WO2025073870A1 - Parg inhibitory compound - Google Patents
Parg inhibitory compound Download PDFInfo
- Publication number
- WO2025073870A1 WO2025073870A1 PCT/EP2024/077899 EP2024077899W WO2025073870A1 WO 2025073870 A1 WO2025073870 A1 WO 2025073870A1 EP 2024077899 W EP2024077899 W EP 2024077899W WO 2025073870 A1 WO2025073870 A1 WO 2025073870A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- crystal form
- pharmaceutically acceptable
- compound
- formula
- acceptable crystal
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
Definitions
- PARG is the dominant cellular PAR degrading enzyme, it cannot act on the terminal protein-ribose bond.
- Additional hydrolases such as terminal ADP-ribose protein glycohydrolase (TARG1) and ADP-ribosylhydrolase 3 (ARH3) are also known to catalyze PAR-degradation.
- TARG1 and ARH3 complete the reversal of PARylation by removing protein-bound mono(ADP-ribose) moieties (a) Fontana et al. Elife 2017, doi: 10.7554/eLife.28533; b) Rack et al. Genes Dev. 2020, 34, 263).
- TARG1 is located in the nucleus and cytoplasm.
- ARH3 is found primarily in the cytoplasm but it can also be found in the mitochondria and in the nucleus (Rack et al. Genes Dev. 2020, 34, 263).
- PARG participates in DNA replication and in various DNA repair mechanisms including singlestrand break (SSB) repair and replication fork restart.
- SSB singlestrand break
- PARG inhibitors have shown synthetic lethal phenotype in cells with high levels of DRS caused by low expression of genes involved in DNA replication and/or replication fork stability (Pillay et al. Cancer Cell. 2019, 35, 519).
- PARG inactivation, depletion or inhibition sensitizes cells to irradiation and to DNA damaging agents such as alkylating agents (e.g. temozolomide and methyl methanesulfonate) (a) Fujihara et al, Curr. Cancer Drug Targets 2009, 9, 953; b) Gogola et al. Cancer Cell 2018, 33, 1078; c) Houl et al, Nat Commun. 2019, 10, 5654).
- alkylating agents e.g. temozolomide and methyl methanesulfonate
- the compound of formula (I) is useful for treating a disease or disorder in which PARG activity is implicated.
- an “aryl” preferably has 6 to 14 ring atoms, more preferably 6 to 10 ring atoms, even more preferably refers to phenyl or naphthyl, and most preferably refers to phenyl.
- heteroaryl refers to an aromatic ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic), wherein said aromatic ring group comprises one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from 0, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group).
- aromatic ring group comprises one or more (such as, e.g., one, two, three
- cycloalkyl refers to a saturated hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings).
- Cycloalkyl may, e.g., refer to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, decalinyl (i.e., decahydronaphthyl), or adamantyl.
- Heterocycloalkyl may, e.g., refer to aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, azepanyl, diazepanyl (e.g., 1 ,4-diazepanyl), oxazolidinyl, isoxazolidinyl, thiazolidinyl, isothiazolidinyl, morpholinyl (e.g., morpholin-4-yl), thiomorpholinyl (e.g., thiomorpholin-4-yl), oxazepanyl, oxiranyl, oxetanyl, tetrahydrofuranyl, 1 ,3-dioxolanyl, tetrahydropyranyl, 1 ,4-dioxanyl, oxepany
- heterocycloalkyl preferably refers to a 3 to 11 membered saturated ring group, which is a monocyclic ring or a fused ring system (e.g., a fused ring system composed of two fused rings), wherein said ring group contains one or more (e.g., one, two, three, or four) ring heteroatoms independently selected from 0, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized; more preferably, “heterocycloalkyl” refers to a 5 to 7 membered saturated monocyclic ring group containing one or more (e.g., one, two, or three) ring heteroatoms independently selected from 0, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms
- each heteroatom-containing ring comprised in said unsaturated alicyclic ring group may contain one or two 0 atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring.
- halogen refers to fluoro (-F), chloro (-CI), bromo (-Br), or iodo (-I).
- compositions comprising “a” compound of formula (I) can be interpreted as referring to a composition comprising “one or more” compounds of formula (I).
- the term “about” is used in connection with the endpoints of a range, it preferably refers to the range from the lower endpoint -10% of its indicated numerical value to the upper endpoint +10% of its indicated numerical value, more preferably to the range from of the lower endpoint -5% to the upper endpoint +5%, and even more preferably to the range defined by the exact numerical values of the lower endpoint and the upper endpoint.
- the term “comprising” (or “comprise”, “comprises”, “contain”, “contains”, or “containing”), unless explicitly indicated otherwise or contradicted by context, has the meaning of “containing, inter alia”, i.e., “containing, among further optional elements, ...”. In addition thereto, this term also includes the narrower meanings of “consisting essentially of’ and “consisting of’. For example, the term “A comprising B and C” has the meaning of “A containing, inter alia, B and C”, wherein A may contain further optional elements (e.g .
- the present invention relates to pharmaceutically acceptable crystal form of a compound of formula (I):
- the compound of formula (I) is present in the crystal form in a free base form (or, in other words, in a non-salt form). It is to be understood that said pharmaceutically acceptable crystal form is obtainable upon recrystallization of the compound of formula (I) from ethanol.
- the particular crystal form of the compound of formula (I), referred to herein, is characterized by outstanding stability, high solubility and negligible hygroscopicity. Thus, these exceptional properties of the claimed crystal form allow for the crystal form to be formulated for both clinical use and the long-term storage.
- the formulation for clinical use is not particularly limited and may refer to, for example, to formulation as tablet.
- the crystal form of the invention is a crystal form of the compound of formula (I).
- said compound of formula (I) is present as a free base, and not as a salt in the crystal form of the invention.
- crystal form 1 of the invention comprises the compound of formula (I) in its free-base form.
- the crystal form consists substantially of the compound of formula (I).
- the compound of formula (I) preferably constitutes at least 95% w/w of the crystal form, more preferably at least 96% w/w of the crystal form, even more preferably at least 97% w/w of the crystal form, still more preferably at least 98% w/w of the crystal form, again more preferably at least 99% w/w of the crystal form.
- the step of complete dissolution in ethanol is performed while stirring, mixing or agitating in any way the solution or the suspension.
- the step of cooling to room temperature of the obtained solution is performed while stirring (or mixing or agitating the so obtained solution in any way known to the skilled person).
- the step of dissolution is performed at elevated temperature.
- said step is performed preferably under reflux.
- dissolution of the compound of formula (I) in ethanol is performed at a temperature of 110 °C.
- temperature of 110 °C refers to a temperature of heating body, such as an oil batch, in which the reaction vessel including said ethanol is heated up.
- ethanol can be brought to boiling under these conditions (such as under reflux conditions). Accordingly, and preferably, the step of dissolution is performed in boiling ethanol.
- the crystal form of the invention is shown in Figure 1.
- the crystal form of the present invention is characterized by the X-ray powder diffraction pattern obtained using CuKa radiation that comprises at least one peak selected from the peaks at 12.24 ⁇ 0.2 °20, 20.04 ⁇ 0.2 °20, 20.40 ⁇ 0.2 °20 and 24.02 ⁇ 0.2 °20. These peaks are characteristic for the currently claimed crystal form of the present invention, and not for other crystal forms, in particular not for form 2 of the compound of formula (I).
- the X-ray powder diffraction pattern obtained using CuKa radiation for the claimed crystal form (also referred to crystal form 1) comprises at least two peaks, or at least three peaks selected from the peaks at 12.24 ⁇ 0.2 °20, 20.04 ⁇ 0.2 °20, 20.40 ⁇ 0.2 °20 and 24.02 ⁇ 0.2 °20.
- the X-ray powder diffraction pattern obtained using CuKa radiation for the claimed crystal form further comprises the peaks at 12.24 ⁇ 0.2 °20, 20.04 ⁇ 0.2 °20, 20.40 ⁇ 0.2 °20 and 24.02 ⁇ 0.2 °20.
- the crystal form is characterized by an X-ray powder diffraction pattern obtained using CuKa radiation additionally comprising a peak at 18.71 ⁇ 0.2 °20, preferably 18.71 ⁇ 0.1 °20, and/or a peak at 6.86 ⁇ 0.2 °20.
- the peak at 18.71 ⁇ 0.2 °20 is the most prominent peak in the X-ray powder diffraction powder for the compound of formula, as clearly and unambiguously seen from Figure 1 .
- the X-ray powder diffraction pattern further comprises at least one peak selected from the peaks at 15.42 ⁇ 0.2 °20, 19.04 ⁇ 0.2 °20, 21 .59 ⁇ 0.2 °20, 23.19 ⁇ 0.2 °20, 23.58 ⁇ 0.2 °20, 28.01 ⁇ 0.2 °20, and 29.18 ⁇ 0.2 °20.
- the X-ray powder diffraction pattern further comprises at least two peaks selected from the peaks at 15.42 ⁇ 0.2 °20, 19.04 ⁇ 0.2 °20, 21 .59 ⁇ 0.2 °20, 23.19 ⁇ 0.2 °20, 23.58 ⁇ 0.2 °20, 28.01 ⁇ 0.2 °20, and 29.18 ⁇ 0.2 °20.
- the X-ray powder diffraction pattern further comprises at least four peaks selected from the peaks at 15.42 ⁇ 0.2 °20, 19.04 ⁇ 0.2 °20, 21 .59 ⁇ 0.2 °20, 23.19 ⁇ 0.2 °20, 23.58 ⁇ 0.2 °20, 28.01 ⁇ 0.2 °20, and 29.18 ⁇ 0.2 °20.
- the pharmaceutically acceptable crystal form of the present invention is characterized by a melting point of 207.5 ⁇ 4.0 °C and by the X-ray powder diffraction pattern obtained using CuKa radiation that comprises at least one peak, at least two peaks, at least three peaks, or four peaks selected from the peaks at 12.24 ⁇ 0.2 °20, 20.04 ⁇ 0.2 °20, 20.40 ⁇ 0.2 °20 and 24.02 ⁇ 0.2 °20.
- the crystal form 1 is characterized by a melting point of 207.5 ⁇ 2.0 °C and by the X-ray powder diffraction pattern obtained using CuKa radiation that comprises at least one peak, at least two peaks, at least three peaks, or four peaks selected from the peaks at 12.24 ⁇ 0.2 °20, 20.04 ⁇ 0.2 °20, 20.40 ⁇ 0.2 °20 and 24.02 ⁇ 0.2 °20.
- the crystal form 1 is characterized by a melting point of 207.5 ⁇ 1 .0 °C and by the X-ray powder diffraction pattern obtained using CuKa radiation that comprises at least one peak, at least two peaks, at least three peaks, or four peaks selected from the peaks at 12.24 ⁇ 0.2 °20, 20.04 ⁇ 0.2 °20, 20.40 ⁇ 0.2 °20 and 24.02 ⁇ 0.2 °20.
- the crystal form 1 is characterized by a melting point of 207.5 ⁇ 0.5 °C and by the X-ray powder diffraction pattern obtained using CuKa radiation that comprises at least one peak, at least two peaks, at least three peaks, or four peaks selected from the peaks at 12.24 ⁇ 0.2 °20, 20.04 ⁇ 0.2 °20, 20.40 ⁇ 0.2 °20 and 24.02 ⁇ 0.2 °20.
- the crystal form is characterized by an X-ray powder diffraction pattern obtained using CuKa radiation additionally comprising a peak at 18.71 ⁇ 0.2 °20, preferably 18.71 ⁇ 0.1 °20, and/or a peak at 6.86 ⁇ 0.2 °20.
- the X-ray powder diffraction pattern further comprises at least three peaks selected from the peaks at 15.42 ⁇ 0.2 °20, 19.04 ⁇ 0.2 °20, 21.59 ⁇ 0.2 °20, 23.19 ⁇ 0.2 °20, 23.58 ⁇ 0.2 °20, 28.01 ⁇ 0.2 °20, and 29.18 ⁇ 0.2 °20.
- the X-ray powder diffraction pattern further comprises at least five peaks selected from the peaks at 15.42 ⁇ 0.2 °20, 19.04 ⁇ 0.2 °20, 21 .59 ⁇ 0.2 °20, 23.19 ⁇ 0.2 °20, 23.58 ⁇ 0.2 °20, 28.01 ⁇ 0.2 °20, and 29.18 ⁇ 0.2 °20.
- the present invention further relates to a method for making the pharmaceutically acceptable crystal form 1 of the present invention.
- the method comprises the step of crystallizing the compound of formula (I) as depicted herein from ethanol or from the mixture of THF/n-heptane.
- the compound is to be crystallized from ethanol.
- the method for making the pharmaceutically acceptable crystal form 1 of the present invention comprises the step of crystallizing the compound of formula (I) from the mixture of THF/n-heptane.
- said crystallization is to be performed with seeding using the crystal form 1 .
- crystal form 2 provided as a reference example, can be obtained as described in the experimental section hereinbelow.
- crystal form 2 is obtainable by trituration of the compound of formula (I) from DCM.
- Detailed experimental protocol is provided herein.
- Dosage forms for nasal administration can be administered via inhalation and insufflation, for example by a metered inhaler.
- Dosage forms for topical administration include creams, gels, ointments, salves, patches and transdermal delivery systems.
- suitable parenteral formulations under sterile conditions is readily accomplished by standard pharmaceutical techniques well known to those skilled in the art.
- Said compounds or pharmaceutical compositions can also be administered orally in the form of tablets, capsules, ovules, elixirs, solutions or suspensions, which may contain flavoring or coloring agents, for immediate-, delayed-, modified-, sustained-, pulsed- or controlled-release applications.
- the compounds or pharmaceutical compositions are preferably administered by oral ingestion, particularly by swallowing.
- the compounds or pharmaceutical compositions can thus be administered to pass through the mouth into the gastrointestinal tract, which can also be referred to as “oral-gastrointestinal” administration.
- sustained-release compositions include semi-permeable polymer matrices in the form of shaped articles, e.g., films, or microcapsules.
- Sustained-release matrices include, e.g., polylactides, copolymers of L-glutamic acid and gamma-ethyl-L-glutamate, poly(2-hydroxyethyl methacrylate), ethylene vinyl acetate, or poly-D-(-)-3-hydroxybutyric acid.
- Sustained-release pharmaceutical compositions also include liposomally entrapped compounds. The present invention thus also relates to liposomes containing a compound of the invention.
- Said compounds or pharmaceutical compositions may also be administered by the pulmonary route, rectal routes, or the ocular route.
- they can be formulated as micronized suspensions in isotonic, pH adjusted, sterile saline, or, preferably, as solutions in isotonic, pH adjusted, sterile saline, optionally in combination with a preservative such as a benzalkonium chloride.
- a preservative such as a benzalkonium chloride.
- they may be formulated in an ointment such as petrolatum. It is also envisaged to prepare dry powder formulations of the compounds of formula (I) for pulmonary administration, particularly inhalation.
- said compounds or pharmaceutical compositions can be formulated as a suitable ointment containing the active compound suspended or dissolved in, for example, a mixture with one or more of the following: mineral oil, liquid petrolatum, white petrolatum, propylene glycol, emulsifying wax and water.
- they can be formulated as a suitable lotion or cream, suspended or dissolved in, for example, a mixture of one or more of the following: mineral oil, sorbitan monostearate, a polyethylene glycol, liquid paraffin, polysorbate 60, cetyl esters wax, 2-octyldodecanol, benzyl alcohol and water.
- a physician will determine the actual dosage which will be most suitable for an individual subject.
- the specific dose level and frequency of dosage for any particular individual subject may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the individual subject undergoing therapy.
- the present invention also provides a method of selectively inhibiting PARG enzyme activity over PARP1 or ARH3 enzyme activity in vitro or in vivo.
- the said method comprises the steps of contacting a cell with an effective amount of a pharmaceutically acceptable crystal form of the compound, as defined herein.
- proliferative disorder are used interchangeably herein and pertain to an unwanted or uncontrolled cellular proliferation of excessive or abnormal cells which is undesired, such as, neoplastic or hyperplastic growth, whether in vitro or in vivo.
- proliferative conditions include, but are not limited to, pre-malignant and malignant cellular proliferation, including but not limited to, malignant neoplasms and tumours, cancers, leukemias, psoriasis, bone diseases, fibroproliferative disorders (e.g . , of connective tissues), and atherosclerosis.
- any type of cell may be treated, including but not limited to, gastric, lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin, preferably lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin.
- the cancer to be treated is selected from gastric, lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin cancer, more preferably lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin cancer.
- the anti-proliferative effects of the compound of formula (I) of the present invention have particular application in the treatment of human cancers (by virtue of their inhibition of PARG enzyme activity).
- the anti-cancer effect may arise through one or more mechanisms, including but not limited to, the regulation of cell proliferation, the inhibition of angiogenesis (the formation of new blood vessels), the inhibition of metastasis (the spread of a tumour from its origin), the inhibition of invasion (the spread of tumour cells into neighbouring normal structures), or the promotion of apoptosis (programmed cell death).
- antiproliferative/antineoplastic drugs and combinations thereof as used in medical oncology, such as alkylating agents (for example cis-platin, oxaliplatin, carboplatin, cyclophosphamide, nitrogen mustard, melphalan, chlorambucil, busulphan, temozolomide and nitrosoureas); antimetabolites (for example gemcitabine and antifolates such as fluoropyrimidines like 5-fluorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside, and hydroxyurea); antitumour antibiotics (for example anthracyclines like adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin); antimitotic agents (for example vinca alkaloids like vincristine, vinblast
- cytostatic agents such as antioestrogens (for example tamoxifen, fulvestrant, toremifene, raloxifene, droloxifene and iodoxyfene), antiandrogens (for example bicalutamide, flutamide, nilutamide and cyproterone acetate), LHRH antagonists or LHRH agonists (for example goserelin, leuprorelin and buserelin), progestagens (for example megestrol acetate), aromatase inhibitors (for example as anastrozole, letrozole, vorazole and exemestane) and inhibitors of 5oc-reductase such as finasteride;
- antioestrogens for example tamoxifen, fulvestrant, toremifene, raloxifene, droloxifene and iodoxyfene
- antiandrogens for example
- the antiproliferative treatment defined hereinbefore may involve, in addition to the compound of formula (I) of the invention, conventional surgery or radiotherapy or chemotherapy.
- Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate dosing of the individual components of the treatment.
- Such combination products employ the compounds of this invention within the dosage range described hereinbefore and the other pharmaceutically-active agent within its approved dosage range.
- the present invention further relates to the pharmaceutically acceptable crystal form of the compound of formula (I), as defined herein, for use in the treatment of a cancer (for example a cancer involving a solid tumour) in combination with another anti-tumour agent.
- the antitumour agent is preferably selected from the anti-tumour agents as listed hereinabove.
- Method 4 SHIMADZU LCMS-2020 Kinetex® EVO C18 2.1X20 mm 2.6 urn at 50°C; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 2.0 mL/min; eluted with the mobile phase over 1.00 min employing UV detection at 220 nm and 254 nm. Gradient information: 0.01-0.60 min, ramped from 95% A-5% B to 5% A-95% B; 0.61-0.78 min, held at 5% A-95% B; 0.78-0.79 min, returned to 95% A-5% B, 0.79-0.80 min, held at 95% A-5% B.
- 1 H NMR spectra were acquired on a Bruker Avance HI spectrometer at 400 MHz using residual undeuterated solvent as reference. 1 H NMR signals are specified with their multiplicity / combined multiplicities as apparent from the spectrum; possible higher-order effects are not considered. Chemical shifts of the signals (6) are specified as ppm (parts per million).
- dichloro compound 1 ,8-dichloro-3- (5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-N-(1-methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide was also formed in this process.
- Vessel 2 was heated to 60°C then N-heptane (0.5 L) was added slowly, followed by 3-[5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl]-N-(1- methylcyclopropyl)-8-[(3S ,5S)-3 , 5-d i methyl pi perazin-1 -yl]imidazo[1 , 5-a]pyrid ine-6-su Ifonamide form 1 seed (1 g, 0.002 mol). The mixture was held at 55-65°C, then additional n-heptane (4 L) was added slowly to the vessel. The mixture was cooled slowly to 5°C, stirred overnight and filtered.
- DSC data was collected on a TA Instruments Discovery DSC equipped with a 54-position sample holder. The instrument was verified for energy and temperature calibration using certified indium. A predefined amount of the sample, 0.5-2.0 mg, was placed in a Tzero Pan with a Tzero Hermetic Lid and heated at 20 °C min- 1 from 30 to 350 °C or varied as experimentation dictated. A purge of dry nitrogen at 50 mL min- 1 was maintained over the sample. The instrument control, data acquisition and analysis were performed with TA Instruments TRIOS software v5.5.0.323. All results feature endotherm up convention. The results of the measurements are shown in Figure 3 for crystal form 1 . Figure 5 shows the reference results for crystal form 2.
- TGA data was collected on a TA Instruments Discovery TGA equipped with a 25-position autosampler.
- the instrument was calibrated using a certified weight and certified Alumel and Nickel for temperature.
- a predefined amount of the sample ca. 5 mg, was loaded into a pre-tared aluminium ACCUPIK sample pan and platinum crucible and was heated at 20 °C min 1 from ambient temperature to 400 °C unless otherwise stated.
- a nitrogen purge at 25 mL min 1 was maintained over the sample.
- the instrument control, data acquisition and analysis were performed with TA Instruments TRIOS software v5.5.0.323. The results of the measurements are shown in Figure 3 for crystal form 1 .
- Figure 5 shows the reference results for crystal form 2.
- Results are summarised in Table 4.
- XRPD data is presented in Figures 7 to 10. Data presented is for solids post drying; XRPD analysis of damp cakes showed no differences to dried solids.
- FaSSGF was ready to use.
- Solubility data of form 1 are reported in the table below.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to a pharmaceutically acceptable crystal form of a compound of formula (I) wherein said pharmaceutically acceptable crystal is obtainable upon recrystallization of the compound of formula (I) from ethanol. The crystal form is characterized by outstanding stability, good solubility and low hygroscopicity, and is particularly suitable for long-term storage and clinical development.
Description
PARG inhibitory compound
Field of the invention
The present invention relates to a pharmaceutically acceptable crystal form of a compound of formula (I), wherein said pharmaceutically acceptable crystal is obtainable upon recrystallization of the compound of formula (I) from ethanol. The crystal form is characterized by outstanding stability, good solubility and low hygroscopicity, and is particularly suitable for long-term storage and clinical development.
Background of the invention
Cancer is a leading cause of death worldwide. Although progression-free survival and overall survival of cancer patients has improved over the past two decades, millions of cancer patients still have few therapeutic options and poor survival outcomes (Jemal et al., J. Natl. Cancer Inst. 2017, 109, 1975).
DNA replication stress (DRS) is a hallmark of cancer cells and a major source of genomic instability (a) Halazonetis et al., Science 2008, 319, 1352; b) Negrini et al., Nat. Rev. Mol. Cell Biol. 2010, 11 , 220). In broad terms, DRS refers to the deregulation of DNA replication and cell cycle progression. DRS can be induced from endogenous or exogenous causes such as oncogene activation and chemotherapeutics, respectively (Zeman and Cimprich, Nat. Cell Biol. 2013, 16, 2). At the level of the replication fork, DRS leads to replication fork stalling, disengagement of the replisome and eventually collapse. Several DNA repair proteins are involved in replication fork stability, protection, and restart under DRS conditions (a) Costantino et al., Science 2014, 343, 88; b) Scully et al., Curr. Opin. Genet. Dev. 2021 71 , 154).
Poly(ADP)ribosylation (PARylation) is a transient and reversible post-translational modification that occurs at DNA damaged sites and is catalyzed by the poly (ADP-ribose) polymerase (PARP) family of proteins (Cohen and Chang, Nat. Chem. Biol. 2018, 14, 236). PARylation of various DNA repair proteins leads to their activation. Degradation of the poly(ADP) ribose chains is mediated primarily by the poly(ADP-ribose) glycohydrolase (PARG) protein. DNA damage dependent PARylation/dePARylation is a rapid and dynamic process which needs to be well regulated since imbalances between the two processes can lead to DNA damage.
Human PARG encodes a 111 kDa protein of 976 amino acids. It contains a N-terminal regulatory domain, a catalytic domain and an ADP-ribose binding macrodomain. Five human PARG transcripts have been identified. Full length PARG is mostly nuclear; the smaller isoforms localize primarily to the cytoplasm. PARG functions primarily as an exo-hydrolase and it releases mainly mono(ADP-ribose) by
hydrolyzing the a-O-glycosidic ribose-ribose bond in PAR. PARG can also act as an endo-hydrolase. PARG preferentially degrades long and linear PAR chains whereas its activity with small and branched PAR chains is significantly reduced (O’Sullivan et al., Nat. Commun. 2019, 10, 1182).
Although PARG is the dominant cellular PAR degrading enzyme, it cannot act on the terminal protein-ribose bond. Additional hydrolases such as terminal ADP-ribose protein glycohydrolase (TARG1) and ADP-ribosylhydrolase 3 (ARH3) are also known to catalyze PAR-degradation. TARG1 and ARH3 complete the reversal of PARylation by removing protein-bound mono(ADP-ribose) moieties (a) Fontana et al. Elife 2017, doi: 10.7554/eLife.28533; b) Rack et al. Genes Dev. 2020, 34, 263). TARG1 is located in the nucleus and cytoplasm. ARH3 is found primarily in the cytoplasm but it can also be found in the mitochondria and in the nucleus (Rack et al. Genes Dev. 2020, 34, 263).
Genomic aberrations targeting tumor suppressor genes or oncogenes, often make cancer cells dependent on specific DNA repair pathways. For instance, it is well known that PARP inhibitors are particularly effective against tumors carrying mutations in the BRCA1 and BRCA2 genes (a) Bryant et al. Nature 2005, 434, 913; b) Farmer et al. Nature 2005, 434, 917). Targeting synthetic lethal interactions like the one between PARP and BRCA is an attractive novel therapeutic approach for cancer treatment.
PARG participates in DNA replication and in various DNA repair mechanisms including singlestrand break (SSB) repair and replication fork restart. PARG inhibitors have shown synthetic lethal phenotype in cells with high levels of DRS caused by low expression of genes involved in DNA replication and/or replication fork stability (Pillay et al. Cancer Cell. 2019, 35, 519). Moreover, PARG inactivation, depletion or inhibition sensitizes cells to irradiation and to DNA damaging agents such as alkylating agents (e.g. temozolomide and methyl methanesulfonate) (a) Fujihara et al, Curr. Cancer Drug Targets 2009, 9, 953; b) Gogola et al. Cancer Cell 2018, 33, 1078; c) Houl et al, Nat Commun. 2019, 10, 5654).
Given the therapeutic potential of PARG inhibitors in cancer treatment, there is an increased need for the development of highly potent and selective PARG inhibitors beyond the ones that have already been described (a) James et al, ACS Chem. Biol. 2016, 11 , 3179; b) Waszkowycz et al, J. Med. Chem. 2018, 61 , 10767).
Certain compounds that are useful as PARG inhibitors are further disclosed in documents WO 2016/092326, WO 2016/097749 and WO 2021/055744.
Document US 2019/233411 discloses certain Gcn2 inhibitors and uses thereof.
Document WO 2009/050183 discloses certain imidazo[1 ,2-a]pyridine derivatives which are useful for treating diseases mediated by the ALK-5 and/or ALK-4 receptor.
Document WO 2023/057389 discloses further potent PARG inhibitors based on imidazo[1 ,5- a]pyridine scaffold.
Summary of the invention
It was an objective technical problem of the present invention to provide compounds that are cell- permeable inhibitors of PARG in a form that enables long term storage and is particularly suitable for future clinical use. The technical problem of the present invention is solved by the embodiments described herein and as characterized by the claims.
In one embodiment, the present invention provides a pharmaceutically acceptable crystal form of a compound of formula (I):
 wherein said pharmaceutically acceptable crystal form is obtainable upon recrystallization of the compound of formula (I) from ethanol.
wherein said pharmaceutically acceptable crystal form is obtainable upon recrystallization of the compound of formula (I) from ethanol.
A further embodiment of the present invention relates to a pharmaceutical composition comprising the pharmaceutically acceptable crystal form of the compound of formula (I) of the invention, and a pharmaceutically acceptable carrier.
In a further embodiment, the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I) of the invention, or a pharmaceutical composition of the present invention, for use in therapy.
The compound of formula (I) is useful for treating a disease or disorder in which PARG activity is implicated.
The compound of formula (I) is useful fora method of treating a proliferative disorder. In a preferred embodiment of the present invention, the proliferative disorder is cancer, preferably a human cancer.
Definitions
The following definitions apply throughout the present specification and the claims, unless specifically indicated otherwise.
The term “hydrogen” is herein used to refer to protium, deuterium and/or tritium, preferably to protium. Accordingly, the term “non-hydrogen atom” refers to any atoms that is not hydrogen, i.e. that is not protium, deuterium or tritium.
The term “hydrocarbon group” refers to a group consisting of carbon atoms and hydrogen atoms.
The term “alicyclic” is used in connection with cyclic groups and denotes that the corresponding cyclic group is non-aromatic.
As used herein, the term “alkyl” refers to a monovalent saturated acyclic (i.e., non-cyclic) hydrocarbon group which may be linear or branched. Accordingly, an “alkyl” group does not comprise any carbon-to-carbon double bond or any carbon-to-carbon triple bond. A “C1-5 alkyl” denotes an alkyl group having 1 to 5 carbon atoms. Preferred exemplary alkyl groups are methyl, ethyl, propyl (e.g., n-propyl or isopropyl), or butyl (e.g., n-butyl, isobutyl, sec-butyl, or tert-butyl). Unless defined otherwise, the term “alkyl” preferably refers to C alkyl, more preferably to methyl or ethyl, and even more preferably to methyl.
As used herein, the term “carbocyclyl” refers to a hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings), wherein said ring group may be saturated, partially unsaturated (i.e., unsaturated but not aromatic) or aromatic. Unless defined otherwise, “carbocyclyl” preferably refers to aryl, cycloalkyl or cycloalkenyl.
As used herein, the term “heterocyclyl” refers to a ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings), wherein said ring group comprises one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from 0, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group), and further wherein said ring group may be saturated, partially unsaturated (i.e., unsaturated but not aromatic) or aromatic. For example, each heteroatom-containing ring comprised in said ring group may contain one or two 0 atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. Unless defined otherwise, “heterocyclyl” preferably refers to heteroaryl, heterocycloalkyl or heterocycloalkenyl.
As used herein, the term “aryl” refers to an aromatic hydrocarbon ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic). “Aryl” may, e.g., refer to phenyl, naphthyl, dialinyl (i.e., 1 ,2-dihydronaphthyl), tetralinyl (i.e., 1 ,2,3,4-tetrahydronaphthyl), indanyl, indenyl (e.g., 1 H-indenyl), anthracenyl, phenanthrenyl, 9H- fluorenyl, or azulenyl. Unless defined otherwise, an “aryl” preferably has 6 to 14 ring atoms, more
preferably 6 to 10 ring atoms, even more preferably refers to phenyl or naphthyl, and most preferably refers to phenyl.
As used herein, the term “heteroaryl” refers to an aromatic ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic), wherein said aromatic ring group comprises one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from 0, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group). For example, each heteroatom-containing ring comprised in said aromatic ring group may contain one or two 0 atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heteroaryl” may, e.g., refer to thienyl (i.e., thiophenyl), benzo[b]thienyl, naphtho[2,3-b]thienyl, thianthrenyl, furyl (i.e., furanyl), benzofuranyl, isobenzofuranyl, chromanyl, chromenyl (e.g., 2H-1- benzopyranyl or 4H-1 -benzopyranyl), isochromenyl (e.g., 1 H-2-benzopyranyl), chromonyl, xanthenyl, phenoxathiinyl, pyrrolyl (e.g., 1 H-pyrrolyl), imidazolyl, pyrazolyl, pyridyl (i.e., pyridinyl; e.g., 2-pyridyl, 3- pyridyl, or 4-pyridyl), pyrazinyl, pyrimidinyl, pyridazinyl, indolyl (e.g., 3H-indolyl), isoindolyl, indazolyl, indolizinyl, purinyl, quinolyl, isoquinolyl, phthalazinyl, naphthyridinyl, quinoxalinyl, cinnolinyl, pteridinyl, carbazolyl, p-carbolinyl, phenanthridinyl, acridinyl, perimidinyl, phenanthrolinyl (e.g., [1 ,10]phenanthrolinyl, [1 ,7]phenanthrolinyl, or [4,7]phenanthrolinyl), phenazinyl, thiazolyl, isothiazolyl, phenothiazinyl, oxazolyl, isoxazolyl, oxadiazolyl (e.g., 1 ,2,4-oxadiazolyl, 1 ,2,5-oxadiazolyl (i.e., furazanyl), or 1 ,3,4-oxadiazolyl), thiadiazolyl (e.g., 1 ,2,4-thiadiazolyl, 1 ,2,5-thiadiazolyl, or 1 ,3,4-thiadiazolyl), phenoxazinyl, pyrazolo[1 ,5-a]pyrimidinyl (e.g., pyrazolo[1 ,5-a]pyrimidin-3-yl), 1 ,2-benzoisoxazol-3-yl, benzothiazolyl, benzothiadiazolyl, benzoxazolyl, benzisoxazolyl, benzimidazolyl, benzo[b]thiophenyl (i.e., benzothienyl), triazolyl (e.g., 1 H-1 ,2,3-triazolyl, 2H-1 ,2,3-triazolyl, 1 H-1 ,2,4-triazolyl, or 4H-1 ,2,4-triazolyl), benzotriazolyl, 1 H-tetrazolyl, 2H-tetrazolyl, triazinyl (e.g., 1 ,2,3-triazinyl, 1 ,2,4-triazinyl, or 1 ,3,5-triazinyl), furo[2,3-c]pyridinyl, dihydrofuropyridinyl (e.g., 2,3-dihydrofuro[2,3-c]pyridinyl or 1 ,3-dihydrofuro[3,4- c]pyridinyl), imidazopyridinyl (e.g., imidazo[1 ,2-a]pyridinyl or imidazo[3,2-a]pyridinyl), quinazolinyl, thienopyridinyl, tetrahydrothienopyridinyl (e.g., 4,5,6,7-tetrahydrothieno[3,2-c]pyridinyl), dibenzofuranyl, 1 ,3-benzodioxolyl, benzodioxanyl (e.g., 1 ,3-benzodioxanyl or 1 ,4-benzodioxanyl), or coumarinyl. Unless defined otherwise, the term “heteroaryl” preferably refers to a 5 to 14 membered (more preferably 5 to 10
membered) monocyclic ring or fused ring system comprising one or more (e.g., one, two, three or four) ring heteroatoms independently selected from 0, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized; even more preferably, a “heteroaryl” refers to a 5 or 6 membered monocyclic ring comprising one or more (e.g., one, two or three) ring heteroatoms independently selected from 0, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized.
As used herein, the term “cycloalkyl” refers to a saturated hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings). “Cycloalkyl” may, e.g., refer to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, decalinyl (i.e., decahydronaphthyl), or adamantyl. Unless defined otherwise, “cycloalkyl” preferably refers to a C3-11 cycloalkyl, and more preferably refers to a C3-7 cycloalkyl. A particularly preferred “cycloalkyl” is a monocyclic saturated hydrocarbon ring having 3 to 7 ring members (e.g., cyclopropyl or cyclohexyl).
As used herein, the term “cycloalkenyl” refers to an unsaturated alicyclic (non-aromatic) hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said hydrocarbon ring group comprises one or more (e.g., one or two) carbon-to-carbon double bonds and does not comprise any carbon-to-carbon triple bond. “Cycloalkenyl” may, e.g., refer to cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cyclohexadienyl, cycloheptenyl, or cycloheptadienyl. Unless defined otherwise, “cycloalkenyl” preferably refers to a C3-11 cycloalkenyl, and more preferably refers to a C3-7 cycloalkenyl. A particularly preferred “cycloalkenyl” is a monocyclic unsaturated alicyclic hydrocarbon ring having 3 to 7 ring members and containing one or more (e.g., one or two; preferably one) carbon-to-carbon double bonds.
As used herein, the term “heterocycloalkyl” refers to a saturated ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said ring group contains one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from 0, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group). For example, each heteroatom-containing ring comprised in said saturated ring group may contain one or two 0 atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the
corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heterocycloalkyl” may, e.g., refer to aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, azepanyl, diazepanyl (e.g., 1 ,4-diazepanyl), oxazolidinyl, isoxazolidinyl, thiazolidinyl, isothiazolidinyl, morpholinyl (e.g., morpholin-4-yl), thiomorpholinyl (e.g., thiomorpholin-4-yl), oxazepanyl, oxiranyl, oxetanyl, tetrahydrofuranyl, 1 ,3-dioxolanyl, tetrahydropyranyl, 1 ,4-dioxanyl, oxepanyl, thiiranyl, thietanyl, tetrahydrothiophenyl (i.e., thiolanyl), 1 ,3-dithiolanyl, thianyl, 1 ,1-dioxothianyl, thiepanyl, decahydroquinolinyl, decahydroisoquinolinyl, or 2-oxa-5-aza-bicyclo[2.2.1]hept-5-yl. Unless defined otherwise, “heterocycloalkyl” preferably refers to a 3 to 11 membered saturated ring group, which is a monocyclic ring or a fused ring system (e.g., a fused ring system composed of two fused rings), wherein said ring group contains one or more (e.g., one, two, three, or four) ring heteroatoms independently selected from 0, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized; more preferably, “heterocycloalkyl” refers to a 5 to 7 membered saturated monocyclic ring group containing one or more (e.g., one, two, or three) ring heteroatoms independently selected from 0, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized.
As used herein, the term “heterocycloalkenyl” refers to an unsaturated alicyclic (non-aromatic) ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said ring group contains one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from 0, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group), and further wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms. For example, each heteroatom-containing ring comprised in said unsaturated alicyclic ring group may contain one or two 0 atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heterocycloalkenyl” may, e.g., refer to imidazolinyl (e.g., 2-imidazolinyl (i.e., 4,5-dihydro-1 H-imidazolyl), 3-imidazolinyl, or 4-imidazolinyl), tetrahydropyridinyl (e.g., 1 ,2,3,6-tetrahydropyridinyl), dihydropyridinyl (e.g., 1 ,2- dihydropyridinyl or 2,3-dihydropyridinyl), pyranyl (e.g., 2H-pyranyl or 4H-pyranyl), thiopyranyl (e.g.,
2H-thiopyranyl or 4H-thiopyranyl), dihydropyranyl, dihydrofuranyl, dihydropyrazolyl, dihydropyrazinyl, dihydroisoindolyl, octahydroquinolinyl (e.g., 1 ,2,3,4,4a,5,6,7-octahydroquinolinyl), or octahydroisoquinolinyl (e.g., 1 ,2,3,4,5,6,7,8-octahydroisoquinolinyl). Unless defined otherwise, “heterocycloalkenyl” preferably refers to a 3 to 11 membered unsaturated alicyclic ring group, which is a monocyclic ring or a fused ring system (e.g., a fused ring system composed of two fused rings), wherein said ring group contains one or more (e.g., one, two, three, or four) ring heteroatoms independently selected from 0, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, wherein one or more carbon ring atoms are optionally oxidized, and wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms; more preferably, “heterocycloalkenyl” refers to a 5 to 7 membered monocyclic unsaturated non-aromatic ring group containing one or more (e.g., one, two, or three) ring heteroatoms independently selected from 0, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, wherein one or more carbon ring atoms are optionally oxidized, and wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms.
As used herein, the term “halogen” refers to fluoro (-F), chloro (-CI), bromo (-Br), or iodo (-I).
As used herein, the term “haloalkyl” refers to an alkyl group substituted with one or more (preferably 1 to 6, more preferably 1 to 3) halogen atoms which are selected independently from fluoro, chloro, bromo and iodo, and are preferably all fluoro atoms. It will be understood that the maximum number of halogen atoms is limited by the number of available attachment sites and, thus, depends on the number of carbon atoms comprised in the alkyl moiety of the haloalkyl group. “Haloalkyl” may, e.g., refer to -CF3, -CHF2, -CH2F, -CF2-CH3, -CH2-CF3, -CH2-CHF2, -CH2-CF2-CH3, -CH2-CF2-CF3, or -CH(CF3)2. A particularly preferred “haloalkyl” group is -CF3.
The terms “bond” and “covalent bond” are used herein synonymously, unless explicitly indicated otherwise or contradicted by context.
As used herein, unless explicitly indicated otherwise or contradicted by context, the terms “a”, “an” and “the” are used interchangeably with “one or more” and “at least one”. Thus, for example, a composition comprising “a” compound of formula (I) can be interpreted as referring to a composition comprising “one or more” compounds of formula (I).
It is to be understood that wherever numerical ranges are provided/disclosed herein, all values and subranges encompassed by the respective numerical range are meant to be encompassed within the scope of the invention. Accordingly, the present invention specifically and individually relates to each value that falls within a numerical range disclosed herein, as well as each subrange encompassed by a numerical range disclosed herein.
As used herein, the term “about” preferably refers to ±10% of the indicated numerical value, more preferably to ±5% of the indicated numerical value, and in particular to the exact numerical value indicated. If the term “about” is used in connection with the endpoints of a range, it preferably refers to the range from the lower endpoint -10% of its indicated numerical value to the upper endpoint +10% of its indicated numerical value, more preferably to the range from of the lower endpoint -5% to the upper endpoint +5%, and even more preferably to the range defined by the exact numerical values of the lower endpoint and the upper endpoint.
As used herein, the term “comprising” (or “comprise”, “comprises”, “contain”, “contains”, or “containing”), unless explicitly indicated otherwise or contradicted by context, has the meaning of “containing, inter alia”, i.e., “containing, among further optional elements, ...”. In addition thereto, this term also includes the narrower meanings of “consisting essentially of’ and “consisting of’. For example, the term “A comprising B and C” has the meaning of “A containing, inter alia, B and C”, wherein A may contain further optional elements (e.g . , “A containing B, C and D” would also be encompassed), but this term also includes the meaning of “A consisting essentially of B and C” and the meaning of “A consisting of B and C” (i.e., no other components than B and C are comprised in A).
Brief description of Figures
The invention is illustrated using the appended Figures, which serve only an illustrative purpose and should not be interpreted as limiting to the scope of the invention in any way.
Figure 1 presents the results of testing the compound of Example 1 in MDA-MB-436 xenograft model. Figure 2 presents an XRPD diffraction pattern obtained for the pharmaceutically acceptable crystal form 1 of the compound of formula (I).
Figure 3 presents DSC and TGA thermographs obtained in the measurements conducted for the pharmaceutically acceptable crystal form 1 of the compound of formula (I).
Figure 4 presents an XRPD diffraction pattern obtained for the pharmaceutically acceptable crystal form 2 of the compound of formula (I).
Figure 5 presents DSC and TGA thermographs obtained in the measurements conducted for the pharmaceutically acceptable crystal form 2 of the compound of formula (I).
Figure 6 presents a comparison of an XRPD diffraction pattern obtained for the pharmaceutically acceptable crystal form 1 of the compound of formula (I) and for the pharmaceutically acceptable crystal form 2 of the compound of formula (I).
Figure ? presents overlay of XRPD diffractograms of form 1 (black) and form 2 (grey) (part 1); Expansion (0 to 17 °20) of overlay of XRPD diffractograms of form 1 (black) and form 2 (grey) (part 2) and Expansion (17 to 35 °20) of overlay of XRPD diffractograms of form 1 (black) and form 2 grey) (part 3.
Figure 8 shows XRPD patterns of solids isolated from competitive equilibrations of form 1 and form 2 in ethanol at various temperatures sampled after 4 and 24 hours compared to form 1 and form 2 references.
Figure 9 shows XRPD patterns of solids isolated from competitive equilibrations of form 1 and form 2 in ethyl acetate at various temperatures sampled after 4 and 24 hours compared to form 1 and form 2 references.
Figure 10 shows XRPD patterns of solids isolated from competitive equilibrations of form 1 and form 2 in acetonitrile at various temperatures sampled after 4 and 24 hours compared form 1 and form 2 references.
Figure 11 presents XRPD patterns of solids isolated from competitive equilibrations of form 1 and form 2 in water at various temperatures sampled after 4 and 24 hours compared to form 1 and form 2 references.
Figure 12 presents slow heating DSC (at 2°C/min) of form 2 resolved the broad, fused endotherm to show a melt-recrystallisation-melt sequence of events.
Figure 13 shows a DSC-recycling experiment starting from crystal form 2.
Detailed description of the invention
The invention is described in detail in the following. It is to be understood that the present invention specifically relates to each and every combination of features and embodiments described herein, including any combination of general and/or preferred features/embodiments.
In a first embodiment, the present invention relates to pharmaceutically acceptable crystal form of a compound of formula (I):

It is to be understood that the compound of formula (I) is present in the crystal form in a free base form (or, in other words, in a non-salt form). It is to be understood that said pharmaceutically acceptable crystal form is obtainable upon recrystallization of the compound of formula (I) from ethanol.
The particular crystal form of the compound of formula (I), referred to herein, is characterized by outstanding stability, high solubility and negligible hygroscopicity. Thus, these exceptional properties of the claimed crystal form allow for the crystal form to be formulated for both clinical use and the long-term storage. It is to be understood that the formulation for clinical use is not particularly limited and may refer to, for example, to formulation as tablet. Importantly, the crystal form referred to herein is not a single (i.e., only) crystal form formed by the compound of formula (I) in its free base form. Accordingly, the compound of formula (I) exhibits polymorphism and other crystal forms have also been discovered and obtained by the present inventors. However, none of these crystal forms exhibits stability, hygroscopicity and/or solubility properties comparable that the properties of the presently claimed crystal form. Furthermore, the crystal form screening has been performed not only for the free base of the compound of formula (I), but also for pharmaceutically acceptable salts of the compound of formula (I), leading to materials with properties inferior to that of the presently claimed crystal form. Thus, accordingly, the crystal form of the present invention exhibits surprisingly improved properties in comparison to other available forms of the compound of formula (I), including other crystal forms of the free base of the compound of formula (I), as well as crystal forms of the salts of the compound of formula (I). It follows that the crystal form provided herein, and referred to as form 1 , is characterized by outstanding properties in comparison to other available solid state forms of the compound of formula (I). Accordingly, crystal form 1 can in no way be considered to be merely arbitrary selection of one crystal form over the other. In particular, the present inventors have identified the second crystal form, being referred to as crystal form 2, which has lower stability than form 1 , and under certain conditions it transforms to a more stable form 1 .
As referred to herein, the expression “crystal form”, “polymorphic form” or, shortly, “crystal” or “polymorph” can be used interchangeably when discussing the particular crystal form of the compound of formula (I) of the present invention.
The compound of formula (I), as shown herein, can also be referred to as 3-(5-(difluoromethyl)- 1 ,3,4-thiadiazol-2-yl)-8-((3S,5S)-3,5-dimethylpiperazin-1-yl)-N-(1-methylcyclopropyl)imidazo[1 ,5- a]pyridine-6-sulfonamide, is a very potent inhibitor of PARG. The compound has shown ICso = 11 nM as determined in PARG enzymatic assay, and has shown low-nanomolar level activity against a number of cancerous cell lines, including NCIH-460 and MDA-MB-436 cells. At the same time, the compound has been shown to be selective and, accordingly, its ECso determined in U2OS cells is >5.0 M, indicating at outstanding selectivity of the compound. Finally, the compound of formula (I) has also been shown to be active in animal xenograft models of cancer, as demonstrated, for example, in Figure 1 .
Accordingly, as understood herein, the crystal form of the invention is a crystal form of the compound of formula (I). To this end, said compound of formula (I) is present as a free base, and not as
a salt in the crystal form of the invention. Thus, crystal form 1 of the invention comprises the compound of formula (I) in its free-base form.
Furthermore, as provided by the present invention, the crystal form of the invention is not a crystal form being a hydrate (or, more generally speaking, a solvate). Accordingly, the crystal form of the invention is free, or substantially free, of solvents, including water. It is to be understood that the term “substantially free of solvent” preferably refers to a situation wherein the crystal includes not more than 2 %w/w of solvent, more preferably not more than 1%w/w of solvent, even more preferably not more than 0.5 %w/w of water. This is apparent upon subjecting the crystal form of the invention, i.e., the crystal form 1 , to thermogravimetric analysis, or in other words TGA, which reveals only marginal loss of weight of the solid substance, i.e., the crystal form 1 , upon heating. This marginal loss of weight indicates that the form is not a solvate form. It is further noted that further loss of weight is observed at temperatures exceeding 230 °C, which is attributed to post-melting decomposition of the compound of formula (I).
Thereby, the crystal form consists substantially of the compound of formula (I). In other words, the compound of formula (I) preferably constitutes at least 95% w/w of the crystal form, more preferably at least 96% w/w of the crystal form, even more preferably at least 97% w/w of the crystal form, still more preferably at least 98% w/w of the crystal form, again more preferably at least 99% w/w of the crystal form.
As described herein, the crystal form of the invention is obtainable by recrystallization of the compound of formula (I) from ethanol. Said recrystallization may involve providing pure (or substantially pure, such as at least 90% pure, or at least 95% pure, or at least 97% pure, wherein purity is defined w/w basis) compound of formula (I), dissolving it in ethanol to obtain an oversaturated solution, and - once crystallized product appears - obtaining the crystal form of the compound of formula (I) of the invention. For example, the experiment described in the experimental procedure for obtaining the crystal form, as provided in the Examples section, necessarily leads to the crystal form of the invention. Therein, the recrystallization from ethanol involves complete dissolution in ethanol, followed by cooling to room temperature over 12 hours. Preferably, the step of complete dissolution in ethanol is performed while stirring, mixing or agitating in any way the solution or the suspension. Furthermore, the step of cooling to room temperature of the obtained solution is performed while stirring (or mixing or agitating the so obtained solution in any way known to the skilled person). It is preferred that the step of dissolution is performed at elevated temperature. Thus, said step is performed preferably under reflux. Alternatively, and preferably, dissolution of the compound of formula (I) in ethanol is performed at a temperature of 110 °C. It is immediately apparent to the skilled person that provided herein temperature of 110 °C refers to a temperature of heating body, such as an oil batch, in which the reaction vessel including said ethanol is heated up. It is immediately apparent to the skilled person that ethanol can be brought to boiling under
these conditions (such as under reflux conditions). Accordingly, and preferably, the step of dissolution is performed in boiling ethanol.
It is preferred that in the process of obtaining the pharmaceutically acceptable crystal form of the present invention, through recrystallization of the compound of formula (I) from ethanol, no crystal seeding is performed.
It is noted that the present definition of the crystal form through the process of obtaining the crystal form does not limit said crystal forms to crystals obtained exactly following the above-mentioned protocol based on recrystallization from ethanol. Instead, a protocol that necessarily leads to the claimed crystal form, thereby defining the said crystal form, is provided. According to the present invention, the same crystal form can also be obtained according to other crystallization protocols, for example crystallization from the mixture of tetrahydrofurane/n-heptane (optionally involving the seeding using the crystal form 1).
Preferably, within the scope of the present invention, the recrystallization of the compound of formula (I) from ethanol, as described in the foregoing, is performed following the deprotection reaction of the Boc group with the use of formic acid (referred to as FA) depicted in the scheme below:

It is to be understood that the reaction is to be performed in neat formic acid.
Preferably, following the deprotection reaction, the formic acid is concentrated under vacuum to give a residue which was diluted with water, wherein the pH was adjusted to 9 with a saturated solution of NaHCCh. The product is then extracted with DCM. The combined organic layer is washed with brine, dried over Na2SO4, filtered and concentrated under vacuum. The so obtained residue is subjected to recrystallization from ethanol, as described herein.
For reference only, obtaining of the reference example - crystal form 1 of the compound of formula (I), is described in the Examples section.
As apparent to the skilled person, the crystal form may be characterized by its structural properties and/or by its physicochemical properties. In particular, the crystal form can be characterized by X-ray diffraction properties, by its melting point and/or by its melting enthalpy.
It is typical to characterize the crystal form by its X-ray powder diffraction pattern. The X-ray powder diffraction pattern of the crystal form of the invention obtained using the CuKa radiation, is shown in Figure 1.
Further preferably, the crystal form of the present invention is characterized by the X-ray powder diffraction pattern obtained using CuKa radiation that comprises at least one peak selected from the peaks at 12.24 ± 0.2 °20, 20.04 ± 0.2 °20, 20.40 ± 0.2 °20 and 24.02 ± 0.2 °20. These peaks are characteristic for the currently claimed crystal form of the present invention, and not for other crystal forms, in particular not for form 2 of the compound of formula (I). Preferably, the X-ray powder diffraction pattern obtained using CuKa radiation for the claimed crystal form (also referred to crystal form 1) comprises at least two peaks, or at least three peaks selected from the peaks at 12.24 ± 0.2 °20, 20.04 ± 0.2 °20, 20.40 ± 0.2 °20 and 24.02 ± 0.2 °20. Even more preferably, the X-ray powder diffraction pattern obtained using CuKa radiation for the claimed crystal form further comprises the peaks at 12.24 ± 0.2 °20, 20.04 ± 0.2 °20, 20.40 ± 0.2 °20 and 24.02 ± 0.2 °20.
It is to be understood that whenever reference is made to an angular value provided as a particular numerical value x ± 0.2 °, preferably said value x ± 0.1 0 is meant, more preferably said exact value x is meant.
Preferably, the crystal form is characterized by an X-ray powder diffraction pattern obtained using CuKa radiation additionally comprising a peak at 18.71 ± 0.2 °20, preferably 18.71 ± 0.1 °20, and/or a peak at 6.86 ± 0.2 °20. The peak at 18.71 ± 0.2 °20 is the most prominent peak in the X-ray powder diffraction powder for the compound of formula, as clearly and unambiguously seen from Figure 1 .
It is further preferred that, in addition to the peaks discussed in the foregoing, the X-ray powder diffraction pattern further comprises at least one peak selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. More preferably, the X-ray powder diffraction pattern further comprises at least two peaks selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. Even more preferably, the X-ray powder diffraction pattern further comprises at least three peaks selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. Even more preferably, the X-ray powder diffraction pattern further comprises at least four peaks selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. Again more preferably, the X-ray powder diffraction pattern further comprises at least five peaks selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. Still more preferably, the X-ray powder diffraction pattern further comprises at least six peaks selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21.59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. Most preferably, the X-ray powder diffraction pattern further comprises the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20.
In one embodiment, the crystal form of the invention preferably has X-ray powder diffraction pattern, obtained using the CuKa radiation, as shown in Figure 2.
The crystal form may also be characterized by its properties relating to melting. One example of such property is melting point. Preferably, the crystal form 1 , as claimed in the present invention, is characterized by a melting point of 207.5 ± 4.0 °C. More preferably, the crystal form 1 is characterized by a melting point of 207.5 ± 2.0 °C. Even more preferably, the crystal form 1 is characterized by a melting point of 207.5 ± 1 .0 °C. Still more preferably, the crystal form 1 is characterized by a melting point of 207.5 ± 0.5 °C. This melting point determined for crystal form 1 is clearly distinct from the melting point determined for other crystal forms, for example and in particular for crystal form 2, which has been measured to be 199 °C (in particular, specific measurements indicate a value of 199 °C ± 2.0 °C). Alternatively or additionally, the crystal form can be characterized by its melting enthalpy. Preferably, the crystal form 1 is characterized by a melting enthalpy of between 115 and 125 J/g. More preferably, the crystal form 1 is characterized by a melting enthalpy of between 118 and 120 J/g. Even more preferably, the crystal form 1 is characterized by a melting enthalpy of 119 J/g.
Experimental data studying melting properties of the crystal form 1 of the compound of formula (I) of the present invention is shown in Figure 3. For comparison purposes only, experimental data summarizing melting properties of the crystal form 2 of the compound of formula (I), as shown in Figure 5.
Accordingly and preferably, the pharmaceutically acceptable crystal form of the present invention is characterized by a melting point of 207.5 ± 4.0 °C and by the X-ray powder diffraction pattern obtained using CuKa radiation that comprises at least one peak, at least two peaks, at least three peaks, or four peaks selected from the peaks at 12.24 ± 0.2 °20, 20.04 ± 0.2 °20, 20.40 ± 0.2 °20 and 24.02 ± 0.2 °20. More preferably, the crystal form 1 is characterized by a melting point of 207.5 ± 2.0 °C and by the X-ray powder diffraction pattern obtained using CuKa radiation that comprises at least one peak, at least two peaks, at least three peaks, or four peaks selected from the peaks at 12.24 ± 0.2 °20, 20.04 ± 0.2 °20, 20.40 ± 0.2 °20 and 24.02 ± 0.2 °20. Even more preferably, the crystal form 1 is characterized by a melting point of 207.5 ± 1 .0 °C and by the X-ray powder diffraction pattern obtained using CuKa radiation that comprises at least one peak, at least two peaks, at least three peaks, or four peaks selected from the peaks at 12.24 ± 0.2 °20, 20.04 ± 0.2 °20, 20.40 ± 0.2 °20 and 24.02 ± 0.2 °20. Still more preferably, the crystal form 1 is characterized by a melting point of 207.5 ± 0.5 °C and by the X-ray powder diffraction pattern obtained using CuKa radiation that comprises at least one peak, at least two peaks, at least three peaks, or four peaks selected from the peaks at 12.24 ± 0.2 °20, 20.04 ± 0.2 °20, 20.40 ± 0.2 °20 and 24.02 ± 0.2 °20. Preferably, the crystal form is characterized by an X-ray powder diffraction pattern obtained using CuKa radiation additionally comprising a peak at 18.71 ± 0.2 °20, preferably 18.71 ± 0.1
°20, and/or a peak at 6.86 ± 0.2 °20. It is further preferred that, in addition to the peaks discussed in the foregoing, the X-ray powder diffraction pattern further comprises at least one peak selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. More preferably, the X-ray powder diffraction pattern further comprises at least two peaks selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. Even more preferably, the X-ray powder diffraction pattern further comprises at least three peaks selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21.59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. Even more preferably, the X-ray powder diffraction pattern further comprises at least four peaks selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. Again more preferably, the X-ray powder diffraction pattern further comprises at least five peaks selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. Still more preferably, the X-ray powder diffraction pattern further comprises at least six peaks selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21.59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20. Most preferably, the X-ray powder diffraction pattern further comprises the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21.59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20.
The present invention further relates to a method for making the pharmaceutically acceptable crystal form 1 of the present invention. The method comprises the step of crystallizing the compound of formula (I) as depicted herein from ethanol or from the mixture of THF/n-heptane. Preferably, the compound is to be crystallized from ethanol. The experimental details of an exemplary embodiment of the method are described herein. However, in one embodiment of the present invention, the method for making the pharmaceutically acceptable crystal form 1 of the present invention comprises the step of crystallizing the compound of formula (I) from the mixture of THF/n-heptane. Optionally, said crystallization is to be performed with seeding using the crystal form 1 .
For reference purposes only, crystal form 2, provided as a reference example, can be obtained as described in the experimental section hereinbelow. In particular, crystal form 2 is obtainable by trituration of the compound of formula (I) from DCM. Detailed experimental protocol is provided herein.
The present description refers to pharmaceutically acceptable salt of the compound of formula (I). This term embraces all pharmaceutically acceptable salt forms of the compound of formula (I) which may be formed, e.g., by protonation of an atom carrying an electron lone pair which is susceptible to protonation, such as an amino group, with an inorganic or organic acid, or as a salt of an acid group (such as a carboxylic acid group) with a physiologically acceptable cation. Exemplary base addition salts comprise, for example: alkali metal salts such as sodium or potassium salts; alkaline earth metal salts
such as calcium or magnesium salts; zinc salts; ammonium salts; aliphatic amine salts such as trimethylamine, triethylamine, dicyclohexylamine, ethanolamine, diethanolamine, triethanolamine, procaine salts, meglumine salts, ethylenediamine salts, or choline salts; aralkyl amine salts such as N,N- dibenzylethylenediamine salts, benzathine salts, benethamine salts; heterocyclic aromatic amine salts such as pyridine salts, picoline salts, quinoline salts or isoquinoline salts; quaternary ammonium salts such as tetramethylammonium salts, tetraethylammonium salts, benzyltrimethylammonium salts, benzyltriethylammonium salts, benzyltributylammonium salts, methyltrioctylammonium salts or tetrabutylammonium salts; and basic amino acid salts such as arginine salts, lysine salts, or histidine salts. Exemplary acid addition salts comprise, for example: mineral acid salts such as hydrochloride, hydrobromide, hydroiodide, sulfate salts (such as, e.g., sulfate or hydrogensulfate salts), nitrate salts, phosphate salts (such as, e.g., phosphate, hydrogenphosphate, or dihydrogenphosphate salts), carbonate salts, hydrogencarbonate salts, perchlorate salts, borate salts, or thiocyanate salts; organic acid salts such as acetate, propionate, butyrate, pentanoate, hexanoate, heptanoate, octanoate, cyclopentanepropionate, decanoate, undecanoate, oleate, stearate, lactate, maleate, oxalate, fumarate, tartrate, malate, citrate, succinate, adipate, gluconate, glycolate, nicotinate, benzoate, salicylate, ascorbate, pamoate (embonate), camphorate, glucoheptanoate, or pivalate salts; sulfonate salts such as methanesulfonate (mesylate), ethanesulfonate (esylate), 2-hydroxyethanesulfonate (isethionate), benzenesulfonate (besylate), p-toluenesulfonate (tosylate), 2-naphthalenesulfonate (napsylate), 3-phenylsulfonate, or camphorsulfonate salts; glycerophosphate salts; and acidic amino acid salts such as aspartate or glutamate salts. Preferred pharmaceutically acceptable salts of the compound of formula (I) include a hydrochloride salt, a hydrobromide salt, a mesylate salt, a sulfate salt, a tartrate salt, a fumarate salt, an acetate salt, a citrate salt, and a phosphate salt. A particularly preferred pharmaceutically acceptable salt of the compound of formula (I) is a hydrochloride salt. Accordingly, it is preferred that the compound of formula (I), including any one of the specific compounds of formula (I) described herein, is in the form of a hydrochloride salt, a hydrobromide salt, a mesylate salt, a sulfate salt, a tartrate salt, a fumarate salt, an acetate salt, a citrate salt, or a phosphate salt, and it is particularly preferred that the compound of formula (I) is in the form of a hydrochloride salt. In one embodiment, the compound of formula (I) may be in the form of a formate salt. In a preferred form, the compound of formula (I) is in a non-salt form. In other words, in the crystal form 1 of the present invention, the compound of formula (I) is in its free base form. It is to be noted that when reference is made to a salt, a specific reference is made to a composition comprising both acid and base that form said salt.
Furthermore, the compound of formula (I) may exist in the form of different isomers, in particular stereoisomers (including, e.g., geometric isomers (or cis/trans isomers), enantiomers and diastereomers) or tautomers (including, in particular, prototropic tautomers, such as keto/enol tautomers or thione/thiol
tautomers). All such isomers of the compounds of formula (I) are contemplated as being part of the present invention, either in admixture or in pure or substantially pure form. As for stereoisomers, the invention embraces the isolated optical isomers of the compounds according to the invention as well as any mixtures thereof (including, in particular, racemic mixtures/racemates). The racemates can be resolved by physical methods, such as, e.g., fractional crystallization, separation or crystallization of diastereomeric derivatives, or separation by chiral column chromatography. The individual optical isomers can also be obtained from the racemates via salt formation with an optically active acid followed by crystallization. The present invention further encompasses any tautomers of the compound of formula (I). It will be understood that some compounds may exhibit tautomerism. In such cases, the formulae provided herein expressly depict only one of the possible tautomeric forms. The formulae and chemical names as provided herein are intended to encompass any tautomeric form of the corresponding compound and not to be limited merely to the specific tautomeric form depicted by the drawing or identified by the name of the compound.
The scope of the invention also embraces compounds of formula (I), in which one or more atoms are replaced by a specific isotope of the corresponding atom. For example, the invention encompasses compounds of formula (I), in which one or more hydrogen atoms (or, e.g., all hydrogen atoms) are replaced by deuterium atoms (i.e., 2H; also referred to as “D”). Accordingly, the invention also embraces compounds of formula (I) which are enriched in deuterium. Naturally occurring hydrogen is an isotopic mixture comprising about 99.98 mol-% hydrogen-1 (1H) and about 0.0156 mol-% deuterium (2H or D). The content of deuterium in one or more hydrogen positions in the compounds of formula (I) can be increased using deuteration techniques known in the art. For example, a compound of formula (I) or a reactant or precursor to be used in the synthesis of the compound of formula (I) can be subjected to an H/D exchange reaction using, e.g., heavy water (D2O). Further suitable deuteration techniques are described in: Atzrodt J et al., Bioorg Med Chem, 20(18), 5658-5667, 2012; William JS et al., Journal of Labelled Compounds and Radiopharmaceuticals, 53(11 -12), 635-644, 2010; Modvig A et al., J Org Chem, 79, 5861-5868, 2014. The content of deuterium can be determined, e.g., using mass spectrometry or NMR spectroscopy. Unless specifically indicated otherwise, it is preferred that the compound of formula (I) is not enriched in deuterium. Accordingly, the presence of naturally occurring hydrogen atoms or 1H hydrogen atoms in the compounds of formula (I) is preferred.
The present invention also embraces compounds of formula (I), in which one or more atoms are replaced by a positron-emitting isotope of the corresponding atom, such as, e.g., 18F, 11C, 13N, 150, 76Br, 77Br, 120l and/or 124L Such compounds can be used as tracers, trackers or imaging probes in positron emission tomography (PET). The invention thus includes (I) compounds of formula (I), in which one or more fluorine atoms (or, e.g., all fluorine atoms) are replaced by 18F atoms, (II) compounds of formula (I), in which one or more carbon atoms (or, e.g., all carbon atoms) are replaced by 11C atoms, (iii) compounds
of formula (I), in which one or more nitrogen atoms (or, e.g., all nitrogen atoms) are replaced by 13N atoms, (iv) compounds of formula (I), in which one or more oxygen atoms (or, e.g., all oxygen atoms) are replaced by 150 atoms, (v) compounds of formula (I), in which one or more bromine atoms (or, e.g., all bromine atoms) are replaced by 76Br atoms, (vi) compounds of formula (I), in which one or more bromine atoms (or, e.g., all bromine atoms) are replaced by 77Br atoms, (vii) compounds of formula (I), in which one or more iodine atoms (or, e.g., all iodine atoms) are replaced by 120l atoms, and (viii) compounds of formula (I), in which one or more iodine atoms (or, e.g., all iodine atoms) are replaced by 124l atoms. In general, it is preferred that none of the atoms in the compounds of formula (I) are replaced by specific isotopes.
Pharmaceutical compositions
The compounds provided herein, in the form of the pharmaceutically acceptable crystal form of the invention, may be administered as compounds per se or may be formulated as medicaments. The medicaments/pharmaceutical compositions may optionally comprise one or more pharmaceutically acceptable excipients, such as carriers, diluents, fillers, disintegrants, lubricating agents, binders, colorants, pigments, stabilizers, preservatives, antioxidants, and/or solubility enhancers. As understood herein, wherever a reference to a compound of formula (I) is made, it is meant as a reference to the claimed pharmaceutically acceptable crystal form of formula (I).
The pharmaceutical compositions may comprise one or more solubility enhancers, such as, e.g., polyethylene glycol), including polyethylene glycol) having a molecular weight in the range of about 200 to about 5,000 Da (e.g., PEG 200, PEG 300, PEG 400, or PEG 600), ethylene glycol, propylene glycol, glycerol, a non-ionic surfactant, tyloxapol, polysorbate 80, macrogol-15-hydroxystearate (e.g., Kolliphor® HS 15, CAS 70142-34-6), a phospholipid, lecithin, dimyristoyl phosphatidylcholine, dipalmitoyl phosphatidylcholine, distearoyl phosphatidylcholine, a cyclodextrin, a-cyclodextrin, p-cyclodextrin, y- cyclodextrin, hydroxyethyl-p-cyclodextrin, hydroxypropyl-p-cyclodextrin, hydroxyethyl-y-cyclodextrin, hydroxypropyl-y-cyclodextrin, dihydroxypropyl-p-cyclodextrin, sulfobutylether-p-cyclodextrin, sulfobutylether-y-cyclodextrin, glucosyl-a-cyclodextrin, glucosyl-p-cyclodextrin, diglucosyl-p-cyclodextrin, maltosyl-a-cyclodextrin, maltosyl-p-cyclodextrin, maltosyl-y-cyclodextrin, maltotriosyl-p-cyclodextrin, maltotriosyl-y-cyclodextrin, dimaltosyl-p-cyclodextrin, methyl-p-cyclodextrin, a carboxyalkyl thioether, hydroxypropyl methylcellulose, hydroxypropylcellulose, polyvinylpyrrolidone, a vinyl acetate copolymer, vinyl pyrrolidone, sodium lauryl sulfate, dioctyl sodium sulfosuccinate, or any combination thereof.
The pharmaceutical compositions may also comprise one or more preservatives, particularly one or more antimicrobial preservatives, such as, e.g., benzyl alcohol, chlorobutanol, 2-ethoxyethanol, m-cresol, chlorocresol (e.g., 2-chloro-3-methyl-phenol or 4-chloro-3-methyl-phenol), benzalkonium chloride, benzethonium chloride, benzoic acid (or a pharmaceutically acceptable salt thereof), sorbic acid (or a pharmaceutically acceptable salt thereof), chlorhexidine, thimerosal, or any combination thereof.
The pharmaceutical compositions can be formulated by techniques known to the person skilled in the art, such as the techniques published in “Remington: The Science and Practice of Pharmacy”, Pharmaceutical Press, 22nd edition. The pharmaceutical compositions can be formulated as dosage forms for oral, parenteral, such as intramuscular, intravenous, subcutaneous, intradermal, intraarterial, intracardial, rectal, nasal, topical, aerosol or vaginal administration. Dosage forms for oral administration include coated and uncoated tablets, soft gelatin capsules, hard gelatin capsules, lozenges, troches, solutions, emulsions, suspensions, syrups, elixirs, powders and granules for reconstitution, dispersible powders and granules, medicated gums, chewing tablets and effervescent tablets. Dosage forms for parenteral administration include solutions, emulsions, suspensions, dispersions and powders and granules for reconstitution. Emulsions are a preferred dosage form for parenteral administration. Dosage forms for rectal and vaginal administration include suppositories and ovula. Dosage forms for nasal administration can be administered via inhalation and insufflation, for example by a metered inhaler. Dosage forms for topical administration include creams, gels, ointments, salves, patches and transdermal delivery systems.
The compounds of formula (I) or the above described pharmaceutical compositions comprising a compound of formula (I) may be administered to a subject by any convenient route of administration, whether systemically/peripherally or at the site of desired action, including but not limited to one or more of: oral (e.g., as a tablet, capsule, or as an ingestible solution), topical (e.g., transdermal, intranasal, ocular, buccal, and sublingual), parenteral (e.g., using injection techniques or infusion techniques, and including, for example, by injection, e.g., subcutaneous, intradermal, intramuscular, intravenous, intraarterial, intracardiac, intrathecal, intraspinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratracheal, subcuticular, intraarticular, subarachnoid, or intrasternal by, e.g., implant of a depot, for example, subcutaneously or intramuscularly), pulmonary (e.g., by inhalation or insufflation therapy using, e.g., an aerosol, e.g., through mouth or nose), gastrointestinal, intrauterine, intraocular, subcutaneous, ophthalmic (including intravitreal or intracameral), rectal, or vaginal administration.
If said compounds or pharmaceutical compositions are administered parenterally, then examples of such administration include one or more of: intravenously, intraarterially, intraperitoneally, intrathecally, intraventricularly, intraurethrally, intrasternally, intracardially, intracranially, intramuscularly or subcutaneously administering the compounds or pharmaceutical compositions, and/or by using infusion techniques. For parenteral administration, the compounds are best used in the form of a sterile aqueous solution which may contain other substances, for example, enough salts or glucose to make the solution isotonic with blood. The aqueous solutions should be suitably buffered (preferably to a pH of from 3 to 9), if necessary. The preparation of suitable parenteral formulations under sterile conditions is readily accomplished by standard pharmaceutical techniques well known to those skilled in the art.
Said compounds or pharmaceutical compositions can also be administered orally in the form of tablets, capsules, ovules, elixirs, solutions or suspensions, which may contain flavoring or coloring agents, for immediate-, delayed-, modified-, sustained-, pulsed- or controlled-release applications.
The tablets may contain excipients such as microcrystalline cellulose, lactose, sodium citrate, calcium carbonate, dibasic calcium phosphate and glycine, disintegrants such as starch (preferably corn, potato or tapioca starch), sodium starch glycolate, croscarmellose sodium and certain complex silicates, and granulation binders such as polyvinylpyrrolidone, hydroxypropylmethylcellulose (HPMC), hydroxypropylcellulose (HPC), sucrose, gelatin and acacia. Additionally, lubricating agents such as magnesium stearate, stearic acid, glyceryl behenate and talc may be included. Solid compositions of a similar type may also be employed as fillers in gelatin capsules. Preferred excipients in this regard include lactose, starch, a cellulose, or high molecular weight polyethylene glycols. For aqueous suspensions and/or elixirs, the agent may be combined with various sweetening or flavoring agents, coloring matter or dyes, with emulsifying and/or suspending agents and with diluents such as water, ethanol, propylene glycol and glycerin, and combinations thereof.
For oral administration, the compounds or pharmaceutical compositions are preferably administered by oral ingestion, particularly by swallowing. The compounds or pharmaceutical compositions can thus be administered to pass through the mouth into the gastrointestinal tract, which can also be referred to as “oral-gastrointestinal” administration.
Alternatively, said compounds or pharmaceutical compositions can be administered in the form of a suppository or pessary, or may be applied topically in the form of a gel, hydrogel, lotion, solution, cream, ointment or dusting powder. The compounds of the present invention may also be dermally or transdermally administered, for example, by the use of a skin patch.
Said compounds or pharmaceutical compositions may also be administered by sustained release systems. Suitable examples of sustained-release compositions include semi-permeable polymer matrices in the form of shaped articles, e.g., films, or microcapsules. Sustained-release matrices include, e.g., polylactides, copolymers of L-glutamic acid and gamma-ethyl-L-glutamate, poly(2-hydroxyethyl methacrylate), ethylene vinyl acetate, or poly-D-(-)-3-hydroxybutyric acid. Sustained-release pharmaceutical compositions also include liposomally entrapped compounds. The present invention thus also relates to liposomes containing a compound of the invention.
Said compounds or pharmaceutical compositions may also be administered by the pulmonary route, rectal routes, or the ocular route. For ophthalmic use, they can be formulated as micronized suspensions in isotonic, pH adjusted, sterile saline, or, preferably, as solutions in isotonic, pH adjusted, sterile saline, optionally in combination with a preservative such as a benzalkonium chloride. Alternatively, they may be formulated in an ointment such as petrolatum.
It is also envisaged to prepare dry powder formulations of the compounds of formula (I) for pulmonary administration, particularly inhalation. Such dry powders may be prepared by spray drying under conditions which result in a substantially amorphous glassy or a substantially crystalline bioactive powder. Accordingly, dry powders of the compounds of the present invention can be made according to an emulsification/spray drying process.
For topical application to the skin, said compounds or pharmaceutical compositions can be formulated as a suitable ointment containing the active compound suspended or dissolved in, for example, a mixture with one or more of the following: mineral oil, liquid petrolatum, white petrolatum, propylene glycol, emulsifying wax and water. Alternatively, they can be formulated as a suitable lotion or cream, suspended or dissolved in, for example, a mixture of one or more of the following: mineral oil, sorbitan monostearate, a polyethylene glycol, liquid paraffin, polysorbate 60, cetyl esters wax, 2-octyldodecanol, benzyl alcohol and water.
The present invention thus relates to the compounds or the pharmaceutical compositions provided herein, wherein the corresponding compound or pharmaceutical composition is to be administered by any one of: an oral route; topical route, including by transdermal, intranasal, ocular, buccal, or sublingual route; parenteral route using injection techniques or infusion techniques, including by subcutaneous, intradermal, intramuscular, intravenous, intraarterial, intracardiac, intrathecal, intraspinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratracheal, subcuticular, intraarticular, subarachnoid, intrasternal, intraventricular, intraurethral, or intracranial route; pulmonary route, including by inhalation or insufflation therapy; gastrointestinal route; intrauterine route; intraocular route; subcutaneous route; ophthalmic route, including by intravitreal, or intracameral route; rectal route; or vaginal route. Preferred routes of administration are oral administration or parenteral administration. For each of the compounds or pharmaceutical compositions provided herein, it is particularly preferred that the respective compound or pharmaceutical composition is to be administered orally (particularly by oral ingestion).
Typically, a physician will determine the actual dosage which will be most suitable for an individual subject. The specific dose level and frequency of dosage for any particular individual subject may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the individual subject undergoing therapy.
A proposed, yet non-limiting dose of the compounds according to the invention for oral administration to a human (of approximately 70 kg body weight) may be 0.05 to 2000 mg, preferably 0.1 mg to 1500 mg, more preferably 0.1 mg to 1000 mg, of the active ingredient per unit dose. The unit dose may be administered, e.g., 1 to 3 times per day. The unit dose may also be administered 1 to 7 times per
week, e.g., with not more than one administration per day. It will be appreciated that it may be necessary to make routine variations to the dosage depending on the age and weight of the patient/su bject as well as the severity of the condition to be treated. The precise dose and also the route of administration will ultimately be at the discretion of the attendant physician or veterinarian.
Therapeutic use
In one embodiment, the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I) or a pharmaceutical composition as defined herein for use in therapy.
The present invention provides pharmaceutically acceptable crystal form of the compound that functions as inhibitors of PARG. Thus, the present invention provides a method of inhibiting PARG enzyme activity in vitro or in vivo, said method comprising contacting a cell with an effective amount of the pharmaceutically acceptable crystal form of the compound of formula (I), as defined herein.
The present invention also provides a method of selectively inhibiting PARG enzyme activity over PARP1 or ARH3 enzyme activity in vitro or in vivo. The said method comprises the steps of contacting a cell with an effective amount of a pharmaceutically acceptable crystal form of the compound, as defined herein.
In a further embodiment, the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I), as disclosed herein, for use in a method of treating a disease or disorder in which PARG activity is implicated in a subject or patient in need of such treatment. Said method of treatment comprises administering to said subject/patient a therapeutically effective amount of a pharmaceutically acceptable crystal form of the compound of formula (I), or a pharmaceutical composition as defined herein. In other words, in one embodiment the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I), as disclosed herein, for use in treating a disease or disorder in which PARG activity is implicated.
In a further embodiment, the present invention relates to a method of inhibiting cell proliferation, in vitro or in vivo, said method comprising contacting a cell with an effective amount of the pharmaceutically acceptable crystal form of the compound of formula (I), as defined herein. Thus, the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I) or a pharmaceutically acceptable salt thereof for use in of inhibiting cell proliferation, in vitro or in vivo.
Thus, in a further embodiment, the present invention relates to a method of treating a proliferative disorder in a subject or patient in need of such treatment. The said method of treating a proliferative disorder in a subject or patient in need thereof comprises administering to said subject/patient a therapeutically effective amount of the pharmaceutically acceptable crystal form of the compound of formula (I), or a pharmaceutical composition as defined herein. Preferably as disclosed herein, the proliferative disorder is cancer. Thus, the present invention relates to a method of treating cancer in a
subject or patient in need thereof. The said method of treating cancer in a subject or patient in need thereof comprises administering to said subject/patient a therapeutically effective amount of the pharmaceutically acceptable crystal form of the compound of formula (I), or a pharmaceutical composition as defined herein. In a particular embodiment, the cancer is human cancer.
In one embodiment, the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I) for use in treating a proliferative disorder. Preferably as disclosed herein, the proliferative disorder is cancer. Therefore, the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I) for use in treating cancer. In a particular embodiment, the cancer is human cancer.
In a further embodiment, the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I), as defined herein, for use in the manufacture of a medicament for the treatment of a proliferative condition. In a preferred embodiment, the proliferative condition is cancer, more preferably a human cancer. Thus, preferably the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I), as defined herein, for use in the manufacture of a medicament for the treatment of cancer, preferably for the treatment of human cancer.
In a further embodiment, the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I), as defined herein, for use in the manufacture of a medicament for the inhibition of PARG enzyme activity. Preferably, the inhibition of PARG enzyme activity is selective inhibition of PARG enzyme activity over PARP1 or ARH3 enzyme activity. Thus, the present invention relates to the pharmaceutically acceptable crystal form of the compound of formula (I), as defined herein, for use in the manufacture of a medicament for the selective inhibition of PARG enzyme activity over PARP1 or ARH3 enzyme activity.
The present invention further provides the pharmaceutically acceptable crystal form of the compound of formula (I), as defined herein for use in the manufacture of a medicament for the treatment of a disease or disorder in which PARG activity is implicated, as defined herein.
As understood herein, the term "proliferative disorder" are used interchangeably herein and pertain to an unwanted or uncontrolled cellular proliferation of excessive or abnormal cells which is undesired, such as, neoplastic or hyperplastic growth, whether in vitro or in vivo. Examples of proliferative conditions include, but are not limited to, pre-malignant and malignant cellular proliferation, including but not limited to, malignant neoplasms and tumours, cancers, leukemias, psoriasis, bone diseases, fibroproliferative disorders (e.g . , of connective tissues), and atherosclerosis. Any type of cell may be treated, including but not limited to, gastric, lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin, preferably lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin.
Preferably, as understood herein, the cancer to be treated is selected from gastric, lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin cancer, more preferably lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin cancer.
The anti-proliferative effects of the compound of formula (I) of the present invention have particular application in the treatment of human cancers (by virtue of their inhibition of PARG enzyme activity). The anti-cancer effect may arise through one or more mechanisms, including but not limited to, the regulation of cell proliferation, the inhibition of angiogenesis (the formation of new blood vessels), the inhibition of metastasis (the spread of a tumour from its origin), the inhibition of invasion (the spread of tumour cells into neighbouring normal structures), or the promotion of apoptosis (programmed cell death).
The antiproliferative treatment with the pharmaceutically acceptable crystal form of the compound of formula (I) as defined hereinbefore, may be applied as a sole therapy or may involve, in addition to the compound of the invention, conventional surgery or radiotherapy or chemotherapy. Such chemotherapy may include one or more of the following categories of anti-tumour agents:-
(i) other antiproliferative/antineoplastic drugs and combinations thereof, as used in medical oncology, such as alkylating agents (for example cis-platin, oxaliplatin, carboplatin, cyclophosphamide, nitrogen mustard, melphalan, chlorambucil, busulphan, temozolomide and nitrosoureas); antimetabolites (for example gemcitabine and antifolates such as fluoropyrimidines like 5-fluorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside, and hydroxyurea); antitumour antibiotics (for example anthracyclines like adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin); antimitotic agents (for example vinca alkaloids like vincristine, vinblastine, vindesine and vinorelbine and taxoids like taxol and taxotere and polokinase inhibitors); and topoisomerase inhibitors (for example epipodophyllotoxins like etoposide and teniposide, amsacrine, topotecan and camptothecin);
(ii) cytostatic agents such as antioestrogens (for example tamoxifen, fulvestrant, toremifene, raloxifene, droloxifene and iodoxyfene), antiandrogens (for example bicalutamide, flutamide, nilutamide and cyproterone acetate), LHRH antagonists or LHRH agonists (for example goserelin, leuprorelin and buserelin), progestagens (for example megestrol acetate), aromatase inhibitors (for example as anastrozole, letrozole, vorazole and exemestane) and inhibitors of 5oc-reductase such as finasteride;
(iii) anti-invasion agents [for example c-Src kinase family inhibitors like 4-(6-chloro-2,3- methylenedioxyanilino)-7-[2-(4-methylpiperazin-1 -yl)ethoxy]-5-tetrahydropyran-4- yloxyquinazoline (AZD0530; International Patent Application WO 01/94341 ), N-(2-chloro-6- methylphenyl)-2-{6-[4-(2- hydroxyethyl)piperazin-1 -yl]-2-methylpyrimidin-4-ylamino}thiazole- 5-carboxamide (dasatinib, BMS- 354825; J. Med. Chem., 2004, 47, 6658-6661 ) and bosutinib (SKI-606), and metalloproteinase inhibitors
like marimastat, inhibitors of urokinase plasminogen activator receptor function or antibodies to Heparanase];
(iv) inhibitors of growth factor function: for example such inhibitors include growth factor antibodies and growth factor receptor antibodies (for example the anti-erbB2 antibody trastuzumab [Herceptin™], the anti-EGFR antibody panitumumab, the anti-erbB 1 antibody cetuximab [Erbitux, C225] and any growth factor or growth factor receptor antibodies disclosed by Stern et al. (Critical reviews in oncology/haematology, 2005, Vol. 54, pp1 1 -29); such inhibitors also include tyrosine kinase inhibitors, for example inhibitors of the epidermal growth factor family (for example EGFR family tyrosine kinase inhibitors such as N-(3-chloro- 4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazolin-4-amine (gefitinib, ZD1839), N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (erlotinib, OSI-774) and 6- acrylamido-N-(3-chloro-4-fluorophenyl)-7-(3-morpholinopropoxy)-quinazolin-4-amine (Cl 1033), erbB2 tyrosine kinase inhibitors such as lapatinib); inhibitors of the hepatocyte growth factor family; inhibitors of the insulin growth factor family; inhibitors of the platelet-derived growth factor family such as imatinib and/or nilotinib (AMN107); inhibitors of serine/threonine kinases (for example Ras/Raf signalling inhibitors such as farnesyl transferase inhibitors, for example sorafenib (BAY 43-9006), tipifarnib (R1 15777) and lonafarnib (SCH66336)), inhibitors of cell signalling through MEK and/or AKT kinases, c-kit inhibitors, abl kinase inhibitors, PI3 kinase inhibitors, Plt3 kinase inhibitors, CSF-1 R kinase inhibitors, IGF receptor (insulin-like growth factor) kinase inhibitors; aurora kinase inhibitors (for example AZD1 152, PH739358, VX-680, MLN8054, R763, MP235, MP529, VX-528 AND AX39459) and cyclin dependent kinase inhibitors such as CDK2 and/or CDK4 inhibitors;
(v) antiangiogenic agents such as those which inhibit the effects of vascular endothelial growth factor, [for example the anti-vascular endothelial cell growth factor antibody bevacizumab (Avastin™) and for example, a VEGF receptor tyrosine kinase inhibitor such as vandetanib (ZD6474), vatalanib (PTK787), sunitinib (SU1 1248), axitinib (AG-013736), pazopanib (GW 786034) and 4-(4-fluoro-2-methylindol-5- yloxy)-6-methoxy-7-(3-pyrrolidin-1 - ylpropoxy)quinazoline (AZD2171 ; Example 240 within WO 00/47212), compounds such as those disclosed in International Patent Applications W097/22596, WO 97/30035, WO 97/32856 and WO 98/13354 and compounds that work by other mechanisms (for example linomide, inhibitors of integrin ov 3 function and angiostatin)];
(vi) vascular damaging agents such as Combretastatin A4 and compounds disclosed in International Patent Applications WO 99/02166, WO 00/40529, WO 00/41669, WO 01 Z92224, WO 02/04434 and WO 02/08213; (vii) an endothelin receptor antagonist, for example zibotentan (ZD4054) or atrasentan;
(viii) antisense therapies, for example those which are directed to the targets listed above, such as ISIS 2503, an anti-ras antisense;
T1
(ix) gene therapy approaches, including for example approaches to replace aberrant genes such as aberrant p53 or aberrant BRCA1 or BRCA2, GDEPT (gene-directed enzyme pro-drug therapy) approaches such as those using cytosine deaminase, thymidine kinase or a bacterial nitroreductase enzyme and approaches to increase patient tolerance to chemotherapy or radiotherapy such as multidrug resistance gene therapy; and
(x) immunotherapy approaches, including for example ex-vivo and in-vivo approaches to increase the immunogenicity of patient tumour cells, such as transfection with cytokines such as interleukin 2, interleukin 4 or granulocyte-macrophage colony stimulating factor, approaches to decrease T-cell anergy, approaches using transfected immune cells such as cytokine-transfected dendritic cells, approaches using cytokine-transfected tumour cell lines and approaches using anti-idiotypic antibodies.
In a particular embodiment, the antiproliferative treatment defined hereinbefore may involve, in addition to the compound of formula (I) of the invention, conventional surgery or radiotherapy or chemotherapy. Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate dosing of the individual components of the treatment. Such combination products employ the compounds of this invention within the dosage range described hereinbefore and the other pharmaceutically-active agent within its approved dosage range.
According to this aspect the present invention further relates to the pharmaceutically acceptable crystal form of the compound of formula (I), as defined herein, for use in the treatment of a cancer (for example a cancer involving a solid tumour) in combination with another anti-tumour agent. The antitumour agent is preferably selected from the anti-tumour agents as listed hereinabove.
As understood herein, the term "combination" refers to simultaneous, separate or sequential administration. In one aspect of the invention "combination" refers to simultaneous administration. In another aspect of the invention "combination" refers to separate administration. In a further aspect of the invention "combination" refers to sequential administration. Where the administration is sequential or separate, the delay in administering the second component should not be such as to lose the beneficial effect of the combination.
Examples
The following examples are merely illustrative of the present invention and should not be construed to limit the scope of the invention which is defined by the appended claims.
Synthesis of the compound of formula (I)
The syntheses of the compounds of formula (I) according to the present invention are preferably carried out as described in, or per analogy to, specific synthetic procedures described in the following synthetic examples.
Preparative examples
General considerations
Abbreviations used in the descriptions that follow are: AcOH (acetic acid); aq. (aqueous); Ar (Argon); Atm (atmosphere); BH3.THF (boran tetrahydrofuran complex); br. (broad, 1H NMR signal); BOC2O (di-tert-butyldicarbonate); (Cataxium APdGs (Mesylate[(di(1-adamantyl)-n-butylphosphine)-2-(2'-amino- 1 ,1'-biphenyl)]palladium(ll)); (CDCI3 (deuterated chloroform); cHex (cyclohexane); CMPB ( Cyanomethylene trimethylphosphorane); CS2CO3 (cesium carbonate); Cui (copper iodide); DABCO ((1 ,4-diazabicyclo[2.2.2]octane)); DAST (diethylaminosulfur trifluoride);DBU (1 ,8- Diazabicyclo(5.4.0)undec-7-ene); DCE (dichloroethane); d (doublet, 1H NMR signal); DCM (dichloromethane); DIBAL-H (diisobutyl aluminium hydride); DIPEA or DIEA (di-zso-propylethylamine); DMAP (4- W-W-dimethylaminopyridine), DME (1 ,2-dimethoxyethane), DMEDA (dimethylethylenediamine ); DMF (W-W-dimethylformamide); DMSO (dimethyl sulfoxide); DPPA (diphenylphosphoride azide);dtbbpy (Bis(1 , 1 -dimethylethyl)-2,2'-bipyridine); ES (electrospray); EtOAc or EA (ethyl acetate); EtOH (ethanol); h (hour(s)); FA (formic acid); HATLI (1-[Bis(dimethylamino)methylene]-1 H-1 ,2,3-triazolo[4,5-b]pyridinium 3- oxide hexafluorophosphate); HFIP ( Hexafluoroisopropanol); 1H NMR (proton nuclear magnetic resonance spectroscopy); HPLC (High Performance Liquid Chromatography), iPrOH (/so-propanol); K3PO4 (tripotassium phosphate); lr[dF(CF3)(dtbbpy)PFe ((4,4'-Di-t-butyl-2,2'-bipyridine)bis[3,5-difluoro-2- [5-trifluoromethyl-2-pyridinyl-kN)phenyl-kC]iridium(l II) hexafluorophosphate); LIOH (lithium hydroxide); m (multiplet, 1H NMR signal); mCPBA (mefa-chloroperoxybenzoic acid), MeCN (acetonitrile), MeOH (methanol); min (minute(s)); Mn02 (Manganese (IV) oxide); MS (mass spectrometry); MTBE (methyl fert- butyl ether); NaBH4 (sodium borohydride); NaHCOs (sodium hydrogenocarbonate); Na2S2O3 (sodium thiosulfate); NCS (N-chlorosuccinimide); NH3 (ammonia); NH4CI (ammonium fluoride); NiCh (nickel dichloride); NIS (N-lodosuccinimide); NMP (N-methylpyrrolidone); NMR (nuclear magnetic resonance); Pd/C (palladium on charcoal); Pd2dba3 (tris(dibenzylideneacetone)dipalladium ); Pd(dppf)Ch (1 ,1 - Bis(diphenylphosphino)ferrocene dichloropalladium ); Pd(Ph3)2Cl2 (Bis(triphenylphosphine)palladium(ll) dichloride ); PE (petroleum ether); Pd-PEPPSI-IPentCI o-picoline ([1 ,3-bis[2,6-bis(1-ethylpropyl)phenyl]- 4 ,5-d ich loro-imidazol-2-yl idene]-d ichloro-(2-methy Ipy rid in-1 -ium-1 -yl)pal lad i um ; Pd(0H)2 (palladium hydroxide); Pd(Phs)4 (Palladium-tetrakis(triphenylphosphine)); Phl(OAc)2 ((Diacetoxyiodo)benzene)); P(tBu)3 (Tri-tert-butylphosphine ); Py (pyridine); q (quartet, 1 H NMR signal); quin (quintet, 1 H NMR signal); rac (racemic); RT (retention time); s (singlet, 1H NMR signal); sat. (saturated); t (triplet, 1H NMR signal); TBAF (tetrabutylammonium fluoride); fert-BuBrettPhos-Pd-G3 ([(2-Di-fert-butylphosphino-3,6-dimethoxy- 2',4',6'-triisopropyl-1 ,1 '-biphenyl)-2-(2'-amino-1 ,1 '-biphenyl)]palladium(ll) methanesulfonate); tBuXPhos Pd G3 (Methanesulfonato(2-di-t-butylphosphino-2',4',6'-tri-i-propyl-1 , T-biphenyl)(2'-amino-1 , T-biphenyl- 2-yl)palladium(ll))TBDMSCI or TBSCI (tert-butyldimethylsilyl chloride); tBuOH (tert-butanol); TEA (triethylamine) ; TFA (trifluoroacetic acid); TFAA (trifluoroacetic anhydride), THF (tetrahydrofuran); TLC
(thin layer chromatography); TMSCHN2 (Trimethylsilyldiazomethane); TMSCN (trimethylsilyl cyanide); TMSOTf (Trimethylsilyl trifluoromethanesulfonate ); TTMSS (trimethylsilane); UPLC (Ultra-High Performance Liquid Chromatography), UV (ultraviolet), wt-% (percent by weight); Xantphos (4,5- Bis(diphenylphosphino)-9,9-dimethylxanthene); Xantphos Pd G4 (Methanesulfonato[9,9-dimethyl-4,5- bis(diphenylphosphino)xanthene](2'-methylamino-1 ,1'-biphenyl-2-yl)palladium(ll)).
General Procedure: All starting materials and solvents were obtained either from commercial sources or prepared according to literature references. Commercially available reagents and anhydrous solvents were used as supplied, without further purification. Unless otherwise stated all reactions were stirred. Organic solutions were routinely dried over anhydrous sodium sulfate. Column chromatography was performed on pre-packed silica (100-1000 mesh, 40-63 pm) cartridges using the amount indicated. All air- and moisture-sensitive reactions were carried out in oven-dried (at 120 °C) glassware under an inert atmosphere of nitrogen or argon. Compound names were generated using ChemDraw Prime (Perkin Elmer). In some cases generally accepted names of commercially available reagents were used in place of ChemDraw generated names.
Reversed Phase HPLC methods for LCMS Analysis of compounds:
Method 1 : SHIMADZU LCMS-2020 Kinetex EVO C18 2.1X30mm,5pm at 50°C; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 1 .55 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1 .20 min, held at 5% A-95% B; 1 .20-1 .21 min, returned to 95% A-5% B, 1 .21-1 .55 min, held at 95% A-5% B.
Method 2: SHIMADZU LCMS-2020 Kinetex EVO C18 2.1X30mm,5pm at 40°C ; Mobile Phase : A: 0.025% NH3-H2O in water (v/v) , B: MeCN; flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.55 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1 .20 min, held at 5% A-95% B; 1 .20-1 .21 min, returned to 95% A-5% B, 1 .21-1 .55 min, held at 95% A-5% B.
Method 3: SHIMADZU LCMS-2020 Kinetex EVO C18 2.1X30mm,5pm at 50°C; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 2.0 mL/min; eluted with the mobile phase over 0.80 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1 .20 min, held at 5% A-95% B; 1 .20-1 .21 min, returned to 95% A-5% B, 1 .21-1 .55 min, held at 95% A-5% B.
Method 4: SHIMADZU LCMS-2020 Kinetex® EVO C18 2.1X20 mm 2.6 urn at 50°C; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 2.0 mL/min; eluted with the mobile phase over 1.00 min employing UV detection at 220 nm and 254 nm. Gradient information:
0.01-0.60 min, ramped from 95% A-5% B to 5% A-95% B; 0.61-0.78 min, held at 5% A-95% B; 0.78-0.79 min, returned to 95% A-5% B, 0.79-0.80 min, held at 95% A-5% B.
Reversed Phase HPLC methods for Analysis of compounds:
Method 1 : Agilent 1100/1200 series liquid chromatograph, Xselect Premier CSH C18, 150 x 4.6 mm, 2.5 pm particle size at 30°C; Mobile Phase: A: De-ionised Water : Trifluoroacetic Acid (100:0.1%); B: Acetonitrile : Trifluoroacetic Acid (100:0.1 %); flow rate held at 1 .0 mL/mn; eluted with the mobile phase over 30.00 min employing UV detection at 268 nm. Gradient information: 0 - 2.00 mn held at 75% A-25% B; 2.00 - 12.00 mn ramped from 75% A-25% B to 60% A-40% B; 12.00 - 25.00 ramped from 60% A-40% B to 5% A-95% B; 25.00 - 29.5 mn held at 5% A-95% B; 29.5 - 30.00 mn return to to 75% A-25% B; held at 75% A-25% B for 5 mn.
1H NMR Spectroscopy:
1H NMR spectra were acquired on a Bruker Avance HI spectrometer at 400 MHz using residual undeuterated solvent as reference. 1H NMR signals are specified with their multiplicity / combined multiplicities as apparent from the spectrum; possible higher-order effects are not considered. Chemical shifts of the signals (6) are specified as ppm (parts per million).
Salt stoichiometry:
In the present text, in particular in the experimental section, for the synthesis of intermediates and of examples of the present invention, when a compound is mentioned as a salt form with the corresponding base or acid, the exact stoichiometric composition of said salt form, as obtained by the respective preparation and/or purification process, is, in most cases, unknown. Unless specified otherwise, suffixes to chemical names or structural formulae such as "hydrochloride", "trifluoroacetate", "sodium salt", or "x HO", "x CF3COOH", "x Na+", for example, are to be understood as not a stoichiometric specification, but solely as a salt form. This applies analogously to cases in which synthesis intermediates or example compounds or salts thereof have been obtained, by the preparation and/or purification processes described, as solvates, such as hydrates with (if defined) unknown stoichiometric composition.
Preparation of Intermediate 1.1
(5-bromo-3-chloropyridin-2-yl)methanamine
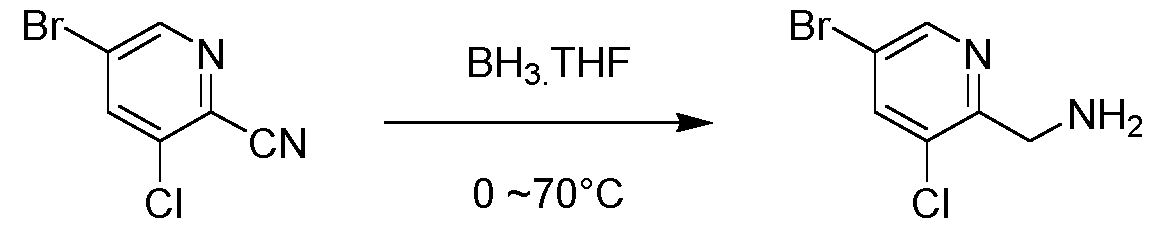
To a mixture of 5-bromo-3-chloropicolinonitrile (2.0 g, 9.20 mmol) in THF (10 mL) under ice-water cooling was added BH3 THF (1 M, 11 .04 mL) over 5 min. The mixture was stirred at 0°C for 30 min before it was warmed to 20°C and stirred for another 30 min at this temperature. The mixture was cooled to 0°C and quenched with dropwise addition of MeOH (10 mL) over 5 min. The mixture was heated to 70 C and
stirred for 30 min at this temperature. The reaction was concentrated under vacuum to give the crude product (2.2 g) as a light brown solid. The crude product was dissolved in HCI (aq. 2M, 20 mL), washed with DCM (20 mL; 2x) and the aqueous phase was finally concentrated under vacuum to give the product (5-bromo-3-chloro-2-pyridyl)methanamine (1 .5 g, 4.07 mmol, 44.26% yield, 70% purity, HCI salt) as a light brown solid.
LC/MS RT 0.18 min (method 2); m/z 222.9 (M+H)+ (ESI+), 1H NMR (400 MHz, DMSO-c/6) 6 = 8.78 (d, J = 2.0 Hz, 1 H), 8.69 (br, 3H), 8.47 (d, J = 2.0 Hz, 1 H), 4.24 (d, J = 6.2 Hz, 2H).
Preparation of Intermediate 1.2
Ethyl 2-(((5-bromo-3-chloropyridin-2-yl)methyl)amino)-2-oxoacetate

To a mixture of (5-bromo-3-chloro-2-pyridyl)methanamine (1 .5 g, 5.82 mmol, HCI salt) in DCM (30 mL) under ice-water cooling was added DIPEA (2.25 g, 17.45 mmol). Then, ethyl 2-chloro-2-oxoacetate (952.77 mg, 6.98 mmol) was added over 5 min and the mixture was stirred at 0°C for 30 min. The mixture was warmed to 20°C and stirred for 30 min at this temperature. The mixture was quenched with aqueous NaHCOa solution (50 mL) and extracted with DCM (50 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated under vacuum. The residue was purified by column chromatography on silica gel (PE: EtOAc=10:1 to 1 :1) to give the product ethyl 2-(((5-bromo-3-chloropyridin-2- yl)methyl)amino)-2-oxoacetate (1300 mg, 3.64 mmol, 62.57% yield, 65.6% purity) as a white solid. RT 0.61min (method 1); m/z 322.8 (M+H)+ (ESI+). The product was used without further purification in the next step.
Preparation of Intermediate 1.3
Ethyl 6-bromo-8-chloroimidazo[1 ,5-a]pyridine-3-carboxylate
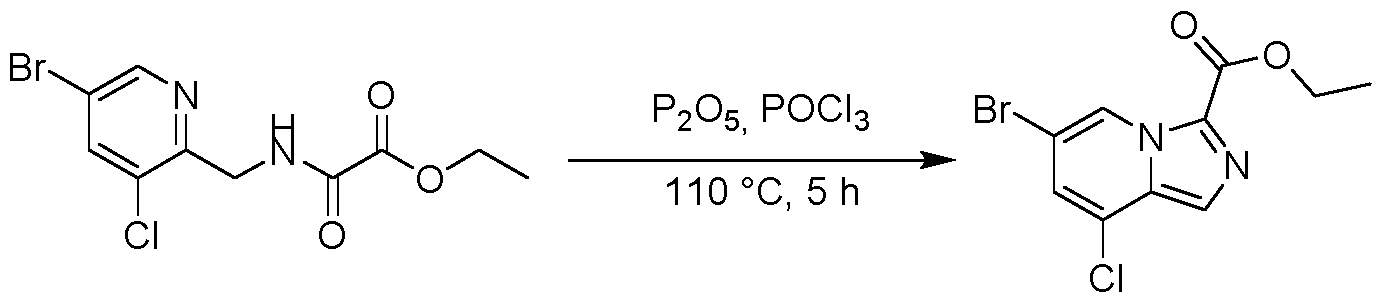
To a mixture of ethyl ethyl 2-(((5-bromo-3-chloropyridin-2-yl)methyl)amino)-2-oxoacetate (1300 mg, 4.04 mmol) in POCh (15 mL) under ice water cooling was added phosphorus pentoxide (2.87 g, 20.21 mmol). The mixture was heated to 110°C and stirred for 5 h at this temperature. The mixture was cooled to 25°C and concentrated under vacuum to give a residue. The residue was dissolved in EtOAc (50 mL) and washed with water (30 mL) and an aqueous NaHCOa solution (30 mL). Then it was was finally concentrated under vacuum to give a residue. The residue was purified by column chromatography on
silica gel (PE: EtOAc=10:1 to 3:1) to give the product ethyl 6-bromo-8-chloroimidazo[1 ,5-a]pyridine-3- carboxylate (900 mg, 2.97 mmol, 73.34% yield) as a white solid.
LC/MS RT 0.718 min (method 1), m/z 304.8(M+H)+ (ESI+), 1H NMR (400 MHz, CHLOROFORM- d) 5 = 9.47 (s, 1 H), 7.77 (s, 1 H), 7.20 (s, 1 H), 4.65-4.42 (m, 2H), 1.57-1.42 (m, 3H)
Preparation of Intermediate 1.4
6-bromo-8-chloroimidazo[1 ,5-a]pyridine-3-carbohydrazide

To a mixture of ethyl 6-bromo-8-chloroimidazo[1 ,5-a]pyridine-3-carboxylate (900 mg, 2.97 mmol) in EtOH (20 mL) was added NH2NH2 H2O (1.48 g, 29.65 mmol, 98%). The mixture was heated to 80°C and stirred for 2 h at this temperature. The reaction was cooled to 25°C and the precipitated solid was separated off. The crude product was triturated with EtOH (5 mL) to give 6-bromo-8-chloroimidazo[1 ,5- a]pyridine-3-carbohydrazide (650 mg, 2.25 mmol, 75.72% yield) as a white solid.
LC/MS RT 0.56 min (method 1); m/z 290.8 (M+H)+ (ESI+); 1H NMR (400 MHz, DMSO-cfe) 5 = 10.02 (s, 1 H), 9.50 (s, 1 H), 7.72 (s, 1 H), 7.51 (s, 1 H), 4.58 (d, J = 4.0 Hz, 2H).
Preparation of Intermediate 1.5
6-bromo-8-chloro-N'-(2,2-difluoroacetyl)imidazo[1 ,5-a]pyridine-3-carbohydrazide

To a mixture of 6-bromo-8-chloroimidazo[1 ,5-a]pyridine-3-carbohydrazide (650 mg, 2.25 mmol) in EtOH (20 mL) was added ethyl 2,2-difluoroacetate (3.10 g, 22.45 mmol) and DBU (683.58 mg, 4.49 mmol). The mixture was heated to 100°C stirred for 16 h at this temperature. The mixture was cooled to 25°C and concentrated under vacuum. The residue was dissolved with DCM (50 mL), washed with an aqueous NH4CI solution (30 mL; 2x) and concentrated under vacuum to give the crude product. The crude product was purified by column chromatography on silica gel (PE/EtOAc=1 : 1 to MeOH: EtOAc=1 : 10) to give the product 6-bromo-8-chloro-N'-(2,2-difluoroacetyl)imidazo[1 ,5-a]pyridine-3-carbohyd razide (650 mg, 1.56 mmol, 69.32% yield, 88% purity) as a white solid.
LC/MS RT 0.62 min (method 1); m/z 368.8 (M+H)+ (ESI+); 1H NMR (400 MHz, DMSO-cfe) 5 = 10.95 (br, 2H), 9.44 (s, 1 H), 7.81 (s, 1 H), 7.59 (s, 1 H), 6.38 (t, J = 53.2, 1H).
Preparation of Intermediate 1.6
2-(6-bromo-8-chloroimidazo[1 ,5-a]pyridin-3-yl)-5-(difluoromethyl)-1 ,3,4-thiadiazole

To a mixture of 6-bromo-8-chloro-N'-(2,2-difluoroacetyl)imidazo[1 ,5-a]pyridine-3-carbohydrazide (550 mg, 1 .50 mmol) in toluene (20 mL) was added Lawessons reagent (665.80 mg, 1 .65 mmol) under a N2 atmosphere. The reaction was heated to 120°C and stirred for 2 h at this temperature. The reaction was cooled to 25°C and concentrated under vacuum. The residue was triturated with MeOH (10 mL) at 70°C for 1 h, filtered and the cake was collected, and dried under vacuum to give the product 2-(6-bromo- 8-chloroimidazo[1 ,5-a]pyridin-3-yl)-5-(difluoromethyl)-1 ,3,4-thiadiazole (530 mg, 1.45 mmol, 96.88% yield) as a light yellow solid.
LC/MS RT 0.806 min (method 1); m/z 366.8 (M+H)+ (ESI+); 1H NMR (400 MHz, DMSO-cfe) 6 = 9.62 (s, 1 H), 8.64 (s, 1 H), 8.09 (s, 1 H), 7.70 (t, J = 53.2, 1 H).
Preparation of Intermediate 1.7
2-(6-(benzylthio)-8-chloroimidazo[1 ,5-a]pyridin-3-yl)-5-(difluoromethyl)-1 ,3,4-thiadiazole
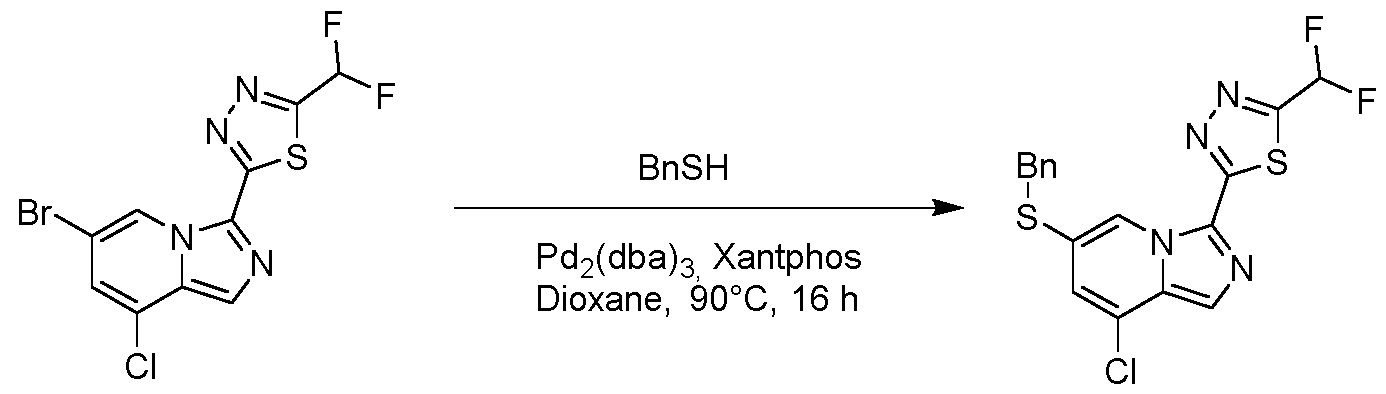
To a mixture of 2-(6-bromo-8-chloroimidazo[1 ,5-a]pyridin-3-yl)-5-(difluoromethyl)-1 ,3,4-thiadiazole (450 mg, 1.23 mmol) and phenylmethanethiol (168.17 mg, 1.35 mmol) in dioxane (10 mL) which was degassed with nitrogen for 2 min was added Pd2(dba)3 (112.72 mg, 123.09 pmol), Xantphos (71 .22 mg, 123.09 pmol) and DIEA (477.26 mg, 3.69 mmol) under nitrogen. The mixture was heated to 90°C and stirred for 16 h at this temperature. The mixture was filtered and concentrated under vacuum. The residue was purified by column chromatography on silica gel (PE: EtOAc=20:1 to 5:1) to give the product 2-(6- (benzylthio)-8-chloroimidazo[1 ,5-a]pyridin-3-yl)-5-(difluoromethyl)-1 ,3,4-thiadiazole (250 mg, 489.15 pmol, 39.74% yield, 80% purity) as a light yellow solid.
LC/MS RT 0.99 min (method 1); m/z 409.0 (M+H)+ (ESI+); 1H NMR (400 MHz, CHLOROFORM-d) 5 = 9.35 (s, 1H), 7.69 - 7.67 (m, 1 H), 7.39 - 7.28 (m, 2H), 7.25 - 7.12 (m, 3H), 7.05 (t, J = 53.2, 1H), 7.00 (s, 1H), 6.90 (s, 1H), 4.10 (s, 2H)
Preparation of Intermediate 1.8
2-(6-(benzylthio)-8-chloro-1-iodoimidazo[1 ,5-a]pyridin-3-yl)-5-(difluoromethyl)-1 ,3,4-thiadiazole
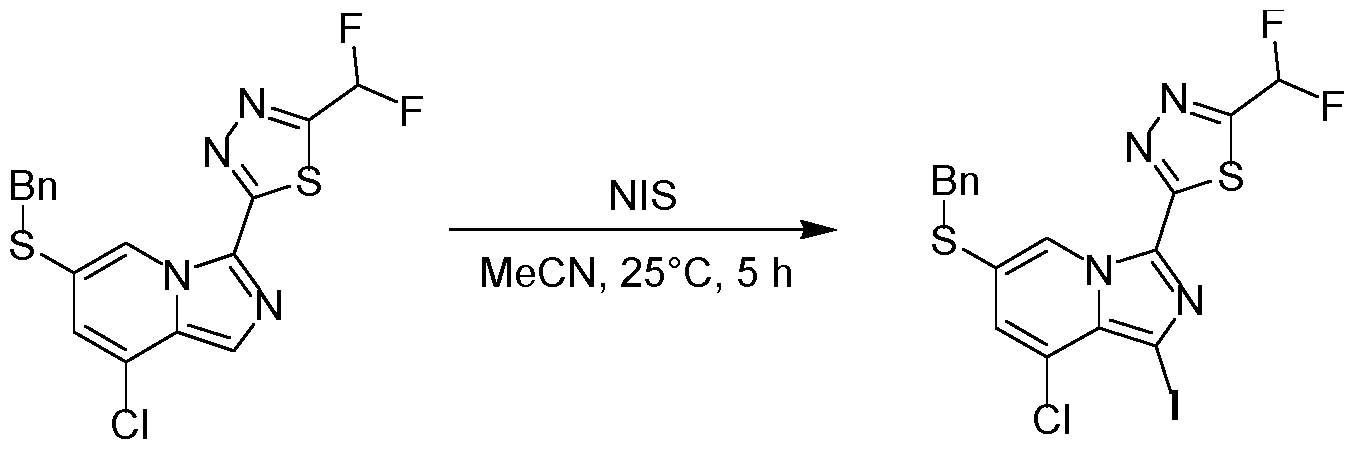
To a mixture of 2-(6-(benzylthio)-8-chloroimidazo[1 ,5-a]pyridin-3-yl)-5-(difluoromethyl)-1 ,3,4- thiadiazole (130 mg, 317.95 pmol) in MeCN (5 mL) at O°C was added NIS (78.68 mg, 349.74 mol). The mixture was stirred at 25°C for 5 h. The reaction mixture was used for the next step directly.
LC/MS RT 0.99 min (method 1); m/z 535.0 (M+H)+ (ESI+)
Preparation of Intermediate 1.9
8-chloro-3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-1-iodoimidazo[1 ,5-a]pyridine-6-sulfonyl chloride

A mixture of 2-(6-(benzylthio)-8-chloro-1 -iodoimidazo[1 ,5-a]pyridin-3-yl)-5-(difluoromethyl)-1 ,3,4- thiadiazole (170 mg, 317.95 pmol) in MeCN (5 mL) was cooled to 0°C before H2O (5.73 mg, 317.95 pmol), AcOH (38.19 mg, 635.89 pmol) and 1 ,3-dichloro-5,5-dimethylimidazolidine-2, 4-dione (125.28 mg, 635.89 pmol) was added. The mixture was stirred at 0°C for 2 h. The mixture was diluted with THF (8 mL), dried over Na2SO4, filtered and concentrated under vacuum to give the crude product 8-chloro-3-(5- (difluoromethyl)-l ,3,4-thiadiazol-2-yl)-1-iodoimidazo[1 ,5-a]pyridine-6-sulfonyl chloride (160 mg, 219.14 pmol, 68.92% yield, 70% purity) as a light brown oil.
It is noted that it cannot be excluded that the dichloro-compound 1 ,8-dichloro-3-(5-(d ifluoromethyl)- 1 ,3,4-thiadiazol-2-yl)imidazo[1 ,5-a]pyridi ne-6-sulfonyl chloride was also formed in this process.
Preparation of Intermediate 1.10
8-ch loro-3-(5-(d ifluoromethyl)- 1 ,3 ,4-th iad iazol-2-yl)-1 -iodo-N-( 1 -methylcyclopropyl)imidazo[1 ,5- a]pyridine-6-sulfonamide

To a mixture of 1-methylcyclopropan-1 -amine (37.80 mg, 531.49 pmol) in pyridine (1 mL) and NMP (N-methyl-2-pyrrol idon) (1 mL) at 0°C was added 8-chloro-3-(5-(d ifluoromethyl)- 1 ,3 ,4-thiad iazol-2- yl)-1-iodoimidazo[1 ,5-a]pyridine-6-sulfonyl chloride (90 mg, 176.09 mol) in MeCN (2 mL). The reaction was stirred at 0°C for 50 min. The reaction mixture was quenched with water (10 mL) and extracted with EtOAc (10 mL; 2x). The organic phase was collected, dried over Na2SO4, filtered and concentrated under vacuum to give a residue which was purified by preparative TLC (PE:EtOAc = 3:1) to give the product 8- chloro-3-(5-(difluoromethyl)-1 ,3 ,4-th iad iazol-2-yl)- 1 -iodo-N-( 1 -methylcyclopropyl)imidazo[1 ,5-a]pyridine- 6-sulfonamide (25 mg, 45.81 pmol, 26.01% yield) as a light yellow solid.
It is noted that it cannot be excluded that be excluded that the dichloro compound 1 ,8-dichloro-3- (5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-N-(1-methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide was also formed in this process.
LC/MS RT 0.510 min (method 3); m/z 545.8 (M+H)+ (ESI+)
Preparation of Intermediate 1.11
8-ch loro-3-(5-(d ifluoromethyl)- 1 ,3 ,4-th iad iazol-2-yl)-N -( 1 -methylcyclopropyl)imidazo[1 ,5- a]pyridine-6-sulfonamide

 of 8-chloro-3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-1 -iodo-N-(1 - methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide (25 mg, 45.81 pmol) in tetrahydrofuran (3 mL) was added Pd/C (5 mg, 10% purity). The reaction was degassed with H2 (15 Psi) three times, then the reaction was stirred at 20°C for 3 h. The reaction mixture was filtered and the filtrate was concentrated under vacuum to give the product 8-chloro-3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-N-(1- methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide (20 mg, 30.96 pmol, 67.59% yield, 65% purity) as a brown solid.
of 8-chloro-3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-1 -iodo-N-(1 - methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide (25 mg, 45.81 pmol) in tetrahydrofuran (3 mL) was added Pd/C (5 mg, 10% purity). The reaction was degassed with H2 (15 Psi) three times, then the reaction was stirred at 20°C for 3 h. The reaction mixture was filtered and the filtrate was concentrated under vacuum to give the product 8-chloro-3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-N-(1- methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide (20 mg, 30.96 pmol, 67.59% yield, 65% purity) as a brown solid.
It is noted that it cannot be excluded that the dichloro compound 1 ,8-dichloro-3-(5-(d ifluoromethy I)- 1 ,3,4-thiadiazol-2-yl)-N-(1-methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide was also formed in this process.
LC/MS RT 0.468 min (method 3); m/z 420.0 (M+H)+ (ESI+)
Preparation of Intermediate 1.12 tert-butyl (2S,6S)-4-(3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-6-(N-(1- methylcyclopropyl)sulfamoyl)imidazo[1 ,5-a]pyridin-8-yl)-2,6-dimethylpiperazine-1 -carboxylate

To a mixture of 8-chloro-3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-N-(1- methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide (30 mg, 0.0715 mmol) in dioxane (0.5mL) was added tert-butyl (2S,6S)-2,6-dimethylpiperazine-1 -carboxylate (15 mg, 0.0715 mmol), CS2CO3 (70 mg, 0.214 mmol) and Pd-PEPPSI-IPentCI o-picoline (7.0 mg, 0.00715 mmol). The reaction mixture was degassed with N2 (3x) and then stirred at 98 °C for 1 h. The mixture was filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative TLC (Petroleum ether: Ethyl acetate = 1 :2) to give the product tert-butyl (2S,6S)-4-(3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-6-(N-(1- methylcyclopropyl)sulfamoyl)imidazo[1 ,5-a] pyrid i n-8-yl)-2 , 6-d i methyl pi perazine-1 -carboxylate (16 mg, 0.0238 mmol, 33.34% yield) as a yellow solid.
LC/MS RT 0.573 min (method 4); m/z 598.1 (M+H+) (ES ); 1H NMR (CDCI3, 400 MHz): 9.74 (s, 1 H), 7.83 (s, 1 H), 7.08 (t, J = 53.6 Hz, 1 H), 6.36 (s, 1 H), 5.06 (s, 1 H), 4.20-4.37 (m, 2H), 4.14-4.11 (m, 2H), 3.67-3.48 (m, 2H), 1.52 (s, 9H), 1.40 (s, 3H), 1.34 (d, J = 6.8 Hz, 6H), 0.98-0.96 (m, 2H), 0.63-0.58 (m, 2H).
Preparation of Example 1
3-(5-(d ifl uoromethyl)-1 ,3 ,4-th iad iazol-2-yl)-8-((3S , 5S)-3 ,5-d i methy Ipiperazi n- 1 -yl)-N -( 1 - methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide (crystal form 1)
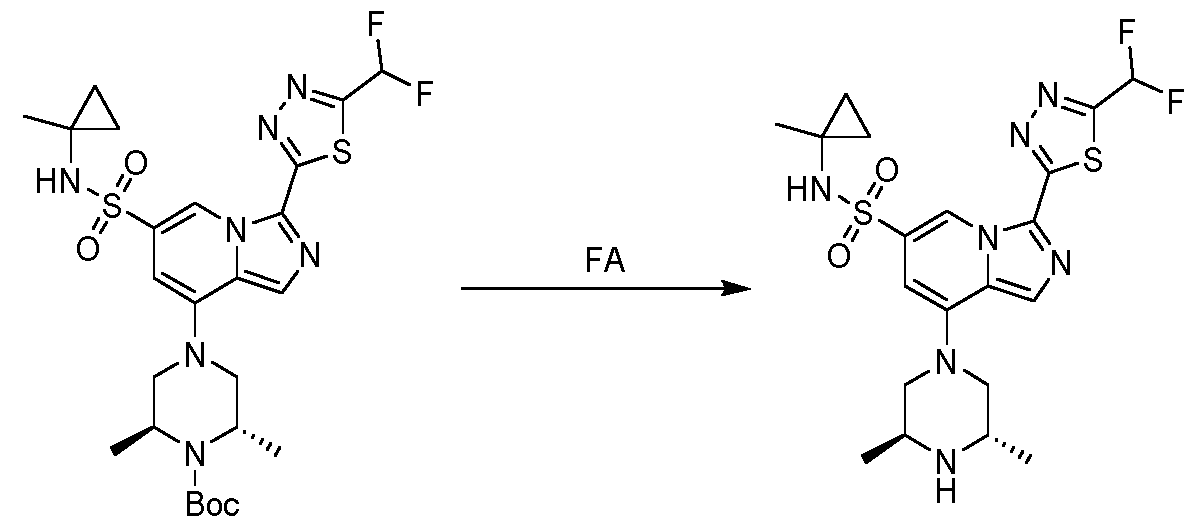 31
31
The reaction was performed twice on 29g of tert-butyl (2S,6S)-4-[3-[5-(difluoromethyl)-1 ,3,4- thiadiazol-2-yl]-6-[(1-methylcyclopropyl)sulfamoyl]imidazo[1 ,5-a]pyridin-8-yl]-2,6-dimethyl-piperazine-1- carboxylate and then, the reaction mixture were combined for the work-up.
A mixture of tert-butyl (2S,6S)-4-[3-[5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl]-6-[(1- methylcyclopropyl)sulfamoyl]imidazo[1 ,5-a]pyridin-8-yl]-2,6-dimethyl-piperazine-1 -carboxylate (29.00 g, 48.5 mmol) in formic acid (290 mL, 7686 mmol, 158 eq) was stirred at 20 °C for 2 h. The both reaction mixtures were combined and the formic acid was concentrated under vacuum to give a residue which was diluted with water (500 mL). The pH was adjusted to 9 with a saturated solution of NaHCCh and the product was extracted with DCM (500 mL*3). The combined organic layer was washed with brine (500 mL), dried over Na2SO4, filtered and concentrated under vacuum. The resulting yellow solid was taken up with EtOH(1.4 L) and stirred at 110 °C until complete dissolution. The resulting clear solution was stirred 12 h cooling to room temperature. The precipitate was filtered off and dried under vacuum to give a fraction A of 3-[5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl]-N-(1 -methylcyclopropyl)-8-[(3S,5S)-3,5- dimethylpiperazin-1-yl]imidazo[1 ,5-a]pyridine-6-sulfonamide (33.00 g, 66.3 mmol, 68.6 % yield) as a yellow solid.
The filtrate was concentrated under vacuum to give a residue (15g) which was recrystallized in EtOH following the same procedure as depicted to get fraction A. The precipitate was filtered off and dried under vacuum to give a fraction B of 3-[5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl]-N-(1-methylcyclopropyl)- 8-[ 3S,5S)-3,5-dimethylpiperazin-1-yl]imidazo[1 ,5-a]pyridine-6-sulfonamide (7.6 g, 15.3 mmol, 15.7 % yield) as a yellow solid.
Both fractions A and B correspond to crystal form 1 .
Fraction A
1H NMR (400 MHz, DMSO-cfe) 5 ppm 9.56 (s, 1 H), 8.43 (s, 1 H), 7.86 (s, 1 H), 7.67 (t, J = 53.2 Hz, 1 H), 6.65 (s, 1 H), 3.25 - 3.31 (m, 2 H), 3.17-3.25 (m, 2 H), 3.00 (m, 2 H), 2.15 (br s, 1 H), 1 .13 - 1 .22 (m, 9 H), 0.66 - 0.77 (m, 2 H), 0.41 - 0.49 (m, 2 H) Fraction B
1H NMR (400 MHz, DMSO-cfe) 5 ppm 9.56 (s, 1 H), 8.43 (s, 1 H), 7.86 (s, 1 H), 7.67 (t, J = 53.2 Hz, 1 H), 6.65 (s, 1 H), 3.25 - 3.31 (m, 2 H), 3.17-3.25 (m, 2 H), 2.95-3.07 (m, 2 H), 2.15 (br s, 1 H), 1 .13 - 1 .22 (m, 9 H), 0.66 - 0.77 (m, 2 H), 0.41 - 0.49 (m, 2 H)
The following Table 1 provides an overview on the compound described in the example section.
Table 1

Alternative recrystallization experiment leading to crystal form 1
Alternative preparation of crystal form 1
3-[5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl]-N-(1-methylcyclopropyl)-8-[(3S,5S)-3,5-dimethylpiperazin-1- yl]imidazo[1 ,5-a]pyridine-6-sulfonamide (1010 g, 2.03 mol) and THF (5.55 L) were charged to Vessel 1 under nitrogen. The mixture was heated to 65°C to obtain a solution and filtered into Vessel 2 at 60-70°C. Vessel 1 was rinsed with THF (0.5 L) which was transferred to Vessel 2. Vessel 2 was heated to 60°C then N-heptane (0.5 L) was added slowly, followed by 3-[5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl]-N-(1- methylcyclopropyl)-8-[(3S ,5S)-3 , 5-d i methyl pi perazin-1 -yl]imidazo[1 , 5-a]pyrid ine-6-su Ifonamide form 1 seed (1 g, 0.002 mol). The mixture was held at 55-65°C, then additional n-heptane (4 L) was added slowly to the vessel. The mixture was cooled slowly to 5°C, stirred overnight and filtered. The vessel and filter cake were washed with the filtrate (1 L). The filter cake was washed with cold 6:4 THF: n-heptane (1 L) and cold n-heptane (1 L). The solid was dried to afford 868 g (85.9% recovery; purity 99% (HPLC method 1)). XRPD indicated the desired crystal form, i.e. , form 1 . HPLC RT 11.9 min (method i)
Reference example - crystal form 2
3-(5-(d ifl uoromethyl)-1 ,3 ,4-th iad iazol-2-yl)-8-((3S , 5S)-3 ,5-d i methy Ipiperazi n- 1 -yl)-N -( 1 - methylcyclopropyl)imidazo[1 ,5-a]pyridi ne-6-sulfonamide (crystal form 2)
 To a solution of tert-butyl (2S,6S)-4-(3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-6-(N-(1- methylcyclopropyl)sulfamoyl)imidazo[1 ,5-a] pyridi n-8-yl)-2 , 6-d i methylpiperazine-1 -carboxylate (13.00 g, 21.8 mmol) in DCM (130mL) was added HCI/dioxane (130 mL, 326 mmol, 2N). The mixture was stirred at 20 °C for 8 hours. The reaction mixture was combined with another reaction performed on 2g tert-butyl (2S,6S)-4-(3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-6-(N-(1-methylcyclopropyl)sulfamoyl)imidazo[1 ,5- a]pyridin-8-yl)-2,6-dimethylpiperazine-1 -carboxylate, then basified with saturated NaHCC (aq.) until pH=10 and extracted with DCM (200 mL, 3x). The combined organic layer was washed with brine (300 mL), dried over anhydrous Na2SC>4, filtered and concentrated under vacuum. The resulting yellow solid (10g) was triturated with DCM (50 mL) at 20 °C for 20 min, filtered and dried under vacuum to give 3-(5- (d ifluoromethyl)- 1 , 3,4-thiad iazol-2-y l)-8-((3S,5S)-3 ,5-d i methyl pi perazin-1 -yl)-N-( 1 - methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide (8.00 g, 16.1 mmol, 80.00 % yield,) as a yellow solid.
To a solution of tert-butyl (2S,6S)-4-(3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-6-(N-(1- methylcyclopropyl)sulfamoyl)imidazo[1 ,5-a] pyridi n-8-yl)-2 , 6-d i methylpiperazine-1 -carboxylate (13.00 g, 21.8 mmol) in DCM (130mL) was added HCI/dioxane (130 mL, 326 mmol, 2N). The mixture was stirred at 20 °C for 8 hours. The reaction mixture was combined with another reaction performed on 2g tert-butyl (2S,6S)-4-(3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-6-(N-(1-methylcyclopropyl)sulfamoyl)imidazo[1 ,5- a]pyridin-8-yl)-2,6-dimethylpiperazine-1 -carboxylate, then basified with saturated NaHCC (aq.) until pH=10 and extracted with DCM (200 mL, 3x). The combined organic layer was washed with brine (300 mL), dried over anhydrous Na2SC>4, filtered and concentrated under vacuum. The resulting yellow solid (10g) was triturated with DCM (50 mL) at 20 °C for 20 min, filtered and dried under vacuum to give 3-(5- (d ifluoromethyl)- 1 , 3,4-thiad iazol-2-y l)-8-((3S,5S)-3 ,5-d i methyl pi perazin-1 -yl)-N-( 1 - methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide (8.00 g, 16.1 mmol, 80.00 % yield,) as a yellow solid.
Then, a mixture of 3-(5-(difluoromethyl)-1 ,3,4-thiadiazol-2-yl)-8-((3S,5S)-3,5-dimethylpiperazin-1- yl)-N-(1-methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide (7.10 g, 14.3 mmol, Pd residue is 95 ppm) and sulfhydryl-resin (4.30 g) in THF (150mL) was stirred at 60 °C for 2 hours. The reaction mixture was cooled to 20 °C and filtered. Sulfhydryl-resin (4.30 g) was added to the filtrate and the mixture was stirred at 60 °C for 16 hours. The reaction mixture was cooled to 20 °C, then filtered and the filtrate was concentrated under reduced pressure.
The resulting 7.0 g of 3-(5-(difluoromethyl)-1 ,3 ,4-thiad iazol-2-yl)-8-((3S ,5S)-3,5-d i methyl pi perazin- 1-yl)-N-(1-methylcyclopropyl)imidazo[1 ,5-a]pyridine-6-sulfonamide were triturated in DCM (50mL) and stirred for 2 hours. Then, the precipitate was filtered and dried under vacuum to give 3-(5-(difluoromethyl)- 1 ,3,4-thiadiazol-2-yl)-8-((3S,5S)-3,5-dimethylpiperazin-1-yl)-N-(1-methylcyclopropyl)imidazo[1 ,5- a]pyridine-6-sulfonamide (4.30 g, 8.64 mmol, 60.5 % yield, form 2). The obtained analytical data (1H NMR and LC/MS) are consistent with the chemical structure of the compound.
X-ray powder diffraction pattern analysis
X-Ray Powder Diffraction patterns were collected on a PANalytical diffractometer using Cu Ka radiation (45 kV, 40 mA), 9 - 0 goniometer, focusing mirror, divergence slit (1/2”), soller slits at both incident and divergent beam (4 mm) and a PIXcel detector. The software used for data collection was X’Pert Data Collector, version 7.4 and Panalytical X’Pert Operator Interface, version1.2. The data was presented using High Score Plus, version 5.2. XRPD patterns were acquired via a transmission foil sample stage (polyimide - Kapton, 12.7 pm thickness film) under ambient conditions using a PANalytical X’Pert PRO. The data collection range was 2.994 - 35 °20 with a continuous scan speed of 0.202004 °s-
1
The measurements for crystal form 1 and crystal form 2 are shown in Table 2 and Table 3, respectively. Furthermore, complete patterns are presented in Figures 2 and 4, and their comparison is shown in Figures 6 and 7. Table 2. Peak list for XRPD measurement performed for the crystal form 1 of the compound of formula (I)-

Table 3. Peak list for XRPD measurement performed for the crystal form 2 of the compound of formula (I)


Differential Scanning Calorimetry (DSC)
DSC data was collected on a TA Instruments Discovery DSC equipped with a 54-position sample holder. The instrument was verified for energy and temperature calibration using certified indium. A predefined amount of the sample, 0.5-2.0 mg, was placed in a Tzero Pan with a Tzero Hermetic Lid and heated at 20 °C min-1 from 30 to 350 °C or varied as experimentation dictated. A purge of dry nitrogen at 50 mL min-1 was maintained over the sample. The instrument control, data acquisition and analysis were performed with TA Instruments TRIOS software v5.5.0.323. All results feature endotherm up convention. The results of the measurements are shown in Figure 3 for crystal form 1 . Figure 5 shows the reference results for crystal form 2.
Further experiments concerning melt-recrystallization-melt sequence starting from form 2 are shown in Figure 12. Slow heat DSC whilst improving resolution has an impact on overall sensitivity and a slight alteration on the relative peak positions (which can alter as a function of how sample is presented as well as sample and particle size variation which can cause a thermal lag type effect). The fused endotherm obtained at higher heating rate (10°C/min) heating was resolved into a melt-recrystallisation- melt sequence. It showed melting of form 2, followed by a recrystallisation from the molten material to give form 1 which then goes on to melt. This data provides further evidence for improved stability of form
1 over form 2. Accordingly, upon melting of form 2, it may, at least in part, recrystallize as form 1 , here identified based on its melting point.
Finally, a DSC recycling experiment, shown in Figure 13, was performed where a sample of form
2 was heated to just beyond the peak melting of form 2. Briefly, the sample was then cooled back down
to ambient, showing the presence of a recrystallisation event, and then reheated showing the presence of a single melting event associated with form 1 . This further confirms that form 1 recrystallises from the melt of form 2, hinting at higher thermodynamic stability of form 1 .
Thermo-Gravimetric Analysis (TGA)
TGA data was collected on a TA Instruments Discovery TGA equipped with a 25-position autosampler. The instrument was calibrated using a certified weight and certified Alumel and Nickel for temperature. A predefined amount of the sample, ca. 5 mg, was loaded into a pre-tared aluminium ACCUPIK sample pan and platinum crucible and was heated at 20 °C min 1 from ambient temperature to 400 °C unless otherwise stated. A nitrogen purge at 25 mL min 1 was maintained over the sample. The instrument control, data acquisition and analysis were performed with TA Instruments TRIOS software v5.5.0.323. The results of the measurements are shown in Figure 3 for crystal form 1 . Figure 5 shows the reference results for crystal form 2.
Competitive equilibrations of crystal form 1 and crystal form 2 of the compound of formula (I)
To investigate the relationship between the crystalline form 1 and crystalline form 2 of the compound of formula (I), a series of solvent based maturations have been conducted to determine whether one form will predominate under various conditions.
Experimental: A saturated solution of the compound of formula (I) was prepared by equilibrating the API (~100 mg) in the relevant solvent (Table 4, 5 mL) for 4 hours. The suspension was filtered and the filtrate used as the media for the competitive equilibrations.
Equal amounts of crystal form 1 and crystal form 2 (10 to 20 mg) were charged to crystallisation tubes and the relevant saturated solutions (1 mL) added. The mixtures were equilibrated at the relevant temperature and were sampled for XPRD analysis following 4 and 24 hours. At the 24 hours timepoint, solids were analysed both as the damp cake and post drying at 45 °C.
Results are summarised in Table 4. XRPD data is presented in Figures 7 to 10. Data presented is for solids post drying; XRPD analysis of damp cakes showed no differences to dried solids.
The data confirms that where some solubility of the API is achieved, form 1 is the predominant form with no evidence of form 2 by XRPD. The conversion of form 2 to form 1 in several entries was noted to be complete within four hours with the level of form 2 remaining visibly diminishing by XRPD as temperature of equilibration is increased. The conversion of form 2 to form 1 at higher temperatures in each of the solvents tested was noted.
For those entries where solubility remained low and thicker suspensions were obtained, a mixture of form 1 and form 2 forms was still visible by XRPD. Solubility was postulated as being very low in these solvents, for example water, at ambient and sub-ambient temperature which did not readily facilitate form conversion.
There were no instances of form 1 having converted to form 2 in any solvent or temperature.
The data is supportive of form 1 being the preferred stable crystalline form of the compound of formula (I).
Table 4.

Solubility of data of crystal form 1
1. Preparation of FaSSIF a. Prepare buffer A
0.420 g of NaOH, 3.438 g of NaFhPO and 6.186 g of NaCI was dissolved into about 900 mL ultrapure water and the pH of the solution was adjusted to 6.5 with 1 N NaOH or 1 N HCI. Then the solution was diluted with ultrapure water to 1000 mL at room temperature. b. Add powder
2.240 g of FaSSIF, FeSSIF and FaSSGF Powder was added to about 500 mL of buffer A. Stir until powder was completely dissolved. Then the solution was diluted with buffer A to 1000 mL at room temperature. c. Ready to use
The solution was stayed for 2 hours. It became slightly opalescent and was ready to use.
Note: Use within 48 hours at room temperature and 24 hours at 37°C.
2. Preparation of FeSSIFvl a. Prepare buffer
4.040 g of NaOH, 8.650 g of glacial acetic acid and 11 .874 g of NaCI were dissolved into about 900 mL ultrapure water and the pH of the solution was adjusted to 5.0 with 1 N NaOH or 1 N HCI. Then the solution was diluted with ultrapure water to 1000 mL at room temperature. b. Add powder
11 .200 g of FaSSIF, FeSSIF & FaSSGF Powder was added to about 500 mL of buffer. Stir until powder was completely dissolved. Then the solution was diluted with buffer to 1000 mL at room temperature. c. Ready to use
FeSSIFVI was ready to use.
Note: Use within 48 hours at room temperature and 24 hours at 37°C.
3. Preparation of FeSSIF v2 a. Prepare buffer
4.040 g of NaOH, 8.650 g of glacial acetic acid and 11 .874 g of NaCI were dissolved into about 900 mL ultrapure water and the pH of the solution was adjusted to 5.8 with 1 N NaOH or 1 N HCI. Then the solution was diluted with ultrapure water to 1000 mL at room temperature. b. Add powder
11 .200 g of FaSSIF, FeSSIF & FaSSGF Powder was added to about 500 mL of buffer. Stir until powder was completely dissolved. Then the solution was diluted with buffer to 1000 mL at room temperature. c. Ready to use
FeSSIFV2 was ready to use.
Note: Use within 48 hours at room temperature and 24 hours at 37°C.
4. Preparation of FaSSGF a. Prepare HCI/NaCI solution.
Dissolve 1 .999 g of NaCI into about 900 mL ultrapure water and the pH of the solution was adjusted to 1.6 with 1 N HCI. Then dilute the solution with ultrapure water to 1000 mL at room temperature. b. Add powder.
Add 0.060 g of FaSSIF, FeSSIF & FaSSGF Powder to about 500 mL of HCI/NaCI solution. Stir until powder is completely dissolved. Then dilute the solution with buffer to 1000 mL at room temperature. c. Ready to use
FaSSGF was ready to use.
Note: Use within 48 hours at room temperature and 24 hours at 37°C.
5. Preparation of Blank FaSSIF pH 6.5
Dissolve 0.420 g of NaOH, 3.438 g of NaH2PO4 and 6.186 g of NaCI into about 900 mL ultrapure water and the pH of the solution was adjusted to 6.5 with 1 N NaOH or 1 N HCI. Then dilute the solution with ultrapure water to 1000 mL at room temperature and store at 4°C for up to 30 days. pH was checked on the day of experiment and was adjusted if outside specification of pH values ± 0.1
6. Preparation of Blank FeSSIF pH 5.0
Dissolve 4.040 g of NaOH, 8.650 g of glacial acetic acid and 11 .874 g of NaCI into about 900 mL ultrapure water and the pH of the solution was adjusted to 5.0 with 1 N NaOH or 1 N HCI. Then dilute the solution with ultrapure water to 1000 mL at room temperature and store at4°C for up to 30 days. pH was checked on the day of experiment and was adjusted if outside specification of pH values ± 0.1
7. Test compound was weighed out for measuring the solubility (about 1.0 mg or 5.0 mg each in three separate 1 .5 mL glass vials; the upper limit was 1 mg/mL or 5 mg/mL). Control compound diclofenac sodium was weighed out for measuring the solubility (about 1 .0 mg each in three separate 1 .5 mL glass vials; the upper limit was 1 mg/mL).
8. For each compound, one vial was used to do the standard while the other two were used to measure the solubility in duplicate.
9. Each compound was placed in order into their proper 96-well racks. One rack for the standard and one rack for the samples.
10. Based on the amount, the proper volume of FaSSGF, Blank FeSSIF, FeSSIFvl , FeSSIFv2, Blank FaSSI or FaSSIF (-1000 pL) was added to each vial of the solubility sample plate using the pipettes. One stir stick was added to each vial and the vials were sealed using a molded PTFE/Silicone plug.
11 . The Solubility Sample plate was transferred to the Eppendorf Thermomixer Comfort plate shaker and shaken at 37°C at 1100 RPM for 0.5, 1 , 4, 8 or 24 hours.
12. After completion of 0.5, 1 , 4, 8 and 24 hours, plugs were removed and the stir sticks were removed using a big magnet. Record the turbidity of each medium. The samples were centrifuged at 2500 g, 37 °C for 30 minutes. Then aliquot of 350 pL was transferred from the supernatant. The tips were placed into acetonitrile for 5 seconds and then into water for another 5 seconds. Then the first 50 pL was disposed into waste and the remaining 300 pL was disposed into another new vial of the cap-less Solubility Sample plate, and then was centrifuged at 2500 g, 37°C for 30 minutes. Then, measured the pH value of each medium. Then, aliquot of 80 pL of the supernatant was transferred from the Solubility Sample plate after second centrifugation. The tips were placed into acetonitrile for 5 seconds and then into water for another 5 seconds. Then the first 30 pL was disposed into waste, and aliquot of 50 pL was transferred into a new 96-well plate followed by addition of 50 pL DMSO and 400 pL of a mixture of H2O and acetonitrile (1 :1 in v/v). Vortex well and then aliquot of 50 pL was taken from the diluent followed by addition of 450 pL of a mixture of H2O and acetonitrile (1 :1 in v/v)). 200 L of diluent was transferred to a new 96-well plate for LC-MS/MS analysis. The dilution factor was changed according to the solubility value and the LC-MS signal response.
Chromatographic conditions
LC system: Shimadzu
MS analysis: Triple Quad™ 5500+ instrument from AB Inc (Canada) with an ESI interface Column temperature: 40°C Injection volume: 1 pL
Column: XSelect Hss T3 2.5 pm (2.1 x 50 mm) Column XP
Mobile phase: 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) Elution rate: 1 .0 mL/min
Solubility data of form 1 are reported in the table below.


The solubility data obtained herein are supportive of a possibility of formulating an oral formulation with the pharmaceutically acceptable crystal form of the invention.
Claims
1 . A pharmaceutically acceptable crystal form of a compound of formula (I):
 wherein said pharmaceutically acceptable crystal is obtainable upon recrystallization of the compound of formula (I) from ethanol.
wherein said pharmaceutically acceptable crystal is obtainable upon recrystallization of the compound of formula (I) from ethanol.
2. The pharmaceutically acceptable crystal form of claim 1 , wherein the recrystallization from ethanol involves complete dissolution in ethanol while stirring, followed by cooling to room temperature over 12 hours, while stirring.
3. The pharmaceutically acceptable crystal form of claim 1 or 2, wherein the X-ray powder diffraction pattern obtained using CuKa radiation comprises at least one peak selected from the peaks at 12.24 ± 0.2 °20, 20.04 ± 0.2 °20, 20.40 ± 0.2 °20 and 24.02 ± 0.2 °20.
4. The pharmaceutically acceptable crystal form of claim 1 or 2, wherein the X-ray powder diffraction pattern obtained using CuKa radiation comprises the peaks at 12.24 ± 0.2 °20, 20.04 ± 0.2 °20, 20.40 ± 0.2 °20 and 24.02 ± 0.2 °20.
5. The pharmaceutically acceptable crystal form of claim 3 or 4, wherein the X-ray powder diffraction pattern obtained using CuKa radiation further comprises a peak at 18.71 ± 0.2 °20 and/or a peak at 6.86 ± 0.2 °20.
6. The pharmaceutically acceptable crystal form of any one of claims 3 to 5, wherein the X-ray powder diffraction pattern obtained using CuKa radiation further comprises at least one peak selected from the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21 .59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20.
7. The pharmaceutically acceptable crystal form of any one of claims 3 to 5, wherein the X-ray powder diffraction pattern obtained using CuKa radiation further comprises the peaks at 15.42 ± 0.2 °20, 19.04 ± 0.2 °20, 21.59 ± 0.2 °20, 23.19 ± 0.2 °20, 23.58 ± 0.2 °20, 28.01 ± 0.2 °20, and 29.18 ± 0.2 °20.
8. The pharmaceutically acceptable crystal form of any one of claims 1 to 7, wherein the crystal form is characterized by a melting point of 207.5 ± 4.0 °C.
9. The pharmaceutically acceptable crystal form of claim 8, wherein the crystal form is characterized by a melting point of 207.5 ± 2.0 °C.
10. The pharmaceutically acceptable crystal form of claim 9, wherein the crystal form is characterized by a melting point of 207.5 ± 1 .0 °C.
11. A pharmaceutical composition comprising the pharmaceutically acceptable crystal of any one of claims 1 to 10 and a pharmaceutically acceptable carrier.
12. The pharmaceutically acceptable crystal of any one of claims 1 to 10, or a pharmaceutical composition of claim 11 , for use in therapy.
13. The pharmaceutically acceptable crystal of any one of claims 1 to 10 or the pharmaceutical composition of claim 11 for use in a method of treating a disease or disorder in which PARG activity is implicated.
14. The pharmaceutically acceptable crystal of any one of claims 1 to 10 or the pharmaceutical composition of claim 11 for use in a method of treating a proliferative disorder.
15. The pharmaceutically acceptable crystal form for use or the pharmaceutical composition for use of claim 14, wherein the proliferative disorder is cancer.
16. The pharmaceutically acceptable crystal form for use or the pharmaceutical composition for use of claim 15, wherein the cancer is selected from gastric, lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin cancer.
17. Use of the pharmaceutically acceptable crystal form of any one of claims 1 to 10 in the manufacture of a medicament for the inhibition of PARG enzyme activity.
18. Use of the pharmaceutically acceptable crystal form of any one of claims 1 to 10 in the manufacture of a medicament for the treatment of a proliferative condition.
19. The use of claim 18, wherein the proliferative condition is cancer.
20. The use of claim 19, wherein the cancer is selected from gastric, lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin cancer.
21 . A method of treating a disease or disorder in which PARG activity is implicated in a subject in need thereof, the method comprising administering to said subject a therapeutically effective amount of the pharmaceutically acceptable crystal form of any one of claims 1 to 10.
22. A method of treating a proliferative disorder in a subject in need thereof, the method comprising administering to said subject a therapeutically effective amount of the pharmaceutically acceptable crystal form of any one of claims 1 to 10.
23. The method of claim 22, wherein the proliferative disorder is cancer.
24. The method of claim 23, wherein the cancer is selected from gastric, lung, colon, breast, ovarian, prostate, liver, pancreas, brain, and skin cancer.
25. A method for making the pharmaceutically acceptable crystal form of any one of claims 1 to 10, the method comprising the step of crystallizing the compound of formula (I) as depicted in claim 1 from ethanol or from the mixture of tetrahydrofurane/n-heptane.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EPPCT/EP2023/077341 | 2023-10-03 | ||
| PCT/EP2023/077341 WO2024074497A1 (en) | 2022-10-03 | 2023-10-03 | Parg inhibitory compound |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025073870A1 true WO2025073870A1 (en) | 2025-04-10 |
Family
ID=92925370
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/077899 Pending WO2025073870A1 (en) | 2023-10-03 | 2024-10-03 | Parg inhibitory compound |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025073870A1 (en) |
Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997022596A1 (en) | 1995-12-18 | 1997-06-26 | Zeneca Limited | Quinazoline derivatives |
| WO1997030035A1 (en) | 1996-02-13 | 1997-08-21 | Zeneca Limited | Quinazoline derivatives as vegf inhibitors |
| WO1997032856A1 (en) | 1996-03-05 | 1997-09-12 | Zeneca Limited | 4-anilinoquinazoline derivatives |
| WO1998013354A1 (en) | 1996-09-25 | 1998-04-02 | Zeneca Limited | Quinazoline derivatives and pharmaceutical compositions containing them |
| WO1999002166A1 (en) | 1997-07-08 | 1999-01-21 | Angiogene Pharmaceuticals Ltd. | Use of colchinol derivatives as vascular damaging agents |
| WO2000040529A1 (en) | 1999-01-07 | 2000-07-13 | Angiogene Pharmaceuticals Ltd. | Colchinol derivatives as vascular damaging agents |
| WO2000041669A2 (en) | 1999-01-15 | 2000-07-20 | Angiogene Pharmaceuticals Ltd. | Benzimidazole vascular damaging agents |
| WO2000047212A1 (en) | 1999-02-10 | 2000-08-17 | Astrazeneca Ab | Quinazoline derivatives as angiogenesis inhibitors |
| WO2001092224A1 (en) | 2000-05-31 | 2001-12-06 | Astrazeneca Ab | Indole derivatives with vascular damaging activity |
| WO2001094341A1 (en) | 2000-06-06 | 2001-12-13 | Astrazeneca Ab | Quinazoline derivatives for the treatment of tumours |
| WO2002004434A1 (en) | 2000-07-07 | 2002-01-17 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as vascular damaging agents |
| WO2002008213A1 (en) | 2000-07-07 | 2002-01-31 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as angiogenesis inhibitors |
| WO2009050183A2 (en) | 2007-10-17 | 2009-04-23 | Novartis Ag | Imidazo [1, 2-a] pyridine derivatives useful as alk inhibitors |
| WO2016092326A1 (en) | 2014-12-12 | 2016-06-16 | Cancer Research Technology Limited | 2,4-dioxo-quinazoline-6-sulfonamide derivatives as inhibitors of parg |
| WO2016097749A1 (en) | 2014-12-19 | 2016-06-23 | Cancer Research Technology Limited | Parg inhibitory compounds |
| US20190233411A1 (en) | 2018-01-29 | 2019-08-01 | Merck Patent Gmbh | Gcn2 inhibitors and uses thereof |
| WO2021055744A1 (en) | 2019-09-20 | 2021-03-25 | Ideaya Biosciences, Inc. | 4-substituted indole and indazole sulfonamido derivatives as parg inhibitors |
| WO2023057389A1 (en) | 2021-10-04 | 2023-04-13 | Forx Therapeutics Ag | Parg inhibitory compounds |
-
2024
- 2024-10-03 WO PCT/EP2024/077899 patent/WO2025073870A1/en active Pending
Patent Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997022596A1 (en) | 1995-12-18 | 1997-06-26 | Zeneca Limited | Quinazoline derivatives |
| WO1997030035A1 (en) | 1996-02-13 | 1997-08-21 | Zeneca Limited | Quinazoline derivatives as vegf inhibitors |
| WO1997032856A1 (en) | 1996-03-05 | 1997-09-12 | Zeneca Limited | 4-anilinoquinazoline derivatives |
| WO1998013354A1 (en) | 1996-09-25 | 1998-04-02 | Zeneca Limited | Quinazoline derivatives and pharmaceutical compositions containing them |
| WO1999002166A1 (en) | 1997-07-08 | 1999-01-21 | Angiogene Pharmaceuticals Ltd. | Use of colchinol derivatives as vascular damaging agents |
| WO2000040529A1 (en) | 1999-01-07 | 2000-07-13 | Angiogene Pharmaceuticals Ltd. | Colchinol derivatives as vascular damaging agents |
| WO2000041669A2 (en) | 1999-01-15 | 2000-07-20 | Angiogene Pharmaceuticals Ltd. | Benzimidazole vascular damaging agents |
| WO2000047212A1 (en) | 1999-02-10 | 2000-08-17 | Astrazeneca Ab | Quinazoline derivatives as angiogenesis inhibitors |
| WO2001092224A1 (en) | 2000-05-31 | 2001-12-06 | Astrazeneca Ab | Indole derivatives with vascular damaging activity |
| WO2001094341A1 (en) | 2000-06-06 | 2001-12-13 | Astrazeneca Ab | Quinazoline derivatives for the treatment of tumours |
| WO2002004434A1 (en) | 2000-07-07 | 2002-01-17 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as vascular damaging agents |
| WO2002008213A1 (en) | 2000-07-07 | 2002-01-31 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as angiogenesis inhibitors |
| WO2009050183A2 (en) | 2007-10-17 | 2009-04-23 | Novartis Ag | Imidazo [1, 2-a] pyridine derivatives useful as alk inhibitors |
| WO2016092326A1 (en) | 2014-12-12 | 2016-06-16 | Cancer Research Technology Limited | 2,4-dioxo-quinazoline-6-sulfonamide derivatives as inhibitors of parg |
| WO2016097749A1 (en) | 2014-12-19 | 2016-06-23 | Cancer Research Technology Limited | Parg inhibitory compounds |
| US20190233411A1 (en) | 2018-01-29 | 2019-08-01 | Merck Patent Gmbh | Gcn2 inhibitors and uses thereof |
| WO2021055744A1 (en) | 2019-09-20 | 2021-03-25 | Ideaya Biosciences, Inc. | 4-substituted indole and indazole sulfonamido derivatives as parg inhibitors |
| WO2023057389A1 (en) | 2021-10-04 | 2023-04-13 | Forx Therapeutics Ag | Parg inhibitory compounds |
Non-Patent Citations (24)
| Title |
|---|
| ATZRODT J ET AL., BIOORG MED CHEM, vol. 20, no. 18, 2012, pages 5658 - 5667 |
| COHENCHANG, NAT. CHEM. BIOL., vol. 14, 2018, pages 236 |
| COSTANTINO ET AL., SCIENCE, vol. 343, 2014, pages 88 |
| FARMER ET AL., NATURE, vol. 434, 2005, pages 917 |
| FONTANA ET AL., ELIFE, 2017 |
| FUJIHARA ET AL., CURR. CANCER DRUG TARGETS, vol. 9, 2009, pages 953 |
| GOGOLA ET AL., CANCER CELL, vol. 33, 2018, pages 1078 |
| HALAZONETIS ET AL., SCIENCE, vol. 319, 2008, pages 1352 |
| HOUL ET AL., NAT COMMUN., vol. 10, 2019, pages 5654 |
| J. MED. CHEM., vol. 47, 2004, pages 6658 - 6661 |
| JAMES ET AL., ACS CHEM. BIOL., vol. 11, 2016, pages 3179 |
| JEMAL ET AL., J. NATL. CANCER INST., vol. 109, 2017, pages 1975 |
| MINO R CAIRA ED - MONTCHAMP JEAN-LUC: "CRYSTALLINE POLYMORPHISM OF ORGANIC COMPOUNDS", TOPICS IN CURRENT CHEMISTRY; [TOPICS IN CURRENT CHEMISTRY], SPRINGER, BERLIN, DE, vol. 198, 1 January 1998 (1998-01-01), pages 163 - 208, XP001156954, ISSN: 0340-1022, [retrieved on 19990226], DOI: 10.1007/3-540-69178-2_5 * |
| MODVIG A ET AL., J ORG CHEM, vol. 79, 2014, pages 5861 - 5868 |
| NEGRINI ET AL., NAT. REV. MOL. CELL BIOL., vol. 11, 2010, pages 220 |
| O'SULLIVAN ET AL., NAT. COMMUN., vol. 10, 2019, pages 1182 |
| PILLAY ET AL., CANCER CELL, vol. 35, 2019, pages 519 |
| RACK ET AL., GENES DEV, vol. 34, 2020, pages 263 |
| RACK ET AL., GENES DEV., vol. 34, 2020, pages 263 |
| SCULLY ET AL., CURR. OPIN. GENET. DEV., vol. 71, 2021, pages 154 |
| STERN ET AL., CRITICAL REVIEWS IN ONCOLOGY/HAEMATOLOGY, vol. 54, 2005, pages 11 - 29 |
| WASZKOWYCZ ET AL., J. MED. CHEM., vol. 61, 2018, pages 10767 |
| WILLIAM JS ET AL., JOURNAL OF LABELLED COMPOUNDS AND RADIOPHARMACEUTICALS, vol. 53, no. 11-12, 2010, pages 635 - 644 |
| ZEMANCIMPRICH, NAT. CELL BIOL., vol. 16, 2013, pages 2 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2023057389A1 (en) | Parg inhibitory compounds | |
| EP3442980B1 (en) | Heterocyclic compounds as ret kinase inhibitors | |
| EP3609898B1 (en) | Compounds useful as ret inhibitors | |
| AU2012228090B2 (en) | Pyrrolopyridineamino derivatives as Mps1 inhibitors | |
| EP3630749B1 (en) | 2-quinolone derived inhibitors of bcl6 | |
| AU2023355735A1 (en) | Parg inhibitory compound | |
| JP2019518059A (en) | Azabenzimidazole derivatives as PI3K beta inhibitors | |
| WO2023083201A1 (en) | Aminopyrazole derivative, and preparation method therefor and use thereof | |
| WO2025073792A1 (en) | Wrn inhibitory compounds | |
| TWI829481B (en) | Bicyclic indazole glucocorticoid receptor antagonists | |
| TW201629060A (en) | Isethionate-dependent protein kinase inhibitor isethionate, crystalline form thereof and preparation method thereof | |
| EP4413000A1 (en) | N,n-dimethyl-4-(7-(n-(1-methylcyclopropyl)sulfamoyl)-imidazo[1,5-a]pyridin-5-yl)piperazine-1-carboxamide derivatives and the corresponding pyrazolo[1,5-a]pyridine derivatives as parg inhibitors for the treatment of cancer | |
| WO2023175185A1 (en) | 2,4-dioxo-1,4-dihydroquinazoline derivatives as parg inhibitors for the treatment of cancer | |
| WO2025133396A1 (en) | Novel bicyclo heteroaryl parg inhibitors | |
| WO2025093755A1 (en) | Novel parc inhibitors | |
| WO2024209035A1 (en) | Parg inhibitory compounds | |
| WO2025133395A1 (en) | Bicyclic (hetero)arylene wrn inhibitory compounds | |
| WO2025073870A1 (en) | Parg inhibitory compound | |
| EP4598922A1 (en) | Parg inhibitory compound | |
| TWI889173B (en) | Pharmaceutical formulations and uses of dihydronaphthalidine compounds containing phenyl substituents | |
| WO2026022150A1 (en) | Parg inhibitory compounds | |
| WO2025191176A1 (en) | Wrn inhibitory compounds | |
| WO2026003380A1 (en) | Wrn inhibitory compounds | |
| EP4413001A1 (en) | Parg inhibitory compounds | |
| WO2025114480A1 (en) | Wrn inhibitory compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24782320 Country of ref document: EP Kind code of ref document: A1 |