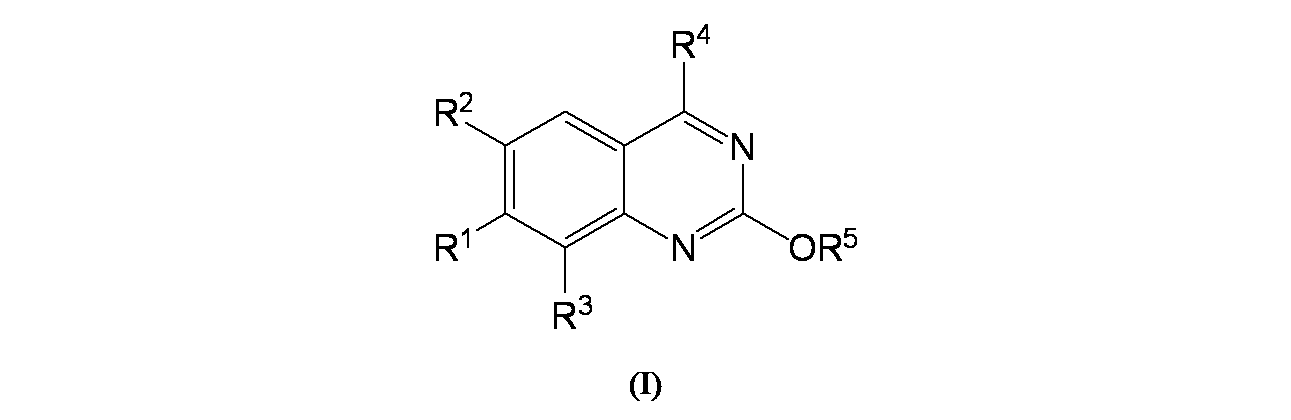
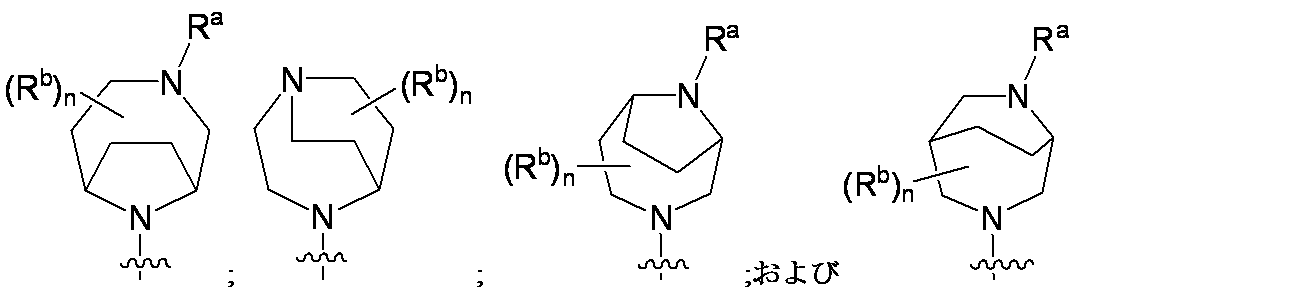
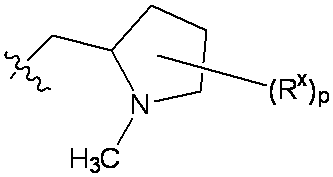
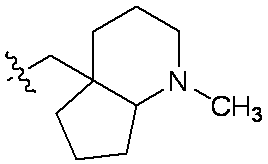
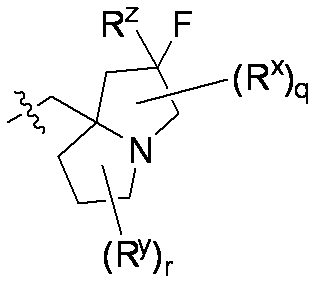
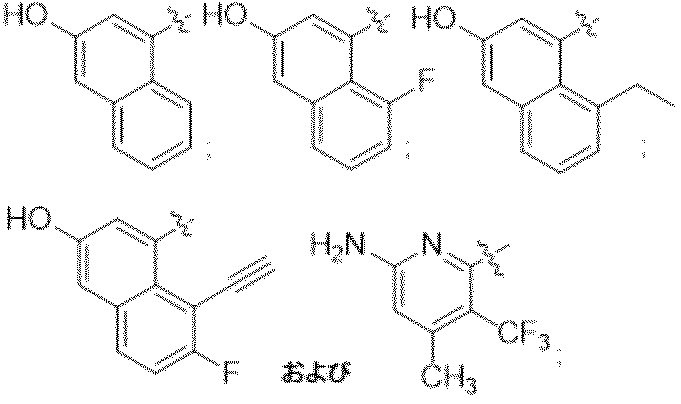
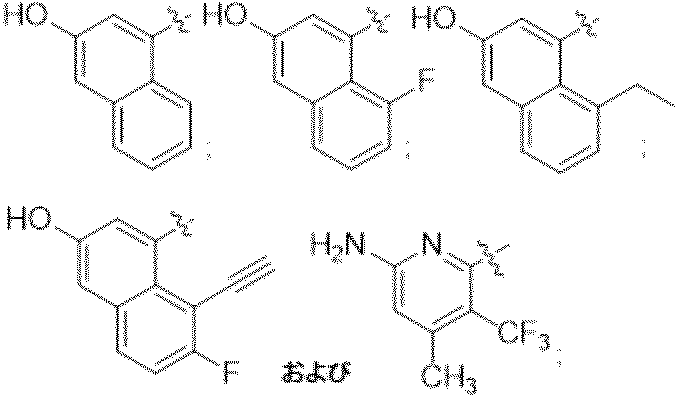
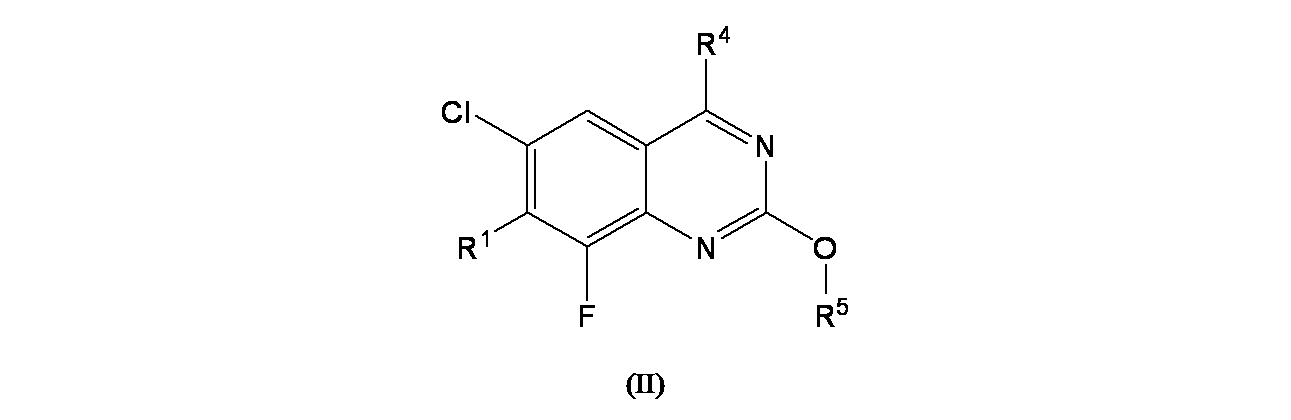
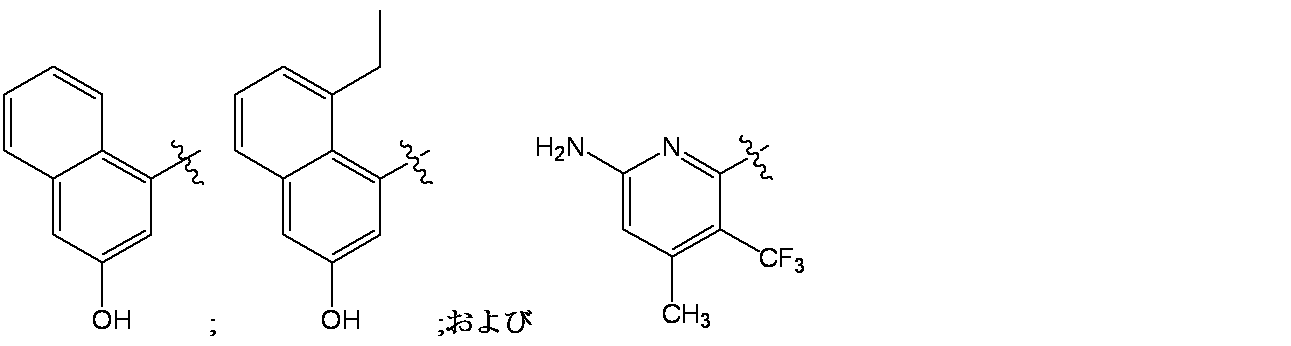
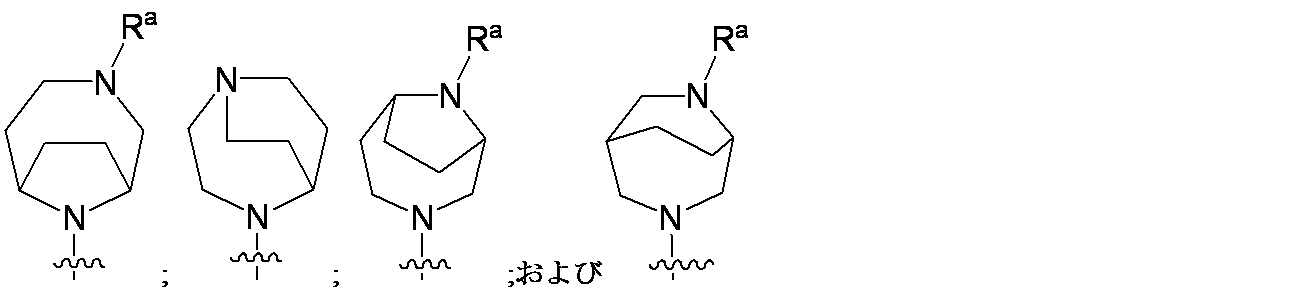
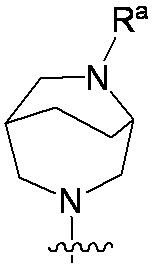
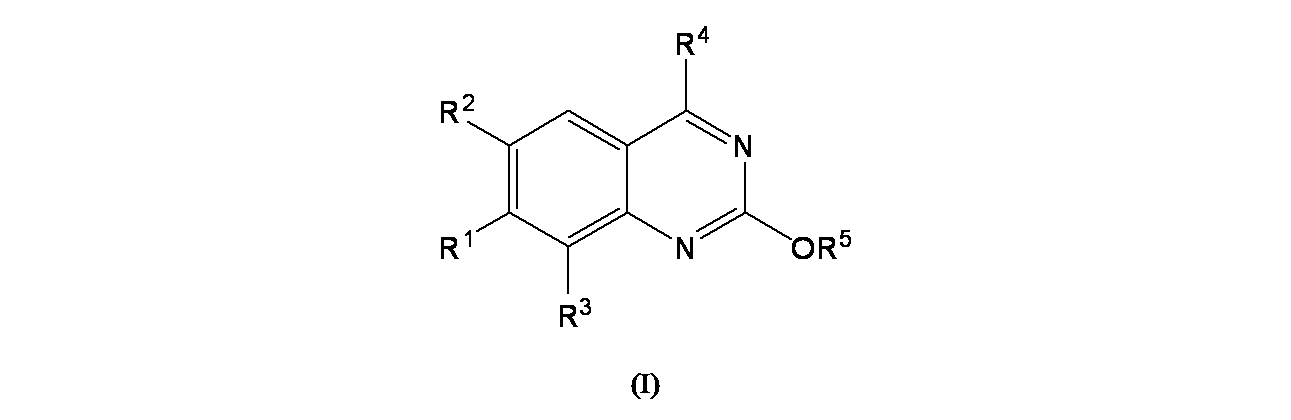
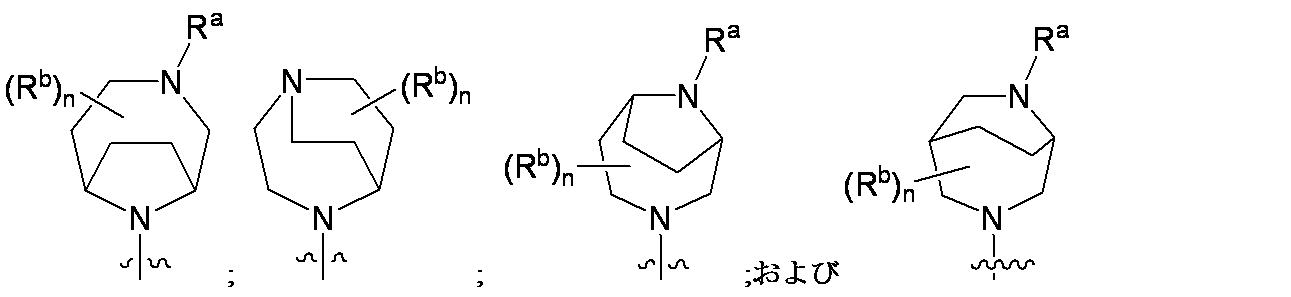
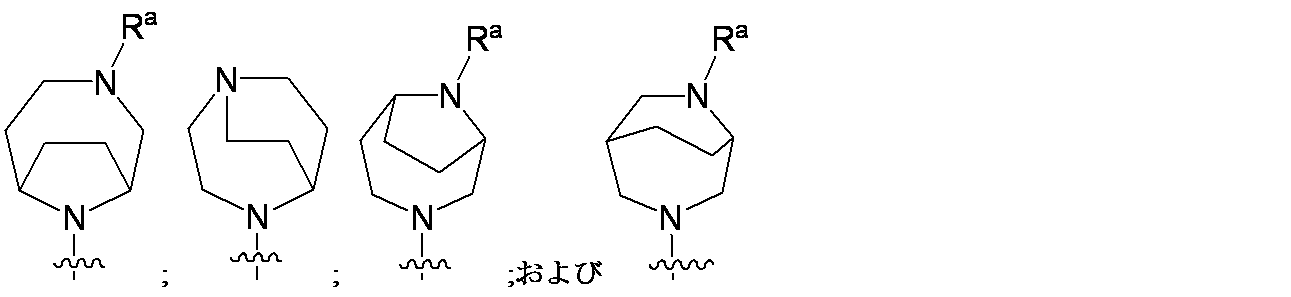
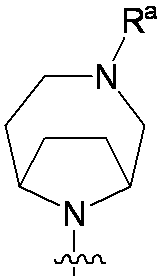
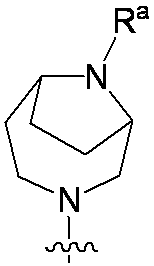
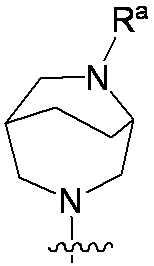
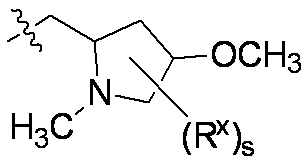
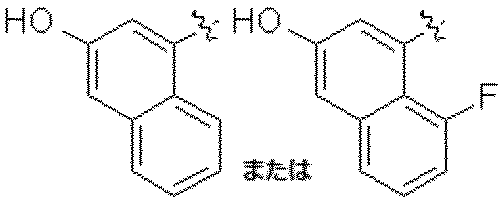
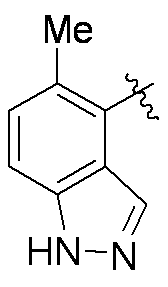
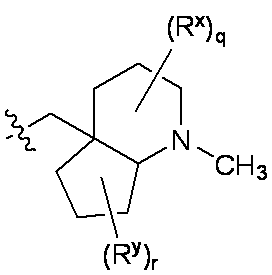
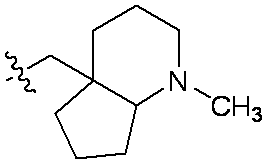
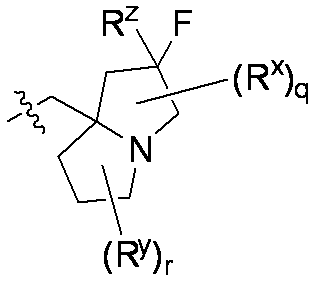
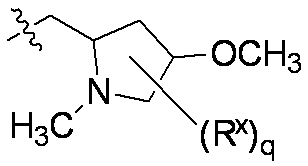
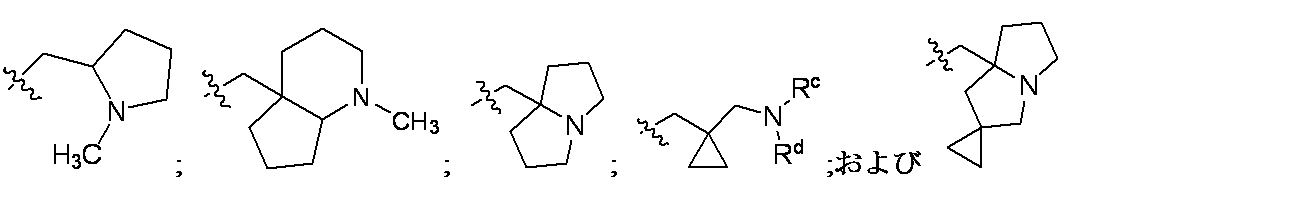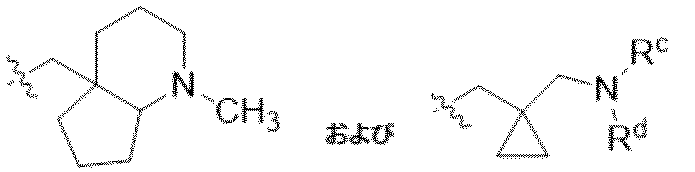特に断りが無い限り、原子価が満たされていないいずれの原子にも、原子価を満たすために十分な水素原子が含まれると見なされる。
単数形の「a」、「an」、および「the」は、文中で明らかに断りが無い限り複数形を含む。
本明細書で用いる用語「or」は論理和(すなわち、および/または)であり、例えば、用語「either」、「unless」、「alternatively」およびこれらと同様の用語で表されない限り排他的論理和を意味しない。
本明細書で用いるフレーズ「またはその医薬的に許容される塩」は、少なくとも1つの化合物、または少なくとも1つの化合物の塩、またはその組み合わせを指す。例えば、「式(I)の化合物またはその医薬的に許容される塩」には、以下に限らないが、1つの式(I)の化合物、2つの式(I)の化合物、1つの式(I)の化合物の医薬的に許容される塩、1つの式(I)の化合物および1以上の式(I)の化合物の医薬的に許容される塩、および2以上の式(I)の化合物の医薬的に許容される塩が含まれる。
本明細書で用いる用語「C2-C4アルケニル」は、2~4つの炭素原子および1つの二重結合を含む、直鎖または分岐鎖の炭化水素由来の基を指す。
本明細書で用いる用語「C1-C3アルコキシ」は、酸素原子を介して親分子に接続するC1-C3アルキル基を指す。
本明細書で用いる用語「C1-C3アルコキシカルボニル」は、カルボニル基を介して親分子に接続するC1-C3アルコキシ基を指す。
本明細書で用いる用語「C1-C3アルキル」は、1~3つの炭素原子を含む直鎖または分岐鎖の飽和炭化水素に由来する基を指す。
本明細書で用いる用語「C1-C6アルキル」は、1~3つの炭素原子を含む直鎖または分岐鎖の飽和炭化水素に由来する基を指す。
本明細書で用いる用語「C1-C3アルキルカルボニル」は、カルボニル基を介して親分子に接続するC1-C3アルキル基を指す。
本明細書で用いる用語「C2-C4アルキニル」は、2~4つの炭素原子および1つの三重結合を含む直鎖または分岐鎖の炭化水素に由来する基を指す。
本明細書で用いる用語「アミノ」は、-NH2を指す。
本明細書で用いる用語「アミノC1-C3アルキル」は、C1-C3アルキル基を介して親分子に接続するアミノ基を指す。
本明細書で用いる用語「アリール」は、フェニル基、または環の片方または両方がフェニル基である二環縮合環を指す。二環縮合環は、4~6員芳香族または非芳香族炭素環と縮合したフェニル基からなる。本開示のアリール基は、基の中の置換可能な任意の炭素原子を介して親分子に接続し得る。代表的なアリール基の例として、以下に限らないが、インダニル、インデニル、ナフチル、フェニル、およびテトラヒドロナフチルが挙げられる。
本明細書で用いる用語「シアノ」は、-CNを指す。
本明細書で用いる用語「C3-C4シクロアルキル」は、3または4つの炭素原子を有し、ヘテロ原子を含まない飽和単環炭化水素環を指す。
本明細書で用いる用語「ハロ」および「ハロゲン」は、F、Cl、Br、またはIをいう。
本明細書で用いる用語「ハロC1-C3アルコキシ」は、酸素原子を介して親分子に接続するハロC1-C3アルキル基を指す。
本明細書で用いる用語「ハロC1-C3アルキル」は、1、2、または3つのハロゲン原子で置換されたC1-C3アルキル基を指す。
本明細書で用いる用語「ヘテロアリール」は、少なくとも1つの原子が、N、O、およびSから選択され、残りの原子が炭素である5または6員の芳香環を指す。用語「ヘテロアリール」には、ヘテロアリール環が0、1、または2つのN、O、およびSから選択されるヘテロ原子をさらに含む4~6員芳香族または非芳香環と縮合した二環式環;および二環式環がN、O、およびSから選択される0、1、または2つのヘテロ原子をさらに含む4~6員芳香族または非芳香環と縮合した三環式環も含まれる。ヘテロアリール基は、基の中の置換可能な任意の炭素原子または窒素原子を介して親分子に接続する。代表的なヘテロアリール基の例として、以下に限らないが、アロキサジン、ベンゾ[1,2-d:4,5-d']ビスチアゾール、ベンゾオキサジアゾリル、ベンゾオキサゾリル、ベンゾフラニル、ベンゾチエニル、フラニル、イミダゾリル、インダゾリル、インドリル、イソオキサゾリル、イソキノリニル、イソチアゾリル、ナフチリジニル、オキサジアゾリル、オキサゾリル、プリン、ピリジニル、ピリダジニル、ピリミジニル、ピラジニル、ピラゾリル、ピロリル、キノリニル、チアゾリル、チエノピリジニル、チエニル、トリアゾリル、チアジアゾリル、およびトリアジニルが挙げられる。
本明細書で用いる用語「ヒドロキシ」は、-OHを指す。
本明細書で用いる用語「ヒドロキシC1-C3アルキル」は、C1-C3アルキル基を介して親分子に接続するヒドロキシ基を指す。
本明細書に記載の対象物の別の態様として、リガンド結合アッセイの開発またはインビボでの吸着、代謝、分布、受容体結合または受容体占有率、または薬物動態のモニターに用いるための、同位体標識リガンドとしての開示化合物の使用が挙げられる。例えば、本明細書に記載の化合物は放射性同位体を用いて製造され得て、得られた同位体標識化合物は結合アッセイの開発または代謝試験のために使用され得る。あるいは、本明細書に記載の化合物は同一の目的で、当業者に公知の方法を用いて、触媒でのトリチウム化により同位体標識化合物に変換され得る。
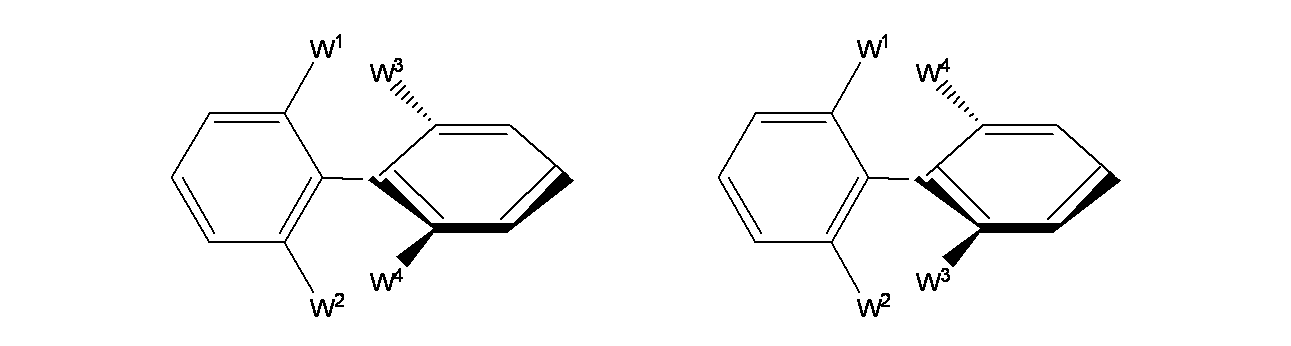
本開示の特定の化合物はアトロプ異性体として存在する。用語「アトロプ異性体」は、分子の他の部分との立体的相互作用の結果、分子内の単結合の回転が妨げられるか、または非常に遅くなり、単結合の両端の置換基が非対称となる場合に生じる立体異性体のことである(すなわち、非対称な炭素中心または立体中心を必要とせずに光学活性が生じる)。単結合の回転障壁が十分に大きく、構造間の相互変換が十分に遅い場合、異性体の分離おおよび単離が可能である。アトロプ異性体は単一の非対称原子を持たないエナンチオマー(またはエピマー)である。
アトロプ異性体は、アトロプ異性体が室温で少なくとも1週間、ほとんどあるいは全く相互変換しないことが認められるほど変換の障壁が十分高い場合、安定と考えられる。一部の態様において、アトロプ異性体は室温で少なくとも1年間、ほとんどあるいは全く相互変換しない。一部の態様において、アトロプ異性体化合物が一般的な固体であり、実質的に純粋な形態である場合、約5%を超える本開示のアトロプ異性体化合物は、室温で1週間他方のアトロプ異性体にほとんどあるいは全く変換されない。一部の態様において、約5%を超える本開示のアトロプ異性体化合物は、室温(おおよそ25℃)で1年間他方のアトロプ異性体にほとんどあるいは全く変換されない。一部の態様において、0℃で少なくとも1週間保管した水溶液の医薬製剤中の本開示のアトロプ異性体化合物の変換は約5%以下に留まるほど、十分安定である。本化学物質、医薬組成物および方法は、ラセミ体混合物、ジアステレオマー混合物、エピマー混合物、単一の光学的に純粋な状態のアトロプ異性体、および中間体混合物など、存在し得る全てのアトロプ異性体を含むことを意図する。
アトロプ異性体の熱によるラセミ化に対するエネルギー障壁は、キラル軸を形成する1つ以上の結合の自由回転に対する立体障害によって決定され得る。特定のビアリール化合物は、C2対称性を欠く環間結合周りの回転が制限されるアトロプ異性体を示す。異性化(エナンチオマー化)の自由エネルギー障壁は回転と関連する環状間結合の安定性の尺度である。電子的因子および立体的因子にもよるが、このような異性体のラセミ化は光励起および熱励起により促進される。
オルト置換ビアリール化合物は、以下のタイプの立体構造および回転異性を示し得る。
置換基がW
1≠W
2およびW
3≠W
4のため非対称分子であるビアリールは、エナンチオマーのキラルアトロプ異性体であり、アリール環間のsp
2炭素-sp
2炭素結合が自由回転を防ぐのに十分高いエネルギー障壁を有している。
W1:W3、W1:W4および/またはW2:W4、W2:W3間の立体的相互作用は、平面的な立体構造のエネルギーを最大にするのに十分である。2つの非平面的な軸不斉キラルエナンチオマーは、相互変換が互いに自由に分離できるほど十分に遅い場合、アトロプ異性体として存在する。上記構造式中の太線および破線は、回転エネルギー障壁のために立体的に制限されている分子の部分、または一部を示す。太線部分はページの平面の上に直交して存在し、破線の部分はページの平面に対して直角に存在する。分子の"平らな"部分(2つのビアリールのそれぞれの左側の環)はページの平面にある。
本開示の医薬組成物は1以上の医薬的に許容される塩を含んでもよい。「医薬的に許容される塩」は、親化合物の所期の生物活性を維持している塩を指し、望ましくない毒性面の影響は与えない(例えばBerge, S.M. et al., J. Pharm. Sci., 66:1-19 (1977)を参照)。当該塩は本明細書に記載の化合物の最後の分離および精製中に得られ得るか、あるいは化合物の遊離塩基官能基と適切な酸との反応または化合物の酸性基と適切な塩基の反応によって個別に得られ得る。酸付加塩には、毒性のない無機酸(例えば塩酸、硝酸、リン酸、硫酸、臭化水素酸、ヨウ化水素酸など)、ならびに毒性のない有機酸(例えば脂肪族モノカルボン酸および脂肪族ジカルボン酸、フェニル置換アルカン酸、ヒドロキシアルカン酸、芳香族酸、脂肪族スルホン酸および芳香族スルホン酸など)から得られるものを含む。塩基付加塩にはアルカリ土類金属(例えばナトリウム、カリウム、マグネシウム、カルシウムなど)、ならびに毒性のない有機アミン(例えばN,N'-ジベンジルエチレンジアミン、N-メチルグルカミン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、プロカインなど)から得られるものを含む。
(医薬組成物)
別の態様において、本開示は、組成物、例えば本開示で記載の1つの化合物またはその組合せを含み、医薬的に許容される担体と共に製剤化された医薬組成物を提供する。また、本開示の医薬組成物は組合せ治療で投与されてもよい(すなわち、本明細書に記載のように、別の薬剤と組み合わせて投与され得る)。
本明細書で用いる「医薬的に許容される担体」には、生理学的に適合するあらゆる溶媒、分散媒体、コーティング剤、抗細菌剤および抗真菌剤、等張化剤および吸収遅延剤などが含まれる。一部の態様において、担体は、例えば注射または点滴による静脈内投与、筋肉内投与、皮下投与、非経口投与、髄腔内投与または上皮投与に適している。投与経路によって、活性化合物は酸の作用および化合物を不活性化させ得るその他の自然条件から保護できる物質でコーティングされ得る。
本開示の医薬組成物は当業者に公知の1以上の様々な方法を用いて、1以上の投与経路で投与され得る。当業者に理解されるように、投与経路および/または投与方法は、所期の結果に応じて変化する。一部の態様において、本開示の化合物の投与経路には、例えば注射または点滴による静脈内、筋肉内、皮内、腹腔内、皮下、髄腔内またはその他非経口投与が挙げられる。本明細書で用いるフレーズ「非経口投与」は、経腸投与および局所投与以外の投与方法を意味し、通常は注射による投与を意味する。これには、以下に限らないが、静脈内、筋肉内、動脈内、髄腔内、関節内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、表皮下、関節内、嚢下、くも膜下、脊髄内、硬膜外および胸骨内の注射および点滴が含まれる。
滅菌注射用溶液は、上記に列挙した1つまたは組合せた成分と共に、適当な溶媒中に所期量の活性化合物を加えた後、必要に応じてマイクロ濾過で滅菌を行うことにより調整される。一般に、分散液は、活性化合物を、基本的な分散溶媒および上記に列挙されたもののうち所期のその他の成分を含む無菌ビークルに加えることで調製される。滅菌注射用溶液の調製に用いる滅菌粉末において、一部の調整方法は、予め滅菌濾過された溶液から活性成分および任意の他の所期の成分の粉末を得る減圧乾燥および凍結乾燥である。
本開示の医薬組成物に用いられ得る適切な水溶性および非水溶性の担体の例として水、エタノール、ポリオール(例えばグリセロール、プロピレングリコール、ポリエチレングリコールなど)、および適切なその混合物、植物性油、および注射可能な有機エステルが挙げられる。適度な流動性は、例えば、コーティング物質(例えばレシチン)の使用、分散液の場合は所期の粒子サイズの維持、および界面活性剤の使用により維持され得る。
医薬的に許容される担体には、滅菌水溶液または分散液、および滅菌注射用溶液または分散液の即時調製用の滅菌粉末が含まれる。医薬活性物質におけるそのような溶媒および薬剤の使用は当業者に公知である。従来の任意の溶媒または薬剤が活性化合物と適合しない場合を除き、それらの使用が本開示の医薬組成物において期待される。追加の活性化合物もまた組成物に包含され得る。
治療用組成物は一般に無菌であり、製造および保存条件下で安定でなければならない。当該組成物は薬物濃度の高い状態に適切な構造を有した溶液または液体として製剤化され得る。担体は溶媒または分散媒体(例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど))、およびその適切な混合物などであり得る。適度な流動性は、例えば、コーティング物質(例えばレシチン)の使用、分散液の場合は所期の粒子サイズの維持、および界面活性剤の使用により維持され得る。多くの場合、組成物には等張化剤(例えば、糖類、ポリアルコール(例えばマンニトール、ソルビトール、または塩化ナトリウム))が含まれることが望ましい。注射用組成物の持続的吸収は、組成物中に吸収遅延化剤(例えばモノステアリン酸塩およびゼラチン)を含むことによりもたらされ得る。
あるいは、本開示の化合物は、非経口投与以外の投与(例えば局所、上皮または粘膜投与(例えば、鼻腔内、経口、腟内、直腸、舌下または局所的投与))がされ得る。
本明細書で検討される医薬組成物はいずれも、例えば、任意に許容される適切ないずれかの経口製剤により経口的に運搬され得る。経口製剤の例として、以下に限らないが、例えば、錠剤、トローチ、ロゼンジ、水性および油性懸濁液、分散性粉末または顆粒、エマルション、ハードおよびソフトカプセル、液体カプセル、シロップ、およびエリキシルが挙げられる。経口投与用の医薬組成物は、経口投与用の医薬組成物を製造する分野で公知の任意の方法に従って製造され得る。医薬的に飲みやすい製剤を提供するために、本開示の医薬組成物は、甘味剤、風味剤、着色剤、粘滑剤、抗酸化剤、および防腐剤から選択される少なくとも1つの薬剤を含み得る。
錠剤は、例えば、少なくとも1つの式(I)の化合物および/または少なくとも1つのその医薬的に許容される塩、錠剤の製造に適切な少なくとも1つの毒性のない医薬的に許容される賦形剤を混合することで製造され得る。
水性懸濁液は、例えば、少なくとも1つの式(I)の化合物および/または少なくとも1つのその医薬的に許容される塩、水性懸濁液の製造に適切な少なくとも1つの添加剤を混合することにより製造され得る。水性懸濁液の製造に適切な添加剤の例としては、以下に限らないが、例えば、懸濁化剤(例えば、ナトリウムカルボキシメチルセルロース、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、アルギン酸、ポリビニルピロリドン、トラガカントガム、およびアラビアガム)、分散剤または湿潤剤(例えば、天然に存在するフォスファチド(例えばレシチン)、アルキレンオキシドと脂肪酸の縮合生成物(例えばポリオキシエチレンステアレート)、エチレンオキシドと長鎖脂肪族アルコールの縮合生成物(例えば、ヘプタデカエチレンオキシセタノール)、エチレンオキシドと、脂肪酸およびヘキシトールから誘導される部分エステルとの縮合生成物(例えばポリオキシエチレンソルビトールモノオレエート)、およびエチレンオキシドと、脂肪酸およびヘキシトール無水物から誘導される部分エステルとの縮合生成物(例えばポリエチレンソルビタンモノオレエート)が挙げられる。また、水性懸濁液は、少なくとも1つの防腐剤(例えば、p-ヒドロキシ安息香酸エチルおよびn-プロピルp-ヒドロキシ安息香酸)、少なくとも1つの着色剤、少なくとも1つの風味剤、および/または少なくとも1つの甘味剤(以下に限らないが、例えば、スクロース、サッカリン、およびアスパルテーム)を包含し得る。
油性懸濁液は、例えば、少なくとも1つの式(I)の化合物および/または少なくとも1つのその医薬的に許容される塩を、植物油(例えば、落花生油、ゴマ油、およびココナッツ油)、または鉱油(例えば液体パラフィン)のいずれかに懸濁することにより製造され得る。また、油性懸濁液は、少なくとも1つの濃化剤(例えば、蜜蝋、固形パラフィン、およびセチルアルコール)を包含し得る。飲みやすい油性懸濁液を提供するために、少なくとも1つの既に上記に記載の甘味剤、および/または少なくとも1つの風味剤が油性懸濁液に添加され得る。油性懸濁液は、さらに少なくとも1つの防腐剤(以下に限らないが、例えば、抗酸化剤(例えば、ブチルヒドロキシアニソール、およびα-トコフェロール)を包含し得る。
分散性粉末および顆粒は、例えば、少なくとも1つの式(I)の化合物および/または少なくとも1つのその医薬的に許容される塩、少なくとも1つの分散剤および/または湿潤剤、少なくとも1つの懸濁化剤、および/または少なくとも1つの防腐剤を混合することにより製造され得る。適切な分散剤、湿潤剤、および懸濁化剤は既に上記に記載されている。防腐剤の例として以下に限らないが、例えば、抗酸化剤(例えば、アスコルビン酸)が挙げられる。さらに、分散性粉末および顆粒は、また、少なくとも1つの賦形剤(以下に限らないが、例えば、甘味剤、風味剤、および着色剤)を包含し得る。
活性化合物は、急速な放出を防ぐ担体(例えば放出制御製剤(インプラント、経皮パッチ、およびマイクロカプセルデリバリーシステムなど))と共に製造され得る。生分解性の生体適合性のあるポリマー(例えばエチレン酢酸ビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸)が使用され得る。そのような製剤の多くの製造方法は、特許されているか、または一般に当業者に公知である。例えば、Robinson, J.R., ed., Sustained and Controlled Release Drug Delivery Systems, Marcel Dekker, Inc., New York (1978)を参照のこと。
治療用組成物は当業者に公知の医療用デバイスで投与され得る。例えば、ある態様において、本開示の治療用組成物は無針皮下注射デバイス(例えば米国特許第5,399,163号、5,383,851号、5,312,335号、5,064,413号、4,941,880号、4,790,824号、または4,596,556号に開示のデバイス)で投与され得る。本開示において有用な公知のインプラントおよびモジュールの例として、米国特許第4,487,603号に開示の一定の速度で投薬するための植え込み型微量注入ポンプ;米国特許第4,486,194号に開示の皮膚を介した投薬のための治療用装置;米国特許第4,447,233号に開示の正確な速度で薬物を送達するための薬物注入ポンプ;米国特許第4,447,224号に開示の継続した薬物送達のための植え込み型変流量注入装置;米国特許第4,439,196号に開示のマルチチャンバーコンパートメントを有する浸透圧を利用した薬物送達システム;および米国特許第4,475,196号に開示の浸透圧を利用した薬物送達システムが挙げられる。これらの特許は参照により本明細書に援用される。その他多くの類似のインプラント、送達システム、およびモジュールが当業者に公知である。
ある態様において、本開示の化合物は非経口投与され得る。すなわち、以下に限らないが、静脈内、筋肉内、動脈内、髄腔内、関節内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、表皮下、関節内、嚢下、くも膜下、脊髄内、硬膜外および胸骨内の注射および/または点滴を含む注入によって投与され得る。
一部の態様において、本開示の化合物は経口投与され得る。すなわち、ゼラチンカプセル剤、錠剤、ハードまたはソフトカプセル剤、または液体カプセル剤で投与され得る。
KRAS阻害剤の使用/治療方法
本明細書に記載の治療剤の投与は、治療上有効量の治療剤の投与を含みうる。本明細書で用いる用語「治療上有効量」は、以下に限らないが、本明細書に記載のKRAS阻害剤を含む組成物の投与により治療可能な症状を治療するための、治療剤の量を指す。この量は、治療または寛解効果が示されるのに十分な量である。当該効果には、例えば、以下に限らないが、本明細書に列記した症状の治療が含まれる。対象に対する正確な有効量は対象の体格および健康状態、治療する状態の性質および程度、治療医の提案、ならびに投与される治療薬または治療薬の組み合わせによる。
本明細書に記載の化合物の投与において、投薬範囲は対象の体重に対して約0.0001~100mg/kg、さらに一般的には0.01~40mg/kgである。治療計画の例として、1日1回、隔週1回、3週に1回、毎週1回、2週間に1回、3週間に1回、4週間に1回、1ヵ月に1回、3ヵ月に1回、または3~6ヵ月に1回の投与が必要となる。
本開示化合物は、足場非依存性細胞増殖を強く阻害するため、腫瘍転移を阻害する可能性を有する。従って、別の態様において本開示は、腫瘍転移を阻害する方法であって、任意の本開示の化合物および医薬的に許容される担体を含む有効量の医薬組成物を必要な対象に投与することを特徴とする方法を提供する。
KRAS変異に限らず、Ras変異もまた血液系腫瘍(例えば、血液、骨髄および/またはリンパ節に影響するがん)において認められている。従って、ある態様は、血液系腫瘍の治療を必要とする患者に対する、(例えば医薬組成物の形態での)開示化合物の投与に関するものである。このような悪性腫瘍には、以下に限らないが、白血病およびリンパ腫が挙げられる。例えば、本開示の化合物は、急性リンパ芽球性白血病(ALL)、急性骨髄性白血病(AML)、慢性リンパ性白血病(CLL)、小リンパ球性リンパ腫(SLL)、慢性骨髄性白血病(CML)、急性単球性白血病(AMoL)および/またはその他の白血病などの疾患の治療に用いられ得る。別の態様において、本化合物はホジキンリンパ腫または非ホジキンリンパ腫などのあらゆる亜型のリンパ腫の治療に有用である。
腫瘍またはがんがKRAS変異を含むか否かの決定は、KRASタンパク質をコードするヌクレオチド配列の評価、KRASタンパク質のアミノ酸配列の評価、またはKRAS突然変異体タンパク質に想定される特徴の評価によって行われ得る。野生型ヒトKRASタンパク質の配列は当業者に公知である。
KRAS変異を検出する方法は当業者に公知である。これらの方法には、以下に限らないが、ポリメラーゼ連鎖反応-制限酵素断片長多型(PCR-RFLP)アッセイ、ポリメラーゼ連鎖反応-一本鎖工事構造多型(PCR-SSCP)アッセイ、リアルタイムPCRアッセイ、PCRシーケンシング、アレル特異的PCR増幅(MASA)アッセイ、ダイレクトシーケンス、プライマー伸長、電気泳動、オリゴヌクレオチドライゲーションアッセイ、混成アッセイ、TaqManアッセイ、SNPジェノタイピングアッセイアッセイ、高分解能融解曲線分析およびマイクロアレイなどが挙げられる。一部の態様において、KRAS変異を含むサンプルをリアルタイムPCR法で評価した。リアルタイムPCR法では、KRAS変異特異的な蛍光プローブが使用される。変異が存在する場合、プローブが結合し、蛍光が検知される。一部の態様において、KRAS遺伝子の特定の領域(例えば、エクソン2および/またはエクソン3)は、ダイレクトシーケンス法を用いてKRAS変異が特定される。この手法により、配列決定された領域に起こり得る全ての変異が同定される。
KRAS変異を検出する方法は当業者に公知である。これらの方法には、以下に限らないが、変異体タンパク質に特異的な結合剤(例えば抗体)を用いたKRAS変異体の検出、タンパク質電気泳動およびウェスタンブロッティング、およびダイレクトペプチドシーケンシングなどが挙げられる。
腫瘍またはがんがKRAS変異を含むか否かの決定方法は、様々なサンプルを使用し得る。一部の態様において、サンプルは腫瘍またはがんを有する対象から採取される。一部の態様において、サンプルは採取したばかりの腫瘍/がんサンプルである。一部の態様において、サンプルは凍結された腫瘍/がんサンプルである。一部の態様において、サンプルは、ホルマリン固定パラフィン包埋サンプルである。一部の態様において、サンプルは、細胞溶解物の状態に処理されている。一部の態様において、サンプルは、DNAまたはRNAの状態に処理されている。また、本開示は、哺乳類における増殖亢進性障害を治療する方法であって、治療上有効量の本開示化合物、またはその医薬的に許容される塩、エステル、プロドラッグ、溶媒和物、水和物または誘導体を哺乳類に投与することを特徴とする方法にも関する。一部の態様において、前記方法は、急性骨髄性白血病、思春期世代のがん、小児副腎皮質がん、AIDS関連がん(例えばリンパ腫およびカポジ肉腫)、肛門がん、虫垂がん、星細胞腫、非定型奇形腫、基底細胞がん、胆管がん、膀胱がん、骨がん、脳幹神経膠腫、脳腫瘍、乳がん、気管支腫瘍、バーキットリンパ腫、カルチノイド腫瘍、非定型奇形腫、胚性腫瘍、胚細胞腫瘍、原発性リンパ腫、子宮頸がん、小児がん、脊索腫、心臓腫瘍、慢性リンパ性白血病(CLL)、慢性骨髄性白血病(CML)、慢性骨髄増殖性疾患、結腸がん、大腸がん、頭蓋咽頭腫、皮膚T細胞リンパ腫、非浸潤性乳管がん(DCIS)、胚性腫瘍、CNSがん、子宮内膜がん、上衣腫、食道がん、伸展性神経芽腫、ユーイング肉腫、頭蓋外胚細胞腫瘍、顎外胚細胞腫瘍、眼がん、骨線維性組織球腫、胆嚢がん、胃がん、消化管カルチノイド腫瘍、消化管間質腫瘍(GIST)、胚細胞腫瘍、妊娠性絨毛腫瘍、毛様細胞白血病、頭頸部癌、心臓がん、肝臓がん、ホジキンリンパ腫、下咽頭がん、眼内黒色腫、膵島細胞腫瘍、膵神経内分泌腫瘍、腎臓がん、喉頭がん、口唇・口腔がん、肝臓がん、非浸潤性小葉がん(LCIS)、肺がん、リンパ腫、原発不明の転移性扁平上皮頸部がん、正中線がん、口のがん、多発性内分泌腫瘍症候群、多発性骨髄腫/形質細胞新生物、菌状息肉症、骨髄異形成症候群、骨髄異形成/骨髄増殖性新生物、多発性骨髄腫、メルケル細胞がん、悪性中皮腫、骨の悪性線維性組織球腫および骨肉腫、鼻腔・副鼻腔がん、上咽頭がん、神経芽腫、非ホジキンリンパ腫、非小細胞肺がん(NSCLC)、口唇がんおよび口腔がん、口腔咽頭癌、卵巣がん、膵臓がん、乳頭腫症、傍神経節腫、副鼻腔がんおよび鼻腔がん、副甲状腺がん、陰茎がん、咽頭がん、胸膜肺芽腫、原発性中枢神経系(CNS)リンパ腫、前立腺がん、直腸がん、移行細胞がん、網膜芽細胞腫、横紋筋肉腫、唾液腺がん、皮膚がん、胃がん、小細胞肺がん、小腸がん、軟部組織肉腫、T細胞リンパ腫、精巣がん、咽頭がん、胸腺腫および胸腺がん、甲状腺がん、腎盂および尿管の移行細胞がん、絨毛腫瘍、小児期の珍しい癌、尿道がん、子宮肉腫、腟がん、外陰がん、またはウイルス性がんなどのがんの治療に関する。一部の態様において、前記方法は、例えば皮膚の良性過形成(例えば乾癬)、再狭窄、または前立腺(例えば良性前立腺肥大症(BPH))などの非がん性増殖亢進性障害の治療に関する。
ある態様において、本開示は、肺がんを治療する方法であって、有効量の任意の上記化合物(または同医薬組成物)を必要な対象に投与することを特徴とする方法に関する。ある態様において、肺がんは非小細胞肺がん(NSCLC)(例えば腺癌、扁平上皮肺癌または大細胞肺がん)である。別の態様において、肺がんは小細胞肺がんである。開示化合物で治療可能なその他の肺がんには、以下に限らないが、腺腫瘍、カルチノイド腫瘍および未分化がんが挙げられる。本開示の方法に従って、本開示の化合物、またはその医薬的に許容される塩、エステル、プロドラッグ、溶媒和物、互変異性体、水和物または誘導体で治療され得る対象は、例えば以下と診断された対象である:急性骨髄性白血病、急性骨髄性白血病、思春期世代のがん、小児副腎皮質がん、AIDS関連がん(例えばリンパ腫およびカポジ肉腫)、肛門がん、虫垂がん、星細胞腫、非定型奇形腫、基底細胞がん、胆管がん、膀胱がん、骨がん、脳幹神経膠腫、脳腫瘍、乳がん、気管支腫瘍、バーキットリンパ腫、カルチノイド腫瘍、非定型奇形腫、胚性腫瘍、胚細胞腫瘍、原発性リンパ腫、子宮頸がん、小児がん、脊索腫、心臓腫瘍、慢性リンパ性白血病(CLL)、慢性骨髄性白血病(CML)、慢性骨髄増殖性疾患、結腸がん、大腸がん、頭蓋咽頭腫、皮膚T細胞リンパ腫、非浸潤性乳管がん(DCIS)、胚性腫瘍、CNSがん、子宮内膜がん、上衣腫、食道がん、伸展性神経芽腫、ユーイング肉腫、頭蓋外胚細胞腫瘍、顎外胚細胞腫瘍、眼がん、骨線維性組織球腫、胆嚢がん、胃がん、消化管カルチノイド腫瘍、消化管間質腫瘍(GIST)、胚細胞腫瘍、妊娠性絨毛腫瘍、毛様細胞白血病、頭頸部癌、心臓がん、肝臓がん、ホジキンリンパ腫、下咽頭がん、眼内黒色腫、膵島細胞腫瘍、膵神経内分泌腫瘍、腎臓がん、喉頭がん、口唇・口腔がん、肝臓がん、非浸潤性小葉がん(LCIS)、肺がん、リンパ腫、原発不明の転移性扁平上皮頸部がん、正中線がん、口のがん、多発性内分泌腫瘍症候群、多発性骨髄腫/形質細胞新生物、菌状息肉症、骨髄異形成症候群、骨髄異形成/骨髄増殖性新生物、多発性骨髄腫、メルケル細胞がん、悪性中皮腫、骨の悪性線維性組織球腫および骨肉腫、鼻腔・副鼻腔がん、上咽頭癌、神経芽腫、非ホジキンリンパ腫、非小細胞肺がん(NSCLC)、口腔癌、口唇がんおよび口腔がん、口腔咽頭癌、卵巣がん、膵臓がん、乳頭腫症、傍神経節腫、副鼻腔がんおよび鼻腔がん、副甲状腺がん、陰茎がん、咽頭がん、胸膜肺芽腫、原発性中枢神経系(CNS)リンパ腫、前立腺がん、直腸がん、移行細胞がん、網膜芽細胞腫、横紋筋肉腫、唾液腺がん、皮膚がん、胃がん、小細胞肺がん、小腸がん、軟部組織肉腫、T細胞リンパ腫、精巣がん、咽頭がん、胸腺腫および胸腺がん、甲状腺がん、腎盂および尿管の移行細胞がん、絨毛腫瘍、小児期の珍しい癌、尿道がん、子宮肉腫、腟がん、外陰がん、またはウイルス性がん。一部の態様において、本開示の化合物で治療する対象は、皮膚の良性過形成(例えば乾癬)、再狭窄、または前立腺(例えば良性前立腺肥大症(BPH))などの非がん性増殖亢進性障害と診断された対象である。
本開示はさらにタンパク質を本開示の化合物の有効量と接触させることによって変異KRASタンパク質の活性を調節する方法を提供する。上記調節とは、タンパク質の阻害または活性化であり得る。一部の態様において、本開示は、変異KRASタンパク質を溶液中の有効量の本開示の化合物と接触させることにより、タンパク質の活性阻害する方法を提供する。一部の態様において、本開示は、目的のタンパク質を発現する細胞、組織、臓器と接触させることで、変異KRASタンパク質の活性を阻害する方法を提供する。一部の態様において、本開示は、有効量の本開示の化合物を投与することにより、以下に限らないが齧歯動物および哺乳動物(例えばヒト)などの対象のタンパク質の活性を阻害する方法を提供する。一部の態様において、調節される割合は25%、30%、40%、50%、60%、70%、80%、または90%を超える。一部の態様において、阻害される割合は25%、30%、40%、50%、60%、70%、80%、または90%を超える。一部の態様において、本開示は、細胞のKRAS変異活性を阻害するのに十分な本開示の化合物の量を細胞と接触させることにより、細胞のKRAS活性を阻害する方法を提供する。一部の態様において、本開示は、組織の変異KRAS活性を阻害するのに十分な本開示の化合物の量を組織と接触させることにより、組織の変異KRASを阻害する方法を提供する。一部の態様において、本開示は、生物のKRAS変異活性を阻害するのに十分な本開示の化合物の量を生物と接触させることにより、生物のKRAS活性を阻害する方法を提供する。一部の態様において、本開示は、動物のKRAS変異活性を阻害するのに十分な本開示の化合物の量を動物と接触させることにより、生物のKRAS活性を阻害する方法を提供する。一部の態様において、本開示は、哺乳類のKRAS変異活性を阻害するのに十分な本開示の化合物の量を哺乳類と接触させることにより、生物のKRAS活性を阻害する方法を提供する。一部の態様において、本開示は、ヒトのKRAS変異活性を阻害するのに十分な本開示の化合物の量をヒトと接触させることにより、生物のKRAS活性を阻害する方法を提供する。本開示は、治療が必要な対象において、KRAS活性が介在する疾患を治療する方法を提供する。本開示は、他の経路を調節することが知られている薬剤、または同経路の別の成分を調節することが知られている薬剤、または標的酵素のセットが重複している薬剤とも、本開示の化合物、またはその医薬的に許容される塩、エステル、プロドラッグ、溶媒和物、互変異性体、水和物または誘導体を組み合わせて用いる、組合せの治療方法も提供する。ある態様において、そのような治療には、以下に限らないが、1以上の本開示の化合物と、化学療法剤、治療用抗体、および放射線治療との組合せが挙げられる。
多くの化学療法は、現在当業者に知られており、本開示の化合物と組み合わせて用いられ得る。一部の態様において、化学療法は、分裂阻害剤、アルキル化剤、代謝拮抗剤、インターカレーション剤、成長因子阻害剤、細胞周期阻害剤、酵素、トポイソメラーゼ阻害剤、生体応答調節物質、抗ホルモン剤、血管新生阻害剤、および抗アンドロゲン剤からなる群から選択される。一部の態様において、化学療法剤は、免疫系を強化、刺激、またはアップレギュレーションする、免疫腫瘍(IO)の薬剤である。
本明細書に記載の化合物は、治療する症状によっては、本開示の薬剤またはその他の適切な薬剤との組合せで用いられ得る。そのため、一部の態様において、1以上の本開示の化合物が上記の他の薬剤と共に投与される。組合せ治療が行われる場合、本明細書に記載の化合物は第2の薬剤と同時、または個別に投与される。この組合せ投与には、同一の剤形の2つの薬剤の同時投与、異なる剤形での同時投与、および個別投与が含まれ得る。
つまり、本明細書に記載の化合物および上述の任意の薬剤は同一の剤形で一緒に製剤化し、同時に投与され得る。あるいは、本開示の化合物および上述の任意の薬剤は異なる剤形で、同時に投与され得る。さらに、本開示の化合物が投与され、それに続いて上述の任意の薬剤が投与され得る。またはその逆も可能である。個別投与での一部の態様において、本開示の化合物および上述の任意の薬剤は、数分後、または数時間後、または数日後に投与される。
化合物は、以下に記載されるものを含む当技術分野で公知の方法によって製造することができ、当業者の技術範囲内で変更され得る。一部の試薬および中間体は当業者に公知である。その他の試薬および中間体は容易に入手可能な化学物質を用いて、当技術分野で公知の方法により製造することができる。任意の変数(番号付けされた「R」置換基など)は、化合物の製造方法を説明することのみを意図しており、特許請求の範囲または本明細書の他のセクションで使用される変数と混同されない。以下の方法は例示を目的としたものであり、本開示の範囲を限定するものではない。
合成
一般スキーム
本明細書に記載の化合物は下記および方法1~4に記載の通り製造され得る。
方法1: ステップ1において、既知化合物A(CAS 1698028-11-3)を適切な溶媒中(例えばTHF)、塩基(例えばジイソプロピルエチルアミン)と共にアミンと反応させ、化合物Bを得た。ステップ2において、化合物Bを溶媒(例えばジメチルアセトアミド)中、フッ化カリウムで処理し、化合物Cを得た。ステップ3において、化合物Cを鈴木カップリング反応条件下でアリールボロン酸またはエステルとカップリングさせ、化合物Dを得た。ステップ4において、化合物Dを溶媒中(例えばTHF)、塩基の存在下、ROHで処理し、化合物Eを得た。
方法2: 化合物Eのアミノ基は、以下の手順に従って別のアミノ基に変換されてもよい。ステップ5において、塩基加水分解により化合物Fを得た。ステップ6において、溶媒(例えばジクロロメタン)中、塩基の存在下、カップリング剤(例えばBOP)を用いてアミン置換基を導入し、化合物Eを得た。
方法3: ステップ7において、化合物Hを塩基の存在下、POCl3で処理し、化合物Gを得た。化合物Gを溶媒(例えばジメチルアセトアミド)中、塩基の存在下、適当なアミンで処理し、化合物Eを得た。
方法4: ステップ9において、化合物Bを塩基の存在下、式R-OHのアルコールで処理し、化合物Hを得た。ステップ10において、化合物Hを鈴木カップリング反応条件下でアリールボロン酸またはエステルとカップリングさせ、化合物Eを得た。当業者により所期の保護基(例えばBoc、PMB、MOMなど)が導入、および除去されてもよく、それらは実施例に記載されている。一般構造Eの化合物を製造するための、アリール、NRR'、およびOR基の官能基化および合成は実施例に記載されている。
実施例
本発明は、次の実施例においてさらに定義される。該実施例は説明によってのみ与えられると理解されるべきである。上記の検討および実施例から、当業者は本発明に本質的な特徴を解明することが出来、発明の本質および範囲から離れることなく、本発明を幅広い条件および用途に適応させるために変更および修正を行うことが出来る。その結果、本発明は以下で説明する該実施例によって制限されず、むしろ本明細書に添付の請求項により定義される。
略語
下記の略語は、以下の実施例の箇所および本明細書のその他の箇所において用いられる。
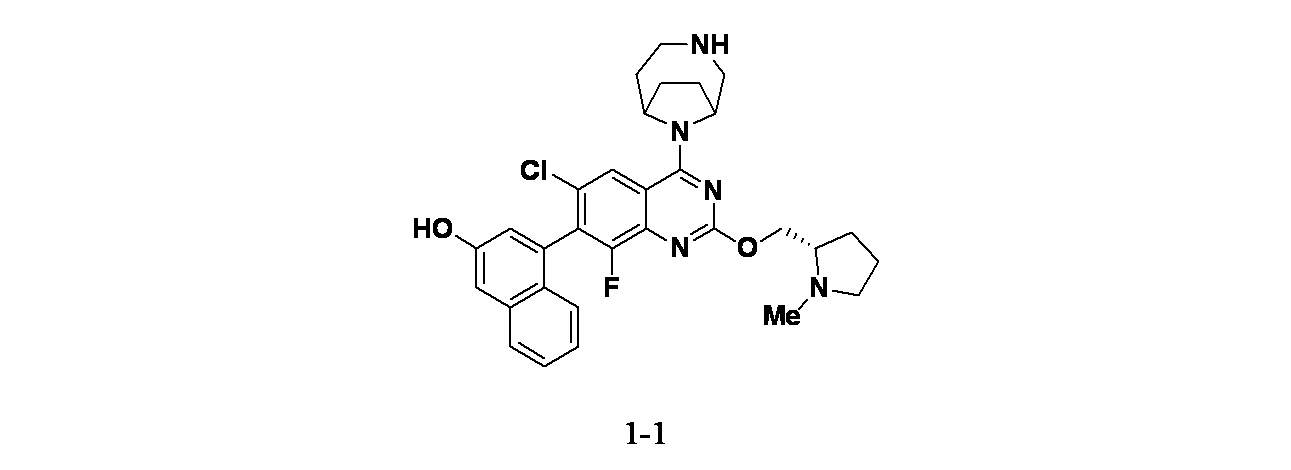
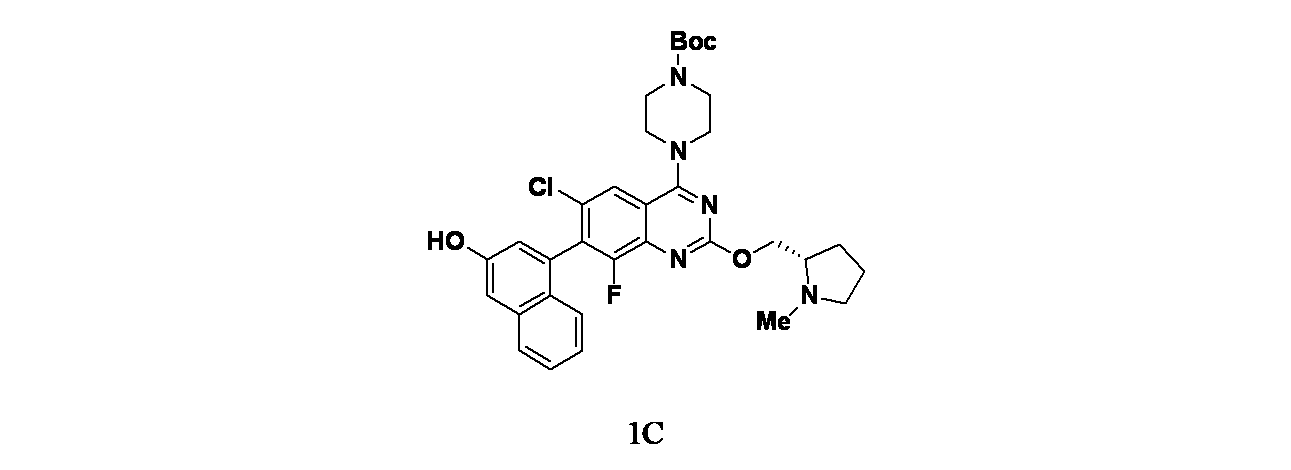
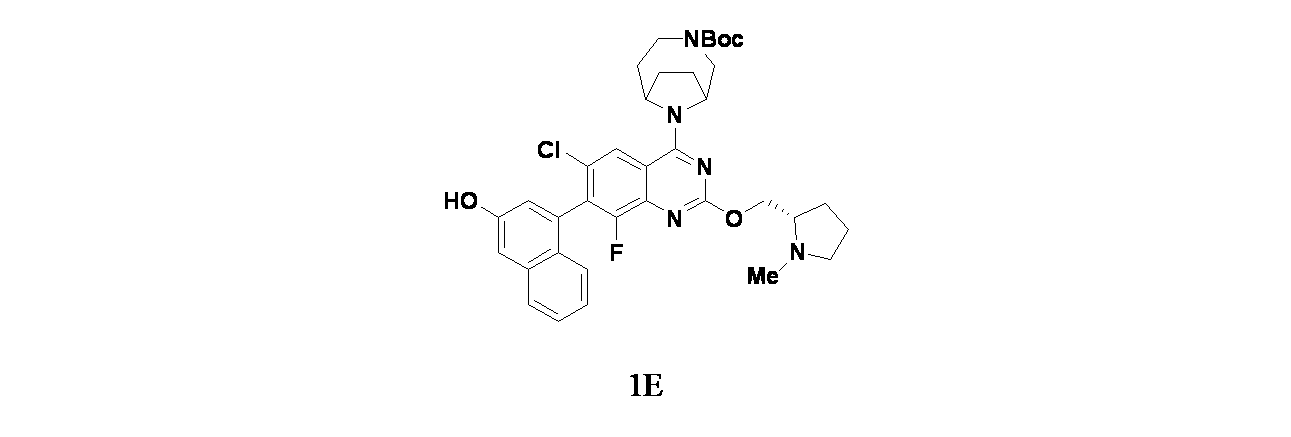
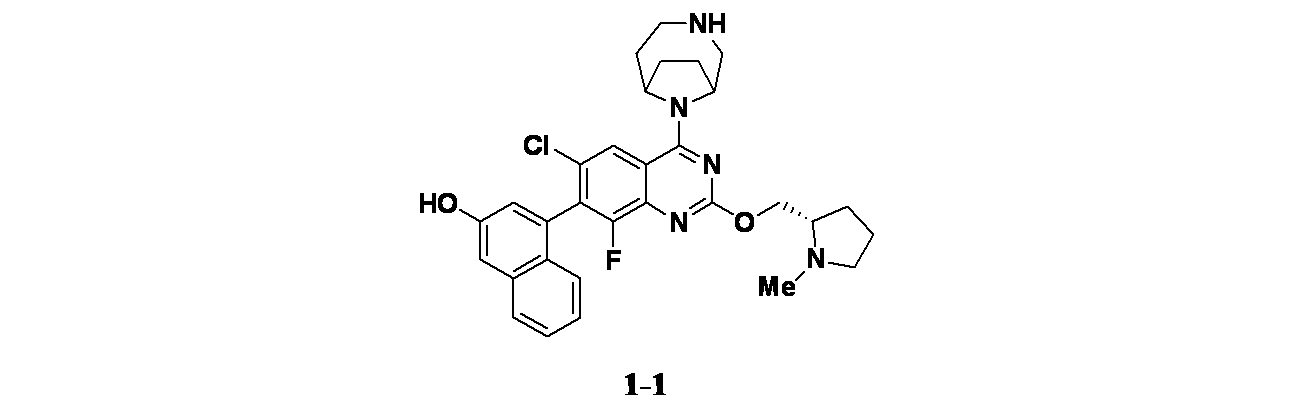
実施例1-1
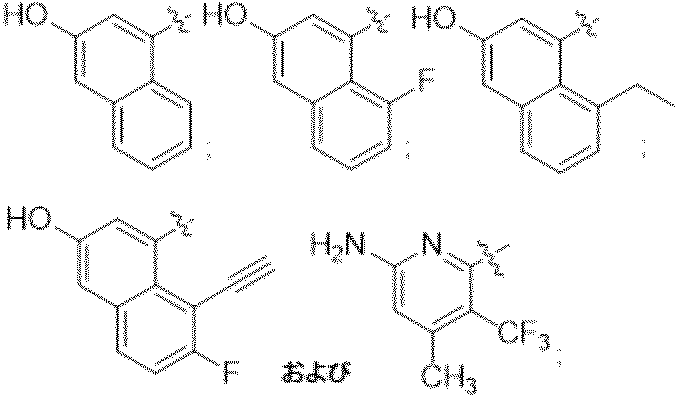
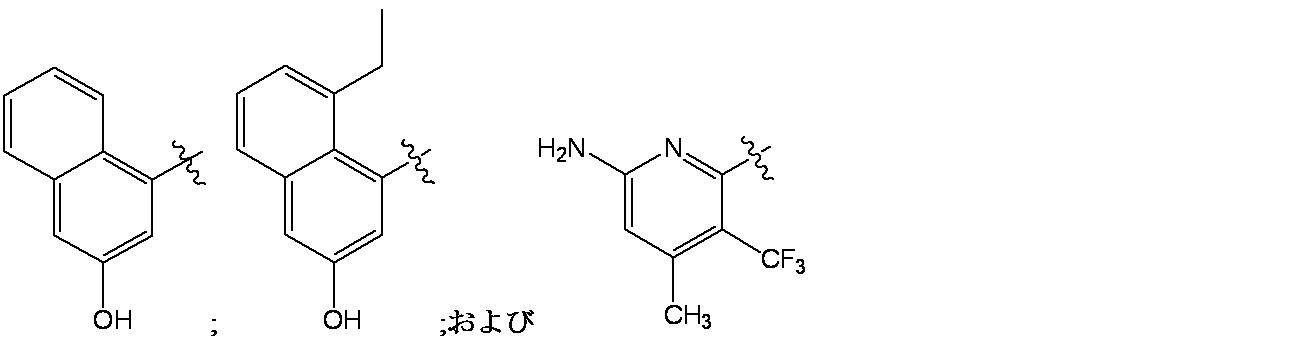
4-(6-クロロ-4-{3,9-ジアザビシクロ[4.2.1]ノナン-9-イル}-8-フルオロ-2-{[(2S)-1-メチルピロリジン-2-イル]メトキシ}キナゾリン-7-イル)ナフタレン-2-オール
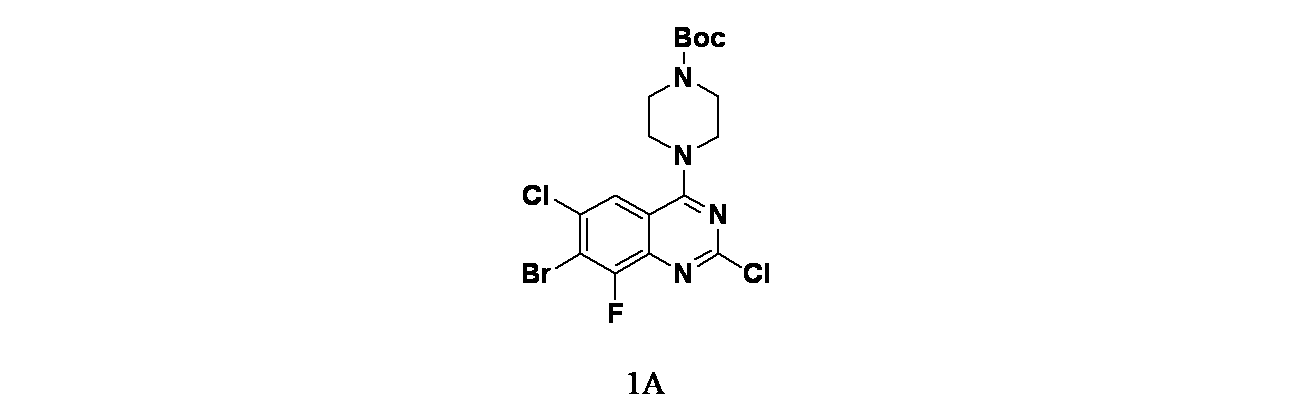
中間体1A: tert-ブチル4-(7-ブロモ-2,6-ジクロロ-8-フルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
7-ブロモ-2,4,6-トリクロロ-8-フルオロキナゾリン(1g、3.03mmol)およびDIPEA(1.32mL、7.57mmol)/THF(15mL)の溶液に、窒素雰囲気下、tert-ブチルピペラジン-1-カルボキシレート(0.56g、3.03mmol)を加え、25℃で2時間撹拌した。この混合物を次いで濃縮した。得られた残渣を酢酸エチル(60mL)で希釈し、水(30mLx2)および食塩水(50mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥し、濾過し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(シリカゲル、石油エーテル:酢酸エチル=10:1~4:1)で精製し、tert-ブチル4-(7-ブロモ-2,6-ジクロロ-8-フルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(1.33g、2.77mmol、収率91.5%)を黄色固体として得た。MS(ESI)m/z 481.0 [M+1]
+;
1H NMR(400MHz、CDCl
3) δ 7.77(d, J=1.6Hz, 1H), 3.93-3.84(m, 4H), 3.72-3.61(m, 4H), 1.50(s, 9H)
中間体1B: tert-ブチル(S)-4-(7-ブロモ-6-クロロ-8-フルオロ-2-((1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
tert-ブチル4-(7-ブロモ-2,6-ジクロロ-8-フルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(540mg、1.125mmol)/DMSO(6mL)の溶液に、フッ化セシウム(342mg、2.249mmol)および(S)-(1-メチルピロリジン-2-イル)メタノール(324mg、2.81mmol)を加え、この混合物を100℃で2時間加熱した。混合物を室温に冷却した後、飽和NaHCO
3(50mL)を加えた。無機層をDCM(50mLx2)で抽出した。抽出したDCM層を合わせて食塩水で洗浄し、乾燥し(Na
2SO
4)、濾過し、次いで濃縮した。得られた残渣をシリカカラム(40g、溶出: 0~10%MeOH/DCM(0.5%TEA含有))で精製し、tert-ブチル(S)-4-(7-ブロモ-6-クロロ-8-フルオロ-2-((1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)ピペラジン-1-カルボキシレート(0.43g、0.769mmol、収率68.4%)を黄色固体として得た。MS(ESI)m/z 558.2/560.2 [M+1]
+
中間体1C: tert-ブチル4-(6-クロロ-8-フルオロ-7-(3-ヒドロキシナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
tert-ブチル(S)-4-(7-ブロモ-6-クロロ-8-フルオロ-2-((1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)ピペラジン-1-カルボキシレート(430mg、0.769mmol)、4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ナフタレン-2-オール(249mg、0.923mmol)、Na
2CO
3(245mg、2.308mmol)/1,4-ジオキサン(10mL)および水(2mL)の懸濁液をN
2で5分間脱気し、次いでテトラキス(トリフェニルホスフィン)パラジウム(0)(178mg、0.154mmol)を1度に加えた。得られた混合物をN
2で脱気し、次いでマイクロ波により95℃で1時間加熱した。反応をRTに冷却し、濾過し、濾過ケーキをジオキサン(2mLx3)で洗浄した。濾液および洗浄液を合わせて濃縮した。得られた残渣をシリカ(24g、溶出: 0~10%MeOH/DCM(0.5%TEA含有))で精製し、tert-ブチル4-(6-クロロ-8-フルオロ-7-(3-ヒドロキシナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)ピペラジン-1-カルボキシレート(174mg、0.280mmol、収率36.4%)を黄色固体として得た。MS(ESI)m/z 622.4 [M+1]
+
中間体1D: 6-クロロ-8-フルオロ-7-(3-ヒドロキシナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4(3H)-オンの製造
tert-ブチル4-(6-クロロ-8-フルオロ-7-(3-ヒドロキシナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)ピペラジン-1-カルボキシレート(400mg、0.643mmol)/EtOH(5mL)および水(0.5mL)の溶液を水酸化ナトリウム(1.286mL、1.286mmol)で処理し、反応を50℃で16時間撹拌した。この混合物を真空濃縮した。得られた残渣をフラッシュシリカゲルクロマトグラフィー(溶出: 0~10%MeOH/DCM(0.5%TEA含有)で精製し、所期の化合物(174mg、0.383mmol、収率59.6%)を白色固体として得た。MS(ESI)m/z: 454.1 [M+H]
+
中間体1E: tert-ブチル9-(6-クロロ-8-フルオロ-7-(3-ヒドロキシナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-3-カルボキシレートの製造
6-クロロ-8-フルオロ-7-(3-ヒドロキシナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-オール(10mg、0.022mmol)、tert-ブチル3,9-ジアザビシクロ[4.2.1]ノナン-3-カルボキシレート(5.98mg、0.026mmol)およびDIEA(11.54μL、0.066mmol)/DCM(2mL)の混合物にBOP(12.18mg、0.028mmol)を加え、反応を16時間室温で撹拌した。反応をNaHCO
3水溶液(4mL)でクエンチし、DCM(5mLx3)で抽出した。有機層を合わせて食塩水で洗浄し、Na
2SO
4で乾燥し、濾過し、減圧濃縮した。得られた残渣をTHF(2mL)に溶解し、TBAF(88μL、0.088mmol)を加えた。この混合物を室温で15分間撹拌し、この反応混合物を濃縮し、得られた残渣をシリカ(4g、溶出: 0~10%MeOH/DCM(0.5%TEA含有))で精製し、1E(12mg、0.018mmol、収率82%)を得た。MS(ESI)m/z: 662.7[M+H]
+
実施例1-1: 4-(4-(3,9-ジアザビシクロ[4.2.1]ノナン-9-イル)-6-クロロ-8-フルオロ-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-7-イル)ナフタレン-2-オールの製造
tert-ブチル9-(6-クロロ-8-フルオロ-7-(3-ヒドロキシナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-3-カルボキシレート(12mg、0.018mmol)を30%TFA/DCM(1mL)を用いて室温で30分間処理した。次いでこの反応混合物を濃縮し、粗製物質を分取HPLC(条件:カラム: XBridge C18、200mmx19mm、粒子径: 5μm; 移動相A: 5:95 アセトニトリル:水(酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル:水(酢酸アンモニウム含有); グラジエント: 14%Bで0分間溶出後、14~54%Bで20分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分;カラム温度: 25℃)で精製した。所期の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥した。得られた物質をさらに分取HPLC(条件:カラム: XBridge C18、200mmx19mm、粒子径: 5μm; 移動相A: 5:95 アセトニトリル:水(0.05%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル:水(0.05%トリフルオロ酢酸含有); グラジエント: 2%Bで0分間溶出後、2~42%Bで20分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分;カラム温度: 25℃)で精製した。所期の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、実施例1-1(1.3mg、1.6μmol、収率9.1%)を得た。MS(ESI)m/z 562.2 [M+1]
+;
1H NMR(500MHz、DMSO-d
6) δ 8.28-7.93(m, 1H), 7.83-7.61(m, 1H), 7.54-7.35(m, 1H), 7.27-7.18(m, 2H), 7.11-6.93(m, 2H), 5.49-5.11(m, 1H), 4.79-4.47(m, 2H), 4.29-4.06(m, 1H), 3.23-3.09(m, 1H), 2.98-2.76(m, 2H), 2.72(s, 3H), 2.65-2.56(m, 1H), 2.33-2.08(m, 2H), 2.01-0.67(m, 11H)
表1の実施例は、実施例1-1に記載の手順に従って適当な出発物質から製造した。
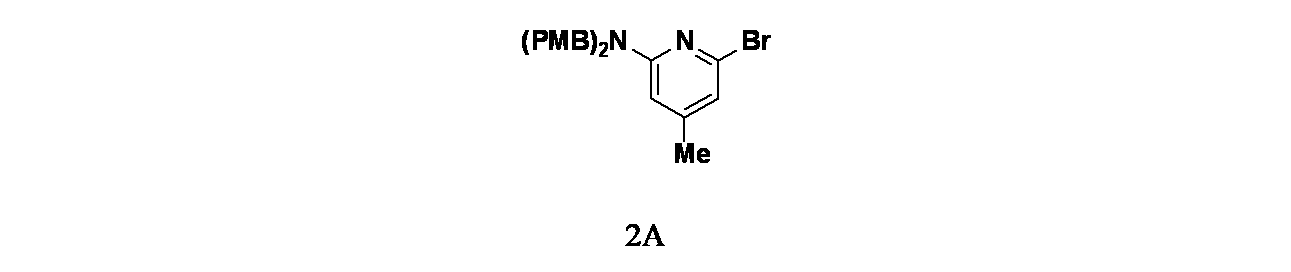
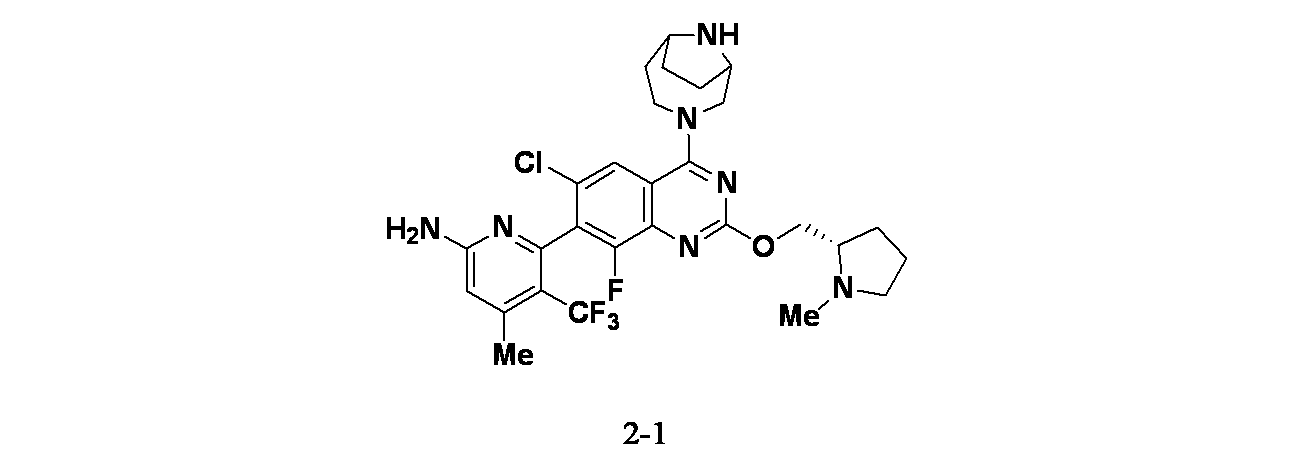
実施例2-1
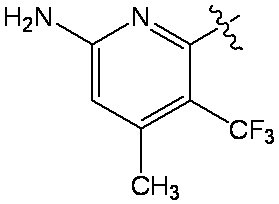
6-(6-クロロ-4-{3,9-ジアザビシクロ[4.2.1]ノナン-3-イル}-8-フルオロ-2-{[(2S)-1-メチルピロリジン-2-イル]メトキシ}キナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
中間体2A: 6-ブロモ-N,N-ビス(4-メトキシベンジル)-4-メチルピリジン-2-アミンの製造
6-ブロモ-4-メチルピリジン-2-アミン(1g、5.35mmol)/DMF(20mL)の溶液に、NaH(0.64g、16mmol、60%)を0℃で加え、0℃で0.5時間撹拌した。次いで1-(クロロメチル)-4-メトキシベンゼン(2.1g、13.4mmol)を加え、この混合物を0℃で1.5時間撹拌した。反応を飽和NH
4Cl(20mL)でクエンチし、酢酸エチル(20mLx3)で抽出し、有機層を合わせて食塩水(50mL)で洗浄し、無水Na
2SO
4で乾燥した。この混合物を濾過し、濾液を減圧濃縮した。得られた残渣をカラムクロマトグラフィー(シリカゲル、石油エーテル:酢酸エチル=5:1)で精製し、6-ブロモ-N,N-ビス(4-メトキシベンジル)-4-メチルピリジン-2-アミン(2g、4.68mmol、収率87.5%)を無色の油状物として得た。
1H NMR(400MHz、CDCl
3) δ 7.16(d, J=8.8Hz, 4H), 6.88-6.84(m, 4H), 6.60(s, 1H), 6.16(s, 1H), 4.64(s, 4H), 3.80(s, 6H), 2.13(s, 3H)
中間体2B: (6-(ビス(4-メトキシベンジル)アミノ)-4-メチルピリジン-2-イル)ボロン酸の製造
6-ブロモ-N,N-ビス(4-メトキシベンジル)-4-メチルピリジン-2-アミン(1000mg、2.34mmol)、ビス(ピナコラト)ジボロン(832.5mg、3.28mmol)、および(1,1'-ビス(ジフェニルホスフィノ)フェロセン)ジクロロパラジウム(II)(171mg、0.23mmol)/1,4-ジオキサン(20mL)の溶液に、KOAc(459.32mg、4.68mmol)を加えた。窒素雰囲気下、この混合物を90℃で5時間撹拌し、反応混合物を濾過した。得られた粗製生成物:(6-(ビス(4-メトキシベンジル)アミノ)-4-メチルピリジン-2-イル)ボロン酸(918mg、2.34mmol、粗製物)/1,4-ジオキサン(20mL)を含む濾液を精製せずに次のステップに用いた。MS(ESI)m/z 393.3 [M+1]
+
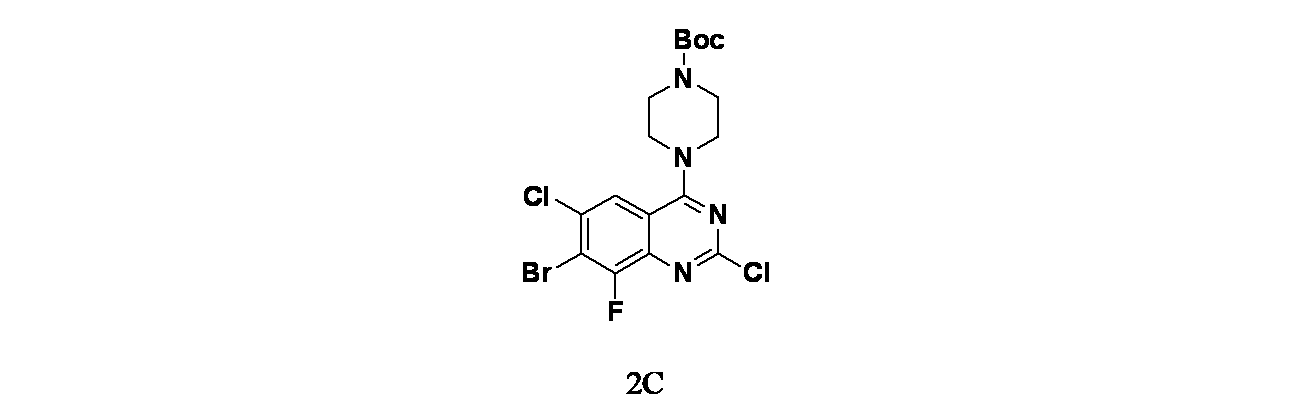
中間体2C: tert-ブチル4-(7-ブロモ-2,6-ジクロロ-8-フルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
7-ブロモ-2,4,6-トリクロロ-8-フルオロキナゾリン(1g、3.03mmol)およびDIPEA(1.32mL、7.57mmol)/THF(15mL)の溶液に、窒素雰囲気下、tert-ブチルピペラジン-1-カルボキシレート(0.56g、3.03mmol)を加えた。この反応混合物を25℃で2時間撹拌し、混合物を次いで濃縮した。得られた残渣を酢酸エチル(60mL)で希釈し、水(30mLx2)および食塩水(50mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥し、濾過し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(シリカゲル、石油エーテル:酢酸エチル=10:1~4:1)で精製し、tert-ブチル4-(7-ブロモ-2,6-ジクロロ-8-フルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(1.33g、2.77mmol、収率91.5%)を黄色固体として得た。MS(ESI)m/z 481.0 [M+3]
+;
1H NMR(400MHz、CDCl
3) δ 7.77(d, J=1.6Hz, 1H), 3.93-3.84(m, 4H), 3.72-3.61(m, 4H), 1.50(s, 9H)
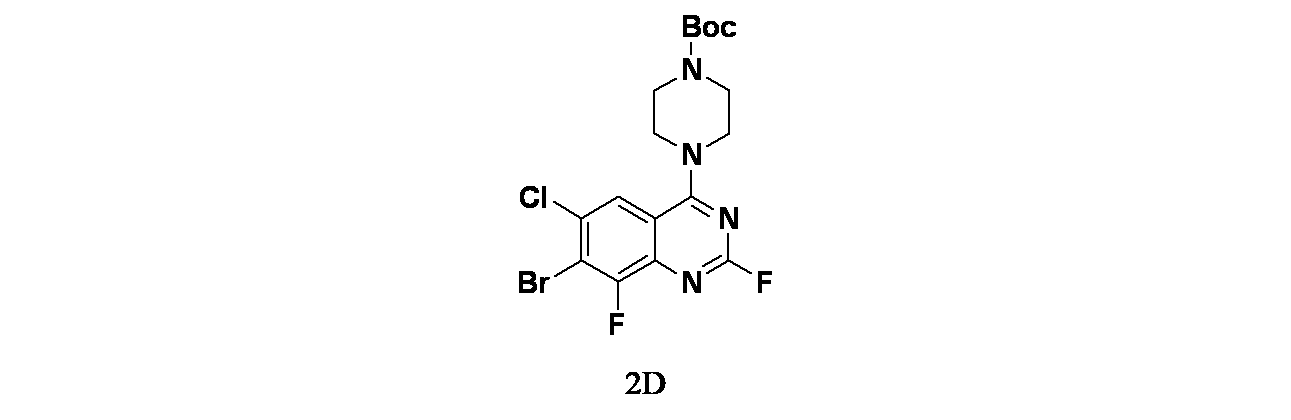
中間体2D: tert-ブチル4-(7-ブロモ-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
tert-ブチル4-(7-ブロモ-2,6-ジクロロ-8-フルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(1000mg、2.08mmol)およびフッ化カリウム(2420mg、41.65mmol)/DMA(10mL)の溶液を窒素雰囲気下、110℃で12時間撹拌した。この反応混合物を水(30mL)でクエンチし、EtOAc(30mLx3)で抽出した。有機層を合わせて食塩水(30mLx3)で洗浄し、無水Na
2SO
4で乾燥した。この混合物を濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(シリカゲル、石油エーテル:酢酸エチル=20:1~3:1)で精製し、tert-ブチル4-(7-ブロモ-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(730mg、1.57mmol、収率75.6%)を黄色固体として得た。MS(ESI)m/z 463.1 [M+1]
+
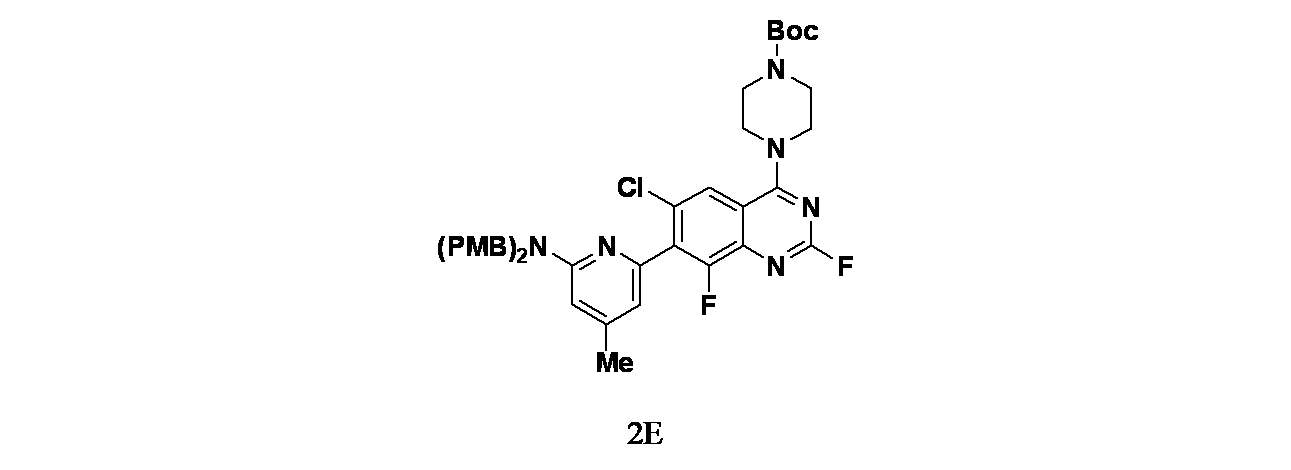
中間体2E: tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
tert-ブチル4-(7-ブロモ-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(600mg、1.29mmol)、(6-(ビス(4-メトキシベンジル)アミノ)-4-メチルピリジン-2-イル)ボロン酸(756mg、1.93mmol)、(1,1'-ビス(ジフェニルホスフィノ)フェロセン)ジクロロパラジウム(II)(104mg、0.14mmol)およびリン酸カリウム(548mg、2.59mmol)/1,4-ジオキサン(20mL)および水(2mL)の溶液を窒素雰囲気下、60℃で12時間撹拌し、混合物を濾過した。濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(シリカゲル、石油エーテル:酢酸エチル=10:1~3:1)で精製し、tert-ブチル4-(7-(4-(ビス(4-メトキシベンジル)アミノ)-6-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(600mg、0.82mmol、収率63.4%)を黄色の油状物として得た。MS(ESI)m/z 731.4 [M+1]
+;
1H NMR(400MHz、CDCl
3) δ 7.76(d, J=1.2Hz, 1 H), 7.18(d, J=8.8Hz, 4 H), 6.85(d, J=8.8Hz, 4H), 6.59(s, 1H), 6.37(s, 1H), 4.69(s, 4H), 3.98-3.87(m, 4H), 3.80(s, 6H), 3.69-3.65(m, 4H), 2.27(s, 3H), 1.51(s, 9H)
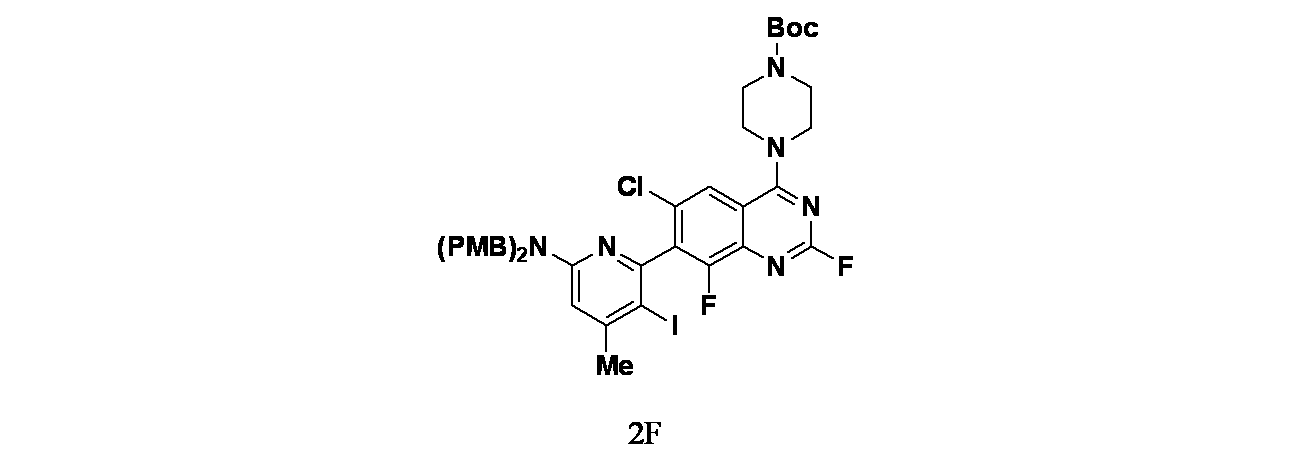
中間体2F: tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-3-ヨード-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
tert-ブチル4-(7-(4-(ビス(4-メトキシベンジル)アミノ)-6-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(800mg、1.09mmol)、TosOH(5mg、0.05mmol)およびNIS(1200mg、5.33mmol)/DMF(10mL)の溶液を25℃で12時間撹拌した。この反応混合物を水(15mL)およびEtOAc(15mL)で希釈し、混合物をEtOAc(30mLx3)で抽出した。有機層を合わせて食塩水(30mLx3)で洗浄し、無水硫酸ナトリウムで乾燥し、濾過し、減圧濃縮した。得られた残渣をカラムクロマトグラフィー(シリカゲル、石油エーテル:酢酸エチル=10:1~3:1)で精製し、tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-3-ヨード-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(400mg、0.467mmol、収率42.7%)を黄色固体として得た。MS(ESI)m/z 857.2 [M+1]
+;
1H NMR(400MHz、CDCl
3) δ 7.82(d, J=1.2Hz, 1H), 7.17(d, J=8.4Hz, 4H), 6.86(d, J=8.4Hz, 4H), 6.48(s, 1H), 4.76-4.65(m, 2H), 4.62-4.50(m, 2H), 4.01-3.92(m, 4H), 3.82(s, 6H), 3.72-3.63(m, 4H), 2.38(s, 3H), 1.52(s, 9H)
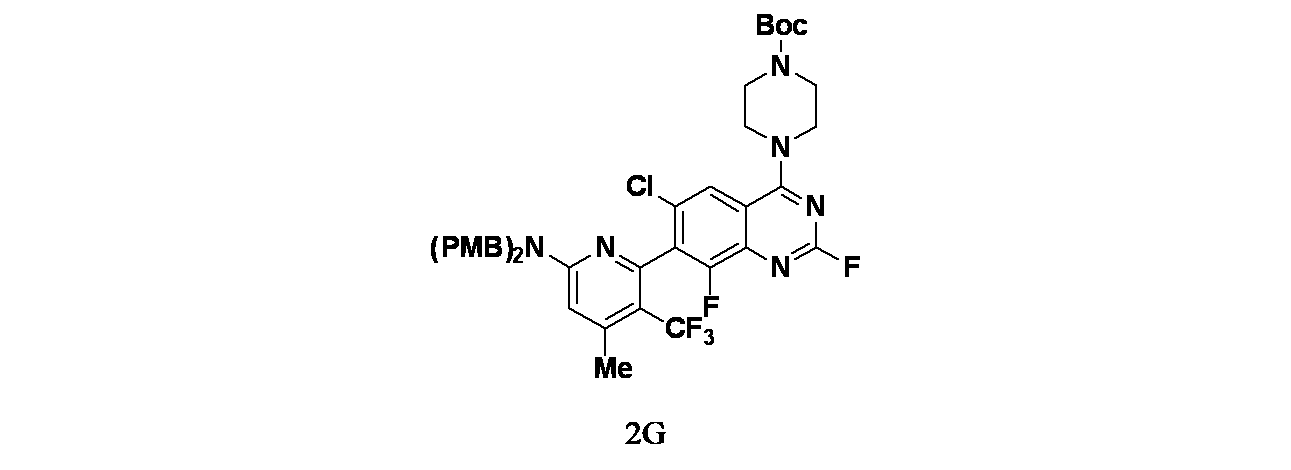
中間体2G: tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-3-ヨード-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(400mg、0.4700mmol)、2,2-ジフルオロ-2-フルオロスルホニル酢酸メチル(1345mg、7mmol)およびCuI(267mg、1.4mmol)/DMA(10mL)の混合物を窒素雰囲気下、80℃で5時間撹拌した。この反応混合物を次いで室温に冷却し、CuI(267mg、1.4mmol)を追加し、2,2-ジフルオロ-2-フルオロスルホニル酢酸メチル(1345mg、7mmol)を混合物に添加した。窒素雰囲気下、この反応混合物を80℃でさらに12時間撹拌した。混合物をEtOAc(50mL)で希釈し、濾過した。濾液を食塩水(30mLx3)で洗浄し、無水Na
2SO
4で乾燥し、濾過した。濾液を減圧濃縮し、得られた残渣をカラムクロマトグラフィー(シリカゲル、石油エーテル:酢酸エチル=10:1~3:1)で精製し、tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(270mg、0.34mmol、収率72.4%)を黄色固体として得た。MS(ESI)m/z 799.0 [M+1]
+
中間体2H: tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
(2S)-1-メチルピロリジン-2-イルメタノール(97.6mg、0.85mmol)/THF(10mL)の溶液に、NaH(81mg、2.03mmol、60%)を0℃で加え、0℃で0.5時間撹拌した。tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(270mg、0.34mmol)/THF(5mL)を加え、この混合物を0℃で1時間撹拌した。この反応混合物を次いで飽和NH
4Cl(20mL)でクエンチし、EtOAc(20mLx3)で抽出した。有機層を合わせて食塩水(30mL)で洗浄し、無水Na
2SO
4で乾燥した。この混合物を濾過し、濾液を減圧濃縮した。得られた残渣をカラムクロマトグラフィー(シリカゲル、DCM:MeOH=10:1)で精製し、tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(250mg、0.28mmol、収率82.7%)を白色固体として得た。MS(ESI)m/z 894.5 [M+1]
+
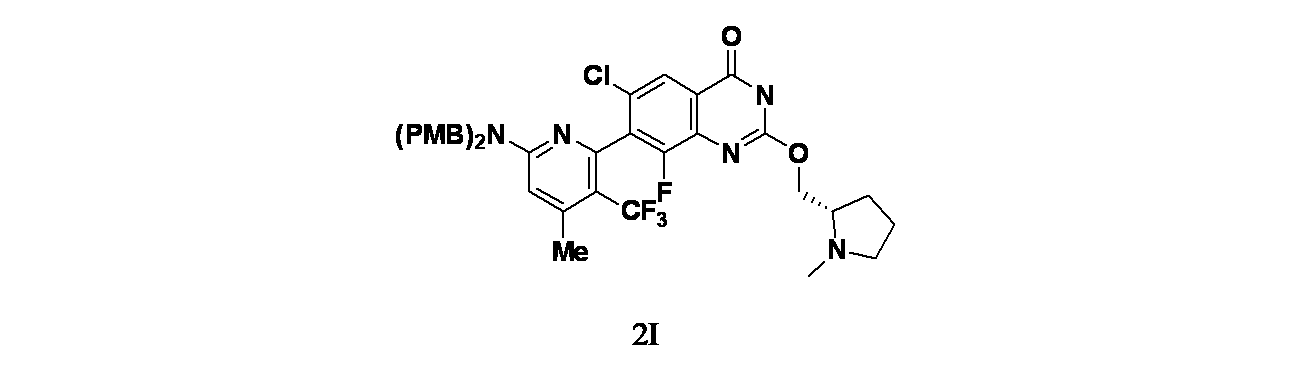
中間体2I: 7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4(3H)-オンの製造
tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(250mg、0.28mmol)およびNaOH(224mg、5.59mmol)/エタノール(30mL)および水(10mL)の混合物を45℃で3日間撹拌した。混合物を2N HClでpH=6~7とし、クエンチした。この混合物を減圧濃縮し、EtOHを除去した。得られた残渣をDCM(20mLx3)で抽出した。有機層を合わせて食塩水(30mL)で洗浄し、無水Na
2SO
4で乾燥した。この混合物を濾過し、濾液を減圧濃縮し、7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4(3H)-オン(200mg、0.28mmol、収率98.5%)を薄黄色固体として得た。MS(ESI)m/z 726.3 [M+1]
+;
1H NMR(400MHz、CDCl
3) δ 8.07(s, 1H), 7.14(d, J=8.4Hz, 4H), 6.85(d, J=8.4Hz, 4H), 6.41(s, 1H), 4.98-4.65(m, 4H), 4.59-4.49(m, 2H), 3.80(s, 6H), 3.55-3.38(m, 1H), 2.90(d, J=8.0Hz, 3H), 2.41(s, 3H), 2.31-2.21(m, 2H), 2.14-2.03(m, 2H), 1.37-1.19(m, 2H)
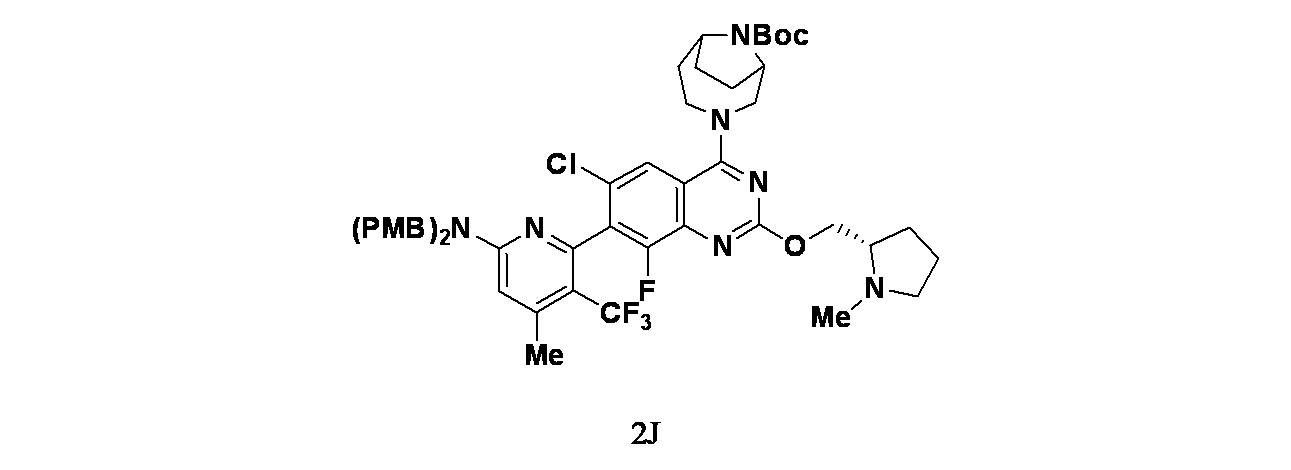
中間体2J: tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4(3H)-オン(50mg、0.0700mmol)およびtert-ブチル3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(75mg、0.3300mmol)/DCM(1mL)の溶液に、DIEA(0.07mL、0.7800mmol)およびBOP(105mg、0.4100mmol)を加え、25℃で12時間撹拌した。この反応混合物を水(20mL)で希釈し、次いでジクロロメタン(15mLx3)で抽出した。有機層を合わせて食塩水(30mL)で洗浄し、無水硫酸ナトリウムで乾燥し、濾過し、減圧濃縮した。得られた残渣を分取HPLC(添加剤: TFA、機器: ACSWH-GX-N;カラム: Phenomenex Synergi C18 150x25mmx10um; 移動相A: H
2O(0.1%TFA)および移動相B: アセトニトリル; グラジエント: 10分かけて直線的にB56%~86%で溶出; 流速: 25mL/分;カラム温度: R.T.; 波長: 220nm、254nm)で精製し、所期の生成物:tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(40mg、0.043mmol)を黄色固体として得た。MS(ESI)m/z: 934.7 [M+H]
+
実施例2-1
6-(6-クロロ-4-{3,9-ジアザビシクロ[4.2.1]ノナン-3-イル}-8-フルオロ-2-{[(2S)-1-メチルピロリジン-2-イル]メトキシ}キナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,6-ジアザビシクロ[3.2.2]ノナン-6-カルボキシレート(45mg、0.05mmol)/TFA(4mL、52.24mmol)の溶液を50℃で4時間撹拌し、この反応混合物を減圧濃縮した。得られた残渣を分取HPLC(添加剤: ギ酸、機器: GX-c;カラム: Phenomenex luna C18 150x25mm、10um; 移動相A: H
2O(0.225%FA)および移動相B: アセトニトリル; グラジエント: 10分かけて直線的にB3%~33%で溶出; 流速: 25mL/分;カラム温度: R.T.; 波長: 220nm、254nm)で精製し、所期の生成物:6-(4-(3,6-ジアザビシクロ[3.2.2]ノナン-3-イル)-6-クロロ-8-フルオロ-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン(17.98mg、0.028mmol)を黄色固体として得た。MS(ESI)m/z: 594.2 [M+H]
+1H NMR(400MHz、CD
3OD) δ 8.51(s, 2H), 7.93(s, 1H), 6.62(s, 1H), 4.76-4.69(m, 1H), 4.66-4.58(m, 1H), 4.54-4.42(m, 1H), 4.13-3.98(m, 2H), 3.97-3.88(m, 1H), 3.65-3.50(m, 4H), 3.49-3.43(m, 1H), 3.05-2.97(m, 1H), 2.92(s, 3H), 2.55-2.48(m, 1H), 2.45(s, 3H), 2.36-2.27(m, 1H), 2.13-2.05(m, 3H), 2.04-1.97(m, 2H), 1.94-1.81(m, 2H)
表2の実施例は、実施例2-1に記載の手順に従って適当な出発物質から製造した。
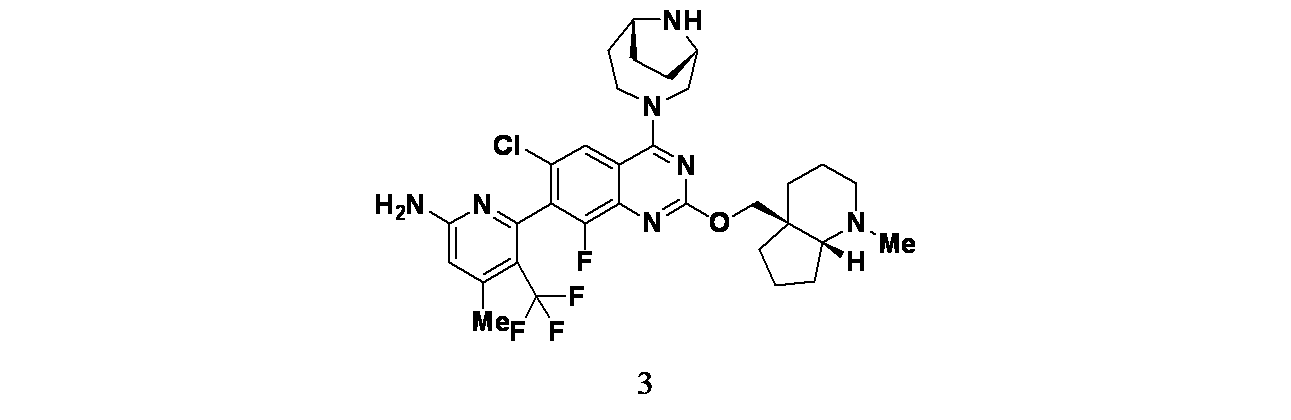
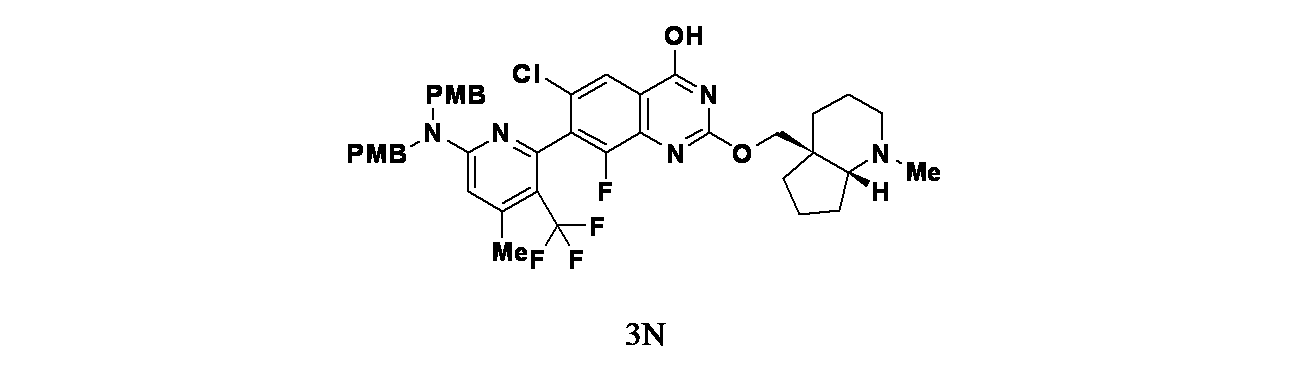
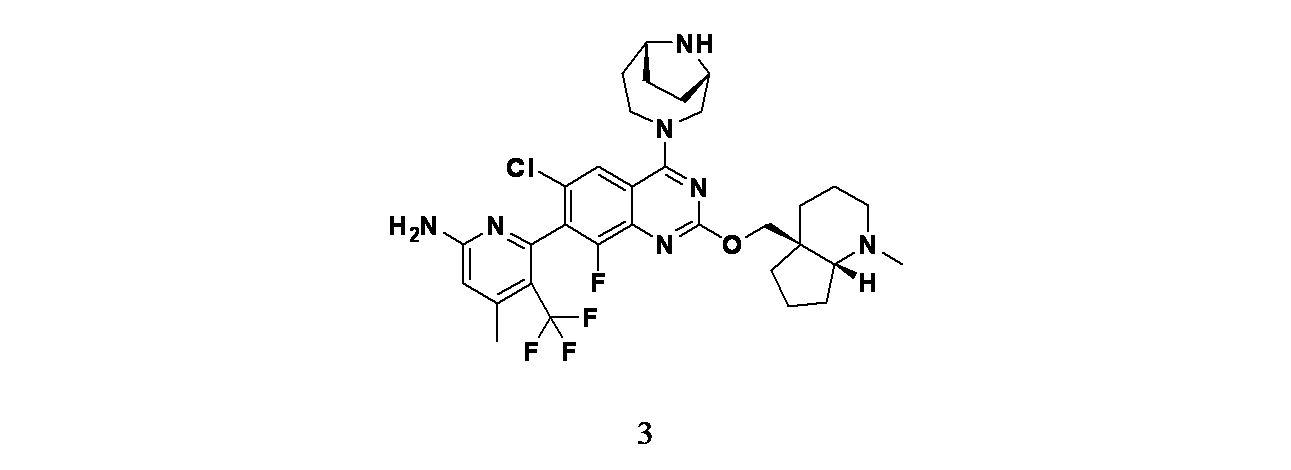
実施例3
6-(2-{[(4aS,7aR)-1-メチル-オクタヒドロ-1H-シクロペンタ[b]ピリジン-4a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
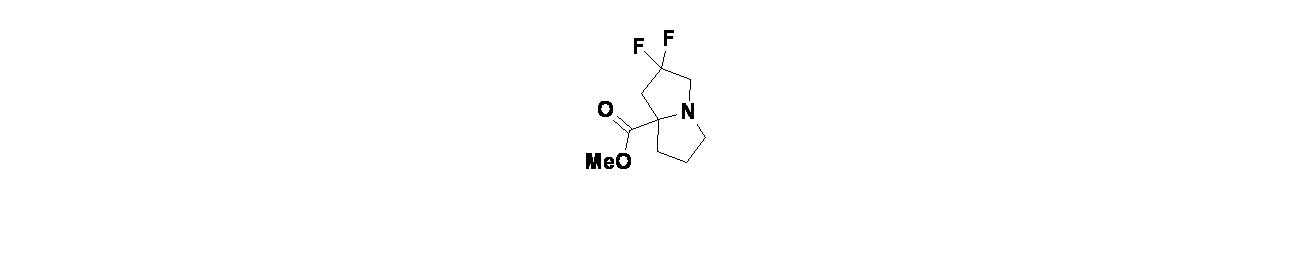
中間体3A: オクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-カルボン酸エチルの製造
中間体3Aを、Molecules 2017, 22, 827に記載の化合物25aの方法に従って製造した。
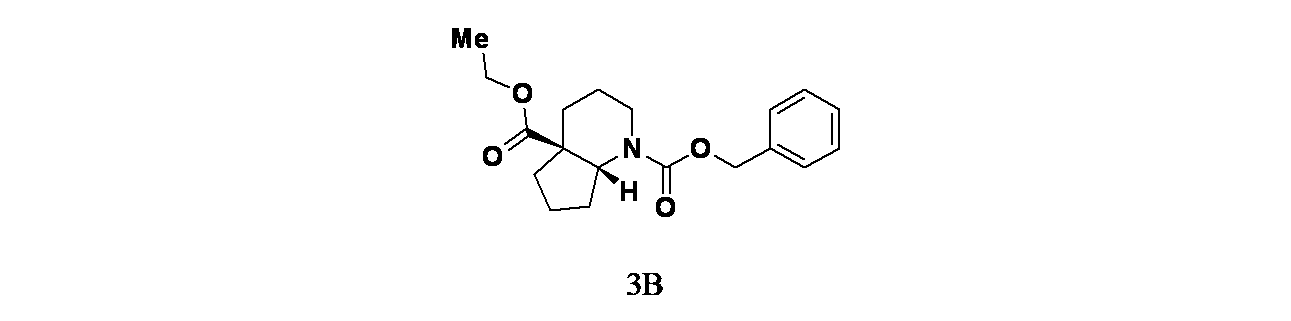
中間体3B: 1-ベンジル4a-エチル(4aS,7aR)-ヘキサヒドロ-1H-シクロペンタ[b]ピリジン-1,4a(2H)-ジカルボキシレートの製造
オクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-カルボン酸エチル(1.7g、8.62mmol)およびTEA(2.40mL、17.23mmol)/THF(10mL)の溶液に、N-(ベンジルオキシカルボニル)スクシンイミド(1.718g、6.89mmol)を加え、混合物を室温で18時間撹拌した。この混合物を次いでEtOAc(15mL)で希釈し、飽和炭酸水素ナトリウム水溶液(2x15mL)で洗浄した。酢酸エチル層を硫酸ナトリウムで乾燥し、濾過し、濃縮した。得られた粗製生成物についてISCOフラッシュクロマトグラフィー(シリカゲル、グラジエント=DCM:20%MeOH/DCM 100:0~50:50)を行い、1-ベンジル4a-エチルヘキサヒドロ-1H-シクロペンタ[b]ピリジン-1,4a(2H)-ジカルボキシレート(2.20g、6.64mmol、収率77%)を得た。1-ベンジル4a-エチルヘキサヒドロ-1H-シクロペンタ[b]ピリジン-1,4a(2H)-ジカルボキシレート(2.20g)についてSFCキラル分離[カラム: Cellulose-4(5*25cm、5μm)、方法=CO
2/IPA:ヘプタン(1:3、0.1%アンモニア水含有)、320mL/分]を行い、1-ベンジル4a-エチル(4aS,7aR)-ヘキサヒドロ-1H-シクロペンタ[b]ピリジン-1,4a(2H)-ジカルボキシレート(640mg、1.835mmol、収率21.29%)を得た。LCMS(ESI)m/z: 332.3 [M+H]
+; LC保持時間: 1.05分(Waters Acquity UPLC BEH C18、2.1x50mm、粒子径:1.7μm; 移動相A: 水(0.05%TFA含有); 移動相B: ACN(0.05%TFA含有); グラジエント: 2~98%Bで1分かけて溶出後、次いで98%Bで0.5分間溶出; 流速: 0.8mL/分; 検出: MSおよびUV(220nm));
1H NMR(499MHz、クロロホルム-d) δ 7.41-7.28(m, 5H), 5.17 (br s, 2H), 4.13 (br d, J=6.4Hz, 2H), 2.86 (br s, 1H), 2.15 (br d, J=10.8Hz, 1H), 2.03-1.90(m, 1H), 1.90-1.74(m, 4H), 1.73-1.63(m, 1H), 1.58-1.42(m, 3H), 1.27-1.14(m, 4H), 0.98-0.68(m, 1H)
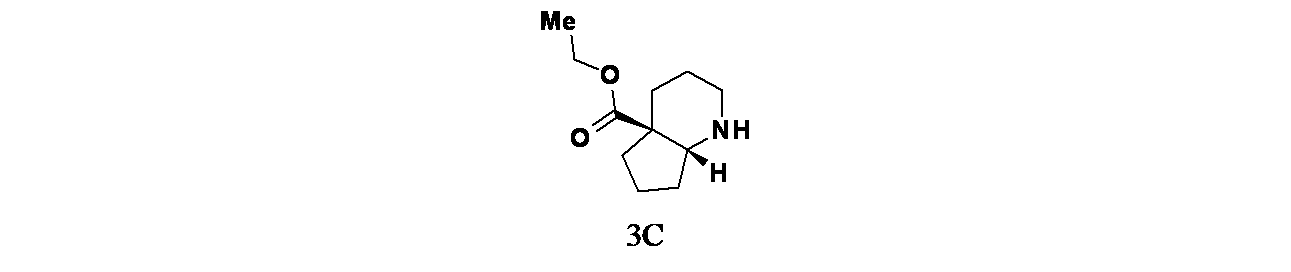
中間体8C: (4aS,7aR)-オクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-カルボン酸エチルの製造
1-ベンジル4a-エチル(4aS,7aR)-ヘキサヒドロ-1H-シクロペンタ[b]ピリジン-1,4a(2H)-ジカルボキシレート(640mg、1.931mmol)および10%Pd-C(103mg、0.097mmol)/MeOH(10mL)の混合物に、水素雰囲気下(1atm)で18時間水素を添加した。Pd/Cを濾過し、濾液を濃縮して、粗製(4aS,7aR)-オクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-カルボン酸エチル(385mg、1.854mmol、収率96%)を透明油状物として得た。
1H NMR(499MHz、CDCl
3) δ 4.17(dtt, J=10.6, 7.1, 3.6Hz, 2H), 3.57(t, J=6.1Hz, 1H), 2.90(ddd, J=13.0, 7.7, 3.7Hz, 1H), 2.71(ddd, J=13.0, 7.0, 3.6Hz, 1H), 2.01-1.92(m, 2H), 1.84-1.62(m, 7H), 1.60-1.40(m, 2H), 1.28(t, J=7.1Hz, 3H)
中間体3D: (4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-カルボン酸エチルの製造
(4aS,7aR)-オクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-カルボン酸エチル(385mg、1.952mmol)およびホルムアルデヒド溶液(37wt%、H
2O(176mg、5.85mmol)/MeOH(5.0mL)の溶液に、シアノ水素化ホウ素ナトリウム(123mg、1.952mmol)を加え、混合物を室温で18時間撹拌した。この混合物を次いで濃縮した。得られた残渣をEtOAc(5mL)で希釈し、飽和炭酸ナトリウム水溶液(2x5mL)で洗浄した。酢酸エチル層を硫酸ナトリウムで乾燥し、濾過し、濃縮し、粗製(4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-カルボン酸エチル(380mg、1.798mmol、収率92%)を得た。
1H NMR(499MHz、CDCl
3) δ 4.24-4.12(m, 2H), 3.29(t, J=6.4Hz, 1H), 2.60-2.51(m, 1H), 2.36-2.28(m, 3H), 2.00-1.88(m, 2H), 1.83-1.60(m, 8H), 1.56-1.40(m, 1H), 1.32-1.26(m, 3H)
中間体3E: ((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メタノールの製造
(4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-カルボン酸エチル(380mg、1.798mmol)/無水THF(2.0mL)の溶液に、リチウムアルミニウムハイドライド(1.0M、THF溶液、4496μL、4.50mmol)を加え、混合物を室温で18時間撹拌した。この混合物に食塩水(0.3mL)を滴下して加えた。EtOAc(5.0mL)を次いで混合物に添加した。この反応混合物を濾過し、濾液を濃縮し、粗製((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メタノール(327mg、1.739mmol、収率97%)を得た。
1H NMR(499MHz、クロロホルム-d) δ 3.69-3.62(m, 2H), 2.87(t, J=7.6Hz, 1H), 2.51 (td, J=11.1, 3.4Hz, 1H), 2.43-2.35(m, 1H), 2.30(s, 3H), 2.01-1.85(m, 2H), 1.82-1.74(m, 1H), 1.68-1.52(m, 6H), 1.47-1.42(m, 1H), 1.39-1.33(m, 1H)
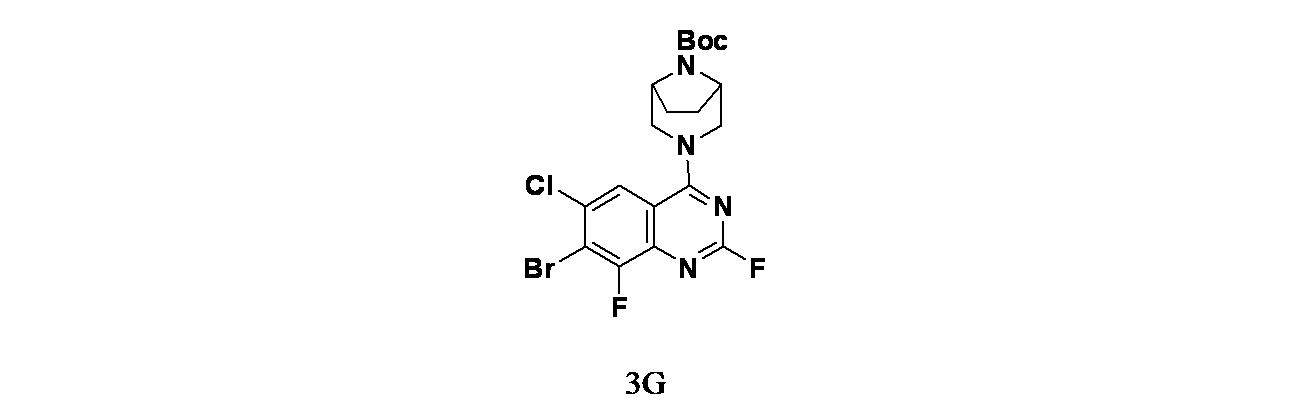
中間体3F: tert-ブチル3-(7-ブロモ-2,6-ジクロロ-8-フルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
7-ブロモ-2,4,6-トリクロロ-8-フルオロキナゾリン(300mg、3.03mmol)/ジオキサン(8mL)の溶液に、DIPEA(0.476mL、2.72mmol)およびtert-ブチル3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(193mg、0.908mmol)を加えた。得られた混合物を25℃で2時間撹拌し、混合物を次いで濃縮した。得られた残渣を酢酸エチル(50mL)で希釈し、水(30mLx2)および食塩水(50mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥し、濾過し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(12g、ISCOカラム、MeOH/DCM、0~5%、20分)で精製し、tert-ブチル3-(7-ブロモ-2,6-ジクロロ-8-フルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(415mg、0.82mmol、収率90%)を白色固体として得た。MS(ESI)m/z 507.0 [M+1]
+;
1H NMR(499MHz、DMSO-d
6) δ 8.10(d, J=1.9Hz, 1H), 4.38 (br d, J=10.6Hz, 2H), 4.25 (br s, 2H), 3.66(m, 2H) 1.79(m, 2H), 1.62(m, 2H), 1.47(s, 9H)
中間体3G: tert-ブチル3-(7-ブロモ-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
tert-ブチル3-(7-ブロモ-2,6-ジクロロ-8-フルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(1g、3.03mmol)/DMA(80mL)の脱気した溶液に、フッ化セシウム(5.25g、34.6mmol)を加えた。この混合物を窒素で10分間脱気し、密封したチューブ中、88℃で5時間を加熱した。水(200mL)および酢酸エチル(150mL)を加え、この混合物を15分間撹拌した。分離した水層を酢酸エチル(2X100mL)で抽出し、有機層を合わせて無水Na
2SO
4で乾燥し、濾過し、濃縮した。得られた残渣をフラッシュカラムクロマトグラフィー(溶離剤: 15~25%酢酸エチル/石油エーテル)で精製し、tert-ブチル3-(7-ブロモ-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(4.7g、8.77mmol、収率63.4%)を淡黄色固体として得た。MS(ESI)m/z 489.0 [M+1]
+
中間体3H: tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
実施例3G(2.00g、4.08mmol)/無水1,4-ジオキサン(20mL)の脱気した溶液に、リン酸カリウム(1.73g、8.17mmol)、N,N-ビス(4-メトキシベンジル)-4-メチル-6-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ピリジン-2-アミン(5.8g、12.25mmol)およびPdCl
2(dppf)(149mg、0.204mmol)を加えた。この混合物を再度脱気し、80℃で48時間加熱した。反応容器を次いで周囲温度に冷却し、酢酸エチル(40mL)で希釈し、セライト
(登録商標)で濾過し、減圧濃縮し、粗製生成物を得た。得られた残渣をシリカゲルカラムクロマトグラフィー(30%酢酸エチル/石油エーテル)で精製し、tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(1.5g、1.74mmol、収率42%)を得た。
1H NMR(400MHz、CDCl
3): δ 7.76(d, J=1.6Hz, 1H), 7.20-7.18(d, J=8.8Hz, 4H), 6.86(dt, J=9.6Hz, 4H), 6.60(s, 1H), 6.38(s, 1H), 4.60(s, 3H), 4.39-4.21(m, 4H), 3.63(s, 6H), 2.29(s, 3H), 1.98-1.96(m, 6H), 1.76-1.63(m, 2H), 1.49(s, 9H)ppm; LCMS(ESI)m/z: 757.2 [M+H]
+
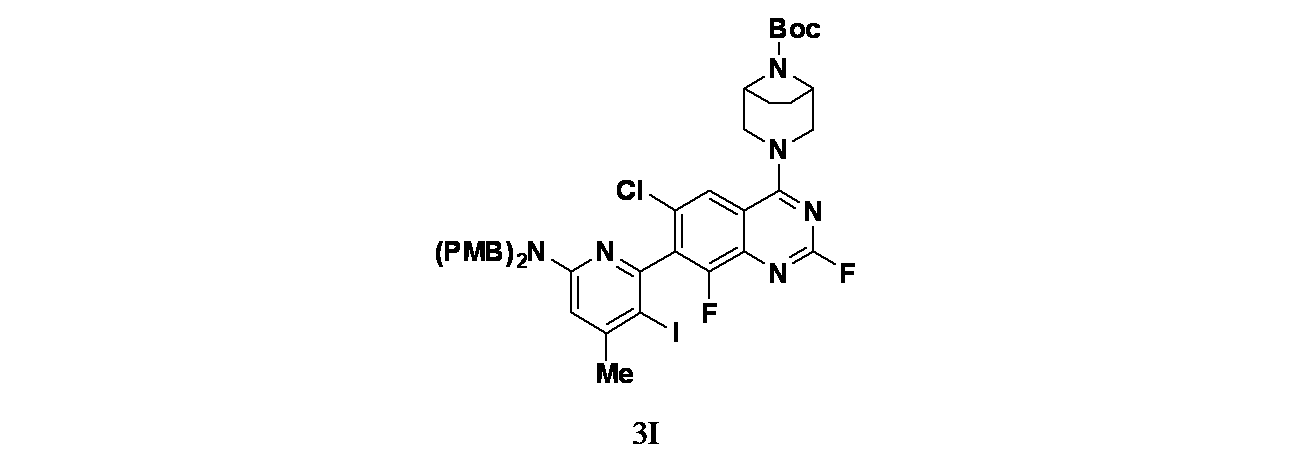
中間体3I: tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-3-ヨード-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(1.40g、1.849mmol)/無水アセトニトリル(15mL)の撹拌溶液に、窒素下0℃でN-ヨードスクシンイミド(0.42g、1.849mmol)およびトリフルオロ酢酸(0.028mL、0.370mmol)を加えた。この反応混合物を1時間かけて室温に戻した。この反応混合物を次いで飽和チオ硫酸ナトリウム水溶液(5mL)および飽和重炭酸ナトリウム水溶液(4mL)でクエンチした。混合物を酢酸エチル(3x20mL)で抽出した。有機層を合わせて無水硫酸ナトリウムで乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。得られた粗製残渣をシリカゲルカラムクロマトグラフィー(30%酢酸エチル/石油エーテル)で精製し、tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-3-ヨード-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(1.42g、1.560mmol、収率84%)を淡黄色固体として得た。LCMS(ESI)m/z: 883.3 [M+H]
+
中間体3J: tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-3-ヨード-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(1.40g、1.585mmol)/無水DMA(10mL)の撹拌溶液に、密封したチューブ中、窒素雰囲気下でヨウ化銅(I)(0.60g、3.17mmol)を加えた。この反応混合物を10分間脱気した後、2,2-ジフルオロ-2-(フルオロスルホニル)酢酸メチル(0.91g、4.76mmol)を添加し、反応混合物を90℃で12時間加熱した。この反応混合物をジエチルエーテル(20mL)および水(10mL)で希釈した。層を分離し、水層をジエチルエーテル(3x20mL)で抽出した。有機層を合わせて無水硫酸ナトリウムで乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。粗製物質をシリカゲルカラムクロマトグラフィー(30%酢酸エチル/石油エーテル)で精製し、tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(0.85g、0.630mmol、収率40%)を淡黄色固体として得た。
1H NMR(400MHz、CDCl
3): δ 7.77(s, 1H), 7.16(d, J=8.8Hz, 4H), 6.87(dt, J=9.6 and 2.8Hz, 4H), 6.43(s, 1H), 4.76-4.72(m, 2H), 4.59-4.55(m, 2H), 3.81(s, 6H), 2.43(s, 3H), 1.97-1.82(m, 4H), 1.97-1.82(m, 4H), 1.53(s, 9H)ppm; LCMS(ESI)m/z: 825.2 [M+H]
+
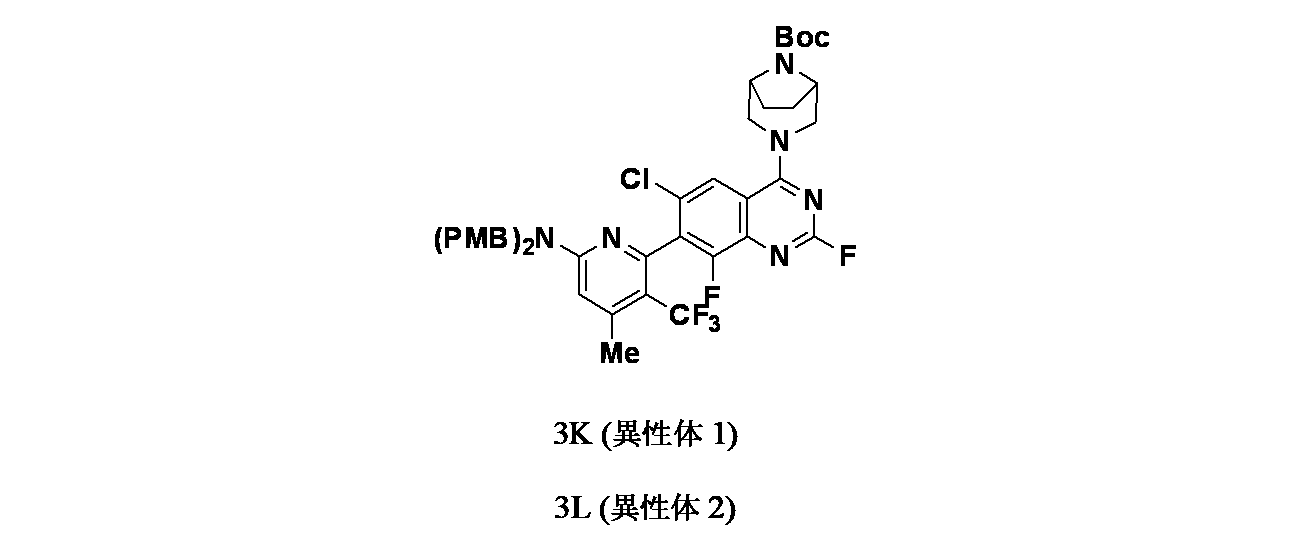
中間体3Kおよび3L: tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート
実施例3J(5.0g、6.06mmol)をSFC(カラム: Chiralpak IH(250mmx4.6x5u)、移動相: 0.25%イソプロパノール)で単一のアトロプ異性体に分離した。ピーク1は保持時間=5.85分(2.4g、2.90mmol、収率40%)、ピーク2は保持時間=9.53分(2.4g、2.90mmol、収率40%)で溶出した。
ピーク1(3K):
1H NMR(400MHz、CDCl
3): δ 7.78(s, 1H), 7.16(d, J=8.8Hz, 4H), 6.87(dt, J=9.6 and 2.8Hz, 4H), 6.43(s, 1H), 4.76-4.72(m, 2H), 4.59-4.55(m, 2H), 3.81(s, 6H), 2.43(s, 3H), 1.97-1.82(m, 4H), 1.97-1.82(m, 4H), 1.53(s, 9H)ppm; LCMS(ESI)m/z: 825.2 [M+H]
+; LCMS(ESI)m/z: 825.2 [M+H]
+; [α]
23.5(MeOH=0.10)=+96.00
ピーク2(3L):
1H NMR(400MHz、CDCl
3): δ 7.78(s, 1H), 7.16(d, J=8.8Hz, 4H), 6.87(dt, J=9.6 and 2.8Hz, 4H), 6.43(s, 1H), 4.76-4.72(m, 2H), 4.59-4.55(m, 2H), 3.81(s, 6H), 2.43(s, 3H), 1.97-1.82(m, 4H), 1.97-1.82(m, 4H), 1.53(s, 9H)ppm; LCMS(ESI)m/z: 825.2 [M+H]
+; [α]
23.3(MeOH=0.10)=-110.00
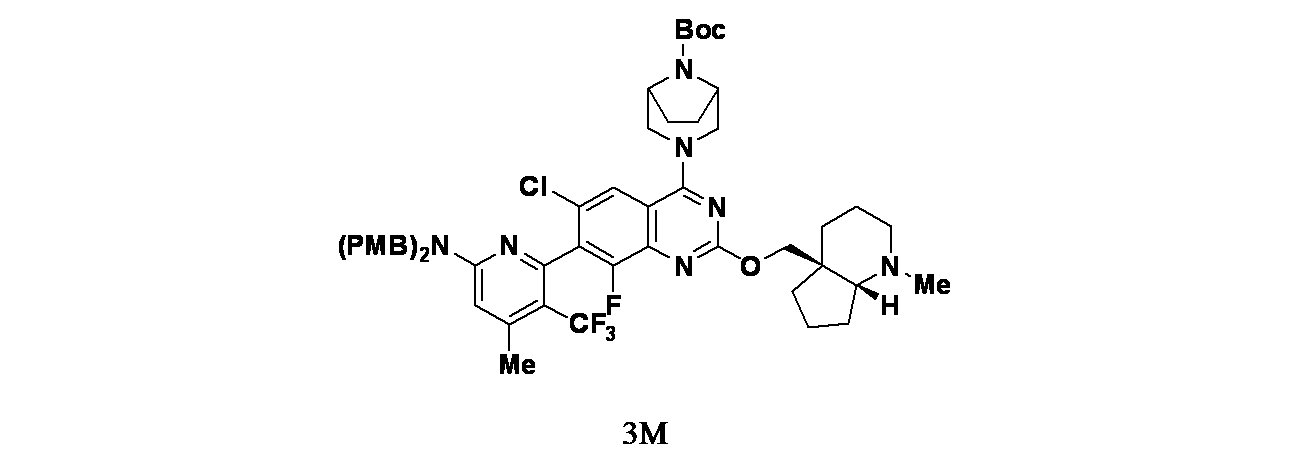
中間体3M: tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
中間体3Lおよび((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メタノール(41.0mg、0.242mmol)/無水THF(1.0mL)の溶液に、窒素下室温でLiHMDS(1.0M、THF溶液、364μL、0.364mmol)を加え、混合物を室温で18時間撹拌した。混合物をDMF(1mL)で希釈し、得られた粗製生成物を分取HPLC(Phenomenex、Luna 5μ、30x250mm、流速=30mL/分、グラジエント=20%A~100%Bを30分で溶出;A=H
2O/ACN/TFA(90:10:0.1)、B=H
2O/ACN/TFA(10:90:0.1))で精製し、tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(267mg、0.233mmol、収率96%)を得た。LCMS(ESI)m/z: 974.5 [M+H]
+ LC保持時間: 1.13分(Waters Acquity UPLC BEH C18、2.1x50mm、粒子径:1.7μm; 移動相A: 水(0.05%TFA含有); 移動相B: ACN(0.05%TFA含有); グラジエント: 2~98%Bで1分かけて溶出後、次いで98%Bで0.5分間溶出; 流速: 0.8mL/分; 検出: MSおよびUV(220nm))
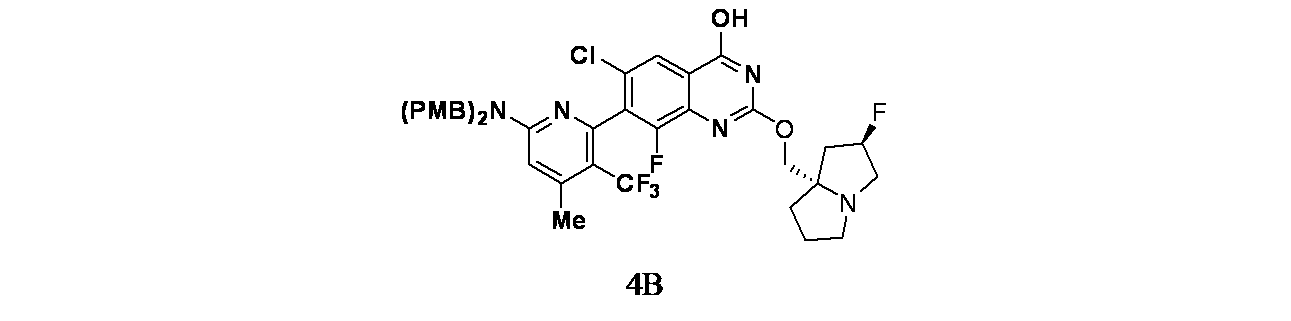
中間体3N: 7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-オールの製造
1.0M水酸化ナトリウム(2453μL、2.453mmol)およびtert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(267mg、0.245mmol)/EtOH(20mL)の混合物を65℃で3日間撹拌した。この混合物を次いで濃縮し、EtOHを除去した。混合物をEtOAc(2x15mL)で抽出し、酢酸エチル層を硫酸ナトリウムで乾燥し、濾過し、濃縮した。得られた粗製生成物を分取HPLC(Phenomenex、Luna 5 μ 30x250mm、流速=30mL/分、グラジエント=20%A~100%Bを30分で溶出;A=H
2O/ACN/TFA(90:10:0.1)、B=H
2O/ACN/TFA(10:90:0.1))で精製した。純粋なフラクションを合わせて濃縮した。次いで純粋な生成物をEtOAc(15mL)で希釈し、飽和炭酸ナトリウム水溶液(2x15mL)で洗浄した。酢酸エチル層を硫酸ナトリウムで乾燥し、濾過し、濃縮し、7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-オール(60mg、0.077mmol、収率31.4%)を黄褐色固体として得た。LCMS(ESI)m/z: 780.3 [M+H]
+; LC保持時間: 1.04分((Waters Acquity UPLC BEH C18、2.1x50mm、粒子径:1.7μm; 移動相A: 水(0.05%TFA含有); 移動相B: ACN(0.05%TFA含有); グラジエント: 2~98%Bで1分かけて溶出後、次いで98%Bで0.5分間溶出; 流速: 0.8mL/分; 検出: MSおよびUV(220nm))
実施例3: 6-(2-{[(4aS,7aR)-1-メチル-オクタヒドロ-1H-シクロペンタ[b]ピリジン-4a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミンの製造
7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-オール(25mg、0.032mmol)、tert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(10.88mg、0.048mmol)およびBOP(21.26mg、0.048mmol)/DCM(1.0mL)の溶液に、DIEA(16.79μL、0.096mmol)を加え、混合物を室温で18時間撹拌した。この混合物を次いで濃縮した。得られた粗製生成物を分取HPLC(Phenomenex、Luna 5μ、30x250mm、流速=30mL/分、グラジエント=20%A~100%Bを30分で溶出;A=H
2O/ACN/TFA(90:10:0.1)、B=H
2O/ACN/TFA(10:90:0.1))で精製し、tert-ブチル(1S,6R)-3-(7-(6-アミノ-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートを得た。tert-ブチル(1S,6R)-3-(7-(6-アミノ-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート/水(1滴)、トリエチルシラン(1滴)およびTFA(1.5mL)の混合物を40℃で18時間撹拌した。この混合物を次いで濃縮した。得られた粗製生成物を分取HPLC(Phenomenex、Luna 5μ、30x250mm、流速=30mL/分、グラジエント=20%A~100%Bを30分で溶出、A=H
2O/ACN/TFA(90:10:0.1)、B=H
2O/ACN/TFA(10:90:0.1))で精製した。純粋なフラクションをOasis MCX カチオンミックスモードポリマーカートリッジ(150mg)にロードし、カートリッジをメタノール(30mL)で洗浄し、生成物を0.1Nアンモニア/メタノール(5.0mL)で溶出した。溶離剤のアンモニアを濃縮し、得られた生成物をACN/H
2O(1:1、5mL)で凍結乾燥し、4 6-(4-((1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル)-6-クロロ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン(10.96mg、0.016mmol、収率50.1%)を白色粉末として得た。LCMS(ESI)m/z: 648.2 [M+H]
+; LC保持時間: 1.27分((Waters Acquity UPLC BEH C18、2.1x50mm、粒子径:1.7μm; 移動相A: 水(0.05%TFA含有); 移動相B: ACN(0.05%TFA含有); グラジエント: 2~98%Bで1分かけて溶出後、次いで98%Bで0.5分間溶出; 流速: 0.8mL/分; 検出: MSおよびUV(220nm));
1H NMR(499MHz、メタノール-d
4) δ 7.96(d, J=1.5Hz, 1H), 6.62(s, 1H), 4.55-4.47(m, 2H), 4.27(d, J=10.7Hz, 1H), 4.05-3.91(m, 2H), 3.84-3.77(m, 2H), 3.68(dd, J=13.5, 3.9Hz, 1H), 2.87 (br s, 1H), 2.73-2.64(m, 1H), 2.47(d, J=1.2Hz, 3H), 2.44-2.35(m, 1H), 2.33(s, 3H), 2.29-2.18(m, 1H), 2.13-1.92(m, 4H), 1.91-1.82(m, 2H), 1.81-1.64(m, 8H), 1.63-1.54(m, 1H)
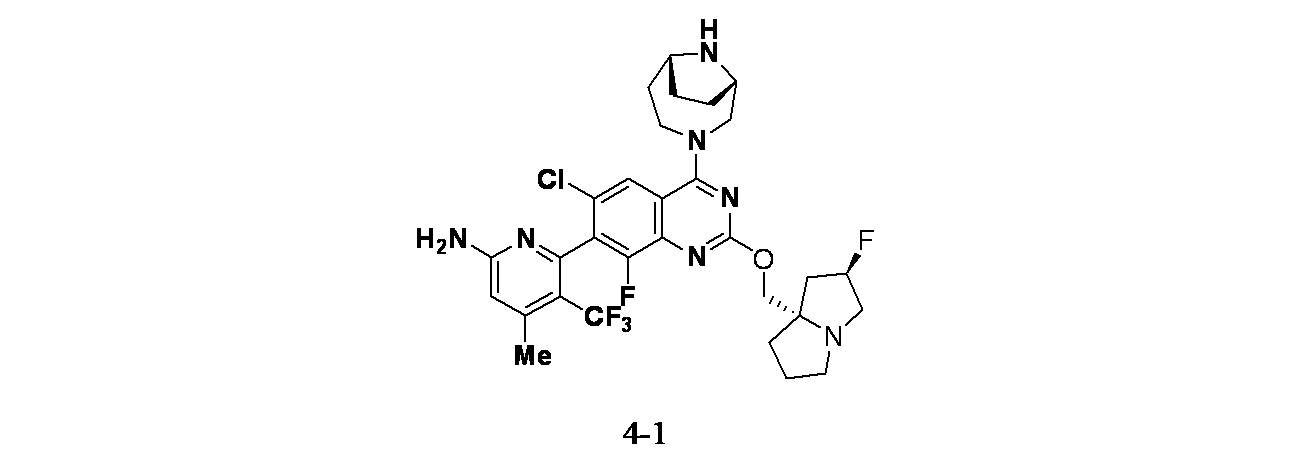
実施例4-1
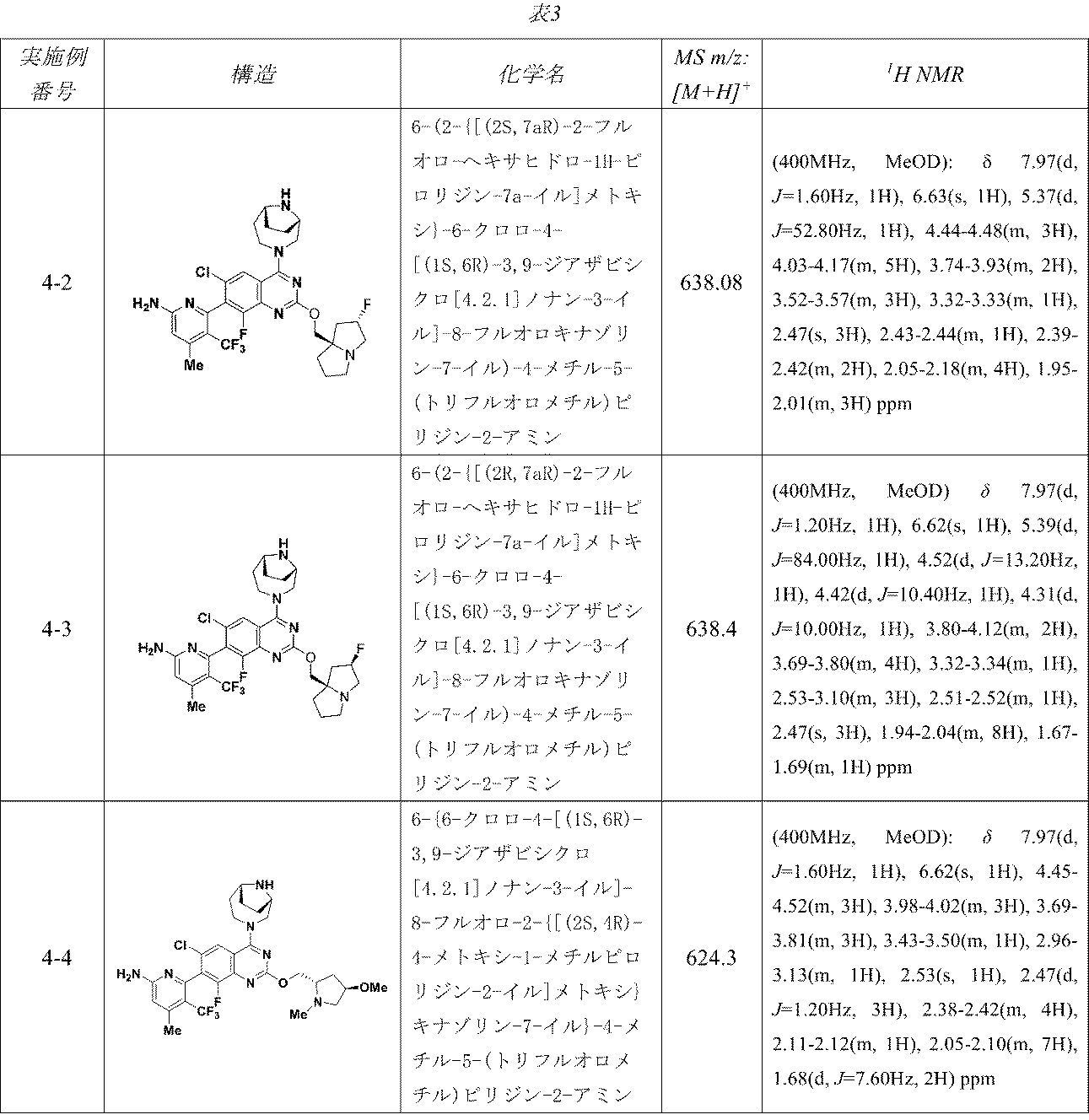
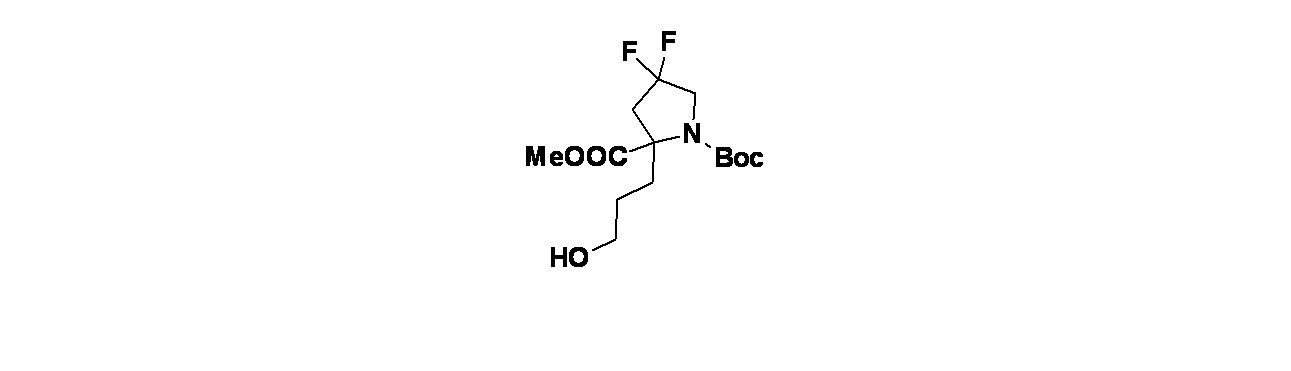
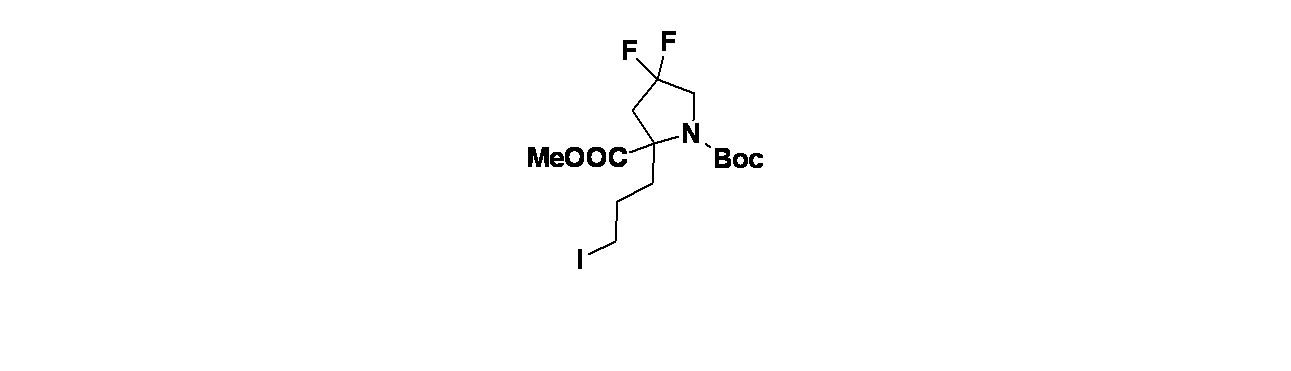
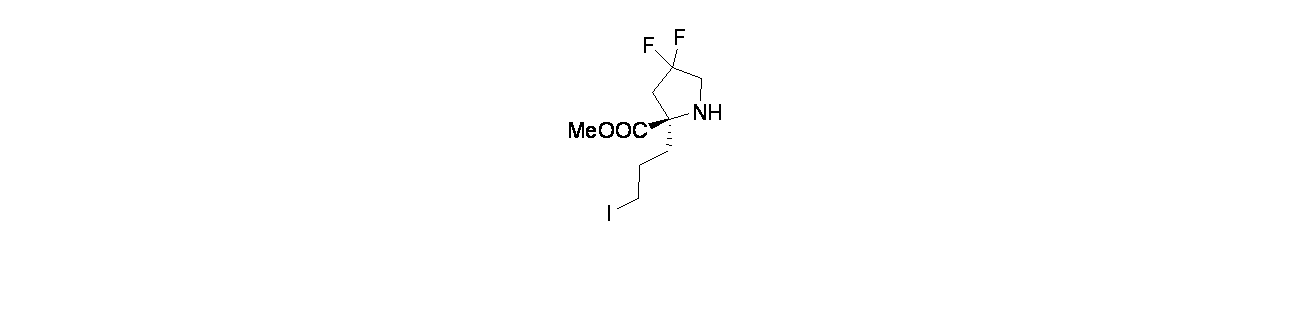
6-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
中間体4A: tert-ブチル-3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メタノールおよびHCl(81mg、0.509mmol)/DCMの撹拌溶液に、炭酸ナトリウム(231mg、2.181mmol)を加え、この反応混合物を10分間ソニケーションし、次いで室温で30分間撹拌した。溶媒をデカンテーションし、減圧濃縮し、((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メタノールを遊離塩基として得た。得られた粗製残渣を無水THF(5mL)に溶解し、0℃に冷却し、次いで水素化ナトリウム(60%、鉱油に分散、12.21mg、0.509mmol)を加えた。この反応混合物を同温度で30分間撹拌した後、中間体3L(280mg、0.339mmol)/THF(1mL)を滴下して加えた。この反応混合物を終夜室温で撹拌した。この反応混合物を次いで氷で冷却し、飽和塩化アンモニウム水溶液(10mL)でクエンチし、酢酸エチル(2x20mL)で抽出した。有機層を合わせて水、次いで食塩水で洗浄し、無水Na
2SO
4で乾燥し、濾過し、減圧濃縮した。得られた粗製生成物をカラムクロマトグラフィー(Grace REVELERIS
(登録商標)、50g snap、乾式充填、中性アルミナ、50~100%酢酸エチル/石油エーテル)で精製した。所期のフラクションを集めて減圧濃縮し、tert-ブチル-3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(240mg、0.241mmol、収率71%)を得た。LCMS(ESI)m/z: 965.8 [M+H]
+
中間体4B: 7-(5-(ビス(4-メトキシベンジル)アミノ)-3-メチル-2-(トリフルオロメチル)フェニル)-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-オールの製造
tert-ブチル-3-(7-(5-(ビス(4-メトキシベンジル)アミノ)-3-メチル-2-(トリフルオロメチル)フェニル)-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(240mg、0.249mmol)/エタノール(5mL)の撹拌溶液に、NaOH(1M水溶液、199mg、0.498mmol)を加え、混合物を70℃で48時間加熱した。この反応混合物を次いで減圧濃縮し、粗製残渣を得た。これをカラムクロマトグラフィー(Biotage、中性アルミナ、30~40%酢酸エチル/石油エーテル)で精製し、7-(5-(ビス(4-メトキシベンジル)アミノ)-3-メチル-2-(トリフルオロメチル)フェニル)-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-オール(60mg、0.077mmol、収率31%)を灰白色固体として得た。LCMS(ESI)m/z: 771.2 [M+H]
+
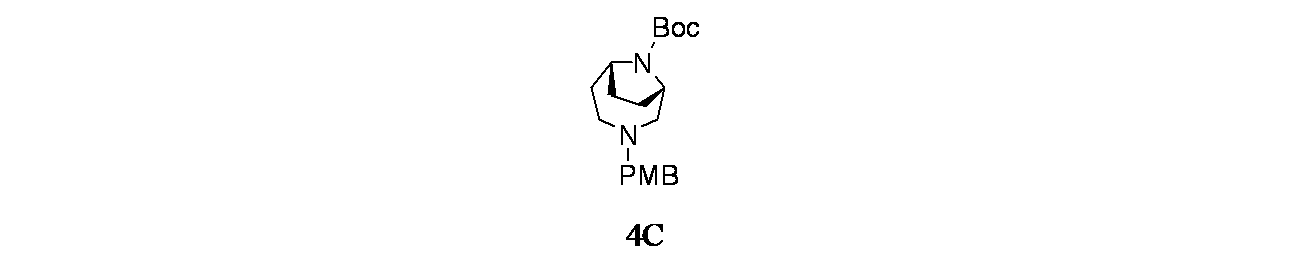
中間体4C: tert-ブチル(1S,6R)-3-(4-メトキシベンジル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
tert-ブチル3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(4.5g、19.88mmol)および4-メトキシベンズアルデヒド(3.25g、23.86mmol)/DCE(50mL)の撹拌溶液に、DIPEA(17.36mL、99mmol)を加え、この反応混合物を15分間25℃で撹拌した後、反応を0℃に冷却した。15分後、温度を5℃未満に保ちながら水素化トリアセトキホウ素ナトリウム(12.64g、59.7mmol)を少しずつ反応混合物に加えた。添加後、この反応混合物を徐々に25℃に加温し、終夜撹拌した。この反応混合物を次いで飽和塩化アンモニウム水溶液(10mL)でクエンチし、ジクロロメタン(3x20mL)で抽出した。有機層を合わせて水および飽和食塩水で洗浄し、次いで無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製生成物を得た。得られた粗製化合物をシリカゲルクロマトグラフィー(60~120mesh、溶出: 10~15%酢酸エチル/石油エーテル)で精製し、tert-ブチル-3-(4-メトキシベンジル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートを得た。純粋なフラクションについてキラルSFC分離[カラム: Chiralpak ADH(250X4.6)mm、5μ、方法=0.5%イソプロピルアミン/MeOH_10、保持時間=5.00分]を行い、tert-ブチル(1R,6S)-3-(4-メトキシベンジル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(1.9g、5.48mmol、収率28%)を薄褐色油状物として、tert-ブチル(1S,6R)-3-(4-メトキシベンジル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(2g、5.77mmol、収率29%)を薄褐色油状物として得た。LCMS(ESI)m/z: 347.2 [M+H]
+;
1H NMR(400MHz、DMSO-d
6、298K) δ 7.31-7.19(m, 2H), 6.88(d, J=8.6Hz, 2H), 5.78-5.71(m, 1H), 4.15-3.89(m, 2H), 3.73(s, 3H), 3.49(s, 2H), 2.78-2.60(m, 1H), 2.48-2.42(m, 1H), 2.40(d, J=2.8Hz, 1H), 2.20-1.99(m, 2H), 1.96-1.55(m, 4H), 1.40(d, J=1.6Hz, 9H)ppm
中間体4D: tert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
tert-ブチル(1S,6R)-3-(4-メトキシベンジル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(1500mg、4.33mmol)/EtOH(20mL)の撹拌溶液に、Pd/C(921mg、8.66mmol)を25℃で加えた。得られた混合物を水素雰囲気(1atm)下、25℃で3時間撹拌した。この反応混合物を次いでセライト濾過し、エタノールで洗浄し、濾液を減圧濃縮し、粗製化合物を得た。得られた粗製残渣をカラムクロマトグラフィー(Biotage、中性アルミナ、MeOH/ジクロロメタン(10%))で精製し、tert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートを得た(700mg、71%)。LCMS(ESI)m/z: 227.1 [M+H]
+
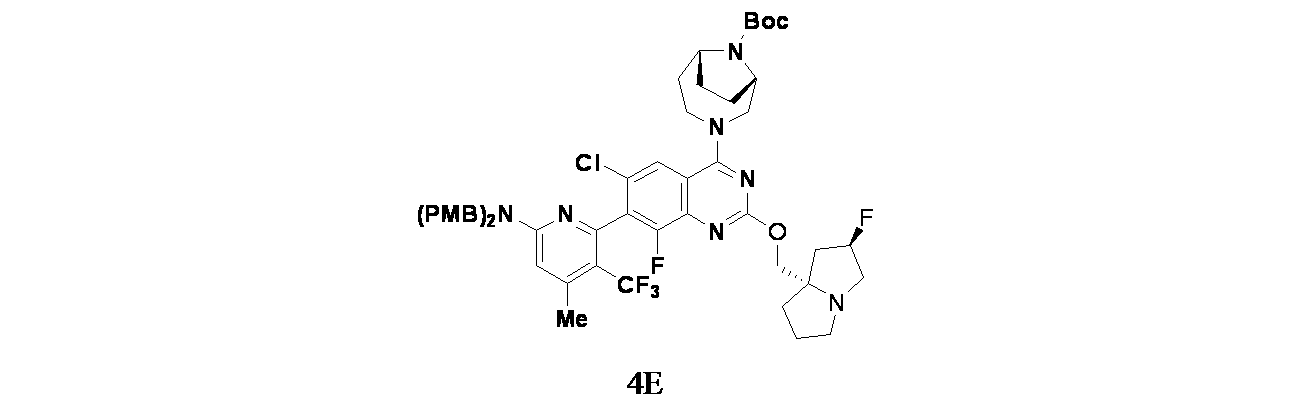
中間体4E: tert-ブチル(1S,6R)-3-(7-(5-(ビス(4-メトキシベンジル)アミノ)-3-メチル-2-(トリフルオロメチル)フェニル)-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-5-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
7-(5-(ビス(4-メトキシベンジル)アミノ)-3-メチル-2-(トリフルオロメチル)フェニル)-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-オール(55mg、0.072mmol)/アセトニトリル(2mL)の撹拌溶液に、BOP(47.4mg、0.107mmol)およびTEA(0.015mL、0.107mmol)を加えた。5分間撹拌後、tert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(20.23mg、0.089mmol)を加え、40℃で16時間混合物を撹拌した。この反応混合物を水(10mL)でクエンチし、酢酸エチル(2x15mL)で抽出した。有機層を合わせて食塩水(10mL)で洗浄し、無水硫酸ナトリウムで乾燥し、濾過し、濾液を減圧濃縮し、残渣を得た。これをカラムクロマトグラフィー(Biotage、中性アルミナ、30~40%酢酸エチル/石油エーテル)で精製し、tert-ブチル(1S,6R)-3-(7-(5-(ビス(4-メトキシベンジル)アミノ)-3-メチル-2-(トリフルオロメチル)フェニル)-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-5-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(55mg、0.052mmol、収率73%)を得た。LCMS(ESI)m/z: 979.51 [M+H]
+
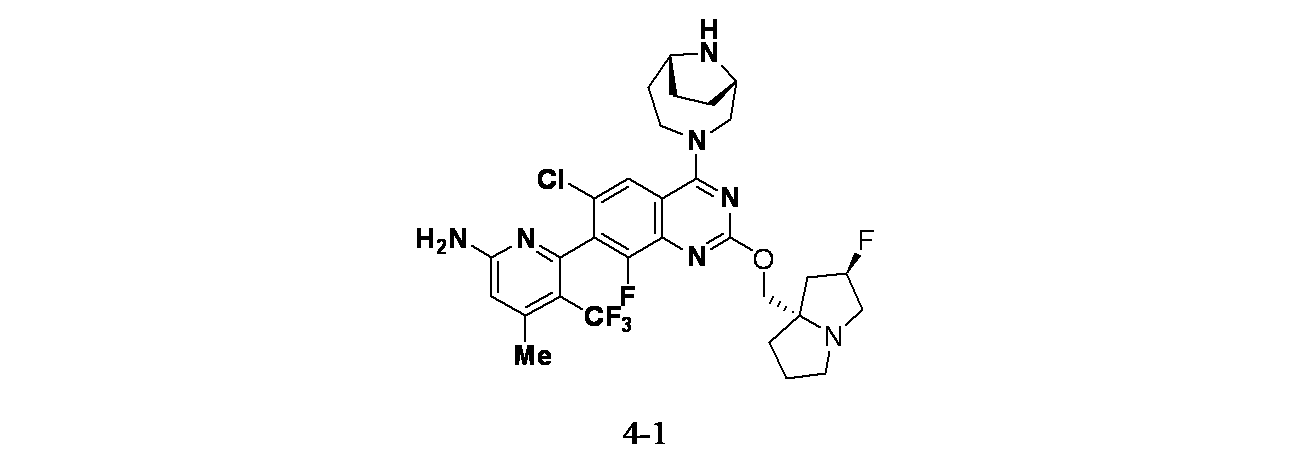
実施例4-1
6-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
tert-ブチル(1S,6R)-3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(55mg、0.056mmol)/TFA(1mL)の撹拌溶液に、水(0.01mL)およびトリエチルシラン(0.898μL、5.62μmol)を加え、40℃で16時間撹拌した。この反応混合物を次いで減圧濃縮し、粗製残渣を得た。これをシリカゲルカラムクロマトグラフィー(5~7%メタノール/DCM)で精製し、6-(4-((1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル)-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン(16mg、0.023mmol、収率41%)を得た。LCMS(ESI)m/z: 638.3 [M+H]
+;
1H-NMR(400MHz、MeOD): δ 7.97(s, 1H), 6.62(s, 1H), 5.33(d, J=54.00Hz, 2H), 4.50(d, J=13.20Hz, 1H), 4.22-4.48(m, 2H), 4.01-4.03(m, 1H), 3.72-3.94(m, 4H), 3.24-3.33(m, 2H), 3.04-3.06(m, 1H), 2.47(s, 3H), 2.14-2.29(m, 9H), 1.98-2.02(m, 1H), 1.70-1.72(m, 2H)ppm
表3の実施例は、実施例4-1に記載の手順に従って適当な出発物質から製造した。
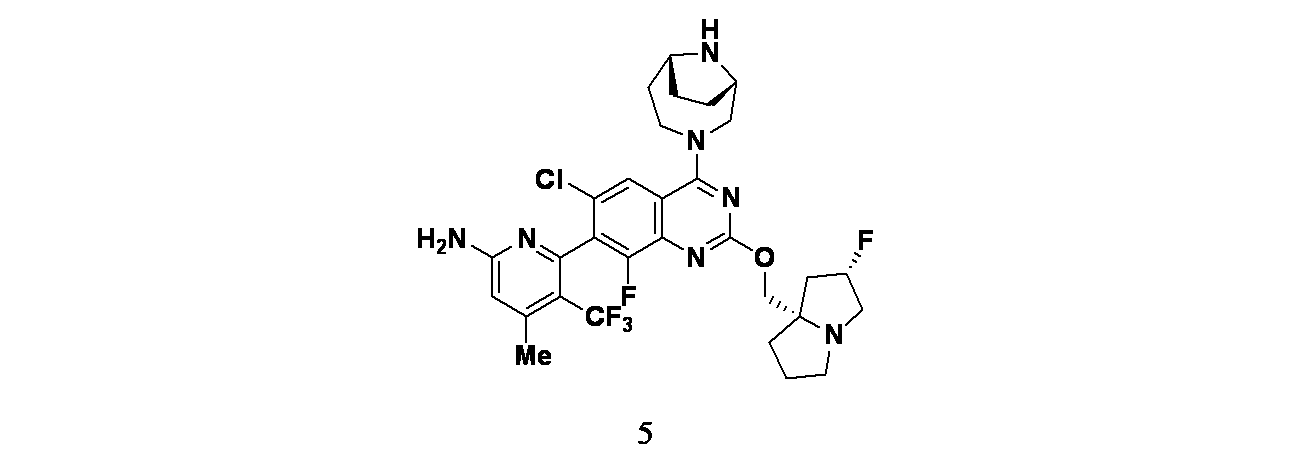
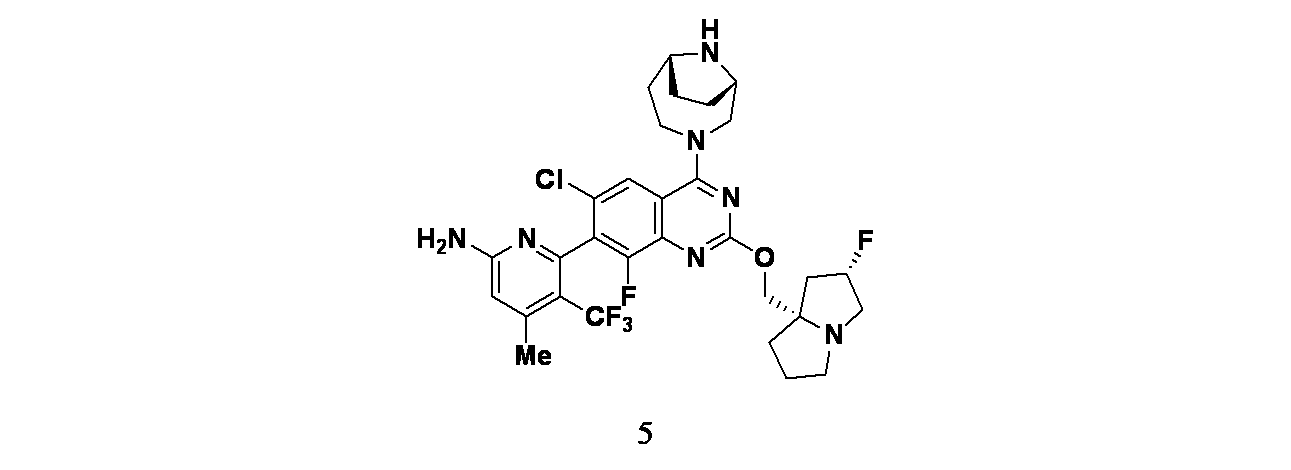
実施例5
6-(2-{[(2S,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
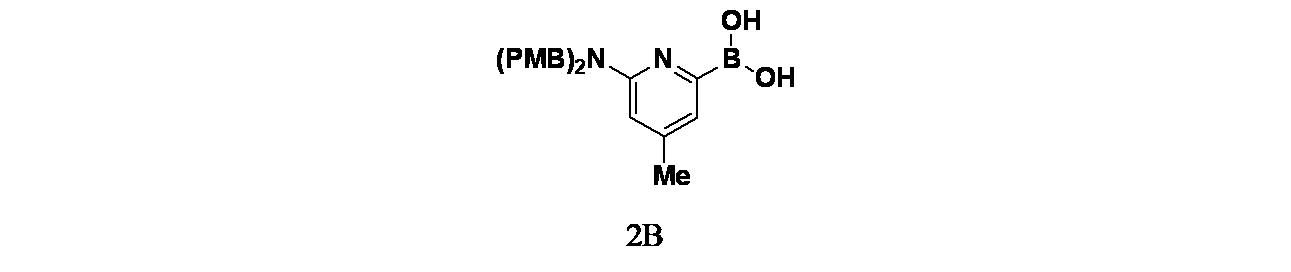
中間体5A(異性体1)および5B(異性体2): tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
密封したチューブ中、tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-3-ヨード-4-メチルピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(21g、24.50mmol)/無水DMA(210mL)の撹拌溶液に、窒素雰囲気下でヨウ化銅(I)(21g、24.50mmol)を加えた。この反応混合物を10分間脱気した後、2,2-ジフルオロ-2-(フルオロスルホニル)酢酸メチル(21g、24.50mmol)を添加し、この反応混合物を90℃で12時間加熱した。室温に冷却後、この反応混合物をジエチルエーテル(200mL)および水(50mL)で希釈した。層を分離し、水層をジエチルエーテル(3x50mL)で抽出した。有機層を合わせて無水硫酸ナトリウムで乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。これをシリカゲルカラムクロマトグラフィー(30%酢酸エチル/石油エーテル)で精製し、tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)ピペラジン-1-カルボキシレート(15.1g、16.44mmol、収率67%)を淡黄色固体として得た。アトロプ異性体をSFC(カラム: Chiralpak IH(250mmx4.6x5μ)、移動相:25%イソプロパノール)を用いて分離した。
ピーク1(中間体5A)は、保持時間=6.00分で溶出した(0.73g、0.98mmol、収率37%)。LCMS(ESI)m/z: 825.2 [M+H]
+
ピーク2(中間体5B)は、保持時間=9.262分で溶出した(0.77g、0.97mmol、38%)。LCMS(ESI)m/z: 825.2 [M+H]
+
中間体5C: tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)ピペラジン-1-カルボキシレートの製造
((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メタノール(100mg、0.626mmol)/THF(5mL)の撹拌溶液に、水素化ナトリウム(60%、鉱油に分散、25.02mg、0.626mmol)を0℃で加え、30分間撹拌した。中間体5B(250mg、0.313mmol)/THF(5mL)を上記反応混合物に0℃で滴下して加え、続いて周囲温度で2時間撹拌した。この反応混合物を飽和塩化アンモニウム水溶液(10mL)でクエンチし、酢酸エチル(2x20mL)で抽出した。有機層を合わせて水および食塩水で洗浄し、次いで無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製生成物を得た。生成物:tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)ピペラジン-1-カルボキシレート(398mg、0.246mmol、収率79%)をさらに精製せずに次の反応に用いた。LCMS(ESI)m/z: 938.3 [M+H]
+
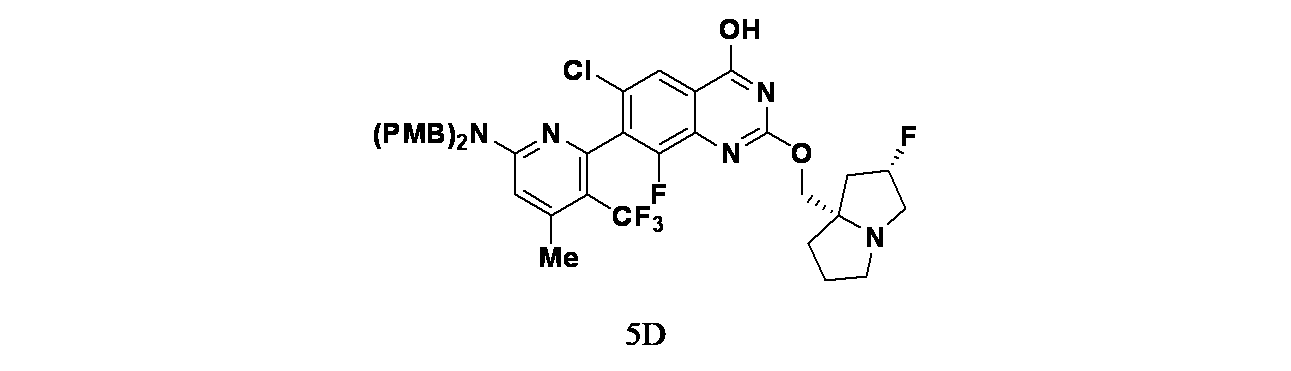
中間体5D: 7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-オールの製造
tert-ブチル4-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)ピペラジン-1-カルボキシレート(398mg、0.424mmol)/EtOH(5mL)およびTHF(1mL)の撹拌溶液に、NaOH(1M水溶液、1696mg、4.98mmol)を加えた。70℃で48時間撹拌後、この反応混合物を減圧濃縮し、粗製残渣を得た。これをカラムクロマトグラフィー(Biotage、中性アルミナ、30~40%酢酸エチル/石油エーテル)で精製し、7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-オール(150mg、0.175mmol、収率41%)を淡黄色油状物として得た。LCMS(ESI)m/z: 770.2 [M+H]
+
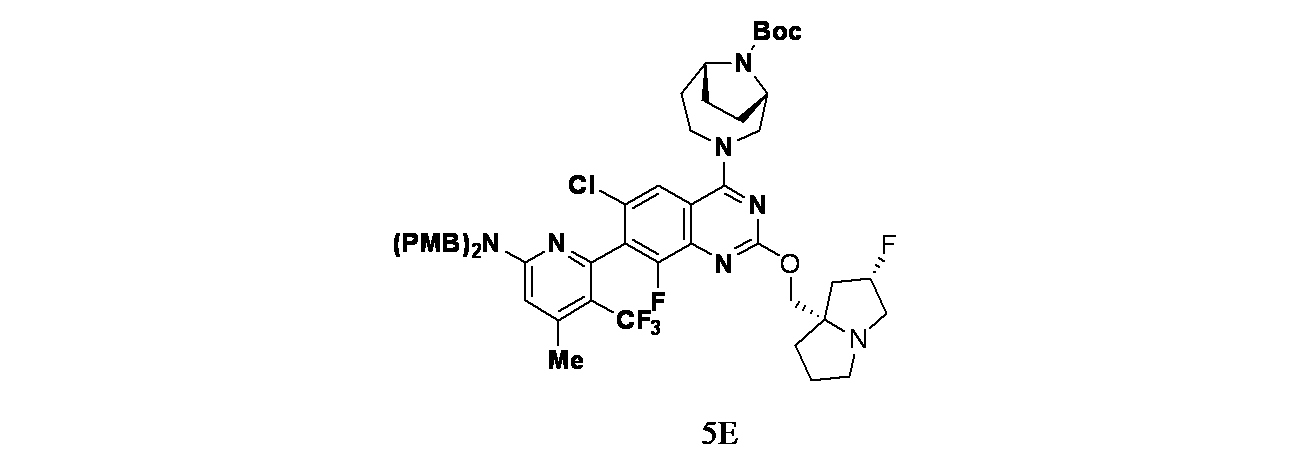
中間体5E: tert-ブチル(1S,6R)-3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-オール(30mg、0.039mmol)/アセトニトリル(1mL)の撹拌溶液に、TEA(8.14μL、0.058mmol)およびBOP(25.8mg、0.058mmol)を加えた。5分間撹拌後、tert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(11.02mg、0.049mmol)をこの反応混合物に加え、混合物を40℃で24時間加熱した。この反応混合物を水(10mL)でクエンチし、酢酸エチル(2x15mL)で抽出した。有機層を合わせて食塩水(10mL)で洗浄し、無水硫酸ナトリウムで乾燥し、濾過し、濾液を減圧濃縮し、粗製化合物を得た。得られた粗製残渣をカラムクロマトグラフィー(Biotage、中性アルミナ、45%酢酸エチル/石油エーテル)で精製し、tert-ブチル(1S,6R)-3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(28mg、0.027mmol、収率69%)を得た。LCMS(ESI)m/z: 979.5 [M+H]
+
実施例5
6-(2-{[(2S,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
tert-ブチル(1S,6R)-3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(28mg、0.029mmol)/TFA(1mL)の撹拌溶液に、トリエチルシラン(0.457μL、2.86μmol)および水(0.05mL、2.78mmol)を加えた。反応を40℃で16時間撹拌した。この反応混合物を減圧濃縮し、得られた残渣をカラムクロマトグラフィー(Biotage、Sfar KP-Amino、溶出: 50~100%酢酸エチル/石油エーテル)で精製し、6-(4-((1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル)-6-クロロ-8-フルオロ-2-(((2S,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン(11.4mg、0.018mmol、収率62%)を得た。LCMS(ESI)m/z: 638.08 [M+H]
+;
1H-NMR(400MHz、MeOD) δ 7.97(d, J=1.60Hz, 1H), 6.63(s, 1H), 5.37(d, J=52.80Hz, 1H), 4.41-4.89(m, 3H), 4.02-4.13(m, 1H), 3.88-3.94(m, 4H), 3.49-3.50(m, 2H), 2.88-3.28(m, 3H), 2.50-2.16(m, 1H), 0.47(s, 3H), 2.19-2.47(m, 1H), 2.12-2.18(m, 4H), 1.91-2.02(m, 3H), 1.79-1.88(m, 2H)ppm
実施例6
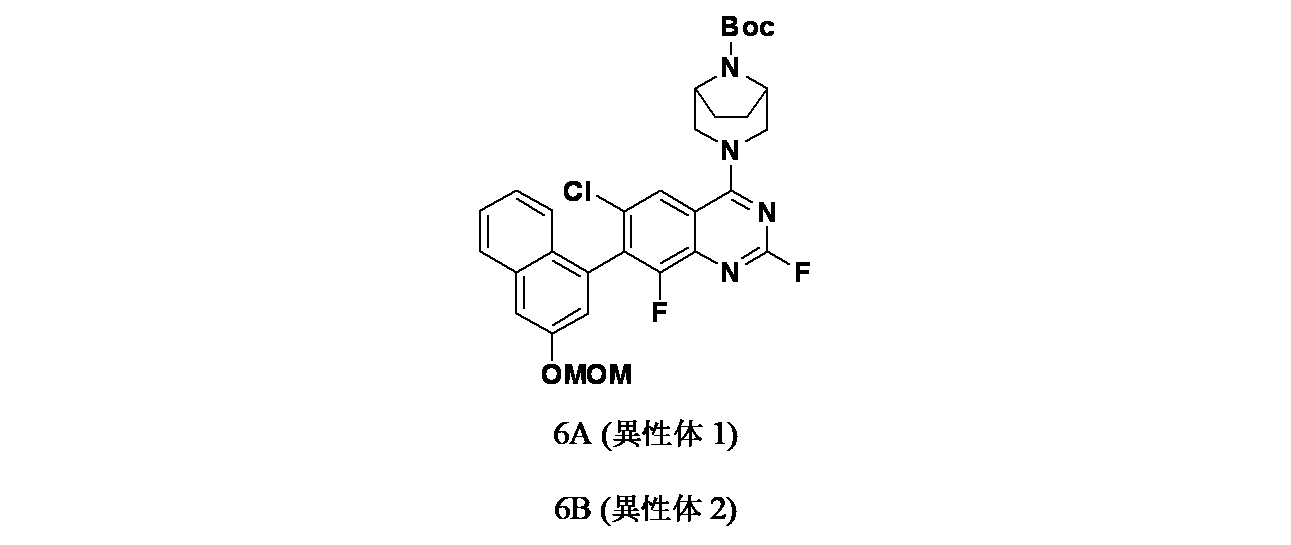
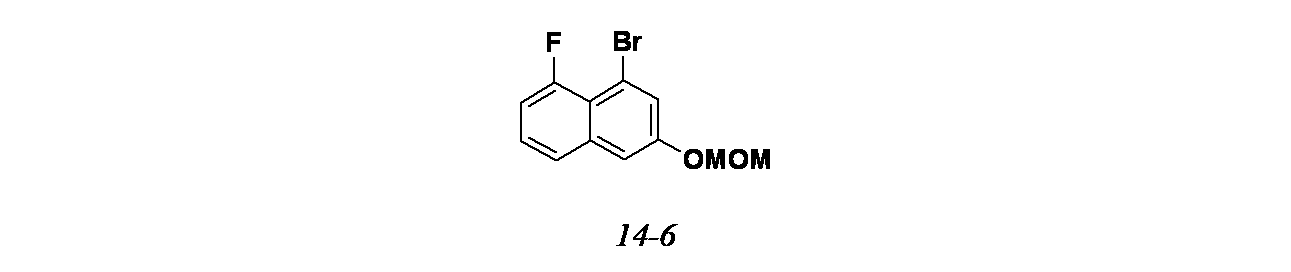
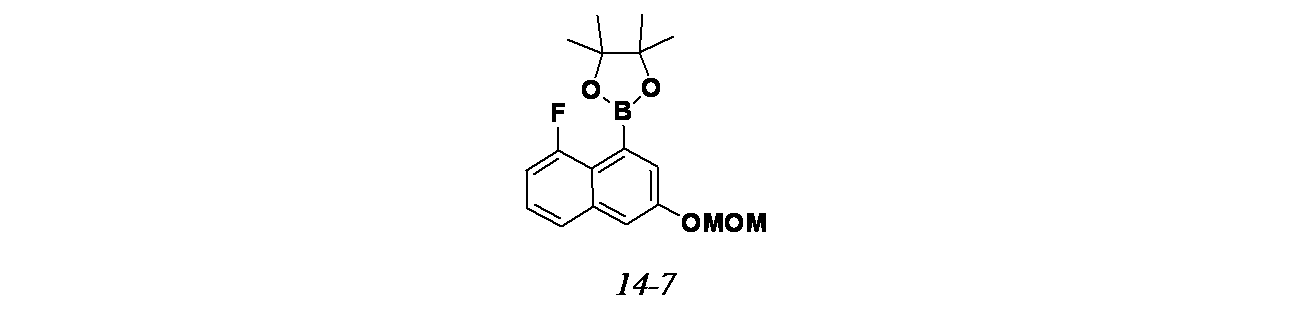
4-{6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロ-2-{[(2S)-1-メチルピロリジン-2-イル]メトキシ}キナゾリン-7-イル}ナフタレン-2-オール
中間体6A(異性体1)および6B(異性体2): tert-ブチル-3-(6-クロロ-2,8-ジフルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
tert-ブチル(1R,5S)-3-(7-ブロモ-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(1.0g、2.042mmol))/1,4-ジオキサン(60mL)の脱気した溶液に、2-(3-(メトキシメトキシ)ナフタレン-1-イル)-4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(1.28g、4.08mmol)、Cs
2CO
3(1.3g、4.08mmol)、Pd
2(dba)
3(93mg、0.102mmol)およびペンタフェニル(ジ-tert-ブチルホスフィノ)フェロセン(0.073g、0.102mmol、QPhos、Cas No: 312959-24-3)を加えた。耐圧バイアル中、この混合物を再度脱気し、70℃で12時間加熱した。この反応混合物を減圧濃縮し、粗製残渣を得た。これをシリカゲルカラムクロマトグラフィー(30%酢酸エチル/石油エーテル)で精製し、tert-ブチル-3-(6-クロロ-2,8-ジフルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(1.0g、1.591mmol、78%)を薄茶色固体として得た。LCMS(ESI)m/z: 597.2 [M+H]
+
精製した化合物についてSFCを行い、アトロプ異性体を分離した(カラム: Chiralpak ADH(250mmx4.6x5μ)、移動相: 30%イソプロパノール)。
ピーク1(中間体6A)は、保持時間=2.72分で溶出した(230mg、46%)。LCMS(ESI)m/z: 597.2[M+H]
+
ピーク2(中間体6B)は、保持時間=5.47分(205mg、41%)で溶出した。LCMS(ESI)m/z: 597.2 [M+H]
+
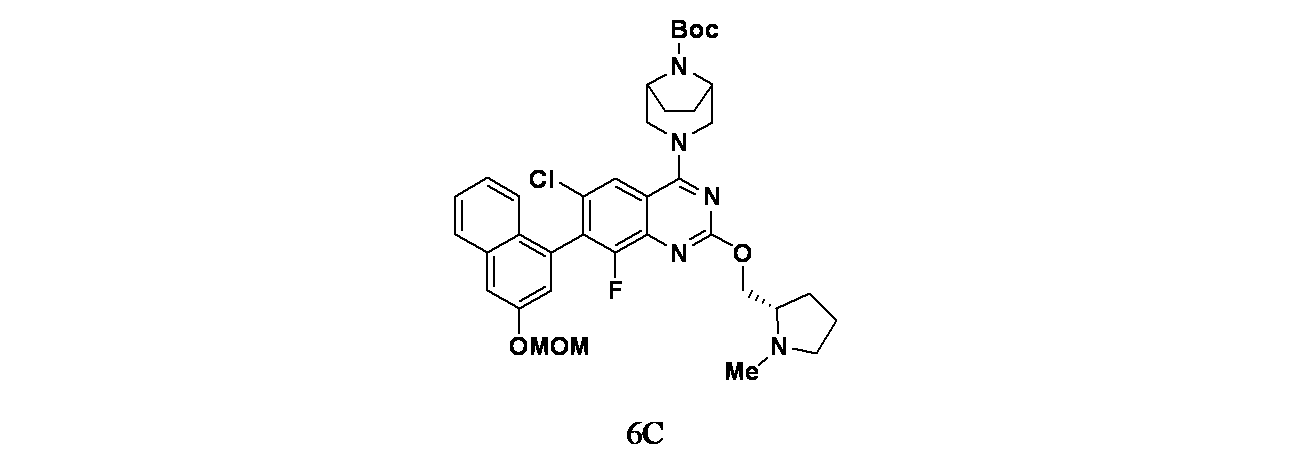
中間体6C: tert-ブチル3-(6-クロロ-8-フルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
(S)-(1-メチルピロリジン-2-イル)メタノール(38.6mg、0.335mmol)/THF(5mL)の撹拌溶液に、0℃でNaH(60%、鉱油に分散、12.06mg、0.301mmol)を加え、この反応混合物を30分間撹拌した。次いで中間体6A(100mg、0.167mmol)/THF(1mL)を滴下して加えた。この反応混合物を周囲温度で2時間撹拌した後、飽和塩化アンモニウム水溶液(10mL)でクエンチし、続いて酢酸エチル(2x20mL)で抽出した。有機層を合わせて水および食塩水で洗浄し、次いで無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製生成物を得た。これをカラムクロマトグラフィー(Grace REVELERIS(登録商標)、50g snap、乾式充填、中性アルミナ、50~100%酢酸エチル/石油エーテル)で精製した。所期のフラクションを集めて減圧濃縮し、tert-ブチル3-(6-クロロ-8-フルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(90mg、0.118mmol、収率71%)を淡黄色油状物として得た。LCMS(ESI)m/z: 691.8 [M+H]
+
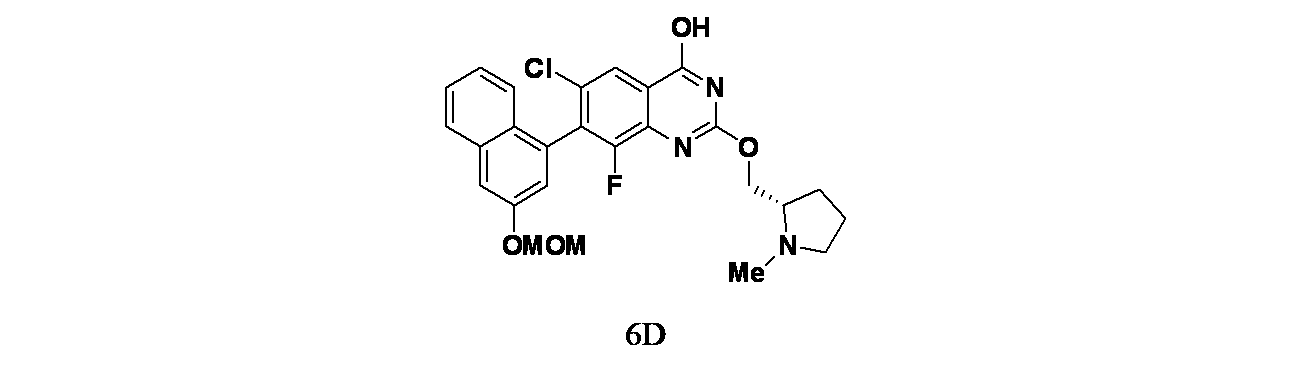
中間体6D: 6-クロロ-8-フルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-オールの製造
tert-ブチル3-(6-クロロ-8-フルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(120mg、0.173mmol)/エタノール(5mL)の撹拌溶液に、NaOH(1M水溶液、199mg、0.498mmol)を加え、70℃で48時間撹拌した。この反応混合物を次いで減圧濃縮し、粗製残渣を得た。これをカラムクロマトグラフィー(Biotage、中性アルミナ、30~40%酢酸エチル/石油エーテル)で精製し、6-クロロ-8-フルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-オール(35mg、0.069mmol、収率40%)を灰白色固体として得た。LCMS(ESI)m/z: 500.1 [M+H]
+
中間体6E: tert-ブチル(1S,6R)-3-(6-クロロ-8-フルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
6-クロロ-8-フルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-オール(35mg、0.070mmol)/アセトニトリル(1mL)の撹拌溶液に、TEA(0.015mL、0.105mmol)およびBOP(46.6mg、0.105mmol)を加えた。5分間撹拌後、tert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(19.88mg、0.088mmol)を加え、続いて40℃で24時間加熱した。この反応混合物を水(10mL)でクエンチし、酢酸エチル(2x15mL)で抽出した。有機層を合わせて食塩水(10mL)で洗浄し、無水硫酸ナトリウムで乾燥し、濾過し、濾液を減圧濃縮し、粗製化合物を得た。得られた残渣をカラムクロマトグラフィー(Biotage、中性アルミナ、40%酢酸エチル/石油エーテル)で精製し、tert-ブチル(1S,6R)-3-(6-クロロ-8-フルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(30mg、0.039mmol、収率56%)を得た。LCMS(ESI)m/z: 707.2 [M+H]
+
実施例6
4-{6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロ-2-{[(2S)-1-メチルピロリジン-2-イル]メトキシ}キナゾリン-7-イル}ナフタレン-2-オール
tert-ブチル(1S,6R)-3-(6-クロロ-8-フルオロ-7-(3-(メトキシメトキシ)ナフタレン-1-イル)-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(30mg、0.042mmol)/メタノール(1mL)の撹拌溶液に、HCl(4.0M、ジオキサン溶液、0.159mL、0.637mmol)を氷冷温度で加えた。反応を室温に戻し、次いで2時間撹拌した。この反応混合物を減圧濃縮し、粗製残渣を得た。これを分取HPLC[カラム: Xselect C18(150x19)mm、5μ、移動相A: 10mM酢酸アンモニウム水溶液、移動相B: アセトニトリル]で精製し、4-(4-((1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル)-6-クロロ-8-フルオロ-2-(((S)-1-メチルピロリジン-2-イル)メトキシ)キナゾリン-7-イル)ナフタレン-2-オール(4mg、6.67μmol、収率16%)を得た。LCMS(ESI)m/z: 707.2 [M+H]
+;
1H-NMR(400MHz、MeOD): δ 8.11(d, J=1.60Hz, 1H), 7.78(d, J=8.40Hz, 1H), 7.42-7.46(m, 1H), 7.21-7.29(m, 4H), 7.05(s, 1H), 4.52-4.67(m, 3H), 3.93-4.00(m, 5H), 3.33-3.34(m, 1H), 2.83(s, 4H), 2.18-2.28(m, 4H), 2.00-2.04(m, 3H), 1.85-1.96(m, 2H)ppm
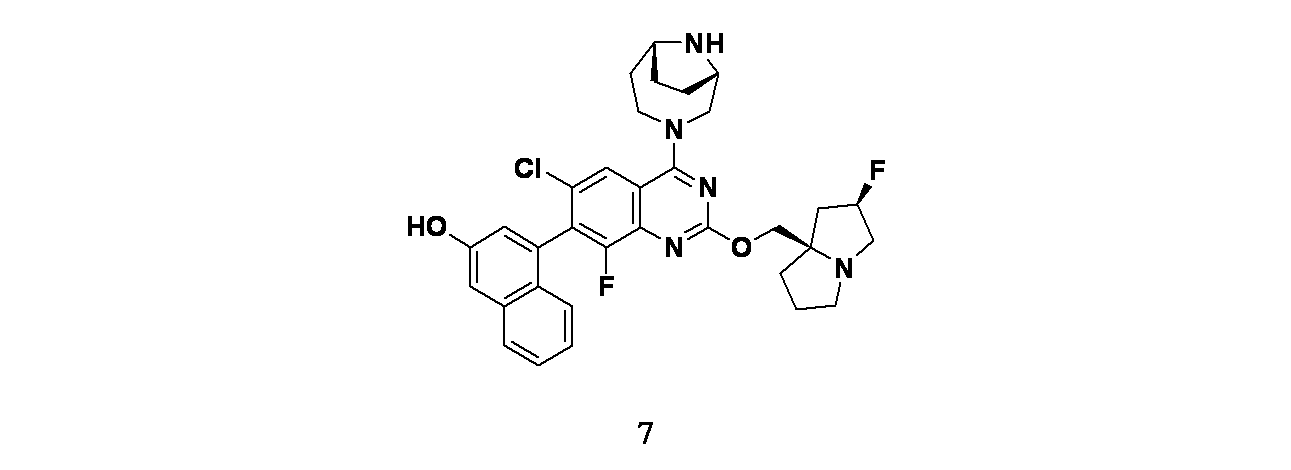
実施例7
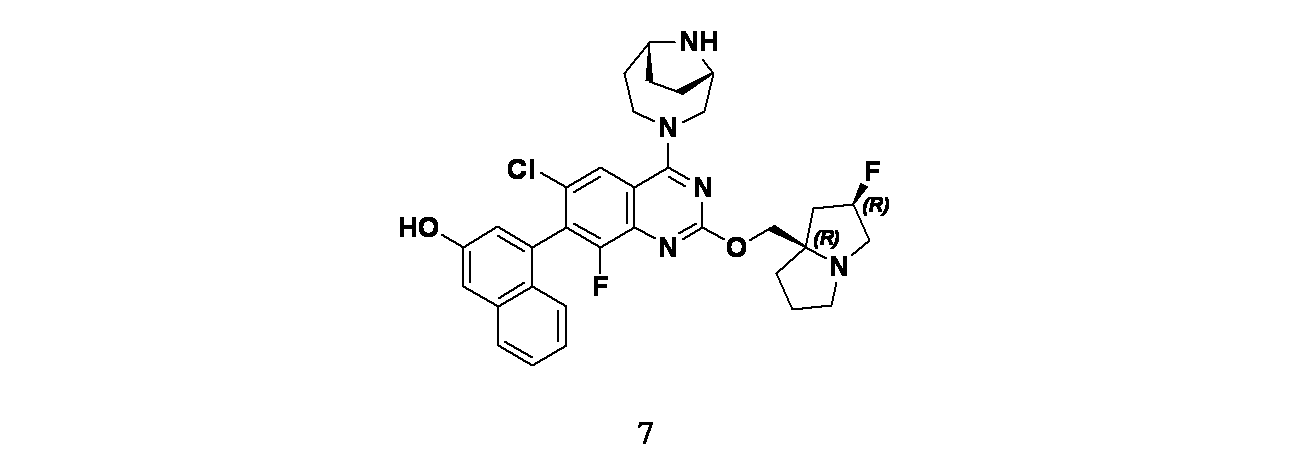
4-(2-{[(2R,7aR)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)ナフタレン-2-オール
中間体7A: tert-ブチル-3-(6-クロロ-8-フルオロ-2-(((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メタノール(53.3mg、0.335mmol)/THF(5mL)の撹拌溶液に、0℃でNaH(60%、鉱油に分散、12.06mg、0.301mmol)を加え、この反応混合物を30分間撹拌した。中間体6A(100mg、0.167mmol)/THF(1mL)を次いで滴下して加えた。この反応混合物を周囲温度で2時間撹拌した後、飽和塩化アンモニウム水溶液(10mL)でクエンチし、続いて酢酸エチル(2x20mL)で抽出した。有機層を合わせて水および食塩水で洗浄し、次いで無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製生成物を得た。これをカラムクロマトグラフィー(Grace REVELERIS
(登録商標)、50g snap、乾式充填、中性アルミナ、50~100%酢酸エチル/石油エーテル)で精製した。所期のフラクションを集めて減圧濃縮し、tert-ブチル-3-(6-クロロ-8-フルオロ-2-(((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(120mg、0.150mmol、収率90%)を淡黄色油状物として得た。LCMS(ESI)m/z: 735.8 [M+H]
+
中間体7B: 6-クロロ-8-フルオロ-2-(((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-オールの製造
tert-ブチル-3-(6-クロロ-8-フルオロ-2-(((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(230mg、0.312mmol)/エタノール(5mL)の撹拌溶液に、NaOH(1M水溶液、1249mg、3.12mmol)を加え、70℃で48時間撹拌した。この反応混合物を次いで減圧濃縮し、粗製残渣を得た。これをカラムクロマトグラフィー(Biotage、中性アルミナ、30~40%酢酸エチル/石油エーテル)で精製し、6-クロロ-8-フルオロ-2-(((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-オール(35mg、0.061mmol、収率19%)を灰白色固体として得た。LCMS(ESI)m/z: 542.2 [M+H]
+
中間体7C: tert-ブチル(1S,6R)-3-(6-クロロ-8-フルオロ-2-(((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
6-クロロ-8-フルオロ-2-(((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-オール(35mg、0.065mmol)/アセトニトリル(1mL)の撹拌溶液に、TEA(0.014mL、0.097mmol)およびBOP(42.8mg、0.097mmol)を加えた。5分間撹拌後、tert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(18.27mg、0.081mmol)を加え、40℃で24時間加熱した。この反応混合物を水(10mL)でクエンチし、酢酸エチル(2x15mL)で抽出した。有機層を食塩水(10mL)で洗浄し、無水硫酸ナトリウムで乾燥し、濾過し、濾液を減圧濃縮し、粗製化合物を得た。得られた残渣をカラムクロマトグラフィー(Biotage、中性アルミナ、30~40%酢酸エチル/石油エーテル)で精製し、tert-ブチル(1S,6R)-3-(6-クロロ-8-フルオロ-2-(((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(38mg、0.048mmol、収率75%)を得た。LCMS(ESI)m/z: 751.2 [M+H]
+
実施例7
4-(2-{[(2R,7aR)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)ナフタレン-2-オール
tert-ブチル(1S,6R)-3-(6-クロロ-8-フルオロ-2-(((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)-7-(3-(メトキシメトキシ)ナフタレン-1-イル)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(30mg、0.042mmol)/メタノール(1mL)の撹拌溶液に、HCl(4.0M、ジオキサン、0.159mL、0.637mmol)を氷冷温度で加えた。反応を室温に戻し、次いで2時間撹拌した。この反応混合物を減圧濃縮し、粗製残渣を得た。これを分取HPLC[カラム: Xselect C18(150x19)mm、5μ、移動相A: 10mM酢酸アンモニウム水溶液、移動相B: アセトニトリル]で精製し、4-(4-((1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル)-6-クロロ-8-フルオロ-2-(((2R,7aR)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-7-イル)ナフタレン-2-オール(7mg、0.01μmol、収率35%)を灰白色固体として得た。LCMS(ESI)m/z: 562.3 [M+H]
+1H-NMR(400MHz、MeOD): δ 8.11(d, J=1.60Hz, 1H), 7.78(d, J=8.40Hz, 1H), 7.42-7.46(m, 1H), 7.21-7.29(m, 4H), 7.05(s, 1H), 4.52-4.67(m, 3H), 3.93-4.00(m, 5H), 3.33-3.34(m, 1H), 2.83(s, 4H), 2.18-2.28(m, 4H), 2.00-2.04(m, 3H), 1.85-1.96(m, 2H)ppm
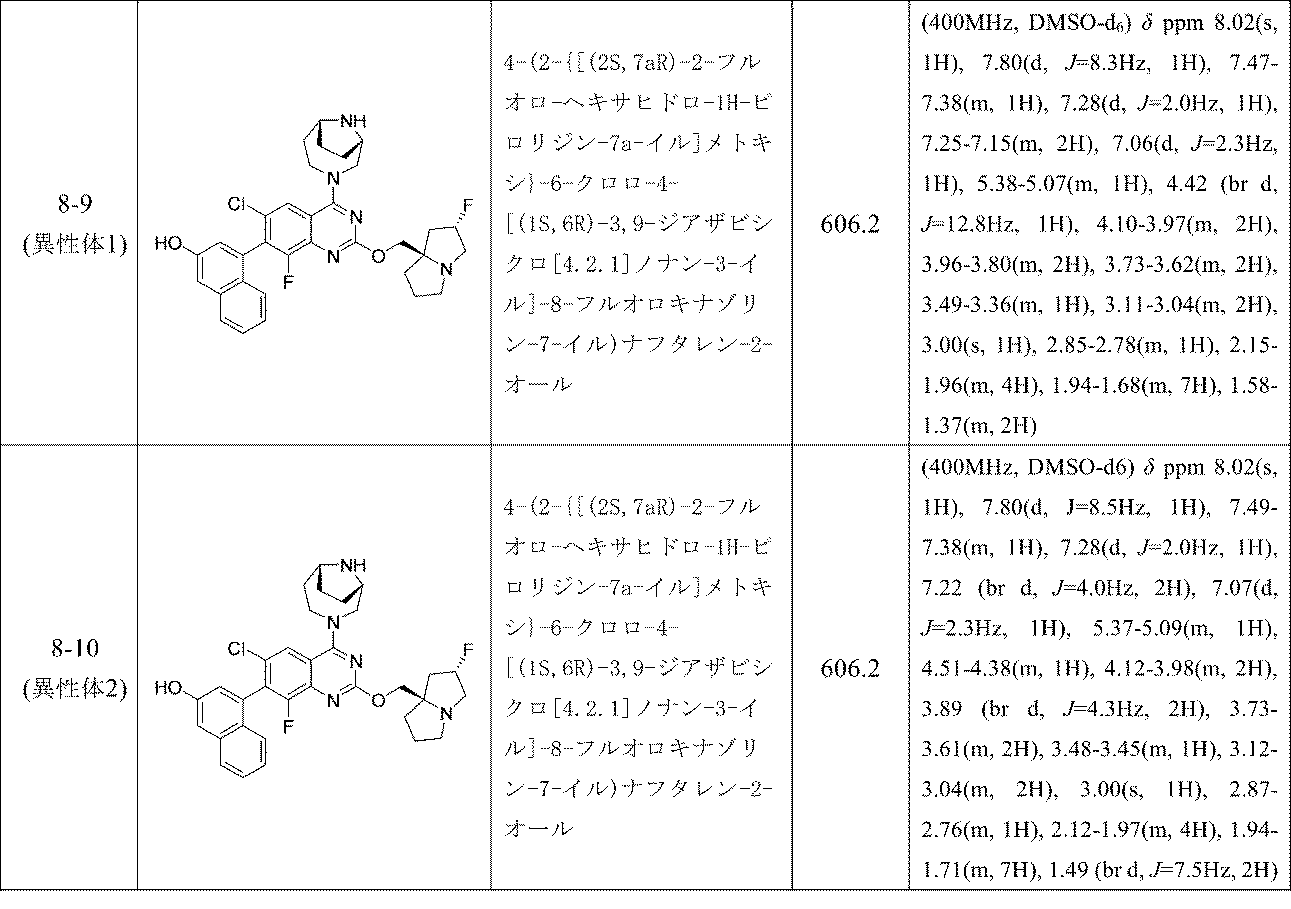
実施例8-1および8-2
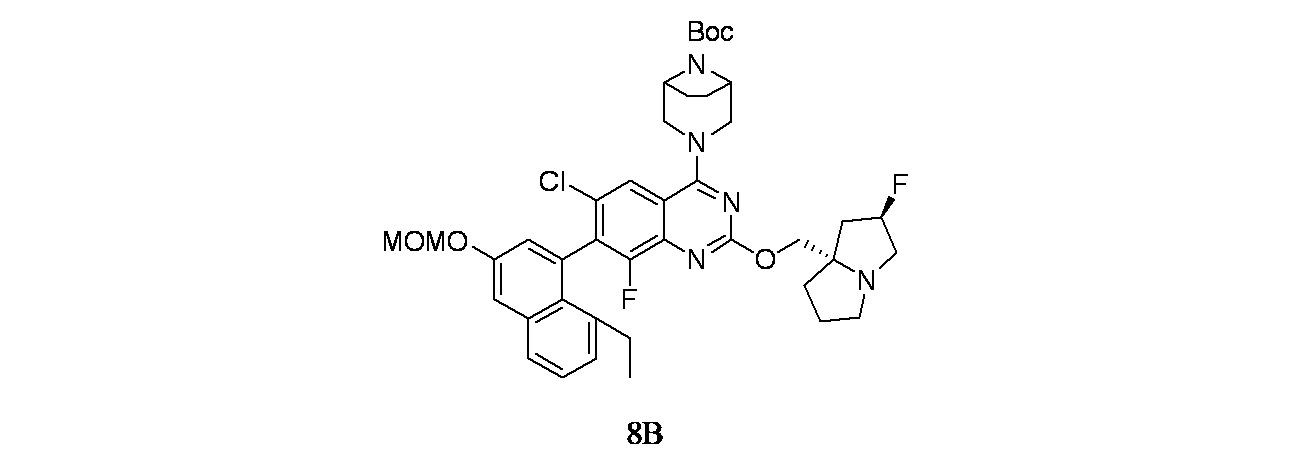
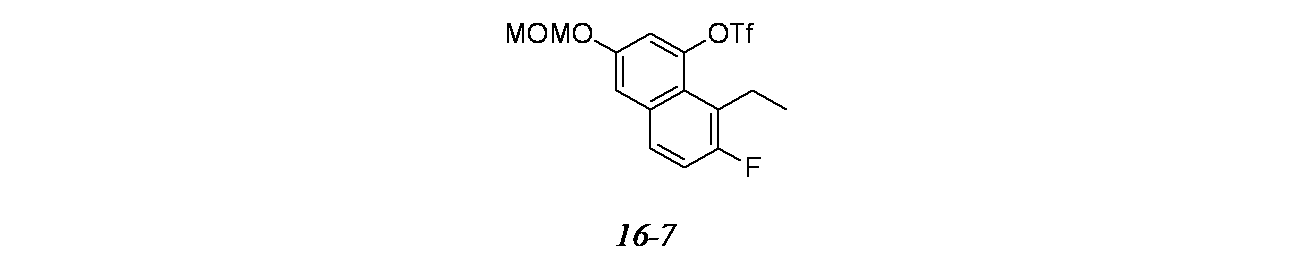
4-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-5-エチルナフタレン-2-オール
中間体8A: tert-ブチル-3-(7-ブロモ-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メタノール(195mg、1.225mmol)/THF(8mL)の撹拌溶液に、0℃でNaH(61.3mg、1.531mmol)を加え、さらに30分間撹拌した。次いで、tert-ブチル-3-(7-ブロモ-6-クロロ-2,8-ジフルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(500mg、1.021mmol)/THF(2mL)の溶液を加え、混合物を1時間かけて徐々に室温に戻した。この反応混合物を飽和塩化アンモニウム水溶液でクエンチし、酢酸エチルで抽出した。有機層を合わせて水および食塩水で洗浄し、次いでNa
2SO
4で乾燥し、濃縮し、粗製残渣を得た。これをシリカゲルカラムクロマトグラフィー(機器: CombiFlash、40g RediSep
(登録商標)カラム、70~80%EtOAc/石油エーテル)で精製し、tert-ブチル-3-(7-ブロモ-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(400mg、0.636mmol、収率62.3%)を得た。MS(ESI)m/z: 628.2 [M+H]
+
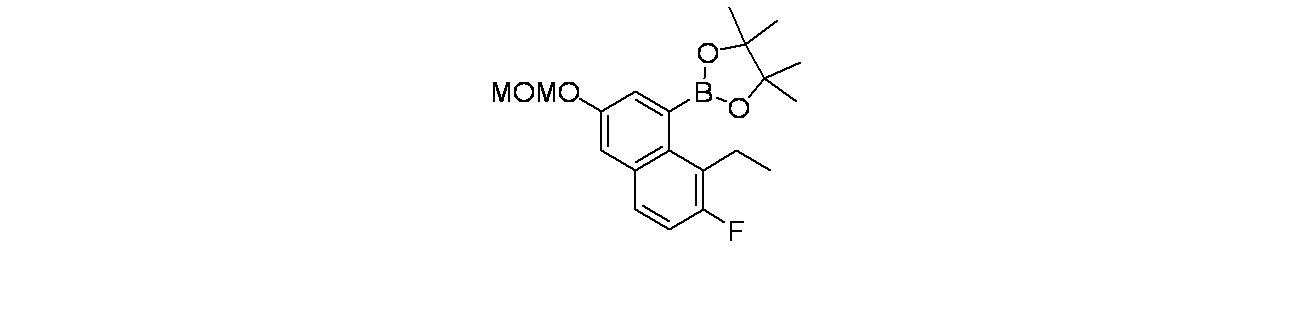
中間体8B: tert-ブチル3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-7-[8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル]-8-フルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
tert-ブチル-3-(7-ブロモ-6-クロロ-8-フルオロ-2-(((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(700mg、1.113mmol)/1,4-ジオキサン(5mL)の撹拌溶液に、室温で2-(8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル)-4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(457mg、1.336mmol)、およびリン酸三カリウム(1.5M水溶液、1.484mL、2.226mmol)を加えた。この反応混合物を窒素で5分間パージし、次いでPd(Ph
3P)
4(129mg、0.111mmol)を加えた。この反応混合物を窒素で3分間再度パージし、85℃で16時間加熱した。この反応混合物を冷却し、セライト濾過し、濾液を減圧濃縮し、粗製化合物を得た。これをシリカゲルカラムクロマトグラフィー(機器: CombiFlash、24g RediSep
(登録商標)カラム; 溶離剤: 石油エーテル/酢酸エチル)で精製した。所期の生成物を60~70%酢酸エチル/石油エーテルで溶出した。純粋なフラクションを合わせて濃縮し、減圧乾燥してtert-ブチル3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-7-[8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル]-8-フルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(350mg、0.458mmol、収率41.1%)を褐色固体として得た。MS(ESI)m/z: 764.3 [M+H]
+
中間体8C: 2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-7-[8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル]-8-フルオロ-3,4-ジヒドロキナゾリン-4-オンの製造
tert-ブチル3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-7-[8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル]-8-フルオロキナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(200mg、0.262mmol)/EtOH(2mL)の懸濁液をNaOH(1M水溶液、2.62mL、2.62mmol)で処理した。この反応混合物を70℃で16時間撹拌した。この反応混合物を次いで減圧濃縮し、得られた残渣を酢酸エチルで希釈し、食塩水で洗浄し、Na
2SO
4で乾燥し、濾過し、濃縮し、粗製化合物:2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-7-[8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル]-8-フルオロ-3,4-ジヒドロキナゾリン-4-オン(160mg)を白色固体として得た。MS(ESI)m/z: 570.2 [M+H]
+
中間体8D: tert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-7-[8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル]-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-7-[8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル]-8-フルオロ-3,4-ジヒドロキナゾリン-4-オン(160mg、0.281mmol)、tert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(76mg、0.337mmol)およびBOP(186mg、0.421mmol)/アセトニトリル(2mL)の溶液をTEA(0.078mL、0.561mmol)で処理し、この反応混合物を80℃で8時間撹拌した。この反応混合物を次いで減圧濃縮し、粗製残渣を得た。得られた粗製化合物をシリカゲルカラムクロマトグラフィー(機器: CombiFlash、0~10%MeOH/DCM(0.5%TEA含有))で精製し、所期の化合物:tert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-7-[8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル]-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(80mg、0.103mmol、収率36.6%)を得た。MS(ESI)m/z: 778.3 [M+H]
+
実施例8-1および8-2
4-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-5-エチルナフタレン-2-オール
tert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-7-[8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル]-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(80mg、0.103mmol)/MeOH(2mL)の溶液に、HCl(4M、1,4-ジオキサン、2mL、65.8mmol)を0℃で滴下して加え、次いで室温で1時間撹拌した。この反応混合物を次いで減圧濃縮し、トルエン(2回)と共沸し、DIPEAで中和し、再度減圧濃縮し、粗製残渣を得た。得られた粗製化合物を分取HPLCで精製し、続いてキラルHPLCを用いてキラル分離し、アトロプ異性体1および2: 4-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-5-エチルナフタレン-2-オールを得た。
分取HPLC条件(カラム: YMC-EXRS(250x21.2mm)、5μ; 移動相A=10mM重炭酸アンモニウム水(pH 9.5)、移動相B=アセトニトリル:MeOH(1:1)、流速: 20mL/分;保持時間=12.6分)
分取キラル条件(カラム: Cellulose-C5(250x21)、5μ; 移動相: 0.1%DEA/MeOH; 流速: 20mL/分; ピーク1の保持時間=5.3分、ピーク2の保持時間=6.3分)
実施例8-1:MS(ESI)m/z: 634.3[M+H]
+;
1H NMR(400MHz、DMSO-d
6) δ=11.0 (bs, 1H), 7.97(s, 1H), 7.68-7.66(m, 1H), 7.40-7.33(m, 1H), 7.28(d, J=2.6Hz, 1H), 7.13-7.11(m, 1H), 6.86(d, J=2.6Hz, 1H), 5.39-5.17(m, 1H), 4.38-4.34(m, 1H), 4.13-4.09(m, 1H), 4.00(d, J=10.4Hz, 1H), 3.95-3.79(m, 3H), 3.72-3.64(m, 3H), 3.12-3.06(m, 4H), 3.04-2.98(m, 2H), 2.88-2.78(m, 2H), 2.36- 2.26(m, 2H), 2.16-2.10(m, 1H), 2.01-1.95(m, 1H), 1.90-1.84(m, 3H), 1.56-1.30(m, 3H), 0.87(t, J=7.4Hz, 3H)
実施例8-2:MS(ESI)m/z: 634.3[M+H]
+;
1H NMR(400MHz、DMSO-d
6) δ=7.97(s, 1H), 7.68-7.66(m, 1H), 7.38-7.34(m, 1H), 7.28(d, J=2.6Hz, 1H), 7.12-7.11(m, 1H), 6.87(d, J=2.6Hz, 1H), 5.38-5.18(m, 1H), 4.47-4.43(m, 1H), 4.11(d, J=10.4Hz, 1H), 3.99(d, J=10.4Hz, 1H), 3.93-3.82(m, 3H), 3.70-3.64(m, 3H), 3.11-3.05(m, 4H), 3.09-2.99(m, 2H), 2.88-2.77(m, 2H), 2.36 -2.30(m, 2H), 2.15-2.07(m, 1H), 2.08-2.00(m, 1H), 1.91-1.81(m, 3H), 1.47-1.26(m, 3H), 0.86(t, J=7.4Hz, 3H)
表4の実施例は、実施例8に記載の手順に従って適当な出発物質から製造した。
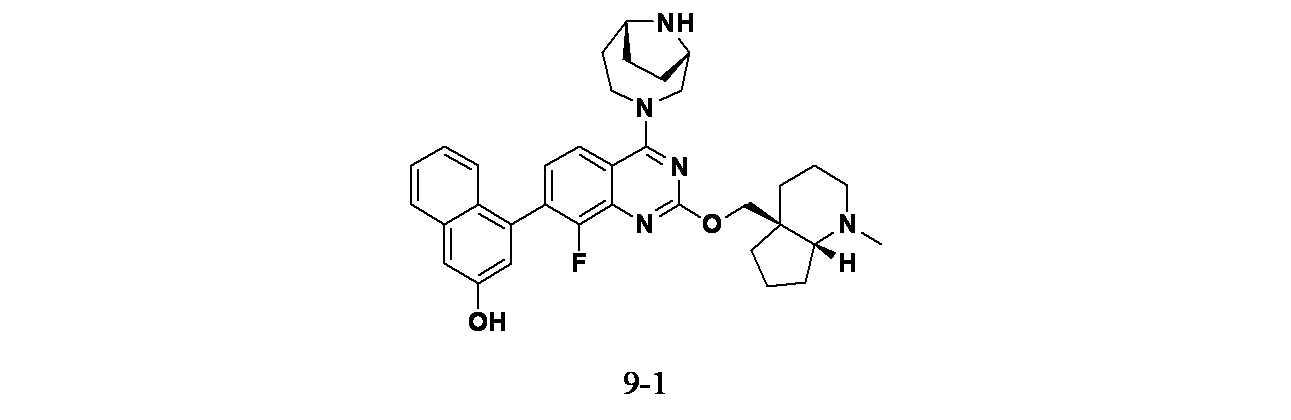
実施例9-1
4-(2-{[(4aS,7aR)-1-メチル-オクタヒドロ-1H-シクロペンタ[b]ピリジン-4a-イル]メトキシ}-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)ナフタレン-2-オール
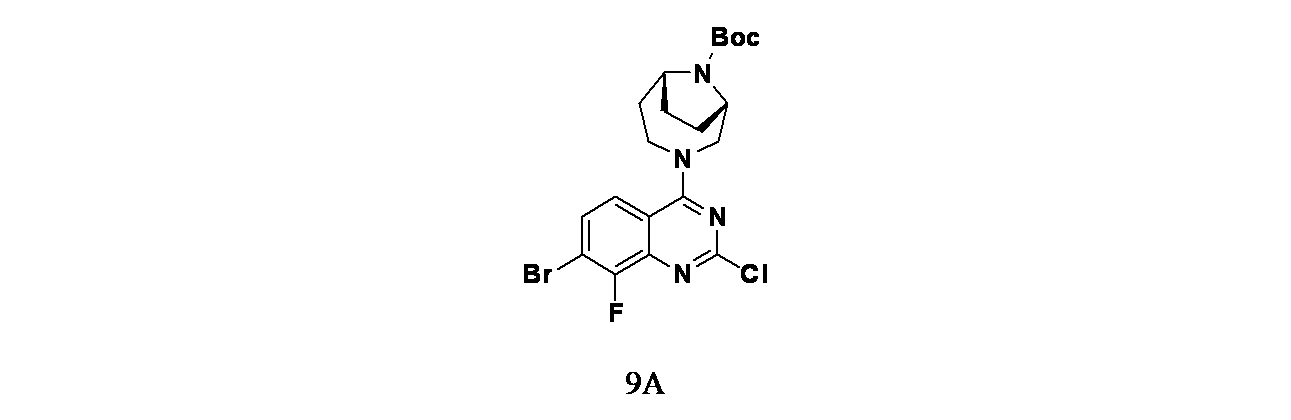
中間体9A: tert-ブチル(1S,6R)-3-(7-ブロモ-2-クロロ-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
7-ブロモ-2,4-ジクロロ-8-フルオロキナゾリン(250mg、0.845mmol)およびDIEA(148μL、0.845mmol)/THF(30mL)の溶液に、窒素下0℃でtert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(191mg、0.845mmol)を加え、混合物を0℃で1時間、および室温で18時間撹拌した。この混合物を次いで濃縮した。次いでこの混合物をEtOAc(35mL)で希釈し、飽和炭酸水素ナトリウム水溶液(2x35mL)で洗浄した。酢酸エチル層を硫酸ナトリウムで乾燥し、濾過し、濃縮した。得られた粗製生成物についてISCOフラッシュクロマトグラフィー(シリカゲル、グラジエント: ヘキサン/EtOAc 100:0~60:40)を行い、tert-ブチル(1S,6R)-3-(7-ブロモ-2-クロロ-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(362mg、0.708mmol、収率84%)を白色発泡体として得た。LCMS(ESI)m/z: 486 [M+H]
+; LC保持時間: 1.14分(Waters Acquity UPLC BEH C18、2.1x50mm、粒子径:1.7μm; 移動相A: 水(0.05%TFA含有); 移動相B: ACN(0.05%TFA含有); グラジエント: 2~98%Bで1分かけて溶出後、次いで98%Bで0.5分間溶出; 流速: 0.8mL/分; 検出: MSおよびUV(220nm))
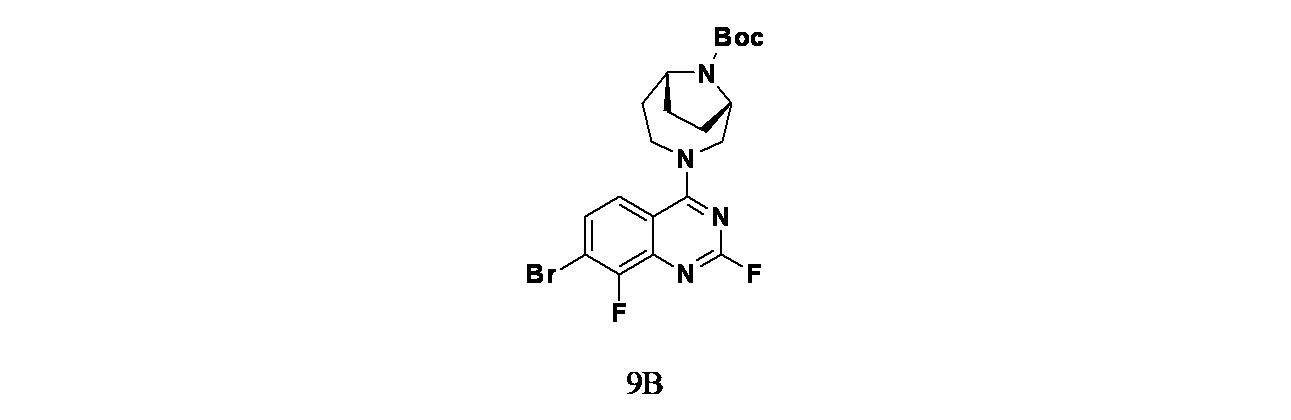
中間体9B: tert-ブチル(1S,6R)-3-(7-ブロモ-2,8-ジフルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
tert-ブチル(1S,6R)-3-(7-ブロモ-2-クロロ-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(362mg、0.745mmol)およびフッ化カリウム(87mg、1.490mmol)/DMSO(5.0mL)の混合物を100℃で2日間撹拌した。この混合物を次いでEtOAc(25mL)で希釈し、飽和炭酸水素ナトリウム水溶液(2x25mL)で洗浄した。酢酸エチル層を硫酸ナトリウムで乾燥し、濾過し、濃縮した。得られた粗製生成物についてISCOフラッシュクロマトグラフィー(シリカゲル、グラジエント: ヘキサン/EtOAc 100:0~40:60)を行い、tert-ブチル(1S,6R)-3-(7-ブロモ-2,8-ジフルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(270mg、0.547mmol、収率73.3%)を白色発泡体として得た。LCMS(ESI)m/z: 470 [M+H]
+; LC保持時間: 1.09分(Waters Acquity UPLC BEH C18、2.1x50mm、粒子径:1.7μm; 移動相A: 水(0.05%TFA含有); 移動相B: ACN(0.05%TFA含有); グラジエント: 2~98%Bで1分かけて溶出後、次いで98%Bで0.5分間溶出; 流速: 0.8mL/分; 検出: MSおよびUV(220nm))
中間体9C: tert-ブチル(1S,6R)-3-(7-ブロモ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メタノール(72.9mg、0.430mmol)およびtert-ブチル(1S,6R)-3-(7-ブロモ-2,8-ジフルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(202mg、0.430mmol)/THF(30mL)の溶液に、窒素下0℃でリチウムビス(トリメチルシリル)アミド(THF溶液、646μL、0.646mmol)を加え、混合物を0℃で1時間撹拌後、室温で18時間撹拌した。この混合物を次いで濃縮した。得られた粗製生成物をクロマトグラフィー(ISCO C18 100gカラム、流速=60mL/分、グラジエント=20%A~100%Bを20分で溶出;A=H
2O/ACN/TFA(90:10:0.1)、B=H
2O/ACN/TFA(10:90:0.1))で精製した。純粋なフラクションを合わせて濃縮し、tert-ブチル(1S,6R)-3-(7-ブロモ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートを白色固体として得た。LCMS(ESI)m/z: 619 [M+H]
+; 保持時間: 0.88分(Waters Acquity UPLC BEH C18、2.1x50mm、粒子径:1.7μm; 移動相A: 水(0.05%TFA含有); 移動相B: ACN(0.05%TFA含有); グラジエント: 2~98%Bで1分かけて溶出後、次いで98%Bで0.5分間溶出; 流速: 0.8mL/分; 検出: MSおよびUV(220nm))
実施例9-1
4-(2-{[(4aS,7aR)-1-メチル-オクタヒドロ-1H-シクロペンタ[b]ピリジン-4a-イル]メトキシ}-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)ナフタレン-2-オール
tert-ブチル(1S,6R)-3-(7-ブロモ-8-フルオロ-2-(((4aS,7aR)-1-メチルオクタヒドロ-4aH-シクロペンタ[b]ピリジン-4a-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(20mg、0.032mmol)、2-(3-(メトキシメトキシ)ナフタレン-1-イル)-4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(10.67mg、0.034mmol)、[1,1'-ビス(ジ-tert-ブチルホスフィノ)フェロセン]ジクロロパラジウム(II)(1.054mg、1.617μmol)および2.0Mリン酸三カリウム(48.5μL、0.097mmol)/1,4-ジオキサン(1mL)の混合物を窒素下50℃で18時間撹拌した。次いでこの混合物にEtOAc(5mL)を加え、酢酸エチル層を硫酸ナトリウムで乾燥し、濾過し、濃縮し、粗製生成物を得た。この粗製物質/DCM(0.6mL)の溶液、TES(1滴)およびTFA(0.4mL)を室温で30分間撹拌した。この混合物を次いで濃縮した。得られた粗製生成物を分取HPLC(Phenomenex、Luna 5μ、30x250mm、流速=30mL/分、グラジエント=20%A~100%Bを12分で溶出、A=H
2O/ACN/TFA(90:10:0.1)、B=H
2O/ACN/TFA(10:90:0.1))で精製した。純粋なフラクションをあわせてOasis MCX カチオンミックスモードポリマーカートリッジ(150mg)にロードし、カートリッジをメタノール(30mL)で洗浄し、生成物を0.1Nアンモニア/メタノール(5.0mL)で溶出した。溶離剤のアンモニアを蒸発させ、純粋な生成物を次いでACN/H
2O(1:1、5mL)から凍結乾燥し、所期の生成物(9.15mg、0.015mmol、収率47.2%)を白色粉末として得た。LCMS(ESI)m/z: 582 [M+H]
+; LC保持時間: 0.69分(Waters Acquity UPLC BEH C18、2.1x50mm、粒子径:1.7μm; 移動相A: 水(0.05%TFA含有); 移動相B: ACN(0.05%TFA含有); グラジエント: 2~98%Bで1分かけて溶出後、次いで98%Bで0.5分間溶出; 流速: 0.8mL/分; 検出: MSおよびUV(220nm));
1H NMR(499MHz、メタノール-d
4) δ 7.99(d, J=8.7Hz, 1H), 7.75(d, J=8.3Hz, 1H), 7.51-7.40(m, 2H), 7.30-7.21(m, 3H), 7.12(d, J=2.3Hz, 1H), 4.77-4.61(m, 1H), 4.50 (br d, J=11.2Hz, 1H), 4.30(d, J=10.7Hz, 1H), 4.13-3.94(m, 2H), 3.86-3.78(m, 2H), 3.72-3.58(m, 1H), 2.90 (br t, J=5.7Hz, 1H), 2.69(ddd, J=11.6, 7.4, 4.2Hz, 1H), 2.41-2.32(m, 4H), 2.26-2.08(m, 3H), 2.07-1.93(m, 2H), 1.90-1.69(m, 10H), 1.67 (br s, 1H), 1.64-1.55(m, 1H)
表5の実施例は、実施例9-1に記載の手順に従って適当な出発物質から製造した。
実施例10-1
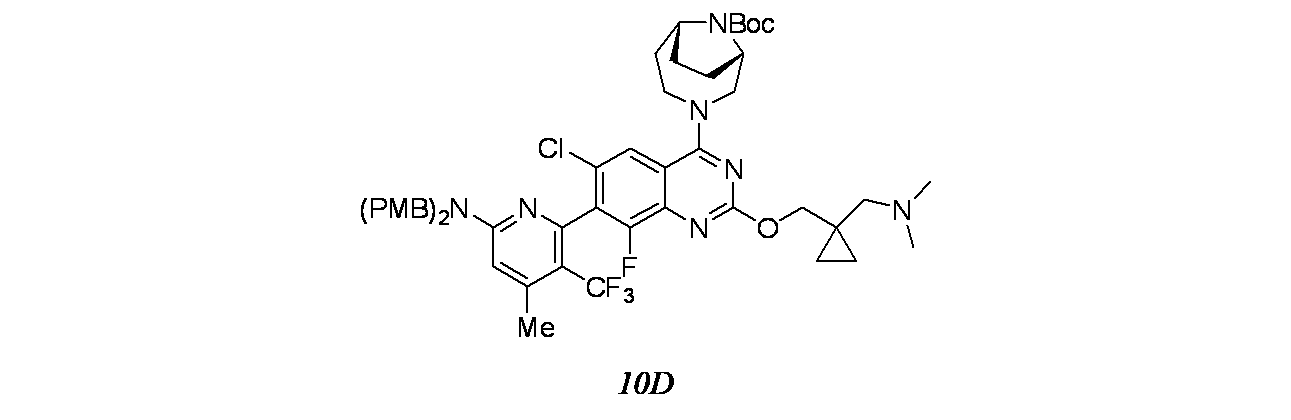
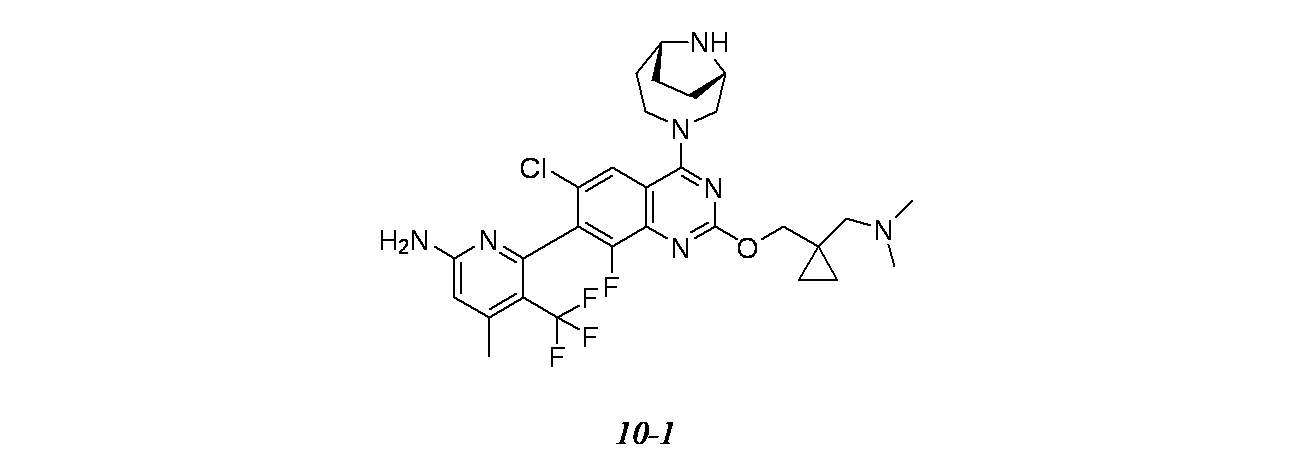
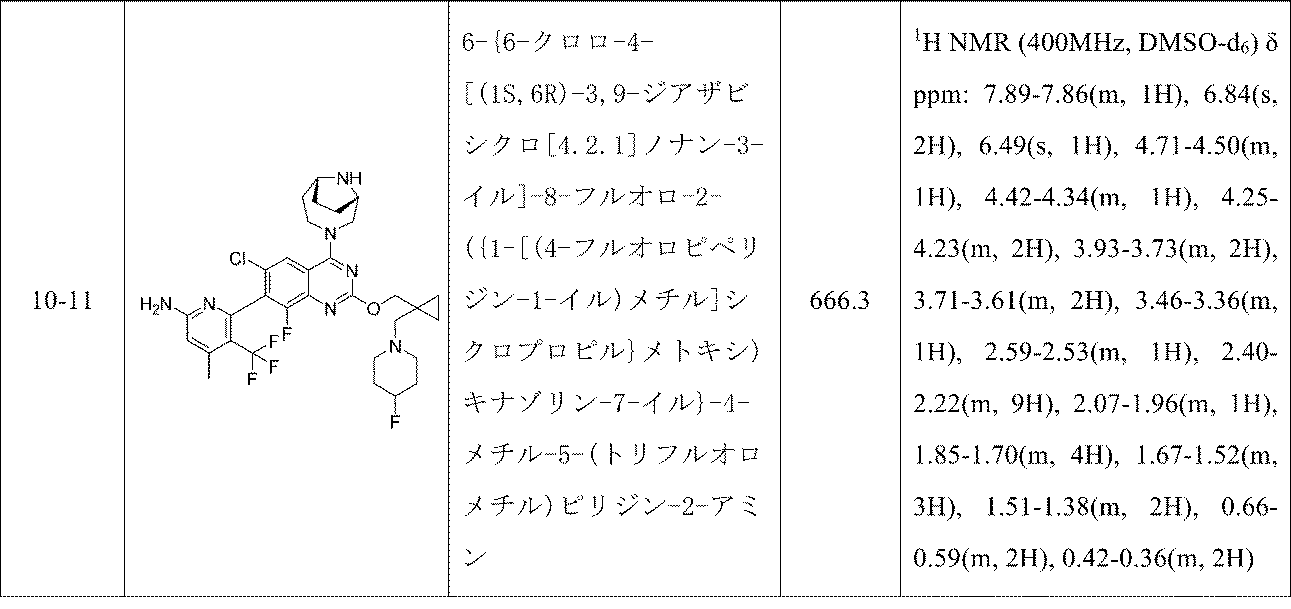
6-{6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-7-イル}-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
中間体10A: {1-[(ジメチルアミノ)メチル]シクロプロピル}メタノールの製造
1-(ジメチルカルバモイル)シクロプロパン-1-カルボン酸メチル(8g、46.7mmol)/THF(150mL)の溶液に、0℃でLiAlH
4(2.4M、THF溶液、38.9mL、93mmol)をゆっくりと加え、この反応混合物を室温で4時間撹拌した。反応を冷却し、水(20mL)、10%NaOH溶液(40mL)および水(40mL)でクエンチし、酢酸エチルで抽出した。有機層を合わせて水、食塩水で洗浄し、無水Na
2SO
4で乾燥し
、濾過し、減圧濃縮し、{1-[(ジメチルアミノ)メチル]シクロプロピル}メタノール(3.7g、28.6mmol、収率61.3%)を黄色液体として得た。
1H NMR(300MHz、CDCl
3) δ ppm 5.30-4.15(m, 1H), 3.55(s, 2H), 2.41(s, 2H), 2.31(s, 6H), 0.54-0.47(m, 2H), 0.39-0.32(m, 2H)
中間体10B: tert-ブチル3-[7-(6-{ビス[(4-メトキシフェニル)-メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}-メトキシ)-8-フルオロキナゾリン-4-イル]-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
{1-[(ジメチルアミノ)メチル]シクロプロピル}メタノール/THF(2mL)の溶液に、0℃でNaH(29.1mg、0.727mmol)を加え、混合物を30分間同温度で撹拌した。次いで、中間体3Kを加え、混合物を2時間かけて徐々に室温に戻した。この反応混合物を次いで飽和塩化アンモニウム水溶液でクエンチし、酢酸エチルで抽出した。有機層を合わせて水、食塩水で洗浄し、無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。これをシリカゲルカラムクロマトグラフィー(CombiFlash、40g RediSep(登録商標)カラム、50~60%EtOAc/石油エーテル)で精製し、tert-ブチル3-[7-(6-{ビス[(4-メトキシフェニル)-メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}-メトキシ)-8-フルオロキナゾリン-4-イル]-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(250mg、0.268mmol、収率73.6%)を淡黄色固体として得た。MS(ESI)m/z: 934.3(M+H)
+
中間体10C: 7-(6-{ビス[(4-メトキシフェニル)メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-4-オールの製造
tert-ブチル3-[7-(6-{ビス[(4-メトキシフェニル)-メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}-メトキシ)-8-フルオロキナゾリン-4-イル]-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(150mg、0.161mmol)/エタノール(2mL)およびTHF(2mL)の溶液に、NaOH(1M水溶液、1.605mL、1.605mmol)を加え、この反応混合物を70℃で16時間撹拌した。この反応混合物を次いで濃縮し、得られた粗製残渣を酢酸エチルに溶解し、食塩水で洗浄し、減圧濃縮し、7-(6-{ビス[(4-メトキシフェニル)メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-4-オールの粗製残渣(70mg、0.095mmol、収率58.9%)を得た。MS(ESI)m/z: 740.2(M+H)
+
中間体10D: tert-ブチル(1S,6R)-3-[7-(6-{ビス[(4-メトキシフェニル)メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-4-イル]-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
7-(6-{ビス[(4-メトキシフェニル)メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-4-オール(70mg、0.095mmol)、tert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(25.7mg、0.113mmol)、およびBOP(62.7mg、0.142mmol)/ACN(3mL)の溶液に、TEA(0.026mL、0.189mmol)を加え、この混合物を80℃で8時間撹拌した。この反応混合物を次いで減圧濃縮した。得られた粗製化合物をシリカゲルカラムクロマトグラフィーで精製し、tert-ブチル(1S,6R)-3-[7-(6-{ビス[(4-メトキシフェニル)メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-4-イル]-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(70mg、0.074mmol、収率78%)を得た。MS(ESI)m/z: 948.2(M+H)
+
実施例10-1
6-{6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-7-イル}-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
TFA(1.5mL、19.47mmol)およびトリエチルシラン(0.5mL、3.13mmol)の撹拌溶液を室温でtert-ブチル(1S,6R)-3-[7-(6-{ビス[(4-メトキシフェニル)メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-4-イル]-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(70mg、0.074mmol)に加え、得られた混合物を40℃で24時間かけて加熱した。次いで、この反応混合物を減圧濃縮し、トルエン(2回)と共沸し、DIPEAで中和し、減圧濃縮し、粗製残渣を得た。これを分取HPLC(カラム/寸法:カラム: Waters XBridge C18(150mmx19mm ID、5μ); 移動相A=10mM酢酸アンモニウム、移動相B=アセトニトリル; 流速: 20mL/分;保持時間=1.748分)で精製し、6-{6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-7-イル}-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン(2.4mg、3.68μmol、収率4.98%)を得た。LCMS(ESI)m/z: 608.2 [M+H]
+;
1H NMR(400MHz、DMSO-d
6) δ ppm 7.87(s, 1H), 6.85(s, 2H), 6.50(s, 1H), 5.36-5.01(m, 1H), 4.40-4.32(m, 1H), 4.28-4.17(m, 2H), 4.13-4.06(m, 1H), 4.03-3.84(m, 2H), 3.79-3.62(m, 2H), 2.38(m, 4H), 2.32-2.27(m, 2H), 2.21(m, 7H), 1.91(m, 3H), 1.72-1.60(m, 1H), 0.64(m, 2H), 0.46-0.38(m, 2H)
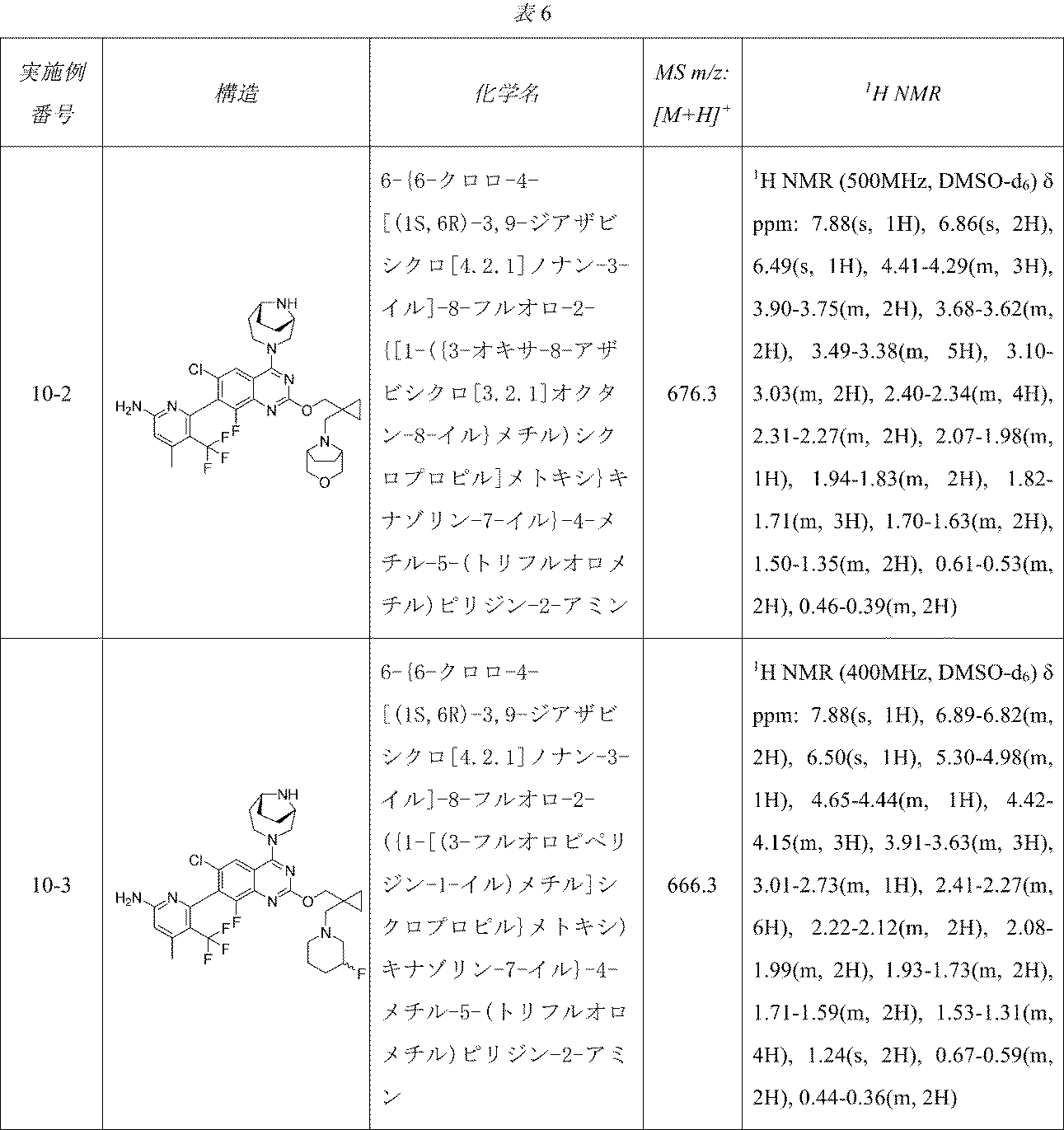
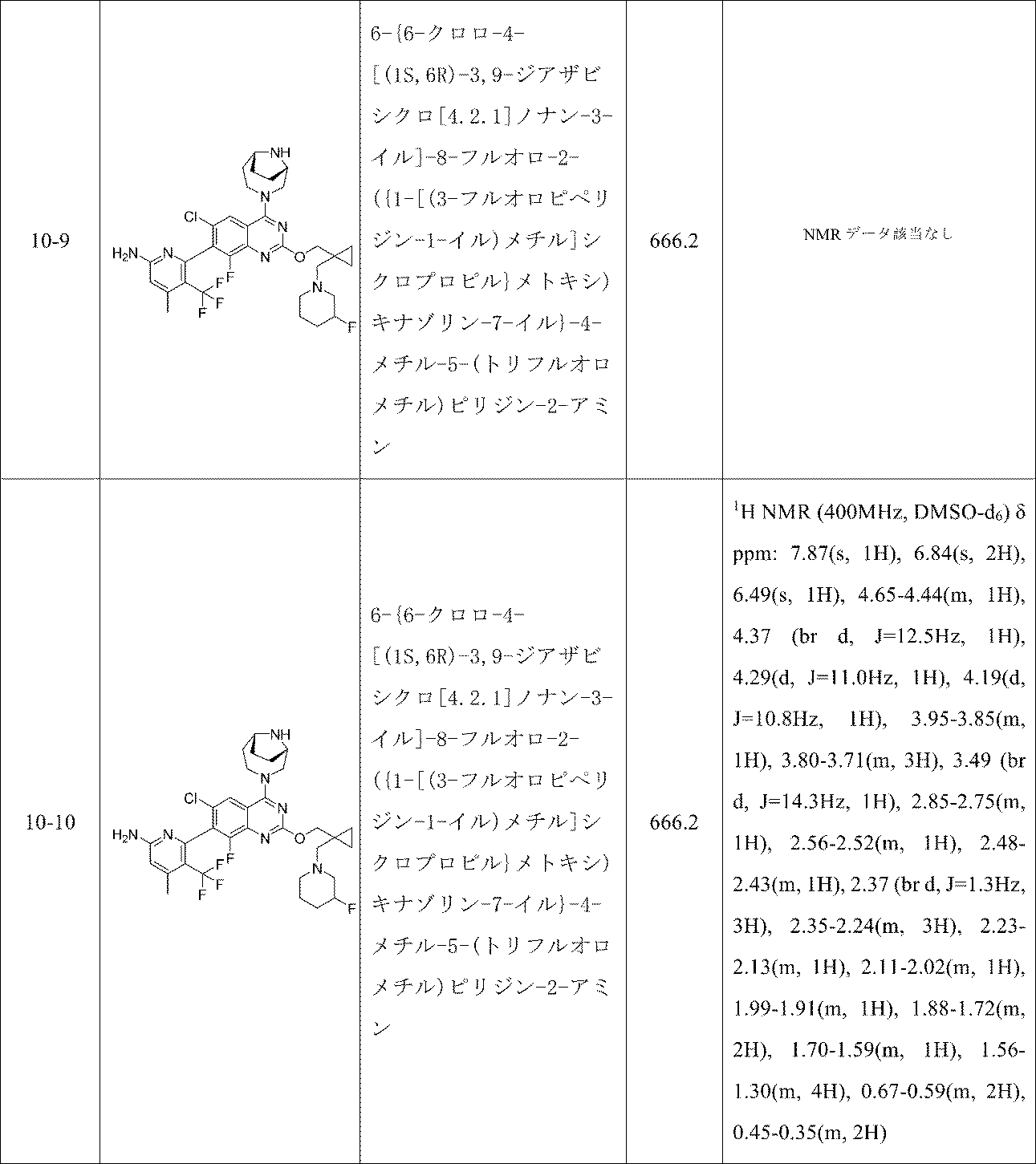
表6の実施例は、実施例10-1に記載の手順に従って適当な出発物質から製造した。
実施例11-1
4-{4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-7-イル}-5-エチニル-6-フルオロナフタレン-2-オール
中間体11A: tert-ブチル(1S,6R)-3-[7-ブロモ-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-4-イル]-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
(1-((ジメチルアミノ)メチル)シクロプロピル)メタノール(186mg、1.441mmol)/THF(2mL)の溶液に、NaH(57.6mg、1.441mmol)を0℃でゆっくりと加え、この混合物を同温度で30分間撹拌した。次いで、tert-ブチル(1S,6R)-3-(7-ブロモ-2-クロロ-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(350mg、0.720mmol)を加え、混合物をゆっくりと室温に加温し、2時間撹拌した。この反応混合物を次いで飽和塩化アンモニウム溶液でクエンチし、酢酸エチルで抽出した。有機層を合わせて水および食塩水で洗浄し、Na
2SO
4で乾燥し、濾過し、濃縮し、粗製生成物:tert-ブチル(1S,6R)-3-(7-ブロモ-2-((1-((ジメチルアミノ)メチル)シクロプロピル)メトキシ)-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(350mg、0.605mmol、収率84%)を得た。MS(ESI)m/z: 580.1(M+H+2)
中間体11B: tert-ブチル(1S,6R)-3-[2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロ-7-[7-フルオロ-3-(メトキシメトキシ)-8-{2-[トリス(プロパン-2-イル)シリル]エチニル}ナフタレン-1-イル]キナゾリン-4-イル]-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
tert-ブチル(1S,6R)-3-(7-ブロモ-2-((1-((ジメチルアミノ)メチル)シクロプロピル)メトキシ)-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(300mg、0.519mmol)、((2-フルオロ-6-(メトキシメトキシ)-8-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ナフタレン-1-イル)エチニル)トリイソプロピルシラン(319mg、0.622mmol)およびNa
2CO
3(0.778mL、1.556mmol)/1,4-ジオキサン(5mL)の脱気した溶液に、ビス(トリフェニルホスフィン)ジクロロパラジウム(II)(36.4mg、0.052mmol)を加え、この混合物を100℃で2時間加熱した。反応を水で次いで希釈し、酢酸エチルで抽出した。有機層を合わせて水および食塩水で洗浄し、Na
2SO
4で乾燥し、濾過し、濃縮し、粗製残渣を得た。これをシリカゲルカラムクロマトグラフィーで精製し、tert-ブチル(1S,6R)-3-(2-((1-((ジメチルアミノ)メチル)シクロプロピル)メトキシ)-8-フルオロ-7-(7-フルオロ-3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-イル)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(130mg、0.147mmol、収率28.4%)を得た。MS(ESI)m/z: 884.5(M+1)
実施例11-1
4-{4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-2-({1-[(ジメチルアミノ)メチル]シクロプロピル}メトキシ)-8-フルオロキナゾリン-7-イル}-5-エチニル-6-フルオロナフタレン-2-オール
4-(4-((1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル)-2-((1-((ジメチルアミノ)メチル)シクロプロピル)メトキシ)-8-フルオロキナゾリン-7-イル)-6-フルオロ-5-((トリイソプロピルシリル)エチニル)ナフタレン-2-オール(120mg、0.162mmol)/DMF(3mL)の溶液に、CsF(123mg、0.811mmol)を室温で加え、この混合物を55℃で24時間撹拌した。この反応混合物を次いで濃縮し、粗製残渣を得た。粗製物質を分取HPLC(条件: カラム: Waters XBridge C18(19x150mm、粒子径: 5μm); 移動相A: 10mM酢酸アンモニウム; 移動相B: アセトニトリル; グラジエント: 10~35%Bで20分かけて溶出後、次いで100%Bで5分間溶出; 流速: 20mL/分)、続いてSFCで精製し、4-(4-((1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル)-2-((1-((ジメチルアミノ)メチル)シクロプロピル)メトキシ)-8-フルオロキナゾリン-7-イル)-5-エチニル-6-フルオロナフタレン-2-オール(0.9mg、1.388μmol、収率0.856%)を得た。MS(ESI)m/z: 584.3 [M+H]
+
表7の実施例は、実施例11-1に記載の手順に従って中間体9Aおよび適当な出発物質から製造した。
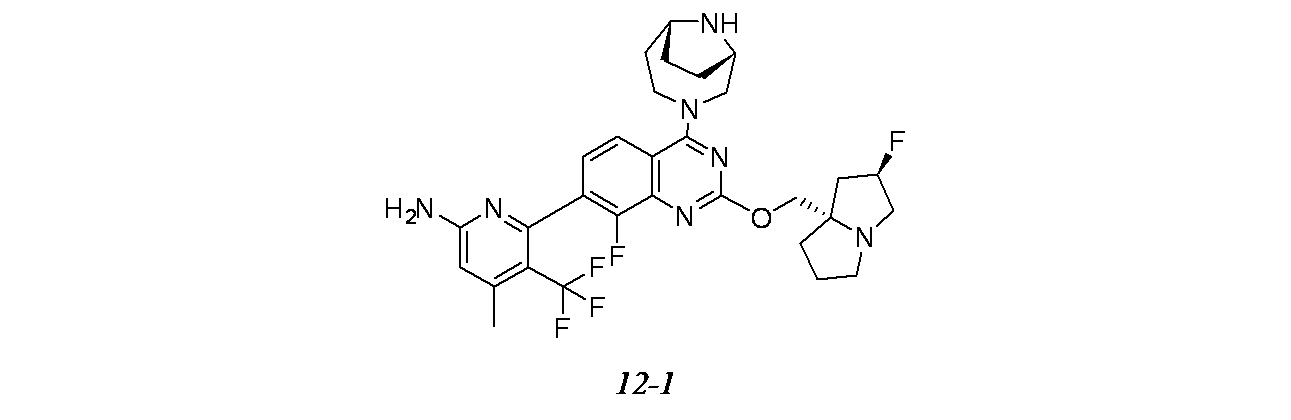
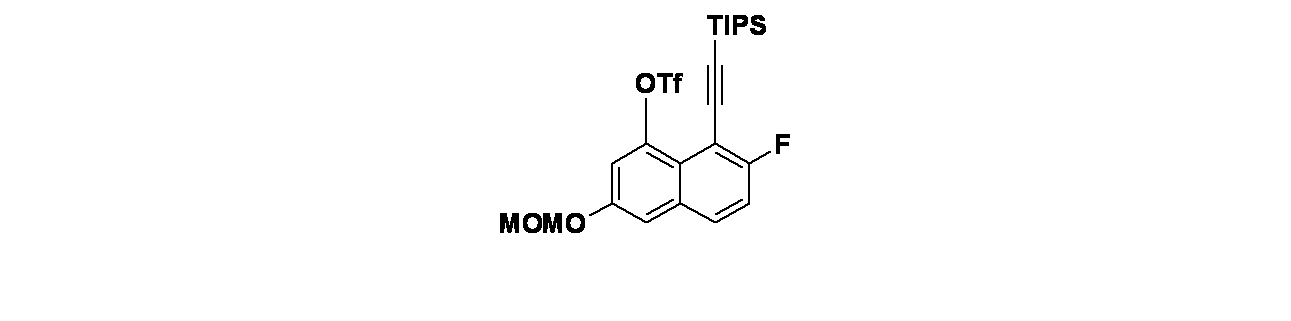
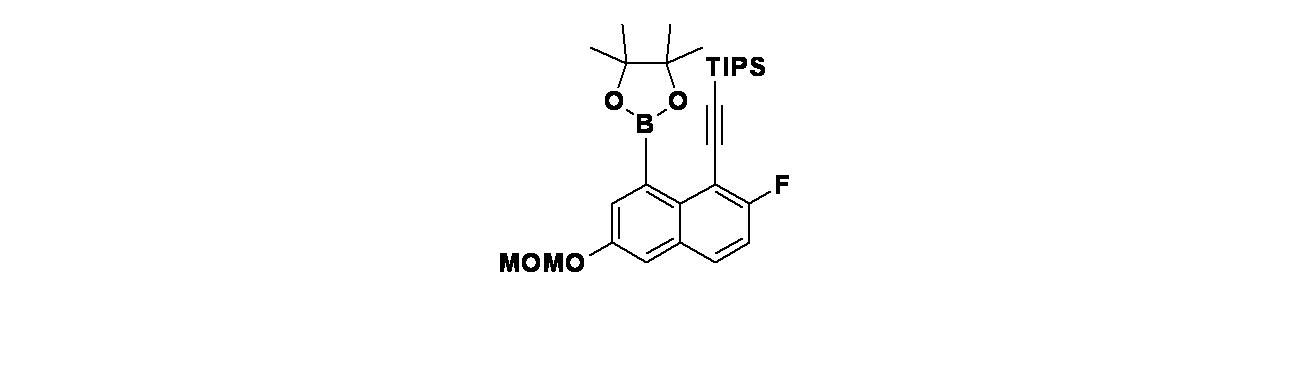
実施例12-1
4-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-5-フルオロナフタレン-2-オール
中間体12A-1:tert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-7-ブロモ-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
((2R,7aS)-2-フルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メタノール(246mg、1.544mmol)/THF(20mL)の撹拌溶液に、水素化ナトリウム(49.4mg、2.059mmol)を加え、混合物を0℃で1時間撹拌した。この反応混合物に、tert-ブチル(1S,6R)-3-(7-ブロモ-2-クロロ-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(500mg、1.029mmol)を加え、ゆっくりと室温に加温し、2時間撹拌した。この反応混合物を水でクエンチし、酢酸エチル(50mLx2)で抽出し、無水硫酸ナトリウムで乾燥し、濾過し、減圧濃縮し、tert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-7-ブロモ-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートを黄色固体として得た。MS(ESI)m/z: 608.2 [M+H]
+
中間体12A: tert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-8-フルオロ-7-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
tert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-7-ブロモ-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(120mg、0.197mmol)、ビス(ピナコラト)ジボロン(75mg、0.296mmol)、酢酸カリウム(38.7mg、0.394mmol)/1,4-ジオキサン(3mL)の脱気した溶液に、PdCl
2(dppf)(14.43mg、0.020mmol)を加え、この混合物を120℃で1.5時間加熱した。この反応混合物を次いでセライト濾過し、濾液をさらにワークアップ、または精製せずに次のステップに用いた。
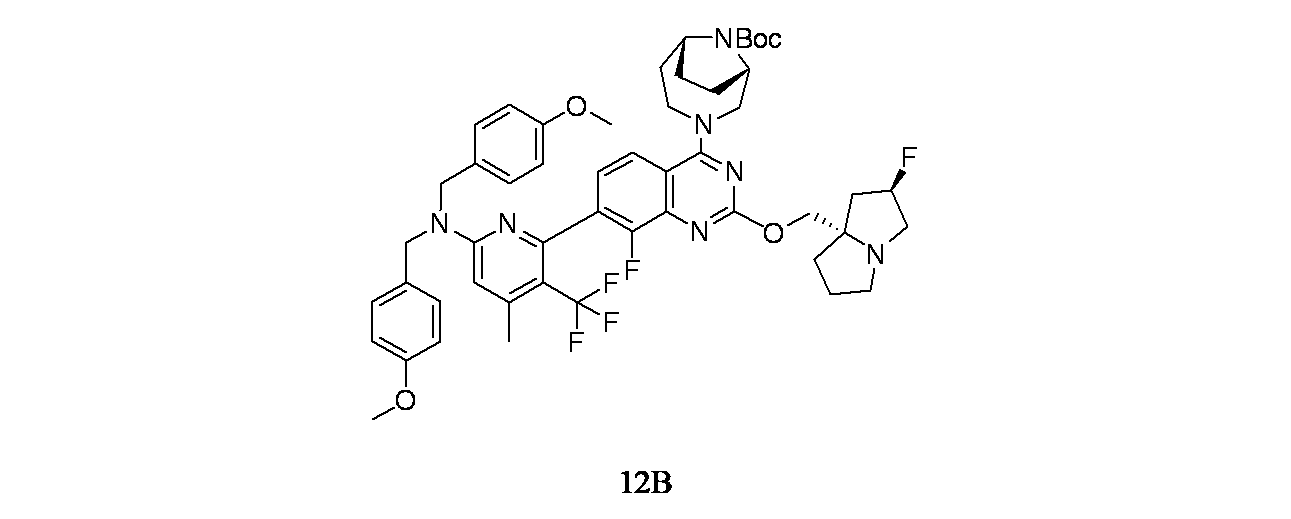
中間体12B: tert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-7-(6-{ビス[(4-メトキシフェニル)メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
tert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-8-フルオロ-7-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(粗製生成物、100mg、0.153mmol)/1,4-ジオキサン(2mL)、6-クロロ-N,N-ビス(4-メトキシベンジル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン(CAS: 2411793-22-9、83mg、0.183mmol)および1.5Mリン酸三カリウム水溶液(0.305mL、0.458mmol)の脱気した溶液に、PdCl
2(dppf)(11.16mg、0.015mmol)を加え、この混合物を100℃で6時間撹拌した。この反応混合物を水で次いで希釈し、酢酸エチルで抽出した。有機層を合わせて水および食塩水で洗浄し、Na
2SO
4で乾燥し、濾過し、濃縮し、粗製残渣を得た。得られた粗製残渣をシリカゲルカラムクロマトグラフィー(24g RediSep(登録商標)カラム、溶出: EtOAc/石油エーテルで60~100%のグラジエント)で精製した。所期の生成物を含むフラクションを濃縮し、tert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-7-(6-{ビス[(4-メトキシフェニル)メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(70mg、0.074mmol、収率48.6%)を得た。LCMS(ESI)m/z: 944.3 [M+H]
+
実施例12-1
4-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-5-フルオロナフタレン-2-オール
TFA(1.5mL、19.47mmol)およびトリエチルシラン(0.5mL、3.13mmol)の撹拌溶液をtert-ブチル(1S,6R)-3-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-7-(6-{ビス[(4-メトキシフェニル)メチル]アミノ}-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-8-フルオロキナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(70mg、0.074mmol)に室温で加え、この混合物を40℃で24時間加熱した。次いで、この反応混合物を減圧濃縮し、トルエン(2回)と共沸し、DIPEAで中和し、減圧濃縮し、粗製残渣を得た。これを分取HPLC[HPLC条件:カラム/寸法: カラム: Kinetex EVO(250mmx21mm ID、5μ); 移動相A=10mM重炭酸アンモニウム水溶液(pH 9.5)、移動相B=アセトニトリル:MeOH(1:1)、流速: 19mL/分;保持時間=11.72分]で精製し、6-(2-{[(2R,7aS)-2-フルオロ-ヘキサヒドロ-1H-ピロリジン-7a-イル]メトキシ}-4-[(1S,6R)-3,9- ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン(18mg、0.030mmol、収率40.2%)を得た。LCMS(ESI)m/z: 604.5 [M+H]
+1H NMR(400MHz、DMSO-d
6) δ ppm 7.86-7.79(m, 1H), 7.18-7.07(m, 1H), 6.82-6.71(m, 2H), 6.47-6.44(m, 1H), 5.39-5.15(m, 1H), 4.53-4.40(m, 1H), 4.13-4.05(m, 1H), 4.02-3.92(m, 1H), 3.88-3.77(m, 2H), 3.70-3.60(m, 2H), 3.46-3.38(m, 2H), 3.12-3.05(m, 2H), 3.04-2.98(m, 1H), 2.86-2.77(m, 1H), 2.37-2.35(m, 3H), 2.16-2.09(m, 1H), 2.01-1.92(m, 3H), 1.84-1.80(s, 3H), 1.79-1.68(m, 3H), 1.50-1.36(m, 2H)
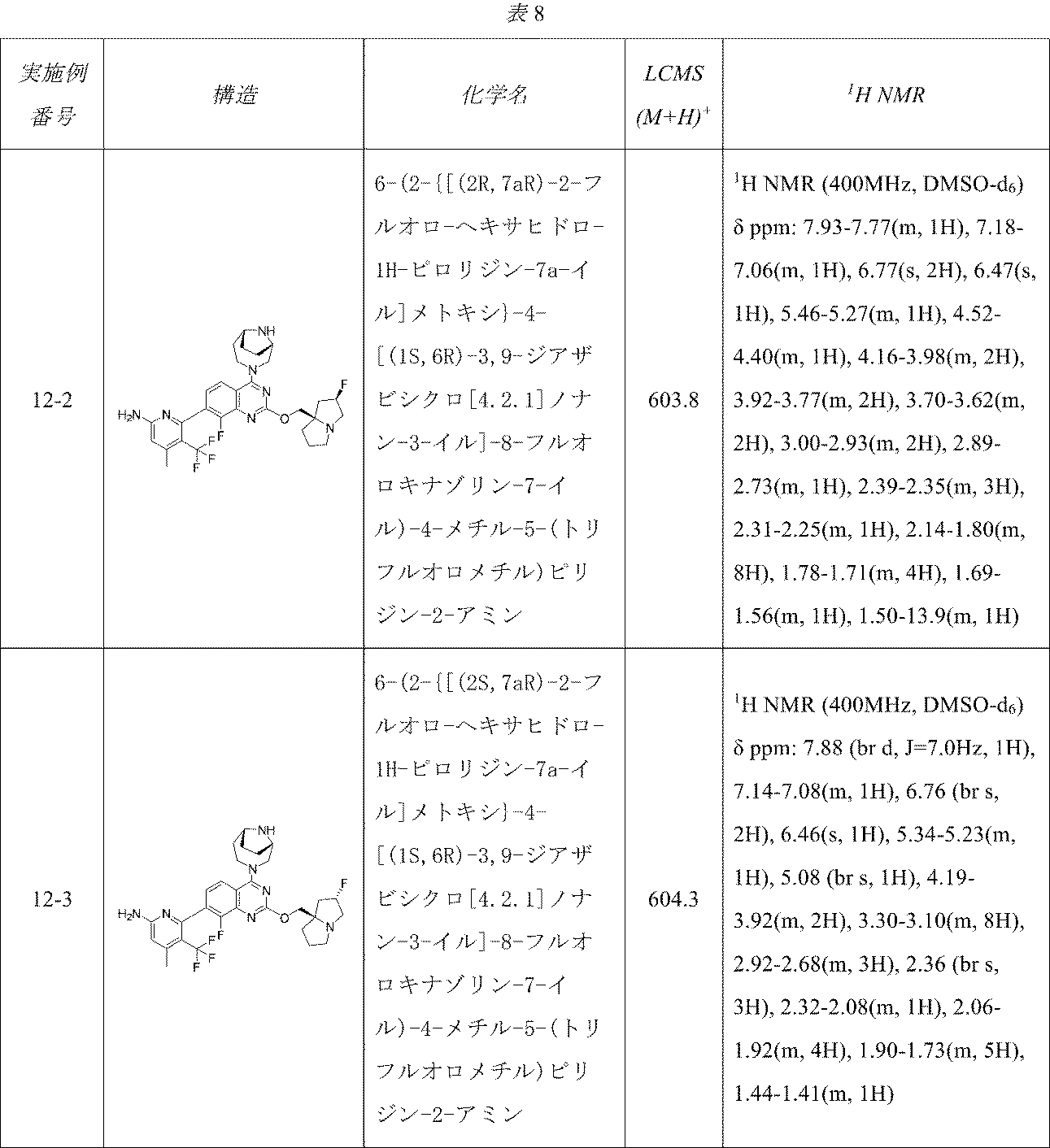
表8の実施例は、実施例12-1に記載の手順に従って適当な出発物質から製造した。
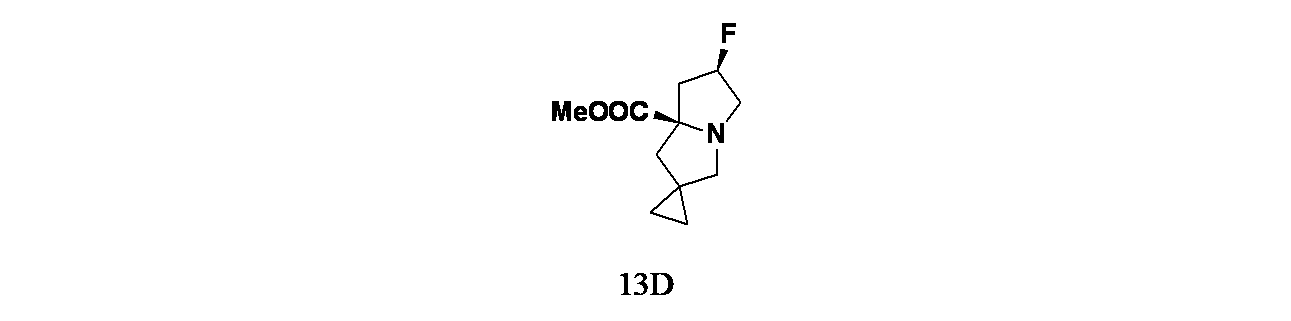
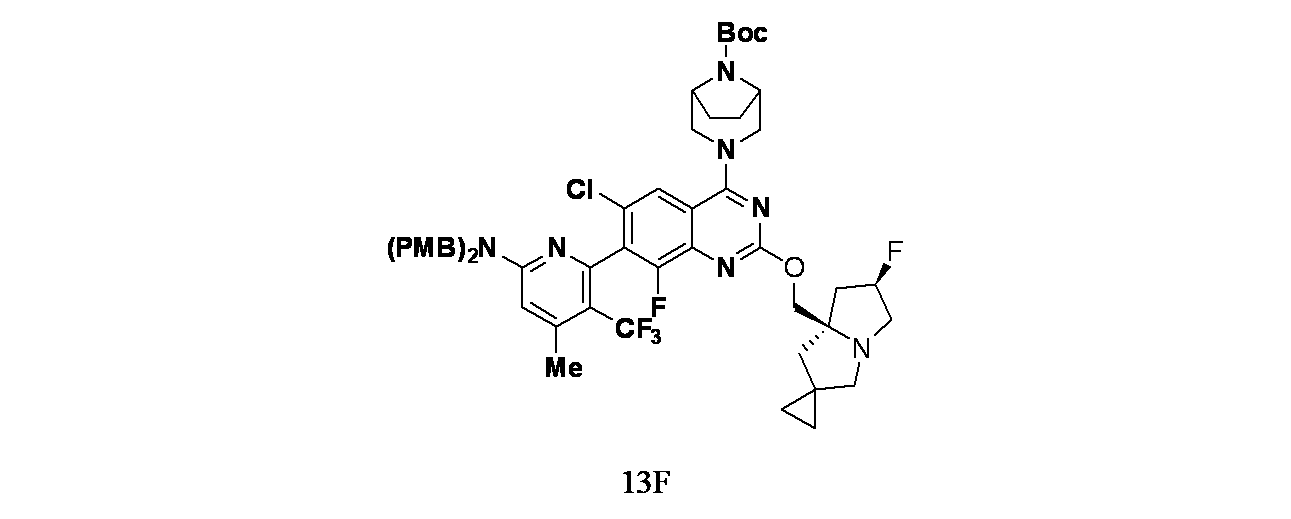
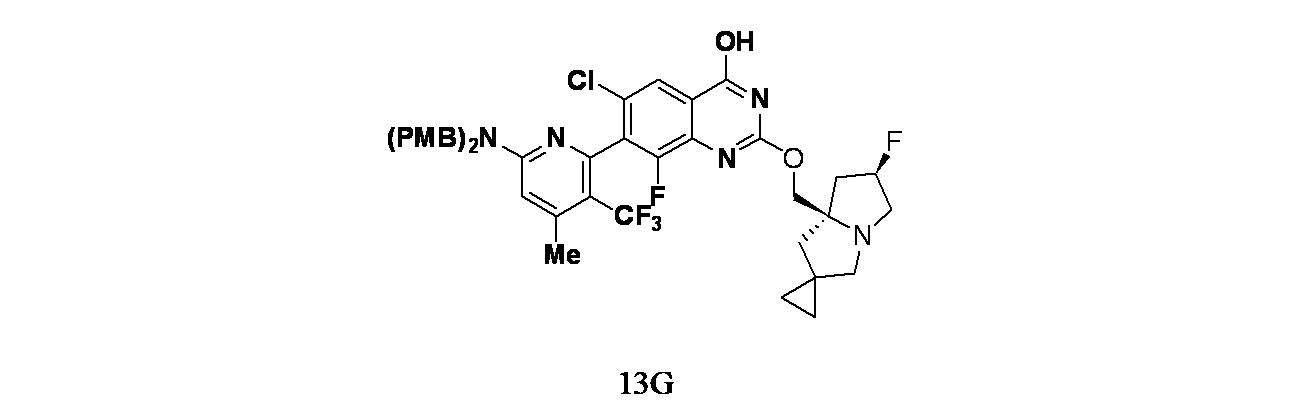
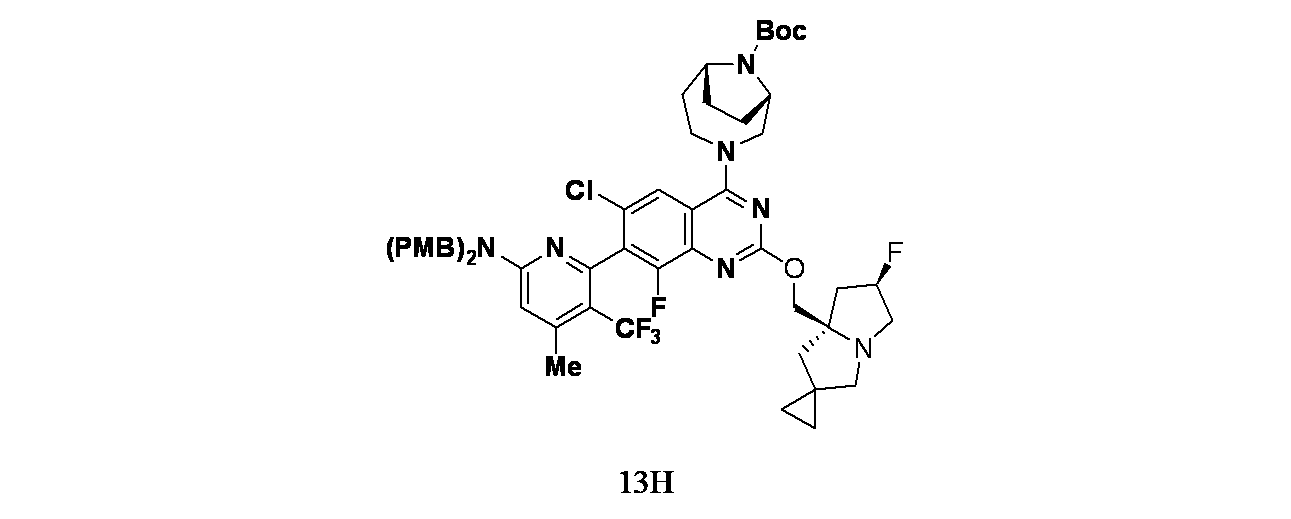
実施例13-1
6-(2-{[(6'R,7'aR)-6'-フルオロ-ヘキサヒドロスピロ[シクロプロパン-1,2'-ピロリジン]-7'a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
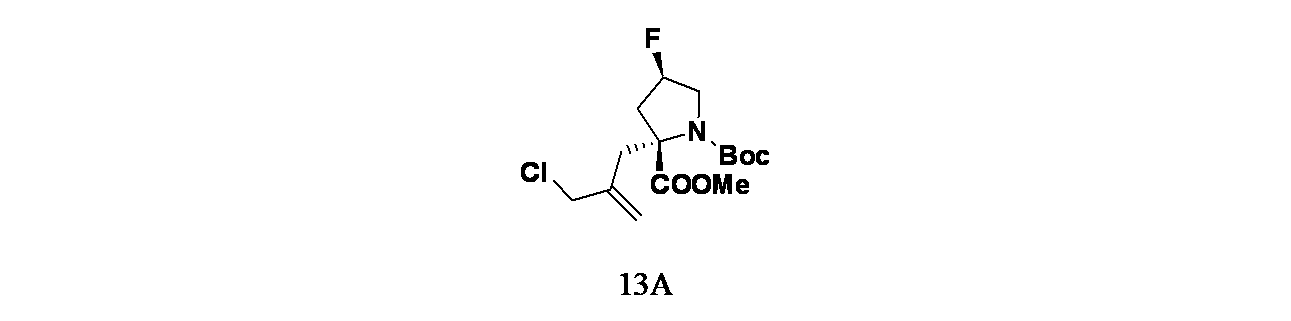
中間体13A: 1-(tert-ブチル)2-メチル(2R,4R)-2-(2-(クロロメチル)アリル)-4-フルオロピロリジン-1,2-ジカルボキシレートの製造
1-(tert-ブチル)2-メチル(2S,4R)-4-フルオロピロリジン-1,2-ジカルボキシレート(50g、202mmol)の撹拌溶液に、窒素雰囲気下、温度を-45℃に保ったままでLiHMDS(1MTHF溶液、303mL、303mmol)を30分かけて滴下して加えた。同温度で1時間撹拌後、3-クロロ-2-(クロロメチル)プロパ-1-エン(30.3g、243mmol)/無水THF(300.0mL)の溶液を滴下して加えた。この反応混合物をゆっくりと2時間かけて室温に戻し、16時間撹拌した。この反応混合物を慎重に飽和塩化アンモニウム溶液(40mL)でクエンチし、酢酸エチル(2x200mL)で抽出した。有機層を合わせて水および飽和食塩水で洗浄し、無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製生成物を得た。得られた粗製残渣をカラムクロマトグラフィー(Grace、350g snap、乾式充填、シリカゲル(230-400mesh、溶出: 10~30%酢酸エチル/石油エーテル)で精製した。所期のフラクションを集めて減圧濃縮し、1-(tert-ブチル)2-メチル(2R,4R)-2-(2-(クロロメチル)アリル)-4-フルオロピロリジン-1,2-ジカルボキシレート(64g、91mmol、収率45%)を無色の液体として得た。LCMS(ESI)m/z: 336.1 [M+H]
+
中間体13B: (2R,4R)-2-(2-(クロロメチル)アリル)-4-フルオロピロリジン-2-カルボン酸メチルの製造
1-(tert-ブチル)2-メチル(2R,4R)-2-(2-(クロロメチル)アリル)-4-フルオロピロリジン-1,2-ジカルボキシレート(64g、191mmol)/無水DCM(600mL)の撹拌溶液に、HCl(4.0M、ジオキサン溶液、119mL、476mmol)を窒素雰囲気下0℃で加えた。この反応混合物を周囲温度で6時間撹拌し、続いて減圧濃縮し、(2R,4R)-2-(2-(クロロメチル)アリル)-4-フルオロピロリジン-2-カルボン酸メチル・HCl(50g、180mmol、収率94%)を薄茶色固体として得た。LCMS(ESI)m/z: 236.2 [M+H]
+
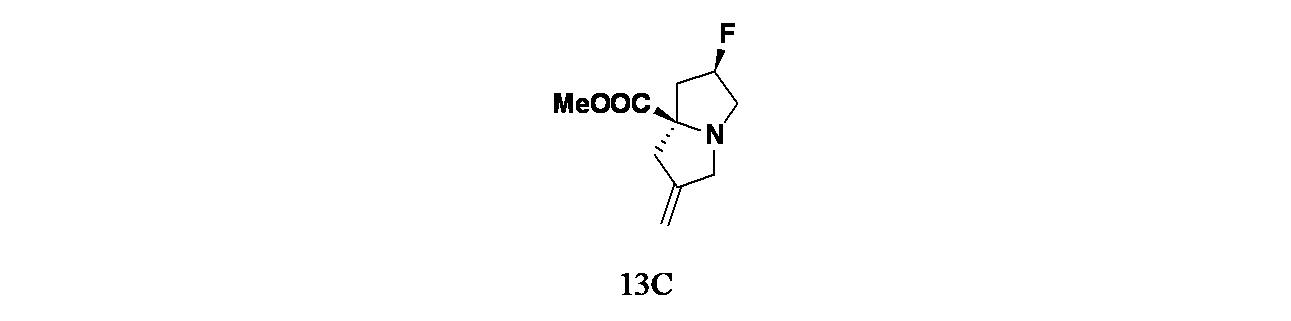
中間体13C: (2R,7aR)-2-フルオロ-6-メチレンテトラヒドロ-1H-ピロリジン-7a(5H)-カルボン酸メチルの製造
(2R,4R)-2-(2-(クロロメチル)アリル)-4-フルオロピロリジン-2-カルボン酸メチル(50g、212mmol)/無水アセトニトリル(500mL)の撹拌溶液に、TEA(44.4mL、318mmol)を加え、この混合物を周囲温度で16時間撹拌した。次いで反応混合物を水(50mL)で希釈し、次いで重炭酸水溶液(70mL)で処理した。得られた溶液をDCM(2x300mL)で抽出し、有機層を合わせて飽和食塩水(50mL)で洗浄し、無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、(2R,7aR)-2-フルオロ-6-メチレンテトラヒドロ-1H-ピロリジン-7a(5H)-カルボン酸メチル(35g、176mmol、収率83%)を褐色液体として得た。LCMS(ESI)m/z: 200.1 [M+H]
+
中間体13D: (6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-カルボン酸メチルの製造
(2R,7aR)-2-フルオロ-6-メチレンテトラヒドロ-1H-ピロリジン-7a(5H)-カルボン酸メチル(2.0g、10.04mmol)/無水トルエン(20mL)の撹拌溶液に、窒素雰囲気下、0℃でジヨードメタン(3.64mL、45.2mmol)を加えた。この反応混合物を0℃で30分間撹拌した後、ジエチル亜鉛(50.2mL、50.2mmol、1Mヘキサン溶液)を滴下して加えた。この反応混合物を室温に加温し、16時間撹拌した後、飽和NH
4Cl水溶液(5mL)でクエンチした。懸濁液を酢酸エチル(2x20mL)で抽出した。有機層を合わせて無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。これをカラムクロマトグラフィー(中性アルミナ、40~50%酢酸エチル/石油エーテル)で精製し、(6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-カルボン酸メチル(2.0g、8.05mmol、収率80%)を無色液体として得た。LCMS-ELSD(ESI)m/z: 204.1 [M+H]
+
中間体13E: ((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メタノールの製造
(6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-カルボン酸メチル(2.0g、9.38mmol)/THF(20mL)の撹拌溶液に、0℃でLiAlH
4(4.69mL、9.38mmol、2M、THF溶液)を加えた。この反応混合物を周囲温度で3時間撹拌し、続いて0℃で飽和塩化アンモニウム水溶液を加えてクエンチした。発泡が収まった後、この反応混合物に無水硫酸ナトリウムを加え、続いてDCM(20mL)を加えた。この反応混合物を20分間撹拌した後、濾過した。濾液を回収し、無水硫酸ナトリウムで乾燥し、減圧濃縮し、((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メタノール(110mg、0.592mmol、収率6%)を無色液体として得た。LCMS-ELSD(ESI)m/z: 186.2 [M+H]
+
中間体13F: tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレートの製造
((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メタノール(0.090g、0.485mmol)/無水THF(5.0mL)の撹拌溶液に、0℃で水素化ナトリウム(60%、鉱油に分散、0.019g、0.485mmol)を加え、この反応混合物を同温度で30分間撹拌した。温度を0℃に維持したまま中間体3K(0.2g、0.242mmol)/THF(2mL)の溶液を滴下して加えた。この混合物を2時間かけて室温に戻し、反応を飽和塩化アンモニウム水溶液(1mL)でクエンチし、酢酸エチル(3x5mL)で抽出した。有機層を合わせて無水硫酸ナトリウムで乾燥し、減圧濃縮し、粗製生成物を得た。得られた粗製生成物をカラムクロマトグラフィー(Grace、50g snap、乾式充填、中性アルミナ、50~100%酢酸エチル/石油エーテル)で精製した。所期のフラクションを集めて減圧濃縮し、tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(0.21g、0.199mmol、収率82%)を淡黄色固体として得た。LCMS(ESI)m/z: 990.4 [M+H]
+
中間体13G: 7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メトキシ)キナゾリン-4-オールの製造
tert-ブチル3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メトキシ)キナゾリン-4-イル)-3,8-ジアザビシクロ[3.2.1]オクタン-8-カルボキシレート(240mg、0.242mmol)/エタノール(5mL)の撹拌溶液に、10%NaOH溶液(2.42mL、2.423mmol)を加え、反応を70℃で48時間加熱した。この反応混合物を次いで減圧濃縮し、粗製残渣を得た。これをカラムクロマトグラフィー(Biotage、中性Al
2O
3、30~40%酢酸エチル/石油エーテル)で精製し、7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メトキシ)キナゾリン-4-オール(172mg、0.110mmol、収率46%)を粘着性の液体として得た。LCMS(ESI)m/z: 796.2 [M+H]
+
中間体13H: tert-ブチル(1S,6R)-3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレートの製造
7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メトキシ)キナゾリン-4-オール(0.17g、0.214mmol)/アセトニトリル(2mL)の撹拌溶液に、BOP(0.142g、0.320mmol)およびTEA(0.045mL、0.320mmol)を加えた。5分間撹拌後、tert-ブチル(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(0.097g、0.427mmol)を加え、混合物を40℃で16時間撹拌した。この反応混合物を次いで水(10mL)でクエンチし、酢酸エチル(2x15mL)で抽出した。有機層を食塩水(10mL)で洗浄し、無水硫酸ナトリウムで乾燥し、濾過し、濾液を濃縮し、粗製生成物(70mg)を得た。得られた残渣をカラムクロマトグラフィー(Biotage、中性アルミナ、30~40%酢酸エチル/石油エーテル)で精製し、tert-ブチル(1S,6R)-3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(150mg、0.103mmol、48%収率)を得た。LCMS(ESI)m/z: 1004.4 [M+H]
+
実施例13-1
6-(2-{[(6'R,7'aR)-6'-フルオロ-ヘキサヒドロスピロ[シクロプロパン-1,2'-ピロリジン]-7'a-イル]メトキシ}-6-クロロ-4-[(1S,6R)-3,9-ジアザビシクロ[4.2.1]ノナン-3-イル]-8-フルオロキナゾリン-7-イル)-4-メチル-5-(トリフルオロメチル)ピリジン-2-アミン
tert-ブチル(1S,6R)-3-(7-(6-(ビス(4-メトキシベンジル)アミノ)-4-メチル-3-(トリフルオロメチル)ピリジン-2-イル)-6-クロロ-8-フルオロ-2-(((6'R,7a'R)-6'-フルオロジヒドロ-1'H,3'H-スピロ[シクロプロパン-1,2'-ピロリジン]-7a'(5'H)-イル)メトキシ)キナゾリン-4-イル)-3,9-ジアザビシクロ[4.2.1]ノナン-9-カルボキシレート(0.15g、0.149mmol)/TFA(1mL)の撹拌溶液に、水(0.01mL)およびトリエチルシラン(0.024mL、0.149mmol)を加えた。反応を40℃で16時間撹拌し、反応混合物を次いで減圧濃縮し、粗製残渣を得た。これを分取HPLC[カラム: Xbridge C18(250x19)mm、5μ、移動相A: 0.1%ギ酸アンモニウム水; 移動相B: アセトニトリル、流速: 15mL/分、グラジエント: 30~70%Bを15分で溶出]で精製し、所期の化合物(10mg、0.012mmol、収率8%)を灰白色固体として得た。LCMS(ESI)m/z: 664.4 [M+H]
+;
1H NMR(400MHz、CD
3SOCD
3、298K) δ: 8.27-8.16(m, 1H), 7.96-7.78(m, 1H), 6.92-6.76(m, 1H), 6.50(s, 1H), 5.54-5.24(m, 1H), 4.43-4.24(m, 1H), 4.19-4.03(m, 1H), 3.95-3.83(m, 1H), 3.82-3.75(m, 2H), 3.74-3.62(m, 2H), 3.34-3.23(m, 3H), 3.08-2.88(m, 1H), 2.86-2.77(m, 5H), 2.68 (td, J=1.8, 3.5Hz, 2H), 2.63(d, J=10.0Hz, 1H), 2.41-2.29(m, 3H), 2.13-1.71(m, 5H), 1.46 (br s, 1H), 0.63-0.36(m, 2H)ppm
中間体14-1: N-(5-ブロモナフタレン-1-イル)-1,1,1-トリメチル-N-(トリメチルシリル)シランアミンの製造
5-ブロモナフタレン-1-アミン(40g、180mmol)/無水THF(650mL)の撹拌溶液に、窒素雰囲気下、-78℃でLiHMDS(1M、THF溶液、396mL、396mmol)を30分かけて滴下して加えた。この反応混合物をゆっくりと30分かけて20℃に加温し、次いで再度-78℃に冷却した。この反応混合物にTMSCl(48.3mL、378mmol)/無水THFの溶液を-78℃で滴下して加え、ゆっくりと1時間かけて20℃に加温した。この反応混合物を減圧濃縮し、粗製残渣を得た。得られた粗製残渣をヘキサン(100mL)に溶解し、セライト濾過し、濾液を減圧濃縮し、粗製生成物を赤色油状物として得た。これをフラッシュカラムクロマトグラフィー(シリカゲル(100-200)、溶離剤: 石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、N-(5-ブロモナフタレン-1-イル)-1,1,1-トリメチル-N-(トリメチルシリル)シランアミン(61g、166mmol、収率92%)を褐色液体として得た。LCMS(ESI)m/z: 366.45 [M+H]
+
中間体14-2: 5-フルオロナフタレン-1-アミンの製造
N-(5-ブロモナフタレン-1-イル)-1,1,1-トリメチル-N-(トリメチルシリル)シランアミン(100g、273mmol)/無水THF(1400mL)の撹拌溶液に、N
2雰囲気下、-78℃でn-ブチルリチウム(164mL、409mmol)を30分かけて滴下して加えた。添加終了後、この反応混合物を10分間撹拌し、次いでN-フルオロベンゼンスルホンイミド(138g、437mmol)/無水THF(400mL)の溶液を20分かけて-78℃で滴下して加えた。得られた反応混合物をゆっくりと1時間20℃で加温し、氷冷水(1000mL)で希釈し、酢酸エチル(3x800mL)で抽出した。抽出した有機層を合わせて無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。得られた残渣をフラッシュカラムクロマトグラフィー(シリカゲル(100-200)、溶離剤: 10~20%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、粗製化合物を得た。これをさらに逆相カラムクロマトグラフィー(80%アセトニトリル/0.01%ギ酸アンモニウム水)で精製した。純粋なフラクションを減圧濃縮し、5-フルオロナフタレン-1-アミン(20g、115mmol、収率42.3%)を褐色固体として得た。LCMS(ESI)m/z: 162.19 [M+H]
+
中間体14-3: 2,4-ジブロモ-5-フルオロナフタレン-1-アミンの製造
5-フルオロナフタレン-1-アミン(40g、228mmol)/酢酸(800mL)の撹拌溶液に、0℃で臭素(25.9mL、502mmol)を30分かけて慎重に加え、得られた混合物を70℃で1時間撹拌した。この反応混合物を濾過し、濾過ケーキを酢酸(2x200mL)で洗浄した。得られた残渣を10%NaOH溶液(600mL)に懸濁し、濾過した。濾過ケーキを水(200mL)で洗浄し、減圧乾燥し、粗製2,4-ジブロモ-5-フルオロナフタレン-1-アミン(66g、170mmol、収率74.3%)を淡黄色固体として得た。得られた粗製化合物はさらに精製せずに次のステップに用いた。LCMS(ESI)m/z: 319.98 [M+H]
+
中間体14-4: 5-ブロモ-6-フルオロナフト[1,2-d][1,2,3]オキサジアゾールの製造
2,4-ジブロモ-5-フルオロナフタレン-1-アミン(66g、170mmol)/酢酸(1000mL)の撹拌溶液に、プロピオン酸(136mL、1815mmol)を0℃で加えた。10分後、この反応混合物に亜硝酸ナトリウム(17.56g、255mmol)を0℃で少しずつ加えた。この反応混合物を30分間0℃で撹拌し、1時間かけて25℃に加温した。この反応混合物を冷水(2000mL)で希釈し、10分間撹拌し、濾過した。得られた固体を水(2X500mL)で洗浄し、減圧乾燥し、粗製5-ブロモ-6-フルオロナフト[1,2-d][1,2,3]オキサジアゾール(40g、118mmol、収率69%)を褐色固体として得た。これをさらに精製せずに次のステップに用いた。LCMS(ESI)m/z: 265.9 [M+H]
+
中間体14-5: 4-ブロモ-5-フルオロナフタレン-2-オールの製造
5-ブロモ-6-フルオロナフト[1,2-d][1,2,3]オキサジアゾール(40g、118mmol))/エタノール(500mL)およびテトラヒドロフラン(250mL)の撹拌溶液に、窒素下0℃でNaBH
4(8.91g、235mmol)を少しずつ30分かけて加えた。反応を同温度で30分間撹拌し、次いで25℃に加温した。この反応混合物を慎重に塩化アンモニウム水溶液(20mL)でクエンチし、減圧濃縮し、EtOHを除去した。懸濁液を酢酸エチル(3X500mL)で抽出し、有機層を合わせて無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、4-ブロモ-5-フルオロナフタレン-2-オール(30g、86mmol、収率73%)を得た。これをさらに精製せずに次のステップに用いた。LCMS(ESI)m/z: 241.08 [M+H]
+
中間体14-6: 1-ブロモ-8-フルオロ-3-(メトキシメトキシ)ナフタレンの製造
4-ブロモ-5-フルオロナフタレン-2-オール(30g、86mmol)およびDIPEA(22.50mL、129mmol)/無水ジクロロメタン(300mL)の撹拌溶液に、窒素雰囲気下、0℃でMOM-Cl(7.83mL、103mmol)を10分かけて滴下して加えた。この混合物を室温に戻し、1時間撹拌した。反応混合物を冷水(500mL)で希釈し、DCM(2X500mL)で抽出した。有機層を合わせて食塩水(50mL)で洗浄し、無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。得られた粗製化合物をフラッシュカラムクロマトグラフィー(シリカゲル(100-200)、溶離剤: 5~10%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、1-ブロモ-8-フルオロ-3-(メトキシメトキシ)ナフタレン(20.5g、68.3mmol、収率80%)を褐色固体として得た。LCMS(ESI)m/z: 285.01 [M+H]
+
中間体14-7: 2-(8-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イル)-4,4,5,5-テトラメチル-1,3,2-ジオキサボロランの製造
1-ブロモ-8-フルオロ-3-(メトキシメトキシ)ナフタレン(15g、50.0mmol)、ビス(ピナコラト)ジボロン(25.4g、100mmol)および酢酸カリウム(14.72g、150mmol)/無水トルエン(300mL)の脱気した溶液に、PdCl
2(dppf)(3.66g、5.00mmol)を不活性雰囲気下、室温で加えた。この混合物を110℃で3時間撹拌した。この反応混合物を水(50mL)で希釈し、酢酸エチル(2x250mL)で抽出した。有機層を合わせて食塩水(50mL)で洗浄し、無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。粗製物質をフラッシュカラムクロマトグラフィー(シリカ(100-200mesh)、溶離剤: 2~4%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、褐色粘着性物質を得た。これをさらに逆相カラムクロマトグラフィー(溶離剤: 80%アセトニトリル/0.01%ギ酸アンモニウム水)で精製した。純粋なフラクションを減圧濃縮し、2-(8-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イル)-4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(9g、26.6mmol、収率53.2%)灰白色固体としてを得た。LCMS(ESI)m/z: 332.1 [M+H]
+1H-NMR(400MHz、DMSO-d
6): δ 7.52(d, J=0.80Hz, 1H), 7.42-7.44(m, 1H), 7.36-7.38(m, 1H), 7.33-7.35(m, 1H), 7.03(dd, J=1.20, 7.60Hz, 1H), 5.31(s, 2H), 3.52(s, 3H), 1.46(s, 12H)ppm
中間体15-1: 8-((トリイソプロピルシリル)エチニル)ナフタレン-1,3-ジオールの製造
ナフタレン-1,3-ジオール(20g、125mmol)、(ブロモエチニル)トリイソプロピルシラン(34.3g、131mmol)、および酢酸カリウム(24.51g、250mmol)/無水ジオキサン(200mL)の撹拌溶液に、窒素雰囲気下でジクロロ(p-シメン)ルテニウム(II)(ダイマー)(7.65g、12.49mmol)を加え、混合物を12時間110℃で撹拌した。この反応混合物を周囲温度に冷却し、セライト濾過した。セライトをEtOAc(2x100mL)で洗浄し、濾液を合わせて減圧濃縮し、粗製残渣を得た。得られた粗製物をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 12~15%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、8-((トリイソプロピルシリル)エチニル)ナフタレン-1,3-ジオール(30g、85mmol、収率67.7%)を得た。LCMS(ESI)m/z: 341.55 [M+H]
+
中間体15-2: 3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-オールの製造
8-((トリイソプロピルシリル)エチニル)ナフタレン-1,3-ジオール(25g、73.4mmol)/無水DCM(300mL)の撹拌溶液に、窒素下でDIPEA(38.5mL、220mmol)を-10℃で加えた。10分後、この反応混合物に窒素雰囲気下、MOM-Cl(6.13mL、81mmol)を20分かけて滴下して加え、2時間同温度で撹拌した。この反応混合物をDCM(100mL)で希釈し、食塩水(200mL)で洗浄した。有機層を合わせて無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。粗製物質をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 5~10%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-オール(21g、53.5mmol、収率72.9%)を淡黄色油状物として得た。LCMS(ESI)m/z: 385.60 [M+H]
+
中間体15-3: ピルビン酸3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-イルの製造
3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-オール(20g、52.0mmol)、TEA(21.75mL、156mmol)、DMAP(1.271g、10.40mmol)/無水DCM(200mL)の撹拌溶液に、窒素雰囲気下-10℃でピバロイルクロライド(12.80mL、104mmol)を10分間滴下して加えた。得られた混合物を窒素下、室温で2時間撹拌した。この反応混合物をDCM(200mL)で次いで希釈し、食塩水(200mL)で洗浄した。有機層を無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。得られた粗製物をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 10~15%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、ピルビン酸3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-イル(23g、49.1mmol、収率94%)を淡黄色油状物として得た。LCMS(ESI)m/z: 469.2 [M+H]
+
中間体15-4: ピルビン酸8-エチニル-3-(メトキシメトキシ)ナフタレン-1-イルの製造
ピルビン酸3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-イル(12g、25.6mmol)/無水DMF(130mL)の撹拌溶液に、窒素雰囲気下、室温で無水CsF(27.2g、179mmol)を加え、混合物を2時間撹拌した。この反応混合物をDCM(200mL)で希釈し、食塩水(200mL)で洗浄した。有機層を無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。得られた粗製物をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 5~8%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、ピルビン酸8-エチニル-3-(メトキシメトキシ)ナフタレン-1-イル(7g、20.62mmol、81%収率)を得た。LCMS(ESI)m/z: 313.3 [M+H]
+
中間体15-5: ピルビン酸8-エチル-3-(メトキシメトキシ)ナフタレン-1-イルの製造
ピルビン酸8-エチニル-3-(メトキシメトキシ)ナフタレン-1-イル(7g、22.41mmol)/無水メタノール(70mL)の撹拌溶液に、25℃でPd/C(1.4g、13.16mmol)を加えた。懸濁液を減圧脱気し、H
2で数回パージした。この混合物をH
2雰囲気下(1atm)、25℃で5時間撹拌した。この反応混合物を濾過し、減圧濃縮し、粗製残渣を得た。粗製物質をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 15~20%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、ピルビン酸8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル(6.56g、17.83mmol、収率80%)を淡黄色油状物として得た。LCMS(ESI)m/z: 316.1 [M+H]
+
中間体15-6: 8-エチル-3-(メトキシメトキシ)ナフタレン-1-オールの製造
ピルビン酸8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル(10g、31.6mmol)/THF:水:MeOH(5:1:5)の撹拌溶液に、無水LiOH(1.135g、47.4mmol)を窒素下室温で加え、混合物を同温度で3時間撹拌した。この反応混合物を減圧濃縮し、MeOHを除去した。この反応混合物をEtOAc(150mL)で希釈し、食塩水(200mL)で洗浄した。有機層を無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。粗製物質をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 25~30%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、8-エチル-3-(メトキシメトキシ)ナフタレン-1-オール(6g、25.8mmol、収率82%)を得た。LCMS(ESI)m/z: 233.2 [M+H]
+
中間体15-7: 8-エチル-3-(メトキシメトキシ)ナフタレン-1-イルトリフルオロメタンスルホナートの製造
8-エチル-3-(メトキシメトキシ)ナフタレン-1-オール(3g、12.92mmol)およびDIPEA(22.50mL、129mmol)/無水ジクロロメタン(50mL)の撹拌溶液に、窒素下-40℃でTf
2O(2.182mL、12.92mmol)を滴下して加えた。得られた反応混合物を室温に戻し、1時間撹拌した。この反応混合物を冷水(500mL)で希釈し、DCM(2X500mL)で抽出した。有機層を合わせて食塩水(50mL)で洗浄し、無水Na
2SO
4で乾燥し、減圧濃縮し、粗製残渣を得た。得られた粗製化合物をフラッシュカラムクロマトグラフィー(シリカゲル: 100-200、溶離剤: 5~10%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、8-エチル-3-(メトキシメトキシ)ナフタレン-1-イルトリフルオロメタンスルホナート(3.5g、9.03mmol、収率69.9%)を淡黄色油状物として得た。LCMS(ESI)m/z: 365.3 [M+H]
+
中間体15-8: 2-(8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル)-4,4,5,5-テトラメチル-1,3,2-ジオキサボロランの製造
8-エチル-3-(メトキシメトキシ)ナフタレン-1-イルトリフルオロメタンスルホナート(7.4g、20.31mmol)/1,4-ジオキサン(80mL)の撹拌溶液に、ビス(ピナコラト)ジボロン(12.89g、50.8mmol)および酢酸カリウム(5.98g、60.9mmol)を加えた。この混合物を脱気し、5分間窒素でパージし、PdCl
2(dppf)(1.659g、2.031mmol)を加えた。得られた混合物を窒素下、3時間100℃で撹拌した。この反応混合物を水(50mL)で希釈し、酢酸エチル(250mL)で抽出した。有機層を合わせて食塩水(50mL)で洗浄し、無水Na
2SO
4で乾燥し、減圧濃縮し、粗製残渣を得た。得られた粗製物をフラッシュカラムクロマトグラフィー(シリカ(100-200mesh)、2~4%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、褐色粘着性物質を得た。これを再度逆相カラムクロマトグラフィー(溶離剤: 80%アセトニトリル/0.01%ギ酸アンモニウム水)で精製した。純粋なフラクションを減圧濃縮し、2-(8-エチル-3-(メトキシメトキシ)ナフタレン-1-イル)-4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(4.5g、13.10mmol、収率64.5%)を得た。LCMS(ESI)m/z: 344.2 [M+H]
+1H-NMR(400MHz、CDCl
3): δ 7.62(dd, J=0.80, 8.00Hz, 1H), 7.36-7.44(m, 3H), 7.27(t, J=0.40Hz, 1H), 5.31(s, 2H), 3.53(s, 3H), 3.21 (q, J=7.20Hz, 2H), 1.46(s, 12H), 1.38(t, J=7.60Hz, 3H)ppm
中間体16-1: 7-フルオロ-8-((トリイソプロピルシリル)エチニル)ナフタレン-1,3-ジオールの製造
7-フルオロナフタレン-1,3-ジオール(20g、112mol)、(ブロモエチニル)トリイソプロピルシラン(30.8g、118mmol)、および酢酸カリウム(22.03g、225mmol)/無水ジオキサン(200mL)の撹拌溶液に、ジクロロ(p-シメン)ルテニウム(II)(ダイマー)(6.87g、11.23mmol)を窒素雰囲気下、室温で加え、この混合物を窒素雰囲気下、110℃で12時間撹拌した。得られた反応混合物をセライト濾過し、セライトをEtOAc(2x50mL)で洗浄した。濾液を回収し、減圧濃縮し、粗製残渣を得た。粗製物質をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 5~10%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、7-フルオロ-8-((トリイソプロピルシリル)エチニル)ナフタレン-1,3-ジオール(32g、88mmol、収率79%)を淡黄色油状物として得た。LCMS(ESI)m/z: 359.1 [M+H]
+
中間体16-2: 7-フルオロ-3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-オールの製造
7-フルオロ-8-((トリイソプロピルシリル)エチニル)ナフタレン-1,3-ジオール(10g、27.9mmol)/無水DCM(100mL)の撹拌溶液に、窒素下、0℃でDIEA(14.61mL、84mmol)を加えた。この反応混合物を同温度で10分間撹拌した後、この反応混合物にMOM-Cl(2.54mL、33.5mmol)を滴下して加えた。得られた混合物を窒素下、室温で1時間撹拌し、反応混合物を冷水(50mL)で希釈し、DCM(2x100mL)で抽出した。有機層を合わせて食塩水(50mL)で洗浄し、無水Na
2SO
4で乾燥し、減圧濃縮し、粗製残渣を得た。粗製物質をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 5~10%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、7-フルオロ-3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-オール(10g、24.84mmol、収率89%)を得た。LCMS(ESI)m/z: 403.1 [M+H]
+
中間体16-3: ピルビン酸7-フルオロ-3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-イルの製造
7-フルオロ-3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-オール(17g、42.2mmol)/無水DCM(170mL)の撹拌溶液に、窒素雰囲気下、0℃でTEA(17.66mL、127mmol)およびDMAP(1.032g、8.45mmol)を加えた。この反応混合物を同温度で10分間撹拌後、ピバロイルクロライド(6.23mL、50.7mmol)を滴下して加えた。得られた反応混合物を室温で2時間撹拌し、この反応混合物を冷水(50mL)で希釈し、DCM(2X100mL)で抽出した。有機層を合わせて食塩水(200mL)で洗浄し、無水Na
2SO
4で乾燥し、減圧濃縮し、粗製残渣を得た。粗製物質をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 20~25%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、ピルビン酸7-フルオロ-3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-イル(19g、35.1mmol、収率83%)を得た。LCMS(ESI)m/z: 487.2 [M+H]
+
中間体16-4: ピルビン酸8-エチニル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イルの製造
7-フルオロ-3-(メトキシメトキシ)-8-((トリイソプロピルシリル)エチニル)ナフタレン-1-イルピルビン酸(20g、41.1mmol)/無水DMF(150mL)の撹拌溶液に、窒素雰囲気下、室温でCsF(43.7g、288mmol)を加え、混合物を5時間室温で撹拌した。この反応混合物をDCM(200mL)で希釈し、食塩水(200mL)で洗浄した。有機層を無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。粗製物質をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 5~8%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、ピルビン酸8-エチニル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イル(12g、32.7mmol、収率80%)を淡黄色油状物として得た。LCMS(ESI)m/z: 331.2 [M+H]
+
中間体16-5: ピルビン酸8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イルの製造
ピルビン酸8-エチニル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イル(15g、45.4mmol)/エタノール(150mL)の撹拌溶液に、10%Pd/C(3g、28.2mmol)を25℃で加えた。懸濁液を減圧脱気し、H
2で数回パージした。この混合物をH
2雰囲気(1atm)下、25℃で5時間撹拌した。この反応混合物を濾過し、減圧濃縮し、粗製残渣を得た。得られた粗製物をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 15-20%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、ピルビン酸8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イル(14g、34.3mmol、収率76%)を淡黄色油状物として得た。LCMS(ESI)m/z: 335.1 [M+H]
+
中間体16-6: 8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-オールの製造
ピルビン酸8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イル(5g、14.95mmol)/MeOH(50mL)の撹拌溶液に、無水LiOH(0.537g、22.43mmol)を窒素下室温で加えた。得られた混合物を同温度で1時間撹拌した。この反応混合物を減圧濃縮し、MeOHを除去した。この反応混合物をEtOAc(150mL)で希釈し、食塩水(200mL)で洗浄した。有機層を無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。粗製物質をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 25~30%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-オール(3.5g、12.59mmol、収率84%)を得た。LCMS(ESI)m/z: 251.2 [M+H]
+
中間体16-7: 8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イルトリフルオロメタンスルホナートの製造
8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-オール(1.5g、5.99mmol)およびDIEA(3.66mL、20.98mmol)/無水ジクロロメタン(50mL)の撹拌溶液に、-10℃でTf
2O(1.215mL、7.19mmol)を滴下して加えた。得られた反応混合物を室温に戻し、1時間撹拌した。この反応混合物を冷水(500mL)で希釈し、次いでDCM(2x500mL)で抽出した。有機層を合わせて食塩水(50mL)で洗浄し、無水Na
2SO
4で乾燥し
、濾過し、減圧濃縮し、粗製残渣を得た。得られた粗製化合物をフラッシュカラムクロマトグラフィー(シリカゲル: 100-200、溶離剤: 5~10%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イルトリフルオロメタンスルホナート(1.8g、4.66mmol、収率78%)を淡黄色油状物として得た。LCMS(ESI)m/z: 383.0 [M+H]
+
中間体16-8: 2-(8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イル)-4,4,5,5-テトラメチル-1,3,2-ジオキサボロランの製造
8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イルトリフルオロメタンスルホナート(2.5g、6.54mmol)/無水1,4-ジオキサン(80mL)の撹拌溶液に、ビス(ピナコラト)ジボロン(4.15g、16.35mmol)および酢酸カリウム(1.925g、19.62mmol)を加え、得られた混合物を脱気し、窒素で5分間パージした。次いで、この反応混合物にPdCl
2(dppf)(0.534g、0.654mmol)を加えた。得られた混合物を窒素雰囲気下、100℃で2時間撹拌した。この反応混合物を水(50mL)で次いで希釈し、酢酸エチル(250mL)で抽出した。有機層を合わせて食塩水(50mL)で洗浄し、無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。得られた粗製物質をフラッシュカラムクロマトグラフィー(シリカ: 100-200mesh、溶離剤: 2~4%酢酸エチル/石油エーテル)で精製した。純粋なフラクションを減圧濃縮し、褐色粘着性物質を得た。これを再度逆相カラムクロマトグラフィー(80%アセトニトリル/0.01%ギ酸アンモニウム水)で精製した。純粋なフラクションを減圧濃縮し、2-(8-エチル-7-フルオロ-3-(メトキシメトキシ)ナフタレン-1-イル)-4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(1.8g、4.99mmol、収率76%)を得た。LCMS(ESI)m/z: 329 [M-OCH
3]
+;
1H-NMR(400MHz、CDCl3): δ 7.77(t, J=8.40Hz, 1H), 7.54(s, 1H), 7.37(t, J=9.60Hz, 1H), 7.28(s, 1H), 5.31(s, 2H), 3.42(s, 3H), 3.03(d, J=6.80Hz, 2H), 1.39(s, 12H), 1.19(t, J=7.20Hz, 3H)ppm
中間体17-1: 1-(tert-ブチル)2-メチル-(S)-4,4-ジフルオロピロリジン-1,2-ジカルボキシレートの製造
1-(tert-ブチル)2-メチル(S)-4-オキソピロリジン-1,2-ジカルボキシレート(30.0g、123mmol)/DCM(20.0mL)の撹拌溶液に、DAST(81mL、617mmol)を0℃で滴下して加え、混合物を周囲温度で16時間撹拌した。この反応混合物を水でクエンチし、ジクロロメタンで抽出した。有機層を合わせて水および飽和食塩水で洗浄し、無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製生成物を得た。得られた粗製生成物をカラムクロマトグラフィー(Grace、340g snap、乾式充填、シリカ(230-400mesh)、溶出: 20~50%酢酸エチル/石油エーテル)で精製した。所期のフラクションを回収して減圧濃縮し、1-(tert-ブチル)2-メチル-(S)-4,4-ジフルオロピロリジン-1,2-ジカルボキシレート(28g、90mmol、収率73.4%)を薄茶色液体として得た。LCMS-ELSD(ESI)m/z: 166.2 [M+H-Boc]
+
中間体17-2: 1-(tert-ブチル)2-メチル-2-(3-((tert-ブチルジメチルシリル)オキシ)プロピル)-4,4-ジフルオロピロリジン-1,2-ジカルボキシレートの製造
1-(tert-ブチル)2-メチル-(S)-4,4-ジフルオロピロリジン-1,2-ジカルボキシレート(28g、106mmol)/THF(300.0mL)の撹拌溶液に、LiHMDS(158mL、158mmol)を-45℃で滴下して加えた。この反応混合物を同温度で30分間撹拌した後、(3-ブロモプロポキシ)(tert-ブチル)ジメチルシラン(40.1g、158mmol)を滴下して加えた。この反応混合物を同温度で30分間撹拌し、次いでゆっくりと室温に戻した。この反応混合物を飽和塩化アンモニウム水溶液(15mL)でクエンチし、次いで水(50mL)で希釈した。混合物を酢酸エチル(3x100mL)で抽出し、有機層を合わせて無水Na
2SO
4で乾燥し、濾過し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(20~30%酢酸エチル/石油エーテル)で精製し、1-(tert-ブチル)2-メチル-2-(3-((tert-ブチルジメチルシリル)オキシ)プロピル)-4,4-ジフルオロピロリジン-1,2-ジカルボキシレート(35g、80mmol、収率76%)を薄茶色液体として得た。LCMS-ELSD(ESI)m/z: 388.2 [M+H-Boc]
+
中間体17-3: 1-(tert-ブチル)2-メチル-4,4-ジフルオロ-2-(3-ヒドロキシプロピル)ピロリジン-1,2-ジカルボキシレートの製造
1-(tert-ブチル)2-メチル-2-(3-((tert-ブチルジメチルシリル)オキシ)プロピル)-4,4-ジフルオロピロリジン-1,2-ジカルボキシレート(35.0g、80mmol)/THF(50mL)の撹拌溶液に、25℃でTBAF(1M、THF溶液、80mL、80mmol)を滴下して加え、同温度で4時間撹拌した。この反応混合物を飽和塩化アンモニウム水溶液(90mL)でクエンチし、酢酸エチル(100mL)で希釈した。混合物を酢酸エチル(3x150mL)で抽出し、有機層を合わせて無水硫酸ナトリウムで乾燥し、減圧濃縮し、粗製残渣を無色の油状物として得た。得られた粗製物をシリカゲルカラムクロマトグラフィー(50%酢酸エチル/石油エーテル)で精製し、1-(tert-ブチル)2-メチル-4,4-ジフルオロ-2-(3-ヒドロキシプロピル)ピロリジン-1,2-ジカルボキシレート(20g、57.9mmol、収率72.4%)を薄茶色液体として得た。LCMS-ELSD(ESI)m/z: 224.2 [M+H-Boc]
+
中間体17-4: 1-(tert-ブチル)2-メチル-4,4-ジフルオロ-2-(3-ヨードプロピル)ピロリジン-1,2-ジカルボキシレートの製造
1-(tert-ブチル)2-メチル-4,4-ジフルオロ-2-(3-ヒドロキシプロピル)ピロリジン-1,2-ジカルボキシレート(20g、61.9mmol)/DCM(100mL)の撹拌溶液に、0℃でトリフェニルホスフィン(48.7g、186mmol)、イミダゾール(8.42g、124mmol)を加え、反応混合物を10分間撹拌後、ヨウ素(62.8g、247mmol)を添加した。この反応混合物を室温で12時間撹拌し、次いで飽和チオ硫酸ナトリウム水溶液(30mL)でクエンチした。懸濁液をジクロロメタン(2x200mL)で抽出し、有機層を合わせて無水硫酸ナトリウムで乾燥し、減圧濃縮し、粗製残渣を得た。この粗製物をシリカゲルカラムクロマトグラフィー(10~20%酢酸エチル/石油エーテル)で精製し、1-(tert-ブチル)2-メチル-4,4-ジフルオロ-2-(3-ヨードプロピル)ピロリジン-1,2-ジカルボキシレート(20g、46.1mmol、収率75%)を無色油状物として得た。LCMS-ELSD(ESI)m/z: 333.0 [M+H-Boc]
+
中間体17-5: 4,4-ジフルオロ-2-(3-ヨードプロピル)ピロリジン-2-カルボン酸メチルの製造
1-(tert-ブチル)2-メチル-4,4-ジフルオロ-2-(3-ヨードプロピル)ピロリジン-1,2-ジカルボキシレート(15g、34.6mmol)/DCM(100mL)の撹拌溶液に、HCl/1,4-ジオキサン(4M、8.66mL、34.6mmol)を0℃で加え、混合物を周囲温度で16時間撹拌した。この反応混合物を次いで減圧濃縮し、室温で(S)-4,4-ジフルオロ-2-(3-ヨードプロピル)ピロリジン-2-カルボン酸メチル・HClの粗製残渣(11.5g、31.1mmol、収率90%)を得た。これをさらに精製せずに次のステップに用いた。LCMS-ELSD(ESI)m/z: 334.1 [M+H]
+
中間体17-6: 2,2-ジフルオロテトラヒドロ-1H-ピロリジン-7a(5H)-カルボン酸メチルの製造
4,4-ジフルオロ-2-(3-ヒドロキシプロピル)ピロリジン-2-カルボン酸メチル・HCl(11.5g、51.5mmol)/THF(120.0mL)の撹拌溶液に、TEA(35.9mL、258mmol)を0℃で加え、続いて45℃で16時間加熱した。この反応混合物を減圧濃縮し、粗製残渣を得た。これをカラムクロマトグラフィー(中性アルミナ、40~50%酢酸エチル/石油エーテル)で精製し、2,2-ジフルオロテトラヒドロ-1H-ピロリジン-7a(5H)-カルボン酸メチル(6g、43.5mmol、収率81%)を褐色液体として得た。
中間体17-7: (2,2-ジフルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メタノールの製造
2,2-ジフルオロテトラヒドロ-1H-ピロリジン-7a(5H)-カルボン酸メチル(12.0g、58.5mmol)/無水THF(20.0mL)の撹拌溶液に、窒素下、0℃でLiAlH
4(117mL、117mmol)を10分かけて滴下して加えた。この混合物を同温度で1時間撹拌した後、飽和塩化アンモニウム水溶液(5mL)を加えて0℃でクエンチした。発泡が収まった後、この反応混合物に無水硫酸ナトリウムを加え、続いてジクロロメタン(20mL)を加えた。この反応混合物を20分間撹拌し、濾過した。濾液を無水硫酸ナトリウムで乾燥し、減圧濃縮し、(2,2-ジフルオロテトラヒドロ-1H-ピロリジン-7a(5H)-イル)メタノールの粗製残渣(4.1g、23.02mmol、収率39.4%)を淡黄色液体として得た。LCMS-ELSD(ESI)m/z: 178.1 [M+H]
+
中間体18-1: 7-フルオロ-3-(メトキシメトキシ)-8-{2-[トリス(プロパン-2-イル)シリル]エチニル}ナフタレン-1-イルトリフルオロメタンスルホナートの製造
7-フルオロ-3-(メトキシメトキシ)-8-{2-[トリス(プロパン-2-イル)シリル]エチニル}ナフタレン-1-オール(中間体16-2、8.6g、22.25mmol)およびDIPEA(11.66mL、66.7mmol)/DCM(35mL)の溶液に、トリフルオロメタンスルホン酸無水物(5.64mL、33.4mmol)を-40℃で滴下して加え、この混合物を同温度で30分間撹拌した。この反応混合物を水で希釈し、DCMで抽出した。有機層を合わせて水、食塩水で洗浄し、無水Na
2SO
4で乾燥し、減圧濃縮し、粗製残渣を得た。これをシリカゲルカラムクロマトグラフィー(機器: CombiFlash、80g RediSep(登録商標)カラム、5~10%EtOAc/石油エーテル)で精製し、7-フルオロ-3-(メトキシメトキシ)-8-{2-[トリス(プロパン-2-イル)シリル]エチニル}ナフタレン-1-イルトリフルオロメタンスルホナート(8.5g、16.39mmol、収率73.7%)を黄色の油状物として得た。
1H NMR(400MHz、DMSO-d
6) δ ppm 8.12(dd, J=9.3, 5.8Hz, 1H), 7.78(d, J=2.5Hz, 1H), 7.65(t, J=9.0Hz, 1H), 7.51(d, J=2.0Hz, 1H), 5.37(s, 2H), 3.43(s, 3H), 1.31-1.08(m, 21H)
中間体18-2: {2-[2-フルオロ-6-(メトキシメトキシ)-8-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ナフタレン-1-イル]エチニル}トリス(プロパン-2-イル)シランの製造
7-フルオロ-3-(メトキシメトキシ)-8-{2-[トリス(プロパン-2-イル)シリル]エチニル}ナフタレン-1-イルトリフルオロメタンスルホナート(4.0g、7.48mmol)、酢酸カリウム(2.203g、22.45mmol)およびビス(ピナコラト)ジボロン(3.80g、14.96mmol)/トルエン(40mL)の脱気した溶液に、1,1-ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)(0.547g、0.748mmol)を加え、混合物を120℃で3時間加熱した。この反応混合物を冷却し、セライト濾過し、酢酸エチルで洗浄した。濾液を水、食塩水で洗浄し、無水Na
2SO
4で乾燥し、濾過し、減圧濃縮し、粗製残渣を得た。これをシリカゲルカラムクロマトグラフィー(機器: CombiFlash、40g RediSep(登録商標)カラム、10%EtOAc/石油エーテル)で精製し、{2-[2-フルオロ-6-(メトキシメトキシ)-8-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ナフタレン-1-イル]エチニル}トリス(プロパン-2-イル)シラン(2.4g、4.68mmol、収率62.6%)を淡黄色固体として得た。MS(ESI)m/z 513.4 [M+1]
+
(生物活性)
KRASG12D RAF破壊アッセイ
アッセイ緩衝液(50mM Tris pH 7.5、100mM NaCl、1mM MgCl2、1mM DTT、100ug/mL BSA)中、組み換えGMPPNP結合KRAS G12D(5nM)を室温下20分間、化合物で処理した。組み換えGST-RAF1 RBD(9nm)を加え、続いてSA-Tb(0.25nm)を添加し、この反応混合物を3時間インキュベートした。HTRFシグナルを測定し(PerkinElmer Envision)、シグナル比率(λem 520/λem 495)を計算し、用量-反応曲線からIC50値を計算した。
KRASG12Dヌクレオチド交換アッセイ
アッセイ緩衝液(10mM Hepes pH 7.4、150mM NaCl、5mM MgCl2、0.0025%Igepal-CA630、0.05%BSA、1mM DTT、0.5nM SA-Tb)中、組み換えGDP結合KRAS G12D(20nm)を室温下20分間、化合物で処理した。BIODIPY標識GDP(400nm)および組み換えSOS(10nm)を添加し、反応を30分間インキュベートした。HTRFシグナルを測定し(PerkinElmer Envision)、シグナル比率(λem 520/λem 495)を計算し、用量-反応曲線からIC50値を計算した。
概略および要約書のセクションではなく、詳細な説明のセクションが請求項を解釈するために用いられることを意図することが理解される。概略および要約書のセクションは、発明者により検討されているような本開示の実施態様の1つ以上の例を示し得るが、すべての例を示し得るものではなく、それ故、本開示および本請求項を制限することを決して意図するものではない。
本開示は、特定の官能基およびその関連性の働きを具体的に示しながら、官能基の構成要素を用いて上記で説明されている。これらの官能基の構成要素の境界線は、説明の便宜上、本明細書で任意に定義される。特定の官能基およびその関連性が適当に機能している間は、別の境界線が定義され得る。
前述の具体的な実施態様の説明により、本開示の一般的性質が完全に明らかになるため、発明者以外が当業者の知識を適用することで、過度の実験をせず、本開示の基本概念から離れることなく、上記の特定の実施態様を容易に様々な応用に改良および/または適応され得る。それ故、そのような応用および改良は、本明細書に提示された内容および指針に基づいて、本開示の実施態様と同等の意義の範囲内であることを意図する。本明細書の表現または専門用語は、説明のための使用であり、内容および指針を踏まえて当業者に解釈される本明細書の表現または専門用語に限定されることはないと理解されるべきである。
本開示の範囲は、上記に記載の任意の例示の実施態様に制限されるべきではなく、次に示す請求項およびそれと同等のもののみに従って定義されるべきである。