and perfuming agents. The liquid dosage form can also be encapsulated in a gelatin capsule, wherein a compound of the present invention can be dissolved in a pharmaceutically acceptable canter containing, for example, one or more soltibilizating agents (e.g., polysorbate 80 and mono and diglycerides), and other suitable excipients (e.g., an antioxidants such as ascorbyl palmitate, or a sweetening or flavoring agent).
- Injectable preparations, for example, sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents. The sterile injectable preparation may also be a sterile injectable solution, suspension or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution, U.S.P. and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil can be employed including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid are used in the preparation of injectables.
- In order to prolong the effect of a drug, it is often desirable to slow the absorption of the drug from subcutaneous or intramuscular injection. This may be accomplished by the use of a liquid suspension of crystalline or amorphous material with poor water solubility. The rate of absorption of the drug then depends upon its rate of dissolution which, in turn, may depend upon crystal size and crystalline form. Alternatively, delayed absorption of a parenterally administered drug form is accomplished by dissolving or suspending the drug in an oil vehicle. Immediate release forms are also contemplated by the present invention.
- Compositions for rectal or vaginal administration are preferably suppositories which can be prepared by mixing the compounds of this invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
- The active compounds can also be in micro-encapsulated form with one or more excipients as noted above
- The solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings, release controlling coatings and other coatings well known in the pharmaceutical formulating art. In such solid dosage forms the active compound may be admixed with at least one inert diluent such as sucrose, lactose or starch. Such dosage forms may also comprise, as is normal practice, additional substances other than inert diluents, e.g., tableting lubricants and other tableting aids such a magnesium stearate and microcrystalline cellulose. In the case of capsules, tablets and pills, the dosage forms may also comprise buffering agents.
- Preferably, a compound of the invention is formulated in a solid dispersion, where the compound can be molecularly dispersed in a matrix which comprises a pharmaceutically acceptable, hydrophilic polymer. The matrix may also contain a pharmaceutically acceptable surfactant. Suitable solid dispersion technology for formulating a compound of the invention includes, but is not limited to, melt extrusion, spray drying, or solvent evaporization.
- Dosage forms for topical or transdermal administration of a compound of this invention include ointments, pastes, creams, lotions, gels, powders, solutions, sprays, inhalants or patches. The active component is admixed under sterile conditions with a pharmaceutically acceptable carrier and any needed preservatives or buffers as may be required. Ophthalmic formulation, ear drops, eye ointments, powders and solutions are also contemplated as being within the scope of this invention.
- The ointments, pastes, creams and gels may contain, in addition to an active compound of this invention, excipients such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- Powders and sprays can contain, in addition to the compounds of this invention, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances. Sprays can additionally contain customary propellants such as chlorafluorohydrocarbons.
- Transdermal patches have the added advantage of providing controlled delivery of a compound to the body. Such dosage forms can be made by dissolving or dispensing the compound in the proper medium. Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate can be controlled by either providing a rate controlling membrane or by dispersing the compound in a polymer matrix or gel.
- According to the methods of treatment of the present invention, viral infections are treated or prevented in a subject, such as a human or another animal, by administering to the subject a therapeutically effective amount of a compound of the invention (or a pharmaceutically acceptable salt, ester or prodrug thereof), in such amounts and for such time as is necessary to achieve the desired result. The term “therapeutically effective amount” of a compound of the invention, as used herein, means a sufficient amount of the compound so as to decrease the viral load in a subject and/or decrease the subject's HCV symptoms. As is well understood in the medical arts a therapeutically effective amount of a compound of this invention will be at a reasonable benefit/risk ratio applicable to any medical treatment.
- Antiviral Activity
- An inhibitory amount or dose of the compounds of the present invention may range from about 0.1 mg/Kg to about 500 mg/Kg, alternatively from about 1 to about 50 mg/Kg. Inhibitory amounts or doses will also vary depending on route of administration, as well as the possibility of co-usage with other agents.
- According to the methods of treatment of the present invention, viral infections are treated or prevented in a subject such as a human or lower mammal by administering to the subject an anti-hepatitis C virally effective amount or an inhibitory amount of a compound of the present invention, in such amounts and for such time as is necessary to achieve the desired result. An additional method of the present invention is the treatment of biological samples with an inhibitory amount of a compound of composition of the present invention in such amounts and for such time as is necessary to achieve the desired result.
- The term “anti-hepatitis C virally effective amount” of a compound of the invention, as used herein, means a sufficient amount of the compound so as to decrease the viral load in a biological sample or in a subject. As well understood in the medical arts, an anti-hepatitis C virally effective amount of a compound of this invention will be at a reasonable benefit/risk ratio applicable to any medical treatment.
- The term “inhibitory amount” of a compound of the present invention means a sufficient amount to decrease the hepatitis C viral load in a biological sample or a subject. It is understood that when said inhibitory amount of a compound of the present invention is administered to a subject it will be at a reasonable benefit/risk ratio applicable to any medical treatment as determined by a physician. The term “biological sample(s),” as used herein, means a substance of biological origin, which may be intended for administration to a subject. Examples of biological samples include, but are not limited to, blood and components thereof such as plasma, platelets, subpopulations of blood cells and the like; organs such as kidney, liver, heart, lung, and the like; sperm and ova; bone marrow and components thereof; or stem cells. Thus, another embodiment of the present invention is a method of treating a biological sample by contacting said biological sample with an inhibitory amount of a compound or pharmaceutical composition of the present invention.
- Upon improvement of a subject's condition, a maintenance dose of a compound, composition or combination of this invention may be administered, if necessary. Subsequently, the dosage or frequency of administration, or both, may be reduced, as a function of the symptoms, to a level at which the improved condition is retained when the symptoms have been alleviated to the desired level, treatment should cease. The subject may, however, require intermittent treatment on a long-term basis upon any recurrence of disease symptoms.
- It will be understood, however, that the total daily usage of the compounds and compositions of the present invention will be decided by the attending physician within the scope of sound medical judgrient. The specific inhibitory dose for any particular patient will depend upon a variety of factors including the disorder being treated and the severity of the disorder; the activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed; and like factors well known in the medical arts.
- The total daily inhibitory dose of the compounds of this invention administered to a subject in single or in divided doses can be in amounts, for example, from 0.01 to 50 mg/kg body weight or more usually from 0.1 to 25 mg/kg body w eight. Single dose compositions may contain such amounts or submultiples thereof to make up the daily dose. In one embodiment, treatment regimens according to the present invention comprise administration to a patient in need of such treatment from about 10 mg to about 1000 mg of the compound(s) of this invention per day in single or multiple doses. In another embodiment, the treatment regimen comprises administration to a patient in need of such treatment from about 25 mg to about 6000 mg of a compound(s) of this invention per day in single or multiple doses, either with or without a cytochrome P450 monooxygenase inhibitor such as ritonavir. The suitable daily dose for the co-administered cytochrorne P450 monooxygenase inhibitor (e.g., ritonavir) can range, without limitation, from 10 to 200 mg. Preferably, a compound(s) of the present invention, or a combination of a compound(s) of the invention and ritonavir, is administered once daily or twice daily to achieve the desired daily dose amount. For instance, when used without ritonavir, a compound of the present invention can be administered to a patient twice a day with a total daily dose of 4000, 4200, 4400, 4600, 4800 or 5000 mg. For another instance, when used in combination with ritonavir, a compound of the present invention can be administered to a patient once or twice a day with a total daily dose of 200, 400, 600 or 800 mg, where the amount of ritonavir can be 25, 50 or 100 mg per administration.
- Synthetic Methods
- The compounds and processes of the present invention will be better understood in connection with the following synthetic examples that illustrate the methods by which the compounds of the invention may be prepared.
- Definitions of variables in the structures in the schemes herein are commensurate with those of corresponding positions in the formulae delineated herein.
- The compounds described herein contain one or more asymmetric centers and thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)— or (S)—, or as (D)- or (L)- for amino acids. The present invention is meant to include all such possible isomers, as well as their racemic and optically pure forms. Optical isomers may be prepared from their respective optically active precursors by the procedures described above, or by resolving the racernic mixtures. The resolution can be carried out in the presence of a resolving agent, by chromatography or by repeated crystallization or by some combination of these techniques which are known to those skilled in the art. Further details regarding resolutions can be found in Jacques, et al., Enantiomers, Racemates, and Resolutions (John Wiley & Sons, 1981). When the compounds described herein contain olefinic double bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers. Likewise, all tautomeric forms are also intended to be included. The configuration of any carbon-carbon double bond appearing herein is selected for convenience only and is not intended to designate a particular configuration unless the text so states; thus a carbon-carbon double bond depicted arbitrarily herein as trans may be cis, trans, or a mixture of the two in any proportion.
- The synthesized compounds can be separated from a reaction mixture and further purified by a method such as column chromatography, high pressure liquid chromatography, or recrystallization. As can be appreciated by the skilled artisan, further methods of synthesizing the compounds of the formulae herein will be evident to those of ordinary skill in the art. Additionally, the various synthetic steps may be performed in an alternate sequence or order to give the desired compounds. In addition, the solvents, temperatures, reaction durations, etc. delineated herein are for purposes of illustration only and one of ordinary skill in the art will recognize that variation of the reaction conditions can produce the desired bridged macrocyclic products of the present invention. Synthetic chemistry transformations and protecting group methodologies (protection and deprotection) useful in synthesizing the compounds described herein are known in the art and include, for example, those such as described in R. Larock, Comprehensive Organic Transformations, VCR Publishers (1989); T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2d. Ed., John Wiley and Sons (1991); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); and L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995), and subsequent editions thereof
- The compounds of this invention may be modified by appending various functionalities via any synthetic means delineated herein to enhance selective biological properties. Such modifications are known in the art and include those which increase biological penetration into a given biological system (e.g., blood, lymphatic system, central nervous system), increase oral availability, increase solubility to allow administration by injection, alter metabolism and alter rate of excretion.
- The recitation of a listing of chemical groups in any definition of a variable herein includes definitions of that variable as any single group or combination of listed groups. The recitation of an embodiment for a variable herein includes that embodiment as any single embodiment or in combination with any other embodiments or portions thereof.
- The compounds and processes of the present invention will be better understood in connection with the following examples, which are intended as an illustration only and not to limit the scope of the invention. The following examples can be prepared according to the schemes as described above, or according to the synthetic steps as described below. Various changes and modifications to the disclosed embodiments will be apparent to those skilled in the art and such changes and modifications including, without limitation, those relating to the chemical structures, substituents, derivatives, formulations and/or methods of the invention may be made without departing from the spirit of the invention and the scope of the appended claims.
- The chemical structures herein contain certain —NH—, —NH2 (amino) and OH (hydroxyl) groups where the corresponding hydrogen atom(s) may not explicitly appear; however they are to be read as —NH—, —NH2 or —OH as the case may be.
- Those of ordinary skill in the art are aware of the biological assays utililized to test the compounds of the invention.
-

- To a solution of (2S,6S,13aS,14aR,16aS,Z)-ethyl 6-(tert-butoxycarbonylamino)-2-hydroxy-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (2.0 g, 4.1 mmol) in dichloromethane (4.1 ml) cooled to 0° C. was added N-ethyl-N-isopropylpropan-2-amine (2.83 ml, 16.2 mmol) and 1-methylimidazole (0.097 ml, 1.2 mmol) followed by benzoic anhydride (1.83 g, 8.10 mmol). The solution was stirred at room temperature for 16 h, DMAP (9.3 mg, 0.076 mmol) was added and the reaction mixture stirred for 2.5 h. The reaction mixture was diluted with ethyl acetate (150 ml) and washed with HCl (1 N, 50 ml×2), water and brine. The organic layer was dried (anhydrous sodium sulfate), concentrated and purified by flash chromatogaphy on silica gel (dichloromethane/methanol=40/1) to provide the title compound (2.06 g, 83% yield) as a foamy white solid.
-
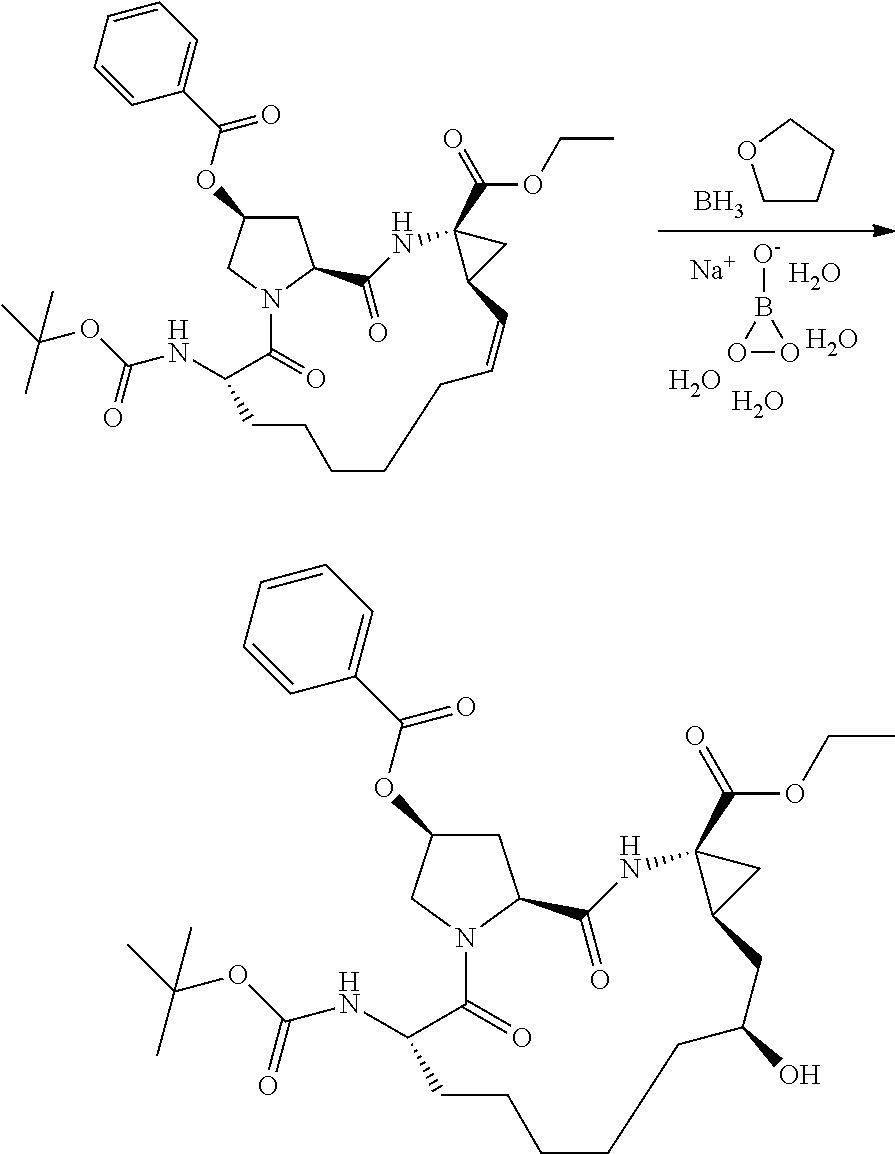
- To a solution of (2S,6S,13aS,14aR,16aS,Z)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonylamino)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocycl opropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (2.06 g, 3.45 mmol) in tetrahydrofuran (34 ml) was added borane-tetrahydrofuran complex (19 ml, 1M, 19.00 mmol) in one portion at 0° C. The mixture was stirred at 0° C. for 20 min and then at room temperature for 20 min. The reaction mixture was quenched by adding water (70 ml) dropwise at 0° C. followed by the addition of sodium perborate tetrahydrate (2.92 g, 19.0 mmol). The mixture was stirred at room temperature overnight. The reaction mixture was diluted with ethyl acetate (300 ml) and washed with water (50 ml×2) and brine (50 ml). The combined aqueous layer was back-extracted with ethyl acetate (75 ml×2). The organic layers were dried (anhydrous sodium sulfate) and concentrated to give a white foamy solid (3.2 g). The residue was purified by flash chromatography on silica gel (dichloromethane/methanol=40/1, 30/1) followed by a second purification by reverse phase RPLC (acetonitrile/water (0.1% TFA)=30/70 to 100/0, 15 min) to provide (2S,6S,12S,13aS,14aR,16aS)-ethyl 2-(benzoyloxy)-6-(tert-butoxyearbonylamino)-12-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (0.43 g, 0.70 mmol, 20% yield).
-
- To a solution of oxalyl chloride (0.092 ml, 1.05 mmol) in dichloromethane (1.75 ml) at −78° C. was added dropwise DMSO (0.149 ml, 2.10 mmol). The reaction mixture was stirred for 10 min and then a solution of (2S,6S,12S,13aS,14aR,16aS)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonylamino)-12-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 1b, 0.43 g, 0.698 mmol) in dichloromethane (4 ml) was added dropwise. The reaction mixture was stirred at −78° C. for 30 min and triethylamine (0.730 ml, 5.24 mmol) was added dropwise. The reaction mixture was stirred at −78° C. for 15 min and warmed to room temperature and stirred for 1.5 hr. The reaction mixture was quenched with water, stirred for 30 min and extracted with dichloromethane and the organic layer dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (dichloromethane/methanol=40/1) to provide the title compound (2S,6S,13aS,14aR,16aS)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonylamino)-5,12,16-trioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (0.36 g, 0.59 mmol, 84% yield).
-
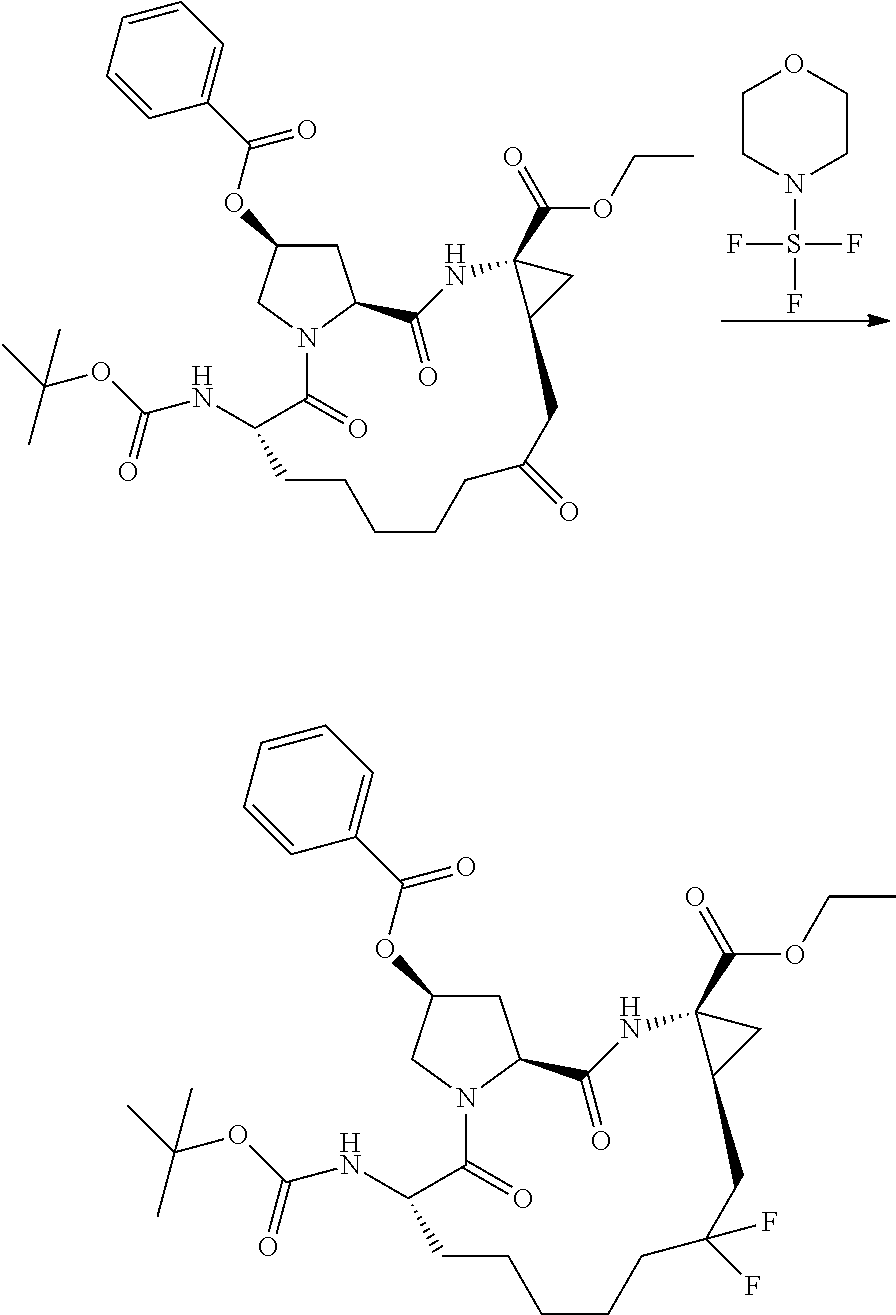
- To (2S,6S,13aS,14aR,16aS)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonylamino)-5,12,16-trioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 1c, 307 mg, 0.500 mmol) was added MORPHOLINOSULFUR TRIFLUORIDE (2.44 rnL, 20.0 mmol). The mixture was sonicated to dissolve all solids. The solution was stirred at room temperature for 16 h., diluted with ethyl acetate, and added dropwise to a mixture of ice and aq. sodium carbonate (10%, 5 ml). The layers were separated, and the aqueous layer was back-extracted with ethyl acetate (10 ml×2). The combined organic layer was washed with 50% brine and dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure to give a light yellow oil (75 mg). This material was purified by reverse phase HPLC (acetonitrile/water (0.1?/WITA)=50/50 to 100/0, 15 min) to provide the title compound (2S,6S,13aS,14aR,16aS)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonyl amino)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (64.9 mg, 0.102 mmol, 20.4% yield).
-
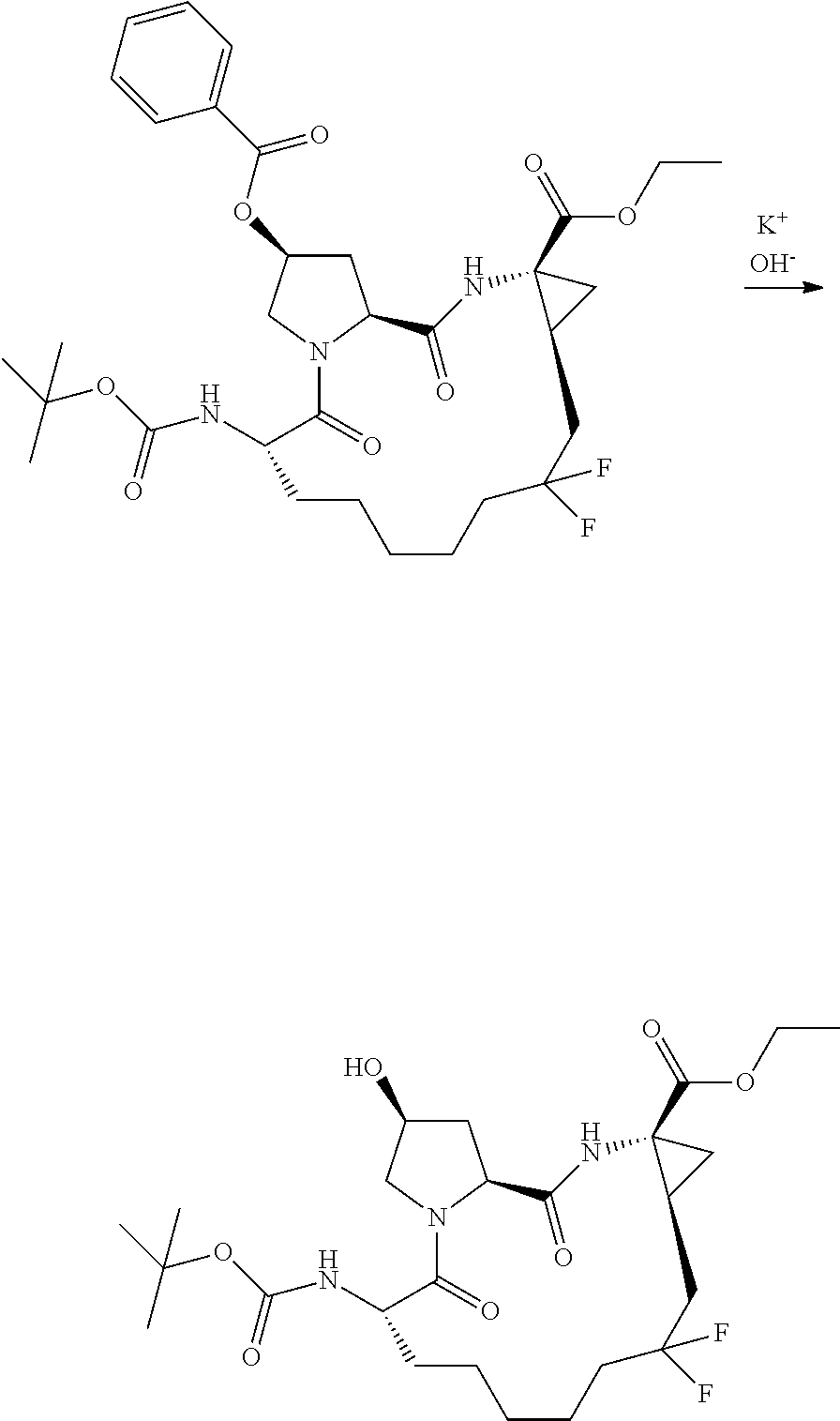
- To a solution of (2S,6S,13aS,14aR,16aS)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonylamino 12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 1d, 94.75 mg, 0.149 mmol) in ethanol (0.414 ml) at 0° C. was added dropwise a solution of KOH (0.25 ml, 0.250 mmol, 1 N in ethanol). The solution was stirred at 0° C. for 50 min and LC/MS showed completion. The reaction mixture was quenched by dropwise addition of HCl (1 N in dioxane/ethyl acetate, 0.25 ml) at 0° C. Another portion of (2 S,6S,13 aS,14aR,16aS)-ethyl 2-(benzoyloxy)-6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (14.36 mg) was reacted under similar condition. Materials from these two reactions were combined and purified by reverse phase HPLC (acetonitrile/water (0.1% TFA)=30/70 to 100/0, 15 min) to provide the title compound (2S,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (91.3 mg) MS (ESI): m/z=532.2 [M+H].
-
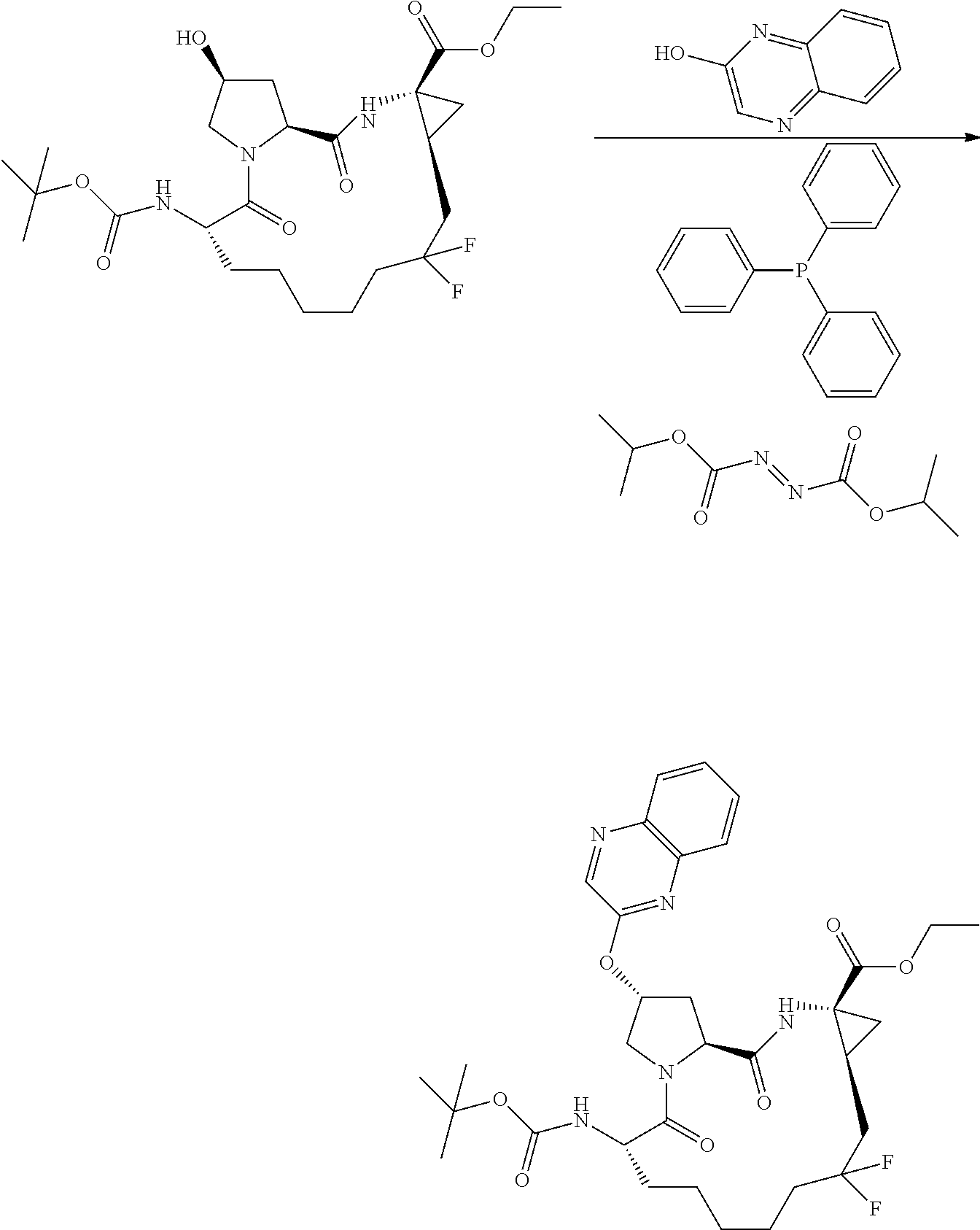
- To (2S,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxooctadecahydrocyclopmpa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 1e, 19 mg, 0.036 mmol) was added 2-hydroxyquinoxaline (8.06 mg, 0.055 mmol) followed by tetrahydrofuran (357 μl). Triphenylphosphine (15.23 mg, 0.058 mmol) was added and the solution was cooled to 0° C. DIAD (10.5 μl, 0.054 mmol) was added dropwise and the mixture was stirred at 0° C. for 15 min and at room temperature for 20 hr. The reaction mixture was diluted with dichloromethane and filtered through Celite. The solvent was evaporated under reduced pressure and the residue was purified by prep-TLC (dichloromethane/ethyl acetate=3/2, 2×) to provide the title compound (2R,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (14.6 mg, 61.8% yield).
-
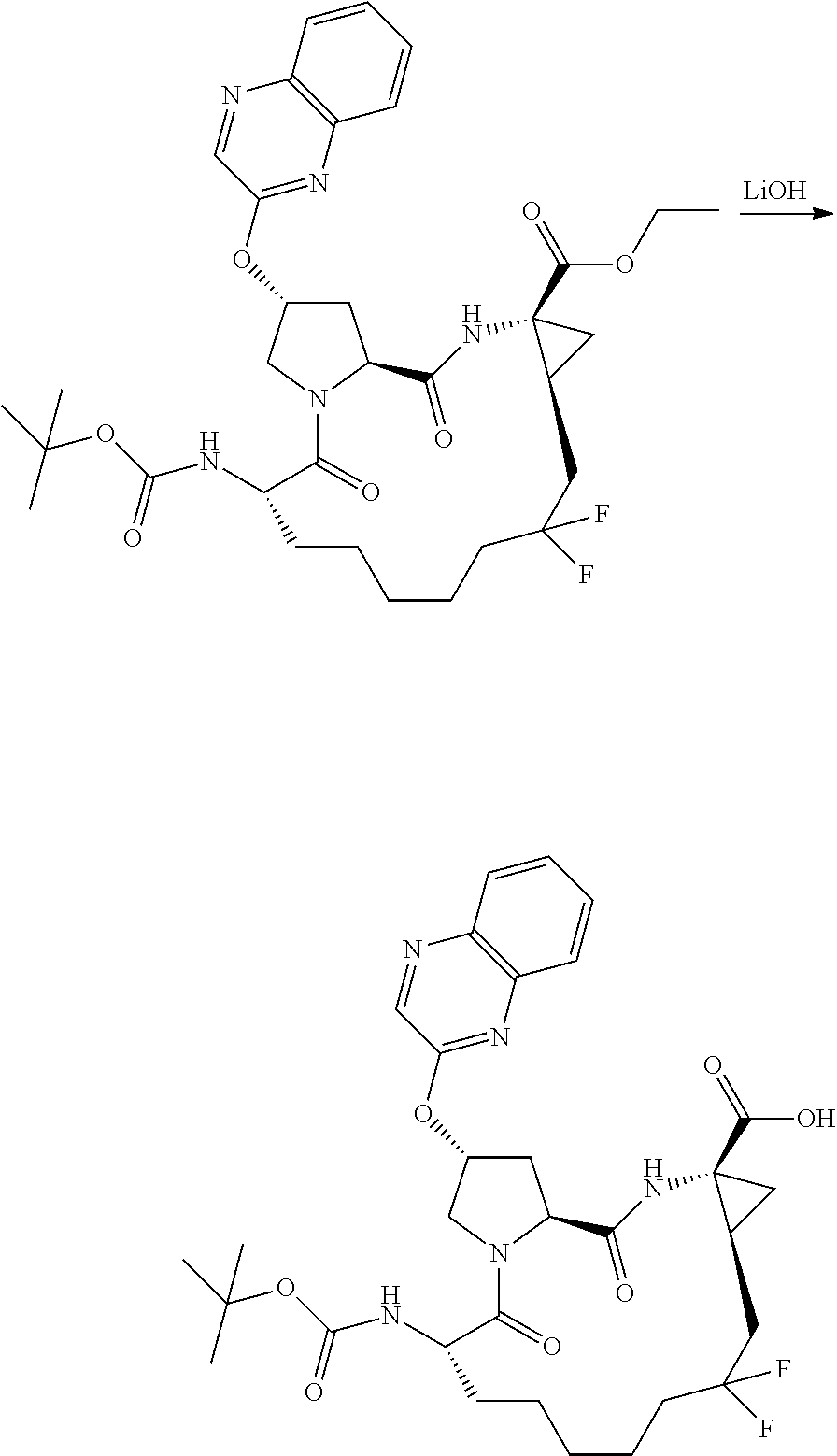
- To a solution of (2R,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 1 h, 14.6 mg, 0.022 mmol) in tetrahydrofuran (55.3 μl), ethanol (27.6 μl) and Water (27.6 μl) was added lithium hydroxide monohydrate (4.19 mg, 0.100 mmol). The reaction mixture was stirred at room temperature overnight, diluted with ethyl acetate and cooled to 0° C. The reaction mixture was neutralized with HCl (4 N in dioxane, 25 μl; diluted with ethyl acetate), stirred at room temperature and dried over anhydrous sodium sulfate. The mixture was filtered and evaporated under reduced pressure and the residue was purified by prep-TLC (elute with dichloromethane/methanol=5/1) to provide the title compound (2R,6S,13aS,14aR,16aS)-6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocycloproparelpyrrolo[1,2-a][1,4]diazacyclopentadeeine-14a-carboxylic acid (I 1.04 mg, 0.017 mmol, 79% yield).
-

- To a solution of (2R,6S,13aS,14aR,16aS)-6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadeeahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopenta decine-14a-carboxylic acid (Example 1 g, 11.0 mg, 0.017 mmol) in dichloroethane (175 μl) was added molecular sieves (3 Å) and carbonyl diimidazole (4.65 mg, 0.029 mmol). The reaction mixture was stirred at 40° C. for 1.5 hr and cooled to room temperature. Cyclopropanesulfonamide (4.24 mg, 0.035 mmol) was added followed by DBU (5.5 μl, 0.036 mmol). The mixture was stirred at room temperature for 2 hr, diluted with dichloromethane, cooled to 0° C., and neutralized with HCl (23.5 μl of 4 N in dioxane and diluted with ethyl acetate). The mixture was filtered to remove sieves and the filtrate was concentrated under reduced pressure. The residue was purified by prep-TLC (dichloromethane/methanol=20/1) to give a white solid (12 mg, 100% yield). This material was repurified by prep-TLC (dichloromethane/ethyl acetate/methanol=1/1/0.02) to provide the title compound tert-butyl (2R,6S,13 aS,14aR,16aS)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydmcyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate (10.3 mg, 0.014 mmol, 80% yield) MS (ESI): m/z=735.4 [M+H]. Example 1 provided an IC50 of >1.0 nM in a 1a enzyme assay; an IC of >1.0 nM in a 1b enzyme assay; and a HLM stability value of between 50-100 μl/min/mg.
-
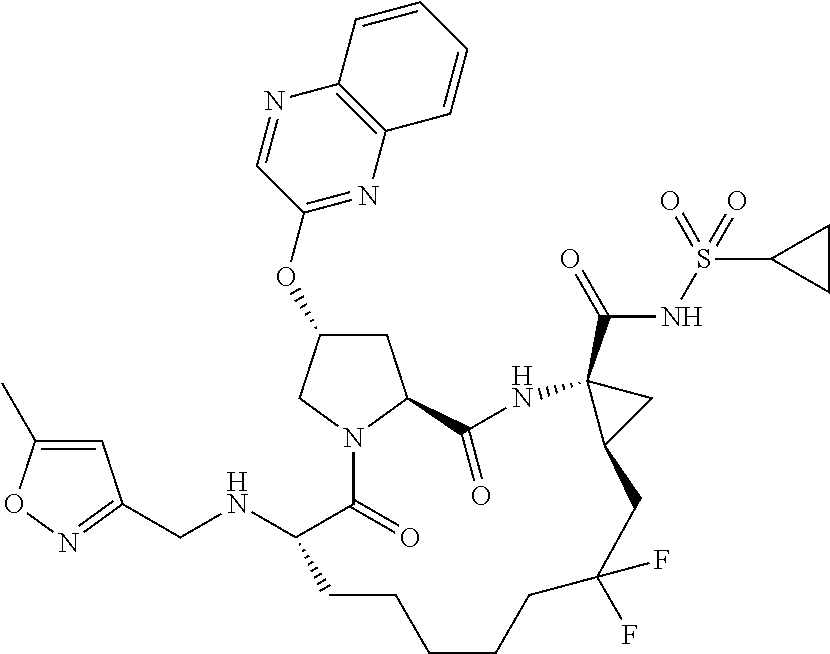
-
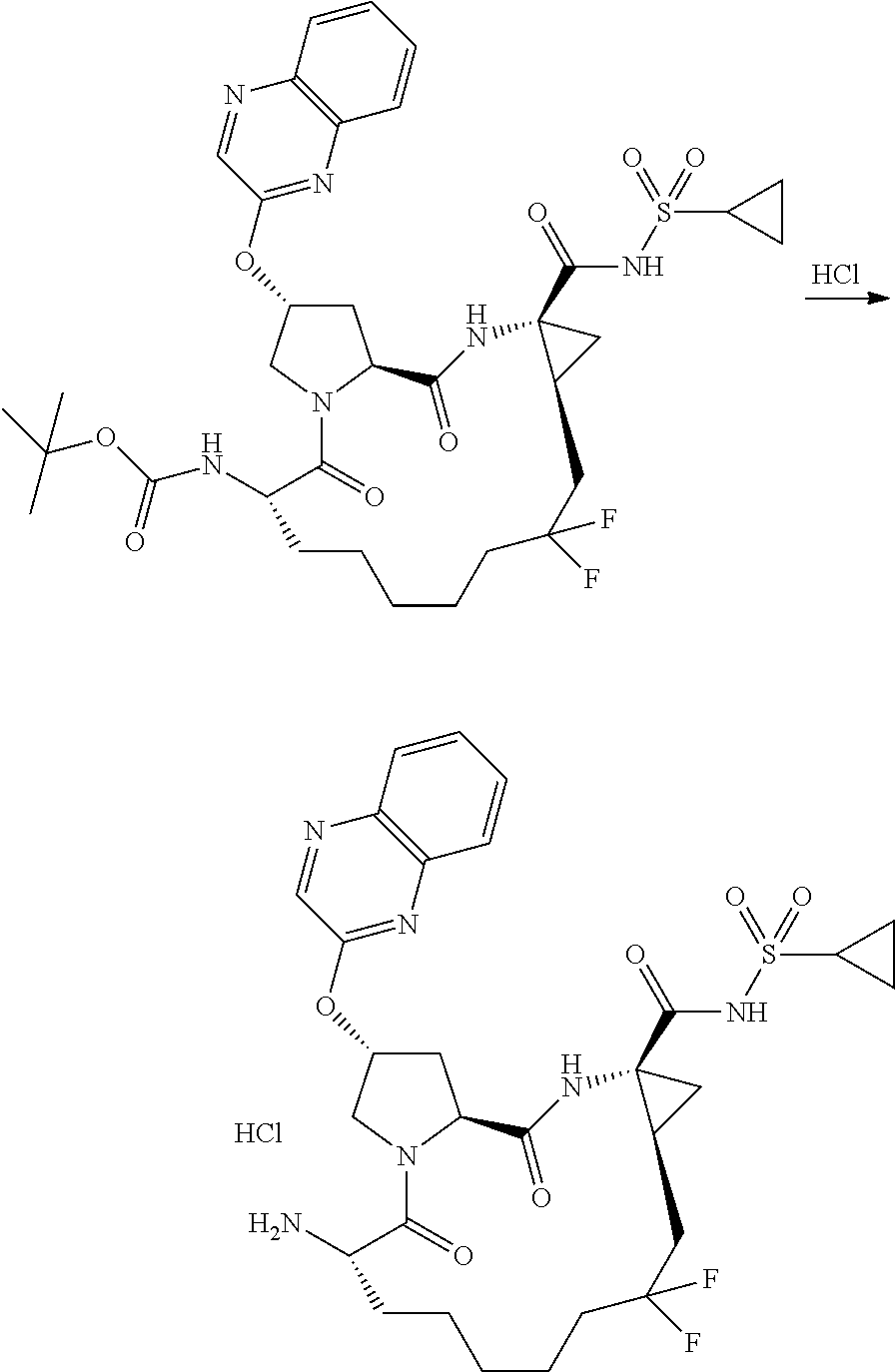
- To tert-butyl (2R,6S,13aS,14aR,16aS)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate (Example 1, 8.81 mg, 0.012 mmol) was added ethyl acetate (60 μl) followed by HCl (4 N in dioxane, 60 μl, 0.240 mmol). The solution was stirred at room temperature for 4 hr (a solid appeared), and LC/MS showed completion. The reaction mixture was concentrated to give the title compound (2R,6S,13aS,14aR,16aS)-6-amino-N-(cyclopropylsulfonyl)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocyclo propa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide, hydrochloric acid (8.05 mg, quantitative yield).
-

- To a solution of (2R,6S,13aS,14aR,16aS)-6-amino-N-(cyclopropylsulfonyl)-12,2-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide, Hydrochloric Acid (Example 2a, 8.05 mg, 0.012 mmol) in DMF (120 μl) was added pyridine (7 μl, 0.087 mmol) followed by 5-methylisoxazole-3-carboxylic acid (2.28 mg, 0.018 mmol) and then HATU (7.04 mg, 0.019 mmol). The reaction mixture was stirred at room temperature overnight, diluted with ethyl acetate, cooled to 0° C. and neutralized with HCl (4 N in dioxane, 22 μl; diluted with ethyl acetate). The mixture was concentrated and purified by prep-TLC (ethyl acetate) to provide the title compound N-((2R,6S,13aS,14aR,16aS)-14a-(cyclopropyisulfonylcarbamoyl)-12,12-difluoro-5,16-dioxo-2-(quinoxalin-2-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-yl)-5-methylisoxazole-3-carboxamide (7.65 mg, 10.29 μmol, 86% yield) MS (ESI): m/z=744.3 [M+H].
- Example 2 provided an IC50 of >1.0 nM in a 1a enzyme assay; an IC50 of >1.0 nM in a 1b enzyme assay; a HLM stability value of <50 μl/min/mg; an EC50 of >5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of >5.0 nM in a replicon cell line assay in a 1b con1 background.
-
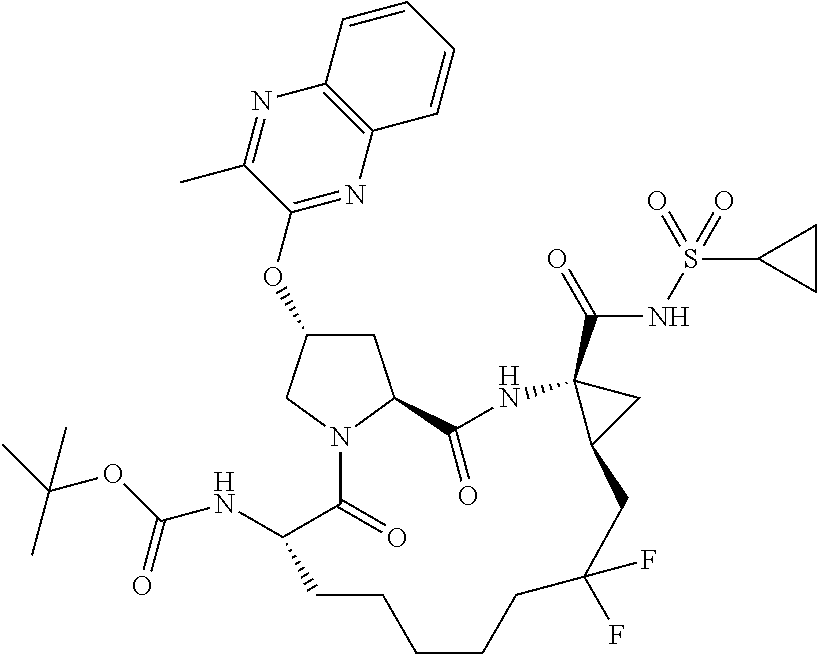
-

- Example 3a was prepared according to the procedure utilized for the preparation of Example if, replacing 2-quinoxalinol with 3-methyl-2-quinoxalinol.
-
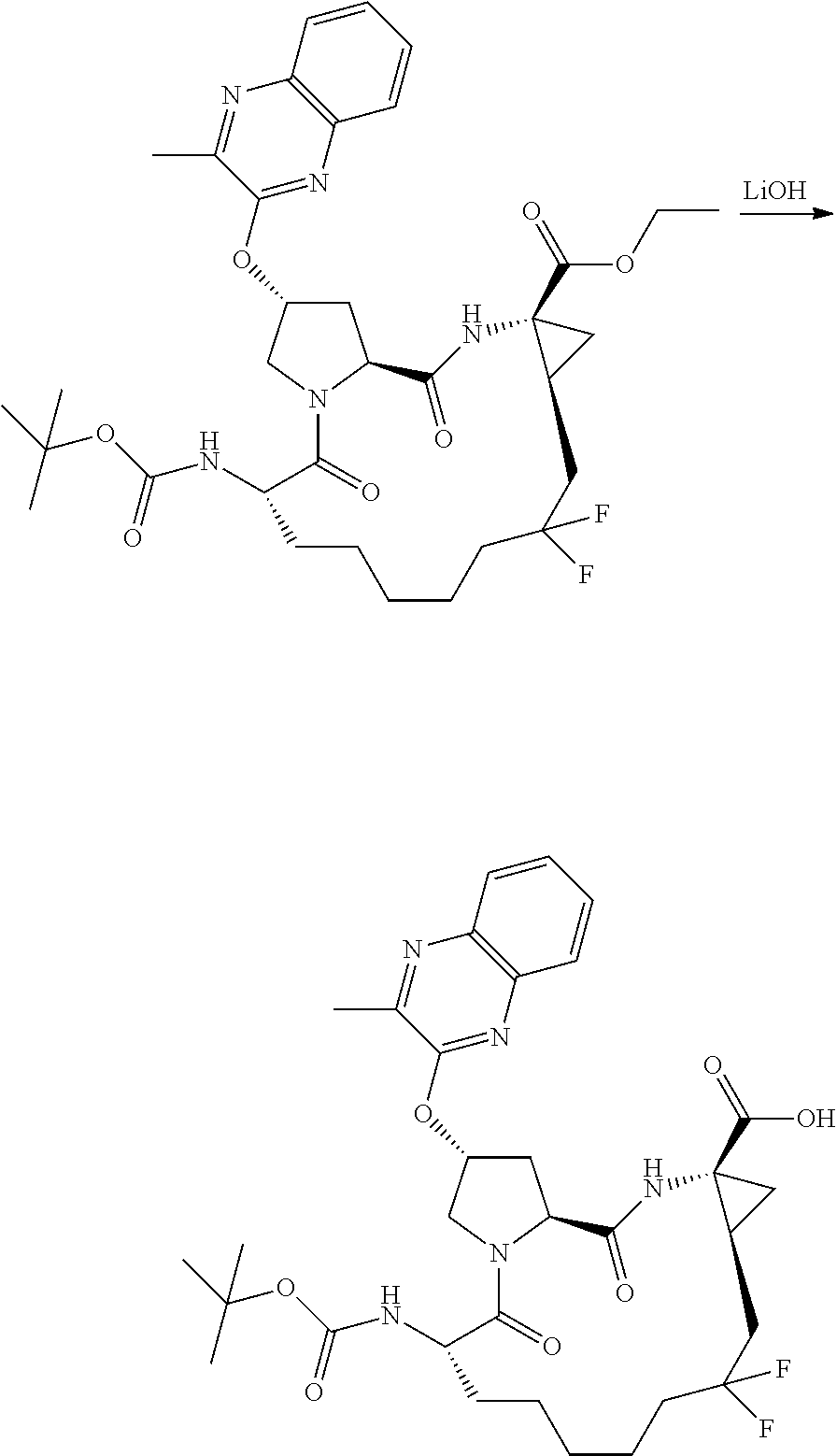
- Example 3b was prepared according to the procedure utilized for the preparation of Example 1 g, replacing the product of Example 1 f with the product of Example 3a.
-

- Example 3 was prepared according to the procedure utilized for the preparation of Example 1, replacing the product of Example 1 g with the product of Example 3b. MS (ESI): m/z=749.3 [M+H].
- Example 3 provided an IC50 of between 0.1 and 0.5 nM in a 1a enzyme assay; and an IC50 of between 0.1 and 0.5 nM in a 1b enzyme assay.
-
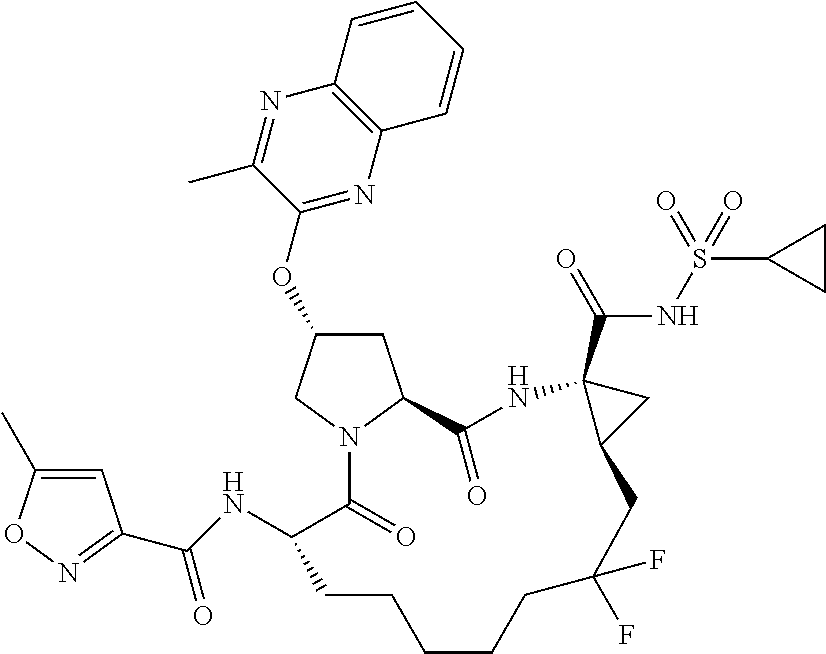
-
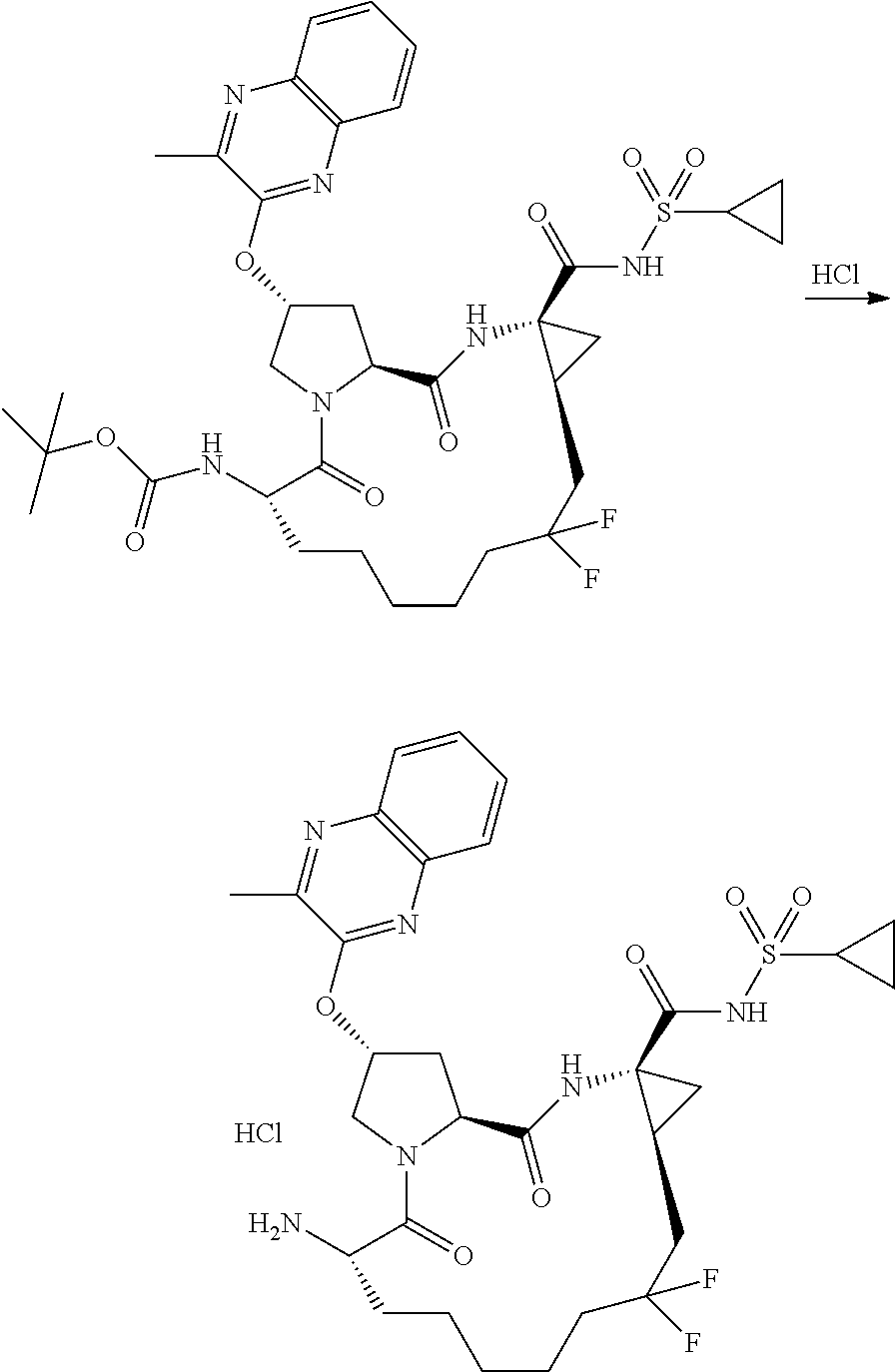
- Example 4a was prepared according to the procedure utilized for the preparation of Example 2a, replacing the product of Example 1 with the product of Example 3.
-
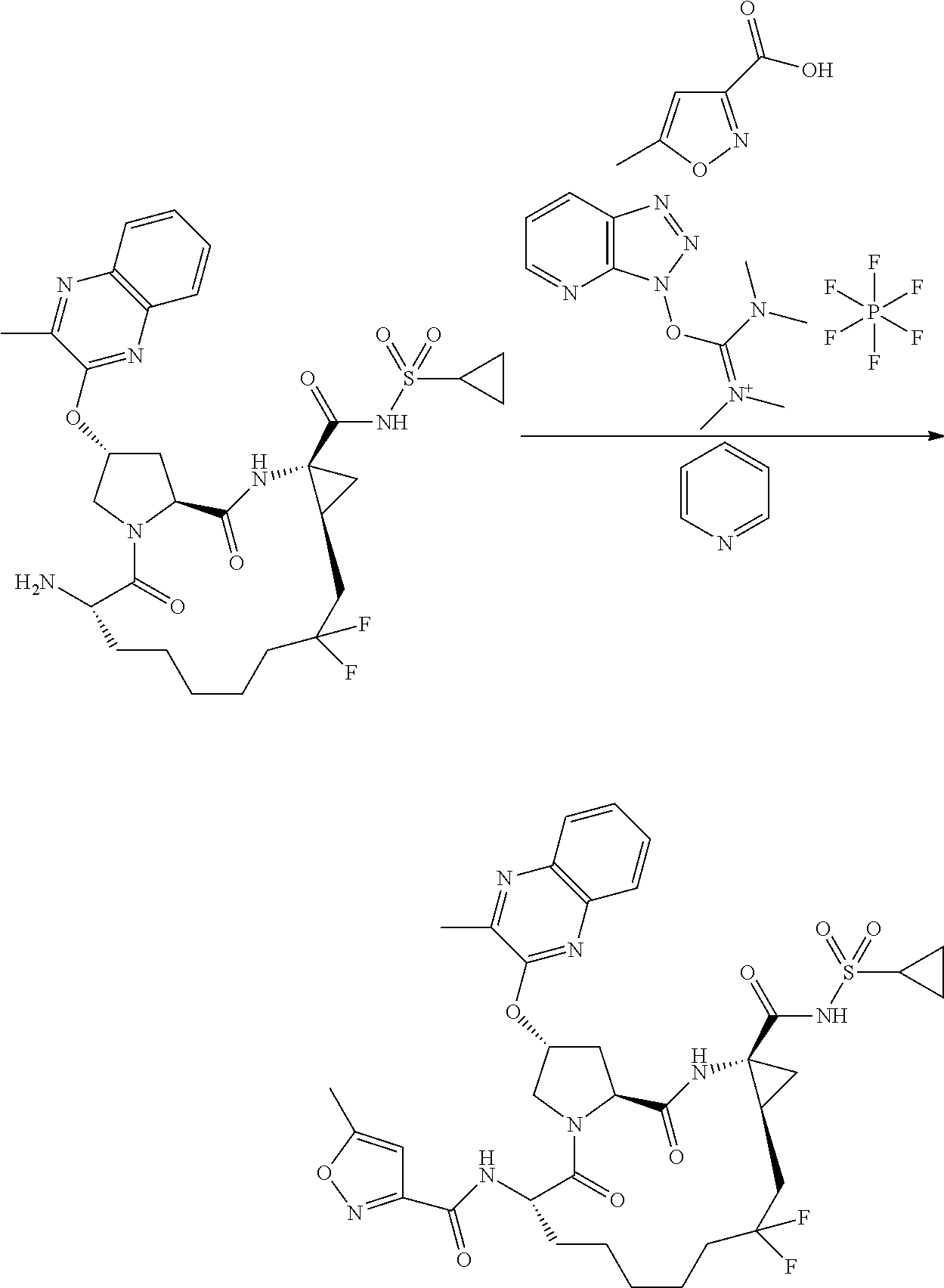
- Example 4 was prepared according to the procedure utilized for the preparation of Example 2, replacing the product of Example 2a with the product of Example 4a. MS (ESD: m/z=758.3 [M+H].
- Example 4 provided an IC50 of between 0.1 and 0.5 nM in a 1a enzyme assay; an IC50 of between 0.5 and 1.0 nM in a 1b enzyme assay; a FILM stability value of <50 μl/min/mg; an EC50 of >5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1 b-con1 background.
-

-
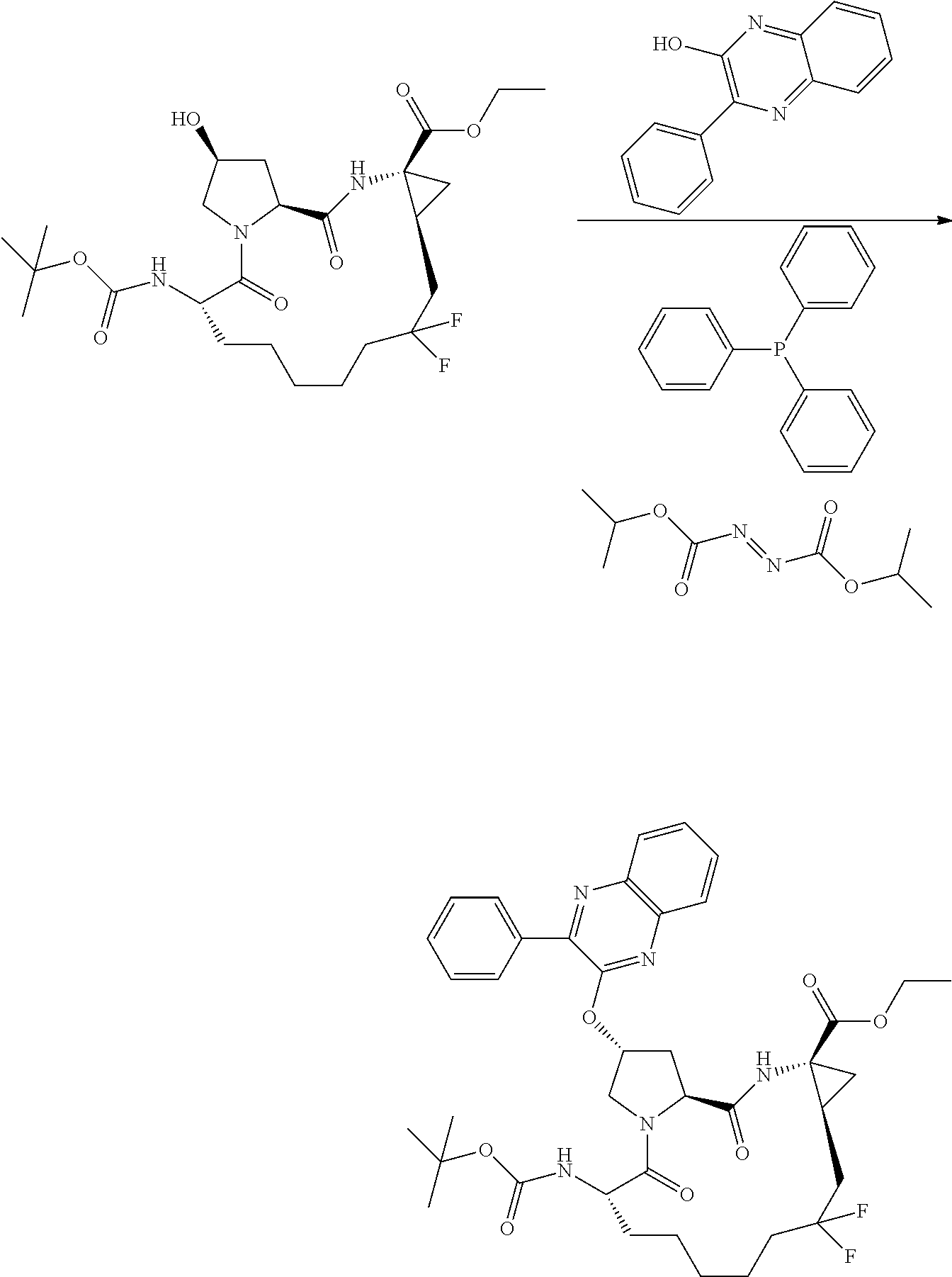
- Example 5a was prepared according to the procedure utilized for the preparation of Example if, replacing 2-quinoxalinol with 3-phenyl-2-quinoxalinol.
-

- Example 5b was prepared according to the procedure utilized for the preparation of Example 1 g, replacing the product of Example if with the product of Example 5a.
-

- Example 5 provided an IC50 of between 0.5 and 1.0 MV in a 1a enzyme assay; and an IC50 of between 0.5 and 1.0 nM in a 1b enzyme assay.
-
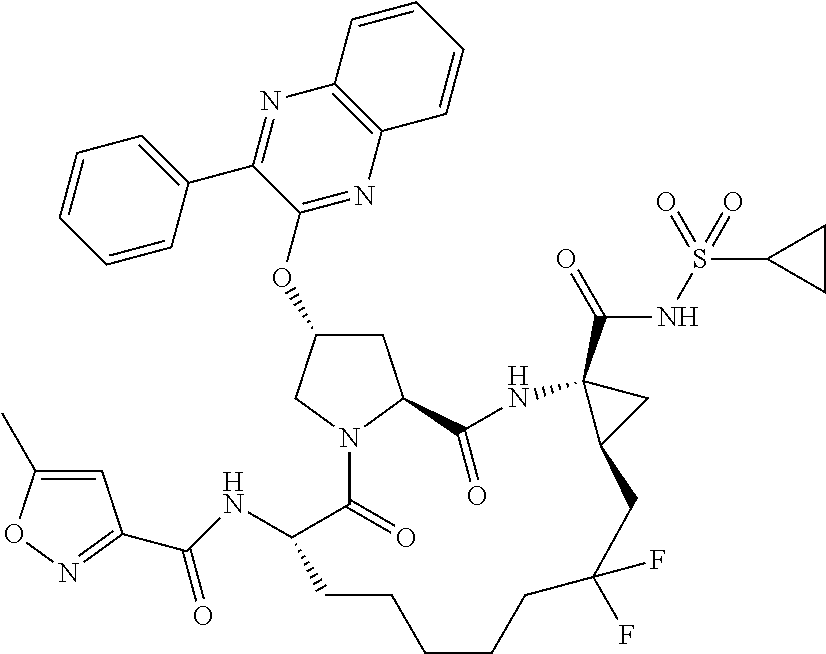
-

- Example 6a was prepared according to the procedure utilized for the preparation of Example 2a, replacing the product of Example 1 with the product of Example 5.
-

- Example 6 was prepared according to the procedure utilized for the preparation of Example 2, replacing the product of Example 2a with the product of Example 6a, MS (ESI): m/z=820.3 [M+H].
- Example 6 provided an IC50 of <0.1 nM in a 1a enzyme assay; an IC50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a HLM stability value of <50 μl/min/mg; an EC50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
-
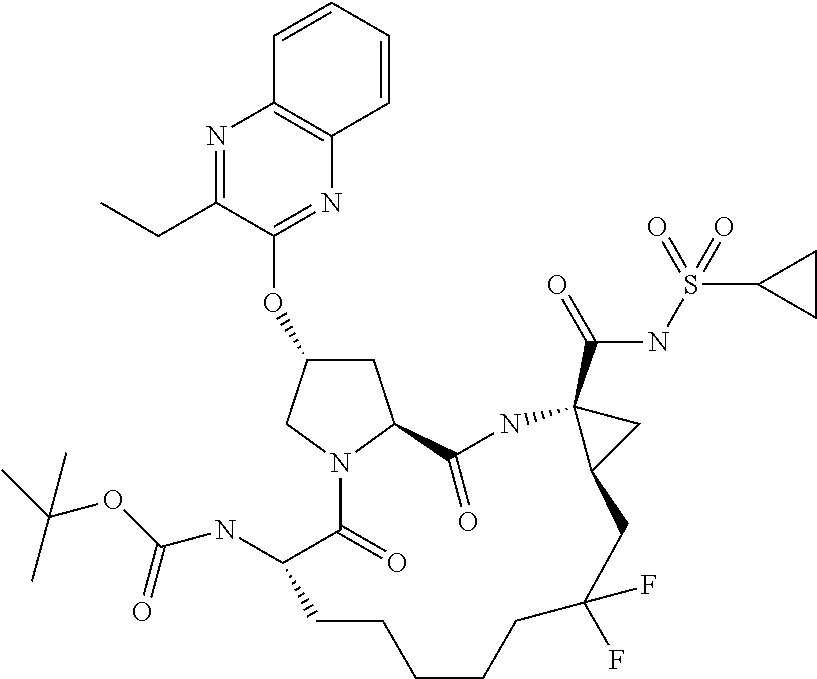
-
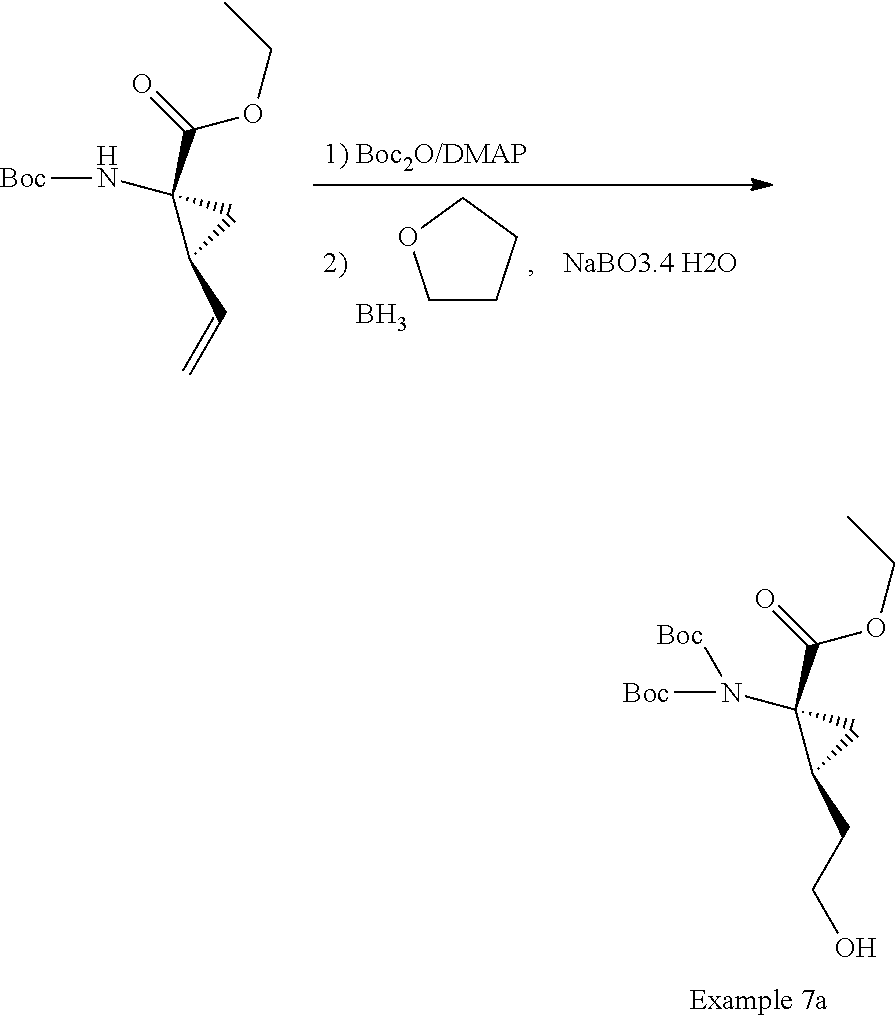
- To (1R,2S)-ethyl 1-(tert-butoxycarbonylamino)-2-vinylcyclopropanecarboxylate (10 g, 39.2 mmol) and Boc anhydride (22.73 mL, 98 mmol) dissolved in tetrahydrofuran (25 mL) at room temperature was added portionwise DMAP (3.83 g, 31.3 mmol) and the mixture was stirred for 3 h. The mixture was quenched with water and extracted with ethyl acetate. The organic layer was washed with 0.1 N HCl followed by saturated aqueous sodium chloride solution, separated, dried over anhydrous magnesium sulfate, filtered, and evaporated under reduced pressure. The residual oil was purified by flash chromatography on silica gel eluting with a 5-15% ethyl acetate/hexane gradient to provide (1R,2S)-ethyl 1-(bis(tert-butoxycarbonyl)amino)-2-vinylcyclopropanecarboxylate (13.2 g, 37.1 mmol, 95% yield) as a clear oil. MS (ESI): m/z=378.0 [M+Na].
- To a solution of (1R,2S)-ethyl 1-(bis(tert-butoxycarbonyl)amino)-2-vinylcyclopropane carboxylate (13.2 g, 37.1 mmol) in tetrahydrofuran (50 mL) at −15° C. was added dropwise 1 N borane tetrahydrofuran complex (74.3 mL, 74.3 mmol) and the mixture was stirred for 2 h at −15° C. To the resulting mixture stirring at 0° C. was added dropwise water (100 mL) followed by sodium perborate tetrahydrate (11.43 g, 74.3 mmol), and the mixture was stirred for 16 hr at rt. The reaction mixture was diluted with ethyl acetate and water, and the resulting organic layer was washed with saturated aqueous sodium chloride solution, separated, dried over anhydrous magnesium sulfate, filtered, and the solvent evaporated under reduced pressure. The residue was purified via flash chromatography on silica gel (10-20% acetone/hex). The resulting impure material was repurified by flash chromatography on silica gel (eluting with a 25%-50% ethyl acetate/hexane gradient) to give (1R,2S)-ethyl 1-(bis(tert-butoxycarbonyl)amino)-2-(2-hydroxyethyl)cyclopropanecarboxylate (9.15 g, 24.50 mmol, 66.0% yield) as an oil. MS (ESI): m/z=396.0 [M+Na].
-
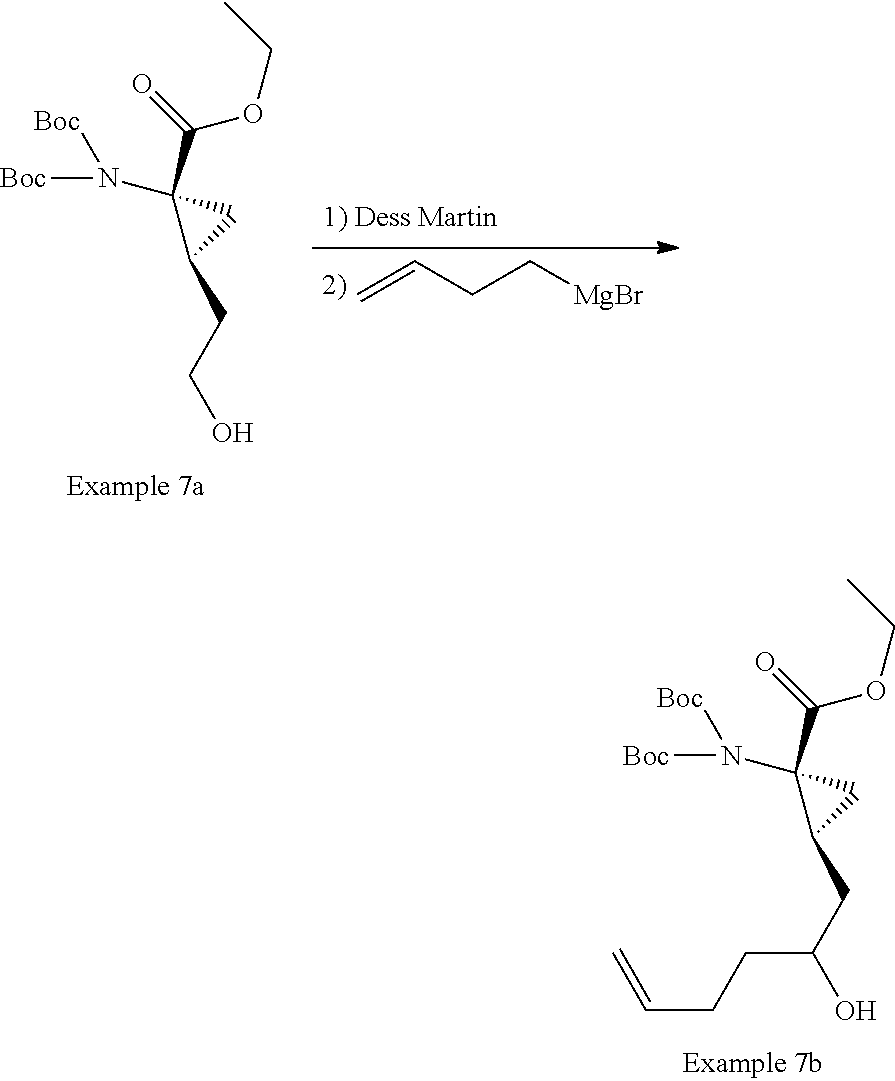
- To a solution of (1R,2S)-ethyl 1-(bis(tert-butoxycarbonyl)amino)-2-(2-hydroxyethyl)cyclopropanecarboxylate (8.65 g, 23.2 mmol) in dichloromethane (116 ml) at 25° C. was added Doss-Martin Periodinane (13.8 g, 32.4 mmol). The mixture was stirred for 3 h and quenched with 1 N sodium thiosulfate (70 mL) and aqueous saturated sodium bicarbonate solution (30 mL), and stirred for 1 h. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. This material was purified by flash chromatography on silica gel (eluted with 25% ethyl acetate/hexane) to yield (1R,2S)-ethyl 1-(bis(tert-butoxycarbonyDamino)-2-(2-oxoethyl)cyclopropanecarboxylate (7.05 g, 82% yield) as an oil. MS (ESI): m/z=394.1 [M+Na].
- To (1R,2S)-ethyl 1-(bis(tert-butoxycarbonyl)amino)-2-(2-oxoethyl)cyclopropanecarboxylate (7.05 g, 19.0 mmol) in tetrahydrofuran (60 mL) at −78° C. was added but-3-enylmagnesium bromide (45.6 mL, 22.78 mmol) and the reaction mixture was stirred for 30 min. The mixture was then quenched with saturated aqueous ammonium chloride and extracted with ethyl acetate. The organic layer was separated, dried over anhydrous magnesium sulfate, filtered, and evaporated under reduced pressure. The residual oil was purified via flash chromatography on silica gel (25% hexane/ethyl acetate) to provide the title compound (2.75 g, 34%) as an oil, along with recovered starting material (2.89 g, 41%). MS (ESI): m/z=450.1 [M+Na].
-
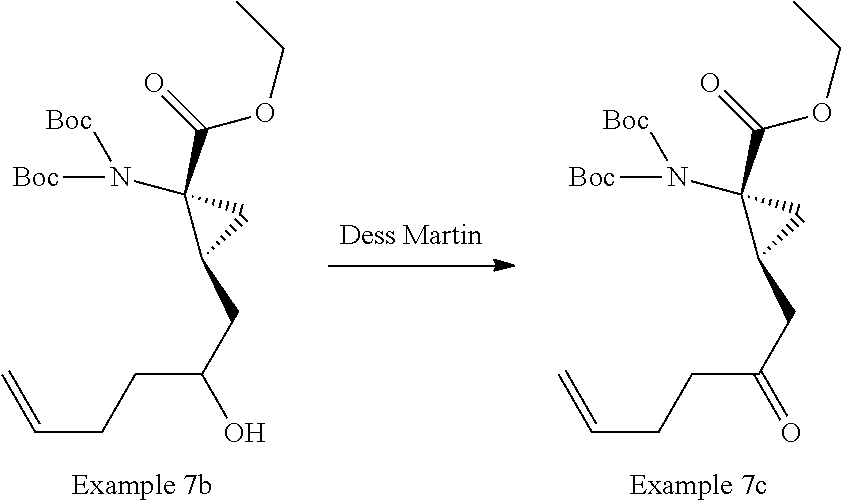
- To a solution of (1R,2S)-ethyl 1-(bis(tert-butoxycarbonyl)amino)-2-(2-hydroxyhex-5-enyl)cyclopropanecarboxylate (4.3 g, 10 mmol) in dichloromethane (50 ml) at 25° C. was added Dess-Martin Periodinane (5.97 g, 14.1 mmol). The reaction mixture was stirred for 3 h and quenched with 1 N aqueous sodium thiosulfate solution (50 mL) and saturated aqueous sodium bicarbonate solution (15 mL), and the reaction mixture stirred for 1 h. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and evaporated under reduced. The crude product was purified by flash chromatography on silica gel (15% ethyl acetate/hexane) to provide (1R,2S)-ethyl 1-(bis(tert-butoxycarbonyl)amino)-2-(2-oxohex-5-enyl)cyclopropane carboxylate (3.8 g, 88% yield) as an oil. MS (ESI): na/z=448.1 [M+Na]
-
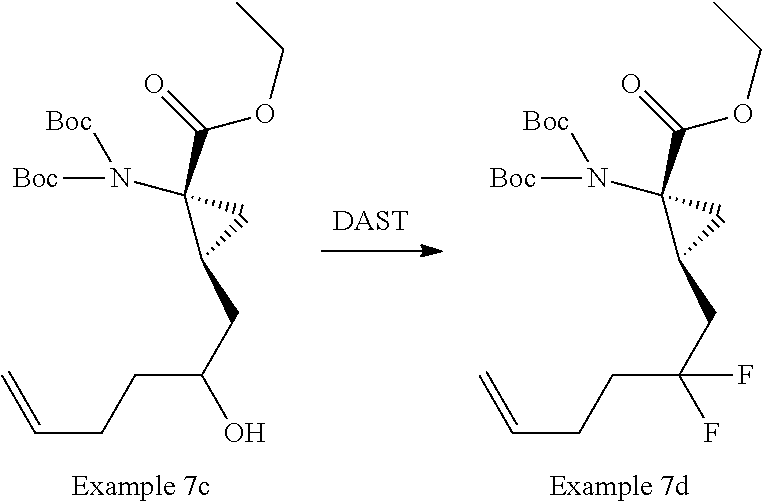
- A solution of (1R,2S)-ethyl 1-(bis(tert-butoxycarbonyl)amino)-2-(2-oxohex-5-enyl)cyclopropanecarboxylate (338 g, 8.88 mmol) in DAST (5.87 ml, 44.4 mmol) was stirred at room temperature for 3 days The reaction mixture was cooled to −78° C., diluted with 50 mL dichloromethane and added in a stream to a 0° C. solution of saturated aqueous sodium bicarbonate solution (200 mL). The reaction mixture was stirred for 30 min and the organic layer was separated, dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The crude material was purified by flash chromatography on silica gel (10-15% ethyl acetate/hexane gradient) to provide the title compound (1.95 g, 49%) as an oil. MS (ESI): m/z=470.1 [M+Na].
-
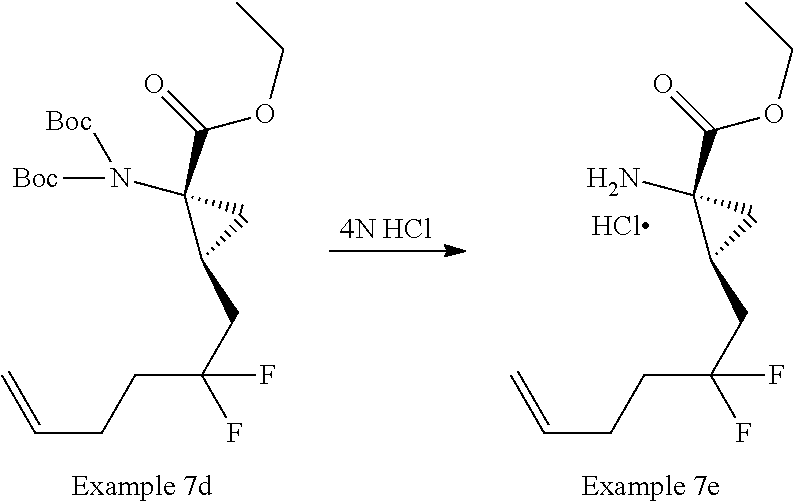
- A solution of (1R,2S)-ethyl 1-(bis(tert-butoxycarbonyl)amino)-2-(2,2-difluorohex-5-enyl)cyclopropanecarboxylate (2.45 g, 5.47 mmol) in 1,4-dioxane (6 mL) and 4 N HCl (11.6 mL, 46.5 mmol) was stirred at room temperature for 3 h. The mixture was evaporated and eluted through a silica gel (50 mL) column with 4-10% methanol/dichloromethane to give (1R,2S)-ethyl 1-amino-2-(2,2-difluorohex-5-enyl)cyclopropanecarboxylate, hydrochloric acid salt (1.4 g, 4.93 mmol, 90% yield) as a yellow oil. MS (ESI): m/z 247.9 [M+H].
-
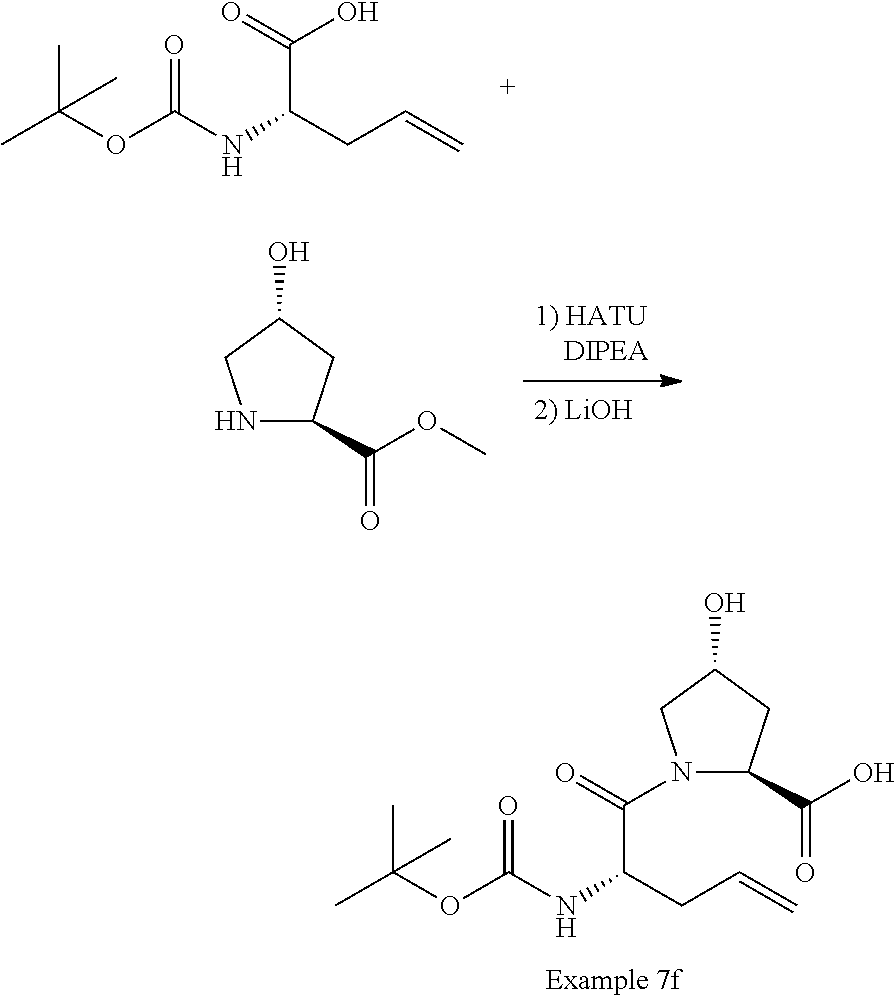
- A mixture of (S)-2-(tert-butoxycarbonylamino)pent-4-enoic acid (3.0 g, 13.9 mmol), and 2-(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yl)-1,1,3,3-tetramethylisouronium hexafluorophosphate(V) (5.83 g, 15.3 mmol) in DMF (15 ml) was stirred for 5 mm at room temperature. To this mixture was added (2S,4R)-methyl 4-hydroxypyrrolidine-2-carboxylate, hydrochloric acid salt (2.78 g, 15.33 mmol) followed by the dropwise addition of N-ethyl-N-isopropylpropan-2-amine (8.52 ml, 48.8 mmol), and the reaction mixture was stirred 16 h at room temperature. The mixture was diluted with ethyl acetate, washed with 1 N aqueous sodium bicarbonate solution followed by 0.1 N HCl, and the organic layer was separated, dried over anhydrous magnesium sulfate, filtered, and evaporated under reduced pressure. The oily residue after was purified by flash chromatography on silica gel (4% methanol/dichloromethane) to provide the title compound as an oil (2S,4R)-methyl 1-((S)-2-(tert-butoxycarbonylamino)pent-4-enoyl)-4-hydroxypyrrolidine-2-carbo xylate (3.0 g, 8.76 mmol, 62.9% yield). MS (ESI): m/z=342.9 [M+H].
- To a solution of (2S,4R)-methyl 1-((S)-2-(tert-butoxycarbonylamino)pent-4-enoyl)-4-hydroxypyrrolidine-2-carboxylate (3.0 g, 8.76 mmol) in tetrahydrofuran (29.2 ml), water (14.6 ml), and ethanol (14.6 ml) stirring at room temperature was added lithium hydroxide monohydrate (2.39 g, 57.0 mmol). The mixture was stirred for 16 h, the organic solvent was evaporated under reduced pressure, and the aqueous layer was extracted once with dichloromethane. The aqueous layer was cooled in an ice bath, acidified with 68 mL of 1N HCl, saturated with sodium chloride, and extracted with ethyl acetate. The organic layer was dried (anhydrous sodium sulfate), filtered, and evaporated to a provide the title compound as a white foam (2.83 g, 8.62 mmol, 98% yield. MS (EST): m/z=329.0 [M+1-1].
-
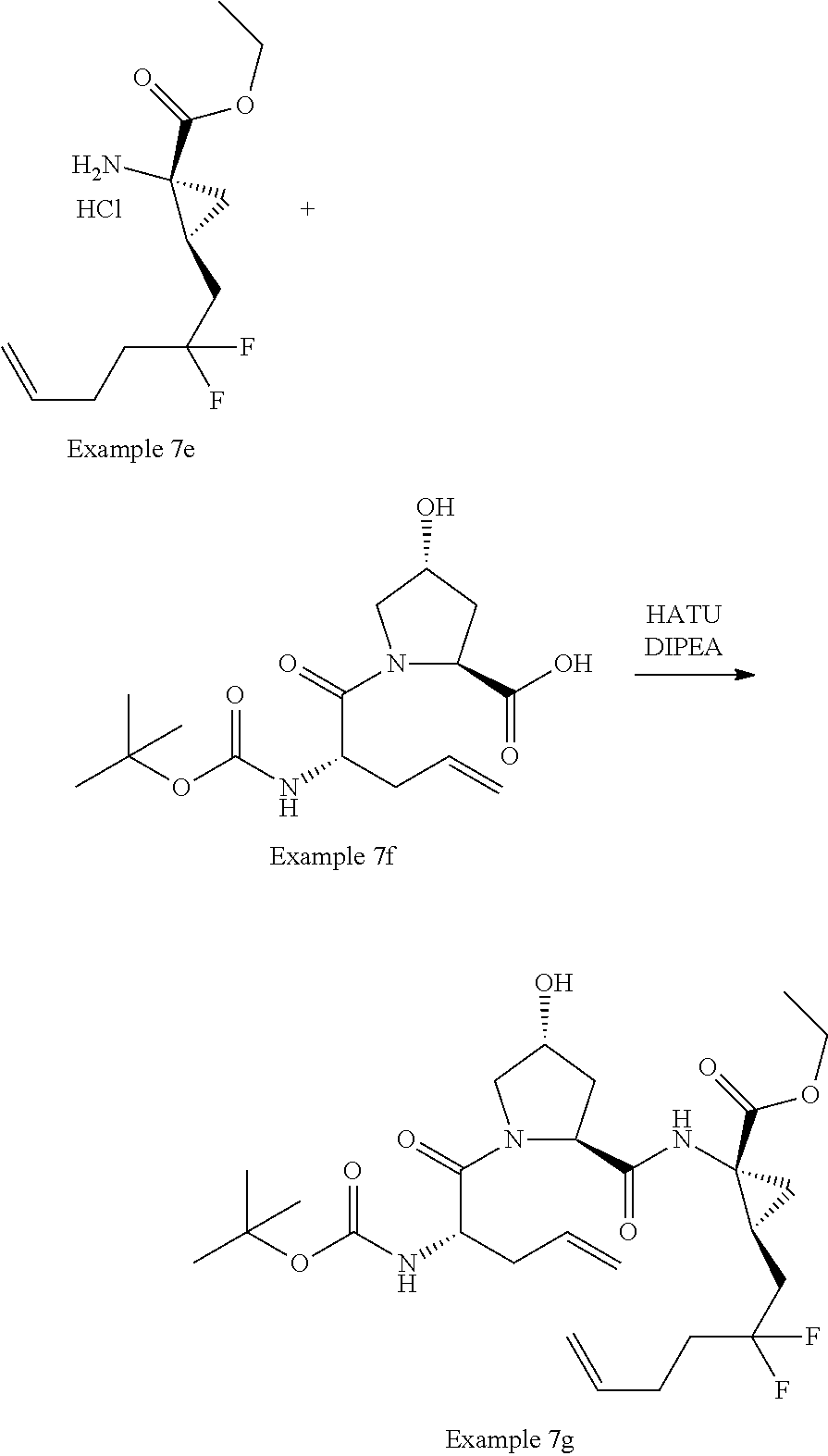
- To a mixture of (2S,4R)-1-((S)-2-(tert-butoxycarbonylamino)pent-4-enoyl)-4-hydroxypyrrolidine-2-carboxylic acid (1.78 g, 5.43 mmol), (1R,2S)-ethyl 1-amino-2-(2,2-difluorohex-5-enyl)cyclopropanecarboxylate (1.4 g, 4.9 mmol) and 2-(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yl)-1,1,3,3-tetramethylisouronium hexafluorophosphate(V) (2.06 g, 5.43 mmol) in dichloromethane (22 mL) was added N-ethyl-N-isopropylpropan-2-amine (3.02 mL, 17.3 mmol), and the mixture was stirred 3 h at room temperature. The reaction mixture was washed with 1 N aqueous sodium bicarbonate solution followed by 1 N HCl and organic layer was separated, dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (5% methanol/dichloromethane). Fractions containing the title compound were evaporated to a yellow oil and repurified by flash chromatogaphy on silica gel (40% acetone/hexane) to give provide the title compound (1R,2S)-ethyl 1-((2S,4R)-1-((S)-2-(tert-butoxycarbonylamino)pent-4-enoyl)-4-hydroxypyrrolidine-2-carboxamido)-2-(2,2-difluorohex-5-enyl)cyclopropanecarboxylate (2.31 g, 84% yield) as a foam. MS (ESI): m/z=558.3 [M+1-1].
-
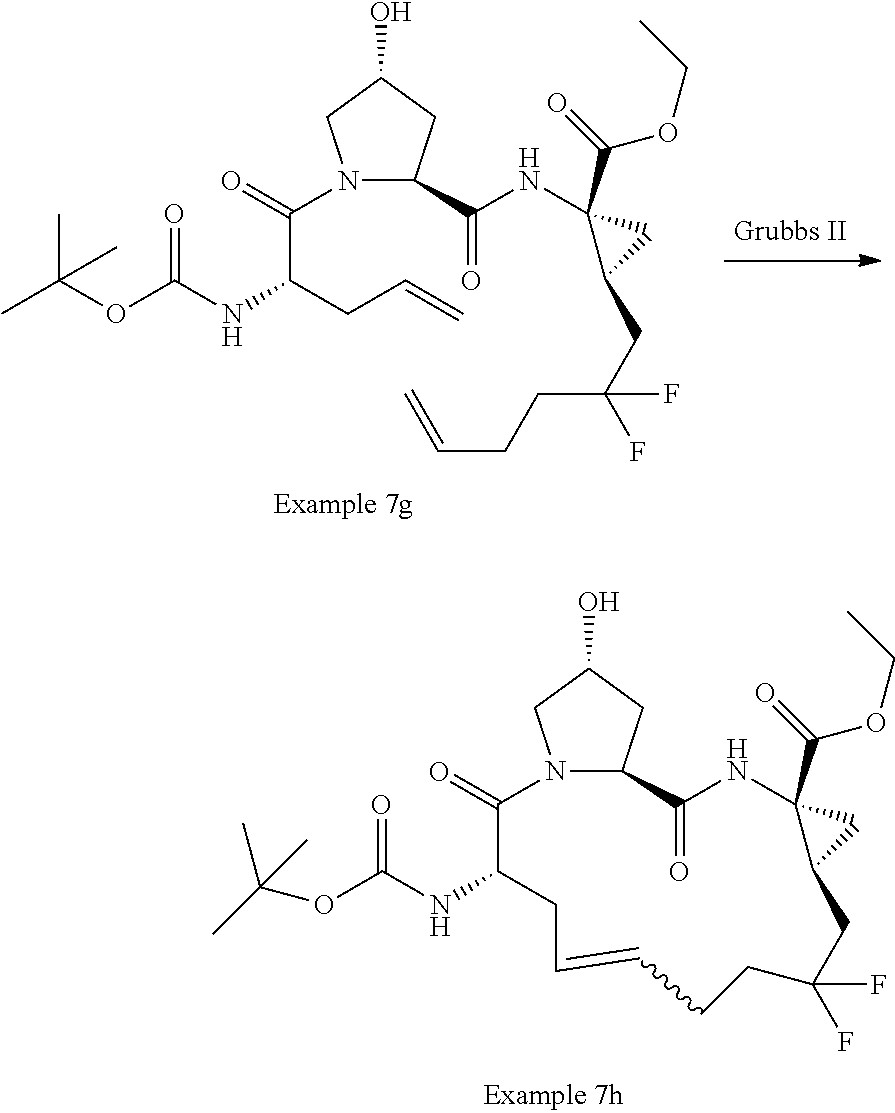
- To a solution of (1R,2S)-ethyl 1-((2S,4R)-1-((S)-2-(tert-butoxycarbonylamino)pent-4-enoyl)-4-hydroxypyrrolidine-2-carboxamido)-2-(2,2-difluorohex-5-enyl)cyclopropanecarboxylate (1.95 g, 3.50 mmol) in dichloroethane (583 ml) which was degassed by bubbling with nitrogen for 45 min was added Grubb 11 catalyst (66 mg, 0.03 eq). The solution was heated at 60° C. for 2 h. Another aliquot of catalyst was added (22 mg, 0.01 eq) and the reaction mixture was heated for an additional 1.5 h. Another aliquot of catalyst (11 mg, 0.005 eq) was added and the reaction mixture was heated for an additional 1 h. The reaction mixture was quenched by the addition of 2-mercaptonicotinic acid (390 mg, 0.74 eq) and stirred at 60° C. for 45 m. The reaction mixture was cooled to 0° C. and 140 mL saturated sodium bicarbonate solution and 280 mL of water was added, and the reaction mixture was stirred for 45 min. The organic layer was separated, dried over anhydrous magnesium sulfate, filtered, and evaporated under reduced pressure. The resulting residue was purified by flash chromatography on silica gel (5% methanol/dichloromethane) to provide (2R,6S,13aS,14aR,16aS,E)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxo-1,2,3,5,6,7,10,11,12,13,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (0.94 g, 1.775 mmol, 50.8% yield) as a white solid. MS (ESI): m/z=530.2 [M+14].
-
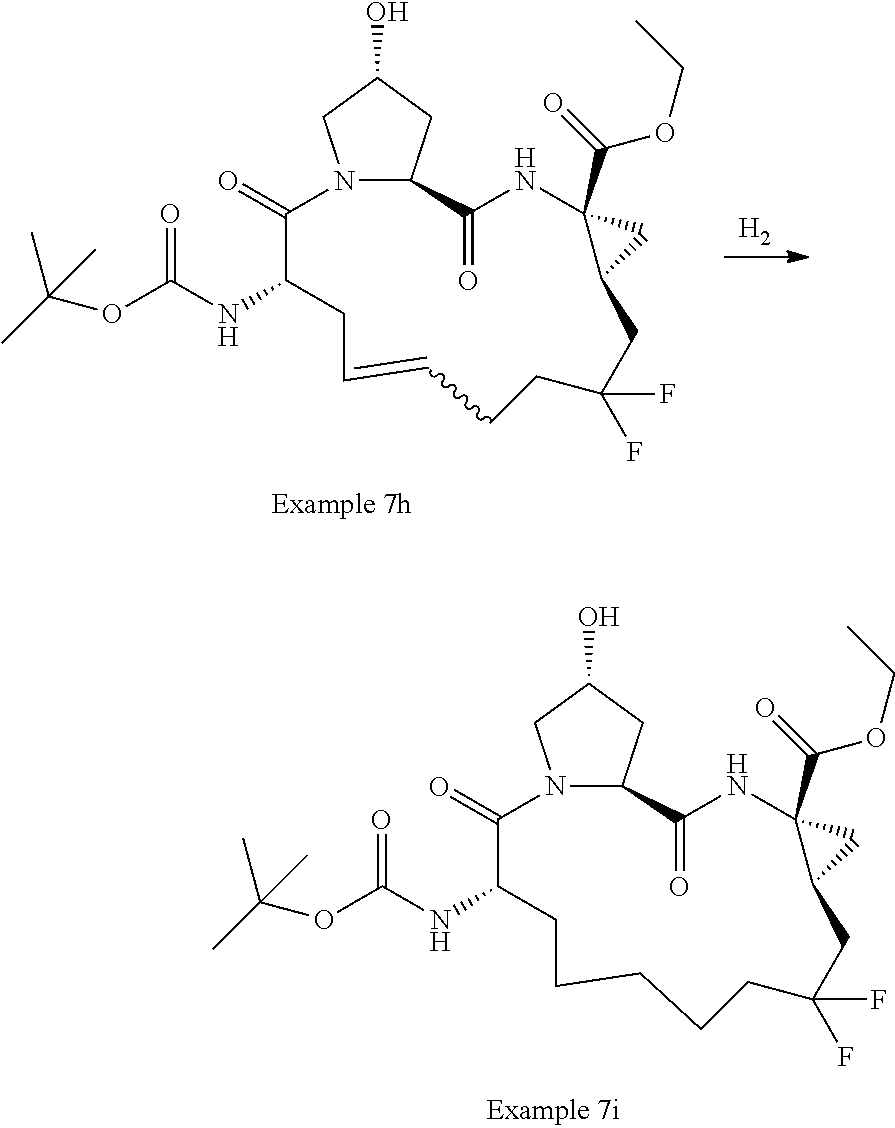
- A solution of (2R,6S,13aS,14aR,16aS,E)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxo-1,2,3,5,6,7,10,11,12,13,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 7 h, 1.06 g, 2.002 mmol) in ethyl acetate (50 ml) at 25° C. was added 10% palladium/carbon (0.170 g, 0.160 mmol). The reaction mixture was stirred under a balloon of hydrogen for 2 days. The reaction mixture was filtered through a small plug of silica gel and eluted with ethyl acetate then methanol. The solvent was evaporated under reduced pressure and the resulting solid was purified by flash chromatography on silica gel (40% ethyl acetate/dichloromethane, then ethyl acetate) to provide (2R,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (1.01 g, 1.900 mmol, 95% yield) as a white solid. MS (ESI): m/z=532.3 [M+H].
-

- To (2R,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonyl amino)-12,12-difluoro-2-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 7i, 900 mg, 1.693 mmol) was added tetrahydrofuran (5.6 ml), methanol (2.80 ml) and water (2.80 ml). Lithium hydroxide monohydrate (145.44 mg, 3.47 mmol) was added at room temperature, and the reaction mixture was stirred at 40° C. for 16 h. The reaction mixture was diluted with CH3CN and neutralized with HCl (2 N, 1.7 ml). The mixture was concentrated under reduced pressure and azeotroped with chloroform followed by ethyl acetate and dried overnight at reduced pressure to provide the title compound (850 mg, quantitative yield). This material was utilized in subsequent reactions without additional purification.
-
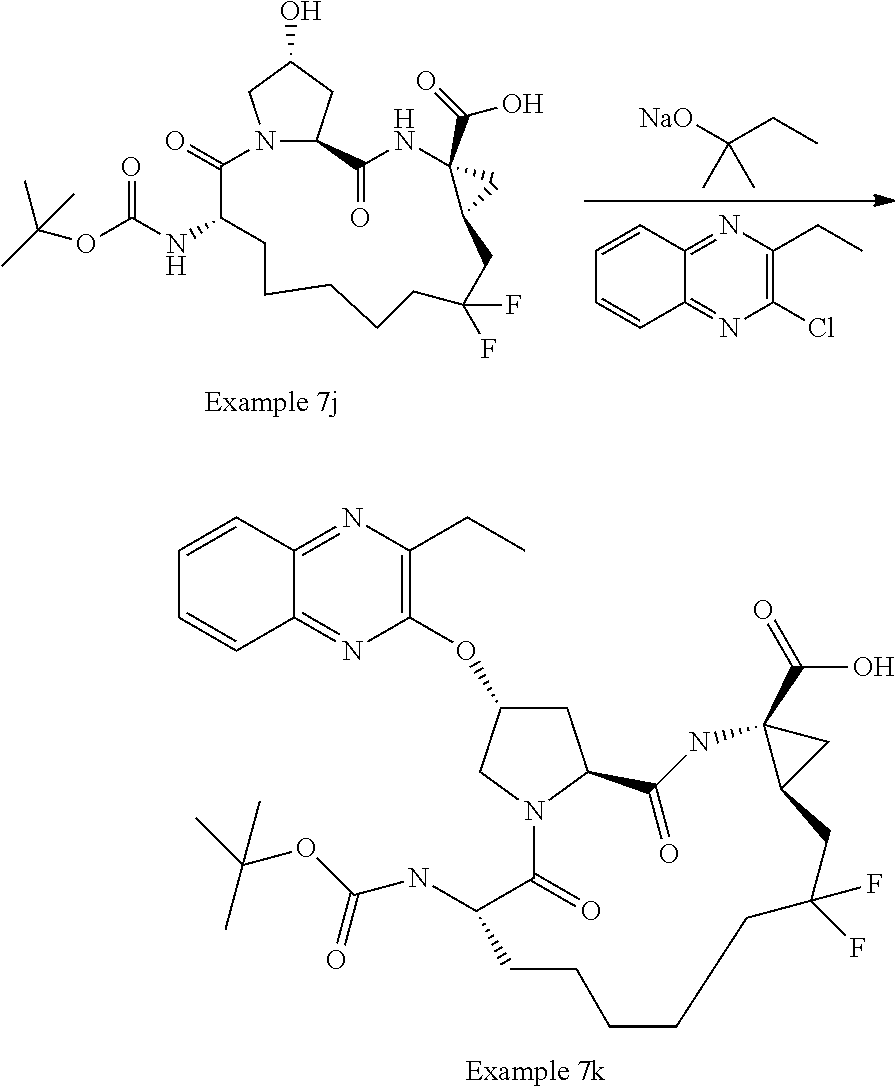
- To (2R,6S,13aS,14aR,166)-6-(tert-butoxycarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid (Example 7j, 87.4 mg, 0.174 mmol) was added 2-chloro-3-ethylquinoxaline (47.7 mg, 0.247 mmol) and DMF (868 μl). To this solution was added sodium tert-pentoxide (48.9 mg, 0.444 mmol) at room temperature, and the mixture turned dark purple (slightly exothermic). The reaction mixture was stirred at room temperature for 17 h. Additional 2-chloro-3-ethylquinoxaline (18.1 mg, 0.094 mmol) and sodium tert-pentoxide (24.2 mg, 0.202 mmol) were added, and the reaction mixture was stirred for 22 h. The reaction mixture was diluted with CU3CN and neutralized with HCl (1 N, 0.7 ml) and evaporated under reduced pressure. The residue was purified by reverse phase HPLC (CH3CN/H2O (0.1% TFA), gradient=20/80-85/15) to provide the title compound as a light brown solid, (2R,6S,13aS,14aR,16aS)-6-(tert-butoxycarbonylamino)-2-(3-ethylquinoxalin-2-yloxy)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,4]diazacyclopentadecine-14a-carboxylic acid (66 mg, 57% yield).
-
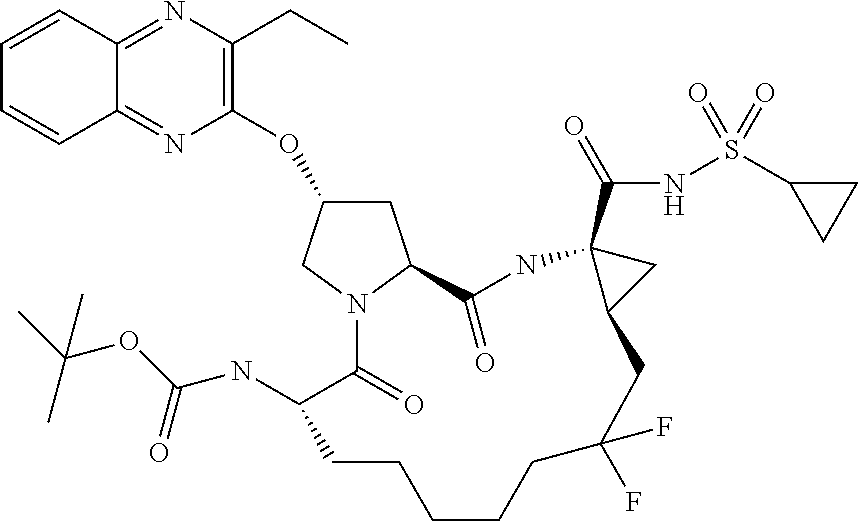
-

- To (2R,6S,13aS,14aR,16aS)-6-(tert-butoxycarbonylamino)-2-(3-ethylquinoxalin-2-yloxy)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid (Example 7k, 26.2 mg, 0.040 mmol) was added sieves (4 A) and dichloroethane (397 μl). To this mixture was added carbonyldiimidazole (13.5 mg, 0.083 mmol), and the reaction mixture was stirred at 40° C. for 2 hr. The reaction mixture was then cooled to room temperature and cyclopropanesulfonamide (12.4 mg, 0.103 mmol) was added followed by DBU (15 μl, 0.100 mmol). The reaction mixture was stirred at room temperature for 16 h, diluted with dichloromethane and cooled to 0° C. This mixture was neutralized with HCl (4 N in dioxane, 67 μl), and the mixture was filtered to remove sieves. The eluant was concentrated under reduced pressure and the residue was purified by reverse phase HPLC (CH3CN/H2O (0.1% TFA) gradient=45/55-85/15) to provide tert-butyl (2R,6S,13aS,14aR,16aS)-14a-(cyclopropylsulfonylcarbamoyl)-2-(3-ethylquinoxalin-2-yloxy)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate (25.8 mg, 85% yield) as a light yellow solid. MS (ESD: m/z=763.0 [M+H], 761.3 [M−H]. Example 7 provided an IC50 of between 0.5 and 1.0 nM in a 1a enzyme assay; an IC50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a HLM stability value of >100 μl/min/mg; an EC50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
-
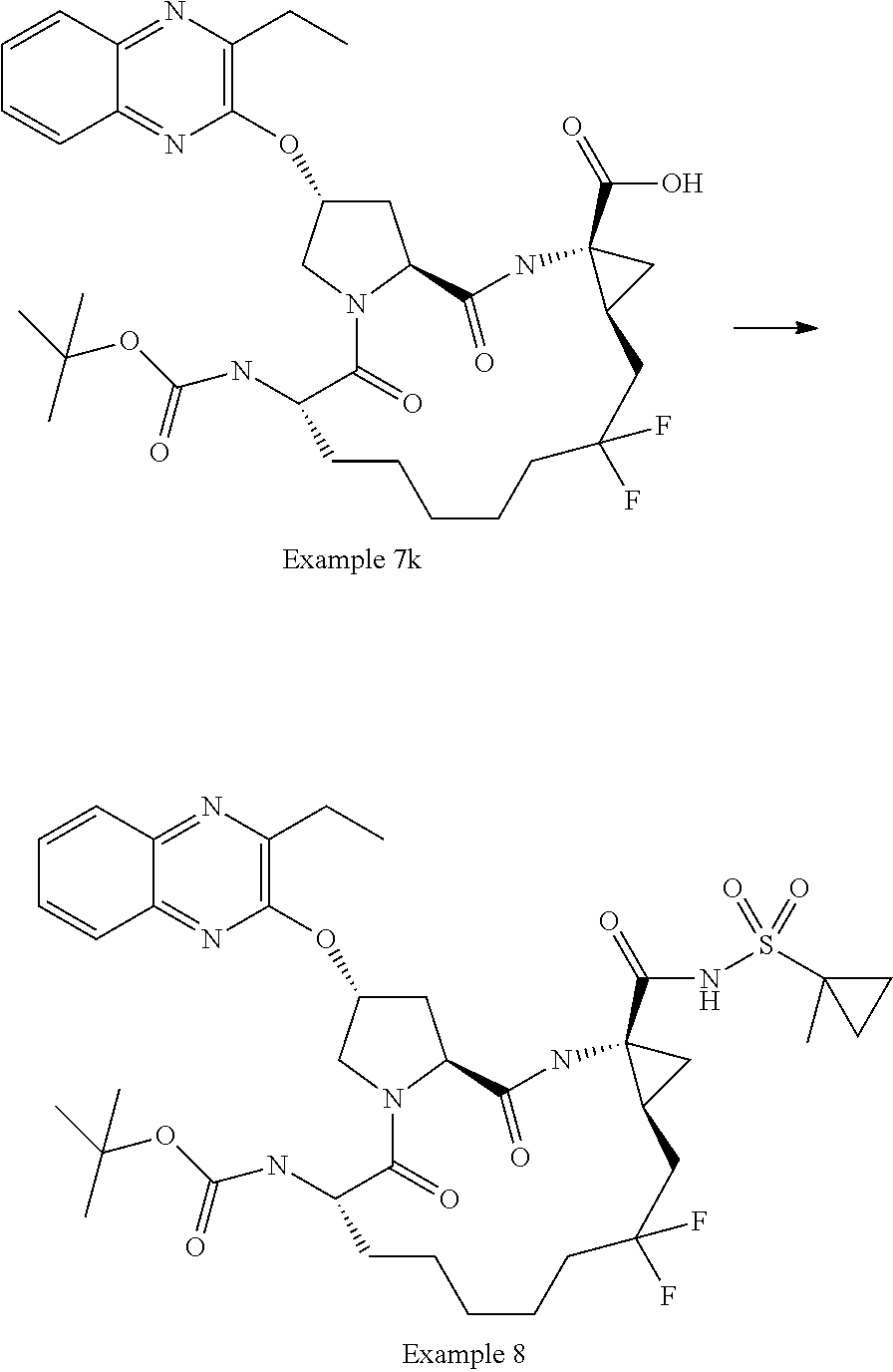
- Example 8 was prepared according to the procedure utilized for the preparation of Example 7 from Example 7k, replacing cyclopropanesulfonamide with 1-methylcyclopropane-1-sulfonamide. Yield=67%. MS (ESI): m/z=777.2 [M+H], 775.4 [M−H].
- Example 8 provided an IC50 of between 0.1 to 0.5 nM in a 1a enzyme assay; an IC50 of between 0.1 to 0.5 nM in a 1b enzyme assay; a HLM stability value of >100 μl/min/mg; an EC50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
-
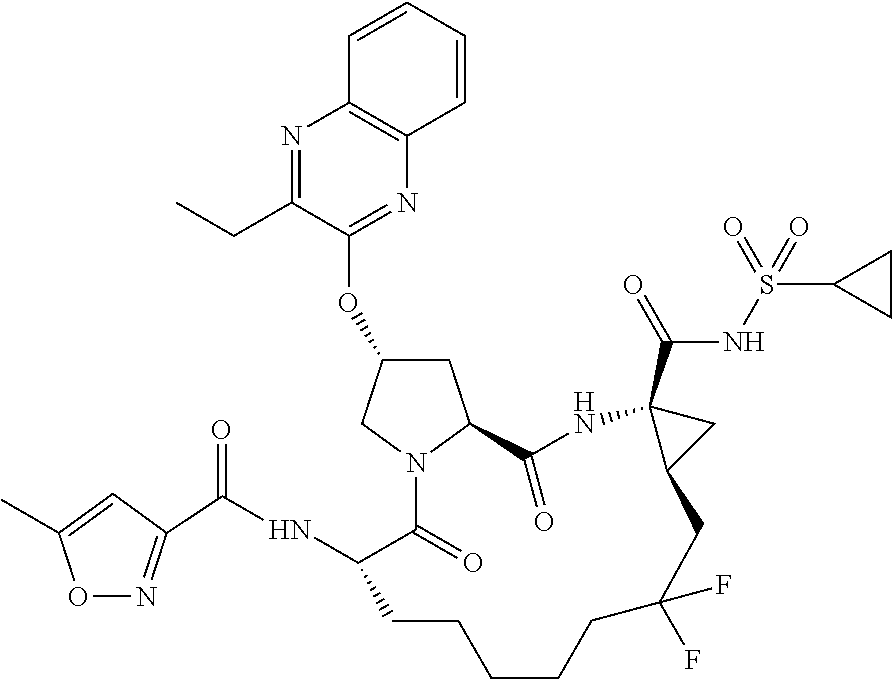
-

- Example 9a was prepared according to the procedure utilized for the preparation of Example 2a, replacing the product of Example 1 with the product of Example 7.
-
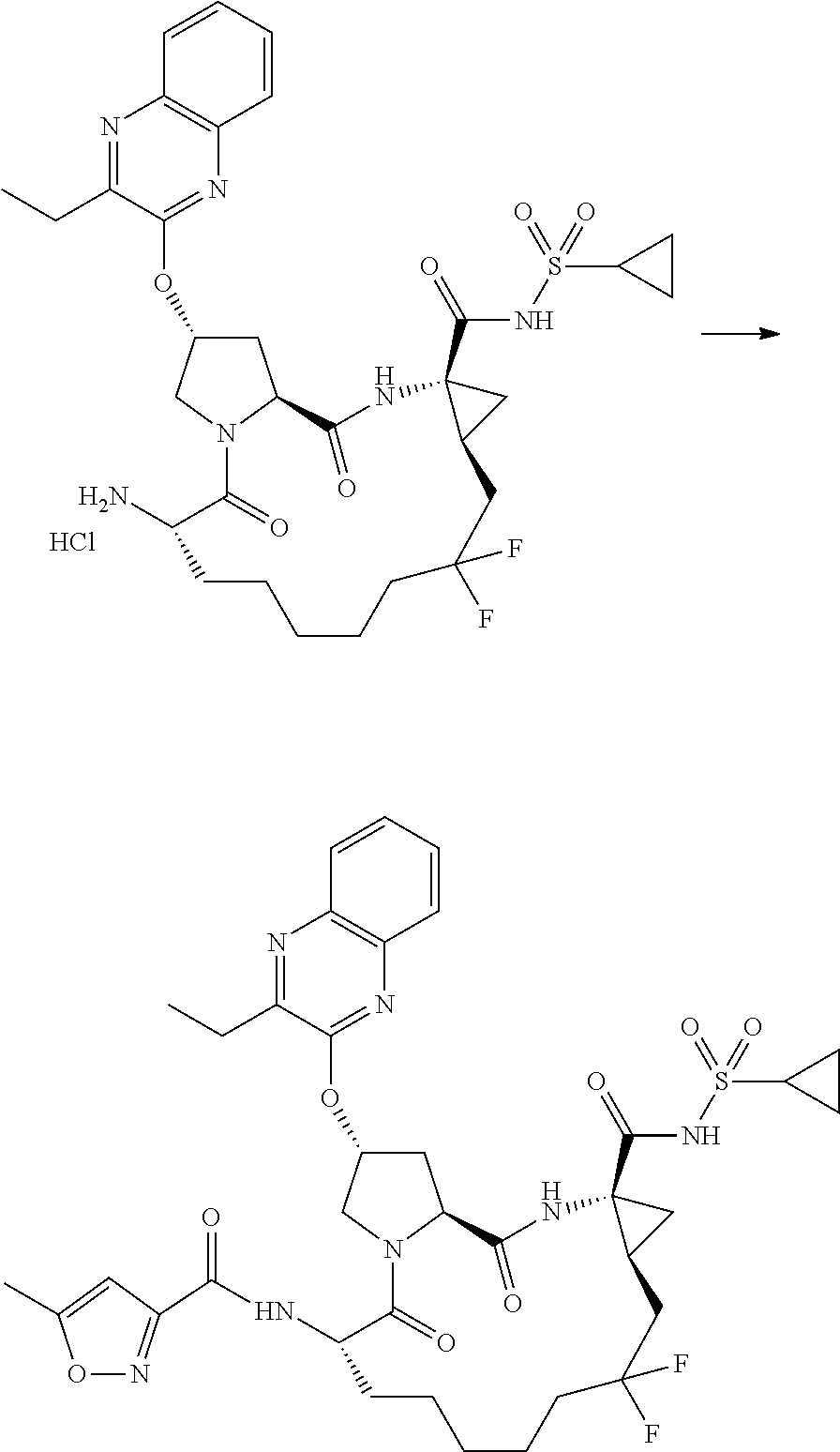
- Example 9 was prepared according to the procedure utilized for the preparation of Example 2, replacing the product of Example 2a with the product of Example 9a. Yield=74%. MS (ESI): m/z=772.3 [M+H], 770.4 [M−H].
- Example 9 provided an IC50 of between 0.5 and 1.0 nM in a 1a enzyme assay; an IC50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a FILM stability value of <50 μl/min/mg; an EC50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
-
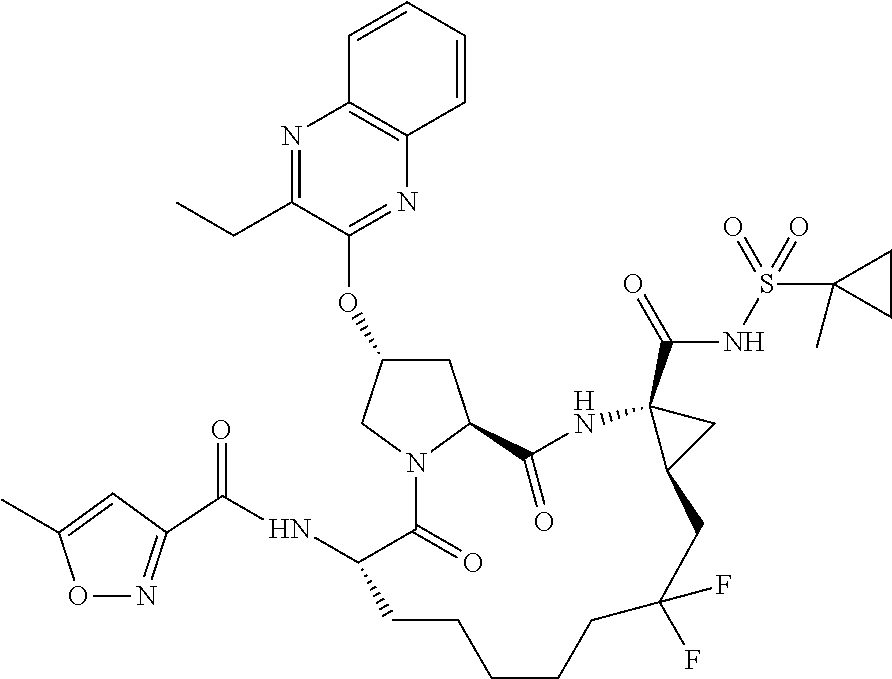
-

- Example 10a was prepared according to the procedure utilized for the preparation of Example 2a, replacing the product of Example 1 with the product of Example 8.
-

- Example 10 was prepared according to the procedure utilized for the preparation of Example 2, replacing the product of Example 2a with the product of Example 10a. Yield=83%. MS (ESI): m/z=786.3 [M±H], 784.3 [M−H].
- Example 10 provided a HLM stability value of <50 μl/min/mg; an EC50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
-
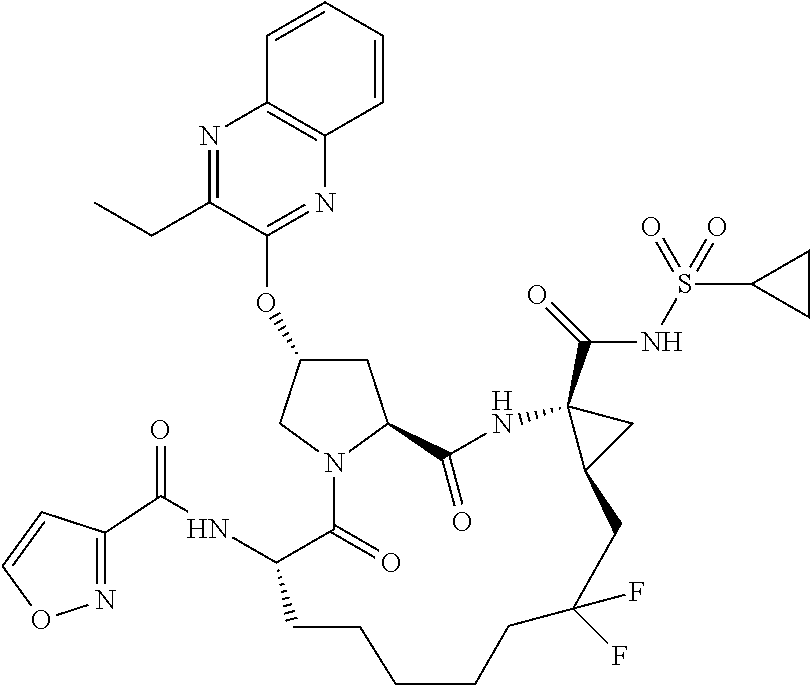
- Example 11 was prepared according to the procedure utilized for the preparation of Example 9, replacing 5-methylisoxazole-3-carboxylic acid with isoxazole-3-carboxylic acid. Yield=74%. MS (ESI): m/z=758.6 [M+H], 756.4 [M−H].
- Example 11 provided an IC50 of between 0.1 and 0.5 nM in a 1a enzyme assay; an IC50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a FILM stability value of <50 μl/min/mg; an EC50 of >5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1b-con1 background.
-
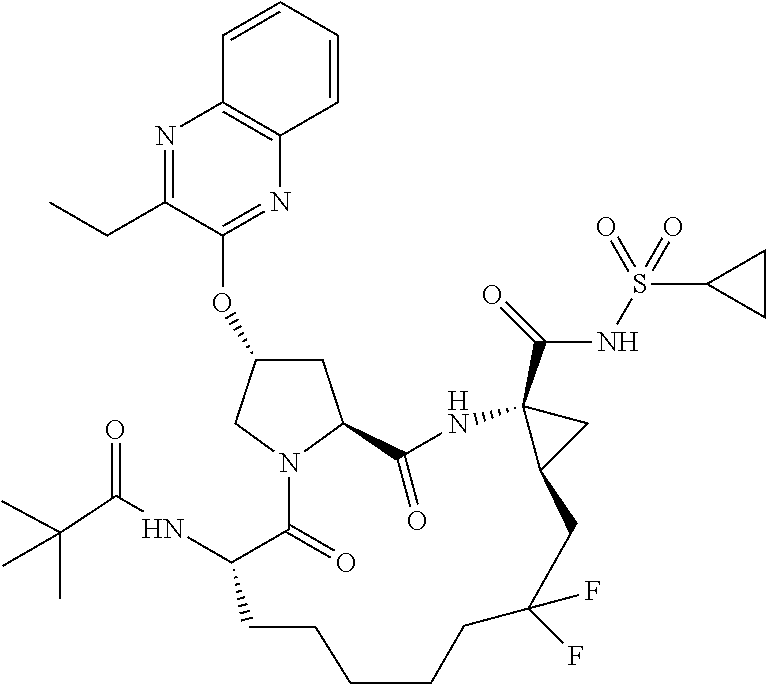
- Example 12 was prepared according to the procedure utilized for the preparation of Example 9, replacing 5-methylisoxazole-3-carboxylic acid with pivalic acid. Yield=85%. MS (ESI): m/z=747.3 [M+H], 745.4 [M−H].
- Example 12 provided an IC50 of between 0.5 and 1.0 nM in a 1a enzyme assay; an IC50 of between 0.5 and 1.0 nM in a 1b enzyme assay; a HLM stability value of <50 μl/min/mg; an EC50 of >5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of >5.0 nM in a replicon cell line assay in a 1b-con1 background.
-
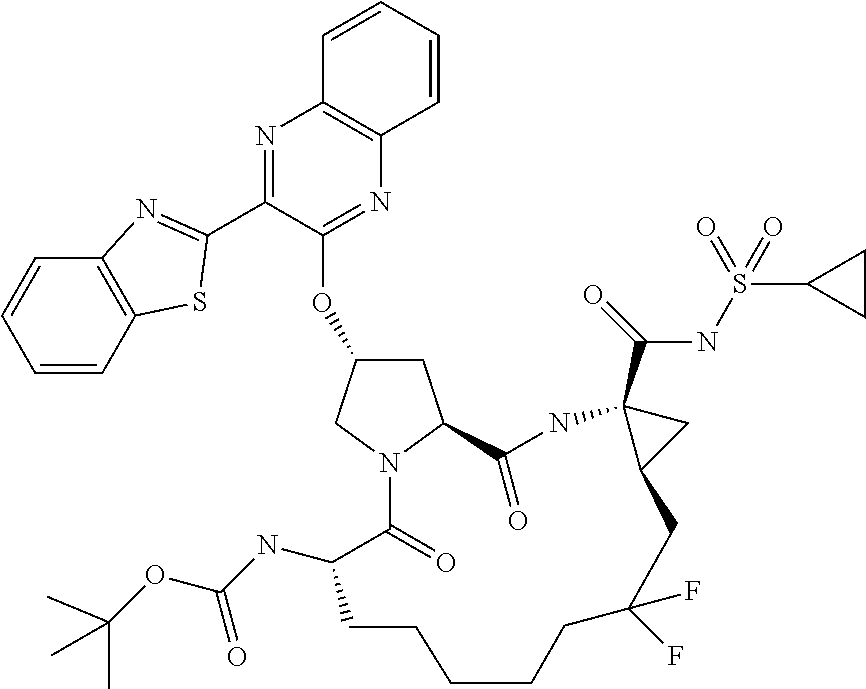
-
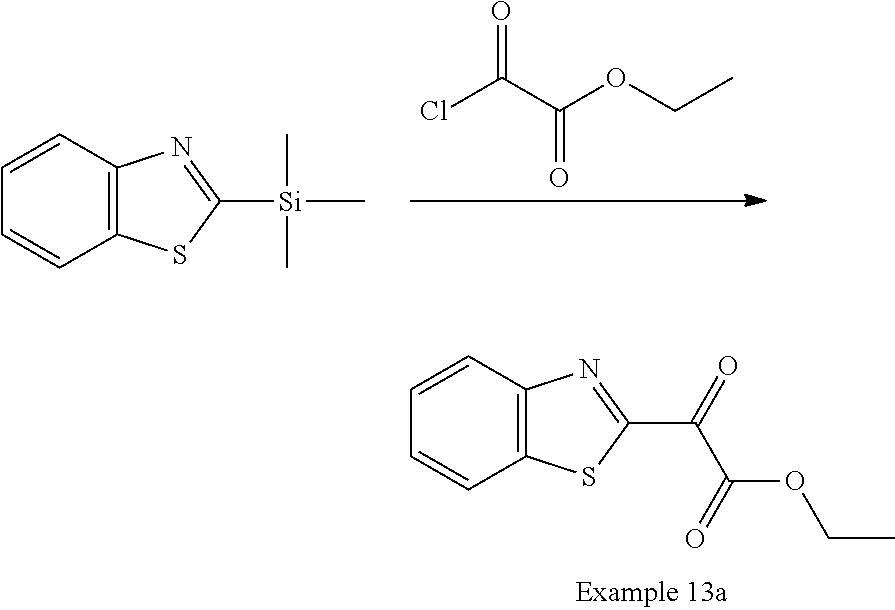
- To a solution of ethyl oxalyl ride (0.537 ml, 4.82 mmol) in dichloromethane (2.411 ml) was added slowly a solution of 2-(trimethylsilyl)benzothiazole (0.945 ml, 4.82 mmol) in dichloromethane (2.4 ml) stirring at 0° C. The reaction mixture was stirred at 0° C. for 30 min allowed to warm slowly to room temperature with stirring over 16 h. The reaction mixture was evaporated under reduced pressure and purified by flash chromatography on silica gel (hex/EA/dichloromethane=8/1/1) to provide the title compound (ethyl 2-(benzo[d]thiazol-2-yl)-2-oxoacetate, 116.2 mg, 10° A yield) as an oil.
-
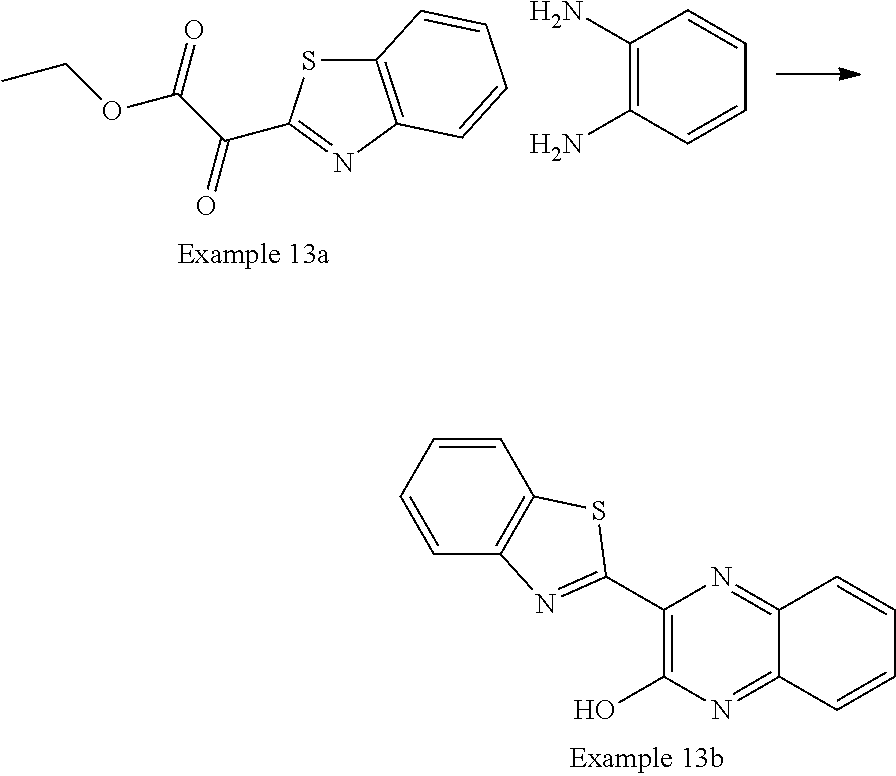
- To a solution of ethyl 2-(benzo[d]thiazol-2-yl)-2-oxoacetate (Example 13a, 655 mg, 2.78 mmol) in EtOH (5.6 mL) was added o-phenylenediamine (300 mg, 2.78 mmol). The reaction mixture was heated with stirring at 90° C. for 1 hr (a yellow/green solid appeared quickly), cooled to 0° C., and the solid filtered and rinsed with cold EtOFI. The solid was dried under vacuum overnight to provide the title compound (3-(benzo[d]thiazol-2-yl)quinoxalin-2-ol, 313 mg, 40% yield) as a yellow green solid.
-
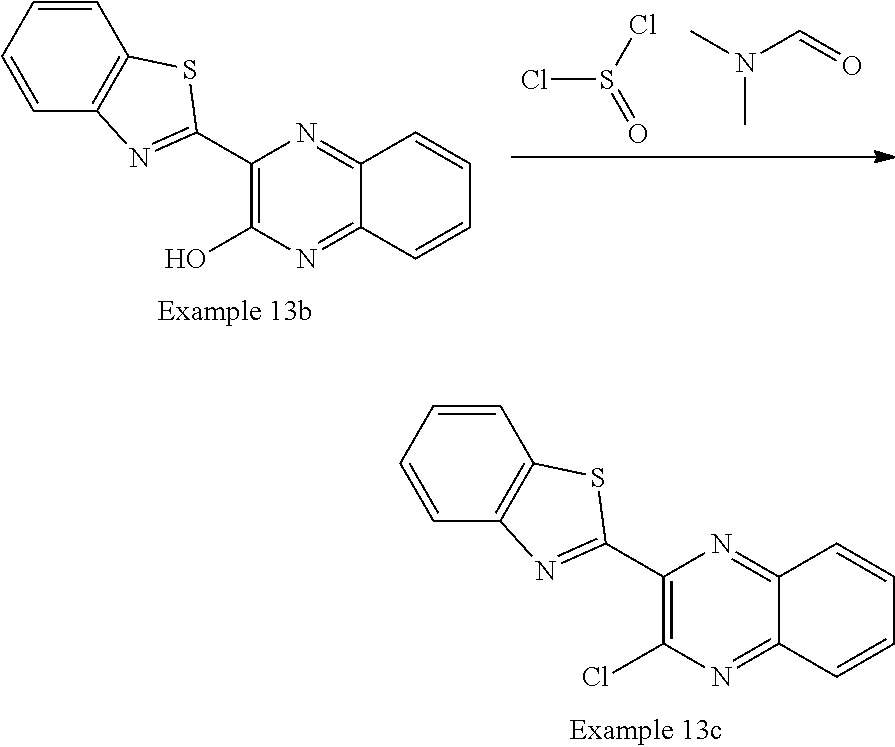
- To a suspension of 3-(benzo[d]thiazol-2-yl)quinoxalin-2-ol (251 mg, 0.900=op in toluene (4500 μl) was added thionyl chloride (131 μl, 1.800 mmol) followed by DMF (69.7 μl, 0.900 mmol). The green mixture was stirred at 110° C. for 3 hr. The reaction mixture was evaporated under reduced pressure and the resulting solid was diluted with CHCl3 (30-40 ml) and the insoluble solid filtered and rinsed with CHCl3. The filtrate was evaporated under reduced pressure and the residue purified by flash chromatography on silica gel (eluted with chloroform, then dichloromethane) to provide the title compound as a light yellow solid.
-
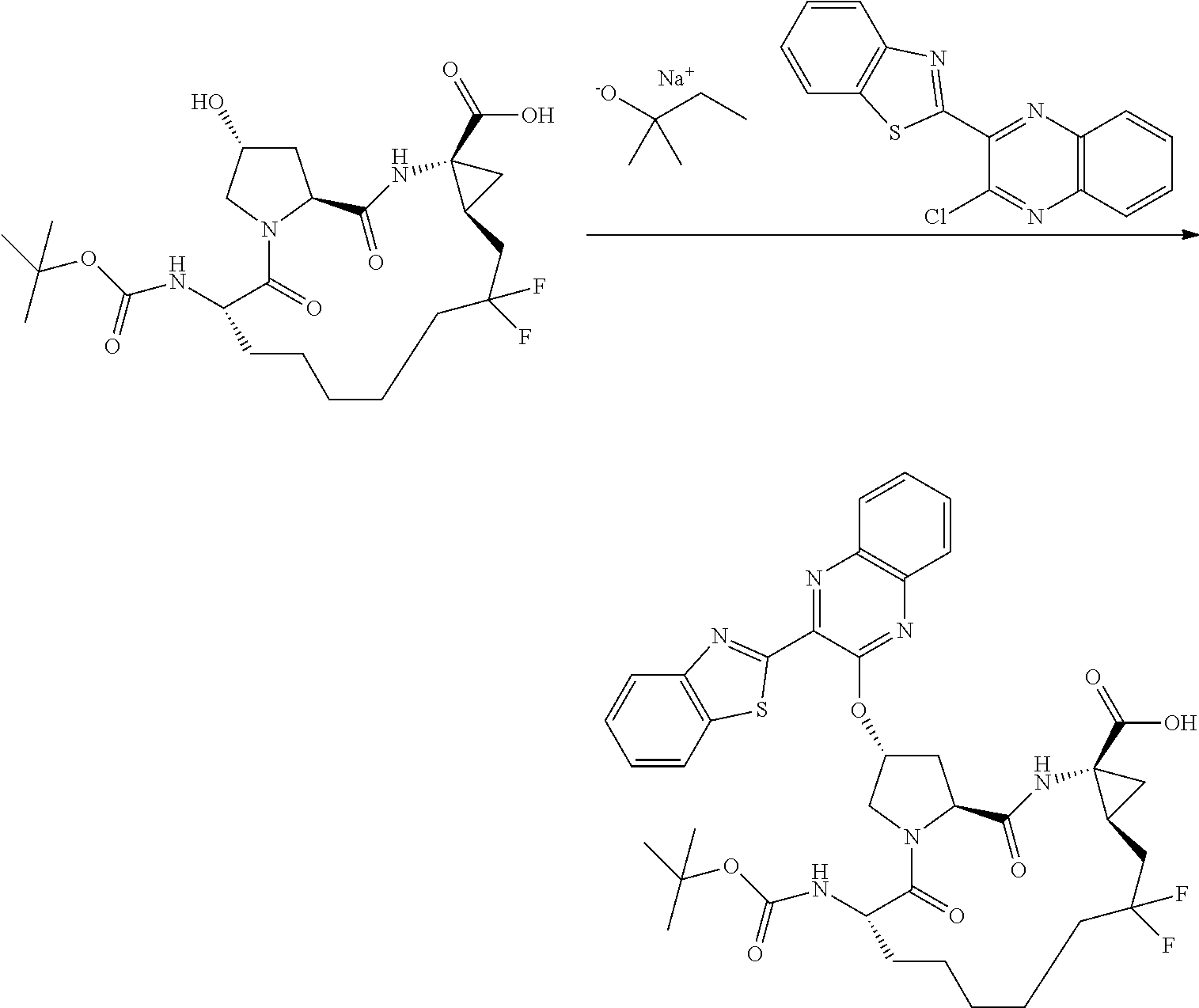
- To (2R,6S,13aS,14aR,16aS)-6-(tert-butoxyrcarbonylamino)-12,12-difluoro-2-hydroxy-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid (Example 13c, 13 mg, 0.026 mmol) was added DMF (129 μl). Sodium tert-pentoxide (6.28 mg, 0.057 mmol) was added in one portion at room temperature and the mixture was stirred at room temperature for 45 minutes. 2-(3-Chloroquinoxalin-2-yl)benzo[d]thiazole (9.13 mg, 0.031 mmol) was added at room temperature. The resulting yellow mixture was stirred at room temperature for 22 hr. The reaction mixture was diluted with CH3CN, neutralized with HCl. (2 N, 15 μl), and evaporated under reduced pressure. The solid was diluted with CH3CN/H2O (0.1% TFA)/DMSO and filtered to remove insoluble yellow solid. The filtrate was purified by reverse phase HPLC (C18, CH3CN/H2O (0.1% TFA) 30/70 to 100/0, 15 min) to provide the title compound (5.2 mg, 26% yield).
-
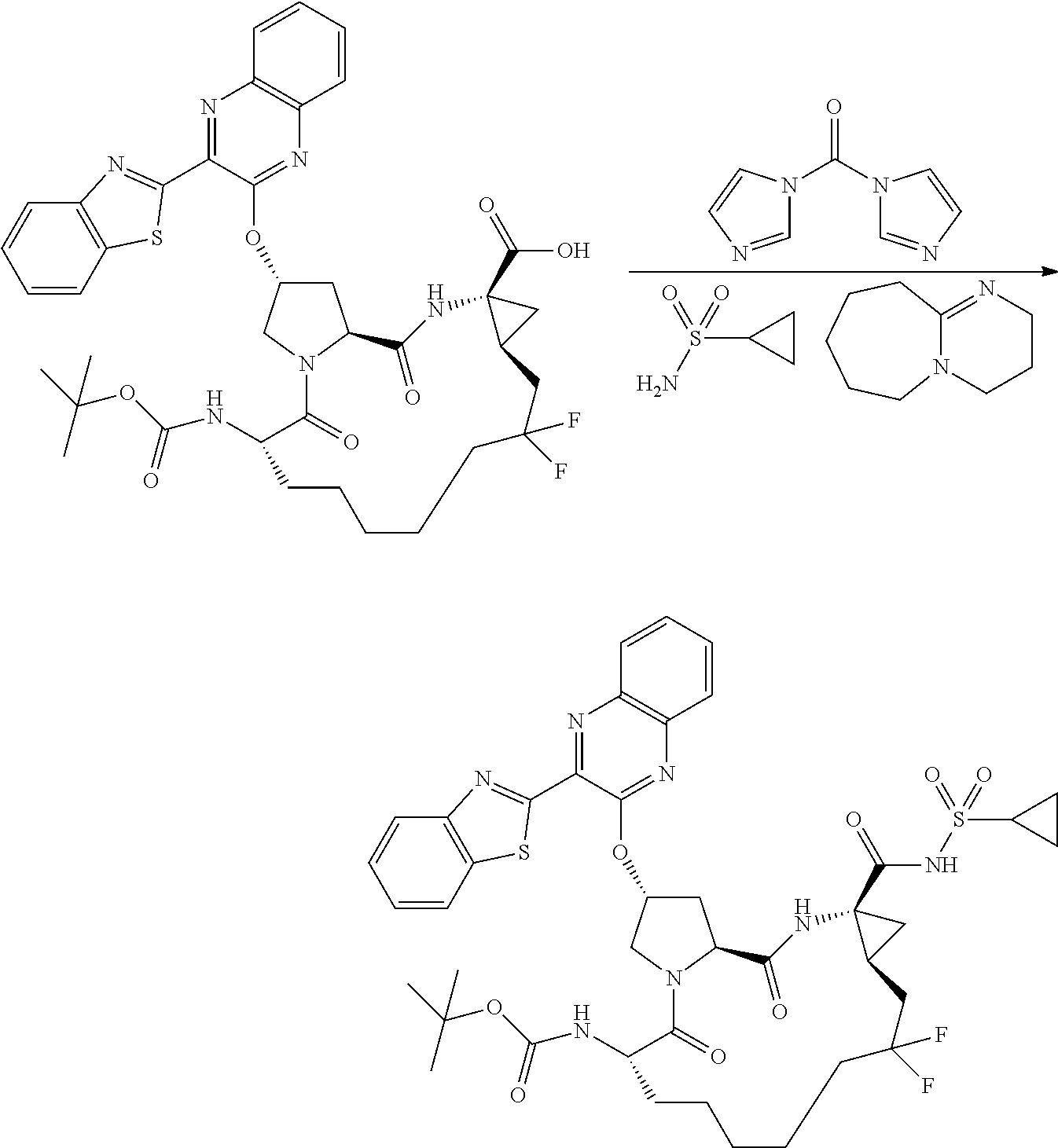
- To a solution of (2R,6S,13aS,14aR,16aS)-2-(3-(benzo[d]thiazol-2-yl)quinoxalin-2-yloxy)-6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid (Example 13d, 6.22 mg, 8.13 μmol) was added molecular sieves and DCE (81 μl). CDI (2.91 mg, 0.018 mmol) was added, and the mixture was stirred at 40° C. for 2 h. The reaction mixture was cooled to rt and cyclopropanesulfonamide (2.4 mg, 0.020 mmol) was added followed by DBU (3 μl, 0.020 mmol). The mixture was stirred at rt for 2.5 h, diluted with dichloromethane and cooled to 0° C. The mixture was neutralized with HCl (14 μl, 4 N in dioxane, diluted with EA) and stirred for 5 min at 0° C., and evaporated under reduced pressure. The residue was purified by silica gel prep-TLC (dichloromethane/EA=1/1) to provide the title compound as a yellow solid, (5.2 mg, 74% yield). MS (ESI): m/z=868.3 [M+11], 866.3 [M−H].
-
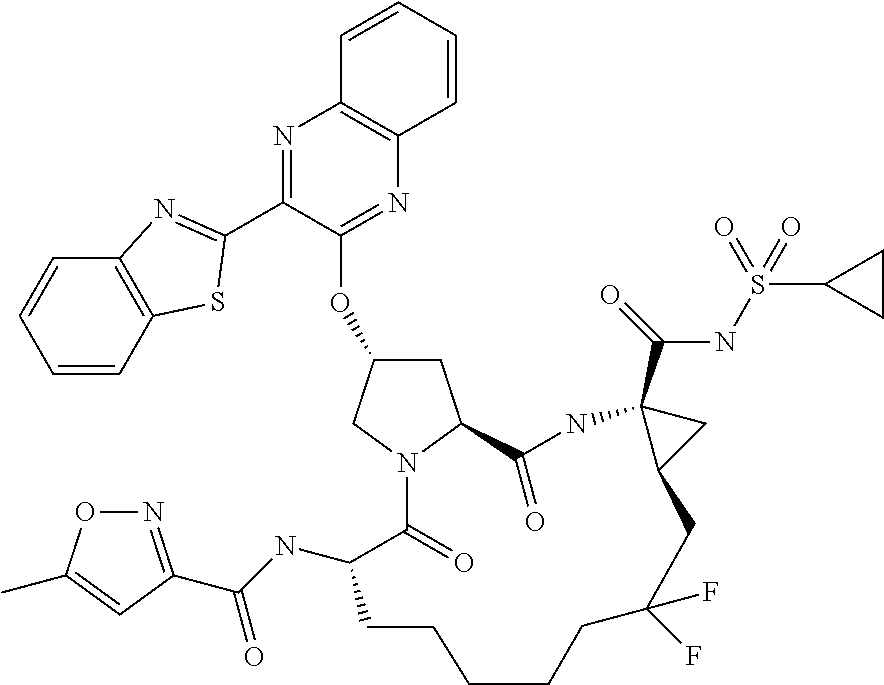
-
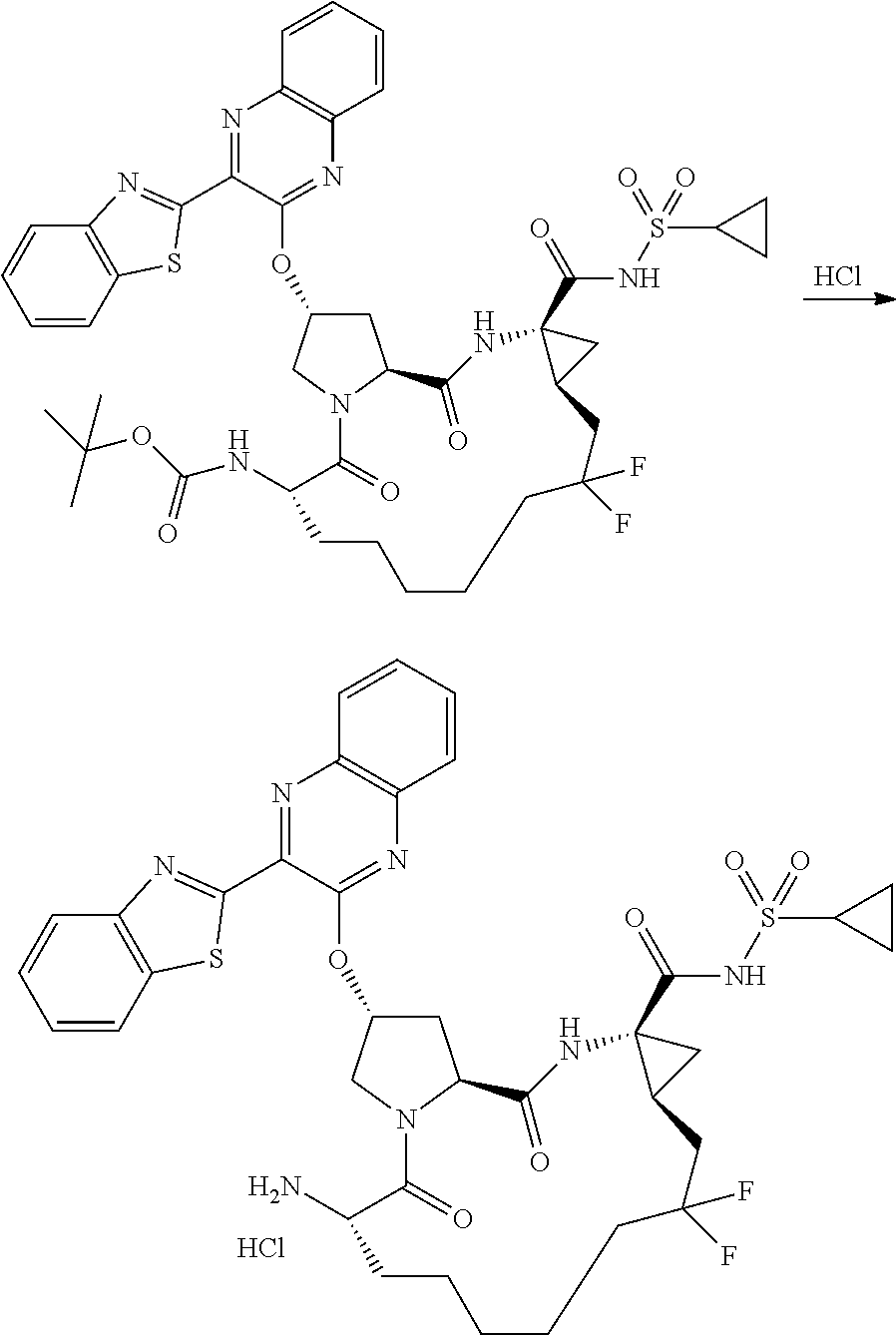
- To a solution of tert-butyl (2R,6S,13aS,14aR,16aS)-2-(3-(benzo[d]thiazol-2-yl)quinoxalin-2-yloxy)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxooctadecahydro cyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate (Example 13, 17.4 mg, 0.020 mmol) in ethyl acetate (100 μl) was added HCl (4 M in dioxane, 100 μl, 0.401 mmol) at rt. The mixture was stirred at rt for 16 h, and more HCl (100 μl, 0.401 mmol) was added, and the reaction mixture stirred for 6 hr. The reaction mixture was concentrated under reduced pressure and azeotroped with EA to provide the title compound as a yellow solid which was used without further purification.
-

- To (2R,6S,13aS,14aR,16aS)-6-amino-2-(3-(benzo[d]thiazol-2-yl)quinoxalin-2-yloxy)-N-(cyclopropylsulfonyl)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide, Hydrochloric Acid (Example 14a, 16.1 mg, 0.020 mmol) was added DMF (200 μl) and diisopropylethylamine (25 μl, 0.14 mmol). To the yellow solution was added 5-methylisoxazole-3-carboxylic acid (3.85 mg, 0.030 mmol) followed by HATU (13.89 mg, 0.037 mmol). The reaction mixture was stirred at rt 2 h, cooled to 0° C. and neutralized with HCl (4 N in dioxane, 36 μl, diluted with EA). The solution was concentrated under reduced pressure and purified by prep-TLC (dichloromethane/EA=1/1, 2×) to provide the title compound N-((2R,6S,13aS,14aR,16aS)-2-(3-(benzo[d]thiazol-2-yl)quinoxalin-2-yloxy)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxooctadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-yl)-5-methylisoxazole-3-carboxamide (14.3 mg, 81% yield). MS (ESI): m/z=877.1 [M+11], 875.2 [M−H].
- Example 14 provided an IC of between 0.1 and 0.5 nM in a 1a enzyme assay; an IC50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a HLM stability value of <50 μl/min/mg; an EC50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1b-con1 background.
-
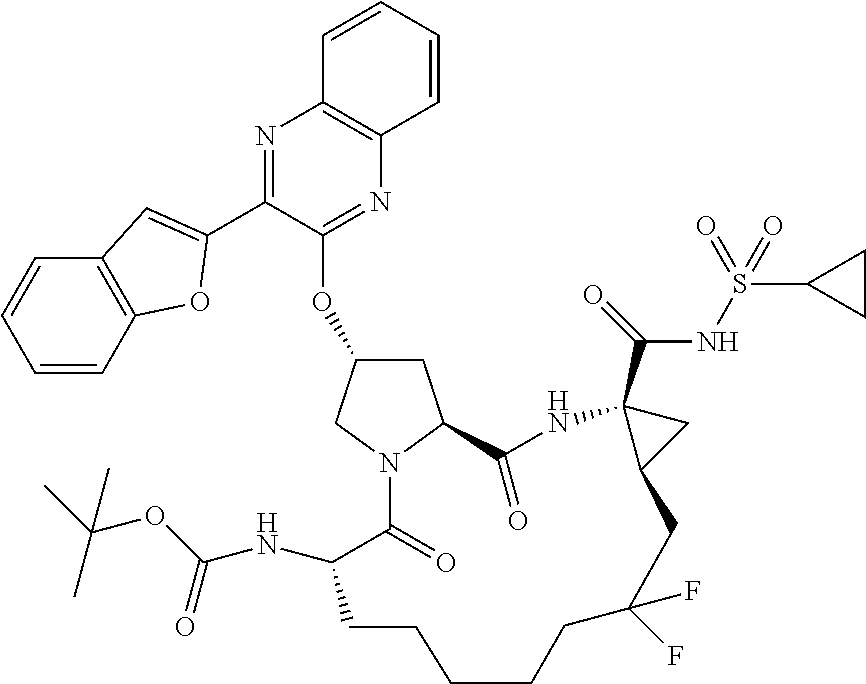
-
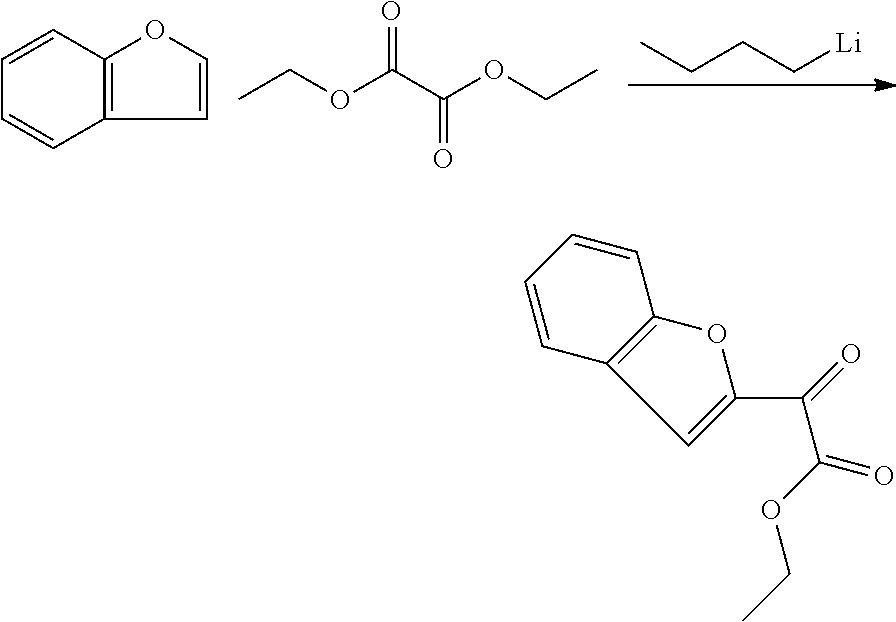
- To a solution of benzofuran (0.250 mL, 2.32 mmol) in tetrahydrofuran (12.5 mL) stirring at −100° C. (CO2, ether bath) was added n-BuLi (1.377 mL, 2.203 mmol, 1.6 M in hexanes). The reaction was stirred for 30 min (yellow color) and added dropwise via a canulated into a solution of diethyl oxalate (0.314 mL, 2.32 mmol) in tetrahydrofuran (7 mL) stirring at −100° C. The reaction mixture was stirred for 1 h and then quenched with saturated aqueous ammonium chloride solution. The mixture was warmed to rt, diluted with dichloromethane, and washed with 0.1 N HCl. The organic layer was separated, dried over anhydrous magnesium sulfate, filtered, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluted with dichloromethane). The fractions containing the title compound were collected and repurified by flash chromatography on silica gel (25% acetone/hexane) to provide the title compound (ethyl 2-(benzofuran-2-yl)-2-oxoacetate, 237 mg, 47% yield).
-
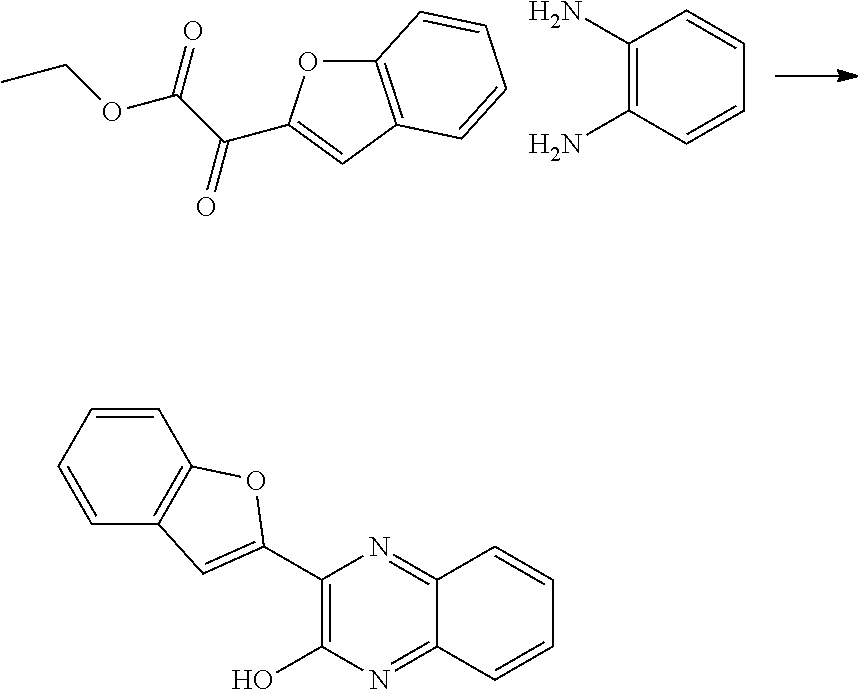
- To a solution of ethyl 2-(benzofuran-2-yl)-2-oxoacetate (Example 15a, 237 mg, 1.09 mmol) in EtOH (2.7 mL) was added o-phenylenediamine (117 mg, 1.09 mmol). The reaction mixture was stirred at 90° C. for 16 h (a yellow solid appeared quickly). The mixture was cooled to 0° C., filtered, and the solid rinsed with cold EtOH. The yellow solid was azeotroped with CHCl3 and benzene and dried to provide the title compound (268 mg, 94% yield).
-

- To a suspension of 3-(benzofuran-2-yl)quinoxalin-2-ol (Example 15b, 268 mg, 1.02 mmol) in Toluene (5.1 mL) was added thionyl chloride (0.30 mL, 4.1 mmol) followed by DMF (238 μl, 3.07 mmol). The yellow reaction mixture was stirred at 110° C. for 2 h, cooled to room temperature, and evaporated under reduced pressure to gave a yellow solid. The solid was suspended in dichloromethane (20 ml) and purified by flash chromatography on silica gel (dichloromethane) to give the title compound, 2-(benzofuran-2-yl)-3-chloroquinoxaline (297 mg, quantitative yield), as a yellow solid.
-
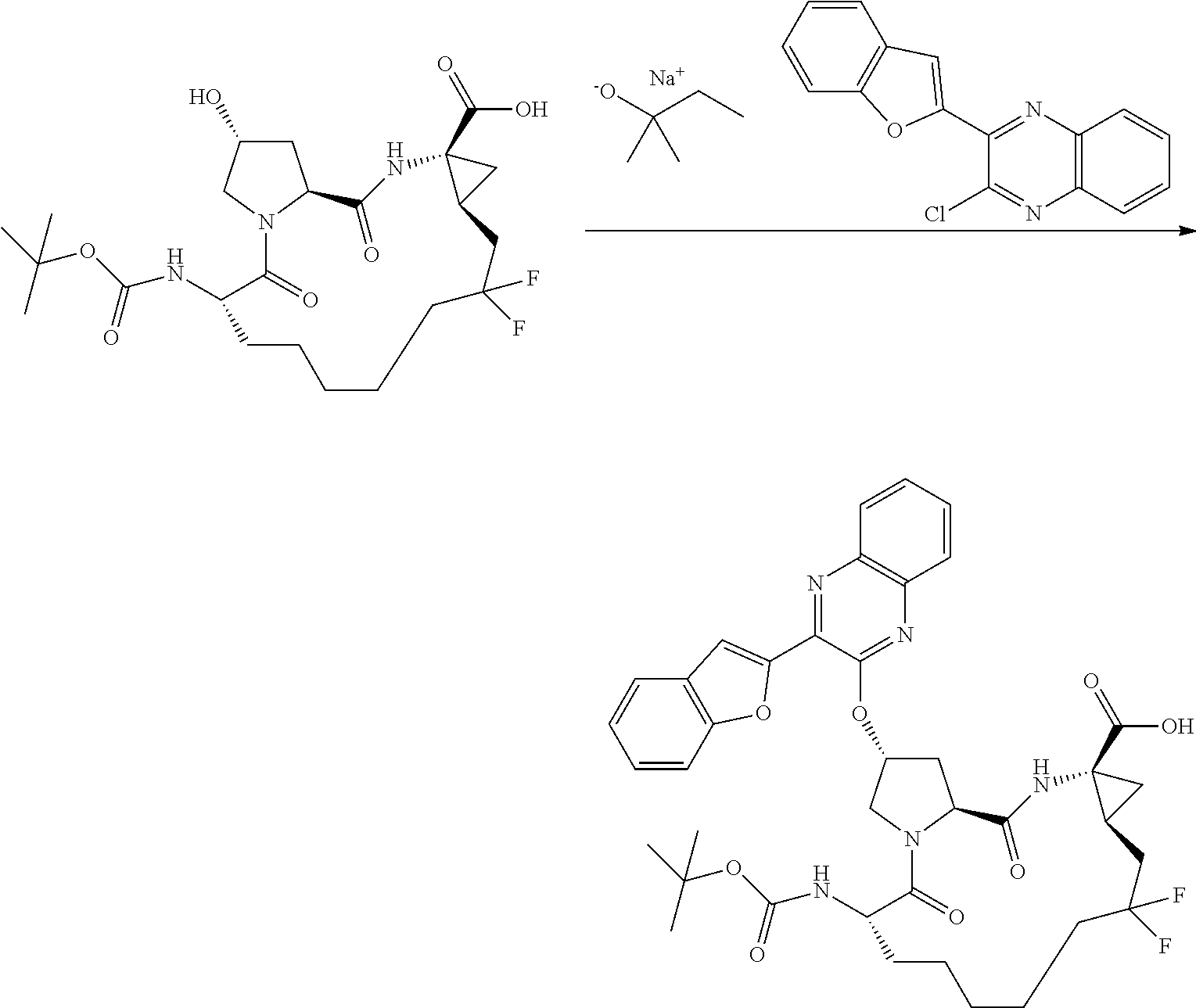
- The title compound was prepared according to the procedure utilized for the preparation of Example 13d, replacing the product of Example 13c with the product of Example 15c. Yield=66%.
-

- The title compound was prepared according to the procedure utilized for the preparation of Example 13, replacing the product of Example 13d with the product of Example 15d. (91% yield).
- MS (ESI): m/z=803.6 [M+H].
-

-

- The title compound was prepared according to the procedure utilized for the preparation of Example 14a, replacing the product of Example 13 with the product of Example 15. (quantitative yield).
-
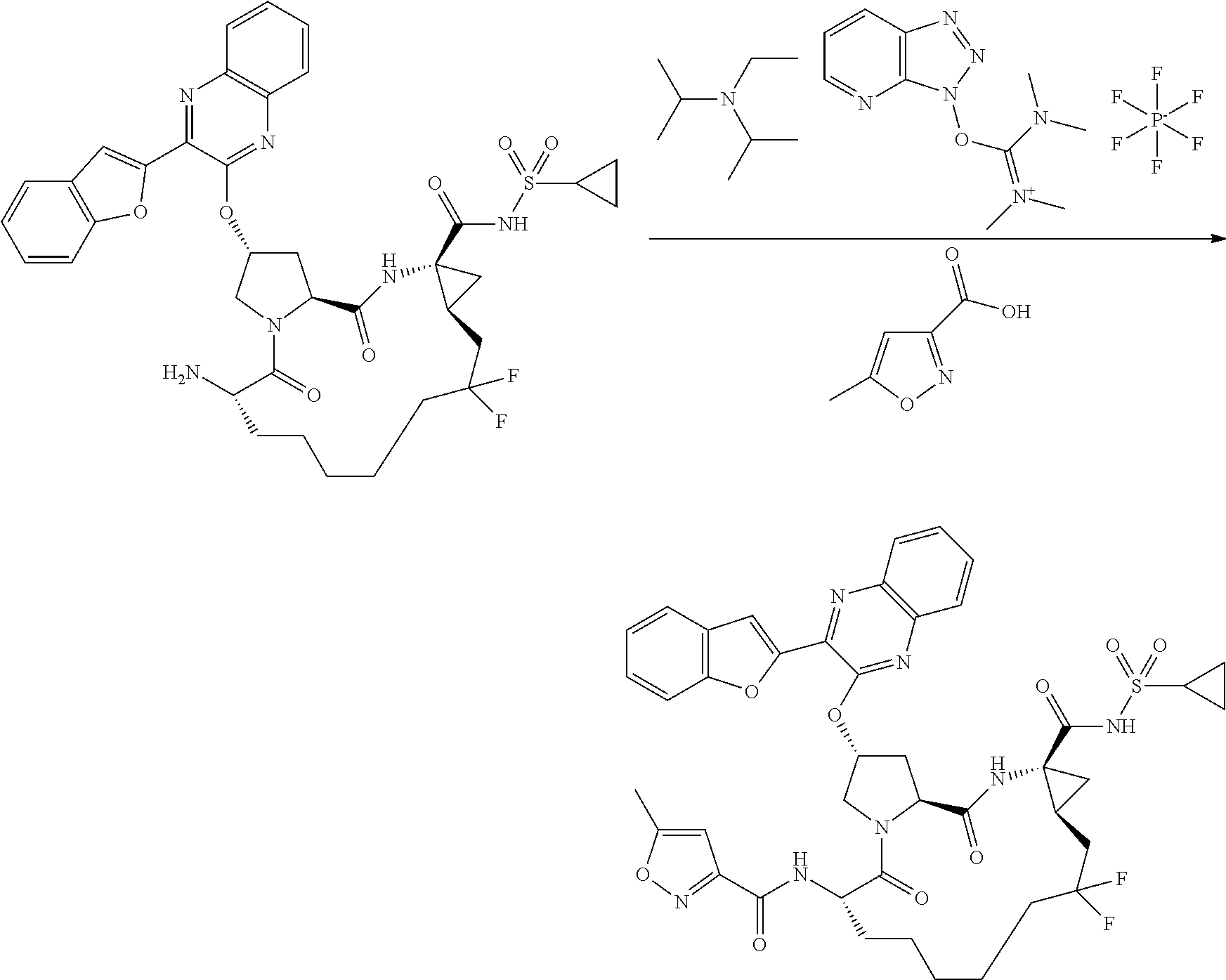
- The title compound was prepared according to the procedure utilized for the preparation of Example 14, replacing the product of Example 14a with the product of Example 16a. (68% yield).
- MS (ESI): m/z=860.3 [M+H], 858.4 [M−H].
- Example 16 provided an IC50 of between 0.1 and 0.5 nM in a 1a enzyme assay; an IC50 of between 0.1 and 0.5 nM in a 1b enzyme assay; a HUM stability value of between 50 and 100 μl/min/mg; an EC50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of <1.0 nM in a replicon cell line assay in a 1b-con1 background.
-
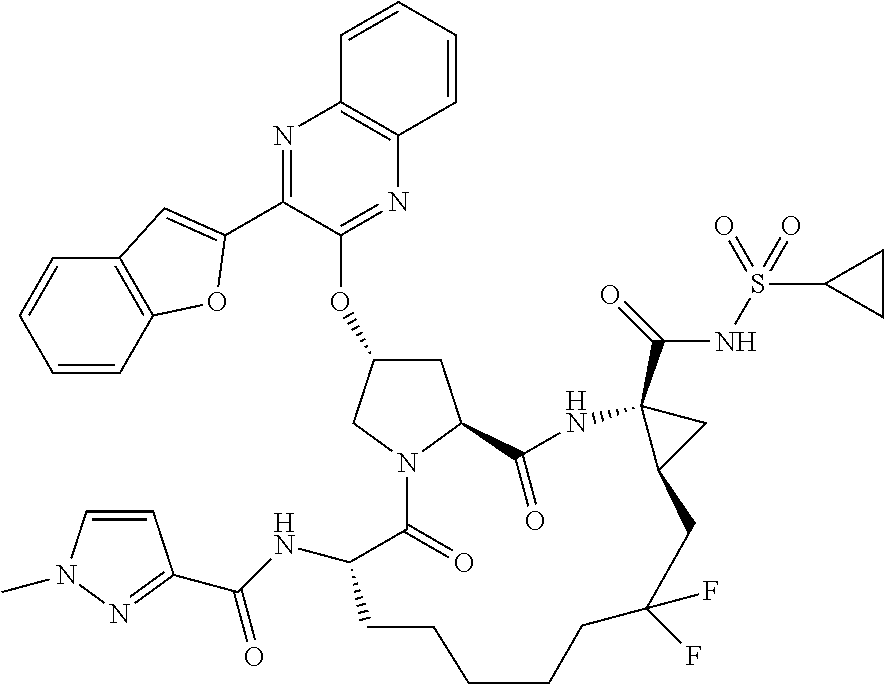
-
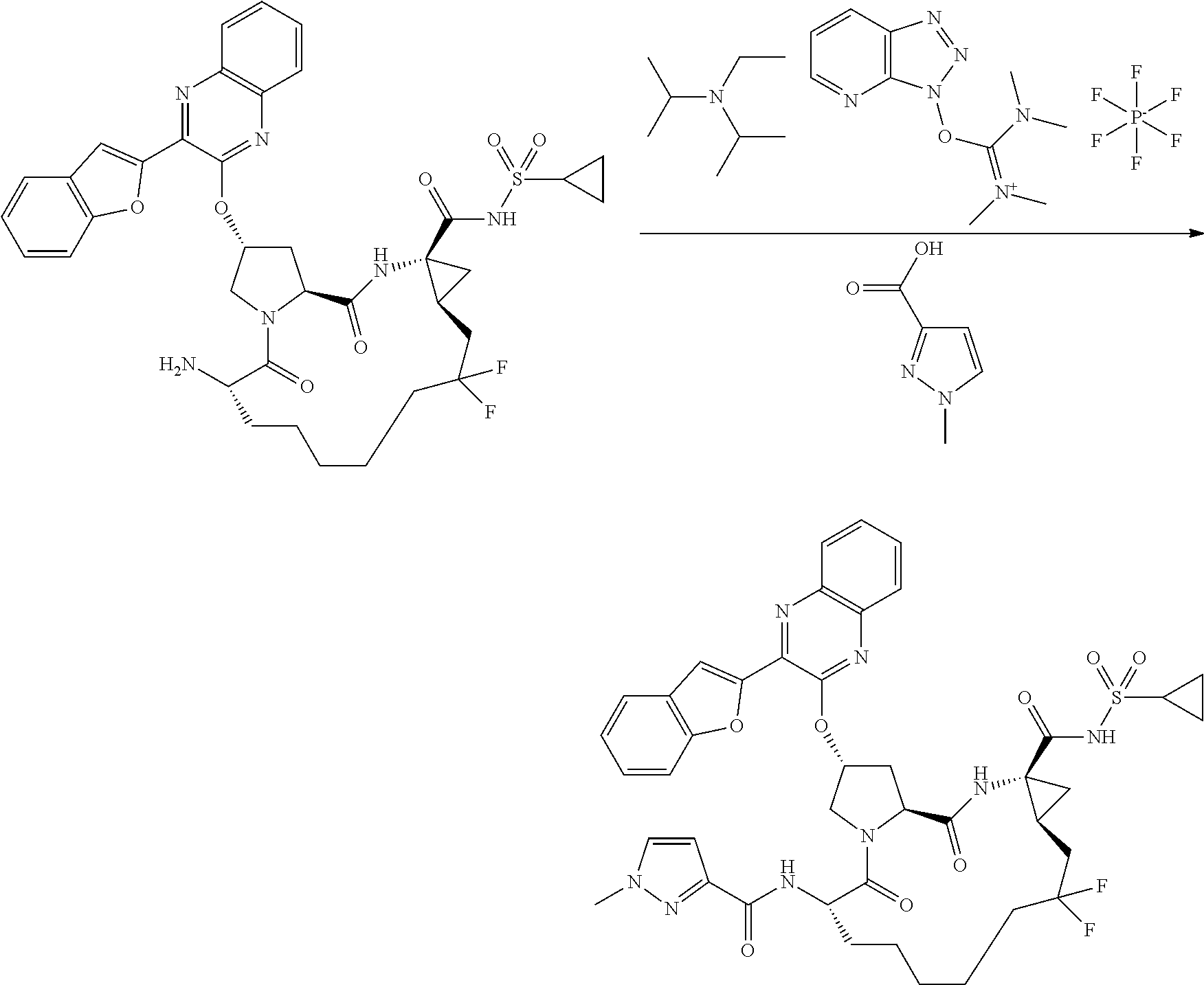
- The title compound was prepared according to the procedure utilized for the preparation of Example 16, replacing 5-methylisoxazole-3-carboxylic acid with 1-methyl-1H-pyrazole-3-carboxylic acid. (96% yield). MS (ESI): m/z=859.0 [M+H], 857.4 [M−H].
- Example 17 provided an IC50 of between 0.1 and 0.5 nM in a 1a enzyme assay; an IC50 of <0.1 nM in a 1b enzyme assay; a FILM stability value of <50 μl/min/mg; an EC50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of <1.0 nM in a replicon cell line assay in a 1b-con1 background.
-
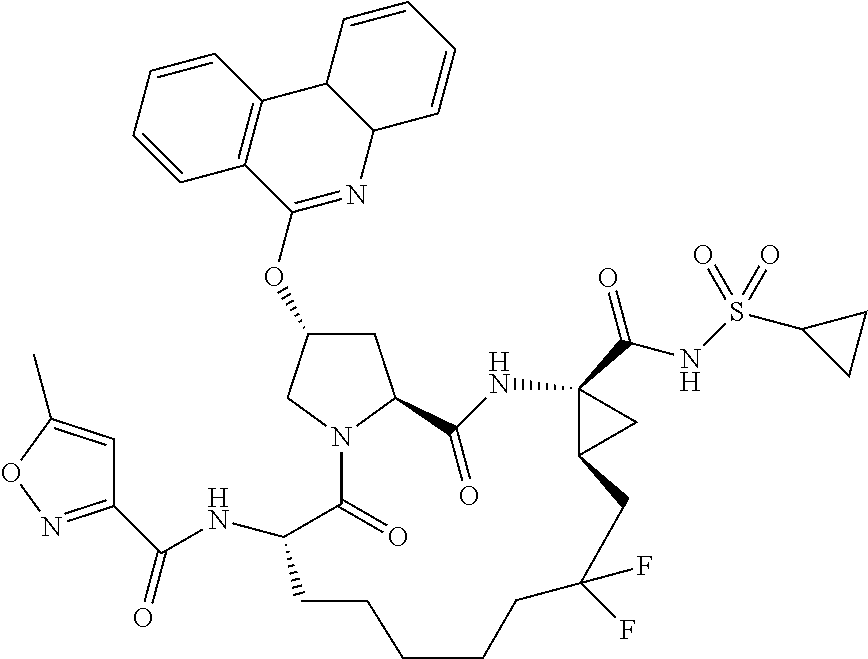
-
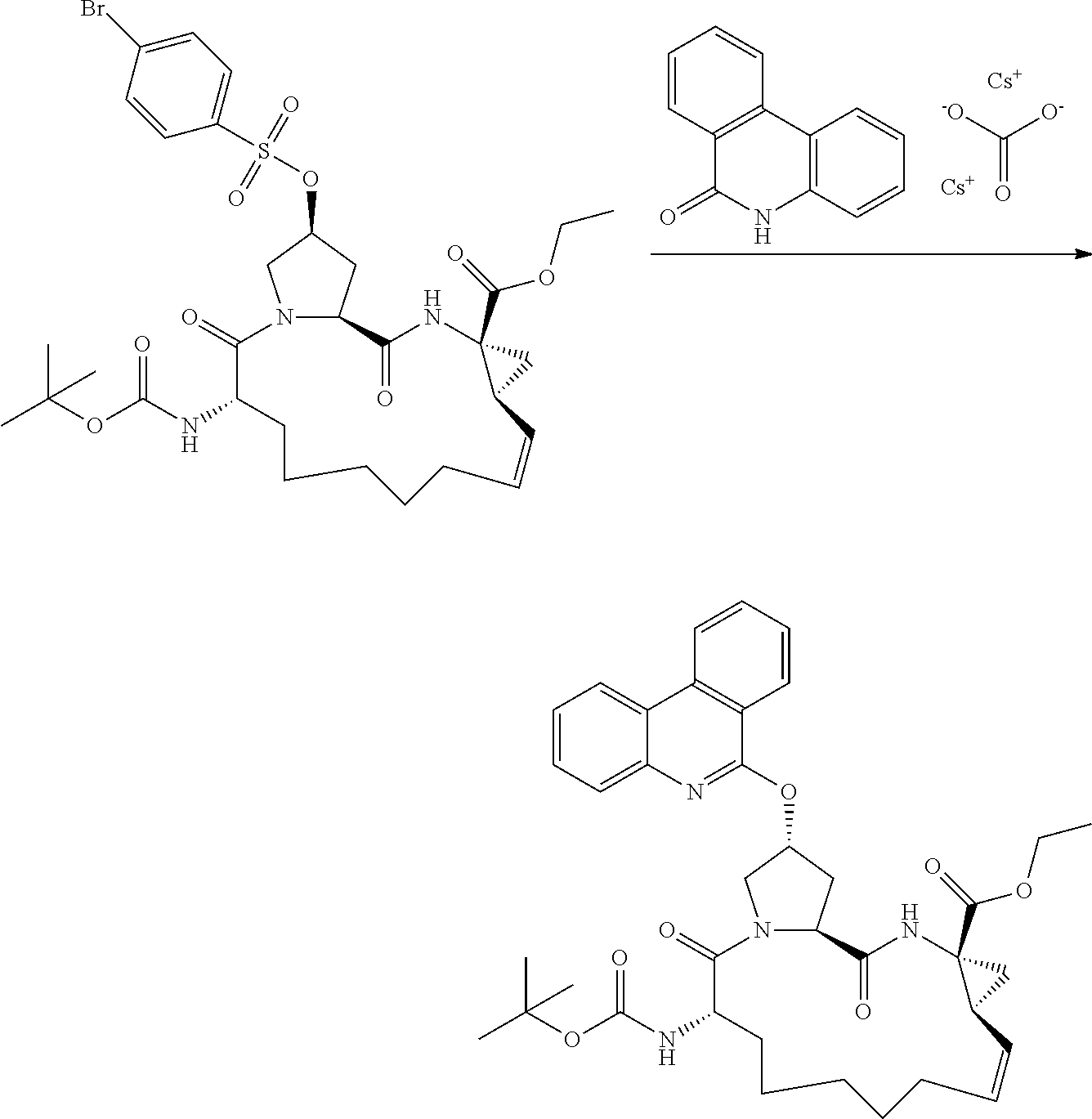
- To (2S,6S,13aS,14aR,16aS,Z)-ethyl 2-(4-bromophenylsulfonyloxy)-6-(tert-butoxycarbonylamino)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (9.00 g, 12.6 mmol) and phenanthridin-6(5H)-one (3.08 g, 15.8 mmol) was added DMSO (50 ml). To this mixture was added cesium carbonate (6.17 g, 18.9 mmol) and the resulting mixture was heated at 75° C. for 3 hr. The reaction mixture was cooled to room temperature, diluted with water (400 ML) and neutralized with HCl (2N, 7.8 mL), The mixture was filtered and the white solid was rinsed with water. The solid was taken up in dichloromethane and filtered to remove insoluble phenanthridinone. The filtrate was concentrated and the residue was purified flash chromatography on silica gel (4:6 hexane/ethyl acetate) to provide the title compound (2R,6S,13aS,14aR,16aS,Z)-ethyl 6-(tert-butoxycarbonylamino)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate as a pale yellow solid (5.07 g, 60% yield).
-

- To a solution of (2R,6S,13aS,14aR,16aS,Z)-ethyl 6-(tert-butoxycarbonylamino)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 18a, 6.25 g, 9.32 mmol) dissolved in Ethyl acetate (50 ml) was added 4 N hydrogen chloride in dioxane (44.3 ml, 177 mmol) and the reaction mixture was stirred at rt for 6 h. The solvent was evaporated under reduced pressure to give a white solid which was rinsed with ethyl acetate. The filtered solid was dried to give (2R,6S,13aS,14aR,16aS,Z)-ethyl 6-amino-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate, Hydrochloric Acid (5.52 g, 98% yield)
-

- To a suspension of (2R,6S,13aS,14 aR,16aS,Z)-ethyl 6-amino-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate, Hydrochloric Acid (Example 18b, 5.00 g, 8.24 mmol), 5-methylisoxazole-3-carboxylic acid (1.15 g, 9.06 mmol) and 2-(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yl)-1,1,3,3-tetramethylisouronium hexafluorophosphate(V) (3.44 g, 9.06 mmol) in dichloromethane (82 ml) was added N-ethyl-N-isopropylpropan-2-amine (5.03 ml, 28.8 mmol). The mixture was stirred at rt for 2 h, and then quenched with saturated aqueous sodium carbonate solution. The organic layer was washed with 1N HCl, separated, filtered, dried over anhydrous magnesium sulfate and evaporated under reduced pressure to provide a white foam.
- This material was purified flash chromatography on silica gel (4:1 dichloromethane/ethyl acetate) to give (2R,6S,13aS,14aR,16aS,Z)-ethyl 6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (4.5 g, 80% yield).
-

- To a solution of (2R,6S,13aS,14aR,16aS,Z)-ethyl 6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 18c, 3.4 g, 5.0 mmol) in tetrahydrofuran (16.7 ml), ethanol (8.4 mL), and water (8.4 mL) was added lithium hydroxide monohydrate (1.36 g, 32.5 mmol). The mixture was stirred at 50° C. for 2 h, the solvent was evaporated under reduced pressure, and the residue partitioned between water and ethyl acetate. The aqueous layer was separated, diluted with ethyl acetate, and washed with 1N HCl. The organic layer was dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure to give (2R,6S,13aS,14aR,16aS,Z)-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid (3.05 g, 94% yield) as a white solid.
-
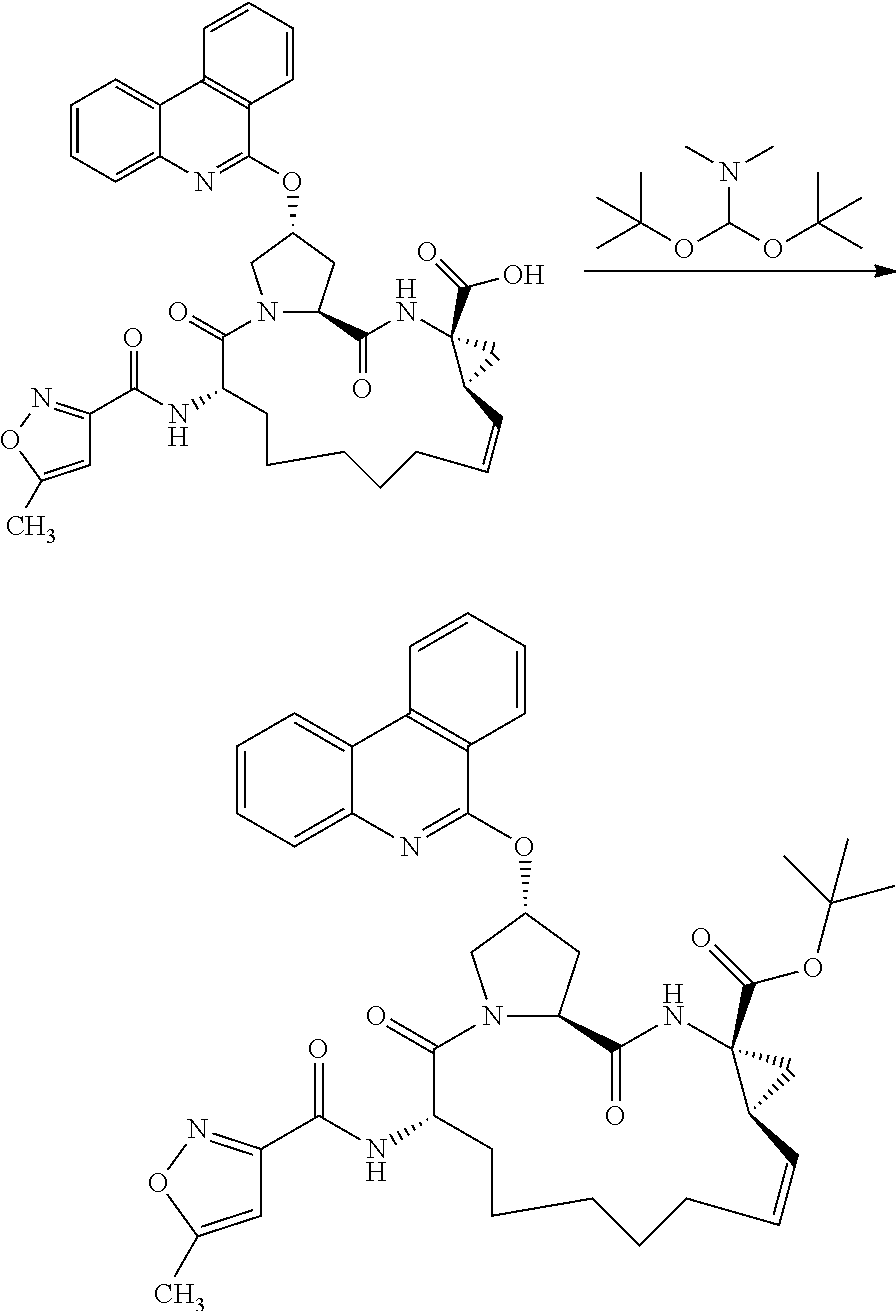
- To a solution of (2R,6S,13aS,14aR,16aS,Z)-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid (Example 18d, 0.84 g, 1.3 mmol) in toluene (3.8 ml) heated at 85° C. was added 1,1-di-tert-butoxy-N,N-dirnethylmethanamine (0.927 ml, 3.87 mmol) dropwise, and the mixture was stirred at 85° C. for 16 hr. The reaction was only 60% complete as shown by LCMS and additional 0.93 mL of 1,1-di-tert-butoxy-N,N-dimethylmethanamine was added in two portions over two hours. The solvent was evaporated and the residue was purified via flash chromatography (1:4 dichloromethane/ethyl acetate) to provide (2R,6S,13aS,14aR,16aS,Z)-tert-butyl 6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrmlo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (720 mg, 1.02 mmol, 79% yield) as a white solid.
-

- To a solution of (2R,6S,13aS,14aR,16aS,Z)-tert-butyl 6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a14,14a,15,16,16a-hexadecabydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 18e, 720 mg, 1.02 mmol) in tetrahydrofuran (10 mL) stirring at 0° C. was added borane tetrahydrofuran complex (1 M in tetrahydrofuran, 6.10 mL, 6.10 mmol). The ice bath was removed after the addition was complete and the reaction mixture was stirred at rt for 0.5 hr. The mixture was cooled to 0° C. and aqueous sodium hydroxide solution (2.5 M, 6.10 mL, 15.26 mmol) was added followed by hydrogen peroxide solution (2.494 rnL, 24.41 mmol). The reaction mixture was stirred at rt for 1 hr. The mixture was then partitioned between ethyl acetate and water, and the organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The solid residue after solvent evaporation was purified via flash chromatography on silica gel (5% methanol/dichloromethane) to give 486 mg of a mixture of products. This material was repurified by preparative thin layer chromatography (5% methanol/dichloromethane) to provide the title compound (2R,6S,13aS,14aR,16aS)-tert-butyl 12-hydroxy-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopenta decine-14a-carboxylate (170 mg, 23%) along with the α-hydroxy isomer (200 mg, 27% yield).
-
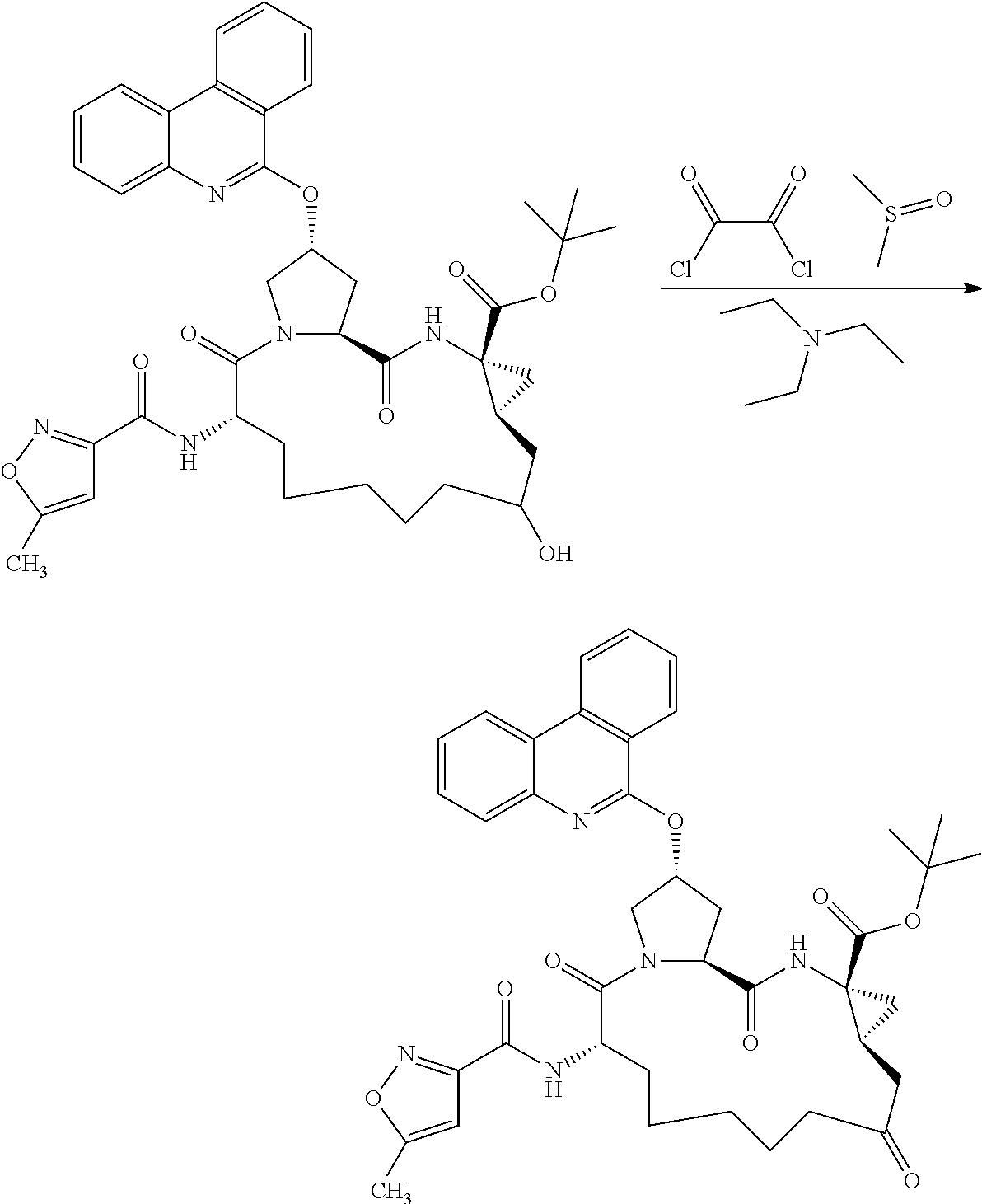
- To a solution of oxalyl chloride (0.017 ml, 0.198 mmol) in dichloromethane (0.6 ml) cooled to −60° C. was added DMSO (0.030 ml, 0.419 mmol) dropwise. The mixture was stirred for 15 min and added dropwise a solution of compound (2R,6S,13aS,14aR,16aS)-tert-butyl 12-hydroxy-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 18 h, 80 mg, 0.11 mmol) in dichloromethane (0.4 ml). The reaction mixture was stirred for 20 min at −60° C., and triethylamine (0.123 ml, 0.882 mmol) was added dropwise. The mixture was stirred at −60° C. for 5 min, and allowed to warm to room temperature with stirring for 10 min. The reaction mixture was quenched with water, stirred for 15 min, and diluted with dichloromethane. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure to give a foamy material. The crude product was purified by flash chromatography on silica gel (5% methanol/dichloromethane) to provide the title compound (2R,6S,13aS,14aR,16aS)-tert-butyl 6-(5-methyl isoxazole-3-carboxamido)-5,12,16-trioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (47 mg, 59° A yield).
-
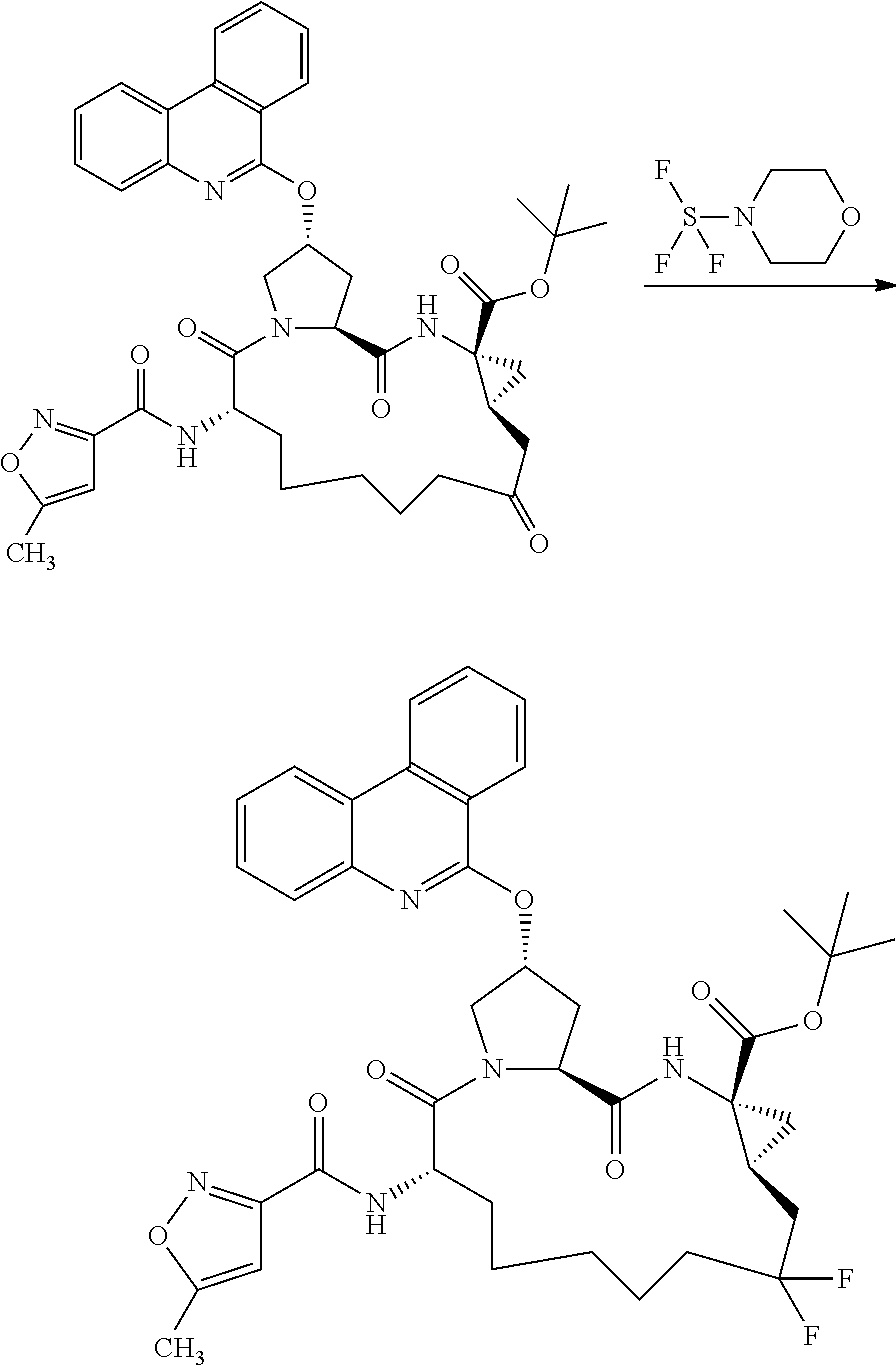
- A solution of (2R,6S,13aS,14aR,16aS)-tert-butyl 6-(5-methylisoxazole-3-carboxamido)-5,12,6-trioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 18 g, 47 mg, 0.065 mmol) in Morpho-DAST (317 μl, 2.60 mmol) was stirred at rt for 40 h. The resulting mixture was dissolved in 1 mL dichloromethane and added dropwise to a cooled solution of saturated aqueous sodium carbonate. The mixture was stirred for 10 min and extracted with dichloromethane. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The crude material was purified by preparative thin layer chromatography (3% methanol/dichloromethane) to provide the title compound (2R,6S,13aS,14aR,16aS)-tert-butyl 12,12-difluoro-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (20 mg, 41% yield) as a white solid.
-
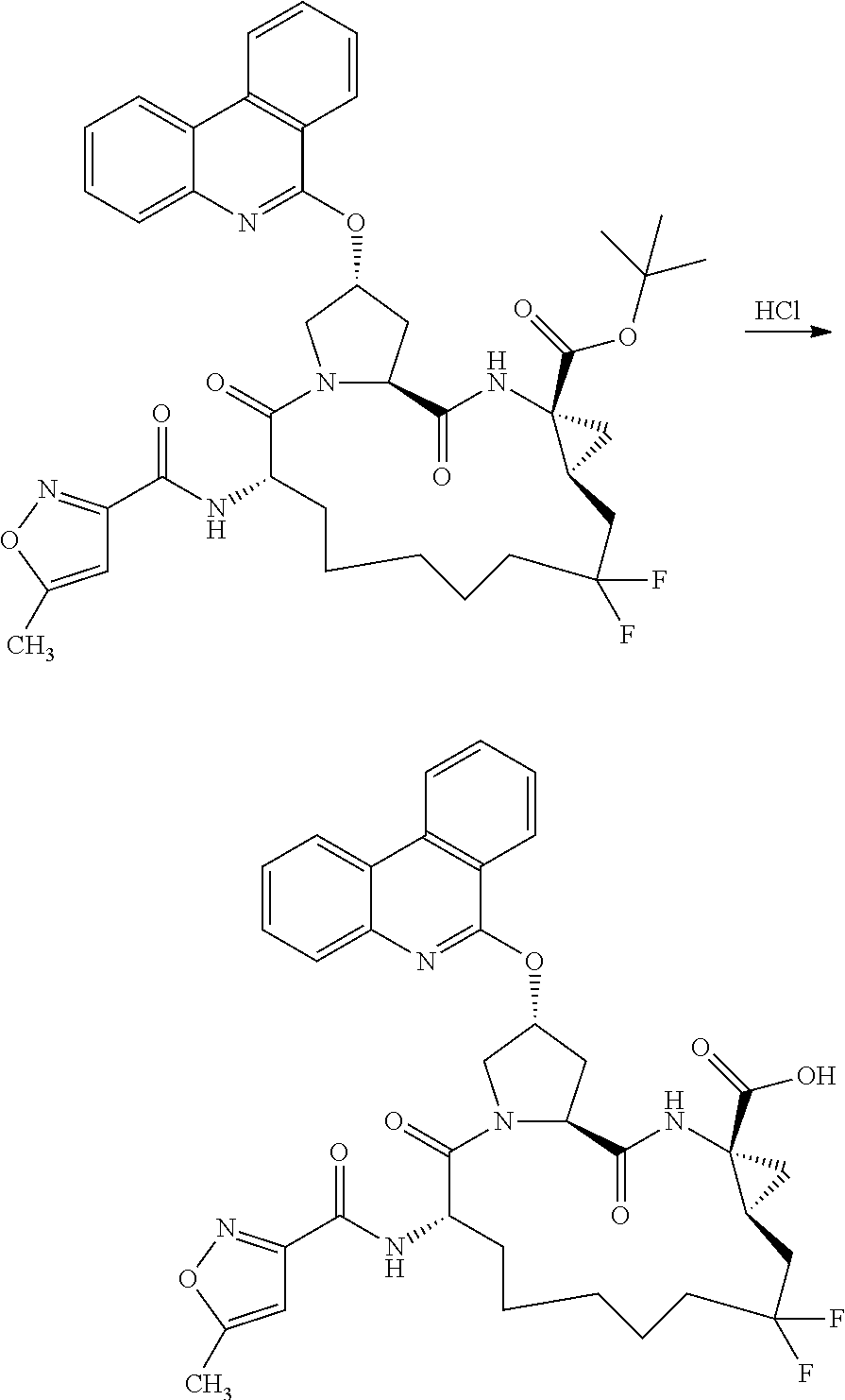
- To a solution of (2R,6S,13aS,14aR,16aS)-tert-butyl 12,12-difluoro-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxyate (Example 18 h, 20 mg, 0.027 mmol) in ethyl acetate (100 μl) stirring at rt was added 4N hydrogen chloride (134 μl, 0.536 mmol) in dioxane. The reaction mixture was stirred for 24 h, and the solvent was evaporated under reduced pressure. The residue was purified by preparative thin layer chromatography (10% methanol/dichloromethane) to provide the title compound (2R,6S,13aS,14aR,16aS)-12,12-difluoro-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid (10.2 mg, 55%)
-
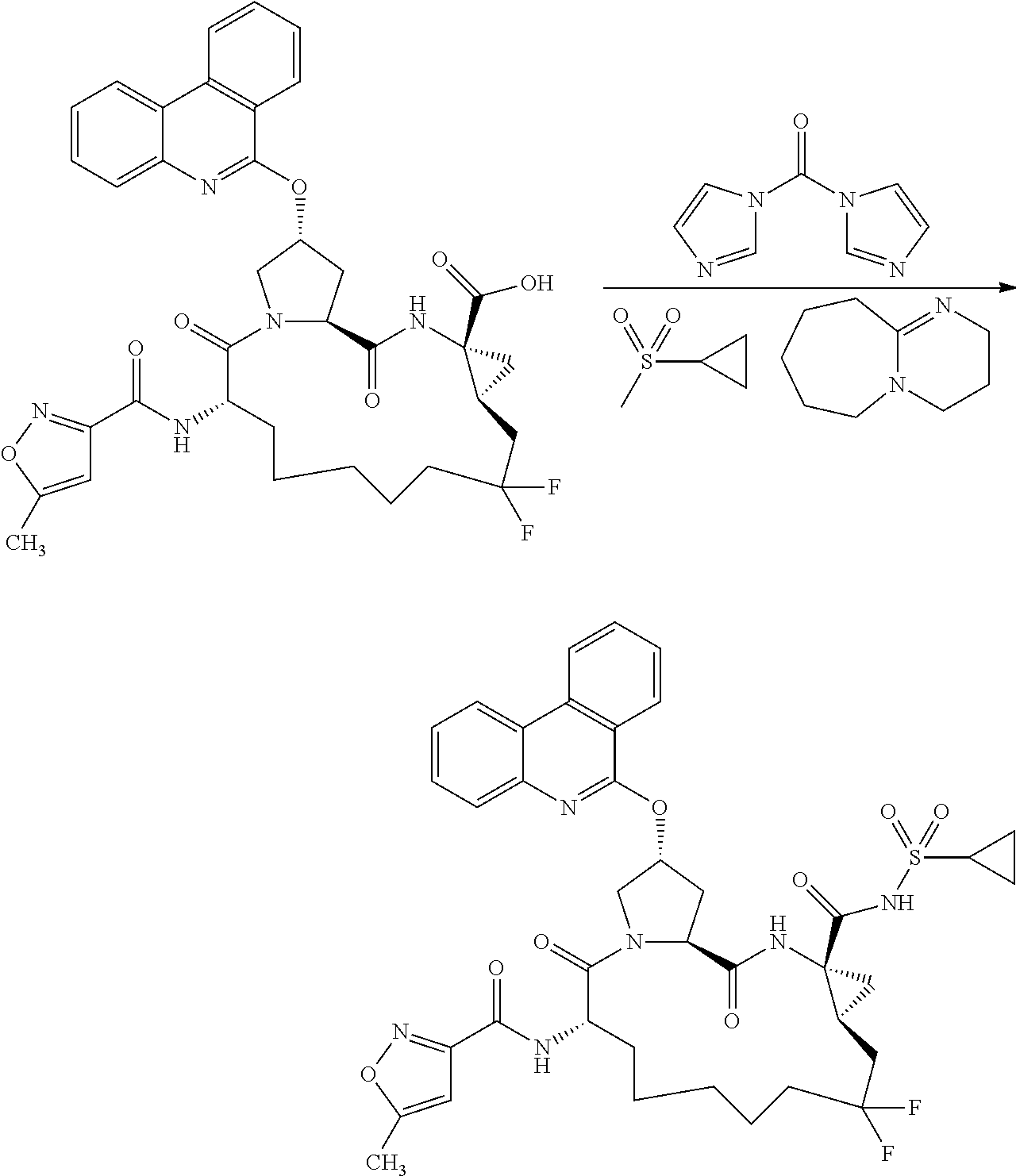
- A solution of 2R,6S,13aS,14aR,16aS)-12,12-difluoro-6-(5-methylisoxazole-3-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclo pentadecine-14a-carboxylic acid (Example 18i, 21 mg, 0.030 mmol) and di(1H-imidazol-1-yl)methanone (10.96 mg, 0.068 mmol) in dichloroethane (400 μl) was stirred for 2 hr at 42° C. To this mixture was added methylsulfonylcyclopropane (11.0 mg, 0.091 mmol) and 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-d]azepine (13.8 μl, 0.091 mmol) and the reaction mixture was stirred for 2 h at 42° C. The reaction mixture was diluted with ethyl acetate (7 mL) and washed with 10 mL of 1 N HCl followed by saturated aqueous sodium chloride solution. The organic layer was dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The resulting residue was purified by flash chromatography on silica gel (3% methanol/dichloromethane). The resulting material was further purified by reverse phase HPLC (40% acetonitrile/water with 0.1% TFA to 100% water) to provide the title compound N-((2R,6S,13aS,14aR,16aS)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-yl)-5-methylisoxazole-3-carboxamide (16 mg, 66.3% yield). MS (EST): nalz=793.1 [1\11-1+1], 791.0 [M−H].
- Example 18 provided a HLM stability value of between 50 and 100 μl/min/mg; an EC50 of <1.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of <1.0 nM in a replicon cell line assay in a 1b-con1 background.
-
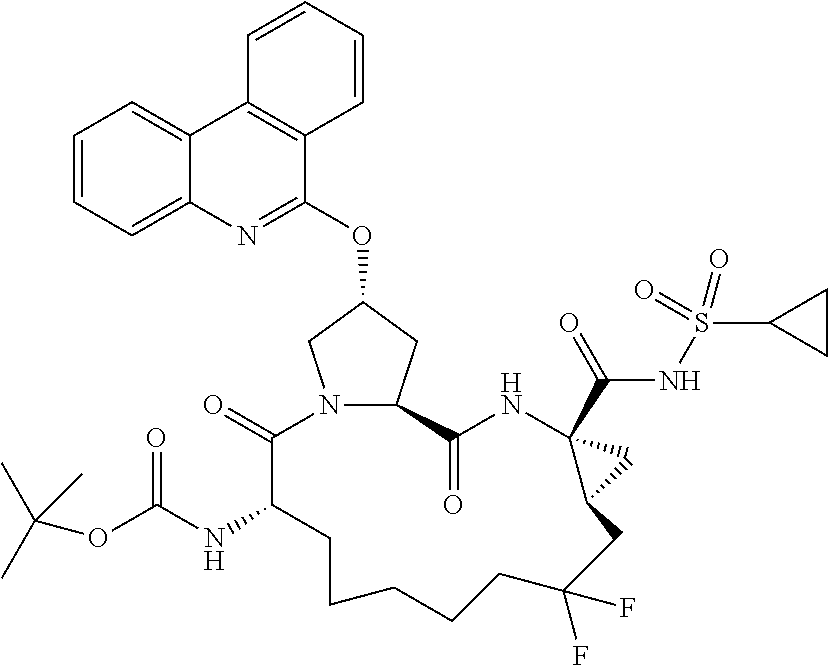
-
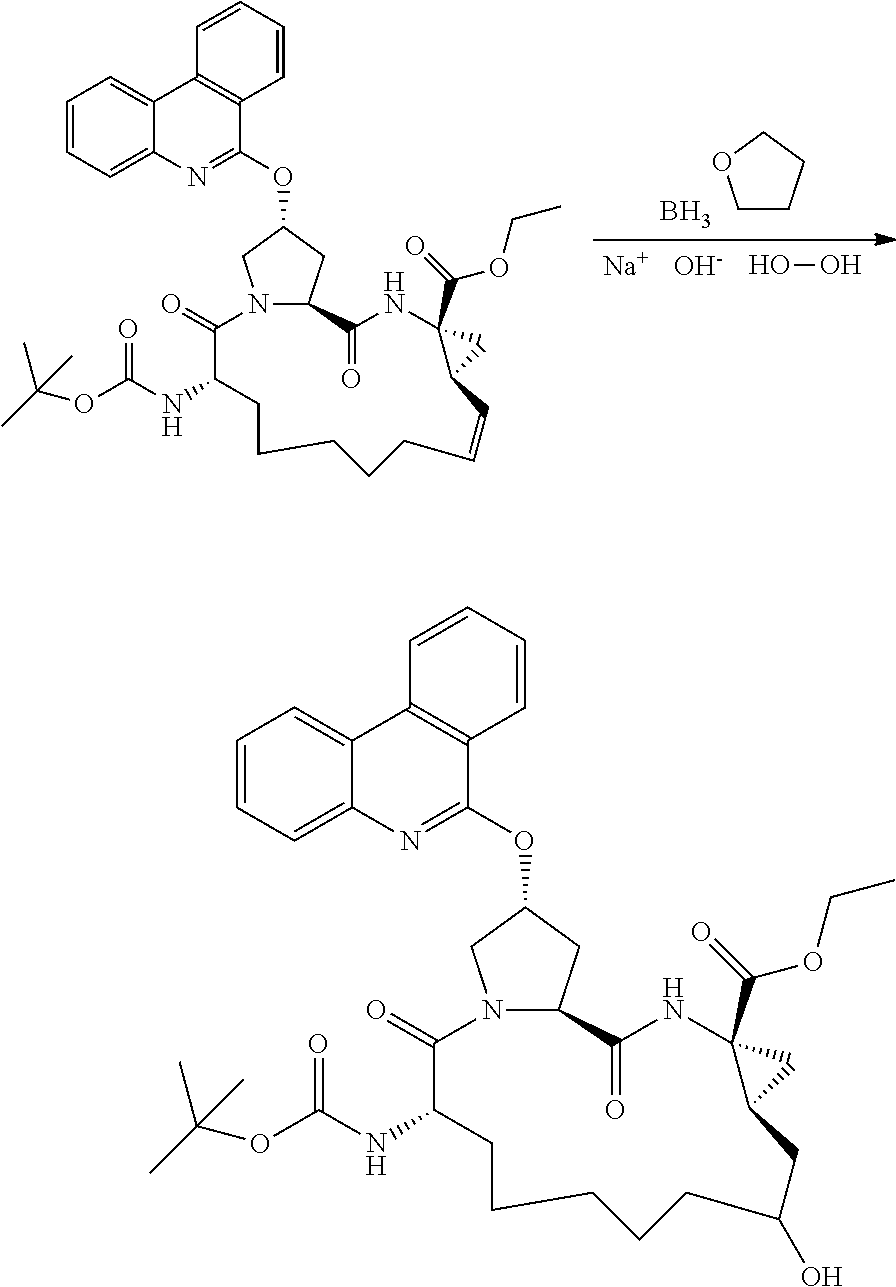
- To a solution of (2R,6S,13aS,14aR,16aS,Z)-ethyl 6-(tert-butoxycarbonylamino)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 18a, 3.2 g, 477 mmol) in tetrahydrofuran (41.5 ml) at rt with a water bath was added dropwise borane tetrahydrofuran complex (1 M in tetrahydrofuran, 14.3 ml, 14.3 mmol) and stirred at room temperature for 1 h. The mixture was cooled to 0° C. and sodium hydroxide (11.5 ml, 28.6 mmol) was added dropwise followed by hydrogen peroxide (4.0 ml, 43 mmol), and the reaction mixture stirred for 1.5 h at 0° C. The mixture was diluted with ethyl acetate and water, and the organic layer was washed with saturated aqueous sodium chloride solution. The resulting solid was purified via flash chromatography on silica gel (5% methanol in dichloromethane) to provide the title compound (2R,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-12-hydroxy-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (1.06 g, 1.539 mmol, 32.3% yield). The α-hydroxy analog (2R,6S,13R,13aR,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-13-hydroxy-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (0.7 g, 21% yield) was also isolated.
-
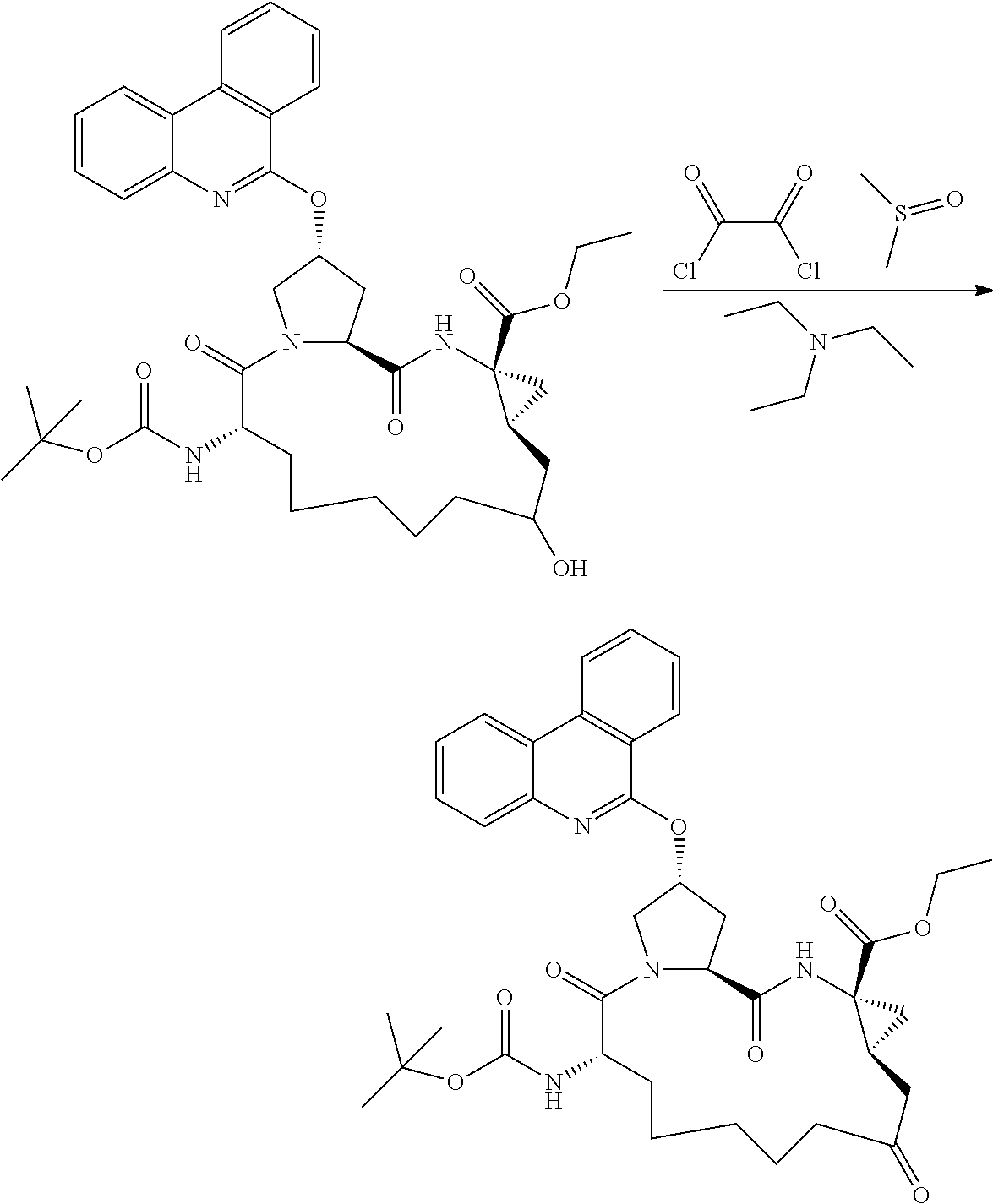
- To a solution of oxalyl chloride (0.242 ml, 2.77 mmol) in dichloromethane (5.13 ml) stirring at 60° C. was added DMSO (0.415 ml, 5.85 mmol) dropwise. The mixture was stirred for 20 min and added dropwise to a solution of (2R,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-12-hydroxy-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydro cyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 19a, 1.06 g, 1.54 mmol) in dichloromethane (5.13 ml). The reaction mixture was stirred for 20 min −60° C., and triethylamine (1.716 ml, 12.31 mmol) was added. The mixture was stirred for 10 min at −60° C., and then stirred for another 20 min at room temperature. The reaction mixture was quenched with water and stirred for 15 mm, and diluted with dichloromethane. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure to give a foamy material. The crude product was purified by flash chromatography on silica gel (5% methanolldichloromethane). The fractions containing the title compound were collected and rechromatographed on a silica gel column (65% ethyl acetate/hexane) to provide the title compound (2R,6S,13 aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-5,12,16-trioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (680 mg, 64.3% yield) as a white solid.
-
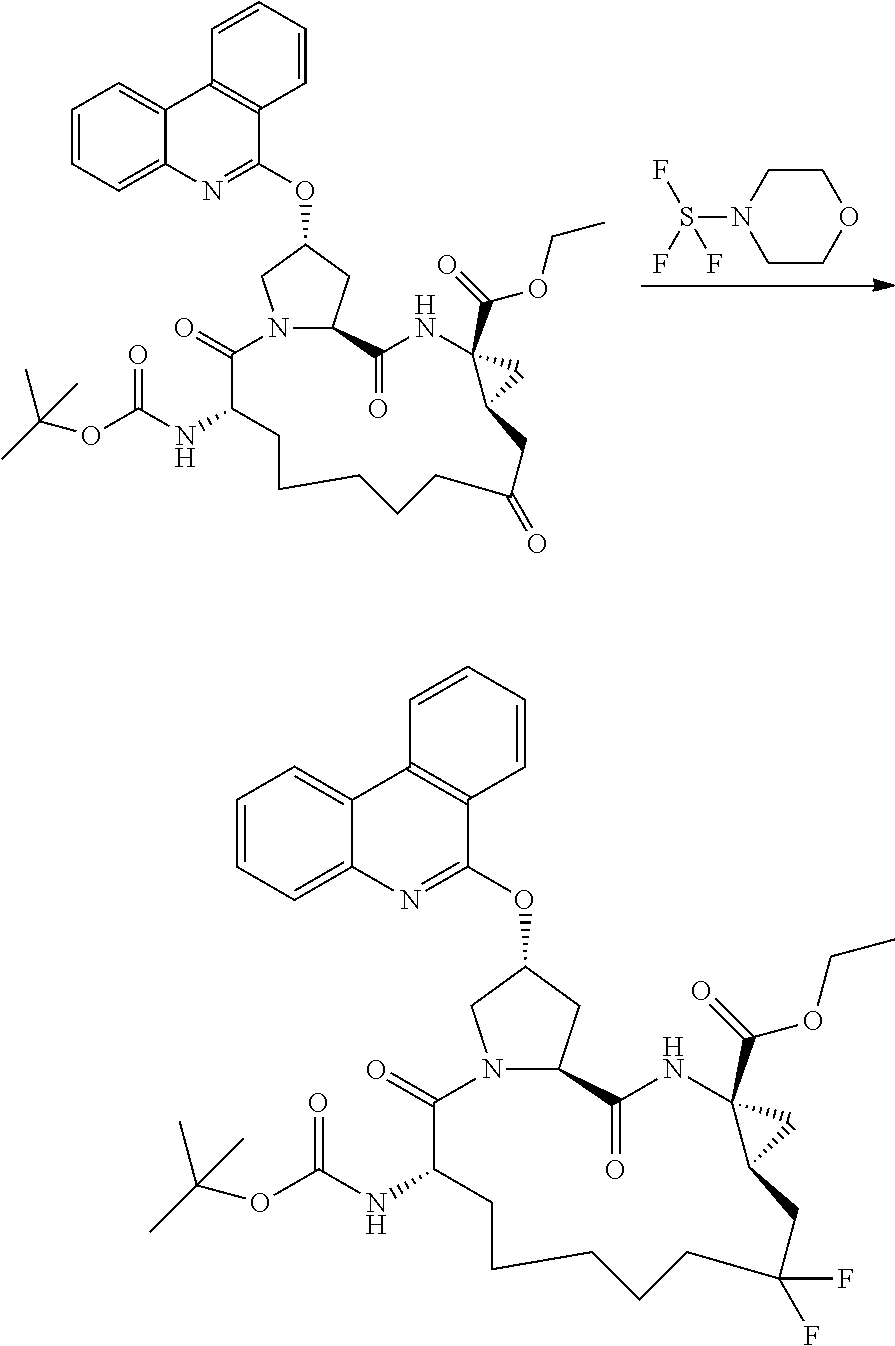
- A solution of (2R,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-5,12,16-trioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (Example 19b, 680 mg, 0.990 mmol) in Morpho-DAST (2.4 mL, 20 mmol) was stirred at room temperature for 48 h. The reaction mixture was dissolved in 2 mL dichloromethane and added dropwise into a cooled solution of saturated aqueous sodium carbonate solution, stirred for 10 min, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The residue was purified by preparative thin layer chromatography on silica gel (3, methanol/dichloromethane) to provide the title compound (2R,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydro cyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylate (320 mg, 45.6% yield) as a white solid.
-
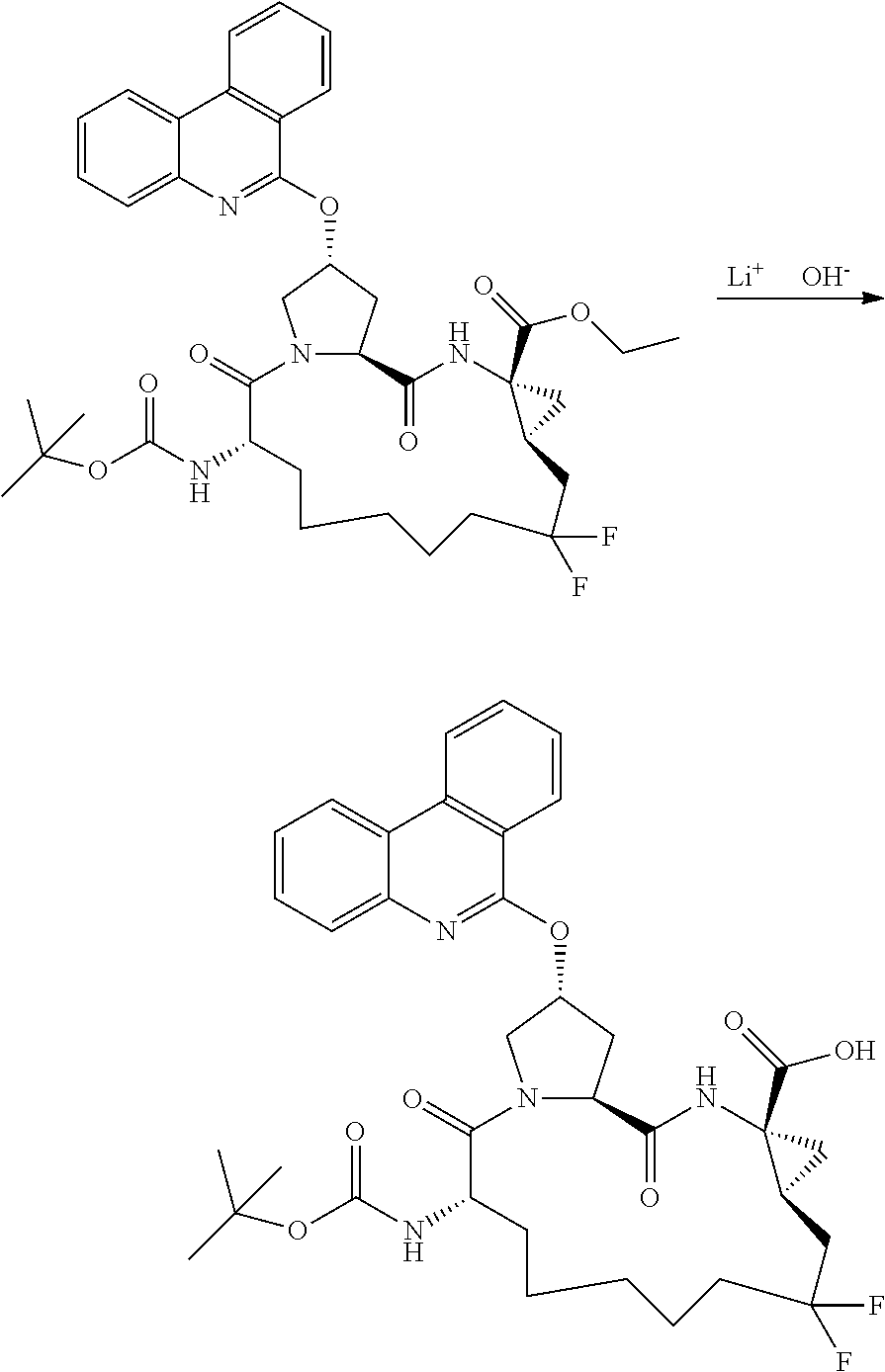
- To a solution of (2R,6S,13aS,14aR,16aS)-ethyl 6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclo pentadecine-14a-carboxylate (Example 19c, 320 mg, 0.451 mmol) in tetrahydrofuran (1505 up, water (752 μl), and ethanol (752 μl) was added lithium hydroxide monohydrate (123 mg, 2.93 mmol) at room temperature The mixture was stirred at room temperature for 16 h and then heated at 37° C. for 6 h. The mixture was cooled to room temperature and then eluted through a pad of silica gel with 10% methanol/dichloromethane, and evaporated under reduced pressure. The crude material was purified by flash chromatography on silica gel (10% methanol/dichloromethane) to provide the title compound (2R,6S,13aS,14aR,16aS)-6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadeca hydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid (215 mg, 70.0° A, yield) as a white solid.
-
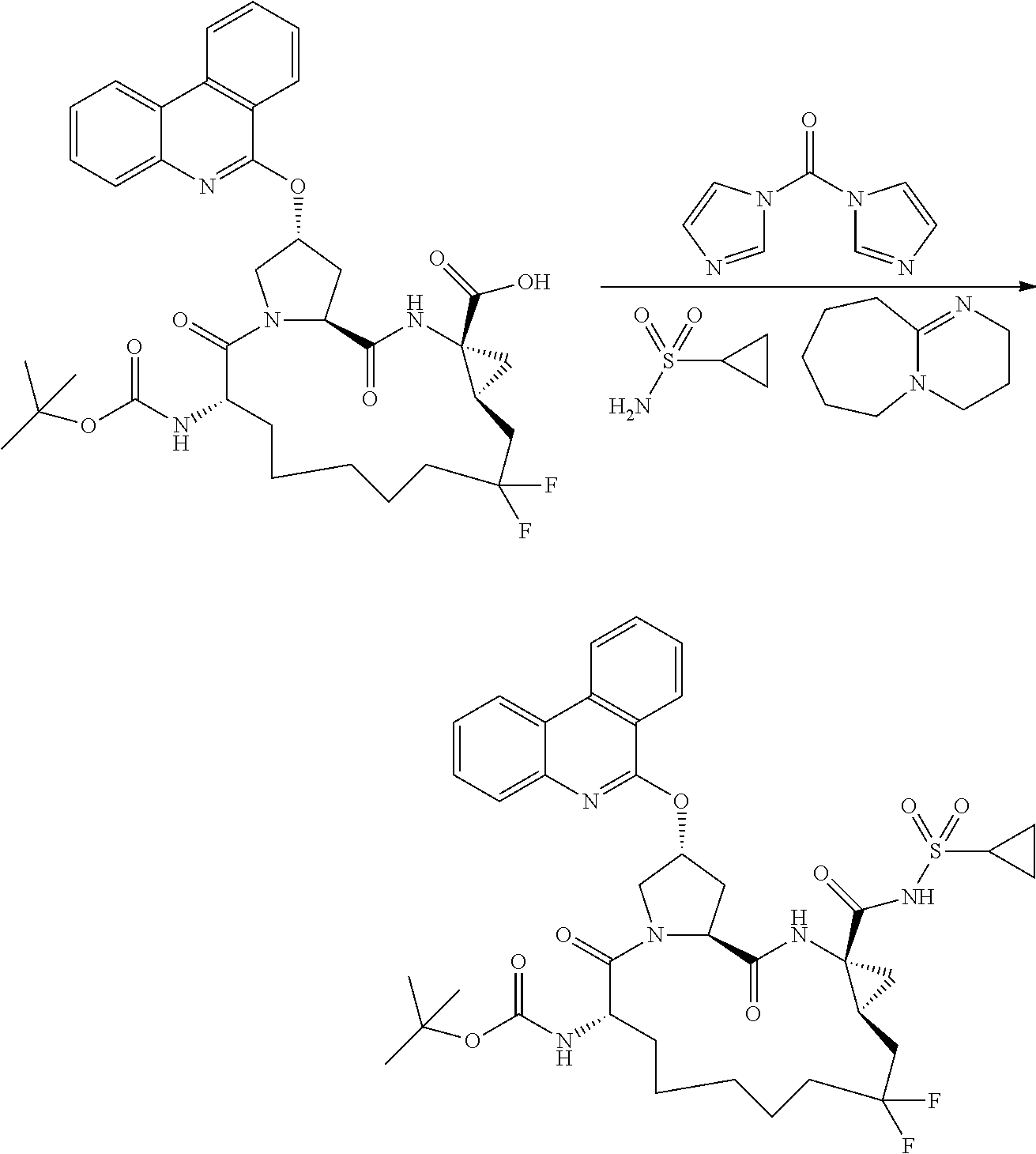
- A solution of (2R,6S,13 aS,14aR,16aS)-6-(tert-butoxycarbonylamino)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydro cyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxylic acid (215 mg, 0.316 mmol) and di(1H-imidazol-1-yl)methanone (Example 19d, 114 mg, 0.701 mmol) in dichloroethane (2 mL) was stirred for 2 h at room temperature. To this mixture was added cyclopropanesulfonamide (115 mg, 0.948 mmol) and 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine (0.143 mL, 0.948 mmol, dried with 4 A sieves powder at 42° C. for 2 h prior to usage), and the solution was stirred for 2 h. The reaction mixture was diluted with ethyl acetate (7 mL) and washed with 10 mL of 0.1 N HCl followed by saturated aqueous sodium chloride solution. The organic layer was dried with anhydrous sodium sulfate, filtered, and evaporated under reduced pressure leaving 0.23 g crude solid which was purified via flash chromatography on silica gel (3% methanol/dichloromethane) to provide the title compound tert-butyl (2R,6S, I 36,14aR,16aS)-14a-(cyclopropylsulfonylcarbamoyl)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolb[1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate (195 mg, 0.249 mmol, 79% yield). MS (ESI): m/z=784.1 [M+H]. Example 19 provided an IC50 of <0.1 nM in a 1a enzyme assay; an IC50 of <0.1 0 nM in a 1b enzyme assay; a HLM stability value of >100 μl/min/mg; an EC50 of <1.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of <1.0 nM in a replicon cell line assay in a 1b-con1 background.
-

- Example 20a. (2R,6S,13aS,14aR,16aS)-6-amino-N-(cyclopropylsulfonyl)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopenta decine-14a-carboxamide
-
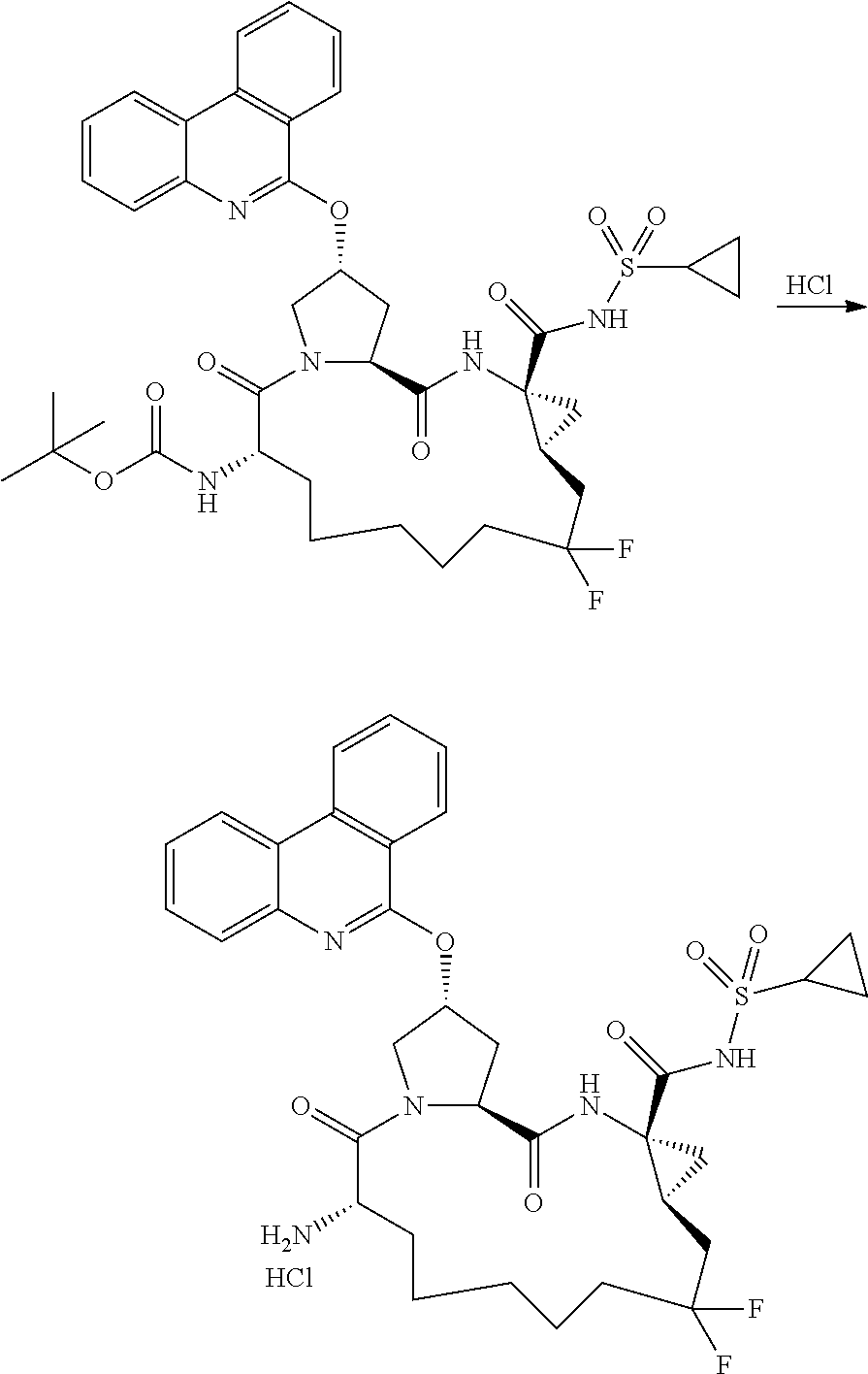
- To a solution of tert-butyl (2R,6S,13aS,14aR,16aS)-14a-(cyclopropylsulthnylcarbamoyl)-12,12-difluoro-5,16-dioxo-2-(phenanthtidin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-6-ylcarbamate (Example 19, 195 mg, 0.249 mmol) in ethyl acetate (1 mL) stirring at room temperature was added 4 N hydrogen chloride (1.24 mL, 5.0 mmol) in dioxane. The reaction mixture was stirred for 4 h, and then the solvent was evaporated under reduced pressure. The residue was triturated with ethyl acetate and the product was filtered as a white solid. The solid was dissolved in methanol and the solvent was evaporated to provide the title compound (2R,6S,13 aS,14aR,16aS)-6-amino-N-(cyclopropylsulfonyl)-12,12-difluoro-5,16-dioxo-2-(phenarithridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide, Hydrochloric Acid (178 mg, 0.247 mmol, 99% yield).
-
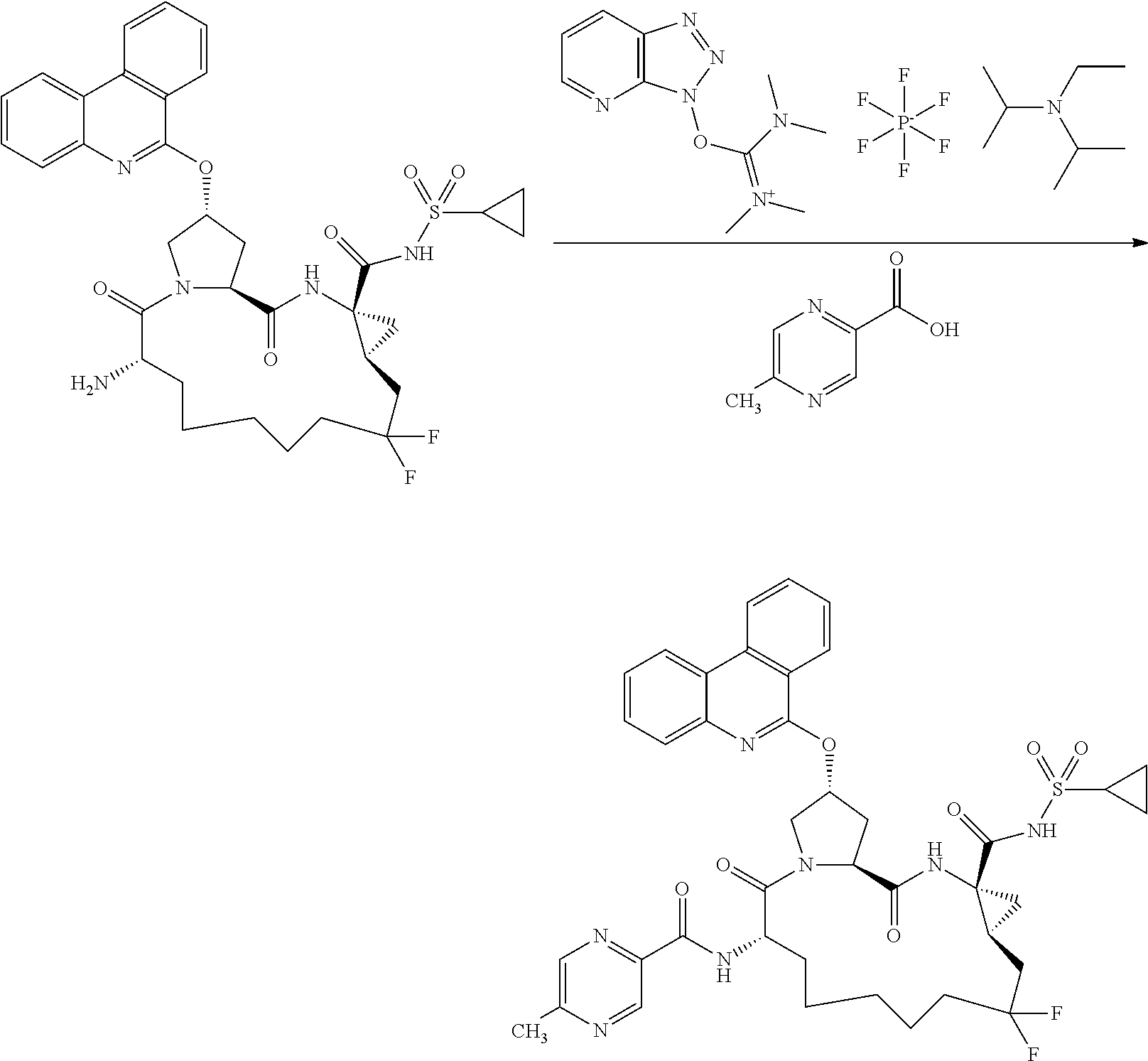
- To a mixture of a (2R,65,13aS,14aR,16aS)-6-amino-N-(cyclopropylsulfonyl)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide, Hydrochloric Acid (Example 20a, 12 mg, 0.017 mmol), 5-methylpyrazine-2-carboxylic acid (2.53 mg, 0.018 mmol) and 2-(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yl)-1,1,3,3-tetramethylisouronium hexafluorophosphate(V) (7.0 mg, 0.018 mmol) in dichloromethane (167 μl) was added N-ethyl-N-isopropylpropan-2-amine (10.18 μl, 0.058 mmol), and the resulting mixture was stirred for 3 h at room temperature. 0.1 N HCl was added and the organic layer was separated, dried over anhydrous sodium sulfate, and filtered. The residue after solvent evaporation was eluted through silica gel with 10% acetone/dichloromethane. The fractions containing the title compound were combined and the resulting crude material was purified by preparative thin layer chromatography on silica gel (20:1 dichloromethane/methanol) to provide the title compound (2R,6S,13aS,14aR,16aS)—N-(cyclopropylsulfonyl)-12,12-difluoro-6-(5-methylpyrazine-2-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide (8 mg, 60% yield) as a white solid. MS (ESI): m/z=804.1 [M+H]. Example 20 provided a HLM stability value of <50 μl/min/mg; an EC50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of between 1.0 and 2.0 nM in a replicon cell line assay in a 1 b-con1 background.
-
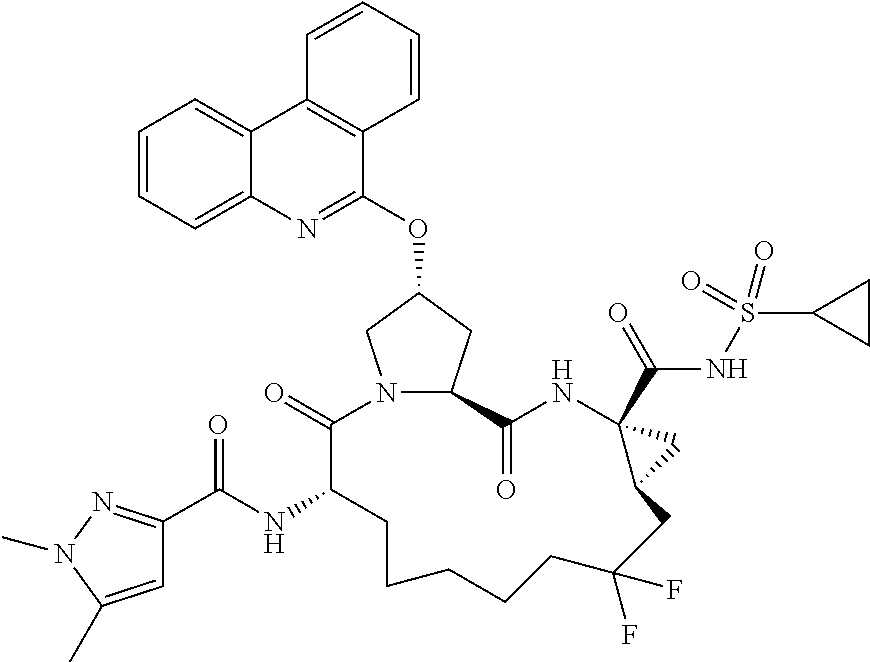
-
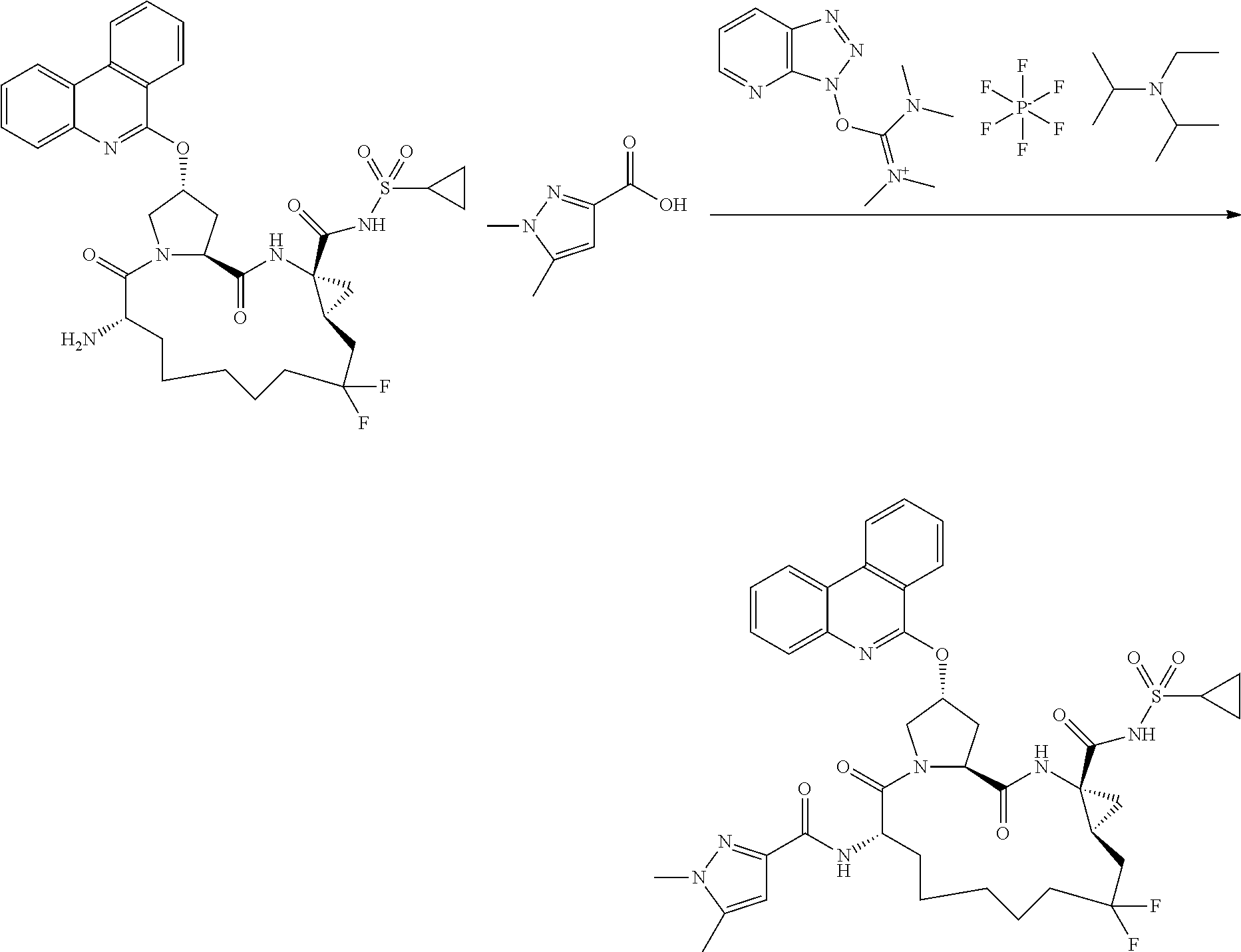
- To a mixture of (2R,6S,13aS,14aR,16aS)-6-amino-N-(cyclopropylsulfonyl)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy-)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide, Hydrochloric Acid (Example 20a, 12 mg, 0.017 mmol), 1,5-dimethyl-1H-pyrazole-3-carboxylic acid (2.57 mg, 0.018 mmol) and 2-(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yl)-1,1,3,3-tetramethylisouronium hexafluorophosphate(V) (7.0 mg, 0.018 mmol) in dichloromethane (167 μl) was added N-ethyl-N-isopropylpropan-2-amine (10.18 μl, 0.058 mmol) and the mixture was stirred for 3 h at room temperature. 0.1 N HCl was added and organic layer was separated, dried over anhydrous sodium sulfate, and filtered. The residue after solvent evaporation under reduced pressure was eluted through silica gel with 10% acetone/dichloromethane. The fractions containing the title compound were combined and the resulting crude material was purified by preparative thin layer chromatogaphy on silica gel (20:1 dichloromethane/methanol) to provide the title compound (2R,6S,13aS,14aR,16aS)—N-(cydopropylsulfonyl)-6-(1,5-dimethyl-1H-pyrazole-3-carboxamido)-12,12-difluoro-5,16-dioxo-2-(phenanthridin-6-yloxy)octadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide (8 mg 60% yield) as a white solid. MS (ESI): m/z=806.1 [M+1-1]. Example 21 provided a FILM stability value of <50 μl/min/mg; an EC50 of between 2.0 and 5.0 nM in a replicon cell line assay in a 1a-H77 background, and an EC50 of <1.0 nM in a replicon cell line assay in a 1b-con1 background.
- The activity of recombinant HCV NS3 proteases derived from isolates representing genotypes 1, 2, 3 or 4 is measured by cleavage of the following peptide substrate:
-
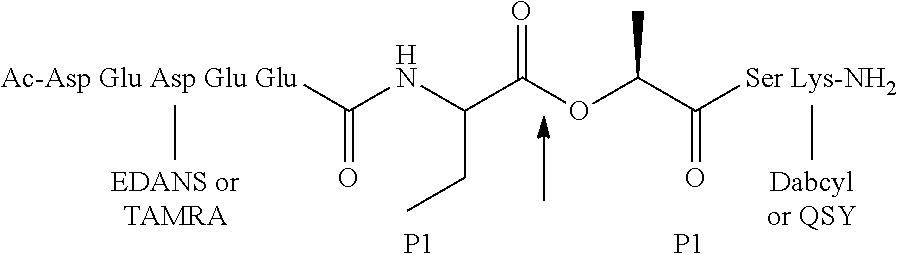
- The substrate is labeled with a fluor and a fluorescence quencher. Cleavage results in release of the quencher and an increase in fluorescence. NS3 protease is incubated with a dilution series of inhibitor in 150 mM NaCl, 10% Glycerol, 5 mM. DTT, with or without 0.01% dodecyl maltoside for either 30 minutes or 300 minutes. Substrate is added at a concentration of 5 uM to initiate the reaction, and fluorescence is measured at 2 minute intervals for 30 minutes. Enzyme concentrations range from 10 to 100 nM in the absence of detergent, or 10-fold lower in the presence of detergent. Substrate peptides are labeled with either EDANS and DABCYL (excitation 355 nm, emission 485 nm) or TAMRA and QSY (excitation 544 nm, emission 590 nm). For routine IC50 determination, 3-fold serial dilutions starting with initial concentrations of 100 μM, 200 μM, or 2 mM are used. For compounds with Ki values approaching or lower than the enzyme concentration, a tight-binding calculation format is used, with 24 dilutions of inhibitor covering a range of 0 to 100 nM inhibitor. Ki values are calculated using the tight binding assay format, according to the following equation:
-
V=A{[(K+I−E)2+4KE])1/2−(K+I−E)}, - where I=total inhibitor concentration, E=active enzyme concentration, K=apparent Ki value and A=[kcat)S/2][Km=(S)].
- Two subgenomic replicon cell lines can be used for compound characterization in cell culture: one derived from genotype 1a and one derived from genotype 1 b. Both replicon constructs are bicistronic subgenomic replicons essentially similar to those described by Bartenschlager and coworkers (Lohmann et al., Science (1999) 285(5424):110-113). The genotype 1a replicon construct contains the NS3-NS5B coding region derived from the H77 strain of HCV (1a-H77) (Blight et al. J Virol (2003) 77(5):3181-3190). The first cistron of the construct consists of the first 36 nucleotides of the HCV1a-H77 core gene fused to a firefly luciferase reporter and a neomycin phosphotransferase (Neo) selectable marker. The luciferase and Neo coding regions are separated by the FMDV 2a protease. The second cistron contains the NS3-NS5B coding region derived from 1a-H77 with the addition of adaptive mutations E1202G in NS3, K1691R in NS4A, and K2040R and S2204I in NS5A. The 1b-Con-1 replicon construct is identical to the 1a-H77 replicon, except that the 5′ and 3′ NTRs and the NS3-NS5B coding region can be derived from the 1b-Con-1 strain (Blight et al., Science (2000) 290(5498):1972-1974), and the adaptive mutations are E1202G and T12801 in NS3 and S2204I in NS5A.
- Replicon cell lines can be maintained in Dulbecco's modified Eagles medium (DMEM) containing 100 IU/ml penicillin, 100 mg/ml streptomycin (Invitrogen), 200 mg/ml G418 (Invitrogen) and 10% (v/v) fetal bovine serum (FBS). Replicon-containing cells can be seeded into 96 well plates at a density of 5000 cells per well in 100 μl DMEM containing 5% FBS. The next day, the compound can be initially diluted in dimethyl sulfoxide (DMSO) to generate a 200× stock of the inhibitor in a series of 8 half-log dilutions. The dilution series can then be diluted 100-fold in the medium containing 5% FBS. One hundred microliters of medium with the inhibitor can be added to each well of the overnight cell culture plate already containing 100 μl of DMEM with 5% FBS. In assays where the protein binding effect on inhibitor potency is assessed, the medium from the overnight cell culture plates can be replaced with 200 μl DMEM containing 40% human plasma (innovative Research) plus 5% FBS as well as compound. The cells can be grown for 4 days in tissue culture incubators. The inhibitory effects of compounds against the replicons can be determined by measuring either the level of luciferase or HCV RNA. The luciferase assay can be conducted using a Luciferase Assay System kit (Promega) following the manufacturer's instructions. Briefly, the cell culture medium is removed and wells are washed with 200 μl of phosphate-buffered saline. To each well Passive Lysis buffer (Promega, WI) is added and the plates are incubated for 30 min with rocking to lyse the cells. Luciferin solution (50 μl, Promega) is added, and luciferase activity is measured with a Victor II luminometer (Perkin-Elmer). To determine HCV RNA levels, RNA extractions can be performed using the CellsDirect kit (Invitrogen), and the HCV RNA copy number can be measured using the SuperScript III Platinum One-Step qRT-PCR system (Invitrogen) and primers specific to the HCV 5′ nontranslated region. Cytotoxicity can be determined by the 3-[4,5-dimethythiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay as follows. Replicon cells is plated in 96-well plates (4000 cells per well), the next day compound dilutions are added as in the activity assay, and the cells are grown in the presence of the inhibitors for 4 days. The MTT solution is diluted in DMEM containing 5% FBS and 60 μl of the solution is added to the cells. After 4 hrs, the cells are solubilized by the addition of 30 □μl SDS (20% in 0.02 N HCl). The plates are incubated overnight and the optical density can be measured at 570 nm. To determine compounds' EC50 and TD50, luciferase, RNA inhibition and MTT data can be analyzed using the GraphPad Prism 4 software (equation: sigmoidal dose-response variable slope).
- Mutations detected in resistance selection studies can be introduced into wild type transient replicon constructs based on genotypes 1a-H77 and 1b-N. Both replicons are bicistronic sub-genomic constructs containing a firefly luciferase reporter similar to those described above, but they do not contain a Neo selectable marker and are therefore only suitable for transient replication assays. The 1a-H77 replicon for transient assays further differs from the replicon in the stable cell line in that it contains NS2 through NS5B in the second cistron. The 1b-N strain replicon contains NS3 through NS5B in the second cistron, with adaptive mutations E1202G in NS3 and S2204I in NS5A. Mutagenesis can be performed using the Stratagene QuikChange XL 11 site-directed mutagenesis kit. Mutants' sequences can be confirmed, plasmids can be linearized with Xba I restriction enzyme and used as template for in vitro transcription reactions to make mutant replicon RNA for transient transfections. In vitro transcription can be performed with the T7 Megascript kit (Ambion).
- Transient replicon transfections can be performed essentially as described by Mo et al. (Antimierob Agents Chemother (2005) 49(10):4305-4314) with slight modifications. Fifteen micrograms of template RNA can be used to electroporate 3×106 cells in a 200 μl volume in a 0.2 cm cuvette. The cells used for transient transfections can be Huh7 cells obtained by curing replicon-containing cells with IFN (Mo et al., supra). Electroporation can be done with a Gene Pulser II (Bio-Rad, CA) at 480V and 25 μF, using two manual pulses. Transfected cells can be diluted to 7.5×10 cells/ml and plated in 96 well plates at 7.5×103 cells per well in DMEM with 5% FBS and 100 IU/ml penicillin, 100 mg/ml streptomycin (Invitrogen). Four hours post-transfection, one plate is harvested for luciferase measurement; this plate may provide a measure of the amount of input RNA that can be translated, and thus of transfection efficiency. To the remaining plates, test compound serial dilutions in DMSO can be added (0.5% DMSO final concentration), and plates are incubated for 4 days.
- Exemplary compounds of the present invention were tested for their anti-HCV activities. Many of the compounds tested showed unexpected anti-HCV activities, including excellent activities in biochemical assays against HCV proteases representing various HCV genotypes, superior activities in standard HCV replicon assays including activity against 1a-H77 and 1b-con1 HCV strains in the absence or presence of 40% human plasma, and/or excellent activities in transient replicon assays against drug-resistant mutants in a number of different HCV genetic backgrounds.
- The contents of all references (including literature references, issued patents, published patent applications, and co-pending patent applications) cited throughout this application are hereby expressly incorporated herein in their entireties by reference. Unless otherwise defined, all technical and scientific terms used herein are accorded the meaning commonly known to one with ordinary skill in the art.
- Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents of the specific embodiments of the invention described herein. Such equivalents are intended with be encompassed by the following claims.
Claims (14)
1. A compound of formula I or formula I′;
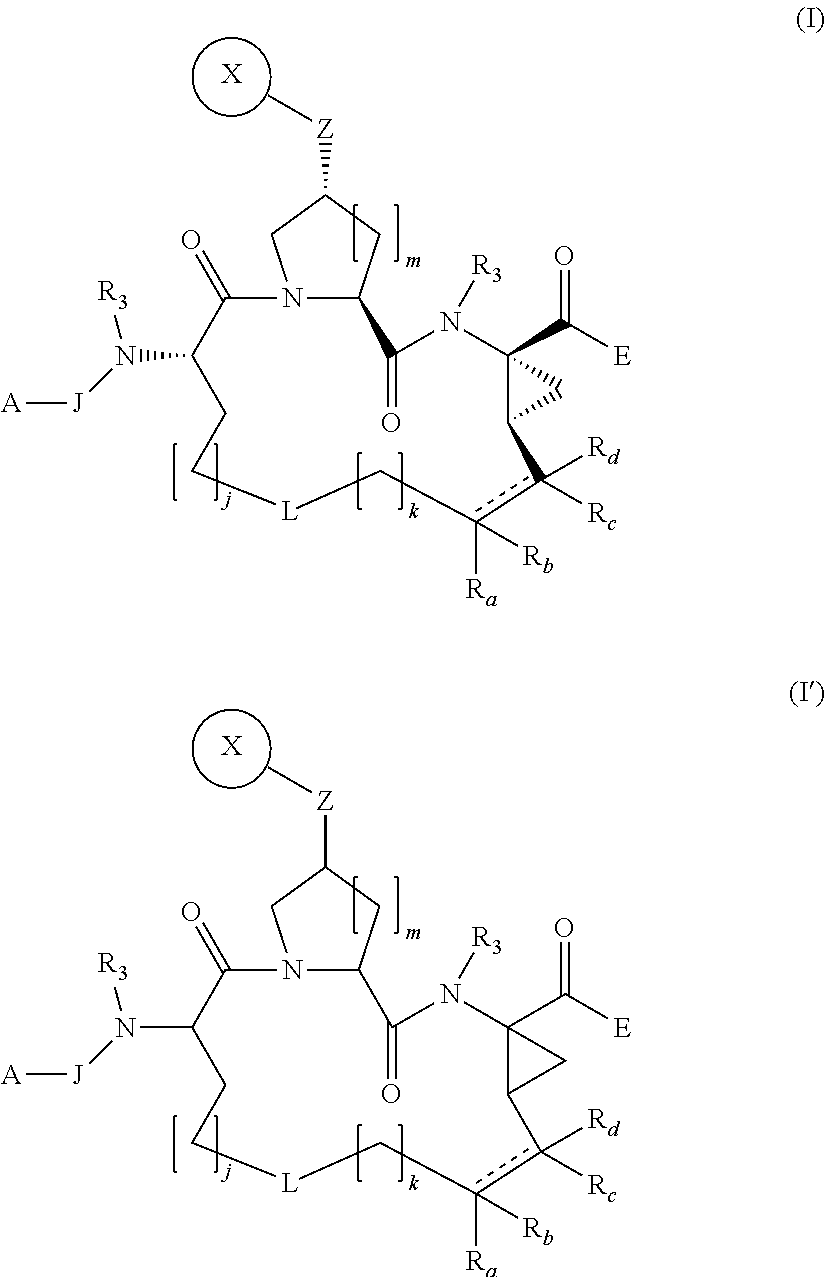
or pharmaceutically acceptable salts, esters, hydrates, solvates, polymorphs, or prodrugs thereof,
wherein:
denotes a carbon-carbon single or double bond (i.e.,
X is an optionally substituted carbocyclic, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl;
Z is O, S(O)m, NRx, OC(O), C(O), C(O)O, NRxC(O), or C(O)NRx;
each R, is independently H or alkyl;
J is —C(O)—, —O—C(O)—, —C(O)O—, —N(R3)—C(O)—, —C(S)—, —C(═NR4)—, —N(R3)—, —S(O)—, or —S(O2)— or absent;
A is selected from the group consisting of the following:
(i) H, aryl; substituted aryl; heteroaryl; substituted heteroaryl;
(ii) heterocyclyl or substituted heterocyclyl; and
(iii) —C1-C8 alkyl, C2-C8 alkenyl or —C2-C8 alkynyl, each containing 0, 1, 2, or 3 heteroatoms independently selected from O, S, and N; substituted —C1-C8 alkyl, substituted —C2-C8 alkenyl or substituted —C2-C8 alkynyl, each containing 0, 1, 2, or 3 heteroatoms independently selected from O, S and N; —C3-C12 carbocyclic, substituted —C3-C12 carbocyclic; —C3-C12 cycloalkenyl, or substituted —C3-C12 cycloalkenyl;
E is -G-R5;
wherein G is absent; optionally substituted alkylene, optionally substituted alkenylene, optionally substituted alkynylene, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N;
or —O—, —S—, —N(R3)—, —N(R3)S(Op), —N(R3)C(O), —N(R3)C(O)S(Op)—, —OS(Op)—, —C(O)S(Op)—, or —C(O)N(R3)S(Op)—;
each p is independently 0, 1, or 2;
R5 is H; optionally substituted alkyl, optionally substituted alkenyl, or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl;
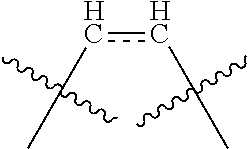
means
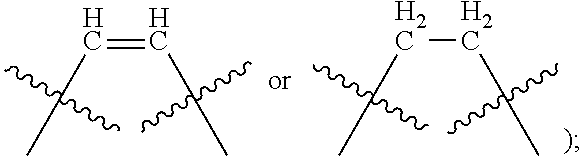
each of Ra, Rb, Rc, and Rd are each independently H or halo, wherein at least two of Ra, Rb, Rc, and Rd are halo; and Rb and Rd are absent if is a double bond
each R3 and R4 is independently selected at each occurrence from the following: H, optionally substituted alkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; and optionally substituted carbocyclic;
L is absent or a C2-C5 saturated or unsaturated chain, optionally containing one to three heteroatoms independently selected from O, N and S(O)q wherein L is optionally substituted with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl and C2-C6alkynyl, wherein each C2-C6alkyl, C2-C6alkenyl and C2-C6alkynyl, group is optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, formyl and cyano;
j=independently 0, 1, 2, 3, or 4;
k=independently 0, 1, 2, or 3;
each m is independently 0, 1, or 2; and
q is independently 0, 1, or 2.
2. The compound of claim 1 , wherein Z is O or OC(O); and ring X is cyciopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, adamantyl, cyclooctyl, [3.3.0]bicyclooctanyl, [4.3.0]bicyclononanyl, [4.4.0]bicyclodecanyl, [2.2.2]bicyclooctanyl, fluorenyl, phenyl, naphtliyi, indanyl, tetrahydronaphthyl, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, NH carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H,6H 1,5,2 dithiazinyl, dihydrofuro[2,3 b]tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, 1H indazolyl, indolenyl, indolinyl indozinyl, indolyl, 3H indolyl, isoindolinyl, isoindolenyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isoxazolyl, morpholinyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, 1,2,3 oxadiazolyl, 1,2,4 oxadiazolyl; 1,2,5-oxadiazolyl, 1,3,4 oxadiazolyl, oxazolidinyl, oxazolyl, oxazolidinyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazdlyl, pyridazinyl, pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyridyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, 2H pyrrolyl, pyrrolyl, quinazolinyl, quinolinyl, 4H quinolizinyl, quinoxalinyl, quinuclidinyl, tetrahydrofuranyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, 6H 1,2,5 thiadiazinyl, 1,2,3-thiadiazolyl, 1,2,4 thiadiazolyl, 1,2,5 thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl, 1,2,3 triazolyl, 1,2,4 triazolyl, 1,2,5 triazolyl, 1,3,4 triazolyl, or xanthenyl; each of which may be optionally substituted.
3. The compound of claim 2 , wherein ring X is phenanthridinyl, quinoxalinyl, quinolinyl, or isoindolinyl.
4. The compound of claim 2 , wherein ring X is
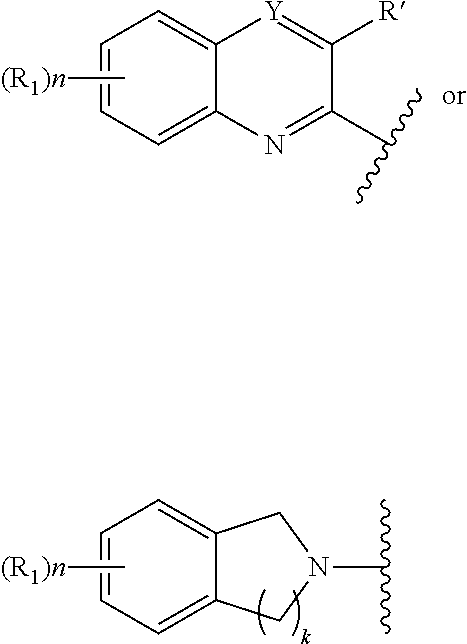
wherein
Y is N or —C(R″)—;
wherein if Y is —C(R″)—, then R′ and R″ taken together with the carbon atoms to which they are attached form a ring, which is optionally substituted;
R′ is H, alkyl, alkenyl, alkynyl, carbocyclic, heterocyclyl, aryl, or heteroaryl, each of which may be optionally substituted;
each R1 is independently selected from halogen, hydroxy, amino, —CN, —CF3, —N3, —NO2, —OR4, —SR4, —SOR4, —SO2R4, N(R3)S(O)2R4, —N(R3)(SO2)NR3R4, —NR3R4, —C(O)—OR4, —C(O)R4, —C(O)NR3R4, —N(R3)C(O)R4; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl, or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N;
each n is independently 0, 1, 2, 3, or 4;
each R3 and R4 are independently selected at each occurrence from the following: optionally substituted alkyl, optionally substituted haloalkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl; optionally substituted heteroaryl; optionally substituted heterocyclic; optionally substituted carbocyclic; or hydrogen; and
k is 0, 1, or 2.
5. (canceled)
6. The compound of claim 1 wherein J is —C(O)—, or —O—C(O)—.
7. The compound of claim 1 , wherein A is methyl, ethyl, propyl, iso-propyl, butyl, i-butyl, t-butyl, pentyl, hexyl, heptyl, phenyl, pyridyl, pyrimidyl, pyrazinyl, triazinyl, furanyl, thiophenyl, pyrrolyl, isoxazolyl, or pyrazolyl; each of which may be optionally substituted.
8. The compound of claim 1 , wherein G is —N(R3)S(O)p— and R5 is cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl, each of which is optionally substituted.
9. The compound of claim 5 , wherein Ra and Rb are F.
10. The compound of claim 5 , wherein Rc and Rd are F.
11-17. (canceled)
18. A pharmaceutical composition comprising a therapeutically effective amount of a compound of claim 1 or a pharmaceutically acceptable salt, ester, or prodrug thereof, in combination with a pharmaceutically acceptable carrier or excipient.
19. A method of treating a viral infection in a subject, comprising administering to the subject a therapeutically effective amount of a compound of claim 1 or a pharmaceutically acceptable salt, ester or prodrug thereof.
20. A method of producing a compound of formula I, comprising the step of reacting a compound of formula I-a:
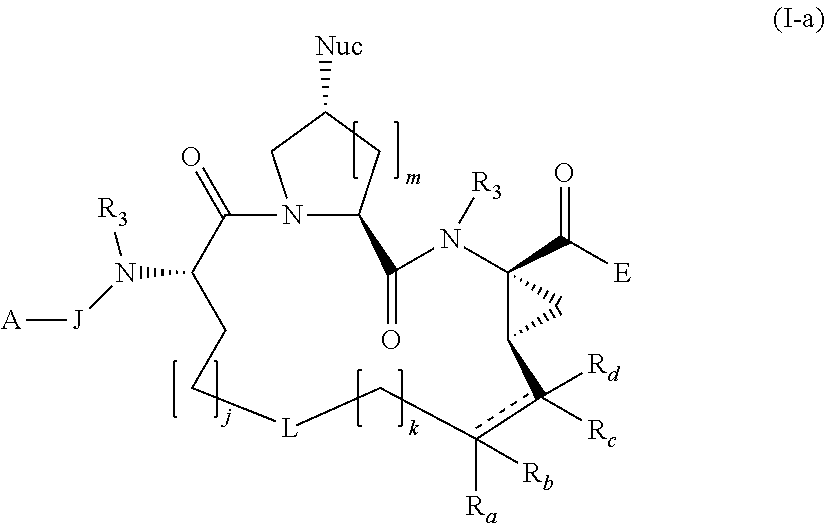
with a compound of formula I-b:
to arrive at a compound of formula I:
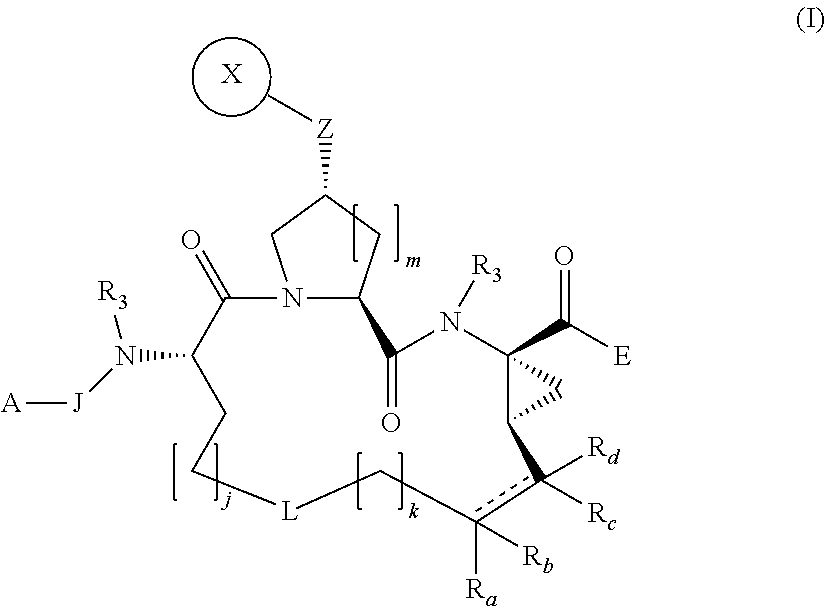
wherein:
denotes a carbon-carbon single or double bond (i.e.,
X is an optionally substituted carbocyclic, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl;
Z is O, S(O)m, NRx, OC(O), C(O), C(O)O, NRxC(O), or C(O)NRx;
each Rx is independently H or alkyl;
Nuc is a nucleophile;
LG is a leaving group;
J is —C(O)—, —O—C(O)—, —C(O)O—, —N(R3)—C(O)—, —C(S)—, —C(═NR4)—, —N(R3)—, —S(O)—, or —S(O2)- or absent;
A is selected from the group consisting of the following:
(i) H, aryl; substituted aryl; heteroaryl; substituted heteroaryl;
(ii) heterocyclyl or substituted beterocyclyl; and
—C1-C8 alkyl, —C2-C8 alkenyl or —C2-C8 alkynyl, each containing 0, 1, 2, or 3 heteroatoms independently selected from O, S, and N; substituted —C1-C8 alkyl, substituted —C2-C8 alkenyl or substituted —C2-C8 alkynyl, each containing 0, 1, 2, or 3 heteroatoms independently selected from O, S and N; —C3-C12 carbocyclic, substituted —C3-C12 carbocyclic; —C3-C12 cycloalkenyl, or substituted —C3-C12 cycloalkenyl;
E is -G-R5;
wherein G is absent; optionally substituted alkylene, optionally substituted alkenylene, optionally substituted alkynylene, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N;
or —O—, —S—, —N(R3)S(Op)—, —N(R3)C(O)—, —N(R3) C(O)S(Op)—, —OS(Op)—, —C(O)S(O)—, or —C(O)N(R3)S(Op)—;
each p is independently 0, 1, or 2;
R5 is H; optionally substituted alkyl, optionally substituted alkenyl, or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted carbocyclic, optionally substituted heterocyclic, optionally substituted aryl, or optionally substituted heteroaryl;
means or);

each of Ra, Rb, Rc, and Rd are each independently H or halo, wherein at least two of Ra, Rb, Rc, and Rd are halo; and Rb and Rd are absent if is a double bond
each R3 and R4 is independently selected at each occurrence from the following; H, optionally substituted alkyl, optionally substituted alkenyl or optionally substituted alkynyl, each containing 0, 1, 2, or 3 heteroatoms selected from O, S, or N; optionally substituted aryl;
optionally substituted heteroaryl; optionally substituted heterocyclic; and optionally substituted carbocyclic;
L is absent or a C2-C5 saturated or unsaturated chain, optionally containing one to three heteroatoms independently selected from O, N and S(O)q, wherein L is optionally substituted with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, cyano, C1-C6alkyl, C2-C6alkenyl and C2-C6alkynyl, wherein each C2-C6alkenyl and C2-C6alkynyl, group is optionally substituted at each occurrence with one or more substituents selected from halogen, hydroxy, mercapto, amino, carboxy, nitro, oxo, phosphonoxy, phosphono, thioxo, formyl and cyano;
j=independently 0, 1, 2, 3, or 4;
k=independently 0, 1, 2, or 3;
each m is independently 0, 1, or 2; and
q is independently 0, 1, or 2.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/588,805 US20150119319A1 (en) | 2010-12-30 | 2015-01-02 | Macrocyclic hepatitis c serine protease inhibitors |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201061428446P | 2010-12-30 | 2010-12-30 | |
| US13/339,440 US8937041B2 (en) | 2010-12-30 | 2011-12-29 | Macrocyclic hepatitis C serine protease inhibitors |
| US14/588,805 US20150119319A1 (en) | 2010-12-30 | 2015-01-02 | Macrocyclic hepatitis c serine protease inhibitors |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/339,440 Continuation US8937041B2 (en) | 2010-12-30 | 2011-12-29 | Macrocyclic hepatitis C serine protease inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20150119319A1 true US20150119319A1 (en) | 2015-04-30 |
Family
ID=46381288
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/339,440 Active 2032-03-21 US8937041B2 (en) | 2010-12-30 | 2011-12-29 | Macrocyclic hepatitis C serine protease inhibitors |
| US14/588,805 Abandoned US20150119319A1 (en) | 2010-12-30 | 2015-01-02 | Macrocyclic hepatitis c serine protease inhibitors |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/339,440 Active 2032-03-21 US8937041B2 (en) | 2010-12-30 | 2011-12-29 | Macrocyclic hepatitis C serine protease inhibitors |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US8937041B2 (en) |
| EP (1) | EP2658859A4 (en) |
| JP (1) | JP2014502620A (en) |
| CN (1) | CN103534256B (en) |
| CA (1) | CA2822556A1 (en) |
| MX (1) | MX2013007677A (en) |
| WO (1) | WO2012092409A2 (en) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201211046A (en) | 2010-06-07 | 2012-03-16 | Enanta Pharm Inc | Macrocyclic hepatitis C serine protease inhibitors |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| UA119315C2 (en) | 2012-07-03 | 2019-06-10 | Гіліад Фармассет Елелсі | HEPATITIS VIRUS INHIBITORS C |
| WO2014011840A1 (en) * | 2012-07-12 | 2014-01-16 | Abbvie Inc. | Crystalline forms of an hcv inhibitor |
| SG11201502802PA (en) | 2012-10-19 | 2015-05-28 | Bristol Myers Squibb Co | Hepatitis c virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| EP2914598B1 (en) | 2012-11-02 | 2017-10-18 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| EP2914614B1 (en) | 2012-11-05 | 2017-08-16 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| CN105164148A (en) | 2013-03-07 | 2015-12-16 | 百时美施贵宝公司 | Hepatitis c virus inhibitors |
| AU2014233390B2 (en) | 2013-03-15 | 2018-03-01 | Gilead Sciences, Inc. | Macrocyclic and bicyclic inhibitors of hepatitis C virus |
| US10118890B2 (en) | 2014-10-10 | 2018-11-06 | The Research Foundation For The State University Of New York | Trifluoromethoxylation of arenes via intramolecular trifluoromethoxy group migration |
| US11040947B2 (en) | 2017-03-22 | 2021-06-22 | The Research Foundation For The State University Of New York | Substituted quinazolines as matrix metalloproteinase-9 hemopexin domain inhibitors |
| CN108467392B (en) * | 2018-03-30 | 2022-01-21 | 广西师范大学 | 2- (omega-dialkyl amino) amine alkyl-3-aryl azole quinoxaline compound and preparation method and application thereof |
| US12083099B2 (en) | 2020-10-28 | 2024-09-10 | Accencio LLC | Methods of treating symptoms of coronavirus infection with viral protease inhibitors |
Family Cites Families (250)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5831002A (en) | 1992-05-20 | 1998-11-03 | Basf Aktiengesellschaft | Antitumor peptides |
| IL134232A0 (en) | 1997-08-11 | 2001-04-30 | Boehringer Ingelheim Ca Ltd | Hepatitis c inhibitor peptides |
| US6767991B1 (en) | 1997-08-11 | 2004-07-27 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis C inhibitor peptides |
| US20040058982A1 (en) | 1999-02-17 | 2004-03-25 | Bioavailability System, Llc | Pharmaceutical compositions |
| AR022061A1 (en) | 1998-08-10 | 2002-09-04 | Boehringer Ingelheim Ca Ltd | INHIBITING PEPTIDES OF HEPATITIS C, A PHARMACEUTICAL COMPOSITION CONTAINING THEM, THE USE OF THE SAME TO PREPARE A PHARMACEUTICAL COMPOSITION, THE USE OF AN INTERMEDIATE PRODUCT FOR THE PREPARATION OF THESE PEPTIDES AND A PROCEDURE FOR THE PREPARATION OF ANOGRAPH . |
| US6323180B1 (en) | 1998-08-10 | 2001-11-27 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| US6608027B1 (en) | 1999-04-06 | 2003-08-19 | Boehringer Ingelheim (Canada) Ltd | Macrocyclic peptides active against the hepatitis C virus |
| UA74546C2 (en) | 1999-04-06 | 2006-01-16 | Boehringer Ingelheim Ca Ltd | Macrocyclic peptides having activity relative to hepatitis c virus, a pharmaceutical composition and use of the pharmaceutical composition |
| US7122627B2 (en) | 1999-07-26 | 2006-10-17 | Bristol-Myers Squibb Company | Lactam inhibitors of Hepatitis C virus NS3 protease |
| EP1252178A1 (en) | 1999-12-03 | 2002-10-30 | Bristol-Myers Squibb Pharma Company | Alpha-ketoamide inhibitors of hepatitis c virus ns3 protease |
| JP2003525294A (en) | 2000-02-29 | 2003-08-26 | ブリストル−マイヤーズ スクイブ ファーマ カンパニー | Hepatitis C virus NS3 protease inhibitor |
| US6846806B2 (en) | 2000-10-23 | 2005-01-25 | Bristol-Myers Squibb Company | Peptide inhibitors of Hepatitis C virus NS3 protein |
| CA2429359A1 (en) | 2000-11-20 | 2002-08-08 | Bristol-Myers Squibb Company | Hepatitis c tripeptide inhibitors |
| US6653295B2 (en) | 2000-12-13 | 2003-11-25 | Bristol-Myers Squibb Company | Inhibitors of hepatitis C virus NS3 protease |
| AU2002230764A1 (en) | 2000-12-13 | 2002-06-24 | Bristol-Myers Squibb Pharma Company | Imidazolidinones and their related derivatives as hepatitis c virus ns3 protease inhibitors |
| FR2821272B1 (en) | 2001-02-23 | 2004-12-17 | Synt Em | COMPOUNDS CONSISTING OF AN ANALGESIC MOLECULE LINKED TO A VECTOR CAPABLE OF VECTORIZING THE SAME MOLECULATED THROUGH THE HEMATOENCEPHALIC BARRIER AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| WO2003025563A1 (en) | 2001-09-16 | 2003-03-27 | Chemometec A/S | Method and a system for detecting and optionally isolating a rare event particle |
| EP1429759A4 (en) | 2001-09-26 | 2004-12-15 | Bristol Myers Squibb Co | Compounds useful for treating hepatitus c virus |
| US6867185B2 (en) | 2001-12-20 | 2005-03-15 | Bristol-Myers Squibb Company | Inhibitors of hepatitis C virus |
| US7119072B2 (en) | 2002-01-30 | 2006-10-10 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis C virus |
| CA2369711A1 (en) | 2002-01-30 | 2003-07-30 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis c virus |
| US7091184B2 (en) | 2002-02-01 | 2006-08-15 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor tri-peptides |
| US6642204B2 (en) | 2002-02-01 | 2003-11-04 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor tri-peptides |
| CA2369970A1 (en) | 2002-02-01 | 2003-08-01 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor tri-peptides |
| CA2370396A1 (en) | 2002-02-01 | 2003-08-01 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor tri-peptides |
| US6828301B2 (en) | 2002-02-07 | 2004-12-07 | Boehringer Ingelheim International Gmbh | Pharmaceutical compositions for hepatitis C viral protease inhibitors |
| MY140680A (en) | 2002-05-20 | 2010-01-15 | Bristol Myers Squibb Co | Hepatitis c virus inhibitors |
| DE60334205D1 (en) | 2002-05-20 | 2010-10-28 | Bristol Myers Squibb Co | Heterocyclische sulfonamid-hepatitis-c-virus-hemmer |
| PL213029B1 (en) | 2002-05-20 | 2012-12-31 | Bristol Myers Squibb Co | Substituted cycloalkyl p1' hepatitis c virus inhibitors |
| ATE503764T1 (en) | 2002-05-20 | 2011-04-15 | Bristol Myers Squibb Co | HEPATITIS C VIRUS INHIBITORS |
| US20060199773A1 (en) | 2002-05-20 | 2006-09-07 | Sausker Justin B | Crystalline forms of (1R,2S)-N-[(1,1-dimethylethoxy)carbonyl]-3-methyl-L-valyl-(4R)-4-[(6-methoxy-1-isoquinolinyl)oxy]-L-prolyl-1-amino-N-(cyclopropylsulfonyl)-2-ethenyl-cyclopropanecarboxamide, monopotassium salt |
| US20030232386A1 (en) | 2002-06-17 | 2003-12-18 | Shah Dinesh O. | Assay conjugate and uses thereof |
| EP1523483A4 (en) | 2002-06-26 | 2006-03-08 | Bristol Myers Squibb Co | Amino-bicyclic pyrazinones and pyridinones as coagulation serine protease inhibitors |
| AU2003264038A1 (en) | 2002-08-12 | 2004-02-25 | Bristol-Myers Squibb Company | Combination pharmaceutical agents as inhibitors of hcv replication |
| US20040138109A1 (en) | 2002-09-30 | 2004-07-15 | Boehringer Ingelheim Pharmaceuticals, Inc. | Potent inhibitor of HCV serine protease |
| US20050075279A1 (en) | 2002-10-25 | 2005-04-07 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US20050159345A1 (en) | 2002-10-29 | 2005-07-21 | Boehringer Ingelheim International Gmbh | Composition for the treatment of infection by Flaviviridae viruses |
| US7601709B2 (en) * | 2003-02-07 | 2009-10-13 | Enanta Pharmaceuticals, Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| US20040180815A1 (en) | 2003-03-07 | 2004-09-16 | Suanne Nakajima | Pyridazinonyl macrocyclic hepatitis C serine protease inhibitors |
| AU2004211637C1 (en) | 2003-02-07 | 2010-08-19 | Enanta Pharmaceuticals, Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| CA2516016C (en) | 2003-03-05 | 2012-05-29 | Boehringer Ingelheim International Gmbh | Hepatitis c inhibiting compounds |
| UY28240A1 (en) | 2003-03-27 | 2004-11-08 | Boehringer Ingelheim Pharma | CRYSTAL PHASES OF A POWERFUL HCV INHIBITOR |
| CN100363055C (en) | 2003-04-02 | 2008-01-23 | 贝林格尔·英格海姆国际有限公司 | Pharmaceutical compositions for use as hepatitis C virus protease inhibitors |
| WO2004092203A2 (en) | 2003-04-10 | 2004-10-28 | Boehringer Ingelheim International, Gmbh | Process for preparing macrocyclic compounds |
| EP1615949A1 (en) | 2003-04-10 | 2006-01-18 | Boehringer Ingelheim International GmbH | Process for the preparation of macrocyclic compounds by ruthenium complex catalysed metathesis reaction |
| JP4733023B2 (en) | 2003-04-16 | 2011-07-27 | ブリストル−マイヤーズ スクイブ カンパニー | Macrocyclic isoquinoline peptide inhibitor of hepatitis C virus |
| PT1615613E (en) | 2003-04-18 | 2010-02-09 | Enanta Pharm Inc | Quinoxalinyl macrocyclic hepatitis c serine protease inhibitors |
| US7176208B2 (en) | 2003-04-18 | 2007-02-13 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl macrocyclic hepatitis C serine protease inhibitors |
| ES2297424T3 (en) | 2003-05-21 | 2008-05-01 | Boehringer Ingelheim International Gmbh | INHIBITING COMPOUNDS OF HEPATITIS C. |
| US7273851B2 (en) | 2003-06-05 | 2007-09-25 | Enanta Pharmaceuticals, Inc. | Tri-peptide hepatitis C serine protease inhibitors |
| US7125845B2 (en) * | 2003-07-03 | 2006-10-24 | Enanta Pharmaceuticals, Inc. | Aza-peptide macrocyclic hepatitis C serine protease inhibitors |
| US7112601B2 (en) | 2003-09-11 | 2006-09-26 | Bristol-Myers Squibb Company | Cycloalkyl heterocycles for treating hepatitis C virus |
| DE602004031645D1 (en) | 2003-09-22 | 2011-04-14 | Boehringer Ingelheim Pharma | MACROCYCLIC PEPTIDES WITH EFFECT AGAINST HEPATITIS C VIRUS |
| DE602004031298D1 (en) | 2003-09-26 | 2011-03-17 | Schering Corp | MACROCYCLIC INHIBITORS OF THE NS3 SERINE PROTEASE OF HEPATITIS C VIRUS |
| RS54573B1 (en) | 2003-10-14 | 2016-06-30 | F. Hoffmann-La Roche Ltd | MACROCYCLIC CARBOXYLIC ACIDS AND ACYLSULPHONAMIDES AS HCV REPLICATION INHIBITORS |
| US7491794B2 (en) | 2003-10-14 | 2009-02-17 | Intermune, Inc. | Macrocyclic compounds as inhibitors of viral replication |
| US7132504B2 (en) | 2003-11-12 | 2006-11-07 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7309708B2 (en) | 2003-11-20 | 2007-12-18 | Birstol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7135462B2 (en) | 2003-11-20 | 2006-11-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2005051980A1 (en) | 2003-11-20 | 2005-06-09 | Schering Corporation | Depeptidized inhibitors of hepatitis c virus ns3 protease |
| FR2863268B1 (en) | 2003-12-09 | 2006-02-24 | Sod Conseils Rech Applic | NOVEL DERIVATIVES OF 2-HYDROXYTETRAHYDROFURAN AND THEIR APPLICATION AS MEDICAMENTS |
| RU2006124594A (en) | 2003-12-11 | 2008-01-27 | Шеринг Корпорейшн (US) | NSA / NS4A SERINE PROTEASE INHIBITORS HEPATITIS C VIRUS |
| EP1730167B1 (en) | 2004-01-21 | 2011-01-12 | Boehringer Ingelheim International GmbH | Macrocyclic peptides active against the hepatitis c virus |
| CA2544886C (en) | 2004-01-28 | 2012-12-04 | Boehringer Ingelheim International Gmbh | Method of removing transition metals from reaction solutions comprising transition metal byproducts |
| CA2552319C (en) | 2004-01-30 | 2012-08-21 | Medivir Ab | Hcv ns-3 serine protease inhibitors |
| ATE459638T1 (en) | 2004-03-15 | 2010-03-15 | Boehringer Ingelheim Int | METHOD FOR PRODUCING MACROCYCLIC DIPEPTIDES SUITABLE FOR THE TREATMENT OF HEPATITIS C VIRUS INFECTIONS |
| BRPI0509467A (en) | 2004-03-30 | 2007-09-11 | Intermune Inc | macrocyclic compounds as viral replication inhibitors |
| US7514557B2 (en) | 2004-05-25 | 2009-04-07 | Boehringer Ingelheim International Gmbh | Process for preparing acyclic HCV protease inhibitors |
| JP4914348B2 (en) | 2004-06-28 | 2012-04-11 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Hepatitis C inhibitor peptide analog |
| DE102004033312A1 (en) | 2004-07-08 | 2006-01-26 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Continuous metathesis process with ruthenium catalysts |
| AP2461A (en) | 2004-07-16 | 2012-09-14 | Gilead Sciences Inc | Antiviral compounds |
| UY29016A1 (en) | 2004-07-20 | 2006-02-24 | Boehringer Ingelheim Int | ANALOGS OF INHIBITING DIPEPTIDES OF HEPATITIS C |
| JP4914355B2 (en) | 2004-07-20 | 2012-04-11 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Hepatitis C inhibitor peptide analog |
| US7153848B2 (en) | 2004-08-09 | 2006-12-26 | Bristol-Myers Squibb Company | Inhibitors of HCV replication |
| US7348425B2 (en) | 2004-08-09 | 2008-03-25 | Bristol-Myers Squibb Company | Inhibitors of HCV replication |
| EP1794179A1 (en) | 2004-09-17 | 2007-06-13 | Boehringer Ingelheim International Gmbh | Ring-closing metathesis process in supercritical fluid |
| CA2577831A1 (en) | 2004-09-17 | 2006-03-30 | Boehringer Ingelheim International Gmbh | Process for preparing macrocyclic hcv protease inhibitors |
| EP1799194B1 (en) | 2004-09-24 | 2011-11-16 | Boehringer Ingelheim Pharmaceuticals Inc. | A new class of surfactant-like materials comprising vitamin e tpgs and a water soluble polymer |
| WO2007001406A2 (en) | 2004-10-05 | 2007-01-04 | Chiron Corporation | Aryl-containing macrocyclic compounds |
| EP1824816A4 (en) | 2004-10-14 | 2008-01-02 | Wuxi Pharma Tech Co Ltd | A NOVEL PROCESS FOR THE PREPARATION OF NONRACEMIC LONG CHAIN alpha-AMINO ACIDS DERIVATIVES |
| MX2007004783A (en) | 2004-10-21 | 2007-05-11 | Pfizer | Inhibitors of hepatitis c virus protease, and compositions and treatments using the same. |
| US7323447B2 (en) | 2005-02-08 | 2008-01-29 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| DK1863833T3 (en) | 2005-03-08 | 2013-12-02 | Boehringer Ingelheim Int | PROCEDURE FOR MAKING MACROCYCLIC COMPOUNDS |
| WO2006114405A2 (en) | 2005-04-25 | 2006-11-02 | Basf Aktiengesellschaft | Use of 5-alkyl-6-phenylalkyl-7-amino-azolopyrimidines, novel azolopyrimidines, methods for the production thereof, and agents containing the same |
| EP1879607B1 (en) | 2005-05-02 | 2014-11-12 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| US7592336B2 (en) | 2005-05-10 | 2009-09-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| AU2006254538A1 (en) | 2005-05-25 | 2006-12-07 | 2Curex Aps | Compounds modifying apoptosis |
| US20060275366A1 (en) | 2005-06-02 | 2006-12-07 | Schering Corporation | Controlled-release formulation |
| AU2006252519B2 (en) | 2005-06-02 | 2012-08-30 | Merck Sharp & Dohme Corp. | HCV protease inhibitors in combination with food |
| WO2006130627A2 (en) | 2005-06-02 | 2006-12-07 | Schering Corporation | Methods for treating hepatitis c |
| WO2006130554A2 (en) | 2005-06-02 | 2006-12-07 | Schering Corporation | Methods of treating hepatitis c virus |
| US20070004635A1 (en) | 2005-06-02 | 2007-01-04 | Schering Corporation | Method of treating interferon non-responders using HCV protease inhibitor |
| AU2006252623A1 (en) | 2005-06-02 | 2006-12-07 | Schering Corporation | Controlled-release formulation useful for treating disorders associated with hepatitis C virus |
| US20060287248A1 (en) | 2005-06-02 | 2006-12-21 | Schering Corporation | Asymmetric dosing methods |
| US20070021351A1 (en) | 2005-06-02 | 2007-01-25 | Schering Corporation | Liver/plasma concentration ratio for dosing hepatitis C virus protease inhibitor |
| WO2006130626A2 (en) | 2005-06-02 | 2006-12-07 | Schering Corporation | Method for modulating activity of hcv protease through use of a novel hcv protease inhibitor to reduce duration of treatment period |
| US20060276406A1 (en) | 2005-06-02 | 2006-12-07 | Schering Corporation | Methods of treating hepatitis C virus |
| ES2572980T3 (en) | 2005-06-02 | 2016-06-03 | Merck Sharp & Dohme Corp. | Combination of HCV protease inhibitors with a surfactant |
| US20070237818A1 (en) | 2005-06-02 | 2007-10-11 | Malcolm Bruce A | Controlled-release formulation of HCV protease inhibitor and methods using the same |
| US20060276404A1 (en) | 2005-06-02 | 2006-12-07 | Anima Ghosal | Medicaments and methods combining a HCV protease inhibitor and an AKR competitor |
| US7608592B2 (en) | 2005-06-30 | 2009-10-27 | Virobay, Inc. | HCV inhibitors |
| US7601686B2 (en) | 2005-07-11 | 2009-10-13 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| TWI389908B (en) | 2005-07-14 | 2013-03-21 | Gilead Sciences Inc | Antiviral compounds |
| TW200738742A (en) | 2005-07-14 | 2007-10-16 | Gilead Sciences Inc | Antiviral compounds |
| WO2007009227A1 (en) | 2005-07-20 | 2007-01-25 | Boehringer Ingelheim International Gmbh | Hepatitis c inhibitor peptide analogs |
| GEP20105124B (en) | 2005-07-25 | 2010-11-25 | Array Biopharma Inc | Novel macrocyclic inhibitors of hepatitis c virus replication |
| DK1919899T3 (en) | 2005-07-29 | 2011-08-01 | Tibotec Pharm Ltd | Macrocyclic inhibitors of hepatitis C virus |
| PE20070343A1 (en) | 2005-07-29 | 2007-05-12 | Medivir Ab | MACRO CYCLIC INHIBITORS OF HEPATITIS C VIRUS |
| ME01318B (en) | 2005-07-29 | 2013-12-20 | Tibotec Pharm Ltd | Macrocylic inhibitors of hepatitis c virus |
| RU2437886C2 (en) | 2005-07-29 | 2011-12-27 | Тиботек Фармасьютикалз Лтд. | Macrocyclic hepatitis c virus inihbitors |
| DK1913015T3 (en) | 2005-07-29 | 2014-03-10 | Janssen R & D Ireland | Macrocyclic inhibitors of hepatitis C virus |
| PE20070211A1 (en) | 2005-07-29 | 2007-05-12 | Medivir Ab | MACROCYCLIC COMPOUNDS AS INHIBITORS OF HEPATITIS C VIRUS |
| JO2768B1 (en) | 2005-07-29 | 2014-03-15 | تيبوتيك فارماسيوتيكالز ليمتد | Macrocylic Inhibitors Hepatitis C Virus |
| WO2007016441A1 (en) | 2005-08-01 | 2007-02-08 | Merck & Co., Inc. | Macrocyclic peptides as hcv ns3 protease inhibitors |
| AR055395A1 (en) | 2005-08-26 | 2007-08-22 | Vertex Pharma | INHIBITING COMPOUNDS OF THE ACTIVITY OF SERINA PROTEASA NS3-NS4A OF HEPATITIS C VIRUS |
| PL1934243T3 (en) | 2005-09-09 | 2011-10-31 | Boehringer Ingelheim Int | Ring-closing metathesis process for the preparation of macrocyclic peptides |
| US7399758B2 (en) | 2005-09-12 | 2008-07-15 | Meanwell Nicholas A | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US7473688B2 (en) | 2005-09-13 | 2009-01-06 | Bristol-Myers Squibb Company | Indolobenzazepine HCV NS5B inhibitors |
| US7723336B2 (en) | 2005-09-22 | 2010-05-25 | Bristol-Myers Squibb Company | Fused heterocyclic compounds useful as kinase modulators |
| CN101415705B (en) | 2005-10-11 | 2011-10-26 | 因特蒙公司 | Compounds and methods for inhibiting hepatitis c viral replication |
| US7772183B2 (en) | 2005-10-12 | 2010-08-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7741281B2 (en) | 2005-11-03 | 2010-06-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7456165B2 (en) | 2006-02-08 | 2008-11-25 | Bristol-Myers Squibb Company | HCV NS5B inhibitors |
| CA2647158C (en) | 2006-03-23 | 2012-07-31 | Schering Corporation | Combinations of hcv protease inhibitor(s) and cyp3a4 inhibitor(s), and methods of treatment related thereto |
| WO2007130592A2 (en) | 2006-05-04 | 2007-11-15 | Crawford Ted C | Head cap for a wrench head, wrench and method utilizing the same |
| GB0609492D0 (en) | 2006-05-15 | 2006-06-21 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| US7521443B2 (en) | 2006-05-17 | 2009-04-21 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US7456166B2 (en) | 2006-05-17 | 2008-11-25 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US7521441B2 (en) | 2006-05-22 | 2009-04-21 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US7521442B2 (en) | 2006-05-25 | 2009-04-21 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US20070281884A1 (en) | 2006-06-06 | 2007-12-06 | Ying Sun | Macrocyclic oximyl hepatitis C protease inhibitors |
| US9526769B2 (en) | 2006-06-06 | 2016-12-27 | Enanta Pharmaceuticals, Inc. | Macrocylic oximyl hepatitis C protease inhibitors |
| US8268776B2 (en) | 2006-06-06 | 2012-09-18 | Enanta Pharmaceuticals, Inc. | Macrocylic oximyl hepatitis C protease inhibitors |
| US7452876B2 (en) | 2006-06-08 | 2008-11-18 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| GB0612423D0 (en) | 2006-06-23 | 2006-08-02 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| PE20080992A1 (en) * | 2006-06-26 | 2008-08-06 | Enanta Pharm Inc | QUINOXALINYL MACROCYCLIC SERINE PROTEASE INHIBITORS OF HEPATITIS C VIRUS |
| US7935670B2 (en) | 2006-07-11 | 2011-05-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| EA017448B1 (en) | 2006-07-13 | 2012-12-28 | Ачиллион Фармасьютикалз, Инк. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| US20090035271A1 (en) | 2007-08-01 | 2009-02-05 | Ying Sun | Tetrazolyl macrocyclic hepatitis c serine protease inhibitors |
| US7635683B2 (en) | 2006-08-04 | 2009-12-22 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl tripeptide hepatitis C virus inhibitors |
| WO2008019303A2 (en) | 2006-08-04 | 2008-02-14 | Enanta Pharmaceuticals, Inc. | Pyridazinonyl macrocyclic hepatitis c serine protease inhibitors |
| CN101674844A (en) | 2006-08-04 | 2010-03-17 | 英安塔制药有限公司 | Tetrazolyl macrocyclic hepatitis C serine protease inhibitors |
| WO2008022006A2 (en) | 2006-08-11 | 2008-02-21 | Enanta Pharmaceuticals, Inc. | Arylalkoxyl hepatitis c virus protease inhibitors |
| US7687459B2 (en) | 2006-08-11 | 2010-03-30 | Enanta Pharmaceuticals, Inc. | Arylalkoxyl hepatitis C virus protease inhibitors |
| US7582605B2 (en) | 2006-08-11 | 2009-09-01 | Enanta Pharmaceuticals, Inc. | Phosphorus-containing hepatitis C serine protease inhibitors |
| US20080038225A1 (en) | 2006-08-11 | 2008-02-14 | Ying Sun | Triazolyl acyclic hepatitis c serine protease inhibitors |
| US7605126B2 (en) | 2006-08-11 | 2009-10-20 | Enanta Pharmaceuticals, Inc. | Acylaminoheteroaryl hepatitis C virus protease inhibitors |
| WO2008021960A2 (en) | 2006-08-11 | 2008-02-21 | Enanta Pharmaceuticals, Inc. | Triazolyl macrocyclic hepatitis c serine protease inhibitors |
| CA2664156A1 (en) | 2006-09-27 | 2008-04-03 | Coley Pharmaceutical Group, Inc. | Compositions of tlr ligands and antivirals |
| WO2008062457A2 (en) | 2006-10-03 | 2008-05-29 | Cadila Healthcare Limited | Antidiabetic compounds |
| US20120220520A1 (en) | 2006-10-17 | 2012-08-30 | Van T Klooster Gerben Albert Eleutherius | Bioavailable combinations for hcv treatment |
| WO2008051475A2 (en) | 2006-10-24 | 2008-05-02 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| CA2667266C (en) | 2006-10-24 | 2015-11-24 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| CA2667031C (en) | 2006-10-27 | 2013-01-22 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| EP2083844B1 (en) | 2006-10-27 | 2013-11-27 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| US8343477B2 (en) | 2006-11-01 | 2013-01-01 | Bristol-Myers Squibb Company | Inhibitors of hepatitis C virus |
| US20080107623A1 (en) | 2006-11-01 | 2008-05-08 | Bristol-Myers Squibb Company | Inhibitors of Hepatitis C Virus |
| US20080107625A1 (en) | 2006-11-01 | 2008-05-08 | Bristol-Myers Squibb Company | Inhibitors of Hepatitis C Virus |
| TW200827364A (en) | 2006-11-02 | 2008-07-01 | Taigen Biotechnology Co Ltd | HCV protease inhibitors |
| US7772180B2 (en) | 2006-11-09 | 2010-08-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7888464B2 (en) | 2006-11-16 | 2011-02-15 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8003604B2 (en) | 2006-11-16 | 2011-08-23 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7763584B2 (en) | 2006-11-16 | 2010-07-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| EA200970493A1 (en) | 2006-11-17 | 2009-10-30 | Тиботек Фармасьютикалз Лтд. | MACROCYCLIC INHIBITORS OF HEPATITIS C VIRUS |
| WO2008070733A2 (en) | 2006-12-06 | 2008-06-12 | Phenomix Corporation | Macrocyclic hepatitis c serine protease inhibitors and uses therefor |
| EA200900969A1 (en) | 2007-01-08 | 2010-02-26 | Феномикс Корпорейшн | MACROCYCLIC INHIBITORS OF HEPATITIS C PROTEASE |
| US7541351B2 (en) | 2007-01-11 | 2009-06-02 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| SI2118098T1 (en) | 2007-02-01 | 2015-01-30 | Janssen R&D Ireland | Polymorphic forms of a macrocyclic inhibitor of hcv |
| US20080207528A1 (en) | 2007-02-01 | 2008-08-28 | Syaulan Yang | Hcv protease inhibitors |
| MX2009008540A (en) | 2007-02-08 | 2009-08-18 | Tibotec Pharm Ltd | Pyrimidine substituted macrocyclic hcv inhibitors. |
| WO2008096001A1 (en) | 2007-02-08 | 2008-08-14 | Tibotec Pharmaceuticals Ltd. | Hcv inhibiting macrocyclic phenylcarbamates |
| CA2676297A1 (en) | 2007-02-16 | 2008-08-21 | Boehringer Ingelheim International Gmbh | Inhibitors of hepatitis c ns3 protease |
| MX2009008872A (en) | 2007-02-20 | 2009-10-30 | Novartis Ag | Macrocyclic compounds as hcv ns3 protease inhibitors. |
| US8435984B2 (en) | 2007-02-26 | 2013-05-07 | Achillion Pharmaceuticals, Inc. | Tertiary amine substituted peptides useful as inhibitors of HCV replication |
| US8063051B2 (en) | 2007-03-19 | 2011-11-22 | Astrazeneca Ab | 9-substituted-8-oxo-adenine compounds as toll-like receptor (TLR7) modulators |
| WO2008124384A2 (en) | 2007-04-03 | 2008-10-16 | Aegerion Pharmaceuticals, Inc. | Combinations of mtp inhibitors with cholesterol absorption inhibitors or interferon for treating hepatitis c |
| ES2355437T3 (en) | 2007-04-24 | 2011-03-25 | F. Hoffmann-La Roche Ag | PROCEDURE FOR AN INTERMEDIARY OF HCV PROTEASE INHIBITORS. |
| US7906513B2 (en) | 2007-04-26 | 2011-03-15 | Enanta Pharmaceuticals, Inc. | Hydrazide-containing hepatitis C serine protease inhibitors |
| US20080267917A1 (en) | 2007-04-26 | 2008-10-30 | Deqiang Niu | N-functionalized amides as hepatitis c serine protease inhibitors |
| US20080279821A1 (en) | 2007-04-26 | 2008-11-13 | Deqiang Niu | Arylpiperidinyl and arylpyrrolidinyl macrocyclic hepatitis c serine protease inhibitors |
| US8377872B2 (en) | 2007-04-26 | 2013-02-19 | Enanta Pharmaceuticals, Inc. | Cyclic P3 tripeptide hepatitis C serine protease inhibitors |
| AP2009005053A0 (en) * | 2007-05-03 | 2009-12-31 | Intermune Inc | Novel macrocyclic inhibitors of hepatitis c virus replication |
| KR20100027134A (en) | 2007-05-10 | 2010-03-10 | 인터뮨, 인크. | Novel peptide inhibitors of hepatitis c virus replication |
| US20090005387A1 (en) | 2007-06-26 | 2009-01-01 | Deqiang Niu | Quinoxalinyl macrocyclic hepatitis c virus serine protease inhibitors |
| US20090047252A1 (en) | 2007-06-29 | 2009-02-19 | Gilead Sciences, Inc. | Antiviral compounds |
| AP2874A (en) | 2007-06-29 | 2014-03-31 | Gilead Sciences Inc | Antiviral compounds |
| EA200971074A1 (en) | 2007-06-29 | 2010-08-30 | Джилид Сайэнс, Инк. | ANTI-VIRUS CONNECTIONS |
| JP5433573B2 (en) | 2007-07-19 | 2014-03-05 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・エルレ・エルレ | Macrocyclic compounds as antiviral agents |
| WO2009014730A1 (en) | 2007-07-26 | 2009-01-29 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| WO2009053828A2 (en) | 2007-10-22 | 2009-04-30 | Enanta Pharmaceuticals, Inc. | P3 hydroxyamino macrocyclic hepatitis c serine protease inhibitors |
| WO2009055335A2 (en) | 2007-10-25 | 2009-04-30 | Taigen Biotechnology Co., Ltd. | Hcv protease inhibitors |
| US8426360B2 (en) | 2007-11-13 | 2013-04-23 | Enanta Pharmaceuticals, Inc. | Carbocyclic oxime hepatitis C virus serine protease inhibitors |
| PT2209789E (en) | 2007-11-20 | 2012-01-16 | Bristol Myers Squibb Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| US8263549B2 (en) | 2007-11-29 | 2012-09-11 | Enanta Pharmaceuticals, Inc. | C5-substituted, proline-derived, macrocyclic hepatitis C serine protease inhibitors |
| US8030307B2 (en) | 2007-11-29 | 2011-10-04 | Enanta Pharmaceuticals, Inc. | Bicyclic, C5-substituted proline derivatives as inhibitors of the hepatitis C virus NS3 protease |
| JP5529036B2 (en) * | 2007-12-05 | 2014-06-25 | エナンタ ファーマシューティカルズ インコーポレイテッド | Fluorinated tripeptide HCV serine protease inhibitor |
| CN101932242A (en) | 2007-12-05 | 2010-12-29 | 益安药业 | Quinoxalinyl derivatives |
| WO2009073713A1 (en) | 2007-12-05 | 2009-06-11 | Enanta Pharmaceuticals, Inc. | Oximyl macrocyclic derivatives |
| US8193346B2 (en) | 2007-12-06 | 2012-06-05 | Enanta Pharmaceuticals, Inc. | Process for making macrocyclic oximyl hepatitis C protease inhibitors |
| RU2523790C2 (en) | 2007-12-21 | 2014-07-27 | Авила Терапьютикс, Инк. | Inhibitors of protease of hepatitis c virus and their application |
| US8309685B2 (en) | 2007-12-21 | 2012-11-13 | Celgene Avilomics Research, Inc. | HCV protease inhibitors and uses thereof |
| RU2515318C2 (en) | 2007-12-21 | 2014-05-10 | Авила Терапьютикс, Инк. | Inhibitors of protease of hepatitis c virus and their application |
| US8202996B2 (en) | 2007-12-21 | 2012-06-19 | Bristol-Myers Squibb Company | Crystalline forms of N-(tert-butoxycarbonyl)-3-methyl-L-valyl-(4R)-4-((7-chloro-4-methoxy-1-isoquinolinyl)oxy)-N- ((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)-L-prolinamide |
| BRPI0820733A2 (en) | 2007-12-21 | 2015-06-16 | Hoffmann La Roche | Process for macrocycle preparation |
| US8293705B2 (en) | 2007-12-21 | 2012-10-23 | Avila Therapeutics, Inc. | HCV protease inhibitors and uses thereof |
| MX2010008109A (en) * | 2008-01-24 | 2010-09-22 | Enanta Pharm Inc | Difluorinated tripeptides as hcv serine protease inhibitors. |
| JP5574982B2 (en) | 2008-02-04 | 2014-08-20 | イデニク プハルマセウティカルス,インコーポレイテッド | Macrocyclic serine protease inhibitor |
| US20100081713A1 (en) | 2008-03-19 | 2010-04-01 | CombinatoRx, (Singapore) Pte. Ltd. | Compositions and methods for treating viral infections |
| WO2009142842A2 (en) | 2008-04-15 | 2009-11-26 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| US8163921B2 (en) | 2008-04-16 | 2012-04-24 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| JP2011519943A (en) | 2008-05-09 | 2011-07-14 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Method for preparing macrocyclic molecules |
| US20090285773A1 (en) | 2008-05-15 | 2009-11-19 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US20090285774A1 (en) | 2008-05-15 | 2009-11-19 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| CN101580535B (en) | 2008-05-16 | 2012-10-03 | 太景生物科技股份有限公司 | Hepatitis C virus protease inhibitors |
| US7964560B2 (en) | 2008-05-29 | 2011-06-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| JP5529120B2 (en) | 2008-05-29 | 2014-06-25 | ブリストル−マイヤーズ スクイブ カンパニー | Hepatitis C virus inhibitor |
| RU2496514C2 (en) | 2008-06-05 | 2013-10-27 | Займоджинетикс, Инк. | Using type iii pegylated interferons for treating hepatitis c |
| TW201012814A (en) | 2008-07-01 | 2010-04-01 | Centocor Ortho Biotech Products L P | Cyclopropyl polymerase inhibitors |
| BRPI0916609A2 (en) | 2008-08-07 | 2015-08-04 | Hoffmann La Roche | Process for the preparation of a macrocycle |
| US8765667B2 (en) | 2008-08-20 | 2014-07-01 | Michael Eissenstat | HCV protease inhibitors |
| MY156789A (en) | 2008-09-05 | 2016-03-31 | Celgene Avilomics Res Inc | Algorithm for designing irreversible inhibitors |
| UY32099A (en) * | 2008-09-11 | 2010-04-30 | Enanta Pharm Inc | HEPATITIS C SERINA PROTEASAS MACROCYCLIC INHIBITORS |
| WO2010033466A1 (en) | 2008-09-16 | 2010-03-25 | Phenomix Corporation | Macrocyclic inhibitors of hepatitis c protease |
| PT2331538E (en) | 2008-09-16 | 2014-05-12 | Boehringer Ingelheim Int | Crystalline forms of a 2-thiazolyl- 4-quinolinyl-oxy derivative, a potent hcv inhibitor |
| KR20110054003A (en) | 2008-09-17 | 2011-05-24 | 베링거 인겔하임 인터내셔날 게엠베하 | Combination of HBC ANS 3 Protease Inhibitor with Interferon and Ribavirin |
| US8603737B2 (en) | 2008-09-19 | 2013-12-10 | Celgene Avilomics Research, Inc. | Methods for identifying HCV protease inhibitors |
| WO2010034105A1 (en) | 2008-09-23 | 2010-04-01 | Boehringer Ingelheim International Gmbh | Hepatitis c inhibitor compounds |
| US20100080770A1 (en) | 2008-09-29 | 2010-04-01 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US8563505B2 (en) | 2008-09-29 | 2013-10-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8044087B2 (en) | 2008-09-29 | 2011-10-25 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2010042834A1 (en) | 2008-10-09 | 2010-04-15 | Anadys Pharmaceuticals, Inc. | A method of inhibiting hepatitis c virus by combination of a 5,6-dihydro-1h-pyridin-2-one and one or more additional antiviral compounds |
| EP2364159A4 (en) | 2008-10-23 | 2012-06-13 | Concert Pharmaceuticals Inc | Deuterated macrocyclic inhibitors of viral ns3 protease |
| US8445430B2 (en) | 2008-11-20 | 2013-05-21 | Achillion Pharmaceuticals, Inc. | Cyclic carboxamide compounds and analogues thereof as of hepatitis C virus |
| KR20110087297A (en) | 2008-11-21 | 2011-08-02 | 베링거 인겔하임 인터내셔날 게엠베하 | Pharmaceutical Compositions of Potent HCC Inhibitors for Oral Administration |
| US20100272674A1 (en) | 2008-12-04 | 2010-10-28 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US8283310B2 (en) | 2008-12-15 | 2012-10-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| BRPI0923184A2 (en) | 2008-12-19 | 2019-09-24 | Gilead Sciences Inc | hcv ns3 protease inhibitors |
| JP2012516351A (en) | 2009-01-30 | 2012-07-19 | アナコール ファーマシューティカルズ,インコーポレーテッド | Compound |
| EP2413955A4 (en) | 2009-04-01 | 2012-12-26 | Amylin Pharmaceuticals Inc | AGONIST COMPOUNDS OF GLP-1 RECEPTORS HAVING CONFORMATIONAL CONSTRAINTS AT THEIR N-TERMINAL END |
| EP2417134B1 (en) | 2009-04-08 | 2017-05-17 | Idenix Pharmaceuticals LLC. | Macrocyclic serine protease inhibitors |
| SG174532A1 (en) | 2009-04-24 | 2011-10-28 | Cadila Healthcare Ltd | Short-chain peptides as parathyroid hormone (pth) receptor agonist |
| WO2010135520A1 (en) | 2009-05-20 | 2010-11-25 | Chimerix, Inc. | Compounds, compositions and methods for treating viral infection |
| US8703700B2 (en) | 2009-05-22 | 2014-04-22 | Sequoia Pharmaceuticals, Inc. | Bimacrocylic HCV NS3 protease inhibitors |
| US8232246B2 (en) | 2009-06-30 | 2012-07-31 | Abbott Laboratories | Anti-viral compounds |
| US20110020272A1 (en) | 2009-07-24 | 2011-01-27 | Ulrich Schubert | Combination therapy for treating hepatitis viral infection |
| TW201117812A (en) | 2009-08-05 | 2011-06-01 | Idenix Pharmaceuticals Inc | Macrocyclic serine protease inhibitors |
| JP5702388B2 (en) | 2009-09-15 | 2015-04-15 | タイゲン バイオテクノロジー カンパニー,リミテッド | HCV protease inhibitor |
| WO2011063501A1 (en) | 2009-11-24 | 2011-06-03 | Boehringer Ingelheim International Gmbh | Hepatitis c inhibitor compounds |
| EP2504344A4 (en) | 2009-11-24 | 2013-06-05 | Boehringer Ingelheim Int | Hepatitis c inhibitor compounds |
| US20110178107A1 (en) | 2010-01-20 | 2011-07-21 | Taigen Biotechnology Co., Ltd. | Hcv protease inhibitors |
-
2011
- 2011-12-29 WO PCT/US2011/067699 patent/WO2012092409A2/en not_active Ceased
- 2011-12-29 CA CA 2822556 patent/CA2822556A1/en not_active Abandoned
- 2011-12-29 JP JP2013547655A patent/JP2014502620A/en active Pending
- 2011-12-29 EP EP11853822.2A patent/EP2658859A4/en not_active Withdrawn
- 2011-12-29 CN CN201180068701.3A patent/CN103534256B/en active Active
- 2011-12-29 MX MX2013007677A patent/MX2013007677A/en unknown
- 2011-12-29 US US13/339,440 patent/US8937041B2/en active Active
-
2015
- 2015-01-02 US US14/588,805 patent/US20150119319A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| EP2658859A4 (en) | 2014-07-30 |
| WO2012092409A2 (en) | 2012-07-05 |
| JP2014502620A (en) | 2014-02-03 |
| WO2012092409A3 (en) | 2012-11-08 |
| US8937041B2 (en) | 2015-01-20 |
| CN103534256A (en) | 2014-01-22 |
| EP2658859A2 (en) | 2013-11-06 |
| US20120172291A1 (en) | 2012-07-05 |
| CA2822556A1 (en) | 2012-07-05 |
| MX2013007677A (en) | 2013-07-30 |
| CN103534256B (en) | 2016-08-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8937041B2 (en) | Macrocyclic hepatitis C serine protease inhibitors | |
| US9309279B2 (en) | Macrocyclic hepatitis C serine protease inhibitors | |
| US9493506B2 (en) | Macrocyclic hepatitis C serine protease inhibitors | |
| US8951964B2 (en) | Phenanthridine macrocyclic hepatitis C serine protease inhibitors | |
| AU2012201327B2 (en) | Macrocyclic hepatitis C serine protease inhibitors | |
| HK1159515B (en) | Macrocyclic hepatitis c serine protease inhibitors | |
| HK1170936B (en) | Macrocyclic hepatitis c serine protease inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: ABBOTT LABORATORIES, ILLINOIS Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:MCDANIEL, KEITH F.;CHEN, HUI-JU;YEUNG, MING C.;AND OTHERS;SIGNING DATES FROM 20120131 TO 20120206;REEL/FRAME:036426/0904 |
|
| AS | Assignment |
Owner name: ABBVIE INC., ILLINOIS Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:ABBOTT LABORATORIES;REEL/FRAME:036735/0490 Effective date: 20120801 |
|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |