WO2025117969A1 - Process for manufacturing lipid nanoparticles - Google Patents
Process for manufacturing lipid nanoparticles Download PDFInfo
- Publication number
- WO2025117969A1 WO2025117969A1 PCT/US2024/058122 US2024058122W WO2025117969A1 WO 2025117969 A1 WO2025117969 A1 WO 2025117969A1 US 2024058122 W US2024058122 W US 2024058122W WO 2025117969 A1 WO2025117969 A1 WO 2025117969A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lipid
- composition
- lnps
- loaded
- less
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/5123—Organic compounds, e.g. fats, sugars
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5192—Processes
Definitions
- nucleic acid therapeutics have rapidly expanded and become the basis for treating a wide variety of diseases.
- Nucleic acid therapies available include, but are not limited to, the use of DNA or viral vectors for insertion of desired genetic information into the host cell, and/or RNA constructed to encode for a therapeutic protein.
- DNA and viral vector deliveries carry their own setbacks and challenges that make them less favorable to RNA therapeutics.
- the introduced DNA in some cases may be unintentionally inserted into an intact gene and result in a mutation that impedes or even wholly eliminates the function of the endogenous gene leading to an elimination or deleteriously reduced production of an essential enzyme or interruption of a gene critical for the regulating cell growth.
- Viral vector-based therapies can result in an adverse immune response.
- RNA is a substantially safer and more effective gene therapy agent due to its ability 7 to encode for the protein outside of the nucleus to perform its function. With this, the RNA does not involve the risk of being stably integrated into the genome of the transfected cell.
- RNA therapeutics conventionally have consisted of engineering linear messenger RNAs (mRNA). Although more effective than DNA or viral vectors, linear mRNAs have their own set of challenges regarding stability, immunogenicity, translation efficiency, and delivery. Some of these challenges may lead to size restraints and/or destruction of the linear mRNA due to the challenges present with linear mRNAs’ caps. To overcome these limitations, circular polynucleotides or circular RNAs may be used. Due to being arranged in covalently closed continuous loops, circular RNAs are useful in the design and production of stable forms of RNA.
- Circular RNA provides an advantage to the study of RNA structure and function, especially in the case of molecules that are prone to folding in an inactive conformation (Wang and Ruffner, 1998). Circular RNA can also be particularly interesting and useful for in vivo applications, especially in the research area of RNA- bascd control of gene expression and therapeutics, including protein replacement therapy and vaccination.
- nanoparticles delivery systems can be used.
- Lipid- containing nanoparticles or lipid nanoparticles, liposomes, and lipoplexes have been used as effective delivery systems to transport into cells and/or intracellular compartments biologically active substances such as small molecule drugs, proteins, and nucleic acids. Though a variety of such lipid nanoparticles delivery systems have been demonstrated, improvements in manufacturing such delivery systems are needed.
- the present application provides methods for manufacturing lipid nanoparticles.
- the present disclosure provides a method of preparing an empty lipid nanoparticle (empty LNP).
- the present disclosure provides a method of preparing a loaded lipid nanoparticle (loaded LNP) associated with a nucleic acid.
- Empty and loaded LNP compositions prepared by the subject methods are also provided.
- a method of preparing an empty lipid nanoparticle (LNP) composition comprising: a) a precipitation step, comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first LNP composition comprising empty’ LNPs; and b) a homogenization step, comprising: homogenizing the first LNP composition to produce a homogeneous LNP composition of empty LNPs.
- a precipitation step comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first LNP composition comprising empty’ LNPs
- a homogenization step comprising: homogenizing the first LNP composition to produce a homogeneous LNP composition of empty LNPs.
- the empty LNPs in the homogeneous LNP composition have an average particle size from 30 nm to 200 nm, such as from 50 nm to 70 nm. In some embodiments, the empty LNPs in the homogeneous LNP composition have a poly dispersity from 0.05 to 0.2, such as 0.1 or less.
- the method further comprises: c) a loading step, comprising: mixing the homogeneous LNP composition with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with the nucleic acid.
- a method of preparing a loaded LNP composition comprising: a) a precipitation step, comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first LNP composition comprising empty LNPs; b) a loading step, comprising: mixing the first LNP composition with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with the nucleic acid; and c) a homogenization step, comprising: homogenizing the loaded LNP composition to produce a homogeneous loaded LNP composition comprising LNPs associated with the nucleic acid.
- an empty LNP composition prepared by a subject method.
- a loaded LNP composition prepared by a subject method.
- FIG. 1 is a schematic outlining the subject distinctive process steps: an LNP formation step (nanoprecipitation step), a homogenization step (particle size reduction step), and an RNA addition step.
- FIG. 2 depicts the average particle size distribution of empty LNPs containing ionizable lipid 89 (Table 1) formed at different temperatures.
- FIG. 3 depicts the average particle size distribution of empty LNPs containing ionizable lipid 144 (Table 1) formed at different mixing speeds.
- FIG. 4 depicts the average particle size and PDI of empty LNPs containing the ionizable lipid 127 (Table 1) after both the LNP formation step and the homogenization step at processing temperatures of 30° C and 50° C.
- FIG. 5 depicts the average particle size of the empty LNPs containing the ionizable lipid 127 (Table 1) with and without cooling after the homogenization step.
- FIG. 6 depicts the average particle size of empty LNPs containing the ionizable lipid 127 (Table 1) after both the LNP formation step and the homogenization step using 6.25 mM sodium acetate buffer at pH 3.0 or 4.5.
- FIG. 7 depicts the zeta potential of empty LNPs containing the ionizable lipid 127 (Tabic 1) after the homogenization step using 6.25 mM sodium acetate buffer at pH 3.0 or 4.5.
- FIG. 9 depicts the average size and PDI of empty LNPs containing the ionizable lipid 123 (Table 1) after both the LNP formation step and the homogenization step using different aqueous buffers.
- FIG. 10 depicts the average particle size of RNA encapsulated LNPs (loaded LNPs) in the different aqueous phase buffer conditions as compared to control empty LNPs.
- FIG. 11 depicts the LNP encapsulation efficiency post RNA encapsulation (i.e., for loaded LNPs) at different pHs in 50 mM sodium acetate.
- FIG. 12 depicts loaded LNP particle size and PDI after the RNA addition step and buffer exchange (TFF step) with phosphate buffered saline at pH 7.4 (PBS. TFF process #1) or ultrapure distilled water (DI water. TFF process #2).
- FIG. 13 depicts the encapsulation efficiency of the loaded LNPs containing the ionizable lipid 127 (Table 1) post RNA addition step and TFF step with phosphate buffered saline at pH 7.4 (PBS, TFF process #1) or ultrapure distilled water (DI water, TFF process #2).
- FIG. 14 depicts the average size and PDI of loaded LNPs containing the ionizable lipid 127 (Table 1) after buffer exchange using different storage buffcrs/tonicity modifiers, water, tris-sucrosc saline (TSS) or PBS.
- FIG. 15 depicts the process for manufacturing the subject loaded LNPs, including the following distinctive process steps: LNP formation step (empty LNPs), a homogenization step (particle size reduction step, empty LNPs), and an RNA addition step (loaded LNPs).
- FIG. 16 depicts the average particle size for RNA encapsulated LNPs (loaded LNPs) formed using the nanoprecipitation (e.g., PNI) process and Process 1.
- PNI nanoprecipitation
- FIG. 17 depicts Cryo-TEM images for loaded LNPs formulated via Process 1 (Panel A) and the nanoprecipitation (e.g., PNI) process (Panel B).
- FIG. 18 depicts the in vivo firefly luciferase expression of the circular RNA encapsulated in the LNPs (loaded LNPs) as measured by ex vivo IVIS as made from Process 1 vs the nanoprecipitation (e.g., PNI) process.
- FIG. 19A-FIG. 19D depict the ex vivo organ imaging for firefly luciferase expression of the loaded LNPs as made from Process 1 vs the nanoprecipitation (e.g., PNI) process.
- PNI nanoprecipitation
- FIG. 20 depicts flux level of loaded LNPs in the quadricep, liver, and spleen.
- the present application provides, among other things, methods for manufacturing lipid nanoparticles.
- the present disclosure provides a method of preparing an empty lipid nanoparticle (empty LNP).
- the present disclosure provides a method of preparing a loaded lipid nanoparticle (loaded LNP) associated with a nucleic acid.
- the loaded lipid nanoparticles can be associated with RNA polynucleotides, particularly circular RNA polynucleotides (aka circRNA or oRNATM)-
- lipid nanoparticles that encapsulate linear mRNA mRNA-LNPs
- precipitation and encapsulation of the RNA occur concurrently via high-energy mixing of aqueous mRNA and a solution of lipids in ethanol.
- the mixing of the ethanol phase and the aqueous phase can generally be controlled by microfluidic chip devices. See for example, Guevara et al., Front. Chem. 2020; 8: 589959; Riley et al., Sci. Adv. 2021; 7: eaba 1028. and Cullis et al., WO2011140627.
- Such concurrent mixing can result in fouling of microfluidic devices and is not amenable to large-scale manufacturing.
- the empty LNPs are prepared by distinctive steps: an LNP formation step (precipitation step to form empty LNPs); and a homogenization step (size reduction step to form homogeneous mixture of empty LNPs).
- the subject methods can further comprise an RNA addition step to form loaded LNPs (LNPs associated with a nucleic acid).
- the homogenization step is carried out after the LNP formation step and before the RNA addition step. In some embodiments, the homogenization step is carried out after the RNA addition step.
- the subject process for preparing empty LNPs is readily scalable. More generally, the subject method for preparing empty LNPs eliminates the need to control the size of the LNPs during the initial LNP formation step, which can be a bottleneck for large scale production in previously known LNP manufacturing processes.
- the present disclosure provides a method of preparing a homogeneous composition of empty LNPs, which can be produced and stored until ready for further use.
- the present disclosure also provides methods in which a nucleotide may be associated with, or encapsulated within, tire preformed empty LNPs to form loaded LNPs.
- This mode of production offers advantages in the context of clinical supply, as empty LNP vesicles may be produced and stored separately prior to recombination with a nucleic acid (e.g., a circular RNA polynucleotide) in a clinical compound setting.
- bedside formulations may promote increased stability since the nucleic acid and empty LNPs can be stored in separately optimized conditions. Process complexity and cost of the products may be reduced since the empty LNP preparation occurs independently of nucleic acid cargo, enabling a platform approach for multiple nucleic acid or active agent constructs.
- the present disclosure provides loaded LNPs associated with a nucleic acid that has an average particle size distribution and poly dispersity that is the same or better than loaded LNPs produced by a previously known LNP manufacturing method.
- the LNP formulation produced by tire method of the present disclosure exhibits a nucleic acid expression (e.g., the RNA expression) higher than the nucleic acid expression (e.g., the RNA expression) of the LNP formulation produced by a previously known LNP manufacturing method. More generally, encapsulating an RNA by combining pre-fonned lipid nanoparticles with mRNA can result in formulated particles that exhibit unexpectedly efficient in vivo delivery of the RNA and surprisingly potent expression of proteins and/or peptides that the RNA encodes. See for example. Karve et al, W02018089801 and Smith et al.. WO2021155274, the disclosure of which is incorporated herein by reference.
- the loaded LNP formulation produced by the method of the present disclosure exhibits a nucleic acid expression (e.g.. a circular RNA expression) higher than the nucleic acid expression (e.g., a circular RNA expression) of the LNP formulation prepared by a previously known LNP manufacturing method by 5% or higher, 10% or more 15% or more, 20% or more. 30% or more. 40% or more, 50% or more, 60% or more, 70% or more, 80% or more. 90% or more, 1 folds or more. 2 folds or more. 3 folds or more. 4 folds or more, 5 folds or more, 10 folds or more, 20 folds or more. 30 folds or more, 40 folds or more.
- a nucleic acid expression e.g. a circular RNA expression
- the nucleic acid expression e.g., a circular RNA expression
- RNA therapy e.g., circular RNA therapy
- the RNA therapy allows for increased RNA stability, expression, and prolonged half-life, among other things.
- provided herein are methods comprising administration of circular RNA polynucleotides provided herein into cells for therapy or production of useful proteins.
- the method is advantageous in providing the production of a desired polypeptide inside eukaryotic cells with a longer half-life than linear RNA, due to the resistance of the circular RNA to ribonucleases.
- the present disclosure provides methods of preparing empty lipid nanoparticles (empty LNPs). and methods of preparing loaded LNPs.
- a method of preparing an empty LNP composition comprising: a) a precipitation step, comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first lipid LNP composition comprising empty LNPs; and b) a homogenization step, comprising: homogenizing the first LNP composition to produce a homogeneous LNP composition of empty LNPs.
- the method further comprises: c) a loading step, comprising: mixing the homogeneous LNP composition with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with the nucleic acid.
- the method further comprises one or more of the following steps: dilution and buffer exchange of the loaded LNP composition; filtration of the loaded LNP composition; and storage of the loaded LNP composition.
- LNP formation step precipitation step
- the subject methods include an LNP formation step (precipitation step), wherein a lipid solution comprising an ionizable lipid, and an aqueous buffer are mixed together to form a lipid nanoparticle composition comprising empty LNPs.
- the precipitation step is performed with a lipid solution further comprising a helper lipid, a structural lipid, a PEG lipid or any combination thereof.
- the precipitation step is performed with a lipid solution further comprising a phospholipid, a structural lipid, and a PEG lipid.
- the precipitation step is performed with a lipid solution further comprising a PEG lipid and a phospholipid.
- the precipitation step is performed with a lipid solution further comprising a PEG lipid and a structural lipid. In some embodiments, the precipitation step is performed with a lipid solution further comprising a phospholipid and a structural lipid. In some embodiments, the precipitation step is performed with a lipid solution further comprising a PEG lipid. In some embodiments, the mixing step is performed with a lipid solution further comprising a phospholipid. In some embodiments, the mixing step is performed with a lipid solution further comprising a structural lipid.
- the precipitation step is performed with a lipid solution comprising a molar ratio of from 40% to 60 % ionizable lipid, a molar ratio of from 3.5% to 14% helper lipid, a molar ratio of from 28% to 50% structural lipid, and a molar ratio of from 0.5% to 5% PEG-lipid.
- the total molar percentage of the ionizable lipid, the helper lipid, the structural lipid, and the PEG-lipid is 100% in the lipid solution.
- the molar ratio of the ionizable lipid in the lipid solution is from 40% to 60% of the total lipid present in the lipid solution. In some embodiments, the molar ratio of the ionizable lipid in the lipid solution is 40%, 41%, 42%, 43%. 44%, 45%, 46%, 47%. 48%, 49%, 50%, 51%. 52%, 53%, 54%, 55%, 56%. 57%, 58%, 59%, or 60% of the total lipid present in the lipid solutions.
- the molar ratio of the helper lipid in the lipid solution is from 3.5% to 14% of the total lipid present in the lipid solution. In some embodiments, the molar ratio of the helper lipid in the lipid solution is 3%, 4%, 5%, 6%, 7%. 8%, 9%, 10%, 11%, 12%, 13%, or 14% of the total lipid present in the lipid solution.
- the helper lipid is DSPC. In some embodiments, the helper lipid is DOPE.
- the molar ratio of the structural lipid in the lipid solution is from 28% to 50% of the total lipid present in the lipid solution. In some embodiments, the molar ratio of the structural lipid in the lipid solution is 28%, 29%, 30%, 31%, 32%, 33%, 34%, 35%, 36%, 37%, 38%, 39%, 40%, 41%, 42%, 43%, 44%, 45%, 46%. 47%, 48%, 49%, or 50% of the total lipid present in the lipid solution. In some embodiments, the structural lipid is cholesterol.
- the molar ratio of the PEG-lipid in the lipid solution is from 0.1% to 5% of the total lipid present in the lipid solution. In some embodiments, the molar ratio of the PEG-lipid in the lipid solution is 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1.0%, 1.1%, 1.2%, 1.3%,
- the PEG-lipid is DMG-PEG2000.
- the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG- lipid in the lipid solution is 45:9:44:2. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the lipid solution is 50: 10:38.5: 1.5. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the lipid solution is 41:12:45:2. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the lipid solution is 62:4:33:1.
- the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the lipid solution is 53:5:41:1. In some embodiments, the molar ratio of each of die ionizable lipid, helper lipid, structural lipid, and PEG-lipid is within 10%, 9%, 8%, 7%, 6%, 5%. 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- the precipitation step is performed with a lipid solution comprising an ionizable lipid as described herein.
- the precipitation step is performed with a lipid solution further comprising a helper lipid, a structural lipid, a PEG lipid or any combination thereof as described herein.
- the helper lipid is 1,2-distearoyl-sn-glvcero- 3phosphocholine (DSPC)
- the structural lipid is cholesterol
- the PEG lipid is DMG-PEG2000.
- the precipitation step is performed with a lipid solution comprising about 30-60 mol % of ionizable lipid (c.g., as described herein); about 5-30 mol % of DSPC; about 15-50 mol % cholesterol; and about 1-2 mol % of DMG-PEG2000.
- a suitable lipid solution may contain a mixture of desired lipids at various concentrations.
- a suitable lipid solution may contain a mixture of desired lipids at a total concentration in a range from 0.1-100 mg/mL, 0.5-90 mg/mL, 1.0-80 mg/mL, 1.0-70 mg/mL, 1.0-60 mg/mL, 1.0-50 mg/mL, 1.0-40 mg/mL, 1.0-30 mg/mL, 1.0-20 mg/mL, 1.0-15 mg/mL, 1.0-10 mg/mL, 1.0-9 mg/mL, 1.0-8 mg/ml, 1.0-7 mg/mL, 1.0-6 mg/mL, or 1.0-5 mg/mL.
- the precipitation step is performed with a total lipid concentration from 5 mg/mL to 80 mg/mL, 6 mg/mL to 70 mg/mL, 7 mg/mL to 60 mg/mL, 8 mg/mL to 50 mg/mL, 9 mg/mL to 40 ing/mL, 10 mg/mL to 30 ing/mL, 15 mg/mL to 25 ing/mL, or 20 mg/mL to 25 mg/mL.
- the precipitation step is performed with a total lipid concentration of 10 mg/mL, 15 mg/mL, 20 mg/mL. 25 mg/mL, 30 mg/mL, 40 mg/mL, 50 mg/mL, 60 mg/mL. 70 mg/mL, or 80 mg/inL.
- the total lipid concentration is within 10%, 9%. 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%. 0.1%, 0.09%. 0.08%, 0.07%, 0.06%, 0.05%. 0.04%, 0.03%, 0.02%.
- the lipid solution comprises an organic solvent that is miscible with the aqueous buffer solution.
- the lipid solution comprises ethanol.
- a suitable lipid solution may contain a mixture of desired lipids dissolved in pure ethanol (i.e., 100% ethanol).
- the lipid solution comprises isopropyl alcohol.
- the lipid solution comprises benzyl alcohol.
- the lipid solution comprises dimethylsulfoxide.
- the lipid solution comprises a mixture of organic solvents including, but not limited to, ethanol, benzyl alcohol, isopropyl alcohol, and dimethylsulfoxide.
- the lipid solution comprises a mixture of ethanol and benzyl alcohol.
- the aqueous buffer solution comprises a buffering agent selected from ammonium sulfate, sodium bicarbonate, sodium citrate, sodium acetate, potassium phosphate. tris(hydroxymethyl)aminomethane (tris). 2-[Bis(2-hydroxyethyl)amino]-2- (hydroxymethyl)propane- 1,3 -diol (bis-tris), sodium phosphate, and HEPES.
- the buffering agent is sodium acetate.
- the precipitation step is performed with an aqueous buffer solution comprising an aqueous buffer at a concentration ranging from 1-500 mM, from 0.1-100 mM, from 0.5- 90 mM, from 1.0-80 mM, from 2-70 mM, from 3-60 mM. from 4-50 mM, from 5-40 mM, from 6-30 mM, from 6-20 mM, from 6-15 mM, or from 6-12 mM.
- the aqueous buffer concentration range is within 10%, 9%. 8%, 7%, 6%, 5%, 4%, 3%. 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%. 0.4%. 0.3%. 0.2%. 0.1%. 0.09%, 0.08%, 0.07%. 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- the precipitation step is performed with an aqueous buffer solution comprising an aqueous buffer at a concentration of or greater than 0.1 mM, 0.5 mM, 1 mM, 2 mM, 4 mM, 6 mM, 8 mM, 10 mM, 15 mM, 20 mM, 25 mM, 30 mM, 35 mM, 40 mM, 45 mM, or 50 mM.
- the precipitation step is performed with a first aqueous buffer comprising an aqueous buffer at a concentration of 6.25 ⁇ 2.0 mM, 6.25 ⁇ 1.5 mM, 6.25 ⁇ 1.0 mM, 6.25 ⁇ 0.9 mM, 6.25 ⁇ 0.8 mM, 6.25 ⁇ 0.7 mM, 6.25 ⁇ 0.6 mM, 6.25 ⁇ 0.5 mM, 6.25 ⁇ 0.4 mM, 6.25 ⁇ 0.3 mM, 6.25 ⁇ 0.2 mM, or 6.25 ⁇ 0.1 mM.
- the precipitation step is performed with a first aqueous buffer comprising an aqueous buffer at a concentration of about 6.25 mM.
- the aqueous buffer solution is at a pH from 2.0 to 9.0. such as from 2.5 to 8.5, from 2.6 to 8.4, from 2.7 to 8.3, from 2.8 to 8.2, from 2.9 to 8.1, from 3.0 to 8.0, from 3.2 to 7.8, from 3.4 to 7.6, from 3.6 to 7.4, from 3.8 to 7.2, from 4.0 to 7.0. from 4.1 to 6.8, from 4.2 to 6.6, from 4.3 to 6.4, from 4.4 to 6.2, from 4.5 to 6.0. from 4.6 to 5.9, from 4.7 to 5.8, from 4.8 to 5.7, from 4.9 to 5.6, from 5.0 to 5.5, from 5.1 to 5.4, or from 5.2 to about 5.3.
- the pH of the aqueous buffer solution is from 2 to 9, such as 2 to 6, or 3 to 5.
- the pH of the aqueous buffer solution is 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5. 7.0. 7.5. 8.0. 8.5. or 9.0.
- the pH of the aqueous buffer solution is within 10%, 9%, 8%, 7%, 6%, 5%. 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%. 0.2%. 0.1%. 0.09%, 0.08%, 0.07%. 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- the precipitation step is performed at a pH of 2.0, 2.1, 2.2, 2.3, 2.4, 2.5,
- the precipitation step is performed at a pH of less than 2.0, less than 2.5, less than 2.6, less than 2.7, less than 2.8. less than 2.9, less than 3.0, less than 3.2, less than 3.4 less than
- the precipitation step is performed at a pH of greater than 2.0. greater than 2.5, greater than 2.6, greater than 2.7, greater than 2.8, greater than 2.9, greater than 3.0. greater than 3.2, greater than 3.4 greater than 3.6, greater than 3.8, greater than 4.0, greater than 4.1. greater than 4.2, greater than 4.3, greater than 4.4, greater than 4.5, greater than 4.6, greater than 4.7. greater than 4.8, greater than 4.9, greater than 5.1, greater than 5.2, greater than 5.3, greater than 5.4. greater than 5.5, greater than 5.6, greater than 5.7, greater than 5.8, greater than 5.9. greater than 6.0.
- the precipitation step is performed at a pH of 3.0 ⁇ 2.0, 3.0 ⁇ 1.5, 3.0 ⁇ 1.0, 3.0 ⁇ 0.9. 3.0 ⁇ 0.8, 3.0 ⁇ 0.7, 3.0 ⁇ 0.6, 3.0 ⁇ 0.5, 3.0 ⁇ 0.4. 3.0 ⁇ 0.3, 3.0 ⁇ 0.2, or 3.0 ⁇ 0.1.
- the precipitation step is performed at a pH of 4.5 ⁇ 2.0, 4.5 ⁇ 1.5, 4.5 ⁇ 1.0, 4.5 ⁇ 0.9. 4.5 ⁇ 0.8, 4.5 ⁇ 0.7, 4.5 ⁇ 0.6, 4.5 ⁇ 0.5, 4.5 ⁇ 0.4. 4.5 ⁇ 0.3, 4.5 ⁇ 0.2, or 4.5 ⁇ 0.1.
- the precipitation step is performed with an aqueous buffer solution having a pH of about 3.0. In some embodiments, the precipitation step is performed with an aqueous buffer solution having a pH of about 4.5. In some embodiments, the mixing step is performed with an aqueous buffer solution comprising an acetate buffer. In some embodiments, the precipitation step is performed with an aqueous buffer solution comprising 6.25 mM sodium acetate. In some embodiments, the precipitation step is performed with an aqueous buffer solution comprising 6.25 mM sodium acetate at pH 3.0. In some embodiments, the precipitation step is performed with an aqueous buffer solution comprising 6.25 mM sodium acetate at pH 4.5.
- the ratio of lipid solution to aqueous buffer solution is within the range of 1 :2 to 1:5 by volume, such as 1 :2.5, 1:3.0, 1 :3.5, 1 :4.0, or 1 :4.5 by volume. In some embodiments, the ratio of lipid solution to aqueous buffer solution is 1 :2. In some embodiments, the ratio of lipid solution to aqueous buffer solution is 1 :3. In some embodiments, the ratio of lipid solution to aqueous buffer solution is 1:4. In some embodiments, the ratio of lipid solution to aqueous buffer solution is 1 :5.
- the ratio of lipid solution to aqueous buffer solution is within 10%, 9%, 8%, 7%, 6%, 5%, 4%. 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%. 0.1%. 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%. or 0.01% of the stated ranges and values.
- the mixing is carried out by simply combining the lipid solution and the aqueous buffer solution together in a vessel. In some embodiments of the precipitation step, the mixing is carried out by in-line mixing. In some embodiments of the precipitation step, the mixing is facilitated by an overhead mixer. In some embodiments of the precipitation step, the mixing is facilitated by a Y -mixer, a T-mixer, a stir bar, or a probe sonicator.
- the mixing in the precipitation step can be conducted at any convenient mixing speed.
- the mixing in the precipitation step is performed at a mixing speed from 100 to 10,000 rpm, such as 200 to 10,000 rpm, 500 to 10,000 rpm, 1000 to 10,000 rpm, 1000 to 9000 rpm, 1000 to 8000 rpm, 1000 to 7000 rpm, 1000 to 6000 rpm, 1000 to 5000 rpm, 1000 to 4000 rpm, 1000 to 4000 rpm, 2000 to 8000 rpm, 2000 to 7000 rpm, 2000 to 6000 rpm, 2000 to 5000 rpm, 2000 to 4000 rpm, or 2000 to 3000 rpm.
- the mixing in the precipitation step is performed at a mixing speed of 1000 rpm, 1500 rpm, 2000 rpm, 2500 rpm, 3000 rpm, 3500 rpm, 4000 rpm, 4500 rpm, 5000 rpm, 5500 rpm, 6000 rpm, 6500 rpm, 7000 rpm, 7500 rpm, or 8000 rpm.
- the mixing speed is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%.
- the mixing is carried out at a temperature of from 2-70 °C, such as 2-60 °C, 2-50 °C, 2-40 °C, 4-50 °C, 4-40 °C, or 4-30 °C.
- the mixing is carried out at a temperature of 4 °C, 5 °C, 6 °C, 7 °C, 8 °C, 9 °C, 10 °C, 11 °C, 12 °C, 13 °C, 14 °C, 15 °C, 16 °C, 17 °C, 18 °C, 19 °C, 20 °C.
- the mixing is carried out at a temperature of 4 °C, 10 °C, 20 °C, 30 °C, or 40 °C. In some embodiments, the mixing is carried out at 30 °C. In some embodiments, the mixing temperature is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%. 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%. 0.2%. 0.1%. 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- the mixing is performed at a temperature of less dian 50 °C, such as less than 45 °C, less than 40 °C, less than 35°C, less than 30°C, less than 25°C, less than 20 °C, less than 15 °C, or less than about 10°C.
- the mixing is performed at ambient temperature.
- the mixing is performed from 1 min to 100 min, such as 1 min to 80 min, 1 min to 70 min, 1 min to 60 min. 1 min to 50 min. 1 min to 45 min, 1 min to
- the mixing step is performed from 1 min to 10 min. such as 1 min to 9 min, 1 min to 8 min. 1 min to 7 min, 1 min to 6 min, 1 min to 5 min. In some embodiments of the precipitation step the mixing is carried out for 5 min.
- the mixing is performed for 5.0 ⁇ 2.0 min, 5.0 ⁇ 1.5 min, 5.0 ⁇ 1.0 min, 5.0 ⁇ 0.9 min. 5.0 ⁇ 0.8 min, 5.0 ⁇ 0.7 min. 5.0 ⁇ 0.6 min, 5.0 ⁇ 0.5 min. 5.0 ⁇ 0.4 min, 5.0 ⁇ 0.3 min, 5.0 ⁇ 0.2 min. or 5.0 ⁇ 0.1 min.
- the first LNP composition formed comprises empty LNPs of a random size distribution.
- the empty LNPs have an average particle size (average particle diameter) of greater than 100 nm. such as greater than 200 nm.
- the empty LNPs have an average particle size of 80-100 nm. such as 50-60 nm.
- the first LNP composition formed comprises empty LNPs having a polydispersity of greater than 0.2, such as 0.21, 0.22, 0.23, 0.24, 0.25, 0.26. 0.27, 0.28, 0.29, 0.30. or even greater. ii. LNP homogenization step
- the subject methods include an LNP homogenization step (particle size reduction step), wherein composition comprising empty LNPs from the precipitation step are homogenized to produce a homogeneous LNP composition of empty’ LNPs.
- the homogenizing step is performed using a microfluidic device.
- the composition is passed through the microfluidic device 1 to 5 times, such as 2 to 5 times, 3 to 5 times, or 4 to 5 times.
- the composition is passed through the microfluidic device two or more times, such as three or more times, four or more times, or five or more times.
- the composition is passed through the microfluidic device up to 5 times, such as up to 4 times, up to 3 times, or up to 2 times.
- the microfluidic device is selected from a high-pressure homogenizer, a high shear homogenizer and a probe homogenizer.
- the homogenizing step is performed using a high-pressure homogenizer. In some embodiments, the homogenizing step is performed using a high-pressure homogenizer at a pressure of from 1000 to 30,000 PSI, such as 1000 to 10,000 PSI, 10,000 to 20,000 PSI, or 20,000 to 30,000 PSI.
- the homogenizing step is performed using a high shear homogenizer.
- the homogenizing step is performed using a probe homogenizer.
- the homogenizing step is carried out at a temperature of 2-70 °C, such as 2-60 °C, 2-50 °C. 2-40 °C, 4-60 °C, 4-50 °C. or 4-40 °C. In some embodiments the homogenizing step is carried out at a temperature of 4 °C, 5 °C, 6 °C. 7 °C, 8 °C, 9 °C. 10 °C, 11 °C, 12 °C.
- the homogenizing step is carried out at a temperature of 4 °C, 10 °C. 20 °C, 30 °C, 40 °C. or 50°C. In some embodiments, the homogenizing step is carried out at 30 °C. such that the temperature is 30 °C in the microfluidic device. In some embodiments, the homogenizing step is carried out at 40 °C, such that the temperature is 40 °C in the microfluidic device.
- the homogenizing step is carried out at 50 °C, such that the temperature is 50 °C in the microfluidic device.
- the homogenizing step temperature is within 10%, 9%, 8%, 7%, 6%, 5%. 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%. 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- the homogenizing step is performed at a temperature of less than 50 °C, less than 45 °C, less than 40 °C, less than 35 °C. less than 30 °C, less than 25 °C, less than 20 °C, less than 15 °C, or less than 10 °C.
- the homogenizing step is performed at ambient temperature.
- the homogeneous LNP composition formed is immediately cooled after the homogenizing step. For example, after the homogenous LNP composition exits the microfluidic device, the composition is cooled. In some embodiments, after exiting the microfluidic device, the homogenous LNP composition is cooled to 10 °C or less, such as 4 °C or less. Without being bound to any particular theory, rapid cooling of the homogeneous LNP composition after it exits the microfluidic device (e.g.. the high-pressure homogenizer) can prevent aggregation of the LNPs in the composition.
- the empty' LNPs in the homogeneous LNP composition formed have an average particle size of from 30 nm to 200 rnn, such as 50 mn to 70 nm. In some embodiments of the homogenization step, the empty LNPs in the homogeneous LNP composition have a poly dispersity of 0.05 to 0.2, such as 0.1 or less.
- the average particle size of the empty LNPs in the homogeneous LNP composition formed is at least 10% less than the average particle size of the empty LNPs of the first LNP composition formed in the precipitation step. In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition is 10-50% less than the average particle size of the empty LNPs of the first LNP composition. In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition is 10-30% less than the average particle size of the empty LNPs of the first LNP composition.
- the average particle size of the empty LNPs in the homogeneous LNP composition formed in the homogenization step is 10% less, such as 15% less, 20% less, 25% less, 30% less. 35% less. 40% less, 45% less, or 50% less than the average particle size of the empty LNPs of the first LNP composition formed in the precipitation step.
- the homogeneous LNP composition obtained after the homogenizing step can be stored at 4 °C, and is storage stable for at least 4 weeks, such as at least 8 weeks, at least 12 weeks, at least 16 weeks, at least 20 weeks, at least 6 months, at least 9 months, or at least a year.
- the subject methods include an LNP loading step, wherein the homogeneous composition of empty LNPs obtained from the homogenization step are mixed with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with the nucleic acid.
- the loaded LNP composition comprises LNPs encapsulating the nucleic acid.
- the amounts of nucleic acid and empty 7 LNPs are selected to provide a specific N:P ratio.
- the N:P ratio of the loaded LNP composition refers to the molar ratio of nitrogen atoms in one or more lipids (e.g., ionizable lipids) to the number of phosphate groups in the nucleic acid.
- the molar ratio of nitrogen atoms in one or more ionizable lipids (i.e., in the empty LNPs) to phosphate groups in tire nucleic acid (N:P) is within the range of 1 :20 to 20: 1.
- the N:P ratio is 1 :20, 1: 19, 1: 18, 1 : 17, 1 : 16, 1:15, 1:14, 1 : 13, 1: 12, 1: 11, 1: 10, 1 :9, 1:8, 1 :7, 1 :6, 1:5, 1:4, 1:3. 1:2 or 1 : 1.
- the N:P ratio is 20: 1, 19:1, 18: 1, 17: 1, 16: 1. 15:1, 14: 1, 13: 1, 12: 1, 11: 1, 10: 1, 9: 1, 8:1, 7: 1. 6: 1, 5: 1, 4: 1, 3:1 or 2:1.
- the N:P ratio is within the range of 4: 1 to 6: 1.
- the N:P ratio is 4.0: 1, 4.1 :1. 4.2: 1. 4.3:1. 4.4: 1, 4.5: 1, 4.6: 1, 4.7: 1, 4.8: 1, 4.9: 1, 5.0: 1, 5.1 : 1, 5.2: 1, 5.3: 1, 5.4: 1, 5.5: 1, 5.6:1, 5.7:1, 5.8:1, 5.9: 1, or 6.0: 1.
- the N:P ratio is within 10%, 9%, 8%, 7%, 6%. 5%, 4%, 3%, 2%. 1%. 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%. 0.1%. 0.09%, 0.08%, 0.07%. 0.06%, 0.05%, 0.04%, 0.03%, 0.02%. or 0.01% of the stated value.
- the nucleic acid solution i.e., nucleic acid in a buffer as described herein
- the process is carried out at ambient temperature.
- the loading step is carried out at a temperature of 2-70 °C, such as 2-60
- the loading step is carried out at a temperature of 4 °C, 10 °C, 20 °C, 30 °C, 40 °C. or 50°C. In some embodiments, the loading step temperature is within 10%, 9%, 8%. 7%, 6%, 5%, 4%. 3%. 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%. 0.4%. 0.3%. 0.2%. 0.1%. 0.09%, 0.08%, 0.07%, 0.06%. 0.05%, 0.04%, 0.03%. 0.02%, or 0.01% of the stated value.
- the loading step is performed at a temperature of less than 50 °C, such as less than 45 °C. less than 40 °C. less than 35 °C. less than 30 °C, less than 25 °C, less than 20 °C, less than 15 °C, or less than 10 °C.
- a suitable nucleic acid solution is an aqueous solution containing a nucleic acid to be encapsulated at various concentrations.
- a suitable nucleic acid solution may contain a nucleic acid at a concentration of or greater than 0.01 mg/mL, such as 0.05 mg/mL. 0.06 mg/mL, 0.07 mg/mL.
- a suitable nucleic acid solution may contain a nucleic acid at a concentration in a range from 0.01-1.0 mg/mL. such as 0.01-0.9 mg/mL, 0.01-0.8 mg/mL.
- the nucleic acid solution comprises a buffer salt selected from an acetate salt, a citrate salt, or a bis-tris salt.
- the buffer salt is selected from ammonium sulfate, sodium bicarbonate, sodium citrate, sodium acetate, potassium phosphate, tris(hydroxymethyl)aminoinethane (tris), 2-[Bis(2-hydroxyethyl)amino]-2- (liydroxymethyl)propane-l,3-diol (bis-tris), sodium phosphate, and HEPES.
- the buffer salt is sodium acetate.
- the loading step is performed with a nucleic acid solution comprising a buffer salt at a concentration ranging from 1-500 mM, such as from 0.1-100 rnM, from 0.5-90 mM, from 1.0-80 mM, from 2-70 mM, from 3-60 mM, from 4-50 mM, from 5-40 mM, from 6-50 mM, from 6-40 mM, from 6-30 mM, or from 6-20 mM.
- 1-500 mM such as from 0.1-100 rnM, from 0.5-90 mM, from 1.0-80 mM, from 2-70 mM, from 3-60 mM, from 4-50 mM, from 5-40 mM, from 6-50 mM, from 6-40 mM, from 6-30 mM, or from 6-20 mM.
- the buffer salt concentration range is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- the loading step is performed with a nucleic acid solution comprising a buffer salt at a concentration of or greater than 0.1 mM, 0.5 mM, 1 mM, 2 mM, 4 mM, 6 mM, 8 mM, 10 mM, 15 mM, 20 mM, 25 mM, 30 mM, 35 mM, 40 mM, 45 mM, or 50 mM.
- the loading step is performed with nucleic acid solution comprising a buffer salt at a concentration of 6.25 ⁇ 2.0 mM, 6.25 ⁇ 1.5 mM, 6.25 ⁇ 1.0 mM, 6.25 ⁇ 0.9 mM. 6.25 ⁇ 0.8 mM, 6.25 ⁇ 0.7 mM, 6.25 ⁇ 0.6 mM, 6.25 ⁇ 0.5 mM, 6.25 ⁇ 0.4 mM, 6.25 ⁇ 0.3 mM, 6.25 ⁇ 0.2 mM, or 6.25 ⁇ 0.1 mM.
- the loading step is performed with nucleic acid solution comprising a buffer salt at a concentration of about 6.25 mM.
- the loading step is performed with nucleic acid solution comprising a buffer salt at a concentration of 50 ⁇ 2.0 mM, 50 ⁇ 1.5 mM, 50 ⁇ 1.0 mM, 50 ⁇ 0.9 mM, 50 ⁇ 0.8 mM. 50 ⁇ 0.7 mM. 50 ⁇ 0.6 mM, 50 ⁇ 0.5 mM, 50 ⁇ 0.4 mM, 50 ⁇ 0.3 mM. 50 ⁇ 0.2 mM. or 5() ⁇ 0.1 mM.
- the loading step is performed with nucleic acid solution comprising a buffer salt at a concentration of about 50 mM.
- the nucleic acid solution is at a pH from 2.0 to 9.0, such as from 2.5 to 8.5, from 2.6 to 8.4, from 2.7 to 8.3, from 2.8 to 8.2, from 2.9 to 8.1. from 3.0 to 8.0. from 3.2 to 7.8, from 3.4 to 7.6, from 3.6 to 7.4, from 3.8 to 7.2, from 4.0 to 7.0. from 4.1 to 6.8, from 4.2 to 6.6, from 4.3 to 6.4, from 4.4 to 6.2, from 4.5 to 6.0, from 4.6 to 5.9, from 4.7 to 5.8, from 4.8 to 5.7, from 4.9 to 5.6, from 5.0 to 5.5, from 5.1 to 5.4, or from 5.2 to 5.3.
- the pH of the nucleic acid solution is from 2 to 9, such as 2 to 7, or 3 to 5. In some embodiments, the pH of the nucleic acid solution is 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, 8.5, or 9.0. In some embodiments, the pH of the nucleic acid solution is within 10%.
- the loading step is performed at a pH of 2.0. 2.1, 2.2, 2.3, 2.4, 2.5, 2.6,
- the loading step is performed at a pH of less than 2.0. less than 2.5, less than 2.6. less than 2.7, less than 2.8, less than 2.9, less than 3.0, less than 3.2. less than 3.4 less than 3.6, less than 3.8, less than 4.0. less than 4.1, less than 4.2, less than 4.3. less than 4.4, less than 4.5, less than 4.6, less than 4.7, less than 4.8, less than 4.9, less than 5.0, less than 5.1, less than 5.2. less than
- the loading step is performed at a pH of greater than 2.0, greater than
- the loading step is performed at a pH of 3.0 ⁇ 2.0, 3.0 ⁇ 1.5, 3.0 ⁇ 1.0, 3.0 ⁇ 0.9, 3.0 ⁇ 0.8, 3.0 ⁇ 0.7, 3.0 ⁇ 0.6, 3.0 ⁇ 0.5, 3.0 ⁇ 0.4, 3.0 ⁇ 0.3, 3.0 ⁇ 0.2, or 3.0 ⁇ 0.1.
- the loading step is performed at a pH of 4.5 ⁇ 2.0, 4.5 ⁇ 1.5, 4.5 ⁇ 1.0, 4.5 ⁇ 0.9, 4.5 ⁇ 0.8, 4.5 ⁇ 0.7, 4.5 ⁇ 0.6, 4.5 ⁇ 0.5, 4.5 ⁇ 0.4, 4.5 ⁇ 0.3, 4.5 ⁇ 0.2, or 4.5 ⁇ 0.1.
- the loading step is performed with a nucleic acid solution comprising a buffer salt having a pH of about 3.0. In some embodiments, the loading step is performed with nucleic acid solution comprising a buffer salt having a pH of about 4.5. In some embodiments, the loading step is performed with a nucleic acid solution comprising an acetate buffer salt.
- the loading step is performed with a nucleic acid solution comprising 6.25 mM sodium acetate. In some embodiments, the loading step is performed with a nucleic acid solution comprising 6.25 mM sodium acetate at pH 3.0. In some embodiments, the loading step is perfonned with a nucleic acid solution comprising 6.25 mM sodium acetate at pH 4.5. In some embodiments, the loading step is performed with a nucleic acid solution comprising 50 mM sodium acetate. In some embodiments, the loading step is performed with a nucleic acid solution comprising 50 mM sodium acetate at pH 3.0. In some embodiments, the loading step is performed with a nucleic acid solution comprising 50 mM sodium acetate at pH 4.5.
- nucleic acid solution suitable for the loading step may be directly dissolved in a buffer solution described herein.
- a nucleic acid solution may be generated by mixing nucleic acid stock solution with a buffer solution prior to mixing with a homogeneous empty LNP composition.
- a nucleic acid solution may be generated by mixing a nucleic acid stock solution with a buffer solution immediately before mixing with a homogeneous empty LNP composition.
- the loading step is performed by mixing the homogeneous LNP composition with a nucleic acid solution (i.e., as described herein) at a flow rate from 50 mL/min to 100 L/min. In some embodiments, the flow rate is from 100 mL/min to 100 L/min. such as 100 mL/min to 95 L/min.
- the flow rate is from 100 mL/min to 50 L/min. such as 100 mL/min to 45 mL/min, 100 mL/min to 40 L/min. 100 mL/min to 35 L/min, 100 mL/min to 30 L/min.
- the flow rate is 100 mL/min to 25 L/min, 100 mL/min to 20 L/min, 100 ml/min to 15 L/min, 100 mL/min to 10 L/min, or 100 mL/min to 5 L/min.
- the flow rate is 100 mL/min to 3L/min. such as 100 mL/min to 2.5 L/min. or 100 mL min to 2 L/min. In some embodiments, the flow rate is 100 inL/inin to 2 L/min.
- L/min such as 150 mL/min to 2 L/min, 200 mL/min to 2 L/min, 300 mL/min to 2 L/min, 400 mL/min to 2 L/min, 500 mL/min to 2 L/min, 600 mL/min to 2L/min, 700 mL/min to 2L/min, 800 mL/inin to 2L/min, 900 mL/min to 2 L/min, 1 L/min to 2 L/min, 1.1 L/min to 2 L/min, 1.2 L/min to 2L/min, 1.3 L/min to 2 L/min, 1.4 L/min to 2 L/min. 1.5 L/min to 2 L/min, 1.6 L/min to 2 L/min, 1.7 L/min to 2 L/min, 1.8 L/min to 2 L/min, or 1.9 L/min to 2 L/min.
- the loading step is performed by mixing the homogenous LNP composition with a nucleic acid solution at a flow rate of at least 100 mL/min, such as at least 150 mL/min, at least 200 mL/min, at least 250 inL/min, at least 300 mL/min, at least 350 mL/min, at least 400 mL/min, at least 450 mL/min, at least 500 mL/min, at least 550 mL/min, at least 600 mL/min, at least 650 mL/min, at least 700 mL/min, at least 750 mL/min, at least 800 mL/min, at least 850 mL/min, at least 900 mL/min, at least 950 mL/min, at least 1,000 mL/min, at least 1,100 mL/min, at least 1,200 mL/min, at least 1,300 mL/min, at least 1,400 mL/min
- the loading step is performed by mixing the homogenous LNP composition with a nucleic acid solution at a flow rate of at least 1 L/min, such as at least 2 L/min, at least 3 L/min, at least 4 L/min, at least 5 L/min, at least 6 L/min, at least 7 L/min, at least 8 L/min, at least 9 L/min, at least 10 L/min.
- a flow rate of at least 1 L/min such as at least 2 L/min, at least 3 L/min, at least 4 L/min, at least 5 L/min, at least 6 L/min, at least 7 L/min, at least 8 L/min, at least 9 L/min, at least 10 L/min.
- the mixing of the homogeneous LNP composition with the nucleic acid solution is performed using a pump.
- the pump is selected from a gear pump, a peristatic pump, or a centrifugal pump.
- the mixing may be performed using microfluidic mixers.
- exemplary microfluidic mixers may include, but are not limited to, a slit interdigital micromixer including, but not limited to, those manufactured by Precision Nanosystems (Vancouver. BC, Canada), Microinnova (Allerheiligen bei Wildon, Austria) and/or a staggered herringbone micromixer (SHM) (Zhigaltsev, I.V. et al. (2012) Langmuir. 28:3633-40; Belliveau, N.M. et al. Mol. Ther. Nucleic. Acids. (2012) Le37; Chen. D. et al. J. Am. Chem. Soc. (2012) 134(22):6948-51; each of which is herein incorporated by reference in its entirety).
- SHM herringbone micromixer
- the mixing may further comprise combining at least two input streams wherein mixing occurs by microstructure-induced chaotic advection (MICA).
- MICA microstructure-induced chaotic advection
- fluid streams flow through channels present in a herringbone pattern causing rotational flow and folding the fluids around each other.
- This method may also comprise a surface for fluid mixing wherein the surface changes orientations during fluid cycling.
- Known methods of generating LNPs using SHM include those disclosed in U.S. Pat. Pub. Nos. US2004/0262223 Al and US2012/0276209 Al, each of which is incorporated herein by reference in their entirety.
- die mixing may be performed using a micromixer such as, but not limited to, a Slit Interdigital Microstructured Mixer (SIMM-V2) or a Standard Slit Interdigital Micro Mixer (SSIMM) or Caterpillar (CPMM) or Impinging-jet (IJMM)from the Institut fur Mikrotechnik Mainz GmbH, Mainz Gennany).
- a micromixer such as, but not limited to, a Slit Interdigital Microstructured Mixer (SIMM-V2) or a Standard Slit Interdigital Micro Mixer (SSIMM) or Caterpillar (CPMM) or Impinging-jet (IJMM)from the Institut fur Mikrotechnik Mainz GmbH, Mainz Gennany).
- the loading step is carried out by utilizing microfluidic technology (see, Whitesides (2006) Nature. 442: 368-373; and Abraham et al. (2002) Science. 295: 647-651; each of which is herein incorporated by reference in its entirety).
- controlled microfluidic formulation includes a passive method for mixing streams of steady pressure-driven flows in micro channels at a low Reynolds number (see, e.g., Abraham et al. (2002) Science. 295: 647651; which is herein incorporated by reference in its entirety).
- the mixing may be performed using a micromixer chip such as. but not limited to, those from Harvard Apparatus (Holliston. MA). Dolomite Microfluidics (Royston, UK), or Precision Nanosystems (Van Couver, BC. Canada).
- a micromixer chip can be used for rapid mixing of two or more fluid streams with a split and recombine mechanism.
- the loading step produces a loaded LNP composition comprising LNPs encapsulating a nucleic acid.
- the nucleic acid is encapsulated in the loaded LNPs with an encapsulation efficiency of from 50 to 100%.
- the nucleic acid is encapsulated in the loaded LNPs with an encapsulation efficiency of 50% or more, such as 60% or more. 70% or more, 80% or more, or 90% or more.
- the nucleic acid is encapsulated in the loaded LNPs with an encapsulation efficiency of at least 50%, for example 50%, 55%, 60%, 65%, 70%. 75%. 80%, 85%, 90%, 91%. 92%. 93%, 94%, 95%, 96%. 97%. 98%, 99%, or 100%.
- the encapsulation efficiency may be at least 80%. In certain embodiments, the encapsulation efficiency may be at least 90%.
- the nucleic acid is an RNA polynucleotide.
- the RNA is a circular RNA polynucleotide (aka circRNA or oRNATM) (as described herein).
- the loaded LNPs formed have an average particle size (average particle diameter) from 50 inn to 300 mn and a polydispersity of 0.3 or less. In some embodiments of the loading step, the loaded LNPs formed have an average particle size from 70 nm to 120 nm and a polydispersity of 0.2 or less.
- the loaded LNP produced is storage stable at a temperature of from 4 °C to -80 °C.
- the loaded LNPs produced are stored in a storage buffer or tonicity modifier.
- the storage buffer or tonicity modifier includes, but is not limited to, tris-sucrose saline, tris-trehalose saline, phosphate buffered saline, or tris buffered saline.
- the loading step (described herein) may be conducted prior to the homogenization step (described herein).
- a method of preparing a loaded LNP composition comprising: a) a precipitation step, comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first LNP composition comprising empty LNPs; b) a loading step, comprising: mixing the first LNP composition with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with tire nucleic acid; and c) a homogenization step, comprising: homogenizing the loaded LNP composition to produce a homogeneous loaded LNP composition comprising LNPs associated with the nucleic acid, wherein each of steps a)-c) above are as described herein above in “Methods of Preparation.” sections i)-iii). iv. Additional processing steps
- the subject methods may optionally include one or more further processing steps.
- one or more further processing steps may be performed on the homogeneous LNP composition of empty LNPs or the loaded LNP composition.
- the further processing steps include one or more of filtering, pH adjusting, buffer exchanging, diluting, concentrating, freezing, lyophilizing, storing, adding a cryoprotectant, and packing.
- one or more further processing steps include a dilution and buffer exchange step of the homogeneous empty LNP composition, filtration of the homogeneous empty LNP composition, and storage of the homogeneous empty LNP composition.
- one or more further processing steps include a dilution and buffer exchange step of the loaded LNP composition, filtration of the loaded LNP composition, and storage of the loaded LNP composition.
- a dilution and buffer exchange step may be performed on the composition of homogeneous empty LNPs.
- the buffer exchange step is performed by filtration.
- the buffer exchange step is performed via tangential flow filtration (TFF).
- a dilution and buffer exchange step may be performed on the composition of loaded LNPs.
- the buffer exchange step is performed by filtration.
- the buffer exchange step is performed via tangential flow filtration (TFF).
- filtration removes an organic solvent (e.g., an alcohol or ethanol) from the composition of empty LNPs or loaded LNPs. In some embodiments, filtration removes substantially all of the organic solvent (e.g., an alcohol or ethanol) from the composition of empty LNPs or loaded LNPs.
- an organic solvent e.g., an alcohol or ethanol
- substantially all of the organic solvent e.g., an alcohol or ethanol
- the additional further processing steps include pH adjustment of the composition comprising the loaded LNPs.
- the pH adjustment may be carried out by adding a second buffering agent.
- the additional further processing steps include concentrating the composition of empty LNPs or loaded LNPs.
- the additional further processing steps includes freezing the composition of empty LNPs or loaded LNPs by. e.g., lyophilization
- the lyophilizing comprises freezing the composition of empty LNPs or loaded LNPs at a temperature from -100 °C to 0 °C, such as -80 °C to -10 °C, -60 ° C to -20 °C, -50 °C to -25 °C, or -40 °C to -30 °C.
- the lyophilizing further comprises drying the frozen composition of empty LNPs or loaded LNPs to form a lyophilized empty LNP or lyophilized loaded LNP.
- the further processing steps include adding a cryoprotectant to the composition of empty’ LNPs or loaded LNPs.
- the further processing steps include storage of the composition of empty LNPs or loaded LNPs.
- the composition of empty LNPs or loaded LNPs are stored at a temperature of 4 °C.
- the composition of empty’ LNPs or loaded LNPs are stored at a temperature from -100 °C to 0 °C, such as -80 °C to -10 °C, -60 0 C to -20 °C, -50 °C to -25 °C, or -40 °C to -30 °C.
- the composition of empty LNPs or loaded LNPs arc stored in a storage buffer or tonicity modifier.
- the storage buffer or tonicity modifier includes, but is not limited to, tris-sucrose saline, tris-trehalose saline, phosphate buffered saline, or tris buffered saline.
- the further processing steps include packing of the composition of empty LNPs or loaded LNPs.
- packing may refer to storing a drug product in its final state or in-process storage of an empty LNP, or loaded LNP before they are placed into final packaging.
- Modes of storage and/or packing include, but are not limited to, refrigeration in sterile bags, refrigerated or frozen formulations in vials, lyophilized formulations in vials and syringes, etc. 2.
- EMPTY LIPID NANOPARTICLES EMPTY LIPID NANOPARTICLES
- the present disclosure provides a homogeneous LNP composition of empty LNPs prepared by a subject method.
- a homogeneous composition of empty LNPs comprising an ionizable lipid, a helper lipid, a cholesterol and a PEG lipid, wherein the empty LNPs have a polydispersity of 0.2 or less, such as 0.1 or less.
- the term “homogeneous” as it applies to a composition of empty 7 LNPs refers to the population of empty LNPs in the composition having a polydispersity 7 of 0.2 or less.
- a homogeneous LNP composition substantially free of loaded LNPs and comprising empty LNPs comprising an ionizable lipid, a helper lipid, a cholesterol and a PEG lipid, wherein the empty LNPs have a poly dispersity of 0.2 or less (e.g., 0. 1 or less), and/or an average particle size from 50 nm to 70 nm.
- the term “substantially 7 free” as it applies to loaded LNPs refers to the homogeneous LNP composition of empty LNPs including few or no loaded LNPs.
- substantially free of loaded LNPs refers to a homogeneous LNP composition including an amount of less than 5%, such as less than 4%, less than 3%, less than 2%, less than 1%, less than 0.9%, less than 0.8%, less than 0.7%, less than 0.6%, less than 0.5%, less than 0.4%, less than 0.3%, less than 0.2% or less than 0.1% of the loaded LNPs.
- the homogeneous LNP composition is at least 99% free of loaded LNPs.
- the homogeneous LNP composition includes no loaded LNPs.
- an empty 7 LNP composition prepared by a method disclosed herein.
- the empty LNPs comprises a molar ratio of from 40% to 60% ionizable lipid, a molar ratio of from 3.5% to 14% helper lipid, a molar ratio of from 28% to 50% structural lipid, and a molar ratio of from 0.5% to 5% PEG-lipid.
- the total molar percentage of the ionizable lipid, the helper lipid, the structural lipid, and the PEG-lipid is 100% in tire empty LNPs.
- the molar ratio of the ionizable lipid in the empty LNPs is from 40 to 60% of the total lipid present in the empty LNPs. In some embodiments, the molar ratio of the ionizable lipid in the empty LNPs is 40%, 41%, 42%. 43%, 44%, 45%. 46%, 47%, 48%, 49%, 50%. 51 %, 52%, 53%, 54%. 55%, 56%, 57%, 58%, 59%. or 60% of the total lipid present in the empty LNPs.
- the molar ratio of the helper lipid in the empty LNPs is from 3.5% to 14% of the total lipid present in the empty LNPs. In some embodiments, the molar ratio of the helper lipid in the lipid solution is 3%, 4%, 5%, 6%. 7%, 8%, 9%, 10%, 11%, 12%, 13%, or 14% of the total lipid present in the empty LNPs.
- the helper lipid is DSPC. In some embodiments, the helper lipid is DOPE.
- the molar ratio of the structural lipid in the empty LNPs is from 28% to 50% of the total lipid present in the empty LNPs.
- tire molar ratio of the structural lipid in the empty LNPs is 28%, 29%, 30%, 31%. 32%, 33%, 34%, 35%, 36%, 37%, 38%, 39%, 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, or 50% of the total lipid present in the empty LNPs.
- the structural lipid is cholesterol.
- the molar ratio of the PEG-lipid in the empty’ LNPs is from 0.1% to 5% of the total lipid present in the empty LNPs. In some embodiments, the molar ratio of the PEG-lipid in the empty LNPs is 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1.0%, 1.1%, 1.2%. 1.3%, 1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, 2.0%, 2.1%, 2.2%, 2.3%, 2.4%, 2.5%, 2.6%, 2.7%, 2.8%. 2.9%, 3.0%, 3.1%, 3.2%, 3.3%, 3.4%, 3.5%, 4.0%, 4.5%, or 5% of the total lipid present in the empty LNPs. In some embodiments, the PEG-lipid is DMG-PEG 2000 .
- the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the empty LNPs is 45:9:44:2. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the empty LNPs is 50:10:38.5:1.5. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the empty LNPs is 41:12:45:2.
- the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the empty' LNPs is 62:4:33:1. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the empty LNPs is 53:5:41:1. In some embodiments, the molar ratio of each of tire ionizable lipid, helper lipid, structural lipid, and PEG-lipid is within 10%, 9%. 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%. 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%. or 0.01% of the stated value.
- the empty LNPs comprise an ionizable lipid as described herein, l,2-distearoyl-sn-glycero-3phosphocholine (DSPC), cholesterol, and DMG- PEG2000.
- the empty LNPs comprise 30-60 mol % ionizable lipid (e.g., as described herein); 5-30 mol % DSPC; 15-50 mol % cholesterol; and 1-2 mol % DMG-PEG2000.
- the LNPs have an average particle size (i.e., an average nanoparticle diameter) from 10 nm to 100 nm such as, but not limited to, 10 nm to 20 nm, 10 nm to 30 nm. 10 nm to 40 nm. 10 mn to 50 nm, 10 nm to 60 nm, 10 nm to 70 nm, 10 nm to 80 mn, 10 nm to 90 nm, 20 nm to 30 nm.
- 10 nm to 100 nm such as, but not limited to, 10 nm to 20 nm, 10 nm to 30 nm.
- the lipid nanoparticles may have a diameter from 30 to 200 nm.
- the homogeneous composition of empty LNPs may have an average particle size of less than 200 nm, less than 180 nm, less than 150 nm, less than 130 nm. less than 120 nm, less than 100 nm, less than 80 nm, less than 60 nm, less than 50 nm, or less than 40 nm. Each possibility represents a separate embodiment of the present disclosure.
- the LNPs have an average particle size of 10-200 mn, 20-200 nm, 30-200 nm, or 50-200 nm. In some embodiments, the empty LNPs have an average particle size of 50-200 nm, 50-150 nm, 50-100 mn, or 50-70 nm. In some embodiments, the empty LNPs have an average particle size of 50 to 70 nm.
- the LNPs have an average particle size from 1 mn to 100 mn, from 1 nm to 10 nm, 1 mn to 20 nm, from 1 nm to 30 mn, from 1 mn to 40 nm.
- nm to 50 nm from 1 mn to 60 mn, from 1 nm to 70 mn, from 1 nm to 80 mn, from 1 nm to 90 mn, from 5 nm to 100 nm, from 5 nm to 10 mn, 5 mn to 20 nm, from 5 mn to 30 mn, from 5 nm to 40 nm.
- the LNPs have an average particle size of 50 mn, 51 nm, 52. nm, 53 nm, 54 nm, 55 nm. 56 nm, 57 mn. 58 mn, 59 nm, 60 mn. 61 nm, 62 nm. 63 nm, 64 nm, 65 nm, 66 nm, 67 mn, 68 nm, 69 mn. or 70 nm. In some embodiments, the average particle size of the homogeneous composition of empty LNPs is within 10%, 9%, 8%, 7%, 6%.
- the average particle size of the empty LNPs in the homogeneous LNP composition is at least 10% less than the average particle size of the empty LNPs of the first LNP composition. In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition is 10-50% less than the average particle size of the empty LNPs of the first LNP composition. In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition is 10-30% less than the average particle size of the empty LNPs of the first LNP composition.
- the average particle size of the empty LNPs in the homogeneous LNP composition is 10% less, such as 15% less, 20% less, 25% less, 30% less, 35% less, 40% less, 45% less, or 50% less than the average particle size of the empty LNPs of the first LNP composition.
- the subject methods provide a homogeneous composition of empty LNPs.
- a poly dispersity index may be used to indicate the homogeneity of the empty LNPs in the composition, e.g., the particle size distribution of the nanoparticle compositions.
- a small (e g., less than 0.2) polydispersity index generally indicates a narrow particle size distribution.
- An empty LNP composition may have a poly dispersity index from about 0.01 to about 0.2, such as 0.01, 0.02, 0.03, 0.04, 0.05. 0.06, 0.07, 0.08, 0.09, 0.10, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, or 0.20.
- the polydispersity index of an empty LNP composition may be from 0.05 to 0.20. In some embodiments, the poly dispersity index of the homogeneous composition of empty LNPs is within 10%, 9%, 8%, 7%, 6%. 5%, 4%, 3%, 2%. 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%. 0.3%. 0.2%, 0.1%, 0.09%. 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- the polydispersity index of the homogeneous composition of empty LNPs is 0.2 or less, such as 0.19 or less, 0.18 or less, 0.17 or less, 0.16 or less, 0.15 or less, 0.14 or less, 0.13 or less, 0.12 or less. 0.11 or less, or 0.10 or less. In some embodiments, the poly dispersity index of the empty LNP composition is 0.1 or less, such as 0.09 or less, 0.08 or less, 0.07 or less, 0.06 or less, or 0.05 less.
- the zeta potential of a nanoparticle composition may be used to indicate the electrokinetic potential of the composition.
- the zeta potential may describe the surface charge of a nanoparticle composition.
- Nanoparticle compositions with relatively low charges, positive or negative, are generally desirable, as more highly charged species may interact undesirably with cells, tissues, and other elements in the body.
- the zeta potential of the subject empty' LNPs may be from -20 mV to +20 mV, from -20 mV to +15 mV, from -20 mV to +10 mV, from -20 mV to +5 mV, from -20 mV to 0 mV, from -20 mV to -5 mV, from -20 mV to -10 mV. from -20 mV to -15 mV from -20 mV to +20 mV, from -20 mV to +15 mV, from -20 mV to +10 mV. from -20 mV to +5 mV, from -20 mV to 0 mV.
- the zeta potential of the subject empty LNPs is within 10%, 9%. 8%. 7%, 6%, 5%, 4%. 3%, 2%. 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%. 0.04%. 0.03%, 0.02%. or 0.01% of the stated value.
- the properties of a homogeneous composition of empty' LNPs may be influenced by factors including, but not limited to. the selection of the cationic lipid component, the degree of cationic lipid saturation, the selection of the helper hpid component, the degree of helper lipid saturation, the selection of the cholesterol lipid component, the selection of the PEG lipid component, the nature of the PEGylation, ratio of all components and biophysical parameters such as size.
- the homogeneous composition of empty LNPs has a pH from 2.0 to 9.0, from 2.5 to 8.5, from 2.6 to 8.4, from 2.7 to 8.3, from 2.8 to 8.2, from 2.9 to 8.1, from 3.0 to 8.0, from 3.2 to 7.8, from 3.4 to 7.6, from 3.6 to 7.4, from 3.8 to 7.2, from 4.0 to 7.0, from 4.1 to 6.8. from 4.2 to 6.6, from 4.3 to 6.4, from 4.4 to 6.2, from 4.5 to 6.0, from 4.6 to 5.9, from 4.7 to 5.8, from 4.8 to 5.7, from 4.9 to 5.6, from 5.0 to 5.5, from 5.1 to 5.4, or from 5.2 to about 5.3.
- the homogeneous composition of empty LNPs has a pH from 2 to 9, such as 2 to 6, or 3 to 5.
- the pH of the homogeneous composition of empty LNPs is 2.0. 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, 8.5, or 9.0.
- the pH of the homogeneous composition of empty LNPs is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- the homogeneous composition of empty LNPs can be stored at 4 °C, and is storage stable for at least 4 weeks, such as at least 8 weeks, at least 12 weeks, at least 16 weeks, at least 20 weeks, at least 6 months, at least 9 months, or at least a year.
- the homogeneous composition of empty LNPs or is stored at a temperature from -100 °C to 0 °C, such as -80 °C to -10 °C, -60 0 C to -20 °C. -50 °C to -25 °C, or -40 °C to -30 °C.
- the homogeneous composition of empty LNPs is stored in a storage buffer or tonicity modifier.
- the storage buffer or tonicity modifier includes, but is not limited to. tris-sucrose saline, tris-trehalose saline, phosphate buffered saline, or tris buffered saline.
- the homogeneous composition of empty LNPs is packed in a vessel or bag for storage. In some embodiments, the homogeneous composition of empty LNPs is packed in sterile bags, vials or syringes and refrigerated or frozen. In some embodiments, the homogeneous composition of empty LNPs is lyophilized in a storage vessel, such as a vial or a syringe.
- the present disclosure provides a loaded LNP composition prepared by a subject method.
- a homogeneous LNP composition of loaded LNPs comprising an ionizable lipid, a helper hpid, a cholesterol, a PEG lipid, and a nucleic acid, wherein the loaded LNPs have an average particle size of 70-120 nm with a polydispersity of 0.3 or less, such as 0.2 or less, or 0.1 or less.
- the term “homogeneous” as it applies to a composition of loaded LNPs refers to the population of empty LNPs in the composition having an average particle size of 70-120 mn with a polydispersity of 0.3 or less, such as 0.2 or less, or 0.1 or less.
- the loaded LNPs comprise a molar ratio of from 40% to 60% ionizable lipid, a molar ratio of from 3.5% to 14% helper lipid, a molar ratio of from 28% to 50% structural lipid, and a molar ratio of from 0.5% to 5% PEG-lipid.
- the total molar percentage of the ionizable lipid, the helper lipid, the structural lipid, and the PEG-lipid is 100% in tire loaded LNPs.
- the molar ratio of the ionizable lipid in the loaded LNP is from 40% to 60% of the total lipid present in the loaded LNPs. In some embodiments, the molar ratio of the ionizable lipid in the loaded LNPs is 40%, 41%, 42%. 43%. 44%, 45%, 46%, 47%, 48%, 49%. 50%. 51%, 52%, 53%, 54%, 55%. 56%. 57%, 58%, 59%, or 60% of the total lipid present in the loaded LNPs.
- the molar ratio of the helper lipid in the loaded LNPs is from 3.5% to 14% of the total lipid present in the loaded LNPs. In some embodiments, the molar ratio of the helper lipid in the loaded LNP is 3%, 4%, 5%, 6%, 7%. 8%. 9%, 10%. 11%, 12%, 13%, or 14% of the total lipid present in the loaded LNPs.
- the helper lipid is DSPC. In some embodiments, the helper lipid is DOPE.
- the molar ratio of the structural lipid in the loaded LNPs is from 28% to 50% of the total lipid present in the loaded LNPs. In some embodiments, the molar ratio of the structural lipid in the loaded LNPs is 28%, 29%. 30%. 31%, 32%, 33%, 34%, 35%. 36%, 37%, 38%, 39%, 40%. 41%, 42%, 43%, 44%. 45%. 46%, 47%, 48%, 49%. or 50% of the total lipid present in the loaded LNPs. In some embodiments, the structural lipid is cholesterol.
- the molar ratio of the PEG-lipid in the loaded LNPs is from 0.1% to 5% of the total lipid present in the loaded LNPs. In some embodiments, the molar ratio of the PEG-lipid in the loaded LNPs is 0.1%, 0.2%, 0.3%, 0.4%. 0.5%. 0.6%, 0.7%, 0.8%, 0.9%, 1.0%, 1.1%, 1.2%. 1.3%. 1.4%. 1.5%. 1.6%. 1.7%. 1.8%, 1.9%, 2.0%, 2.1%, 2.2%, 2.3%, 2.4%, 2.5%, 2.6%, 2.7%, 2.8%. 2.9%. 3.0%. 3.1%. 3.2%. 3.3%, 3.4%, 3.5%, 4.0%, 4.5%, or 5% of the total lipid present in the loaded LNPs. In some embodiments, the PEG-lipid is DMG-PEG 2000 .
- the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the loaded LNPs is 45:9:44:2. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the loaded LNPs is 50:10:38.5: 1.5. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the loaded LNPs is 41:12:45:2.
- the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the loaded LNPs is 62:4:33:1. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the loaded LNPs is 53:5:41 : 1. In some embodiments, the molar ratio of each of the ionizable lipid, helper lipid, structural lipid, and PEG-lipid is within 10%, 9%.
- the loaded LNPs comprise an ionizable lipid as described herein, l,2-distearoyl-sn-glycero-3phosphocholine (DSPC), cholesterol, and DMG- PEG 2000 .
- DSPC l,2-distearoyl-sn-glycero-3phosphocholine
- the loaded LNPs comprise 30-60 mol % of ionizable lipid (e.g., as described herein); 5-30 mol % of DSPC; 15-50 mol % of cholesterol; and 1-2 mol % of DMG-PEG 200U .
- the loaded LNPs have an average particle size (i.e., an average nanoparticle diameter) from 50 mn to 300 mn such as, but not limited to, 50 mn to 60 nm, 50 nm to 70 nm, 50 mn to 80 mn, 50 mn to 90 mn, 50 mn to 100 run, 50 nm to 120 nm, 50 mn to 150 nm, 50 mn to 200 nm, 70 nm to 80 mn, 70 nm to 90 nm, 70 nm to 100 nm, 70 nm to 110 nm, 70 nm to 120 nm, 70 nm to 130 nm, 70 mn to 140 nm, 70 nm to 150 nm, 80 nm to 90 nm, 80 nm to 100 nm, 80 nm to 110 nm, 80 nm to 120 nm, 80 nm to 100 nm, 80 nm to
- the loaded LNPs may have an average particle size from 70 nm to 120 nm. In one embodiment, the loaded LNPs may have an average particle size of less than 300 nm, less than 250 nm, less than 200 mn, less than 180 nm, less than 150 nm, less than 120 nm, less than 100 mn, less than 90 mn, less than 80 nm, or less than 75 nm. Each possibility represents a separate embodiment of the present disclosure.
- the loaded LNPs have an average particle size of 50-300 nm, 60-300 nm, 70-300 nm, or 80-300 mn. In some embodiments, the loaded LNPs have an average particle size of 50-200 nm, 50-180 mn, 50-150 nm, or 50-120 nm.
- the loaded LNPs have an average particle size of from 1 nm to 100 mn, from 1 nm to 10 mn, 1 nm to 20 nm, from 1 nm to 30 nm, from 1 nm to 40 nm, from 1 mn to 50 nm, from 1 mn to 60 nm, from 1 mn to 70 nm, from 1 mn to 80 nm, from 1 mn to 90 nm, from 5 nm to 100 nm, from 5 mn to 10 nm, 5 nm to 20 mn, from 5 nm to 30 nm, from 5 nm to 40 nm, from 5 nm to 50 mn, from 5 mn to 60 nm, from 5 nm to 70 nm, from 5 nm to 80 nm, from 5 nm to 90 mn, 10 mn to 50 nm, from 1 mn to 50 nm, from 1 m
- the loaded LNPs have an average particle size of 70 nm, 71 nm, 72, nm, 73 mn, 74 nm, 75 nm, 76 mn, 77 nm, 78 nm, 79 nm, 80 nm, 81 nm, 82 nm, 83 mn, 84 mn, 85 nm, 86 mn, 87 nm, 88 nm, 89 nm, 90 nm, 91 mn, 92 nm, 93 nm, 94 nm, 95 nm, 96 nm, 97 mn, 98 nm, 99 mn, 100 nm, 101 mn, 102 nm, 103 nm, 104 nm, 105 nm, 106 mn, 107 nm, 108 nm, 109 n, 100 nm, 101 mn, 102
- the average particle size of the loaded LNPs is within 10%, 9%. 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- a loaded LNP composition may have a poly dispersity index from about 0.01 to about 0.3, such as 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.10, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, or 0.30.
- the polydispersity index of a loaded LNP composition may be from 0.01 to 0.20.
- the poly dispersity index of the loaded LNPs is within 10%, 9%, 8%, 7%, 6%, 5%, 4%.
- the polydispersity index of the loaded LNP composition is 0.3 or less, such as 0.25 or less, 0.20 or less, 0.15 or less, or 0.1 or less.
- the LNPs in the loaded LNP composition have an average particle size of 50 nm to 300 nm and a polydispersity of 0.3 or less. In some embodiments, the LNPs in the loaded LNP composition have an average particle size of 70 mn to 120 nm and a polydispersity of 0.2 or less.
- the zeta potential of the subject loaded LNPs may be from -20 Mv to +20 Mv, from -20 Mv to +15 Mv, from -20 Mv to +10 Mv, from -20 Mv to +5 Mv, from -20 Mv to 0 Mv, from -20 Mv to -5 Mv.
- the zeta potential of the subject loaded LNPs is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- the amounts of nucleic acid and empty LNPs are selected to provide a specific N:P ratio.
- the molar ratio of nitrogen atoms in one or more ionizable lipids (i.e., in the empty LNPs) to phosphate groups in the nucleic acid (N:P) is within the range of 1:20 to 20: 1.
- the N:P ratio is 1:20, 1: 19. 1:18, 1 :17, 1 :16, 1: 15, 1: 14, 1:13, 1 : 12, 1: 11, 1: 10, 1 :9, 1:8. 1:7, 1:6, 1 :5, 1 :4, 1:3, 1:2 or 1: 1.
- the N:P ratio is 20:1. 19: 1, 18: 1, 17: 1. 16:1, 15: 1, 14: 1, 13: 1.
- the N:P ratio is within the range of 4:1 to 6: 1.
- the N:P ratio is 4.0: 1, 4.1: 1, 4.2: 1, 4.3: 1. 4.4: 1. 4.5: 1. 4.6: 1. 4.7:1. 4.8: 1. 4.9: 1. 5.0: 1. 5.1: 1. 5.2: 1. 5.3: 1, 5.4: 1. 5.5: 1. 5.6: 1. 5.7: 1. 5.8: 1. 5.9: 1. or 6.0: 1.
- the N:P ratio is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
- the loaded LNP is associated with a nucleic acid.
- the nucleic acid is an RNA polynucleotide.
- the RNA is a circular RNA polynucleotide (aka circRNA or OmaTM) (as described herein).
- the nucleic acid is encapsulated in the loaded LNP.
- the nucleic acid is encapsulated in the loaded LNP with an encapsulation efficiency of from 50% to 100%, such as 60% to 100%, 70% to 100%, 80% to 100%. or 90% to 100%.
- the efficiency of encapsulation of a therapeutic agent describes the amount of therapeutic agent that is encapsulated or otherwise associated with a nanoparticle composition after preparation, relative to the initial amount provided.
- the encapsulation efficiency is desirably high (e.g.. close to 100%).
- the encapsulation efficiency may be measured, for example, by comparing the amount of therapeutic agent in a solution containing the nanoparticle composition before and after breaking up the nanoparticle composition with one or more organic solvents or detergents. Fluorescence may be used to measure the amount of free therapeutic agent (e.g., nucleic acids) in a solution.
- the encapsulation efficiency of a therapeutic agent may be at least 50%. for example 50%. 55%. 60%, 65%, 70%, 75%, 80%, 85%, 90%. 91%, 92%, 93%, 94%, 95%, 96%, 97%. 98%, 99%, or 100%.
- the encapsulation efficiency may be at least 80%.
- the encapsulation efficiency may be at least 90%.
- the properties of a loaded LNP may be influenced by factors including, but not limited to, the selection of the cationic lipid component, the degree of cationic lipid saturation, the selection of the helper lipid component, the degree of helper lipid saturation, the selection of the cholesterol lipid component, the selection of the PEG-lipid component, the nature of the PEGylation, ratio of all components, and biophysical parameters such as size.
- the loaded LNP composition is storage stable at a temperature from 4 °C to -80 °C. In some embodiments, the loaded LNP composition is stored at a temperature from -80 °C to 0 °C, such as -80 °C to -10 °C, -60 °C to -20 °C, -50 °C to -25 °C, or -40 °C to -30 °C. In some embodiments, the loaded LNP composition is stored in a storage buffer or tonicity’ modifier.
- the storage buffer or tonicity modifier includes, but is not limited to, tris-sucrosc saline, tris-trehalose saline, phosphate buffered saline, or tris buffered saline.
- the loaded LNP composition is packed in a vessel or a bag for storage. In some embodiments, the loaded LNP composition is packed in a sterile bag, vial or syringe and refrigerated or frozen. In some embodiments, the loaded LNP composition is lyophilized in a storage vessel, such as a vial or a syringe.
- the loaded LNP composition can be stored at 4 °C, and is storage stable for at least 4 weeks, such as at least 8 weeks, at least 12 weeks, at least 16 weeks, at least 20 weeks, at least 6 months, at least 9 months, or at least a year.
- the present disclosure provides lipid solutions and lipid nanoparticles comprising one or more ionizable lipids.
- the subject lipid solutions and lipid nanoparticles can also include one or more helper lipids, structural lipids (e.g., cholesterol), and PEG-lipids (as described herein below).
- the molar ratio of the ionizable lipid, the helper lipid, the structural lipid (e.g., cholesterol) and PEG-lipid in the lipid solution and lipid nanoparticles is as described herein above.
- the lipid solutions and lipid nanoparticles disclosed herein comprise ionizable lipids.
- the subject ionizable lipids may be used as a component of a composition to facilitate encapsulation and release of nucleic acid cargo (e.g., circular RNA) to one or more target cells.
- an ionizable lipid comprises one or more cleavable functional groups (e.g., a disulfide) tiiat allow, for example, a hydrophilic functional head-group to dissociate from a lipophilic functional tail-group of the compound (e.g., upon exposure to oxidative, reducing, or acidic conditions).
- the ionizable lipid has a pKa from 6 to 12. In some embodiments, the ionizable lipid has a pKa from 7 to 9. In some embodiments, the ionizable lipid has a pKa of 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, 7.0, 7.1, 7.2, 7.3, 7.4, 7.5, 7.6, 7.7, 7.8, 7.9, 8.0, 8.1, 8.2, 8.3, 8.4, 8.5, 8.6, 8.7, 8.8, 8.9, or 9.0 or any ranges created by these.
- the ionizable lipid comprises an amino group.
- the ionizable lipid comprises a divalent headgroup and one or more straight hydrocarbon lipid tails.
- the straight hydrocarbon lipid tails are from 3- 25 carbon atoms in length, such as 5 to 25. 5 to 20. 5 to 15, 5 to 10, 10 to 15. 10 to 20, or 10 to 25 carbon atoms in length.
- the ionizable lipid comprises a divalent headgroup and one or more branched hydrocarbon lipid tails.
- the branched hydrocarbon lipid tails are from 3-25 carbon atoms in length, such as 5 to 25, 5 to 20, 5 to 15, 5 to 10, 10 to 15. 10 to 20, or 10 to 25 carbon atoms in length.
- the divalent headgroup is selected from guanidine and squaramide.
- the squaramide headgroup is of the following formula: wherein R A and R B are each independently a C 1 -C 6 alkyl group or H; and represents the point of attachment of the headgroup to a straight or branched hydrocarbon lipid tail.
- the ionizable lipid comprises a headgroup selected from: wherein represents the point of attachment of the headgroup to a straight or branched hydrocarbon lipid tail.
- the ionizable lipid comprises a head group selected from: wherein ⁇ vwvw represents the point of attachment of the headgroup to a straight or branched hydrocarbon lipid tail.
- the ionizable lipid comprises a hydrophilic headgroup as disclosed in Jayaraman et al. Angew. Chem. hit. Ed. (2012), 51, 8529-8533.
- the ionizable lipid is ethyl lauryl arginate (EL A).
- die ionizable lipid is ionizable lipid 1, wherein ionizable lipid 1 comprises:
- the one or more of the cationic or ionizable lipids are represented by Fonnula (LI):
- n is an integer between 1 and 4;
- R is hydrogen or hydroxyl
- R 1 and R 2 are each independently a linear or branched Ce-C 30 alkyl, C6-C 30 alkenyl, or Ce-C 30 heteroalkyl, optionally substituted by one or more substituents selected from oxo, halo, hydroxy, cyano, alkyl, alkenyl, aldehyde, heterocyclylalkyl, hydroxyalkyl, dihydroxyalkyl, hydroxyalkylaminoalkyl. aminoalkyl.
- alkylaminoalkyl dialkylaminoalkyl, (heterocyclyl)(alkyl)aminoalkyl, heterocyclyl, heteroaryl, alkylheteroaryl, alkynyl, alkoxy, amino, dialkylamino. aminoalkylcarbonylamino. aminocarbonylalkylamino, (aminocarbonylalky l)(alkyl)amino. alkenylcarbonylamino.
- alkylaminoalkyl (alkyl)aminocarbonyl, alkylaminoalkylcarbonyl, dialkylaminoalkylcarbonyl, heterocyclylcarbonyl. alkenylcarbonyl, alkynylcarbonyl. alkylsulfoxide, alkylsulfoxidealkyl, alkylsulfonyl, and alkylsulfonealkyl.
- R a is hydrogen. In some embodiments, R a is hydroxyl.
- the ionizable lipid is represented by Fonnula (Lla -1), Fonnula (LIa-2), or Fonnula (LIa-3):
- the ionizable lipid is represented by Formula (LIb-1), Fonnula (LIb-2), or Fonnula (LIb-3):
- the ionizable lipid is represented by Fonnula (LIb-4), Formula (LIb-5), Fonnula (LIb-6), Formula (Lib- 7), Formula (LIb-8). or Formula (LIb-9):
- the one or more of the cationic or ionizable lipids arc represented byFormula (LI), wherein R 1 and R 2 are each independently selected from:
- R 1 and R 2 are the same. In some embodiments, R 1 and R 2 are different.
- the one or more of the cationic or ionizable lipids are represented by Fonnula (LI*):
- n* is an integer between 1 to 7
- R a is hydrogen or hydroxyl.
- alkenylcarbonyloxy alkenylcarbonate, alkynyloxycarbonyl, alkynylcarbonyloxy, alkynylcarbonate, aminocarbonyl, aminoalkylaminocarbonyl, alkylaminoalkylaminocarbonyl, dialkylaminoalkylaminocarbonyl, heterocyclylalkylaminocarbonyl,
- alkylaminoalkyl (alkyl)aminocarbonyl, alkylaminoalkylcarbonyl, dialkylaminoalkylcarbonyl, heterocyclylcarbonyl, alkenylcarbonyl, alkynylcarbonyl, alkylsulfoxide, alkylsulfoxidealkyl, alkylsulfonyl, and alkylsulfonealkyl.
- the one or more of the cationic or ionizable lipids are represented by Formula (LII):
- each 11 is independently an integer from 2 to 15;
- L 1 and L 3 are each independently -OC(O)-* or -C(O)O-*, wherein indicates the attachment point to R 1 or R 3 ;
- R 1 and R 3 are each independently a linear or branched C 9 -C 20 alkyl or C 9 -C 20 alkenyl, optionally substituted by one or more substituents selected from oxo, halo, hydroxy', cyano, alkyl, alkenyl, aldehyde, heterocyclylalkyl, hydroxyalkyl, dihydroxyalkyl, hydroxyalkylaminoalkyl, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl.
- heterocyclyl (alkyl)aminoalkyl, heterocyclyl, heteroaryl, alkylheteroaryl, alkynyl, alkoxy, amino, dialkylamino, aminoalkylcarbonylamino, aminocarbonylalky' lamino, (aminocarbonylalky 1) (alkyl) amino, alkenylcarbonylamino, hydroxy carbonyl, alkyloxycarbonyl, aminocarbonyl, aminoalkylaminocarbonyl, alkylaminoalkylaminocarbonyl, dialkylaminoalkylaminocarbonyl, heterocyclylalkylaminocarbonyl,
- alkylaminoalkyl (alkyl)aminocarbonyl, alkylaminoalkylcarbonyl, dialkylaminoalkylcarbonyl, heterocyclylcarbonyl, alkenylcarbonyl, alkynylcarbonyl, alkylsulfoxide, alkylsulfoxidealkyl, alkylsulfonyl, and alkylsulfonealkyl; and
- R 2 is selected from:
- the ionizable lipid is selected from an ionizable lipid of Formula LII, wherein R 1 and R 3 are each independently selected from:
- R 1 and R 3 are the same. In some embodiments, R 1 and R 3 are different.
- the one or more of the cationic or ionizable lipids 1 re represented by Formula (LII-1) or Formula (LII-2):
- the ionizable lipid is selected from an ionizable lipid of WO2015/095340. In some embodiments, the ionizable lipid is selected from an ionizable lipid of WO2021/021634, WO2020/237227, or WO2019/236673. In some embodiments, the ionizable lipid is selected from an ionizable lipid of WO2021226597 and WO2021113777. In some embodiments, the ionizable lipid is selected from an ionizable lipid of W02023056033. In some embodiments, the ionizable lipid is selected from an ionizable lipid of WO2023081526.
- L 1 is C 2 -C 11 alkylene.
- R 2 and R 3 are each independently C 6 -C 30 -alky 1, C 6 -C 30 -alkenyl, or C 6 -C 30 -alkynyl.
- X 1 is OR 1 , SR 1 , or N(R') 2 , where R 1 is independently H or unsubstituted C 1 -C 6 alkyl;
- R 2 and R 3 are each independently a linear or branched C 1 -C 30 alkyl, C 2 -C 30 alkenyl, or C 1 -C 30 heteroalkyl, optionally substituted by one or more substituents selected from oxo, halo, hydroxy, cyano, alkyl, alkenyl, aldehyde, heterocyclylalkyl, hydroxyalkyl, dihydroxyalkyl, hydroxyalkylaminoalkyl, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl. (heterocyclyl)(alkyl)aminoalkyl, heterocyclyl. heteroaryl, alkvlhctcroarvl.
- alkynylcarbonyloxy alkynylcarbonate, aminocarbonyl, aminoalkylaminocarbonyl, alkylaminoalkylaminocarbonyl, dialkylaminoalkylaminocarbonyl. heterocyclylalkylaminocarbonyl,
- alkylaminoalkyl (alkyl)aminocarbonyl.
- alkylaminoalkylcarbonyl dialkylaminoalkylcarbonyl, heterocyclylcarbonyl, alkenylcarbonyl, alkyny Icarbonyl, alkylsulfoxide, alkylsulfoxidealkyl, alkydsulfonyl, and alkylsulfonealkyl.
- n’ is an integer from 1 to 7;
- R a is hydrogen or hydroxyl
- R 11 is hydrogen or C 1 -Ce alkyl
- R 1 is C 1 -C 30 alkyl or R ] ’ ;
- R 2 is C 1 -C 30 alkyl or R 2 *;
- R 1 * and R 2 * are independently selected from:
- R 8 is H or R n ;
- R 9 , R 10 , and R 11 are each independently C 1 -C 2 o alky l or C 2 -C 2 o-alkenyl; and wherein (i) R 1 is R 1 *, (ii) R 2 is R 2 ’, or (iii) R 1 is R 1 ’ and R 2 is R 2 ’.
- an ionizable lipid of the present disclosure is represented by Formula
- Formula (LV) or is a pharmaceutically acceptable salt thereof, wherein:
- R a is hydrogen or hydroxyl
- R 1 is C 1 -C 30 alkyl or R’’ ;
- R 2 is C 1 -C 30 alkyl or R 2 *;
- R 1 * and R 2 * are independently selected from:
- R 4 is hydrogen or R 7 ;
- R ⁇ R 6 . and R 7 are each independently C 1 -C 20 alkyl or C 2 -C 2 o-alkenyl; wherein (i) R 1 is R 1 *, (ii) R 2 is R 2 ’, or (iii) R 1 is R 1 ’ and R 2 is R 2 *; and
- R 3 is L-R’, wherein L is linear or branched C 1 -C 10 alkylene, and R’ is (i) mono- or bicyclic heterocyclyl or heteroaryl, such as imida1olyl, pyrazolyl, 1.2,4-triazolyl. or benzimidazolyl, each optionally substituted at one or more available carbon and/or nitrogen atoms by C 1 -C 6 alky l, or (ii) R A , R B , or R c , wherein: R A is selected from:
- the ionizable lipid is selected from a lipid of Table 1:
- the lipid solutions and lipid nanoparticles described herein comprise one or more non-cationic helper lipids.
- the helper lipid is a phospholipid.
- the helper lipid is a phospholipid substitute or replacement.
- the phospholipid or phospholipid substitute can be, for example, one or more saturated or (poly)unsaturatcd phospholipids, or phospholipid substitutes, or a combination thereof.
- phospholipids comprise a phospholipid moiety and one or more fatty acid moieties.
- a phospholipid moiety can be selected, for example, from the non-limiting group of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and sphingomyelin.
- a fatty acid moiety can be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid.
- Phospholipids include, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin.
- the helper lipid is a l,2-distearoyl-177-glycero-3-phosphocholine (DSPC) analog, a DSPC substitute, oleic acid, or an oleic acid analog.
- DSPC l,2-distearoyl-177-glycero-3-phosphocholine
- a helper lipid is a non-phosphatidyl choline (PC) zwitterionic lipid, a DSPC analog, oleic acid, an oleic acid analog, or a DSPC substitute.
- PC non-phosphatidyl choline
- helper lipid is described in PCT/US2018/053569.
- Helper lipids suitable for use in a lipid composition of the disclosure include, for example, a variety of neutral, uncharged or zwitterionic lipids. Such helper lipids are preferably used in combination with one or more of the compounds and lipids disclosed herein.
- helper lipids include, but are not limited to, 5-heptadecylbenzene-l,3-diol (resorcinol), dipalmitoylphosphatidylcholine (DPPC), distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dimyristoylphosphatidylcholine (DMPC), phosphatidylcholine (PLPC). l,2-distearoylsn-glycero-3- phosphocholine (DAPC).
- DPPC dipalmitoylphosphatidylcholine
- DSPC distearoylphosphatidylcholine
- DOPC dioleoylphosphatidylcholine
- DMPC dimyristoylphosphatidylcholine
- PLPC phosphatidylcholine
- DAPC l,2-distearoylsn-glycero-3- phosphocho
- PE phosphatidylethanolamine
- EPC egg phosphatidylcholine
- DLPC dilauryloylphosphatidylcholine
- DMPC dimyristoylphosphatidylcholine
- PMPC 1-paimitoy 1-2 -myristoyl phosphatidylcholine
- PSPC 1- pahnitoy 1-2-stearoy 1 phosphatidylcholine
- DBPC 1-stearoy 1-2 -palmitoyl phosphatidylcholine
- SPPC l,2-dieicosenoyl-sn-glycero-3- phosphocholinc
- POPC paimitoyioicoyl phosphatidylcholine
- POPC paimitoyioicoyl phosphatidylcholine
- tire helper lipid may be distearoylphosphatidylcholine (DSPC) or dimyristoyl phosphatidyl ethanolamine (DMPE).
- the helper lipid may be distearoylphosphatidylcholine (DSPC).
- Helper lipids function to stabilize and improve processing of the lipid solutions and lipid nanoparticles.
- Such helper lipids are preferably used in combination with other excipients, for example, one or more of the ionizable lipids disclosed herein.
- the lipid solutions and lipid nanoparticles described herein comprise one or more structural lipids. Incorporation of structural lipid(s) in the lipid nanoparticle may help mitigate aggregation of other lipids in the particle.
- Structural lipids can include, but are not limited to, cholesterol, fecosterol, ergosterol, bassicasterol, tomatidine, tomatine, ursolic, alpha-tocopherol, and mixtures thereof.
- the structural lipid is cholesterol.
- die structural lipid includes cholesterol and a corticosteroid (such as, for example, prednisolone, dexamethasone, prednisone, and hydrocortisone), or a combination thereof.
- a structural lipid is described in international patent application PCT/US2019/015913, which is incorporated by reference herein in its entirety.
- the structural lipid is a sterol. In certain embodiments, the structural lipid is a steroid. In certain embodiments, the structural lipid is cholesterol. In certain embodiments, the structural lipid is an analog of cholesterol. In certain embodiments, the structural lipid is alphatocopherol.
- the lipid solutions and lipid nanoparticles described herein comprise one or more structural lipids. Incorporation of structural lipids in a transfer vehicle, e.g., a lipid nanoparticle, may help mitigate aggregation of other lipids in the particle.
- the structural lipid includes cholesterol, a corticosteroid (such as, for example, prednisolone, dexamethasone, prednisone, and hydrocortisone), or a combination thereof.
- the structural lipid is a sterol.
- Structural lipids can include, but are not limited to, sterols (e.g.. phytosterols or zoosterols).
- the structural lipid is a steroid.
- sterols can include, but are not limited to, cholesterol, p-sitosterol, fecosterol. ergosterol, sitosterol, campesterol, stigmasterol, brassicasterol. ergosterol, tomatidine. tomatine, ursolic acid, or alpha-tocopherol.
- the lipid solutions and lipid nanoparticles include an effective amount of an immune cell delivery potentiating lipid, e.g., a cholesterol analog or an amino lipid or combination thereof, that, when present in a transfer vehicle, e.g., an lipid nanoparticle, may function by enhancing cellular association and/or uptake, internalization, intracellular trafficking and/or processing, and/or endosomal escape and/or may enhance recognition by and/or binding to immune cells, relative to a transfer vehicle lacking the immune cell delivery potentiating lipid.
- an immune cell delivery potentiating lipid e.g., a cholesterol analog or an amino lipid or combination thereof
- a structural lipid or other immune cell delivery potentiating lipid of the disclosure binds to Clq or promotes the binding of a transfer vehicle comprising such lipid to Clq.
- culture conditions that include Clq are used (e.g.. use of culture media that includes serum or addition of exogenous Clq to serum- free media).
- the requirement for Cl q is supplied by endogenous Clq.
- the structural lipid is cholesterol. In some embodiments, the structural lipid is an analog of cholesterol. In some embodiments, the structural lipid is a lipid in Table 2 below: Table 2: Example Structural lipids
- PEG polyethylene glycol
- PEG-CER derivatized ceramides
- N-Octanoyl-Sphingosine-1- [Succinyl(Methoxy Polyethylene Glycol)-2000] (C 6 PEG-2000 ceramide) in the lipid solutions and lipid nanoparticlcs described herein is contemplated, preferably in combination with one or more of the compounds and lipids disclosed herein.
- Contemplated PEG-modified lipids include, but are not limited to, a polyethylene glycol chain of up to 5 kDa in length covalently attached to a lipid with alkyl chain(s) of C6-C 2 0 length.
- the PEG-modified lipid employed in the compositions and methods of the disclosure is 1,2-dimyristoyl-sn-glycerol, methoxypolyethylene Glycol (2000 MW PEG) “DMG-PEG2000.”
- the addition of PEG-modified lipids to the lipid delivery vehicle may prevent complex aggregation and may also provide a means for increasing circulation lifetime and increasing the delivery of the lipid-polynucleotide composition to the target tissues, (Klibanov et al.
- PEG-ceramides having shorter acyl chains e.g., C14 or Cl 8
- the PEG-modified phospholipid and derivatized lipids of the present disclosure may comprise a molar ratio from about 0% to about 20%, about 0.5% to about 20%, about 1% to about 15%, about 4% to about 10%, or about 2% of the total lipid present in the lipid solution or lipid nanoparticle.
- a PEG-modified lipid is described in International Pat. Appl. No. PCT/US2019/015913 or PCT/US2020/046407, which arc incorporated herein by reference in their entirety .
- a transfer vehicle comprises one or more PEG-modified lipids.
- Non-limiting examples of PEG-modified lipids include PEG-modified phosphatidylethanolamines and phosphatidic acids, PEG-ceramide conjugates (e.g, PEG-CerC14 or PEG-CerC 2 0), PEG-modified dialkylamines and PEG-modified l .2-diacyloxypropan-3-amines.
- a PEG-modified lipid may be, e.g., PEG-c-DOMG, PEG-DMG, PEG- DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE.
- the PEG-modified lipid includes, but is not limited to 1,2-dimyristoyl- sn-glycerol mcthoxypolyethylenc glycol (PEG-DMG), l,2-distearoyl-sn-glycero-3- phosphoethanolamine-N-[amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG- DSG), PEG-dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG-DAG), PEG- dipahnitoyl phosphatidy lethanolamine (PEG-DPPE), PEG-1, 2-dimyristyloxlpropy 1-3 -amine (PEG-c- DMA).
- PEG-DMG 1,2-dimyristoyl- sn-glycerol mcthoxypolyeth
- the PEG-modified lipid is DSPE-PEG, DMG-PEG, PEG-DAG, PEG- S-DAG, PEG-PE, PEG-S-DMG, PEG-cer, PEG-dialkoxypropylcarbamate, PEG-OR, PEG-OH, PEG- c-DOMG, or PEG-1.
- the PEG-modified lipid is DSPE-PEG(2000).
- the PEG-modified lipid comprises a PEG moiety comprising 10-70 (e.g., 30-60) oxycthylcne (-O-CH 2 -CH 2 -) units or portions thereof.
- the PEG- modified lipid comprises (OCH 2 CH2) V -OR W , and v is an integer between 0 and 70 (inclusive) (e.g.. an integer between 30 and 60), w is hydrogen or alky l.
- a PEG-modified lipid may also be referred to as “PEGylated lipid” or “PEG-lipid.”
- the PEG-lipid is selected from a PEG-modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG-modified ceramide, a PEG-modified dialkylamine, a PEG- modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof.
- the lipid moiety of the PEG-lipids includes those having lengths of from about Cu to about C 2 2. such as from about C14 to about C 1 s.
- a PEG moiety for example a mPEG-NH 2 , has a size of about 1000, about 2000, about 5000, about 10,000, about 15,000 or about 20,000 daltons.
- the PEG-lipid is PEG2k-DMG.
- the lipid nanoparticles described herein can comprise a lipid modified with a non-diffusible PEG.
- non-diffusible PEGs include PEG-DSG and PEG- DSPE.
- PEG-lipids are known in the art, such as those described in U.S. Pat. No. 8,158,601 and International Pat. Publ. No. WO2015/130584 A2, which are incorporated herein by reference in their entirety 7 .
- lipids e.g., PEG-lipids
- described herein may be synthesized as described International Pat. Publ. No. PCT/US2016/000129, which is incorporated by reference in its entirety.
- the lipid component of a lipid solution or lipid nanoparticle composition may include one or more molecules comprising polyethylene glycol, such as PEG or PEG-modified lipids. Such species may be alternately referred to as PEGylated lipids.
- a PEG lipid is a lipid modified with polyethylene glycol.
- a PEG lipid may be selected from the non-limiting group including PEG-modified phosphatidylethanolamines. PEG-modified phosphatidic acids, PEG-modified ceramides. PEG- modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkyl glycerols, and mixtures thereof.
- a PEG lipid may be PEG-c-DOMG.
- the PEG-modified lipids are a modified form of PEG-DMG.
- the PEG-modified lipids are a modified form of PEG-C18, or PEG-1.
- PEG-1 has the following structure:
- PEG lipids useful in the present disclosure can be PEGylated lipids described in International Publication No. WO2012099755, the contents of which is herein incorporated by reference in its entirety . Any of these exemplary PEG lipids described herein may be modified to comprise a hydroxyl group on the PEG chain.
- the PEG lipid is a PEG-OH lipid.
- the PEG-OH lipid includes one or more hydroxyl groups on the PEG chain.
- a PEG-OH or hydroxy -PEGylated lipid comprises an -OH group at the terminus of the PEG chain.
- the PEG lipid is a compound of Formula (Pl): or a salt or isomer thereof, wherein: r is an integer between 1 and 100;
- R is C 10 -40 alkyl, C 10 -ro alkenyl, or C 10 -4o alkynyl; and optionally one or more methylene groups of R are independently replaced with C3-10 carbocyclylene, 4 to 10 membered heterocyclylene, Ce io arylene.
- -NR N C( NR N )N(R N )- ,-C(S)-, -C(S)N(R N )-, -NR N C(S)-.
- R is C17 alkyl.
- PEG lipid is a compound of Formula (Pl-a):
- the PEG lipid is a compound of the following formula: 5.
- the present disclosure provides loaded lipid nanoparticles comprising a polynucleotide.
- the polynucleotide is DNA.
- the polynucleotide is RNA.
- the polynucleotide is linear RNA.
- the polynucleotide is circular RNA.
- a DNA template e.g., comprising a 3’ intron element, 3’ exon element, a core functional element, a 5’ exon element, and a 5’ intron element
- this DNA template comprises a vector, a PCR product, a plasmid, a minicircle DNA, a cosmid, an artificial chromosome, a complementary DNA (cDNA), an extrachromosomal DNA (ecDNA). or a fragment therein.
- the minicircle DNA may be linearized or non-linearized.
- the plasmid may be linearized or non-linearized.
- the DNA template may be singlestranded. In other embodiments, the DNA template may be double-stranded. In some embodiments, the DNA template comprises in whole or in part from a viral, bacterial, or eukaryotic vector.
- the present disclosure comprises a DNA template that shares the same sequence as the precursor linear RNA polynucleotide prior to splicing of the precursor linear RNA polynucleotide (e.g., a 3’ intron element, a 3’ exon element, a core functional element, and a 5‘ exon element, a 5’ intron element).
- said linear precursor RNA polynucleotide undergoes splicing leading to the removal of the 3’ intron element and 5‘ intron element during the process of circularization.
- the resulting circular RNA polynucleotide lacks a 3 ’ intron fragment and a 5’ intron fragment, but maintains a 3’ exon fragment, a core functional element, and a 5‘ exon element.
- the precursor linear RNA polynucleotide circularizes when incubated in the presence of one or more guanosine nucleotides or nucleoside (e.g., GTP) and a divalent cation (e.g., Mg 2+ ).
- the 3’ exon element, 5’ exon element, and/or core functional element in whole or in part promotes the circularization of the precursor linear RNA polynucleotide to form the circular RNA polynucleotide provided herein.
- the circular RNA provided herein is produced inside a cell.
- the precursor RNA is transcribed using a DNA template (e.g., in some embodiments, using a vector provided herein) in the cytoplasm by a bacteriophage RNA polymerase, or in the nucleus by host RNA polymerase II and then circularized.
- the circular RNA provided herein is injected into an animal (e.g., a human), such that a polypeptide encoded by the circular RNA molecule is expressed inside the animal.
- the DNA template (e.g., vector), linear RNA (e.g., precursor RNA), and/or circular RNA polynucleotide provided herein is between 300 and 10000, 400 and 9000, 500 and 8000, 600 and 7000, 700 and 6000, 800 and 5000, 900 and 5000, 1000 and 5000, 1100 and 5000. 1200 and 5000, 1300 and 5000, 1400 and 5000, and/or 1500 and 5000 nucleotides (nt) in length.
- the polynucleotide is at least 300 nt, 400 nt. 500 nt, 600 nt, 700 nt, 800 nt, 900 nt. 1000 nt, 1100 nt, 1200 nt, 1300 nt. 1400 nt, 1500 nt, 2000 nt, 2500 nt, 3000 nt, 3500 nt. 4000 nt, 4500 nt, or 5000 nt in length. In some embodiments, the polynucleotide is no more than 3000 nt, 3500 nt, 4000 nt, 4500 nt.
- the length of a DNA, linear RNA, and/or circular RNA polynucleotide provided herein is 300 nt. 400 nt, 500 nt, 600 nt, 700 nt. 800 nt. 900 nt, 1000 nt. 1100 nt, 1200 nt, 1300 nt. 1400 nt, 1500 nt, 2000 nt.
- the circular RNA provided herein has higher functional stability than mRNA comprising the same expression sequence. In some embodiments, the circular RNA provided herein has higher functional stability than mRNA comprising the same expression sequence, 5moU modifications, an optimized UTR, a cap, and/or a polyA tail.
- the circular RNA polynucleotide provided herein has a functional halflife of at least 5 hours, 10 hours, 15 hours, 20 hours. 30 hours. 40 hours, 50 hours, 60 hours, 70 hours or 80 hours. In some embodiments, the circular RNA polynucleotide provided herein has a functional half-life of 5-80. 10-70, 15-60, or 20-50 hours. In some embodiments, the circular RNA polynucleotide provided herein has a functional half-life greater than (e.g.. at least 1.5-fold greater than, at least 2-fold greater than) that of an equivalent linear RNA polynucleotide encoding the same protein. In some embodiments, functional half-life can be assessed through the detection of functional protein synthesis.
- the circular RNA polynucleotide, or pharmaceutical composition thereof has a functional half-life in a human cell greater than or equal to that of a pre-determined threshold value.
- the functional half-life is determined by a functional protein assay.
- the functional half-life is determined by an in vitro luciferase assay, wherein the activity of Gaussia luciferase (GLuc) is measured in the media of human cells (e.g. HepG2) expressing the circular RNA polynucleotide every 1, 2, 6, 12, or 24 hours over 1. 2. 3, 4, 5, 6. 7. or 14 days.
- the functional half-life is determined by an in vivo assay, wherein levels of a protein encoded by the expression sequence of the circular RNA polynucleotide are measured in patient serum or tissue samples every 1, 2, 6, 12, or 24 hours over 1. 2. 3, 4, 5, 6. 7. or 14 days.
- the pre-determined threshold value is the functional half-life of a reference linear RNA polynucleotide comprising the same expression sequence as the circular RNA polynucleotide.
- the circular RNA provided herein may have a higher magnitude of expression than equivalent linear mRNA, e.g., a higher magnitude of expression 24 hours after administration of RNA to cells.
- the circular RNA provided herein has a higher magnitude of expression than mRNA comprising the same expression sequence, 5moU modifications, an optimized UTR, a cap, and/or a polyA tail.
- the circular RNA provided herein may be less immunogenic than an equivalent mRNA when exposed to an immune system of an organism or a certain type of immune cell.
- the circular RNA provided herein is associated with modulated production of cytokines when exposed to an immune system of an organism or a certain type of immune cell.
- the circular RNA provided herein is associated with reduced production of IFN-(31, RIG-I, IL-2, IL-6, IFNy, and/or TNFa when exposed to an immune system of an organism or a certain type of immune cell as compared to mRNA comprising the same expression sequence.
- the circular RNA provided herein is associated with less IFN-(31, RIG-I, IL-2. IL-6, IFNy, and/or TNFa transcript induction when exposed to an immune system of an organism or a certain type of immune cell as compared to mRNA comprising the same expression sequence.
- the circular RNA provided herein is less immunogenic than mRNA comprising the same expression sequence.
- the circular RNA provided herein is less immunogenic than mRNA comprising the same expression sequence, 5moU modifications, an optimized UTR. a cap. and/or a polyA tail.
- the circular RNA provided herein can be transfected into a cell as is or can be transfected in DNA vector form and transcribed in the cell. Transcription of circular RNA from a transfected DNA vector can be via added polymerases or polymerases encoded by nucleic acids transfected into the cell, or preferably via endogenous polymerases. i. Enhanced Intron Elements & Enhanced Exon Elements
- the DNA template e.g., vector
- linear RNA e.g., precursor RNA
- the DNA template comprises an enhanced intron element and/or an enhanced exon element.
- the enhanced intron elements and enhanced exon elements may comprise spacers, duplex regions, affinity sequences, intron fragments, exon fragments and various untranslated elements. These sequences within the enhanced intron elements or enhanced exon elements are arranged to optimize circularization or protein expression.
- the DNA template, precursor linear RNA polynucleotide and circular RNA provided herein comprise a first (5 ’) and/or a second (3 ) spacer.
- the DNA template or precursor linear RNA polynucleotide comprises one or more spacers in the enhanced intron elements.
- the DNA template, precursor linear RNA polynucleotide comprises one or more spacers in the enhanced exon elements.
- the DNA template or linear RNA polynucleotide comprises a spacer in the 3 ’ enhanced intron fragment and a spacer in the 5’ enhanced intron fragment.
- DNA template, precursor linear RNA polynucleotide, or circular RNA comprises a spacer in the 3’ enhanced exon fragment and another spacer in the 5' enhanced exon fragment to aid with circularization or protein expression due to symmetry created in the overall sequence.
- including a spacer between the 3 ‘ group I intron fragment and the core functional element may conserve secondary structures in those regions by preventing them from interacting, thus increasing splicing efficiency.
- the first (between 3’ group I intron fragment and core functional element) and the second (between the two expression sequences and core functional element) spacers comprise additional base pairing regions that are predicted to base pair with each odier and not to the first and second duplex regions.
- the first (between 3’ group I intron fragment and core functional element) and the second (between the one of the core functional element and 5 ’ group I intron fragment) spacers comprise additional base pairing regions that are predicted to base pair with each other and not to the first and second duplex regions.
- such spacer base pairing brings the group I intron fragments in close proximity to each other, further increasing splicing efficiency. Additionally, in some embodiments, the combination of base pairing between the first and second duplex regions, and separately, base pairing between the first and second spacers, promotes the formation of a splicing bubble containing the group I intron fragments flanked by adjacent regions of base pairing.
- Typical spacers are contiguous sequences with one or more of the following qualities: 1) predicted to avoid interfering with proximal structures, for example, the IRES, expression sequence, aptamer, or intron; 2) is at least 7 nt long and no longer than 100 nt; 3) is located after and adjacent to the 3’ intron fragment and/or before and adjacent to the 5’ intron fragment; and 4) contains one or more of the following: a) an unstructured region at least 5 nt long, b) a region of base pairing at least 5 nt long to a distal sequence, including another spacer, and c) a structured region at least 7 nt long limited in scope to the sequence of the spacer.
- Spacers may have several regions, including an unstructured region, a base pairing region, a hairpin/structured region, and combinations thereof.
- the spacer has a structured region with high GC content.
- a spacer comprises one or more hairpin structures.
- a spacer comprises one or more hairpin structures with a stem of 4 to 12 nucleotides and a loop of 2 to 10 nucleotides.
- tiiere is an additional spacer betw een the 3 ’ group I intron fragment and the core functional element.
- this additional spacer prevents the structured regions of the IRES or aptamer of a TIE from interfering widr the folding of the 3’ group I intron fragment or reduces the extent to which this occurs.
- the 5’ spacer sequence is at least 7, 8, 9, 10. 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25 or 30 nucleotides in length. In some embodiments, the 5‘ spacer sequence is no more than 100, 90, 80, 70, 60, 50, 45, 40, 35 or 30 nucleotides in length. In some embodiments the 5’ spacer sequence is between 5 and 50, 10 and 50, 20 and 50, 20 and 40, and/or 25 and 35 nucleotides in length.
- the 5’ spacer sequence is 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 or 50 nucleotides in length.
- the 5’ spacer sequence is a poly A sequence.
- the 5’ spacer sequence is a poly AC sequence.
- a spacer comprises 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or 100% poly AC content.
- a spacer comprises 10%. 20%, 30%, 40%, 50%. 60%, 70%, 80%, 90%. or 100% polypyrimidine (C/T or C/U) content.
- the DNA template and precursor linear RNA polynucleotides and circular RNA polynucleotide provided herein comprise a first (5’) duplex region and a second (3’) duplex region.
- the DNA template and precursor linear RNA polynucleotide comprises a 5' external duplex region located within the 3‘ enhanced intron fragment and a 3’ external duplex region located within the 5’ enhanced intron fragment.
- the DNA template, precursor linear RNA polynucleotide and circular RNA polynucleotide comprise a 5 ‘ internal duplex region located within the 3’ enhanced exon fragment and a 3’ internal duplex region located within the 5’ enhanced exon fragment.
- the DNA polynucleotide and precursor linear RN A polynucleotide comprises a 5 ’ external duplex region. 5 ’ internal duplex region, a 3 ’ internal duplex region, and a 3‘ external duplex region.
- the first and second duplex regions may form perfect or imperfect duplexes. Thus, in certain embodiments at least 75%. 80%, 85%, 90%, 91%, 92%, 93%. 94%, 95%, 96%, 97%, 98%, 99%, or 100% of the first and second duplex regions may be base paired with one another. In some embodiments, the duplex regions are predicted to have less than 50% (e.g., less than 45%, less than 40%, less than 35%, less than 30%. less than 25%) base pairing with unintended sequences in the RNA (e.g., non-duplex region sequences).
- the duplex regions are 3 to 100 nucleotides in length (e.g., 3-75 nucleotides in length, 3-50 nucleotides in length. 20-50 nucleotides in length. 35-50 nucleotides in length. 5-25 nucleotides in length, 9-19 nucleotides in length). In some embodiments, the duplex regions are 3, 4. 5. 6, 7, 8, 9, 10, 11, 12. 13, 14, 15, 16. 17, 18, 19, 20. 21. 22, 23, 24. 25. 26, 27, 28, 29. 30, 31, 32, 33, 34.
- the duplex regions have a length of 9 to 50 nucleotides. In one embodiment, the duplex regions have a length of 9 to 19 nucleotides. In some embodiments, the duplex regions have a length of 20 to 40 nucleotides. In certain embodiments, the duplex regions have a length of 30 nucleotides.
- the DNA template, precursor linear RNA polynucleotide, or circular RNA polynucleotide does not comprise of any duplex regions to optimize translation or circularization.
- the DNA template or precursor linear RNA polynucleotide may comprise an affinity tag.
- the affinity tag is located in the 3 ‘ enhanced intron element.
- the affinity tag is located in the 5’ enhanced intron element.
- both (3 ' and 5 ') enhanced intron elements each comprise an affinity tag.
- an affinity tag of the 3 ‘ enhanced intron element is the length as an affinity tag in the 5 ’ enhanced intron element.
- an affinity tag of the 3 ’ enhanced intron element is the same sequence as an affinity tag in the 5 ’ enhanced intron element.
- the affinity sequence is placed to optimize oligo-dT purification.
- an affinity tag comprises a polyA region.
- the polyA region is at least 15, 30, or 60 nucleotides long.
- one or both polyA regions is 15-50 nucleotides long.
- one or both polyA regions is 20-25 nucleotides long.
- the polyA sequence is removed upon circularization.
- an oligonucleotide hybridizing with the polyA sequence such as a deoxythymine oligonucleotide (oligo(dT)) conjugated to a solid surface (e.g., a resin), can be used to separate circular RNA from its precursor RNA.
- the 3’ enhanced intron element comprises a leading untranslated sequence.
- the leading untranslated sequence is a the 5’ end of the 3‘ enhanced intron fragment.
- the leading untranslated sequence comprises of the last nucleotide of a transcription start site (TSS).
- TSS transcription start site
- the TSS is chosen from a viral, bacterial, or eukaryotic DNA template.
- the leading untranslated sequence comprise the last nucleotide of a TSS and 0 to 100 additional nucleotides.
- the TSS is a terminal spacer.
- the leading untranslated sequence contains a guanosine at the 5’ end upon translation of an RNA T7 polymerase.
- the 5’ enhanced intron element comprises a trailing untranslated sequence.
- the 5' trailing untranslated sequence is located at the 3’ end of the 5’ enhanced intron element.
- the trailing untranslated sequence is a partial restriction digest sequence.
- the trailing untranslated sequence is in whole or in part a restriction digest site used to linearize the DNA template.
- the restriction digest site is in whole or in part from a natural viral, bacterial or eukaryotic DNA template.
- the trailing untranslated sequence is a terminal restriction site fragment.
- the 3’ enhanced intron element and 5’ enhanced intron element each comprise an intron fragment.
- a 3 ‘ intron fragment is a contiguous sequence at least 75% homologous (e.g., at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% homologous) to a 3’ proximal fragment of a natural group I or II intron including the 3’ splice site dinucleotide.
- a 5' intron fragment is a contiguous sequence at least 75% homologous (e.g., at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% homologous) to a 5 ' proximal fragment of a natural group I or II intron including the 5 ’ splice site dinucleotide.
- the 3’ intron fragment includes the first nucleotide of a 3‘ group I or II splice site dinucleotide.
- the 5’ intron fragment includes the first nucleotide of a 5’ group I or II splice site dinucleotide.
- the 3‘ intron fragment includes the first and second nucleotides of a 3’ group I or II intron fragment splice site dinucleotide; and the 5’ intron fragment includes the first and second nucleotides of a 3’ group I or II intron fragment dinucleotide.
- the 3’ enhanced intron element and 5’ enhanced intron element comprises a synthetic intron fragment.
- the DNA template, linear precursor RNA polynucleotide, and circular RNA polynucleotide each comprise an enhanced exon fragment.
- the 3’ enhanced exon element is located upstream to core functional element.
- the 5’ enhanced intron element is located downstream to the core functional element.
- the 3’ enhanced exon element and 5’ enhanced exon element each comprise an exon fragment.
- the 3’ enhanced exon element comprises a 3‘ exon fragment.
- the 5’ enhanced exon element comprises a 5’ exon fragment.
- the 3 ’ exon fragment and 5 ’ exon fragment each comprises a group I or II intron fragment and 1 to 100 nucleotides of an exon sequence.
- a 3’ intron fragment is a contiguous sequence at least 75% homologous (e.g., at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% homologous) to a 3’ proximal fragment of a natural group I or II intron including the 3' splice site dinucleotide.
- a 5’ group I or II intron fragment is a contiguous sequence at least 75% homologous (e.g., at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%.
- the 3 ’ exon fragment comprises a second nucleotide of a 3’ group I or II intron splice site dinucleotide and 1 to 100 nucleotides of an exon sequence.
- the 5‘ exon fragment comprises the first nucleotide of a 5’ group I or II intron splice site dinucleotide and 1 to 100 nucleotides of an exon sequence.
- the exon sequence comprises in part or in whole from a naturally occurring exon sequence from a virus, bacterium or eukaryotic DNA vector.
- the exon sequence further comprises a synthetic, genetically modified (e.g., containing modified nucleotide), or other engineered exon sequence.
- the exon fragments located within the 5’ enhanced exon element and 3’ enhanced exon element does not comprise of a group I or II splice site dinucleotide.
- a 3’ enhanced intron element comprises in the following 5’ to 3’ order: a leading untranslated sequence, a 5’ affinity tag, an optional 5’ external duplex region, a 5’ external spacer, and a 3' intron fragment.
- the 3’ enhanced exon element comprises in the following 5’ to 3’ order: a 3’ exon fragment, an optional 5’ internal duplex region, an optional 5’ internal duplex region, and a 5’ internal spacer.
- the 5‘ enhanced exon element comprises in the following 5' to 3’ order: a 3’ internal spacer, an optional 3‘ internal duplex region, and a 5’ exon fragment.
- the 3 ’ enhanced intron element comprises in the following 5 ’ to 3 ’ order: a 5 ’ intron fragment, a 3‘ external spacer, an optional 3' external duplex region, a 3‘ affinity tag, and a trailing untranslated sequence.
- the DNA template, linear precursor RNA polynucleotide, and circular RNA polynucleotide comprise a core functional element.
- the core functional element comprises a coding or a noncoding element.
- the core functional element may contain both a coding and a noncoding element.
- the core functional element further comprises a translation initiation element (TIE) upstream to the coding or noncoding element.
- the core functional element comprises a termination element.
- the termination element is located downstream to the TIE and coding element.
- the termination element is located downstream to the coding element but upstream to the TIE.
- a core functional element lacks a TIE and/or a termination element.
- the polynucleotides herein comprise a coding element, a noncoding element, or a combination of both.
- the coding element comprises an expression sequence.
- the coding element encodes at least one therapeutic protein.
- the circular RNA encodes two or more polypeptides.
- the circular RNA is a bicistronic RNA.
- the sequences encoding the two or more polypeptides can be separated by a ribosomal skipping element or a nucleotide sequence encoding a protease cleavage site.
- the ribosomal skipping element encodes thosea-asigna virus 2A peptide (T2A), porcine teschovirus-1 2 A peptide (P2A). foot-and-mouth disease virus 2 A peptide (F2A).
- E2A equine rhinitis A vims 2A peptide
- BmCPV 2A cytoplasmic polyhedrosis vims 2A peptide
- BmIFV 2A flacherie vims of B. mori 2A peptide
- TIE Translation Initiation Element
- the core functional element comprises at least one translation initiation element (TIE).
- TIEs are designed to allow translation efficiency of an encoded protein.
- optimal core functional elements comprising only of noncoding elements lack any TIEs.
- core functional elements comprising one or more coding element will further comprise one or more TIEs.
- a TIE comprises an untranslated region (UTR).
- the TIE provided herein comprise an internal ribosome entry site (IRES).
- IRES internal ribosome entry site
- IRES pennits the translation of one or more open reading frames from a circular RNA (e.g., open reading frames that form the expression sequences).
- the IRES element attracts a eukaryotic ribosomal translation initiation complex and promotes translation initiation. See. e.g., Kaufman et al., Nuc. Acids Res. (1991) 19:4485- 4490; Gurtu et al., Biochem. Biophys. Res. Comm.
- the IRES element is selected from those disclosed in international publication WO/2022/261490, the contents of which are hereby incorporated in their entireties.
- the circular RNA polynucleotide, linear RNA polynucleotide, and/or DNA template may further comprise accessory elements.
- these accessory elements may be included within the sequences of the circular RNA, linear RNA polynucleotide and/or DNA template for enhancing circularization, translation or both.
- Accessory elements are sequences, in certain embodiments that are located with specificity between or within the enhanced intron elements, enhanced exon elements, or core functional element of the respective polynucleotide.
- an accessory element includes an IRES transacting factor region, a miRNA binding site, a restriction site, an RNA editing region, a structural or sequence element, a granule site, a zip code element, an RNA trafficking element or another specialized sequence as found in the art that enhances circularization and/or translation of the protein encoded within the circular RNA polynucleotide.
- the accessory element comprises an IRES transacting factor (ITAF) region.
- IRES transacting factor region modulates the initiation of translation through binding to PCBP1 - PCBP4 (polyC binding protein), PABP1 (poly A binding protein). PTB (polyprimidine tract binding), Argonaute protein family, HNRNPK (Heterogeneous nuclear ribonucleoprotein K protein), or La protein.
- the IRES transacting factor region comprises a poly A, polyC, poly AC, or polyprimidine track.
- the ITAF region is located within the core functional element. In some embodiments, the ITAF region is located within the TIE.
- the accessory’ element comprises a miRNA binding site.
- the miRNA binding site is located within the 5’ intron clement, 5’ exon clement, core functional element, 3’ exon element, and/or 3’ intron element.
- the miRNA binding site is located within the spacer within the intron element or exon element. In certain embodiments, the miRNA binding site comprises the entire spacer regions.
- the 5’ intron element and 3’ intron elements each comprise identical miRNA binding sites.
- the miRNA binding site of the 5’ intron element comprises a different, in length or nucleotides. miRNA binding site than the 3’ intron element.
- the 5’ exon element and 3’ exon element comprise identical miRNA binding sites.
- the 5’ exon element and 3’ exon element comprises different, in length or nucleotides, miRNA binding sites.
- the miRNA binding sites are located adjacent to each other within the circular RNA polynucleotide, linear RNA polynucleotide precursor, and/or DNA template.
- the first nucleotide of one of the miRNA binding sites follows the first nucleotide last nucleotide of the second miRNA binding site.
- the miRNA binding site is located within a translation initiation element (TIE) of a core functional element. In one embodiment, the miRNA binding site is located before, trailing or within an internal ribosome entry site (IRES). In another embodiment, the miRNA binding site is located before, trailing, or within an aptamer complex.
- TIE translation initiation element
- IRS internal ribosome entry site
- IRES sequences include sequences derived from a wide variety of viruses, such as from leader sequences of piccircRNAviruses such as the encephalomyocarditis virus (EMCV) UTR (Jang et al., J. Virol. (1989) 63: 1651-1660), the polio leader sequence, the hepatitis A virus leader, the hepatitis C virus IRES, human rhinovirus type 2 IRES (Dobrikova et al., Proc. Natl. Acad. Sci. (2003) 100(25): 15125- 15130). an IRES element from the foot and mouth disease virus (Ramesh et al., Nucl. Acid Res.
- EMCV encephalomyocarditis virus
- UTR the polio leader sequence
- the hepatitis A virus leader the hepatitis C virus IRES
- human rhinovirus type 2 IRES Dobrikova et al., Proc. Natl. Acad. Sci. (2003) 100
- tire circular RNA comprises an IRES operably linked to a protein coding sequence.
- Modifications of IRES and accessory sequences are disclosed herein to increase or reduce IRES activities, for example, by truncating the 5’ and/or 3’ ends of the IRES, adding a spacer 5‘ to the IRES, modifying the 6 nucleotides 5’ to the translation initiation site (Kozak sequence), modification of alternative translation initiation sites, and creating chimeric/hybrid IRES sequences.
- the IRES sequence in the circular RNA disclosed herein comprises one or more of these modifications relative to a native IRES.
- the IRES is an IRES sequence of Taura syndrome virus, Triatoma virus, Theiler's encephalomyelitis virus, Simian Virus 40, Solenopsis invicta virus 1, Rhopalosiphum padi virus, Reticuloendotheliosis virus, Human poliovirus 1, Plautia stali intestine virus, Kashmir bee virus, Human rhinovirus 2, Homalodisca coagulata virus- 1, Human Immunodeficiency Virus type 1, Himetobi P virus, Hepatitis C virus, Hepatitis A virus, Hepatitis GB virus , Foot and mouth disease virus, Human enterovirus 71, Equine rhinitis virus, Ectropis obliqua piccircRNA-like virus, Encephalomyocarditis virus.
- Drosophila C Virus Human coxsackievirus B3, Crucifer tobamovirus, Cricket paralysis virus, Bovine viral diarrhea virus 1, Black Queen Cell Virus. Aphid lethal paralysis virus. Avian encephalomyelitis virus, Acute bee paralysis virus. Hibiscus chlorotic ringspot virus. Classical swine fever virus. Human FGF2, Human SFTPA1, Human AML1/RUNX1. Drosophila antennapedia. Human AQP4, Human AT1R, Human BAG-1, Human BCL2. Human BiP, Human c- IAP1, Human c-myc. Human eIF4G. Mouse NDST4L. Human LEF1, Mouse HIF1 alpha, Human n.myc. Mouse Gtx.
- the IRES comprises in whole or in part a eukary otic or cellular IRES.
- the IRES is from a human gene, wherein the human gene is ABCF1, ABCG1, ACAD10, ACOT7, ACSS3, ACTG2, ADCYAP1, ADK, AGTR1, AHCYL2, AHI1, AKAP8L, AKR1A1, ALDH3A1, ALDOA, ALG13, AMMECR1L, ANGPTL4, ANK3, AOC3, AP4B1, AP4E1, APAF1.
- CSNK2A 1.
- CYP3A5. DAG1.
- DAXX. DCAF4, DCAF7, DCLRE1A, DCP1A.
- DMKN DNAH6.
- ECN ENO1, EPB41, ERMN. ERVV-1, ESRRG, ETFB, ETFBKMT, ETV1, ETV4, EXD1, EXT1, EZH2, FAM111B, FAM157A, FAM213A, FBXO25, FBXO9. FBXW7, FCMR, FGF1, FGF1. FGF1A, FGF2, FGF2. FGF-9, FHL5, FMRI, FN1, FOXP1, FTH1. FUBP1, G3BP1, GABBR1, GALC, GART.
- GAS7 gastrin, GATA1, GATA4, GFM2, GHR, GJB2, GLI1, GLRA2, GMNN, GPAT3, GPATCH3, GPR137, GPR 3 4, GPR55, GPR89A, GPRASP1, GRAP2, GSDMB, GSTO2.
- LYRM2 MAGEA11, MAGEA8, MAGEB1, MAGEB16. MAGEB3, MAPT.
- MITF MKLN1.
- MNT MORF4L2.
- MPD6 MRFAP1, MRPL21, MRPS12, MSI2, MSLN, MSN, MT2A, MTFR1L, MTMR 2 , MTRR, MTUS1, MYB. MYC, MYCL, MYCN, MYL10, MYL3. MYLK, MYO1 A. MYT2, MZB1, NAP1L1, NAVI. NBAS, NCF2. NDRG1, NDST2, NDUFA7, NDUFB11. NDUFC1, NDUFS1, NEDD4L. NFAT5, NFE2L2, NFE2L2. NFIA, NHEJ1.
- RAB7B RABGGTB.
- RAET1E RALGDS, RALYL, RARB, RCVRN, REG3G, RFC5, RGL4, RGS19, RGS3, RHD, RINL, RIPOR 2 , RITA1, RMDN2, RNASE1, RNASE4, RNF4, RPA2, RPL17.
- RPL26L 1, RPL28, RPL29, RPL41, RPL9.
- RPS11 RPS13.
- RRBP1 RSU1, RTP2, RUNX1, RUNX1T1, RUNX1T1, RUNX2, RUSC1, RXRG, S100A13. S100A4.
- TMEM126A TMEM159, TMEM208, TMEM230, TMEM67.
- TMPRSS13 TMUB2, TNFSF4, TNIP3.
- TTC39B TTLL11, TUBB6, TXLNB, TXNIP, TXNL1, TXNRD1, TYROBP, U2AF1, UBA1, UBE2D3, UBE2I, UBE2L3, UBE2V1, UBE2V2, UMPS, UNG, UPP2, USMG5, USP18, UTP14A, UTRN, UTS2, VDR, VEGFA, VEGFA, VEPH1, VIPAS39, VPS29, VSIG10L, WDHD1, WDR12, WDR4, WDR45, WDYHV1, WRAP53, XIAP, XPNPEP3, YAP1, YWHAZ, YY1AP1, ZBTB32, ZNF146, ZNF250, ZNF385A, ZNF408, ZNF410, ZNF423.
- a translation initiation element comprises a synthetic TIE.
- a synthetic TIE comprises aptamer complexes, synthetic IRES or other engineered TIES capable of initiating translation of a linear RNA or circular RNA polynucleotide.
- one or more aptamer sequences is capable of binding to a component of a eukaryotic initiation factor to either enhance or initiate translation.
- an aptamer may be used to enhance translation in vivo and in vitro by promoting specific eukary otic initiation factors (elF) (e.g., aptamer in WO2019081383A1 is capable of binding to eukary otic initiation factor 4F (eIF4F).
- the aptamer or a complex of aptamers may be capable of binding to EIF4G, EIF4E, EIF4A, EIF4B, EIF3, EIF2, EIF5, EIF1, EIF1A, 40S ribosome, PCBP1 (polyC binding protein), PCBP2, PCBP3, PCBP4, PABP1 (polyA binding protein).
- PTB Argonaute protein family, HNRNPK (heterogeneous nuclear ribonucleoprotein K). or La protein. viii. Termination Sequence
- the core functional element comprises a termination sequence.
- the termmation sequence comprises a stop codon.
- the tennination sequence comprises a stop cassette.
- the stop cassette comprises at least 2 stop codons.
- the stop cassette comprises at least 2 frames of stop codons.
- the frames of the stop codons in a stop cassette each comprise 1, 2, or more stop codons.
- the stop cassette comprises a LoxP or a RoxStopRox, or frt-flanked stop cassette.
- the stop cassette comprises a lox-stop-lox stop cassette. ix.
- a circular RNA polynucleotide provided herein comprises modified RNA nucleotides and/or modified nucleosides.
- the modified nucleoside is m 5 C (5-methylcytidine).
- the modified nucleoside is m 5 U (5 -methyluridine).
- the modified nucleoside is m 6 A (N 6 -methyladenosine).
- the modified nucleoside is s 2 U (2-thiouridine).
- the modified nucleoside is (pseudouridine).
- the modified nucleoside is Um (2'-O-methyluridine).
- the modified nucleoside is m' A (1 -methyladenosine); m 2 A (2 -methyladenosine); Am (2’-O-methyladenosine); ms 2 m 6 A (2-methylthio-N 6 -methyladenosine); i 6 A (N 6 -isopentenyladenosine); ms 2 i6A (2-methylthio-N 6 isopentenyladenosine); io 6 A (N 6 -(cis-hydroxyisopentenyl)adenosine); ms 2 io 6 A (2-methylthio-N 6 -(cis-hydroxyisopentenyl)adenosine); g 6 A (N 6 - glycinylcarbamoy ladenosine); t 6 A (N 6 -threonylcarbamoy ladenosine); ms 2 t 6 A (2-methylthio-N 6 - dire
- the modified nucleoside may include a compound selected from: pyridin-4-one ribonucleoside. 5 -aza-uridine. 2-thio-5 -aza-uridine, 2-thiouridine. 4-thio-pseudouridine, 2-thio-pseudouridine, 5 -hydroxy uridine, 3 -methyluridine, 5-carboxymethyl-uridine, 1 -carboxy methyl- pseudouridine.
- the modifications are independently selected from 5-methylcytosine, pseudouridine and 1 -methylpseudouridine.
- the modified ribonucleosides include 5-methylcytidine, 5- methoxyuridine. 1-methyl-pseudouridine, N6-methyladenosine, and/or pscudoundinc. In some embodiments, such modified nucleosides provide additional stability and resistance to immune activation.
- polynucleotides may be codon-optimized.
- a codon optimized sequence may be one in which codons in a polynucleotide encoding a polypeptide have been substituted in order to increase the expression, stability and/or activity of the polypeptide.
- Factors that influence codon optimization include, but are not limited to one or more of: (i) variation of codon biases betw een two or more organisms or genes or synthetically constructed bias tables, (ii) variation in the degree of codon bias within an organism, gene, or set of genes, (iii) systematic variation of codons including context, (iv) variation of codons according to their decoding tRNAs, (v) variation of codons according to GC %, either overall or in one position of the triplet, (vi) variation in degree of similarity to a reference sequence for example a naturally occurring sequence, (vii) variation in the codon frequency cutoff, (viii) structural properties of mRNAs transcribed from the DNA sequence, (ix) prior knowledge the function of the DNA sequences upon which design of the codon substitution set is to be based, and/or (x) systematic variation of codon sets for each amino acid.
- a codon optimized polynucleotide may minimize ribozyme collisions
- the polynucleotide (e.g., circRNA) expression sequence encodes a therapeutic protein.
- the therapeutic protein is selected from the proteins listed in the following Table 3.
- the expression sequence encodes an inhibitory receptor agonist (e.g., PDL1, PDL2, Galectin-9, VISTA, B7H4, or MHCII) or inhibitory receptor (e.g., PD1, CTLA4, TIGIT, LAG3, or TIM3).
- the expression sequence encodes an inhibitor ⁇ ' receptor antagonist.
- the expression sequence encodes one or more TCR chains (alpha and beta chains or gamma and delta chains).
- the expression sequence encodes a secreted T cell or immune cell engager (e.g., a bispecific antibody such as BiTE, targeting, e.g., CD3, CD137, or CD28 and a tumor-expressed protein e.g., CD19, CD20, or BCMA etc.).
- the expression sequence encodes a transcription factor (e.g., F0XP3, HELIOS, TOX1, or T0X2).
- the expression sequence encodes an immunosuppressive enzyme (e.g., IDO or CD39/CD73).
- the expression sequence encodes a GvHD (e.g., anti-HLA- A2 CAR-Tregs).
- the precursor RNA polynucleotide and circular RNA constructs comprise at least one expression sequence encoding an antigen, adjuvant, or adjuvant-like protein, e.g., from an infectious agent.
- the circular RNA construct may be used as a vaccine.
- the one or more circular RNA polynucleotide encodes an antigen or adjuvant derived from an infectious agent.
- the infectious agent from which the antigen or adjuvant is derived or engineered includes, but is not limited to a virus, bacterium, fungus, protozoan, and/or parasite.
- the antigen is a viral antigen or viral antigenic polypeptide.
- the antigen is selected from or derived from the group consisting of rotavirus, foot and mouth disease virus, influenza A virus, influenza B virus, influenza C virus, H1N1, H2N2, H3N2, H5N1, H7N7, H1N2, H9N2, H7N2, H7N3, H10N7, human parainfluenza type 2, herpes simplex virus.
- distemper virus Venezuelan equine encephalomyelitis, feline leukemia virus, reovirus, respiratory syncytial virus. Lassa fever virus, polyoma tumor virus, canine parvovirus, papilloma virus, tick borne encephalitis virus, rinderpest virus, human rhinovirus species. Enterovirus species.
- Mengovirus paramyxovirus, avian infectious bronchitis virus, human T-cell leukemia-lymphoma virus 1, human immunodeficiency virus- 1, human immunodeficiency virus-2, lymphocytic choriomeningitis virus, parvovirus Bl 9, adenovirus, rubella virus, yellow fever virus, dengue virus, bovine respiratory syncitial virus, corona virus, Bordetella pertussis, Bordetella bronchiseptica, Bordetella parapertussis, Brucella abortis, Brucella melitensis, Brucella suis, Brucella ovis, Brucella species, Escherichia coli, Salmonella species, Salmonella typhi, Streptococci, Vibrio cholera, Vibrio parahaemolyticus.
- Chlamydia trachomatis Chlamydia psittaci, Lymphogranuloma venereum, Treponema pallidum, Haemophilus species, Mycoplasma bovigenitalium, Mycoplasma pulmonis, Mycoplasma species, Borrelia burgdorferi, Legionalla pneumophila, Colstridium botulinum, Corynebacterium diphtheriae, Yersinia entercolitica, Ricketsia ricketsii, Ricketsia tv phi.
- Neisseria gonorrhoeae malaria circumsporozoite protein, malaria merozoite protein, trypanosome surface antigen protein, pertussis, alphaviruses, adenovirus, diphtheria toxoid, tetanus toxoid, meningococcal outer membrane protein, streptococcal M protein, Influenza hemagglutinin, cancer antigen, tumor antigens, toxins, Clostridium perfringens epsilon toxin, ricin toxin, pseudomonas exotoxin, exotoxins, neurotoxins, cytokines, cytokine receptors, monokines, monokine receptors, plant pollens, animal dander, and dust mites.
- the antigenic polypeptide is a viral polypeptide from an adenovirus; Herpes simplex, type 1; Herpes simplex, type 2; encephalitis virus, papillomavirus, Varicella-zoster virus; Epstein-barr virus; Human cytomegalovirus: Human herpes virus, type 8; Human papillomavirus; BK virus; JC virus; Smallpox; polio virus; Hepatitis B virus; Human bocavirus; Parvovirus B19: Human astrovirus; Norwalk virus; coxsackievirus; hepatitis A virus; poliovirus; rhinovirus; Severe acute respiratory syndrome virus; Hepatitis C virus; Yellow Fever virus; Dengue virus; West Nile virus; Rubella virus; Hepatitis E virus; Human Immunodeficiency virus (HIV); Influenza virus; Guanarito virus; Junin virus; Lassa virus; Machupo virus; Sabia virus; Crime
- the adjuvant is selected from or derived from the group consisting of BCSP31, MOMP, FomA, MymA, ESAT6, PorB, PVL, Porin, OmpA, PepO, OmpU, Lumazine synthase, 0mpl6, Ompl9, CobT. RpfE. Rv0652, HBHA, NhhA. DnaJ. Pneumolysin. Falgellin, IFN- alpha, IFN-gamma, IL-2. IL-12, IL-15, IL-18. IL-21, GM-CSF, IL-lb, IL-6, TNF-a, IL-7. IL-17, IL- IBeta. anti-CTLA4, anti-PDl, anti-41BB, PD-L1, Tim-3, Lag-3, TIGIT, GITR, and andti-CD3.
- a polynucleotide encodes a protein that is made up of subunits that are encoded by more than one gene.
- the protein may be a heterodimer, wherein each chain or subunit of the protein is encoded by a separate gene. It is possible that more than one circRNA molecule is delivered in the transfer vehicle and each circRNA encodes a separate subunit of the protein. Alternatively, a single circRNA may be engineered to encode more than one subunit. In certain embodiments, separate circRNA molecules encoding the individual submits may be administered in separate transfer vehicles.
- Additional polynucleotides including but not limited to intron elements, exon elements, translation initiation elements, expression sequences, and lipids are in WO2019236673; WO2020237227; WO2021113777; WO2021226597; WO2021189059; WO2021236855;
- Chimeric antigen receptors are genetically engineered receptors. These engineered receptors may be inserted into and expressed by immune cells, including T cells via circular RNA as described herein. With a CAR, a single receptor may be programmed to both recognize a specific antigen and, when bound to that antigen, activate the immune cell to attack and destroy the cell bearing that antigen. When these antigens exist on tumor cells, an immune cell that expresses the CAR may target and kill the tumor cell.
- the CAR encoded by the polynucleotide comprises (i) an antigen-binding molecule that specifically binds to a target antigen, (ii) a hinge domain, a transmembrane domain, and an intracellular domain, and (iii) an activating domain.
- an orientation of the CARs in accordance with the disclosure comprises an antigen binding domain (such as an scFv) in tandem with a costimulatory domain and an activating domain.
- the costimulatory domain may comprise one or more of an extracellular portion, a transmembrane portion, and an intracellular portion. In other embodiments, multiple costimulatory domains may be utilized in tandem.
- CARs may be engineered to bind to an antigen (such as a cell-surface antigen) by incorporating an antigen binding molecule that interacts with that targeted antigen.
- the antigen binding molecule is an antibody fragment thereof, e.g.. one or more single chain antibody fragment (scFv).
- scFv is a single chain antibody fragment having the variable regions of the heavy and light chains of an antibody linked together. See U.S. Patent Nos. 7,741,465, and 6,319,494 as well as Eshhar et al.. Cancer Immunol Immunotherapy (1997) 45: 131-136.
- An scFv retains the parent antibody's ability to specifically interact with target antigen.
- scFvs are useful in chimeric antigen receptors because they may be engineered to be expressed as part of a single chain along with the other CAR components. Id. See also Krause et al., J. Exp. Med.. Volume 188, No. 4. 1998 (619-626); Finney et al., Journal of Immunology, 1998, 161: 2791-2797. It will be appreciated that the antigen binding molecule is typically contained within the extracellular portion of the CAR such that it is capable of recognizing and binding to the antigen of interest. Bispecific and multispecific CARs are contemplated within the scope of the present disclosure, with specificity to more than one target of interest.
- the antigen binding molecule comprises a single chain, wherein the heavy chain variable region and the light chain variable region are comiected by a linker.
- the VH is located at the N terminus of die linker and the VL is located at the C terminus of the linker.
- the VL is located at the N terminus of the linker and the VH is located at the C terminus of the linker.
- the linker comprises at least 5. at least 8, at least 10, at least 13, at least 15, at least 18, at least 20. at least 25, at least 30, at least 35, at least 40, at least 45, at least 50, at least 60. at least 70, at least 80, at least 90, or at least 100 amino acids.
- the antigen binding molecule comprises a nanobody. In some embodiments, the antigen binding molecule comprises a DARPin. In some embodiments, the antigen binding molecule comprises an anticalin or another synthetic protein capable of specific binding to target protein.
- the CAR comprises an antigen binding domain specific for an antigen selected from CD19, CD123, CD22, CD30, CD171, CS-1, C-type lectin-like molecule-1, CD33, epidermal growth factor receptor variant III (EGFRvIII), ganglioside G2 (GD2), ganglioside GD3, TNF receptor family member B cell maturation (BCMA).
- an antigen selected from CD19, CD123, CD22, CD30, CD171, CS-1, C-type lectin-like molecule-1, CD33, epidermal growth factor receptor variant III (EGFRvIII), ganglioside G2 (GD2), ganglioside GD3, TNF receptor family member B cell maturation (BCMA).
- Tn antigen (Tn Ag) or (GalNAca-Ser/Thr)
- PSMA prostate-specific membrane antigen
- ROR1 Receptor tyrosine kinase-like orphan receptor 1
- FLT3 Fms-Like Tyrosine Kinase 3
- TAG72 Tumor-associated glycoprotein 72
- CD38 CD44v6, Carcinoembryonic antigen (CEA), Epithelial cell adhesion molecule (EPCAM), B7H3 (CD276), KIT (CD117), Interleukin- 13 receptor subunit alpha-2, mesothelin, Interleukin 11 receptor alpha (IL-1 IRa), prostate stem cell antigen (PSCA).
- MUC1 cell surface associated
- EGFR epidermal growth factor receptor
- NCAM neural cell adhesion molecule
- Prostase prostatic acid phosphatase (PAP), elongation factor 2 mutated (ELF2M), Ephrin B2, fibroblast activation protein alpha (FAP), insulin-like growth factor 1 receptor (IGF-I receptor), carbonic anhydrase IX (CAIX), Proteasome (Prosome, Macropain) Subunit, Beta Type, 9 (LMP2). glycoprotein 100 (gplOO).
- oncogene fusion protein consisting of breakpoint cluster region (BCR) and Abelson murine leukemia viral oncogene homolog 1 (Abl) (bcr-abl), tyrosinase, ephrin tvpe- A receptor 2 (EphA2), Fucosyl GM1, sialyl Lewis adhesion molecule (sLe). ganglioside GM3, transglutaminase 5 (TGS5), high molecular weight-melanoma-associated antigen (HMWMAA).
- OAcGD2 o- acetyl-GD2 ganglioside
- Folate receptor beta tumor endothelial marker 1 (TEM1/CD248), tumor endothelial marker 7-related (TEM7R).
- Claudin 6 CLDN6
- TSHR thyroid stimulating hormone receptor
- GPRC5D G protein-coupled receptor class C group 5, member D
- CXORF61 chromosome X open reading frame 61
- CD97 CD179a
- ALK anaplastic lymphoma kinase
- Polysialic acid placenta-specific 1 (PLAC1), hcxasaccharidc portion of globoH glycoccramidc (GloboH), mammary gland differentiation antigen (NY-BR-1), uroplakin 2 (UPK2), Hepatitis A virus cellular receptor 1 (HAVCR1), adrenoceptor beta 3 (ADRB3), pannexin 3 (PANX3), G protein-coupled receptor 20
- chromosome 12p located on chromosome 12p (ETV6-AML), sperm protein 17 (SPA17), X Antigen Family, Member 1A (XAGE1), angiopoietin-binding cell surface receptor 2 (Tie 2), melanoma cancer testis antigen-1 (MAD-CT-1), melanoma cancer testis antigen-2 (MAD-CT-2), Fos-related antigen 1, tumor protein p53 (p53), p53 mutant, prostein, surviving, telomerase, prostate carcinoma tumor antigen- 1.
- melanoma antigen recognized by T cells 1 Rat sarcoma (Ras) mutant, human Telomerase reverse transcriptase (hTERT), sarcoma translocation breakpoints, melanoma inhibitor of apoptosis (ML-IAP).
- ERG transmembrane protease, serine 2 (TMPRSS2) ETS fusion gene), N-Acetyl glucosaminyl-transferase V (NA17), paired box protein Pax-3 (PAX3). Androgen receptor. Cyclin Bl.
- v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog MYCN
- Ras Homolog Family Member C RhoC
- Tyrosinase-related protein 2 TRP-2
- Cytochrome P450 1B1 CYP1B1
- CCCTC-Binding Factor Zinc Finger Protein-Like. Squamous Cell Carcinoma Antigen Recognized By T Cells 3 (SART3). Paired box protein Pax-5 (PAX5).
- proacrosin binding protein sp32 OY-TES1
- LCK lymphocyte-specific protein tyrosine kinase
- AKAP-4 A kinase anchor protein 4
- SSX2 synovial sarcoma
- RAGE-1 Receptor for Advanced Glycation Endproducts
- RU1 renal ubiquitous 1
- RU2 renal ubiquitous 2
- legumain human papilloma virus E6
- human papilloma virus E7 HPV E7
- intestinal carboxyl esterase heat shock protein 70-2 mutated (mut hsp70-2), CD79a, CD79b.
- CD72 Leukocyte-associated immunoglobulin-like receptor 1 (LAIR1), Fc fragment of IgA receptor (FCAR or CD89), Leukocyte immunoglobulin-like receptor subfamily A member 2 (LILRA2), CD300 molecule-like family member f (CD300LF), C-type lectin domain family 12 member A (CLEC12A), bone marrow stromal cell antigen 2 (BST2), EGF-likc module-containing mucin-likc hormone rcccptor- like 2 (EMR 2 ), lymphocyte antigen 75 (LY75), Glypican-3 (GPC3), Fc receptor-like 5 (FCRL5), MUC16, 5T4, 8H9, av
- MART-1 immuno globulin lambda-like polypeptide 1 (IGLL1), Hepatitis B Surface Antigen Binding Protein (HBsAg), viral capsid antigen (VCA), early antigen (EA), EBV nuclear antigen (EBNA), HHV-6 p41 early antigen, HHV-6B U94 latent antigen, HHV-6B p98 late antigen , cytomegalovirus (CMV) antigen, large T antigen, small T antigen, adenovirus antigen, respiratory syncytial virus (RSV) antigen, haemagglutinin (HA), neuraminidase (NA), parainfluenza type 1 antigen, parainfluenza type 2 antigen, parainfluenza ty pe 3 antigen, parainfluenza ty pe 4 antigen, Human Metapneum
- a CAR comprises a hinge or spacer domain.
- the hinge/spacer domain may comprise a truncated hinge/spacer domain (THD) the THD domain is a truncated version of a complete hinge/spacer domain (“CHD”).
- THD truncated hinge/spacer domain
- CHD complete hinge/spacer domain
- an extracellular domain is from or derived from (e g., comprises all or a fragment of) ErbB2, glycophorin A (GpA), CD2, CD3 delta, CD3 epsilon, CD3 gamma, CD4, CD7, CD8a, CD8[T CD1 la (IT GAL), CD1 lb (IT GAM), CD1 1c (ITGAX), CD1 Id (IT GAD), CD18 (ITGB2), CD19 (B4), CD27 (TNFRSF7), CD28, CD28T. CD29 (ITGB1). CD30 (TNFRSF8), CD40 (TNFRSF5).
- GpA glycophorin A
- CD2 CD3 delta
- CD3 epsilon CD3 gamma
- CD48 SLAMF2
- CD49a IGA1
- CD49d IGA4
- CD49f IGA6
- CD66a CEACAM1
- CD66b CEACAM8
- CD66c CEACAM6
- CD66d CEACAM3
- CD66e CEACAM5
- CD69 CLEC 2
- CD79A B-cell antigen receptor complex-associated alpha chain
- CD79B B-cell antigen receptor complex-associated beta chain
- CD84 SLAMF5
- CD96 Tactile).
- CD100 SEMA4D
- CD103 1TGAE
- CD134 (0X40
- CD137 4- 1BB
- CD150 SLAMF1
- CD158A KIR 2 DL1
- CD158B1 KIR 2 DL2
- CD158B1 KIR 2 DL2
- CD158B2 KIR 2 DL3), CD158C (KIR 3 DP1), CD158D (KIRDL4), CD158F1 (KIR 2 DL5A), CD158F2 (KIR 2 DL5B), CD158K (KIR 3 DL2), CD160 (BY55).
- CD162 SELPLG
- CD226 DNAM1
- CD229 SLAMF3
- CD244 SLAMF4
- CD247 CD3-zeta
- CD258 LIGHT
- CD268 BAFFR
- CD270 TNFSF14
- BTLA CD272
- CD276 (B7-H3), CD279 (PD-1), CD314 (NKG2D), CD319 (SLAMF7), CD335 (NK-p46), CD336 (NK-p44), CD337 (NK-p30), CD352 (SLAMF6), CD353 (SLAMF8), CD355 (CRT AM), CD357 (TNFRSF18), inducible T cell co-stimulator (ICOS), LFA-1 (CD1 la/CD18), NKG2C. DAP-10, ICAM-1, NKp80 (KLRF1), IL-2R beta, IL-2R gamma, IL-7R alpha. LFA-1, SLAMF9. LAT.
- GrpL GrpL
- SLP-76 LCP2
- PAG1/CBP PAG1/CBP
- CD83 ligand Fc gamma receptor, MHC class 1 molecule, MHC class 2 molecule, a TNF receptor protein, an immunoglobulin protein, a cytokine receptor, an integrin, activating NK cell receptors, a Toll ligand receptor, and fragments or combinations thereof.
- a hinge or spacer domain may be derived either from a natural or from a synthetic source.
- a hinge or spacer domain is positioned between an antigen binding molecule (e.g., an scFv) and a transmembrane domain. In this orientation, the hinge/spacer domain provides distance between the antigen binding molecule and the surface of a cell membrane on which the CAR is expressed.
- a hinge or spacer domain is from or is derived from an immunoglobulin.
- a hinge or spacer domain is selected from the hinge/spacer regions of IgGl, IgG2, IgG3, IgG4, IgA, IgD, IgE, IgM, or a fragment thereof.
- a hinge or spacer domain comprises, is from, or is derived from the hinge/spacer region of CD8 alpha. In some embodiments, a hinge or spacer domain comprises, is from, or is derived from the hinge/spacer region of CD28. In some embodiments, a hinge or spacer domain comprises a fragment of the hinge/spacer region of CD8 alpha or a fragment of the hinge/spacer region of CD28, wherein the fragment is any tiring less than the whole hinge/spacer region. In some embodiments, the fragment of the CD8 alpha hinge/spacer region or the fragment of the CD28 hinge/spacer region comprises an amino acid sequence that excludes at least 1, at least 2, at least 3.
- the CAR may further comprise a transmembrane domain and/or an intracellular signaling domain.
- the transmembrane domain may be designed to be fused to the extracellular domain of the CAR. It may similarly be fused to the intracellular domain of the CAR.
- the transmembrane domain that naturally is associated with one of the domains in a CAR is used.
- the transmembrane domain may be selected or modified (e.g., by an amino acid substitution) to avoid binding of such domains to the transmembrane domains of die same or different surface membrane proteins to minimize interactions with other members of the receptor complex.
- the transmembrane domain may be derived either from a natural or from a synthetic source. Where the source is natural, the domain may be derived from any membrane -bound or transmembrane protein.
- Transmembrane regions may be derived from (i.e., comprise) a receptor tyrosine kinase (e.g., ErbB2). glycophorin A (GpA), 4-1BB/CD137, activating NK cell receptors, an immunoglobulin protein, B7-H3, BAFFR, BFAME (SEAMF8), BTEA, CD100 (SEMA4D), CD103, CD160 (BY55), CD 18, CD 19, CD 19a, CD2, CD247, CD27, CD276 (B7-H3), CD28. CD29, CD3 delta. CD3 epsilon, CD3 gamma, CD30. CD4. CD40, CD49a, CD49D, CD49f, CD69.
- GpA glycophorin A
- 4-1BB/CD137 activating NK cell receptors
- B7-H3, BAFFR BFAME
- SEAMF8 BFAME
- BTEA BTEA
- CD100 SEMA4D
- CD103
- CDS. CEACAM1, CRT AM, cytokine receptor, DAP-10, DNAM1 (CD226). Fc gamma receptor, GADS. GITR, HVEM (EIGHTR), IA4.
- PAG/Cbp programmed death-1 (PD-1), PSGL1, SELPLG (CD162).
- SLAM proteins Signaling Lymphocytic Activation Molecules
- SLAMFL CD150; IPO-3
- SLAMF4 CD244; 2B4
- SLAMF6 NTB-A; Lyl08
- SLAMF7 SLP-76
- TNF receptor proteins TNFR 2 , TNFSF14, a Toll ligand receptor, TRANCE/RANKL, VLA1, or VLA-6, or a fragment, truncation, or a combination thereof.
- suitable intracellular signaling domain include, but are not limited to, activating Macrophage/Myeloid cell receptors CSFR1, MYD88, CD14, TIE2, TLR4, CR 3 .
- a receptor tyrosine kinase may be derived from (e.g., comprise) Insulin receptor (InsR), Insulin-like growth factor I receptor (IGF1R), Insulin receptor-related receptor (IRR), platelet derived growth factor receptor alpha (PDGFRa), platelet derived growth factor receptor beta (PDGFRfi).
- Insulin receptor Insulin receptor
- IGF1R Insulin-like growth factor I receptor
- IRR Insulin receptor-related receptor
- PDGFRa platelet derived growth factor receptor alpha
- PDGFRfi platelet derived growth factor receptor beta
- KIT proto-oncogene receptor tyrosine kinase Kit
- colony stimulating factor 1 receptor CSFR
- FLT3 fms related tyrosine kinase 3
- VGFR-1 kinase insert domain receptor
- VGFR-2 kinase insert domain receptor
- VFGFR-3 fibroblast growth factor receptor 1
- fibroblast growth factor receptor 2 FGFR 2
- fibroblast growth factor receptor 3 FGFR 3
- fibroblast growth factor receptor 4 FGFR4
- protein tyrosine kinase 7 CCK4
- CCK4 protein tyrosine kinase 7
- trkA neurotrophic receptor tyrosine kinase 1
- trkB neurotrophic receptor tyrosine kinase 2
- trkC neurotrophic receptor tyrosine kinase 3
- receptor tyrosine kinase like orphan receptor 1 ROR1
- receptor tyrosine kinase like orphan receptor 2 ROR 2
- MusSK muscle associated receptor tyrosine kinase
- MET receptor tyrosine kinase
- Rh macrophage stimulating 1 receptor
- Axl AXL receptor tyrosine kinase
- Tyro3 TYR03 protein tyrosine kinase
- TIE1 TEK receptor ty rosine kinase with immunoglobulin like and EGF like domains 1
- TIE2 EGF like domains 1
- EphA2 EPH receptor A2
- EPH receptor A3 Eph A3, EPH receptor A4 (EphA4)
- EPH receptor A5 EphA5
- EPH receptor A6 EphA6
- EPH receptor A7 EphA7
- EPH receptor A8 EphA8
- EPH receptor Alt EphAlO
- EPH receptor Bl EphBl
- EPH receptor B2 EphB2
- EPH receptor B3 EPH receptor B3
- EPH receptor B4 EphB4
- EPH receptor B6 EphB6
- Ret ret proto oncogene
- RYK receptor-like ty rosine kinase
- discoidin domain receptor tyrosine kinase 1 DDR1
- discoidin domain receptor tyrosine kinase 2 DDR 2
- ROS receptor tyrosine kinase
- Lmrl apoptosis associated ty rosine kinase
- Lmr2 lemur ty rosine kinase 2
- Lmr3 lemur tyrosine kinase 3
- Lmr3 leukocyte receptor tyrosine kinase
- LTK leukocyte receptor tyrosine kinase
- ALK receptor ty rosine kinase ALK receptor ty rosine kinase
- STYK serine/threonine/tyrosine kinase 1
- the CAR comprises a costimulatoiy domain.
- the costimulatoiy domain comprises 4-1BB (CD137), CD28, or both, and/or an intracellular T cell signaling domain.
- the costimulatoiy domain is human CD28, human 4-1BB, or both, and the intracellular T cell signaling domain is human CD3 zeta (Q. 4- IBB, CD28, CD3 zeta may comprise less than the whole 4-1BB, CD28 or CD3 zeta, respectively.
- Chimeric antigen receptors may incorporate costimulatory (signaling) domains to increase their potency. See U.S. Patent Nos.
- the intracellular (signaling) domain of the engineered T cells disclosed herein may provide signaling to an activating domain, which then activates at least one of the normal effector functions of die immune cell.
- Effector function of a T cell for example, may be cytolytic activity or helper activity including the secretion of cytokines.
- suitable intracellular signaling domain include (e.g., comprise), but are not limited to 4-1BB/CD137, activating NK cell receptors, an Immunoglobulin protein, B7-H3, BAFFR, BLAME (SLAMF8), BTLA, CD100 (SEMA4D), CD103, CD160 (BY55), CD18, CD19, CD 19a, CD2, CD247, CD27, CD276 (B7-H3), CD28, CD29, CD3 delta, CD3 epsilon, CD3 gamma, CD30, CD4, CD40, CD49a, CD49D, CD49f, CD69, CD7, CD84, CD8alpha, CD8beta, CD96 (Tactile), CD1 la, CD1 lb, CD1 1c, CD1 Id, CDS, CEACAM1, CRT AM, cytokine receptor, DAP-10, DNAM1 (CD226), Fc gamma receptor, GADS,
- Ig alpha (CD79a), IL- 2R beta, IL-2R gamma, IL-7R alpha, inducible T cell costimulator (ICOS), integrins, ITGA4, ITGA6, ITGAD, ITGAE, ITGAL, ITGAM, ITGAX, ITGB2, ITGB7, ITGB1, KIRDS2, LAT, LFA-1, ligand that specifically binds with CD83, LIGHT, LTBR, Ly9 (CD229), Lyl08, lymphocyte function- associated antigen- 1 (LFA-1; CDl-la/CD18), MHC class 1 molecule, NKG2C, NKG2D, NKp30, NKp44. NKp46. NKp80 (KLRF1).
- OX-40 PAG/Cbp. programmed death-1 (PD-1). PSGL1.
- SELPLG CD162
- SLAMF1 Signaling Lymphocytic Activation Molecules
- SLAMF1 SLAMF1; CD150; IPO-3
- SLAMF4 CD244; 2B4
- SLAMF6 NTB-A
- SLAMF7 SLP-76. TNF receptor proteins. TNFR 2 . TNFSF14. a Toll ligand receptor, TRANCE/RANKL, VLA1, or VLA-6. or a fragment, truncation, or a combination thereof.
- CD3 is an element of the T cell receptor on native T cells and has been shown to be an important intracellular activating element in CARs.
- the CD3 is CD3 zeta.
- the sequence encoding the CAR comprises a sequence from Table 4.
- TCR T-Cell Receptors
- TCRs are described using the International Immunogenetics (IMGT) TCR nomenclature, and links to the IMGT public database of TCR sequences.
- Native alpha-beta heterodimeric TCRs have an alpha chain and a beta chain.
- each chain may comprise variable, joining and constant regions, and the beta chain also usually contains a short diversity region between the variable and joining regions, but this diversity region is often considered as part of the joining region.
- Each variable region may comprise three CDRs (Complementarity Determining Regions) embedded in a framework sequence, one being the hypervariable region named CDR 3 .
- V ⁇ alpha chain variable
- VP beta chain variable
- the Va types are referred to in IMGT nomenclature by a unique TRAV number.
- TRAV21 defines a TCR Va region having unique framework and CDR1 and CDR 2 sequences, and a CDR 3 sequence which is partly defined by an amino acid sequence which is preserved from TCR to TCR, but which also includes an amino acid sequence which varies from TCR to TCR.
- TRBV5-1 defines a TCR VP region having unique framework and CDR1 and CDR 2 sequences, but with only a partly defined CDR 3 sequence.
- the joining regions of the TCR are similarly defined by the unique IMGT TRAJ and TRBJ nomenclature, and the constant regions by the IMGT TRAC and TRBC nomenclature.
- the beta chain diversity region is referred to in IMGT nomenclature by the abbreviation TRBD, and, as mentioned, the concatenated TRBD/TRBJ regions are often considered together as the joining region.
- TCRs exist in heterodimeric aP or y5 forms. However, recombinant TCRs consisting of aa or pp homodimers have previously been shown to bind to peptide MHC molecules. Therefore, the TCR of the present disclosure may be a heterodimeric aP TCR or may be an aa or pp homodimeric TCR.
- an aP heterodimeric TCR may, for example, be transfected as full- length chains having both cytoplasmic and transmembrane domains.
- TCRs of die present disclosure may have an introduced disulfide bond between residues of the respective constant domains, as described, for example, in WO 2006/000830.
- TCRs of the present disclosure may comprise an alpha chain TRAC constant domain sequence and/or a beta chain TRBC 1 or TRBC 2 constant domain sequence.
- the alpha and beta chain constant domain sequences may be modified by truncation or substitution to delete the native disulfide bond betw een Cys4 of exon 2 of TRAC and Cys2 of exon 2 of TRBC1 or TRBC 2 .
- the alpha and/or beta chain constant domain sequence(s) may also be modified by substitution of cysteine residues for Thr 48 of TRAC and Ser 57 of TRBC1 or TRBC 2 , the said cysteines forming a disulfide bond betw een the alpha and beta constant domains of the TCR.
- Binding affinity (inversely proportional to the equilibrium constant K D ) and binding half-life (expressed as T1 ⁇ 2) can be determined by any appropriate method. It will be appreciated that doubling die affinity of a TCR results in halving the K D . T!4 is calculated as In 2 divided by the off-rate (koff). Therefore, doubling of T!4 results in a halving in koff. Ku and koff values for TCRs are usually measured for soluble forms of the TCR, i.e., those forms which are truncated to remove cytoplasmic and transmembrane domain residues.
- a given TCR has an improved binding affinity for, and/or a binding half-life for the parental TCR if a soluble form of that TCR has the said characteristics.
- the binding affinity or binding half-life of a given TCR is measured several times, for example 3 or more times, using the same assay protocol, and an average of the results is taken.
- the present disclosure includes a non-naturally occurring and/or purified and/or or engineered cell, especially a T-cell, presenting a TCR of the present disclosure.
- nucleic acid such as DNA, cDNA or RNA
- T cells expressing the TCRs of the present disclosure will be suitable for use in adoptive therapy-based treatment of cancers such as those of the pancreas and liver.
- suitable methods by which adoptive therapy can be carried out see for example Rosenberg et al., (2008) Nat Rev Cancer 8(4): 299-308).
- TCRs of the present disclosure may be subject to post-translational modifications when expressed by transfected cells.
- Glycosylation is one such modification, which may comprise the covalent attachment of oligosaccharide moieties to defined amino acids in the TCR chain.
- asparagine residues, or serine/threonine residues are well-known locations for oligosaccharide attachment.
- the glycosylation status of a particular protein depends on a number of factors, including protein sequence, protein conformation and the availability of certain enzymes. Furthermore, glycosylation status (i.e.. oligosaccharide type, covalent linkage and total number of attachments) can influence protein function.
- Glycosylation of transfected TCRs may be controlled by mutations of the transfected gene (Kuball J et al. (2009). J Exp Med 206(2):463-475). Such mutations are also encompassed in this disclosure.
- a TCR may be specific for an antigen in tire group MAGE-A1, MAGE-A2, MAGE -A3, MAGE-A4, MAGE-A5, MAGE-A6, MAGE-A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, MAGE-A12, MAGE-A13, GAGE-1, GAGE-2, GAGE-3, GAGE-4, GAGE-5, GAGE-6, GAGE-7, GAGE-8, BAGE-1, RAGE-1, LB33/MUM-1, PRAME, NAG, MAGE-Xp2 (MAGE-B2), MAGE-Xp3 (MAGE-B3), MAGE-Xp4 (AGE-B4), tyrosinase, brain glycogen phosphorylase, Melan-A, MAGE-CI, MAGE-C 2 , NY-ESO-1, LAGE-1, SSX-1.
- SSX-2 (HOM-MEL-40), SSX-1, SSX-4, SSX-5.
- SCP-1 CT- 7. alpha-actinin-4.
- Bcr-Abl fusion protein Casp-8, beta-catenin, cdc27, cdk4, cdkn2a, coa-1, dek-can fusion protein, EF2.
- HLA-A11, hsp70-2, KIAAO205 HLA-A11, hsp70-2, KIAAO205.
- MYL-RAR Epstein Barr virus antigens
- EBNA human papillomavirus
- HPV human papillomavirus
- c-met nm-23Hl, PSA, TAG-72-4.
- a- fetoprotein 13HCG, BCA225, BTAA, CA 125.
- CA 15-3 CA 27.29 ⁇ BCAA
- CA 195 CA 242, CA- 50, CAM43.
- BCR B-Cell Receptors
- B-cell receptors or B-cell antigen receptors are immunoglobulin molecules that form a type I transmembrane protein on the surface of a B cell.
- a BCR is capable of transmitting an activatory signal into a B cell following recognition of a specific antigen. Prior to binding of a B cell to an antigen, the BCR will remain in an unstimulated or “resting” stage. Binding of an antigen to a BCR leads to signaling that initiates a humoral immune response.
- a BCR is expressed by mature B cells. These B cells work with immunoglobulins (Igs) in recognizing and tagging pathogens.
- the typical BCR comprises a membrane-bound immunoglobulin (e.g., mlgA, mlgD, mlgE, mlgG, and mlgM), along with associated and Iga/IgP (CD79a/CD79b) heterodimers (a/p).
- membrane-bound immunoglobulins are tetramers consisting of two identical heavy and two light chains.
- the membrane bound immunoglobulins is capable of responding to antigen binding by signal transmission across the plasma membrane leading to B cell activation and consequently clonal expansion and specific antibody production (Friess M el al. (2016), Front. Immunol. 2947(9)).
- the Iga/IgP heterodimers is responsible for transducing signals to the cell interior.
- a Igot/IgP heterodimer signaling relies on the presence of immunoreceptor tyrosine-based activation motifs (ITAMs) located on each of the cytosolic tails of the heterodimers.
- ITAMs immunoreceptor tyrosine-based activation motifs
- ITAMs comprise two tyrosine residues separated by 9-12 amino acids (e.g., tyrosine, leucine, and/or valine).
- tyrosine of the BCR’s ITAMs Upon binding of an antigen, the tyrosine of the BCR’s ITAMs become phosphorylated by Src-family tyrosine kinases Blk, Fyn, or Lyn (Janeway C etal., Immunobiology: The Immune System in Health and Disease (Garland Science, 5th ed. 2001)).
- the circular RNA polynucleotide may encode for a various number of other chimeric proteins available in the art.
- the chimeric proteins may include recombinant fusion proteins, chimeric mutant protein, or other fusion proteins.
- the circular RNA polynucleotide encodes for an immune modulatory ligand.
- the immune modulatory ligand may be immuno stimulatory; while in other embodiments, the immune modulatory ligand may be immunosuppressive.
- the circular RNA polynucleotide encodes for a cytokine.
- the cytokine comprises a chemokine, interferon, interleukin, lymphokine, and tumor necrosis factor.
- Chemokines are chemotactic cytokine produced by a variety of cell types in acute and chronic inflammation that mobilizes and activates white blood cells.
- An interferon comprises a family of secreted a-helical cytokines induced in response to specific extracellular molecules through stimulation of TLRs (Borden, Molecular Basis of Cancer (Fourth Edition) 2015).
- Interleukins are cytokines expressed by leukocytes.
- the circular RNA polynucleotide may encode for a transcription factor.
- Regulatory T cells are important in maintaining homeostasis, controlling the magnitude and duration of the inflammatory response, and in preventing autoimmune and allergic responses.
- Tregs are thought to be mainly involved in suppressing immune responses, functioning in part as a “self-check” for the immrme system to prevent excessive reactions.
- Tregs are involved in maintaining tolerance to self-antigens, harmless agents such as pollen or food, and abrogating autoimmune disease.
- Tregs are found throughout the body including, without limitation, the gut, skin, lung, and liver. Additionally, Treg cells may also be found in certain compartments of the body that are not directly exposed to the external environment such as the spleen, lymph nodes, and even adipose tissue. Each of these Treg cell populations is known or suspected to have one or more unique features and additional information may be found in Lehtimaki and Lahesmaa, Regulatory' T cells control immune responses through their non-redundant tissue specific features, 2013, Frontiers in Immunol., 4(294): 1-10, the disclosure of which is hereby incorporated in its entirety.
- Tregs are knoyvn to require TGF- ⁇ and IL-2 for proper activation and development.
- Tregs expressing abundant amounts of the IL-2 receptor (IL-2R), arc reliant on IL -2 produced byactivated T cells.
- Tregs are known to produce both IL- 10 and TGF-P, both potent immune suppressive cytokines.
- Tregs are knoyvn to inhibit the ability- of antigen presenting cells (APCs) to stimulate T cells.
- APCs antigen presenting cells
- CTLA-4 may bind to B7 molecules on APCs and either block these molecules or remove them by causing internalization resulting in reduced availability of B7 and an inability to provide adequate co-stimulation for immune responses. Additional discussion regarding the origin, differentiation and function of Tregs may be found in Dhamne et al., Peripheral and thymic Foxp3+ regulatory T cells in search of origin, distinction, and function. 2013, Frontiers in Immunol., 4 (253): 1-11. the disclosure of which is hereby incorporated in its entirety.
- the coding element of the circular RNA polynucleotide encodes for one or more checkpoint inhibitors or agonists.
- the immune checkpoint inhibitor is an inhibitor of Programmed Death- Ligand 1 (PD-L1, also known as B7-H1, CD274), Programmed Death 1 (PD-1). CTLA-4, PD-L2 (B7- DC.
- PD-L1 Programmed Death- Ligand 1
- B7-H1, CD274 Programmed Death 1
- CTLA-4 PD-L2
- B7- DC PD-L2
- CD273 LAG3, TIM3, 2B4, A2aR, B7H1, B7H3, B7H4, BTLA, CD2, CD27, CD28, CD30, CD40, CD70, CD80, CD86, CD137, CD160, CD226, CD276, DR 3 , GAL9, GITR, HAVCR 2 , HVEM, IDO1, IDO2, ICOS (inducible T cell co-stimulator), KIR, LAIR1, LIGHT, MARCO (macrophage receptor yvith a collagenous structure), PS (phosphatidylserine), OX-40, SLAM. TIGHT, VISTA, VTCN1, or any combinations thereof.
- the immune checkpoint inhibitor is an inhibitor of IDO1, CTLA4, PD-1, LAG3, PD-L1, TIM3, or combinations thereof. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-L1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of CTLA-4. In some embodiments, the immune checkpoint inhibitor is an inhibitor of L AG3. In some embodiments, the immune checkpoint inhibitor is an inhibitor of TIM3. In some embodiments, the immune checkpoint inhibitor is an inhibitor of IDOL
- the present disclosure encompasses the use of immune checkpoint antagonists.
- immune checkpoint antagonists include antagonists of immune checkpoint molecules such as Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4), Programmed Cell Death Protein 1 (PD-1), Programmed Death-Ligand 1 (PDL-1), Lymphocyte- activation gene 3 (LAG-3), and T-cell immunoglobulin and mucin domain 3 (TIM-3).
- CTLA-4 Cytotoxic T-Lymphocyte Antigen 4
- PD-1 Programmed Cell Death Protein 1
- PDL-1 Programmed Death-Ligand 1
- LAG-3 Lymphocyte- activation gene 3
- TIM-3 T-cell immunoglobulin and mucin domain 3
- An antagonist of CTLA-4, PD-1, PDL-1, LAG-3, or TIM-3 interferes with CTLA-4, PD-1, PDL-1, LAG-3, or TIM-3 function, respectively.
- Such antagonists of CTLA-4, PD-1, PDL-1, LAG-3, and TIM -3 can include antibodies which specifically bind to CTLA-4, PD-1, PDL-1, LAG-3, and TIM-3, respectively and inhibit and/or block biological activity and function.
- the pay load encoded within one or more of the coding elements is a hormone, FC fusion protein, anticoagulant, blood clotting factor, protein associated with deficiencies and genetic disease, a chaperone protein, an antimicrobial protein, an enzy me (e.g., metabolic enzyme), a structural protein (e.g., a channel or nuclear pore protein), protein variant, small molecule, antibody, nanobody, an engineered non-body antibody, or a combination thereof.
- DNA templates can be made using standard techniques of molecular biology .
- the various elements of the vectors provided herein can be obtained using recombinant methods, such as by screening cDNA and genomic libraries from cells, or by deriving the polynucleotides from a DNA template known to include the same.
- the various elements of the DNA template can also be produced synthetically, rather than cloned, based on the known sequences.
- the complete sequence can be assembled from overlapping oligonucleotides prepared by standard methods and assembled into the complete sequence. See. e.g., Edge, Nature (1981) 292:756; Nambair et al., Science (1984) 223: 1299; and Jay et al., J. Biol. Chem. (1984) 259:631 1.
- nucleotide sequences can be obtained from DNA template harboring the desired sequences or synthesized completely, or in part, using various oligonucleotide synthesis techniques known in the art, such as site-directed mutagenesis and polymerase chain reaction (PCR) techniques where appropriate.
- oligonucleotide synthesis techniques known in the art, such as site-directed mutagenesis and polymerase chain reaction (PCR) techniques where appropriate.
- PCR polymerase chain reaction
- One method of obtaining nucleotide sequences encoding the desired DNA template elements is by annealing complementary sets of overlapping synthetic oligonucleotides produced in a conventional, automated polynucleotide synthesizer, followed by ligation with an appropriate DNA ligase and amplification of the ligated nucleotide sequence via PCR. See. e.g., Jayaraman et al., Proc. Natl.
- oligonucleotide- directed synthesis Jones et al., Nature (1986) 54:75-82
- oligonucleotide directed mutagenesis of preexisting nucleotide regions Riechmann et al., Nature (1988) 332:323-327 and Verhoeyen et al., Science (1988) 239: 1534-1536
- enzymatic filling-in of gapped oligonucleotides using T4 DNA polymerase Queen et al., Proc. Natl. Acad. Sci. USA (1989) 86: 10029-10033
- the precursor RNA can be generated by incubating a DNA template under conditions permissive of transcription of the precursor RNA encoded by the DNA template.
- a precursor RNA is synthesized by incubating a DNA template provided herein that comprises an RNA polymerase promoter upstream of its 5’ duplex sequence and/or expression sequences with a compatible RNA polymerase enzyme under conditions permissive of in vitro transcription.
- the DNA template is incubated inside of a cell by a bacteriophage RNA polymerase or in the nucleus of a cell by host RNA polymerase II.
- compositions comprising a therapeutic agent provided herein.
- the therapeutic agent is a circular RNA polynucleotide.
- the therapeutic agent is a vector.
- the therapeutic agent is a cell comprising a circular RNA or vector (e.g., a human cell, such as a human T cell).
- the composition further comprises a pharmaceutically acceptable carrier.
- compositions provided herein comprise a therapeutic agent provided herein in combination with other pharmaceutically active agents or drugs, such as antiinflammatory drugs or antibodies capable of targeting B cell antigens, e.g., anti-CD20 antibodies, e.g., rituximab.
- other pharmaceutically active agents or drugs such as antiinflammatory drugs or antibodies capable of targeting B cell antigens, e.g., anti-CD20 antibodies, e.g., rituximab.
- the pharmaceutically acceptable carrier can be any of those conventionally used and is limited only by chemical-physical considerations, such as solubility and lack of reactivity with the active agent(s), and by the route of administration.
- the pharmaceutically acceptable carriers described herein, for example, vehicles, adjuvants, excipients, and diluents, are well-known to those skilled in the art and are readily available to the public. It is preferred drat the pharmaceutically acceptable carrier be one which is chemically inert to the therapeutic agent(s) and one which has no detrimental side effects or toxicity under the conditions of use.
- the pharmaceutical composition comprises a preservative.
- suitable preservatives may include, for example, methylparaben, propylparaben, sodium benzoate, and benzalkonium chloride.
- a mixture of two or more preservatives may be used.
- the preservative or mixtures thereof are typically present in an amount of 0.0001% to 2% by weight of the total composition.
- the pharmaceutical composition comprises a buffering agent.
- suitable buffering agents may include, for example, citric acid, sodium citrate, phosphoric acid, potassium phosphate, and various other acids and salts.
- a mixture of tw o or more buffering agents optionally may be used.
- the buffering agent or mixtures thereof are ty pically present in an amount of 0.001% to 4% by weight of the total composition.
- the concentration of therapeutic agent in the pharmaceutical composition can vary, e.g., less than 1%, or at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9% 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, or 50% or more by weight, and can be selected primarily by fluid volumes, and viscosities, in accordance with the particular mode of administration selected.
- compositions for oral, aerosol, parenteral (e.g., subcutaneous, intravenous, intraarterial, intramuscular, intradermal, intraperitoneal, and intrathecal), and topical administration are merely exemplary' and are in no way limiting. More than one route can be used to administer the therapeutic agents provided herein, and in certain instances, a particular route can provide a more immediate and more effective response than another route.
- Formulations suitable for oral administration can comprise or consist of (a) liquid solutions, such as an effective amount of the therapeutic agent dissolved in diluents, such as water, saline, or orange juice; (b) capsules, sachets, tablets, lozenges, and troches, each containing a predetermined amount of the active ingredient, as solids or granules; (c) powders; (d) suspensions in an appropriate liquid; and (e) suitable emulsions.
- Liquid formulations may include diluents, such as water and alcohols, for example, ethanol, benzy l alcohol and the polyethylene alcohols, either with or without the addition of a pharmaceutically acceptable surfactant.
- Capsule forms can be of the ordinary' hard- or soft-shelled gelatin type containing, for example, surfactants, lubricants, and inert fillers, such as lactose, sucrose, calcium phosphate, and com starch.
- Tablet forms can include one or more of lactose, sucrose, mannitol, corn starch, potato starch, alginic acid, microcrystalline cellulose, acacia, gelatin, guar gum, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, calcium stearate, zinc stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, disintegrating agents, moistening agents, preservatives, flavoring agents, and other pharmacologically compatible excipients.
- Lozenge forms can comprise the therapeutic agent with a flavorant, usually sucrose, acacia or tragacanth.
- Pastilles can comprise the therapeutic agent with an inert base, such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to, such excipients as are known in the art.
- an inert base such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to, such excipients as are known in the art.
- Formulations suitable for parenteral administration include aqueous and nonaqueous isotonic sterile injection solutions, which can contain antioxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the intended recipient, and aqueous and nonaqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives.
- the therapeutic agents provided herein can be administered in a physiologically acceptable diluent in a pharmaceutical carrier, such as a sterile liquid or mixture of liquids including water, saline, aqueous dextrose and related sugar solutions, an alcohol such as ethanol or hexadecyl alcohol, a glycol such as propylene glycol or polyethylene glycol, dimethylsulfoxide, glycerol, ketals such as 2,2-dimethyl-l,3-dioxolane-4-methanol, ethers, poly(ethyleneglycol) 400, oils, fatty acids, fatty acid esters or glycerides, or acety lated fatty acid glycerides with or without the addition of a pharmaceutically acceptable surfactant such as a soap or a detergent, suspending agent such as pectin, carbomers, methylcellulose, hydroxypropylmethylcellulose, or carboxymethylcellulose, or emulsifying agents and other pharmaceutical adjuvants.
- Oils which can be used in parenteral formulations in some embodiments, include petroleum, animal oils, vegetable oils, or synthetic oils. Specific examples of oils include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral oil.
- Suitable fatty' acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters.
- Suitable soaps for use in certain embodiments of parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts
- suitable detergents include (a) cationic detergents such as, for example, dimethyl dialkyl ammonium halides, and alkyl pyridinium halides, (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyd, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine oxides, fatty acid alkanolamides, and polyoxyethylenepolypropylene copolymers, (d) amphoteric detergents such as, for example, alkyl-p-aminopropionates, and 2-alkyl-imidazoline quaternary ammonium salts, and (e) mixtures thereof.
- the parenteral formulations will contain, for example, from 0.5% to 25% by weight of the therapeutic agent in solution. Preservatives and buffers may be used. In order to minimize or eliminate irritation at the site of injection, such compositions may contain one or more nonionic surfactants having, for example, a hydrophile-lipophile balance (HLB) of from 12 to 17. The quantity of surfactant in such formulations will ty pically range, for example, from 5% to 15% by weight.
- HLB hydrophile-lipophile balance
- Suitable surfactants include polyethylene glycol, sorbitan, fatty acid esters such as sorbitan monooleate, and high molecular weight adducts of ethylene oxide with a hydrophobic base formed by the condensation of propylene oxide with propylene glycol.
- the parenteral formulations can be presented in unit-dose or multi-dose sealed containers, such as ampoules or vials, and can be stored in a freeze- dried (lyophilized) condition requiring only the addition of a sterile liquid excipient, for example, water for injections, immediately prior to use. Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets of the kind previously described.
- injectable formulations are provided herein.
- the requirements for effective pharmaceutical carriers for injectable compositions are well-known to those of ordinary skill in the art (see, e.g., Pharmaceutics and Pharmacy Practice, I.B. Lippincott Company, Philadelphia, PA, Banker and Chalmers, eds., pages 238-250 (1982), and ASHP Handbook on Injectable Drugs, Toissel, 4th ed, pages 622-630 (1986)).
- the therapeutic agents provided herein are formulated in tune-released, delayed release, or sustained release delivery systems such that the delivery of the composition occurs prior to, and with sufficient time to, cause sensitization of the site to be treated. Such systems can avoid repeated administrations of the therapeutic agent, thereby increasing convenience to the subject and the physician, and may be particularly suitable for certain composition embodiments provided herein.
- the compositions of the invention are formulated such that they are suitable for extended release of the circRNA contained therein. Such extended-release compositions may be conveniently administered to a subject at extended dosing intervals. For example, in one embodiment, the compositions of the present invention are administered to a subject twice a day, daily or every other day.
- compositions of the present invention are administered to a subject twice a week, once a week, every ten days, every two weeks, every three weeks, every four weeks, once a month, every six weeks, every eight weeks, every three months, every four months, every six months, every eight months, every nine months or annually.
- a protein encoded by a circRNA is produced by a target cell for sustained amounts of time.
- the protein may be produced for more than one hour, more than four, more than six, more than 12, more than 24, more than 48 hours, or more than 72 hours after administration.
- the polypeptide is expressed at a peak level six hours after administration.
- the expression of the polypeptide is sustained at least at a therapeutic level.
- the polypeptide is expressed at least at a therapeutic level for more than one, more than four, more than six. more than 12, more than 24, more than 48, or more than 72 hours after administration.
- the polypeptide is detectable at a therapeutic level in patient tissue (e.g., liver or lung).
- the level of detectable polypeptide is from continuous expression from the circRNA composition over periods of time of more than one. more than four, more than six, more than 12, more than 24, more than 48, or more than 72 hours after administration.
- a protein encoded by circRNA is produced at levels above normal physiological levels.
- the level of protein may be increased as compared to a control.
- the control is the baseline physiological level of the polypeptide in a nonnal individual or in a population of normal individuals.
- the control is the baseline physiological level of the polypeptide in an individual having a deficiency in the relevant protein or polypeptide or in a population of individuals having a deficiency in the relevant protein or polypeptide.
- the control can be the normal level of the relevant protein or polypeptide in the individual to whom the composition is administered.
- the control is the expression level of die polypeptide upon other therapeutic intervention, e.g., upon direct injection of the corresponding polypeptide, at one or more comparable time points.
- the method yields a sustained circulation half-life of a protein encoded by a circRNA.
- the protein may be detected for hours or days longer than the half-life observed via subcutaneous injection of the protein or mRNA encoding the protein.
- the half-life of the protein is 1 day, 2 days, 3 days, 4 days, 5 days, or 1 week or more.
- release delivery systems are available and known to those of ordinary skill in the art. They include polymer-based systems such as poly(lactide-glycolide), copolyoxalates, polycaprolactones, polyesteramides, polyorthoesters, polyhydroxybutyric acid, and polyanhydrides. Microcapsules of the foregoing polymers containing drugs are described in, for example, U.S. Patent 5,075.109.
- Delivery systems also include non-polymer systems that are lipids including sterols such as cholesterol, cholesterol esters, and fatty' acids or neutral fats such as mono-di-and tri-glycerides; hydrogel release systems; sylastic systems; peptide based systems: wax coatings; compressed tablets using conventional binders and excipients; partially fused implants; and the like.
- lipids including sterols such as cholesterol, cholesterol esters, and fatty' acids or neutral fats such as mono-di-and tri-glycerides
- hydrogel release systems such as sterols such as cholesterol, cholesterol esters, and fatty' acids or neutral fats such as mono-di-and tri-glycerides
- sylastic systems such as sterols such as cholesterol, cholesterol esters, and fatty' acids or neutral fats such as mono-di-and tri-glycerides
- sylastic systems such as sterols such as cholesterol, cholesterol esters, and fatty' acids or
- pump-based hardware delivery systems can be used, some of which are adapted for implantation.
- the therapeutic agent can be conjugated either directly or indirectly through a linking moiety to a targeting moiety.
- Methods for conjugating therapeutic agents to targeting moictics is known in the art. See, for instance, Wadwa ct al., J. Drug Targeting 3:111 (1995) and U.S. Patent 5,087,616.
- the therapeutic agents provided herein are formulated into a depot form, such that the manner in which the therapeutic agent is released into the body to which it is administered is controlled with respect to time and location within the body (see, for example, U.S. Patent 4.450, 150).
- Depot forms of therapeutic agents can be, for example, an implantable composition comprising the therapeutic agents and a porous or non-porous material, such as a poly mer, wherein the therapeutic agents are encapsulated by or diffused throughout the material and/or degradation of the non-porous material.
- the depot is then implanted into the desired location within the body and the therapeutic agents are released from the implant at a predetermined rate.
- the present disclosure also contemplates the discriminatory targeting of target cells and tissues by both passive and active targeting means.
- the phenomenon of passive targeting exploits tire natural distributions patterns of a transfer vehicle in vivo without relying upon the use of additional excipients or means to enhance recognition of the transfer vehicle by target cells.
- transfer vehicles which are subject to phagocytosis by the cells of the reticulo-endothelial system are likely to accumulate in the liver or spleen, and accordingly may provide a means to passively direct the delivery of the subject compositions to such target cells.
- the present disclosure contemplates active targeting, which involves the use of targeting moieties that may be bound (either covalently or non-covalently) to the lipid nanoparticle to encourage localization of such at certain target cells or target tissues.
- targeting may be mediated by the inclusion of one or more endogenous targeting moieties in or on the lipid nanoparticle to encourage distribution to the target cells or tissues.
- the composition can comprise a moiety capable of enhancing affinity of the composition to the target cell.
- Targeting moieties may be linked to the outer layer of the lipid nanoparticle during formulation or post-formulation.
- some lipid nanoparticle formulations may employ fusogenic polymers such as PEAA. hemagluttinin, other lipopeptides (see U.S. patent application Ser.
- compositions of the present disclosure demonstrate improved transfection efficacies, and/or demonstrate enhanced selectivity’ towards target cells or tissues of interest.
- Contemplated therefore are compositions which comprise one or more moieties (e.g., peptides, aptamers, oligonucleotides, vitamins or other molecules) that are capable of enhancing the affinity of the compositions and their nucleic acid contents for the target cells or tissues.
- moieties may optionally be bound or linked to the surface of the nanoparticle.
- the targeting moiety may span the surface of a nanoparticle or be encapsulated within tire nanoparticle.
- Suitable moieties are selected based upon their physical, chemical or biological properties (e.g., selective affinity and/or recognition of target cell surface markers or features). Cell-specific target sites and their corresponding targeting ligand can vary’ widely. Suitable targeting moieties are selected such that the unique characteristics of a target cell are exploited, thus allowing the composition to discriminate between target and non-target cells.
- compositions of the present disclosure may include surface markers (e.g., apolipoprotein-B (APOB) or apolipoprotein-E (APOE)) that selectively enhance recognition of, or affinity' to hepatocytes (e.g., by receptor-mediated recognition of and binding to such surface markers).
- surface markers e.g., apolipoprotein-B (APOB) or apolipoprotein-E (APOE)
- APOB apolipoprotein-B
- APOE apolipoprotein-E
- the use of galactose as a targeting moiety would be expected to direct the compositions of the present disclosure to parenchymal hepatocytes, or alternatively the use of mannose containing sugar residues as a targeting ligand would be expected to direct the compositions of the present disclosure to liver endothelial cells (e.g., mannose containing sugar residues that may bind preferentially to the asialoglycoprotein receptor present in hepatocytes).
- liver endothelial cells e.g., mannose containing sugar residues that may bind preferentially to the asialoglycoprotein receptor present in hepatocytes.
- targeting moieties that have been conjugated to moieties present in the lipid nanoparticle composition therefore facilitate recognition and uptake of the compositions of the present disclosure in target cells and tissues.
- suitable targeting moieties include one or more peptides, proteins, aptamers, vitamins and oligonucleotides.
- a LNP composition comprises a targeting moiety.
- the targeting moiety mediates receptor-mediated endocytosis selectively into a specific population of cells.
- the targeting moiety is operably connected, or linked, to the transfer vehicle.
- the targeting moiety is capable of binding to an immune cell antigen.
- the targeting moiety is capable of binding to a T cell antigen.
- Exemplary T cell antigens include, but are not limited to, CD2, CD3, CD5, CD7, CD8, CD4, beta7 integrin, beta2ingetrin, and ClqR.
- the targeting moiety is capable of binding to a NK, NKT. or macrophage antigen.
- the targeting moiety is capable of binding to a protein selected from CD3, CD4, CD8. PD-1, 4-1BB. and CD2.
- the targeting moiety is a single chain Fv (scFv) fragment, nanobody, peptide, peptide-based macrocycle, minibody, heavy chain variable region, light chain variable region or fragment thereof.
- the targeting moiety is selected from T-cell receptor motif antibodies, T-cell a chain antibodies, T-cell ⁇ chain antibodies. T-cell y chain antibodies. T-cell 5 chain antibodies.
- CD5 antibodies, CD7 antibodies, CD8 antibodies, CDl lb antibodies, CDl lc antibodies are examples of T-cell receptor motif antibodies.
- the targeting moiety is a small molecule binder of an ectoenzy me on lymphocytcs.
- Small molecule binders of ectoenzy mes include A2A inhibitors CD73 inhibitors, CD39 or adesine receptors A2aR and A2bR.
- Potential small molecules include AB928.
- the immune cell represents the target cell.
- the compositions of the present disclosure transfect the target cells on a discriminatory basis (i.e., do not transfect non-target cells).
- the compositions of the present disclosure may also be prepared to preferentially target a variety of target cells, which include, but are not limited to, T cells, B cells, macrophages, and dendritic cells.
- the target cells are deficient in a protein or enzyme of interest.
- the hepatocyte represents the target cell.
- the compositions of the present disclosure transfect the target cells on a discriminatory basis (i.e., do not transfect non-target cells).
- compositions of the present disclosure may also be prepared to preferentially target a variety of target cells, which include, but arc not limited to, hepatocytes, epithelial cells, hematopoietic cells, epithelial cells, endothelial cells, lung cells, bone cells, stem cells, mesenchymal cells, neural cells (e.g., meninges, astrocytes, motor neurons, cells of the dorsal root ganglia and anterior hom motor neurons), photoreceptor cells (e.g., rods and cones), retinal pigmented epithelial cells, secretory cells, cardiac cells, adipocytes, vascular smooth muscle cells, cardiomyocytes, skeletal muscle cells, beta cells, pituitary cells, synovial lining cells, ovarian cells, testicular cells, fibroblasts, B cells, T cells, reticulocytes, leukocytes, granulocytes and tumor cells.
- target cells include, but arc not limited to, hepatocyte
- compositions of the present disclosure may be prepared to preferentially distribute to target cells such as in the heart, lungs, kidneys, liver, and spleen.
- the compositions of the present disclosure distribute into the cells of the liver or spleen to facilitate the delivery and the subsequent expression of the circRNA comprised therein by the cells of the liver (e.g., hepatocytes) or the cells of spleen (e.g., immune cells).
- the targeted cells may function as a biological “reservoir” or “depot” capable of producing, and systemically excreting a functional protein or enzyme.
- the transfer vehicle may target hepatocytes or immune cells and/or preferentially distribute to the cells of the liver or spleen upon deliver,'.
- the circRNA loaded in the nanoparticle are translated and a functional protein product is produced, excreted and systemically distributed.
- cells other than hepatocytes e.g., lung, spleen, heart, ocular, or cells of the central nervous system
- the compositions of the present disclosure facilitate a subject's endogenous production of one or more functional proteins and/or enzymes.
- the lipid nanoparticles comprise circRNA which encode a deficient protein or enzyme.
- the exogenous circRNA loaded into the nanoparticle may be translated in vivo to produce a functional protein or enzyme encoded by the exogenously administered circRNA (e.g., a protein or enzyme in which the subject is deficient).
- compositions of the present disclosure exploit a subject's ability to translate exogenously- or recombinantly-prepared circRNA to produce an endogenously-translated protein or enzyme, and thereby produce (and where applicable excrete) a functional protein or enzyme.
- the expressed or translated proteins or enzymes may also be characterized by the in vivo inclusion of native post-translational modifications which may often be absent in recomb inantly -prepared proteins or enzymes, thereby further reducing the immunogenicity of the translated protein or enzyme.
- circRNA encoding a deficient protein or enzyme avoids the need to deliver the nucleic acids to specific organelles within a target cell. Rather, upon transfection of a target cell and deliver ⁇ ' of the nucleic acids to the cytoplasm of the target cell, the circRNA contents of a transfer vehicle may be translated and a functional protein or enzy me expressed.
- a circular RNA comprises one or more miRNA binding sites.
- a circular RNA comprises one or more miRNA binding sites recognized by miRNA present in one or more non-target cells or non-target cell types (e.g., Kupffer cells or hepatic cells) and not present in one or more target cells or target cell types (e.g., hepatocytes or T cells).
- a circular RNA comprises one or more miRNA binding sites recognized by miRNA present in an increased concentration in one or more non-target cells or non-target cell types (e.g., Kupffer cells or hepatic cells) compared to one or more target cells or target cell types (e.g., hepatocytes or T cells). miRNAs are thought to function by pairing with complementary' sequences within RNA molecules, resulting in gene silencing.
- the compositions of the present disclosure transfect or distribute to target cells on a discriminatory basis (i.e., do not transfect non-target cells).
- the compositions of the present disclosure may also be prepared to preferentially target a variety of target cells, which include, but arc not limited to, hepatocytes, epithelial cells, hematopoietic cells, epithelial cells, endothelial cells, lung cells, bone cells, stem cells, mesenchymal cells, neural cells (e.g., meninges, astrocytes, motor neurons, cells of the dorsal root ganglia and anterior horn motor neurons), photoreceptor cells (e.g., rods and cones), retinal pigmented epithelial cells, secretory cells, cardiac cells, adipocytes, vascular smooth muscle cells, cardiomyocytes, skeletal muscle cells, beta cells, pituitary' cells, synovial lining cells, ovarian cells, testicular cells, fibroblast
- provided herein is a method of producing a protein of interest in a subject in need thereof by' introducing or administering an effective amount of a pharmaceutical composition described herein comprising at least one lipid nanoparticle described herein.
- provided herein is a method of treating and/or preventing a condition comprising administering an effective amount of a pharmaceutical composition described herein comprising at least one LNP as described herein.
- the pharmaceutical compositions described herein are co-administered with one or more additional therapeutic agents (e.g., in the same pharmaceutical composition or in separate pharmaceutical compositions).
- die pharmaceutical compositions provided herein can be administered first and the one or more additional therapeutic agents can be administered second, or vice versa.
- die pharmaceutical compositions provided herein and the one or more additional therapeutic agents can be administered simultaneously.
- the subject is a mammal.
- the mammal referred to herein can be any mammal, including, but not limited to, mammals of the order Rodentia, such as mice and hamsters, or mammals of the order Logomorpha, such as rabbits.
- the mammals may be from the order Carnivora, including Felines (cats) and Canines (dogs).
- the mammals may be from the order Artiodactyla, including Bovines (cows) and Swines (pigs), or of the order Perssodactyla, including Equines (horses).
- the mammals may be of the order Primates, Ceboids, or Simoids (monkeys) or of the order Anthropoids (humans and apes).
- the mammal is a human.
- provided herein is a method of vaccinating a subject by introducing or administering an effective amount of a pharmaceutical composition described herein comprising at least one lipid nanoparticle described herein.
- the method of vaccinating comprises administering an effective amount of an antigen comprising a viral polypeptide from an adenovirus; Herpes simplex, type 1; Herpes simplex, type 2; encephalitis virus, papillomavirus, Varicella-zoster virus; Epstein-barr virus; Human cytomegalovirus; Human herpes virus, type 8; Human papillomavirus; BK virus; JC virus; Smallpox; polio virus; Hepatitis B virus; Human bocavirus; Parvovirus B19; Human astrovirus; Norwalk virus; coxsackievirus; hepatitis A virus; poliovirus; rhinovirus; Severe acute respiratory syndrome virus; Hepatitis C virus; Yellow Fever virus; Dengue virus; West Nile virus; Rubella virus; Hepatitis E virus; Human Immunodeficiency virus (HIV); Influenza virus; Guanarito virus; Junin virus; Lassa
- provided herein is a method of treating an autoimmune disorder in a subject by introducing or administering an effective amount of a pharmaceutical composition described herein comprising at least one lipid nanoparticle described herein.
- a method of treating cancer in a subject by introducing or administering an effective amount of a pharmaceutical composition described herein comprising at least one lipid nanoparticle described herein.
- the circular RNA construct encodes a CAR.
- the CARs have biological activity, e.g., ability to recognize an antigen, e.g., CD19, HER 2 , or BCMA, such that the CAR, when expressed by a cell, is able to mediate an immune response against the cell expressing the antigen, e.g., CD19, HER 2 , or BCMA, for which the CAR is specific.
- CAR-T chimeric antigen receptor
- CRS cytokine release syndrome
- CRES CAR-T cell-related encephalopathy syndrome
- CRS is the most common and well-described toxicity' associated yvith CAR-T therapy, occurring in over 90% of patients at any grade and is characterized by high fever, hypotension, hypoxia and/ or multiple organ toxicity and can lead to death.
- Neurotoxicity is characterized by damage to nervous tissue that can cause tremors, encephalopathy, dizziness or seizures.
- lymphodepletion is known to increase CAR- T cell expansion and enhanced efficacy of infused CAR-T cells by, for example, altering the tumor phenotype and microenvironment.
- lymphodepletion agents often cause side effects to the patients.
- lymphodepletion can cause neutropenia, anemia, thrombocytopenia, and immunosuppression, leading to a greater risk of infection, along with other toxicities.
- CAR-T therapies require an assortment of protocols to isolate, genetically modify, and selectively' expand the redirected cells before infusing them back into the patient.
- the subject has a cancer selected from the group consisting of: acute myeloid leukemia (AML); alveolar rhabdomyosarcoma; B cell malignancies; bladder cancer (e.g.. bladder carcinoma); bone cancer; brain cancer (e.g..
- breast cancer cancer; cancer of the anus, anal canal, or anorectum; cancer of the eye; cancer of the intrahepatic bile duct; cancer of the joints; cancer of the neck; gallbladder cancer; cancer of the pleura; cancer of the nose, nasal cavity, or middle ear; cancer of the oral cavity; cancer of the vulva; chronic lymphocytic leukemia; chronic myeloid cancer; colon cancer; esophageal cancer, cervical cancer; fibrosarcoma; gastrointestinal carcinoid tumor; head and neck cancer (e.g., head and neck squamous cell carcinoma); Hodgkin lymphoma; hypopharynx cancer; kidney cancer; larynx cancer; leukemia; liquid tumors; lipoma; liver cancer; lung cancer (e g., non-small cell lung carcinoma, lung adenocarcinoma, and small cell lung carcinoma); lymphoma; mesothelio
- the subject has an autoimmune disorder selected from scleroderma, Grave's disease, Crohn's disease, Sjogren's disease, multiple sclerosis, Hashimoto's disease, psoriasis, myasthenia gravis, autoimmune polyendocrinopathy syndromes, Type I diabetes mellitus (TIDM), autoimmune gastritis, autoimmune uveoretinitis, polymyositis, colitis, thyroiditis, and the generalized autoimmune diseases typified by human Lupus.
- TIDM Type I diabetes mellitus
- the term “storage stable” refers to a composition of empty LNPs or a composition of loaded LNPs (as described herein), having a polydispersity’ index (PDI) that increases by less than 25%, such as less than 20%, less than 15%, less than 10%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% after storage of the composition at 4 °C or -80 °C for at least 4 weeks, such as at least 1 month, at least 2 months, at least 3 months, at least 6 months, at least 8 months, or at least 1 year.
- PDI polydispersity’ index
- storage stable also refers to a composition of loaded LNPs (as described herein), having less than 25% decrease in encapsulation efficiency, such as less than 20%, less than 15%, less than 10%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% after storage of the composition at 4 °C or -80 °C for at least 4 weeks, such as at least 1 month, at least 2 months, at least 3 months, at least 6 months, at least 8 months, or at least 1 year.
- encapsulation may refer to complete, substantial, or partial enclosure, confinement, surrounding, or encasement.
- encapsulation or “association with” may refer to the process of at least partially confining an individual nucleic acid molecule within a nanoparticle and/or establishing a physiochemical relationship betw een an individual nucleic acid molecule and a nanoparticle.
- an “empty lipid nanoparticle” or “empty’ LNPs” may refer to a LNP that is substantially free of a therapeutic or prophylactic agent. As used herein, an “empty’ LNPs” may refer to a LNP that is substantially free of a nucleic acid. As used herein, an “empty LNPs” may refer to a nanoparticle that consists substantially of only lipid components.
- RNA refers to a single-stranded RNA polynucleotide wherein the 3’ and 5’ ends that are normally present in a linear RNA polynucleotide have been joined together.
- DNA template refers to a DNA sequence capable of transcribing a linear RNA polynucleotide.
- a DNA template may include a DNA vector, PCR product or plasmid.
- a circular RNA comprises a post splicing 3’ group I intron fragment.
- the post splicing 3’ group I intron fragment in the circular RNA is a post splicing stretch of exon sequence.
- the circular RNA further comprises a desired expression sequence
- tire post splicing stretch of exon sequence is (e.g., designed) to be a portion of the desired expression sequence, contiguous with die desired expression sequence, and/or in frame with the desired expression sequence.
- a circular RNA comprises a post splicing 3’ group II intron fragment.
- the post splicing 3' group II intron fragment in the circular RNA is a post splicing stretch of exon sequence.
- the circular RNA further comprises a desired expression sequence
- the post splicing stretch of exon sequence is (e.g., designed) to be a portion of the desired expression sequence, contiguous with the desired expression sequence, and/or in frame with the desired expression sequence.
- a circular RNA comprises a post splicing 5’ group I intron fragment.
- the post splicing 5’ group I intron fragment in the circular RNA is a post splicing stretch of exon sequence.
- tire circular RNA further comprises a desired expression sequence, and die post splicing stretch of exon sequence is (e.g., designed) to be a portion of the desired expression sequence, contiguous with the desired expression sequence, and/or in frame with the desired expression sequence.
- a circular RNA comprises a post splicing 5’ group II intron fragment.
- the post splicing 5' group II intron fragment in the circular RNA is a post splicing stretch of exon sequence.
- the circular RNA further comprises a desired expression sequence
- tire post splicing stretch of exon sequence is (e.g., designed) to be a portion of the desired expression sequence, contiguous with die desired expression sequence, and/or in frame with the desired expression sequence.
- RNA polynucleotides comprising a post splicing 3’ group I or II intron fragment (e.g., a stretch of exon sequence), optionally a first spacer, an IRES, an expression sequence, optionally a second spacer, and a post splicing 5’ group I or II intron fragment (e.g., a stretch of exon sequence).
- permutation site refers to the site in a group I or II intron where a cut is made prior to permutation of the intron. This cut generates 3 ’ and 5 ’ group I or II intron fragments that are permuted to be on either side of a stretch of precursor RNA to be circularized.
- splice site refers to a dinucleotide that is partially or fully included in a group I intron and between which a phosphodiester bond is cleaved during RNA circularization.
- splice site refers to the dinucleotide or dinucleotides between which cleavage of the phosphodiester bond occurs during a splicing reaction.
- a “5’ splice site” refers to the natural 5’ dinucleotide of the intron e.g., group I intron, while a “3’ splice site” refers to the natural 3’ dinucleotide of the intron).
- expression sequence refers to a nucleic acid sequence that encodes a product, e.g., a peptide or polypeptide, regulatory nucleic acid, or non-coding nucleic acid.
- An exemplary expression sequence that codes for a peptide or polypeptide can comprise a plurality of nucleotide triads, each of which can code for an amino acid and is termed as a “codon.”
- coding element or “coding region” is region located within the expression sequence and encodings for one or more proteins or polypeptides (e.g., therapeutic protein).
- a "noncoding element” or “non-coding nucleic acid” is a region located within the expression sequence. This sequence, but itself does not encode for a protein or polypeptide, but may have other regulatory functions, including but not limited, allow the overall polynucleotide to act as a biomarker or adjuvant to a specific cell.
- therapeutic protein refers to any protein that, when administered to a subject directly or indirectly in the form of a translated nucleic acid, has a therapeutic, diagnostic, and/or prophylactic effect and/or elicits a desired biological and/or pharmacological effect.
- the term “immunogenic” refers to a potential to induce an immune response to a substance.
- An immune response may be induced when an immune system of an organism or a certain type of immune cells is exposed to an immunogenic substance.
- the term “non-immunogenic” refers to a lack of or absence of an immune response above a detectable threshold to a substance. No immune response is detected when an immune system of an organism or a certain type of immune cells is exposed to a non-immunogenic substance.
- a non-immunogenic circular polyribonucleotide as provided herein does not induce an immune response above a pre-determined threshold when measured by an immunogenicity assay.
- no innate immune response is detected when an immune system of an organism or a certain type of immune cells is exposed to a non-immunogenic circular polyribonucleotide as provided herein.
- no adaptive immune response is detected when an immune system of an organism or a certain ty pe of immune cell is exposed to a non-immunogenic circular polyribonucleotide as provided herein.
- circularization efficiency refers to a measurement of resultant circular polyribonucleotide as compared to its linear starting material.
- translation efficiency refers to a rate or amount of protein or peptide production from a ribonucleotide transcript. In some embodiments, translation efficiency can be expressed as amount of protein or peptide produced per given amount of transcript that codes for the protein or peptide.
- nucleotide refers to a ribonucleotide, a deoxyribonucleotide, a modified form thereof, or an analog thereof.
- Nucleotides include species that comprise purines, e.g., adenine, hypoxanthine, guanine, and their derivatives and analogs, as well as pyrimidines, e.g., cytosine, uracil, thymine, and their derivatives and analogs.
- Nucleotide analogs include nucleotides having modifications in the chemical structure of the base, sugar and/or phosphate, including, but not limited to, 5 ’-position pyrimidine modifications, 8’ -position purinc modifications, modifications at cytosine exocyclic amines, and substitution of 5-bromo-uracil; and 2’-position sugar modifications, including but not limited to, sugar-modified ribonucleotides in which the 2’-OH is replaced by a group such as an H, OR, R, halo, SH, SR, NH2, NHR, NR 2 , or CN, wherein R is an alkyl moiety as defined herein.
- Nucleotide analogs are also meant to include nucleotides with bases such as inosine, queuosine, xanthine; sugars such as 2’-methyl ribose; non-natural phosphodiester linkages such as methylphosphonate, phosphorothioate and peptide linkages. Nucleotide analogs include 5- methoxyuridine. 1 -methylpseudouridine, and 6-methyladenosine.
- nucleic acid and “polynucleotide” are used interchangeably herein to describe a polymer of any length, e.g., greater than 2 bases, greater than 10 bases, greater than 100 bases, greater than 500 bases, greater than 1000 bases, or up to 10.000 or more bases, composed of nucleotides, e.g., deoxyribonucleotides or ribonucleotides, and may be produced enzymatically or synthetically (e.g., as described in U.S. Pat. No.
- Naturally occurring nucleic acids are comprised of nucleotides including guanine, cytosine, adenine, thymine, and uracil (G, C. A, T. and U respectively).
- ribonucleic acid and RNA as used herein mean a polymer composed of ribonucleotides.
- deoxyribonucleic acid and “DNA” as used herein mean a polymer composed of deoxy ribonucleotides.
- isolated or “purified” generally refer to isolation of a substance (for example, in some embodiments, a compound, a polynucleotide, a protein, a polypeptide, a polynucleotide composition, or a polypeptide composition) such that the substance comprises a significant percent (e.g., greater than 1%, greater than 2%. greater than 5%, greater than 10%, greater than 20%, greater than 50%, or more, usually up to 90%-100%) of the sample in which it resides.
- a substantially purified component comprises at least 50%, 80%-85%, or 90%-95% of the sample.
- Techniques for purifying polynucleotides and polypeptides of interest include, for example, ion-exchange chromatography, affinity chromatography and sedimentation according to density.
- a substance is purified when it exists in a sample in an amount, relative to other components of the sample, that is more than as it is found naturally.
- duplexed double-stranded
- hybridized refers to nucleic acids formed by hybridization of tw o single strands of nucleic acids containing complementary sequences. In most cases, genomic DNA is double-stranded. Sequences can be fully complementary or partially complementary.
- unstructured with regard to RNA refers to an RNA sequence that is not predicted by the RNAFold software or similar predictive tools to form a structure (e.g., a hairpin loop) with itself or other sequences in the same RNA molecule.
- unstructured RNA can be functionally characterized using nuclease protection assay s.
- RNA refers to an RNA sequence that is predicted by the RNAFold software or similar predictive tools to form a structure (e.g., a hairpin loop) w ith itself or other sequences in the same RNA molecule.
- two “duplex sequences,” “duplex region,” “duplex regions.” “homology arms.” or “homology regions” may be any’ two regions that are thermodynamically favored to cross-pair in a sequence specific interaction.
- two duplex sequences, duplex regions, homology arms, or homology regions share a sufficient level of sequence identity’ to one another’s reverse complement to act as substrates for a hybridization reaction.
- polynucleotide sequences have “homology” when they are either identical or share sequence identity to a reverse complement or “complementary” sequence.
- the percent sequence identity between a homology region and a counterpart homology’ region’s reverse complement can be any percent of sequence identity’ that allows for hybridization to occur.
- an internal duplex region of an inventive polynucleotide is capable of forming a duplex with another internal duplex region and does not form a duplex with an external duplex region.
- an “affinity sequence” or “affinity tag” is a region of polynucleotide sequences polynucleotide sequence ranging from 1 nucleotide to hundreds or thousands of nucleotides containing a repeated set of nucleotides for the purposes of aiding purification of a polynucleotide sequence.
- an affinity sequence may comprise, but is not limited to, a poly A or poly AC sequence.
- a “spacer” refers to a region of a polynucleotide sequence ranging from 1 nucleotide to hundreds or thousands of nucleotides separating two other elements along a polynucleotide sequence.
- the sequences can be defined or can be random.
- a spacer is typically noncoding. In some embodiments, spacers include duplex regions.
- Linear nucleic acid molecules are said to have a “5’-terminus” (5’ end) and a “3’-terminus” (3’ end) because nucleic acid phosphodiester linkages occur at the 5’ carbon and 3’ carbon of the sugar moieties of the substituent mononucleotides.
- the end nucleotide of a polynucleotide at which a new linkage would be to a 5’ carbon is its 5’ terminal nucleotide.
- the end nucleotide of a polynucleotide at which a new linkage would be to a 3’ carbon is its 3’ terminal nucleotide.
- a terminal nucleotide, as used herein, is the nucleotide at the end position of the 3’- or 5 ’-terminus.
- a “leading untranslated sequence” is a region of polynucleotide sequences ranging from 1 nucleotide to hundreds of nucleotides located at the upmost 5' end of a polynucleotide sequence.
- the sequences can be defined or can be random.
- a leading untranslated sequence is noncoding.
- a “leading untranslated sequence” is a region of polynucleotide sequences ranging from 1 nucleotide to hundreds of nucleotides located at the downmost 3' end of a polynucleotide sequence. The sequences can be defined or can be random. A leading untranslated sequence is noncoding.
- Transcription means the formation or synthesis of an RNA molecule by an RNA polymerase using a DNA molecule as a template.
- the invention is not limited with respect to the RNA polymerase that is used for transcription.
- a T7-type RNA polymerase can be used.
- Translation means the formation of a polypeptide molecule by a ribosome based upon an RNA template.
- the term “encode” refers broadly to any process whereby die information in a polymeric macromolecule is used to direct the production of a second molecule that is different from die first.
- the second molecule may have a chemical structure diat is different from the chemical nature of the first molecule.
- co-administering is meant administering a therapeutic agent provided herein in conjunction with one or more additional therapeutic agents sufficiently close in time such that the therapeutic agent provided herein can enhance the effect of the one or more additional therapeutic agents, or vice versa.
- treatment or prevention can include treatment or prevention of one or more conditions or symptoms of the disease. Also, for purposes herein, “prevention” can encompass delaying the onset of the disease, or a symptom or condition thereof.
- an “internal ribosome entry site” or “IRES” refers to an RNA sequence or structural element ranging in size from 10 nt to 1000 nt or more, capable of initiating translation of a polypeptide in the absence of a typical RNA cap structure.
- An IRES is typically 500 nt to 700 nt in length.
- aptamer refers in general to either an oligonucleotide of a single defined sequence or a mixture of said nucleotides, wherein the mixture retains the properties of binding specifically to the target molecule (e.g., eukaryotic initiation factor, 40S ribosome.
- target molecule e.g., eukaryotic initiation factor, 40S ribosome.
- aptamer denotes both singular and plural sequences of nucleotides, as defined hereinabove.
- aptamer is meant to refer to a single- or double-stranded nucleic acid which is capable of binding to a protein or other molecule.
- aptamers preferably comprise 10 to 100 nucleotides, preferably 15 to 40 nucleotides, more preferably 20 to 40 nucleotides, in that oligonucleotides of a length that falls within these ranges are readily prepared by conventional techniques.
- aptamers can further comprise a minim um of approximately 6 nucleotides, preferably 10, and more preferably 14 or 15 nucleotides, that are necessary to effect specific binding.
- Aii “eukaryotic initiation factor” or “elF” refers to a protein or protein complex used in assembling an initiator tRNA, 40S and 60S ribosomal subimits required for initiating eukaryotic translation.
- an “internal ribosome entry site” or “IRES” refers to an RNA sequence or structural element ranging in size from 10 nt to 1000 nt or more, capable of initiating translation of a polypeptide in the absence of a ty pical RNA cap structure.
- An IRES is typically 500 nt to 700 nt in length.
- a “miRNA site” refers to a stretch of nucleotides within a polynucleotide that is capable of forming a duplex with at least 8 nucleotides of a natural miRNA sequence.
- an “endonuclease site” refers to a stretch of nucleotides within a polynucleotide that is capable of being recognized and cleaved by an endonuclease protein.
- bicistronic RNA refers to a polynucleotide that includes two expression sequences coding for two distinct proteins. These expression sequences can be separated by a nucleotide sequence encoding a cleavable peptide such as a protease cleavage site. They can also be separated by a ribosomal skipping element.
- ribosomal skipping element refers to a nucleotide sequence encoding a short peptide sequence capable of causing generation of two peptide chains from translation of one RNA molecule. While not wishing to be bound by theory', it is hypothesized that ribosomal skipping elements function by (1) terminating translation of the first peptide chain and re-initiating translation of the second peptide chain; or (2) cleavage of a peptide bond in the peptide sequence encoded by the ribosomal skipping element by an intrinsic protease activity of the encoded peptide, or by another protease in the environment (e.g., cytosol).
- the term “co-formulate” refers to a nanoparticle formulation comprising two or more nucleic acids or a nucleic acid and other active drug substance. Typically, the ratios are equimolar or defined in the ratiometric amount of the two or more nucleic acids or the nucleic acid and other active drug substance.
- the phrase “ionizable lipid” refers to any of a number of lipid species that carry a net positive charge at a selected pH. such as physiological pH 4 and a neutral charge at other pHs such as physiological pH 7.
- a lipid or compound described herein comprises one or more cleavable groups.
- cleave and cleavable are used herein to mean that one or more chemical bonds (e.g., one or more of covalent bonds, hydrogen-bonds, van der Waals' forces and/or ionic interactions) between atoms in or adjacent to the subject functional group are broken (e.g.. hydrolyzed) or are capable of being broken upon exposure to selected conditions (e.g., upon exposure to enzy matic conditions).
- the cleavable group is a disulfide functional group, and in particular embodiments is a disulfide group that is capable of being cleaved upon exposure to selected biological conditions (e.g., intracellular conditions).
- the cleavable group is an ester functional group that is capable of being cleaved upon exposure to selected biological conditions.
- the disulfide groups may be cleaved enzymatically or by a hydrolysis, oxidation or reduction reaction. Upon cleavage of such disulfide functional group, the one or more functional moieties or groups (e.g., one or more of a head-group and/or a tail-group) that are bound thereto may be liberated.
- a cleavable group is bound (e.g., bound by one or more of hydrogen-bonds, van der Waals' forces, ionic interactions and covalent bonds) to one or more functional moieties or groups.
- Compound described herein may also comprise one or more isotopic substitutions.
- H may be in any isotopic form, including 1H, 2H (D or deuterium), and 3H (T or tritium);
- C may be in any isotopic form, including 12C, 13C, and 14C;
- O may be in any isotopic form, including 160 and 180;
- F may be in any isotopic form, including 18F and 19F; and the like.
- C 1 -6 alkyl is intended to encompass, C 1 , C 2 , C3, C 4 , C5, Ce, C 1 -g, C1-5, C 1 ⁇ i, C1-3, C1-2, C 2 -6, C 2 -5, C 2 -4, C 2 -3, C3-6, C3-5. C3-4, C 4 -6. C 4 -5, and C5-6 alkyl.
- head-group as used describe the compounds of the present invention, and in particular functional groups that comprise such compounds, are used for ease of reference to describe the orientation of one or more functional groups relative to other functional groups.
- a hydrophilic head-group e.g., an amino group
- a hydrophobic tail-group e.g., cholesterol
- the present invention is intended to encompass the compounds disclosed herein, and the pharmaceutically acceptable salts, pharmaceutically acceptable esters, tautomeric forms, polymorphs, and prodrugs of such compounds.
- the present invention includes a pharmaceutically acceptable addition salt, a pharmaceutically acceptable ester, a solvate (e.g., hydrate) of an addition salt, a tautomeric form, a poly morph, an enantiomer, a mixture of enantiomers, a stereoisomer or mixture of stereoisomers (pure or as a racemic or non-racemic mixture) of a compound described herein.
- Compounds described herein can comprise one or more asymmetric centers, and thus can exist in various isomeric forms, e.g., enantiomers and/or diastereomers.
- the compounds described herein can be in the form of an individual enantiomer, diastereomer or geometric isomer, or can be in the form of a mixture of stereoisomers, including racemic mixtures and mixtures enriched in one or more stereoisomer.
- Isomers can be isolated from mixtures by methods known to those skilled in the art, including chiral high pressure liquid chromatography (HPLC) and the formation and crystallization of chiral salts; or preferred isomers can be prepared by asymmetric syntheses.
- HPLC high pressure liquid chromatography
- the subject compositions exhibit an enhanced (e.g., increased) ability to transfect one or more target cells.
- methods of transfecting one or more target cells generally comprise the step of contacting the one or more target cells with the compositions disclosed herein such that the one or more target cells are transfected with the circular RNA encapsulated therein.
- transfect or “transfection” refer to the intracellular introduction of one or more encapsulated materials (e.g.. nucleic acids and/or polynucleotides) into a cell, or preferably into a target cell.
- transfection efficiency refers to the relative amount of such encapsulated material (e.g., polynucleotides) uptaken by. introduced into and/or expressed by the target cell which is subject to transfection.
- nucleotide sequences disclosed herein can represent an RNA sequence or a corresponding DNA sequence. It is understood that deoxythymidine (dT or T) in a DNA is transcribed into a uridine (U) in an RNA. As such, "T” and “U” are used interchangeably herein in nucleotide sequences.
- sequence identity or, for example, comprising a “sequence 50% identical to,” as used herein, refer to the extent that sequences are identical on a nucleotide-by -nucleotide basis or an amino acid-by-amino acid basis over a window of comparison.
- a “percentage of sequence identity” may be calculated by comparing two optimally aligned sequences over the window of comparison, determining the number of positions at which the identical nucleic acid base (e.g., A. T, C, G, I) or the identical amino acid residue (e.g., Ala. Pro, Ser, Thr, Gly. Vai, Leu, He, Phe, Tyr.
- Trp, Lys, Arg, His, Asp, Glu, Asn. Gin, Cys and Met occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison (i.e., the window size), and multiplying the result by 100 to yield the percentage of sequence identity.
- nucleotides and polypeptides having at least 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% sequence identity to any of the reference sequences described herein, typically where the polypeptide variant maintains at least one biological activity of the reference polypeptide.
- the expression sequences in the polynucleotide construct may be separated by a “cleavage site” sequence which enables polypeptides encoded by the expression sequences, once translated, to be expressed separately by tire cell.
- a “self-cleaving peptide” refers to a peptide which is translated without a peptide bond between two adjacent amino acids, or functions such that when the polypeptide comprising tire proteins and the self-cleaving peptide is produced, it is immediately cleaved or separated into distinct and discrete first and second polypeptides without the need for any external cleavage activity .
- the a and (3 chains of a(3 TCR's are generally regarded as each having two domains or regions, namely variable and constant domains/regions.
- the variable domain consists of a concatenation of variable regions and joining regions.
- TCR alpha variable domain therefore refers to the concatenation of TRAV and TRAJ regions
- TCR alpha constant domain refers to the extracellular TRAC region, or to a C-tenninal truncated TRAC sequence.
- TCR beta variable domain refers to the concatenation of TRBV and TRBD/TRBJ regions
- TCR beta constant domain refers to the extracellular TRBC region, or to a C-terminal truncated TRBC sequence.
- duplexed double-stranded
- hybridized refer to nucleic acids formed by hybridization of two single strands of nucleic acids containing complementary sequences. In most cases, genomic DNA is double-stranded. Sequences can be fully complementary or partially complementary’.
- a “vaccine” refers to a composition for generating immunity for the prophylaxis and/or treatment of diseases. Accordingly, vaccines are medicaments which comprise antigens and are intended to be used in humans or animals for generating specific defense and protective substances upon administration to the human or animal.
- Organic phase lipids dissolved in ethanol
- aqueous phase buffer
- Lipids used to form the organic phase included ionizable lipids, helper lipids, a cholesterol lipid, and lipid anchored PEGylated lipids.
- the selected lipids were solubilized in ethanol in exemplary molar ratios provided in Table 5.
- salt solution e.g., comprising citrate, sodium acetate, malic acid, and/or bis-tris methane
- Table 6 depicts exemplary aqueous buffer compositions for each of the ionizable lipids. Optimum buffer compositions were analyzed for the smallest and most stable LNPs using screening experiments of various aqueous buffer compositions.
- both the aqueous and the lipid ethanol phase were mixed using a commercially available mixer (e.g.. Silverson L5M-A) for 5 minutes at 8000 RPM using the standard mixer head mesh at a processing temperature from 4 to 30 ° C.
- a commercially available mixer e.g.. Silverson L5M-A
- the first composition of empty LNPs were then fed into a commercially available high shear microfluidizer (e.g., Microfluidics LM20 high shear microfluidizer) at 10,000 - 30.000 psi for further particle size reduction.
- the processing temperature at this step was kept the same or close to the mixing processing temperature (e.g., 4° C to 30° C).
- Empty LNPs were then stored at 4° C until RNA addition step.
- Ionizable lipid 89 (Table 1), 1.2-distearoyl-sn-giycero-3phosphochoiine (DSPC), cholesterol (Choi), and DMG-PEG2000 were mixed at a 50: 10:38.5: 1.5 molar ratio and were used to evaluate LNP sizing using overhead mixing at different temperatures.
- Lipids dissolved in ethanol organic phase
- an aqueous phase buffer solution e.g., comprising citrate, sodium acetate, malic acid, and/or bis-tris methane
- Organic phase and aqueous phase were mixed using an overhead mixer for 5 minutes at temperatures of 4° C, 10° C, 20° C, 30° C, and 40° C.
- FIG. 2 shows die particle size distribution of LNPs processed in different temperatures. Based on the data provided in FIG. 2, 30° C provides the smallest particle size (104 nm) and lowest PDI at 0.09.
- Ionizable lipid 144 (Table 1), DSPC, Cholesterol, and DMG-PEG2000 at 50: 10:38.5:1.5 molar ratio were dissolved in ethanol (organic phase) and mixed with aqueous buffer. The duration of mixing was set at 5 minutes, and different RPMs were tested at 1000, 2000, 4000, and 8000 RPM.
- FIG. 3 represents ionizable lipid 144 (Table 1) LNP post LNP formation and post homogenization step. The slowest rotor speed at 1000 RPM resulted in an average particle size of 101 nm, and the highest rotor speed resulted in average particle sizes of 89 nm (see FIG. 3).
- Ionizable lipid 127 (Table 1), DSPC. Cholesterol, and DMG-PEG2000 at 50: 10:38.5.1.5 molar ratio were dissolved in ethanol (organic phase) and mixed with an aqueous phase of 6.25 mM sodium acetate at pH 3.0, using an overhead mixer to form empty LNPs (LNP formation step). The formed empty LNPs were then fed through a homogenizer for particle size reduction (homogenization step). To elucidate the effect of temperature for the homogenization step using a high-pressure homogenizer, empty lipid 127 containing LNPs were processed through the homogenizer at two processing temperatures, 30° C and 50° C.
- FIG. 4 provides particle size and PDI after both the LNP formation step and the homogenization step. Particle size reduction after homogenization was observed for both temperatures compared to the LNP formation step in FIG 4.
- a cooling step was implemented using a heat exchanger immediately after the homogenization step.
- FIG. 5 shows the particle size of the empty lipid 127 LNPs with and without cooling after the homogenization step.
- the buffer composition at both the first LNP formation step and tire homogenization step were evaluated.
- Ionizable lipid 127 (Table 1), DSPC, Cholesterol, and DMG-PEG2000 at 50: 10:38.5.1.5 molar ratio were dissolved in ethanol (organic phase) and mixed with an aqueous phase of 6.25 mM sodium acetate at pH 3.0 or 4.5. using an overhead mixer and then fed through a homogenizer for particle size reduction.
- FIG. 6 represents the particle size and PDI relationship with pH of the aqueous phase over multiple batches. As seen in FIG. 6.
- LNPs in pH 3.0 sodium acetate buffer not only resulted in smaller particle size post LNP formation than the LNPs in pH 4.5 sodium acetate buffer, but also resulted in smaller particle size post homogenization step than the LNPs in pH 4.5 sodium acetate. More generally, as also depicted in FIG. 6 particle size reduction was observed for both buffers after homogenization. Zeta potential was also measured for each of the LNPs post homogenization, as depicted in FIG. 7. In some embodiments, various buffer salts can be tested at different concentrations and pH ranges as provided in Table 7.
- Ionizable lipids with different tails and/or headgroups such as lipid 144 and lipid 123 (Table 1) were investigated (in addition to lipid 127; see Examples 4 and 5 for lipid 127). The following lipid solutions were used:
- Lipid 144 • Lipid 144. DSPC, Cholesterol, and DMG-PEG2000 were formulated at 50:10:38.5.1.5 molar ratio.
- Lipid 123 Lipid 123.
- DSPC, cholesterol, and DMG-PEG 2000 were formulated at standard molar ratio of 45 :9:44:2 and with RN A.
- the N :P ratio was kept at 4.5 : 1 ratio.
- the lipid solution made using ionizable lipid 144 was dissolved in ethanol (organic phase), mixed with an aqueous buffer phase (6.25 mM Sodium Acetate at pH 3.0; 50mM Na Acetate at pH 3.0; or 6.25 mM Bis-Tris at pH 7.0) using an overhead mixer, and then fed through a homogenizer for particle size reduction. Results are depicted in FIG. 8 for empty LNPs formed containing lipid 144.
- the lipid solution made using ionizable lipid 123 was dissolved in ethanol (organic phase), mixed with an aqueous buffer phase (6.25 mM Citrate buffer at pH 2.0; 6.25 mM citrate buffer at pH 3.0; 6.25 mM sodium acetate at pH 3.0; or 6.25 mM sodium acetate at pH 4.5) using an overhead mixer, and then fed through a homogenizer for particle size reduction. Results are depicted in FIG. 9 for LNPs formed containing lipid 123.
- a composition of empty LNPs was formed using ionizable lipid 127 (Table 1) according to the method of Example 1. Encapsulation of circular RNA within the empty LNPs was then investigated to form a composition of loaded LNPs. Numerous aqueous buffers were screened on a plate-based assay first in a high-throughput fashion ID lead buffer conditions that would produce high encapsulation and small particle size, after the RNA addition step. Table 7 depicts the full range of buffers tested. Initial results showed that 50 mM sodium acetate resulted in high RNA encapsulation while keeping the particles small within 70 - 90 nm particle size range.
- FIG. 10 represents the average particle size of RNA encapsulated LNP (loaded LNPs) in the different aqueous phase buffer conditions.
- FIG. 11 provides the LNP encapsulation efficiency post RNA encapsulation (i.e., for loaded LNPs) at different pHs in 50 mM sodium acetate.
- a composition of empty LNPs were formed using ionizable lipid 127 (Table 1) according to the method of Example 1. Circular RNA was encapsulated within the empty LNPs to form a composition of loaded LNPs according to the method of Example 7 (RNA addition step). Following the RNA addition step, buffer exchange was performed via tangential flow filtration (TFF). The composition of loaded LNPs underwent buffer exchange either with phosphate buffered saline at pH 7.4 (PBS, TFF process #1) or ultrapure distilled water (DI water. TFF process #2). The resulting loaded LNP particle size and PDI after the RNA addition step and buffer exchange (TFF step) are shown in FIG. 12.
- Buffer exchange step (or TFF step) performed using PBS caused loaded LNP particle size to grow in average particle size to 142 nm with PDI at 0.22.
- buffer exchange (TFF step) performed using DI water, resulted in significantly smaller particle sizes of approximately 69 nm with PDI 0.08.
- FIG. 13 provides the encapsulation efficiency of the loaded LNPs post RNA addition step and TFF step with PBS or DI water.
- TSS tris-sucrose saline
- PBS Phosphate Buffered Saline
- Tris Buffered Saline Tris Buffered Saline
- a composition of empty’ LNPs was formed using ionizable lipid 127 (Tabic 1) according to the method of Example 1.
- Circular RNA encoding firefly luciferase was encapsulated within the empty LNPs to form a composition of loaded LNPs according to the method of Example 7 (RNA addition step).
- buffer exchange was performed via tangential flow filtration (TFF).
- TFF tangential flow filtration
- Process 1 also depicts a schematic outlining the subject distinctive process steps: an LNP formation step (nanoprecipitation step), a homogenization step (particle size reduction step), and an RNA addition step.
- LNP formation step nanoprecipitation step
- homogenization step particle size reduction step
- RNA addition step an RNA addition step.
- Process 1 was evaluated for in vivo performance via intravenous (IV) and intramuscular (IM) route of administration.
- LNP particle sizes were evaluated for loaded LNPs prepared using the nanoprecipitation process (e.g., Precision Nanosystem Ignite (PNI) nanoprecipitation process, see e.g., https://www.precisionnanosystems.com/platform-teclmologies/product-comparison/ignite) and Process 1.
- PNI Precision Nanosystem Ignite
- FIG. 18 shows 6-hour ex vivo circular RNA encoding firefly luciferase expression for IV injected loaded LNPs.
- the organs of the mice were extracted and ex vivo I VIS was performed on the organs.
- FIG. 18 provides the in vivo firefly luciferase expression of the circular RNA encapsulated in the LNPs as measured by ex vivo IVIS as made from Process 1.
- FIG. 19D provides the ex vivo organ imaging for firefly luciferase expression of the loaded LNPs as made from Process 1, wherein Process 1 used PBS (FIG. 19C) or TSS (FIG. 19D).
- FIG. 19A-FIG. 19B provides the ex vivo organ imaging for firefly luciferase expression of the loaded LNPs as made from nanoprecipitation (e.g., PNI) process, wherein the nanoprecipitation (e.g.. PNI) process used PBS (FIG. 19A) or TSS (FIG. 19B).
- nanoprecipitation e.g., PNI
- Intramuscular injected LNPs were also characterized for protein expression using IVIS at 24 hours post injection.
- FIG. 20 shows flux level of LNPs in the quadricep, liver, and spleen.
- PCT/US2021/031629 PCT/US2021/023540, PCT/US2021/033276.
- PCT/US2022/033091 PCT/US2021/031629, PCT/US2021/023540, PCT/US2021/033276.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Physics & Mathematics (AREA)
- Optics & Photonics (AREA)
- Biomedical Technology (AREA)
- Nanotechnology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present application provides methods for manufacturing lipid nanoparticles. The present disclosure provides methods of preparing empty lipid nanoparticles (empty LNPs). The present disclosure provides methods of preparing loaded lipid nanoparticles (loaded LNPs) associated with a nucleic acid. Empty and loaded LNP compositions prepared by the subject methods are also provided. There is provided a homogeneous LNP composition of empty or loaded LNPs of particularly favorable average particle size and poly dispersity index.
Description
PROCESS FOR MANUFACTURING LIPID NANOPARTICLES
BACKGROUND
[0001] In the past few decades, nucleic acid therapeutics have rapidly expanded and become the basis for treating a wide variety of diseases. Nucleic acid therapies available include, but are not limited to, the use of DNA or viral vectors for insertion of desired genetic information into the host cell, and/or RNA constructed to encode for a therapeutic protein. DNA and viral vector deliveries carry their own setbacks and challenges that make them less favorable to RNA therapeutics. For example, the introduced DNA in some cases may be unintentionally inserted into an intact gene and result in a mutation that impedes or even wholly eliminates the function of the endogenous gene leading to an elimination or deleteriously reduced production of an essential enzyme or interruption of a gene critical for the regulating cell growth. Viral vector-based therapies can result in an adverse immune response. Compared to DNA or viral vectors, RNA is a substantially safer and more effective gene therapy agent due to its ability7 to encode for the protein outside of the nucleus to perform its function. With this, the RNA does not involve the risk of being stably integrated into the genome of the transfected cell.
[0002] RNA therapeutics conventionally have consisted of engineering linear messenger RNAs (mRNA). Although more effective than DNA or viral vectors, linear mRNAs have their own set of challenges regarding stability, immunogenicity, translation efficiency, and delivery. Some of these challenges may lead to size restraints and/or destruction of the linear mRNA due to the challenges present with linear mRNAs’ caps. To overcome these limitations, circular polynucleotides or circular RNAs may be used. Due to being arranged in covalently closed continuous loops, circular RNAs are useful in the design and production of stable forms of RNA. The circularization of an RNA molecule provides an advantage to the study of RNA structure and function, especially in the case of molecules that are prone to folding in an inactive conformation (Wang and Ruffner, 1998). Circular RNA can also be particularly interesting and useful for in vivo applications, especially in the research area of RNA- bascd control of gene expression and therapeutics, including protein replacement therapy and vaccination.
[0003] To promote effective delivery of the RNA, nanoparticles delivery systems can be used. Lipid- containing nanoparticles or lipid nanoparticles, liposomes, and lipoplexes have been used as effective delivery systems to transport into cells and/or intracellular compartments biologically active substances such as small molecule drugs, proteins, and nucleic acids. Though a variety of such lipid nanoparticles delivery systems have been demonstrated, improvements in manufacturing such delivery systems are needed.
SUMMARY
[0004] The present application provides methods for manufacturing lipid nanoparticles. In some embodiments, the present disclosure provides a method of preparing an empty lipid nanoparticle (empty LNP). In some embodiments, the present disclosure provides a method of preparing a loaded lipid nanoparticle (loaded LNP) associated with a nucleic acid. Empty and loaded LNP compositions prepared by the subject methods are also provided. In some embodiments, there is provided a homogeneous LNP composition of empty or loaded LNPs of particularly favorable average particle size and polydispersity index.
[0005] In one aspect, provided herein is a method of preparing an empty lipid nanoparticle (LNP) composition, comprising: a) a precipitation step, comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first LNP composition comprising empty’ LNPs; and b) a homogenization step, comprising: homogenizing the first LNP composition to produce a homogeneous LNP composition of empty LNPs.
[0006] In some embodiments, the empty LNPs in the homogeneous LNP composition have an average particle size from 30 nm to 200 nm, such as from 50 nm to 70 nm. In some embodiments, the empty LNPs in the homogeneous LNP composition have a poly dispersity from 0.05 to 0.2, such as 0.1 or less.
[0007] In some embodiments, the method further comprises: c) a loading step, comprising: mixing the homogeneous LNP composition with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with the nucleic acid.
[0008] In another aspect, provided herein is a method of preparing a loaded LNP composition, the method comprising: a) a precipitation step, comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first LNP composition comprising empty LNPs; b) a loading step, comprising:
mixing the first LNP composition with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with the nucleic acid; and c) a homogenization step, comprising: homogenizing the loaded LNP composition to produce a homogeneous loaded LNP composition comprising LNPs associated with the nucleic acid.
[0009] In another aspect, provided herein is an empty LNP composition prepared by a subject method.
[0010] In another aspect, provided herein is a loaded LNP composition prepared by a subject method.
[0011] In another aspect, provided herein is a homogeneous LNP composition substantially free of loaded LNPs and comprising empty LNPs comprising: an ionizable lipid; a helper lipid; a cholesterol; and a PEG-lipid, wherein the empty’ LNPs have
(a) a polydispersity' of 0.2 or less (e.g., 0.1 or less); and / or
(b) an average particle size from 50 nm to 70 nm.
BRIEF DESCRIPTION OF THE DRAWINGS
[0012] FIG. 1 is a schematic outlining the subject distinctive process steps: an LNP formation step (nanoprecipitation step), a homogenization step (particle size reduction step), and an RNA addition step.
[0013] FIG. 2 depicts the average particle size distribution of empty LNPs containing ionizable lipid 89 (Table 1) formed at different temperatures.
[0014] FIG. 3 depicts the average particle size distribution of empty LNPs containing ionizable lipid 144 (Table 1) formed at different mixing speeds.
[0015] FIG. 4 depicts the average particle size and PDI of empty LNPs containing the ionizable lipid 127 (Table 1) after both the LNP formation step and the homogenization step at processing temperatures of 30° C and 50° C.
[0016] FIG. 5 depicts the average particle size of the empty LNPs containing the ionizable lipid 127 (Table 1) with and without cooling after the homogenization step.
[0017] FIG. 6 depicts the average particle size of empty LNPs containing the ionizable lipid 127 (Table 1) after both the LNP formation step and the homogenization step using 6.25 mM sodium acetate buffer at pH 3.0 or 4.5.
[0018] FIG. 7 depicts the zeta potential of empty LNPs containing the ionizable lipid 127 (Tabic 1) after the homogenization step using 6.25 mM sodium acetate buffer at pH 3.0 or 4.5.
[0019] FIG. 8 depicts the average size and PDI of empty LNPs containing the ionizable lipid 144 (Table 1) after both the LNP formation step and the homogenization step using different aqueous buffers.
[0020] FIG. 9 depicts the average size and PDI of empty LNPs containing the ionizable lipid 123 (Table 1) after both the LNP formation step and the homogenization step using different aqueous buffers.
[0021] FIG. 10 depicts the average particle size of RNA encapsulated LNPs (loaded LNPs) in the different aqueous phase buffer conditions as compared to control empty LNPs.
[0022] FIG. 11 depicts the LNP encapsulation efficiency post RNA encapsulation (i.e., for loaded LNPs) at different pHs in 50 mM sodium acetate.
[0023] FIG. 12 depicts loaded LNP particle size and PDI after the RNA addition step and buffer exchange (TFF step) with phosphate buffered saline at pH 7.4 (PBS. TFF process #1) or ultrapure distilled water (DI water. TFF process #2).
[0024] FIG. 13 depicts the encapsulation efficiency of the loaded LNPs containing the ionizable lipid 127 (Table 1) post RNA addition step and TFF step with phosphate buffered saline at pH 7.4 (PBS, TFF process #1) or ultrapure distilled water (DI water, TFF process #2).
[0025] FIG. 14 depicts the average size and PDI of loaded LNPs containing the ionizable lipid 127 (Table 1) after buffer exchange using different storage buffcrs/tonicity modifiers, water, tris-sucrosc saline (TSS) or PBS.
[0026] FIG. 15 depicts the process for manufacturing the subject loaded LNPs, including the following distinctive process steps: LNP formation step (empty LNPs), a homogenization step (particle size reduction step, empty LNPs), and an RNA addition step (loaded LNPs).
[0027] FIG. 16 depicts the average particle size for RNA encapsulated LNPs (loaded LNPs) formed using the nanoprecipitation (e.g., PNI) process and Process 1.
[0028] FIG. 17 depicts Cryo-TEM images for loaded LNPs formulated via Process 1 (Panel A) and the nanoprecipitation (e.g., PNI) process (Panel B).
[0029] FIG. 18 depicts the in vivo firefly luciferase expression of the circular RNA encapsulated in the LNPs (loaded LNPs) as measured by ex vivo IVIS as made from Process 1 vs the nanoprecipitation (e.g., PNI) process.
[0030] FIG. 19A-FIG. 19D depict the ex vivo organ imaging for firefly luciferase expression of the loaded LNPs as made from Process 1 vs the nanoprecipitation (e.g., PNI) process.
[0031] FIG. 20 depicts flux level of loaded LNPs in the quadricep, liver, and spleen.
DETAILED DESCRIPTION
[0032] The present application provides, among other things, methods for manufacturing lipid nanoparticles. In some embodiments, the present disclosure provides a method of preparing an empty lipid nanoparticle (empty LNP). In some embodiments, the present disclosure provides a method of preparing a loaded lipid nanoparticle (loaded LNP) associated with a nucleic acid. As disclosed herein, the loaded lipid nanoparticles can be associated with RNA polynucleotides, particularly circular RNA polynucleotides (aka circRNA or oRNA™)-
[0033] Traditionally, methods for producing lipid nanoparticles that encapsulate linear mRNA (mRNA-LNPs), precipitation and encapsulation of the RNA occur concurrently via high-energy mixing of aqueous mRNA and a solution of lipids in ethanol. The mixing of the ethanol phase and the aqueous phase can generally be controlled by microfluidic chip devices. See for example, Guevara et al., Front. Chem. 2020; 8: 589959; Riley et al., Sci. Adv. 2021; 7: eaba 1028. and Cullis et al., WO2011140627. Such concurrent mixing can result in fouling of microfluidic devices and is not amenable to large-scale manufacturing.
[0034] As disclosed herein, the empty LNPs are prepared by distinctive steps: an LNP formation step (precipitation step to form empty LNPs); and a homogenization step (size reduction step to form homogeneous mixture of empty LNPs). The subject methods can further comprise an RNA addition step to form loaded LNPs (LNPs associated with a nucleic acid). In some embodiments, the homogenization step is carried out after the LNP formation step and before the RNA addition step. In some embodiments, the homogenization step is carried out after the RNA addition step.
[0035] Without being bound to any particular theory, by separating the LNP formation, and LNP homogenization steps (i.e., as seen in the subject process for preparing empty LNPs), the need to control a kinetic precipitation step (for example, with a microfluidic chip) is eliminated. As such, the subject process for preparing empty LNPs is readily scalable. More generally, the subject method for preparing empty LNPs eliminates the need to control the size of the LNPs during the initial LNP formation step, which can be a bottleneck for large scale production in previously known LNP manufacturing processes.
[0036] The present disclosure provides a method of preparing a homogeneous composition of empty LNPs, which can be produced and stored until ready for further use. The present disclosure also provides methods in which a nucleotide may be associated with, or encapsulated within, tire preformed empty LNPs to form loaded LNPs. This mode of production offers advantages in the context of clinical supply, as empty LNP vesicles may be produced and stored separately prior to recombination with a nucleic acid (e.g., a circular RNA polynucleotide) in a clinical compound setting. Specifically, bedside formulations may promote increased stability since the nucleic acid and empty LNPs can be stored in separately optimized conditions. Process complexity and cost of the products may be reduced since the empty LNP preparation occurs independently of nucleic acid cargo, enabling a platform approach for multiple nucleic acid or active agent constructs.
[0037] In some embodiments, the present disclosure provides loaded LNPs associated with a nucleic acid that has an average particle size distribution and poly dispersity that is the same or better than loaded LNPs produced by a previously known LNP manufacturing method.
[0038] In some embodiments, the LNP formulation produced by tire method of the present disclosure exhibits a nucleic acid expression (e.g., the RNA expression) higher than the nucleic acid expression (e.g., the RNA expression) of the LNP formulation produced by a previously known LNP manufacturing method. More generally, encapsulating an RNA by combining pre-fonned lipid nanoparticles with mRNA can result in formulated particles that exhibit unexpectedly efficient in vivo delivery of the RNA and surprisingly potent expression of proteins and/or peptides that the RNA encodes. See for example. Karve et al, W02018089801 and Smith et al.. WO2021155274, the disclosure of which is incorporated herein by reference.
[0039] In some embodiments, the loaded LNP formulation produced by the method of the present disclosure exhibits a nucleic acid expression (e.g.. a circular RNA expression) higher than the nucleic acid expression (e.g., a circular RNA expression) of the LNP formulation prepared by a previously known LNP manufacturing method by 5% or higher, 10% or more 15% or more, 20% or more. 30% or more. 40% or more, 50% or more, 60% or more, 70% or more, 80% or more. 90% or more, 1 folds or more. 2 folds or more. 3 folds or more. 4 folds or more, 5 folds or more, 10 folds or more, 20 folds or more. 30 folds or more, 40 folds or more. 50 folds or more, 100 folds or more, 200 folds or more, 300 folds or more. 400 folds or more, 500 folds or more, 1000 folds or more. 2000 folds or more, 3000 folds or more. 4000 folds or more, 5000 folds or more, or 10000 folds or more.
[0040] Also disclosed herein is RNA therapy (e.g., circular RNA therapy), along with associated compositions and methods. In some embodiments, the RNA therapy allows for increased RNA stability, expression, and prolonged half-life, among other things.
[0041] In some embodiments, provided herein are methods comprising administration of circular RNA polynucleotides provided herein into cells for therapy or production of useful proteins. In some
embodiments, the method is advantageous in providing the production of a desired polypeptide inside eukaryotic cells with a longer half-life than linear RNA, due to the resistance of the circular RNA to ribonucleases.
[0042] Various aspects of the disclosure are described in detail in the following sections. The use of sections is not meant to limit the disclosure. Each section can apply to any aspect of the disclosure. In diis application, the use of “or” means “and/or” unless stated otherwise.
1. METHODS OF PREPARATION
[0043] As summarized herein, the present disclosure provides methods of preparing empty lipid nanoparticles (empty LNPs). and methods of preparing loaded LNPs.
[0044] In one embodiment, provided herein is a method of preparing an empty LNP composition, the method comprising: a) a precipitation step, comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first lipid LNP composition comprising empty LNPs; and b) a homogenization step, comprising: homogenizing the first LNP composition to produce a homogeneous LNP composition of empty LNPs.
[0045] In some embodiments, the method further comprises: c) a loading step, comprising: mixing the homogeneous LNP composition with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with the nucleic acid.
[0046] In some embodiments, the method further comprises one or more of the following steps: dilution and buffer exchange of the loaded LNP composition; filtration of the loaded LNP composition; and storage of the loaded LNP composition. i. LNP formation step (precipitation step)
[0047] The subject methods include an LNP formation step (precipitation step), wherein a lipid solution comprising an ionizable lipid, and an aqueous buffer are mixed together to form a lipid nanoparticle composition comprising empty LNPs.
[0048] In some embodiments, the precipitation step is performed with a lipid solution further comprising a helper lipid, a structural lipid, a PEG lipid or any combination thereof. In some embodiments, the precipitation step is performed with a lipid solution further comprising a phospholipid, a structural lipid, and a PEG lipid. In some embodiments, the precipitation step is performed with a lipid solution further comprising a PEG lipid and a phospholipid. In some embodiments, the precipitation step is performed with a lipid solution further comprising a PEG lipid and a structural lipid. In some embodiments, the precipitation step is performed with a lipid solution further comprising a phospholipid and a structural lipid. In some embodiments, the precipitation step is performed with a lipid solution further comprising a PEG lipid. In some embodiments, the mixing step is performed with a lipid solution further comprising a phospholipid. In some embodiments, the mixing step is performed with a lipid solution further comprising a structural lipid.
[0049] In some embodiments, the precipitation step is performed with a lipid solution comprising a molar ratio of from 40% to 60 % ionizable lipid, a molar ratio of from 3.5% to 14% helper lipid, a molar ratio of from 28% to 50% structural lipid, and a molar ratio of from 0.5% to 5% PEG-lipid. In some embodiments, the total molar percentage of the ionizable lipid, the helper lipid, the structural lipid, and the PEG-lipid is 100% in the lipid solution.
[0050] In some embodiments, the molar ratio of the ionizable lipid in the lipid solution is from 40% to 60% of the total lipid present in the lipid solution. In some embodiments, the molar ratio of the ionizable lipid in the lipid solution is 40%, 41%, 42%, 43%. 44%, 45%, 46%, 47%. 48%, 49%, 50%, 51%. 52%, 53%, 54%, 55%, 56%. 57%, 58%, 59%, or 60% of the total lipid present in the lipid solutions.
[0051] In some embodiments, the molar ratio of the helper lipid in the lipid solution is from 3.5% to 14% of the total lipid present in the lipid solution. In some embodiments, the molar ratio of the helper lipid in the lipid solution is 3%, 4%, 5%, 6%, 7%. 8%, 9%, 10%, 11%, 12%, 13%, or 14% of the total lipid present in the lipid solution. In some embodiments, the helper lipid is DSPC. In some embodiments, the helper lipid is DOPE.
[0052] In some embodiments, the molar ratio of the structural lipid in the lipid solution is from 28% to 50% of the total lipid present in the lipid solution. In some embodiments, the molar ratio of the structural lipid in the lipid solution is 28%, 29%, 30%, 31%, 32%, 33%, 34%, 35%, 36%, 37%, 38%, 39%, 40%, 41%, 42%, 43%, 44%, 45%, 46%. 47%, 48%, 49%, or 50% of the total lipid present in the lipid solution. In some embodiments, the structural lipid is cholesterol.
[0053] In some embodiments, the molar ratio of the PEG-lipid in the lipid solution is from 0.1% to 5% of the total lipid present in the lipid solution. In some embodiments, the molar ratio of the PEG-lipid in the lipid solution is 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1.0%, 1.1%, 1.2%, 1.3%,
1.4%, 1.5%, 1.6%. 1.7%, 1.8%, 1.9%, 2.0%, 2.1%, 2.2%, 2.3%, 2.4%, 2.5%, 2.6%, 2.7%, 2.8%, 2.9%,
3.0%, 3.1%, 3.2%, 3.3%, 3.4%. 3.5%, 4.0%, 4.5%, or 5% of the total lipid present in the lipid solution. In some embodiments, the PEG-lipid is DMG-PEG2000.
[0054] In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG- lipid in the lipid solution is 45:9:44:2. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the lipid solution is 50: 10:38.5: 1.5. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the lipid solution is 41:12:45:2. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the lipid solution is 62:4:33:1. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the lipid solution is 53:5:41:1. In some embodiments, the molar ratio of each of die ionizable lipid, helper lipid, structural lipid, and PEG-lipid is within 10%, 9%, 8%, 7%, 6%, 5%. 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0055] In some embodiments, the precipitation step is performed with a lipid solution comprising an ionizable lipid as described herein. In some embodiments, the precipitation step is performed with a lipid solution further comprising a helper lipid, a structural lipid, a PEG lipid or any combination thereof as described herein. In some embodiments, the helper lipid is 1,2-distearoyl-sn-glvcero- 3phosphocholine (DSPC), the structural lipid is cholesterol, and the PEG lipid is DMG-PEG2000.
[0056] In some embodiments, the precipitation step is performed with a lipid solution comprising about 30-60 mol % of ionizable lipid (c.g., as described herein); about 5-30 mol % of DSPC; about 15-50 mol % cholesterol; and about 1-2 mol % of DMG-PEG2000.
[0057] In some embodiments of the precipitation step, a suitable lipid solution may contain a mixture of desired lipids at various concentrations. In some embodiments, a suitable lipid solution may contain a mixture of desired lipids at a total concentration in a range from 0.1-100 mg/mL, 0.5-90 mg/mL, 1.0-80 mg/mL, 1.0-70 mg/mL, 1.0-60 mg/mL, 1.0-50 mg/mL, 1.0-40 mg/mL, 1.0-30 mg/mL, 1.0-20 mg/mL, 1.0-15 mg/mL, 1.0-10 mg/mL, 1.0-9 mg/mL, 1.0-8 mg/ml, 1.0-7 mg/mL, 1.0-6 mg/mL, or 1.0-5 mg/mL.
[0058] In some embodiments, the precipitation step is performed with a total lipid concentration from 5 mg/mL to 80 mg/mL, 6 mg/mL to 70 mg/mL, 7 mg/mL to 60 mg/mL, 8 mg/mL to 50 mg/mL, 9 mg/mL to 40 ing/mL, 10 mg/mL to 30 ing/mL, 15 mg/mL to 25 ing/mL, or 20 mg/mL to 25 mg/mL.
[0059] In some embodiments, the precipitation step is performed with a total lipid concentration of 10 mg/mL, 15 mg/mL, 20 mg/mL. 25 mg/mL, 30 mg/mL, 40 mg/mL, 50 mg/mL, 60 mg/mL. 70 mg/mL, or 80 mg/inL. In some embodiments, the total lipid concentration is within 10%, 9%. 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%. 0.1%, 0.09%. 0.08%, 0.07%, 0.06%, 0.05%. 0.04%, 0.03%, 0.02%. or 0.01% of the stated value.
[0060] In some embodiments of the precipitation step, the lipid solution comprises an organic solvent that is miscible with the aqueous buffer solution. In some embodiments, the lipid solution comprises ethanol. For example, a suitable lipid solution may contain a mixture of desired lipids dissolved in pure ethanol (i.e., 100% ethanol). In some embodiments the lipid solution comprises isopropyl alcohol. In some embodiments the lipid solution comprises benzyl alcohol. In some embodiments the lipid solution comprises dimethylsulfoxide. In some embodiments, the lipid solution comprises a mixture of organic solvents including, but not limited to, ethanol, benzyl alcohol, isopropyl alcohol, and dimethylsulfoxide. In some embodiments, the lipid solution comprises a mixture of ethanol and benzyl alcohol.
[0061] In some embodiments of the precipitation step, the aqueous buffer solution comprises a buffering agent selected from ammonium sulfate, sodium bicarbonate, sodium citrate, sodium acetate, potassium phosphate. tris(hydroxymethyl)aminomethane (tris). 2-[Bis(2-hydroxyethyl)amino]-2- (hydroxymethyl)propane- 1,3 -diol (bis-tris), sodium phosphate, and HEPES. In some embodiments, the buffering agent is sodium acetate.
[0062] In some embodiments, the precipitation step is performed with an aqueous buffer solution comprising an aqueous buffer at a concentration ranging from 1-500 mM, from 0.1-100 mM, from 0.5- 90 mM, from 1.0-80 mM, from 2-70 mM, from 3-60 mM. from 4-50 mM, from 5-40 mM, from 6-30 mM, from 6-20 mM, from 6-15 mM, or from 6-12 mM. In some embodiments, the aqueous buffer concentration range is within 10%, 9%. 8%, 7%, 6%, 5%, 4%, 3%. 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%. 0.4%. 0.3%. 0.2%. 0.1%. 0.09%, 0.08%, 0.07%. 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0063] In some embodiments, the precipitation step is performed with an aqueous buffer solution comprising an aqueous buffer at a concentration of or greater than 0.1 mM, 0.5 mM, 1 mM, 2 mM, 4 mM, 6 mM, 8 mM, 10 mM, 15 mM, 20 mM, 25 mM, 30 mM, 35 mM, 40 mM, 45 mM, or 50 mM.
[0064] In some embodiments, the precipitation step is performed with a first aqueous buffer comprising an aqueous buffer at a concentration of 6.25±2.0 mM, 6.25±1.5 mM, 6.25±1.0 mM, 6.25±0.9 mM, 6.25±0.8 mM, 6.25±0.7 mM, 6.25±0.6 mM, 6.25±0.5 mM, 6.25±0.4 mM, 6.25±0.3 mM, 6.25±0.2 mM, or 6.25±0.1 mM.
[0065] In some embodiments, the precipitation step is performed with a first aqueous buffer comprising an aqueous buffer at a concentration of about 6.25 mM.
[0066] In some embodiments of the precipitation step, the aqueous buffer solution is at a pH from 2.0 to 9.0. such as from 2.5 to 8.5, from 2.6 to 8.4, from 2.7 to 8.3, from 2.8 to 8.2, from 2.9 to 8.1, from 3.0 to 8.0, from 3.2 to 7.8, from 3.4 to 7.6, from 3.6 to 7.4, from 3.8 to 7.2, from 4.0 to 7.0. from 4.1 to 6.8, from 4.2 to 6.6, from 4.3 to 6.4, from 4.4 to 6.2, from 4.5 to 6.0. from 4.6 to 5.9, from 4.7 to 5.8, from 4.8 to 5.7, from 4.9 to 5.6, from 5.0 to 5.5, from 5.1 to 5.4, or from 5.2 to about 5.3.
[0067] In some embodiments of the precipitation step, the pH of the aqueous buffer solution is from 2 to 9, such as 2 to 6, or 3 to 5. In some embodiments, the pH of the aqueous buffer solution is 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5. 7.0. 7.5. 8.0. 8.5. or 9.0. In some embodiments, the pH of the aqueous buffer solution is within 10%, 9%, 8%, 7%, 6%, 5%. 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%. 0.2%. 0.1%. 0.09%, 0.08%, 0.07%. 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0068] In some embodiments, the precipitation step is performed at a pH of 2.0, 2.1, 2.2, 2.3, 2.4, 2.5,
2.6, 2.7. 2.8, 2.9, 3.0, 3.1, 3.2, 3.3, 3.4, 3.5, 3.6, 3.7, 3.8, 3.9, 4.0, 4.1, 4.2, 4.3. 4.4, 4.5, 4.6, 4.7, 4.8, 4.9, 5.0. 5.1, 5.2, 5.3, 5.4, 5.5, 5.6, 5.7, 5.8, 5.9, 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6. 6.7, 6.8, 6.9, 7.0, 7.1,
7.2, 7.3. 7.4. 7.5, 7.6, 7.7, 7.8, 7.9, 8.0, 8.1, 8.2, 8.3, 8.4, 8.5, 8.6, 8.7. 8.8. 8.9, or 9.
[0069] In some embodiments, the precipitation step is performed at a pH of less than 2.0, less than 2.5, less than 2.6, less than 2.7, less than 2.8. less than 2.9, less than 3.0, less than 3.2, less than 3.4 less than
3.6, less than 3.8, less than 4.0, less than 4.1, less than 4.2, less than 4.3, less than 4.4, less than 4.5, less than 4.6, less than 4.7, less than 4.8, less than 4.9, less than 5.0, less than 5.1, less than 5.2, less than
5.3, less than 5.4, less than 5.5, less than 5.6, less than 5.7, less than 5.8, less than 5.9, less than 6.0, less than 6.2, less than 6.4. less than 6.6, less than 6.8, less than 7.0, less than 7.2, less than 7.4, less than
7.6, less than 7.8, less than 8.0, less than 8.1, less than 8.2. less than 8.3, less than 8.4. less than 8.5, or less than 9.0.
[0070] In some embodiments, the precipitation step is performed at a pH of greater than 2.0. greater than 2.5, greater than 2.6, greater than 2.7, greater than 2.8, greater than 2.9, greater than 3.0. greater than 3.2, greater than 3.4 greater than 3.6, greater than 3.8, greater than 4.0, greater than 4.1. greater than 4.2, greater than 4.3, greater than 4.4, greater than 4.5, greater than 4.6, greater than 4.7. greater than 4.8, greater than 4.9, greater than 5.1, greater than 5.2, greater than 5.3, greater than 5.4. greater than 5.5, greater than 5.6, greater than 5.7, greater than 5.8, greater than 5.9. greater than 6.0. greater than 6.2, greater than 6.4, greater than 6.6, greater than 6.8, greater than 7.0. greater than 7.2. greater than 7.4, greater than 7.6, greater than 7.8, greater than 8.0, greater than 8.1, greater than 8.2, greater than 8.3, greater than 8.4, greater than 8.5, or greater than 8.6.
[0071] In some embodiments, the precipitation step is performed at a pH of 3.0±2.0, 3.0±1.5, 3.0±1.0, 3.0±0.9. 3.0±0.8, 3.0±0.7, 3.0±0.6, 3.0±0.5, 3.0±0.4. 3.0±0.3, 3.0±0.2, or 3.0±0.1.
[0072] In some embodiments, the precipitation step is performed at a pH of 4.5±2.0, 4.5±1.5, 4.5±1.0, 4.5±0.9. 4.5±0.8, 4.5±0.7, 4.5±0.6, 4.5±0.5, 4.5±0.4. 4.5±0.3, 4.5±0.2, or 4.5±0.1.
[0073] In some embodiments, the precipitation step is performed with an aqueous buffer solution having a pH of about 3.0. In some embodiments, the precipitation step is performed with an aqueous buffer solution having a pH of about 4.5. In some embodiments, the mixing step is performed with an
aqueous buffer solution comprising an acetate buffer. In some embodiments, the precipitation step is performed with an aqueous buffer solution comprising 6.25 mM sodium acetate. In some embodiments, the precipitation step is performed with an aqueous buffer solution comprising 6.25 mM sodium acetate at pH 3.0. In some embodiments, the precipitation step is performed with an aqueous buffer solution comprising 6.25 mM sodium acetate at pH 4.5.
[0074] In some embodiments of the precipitation step, the ratio of lipid solution to aqueous buffer solution is within the range of 1 :2 to 1:5 by volume, such as 1 :2.5, 1:3.0, 1 :3.5, 1 :4.0, or 1 :4.5 by volume. In some embodiments, the ratio of lipid solution to aqueous buffer solution is 1 :2. In some embodiments, the ratio of lipid solution to aqueous buffer solution is 1 :3. In some embodiments, the ratio of lipid solution to aqueous buffer solution is 1:4. In some embodiments, the ratio of lipid solution to aqueous buffer solution is 1 :5. In some embodiments, the ratio of lipid solution to aqueous buffer solution is within 10%, 9%, 8%, 7%, 6%, 5%, 4%. 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%. 0.1%. 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%. or 0.01% of the stated ranges and values.
[0075] In some embodiments of the precipitation step, the mixing is carried out by simply combining the lipid solution and the aqueous buffer solution together in a vessel. In some embodiments of the precipitation step, the mixing is carried out by in-line mixing. In some embodiments of the precipitation step, the mixing is facilitated by an overhead mixer. In some embodiments of the precipitation step, the mixing is facilitated by a Y -mixer, a T-mixer, a stir bar, or a probe sonicator.
[0076] The mixing in the precipitation step can be conducted at any convenient mixing speed. In some embodiments, the mixing in the precipitation step is performed at a mixing speed from 100 to 10,000 rpm, such as 200 to 10,000 rpm, 500 to 10,000 rpm, 1000 to 10,000 rpm, 1000 to 9000 rpm, 1000 to 8000 rpm, 1000 to 7000 rpm, 1000 to 6000 rpm, 1000 to 5000 rpm, 1000 to 4000 rpm, 1000 to 4000 rpm, 2000 to 8000 rpm, 2000 to 7000 rpm, 2000 to 6000 rpm, 2000 to 5000 rpm, 2000 to 4000 rpm, or 2000 to 3000 rpm.
[0077] In some embodiments, the mixing in the precipitation step is performed at a mixing speed of 1000 rpm, 1500 rpm, 2000 rpm, 2500 rpm, 3000 rpm, 3500 rpm, 4000 rpm, 4500 rpm, 5000 rpm, 5500 rpm, 6000 rpm, 6500 rpm, 7000 rpm, 7500 rpm, or 8000 rpm. In some embodiments, the mixing speed is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%. 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%. or 0.01% of the stated value.
[0078] In some embodiments of the precipitation step, the mixing is carried out at a temperature of from 2-70 °C, such as 2-60 °C, 2-50 °C, 2-40 °C, 4-50 °C, 4-40 °C, or 4-30 °C. In some embodiments of the precipitation step, the mixing is carried out at a temperature of 4 °C, 5 °C, 6 °C, 7 °C, 8 °C, 9 °C, 10 °C, 11 °C, 12 °C, 13 °C, 14 °C, 15 °C, 16 °C, 17 °C, 18 °C, 19 °C, 20 °C. 21 °C, 22 °C, 23 °C, 24 °C, 25 °C, 26 °C, 27 °C, 28 °C, 29 °C, 30 °C, 31 °C, 32 °C, 33 °C, 34 °C, 35 °C, 36 °C, 37 °C, 38 °C,
39 °C, 40 °C, 41 °C, 42 °C, 43 °C, 44 °C, 45 °C, 46 °C, 47 °C, 48 °C, 49 °C, or 50 °C. In some embodiments of the precipitation step, the mixing is carried out at a temperature of 4 °C, 10 °C, 20 °C, 30 °C, or 40 °C. In some embodiments, the mixing is carried out at 30 °C. In some embodiments, the mixing temperature is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%. 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%. 0.2%. 0.1%. 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0079] In some embodiments of the precipitation step, the mixing is performed at a temperature of less dian 50 °C, such as less than 45 °C, less than 40 °C, less than 35°C, less than 30°C, less than 25°C, less than 20 °C, less than 15 °C, or less than about 10°C.
[0080] In some embodiments of the precipitation step, the mixing is performed at ambient temperature.
[0081] In some embodiments of the precipitation step, the mixing is performed from 1 min to 100 min, such as 1 min to 80 min, 1 min to 70 min, 1 min to 60 min. 1 min to 50 min. 1 min to 45 min, 1 min to
40 min, 1 min to 35 min, 1 min to 30 min, 1 min to 20 min. 1 min to 10 min, or 1 min to 5 min.
[0082] In some embodiments of the precipitation step, the mixing step is performed from 1 min to 10 min. such as 1 min to 9 min, 1 min to 8 min. 1 min to 7 min, 1 min to 6 min, 1 min to 5 min. In some embodiments of the precipitation step the mixing is carried out for 5 min.
[0083] In some embodiments of the precipitation step, the mixing is performed for 5.0±2.0 min, 5.0±1.5 min, 5.0±1.0 min, 5.0±0.9 min. 5.0±0.8 min, 5.0±0.7 min. 5.0±0.6 min, 5.0±0.5 min. 5.0±0.4 min, 5.0±0.3 min, 5.0±0.2 min. or 5.0±0.1 min.
[0084] In some embodiments of die precipitation step, the first LNP composition formed comprises empty LNPs of a random size distribution. In some embodiments of the LNP composition formed in the precipitations step, the empty LNPs have an average particle size (average particle diameter) of greater than 100 nm. such as greater than 200 nm. In some embodiments of the first LNP composition formed in the precipitation step, the empty LNPs have an average particle size of 80-100 nm. such as 50-60 nm.
[0085] In some embodiments of the precipitation step, the first LNP composition formed comprises empty LNPs having a polydispersity of greater than 0.2, such as 0.21, 0.22, 0.23, 0.24, 0.25, 0.26. 0.27, 0.28, 0.29, 0.30. or even greater. ii. LNP homogenization step
[0086] The subject methods include an LNP homogenization step (particle size reduction step), wherein composition comprising empty LNPs from the precipitation step are homogenized to produce a homogeneous LNP composition of empty’ LNPs.
[0087] In some embodiments, the homogenizing step is performed using a microfluidic device. In some embodiments of the homogenizing step, the composition is passed through the microfluidic device 1 to 5 times, such as 2 to 5 times, 3 to 5 times, or 4 to 5 times. In some embodiments, the composition is passed through the microfluidic device two or more times, such as three or more times, four or more times, or five or more times. In some embodiments, the composition is passed through the microfluidic device up to 5 times, such as up to 4 times, up to 3 times, or up to 2 times.
[0088] In some embodiments, the microfluidic device is selected from a high-pressure homogenizer, a high shear homogenizer and a probe homogenizer.
[0089] In some embodiments, the homogenizing step is performed using a high-pressure homogenizer. In some embodiments, the homogenizing step is performed using a high-pressure homogenizer at a pressure of from 1000 to 30,000 PSI, such as 1000 to 10,000 PSI, 10,000 to 20,000 PSI, or 20,000 to 30,000 PSI.
[0090] In some embodiments, the homogenizing step is performed using a high shear homogenizer.
[0091] In some embodiments, the homogenizing step is performed using a probe homogenizer.
[0092] In some embodiments, the homogenizing step is carried out at a temperature of 2-70 °C, such as 2-60 °C, 2-50 °C. 2-40 °C, 4-60 °C, 4-50 °C. or 4-40 °C. In some embodiments the homogenizing step is carried out at a temperature of 4 °C, 5 °C, 6 °C. 7 °C, 8 °C, 9 °C. 10 °C, 11 °C, 12 °C. 13 °C, 14 °C, 15 °C, 16 °C, 17 °C, 18 °C, 19 °C, 20 °C, 21 °C, 22 °C, 23 °C, 24 °C, 25 °C, 26 °C, 27 °C, 28 °C, 29 °C. 30 °C, 31 °C, 32 °C. 33 °C, 34 °C. 35 °C, 36 °C, 37 °C. 38 °C, 39 °C, 40 °C. 41 °C, 42 °C. 43 °C, 44 °C, 45 °C. 46 °C, 47 °C, 48 °C. 49 °C, 50 °C, 51 °C. 52 °C, 53 °C, 54 °C. 55 °C, 56. °C, 57, °C, 58 °C. 59 °C, or 60 °C. In some embodiments, the homogenizing step is carried out at a temperature of 4 °C, 10 °C. 20 °C, 30 °C, 40 °C. or 50°C. In some embodiments, the homogenizing step is carried out at 30 °C. such that the temperature is 30 °C in the microfluidic device. In some embodiments, the homogenizing step is carried out at 40 °C, such that the temperature is 40 °C in the microfluidic device. In some embodiments, the homogenizing step is carried out at 50 °C, such that the temperature is 50 °C in the microfluidic device. In some embodiments, the homogenizing step temperature is within 10%, 9%, 8%, 7%, 6%, 5%. 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%. 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0093] In some embodiments, the homogenizing step is performed at a temperature of less than 50 °C, less than 45 °C, less than 40 °C, less than 35 °C. less than 30 °C, less than 25 °C, less than 20 °C, less than 15 °C, or less than 10 °C.
[0094] In some embodiments, the homogenizing step is performed at ambient temperature.
[0095] In some embodiments of die homogenization step, the homogeneous LNP composition formed is immediately cooled after the homogenizing step. For example, after the homogenous LNP composition exits the microfluidic device, the composition is cooled. In some embodiments, after exiting the microfluidic device, the homogenous LNP composition is cooled to 10 °C or less, such as 4 °C or less. Without being bound to any particular theory, rapid cooling of the homogeneous LNP composition after it exits the microfluidic device (e.g.. the high-pressure homogenizer) can prevent aggregation of the LNPs in the composition.
[0096] In some embodiments of the homogenization step, the empty' LNPs in the homogeneous LNP composition formed have an average particle size of from 30 nm to 200 rnn, such as 50 mn to 70 nm. In some embodiments of the homogenization step, the empty LNPs in the homogeneous LNP composition have a poly dispersity of 0.05 to 0.2, such as 0.1 or less.
[0097] In some embodiments of the homogenization step, the average particle size of the empty LNPs in the homogeneous LNP composition formed is at least 10% less than the average particle size of the empty LNPs of the first LNP composition formed in the precipitation step. In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition is 10-50% less than the average particle size of the empty LNPs of the first LNP composition. In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition is 10-30% less than the average particle size of the empty LNPs of the first LNP composition. In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition formed in the homogenization step is 10% less, such as 15% less, 20% less, 25% less, 30% less. 35% less. 40% less, 45% less, or 50% less than the average particle size of the empty LNPs of the first LNP composition formed in the precipitation step.
[0098] In some embodiments, the homogeneous LNP composition obtained after the homogenizing step can be stored at 4 °C, and is storage stable for at least 4 weeks, such as at least 8 weeks, at least 12 weeks, at least 16 weeks, at least 20 weeks, at least 6 months, at least 9 months, or at least a year.
Hi. LNP loading step
[0099] The subject methods include an LNP loading step, wherein the homogeneous composition of empty LNPs obtained from the homogenization step are mixed with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with the nucleic acid. In some embodiments, the loaded LNP composition comprises LNPs encapsulating the nucleic acid.
[0100] In some embodiments of the loading step, the amounts of nucleic acid and empty7 LNPs are selected to provide a specific N:P ratio. The N:P ratio of the loaded LNP composition refers to the molar ratio of nitrogen atoms in one or more lipids (e.g., ionizable lipids) to the number of phosphate
groups in the nucleic acid. In some embodiments of the loaded LNP composition, the molar ratio of nitrogen atoms in one or more ionizable lipids (i.e., in the empty LNPs) to phosphate groups in tire nucleic acid (N:P) is within the range of 1 :20 to 20: 1. In some embodiments, the N:P ratio is 1 :20, 1: 19, 1: 18, 1 : 17, 1 : 16, 1:15, 1:14, 1 : 13, 1: 12, 1: 11, 1: 10, 1 :9, 1:8, 1 :7, 1 :6, 1:5, 1:4, 1:3. 1:2 or 1 : 1. In some embodiments, the N:P ratio is 20: 1, 19:1, 18: 1, 17: 1, 16: 1. 15:1, 14: 1, 13: 1, 12: 1, 11: 1, 10: 1, 9: 1, 8:1, 7: 1. 6: 1, 5: 1, 4: 1, 3:1 or 2:1. In some embodiments of the loaded LNP composition, the N:P ratio is within the range of 4: 1 to 6: 1. In some embodiments of the loaded LNP composition, the N:P ratio is 4.0: 1, 4.1 :1. 4.2: 1. 4.3:1. 4.4: 1, 4.5: 1, 4.6: 1, 4.7: 1, 4.8: 1, 4.9: 1, 5.0: 1, 5.1 : 1, 5.2: 1, 5.3: 1, 5.4: 1, 5.5: 1, 5.6:1, 5.7:1, 5.8:1, 5.9: 1, or 6.0: 1. In some embodiments of the loaded LNP composition, the N:P ratio is within 10%, 9%, 8%, 7%, 6%. 5%, 4%, 3%, 2%. 1%. 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%. 0.1%. 0.09%, 0.08%, 0.07%. 0.06%, 0.05%, 0.04%, 0.03%, 0.02%. or 0.01% of the stated value.
[0101] In some embodiments of the loading step, in order to achieve high encapsulation of the nucleic acid, the nucleic acid solution (i.e., nucleic acid in a buffer as described herein) is heated. In some embodiments of the loading step, no heating is needed (i.e.. the process is carried out at ambient temperature).
[0102] In some embodiments, the loading step is carried out at a temperature of 2-70 °C, such as 2-60
°C, 2-50 °C. 2-40 °C, 4-60 °C, 4-50 °C, 4-40 °C, or 4-25 °C. In some embodiments the loading step is carried out at a temperature of 4 °C. 5 °C, 6 °C, 7 °C, 8 °C, 9 °C, 10 °C. 11 °C, 12 °C, 13 °C, 14 °C, 15
C, 16 °C. 17 °C, 18 °C, 19 °C, 20 °C, 21 °C, 22 °C, 23 °C, 24 °C, 25 °C, 26 °C, 27 °C. 28 °C, 29 °C,
30 °C, 31 °C, 32 °C. 33 °C, 34 °C, 35 °C, 36 °C, 37 °C, 38 °C, 39 °C, 40 °C. 41 °C, 42 °C, 43 °C, 44
°C, 45 °C, 46 °C, 47 °C, 48 °C, 49 °C, 50 °C, 51 °C, 52 °C, 53 °C. 54 °C, 55 °C, 56 °C. 57 °C, 58 °C,
59 °C, or 60 °C. In some embodiments, the loading step is carried out at a temperature of 4 °C, 10 °C, 20 °C, 30 °C, 40 °C. or 50°C. In some embodiments, the loading step temperature is within 10%, 9%, 8%. 7%, 6%, 5%, 4%. 3%. 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%. 0.4%. 0.3%. 0.2%. 0.1%. 0.09%, 0.08%, 0.07%, 0.06%. 0.05%, 0.04%, 0.03%. 0.02%, or 0.01% of the stated value.
[0103] In some embodiments, the loading step is performed at a temperature of less than 50 °C, such as less than 45 °C. less than 40 °C. less than 35 °C. less than 30 °C, less than 25 °C, less than 20 °C, less than 15 °C, or less than 10 °C.
[0104] In some embodiments of the loading step, a suitable nucleic acid solution is an aqueous solution containing a nucleic acid to be encapsulated at various concentrations. For example, a suitable nucleic acid solution may contain a nucleic acid at a concentration of or greater than 0.01 mg/mL, such as 0.05 mg/mL. 0.06 mg/mL, 0.07 mg/mL. 0.08 mg/mL, 0.09 mg/mL, 0.1 mg/mL, 0.15 mg/mL, 0.2 mg/mL, 0.3 mg/mL, 0.4 mg/mL, 0.5 mg/mL, 0.6 mg/mL, 0.7 mg/mL, 0.8 mg/mL, 0.9 mg/mL, or 1.0 mg/mL. In some embodiments, a suitable nucleic acid solution may contain a nucleic acid at a concentration in a range from 0.01-1.0 mg/mL. such as 0.01-0.9 mg/mL, 0.01-0.8 mg/mL. 0.01-0.7 mg/mL, 0.01-0.6
mg/mL, 0.01-0.5 mg/mL, 0.01-0.4 mg/mL, 0.01-0.3 mg/mL, 0.01-0.2 mg/mL, 0.01-0.1 mg/mL, 0.05- 1.0 mg/mL, 0.05-0.9 mg/mL, 0.05-0.8 mg/mL, 0.05-0.7 mg/mL, 0.05-0.6 mg/mL, 0.05-0.5 mg/mL, 0.05-0.4 mg/mL, 0.05-0.3 mg/mL, 0.05-0.2 mg/mL, 0.05-0.1 mg/mL, 0.1-1.0 mg/mL, 0.2-0.9 mg/mL, 0.3-0.8 mg/mL, 0.4-0.7 mg/mL, or 0.5-0.6 mg/mL.
[0105] In some embodiments of the loading step, the nucleic acid solution comprises a buffer salt selected from an acetate salt, a citrate salt, or a bis-tris salt. In some embodiments, the buffer salt is selected from ammonium sulfate, sodium bicarbonate, sodium citrate, sodium acetate, potassium phosphate, tris(hydroxymethyl)aminoinethane (tris), 2-[Bis(2-hydroxyethyl)amino]-2- (liydroxymethyl)propane-l,3-diol (bis-tris), sodium phosphate, and HEPES. In some embodiments, the buffer salt is sodium acetate.
[0106] In some embodiments, the loading step is performed with a nucleic acid solution comprising a buffer salt at a concentration ranging from 1-500 mM, such as from 0.1-100 rnM, from 0.5-90 mM, from 1.0-80 mM, from 2-70 mM, from 3-60 mM, from 4-50 mM, from 5-40 mM, from 6-50 mM, from 6-40 mM, from 6-30 mM, or from 6-20 mM. In some embodiments, the buffer salt concentration range is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0107] In some embodiments, the loading step is performed with a nucleic acid solution comprising a buffer salt at a concentration of or greater than 0.1 mM, 0.5 mM, 1 mM, 2 mM, 4 mM, 6 mM, 8 mM, 10 mM, 15 mM, 20 mM, 25 mM, 30 mM, 35 mM, 40 mM, 45 mM, or 50 mM.
[0108] In some embodiments, the loading step is performed with nucleic acid solution comprising a buffer salt at a concentration of 6.25±2.0 mM, 6.25±1.5 mM, 6.25±1.0 mM, 6.25±0.9 mM. 6.25±0.8 mM, 6.25±0.7 mM, 6.25±0.6 mM, 6.25±0.5 mM, 6.25±0.4 mM, 6.25±0.3 mM, 6.25±0.2 mM, or 6.25±0.1 mM.
[0109] In some embodiments, the loading step is performed with nucleic acid solution comprising a buffer salt at a concentration of about 6.25 mM.
[0110] In some embodiments, the loading step is performed with nucleic acid solution comprising a buffer salt at a concentration of 50±2.0 mM, 50±1.5 mM, 50±1.0 mM, 50±0.9 mM, 50±0.8 mM. 50±0.7 mM. 50±0.6 mM, 50±0.5 mM, 50±0.4 mM, 50±0.3 mM. 50±0.2 mM. or 5()±0.1 mM.
[0111] In some embodiments, the loading step is performed with nucleic acid solution comprising a buffer salt at a concentration of about 50 mM.
[0112] In some embodiments of the loading step, the nucleic acid solution is at a pH from 2.0 to 9.0, such as from 2.5 to 8.5, from 2.6 to 8.4, from 2.7 to 8.3, from 2.8 to 8.2, from 2.9 to 8.1. from 3.0 to 8.0. from 3.2 to 7.8, from 3.4 to 7.6, from 3.6 to 7.4, from 3.8 to 7.2, from 4.0 to 7.0. from 4.1 to 6.8,
from 4.2 to 6.6, from 4.3 to 6.4, from 4.4 to 6.2, from 4.5 to 6.0, from 4.6 to 5.9, from 4.7 to 5.8, from 4.8 to 5.7, from 4.9 to 5.6, from 5.0 to 5.5, from 5.1 to 5.4, or from 5.2 to 5.3.
[0113] In some embodiments of the loading step, the pH of the nucleic acid solution is from 2 to 9, such as 2 to 7, or 3 to 5. In some embodiments, the pH of the nucleic acid solution is 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, 8.5, or 9.0. In some embodiments, the pH of the nucleic acid solution is within 10%. 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0114] In some embodiments, the loading step is performed at a pH of 2.0. 2.1, 2.2, 2.3, 2.4, 2.5, 2.6,
2.7, 2.8, 2.9. 3.0, 3.1, 3.2, 3.3, 3.4, 3.5, 3.6, 3.7. 3.8, 3.9, 4.0, 4.1, 4.2, 4.3, 4.4, 4.5, 4.6, 4.7, 4.8, 4.9, 5.0, 5.1, 5.2, 5.3, 5.4, 5.5, 5.6, 5.7, 5.8, 5.9, 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, 7.0, 7.1, 7.2,
7.3, 7.4, 7.5, 7.6, 7.7, 7.8, 7.9, 8.0, 8.1, 8.2, 8.3, 8.4, 8.5, 8.6, 8.7, 8.8, 8.9, or 9.
[0115] In some embodiments, the loading step is performed at a pH of less than 2.0. less than 2.5, less than 2.6. less than 2.7, less than 2.8, less than 2.9, less than 3.0, less than 3.2. less than 3.4 less than 3.6, less than 3.8, less than 4.0. less than 4.1, less than 4.2, less than 4.3. less than 4.4, less than 4.5, less than 4.6, less than 4.7, less than 4.8, less than 4.9, less than 5.0, less than 5.1, less than 5.2. less than
5.3, less than 5.4, less than 5.5, less than 5.6. less than 5.7, less than 5.8, less than 5.9, less than 6.0, less than 6.2, less than 6.4, less than 6.6, less than 6.8, less than 7.0, less than 7.2, less than 7.4, less than 7.6, less than 7.8, less than 8.0, less than 8.1, less than 8.2, less than 8.3, less than 8.4, less than 8.5, or less than 9.0.
[0116] In some embodiments, the loading step is performed at a pH of greater than 2.0, greater than
2.5, greater than 2.6, greater than 2.7, greater than 2.8, greater than 2.9, greater than 3.0, greater than
3.2, greater than 3.4 greater than 3.6, greater than 3.8, greater than 4.0, greater than 4.1, greater than
4.2, greater than 4.3, greater than 4.4, greater than 4.5, greater than 4.6, greater than 4.7, greater than
4.8, greater than 4.9, greater than 5.1, greater than 5.2, greater than 5.3, greater than 5.4, greater than
5.5, greater than 5.6, greater than 5.7, greater than 5.8, greater than 5.9, greater than 6.0, greater than
6.2, greater than 6.4, greater than 6.6, greater than 6.8, greater than 7.0, greater than 7.2, greater than
7.4, greater than 7.6, greater than 7.8, greater than 8.0. greater than 8.1, greater than 8.2, greater than
8.3, greater than 8.4, greater than 8.5, or greater than 8.6.
[0117] In some embodiments, the loading step is performed at a pH of 3.0±2.0, 3.0±1.5, 3.0±1.0, 3.0±0.9, 3.0±0.8, 3.0±0.7, 3.0±0.6, 3.0±0.5, 3.0±0.4, 3.0±0.3, 3.0±0.2, or 3.0±0.1.
[0118] In some embodiments, the loading step is performed at a pH of 4.5±2.0, 4.5±1.5, 4.5±1.0, 4.5±0.9, 4.5±0.8, 4.5±0.7, 4.5±0.6, 4.5±0.5, 4.5±0.4, 4.5±0.3, 4.5±0.2, or 4.5±0.1.
[0119] In some embodiments, the loading step is performed with a nucleic acid solution comprising a buffer salt having a pH of about 3.0. In some embodiments, the loading step is performed with nucleic acid solution comprising a buffer salt having a pH of about 4.5. In some embodiments, the loading step is performed with a nucleic acid solution comprising an acetate buffer salt. In some embodiments, the loading step is performed with a nucleic acid solution comprising 6.25 mM sodium acetate. In some embodiments, the loading step is performed with a nucleic acid solution comprising 6.25 mM sodium acetate at pH 3.0. In some embodiments, the loading step is perfonned with a nucleic acid solution comprising 6.25 mM sodium acetate at pH 4.5. In some embodiments, the loading step is performed with a nucleic acid solution comprising 50 mM sodium acetate. In some embodiments, the loading step is performed with a nucleic acid solution comprising 50 mM sodium acetate at pH 3.0. In some embodiments, the loading step is performed with a nucleic acid solution comprising 50 mM sodium acetate at pH 4.5.
[0120] Various methods may be used to prepare a nucleic acid solution suitable for the loading step. In some embodiments, the nucleic acid may be directly dissolved in a buffer solution described herein. In some embodiments, a nucleic acid solution may be generated by mixing nucleic acid stock solution with a buffer solution prior to mixing with a homogeneous empty LNP composition. In some embodiments, a nucleic acid solution may be generated by mixing a nucleic acid stock solution with a buffer solution immediately before mixing with a homogeneous empty LNP composition.
[0121] In some embodiments, the loading step is performed by mixing the homogeneous LNP composition with a nucleic acid solution (i.e., as described herein) at a flow rate from 50 mL/min to 100 L/min. In some embodiments, the flow rate is from 100 mL/min to 100 L/min. such as 100 mL/min to 95 L/min. 150 mL/min to 90 L/min, 200 mL/min to 85 L/min, 250 mL/min to 80 L/min, 300 mL/min to 75 L/min, 350 mL/min to 70 L/min, 400 mL/min to 65 L/min, 450 mL/min to 60 L/min, or 500 mL/min to 55 L/min, or 500 mL to 50 L/min. In some embodiments, the flow rate is from 100 mL/min to 50 L/min. such as 100 mL/min to 45 mL/min, 100 mL/min to 40 L/min. 100 mL/min to 35 L/min, 100 mL/min to 30 L/min. 100 mL/min to 25 L/min, 100 mL/min to 20 L/min, 100 ml/min to 15 L/min, 100 mL/min to 10 L/min, or 100 mL/min to 5 L/min. In some embodiments, the flow rate is 100 mL/min to 3L/min. such as 100 mL/min to 2.5 L/min. or 100 mL min to 2 L/min. In some embodiments, the flow rate is 100 inL/inin to 2 L/min. such as 150 mL/min to 2 L/min, 200 mL/min to 2 L/min, 300 mL/min to 2 L/min, 400 mL/min to 2 L/min, 500 mL/min to 2 L/min, 600 mL/min to 2L/min, 700 mL/min to 2L/min, 800 mL/inin to 2L/min, 900 mL/min to 2 L/min, 1 L/min to 2 L/min, 1.1 L/min to 2 L/min, 1.2 L/min to 2L/min, 1.3 L/min to 2 L/min, 1.4 L/min to 2 L/min. 1.5 L/min to 2 L/min, 1.6 L/min to 2 L/min, 1.7 L/min to 2 L/min, 1.8 L/min to 2 L/min, or 1.9 L/min to 2 L/min.
[0122] In some embodiments, the loading step is performed by mixing the homogenous LNP composition with a nucleic acid solution at a flow rate of at least 100 mL/min, such as at least 150
mL/min, at least 200 mL/min, at least 250 inL/min, at least 300 mL/min, at least 350 mL/min, at least 400 mL/min, at least 450 mL/min, at least 500 mL/min, at least 550 mL/min, at least 600 mL/min, at least 650 mL/min, at least 700 mL/min, at least 750 mL/min, at least 800 mL/min, at least 850 mL/min, at least 900 mL/min, at least 950 mL/min, at least 1,000 mL/min, at least 1,100 mL/min, at least 1,200 mL/min, at least 1,300 mL/min, at least 1,400 mL/min, at least 1,500 mL/min, at least 1,600 mL/min, at least 1,700 mL/min, at least 1,800 mL/min, at least 1,900 mL/min, or at least 2,000 mL/min.
[0123] In some embodiments, the loading step is performed by mixing the homogenous LNP composition with a nucleic acid solution at a flow rate of at least 1 L/min, such as at least 2 L/min, at least 3 L/min, at least 4 L/min, at least 5 L/min, at least 6 L/min, at least 7 L/min, at least 8 L/min, at least 9 L/min, at least 10 L/min. at least 11 L/min, at least 12 L/min, at least 13 L/min, at least 14 L/min, at least 15 L/min, at least 16 L/min, at least 17 L/min, at least 18 L/min, at least 19 L/min, at least 20 L/min, at least 21 L/min, at least 22 L /min, at least 23 L/min. at least 24 L/min, at least 25 L/min, at least 26 L/min, at least 27 L/min, at least 28 L/min, at least 29 L/min, at least 30 L/min, at least 31 L/min, at least 32 L/min, at least 33 L/min. at least 34 L/min, at least 35 L/min, at least 36 L/min, at least 37 L/min, at least 38 L/min, at least 39 L/min, at least 40 L/min, at least 41 L/min, at least 42 L/min, at least 43 L/min, at least 44 L/min. at least 45 L/min. at least 46 L/min, at least 47 L/min, at least 48 L/min, at least 49 L/min. or at least 50 L/min.
[0124] In some embodiments of the loading step, the mixing of the homogeneous LNP composition with the nucleic acid solution is performed using a pump. In some embodiments, the pump is selected from a gear pump, a peristatic pump, or a centrifugal pump.
[0125] In some embodiments of the loading step, the mixing may be performed using microfluidic mixers. Exemplary microfluidic mixers may include, but are not limited to, a slit interdigital micromixer including, but not limited to, those manufactured by Precision Nanosystems (Vancouver. BC, Canada), Microinnova (Allerheiligen bei Wildon, Austria) and/or a staggered herringbone micromixer (SHM) (Zhigaltsev, I.V. et al. (2012) Langmuir. 28:3633-40; Belliveau, N.M. et al. Mol. Ther. Nucleic. Acids. (2012) Le37; Chen. D. et al. J. Am. Chem. Soc. (2012) 134(22):6948-51; each of which is herein incorporated by reference in its entirety).
[0126] In some embodiments of the loading step, the mixing may further comprise combining at least two input streams wherein mixing occurs by microstructure-induced chaotic advection (MICA). According to this mixing method, fluid streams flow through channels present in a herringbone pattern causing rotational flow and folding the fluids around each other. This method may also comprise a surface for fluid mixing wherein the surface changes orientations during fluid cycling. Known methods of generating LNPs using SHM include those disclosed in U.S. Pat. Pub. Nos. US2004/0262223 Al and US2012/0276209 Al, each of which is incorporated herein by reference in their entirety.
[0127] In one embodiment of the loading step, die mixing may be performed using a micromixer such as, but not limited to, a Slit Interdigital Microstructured Mixer (SIMM-V2) or a Standard Slit Interdigital Micro Mixer (SSIMM) or Caterpillar (CPMM) or Impinging-jet (IJMM)from the Institut fur Mikrotechnik Mainz GmbH, Mainz Gennany). In one embodiment, the loading step is carried out by utilizing microfluidic technology (see, Whitesides (2006) Nature. 442: 368-373; and Abraham et al. (2002) Science. 295: 647-651; each of which is herein incorporated by reference in its entirety). As a non-limiting example, controlled microfluidic formulation includes a passive method for mixing streams of steady pressure-driven flows in micro channels at a low Reynolds number (see, e.g., Abraham et al. (2002) Science. 295: 647651; which is herein incorporated by reference in its entirety).
[0128] In one embodiment of the loading step, the mixing may be performed using a micromixer chip such as. but not limited to, those from Harvard Apparatus (Holliston. MA). Dolomite Microfluidics (Royston, UK), or Precision Nanosystems (Van Couver, BC. Canada). A micromixer chip can be used for rapid mixing of two or more fluid streams with a split and recombine mechanism.
[0129] In some embodiments the loading step produces a loaded LNP composition comprising LNPs encapsulating a nucleic acid. In some embodiments, the nucleic acid is encapsulated in the loaded LNPs with an encapsulation efficiency of from 50 to 100%. In some embodiments, the nucleic acid is encapsulated in the loaded LNPs with an encapsulation efficiency of 50% or more, such as 60% or more. 70% or more, 80% or more, or 90% or more. In some embodiments, the nucleic acid is encapsulated in the loaded LNPs with an encapsulation efficiency of at least 50%, for example 50%, 55%, 60%, 65%, 70%. 75%. 80%, 85%, 90%, 91%. 92%. 93%, 94%, 95%, 96%. 97%. 98%, 99%, or 100%. In some embodiments, the encapsulation efficiency may be at least 80%. In certain embodiments, the encapsulation efficiency may be at least 90%.
[0130] In some embodiments of the loading step, the nucleic acid is an RNA polynucleotide. In some embodiments, the RNA is a circular RNA polynucleotide (aka circRNA or oRNA™) (as described herein).
[0131] In some embodiments of the loading step, the loaded LNPs formed have an average particle size (average particle diameter) from 50 inn to 300 mn and a polydispersity of 0.3 or less. In some embodiments of the loading step, the loaded LNPs formed have an average particle size from 70 nm to 120 nm and a polydispersity of 0.2 or less.
[0132] In some embodiments of the loading step, the loaded LNP produced is storage stable at a temperature of from 4 °C to -80 °C. In some embodiments, the loaded LNPs produced are stored in a storage buffer or tonicity modifier. In some embodiments, the storage buffer or tonicity modifier includes, but is not limited to, tris-sucrose saline, tris-trehalose saline, phosphate buffered saline, or tris buffered saline.
[0133] In some embodiments, the loading step (described herein) may be conducted prior to the homogenization step (described herein). Accordingly, provided herein is a method of preparing a loaded LNP composition, the method comprising: a) a precipitation step, comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first LNP composition comprising empty LNPs; b) a loading step, comprising: mixing the first LNP composition with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with tire nucleic acid; and c) a homogenization step, comprising: homogenizing the loaded LNP composition to produce a homogeneous loaded LNP composition comprising LNPs associated with the nucleic acid, wherein each of steps a)-c) above are as described herein above in “Methods of Preparation.” sections i)-iii). iv. Additional processing steps
[0134] The subject methods may optionally include one or more further processing steps. In some embodiments, one or more further processing steps may be performed on the homogeneous LNP composition of empty LNPs or the loaded LNP composition. In some embodiments, the further processing steps include one or more of filtering, pH adjusting, buffer exchanging, diluting, concentrating, freezing, lyophilizing, storing, adding a cryoprotectant, and packing.
[0135] In some embodiments, one or more further processing steps include a dilution and buffer exchange step of the homogeneous empty LNP composition, filtration of the homogeneous empty LNP composition, and storage of the homogeneous empty LNP composition.
[0136] In some embodiments, one or more further processing steps include a dilution and buffer exchange step of the loaded LNP composition, filtration of the loaded LNP composition, and storage of the loaded LNP composition.
[0137] In some embodiments after the homogenizing step, a dilution and buffer exchange step may be performed on the composition of homogeneous empty LNPs. In some embodiments, the buffer exchange step is performed by filtration. In some embodiments, the buffer exchange step is performed via tangential flow filtration (TFF).
[0138] In some embodiments after the loading step, a dilution and buffer exchange step may be performed on the composition of loaded LNPs. In some embodiments, the buffer exchange step is
performed by filtration. In some embodiments, the buffer exchange step is performed via tangential flow filtration (TFF).
[0139] In some embodiments, filtration removes an organic solvent (e.g., an alcohol or ethanol) from the composition of empty LNPs or loaded LNPs. In some embodiments, filtration removes substantially all of the organic solvent (e.g., an alcohol or ethanol) from the composition of empty LNPs or loaded LNPs.
[0140] In some embodiments, the additional further processing steps include pH adjustment of the composition comprising the loaded LNPs. The pH adjustment may be carried out by adding a second buffering agent.
[0141] In some embodiments, the additional further processing steps include concentrating the composition of empty LNPs or loaded LNPs.
[0142] In some embodiments, the additional further processing steps includes freezing the composition of empty LNPs or loaded LNPs by. e.g., lyophilization
[0143] In some embodiments, the lyophilizing comprises freezing the composition of empty LNPs or loaded LNPs at a temperature from -100 °C to 0 °C, such as -80 °C to -10 °C, -60 ° C to -20 °C, -50 °C to -25 °C, or -40 °C to -30 °C.
[0144] In some embodiments, the lyophilizing further comprises drying the frozen composition of empty LNPs or loaded LNPs to form a lyophilized empty LNP or lyophilized loaded LNP.
[0145] In some embodiments, the further processing steps include adding a cryoprotectant to the composition of empty’ LNPs or loaded LNPs.
[0146] In some embodiments, the further processing steps include storage of the composition of empty LNPs or loaded LNPs. In some embodiments, the composition of empty LNPs or loaded LNPs are stored at a temperature of 4 °C. In some embodiments, the composition of empty’ LNPs or loaded LNPs are stored at a temperature from -100 °C to 0 °C, such as -80 °C to -10 °C, -60 0 C to -20 °C, -50 °C to -25 °C, or -40 °C to -30 °C. In some embodiments, the composition of empty LNPs or loaded LNPs arc stored in a storage buffer or tonicity modifier. In some embodiments, the storage buffer or tonicity modifier includes, but is not limited to, tris-sucrose saline, tris-trehalose saline, phosphate buffered saline, or tris buffered saline.
[0147] In some embodiments, the further processing steps include packing of the composition of empty LNPs or loaded LNPs. As used herein, “packing” may refer to storing a drug product in its final state or in-process storage of an empty LNP, or loaded LNP before they are placed into final packaging. Modes of storage and/or packing include, but are not limited to, refrigeration in sterile bags, refrigerated or frozen formulations in vials, lyophilized formulations in vials and syringes, etc.
2. EMPTY LIPID NANOPARTICLES (EMPTY LNPs)
[0148] As summarized herein, the present disclosure provides a homogeneous LNP composition of empty LNPs prepared by a subject method.
[0149] In some embodiments, there is provided a homogeneous composition of empty LNPs comprising an ionizable lipid, a helper lipid, a cholesterol and a PEG lipid, wherein the empty LNPs have a polydispersity of 0.2 or less, such as 0.1 or less. The term “homogeneous” as it applies to a composition of empty7 LNPs refers to the population of empty LNPs in the composition having a polydispersity7 of 0.2 or less.
[0150] In some embodiments, there is provided a homogeneous LNP composition substantially free of loaded LNPs and comprising empty LNPs comprising an ionizable lipid, a helper lipid, a cholesterol and a PEG lipid, wherein the empty LNPs have a poly dispersity of 0.2 or less (e.g., 0. 1 or less), and/or an average particle size from 50 nm to 70 nm. The term “substantially7 free” as it applies to loaded LNPs refers to the homogeneous LNP composition of empty LNPs including few or no loaded LNPs. In some embodiments, substantially free of loaded LNPs refers to a homogeneous LNP composition including an amount of less than 5%, such as less than 4%, less than 3%, less than 2%, less than 1%, less than 0.9%, less than 0.8%, less than 0.7%, less than 0.6%, less than 0.5%, less than 0.4%, less than 0.3%, less than 0.2% or less than 0.1% of the loaded LNPs. In some embodiments, the homogeneous LNP composition is at least 99% free of loaded LNPs. In some embodiments, the homogeneous LNP composition includes no loaded LNPs.
[0151] In some aspects of the present disclosure, there is provided an empty7 LNP composition prepared by a method disclosed herein.
[0152] In some embodiments of the empty LNP composition, the empty LNPs comprises a molar ratio of from 40% to 60% ionizable lipid, a molar ratio of from 3.5% to 14% helper lipid, a molar ratio of from 28% to 50% structural lipid, and a molar ratio of from 0.5% to 5% PEG-lipid. In some embodiments, the total molar percentage of the ionizable lipid, the helper lipid, the structural lipid, and the PEG-lipid is 100% in tire empty LNPs.
[0153] In some embodiments of the empty LNP composition, the molar ratio of the ionizable lipid in the empty LNPs is from 40 to 60% of the total lipid present in the empty LNPs. In some embodiments, the molar ratio of the ionizable lipid in the empty LNPs is 40%, 41%, 42%. 43%, 44%, 45%. 46%, 47%, 48%, 49%, 50%. 51 %, 52%, 53%, 54%. 55%, 56%, 57%, 58%, 59%. or 60% of the total lipid present in the empty LNPs.
[0154] In some embodiments of the empty LNP composition, the molar ratio of the helper lipid in the empty LNPs is from 3.5% to 14% of the total lipid present in the empty LNPs. In some embodiments, the molar ratio of the helper lipid in the lipid solution is 3%, 4%, 5%, 6%. 7%, 8%, 9%, 10%, 11%,
12%, 13%, or 14% of the total lipid present in the empty LNPs. In some embodiments, the helper lipid is DSPC. In some embodiments, the helper lipid is DOPE.
[0155] In some embodiments of the empty LNP composition, the molar ratio of the structural lipid in the empty LNPs is from 28% to 50% of the total lipid present in the empty LNPs. In some embodiments, tire molar ratio of the structural lipid in the empty LNPs is 28%, 29%, 30%, 31%. 32%, 33%, 34%, 35%, 36%, 37%, 38%, 39%, 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, or 50% of the total lipid present in the empty LNPs. In some embodiments, the structural lipid is cholesterol.
[0156] In some embodiments of the empty' LNP composition, the molar ratio of the PEG-lipid in the empty’ LNPs is from 0.1% to 5% of the total lipid present in the empty LNPs. In some embodiments, the molar ratio of the PEG-lipid in the empty LNPs is 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1.0%, 1.1%, 1.2%. 1.3%, 1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, 2.0%, 2.1%, 2.2%, 2.3%, 2.4%, 2.5%, 2.6%, 2.7%, 2.8%. 2.9%, 3.0%, 3.1%, 3.2%, 3.3%, 3.4%, 3.5%, 4.0%, 4.5%, or 5% of the total lipid present in the empty LNPs. In some embodiments, the PEG-lipid is DMG-PEG2000.
[0157] In some embodiments of the empty LNP composition, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the empty LNPs is 45:9:44:2. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the empty LNPs is 50:10:38.5:1.5. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the empty LNPs is 41:12:45:2. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the empty' LNPs is 62:4:33:1. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the empty LNPs is 53:5:41:1. In some embodiments, the molar ratio of each of tire ionizable lipid, helper lipid, structural lipid, and PEG-lipid is within 10%, 9%. 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%. 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%. or 0.01% of the stated value.
[0158] In some embodiments of the empty LNP composition, the empty LNPs comprise an ionizable lipid as described herein, l,2-distearoyl-sn-glycero-3phosphocholine (DSPC), cholesterol, and DMG- PEG2000.
[0159] In some embodiments of the empty LNP composition, the empty LNPs comprise 30-60 mol % ionizable lipid (e.g., as described herein); 5-30 mol % DSPC; 15-50 mol % cholesterol; and 1-2 mol % DMG-PEG2000.
[0160] In one embodiment of the homogeneous composition of empty LNPs. the LNPs have an average particle size (i.e., an average nanoparticle diameter) from 10 nm to 100 nm such as, but not limited to, 10 nm to 20 nm, 10 nm to 30 nm. 10 nm to 40 nm. 10 mn to 50 nm, 10 nm to 60 nm, 10 nm to 70 nm, 10 nm to 80 mn, 10 nm to 90 nm, 20 nm to 30 nm. 20 nm to 40 nm, 20 nm to 50 nm, 20 nm to 60 nm, 20 nm to 70 nm, 20 nm to 80 nm, 20 nm to 90 nm, 20 nm to 100 nm, 30 nm to 40 nm, 30 nm
to 50 nm, 30 nm to 60 nm, 30 nm to 70 nm, 30 nm to 80 nm, 30 nm to 90 nm, 30 nm to 100 nm, 40 nm to 50 nm, 40 nm to 60 nm, 40 nm to 70 nm, 40 nm to 80 nm, 40 nm to 90 nm, 40 nm to 100 nm, 50 nm to 60 nm, 50 nm to 70 nm 50 nm to 80 mn, 50 nm to 90 mn, 50 nm to 100 nm, 60 mn to 70 nm, 60 nm to 80 nm, 60 nm to 90 nm, 60 nm to 100 mn, 70 nm to 80 mn, 70 mn to 90 nm, 70 mn to 100 nm, 80 mn to 90 nm, 80 nm to 100 nm, or 90 nm to 100 mn. In one embodiment, the lipid nanoparticles may have a diameter from 30 to 200 nm. In one embodiment, the homogeneous composition of empty LNPs may have an average particle size of less than 200 nm, less than 180 nm, less than 150 nm, less than 130 nm. less than 120 nm, less than 100 nm, less than 80 nm, less than 60 nm, less than 50 nm, or less than 40 nm. Each possibility represents a separate embodiment of the present disclosure.
[0161] In some embodiments of the homogeneous composition of empty LNPs, the LNPs have an average particle size of 10-200 mn, 20-200 nm, 30-200 nm, or 50-200 nm. In some embodiments, the empty LNPs have an average particle size of 50-200 nm, 50-150 nm, 50-100 mn, or 50-70 nm. In some embodiments, the empty LNPs have an average particle size of 50 to 70 nm.
[0162] In some embodiments of the homogeneous composition of empty LNPs, the LNPs have an average particle size from 1 mn to 100 mn, from 1 nm to 10 nm, 1 mn to 20 nm, from 1 nm to 30 mn, from 1 mn to 40 nm. from 1 nm to 50 nm, from 1 mn to 60 mn, from 1 nm to 70 mn, from 1 nm to 80 mn, from 1 nm to 90 mn, from 5 nm to 100 nm, from 5 nm to 10 mn, 5 mn to 20 nm, from 5 mn to 30 mn, from 5 nm to 40 nm. from 5 nm to 50 mn, from 5 nm to 60 nm, from 5 nm to 70 nm, from 5 nm to 80 nm, from 5 mn to 90 nm, 10 mn to 50 nm, from 20 nm to 50 mn, from 30 nm to 50 nm, from 40 mn to 50 nm, from 20 nm to 60 nm, from 30 nm to 60 mn, from 40 nm to 60 nm, from 20 mn to 70 nm, from 30 mn to 70 nm, from 40 nm to 70 nm, from 50 nm to 70 nm, from 60 mn to about 70 mn, from 20 nm to 80 mn. from 30 nm to 80 nm, from 40 nm to 80 nm, from 50 nm to 80 nm, from 60 nm to 80 mn. from 20 nm to 90 mn, from 30 nm to 90 nm, from 40 nm to 90 nm. from 50 nm to 90 nm, from 60 mn to 90 nm, or from 70 to 90 nm. Each possibility represents a separate embodiment of the present disclosure.
[0163] In some embodiments of the homogeneous composition of empty LNPs, the LNPs have an average particle size of 50 mn, 51 nm, 52. nm, 53 nm, 54 nm, 55 nm. 56 nm, 57 mn. 58 mn, 59 nm, 60 mn. 61 nm, 62 nm. 63 nm, 64 nm, 65 nm, 66 nm, 67 mn, 68 nm, 69 mn. or 70 nm. In some embodiments, the average particle size of the homogeneous composition of empty LNPs is within 10%, 9%, 8%, 7%, 6%. 5%, 4%, 3%. 2%. 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%. 0.4%. 0.3%. 0.2%, 0.1%, 0.09%. 0.08%, 0.07%, 0.06%, 0.05%. 0.04%, 0.03%, 0.02%. or 0.01% of the stated value.
[0164] In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition is at least 10% less than the average particle size of the empty LNPs of the first LNP composition. In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition is 10-50% less than the average particle size of the empty LNPs of the first LNP
composition. In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition is 10-30% less than the average particle size of the empty LNPs of the first LNP composition. In some embodiments, the average particle size of the empty LNPs in the homogeneous LNP composition is 10% less, such as 15% less, 20% less, 25% less, 30% less, 35% less, 40% less, 45% less, or 50% less than the average particle size of the empty LNPs of the first LNP composition.
[0165] As disclosed herein, the subject methods provide a homogeneous composition of empty LNPs. A poly dispersity index may be used to indicate the homogeneity of the empty LNPs in the composition, e.g., the particle size distribution of the nanoparticle compositions. A small (e g., less than 0.2) polydispersity index generally indicates a narrow particle size distribution. An empty LNP composition may have a poly dispersity index from about 0.01 to about 0.2, such as 0.01, 0.02, 0.03, 0.04, 0.05. 0.06, 0.07, 0.08, 0.09, 0.10, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, or 0.20. In some embodiments, the polydispersity index of an empty LNP composition may be from 0.05 to 0.20. In some embodiments, the poly dispersity index of the homogeneous composition of empty LNPs is within 10%, 9%, 8%, 7%, 6%. 5%, 4%, 3%, 2%. 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%. 0.3%. 0.2%, 0.1%, 0.09%. 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0166] In some embodiments, the polydispersity index of the homogeneous composition of empty LNPs is 0.2 or less, such as 0.19 or less, 0.18 or less, 0.17 or less, 0.16 or less, 0.15 or less, 0.14 or less, 0.13 or less, 0.12 or less. 0.11 or less, or 0.10 or less. In some embodiments, the poly dispersity index of the empty LNP composition is 0.1 or less, such as 0.09 or less, 0.08 or less, 0.07 or less, 0.06 or less, or 0.05 less.
[0167] The zeta potential of a nanoparticle composition may be used to indicate the electrokinetic potential of the composition. For example, the zeta potential may describe the surface charge of a nanoparticle composition. Nanoparticle compositions with relatively low charges, positive or negative, are generally desirable, as more highly charged species may interact undesirably with cells, tissues, and other elements in the body. In some embodiments, the zeta potential of the subject empty' LNPs may be from -20 mV to +20 mV, from -20 mV to +15 mV, from -20 mV to +10 mV, from -20 mV to +5 mV, from -20 mV to 0 mV, from -20 mV to -5 mV, from -20 mV to -10 mV. from -20 mV to -15 mV from -20 mV to +20 mV, from -20 mV to +15 mV, from -20 mV to +10 mV. from -20 mV to +5 mV, from -20 mV to 0 mV. from 0 mV to +20 mV, from 0 mV to +15 mV. from 0 mV to +10 mV, from 0 mV to +5 mV, from +5 mV to +20 mV. from +5 mV to +15 mV. or from +5 mV to +10 mV. In some embodiments, the zeta potential of the subject empty LNPs is within 10%, 9%. 8%. 7%, 6%, 5%, 4%. 3%, 2%. 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%. 0.04%. 0.03%, 0.02%. or 0.01% of the stated value.
[0168] The properties of a homogeneous composition of empty' LNPs may be influenced by factors including, but not limited to. the selection of the cationic lipid component, the degree of cationic lipid
saturation, the selection of the helper hpid component, the degree of helper lipid saturation, the selection of the cholesterol lipid component, the selection of the PEG lipid component, the nature of the PEGylation, ratio of all components and biophysical parameters such as size.
[0169] In some embodiments, the homogeneous composition of empty LNPs has a pH from 2.0 to 9.0, from 2.5 to 8.5, from 2.6 to 8.4, from 2.7 to 8.3, from 2.8 to 8.2, from 2.9 to 8.1, from 3.0 to 8.0, from 3.2 to 7.8, from 3.4 to 7.6, from 3.6 to 7.4, from 3.8 to 7.2, from 4.0 to 7.0, from 4.1 to 6.8. from 4.2 to 6.6, from 4.3 to 6.4, from 4.4 to 6.2, from 4.5 to 6.0, from 4.6 to 5.9, from 4.7 to 5.8, from 4.8 to 5.7, from 4.9 to 5.6, from 5.0 to 5.5, from 5.1 to 5.4, or from 5.2 to about 5.3.
[0170] In some embodiments, the homogeneous composition of empty LNPs has a pH from 2 to 9, such as 2 to 6, or 3 to 5. In some embodiments, the pH of the homogeneous composition of empty LNPs is 2.0. 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, 8.5, or 9.0. In some embodiments, the pH of the homogeneous composition of empty LNPs is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0171] In some embodiments, the homogeneous composition of empty LNPs can be stored at 4 °C, and is storage stable for at least 4 weeks, such as at least 8 weeks, at least 12 weeks, at least 16 weeks, at least 20 weeks, at least 6 months, at least 9 months, or at least a year.
[0172] In some embodiments, the homogeneous composition of empty LNPs or is stored at a temperature from -100 °C to 0 °C, such as -80 °C to -10 °C, -60 0 C to -20 °C. -50 °C to -25 °C, or -40 °C to -30 °C. In some embodiments, the homogeneous composition of empty LNPs is stored in a storage buffer or tonicity modifier. In some embodiments, the storage buffer or tonicity modifier includes, but is not limited to. tris-sucrose saline, tris-trehalose saline, phosphate buffered saline, or tris buffered saline.
[0173] In some embodiments, the homogeneous composition of empty LNPs is packed in a vessel or bag for storage. In some embodiments, the homogeneous composition of empty LNPs is packed in sterile bags, vials or syringes and refrigerated or frozen. In some embodiments, the homogeneous composition of empty LNPs is lyophilized in a storage vessel, such as a vial or a syringe.
3. LOADED LIPID NANOPARTICLES (LOADED LNPs)
[0174] As summarized herein, the present disclosure provides a loaded LNP composition prepared by a subject method.
[0175] In some embodiments, there is provided a homogeneous LNP composition of loaded LNPs comprising an ionizable lipid, a helper hpid, a cholesterol, a PEG lipid, and a nucleic acid, wherein the loaded LNPs have an average particle size of 70-120 nm with a polydispersity of 0.3 or less, such as 0.2 or less, or 0.1 or less. The term “homogeneous” as it applies to a composition of loaded LNPs refers
to the population of empty LNPs in the composition having an average particle size of 70-120 mn with a polydispersity of 0.3 or less, such as 0.2 or less, or 0.1 or less.
[0176] In some aspects of the present disclosure, there is provided a loaded LNP composition prepared by a method disclosed herein.
[0177] In some embodiments of the loaded LNP composition, the loaded LNPs comprise a molar ratio of from 40% to 60% ionizable lipid, a molar ratio of from 3.5% to 14% helper lipid, a molar ratio of from 28% to 50% structural lipid, and a molar ratio of from 0.5% to 5% PEG-lipid. In some embodiments, the total molar percentage of the ionizable lipid, the helper lipid, the structural lipid, and the PEG-lipid is 100% in tire loaded LNPs.
[0178] In some embodiments of the loaded LNP composition, the molar ratio of the ionizable lipid in the loaded LNP is from 40% to 60% of the total lipid present in the loaded LNPs. In some embodiments, the molar ratio of the ionizable lipid in the loaded LNPs is 40%, 41%, 42%. 43%. 44%, 45%, 46%, 47%, 48%, 49%. 50%. 51%, 52%, 53%, 54%, 55%. 56%. 57%, 58%, 59%, or 60% of the total lipid present in the loaded LNPs.
[0179] In some embodiments of the loaded LNP composition, the molar ratio of the helper lipid in the loaded LNPs is from 3.5% to 14% of the total lipid present in the loaded LNPs. In some embodiments, the molar ratio of the helper lipid in the loaded LNP is 3%, 4%, 5%, 6%, 7%. 8%. 9%, 10%. 11%, 12%, 13%, or 14% of the total lipid present in the loaded LNPs. In some embodiments, the helper lipid is DSPC. In some embodiments, the helper lipid is DOPE.
[0180] In some embodiments of the loaded LNP composition, the molar ratio of the structural lipid in the loaded LNPs is from 28% to 50% of the total lipid present in the loaded LNPs. In some embodiments, the molar ratio of the structural lipid in the loaded LNPs is 28%, 29%. 30%. 31%, 32%, 33%, 34%, 35%. 36%, 37%, 38%, 39%, 40%. 41%, 42%, 43%, 44%. 45%. 46%, 47%, 48%, 49%. or 50% of the total lipid present in the loaded LNPs. In some embodiments, the structural lipid is cholesterol.
[0181] In some embodiments of the loaded LNP composition, the molar ratio of the PEG-lipid in the loaded LNPs is from 0.1% to 5% of the total lipid present in the loaded LNPs. In some embodiments, the molar ratio of the PEG-lipid in the loaded LNPs is 0.1%, 0.2%, 0.3%, 0.4%. 0.5%. 0.6%, 0.7%, 0.8%, 0.9%, 1.0%, 1.1%, 1.2%. 1.3%. 1.4%. 1.5%. 1.6%. 1.7%. 1.8%, 1.9%, 2.0%, 2.1%, 2.2%, 2.3%, 2.4%, 2.5%, 2.6%, 2.7%, 2.8%. 2.9%. 3.0%. 3.1%. 3.2%. 3.3%, 3.4%, 3.5%, 4.0%, 4.5%, or 5% of the total lipid present in the loaded LNPs. In some embodiments, the PEG-lipid is DMG-PEG2000.
[0182] In some embodiments of the loaded LNP composition, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the loaded LNPs is 45:9:44:2. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the loaded LNPs is 50:10:38.5: 1.5. In some
embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the loaded LNPs is 41:12:45:2. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the loaded LNPs is 62:4:33:1. In some embodiments, the molar ratio of ionizable lipid: helper lipid: structural lipid: PEG-lipid in the loaded LNPs is 53:5:41 : 1. In some embodiments, the molar ratio of each of the ionizable lipid, helper lipid, structural lipid, and PEG-lipid is within 10%, 9%. 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0183] In some embodiments of the loaded LNP composition, the loaded LNPs comprise an ionizable lipid as described herein, l,2-distearoyl-sn-glycero-3phosphocholine (DSPC), cholesterol, and DMG- PEG2000.
[0184] In some embodiments of the loaded LNP composition, the loaded LNPs comprise 30-60 mol % of ionizable lipid (e.g., as described herein); 5-30 mol % of DSPC; 15-50 mol % of cholesterol; and 1-2 mol % of DMG-PEG200U.
[0185] In one embodiment of the loaded LNP composition, the loaded LNPs have an average particle size (i.e., an average nanoparticle diameter) from 50 mn to 300 mn such as, but not limited to, 50 mn to 60 nm, 50 nm to 70 nm, 50 mn to 80 mn, 50 mn to 90 mn, 50 mn to 100 run, 50 nm to 120 nm, 50 mn to 150 nm, 50 mn to 200 nm, 70 nm to 80 mn, 70 nm to 90 nm, 70 nm to 100 nm, 70 nm to 110 nm, 70 nm to 120 nm, 70 nm to 130 nm, 70 mn to 140 nm, 70 nm to 150 nm, 80 nm to 90 nm, 80 nm to 100 nm, 80 nm to 110 nm, 80 nm to 120 nm, 80 mn to 130 nm, 80 nm to 140 nm, 80 nm to 150 nm, 90 nm to 100 nm, 90 nm to 110 nm, 90 nm to 120 nm, 90 nm to 130 nm, 90 nm to 140 nm, 90 nm to 150 nm, 100 nm to 110 mn, 100 nm to 120 nm. In one embodiment, the loaded LNPs may have an average particle size from 70 nm to 120 nm. In one embodiment, the loaded LNPs may have an average particle size of less than 300 nm, less than 250 nm, less than 200 mn, less than 180 nm, less than 150 nm, less than 120 nm, less than 100 mn, less than 90 mn, less than 80 nm, or less than 75 nm. Each possibility represents a separate embodiment of the present disclosure.
[0186] In some embodiments of the loaded LNP composition, the loaded LNPs have an average particle size of 50-300 nm, 60-300 nm, 70-300 nm, or 80-300 mn. In some embodiments, the loaded LNPs have an average particle size of 50-200 nm, 50-180 mn, 50-150 nm, or 50-120 nm.
[0187] In some embodiments of the loaded LNP composition, the loaded LNPs have an average particle size of from 1 nm to 100 mn, from 1 nm to 10 mn, 1 nm to 20 nm, from 1 nm to 30 nm, from 1 nm to 40 nm, from 1 mn to 50 nm, from 1 mn to 60 nm, from 1 mn to 70 nm, from 1 mn to 80 nm, from 1 mn to 90 nm, from 5 nm to 100 nm, from 5 mn to 10 nm, 5 nm to 20 mn, from 5 nm to 30 nm, from 5 nm to 40 nm, from 5 nm to 50 mn, from 5 mn to 60 nm, from 5 nm to 70 nm, from 5 nm to 80 nm, from 5 nm to 90 mn, 10 mn to 50 nm, from 20 nm to 50 nm, from 30 nm to 50 nm, from 40 nm to 50 mn, from 20 nm to 60 nm, from 30 nm to 60 nm, from 40 run to 60 mn, from 20 nm to 70 nm, from 30
nm to 70 nm, from 40 nm to 70 nm, from 50 nm to 70 nm, from 60 nm to about 70 mu, from 20 nm to 80 nm, from 30 nm to 80 nm, from 40 nm to 80 nm, from 50 nm to 80 nm, from 60 mn to 80 nm, from 20 nm to 90 mn, from 30 nm to 90 nm, from 40 nm to 90 nm, from 50 nm to 90 mn, from 60 nm to 90 mn, or from 70 to 90 nm. Each possibility represents a separate embodiment of the present disclosure.
[0188] In some embodiments of the loaded LNP composition, the loaded LNPs have an average particle size of 70 nm, 71 nm, 72, nm, 73 mn, 74 nm, 75 nm, 76 mn, 77 nm, 78 nm, 79 nm, 80 nm, 81 nm, 82 nm, 83 mn, 84 mn, 85 nm, 86 mn, 87 nm, 88 nm, 89 nm, 90 nm, 91 mn, 92 nm, 93 nm, 94 nm, 95 nm, 96 nm, 97 mn, 98 nm, 99 mn, 100 nm, 101 mn, 102 nm, 103 nm, 104 nm, 105 nm, 106 mn, 107 nm, 108 nm, 109 nm, 110 nm, 111 nm, 112 nm, 113 mn, 114 mn, 115 nm, 116 nm, 117 nm, 118 nm, 119 nm, or 120 mn. In some embodiments, the average particle size of the loaded LNPs is within 10%, 9%. 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0189] A loaded LNP composition may have a poly dispersity index from about 0.01 to about 0.3, such as 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.10, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, or 0.30. In some embodiments, the polydispersity index of a loaded LNP composition may be from 0.01 to 0.20. In some embodiments, the poly dispersity index of the loaded LNPs is within 10%, 9%, 8%, 7%, 6%, 5%, 4%. 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%. 0.4%. 0.3%. 0.2%. 0.1%. 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0190] In some embodiments, the polydispersity index of the loaded LNP composition is 0.3 or less, such as 0.25 or less, 0.20 or less, 0.15 or less, or 0.1 or less.
[0191] In some embodiments, the LNPs in the loaded LNP composition have an average particle size of 50 nm to 300 nm and a polydispersity of 0.3 or less. In some embodiments, the LNPs in the loaded LNP composition have an average particle size of 70 mn to 120 nm and a polydispersity of 0.2 or less.
[0192] In some embodiments of the loaded LNP composition, the zeta potential of the subject loaded LNPs may be from -20 Mv to +20 Mv, from -20 Mv to +15 Mv, from -20 Mv to +10 Mv, from -20 Mv to +5 Mv, from -20 Mv to 0 Mv, from -20 Mv to -5 Mv. from -20 Mv to -10 Mv, from -20 Mv to -15 Mv from -20 Mv to +20 Mv, from -20 Mv to +15 Mv, from -20 Mv to +10 Mv, from -20 Mv to +5 Mv, from -20 Mv to 0 Mv, from 0 Mv to +20 Mv, from 0 Mv to +15 Mv, from 0 Mv to +10 Mv, from 0 Mv to +5 Mv. from +5 Mv to +20 Mv, from +5 Mv to +15 Mv, or from +5 Mv to +10 Mv. In some embodiments, the zeta potential of the subject loaded LNPs is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0193] In some embodiments of the loaded LNP composition, the amounts of nucleic acid and empty LNPs are selected to provide a specific N:P ratio. In some embodiments of the loaded LNP composition, the molar ratio of nitrogen atoms in one or more ionizable lipids (i.e., in the empty LNPs) to phosphate groups in the nucleic acid (N:P) is within the range of 1:20 to 20: 1. In some embodiments, the N:P ratio is 1:20, 1: 19. 1:18, 1 :17, 1 :16, 1: 15, 1: 14, 1:13, 1 : 12, 1: 11, 1: 10, 1 :9, 1:8. 1:7, 1:6, 1 :5, 1 :4, 1:3, 1:2 or 1: 1. In some embodiments, the N:P ratio is 20:1. 19: 1, 18: 1, 17: 1. 16:1, 15: 1, 14: 1, 13: 1. 12: 1, 11 : 1, 10: 1, 9:1, 8: 1, 7: 1, 6: 1, 5: 1, 4: 1, 3: 1 or 2: 1. In some embodiments of the loaded LNP composition, the N:P ratio is within the range of 4:1 to 6: 1. In some embodiments of the loaded LNP composition, the N:P ratio is 4.0: 1, 4.1: 1, 4.2: 1, 4.3: 1. 4.4: 1. 4.5: 1. 4.6: 1. 4.7:1. 4.8: 1. 4.9: 1. 5.0: 1. 5.1: 1. 5.2: 1. 5.3: 1, 5.4: 1. 5.5: 1. 5.6: 1. 5.7: 1. 5.8: 1. 5.9: 1. or 6.0: 1. In some embodiments of the loaded LNP composition, the N:P ratio is within 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, or 0.01% of the stated value.
[0194] As disclosed herein, the loaded LNP is associated with a nucleic acid. In some embodiments, the nucleic acid is an RNA polynucleotide. In some embodiments, the RNA is a circular RNA polynucleotide (aka circRNA or Oma™) (as described herein). In some embodiments, the nucleic acid is encapsulated in the loaded LNP. In some embodiments, the nucleic acid is encapsulated in the loaded LNP with an encapsulation efficiency of from 50% to 100%, such as 60% to 100%, 70% to 100%, 80% to 100%. or 90% to 100%.
[0195] The efficiency of encapsulation of a therapeutic agent describes the amount of therapeutic agent that is encapsulated or otherwise associated with a nanoparticle composition after preparation, relative to the initial amount provided. The encapsulation efficiency is desirably high (e.g.. close to 100%). The encapsulation efficiency may be measured, for example, by comparing the amount of therapeutic agent in a solution containing the nanoparticle composition before and after breaking up the nanoparticle composition with one or more organic solvents or detergents. Fluorescence may be used to measure the amount of free therapeutic agent (e.g., nucleic acids) in a solution. For the nanoparticle compositions described herein, the encapsulation efficiency of a therapeutic agent (e.g., a nucleic acid) may be at least 50%. for example 50%. 55%. 60%, 65%, 70%, 75%, 80%, 85%, 90%. 91%, 92%, 93%, 94%, 95%, 96%, 97%. 98%, 99%, or 100%. In some embodiments, the encapsulation efficiency may be at least 80%. In certain embodiments, the encapsulation efficiency may be at least 90%. Each possibility represents a separate embodiment of the present disclosure.
[0196] The properties of a loaded LNP may be influenced by factors including, but not limited to, the selection of the cationic lipid component, the degree of cationic lipid saturation, the selection of the helper lipid component, the degree of helper lipid saturation, the selection of the cholesterol lipid
component, the selection of the PEG-lipid component, the nature of the PEGylation, ratio of all components, and biophysical parameters such as size.
[0197] In some embodiments, the loaded LNP composition is storage stable at a temperature from 4 °C to -80 °C. In some embodiments, the loaded LNP composition is stored at a temperature from -80 °C to 0 °C, such as -80 °C to -10 °C, -60 °C to -20 °C, -50 °C to -25 °C, or -40 °C to -30 °C. In some embodiments, the loaded LNP composition is stored in a storage buffer or tonicity’ modifier. In some embodiments, the storage buffer or tonicity modifier includes, but is not limited to, tris-sucrosc saline, tris-trehalose saline, phosphate buffered saline, or tris buffered saline.
[0198] In some embodiments, the loaded LNP composition is packed in a vessel or a bag for storage. In some embodiments, the loaded LNP composition is packed in a sterile bag, vial or syringe and refrigerated or frozen. In some embodiments, the loaded LNP composition is lyophilized in a storage vessel, such as a vial or a syringe.
[0199] In some embodiments, the loaded LNP composition can be stored at 4 °C, and is storage stable for at least 4 weeks, such as at least 8 weeks, at least 12 weeks, at least 16 weeks, at least 20 weeks, at least 6 months, at least 9 months, or at least a year.
4. LIPID SOLUTIONS AND LIPID NANOPARTICLE COMPONENTS
[0200] In some embodiments, the present disclosure provides lipid solutions and lipid nanoparticles comprising one or more ionizable lipids. The subject lipid solutions and lipid nanoparticles can also include one or more helper lipids, structural lipids (e.g., cholesterol), and PEG-lipids (as described herein below).
[0201] In some embodiments, the molar ratio of the ionizable lipid, the helper lipid, the structural lipid (e.g., cholesterol) and PEG-lipid in the lipid solution and lipid nanoparticles is as described herein above.
A. IONIZABLE LIPIDS
[0202] In some embodiments, the lipid solutions and lipid nanoparticles disclosed herein comprise ionizable lipids. The subject ionizable lipids may be used as a component of a composition to facilitate encapsulation and release of nucleic acid cargo (e.g., circular RNA) to one or more target cells. In some embodiments, an ionizable lipid comprises one or more cleavable functional groups (e.g., a disulfide) tiiat allow, for example, a hydrophilic functional head-group to dissociate from a lipophilic functional tail-group of the compound (e.g., upon exposure to oxidative, reducing, or acidic conditions).
[0203] In some embodiments, the ionizable lipid has a pKa from 6 to 12. In some embodiments, the ionizable lipid has a pKa from 7 to 9. In some embodiments, the ionizable lipid has a pKa of 6.0, 6.1,
6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, 7.0, 7.1, 7.2, 7.3, 7.4, 7.5, 7.6, 7.7, 7.8, 7.9, 8.0, 8.1, 8.2, 8.3, 8.4, 8.5, 8.6, 8.7, 8.8, 8.9, or 9.0 or any ranges created by these.
[0204] In some embodiments, the ionizable lipid comprises an amino group.
[0205] In some embodiments, the ionizable lipid comprises a divalent headgroup and one or more straight hydrocarbon lipid tails. In some embodiments, the straight hydrocarbon lipid tails are from 3- 25 carbon atoms in length, such as 5 to 25. 5 to 20. 5 to 15, 5 to 10, 10 to 15. 10 to 20, or 10 to 25 carbon atoms in length.
[0206] In some embodiments, the ionizable lipid comprises a divalent headgroup and one or more branched hydrocarbon lipid tails. In some embodiments, the branched hydrocarbon lipid tails are from 3-25 carbon atoms in length, such as 5 to 25, 5 to 20, 5 to 15, 5 to 10, 10 to 15. 10 to 20, or 10 to 25 carbon atoms in length.
[0207] In some embodiments, the divalent headgroup is selected from guanidine and squaramide.
[0208] In some embodiments, the squaramide headgroup is of the following formula:
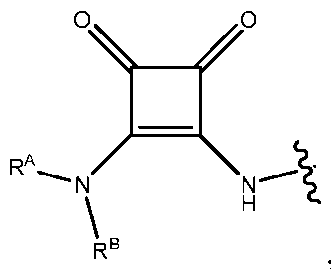 wherein RA and RB are each independently a C1-C6 alkyl group or H; and represents the point of attachment of the headgroup to a straight or branched hydrocarbon lipid tail.
wherein RA and RB are each independently a C1-C6 alkyl group or H; and represents the point of attachment of the headgroup to a straight or branched hydrocarbon lipid tail.
[0209] In some embodiments, the ionizable lipid comprises a headgroup selected from:
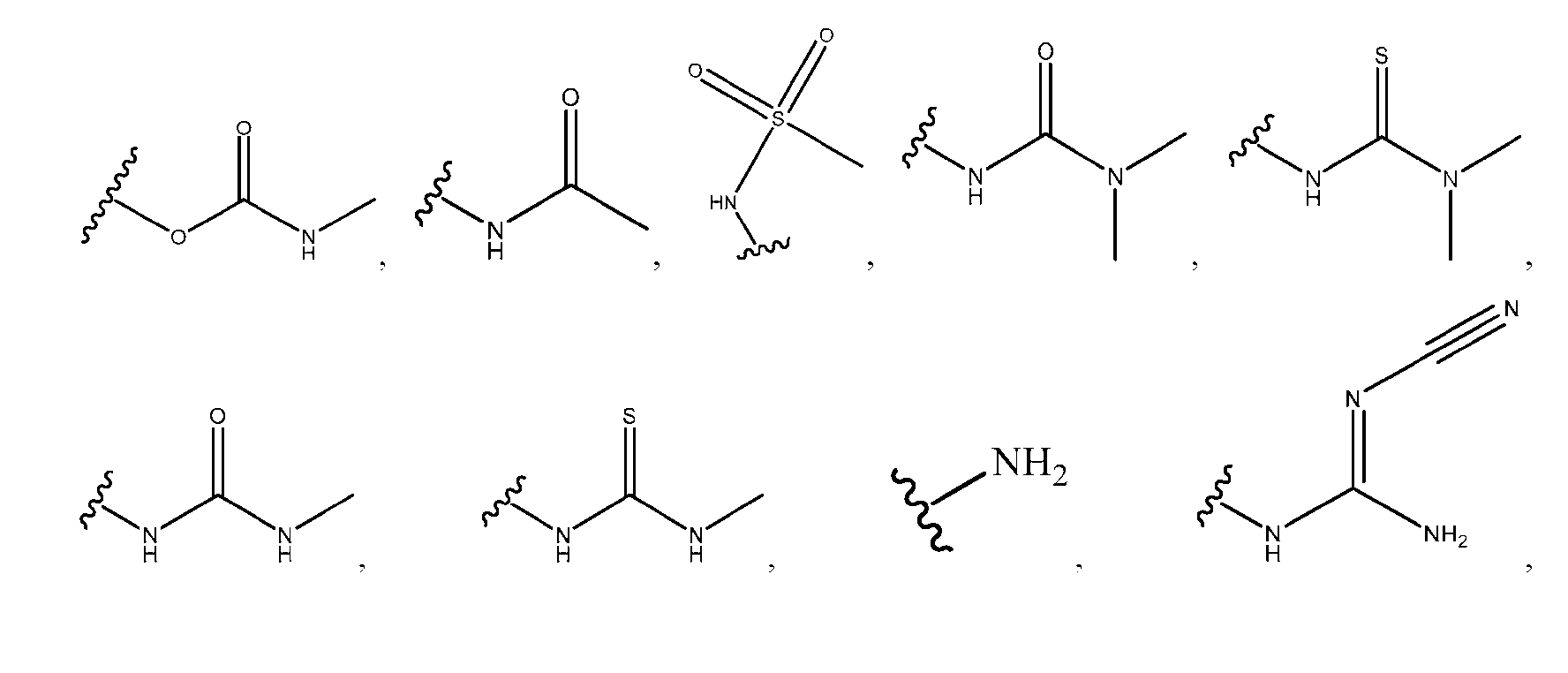
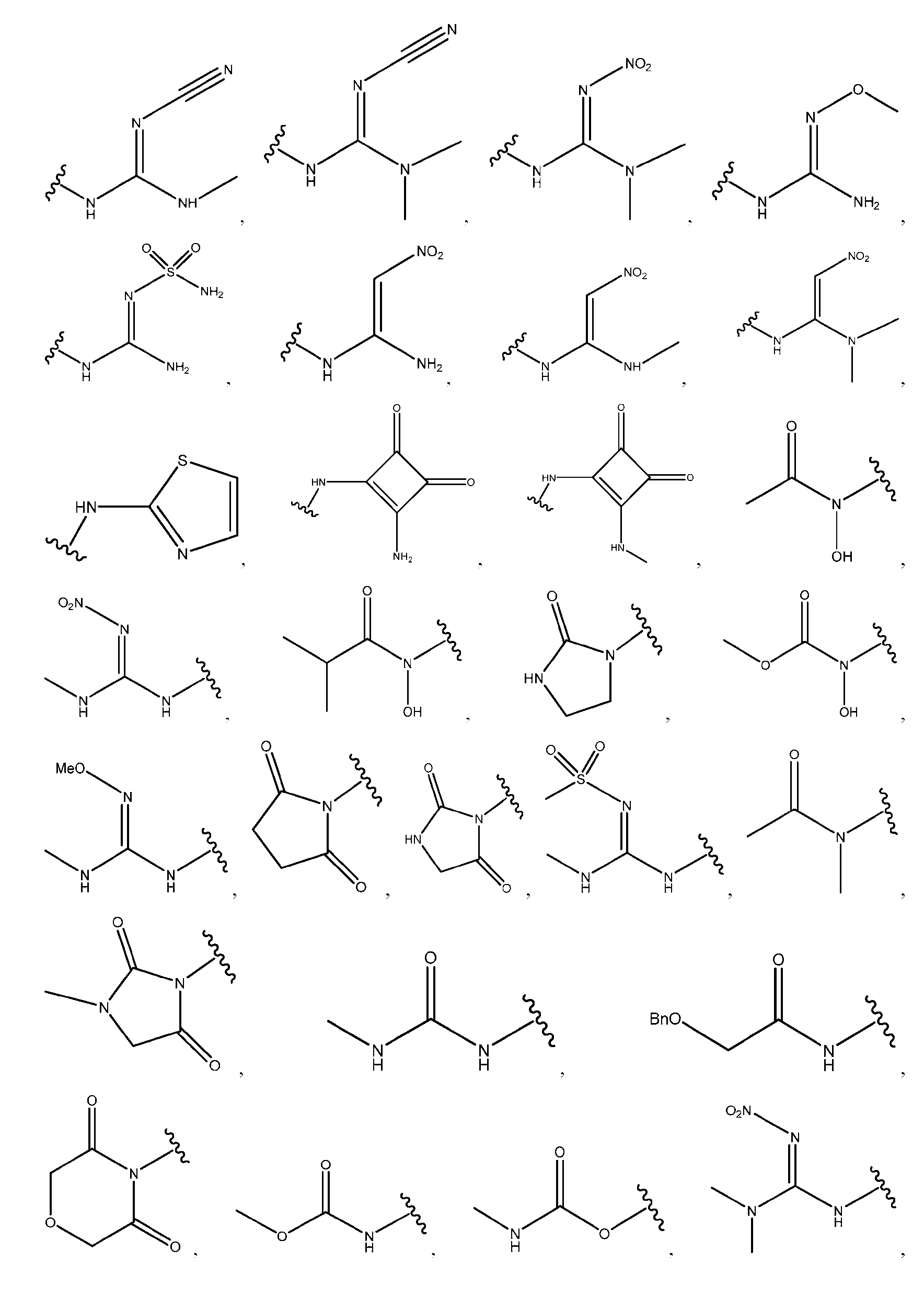
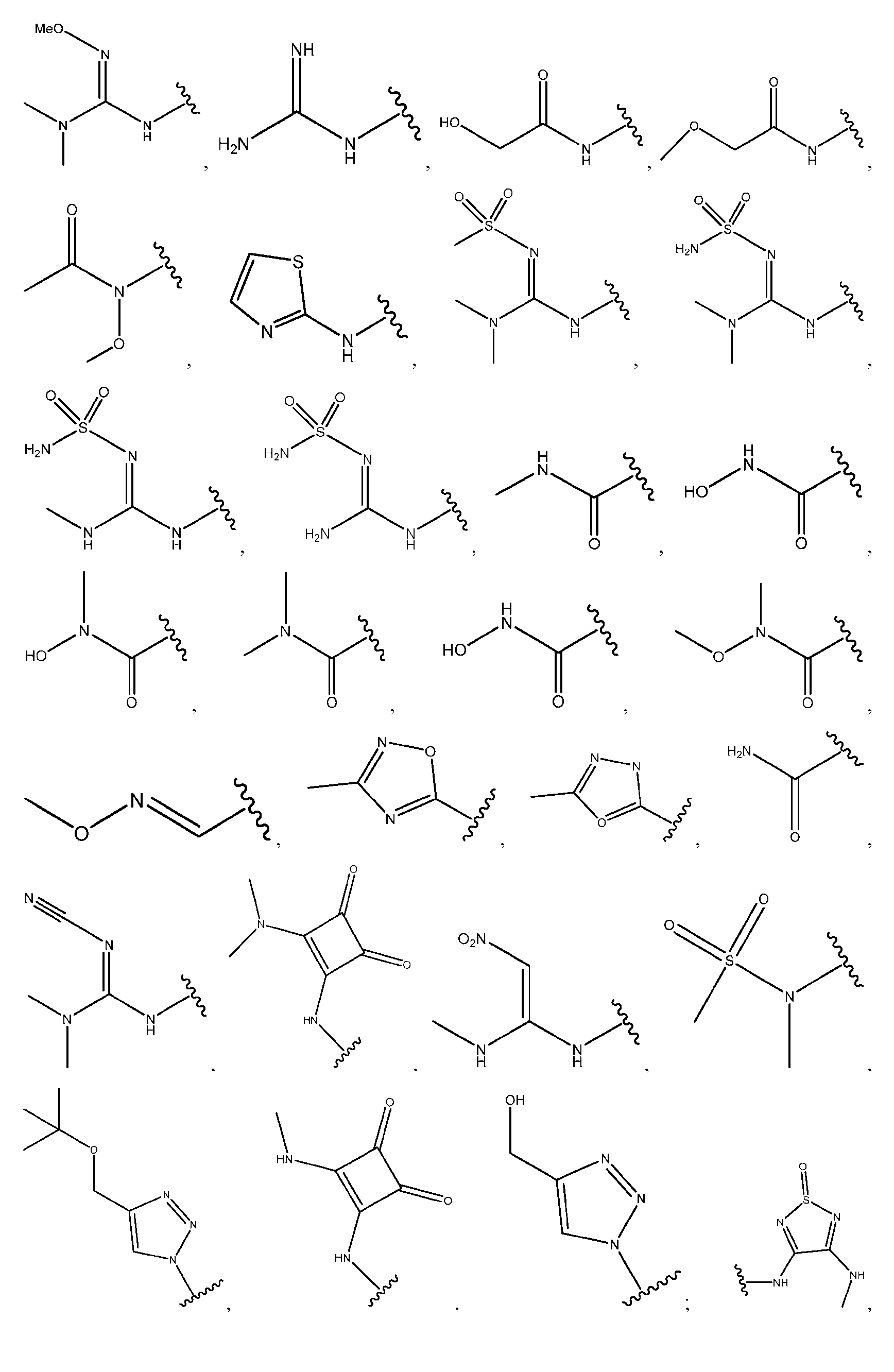
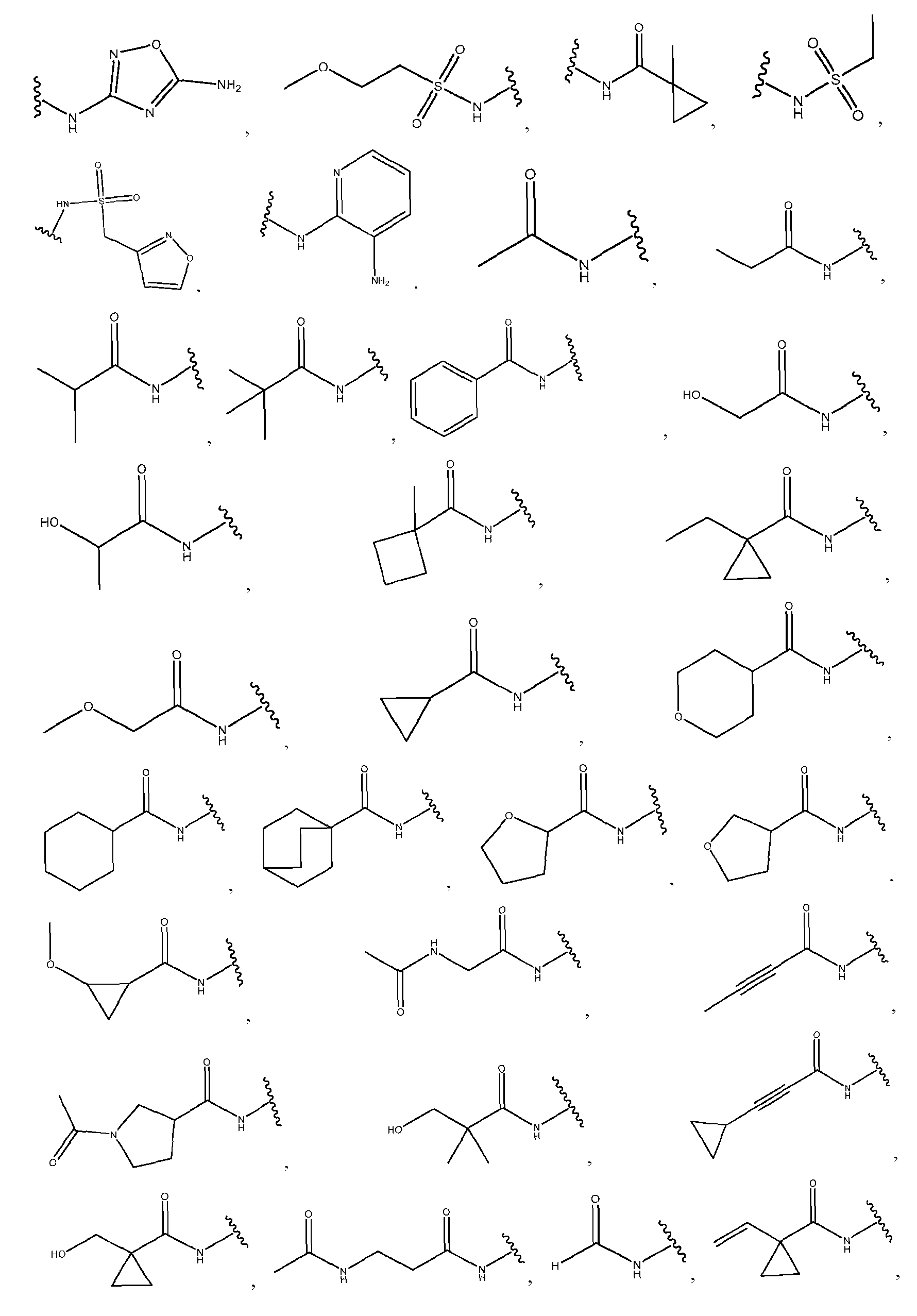
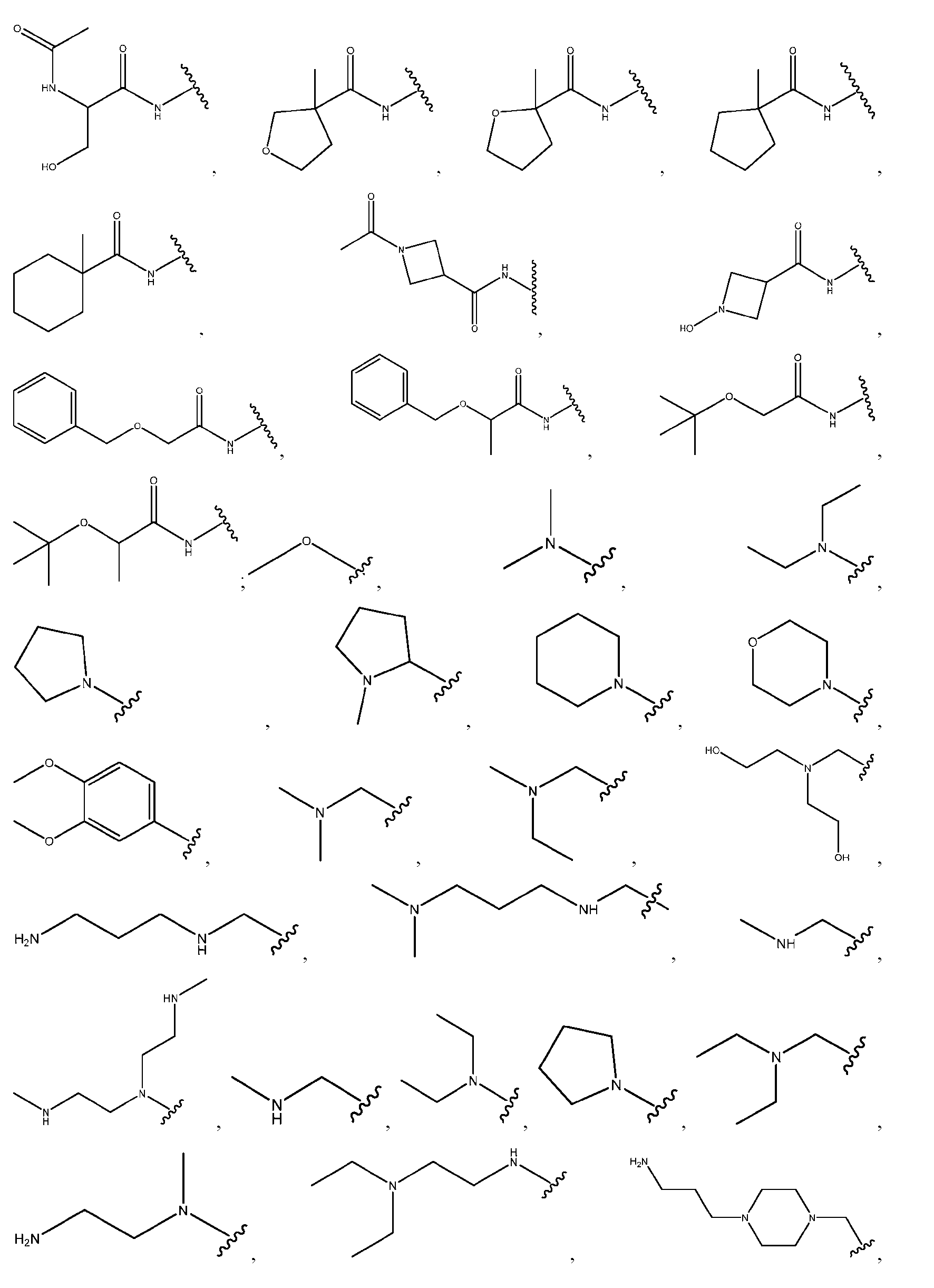
 wherein represents the point of attachment of the headgroup to a straight or branched hydrocarbon lipid tail.
wherein represents the point of attachment of the headgroup to a straight or branched hydrocarbon lipid tail.
[0210] In some embodiments, the ionizable lipid comprises a head group selected from:
 wherein ^vwvw represents the point of attachment of the headgroup to a straight or branched hydrocarbon lipid tail.
[0211] In some embodiments, the ionizable lipid comprises a hydrophilic headgroup as disclosed in Jayaraman et al. Angew. Chem. hit. Ed. (2012), 51, 8529-8533.
wherein ^vwvw represents the point of attachment of the headgroup to a straight or branched hydrocarbon lipid tail.
[0211] In some embodiments, the ionizable lipid comprises a hydrophilic headgroup as disclosed in Jayaraman et al. Angew. Chem. hit. Ed. (2012), 51, 8529-8533.
[0212] In some embodiments, the ionizable lipid is ethyl lauryl arginate (EL A). In some embodiments, die ionizable lipid is ionizable lipid 1, wherein ionizable lipid 1 comprises:
[0213] In some embodiments, the one or more of the cationic or ionizable lipids are represented by Fonnula (LI):

Formula (LI) wherein: n is an integer between 1 and 4;
R, is hydrogen or hydroxyl; and R1 and R2 are each independently a linear or branched Ce-C30 alkyl, C6-C30 alkenyl, or Ce-C30 heteroalkyl, optionally substituted by one or more substituents selected from oxo, halo, hydroxy, cyano, alkyl, alkenyl, aldehyde, heterocyclylalkyl, hydroxyalkyl, dihydroxyalkyl, hydroxyalkylaminoalkyl. aminoalkyl. alkylaminoalkyl, dialkylaminoalkyl, (heterocyclyl)(alkyl)aminoalkyl, heterocyclyl, heteroaryl, alkylheteroaryl, alkynyl, alkoxy, amino, dialkylamino. aminoalkylcarbonylamino. aminocarbonylalkylamino, (aminocarbonylalky l)(alkyl)amino. alkenylcarbonylamino. hydroxycarbonyl, alkyloxycarbonyl, aminocarbonyl, aminoalkylaminocarbonyl, alkylaminoalkylaminocarbonyl, dialkylaminoalkylaminocarbonyl, heterocyclylalkylaminocarbonyl,
(alkylaminoalkyl)(alkyl)aminocarbonyl, alkylaminoalkylcarbonyl, dialkylaminoalkylcarbonyl, heterocyclylcarbonyl. alkenylcarbonyl, alkynylcarbonyl. alkylsulfoxide, alkylsulfoxidealkyl, alkylsulfonyl, and alkylsulfonealkyl.
[0214] In some embodiments. Ra is hydrogen. In some embodiments, Ra is hydroxyl.
[0215] In some embodiments, the ionizable lipid is represented by Fonnula (Lla -1), Fonnula (LIa-2), or Fonnula (LIa-3):
Fonnula (LIa-1) Formula (Lla -2) Formula (LIa-3)
[0216] In some embodiments, the ionizable lipid is represented by Formula (LIb-1), Fonnula (LIb-2), or Fonnula (LIb-3):

Formula (LIb-1) Formula (LIb-2) Formula (LIb-3).
[0217] In some embodiments, the ionizable lipid is represented by Fonnula (LIb-4), Formula (LIb-5), Fonnula (LIb-6), Formula (Lib- 7), Formula (LIb-8). or Formula (LIb-9):
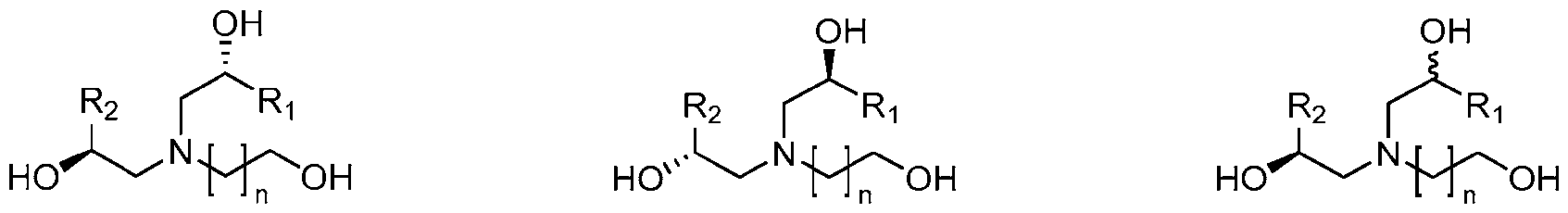
Formula (LIb-4) Formula (LIb-5) Formula (LIb-6)

Fonnula (LIb-7) Formula (LIb-8) Formula (LIb-9).
[0218] In some embodiments, the one or more of the cationic or ionizable lipids arc represented byFormula (LI), wherein R1 and R2 are each independently selected from:


[0219] In some embodiments. R1 and R2 are the same. In some embodiments, R1 and R2 are different.
[0220] In various embodiments, the one or more of the cationic or ionizable lipids are represented by Fonnula (LI*):
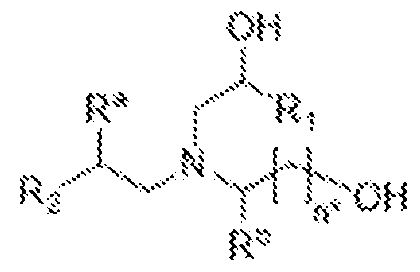
Formula (LI*) wherein: n* is an integer between 1 to 7,
Ra is hydrogen or hydroxyl.
Rb is hydrogen or C1-C6 alkyl, and R1 and R2 are each independently a linear or branched C1-C30 alkyl, C2-C30 alkenyl, or C1-C30 heteroalkyl, optionally substituted by one or more substituents selected from oxo, halo, hydroxy, cyano, alkyl, alkenyl, aldehyde, heterocyclylalkyl, hydroxyalkyl, dihydroxy alkyl, hydroxy alkylaminoalkyl, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl, (heterocyclyl)(alkyl)aminoalkyl, heterocyclyl, heteroaryl, alkylheteroaryl, alkynyl, alkoxy, amino, dialkylamino, aminoalkylcarbonylamino, aminocarbonylalkylamino, (aminocarbonylalky l)(alkyl)amino, alkenylcarbonylamino, hydroxycarbonyl, alkyloxycarbonyl, alkylcarbonyloxy, alkylcarbonate, alkcnyloxycarbonyl. alkenylcarbonyloxy, alkenylcarbonate, alkynyloxycarbonyl, alkynylcarbonyloxy, alkynylcarbonate, aminocarbonyl, aminoalkylaminocarbonyl, alkylaminoalkylaminocarbonyl, dialkylaminoalkylaminocarbonyl, heterocyclylalkylaminocarbonyl,
(alkylaminoalkyl)(alkyl)aminocarbonyl, alkylaminoalkylcarbonyl, dialkylaminoalkylcarbonyl, heterocyclylcarbonyl, alkenylcarbonyl, alkynylcarbonyl, alkylsulfoxide, alkylsulfoxidealkyl, alkylsulfonyl, and alkylsulfonealkyl.
[0221] In some embodiments, the one or more of the cationic or ionizable lipids are represented by Formula (LII):
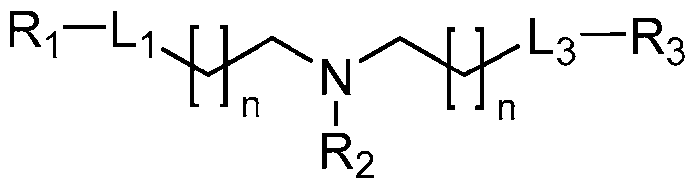
Formula (LII) wherein: each 11 is independently an integer from 2 to 15;
L1 and L3 are each independently -OC(O)-* or -C(O)O-*, wherein indicates the attachment point to R1 or R3;
R1 and R3 are each independently a linear or branched C9-C20 alkyl or C9-C20 alkenyl, optionally substituted by one or more substituents selected from oxo, halo, hydroxy', cyano, alkyl, alkenyl, aldehyde, heterocyclylalkyl, hydroxyalkyl, dihydroxyalkyl, hydroxyalkylaminoalkyl, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl. (heterocyclyl)(alkyl)aminoalkyl, heterocyclyl, heteroaryl, alkylheteroaryl, alkynyl, alkoxy, amino, dialkylamino, aminoalkylcarbonylamino, aminocarbonylalky' lamino, (aminocarbonylalky 1) (alkyl) amino, alkenylcarbonylamino, hydroxy carbonyl, alkyloxycarbonyl, aminocarbonyl, aminoalkylaminocarbonyl, alkylaminoalkylaminocarbonyl, dialkylaminoalkylaminocarbonyl, heterocyclylalkylaminocarbonyl,
(alkylaminoalkyl)(alkyl)aminocarbonyl, alkylaminoalkylcarbonyl, dialkylaminoalkylcarbonyl, heterocyclylcarbonyl, alkenylcarbonyl, alkynylcarbonyl, alkylsulfoxide, alkylsulfoxidealkyl, alkylsulfonyl, and alkylsulfonealkyl; and
R2 is selected from:

[0222] In some embodiments, the ionizable lipid is selected from an ionizable lipid of Formula LII, wherein R1 and R3 are each independently selected from:

[0223] In some embodiments, R1 and R3 are the same. In some embodiments, R1 and R3 are different.
[0224] In some embodiments, the one or more of the cationic or ionizable lipids 1re represented by Formula (LII-1) or Formula (LII-2):
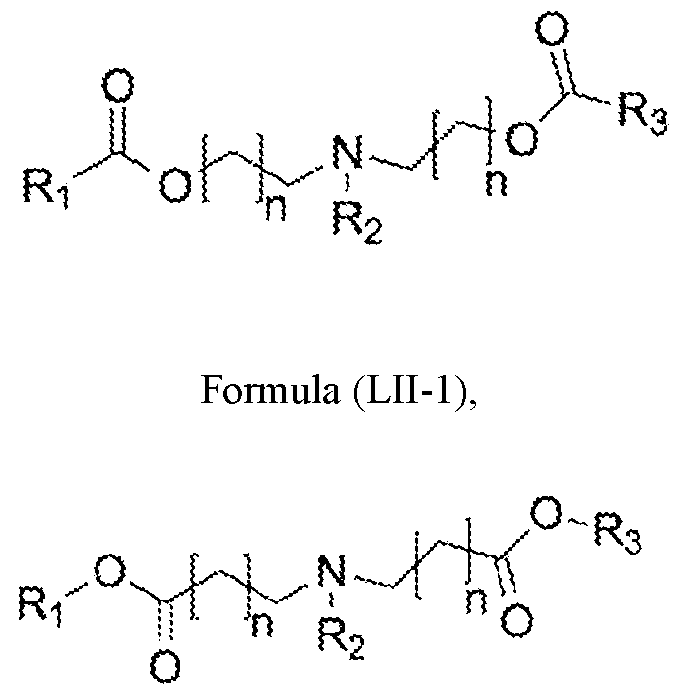
Formula (LII-2).
[0225] In some embodiments, the ionizable lipid is selected from an ionizable lipid of WO2015/095340. In some embodiments, the ionizable lipid is selected from an ionizable lipid of WO2021/021634, WO2020/237227, or WO2019/236673. In some embodiments, the ionizable lipid is selected from an ionizable lipid of WO2021226597 and WO2021113777. In some embodiments, the ionizable lipid is selected from an ionizable lipid of W02023056033. In some embodiments, the ionizable lipid is selected from an ionizable lipid of WO2023081526.
[0226] In some embodiments, tire one or more of the cationic or ionizable lipids are represented by Formula (LIII):
Formula (T..ITI) or a pharmaceutically acceptable salt thereof, wherein:
L1 is C2-C11 alkylene. C4-C10-alkenylene, or C4-C10-alkynylene;
X1 is OR1. SR1, or N(R')2, where R1 is independently H or unsubstituted C1-C6 alkyl; and
R2 and R3 are each independently C6-C30-alky 1, C6-C30-alkenyl, or C6-C30-alkynyl.
[0227] In some embodiments, the one or more of the cationic or ionizable lipids are represented by Formula (LIII*):

Formula (1,1 II*) or a pharmaceutically acceptable salt thereof, wherein:
L1 is C2-C11 alky lene, C4-C10-alkenylene, or C4-Cw-alkynylene;
X1 is OR1, SR1, or N(R')2, where R1 is independently H or unsubstituted C1 -C6 alkyl; and
R2 and R3 are each independently a linear or branched C1-C30 alkyl, C2-C30 alkenyl, or C1-C30 heteroalkyl, optionally substituted by one or more substituents selected from oxo, halo, hydroxy, cyano, alkyl, alkenyl, aldehyde, heterocyclylalkyl, hydroxyalkyl, dihydroxyalkyl, hydroxyalkylaminoalkyl, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl. (heterocyclyl)(alkyl)aminoalkyl, heterocyclyl. heteroaryl, alkvlhctcroarvl. alkynyl, alkoxy, amino, dialkylamino, aminoalkylcarbonylamino, aminocarbonylalkylamino, (aminocarbonylalky l)(alkyl)amino, alkenylcarbonylamino, hydroxycarbonyl, alkyloxycarbonyl, alkylcarbonyloxy, alkylcarbonate, alkenyloxycarbonyl, alkenylcarbonyloxy, alkenylcarbonate, alkynyloxycarbonyl. alkynylcarbonyloxy , alkynylcarbonate, aminocarbonyl, aminoalkylaminocarbonyl, alkylaminoalkylaminocarbonyl, dialkylaminoalkylaminocarbonyl. heterocyclylalkylaminocarbonyl,
(alkylaminoalkyl)(alkyl)aminocarbonyl. alkylaminoalkylcarbonyl, dialkylaminoalkylcarbonyl,
heterocyclylcarbonyl, alkenylcarbonyl, alkyny Icarbonyl, alkylsulfoxide, alkylsulfoxidealkyl, alkydsulfonyl, and alkylsulfonealkyl.
[0228] In some embodiments, an ionizable lipid is a compound of Formula (LIV):

Formula (LIV) or is a pharmaceutically acceptable salt thereof, wherein: n’ is an integer from 1 to 7;
Ra is hydrogen or hydroxyl;
R11 is hydrogen or C1 -Ce alkyl;
R1 is C1-C30 alkyl or R]’;
R2 is C1-C30 alkyl or R2*;
R1* and R2* are independently selected from:
-(CH2)qC(O)O(CH2)rC(R8)(R9)(R10),
-(CH2)qOC(O)(CH2)rC(R8)(R9)(R10), and
-(CH2)qOC(O)O(CH2),C(R8)(R9)(R10); wherein: q is an integer from 0 to 12, r is an integer from 0 to 6. wherein at least one occurrence of r is not 0;
R8 is H or Rn;
R9, R10, and R11 are each independently C1-C2o alky l or C2-C2o-alkenyl; and wherein (i) R1 is R1*, (ii) R2 is R2’, or (iii) R1 is R1’ and R2 is R2’.
[0229] In some embodiments, an ionizable lipid of the present disclosure is represented by Formula
(LV):
Formula (LV) or is a pharmaceutically acceptable salt thereof, wherein:
Ra is hydrogen or hydroxyl;
R1 is C1-C30 alkyl or R’’;
R2 is C1-C30 alkyl or R2*;
R1* and R2* are independently selected from:
-(CH2)qC(O)O(CH2)rC(R4)(R5)(R6),
-(CH2)qOC(O)(CH2)iC(R4)(R5)(R6), and
-(CH2)qOC(O)O(CH2),C(R4)(R5)(R6); wherein: q is an integer from 0 to 12, r is an integer from 0 to 6. wherein at least one occurrence of r is not 0;
R4 is hydrogen or R7;
R\ R6. and R7 are each independently C1-C20 alkyl or C2-C2o-alkenyl; wherein (i) R1 is R1*, (ii) R2 is R2’, or (iii) R1 is R1’ and R2 is R2*; and
R3 is L-R’, wherein L is linear or branched C1-C10 alkylene, and R’ is (i) mono- or bicyclic heterocyclyl or heteroaryl, such as imida1olyl, pyrazolyl, 1.2,4-triazolyl. or benzimidazolyl, each optionally substituted at one or more available carbon and/or nitrogen atoms by C1-C6 alky l, or (ii) RA, RB, or Rc, wherein:
RA is selected from:




[0230] In some embodiments, the ionizable lipid is selected from a lipid of Table 1:
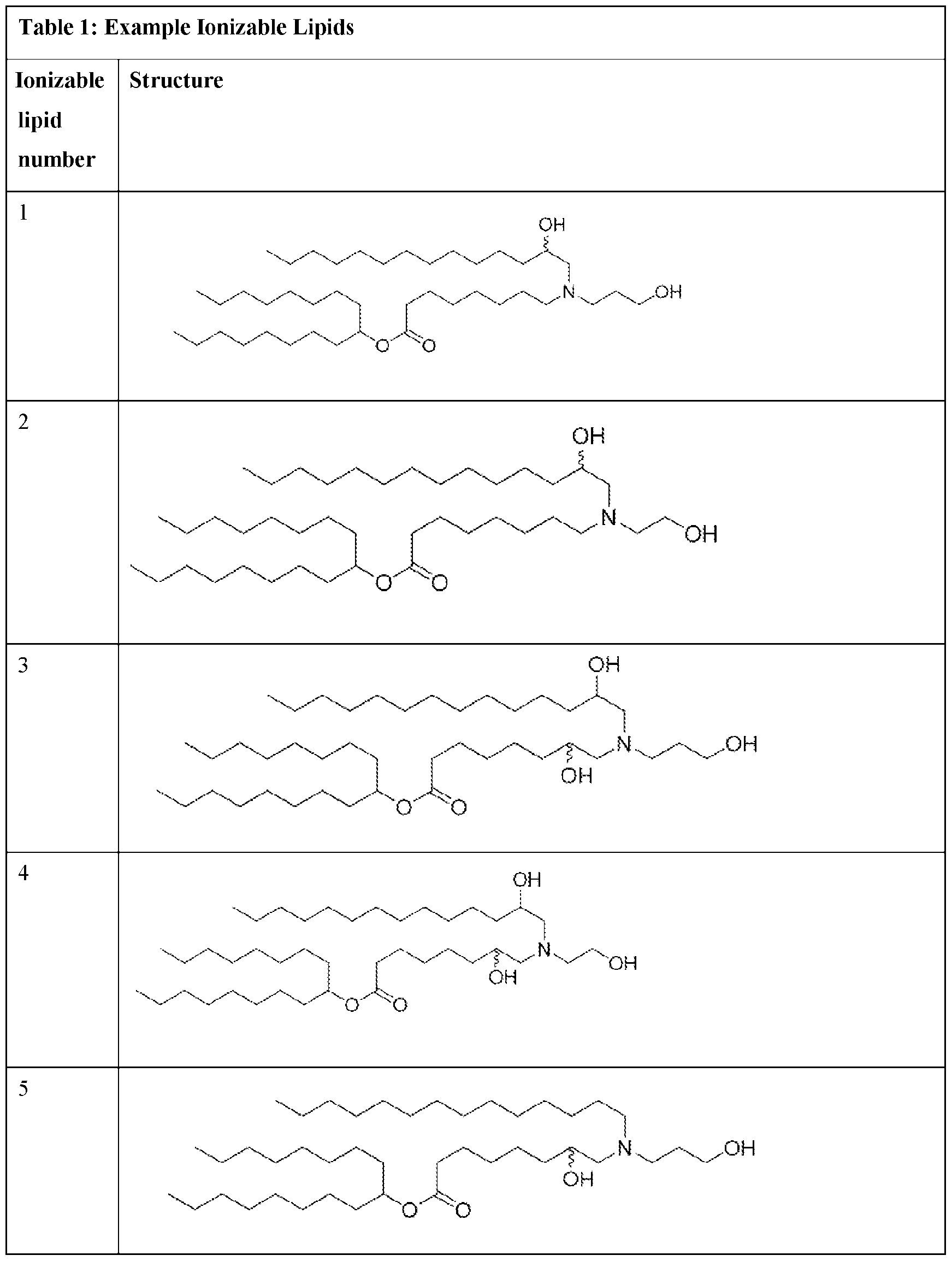
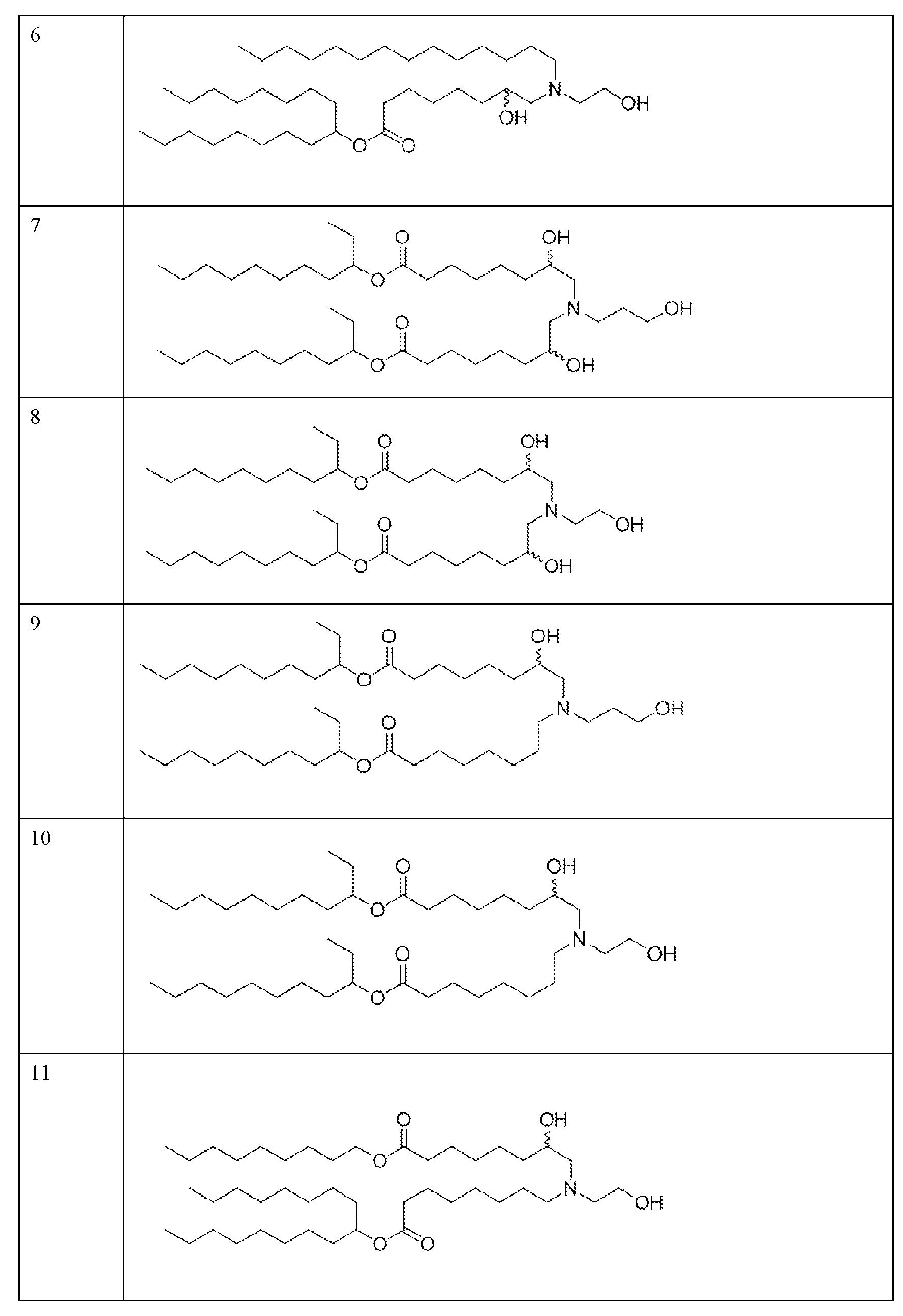


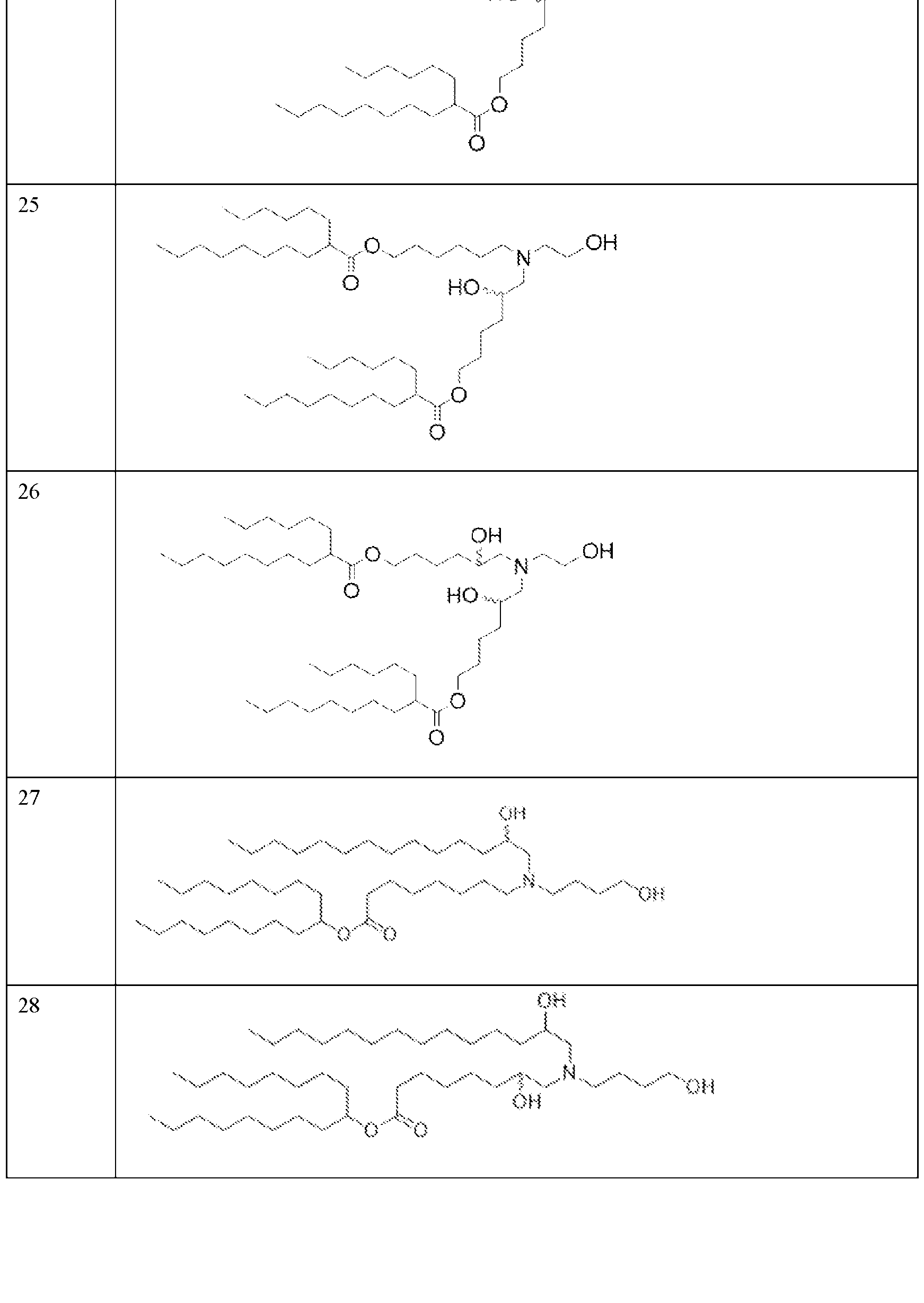
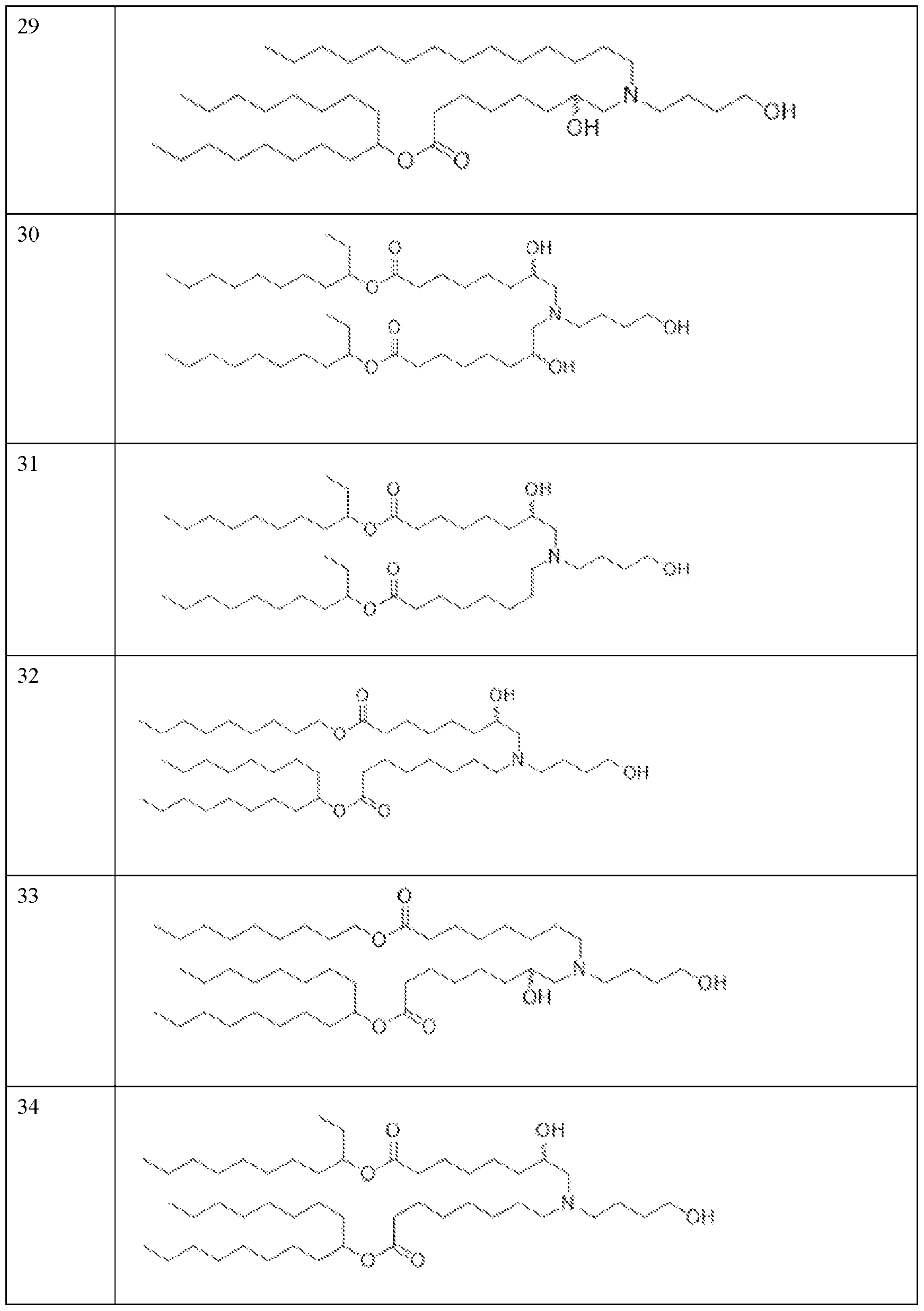

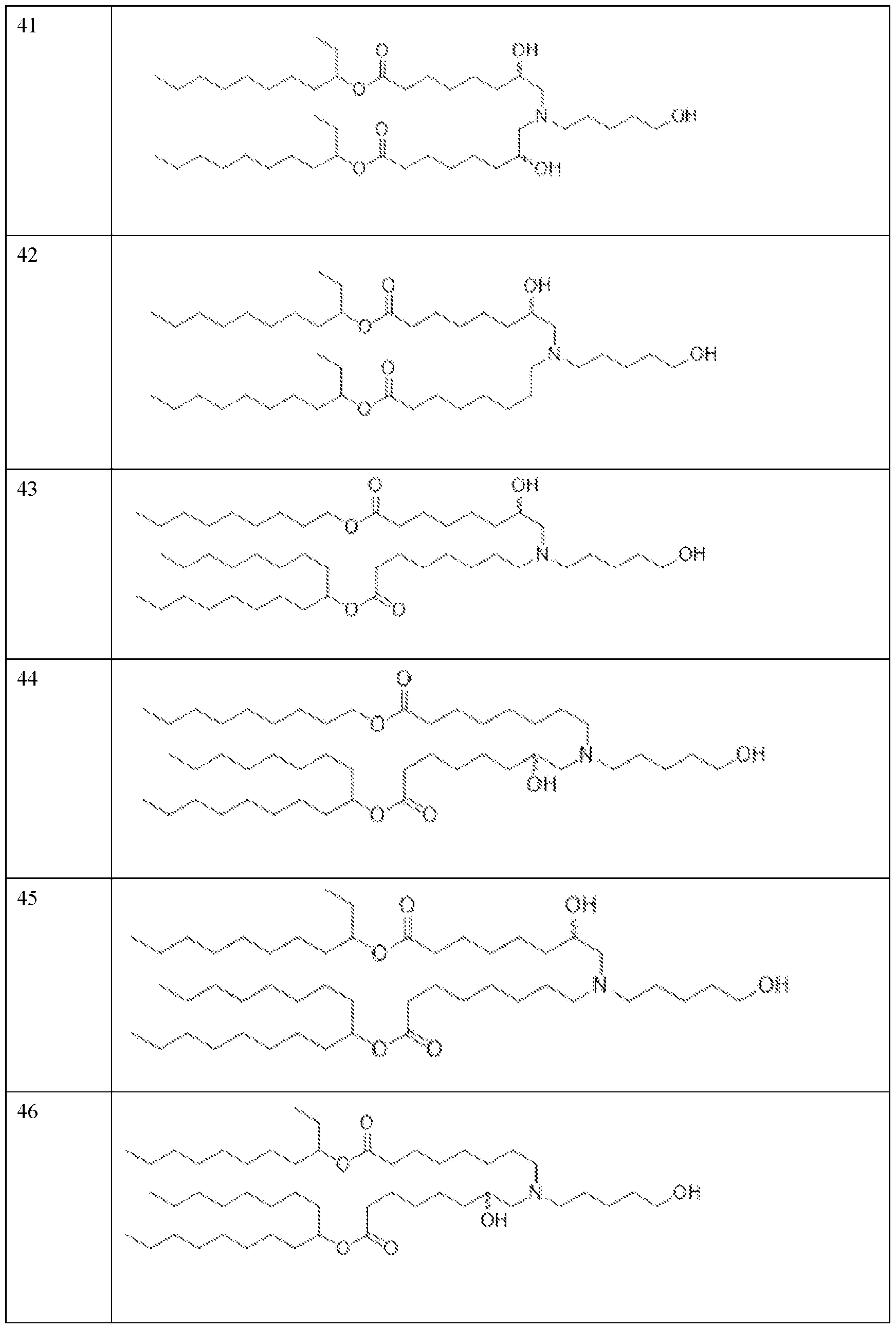
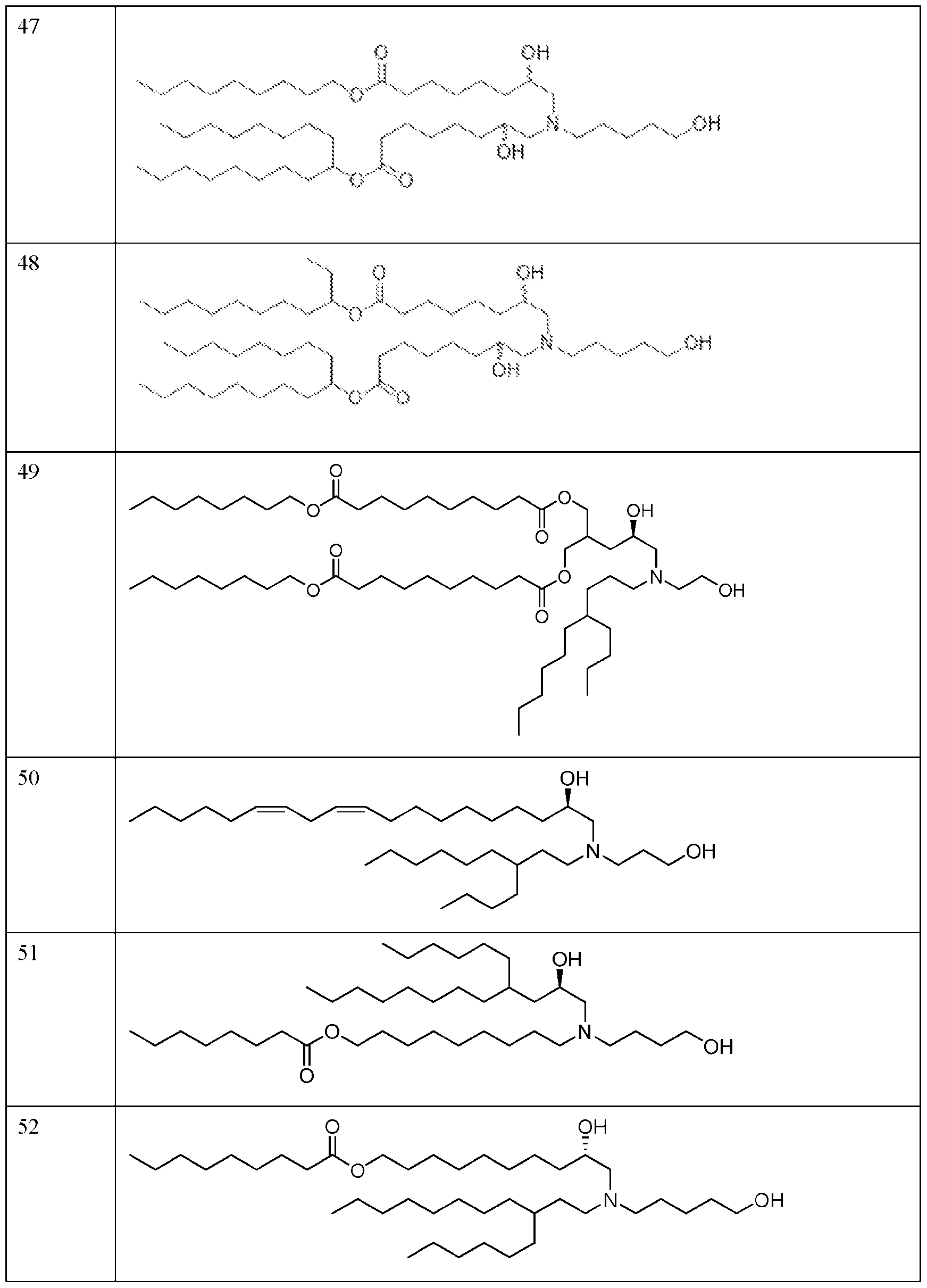
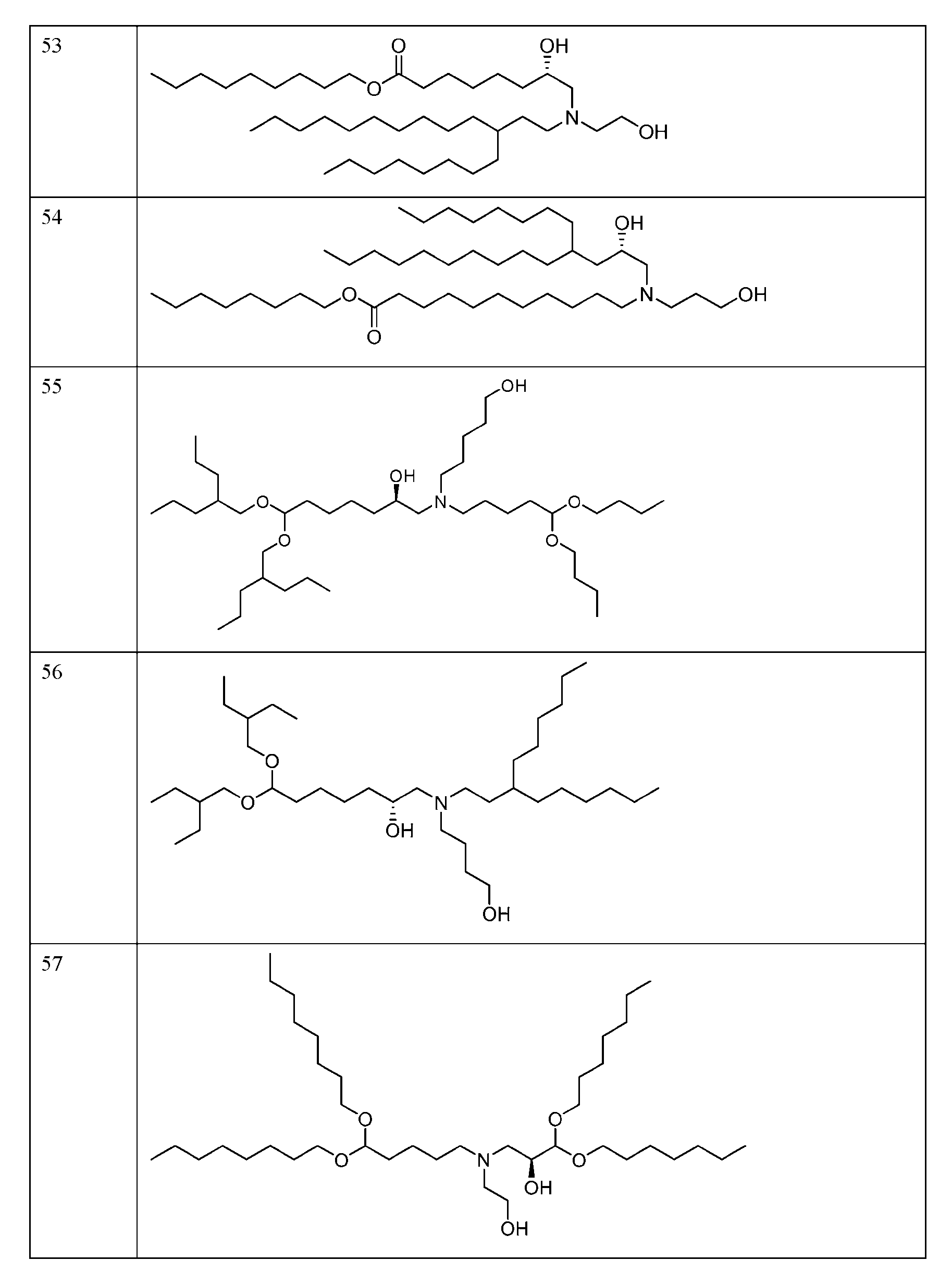
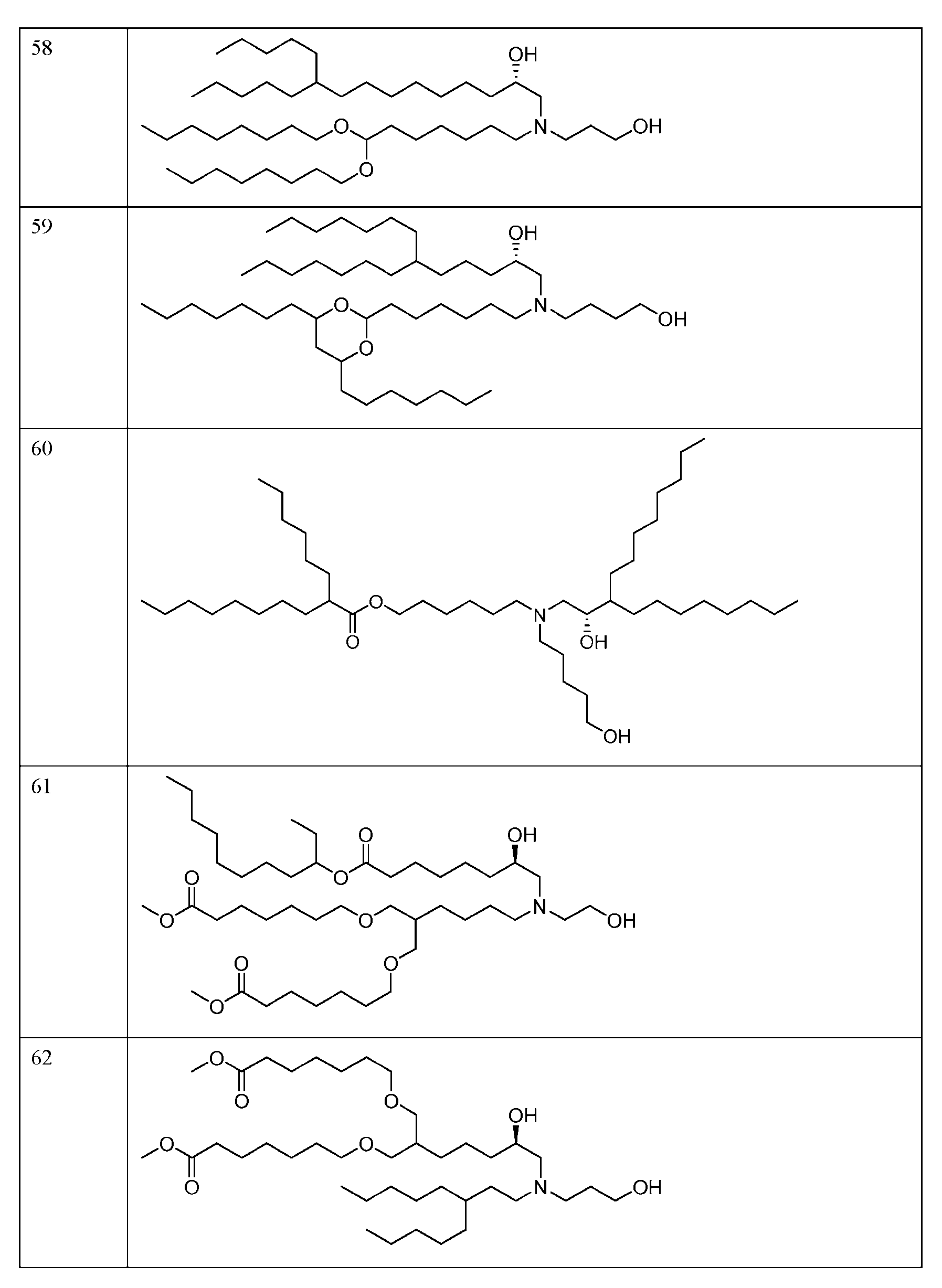
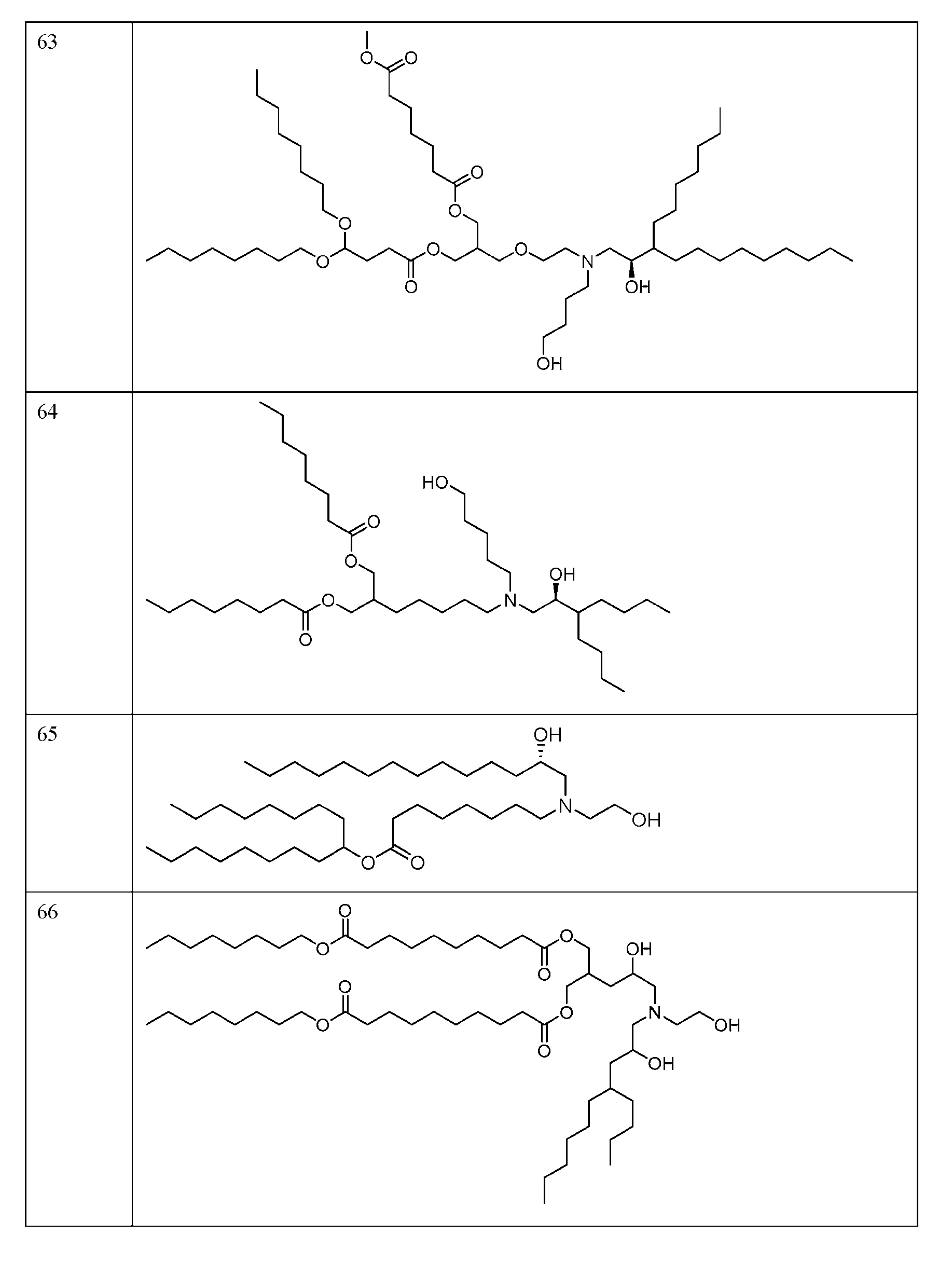
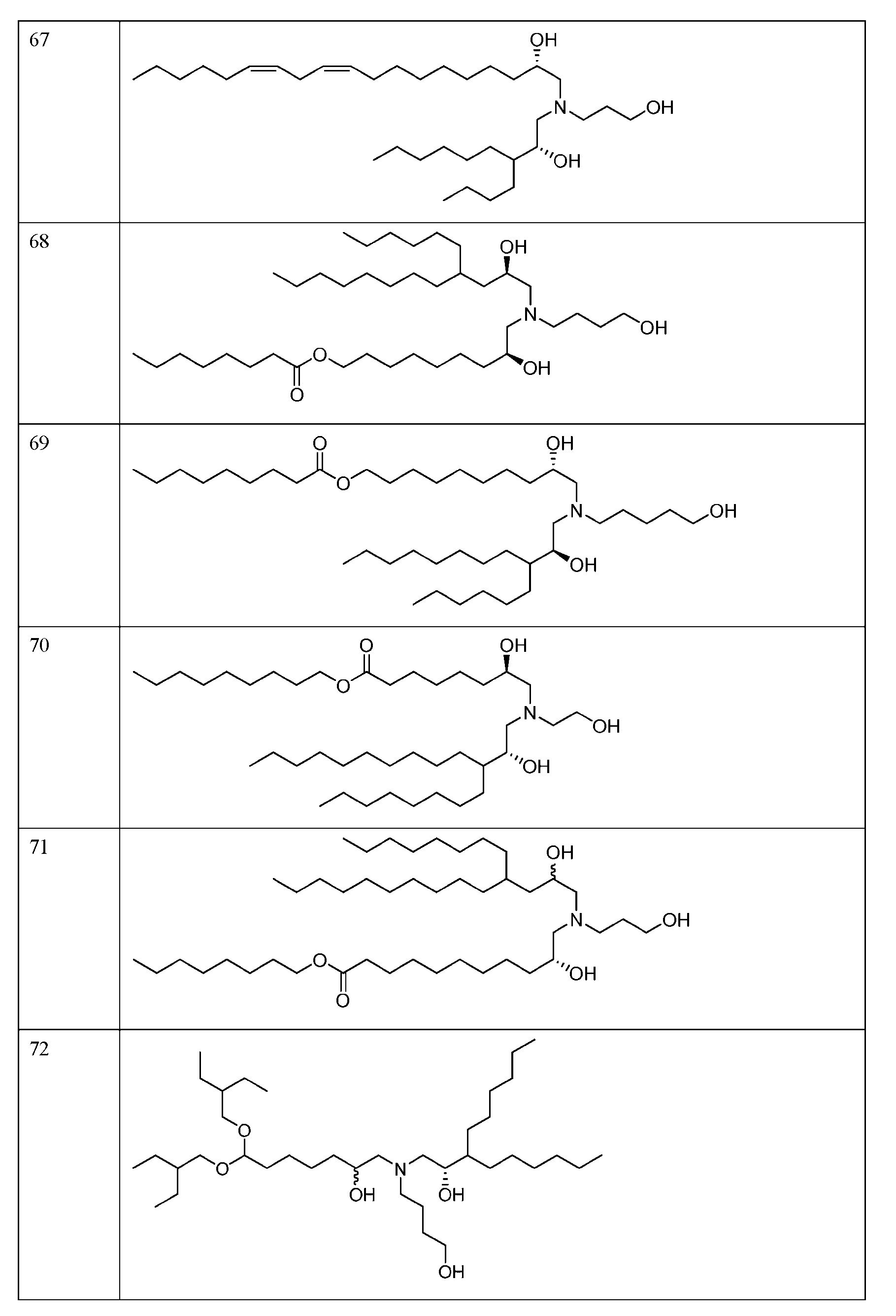

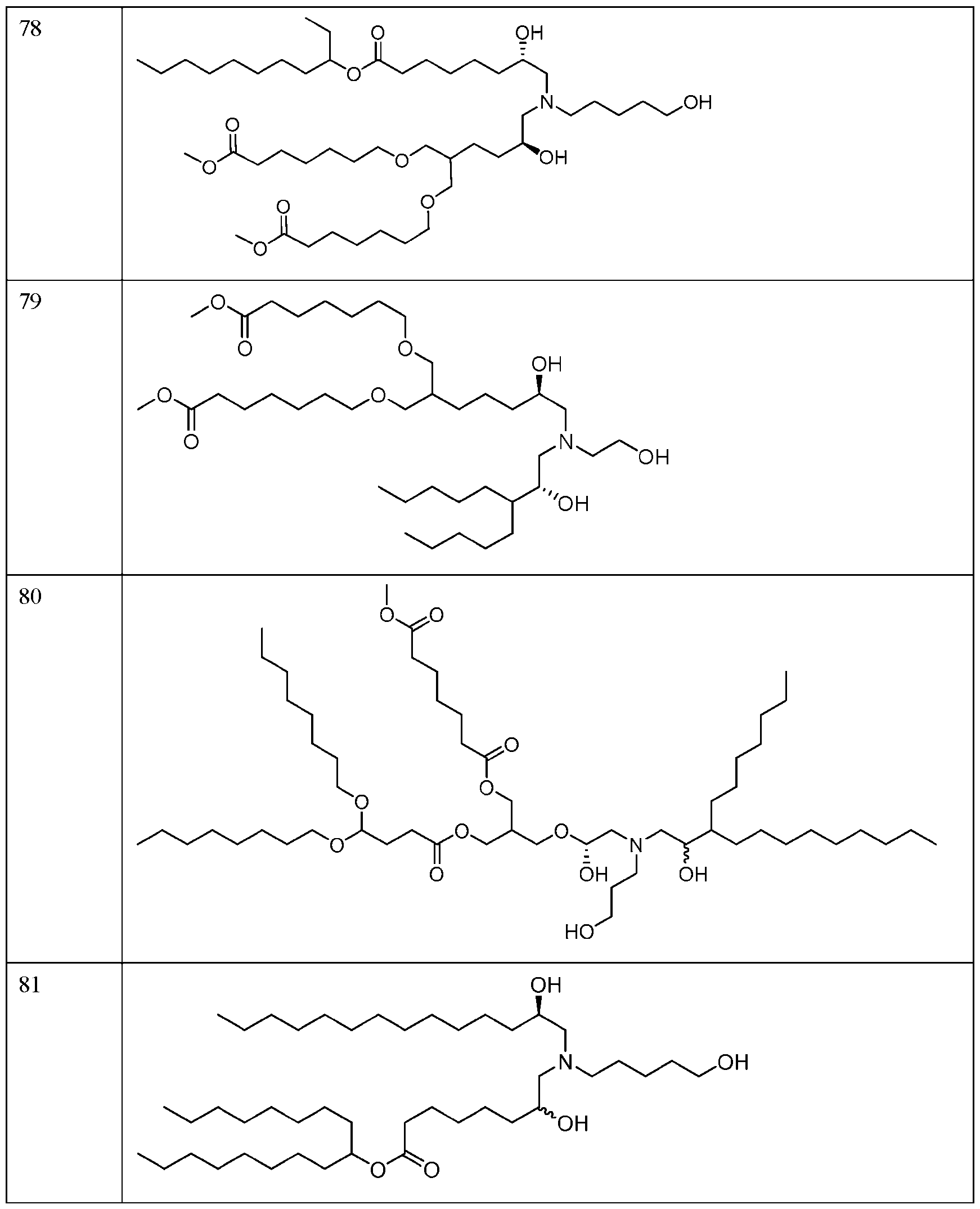


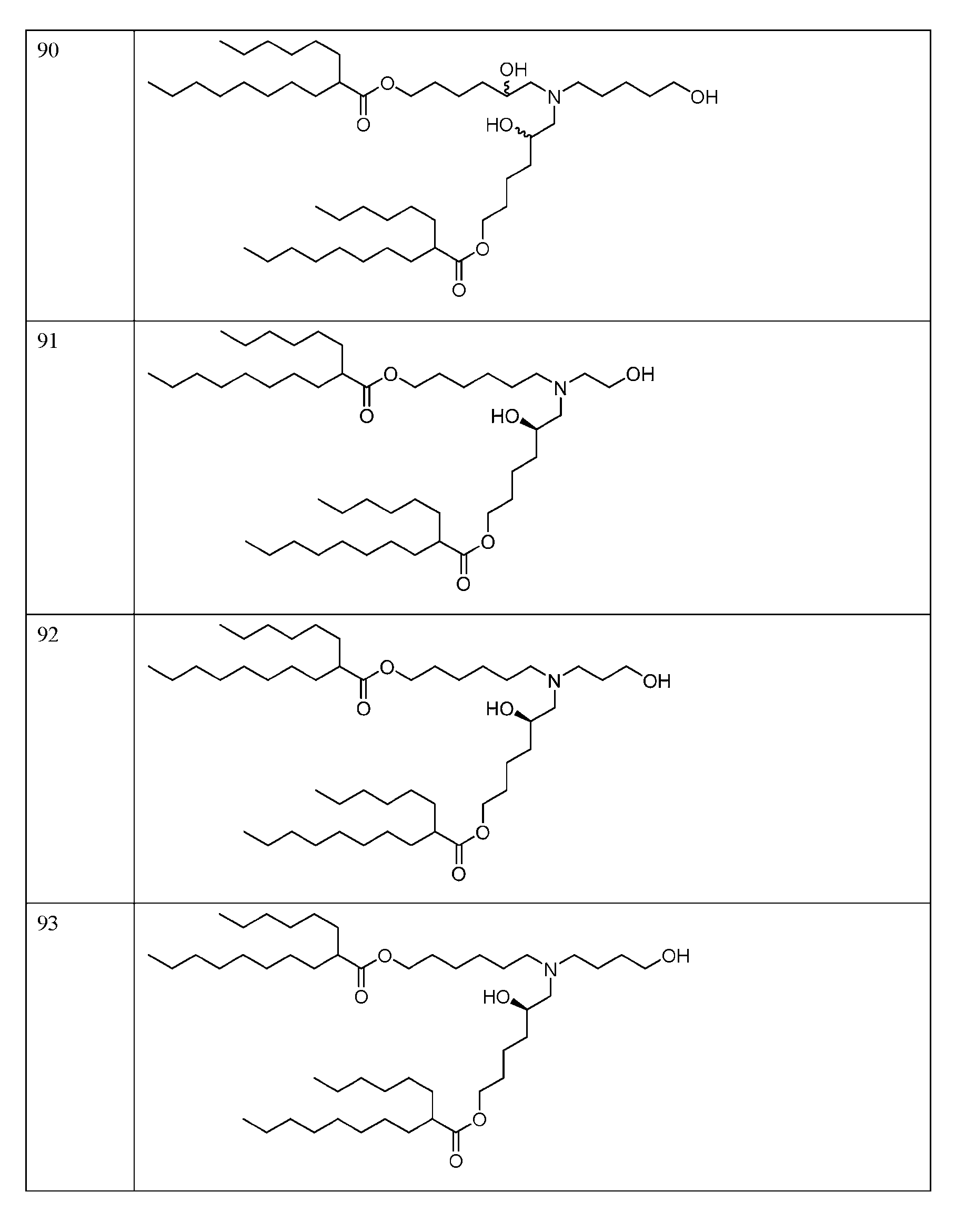
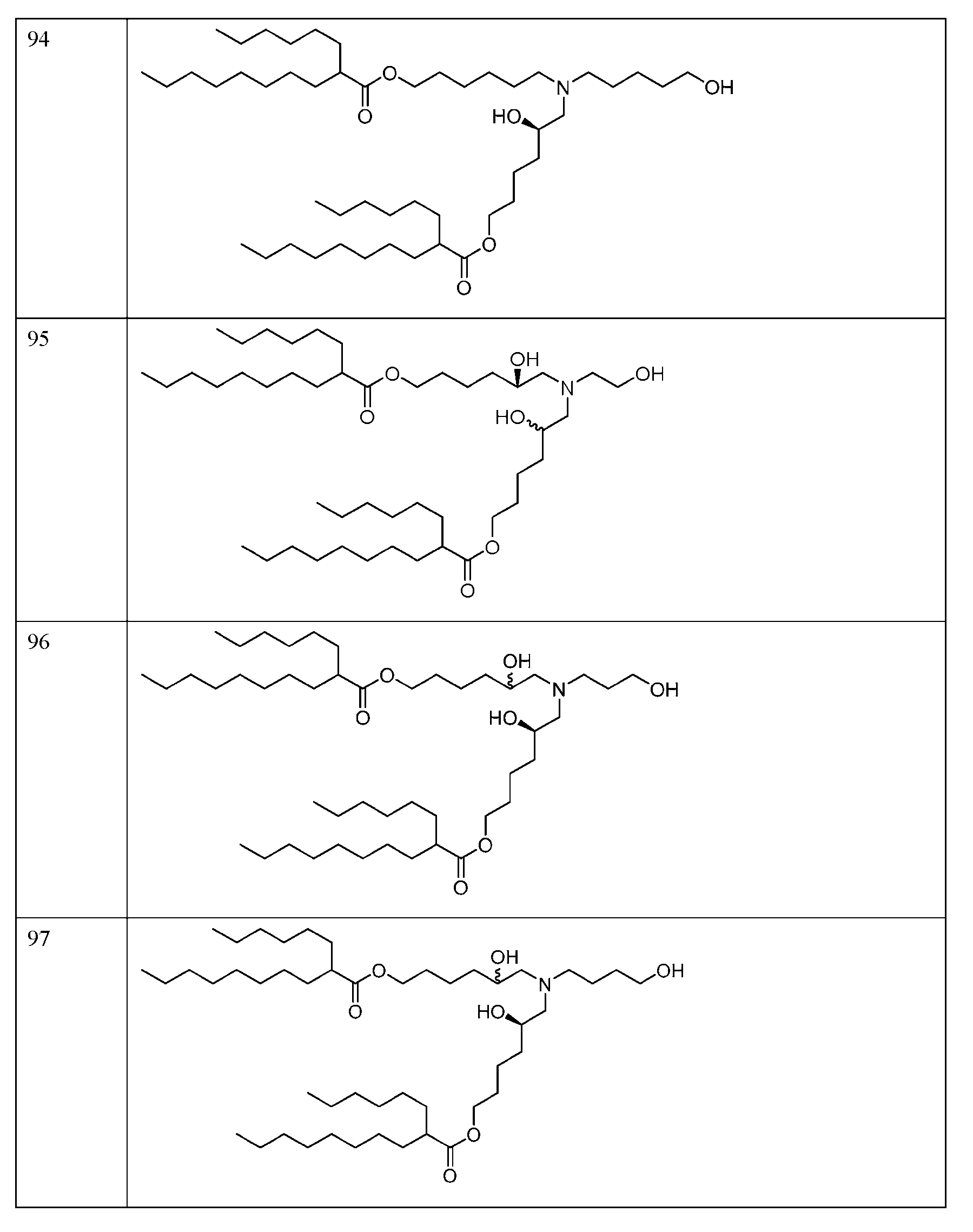
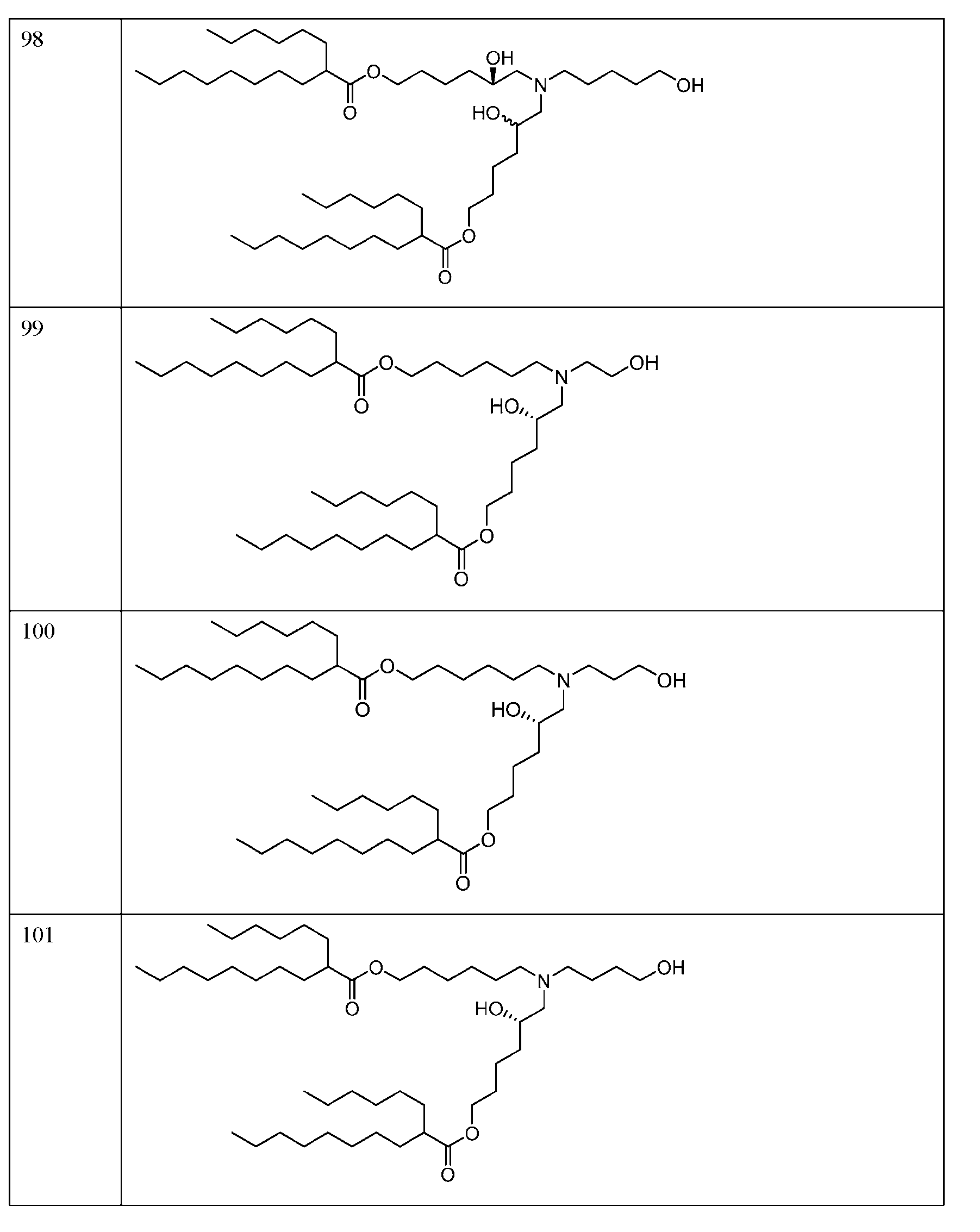
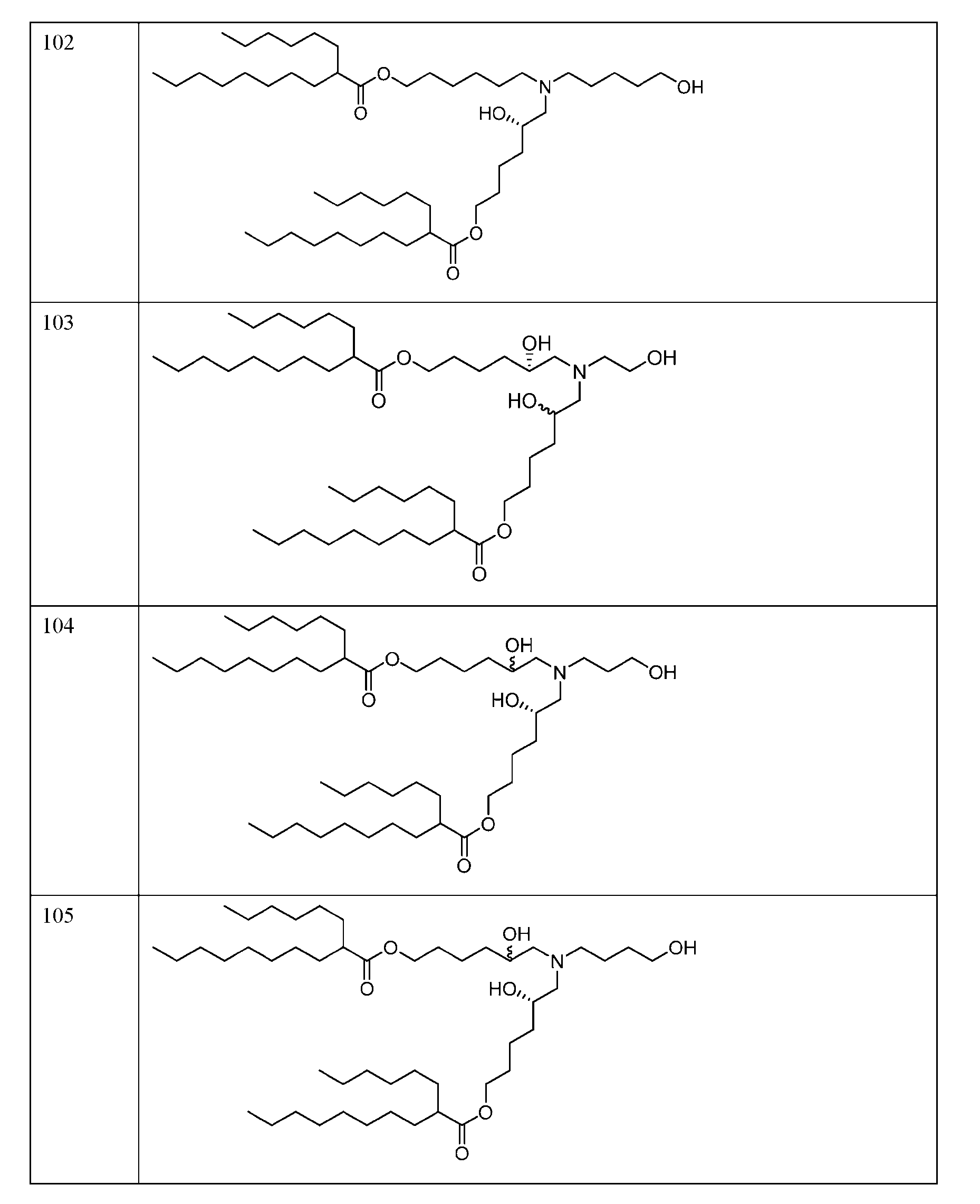
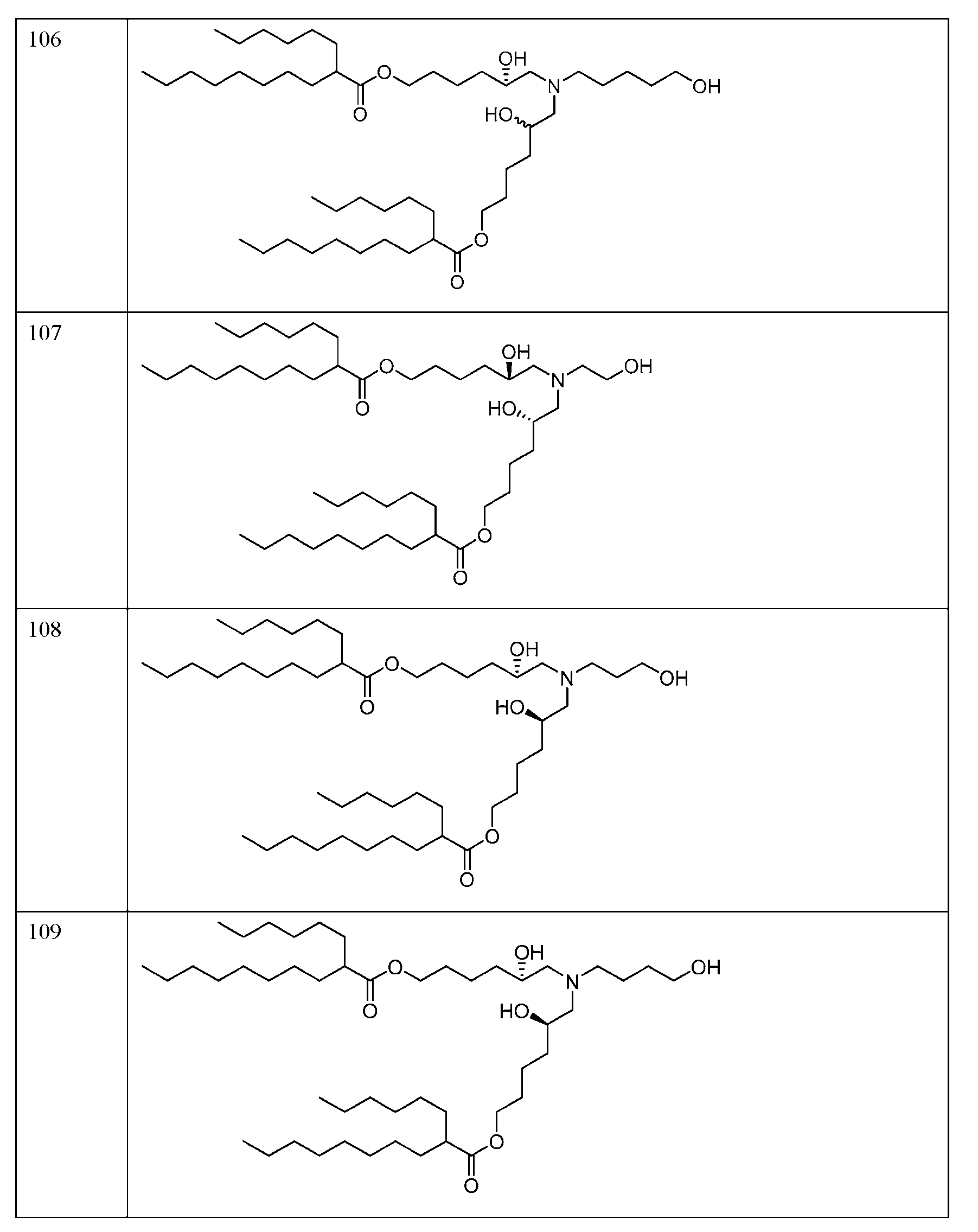



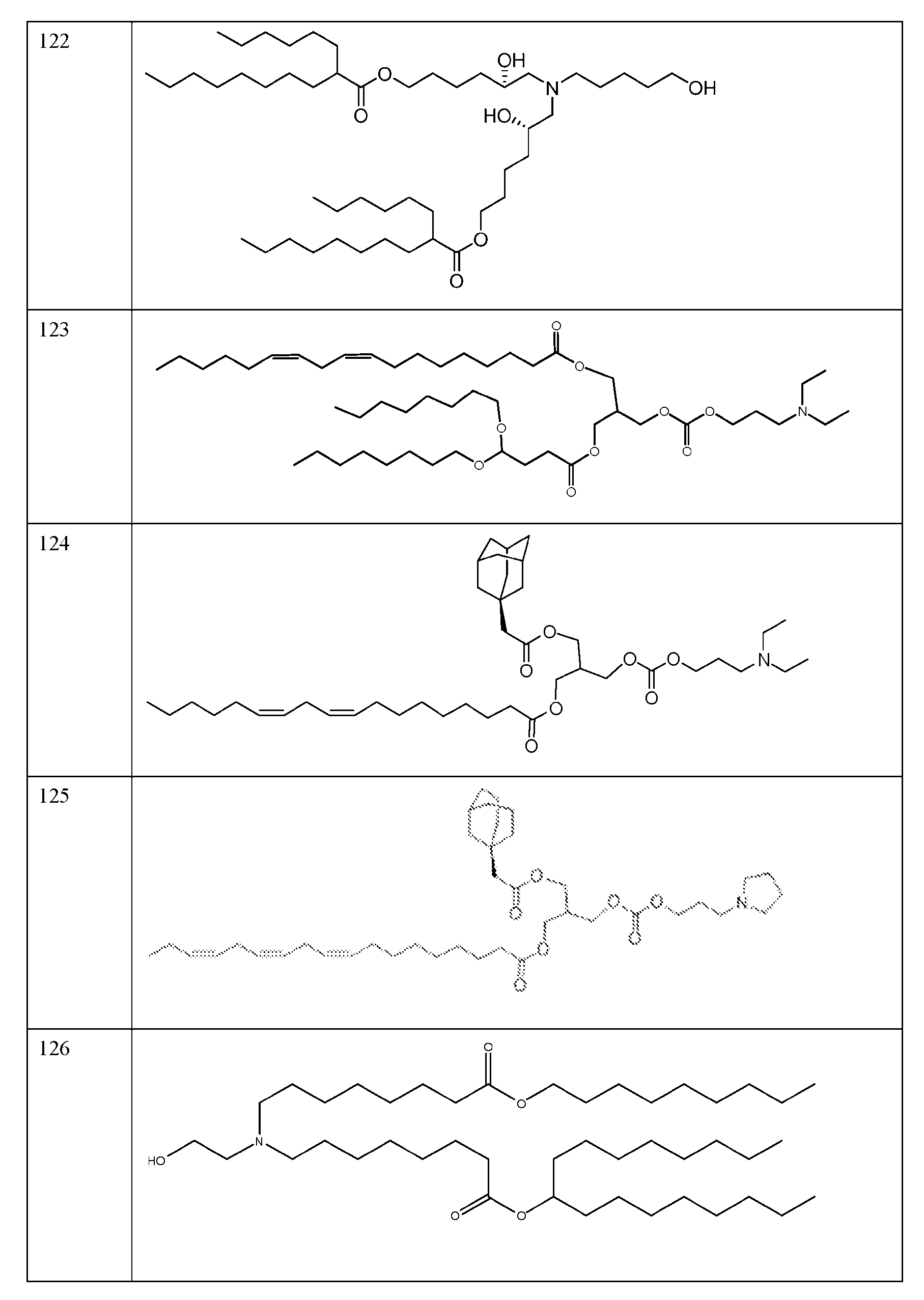

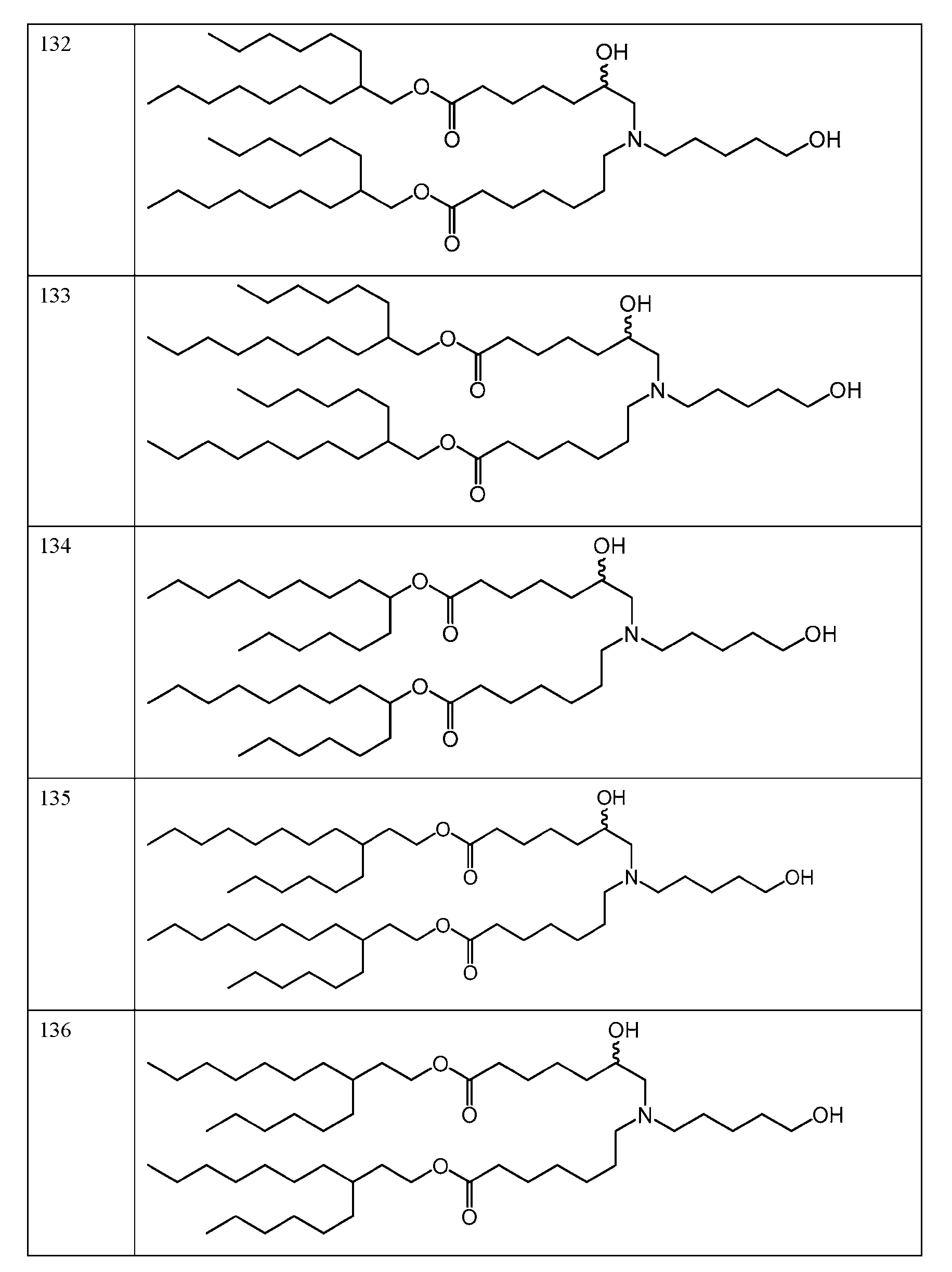


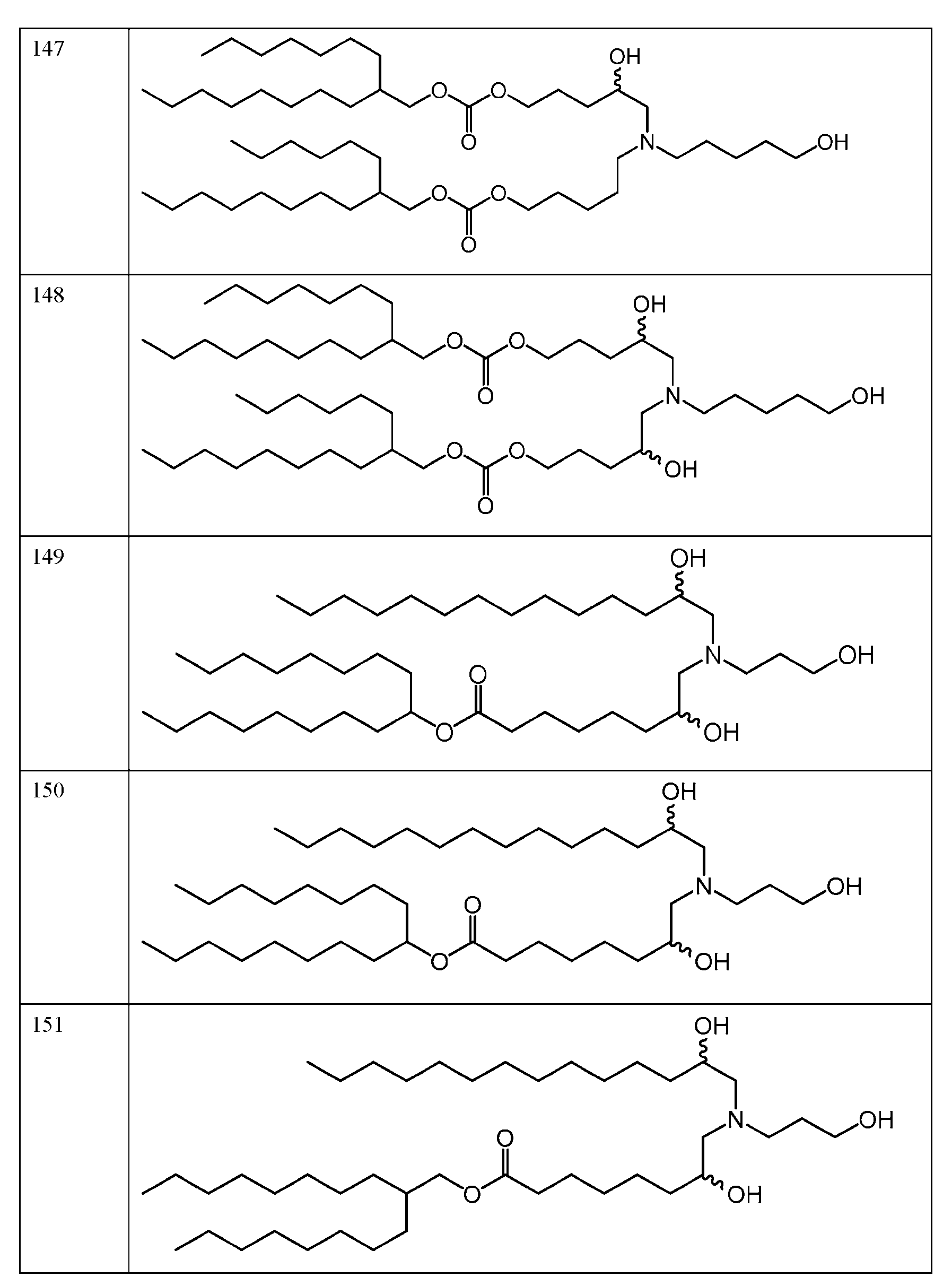



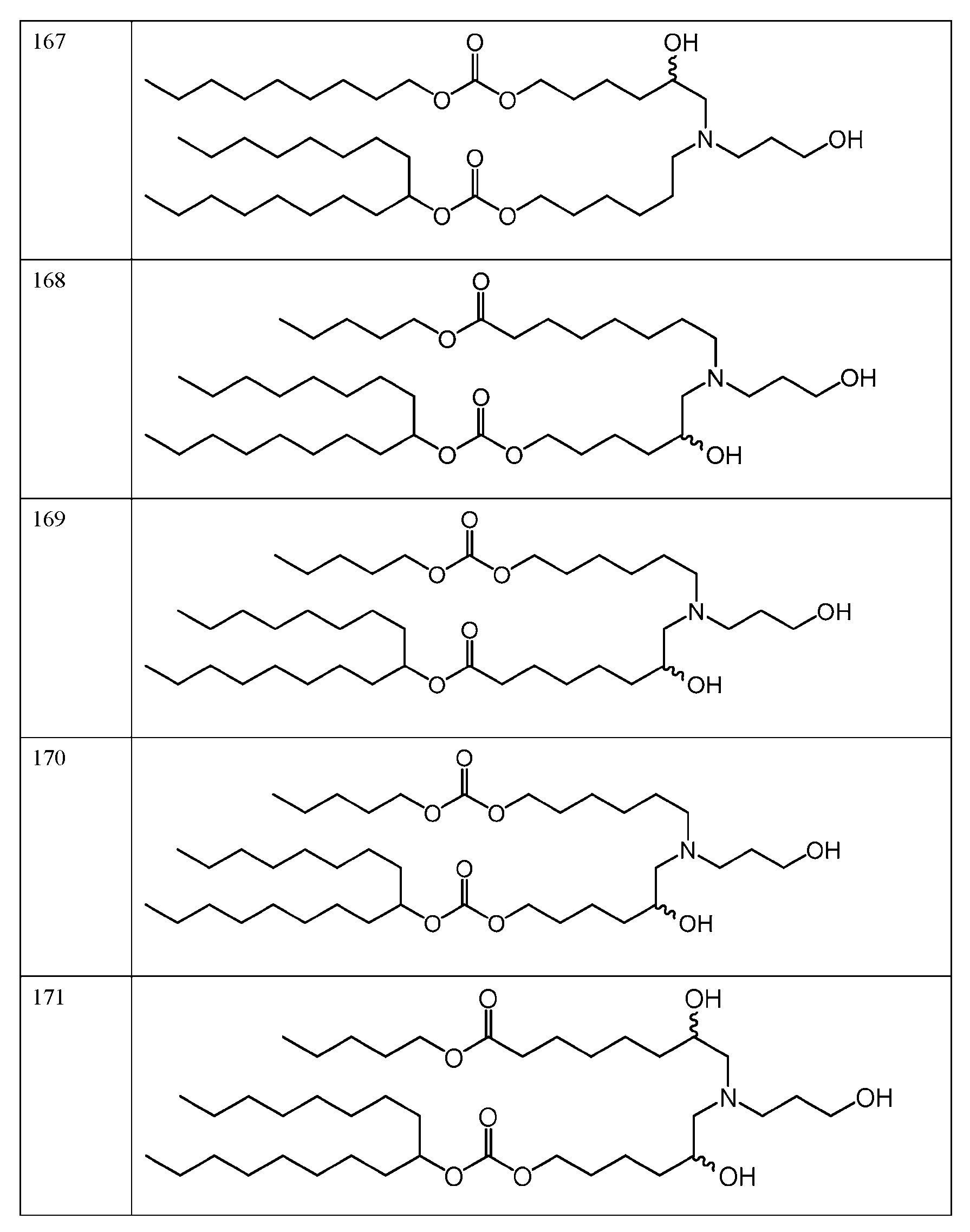
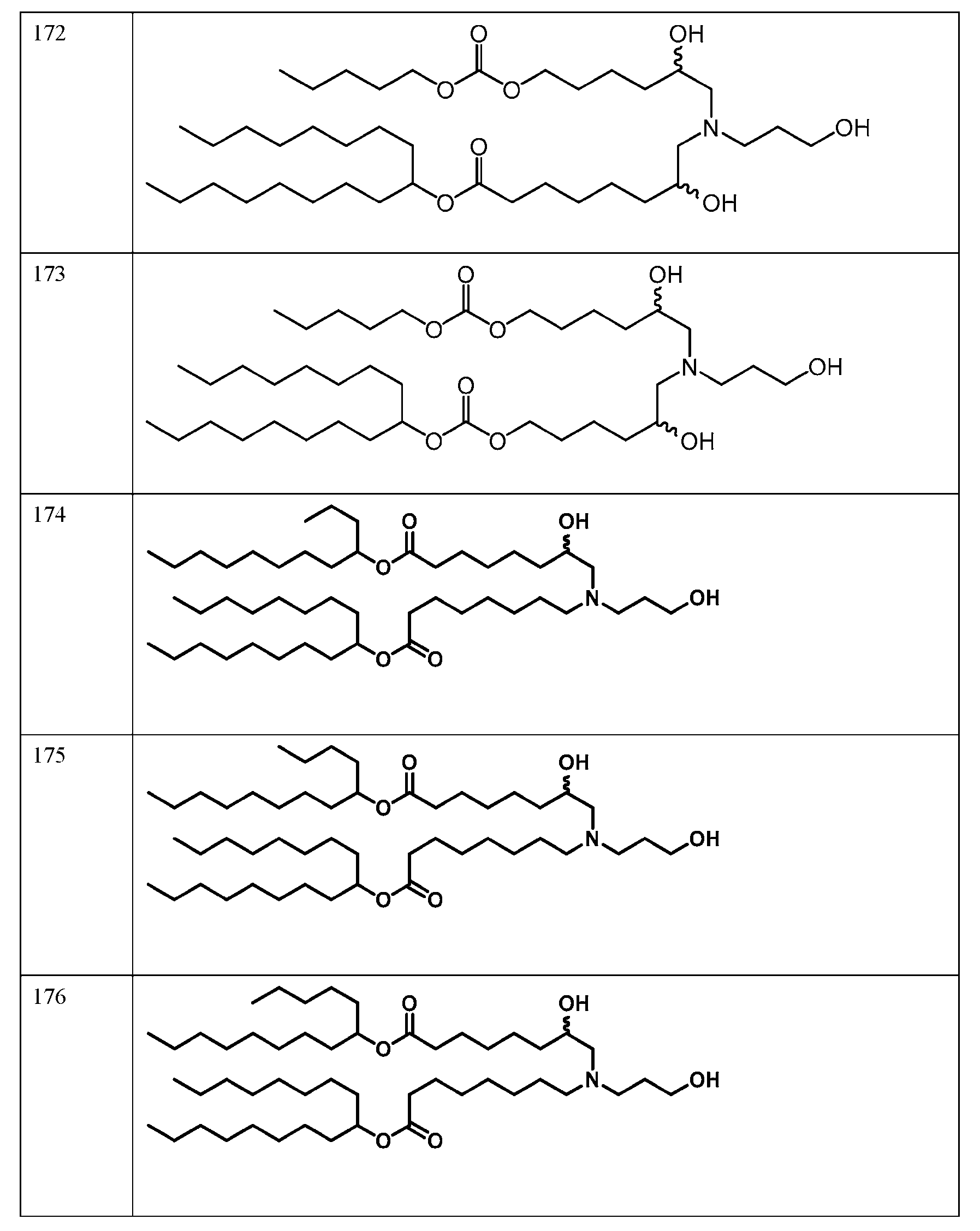
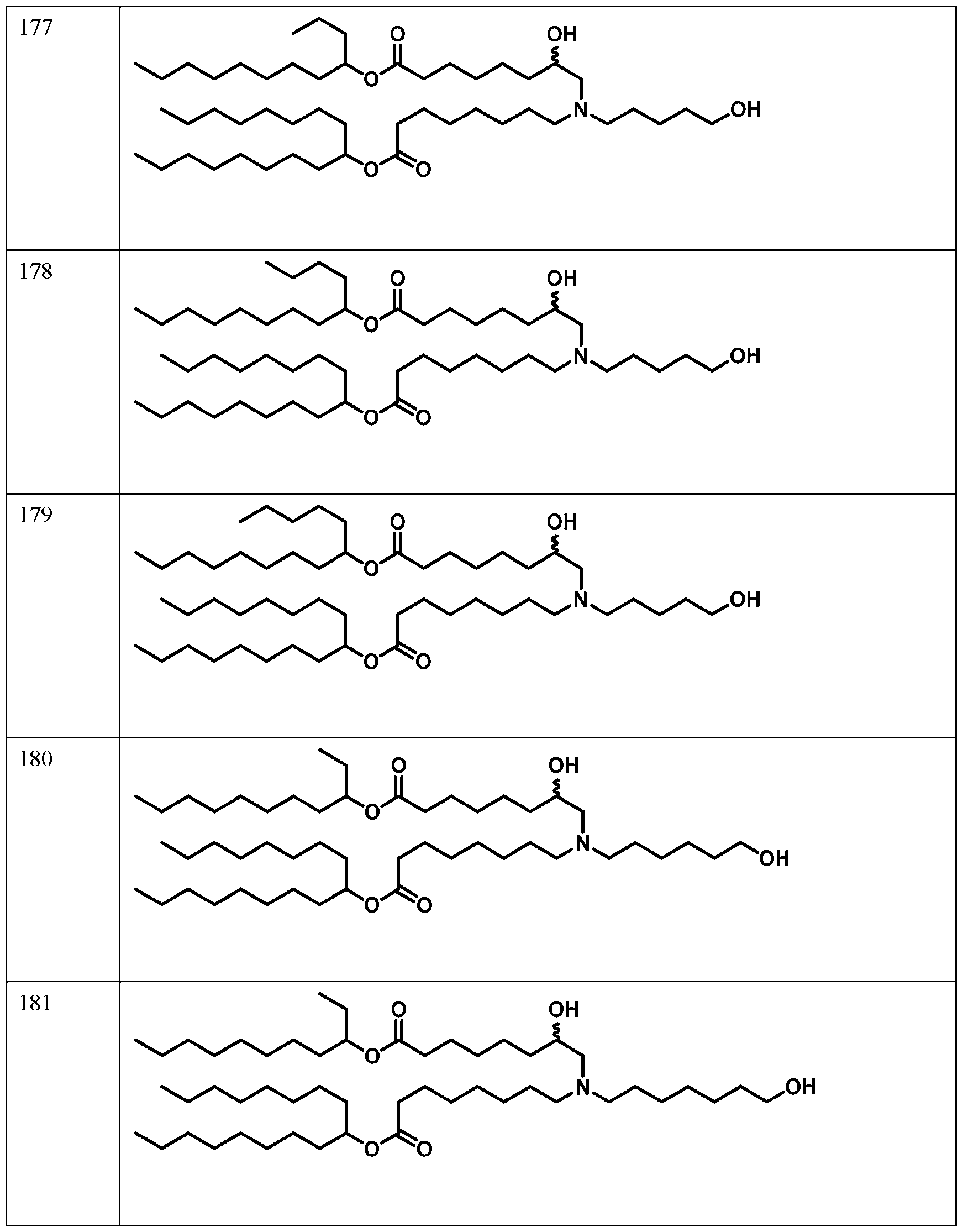

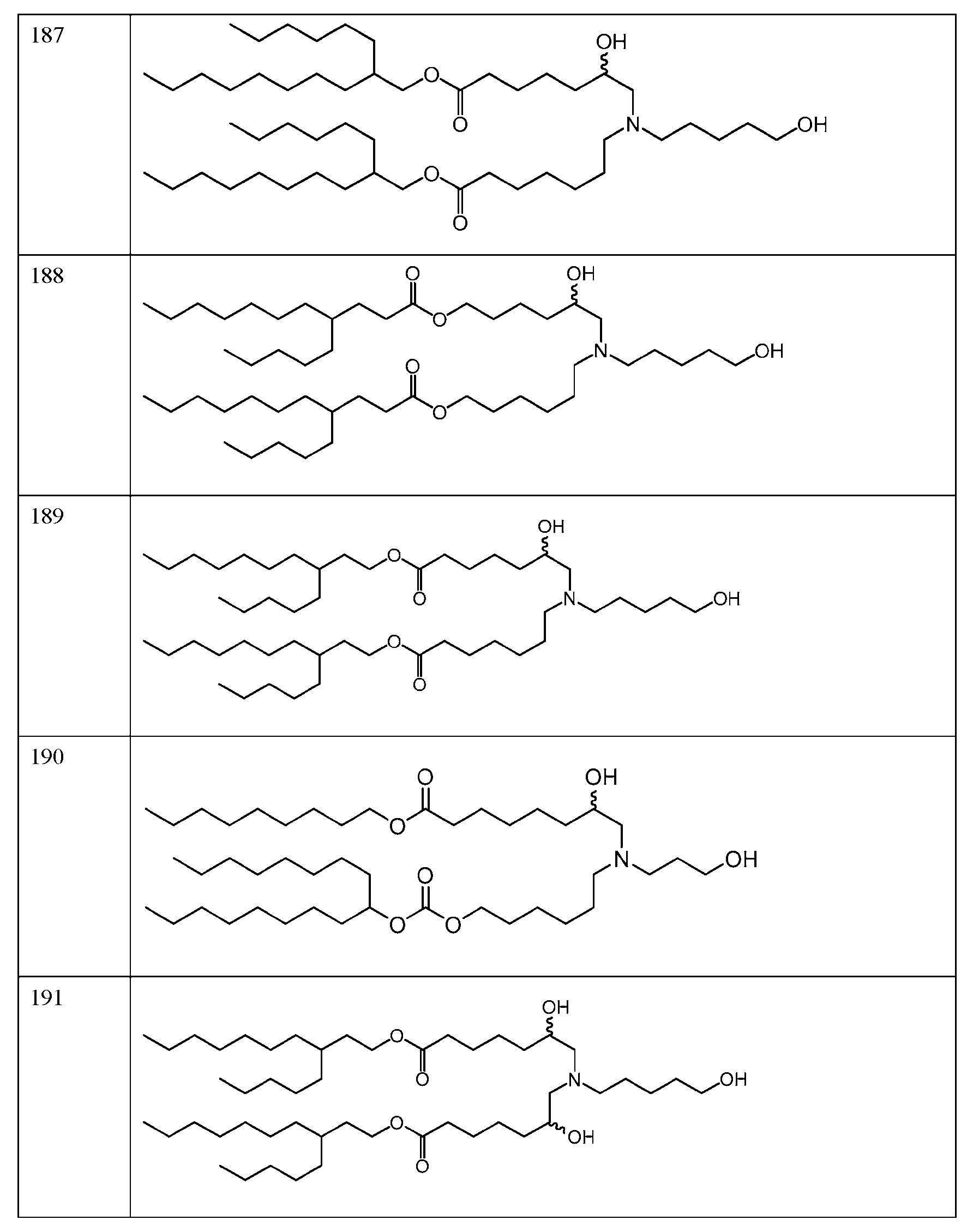
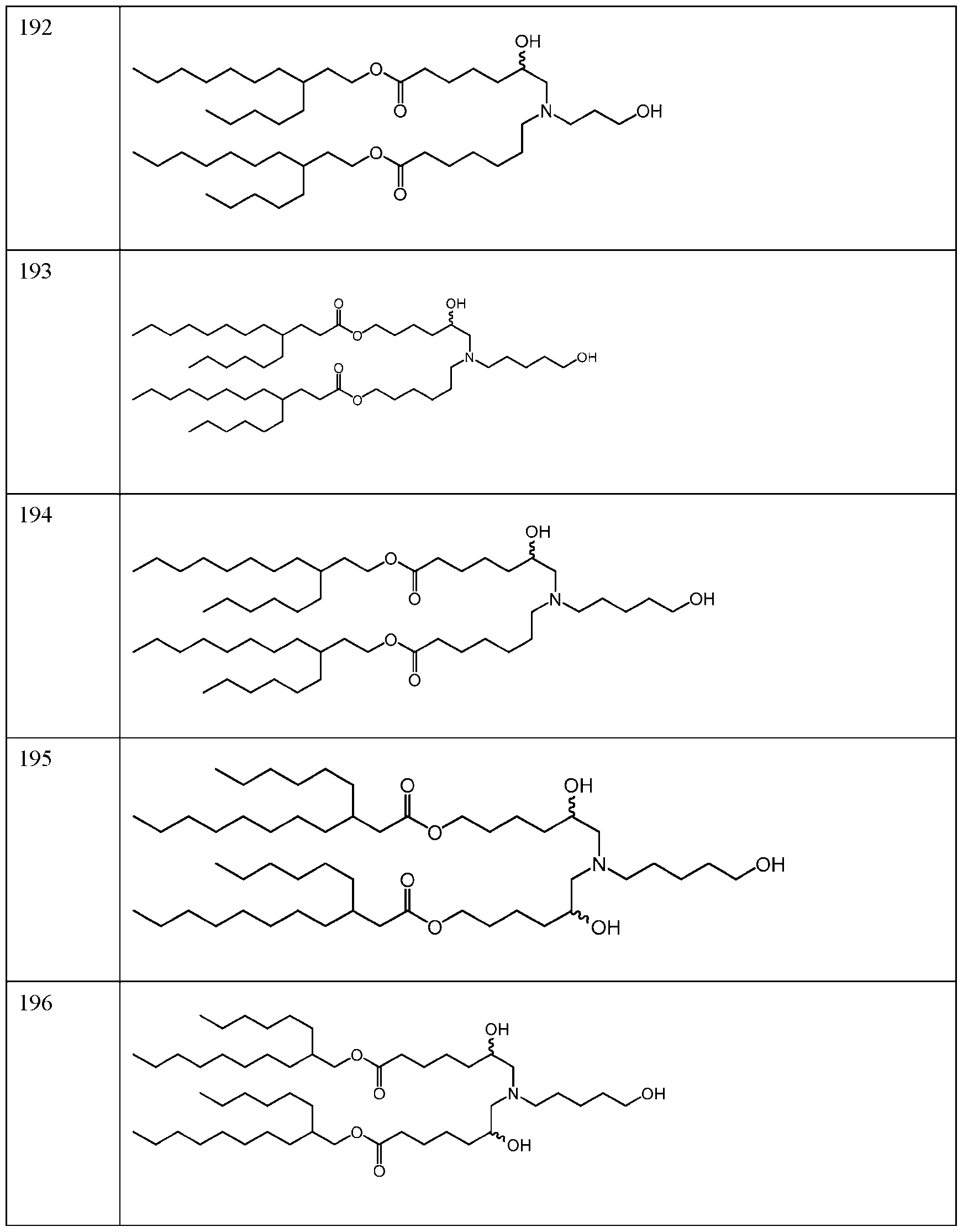


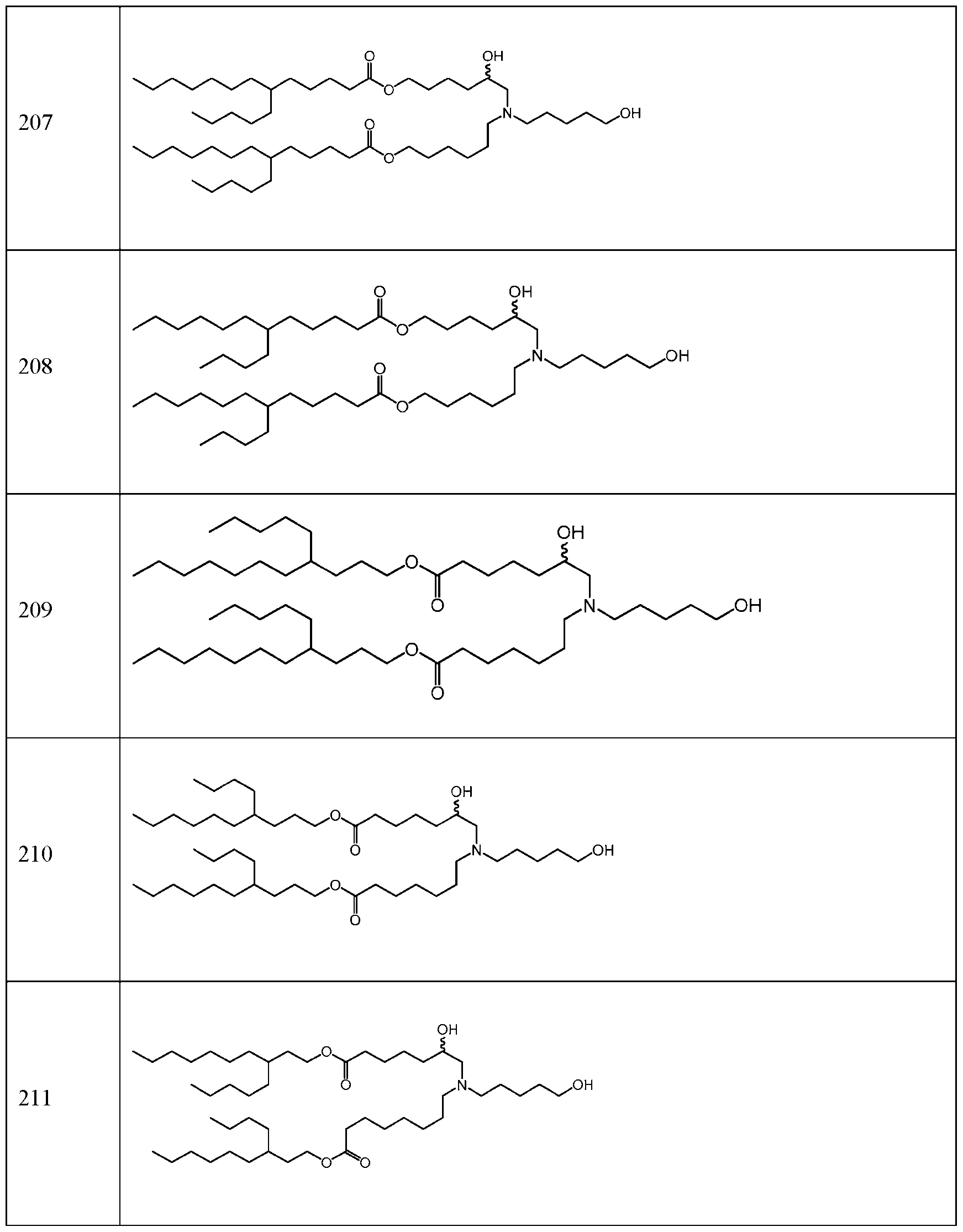

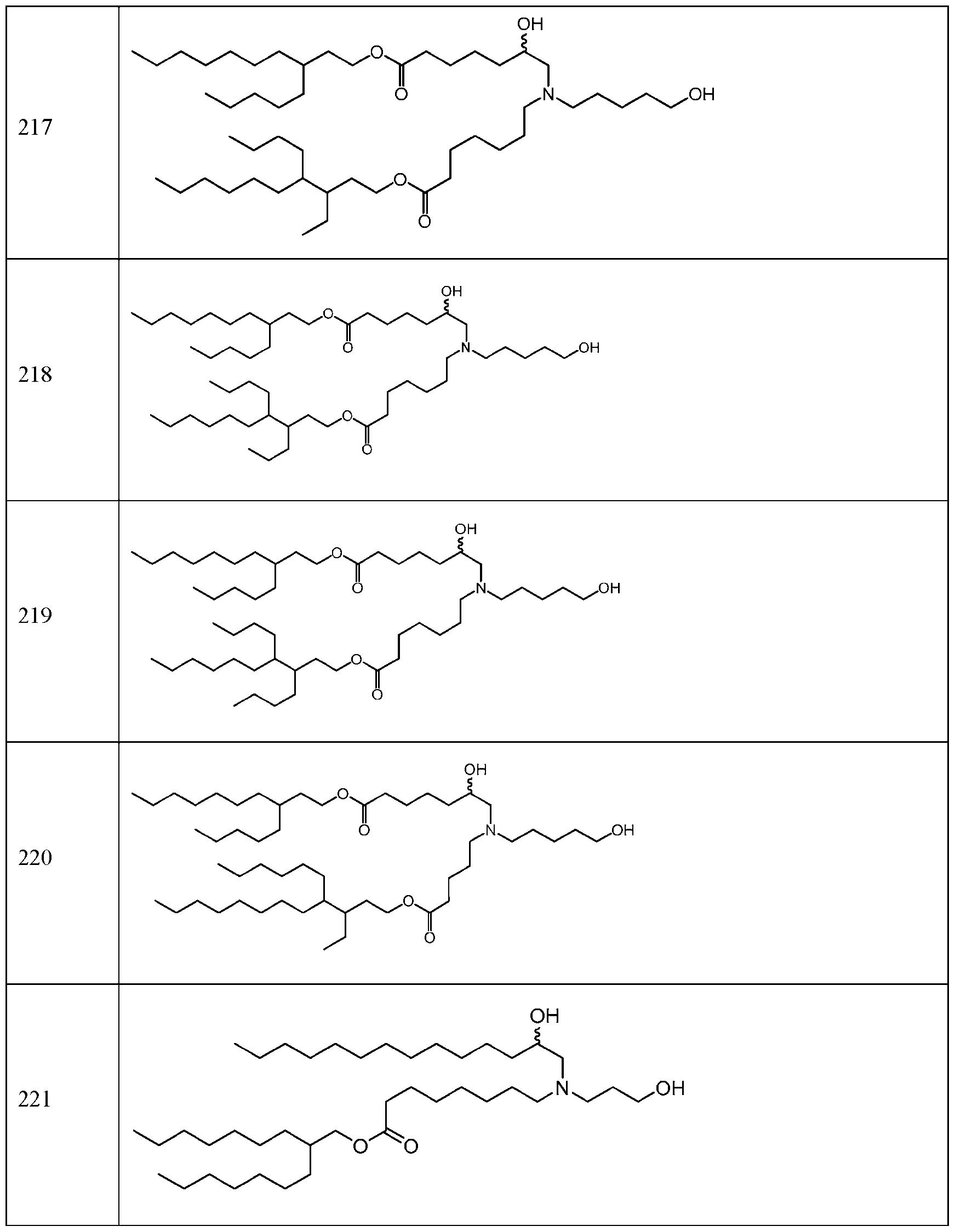
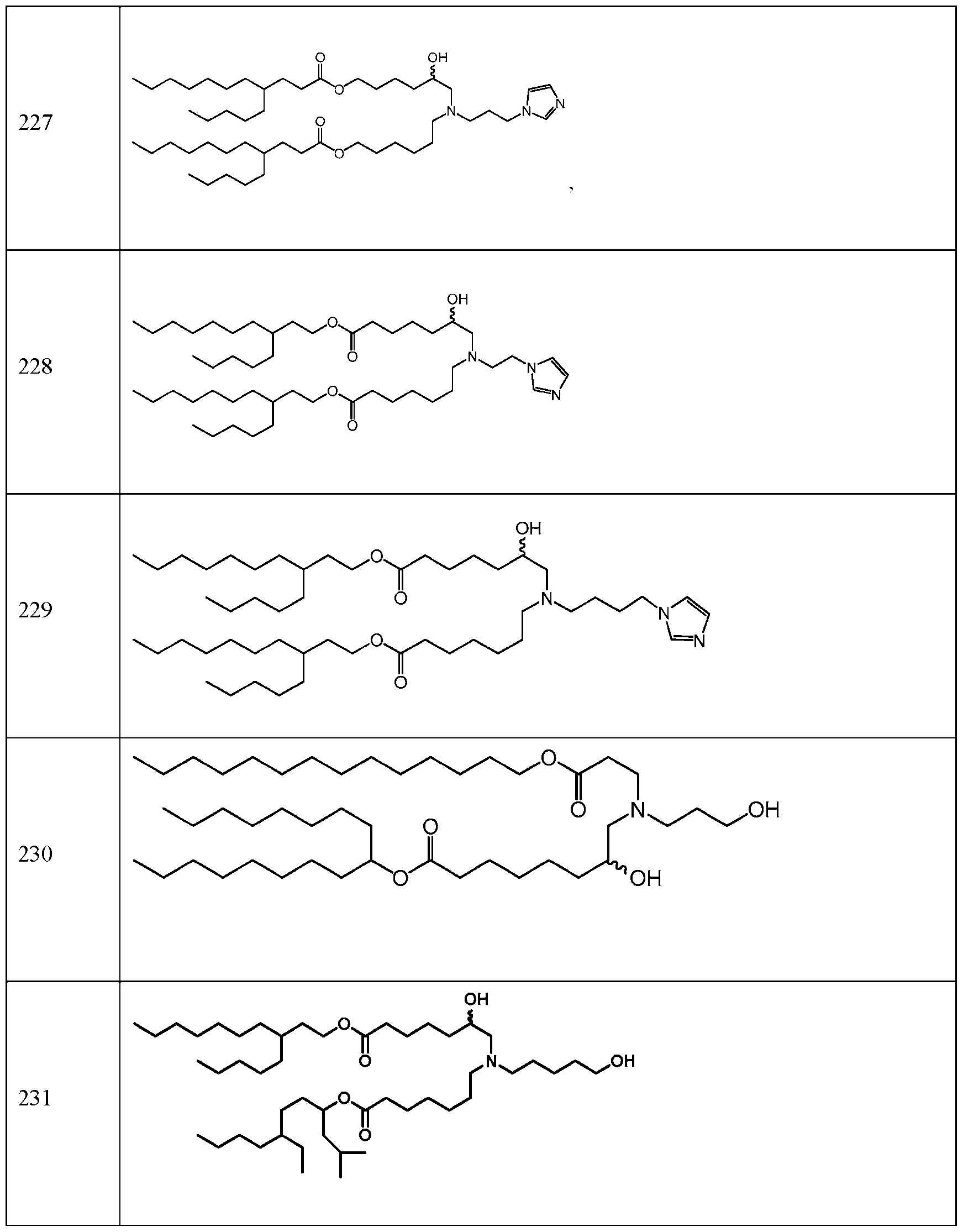

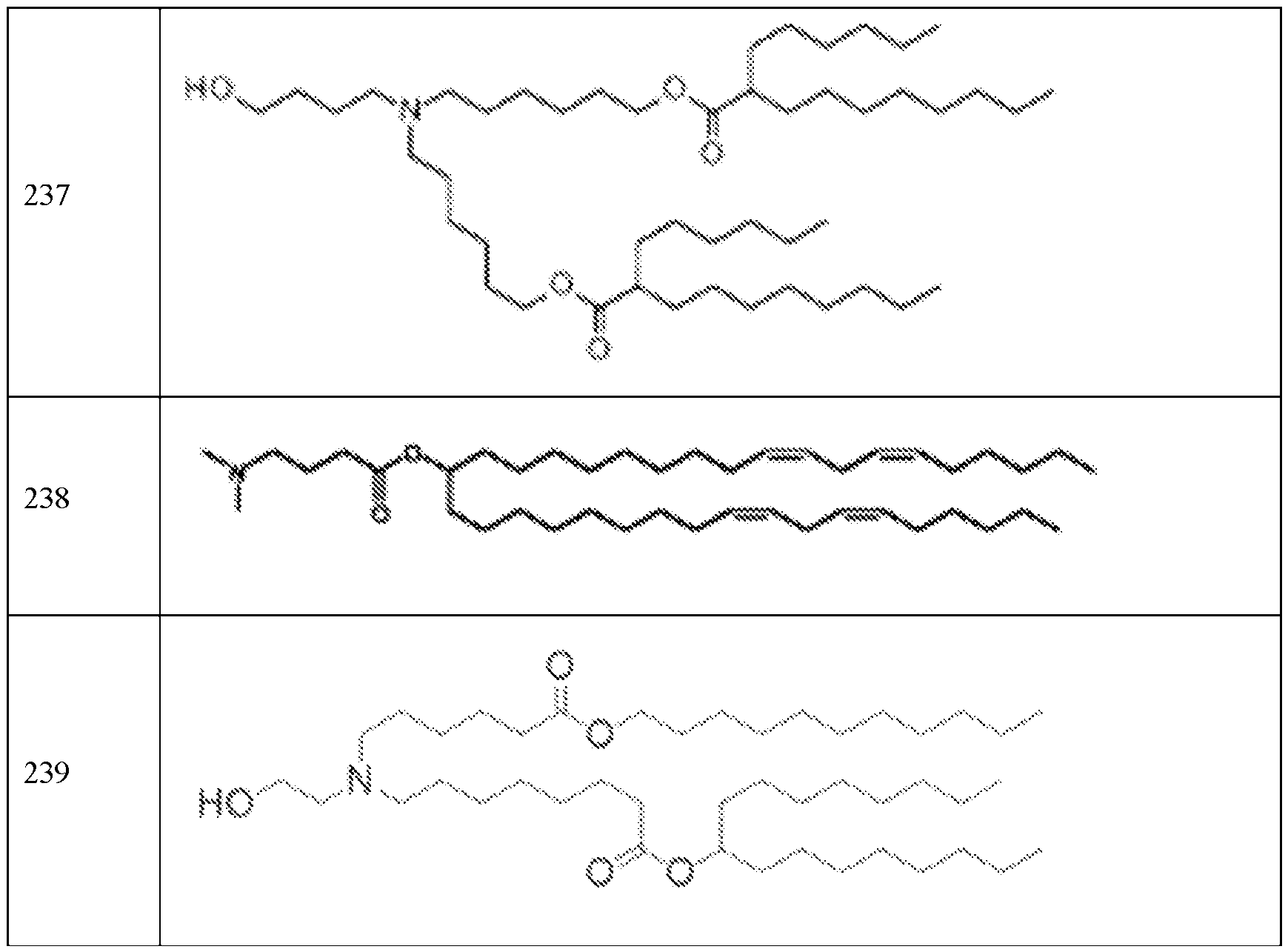
[0231] In some embodiments, the lipid solutions and lipid nanoparticles described herein comprise one or more non-cationic helper lipids. In some embodiments, the helper lipid is a phospholipid. In some embodiments, the helper lipid is a phospholipid substitute or replacement. In some embodiments, the phospholipid or phospholipid substitute can be, for example, one or more saturated or (poly)unsaturatcd phospholipids, or phospholipid substitutes, or a combination thereof. In general, phospholipids comprise a phospholipid moiety and one or more fatty acid moieties.
[0232] A phospholipid moiety can be selected, for example, from the non-limiting group of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and sphingomyelin.
[0233] A fatty acid moiety can be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid.
[0234] Phospholipids include, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols,
phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin.
[0235] In some embodiments, the helper lipid is a l,2-distearoyl-177-glycero-3-phosphocholine (DSPC) analog, a DSPC substitute, oleic acid, or an oleic acid analog.
[0236] In some embodiments, a helper lipid is a non-phosphatidyl choline (PC) zwitterionic lipid, a DSPC analog, oleic acid, an oleic acid analog, or a DSPC substitute.
[0237] In some embodiments, a helper lipid is described in PCT/US2018/053569. Helper lipids suitable for use in a lipid composition of the disclosure include, for example, a variety of neutral, uncharged or zwitterionic lipids. Such helper lipids are preferably used in combination with one or more of the compounds and lipids disclosed herein. Examples of helper lipids include, but are not limited to, 5-heptadecylbenzene-l,3-diol (resorcinol), dipalmitoylphosphatidylcholine (DPPC), distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dimyristoylphosphatidylcholine (DMPC), phosphatidylcholine (PLPC). l,2-distearoylsn-glycero-3- phosphocholine (DAPC). phosphatidylethanolamine (PE), egg phosphatidylcholine (EPC), dilauryloylphosphatidylcholine (DLPC), dimyristoylphosphatidylcholine (DMPC), 1 -myristoyl-2- pahnitoyl phosphatidylcholine (MPPC), 1-paimitoy 1-2 -myristoyl phosphatidylcholine (PMPC), 1- pahnitoy 1-2-stearoy 1 phosphatidylcholine (PSPC), 1 ,2-diarachidoyl-sn-glycero-3 -phosphocholine (DBPC), 1-stearoy 1-2 -palmitoyl phosphatidylcholine (SPPC), l,2-dieicosenoyl-sn-glycero-3- phosphocholinc (DEPC), paimitoyioicoyl phosphatidylcholine (POPC), lysophosphatidyl choline, dioleoyl phosphatidylethanol amine (DOPE) dilinoleoylphosphatidylcholine distearoylphosphatidylethanolamine (DSPE), dimyristoyl phosphatidylethanolamine (DMPE), dipalmitoyl phosphatidylethanolamine (DPPE), palmitoyloleoyl phosphatidylethanolamine (POPE), lysophosphatidylethanolamine and combmations thereof. In one embodiment, tire helper lipid may be distearoylphosphatidylcholine (DSPC) or dimyristoyl phosphatidyl ethanolamine (DMPE). In another embodiment, the helper lipid may be distearoylphosphatidylcholine (DSPC). Helper lipids function to stabilize and improve processing of the lipid solutions and lipid nanoparticles. Such helper lipids are preferably used in combination with other excipients, for example, one or more of the ionizable lipids disclosed herein.
C. STRUCTURAL LIPIDS
[0238] In some embodiments, the lipid solutions and lipid nanoparticles described herein comprise one or more structural lipids. Incorporation of structural lipid(s) in the lipid nanoparticle may help mitigate aggregation of other lipids in the particle. Structural lipids can include, but are not limited to, cholesterol, fecosterol, ergosterol, bassicasterol, tomatidine, tomatine, ursolic, alpha-tocopherol, and mixtures thereof. In certain embodiments, the structural lipid is cholesterol. In certain embodiments,
die structural lipid includes cholesterol and a corticosteroid (such as, for example, prednisolone, dexamethasone, prednisone, and hydrocortisone), or a combination thereof.
[0239] In some embodiments, a structural lipid is described in international patent application PCT/US2019/015913, which is incorporated by reference herein in its entirety.
[0240] In some embodiments, the structural lipid is a sterol. In certain embodiments, the structural lipid is a steroid. In certain embodiments, the structural lipid is cholesterol. In certain embodiments, the structural lipid is an analog of cholesterol. In certain embodiments, the structural lipid is alphatocopherol.
[0241] The lipid solutions and lipid nanoparticles described herein comprise one or more structural lipids. Incorporation of structural lipids in a transfer vehicle, e.g., a lipid nanoparticle, may help mitigate aggregation of other lipids in the particle. In some embodiments, the structural lipid includes cholesterol, a corticosteroid (such as, for example, prednisolone, dexamethasone, prednisone, and hydrocortisone), or a combination thereof.
[0242] In some embodiments, the structural lipid is a sterol. Structural lipids can include, but are not limited to, sterols (e.g.. phytosterols or zoosterols).
[0243] In some embodiments, the structural lipid is a steroid. For example, sterols can include, but are not limited to, cholesterol, p-sitosterol, fecosterol. ergosterol, sitosterol, campesterol, stigmasterol, brassicasterol. ergosterol, tomatidine. tomatine, ursolic acid, or alpha-tocopherol.
[0244] In some embodiments, the lipid solutions and lipid nanoparticles include an effective amount of an immune cell delivery potentiating lipid, e.g., a cholesterol analog or an amino lipid or combination thereof, that, when present in a transfer vehicle, e.g., an lipid nanoparticle, may function by enhancing cellular association and/or uptake, internalization, intracellular trafficking and/or processing, and/or endosomal escape and/or may enhance recognition by and/or binding to immune cells, relative to a transfer vehicle lacking the immune cell delivery potentiating lipid. Accordingly, while not intending to be bound by any particular mechanism or theory, in one embodiment, a structural lipid or other immune cell delivery potentiating lipid of the disclosure binds to Clq or promotes the binding of a transfer vehicle comprising such lipid to Clq. Thus, for in vitro use of the transfer vehicles of the disclosure for deliver of a nucleic acid molecule to an immune cell, culture conditions that include Clq are used (e.g.. use of culture media that includes serum or addition of exogenous Clq to serum- free media). For in vivo use of the transfer vehicles of the disclosure, the requirement for Cl q is supplied by endogenous Clq.
[0245] In some embodiments, the structural lipid is cholesterol. In some embodiments, the structural lipid is an analog of cholesterol. In some embodiments, the structural lipid is a lipid in Table 2 below:
Table 2: Example Structural lipids

901

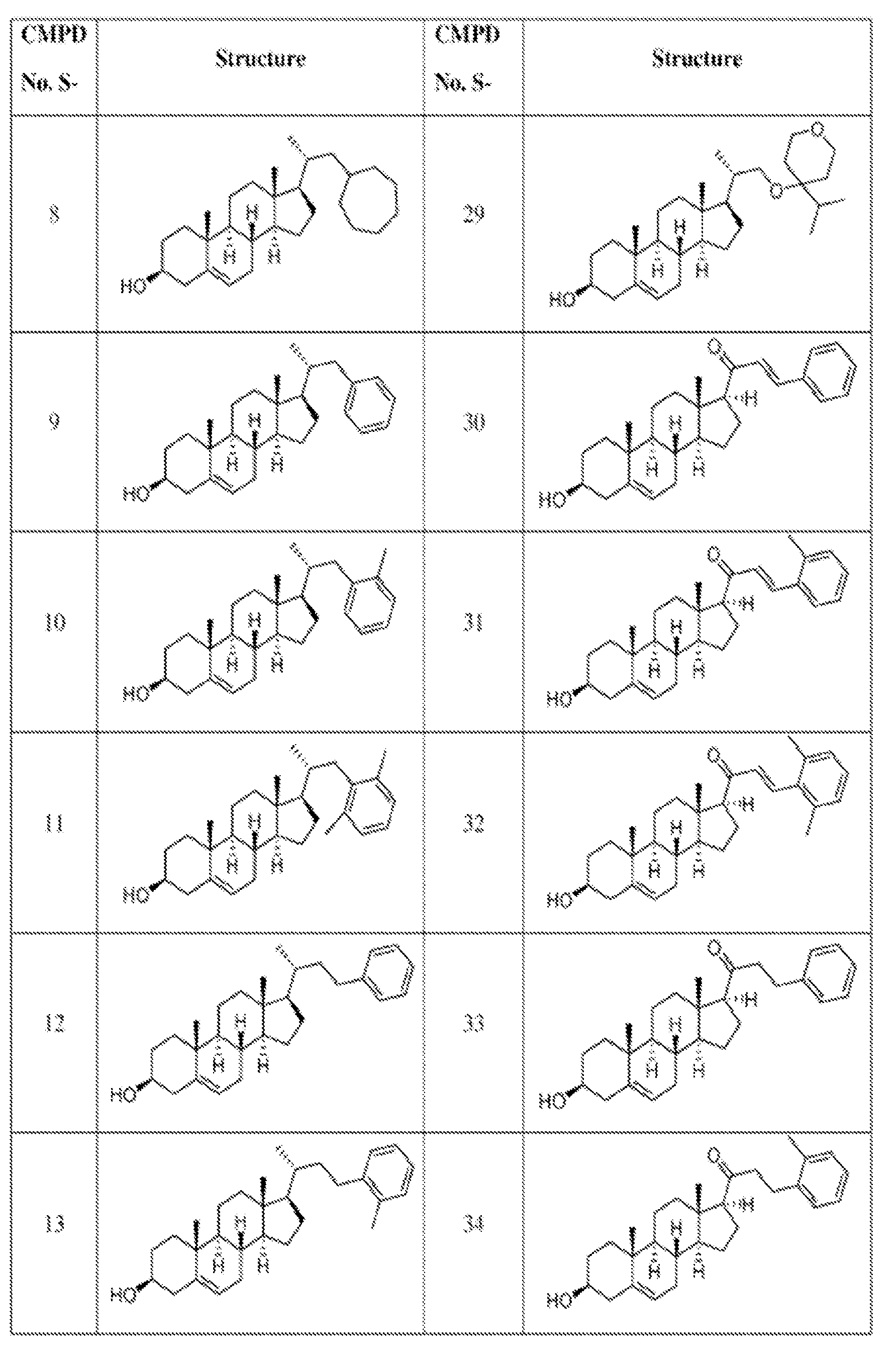





ĶTI



[0246] The use and inclusion of polyethylene glycol (PEG)-modified phospholipids and derivatized lipids such as derivatized ceramides (PEG-CER), including N-Octanoyl-Sphingosine-1- [Succinyl(Methoxy Polyethylene Glycol)-2000] (C6 PEG-2000 ceramide) in the lipid solutions and lipid nanoparticlcs described herein is contemplated, preferably in combination with one or more of the compounds and lipids disclosed herein. Contemplated PEG-modified lipids include, but are not limited to, a polyethylene glycol chain of up to 5 kDa in length covalently attached to a lipid with alkyl chain(s) of C6-C20 length. In some embodiments, the PEG-modified lipid employed in the compositions and methods of the disclosure is 1,2-dimyristoyl-sn-glycerol, methoxypolyethylene Glycol (2000 MW PEG) “DMG-PEG2000.” The addition of PEG-modified lipids to the lipid delivery vehicle may prevent complex aggregation and may also provide a means for increasing circulation lifetime and increasing the delivery of the lipid-polynucleotide composition to the target tissues, (Klibanov et al. (1990) FEBS Letters, 268 (1): 235-237), or they may be selected to rapidly exchange out of the formulation in vivo
(see U.S. Pat. No. 5,885,613). Particularly useful exchangeable lipids are PEG-ceramides having shorter acyl chains (e.g., C14 or Cl 8). The PEG-modified phospholipid and derivatized lipids of the present disclosure may comprise a molar ratio from about 0% to about 20%, about 0.5% to about 20%, about 1% to about 15%, about 4% to about 10%, or about 2% of the total lipid present in the lipid solution or lipid nanoparticle.
[0247] In one embodiment, a PEG-modified lipid is described in International Pat. Appl. No. PCT/US2019/015913 or PCT/US2020/046407, which arc incorporated herein by reference in their entirety . In one embodiment, a transfer vehicle comprises one or more PEG-modified lipids.
[0248] Non-limiting examples of PEG-modified lipids include PEG-modified phosphatidylethanolamines and phosphatidic acids, PEG-ceramide conjugates (e.g, PEG-CerC14 or PEG-CerC2 0), PEG-modified dialkylamines and PEG-modified l .2-diacyloxypropan-3-amines. In some further embodiments, a PEG-modified lipid may be, e.g., PEG-c-DOMG, PEG-DMG, PEG- DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE.
[0249] In some embodiments, the PEG-modified lipid includes, but is not limited to 1,2-dimyristoyl- sn-glycerol mcthoxypolyethylenc glycol (PEG-DMG), l,2-distearoyl-sn-glycero-3- phosphoethanolamine-N-[amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG- DSG), PEG-dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG-DAG), PEG- dipahnitoyl phosphatidy lethanolamine (PEG-DPPE), PEG-1, 2-dimyristyloxlpropy 1-3 -amine (PEG-c- DMA).
[0250] In some embodiments, the PEG-modified lipid is DSPE-PEG, DMG-PEG, PEG-DAG, PEG- S-DAG, PEG-PE, PEG-S-DMG, PEG-cer, PEG-dialkoxypropylcarbamate, PEG-OR, PEG-OH, PEG- c-DOMG, or PEG-1. In some embodiments, the PEG-modified lipid is DSPE-PEG(2000).
[0251] In some embodiments, the PEG-modified lipid comprises a PEG moiety comprising 10-70 (e.g., 30-60) oxycthylcne (-O-CH2-CH2-) units or portions thereof. In some embodiments, the PEG- modified lipid comprises (OCH2CH2)V-ORW, and v is an integer between 0 and 70 (inclusive) (e.g.. an integer between 30 and 60), w is hydrogen or alky l.
[0252] In some embodiments, a PEG-modified lipid may also be referred to as “PEGylated lipid” or “PEG-lipid.”
[0253] In one embodiment, the PEG-lipid is selected from a PEG-modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG-modified ceramide, a PEG-modified dialkylamine, a PEG- modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof.
[0254] In some embodiments, the lipid moiety of the PEG-lipids includes those having lengths of from about Cu to about C22. such as from about C14 to about C1s. In some embodiments, a PEG moiety, for
example a mPEG-NH2, has a size of about 1000, about 2000, about 5000, about 10,000, about 15,000 or about 20,000 daltons. In one embodiment, the PEG-lipid is PEG2k-DMG.
[0255] In one embodiment, the lipid nanoparticles described herein can comprise a lipid modified with a non-diffusible PEG. Non-limiting examples of non-diffusible PEGs include PEG-DSG and PEG- DSPE.
[0256] PEG-lipids are known in the art, such as those described in U.S. Pat. No. 8,158,601 and International Pat. Publ. No. WO2015/130584 A2, which are incorporated herein by reference in their entirety7.
[0257] In some embodiments, lipids (e.g., PEG-lipids), described herein may be synthesized as described International Pat. Publ. No. PCT/US2016/000129, which is incorporated by reference in its entirety.
[0258] The lipid component of a lipid solution or lipid nanoparticle composition may include one or more molecules comprising polyethylene glycol, such as PEG or PEG-modified lipids. Such species may be alternately referred to as PEGylated lipids. A PEG lipid is a lipid modified with polyethylene glycol. A PEG lipid may be selected from the non-limiting group including PEG-modified phosphatidylethanolamines. PEG-modified phosphatidic acids, PEG-modified ceramides. PEG- modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkyl glycerols, and mixtures thereof. For example, a PEG lipid may be PEG-c-DOMG. PEG-DMG. PEG-DLPE, PEG-DMPE, PEG- DPPC, or a PEG-DSPE lipid.
[0259] In some embodiments the PEG-modified lipids are a modified form of PEG-DMG. PEG-DMG
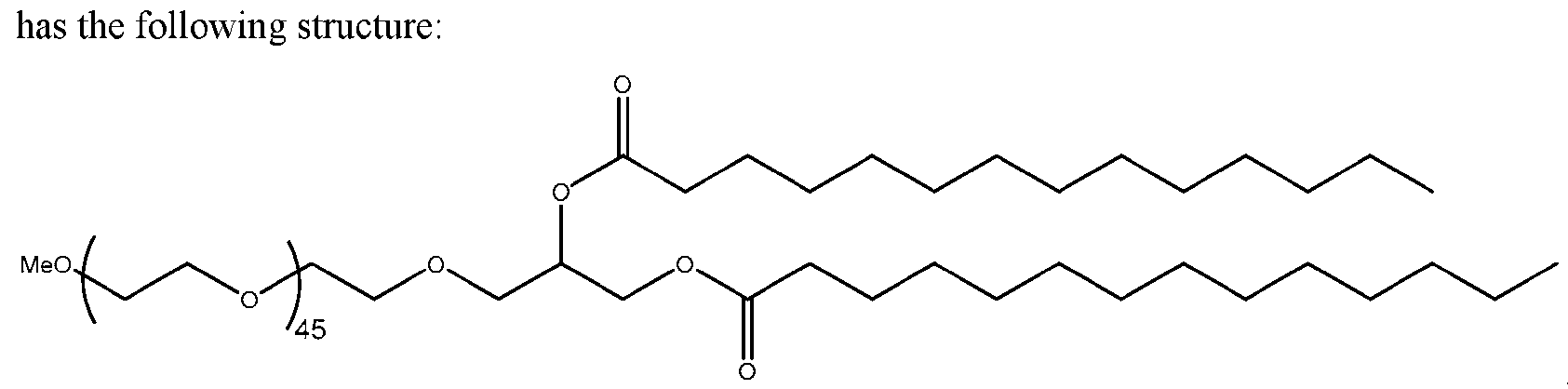
[0260] In some embodiments the PEG-modified lipids are a modified form of PEG-C18, or PEG-1.
PEG-1 has the following structure:

[0261] In one embodiment, PEG lipids useful in the present disclosure can be PEGylated lipids described in International Publication No. WO2012099755, the contents of which is herein incorporated
by reference in its entirety . Any of these exemplary PEG lipids described herein may be modified to comprise a hydroxyl group on the PEG chain. In certain embodiments, the PEG lipid is a PEG-OH lipid. In certain embodiments, the PEG-OH lipid includes one or more hydroxyl groups on the PEG chain. In certain embodiments, a PEG-OH or hydroxy -PEGylated lipid comprises an -OH group at the terminus of the PEG chain. Each possibility represents a separate embodiment of the present disclosure.
[0262] In some embodiments, the PEG lipid is a compound of Formula (Pl):
 or a salt or isomer thereof, wherein: r is an integer between 1 and 100;
or a salt or isomer thereof, wherein: r is an integer between 1 and 100;
R is C10-40 alkyl, C10-ro alkenyl, or C10-4o alkynyl; and optionally one or more methylene groups of R are independently replaced with C3-10 carbocyclylene, 4 to 10 membered heterocyclylene, Ce io arylene. 4 to 10 membered heteroarylene, -N(RN)-, -O-, -S-, -C(O)- ,-C(O)N(RN)-, - NRNC(O)-, -NRNC(O)N(RN)-, -C(O)O- -OC(O)-, -OC(O)O- ,-OC(O)N(RN)-, - NRNC(O)O-, -C(O)S-, -SC(O)-, -C(=NRN)-, -C(=NRN)N(RN)-. -NRNC(=NRN)-. - NRNC(=NRN)N(RN)- ,-C(S)-, -C(S)N(RN)-, -NRNC(S)-. -NRNC(S)N(RN)-, -S(O)-, - OS(O)-, -S(O)O-, -OS(O)O-, -OS(O)2- -S(O)2O-, -OS(O)2O-, -N(RN)S(O)-, - S(O)N(RN)-, -N(RN)S(O)N(RN)-, -OS(O)N(RN)-, -N(RN)S(O)O-, -S(O)2- -N(RN)S(O)2- -S(O)2N(RN)-, -N(RN)S(O)2N(RN)-, -OS(O)2N(RN)-, or -N(RN)S(O)2O-; and each instance of RN is independently hydrogen. C1.g alkyl, or a nitrogen protecting group.
[0263] For example, R is C17 alkyl. For example, the PEG lipid is a compound of Formula (Pl-a):

(Pl-a) or a salt or isomer thereof, wherein r is an integer between 1 and 100.
[0264] For example, the PEG lipid is a compound of the following formula:
 5. POLYNUCLEOTIDES
5. POLYNUCLEOTIDES
[0265] In some embodiments, the present disclosure provides loaded lipid nanoparticles comprising a polynucleotide. In some embodiments, the polynucleotide is DNA. In some embodiments, the polynucleotide is RNA. In some embodiments, the polynucleotide is linear RNA. In preferred embodiments, the polynucleotide is circular RNA.
[0266] Transcription of a DNA template (e.g., comprising a 3’ intron element, 3’ exon element, a core functional element, a 5’ exon element, and a 5’ intron element) results in formation of a precursor linear RNA polynucleotide capable of circularizing. In some embodiments, this DNA template comprises a vector, a PCR product, a plasmid, a minicircle DNA, a cosmid, an artificial chromosome, a complementary DNA (cDNA), an extrachromosomal DNA (ecDNA). or a fragment therein. In certain embodiments, the minicircle DNA may be linearized or non-linearized. In certain embodiments, the plasmid may be linearized or non-linearized. In some embodiments, the DNA template may be singlestranded. In other embodiments, the DNA template may be double-stranded. In some embodiments, the DNA template comprises in whole or in part from a viral, bacterial, or eukaryotic vector.
[0267] The present disclosure, as provided herein, comprises a DNA template that shares the same sequence as the precursor linear RNA polynucleotide prior to splicing of the precursor linear RNA polynucleotide (e.g., a 3’ intron element, a 3’ exon element, a core functional element, and a 5‘ exon element, a 5’ intron element). In some embodiments, said linear precursor RNA polynucleotide undergoes splicing leading to the removal of the 3’ intron element and 5‘ intron element during the process of circularization. In some embodiments, the resulting circular RNA polynucleotide lacks a 3 ’ intron fragment and a 5’ intron fragment, but maintains a 3’ exon fragment, a core functional element, and a 5‘ exon element.
[0268] In some embodiments, the precursor linear RNA polynucleotide circularizes when incubated in the presence of one or more guanosine nucleotides or nucleoside (e.g., GTP) and a divalent cation (e.g., Mg2+). In some embodiments, the 3’ exon element, 5’ exon element, and/or core functional element in whole or in part promotes the circularization of the precursor linear RNA polynucleotide to form the circular RNA polynucleotide provided herein.
[0269] In certain embodiments the circular RNA provided herein is produced inside a cell. In some embodiments, the precursor RNA is transcribed using a DNA template (e.g., in some embodiments, using a vector provided herein) in the cytoplasm by a bacteriophage RNA polymerase, or in the nucleus by host RNA polymerase II and then circularized.
[0270] In certain embodiments, the circular RNA provided herein is injected into an animal (e.g., a human), such that a polypeptide encoded by the circular RNA molecule is expressed inside the animal.
[0271] In some embodiments, the DNA template (e.g., vector), linear RNA (e.g., precursor RNA), and/or circular RNA polynucleotide provided herein is between 300 and 10000, 400 and 9000, 500 and 8000, 600 and 7000, 700 and 6000, 800 and 5000, 900 and 5000, 1000 and 5000, 1100 and 5000. 1200 and 5000, 1300 and 5000, 1400 and 5000, and/or 1500 and 5000 nucleotides (nt) in length. In some embodiments, the polynucleotide is at least 300 nt, 400 nt. 500 nt, 600 nt, 700 nt, 800 nt, 900 nt. 1000 nt, 1100 nt, 1200 nt, 1300 nt. 1400 nt, 1500 nt, 2000 nt, 2500 nt, 3000 nt, 3500 nt. 4000 nt, 4500 nt, or 5000 nt in length. In some embodiments, the polynucleotide is no more than 3000 nt, 3500 nt, 4000 nt, 4500 nt. 5000 nt, 6000 nt, 7000 nt, 8000 nt. 9000 nt, or 10000 nt in length. In some embodiments, the length of a DNA, linear RNA, and/or circular RNA polynucleotide provided herein is 300 nt. 400 nt, 500 nt, 600 nt, 700 nt. 800 nt. 900 nt, 1000 nt. 1100 nt, 1200 nt, 1300 nt. 1400 nt, 1500 nt, 2000 nt. 2500 nt, 3000 nt, 3500 nt, 4000 nt, 4500 nt, 5000 nt, 6000 nt, 7000 nt, 8000 nt, 9000 nt, or 10000 nt.
[0272] In some embodiments, the circular RNA provided herein has higher functional stability than mRNA comprising the same expression sequence. In some embodiments, the circular RNA provided herein has higher functional stability than mRNA comprising the same expression sequence, 5moU modifications, an optimized UTR, a cap, and/or a polyA tail.
[0273] In some embodiments, the circular RNA polynucleotide provided herein has a functional halflife of at least 5 hours, 10 hours, 15 hours, 20 hours. 30 hours. 40 hours, 50 hours, 60 hours, 70 hours or 80 hours. In some embodiments, the circular RNA polynucleotide provided herein has a functional half-life of 5-80. 10-70, 15-60, or 20-50 hours. In some embodiments, the circular RNA polynucleotide provided herein has a functional half-life greater than (e.g.. at least 1.5-fold greater than, at least 2-fold greater than) that of an equivalent linear RNA polynucleotide encoding the same protein. In some embodiments, functional half-life can be assessed through the detection of functional protein synthesis.
[0274] In some embodiments, the circular RNA polynucleotide, or pharmaceutical composition thereof, has a functional half-life in a human cell greater than or equal to that of a pre-determined threshold value. In some embodiments, the functional half-life is determined by a functional protein assay. For example, in some embodiments, the functional half-life is determined by an in vitro luciferase assay, wherein the activity of Gaussia luciferase (GLuc) is measured in the media of human cells (e.g. HepG2) expressing the circular RNA polynucleotide every 1, 2, 6, 12, or 24 hours over 1. 2. 3, 4, 5, 6. 7. or 14 days. In other embodiments, the functional half-life is determined by an in vivo assay, wherein levels of a protein encoded by the expression sequence of the circular RNA polynucleotide are measured in patient serum or tissue samples every 1, 2, 6, 12, or 24 hours over 1. 2. 3, 4, 5, 6. 7. or 14 days. In some embodiments, the pre-determined threshold value is the functional half-life of a reference linear RNA polynucleotide comprising the same expression sequence as the circular RNA polynucleotide.
[0275] In some embodiments, the circular RNA provided herein may have a higher magnitude of expression than equivalent linear mRNA, e.g., a higher magnitude of expression 24 hours after
administration of RNA to cells. In some embodiments, the circular RNA provided herein has a higher magnitude of expression than mRNA comprising the same expression sequence, 5moU modifications, an optimized UTR, a cap, and/or a polyA tail.
[0276] In some embodiments, the circular RNA provided herein may be less immunogenic than an equivalent mRNA when exposed to an immune system of an organism or a certain type of immune cell. In some embodiments, the circular RNA provided herein is associated with modulated production of cytokines when exposed to an immune system of an organism or a certain type of immune cell. For example, in some embodiments, the circular RNA provided herein is associated with reduced production of IFN-(31, RIG-I, IL-2, IL-6, IFNy, and/or TNFa when exposed to an immune system of an organism or a certain type of immune cell as compared to mRNA comprising the same expression sequence. In some embodiments, the circular RNA provided herein is associated with less IFN-(31, RIG-I, IL-2. IL-6, IFNy, and/or TNFa transcript induction when exposed to an immune system of an organism or a certain type of immune cell as compared to mRNA comprising the same expression sequence. In some embodiments, the circular RNA provided herein is less immunogenic than mRNA comprising the same expression sequence. In some embodiments, the circular RNA provided herein is less immunogenic than mRNA comprising the same expression sequence, 5moU modifications, an optimized UTR. a cap. and/or a polyA tail.
[0277] In certain embodiments, the circular RNA provided herein can be transfected into a cell as is or can be transfected in DNA vector form and transcribed in the cell. Transcription of circular RNA from a transfected DNA vector can be via added polymerases or polymerases encoded by nucleic acids transfected into the cell, or preferably via endogenous polymerases. i. Enhanced Intron Elements & Enhanced Exon Elements
[0278] In some embodiments, the DNA template (e.g., vector) or linear RNA (e.g., precursor RNA) comprises an enhanced intron element and/or an enhanced exon element. The enhanced intron elements and enhanced exon elements may comprise spacers, duplex regions, affinity sequences, intron fragments, exon fragments and various untranslated elements. These sequences within the enhanced intron elements or enhanced exon elements are arranged to optimize circularization or protein expression.
[0279] In certain embodiments, the DNA template, precursor linear RNA polynucleotide and circular RNA provided herein comprise a first (5 ’) and/or a second (3 ) spacer. In some embodiments, the DNA template or precursor linear RNA polynucleotide comprises one or more spacers in the enhanced intron elements. In some embodiments, the DNA template, precursor linear RNA polynucleotide comprises one or more spacers in the enhanced exon elements. In certain embodiments, the DNA template or linear RNA polynucleotide comprises a spacer in the 3 ’ enhanced intron fragment and a spacer in the 5’ enhanced intron fragment. In certain embodiments, DNA template, precursor linear RNA
polynucleotide, or circular RNA comprises a spacer in the 3’ enhanced exon fragment and another spacer in the 5' enhanced exon fragment to aid with circularization or protein expression due to symmetry created in the overall sequence.
[0280] In some embodiments, including a spacer between the 3 ‘ group I intron fragment and the core functional element may conserve secondary structures in those regions by preventing them from interacting, thus increasing splicing efficiency. In some embodiments, the first (between 3’ group I intron fragment and core functional element) and the second (between the two expression sequences and core functional element) spacers comprise additional base pairing regions that are predicted to base pair with each odier and not to the first and second duplex regions. In other embodiments, the first (between 3’ group I intron fragment and core functional element) and the second (between the one of the core functional element and 5 ’ group I intron fragment) spacers comprise additional base pairing regions that are predicted to base pair with each other and not to the first and second duplex regions. In some embodiments, such spacer base pairing brings the group I intron fragments in close proximity to each other, further increasing splicing efficiency. Additionally, in some embodiments, the combination of base pairing between the first and second duplex regions, and separately, base pairing between the first and second spacers, promotes the formation of a splicing bubble containing the group I intron fragments flanked by adjacent regions of base pairing. Typical spacers are contiguous sequences with one or more of the following qualities: 1) predicted to avoid interfering with proximal structures, for example, the IRES, expression sequence, aptamer, or intron; 2) is at least 7 nt long and no longer than 100 nt; 3) is located after and adjacent to the 3’ intron fragment and/or before and adjacent to the 5’ intron fragment; and 4) contains one or more of the following: a) an unstructured region at least 5 nt long, b) a region of base pairing at least 5 nt long to a distal sequence, including another spacer, and c) a structured region at least 7 nt long limited in scope to the sequence of the spacer. Spacers may have several regions, including an unstructured region, a base pairing region, a hairpin/structured region, and combinations thereof. In an embodiment, the spacer has a structured region with high GC content. In an embodiment, a region within a spacer base pairs with another region within the same spacer. In an embodiment, a region within a spacer base pairs with a region within another spacer. In an embodiment, a spacer comprises one or more hairpin structures. In an embodiment, a spacer comprises one or more hairpin structures with a stem of 4 to 12 nucleotides and a loop of 2 to 10 nucleotides. In an embodiment, tiiere is an additional spacer betw een the 3 ’ group I intron fragment and the core functional element. In an embodiment, this additional spacer prevents the structured regions of the IRES or aptamer of a TIE from interfering widr the folding of the 3’ group I intron fragment or reduces the extent to which this occurs. In some embodiments, the 5’ spacer sequence is at least 7, 8, 9, 10. 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25 or 30 nucleotides in length. In some embodiments, the 5‘ spacer sequence is no more than 100, 90, 80, 70, 60, 50, 45, 40, 35 or 30 nucleotides in length. In some embodiments the 5’ spacer sequence is between 5 and 50, 10 and 50, 20 and 50, 20 and 40, and/or 25 and 35 nucleotides in length.
In certain embodiments, the 5’ spacer sequence is 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 or 50 nucleotides in length. In one embodiment, the 5’ spacer sequence is a poly A sequence. In another embodiment, the 5’ spacer sequence is a poly AC sequence. In one embodiment, a spacer comprises 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or 100% poly AC content. In one embodiment, a spacer comprises 10%. 20%, 30%, 40%, 50%. 60%, 70%, 80%, 90%. or 100% polypyrimidine (C/T or C/U) content.
[0281] In some embodiments, the DNA template and precursor linear RNA polynucleotides and circular RNA polynucleotide provided herein comprise a first (5’) duplex region and a second (3’) duplex region. In certain embodiments, the DNA template and precursor linear RNA polynucleotide comprises a 5' external duplex region located within the 3‘ enhanced intron fragment and a 3’ external duplex region located within the 5’ enhanced intron fragment. In some embodiments, the DNA template, precursor linear RNA polynucleotide and circular RNA polynucleotide comprise a 5 ‘ internal duplex region located within the 3’ enhanced exon fragment and a 3’ internal duplex region located within the 5’ enhanced exon fragment. In some embodiments, the DNA polynucleotide and precursor linear RN A polynucleotide comprises a 5 ’ external duplex region. 5 ’ internal duplex region, a 3 ’ internal duplex region, and a 3‘ external duplex region.
[0282] In certain embodiments, the first and second duplex regions may form perfect or imperfect duplexes. Thus, in certain embodiments at least 75%. 80%, 85%, 90%, 91%, 92%, 93%. 94%, 95%, 96%, 97%, 98%, 99%, or 100% of the first and second duplex regions may be base paired with one another. In some embodiments, the duplex regions are predicted to have less than 50% (e.g., less than 45%, less than 40%, less than 35%, less than 30%. less than 25%) base pairing with unintended sequences in the RNA (e.g., non-duplex region sequences). In some embodiments, including such duplex regions on the ends of the precursor RNA strand, and adjacent or very close to the group 1 intron fragment, bring the group I intron fragments in close proximity to each other, increasing splicing efficiency. In some embodiments, the duplex regions are 3 to 100 nucleotides in length (e.g., 3-75 nucleotides in length, 3-50 nucleotides in length. 20-50 nucleotides in length. 35-50 nucleotides in length. 5-25 nucleotides in length, 9-19 nucleotides in length). In some embodiments, the duplex regions are 3, 4. 5. 6, 7, 8, 9, 10, 11, 12. 13, 14, 15, 16. 17, 18, 19, 20. 21. 22, 23, 24. 25. 26, 27, 28, 29. 30, 31, 32, 33, 34. 35, 36, 37, 38. 39, 40, 41, 42. 43, 44, 45, 46. 47, 48, 49, or 50 nucleotides in length. In some embodiments, the duplex regions have a length of 9 to 50 nucleotides. In one embodiment, the duplex regions have a length of 9 to 19 nucleotides. In some embodiments, the duplex regions have a length of 20 to 40 nucleotides. In certain embodiments, the duplex regions have a length of 30 nucleotides.
[0283] In other embodiments, the DNA template, precursor linear RNA polynucleotide, or circular RNA polynucleotide does not comprise of any duplex regions to optimize translation or circularization.
[0284] As provided herein, the DNA template or precursor linear RNA polynucleotide may comprise an affinity tag. In some embodiments, the affinity tag is located in the 3 ‘ enhanced intron element. In some embodiments, the affinity tag is located in the 5’ enhanced intron element. In some embodiments, both (3 ' and 5 ') enhanced intron elements each comprise an affinity tag. In one embodiment, an affinity tag of the 3 ‘ enhanced intron element is the length as an affinity tag in the 5 ’ enhanced intron element. In some embodiments, an affinity tag of the 3 ’ enhanced intron element is the same sequence as an affinity tag in the 5 ’ enhanced intron element. In some embodiments, the affinity sequence is placed to optimize oligo-dT purification.
[0285] In some embodiments, an affinity tag comprises a polyA region. In some embodiments the polyA region is at least 15, 30, or 60 nucleotides long. In some embodiments, one or both polyA regions is 15-50 nucleotides long. In some embodiments, one or both polyA regions is 20-25 nucleotides long. The polyA sequence is removed upon circularization. Thus, an oligonucleotide hybridizing with the polyA sequence, such as a deoxythymine oligonucleotide (oligo(dT)) conjugated to a solid surface (e.g., a resin), can be used to separate circular RNA from its precursor RNA.
[0286] In certain embodiments, the 3’ enhanced intron element comprises a leading untranslated sequence. In some embodiments, the leading untranslated sequence is a the 5’ end of the 3‘ enhanced intron fragment. In some embodiments, the leading untranslated sequence comprises of the last nucleotide of a transcription start site (TSS). In some embodiments, the TSS is chosen from a viral, bacterial, or eukaryotic DNA template. In one embodiment, the leading untranslated sequence comprise the last nucleotide of a TSS and 0 to 100 additional nucleotides. In some embodiments, the TSS is a terminal spacer. In one embodiment, the leading untranslated sequence contains a guanosine at the 5’ end upon translation of an RNA T7 polymerase.
[0287] In certain embodiments, the 5’ enhanced intron element comprises a trailing untranslated sequence. In some embodiments, the 5' trailing untranslated sequence is located at the 3’ end of the 5’ enhanced intron element. In some embodiments, the trailing untranslated sequence is a partial restriction digest sequence. In one embodiment, the trailing untranslated sequence is in whole or in part a restriction digest site used to linearize the DNA template. In some embodiments, the restriction digest site is in whole or in part from a natural viral, bacterial or eukaryotic DNA template. In some embodiments, the trailing untranslated sequence is a terminal restriction site fragment. a. Enhanced Intron Fragments
[0288] In some embodiments, the 3’ enhanced intron element and 5’ enhanced intron element each comprise an intron fragment. In certain embodiments, a 3 ‘ intron fragment is a contiguous sequence at least 75% homologous (e.g., at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% homologous) to a 3’ proximal fragment of a natural group I or II intron including the 3’ splice site dinucleotide. Typically, a 5' intron fragment is a contiguous sequence at least 75%
homologous (e.g., at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% homologous) to a 5 ' proximal fragment of a natural group I or II intron including the 5 ’ splice site dinucleotide. In some embodiments, the 3’ intron fragment includes the first nucleotide of a 3‘ group I or II splice site dinucleotide. In some embodiments, the 5’ intron fragment includes the first nucleotide of a 5’ group I or II splice site dinucleotide. In other embodiments, the 3‘ intron fragment includes the first and second nucleotides of a 3’ group I or II intron fragment splice site dinucleotide; and the 5’ intron fragment includes the first and second nucleotides of a 3’ group I or II intron fragment dinucleotide. In some embodiments the 3’ enhanced intron element and 5’ enhanced intron element comprises a synthetic intron fragment. b. Enhanced Exon Fragments
[0289] In certain embodiments, as provided herein, the DNA template, linear precursor RNA polynucleotide, and circular RNA polynucleotide each comprise an enhanced exon fragment. In some embodiments, following a 5’ to 3’ order, the 3’ enhanced exon element is located upstream to core functional element. In some embodiments, following a 5’ to 3’ order, the 5’ enhanced intron element is located downstream to the core functional element.
[0290] In some embodiments, the 3’ enhanced exon element and 5’ enhanced exon element each comprise an exon fragment. In some embodiments, the 3’ enhanced exon element comprises a 3‘ exon fragment. In some embodiments, the 5’ enhanced exon element comprises a 5’ exon fragment. In certain embodiments, as provided herein, the 3 ’ exon fragment and 5 ’ exon fragment each comprises a group I or II intron fragment and 1 to 100 nucleotides of an exon sequence. In certain embodiments, a 3’ intron fragment is a contiguous sequence at least 75% homologous (e.g., at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% homologous) to a 3’ proximal fragment of a natural group I or II intron including the 3' splice site dinucleotide. Typically, a 5’ group I or II intron fragment is a contiguous sequence at least 75% homologous (e.g., at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%. 97%, 98%, 99% or 100% homologous) to a 5’ proximal fragment of a natural group I or II intron including the 5 ’ splice site dinucleotide. In some embodiments, the 3 ’ exon fragment comprises a second nucleotide of a 3’ group I or II intron splice site dinucleotide and 1 to 100 nucleotides of an exon sequence. In some embodiments, the 5‘ exon fragment comprises the first nucleotide of a 5’ group I or II intron splice site dinucleotide and 1 to 100 nucleotides of an exon sequence. In some embodiments, the exon sequence comprises in part or in whole from a naturally occurring exon sequence from a virus, bacterium or eukaryotic DNA vector. In other embodiments, the exon sequence further comprises a synthetic, genetically modified (e.g., containing modified nucleotide), or other engineered exon sequence.
[0291] In one embodiment, where the 3’ intron fragment comprises both nucleotides of a 3’ group I or II splice site dinucleotide and the 5’ intron fragment comprises both nucleotides of a 5’ group I or II
splice site dinucleotide, the exon fragments located within the 5’ enhanced exon element and 3’ enhanced exon element does not comprise of a group I or II splice site dinucleotide.
[0292] For means of example and not intended to be limiting, in some embodiment, a 3’ enhanced intron element comprises in the following 5’ to 3’ order: a leading untranslated sequence, a 5’ affinity tag, an optional 5’ external duplex region, a 5’ external spacer, and a 3' intron fragment. In same embodiments, the 3’ enhanced exon element comprises in the following 5’ to 3’ order: a 3’ exon fragment, an optional 5’ internal duplex region, an optional 5’ internal duplex region, and a 5’ internal spacer. In the same embodiments, the 5‘ enhanced exon element comprises in the following 5' to 3’ order: a 3’ internal spacer, an optional 3‘ internal duplex region, and a 5’ exon fragment. In still the same embodiments, the 3 ’ enhanced intron element comprises in the following 5 ’ to 3 ’ order: a 5 ’ intron fragment, a 3‘ external spacer, an optional 3' external duplex region, a 3‘ affinity tag, and a trailing untranslated sequence. ii. Core Functional Element
[0293] In some embodiments, the DNA template, linear precursor RNA polynucleotide, and circular RNA polynucleotide comprise a core functional element. In some embodiments, the core functional element comprises a coding or a noncoding element. In certain embodiments, the core functional element may contain both a coding and a noncoding element. In some embodiments, the core functional element further comprises a translation initiation element (TIE) upstream to the coding or noncoding element. In some embodiments, the core functional element comprises a termination element. In some embodiments, the termination element is located downstream to the TIE and coding element. In some embodiments, the termination element is located downstream to the coding element but upstream to the TIE. In certain embodiments, where the coding element comprises a noncoding region, a core functional element lacks a TIE and/or a termination element.
Hi. Coding or Noncoding Element
[0294] In some embodiments, the polynucleotides herein comprise a coding element, a noncoding element, or a combination of both. In some embodiments, the coding element comprises an expression sequence. In some embodiments, the coding element encodes at least one therapeutic protein.
[0295] In some embodiments, the circular RNA encodes two or more polypeptides. In some embodiments, the circular RNA is a bicistronic RNA. The sequences encoding the two or more polypeptides can be separated by a ribosomal skipping element or a nucleotide sequence encoding a protease cleavage site. In certain embodiments, the ribosomal skipping element encodes thosea-asigna virus 2A peptide (T2A), porcine teschovirus-1 2 A peptide (P2A). foot-and-mouth disease virus 2 A peptide (F2A). equine rhinitis A vims 2A peptide (E2A), cytoplasmic polyhedrosis vims 2A peptide (BmCPV 2A), or flacherie vims of B. mori 2A peptide (BmIFV 2A).
iv. Translation Initiation Element (TIE)
[0296] As provided herein in some embodiments, the core functional element comprises at least one translation initiation element (TIE). TIEs are designed to allow translation efficiency of an encoded protein. Thus, optimal core functional elements comprising only of noncoding elements lack any TIEs. In some embodiments, core functional elements comprising one or more coding element will further comprise one or more TIEs.
[0297] In some embodiments, a TIE comprises an untranslated region (UTR). In certain embodiments, the TIE provided herein comprise an internal ribosome entry site (IRES). Inclusion of an IRES pennits the translation of one or more open reading frames from a circular RNA (e.g., open reading frames that form the expression sequences). The IRES element attracts a eukaryotic ribosomal translation initiation complex and promotes translation initiation. See. e.g., Kaufman et al., Nuc. Acids Res. (1991) 19:4485- 4490; Gurtu et al., Biochem. Biophys. Res. Comm. (1996) 229:295-298; Rees et al., BioTechniques (1996) 20: 102-110; Kobayashi et al., BioTcchniqucs (1996) 21 :399-402; and Mosscr et al., BioTechniques 1997 22 150-161. In some embodiments, the IRES element is selected from those disclosed in international publication WO/2022/261490, the contents of which are hereby incorporated in their entireties. v. Additional Accessory Elements (Sequence Elements)
[0298] As described in this disclosure, the circular RNA polynucleotide, linear RNA polynucleotide, and/or DNA template may further comprise accessory elements. In certain embodiments, these accessory elements may be included within the sequences of the circular RNA, linear RNA polynucleotide and/or DNA template for enhancing circularization, translation or both. Accessory elements are sequences, in certain embodiments that are located with specificity between or within the enhanced intron elements, enhanced exon elements, or core functional element of the respective polynucleotide. As an example, but not intended to be limiting, an accessory element includes an IRES transacting factor region, a miRNA binding site, a restriction site, an RNA editing region, a structural or sequence element, a granule site, a zip code element, an RNA trafficking element or another specialized sequence as found in the art that enhances circularization and/or translation of the protein encoded within the circular RNA polynucleotide.
[0299] In certain embodiments, the accessory element comprises an IRES transacting factor (ITAF) region. In some embodiments, the IRES transacting factor region modulates the initiation of translation through binding to PCBP1 - PCBP4 (polyC binding protein), PABP1 (poly A binding protein). PTB (polyprimidine tract binding), Argonaute protein family, HNRNPK (Heterogeneous nuclear ribonucleoprotein K protein), or La protein. In some embodiments, the IRES transacting factor region comprises a poly A, polyC, poly AC, or polyprimidine track.
[0300] In some embodiments, the ITAF region is located within the core functional element. In some embodiments, the ITAF region is located within the TIE.
[0301] In certain embodiments, the accessory’ element comprises a miRNA binding site. In some embodiments the miRNA binding site is located within the 5’ intron clement, 5’ exon clement, core functional element, 3’ exon element, and/or 3’ intron element.
[0302] In some embodiments, wherein the miRNA binding site is located within the spacer within the intron element or exon element. In certain embodiments, the miRNA binding site comprises the entire spacer regions.
[0303] In some embodiments, the 5’ intron element and 3’ intron elements each comprise identical miRNA binding sites. In another embodiment, the miRNA binding site of the 5’ intron element comprises a different, in length or nucleotides. miRNA binding site than the 3’ intron element. In one embodiment, the 5’ exon element and 3’ exon element comprise identical miRNA binding sites. In other embodiments, the 5’ exon element and 3’ exon element comprises different, in length or nucleotides, miRNA binding sites.
[0304] In some embodiments, the miRNA binding sites are located adjacent to each other within the circular RNA polynucleotide, linear RNA polynucleotide precursor, and/or DNA template. In certain embodiments, the first nucleotide of one of the miRNA binding sites follows the first nucleotide last nucleotide of the second miRNA binding site.
[0305] In some embodiments, the miRNA binding site is located within a translation initiation element (TIE) of a core functional element. In one embodiment, the miRNA binding site is located before, trailing or within an internal ribosome entry site (IRES). In another embodiment, the miRNA binding site is located before, trailing, or within an aptamer complex.
[0306] The unique sequences defined by the miRNA nomenclature are widely known and accessible to those working in the micrcircRNA field. For example, they can be found in the miRDB public database. vi. Natural Ties: Viral & Eukaryotic/ Cellular Internal Ribosome Entry Sites (IRES)
[0307] A multitude of IRES sequences are available and include sequences derived from a wide variety of viruses, such as from leader sequences of piccircRNAviruses such as the encephalomyocarditis virus (EMCV) UTR (Jang et al., J. Virol. (1989) 63: 1651-1660), the polio leader sequence, the hepatitis A virus leader, the hepatitis C virus IRES, human rhinovirus type 2 IRES (Dobrikova et al., Proc. Natl. Acad. Sci. (2003) 100(25): 15125- 15130). an IRES element from the foot and mouth disease virus (Ramesh et al., Nucl. Acid Res. (1996) 24:2697-2700), a giardiavirus IRES (Garlapati et al., J. Biol. Chem. (2004) 279(5):3389-3397).
[0308] For driving protein expression, tire circular RNA comprises an IRES operably linked to a protein coding sequence. Modifications of IRES and accessory sequences are disclosed herein to increase or reduce IRES activities, for example, by truncating the 5’ and/or 3’ ends of the IRES, adding a spacer 5‘ to the IRES, modifying the 6 nucleotides 5’ to the translation initiation site (Kozak sequence), modification of alternative translation initiation sites, and creating chimeric/hybrid IRES sequences. In some embodiments, the IRES sequence in the circular RNA disclosed herein comprises one or more of these modifications relative to a native IRES.
[0309] In some embodiments, the IRES is an IRES sequence of Taura syndrome virus, Triatoma virus, Theiler's encephalomyelitis virus, Simian Virus 40, Solenopsis invicta virus 1, Rhopalosiphum padi virus, Reticuloendotheliosis virus, Human poliovirus 1, Plautia stali intestine virus, Kashmir bee virus, Human rhinovirus 2, Homalodisca coagulata virus- 1, Human Immunodeficiency Virus type 1, Himetobi P virus, Hepatitis C virus, Hepatitis A virus, Hepatitis GB virus , Foot and mouth disease virus, Human enterovirus 71, Equine rhinitis virus, Ectropis obliqua piccircRNA-like virus, Encephalomyocarditis virus. Drosophila C Virus, Human coxsackievirus B3, Crucifer tobamovirus, Cricket paralysis virus, Bovine viral diarrhea virus 1, Black Queen Cell Virus. Aphid lethal paralysis virus. Avian encephalomyelitis virus, Acute bee paralysis virus. Hibiscus chlorotic ringspot virus. Classical swine fever virus. Human FGF2, Human SFTPA1, Human AML1/RUNX1. Drosophila antennapedia. Human AQP4, Human AT1R, Human BAG-1, Human BCL2. Human BiP, Human c- IAP1, Human c-myc. Human eIF4G. Mouse NDST4L. Human LEF1, Mouse HIF1 alpha, Human n.myc. Mouse Gtx. Human p27kipl. Human PDGF2/c-sis, Human p53. Human Pim-1, Mouse Rbm3, Drosophila reaper, Canine Scamper, Drosophila Ubx, Human UNR, Mouse UtrA, Human VEGF-A, Human XIAP, Drosophila hairless, S. cerevisiae TFIID, S. cerevisiae YAP1, tobacco etch virus, turnip crinkle virus, EMCV-A, EMCV-B, EMCV-Bf, EMCV-Cf, EMCV pEC9, Picobirnavirus, HCV QC64, Human Cosavirus E/D, Human Cosavirus F, Human Cosavirus JMY, Rhinovirus NAT001, HRV14, HRV89. HRVC-02, HRV-A21, Salivirus A SHI. Salivirus FHB, Salivirus NG-J1, Human Parechovirus 1, Crohivirus B, Yc-3, Rosavirus M-7. Shanbavirus A, Pasivirus A, Pasivirus A 2, Echovirus E14, Human Parechovirus 5, Aichi Virus, Hepatitis A Virus HA16, Phopivirus, CVA10, Enterovirus C, Enterovirus D, Enterovirus J, Human Pegivirus 2, GBV-C GT110, GBV-C K1737, GBV-C Iowa, Pegivirus A 1220, Pasivirus A 3, Sapelovirus, Rosavirus B, Bakunsa Virus, Tremovirus A, Swine Pasivirus 1, PLV-CHN, Pasivirus A, Sicinivirus, Hepacivirus K, Hepacivirus A, BVDV1, Border Disease Virus, BVDV2, CSFV-PK15C, SF573 Dicistrovirus, Hubei PiccircRNA-like Virus, CRPV, Salivirus A BN5, Salivirus A BN2, Salivirus A 02394, Salivirus A GUT, Salivirus A CH, Salivirus A SZ1, Salivirus FHB, CVB3, CVB1, Echovirus 7, CVB5, EVA71. CVA3, CVA12, EV24 or an aptamer to eIF4G.
[0310] In some embodiments, the IRES comprises in whole or in part a eukary otic or cellular IRES. In certain embodiments, the IRES is from a human gene, wherein the human gene is ABCF1, ABCG1,
ACAD10, ACOT7, ACSS3, ACTG2, ADCYAP1, ADK, AGTR1, AHCYL2, AHI1, AKAP8L, AKR1A1, ALDH3A1, ALDOA, ALG13, AMMECR1L, ANGPTL4, ANK3, AOC3, AP4B1, AP4E1, APAF1. APBB1, APC, APH1A, APOBEC3D, APOM, APP, AQP4, ARHGAP36, ARL13B, ARMC8, ARMCX6, ARPC1A, ARPC2 , ARRDC3, ASAP1, ASB3, ASB5, ASCL1, ASMTL, ATF2, ATF3, ATG4A, ATP5B. ATP6V0A1, ATXN3, AURKA, AURKA, AURKA, AURKA, B3GALNT1, B3GNTL1, B4GALT3, BAAT. BAG1, BAIAP2, BAIAP2L2, BAZ2A, BBX, BCAR1, BCL2, BCS1L, BET1, BID, BIRC2 , BPGM, BPIFA2, BRINP2, BSG, BTN3A2, C12orf43. C14orf93, C17orf62, Clorf226. C2 1orf62, C2 orfl5, C4 BPB. C4 orf22, C9orf84, CACNA1A, CALCOCO2, CAPN11, CASP12, CASP8AP2. CAV1, CBX5, CCDC120, CCDC17, CCDC186, CCDC51, CCN1, CCND1, CCNT1, CD2BP2, CD9, CDC25C, CDC42, CDC7, CDCA7L, CDIP1, CDK1, CDK11A, CDKN1B, CEACAM7, CEP295NL, CFLAR, CHCHD7, CHIA, CHICI, CHMP2A. CHRNA2. CLCN3, CLEC12A, CLEC7A. CLECL1, CLRN1. CMSS1, CNIH1. CNR1. CNTN5, COG4, COMMD1, COMMD5. CPEB1. CPS1, CRACR2B, CRBN, CREM. CRYBG1, CSDE1, CSF2RA. CSNK2A1. CSTF3, CTCFL, CTH, CTNNA3, CTNNB1, CTNNBL CTNND1, CTSL, CUTA, CXCR5, CYB5R3 , CYP24A1. CYP3A5. DAG1. DAP3, DAP5. DAXX. DCAF4, DCAF7, DCLRE1A, DCP1A. DCTN1, DCTN2. DDX19B. DDX46, DEFB123, DGKA, DGKD, DHRS4. DHX15, DIO3, DLG1. DLL4, DMD UTR, DMD ex5. DMKN, DNAH6. DNAL4, DUSP13, DUSP19, DYNC1I2, DYNLRB2. DYRK1A, ECI2, ECT2, EIF1 AD, EIF2B4. EIF4G1, EIF4G2, EIF4G3, ELANE, ELOVL6, ELP5. EMCN. ENO1, EPB41, ERMN. ERVV-1, ESRRG, ETFB, ETFBKMT, ETV1, ETV4, EXD1, EXT1, EZH2, FAM111B, FAM157A, FAM213A, FBXO25, FBXO9. FBXW7, FCMR, FGF1, FGF1. FGF1A, FGF2, FGF2. FGF-9, FHL5, FMRI, FN1, FOXP1, FTH1. FUBP1, G3BP1, GABBR1, GALC, GART. GAS7, gastrin, GATA1, GATA4, GFM2, GHR, GJB2, GLI1, GLRA2, GMNN, GPAT3, GPATCH3, GPR137, GPR34, GPR55, GPR89A, GPRASP1, GRAP2, GSDMB, GSTO2. GTF2B, GTF2H4, GUCY1B2, HAX1, HCST, HIGD1A, HIGD1B, HIPK1, HIST1H1C, HIST1H3H, HK1, HLA-DRB4, HMBS, HMGA1, HNRNPC, HOPX, HOXA2, HOXA3, HPCAL1, HR, HSP90AB1, HSPA1A, HSPA4L, HSPA5, HYPK, IFFO1, IFT74, IFT81, IGF1, IGF1R, IGF1R, IGF2, IL11, IL17RE, IL1RL1, IL1RN, IL32, IL6, ILF2, ILVBL, INSR, INTS13, IP6K1, ITGA4, ITGAE, KCNE4, KERA, KIAA0355, KIAA0895L, KIAA1324, KIAA1522, KIAA1683, KIF2C, KIZ, KLHL31, KLK7, KRR1, KRT14, KRT17. KRT33A, KRT6A, KRTAP10-2, KRTAP13-3, KRTAP13-4, KRTAP5-11, KRTCAP2, LACRT, LAMB1, LAMB3, LANCL1, LBX2. LCAT, LDHA, LDHAL6A, LEF1, LINC-PINT, LMO3, LRRC4 C, LRRC7, LRTOMT, LSM5, LTB4R, LYRM1. LYRM2, MAGEA11, MAGEA8, MAGEB1, MAGEB16. MAGEB3, MAPT. MARS, MC1R, MCCC1, METTL12, METTL7A. MGC 16025, MGC16025, MIA2, MIA2. MITF, MKLN1. MNT, MORF4L2. MPD6, MRFAP1, MRPL21, MRPS12, MSI2, MSLN, MSN, MT2A, MTFR1L, MTMR2 , MTRR, MTUS1, MYB. MYC, MYCL, MYCN, MYL10, MYL3. MYLK, MYO1 A. MYT2, MZB1, NAP1L1, NAVI. NBAS, NCF2. NDRG1, NDST2, NDUFA7, NDUFB11. NDUFC1, NDUFS1, NEDD4L. NFAT5, NFE2L2, NFE2L2. NFIA, NHEJ1.
NHP2, NITI, NKRF, NME1-NME2, NPAT, NR3C1, NRBF2, NRF1, NTRK2, NUDCD1, NXF2, NXT2, ODC1, ODF2, OPTN, OR10R2 , OR11L1, OR2M2, OR2M3, OR2M5, OR2T10, OR4C15, OR4F17, OR4F5, OR5H1, OR5K1, OR6C3, OR6C75, 0R6N1, OR7G2, p53, P2RY4, PAN2, PAQR6, PARP4, PARP9, PC, PCBP4, PCDHGC3, PCLAF, PDGFB. PDZRN4, PELO, PEMT, PEX2, PFKM, PGBD4, PGLYRP3, PHLDA2, PHTF1, PI4KB, PIGC. PIM1, PKD2L1, PKM, PLCB4, PLD3, PLEKHA1, PLEKHB1, PLS3. PML, PNMA5, PNN. POC1A, POC1B, POLD2, POLD4, POU5F1, PPIG. PQBP1, PRAME, PRPF4, PRR11, PRRT1, PRSS8, PSMA2, PSMA3, PSMA4, PSMD11, PSMD4, PSMD6. PSME3, PSMG3, PTBP3, PTCHI, PTHLH, PTPRD, PUS7L, PVRIG, QPRT, RAB27A. RAB7B, RABGGTB. RAET1E, RALGDS, RALYL, RARB, RCVRN, REG3G, RFC5, RGL4, RGS19, RGS3, RHD, RINL, RIPOR2 , RITA1, RMDN2, RNASE1, RNASE4, RNF4, RPA2, RPL17. RPL21. RPL26L1, RPL28, RPL29, RPL41, RPL9. RPS11. RPS13. RPS14. RRBP1, RSU1, RTP2, RUNX1, RUNX1T1, RUNX1T1, RUNX2, RUSC1, RXRG, S100A13. S100A4. SAT1, SCHIP1, SCMH1, SEC14L1, SEMA4A, SERPINA1, SERPINB4, SERTAD3, SFTPD. SH3D19. SHC1, SHMT1, SHPRH, SIM1, SIRT5, SLC11A2. SLC12A4, SLC16A1, SLC25A3. SLC26A9, SLC5A11. SLC6A12, SLC6A19. SLC7A1, SLFN11, SLIRP. SMAD5. SMARCAD1. SMN1, SNCA, SNRNP200, SNRPB2. SNX12, SOD1. SOX13, SOX5. SP8, SPARCL1, SPATA12, SPATA31C2 , SPN, SPOP. SQSTM1, SRBD1. SRC. SREBF1. SRPK2, SSB, SSB. SSBP1, ST3GAL6. STAB1, STAMBP. STAU1, STAU1. STAU1, STAU1. STAU1, STK16, STK24. STK38, STMN1, STX7, SULT2B1, SYK, SYNPR, TAF1C, TAGLN, TANK. TAS2R40, TBC1D15. TBXAS1. TCF4, TDGF1, TDP2, TDRD3, TDRD5, TESK2, THAP6, THBD, THTPA, TIAM2, TKFC, TKTL1, TLR10, TM9SF2, TMC6, TMCO2, TMED10, TMEM116. TMEM126A, TMEM159, TMEM208, TMEM230, TMEM67. TMPRSS13, TMUB2, TNFSF4, TNIP3. TP53, TP53, TP73, TRAF1, TRAK1, TRIM31, TRIM6, TRMT1, TRMT2B, TRPM7, TRPM8, TSPEAR. TTC39B, TTLL11, TUBB6, TXLNB, TXNIP, TXNL1, TXNRD1, TYROBP, U2AF1, UBA1, UBE2D3, UBE2I, UBE2L3, UBE2V1, UBE2V2, UMPS, UNG, UPP2, USMG5, USP18, UTP14A, UTRN, UTS2, VDR, VEGFA, VEGFA, VEPH1, VIPAS39, VPS29, VSIG10L, WDHD1, WDR12, WDR4, WDR45, WDYHV1, WRAP53, XIAP, XPNPEP3, YAP1, YWHAZ, YY1AP1, ZBTB32, ZNF146, ZNF250, ZNF385A, ZNF408, ZNF410, ZNF423. ZNF43. ZNF502, ZNF512, ZNF513, ZNF580, ZNF609, ZNF707, or ZNRD1. vii. Synthetic Ties: Aptamer Complexes, Modified Nucleotides, IRES Variants & Other Engineered Ties
[0311] As contemplated herein, in some embodiments, a translation initiation element (TIE) comprises a synthetic TIE. In some embodiments, a synthetic TIE comprises aptamer complexes, synthetic IRES or other engineered TIES capable of initiating translation of a linear RNA or circular RNA polynucleotide.
[0312] In some embodiments, one or more aptamer sequences is capable of binding to a component of a eukaryotic initiation factor to either enhance or initiate translation. In some embodiments, an aptamer may be used to enhance translation in vivo and in vitro by promoting specific eukary otic initiation factors (elF) (e.g., aptamer in WO2019081383A1 is capable of binding to eukary otic initiation factor 4F (eIF4F). In some embodiments, the aptamer or a complex of aptamers may be capable of binding to EIF4G, EIF4E, EIF4A, EIF4B, EIF3, EIF2, EIF5, EIF1, EIF1A, 40S ribosome, PCBP1 (polyC binding protein), PCBP2, PCBP3, PCBP4, PABP1 (polyA binding protein). PTB, Argonaute protein family, HNRNPK (heterogeneous nuclear ribonucleoprotein K). or La protein. viii. Termination Sequence
[0313] In some embodiments, the core functional element comprises a termination sequence. In some embodiments, the termmation sequence comprises a stop codon. In one embodiment, the tennination sequence comprises a stop cassette. In some embodiments, the stop cassette comprises at least 2 stop codons. In some embodiments, the stop cassette comprises at least 2 frames of stop codons. In the same embodiment, the frames of the stop codons in a stop cassette each comprise 1, 2, or more stop codons. In some embodiments, the stop cassette comprises a LoxP or a RoxStopRox, or frt-flanked stop cassette. In the same embodiment, the stop cassette comprises a lox-stop-lox stop cassette. ix. Variants
[0314] In some embodiments, a circular RNA polynucleotide provided herein comprises modified RNA nucleotides and/or modified nucleosides. In some embodiments, the modified nucleoside is m5C (5-methylcytidine). In one embodiment, the modified nucleoside is m5U (5 -methyluridine). In another embodiment, the modified nucleoside is m6A (N6-methyladenosine). In another embodiment, the modified nucleoside is s2U (2-thiouridine). In another embodiment, the modified nucleoside is
 (pseudouridine). In another embodiment, the modified nucleoside is Um (2'-O-methyluridine). In other embodiments, the modified nucleoside is m' A (1 -methyladenosine); m2A (2 -methyladenosine); Am (2’-O-methyladenosine); ms2m6A (2-methylthio-N6-methyladenosine); i6A (N6-isopentenyladenosine); ms2i6A (2-methylthio-N6 isopentenyladenosine); io6A (N6-(cis-hydroxyisopentenyl)adenosine); ms2io6A (2-methylthio-N6-(cis-hydroxyisopentenyl)adenosine); g6A (N6- glycinylcarbamoy ladenosine); t6A (N6-threonylcarbamoy ladenosine); ms2t6A (2-methylthio-N6- direonyl carbamoyladenosine); m6t6A (N6-methyl-N6-threonylcarbamoyladenosine); hn6A(N6- hydroxy norvalylcarbamoyladenosine); ms2hn6A (2-methylthio-N6-hydroxynorvalyl carbamoyladenosine); Ar(p) (2’ -O-ribosy ladenosine (phosphate)); I (inosine); m1! (1-methylinosine); in' lm (1.2'-O-dimethylinosine): m3C (3-methylcytidine); Cm (2’-O-methylcytidine); s2C (2- thiocytidine); ac4C (N4 -acetylcytidine); FC (5 -formylcytidine); m5Cm (5,2'-O-dimethylcytidine); ac4Cm (N4-acetyl-2’-O-methylcytidine); k2C (lysidine); m'G (1-methylguanosine); m2G (N2- methylguanosine); m7G (7-methylguanosine); Gm (2'-O-methylguanosine); nr 2G (N2,N2-
dimethylguanosine); m2Gm (N2,2’-O-dimethylguanosine); m2 2Gm (N2,N2,2’-O-trimethylguanosine); Gr(p) (2’-O-ribosylguanosine(phosphate)); yW (wybutosine); o2yW (peroxywybutosine); OHyW (hydroxy wybutosine): OHyW* (undermodified hydroxy wybutosine): imG (wyosine); inimG (methylwyosine); Q (queuosine); oQ (epoxy queuosine); galQ (galactosyl-queuosine); manQ (mannosyl-queuosine); preQo (7-cyano-7-deazaguanosine); preQi (7-aminomethyl-7-deazaguanosine); G+ (archaeosine); D (dihydrouridine); m5Um (5,2’-O-dimethyluridine): s4U (4-thiouridine); m5s2U (5- methyl-2-thiouridine); s2Um (2-thio-2’-O-methyluridine); acp3U (3-(3-amino-3- carboxypropyl)uridine); ho5U (5-hydroxyuridine); mo5U (5-methoxyuridine); cmo5U (uridine 5- oxyacetic acid); mcmo’U (uridine 5-oxyacetic acid methyl ester); chm5U (5- (carboxyhydroxymethyl)uridine)); mchm’U (5-(carboxyhydroxymethyl)uridine methyl ester); mcm5U (5-methoxy carbonylmethyluridine); mcm5Um (5-methoxycarbonylmethyl-2’-O-methyluridine); mcmVU (5-methoxycarbonylmethyl-2-thiouridine); nm5S2U (5-aminomethyl-2-thiouridine); mnm 'U (5 -methylaminomethyluridine); mnm5s2U (5-methylaminomethyl-2-thiouridine); mnm5se2U (5- methylaminomethyl-2-selenouridine); ncnfU (5-carbamoylmethyluridine); ncnfUm (5- carbamoylmethyl-2'-O-methyluridine); cmnm'U (5-carboxymethylaminomethyluridine); cmnm5Um (5-carboxymethylaminomethyl-2'-O-methyluridine); cmnm5s2U (5-carboxymethylaminomethyl-2- thiouridine); m6 2A (N6,N6-dimethyladenosine); Im (2’-O-methylinosine); m4C (N4-methylcytidine); m4Cm (N4,2’-O-dimethylcytidine); hnfC (5-hydroxymethylcytidine); m3U (3 -methyluridine); cm5U (5 -carboxy methyluridine); m6Am (N6.2’-O-dimethyladenosine); m6 2Am (N6,N6,O-2’- trimethyladenosine); m27G (N2,7-dimethylguanosine); m2,27G (N2,N2,7-trimethylguanosine); m3Um (3,2’-O-dimethyluridine); m5D (5-methyldihydrouridine); fCm (5-formyl-2’-O-methylcytidine); m'Gni (l,2’-O-dimethylguanosine); m’Am (l,2’-O-dimethyladenosine); Tin 3U (5- taurinomethyluridine); rm5s2U (5-taurinomethyl-2-thiouridine)); imG-14 (4-demethyhvyosine); imG2 (isowyosinc): or ac6A (N6-acetyladenosine).
(pseudouridine). In another embodiment, the modified nucleoside is Um (2'-O-methyluridine). In other embodiments, the modified nucleoside is m' A (1 -methyladenosine); m2A (2 -methyladenosine); Am (2’-O-methyladenosine); ms2m6A (2-methylthio-N6-methyladenosine); i6A (N6-isopentenyladenosine); ms2i6A (2-methylthio-N6 isopentenyladenosine); io6A (N6-(cis-hydroxyisopentenyl)adenosine); ms2io6A (2-methylthio-N6-(cis-hydroxyisopentenyl)adenosine); g6A (N6- glycinylcarbamoy ladenosine); t6A (N6-threonylcarbamoy ladenosine); ms2t6A (2-methylthio-N6- direonyl carbamoyladenosine); m6t6A (N6-methyl-N6-threonylcarbamoyladenosine); hn6A(N6- hydroxy norvalylcarbamoyladenosine); ms2hn6A (2-methylthio-N6-hydroxynorvalyl carbamoyladenosine); Ar(p) (2’ -O-ribosy ladenosine (phosphate)); I (inosine); m1! (1-methylinosine); in' lm (1.2'-O-dimethylinosine): m3C (3-methylcytidine); Cm (2’-O-methylcytidine); s2C (2- thiocytidine); ac4C (N4 -acetylcytidine); FC (5 -formylcytidine); m5Cm (5,2'-O-dimethylcytidine); ac4Cm (N4-acetyl-2’-O-methylcytidine); k2C (lysidine); m'G (1-methylguanosine); m2G (N2- methylguanosine); m7G (7-methylguanosine); Gm (2'-O-methylguanosine); nr 2G (N2,N2-
dimethylguanosine); m2Gm (N2,2’-O-dimethylguanosine); m2 2Gm (N2,N2,2’-O-trimethylguanosine); Gr(p) (2’-O-ribosylguanosine(phosphate)); yW (wybutosine); o2yW (peroxywybutosine); OHyW (hydroxy wybutosine): OHyW* (undermodified hydroxy wybutosine): imG (wyosine); inimG (methylwyosine); Q (queuosine); oQ (epoxy queuosine); galQ (galactosyl-queuosine); manQ (mannosyl-queuosine); preQo (7-cyano-7-deazaguanosine); preQi (7-aminomethyl-7-deazaguanosine); G+ (archaeosine); D (dihydrouridine); m5Um (5,2’-O-dimethyluridine): s4U (4-thiouridine); m5s2U (5- methyl-2-thiouridine); s2Um (2-thio-2’-O-methyluridine); acp3U (3-(3-amino-3- carboxypropyl)uridine); ho5U (5-hydroxyuridine); mo5U (5-methoxyuridine); cmo5U (uridine 5- oxyacetic acid); mcmo’U (uridine 5-oxyacetic acid methyl ester); chm5U (5- (carboxyhydroxymethyl)uridine)); mchm’U (5-(carboxyhydroxymethyl)uridine methyl ester); mcm5U (5-methoxy carbonylmethyluridine); mcm5Um (5-methoxycarbonylmethyl-2’-O-methyluridine); mcmVU (5-methoxycarbonylmethyl-2-thiouridine); nm5S2U (5-aminomethyl-2-thiouridine); mnm 'U (5 -methylaminomethyluridine); mnm5s2U (5-methylaminomethyl-2-thiouridine); mnm5se2U (5- methylaminomethyl-2-selenouridine); ncnfU (5-carbamoylmethyluridine); ncnfUm (5- carbamoylmethyl-2'-O-methyluridine); cmnm'U (5-carboxymethylaminomethyluridine); cmnm5Um (5-carboxymethylaminomethyl-2'-O-methyluridine); cmnm5s2U (5-carboxymethylaminomethyl-2- thiouridine); m6 2A (N6,N6-dimethyladenosine); Im (2’-O-methylinosine); m4C (N4-methylcytidine); m4Cm (N4,2’-O-dimethylcytidine); hnfC (5-hydroxymethylcytidine); m3U (3 -methyluridine); cm5U (5 -carboxy methyluridine); m6Am (N6.2’-O-dimethyladenosine); m6 2Am (N6,N6,O-2’- trimethyladenosine); m27G (N2,7-dimethylguanosine); m2,27G (N2,N2,7-trimethylguanosine); m3Um (3,2’-O-dimethyluridine); m5D (5-methyldihydrouridine); fCm (5-formyl-2’-O-methylcytidine); m'Gni (l,2’-O-dimethylguanosine); m’Am (l,2’-O-dimethyladenosine); Tin 3U (5- taurinomethyluridine); rm5s2U (5-taurinomethyl-2-thiouridine)); imG-14 (4-demethyhvyosine); imG2 (isowyosinc): or ac6A (N6-acetyladenosine).
[0315] In some embodiments, the modified nucleoside may include a compound selected from: pyridin-4-one ribonucleoside. 5 -aza-uridine. 2-thio-5 -aza-uridine, 2-thiouridine. 4-thio-pseudouridine, 2-thio-pseudouridine, 5 -hydroxy uridine, 3 -methyluridine, 5-carboxymethyl-uridine, 1 -carboxy methyl- pseudouridine. 5-propynyl-uridine, 1-propynyl-pseudouridine, 5 -taurinom ethyluridine, 1- taurinomethyl-pseudouridine, 5-taurinomethy 1-2 -thio -uridine, l-taurinomethyl-4-thio-uridine, 5- methyl-uridine, 1-methyl-pseudouridine, 4-thio-l-methyl-pseudouridine, 2-thio-l-methyl- pscudouridinc, 1 -methyl- 1-dcaza-pscudouridinc, 2-thio-l-mcthyl-l-dcaza-pscudouridinc, dihydrouridine, dihydropseudouridine, 2-thio-dihydrouridine, 2-thio-dihydropseudouridine, 2- methoxyuridine, 2-methoxy-4-thio-uridine, 4-methoxy-pseudouridine, 4-m ethoxy-2-thio- pseudouridine, 5-aza-cytidine, pseudoisocytidine, 3-methyl-cytidine, N4-acetylcytidine, 5- formylcytidine, N4-methylcytidine, 5-hydroxymethylcytidine, 1-methyl-pseudoisocytidine, pyrrolo- cytidine, pyrrolo-pseudoisocytidine, 2-thio-cytidine, 2-thio-5-methyl-cytidine, 4-thio-
pseudoisocytidine, 4-thio- 1 -inethy 1-pseudoisocy tidine, 4-thio- 1 -methyl- 1 -deaza-pseudoisocytidine, 1 - methyl- 1-deaza-pseudoisocytidine, zebularine, 5-aza-zebularine, 5-methyl-zebularine, 5-aza-2-thio- zebularine, 2-thio-zebularine, 2-methoxy -cytidine, 2 -methoxy -5 -methyl-cy tidine, 4-methoxy- pseudoisocytidine, 4-methoxy-l-methyl-pseudoisocytidine, 2-aminopurine, 2, 6 -diaminopurine. 7- deaza-adenine, 7-deaza-8-aza-adenine, 7-deaza-2-aminopurine, 7-deaza-8-aza-2-aminopurine, 7- deaza-2,6-diaminopurine, 7-deaza-8-aza-2,6-diaminopurine, 1 -methyladenosine, N6-methyladenosine, N6-isopentenyladenosine, N6-(cis-hydroxyisopentenyl)adenosine, 2-methylthio-N6-(cis- hydroxyisopentenyl) adenosine, N6-glycinylcarbamoyladenosine, N6-threonylcarbamoyladenosine, 2- methylthio-N6-threonyl carbamoyladenosine, N6,N6-dimethyladenosine. 7-methyladenine, 2- methylthio-adenine, 2-methoxy-adenine, inosine. 1-methyl-inosine. wyosine, wybutosine. 7-deaza- guanosine, 7-deaza-8-aza-guanosine. 6-thio-guanosine. 6-thio-7-deaza-guanosine. 6-thio-7-deaza-8- aza-guanosine, 7-methyl-guanosine, 6-thio-7-methyl-guanosine, 7-methylinosine. 6-methoxy- guanosine, 1 -methylguanosine. N2-methylguanosine, N2,N2-dimethylguanosine. 8-oxo-guanosine, 7- methyl-8-oxo-guanosine, 1 -methyl-6-thio-guanosine, N2-methyl-6-thio-guanosine, and N2,N2- dimethyl-6-thio-guanosine. In another embodiment, the modifications are independently selected from 5-methylcytosine, pseudouridine and 1 -methylpseudouridine.
[0316] In some embodiments, the modified ribonucleosides include 5-methylcytidine, 5- methoxyuridine. 1-methyl-pseudouridine, N6-methyladenosine, and/or pscudoundinc. In some embodiments, such modified nucleosides provide additional stability and resistance to immune activation.
[0317] In particular embodiments, polynucleotides may be codon-optimized. A codon optimized sequence may be one in which codons in a polynucleotide encoding a polypeptide have been substituted in order to increase the expression, stability and/or activity of the polypeptide. Factors that influence codon optimization include, but are not limited to one or more of: (i) variation of codon biases betw een two or more organisms or genes or synthetically constructed bias tables, (ii) variation in the degree of codon bias within an organism, gene, or set of genes, (iii) systematic variation of codons including context, (iv) variation of codons according to their decoding tRNAs, (v) variation of codons according to GC %, either overall or in one position of the triplet, (vi) variation in degree of similarity to a reference sequence for example a naturally occurring sequence, (vii) variation in the codon frequency cutoff, (viii) structural properties of mRNAs transcribed from the DNA sequence, (ix) prior knowledge the function of the DNA sequences upon which design of the codon substitution set is to be based, and/or (x) systematic variation of codon sets for each amino acid. In some embodiments, a codon optimized polynucleotide may minimize ribozyme collisions and/or limit structural interference between the expression sequence and the core functional element.
x. Payloads
[0318] In some embodiments, the polynucleotide (e.g., circRNA) expression sequence encodes a therapeutic protein. In some embodiments, the therapeutic protein is selected from the proteins listed in the following Table 3.



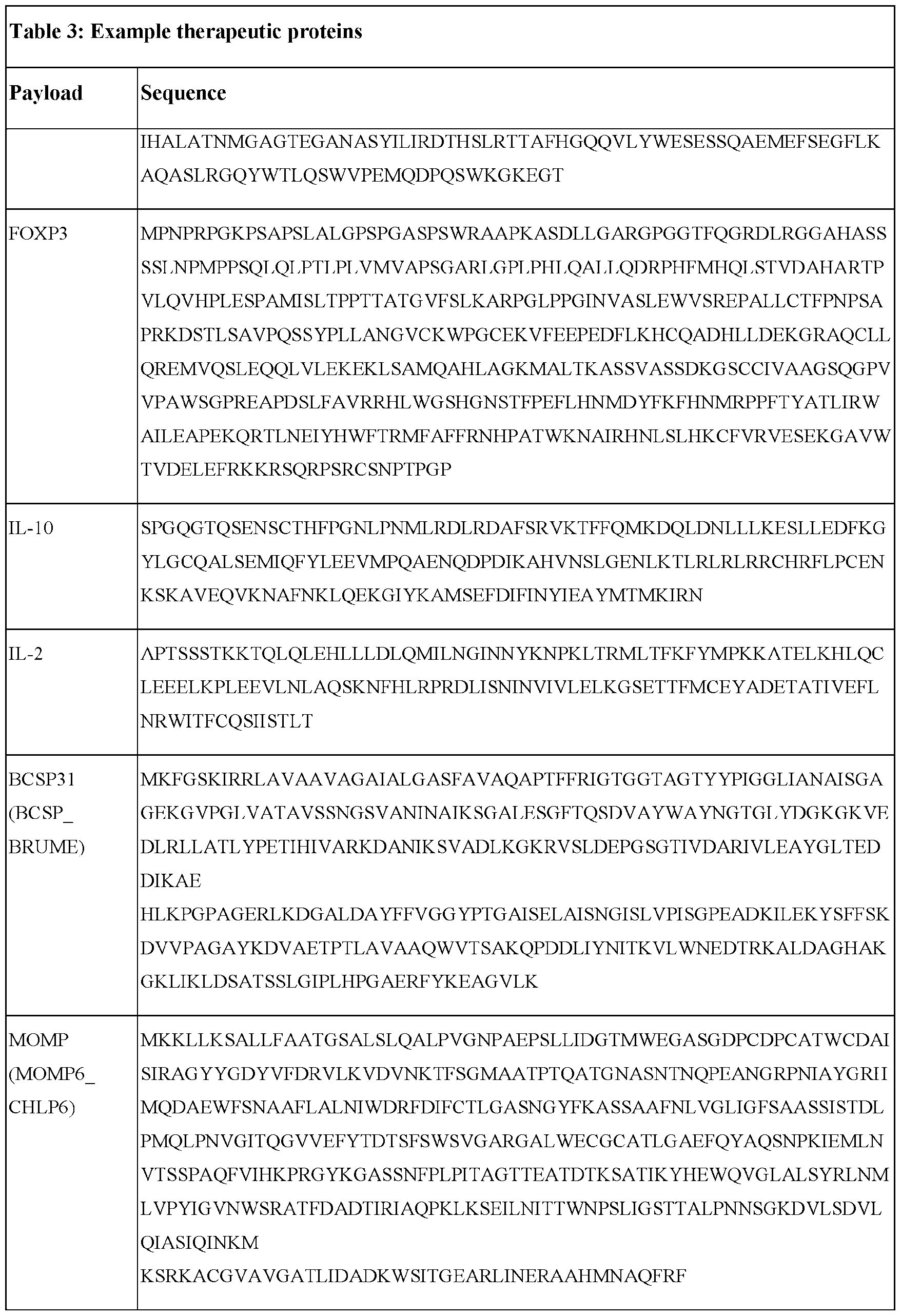
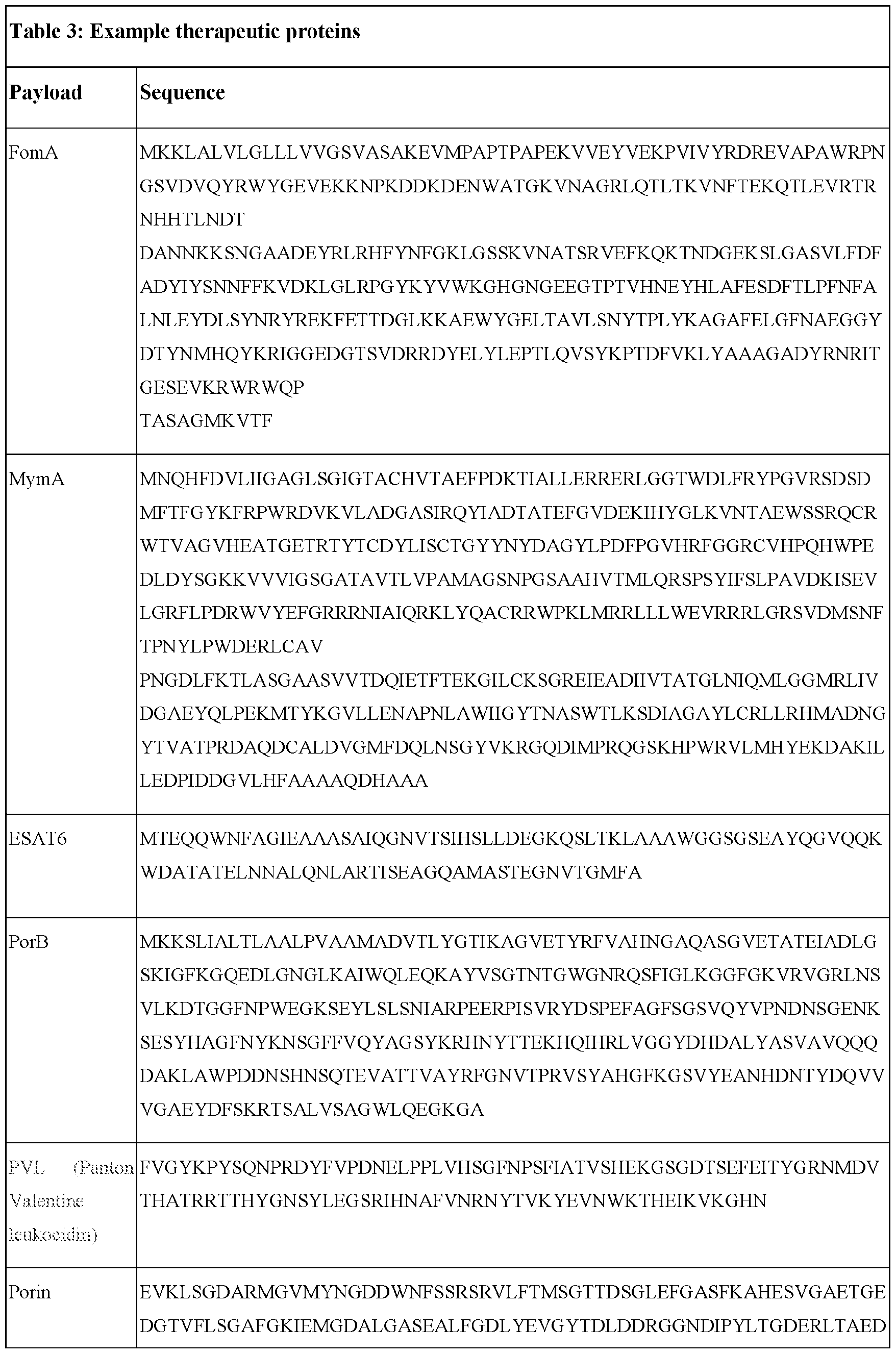
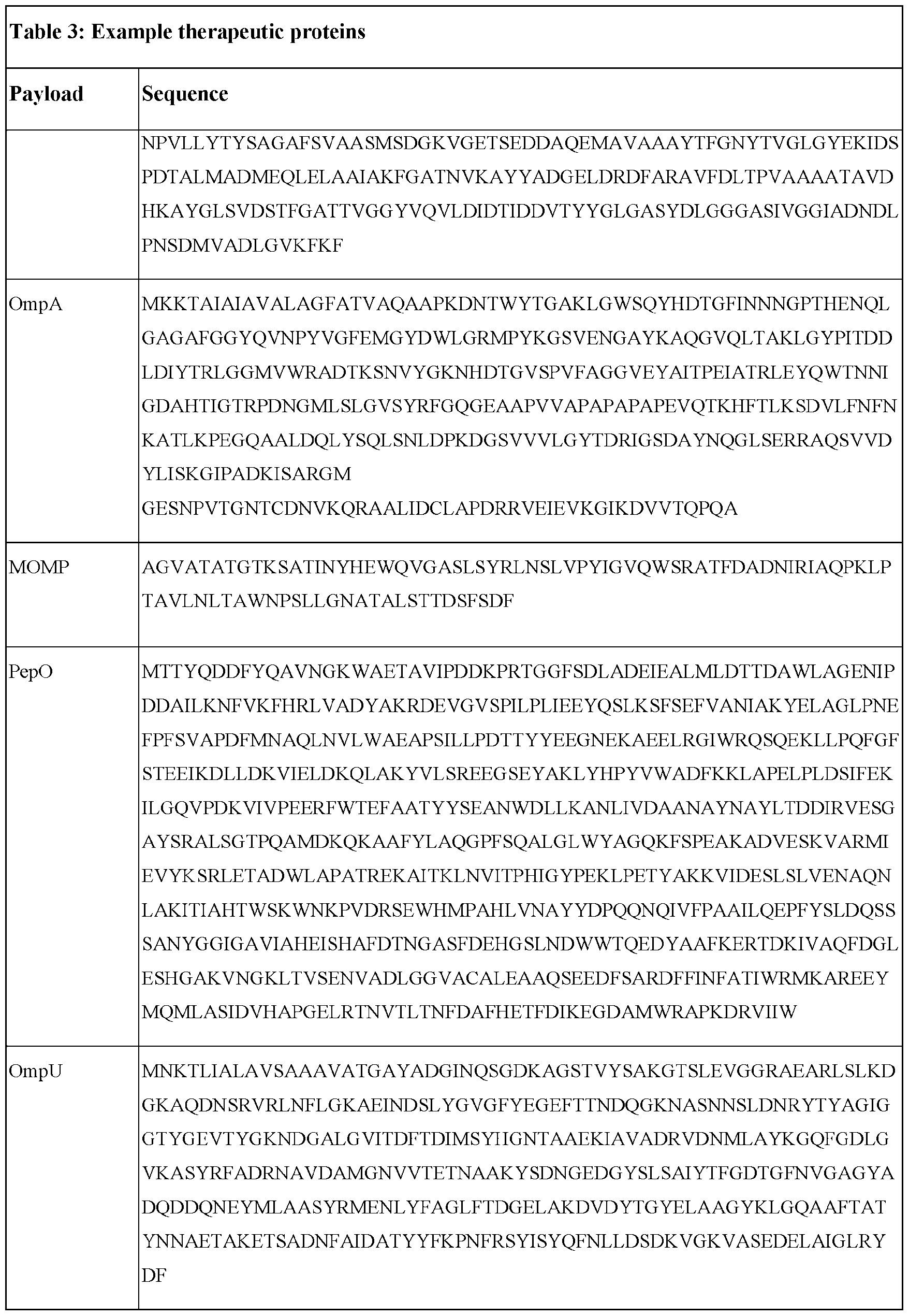
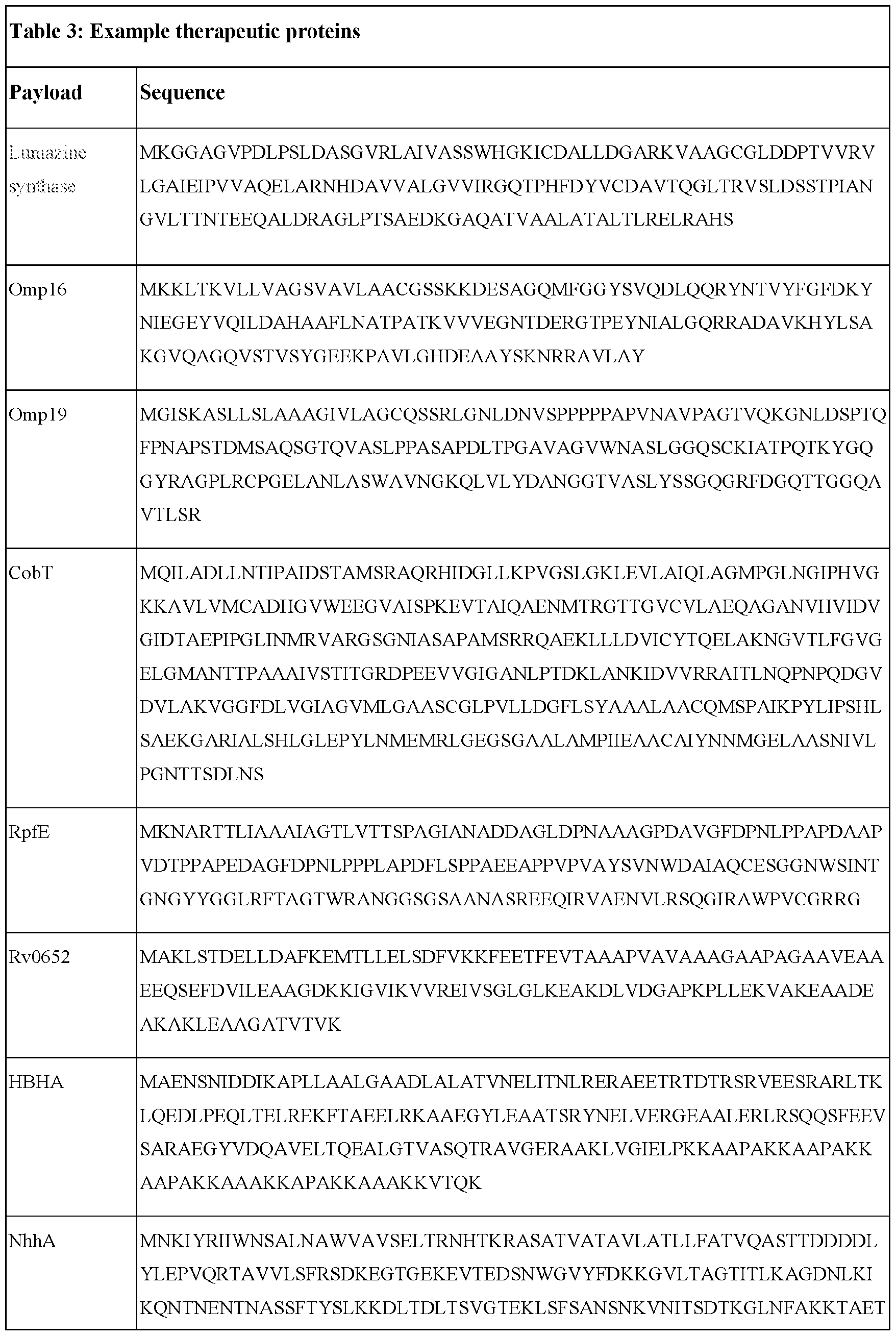
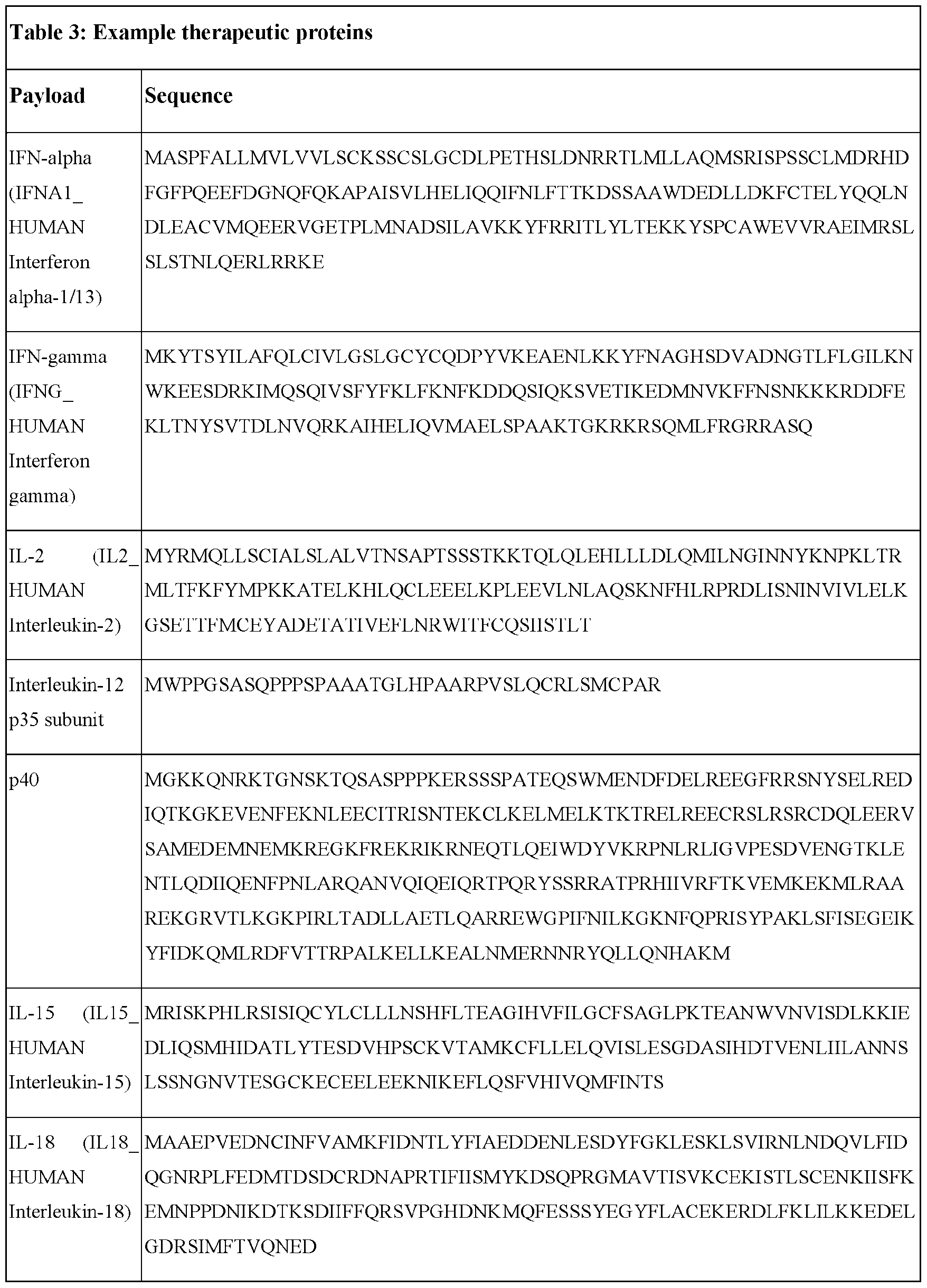

[0319] In some embodiments, the expression sequence encodes a therapeutic protein. In some embodiments, the expression sequence encodes a cytokine, e.g., IL-12p70, IL-15, IL -2, IL-18, IL-21, IFN-a, IFN- P, IL-10, TGF-beta, IL-4, or IL -35, or a functional fragment thereof. In some embodiments, the expression sequence encodes an immune checkpoint inhibitor. In some embodiments, the expression sequence encodes an agonist (e.g., a TNFR family member such as CD137L, OX40L, ICOSL, LIGHT, or CD70). In some embodiments, the expression sequence encodes a chimeric antigen receptor. In some embodiments, the expression sequence encodes an inhibitory receptor agonist (e.g.,
PDL1, PDL2, Galectin-9, VISTA, B7H4, or MHCII) or inhibitory receptor (e.g., PD1, CTLA4, TIGIT, LAG3, or TIM3). In some embodiments, the expression sequence encodes an inhibitor}' receptor antagonist. In some embodiments, the expression sequence encodes one or more TCR chains (alpha and beta chains or gamma and delta chains). In some embodiments, the expression sequence encodes a secreted T cell or immune cell engager (e.g., a bispecific antibody such as BiTE, targeting, e.g., CD3, CD137, or CD28 and a tumor-expressed protein e.g., CD19, CD20, or BCMA etc.). In some embodiments, the expression sequence encodes a transcription factor (e.g., F0XP3, HELIOS, TOX1, or T0X2). In some embodiments, the expression sequence encodes an immunosuppressive enzyme (e.g., IDO or CD39/CD73). In some embodiments, the expression sequence encodes a GvHD (e.g., anti-HLA- A2 CAR-Tregs).
[0320] In some embodiments, the precursor RNA polynucleotide and circular RNA constructs comprise at least one expression sequence encoding an antigen, adjuvant, or adjuvant-like protein, e.g., from an infectious agent. In these embodiments, the circular RNA construct may be used as a vaccine. In some embodiments, the one or more circular RNA polynucleotide encodes an antigen or adjuvant derived from an infectious agent. In some embodiments the infectious agent from which the antigen or adjuvant is derived or engineered includes, but is not limited to a virus, bacterium, fungus, protozoan, and/or parasite. In some embodiments, the antigen is a viral antigen or viral antigenic polypeptide.
[0321] In an embodiment, the antigen is selected from or derived from the group consisting of rotavirus, foot and mouth disease virus, influenza A virus, influenza B virus, influenza C virus, H1N1, H2N2, H3N2, H5N1, H7N7, H1N2, H9N2, H7N2, H7N3, H10N7, human parainfluenza type 2, herpes simplex virus. Epstein-Barr virus, varicella virus, porcine herpesvirus 1, cytomegalovirus, lyssavirus, Bacillus anthracis, anthrax PA and derivatives, poliovirus, Hepatitis A, Hepatitis B, Hepatitis C, Hepatitis E. distemper virus, Venezuelan equine encephalomyelitis, feline leukemia virus, reovirus, respiratory syncytial virus. Lassa fever virus, polyoma tumor virus, canine parvovirus, papilloma virus, tick borne encephalitis virus, rinderpest virus, human rhinovirus species. Enterovirus species. Mengovirus, paramyxovirus, avian infectious bronchitis virus, human T-cell leukemia-lymphoma virus 1, human immunodeficiency virus- 1, human immunodeficiency virus-2, lymphocytic choriomeningitis virus, parvovirus Bl 9, adenovirus, rubella virus, yellow fever virus, dengue virus, bovine respiratory syncitial virus, corona virus, Bordetella pertussis, Bordetella bronchiseptica, Bordetella parapertussis, Brucella abortis, Brucella melitensis, Brucella suis, Brucella ovis, Brucella species, Escherichia coli, Salmonella species, Salmonella typhi, Streptococci, Vibrio cholera, Vibrio parahaemolyticus. Shigella, Pseudomonas, tuberculosis, avium. Bacille Calmette Guerin, Mycobacterium leprae, Pneumococci, Staphlylococci, Enterobacter species, Rochalimaia henselae, Pasteurella haemolytica, Pasteurella multocida. Chlamydia trachomatis, Chlamydia psittaci, Lymphogranuloma venereum, Treponema pallidum, Haemophilus species, Mycoplasma bovigenitalium, Mycoplasma pulmonis, Mycoplasma species, Borrelia burgdorferi, Legionalla pneumophila, Colstridium botulinum, Corynebacterium
diphtheriae, Yersinia entercolitica, Ricketsia ricketsii, Ricketsia tv phi. Ricketsia prowsaekii, Ehrlichia chaffeensis, Anaplasma phagocytophilum, Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, Schistosomes, trypanosomes, Leishmania species, Filarial nematodes, trichomoniasis, sarcosporidiasis, Taenia saginata, Taenia solium, Leishmania, Toxoplasma gondii, Trichinella spiralis, coccidiosis, Eimeria tenella, Cryptococcus neoformans, Candida albican, Aspergillus fumigatus. coccidioidomycosis. Neisseria gonorrhoeae, malaria circumsporozoite protein, malaria merozoite protein, trypanosome surface antigen protein, pertussis, alphaviruses, adenovirus, diphtheria toxoid, tetanus toxoid, meningococcal outer membrane protein, streptococcal M protein, Influenza hemagglutinin, cancer antigen, tumor antigens, toxins, Clostridium perfringens epsilon toxin, ricin toxin, pseudomonas exotoxin, exotoxins, neurotoxins, cytokines, cytokine receptors, monokines, monokine receptors, plant pollens, animal dander, and dust mites.
[0322] In some embodiments, the antigenic polypeptide is a viral polypeptide from an adenovirus; Herpes simplex, type 1; Herpes simplex, type 2; encephalitis virus, papillomavirus, Varicella-zoster virus; Epstein-barr virus; Human cytomegalovirus: Human herpes virus, type 8; Human papillomavirus; BK virus; JC virus; Smallpox; polio virus; Hepatitis B virus; Human bocavirus; Parvovirus B19: Human astrovirus; Norwalk virus; coxsackievirus; hepatitis A virus; poliovirus; rhinovirus; Severe acute respiratory syndrome virus; Hepatitis C virus; Yellow Fever virus; Dengue virus; West Nile virus; Rubella virus; Hepatitis E virus; Human Immunodeficiency virus (HIV); Influenza virus; Guanarito virus; Junin virus; Lassa virus; Machupo virus; Sabia virus; Crimean-Congo hemorrhagic fever virus; Ebola virus; Marburg virus; Measles virus; Mumps virus; Parainfluenza virus; Respiratory syncytial virus; Human metapneumo virus; Hendra virus; Nipah virus; Rabies virus; Hepatitis D; Rotavirus; Orbivirus; Coltivirus; Banna virus; Human Enterovirus; Hanta virus; West Nile virus; Middle East Respiratory Syndrome Corona Virus; Japanese encephalitis virus; Vesicular exanthemavirus; SARS- CoV-2; Eastern equine encephalitis, or a combination of any two or more of the foregoing.
[0323] In some embodiments, the adjuvant is selected from or derived from the group consisting of BCSP31, MOMP, FomA, MymA, ESAT6, PorB, PVL, Porin, OmpA, PepO, OmpU, Lumazine synthase, 0mpl6, Ompl9, CobT. RpfE. Rv0652, HBHA, NhhA. DnaJ. Pneumolysin. Falgellin, IFN- alpha, IFN-gamma, IL-2. IL-12, IL-15, IL-18. IL-21, GM-CSF, IL-lb, IL-6, TNF-a, IL-7. IL-17, IL- IBeta. anti-CTLA4, anti-PDl, anti-41BB, PD-L1, Tim-3, Lag-3, TIGIT, GITR, and andti-CD3.
[0324] In some embodiments, a polynucleotide encodes a protein that is made up of subunits that are encoded by more than one gene. For example, the protein may be a heterodimer, wherein each chain or subunit of the protein is encoded by a separate gene. It is possible that more than one circRNA molecule is delivered in the transfer vehicle and each circRNA encodes a separate subunit of the protein. Alternatively, a single circRNA may be engineered to encode more than one subunit. In certain
embodiments, separate circRNA molecules encoding the individual submits may be administered in separate transfer vehicles.
[0325] Additional polynucleotides, including but not limited to intron elements, exon elements, translation initiation elements, expression sequences, and lipids are in WO2019236673; WO2020237227; WO2021113777; WO2021226597; WO2021189059; WO2021236855;
WO2022261490; W02023056033; WO2023081526; the contents of which are hereby incorporated by reference in their entireties.
(1) Chimeric Antigen Receptors (CARs)
[0326] Chimeric antigen receptors (CARs or CAR-Ts) are genetically engineered receptors. These engineered receptors may be inserted into and expressed by immune cells, including T cells via circular RNA as described herein. With a CAR, a single receptor may be programmed to both recognize a specific antigen and, when bound to that antigen, activate the immune cell to attack and destroy the cell bearing that antigen. When these antigens exist on tumor cells, an immune cell that expresses the CAR may target and kill the tumor cell. In some embodiments, the CAR encoded by the polynucleotide comprises (i) an antigen-binding molecule that specifically binds to a target antigen, (ii) a hinge domain, a transmembrane domain, and an intracellular domain, and (iii) an activating domain.
[0327] In some embodiments, an orientation of the CARs in accordance with the disclosure comprises an antigen binding domain (such as an scFv) in tandem with a costimulatory domain and an activating domain. The costimulatory domain may comprise one or more of an extracellular portion, a transmembrane portion, and an intracellular portion. In other embodiments, multiple costimulatory domains may be utilized in tandem.
[0328] CARs may be engineered to bind to an antigen (such as a cell-surface antigen) by incorporating an antigen binding molecule that interacts with that targeted antigen. In some embodiments, the antigen binding molecule is an antibody fragment thereof, e.g.. one or more single chain antibody fragment (scFv). An scFv is a single chain antibody fragment having the variable regions of the heavy and light chains of an antibody linked together. See U.S. Patent Nos. 7,741,465, and 6,319,494 as well as Eshhar et al.. Cancer Immunol Immunotherapy (1997) 45: 131-136. An scFv retains the parent antibody's ability to specifically interact with target antigen. scFvs are useful in chimeric antigen receptors because they may be engineered to be expressed as part of a single chain along with the other CAR components. Id. See also Krause et al., J. Exp. Med.. Volume 188, No. 4. 1998 (619-626); Finney et al., Journal of Immunology, 1998, 161: 2791-2797. It will be appreciated that the antigen binding molecule is typically contained within the extracellular portion of the CAR such that it is capable of recognizing and binding to the antigen of interest. Bispecific and multispecific CARs are contemplated within the scope of the present disclosure, with specificity to more than one target of interest.
[0329] In some embodiments, the antigen binding molecule comprises a single chain, wherein the heavy chain variable region and the light chain variable region are comiected by a linker. In some embodiments, the VH is located at the N terminus of die linker and the VL is located at the C terminus of the linker. In other embodiments, the VL is located at the N terminus of the linker and the VH is located at the C terminus of the linker. In some embodiments, the linker comprises at least 5. at least 8, at least 10, at least 13, at least 15, at least 18, at least 20. at least 25, at least 30, at least 35, at least 40, at least 45, at least 50, at least 60. at least 70, at least 80, at least 90, or at least 100 amino acids.
[0330] In some embodiments, the antigen binding molecule comprises a nanobody. In some embodiments, the antigen binding molecule comprises a DARPin. In some embodiments, the antigen binding molecule comprises an anticalin or another synthetic protein capable of specific binding to target protein.
[0331] In some embodiments, the CAR comprises an antigen binding domain specific for an antigen selected from CD19, CD123, CD22, CD30, CD171, CS-1, C-type lectin-like molecule-1, CD33, epidermal growth factor receptor variant III (EGFRvIII), ganglioside G2 (GD2), ganglioside GD3, TNF receptor family member B cell maturation (BCMA). Tn antigen ((Tn Ag) or (GalNAca-Ser/Thr)), prostate-specific membrane antigen (PSMA), Receptor tyrosine kinase-like orphan receptor 1 (ROR1), Fms-Like Tyrosine Kinase 3 (FLT3), Tumor-associated glycoprotein 72 (TAG72), CD38. CD44v6, Carcinoembryonic antigen (CEA), Epithelial cell adhesion molecule (EPCAM), B7H3 (CD276), KIT (CD117), Interleukin- 13 receptor subunit alpha-2, mesothelin, Interleukin 11 receptor alpha (IL-1 IRa), prostate stem cell antigen (PSCA). Protease Serine 21, vascular endothelial growth factor receptor 2 (VEGFR2), Lewis(Y) antigen, CD24. Platelet-derived growth factor receptor beta (PDGFR-beta), Stage-specific embryonic antigen-4 (SSEA-4), CD20. Folate receptor alpha. HER2, HER3. Mucin 1. cell surface associated (MUC1), epidermal growth factor receptor (EGFR), neural cell adhesion molecule (NCAM). Prostase, prostatic acid phosphatase (PAP), elongation factor 2 mutated (ELF2M), Ephrin B2, fibroblast activation protein alpha (FAP), insulin-like growth factor 1 receptor (IGF-I receptor), carbonic anhydrase IX (CAIX), Proteasome (Prosome, Macropain) Subunit, Beta Type, 9 (LMP2). glycoprotein 100 (gplOO). oncogene fusion protein consisting of breakpoint cluster region (BCR) and Abelson murine leukemia viral oncogene homolog 1 (Abl) (bcr-abl), tyrosinase, ephrin tvpe- A receptor 2 (EphA2), Fucosyl GM1, sialyl Lewis adhesion molecule (sLe). ganglioside GM3, transglutaminase 5 (TGS5), high molecular weight-melanoma-associated antigen (HMWMAA). o- acetyl-GD2 ganglioside (OAcGD2), Folate receptor beta, tumor endothelial marker 1 (TEM1/CD248), tumor endothelial marker 7-related (TEM7R). claudin 6 (CLDN6), thyroid stimulating hormone receptor (TSHR), G protein-coupled receptor class C group 5, member D (GPRC5D), chromosome X open reading frame 61 (CXORF61), CD97, CD179a, anaplastic lymphoma kinase (ALK), Polysialic acid, placenta-specific 1 (PLAC1), hcxasaccharidc portion of globoH glycoccramidc (GloboH), mammary gland differentiation antigen (NY-BR-1), uroplakin 2 (UPK2), Hepatitis A virus cellular
receptor 1 (HAVCR1), adrenoceptor beta 3 (ADRB3), pannexin 3 (PANX3), G protein-coupled receptor 20 (GPR20), lymphocyte antigen 6 complex, locus K 9 (LY6K), Olfactory receptor 51E2 (OR51E2), TCR Gamma Alternate Reading Frame Protein (TARP), Wilms tumor protein (WT1), Cancer/testis antigen 1 (NY-ESO-1), Cancer/testis antigen 2 (LAGE-la), MAGE family members (including MAGE-A1, MAGE-A3 and MAGE-A4), ETS translocation-variant gene 6. located on chromosome 12p (ETV6-AML), sperm protein 17 (SPA17), X Antigen Family, Member 1A (XAGE1), angiopoietin-binding cell surface receptor 2 (Tie 2), melanoma cancer testis antigen-1 (MAD-CT-1), melanoma cancer testis antigen-2 (MAD-CT-2), Fos-related antigen 1, tumor protein p53 (p53), p53 mutant, prostein, surviving, telomerase, prostate carcinoma tumor antigen- 1. melanoma antigen recognized by T cells 1, Rat sarcoma (Ras) mutant, human Telomerase reverse transcriptase (hTERT), sarcoma translocation breakpoints, melanoma inhibitor of apoptosis (ML-IAP). ERG (transmembrane protease, serine 2 (TMPRSS2) ETS fusion gene), N-Acetyl glucosaminyl-transferase V (NA17), paired box protein Pax-3 (PAX3). Androgen receptor. Cyclin Bl. v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog (MYCN), Ras Homolog Family Member C (RhoC), Tyrosinase-related protein 2 (TRP-2), Cytochrome P450 1B1 (CYP1B1). CCCTC-Binding Factor (Zinc Finger Protein)-Like. Squamous Cell Carcinoma Antigen Recognized By T Cells 3 (SART3). Paired box protein Pax-5 (PAX5). proacrosin binding protein sp32 (OY-TES1), lymphocyte-specific protein tyrosine kinase (LCK), A kinase anchor protein 4 (AKAP-4), synovial sarcoma, X breakpoint 2 (SSX2), Receptor for Advanced Glycation Endproducts (RAGE-1), renal ubiquitous 1 (RU1), renal ubiquitous 2 (RU2), legumain, human papilloma virus E6 (HPV E6). human papilloma virus E7 (HPV E7), intestinal carboxyl esterase, heat shock protein 70-2 mutated (mut hsp70-2), CD79a, CD79b. CD72, Leukocyte-associated immunoglobulin-like receptor 1 (LAIR1), Fc fragment of IgA receptor (FCAR or CD89), Leukocyte immunoglobulin-like receptor subfamily A member 2 (LILRA2), CD300 molecule-like family member f (CD300LF), C-type lectin domain family 12 member A (CLEC12A), bone marrow stromal cell antigen 2 (BST2), EGF-likc module-containing mucin-likc hormone rcccptor- like 2 (EMR2), lymphocyte antigen 75 (LY75), Glypican-3 (GPC3), Fc receptor-like 5 (FCRL5), MUC16, 5T4, 8H9, av|30 integrin, av06 integrin, alphafetoprotein (AFP), B7-H6, ca-125, CA9, CD44, CD44v7/8, CD52, E-cadherin, EMA (epithelial membrane antigen), epithelial glycoprotein-2 (EGP-2), epithelial glycoprotein-40 (EGP-40), ErbB4, epithelial tumor antigen (ETA), folate binding protein (FBP), kinase insert domain receptor (KDR), k-light chain, LI cell adhesion molecule, MUC18, NKG2D, oncofetal antigen (h5T4). tumor/testis-antigen IB, GAGE, GAGE-1, BAGE, SCP-1, CTZ9, SAGE, CAGE, CT10. MART-1, immuno globulin lambda-like polypeptide 1 (IGLL1), Hepatitis B Surface Antigen Binding Protein (HBsAg), viral capsid antigen (VCA), early antigen (EA), EBV nuclear antigen (EBNA), HHV-6 p41 early antigen, HHV-6B U94 latent antigen, HHV-6B p98 late antigen , cytomegalovirus (CMV) antigen, large T antigen, small T antigen, adenovirus antigen, respiratory syncytial virus (RSV) antigen, haemagglutinin (HA), neuraminidase (NA), parainfluenza
type 1 antigen, parainfluenza type 2 antigen, parainfluenza ty pe 3 antigen, parainfluenza ty pe 4 antigen, Human Metapneumovirus (HMPV) antigen, hepatitis C virus (HCV) core antigen, HIV p24 antigen, human T-cell lympotrophic virus (HTLV-1) antigen, Merkel cell polyoma virus small T antigen, Merkel cell polyoma virus large T antigen, Kaposi sarcoma-associated herpesvirus (KSHV) lytic nuclear antigen and KSHV latent nuclear antigen. In some embodiments, an antigen binding domain comprises SEQ ID NO: 321 and/or 322.
[0332] In some embodiments, a CAR comprises a hinge or spacer domain. In some embodiments, the hinge/spacer domain may comprise a truncated hinge/spacer domain (THD) the THD domain is a truncated version of a complete hinge/spacer domain (“CHD”). In some embodiments, an extracellular domain is from or derived from (e g., comprises all or a fragment of) ErbB2, glycophorin A (GpA), CD2, CD3 delta, CD3 epsilon, CD3 gamma, CD4, CD7, CD8a, CD8[T CD1 la (IT GAL), CD1 lb (IT GAM), CD1 1c (ITGAX), CD1 Id (IT GAD), CD18 (ITGB2), CD19 (B4), CD27 (TNFRSF7), CD28, CD28T. CD29 (ITGB1). CD30 (TNFRSF8), CD40 (TNFRSF5). CD48 (SLAMF2), CD49a (ITGA1), CD49d (ITGA4), CD49f (ITGA6), CD66a (CEACAM1), CD66b (CEACAM8). CD66c (CEACAM6), CD66d (CEACAM3). CD66e (CEACAM5), CD69 (CLEC2 ), CD79A (B-cell antigen receptor complex-associated alpha chain), CD79B (B-cell antigen receptor complex-associated beta chain), CD84 (SLAMF5). CD96 (Tactile). CD100 (SEMA4D), CD103 (1TGAE), CD134 (0X40), CD137 (4- 1BB). CD150 (SLAMF1), CD158A (KIR2 DL1). CD158B1 (KIR2 DL2). CD158B2 (KIR2 DL3), CD158C (KIR3 DP1), CD158D (KIRDL4), CD158F1 (KIR2 DL5A), CD158F2 (KIR2 DL5B), CD158K (KIR3 DL2), CD160 (BY55). CD162 (SELPLG), CD226 (DNAM1). CD229 (SLAMF3), CD244 (SLAMF4), CD247 (CD3-zeta), CD258 (LIGHT), CD268 (BAFFR). CD270 (TNFSF14), CD272 (BTLA). CD276 (B7-H3), CD279 (PD-1), CD314 (NKG2D), CD319 (SLAMF7), CD335 (NK-p46), CD336 (NK-p44), CD337 (NK-p30), CD352 (SLAMF6), CD353 (SLAMF8), CD355 (CRT AM), CD357 (TNFRSF18), inducible T cell co-stimulator (ICOS), LFA-1 (CD1 la/CD18), NKG2C. DAP-10, ICAM-1, NKp80 (KLRF1), IL-2R beta, IL-2R gamma, IL-7R alpha. LFA-1, SLAMF9. LAT. GADS (GrpL), SLP-76 (LCP2), PAG1/CBP, a CD83 ligand. Fc gamma receptor, MHC class 1 molecule, MHC class 2 molecule, a TNF receptor protein, an immunoglobulin protein, a cytokine receptor, an integrin, activating NK cell receptors, a Toll ligand receptor, and fragments or combinations thereof. A hinge or spacer domain may be derived either from a natural or from a synthetic source.
[0333] In some embodiments, a hinge or spacer domain is positioned between an antigen binding molecule (e.g., an scFv) and a transmembrane domain. In this orientation, the hinge/spacer domain provides distance between the antigen binding molecule and the surface of a cell membrane on which the CAR is expressed. In some embodiments, a hinge or spacer domain is from or is derived from an immunoglobulin. In some embodiments, a hinge or spacer domain is selected from the hinge/spacer regions of IgGl, IgG2, IgG3, IgG4, IgA, IgD, IgE, IgM, or a fragment thereof. In some embodiments, a hinge or spacer domain comprises, is from, or is derived from the hinge/spacer region of CD8 alpha.
In some embodiments, a hinge or spacer domain comprises, is from, or is derived from the hinge/spacer region of CD28. In some embodiments, a hinge or spacer domain comprises a fragment of the hinge/spacer region of CD8 alpha or a fragment of the hinge/spacer region of CD28, wherein the fragment is any tiring less than the whole hinge/spacer region. In some embodiments, the fragment of the CD8 alpha hinge/spacer region or the fragment of the CD28 hinge/spacer region comprises an amino acid sequence that excludes at least 1, at least 2, at least 3. at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least 14, at least 15. at least 16, at least 17, at least 18, at least 19. or at least 20 amino acids at the N-tenninus or C-Terminus, or both, of the CD8 alpha hinge/spacer region, or of the CD28 hinge/spacer region.
[0334] The CAR may further comprise a transmembrane domain and/or an intracellular signaling domain. The transmembrane domain may be designed to be fused to the extracellular domain of the CAR. It may similarly be fused to the intracellular domain of the CAR. In some embodiments, the transmembrane domain that naturally is associated with one of the domains in a CAR is used. In some instances, the transmembrane domain may be selected or modified (e.g., by an amino acid substitution) to avoid binding of such domains to the transmembrane domains of die same or different surface membrane proteins to minimize interactions with other members of the receptor complex. The transmembrane domain may be derived either from a natural or from a synthetic source. Where the source is natural, the domain may be derived from any membrane -bound or transmembrane protein.
[0335] Transmembrane regions may be derived from (i.e., comprise) a receptor tyrosine kinase (e.g., ErbB2). glycophorin A (GpA), 4-1BB/CD137, activating NK cell receptors, an immunoglobulin protein, B7-H3, BAFFR, BFAME (SEAMF8), BTEA, CD100 (SEMA4D), CD103, CD160 (BY55), CD 18, CD 19, CD 19a, CD2, CD247, CD27, CD276 (B7-H3), CD28. CD29, CD3 delta. CD3 epsilon, CD3 gamma, CD30. CD4. CD40, CD49a, CD49D, CD49f, CD69. CD7. CD84, CD8alpha. CDSbeta, CD96 (Tactile). CD1 la. CD1 lb, CD1 1c, CD1 Id. CDS. CEACAM1, CRT AM, cytokine receptor, DAP-10, DNAM1 (CD226). Fc gamma receptor, GADS. GITR, HVEM (EIGHTR), IA4. ICAM-1. ICAM-1, Ig alpha (CD79a), IE-2R beta, IE-2R gamma, IE-7R alpha, inducible T cell costimulator (ICOS), integrins, ITGA4, ITGA4. ITGA6. IT GAD. ITGAE, ITGAE, IT GAM. ITGAX, ITGB2, ITGB7, ITGB1, KIRDS2, EAT, LFA-1, LFA-1. a ligand that specifically binds with CD83, LIGHT, LIGHT, LTBR, Ly9 (CD229), lymphocyte function-associated antigen-1 (LFA-1 ; CDl-la/CD 18), MHC class 1 molecule, NKG2C. NKG2D, NKp30, NKp44. NKp46, NKp80 (KLRF1), OX-40. PAG/Cbp, programmed death-1 (PD-1), PSGL1, SELPLG (CD162). Signaling Lymphocytic Activation Molecules (SLAM proteins), SLAM (SLAMFL CD150; IPO-3), SLAMF4 (CD244; 2B4), SLAMF6 (NTB-A; Lyl08), SLAMF7, SLP-76, TNF receptor proteins, TNFR2 , TNFSF14, a Toll ligand receptor, TRANCE/RANKL, VLA1, or VLA-6, or a fragment, truncation, or a combination thereof.
[0336] In some embodiments, suitable intracellular signaling domain include, but are not limited to, activating Macrophage/Myeloid cell receptors CSFR1, MYD88, CD14, TIE2, TLR4, CR3. CD64, TREM2, DAP10, DAP12, CD169, DECTIN1, CD206, CD47, CD163, CD36, MARCO. TIM4, MERTK, F4/80. CD91, C1QR, LOX-1, CD68, SRA, BAI-1, ABCA7, CD36, CD31, Lactoferrin, or a fragment, truncation, or combination thereof.
[0337] In some embodiments, a receptor tyrosine kinase may be derived from (e.g., comprise) Insulin receptor (InsR), Insulin-like growth factor I receptor (IGF1R), Insulin receptor-related receptor (IRR), platelet derived growth factor receptor alpha (PDGFRa), platelet derived growth factor receptor beta (PDGFRfi). KIT proto-oncogene receptor tyrosine kinase (Kit), colony stimulating factor 1 receptor (CSFR), fms related tyrosine kinase 3 (FLT3), fins related tyrosine kinase 1 (VEGFR-1), kinase insert domain receptor (VEGFR-2), fms related tyrosine kinase 4 (VEGFR-3), fibroblast growth factor receptor 1 (FGFR1). fibroblast growth factor receptor 2 (FGFR2), fibroblast growth factor receptor 3 (FGFR3 ), fibroblast growth factor receptor 4 (FGFR4), protein tyrosine kinase 7 (CCK4), neurotrophic receptor tyrosine kinase 1 (trkA). neurotrophic receptor tyrosine kinase 2 (trkB), neurotrophic receptor tyrosine kinase 3 (trkC), receptor tyrosine kinase like orphan receptor 1 (ROR1), receptor tyrosine kinase like orphan receptor 2 (ROR2), muscle associated receptor tyrosine kinase (MuSK). MET protooncogene. receptor tyrosine kinase (MET), macrophage stimulating 1 receptor (Ron), AXL receptor tyrosine kinase (Axl), TYR03 protein tyrosine kinase (Tyro3). MER proto-oncogene, tyrosine kinase (Mer). tyrosine kinase with immunoglobulin like and EGF like domains 1 (TIE1), TEK receptor ty rosine kinase (TIE2), EPH receptor Al (Eph Al), EPH receptor A2 (EphA2), (EPH receptor A3) Eph A3, EPH receptor A4 (EphA4), EPH receptor A5 (EphA5), EPH receptor A6 (EphA6), EPH receptor A7 (EphA7), EPH receptor A8 (EphA8), EPH receptor Alt) (EphAlO), EPH receptor Bl (EphBl). EPH receptor B2 (EphB2). EPH receptor B3 (EphB3), EPH receptor B4 (EphB4), EPH receptor B6 (EphB6), ret proto oncogene (Ret), receptor-like ty rosine kinase (RYK). discoidin domain receptor tyrosine kinase 1 (DDR1), discoidin domain receptor tyrosine kinase 2 (DDR2), c-ros oncogene 1, receptor tyrosine kinase (ROS), apoptosis associated ty rosine kinase (Lmrl), lemur ty rosine kinase 2 (Lmr2), lemur tyrosine kinase 3 (Lmr3), leukocyte receptor tyrosine kinase (LTK), ALK receptor ty rosine kinase (ALK), or serine/threonine/tyrosine kinase 1 (STYK1).
[0338] In some embodiments, the CAR comprises a costimulatoiy domain. In some embodiments, the costimulatoiy domain comprises 4-1BB (CD137), CD28, or both, and/or an intracellular T cell signaling domain. In a preferred embodiment, the costimulatoiy domain is human CD28, human 4-1BB, or both, and the intracellular T cell signaling domain is human CD3 zeta (Q. 4- IBB, CD28, CD3 zeta may comprise less than the whole 4-1BB, CD28 or CD3 zeta, respectively. Chimeric antigen receptors may incorporate costimulatory (signaling) domains to increase their potency. See U.S. Patent Nos. 7.741,465, and 6,319,494, as well as Krause et al. and Fimrey et al. (supra), Song et al.. Blood
119:696-706 (2012); Kalos et al., Sci Transl. Med. 3:95 (2011); Porter et al., N. Engl. J. Med.
365:725-33 (2011), and Gross et al., Amur. Rev. Pharmacol. Toxicol. 56:59-83 (2016).
[0339] The intracellular (signaling) domain of the engineered T cells disclosed herein may provide signaling to an activating domain, which then activates at least one of the normal effector functions of die immune cell. Effector function of a T cell, for example, may be cytolytic activity or helper activity including the secretion of cytokines.
[0340] In some embodiments, suitable intracellular signaling domain include (e.g., comprise), but are not limited to 4-1BB/CD137, activating NK cell receptors, an Immunoglobulin protein, B7-H3, BAFFR, BLAME (SLAMF8), BTLA, CD100 (SEMA4D), CD103, CD160 (BY55), CD18, CD19, CD 19a, CD2, CD247, CD27, CD276 (B7-H3), CD28, CD29, CD3 delta, CD3 epsilon, CD3 gamma, CD30, CD4, CD40, CD49a, CD49D, CD49f, CD69, CD7, CD84, CD8alpha, CD8beta, CD96 (Tactile), CD1 la, CD1 lb, CD1 1c, CD1 Id, CDS, CEACAM1, CRT AM, cytokine receptor, DAP-10, DNAM1 (CD226), Fc gamma receptor, GADS, GITR, HVEM (LIGHTR), IA4, ICAM-1. Ig alpha (CD79a), IL- 2R beta, IL-2R gamma, IL-7R alpha, inducible T cell costimulator (ICOS), integrins, ITGA4, ITGA6, ITGAD, ITGAE, ITGAL, ITGAM, ITGAX, ITGB2, ITGB7, ITGB1, KIRDS2, LAT, LFA-1, ligand that specifically binds with CD83, LIGHT, LTBR, Ly9 (CD229), Lyl08, lymphocyte function- associated antigen- 1 (LFA-1; CDl-la/CD18), MHC class 1 molecule, NKG2C, NKG2D, NKp30, NKp44. NKp46. NKp80 (KLRF1). OX-40, PAG/Cbp. programmed death-1 (PD-1). PSGL1. SELPLG (CD162), Signaling Lymphocytic Activation Molecules (SLAM proteins), SLAM (SLAMF1; CD150; IPO-3). SLAMF4 (CD244; 2B4). SLAMF6 (NTB-A). SLAMF7, SLP-76. TNF receptor proteins. TNFR2. TNFSF14. a Toll ligand receptor, TRANCE/RANKL, VLA1, or VLA-6. or a fragment, truncation, or a combination thereof.
[0341] CD3 is an element of the T cell receptor on native T cells and has been shown to be an important intracellular activating element in CARs. In some embodiments, the CD3 is CD3 zeta.
[0342] In some embodiments, the sequence encoding the CAR comprises a sequence from Table 4.
Table 4










(2) T-Cell Receptors (TCR)
[0343] TCRs are described using the International Immunogenetics (IMGT) TCR nomenclature, and links to the IMGT public database of TCR sequences. Native alpha-beta heterodimeric TCRs have an alpha chain and a beta chain. Broadly, each chain may comprise variable, joining and constant regions,
and the beta chain also usually contains a short diversity region between the variable and joining regions, but this diversity region is often considered as part of the joining region. Each variable region may comprise three CDRs (Complementarity Determining Regions) embedded in a framework sequence, one being the hypervariable region named CDR3. There are several types of alpha chain variable (Vα) regions and several types of beta chain variable (VP) regions distinguished by their framework, CDR1 and CDR2 sequences, and by a partly defined CDR3 sequence. The Va types are referred to in IMGT nomenclature by a unique TRAV number. Thus “TRAV21” defines a TCR Va region having unique framework and CDR1 and CDR2 sequences, and a CDR3 sequence which is partly defined by an amino acid sequence which is preserved from TCR to TCR, but which also includes an amino acid sequence which varies from TCR to TCR. In the same way, “TRBV5-1” defines a TCR VP region having unique framework and CDR1 and CDR2 sequences, but with only a partly defined CDR3 sequence.
[0344] The joining regions of the TCR are similarly defined by the unique IMGT TRAJ and TRBJ nomenclature, and the constant regions by the IMGT TRAC and TRBC nomenclature.
[0345] The beta chain diversity region is referred to in IMGT nomenclature by the abbreviation TRBD, and, as mentioned, the concatenated TRBD/TRBJ regions are often considered together as the joining region.
[0346] The unique sequences defined by the IMGT nomenclature are widely known and accessible to those working in the TCR field. For example, they can be found in the IMGT public database. The “T cell Receptor Factsbook”, (2001) LeFranc and LeFranc, Academic Press, ISBN 0-12-441352-8 also discloses sequences defined by the IMGT nomenclature, but because of its publication date and consequent time-lag, the information therein sometimes needs to be confirmed by reference to the IMGT database.
[0347] Native TCRs exist in heterodimeric aP or y5 forms. However, recombinant TCRs consisting of aa or pp homodimers have previously been shown to bind to peptide MHC molecules. Therefore, the TCR of the present disclosure may be a heterodimeric aP TCR or may be an aa or pp homodimeric TCR.
[0348] For use in adoptive therapy, an aP heterodimeric TCR may, for example, be transfected as full- length chains having both cytoplasmic and transmembrane domains. In certain embodiments TCRs of die present disclosure may have an introduced disulfide bond between residues of the respective constant domains, as described, for example, in WO 2006/000830.
[0349] TCRs of the present disclosure, particularly alpha-beta heterodimeric TCRs, may comprise an alpha chain TRAC constant domain sequence and/or a beta chain TRBC 1 or TRBC2 constant domain sequence. The alpha and beta chain constant domain sequences may be modified by truncation or
substitution to delete the native disulfide bond betw een Cys4 of exon 2 of TRAC and Cys2 of exon 2 of TRBC1 or TRBC2 . The alpha and/or beta chain constant domain sequence(s) may also be modified by substitution of cysteine residues for Thr 48 of TRAC and Ser 57 of TRBC1 or TRBC2 , the said cysteines forming a disulfide bond betw een the alpha and beta constant domains of the TCR.
[0350] Binding affinity (inversely proportional to the equilibrium constant KD) and binding half-life (expressed as T½) can be determined by any appropriate method. It will be appreciated that doubling die affinity of a TCR results in halving the KD. T!4 is calculated as In 2 divided by the off-rate (koff). Therefore, doubling of T!4 results in a halving in koff. Ku and koff values for TCRs are usually measured for soluble forms of the TCR, i.e., those forms which are truncated to remove cytoplasmic and transmembrane domain residues. Therefore, it is to be understood that a given TCR has an improved binding affinity for, and/or a binding half-life for the parental TCR if a soluble form of that TCR has the said characteristics. Preferably the binding affinity or binding half-life of a given TCR is measured several times, for example 3 or more times, using the same assay protocol, and an average of the results is taken.
[0351] Since the TCRs of the present disclosure have utility in adoptive therapy, the present disclosure includes a non-naturally occurring and/or purified and/or or engineered cell, especially a T-cell, presenting a TCR of the present disclosure. There are a number of methods suitable for the transfection of T cells with nucleic acid (such as DNA, cDNA or RNA) encoding the TCRs of the present disclosure (see for example Robbins et al., (2008) J Immunol. 180: 6116-6131). T cells expressing the TCRs of the present disclosure will be suitable for use in adoptive therapy-based treatment of cancers such as those of the pancreas and liver. As will be known to those skilled in the art, there is a number of suitable methods by which adoptive therapy can be carried out (see for example Rosenberg et al., (2008) Nat Rev Cancer 8(4): 299-308).
[0352] As is well-known in the art TCRs of the present disclosure may be subject to post-translational modifications when expressed by transfected cells. Glycosylation is one such modification, which may comprise the covalent attachment of oligosaccharide moieties to defined amino acids in the TCR chain. For example, asparagine residues, or serine/threonine residues are well-known locations for oligosaccharide attachment. The glycosylation status of a particular protein depends on a number of factors, including protein sequence, protein conformation and the availability of certain enzymes. Furthermore, glycosylation status (i.e.. oligosaccharide type, covalent linkage and total number of attachments) can influence protein function. Therefore, when producing recombinant proteins, controlling glycosylation is often desirable. Glycosylation of transfected TCRs may be controlled by mutations of the transfected gene (Kuball J et al. (2009). J Exp Med 206(2):463-475). Such mutations are also encompassed in this disclosure.
[0353] A TCR may be specific for an antigen in tire group MAGE-A1, MAGE-A2, MAGE -A3, MAGE-A4, MAGE-A5, MAGE-A6, MAGE-A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, MAGE-A12, MAGE-A13, GAGE-1, GAGE-2, GAGE-3, GAGE-4, GAGE-5, GAGE-6, GAGE-7, GAGE-8, BAGE-1, RAGE-1, LB33/MUM-1, PRAME, NAG, MAGE-Xp2 (MAGE-B2), MAGE-Xp3 (MAGE-B3), MAGE-Xp4 (AGE-B4), tyrosinase, brain glycogen phosphorylase, Melan-A, MAGE-CI, MAGE-C2 , NY-ESO-1, LAGE-1, SSX-1. SSX-2(HOM-MEL-40), SSX-1, SSX-4, SSX-5. SCP-1, CT- 7. alpha-actinin-4. Bcr-Abl fusion protein, Casp-8, beta-catenin, cdc27, cdk4, cdkn2a, coa-1, dek-can fusion protein, EF2. ETV6-AML1 fusion protein, LDLR-fucosyltransferaseAS fusion protein, HLA- A2. HLA-A11, hsp70-2, KIAAO205. Mart2, Mum-2, and 3, neo-PAP, myosin class 1, OS-9, pml-RARa fusion protein, PTPRK, K-ras. N-ras. Triosephosphate isomeras, GnTV. Herv-K-mel, Lage-1, Mage- C2 , NA-88, Lage-2, SP17, and TRP2-Int2, (MART-I), gplOO (Pmel 17), TRP-1, TRP-2, MAGE-1, MAGE-3, pl5(58). CEA, NY-ESO (LAGE), SCP-1, Hom/Mel-40, p53. H-Ras. HER-2/neu. BCR- ABL, E2A-PRL, H4-RET, IGH-IGK. MYL-RAR, Epstein Barr virus antigens, EBNA, human papillomavirus (HPV) antigens E6 and E7, TSP-180, MAGE-4, MAGE-5. MAGE-6, pl85erbB2, pl80erbB-3. c-met, nm-23Hl, PSA, TAG-72-4. CA 19-9. CA 72-4, CAM 17.1, NuMa, K-ras, beta- catenin, CDK4, Mum-1. pl6, TAGE, PSMA, PSCA, CT7. telomerase. 43-9F, 5T4, 791Tgp72. a- fetoprotein, 13HCG, BCA225, BTAA, CA 125. CA 15-3 (CA 27.29\BCAA), CA 195, CA 242, CA- 50, CAM43. CD68\KP1, CO-029, FGF-5. G250, Ga733 (EpCAM), HTgp-175, M344, MA-50, MG7- Ag, M0V18, NBM70K, NY-CO-1, RCAS1, SDCCAG16, TA-90 (Mac-2 binding protein\cyclophilin C-associated protein), TAAL6, TAG72, TLP, and TPS.
(3) B-Cell Receptors (BCR)
[0354] B-cell receptors (BCRs) or B-cell antigen receptors are immunoglobulin molecules that form a type I transmembrane protein on the surface of a B cell. A BCR is capable of transmitting an activatory signal into a B cell following recognition of a specific antigen. Prior to binding of a B cell to an antigen, the BCR will remain in an unstimulated or “resting” stage. Binding of an antigen to a BCR leads to signaling that initiates a humoral immune response.
[0355] A BCR is expressed by mature B cells. These B cells work with immunoglobulins (Igs) in recognizing and tagging pathogens. The typical BCR comprises a membrane-bound immunoglobulin (e.g., mlgA, mlgD, mlgE, mlgG, and mlgM), along with associated and Iga/IgP (CD79a/CD79b) heterodimers (a/p). These membrane -bound immunoglobulins are tetramers consisting of two identical heavy and two light chains. Within the BCR, the membrane bound immunoglobulins is capable of responding to antigen binding by signal transmission across the plasma membrane leading to B cell activation and consequently clonal expansion and specific antibody production (Friess M el al. (2018), Front. Immunol. 2947(9)). The Iga/IgP heterodimers is responsible for transducing signals to the cell interior.
[0356] A Igot/IgP heterodimer signaling relies on the presence of immunoreceptor tyrosine-based activation motifs (ITAMs) located on each of the cytosolic tails of the heterodimers. ITAMs comprise two tyrosine residues separated by 9-12 amino acids (e.g., tyrosine, leucine, and/or valine). Upon binding of an antigen, the tyrosine of the BCR’s ITAMs become phosphorylated by Src-family tyrosine kinases Blk, Fyn, or Lyn (Janeway C etal., Immunobiology: The Immune System in Health and Disease (Garland Science, 5th ed. 2001)).
(4) Other Chimeric Proteins
[0357] In addition to the chimeric proteins provided above, the circular RNA polynucleotide may encode for a various number of other chimeric proteins available in the art. The chimeric proteins may include recombinant fusion proteins, chimeric mutant protein, or other fusion proteins.
[0358] In some embodiments, the circular RNA polynucleotide encodes for an immune modulatory ligand. In certain embodiments, the immune modulatory ligand may be immuno stimulatory; while in other embodiments, the immune modulatory ligand may be immunosuppressive.
[0359] In some embodiments, the circular RNA polynucleotide encodes for a cytokine. In some embodiments, the cytokine comprises a chemokine, interferon, interleukin, lymphokine, and tumor necrosis factor. Chemokines are chemotactic cytokine produced by a variety of cell types in acute and chronic inflammation that mobilizes and activates white blood cells. An interferon comprises a family of secreted a-helical cytokines induced in response to specific extracellular molecules through stimulation of TLRs (Borden, Molecular Basis of Cancer (Fourth Edition) 2015). Interleukins are cytokines expressed by leukocytes.
[0360] Descriptions and/or amino acid sequences of IL-2, IL-7, IL-10, IL-12. IL-15, IL-18, IL-270, IFNy, and/or TGF0 I are provided herein and at the www.uniprot.org database at accession numbers: P60568 (IL-2), P29459 (IL-12A), P29460 (IL-12B), P13232 (IL-7), P22301 (IL-10), P40933 (IL-15), Q14116 (IL-18). Q14213 (IL-270), P01579 (IFNy), and/or P01137 (TGF01).
[0361] In some embodiments, the circular RNA polynucleotide may encode for a transcription factor. Regulatory T cells (Treg) are important in maintaining homeostasis, controlling the magnitude and duration of the inflammatory response, and in preventing autoimmune and allergic responses.
[0362] In general, Tregs are thought to be mainly involved in suppressing immune responses, functioning in part as a “self-check” for the immrme system to prevent excessive reactions. In particular, Tregs are involved in maintaining tolerance to self-antigens, harmless agents such as pollen or food, and abrogating autoimmune disease.
[0363] Tregs are found throughout the body including, without limitation, the gut, skin, lung, and liver. Additionally, Treg cells may also be found in certain compartments of the body that are not directly
exposed to the external environment such as the spleen, lymph nodes, and even adipose tissue. Each of these Treg cell populations is known or suspected to have one or more unique features and additional information may be found in Lehtimaki and Lahesmaa, Regulatory' T cells control immune responses through their non-redundant tissue specific features, 2013, Frontiers in Immunol., 4(294): 1-10, the disclosure of which is hereby incorporated in its entirety.
[0364] Typically, Tregs are knoyvn to require TGF-β and IL-2 for proper activation and development. Tregs, expressing abundant amounts of the IL-2 receptor (IL-2R), arc reliant on IL -2 produced byactivated T cells. Tregs are known to produce both IL- 10 and TGF-P, both potent immune suppressive cytokines. Additionally, Tregs are knoyvn to inhibit the ability- of antigen presenting cells (APCs) to stimulate T cells. One proposed mechanism for APC inhibition is via CTLA-4, which is expressed by Foxp3+ Tregs. It is thought that CTLA-4 may bind to B7 molecules on APCs and either block these molecules or remove them by causing internalization resulting in reduced availability of B7 and an inability to provide adequate co-stimulation for immune responses. Additional discussion regarding the origin, differentiation and function of Tregs may be found in Dhamne et al., Peripheral and thymic Foxp3+ regulatory T cells in search of origin, distinction, and function. 2013, Frontiers in Immunol., 4 (253): 1-11. the disclosure of which is hereby incorporated in its entirety.
[0365] As provided herein, in certain embodiments, the coding element of the circular RNA polynucleotide encodes for one or more checkpoint inhibitors or agonists.
[0366] In some embodiments, the immune checkpoint inhibitor is an inhibitor of Programmed Death- Ligand 1 (PD-L1, also known as B7-H1, CD274), Programmed Death 1 (PD-1). CTLA-4, PD-L2 (B7- DC. CD273), LAG3, TIM3, 2B4, A2aR, B7H1, B7H3, B7H4, BTLA, CD2, CD27, CD28, CD30, CD40, CD70, CD80, CD86, CD137, CD160, CD226, CD276, DR3 , GAL9, GITR, HAVCR2 , HVEM, IDO1, IDO2, ICOS (inducible T cell co-stimulator), KIR, LAIR1, LIGHT, MARCO (macrophage receptor yvith a collagenous structure), PS (phosphatidylserine), OX-40, SLAM. TIGHT, VISTA, VTCN1, or any combinations thereof. In some embodiments, the immune checkpoint inhibitor is an inhibitor of IDO1, CTLA4, PD-1, LAG3, PD-L1, TIM3, or combinations thereof. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-L1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of CTLA-4. In some embodiments, the immune checkpoint inhibitor is an inhibitor of L AG3. In some embodiments, the immune checkpoint inhibitor is an inhibitor of TIM3. In some embodiments, the immune checkpoint inhibitor is an inhibitor of IDOL
[0367] As described herein, at least in one aspect, the present disclosure encompasses the use of immune checkpoint antagonists. Such immune checkpoint antagonists include antagonists of immune checkpoint molecules such as Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4), Programmed Cell Death Protein 1 (PD-1), Programmed Death-Ligand 1 (PDL-1), Lymphocyte- activation gene 3
(LAG-3), and T-cell immunoglobulin and mucin domain 3 (TIM-3). An antagonist of CTLA-4, PD-1, PDL-1, LAG-3, or TIM-3 interferes with CTLA-4, PD-1, PDL-1, LAG-3, or TIM-3 function, respectively. Such antagonists of CTLA-4, PD-1, PDL-1, LAG-3, and TIM -3 can include antibodies which specifically bind to CTLA-4, PD-1, PDL-1, LAG-3, and TIM-3, respectively and inhibit and/or block biological activity and function.
[0368] In some embodiments, the pay load encoded within one or more of the coding elements is a hormone, FC fusion protein, anticoagulant, blood clotting factor, protein associated with deficiencies and genetic disease, a chaperone protein, an antimicrobial protein, an enzy me (e.g., metabolic enzyme), a structural protein (e.g., a channel or nuclear pore protein), protein variant, small molecule, antibody, nanobody, an engineered non-body antibody, or a combination thereof. xi. Production of polynucleotides
[0369] DNA templates can be made using standard techniques of molecular biology . For example, the various elements of the vectors provided herein can be obtained using recombinant methods, such as by screening cDNA and genomic libraries from cells, or by deriving the polynucleotides from a DNA template known to include the same.
[0370] The various elements of the DNA template can also be produced synthetically, rather than cloned, based on the known sequences. The complete sequence can be assembled from overlapping oligonucleotides prepared by standard methods and assembled into the complete sequence. See. e.g., Edge, Nature (1981) 292:756; Nambair et al., Science (1984) 223: 1299; and Jay et al., J. Biol. Chem. (1984) 259:631 1.
[0371] Thus, particular nucleotide sequences can be obtained from DNA template harboring the desired sequences or synthesized completely, or in part, using various oligonucleotide synthesis techniques known in the art, such as site-directed mutagenesis and polymerase chain reaction (PCR) techniques where appropriate. One method of obtaining nucleotide sequences encoding the desired DNA template elements is by annealing complementary sets of overlapping synthetic oligonucleotides produced in a conventional, automated polynucleotide synthesizer, followed by ligation with an appropriate DNA ligase and amplification of the ligated nucleotide sequence via PCR. See. e.g., Jayaraman et al., Proc. Natl. Acad. Sci. USA (1991) 88:4084-4088. Additionally, oligonucleotide- directed synthesis (Jones et al., Nature (1986) 54:75-82), oligonucleotide directed mutagenesis of preexisting nucleotide regions (Riechmann et al., Nature (1988) 332:323-327 and Verhoeyen et al., Science (1988) 239: 1534-1536), and enzymatic filling-in of gapped oligonucleotides using T4 DNA polymerase (Queen et al., Proc. Natl. Acad. Sci. USA (1989) 86: 10029-10033) can be used.
[0372] The precursor RNA can be generated by incubating a DNA template under conditions permissive of transcription of the precursor RNA encoded by the DNA template. For example, in some
embodiments a precursor RNA is synthesized by incubating a DNA template provided herein that comprises an RNA polymerase promoter upstream of its 5’ duplex sequence and/or expression sequences with a compatible RNA polymerase enzyme under conditions permissive of in vitro transcription. In some embodiments, the DNA template is incubated inside of a cell by a bacteriophage RNA polymerase or in the nucleus of a cell by host RNA polymerase II.
6. PHARMACEUTICAL COMPOSITIONS
[0373] In certain embodiments, provided herein are compositions (e.g.. pharmaceutical compositions) comprising a therapeutic agent provided herein. In some embodiments, the therapeutic agent is a circular RNA polynucleotide. In some embodiments the therapeutic agent is a vector. In some embodiments, the therapeutic agent is a cell comprising a circular RNA or vector (e.g., a human cell, such as a human T cell). In certain embodiments, the composition further comprises a pharmaceutically acceptable carrier. In some embodiments, the compositions provided herein comprise a therapeutic agent provided herein in combination with other pharmaceutically active agents or drugs, such as antiinflammatory drugs or antibodies capable of targeting B cell antigens, e.g., anti-CD20 antibodies, e.g., rituximab.
[0374] With respect to pharmaceutical compositions, the pharmaceutically acceptable carrier can be any of those conventionally used and is limited only by chemical-physical considerations, such as solubility and lack of reactivity with the active agent(s), and by the route of administration. The pharmaceutically acceptable carriers described herein, for example, vehicles, adjuvants, excipients, and diluents, are well-known to those skilled in the art and are readily available to the public. It is preferred drat the pharmaceutically acceptable carrier be one which is chemically inert to the therapeutic agent(s) and one which has no detrimental side effects or toxicity under the conditions of use.
[0375] The choice of carrier will be determined in part by the particular therapeutic agent, as well as by the particular method used to administer the therapeutic agent. Accordingly, there are a variety of suitable formulations of the pharmaceutical compositions provided herein.
[0376] In certain embodiments, the pharmaceutical composition comprises a preservative. In certain embodiments, suitable preservatives may include, for example, methylparaben, propylparaben, sodium benzoate, and benzalkonium chloride. Optionally, a mixture of two or more preservatives may be used. The preservative or mixtures thereof are typically present in an amount of 0.0001% to 2% by weight of the total composition.
[0377] In some embodiments, the pharmaceutical composition comprises a buffering agent. In some embodiments, suitable buffering agents may include, for example, citric acid, sodium citrate, phosphoric acid, potassium phosphate, and various other acids and salts. A mixture of tw o or more
buffering agents optionally may be used. The buffering agent or mixtures thereof are ty pically present in an amount of 0.001% to 4% by weight of the total composition.
[0378] In some embodiments, the concentration of therapeutic agent in the pharmaceutical composition can vary, e.g., less than 1%, or at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9% 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, or 50% or more by weight, and can be selected primarily by fluid volumes, and viscosities, in accordance with the particular mode of administration selected.
[0379] The following formulations for oral, aerosol, parenteral (e.g., subcutaneous, intravenous, intraarterial, intramuscular, intradermal, intraperitoneal, and intrathecal), and topical administration are merely exemplary' and are in no way limiting. More than one route can be used to administer the therapeutic agents provided herein, and in certain instances, a particular route can provide a more immediate and more effective response than another route.
[0380] Formulations suitable for oral administration can comprise or consist of (a) liquid solutions, such as an effective amount of the therapeutic agent dissolved in diluents, such as water, saline, or orange juice; (b) capsules, sachets, tablets, lozenges, and troches, each containing a predetermined amount of the active ingredient, as solids or granules; (c) powders; (d) suspensions in an appropriate liquid; and (e) suitable emulsions. Liquid formulations may include diluents, such as water and alcohols, for example, ethanol, benzy l alcohol and the polyethylene alcohols, either with or without the addition of a pharmaceutically acceptable surfactant. Capsule forms can be of the ordinary' hard- or soft-shelled gelatin type containing, for example, surfactants, lubricants, and inert fillers, such as lactose, sucrose, calcium phosphate, and com starch. Tablet forms can include one or more of lactose, sucrose, mannitol, corn starch, potato starch, alginic acid, microcrystalline cellulose, acacia, gelatin, guar gum, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, calcium stearate, zinc stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, disintegrating agents, moistening agents, preservatives, flavoring agents, and other pharmacologically compatible excipients. Lozenge forms can comprise the therapeutic agent with a flavorant, usually sucrose, acacia or tragacanth. Pastilles can comprise the therapeutic agent with an inert base, such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to, such excipients as are known in the art.
[0381] Formulations suitable for parenteral administration include aqueous and nonaqueous isotonic sterile injection solutions, which can contain antioxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the intended recipient, and aqueous and nonaqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives. In some embodiments, the therapeutic agents provided herein can be administered in a physiologically acceptable diluent in a pharmaceutical carrier, such as a sterile liquid or mixture of liquids including water, saline, aqueous dextrose and related sugar solutions, an alcohol such as ethanol
or hexadecyl alcohol, a glycol such as propylene glycol or polyethylene glycol, dimethylsulfoxide, glycerol, ketals such as 2,2-dimethyl-l,3-dioxolane-4-methanol, ethers, poly(ethyleneglycol) 400, oils, fatty acids, fatty acid esters or glycerides, or acety lated fatty acid glycerides with or without the addition of a pharmaceutically acceptable surfactant such as a soap or a detergent, suspending agent such as pectin, carbomers, methylcellulose, hydroxypropylmethylcellulose, or carboxymethylcellulose, or emulsifying agents and other pharmaceutical adjuvants.
[0382] Oils, which can be used in parenteral formulations in some embodiments, include petroleum, animal oils, vegetable oils, or synthetic oils. Specific examples of oils include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral oil. Suitable fatty' acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters.
[0383] Suitable soaps for use in certain embodiments of parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts, and suitable detergents include (a) cationic detergents such as, for example, dimethyl dialkyl ammonium halides, and alkyl pyridinium halides, (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyd, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine oxides, fatty acid alkanolamides, and polyoxyethylenepolypropylene copolymers, (d) amphoteric detergents such as, for example, alkyl-p-aminopropionates, and 2-alkyl-imidazoline quaternary ammonium salts, and (e) mixtures thereof.
[0384] In some embodiments, the parenteral formulations will contain, for example, from 0.5% to 25% by weight of the therapeutic agent in solution. Preservatives and buffers may be used. In order to minimize or eliminate irritation at the site of injection, such compositions may contain one or more nonionic surfactants having, for example, a hydrophile-lipophile balance (HLB) of from 12 to 17. The quantity of surfactant in such formulations will ty pically range, for example, from 5% to 15% by weight. Suitable surfactants include polyethylene glycol, sorbitan, fatty acid esters such as sorbitan monooleate, and high molecular weight adducts of ethylene oxide with a hydrophobic base formed by the condensation of propylene oxide with propylene glycol. The parenteral formulations can be presented in unit-dose or multi-dose sealed containers, such as ampoules or vials, and can be stored in a freeze- dried (lyophilized) condition requiring only the addition of a sterile liquid excipient, for example, water for injections, immediately prior to use. Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets of the kind previously described.
[0385] In certain embodiments, injectable formulations are provided herein. The requirements for effective pharmaceutical carriers for injectable compositions are well-known to those of ordinary skill in the art (see, e.g., Pharmaceutics and Pharmacy Practice, I.B. Lippincott Company, Philadelphia, PA,
Banker and Chalmers, eds., pages 238-250 (1982), and ASHP Handbook on Injectable Drugs, Toissel, 4th ed, pages 622-630 (1986)).
[0386] In some embodiments, the therapeutic agents provided herein are formulated in tune-released, delayed release, or sustained release delivery systems such that the delivery of the composition occurs prior to, and with sufficient time to, cause sensitization of the site to be treated. Such systems can avoid repeated administrations of the therapeutic agent, thereby increasing convenience to the subject and the physician, and may be particularly suitable for certain composition embodiments provided herein. In one embodiment, the compositions of the invention are formulated such that they are suitable for extended release of the circRNA contained therein. Such extended-release compositions may be conveniently administered to a subject at extended dosing intervals. For example, in one embodiment, the compositions of the present invention are administered to a subject twice a day, daily or every other day. In an embodiment, the compositions of the present invention are administered to a subject twice a week, once a week, every ten days, every two weeks, every three weeks, every four weeks, once a month, every six weeks, every eight weeks, every three months, every four months, every six months, every eight months, every nine months or annually.
[0387] In some embodiments, a protein encoded by a circRNA is produced by a target cell for sustained amounts of time. For example, the protein may be produced for more than one hour, more than four, more than six, more than 12, more than 24, more than 48 hours, or more than 72 hours after administration. In some embodiments the polypeptide is expressed at a peak level six hours after administration. In some embodiments the expression of the polypeptide is sustained at least at a therapeutic level. In some embodiments, the polypeptide is expressed at least at a therapeutic level for more than one, more than four, more than six. more than 12, more than 24, more than 48, or more than 72 hours after administration. In some embodiments, the polypeptide is detectable at a therapeutic level in patient tissue (e.g., liver or lung). In some embodiments, the level of detectable polypeptide is from continuous expression from the circRNA composition over periods of time of more than one. more than four, more than six, more than 12, more than 24, more than 48, or more than 72 hours after administration.
[0388] In certain embodiments, a protein encoded by circRNA is produced at levels above normal physiological levels. The level of protein may be increased as compared to a control. In some embodiments, the control is the baseline physiological level of the polypeptide in a nonnal individual or in a population of normal individuals. In other embodiments, the control is the baseline physiological level of the polypeptide in an individual having a deficiency in the relevant protein or polypeptide or in a population of individuals having a deficiency in the relevant protein or polypeptide. In some embodiments, the control can be the normal level of the relevant protein or polypeptide in the individual to whom the composition is administered. In other embodiments, the control is the expression level of
die polypeptide upon other therapeutic intervention, e.g., upon direct injection of the corresponding polypeptide, at one or more comparable time points.
[0389] In certain embodiments, the levels of a protein encoded by a circRNA are detectable at 3 days, 4 days, 5 days, or 1 week or more after administration. Increased levels of protein may be observed in a tissue (e.g., liver or lung).
[0390] In some embodiments, the method yields a sustained circulation half-life of a protein encoded by a circRNA. For example, the protein may be detected for hours or days longer than the half-life observed via subcutaneous injection of the protein or mRNA encoding the protein. In some embodiments, the half-life of the protein is 1 day, 2 days, 3 days, 4 days, 5 days, or 1 week or more.
[0391] Many types of release delivery systems are available and known to those of ordinary skill in the art. They include polymer-based systems such as poly(lactide-glycolide), copolyoxalates, polycaprolactones, polyesteramides, polyorthoesters, polyhydroxybutyric acid, and polyanhydrides. Microcapsules of the foregoing polymers containing drugs are described in, for example, U.S. Patent 5,075.109. Delivery systems also include non-polymer systems that are lipids including sterols such as cholesterol, cholesterol esters, and fatty' acids or neutral fats such as mono-di-and tri-glycerides; hydrogel release systems; sylastic systems; peptide based systems: wax coatings; compressed tablets using conventional binders and excipients; partially fused implants; and the like. Specific examples include but are not limited to: (a) erosional systems in which the active composition is contained in a form within a matrix such as those described in U.S. Patents 4,452,775, 4,667,014, 4,748,034, and 5,239,660 and (b) diffusional systems in which an active component permeates at a controlled rate from a polymer such as described in U.S. Patents 3,832,253 and 3,854,480. In addition, pump-based hardware delivery systems can be used, some of which are adapted for implantation.
[0392] In some embodiments, the therapeutic agent can be conjugated either directly or indirectly through a linking moiety to a targeting moiety. Methods for conjugating therapeutic agents to targeting moictics is known in the art. See, for instance, Wadwa ct al., J. Drug Targeting 3:111 (1995) and U.S. Patent 5,087,616.
[0393] In some embodiments, the therapeutic agents provided herein are formulated into a depot form, such that the manner in which the therapeutic agent is released into the body to which it is administered is controlled with respect to time and location within the body (see, for example, U.S. Patent 4.450, 150). Depot forms of therapeutic agents can be, for example, an implantable composition comprising the therapeutic agents and a porous or non-porous material, such as a poly mer, wherein the therapeutic agents are encapsulated by or diffused throughout the material and/or degradation of the non-porous material. The depot is then implanted into the desired location within the body and the therapeutic agents are released from the implant at a predetermined rate.
[0394] The present disclosure also contemplates the discriminatory targeting of target cells and tissues by both passive and active targeting means. The phenomenon of passive targeting exploits tire natural distributions patterns of a transfer vehicle in vivo without relying upon the use of additional excipients or means to enhance recognition of the transfer vehicle by target cells. For example, transfer vehicles which are subject to phagocytosis by the cells of the reticulo-endothelial system are likely to accumulate in the liver or spleen, and accordingly may provide a means to passively direct the delivery of the subject compositions to such target cells.
[0395] Alternatively, the present disclosure contemplates active targeting, which involves the use of targeting moieties that may be bound (either covalently or non-covalently) to the lipid nanoparticle to encourage localization of such at certain target cells or target tissues. For example, targeting may be mediated by the inclusion of one or more endogenous targeting moieties in or on the lipid nanoparticle to encourage distribution to the target cells or tissues. Recognition of the targeting moiety by the target tissues actively facilitates tissue distribution and cellular uptake of the lipid nanoparticle and/or its contents in the target cells and tissues (e.g., the inclusion of an apolipoprotein-E targeting ligand encourages recognition and binding of the lipid nanoparticle o endogenous low density lipoprotein receptors expressed by hepatocytes). As provided herein, the composition can comprise a moiety capable of enhancing affinity of the composition to the target cell. Targeting moieties may be linked to the outer layer of the lipid nanoparticle during formulation or post-formulation. In addition, some lipid nanoparticle formulations may employ fusogenic polymers such as PEAA. hemagluttinin, other lipopeptides (see U.S. patent application Ser. No. 08/835,281, and 60/083.294, which are incorporated herein by reference) and other features useful for in vivo and/or intracellular delivery. In some embodiments, the compositions of the present disclosure demonstrate improved transfection efficacies, and/or demonstrate enhanced selectivity’ towards target cells or tissues of interest. Contemplated therefore are compositions which comprise one or more moieties (e.g., peptides, aptamers, oligonucleotides, vitamins or other molecules) that are capable of enhancing the affinity of the compositions and their nucleic acid contents for the target cells or tissues. Suitable moieties may optionally be bound or linked to the surface of the nanoparticle. In some embodiments, the targeting moiety’ may span the surface of a nanoparticle or be encapsulated within tire nanoparticle. Suitable moieties and are selected based upon their physical, chemical or biological properties (e.g., selective affinity and/or recognition of target cell surface markers or features). Cell-specific target sites and their corresponding targeting ligand can vary’ widely. Suitable targeting moieties are selected such that the unique characteristics of a target cell are exploited, thus allowing the composition to discriminate between target and non-target cells. For example, compositions of the present disclosure may include surface markers (e.g., apolipoprotein-B (APOB) or apolipoprotein-E (APOE)) that selectively enhance recognition of, or affinity' to hepatocytes (e.g., by receptor-mediated recognition of and binding to such surface markers). As an example, the use of galactose as a targeting moiety would be expected to direct
the compositions of the present disclosure to parenchymal hepatocytes, or alternatively the use of mannose containing sugar residues as a targeting ligand would be expected to direct the compositions of the present disclosure to liver endothelial cells (e.g., mannose containing sugar residues that may bind preferentially to the asialoglycoprotein receptor present in hepatocytes). (See Hilleiy A. M., et al. “Drug Delivery and Targeting: For Pharmacists and Pharmaceutical Scientists” (2002) Taylor & Francis, Inc.) The presentation of such targeting moieties that have been conjugated to moieties present in the lipid nanoparticle composition therefore facilitate recognition and uptake of the compositions of the present disclosure in target cells and tissues. Examples of suitable targeting moieties include one or more peptides, proteins, aptamers, vitamins and oligonucleotides.
[0396] In particular embodiments, a LNP composition comprises a targeting moiety. In some embodiments, the targeting moiety mediates receptor-mediated endocytosis selectively into a specific population of cells. In some embodiments, the targeting moiety is operably connected, or linked, to the transfer vehicle. In some embodiments, the targeting moiety is capable of binding to an immune cell antigen. In some embodiments, the targeting moiety is capable of binding to a T cell antigen. Exemplary T cell antigens include, but are not limited to, CD2, CD3, CD5, CD7, CD8, CD4, beta7 integrin, beta2ingetrin, and ClqR. In some embodiments, the targeting moiety is capable of binding to a NK, NKT. or macrophage antigen. In some embodiments, the targeting moiety is capable of binding to a protein selected from CD3, CD4, CD8. PD-1, 4-1BB. and CD2. In some embodiments, the targeting moiety is a single chain Fv (scFv) fragment, nanobody, peptide, peptide-based macrocycle, minibody, heavy chain variable region, light chain variable region or fragment thereof. In some embodiments, the targeting moiety is selected from T-cell receptor motif antibodies, T-cell a chain antibodies, T-cell β chain antibodies. T-cell y chain antibodies. T-cell 5 chain antibodies. CCR7 antibodies, CD3 antibodies, CD4 antibodies. CD5 antibodies, CD7 antibodies, CD8 antibodies, CDl lb antibodies, CDl lc antibodies. CD 16 antibodies, CD 19 antibodies, CD20 antibodies. CD21 antibodies, CD22 antibodies, CD25 antibodies, CD28 antibodies, CD34 antibodies. CD35 antibodies, CD40 antibodies, CD45RA antibodies, CD45RO antibodies, CD52 antibodies, CD56 antibodies, CD62L antibodies. CD68 antibodies, CD80 antibodies, CD95 antibodies, CD117 antibodies, CD127 antibodies, CD133 antibodies, CD137 (4-1BB) antibodies, CD163 antibodies, F4/80 antibodies, IL-4Ra antibodies, Sca-1 antibodies, CTLA-4 antibodies, GITR antibodies GARP antibodies, LAP antibodies, granzyme B antibodies, LFA-1 antibodies, transferrin receptor antibodies, and fragments thereof. In some embodiments, the targeting moiety is a small molecule binder of an ectoenzy me on lymphocytcs. Small molecule binders of ectoenzy mes include A2A inhibitors CD73 inhibitors, CD39 or adesine receptors A2aR and A2bR. Potential small molecules include AB928.
[0397] Where it is desired to deliver a nucleic acid to an immune cell, the immune cell represents the target cell. In some embodiments, the compositions of the present disclosure transfect the target cells on a discriminatory basis (i.e., do not transfect non-target cells). The compositions of the present
disclosure may also be prepared to preferentially target a variety of target cells, which include, but are not limited to, T cells, B cells, macrophages, and dendritic cells.
[0398] In some embodiments, the target cells are deficient in a protein or enzyme of interest. For example, where it is desired to deliver a nucleic acid to a hepatocyte, the hepatocyte represents the target cell. In some embodiments, the compositions of the present disclosure transfect the target cells on a discriminatory basis (i.e., do not transfect non-target cells). The compositions of the present disclosure may also be prepared to preferentially target a variety of target cells, which include, but arc not limited to, hepatocytes, epithelial cells, hematopoietic cells, epithelial cells, endothelial cells, lung cells, bone cells, stem cells, mesenchymal cells, neural cells (e.g., meninges, astrocytes, motor neurons, cells of the dorsal root ganglia and anterior hom motor neurons), photoreceptor cells (e.g., rods and cones), retinal pigmented epithelial cells, secretory cells, cardiac cells, adipocytes, vascular smooth muscle cells, cardiomyocytes, skeletal muscle cells, beta cells, pituitary cells, synovial lining cells, ovarian cells, testicular cells, fibroblasts, B cells, T cells, reticulocytes, leukocytes, granulocytes and tumor cells.
[0399] The compositions of the present disclosure may be prepared to preferentially distribute to target cells such as in the heart, lungs, kidneys, liver, and spleen. In some embodiments, the compositions of the present disclosure distribute into the cells of the liver or spleen to facilitate the delivery and the subsequent expression of the circRNA comprised therein by the cells of the liver (e.g., hepatocytes) or the cells of spleen (e.g., immune cells). The targeted cells may function as a biological “reservoir” or “depot” capable of producing, and systemically excreting a functional protein or enzyme. Accordingly, in one embodiment of the present disclosure the transfer vehicle may target hepatocytes or immune cells and/or preferentially distribute to the cells of the liver or spleen upon deliver,'. In an embodiment, following transfection of the target hepatocytes or immune cells, the circRNA loaded in the nanoparticle are translated and a functional protein product is produced, excreted and systemically distributed. In other embodiments, cells other than hepatocytes (e.g., lung, spleen, heart, ocular, or cells of the central nervous system) can serve as a depot location for protein production.
[0400] In one embodiment, the compositions of the present disclosure facilitate a subject's endogenous production of one or more functional proteins and/or enzymes. In an embodiment of the present disclosure, the lipid nanoparticles comprise circRNA which encode a deficient protein or enzyme. Upon distribution of such compositions to the target tissues and the subsequent transfection of such target cells, the exogenous circRNA loaded into the nanoparticle may be translated in vivo to produce a functional protein or enzyme encoded by the exogenously administered circRNA (e.g., a protein or enzyme in which the subject is deficient). Accordingly, the compositions of the present disclosure exploit a subject's ability to translate exogenously- or recombinantly-prepared circRNA to produce an endogenously-translated protein or enzyme, and thereby produce (and where applicable excrete) a functional protein or enzyme. The expressed or translated proteins or enzymes may also be
characterized by the in vivo inclusion of native post-translational modifications which may often be absent in recomb inantly -prepared proteins or enzymes, thereby further reducing the immunogenicity of the translated protein or enzyme.
[0401] The administration of circRNA encoding a deficient protein or enzyme avoids the need to deliver the nucleic acids to specific organelles within a target cell. Rather, upon transfection of a target cell and deliver}' of the nucleic acids to the cytoplasm of the target cell, the circRNA contents of a transfer vehicle may be translated and a functional protein or enzy me expressed.
[0402] In some embodiments, a circular RNA comprises one or more miRNA binding sites. In some embodiments, a circular RNA comprises one or more miRNA binding sites recognized by miRNA present in one or more non-target cells or non-target cell types (e.g., Kupffer cells or hepatic cells) and not present in one or more target cells or target cell types (e.g., hepatocytes or T cells). In some embodiments, a circular RNA comprises one or more miRNA binding sites recognized by miRNA present in an increased concentration in one or more non-target cells or non-target cell types (e.g., Kupffer cells or hepatic cells) compared to one or more target cells or target cell types (e.g., hepatocytes or T cells). miRNAs are thought to function by pairing with complementary' sequences within RNA molecules, resulting in gene silencing.
[0403] In some embodiments, the compositions of the present disclosure transfect or distribute to target cells on a discriminatory basis (i.e., do not transfect non-target cells). The compositions of the present disclosure may also be prepared to preferentially target a variety of target cells, which include, but arc not limited to, hepatocytes, epithelial cells, hematopoietic cells, epithelial cells, endothelial cells, lung cells, bone cells, stem cells, mesenchymal cells, neural cells (e.g., meninges, astrocytes, motor neurons, cells of the dorsal root ganglia and anterior horn motor neurons), photoreceptor cells (e.g., rods and cones), retinal pigmented epithelial cells, secretory cells, cardiac cells, adipocytes, vascular smooth muscle cells, cardiomyocytes, skeletal muscle cells, beta cells, pituitary' cells, synovial lining cells, ovarian cells, testicular cells, fibroblasts, B cells, T cells, reticulocytes, leukocytes, granulocytes and tumor cells.
7. THERAPEUTIC METHODS
[0404] In certain aspects, provided herein is a method of producing a protein of interest in a subject in need thereof by' introducing or administering an effective amount of a pharmaceutical composition described herein comprising at least one lipid nanoparticle described herein.
[0405] In certain aspects, provided herein is a method of treating and/or preventing a condition comprising administering an effective amount of a pharmaceutical composition described herein comprising at least one LNP as described herein.
[0406] In certain embodiments, the pharmaceutical compositions described herein are co-administered with one or more additional therapeutic agents (e.g., in the same pharmaceutical composition or in separate pharmaceutical compositions). In some embodiments, die pharmaceutical compositions provided herein can be administered first and the one or more additional therapeutic agents can be administered second, or vice versa. Alternatively, die pharmaceutical compositions provided herein and the one or more additional therapeutic agents can be administered simultaneously.
[0407] In some embodiments, the subject is a mammal. In some embodiments, the mammal referred to herein can be any mammal, including, but not limited to, mammals of the order Rodentia, such as mice and hamsters, or mammals of the order Logomorpha, such as rabbits. The mammals may be from the order Carnivora, including Felines (cats) and Canines (dogs). The mammals may be from the order Artiodactyla, including Bovines (cows) and Swines (pigs), or of the order Perssodactyla, including Equines (horses). The mammals may be of the order Primates, Ceboids, or Simoids (monkeys) or of the order Anthropoids (humans and apes). In some embodiments, the mammal is a human.
[0408] In some embodiments, provided herein is a method of vaccinating a subject by introducing or administering an effective amount of a pharmaceutical composition described herein comprising at least one lipid nanoparticle described herein.
[0409] In some embodiments, the method of vaccinating comprises administering an effective amount of an antigen comprising a viral polypeptide from an adenovirus; Herpes simplex, type 1; Herpes simplex, type 2; encephalitis virus, papillomavirus, Varicella-zoster virus; Epstein-barr virus; Human cytomegalovirus; Human herpes virus, type 8; Human papillomavirus; BK virus; JC virus; Smallpox; polio virus; Hepatitis B virus; Human bocavirus; Parvovirus B19; Human astrovirus; Norwalk virus; coxsackievirus; hepatitis A virus; poliovirus; rhinovirus; Severe acute respiratory syndrome virus; Hepatitis C virus; Yellow Fever virus; Dengue virus; West Nile virus; Rubella virus; Hepatitis E virus; Human Immunodeficiency virus (HIV); Influenza virus; Guanarito virus; Junin virus; Lassa virus; Machupo virus; Sabia virus: Crimean-Congo hemorrhagic fever virus; Ebola virus; Marburg virus; Measles virus; Mumps virus; Parainfluenza virus; Respiratory syncytial virus; Human metapneumo virus; Hendra virus; Nipah virus; Rabies virus; Hepatitis D; Rotavirus; Orbivirus; Coltivirus; Banna virus; Human Enterovirus; Hanta virus; West Nile virus; Middle East Respiratory Syndrome Corona Virus; Japanese encephalitis virus; Vesicular exanthemavirus; SARS-CoV-2; Eastern equine encephalitis, or a combination of any two or more of the foregoing.
[0410] In some embodiments, provided herein is a method of treating an autoimmune disorder in a subject by introducing or administering an effective amount of a pharmaceutical composition described herein comprising at least one lipid nanoparticle described herein.
[0411] In some embodiments, provided herein is a method of treating cancer in a subject by introducing or administering an effective amount of a pharmaceutical composition described herein comprising at least one lipid nanoparticle described herein.
[0412] In some embodiments, the circular RNA construct encodes a CAR. The CARs have biological activity, e.g., ability to recognize an antigen, e.g., CD19, HER2 , or BCMA, such that the CAR, when expressed by a cell, is able to mediate an immune response against the cell expressing the antigen, e.g., CD19, HER2 , or BCMA, for which the CAR is specific.
[0413] Adoptive T-cell immunotherapy is a rapidly growing field, in particular in cancer treatments. In general, chimeric antigen receptor (CAR) T cell or “CAR-T” engagement of CD19-expressing cancer cells results in T-cell activation, proliferation and secretion of inflammatory cytokines and chemokines resulting in tumor cell lysis. However, while CAR-T therapies have become an important tool in cancer treatments, they have toxic side effects and involve complex procedures. Treatment with CAR-T can lead to a large and rapid release of cytokines into the blood and can cause cytokine release syndrome (CRS) or CAR-T cell-related encephalopathy syndrome (CRES), also referred to as nemotoxicity associated with CAR-T. CRS is the most common and well-described toxicity' associated yvith CAR-T therapy, occurring in over 90% of patients at any grade and is characterized by high fever, hypotension, hypoxia and/ or multiple organ toxicity and can lead to death. Neurotoxicity is characterized by damage to nervous tissue that can cause tremors, encephalopathy, dizziness or seizures. Additionally, prior to infusion, the patients generally undergo lymphodepletion. Lymphodepletion is known to increase CAR- T cell expansion and enhanced efficacy of infused CAR-T cells by, for example, altering the tumor phenotype and microenvironment. However, lymphodepletion agents often cause side effects to the patients. For example, lymphodepletion can cause neutropenia, anemia, thrombocytopenia, and immunosuppression, leading to a greater risk of infection, along with other toxicities. In addition to the toxicities associated with targeted CAR-T therapies, there are procedures, specialized equipment, and costs involved in producing the modified lymphocytes. CAR-T therapies require an assortment of protocols to isolate, genetically modify, and selectively' expand the redirected cells before infusing them back into the patient.
[0414] In some embodiments, the subject has a cancer selected from the group consisting of: acute myeloid leukemia (AML); alveolar rhabdomyosarcoma; B cell malignancies; bladder cancer (e.g.. bladder carcinoma); bone cancer; brain cancer (e.g.. medulloblastoma and glioblastoma multiforme); breast cancer; cancer of the anus, anal canal, or anorectum; cancer of the eye; cancer of the intrahepatic bile duct; cancer of the joints; cancer of the neck; gallbladder cancer; cancer of the pleura; cancer of the nose, nasal cavity, or middle ear; cancer of the oral cavity; cancer of the vulva; chronic lymphocytic leukemia; chronic myeloid cancer; colon cancer; esophageal cancer, cervical cancer; fibrosarcoma; gastrointestinal carcinoid tumor; head and neck cancer (e.g., head and neck squamous cell carcinoma);
Hodgkin lymphoma; hypopharynx cancer; kidney cancer; larynx cancer; leukemia; liquid tumors; lipoma; liver cancer; lung cancer (e g., non-small cell lung carcinoma, lung adenocarcinoma, and small cell lung carcinoma); lymphoma; mesothelioma; mastocytoma; melanoma; multiple myeloma; nasopharynx cancer; non-Hodgkin lymphoma; B-chronic lymphocytic leukemia; hairy cell leukemia; Burkitt's lymphoma; ovarian cancer; pancreatic cancer; cancer of tire peritoneum; cancer of the omentum; mesentery cancer; pharynx cancer; prostate cancer; rectal cancer; renal cancer; skin cancer; small intestine cancer: soft tissue cancer; solid tumors; synovial sarcoma; gastric cancer; teratoma; testicular cancer; thyroid cancer; and ureter cancer.
[0415] In some embodiments, the subject has an autoimmune disorder selected from scleroderma, Grave's disease, Crohn's disease, Sjogren's disease, multiple sclerosis, Hashimoto's disease, psoriasis, myasthenia gravis, autoimmune polyendocrinopathy syndromes, Type I diabetes mellitus (TIDM), autoimmune gastritis, autoimmune uveoretinitis, polymyositis, colitis, thyroiditis, and the generalized autoimmune diseases typified by human Lupus.
8. DEFINITIONS
[0416] As used herein the term “storage stable” refers to a composition of empty LNPs or a composition of loaded LNPs (as described herein), having a polydispersity’ index (PDI) that increases by less than 25%, such as less than 20%, less than 15%, less than 10%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% after storage of the composition at 4 °C or -80 °C for at least 4 weeks, such as at least 1 month, at least 2 months, at least 3 months, at least 6 months, at least 8 months, or at least 1 year. The term “storage stable” also refers to a composition of loaded LNPs (as described herein), having less than 25% decrease in encapsulation efficiency, such as less than 20%, less than 15%, less than 10%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% after storage of the composition at 4 °C or -80 °C for at least 4 weeks, such as at least 1 month, at least 2 months, at least 3 months, at least 6 months, at least 8 months, or at least 1 year.
[0417] As used herein, “encapsulation”, “encapsulated”, “loaded”, and “associated with " may refer to complete, substantial, or partial enclosure, confinement, surrounding, or encasement. As used herein, “encapsulation” or “association with” may refer to the process of at least partially confining an individual nucleic acid molecule within a nanoparticle and/or establishing a physiochemical relationship betw een an individual nucleic acid molecule and a nanoparticle.
[0418] As used herein, an “empty lipid nanoparticle” or “empty’ LNPs” may refer to a LNP that is substantially free of a therapeutic or prophylactic agent. As used herein, an “empty’ LNPs” may refer to a LNP that is substantially free of a nucleic acid. As used herein, an “empty LNPs” may refer to a nanoparticle that consists substantially of only lipid components.
[0419] As used herein, the terms “circRNA” or “circular polyribonucleotide” or “circular RNA” or “oRNA” are used interchangeably and refers to a single-stranded RNA polynucleotide wherein the 3’ and 5’ ends that are normally present in a linear RNA polynucleotide have been joined together.
[0420] As used herein, the term “DNA template” refers to a DNA sequence capable of transcribing a linear RNA polynucleotide. For example, but not intending to be limiting, a DNA template may include a DNA vector, PCR product or plasmid.
[0421] As used herein, the term “3’ group I intron fragment” refers to a sequence with 75% or higher similarity to the 3 ’-proximal end of a natural group I intron including the splice site dinucleotide and optionally a stretch of natural exon sequence. In some embodiments, a circular RNA comprises a post splicing 3’ group I intron fragment. In some embodiments, the post splicing 3’ group I intron fragment in the circular RNA is a post splicing stretch of exon sequence. In some embodiments, the circular RNA further comprises a desired expression sequence, and tire post splicing stretch of exon sequence is (e.g., designed) to be a portion of the desired expression sequence, contiguous with die desired expression sequence, and/or in frame with the desired expression sequence.
[0422] As used herein, the term “3’ group II intron fragment” refers to a sequence with 75% or higher similarity to the 3 ‘-proximal end of a natural group II intron including the splice site dinuclcotidc and optionally a stretch of natural exon sequence. In some embodiments, a circular RNA comprises a post splicing 3’ group II intron fragment. In some embodiments, the post splicing 3' group II intron fragment in the circular RNA is a post splicing stretch of exon sequence. In some embodiments, the circular RNA further comprises a desired expression sequence, and the post splicing stretch of exon sequence is (e.g., designed) to be a portion of the desired expression sequence, contiguous with the desired expression sequence, and/or in frame with the desired expression sequence.
[0423] As used herein, the term “5’ group I intron fragment” refers to a sequence with 75% or higher similarity to the 5 ’-proximal end of a natural group I intron including the splice site dinucleotide and optionally a stretch of natural exon sequence. In some embodiments, a circular RNA comprises a post splicing 5’ group I intron fragment. In some embodiments, the post splicing 5’ group I intron fragment in the circular RNA is a post splicing stretch of exon sequence. In some embodiments, tire circular RNA further comprises a desired expression sequence, and die post splicing stretch of exon sequence is (e.g., designed) to be a portion of the desired expression sequence, contiguous with the desired expression sequence, and/or in frame with the desired expression sequence.
[0424] As used herein, the term “5’ group II intron fragment” refers to a sequence with 75% or higher similarity to the 5 ‘-proximal end of a natural group II intron including the splice site dinucleotide and optionally a stretch of natural exon sequence. In some embodiments, a circular RNA comprises a post splicing 5’ group II intron fragment. In some embodiments, the post splicing 5' group II intron fragment in the circular RNA is a post splicing stretch of exon sequence. In some embodiments, the circular RNA
further comprises a desired expression sequence, and tire post splicing stretch of exon sequence is (e.g., designed) to be a portion of the desired expression sequence, contiguous with die desired expression sequence, and/or in frame with the desired expression sequence.
[0425] In some embodiments, provided herein are circular RNA polynucleotides comprising a post splicing 3’ group I or II intron fragment (e.g., a stretch of exon sequence), optionally a first spacer, an IRES, an expression sequence, optionally a second spacer, and a post splicing 5’ group I or II intron fragment (e.g., a stretch of exon sequence).
[0426] As used herein, the term “permutation site” refers to the site in a group I or II intron where a cut is made prior to permutation of the intron. This cut generates 3 ’ and 5 ’ group I or II intron fragments that are permuted to be on either side of a stretch of precursor RNA to be circularized.
[0427] As used herein, the term “splice site” refers to a dinucleotide that is partially or fully included in a group I intron and between which a phosphodiester bond is cleaved during RNA circularization. (As used herein, “splice site” refers to the dinucleotide or dinucleotides between which cleavage of the phosphodiester bond occurs during a splicing reaction. A “5’ splice site” refers to the natural 5’ dinucleotide of the intron e.g., group I intron, while a “3’ splice site” refers to the natural 3’ dinucleotide of the intron).
[0428] As used herein, the term “expression sequence” refers to a nucleic acid sequence that encodes a product, e.g., a peptide or polypeptide, regulatory nucleic acid, or non-coding nucleic acid. An exemplary expression sequence that codes for a peptide or polypeptide can comprise a plurality of nucleotide triads, each of which can code for an amino acid and is termed as a “codon.”
[0429] As used herein, “coding element” or “coding region” is region located within the expression sequence and encodings for one or more proteins or polypeptides (e.g., therapeutic protein).
[0430] As used herein, a "noncoding element” or “non-coding nucleic acid” is a region located within the expression sequence. This sequence, but itself does not encode for a protein or polypeptide, but may have other regulatory functions, including but not limited, allow the overall polynucleotide to act as a biomarker or adjuvant to a specific cell.
[0431] As used herein, the term “therapeutic protein” refers to any protein that, when administered to a subject directly or indirectly in the form of a translated nucleic acid, has a therapeutic, diagnostic, and/or prophylactic effect and/or elicits a desired biological and/or pharmacological effect.
[0432] As used herein, the term “immunogenic” refers to a potential to induce an immune response to a substance. An immune response may be induced when an immune system of an organism or a certain type of immune cells is exposed to an immunogenic substance. The term “non-immunogenic” refers to a lack of or absence of an immune response above a detectable threshold to a substance. No immune response is detected when an immune system of an organism or a certain type of immune cells is
exposed to a non-immunogenic substance. In some embodiments, a non-immunogenic circular polyribonucleotide as provided herein, does not induce an immune response above a pre-determined threshold when measured by an immunogenicity assay. In some embodiments, no innate immune response is detected when an immune system of an organism or a certain type of immune cells is exposed to a non-immunogenic circular polyribonucleotide as provided herein. In some embodiments, no adaptive immune response is detected when an immune system of an organism or a certain ty pe of immune cell is exposed to a non-immunogenic circular polyribonucleotide as provided herein.
[0433] As used herein, the term “circularization efficiency” refers to a measurement of resultant circular polyribonucleotide as compared to its linear starting material.
[0434] As used herein, the term “translation efficiency” refers to a rate or amount of protein or peptide production from a ribonucleotide transcript. In some embodiments, translation efficiency can be expressed as amount of protein or peptide produced per given amount of transcript that codes for the protein or peptide.
[0435] The term “nucleotide” refers to a ribonucleotide, a deoxyribonucleotide, a modified form thereof, or an analog thereof. Nucleotides include species that comprise purines, e.g., adenine, hypoxanthine, guanine, and their derivatives and analogs, as well as pyrimidines, e.g., cytosine, uracil, thymine, and their derivatives and analogs. Nucleotide analogs include nucleotides having modifications in the chemical structure of the base, sugar and/or phosphate, including, but not limited to, 5 ’-position pyrimidine modifications, 8’ -position purinc modifications, modifications at cytosine exocyclic amines, and substitution of 5-bromo-uracil; and 2’-position sugar modifications, including but not limited to, sugar-modified ribonucleotides in which the 2’-OH is replaced by a group such as an H, OR, R, halo, SH, SR, NH2, NHR, NR2, or CN, wherein R is an alkyl moiety as defined herein. Nucleotide analogs are also meant to include nucleotides with bases such as inosine, queuosine, xanthine; sugars such as 2’-methyl ribose; non-natural phosphodiester linkages such as methylphosphonate, phosphorothioate and peptide linkages. Nucleotide analogs include 5- methoxyuridine. 1 -methylpseudouridine, and 6-methyladenosine.
[0436] The term “nucleic acid” and “polynucleotide” are used interchangeably herein to describe a polymer of any length, e.g., greater than 2 bases, greater than 10 bases, greater than 100 bases, greater than 500 bases, greater than 1000 bases, or up to 10.000 or more bases, composed of nucleotides, e.g., deoxyribonucleotides or ribonucleotides, and may be produced enzymatically or synthetically (e.g., as described in U.S. Pat. No. 5,948,902 and the references cited therein), which can hybridize with naturally occurring nucleic acids in a sequence specific manner analogous to that of two naturally occurring nucleic acids, e.g., can participate in Watson-Crick base pairing interactions. Naturally occurring nucleic acids are comprised of nucleotides including guanine, cytosine, adenine, thymine, and uracil (G, C. A, T. and U respectively).
[0437] The terms ‘’ribonucleic acid” and “RNA” as used herein mean a polymer composed of ribonucleotides.
[0438] The tenns “deoxyribonucleic acid” and “DNA” as used herein mean a polymer composed of deoxy ribonucleotides.
[0439] “Isolated” or “purified” generally refer to isolation of a substance (for example, in some embodiments, a compound, a polynucleotide, a protein, a polypeptide, a polynucleotide composition, or a polypeptide composition) such that the substance comprises a significant percent (e.g., greater than 1%, greater than 2%. greater than 5%, greater than 10%, greater than 20%, greater than 50%, or more, usually up to 90%-100%) of the sample in which it resides. In certain embodiments, a substantially purified component comprises at least 50%, 80%-85%, or 90%-95% of the sample. Techniques for purifying polynucleotides and polypeptides of interest arc well-known in die art and include, for example, ion-exchange chromatography, affinity chromatography and sedimentation according to density. Generally, a substance is purified when it exists in a sample in an amount, relative to other components of the sample, that is more than as it is found naturally.
[0440] The terms “duplexed,” “double-stranded,” or “hybridized” as used herein refer to nucleic acids formed by hybridization of tw o single strands of nucleic acids containing complementary sequences. In most cases, genomic DNA is double-stranded. Sequences can be fully complementary or partially complementary.
[0441] As used herein, “unstructured” with regard to RNA refers to an RNA sequence that is not predicted by the RNAFold software or similar predictive tools to form a structure (e.g., a hairpin loop) with itself or other sequences in the same RNA molecule. In some embodiments, unstructured RNA can be functionally characterized using nuclease protection assay s.
[0442] As used herein, “structured” with regard to RNA refers to an RNA sequence that is predicted by the RNAFold software or similar predictive tools to form a structure (e.g., a hairpin loop) w ith itself or other sequences in the same RNA molecule.
[0443] As used herein, two “duplex sequences,” “duplex region,” “duplex regions.” “homology arms.” or “homology regions” may be any’ two regions that are thermodynamically favored to cross-pair in a sequence specific interaction. In some embodiments, two duplex sequences, duplex regions, homology arms, or homology regions, share a sufficient level of sequence identity’ to one another’s reverse complement to act as substrates for a hybridization reaction. As used herein polynucleotide sequences have “homology” when they are either identical or share sequence identity to a reverse complement or “complementary” sequence. The percent sequence identity between a homology region and a counterpart homology’ region’s reverse complement can be any percent of sequence identity’ that allows for hybridization to occur. In some embodiments, an internal duplex region of an inventive
polynucleotide is capable of forming a duplex with another internal duplex region and does not form a duplex with an external duplex region.
[0444] As used herein, an “affinity sequence” or “affinity tag” is a region of polynucleotide sequences polynucleotide sequence ranging from 1 nucleotide to hundreds or thousands of nucleotides containing a repeated set of nucleotides for the purposes of aiding purification of a polynucleotide sequence. For example, an affinity sequence may comprise, but is not limited to, a poly A or poly AC sequence.
[0445] As used herein, a “spacer” refers to a region of a polynucleotide sequence ranging from 1 nucleotide to hundreds or thousands of nucleotides separating two other elements along a polynucleotide sequence. The sequences can be defined or can be random. A spacer is typically noncoding. In some embodiments, spacers include duplex regions.
[0446] Linear nucleic acid molecules are said to have a “5’-terminus” (5’ end) and a “3’-terminus” (3’ end) because nucleic acid phosphodiester linkages occur at the 5’ carbon and 3’ carbon of the sugar moieties of the substituent mononucleotides. The end nucleotide of a polynucleotide at which a new linkage would be to a 5’ carbon is its 5’ terminal nucleotide. The end nucleotide of a polynucleotide at which a new linkage would be to a 3’ carbon is its 3’ terminal nucleotide. A terminal nucleotide, as used herein, is the nucleotide at the end position of the 3’- or 5 ’-terminus.
[0447] As used herein, a “leading untranslated sequence” is a region of polynucleotide sequences ranging from 1 nucleotide to hundreds of nucleotides located at the upmost 5' end of a polynucleotide sequence. The sequences can be defined or can be random. A leading untranslated sequence is noncoding.
[0448] As used herein, a “leading untranslated sequence” is a region of polynucleotide sequences ranging from 1 nucleotide to hundreds of nucleotides located at the downmost 3' end of a polynucleotide sequence. The sequences can be defined or can be random. A leading untranslated sequence is noncoding.
[0449] “Transcription” means the formation or synthesis of an RNA molecule by an RNA polymerase using a DNA molecule as a template. The invention is not limited with respect to the RNA polymerase that is used for transcription. For example, in some embodiments, a T7-type RNA polymerase can be used.
[0450] “Translation” means the formation of a polypeptide molecule by a ribosome based upon an RNA template.
[0451] It is to be understood that the terminology used herein is for the purpose of describing particular embodiments only and is not intended to be limiting. As used in this specification and the appended claims, the singular fonns “a,” “an,” and “the” include plural referents unless the content clearly dictates otherwise. Thus, for example, reference to “a cell” includes combinations of two or more cells, or entire
cultures of cells; reference to “a polynucleotide” includes, as a practical matter, many copies of that polynucleotide. Unless specifically stated or obvious from context, as used herein, the term “or” is understood to be inclusive. Unless defined herein and below in the reminder of die specification, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which the invention pertains.
[0452] As used herein, the term “encode” refers broadly to any process whereby die information in a polymeric macromolecule is used to direct the production of a second molecule that is different from die first. The second molecule may have a chemical structure diat is different from the chemical nature of the first molecule.
[0453] By “co-administering” is meant administering a therapeutic agent provided herein in conjunction with one or more additional therapeutic agents sufficiently close in time such that the therapeutic agent provided herein can enhance the effect of the one or more additional therapeutic agents, or vice versa.
[0454] The terms “treat,” and “prevent” as well as words stemming therefrom, as used herein, do not necessarily imply 100% or complete treatment or prevention. Rather, there are varying degrees of treatment or prevention of which one of ordinary' skill in the art recognizes as having a potential benefit or therapeutic effect. The treatment or prevention provided by the method disclosed herein can include treatment or prevention of one or more conditions or symptoms of the disease. Also, for purposes herein, “prevention” can encompass delaying the onset of the disease, or a symptom or condition thereof.
[0455] As used herein, an “internal ribosome entry site” or “IRES” refers to an RNA sequence or structural element ranging in size from 10 nt to 1000 nt or more, capable of initiating translation of a polypeptide in the absence of a typical RNA cap structure. An IRES is typically 500 nt to 700 nt in length.
[0456] As used herein, “aptamer” refers in general to either an oligonucleotide of a single defined sequence or a mixture of said nucleotides, wherein the mixture retains the properties of binding specifically to the target molecule (e.g., eukaryotic initiation factor, 40S ribosome. polyC binding protein, polyA binding protein, polypyrimidine tract-binding protein, argonaute protein family, Heterogeneous nuclear ribonucleoprotein K and La and related RNA-binding protein). Thus, as used herein “aptamer” denotes both singular and plural sequences of nucleotides, as defined hereinabove. The term “aptamer” is meant to refer to a single- or double-stranded nucleic acid which is capable of binding to a protein or other molecule. In general, aptamers preferably comprise 10 to 100 nucleotides, preferably 15 to 40 nucleotides, more preferably 20 to 40 nucleotides, in that oligonucleotides of a length that falls within these ranges are readily prepared by conventional techniques. Optionally, aptamers can further comprise a minim um of approximately 6 nucleotides, preferably 10, and more preferably 14 or 15 nucleotides, that are necessary to effect specific binding.
[0457] Aii “eukaryotic initiation factor” or “elF” refers to a protein or protein complex used in assembling an initiator tRNA, 40S and 60S ribosomal subimits required for initiating eukaryotic translation.
[0458] As used herein, an “internal ribosome entry site” or “IRES” refers to an RNA sequence or structural element ranging in size from 10 nt to 1000 nt or more, capable of initiating translation of a polypeptide in the absence of a ty pical RNA cap structure. An IRES is typically 500 nt to 700 nt in length.
[0459] As used herein, a “miRNA site” refers to a stretch of nucleotides within a polynucleotide that is capable of forming a duplex with at least 8 nucleotides of a natural miRNA sequence.
[0460] As used herein, an “endonuclease site” refers to a stretch of nucleotides within a polynucleotide that is capable of being recognized and cleaved by an endonuclease protein.
[0461] As used herein, “bicistronic RNA” refers to a polynucleotide that includes two expression sequences coding for two distinct proteins. These expression sequences can be separated by a nucleotide sequence encoding a cleavable peptide such as a protease cleavage site. They can also be separated by a ribosomal skipping element.
[0462] As used herein, the term “ribosomal skipping element” refers to a nucleotide sequence encoding a short peptide sequence capable of causing generation of two peptide chains from translation of one RNA molecule. While not wishing to be bound by theory', it is hypothesized that ribosomal skipping elements function by (1) terminating translation of the first peptide chain and re-initiating translation of the second peptide chain; or (2) cleavage of a peptide bond in the peptide sequence encoded by the ribosomal skipping element by an intrinsic protease activity of the encoded peptide, or by another protease in the environment (e.g., cytosol).
[0463] As used herein, the term “co-formulate” refers to a nanoparticle formulation comprising two or more nucleic acids or a nucleic acid and other active drug substance. Typically, the ratios are equimolar or defined in the ratiometric amount of the two or more nucleic acids or the nucleic acid and other active drug substance.
[0464] As used herein, the phrase “ionizable lipid” refers to any of a number of lipid species that carry a net positive charge at a selected pH. such as physiological pH 4 and a neutral charge at other pHs such as physiological pH 7.
[0465] In some embodiments, a lipid or compound described herein comprises one or more cleavable groups. The terms “cleave” and “cleavable” are used herein to mean that one or more chemical bonds (e.g., one or more of covalent bonds, hydrogen-bonds, van der Waals' forces and/or ionic interactions) between atoms in or adjacent to the subject functional group are broken (e.g.. hydrolyzed) or are capable
of being broken upon exposure to selected conditions (e.g., upon exposure to enzy matic conditions). In certain embodiments, the cleavable group is a disulfide functional group, and in particular embodiments is a disulfide group that is capable of being cleaved upon exposure to selected biological conditions (e.g., intracellular conditions). In certain embodiments, the cleavable group is an ester functional group that is capable of being cleaved upon exposure to selected biological conditions. For example, the disulfide groups may be cleaved enzymatically or by a hydrolysis, oxidation or reduction reaction. Upon cleavage of such disulfide functional group, the one or more functional moieties or groups (e.g., one or more of a head-group and/or a tail-group) that are bound thereto may be liberated. In some embodiments, a cleavable group is bound (e.g., bound by one or more of hydrogen-bonds, van der Waals' forces, ionic interactions and covalent bonds) to one or more functional moieties or groups.
[0466] Compound described herein may also comprise one or more isotopic substitutions. For example, H may be in any isotopic form, including 1H, 2H (D or deuterium), and 3H (T or tritium); C may be in any isotopic form, including 12C, 13C, and 14C; O may be in any isotopic form, including 160 and 180; F may be in any isotopic form, including 18F and 19F; and the like.
[0467] When describing the invention, which may include compounds and pharmaceutically acceptable salts thereof, pharmaceutical compositions containing such compounds and methods of using such compounds and compositions, the following terms, if present, have the following meanings unless otherwise indicated. It should also be understood that when described herein any of the moieties defined forth below may be substituted with a variety' of substituents, and that the respective definitions are intended to include such substituted moieties within their scope as set out below. Unless otherwise stated, the term “substituted” is to be defined as set out below. It should be further understood that the terms “groups” and “radicals” can be considered interchangeable when used herein.
[0468] When a range of values is listed, it is intended to encompass each value and sub-range within the range. For example, “C1-6 alkyl” is intended to encompass, C1, C2, C3, C4, C5, Ce, C1-g, C1-5, C1^i, C1-3, C1-2, C2-6, C2-5, C2-4, C2-3, C3-6, C3-5. C3-4, C4-6. C4-5, and C5-6 alkyl.
[0469] It should be noted that the term “head-group” as used describe the compounds of the present invention, and in particular functional groups that comprise such compounds, are used for ease of reference to describe the orientation of one or more functional groups relative to other functional groups. For example, in certain embodiments a hydrophilic head-group (e.g., an amino group) is bound (e.g., by one or more of hydrogen-bonds, van der Waals' forces, ionic interactions and covalent bonds) to a hydrophobic tail-group (e.g., cholesterol).
[0470] In typical embodiments, the present invention is intended to encompass the compounds disclosed herein, and the pharmaceutically acceptable salts, pharmaceutically acceptable esters, tautomeric forms, polymorphs, and prodrugs of such compounds. In some embodiments, the present invention includes a pharmaceutically acceptable addition salt, a pharmaceutically acceptable ester, a
solvate (e.g., hydrate) of an addition salt, a tautomeric form, a poly morph, an enantiomer, a mixture of enantiomers, a stereoisomer or mixture of stereoisomers (pure or as a racemic or non-racemic mixture) of a compound described herein.
[0471] Compounds described herein can comprise one or more asymmetric centers, and thus can exist in various isomeric forms, e.g., enantiomers and/or diastereomers. For example, the compounds described herein can be in the form of an individual enantiomer, diastereomer or geometric isomer, or can be in the form of a mixture of stereoisomers, including racemic mixtures and mixtures enriched in one or more stereoisomer. Isomers can be isolated from mixtures by methods known to those skilled in the art, including chiral high pressure liquid chromatography (HPLC) and the formation and crystallization of chiral salts; or preferred isomers can be prepared by asymmetric syntheses. See, for example, Jacques et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen et al., Tetrahedron 33:2725 (1977); Eliel, Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962); and Wilen, Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed.. Univ, of Notre Dame Press, Notre Dame, IN 1972). The invention additionally encompasses compounds described herein as individual isomers substantially free of other isomers, and alternatively, as mixtures of various isomers.
[0472] In certain embodiments the subject compositions (e.g., loaded LNPs) exhibit an enhanced (e.g., increased) ability to transfect one or more target cells. Accordingly, also provided herein are methods of transfecting one or more target cells. Such methods generally comprise the step of contacting the one or more target cells with the compositions disclosed herein such that the one or more target cells are transfected with the circular RNA encapsulated therein. As used herein, the terms “transfect” or “transfection” refer to the intracellular introduction of one or more encapsulated materials (e.g.. nucleic acids and/or polynucleotides) into a cell, or preferably into a target cell. The term “transfection efficiency” refers to the relative amount of such encapsulated material (e.g., polynucleotides) uptaken by. introduced into and/or expressed by the target cell which is subject to transfection.
[0473] All nucleotide sequences disclosed herein can represent an RNA sequence or a corresponding DNA sequence. It is understood that deoxythymidine (dT or T) in a DNA is transcribed into a uridine (U) in an RNA. As such, "T” and “U” are used interchangeably herein in nucleotide sequences.
[0474] The recitations “sequence identity” or, for example, comprising a “sequence 50% identical to,” as used herein, refer to the extent that sequences are identical on a nucleotide-by -nucleotide basis or an amino acid-by-amino acid basis over a window of comparison. Thus, a “percentage of sequence identity” may be calculated by comparing two optimally aligned sequences over the window of comparison, determining the number of positions at which the identical nucleic acid base (e.g., A. T, C, G, I) or the identical amino acid residue (e.g., Ala. Pro, Ser, Thr, Gly. Vai, Leu, He, Phe, Tyr. Trp, Lys, Arg, His, Asp, Glu, Asn. Gin, Cys and Met) occurs in both sequences to yield the number of matched
positions, dividing the number of matched positions by the total number of positions in the window of comparison (i.e., the window size), and multiplying the result by 100 to yield the percentage of sequence identity. Included are nucleotides and polypeptides having at least 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% sequence identity to any of the reference sequences described herein, typically where the polypeptide variant maintains at least one biological activity of the reference polypeptide.
[0475] The expression sequences in the polynucleotide construct may be separated by a “cleavage site” sequence which enables polypeptides encoded by the expression sequences, once translated, to be expressed separately by tire cell.
[0476] A “self-cleaving peptide” refers to a peptide which is translated without a peptide bond between two adjacent amino acids, or functions such that when the polypeptide comprising tire proteins and the self-cleaving peptide is produced, it is immediately cleaved or separated into distinct and discrete first and second polypeptides without the need for any external cleavage activity .
[0477] The a and (3 chains of a(3 TCR's are generally regarded as each having two domains or regions, namely variable and constant domains/regions. The variable domain consists of a concatenation of variable regions and joining regions. In the present specification and claims, the term “TCR alpha variable domain” therefore refers to the concatenation of TRAV and TRAJ regions, and the term TCR alpha constant domain refers to the extracellular TRAC region, or to a C-tenninal truncated TRAC sequence. Likewise, the term “TCR beta variable domain” refers to the concatenation of TRBV and TRBD/TRBJ regions, and the term TCR beta constant domain refers to the extracellular TRBC region, or to a C-terminal truncated TRBC sequence.
[0478] The terms “duplexed,” “double-stranded,” or “hybridized” as used herein refer to nucleic acids formed by hybridization of two single strands of nucleic acids containing complementary sequences. In most cases, genomic DNA is double-stranded. Sequences can be fully complementary or partially complementary’.
[0479] As used herein, a “vaccine” refers to a composition for generating immunity for the prophylaxis and/or treatment of diseases. Accordingly, vaccines are medicaments which comprise antigens and are intended to be used in humans or animals for generating specific defense and protective substances upon administration to the human or animal.
EXAMPLES
[0480] Wesselhoeft et al.. (2019) RNA Circularization Diminishes Immunogenicity and Can Extend Translation Duration In vivo. Molecular Cell. 74(3), 508-520 and Wesselhoeft et al.. (2018) Engineering circular RNA for Potent and Stable Translation in Eukaryotic Cells. Nature Communications, 9, 2629 are incorporated by reference in their entirety’.
[0481] The present disclosure further includes the following examples that provide those of ordinary skill in the art with a description of how to make and use the various embodiments of the present disclosure. These examples are not intended to limit tire scope of what is regarded as the claimed invention.
EXAMPLE 1
General procedure for the preparation of empty LNPs with various ionizable lipids
[0482] Organic phase (lipids dissolved in ethanol) and aqueous phase (buffer) were mixed at 1:3 ethanol to aqueous phase ratios to prepare the empty LNP formation as depicted in the schematic of FIG. 1. Lipids used to form the organic phase included ionizable lipids, helper lipids, a cholesterol lipid, and lipid anchored PEGylated lipids. The selected lipids were solubilized in ethanol in exemplary molar ratios provided in Table 5. For the aqueous phase, salt solution (e.g., comprising citrate, sodium acetate, malic acid, and/or bis-tris methane) at a defined molarity and pH was mixed to effectively protonate the ionizable lipid. Table 6 depicts exemplary aqueous buffer compositions for each of the ionizable lipids. Optimum buffer compositions were analyzed for the smallest and most stable LNPs using screening experiments of various aqueous buffer compositions.
[0483] To undergo formation of the first empty LNP composition, both the aqueous and the lipid ethanol phase were mixed using a commercially available mixer (e.g.. Silverson L5M-A) for 5 minutes at 8000 RPM using the standard mixer head mesh at a processing temperature from 4 to 30 ° C.
[0484] The first composition of empty LNPs were then fed into a commercially available high shear microfluidizer (e.g., Microfluidics LM20 high shear microfluidizer) at 10,000 - 30.000 psi for further particle size reduction. The processing temperature at this step was kept the same or close to the mixing processing temperature (e.g., 4° C to 30° C). The output temperature, as the size reduced empty LNPs exit the microfluidizer, was controlled at 4°C for all lipids. Empty LNPs were then stored at 4° C until RNA addition step.
Table 5:
 Table 6:
Table 6:

EXAMPLE 2
Empty! LNP formulation using various mixing temperatures
[0485] Ionizable lipid 89 (Table 1), 1.2-distearoyl-sn-giycero-3phosphochoiine (DSPC), cholesterol (Choi), and DMG-PEG2000 were mixed at a 50: 10:38.5: 1.5 molar ratio and were used to evaluate LNP sizing using overhead mixing at different temperatures. Lipids dissolved in ethanol (organic phase), were mixed with an aqueous phase buffer solution (e.g., comprising citrate, sodium acetate, malic acid, and/or bis-tris methane) at a fixed ratio. Organic phase and aqueous phase were mixed using an overhead mixer for 5 minutes at temperatures of 4° C, 10° C, 20° C, 30° C, and 40° C. FIG. 2 shows die particle size distribution of LNPs processed in different temperatures. Based on the data provided in FIG. 2, 30° C provides the smallest particle size (104 nm) and lowest PDI at 0.09.
EXAMPLE 3
Empty LNP formulation processed at various speeds in a high-pressure homogenizer
[0486] Ionizable lipid 144 (Table 1), DSPC, Cholesterol, and DMG-PEG2000 at 50: 10:38.5:1.5 molar ratio were dissolved in ethanol (organic phase) and mixed with aqueous buffer. The duration of mixing was set at 5 minutes, and different RPMs were tested at 1000, 2000, 4000, and 8000 RPM. FIG. 3 represents ionizable lipid 144 (Table 1) LNP post LNP formation and post homogenization step. The slowest rotor speed at 1000 RPM resulted in an average particle size of 101 nm, and the highest rotor speed resulted in average particle sizes of 89 nm (see FIG. 3). The intermediate speed of 2000 RPM resulted in an average particle size of 77 nm. The formulations were further processed for particle size reduction using a high-pressure homogenizer. All the particles post-LNP formation resulted in similar average particle size reduction post homogenization (i.e., 60 nm) at speeds of 2000. 4000. or 8000 RPM (See FIG. 3).
EXAMPLE 4
Empty LNP particle size reduction at various temperatures
[0487] Ionizable lipid 127 (Table 1), DSPC. Cholesterol, and DMG-PEG2000 at 50: 10:38.5.1.5 molar ratio were dissolved in ethanol (organic phase) and mixed with an aqueous phase of 6.25 mM sodium acetate at pH 3.0, using an overhead mixer to form empty LNPs (LNP formation step). The formed empty LNPs were then fed through a homogenizer for particle size reduction (homogenization step). To elucidate the effect of temperature for the homogenization step using a high-pressure homogenizer, empty lipid 127 containing LNPs were processed through the homogenizer at two processing temperatures, 30° C and 50° C. Note that, the full range of temperature that can be tested ranges from 4° C to 60° C. FIG. 4 provides particle size and PDI after both the LNP formation step and the homogenization step. Particle size reduction after homogenization was observed for both temperatures compared to the LNP formation step in FIG 4. To minimize particle aggregation post homogenization step, a cooling step was implemented using a heat exchanger immediately after the homogenization step. FIG. 5 shows the particle size of the empty lipid 127 LNPs with and without cooling after the homogenization step.
EXAMPLE 5
Empty LNP particle size reduction in the presence of a cooling step
[0488] To further optimize empty LNP formation, the buffer composition at both the first LNP formation step and tire homogenization step (particle size reduction step) were evaluated. Ionizable lipid 127 (Table 1), DSPC, Cholesterol, and DMG-PEG2000 at 50: 10:38.5.1.5 molar ratio were dissolved in ethanol (organic phase) and mixed with an aqueous phase of 6.25 mM sodium acetate at pH 3.0 or 4.5. using an overhead mixer and then fed through a homogenizer for particle size reduction. FIG. 6 represents the particle size and PDI relationship with pH of the aqueous phase over multiple batches. As seen in FIG. 6. LNPs in pH 3.0 sodium acetate buffer not only resulted in smaller particle size post LNP formation than the LNPs in pH 4.5 sodium acetate buffer, but also resulted in smaller particle size post homogenization step than the LNPs in pH 4.5 sodium acetate. More generally, as also depicted in FIG. 6 particle size reduction was observed for both buffers after homogenization. Zeta potential was also measured for each of the LNPs post homogenization, as depicted in FIG. 7. In some embodiments, various buffer salts can be tested at different concentrations and pH ranges as provided in Table 7.
[0489] Further to the experimental results discussed above, various buffer salts can be tested at different concentrations and pH ranges, as provided in Table 7.
Table 7:


EXAMPLE 6
Particle size reduction for various empty LNPs
[0490] Ionizable lipids with different tails and/or headgroups such as lipid 144 and lipid 123 (Table 1) were investigated (in addition to lipid 127; see Examples 4 and 5 for lipid 127). The following lipid solutions were used:
• Lipid 144. DSPC, Cholesterol, and DMG-PEG2000 were formulated at 50:10:38.5.1.5 molar ratio.
• Lipid 123. DSPC, cholesterol, and DMG-PEG 2000 were formulated at standard molar ratio of 45 :9:44:2 and with RN A. The N :P ratio was kept at 4.5 : 1 ratio.
[0491] The lipid solution made using ionizable lipid 144 was dissolved in ethanol (organic phase), mixed with an aqueous buffer phase (6.25 mM Sodium Acetate at pH 3.0; 50mM Na Acetate at pH 3.0; or 6.25 mM Bis-Tris at pH 7.0) using an overhead mixer, and then fed through a homogenizer for particle size reduction. Results are depicted in FIG. 8 for empty LNPs formed containing lipid 144.
[0492] The lipid solution made using ionizable lipid 123 was dissolved in ethanol (organic phase), mixed with an aqueous buffer phase (6.25 mM Citrate buffer at pH 2.0; 6.25 mM citrate buffer at pH 3.0; 6.25 mM sodium acetate at pH 3.0; or 6.25 mM sodium acetate at pH 4.5) using an overhead mixer, and then fed through a homogenizer for particle size reduction. Results are depicted in FIG. 9 for LNPs formed containing lipid 123.
EXAMPLE 7
Aqueous buffers used post RNA addition and encapsulation
[0493] A composition of empty LNPs was formed using ionizable lipid 127 (Table 1) according to the method of Example 1. Encapsulation of circular RNA within the empty LNPs was then investigated to form a composition of loaded LNPs. Numerous aqueous buffers were screened on a plate-based assay first in a high-throughput fashion ID lead buffer conditions that would produce high encapsulation and small particle size, after the RNA addition step. Table 7 depicts the full range of buffers tested. Initial results showed that 50 mM sodium acetate resulted in high RNA encapsulation while keeping the
particles small within 70 - 90 nm particle size range. The lead aqueous phase conditions from tire platebased assay were scaled up to a larger scale (2-5mL) to confirm the results. Aqueous phases including 50 mM sodium acetate pH 3.0, 4.0 and 4.5 were evaluated. FIG. 10 represents the average particle size of RNA encapsulated LNP (loaded LNPs) in the different aqueous phase buffer conditions. FIG. 11 provides the LNP encapsulation efficiency post RNA encapsulation (i.e., for loaded LNPs) at different pHs in 50 mM sodium acetate.
EXAMPLE 8
Aqueous buffers, sterile filtration and storage of loaded LNPs (post RNA addition encapsulation)
[0494] A composition of empty LNPs were formed using ionizable lipid 127 (Table 1) according to the method of Example 1. Circular RNA was encapsulated within the empty LNPs to form a composition of loaded LNPs according to the method of Example 7 (RNA addition step). Following the RNA addition step, buffer exchange was performed via tangential flow filtration (TFF). The composition of loaded LNPs underwent buffer exchange either with phosphate buffered saline at pH 7.4 (PBS, TFF process #1) or ultrapure distilled water (DI water. TFF process #2). The resulting loaded LNP particle size and PDI after the RNA addition step and buffer exchange (TFF step) are shown in FIG. 12. Buffer exchange step (or TFF step) performed using PBS caused loaded LNP particle size to grow in average particle size to 142 nm with PDI at 0.22. However, buffer exchange (TFF step) performed using DI water, resulted in significantly smaller particle sizes of approximately 69 nm with PDI 0.08. FIG. 13 provides the encapsulation efficiency of the loaded LNPs post RNA addition step and TFF step with PBS or DI water.
[0495] After buffer exchange (TFF step), the loaded LNPs were stored at 4°C or frozen (at -80°C). Storage buffers (and tonicity modifiers) tris-sucrose saline (TSS) or PBS were evaluated. Results for the TSS and PBS buffers/tonicity modifiers used are shown in FIG. 14. Further storage buffers may include but are not limited to, Tris-Sucrose Saline, Tris-Trehalose Saline, Phosphate Buffered Saline, and Tris Buffered Saline.
EXAMPLE 9
In vivo performance of LNPs formed from distinctive LNP formation, homogenization (LNP size reduction), and RNA addition steps
[0496] A composition of empty’ LNPs was formed using ionizable lipid 127 (Tabic 1) according to the method of Example 1. Circular RNA encoding firefly luciferase was encapsulated within the empty LNPs to form a composition of loaded LNPs according to the method of Example 7 (RNA addition step). Following the RNA addition step, buffer exchange was performed via tangential flow filtration (TFF). The process for preparing a composition of loaded LNPs using distinctive LNP formation, homogenization (LNP size reduction) and RNA addition steps (“Process 1”) is summarized in the
schematic of FIG. 15. FIG. 1 also depicts a schematic outlining the subject distinctive process steps: an LNP formation step (nanoprecipitation step), a homogenization step (particle size reduction step), and an RNA addition step. Process 1 was evaluated for in vivo performance via intravenous (IV) and intramuscular (IM) route of administration. LNP particle sizes were evaluated for loaded LNPs prepared using the nanoprecipitation process (e.g., Precision Nanosystem Ignite (PNI) nanoprecipitation process, see e.g., https://www.precisionnanosystems.com/platform-teclmologies/product-comparison/ignite) and Process 1. Both the nanoprecipitation (e.g., PNI) process loaded LNPs and Process 1 loaded LNPs demonstrated similar particle sizes as shown in FIG. 16. Loaded LNPs formulated via Process 1 and nanoprecipitation (e.g., PNI) process were imaged via Cryo-TEM. Particle morphology for both LNPs, made using different processes, appeared similar as shown in FIG. 17 (Panel A = Process 1 LNPs; Panel B = nanoprecipitation (e.g., PNI) process). Within the particles, there was uniform spread of punctate dots, suggesting uniform circular RNA encapsulation throughout the loaded LNPs. Particle size ranged approximately from 50 - 100 nm for both processes.
[0497] The loaded LNPs (LNP -circular RNAs) were injected intravenously into mice at RNA dose of 0.5 mpk or via IM at RNA dose of 0.4 mpk. FIG. 18 shows 6-hour ex vivo circular RNA encoding firefly luciferase expression for IV injected loaded LNPs. The organs of the mice were extracted and ex vivo I VIS was performed on the organs. FIG. 18 provides the in vivo firefly luciferase expression of the circular RNA encapsulated in the LNPs as measured by ex vivo IVIS as made from Process 1. FIG. 19C-FIG. 19D provides the ex vivo organ imaging for firefly luciferase expression of the loaded LNPs as made from Process 1, wherein Process 1 used PBS (FIG. 19C) or TSS (FIG. 19D). FIG. 19A-FIG. 19B provides the ex vivo organ imaging for firefly luciferase expression of the loaded LNPs as made from nanoprecipitation (e.g., PNI) process, wherein the nanoprecipitation (e.g.. PNI) process used PBS (FIG. 19A) or TSS (FIG. 19B).
[0498] Intramuscular injected LNPs were also characterized for protein expression using IVIS at 24 hours post injection. FIG. 20 shows flux level of LNPs in the quadricep, liver, and spleen.
INCORPORATION BY REFERENCE
[0499] All publications, patents, and patent applications mentioned in this specification arc herein incorporated by reference to the same extent as if each individual publication, patent, or patent application was specifically and individually indicated as being incorporated by reference herein, including, for example, U.S. provisional patent application no. 63/605,295, International patent application nos. PCT/US2019/035531, PCT/US2020/034418, PCT/US2020/063494,
PCT/US2021/031629, PCT/US2021/023540, PCT/US2021/033276. PCT/US2022/033091,
PCT/US2022/045408, and PCT/US2022/049313.
Claims
1. A method of preparing an empty’ lipid nanoparticle (LNP) composition, the method comprising: a) a precipitation step, comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first LNP composition comprising empty LNPs; and b) a homogenization step, comprising: homogenizing the first LNP composition to produce a homogeneous LNP composition of empty LNPs.
2. The method of claim 1, wherein the homogeneous LNP composition comprises empty LNPs having an average particle size of from 30 nm to 200 nm.
3. The method of claim 2, wherein the average particle size is from 50 nm to 70 nm.
4. The method of any one of claims 1 to 3, wherein the homogeneous LNP composition has a polydispersity of 0.05 to 0.2.
5. The method of claim 4, wherein the homogeneous LNP composition has a polydispersity’ of 0.1 or less.
6. The method of any one of claims 1 to 5, wherein the average particle size of empty LNPs in the homogeneous LNP composition is at least 10% less than the average particle size of the empty LNPs of the first lipid nanoparticle (LNP) composition (e.g.. average particle size of 10-50% less, or 10-30% less).
7. The method of any’ one of claims 1 to 6, wherein the homogeneous LNP composition is storage stable at 4 °C for at least 4 weeks.
8. The method of any one of claims 1 to 7, further comprising: c) a loading step, comprising: mixing the homogeneous LNP composition with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with the nucleic acid.
9. The method of claim 8, wherein the LNPs in the loaded LNP composition has an average particle size of 50 nm to 300 nm and a polydispersity of 0.3 or less.
10. The method of claim 9, wherein the LNPs in the loaded LNP composition has an average particle size of 70 nm to 120 mn and a polydispersity of 0.2 or less.
11. The method of any one of claims 1 to 10. wherein in step a), the ratio of lipid solution to aqueous buffer solution is from 1:2 to 1:5 by volume.
12. The method of claim 11, wherein the ratio of lipid solution to aqueous buffer solution is 1 :3 by volume.
13. The method of claim 11, wherein the lipid solution comprises an organic solvent (e.g., ethanol) that is miscible with the aqueous buffer solution.
14. The method of any one of claims 1 to 13, wherein in step a), the pH of the aqueous buffer solution is from 2 to 9 (e.g., pH of 2 to 6, or 3 to 5, such as 3.0 or 4.5).
15. The method of any one of claims 1 to 14, wherein the mixing of step a) is carried out at a temperature of from 2-70 °C (e.g., 30 or 50 °C).
16. The method of claim 15, wherein the mixing of step a) is carried out at a temperature of from 4 to 40 °C.
17. The method of any one of claims 1 to 16. wherein the homogenizing of step b) is performed using a microfluidic device.
18. The method of claim 17, wherein the microfluidic device is selected from a high-pressure homogenizer, a high shear homogenizer and a probe homogenizer.
19. The method of claim 18, wherein the homogenizing of step b) is performed using a high- pressure homogenizer at a pressure of from 1000 to 30,000 PSI.
20. The method of any one of claims 17 to 19. wherein the homogenizing of step b) comprises processing the composition through the microfluidic device two or more times.
21. The method of claim 20, wherein the homogenizing of step b) comprises processing the composition through the microfluidic device up to 5 tunes.
22. The method of any one of claims 1 to 21, wherein the homogenizing of step b) is carried out at a temperature of from 4 to 60 °C (e.g., 30 to 50 °C, such as 30 °C or 50 °C in the microfluidic device).
23. The method of claim 22, further comprising cooling the homogeneous LNP composition immediately after the homogenizing of step b) (e.g., after the composition exits the microfluidic device cooling the composition to e.g., 4 °C, to prevent LNP aggregation).
24. The method of claim 22. wherein the homogeneous LNP composition is storage stable at 4 °C for at least 4 weeks.
25. The method of any one of claims 1 to 24, wherein the lipid solution comprises: an ionizable lipid; a helper lipid; a cholesterol; and a PEG-lipid.
26. The method of claim 25, wherein the molar ratio of ionizable lipid: helper lipid: cholesterol: PEG-lipid is 45:9:44:2, 50:10:38.5:1.5, 41:12:45:2, 62:4:33:1, or 53:5:41:1.
27. The method of any one of claims 8 to 26, wherein in step c) the molar ratio of nitrogen: phosphate (N:P) in the loaded LNP composition is from 1:20 to 20:1 (e.g., 4.0. 4.1, 4.2, 4.3, 4.4, 4.5, 4.6. 4.7. 4.8. 4.9, 5.0, 5.1, 5.2, 5.3, 5.4, 5.5, 5.6, 5.7, 5.8, 5.9, 6.0).
28. The method of any one of claims 8 to 27 wherein the nucleic acid solution comprises a buffer salt selected from an acetate salt, a citrate salt, or a bis-tris salt.
29. The method of claim 28, wherein the concentration of the buffer salt in the nucleic acid solution is from 1 mM to 500 mM (e.g., 6.25-50 mM).
30. The method of any one of claims 8 to 29. wherein the pH of the nucleic acid solution is from 2-9 (e.g., pH 2-7)
31. The method of any one of claims 8 to 30. wherein the loading step c) is carried out at a temperature of from 2-70 °C (e.g., 4-25 °C).
32. The method of any one of claims 8 to 31, wherein the loading step c) is performed by mixing the homogeneous LNP composition with a nucleic acid solution at a flow rate from 100 mL/min to 100 L/min (e.g., at least 2L/min).
33. The method of claim 32, wherein the mixing is performed using a pump.
34. The method of claim 33, wherein the pump is selected from a gear pump, a peristatic pump, and a centrifugal pump.
35. The method of any one of claims 8 to 34, wherein the nucleic acid is encapsulated in the loaded LNPs with an encapsulation efficiency of from 50 to 100% (e.g.. 90% or more).
36. The method of any one of claims 8 to 35, wherein the nucleic acid is an RNA.
37. The method of claim 36, wherein the RNA is a circular RNA.
38. The method of any one of claims 8 to 37, further comprising one or more of the following steps: dilution and buffer exchange of the loaded LNP composition; filtration of the loaded LNP composition; and storage of the loaded LNP composition.
39. The method of claim 38, wherein the loaded LNP composition is storage stable at a temperature from 4 °C to -80 °C (e.g., in a storage buffer or tonicity modifier).
40. A method of preparing a loaded lipid nanoparticle (LNP) composition, the method comprising: a) a precipitation step, comprising: mixing a lipid solution comprising an ionizable lipid with an aqueous buffer solution, thereby forming a first LNP composition comprising empty LNPs; b) a loading step, comprising: mixing the first LNP composition with a nucleic acid solution comprising a nucleic acid, thereby producing a loaded LNP composition comprising LNPs associated with the nucleic acid; and c) a homogenization step, comprising: homogenizing the loaded LNP composition to produce a homogeneous loaded LNP composition comprising LNPs associated with the nucleic acid.
41. The method of claim 40, wherein the LNPs in the homogeneous loaded LNP composition have an average particle size of 50 nm to 300 nm and a poly dispersity of 0.3 or less.
42. The method of claim 41, wherein the LNPs in the homogeneous loaded LNP composition have an average particle size of 70 nm to 120 nm and a poly dispersity of 0.2 or less.
43. The method of any one of claims 40 to 42, wherein in step b) the molar ratio of nitrogen: phosphate (N:P) in the loaded LNP composition is from 1:20 to 20:1 (e.g.,4.0, 4.1, 4.2, 4.3, 4.4, 4.5, 4.6, 4.7, 4.8, 4.9, 5.0, 5.1, 5.2, 5.3, 5.4, 5.5, 5.6, 5.7, 5.8, 5.9, 6.0).
44. The method of any one of claims 40 to 43. wherein the nucleic acid solution comprises a buffer salt selected from an acetate salt, a citrate salt, or a bis-tris salt.
45. The method of claim 44, wherein the concentration of the buffer salt in the nucleic acid solution is from 1 mM to 500 mM (e.g., 6.25-50 mM).
46. The method of any one of claims 40 to 45, wherein tire pH of the nucleic acid solution is from 2-9 (e.g., pH 2-7)
47. The method of any one of claims 40 to 46, wherein the loading step b) is carried out at a temperature of from 2-70 °C (e.g.. 4-25 °C).
48. The method of any one of claims 40 to 47. w herein the loading step b) is performed by mixing the first LNP composition with a nucleic acid solution at a flow rate from 100 mL/min to 100 L/min (e.g., at least 2L/min).
49. The method of claim 48, wherein the mixing is performed using a pump.
50. The method of claim 49, wherein the pump is selected from a gear pump, a peristatic pump, and a centrifugal pump.
51. The method of any one of claims 40 to 50, wherein the nucleic acid is encapsulated in the loaded LNPs with an encapsulation efficiency of from 50 to 100% (e.g., 90% or more).
52. The method of any one of claims 40 to 51, wherein the homogenizing of step c) is performed using a microfluidic device.
53. The method of claim 52, wherein the microfluidic device is selected from a high-pressure homogenizer, a high shear homogenizer and a probe homogenizer.
54. The method of claim 53, wherein the homogenizing of step c) is performed using a high- pressure homogenizer at a pressure of from 1000 to 30,000 PSI.
55. The method of any one of claims 52 to 54, wherein the homogenizing of step c) comprises processing the loaded LNP composition through the microfluidic device two or more times.
56. The method of claim 55, wherein the homogenizing of step c) comprises processing the loaded LNP composition through the microfluidic device up to 5 times.
57. The method of any one of claims 40 to 56, wherein the homogenizing of step c) is carried out at a temperature of from 4 to 60 °C (e.g.. 30 to 50 °C. such as 30 °C or 50 °C in the microfluidic device).
58. The method of claim 57, further comprising cooling the loaded LNP composition immediately after the homogenizing of step c).
59. The method of claim 58, wherein the loaded LNP composition is storage stable at 4 °C for at least 4 weeks.
60. The method of any one of claims 40 to 59. further comprising one or more of the following steps: dilution and buffer exchange of the loaded LNP composition; filtration of the loaded LNP composition; and storage of the loaded LNP composition.
61. The method of claim 60, wherein the loaded LNP composition is storage stable at a temperature from 4 °C to -80 °C (e.g., in a storage buffer or tonicity modifier).
62. An empty LNP composition prepared by a method of any one of claims 1 to 61.
63. A loaded LNP composition prepared by a method of any one of claims 8 to 62.
64. A homogeneous LNP composition substantially free of loaded LNPs and comprising empty LNPs comprising: an ionizable lipid; a helper lipid; a cholesterol; and a PEG-lipid, wherein the empty LNPs have
(a) a poly dispersity of 0.2 or less (e.g., 0.1 or less); and/or
(b) an average particle size from 50 mn to 70 nm.
65. The composition of claim 64, wherein the empty LNPs have a poly dispersity of 0.2 or less.
66. The composition of claim 64, wherein the empty LNPs have a poly dispersity' of 0.1 or less.
67. The composition of claim 64. wherein the empty LNPs have a poly dispersity of 0.05 to 0.2.
68. The composition of claim 64, wherein the empty LNPs have an average particle size from 50 nm to 70 nm.
69. The composition of any one of claims 64 to 68, wherein the empty LNPs have a polydispersity of 0.2 or less and an average particle size from 50 nm to 70 nm.
70. The composition of any one of claims 64 to 69, wherein the composition is storage stable at 4 °C for at least 4 weeks.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363605295P | 2023-12-01 | 2023-12-01 | |
| US63/605,295 | 2023-12-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025117969A1 true WO2025117969A1 (en) | 2025-06-05 |
Family
ID=93924884
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/058122 Pending WO2025117969A1 (en) | 2023-12-01 | 2024-12-02 | Process for manufacturing lipid nanoparticles |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025117969A1 (en) |
Citations (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3832253A (en) | 1973-03-21 | 1974-08-27 | Baxter Laboratories Inc | Method of making an inflatable balloon catheter |
| US3854480A (en) | 1969-04-01 | 1974-12-17 | Alza Corp | Drug-delivery system |
| US4450150A (en) | 1973-05-17 | 1984-05-22 | Arthur D. Little, Inc. | Biodegradable, implantable drug delivery depots, and method for preparing and using the same |
| US4452775A (en) | 1982-12-03 | 1984-06-05 | Syntex (U.S.A.) Inc. | Cholesterol matrix delivery system for sustained release of macromolecules |
| US4667014A (en) | 1983-03-07 | 1987-05-19 | Syntex (U.S.A.) Inc. | Nonapeptide and decapeptide analogs of LHRH, useful as LHRH antagonists |
| US4748034A (en) | 1983-05-13 | 1988-05-31 | Nestec S.A. | Preparing a heat stable aqueous solution of whey proteins |
| US5075109A (en) | 1986-10-24 | 1991-12-24 | Southern Research Institute | Method of potentiating an immune response |
| US5087616A (en) | 1986-08-07 | 1992-02-11 | Battelle Memorial Institute | Cytotoxic drug conjugates and their delivery to tumor cells |
| US5239660A (en) | 1990-10-31 | 1993-08-24 | Nec Corporation | Vector processor which can be formed by an integrated circuit of a small size |
| US5885613A (en) | 1994-09-30 | 1999-03-23 | The University Of British Columbia | Bilayer stabilizing components and their use in forming programmable fusogenic liposomes |
| US5948902A (en) | 1997-11-20 | 1999-09-07 | South Alabama Medical Science Foundation | Antisense oligonucleotides to human serine/threonine protein phosphatase genes |
| US6319494B1 (en) | 1990-12-14 | 2001-11-20 | Cell Genesys, Inc. | Chimeric chains for receptor-associated signal transduction pathways |
| US20040262223A1 (en) | 2001-07-27 | 2004-12-30 | President And Fellows Of Harvard College | Laminar mixing apparatus and methods |
| WO2006000830A2 (en) | 2004-06-29 | 2006-01-05 | Avidex Ltd | Cells expressing a modified t cell receptor |
| US7741465B1 (en) | 1992-03-18 | 2010-06-22 | Zelig Eshhar | Chimeric receptor genes and cells transformed therewith |
| WO2011140627A1 (en) | 2009-11-04 | 2011-11-17 | The University Of British Columbia | Nucleic acid-containing lipid particles and related methods |
| US8158601B2 (en) | 2009-06-10 | 2012-04-17 | Alnylam Pharmaceuticals, Inc. | Lipid formulation |
| WO2012099755A1 (en) | 2011-01-11 | 2012-07-26 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| WO2015095340A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| WO2015130584A2 (en) | 2014-02-25 | 2015-09-03 | Merck Sharp & Dohme Corp. | Lipid nanoparticle vaccine adjuvants and antigen delivery systems |
| WO2018089801A1 (en) | 2016-11-10 | 2018-05-17 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| WO2019081383A1 (en) | 2017-10-25 | 2019-05-02 | Universität Zürich | Eukaryotic initiation factor 4 recruiting aptamers for enhancing translation |
| JP2019516351A (en) * | 2016-03-30 | 2019-06-20 | インテリア セラピューティクス,インコーポレイテッド | Lipid Nanoparticle Formulations for CRISPR / CAS Components |
| WO2019236673A1 (en) | 2018-06-06 | 2019-12-12 | Massachusetts Institute Of Technology | Circular rna for translation in eukaryotic cells |
| WO2020237227A1 (en) | 2019-05-22 | 2020-11-26 | Massachusetts Institute Of Technology | Circular rna compositions and methods |
| WO2021021634A1 (en) | 2019-07-29 | 2021-02-04 | Georgia Tech Research Corporation | Nanomaterials containing constrained lipids and uses thereof |
| WO2021113777A2 (en) | 2019-12-04 | 2021-06-10 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2021155274A1 (en) | 2020-01-31 | 2021-08-05 | Modernatx, Inc. | Methods of preparing lipid nanoparticles |
| WO2021189059A2 (en) | 2020-03-20 | 2021-09-23 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2021226597A2 (en) | 2020-05-08 | 2021-11-11 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2021236855A1 (en) | 2020-05-19 | 2021-11-25 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| TW202228725A (en) * | 2020-10-14 | 2022-08-01 | 喬治梅森研究基金會 | Methods of lipid nanoparticle manufacture and compositions derived therefrom |
| WO2022261490A2 (en) | 2021-06-10 | 2022-12-15 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| US20230027864A1 (en) * | 2018-01-30 | 2023-01-26 | Modernatx, Inc. | Compositions and methods for delivery of agents to immune cells |
| WO2023056033A1 (en) | 2021-09-30 | 2023-04-06 | Orna Therapeutics, Inc. | Lipid nanoparticle compositions for delivering circular polynucleotides |
| WO2023081526A1 (en) | 2021-11-08 | 2023-05-11 | Orna Therapeutics, Inc. | Lipid nanoparticle compositions for delivering circular polynucleotides |
-
2024
- 2024-12-02 WO PCT/US2024/058122 patent/WO2025117969A1/en active Pending
Patent Citations (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3854480A (en) | 1969-04-01 | 1974-12-17 | Alza Corp | Drug-delivery system |
| US3832253A (en) | 1973-03-21 | 1974-08-27 | Baxter Laboratories Inc | Method of making an inflatable balloon catheter |
| US4450150A (en) | 1973-05-17 | 1984-05-22 | Arthur D. Little, Inc. | Biodegradable, implantable drug delivery depots, and method for preparing and using the same |
| US4452775A (en) | 1982-12-03 | 1984-06-05 | Syntex (U.S.A.) Inc. | Cholesterol matrix delivery system for sustained release of macromolecules |
| US4667014A (en) | 1983-03-07 | 1987-05-19 | Syntex (U.S.A.) Inc. | Nonapeptide and decapeptide analogs of LHRH, useful as LHRH antagonists |
| US4748034A (en) | 1983-05-13 | 1988-05-31 | Nestec S.A. | Preparing a heat stable aqueous solution of whey proteins |
| US5087616A (en) | 1986-08-07 | 1992-02-11 | Battelle Memorial Institute | Cytotoxic drug conjugates and their delivery to tumor cells |
| US5075109A (en) | 1986-10-24 | 1991-12-24 | Southern Research Institute | Method of potentiating an immune response |
| US5239660A (en) | 1990-10-31 | 1993-08-24 | Nec Corporation | Vector processor which can be formed by an integrated circuit of a small size |
| US6319494B1 (en) | 1990-12-14 | 2001-11-20 | Cell Genesys, Inc. | Chimeric chains for receptor-associated signal transduction pathways |
| US7741465B1 (en) | 1992-03-18 | 2010-06-22 | Zelig Eshhar | Chimeric receptor genes and cells transformed therewith |
| US5885613A (en) | 1994-09-30 | 1999-03-23 | The University Of British Columbia | Bilayer stabilizing components and their use in forming programmable fusogenic liposomes |
| US5948902A (en) | 1997-11-20 | 1999-09-07 | South Alabama Medical Science Foundation | Antisense oligonucleotides to human serine/threonine protein phosphatase genes |
| US20040262223A1 (en) | 2001-07-27 | 2004-12-30 | President And Fellows Of Harvard College | Laminar mixing apparatus and methods |
| WO2006000830A2 (en) | 2004-06-29 | 2006-01-05 | Avidex Ltd | Cells expressing a modified t cell receptor |
| US8158601B2 (en) | 2009-06-10 | 2012-04-17 | Alnylam Pharmaceuticals, Inc. | Lipid formulation |
| US20120276209A1 (en) | 2009-11-04 | 2012-11-01 | The University Of British Columbia | Nucleic acid-containing lipid particles and related methods |
| WO2011140627A1 (en) | 2009-11-04 | 2011-11-17 | The University Of British Columbia | Nucleic acid-containing lipid particles and related methods |
| WO2012099755A1 (en) | 2011-01-11 | 2012-07-26 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| WO2015095340A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| WO2015130584A2 (en) | 2014-02-25 | 2015-09-03 | Merck Sharp & Dohme Corp. | Lipid nanoparticle vaccine adjuvants and antigen delivery systems |
| JP2019516351A (en) * | 2016-03-30 | 2019-06-20 | インテリア セラピューティクス,インコーポレイテッド | Lipid Nanoparticle Formulations for CRISPR / CAS Components |
| WO2018089801A1 (en) | 2016-11-10 | 2018-05-17 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| WO2019081383A1 (en) | 2017-10-25 | 2019-05-02 | Universität Zürich | Eukaryotic initiation factor 4 recruiting aptamers for enhancing translation |
| US20230027864A1 (en) * | 2018-01-30 | 2023-01-26 | Modernatx, Inc. | Compositions and methods for delivery of agents to immune cells |
| WO2019236673A1 (en) | 2018-06-06 | 2019-12-12 | Massachusetts Institute Of Technology | Circular rna for translation in eukaryotic cells |
| WO2020237227A1 (en) | 2019-05-22 | 2020-11-26 | Massachusetts Institute Of Technology | Circular rna compositions and methods |
| WO2021021634A1 (en) | 2019-07-29 | 2021-02-04 | Georgia Tech Research Corporation | Nanomaterials containing constrained lipids and uses thereof |
| WO2021113777A2 (en) | 2019-12-04 | 2021-06-10 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2021155274A1 (en) | 2020-01-31 | 2021-08-05 | Modernatx, Inc. | Methods of preparing lipid nanoparticles |
| WO2021189059A2 (en) | 2020-03-20 | 2021-09-23 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2021226597A2 (en) | 2020-05-08 | 2021-11-11 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2021236855A1 (en) | 2020-05-19 | 2021-11-25 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| TW202228725A (en) * | 2020-10-14 | 2022-08-01 | 喬治梅森研究基金會 | Methods of lipid nanoparticle manufacture and compositions derived therefrom |
| WO2022261490A2 (en) | 2021-06-10 | 2022-12-15 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2023056033A1 (en) | 2021-09-30 | 2023-04-06 | Orna Therapeutics, Inc. | Lipid nanoparticle compositions for delivering circular polynucleotides |
| WO2023081526A1 (en) | 2021-11-08 | 2023-05-11 | Orna Therapeutics, Inc. | Lipid nanoparticle compositions for delivering circular polynucleotides |
Non-Patent Citations (50)
| Title |
|---|
| "ASHP Handbook on Injectable Drugs", 1986, TOISSEL, pages: 622 - 630 |
| "Pharmaceutics and Pharmacy Practice", 1982, J.B. LIPPINCOTT COMPANY, pages: 238 - 250 |
| ABRAHAM ET AL., SCIENCE, vol. 295, 2002, pages 64765 1 - 651 |
| BELLIVEAU, N.M. ET AL., MOL. THER. NUCLEIC. ACIDS. |
| BORDEN: "Molecular Basis of Cancer", 2015 |
| CHEN. D. ET AL., J. AM. CHEM. SOC., vol. 134, no. 22, 2012, pages 6948 - 51 |
| DHAMNE ET AL.: "Peripheral and thymic Foxp3+ regulatory T cells in search of origin, distinction, and function", FRONTIERS IN IMMUNOL., vol. 4, no. 253, 2013, pages 1 - 11 |
| DOBRIKOVA ET AL., PROC. NATL. ACAD. SCI., vol. 100, no. 25, 2003, pages 15125 - 15130 |
| EDGE, NATURE, vol. 292, 1981, pages 756 |
| ELIEL: "Stereochemistry of Carbon Compounds", 1962, MCGRAW-HILL |
| ESHHAR ET AL., CANCER IMMUNOL IMMUNOTHERAPY, vol. 45, 1997, pages 131 - 136 |
| FINNEY ET AL., JOURNAL OF IMMUNOLOGY, vol. 161, 1998, pages 2791 - 2797 |
| FRIESS M ET AL., FRONT. IMMUNOL., vol. 2947, no. 9, 2018 |
| GARLAPATI ET AL., J. BIOL. CHEM., vol. 279, no. 5, 2004, pages 3389 - 3397 |
| GROSS ET AL., AMUR. REV. PHARMACOL. TOXICOL., vol. 56, 2016, pages 59 - 83 |
| GUEVARA ET AL., FRONT. CHEM., vol. 8, 2020, pages 589959 |
| GURTU ET AL., BIOCHEM. BIOPHYS. RES. COMM., vol. 229, 1996, pages 295 - 298 |
| HILLERY A. M. ET AL.: "Drug Delivery and Targeting: For Pharmacists and Pharmaceutical Scientists", 2002, TAYLOR & FRANCIS, INC |
| JACQUES ET AL.: "Enantiomers, Racemates and Resolutions", 1981, WILEY INTERSCIENCE |
| JANEWAY C ET AL.: "Immunobiology: The Immune System in Health and Disease", 2001, GARLAND SCIENCE |
| JANG ET AL., J. VIROL., vol. 63, 1989, pages 1651 - 1660 |
| JAY ET AL., J. BIOL. CHEM. |
| JAYARAMAN ET AL., ANGEW. CHEM. INT. ED., vol. 51, 2012, pages 8529 - 8533 |
| JAYARAMAN ET AL., PROC. NATL. ACAD. SCI. USA, vol. 88, 1991, pages 4084 - 4088 |
| JONES ET AL., NATURE, vol. 54, 1986, pages 75 - 82 |
| KALOS ET AL., SCI TRANSL. MED., vol. 3, 2011, pages 95 |
| KAUFMAN ET AL., NUC. ACIDS RES., vol. 19, 1991, pages 4485 - 4490 |
| KLIBANOV ET AL., FEBS LETTERS, vol. 268, no. 1, 1990, pages 235 - 237 |
| KOBAYASHI ET AL., BIOTECHNIQUES, vol. 21, 1996, pages 399 - 402 |
| KRAUSE ET AL., J. EXP. MED., vol. 188, no. 4, 1998, pages 619 - 626 |
| KUBALL J ET AL., J EXP MED, vol. 206, no. 2, 2009, pages 463 - 475 |
| LEHTIMAKILAHESMAA: "Regulatory T cells control immune responses through their non-redundant tissue specific features", FRONTIERS IN IMMUNOL., vol. 4, no. 294, 2013, pages 1 - 10 |
| MOSSER ET AL., BIOTECHNIQUES, vol. 22, 1997, pages 150 - 161 |
| NAMBAIR ET AL., SCIENCE, vol. 223, 1984, pages 1299 |
| PORTER ET AL., N. ENGL. J. MED., vol. 365, 2011, pages 725 - 33 |
| QUEEN ET AL., PROC. NATL. ACAD. SCI. USA, vol. 86, 1989, pages 10029 - 10033 |
| RAMESH ET AL., NUCL. ACID RES., vol. 24, 1996, pages 2697 - 2700 |
| RIECHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 327 |
| RILEY ET AL., SCI. ADV., vol. 7, 2021, pages 1028 |
| ROBBINS ET AL., J IMMUNOL., vol. 180, 2008, pages 6116 - 6131 |
| ROSENBERG ET AL., NAT REV CANCER, vol. 8, no. 4, 2008, pages 299 - 308 |
| SONG ET AL., BLOOD, vol. 119, 2012, pages 696 - 706 |
| VERHOEYEN ET AL., SCIENCE, vol. 239, 1988, pages 1534 - 1536 |
| WADWA ET AL., J. DRUG TARGETING, vol. 3, 1995, pages 111 |
| WESSELHOEFT ET AL.: "Engineering circular RNA for Potent and Stable Translation in Eukaryotic Cells", NATURE COMMUNICATIONS, vol. 9, 2018, pages 2629, XP093230636, DOI: 10.1038/s41467-018-05096-6 |
| WESSELHOEFT ET AL.: "RNA Circularization Diminishes Immunogenicity and Can Extend Translation Duration In vivo", MOLECULAR CELL., vol. 74, no. 3, 2019, pages 508 - 520 |
| WHITESIDES, NATURE, vol. 442, 2006, pages 368 - 373 |
| WILEN ET AL.: "Tetrahedron", vol. 33, 1977, pages: 2725 |
| WILEN: "Tables of Resolving Agents and Optical Resolutions", 1972, UNIV. OF NOTRE DAME PRESS, pages: 268 |
| ZHIGALTSEV, I.V. ET AL., LANGMUIR, vol. 28, 2012, pages 3633 - 40 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12384741B2 (en) | Lipid nanoparticle compositions for delivering circular polynucleotides | |
| US20250283073A1 (en) | Circular RNA Compositions and Methods | |
| AU2022353839A1 (en) | Lipid nanoparticle compositions for delivering circular polynucleotides | |
| US12297285B2 (en) | Circular RNA encoding chimeric antigen receptors targeting BCMA | |
| US20250304994A1 (en) | Circular RNA Compositions and Methods | |
| WO2024102762A1 (en) | Lipids and lipid nanoparticle compositions for delivering polynucleotides | |
| AU2024269222A1 (en) | Circular rna compositions and methods | |
| WO2024205657A2 (en) | Lipids and lipid nanoparticle compositions for delivering polynucleotides | |
| WO2025117969A1 (en) | Process for manufacturing lipid nanoparticles | |
| WO2025049690A1 (en) | Circular polyethylene glycol lipids | |
| WO2025007148A1 (en) | Polymer lipid nanoparticle compositions for delivering circular polynucleotides | |
| WO2026006593A1 (en) | Synthetic internal ribosome entry sites | |
| TW202425959A (en) | Lipids and nanoparticle compositions for delivering polynucleotides | |
| WO2025166238A1 (en) | Fast-shedding polyethylene glycol lipids | |
| EA049788B1 (en) | CIRCULAR RNA COMPOSITIONS AND METHODS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24827593 Country of ref document: EP Kind code of ref document: A1 |