WO2025073792A1 - Wrn inhibitory compounds - Google Patents
Wrn inhibitory compounds Download PDFInfo
- Publication number
- WO2025073792A1 WO2025073792A1 PCT/EP2024/077789 EP2024077789W WO2025073792A1 WO 2025073792 A1 WO2025073792 A1 WO 2025073792A1 EP 2024077789 W EP2024077789 W EP 2024077789W WO 2025073792 A1 WO2025073792 A1 WO 2025073792A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- alkylene
- dihydrothiophen
- dioxido
- dimethylphenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- MSI status can be assessed by molecular testing of certain microsatellites, next-generation sequencing of patient genome or by immunohistochemical evaluation of expression of certain MMR proteins. Tumors can be categorized into MSI high (MSI-H), MSI low (MSI-L) and MSS depending on the number of tested microsatellite showing instability. Based on a consensus NCI- Reference Panel (Bethesda, 1998), MSI can be assessed by molecular testing of five microsatellites - including two mononucleotides (BAT25 and BAT26) and three dinucleotides (D2S123, D5S346, D17S250).
- Tumors are denoted as MSI-high (MSI-H) if two or more of the microsatellite markers show instability, MSI-low (MSI-L) if only one microsatellite marker shows instability, and MS-stable (MSS) if none of the five microsatellite markers show instability.
- MSI-H MSI-high
- MSI-L MSI-low
- MSS MS-stable
- WRN WRN RecQ helicase
- WRN RecQ helicase has been identified as a synthetic lethality vulnerability to cancer cells with high microsatellite instability status (MSI-H).
- WRN contains an exonuclease domain and an ATP - dependent helicase domain. It is localized to the nucleus and unwinds double strand DNA, particularly secondary structures (fork DNA, holliday junction, G4-quadruaplex, DNA hairpin and cruciform etc.) during DNA replication, damage and repair processes. Its helicase activity has been shown to be indispensable to the survival of MSI cell lines as helicase-deficient WRN mutant is insufficient to rescue impaired cell viability from WRN knockout or knockdown. The absence of either the WRN protein or inhibition of its helicase activity prevents normal DNA damage and repair processes, leading to increased DNA double- strand breaks (DSB) and subsequent growth arrest and cell death.
- DSB DNA double- strand breaks
- Covalent inhibitors represent a class of small molecules which form covalent bonds with their biological targets to inhibit activities of these targets in physiological or pathological conditions.
- covalent inhibitors engage with nucleophilic residues (e.g. Cysteine, Serine, Threonine, Histidine, Arginine, Tyrosine) lining specific binding pockets on target proteins, in a nucleophilic addition or substitution reaction, with their reactive electrophilic warhead.
- nucleophilic residues e.g. Cysteine, Serine, Threonine, Histidine, Arginine, Tyrosine
- reactive warheads include epoxide, aziridine, ester, ketone, a, -unsaturated carbonyl, nitrile, etc.
- Covalent inhibitors have been discovered as medicines for more than a century, starting with Aspirin being manufactured and marketed as painkillers and anti-inflammatory drug, although its mechanism of action was not revealed until 1970s to be an irreversible inhibitor of cyclooxygenase- 1 (COX- 1).
- Other notable covalent inhibitors used as medicine include antibiotics Penicillin, proton pump inhibitor Omeprazole and Lansoprazole, anticoagulant Clopidogrel.
- Document WO 2023/062575 discloses certain cyclic vinyl sulfone compounds as WRN inhibitors.
- Documents WO 2024/010782 and WO 2024/010784 disclose certain covalent WRN inhibitors. Further covalent inhibitors of WRN are disclosed in document WO 2024/028169.
- the present invention relates to a compound of formula (I) for use as a medicament.
- the present invention relates to a compound of formula (I) for use in the treatment of cancer. It is preferred that the cancer is characterized by high microsatellite instability (MSI) and/or by defective DNA mismatch repair system (dMMR) in a patient.
- MSI microsatellite instability
- dMMR defective DNA mismatch repair system
- the present invention relates to use of a compound of formula (I) in a manufacture of a medicament.
- the present invention relates to use of a compound of formula (I) in a manufacture of a medicament for the treatment of cancer.
- Preferred exemplary alkenyl groups are ethenyl, propenyl (e.g., prop-1-en-1-yl, prop-1-en-2-yl, or prop-2-en-1-yl), butenyl, butadienyl (e.g., buta-1,3-dien-1-yl or buta-1,3- dien-2-yl), pentenyl, or pentadienyl (e.g., isoprenyl).
- alkenyl preferably refers to C2-4 alkenyl.
- alkynyl refers to a monovalent unsaturated acyclic hydrocarbon group which may be linear or branched and comprises one or more (e.g., one or two) carbon-to-carbon triple bonds and optionally one or more (e.g., one or two) carbon-to-carbon double bonds.
- C2-5 alkynyl denotes an alkynyl group having 2 to 5 carbon atoms.
- Preferred exemplary alkynyl groups are ethynyl, propynyl (e.g., propargyl), or butynyl.
- alkynyl preferably refers to C2-4 alkynyl.
- carbocyclyl refers to a hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings), wherein said ring group may be saturated, partially unsaturated (i.e., unsaturated but not aromatic) or aromatic.
- “carbocyclyl” preferably refers to aryl, cycloalkyl or cycloalkenyl.
- each heteroatom-containing ring comprised in said ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring.
- heterocyclyl preferably refers to heteroaryl, heterocycloalkyl or heterocycloalkenyl.
- aryl refers to an aromatic hydrocarbon ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic).
- an “aryl” preferably has 6 to 14 ring atoms, more preferably 6 to 10 ring atoms, even more preferably refers to phenyl or naphthyl, and most preferably refers to phenyl.
- the term “arylene” refers to an aryl group, as defined herein above, but having two points of attachment, i.e.
- a divalent aromatic hydrocarbon ring group including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic).
- an “arylene” preferably has 6 to 14 ring atoms, more preferably 6 to 10 ring atoms, even more preferably refers to phenylene or naphthylene, and most preferably refers to phenylene (particularly phen- 1,4-diyl).
- heteroaryl refers to an aromatic ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic), wherein said aromatic ring group comprises one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group).
- aromatic ring group comprises one or more (such as, e.g., one, two,
- Heteroarylene may, e.g., refer to thienylene (i.e., thiophenylene; e.g., thien-2,3-diyl, thien-2,4-diyl, or thien-2,5-diyl), benzo[b]thienylene, naphtho[2,3-b]thienylene, thianthrenylene, furylene (i.e., furanylene; e.g., furan-2,3-diyl, furan-2,4-diyl, or furan-2,5-diyl), benzofuranylene, isobenzofuranylene, chromanylene, chromenylene, isochromenylene, chromonylene, xanthenylene, phenoxathiinylene, pyrrolylene, imidazolylene, pyrazolylene, pyridylene (i.e., pyridinylene),
- a “heteroarylene”, including any of the specific heteroarylene groups described herein, may be attached through two carbon ring atoms, particularly through those two carbon ring atoms that have the greatest distance from one another (in terms of the number of ring atoms separating them by the shortest possible connection) within one single ring or within the entire ring system of the corresponding heteroarylene.
- the term “cycloalkyl” refers to a saturated hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings).
- Cycloalkylene may, e.g., refer to cyclopropylene (e.g., cyclopropan-1,1-diyl or cyclopropan-1,2-diyl), cyclobutylene (e.g., cyclobutan-1,1-diyl, cyclobutan-1,2-diyl, or cyclobutan-1,3-diyl), cyclopentylene (e.g., cyclopentan-1,1-diyl, cyclopentan-1,2-diyl, or cyclopentan-1,3-diyl), cyclohexylene (e.g., cyclohexan-1,1-diyl, cyclohexan-1,2-diyl, cyclohexan-1,3-diyl, or cyclohexan-1,4-diyl), cycloheptylene, decalinylene (i.e
- each heteroatom-containing ring comprised in said saturated ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring.
- Heterocycloalkyl may, e.g., refer to aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, azepanyl, diazepanyl (e.g., 1,4-diazepanyl), oxazolidinyl, isoxazolidinyl, thiazolidinyl, isothiazolidinyl, morpholinyl (e.g., morpholin-4-yl), thiomorpholinyl (e.g., thiomorpholin-4-yl), oxazepanyl, oxiranyl, oxetanyl, tetrahydrofuranyl, 1,3-dioxolanyl, tetrahydropyranyl, 1,4-dioxanyl, oxepanyl, thiiran
- each heteroatom-containing ring comprised in said saturated ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring.
- cycloalkenyl refers to an unsaturated alicyclic (non-aromatic) hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said hydrocarbon ring group comprises one or more (e.g., one or two) carbon-to-carbon double bonds and does not comprise any carbon-to-carbon triple bond.
- Cycloalkenyl may, e.g., refer to cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cyclohexadienyl, cycloheptenyl, or cycloheptadienyl. Unless defined otherwise, “cycloalkenyl” preferably refers to a C3-11 cycloalkenyl, and more preferably refers to a C3-7 cycloalkenyl.
- a divalent unsaturated alicyclic (non-aromatic) hydrocarbon ring group including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said hydrocarbon ring group comprises one or more (e.g., one or two) carbon-to- carbon double bonds and does not comprise any carbon-to-carbon triple bond.
- each heteroatom-containing ring comprised in said unsaturated alicyclic ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring.
- the term “halogen” (or Hal) refers to fluoro (-F), chloro (-Cl), bromo (-Br), or iodo (-I).
- haloalkyl refers to an alkyl group substituted with one or more (preferably 1 to 6, more preferably 1 to 3) halogen atoms which are selected independently from fluoro, chloro, bromo and iodo, and are preferably all fluoro atoms. It will be understood that the maximum number of halogen atoms is limited by the number of available attachment sites and, thus, depends on the number of carbon atoms comprised in the alkyl moiety of the haloalkyl group.
- Haloalkyl may, e.g., refer to -CF3, -CHF2, -CH2F, -CF2-CH3, -CH2-CF3, -CH2-CHF2, -CH2-CF2-CH3, -CH2-CF2-CF3, or -CH(CF3)2.
- a particularly preferred “haloalkyl” group is -CF3.
- haloalkyl may also be a perhaloalkyl.
- perhaloalkyl refers to a haloalkyl wherein every -H atom has been substituted with a halo atom.
- said perhaloalkyl is perfluoroalkyl.
- the present invention specifically relates to each and every combination of features and embodiments described herein, including any combination of general and/or preferred features/embodiments.
- the present invention relates to a compound of formula (I): or a pharmaceutically acceptable salt thereof.
- Particularly suitable aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl and cycloalkenyl in A are each a single-ring system.
- A is selected from a single ring aryl, a single ring heteroaryl, a single ring heterocycloalkyl, a single ring heterocycloalkenyl, a single ring cycloalkyl, a single ring cycloalkenyl and C2 alkynyl, wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R 1 , and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl or C1- 6 haloalkyl.
- each R 1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1
- each R 1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkyl, C2
- said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X.
- B is heteroarylene, optionally substituted with one or more R 2 .
- Particularly preferred are 5 or 6-membered heteroarylene moieties.
- the heteroarylene may be a 5-membered heteroarylene, optionally substituted with one or more R 2 .
- said 5-membered heteroarylene may be a heteroarylene comprising at least one N atom in the ring, more preferably at least two N atoms in the ring.
- Exemplary suitable 5-membered heteroarylenes Alternatively, B may also be a 6-membered heteroarylene, optionally substituted with one or more R 2 .
- said 6-membered heteroarylene is pyridinylene, preferably a 2,5-pyridinylene, optionally substituted with one or more R 2 .
- B is a 2,5-pyridinylene moiety, as disclosed herein, include .
- the heteroarylene may also be optionally oxidized in the ring.
- One suitable example of such heteroarylene moiety including an oxo group in the ring wherein the heteroarylene is optionally substituted with one or more R 2 , and wherein R D1 is selected from H and C1-6 alkyl (such as methyl).
- the heteroarylene optionally substituted with one or more R 2 .
- each heteroarylene moiety discussed herein, including each specific example, is optionally substituted with one or more R 2 .
- Particularly preferred B being As it is apparent to the skilled person, B is a bivalent moiety and accordingly, as understood herein, preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X.
- B is 2,5-pyridinylene
- B is substituted with -O-(C0-3 alkylene)-carbocyclyl, or - O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or -O-heterocyclyl, more preferably with - O-carbocyclyl, in particular -O-phenyl.
- said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X.
- each R 2 is independently selected from C 1-5 alkyl, C 2-5 alkenyl, C 2-5 alkynyl, -(C 0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(
- each R 2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), and -(C0-3 alkylene)-halogen.
- each R 2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -OH, -O(C1-5 alkyl), -SH, -NH2, -NH(C1-5 alkyl), -N(C1-5 alkyl)(C1-5 alkyl), -NH-OH, -N(C1-5 alkyl)-OH, -NH-O(C1-5 alkyl), -N(C1-5 alkyl)-O(C1-5 alkyl), and halogen.
- heterocycloalkylene is 3,3-oxetanylene.
- Particularly suitable cycloalkylene is 1,2- cyclopopylene.
- Particularly suitable heteroarylene is 1,2,3-triazolyl-1,4-ene.
- X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, - SO2N(C1-6 alkyl)-, -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, -CH(CF3)N(C1-6 haloalkyl)-, C2 alkenylene optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl, cycloalkylene (preferably cyclopropylene) optionally substituted with one or more C1-6 alkyl, -(heterocycloalkylene)
- said heterocycloalkylene is a nitrogen-containing five-membered heterocycloalkylene
- said heterocycloalkenylene is a nitrogen-containing five-membered heterocycloalkenylene
- said heteroarylene is a nitrogen-containing five-membered heteroarylene.
- Particularly suitable heterocycloalkenylene particularly suitable heteroarylene is selected from understood that, preferably, the left side of the bivalent formula embodying X, as shown herein, is connected to B, and the right side of said bivalent formula embodying X is connected to Y.
- the compound of formula (I) is not a compound wherein: A is selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R 1 ; B is according to formula wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein R D1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; X is -CONH- wherein the C atom in said -CON
- B is a 5-membered heteroarylene, optionally substituted with one or more R 2 .
- B is a 6-membered heteroarylene, oxidised in the ring and optionally substituted with one or more R 2 , preferably selected from , optionally substituted with one or more optionally substituted with one or more R 2 , wherein R D1 is selected from H and C1-6 alkyl (such as methyl), wherein the left empty valence is connected to A, and the right empty valence is connected to X.
- B is a 6-membered heteroarylene or phenylene, wherein said heteroarylene and said phenylene are attached to A and X respectively at the opposite positions of the ring (i.e., 1,4 positions), wherein: said heteroarylene and said phenylene are each optionally substituted with at least one group selected from -OH, -COOH, -COO(C1-5 alkyl), -CONH2, -CONH(C1-5 alkyl) and -CON(C1-5 alkyl)(C1-5 alkyl) and/or said heteroarylene and said phenylene are each optionally substituted at a ring position adjacent to the point of attachment of said heteroarylene or said phenylene to X with at least one group selected from halo and -CN, and/or said heteroarylene and said phenylene are each optionally substituted at a ring position adjacent to the point of attachment of said heteroarylene or said phenylene to A with at least one group selected from
- X is -CONH-, wherein the C atom is connected to B and the N atom is connected to Y, C2 alkenylene optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl, or 1,2-cyclopropylene optionally substituted with one or more C1-6 alkyl.
- m is 1 or 2
- m is 1.
- Y is –(C1-2 alkylene)CN.
- Y is -CH2CN or -CH(CH3)CN.
- a and B is as defined for formula (I), and X and Y form a two ring spiro system, defined as: wherein D is a nitrogen-containing heterocycloalkyl or heterocycloalkenyl, m is 1 or 2, and the optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F).
- Y is a moiety according to formula: wherein R y1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, R y2 is selected from -H, -F and -CN (preferably R y2 is -H), and R y3 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible.
- Y is preferably selected from:
- Y is according to formula: or according to formula: wherein R y1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and R y2 is selected from -H, -F and -CN (preferably R y2 is H), and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible.
- A is a cycloalkenyl.
- A may be a moiety according to formula: .
- A is preferably -CF2CH3.
- B is 2,5-pyridinylene, optionally substituted with -O-(C0-3 alkylene)-carbocyclyl, or -O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or -O-heterocyclyl, more preferably with -O-carbocyclyl, in particular -O-phenyl.
- said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X.
- the compound of formula (I) is a compound of formula (Ib): wherein A and Y are as defined for formula (I), including any preferred definition and definition in any of the specific embodiments of the compound of formula (I).
- the compound of formula (I) is a compound of formula (Ic): wherein B and Y are as defined for formula (I), including any preferred definition and definition in any of the specific embodiments of the compound of formula (I).
- Particularly preferred compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts: , , , , ,5 , , , , ,5 , , , 5 , , , , 5 , , , , 5 , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , ,
- the compound of formula (I) is preferably a compound selected from the following compounds: 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3S or R)-1-imino-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R or S,3S or R)-1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R
- a compound described as 33c i.e., the compound of formula: as defined in the foregoing.
- the scope of the invention embraces all pharmaceutically acceptable salt forms of the compounds of formula (I) which may be formed, e.g., by protonation of an atom carrying an electron lone pair which is susceptible to protonation, such as an amino group, with an inorganic or organic acid, or as a salt of an acid group (such as a carboxylic acid group) with a physiologically acceptable cation.
- Exemplary acid addition salts comprise, for example: mineral acid salts such as hydrochloride, hydrobromide, hydroiodide, sulfate salts (such as, e.g., sulfate or hydrogensulfate salts), nitrate salts, phosphate salts (such as, e.g., phosphate, hydrogenphosphate, or dihydrogenphosphate salts), carbonate salts, hydrogencarbonate salts, perchlorate salts, borate salts, or thiocyanate salts; organic acid salts such as acetate, propionate, butyrate, pentanoate, hexanoate, heptanoate, octanoate, cyclopentanepropionate, decanoate, undecanoate, oleate, stearate, lactate, maleate, oxalate, fumarate, tartrate, malate, citrate, succinate, adipate, gluconate, glycolate, nic
- Preferred pharmaceutically acceptable salts of the compounds of formula (I) include a hydrochloride salt, a hydrobromide salt, a mesylate salt, a sulfate salt, a tartrate salt, a fumarate salt, an acetate salt, a citrate salt, and a phosphate salt.
- a particularly preferred pharmaceutically acceptable salt of the compound of formula (I) is a hydrochloride salt.
- the compound of formula (I), including any one of the specific compounds of formula (I) described herein is in the form of a hydrochloride salt, a hydrobromide salt, a mesylate salt, a sulfate salt, a tartrate salt, a fumarate salt, an acetate salt, a citrate salt, or a phosphate salt, and it is particularly preferred that the compound of formula (I) is in the form of a hydrochloride salt.
- the present invention also specifically relates to the compound of formula (I), including any one of the specific compounds of formula (I) described herein, in non-salt form.
- the scope of the invention embraces the compounds of formula (I) in any solvated form, including, e.g., solvates with water (i.e., as a hydrate) or solvates with organic solvents such as, e.g., methanol, ethanol, isopropanol, acetic acid, ethyl acetate, ethanolamine, DMSO, or acetonitrile. All physical forms, including any amorphous or crystalline forms (i.e., polymorphs), of the compounds of formula (I) are also encompassed within the scope of the invention. It is to be understood that such solvates and physical forms of pharmaceutically acceptable salts of the compounds of the formula (I) are likewise embraced by the invention.
- the compounds of formula (I) may exist in the form of different isomers, in particular stereoisomers (including, e.g., geometric isomers (or cis/trans isomers), enantiomers and diastereomers) or tautomers (including, in particular, prototropic tautomers, such as keto/enol tautomers or thione/thiol tautomers). All such isomers of the compounds of formula (I) are contemplated as being part of the present invention, either in admixture or in pure or substantially pure form.
- stereoisomers the invention embraces the isolated optical isomers of the compounds according to the invention as well as any mixtures thereof (including, in particular, racemic mixtures/racemates).
- racemates can be resolved by physical methods, such as, e.g., fractional crystallization, separation or crystallization of diastereomeric derivatives, or separation by chiral column chromatography.
- the individual optical isomers can also be obtained from the racemates via salt formation with an optically active acid followed by crystallization.
- the present invention further encompasses any tautomers of the compounds of formula (I). It will be understood that some compounds may exhibit tautomerism. In such cases, the formulae provided herein expressly depict only one of the possible tautomeric forms.
- the formulae and chemical names as provided herein are intended to encompass any tautomeric form of the corresponding compound and not to be limited merely to the specific tautomeric form depicted by the drawing or identified by the name of the compound.
- the scope of the invention also embraces compounds of formula (I), in which one or more atoms are replaced by a specific isotope of the corresponding atom.
- the invention encompasses compounds of formula (I), in which one or more hydrogen atoms (or, e.g., all hydrogen atoms) are replaced by deuterium atoms (i.e., 2 H; also referred to as “D”).
- the invention also embraces compounds of formula (I) which are enriched in deuterium.
- Naturally occurring hydrogen is an isotopic mixture comprising about 99.98 mol-% hydrogen-1 ( 1 H) and about 0.0156 mol-% deuterium ( 2 H or D).
- the content of deuterium in one or more hydrogen positions in the compounds of formula (I) can be increased using deuteration techniques known in the art.
- a compound of formula (I) or a reactant or precursor to be used in the synthesis of the compound of formula (I) can be subjected to an H/D exchange reaction using, e.g., heavy water (D2O).
- deuteration techniques are described in: Atzrodt J et al., Bioorg Med Chem, 20(18), 5658-5667, 2012; William JS et al., Journal of Labelled Compounds and Radiopharmaceuticals, 53(11-12), 635-644, 2010; Modvig A et al., J Org Chem, 79, 5861-5868, 2014.
- the content of deuterium can be determined, e.g., using mass spectrometry or NMR spectroscopy.
- it is preferred that the compound of formula (I) is not enriched in deuterium. Accordingly, the presence of naturally occurring hydrogen atoms or 1 H hydrogen atoms in the compounds of formula (I) is preferred.
- the invention thus includes (i) compounds of formula (I), in which one or more fluorine atoms (or, e.g., all fluorine atoms) are replaced by 18 F atoms, (ii) compounds of formula (I), in which one or more carbon atoms (or, e.g., all carbon atoms) are replaced by 11 C atoms, (iii) compounds of formula (I), in which one or more nitrogen atoms (or, e.g., all nitrogen atoms) are replaced by 13 N atoms, (iv) compounds of formula (I), in which one or more oxygen atoms (or, e.g., all oxygen atoms) are replaced by 15 O atoms, (v) compounds of formula (I), in which one or more bromine atoms (or, e.g., all bromine atoms) are replaced by 76 Br atoms, (vi) compounds of formula (I), in which one or more bromine atoms (or, e.g., all
- the tablets may contain excipients such as microcrystalline cellulose, lactose, sodium citrate, calcium carbonate, dibasic calcium phosphate and glycine, disintegrants such as starch (preferably corn, potato or tapioca starch), sodium starch glycolate, croscarmellose sodium and certain complex silicates, and granulation binders such as polyvinylpyrrolidone, hydroxypropylmethylcellulose (HPMC), hydroxypropylcellulose (HPC), sucrose, gelatin and acacia. Additionally, lubricating agents such as magnesium stearate, stearic acid, glyceryl behenate and talc may be included. Solid compositions of a similar type may also be employed as fillers in gelatin capsules.
- excipients such as microcrystalline cellulose, lactose, sodium citrate, calcium carbonate, dibasic calcium phosphate and glycine
- disintegrants such as starch (preferably corn, potato or tapioca starch), sodium starch glyco
- the cancer to be treated may also be a chemoresistant and/or a metastatic cancer.
- the antiproliferative treatment i.e. the treatment of cancer
- the compound of formula (I) or a pharmaceutically acceptable salt thereof, as defined hereinbefore may be applied as a sole therapy or may involve, in addition to the compound of the invention, conventional surgery or radiotherapy or chemotherapy.
- Such chemotherapy may include one or more of the following categories of anti-tumour agents:- (i) other antiproliferative/antineoplastic drugs and combinations thereof, as used in medical oncology, such as alkylating agents (for example cis-platin, oxaliplatin, carboplatin, cyclophosphamide, nitrogen mustard, melphalan, chlorambucil, busulphan, temozolamide and nitrosoureas); antimetabolites (for example gemcitabine and antifolates such as fluoropyrimidines like 5-fluorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside, and hydroxyurea); antitumour antibiotics (for example anthracyclines like adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin
- inhibitors also include tyrosine kinase inhibitors, for example inhibitors of the epidermal growth factor family (for example EGFR family tyrosine kinase inhibitors such as N-(3-chloro- 4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazolin-4-amine (gefitinib, ZD1839), N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (erlotinib, OSI-774) and 6- acrylamido-N-(3-chloro-4-fluorophenyl)-7-(3-morpholinopropoxy)-quinazolin-4-amine (CI 1033), erbB2 tyrosine kinase inhibitors such as lapatinib
- EGFR family tyrosine kinase inhibitors such as N-(3-chloro
- the antiproliferative treatment defined hereinbefore may involve, in addition to the compound of formula (I) of the invention, conventional surgery or radiotherapy or chemotherapy.
- Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate dosing of the individual components of the treatment.
- Such combination products employ the compounds of this invention within the dosage range described hereinbefore and the other pharmaceutically-active agent within its approved dosage range.
- the present invention further relates to the compound of formula (I) or a pharmaceutically acceptable salt thereof, as defined herein, for use in the treatment of a cancer (for example a cancer involving a solid tumour) in combination with another anti-tumour agent.
- the anti- tumour agent is preferably selected from the anti-tumour agents as listed hereinabove.
- tert-BuBrettPhos-Pd-G3 [(2-di-tert- butylphosphino-3,6-dimethoxy-2 ,4 ,6 -triisopropyl-1,1 -biphenyl)-2-(2 -amino-1,1 -biphenyl)]palladium(II) methanesulfonate); tBuXPhos Pd G3 (methanesulfonato(2-di-t-butylphosphino-2',4',6'-tri-i-propyl-1,1'- biphenyl)(2'-amino-1,1'-biphenyl-2-yl)palladium(II)); TBDMSCl or TBSCl (tert-buty
- Methods for MS Analysis of compounds Method 1: SHIMADZU LCMS-2020 Kinetex® EVO C182.1X30 mm, 5 m at 40 Mobile Phase: A: 0.025% NH3 ⁇ H2O in water (v/v); B: MeCN; flow rate held at 1.5 mL/min; eluted with the mobile phase over 0.40 min employing UV detection at 220 nm.
- SFC Method 10 Column: Chiralpak AD-350*4.6mm I.D.,3 m
- Mobile phase Phase A for CO2,and Phase B for IPA+ACN(0.05%DEA); Gradient elution: IPA+ACN(0.05%DEA) in CO2 from 20% to 60%, Flow rate:3mL/min; Detector:PDA; Column Temp:35C;Back Pressure:100Bar
- SFC Method 11 Column: (S,S)Whelk-O150*4.6mm I.D., 3.5 m; Mobile phase:Phase A for CO2,and Phase B for IPA(0.05%DEA); Gradient elution: IPA (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100 Bar.
- SFC Method 17 Column: Chiralpak IG-350*4.6 mm I.D., 3 ⁇ m; Mobile phase: Phase A for CO2, Phase B for EtOH+ACN (0.05% DEA); Gradient elution: 60% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar.
- SFC Method 18 Column: Chiralpak IG-350*4.6 mm I.D., 3 ⁇ m; Mobile phase: Phase A for CO2, Phase B for IPA (0.05% DEA); Gradient elution: 40% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar.
- SFC Method 20 Column: Chiralpak AD-350*4.6 mm I.D., 3 ⁇ m; Mobile phase: Phase A for CO2, Phase B for IPA (0.05% DEA); Gradient elution: IPA (0.05% DEA) in CO2 from 20% to 60%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar SFC Method 21: Column:Chiralcel OJ-350*4.6 mm I.D., 3 ⁇ m; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: IPA+ACN (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar.
- the pH of the reaction mixture was adjusted to a value ranged between 1 to 6 by addition of an aqueous solution of hydrochloric acid (1N).
- Work up procedure 1 The resulting precipitate was filtered, collected and dried under reduced pressure to give the desired product.
- Work up procedure 2 The aqueous layer was extracted with EtOAc or DCM (2x or 3x). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure give the desired product.
- Example 2 6-(3,4-dimethylphenyl)-N-((1S or R, 3S)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide
- Example 3 6-(3,4-dimethylphenyl)-N-((1R or S, 3S)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide
- Example 4 6-(3,4-dimethylphenyl)-N-((1S or R, 3R)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide
- Example 5 6-(3,4-dimethylphenyl)-N-(((
- Peak B (SFC Method 1, RT: 1.606 min) (35 mg, 0.0979 mmol) was further purified by preparative SFC (Column:DAICEL CHIRALPAK IG 250 mm*30 mm,10 ⁇ m; Mobile phase: A for CO2, B for MeOH+ACN (with 0.1% NH3 . H2O additive); Gradient elution: B%: 75%-75%,10 min).
- the pH of the two resulting solutions were adjusted to 7 with formic acid and concentrated separately under vacuum (30 °C) until a volume of ⁇ 20 mL was left. Then, to the two solutions was added DCM (40 mL) and pure water (20 mL).
- reaction mixture was stirred at 20 ° C for 2 h.
- the mixture was poured into water (5 mL) and extracted with EtOAc (5 mL, 3x).
- EtOAc 5 mL, 3x
- the combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure.
- Example 7a The solution containing Example 7a was concentrated under vacuum (40 °C) to give (R or S)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (10 mg, 0.0289 mmol, 34.53 % yield, ee.100 % based on SFC method 4) as off-white solid.
- reaction mixture was stirred at 20 o C for 2 h before it was poured into water (5 mL) and extracted with EtOAc (5 mL, 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure.
- the reaction mixture was stirred at 80 °C for 2 h under N2 atmosphere.
- the resulting mixture was filtered and the filtrate was purified by flash silica gel chromatography (ISCO ® ; 20 g SepaFlash ® Silica Flash Column, Mobile phase: A: Petroleum ether, B: Ethyl acetate; B%: 0% to 0%; 50 mL/min) to give the product 3- chloro-6-(3,4-dimethylphenyl)-2-methoxypyridine (920 mg, 2.97 mmol, 66.10 % yield) as a white solid.
- the mixture was stirred at 25 °C for 2 h.
- the mixture was filtered and the filtrate was purified by reversed phase flash (ISCO®; 40 g SepaFlash® column: Spherical C1820-45 ⁇ m, 100 A, mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 80%-95%, 50 mL/min).
- the resulting solution was lyophilized to give the product 3-(benzylthio)-6-(3,4-dimethylphenyl)pyridin-2(1H)-one (100 mg, 0.280 mmol, 62.99 % yield) as a black solid.
- the reaction mixture was degassed and purged with N2 (3x) before prop-1-yne (0.83 mL, 0.826 mmol) was added at 20°C.
- the reaction mixture was stirred at 60 °C for 16 h under N2 atmosphere and then filtered.
- the pH of the filtrate was adjusted with HCl (aq., 6N) to around 4-5.
- the resulting impure product was further purified by preparative HPLC (column: Waters Xbridge 150*25 mm* 5 ⁇ m; mobile phase: A: 0.05% Ammonia in water, B: MeCN; B%: 30%-60%, 10 min) and lyophilized directly to give the product N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6- (prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxamide (0.66 mg, 0.00219 mmol, 3.88 % yield, 97.68% purity) as a grey solid .
- the mixture was stirred at 20 oC for 2 h.
- the reaction mixture was purified by preparative HPLC (column: Phenomenex Synergi C18150*25 mm*10 ⁇ m; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 45%-75%, 15 min) and lyophilized to give the product N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoro-3',4'- dimethyl-[1,1'-biphenyl]-4-carboxamide (20 mg, 0.0543 mmol, 66.26 % yield) as a white solid.
- Example 26 3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide
- Example 25a&25b (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide and (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H- 1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide (85 mg, 0.210 mmol) was separated by preparative SFC (column: Daicel Chiralpak IC 250x30mm I.D.
- reaction mixture was stirred at 0 °C for 3 h, then diluted with water(20 mL) and extracted with ethyl acetate (30 mL;3x). The combined organic layer was washed with aq. Solution of Na2SO3 (sat., 30 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent DCM/MeOH gradient from 100/0 to ⁇ 90/ ⁇ 10 @ 60 mL/min) to give the product diethyl ((methylsulfinyl)methyl)phosphonate (870 mg, 4.06 mmol, 40.25 % yield) as colorless oil.
- reaction mixture was stirred at 80 °C for 1 h, then cooled to room temperature, diluted with water (20 mL) and extracted with ethyl acetate (30 mL;3x). The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated.
- Example 32a N-((S or R, E)-1-cyclopropyl-3-((R or S)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide
- Example 32b N-((S or R, E)-1-cyclopropyl-3-((S or R)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide
- Example 32c N-((R or S, E)-1-cyclopropyl-3-((R or S)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide
- Example 32d N-((R or S, E)-1-cyclopropyl-3-((R or S)
- Example 32b N-((S or R,E)-1-cyclopropyl-3-((S or R)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (51 mg, 0.115 mmol, 99.07% purity,) as colorless gum.
- a typical reaction consists of 30 ⁇ L of 1 nM WRN in 25 mM Tris/HCl pH 7.5, 100 mM NaCl, 1 mM DTT, 0.01 % TWEEN-20, 0.025 mg/mL BSA, 0.1 mM forked DNA substrate, 1 mM Oligo-BLOCK and 0.5 mM Mg-ATP in a 384 well black plate (Thermo Scientific).
- IC50s were determined by pre-incubating compounds (10 mM DMSO stock solutions) with WRN in a 12-point custom dilution series to a final DMSO concentration of 1 %.
- HCT116 and SW48 both microsatellite instability- high and WRN- inhibition sensitive cell lines
- SW620 microsatellite stable and WRN-inhibition insensitive cell line
- HCT116 were cultured in growth medium composed of McCoy's 5A (Modified) Medium (ThermoFisher Scientific Cat# 16600082), 1x Penicillin-Streptomycin (ThermoFisher Scientific Cat# 15140163), and 10% fetal bovine serum (ThermoFisher Scientific Cat#A5256801).
- SW48 and SW620 were cultured in growth medium composed of Leibovitz's L-15 Medium (ThermoFisher Scientific Cat#11415056), 1x Penicillin-Streptomycin and 10% fetal bovine serum.
- HCT116, SW48 and SW620 were plated at 1500 cells/well, 2000 cells/well and 2000 cells/well, respectively, in 96-well black plates with clear flat bottom (Huberlab #.655983), in a volume of 200 ⁇ L per well.
- the outer wells of the plate were filled with DPBS (ThermoFisher Scientific Cat#14190250) to compensate evaporation mediated effects in the plate periphery.
- the compounds were dispensed with the Tecan digital dispenser (D300e), starting at 20 ⁇ M for a 8-point dose curve at 1:4 dilution, in duplicates.
- the final concentration of DMSO was normalized to the highest compound concentration and a maximum of 0.1%.
- 145 ⁇ l of the growth medium were removed and 50 ⁇ L of Cell Titer-Glo (Promega Cat#G9243) were added per well. Plates were shaken for 2 minutes and after an incubation of 10 minutes, luminescence was read using a plate reader (Tecan Spark). For data analysis, the assay background signal which was determined in wells that contained medium only was subtracted from all the data points.
- Sample Processing and Extraction Procedure At each corresponding time point, all samples were extracted with a protein precipitation method by adding 500 ⁇ L of stop solution.
- the kinetic solubility was measured according to the following protocol: 1. Samples were weighed and dissolved in 100% DMSO to make a stock solution of 10 mM. About 100 L of stock solution is needed to cover this assay. 2. Test compounds and controls (10 mM in DMSO, 10 L/tube) were added into the buffer (490 L/well) which were placed in a Mini-UniPrep filter. The buffer was prepared as the customer’s requirement. 3. The kinetic solubility samples were vortexed for 2 minutes. 4. The solubility solutions were shaken in an orbital shaker for 24 hr at room temperature .
- Caco-2 cells purchased from ATCC were seeded onto polyethylene membranes (PET) in 96- well BD insert plates at 1 x 105 cells/cm 2 , and refreshed medium every 4 ⁇ 5 days until to the 21 st to 28 th day for confluent cell monolayer formation.
- the integrity of the monolayer was verified by performing Lucifer yellow rejection assay.
- the quality of the monolayer was verified by measuring the unidirectional (A B) permeability of fenoterol/nadolol (low permeability marker), propranolol/metopronolol (high permeability marker) and bi-directional permeability of digoxin (a P-glycoprotein substrate marker) in duplicate wells. 4.
- Dosing solution were spiked and mixed with transport buffer and stop solution (containing an appropriate internal standard (IS)) as T0 sample. 6.
- sample solutions from both donor and receiver wells were mixed with stop solution immediately. 7. All samples including T0 samples, donor samples and receiver samples were analyzed using LC/MS/MS.
- Liver Microsomes Stability Assay 1. Materials 1.1 Liver microsomes Animal or human liver microsomes were purchased from Xenotech or Corning and stored in a freezer (lower than -60°C) before use. 1.2 -nicotinamide adenine dinucleotide phosphate reduced form, tetrasodium salt, Vendor: Chem-Impex International, Cat.No.00616 1.3 Control compounds: Testosterone, diclofenac and propafenone. 2. Preparation of Working Solution Stock Solution: 10 mM test compound in DMSO.
- Microsome working solutions (0.56 mg/mL) were transferred (445 uL) into pre-warmed 'Incubation' T60 and NCF60 plates, followed by incubation for 10 min at 37°C with constant shaking. Liver microsomes (54 ⁇ L) were transferred to a Blank60 plate, followed by the addition of 6 ⁇ L NAPDH cofactor and 180 ⁇ L stop solution (acetonitrile containing internal standards) into each well. An aliquot (5 ⁇ L) of compound working solution (100 M) was added into the 'incubation' plates (T60 and NCF60) containing microsomes and mixed 3 times thoroughly. For the 'Incubation' NCF60 plate, 50 uL of buffer was added and mixed 3 times thoroughly.
- the plates were incubated at 37°C for 60 min while shaking. samples were mixed once and 60 ⁇ L was transferred from the NCF60 incubation plate to the stop plate containing stop solution after the 60- min incubation. Stop solution (180 ⁇ L) and NAPDH cofactor (6 ⁇ L) were added to 'Quenching' plate T0. Plates were chilled to prevent evaporation. For the 'Incubation' T60 plate: mixed 3 times thoroughly, and immediately removed 54 ⁇ L mixture for the 0-min time point to stop plate ('Quenching' plate T0). NAPDH cofactor (44 ⁇ L) was added to the 'Incubation' T60 plate. The plate was incubated at 37°C for 60 min while shaking.
- test compound and positive controls were added into plate except for the blank plate. Incubate all plates at 37.0°C in a 95.0% humidified incubator at 5.0% CO2 to start the reactions with constant shaking.
- a corresponding quenching plate was prepared by adding 125 ⁇ L/well of acetonitrile containing 200 ng/mL tolbutamide and 200 ng/mL labetalol as internal standards (stop solution), and 25 ⁇ L/well of the incubation sample were transferred to this plate after shaking for 1 minute to ensure homogeneity.
- the corresponding plate was removed from the incubator, and 25 ⁇ L/well of the corresponding sample was transferred to its corresponding quenching plate containing 125 ⁇ L/well of stop solution.
- Medium control (MC) plates (T0-MC and T90-MC) were prepared by adding everything except for Williams’ Medium E at the corresponding time-points. The plates were then sealed and shaken for 10 minutes prior to centrifugation at 4000 rpm and 4°C for 20 minutes. 80 ⁇ L/well of the resulting supernatant were diluted with 240 ⁇ L/well of pure water and sealed and shaken for 10 minutes prior to LC-MS/MS analysis. 4.
- Matrix Sampling time point (hr) 0 1 1 M01, M02, Plasm 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M 03 a post dose 0 2 10 M04, M05, Pl 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M 06 asma post dose 0 3 1 M07, M08, 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M 09 Plasma post dose 0 4 10 M10, M11, 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M 12 Plasma post dose ANIMAL SPECIFICATIONS Strain: BALB/c nude Species: Mouse Vender: LC or other qualified vendors Gender: Female Age: 6-9 W Total Number of Animals: 6 Overnight Fast of Animals: True Comments: Fast for PO, fed for IV Food Returned: 4 hours post dose.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The present invention relates to a compound of formula (I) or a pharmaceutically acceptable salt thereof. The present invention further relates to the compound of formula (I) of the present invention for use in therapy. Instant compounds are particularly useful as WRN inhibitors, and can be used in a method of treatment of cancer, in particular, the cancer is treatable by inhibition of WRN, and/or the cancer characterized by MSI-H and/or dMMR
Description
New PCT patent application based on US 63/587,354 and US 63/665,751 FoRx Therapeutics AG Vossius Ref.: AE3393 PCT BS WRN inhibitory compounds Field of the invention The present invention relates to a compound of formula (I):
 or a pharmaceutically acceptable salt thereof. The present invention further relates to the compound of formula (I) of the present invention for use in therapy. Instant compounds are particularly useful as WRN inhibitors, and can be used in a method of treatment of cancer, in particular, the cancer is treatable by inhibition of WRN, and/or the cancer characterized by MSI-H and/or dMMR. Background of the invention Cancer is a leading cause of death worldwide. A limitation of prevailing therapeutic approaches, e.g. chemotherapy is that their cytotoxic effects are not restricted to cancer cells and adverse side effects can occur within normal tissues. Consequently, novel strategies are needed to better target cancer cells. Synthetic lethality (SL) arises when a combination of genetic deficiencies (e.g. gene mutations, silencing or global genomic lesions) and/or molecular perturbations (e.g. gene expression knockout/knockdown, pharmacological inhibition/activation) corresponding to two or more genes impaired cell wellbeing, whereas presence of single deficiency/perturbation does not (Dobzhansky, T., Genetics 1946; 31, 269-290, Huang et al., Nature Reviews Drug Discovery 2020; volume 19, pages 23-38). Microsatellite instability is a genomic lesion caused by defects in mismatch repair machinery (dMMR). MSI status is present in colorectal cancer, endometrial cancer, gastric cancer and other cancer types. Mutation or silencing of MMR genes, including MLH1, MSH2, MSH6 and PMS2, abrogates cell’s ability to repair DNA mismatch mutations (Baudrin et al., Front. Oncol.2018). As a consequence, tumor with MSI-H status carries higher mutation burden, disrupted microsatellite repeat sequences and extended TA dinucleotide repeat sequences across the genome (van Wietmarschen N. et al., Nature 2020; 586, pages 292-298). MSI status can be assessed by molecular testing of certain microsatellites, next-generation sequencing of patient genome or by immunohistochemical evaluation of expression of certain MMR proteins. Tumors can be categorized into MSI high (MSI-H), MSI low (MSI-L) and MSS depending on the number of tested microsatellite showing instability. Based on a consensus NCI- Reference Panel (Bethesda, 1998), MSI can be assessed by molecular testing of five microsatellites -
including two mononucleotides (BAT25 and BAT26) and three dinucleotides (D2S123, D5S346, D17S250). Tumors are denoted as MSI-high (MSI-H) if two or more of the microsatellite markers show instability, MSI-low (MSI-L) if only one microsatellite marker shows instability, and MS-stable (MSS) if none of the five microsatellite markers show instability. In some instances, for example where molecular testing or immunohistochemical evaluation is not able to distinguish between MSI-L and general chromosomal instability, tumors can be classified as a MSS neoplasms. WRN (WRN RecQ helicase) has been identified as a synthetic lethality vulnerability to cancer cells with high microsatellite instability status (MSI-H). WRN contains an exonuclease domain and an ATP - dependent helicase domain. It is localized to the nucleus and unwinds double strand DNA, particularly secondary structures (fork DNA, holliday junction, G4-quadruaplex, DNA hairpin and cruciform etc.) during DNA replication, damage and repair processes. Its helicase activity has been shown to be indispensable to the survival of MSI cell lines as helicase-deficient WRN mutant is insufficient to rescue impaired cell viability from WRN knockout or knockdown. The absence of either the WRN protein or inhibition of its helicase activity prevents normal DNA damage and repair processes, leading to increased DNA double- strand breaks (DSB) and subsequent growth arrest and cell death. Covalent inhibitors represent a class of small molecules which form covalent bonds with their biological targets to inhibit activities of these targets in physiological or pathological conditions. In general, covalent inhibitors engage with nucleophilic residues (e.g. Cysteine, Serine, Threonine, Histidine, Arginine, Tyrosine) lining specific binding pockets on target proteins, in a nucleophilic addition or substitution reaction, with their reactive electrophilic warhead. To date, a variety of reactive warheads have been identified, including epoxide, aziridine, ester, ketone, a, -unsaturated carbonyl, nitrile, etc. Covalent inhibitors have been discovered as medicines for more than a century, starting with Aspirin being manufactured and marketed as painkillers and anti-inflammatory drug, although its mechanism of action was not revealed until 1970s to be an irreversible inhibitor of cyclooxygenase- 1 (COX- 1). Other notable covalent inhibitors used as medicine include antibiotics Penicillin, proton pump inhibitor Omeprazole and Lansoprazole, anticoagulant Clopidogrel. Document WO 2023/062575 discloses certain cyclic vinyl sulfone compounds as WRN inhibitors. Documents WO 2024/010782 and WO 2024/010784 disclose certain covalent WRN inhibitors. Further covalent inhibitors of WRN are disclosed in document WO 2024/028169. Document WO 2022/249060 discloses certain compounds as WRN reversible inhibitors. Similar compounds are disclosed in WO 2024/079623. Further reversible WRN inhibitors are disclosed in WO 2024/120378. Summary of the invention It was an objective technical problem of the present invention to provide compounds that are cell-
permeable inhibitors of WRN. The technical problem of the present invention is solved by the embodiments described herein and as characterized by the claims. In a first embodiment, the present invention relates to a compound of formula (I)
or a pharmaceutically acceptable salt thereof. The present invention further relates to the compound of formula (I) of the present invention for use in therapy. Instant compounds are particularly useful as WRN inhibitors, and can be used in a method of treatment of cancer, in particular, the cancer is treatable by inhibition of WRN, and/or the cancer characterized by MSI-H and/or dMMR. Background of the invention Cancer is a leading cause of death worldwide. A limitation of prevailing therapeutic approaches, e.g. chemotherapy is that their cytotoxic effects are not restricted to cancer cells and adverse side effects can occur within normal tissues. Consequently, novel strategies are needed to better target cancer cells. Synthetic lethality (SL) arises when a combination of genetic deficiencies (e.g. gene mutations, silencing or global genomic lesions) and/or molecular perturbations (e.g. gene expression knockout/knockdown, pharmacological inhibition/activation) corresponding to two or more genes impaired cell wellbeing, whereas presence of single deficiency/perturbation does not (Dobzhansky, T., Genetics 1946; 31, 269-290, Huang et al., Nature Reviews Drug Discovery 2020; volume 19, pages 23-38). Microsatellite instability is a genomic lesion caused by defects in mismatch repair machinery (dMMR). MSI status is present in colorectal cancer, endometrial cancer, gastric cancer and other cancer types. Mutation or silencing of MMR genes, including MLH1, MSH2, MSH6 and PMS2, abrogates cell’s ability to repair DNA mismatch mutations (Baudrin et al., Front. Oncol.2018). As a consequence, tumor with MSI-H status carries higher mutation burden, disrupted microsatellite repeat sequences and extended TA dinucleotide repeat sequences across the genome (van Wietmarschen N. et al., Nature 2020; 586, pages 292-298). MSI status can be assessed by molecular testing of certain microsatellites, next-generation sequencing of patient genome or by immunohistochemical evaluation of expression of certain MMR proteins. Tumors can be categorized into MSI high (MSI-H), MSI low (MSI-L) and MSS depending on the number of tested microsatellite showing instability. Based on a consensus NCI- Reference Panel (Bethesda, 1998), MSI can be assessed by molecular testing of five microsatellites -
including two mononucleotides (BAT25 and BAT26) and three dinucleotides (D2S123, D5S346, D17S250). Tumors are denoted as MSI-high (MSI-H) if two or more of the microsatellite markers show instability, MSI-low (MSI-L) if only one microsatellite marker shows instability, and MS-stable (MSS) if none of the five microsatellite markers show instability. In some instances, for example where molecular testing or immunohistochemical evaluation is not able to distinguish between MSI-L and general chromosomal instability, tumors can be classified as a MSS neoplasms. WRN (WRN RecQ helicase) has been identified as a synthetic lethality vulnerability to cancer cells with high microsatellite instability status (MSI-H). WRN contains an exonuclease domain and an ATP - dependent helicase domain. It is localized to the nucleus and unwinds double strand DNA, particularly secondary structures (fork DNA, holliday junction, G4-quadruaplex, DNA hairpin and cruciform etc.) during DNA replication, damage and repair processes. Its helicase activity has been shown to be indispensable to the survival of MSI cell lines as helicase-deficient WRN mutant is insufficient to rescue impaired cell viability from WRN knockout or knockdown. The absence of either the WRN protein or inhibition of its helicase activity prevents normal DNA damage and repair processes, leading to increased DNA double- strand breaks (DSB) and subsequent growth arrest and cell death. Covalent inhibitors represent a class of small molecules which form covalent bonds with their biological targets to inhibit activities of these targets in physiological or pathological conditions. In general, covalent inhibitors engage with nucleophilic residues (e.g. Cysteine, Serine, Threonine, Histidine, Arginine, Tyrosine) lining specific binding pockets on target proteins, in a nucleophilic addition or substitution reaction, with their reactive electrophilic warhead. To date, a variety of reactive warheads have been identified, including epoxide, aziridine, ester, ketone, a, -unsaturated carbonyl, nitrile, etc. Covalent inhibitors have been discovered as medicines for more than a century, starting with Aspirin being manufactured and marketed as painkillers and anti-inflammatory drug, although its mechanism of action was not revealed until 1970s to be an irreversible inhibitor of cyclooxygenase- 1 (COX- 1). Other notable covalent inhibitors used as medicine include antibiotics Penicillin, proton pump inhibitor Omeprazole and Lansoprazole, anticoagulant Clopidogrel. Document WO 2023/062575 discloses certain cyclic vinyl sulfone compounds as WRN inhibitors. Documents WO 2024/010782 and WO 2024/010784 disclose certain covalent WRN inhibitors. Further covalent inhibitors of WRN are disclosed in document WO 2024/028169. Document WO 2022/249060 discloses certain compounds as WRN reversible inhibitors. Similar compounds are disclosed in WO 2024/079623. Further reversible WRN inhibitors are disclosed in WO 2024/120378. Summary of the invention It was an objective technical problem of the present invention to provide compounds that are cell-
permeable inhibitors of WRN. The technical problem of the present invention is solved by the embodiments described herein and as characterized by the claims. In a first embodiment, the present invention relates to a compound of formula (I)
 or a pharmaceutically acceptable salt thereof. In a second embodiment, the present invention relates to a pharmaceutical composition comprising a compound of formula (I). In a third embodiment, the present invention relates to a compound of formula (I) for use as a medicament. In a fourth embodiment, the present invention relates to a compound of formula (I) for use in the treatment of cancer. It is preferred that the cancer is characterized by high microsatellite instability (MSI) and/or by defective DNA mismatch repair system (dMMR) in a patient. In a fifth embodiment, the present invention relates to use of a compound of formula (I) in a manufacture of a medicament. In a sixth embodiment, the present invention relates to use of a compound of formula (I) in a manufacture of a medicament for the treatment of cancer. It is preferred that the cancer is characterized by high microsatellite instability (MSI) and/or by defective DNA mismatch repair system (dMMR) in a patient In a seventh embodiment, the present invention relates to a method of treatment of cancer in a subject in need thereof, the method comprising the step of administering the compound of formula (I) to said subject. Typically, a therapeutically effective amount of the compound of formula (I) is administered. It is preferred that the cancer is characterized by high microsatellite instability (MSI) and/or by defective DNA mismatch repair system (dMMR) in a patient Definitions The following definitions apply throughout the present specification and the claims, unless specifically indicated otherwise. The term “hydrogen” is herein used to refer to protium, deuterium and/or tritium, preferably to protium. Accordingly, the term “non-hydrogen atom” refers to any atoms that is not hydrogen, i.e. that is not protium, deuterium or tritium. The term “hydrocarbon group” refers to a group consisting of carbon atoms and hydrogen atoms. The term “alicyclic” is used in connection with cyclic groups and denotes that the corresponding cyclic group is non-aromatic.
As used herein, the term “alkyl” refers to a monovalent saturated acyclic (i.e., non-cyclic) hydrocarbon group which may be linear or branched. Accordingly, an “alkyl” group does not comprise any carbon-to-carbon double bond or any carbon-to-carbon triple bond. A “C1-5 alkyl” denotes an alkyl group having 1 to 5 carbon atoms. Preferred exemplary alkyl groups are methyl, ethyl, propyl (e.g., n-propyl or isopropyl), or butyl (e.g., n-butyl, isobutyl, sec-butyl, or tert-butyl). Unless defined otherwise, the term “alkyl” preferably refers to C1-4 alkyl, more preferably to methyl or ethyl, and even more preferably to methyl. As used herein, the term “alkenyl” refers to a monovalent unsaturated acyclic hydrocarbon group which may be linear or branched and comprises one or more (e.g., one or two) carbon-to-carbon double bonds while it does not comprise any carbon-to-carbon triple bond. The term “C2-5 alkenyl” denotes an alkenyl group having 2 to 5 carbon atoms. Preferred exemplary alkenyl groups are ethenyl, propenyl (e.g., prop-1-en-1-yl, prop-1-en-2-yl, or prop-2-en-1-yl), butenyl, butadienyl (e.g., buta-1,3-dien-1-yl or buta-1,3- dien-2-yl), pentenyl, or pentadienyl (e.g., isoprenyl). Unless defined otherwise, the term “alkenyl” preferably refers to C2-4 alkenyl. As used herein, the term “alkynyl” refers to a monovalent unsaturated acyclic hydrocarbon group which may be linear or branched and comprises one or more (e.g., one or two) carbon-to-carbon triple bonds and optionally one or more (e.g., one or two) carbon-to-carbon double bonds. The term “C2-5 alkynyl” denotes an alkynyl group having 2 to 5 carbon atoms. Preferred exemplary alkynyl groups are ethynyl, propynyl (e.g., propargyl), or butynyl. Unless defined otherwise, the term “alkynyl” preferably refers to C2-4 alkynyl. As used herein, the term “alkylene” refers to an alkanediyl group, i.e. a divalent saturated acyclic hydrocarbon group which may be linear or branched. A “C1-5 alkylene” denotes an alkylene group having 1 to 5 carbon atoms, and the term “C0-3 alkylene” indicates that a covalent bond (corresponding to the option “C0 alkylene”) or a C1-3 alkylene is present. Preferred exemplary alkylene groups are methylene (- CH2-), ethylene (e.g., -CH2-CH2- or -CH(-CH3)-), propylene (e.g., -CH2-CH2-CH2-, -CH(-CH2-CH3)-, -CH2- CH(-CH3)-, or -CH(-CH3)-CH2-), or butylene (e.g., -CH2-CH2-CH2-CH2-). Unless defined otherwise, the term “alkylene” preferably refers to C1-4 alkylene (including, in particular, linear C1-4 alkylene), more preferably to methylene or ethylene, and even more preferably to methylene. As used herein, the term “alkenylene” refers to an alkenediyl group, i.e. a divalent unsaturated acyclic hydrocarbon group which may be linear or branched and comprises one or more (e.g., one or two) carbon-to-carbon double bonds while it does not comprise any carbon-to-carbon triple bond. A “C2- 5 alkenylene” denotes an alkenylene group having 2 to 5 carbon atoms. Unless defined otherwise, the term “alkenylene” preferably refers to C2-4 alkenylene (including, in particular, linear C2-4 alkenylene). As used herein, the term “alkynylene” refers to an alkynediyl group, i.e. a divalent unsaturated
acyclic hydrocarbon group which may be linear or branched and comprises one or more (e.g., one or two) carbon-to-carbon triple bonds and optionally one or more (e.g., one or two) carbon-to-carbon double bonds. A “C2-5 alkynylene” denotes an alkynylene group having 2 to 5 carbon atoms. Unless defined otherwise, the term “alkynylene” preferably refers to C2-4 alkynylene (including, in particular, linear C2-4 alkynylene). As used herein, the term “carbocyclyl” refers to a hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings), wherein said ring group may be saturated, partially unsaturated (i.e., unsaturated but not aromatic) or aromatic. Unless defined otherwise, “carbocyclyl” preferably refers to aryl, cycloalkyl or cycloalkenyl. As used herein, the term “heterocyclyl” refers to a ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings), wherein said ring group comprises one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group), and further wherein said ring group may be saturated, partially unsaturated (i.e., unsaturated but not aromatic) or aromatic. For example, each heteroatom-containing ring comprised in said ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. Unless defined otherwise, “heterocyclyl” preferably refers to heteroaryl, heterocycloalkyl or heterocycloalkenyl. As used herein, the term “aryl” refers to an aromatic hydrocarbon ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic). “Aryl” may, e.g., refer to phenyl, naphthyl, dialinyl (i.e., 1,2-dihydronaphthyl), tetralinyl (i.e., 1,2,3,4-tetrahydronaphthyl), indanyl, indenyl (e.g., 1H-indenyl), anthracenyl, phenanthrenyl, 9H- fluorenyl, or azulenyl. Unless defined otherwise, an “aryl” preferably has 6 to 14 ring atoms, more preferably 6 to 10 ring atoms, even more preferably refers to phenyl or naphthyl, and most preferably refers to phenyl. As used herein, the term “arylene” refers to an aryl group, as defined herein above, but having two points of attachment, i.e. a divalent aromatic hydrocarbon ring group, including monocyclic aromatic rings
as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic). “Arylene” may, e.g., refer to phenylene (e.g., phen-1,2-diyl, phen-1,3-diyl, or phen-1,4-diyl), naphthylene (e.g., naphthalen-1,2-diyl, naphthalen-1,3-diyl, naphthalen-1,4-diyl, naphthalen-1,5-diyl, naphthalen-1,6- diyl, naphthalen-1,7-diyl, naphthalen-2,3-diyl, naphthalen-2,5-diyl, naphthalen-2,6-diyl, naphthalen-2,7- diyl, or naphthalen-2,8-diyl), 1,2-dihydronaphthylene, 1,2,3,4-tetrahydronaphthylene, indanylene, indenylene, anthracenylene, phenanthrenylene, 9H-fluorenylene, or azulenylene. Unless defined otherwise, an “arylene” preferably has 6 to 14 ring atoms, more preferably 6 to 10 ring atoms, even more preferably refers to phenylene or naphthylene, and most preferably refers to phenylene (particularly phen- 1,4-diyl). As used herein, the term “heteroaryl” refers to an aromatic ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic), wherein said aromatic ring group comprises one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group). For example, each heteroatom-containing ring comprised in said aromatic ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heteroaryl” may, e.g., refer to thienyl (i.e., thiophenyl), benzo[b]thienyl, naphtho[2,3-b]thienyl, thianthrenyl, furyl (i.e., furanyl), benzofuranyl, isobenzofuranyl, chromanyl, chromenyl (e.g., 2H-1- benzopyranyl or 4H-1-benzopyranyl), isochromenyl (e.g., 1H-2-benzopyranyl), chromonyl, xanthenyl, phenoxathiinyl, pyrrolyl (e.g., 1H-pyrrolyl), imidazolyl, pyrazolyl, pyridyl (i.e., pyridinyl; e.g., 2-pyridyl, 3- pyridyl, or 4-pyridyl), pyrazinyl, pyrimidinyl, pyridazinyl, indolyl (e.g., 3H-indolyl), isoindolyl, indazolyl, indolizinyl, purinyl, quinolyl, isoquinolyl, phthalazinyl, naphthyridinyl, quinoxalinyl, cinnolinyl, pteridinyl, carbazolyl, -carbolinyl, phenanthridinyl, acridinyl, perimidinyl, phenanthrolinyl (e.g., [1,10]phenanthrolinyl, [1,7]phenanthrolinyl, or [4,7]phenanthrolinyl), phenazinyl, thiazolyl, isothiazolyl, phenothiazinyl, oxazolyl, isoxazolyl, oxadiazolyl (e.g., 1,2,4-oxadiazolyl, 1,2,5-oxadiazolyl (i.e., furazanyl), or 1,3,4-oxadiazolyl), thiadiazolyl (e.g., 1,2,4-thiadiazolyl, 1,2,5-thiadiazolyl, or 1,3,4-thiadiazolyl),
phenoxazinyl, pyrazolo[1,5-a]pyrimidinyl (e.g., pyrazolo[1,5-a]pyrimidin-3-yl), 1,2-benzoisoxazol-3-yl, benzothiazolyl, benzothiadiazolyl, benzoxazolyl, benzisoxazolyl, benzimidazolyl, benzo[b]thiophenyl (i.e., benzothienyl), triazolyl (e.g., 1H-1,2,3-triazolyl, 2H-1,2,3-triazolyl, 1H-1,2,4-triazolyl, or 4H-1,2,4-triazolyl), benzotriazolyl, 1H-tetrazolyl, 2H-tetrazolyl, triazinyl (e.g., 1,2,3-triazinyl, 1,2,4-triazinyl, or 1,3,5-triazinyl), furo[2,3-c]pyridinyl, dihydrofuropyridinyl (e.g., 2,3-dihydrofuro[2,3-c]pyridinyl or 1,3-dihydrofuro[3,4- c]pyridinyl), imidazopyridinyl (e.g., imidazo[1,2-a]pyridinyl or imidazo[3,2-a]pyridinyl), quinazolinyl, thienopyridinyl, tetrahydrothienopyridinyl (e.g., 4,5,6,7-tetrahydrothieno[3,2-c]pyridinyl), dibenzofuranyl, 1,3-benzodioxolyl, benzodioxanyl (e.g., 1,3-benzodioxanyl or 1,4-benzodioxanyl), or coumarinyl. Unless defined otherwise, the term “heteroaryl” preferably refers to a 5 to 14 membered (more preferably 5 to 10 membered) monocyclic ring or fused ring system comprising one or more (e.g., one, two, three or four) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized; even more preferably, a “heteroaryl” refers to a 5 or 6 membered monocyclic ring comprising one or more (e.g., one, two or three) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized. As used herein, the term “heteroarylene” refers to a heteroaryl group, as defined herein above, but having two points of attachment, i.e. a divalent aromatic ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic), wherein said aromatic ring group comprises one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group). For example, each heteroatom-containing ring comprised in said aromatic ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three, or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heteroarylene” may, e.g., refer to thienylene (i.e., thiophenylene; e.g., thien-2,3-diyl, thien-2,4-diyl, or thien-2,5-diyl), benzo[b]thienylene, naphtho[2,3-b]thienylene, thianthrenylene, furylene (i.e., furanylene; e.g., furan-2,3-diyl, furan-2,4-diyl, or furan-2,5-diyl), benzofuranylene, isobenzofuranylene, chromanylene, chromenylene, isochromenylene, chromonylene, xanthenylene, phenoxathiinylene, pyrrolylene,
imidazolylene, pyrazolylene, pyridylene (i.e., pyridinylene), pyrazinylene, pyrimidinylene, pyridazinylene, indolylene, isoindolylene, indazolylene, indolizinylene, purinylene, quinolylene, isoquinolylene, phthalazinylene, naphthyridinylene, quinoxalinylene, cinnolinylene, pteridinylene, carbazolylene, -carbolinylene, phenanthridinylene, acridinylene, perimidinylene, phenanthrolinylene, phenazinylene, thiazolylene (e.g., thiazol-2,4-diyl, thiazol-2,5-diyl, or thiazol-4,5-diyl), isothiazolylene (e.g., isothiazol-3,4- diyl, isothiazol-3,5-diyl, or isothiazol-4,5-diyl), phenothiazinylene, oxazolylene (e.g., oxazol-2,4-diyl, oxazol-2,5-diyl, or oxazol-4,5-diyl), isoxazolylene (e.g., isoxazol-3,4-diyl, isoxazol-3,5-diyl, or isoxazol-4,5- diyl), oxadiazolylene (e.g., 1,2,4-oxadiazol-3,5-diyl, 1,2,5-oxadiazol-3,4-diyl, or 1,3,4-oxadiazol-2,5-diyl), thiadiazolylene (e.g., 1,2,4-thiadiazol-3,5-diyl, 1,2,5-thiadiazol-3,4-diyl, or 1,3,4-thiadiazol-2,5-diyl), phenoxazinylene, pyrazolo[1,5-a]pyrimidinylene, 1,2-benzoisoxazolylene, benzothiazolylene, benzothiadiazolylene, benzoxazolylene, benzisoxazolylene, benzimidazolylene, benzo[b]thiophenylene (i.e., benzothienylene), triazolylene (e.g., 1H-1,2,3-triazolylene, 2H-1,2,3-triazolylene, 1H-1,2,4- triazolylene, or 4H-1,2,4-triazolylene), benzotriazolylene, 1H-tetrazolylene, 2H-tetrazolylene, triazinylene (e.g., 1,2,3-triazinylene, 1,2,4-triazinylene, or 1,3,5-triazinylene), furo[2,3-c]pyridinylene, dihydrofuropyridinylene (e.g., 2,3-dihydrofuro[2,3-c]pyridinylene or 1,3-dihydrofuro[3,4-c]pyridinylene), imidazopyridinylene (e.g., imidazo[1,2-a]pyridinylene or imidazo[3,2-a]pyridinylene), quinazolinylene, thienopyridinylene, tetrahydrothienopyridinylene (e.g., 4,5,6,7-tetrahydrothieno[3,2-c]pyridinylene), dibenzofuranylene, 1,3-benzodioxolylene, benzodioxanylene (e.g., 1,3-benzodioxanylene or 1,4-benzodioxanylene), or coumarinylene. Unless defined otherwise, the term “heteroarylene” preferably refers to a divalent 5 to 14 membered (more preferably 5 to 10 membered) monocyclic ring or fused ring system comprising one or more (e.g., one, two, three or four) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized; even more preferably, a “heteroarylene” refers to a divalent 5 or 6 membered monocyclic ring comprising one or more (e.g., one, two or three) ring heteroatoms independently selected from O, S, and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized. A “heteroarylene”, including any of the specific heteroarylene groups described herein, may be attached through two carbon ring atoms, particularly through those two carbon ring atoms that have the greatest distance from one another (in terms of the number of ring atoms separating them by the shortest possible connection) within one single ring or within the entire ring system of the corresponding heteroarylene. As used herein, the term “cycloalkyl” refers to a saturated hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings).
“Cycloalkyl” may, e.g., refer to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, decalinyl (i.e., decahydronaphthyl), or adamantyl. Unless defined otherwise, “cycloalkyl” preferably refers to a C3-11 cycloalkyl, and more preferably refers to a C3-7 cycloalkyl. A particularly preferred “cycloalkyl” is a monocyclic saturated hydrocarbon ring having 3 to 7 ring members (e.g., cyclopropyl or cyclohexyl). As used herein, the term “cycloalkylene” refers to a cycloalkyl group, as defined herein above, but having two points of attachment, i.e. a divalent saturated hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings). “Cycloalkylene” may, e.g., refer to cyclopropylene (e.g., cyclopropan-1,1-diyl or cyclopropan-1,2-diyl), cyclobutylene (e.g., cyclobutan-1,1-diyl, cyclobutan-1,2-diyl, or cyclobutan-1,3-diyl), cyclopentylene (e.g., cyclopentan-1,1-diyl, cyclopentan-1,2-diyl, or cyclopentan-1,3-diyl), cyclohexylene (e.g., cyclohexan-1,1-diyl, cyclohexan-1,2-diyl, cyclohexan-1,3-diyl, or cyclohexan-1,4-diyl), cycloheptylene, decalinylene (i.e., decahydronaphthylene), or adamantylene. Unless defined otherwise, “cycloalkylene” preferably refers to a C3-11 cycloalkylene, and more preferably refers to a C3-7 cycloalkylene. A particularly preferred “cycloalkylene” is a divalent monocyclic saturated hydrocarbon ring having 3 to 7 ring members (e.g., cyclopropylene or cyclohexylene). As used herein, the term “heterocycloalkyl” refers to a saturated ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said ring group contains one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group). For example, each heteroatom-containing ring comprised in said saturated ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heterocycloalkyl” may, e.g., refer to aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, azepanyl, diazepanyl (e.g., 1,4-diazepanyl), oxazolidinyl, isoxazolidinyl, thiazolidinyl, isothiazolidinyl, morpholinyl (e.g., morpholin-4-yl), thiomorpholinyl (e.g., thiomorpholin-4-yl), oxazepanyl, oxiranyl, oxetanyl, tetrahydrofuranyl, 1,3-dioxolanyl, tetrahydropyranyl, 1,4-dioxanyl, oxepanyl, thiiranyl, thietanyl, tetrahydrothiophenyl (i.e., thiolanyl), 1,3-dithiolanyl, thianyl, 1,1-dioxothianyl, thiepanyl, decahydroquinolinyl, decahydroisoquinolinyl, or 2-oxa-5-aza-bicyclo[2.2.1]hept-5-yl. Unless defined
otherwise, “heterocycloalkyl” preferably refers to a 3 to 11 membered saturated ring group, which is a monocyclic ring or a fused ring system (e.g., a fused ring system composed of two fused rings), wherein said ring group contains one or more (e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized; more preferably, “heterocycloalkyl” refers to a 5 to 7 membered saturated monocyclic ring group containing one or more (e.g., one, two, or three) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized. As used herein, the term “heterocycloalkylene” refers to a heterocycloalkyl group, as defined herein above, but having two points of attachment, i.e. a divalent saturated ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said ring group contains one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group). For example, each heteroatom-containing ring comprised in said saturated ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heterocycloalkylene” may, e.g., refer to aziridinylene, azetidinylene, pyrrolidinylene, imidazolidinylene, pyrazolidinylene, piperidinylene, piperazinylene, azepanylene, diazepanylene (e.g., 1,4-diazepanylene), oxazolidinylene, isoxazolidinylene, thiazolidinylene, isothiazolidinylene, morpholinylene, thiomorpholinylene, oxazepanylene, oxiranylene, oxetanylene, tetrahydrofuranylene, 1,3-dioxolanylene, tetrahydropyranylene, 1,4-dioxanylene, oxepanylene, thiiranylene, thietanylene, tetrahydrothiophenylene (i.e., thiolanylene), 1,3-dithiolanylene, thianylene, 1,1-dioxothianylene, thiepanylene, decahydroquinolinylene, decahydroisoquinolinylene, or 2-oxa-5-aza-bicyclo[2.2.1]hept-5-ylene. Unless defined otherwise, “heterocycloalkylene” preferably refers to a divalent 3 to 11 membered saturated ring group, which is a monocyclic ring or a fused ring system (e.g., a fused ring system composed of two fused rings), wherein said ring group contains one or more (e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are
optionally oxidized; more preferably, “heterocycloalkylene” refers to a divalent 5 to 7 membered saturated monocyclic ring group containing one or more (e.g., one, two, or three) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized. As used herein, the term “N-heterocycloalkyl” refers to the heterocycloalkyl groups as defined hereinabove wherein said heterocycloalkyl includes at least one nitrogen atom which serves as an attachment point of said heterocycloalkyl. As used herein, the term “cycloalkenyl” refers to an unsaturated alicyclic (non-aromatic) hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said hydrocarbon ring group comprises one or more (e.g., one or two) carbon-to-carbon double bonds and does not comprise any carbon-to-carbon triple bond. “Cycloalkenyl” may, e.g., refer to cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cyclohexadienyl, cycloheptenyl, or cycloheptadienyl. Unless defined otherwise, “cycloalkenyl” preferably refers to a C3-11 cycloalkenyl, and more preferably refers to a C3-7 cycloalkenyl. A particularly preferred “cycloalkenyl” is a monocyclic unsaturated alicyclic hydrocarbon ring having 3 to 7 ring members and containing one or more (e.g., one or two; preferably one) carbon-to-carbon double bonds. As used herein, the term “cycloalkenylene” refers to a cycloalkenyl group, as defined hereinabove, but having two points of attachment, i.e. a divalent unsaturated alicyclic (non-aromatic) hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said hydrocarbon ring group comprises one or more (e.g., one or two) carbon-to- carbon double bonds and does not comprise any carbon-to-carbon triple bond. As used herein, the term “heterocycloalkenyl” refers to an unsaturated alicyclic (non-aromatic) ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said ring group contains one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group), and further wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms. For example, each heteroatom-containing ring comprised in said unsaturated alicyclic ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N
atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heterocycloalkenyl” may, e.g., refer to imidazolinyl (e.g., 2-imidazolinyl (i.e., 4,5-dihydro-1H-imidazolyl), 3-imidazolinyl, or 4-imidazolinyl), tetrahydropyridinyl (e.g., 1,2,3,6-tetrahydropyridinyl), dihydropyridinyl (e.g., 1,2- dihydropyridinyl or 2,3-dihydropyridinyl), pyranyl (e.g., 2H-pyranyl or 4H-pyranyl), thiopyranyl (e.g., 2H-thiopyranyl or 4H-thiopyranyl), dihydropyranyl, dihydrofuranyl, dihydropyrazolyl, dihydropyrazinyl, dihydroisoindolyl, octahydroquinolinyl (e.g., 1,2,3,4,4a,5,6,7-octahydroquinolinyl), or octahydroisoquinolinyl (e.g., 1,2,3,4,5,6,7,8-octahydroisoquinolinyl). Unless defined otherwise, “heterocycloalkenyl” preferably refers to a 3 to 11 membered unsaturated alicyclic ring group, which is a monocyclic ring or a fused ring system (e.g., a fused ring system composed of two fused rings), wherein said ring group contains one or more (e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, wherein one or more carbon ring atoms are optionally oxidized, and wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms; more preferably, “heterocycloalkenyl” refers to a 5 to 7 membered monocyclic unsaturated non-aromatic ring group containing one or more (e.g., one, two, or three) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, wherein one or more carbon ring atoms are optionally oxidized, and wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms. As used herein, the term “heterocycloalkenylene” refers to a heterocycloalkenyl group, as defined hereinabove, as defined hereinabove, but having two points of attachment, i.e. a divalent unsaturated alicyclic (non-aromatic) ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said ring group contains one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group), and further wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms. For example, each heteroatom-containing ring comprised in said unsaturated alicyclic ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the
corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. As used herein, the term “halogen” (or Hal) refers to fluoro (-F), chloro (-Cl), bromo (-Br), or iodo (-I). Preferred halogen is fluoro. As used herein, the term “haloalkyl” refers to an alkyl group substituted with one or more (preferably 1 to 6, more preferably 1 to 3) halogen atoms which are selected independently from fluoro, chloro, bromo and iodo, and are preferably all fluoro atoms. It will be understood that the maximum number of halogen atoms is limited by the number of available attachment sites and, thus, depends on the number of carbon atoms comprised in the alkyl moiety of the haloalkyl group. “Haloalkyl” may, e.g., refer to -CF3, -CHF2, -CH2F, -CF2-CH3, -CH2-CF3, -CH2-CHF2, -CH2-CF2-CH3, -CH2-CF2-CF3, or -CH(CF3)2. A particularly preferred “haloalkyl” group is -CF3. In one embodiment, haloalkyl may also be a perhaloalkyl. The term “perhaloalkyl” refers to a haloalkyl wherein every -H atom has been substituted with a halo atom. Preferably, said perhaloalkyl is perfluoroalkyl. The terms “bond” and “covalent bond” are used herein synonymously, unless explicitly indicated otherwise or contradicted by context. As used herein, the terms “optional”, “optionally” and “may” denote that the indicated feature may be present but can also be absent. Whenever the term “optional”, “optionally” or “may” is used, the present invention specifically relates to both possibilities, i.e., that the corresponding feature is present or, alternatively, that the corresponding feature is absent. For example, the expression “X is optionally substituted with Y” (or “X may be substituted with Y”) means that X is either substituted with Y or is unsubstituted. Likewise, if a component of a composition is indicated to be “optional”, the invention specifically relates to both possibilities, i.e., that the corresponding component is present (contained in the composition) or that the corresponding component is absent from the composition. Various groups are referred to as being “optionally substituted” in this specification. Generally, these groups may carry one or more substituents, such as, e.g., one, two, three or four substituents. It will be understood that the maximum number of substituents is limited by the number of attachment sites available on the substituted moiety. Unless defined otherwise, the “optionally substituted” groups referred to in this specification carry preferably not more than two substituents and may, in particular, carry only one substituent. Moreover, unless defined otherwise, it is preferred that the optional substituents are absent, i.e. that the corresponding groups are unsubstituted. A skilled person will appreciate that the substituent groups comprised in the compounds of the present invention may be attached to the remainder of the respective compound via a number of different positions of the corresponding specific substituent group. Unless defined otherwise, the preferred attachment positions for the various specific substituent groups are as illustrated in the examples.
As used herein, unless explicitly indicated otherwise or contradicted by context, the terms “a”, “an” and “the” are used interchangeably with “one or more” and “at least one”. Thus, for example, a composition comprising “a” compound of formula (I) can be interpreted as referring to a composition comprising “one or more” compounds of formula (I). It is to be understood that wherever numerical ranges are provided/disclosed herein, all values and subranges encompassed by the respective numerical range are meant to be encompassed within the scope of the invention. Accordingly, the present invention specifically and individually relates to each value that falls within a numerical range disclosed herein, as well as each subrange encompassed by a numerical range disclosed herein. As used herein, the term “about” preferably refers to ±10% of the indicated numerical value, more preferably to ±5% of the indicated numerical value, and in particular to the exact numerical value indicated. If the term “about” is used in connection with the endpoints of a range, it preferably refers to the range from the lower endpoint -10% of its indicated numerical value to the upper endpoint +10% of its indicated numerical value, more preferably to the range from of the lower endpoint -5% to the upper endpoint +5%, and even more preferably to the range defined by the exact numerical values of the lower endpoint and the upper endpoint. As used herein, the term “comprising” (or “comprise”, “comprises”, “contain”, “contains”, or “containing”), unless explicitly indicated otherwise or contradicted by context, has the meaning of “containing, inter alia”, i.e., “containing, among further optional elements, …”. In addition thereto, this term also includes the narrower meanings of “consisting essentially of” and “consisting of”. For example, the term “A comprising B and C” has the meaning of “A containing, inter alia, B and C”, wherein A may contain further optional elements (e.g., “A containing B, C and D” would also be encompassed), but this term also includes the meaning of “A consisting essentially of B and C” and the meaning of “A consisting of B and C” (i.e., no other components than B and C are comprised in A). Detailed description of the invention The invention is described in detail in the following. It is to be understood that the present invention specifically relates to each and every combination of features and embodiments described herein, including any combination of general and/or preferred features/embodiments. In a first embodiment, the present invention relates to a compound of formula (I):
or a pharmaceutically acceptable salt thereof. In a second embodiment, the present invention relates to a pharmaceutical composition comprising a compound of formula (I). In a third embodiment, the present invention relates to a compound of formula (I) for use as a medicament. In a fourth embodiment, the present invention relates to a compound of formula (I) for use in the treatment of cancer. It is preferred that the cancer is characterized by high microsatellite instability (MSI) and/or by defective DNA mismatch repair system (dMMR) in a patient. In a fifth embodiment, the present invention relates to use of a compound of formula (I) in a manufacture of a medicament. In a sixth embodiment, the present invention relates to use of a compound of formula (I) in a manufacture of a medicament for the treatment of cancer. It is preferred that the cancer is characterized by high microsatellite instability (MSI) and/or by defective DNA mismatch repair system (dMMR) in a patient In a seventh embodiment, the present invention relates to a method of treatment of cancer in a subject in need thereof, the method comprising the step of administering the compound of formula (I) to said subject. Typically, a therapeutically effective amount of the compound of formula (I) is administered. It is preferred that the cancer is characterized by high microsatellite instability (MSI) and/or by defective DNA mismatch repair system (dMMR) in a patient Definitions The following definitions apply throughout the present specification and the claims, unless specifically indicated otherwise. The term “hydrogen” is herein used to refer to protium, deuterium and/or tritium, preferably to protium. Accordingly, the term “non-hydrogen atom” refers to any atoms that is not hydrogen, i.e. that is not protium, deuterium or tritium. The term “hydrocarbon group” refers to a group consisting of carbon atoms and hydrogen atoms. The term “alicyclic” is used in connection with cyclic groups and denotes that the corresponding cyclic group is non-aromatic.
As used herein, the term “alkyl” refers to a monovalent saturated acyclic (i.e., non-cyclic) hydrocarbon group which may be linear or branched. Accordingly, an “alkyl” group does not comprise any carbon-to-carbon double bond or any carbon-to-carbon triple bond. A “C1-5 alkyl” denotes an alkyl group having 1 to 5 carbon atoms. Preferred exemplary alkyl groups are methyl, ethyl, propyl (e.g., n-propyl or isopropyl), or butyl (e.g., n-butyl, isobutyl, sec-butyl, or tert-butyl). Unless defined otherwise, the term “alkyl” preferably refers to C1-4 alkyl, more preferably to methyl or ethyl, and even more preferably to methyl. As used herein, the term “alkenyl” refers to a monovalent unsaturated acyclic hydrocarbon group which may be linear or branched and comprises one or more (e.g., one or two) carbon-to-carbon double bonds while it does not comprise any carbon-to-carbon triple bond. The term “C2-5 alkenyl” denotes an alkenyl group having 2 to 5 carbon atoms. Preferred exemplary alkenyl groups are ethenyl, propenyl (e.g., prop-1-en-1-yl, prop-1-en-2-yl, or prop-2-en-1-yl), butenyl, butadienyl (e.g., buta-1,3-dien-1-yl or buta-1,3- dien-2-yl), pentenyl, or pentadienyl (e.g., isoprenyl). Unless defined otherwise, the term “alkenyl” preferably refers to C2-4 alkenyl. As used herein, the term “alkynyl” refers to a monovalent unsaturated acyclic hydrocarbon group which may be linear or branched and comprises one or more (e.g., one or two) carbon-to-carbon triple bonds and optionally one or more (e.g., one or two) carbon-to-carbon double bonds. The term “C2-5 alkynyl” denotes an alkynyl group having 2 to 5 carbon atoms. Preferred exemplary alkynyl groups are ethynyl, propynyl (e.g., propargyl), or butynyl. Unless defined otherwise, the term “alkynyl” preferably refers to C2-4 alkynyl. As used herein, the term “alkylene” refers to an alkanediyl group, i.e. a divalent saturated acyclic hydrocarbon group which may be linear or branched. A “C1-5 alkylene” denotes an alkylene group having 1 to 5 carbon atoms, and the term “C0-3 alkylene” indicates that a covalent bond (corresponding to the option “C0 alkylene”) or a C1-3 alkylene is present. Preferred exemplary alkylene groups are methylene (- CH2-), ethylene (e.g., -CH2-CH2- or -CH(-CH3)-), propylene (e.g., -CH2-CH2-CH2-, -CH(-CH2-CH3)-, -CH2- CH(-CH3)-, or -CH(-CH3)-CH2-), or butylene (e.g., -CH2-CH2-CH2-CH2-). Unless defined otherwise, the term “alkylene” preferably refers to C1-4 alkylene (including, in particular, linear C1-4 alkylene), more preferably to methylene or ethylene, and even more preferably to methylene. As used herein, the term “alkenylene” refers to an alkenediyl group, i.e. a divalent unsaturated acyclic hydrocarbon group which may be linear or branched and comprises one or more (e.g., one or two) carbon-to-carbon double bonds while it does not comprise any carbon-to-carbon triple bond. A “C2- 5 alkenylene” denotes an alkenylene group having 2 to 5 carbon atoms. Unless defined otherwise, the term “alkenylene” preferably refers to C2-4 alkenylene (including, in particular, linear C2-4 alkenylene). As used herein, the term “alkynylene” refers to an alkynediyl group, i.e. a divalent unsaturated
acyclic hydrocarbon group which may be linear or branched and comprises one or more (e.g., one or two) carbon-to-carbon triple bonds and optionally one or more (e.g., one or two) carbon-to-carbon double bonds. A “C2-5 alkynylene” denotes an alkynylene group having 2 to 5 carbon atoms. Unless defined otherwise, the term “alkynylene” preferably refers to C2-4 alkynylene (including, in particular, linear C2-4 alkynylene). As used herein, the term “carbocyclyl” refers to a hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings), wherein said ring group may be saturated, partially unsaturated (i.e., unsaturated but not aromatic) or aromatic. Unless defined otherwise, “carbocyclyl” preferably refers to aryl, cycloalkyl or cycloalkenyl. As used herein, the term “heterocyclyl” refers to a ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings), wherein said ring group comprises one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group), and further wherein said ring group may be saturated, partially unsaturated (i.e., unsaturated but not aromatic) or aromatic. For example, each heteroatom-containing ring comprised in said ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. Unless defined otherwise, “heterocyclyl” preferably refers to heteroaryl, heterocycloalkyl or heterocycloalkenyl. As used herein, the term “aryl” refers to an aromatic hydrocarbon ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic). “Aryl” may, e.g., refer to phenyl, naphthyl, dialinyl (i.e., 1,2-dihydronaphthyl), tetralinyl (i.e., 1,2,3,4-tetrahydronaphthyl), indanyl, indenyl (e.g., 1H-indenyl), anthracenyl, phenanthrenyl, 9H- fluorenyl, or azulenyl. Unless defined otherwise, an “aryl” preferably has 6 to 14 ring atoms, more preferably 6 to 10 ring atoms, even more preferably refers to phenyl or naphthyl, and most preferably refers to phenyl. As used herein, the term “arylene” refers to an aryl group, as defined herein above, but having two points of attachment, i.e. a divalent aromatic hydrocarbon ring group, including monocyclic aromatic rings
as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic). “Arylene” may, e.g., refer to phenylene (e.g., phen-1,2-diyl, phen-1,3-diyl, or phen-1,4-diyl), naphthylene (e.g., naphthalen-1,2-diyl, naphthalen-1,3-diyl, naphthalen-1,4-diyl, naphthalen-1,5-diyl, naphthalen-1,6- diyl, naphthalen-1,7-diyl, naphthalen-2,3-diyl, naphthalen-2,5-diyl, naphthalen-2,6-diyl, naphthalen-2,7- diyl, or naphthalen-2,8-diyl), 1,2-dihydronaphthylene, 1,2,3,4-tetrahydronaphthylene, indanylene, indenylene, anthracenylene, phenanthrenylene, 9H-fluorenylene, or azulenylene. Unless defined otherwise, an “arylene” preferably has 6 to 14 ring atoms, more preferably 6 to 10 ring atoms, even more preferably refers to phenylene or naphthylene, and most preferably refers to phenylene (particularly phen- 1,4-diyl). As used herein, the term “heteroaryl” refers to an aromatic ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic), wherein said aromatic ring group comprises one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group). For example, each heteroatom-containing ring comprised in said aromatic ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heteroaryl” may, e.g., refer to thienyl (i.e., thiophenyl), benzo[b]thienyl, naphtho[2,3-b]thienyl, thianthrenyl, furyl (i.e., furanyl), benzofuranyl, isobenzofuranyl, chromanyl, chromenyl (e.g., 2H-1- benzopyranyl or 4H-1-benzopyranyl), isochromenyl (e.g., 1H-2-benzopyranyl), chromonyl, xanthenyl, phenoxathiinyl, pyrrolyl (e.g., 1H-pyrrolyl), imidazolyl, pyrazolyl, pyridyl (i.e., pyridinyl; e.g., 2-pyridyl, 3- pyridyl, or 4-pyridyl), pyrazinyl, pyrimidinyl, pyridazinyl, indolyl (e.g., 3H-indolyl), isoindolyl, indazolyl, indolizinyl, purinyl, quinolyl, isoquinolyl, phthalazinyl, naphthyridinyl, quinoxalinyl, cinnolinyl, pteridinyl, carbazolyl, -carbolinyl, phenanthridinyl, acridinyl, perimidinyl, phenanthrolinyl (e.g., [1,10]phenanthrolinyl, [1,7]phenanthrolinyl, or [4,7]phenanthrolinyl), phenazinyl, thiazolyl, isothiazolyl, phenothiazinyl, oxazolyl, isoxazolyl, oxadiazolyl (e.g., 1,2,4-oxadiazolyl, 1,2,5-oxadiazolyl (i.e., furazanyl), or 1,3,4-oxadiazolyl), thiadiazolyl (e.g., 1,2,4-thiadiazolyl, 1,2,5-thiadiazolyl, or 1,3,4-thiadiazolyl),
phenoxazinyl, pyrazolo[1,5-a]pyrimidinyl (e.g., pyrazolo[1,5-a]pyrimidin-3-yl), 1,2-benzoisoxazol-3-yl, benzothiazolyl, benzothiadiazolyl, benzoxazolyl, benzisoxazolyl, benzimidazolyl, benzo[b]thiophenyl (i.e., benzothienyl), triazolyl (e.g., 1H-1,2,3-triazolyl, 2H-1,2,3-triazolyl, 1H-1,2,4-triazolyl, or 4H-1,2,4-triazolyl), benzotriazolyl, 1H-tetrazolyl, 2H-tetrazolyl, triazinyl (e.g., 1,2,3-triazinyl, 1,2,4-triazinyl, or 1,3,5-triazinyl), furo[2,3-c]pyridinyl, dihydrofuropyridinyl (e.g., 2,3-dihydrofuro[2,3-c]pyridinyl or 1,3-dihydrofuro[3,4- c]pyridinyl), imidazopyridinyl (e.g., imidazo[1,2-a]pyridinyl or imidazo[3,2-a]pyridinyl), quinazolinyl, thienopyridinyl, tetrahydrothienopyridinyl (e.g., 4,5,6,7-tetrahydrothieno[3,2-c]pyridinyl), dibenzofuranyl, 1,3-benzodioxolyl, benzodioxanyl (e.g., 1,3-benzodioxanyl or 1,4-benzodioxanyl), or coumarinyl. Unless defined otherwise, the term “heteroaryl” preferably refers to a 5 to 14 membered (more preferably 5 to 10 membered) monocyclic ring or fused ring system comprising one or more (e.g., one, two, three or four) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized; even more preferably, a “heteroaryl” refers to a 5 or 6 membered monocyclic ring comprising one or more (e.g., one, two or three) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized. As used herein, the term “heteroarylene” refers to a heteroaryl group, as defined herein above, but having two points of attachment, i.e. a divalent aromatic ring group, including monocyclic aromatic rings as well as bridged ring and/or fused ring systems containing at least one aromatic ring (e.g., ring systems composed of two or three fused rings, wherein at least one of these fused rings is aromatic; or bridged ring systems composed of two or three rings, wherein at least one of these bridged rings is aromatic), wherein said aromatic ring group comprises one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group). For example, each heteroatom-containing ring comprised in said aromatic ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three, or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heteroarylene” may, e.g., refer to thienylene (i.e., thiophenylene; e.g., thien-2,3-diyl, thien-2,4-diyl, or thien-2,5-diyl), benzo[b]thienylene, naphtho[2,3-b]thienylene, thianthrenylene, furylene (i.e., furanylene; e.g., furan-2,3-diyl, furan-2,4-diyl, or furan-2,5-diyl), benzofuranylene, isobenzofuranylene, chromanylene, chromenylene, isochromenylene, chromonylene, xanthenylene, phenoxathiinylene, pyrrolylene,
imidazolylene, pyrazolylene, pyridylene (i.e., pyridinylene), pyrazinylene, pyrimidinylene, pyridazinylene, indolylene, isoindolylene, indazolylene, indolizinylene, purinylene, quinolylene, isoquinolylene, phthalazinylene, naphthyridinylene, quinoxalinylene, cinnolinylene, pteridinylene, carbazolylene, -carbolinylene, phenanthridinylene, acridinylene, perimidinylene, phenanthrolinylene, phenazinylene, thiazolylene (e.g., thiazol-2,4-diyl, thiazol-2,5-diyl, or thiazol-4,5-diyl), isothiazolylene (e.g., isothiazol-3,4- diyl, isothiazol-3,5-diyl, or isothiazol-4,5-diyl), phenothiazinylene, oxazolylene (e.g., oxazol-2,4-diyl, oxazol-2,5-diyl, or oxazol-4,5-diyl), isoxazolylene (e.g., isoxazol-3,4-diyl, isoxazol-3,5-diyl, or isoxazol-4,5- diyl), oxadiazolylene (e.g., 1,2,4-oxadiazol-3,5-diyl, 1,2,5-oxadiazol-3,4-diyl, or 1,3,4-oxadiazol-2,5-diyl), thiadiazolylene (e.g., 1,2,4-thiadiazol-3,5-diyl, 1,2,5-thiadiazol-3,4-diyl, or 1,3,4-thiadiazol-2,5-diyl), phenoxazinylene, pyrazolo[1,5-a]pyrimidinylene, 1,2-benzoisoxazolylene, benzothiazolylene, benzothiadiazolylene, benzoxazolylene, benzisoxazolylene, benzimidazolylene, benzo[b]thiophenylene (i.e., benzothienylene), triazolylene (e.g., 1H-1,2,3-triazolylene, 2H-1,2,3-triazolylene, 1H-1,2,4- triazolylene, or 4H-1,2,4-triazolylene), benzotriazolylene, 1H-tetrazolylene, 2H-tetrazolylene, triazinylene (e.g., 1,2,3-triazinylene, 1,2,4-triazinylene, or 1,3,5-triazinylene), furo[2,3-c]pyridinylene, dihydrofuropyridinylene (e.g., 2,3-dihydrofuro[2,3-c]pyridinylene or 1,3-dihydrofuro[3,4-c]pyridinylene), imidazopyridinylene (e.g., imidazo[1,2-a]pyridinylene or imidazo[3,2-a]pyridinylene), quinazolinylene, thienopyridinylene, tetrahydrothienopyridinylene (e.g., 4,5,6,7-tetrahydrothieno[3,2-c]pyridinylene), dibenzofuranylene, 1,3-benzodioxolylene, benzodioxanylene (e.g., 1,3-benzodioxanylene or 1,4-benzodioxanylene), or coumarinylene. Unless defined otherwise, the term “heteroarylene” preferably refers to a divalent 5 to 14 membered (more preferably 5 to 10 membered) monocyclic ring or fused ring system comprising one or more (e.g., one, two, three or four) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized; even more preferably, a “heteroarylene” refers to a divalent 5 or 6 membered monocyclic ring comprising one or more (e.g., one, two or three) ring heteroatoms independently selected from O, S, and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized. A “heteroarylene”, including any of the specific heteroarylene groups described herein, may be attached through two carbon ring atoms, particularly through those two carbon ring atoms that have the greatest distance from one another (in terms of the number of ring atoms separating them by the shortest possible connection) within one single ring or within the entire ring system of the corresponding heteroarylene. As used herein, the term “cycloalkyl” refers to a saturated hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings).
“Cycloalkyl” may, e.g., refer to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, decalinyl (i.e., decahydronaphthyl), or adamantyl. Unless defined otherwise, “cycloalkyl” preferably refers to a C3-11 cycloalkyl, and more preferably refers to a C3-7 cycloalkyl. A particularly preferred “cycloalkyl” is a monocyclic saturated hydrocarbon ring having 3 to 7 ring members (e.g., cyclopropyl or cyclohexyl). As used herein, the term “cycloalkylene” refers to a cycloalkyl group, as defined herein above, but having two points of attachment, i.e. a divalent saturated hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings). “Cycloalkylene” may, e.g., refer to cyclopropylene (e.g., cyclopropan-1,1-diyl or cyclopropan-1,2-diyl), cyclobutylene (e.g., cyclobutan-1,1-diyl, cyclobutan-1,2-diyl, or cyclobutan-1,3-diyl), cyclopentylene (e.g., cyclopentan-1,1-diyl, cyclopentan-1,2-diyl, or cyclopentan-1,3-diyl), cyclohexylene (e.g., cyclohexan-1,1-diyl, cyclohexan-1,2-diyl, cyclohexan-1,3-diyl, or cyclohexan-1,4-diyl), cycloheptylene, decalinylene (i.e., decahydronaphthylene), or adamantylene. Unless defined otherwise, “cycloalkylene” preferably refers to a C3-11 cycloalkylene, and more preferably refers to a C3-7 cycloalkylene. A particularly preferred “cycloalkylene” is a divalent monocyclic saturated hydrocarbon ring having 3 to 7 ring members (e.g., cyclopropylene or cyclohexylene). As used herein, the term “heterocycloalkyl” refers to a saturated ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said ring group contains one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group). For example, each heteroatom-containing ring comprised in said saturated ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heterocycloalkyl” may, e.g., refer to aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, azepanyl, diazepanyl (e.g., 1,4-diazepanyl), oxazolidinyl, isoxazolidinyl, thiazolidinyl, isothiazolidinyl, morpholinyl (e.g., morpholin-4-yl), thiomorpholinyl (e.g., thiomorpholin-4-yl), oxazepanyl, oxiranyl, oxetanyl, tetrahydrofuranyl, 1,3-dioxolanyl, tetrahydropyranyl, 1,4-dioxanyl, oxepanyl, thiiranyl, thietanyl, tetrahydrothiophenyl (i.e., thiolanyl), 1,3-dithiolanyl, thianyl, 1,1-dioxothianyl, thiepanyl, decahydroquinolinyl, decahydroisoquinolinyl, or 2-oxa-5-aza-bicyclo[2.2.1]hept-5-yl. Unless defined
otherwise, “heterocycloalkyl” preferably refers to a 3 to 11 membered saturated ring group, which is a monocyclic ring or a fused ring system (e.g., a fused ring system composed of two fused rings), wherein said ring group contains one or more (e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized; more preferably, “heterocycloalkyl” refers to a 5 to 7 membered saturated monocyclic ring group containing one or more (e.g., one, two, or three) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized. As used herein, the term “heterocycloalkylene” refers to a heterocycloalkyl group, as defined herein above, but having two points of attachment, i.e. a divalent saturated ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said ring group contains one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, and further wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group). For example, each heteroatom-containing ring comprised in said saturated ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heterocycloalkylene” may, e.g., refer to aziridinylene, azetidinylene, pyrrolidinylene, imidazolidinylene, pyrazolidinylene, piperidinylene, piperazinylene, azepanylene, diazepanylene (e.g., 1,4-diazepanylene), oxazolidinylene, isoxazolidinylene, thiazolidinylene, isothiazolidinylene, morpholinylene, thiomorpholinylene, oxazepanylene, oxiranylene, oxetanylene, tetrahydrofuranylene, 1,3-dioxolanylene, tetrahydropyranylene, 1,4-dioxanylene, oxepanylene, thiiranylene, thietanylene, tetrahydrothiophenylene (i.e., thiolanylene), 1,3-dithiolanylene, thianylene, 1,1-dioxothianylene, thiepanylene, decahydroquinolinylene, decahydroisoquinolinylene, or 2-oxa-5-aza-bicyclo[2.2.1]hept-5-ylene. Unless defined otherwise, “heterocycloalkylene” preferably refers to a divalent 3 to 11 membered saturated ring group, which is a monocyclic ring or a fused ring system (e.g., a fused ring system composed of two fused rings), wherein said ring group contains one or more (e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are
optionally oxidized; more preferably, “heterocycloalkylene” refers to a divalent 5 to 7 membered saturated monocyclic ring group containing one or more (e.g., one, two, or three) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, and wherein one or more carbon ring atoms are optionally oxidized. As used herein, the term “N-heterocycloalkyl” refers to the heterocycloalkyl groups as defined hereinabove wherein said heterocycloalkyl includes at least one nitrogen atom which serves as an attachment point of said heterocycloalkyl. As used herein, the term “cycloalkenyl” refers to an unsaturated alicyclic (non-aromatic) hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said hydrocarbon ring group comprises one or more (e.g., one or two) carbon-to-carbon double bonds and does not comprise any carbon-to-carbon triple bond. “Cycloalkenyl” may, e.g., refer to cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cyclohexadienyl, cycloheptenyl, or cycloheptadienyl. Unless defined otherwise, “cycloalkenyl” preferably refers to a C3-11 cycloalkenyl, and more preferably refers to a C3-7 cycloalkenyl. A particularly preferred “cycloalkenyl” is a monocyclic unsaturated alicyclic hydrocarbon ring having 3 to 7 ring members and containing one or more (e.g., one or two; preferably one) carbon-to-carbon double bonds. As used herein, the term “cycloalkenylene” refers to a cycloalkenyl group, as defined hereinabove, but having two points of attachment, i.e. a divalent unsaturated alicyclic (non-aromatic) hydrocarbon ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said hydrocarbon ring group comprises one or more (e.g., one or two) carbon-to- carbon double bonds and does not comprise any carbon-to-carbon triple bond. As used herein, the term “heterocycloalkenyl” refers to an unsaturated alicyclic (non-aromatic) ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said ring group contains one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group), and further wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms. For example, each heteroatom-containing ring comprised in said unsaturated alicyclic ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N
atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. “Heterocycloalkenyl” may, e.g., refer to imidazolinyl (e.g., 2-imidazolinyl (i.e., 4,5-dihydro-1H-imidazolyl), 3-imidazolinyl, or 4-imidazolinyl), tetrahydropyridinyl (e.g., 1,2,3,6-tetrahydropyridinyl), dihydropyridinyl (e.g., 1,2- dihydropyridinyl or 2,3-dihydropyridinyl), pyranyl (e.g., 2H-pyranyl or 4H-pyranyl), thiopyranyl (e.g., 2H-thiopyranyl or 4H-thiopyranyl), dihydropyranyl, dihydrofuranyl, dihydropyrazolyl, dihydropyrazinyl, dihydroisoindolyl, octahydroquinolinyl (e.g., 1,2,3,4,4a,5,6,7-octahydroquinolinyl), or octahydroisoquinolinyl (e.g., 1,2,3,4,5,6,7,8-octahydroisoquinolinyl). Unless defined otherwise, “heterocycloalkenyl” preferably refers to a 3 to 11 membered unsaturated alicyclic ring group, which is a monocyclic ring or a fused ring system (e.g., a fused ring system composed of two fused rings), wherein said ring group contains one or more (e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, wherein one or more carbon ring atoms are optionally oxidized, and wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms; more preferably, “heterocycloalkenyl” refers to a 5 to 7 membered monocyclic unsaturated non-aromatic ring group containing one or more (e.g., one, two, or three) ring heteroatoms independently selected from O, S and N, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) are optionally oxidized, wherein one or more carbon ring atoms are optionally oxidized, and wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms. As used herein, the term “heterocycloalkenylene” refers to a heterocycloalkenyl group, as defined hereinabove, as defined hereinabove, but having two points of attachment, i.e. a divalent unsaturated alicyclic (non-aromatic) ring group, including monocyclic rings as well as bridged ring, spiro ring and/or fused ring systems (which may be composed, e.g., of two or three rings; such as, e.g., a fused ring system composed of two or three fused rings), wherein said ring group contains one or more (such as, e.g., one, two, three, or four) ring heteroatoms independently selected from O, S and N, and the remaining ring atoms are carbon atoms, wherein one or more S ring atoms (if present) and/or one or more N ring atoms (if present) may optionally be oxidized, wherein one or more carbon ring atoms may optionally be oxidized (i.e., to form an oxo group), and further wherein said ring group comprises at least one double bond between adjacent ring atoms and does not comprise any triple bond between adjacent ring atoms. For example, each heteroatom-containing ring comprised in said unsaturated alicyclic ring group may contain one or two O atoms and/or one or two S atoms (which may optionally be oxidized) and/or one, two, three or four N atoms (which may optionally be oxidized), provided that the total number of heteroatoms in the
corresponding heteroatom-containing ring is 1 to 4 and that there is at least one carbon ring atom (which may optionally be oxidized) in the corresponding heteroatom-containing ring. As used herein, the term “halogen” (or Hal) refers to fluoro (-F), chloro (-Cl), bromo (-Br), or iodo (-I). Preferred halogen is fluoro. As used herein, the term “haloalkyl” refers to an alkyl group substituted with one or more (preferably 1 to 6, more preferably 1 to 3) halogen atoms which are selected independently from fluoro, chloro, bromo and iodo, and are preferably all fluoro atoms. It will be understood that the maximum number of halogen atoms is limited by the number of available attachment sites and, thus, depends on the number of carbon atoms comprised in the alkyl moiety of the haloalkyl group. “Haloalkyl” may, e.g., refer to -CF3, -CHF2, -CH2F, -CF2-CH3, -CH2-CF3, -CH2-CHF2, -CH2-CF2-CH3, -CH2-CF2-CF3, or -CH(CF3)2. A particularly preferred “haloalkyl” group is -CF3. In one embodiment, haloalkyl may also be a perhaloalkyl. The term “perhaloalkyl” refers to a haloalkyl wherein every -H atom has been substituted with a halo atom. Preferably, said perhaloalkyl is perfluoroalkyl. The terms “bond” and “covalent bond” are used herein synonymously, unless explicitly indicated otherwise or contradicted by context. As used herein, the terms “optional”, “optionally” and “may” denote that the indicated feature may be present but can also be absent. Whenever the term “optional”, “optionally” or “may” is used, the present invention specifically relates to both possibilities, i.e., that the corresponding feature is present or, alternatively, that the corresponding feature is absent. For example, the expression “X is optionally substituted with Y” (or “X may be substituted with Y”) means that X is either substituted with Y or is unsubstituted. Likewise, if a component of a composition is indicated to be “optional”, the invention specifically relates to both possibilities, i.e., that the corresponding component is present (contained in the composition) or that the corresponding component is absent from the composition. Various groups are referred to as being “optionally substituted” in this specification. Generally, these groups may carry one or more substituents, such as, e.g., one, two, three or four substituents. It will be understood that the maximum number of substituents is limited by the number of attachment sites available on the substituted moiety. Unless defined otherwise, the “optionally substituted” groups referred to in this specification carry preferably not more than two substituents and may, in particular, carry only one substituent. Moreover, unless defined otherwise, it is preferred that the optional substituents are absent, i.e. that the corresponding groups are unsubstituted. A skilled person will appreciate that the substituent groups comprised in the compounds of the present invention may be attached to the remainder of the respective compound via a number of different positions of the corresponding specific substituent group. Unless defined otherwise, the preferred attachment positions for the various specific substituent groups are as illustrated in the examples.
As used herein, unless explicitly indicated otherwise or contradicted by context, the terms “a”, “an” and “the” are used interchangeably with “one or more” and “at least one”. Thus, for example, a composition comprising “a” compound of formula (I) can be interpreted as referring to a composition comprising “one or more” compounds of formula (I). It is to be understood that wherever numerical ranges are provided/disclosed herein, all values and subranges encompassed by the respective numerical range are meant to be encompassed within the scope of the invention. Accordingly, the present invention specifically and individually relates to each value that falls within a numerical range disclosed herein, as well as each subrange encompassed by a numerical range disclosed herein. As used herein, the term “about” preferably refers to ±10% of the indicated numerical value, more preferably to ±5% of the indicated numerical value, and in particular to the exact numerical value indicated. If the term “about” is used in connection with the endpoints of a range, it preferably refers to the range from the lower endpoint -10% of its indicated numerical value to the upper endpoint +10% of its indicated numerical value, more preferably to the range from of the lower endpoint -5% to the upper endpoint +5%, and even more preferably to the range defined by the exact numerical values of the lower endpoint and the upper endpoint. As used herein, the term “comprising” (or “comprise”, “comprises”, “contain”, “contains”, or “containing”), unless explicitly indicated otherwise or contradicted by context, has the meaning of “containing, inter alia”, i.e., “containing, among further optional elements, …”. In addition thereto, this term also includes the narrower meanings of “consisting essentially of” and “consisting of”. For example, the term “A comprising B and C” has the meaning of “A containing, inter alia, B and C”, wherein A may contain further optional elements (e.g., “A containing B, C and D” would also be encompassed), but this term also includes the meaning of “A consisting essentially of B and C” and the meaning of “A consisting of B and C” (i.e., no other components than B and C are comprised in A). Detailed description of the invention The invention is described in detail in the following. It is to be understood that the present invention specifically relates to each and every combination of features and embodiments described herein, including any combination of general and/or preferred features/embodiments. In a first embodiment, the present invention relates to a compound of formula (I):
 or a pharmaceutically acceptable salt thereof.
In formula (I), A is selected from aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl, cycloalkenyl, C2 alkynyl, -N(C1-5 alkyl)(C1-5 alkyl) (such as -N(CH3)(CH(CH3)2)), C2-haloalkyl (such as - CF2CH3) and –(C1-2 haloalkylene)-cycloalkyl (such as -CF2-cyclopropyl), wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl, C1-6 haloalkyl, aryl (such as phenyl) or heteroaryl (such as thien-2-yl), preferably with C1-6 alkyl, C1- 6 haloalkyl, or aryl (such as phenyl), more preferably with C1-6 alkyl or C1-6 haloalkyl, such as C1-6 alkyl. Preferably, A is selected from aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl, cycloalkenyl and C2 alkynyl, wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl or C1-6 haloalkyl, such as C1-6 alkyl. It is to be understood that said cyclopropyl in -CF2-cyclopropyl is not substituted. Particularly suitable aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl and cycloalkenyl in A are each a single-ring system. Thus, preferably, A is selected from a single ring aryl, a single ring heteroaryl, a single ring heterocycloalkyl, a single ring heterocycloalkenyl, a single ring cycloalkyl, a single ring cycloalkenyl and C2 alkynyl, wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl or C1- 6 haloalkyl. If A is aryl, heteroaryl, heterocycloalkyl, heterocycloalkyl, cycloalkyl or cycloalkenyl that is optionally substituted with one or more R1, then said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl or said cycloalkenyl may preferably be substituted with 0, 1, 2, 3 or 4 groups R1. Preferably, said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl or said cycloalkenyl is substituted with 0, 1 or 2 groups R1. Preferably, A is selected from heterocycloalkyl, heterocycloalkenyl and C2 alkynyl, wherein said heterocycloalkyl and said heterocycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl. More preferably, A is heterocycloalkyl, or heterocycloalkenyl wherein said heterocycloalkyl and said heterocycloalkenyl are each optionally substituted with one or more R1. Even more preferably, A is heterocycloalkyl optionally substituted with one or more R1. In formula (I), each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5
alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl, -NH-(C0-3 alkylene)-carbocyclyl, -NH-(C0-3 alkylene)-heterocyclyl, -N(C1-5 alkyl)-(C0-3 alkylene)-carbocyclyl, -N(C1-5 alkyl)-(C0-3 alkylene)-heterocyclyl, -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -NH-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -N(C1-5 alkyl)-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl, the heterocyclyl moiety in said -NH-(C0-3 alkylene)-heterocyclyl, the heterocyclyl moiety in said -N(C1-5 alkyl)-(C0-3 alkylene)-heterocyclyl, and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1- 4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), -CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), - NHCO(C1-4 alkyl) and -N(C1-4 alkyl)-CO(C1-4 alkyl). Preferably, R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3
alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl, -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O- (C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), -CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)-CO(C1-4 alkyl). More preferably, each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), and -(C0-3 alkylene)-SO-(C1-5 alkyl). Even more preferably, each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5
alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), and -(C0-3 alkylene)-O-(C1-5 haloalkyl), preferably each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), and -(C0-3 alkylene)-halogen. Even more preferably, each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -OH, -O(C1-5 alkyl), -SH, -NH2, -NH(C1-5 alkyl), -N(C1-5 alkyl)(C1-5 alkyl), -NH-OH, -N(C1-5 alkyl)-OH, -NH-O(C1-5 alkyl), -N(C1-5 alkyl)-O(C1-5 alkyl), C1-5 haloalkyl and halogen, preferably each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -OH, -O(C1-5 alkyl), -SH, -NH2, -NH(C1-5 alkyl), -N(C1-5 alkyl)(C1-5 alkyl), -NH-OH, -N(C1-5 alkyl)-OH, -NH-O(C1-5 alkyl), -N(C1-5 alkyl)-O(C1-5 alkyl), and halogen. Even more preferably, each R1 is independently selected from -OH and halogen. Particularly suitable A are selected from:
or a pharmaceutically acceptable salt thereof.
In formula (I), A is selected from aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl, cycloalkenyl, C2 alkynyl, -N(C1-5 alkyl)(C1-5 alkyl) (such as -N(CH3)(CH(CH3)2)), C2-haloalkyl (such as - CF2CH3) and –(C1-2 haloalkylene)-cycloalkyl (such as -CF2-cyclopropyl), wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl, C1-6 haloalkyl, aryl (such as phenyl) or heteroaryl (such as thien-2-yl), preferably with C1-6 alkyl, C1- 6 haloalkyl, or aryl (such as phenyl), more preferably with C1-6 alkyl or C1-6 haloalkyl, such as C1-6 alkyl. Preferably, A is selected from aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl, cycloalkenyl and C2 alkynyl, wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl or C1-6 haloalkyl, such as C1-6 alkyl. It is to be understood that said cyclopropyl in -CF2-cyclopropyl is not substituted. Particularly suitable aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl and cycloalkenyl in A are each a single-ring system. Thus, preferably, A is selected from a single ring aryl, a single ring heteroaryl, a single ring heterocycloalkyl, a single ring heterocycloalkenyl, a single ring cycloalkyl, a single ring cycloalkenyl and C2 alkynyl, wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl or C1- 6 haloalkyl. If A is aryl, heteroaryl, heterocycloalkyl, heterocycloalkyl, cycloalkyl or cycloalkenyl that is optionally substituted with one or more R1, then said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl or said cycloalkenyl may preferably be substituted with 0, 1, 2, 3 or 4 groups R1. Preferably, said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl or said cycloalkenyl is substituted with 0, 1 or 2 groups R1. Preferably, A is selected from heterocycloalkyl, heterocycloalkenyl and C2 alkynyl, wherein said heterocycloalkyl and said heterocycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl. More preferably, A is heterocycloalkyl, or heterocycloalkenyl wherein said heterocycloalkyl and said heterocycloalkenyl are each optionally substituted with one or more R1. Even more preferably, A is heterocycloalkyl optionally substituted with one or more R1. In formula (I), each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5
alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl, -NH-(C0-3 alkylene)-carbocyclyl, -NH-(C0-3 alkylene)-heterocyclyl, -N(C1-5 alkyl)-(C0-3 alkylene)-carbocyclyl, -N(C1-5 alkyl)-(C0-3 alkylene)-heterocyclyl, -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -NH-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -N(C1-5 alkyl)-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl, the heterocyclyl moiety in said -NH-(C0-3 alkylene)-heterocyclyl, the heterocyclyl moiety in said -N(C1-5 alkyl)-(C0-3 alkylene)-heterocyclyl, and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1- 4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), -CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), - NHCO(C1-4 alkyl) and -N(C1-4 alkyl)-CO(C1-4 alkyl). Preferably, R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3
alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl, -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O- (C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), -CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)-CO(C1-4 alkyl). More preferably, each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), and -(C0-3 alkylene)-SO-(C1-5 alkyl). Even more preferably, each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5
alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), and -(C0-3 alkylene)-O-(C1-5 haloalkyl), preferably each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), and -(C0-3 alkylene)-halogen. Even more preferably, each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -OH, -O(C1-5 alkyl), -SH, -NH2, -NH(C1-5 alkyl), -N(C1-5 alkyl)(C1-5 alkyl), -NH-OH, -N(C1-5 alkyl)-OH, -NH-O(C1-5 alkyl), -N(C1-5 alkyl)-O(C1-5 alkyl), C1-5 haloalkyl and halogen, preferably each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -OH, -O(C1-5 alkyl), -SH, -NH2, -NH(C1-5 alkyl), -N(C1-5 alkyl)(C1-5 alkyl), -NH-OH, -N(C1-5 alkyl)-OH, -NH-O(C1-5 alkyl), -N(C1-5 alkyl)-O(C1-5 alkyl), and halogen. Even more preferably, each R1 is independently selected from -OH and halogen. Particularly suitable A are selected from:
 , ,
, ,
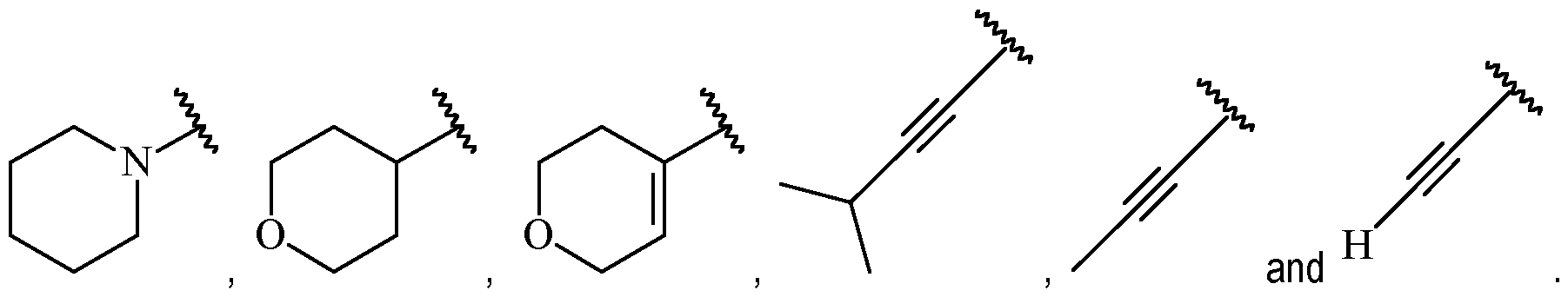 In one embodiment, A is -CF2-cyclopropyl. In one embodiment, A is -CF2CH3. In one embodiment, A is -N(C1-5 alkyl)(C1-5 alkyl), preferably -N(CH3)(CH(CH3)2. In one embodiment, A is -C C-C6H5. In formula (I), B is selected from arylene and heteroarylene, wherein said arylene and said hereroarylene are each optionally substituted with one or more R2. Preferably, B is arylene, optionally substituted with one or more R2. Particularly preferred arylene in B is phenylene, optionally substituted with one or more R2, in particular 1,4-phenylene optionally
substituted with one or more R2. Accordingly, in one embodiment, another embodiment,
In one embodiment, A is -CF2-cyclopropyl. In one embodiment, A is -CF2CH3. In one embodiment, A is -N(C1-5 alkyl)(C1-5 alkyl), preferably -N(CH3)(CH(CH3)2. In one embodiment, A is -C C-C6H5. In formula (I), B is selected from arylene and heteroarylene, wherein said arylene and said hereroarylene are each optionally substituted with one or more R2. Preferably, B is arylene, optionally substituted with one or more R2. Particularly preferred arylene in B is phenylene, optionally substituted with one or more R2, in particular 1,4-phenylene optionally
substituted with one or more R2. Accordingly, in one embodiment, another embodiment,
 one particularly preferred embodiment,
one particularly preferred embodiment,
 It is preferred that if B is 1,4-phenylene optionally substituted with one or more R2, for example as recited in the foregoing, B is (further) substituted with -O-(C0-3 alkylene)-carbocyclyl, or -O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or -O-heterocyclyl, more preferably with - O-carbocyclyl, in particular -O-phenyl. Preferably, said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X. In one embodiment, B is heteroarylene, optionally substituted with one or more R2. Particularly preferred are 5 or 6-membered heteroarylene moieties. Accordingly, the heteroarylene may be a 5-membered heteroarylene, optionally substituted with one or more R2. Preferably, said 5-membered heteroarylene may be a heteroarylene comprising at least one N atom in the ring, more preferably at least two N atoms in the ring. Exemplary suitable 5-membered heteroarylenes
It is preferred that if B is 1,4-phenylene optionally substituted with one or more R2, for example as recited in the foregoing, B is (further) substituted with -O-(C0-3 alkylene)-carbocyclyl, or -O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or -O-heterocyclyl, more preferably with - O-carbocyclyl, in particular -O-phenyl. Preferably, said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X. In one embodiment, B is heteroarylene, optionally substituted with one or more R2. Particularly preferred are 5 or 6-membered heteroarylene moieties. Accordingly, the heteroarylene may be a 5-membered heteroarylene, optionally substituted with one or more R2. Preferably, said 5-membered heteroarylene may be a heteroarylene comprising at least one N atom in the ring, more preferably at least two N atoms in the ring. Exemplary suitable 5-membered heteroarylenes
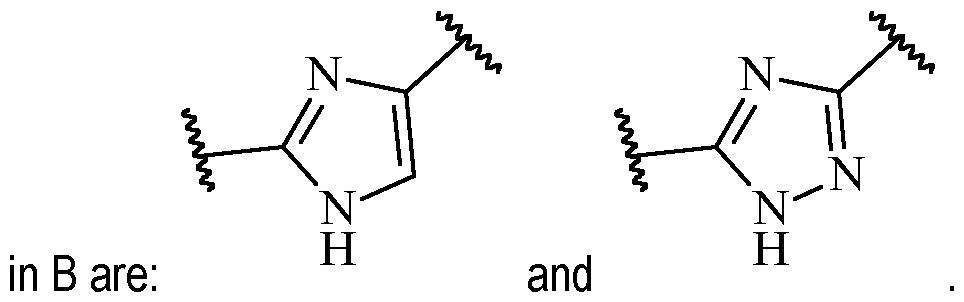 Alternatively, B may also be a 6-membered heteroarylene, optionally substituted with one or more R2. Preferably, said 6-membered heteroarylene is pyridinylene, preferably a 2,5-pyridinylene, optionally substituted with one or more R2. Suitable examples wherein B is a 2,5-pyridinylene moiety, as disclosed herein, include
Alternatively, B may also be a 6-membered heteroarylene, optionally substituted with one or more R2. Preferably, said 6-membered heteroarylene is pyridinylene, preferably a 2,5-pyridinylene, optionally substituted with one or more R2. Suitable examples wherein B is a 2,5-pyridinylene moiety, as disclosed herein, include
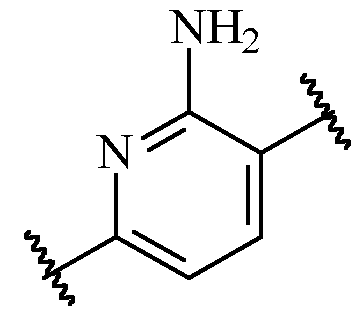 . The heteroarylene may also be optionally oxidized in the ring. One suitable example of such heteroarylene moiety including an oxo group in the ring
. The heteroarylene may also be optionally oxidized in the ring. One suitable example of such heteroarylene moiety including an oxo group in the ring
 wherein the heteroarylene is optionally substituted with one or more R2, and wherein RD1 is selected from H and C1-6 alkyl (such as methyl). In one preferred embodiment, the heteroarylene
wherein the heteroarylene is optionally substituted with one or more R2, and wherein RD1 is selected from H and C1-6 alkyl (such as methyl). In one preferred embodiment, the heteroarylene
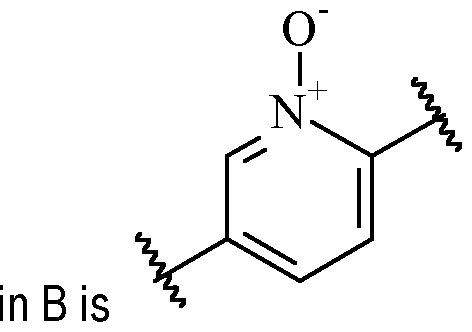 , optionally substituted with one or more R2. As it is to be understood herein, each heteroarylene moiety discussed herein, including each specific example, is optionally substituted with one or more R2. Particularly preferred B being
, optionally substituted with one or more R2. As it is to be understood herein, each heteroarylene moiety discussed herein, including each specific example, is optionally substituted with one or more R2. Particularly preferred B being
 As it is apparent to the skilled person, B is a bivalent moiety and accordingly, as understood herein, preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X. It is preferred that if B is 2,5-pyridinylene, B is substituted with -O-(C0-3 alkylene)-carbocyclyl, or - O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or -O-heterocyclyl, more preferably with - O-carbocyclyl, in particular -O-phenyl. Preferably, said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X. It is further preferred that if B is 2,5-pyrimidinylene, B is substituted with -O-(C0-3 alkylene)-carbocyclyl, or -O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or - O-heterocyclyl, more preferably with -O-carbocyclyl, in particular -O-phenyl. Preferably, said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X, or, in other words, at position 4 in the 2,5-pyrimidinylene. In formula (I), each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3
alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O- (C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), -CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)-CO(C1-4 alkyl). Preferably, each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), and -(C0-3 alkylene)-SO-(C1-5 alkyl).
More preferably, each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), and -(C0-3 alkylene)-halogen. Even more preferably, each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -OH, -O(C1-5 alkyl), -SH, -NH2, -NH(C1-5 alkyl), -N(C1-5 alkyl)(C1-5 alkyl), -NH-OH, -N(C1-5 alkyl)-OH, -NH-O(C1-5 alkyl), -N(C1-5 alkyl)-O(C1-5 alkyl), and halogen. In formula (I), X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, - SO2N(C1-6 alkyl)-, -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, -CH(CF3)N(C1-6 haloalkyl)-, C2 alkenylene optionally substituted with one or more halogen, C1-6 alkyl or C1-6 haloalkyl (preferably optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl), cycloalkylene (preferably cyclopropylene) optionally substituted with one or more C1-6 alkyl, -(heterocycloalkylene)-NH-, - (heterocycloalkylene)-N(C1-6 alkyl)-, heterocycloalkylene, heterocycloalkenylene and heteroarylene. Particularly suitable heterocycloalkylene is 3,3-oxetanylene. Particularly suitable cycloalkylene is 1,2- cyclopopylene. Particularly suitable heteroarylene is 1,2,3-triazolyl-1,4-ene. Preferably, X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, - SO2N(C1-6 alkyl)-, -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, -CH(CF3)N(C1-6 haloalkyl)-, C2 alkenylene optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl, cycloalkylene (preferably cyclopropylene) optionally substituted with one or more C1-6 alkyl, -(heterocycloalkylene)-NH- , and -(heterocycloalkylene)-N(C1-6 alkyl)- Preferably, X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, - SO2N(C1-6 alkyl)-, -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, and -CH(CF3)N(C1-6 haloalkyl)-. More preferably, X is selected from -CONH-, and -CON(C1-6 alkyl)-. Even more preferably, X is - CONH-. It is to be understood that, preferably, the left side of the bivalent formula embodying X, as shown herein, is connected to B, and the right side of said bivalent formula embodying X is connected to Y. Alternatively, X is selected from heterocycloalkylene, heterocycloalkenylene and heteroarylene. Preferably, said heterocycloalkylene is a nitrogen-containing five-membered heterocycloalkylene, said heterocycloalkenylene is a nitrogen-containing five-membered heterocycloalkenylene, and said heteroarylene is a nitrogen-containing five-membered heteroarylene. Particularly suitable heterocycloalkenylene
As it is apparent to the skilled person, B is a bivalent moiety and accordingly, as understood herein, preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X. It is preferred that if B is 2,5-pyridinylene, B is substituted with -O-(C0-3 alkylene)-carbocyclyl, or - O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or -O-heterocyclyl, more preferably with - O-carbocyclyl, in particular -O-phenyl. Preferably, said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X. It is further preferred that if B is 2,5-pyrimidinylene, B is substituted with -O-(C0-3 alkylene)-carbocyclyl, or -O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or - O-heterocyclyl, more preferably with -O-carbocyclyl, in particular -O-phenyl. Preferably, said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X, or, in other words, at position 4 in the 2,5-pyrimidinylene. In formula (I), each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3
alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O- (C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), -CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)-CO(C1-4 alkyl). Preferably, each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), and -(C0-3 alkylene)-SO-(C1-5 alkyl).
More preferably, each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), and -(C0-3 alkylene)-halogen. Even more preferably, each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -OH, -O(C1-5 alkyl), -SH, -NH2, -NH(C1-5 alkyl), -N(C1-5 alkyl)(C1-5 alkyl), -NH-OH, -N(C1-5 alkyl)-OH, -NH-O(C1-5 alkyl), -N(C1-5 alkyl)-O(C1-5 alkyl), and halogen. In formula (I), X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, - SO2N(C1-6 alkyl)-, -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, -CH(CF3)N(C1-6 haloalkyl)-, C2 alkenylene optionally substituted with one or more halogen, C1-6 alkyl or C1-6 haloalkyl (preferably optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl), cycloalkylene (preferably cyclopropylene) optionally substituted with one or more C1-6 alkyl, -(heterocycloalkylene)-NH-, - (heterocycloalkylene)-N(C1-6 alkyl)-, heterocycloalkylene, heterocycloalkenylene and heteroarylene. Particularly suitable heterocycloalkylene is 3,3-oxetanylene. Particularly suitable cycloalkylene is 1,2- cyclopopylene. Particularly suitable heteroarylene is 1,2,3-triazolyl-1,4-ene. Preferably, X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, - SO2N(C1-6 alkyl)-, -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, -CH(CF3)N(C1-6 haloalkyl)-, C2 alkenylene optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl, cycloalkylene (preferably cyclopropylene) optionally substituted with one or more C1-6 alkyl, -(heterocycloalkylene)-NH- , and -(heterocycloalkylene)-N(C1-6 alkyl)- Preferably, X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, - SO2N(C1-6 alkyl)-, -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, and -CH(CF3)N(C1-6 haloalkyl)-. More preferably, X is selected from -CONH-, and -CON(C1-6 alkyl)-. Even more preferably, X is - CONH-. It is to be understood that, preferably, the left side of the bivalent formula embodying X, as shown herein, is connected to B, and the right side of said bivalent formula embodying X is connected to Y. Alternatively, X is selected from heterocycloalkylene, heterocycloalkenylene and heteroarylene. Preferably, said heterocycloalkylene is a nitrogen-containing five-membered heterocycloalkylene, said heterocycloalkenylene is a nitrogen-containing five-membered heterocycloalkenylene, and said heteroarylene is a nitrogen-containing five-membered heteroarylene. Particularly suitable heterocycloalkenylene
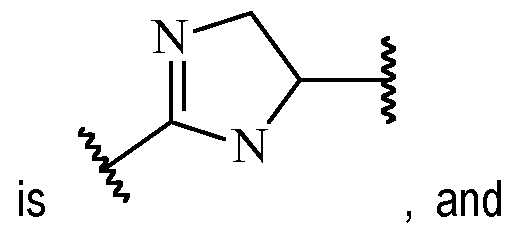 particularly suitable heteroarylene is selected from
understood that, preferably, the left side of the bivalent formula embodying X, as shown herein, is connected to B, and the right side of said bivalent formula embodying X is connected to Y.
particularly suitable heteroarylene is selected from
understood that, preferably, the left side of the bivalent formula embodying X, as shown herein, is connected to B, and the right side of said bivalent formula embodying X is connected to Y.
 CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, C1-4 haloalkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO3H, -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), -SO2NH2, - SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, -SO2-cycloalkyl, -SO(=NH)-cycloalkyl, -SO(=N-(C1-4 alkyl))-cycloalkyl, heterocycloalkyl, -SO2-heterocycloalkyl, -SO(=NH)-heterocycloalkyl, -SO(=N-(C1-4 alkyl))- heterocycloalkyl, aryl, -SO2-aryl, -SO(=NH)-aryl, -SO(=N-(C1-4 alkyl))-aryl, heteroaryl, -SO2-heteroaryl, - SO(=NH)-heteroaryl, -SO(=N-(C1-4 alkyl))-heteroaryl,-Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), - SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), - CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, still more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2- (C1-4 alkyl), even more preferably substituted with one or more optional substituents selected from -CONH-
alkyl)
CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, C1-4 haloalkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO3H, -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), -SO2NH2, - SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, -SO2-cycloalkyl, -SO(=NH)-cycloalkyl, -SO(=N-(C1-4 alkyl))-cycloalkyl, heterocycloalkyl, -SO2-heterocycloalkyl, -SO(=NH)-heterocycloalkyl, -SO(=N-(C1-4 alkyl))- heterocycloalkyl, aryl, -SO2-aryl, -SO(=NH)-aryl, -SO(=N-(C1-4 alkyl))-aryl, heteroaryl, -SO2-heteroaryl, - SO(=NH)-heteroaryl, -SO(=N-(C1-4 alkyl))-heteroaryl,-Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), - SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), - CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, still more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2- (C1-4 alkyl), even more preferably substituted with one or more optional substituents selected from -CONH-
alkyl)
 are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, m is 1.
are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, m is 1.
 -SO2(C2 alkenyl), -CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, C1-4 haloalkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), - CON(C1-4 alkyl)(C1-4 alkyl), -SO3H, -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, -SO2-cycloalkyl, -SO(=NH)-cycloalkyl, -SO(=N-(C1-4 alkyl))- cycloalkyl, heterocycloalkyl, -SO2-heterocycloalkyl, -SO(=NH)-heterocycloalkyl, -SO(=N-(C1-4 alkyl))- heterocycloalkyl, aryl, -SO2-aryl, -SO(=NH)-aryl, -SO(=N-(C1-4 alkyl))-aryl, heteroaryl, -SO2-heteroaryl, - SO(=NH)-heteroaryl, -SO(=N-(C1-4 alkyl))-heteroaryl,-Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), - SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), - CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-
heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, still more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2- (C1-4 alkyl), even more preferably substituted with one or more optional substituents selected from -CONH- (C1-4 alkyl), and -SO2-(C1-4 alkyl), and wherein said
-SO2(C2 alkenyl), -CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, C1-4 haloalkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), - CON(C1-4 alkyl)(C1-4 alkyl), -SO3H, -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, -SO2-cycloalkyl, -SO(=NH)-cycloalkyl, -SO(=N-(C1-4 alkyl))- cycloalkyl, heterocycloalkyl, -SO2-heterocycloalkyl, -SO(=NH)-heterocycloalkyl, -SO(=N-(C1-4 alkyl))- heterocycloalkyl, aryl, -SO2-aryl, -SO(=NH)-aryl, -SO(=N-(C1-4 alkyl))-aryl, heteroaryl, -SO2-heteroaryl, - SO(=NH)-heteroaryl, -SO(=N-(C1-4 alkyl))-heteroaryl,-Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), - SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), - CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-
heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, still more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2- (C1-4 alkyl), even more preferably substituted with one or more optional substituents selected from -CONH- (C1-4 alkyl), and -SO2-(C1-4 alkyl), and wherein said
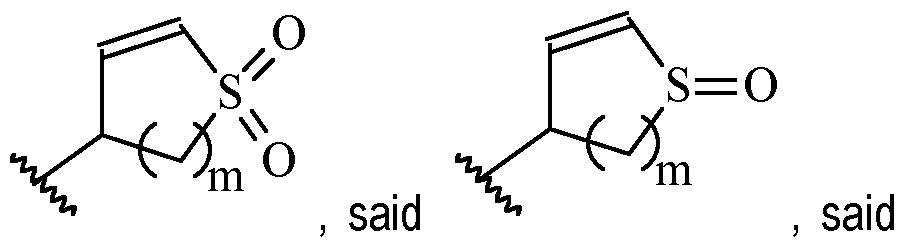
 , , , and said
, , , and said
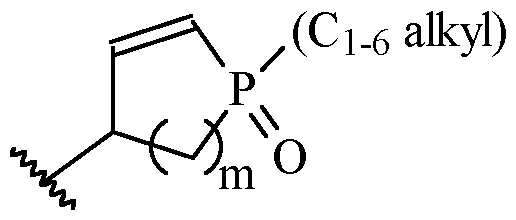 are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, m is 1. More preferably, Y is selected from:
are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, m is 1. More preferably, Y is selected from:
 , ,
, ,
 -SO2(C2 alkenyl), -CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, C1-4 haloalkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), - CON(C1-4 alkyl)(C1-4 alkyl), -SO3H, -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, -SO2- cycloalkyl, heterocycloalkyl, -SO2-heterocycloalkyl, aryl, -SO2-aryl, heteroaryl, -SO2-heteroaryl, -Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1- 4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, - SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2- (C1-4 alkyl), even more preferably substituted with one or more optional substituents selected from -CONH-
-SO2(C2 alkenyl), -CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, C1-4 haloalkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), - CON(C1-4 alkyl)(C1-4 alkyl), -SO3H, -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, -SO2- cycloalkyl, heterocycloalkyl, -SO2-heterocycloalkyl, aryl, -SO2-aryl, heteroaryl, -SO2-heteroaryl, -Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1- 4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, - SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2- (C1-4 alkyl), even more preferably substituted with one or more optional substituents selected from -CONH-
 are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, m is 1.
are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, m is 1.
 -SO2(C2 alkenyl), -CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, - CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, - CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl), even more preferably substituted with one or more optional substituents selected from -CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl), and wherein said
-SO2(C2 alkenyl), -CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, - CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, - CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl), even more preferably substituted with one or more optional substituents selected from -CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl), and wherein said
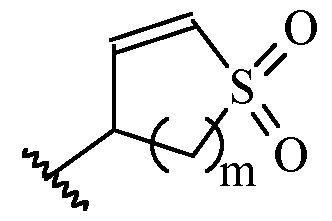 , said
, said
 , and said
, and said
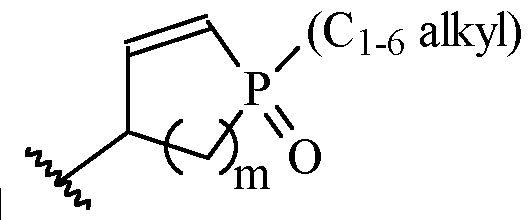 are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, m is 1. Even more preferably, Y is selected from:
are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, m is 1. Even more preferably, Y is selected from:
 , ,
, ,
 -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), - CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl), preferably selected from -CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl), and wherein said
-CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), - CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl), preferably selected from -CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl), and wherein said
 ,
,
 , and said
, and said
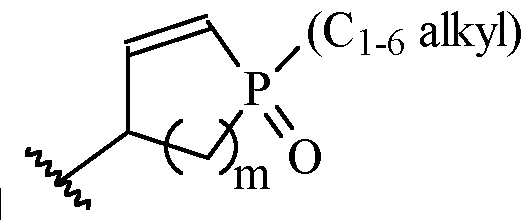 are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, m is 1.
Again more preferably, Y is selected from:
are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, m is 1.
Again more preferably, Y is selected from:
 , ,
, ,
 , , , and
, , , and
 C1-6 alkyl (such as methyl) or Hal (such as F). Still more preferably, Y is selected from:
C1-6 alkyl (such as methyl) or Hal (such as F). Still more preferably, Y is selected from:


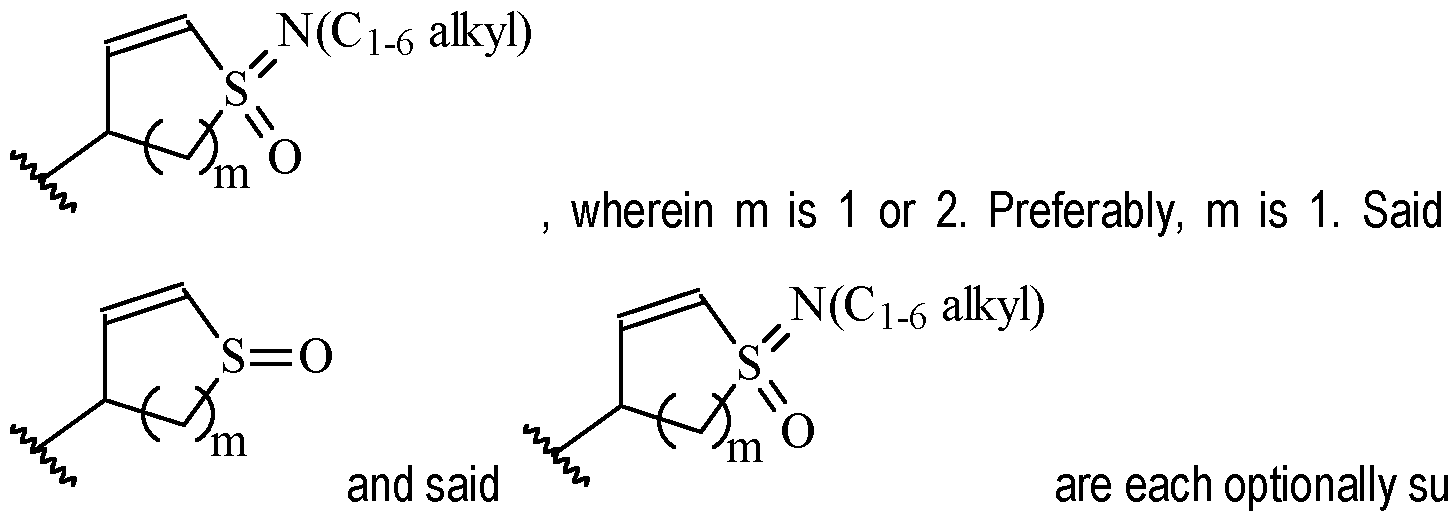 by one or more C1-6 alkyl (such as methyl) or Hal (such as F).
Even more preferably, , wherein m is 1 or 2. It is particularly preferred that m is 1. Said
by one or more C1-6 alkyl (such as methyl) or Hal (such as F).
Even more preferably, , wherein m is 1 or 2. It is particularly preferred that m is 1. Said
 optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). As recognized by the skilled person, Y may include chiral sulfur atom, and accordingly the presented formulae, which are drawn without indication of stereochemistry, may represent any of stereoisomers, in particular diastereoisomers or enantiomers embraced by the formula, including each specific possible stereoisomer or diastereoisomer, as well as it may refer to racemic mixtures or mixtures including an excess of one specific stereoisomer, enantiomer or diastereoisomer over another stereoisomer, enantiomer or diastereoisomer differing from the first one by configuration of at least one chiral center. For example, a formula
optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). As recognized by the skilled person, Y may include chiral sulfur atom, and accordingly the presented formulae, which are drawn without indication of stereochemistry, may represent any of stereoisomers, in particular diastereoisomers or enantiomers embraced by the formula, including each specific possible stereoisomer or diastereoisomer, as well as it may refer to racemic mixtures or mixtures including an excess of one specific stereoisomer, enantiomer or diastereoisomer over another stereoisomer, enantiomer or diastereoisomer differing from the first one by configuration of at least one chiral center. For example, a formula
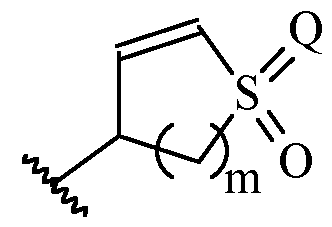 wherein Q is =NH, =NCN, =N(C1-6 alkyl) or is absent may refer to each single one of the following formulae:
wherein Q is =NH, =NCN, =N(C1-6 alkyl) or is absent may refer to each single one of the following formulae:
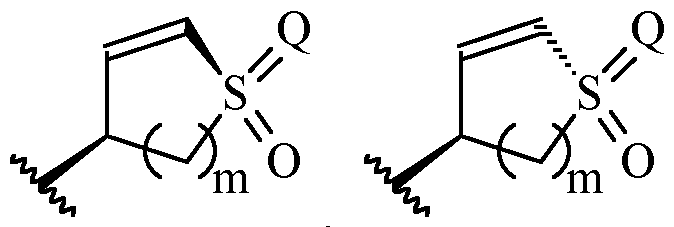 , ,
, ,
 well as it may refer to racemic mixtures or mixtures including an excess of one specific stereoisomer, enantiomer or diastereoisomer over another stereoisomer, enantiomer or diastereoisomer differing from the first one by configuration of at least one chiral center. Similar reasoning applies to the compounds of the present invention including a chiral phosphorus atom. Alternatively, in formula (I) B and X are combined to form a two-ring system according to formula:
well as it may refer to racemic mixtures or mixtures including an excess of one specific stereoisomer, enantiomer or diastereoisomer over another stereoisomer, enantiomer or diastereoisomer differing from the first one by configuration of at least one chiral center. Similar reasoning applies to the compounds of the present invention including a chiral phosphorus atom. Alternatively, in formula (I) B and X are combined to form a two-ring system according to formula:
 wherein C is aryl or heteroaryl ring, optionally substituted with one or more R2, Z is selected from C1-2 alkylene, -CH=CH-, -N=CH-, -N=C(C1-6 alkyl)-, -CH=N-, -C(C1-6 alkyl)=N-, -CO-, -SO-, -SO2-, -CONH-
, -NHCO-, -CON(C1-6 alkyl)-, -N(C1-6 alkyl)CO-, -NH-, -N(C1-6 alkyl)- and -O-, and A and Y are as defined hereinabove. Preferably, C is benzene ring or pyridine ring, wherein each of these rings is optionally substituted with one or more R2. More preferably C is selected from:
wherein C is aryl or heteroaryl ring, optionally substituted with one or more R2, Z is selected from C1-2 alkylene, -CH=CH-, -N=CH-, -N=C(C1-6 alkyl)-, -CH=N-, -C(C1-6 alkyl)=N-, -CO-, -SO-, -SO2-, -CONH-
, -NHCO-, -CON(C1-6 alkyl)-, -N(C1-6 alkyl)CO-, -NH-, -N(C1-6 alkyl)- and -O-, and A and Y are as defined hereinabove. Preferably, C is benzene ring or pyridine ring, wherein each of these rings is optionally substituted with one or more R2. More preferably C is selected from:
 wherein each of these rings is optionally substituted with one or more R2. It is to be understood that, preferably, the empty valence on the left side of the molecule is directed to A ring, the empty valence on the upper side of the molecule connects to -CO- moiety of -CONZ-, and the empty valence on the right-lower side of the molecule is connected to -Z- of -CONZ-. Preferably, Z is selected from C1-2 alkylene, -CH=CH-, -N=CH-, -SO2-, and -NHCO-.
wherein each of these rings is optionally substituted with one or more R2. It is to be understood that, preferably, the empty valence on the left side of the molecule is directed to A ring, the empty valence on the upper side of the molecule connects to -CO- moiety of -CONZ-, and the empty valence on the right-lower side of the molecule is connected to -Z- of -CONZ-. Preferably, Z is selected from C1-2 alkylene, -CH=CH-, -N=CH-, -SO2-, and -NHCO-.
 Alternatively, A and B are as defined hereinabove, and X and Y form a two ring spiro system, defined as:
Alternatively, A and B are as defined hereinabove, and X and Y form a two ring spiro system, defined as:
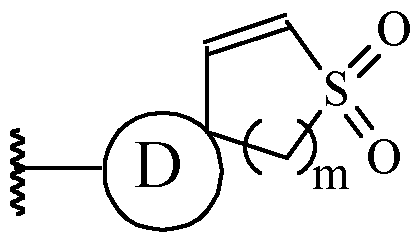 wherein D is a nitrogen-containing heterocycloalkyl or heterocycloalkenyl, m is 1 or 2, and the
wherein D is a nitrogen-containing heterocycloalkyl or heterocycloalkenyl, m is 1 or 2, and the
 optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). By X and Y forming a two-ring spiro system it is understood that X and Y are replaced with a two ring spiro system, as defined herein. It is to be understood that such compounds are also encompassed by formula (I). Nitrogen containing heterocycloalkyl or heterocycloalkenyl is defined as heterocycloalkyl or heterocycloalkenyl, respectively, including at least one N atom in the ring. Preferably, D is 4, 5 or 6 membered nitrogen-containing heterocycloalkyl or heterocycloalkenyl, preferably connected to B through its nitrogen atom. Accordingly and preferably, D is an N- heterocycloalkyl. Exemplary heterocycloalkyl and heterocycloalkenyl moieties suitable as D are selected from
optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). By X and Y forming a two-ring spiro system it is understood that X and Y are replaced with a two ring spiro system, as defined herein. It is to be understood that such compounds are also encompassed by formula (I). Nitrogen containing heterocycloalkyl or heterocycloalkenyl is defined as heterocycloalkyl or heterocycloalkenyl, respectively, including at least one N atom in the ring. Preferably, D is 4, 5 or 6 membered nitrogen-containing heterocycloalkyl or heterocycloalkenyl, preferably connected to B through its nitrogen atom. Accordingly and preferably, D is an N- heterocycloalkyl. Exemplary heterocycloalkyl and heterocycloalkenyl moieties suitable as D are selected from
 or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, the following provisions apply to formula (I):
or more C1-6 alkyl (such as methyl) or Hal (such as F). Preferably, the following provisions apply to formula (I):
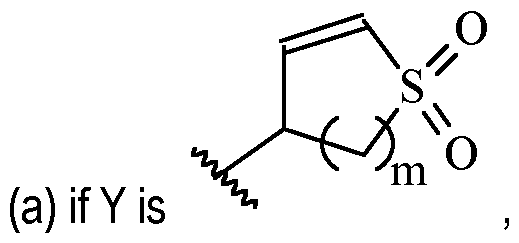 wherein m is 1 or 2, and wherein said
wherein m is 1 or 2, and wherein said
 optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F); then: (a1) A is not selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; and/or (a2) B is not according to formula
optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F); then: (a1) A is not selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; and/or (a2) B is not according to formula
 wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; and/or (a3) X is not -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; (b) if X is -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y, then: (b1) A is not selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; and/or
(b2) B is not according to formula wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; and/or (b3) Y is not
wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; and/or (a3) X is not -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; (b) if X is -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y, then: (b1) A is not selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; and/or
(b2) B is not according to formula wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; and/or (b3) Y is not
 , wherein m is 1 or 2, and wherein said
, wherein m is 1 or 2, and wherein said
 is optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F); (c) if B is according to formula :
is optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F); (c) if B is according to formula :
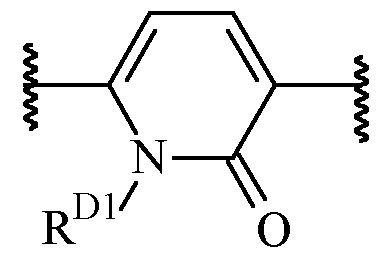 , wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-( C1-6 alkyl), - NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; then (c1) A is not selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; and/or (c2) X is not -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and/or (c3) Y is not
, wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-( C1-6 alkyl), - NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; then (c1) A is not selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; and/or (c2) X is not -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and/or (c3) Y is not
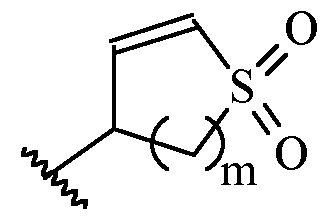 , wherein m is 1 or 2, and wherein said
, wherein m is 1 or 2, and wherein said
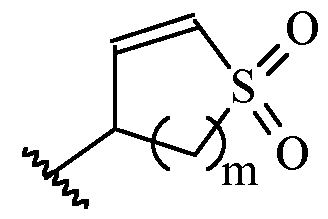 is optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). ; (d) if A is selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, then:
(d1) B is not according to formula wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; and/or (d2) X is not -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and/or (d3) Y is not
is optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). ; (d) if A is selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, then:
(d1) B is not according to formula wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; and/or (d2) X is not -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and/or (d3) Y is not
 , wherein m is 1 or 2, and wherein said
, wherein m is 1 or 2, and wherein said
 is optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Alternatively, the following provisions apply to formula (I): (e1) A is not selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; and/or (e2) B is not according to formula
is optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). Alternatively, the following provisions apply to formula (I): (e1) A is not selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; and/or (e2) B is not according to formula
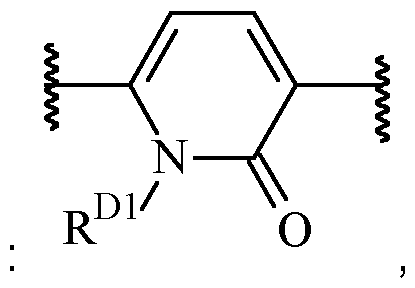 wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; and/or (e3) X is not -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and/or (e4) Y is not
wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; and/or (e3) X is not -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and/or (e4) Y is not
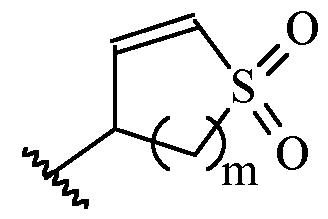 , wherein m is 1 or 2, and wherein said
, wherein m is 1 or 2, and wherein said
 is optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F).
It is to be understood that at least one of conditions (e1) to (e4) must preferably be fulfilled in the compound of formula (I). Accordingly and preferably, from formula (I) excluded is a compound of formula (I), wherein: A is selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; B is according to formula
is optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F).
It is to be understood that at least one of conditions (e1) to (e4) must preferably be fulfilled in the compound of formula (I). Accordingly and preferably, from formula (I) excluded is a compound of formula (I), wherein: A is selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; B is according to formula
 wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; X is -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and Y is not
wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; X is -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and Y is not
 , wherein m is 1 or 2, and wherein said
, wherein m is 1 or 2, and wherein said
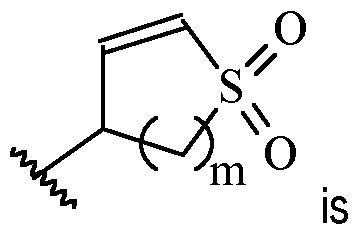 optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). In other words, it is preferred that the compound of formula (I) is not a compound wherein: A is selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; B is according to formula
optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). In other words, it is preferred that the compound of formula (I) is not a compound wherein: A is selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; B is according to formula
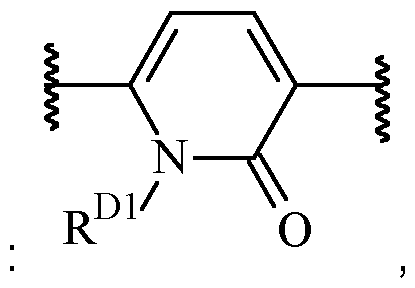 wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; X is -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and
Y is not , wherein m is 1 or 2, and wherein said optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F), or its pharmaceutically acceptable salt. In addition to the provisions recited hereinabove, or alternatively, the following provisions preferably apply to formula (I): (f1) if Y is a moiety according to formula:
wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; X is -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and
Y is not , wherein m is 1 or 2, and wherein said optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F), or its pharmaceutically acceptable salt. In addition to the provisions recited hereinabove, or alternatively, the following provisions preferably apply to formula (I): (f1) if Y is a moiety according to formula:
 wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, Ry2 is -H and Ry3 is selected from C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible, then B is not a moiety according to formula:
wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, Ry2 is -H and Ry3 is selected from C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible, then B is not a moiety according to formula:
 wherein the left empty valence is connected to A, and the right empty valence is connected to X, wherein: Xv is CRv2 or N; Yv is CRv4 or N; Zv is CRv5 or N; or Yv and Zv taken together form an optionally substituted five- to six-membered heteroaryl, or an optionally substituted five- to six-membered heterocyclyl; with the proviso that Xv, Yv, and Zv are not simultaneously N; Rv is H, -O-(optionally substituted C3-C8 cycloalkyl), -O-(optionally substituted C1-C6 alkyl), -O- (optionally substituted C6-C10 aryl), -O-(optionally substituted five- to six-membered heteroaryl), -O- (optionally substituted five- to six-membered heterocyclyl), or optionally substituted C3-C8 cycloalkyl; or Rv together with the carbon atoms to which it is shown attached and Xv form an optionally substituted five- to six-membered heterocyclyl;
Rv2 is H, optionally substituted C1-C6 alkyl, or halo; Rv4 is H, C1-C6 alkyl, cyano, or halo; or Rv4 together with the carbon atom to which it is shown attached and Zv form an optionally substituted five- to six-membered heteroaryl; Rv5 is H, C1-C6 alkyl, -NR2, or -N(R)-C(=O)-(C1-C6 alkyl); each R independently is H, or optionally substituted C1-C6 alkyl. Said optional substituents of alkyl, alkenyl, cycloalkenyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl groups in Rv, Rv2, Rv4, and Rv5, are selected from the group consisting of halogen, –CN, – NO2, –N3, –SO2H, –SO3H, –OH, –ORaa, –N(Rbb)2, –N(ORcc)Rbb, –SH, –SRaa, –C(=O)Raa, –CO2H, –CHO, – CO2Raa, –OC(=O)Raa, –OCO2Raa, –C(=O)N(Rbb)2, –OC(=O)N(Rbb)2, –NRbbC(=O)Raa, –NRbbCO2Raa, – NRbbC(=O)N(Rbb)2, –C(=NRbb)Raa, –C(=NRbb)ORaa, –OC(=NRbb)Raa, –OC(=NRbb)ORaa, – C(=NRbb)N(Rbb)2, – OC(=NRbb)N(Rbb)2, –NRbbC(=NRbb)N(Rbb)2, –C(=O)NRbbSO2Raa, –NRbbSO2Raa, – SO2N(Rbb)2, –SO2Raa, – S(=O)Raa, –OS(=O)Raa, -B(ORcc)2, C1–10 alkyl, C2–10 alkenyl, C2–10 alkynyl, C3– 14 cycloalkyl, 3– to 14- membered heterocyclyl, C6–14 aryl, and 5– to 14- membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups, or two geminal hydrogens on a carbon atom are replaced with the group =O; Each instance of Raa is, independently, selected from the group consisting of C1–10 alkyl, C1– 10 perhaloalkyl, C2–10 alkenyl, C2–10 alkynyl, C3–14 carbocyclyl, 3– to 14- membered heterocyclyl, C6–14 aryl, and 5– to 14- membered heteroaryl, or two Raa groups are joined to form a 3– to 14- membered heterocyclyl or 5– to 14- membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups; each instance of Rbb is, independently, selected from the group consisting of hydrogen, –OH, – ORaa, –N(Rcc)2, –CN, –C(=O)Raa, –C(=O)N(Rcc)2, –CO2Raa, –SO2Raa, –SO2N(Rcc)2, –SORaa, C1–10 alkyl, C1–10 perhaloalkyl, C2–10 alkenyl, C2–10 alkynyl, C3–14 carbocyclyl, 3– to 14- membered heterocyclyl, C6– 14 aryl, and 5– to 14- membered heteroaryl, or two Rbb groups are joined to form a 3– to 14- membered heterocyclyl or 5– to 14- membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups; each instance of Rcc is, independently, selected from the group consisting of hydrogen, C1– 10 alkyl, C1– 10 perhaloalkyl, C2–10 alkenyl, C2–10 alkynyl, C3–14 carbocyclyl, 3– to 14- membered heterocyclyl, C6–14 aryl, and 5– to 14- membered heteroaryl, or two Rcc groups are joined to form a 3– to 14- membered heterocyclyl or 5– to 14- membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups; and each instance of Rdd is, independently, selected from the group consisting of halogen, –CN, – NO2, –N3, –SO2H, –SO3H, –OH, –OC1–6 alkyl, –ON(C1–6 alkyl)2, –N(C1–6 alkyl)2, –N(OC1–6 alkyl)(C1–
6 alkyl), – N(OH)(C1–6 alkyl), –NH(OH), –SH, –SC1–6 alkyl, –C(=O)(C1–6 alkyl), –CO2H, –CO2(C1–6 alkyl), – OC(=O)(C1–6 alkyl), –OCO2(C1–6 alkyl), –C(=O)NH2, –C(=O)N(C1–6 alkyl)2, –OC(=O)NH(C1–6 alkyl), – NHC(=O)(C1–6 alkyl), –N(C1–6 alkyl)C(=O)( C1–6 alkyl), –NHCO2(C1–6 alkyl), –NHC(=O)N(C1–6 alkyl)2, – NHC(=O)NH(C1–6 alkyl), –NHC(=O)NH2, –C(=NH)O(C1–6 alkyl),–OC(=NH)(C1–6 alkyl), –OC(=NH)OC1– 6 alkyl, –C(=NH)N(C1–6 alkyl)2, –C(=NH)NH(C1–6 alkyl), –C(=NH)NH2, –OC(=NH)N(C1–6 alkyl)2, – OC(NH)NH(C1–6 alkyl), –OC(NH)NH2, –NHC(NH)N(C1–6 alkyl)2, –NHC(=NH)NH2, –NHSO2(C1–6 alkyl), – SO2N(C1–6 alkyl)2, –SO2NH(C1–6 alkyl), –SO2NH2,–SO2C1–6 alkyl, -B(OH)2, -B(OC1–6 alkyl)2,C1–6 alkyl, C1– 6 perhaloalkyl, C2–6 alkenyl, C2–6 alkynyl, C3–10 carbocyclyl, C6–10 aryl, 3- to 10- membered heterocyclyl, and 5- to 10- membered heteroaryl; or two geminal Rdd substituents on a carbon atom may be joined to form =O. (f2) if Y is a moiety according to formula:
wherein the left empty valence is connected to A, and the right empty valence is connected to X, wherein: Xv is CRv2 or N; Yv is CRv4 or N; Zv is CRv5 or N; or Yv and Zv taken together form an optionally substituted five- to six-membered heteroaryl, or an optionally substituted five- to six-membered heterocyclyl; with the proviso that Xv, Yv, and Zv are not simultaneously N; Rv is H, -O-(optionally substituted C3-C8 cycloalkyl), -O-(optionally substituted C1-C6 alkyl), -O- (optionally substituted C6-C10 aryl), -O-(optionally substituted five- to six-membered heteroaryl), -O- (optionally substituted five- to six-membered heterocyclyl), or optionally substituted C3-C8 cycloalkyl; or Rv together with the carbon atoms to which it is shown attached and Xv form an optionally substituted five- to six-membered heterocyclyl;
Rv2 is H, optionally substituted C1-C6 alkyl, or halo; Rv4 is H, C1-C6 alkyl, cyano, or halo; or Rv4 together with the carbon atom to which it is shown attached and Zv form an optionally substituted five- to six-membered heteroaryl; Rv5 is H, C1-C6 alkyl, -NR2, or -N(R)-C(=O)-(C1-C6 alkyl); each R independently is H, or optionally substituted C1-C6 alkyl. Said optional substituents of alkyl, alkenyl, cycloalkenyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl groups in Rv, Rv2, Rv4, and Rv5, are selected from the group consisting of halogen, –CN, – NO2, –N3, –SO2H, –SO3H, –OH, –ORaa, –N(Rbb)2, –N(ORcc)Rbb, –SH, –SRaa, –C(=O)Raa, –CO2H, –CHO, – CO2Raa, –OC(=O)Raa, –OCO2Raa, –C(=O)N(Rbb)2, –OC(=O)N(Rbb)2, –NRbbC(=O)Raa, –NRbbCO2Raa, – NRbbC(=O)N(Rbb)2, –C(=NRbb)Raa, –C(=NRbb)ORaa, –OC(=NRbb)Raa, –OC(=NRbb)ORaa, – C(=NRbb)N(Rbb)2, – OC(=NRbb)N(Rbb)2, –NRbbC(=NRbb)N(Rbb)2, –C(=O)NRbbSO2Raa, –NRbbSO2Raa, – SO2N(Rbb)2, –SO2Raa, – S(=O)Raa, –OS(=O)Raa, -B(ORcc)2, C1–10 alkyl, C2–10 alkenyl, C2–10 alkynyl, C3– 14 cycloalkyl, 3– to 14- membered heterocyclyl, C6–14 aryl, and 5– to 14- membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups, or two geminal hydrogens on a carbon atom are replaced with the group =O; Each instance of Raa is, independently, selected from the group consisting of C1–10 alkyl, C1– 10 perhaloalkyl, C2–10 alkenyl, C2–10 alkynyl, C3–14 carbocyclyl, 3– to 14- membered heterocyclyl, C6–14 aryl, and 5– to 14- membered heteroaryl, or two Raa groups are joined to form a 3– to 14- membered heterocyclyl or 5– to 14- membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups; each instance of Rbb is, independently, selected from the group consisting of hydrogen, –OH, – ORaa, –N(Rcc)2, –CN, –C(=O)Raa, –C(=O)N(Rcc)2, –CO2Raa, –SO2Raa, –SO2N(Rcc)2, –SORaa, C1–10 alkyl, C1–10 perhaloalkyl, C2–10 alkenyl, C2–10 alkynyl, C3–14 carbocyclyl, 3– to 14- membered heterocyclyl, C6– 14 aryl, and 5– to 14- membered heteroaryl, or two Rbb groups are joined to form a 3– to 14- membered heterocyclyl or 5– to 14- membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups; each instance of Rcc is, independently, selected from the group consisting of hydrogen, C1– 10 alkyl, C1– 10 perhaloalkyl, C2–10 alkenyl, C2–10 alkynyl, C3–14 carbocyclyl, 3– to 14- membered heterocyclyl, C6–14 aryl, and 5– to 14- membered heteroaryl, or two Rcc groups are joined to form a 3– to 14- membered heterocyclyl or 5– to 14- membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups; and each instance of Rdd is, independently, selected from the group consisting of halogen, –CN, – NO2, –N3, –SO2H, –SO3H, –OH, –OC1–6 alkyl, –ON(C1–6 alkyl)2, –N(C1–6 alkyl)2, –N(OC1–6 alkyl)(C1–
6 alkyl), – N(OH)(C1–6 alkyl), –NH(OH), –SH, –SC1–6 alkyl, –C(=O)(C1–6 alkyl), –CO2H, –CO2(C1–6 alkyl), – OC(=O)(C1–6 alkyl), –OCO2(C1–6 alkyl), –C(=O)NH2, –C(=O)N(C1–6 alkyl)2, –OC(=O)NH(C1–6 alkyl), – NHC(=O)(C1–6 alkyl), –N(C1–6 alkyl)C(=O)( C1–6 alkyl), –NHCO2(C1–6 alkyl), –NHC(=O)N(C1–6 alkyl)2, – NHC(=O)NH(C1–6 alkyl), –NHC(=O)NH2, –C(=NH)O(C1–6 alkyl),–OC(=NH)(C1–6 alkyl), –OC(=NH)OC1– 6 alkyl, –C(=NH)N(C1–6 alkyl)2, –C(=NH)NH(C1–6 alkyl), –C(=NH)NH2, –OC(=NH)N(C1–6 alkyl)2, – OC(NH)NH(C1–6 alkyl), –OC(NH)NH2, –NHC(NH)N(C1–6 alkyl)2, –NHC(=NH)NH2, –NHSO2(C1–6 alkyl), – SO2N(C1–6 alkyl)2, –SO2NH(C1–6 alkyl), –SO2NH2,–SO2C1–6 alkyl, -B(OH)2, -B(OC1–6 alkyl)2,C1–6 alkyl, C1– 6 perhaloalkyl, C2–6 alkenyl, C2–6 alkynyl, C3–10 carbocyclyl, C6–10 aryl, 3- to 10- membered heterocyclyl, and 5- to 10- membered heteroaryl; or two geminal Rdd substituents on a carbon atom may be joined to form =O. (f2) if Y is a moiety according to formula:
 wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, Ry2 is -H, and Ry3 is selected from C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible, then B is selected from: a 5-membered heteroarylene, optionally substituted with one or more R2, a 6-membered heteroarylene, oxidised in the ring and optionally substituted with one or more R2, preferably selected from
wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, Ry2 is -H, and Ry3 is selected from C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible, then B is selected from: a 5-membered heteroarylene, optionally substituted with one or more R2, a 6-membered heteroarylene, oxidised in the ring and optionally substituted with one or more R2, preferably selected from
 , optionally substituted with one or more R2, and
, optionally substituted with one or more R2, and
 , optionally substituted with one or more R2, wherein RD1 is selected from H and C1-6 alkyl (such as methyl), wherein the left empty valence is connected to A, and the right empty valence is connected to X, a 6-membered heteroarylene or phenylene, wherein said heteroarylene and said phenylene are attached to A and X respectively at the opposite positions of the ring (i.e., 1,4 positions), wherein:
said heteroarylene and said phenylene are each optionally substituted with at least one group selected from -OH, -COOH, -COO(C1-5 alkyl), -CONH2, -CONH(C1-5 alkyl) and -CON(C1-5 alkyl)(C1-5 alkyl) and/or said heteroarylene and said phenylene are each optionally substituted at a ring position adjacent to the point of attachment of said heteroarylene or said phenylene to X with at least one group selected from halo and -CN, and/or said heteroarylene and said phenylene are each optionally substituted at a ring position adjacent to the point of attachment of said heteroarylene or said phenylene to A with at least one group selected from -NH2, NH(C1-5 alkyl), and -N(C1-5 alkyl)(C1-5 alkyl). Accordingly, in one embodiment, B is a 5-membered heteroarylene, optionally substituted with one or more R2. In one embodiment, B is a 6-membered heteroarylene, oxidised in the ring and optionally substituted with one or more R2, preferably selected from
, optionally substituted with one or more R2, wherein RD1 is selected from H and C1-6 alkyl (such as methyl), wherein the left empty valence is connected to A, and the right empty valence is connected to X, a 6-membered heteroarylene or phenylene, wherein said heteroarylene and said phenylene are attached to A and X respectively at the opposite positions of the ring (i.e., 1,4 positions), wherein:
said heteroarylene and said phenylene are each optionally substituted with at least one group selected from -OH, -COOH, -COO(C1-5 alkyl), -CONH2, -CONH(C1-5 alkyl) and -CON(C1-5 alkyl)(C1-5 alkyl) and/or said heteroarylene and said phenylene are each optionally substituted at a ring position adjacent to the point of attachment of said heteroarylene or said phenylene to X with at least one group selected from halo and -CN, and/or said heteroarylene and said phenylene are each optionally substituted at a ring position adjacent to the point of attachment of said heteroarylene or said phenylene to A with at least one group selected from -NH2, NH(C1-5 alkyl), and -N(C1-5 alkyl)(C1-5 alkyl). Accordingly, in one embodiment, B is a 5-membered heteroarylene, optionally substituted with one or more R2. In one embodiment, B is a 6-membered heteroarylene, oxidised in the ring and optionally substituted with one or more R2, preferably selected from
 , optionally substituted with one or more
, optionally substituted with one or more
 optionally substituted with one or more R2, wherein RD1 is selected from H and C1-6 alkyl (such as methyl), wherein the left empty valence is connected to A, and the right empty valence is connected to X. In one embodiment, B is a 6-membered heteroarylene or phenylene, wherein said heteroarylene and said phenylene are attached to A and X respectively at the opposite positions of the ring (i.e., 1,4 positions), wherein: said heteroarylene and said phenylene are each optionally substituted with at least one group selected from -OH, -COOH, -COO(C1-5 alkyl), -CONH2, -CONH(C1-5 alkyl) and -CON(C1-5 alkyl)(C1-5 alkyl) and/or said heteroarylene and said phenylene are each optionally substituted at a ring position adjacent to the point of attachment of said heteroarylene or said phenylene to X with at least one group selected from halo and -CN, and/or said heteroarylene and said phenylene are each optionally substituted at a ring position adjacent to the point of attachment of said heteroarylene or said phenylene to A with at least one group selected from -NH2, NH(C1-5 alkyl), and -N(C1-5 alkyl)(C1-5 alkyl). The compound of formula (I) may be a compound according to any one of the following specific embodiments. In the following description of each embodiment, not every variable group of formula (I) is
explicitly discussed. It is to be understood that all the remaining variable groups are as in the definition of the compound of formula (I), including any preferred definitions and embodiments. Accordingly, for example, while in a first specific embodiment of the compound of formula (I), as discussed hereinbelow, only A is discussed in detail, it is to be understood that in this first specific embodiment of the compound of formula (I) B, X and Y are as in formula (I), including any of provided preferred definitions as well as any of the specific embodiments of the compound pf formula (I). In a first specific embodiment of the compound of formula (I), A is selected from aryl, heteroaryl, cycloalkyl, and cycloalkenyl wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1. In this first specific embodiment, A is preferably selected from aryl and heteroaryl, wherein said aryl and said heteroaryl are each optionally substituted with one or more R1. More preferably, in this first specific embodiment of the compound of formula (I), A is aryl, optionally substituted with one or more R1. Particularly suitable aryl group is phenyl. Accordingly, even more preferably A is phenyl, optionally substituted with one or more R1 (such as C1-6 alkyl and C1-6 haloalkyl). Particularly preferred A in this first specific embodiment of the present invention are selected
optionally substituted with one or more R2, wherein RD1 is selected from H and C1-6 alkyl (such as methyl), wherein the left empty valence is connected to A, and the right empty valence is connected to X. In one embodiment, B is a 6-membered heteroarylene or phenylene, wherein said heteroarylene and said phenylene are attached to A and X respectively at the opposite positions of the ring (i.e., 1,4 positions), wherein: said heteroarylene and said phenylene are each optionally substituted with at least one group selected from -OH, -COOH, -COO(C1-5 alkyl), -CONH2, -CONH(C1-5 alkyl) and -CON(C1-5 alkyl)(C1-5 alkyl) and/or said heteroarylene and said phenylene are each optionally substituted at a ring position adjacent to the point of attachment of said heteroarylene or said phenylene to X with at least one group selected from halo and -CN, and/or said heteroarylene and said phenylene are each optionally substituted at a ring position adjacent to the point of attachment of said heteroarylene or said phenylene to A with at least one group selected from -NH2, NH(C1-5 alkyl), and -N(C1-5 alkyl)(C1-5 alkyl). The compound of formula (I) may be a compound according to any one of the following specific embodiments. In the following description of each embodiment, not every variable group of formula (I) is
explicitly discussed. It is to be understood that all the remaining variable groups are as in the definition of the compound of formula (I), including any preferred definitions and embodiments. Accordingly, for example, while in a first specific embodiment of the compound of formula (I), as discussed hereinbelow, only A is discussed in detail, it is to be understood that in this first specific embodiment of the compound of formula (I) B, X and Y are as in formula (I), including any of provided preferred definitions as well as any of the specific embodiments of the compound pf formula (I). In a first specific embodiment of the compound of formula (I), A is selected from aryl, heteroaryl, cycloalkyl, and cycloalkenyl wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1. In this first specific embodiment, A is preferably selected from aryl and heteroaryl, wherein said aryl and said heteroaryl are each optionally substituted with one or more R1. More preferably, in this first specific embodiment of the compound of formula (I), A is aryl, optionally substituted with one or more R1. Particularly suitable aryl group is phenyl. Accordingly, even more preferably A is phenyl, optionally substituted with one or more R1 (such as C1-6 alkyl and C1-6 haloalkyl). Particularly preferred A in this first specific embodiment of the present invention are selected
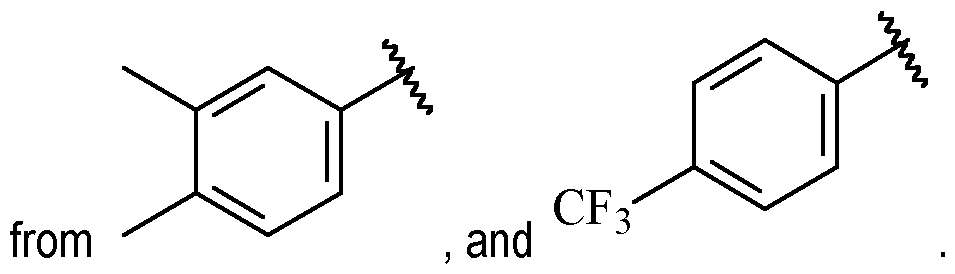 In a second specific embodiment of the compound of formula (I), A is selected from heterocycloalkyl and heterocycloalkenyl, wherein said heterocycloalkyl and said heterocycloalkenyl are each optionally substituted with one or more R1. Particularly suitable heterocycloalkyl and heterocycloalkenyl are heterocycloalkyl and heterocycloalkenyl consisting of a single 5 or 6-membered ring (preferably 6-membered), and optionally substituted with one or more R1. Examples of particularly preferred A in this second specific embodiment include
In a second specific embodiment of the compound of formula (I), A is selected from heterocycloalkyl and heterocycloalkenyl, wherein said heterocycloalkyl and said heterocycloalkenyl are each optionally substituted with one or more R1. Particularly suitable heterocycloalkyl and heterocycloalkenyl are heterocycloalkyl and heterocycloalkenyl consisting of a single 5 or 6-membered ring (preferably 6-membered), and optionally substituted with one or more R1. Examples of particularly preferred A in this second specific embodiment include
 . In a third specific embodiment of the compound of formula (I), A is C2 alkynyl, optionally substituted with C1-6 alkyl or C1-6 haloalkyl, such as C1-6 alkyl. Particularly suitable C1-6 alkyl are methyl
and isopropyl. Thus, in this third specific embodiment of the compound of formula (I), A is selected from
. In a third specific embodiment of the compound of formula (I), A is C2 alkynyl, optionally substituted with C1-6 alkyl or C1-6 haloalkyl, such as C1-6 alkyl. Particularly suitable C1-6 alkyl are methyl
and isopropyl. Thus, in this third specific embodiment of the compound of formula (I), A is selected from
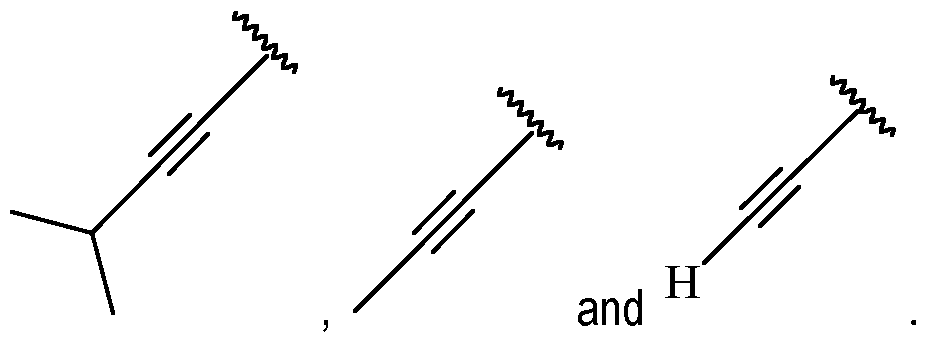 In a fourth specific embodiment of the compound of formula
In a fourth specific embodiment of the compound of formula
 optionally substituted with one or more R2, wherein RD1 is selected from H and C1-6 alkyl (such as methyl). Preferably, RD1 is H. In this fourth specific embodiment, it is preferred that the heteroarylene moiety bears no further substitutions. Preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X. In a fifth specific embodiment of the compound of formula
optionally substituted with one or more R2, wherein RD1 is selected from H and C1-6 alkyl (such as methyl). Preferably, RD1 is H. In this fourth specific embodiment, it is preferred that the heteroarylene moiety bears no further substitutions. Preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X. In a fifth specific embodiment of the compound of formula
 , optionally substituted with one or more R2. In this fifth specific embodiment, it is preferred that the heteroarylene moiety bears no further substitutions. Alternatively, is optionally substituted with methyl, -OH or F. Particularly preferred B bearing such substitution in this fifth specific embodiment are selected from
, optionally substituted with one or more R2. In this fifth specific embodiment, it is preferred that the heteroarylene moiety bears no further substitutions. Alternatively, is optionally substituted with methyl, -OH or F. Particularly preferred B bearing such substitution in this fifth specific embodiment are selected from

 . Preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X.
In a sixth specific embodiment of the compound of formula , optionally substituted with one or more R2. In this sixth specific embodiment, it is preferred that the heteroarylene moiety bears no further substitutions. Preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X. In a seventh specific embodiment of the compound of formula
. Preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X.
In a sixth specific embodiment of the compound of formula , optionally substituted with one or more R2. In this sixth specific embodiment, it is preferred that the heteroarylene moiety bears no further substitutions. Preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X. In a seventh specific embodiment of the compound of formula
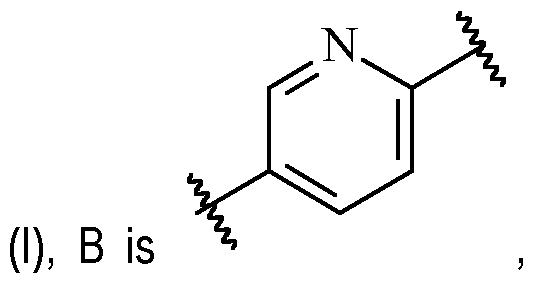 optionally substituted with one or more R2. In this seventh specific embodiment, it is preferred that the heteroarylene moiety bears no further substitutions. Preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X. In an eighth specific embodiment of the compound of formula
optionally substituted with one or more R2. In this seventh specific embodiment, it is preferred that the heteroarylene moiety bears no further substitutions. Preferably the left side of B as shown in the specific structural formulas, is connected to A, and the rights side of B is connected to X. In an eighth specific embodiment of the compound of formula
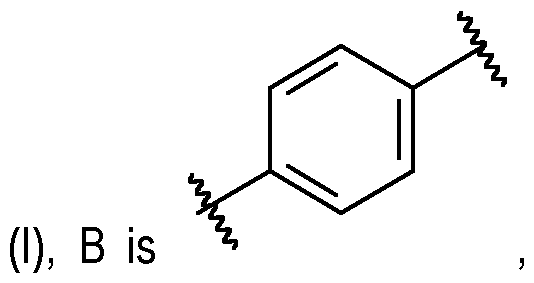 optionally substituted with one or more R2. In this eighth specific embodiment, it is preferred that the heteroarylene moiety bears no further substitutions. In a ninth specific embodiment of the compound of formula (I), X is -CONH-, wherein the C atom is connected to B and the N atom is connected to Y, C2 alkenylene optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl, or 1,2-cyclopropylene optionally substituted with one or more C1-6 alkyl. It is to be understood that, preferably, the left side of the bivalent formula embodying X, as shown herein, is connected to B, and the right side of said bivalent formula embodying X is connected to Y. In a tenth specific embodiment of the compound of formula
optionally substituted with one or more R2. In this eighth specific embodiment, it is preferred that the heteroarylene moiety bears no further substitutions. In a ninth specific embodiment of the compound of formula (I), X is -CONH-, wherein the C atom is connected to B and the N atom is connected to Y, C2 alkenylene optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl, or 1,2-cyclopropylene optionally substituted with one or more C1-6 alkyl. It is to be understood that, preferably, the left side of the bivalent formula embodying X, as shown herein, is connected to B, and the right side of said bivalent formula embodying X is connected to Y. In a tenth specific embodiment of the compound of formula
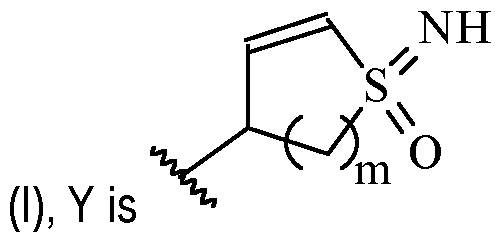 , wherein m is 1 or 2, preferably wherein m is 1.
In an eleventh specific embodiment of the compound of formula
, wherein m is 1 or 2, preferably wherein m is 1.
In an eleventh specific embodiment of the compound of formula
 alkyl)
alkyl)
 , wherein m is 1 or 2, preferably wherein m is 1. Preferably, in this eleventh alkyl) specific embodiment,
, wherein m is 1 or 2, preferably wherein m is 1. Preferably, in this eleventh alkyl) specific embodiment,
 . In a twelfth specific embodiment of the compound of formula
. In a twelfth specific embodiment of the compound of formula

 In a thirteenth specific embodiment of the compound of formula (I), Y is selected from -SO2(C2 alkenyl), -CO-(C2 alkenyl), and -CH2-(C2 alkenyl), wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH(C1-4 alkyl), and -SO2(C1-4 alkyl). In a thirteenth specific embodiment of the compound of formula (I), Y is –(C1-2 alkylene)CN. Preferably, Y is -CH2CN or -CH(CH3)CN. In a fourteenth specific embodiment of the compound of formula (I), A and B is as defined for formula (I), and X and Y form a two ring spiro system, defined as:
In a thirteenth specific embodiment of the compound of formula (I), Y is selected from -SO2(C2 alkenyl), -CO-(C2 alkenyl), and -CH2-(C2 alkenyl), wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH(C1-4 alkyl), and -SO2(C1-4 alkyl). In a thirteenth specific embodiment of the compound of formula (I), Y is –(C1-2 alkylene)CN. Preferably, Y is -CH2CN or -CH(CH3)CN. In a fourteenth specific embodiment of the compound of formula (I), A and B is as defined for formula (I), and X and Y form a two ring spiro system, defined as:
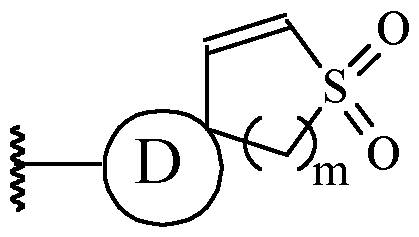 wherein D is a nitrogen-containing heterocycloalkyl or heterocycloalkenyl, m is 1 or 2, and the
wherein D is a nitrogen-containing heterocycloalkyl or heterocycloalkenyl, m is 1 or 2, and the
 optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). In a fifteenth specific embodiment of the compound of formula (I), A and Y are as defined for formula (I), B and X are combined to form a two-ring system according to formula:
optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F). In a fifteenth specific embodiment of the compound of formula (I), A and Y are as defined for formula (I), B and X are combined to form a two-ring system according to formula:
 wherein C is aryl or heteroaryl ring, optionally substituted with one or more R2, Z is selected from C1-2 alkylene, -CH=CH-, -N=CH-, -CH=N-, -CO-, -SO-, -SO2-, -CONH-, -NHCO-, -CONH(C1-6 alkyl)-, - NH(C1-6 alkyl)CO-, -NH-, -N(C1-6 alkyl)- and -O-, and Y is as defined for formula (I). In a sixteenth specific embodiment of the compound of formula (I), Y is selected from -SO2(C2 alkenyl), -CO(C2 alkenyl), -CH2(C2 alkenyl), and –(C1-2 alkylene)CN, wherein said -CH2- is optionally substituted with C1-4 alkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2H, -SO2(C1-4 alkyl), -SO2NH2, - SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, -SO2-cycloalkyl, heterocycloalkyl, -SO2-heterocycloalkyl, aryl, -SO2-aryl, heteroaryl, -SO2-heteroaryl, -Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl). In a seventeenth specific embodiment of the compound of formula (I), Y is -CH2(C2 alkenyl), wherein said -CH2- is optionally substituted with C1-4 alkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1- 4 alkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2H, -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, -SO2-cycloalkyl, heterocycloalkyl, -SO2-
heterocycloalkyl, aryl, -SO2-aryl, heteroaryl, -SO2-heteroaryl, -Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), - CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl). Preferably, in this seventeenth specific embodiment, Y is a moiety according to formula:
wherein C is aryl or heteroaryl ring, optionally substituted with one or more R2, Z is selected from C1-2 alkylene, -CH=CH-, -N=CH-, -CH=N-, -CO-, -SO-, -SO2-, -CONH-, -NHCO-, -CONH(C1-6 alkyl)-, - NH(C1-6 alkyl)CO-, -NH-, -N(C1-6 alkyl)- and -O-, and Y is as defined for formula (I). In a sixteenth specific embodiment of the compound of formula (I), Y is selected from -SO2(C2 alkenyl), -CO(C2 alkenyl), -CH2(C2 alkenyl), and –(C1-2 alkylene)CN, wherein said -CH2- is optionally substituted with C1-4 alkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2H, -SO2(C1-4 alkyl), -SO2NH2, - SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, -SO2-cycloalkyl, heterocycloalkyl, -SO2-heterocycloalkyl, aryl, -SO2-aryl, heteroaryl, -SO2-heteroaryl, -Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl). In a seventeenth specific embodiment of the compound of formula (I), Y is -CH2(C2 alkenyl), wherein said -CH2- is optionally substituted with C1-4 alkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1- 4 alkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2H, -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, -SO2-cycloalkyl, heterocycloalkyl, -SO2-
heterocycloalkyl, aryl, -SO2-aryl, heteroaryl, -SO2-heteroaryl, -Hal, -CN and -CF3, preferably substituted with one or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), - CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from C1-4 alkyl, -CONH-(C1-4 alkyl), and -SO2-(C1-4 alkyl). Preferably, in this seventeenth specific embodiment, Y is a moiety according to formula:
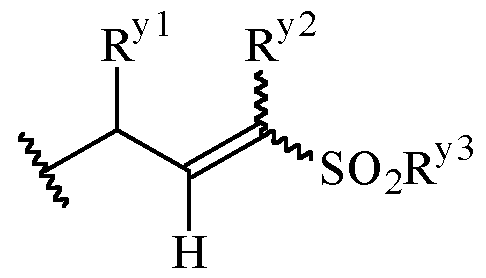 wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, Ry2 is selected from -H, -F and -CN (preferably Ry2 is -H), and Ry3 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible. In this specific embodiment, Y is preferably selected from:
wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, Ry2 is selected from -H, -F and -CN (preferably Ry2 is -H), and Ry3 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible. In this specific embodiment, Y is preferably selected from:
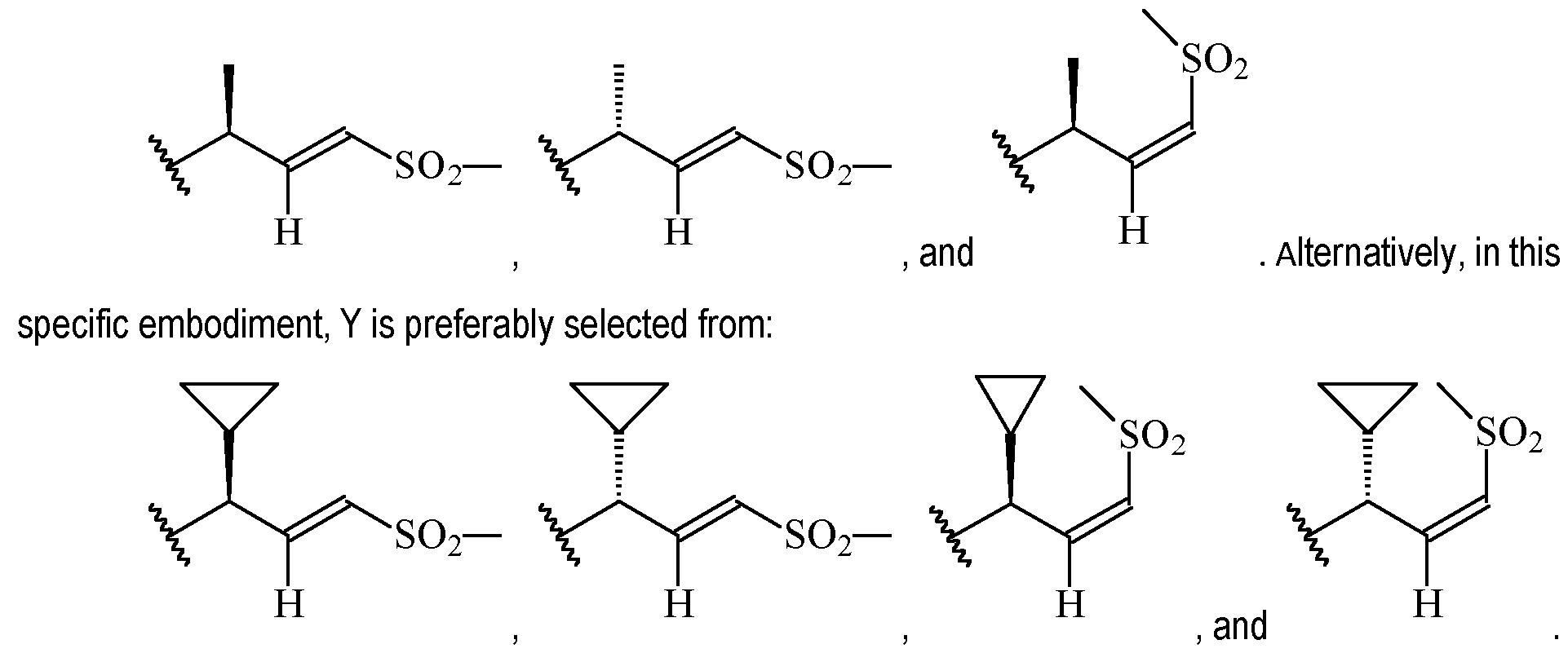 In eighteenth specific embodiment of the compound of formula (I), Y is according to formula:
In eighteenth specific embodiment of the compound of formula (I), Y is according to formula:
 or according to formula:
wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and Ry2 is selected from -H, -F and -CN (preferably Ry2 is H), and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible. In a nineteenth specific embodiment of the compound of formula (I), A is a cycloalkenyl. For example, A may be a moiety according to formula:
or according to formula:
wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and Ry2 is selected from -H, -F and -CN (preferably Ry2 is H), and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible. In a nineteenth specific embodiment of the compound of formula (I), A is a cycloalkenyl. For example, A may be a moiety according to formula:
 . In a twentieth specific embodiment of the compound of formula (I), A is heteroaryl, optionally substituted with one or more R1. Preferably, in this twentieth specific embodiment, A is selected from
. In a twentieth specific embodiment of the compound of formula (I), A is heteroaryl, optionally substituted with one or more R1. Preferably, in this twentieth specific embodiment, A is selected from
 In a twenty-first specific embodiment of the compound of formula (I), Y is a moiety according to formula:
In a twenty-first specific embodiment of the compound of formula (I), Y is a moiety according to formula:
 Wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, Ry2 is -F, and Ry3 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible. In this twenty-first specific
In a twenty-second specific embodiment of the compound of formula (I), Y is a moiety according to formula:
Wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, Ry2 is -F, and Ry3 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible. In this twenty-first specific
In a twenty-second specific embodiment of the compound of formula (I), Y is a moiety according to formula:
 wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and Ry2 is selected from -H, -F and -CN (preferably Ry2 is -H), and Ry3 is selected from C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, preferably wherein Ry3 is C1-4 alkyl, preferably methyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible. In this twenty-second
wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and Ry2 is selected from -H, -F and -CN (preferably Ry2 is -H), and Ry3 is selected from C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, preferably wherein Ry3 is C1-4 alkyl, preferably methyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible. In this twenty-second
 second specific embodiment of the compound of formula (I), A is preferably -CF2CH3. In a twenty-third specific embodiment of the compound of formula (I), Y is a moiety according to formula:
second specific embodiment of the compound of formula (I), A is preferably -CF2CH3. In a twenty-third specific embodiment of the compound of formula (I), Y is a moiety according to formula:
 wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and Ry2 is selected from -H, -F and -CN (preferably Ry2 is -H), and Ry3 is selected from C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, preferably wherein Ry3 is C1-4 alkyl, preferably methyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible. In this twenty-second
wherein Ry1 is selected from H, C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, and Ry2 is selected from -H, -F and -CN (preferably Ry2 is -H), and Ry3 is selected from C1-4 alkyl (such as methyl), C3-8 cycloalkyl (such as cyclopropyl), and 5 or 6-membered heterocyclyl, preferably wherein Ry3 is C1-4 alkyl, preferably methyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible. In this twenty-second
 (such as
(such as
 (selected from
(selected from

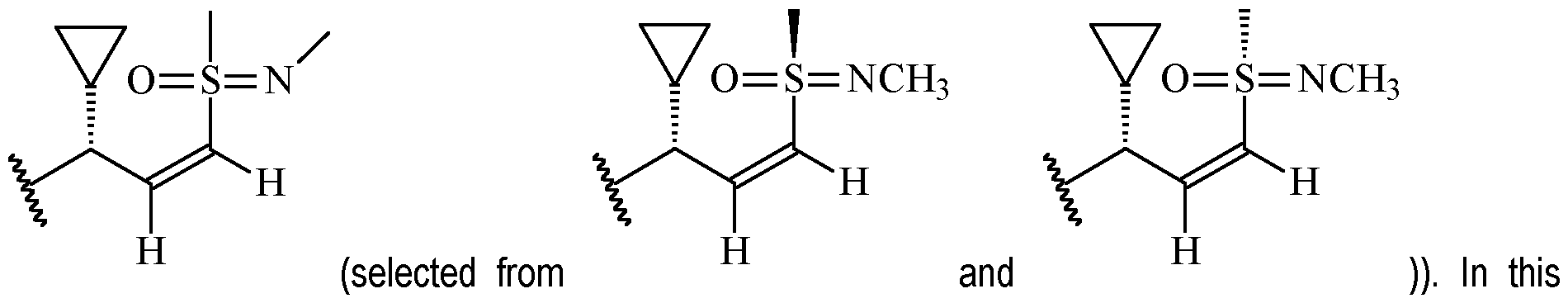 twenty third specific embodiment of the compound of formula (I), A is preferably -CF2CH3. In a twenty-fourth specific embodiment of the compound of formula (I), B is 2,5-pyridinylene, optionally substituted with -O-(C0-3 alkylene)-carbocyclyl, or -O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or -O-heterocyclyl, more preferably with -O-carbocyclyl, in particular -O-phenyl. Preferably, said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X. In a twenty-fifth specific embodiment of the compound of formula (I), B is 2,5-pyrimidinylene, optionally substituted with -O-(C0-3 alkylene)-carbocyclyl, or -O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or -O-heterocyclyl, more preferably with -O-carbocyclyl, in particular -O-phenyl. Preferably, said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X, or, in other words, at position 4 in the 2,5-pyrimidinylene. In a twenty-sixth specific embodiment of the compound of formula (I), Y is -CH2(C2 alkenyl), wherein said -CH2- is optionally substituted with C1-4 alkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1- 4 alkyl, C1-4 haloalkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO3H, -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), -SO2NH2, - SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, -SO2-cycloalkyl, -SO(=NH)-cycloalkyl, -SO(=N-(C1-4 alkyl))-cycloalkyl, heterocycloalkyl, -SO2-heterocycloalkyl, -SO(=NH)-heterocycloalkyl, -SO(=N-(C1-4 alkyl))- heterocycloalkyl, aryl, -SO2-aryl, -SO(=NH)-aryl, -SO(=N-(C1-4 alkyl))-aryl, heteroaryl, -SO2-heteroaryl, - SO(=NH)-heteroaryl, -SO(=N-(C1-4 alkyl))-heteroaryl,-Hal, -CN and -CF3, preferably substituted with one
or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), - SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from -SO2(C1-4 alkyl), -SO(=NH)- (C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), even more preferably substituted with one or more optional substituents selected from -SO(=NH)-(C1-4 alkyl) and -SO(=N-(C1-4 alkyl))-(C1-4 alkyl). Particularly suitable C1-4 alkyl is methyl. In a twenty-seventh specific embodiment of the compound of formula (I), X is selected from heterocycloalkylene, heterocycloalkenylene and heteroarylene, and Y is -CH=CH-SO2-(C1-4 alkyl). Preferably, in this twenty-seventh specific embodiment, said heterocycloalkylene is a nitrogen-containing five-membered heterocycloalkylene, said heterocycloalkenylene is a nitrogen-containing five-membered heterocycloalkenylene, and said heteroarylene is a nitrogen-containing five-membered heteroarylene. Particularly suitable heterocycloalkenylene
twenty third specific embodiment of the compound of formula (I), A is preferably -CF2CH3. In a twenty-fourth specific embodiment of the compound of formula (I), B is 2,5-pyridinylene, optionally substituted with -O-(C0-3 alkylene)-carbocyclyl, or -O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or -O-heterocyclyl, more preferably with -O-carbocyclyl, in particular -O-phenyl. Preferably, said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X. In a twenty-fifth specific embodiment of the compound of formula (I), B is 2,5-pyrimidinylene, optionally substituted with -O-(C0-3 alkylene)-carbocyclyl, or -O-(C0-3 alkylene)-heterocyclyl, preferably with -O-carbocyclyl, or -O-heterocyclyl, more preferably with -O-carbocyclyl, in particular -O-phenyl. Preferably, said substitution is at the carbon atom in the ring adjacent to the carbon atom connected to X, or, in other words, at position 4 in the 2,5-pyrimidinylene. In a twenty-sixth specific embodiment of the compound of formula (I), Y is -CH2(C2 alkenyl), wherein said -CH2- is optionally substituted with C1-4 alkyl, C3-8 cycloalkyl or 5 or 6-membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1- 4 alkyl, C1-4 haloalkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO3H, -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), -SO2NH2, - SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N- heterocycloalkyl), cycloalkyl, -SO2-cycloalkyl, -SO(=NH)-cycloalkyl, -SO(=N-(C1-4 alkyl))-cycloalkyl, heterocycloalkyl, -SO2-heterocycloalkyl, -SO(=NH)-heterocycloalkyl, -SO(=N-(C1-4 alkyl))- heterocycloalkyl, aryl, -SO2-aryl, -SO(=NH)-aryl, -SO(=N-(C1-4 alkyl))-aryl, heteroaryl, -SO2-heteroaryl, - SO(=NH)-heteroaryl, -SO(=N-(C1-4 alkyl))-heteroaryl,-Hal, -CN and -CF3, preferably substituted with one
or more optional substituents selected from C1-4 alkyl, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), - SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and -CF3, more preferably substituted with one or more optional substituents selected from -SO2(C1-4 alkyl), -SO(=NH)- (C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), even more preferably substituted with one or more optional substituents selected from -SO(=NH)-(C1-4 alkyl) and -SO(=N-(C1-4 alkyl))-(C1-4 alkyl). Particularly suitable C1-4 alkyl is methyl. In a twenty-seventh specific embodiment of the compound of formula (I), X is selected from heterocycloalkylene, heterocycloalkenylene and heteroarylene, and Y is -CH=CH-SO2-(C1-4 alkyl). Preferably, in this twenty-seventh specific embodiment, said heterocycloalkylene is a nitrogen-containing five-membered heterocycloalkylene, said heterocycloalkenylene is a nitrogen-containing five-membered heterocycloalkenylene, and said heteroarylene is a nitrogen-containing five-membered heteroarylene. Particularly suitable heterocycloalkenylene
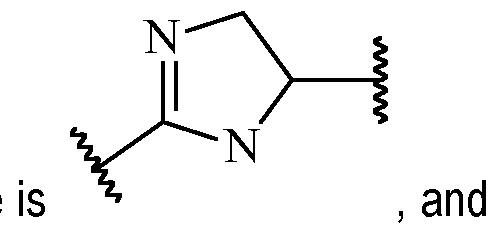 particularly suitable heteroarylene is selected from
particularly suitable heteroarylene is selected from
 understood that, preferably, the left side of the bivalent formula embodying X, as shown herein, is connected to B, and the right side of said bivalent formula embodying X is connected to Y. Further in this twenty-seventh specific embodiment, particularly suitable C1-4 alkyl in Y is methyl. In one particular embodiment, the compound of formula (I) is a compound of formula (Ia):
understood that, preferably, the left side of the bivalent formula embodying X, as shown herein, is connected to B, and the right side of said bivalent formula embodying X is connected to Y. Further in this twenty-seventh specific embodiment, particularly suitable C1-4 alkyl in Y is methyl. In one particular embodiment, the compound of formula (I) is a compound of formula (Ia):
 wherein A and B are as defined for formula (I), including any preferred definition and definition in any of the specific embodiments of the compound of formula (I). In one particular embodiment, the compound of formula (I) is a compound of formula (Ib):
wherein A and Y are as defined for formula (I), including any preferred definition and definition in any of the specific embodiments of the compound of formula (I). In one particular embodiment, the compound of formula (I) is a compound of formula (Ic):
wherein A and B are as defined for formula (I), including any preferred definition and definition in any of the specific embodiments of the compound of formula (I). In one particular embodiment, the compound of formula (I) is a compound of formula (Ib):
wherein A and Y are as defined for formula (I), including any preferred definition and definition in any of the specific embodiments of the compound of formula (I). In one particular embodiment, the compound of formula (I) is a compound of formula (Ic):
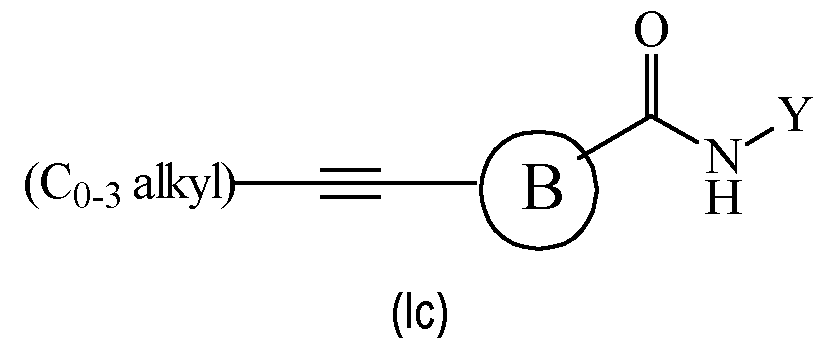 wherein B and Y are as defined for formula (I), including any preferred definition and definition in any of the specific embodiments of the compound of formula (I). Particularly preferred compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts:
wherein B and Y are as defined for formula (I), including any preferred definition and definition in any of the specific embodiments of the compound of formula (I). Particularly preferred compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts:
 , ,
, , , ,5 ,
, , , ,5 , , ,
5 , , , ,
5 , , , ,
5
, ,
, , , ,5 ,
, , , ,5 , , ,
5 , , , ,
5 , , , ,
5
 , , , Further preferred compounds of formula (I) are selected from the following compounds or their pharmaceutically suitable salts: 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3 S)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide 6-(3,4-dimethylphenyl)-N-((1R or S,3 S)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3 R)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R or S,3 R)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-(1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide;
5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- sulfonamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3- carboxamide; and 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-thioxo-1,2-dihydropyridine-3- carboxamide. Further suitable compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts:
, , , Further preferred compounds of formula (I) are selected from the following compounds or their pharmaceutically suitable salts: 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3 S)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide 6-(3,4-dimethylphenyl)-N-((1R or S,3 S)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3 R)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R or S,3 R)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-(1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide;
5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- sulfonamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3- carboxamide; and 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-thioxo-1,2-dihydropyridine-3- carboxamide. Further suitable compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts:
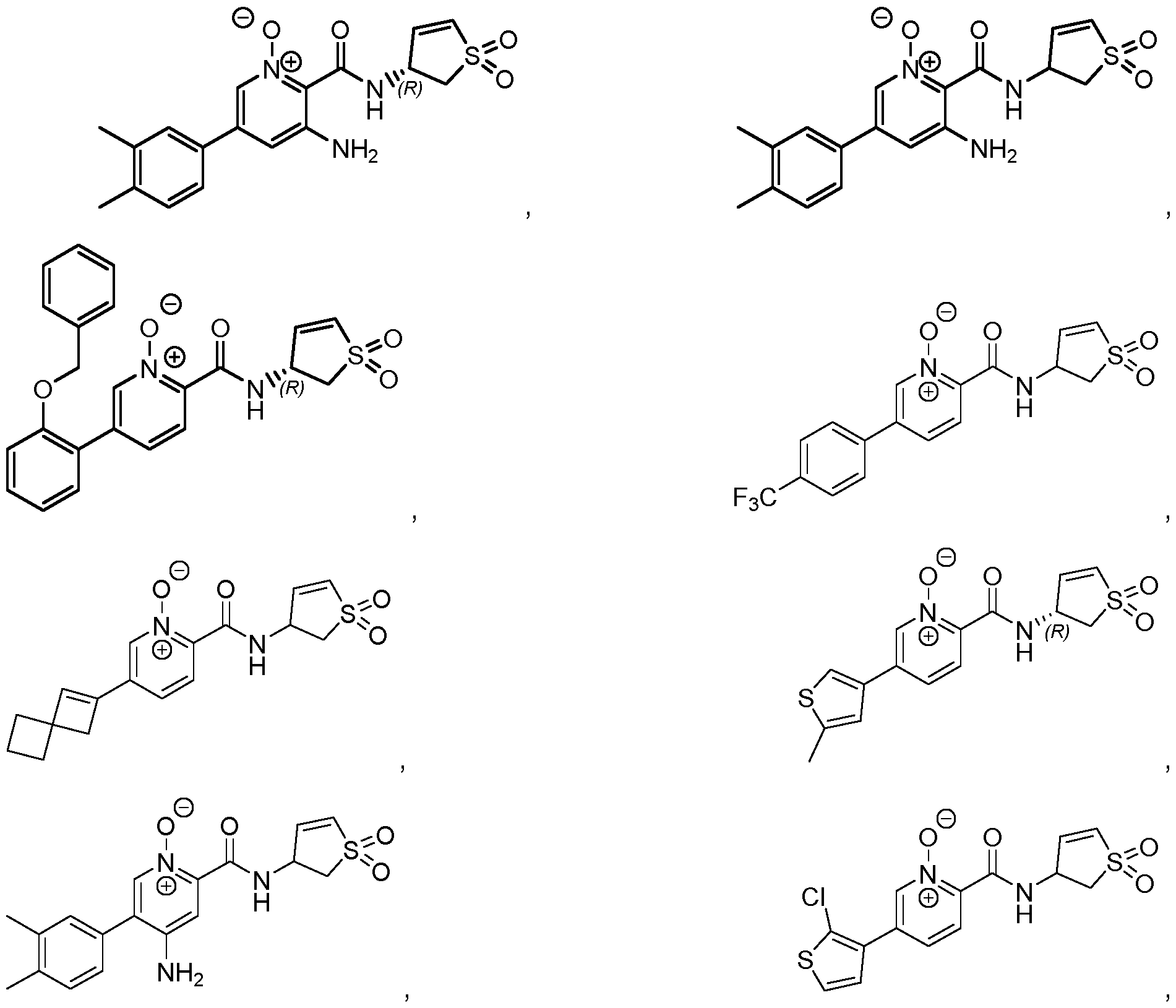 , , , 5 Further suitable compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts:
, , , 5 Further suitable compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts:
 ,
, , , Further suitable compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts:
,
, , , Further suitable compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts:
 Further suitable compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts:
Further suitable compounds of formula (I) are selected from the following compounds, or their pharmaceutically acceptable salts:

 In one embodiment, particularly suitable compound of formula
In one embodiment, particularly suitable compound of formula
 is
is
 its pharmaceutically acceptable salt.
In one embodiment, particularly suitable compound of formula (I) is
its pharmaceutically acceptable salt.
In one embodiment, particularly suitable compound of formula (I) is
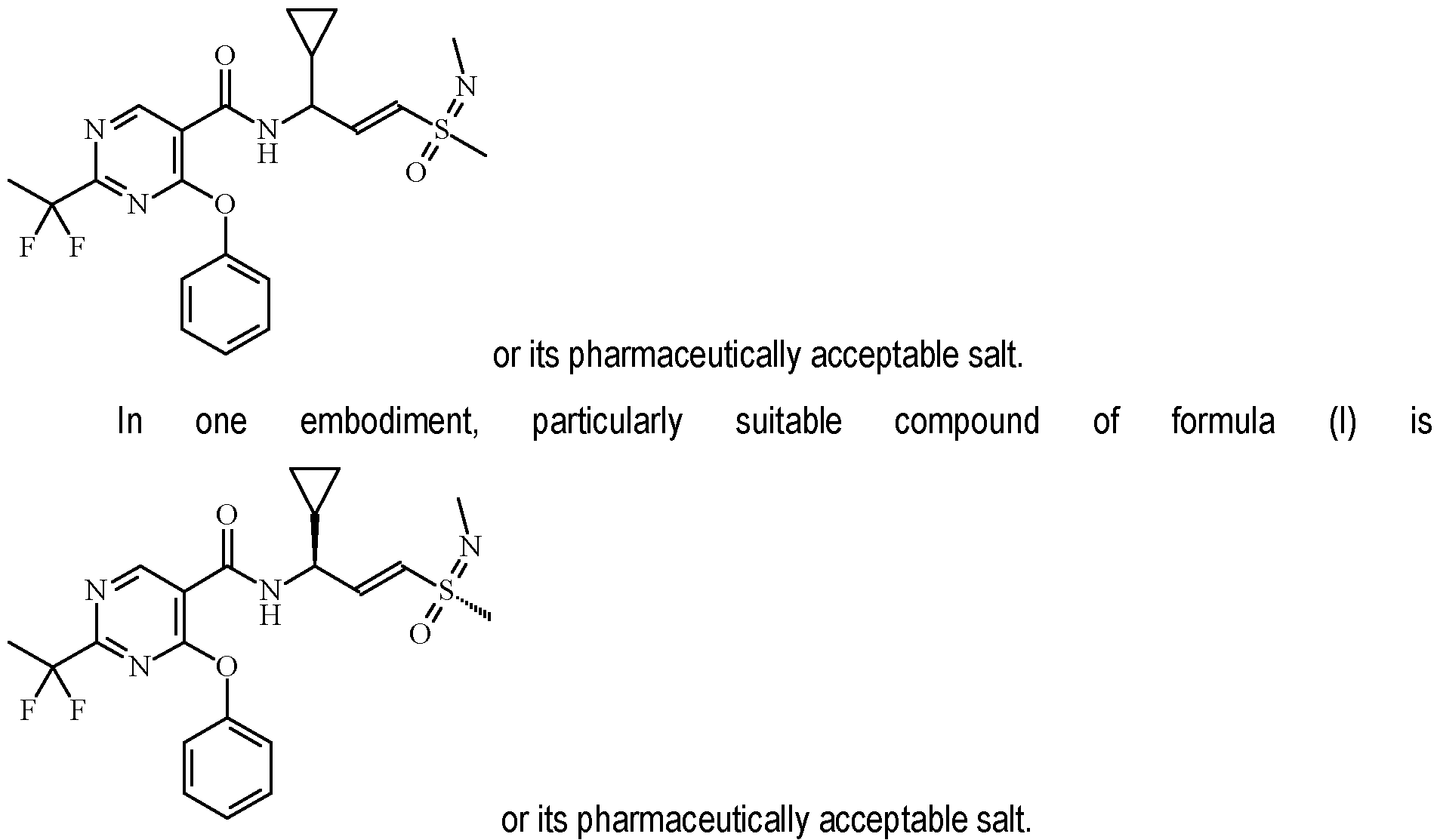 In one embodiment, particularly suitable compound of formula (I) is
In one embodiment, particularly suitable compound of formula (I) is
 In one embodiment, particularly suitable compound of formula (I) is
its pharmaceutically acceptable salt. The compound of formula (I) is preferably a compound selected from the following compounds: 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3S or R)-1-imino-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R or S,3S or R)-1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3R or S)-1-imino-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R or S,3R or S)-1-imino-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-(1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (R or S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (S or R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- sulfonamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-thioxo-1,2-dihydropyridine-3- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide;
3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-(trifluoromethyl)phenyl)pyridine 1- oxide; 4-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-5-(2-(benzyloxy)phenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-methylthiophen-3-yl)pyridine 1- oxide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(spiro[3.3]hept-1-en-2-yl)pyridine 1-oxide; 5-(2-chlorothiophen-3-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide; 3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide; (R)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide; (S)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide; (R)-3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride; (S)-3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride; (S,Z)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; (R)-5-(4,5-dimethylthiophen-2-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen-2- yl)pyridine 1-oxide; (E)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide ; (Z)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide;
(E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide; (E)-N-(1-cyclopropyl-3-(N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-ethynyl-2-oxo-1,2-dihydropyridine-3-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(pyrrolidin-1-yl)-1,2-dihydropyridine-3- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-(3-methylbut-1-yn-1-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-(3-methylbut-1-yn-1-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-5-(spiro[3.3]heptan-2-yl)picolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-1,2,4-triazole-3- carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4H-1,2,4-triazole-3- carboxamide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-methylthiophen-2-yl)pyridine 1- oxide; (R)-5-(cyclohex-1-en-1-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-cyclohexyl-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(tetrahydro-2H-pyran-3-yl)-1,2- dihydropyridine-3-carboxamide; 6-(3,4-dihydro-2H-pyran-6-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(tetrahydro-2H-pyran-2-yl)-1,2- dihydropyridine-3-carboxamide; 6-(3,6-dihydro-2H-pyran-4-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(tetrahydro-2H-pyran-4-yl)-1,2- dihydropyridine-3-carboxamide; 6-(5,6-dihydro-2H-pyran-3-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)thiazole-2-carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-phenyl-2H-pyran-3-carboxamide;
2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(piperidin-1-yl)pyridine 1-oxide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1,3,4-thiadiazole-2- carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-imidazole-2- carboxamide; (S)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1-methyl-2-(p-tolyl)-1H-imidazole-4-carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1-methyl-2-(p-tolyl)-1H-imidazole-4-carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-imidazole-4-carboxamide; (R)-5-(4-chlorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxy-6-(tetrahydro-2H-pyran-4- yl)nicotinamide; 6-(3,4-dimethylphenyl)-N-((3R)-1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)nicotinamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluorothiophene-2- carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrimidine-5-carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrimidine-2-carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyridazine-3-carboxamide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(spiro[3.3]heptan-2-yl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-3-(6,6-dioxido-6-thia-2-azaspiro[3.4]oct-7-en-2-yl)pyridin-2(1H)-one; 3-((6-(3,4-dimethylphenyl)-1H-indazol-3-yl)oxy)-2,3-dihydrothiophene 1,1-dioxide; 6-(3,4-dimethylphenyl)-1-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1,2-dihydro-3H-indazol-3-one; 5-(3,4-dimethylphenyl)-2-(1,1-dioxido-2,3-dihydrothiophen-3-yl)isoindolin-1-one; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(phenylethynyl)-1,2-dihydropyridine-3- carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-hydroxypicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-hydroxypicolinamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-oxo-1,4-dihydropyridine-3- carboxamide; (R)-5-(2,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-(3,4-dichlorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-(3,4-difluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide;
(R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-methoxyphenyl)pyridine 1-oxide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoropicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-fluoropicolinamide; 6-(3,4-dimethylphenyl)-N-(1-methyl-1-oxido-2,3-dihydrophosphol-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 3-amino-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-methylpicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-methylpicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methylpicolinamide; 2-amino-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; (S)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-(4-(trifluoromethyl)phenyl)thiazole-4- carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-(4-(trifluoromethyl)phenyl)thiazole-4- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; 3-chloro-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2,3',4'-trimethyl-[1,1'-biphenyl]-4-carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-oxo-1,6-dihydropyrimidine-5- carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-oxo-3,4-dihydropyrazine-2- carboxamide; 2-amino-6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)nicotinamide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(2,4,6-trifluorophenyl)pyridine 1- oxide; 3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-fluoropyridine 1- oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-4-fluoropyridine 1- oxide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxynicotinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxypicolinamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1-methyl-4-oxo-1,4- dihydropyridine-3-carboxamide; 3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-methylpyridine 1-
oxide; 3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-hydroxypyridine 1- oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-4-hydroxypyridine 1- oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-3-hydroxypyridine 1- oxide; (R)-5-chloro-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; (S)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-5-fluoro-2-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-5-fluoro-2-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-methoxypyrimidine-5- carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxypyrazine-2- carboxamide; 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; (E)-6-(3,4-dimethylphenyl)-3-(5-(2-(methylsulfonyl)vinyl)-1H-imidazol-2-yl)pyridin-2-ol; (R)-5-(4-cyclopropylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-5-(benzofuran-5-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-(2,3-dihydrobenzofuran-5-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-6-thia-2-azaspiro[3.4]oct-7-ene 6,6-dioxide; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)quinazolin-4(3H)-one; 2-(3,4-dimethylphenyl)-6-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-7,8-dihydro-1,6-naphthyridin- 5(6H)-one; 6-(3,4-dimethylphenyl)-2-(1,1-dioxido-2,3-dihydrothiophen-3-yl)isoquinolin-1(2H)-one;
6-(3,4-dimethylphenyl)-2-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3,4-dihydroisoquinolin-1(2H)- one; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrido[2,3-d]pyrimidin-4(3H)- one; 2-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7- dioxide; (R)-2-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (S)-2-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (R,Z)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; (R,E)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; (S,E)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; 7-(5-(4-(trifluoromethyl)phenyl)pyridin-2-yl)-2-thia-7-azaspiro[4.4]non-3-ene 2,2-dioxide; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-8-fluoroquinazolin-4(3H)-one; 7-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-2-thia-7-azaspiro[4.4]non-3-ene 2,2-dioxide; (S)-4-(3-cyclopropyl-1H-pyrazol-1-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxy-6- methylbenzamide; (R)-4-(3-cyclopropyl-1H-pyrazol-1-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxy-6- methylbenzamide; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrido[2,3-d]pyrimidine- 2,4(1H,3H)-dione; 2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7- dioxide; (R)-2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (S)-2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (R)-7-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-benzo[d]imidazole-4- carboxamide; N-(1-(cyanoimino)-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)-6-(3,4-dimethylphenyl)-2-oxo-1,2- dihydropyridine-3-carboxamide; N-(1-(cyanoimino)-1-oxido-4,5-dihydro-1H-16-thiophen-3-yl)-6-(3,4-dimethylphenyl)-2-oxo-1,2- dihydropyridine-3-carboxamide;
3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-methoxypyridine 1-oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-4-methoxypyridine 1-oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-3-methoxypyridine 1-oxide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(4-(trifluoromethyl)piperidin-1-yl)-1,2- dihydropyridine-3-carboxamide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-(trifluoromethyl)piperidin-1-yl)pyridine 1-oxide; (R)-3-amino-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen- 3-yl)pyridine 1-oxide; (R)-3-amino-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen- 2-yl)pyridine 1-oxide; (R)-3-amino-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-(trifluoromethyl)thiophen- 2-yl)pyridine 1-oxide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(3-methyl-4- (trifluoromethyl)phenyl)pyridine 1-oxide; (S,Z)-N-(1-cyclopropyl-3-(methylsulfonyl)allyl)-6-(3,4-dimethylphenyl)-2-oxo-1,2-dihydropyridine- 3-carboxamide; (S,E)-N-(1-cyclopropyl-3-(methylsulfonyl)allyl)-6-(3,4-dimethylphenyl)-2-oxo-1,2-dihydropyridine- 3-carboxamide; (S,Z)-2-((1-cyclopropyl-3-(methylsulfonyl)allyl)carbamoyl)-5-(3,4-dimethylphenyl)pyridine 1- oxide; (S,E)-2-((1-cyclopropyl-3-(methylsulfonyl)allyl)carbamoyl)-5-(3,4-dimethylphenyl)pyridine 1- oxide; (R)-5-(2-chloro-4-(trifluoromethyl)phenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-3-(1-((1,1-dioxido-2,3-dihydrothiophen-3-yl)amino)-2,2,2- trifluoroethyl)pyridin-2(1H)-one; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-8-fluoro-2-hydroxy-2,3- dihydroquinazolin-4(1H)-one; 8-(5-(4-(trifluoromethyl)phenyl)pyridin-2-yl)-2-thia-8-azaspiro[4.5]dec-3-ene 2,2-dioxide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)quinoline-8-carboxamide;
2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-8-thia-1,3-diazaspiro[4.5]deca-2,6-diene 8,8- dioxide; 3-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-9-thia-2,4-diazaspiro[5.5]undeca-2,7-diene 9,9- dioxide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)quinazoline-8-carboxamide; 3-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-9-thia-2,4-diazaspiro[5.5]undeca-2,7-diene 9,9-dioxide; 3-(4-(6-cyclopentyl-2-phenoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1- dioxide; 2-(2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidin-5-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8- diene 7,7-dioxide; (E)-2-(cyclopropyldifluoromethyl)-5-(5-(2-(methylsulfonyl)vinyl)-1H-imidazol-2-yl)-4- phenoxypyrimidine; 3-(4-(6-(isopropyl(methyl)amino)-2-phenoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide; (R)-5-(2-(benzylamino)phenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; 2-(6-(3,4-dimethylphenyl)-2-phenoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7- dioxide; (S)-2-(6-(3,4-dimethylphenyl)-2-phenoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (R)-5-(2-(benzyloxy)-3-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-4-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-5-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-6-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-5-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-3-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-4-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-
yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-6-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-fluoro-2-(pyridin-4- ylmethoxy)phenyl)pyridine 1-oxide; or a pharmaceutically acceptable salt thereof. As further provided herein, particularly preferred is a compound selected from: N-((S,E)-1-cyclopropyl-3-((R)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide, -((S,E)-1-cyclopropyl-3-((S)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, N-((R,E)-1-cyclopropyl-3-((R)-S- methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, and N-((R,E)-1- cyclopropyl-3-((S)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, characterized by the highest retention time of these compounds in an SFC experiment according to method 13, or a pharmaceutically acceptable salt thereof. Accordingly, particularly suitable is the compound described as compound 32d, i.e., the compound of formula:
In one embodiment, particularly suitable compound of formula (I) is
its pharmaceutically acceptable salt. The compound of formula (I) is preferably a compound selected from the following compounds: 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3S or R)-1-imino-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R or S,3S or R)-1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3R or S)-1-imino-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R or S,3R or S)-1-imino-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-(1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (R or S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (S or R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- sulfonamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-thioxo-1,2-dihydropyridine-3- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide;
3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-(trifluoromethyl)phenyl)pyridine 1- oxide; 4-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-5-(2-(benzyloxy)phenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-methylthiophen-3-yl)pyridine 1- oxide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(spiro[3.3]hept-1-en-2-yl)pyridine 1-oxide; 5-(2-chlorothiophen-3-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide; 3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide; (R)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide; (S)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide; (R)-3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride; (S)-3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride; (S,Z)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; (R)-5-(4,5-dimethylthiophen-2-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen-2- yl)pyridine 1-oxide; (E)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide ; (Z)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide;
(E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide; (E)-N-(1-cyclopropyl-3-(N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-ethynyl-2-oxo-1,2-dihydropyridine-3-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(pyrrolidin-1-yl)-1,2-dihydropyridine-3- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-(3-methylbut-1-yn-1-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-(3-methylbut-1-yn-1-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-5-(spiro[3.3]heptan-2-yl)picolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-1,2,4-triazole-3- carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4H-1,2,4-triazole-3- carboxamide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-methylthiophen-2-yl)pyridine 1- oxide; (R)-5-(cyclohex-1-en-1-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-cyclohexyl-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(tetrahydro-2H-pyran-3-yl)-1,2- dihydropyridine-3-carboxamide; 6-(3,4-dihydro-2H-pyran-6-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(tetrahydro-2H-pyran-2-yl)-1,2- dihydropyridine-3-carboxamide; 6-(3,6-dihydro-2H-pyran-4-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(tetrahydro-2H-pyran-4-yl)-1,2- dihydropyridine-3-carboxamide; 6-(5,6-dihydro-2H-pyran-3-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)thiazole-2-carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-phenyl-2H-pyran-3-carboxamide;
2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(piperidin-1-yl)pyridine 1-oxide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1,3,4-thiadiazole-2- carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-imidazole-2- carboxamide; (S)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1-methyl-2-(p-tolyl)-1H-imidazole-4-carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1-methyl-2-(p-tolyl)-1H-imidazole-4-carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-imidazole-4-carboxamide; (R)-5-(4-chlorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxy-6-(tetrahydro-2H-pyran-4- yl)nicotinamide; 6-(3,4-dimethylphenyl)-N-((3R)-1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)nicotinamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluorothiophene-2- carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrimidine-5-carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrimidine-2-carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyridazine-3-carboxamide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(spiro[3.3]heptan-2-yl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-3-(6,6-dioxido-6-thia-2-azaspiro[3.4]oct-7-en-2-yl)pyridin-2(1H)-one; 3-((6-(3,4-dimethylphenyl)-1H-indazol-3-yl)oxy)-2,3-dihydrothiophene 1,1-dioxide; 6-(3,4-dimethylphenyl)-1-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1,2-dihydro-3H-indazol-3-one; 5-(3,4-dimethylphenyl)-2-(1,1-dioxido-2,3-dihydrothiophen-3-yl)isoindolin-1-one; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(phenylethynyl)-1,2-dihydropyridine-3- carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-hydroxypicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-hydroxypicolinamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-oxo-1,4-dihydropyridine-3- carboxamide; (R)-5-(2,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-(3,4-dichlorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-(3,4-difluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide;
(R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-methoxyphenyl)pyridine 1-oxide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoropicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-fluoropicolinamide; 6-(3,4-dimethylphenyl)-N-(1-methyl-1-oxido-2,3-dihydrophosphol-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 3-amino-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-methylpicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-methylpicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methylpicolinamide; 2-amino-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; (S)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-(4-(trifluoromethyl)phenyl)thiazole-4- carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-(4-(trifluoromethyl)phenyl)thiazole-4- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; 3-chloro-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2,3',4'-trimethyl-[1,1'-biphenyl]-4-carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-oxo-1,6-dihydropyrimidine-5- carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-oxo-3,4-dihydropyrazine-2- carboxamide; 2-amino-6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)nicotinamide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(2,4,6-trifluorophenyl)pyridine 1- oxide; 3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-fluoropyridine 1- oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-4-fluoropyridine 1- oxide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxynicotinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxypicolinamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1-methyl-4-oxo-1,4- dihydropyridine-3-carboxamide; 3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-methylpyridine 1-
oxide; 3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-hydroxypyridine 1- oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-4-hydroxypyridine 1- oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-3-hydroxypyridine 1- oxide; (R)-5-chloro-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; (S)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-5-fluoro-2-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-5-fluoro-2-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-methoxypyrimidine-5- carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxypyrazine-2- carboxamide; 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; (E)-6-(3,4-dimethylphenyl)-3-(5-(2-(methylsulfonyl)vinyl)-1H-imidazol-2-yl)pyridin-2-ol; (R)-5-(4-cyclopropylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-5-(benzofuran-5-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-(2,3-dihydrobenzofuran-5-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-6-thia-2-azaspiro[3.4]oct-7-ene 6,6-dioxide; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)quinazolin-4(3H)-one; 2-(3,4-dimethylphenyl)-6-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-7,8-dihydro-1,6-naphthyridin- 5(6H)-one; 6-(3,4-dimethylphenyl)-2-(1,1-dioxido-2,3-dihydrothiophen-3-yl)isoquinolin-1(2H)-one;
6-(3,4-dimethylphenyl)-2-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3,4-dihydroisoquinolin-1(2H)- one; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrido[2,3-d]pyrimidin-4(3H)- one; 2-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7- dioxide; (R)-2-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (S)-2-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (R,Z)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; (R,E)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; (S,E)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; 7-(5-(4-(trifluoromethyl)phenyl)pyridin-2-yl)-2-thia-7-azaspiro[4.4]non-3-ene 2,2-dioxide; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-8-fluoroquinazolin-4(3H)-one; 7-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-2-thia-7-azaspiro[4.4]non-3-ene 2,2-dioxide; (S)-4-(3-cyclopropyl-1H-pyrazol-1-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxy-6- methylbenzamide; (R)-4-(3-cyclopropyl-1H-pyrazol-1-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxy-6- methylbenzamide; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrido[2,3-d]pyrimidine- 2,4(1H,3H)-dione; 2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7- dioxide; (R)-2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (S)-2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (R)-7-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-benzo[d]imidazole-4- carboxamide; N-(1-(cyanoimino)-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)-6-(3,4-dimethylphenyl)-2-oxo-1,2- dihydropyridine-3-carboxamide; N-(1-(cyanoimino)-1-oxido-4,5-dihydro-1H-16-thiophen-3-yl)-6-(3,4-dimethylphenyl)-2-oxo-1,2- dihydropyridine-3-carboxamide;
3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-methoxypyridine 1-oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-4-methoxypyridine 1-oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-3-methoxypyridine 1-oxide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(4-(trifluoromethyl)piperidin-1-yl)-1,2- dihydropyridine-3-carboxamide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-(trifluoromethyl)piperidin-1-yl)pyridine 1-oxide; (R)-3-amino-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen- 3-yl)pyridine 1-oxide; (R)-3-amino-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen- 2-yl)pyridine 1-oxide; (R)-3-amino-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-(trifluoromethyl)thiophen- 2-yl)pyridine 1-oxide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(3-methyl-4- (trifluoromethyl)phenyl)pyridine 1-oxide; (S,Z)-N-(1-cyclopropyl-3-(methylsulfonyl)allyl)-6-(3,4-dimethylphenyl)-2-oxo-1,2-dihydropyridine- 3-carboxamide; (S,E)-N-(1-cyclopropyl-3-(methylsulfonyl)allyl)-6-(3,4-dimethylphenyl)-2-oxo-1,2-dihydropyridine- 3-carboxamide; (S,Z)-2-((1-cyclopropyl-3-(methylsulfonyl)allyl)carbamoyl)-5-(3,4-dimethylphenyl)pyridine 1- oxide; (S,E)-2-((1-cyclopropyl-3-(methylsulfonyl)allyl)carbamoyl)-5-(3,4-dimethylphenyl)pyridine 1- oxide; (R)-5-(2-chloro-4-(trifluoromethyl)phenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-3-(1-((1,1-dioxido-2,3-dihydrothiophen-3-yl)amino)-2,2,2- trifluoroethyl)pyridin-2(1H)-one; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-8-fluoro-2-hydroxy-2,3- dihydroquinazolin-4(1H)-one; 8-(5-(4-(trifluoromethyl)phenyl)pyridin-2-yl)-2-thia-8-azaspiro[4.5]dec-3-ene 2,2-dioxide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)quinoline-8-carboxamide;
2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-8-thia-1,3-diazaspiro[4.5]deca-2,6-diene 8,8- dioxide; 3-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-9-thia-2,4-diazaspiro[5.5]undeca-2,7-diene 9,9- dioxide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)quinazoline-8-carboxamide; 3-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-9-thia-2,4-diazaspiro[5.5]undeca-2,7-diene 9,9-dioxide; 3-(4-(6-cyclopentyl-2-phenoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1- dioxide; 2-(2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidin-5-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8- diene 7,7-dioxide; (E)-2-(cyclopropyldifluoromethyl)-5-(5-(2-(methylsulfonyl)vinyl)-1H-imidazol-2-yl)-4- phenoxypyrimidine; 3-(4-(6-(isopropyl(methyl)amino)-2-phenoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide; (R)-5-(2-(benzylamino)phenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; 2-(6-(3,4-dimethylphenyl)-2-phenoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7- dioxide; (S)-2-(6-(3,4-dimethylphenyl)-2-phenoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (R)-5-(2-(benzyloxy)-3-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-4-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-5-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-6-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-5-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-3-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-4-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-
yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-6-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-fluoro-2-(pyridin-4- ylmethoxy)phenyl)pyridine 1-oxide; or a pharmaceutically acceptable salt thereof. As further provided herein, particularly preferred is a compound selected from: N-((S,E)-1-cyclopropyl-3-((R)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide, -((S,E)-1-cyclopropyl-3-((S)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, N-((R,E)-1-cyclopropyl-3-((R)-S- methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, and N-((R,E)-1- cyclopropyl-3-((S)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, characterized by the highest retention time of these compounds in an SFC experiment according to method 13, or a pharmaceutically acceptable salt thereof. Accordingly, particularly suitable is the compound described as compound 32d, i.e., the compound of formula:
 as defined in the foregoing. As further provided herein, particularly preferred is a compound selected from: N-((S,E)-1-cyclopropyl-3-((R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide, N-((S,E)-1-cyclopropyl-3-((S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, N-((R,E)-1-cyclopropyl-3-((R)-N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, and N-((R,E)-1- cyclopropyl-3-((S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide, characterized by the second highest retention time of these compounds in an SFC experiment according to method 14. Accordingly, particularly suitable is a compound described as 33c , i.e., the compound of formula:
as defined in the foregoing. Alternatively, particularly preferred is a compound selected from: N-((S,E)-1-cyclopropyl-3-((R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide, N-((S,E)-1-cyclopropyl-3-((S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, N-((R,E)-1-cyclopropyl-3-((R)-N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, and N-((R,E)-1- cyclopropyl-3-((S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide, characterized by the highest retention time of these compounds in an SFC experiment according to method 14. Accordingly, particularly suitable is a compound described as 33d, i.e., the compound of formula:
as defined in the foregoing. As further provided herein, particularly preferred is a compound selected from: N-((S,E)-1-cyclopropyl-3-((R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide, N-((S,E)-1-cyclopropyl-3-((S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, N-((R,E)-1-cyclopropyl-3-((R)-N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, and N-((R,E)-1- cyclopropyl-3-((S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide, characterized by the second highest retention time of these compounds in an SFC experiment according to method 14. Accordingly, particularly suitable is a compound described as 33c , i.e., the compound of formula:
as defined in the foregoing. Alternatively, particularly preferred is a compound selected from: N-((S,E)-1-cyclopropyl-3-((R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide, N-((S,E)-1-cyclopropyl-3-((S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, N-((R,E)-1-cyclopropyl-3-((R)-N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxamide, and N-((R,E)-1- cyclopropyl-3-((S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide, characterized by the highest retention time of these compounds in an SFC experiment according to method 14. Accordingly, particularly suitable is a compound described as 33d, i.e., the compound of formula:
 as defined in the foregoing. For a number of compounds disclosed herein, enantiomeric forms may exist. The present invention further refers to each and every enantiomeric form of such compound individually. The present invention also relates to each of the intermediates described further below in the examples section of this specification, including any one of these intermediates in non-salt form or in the form of a salt (e.g., a pharmaceutically acceptable salt) of the respective compound. Such intermediates can be used, in particular, in the synthesis of the compounds of formula (I). The scope of the invention embraces all pharmaceutically acceptable salt forms of the compounds of formula (I) which may be formed, e.g., by protonation of an atom carrying an electron lone pair which is susceptible to protonation, such as an amino group, with an inorganic or organic acid, or as a salt of an acid group (such as a carboxylic acid group) with a physiologically acceptable cation. Exemplary base addition salts comprise, for example: alkali metal salts such as sodium or potassium salts; alkaline earth
metal salts such as calcium or magnesium salts; zinc salts; ammonium salts; aliphatic amine salts such as trimethylamine, triethylamine, dicyclohexylamine, ethanolamine, diethanolamine, triethanolamine, procaine salts, meglumine salts, ethylenediamine salts, or choline salts; aralkyl amine salts such as N,N- dibenzylethylenediamine salts, benzathine salts, benethamine salts; heterocyclic aromatic amine salts such as pyridine salts, picoline salts, quinoline salts or isoquinoline salts; quaternary ammonium salts such as tetramethylammonium salts, tetraethylammonium salts, benzyltrimethylammonium salts, benzyltriethylammonium salts, benzyltributylammonium salts, methyltrioctylammonium salts or tetrabutylammonium salts; and basic amino acid salts such as arginine salts, lysine salts, or histidine salts. Exemplary acid addition salts comprise, for example: mineral acid salts such as hydrochloride, hydrobromide, hydroiodide, sulfate salts (such as, e.g., sulfate or hydrogensulfate salts), nitrate salts, phosphate salts (such as, e.g., phosphate, hydrogenphosphate, or dihydrogenphosphate salts), carbonate salts, hydrogencarbonate salts, perchlorate salts, borate salts, or thiocyanate salts; organic acid salts such as acetate, propionate, butyrate, pentanoate, hexanoate, heptanoate, octanoate, cyclopentanepropionate, decanoate, undecanoate, oleate, stearate, lactate, maleate, oxalate, fumarate, tartrate, malate, citrate, succinate, adipate, gluconate, glycolate, nicotinate, benzoate, salicylate, ascorbate, pamoate (embonate), camphorate, glucoheptanoate, or pivalate salts; sulfonate salts such as methanesulfonate (mesylate), ethanesulfonate (esylate), 2-hydroxyethanesulfonate (isethionate), benzenesulfonate (besylate), p-toluenesulfonate (tosylate), 2-naphthalenesulfonate (napsylate), 3-phenylsulfonate, or camphorsulfonate salts; glycerophosphate salts; and acidic amino acid salts such as aspartate or glutamate salts. Preferred pharmaceutically acceptable salts of the compounds of formula (I) include a hydrochloride salt, a hydrobromide salt, a mesylate salt, a sulfate salt, a tartrate salt, a fumarate salt, an acetate salt, a citrate salt, and a phosphate salt. A particularly preferred pharmaceutically acceptable salt of the compound of formula (I) is a hydrochloride salt. Accordingly, it is preferred that the compound of formula (I), including any one of the specific compounds of formula (I) described herein, is in the form of a hydrochloride salt, a hydrobromide salt, a mesylate salt, a sulfate salt, a tartrate salt, a fumarate salt, an acetate salt, a citrate salt, or a phosphate salt, and it is particularly preferred that the compound of formula (I) is in the form of a hydrochloride salt. The present invention also specifically relates to the compound of formula (I), including any one of the specific compounds of formula (I) described herein, in non-salt form. Moreover, the scope of the invention embraces the compounds of formula (I) in any solvated form, including, e.g., solvates with water (i.e., as a hydrate) or solvates with organic solvents such as, e.g., methanol, ethanol, isopropanol, acetic acid, ethyl acetate, ethanolamine, DMSO, or acetonitrile. All physical forms, including any amorphous or crystalline forms (i.e., polymorphs), of the compounds of formula (I) are also encompassed within the scope of the invention. It is to be understood that such
solvates and physical forms of pharmaceutically acceptable salts of the compounds of the formula (I) are likewise embraced by the invention. Furthermore, the compounds of formula (I) may exist in the form of different isomers, in particular stereoisomers (including, e.g., geometric isomers (or cis/trans isomers), enantiomers and diastereomers) or tautomers (including, in particular, prototropic tautomers, such as keto/enol tautomers or thione/thiol tautomers). All such isomers of the compounds of formula (I) are contemplated as being part of the present invention, either in admixture or in pure or substantially pure form. As for stereoisomers, the invention embraces the isolated optical isomers of the compounds according to the invention as well as any mixtures thereof (including, in particular, racemic mixtures/racemates). The racemates can be resolved by physical methods, such as, e.g., fractional crystallization, separation or crystallization of diastereomeric derivatives, or separation by chiral column chromatography. The individual optical isomers can also be obtained from the racemates via salt formation with an optically active acid followed by crystallization. The present invention further encompasses any tautomers of the compounds of formula (I). It will be understood that some compounds may exhibit tautomerism. In such cases, the formulae provided herein expressly depict only one of the possible tautomeric forms. The formulae and chemical names as provided herein are intended to encompass any tautomeric form of the corresponding compound and not to be limited merely to the specific tautomeric form depicted by the drawing or identified by the name of the compound. The scope of the invention also embraces compounds of formula (I), in which one or more atoms are replaced by a specific isotope of the corresponding atom. For example, the invention encompasses compounds of formula (I), in which one or more hydrogen atoms (or, e.g., all hydrogen atoms) are replaced by deuterium atoms (i.e., 2H; also referred to as “D”). Accordingly, the invention also embraces compounds of formula (I) which are enriched in deuterium. Naturally occurring hydrogen is an isotopic mixture comprising about 99.98 mol-% hydrogen-1 (1H) and about 0.0156 mol-% deuterium (2H or D). The content of deuterium in one or more hydrogen positions in the compounds of formula (I) can be increased using deuteration techniques known in the art. For example, a compound of formula (I) or a reactant or precursor to be used in the synthesis of the compound of formula (I) can be subjected to an H/D exchange reaction using, e.g., heavy water (D2O). Further suitable deuteration techniques are described in: Atzrodt J et al., Bioorg Med Chem, 20(18), 5658-5667, 2012; William JS et al., Journal of Labelled Compounds and Radiopharmaceuticals, 53(11-12), 635-644, 2010; Modvig A et al., J Org Chem, 79, 5861-5868, 2014. The content of deuterium can be determined, e.g., using mass spectrometry or NMR spectroscopy. Unless specifically indicated otherwise, it is preferred that the compound of formula (I) is not enriched in deuterium. Accordingly, the presence of naturally occurring hydrogen atoms or 1H hydrogen atoms in the compounds of formula (I) is preferred. The present invention also embraces compounds of formula (I), in which one or more atoms are
replaced by a positron-emitting isotope of the corresponding atom, such as, e.g., 18F, 11C, 13N, 15O, 76Br, 77Br, 120I and/or 124I. Such compounds can be used as tracers, trackers or imaging probes in positron emission tomography (PET). The invention thus includes (i) compounds of formula (I), in which one or more fluorine atoms (or, e.g., all fluorine atoms) are replaced by 18F atoms, (ii) compounds of formula (I), in which one or more carbon atoms (or, e.g., all carbon atoms) are replaced by 11C atoms, (iii) compounds of formula (I), in which one or more nitrogen atoms (or, e.g., all nitrogen atoms) are replaced by 13N atoms, (iv) compounds of formula (I), in which one or more oxygen atoms (or, e.g., all oxygen atoms) are replaced by 15O atoms, (v) compounds of formula (I), in which one or more bromine atoms (or, e.g., all bromine atoms) are replaced by 76Br atoms, (vi) compounds of formula (I), in which one or more bromine atoms (or, e.g., all bromine atoms) are replaced by 77Br atoms, (vii) compounds of formula (I), in which one or more iodine atoms (or, e.g., all iodine atoms) are replaced by 120I atoms, and (viii) compounds of formula (I), in which one or more iodine atoms (or, e.g., all iodine atoms) are replaced by 124I atoms. In general, it is preferred that none of the atoms in the compounds of formula (I) are replaced by specific isotopes. The present invention further embraces the prodrugs of the compounds of formula (I). As preferably understood herein, the term “prodrug” of the compound of formula (I) refers to a derivative of the compounds of formula (I) that upon administration to a subject becomes metabolized to the said compound of formula (I). Said prodrugs of the compound of formula (I) may include modifications of -OH, -NH2, or -COOH group if present in the compound of formula (I), which preferably can be hydrolyzed to - OH, -NH2, or -COOH groups, respectively, e.g. upon administration to the subject. For example, as known to the skilled person, such prodrugs may preferably include for the compounds of formula (I) which comprise -OH moiety derivatives wherein said -OH moiety is turned into an -ORx moiety, wherein Rx preferably comprises a moiety selected from -CO-, -CH2-O-CO, -CH2-O-CO-O-, and -CH(CH3)-O-COO-, more preferably wherein Rx is selected from -CO-Ry, -CH2-O-CO-Ry, -CH2-O-CO-O-Ry, and -CH(CH3)-O- COO-Ry, wherein Ry is preferably carbocyclyl, heterocyclyl, C1-5 alkyl, -NH-(C1-5 alkyl) or -S-(C1-5 alkyl), wherein the said alkyl is optionally substituted with a group selected from halogen, -CN, -OH, C1-5 alkyl, C1-5 haloalkyl, -O(C1-5 alkyl), -O(C1-5 haloalkyl), -SH, -S(C1-5 alkyl), -S(C1-5 haloalkyl), -NH2, -NH(C1-5 alkyl), -NH(C1-5 haloalkyl), -N(C1-5 alkyl)(C1-5 alkyl), -N(C1-5 haloalkyl)(C1-5 alkyl), -CONH2, -CONH(C1-5 alkyl), and -CON(C1-5 alkyl)(C1-5 alkyl), and wherein the said carbocyclyl and heterocyclyl are each optionally substituted with a group selected from halogen, -CN, -OH, C1-5 alkyl, C1-5 haloalkyl, -O(C1-5 alkyl), -O(C1-5 haloalkyl), -SH, -S(C1-5 alkyl), -S(C1-5 haloalkyl), -NH2, -NH(C1-5 alkyl), -NH(C1-5 haloalkyl), -N(C1-5 alkyl)(C1-5 alkyl), -N(C1-5 haloalkyl)(C1-5 alkyl), -CONH2, -CONH(C1-5 alkyl), and -CON(C1-5 alkyl)(C1-5 alkyl). Furthermore, for example, as known to the skilled person, such prodrugs may preferably include for the compounds of formula (I) which comprise -NH2 moiety derivatives wherein said -NH2 moiety is turned into -NHCOO-Ry moiety, wherein Ry is as defined hereinabove. Furthermore,
for examples, as known to the skilled person, such prodrugs may preferably include for the compounds of formula (I) which comprise -COOH moiety derivatives wherein said -COOH group is turned into -COORy moiety, wherein Ry is as defined hereinabove. Further examples of groups that can be derivatized to yield prodrugs are known to the skilled person. Pharmaceutical compositions The compounds provided herein may be administered as compounds per se or may be formulated as medicaments. The medicaments/pharmaceutical compositions may optionally comprise one or more pharmaceutically acceptable excipients, such as carriers, diluents, fillers, disintegrants, lubricating agents, binders, colorants, pigments, stabilizers, preservatives, antioxidants, and/or solubility enhancers. The pharmaceutical compositions may comprise one or more solubility enhancers, such as, e.g., poly(ethylene glycol), including poly(ethylene glycol) having a molecular weight in the range of about 200 to about 5,000 Da (e.g., PEG 200, PEG 300, PEG 400, or PEG 600), ethylene glycol, propylene glycol, glycerol, a non-ionic surfactant, tyloxapol, polysorbate 80, macrogol-15-hydroxystearate (e.g., Kolliphor® HS 15, CAS 70142-34-6), a phospholipid, lecithin, dimyristoyl phosphatidylcholine, dipalmitoyl phosphatidylcholine, distearoyl phosphatidylcholine, a cyclodextrin, -cyclodextrin, -cyclodextrin, - cyclodextrin, hydroxyethyl- -cyclodextrin, hydroxypropyl- -cyclodextrin, hydroxyethyl- -cyclodextrin, hydroxypropyl- -cyclodextrin, dihydroxypropyl- -cyclodextrin, sulfobutylether- -cyclodextrin, sulfobutylether- -cyclodextrin, glucosyl- -cyclodextrin, glucosyl- -cyclodextrin, diglucosyl- -cyclodextrin, maltosyl- -cyclodextrin, maltosyl- -cyclodextrin, maltosyl- -cyclodextrin, maltotriosyl- -cyclodextrin, maltotriosyl- -cyclodextrin, dimaltosyl- -cyclodextrin, methyl- -cyclodextrin, a carboxyalkyl thioether, hydroxypropyl methylcellulose, hydroxypropylcellulose, polyvinylpyrrolidone, a vinyl acetate copolymer, vinyl pyrrolidone, sodium lauryl sulfate, dioctyl sodium sulfosuccinate, or any combination thereof. The pharmaceutical compositions may also comprise one or more preservatives, particularly one or more antimicrobial preservatives, such as, e.g., benzyl alcohol, chlorobutanol, 2-ethoxyethanol, m-cresol, chlorocresol (e.g., 2-chloro-3-methyl-phenol or 4-chloro-3-methyl-phenol), benzalkonium chloride, benzethonium chloride, benzoic acid (or a pharmaceutically acceptable salt thereof), sorbic acid (or a pharmaceutically acceptable salt thereof), chlorhexidine, thimerosal, or any combination thereof. The pharmaceutical compositions can be formulated by techniques known to the person skilled in the art, such as the techniques published in “Remington: The Science and Practice of Pharmacy”, Pharmaceutical Press, 22nd edition. The pharmaceutical compositions can be formulated as dosage forms for oral, parenteral, such as intramuscular, intravenous, subcutaneous, intradermal, intraarterial, intracardial, rectal, nasal, topical, aerosol or vaginal administration. Dosage forms for oral administration include coated and uncoated tablets, soft gelatin capsules, hard gelatin capsules, lozenges, troches, solutions, emulsions, suspensions, syrups, elixirs, powders and granules for reconstitution, dispersible
powders and granules, medicated gums, chewing tablets and effervescent tablets. Dosage forms for parenteral administration include solutions, emulsions, suspensions, dispersions and powders and granules for reconstitution. Emulsions are a preferred dosage form for parenteral administration. Dosage forms for rectal and vaginal administration include suppositories and ovula. Dosage forms for nasal administration can be administered via inhalation and insufflation, for example by a metered inhaler. Dosage forms for topical administration include creams, gels, ointments, salves, patches and transdermal delivery systems. The compounds of formula (I) or the above described pharmaceutical compositions comprising a compound of formula (I) may be administered to a subject by any convenient route of administration, whether systemically/peripherally or at the site of desired action, including but not limited to one or more of: oral (e.g., as a tablet, capsule, or as an ingestible solution), topical (e.g., transdermal, intranasal, ocular, buccal, and sublingual), parenteral (e.g., using injection techniques or infusion techniques, and including, for example, by injection, e.g., subcutaneous, intradermal, intramuscular, intravenous, intraarterial, intracardiac, intrathecal, intraspinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratracheal, subcuticular, intraarticular, subarachnoid, or intrasternal by, e.g., implant of a depot, for example, subcutaneously or intramuscularly), pulmonary (e.g., by inhalation or insufflation therapy using, e.g., an aerosol, e.g., through mouth or nose), gastrointestinal, intrauterine, intraocular, subcutaneous, ophthalmic (including intravitreal or intracameral), rectal, or vaginal administration. If said compounds or pharmaceutical compositions are administered parenterally, then examples of such administration include one or more of: intravenously, intraarterially, intraperitoneally, intrathecally, intraventricularly, intraurethrally, intrasternally, intracardially, intracranially, intramuscularly or subcutaneously administering the compounds or pharmaceutical compositions, and/or by using infusion techniques. For parenteral administration, the compounds are best used in the form of a sterile aqueous solution which may contain other substances, for example, enough salts or glucose to make the solution isotonic with blood. The aqueous solutions should be suitably buffered (preferably to a pH of from 3 to 9), if necessary. The preparation of suitable parenteral formulations under sterile conditions is readily accomplished by standard pharmaceutical techniques well known to those skilled in the art. Said compounds or pharmaceutical compositions can also be administered orally in the form of tablets, capsules, ovules, elixirs, solutions or suspensions, which may contain flavoring or coloring agents, for immediate-, delayed-, modified-, sustained-, pulsed- or controlled-release applications. The tablets may contain excipients such as microcrystalline cellulose, lactose, sodium citrate, calcium carbonate, dibasic calcium phosphate and glycine, disintegrants such as starch (preferably corn, potato or tapioca starch), sodium starch glycolate, croscarmellose sodium and certain complex silicates, and granulation binders such as polyvinylpyrrolidone, hydroxypropylmethylcellulose (HPMC),
hydroxypropylcellulose (HPC), sucrose, gelatin and acacia. Additionally, lubricating agents such as magnesium stearate, stearic acid, glyceryl behenate and talc may be included. Solid compositions of a similar type may also be employed as fillers in gelatin capsules. Preferred excipients in this regard include lactose, starch, a cellulose, or high molecular weight polyethylene glycols. For aqueous suspensions and/or elixirs, the agent may be combined with various sweetening or flavoring agents, coloring matter or dyes, with emulsifying and/or suspending agents and with diluents such as water, ethanol, propylene glycol and glycerin, and combinations thereof. For oral administration, the compounds or pharmaceutical compositions are preferably administered by oral ingestion, particularly by swallowing. The compounds or pharmaceutical compositions can thus be administered to pass through the mouth into the gastrointestinal tract, which can also be referred to as “oral-gastrointestinal” administration. Alternatively, said compounds or pharmaceutical compositions can be administered in the form of a suppository or pessary, or may be applied topically in the form of a gel, hydrogel, lotion, solution, cream, ointment or dusting powder. The compounds of the present invention may also be dermally or transdermally administered, for example, by the use of a skin patch. Said compounds or pharmaceutical compositions may also be administered by sustained release systems. Suitable examples of sustained-release compositions include semi-permeable polymer matrices in the form of shaped articles, e.g., films, or microcapsules. Sustained-release matrices include, e.g., polylactides, copolymers of L-glutamic acid and gamma-ethyl-L-glutamate, poly(2-hydroxyethyl methacrylate), ethylene vinyl acetate, or poly-D-(–)-3-hydroxybutyric acid. Sustained-release pharmaceutical compositions also include liposomally entrapped compounds. The present invention thus also relates to liposomes containing a compound of the invention. Said compounds or pharmaceutical compositions may also be administered by the pulmonary route, rectal routes, or the ocular route. For ophthalmic use, they can be formulated as micronized suspensions in isotonic, pH adjusted, sterile saline, or, preferably, as solutions in isotonic, pH adjusted, sterile saline, optionally in combination with a preservative such as a benzalkonium chloride. Alternatively, they may be formulated in an ointment such as petrolatum. It is also envisaged to prepare dry powder formulations of the compounds of formula (I) for pulmonary administration, particularly inhalation. Such dry powders may be prepared by spray drying under conditions which result in a substantially amorphous glassy or a substantially crystalline bioactive powder. Accordingly, dry powders of the compounds of the present invention can be made according to an emulsification/spray drying process. For topical application to the skin, said compounds or pharmaceutical compositions can be formulated as a suitable ointment containing the active compound suspended or dissolved in, for example,
a mixture with one or more of the following: mineral oil, liquid petrolatum, white petrolatum, propylene glycol, emulsifying wax and water. Alternatively, they can be formulated as a suitable lotion or cream, suspended or dissolved in, for example, a mixture of one or more of the following: mineral oil, sorbitan monostearate, a polyethylene glycol, liquid paraffin, polysorbate 60, cetyl esters wax, 2-octyldodecanol, benzyl alcohol and water. The present invention thus relates to the compounds or the pharmaceutical compositions provided herein, wherein the corresponding compound or pharmaceutical composition is to be administered by any one of: an oral route; topical route, including by transdermal, intranasal, ocular, buccal, or sublingual route; parenteral route using injection techniques or infusion techniques, including by subcutaneous, intradermal, intramuscular, intravenous, intraarterial, intracardiac, intrathecal, intraspinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratracheal, subcuticular, intraarticular, subarachnoid, intrasternal, intraventricular, intraurethral, or intracranial route; pulmonary route, including by inhalation or insufflation therapy; gastrointestinal route; intrauterine route; intraocular route; subcutaneous route; ophthalmic route, including by intravitreal, or intracameral route; rectal route; or vaginal route. Preferred routes of administration are oral administration or parenteral administration. For each of the compounds or pharmaceutical compositions provided herein, it is particularly preferred that the respective compound or pharmaceutical composition is to be administered orally (particularly by oral ingestion). Typically, a physician will determine the actual dosage which will be most suitable for an individual subject. The specific dose level and frequency of dosage for any particular individual subject may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the individual subject undergoing therapy. A proposed, yet non-limiting dose of the compounds according to the invention for oral administration to a human (of approximately 70 kg body weight) may be 0.05 to 2000 mg, preferably 0.1 mg to 1000 mg, of the active ingredient per unit dose. The unit dose may be administered, e.g., 1 to 3 times per day. The unit dose may also be administered 1 to 7 times per week, e.g., with not more than one administration per day. It will be appreciated that it may be necessary to make routine variations to the dosage depending on the age and weight of the patient/subject as well as the severity of the condition to be treated. The precise dose and also the route of administration will ultimately be at the discretion of the attendant physician or veterinarian. Therapeutic use In one embodiment, the present invention relates to the compound of formula (I), or a pharmaceutically acceptable salt, or a pharmaceutical composition as defined herein for use in therapy.
The present invention provides compounds that have activity of inhibitors of Werner syndrome ATP- dependent helicase (WRN). Thus, accordingly, the present invention provides a method of inhibiting WRN enzyme activity in vitro and in vivo, said method comprising contacting a cell with an effective amount of the compound of formula (I), or a pharmaceutically acceptable salt thereof, as defined herein. In one embodiment, the present invention provides a method for decreasing proliferation in a proliferative cell having a microsatellite instability (MSI), comprising decreasing the helicase activity of Werner syndrome ATP-dependent helicase (WRN) in the proliferative cell. In some embodiments, decreasing the helicase activity of Wemer syndrome ATP-dependent helicase (WRN) in the proliferative cell is achieved by administering a compound of Formula of (I) (or any embodiment thereof disclosed herein) or a pharmaceutically acceptable salt thereof. In some embodiments, the proliferative cell is characterized as having MSI low (MSI-L). In some embodiments, the proliferative cell is characterized as having high MSI (MSI-H), used interchangeably with MSI-high. Cells can be characterized as MSI, including MSI-L or MSI- H, or as MSS (MS-stable), according to the method known in the art (see, for example, Dudley, Jonathan C., et al., Clinical Cancer Research, 22(4): 813-820, 2016.). MSI-H is used to classify tumors as having a high frequency of MSI. A tumor can be classified as MSI, including MSI-low or MSI-high, using polymerase chain reaction (PCR) and/or immunohistochemistry (IHC) assays. As stated in Dudley et al., a tumor is classified as MSI-H by PCR if (i) there is a shift (usually downward) in the size of at least two microsatellite loci from a reference panel of five microsatellite loci in tumor relative to normal, where the reference panel can be the “Bethesda Panel,” also referred to herein as the “NCI- Reference Panel (Bethesda, 1998)”, which includes two mononucleotide loci (BAT-25 and BAT-26) and three dinucleotide loci (D2S123, D5S346, and D17S250), or alternatively, the reference panel can be Promega Corporation’s MSI Analysis System, which includes five mononucleotide loci (BAT-25, BAT-26, NR-21, NR-24, and MONO-27); or (ii) there is a shift in the size of 30% or more microsatellite loci from a reference panel of more than five microsatellite loci in tumor relative to normal. The MSI-H phenotype is associated with germline defects in the mismatch repair genes MLH1, MSH2, MSH6, and PMS2, and is the primary phenotype observed in tumors from patients with HNPCC/Lynch syndrome. A tumor is classified as MSI-H in IHC test if it shows a loss of protein expression for at least 1 of the above 4 mismatch repair genes. Cells can be similarly classified as MSI-H using the tests described herein for tumors. In some embodiments, a tumor or cell is classified as MSI-H using PCR to amplify the five microsatellite loci of the “Bethesda Panel” (BAT-25, BAT-26, D2S123, D5S346, and D17S250) from both tumor tissue or cells and normal tissue or cells, wherein the tumor or cell is classified as MSI-H if there is a shift in the size of at least two of the microsatellite loci from the tumor tissue or cells relative to the normal tissue or cells. In some embodiments, the shift in size of the microsatellite loci is a downward shift. In some embodiments, a tumor or cell is classified as MSI-H using PCR to amplify the five
microsatellite loci of Promega Corporation’s MSI Analysis System (BAT-25, BAT-26, NR- 21, NR-24, and MONO-27) from both tumor tissue or cells and normal tissue or cells, wherein the tumor or cell is classified as MSI-H if there is a shift in the size of at least two of the microsatellite loci from the tumor tissue or cells relative to the normal tissue or cells. In some embodiments, the shift in size of the microsatellite loci is a downward shift. In some embodiments, a tumor is classified as MSI-H using IHC to determine the expression level of the MMR proteins MLH1, MSH2, MSH6, and/or PMS2 in both tumor tissue and normal tissue, wherein the tumor is classified as MSI-H if there is a loss of protein expression for at least one of the MMR proteins in the tumor tissue relative to the normal tissue. In some embodiments, the loss of protein expression is a decrease of at least 20% (such as a decrease of 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 99%, or more). In contrast, a tumor is classified as MSI-L by PCR if (i) there is a shift in the size of one microsatellite locus from a reference panel of five microsatellite loci in tumor relative to normal, where the reference panel can be the “Bethesda Panel” or Promega Corporation’s MSI Analysis System; or (ii) there is a shift in the size of less than 30% microsatellite loci from a reference panel of more than five microsatellite loci in tumor relative to normal. MSI-L tumors are thought to represent a distinct mutator phenotype with potentially different molecular etiology than MSI-H tumors (Thibodeau, 1998; Wu et al., 1999, Am J Hum Genetics 65: 1291-1298). Cells can be similarly classified as MSI-L using the tests described herein for tumors. Cancers classified as MSI-H include, but not limited to, uterine corpus endometrial carcinoma, colon adenocarcinoma, stomach adenocarcinoma, rectal adenocarcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cervical squamous cell carcinoma, and endocervical adenocarcinoma. In one embodiment, the present invention relates to a method for treating a cancer treatable by inhibition of WRN in a subject in need thereof, the method comprising the step of administering to the subject the compound of formula (I) or a pharmaceutically acceptable salt thereof. It is to be understood that, typically, a therapeutically effective amount is to be administered. In one embodiment, the present invention relates to a method for treating a cancer characterized by MSI-H and/or dMMR in a subject in need thereof, the method comprising the step of administering to the subject the compound of formula (I) or a pharmaceutically acceptable salt thereof. It is to be understood that, typically, a therapeutically effective amount is to be administered. In one embodiment, the present invention relates to use of a compound of formula (I) or pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer. Preferably, the cancer is treatable by inhibition of WRN. In one embodiment, the cancer is characterized by MSI-H and/or dMMR.
In one embodiment, the present invention relates to the compound of formula (I) or a pharmaceutically acceptable salt thereof, for use in treating cancer. Preferably, the cancer is treatable by inhibition of WRN. In one embodiment, the cancer is characterized by MSI-H and/or dMMR. The cancer to be treated in accordance with the present invention may be a solid cancer or a hematological cancer. Preferably, the cancer is selected from lung cancer (e.g., small cell lung cancer, non-small cell lung cancer, large cell lung carcinoma, lung adenocarcinoma, including also lung adenocarcinoma with EGFR mutation E746-A750, or squamous cell carcinoma of the lung), renal cancer (or kidney cancer; e.g., renal carcinoma), gastrointestinal cancer, stomach cancer, colorectal cancer (e.g., colorectal carcinoma), colon cancer, anal cancer, genitourinary cancer, bladder cancer, liver cancer (e.g., hepatocellular carcinoma), pancreatic cancer (e.g., pancreatic adenocarcinoma or pancreatic ductal adenocarcinoma), cervical cancer, endometrial cancer, vaginal cancer, vulvar cancer, ovarian cancer (e.g., ovarian carcinoma), uterine cancer, prostate cancer (e.g., hormone-refractory prostate cancer), testicular cancer, biliary tract cancer, hepatobiliary cancer, neuroblastoma, brain cancer (e.g., glioblastoma), breast cancer (e.g., triple-negative breast cancer, breast cancer having a BRCA1 and/or BRCA2 gene mutation, or breast adenocarcinoma), head and/or neck cancer (e.g., head and neck squamous cell carcinoma), skin cancer, melanoma, Merkel-cell cancer (e.g., Merkel-cell carcinoma), epidermoid cancer, squamous cell cancer (or squamous cell carcinoma; including, e.g., oral squamous cell carcinoma/squamous-cell mouth carcinoma, squamous-cell skin cancer, squamous-cell lung carcinoma, squamous-cell thyroid carcinoma, squamous-cell esophageal carcinoma, or squamous-cell vaginal carcinoma), bone cancer (e.g., osteosarcoma or osteogenic sarcoma), fibrosarcoma, Ewing’s sarcoma, malignant mesothelioma, esophageal cancer, laryngeal cancer, mouth cancer, thymoma, neuroendocrine cancer (e.g., neuroendocrine carcinoma), goblet cell cancer (e.g., goblet cell carcinoid), hematological cancer, leukemia (e.g., acute lymphoblastic leukemia, acute myeloid leukemia, chronic lymphocytic leukemia, or chronic myeloid leukemia), lymphoma (e.g., Hodgkin lymphoma or non-Hodgkin lymphoma, such as, e.g., follicular lymphoma or diffuse large B-cell lymphoma), and multiple myeloma. Moreover, the cancer to be treated (including any one of the aforementioned specific types of cancer) may also be a chemoresistant and/or a metastatic cancer. The antiproliferative treatment (i.e. the treatment of cancer) with the compound of formula (I) or a pharmaceutically acceptable salt thereof, as defined hereinbefore, may be applied as a sole therapy or may involve, in addition to the compound of the invention, conventional surgery or radiotherapy or chemotherapy. Such chemotherapy may include one or more of the following categories of anti-tumour agents:- (i) other antiproliferative/antineoplastic drugs and combinations thereof, as used in medical oncology, such as alkylating agents (for example cis-platin, oxaliplatin, carboplatin, cyclophosphamide,
nitrogen mustard, melphalan, chlorambucil, busulphan, temozolamide and nitrosoureas); antimetabolites (for example gemcitabine and antifolates such as fluoropyrimidines like 5-fluorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside, and hydroxyurea); antitumour antibiotics (for example anthracyclines like adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin); antimitotic agents (for example vinca alkaloids like vincristine, vinblastine, vindesine and vinorelbine and taxoids like taxol and taxotere and polokinase inhibitors); and topoisomerase inhibitors (for example epipodophyllotoxins like etoposide and teniposide, amsacrine, topotecan and camptothecin); (ii) cytostatic agents such as antioestrogens (for example tamoxifen, fulvestrant, toremifene, raloxifene, droloxifene and iodoxyfene), antiandrogens (for example bicalutamide, flutamide, nilutamide and cyproterone acetate), LHRH antagonists or LHRH agonists (for example goserelin, leuprorelin and buserelin), progestogens (for example megestrol acetate), aromatase inhibitors (for example as anastrozole, letrozole, vorazole and exemestane) and inhibitors of 5oc-reductase such as finasteride; (iii) anti-invasion agents [for example c-Src kinase family inhibitors like 4-(6-chloro-2,3- methylenedioxyanilino)-7-[2-(4-methylpiperazin-1 -yl)ethoxy]-5-tetrahydropyran-4- yloxyquinazoline (AZD0530; International Patent Application WO 01/94341 ), N-(2-chloro-6- methylphenyl)-2-{6-[4-(2- hydroxyethyl)piperazin-1 -yl]-2-methylpyrimidin-4-ylamino}thiazole- 5-carboxamide (dasatinib, BMS- 354825; J. Med. Chem., 2004, 47, 6658-6661 ) and bosutinib (SKI-606), and metalloproteinase inhibitors like marimastat, inhibitors of urokinase plasminogen activator receptor function or antibodies to Heparanase]; (iv) inhibitors of growth factor function: for example such inhibitors include growth factor antibodies and growth factor receptor antibodies (for example the anti-erbB2 antibody trastuzumab [Herceptin™], the anti-EGFR antibody panitumumab, the anti-erbB1 antibody cetuximab [Erbitux, C225] and any growth factor or growth factor receptor antibodies disclosed by Stern et al. (Critical reviews in oncology/haematology, 2005, Vol.54, pp11 -29); such inhibitors also include tyrosine kinase inhibitors, for example inhibitors of the epidermal growth factor family (for example EGFR family tyrosine kinase inhibitors such as N-(3-chloro- 4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazolin-4-amine (gefitinib, ZD1839), N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (erlotinib, OSI-774) and 6- acrylamido-N-(3-chloro-4-fluorophenyl)-7-(3-morpholinopropoxy)-quinazolin-4-amine (CI 1033), erbB2 tyrosine kinase inhibitors such as lapatinib); inhibitors of the hepatocyte growth factor family; inhibitors of the insulin growth factor family; inhibitors of the platelet-derived growth factor family such as imatinib and/or nilotinib (AMN107); inhibitors of serine/threonine kinases (for example Ras/Raf signalling inhibitors such as farnesyl transferase inhibitors, for example sorafenib (BAY 43-9006), tipifarnib (R1 15777) and lonafarnib (SCH66336)), inhibitors of cell signalling through MEK and/or AKT kinases, c-kit
inhibitors, abl kinase inhibitors, PI3 kinase inhibitors, Plt3 kinase inhibitors, CSF-1 R kinase inhibitors, IGF receptor (insulin-like growth factor) kinase inhibitors; aurora kinase inhibitors (for example AZD1152, PH739358, VX-680, MLN8054, R763, MP235, MP529, VX-528 AND AX39459) and cyclin dependent kinase inhibitors such as CDK2 and/or CDK4 inhibitors; (v) antiangiogenic agents such as those which inhibit the effects of vascular endothelial growth factor, [for example the anti-vascular endothelial cell growth factor antibody bevacizumab (Avastin™) and for example, a VEGF receptor tyrosine kinase inhibitor such as vandetanib (ZD6474), vatalanib (PTK787), sunitinib (SU11248), axitinib (AG-013736), pazopanib (GW 786034) and 4-(4-fluoro-2-methylindol-5- yloxy)-6-methoxy-7-(3-pyrrolidin-1 - ylpropoxy)quinazoline (AZD2171 ; Example 240 within WO 00/47212), compounds such as those disclosed in International Patent Applications W097/22596, WO 97/30035, WO 97/32856 and WO 98/13354 and compounds that work by other mechanisms (for example linomide, inhibitors of integrin 3 function and angiostatin)]; (vi) vascular damaging agents such as Combretastatin A4 and compounds disclosed in International Patent Applications WO 99/02166, WO 00/40529, WO 00/41669, WO 01 /92224, WO 02/04434 and WO 02/08213; (vii) an endothelin receptor antagonist, for example zibotentan (ZD4054) or atrasentan; (viii) antisense therapies, for example those which are directed to the targets listed above, such as ISIS 2503, an anti-ras antisense; (ix) gene therapy approaches, including for example approaches to replace aberrant genes such as aberrant p53 or aberrant BRCA1 or BRCA2, GDEPT (gene-directed enzyme pro-drug therapy) approaches such as those using cytosine deaminase, thymidine kinase or a bacterial nitroreductase enzyme and approaches to increase patient tolerance to chemotherapy or radiotherapy such as multi- drug resistance gene therapy; and (x) immunotherapy approaches, including for example ex-vivo and in-vivo approaches to increase the immunogenicity of patient tumour cells, such as transfection with cytokines such as interleukin 2, interleukin 4 or granulocyte-macrophage colony stimulating factor, approaches to decrease T-cell anergy, approaches using transfected immune cells such as cytokine-transfected dendritic cells, approaches using cytokine-transfected tumour cell lines and approaches using anti-idiotypic antibodies. In a particular embodiment, the antiproliferative treatment defined hereinbefore may involve, in addition to the compound of formula (I) of the invention, conventional surgery or radiotherapy or chemotherapy. Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate dosing of the individual components of the treatment. Such combination products employ the compounds of this invention within the dosage range described hereinbefore and the other pharmaceutically-active agent within its approved dosage range.
According to this aspect the present invention further relates to the compound of formula (I) or a pharmaceutically acceptable salt thereof, as defined herein, for use in the treatment of a cancer (for example a cancer involving a solid tumour) in combination with another anti-tumour agent. The anti- tumour agent is preferably selected from the anti-tumour agents as listed hereinabove. As understood herein, the term "combination" refers to simultaneous, separate or sequential administration. In one aspect of the invention "combination" refers to simultaneous administration. In another aspect of the invention "combination" refers to separate administration. In a further aspect of the invention "combination" refers to sequential administration. Where the administration is sequential or separate, the delay in administering the second component should not be such as to lose the beneficial effect of the combination. Examples The following examples are merely illustrative of the present invention and should not be construed to limit the scope of the invention which is defined by the appended claims. Synthesis of the compounds of formula (I) The syntheses of the compounds of formula (I) according to the present invention are preferably carried out according to the general synthetic sequences as shown in Scheme 1. In addition to said routes described below, also other routes may be used to synthesize the target compounds, in accordance with common general knowledge of a person skilled in the art of organic synthesis. The order of transformations exemplified in the following Schemes is therefore not intended to be limiting, and suitable synthesis steps from various schemes can be combined to form additional synthesis sequences. In addition, modification of any of the substituents can be achieved before and/or after the exemplified transformations. These modifications can be such as the introduction of protective groups, cleavage of protective groups, reduction or oxidation of functional groups, halogenation, metallation, metal-catalyzed coupling reactions, substitution or other reactions known to a person skilled in the art. These transformations include those which introduce a functionality allowing for further interconversion of substituents. Appropriate protective groups and their introduction and cleavage are well-known to a person skilled in the art (see for example: Greene's Protective Groups in Organic Synthesis; Editor: P.G.M. Wuts, 5th edition, Wiley 2014). Specific examples are described in the subsequent paragraphs. Further, it is possible that two or more successive steps may be performed without work-up being performed between said steps, e.g. a “one-pot” reaction, as it is well-known to a person skilled in the art. It is further understood to the skilled person that a reaction can lead to side product(s) which, when appropriate, can be used for the preparation of compounds of formula (I) using similar procedures as reported in the general schemes hereinbelow. Scheme 1
Scheme 1 illustrates a preferred synthetic approach to compounds of the general formula (I). In the first step, compound 3 is prepared via cross-coupling reactions of suitably functionalized building blocks 1 and 2. Formation of a C-C bond between A (1) and B-X (2) can be e.g. achieved by the cross-coupling of an aryl, heteroaryl, alkyl, or vinyl boronic acid, borate ester, or borane 1 with an aryl or heteroaryl halide or triflate 2 using a variety of palladium catalysts (Suzuki reaction; see e.g. B. S. Kadu, Catal. Sci. Technol., 2021,11, 1186-1221). The formation of a carbon–carbon bond between a terminal alkyne 1 and an aryl or heteroaryl halide 2 can be achieved employing palladium catalysts as well as copper co-catalysts (Sonogashira coupling, see e.g. I. Kanwal et al, Catalysts 2020, 10(4), 443). Moreover, formation of a C-N can be achieved via the palladium-catalyzed coupling reactions of amines 1 with aryl and heteroaryl halides 2 (Buchwald–Hartwig coupling, see e.g. R. Dorel et al, Angew. Chem. Int. Ed.2019, 58, 17118). In the second step, cross-coupling of A-B-X (3) and Y (4) can be achieved by various methods known of a person skilled in the art of organic synthesis, including e.g. a) coupling of carboxylic acids (3) and amines (4) (amide bond formation; see e.g. E. Massolo et al, Eur. J. Org. Chem.2020, 4641), b) coupling of sulfonyl chlorides (3) and amines (4) (sulfonamide bond formation: see e.g. A. Kolaczek et al, CHEMIK 2014, 68, 620) or c) coupling of amines (3) and halides (4) (amine alkylation: see e.g. R. N. Salvatoreet al, Tetrahedron 2001, 7785). For the synthesis of compounds of formula (I) in which B and X are combined to form a two ring system:
as defined in the foregoing. For a number of compounds disclosed herein, enantiomeric forms may exist. The present invention further refers to each and every enantiomeric form of such compound individually. The present invention also relates to each of the intermediates described further below in the examples section of this specification, including any one of these intermediates in non-salt form or in the form of a salt (e.g., a pharmaceutically acceptable salt) of the respective compound. Such intermediates can be used, in particular, in the synthesis of the compounds of formula (I). The scope of the invention embraces all pharmaceutically acceptable salt forms of the compounds of formula (I) which may be formed, e.g., by protonation of an atom carrying an electron lone pair which is susceptible to protonation, such as an amino group, with an inorganic or organic acid, or as a salt of an acid group (such as a carboxylic acid group) with a physiologically acceptable cation. Exemplary base addition salts comprise, for example: alkali metal salts such as sodium or potassium salts; alkaline earth
metal salts such as calcium or magnesium salts; zinc salts; ammonium salts; aliphatic amine salts such as trimethylamine, triethylamine, dicyclohexylamine, ethanolamine, diethanolamine, triethanolamine, procaine salts, meglumine salts, ethylenediamine salts, or choline salts; aralkyl amine salts such as N,N- dibenzylethylenediamine salts, benzathine salts, benethamine salts; heterocyclic aromatic amine salts such as pyridine salts, picoline salts, quinoline salts or isoquinoline salts; quaternary ammonium salts such as tetramethylammonium salts, tetraethylammonium salts, benzyltrimethylammonium salts, benzyltriethylammonium salts, benzyltributylammonium salts, methyltrioctylammonium salts or tetrabutylammonium salts; and basic amino acid salts such as arginine salts, lysine salts, or histidine salts. Exemplary acid addition salts comprise, for example: mineral acid salts such as hydrochloride, hydrobromide, hydroiodide, sulfate salts (such as, e.g., sulfate or hydrogensulfate salts), nitrate salts, phosphate salts (such as, e.g., phosphate, hydrogenphosphate, or dihydrogenphosphate salts), carbonate salts, hydrogencarbonate salts, perchlorate salts, borate salts, or thiocyanate salts; organic acid salts such as acetate, propionate, butyrate, pentanoate, hexanoate, heptanoate, octanoate, cyclopentanepropionate, decanoate, undecanoate, oleate, stearate, lactate, maleate, oxalate, fumarate, tartrate, malate, citrate, succinate, adipate, gluconate, glycolate, nicotinate, benzoate, salicylate, ascorbate, pamoate (embonate), camphorate, glucoheptanoate, or pivalate salts; sulfonate salts such as methanesulfonate (mesylate), ethanesulfonate (esylate), 2-hydroxyethanesulfonate (isethionate), benzenesulfonate (besylate), p-toluenesulfonate (tosylate), 2-naphthalenesulfonate (napsylate), 3-phenylsulfonate, or camphorsulfonate salts; glycerophosphate salts; and acidic amino acid salts such as aspartate or glutamate salts. Preferred pharmaceutically acceptable salts of the compounds of formula (I) include a hydrochloride salt, a hydrobromide salt, a mesylate salt, a sulfate salt, a tartrate salt, a fumarate salt, an acetate salt, a citrate salt, and a phosphate salt. A particularly preferred pharmaceutically acceptable salt of the compound of formula (I) is a hydrochloride salt. Accordingly, it is preferred that the compound of formula (I), including any one of the specific compounds of formula (I) described herein, is in the form of a hydrochloride salt, a hydrobromide salt, a mesylate salt, a sulfate salt, a tartrate salt, a fumarate salt, an acetate salt, a citrate salt, or a phosphate salt, and it is particularly preferred that the compound of formula (I) is in the form of a hydrochloride salt. The present invention also specifically relates to the compound of formula (I), including any one of the specific compounds of formula (I) described herein, in non-salt form. Moreover, the scope of the invention embraces the compounds of formula (I) in any solvated form, including, e.g., solvates with water (i.e., as a hydrate) or solvates with organic solvents such as, e.g., methanol, ethanol, isopropanol, acetic acid, ethyl acetate, ethanolamine, DMSO, or acetonitrile. All physical forms, including any amorphous or crystalline forms (i.e., polymorphs), of the compounds of formula (I) are also encompassed within the scope of the invention. It is to be understood that such
solvates and physical forms of pharmaceutically acceptable salts of the compounds of the formula (I) are likewise embraced by the invention. Furthermore, the compounds of formula (I) may exist in the form of different isomers, in particular stereoisomers (including, e.g., geometric isomers (or cis/trans isomers), enantiomers and diastereomers) or tautomers (including, in particular, prototropic tautomers, such as keto/enol tautomers or thione/thiol tautomers). All such isomers of the compounds of formula (I) are contemplated as being part of the present invention, either in admixture or in pure or substantially pure form. As for stereoisomers, the invention embraces the isolated optical isomers of the compounds according to the invention as well as any mixtures thereof (including, in particular, racemic mixtures/racemates). The racemates can be resolved by physical methods, such as, e.g., fractional crystallization, separation or crystallization of diastereomeric derivatives, or separation by chiral column chromatography. The individual optical isomers can also be obtained from the racemates via salt formation with an optically active acid followed by crystallization. The present invention further encompasses any tautomers of the compounds of formula (I). It will be understood that some compounds may exhibit tautomerism. In such cases, the formulae provided herein expressly depict only one of the possible tautomeric forms. The formulae and chemical names as provided herein are intended to encompass any tautomeric form of the corresponding compound and not to be limited merely to the specific tautomeric form depicted by the drawing or identified by the name of the compound. The scope of the invention also embraces compounds of formula (I), in which one or more atoms are replaced by a specific isotope of the corresponding atom. For example, the invention encompasses compounds of formula (I), in which one or more hydrogen atoms (or, e.g., all hydrogen atoms) are replaced by deuterium atoms (i.e., 2H; also referred to as “D”). Accordingly, the invention also embraces compounds of formula (I) which are enriched in deuterium. Naturally occurring hydrogen is an isotopic mixture comprising about 99.98 mol-% hydrogen-1 (1H) and about 0.0156 mol-% deuterium (2H or D). The content of deuterium in one or more hydrogen positions in the compounds of formula (I) can be increased using deuteration techniques known in the art. For example, a compound of formula (I) or a reactant or precursor to be used in the synthesis of the compound of formula (I) can be subjected to an H/D exchange reaction using, e.g., heavy water (D2O). Further suitable deuteration techniques are described in: Atzrodt J et al., Bioorg Med Chem, 20(18), 5658-5667, 2012; William JS et al., Journal of Labelled Compounds and Radiopharmaceuticals, 53(11-12), 635-644, 2010; Modvig A et al., J Org Chem, 79, 5861-5868, 2014. The content of deuterium can be determined, e.g., using mass spectrometry or NMR spectroscopy. Unless specifically indicated otherwise, it is preferred that the compound of formula (I) is not enriched in deuterium. Accordingly, the presence of naturally occurring hydrogen atoms or 1H hydrogen atoms in the compounds of formula (I) is preferred. The present invention also embraces compounds of formula (I), in which one or more atoms are
replaced by a positron-emitting isotope of the corresponding atom, such as, e.g., 18F, 11C, 13N, 15O, 76Br, 77Br, 120I and/or 124I. Such compounds can be used as tracers, trackers or imaging probes in positron emission tomography (PET). The invention thus includes (i) compounds of formula (I), in which one or more fluorine atoms (or, e.g., all fluorine atoms) are replaced by 18F atoms, (ii) compounds of formula (I), in which one or more carbon atoms (or, e.g., all carbon atoms) are replaced by 11C atoms, (iii) compounds of formula (I), in which one or more nitrogen atoms (or, e.g., all nitrogen atoms) are replaced by 13N atoms, (iv) compounds of formula (I), in which one or more oxygen atoms (or, e.g., all oxygen atoms) are replaced by 15O atoms, (v) compounds of formula (I), in which one or more bromine atoms (or, e.g., all bromine atoms) are replaced by 76Br atoms, (vi) compounds of formula (I), in which one or more bromine atoms (or, e.g., all bromine atoms) are replaced by 77Br atoms, (vii) compounds of formula (I), in which one or more iodine atoms (or, e.g., all iodine atoms) are replaced by 120I atoms, and (viii) compounds of formula (I), in which one or more iodine atoms (or, e.g., all iodine atoms) are replaced by 124I atoms. In general, it is preferred that none of the atoms in the compounds of formula (I) are replaced by specific isotopes. The present invention further embraces the prodrugs of the compounds of formula (I). As preferably understood herein, the term “prodrug” of the compound of formula (I) refers to a derivative of the compounds of formula (I) that upon administration to a subject becomes metabolized to the said compound of formula (I). Said prodrugs of the compound of formula (I) may include modifications of -OH, -NH2, or -COOH group if present in the compound of formula (I), which preferably can be hydrolyzed to - OH, -NH2, or -COOH groups, respectively, e.g. upon administration to the subject. For example, as known to the skilled person, such prodrugs may preferably include for the compounds of formula (I) which comprise -OH moiety derivatives wherein said -OH moiety is turned into an -ORx moiety, wherein Rx preferably comprises a moiety selected from -CO-, -CH2-O-CO, -CH2-O-CO-O-, and -CH(CH3)-O-COO-, more preferably wherein Rx is selected from -CO-Ry, -CH2-O-CO-Ry, -CH2-O-CO-O-Ry, and -CH(CH3)-O- COO-Ry, wherein Ry is preferably carbocyclyl, heterocyclyl, C1-5 alkyl, -NH-(C1-5 alkyl) or -S-(C1-5 alkyl), wherein the said alkyl is optionally substituted with a group selected from halogen, -CN, -OH, C1-5 alkyl, C1-5 haloalkyl, -O(C1-5 alkyl), -O(C1-5 haloalkyl), -SH, -S(C1-5 alkyl), -S(C1-5 haloalkyl), -NH2, -NH(C1-5 alkyl), -NH(C1-5 haloalkyl), -N(C1-5 alkyl)(C1-5 alkyl), -N(C1-5 haloalkyl)(C1-5 alkyl), -CONH2, -CONH(C1-5 alkyl), and -CON(C1-5 alkyl)(C1-5 alkyl), and wherein the said carbocyclyl and heterocyclyl are each optionally substituted with a group selected from halogen, -CN, -OH, C1-5 alkyl, C1-5 haloalkyl, -O(C1-5 alkyl), -O(C1-5 haloalkyl), -SH, -S(C1-5 alkyl), -S(C1-5 haloalkyl), -NH2, -NH(C1-5 alkyl), -NH(C1-5 haloalkyl), -N(C1-5 alkyl)(C1-5 alkyl), -N(C1-5 haloalkyl)(C1-5 alkyl), -CONH2, -CONH(C1-5 alkyl), and -CON(C1-5 alkyl)(C1-5 alkyl). Furthermore, for example, as known to the skilled person, such prodrugs may preferably include for the compounds of formula (I) which comprise -NH2 moiety derivatives wherein said -NH2 moiety is turned into -NHCOO-Ry moiety, wherein Ry is as defined hereinabove. Furthermore,
for examples, as known to the skilled person, such prodrugs may preferably include for the compounds of formula (I) which comprise -COOH moiety derivatives wherein said -COOH group is turned into -COORy moiety, wherein Ry is as defined hereinabove. Further examples of groups that can be derivatized to yield prodrugs are known to the skilled person. Pharmaceutical compositions The compounds provided herein may be administered as compounds per se or may be formulated as medicaments. The medicaments/pharmaceutical compositions may optionally comprise one or more pharmaceutically acceptable excipients, such as carriers, diluents, fillers, disintegrants, lubricating agents, binders, colorants, pigments, stabilizers, preservatives, antioxidants, and/or solubility enhancers. The pharmaceutical compositions may comprise one or more solubility enhancers, such as, e.g., poly(ethylene glycol), including poly(ethylene glycol) having a molecular weight in the range of about 200 to about 5,000 Da (e.g., PEG 200, PEG 300, PEG 400, or PEG 600), ethylene glycol, propylene glycol, glycerol, a non-ionic surfactant, tyloxapol, polysorbate 80, macrogol-15-hydroxystearate (e.g., Kolliphor® HS 15, CAS 70142-34-6), a phospholipid, lecithin, dimyristoyl phosphatidylcholine, dipalmitoyl phosphatidylcholine, distearoyl phosphatidylcholine, a cyclodextrin, -cyclodextrin, -cyclodextrin, - cyclodextrin, hydroxyethyl- -cyclodextrin, hydroxypropyl- -cyclodextrin, hydroxyethyl- -cyclodextrin, hydroxypropyl- -cyclodextrin, dihydroxypropyl- -cyclodextrin, sulfobutylether- -cyclodextrin, sulfobutylether- -cyclodextrin, glucosyl- -cyclodextrin, glucosyl- -cyclodextrin, diglucosyl- -cyclodextrin, maltosyl- -cyclodextrin, maltosyl- -cyclodextrin, maltosyl- -cyclodextrin, maltotriosyl- -cyclodextrin, maltotriosyl- -cyclodextrin, dimaltosyl- -cyclodextrin, methyl- -cyclodextrin, a carboxyalkyl thioether, hydroxypropyl methylcellulose, hydroxypropylcellulose, polyvinylpyrrolidone, a vinyl acetate copolymer, vinyl pyrrolidone, sodium lauryl sulfate, dioctyl sodium sulfosuccinate, or any combination thereof. The pharmaceutical compositions may also comprise one or more preservatives, particularly one or more antimicrobial preservatives, such as, e.g., benzyl alcohol, chlorobutanol, 2-ethoxyethanol, m-cresol, chlorocresol (e.g., 2-chloro-3-methyl-phenol or 4-chloro-3-methyl-phenol), benzalkonium chloride, benzethonium chloride, benzoic acid (or a pharmaceutically acceptable salt thereof), sorbic acid (or a pharmaceutically acceptable salt thereof), chlorhexidine, thimerosal, or any combination thereof. The pharmaceutical compositions can be formulated by techniques known to the person skilled in the art, such as the techniques published in “Remington: The Science and Practice of Pharmacy”, Pharmaceutical Press, 22nd edition. The pharmaceutical compositions can be formulated as dosage forms for oral, parenteral, such as intramuscular, intravenous, subcutaneous, intradermal, intraarterial, intracardial, rectal, nasal, topical, aerosol or vaginal administration. Dosage forms for oral administration include coated and uncoated tablets, soft gelatin capsules, hard gelatin capsules, lozenges, troches, solutions, emulsions, suspensions, syrups, elixirs, powders and granules for reconstitution, dispersible
powders and granules, medicated gums, chewing tablets and effervescent tablets. Dosage forms for parenteral administration include solutions, emulsions, suspensions, dispersions and powders and granules for reconstitution. Emulsions are a preferred dosage form for parenteral administration. Dosage forms for rectal and vaginal administration include suppositories and ovula. Dosage forms for nasal administration can be administered via inhalation and insufflation, for example by a metered inhaler. Dosage forms for topical administration include creams, gels, ointments, salves, patches and transdermal delivery systems. The compounds of formula (I) or the above described pharmaceutical compositions comprising a compound of formula (I) may be administered to a subject by any convenient route of administration, whether systemically/peripherally or at the site of desired action, including but not limited to one or more of: oral (e.g., as a tablet, capsule, or as an ingestible solution), topical (e.g., transdermal, intranasal, ocular, buccal, and sublingual), parenteral (e.g., using injection techniques or infusion techniques, and including, for example, by injection, e.g., subcutaneous, intradermal, intramuscular, intravenous, intraarterial, intracardiac, intrathecal, intraspinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratracheal, subcuticular, intraarticular, subarachnoid, or intrasternal by, e.g., implant of a depot, for example, subcutaneously or intramuscularly), pulmonary (e.g., by inhalation or insufflation therapy using, e.g., an aerosol, e.g., through mouth or nose), gastrointestinal, intrauterine, intraocular, subcutaneous, ophthalmic (including intravitreal or intracameral), rectal, or vaginal administration. If said compounds or pharmaceutical compositions are administered parenterally, then examples of such administration include one or more of: intravenously, intraarterially, intraperitoneally, intrathecally, intraventricularly, intraurethrally, intrasternally, intracardially, intracranially, intramuscularly or subcutaneously administering the compounds or pharmaceutical compositions, and/or by using infusion techniques. For parenteral administration, the compounds are best used in the form of a sterile aqueous solution which may contain other substances, for example, enough salts or glucose to make the solution isotonic with blood. The aqueous solutions should be suitably buffered (preferably to a pH of from 3 to 9), if necessary. The preparation of suitable parenteral formulations under sterile conditions is readily accomplished by standard pharmaceutical techniques well known to those skilled in the art. Said compounds or pharmaceutical compositions can also be administered orally in the form of tablets, capsules, ovules, elixirs, solutions or suspensions, which may contain flavoring or coloring agents, for immediate-, delayed-, modified-, sustained-, pulsed- or controlled-release applications. The tablets may contain excipients such as microcrystalline cellulose, lactose, sodium citrate, calcium carbonate, dibasic calcium phosphate and glycine, disintegrants such as starch (preferably corn, potato or tapioca starch), sodium starch glycolate, croscarmellose sodium and certain complex silicates, and granulation binders such as polyvinylpyrrolidone, hydroxypropylmethylcellulose (HPMC),
hydroxypropylcellulose (HPC), sucrose, gelatin and acacia. Additionally, lubricating agents such as magnesium stearate, stearic acid, glyceryl behenate and talc may be included. Solid compositions of a similar type may also be employed as fillers in gelatin capsules. Preferred excipients in this regard include lactose, starch, a cellulose, or high molecular weight polyethylene glycols. For aqueous suspensions and/or elixirs, the agent may be combined with various sweetening or flavoring agents, coloring matter or dyes, with emulsifying and/or suspending agents and with diluents such as water, ethanol, propylene glycol and glycerin, and combinations thereof. For oral administration, the compounds or pharmaceutical compositions are preferably administered by oral ingestion, particularly by swallowing. The compounds or pharmaceutical compositions can thus be administered to pass through the mouth into the gastrointestinal tract, which can also be referred to as “oral-gastrointestinal” administration. Alternatively, said compounds or pharmaceutical compositions can be administered in the form of a suppository or pessary, or may be applied topically in the form of a gel, hydrogel, lotion, solution, cream, ointment or dusting powder. The compounds of the present invention may also be dermally or transdermally administered, for example, by the use of a skin patch. Said compounds or pharmaceutical compositions may also be administered by sustained release systems. Suitable examples of sustained-release compositions include semi-permeable polymer matrices in the form of shaped articles, e.g., films, or microcapsules. Sustained-release matrices include, e.g., polylactides, copolymers of L-glutamic acid and gamma-ethyl-L-glutamate, poly(2-hydroxyethyl methacrylate), ethylene vinyl acetate, or poly-D-(–)-3-hydroxybutyric acid. Sustained-release pharmaceutical compositions also include liposomally entrapped compounds. The present invention thus also relates to liposomes containing a compound of the invention. Said compounds or pharmaceutical compositions may also be administered by the pulmonary route, rectal routes, or the ocular route. For ophthalmic use, they can be formulated as micronized suspensions in isotonic, pH adjusted, sterile saline, or, preferably, as solutions in isotonic, pH adjusted, sterile saline, optionally in combination with a preservative such as a benzalkonium chloride. Alternatively, they may be formulated in an ointment such as petrolatum. It is also envisaged to prepare dry powder formulations of the compounds of formula (I) for pulmonary administration, particularly inhalation. Such dry powders may be prepared by spray drying under conditions which result in a substantially amorphous glassy or a substantially crystalline bioactive powder. Accordingly, dry powders of the compounds of the present invention can be made according to an emulsification/spray drying process. For topical application to the skin, said compounds or pharmaceutical compositions can be formulated as a suitable ointment containing the active compound suspended or dissolved in, for example,
a mixture with one or more of the following: mineral oil, liquid petrolatum, white petrolatum, propylene glycol, emulsifying wax and water. Alternatively, they can be formulated as a suitable lotion or cream, suspended or dissolved in, for example, a mixture of one or more of the following: mineral oil, sorbitan monostearate, a polyethylene glycol, liquid paraffin, polysorbate 60, cetyl esters wax, 2-octyldodecanol, benzyl alcohol and water. The present invention thus relates to the compounds or the pharmaceutical compositions provided herein, wherein the corresponding compound or pharmaceutical composition is to be administered by any one of: an oral route; topical route, including by transdermal, intranasal, ocular, buccal, or sublingual route; parenteral route using injection techniques or infusion techniques, including by subcutaneous, intradermal, intramuscular, intravenous, intraarterial, intracardiac, intrathecal, intraspinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratracheal, subcuticular, intraarticular, subarachnoid, intrasternal, intraventricular, intraurethral, or intracranial route; pulmonary route, including by inhalation or insufflation therapy; gastrointestinal route; intrauterine route; intraocular route; subcutaneous route; ophthalmic route, including by intravitreal, or intracameral route; rectal route; or vaginal route. Preferred routes of administration are oral administration or parenteral administration. For each of the compounds or pharmaceutical compositions provided herein, it is particularly preferred that the respective compound or pharmaceutical composition is to be administered orally (particularly by oral ingestion). Typically, a physician will determine the actual dosage which will be most suitable for an individual subject. The specific dose level and frequency of dosage for any particular individual subject may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the individual subject undergoing therapy. A proposed, yet non-limiting dose of the compounds according to the invention for oral administration to a human (of approximately 70 kg body weight) may be 0.05 to 2000 mg, preferably 0.1 mg to 1000 mg, of the active ingredient per unit dose. The unit dose may be administered, e.g., 1 to 3 times per day. The unit dose may also be administered 1 to 7 times per week, e.g., with not more than one administration per day. It will be appreciated that it may be necessary to make routine variations to the dosage depending on the age and weight of the patient/subject as well as the severity of the condition to be treated. The precise dose and also the route of administration will ultimately be at the discretion of the attendant physician or veterinarian. Therapeutic use In one embodiment, the present invention relates to the compound of formula (I), or a pharmaceutically acceptable salt, or a pharmaceutical composition as defined herein for use in therapy.
The present invention provides compounds that have activity of inhibitors of Werner syndrome ATP- dependent helicase (WRN). Thus, accordingly, the present invention provides a method of inhibiting WRN enzyme activity in vitro and in vivo, said method comprising contacting a cell with an effective amount of the compound of formula (I), or a pharmaceutically acceptable salt thereof, as defined herein. In one embodiment, the present invention provides a method for decreasing proliferation in a proliferative cell having a microsatellite instability (MSI), comprising decreasing the helicase activity of Werner syndrome ATP-dependent helicase (WRN) in the proliferative cell. In some embodiments, decreasing the helicase activity of Wemer syndrome ATP-dependent helicase (WRN) in the proliferative cell is achieved by administering a compound of Formula of (I) (or any embodiment thereof disclosed herein) or a pharmaceutically acceptable salt thereof. In some embodiments, the proliferative cell is characterized as having MSI low (MSI-L). In some embodiments, the proliferative cell is characterized as having high MSI (MSI-H), used interchangeably with MSI-high. Cells can be characterized as MSI, including MSI-L or MSI- H, or as MSS (MS-stable), according to the method known in the art (see, for example, Dudley, Jonathan C., et al., Clinical Cancer Research, 22(4): 813-820, 2016.). MSI-H is used to classify tumors as having a high frequency of MSI. A tumor can be classified as MSI, including MSI-low or MSI-high, using polymerase chain reaction (PCR) and/or immunohistochemistry (IHC) assays. As stated in Dudley et al., a tumor is classified as MSI-H by PCR if (i) there is a shift (usually downward) in the size of at least two microsatellite loci from a reference panel of five microsatellite loci in tumor relative to normal, where the reference panel can be the “Bethesda Panel,” also referred to herein as the “NCI- Reference Panel (Bethesda, 1998)”, which includes two mononucleotide loci (BAT-25 and BAT-26) and three dinucleotide loci (D2S123, D5S346, and D17S250), or alternatively, the reference panel can be Promega Corporation’s MSI Analysis System, which includes five mononucleotide loci (BAT-25, BAT-26, NR-21, NR-24, and MONO-27); or (ii) there is a shift in the size of 30% or more microsatellite loci from a reference panel of more than five microsatellite loci in tumor relative to normal. The MSI-H phenotype is associated with germline defects in the mismatch repair genes MLH1, MSH2, MSH6, and PMS2, and is the primary phenotype observed in tumors from patients with HNPCC/Lynch syndrome. A tumor is classified as MSI-H in IHC test if it shows a loss of protein expression for at least 1 of the above 4 mismatch repair genes. Cells can be similarly classified as MSI-H using the tests described herein for tumors. In some embodiments, a tumor or cell is classified as MSI-H using PCR to amplify the five microsatellite loci of the “Bethesda Panel” (BAT-25, BAT-26, D2S123, D5S346, and D17S250) from both tumor tissue or cells and normal tissue or cells, wherein the tumor or cell is classified as MSI-H if there is a shift in the size of at least two of the microsatellite loci from the tumor tissue or cells relative to the normal tissue or cells. In some embodiments, the shift in size of the microsatellite loci is a downward shift. In some embodiments, a tumor or cell is classified as MSI-H using PCR to amplify the five
microsatellite loci of Promega Corporation’s MSI Analysis System (BAT-25, BAT-26, NR- 21, NR-24, and MONO-27) from both tumor tissue or cells and normal tissue or cells, wherein the tumor or cell is classified as MSI-H if there is a shift in the size of at least two of the microsatellite loci from the tumor tissue or cells relative to the normal tissue or cells. In some embodiments, the shift in size of the microsatellite loci is a downward shift. In some embodiments, a tumor is classified as MSI-H using IHC to determine the expression level of the MMR proteins MLH1, MSH2, MSH6, and/or PMS2 in both tumor tissue and normal tissue, wherein the tumor is classified as MSI-H if there is a loss of protein expression for at least one of the MMR proteins in the tumor tissue relative to the normal tissue. In some embodiments, the loss of protein expression is a decrease of at least 20% (such as a decrease of 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 99%, or more). In contrast, a tumor is classified as MSI-L by PCR if (i) there is a shift in the size of one microsatellite locus from a reference panel of five microsatellite loci in tumor relative to normal, where the reference panel can be the “Bethesda Panel” or Promega Corporation’s MSI Analysis System; or (ii) there is a shift in the size of less than 30% microsatellite loci from a reference panel of more than five microsatellite loci in tumor relative to normal. MSI-L tumors are thought to represent a distinct mutator phenotype with potentially different molecular etiology than MSI-H tumors (Thibodeau, 1998; Wu et al., 1999, Am J Hum Genetics 65: 1291-1298). Cells can be similarly classified as MSI-L using the tests described herein for tumors. Cancers classified as MSI-H include, but not limited to, uterine corpus endometrial carcinoma, colon adenocarcinoma, stomach adenocarcinoma, rectal adenocarcinoma, adenoid cystic carcinoma, uterine carcinosarcoma, cervical squamous cell carcinoma, and endocervical adenocarcinoma. In one embodiment, the present invention relates to a method for treating a cancer treatable by inhibition of WRN in a subject in need thereof, the method comprising the step of administering to the subject the compound of formula (I) or a pharmaceutically acceptable salt thereof. It is to be understood that, typically, a therapeutically effective amount is to be administered. In one embodiment, the present invention relates to a method for treating a cancer characterized by MSI-H and/or dMMR in a subject in need thereof, the method comprising the step of administering to the subject the compound of formula (I) or a pharmaceutically acceptable salt thereof. It is to be understood that, typically, a therapeutically effective amount is to be administered. In one embodiment, the present invention relates to use of a compound of formula (I) or pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer. Preferably, the cancer is treatable by inhibition of WRN. In one embodiment, the cancer is characterized by MSI-H and/or dMMR.
In one embodiment, the present invention relates to the compound of formula (I) or a pharmaceutically acceptable salt thereof, for use in treating cancer. Preferably, the cancer is treatable by inhibition of WRN. In one embodiment, the cancer is characterized by MSI-H and/or dMMR. The cancer to be treated in accordance with the present invention may be a solid cancer or a hematological cancer. Preferably, the cancer is selected from lung cancer (e.g., small cell lung cancer, non-small cell lung cancer, large cell lung carcinoma, lung adenocarcinoma, including also lung adenocarcinoma with EGFR mutation E746-A750, or squamous cell carcinoma of the lung), renal cancer (or kidney cancer; e.g., renal carcinoma), gastrointestinal cancer, stomach cancer, colorectal cancer (e.g., colorectal carcinoma), colon cancer, anal cancer, genitourinary cancer, bladder cancer, liver cancer (e.g., hepatocellular carcinoma), pancreatic cancer (e.g., pancreatic adenocarcinoma or pancreatic ductal adenocarcinoma), cervical cancer, endometrial cancer, vaginal cancer, vulvar cancer, ovarian cancer (e.g., ovarian carcinoma), uterine cancer, prostate cancer (e.g., hormone-refractory prostate cancer), testicular cancer, biliary tract cancer, hepatobiliary cancer, neuroblastoma, brain cancer (e.g., glioblastoma), breast cancer (e.g., triple-negative breast cancer, breast cancer having a BRCA1 and/or BRCA2 gene mutation, or breast adenocarcinoma), head and/or neck cancer (e.g., head and neck squamous cell carcinoma), skin cancer, melanoma, Merkel-cell cancer (e.g., Merkel-cell carcinoma), epidermoid cancer, squamous cell cancer (or squamous cell carcinoma; including, e.g., oral squamous cell carcinoma/squamous-cell mouth carcinoma, squamous-cell skin cancer, squamous-cell lung carcinoma, squamous-cell thyroid carcinoma, squamous-cell esophageal carcinoma, or squamous-cell vaginal carcinoma), bone cancer (e.g., osteosarcoma or osteogenic sarcoma), fibrosarcoma, Ewing’s sarcoma, malignant mesothelioma, esophageal cancer, laryngeal cancer, mouth cancer, thymoma, neuroendocrine cancer (e.g., neuroendocrine carcinoma), goblet cell cancer (e.g., goblet cell carcinoid), hematological cancer, leukemia (e.g., acute lymphoblastic leukemia, acute myeloid leukemia, chronic lymphocytic leukemia, or chronic myeloid leukemia), lymphoma (e.g., Hodgkin lymphoma or non-Hodgkin lymphoma, such as, e.g., follicular lymphoma or diffuse large B-cell lymphoma), and multiple myeloma. Moreover, the cancer to be treated (including any one of the aforementioned specific types of cancer) may also be a chemoresistant and/or a metastatic cancer. The antiproliferative treatment (i.e. the treatment of cancer) with the compound of formula (I) or a pharmaceutically acceptable salt thereof, as defined hereinbefore, may be applied as a sole therapy or may involve, in addition to the compound of the invention, conventional surgery or radiotherapy or chemotherapy. Such chemotherapy may include one or more of the following categories of anti-tumour agents:- (i) other antiproliferative/antineoplastic drugs and combinations thereof, as used in medical oncology, such as alkylating agents (for example cis-platin, oxaliplatin, carboplatin, cyclophosphamide,
nitrogen mustard, melphalan, chlorambucil, busulphan, temozolamide and nitrosoureas); antimetabolites (for example gemcitabine and antifolates such as fluoropyrimidines like 5-fluorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside, and hydroxyurea); antitumour antibiotics (for example anthracyclines like adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin); antimitotic agents (for example vinca alkaloids like vincristine, vinblastine, vindesine and vinorelbine and taxoids like taxol and taxotere and polokinase inhibitors); and topoisomerase inhibitors (for example epipodophyllotoxins like etoposide and teniposide, amsacrine, topotecan and camptothecin); (ii) cytostatic agents such as antioestrogens (for example tamoxifen, fulvestrant, toremifene, raloxifene, droloxifene and iodoxyfene), antiandrogens (for example bicalutamide, flutamide, nilutamide and cyproterone acetate), LHRH antagonists or LHRH agonists (for example goserelin, leuprorelin and buserelin), progestogens (for example megestrol acetate), aromatase inhibitors (for example as anastrozole, letrozole, vorazole and exemestane) and inhibitors of 5oc-reductase such as finasteride; (iii) anti-invasion agents [for example c-Src kinase family inhibitors like 4-(6-chloro-2,3- methylenedioxyanilino)-7-[2-(4-methylpiperazin-1 -yl)ethoxy]-5-tetrahydropyran-4- yloxyquinazoline (AZD0530; International Patent Application WO 01/94341 ), N-(2-chloro-6- methylphenyl)-2-{6-[4-(2- hydroxyethyl)piperazin-1 -yl]-2-methylpyrimidin-4-ylamino}thiazole- 5-carboxamide (dasatinib, BMS- 354825; J. Med. Chem., 2004, 47, 6658-6661 ) and bosutinib (SKI-606), and metalloproteinase inhibitors like marimastat, inhibitors of urokinase plasminogen activator receptor function or antibodies to Heparanase]; (iv) inhibitors of growth factor function: for example such inhibitors include growth factor antibodies and growth factor receptor antibodies (for example the anti-erbB2 antibody trastuzumab [Herceptin™], the anti-EGFR antibody panitumumab, the anti-erbB1 antibody cetuximab [Erbitux, C225] and any growth factor or growth factor receptor antibodies disclosed by Stern et al. (Critical reviews in oncology/haematology, 2005, Vol.54, pp11 -29); such inhibitors also include tyrosine kinase inhibitors, for example inhibitors of the epidermal growth factor family (for example EGFR family tyrosine kinase inhibitors such as N-(3-chloro- 4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazolin-4-amine (gefitinib, ZD1839), N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (erlotinib, OSI-774) and 6- acrylamido-N-(3-chloro-4-fluorophenyl)-7-(3-morpholinopropoxy)-quinazolin-4-amine (CI 1033), erbB2 tyrosine kinase inhibitors such as lapatinib); inhibitors of the hepatocyte growth factor family; inhibitors of the insulin growth factor family; inhibitors of the platelet-derived growth factor family such as imatinib and/or nilotinib (AMN107); inhibitors of serine/threonine kinases (for example Ras/Raf signalling inhibitors such as farnesyl transferase inhibitors, for example sorafenib (BAY 43-9006), tipifarnib (R1 15777) and lonafarnib (SCH66336)), inhibitors of cell signalling through MEK and/or AKT kinases, c-kit
inhibitors, abl kinase inhibitors, PI3 kinase inhibitors, Plt3 kinase inhibitors, CSF-1 R kinase inhibitors, IGF receptor (insulin-like growth factor) kinase inhibitors; aurora kinase inhibitors (for example AZD1152, PH739358, VX-680, MLN8054, R763, MP235, MP529, VX-528 AND AX39459) and cyclin dependent kinase inhibitors such as CDK2 and/or CDK4 inhibitors; (v) antiangiogenic agents such as those which inhibit the effects of vascular endothelial growth factor, [for example the anti-vascular endothelial cell growth factor antibody bevacizumab (Avastin™) and for example, a VEGF receptor tyrosine kinase inhibitor such as vandetanib (ZD6474), vatalanib (PTK787), sunitinib (SU11248), axitinib (AG-013736), pazopanib (GW 786034) and 4-(4-fluoro-2-methylindol-5- yloxy)-6-methoxy-7-(3-pyrrolidin-1 - ylpropoxy)quinazoline (AZD2171 ; Example 240 within WO 00/47212), compounds such as those disclosed in International Patent Applications W097/22596, WO 97/30035, WO 97/32856 and WO 98/13354 and compounds that work by other mechanisms (for example linomide, inhibitors of integrin 3 function and angiostatin)]; (vi) vascular damaging agents such as Combretastatin A4 and compounds disclosed in International Patent Applications WO 99/02166, WO 00/40529, WO 00/41669, WO 01 /92224, WO 02/04434 and WO 02/08213; (vii) an endothelin receptor antagonist, for example zibotentan (ZD4054) or atrasentan; (viii) antisense therapies, for example those which are directed to the targets listed above, such as ISIS 2503, an anti-ras antisense; (ix) gene therapy approaches, including for example approaches to replace aberrant genes such as aberrant p53 or aberrant BRCA1 or BRCA2, GDEPT (gene-directed enzyme pro-drug therapy) approaches such as those using cytosine deaminase, thymidine kinase or a bacterial nitroreductase enzyme and approaches to increase patient tolerance to chemotherapy or radiotherapy such as multi- drug resistance gene therapy; and (x) immunotherapy approaches, including for example ex-vivo and in-vivo approaches to increase the immunogenicity of patient tumour cells, such as transfection with cytokines such as interleukin 2, interleukin 4 or granulocyte-macrophage colony stimulating factor, approaches to decrease T-cell anergy, approaches using transfected immune cells such as cytokine-transfected dendritic cells, approaches using cytokine-transfected tumour cell lines and approaches using anti-idiotypic antibodies. In a particular embodiment, the antiproliferative treatment defined hereinbefore may involve, in addition to the compound of formula (I) of the invention, conventional surgery or radiotherapy or chemotherapy. Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate dosing of the individual components of the treatment. Such combination products employ the compounds of this invention within the dosage range described hereinbefore and the other pharmaceutically-active agent within its approved dosage range.
According to this aspect the present invention further relates to the compound of formula (I) or a pharmaceutically acceptable salt thereof, as defined herein, for use in the treatment of a cancer (for example a cancer involving a solid tumour) in combination with another anti-tumour agent. The anti- tumour agent is preferably selected from the anti-tumour agents as listed hereinabove. As understood herein, the term "combination" refers to simultaneous, separate or sequential administration. In one aspect of the invention "combination" refers to simultaneous administration. In another aspect of the invention "combination" refers to separate administration. In a further aspect of the invention "combination" refers to sequential administration. Where the administration is sequential or separate, the delay in administering the second component should not be such as to lose the beneficial effect of the combination. Examples The following examples are merely illustrative of the present invention and should not be construed to limit the scope of the invention which is defined by the appended claims. Synthesis of the compounds of formula (I) The syntheses of the compounds of formula (I) according to the present invention are preferably carried out according to the general synthetic sequences as shown in Scheme 1. In addition to said routes described below, also other routes may be used to synthesize the target compounds, in accordance with common general knowledge of a person skilled in the art of organic synthesis. The order of transformations exemplified in the following Schemes is therefore not intended to be limiting, and suitable synthesis steps from various schemes can be combined to form additional synthesis sequences. In addition, modification of any of the substituents can be achieved before and/or after the exemplified transformations. These modifications can be such as the introduction of protective groups, cleavage of protective groups, reduction or oxidation of functional groups, halogenation, metallation, metal-catalyzed coupling reactions, substitution or other reactions known to a person skilled in the art. These transformations include those which introduce a functionality allowing for further interconversion of substituents. Appropriate protective groups and their introduction and cleavage are well-known to a person skilled in the art (see for example: Greene's Protective Groups in Organic Synthesis; Editor: P.G.M. Wuts, 5th edition, Wiley 2014). Specific examples are described in the subsequent paragraphs. Further, it is possible that two or more successive steps may be performed without work-up being performed between said steps, e.g. a “one-pot” reaction, as it is well-known to a person skilled in the art. It is further understood to the skilled person that a reaction can lead to side product(s) which, when appropriate, can be used for the preparation of compounds of formula (I) using similar procedures as reported in the general schemes hereinbelow. Scheme 1
Scheme 1 illustrates a preferred synthetic approach to compounds of the general formula (I). In the first step, compound 3 is prepared via cross-coupling reactions of suitably functionalized building blocks 1 and 2. Formation of a C-C bond between A (1) and B-X (2) can be e.g. achieved by the cross-coupling of an aryl, heteroaryl, alkyl, or vinyl boronic acid, borate ester, or borane 1 with an aryl or heteroaryl halide or triflate 2 using a variety of palladium catalysts (Suzuki reaction; see e.g. B. S. Kadu, Catal. Sci. Technol., 2021,11, 1186-1221). The formation of a carbon–carbon bond between a terminal alkyne 1 and an aryl or heteroaryl halide 2 can be achieved employing palladium catalysts as well as copper co-catalysts (Sonogashira coupling, see e.g. I. Kanwal et al, Catalysts 2020, 10(4), 443). Moreover, formation of a C-N can be achieved via the palladium-catalyzed coupling reactions of amines 1 with aryl and heteroaryl halides 2 (Buchwald–Hartwig coupling, see e.g. R. Dorel et al, Angew. Chem. Int. Ed.2019, 58, 17118). In the second step, cross-coupling of A-B-X (3) and Y (4) can be achieved by various methods known of a person skilled in the art of organic synthesis, including e.g. a) coupling of carboxylic acids (3) and amines (4) (amide bond formation; see e.g. E. Massolo et al, Eur. J. Org. Chem.2020, 4641), b) coupling of sulfonyl chlorides (3) and amines (4) (sulfonamide bond formation: see e.g. A. Kolaczek et al, CHEMIK 2014, 68, 620) or c) coupling of amines (3) and halides (4) (amine alkylation: see e.g. R. N. Salvatoreet al, Tetrahedron 2001, 7785). For the synthesis of compounds of formula (I) in which B and X are combined to form a two ring system:
 similar cross-coupling reactions can be employed. The synthesis of the compounds of formula (I) bearing a spiro moiety:
similar cross-coupling reactions can be employed. The synthesis of the compounds of formula (I) bearing a spiro moiety:
 can be achieved utilizing the described cross-coupling reactions, too. Suitable building block containing the spiro motive are either commercial or can be synthesized by various methods known of a person skilled in the art of organic synthesis (see e.g. Spiro Compounds: Synthesis and Applications, ISBN:9781119567646, Editor: R. R. Torres, John Wiley & Sons). Preparative examples General considerations Abbreviations used in the descriptions that follow are: AcOH (acetic acid); aq. (aqueous); Ar (argon); Atm (atmosphere); BH3.THF (boran tetrahydrofuran complex); br (broad, 1H NMR signal); Boc2O (di-tert- butyldicarbonate); BuLi (Butyl Lithium), Cataxium APdG3 (mesylate[(di(1-adamantyl)-n-butylphosphine)- 2-(2 -amino-1,1 -biphenyl)]palladium(II)); CDCl3 (deuterated chloroform); cHex (cyclohexane); CMPB (cyanomethylene trimethylphosphorane); Cs2CO3 (cesium carbonate); CuI (copper iodide); CuSO4 (cupric sulfate), DABCO (1,4-diazabicyclo[2.2.2]octane); DAST (diethylaminosulfur trifluoride); DBU (1,8-diazabicyclo(5.4.0)undec-7-ene); DCE (dichloroethane); d (doublet, 1H NMR signal); DCM (dichloromethane); DEA (diethylamine); DIBAL-H (diisobutyl aluminium hydride); DIPEA or DIEA (di-iso- propylethylamine); DMA (dimethylacetamide); (DMAP (4- N-N-dimethylaminopyridine), DME (1,2- dimethoxyethane), DMEDA (dimethylethylenediamine ); DMF (N-N-dimethylformamide); DMSO (dimethyl sulfoxide); DPPA (diphenylphosphoride azide); dtbbpy (bis(1,1-dimethylethyl)-2,2 -bipyridine); ee (enantiomeric excess); ES (electrospray); EtOAc or EA (ethyl acetate); EtOH (ethanol); h (hour(s)); FA (formic acid); MgO (magnesium oxide); HATU (1-[bis(dimethylamino)methylene]-1H-1,2,3- triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate); HBr (hydrobromic acid); HFIP ( hexafluoroisopropanol); HI (hydrogen iodide); 1H NMR (proton nuclear magnetic resonance spectroscopy); HPLC (high performance liquid chromatography); iPrMgBr (isopropylmagnesium bromide); iPrOH (iso-propanol); K2CO3 (potassium carbonate); K3PO4 (tripotassium phosphate); Ir[dF(CF3)(dtbbpy)PF6 ((4,4'-di-t-butyl-2,2'-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl- kN)phenyl-kC]iridium(III) hexafluorophosphate); LiOH (lithium hydroxide); m (multiplet, 1H NMR signal); mCPBA (meta-chloroperoxybenzoic acid), MeCN (acetonitrile), MeOH (methanol); MeOK (potassium methanolate); min (minute(s)); MnO2 (manganese (IV) oxide); MS (mass spectrometry); MTBE (methyl tert-butyl ether); NaBH4 (sodium borohydride); NaHCO3 (sodium hydrogenocarbonate); NaI (sodium iodide); NaIO4 (sodium periodate); Na2SO3 (sodium sulfite); Na2S2O3 (sodium thiosulfate); Na2SO4 (sodium sulfate); NCS (N-chlorosuccinimide); NH3 (ammonia); NH4Cl (ammonium chloride); NiCl2 (nickel dichloride); NIS (N-iodosuccinimide); NMP (N-methylpyrrolidone); NMR (nuclear magnetic resonance); Pd/C (palladium on charcoal); Pd2dba3 (tris(dibenzylideneacetone)dipalladium); Pd(dppf)Cl2 (1,1- bis(diphenylphosphino)ferrocene dichloropalladium); Pd(Ph3)4 (tetrakis(triphenylphosphine) palladium (0)); Pd(Ph3)2Cl2 (bis(triphenylphosphine)palladium(II) dichloride ); PE (petroleum ether); Pd-PEPPSI-
IPentCl o-picoline ([1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5-dichloro-imidazol-2-ylidene]-dichloro-(2- methylpyridin-1-ium-1-yl)palladiµm; Pd(OH)2 (palladium hydroxide); Pd(Ph3)4 (palladium- tetrakis(triphenylphosphine)); PhI(OAc)2 ((diacetoxyiodo)benzene)); P(tBu)3 (tri-tert-butylphosphine ); Py (pyridine); q (quartet, 1H NMR signal); quin (quintet, 1H NMR signal); rac (racemic); RT (retention time); s (singlet, 1H NMR signal); sat. (saturated); t (triplet, 1H NMR signal); SFC (supercritical fluid chromatography); TBAF (tetrabutylammonium fluoride); tert-BuBrettPhos-Pd-G3 ([(2-di-tert- butylphosphino-3,6-dimethoxy-2 ,4 ,6 -triisopropyl-1,1 -biphenyl)-2-(2 -amino-1,1 -biphenyl)]palladium(II) methanesulfonate); tBuXPhos Pd G3 (methanesulfonato(2-di-t-butylphosphino-2',4',6'-tri-i-propyl-1,1'- biphenyl)(2'-amino-1,1'-biphenyl-2-yl)palladium(II)); TBDMSCl or TBSCl (tert-butyldimethylsilyl chloride); tBuOH (tert-butanol); TEA (triethylamine); TFA (trifluoroacetic acid); TFAA (trifluoroacetic anhydride); THF (tetrahydrofuran); TLC (thin layer chromatography); TMSCHN2 (trimethylsilyldiazomethane); TMSCN (trimethylsilyl cyanide); TMSI (trimethylsilyl iodide); TMSOTf (trimethylsilyl trifluoromethanesulfonate); TTMSS (trimethylsilane); TsOH (p-Toluenesulfonic acid); T4P (2,4,6-tributyl- 1,3,5,2,4,6-trioxatriphosphinane 2,4,6-trioxide); UPLC (ultra-high performance liquid chromatography), UV (ultraviolet), wt-% (percent by weight); Xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene); Xantphos Pd G4 (methanesulfonato[9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene](2'- methylamino-1,1'-biphenyl-2-yl)palladium(II)), Xphos (2-Dicyclohexylphosphin-2 ,4 ,6 - triisopropylbiphenyl). General Procedure: All starting materials and solvents were obtained either from commercial sources or prepared according to literature references. Commercially available reagents and anhydrous solvents were used as supplied, without further purification. Unless otherwise stated all reactions were stirred. Organic solutions were routinely dried over anhydrous sodium sulfate. Column chromatography was performed on pre-packed silica (100-1000 mesh, 40-63 µm) cartridges using the amount indicated. All air- and moisture-sensitive reactions were carried out in oven-dried (at 120 °C) glassware under an inert atmosphere of nitrogen or argon. Compound names were generated using ChemDraw Professional (Perkin Elmer, Version 16.0.1.4 (77)). In some cases generally accepted names of commercially available reagents were used in place of ChemDraw generated names. Methods for Reversed Phase HPLC conditions for LCMS Analysis of compounds: Method 1: SHIMADZU LCMS-2020 Kinetex EVO C182.1X30mm, 5 m at 50 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.55 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1.20 min, held at 5% A-95% B; 1.20-1.21 min, returned to 95% A-5% B, 1.21-1.55 min, held at 95% A-5% B.
Method 2: SHIMADZU LCMS-2020 Kinetex EVO C182.1X30mm, 5 m at 40 Mobile Phase A: 0.025% NH3·H2O in water (v/v); B: MeCN; flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.55 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1.20 min, held at 5% A-95% B; 1.20-1.21 min, returned to 95% A-5% B, 1.21-1.55 min, held at 95% A-5% B. Method 3: SHIMADZU LCMS-2020 Kinetex EVO C182.1X30mm,5 m at 50 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 2.0 mL/min; eluted with the mobile phase over 0.80 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1.20 min, held at 5% A-95% B; 1.20-1.21 min, returned to 95% A-5% B, 1.21-1.55 min, held at 95% A-5% B. Method 4: SHIMADZU LCMS-2020 Kinetex® EVO C182.1X20 mm 2.6 m at 50°C; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 2.0 mL/min; eluted with the mobile phase over 1.00 min employing UV detection at 220 nm and 254 nm. Gradient information: 0.01-0.60 min, ramped from 95% A-5% B to 5% A-95% B; 0.61-0.78 min, held at 5% A-95% B; 0.78-0.79 min, returned to 95% A-5% B, 0.79-0.80 min, held at 95% A-5% B. Method 5: SHIMADZU LCMS-2020 Kinetex® EVO C182.1X30 mm 5 m at 50°C Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.00 min employing UV detection at 220 nm and 254 nm. Gradient information: 0.01-0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-0.95 min, held at 5% A-95% B; 0.95-0.96 min, returned to 95% A-5% B, 0.96-1.00 min, held at 95% A-5% B. Method 6: SHIMADZU LCMS-2020 Kinetex® HALO C183.0X30mm, 5 m at 50 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.05 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 0.80 min, ramped from 50% A-50% B to 0% A-100% B; 0.80-1.05 min, held at 50% A-50% B. Method 7: SHIMADZU LCMS-2020 HALO C183.0X30 mm, 5 m at 50 , Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in Acetonitrile (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.05 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-0.50 min, ramped from 95% A-5% B to 5% A-95% B; 0.50-0.80 min, held at 5% A-95% B; 0.80-0.81 min, returned to 95% A-5% B, 0.81-1.05 min, held at 95% A-5% B. Method 8: SHIMADZU LCMS-2020 HALO C183.0X30mm, 5 m at 50 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with
the mobile phase over 0.80 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 0.45 min, ramped from 95% A-5% B to 5% A-95% B; 0.45-0.70 min, held at 5% A-95% B; 0.70-0.71 min, returned to 95% A-5% B, 0.71-0.80 min, held at 95% A-5% B. Method 9: SHIMADZU LCMS-2020 Kinetex® EVO C182.1X30 mm, 5 m at 40 Mobile Phase: A: water; B: MeCN; flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.55 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1.20 min, held at 5% A-95% B; 1.20-1.21 min, returned to 95% A-5% B, 1.21-1.55 min, held at 95% A-5% B. Method 10: SHIMADZU LCMS-2020 Waters Xselect HSS T3, 3.5 µm, 4.6*50mm at 40 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 5.20 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-3.50 min, ramped from 95% A-5% B to 5% A-95% B; 3.50-4.80 min, held at 5% A-95% B; 4.80-4.81 min, returned to 95% A-5% B; 4.81-5.20 min, held at 95% A-5% B Method 11: SHIMADZU LCMS-2020 HALO C183.0X30mm,5 m at 50 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 3.00 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 2.10 min, ramped from 95% A-5% B to 5% A-95% B; 2.10-2.80 min, held at 5% A-95% B; 2.80-2.81 min, returned to 95% A-5% B, 2.81-3.00 min, held at 95% A-5% B. Method 12: SHIMADZU LCMS-2020 HALO C183.0X30 mm, 5 m at 50 , Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in Acetonitrile (v/v); flow rate held at 2 mL/min; eluted with the mobile phase over 1.05 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-0.40 min, ramped from 95% A-5% B to 5% A-95% B; 0.40-0.75 min, held at 5% A-95% B; 0.75-0.76 min, returned to 95% A-5% B, 0.76-1.05 min, held at 95% A-5% B. Methods for MS Analysis of compounds: Method 1: SHIMADZU LCMS-2020 Kinetex® EVO C182.1X30 mm, 5 m at 40 Mobile Phase: A: 0.025% NH3·H2O in water (v/v); B: MeCN; flow rate held at 1.5 mL/min; eluted with the mobile phase over 0.40 min employing UV detection at 220 nm. Gradient information: 0-0.40 min, ramped from 10% A-90% B to 10% A-90% B; Methods for SFC analysis of compounds: SFC Method 1: Column: Chiralcel OX-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH+ACN with 0.05% DEA additive; Gradient elution: 50% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar.
SFC Method 2: Column:Chiralpak AD-350*4.6 mm I.D., 3 µm Mobile phase: Phase A for CO2, Phase B for EtOH+ACN with 0.05% DEA additive; Gradient elution: 60% B in CO2 , Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 3: Column:Chiralpak IG-350*4.6 mm I.D., 3 m; Mobile phase: Phase A for CO2, and Phase B for MeOH+ACN with 0.05% DEA additive; Gradient elution: 60% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 4: Column:Chiralcel OJ-350*4.6mm I.D.,3 m; Mobile phase: Phase A for CO2,and Phase B for MeOH(0.05% DEA); Gradient elution: MeOH(0.05% DEA) in CO2 from 5% to 40%, Flow rate:3 mL/min; Detector: PDA; Column Temp: 35C; Back Pressure:100Bar. SFC Method 5: Column:Chiralpak IG-350*4.6mm I.D.,3 m; Mobile phase: Phase A for CO2, and Phase B for IPA+ACN(0.05% DEA); Gradient elution: IPA+ACN(0.05% DEA) from 20% to 60% in CO2; Flow rate:3 mL/min; Detector :PDA; Column Temp: 35C; Back Pressure:100Bar. SFC Method 6: Column: Chiralcel OJ-350*4.6mm I.D.,3 m; Mobile phase: Phase A for CO2, and Phase B for IPA(0.05% DEA); Gradient elution: IPA(0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35C; Back Pressure: 100Bar. SFC Method 7: Column:Chiralcel OJ-350*4.6 mm I.D., 3 m; Mobile phase:Phase A for CO2, and Phase B for EtOH(0.05% DEA); Gradient elution: EtOH(0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min;Detector: PDA; Column Temp: 35C; Back Pressure: 100Bar. SFC Method 8: Column: Chiralpak IC-350*4.6mm I.D., 3 m Mobile phase: Phase A for CO2 and Phase B for EtOH(0.05%DEA); Gradient elution: 60% EtOH(0.05%DEA) in CO2 ,Flow rate:3mL/min; Detector:PDA; Column Temp:35C; Back Pressure:100Bar" SFC Method 9: Column: Chiralpak IC-350*4.6mm I.D., 3 m Mobile phase: Phase A for CO2and Phase B for IPA+ACN(0.05%DEA); Gradient elution: IPA+ACN(0.05%DEA) in CO2 from 20% to 60%, Flow rate:3mL/min; Detector:PDA; Column Temp:35C; Back Pressure:100Bar". SFC Method 10: Column: Chiralpak AD-350*4.6mm I.D.,3 m Mobile phase: Phase A for CO2,and Phase B for IPA+ACN(0.05%DEA); Gradient elution: IPA+ACN(0.05%DEA) in CO2 from 20% to 60%, Flow rate:3mL/min; Detector:PDA; Column Temp:35C;Back Pressure:100Bar SFC Method 11: Column: (S,S)Whelk-O150*4.6mm I.D., 3.5 m; Mobile phase:Phase A for CO2,and Phase B for IPA(0.05%DEA); Gradient elution: IPA (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100 Bar. SFC Method 12: Column: Chiralpak AD-350*4.6mm I.D., 3 m; Mobile phase: Phase A for CO2, and Phase B for MeOH(0.05%DEA); Gradient elution: MeOH (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100 Bar.
SFC Method 13: Column: Chiralpak AD-350*4.6mm I.D., 3 m Mobile phase: Phase A for CO2, and Phase B for IPA (0.05%DEA); Gradient elution: IPA (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100 Bar. SFC Method 14: Column: Chiralpak IG-350*4.6 mm I.D., 3 m; Mobile phase: Phase A for CO2, and Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 20% to 60% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100 Bar. SFC Method 15: Column: Chiralcel OX-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH (0.05% DEA), Gradient elution: MeOH (0.05% DEA) from 10% to 60% in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 16: Column: Chiralcel OD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA), Gradient elution: EtOH (0.05% DEA) from 5% to 40% in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 17: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH+ACN (0.05% DEA); Gradient elution: 60% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 18: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA (0.05% DEA); Gradient elution: 40% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 19: Column: Chiralcel OZ -350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH+ACN (0.05% DEA); Gradient elution: 60% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 20: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA (0.05% DEA); Gradient elution: IPA (0.05% DEA) in CO2 from 20% to 60%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar SFC Method 21: Column:Chiralcel OJ-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: IPA+ACN (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 22: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: 60% EtOH (0.05% DEA) in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 23: Column: Lux 3um Cellulose-250*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH (0.05% DEA); Gradient elution: 60% MeOH (0.05% DEA) in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar.
SFC Method 24: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 25: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: 40% EtOH (0.05% DEA) in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 26: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 20% to 60% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 27: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH (0.05% DEA); Gradient elution: 40% MeOH (0.05% DEA) in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 28: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: 40% IPA+ACN in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar SFC Method 29: Column: Chiralpak ID-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: 60% IPA+ACN (0.05% DEA) in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 30: Column: Chiralpak IC-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) in CO2 from 10% to 60%, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 31: Column: Chiralcel OJ-350*4.6mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH+ACN (0.05% DEA); Gradient elution: from 20% to 60% of EtOH+ACN (0.05% DEA) in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar SFC Method 32: Column: Chiralpak IE-3100*4.6mm I.D., 3 µm; Mobile phase: Phase A for Water with 0.0375%TFA, Phase B for ACN (0.01875% TFA); Gradient elution: 80% B in A; Flow rate: 1mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar SFC Method 33: Column: Chiralpak IK-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: 50% IPA+ACN (0.05% DEA) in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar SFC Method 34: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 20% to 40% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar.
SFC Method 35: Column: Chiralpak IH-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 5% to 40% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 36: Column: Chiralpak AS-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 5% to 40% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar SFC Method 37: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH (0.05% DEA); Gradient elution: 60% MeOH (0.05% DEA) in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 38: Column: Chiralcel Cellulose-250*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 20% to 60% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 39: Column: Chiralpak AY-350*4.6mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: 40% EtOH (0.05%DEA) in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 40: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 5% to 40% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 41: Column: Chiralpak AS-350*4.6mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH (0.05% DEA); Gradient elution: MeOH (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 42: Column: Lux 3um Cellulose-450*4.6mm I.D, 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: IPA+ACN (0.05% DEA) from 20% to 60% in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar SFC Method 43: Column: Chiralcel OX-350*4.6 mm I.D., 3 m; Mobile phase: Phase A for CO2, and Phase B for IPA (0.05% DEA); Gradient elution: IPA (0.05 % DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100Bar. SFC Method 44: Column: Chiralpak IC-350*4.6mm I.D.,3 m Mobile phase: Phase A for CO2 and Phase B for EtOH (0.05%DEA); Gradient elution: EtOH(0.05%DEA) in CO2 from 5% to 40%, Flow rate:3mL/min; Detector: PDA; Column Temp:35 ; Back Pressure:100Bar 1H NMR Spectroscopy: 1H NMR spectra were acquired on a Bruker Avance spectrometer at 400 MHz using residual undeuterated solvent as reference. 1H NMR signals are specified with their multiplicity / combined
multiplicities as apparent from the spectrµm; possible higher-order effects are not considered. Chemical shifts of the signals ( ) are specified as ppm (parts per million). Salt stoichiometry: In the present text, in particular in the experimental section, for the synthesis of intermediates and of examples of the present invention, when a compound is mentioned as a salt form with the corresponding base or acid, the exact stoichiometric composition of said salt form, as obtained by the respective preparation and/or purification process, is, in most cases, unknown. Unless specified otherwise, suffixes to chemical names or structural formulae such as "hydrochloride", "trifluoroacetate", "sodium salt", or "x HO", "x CF3COOH", "x Na+", for example, are to be understood as not a stoichiometric specification, but solely as a salt form. This applies analogously to cases in which synthesis intermediates or example compounds or salts thereof have been obtained, by the preparation and/or purification processes described, as solvates, such as hydrates with (if defined) unknown stoichiometric composition. General procedure 1 (Suzuki coupling): To a solution of the bromide compound (1 eq), the borate ester or borate acid substrate (1 to 2 eq) and Na2CO3 or K2CO3 (2 to 3 eq) in dioxane (20 to 100 mg/mL) and H2O (1/10 to 1/5 of the dioxane volume) was added Pd(dppf)Cl2 or Pd(PPh3)4 (0.05 to 0.1 eq). The reaction mixture was degassed and purged with N2 (3x), stirred at 80 to 100 °C for 2 to16 h and concentrated under reduced pressure. The resulting residue was purified by preparative-HPLC or phase chromatographic column to give the corresponding product. Alternatively, after stirring at 80 to 100 °C for 2 to 16 h, the reaction mixture was cooled to room temperature, diluted with H2O and extracted with EtOAc or DCM (3x). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by preparative-HPLC or phase chromatographic column to give the corresponding product. General procedure 2 (oxidation, via m-CPBA) To a solution of the compound (1.0 eq) in DCM (50 to 100 mg/mL) was added m-CPBA (2.0 to 3.0 eq). The reaction mixture was stirred at 25 °C for 2 to 12 h. The mixture was poured into saturated sodium sulfite aqueous solution. The aqueous layer was extracted with EtOAc or DCM (2x or 3x). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by preparative-HPLC or chromatographic column to give the corresponding product. General procedure 3 (Saponification): To the mixture of the ester substrate (1 eq) in MeOH or THF (50 to 100 mg/mL) and H2O (1/2 to 1/1 of the MeOH or THF volume) was added LiOH or LiOH.H2O (3.0 to 5.0 eq). The mixture was stirred at 20 °C for 1 to 2 h. The pH of the reaction mixture was adjusted to a value ranged between 1 to 6 by addition of an aqueous solution of hydrochloric acid (1N). Work up procedure 1: The resulting precipitate was filtered, collected and dried under reduced pressure to give the
desired product. Work up procedure 2: The aqueous layer was extracted with EtOAc or DCM (2x or 3x). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure give the desired product. General procedure 4 (amidation): To a solution of the carboxylic acid compound (1.0 eq), the amino compound (1.3 to 2.0 eq), DIEA (3.0 to 4.0 eq,) in DMF or DCM (15 to 70 mg of carboxylic acid compound/mL) was added T4P in EtOAc (1.3 to 2.5 eq, 50% in EtOAc) or HATU (1.3 to 2.5 eq). The mixture was stirred at 25 °C for 1 to 2 h. Work up procedure 1: The resulting mixture was diluted with H2O, followed by extraction with EtOAc (3x). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by preparative-HPLC to give the corresponding product. Work up 2: The resulting mixture was directly purified by preparative-HPLC or Reversed-phase flash purification to give the corresponding product. General procedure 5 (Boc cleavage): To a solution of the Boc-protected compound in DCM (0.1 g/mL) was added TFA (1/5 to1/3 of the DCM volume) at 25 °C. The mixture was stirred at 25 °C for 1 to 2 h and, then, concentrated under reduce pressure. The resulting residue was purified by preparative- HPLC or reversed-phase flash purification to give the corresponding product. Preparation of Intermediate 1.1 S-(2-(((benzyloxy)carbonyl)amino)but-3-yn-1-yl) benzothioate
can be achieved utilizing the described cross-coupling reactions, too. Suitable building block containing the spiro motive are either commercial or can be synthesized by various methods known of a person skilled in the art of organic synthesis (see e.g. Spiro Compounds: Synthesis and Applications, ISBN:9781119567646, Editor: R. R. Torres, John Wiley & Sons). Preparative examples General considerations Abbreviations used in the descriptions that follow are: AcOH (acetic acid); aq. (aqueous); Ar (argon); Atm (atmosphere); BH3.THF (boran tetrahydrofuran complex); br (broad, 1H NMR signal); Boc2O (di-tert- butyldicarbonate); BuLi (Butyl Lithium), Cataxium APdG3 (mesylate[(di(1-adamantyl)-n-butylphosphine)- 2-(2 -amino-1,1 -biphenyl)]palladium(II)); CDCl3 (deuterated chloroform); cHex (cyclohexane); CMPB (cyanomethylene trimethylphosphorane); Cs2CO3 (cesium carbonate); CuI (copper iodide); CuSO4 (cupric sulfate), DABCO (1,4-diazabicyclo[2.2.2]octane); DAST (diethylaminosulfur trifluoride); DBU (1,8-diazabicyclo(5.4.0)undec-7-ene); DCE (dichloroethane); d (doublet, 1H NMR signal); DCM (dichloromethane); DEA (diethylamine); DIBAL-H (diisobutyl aluminium hydride); DIPEA or DIEA (di-iso- propylethylamine); DMA (dimethylacetamide); (DMAP (4- N-N-dimethylaminopyridine), DME (1,2- dimethoxyethane), DMEDA (dimethylethylenediamine ); DMF (N-N-dimethylformamide); DMSO (dimethyl sulfoxide); DPPA (diphenylphosphoride azide); dtbbpy (bis(1,1-dimethylethyl)-2,2 -bipyridine); ee (enantiomeric excess); ES (electrospray); EtOAc or EA (ethyl acetate); EtOH (ethanol); h (hour(s)); FA (formic acid); MgO (magnesium oxide); HATU (1-[bis(dimethylamino)methylene]-1H-1,2,3- triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate); HBr (hydrobromic acid); HFIP ( hexafluoroisopropanol); HI (hydrogen iodide); 1H NMR (proton nuclear magnetic resonance spectroscopy); HPLC (high performance liquid chromatography); iPrMgBr (isopropylmagnesium bromide); iPrOH (iso-propanol); K2CO3 (potassium carbonate); K3PO4 (tripotassium phosphate); Ir[dF(CF3)(dtbbpy)PF6 ((4,4'-di-t-butyl-2,2'-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl- kN)phenyl-kC]iridium(III) hexafluorophosphate); LiOH (lithium hydroxide); m (multiplet, 1H NMR signal); mCPBA (meta-chloroperoxybenzoic acid), MeCN (acetonitrile), MeOH (methanol); MeOK (potassium methanolate); min (minute(s)); MnO2 (manganese (IV) oxide); MS (mass spectrometry); MTBE (methyl tert-butyl ether); NaBH4 (sodium borohydride); NaHCO3 (sodium hydrogenocarbonate); NaI (sodium iodide); NaIO4 (sodium periodate); Na2SO3 (sodium sulfite); Na2S2O3 (sodium thiosulfate); Na2SO4 (sodium sulfate); NCS (N-chlorosuccinimide); NH3 (ammonia); NH4Cl (ammonium chloride); NiCl2 (nickel dichloride); NIS (N-iodosuccinimide); NMP (N-methylpyrrolidone); NMR (nuclear magnetic resonance); Pd/C (palladium on charcoal); Pd2dba3 (tris(dibenzylideneacetone)dipalladium); Pd(dppf)Cl2 (1,1- bis(diphenylphosphino)ferrocene dichloropalladium); Pd(Ph3)4 (tetrakis(triphenylphosphine) palladium (0)); Pd(Ph3)2Cl2 (bis(triphenylphosphine)palladium(II) dichloride ); PE (petroleum ether); Pd-PEPPSI-
IPentCl o-picoline ([1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5-dichloro-imidazol-2-ylidene]-dichloro-(2- methylpyridin-1-ium-1-yl)palladiµm; Pd(OH)2 (palladium hydroxide); Pd(Ph3)4 (palladium- tetrakis(triphenylphosphine)); PhI(OAc)2 ((diacetoxyiodo)benzene)); P(tBu)3 (tri-tert-butylphosphine ); Py (pyridine); q (quartet, 1H NMR signal); quin (quintet, 1H NMR signal); rac (racemic); RT (retention time); s (singlet, 1H NMR signal); sat. (saturated); t (triplet, 1H NMR signal); SFC (supercritical fluid chromatography); TBAF (tetrabutylammonium fluoride); tert-BuBrettPhos-Pd-G3 ([(2-di-tert- butylphosphino-3,6-dimethoxy-2 ,4 ,6 -triisopropyl-1,1 -biphenyl)-2-(2 -amino-1,1 -biphenyl)]palladium(II) methanesulfonate); tBuXPhos Pd G3 (methanesulfonato(2-di-t-butylphosphino-2',4',6'-tri-i-propyl-1,1'- biphenyl)(2'-amino-1,1'-biphenyl-2-yl)palladium(II)); TBDMSCl or TBSCl (tert-butyldimethylsilyl chloride); tBuOH (tert-butanol); TEA (triethylamine); TFA (trifluoroacetic acid); TFAA (trifluoroacetic anhydride); THF (tetrahydrofuran); TLC (thin layer chromatography); TMSCHN2 (trimethylsilyldiazomethane); TMSCN (trimethylsilyl cyanide); TMSI (trimethylsilyl iodide); TMSOTf (trimethylsilyl trifluoromethanesulfonate); TTMSS (trimethylsilane); TsOH (p-Toluenesulfonic acid); T4P (2,4,6-tributyl- 1,3,5,2,4,6-trioxatriphosphinane 2,4,6-trioxide); UPLC (ultra-high performance liquid chromatography), UV (ultraviolet), wt-% (percent by weight); Xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene); Xantphos Pd G4 (methanesulfonato[9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene](2'- methylamino-1,1'-biphenyl-2-yl)palladium(II)), Xphos (2-Dicyclohexylphosphin-2 ,4 ,6 - triisopropylbiphenyl). General Procedure: All starting materials and solvents were obtained either from commercial sources or prepared according to literature references. Commercially available reagents and anhydrous solvents were used as supplied, without further purification. Unless otherwise stated all reactions were stirred. Organic solutions were routinely dried over anhydrous sodium sulfate. Column chromatography was performed on pre-packed silica (100-1000 mesh, 40-63 µm) cartridges using the amount indicated. All air- and moisture-sensitive reactions were carried out in oven-dried (at 120 °C) glassware under an inert atmosphere of nitrogen or argon. Compound names were generated using ChemDraw Professional (Perkin Elmer, Version 16.0.1.4 (77)). In some cases generally accepted names of commercially available reagents were used in place of ChemDraw generated names. Methods for Reversed Phase HPLC conditions for LCMS Analysis of compounds: Method 1: SHIMADZU LCMS-2020 Kinetex EVO C182.1X30mm, 5 m at 50 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.55 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1.20 min, held at 5% A-95% B; 1.20-1.21 min, returned to 95% A-5% B, 1.21-1.55 min, held at 95% A-5% B.
Method 2: SHIMADZU LCMS-2020 Kinetex EVO C182.1X30mm, 5 m at 40 Mobile Phase A: 0.025% NH3·H2O in water (v/v); B: MeCN; flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.55 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1.20 min, held at 5% A-95% B; 1.20-1.21 min, returned to 95% A-5% B, 1.21-1.55 min, held at 95% A-5% B. Method 3: SHIMADZU LCMS-2020 Kinetex EVO C182.1X30mm,5 m at 50 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 2.0 mL/min; eluted with the mobile phase over 0.80 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1.20 min, held at 5% A-95% B; 1.20-1.21 min, returned to 95% A-5% B, 1.21-1.55 min, held at 95% A-5% B. Method 4: SHIMADZU LCMS-2020 Kinetex® EVO C182.1X20 mm 2.6 m at 50°C; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 2.0 mL/min; eluted with the mobile phase over 1.00 min employing UV detection at 220 nm and 254 nm. Gradient information: 0.01-0.60 min, ramped from 95% A-5% B to 5% A-95% B; 0.61-0.78 min, held at 5% A-95% B; 0.78-0.79 min, returned to 95% A-5% B, 0.79-0.80 min, held at 95% A-5% B. Method 5: SHIMADZU LCMS-2020 Kinetex® EVO C182.1X30 mm 5 m at 50°C Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.00 min employing UV detection at 220 nm and 254 nm. Gradient information: 0.01-0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-0.95 min, held at 5% A-95% B; 0.95-0.96 min, returned to 95% A-5% B, 0.96-1.00 min, held at 95% A-5% B. Method 6: SHIMADZU LCMS-2020 Kinetex® HALO C183.0X30mm, 5 m at 50 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.05 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 0.80 min, ramped from 50% A-50% B to 0% A-100% B; 0.80-1.05 min, held at 50% A-50% B. Method 7: SHIMADZU LCMS-2020 HALO C183.0X30 mm, 5 m at 50 , Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in Acetonitrile (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.05 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-0.50 min, ramped from 95% A-5% B to 5% A-95% B; 0.50-0.80 min, held at 5% A-95% B; 0.80-0.81 min, returned to 95% A-5% B, 0.81-1.05 min, held at 95% A-5% B. Method 8: SHIMADZU LCMS-2020 HALO C183.0X30mm, 5 m at 50 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with
the mobile phase over 0.80 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 0.45 min, ramped from 95% A-5% B to 5% A-95% B; 0.45-0.70 min, held at 5% A-95% B; 0.70-0.71 min, returned to 95% A-5% B, 0.71-0.80 min, held at 95% A-5% B. Method 9: SHIMADZU LCMS-2020 Kinetex® EVO C182.1X30 mm, 5 m at 40 Mobile Phase: A: water; B: MeCN; flow rate held at 1.5 mL/min; eluted with the mobile phase over 1.55 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-0.80 min, ramped from 95% A-5% B to 5% A-95% B; 0.80-1.20 min, held at 5% A-95% B; 1.20-1.21 min, returned to 95% A-5% B, 1.21-1.55 min, held at 95% A-5% B. Method 10: SHIMADZU LCMS-2020 Waters Xselect HSS T3, 3.5 µm, 4.6*50mm at 40 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 5.20 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-3.50 min, ramped from 95% A-5% B to 5% A-95% B; 3.50-4.80 min, held at 5% A-95% B; 4.80-4.81 min, returned to 95% A-5% B; 4.81-5.20 min, held at 95% A-5% B Method 11: SHIMADZU LCMS-2020 HALO C183.0X30mm,5 m at 50 ; Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in MeCN (v/v); flow rate held at 1.5 mL/min; eluted with the mobile phase over 3.00 min employing UV detection at 220 nm and 254 nm. Gradient information: 0- 2.10 min, ramped from 95% A-5% B to 5% A-95% B; 2.10-2.80 min, held at 5% A-95% B; 2.80-2.81 min, returned to 95% A-5% B, 2.81-3.00 min, held at 95% A-5% B. Method 12: SHIMADZU LCMS-2020 HALO C183.0X30 mm, 5 m at 50 , Mobile Phase: A: 0.0375% TFA in water (v/v); B: 0.01875% TFA in Acetonitrile (v/v); flow rate held at 2 mL/min; eluted with the mobile phase over 1.05 min employing UV detection at 220 nm and 254 nm. Gradient information: 0-0.40 min, ramped from 95% A-5% B to 5% A-95% B; 0.40-0.75 min, held at 5% A-95% B; 0.75-0.76 min, returned to 95% A-5% B, 0.76-1.05 min, held at 95% A-5% B. Methods for MS Analysis of compounds: Method 1: SHIMADZU LCMS-2020 Kinetex® EVO C182.1X30 mm, 5 m at 40 Mobile Phase: A: 0.025% NH3·H2O in water (v/v); B: MeCN; flow rate held at 1.5 mL/min; eluted with the mobile phase over 0.40 min employing UV detection at 220 nm. Gradient information: 0-0.40 min, ramped from 10% A-90% B to 10% A-90% B; Methods for SFC analysis of compounds: SFC Method 1: Column: Chiralcel OX-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH+ACN with 0.05% DEA additive; Gradient elution: 50% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar.
SFC Method 2: Column:Chiralpak AD-350*4.6 mm I.D., 3 µm Mobile phase: Phase A for CO2, Phase B for EtOH+ACN with 0.05% DEA additive; Gradient elution: 60% B in CO2 , Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 3: Column:Chiralpak IG-350*4.6 mm I.D., 3 m; Mobile phase: Phase A for CO2, and Phase B for MeOH+ACN with 0.05% DEA additive; Gradient elution: 60% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 4: Column:Chiralcel OJ-350*4.6mm I.D.,3 m; Mobile phase: Phase A for CO2,and Phase B for MeOH(0.05% DEA); Gradient elution: MeOH(0.05% DEA) in CO2 from 5% to 40%, Flow rate:3 mL/min; Detector: PDA; Column Temp: 35C; Back Pressure:100Bar. SFC Method 5: Column:Chiralpak IG-350*4.6mm I.D.,3 m; Mobile phase: Phase A for CO2, and Phase B for IPA+ACN(0.05% DEA); Gradient elution: IPA+ACN(0.05% DEA) from 20% to 60% in CO2; Flow rate:3 mL/min; Detector :PDA; Column Temp: 35C; Back Pressure:100Bar. SFC Method 6: Column: Chiralcel OJ-350*4.6mm I.D.,3 m; Mobile phase: Phase A for CO2, and Phase B for IPA(0.05% DEA); Gradient elution: IPA(0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35C; Back Pressure: 100Bar. SFC Method 7: Column:Chiralcel OJ-350*4.6 mm I.D., 3 m; Mobile phase:Phase A for CO2, and Phase B for EtOH(0.05% DEA); Gradient elution: EtOH(0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min;Detector: PDA; Column Temp: 35C; Back Pressure: 100Bar. SFC Method 8: Column: Chiralpak IC-350*4.6mm I.D., 3 m Mobile phase: Phase A for CO2 and Phase B for EtOH(0.05%DEA); Gradient elution: 60% EtOH(0.05%DEA) in CO2 ,Flow rate:3mL/min; Detector:PDA; Column Temp:35C; Back Pressure:100Bar" SFC Method 9: Column: Chiralpak IC-350*4.6mm I.D., 3 m Mobile phase: Phase A for CO2and Phase B for IPA+ACN(0.05%DEA); Gradient elution: IPA+ACN(0.05%DEA) in CO2 from 20% to 60%, Flow rate:3mL/min; Detector:PDA; Column Temp:35C; Back Pressure:100Bar". SFC Method 10: Column: Chiralpak AD-350*4.6mm I.D.,3 m Mobile phase: Phase A for CO2,and Phase B for IPA+ACN(0.05%DEA); Gradient elution: IPA+ACN(0.05%DEA) in CO2 from 20% to 60%, Flow rate:3mL/min; Detector:PDA; Column Temp:35C;Back Pressure:100Bar SFC Method 11: Column: (S,S)Whelk-O150*4.6mm I.D., 3.5 m; Mobile phase:Phase A for CO2,and Phase B for IPA(0.05%DEA); Gradient elution: IPA (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100 Bar. SFC Method 12: Column: Chiralpak AD-350*4.6mm I.D., 3 m; Mobile phase: Phase A for CO2, and Phase B for MeOH(0.05%DEA); Gradient elution: MeOH (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100 Bar.
SFC Method 13: Column: Chiralpak AD-350*4.6mm I.D., 3 m Mobile phase: Phase A for CO2, and Phase B for IPA (0.05%DEA); Gradient elution: IPA (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100 Bar. SFC Method 14: Column: Chiralpak IG-350*4.6 mm I.D., 3 m; Mobile phase: Phase A for CO2, and Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 20% to 60% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100 Bar. SFC Method 15: Column: Chiralcel OX-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH (0.05% DEA), Gradient elution: MeOH (0.05% DEA) from 10% to 60% in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 16: Column: Chiralcel OD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA), Gradient elution: EtOH (0.05% DEA) from 5% to 40% in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 17: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH+ACN (0.05% DEA); Gradient elution: 60% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 18: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA (0.05% DEA); Gradient elution: 40% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 19: Column: Chiralcel OZ -350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH+ACN (0.05% DEA); Gradient elution: 60% B in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 20: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA (0.05% DEA); Gradient elution: IPA (0.05% DEA) in CO2 from 20% to 60%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar SFC Method 21: Column:Chiralcel OJ-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: IPA+ACN (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 22: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: 60% EtOH (0.05% DEA) in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 23: Column: Lux 3um Cellulose-250*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH (0.05% DEA); Gradient elution: 60% MeOH (0.05% DEA) in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar.
SFC Method 24: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 25: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: 40% EtOH (0.05% DEA) in CO2, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 26: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 20% to 60% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 27: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH (0.05% DEA); Gradient elution: 40% MeOH (0.05% DEA) in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 28: Column: Chiralpak AD-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: 40% IPA+ACN in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar SFC Method 29: Column: Chiralpak ID-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: 60% IPA+ACN (0.05% DEA) in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 30: Column: Chiralpak IC-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) in CO2 from 10% to 60%, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar. SFC Method 31: Column: Chiralcel OJ-350*4.6mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH+ACN (0.05% DEA); Gradient elution: from 20% to 60% of EtOH+ACN (0.05% DEA) in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar SFC Method 32: Column: Chiralpak IE-3100*4.6mm I.D., 3 µm; Mobile phase: Phase A for Water with 0.0375%TFA, Phase B for ACN (0.01875% TFA); Gradient elution: 80% B in A; Flow rate: 1mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar SFC Method 33: Column: Chiralpak IK-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: 50% IPA+ACN (0.05% DEA) in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar SFC Method 34: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 20% to 40% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar.
SFC Method 35: Column: Chiralpak IH-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 5% to 40% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 36: Column: Chiralpak AS-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 5% to 40% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar SFC Method 37: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH (0.05% DEA); Gradient elution: 60% MeOH (0.05% DEA) in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 38: Column: Chiralcel Cellulose-250*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 20% to 60% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 39: Column: Chiralpak AY-350*4.6mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: 40% EtOH (0.05%DEA) in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 40: Column: Chiralpak IG-350*4.6 mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for EtOH (0.05% DEA); Gradient elution: EtOH (0.05% DEA) from 5% to 40% in CO2; Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 41: Column: Chiralpak AS-350*4.6mm I.D., 3 µm; Mobile phase: Phase A for CO2, Phase B for MeOH (0.05% DEA); Gradient elution: MeOH (0.05% DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100 Bar. SFC Method 42: Column: Lux 3um Cellulose-450*4.6mm I.D, 3 µm; Mobile phase: Phase A for CO2, Phase B for IPA+ACN (0.05% DEA); Gradient elution: IPA+ACN (0.05% DEA) from 20% to 60% in CO2, Flow rate: 3mL/min; Detector: PDA; Column Temp: 35 °C; Back Pressure: 100Bar SFC Method 43: Column: Chiralcel OX-350*4.6 mm I.D., 3 m; Mobile phase: Phase A for CO2, and Phase B for IPA (0.05% DEA); Gradient elution: IPA (0.05 % DEA) in CO2 from 5% to 40%, Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35 ; Back Pressure: 100Bar. SFC Method 44: Column: Chiralpak IC-350*4.6mm I.D.,3 m Mobile phase: Phase A for CO2 and Phase B for EtOH (0.05%DEA); Gradient elution: EtOH(0.05%DEA) in CO2 from 5% to 40%, Flow rate:3mL/min; Detector: PDA; Column Temp:35 ; Back Pressure:100Bar 1H NMR Spectroscopy: 1H NMR spectra were acquired on a Bruker Avance spectrometer at 400 MHz using residual undeuterated solvent as reference. 1H NMR signals are specified with their multiplicity / combined
multiplicities as apparent from the spectrµm; possible higher-order effects are not considered. Chemical shifts of the signals ( ) are specified as ppm (parts per million). Salt stoichiometry: In the present text, in particular in the experimental section, for the synthesis of intermediates and of examples of the present invention, when a compound is mentioned as a salt form with the corresponding base or acid, the exact stoichiometric composition of said salt form, as obtained by the respective preparation and/or purification process, is, in most cases, unknown. Unless specified otherwise, suffixes to chemical names or structural formulae such as "hydrochloride", "trifluoroacetate", "sodium salt", or "x HO", "x CF3COOH", "x Na+", for example, are to be understood as not a stoichiometric specification, but solely as a salt form. This applies analogously to cases in which synthesis intermediates or example compounds or salts thereof have been obtained, by the preparation and/or purification processes described, as solvates, such as hydrates with (if defined) unknown stoichiometric composition. General procedure 1 (Suzuki coupling): To a solution of the bromide compound (1 eq), the borate ester or borate acid substrate (1 to 2 eq) and Na2CO3 or K2CO3 (2 to 3 eq) in dioxane (20 to 100 mg/mL) and H2O (1/10 to 1/5 of the dioxane volume) was added Pd(dppf)Cl2 or Pd(PPh3)4 (0.05 to 0.1 eq). The reaction mixture was degassed and purged with N2 (3x), stirred at 80 to 100 °C for 2 to16 h and concentrated under reduced pressure. The resulting residue was purified by preparative-HPLC or phase chromatographic column to give the corresponding product. Alternatively, after stirring at 80 to 100 °C for 2 to 16 h, the reaction mixture was cooled to room temperature, diluted with H2O and extracted with EtOAc or DCM (3x). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by preparative-HPLC or phase chromatographic column to give the corresponding product. General procedure 2 (oxidation, via m-CPBA) To a solution of the compound (1.0 eq) in DCM (50 to 100 mg/mL) was added m-CPBA (2.0 to 3.0 eq). The reaction mixture was stirred at 25 °C for 2 to 12 h. The mixture was poured into saturated sodium sulfite aqueous solution. The aqueous layer was extracted with EtOAc or DCM (2x or 3x). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by preparative-HPLC or chromatographic column to give the corresponding product. General procedure 3 (Saponification): To the mixture of the ester substrate (1 eq) in MeOH or THF (50 to 100 mg/mL) and H2O (1/2 to 1/1 of the MeOH or THF volume) was added LiOH or LiOH.H2O (3.0 to 5.0 eq). The mixture was stirred at 20 °C for 1 to 2 h. The pH of the reaction mixture was adjusted to a value ranged between 1 to 6 by addition of an aqueous solution of hydrochloric acid (1N). Work up procedure 1: The resulting precipitate was filtered, collected and dried under reduced pressure to give the
desired product. Work up procedure 2: The aqueous layer was extracted with EtOAc or DCM (2x or 3x). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure give the desired product. General procedure 4 (amidation): To a solution of the carboxylic acid compound (1.0 eq), the amino compound (1.3 to 2.0 eq), DIEA (3.0 to 4.0 eq,) in DMF or DCM (15 to 70 mg of carboxylic acid compound/mL) was added T4P in EtOAc (1.3 to 2.5 eq, 50% in EtOAc) or HATU (1.3 to 2.5 eq). The mixture was stirred at 25 °C for 1 to 2 h. Work up procedure 1: The resulting mixture was diluted with H2O, followed by extraction with EtOAc (3x). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by preparative-HPLC to give the corresponding product. Work up 2: The resulting mixture was directly purified by preparative-HPLC or Reversed-phase flash purification to give the corresponding product. General procedure 5 (Boc cleavage): To a solution of the Boc-protected compound in DCM (0.1 g/mL) was added TFA (1/5 to1/3 of the DCM volume) at 25 °C. The mixture was stirred at 25 °C for 1 to 2 h and, then, concentrated under reduce pressure. The resulting residue was purified by preparative- HPLC or reversed-phase flash purification to give the corresponding product. Preparation of Intermediate 1.1 S-(2-(((benzyloxy)carbonyl)amino)but-3-yn-1-yl) benzothioate
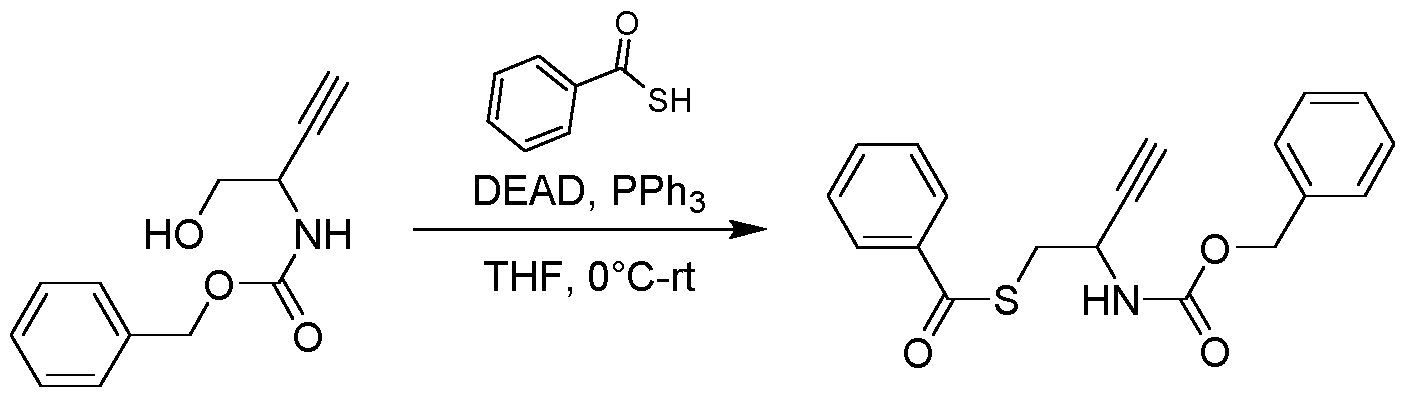 To a solution of triphenylphosphane (44.74 g, 171 mmol) in THF (170 mL) under N2 was added diethylazodicarboxylate (25 mL, 171 mmol) at 0 °C and the mixture was stirred at 0 °C for 10 min under N2. Benzyl (1-hydroxybut-3-yn-2-yl)carbamate (17.00 g, 77.5 mmol) in THF (170 mL) was added and the mixture was stirred at 0 °C for 10 min under N2. Benzothioic S-acid (21 mL, 171 mmol) was added and the mixture was stirred at 0 °C for 1 h under N2. The mixture was poured into Na2HCO3 (aq., sat., 150 mL). The aqueous phase was extracted with ethyl acetate (150 mL, 3x). The combined organic layer was washed with brine (150 mL), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum. The residue was purified by silica gel column flash chromatography (ISCO®; 400 g SepaFlash® Silica Flash Column, Eluent of 0~12% Ethyl acetate/Petroleum ether gradient, 80 mL/min) to give the product S-(2-(((benzyloxy)carbonyl)amino)but-3-yn-1-yl) benzothioate (39.00 g,115 mmol) as a yellow solid.
RT 0.585 min (method 4); m/z 362.0 (M+Na)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 8.02-7.95 (m, 2H), 7.62-7.57 (m, 1H), 7.49-7.45 (m, 2H), 7.35-7.30 (m, 5H), 5.31 (br, 1H), 5.12 (s, 2H), 4.84-4.74 (m, 1H), 3.49 (s, 2H), 2.37 (s, 1H). Preparation of Intermediate 1.2 Benzyl (2,3-dihydrothiophen-3-yl)carbamate
To a solution of triphenylphosphane (44.74 g, 171 mmol) in THF (170 mL) under N2 was added diethylazodicarboxylate (25 mL, 171 mmol) at 0 °C and the mixture was stirred at 0 °C for 10 min under N2. Benzyl (1-hydroxybut-3-yn-2-yl)carbamate (17.00 g, 77.5 mmol) in THF (170 mL) was added and the mixture was stirred at 0 °C for 10 min under N2. Benzothioic S-acid (21 mL, 171 mmol) was added and the mixture was stirred at 0 °C for 1 h under N2. The mixture was poured into Na2HCO3 (aq., sat., 150 mL). The aqueous phase was extracted with ethyl acetate (150 mL, 3x). The combined organic layer was washed with brine (150 mL), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum. The residue was purified by silica gel column flash chromatography (ISCO®; 400 g SepaFlash® Silica Flash Column, Eluent of 0~12% Ethyl acetate/Petroleum ether gradient, 80 mL/min) to give the product S-(2-(((benzyloxy)carbonyl)amino)but-3-yn-1-yl) benzothioate (39.00 g,115 mmol) as a yellow solid.
RT 0.585 min (method 4); m/z 362.0 (M+Na)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 8.02-7.95 (m, 2H), 7.62-7.57 (m, 1H), 7.49-7.45 (m, 2H), 7.35-7.30 (m, 5H), 5.31 (br, 1H), 5.12 (s, 2H), 4.84-4.74 (m, 1H), 3.49 (s, 2H), 2.37 (s, 1H). Preparation of Intermediate 1.2 Benzyl (2,3-dihydrothiophen-3-yl)carbamate
 To a solution of S-(2-(((benzyloxy)carbonyl)amino)but-3-yn-1-yl) benzothioate (10.00 g, 29.5 mmol) in methanol (1 L) was added potassium methanolate (4.07 g, 29.5 mmol). The mixture was stirred at 20 °C for 2 h under N2. Water (100 mL) was added and the mixture was stirred at 20 °C for 15 min before NaOH (1M, 60 mL) was added. The mixture was stirred at 20 °C for 0.5 h and a white solid precipitated. After filtration, the precipitate was washed with water (100 mL, 2x) and collected. The precipitate was dried by lyophilization to give the product benzyl (2,3-dihydrothiophen-3-yl)carbamate (8.50 g, 36.1 mmol) as a yellow solid. 1H NMR (DMSO-d6, 400 MHz): 7.71-7.61 (m, 1H), 7.42-7.28 (m, 5H), 6.48 (dd, J = 1.6, 5.6 Hz, 1H), 5.58 (dd, J = 2.4, 5.6 Hz, 1H), 5.02 (s, 2H), 4.97-4.85 (m, 1H), 3.46-3.42 (m, 1H), 3.01-2.93 (m, 1H) Preparation of Intermediate 1.3 Benzyl (1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)carbamate
To a solution of S-(2-(((benzyloxy)carbonyl)amino)but-3-yn-1-yl) benzothioate (10.00 g, 29.5 mmol) in methanol (1 L) was added potassium methanolate (4.07 g, 29.5 mmol). The mixture was stirred at 20 °C for 2 h under N2. Water (100 mL) was added and the mixture was stirred at 20 °C for 15 min before NaOH (1M, 60 mL) was added. The mixture was stirred at 20 °C for 0.5 h and a white solid precipitated. After filtration, the precipitate was washed with water (100 mL, 2x) and collected. The precipitate was dried by lyophilization to give the product benzyl (2,3-dihydrothiophen-3-yl)carbamate (8.50 g, 36.1 mmol) as a yellow solid. 1H NMR (DMSO-d6, 400 MHz): 7.71-7.61 (m, 1H), 7.42-7.28 (m, 5H), 6.48 (dd, J = 1.6, 5.6 Hz, 1H), 5.58 (dd, J = 2.4, 5.6 Hz, 1H), 5.02 (s, 2H), 4.97-4.85 (m, 1H), 3.46-3.42 (m, 1H), 3.01-2.93 (m, 1H) Preparation of Intermediate 1.3 Benzyl (1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)carbamate
 To a solution of benzyl (2,3-dihydrothiophen-3-yl)carbamate (7.50 g, 31.9 mmol) and PhI(OAc)2 (20.53 g, 63.7 mmol) in methanol (150 mL) was added ammonium carbamate (7.47 g, 95.6 mmol) at room temperature. The mixture was stirred at 20 °C for 2 h. The crude was directly purified by preparative normal phase HPLC (column: Welch Ultimate XB-CN 250 mm*100 mm*10 µm; mobile phase: A: Hexane, B: EtOH; B%: 5%-45%, 10 min) and concentrated under vacuum to give benzyl (1-imino-1-oxido-2,3- dihydro-1H-1 -thiophen-3-yl)carbamate (4.80 g,18.0 mmol) as a yellow solid.
RT 0.388 min (method 4); m/z 267.0 (M+H)+ (ESI+).1H NMR (CDCl3, 400 MHz): 7.42-7.30 (m, 5H), 6.76-6.66 (m, 1H), 6.56-6.45 (m, 1H), 5.96-5.57 (m, 1H), 5.15 (br, 1H), 5.12 (s, 2H), 3.69-3.59 (m, 1H), 3.24-3.17 (m, 1H), 3.01 (br, 1H). Preparation of Intermediate 1.4 3-amino-1-imino-2,3-dihydro-1H-1 -thiophene 1-oxide hydrobromide
To a solution of benzyl (2,3-dihydrothiophen-3-yl)carbamate (7.50 g, 31.9 mmol) and PhI(OAc)2 (20.53 g, 63.7 mmol) in methanol (150 mL) was added ammonium carbamate (7.47 g, 95.6 mmol) at room temperature. The mixture was stirred at 20 °C for 2 h. The crude was directly purified by preparative normal phase HPLC (column: Welch Ultimate XB-CN 250 mm*100 mm*10 µm; mobile phase: A: Hexane, B: EtOH; B%: 5%-45%, 10 min) and concentrated under vacuum to give benzyl (1-imino-1-oxido-2,3- dihydro-1H-1 -thiophen-3-yl)carbamate (4.80 g,18.0 mmol) as a yellow solid.
RT 0.388 min (method 4); m/z 267.0 (M+H)+ (ESI+).1H NMR (CDCl3, 400 MHz): 7.42-7.30 (m, 5H), 6.76-6.66 (m, 1H), 6.56-6.45 (m, 1H), 5.96-5.57 (m, 1H), 5.15 (br, 1H), 5.12 (s, 2H), 3.69-3.59 (m, 1H), 3.24-3.17 (m, 1H), 3.01 (br, 1H). Preparation of Intermediate 1.4 3-amino-1-imino-2,3-dihydro-1H-1 -thiophene 1-oxide hydrobromide
 A solution of benzyl (1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)carbamate (500 mg, 1.88 mmol) in HBr/AcOH (5.79 g, 33% wt/wt, 17.8 mmol) was stirred at 25 °C for 1 h. MTBE (10 mL) was added and a yellow solid precipitated. The suspension was shaken, and then, it was left to stand. The supernatant was removed after static settlement. The lower layer precipitation was used to repeat the work up process 6 times. Then, the lower layer precipitation was directly dried under high reduced pressure at room temperature to give the product 3-amino-1-imino-2,3-dihydro-1H-1 -thiophene 1-oxide hydrobromide (1.10 g, 5.16 mmol, HBr salt) as a yellow solid. 1H NMR (DMSO-d6, 400 MHz): 8.61 (br, 3 H), 7.36-7.69 (m, 1 H), 6.83-7.15 (m, 1 H), 4.82 (m, 1 H), 4.14-4.21 (m, 1 H), 3.42-3.51 (m, 1 H) Preparation of Example 1 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide
A solution of benzyl (1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)carbamate (500 mg, 1.88 mmol) in HBr/AcOH (5.79 g, 33% wt/wt, 17.8 mmol) was stirred at 25 °C for 1 h. MTBE (10 mL) was added and a yellow solid precipitated. The suspension was shaken, and then, it was left to stand. The supernatant was removed after static settlement. The lower layer precipitation was used to repeat the work up process 6 times. Then, the lower layer precipitation was directly dried under high reduced pressure at room temperature to give the product 3-amino-1-imino-2,3-dihydro-1H-1 -thiophene 1-oxide hydrobromide (1.10 g, 5.16 mmol, HBr salt) as a yellow solid. 1H NMR (DMSO-d6, 400 MHz): 8.61 (br, 3 H), 7.36-7.69 (m, 1 H), 6.83-7.15 (m, 1 H), 4.82 (m, 1 H), 4.14-4.21 (m, 1 H), 3.42-3.51 (m, 1 H) Preparation of Example 1 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide
 To a solution of 6-(3,4-dimethylphenyl)-2-oxo-1H-pyridine-3-carboxylic acid (60 mg, 0.247 mmol), 1-imino-1-oxo-2,3-dihydrothiophen-3-amine hydrobromide (105 mg, 0.493 mmol, HBr salt) in DMA (18 mL) was added DIEA (0.31 mL, 1.73 mmol) and T4P in EtOAc (355 mg, 50% purity, 0.493 mmol) at 0 °C . The mixture was stirred at 25 °C for 10 mins and then, directly purified by preparative HPLC (column: Unisil 3-100 C18 Ultra 150*50 mm*10 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 20%-50%, 10 min). To the resulting solution was added NaHCO3 (aq., sat.) to adjust the pH to 7-8. Then,
the solution was extracted with DCM (60 mL, 6x) and washed with pure water (40 mL, 2x). The organic layer was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated at 20 °C to give the product 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide (46 mg, 0.129 mmol, 97.72% purity) as a brown solid. RT 0.427 min (method 4); m/z 358.0 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 10.22-10.08 (m, 1H), 10.06-9.99 (m, 1H), 8.12-8.05 (m, 1H), 7.45-7.36 (m, 2H), 7.34-7.29 (m, 1H), 6.80-6.71 (m, 2H), 6.61- 6.55 (m, 1H), 5.63-5.50 (m, 1H), 3.92-3.77 (m, 1H), 3.36-3.22 (m, 1H), 2.36 (s, 6H); SFC Method 2 showed four peaks. Preparation of: Example 2 6-(3,4-dimethylphenyl)-N-((1S or R, 3S)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide, Example 3 6-(3,4-dimethylphenyl)-N-((1R or S, 3S)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide, Example 4 6-(3,4-dimethylphenyl)-N-((1S or R, 3R)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide and Example 5 6-(3,4-dimethylphenyl)-N-((1R or S, 3R)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide
To a solution of 6-(3,4-dimethylphenyl)-2-oxo-1H-pyridine-3-carboxylic acid (60 mg, 0.247 mmol), 1-imino-1-oxo-2,3-dihydrothiophen-3-amine hydrobromide (105 mg, 0.493 mmol, HBr salt) in DMA (18 mL) was added DIEA (0.31 mL, 1.73 mmol) and T4P in EtOAc (355 mg, 50% purity, 0.493 mmol) at 0 °C . The mixture was stirred at 25 °C for 10 mins and then, directly purified by preparative HPLC (column: Unisil 3-100 C18 Ultra 150*50 mm*10 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 20%-50%, 10 min). To the resulting solution was added NaHCO3 (aq., sat.) to adjust the pH to 7-8. Then,
the solution was extracted with DCM (60 mL, 6x) and washed with pure water (40 mL, 2x). The organic layer was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated at 20 °C to give the product 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide (46 mg, 0.129 mmol, 97.72% purity) as a brown solid. RT 0.427 min (method 4); m/z 358.0 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 10.22-10.08 (m, 1H), 10.06-9.99 (m, 1H), 8.12-8.05 (m, 1H), 7.45-7.36 (m, 2H), 7.34-7.29 (m, 1H), 6.80-6.71 (m, 2H), 6.61- 6.55 (m, 1H), 5.63-5.50 (m, 1H), 3.92-3.77 (m, 1H), 3.36-3.22 (m, 1H), 2.36 (s, 6H); SFC Method 2 showed four peaks. Preparation of: Example 2 6-(3,4-dimethylphenyl)-N-((1S or R, 3S)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide, Example 3 6-(3,4-dimethylphenyl)-N-((1R or S, 3S)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide, Example 4 6-(3,4-dimethylphenyl)-N-((1S or R, 3R)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide and Example 5 6-(3,4-dimethylphenyl)-N-((1R or S, 3R)-1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo- 1,2-dihydropyridine-3-carboxamide
 The 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide (80 mg, 0.224 mmol) was purified by preparative SFC (Column: DAICEL CHIRALCEL OX 250 mm*30 mm,10 µm; Mobile phase: A for CO2, B for MeOH+ACN (with 0.1% NH3.H2O additive); Gradient elution: 60% B in CO2,10 min) to give two product solutions. The solution containing peak 1 was concentrated under vacuum (30 °C) until a remaining volume of ~20 mL. The resulting solution was diluted with pure water (20 mL) and extracted with DCM (40 mL, 2x). The combined organic layer was washed with pure water (20 mL, 2x), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum to give peak A (SFC Method 1, RT: 1.156 min). The solution containing peak 2 was concentrated under vacuum (30 °C) until a remaining volume of ~20 mL. The resulting solution was diluted with pure water (20 mL) and extracted with DCM (40 mL, 2x). The combined organic layers
were washed with pure water (20 mL, 2x), dried over anhydrous Na2SO4, filtered, and the filtrate was concentrated under vacuum to give peak B (SFC Method 1, RT: 1.606 min). Peak A (SFC Method 1, :1.156 min) (20 mg, 0.0560 mmol) was further purified by preparative SFC (Column: DAICEL CHIRALCEL OJ 250 mm*30 mm, 10 µm; Mobile phase: Mobile phase: A for CO2, B for i-PrOH (with 0.1% NH3.H2O additive); B%: 25%-25%,10 min) to afford 2 solutions which were concentrated separately under vacuum (30 °C) to give: - Example 2 (SFC Method 2, RT: 1.208 min), which was further purified by preparative HPLC (column: Welch Xtimate C18150*25 mm*5 µm; mobile phase: A: 0.225% formic acid in water; B%: 18%-48%,10 min) and lyophilized to give the product 6-(3,4-dimethylphenyl)-N-((1S or R,3R)-1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide (1.16 mg, 0.00321 mmol, 99.04% purity, 83.23%ee) as an off-white solid. RT 0.447 min (method 2); m/z 358.2 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 10.51-10.30 (m, 1H), 10.06 (d, J = 7.6 Hz, 1H), 8.57 (d, J = 7.6 Hz, 1H), 7.45-7.36 (m, 2H), 7.40-7.32 (m, 1H), 6.77-6.70 (m, 2H), 6.64-6.55 (m, 1H), 5.64-5.48 (m, 1H), 3.80 (dd, J = 7.6, 13.6 Hz, 1H), 3.39- 3.29 (m, 1H), 2.96 (br, 1H), 2.36 (s, 6H). - Example 3 (SFC Method 2, RT: 1.457 min), which was further purified by preparative HPLC (column: Welch Xtimate C18150*25 mm*5 µm; mobile phase: A: 0.225% formic acid in water; B%: 18%-48%,10 min) and lyophilized to give the product 6-(3,4-dimethylphenyl)-N-((1R or S,3S)-1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide (1.66 mg, 0.0044 mmol, 94.7% purity, 93.06%ee) as an off-white solid. RT 0.437 min (method 2); m/z 358.2 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 10.40-10.15 (m, 1H), 10.10-10.00 (m, 1H), 8.58 (d, J = 7.6 Hz, 1H), 7.44-7.37 (m, 2H), 7.40-7.32 (m, 1H), 6.80-6.70 (m, 2H), 6.63-6.55 (m, 1H), 5.65-5.50 (m, 1H), 3.86 (dd, J = 7.6, 13.6 Hz, 1H), 3.27 (dd, J = 5.2, 13.6 Hz, 1H), , 2.36 (s, 6H). Peak B (SFC Method 1, RT: 1.606 min) (35 mg, 0.0979 mmol) was further purified by preparative SFC (Column:DAICEL CHIRALPAK IG 250 mm*30 mm,10 µm; Mobile phase: A for CO2, B for MeOH+ACN (with 0.1% NH3.H2O additive); Gradient elution: B%: 75%-75%,10 min). The pH of the two resulting solutions were adjusted to 7 with formic acid and concentrated separately under vacuum (30 °C) until a volume of ~ 20 mL was left. Then, to the two solutions was added DCM (40 mL) and pure water (20 mL). The organic layer was washed with pure water (20 mL, 2x), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated separately under vacuum to give : - Example 4 (SFC Method 2, RT: 1.611 min) 6-(3,4-dimethylphenyl)-N-((1S or R,3R)-1-imino- 1-oxido-2,3-dihydro-1H- 66-thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-carboxamide (6.6 mg, 0.0173 mmol, 93.93% purity, 100% ee) as a brown solid. (We proceed arbitrarily with the
attribution of the stereochemistry). RT 0.433 min (method 2); m/z 358.1 (M+H)+ (ESI+);; 1H NMR (CDCl3, 400 MHz): 10.23-9.92 (m, 2H), 8.57 (d, J = 7.6 Hz, 1H), 7.42-7.36 (m, 2H), 7.33-7.29 (m, 1H), 6.79-6.70 (m, 2H), 6.61 (dd, J = 2.8, 6.4 Hz, 1H), 5.63-5.50 (m, 1H), 3.81 (dd, J = 7.6, 13.6 Hz, 1H), 3.36-3.30 (m, 1H), 2.94 (br, 1H), 2.36 (s, 6H). - Example 5 (SFC Method 2, RT: 1.834 min) 6-(3,4-dimethylphenyl)-N-((1R or S,3R)-1-imino-1- oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-carboxamide (1.9 mg, 0.00487 mmol, 94.13% purity, 100%ee) as a brown solid. RT 0.436 min (method 2); m/z 358.1 (M+H)+ (ESI+);; 1H NMR (CDCl3, 400 MHz): 10.23-9.90 (m, 2H), 8.60-8.56 (m, 1H), 7.43-7.37 (m, 2H), 7.33-7.30 (m, 1H), 6.79-6.71 (m, 2H), 6.60 (dd, J = 2.8, 6.4 Hz, 1H), 5.65-5.55 (m, 1H), 3.86 (dd, J = 7.6,13.6 Hz, 1H), 3.3-3.24 (m, 1H), 3.06-2.81 (m, 1H), 2.36 (s, 6H) Preparation of Example 6 6-(3,4-dimethylphenyl)-N-(1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide
The 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1 -thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide (80 mg, 0.224 mmol) was purified by preparative SFC (Column: DAICEL CHIRALCEL OX 250 mm*30 mm,10 µm; Mobile phase: A for CO2, B for MeOH+ACN (with 0.1% NH3.H2O additive); Gradient elution: 60% B in CO2,10 min) to give two product solutions. The solution containing peak 1 was concentrated under vacuum (30 °C) until a remaining volume of ~20 mL. The resulting solution was diluted with pure water (20 mL) and extracted with DCM (40 mL, 2x). The combined organic layer was washed with pure water (20 mL, 2x), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum to give peak A (SFC Method 1, RT: 1.156 min). The solution containing peak 2 was concentrated under vacuum (30 °C) until a remaining volume of ~20 mL. The resulting solution was diluted with pure water (20 mL) and extracted with DCM (40 mL, 2x). The combined organic layers
were washed with pure water (20 mL, 2x), dried over anhydrous Na2SO4, filtered, and the filtrate was concentrated under vacuum to give peak B (SFC Method 1, RT: 1.606 min). Peak A (SFC Method 1, :1.156 min) (20 mg, 0.0560 mmol) was further purified by preparative SFC (Column: DAICEL CHIRALCEL OJ 250 mm*30 mm, 10 µm; Mobile phase: Mobile phase: A for CO2, B for i-PrOH (with 0.1% NH3.H2O additive); B%: 25%-25%,10 min) to afford 2 solutions which were concentrated separately under vacuum (30 °C) to give: - Example 2 (SFC Method 2, RT: 1.208 min), which was further purified by preparative HPLC (column: Welch Xtimate C18150*25 mm*5 µm; mobile phase: A: 0.225% formic acid in water; B%: 18%-48%,10 min) and lyophilized to give the product 6-(3,4-dimethylphenyl)-N-((1S or R,3R)-1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide (1.16 mg, 0.00321 mmol, 99.04% purity, 83.23%ee) as an off-white solid. RT 0.447 min (method 2); m/z 358.2 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 10.51-10.30 (m, 1H), 10.06 (d, J = 7.6 Hz, 1H), 8.57 (d, J = 7.6 Hz, 1H), 7.45-7.36 (m, 2H), 7.40-7.32 (m, 1H), 6.77-6.70 (m, 2H), 6.64-6.55 (m, 1H), 5.64-5.48 (m, 1H), 3.80 (dd, J = 7.6, 13.6 Hz, 1H), 3.39- 3.29 (m, 1H), 2.96 (br, 1H), 2.36 (s, 6H). - Example 3 (SFC Method 2, RT: 1.457 min), which was further purified by preparative HPLC (column: Welch Xtimate C18150*25 mm*5 µm; mobile phase: A: 0.225% formic acid in water; B%: 18%-48%,10 min) and lyophilized to give the product 6-(3,4-dimethylphenyl)-N-((1R or S,3S)-1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide (1.66 mg, 0.0044 mmol, 94.7% purity, 93.06%ee) as an off-white solid. RT 0.437 min (method 2); m/z 358.2 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 10.40-10.15 (m, 1H), 10.10-10.00 (m, 1H), 8.58 (d, J = 7.6 Hz, 1H), 7.44-7.37 (m, 2H), 7.40-7.32 (m, 1H), 6.80-6.70 (m, 2H), 6.63-6.55 (m, 1H), 5.65-5.50 (m, 1H), 3.86 (dd, J = 7.6, 13.6 Hz, 1H), 3.27 (dd, J = 5.2, 13.6 Hz, 1H), , 2.36 (s, 6H). Peak B (SFC Method 1, RT: 1.606 min) (35 mg, 0.0979 mmol) was further purified by preparative SFC (Column:DAICEL CHIRALPAK IG 250 mm*30 mm,10 µm; Mobile phase: A for CO2, B for MeOH+ACN (with 0.1% NH3.H2O additive); Gradient elution: B%: 75%-75%,10 min). The pH of the two resulting solutions were adjusted to 7 with formic acid and concentrated separately under vacuum (30 °C) until a volume of ~ 20 mL was left. Then, to the two solutions was added DCM (40 mL) and pure water (20 mL). The organic layer was washed with pure water (20 mL, 2x), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated separately under vacuum to give : - Example 4 (SFC Method 2, RT: 1.611 min) 6-(3,4-dimethylphenyl)-N-((1S or R,3R)-1-imino- 1-oxido-2,3-dihydro-1H- 66-thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-carboxamide (6.6 mg, 0.0173 mmol, 93.93% purity, 100% ee) as a brown solid. (We proceed arbitrarily with the
attribution of the stereochemistry). RT 0.433 min (method 2); m/z 358.1 (M+H)+ (ESI+);; 1H NMR (CDCl3, 400 MHz): 10.23-9.92 (m, 2H), 8.57 (d, J = 7.6 Hz, 1H), 7.42-7.36 (m, 2H), 7.33-7.29 (m, 1H), 6.79-6.70 (m, 2H), 6.61 (dd, J = 2.8, 6.4 Hz, 1H), 5.63-5.50 (m, 1H), 3.81 (dd, J = 7.6, 13.6 Hz, 1H), 3.36-3.30 (m, 1H), 2.94 (br, 1H), 2.36 (s, 6H). - Example 5 (SFC Method 2, RT: 1.834 min) 6-(3,4-dimethylphenyl)-N-((1R or S,3R)-1-imino-1- oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-carboxamide (1.9 mg, 0.00487 mmol, 94.13% purity, 100%ee) as a brown solid. RT 0.436 min (method 2); m/z 358.1 (M+H)+ (ESI+);; 1H NMR (CDCl3, 400 MHz): 10.23-9.90 (m, 2H), 8.60-8.56 (m, 1H), 7.43-7.37 (m, 2H), 7.33-7.30 (m, 1H), 6.79-6.71 (m, 2H), 6.60 (dd, J = 2.8, 6.4 Hz, 1H), 5.65-5.55 (m, 1H), 3.86 (dd, J = 7.6,13.6 Hz, 1H), 3.3-3.24 (m, 1H), 3.06-2.81 (m, 1H), 2.36 (s, 6H) Preparation of Example 6 6-(3,4-dimethylphenyl)-N-(1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide
 To tert-butyl nitrite (30 mg, 0.294 mmol) was added a solution of 6-(3,4-dimethylphenyl)-N-(1-imino- 1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-carboxamide (30 mg, 0.0839 mmol) in chloroform (1.0 mL) at 25 °C. The mixture was stirred at 25 °C for 2 h and then, directly purified by preparative HPLC (column: Waters xbridge 150*25 mm 10 µm; mobile phase: A: 10 mmol/L NH4HCO3 in water, B: MeCN; B%: 18%-55%, 10 min). The resulting fraction was lyophilized to give the product 6- (3,4-dimethylphenyl)-N-(1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-carboxamide (6.0 mg,0.0166 mmol, 97.86% purity) as an off-white gum. RT 0.596 min (method 2); m/z 343.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 10.60-10.21 (m, 1H), 10.13-9.70 (m, 1H), 8.63-8.50 (m, 1H), 7.44-7.37 (m, 2H), 7.30 (d, J = 7.6 Hz, 1H), 6.84-6.58 (m, 3H), 5.93-5.42 (m, 1H), 4.25-3.28 (m, 1H), 3.23-2.76 (m, 1H), 2.37 (s, 6H); SFC (Column:Chiralpak IG-3 50*4.6mm I.D., 3µm; Mobile phase: Phase A for CO2, Phase B for EtOH+ACN (with 0.05% DEA additive); Gradient elution: 60% B in CO2, Flow rate: 3mL/min) showed four peaks. Preparation of Intermediate 7.1 5-(3,4-dimethylphenyl)picolinic acid
To a solution of 5-bromopyridine-2-carboxylic acid (500 mg, 2.48 mmol) in 1,4-dioxane (10 mL) and water (2 mL) was added 2-(3,4-dimethylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (445 mg, 2.97 mmol), Na2CO3 (525 mg, 4.95 mmol) and Pd(dppf)Cl2 (181 mg, 0.248 mmol). The reaction mixture was degassed and purged with N2 (3x) and stirred at 100 °C for 16 h. The reaction mixture was filtered and the cake was washed with MeOH (5 mL, 3x). The filtrate was acidified with HCl (aq., 1N) until pH=1 and the mixture was concentrated under reduced pressure. The residue was purified by preparative HPLC (column: Phenomenex luna C18150*40 mm* 15 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 28%-58%, 15 min) and lyophilized to give the product 5-(3,4-dimethylphenyl)pyridine-2- carboxylic acid (240 mg, 0.444 mmol, 17.92 % yield) as a grey solid. RT 0.471 min (method 4); m/z 228.1 (M+H)+ (ESI+); Preparation of Intermediate 7.2 2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide
To tert-butyl nitrite (30 mg, 0.294 mmol) was added a solution of 6-(3,4-dimethylphenyl)-N-(1-imino- 1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-carboxamide (30 mg, 0.0839 mmol) in chloroform (1.0 mL) at 25 °C. The mixture was stirred at 25 °C for 2 h and then, directly purified by preparative HPLC (column: Waters xbridge 150*25 mm 10 µm; mobile phase: A: 10 mmol/L NH4HCO3 in water, B: MeCN; B%: 18%-55%, 10 min). The resulting fraction was lyophilized to give the product 6- (3,4-dimethylphenyl)-N-(1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-carboxamide (6.0 mg,0.0166 mmol, 97.86% purity) as an off-white gum. RT 0.596 min (method 2); m/z 343.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 10.60-10.21 (m, 1H), 10.13-9.70 (m, 1H), 8.63-8.50 (m, 1H), 7.44-7.37 (m, 2H), 7.30 (d, J = 7.6 Hz, 1H), 6.84-6.58 (m, 3H), 5.93-5.42 (m, 1H), 4.25-3.28 (m, 1H), 3.23-2.76 (m, 1H), 2.37 (s, 6H); SFC (Column:Chiralpak IG-3 50*4.6mm I.D., 3µm; Mobile phase: Phase A for CO2, Phase B for EtOH+ACN (with 0.05% DEA additive); Gradient elution: 60% B in CO2, Flow rate: 3mL/min) showed four peaks. Preparation of Intermediate 7.1 5-(3,4-dimethylphenyl)picolinic acid
To a solution of 5-bromopyridine-2-carboxylic acid (500 mg, 2.48 mmol) in 1,4-dioxane (10 mL) and water (2 mL) was added 2-(3,4-dimethylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (445 mg, 2.97 mmol), Na2CO3 (525 mg, 4.95 mmol) and Pd(dppf)Cl2 (181 mg, 0.248 mmol). The reaction mixture was degassed and purged with N2 (3x) and stirred at 100 °C for 16 h. The reaction mixture was filtered and the cake was washed with MeOH (5 mL, 3x). The filtrate was acidified with HCl (aq., 1N) until pH=1 and the mixture was concentrated under reduced pressure. The residue was purified by preparative HPLC (column: Phenomenex luna C18150*40 mm* 15 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 28%-58%, 15 min) and lyophilized to give the product 5-(3,4-dimethylphenyl)pyridine-2- carboxylic acid (240 mg, 0.444 mmol, 17.92 % yield) as a grey solid. RT 0.471 min (method 4); m/z 228.1 (M+H)+ (ESI+); Preparation of Intermediate 7.2 2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide
 To a solution of 5-(3,4-dimethylphenyl)pyridine-2-carboxylic acid (80 mg, 0.352 mmol) in MeCN (4 mL) was added urea-H2O2 (70 mg, 0.739 mmol) at 0 °C. TFAA (0.098 mL, 0.704 mmol) was added dropwise and the mixture was stirred at 25 °C for 3 h. The mixture was poured into Na2SO3 (aq., sat., 6 mL) and extracted with DCM (10 mL, 3x). The combined organic layers were washed with brine (10 mL, 3x), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (column: YMC-Actus Triart C18150*30 mm*7 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 40%-70%, 10 min) and lyophilized to give the product 5-(3,4- dimethylphenyl)-1-oxido-pyridin-1-ium-2-carboxylic acid (25 mg,0.103 mmol, 29.19 % yield) as a white solid. RT 0.507 min (method 4); m/z 244.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz) 8.57 (d, J = 1.2 Hz, 1H), 8.45 (d, J = 8.4 Hz, 1H), 7.86 (dd, J = 1.6, 8.4 Hz, 1H), 7.39-7.27 (m, 3H), 2.39-2.35 (m, 6H). Preparation of Example 7
5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide
To a solution of 5-(3,4-dimethylphenyl)pyridine-2-carboxylic acid (80 mg, 0.352 mmol) in MeCN (4 mL) was added urea-H2O2 (70 mg, 0.739 mmol) at 0 °C. TFAA (0.098 mL, 0.704 mmol) was added dropwise and the mixture was stirred at 25 °C for 3 h. The mixture was poured into Na2SO3 (aq., sat., 6 mL) and extracted with DCM (10 mL, 3x). The combined organic layers were washed with brine (10 mL, 3x), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (column: YMC-Actus Triart C18150*30 mm*7 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 40%-70%, 10 min) and lyophilized to give the product 5-(3,4- dimethylphenyl)-1-oxido-pyridin-1-ium-2-carboxylic acid (25 mg,0.103 mmol, 29.19 % yield) as a white solid. RT 0.507 min (method 4); m/z 244.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz) 8.57 (d, J = 1.2 Hz, 1H), 8.45 (d, J = 8.4 Hz, 1H), 7.86 (dd, J = 1.6, 8.4 Hz, 1H), 7.39-7.27 (m, 3H), 2.39-2.35 (m, 6H). Preparation of Example 7
5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide
 To a solution of 5-(3,4-dimethylphenyl)-1-oxido-pyridin-1-ium-2-carboxylic acid (12 mg, 0.0493 mmol), DIEA (0.044 mL, 0.247 mmol) and 1,1-dioxo-2,3-dihydrothiophen-3-amine hydrochloride (11 mg, 0.0641 mmol, HCl salt) in DMF (2 mL) was added T4P in EtOAc (46 mg, 50% purity, 0.0641 mmol) at 0 °C. The reaction mixture was stirred at 20 °C for 2 h. The mixture was poured into water (5 mL) and extracted with EtOAc (5 mL, 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (column: YMC- Actus Triart C18150*30 mm*7 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 38%- 68%, 10 min) and lyophilized to give the product 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (8.9 mg, 0.0248 mmol, 50.34 % yield) as an off-white solid. RT 0.486 min (method 4); m/z 359.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz) 11.61 (d, J = 7.6 Hz, 1H), 8.81 (d, J = 1.6 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 7.95 (dd, J = 1.6, 8.4 Hz, 1H), 7.66 (s, 1H), 7.59-7.56 (m, 1H), 7.32-7.28 (m, 2H), 7.04-7.00 (m, 1H), 5.45-5.35 (m, 1H), 3.78-3.74 (m, 1H), 3.40-3.36 (m, 1H), 2.30 (s, 3H), 2.28 (s, 3H); Preparation of Example 7a &7b (R or S)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide and (S or R)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide
To a solution of 5-(3,4-dimethylphenyl)-1-oxido-pyridin-1-ium-2-carboxylic acid (12 mg, 0.0493 mmol), DIEA (0.044 mL, 0.247 mmol) and 1,1-dioxo-2,3-dihydrothiophen-3-amine hydrochloride (11 mg, 0.0641 mmol, HCl salt) in DMF (2 mL) was added T4P in EtOAc (46 mg, 50% purity, 0.0641 mmol) at 0 °C. The reaction mixture was stirred at 20 °C for 2 h. The mixture was poured into water (5 mL) and extracted with EtOAc (5 mL, 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (column: YMC- Actus Triart C18150*30 mm*7 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 38%- 68%, 10 min) and lyophilized to give the product 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (8.9 mg, 0.0248 mmol, 50.34 % yield) as an off-white solid. RT 0.486 min (method 4); m/z 359.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz) 11.61 (d, J = 7.6 Hz, 1H), 8.81 (d, J = 1.6 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 7.95 (dd, J = 1.6, 8.4 Hz, 1H), 7.66 (s, 1H), 7.59-7.56 (m, 1H), 7.32-7.28 (m, 2H), 7.04-7.00 (m, 1H), 5.45-5.35 (m, 1H), 3.78-3.74 (m, 1H), 3.40-3.36 (m, 1H), 2.30 (s, 3H), 2.28 (s, 3H); Preparation of Example 7a &7b (R or S)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide and (S or R)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide
 The compound 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (30 mg, 0.0837 mmol) was purified by preparative SFC (Column: DAICEL CHIRALCEL OJ 250 mm*30 mm,10 µm; Mobile phase: A for CO2, B for MeOH+ACN (neutral; Gradient elution: 40% B in CO2, 2.56 min) to give two product solutions. The solution containing Example 7a was concentrated under vacuum (40 °C) to give (R or S)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-
dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (10 mg, 0.0289 mmol, 34.53 % yield, ee.100 % based on SFC method 4) as off-white solid. The solution containing Example 7b was concentrated under vacuum (40 °C) to give (S or R)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (9.5 mg, 0.0263 mmol, 31.48 % yield, ee.99.57 % based on SFC method 4) as off-white solid. Example 7a: LCMS: RT 0.489 min (method 7); m/z 359.1 (M+H)+ (ESI+); SFC: RT: 1.880 min, 100% ee (Method 4); 1H NMR (CDCl3, 400 MHz): 11.95 (d, J = 7.2 Hz, 1H), 8.49 (d, J = 1.6 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 7.68 (dd, J = 1.6, 8.4 Hz, 1H), 7.40-7.26 (m, 3H), 6.86-6.78 (m, 2H), 5.60-5.53 (m, 1H), 3.81 (dd, J = 8.0, 13.6 Hz, 1H), 3.28 (dd, J = 4.8, 13.6 Hz, 1H), 2.36 (s, 3H), 2.35 (s, 3H) Example 7b: LCMS RT 0.488 min (method 7); m/z 359.1 (M+H)+ (ESI+); SFC: RT: 2.121 min, 99.57% ee (Method 4); 1H NMR (CDCl3, 400 MHz): 11.95 (d, J = 7.6 Hz, 1H), 8.49 (d, J = 1.6 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 7.68 (dd, J = 1.6, 8.4 Hz, 1H), 7.40-7.26 (m, 3H), 6.86-6.78 (m, 2H), 5.60-5.53 (m, 1H), 3.81 (dd, J = 8.0, 13.6 Hz, 1H), 3.28 (dd, J = 4.8, 13.6 Hz, 1H), 2.36 (s, 3H), 2.35(s, 3H) Preparation of Intermediate 8.1 Benzyl (1-(methylimino)-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)carbamate
The compound 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (30 mg, 0.0837 mmol) was purified by preparative SFC (Column: DAICEL CHIRALCEL OJ 250 mm*30 mm,10 µm; Mobile phase: A for CO2, B for MeOH+ACN (neutral; Gradient elution: 40% B in CO2, 2.56 min) to give two product solutions. The solution containing Example 7a was concentrated under vacuum (40 °C) to give (R or S)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-
dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (10 mg, 0.0289 mmol, 34.53 % yield, ee.100 % based on SFC method 4) as off-white solid. The solution containing Example 7b was concentrated under vacuum (40 °C) to give (S or R)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (9.5 mg, 0.0263 mmol, 31.48 % yield, ee.99.57 % based on SFC method 4) as off-white solid. Example 7a: LCMS: RT 0.489 min (method 7); m/z 359.1 (M+H)+ (ESI+); SFC: RT: 1.880 min, 100% ee (Method 4); 1H NMR (CDCl3, 400 MHz): 11.95 (d, J = 7.2 Hz, 1H), 8.49 (d, J = 1.6 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 7.68 (dd, J = 1.6, 8.4 Hz, 1H), 7.40-7.26 (m, 3H), 6.86-6.78 (m, 2H), 5.60-5.53 (m, 1H), 3.81 (dd, J = 8.0, 13.6 Hz, 1H), 3.28 (dd, J = 4.8, 13.6 Hz, 1H), 2.36 (s, 3H), 2.35 (s, 3H) Example 7b: LCMS RT 0.488 min (method 7); m/z 359.1 (M+H)+ (ESI+); SFC: RT: 2.121 min, 99.57% ee (Method 4); 1H NMR (CDCl3, 400 MHz): 11.95 (d, J = 7.6 Hz, 1H), 8.49 (d, J = 1.6 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 7.68 (dd, J = 1.6, 8.4 Hz, 1H), 7.40-7.26 (m, 3H), 6.86-6.78 (m, 2H), 5.60-5.53 (m, 1H), 3.81 (dd, J = 8.0, 13.6 Hz, 1H), 3.28 (dd, J = 4.8, 13.6 Hz, 1H), 2.36 (s, 3H), 2.35(s, 3H) Preparation of Intermediate 8.1 Benzyl (1-(methylimino)-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)carbamate
 To the mixture of benzyl (1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)carbamate (200 mg, 0.751 mmol) in DCM (4mL) was added trimethyloxonium tetrafluoroborate (133 mg, 0.901 mmol). The mixture was stirred at 20 °C for 2 h and then, concentrated under vacuum. The resulting residue was purified by preparative HPLC (column: Unisil 3-100 C18 Ultra 150*50 mm*10 m; mobile phase: A: 1 mM aqueous solution of NH4HCO3, B: MeCN; B%: 15%-45%, 9 min) and the resulting fraction was extracted with DCM (30 mL, 3x). The combined organic layer was washed with brine (50 mL), dried over Na2SO4, filtered and concentrated in vacuo to give benzyl (1-(methylimino)-1-oxido-2,3-dihydro-1H-1 6-thiophen- 3-yl)carbamate (24 mg, 0.0856 mmol, 11.40 % yield) as a yellow oil. RT 0.577 min (method 2); m/z 281.1 (M+H)+ (ESI+); 1H NMR (400 MHz, DMSO-d6) 7.88-8.04 (m, 1 H), 7.30-7.39 (m, 5 H), 7.07-7.16 (m, 1 H), 6.5-6.69 (m, 1 H), 5.05 (s, 2 H), 4.78-4.94 (m, 1 H), 3.50- 3.80 (m, 1 H), 2.90-3.08 (m, 1 H) Preparation of Intermediate 8.2 3-amino-1-(methylimino)-2,3-dihydro-1H-1 6-thiophene 1-oxide hydrobromide
A solution of benzyl (1-(methylimino)-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)carbamate (1.00 eq, 23 mg, 0.0820 mmol) in HBr/AcOH (253 mg, 33% wt/wt, 0.779 mmol) was stirred at 25 °C for 1 h. MTBE (10 mL) was added to precipitate a solid and the supernatant was removed after static settlement. This process was repeated six times to give a residue. The resulting residue was dried under vacuum to give 3-amino-1-(methylimino)-2,3-dihydro-1H-1 6-thiophene 1-oxide hydrobromide (18 mg, 0.0793 mmol, 96.60 % yield, HBr salt) as a yellow oil. Preparation of Example 8 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2-
To the mixture of benzyl (1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)carbamate (200 mg, 0.751 mmol) in DCM (4mL) was added trimethyloxonium tetrafluoroborate (133 mg, 0.901 mmol). The mixture was stirred at 20 °C for 2 h and then, concentrated under vacuum. The resulting residue was purified by preparative HPLC (column: Unisil 3-100 C18 Ultra 150*50 mm*10 m; mobile phase: A: 1 mM aqueous solution of NH4HCO3, B: MeCN; B%: 15%-45%, 9 min) and the resulting fraction was extracted with DCM (30 mL, 3x). The combined organic layer was washed with brine (50 mL), dried over Na2SO4, filtered and concentrated in vacuo to give benzyl (1-(methylimino)-1-oxido-2,3-dihydro-1H-1 6-thiophen- 3-yl)carbamate (24 mg, 0.0856 mmol, 11.40 % yield) as a yellow oil. RT 0.577 min (method 2); m/z 281.1 (M+H)+ (ESI+); 1H NMR (400 MHz, DMSO-d6) 7.88-8.04 (m, 1 H), 7.30-7.39 (m, 5 H), 7.07-7.16 (m, 1 H), 6.5-6.69 (m, 1 H), 5.05 (s, 2 H), 4.78-4.94 (m, 1 H), 3.50- 3.80 (m, 1 H), 2.90-3.08 (m, 1 H) Preparation of Intermediate 8.2 3-amino-1-(methylimino)-2,3-dihydro-1H-1 6-thiophene 1-oxide hydrobromide
A solution of benzyl (1-(methylimino)-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)carbamate (1.00 eq, 23 mg, 0.0820 mmol) in HBr/AcOH (253 mg, 33% wt/wt, 0.779 mmol) was stirred at 25 °C for 1 h. MTBE (10 mL) was added to precipitate a solid and the supernatant was removed after static settlement. This process was repeated six times to give a residue. The resulting residue was dried under vacuum to give 3-amino-1-(methylimino)-2,3-dihydro-1H-1 6-thiophene 1-oxide hydrobromide (18 mg, 0.0793 mmol, 96.60 % yield, HBr salt) as a yellow oil. Preparation of Example 8 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2-
 To a solution of 6-(3,4-dimethylphenyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (10 mg, 0.0411 mmol) and 3-amino-1-(methylimino)-2,3-dihydro-1H-1 6-thiophene 1-oxide (19 mg, 0.0822 mmol, HBr salt) in DMA (0.30 mL) was added DIEA (51 µL, 0.288 mmol) and T4P in EtOAc (59 mg, 50% purity, 0.0822 mmol) at 0 °C. The mixture was stirred at 25 °C for 1 h. The reaction mixture was poured into water (5 mL) and extracted with DCM (5 mL, 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative normal phase HPLC (column: Welch Ultimate XB-CN 250 mm*100 mm*10 µm; mobile phase: A: Hexane, B: EtOH; B%: 15%-55%, 15 min) and concentrated under vacuum to give a crude product, which was further purified by preparative HPLC (column: Unisil 3-100 C18 Ultra 150*25 mm*10 µm; mobile phase: A: 10 mM aqueous solution of NH4HCO3, B: MeCN; B%: 20%-50%, 9 min) and lyophilized directly to give the product 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-1 6- thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-carboxamide (1.0 mg, 0.00269 mmol, 6.55 % yield) as a white solid.
RT 0.428 min (method 2); m/z 372.2 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 10.00-10.24 (m, 1 H), 9.71-9.99 (m, 1 H), 8.53-8.61 (m, 1 H), 7.36-7.44 (m, 2 H), 7.29-7.34 (m, 1 H), 6.64-6.90 (m, 3 H), 5.54-5.68 (m, 1 H), 3.67-3.77 (m, 1 H), 3.24-3.42 (m, 1 H), 2.88-2.95 (m, 3 H), 2.36 (s, 6 H). Preparation of Intermediate 9.1 5-(3,4-dimethylphenyl)picolinic acid
To a solution of 6-(3,4-dimethylphenyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (10 mg, 0.0411 mmol) and 3-amino-1-(methylimino)-2,3-dihydro-1H-1 6-thiophene 1-oxide (19 mg, 0.0822 mmol, HBr salt) in DMA (0.30 mL) was added DIEA (51 µL, 0.288 mmol) and T4P in EtOAc (59 mg, 50% purity, 0.0822 mmol) at 0 °C. The mixture was stirred at 25 °C for 1 h. The reaction mixture was poured into water (5 mL) and extracted with DCM (5 mL, 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative normal phase HPLC (column: Welch Ultimate XB-CN 250 mm*100 mm*10 µm; mobile phase: A: Hexane, B: EtOH; B%: 15%-55%, 15 min) and concentrated under vacuum to give a crude product, which was further purified by preparative HPLC (column: Unisil 3-100 C18 Ultra 150*25 mm*10 µm; mobile phase: A: 10 mM aqueous solution of NH4HCO3, B: MeCN; B%: 20%-50%, 9 min) and lyophilized directly to give the product 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-1 6- thiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-carboxamide (1.0 mg, 0.00269 mmol, 6.55 % yield) as a white solid.
RT 0.428 min (method 2); m/z 372.2 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 10.00-10.24 (m, 1 H), 9.71-9.99 (m, 1 H), 8.53-8.61 (m, 1 H), 7.36-7.44 (m, 2 H), 7.29-7.34 (m, 1 H), 6.64-6.90 (m, 3 H), 5.54-5.68 (m, 1 H), 3.67-3.77 (m, 1 H), 3.24-3.42 (m, 1 H), 2.88-2.95 (m, 3 H), 2.36 (s, 6 H). Preparation of Intermediate 9.1 5-(3,4-dimethylphenyl)picolinic acid
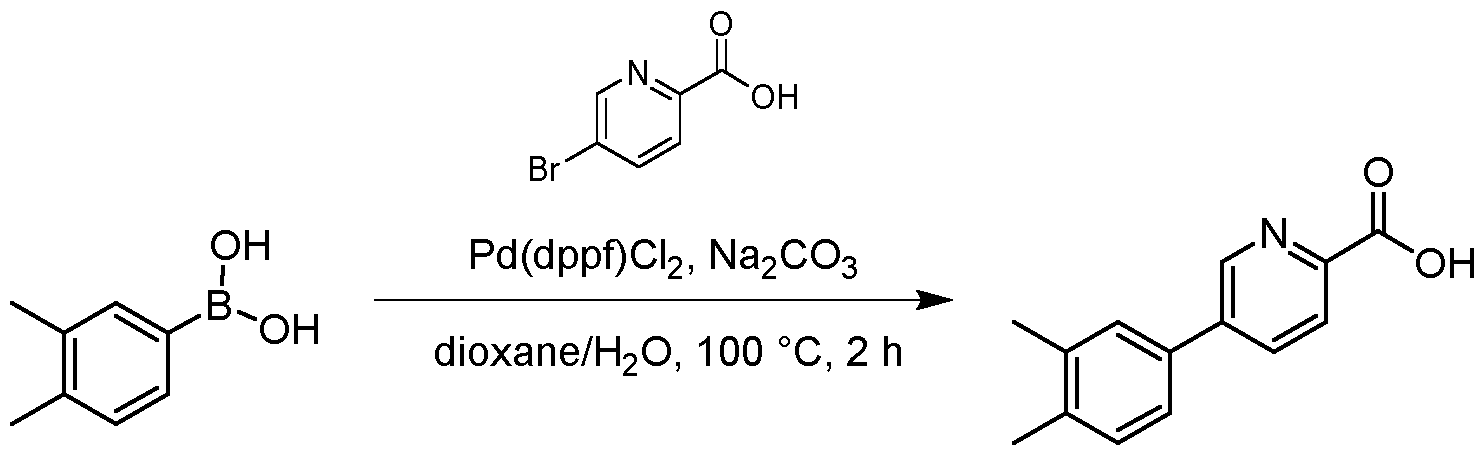 To a solution of 5-bromopicolinic acid (500 mg, 2.48 mmol) in 1,4-dioxane (10 mL) and water (2 mL) was added (3,4-dimethylphenyl)boronic acid (445 mg, 2.97 mmol), Na2CO3 (525 mg, 4.95 mmol) and Pd(dppf)Cl2 (181 mg, 0.248 mmol). The reaction mixture was degassed and purged with N2 (3x) before it was stirred at 100 °C for 2 h and then filtered. The cake was washed with MeOH (5 mL, 3x) and the filtrate was acidified with 1 M HCl until pH = 1. The resulting solution was concentrated under reduced pressure. The residue was purified by reversed-phase flash (ISCO®; 20 g Flash column Welch Ultimate XB_C18 20-40 µm; 120 A; Mobile phase: A: 0.1% formic acid in water, B: MeCN; B%: 5%-95%, 50 mL/min) and lyophilized to give the product 5-(3,4-dimethylphenyl)picolinic acid (120 mg, 0.296 mmol, 11.95 % yield) as a gray solid. RT 0.453 min (method 5); m/z 228.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.99 (d, J = 2.0 Hz, 1H), 8.23 (dd, J = 8.0, 2.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.61 (s, 1H), 7.49 (d, J = 7.6 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 2.31 (s, 3H), 2.28 (s, 3H). Preparation of Example 9 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide
To a solution of 5-bromopicolinic acid (500 mg, 2.48 mmol) in 1,4-dioxane (10 mL) and water (2 mL) was added (3,4-dimethylphenyl)boronic acid (445 mg, 2.97 mmol), Na2CO3 (525 mg, 4.95 mmol) and Pd(dppf)Cl2 (181 mg, 0.248 mmol). The reaction mixture was degassed and purged with N2 (3x) before it was stirred at 100 °C for 2 h and then filtered. The cake was washed with MeOH (5 mL, 3x) and the filtrate was acidified with 1 M HCl until pH = 1. The resulting solution was concentrated under reduced pressure. The residue was purified by reversed-phase flash (ISCO®; 20 g Flash column Welch Ultimate XB_C18 20-40 µm; 120 A; Mobile phase: A: 0.1% formic acid in water, B: MeCN; B%: 5%-95%, 50 mL/min) and lyophilized to give the product 5-(3,4-dimethylphenyl)picolinic acid (120 mg, 0.296 mmol, 11.95 % yield) as a gray solid. RT 0.453 min (method 5); m/z 228.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.99 (d, J = 2.0 Hz, 1H), 8.23 (dd, J = 8.0, 2.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.61 (s, 1H), 7.49 (d, J = 7.6 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 2.31 (s, 3H), 2.28 (s, 3H). Preparation of Example 9 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide
 To a solution of 5-(3,4-dimethylphenyl)picolinic acid (80 mg, 0.352 mmol), DIEA (0.31 mL, 1.76 mmol) and 3-amino-2,3-dihydrothiophene 1,1-dioxide (78 mg, 0.458 mmol, HCl salt) in DMF (2 mL) was added T4P in EtOAc (329 mg, 50% purity, 0.458 mmol). The reaction mixture was stirred at 20 oC for 2 h
before it was poured into water (5 mL) and extracted with EtOAc (5 mL, 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (column: YMC-Actus Triart C18150*30 mm*7 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 43%-73%, 10 min) and lyophilized directly to give the product 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide (28 mg,0.0801 mmol, 22.77 % yield) as an off-white solid. RT 0.519 min (method 5); m/z 343.1 (M+H)+ (ESI+) 1H NMR (DMSO-d6, 400 MHz): 9.41 (d, J = 8.0 Hz, 1H), 8.93 (d, J = 2.0 Hz, 1H), 8.26 (dd, J = 8.0, 2.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.60 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.22 (dd, J = 6.4, 2.4 Hz, 1H), 6.94 (dd, J = 6.4, 2.4 Hz, 1H), 5.54-5.26 (m, 1H), 3.76 (dd, J = 13.6, 6.0 Hz, 1H), 3.44 (dd, J = 13.6, 6.0 Hz, 1H), 2.32 (s, 3H), 2.28 (s, 3H); Preparation of Example 10 (R or S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3- yl)picolinamide and Example 11 (S or R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3- yl)picolinamide
To a solution of 5-(3,4-dimethylphenyl)picolinic acid (80 mg, 0.352 mmol), DIEA (0.31 mL, 1.76 mmol) and 3-amino-2,3-dihydrothiophene 1,1-dioxide (78 mg, 0.458 mmol, HCl salt) in DMF (2 mL) was added T4P in EtOAc (329 mg, 50% purity, 0.458 mmol). The reaction mixture was stirred at 20 oC for 2 h
before it was poured into water (5 mL) and extracted with EtOAc (5 mL, 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (column: YMC-Actus Triart C18150*30 mm*7 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 43%-73%, 10 min) and lyophilized directly to give the product 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide (28 mg,0.0801 mmol, 22.77 % yield) as an off-white solid. RT 0.519 min (method 5); m/z 343.1 (M+H)+ (ESI+) 1H NMR (DMSO-d6, 400 MHz): 9.41 (d, J = 8.0 Hz, 1H), 8.93 (d, J = 2.0 Hz, 1H), 8.26 (dd, J = 8.0, 2.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.60 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.22 (dd, J = 6.4, 2.4 Hz, 1H), 6.94 (dd, J = 6.4, 2.4 Hz, 1H), 5.54-5.26 (m, 1H), 3.76 (dd, J = 13.6, 6.0 Hz, 1H), 3.44 (dd, J = 13.6, 6.0 Hz, 1H), 2.32 (s, 3H), 2.28 (s, 3H); Preparation of Example 10 (R or S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3- yl)picolinamide and Example 11 (S or R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3- yl)picolinamide
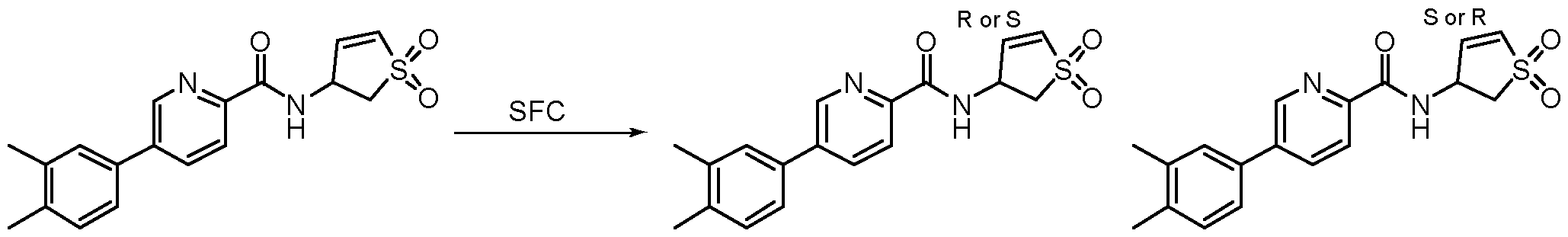 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide (28 mg, 0.0818 mmol) was separated by SFC (column: DAICEL CHIRALPAK IG 250 mm*30 mm, 10 µm; mobile phase: CO2-MeOH/ACN; B%: 75%-75%,12 min) and concentrated separately to give: - Example 10 (SFC Method 3, Rt 1.093 min) (R or S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)picolinamide (13 mg, 0.0376 mmol, 45.94 % yield, 98.56% purity, 100% ee) as an brown solid. RT 0.516min (method 5); m/z 343.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 9.41 (d, J = 8.0 Hz, 1H), 8.93 (d, J = 2.0 Hz, 1H), 8.26 (dd, J = 8.0, 2.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.60 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.22 (dd, J = 6.4, 2.4 Hz, 1H), 6.94 (dd, J = 6.4, 2.4 Hz, 1H), 5.54-5.26 (m, 1H), 3.76 (dd, J = 13.6, 6.0 Hz, 1H), 3.44 (dd, J = 13.6, 6.0 Hz, 1H), 2.32 (s, 3H), 2.28 (s, 3H); - Example 11 (SFC Method 3, Rt 3.075 min) (S or R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)picolinamide (14 mg, 0.0394 mmol, 48.17 % yield, 98.03% purity, 100% ee) as an off-white solid. RT 0.516min (method 5); m/z 343.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 9.41 (d, J = 8.0 Hz,
1H), 8.93 (d, J = 2.0 Hz, 1H), 8.26 (dd, J = 8.0, 2.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.60 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.22 (dd, J = 6.4, 2.4 Hz, 1H), 6.94 (dd, J = 6.4, 2.4 Hz, 1H), 5.54-5.26 (m, 1H), 3.76 (dd, J = 13.2, 6.0 Hz, 1H), 3.44 (dd, J = 13.2, 6.0 Hz, 1H), 2.32 (s, 3H), 2.28 (s, 3H); Preparation of Intermediate 12.1 Methyl 2-methoxy-6-(piperidin-1-yl)nicotinate
5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide (28 mg, 0.0818 mmol) was separated by SFC (column: DAICEL CHIRALPAK IG 250 mm*30 mm, 10 µm; mobile phase: CO2-MeOH/ACN; B%: 75%-75%,12 min) and concentrated separately to give: - Example 10 (SFC Method 3, Rt 1.093 min) (R or S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)picolinamide (13 mg, 0.0376 mmol, 45.94 % yield, 98.56% purity, 100% ee) as an brown solid. RT 0.516min (method 5); m/z 343.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 9.41 (d, J = 8.0 Hz, 1H), 8.93 (d, J = 2.0 Hz, 1H), 8.26 (dd, J = 8.0, 2.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.60 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.22 (dd, J = 6.4, 2.4 Hz, 1H), 6.94 (dd, J = 6.4, 2.4 Hz, 1H), 5.54-5.26 (m, 1H), 3.76 (dd, J = 13.6, 6.0 Hz, 1H), 3.44 (dd, J = 13.6, 6.0 Hz, 1H), 2.32 (s, 3H), 2.28 (s, 3H); - Example 11 (SFC Method 3, Rt 3.075 min) (S or R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)picolinamide (14 mg, 0.0394 mmol, 48.17 % yield, 98.03% purity, 100% ee) as an off-white solid. RT 0.516min (method 5); m/z 343.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 9.41 (d, J = 8.0 Hz,
1H), 8.93 (d, J = 2.0 Hz, 1H), 8.26 (dd, J = 8.0, 2.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.60 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.22 (dd, J = 6.4, 2.4 Hz, 1H), 6.94 (dd, J = 6.4, 2.4 Hz, 1H), 5.54-5.26 (m, 1H), 3.76 (dd, J = 13.2, 6.0 Hz, 1H), 3.44 (dd, J = 13.2, 6.0 Hz, 1H), 2.32 (s, 3H), 2.28 (s, 3H); Preparation of Intermediate 12.1 Methyl 2-methoxy-6-(piperidin-1-yl)nicotinate
 To a solution of methyl 6-bromo-2-methoxynicotinate (500 mg, 2.03 mmol) in DMF (5 mL) was added piperidine hydrochloride (494 mg, 4.06 mmol, HCl salt) and DIEA (1.8 mL, 10.2 mmol). The mixture was stirred at 100 °C for 2 h. The resulting mixture was poured into water (10 mL) and extracted with ethyl acetate (10 mL, 2x). The combined organic layer was washed with brine (5 ml, 3x), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum to give the product methyl 2-methoxy-6- (piperidin-1-yl)nicotinate (600 mg, crude) as a yellow oil. RT 0.565 min (method 5); m/z 251.0 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 8.01 (d, J = 8.8 Hz, 1 H), 6.14 (d, J = 8.8 Hz, 1 H), 3.96 (s, 3 H), 3.81 (s, 3 H), 3.57-3.67 (m, 4 H), 1.55-1.72 (m, 6 H). Preparation of Intermediate 12.2 Methyl 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylate
To a solution of methyl 6-bromo-2-methoxynicotinate (500 mg, 2.03 mmol) in DMF (5 mL) was added piperidine hydrochloride (494 mg, 4.06 mmol, HCl salt) and DIEA (1.8 mL, 10.2 mmol). The mixture was stirred at 100 °C for 2 h. The resulting mixture was poured into water (10 mL) and extracted with ethyl acetate (10 mL, 2x). The combined organic layer was washed with brine (5 ml, 3x), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum to give the product methyl 2-methoxy-6- (piperidin-1-yl)nicotinate (600 mg, crude) as a yellow oil. RT 0.565 min (method 5); m/z 251.0 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 8.01 (d, J = 8.8 Hz, 1 H), 6.14 (d, J = 8.8 Hz, 1 H), 3.96 (s, 3 H), 3.81 (s, 3 H), 3.57-3.67 (m, 4 H), 1.55-1.72 (m, 6 H). Preparation of Intermediate 12.2 Methyl 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylate
 A solution of ethyl 2-methoxy-6-(piperidin-1-yl)nicotinate (300 mg, 1.20 mmol) and sodium iodide (719 mg, 4.79 mmol) in MeCN (3 mL) was cooled to 0°C and TMSI ( 0.68 mL, 4.79 mmol) was added. The mixture was stirred at 0 °C for 20 min and then for 16h at 25 °C. The resulting mixture was poured into water (10 mL) and extracted with ethyl acetate (10 mL, 2x). The combined organic layer was washed with brine (5 ml, 3x), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum to give the crude product methyl 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylate (300 mg, crude), which was used directly in the next step without further purification. RT 0.484 min (method 5); m/z 259.0 (M+Na)+ (ESI+)
Preparation of Intermediate 12.3 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylic acid
A solution of ethyl 2-methoxy-6-(piperidin-1-yl)nicotinate (300 mg, 1.20 mmol) and sodium iodide (719 mg, 4.79 mmol) in MeCN (3 mL) was cooled to 0°C and TMSI ( 0.68 mL, 4.79 mmol) was added. The mixture was stirred at 0 °C for 20 min and then for 16h at 25 °C. The resulting mixture was poured into water (10 mL) and extracted with ethyl acetate (10 mL, 2x). The combined organic layer was washed with brine (5 ml, 3x), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum to give the crude product methyl 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylate (300 mg, crude), which was used directly in the next step without further purification. RT 0.484 min (method 5); m/z 259.0 (M+Na)+ (ESI+)
Preparation of Intermediate 12.3 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylic acid
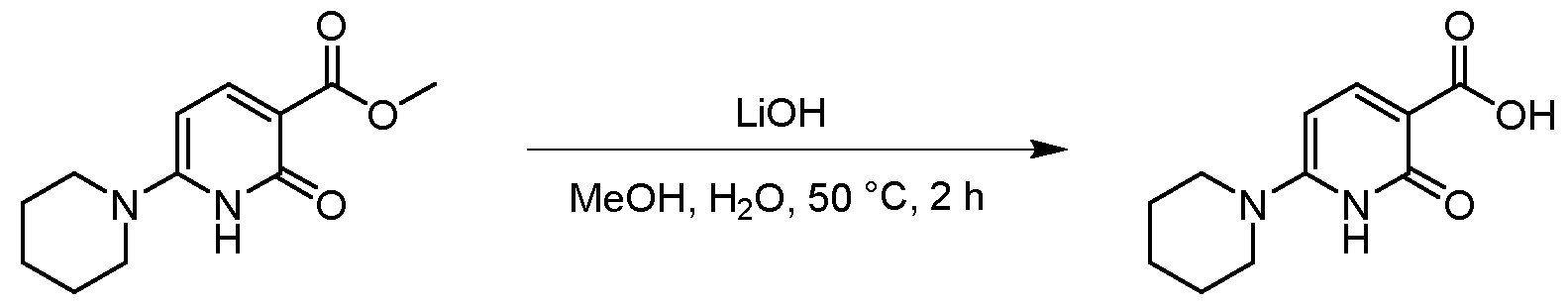 To a solution of methyl 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylate (150 mg, 0.635 mmol) in methanol (1.5 mL) and water (0.30 mL) was added LiOH.H2O (76 mg, 3.17 mmol). The mixture was stirred at 50 °C for 16 h. Then pH was adjusted to 3 with HCl (aq., 1N). The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (5 mL, 3x). The combined organic layer was washed with brine (5 mL, 2x), dried over anhydrous Na2SO4, filtered, and the filtrate was concentrated under vacuum to give the product 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylic acid (150 mg, crude) as a brown solid which was used in the next step without further purification. 1H NMR (DMSO-d6, 400 MHz): 13.50-14.75 (m, 1 H), 11.49-12.13 (m, 1 H), 7.91 (d, J = 8.8 Hz, 1 H), 6.10 (d, J = 8.4 Hz, 1 H), 3.54-3.58 (m, 4 H), 1.59-1.66 (m, 2 H), 1.55 (d, J = 4.4 Hz, 4 H). Preparation of Example 12 N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3- carboxamide
To a solution of methyl 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylate (150 mg, 0.635 mmol) in methanol (1.5 mL) and water (0.30 mL) was added LiOH.H2O (76 mg, 3.17 mmol). The mixture was stirred at 50 °C for 16 h. Then pH was adjusted to 3 with HCl (aq., 1N). The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (5 mL, 3x). The combined organic layer was washed with brine (5 mL, 2x), dried over anhydrous Na2SO4, filtered, and the filtrate was concentrated under vacuum to give the product 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylic acid (150 mg, crude) as a brown solid which was used in the next step without further purification. 1H NMR (DMSO-d6, 400 MHz): 13.50-14.75 (m, 1 H), 11.49-12.13 (m, 1 H), 7.91 (d, J = 8.8 Hz, 1 H), 6.10 (d, J = 8.4 Hz, 1 H), 3.54-3.58 (m, 4 H), 1.59-1.66 (m, 2 H), 1.55 (d, J = 4.4 Hz, 4 H). Preparation of Example 12 N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3- carboxamide
 To a solution of 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylic acid (50 mg, 0.225 mmol) in DMF (0.50 mL) was added 1,1-dioxo-2,3-dihydrothiophen-3-amine hydrochloride (46 mg, 0.270 mmol, HCl salt), DIEA (0.12 mL, 0.675 mmol) and T4P in EtOAc (211 mg, 50% purity, 0.292 mmol). The mixture was stirred at 20 °C for 2 h and purified by preparative HPLC (column: Phenomenex luna C18150*25 mm*10 µm; Mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 15%-45%; 10 min). The product solution was lyophilized to give the product N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(piperidin-1- yl)-1,2-dihydropyridine-3-carboxamide (1.8 mg, 0.00534 mmol, 2.37 % yield) as a white solid. RT 0.401 min (method 1); m/z 338.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz) 11.24 (s, 1 H), 9.69-9.92 (m, 1 H), 8.00 (d, J = 8.8 Hz, 1 H), 7.20 (m, 1 H), 6.93 (m, 1 H), 5.77-6.00 (m, 1 H), 5.21-5.40 (m, 1 H), 3.64-3.75 (m, 1 H), 3.46-3.53 (m, 4 H), 3.13-3.18 (m, 1 H), 1.58-1.64 (m, 2 H), 1.51-1.57 (m, 4 H)
Preparation of Intermediate 13.1 3-chloro-6-(3,4-dimethylphenyl)-2-methoxypyridine
To a solution of 2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3-carboxylic acid (50 mg, 0.225 mmol) in DMF (0.50 mL) was added 1,1-dioxo-2,3-dihydrothiophen-3-amine hydrochloride (46 mg, 0.270 mmol, HCl salt), DIEA (0.12 mL, 0.675 mmol) and T4P in EtOAc (211 mg, 50% purity, 0.292 mmol). The mixture was stirred at 20 °C for 2 h and purified by preparative HPLC (column: Phenomenex luna C18150*25 mm*10 µm; Mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 15%-45%; 10 min). The product solution was lyophilized to give the product N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(piperidin-1- yl)-1,2-dihydropyridine-3-carboxamide (1.8 mg, 0.00534 mmol, 2.37 % yield) as a white solid. RT 0.401 min (method 1); m/z 338.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz) 11.24 (s, 1 H), 9.69-9.92 (m, 1 H), 8.00 (d, J = 8.8 Hz, 1 H), 7.20 (m, 1 H), 6.93 (m, 1 H), 5.77-6.00 (m, 1 H), 5.21-5.40 (m, 1 H), 3.64-3.75 (m, 1 H), 3.46-3.53 (m, 4 H), 3.13-3.18 (m, 1 H), 1.58-1.64 (m, 2 H), 1.51-1.57 (m, 4 H)
Preparation of Intermediate 13.1 3-chloro-6-(3,4-dimethylphenyl)-2-methoxypyridine
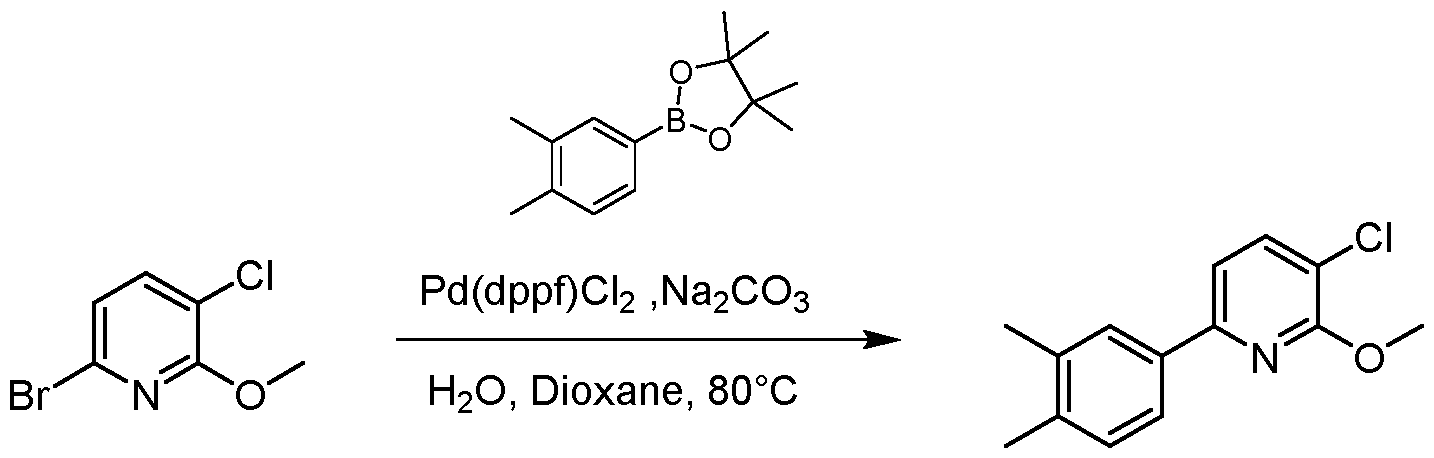 To a solution of 6-bromo-3-chloro-2-methoxypyridine (1.00 g, 4.49 mmol) and 2-(3,4- dimethylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.04 g, 4.50 mmol) in 1,4-dioxane (9 mL) and water (1.5 mL) was added Na2CO3 (953 mg, 8.99 mmol)) and Pd(dppf)Cl2 (367 mg, 0.449 mmol). The reaction mixture was stirred at 80 °C for 2 h under N2 atmosphere. The resulting mixture was filtered and the filtrate was purified by flash silica gel chromatography (ISCO®; 20 g SepaFlash® Silica Flash Column, Mobile phase: A: Petroleum ether, B: Ethyl acetate; B%: 0% to 0%; 50 mL/min) to give the product 3- chloro-6-(3,4-dimethylphenyl)-2-methoxypyridine (920 mg, 2.97 mmol, 66.10 % yield) as a white solid. RT 0.754 min (method 5); m/z 248.0 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.9-8.0 (m, 1H), 7.35-7.44 (m, 3H), 7.16 (d, 1H, J = 7.6 Hz), 3.88 (s, 3H), 2.27 (s, 3H), 2.25 (s, 3H). Preparation of Intermediate 13.2 3-(benzylthio)-6-(3,4-dimethylphenyl)-2-methoxypyridine
To a solution of 6-bromo-3-chloro-2-methoxypyridine (1.00 g, 4.49 mmol) and 2-(3,4- dimethylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.04 g, 4.50 mmol) in 1,4-dioxane (9 mL) and water (1.5 mL) was added Na2CO3 (953 mg, 8.99 mmol)) and Pd(dppf)Cl2 (367 mg, 0.449 mmol). The reaction mixture was stirred at 80 °C for 2 h under N2 atmosphere. The resulting mixture was filtered and the filtrate was purified by flash silica gel chromatography (ISCO®; 20 g SepaFlash® Silica Flash Column, Mobile phase: A: Petroleum ether, B: Ethyl acetate; B%: 0% to 0%; 50 mL/min) to give the product 3- chloro-6-(3,4-dimethylphenyl)-2-methoxypyridine (920 mg, 2.97 mmol, 66.10 % yield) as a white solid. RT 0.754 min (method 5); m/z 248.0 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.9-8.0 (m, 1H), 7.35-7.44 (m, 3H), 7.16 (d, 1H, J = 7.6 Hz), 3.88 (s, 3H), 2.27 (s, 3H), 2.25 (s, 3H). Preparation of Intermediate 13.2 3-(benzylthio)-6-(3,4-dimethylphenyl)-2-methoxypyridine
 To a solution of 3-chloro-6-(3,4-dimethylphenyl)-2-methoxypyridine (500 mg, 2.02 mmol) in 1,4- dioxane (8 mL) was added Cs2CO3 (837 mg, 6.06 mmol) and BrettPhos-Pd-G4 (186 mg, 0.202 mmol). The reaction mixture was degassed and purged with N2 (3x). Phenylmethanethiol (376 mg, 3.03 mmol) was added and the reaction mixture was stirred at 90 °C for 2 h under N2 atmosphere. The mixture was filtered and the filtrate was purified by flash chromatography (ISCO®; 12 g SepaFlash® Silica Flash Column, Mobile phase: A: Petroleum ether, B: Ethyl acetate; B%: 5% to 5%; 50 mL/min) to give the product (3-(benzylthio)-6-(3,4-dimethylphenyl)-2-methoxypyridine (180 mg,0.447 mmol, 22.15 % yield) as a white solid.
RT 0.772 min (method 5); m/z 336.2 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.79 (s, 1H), 7.71- 7.81 (m, 1H), 7.46 (d, 1H, J = 7.8 Hz), 7.31-7.34 (m, 4H), 7.22-7.24 (m, 3H), 4.14 (s, 3H), 4.13 (s, 2H), 2.35 (s, 3H), 2.33 (s, 3H) Preparation of Intermediate 13.3 3-(benzylthio)-6-(3,4-dimethylphenyl)pyridin-2(1H)-one
To a solution of 3-chloro-6-(3,4-dimethylphenyl)-2-methoxypyridine (500 mg, 2.02 mmol) in 1,4- dioxane (8 mL) was added Cs2CO3 (837 mg, 6.06 mmol) and BrettPhos-Pd-G4 (186 mg, 0.202 mmol). The reaction mixture was degassed and purged with N2 (3x). Phenylmethanethiol (376 mg, 3.03 mmol) was added and the reaction mixture was stirred at 90 °C for 2 h under N2 atmosphere. The mixture was filtered and the filtrate was purified by flash chromatography (ISCO®; 12 g SepaFlash® Silica Flash Column, Mobile phase: A: Petroleum ether, B: Ethyl acetate; B%: 5% to 5%; 50 mL/min) to give the product (3-(benzylthio)-6-(3,4-dimethylphenyl)-2-methoxypyridine (180 mg,0.447 mmol, 22.15 % yield) as a white solid.
RT 0.772 min (method 5); m/z 336.2 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.79 (s, 1H), 7.71- 7.81 (m, 1H), 7.46 (d, 1H, J = 7.8 Hz), 7.31-7.34 (m, 4H), 7.22-7.24 (m, 3H), 4.14 (s, 3H), 4.13 (s, 2H), 2.35 (s, 3H), 2.33 (s, 3H) Preparation of Intermediate 13.3 3-(benzylthio)-6-(3,4-dimethylphenyl)pyridin-2(1H)-one
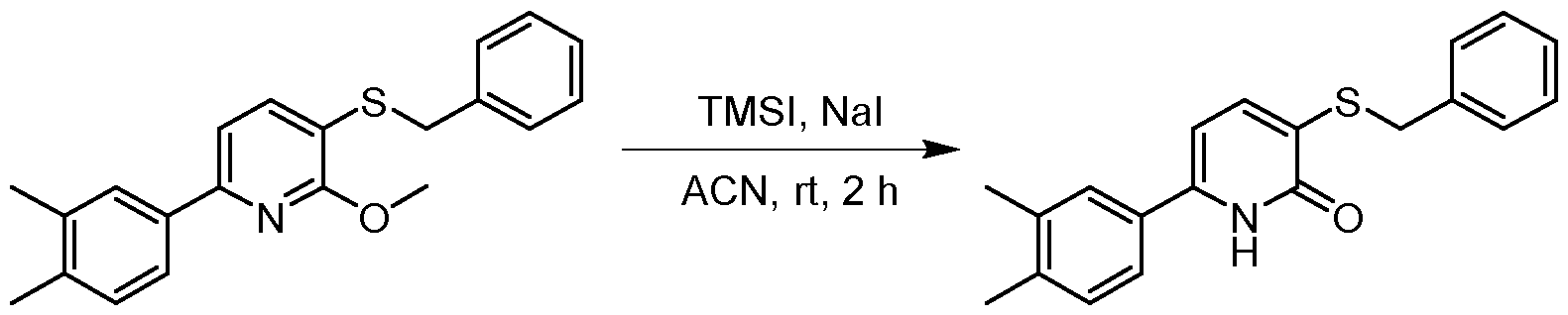 To a solution of 3-(benzylthio)-6-(3,4-dimethylphenyl)-2-methoxypyridine (149 mg, 0.444 mmol) in MeCN (2 mL) was added sodium iodide (333 mg, 2.22 mmol) and TMSI (445 mg, 2.22 mmol) at 0 °C. The mixture was stirred at 25 °C for 2 h. The mixture was filtered and the filtrate was purified by reversed phase flash (ISCO®; 40 g SepaFlash® column: Spherical C1820-45 µm, 100 A, mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 80%-95%, 50 mL/min). The resulting solution was lyophilized to give the product 3-(benzylthio)-6-(3,4-dimethylphenyl)pyridin-2(1H)-one (100 mg, 0.280 mmol, 62.99 % yield) as a black solid. 1H NMR (CDCl3, 400 MHz): 10.07 (br, 1H), 7.30-7.38 (m, 6H), 7.30-7.23 (m, 3H), 6.38 (d, 1H, J = 7.2 Hz), 4.18 (s, 2H), 2.33 (s, 3H), 2.31 (s, 3H) Preparation of Intermediate 13.4 6-(3,4-dimethylphenyl)-2-hydroxy-pyridine-3-sulfonyl chloride
To a solution of 3-(benzylthio)-6-(3,4-dimethylphenyl)-2-methoxypyridine (149 mg, 0.444 mmol) in MeCN (2 mL) was added sodium iodide (333 mg, 2.22 mmol) and TMSI (445 mg, 2.22 mmol) at 0 °C. The mixture was stirred at 25 °C for 2 h. The mixture was filtered and the filtrate was purified by reversed phase flash (ISCO®; 40 g SepaFlash® column: Spherical C1820-45 µm, 100 A, mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 80%-95%, 50 mL/min). The resulting solution was lyophilized to give the product 3-(benzylthio)-6-(3,4-dimethylphenyl)pyridin-2(1H)-one (100 mg, 0.280 mmol, 62.99 % yield) as a black solid. 1H NMR (CDCl3, 400 MHz): 10.07 (br, 1H), 7.30-7.38 (m, 6H), 7.30-7.23 (m, 3H), 6.38 (d, 1H, J = 7.2 Hz), 4.18 (s, 2H), 2.33 (s, 3H), 2.31 (s, 3H) Preparation of Intermediate 13.4 6-(3,4-dimethylphenyl)-2-hydroxy-pyridine-3-sulfonyl chloride
 To a solution of 3-(benzylthio)-6-(3,4-dimethylphenyl)pyridin-2(1H)-one (35 mg, 0.109 mmol) in acetic acid (1 mL) was added 1-chloropyrrolidine-2,5-dione (58 mg, 0.436 mmol) and H2O (0.3 mL) at 0 °C .The mixture was stirred at 0 °C for 18 mins before it was filtered and concentrated to give the crude product 6-(3,4-dimethylphenyl)-2-hydroxy-pyridine-3-sulfonyl chloride (20 mg,0.0672 mmol, 61.69 % yield) as a colorless oil. RT 0.577min (method 5); m/z 298.0 (M+H)+ (ESI+); Preparation of Example 13 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- sulfonamide
To a solution of 6-(3,4-dimethylphenyl)-2-oxo-1,2-dihydropyridine-3-sulfonyl chloride (20 mg, 0.0672 mmol) in THF (0.50 mL) and water (0.10 mL) was added NaHCO3 (5.6 mg, 0.0672 mmol) and 3-amino- 2,3-dihydrothiophene 1,1-dioxide hydrochloride (9.1 mg, 0.0536 mmol, HCl salt) at 0 °C. The mixture was stirred at 0 °C for 6 min and then filtered. The filtrate was purified by preparative HPLC (Column Phenomenex luna C18150*25 mm* 10 µm; Mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 29%-59%; 10 min). The resulting solution was lyophilized to give the product 6-(3,4-dimethylphenyl)-N- (1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-sulfonamide (6.3 mg, 96.70% purity, 0.0161 mmol, 23.93 % yield) as a white solid. RT 0.498 min (method 5); m/z 395.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.06 (d, J = 7.6 Hz, 1H), 7.63 (s, 1H), 7.55 (d, J = 7.6 Hz, 1H ), 7.29 (d, 1H, J = 8.0 Hz), 7.16 (dd, J = 2.4, 6.8 Hz,1H), 6.71-6.74 (m, 2H), 4.99-5.02 (m, 1H), 3.66 (dd, J = 8.0, 13.6 Hz, 1H), 3.06 (dd, J = 5.2, 13.6 Hz, 1H), 2.29 (s, 6H) Preparation of Intermediate 14.1 2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxylic acid
To a solution of 3-(benzylthio)-6-(3,4-dimethylphenyl)pyridin-2(1H)-one (35 mg, 0.109 mmol) in acetic acid (1 mL) was added 1-chloropyrrolidine-2,5-dione (58 mg, 0.436 mmol) and H2O (0.3 mL) at 0 °C .The mixture was stirred at 0 °C for 18 mins before it was filtered and concentrated to give the crude product 6-(3,4-dimethylphenyl)-2-hydroxy-pyridine-3-sulfonyl chloride (20 mg,0.0672 mmol, 61.69 % yield) as a colorless oil. RT 0.577min (method 5); m/z 298.0 (M+H)+ (ESI+); Preparation of Example 13 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- sulfonamide
To a solution of 6-(3,4-dimethylphenyl)-2-oxo-1,2-dihydropyridine-3-sulfonyl chloride (20 mg, 0.0672 mmol) in THF (0.50 mL) and water (0.10 mL) was added NaHCO3 (5.6 mg, 0.0672 mmol) and 3-amino- 2,3-dihydrothiophene 1,1-dioxide hydrochloride (9.1 mg, 0.0536 mmol, HCl salt) at 0 °C. The mixture was stirred at 0 °C for 6 min and then filtered. The filtrate was purified by preparative HPLC (Column Phenomenex luna C18150*25 mm* 10 µm; Mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 29%-59%; 10 min). The resulting solution was lyophilized to give the product 6-(3,4-dimethylphenyl)-N- (1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3-sulfonamide (6.3 mg, 96.70% purity, 0.0161 mmol, 23.93 % yield) as a white solid. RT 0.498 min (method 5); m/z 395.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.06 (d, J = 7.6 Hz, 1H), 7.63 (s, 1H), 7.55 (d, J = 7.6 Hz, 1H ), 7.29 (d, 1H, J = 8.0 Hz), 7.16 (dd, J = 2.4, 6.8 Hz,1H), 6.71-6.74 (m, 2H), 4.99-5.02 (m, 1H), 3.66 (dd, J = 8.0, 13.6 Hz, 1H), 3.06 (dd, J = 5.2, 13.6 Hz, 1H), 2.29 (s, 6H) Preparation of Intermediate 14.1 2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxylic acid
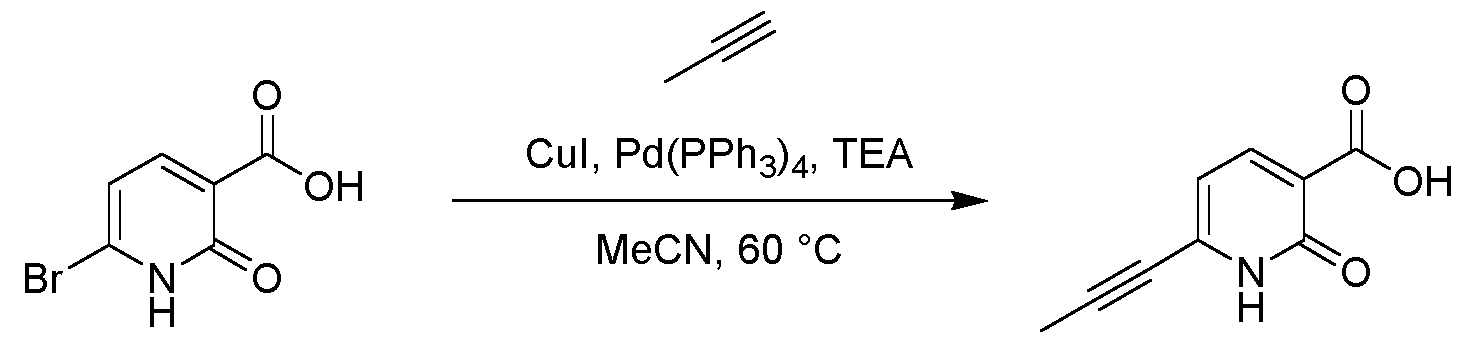 To a solution of 6-bromo-2-oxo-1,2-dihydropyridine-3-carboxylic acid (60 mg, 0.275 mmol) in MeCN (1.5 mL) was added CuI (21 mg, 0.110 mmol), Pd(PPh3)4 (32 mg, 0.0275 mmol) and TEA (0.11 mL, 0.826 mmol). The reaction mixture was degassed and purged with N2 (3x) before prop-1-yne (0.83 mL, 0.826 mmol) was added at 20°C. The reaction mixture was stirred at 60 °C for 16 h under N2 atmosphere and then filtered. The pH of the filtrate was adjusted with HCl (aq., 6N) to around 4-5. The resulting solution was purified by preparative HPLC (column: Phenomenex luna C18150*25 mm* 10 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 8%-38%, 10 min) and lyophilized directly to give the product 2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxylic acid (30 mg, 0.169 mmol, 61.53 % yield) as a white solid.
RT 0.351 min (method 5); m/z 199.9 (M+Na)+ (ESI+);1H NMR (400MHz, DMSO-d6): 14.60 (br, 1H), 13.66 (br, 1H), 8.27 (d, J = 7.6 Hz, 1H), 6.75 (d, J =7.6 Hz, 1H), 2.18 (s, 3H). Preparation of Example 14 N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3- carboxamide
To a solution of 6-bromo-2-oxo-1,2-dihydropyridine-3-carboxylic acid (60 mg, 0.275 mmol) in MeCN (1.5 mL) was added CuI (21 mg, 0.110 mmol), Pd(PPh3)4 (32 mg, 0.0275 mmol) and TEA (0.11 mL, 0.826 mmol). The reaction mixture was degassed and purged with N2 (3x) before prop-1-yne (0.83 mL, 0.826 mmol) was added at 20°C. The reaction mixture was stirred at 60 °C for 16 h under N2 atmosphere and then filtered. The pH of the filtrate was adjusted with HCl (aq., 6N) to around 4-5. The resulting solution was purified by preparative HPLC (column: Phenomenex luna C18150*25 mm* 10 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 8%-38%, 10 min) and lyophilized directly to give the product 2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxylic acid (30 mg, 0.169 mmol, 61.53 % yield) as a white solid.
RT 0.351 min (method 5); m/z 199.9 (M+Na)+ (ESI+);1H NMR (400MHz, DMSO-d6): 14.60 (br, 1H), 13.66 (br, 1H), 8.27 (d, J = 7.6 Hz, 1H), 6.75 (d, J =7.6 Hz, 1H), 2.18 (s, 3H). Preparation of Example 14 N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3- carboxamide
 To a solution of 2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxylic acid (10 mg, 0.0564 mmol) in DMF (1mL) was added at 0°C 3-amino-2,3-dihydrothiophene 1,1-dioxide hydrochloride (12 mg, 0.0734 mmol, HCl salt), DIEA (0.050 mL, 0.282 mmol) followed by T4P in EtOAc (47 mg, 50% purity, 0.0734 mmol). The mixture was then stirred at 20 °C for 2 h and directly purified by prep-HPLC (column: Phenomenex luna C18150*25 mm* 10 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 10%-40%, 10min). The resulting impure product was further purified by preparative HPLC (column: Waters Xbridge 150*25 mm* 5 µm; mobile phase: A: 0.05% Ammonia in water, B: MeCN; B%: 30%-60%, 10 min) and lyophilized directly to give the product N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6- (prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxamide (0.66 mg, 0.00219 mmol, 3.88 % yield, 97.68% purity) as a grey solid . RT 0.391 min (method 1); m/z 293.2 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 12.93 (br, 1H), 10.16 (br, 1H), 8.21 (d, J = 7.6 Hz, 1H), 7.24 (dd, J = 6.8, 2.0 Hz, 1H), 6.95 (dd, J = 6.8, 2.0 Hz, 1H), 6.57 (d, J = 7.6 Hz, 1H), 5.32 (s, 1H), 3.73-3.67 (m ,1H), 3.25-3.21 (m ,1H), 2.13 (s, 3H). Preparation of Intermediate 15.1 Methyl 2-chloro-6-(3,4-dimethylphenyl)nicotinate
To a solution of 2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxylic acid (10 mg, 0.0564 mmol) in DMF (1mL) was added at 0°C 3-amino-2,3-dihydrothiophene 1,1-dioxide hydrochloride (12 mg, 0.0734 mmol, HCl salt), DIEA (0.050 mL, 0.282 mmol) followed by T4P in EtOAc (47 mg, 50% purity, 0.0734 mmol). The mixture was then stirred at 20 °C for 2 h and directly purified by prep-HPLC (column: Phenomenex luna C18150*25 mm* 10 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 10%-40%, 10min). The resulting impure product was further purified by preparative HPLC (column: Waters Xbridge 150*25 mm* 5 µm; mobile phase: A: 0.05% Ammonia in water, B: MeCN; B%: 30%-60%, 10 min) and lyophilized directly to give the product N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6- (prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxamide (0.66 mg, 0.00219 mmol, 3.88 % yield, 97.68% purity) as a grey solid . RT 0.391 min (method 1); m/z 293.2 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 12.93 (br, 1H), 10.16 (br, 1H), 8.21 (d, J = 7.6 Hz, 1H), 7.24 (dd, J = 6.8, 2.0 Hz, 1H), 6.95 (dd, J = 6.8, 2.0 Hz, 1H), 6.57 (d, J = 7.6 Hz, 1H), 5.32 (s, 1H), 3.73-3.67 (m ,1H), 3.25-3.21 (m ,1H), 2.13 (s, 3H). Preparation of Intermediate 15.1 Methyl 2-chloro-6-(3,4-dimethylphenyl)nicotinate
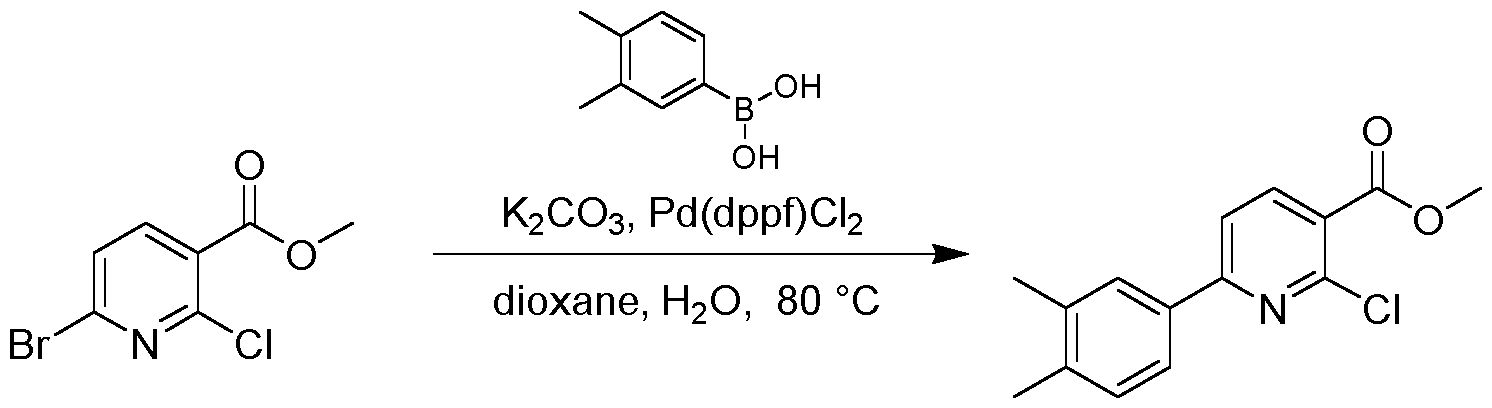 To a solution of methyl 6-bromo-2-chloronicotinate (1.0 g, 3.99 mmol) in 1,4-dioxane (10 mL) and water (2 mL) was added (3,4-dimethylphenyl)boronic acid (659 mg, 4.39 mmol), Na2CO3 (846 mg, 7.98 mmol) and Pd(dppf)Cl2 (163 mg, 0.200 mmol). The mixture was stirred at 80 °C for 2 h under N2. The
reaction mixture was poured into water (20 mL) and extracted with ethyl acetate (20 mL, 2x). The combined organic layer was washed with brine (20 mL), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated. The residue was purified by preparative normal phase HPLC (Welch Ultimate XB-CN 250*70*10 µm, mobile phase: A: Hexane, B: EtOH; B%: 0%-1%, 15 min) to give methyl 2-chloro- 6-(3,4-dimethylphenyl)nicotinate (950 mg, 3.45 mmol, 86.30 % yield) as a yellow solid. RT 0.617 min (method 5); m/z 276.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400MHz): 8.30 (d, J = 8.0 Hz, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.92 (s, 1H), 7.85 (dd, J = 8.0, 1.6 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 3.89 (s, 3H), 2.32 (s, 3H), 2.29 (s, 3H) Preparation of Intermediate 15.2 Methyl 6-(3,4-dimethylphenyl)-2-((3-((2-ethylhexyl)oxy)-3-oxopropyl)thio)nicotinate
To a solution of methyl 6-bromo-2-chloronicotinate (1.0 g, 3.99 mmol) in 1,4-dioxane (10 mL) and water (2 mL) was added (3,4-dimethylphenyl)boronic acid (659 mg, 4.39 mmol), Na2CO3 (846 mg, 7.98 mmol) and Pd(dppf)Cl2 (163 mg, 0.200 mmol). The mixture was stirred at 80 °C for 2 h under N2. The
reaction mixture was poured into water (20 mL) and extracted with ethyl acetate (20 mL, 2x). The combined organic layer was washed with brine (20 mL), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated. The residue was purified by preparative normal phase HPLC (Welch Ultimate XB-CN 250*70*10 µm, mobile phase: A: Hexane, B: EtOH; B%: 0%-1%, 15 min) to give methyl 2-chloro- 6-(3,4-dimethylphenyl)nicotinate (950 mg, 3.45 mmol, 86.30 % yield) as a yellow solid. RT 0.617 min (method 5); m/z 276.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400MHz): 8.30 (d, J = 8.0 Hz, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.92 (s, 1H), 7.85 (dd, J = 8.0, 1.6 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 3.89 (s, 3H), 2.32 (s, 3H), 2.29 (s, 3H) Preparation of Intermediate 15.2 Methyl 6-(3,4-dimethylphenyl)-2-((3-((2-ethylhexyl)oxy)-3-oxopropyl)thio)nicotinate
 To a solution of methyl 2-chloro-6-(3,4-dimethylphenyl)nicotinate (950 mg, 3.45 mmol) and 2- ethylhexyl 3-mercaptopropanoate (1505 mg, 6.89 mmol) in 1,4-dioxane (12 mL) was added DIEA (1.8 mL, 10.3 mmol), Xantphos (399 mg, 0.689 mmol) and Pd2(dba)3 (316 mg, 0.345 mmol). The reaction mixture was degassed and purged with N2 (3x) before it was stirred at 90 °C for 16 h. The mixture was filtered and the filtrate was purified by preparative normal phase HPLC (Welch Ultimate XB-CN 250*70*10 µm, mobile phase: A: Hexane, B: EtOH; B%: 0%-0%, 15 min) to give the product methyl 6-(3,4- dimethylphenyl)-2-((3-((2-ethylhexyl)oxy)-3-oxopropyl)thio)nicotinate (1.3 g, 2.84 mmol, 82.45 % yield) as a yellow oil RT 0.805 min (method 5); m/z 458.2 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz ): 8.25 (d, J = 8.0 Hz, 1H), 7.88 (s, 1H), 7.84 (dd, J = 8.0, 1.6 Hz, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 4.06-4.02 (m, 2H), 3.94 (s, 3H), 3.57 (t, J = 7.6 Hz, 2H), 2.88 (t, J = 7.6 Hz, 2H), 2.35 (d, J = 10.4 Hz, 6H), 1.59-1.55 (m, 2H), 1.42-1.28 (m, 6H), 0.91-0.84 (m, 7H). Preparation of Intermediate 15.3 Methyl 6-(3,4-dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylate
To a solution of methyl 2-chloro-6-(3,4-dimethylphenyl)nicotinate (950 mg, 3.45 mmol) and 2- ethylhexyl 3-mercaptopropanoate (1505 mg, 6.89 mmol) in 1,4-dioxane (12 mL) was added DIEA (1.8 mL, 10.3 mmol), Xantphos (399 mg, 0.689 mmol) and Pd2(dba)3 (316 mg, 0.345 mmol). The reaction mixture was degassed and purged with N2 (3x) before it was stirred at 90 °C for 16 h. The mixture was filtered and the filtrate was purified by preparative normal phase HPLC (Welch Ultimate XB-CN 250*70*10 µm, mobile phase: A: Hexane, B: EtOH; B%: 0%-0%, 15 min) to give the product methyl 6-(3,4- dimethylphenyl)-2-((3-((2-ethylhexyl)oxy)-3-oxopropyl)thio)nicotinate (1.3 g, 2.84 mmol, 82.45 % yield) as a yellow oil RT 0.805 min (method 5); m/z 458.2 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz ): 8.25 (d, J = 8.0 Hz, 1H), 7.88 (s, 1H), 7.84 (dd, J = 8.0, 1.6 Hz, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 4.06-4.02 (m, 2H), 3.94 (s, 3H), 3.57 (t, J = 7.6 Hz, 2H), 2.88 (t, J = 7.6 Hz, 2H), 2.35 (d, J = 10.4 Hz, 6H), 1.59-1.55 (m, 2H), 1.42-1.28 (m, 6H), 0.91-0.84 (m, 7H). Preparation of Intermediate 15.3 Methyl 6-(3,4-dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylate
 A solution of methyl 6-(3,4-dimethylphenyl)-2-((3-((2-ethylhexyl)oxy)-3-oxopropyl)thio)nicotinate (300 mg, 0.656 mmol) in THF (3 mL) was cooled to -60 °C. A solution of t-BuOK in THF (0.74 mL, 6.56 mmol, 1M) was added dropwise and the solution was stirred at -60 °C for 2 h. The resulting solution was
quenched with water (5 mL), the pH was adjusted to 3 with HCl (aq., 1N) and the mixture was extracted with ethyl acetate (5 mL, 2x). The combined organic layer was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum to give methyl 6-(3,4- dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylate (100 mg, 0.366 mmol, 55.81% yield) as a yellow solid. RT 0.530min (method 5); m/z 274.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 8.18 (d, J = 8.0 Hz, 1H), 7.74 (s, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 3.96 (s, 3H), 2.36 (s, 3H), 2.34 (s, 3H) Preparation of Intermediate 15.4 6-(3,4-dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylic acid
A solution of methyl 6-(3,4-dimethylphenyl)-2-((3-((2-ethylhexyl)oxy)-3-oxopropyl)thio)nicotinate (300 mg, 0.656 mmol) in THF (3 mL) was cooled to -60 °C. A solution of t-BuOK in THF (0.74 mL, 6.56 mmol, 1M) was added dropwise and the solution was stirred at -60 °C for 2 h. The resulting solution was
quenched with water (5 mL), the pH was adjusted to 3 with HCl (aq., 1N) and the mixture was extracted with ethyl acetate (5 mL, 2x). The combined organic layer was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum to give methyl 6-(3,4- dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylate (100 mg, 0.366 mmol, 55.81% yield) as a yellow solid. RT 0.530min (method 5); m/z 274.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 8.18 (d, J = 8.0 Hz, 1H), 7.74 (s, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 3.96 (s, 3H), 2.36 (s, 3H), 2.34 (s, 3H) Preparation of Intermediate 15.4 6-(3,4-dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylic acid
 To a solution of methyl 6-(3,4-dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylate (100 mg, 0.366 mmol) in methanol (0.50 mL), THF (0.50 mL) and water (0.50 mL) was added LiOH.H2O (77 mg, 1.83 mmol) and the mixture was stirred at 20 °C for 12 h. The pH of the mixture was adjusted to 3 with 1M HCl. The resulting solid was filtered and dried under vacuum to give the product 6-(3,4- dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylic acid (65 mg, 0.251 mmol, 68.52 % yield) as a yellow solid. RT 0.493 min (method 1); m/z 260.0 (M+H)+ (ESI+); 1H NMR (400 MHz, DMSO-d6): 14.85 (br, 1H), 14.40 (br, 1H), 8.54 (d, J = 8.0 Hz, 1H), 7.67 (s, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.30-7.35 (m, 2H), 2.31 (s, 6H) Preparation of Example 15 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-thioxo-1,2-dihydropyridine-3- carboxamide
To a solution of methyl 6-(3,4-dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylate (100 mg, 0.366 mmol) in methanol (0.50 mL), THF (0.50 mL) and water (0.50 mL) was added LiOH.H2O (77 mg, 1.83 mmol) and the mixture was stirred at 20 °C for 12 h. The pH of the mixture was adjusted to 3 with 1M HCl. The resulting solid was filtered and dried under vacuum to give the product 6-(3,4- dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylic acid (65 mg, 0.251 mmol, 68.52 % yield) as a yellow solid. RT 0.493 min (method 1); m/z 260.0 (M+H)+ (ESI+); 1H NMR (400 MHz, DMSO-d6): 14.85 (br, 1H), 14.40 (br, 1H), 8.54 (d, J = 8.0 Hz, 1H), 7.67 (s, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.30-7.35 (m, 2H), 2.31 (s, 6H) Preparation of Example 15 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-thioxo-1,2-dihydropyridine-3- carboxamide
 To a solution of 6-(3,4-dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylic acid (20 mg, 0.0771 mmol) in DMF (1 mL) was added HATU (26 mg, 0.100 mmol) and DIEA (0.041 mL, 0.231 mmol)
at 0 °C. The mixture was stirred at 0 °C for 6 min under N2.3-amino-2,3-dihydrothiophene 1,1-dioxide hydrochloride (17 mg, 0.100 mmol, HCl salt) was added and the mixture was stirred at 0 °C for 1 h under N2. The resulting mixture was diluted with water (2 mL) and EtOAc (2 mL) leading to formation of a precipitate. After filtration, the solid was collected, dissolved in DMSO, purified by preparative HPLC (column: Welch Ultimate C18150*25 mm*10 µm; mobile phase: A: 10 mM aqueous solution of NH4HCO3, B: MeCN; B%: 24%-54%, 9 min) and lyophilized to give the product 6-(3,4-dimethylphenyl)-N-(1,1-dioxido- 2,3-dihydrothiophen-3-yl)-2-thioxo-1,2-dihydropyridine-3-carboxamide (6.0 mg, 0.0158 mmol, 20.48 % yield, 98.56% purity ) as a yellow solid. RT 0.460 min (method 4); m/z 375.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 11.48 (d, J = 0.64 Hz, 1H), 10.81 (br, 1H), 8.76 (d, J = 8.0 Hz, 1H), 7.45-7.40 (m, 2H), 7.37-7.33 (m, 1H), 7.14 (d, J = 8.0 Hz, 1H), 6.88-6.80 (m, 2H), 5.58-5.50 (m, 1H), 3.84- 3.78 (m, 1H), 3.34-3.26 (m, 1H), 2.38 (s, 6H). Preparation of Intermediate 16.1 Methyl 3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxylate
To a solution of 6-(3,4-dimethylphenyl)-2-thioxo-1,2-dihydropyridine-3-carboxylic acid (20 mg, 0.0771 mmol) in DMF (1 mL) was added HATU (26 mg, 0.100 mmol) and DIEA (0.041 mL, 0.231 mmol)
at 0 °C. The mixture was stirred at 0 °C for 6 min under N2.3-amino-2,3-dihydrothiophene 1,1-dioxide hydrochloride (17 mg, 0.100 mmol, HCl salt) was added and the mixture was stirred at 0 °C for 1 h under N2. The resulting mixture was diluted with water (2 mL) and EtOAc (2 mL) leading to formation of a precipitate. After filtration, the solid was collected, dissolved in DMSO, purified by preparative HPLC (column: Welch Ultimate C18150*25 mm*10 µm; mobile phase: A: 10 mM aqueous solution of NH4HCO3, B: MeCN; B%: 24%-54%, 9 min) and lyophilized to give the product 6-(3,4-dimethylphenyl)-N-(1,1-dioxido- 2,3-dihydrothiophen-3-yl)-2-thioxo-1,2-dihydropyridine-3-carboxamide (6.0 mg, 0.0158 mmol, 20.48 % yield, 98.56% purity ) as a yellow solid. RT 0.460 min (method 4); m/z 375.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 11.48 (d, J = 0.64 Hz, 1H), 10.81 (br, 1H), 8.76 (d, J = 8.0 Hz, 1H), 7.45-7.40 (m, 2H), 7.37-7.33 (m, 1H), 7.14 (d, J = 8.0 Hz, 1H), 6.88-6.80 (m, 2H), 5.58-5.50 (m, 1H), 3.84- 3.78 (m, 1H), 3.34-3.26 (m, 1H), 2.38 (s, 6H). Preparation of Intermediate 16.1 Methyl 3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxylate
 To a solution of methyl 4-bromo-2-fluorobenzoate (1000 mg, 4.29 mmol) in 1,4-dioxane (10 mL) and water (2 mL) was added (3,4-dimethylphenyl)boronic acid (708 mg, 4.72 mmol), K2CO3 (1186 mg, 8.58 mmol) and Pd(dppf)Cl2 (157 mg, 0.215 mmol). The mixture was stirred at 80 °C for 2 h under N2. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (ISCO; 20 g SepaFlash Silica Flash Column, Eluent of 0~20% Ethyl acetate/Petroleum ether gradient, 60 mL/min) to give the product methyl 3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxylate (1.00 g, 3.76 mmol, 87.51 % yield) as a white solid. RT 0.634 min (method 1); m/z 259.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.99 (t, J = 8.0 Hz, 1H), 7.4-7.35 (m, 4H), 7.24 (d, J = 7.6 Hz, 1H), 3.96 (s, 3H), 2.35-2.33 (m, 6H) Preparation of Intermediate 16.2 3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxylic acid
To a solution of methyl 3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxylate (100 mg, 0.387 mmol) in THF (0.50 mL) and water (0.50 mL) was added LiOH.H2O (81 mg, 1.94 mmol) at 25 °C. The mixture was stirred at 25 °C for 1 h. Methanol (0.50 mL) was added and the reaction mixture was stirred at 25 °C for 1 h. The resulting mixture was concentrated under reduced pressure. The residue was diluted with water (0.5 m L) and then, HCl (aq., 1N) was added to the mixture until pH = 3~4. The aqueous layer was extracted with ethyl acetate (5 mL, 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure to give the product 3-fluoro-3',4'- dimethyl-[1,1'-biphenyl]-4-carboxylic acid (90 mg, 0.361 mmol, 93.27 % yield) as a white solid. RT 0.553 min (method 1); m/z 244.9 (M+H)+ (ESI+); Preparation of Example 16 N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide
To a solution of methyl 4-bromo-2-fluorobenzoate (1000 mg, 4.29 mmol) in 1,4-dioxane (10 mL) and water (2 mL) was added (3,4-dimethylphenyl)boronic acid (708 mg, 4.72 mmol), K2CO3 (1186 mg, 8.58 mmol) and Pd(dppf)Cl2 (157 mg, 0.215 mmol). The mixture was stirred at 80 °C for 2 h under N2. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (ISCO; 20 g SepaFlash Silica Flash Column, Eluent of 0~20% Ethyl acetate/Petroleum ether gradient, 60 mL/min) to give the product methyl 3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxylate (1.00 g, 3.76 mmol, 87.51 % yield) as a white solid. RT 0.634 min (method 1); m/z 259.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.99 (t, J = 8.0 Hz, 1H), 7.4-7.35 (m, 4H), 7.24 (d, J = 7.6 Hz, 1H), 3.96 (s, 3H), 2.35-2.33 (m, 6H) Preparation of Intermediate 16.2 3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxylic acid
To a solution of methyl 3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxylate (100 mg, 0.387 mmol) in THF (0.50 mL) and water (0.50 mL) was added LiOH.H2O (81 mg, 1.94 mmol) at 25 °C. The mixture was stirred at 25 °C for 1 h. Methanol (0.50 mL) was added and the reaction mixture was stirred at 25 °C for 1 h. The resulting mixture was concentrated under reduced pressure. The residue was diluted with water (0.5 m L) and then, HCl (aq., 1N) was added to the mixture until pH = 3~4. The aqueous layer was extracted with ethyl acetate (5 mL, 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure to give the product 3-fluoro-3',4'- dimethyl-[1,1'-biphenyl]-4-carboxylic acid (90 mg, 0.361 mmol, 93.27 % yield) as a white solid. RT 0.553 min (method 1); m/z 244.9 (M+H)+ (ESI+); Preparation of Example 16 N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide
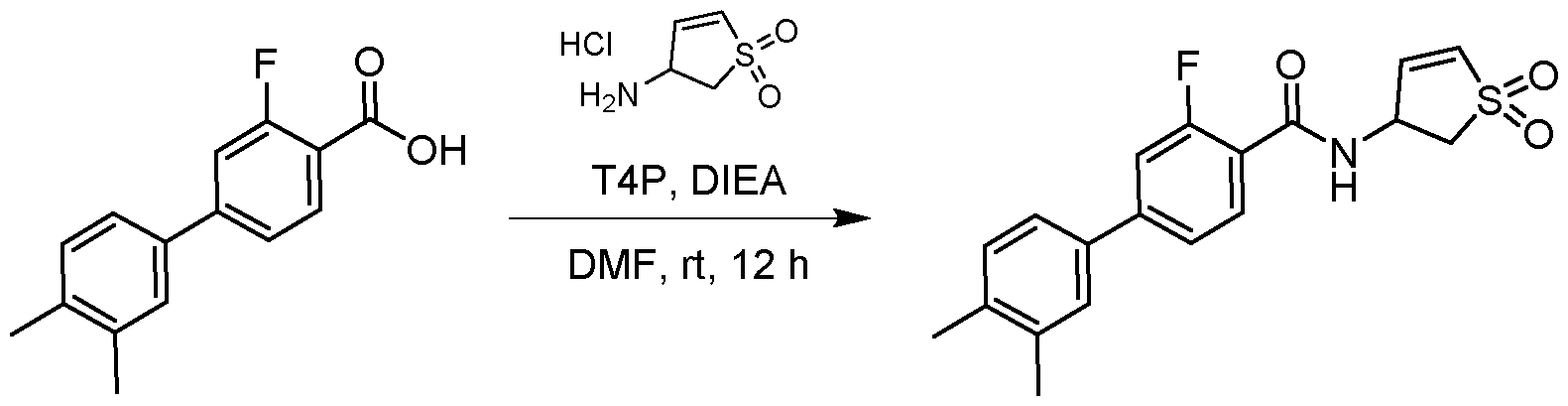 To a solution of 3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxylic acid (20 mg, 0.0819 mmol), DIEA (3.00 eq, 0.044 mL, 0.246 mmol), 3-amino-2,3-dihydrothiophene 1,1-dioxide hydrochloride (18 mg, 0.106 mmol) in DMF (1.5 mL) was added T4P in EtOAc (77 mg, 50% purity, 0.106 mmol) at 0 ºC. The mixture was stirred at 20 ºC for 2 h. The reaction mixture was purified by preparative HPLC (column: Phenomenex Synergi C18150*25 mm*10 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 45%-75%, 15 min) and lyophilized to give the product N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoro-3',4'- dimethyl-[1,1'-biphenyl]-4-carboxamide (20 mg, 0.0543 mmol, 66.26 % yield) as a white solid. RT 360.1 min (method 1); m/z 360.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.98 (d, J = 7.6 Hz, 1H), 7.74-7.67 (m, 1H), 7.65-7.58 (m, 2H), 7.56 (s, 1H), 7.51-7.46 (m, 1H), 7.29-7.20 (m, 2H), 6.92 (dd, J = 2.5, 6.7 Hz, 1H), 5.38-5.34 (m, 1H), 3.82 (dd, J = 8.1, 13.7 Hz, 1H), 3.23 (dd, J = 5.4, 13.7 Hz, 1H), 2.30 (s, 3H), 2.26 (s, 3H). Preparation of Intermediate 17.1 Methyl 3-amino-5-(3,4-dimethylphenyl)picolinate
To a solution of methyl 3-amino-5-bromopicolinate (1 g, 4.33 mmol) in dioxane (10 mL) and water (2 mL) was added (3,4-dimethylphenyl)boronic acid (714 mg, 4.76 mmol) K2CO3 (1196 mg, 8.66 mmol) and Pd(dppf)Cl2 (158 mg, 0.216 mmol). The mixture was degassed with N2 three times and then, stirred at 80 °C for 2 h. The resulting reaction mixture was filtered and the filtrate was concentrated under vacuum. The residue was diluted with water (20 mL) and then, extracted with ethyl acetate (20 mL, 3x). The combined organic phase was washed with brine (20 mL, 2x) and dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent of 0~36% Ethyl acetate/Petroleum ether, gradient 60 mL/min) and concentrated under vacuum to give the product methyl 3-amino-5-(3,4- dimethylphenyl)picolinate (800 mg, 3.09 mmol, 71.40 % yield) as a red solid. RT 0.446 min (method 1); m/z 257.1 (M+H)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 8.14 (s, 1H), 7.45 (s, 1H), 7.41 (s, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.26 (d, J = 8.0 Hz, 1H), 6.72 (s, 2H), 3.82 (s, 3H), 2.29 (s, 3H), 2.26 (s, 3H) Preparation of Intermediate 17.2 Methyl 3-(bis-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)picolinate
To a solution of 3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxylic acid (20 mg, 0.0819 mmol), DIEA (3.00 eq, 0.044 mL, 0.246 mmol), 3-amino-2,3-dihydrothiophene 1,1-dioxide hydrochloride (18 mg, 0.106 mmol) in DMF (1.5 mL) was added T4P in EtOAc (77 mg, 50% purity, 0.106 mmol) at 0 ºC. The mixture was stirred at 20 ºC for 2 h. The reaction mixture was purified by preparative HPLC (column: Phenomenex Synergi C18150*25 mm*10 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 45%-75%, 15 min) and lyophilized to give the product N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoro-3',4'- dimethyl-[1,1'-biphenyl]-4-carboxamide (20 mg, 0.0543 mmol, 66.26 % yield) as a white solid. RT 360.1 min (method 1); m/z 360.1 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.98 (d, J = 7.6 Hz, 1H), 7.74-7.67 (m, 1H), 7.65-7.58 (m, 2H), 7.56 (s, 1H), 7.51-7.46 (m, 1H), 7.29-7.20 (m, 2H), 6.92 (dd, J = 2.5, 6.7 Hz, 1H), 5.38-5.34 (m, 1H), 3.82 (dd, J = 8.1, 13.7 Hz, 1H), 3.23 (dd, J = 5.4, 13.7 Hz, 1H), 2.30 (s, 3H), 2.26 (s, 3H). Preparation of Intermediate 17.1 Methyl 3-amino-5-(3,4-dimethylphenyl)picolinate
To a solution of methyl 3-amino-5-bromopicolinate (1 g, 4.33 mmol) in dioxane (10 mL) and water (2 mL) was added (3,4-dimethylphenyl)boronic acid (714 mg, 4.76 mmol) K2CO3 (1196 mg, 8.66 mmol) and Pd(dppf)Cl2 (158 mg, 0.216 mmol). The mixture was degassed with N2 three times and then, stirred at 80 °C for 2 h. The resulting reaction mixture was filtered and the filtrate was concentrated under vacuum. The residue was diluted with water (20 mL) and then, extracted with ethyl acetate (20 mL, 3x). The combined organic phase was washed with brine (20 mL, 2x) and dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent of 0~36% Ethyl acetate/Petroleum ether, gradient 60 mL/min) and concentrated under vacuum to give the product methyl 3-amino-5-(3,4- dimethylphenyl)picolinate (800 mg, 3.09 mmol, 71.40 % yield) as a red solid. RT 0.446 min (method 1); m/z 257.1 (M+H)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 8.14 (s, 1H), 7.45 (s, 1H), 7.41 (s, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.26 (d, J = 8.0 Hz, 1H), 6.72 (s, 2H), 3.82 (s, 3H), 2.29 (s, 3H), 2.26 (s, 3H) Preparation of Intermediate 17.2 Methyl 3-(bis-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)picolinate
 To a solution of methyl 3-amino-5-(3,4-dimethylphenyl)picolinate (400 mg, 1.56 mmol) and (Boc)2O (1362 mg, 6.24 mmol) in DCM (4 mL) was added DIEA (0.83 mL, 4.68 mmol) and DMAP (19 mg, 0.156 mmol) at 0 °C. The mixture was stirred at °C for 12 h and then, concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent of 0~16% Ethyl acetate/Petroleum ether, gradient 60 mL/min) and concentrated under vacuum to give the product methyl 3-(bis-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)picolinate (400 mg, 0.850 mmol, 54.46 % yield) as a yellow solid.
RT 0.626 min (method 1); m/z 457.2 (M+H)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 8.98 (s, 1H), 8.24 (s, 1H), 7.68 (s, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 3.84 (s, 3H), 2.31 (s, 3H), 2.28 (s, 3H), 1.34 (s, 18H) Preparation of Intermediate 17.3 3-(bis-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-(methoxycarbonyl)pyridine 1-oxide
To a solution of methyl 3-amino-5-(3,4-dimethylphenyl)picolinate (400 mg, 1.56 mmol) and (Boc)2O (1362 mg, 6.24 mmol) in DCM (4 mL) was added DIEA (0.83 mL, 4.68 mmol) and DMAP (19 mg, 0.156 mmol) at 0 °C. The mixture was stirred at °C for 12 h and then, concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent of 0~16% Ethyl acetate/Petroleum ether, gradient 60 mL/min) and concentrated under vacuum to give the product methyl 3-(bis-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)picolinate (400 mg, 0.850 mmol, 54.46 % yield) as a yellow solid.
RT 0.626 min (method 1); m/z 457.2 (M+H)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 8.98 (s, 1H), 8.24 (s, 1H), 7.68 (s, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 3.84 (s, 3H), 2.31 (s, 3H), 2.28 (s, 3H), 1.34 (s, 18H) Preparation of Intermediate 17.3 3-(bis-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-(methoxycarbonyl)pyridine 1-oxide
 To a solution of methyl 3-(di-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)picolinate (400 mg, 0.876 mmol) in DCM (5 mL) was added m-CPBA (454 mg, 2.24 mmol, 85% purity) at 0°C. The reaction mixture was stirred at 25 °C for 12 h, then quenched by addition of 80 mL of saturated Na2SO3 aqueous solution and extracted with EtOAc (60 mL; 3x). The combined organic layer was washed with brine (60 mL; 3x), separated, dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent of 0~36% Ethyl acetate/Petroleum ether, gradient 60 mL/min) and concentrated under vacuum to give the product 3-(bis-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-(methoxycarbonyl)pyridine 1-oxide (220 mg, 0.466 mmol, 53.14 % yield) as colorless oil. RT 0.581 min (method 1); m/z 473.2 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.78 (s, 1H), 8.02 (s, 1H), 7.65 (s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 8.0 Hz, 1H), 3.85 (s, 3H), 2.29 (s, 3H), 2.27 (s, 3H), 1.37 (s, 18H) Preparation of Intermediate 17.4 3-((tert-butoxycarbonyl)amino)-2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide
To a solution of methyl 3-(di-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)picolinate (400 mg, 0.876 mmol) in DCM (5 mL) was added m-CPBA (454 mg, 2.24 mmol, 85% purity) at 0°C. The reaction mixture was stirred at 25 °C for 12 h, then quenched by addition of 80 mL of saturated Na2SO3 aqueous solution and extracted with EtOAc (60 mL; 3x). The combined organic layer was washed with brine (60 mL; 3x), separated, dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent of 0~36% Ethyl acetate/Petroleum ether, gradient 60 mL/min) and concentrated under vacuum to give the product 3-(bis-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-(methoxycarbonyl)pyridine 1-oxide (220 mg, 0.466 mmol, 53.14 % yield) as colorless oil. RT 0.581 min (method 1); m/z 473.2 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.78 (s, 1H), 8.02 (s, 1H), 7.65 (s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 8.0 Hz, 1H), 3.85 (s, 3H), 2.29 (s, 3H), 2.27 (s, 3H), 1.37 (s, 18H) Preparation of Intermediate 17.4 3-((tert-butoxycarbonyl)amino)-2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide
 To a solution of 3-(bis-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2- (methoxycarbonyl)pyridine 1-oxide (220 mg, 0.466 mmol) in THF (1 mL), water (1 mL) and methanol (1 mL) was added LiOH·H2O (98 mg, 2.33 mmol) at 25 °C. The mixture was stirred at 25 °C for 12 h and then, concentrated under reduced pressure. The residue was diluted with water (2 mL). Then, the pH was adjusted to pH 3~4 by adding HCl (aq., 1N) . The resulting suspension was filtered and the filter cake was washed with MeCN (3 mL). The solid was collected and dried under reduced pressure to give the
product 3-((tert-butoxycarbonyl)amino)-2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide (160 mg, 0.420 mmol, 90.14 % yield) as a white solid. RT 0.634 min (method 1); m/z 359.1 (M +H)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 11.46 (br s, 1H), 8.95 (s, 1H), 8.84 (s, 1H), 7.59 (s, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 7.6 Hz, 1H), 2.31 (s, 3H), 2.29 (s, 3H), 1.52 (s, 9H) Preparation of Intermediate 17.5 3-((tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide
To a solution of 3-(bis-(tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2- (methoxycarbonyl)pyridine 1-oxide (220 mg, 0.466 mmol) in THF (1 mL), water (1 mL) and methanol (1 mL) was added LiOH·H2O (98 mg, 2.33 mmol) at 25 °C. The mixture was stirred at 25 °C for 12 h and then, concentrated under reduced pressure. The residue was diluted with water (2 mL). Then, the pH was adjusted to pH 3~4 by adding HCl (aq., 1N) . The resulting suspension was filtered and the filter cake was washed with MeCN (3 mL). The solid was collected and dried under reduced pressure to give the
product 3-((tert-butoxycarbonyl)amino)-2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide (160 mg, 0.420 mmol, 90.14 % yield) as a white solid. RT 0.634 min (method 1); m/z 359.1 (M +H)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 11.46 (br s, 1H), 8.95 (s, 1H), 8.84 (s, 1H), 7.59 (s, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 7.6 Hz, 1H), 2.31 (s, 3H), 2.29 (s, 3H), 1.52 (s, 9H) Preparation of Intermediate 17.5 3-((tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide
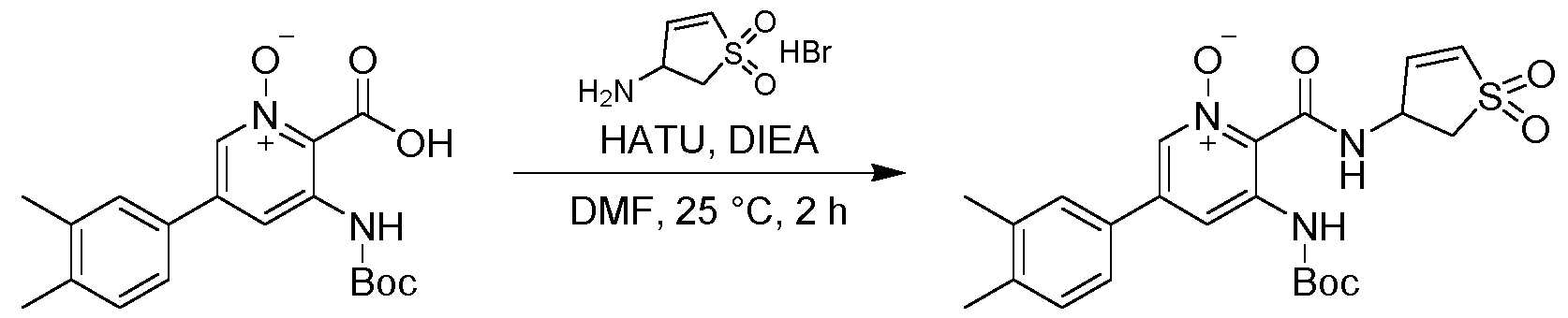 To a solution of 3-((tert-butoxycarbonyl)amino)-2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide (40 mg, 0.112 mmol) in DMF (0.5 mL) was added 3-amino-2,3-dihydrothiophene 1,1-dioxide hydrogen bromide (31 mg, 0.145 mmol) DIEA (0.060 mL, 0.335 mmol) and HATU (55 mg, 0.145 mmol) at 25 °C. The mixture was stirred at 25 °C for 2 h, then diluted with water (5 mL) and extracted with ethyl acetate (5 mL, 3x). The combined organic layer was washed with brine (5 mL, 2x), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by reversed-phase flash (ISCO; 20 g Flash Column Welch Ultimate XB_C1820-40 m; 120 A, mobile phase: A: 0.1% formic acid in water, B: MeCN; B%: 0%-60%, 40mL/min) and lyophilized directly to give the product 3-((tert- butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (40 mg, 0.0667 mmol, 59.79 % yield) as a yellow solid. RT 0.607 min (method 1); m/z 474.1 (M+H)+ (ESI+). Preparation of Example 17 3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl) pyridine 1- oxide
To a solution of 3-((tert-butoxycarbonyl)amino)-2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide (40 mg, 0.112 mmol) in DMF (0.5 mL) was added 3-amino-2,3-dihydrothiophene 1,1-dioxide hydrogen bromide (31 mg, 0.145 mmol) DIEA (0.060 mL, 0.335 mmol) and HATU (55 mg, 0.145 mmol) at 25 °C. The mixture was stirred at 25 °C for 2 h, then diluted with water (5 mL) and extracted with ethyl acetate (5 mL, 3x). The combined organic layer was washed with brine (5 mL, 2x), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by reversed-phase flash (ISCO; 20 g Flash Column Welch Ultimate XB_C1820-40 m; 120 A, mobile phase: A: 0.1% formic acid in water, B: MeCN; B%: 0%-60%, 40mL/min) and lyophilized directly to give the product 3-((tert- butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (40 mg, 0.0667 mmol, 59.79 % yield) as a yellow solid. RT 0.607 min (method 1); m/z 474.1 (M+H)+ (ESI+). Preparation of Example 17 3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl) pyridine 1- oxide
 A solution of 3-((tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (20 mg, 0.0422 mmol) in TFA (0.1 mL) and DCM (0.5 mL) was stirred at 25 °C for 2 h. The resulting mixture was concentrated under reduced pressure. The
resulting residue was purified by preparative HPLC (column: Phenomenex Synergi C18150*25 mm*10 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 32%-62%, 9 min) and lyophilized directly to give the product 3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (6.5 mg, 0.0169 mmol, 46.03 % yield) as an off-white solid. RT 0.475 min (LCMS: method 1); m/z 374.3 (M+H)+ (ESI+); RT 2.149, 2.621 min (SFC: Method 4); 1H NMR (DMSO-d6, 400 MHz):12.13 (d, J = 7.2 Hz, 1H), 8.03 (d, J = 1.6 Hz, 1H), 7.51-7.48 (m, 3H), 7.40 (dd, J = 1.6, 8.0 Hz, 1H), 7.29-7.24 (m, 2H), 7.20 (d, J = 1.6 Hz, 1H), 7.00 (dd, J = 1.6, 8.0 Hz, 1H), 5.38- 5.30 (m, 1H), 3.74 (dd, J = 7.2, 13.6 Hz, 1H), 3.29 (J = 4.8, 13.6 Hz, 1H), 2.29 (s, 3H), 2.27 (s, 3H) Preparation of Intermediate 18.1 (R)-3-((tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide
A solution of 3-((tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (20 mg, 0.0422 mmol) in TFA (0.1 mL) and DCM (0.5 mL) was stirred at 25 °C for 2 h. The resulting mixture was concentrated under reduced pressure. The
resulting residue was purified by preparative HPLC (column: Phenomenex Synergi C18150*25 mm*10 µm; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 32%-62%, 9 min) and lyophilized directly to give the product 3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (6.5 mg, 0.0169 mmol, 46.03 % yield) as an off-white solid. RT 0.475 min (LCMS: method 1); m/z 374.3 (M+H)+ (ESI+); RT 2.149, 2.621 min (SFC: Method 4); 1H NMR (DMSO-d6, 400 MHz):12.13 (d, J = 7.2 Hz, 1H), 8.03 (d, J = 1.6 Hz, 1H), 7.51-7.48 (m, 3H), 7.40 (dd, J = 1.6, 8.0 Hz, 1H), 7.29-7.24 (m, 2H), 7.20 (d, J = 1.6 Hz, 1H), 7.00 (dd, J = 1.6, 8.0 Hz, 1H), 5.38- 5.30 (m, 1H), 3.74 (dd, J = 7.2, 13.6 Hz, 1H), 3.29 (J = 4.8, 13.6 Hz, 1H), 2.29 (s, 3H), 2.27 (s, 3H) Preparation of Intermediate 18.1 (R)-3-((tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide
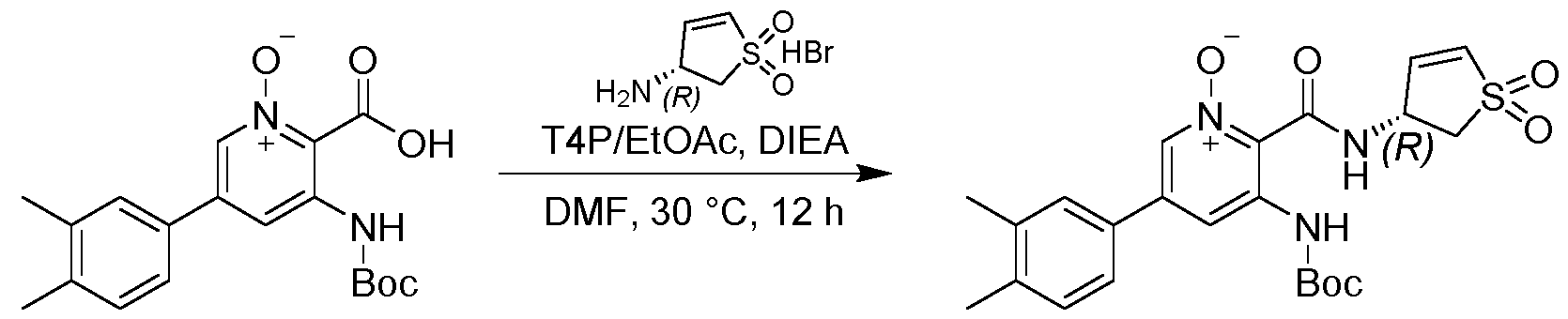 To a solution of 3-((tert-butoxycarbonyl)amino)-2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide (260 mg, 0.725 mmol) DIEA (0.65 mL, 3.63 mmol) and (R)-3-amino-2,3-dihydrothiophene 1,1-dioxide hydrogen bromide (202 mg, 0.943 mmol, 91.5% ee) in DMF (6 mL) was added T4P in EtOAc (1567 mg, 2.18 mmol, wt%: 50%).The mixture was stirred at 30 °C for 12 h, then diluted with water (20 mL) and extracted with ethyl acetate (20 mL, 3x). The combined organic layer was washed with brine (20 mL, 2x), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent of 0~20% Ethyl acetate/Petroleum ether; gradient, 60 mL/min) and concentrated under vacuum to give the product (R)-3- ((tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (130 mg, 0.187 mmol, 25.73% yield) as a yellow solid. RT 0.604 min (method 1); m/z 474.1 (M+H)+ (ESI+). Preparation of Example 18 (R)-3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide
To a solution of 3-((tert-butoxycarbonyl)amino)-2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide (260 mg, 0.725 mmol) DIEA (0.65 mL, 3.63 mmol) and (R)-3-amino-2,3-dihydrothiophene 1,1-dioxide hydrogen bromide (202 mg, 0.943 mmol, 91.5% ee) in DMF (6 mL) was added T4P in EtOAc (1567 mg, 2.18 mmol, wt%: 50%).The mixture was stirred at 30 °C for 12 h, then diluted with water (20 mL) and extracted with ethyl acetate (20 mL, 3x). The combined organic layer was washed with brine (20 mL, 2x), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent of 0~20% Ethyl acetate/Petroleum ether; gradient, 60 mL/min) and concentrated under vacuum to give the product (R)-3- ((tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (130 mg, 0.187 mmol, 25.73% yield) as a yellow solid. RT 0.604 min (method 1); m/z 474.1 (M+H)+ (ESI+). Preparation of Example 18 (R)-3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide
 A solution of (R)-3-((tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (107 mg, 0.227 mmol) in TFA (0.3 mL) and DCM (0.9 mL) was stirred at 25 °C for 1 h. The resulting mixture was concentrated under reduced pressure. The residue was purified by reversed-phase flash (ISCO®; 40 g Flash Column Welch Ultimate XB_C1820-40 m; 120 A, mobile phase: A: 0.1% TFA in water, B: MeCN; B%: 0%-40%, 40mL/min) and lyophilized directly to give the product (R)-3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (47 mg, 0.125 mmol, 55.14 % yield, ee%: 90.78% based on SFC) as a yellow solid. RT 0.469 min (LCMS: method 1); m/z 374.3 (M+H)+ (ESI+); RT 2.588 min, ee%: 90.78% (SFC: Method 4); 1H NMR (DMSO-d6, 400 MHz): 12.12 (d, J = 7.6 Hz, 1H), 8.03 (d, J = 1.6 Hz, 1H), 7.60-7.50 (m, 3H), 7.40 (dd, J = 1.2, 7.2 Hz, 1H), 7.30-7.24 (m, 2H), 7.20 (d, J = 1.6 Hz, 1H), 7.00 (dd, J = 1.2, 7.2 Hz, 1H), 5.38-5.30 (m, 1H), 3.74 (dd, J = 7.6, 13.6 Hz, 1H), 3.30 (dd, J = 4.4, 13.6 Hz, 1H), 2.29 (s, 3H), 2.27 (s, 3H) Examples listed in the table below were prepared according to the corresponding general procedures and starting from the corresponding intermediates or examples. Cpd Structure Yield Procedures LC/MS and SFC 1H NMR number (%) information 1H NMR (DMSO-d6, 400 MHz):11.55 (d, J = 8.0 Hz, Procedure1, LCMS: RT 0.484 min 1H), 8.95 (s, Procedure 3, ((Method 1), m/z 399.0 + + 1H), 8.30 (d, 19 50.43 Procedure 2, (M+H) (ESI); J = 8.4 Hz, then SFC: RT 1.549 min, 1H), 8.07 (d, Procedure 4 2.068 min (Method 5) J = 8.0 Hz, 2H), 8.02 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 8.0 Hz,
2H), 7.31 (dd, J = 1.6, 6.8Hz, 1H), 7.02 (dd, J = 1.6, 6.8 Hz, 1H), 5.41- 5.40 (m, 1H), 3.77 (dd, J = 8.0, 13.6 Hz, 1H), 3.39 (dd, J = 4.0, 13.6 Hz, 1H) 1H NMR (CDCl3 , 400 MHz): 11.36 (s, 1H), 8.04 (br s, 1H), 7.70 (br s, 1H), 7.33 Procedure 1, LCMS: RT 0.427 min (d, J = 7.2 Hz, Procedure 2, (Method 1), m/z 374.1 1H), 7.20(s, Procedure 3, (M+H)+ (ESI+); 45.35 1H), 7.15 (d, Procedure 4, SFC: Two peaks, RT J = 7.2 Hz, then 1.764 min, 2.076 min 1H), 6.84 (br Procedure 5 (Method 5) s, 2H), 5.46 (s, 1H) , 3.82- 3.77 (m, 1H), 3.35-3.32 (m, 1H), 2.36 (s, 3H), 2.35 (s, 3H)
1H NMR (CDCl3, 400 MHz): 12.00 (d, J = 6.4 Hz, 1H), 8.53 (d, J = 1.6 Hz, 1H), 8.35 (d, J = 8.4 Hz, 1H), 7.73 (dd, LCMS: RT 0.507 min J = 1.6, 8.4 Procedure 1, (Method 1), m/z 437.0 Hz, 1H), Procedure 2, (M+H)+ (ESI+); 7.47-7.42 (m, 42.35 Procedure 3 SFC: RT 2.462 min, 1H), 7.39- then ee%: 90.75% (Method 7.31 (m, 6H), Procedure 4 6) 7.15-7.09 (m, 2H), 6.90- 6.84 (m, 2H), 5.58-5.53 (m, 1H), 5.16 (s, 2H), 3.81 (dd, J = 6.4, 13.6 Hz, 1H), 3.28 (dd, J = 4.8, 13.6 Hz, 1H). 1H NMR (DMSO-d6, LCMS:RT 0. 503 min 400 MHz): Procedure 1 (method 1), m/z 351.1 11.52 (d, J = then (M+H)+ (ESI+); 7.6 Hz, 1H), 5.87 Procedure 4 SFC: RT 1.950 min, 8.78 (s, 1H), ee%=87.32% (Method 8.21 (d, J = 5) 8.4 Hz, 1H), 8.10 (s, 1H), 7.90 (d, J =
8.4 Hz, 1H), 7.40 (s, 1H), 7.20 (dd, J = 2.8, 6.8 Hz, 1H), 6.98 (dd, J = 2.8, 6.8 Hz, 1H), 5.47-5.37 (m, 1H), 3.79- 3.73 (dd, J = 7.6, 13.6 Hz, 1H), 3.28 (dd, J = 4.4, 13.6 Hz, 1H), 2.50 (s, 3H) 1H NMR (DMSO-d6, 400 MHz):11.55 (d, J = 8.0 Hz, 1H), 8.46 (s, 1H), 8.17 (d, LCMS: RT 0.499 min J = 8.4 Hz, (Method 1), m/z 347.2 Procedure 1 1H), 7.58 (d, (M+H)+ (ESI+); 56.13 then 8.4 Hz, 1H), SFC: Two peaks, RT Procedure 4 7.28 (dd, J = 0.970 min, 2.048 min 3.2, 6.8 Hz, (Method 5) 1H), 7.00 (dd, J = 3.2, 6.8 Hz, 1H), 6– 96 (s, 1H), 5.39 - 5.33 (m, 1H), 3.74 (dd, J = 8.0,
13.6 Hz, 1H), 3.36 (dd, J = 4.4, 13.6 Hz, 1H), 2–78 (s, 2H), 2.21 - 2– 12 (m, 4H), 1.96 - 1.80 (m, 2H) 1H NMR (DMSO-d6, 400 MHz): 11.49 (d, J = 8.0 Hz, 1H), 8.72 (d, J = 1.2 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 7.89 (dd, J = 1.2, 8.4 Hz, Procedure 1, LCMS: RT 0.442 min ocedure 2 (Meth 1H), 7.68 (d, Pr od 1), m/z 370.9 (M+H)+ (ESI+) J = 5.6 Hz, 29.74 Procedure 3 SFC: Two peaks, RT 1H), 7.42 (d, and then 2.056 min 2.257 min J = 5.6 Hz, Procedure 4 (Method 7) 1H), 7.30 (dd, J = 2.8, 6.8 Hz, 1H), 7.01 (dd, J = 2.8, 6.8 Hz, 1H), 5.46-5.35 (m, 1H), 3.76 (dd, J = 8.0, 13.6 Hz, 1H), 3.38 (dd, J = 4.0, 13.6 Hz, 1H)
Preparation of Intermediate 25.1 3-chloro-6-(3,4-dimethylphenyl)-2-methoxypyridine
A solution of (R)-3-((tert-butoxycarbonyl)amino)-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (107 mg, 0.227 mmol) in TFA (0.3 mL) and DCM (0.9 mL) was stirred at 25 °C for 1 h. The resulting mixture was concentrated under reduced pressure. The residue was purified by reversed-phase flash (ISCO®; 40 g Flash Column Welch Ultimate XB_C1820-40 m; 120 A, mobile phase: A: 0.1% TFA in water, B: MeCN; B%: 0%-40%, 40mL/min) and lyophilized directly to give the product (R)-3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide (47 mg, 0.125 mmol, 55.14 % yield, ee%: 90.78% based on SFC) as a yellow solid. RT 0.469 min (LCMS: method 1); m/z 374.3 (M+H)+ (ESI+); RT 2.588 min, ee%: 90.78% (SFC: Method 4); 1H NMR (DMSO-d6, 400 MHz): 12.12 (d, J = 7.6 Hz, 1H), 8.03 (d, J = 1.6 Hz, 1H), 7.60-7.50 (m, 3H), 7.40 (dd, J = 1.2, 7.2 Hz, 1H), 7.30-7.24 (m, 2H), 7.20 (d, J = 1.6 Hz, 1H), 7.00 (dd, J = 1.2, 7.2 Hz, 1H), 5.38-5.30 (m, 1H), 3.74 (dd, J = 7.6, 13.6 Hz, 1H), 3.30 (dd, J = 4.4, 13.6 Hz, 1H), 2.29 (s, 3H), 2.27 (s, 3H) Examples listed in the table below were prepared according to the corresponding general procedures and starting from the corresponding intermediates or examples. Cpd Structure Yield Procedures LC/MS and SFC 1H NMR number (%) information 1H NMR (DMSO-d6, 400 MHz):11.55 (d, J = 8.0 Hz, Procedure1, LCMS: RT 0.484 min 1H), 8.95 (s, Procedure 3, ((Method 1), m/z 399.0 + + 1H), 8.30 (d, 19 50.43 Procedure 2, (M+H) (ESI); J = 8.4 Hz, then SFC: RT 1.549 min, 1H), 8.07 (d, Procedure 4 2.068 min (Method 5) J = 8.0 Hz, 2H), 8.02 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 8.0 Hz,
2H), 7.31 (dd, J = 1.6, 6.8Hz, 1H), 7.02 (dd, J = 1.6, 6.8 Hz, 1H), 5.41- 5.40 (m, 1H), 3.77 (dd, J = 8.0, 13.6 Hz, 1H), 3.39 (dd, J = 4.0, 13.6 Hz, 1H) 1H NMR (CDCl3 , 400 MHz): 11.36 (s, 1H), 8.04 (br s, 1H), 7.70 (br s, 1H), 7.33 Procedure 1, LCMS: RT 0.427 min (d, J = 7.2 Hz, Procedure 2, (Method 1), m/z 374.1 1H), 7.20(s, Procedure 3, (M+H)+ (ESI+); 45.35 1H), 7.15 (d, Procedure 4, SFC: Two peaks, RT J = 7.2 Hz, then 1.764 min, 2.076 min 1H), 6.84 (br Procedure 5 (Method 5) s, 2H), 5.46 (s, 1H) , 3.82- 3.77 (m, 1H), 3.35-3.32 (m, 1H), 2.36 (s, 3H), 2.35 (s, 3H)
1H NMR (CDCl3, 400 MHz): 12.00 (d, J = 6.4 Hz, 1H), 8.53 (d, J = 1.6 Hz, 1H), 8.35 (d, J = 8.4 Hz, 1H), 7.73 (dd, LCMS: RT 0.507 min J = 1.6, 8.4 Procedure 1, (Method 1), m/z 437.0 Hz, 1H), Procedure 2, (M+H)+ (ESI+); 7.47-7.42 (m, 42.35 Procedure 3 SFC: RT 2.462 min, 1H), 7.39- then ee%: 90.75% (Method 7.31 (m, 6H), Procedure 4 6) 7.15-7.09 (m, 2H), 6.90- 6.84 (m, 2H), 5.58-5.53 (m, 1H), 5.16 (s, 2H), 3.81 (dd, J = 6.4, 13.6 Hz, 1H), 3.28 (dd, J = 4.8, 13.6 Hz, 1H). 1H NMR (DMSO-d6, LCMS:RT 0. 503 min 400 MHz): Procedure 1 (method 1), m/z 351.1 11.52 (d, J = then (M+H)+ (ESI+); 7.6 Hz, 1H), 5.87 Procedure 4 SFC: RT 1.950 min, 8.78 (s, 1H), ee%=87.32% (Method 8.21 (d, J = 5) 8.4 Hz, 1H), 8.10 (s, 1H), 7.90 (d, J =
8.4 Hz, 1H), 7.40 (s, 1H), 7.20 (dd, J = 2.8, 6.8 Hz, 1H), 6.98 (dd, J = 2.8, 6.8 Hz, 1H), 5.47-5.37 (m, 1H), 3.79- 3.73 (dd, J = 7.6, 13.6 Hz, 1H), 3.28 (dd, J = 4.4, 13.6 Hz, 1H), 2.50 (s, 3H) 1H NMR (DMSO-d6, 400 MHz):11.55 (d, J = 8.0 Hz, 1H), 8.46 (s, 1H), 8.17 (d, LCMS: RT 0.499 min J = 8.4 Hz, (Method 1), m/z 347.2 Procedure 1 1H), 7.58 (d, (M+H)+ (ESI+); 56.13 then 8.4 Hz, 1H), SFC: Two peaks, RT Procedure 4 7.28 (dd, J = 0.970 min, 2.048 min 3.2, 6.8 Hz, (Method 5) 1H), 7.00 (dd, J = 3.2, 6.8 Hz, 1H), 6– 96 (s, 1H), 5.39 - 5.33 (m, 1H), 3.74 (dd, J = 8.0,
13.6 Hz, 1H), 3.36 (dd, J = 4.4, 13.6 Hz, 1H), 2–78 (s, 2H), 2.21 - 2– 12 (m, 4H), 1.96 - 1.80 (m, 2H) 1H NMR (DMSO-d6, 400 MHz): 11.49 (d, J = 8.0 Hz, 1H), 8.72 (d, J = 1.2 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 7.89 (dd, J = 1.2, 8.4 Hz, Procedure 1, LCMS: RT 0.442 min ocedure 2 (Meth 1H), 7.68 (d, Pr od 1), m/z 370.9 (M+H)+ (ESI+) J = 5.6 Hz, 29.74 Procedure 3 SFC: Two peaks, RT 1H), 7.42 (d, and then 2.056 min 2.257 min J = 5.6 Hz, Procedure 4 (Method 7) 1H), 7.30 (dd, J = 2.8, 6.8 Hz, 1H), 7.01 (dd, J = 2.8, 6.8 Hz, 1H), 5.46-5.35 (m, 1H), 3.76 (dd, J = 8.0, 13.6 Hz, 1H), 3.38 (dd, J = 4.0, 13.6 Hz, 1H)
Preparation of Intermediate 25.1 3-chloro-6-(3,4-dimethylphenyl)-2-methoxypyridine
 To a solution of 6-bromo-3-chloro-2-methoxypyridine (1.5 g, 6.74 mmol) in 1,4-Dioxane (15 mL) and water (3 mL) were added (3,4-dimethylphenyl)boronic acid (1213 mg, 8.09 mmol), K2CO3 (1864 mg, 13.5 mmol) and Pd(dppf)Cl2·DCM (275 mg, 0.337 mmol). The mixture was degassed with N2 (3x) and, then stirred at 80 °C for 12 h. The resulting reaction mixture was filtered and the cake was washed with EtOAc (50 mL). The combined filtrate was concentrated to give a residue which was purified by flash silica gel chromatography (ISCO®; 20 g SepaFlash® Silica Flash Column, Eluent of 0~10% Ethyl acetate/Petroleum ether gradient, 20 mL/min) to give the product 3-chloro-6-(3,4-dimethylphenyl)-2- methoxy-pyridine (1.80 g, 5.96 mmol, 88.42 % yield) as a yellow solid. RT 0.645 min (method 1); m/z 247.9 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.77 (s, 1H), 7.76 - 7.72 (, J = 8.0 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.24 - 7.18 (J = 8.0 Hz, 1H), 4.13 (s, 3H), 2.35 (s, 3H), 2.31 (s, 3H) Preparation of Intermediate 25.2 6-(3,4-dimethylphenyl)-2-methoxy-3-((triisopropylsilyl)ethynyl)pyridine
To a solution of 6-bromo-3-chloro-2-methoxypyridine (1.5 g, 6.74 mmol) in 1,4-Dioxane (15 mL) and water (3 mL) were added (3,4-dimethylphenyl)boronic acid (1213 mg, 8.09 mmol), K2CO3 (1864 mg, 13.5 mmol) and Pd(dppf)Cl2·DCM (275 mg, 0.337 mmol). The mixture was degassed with N2 (3x) and, then stirred at 80 °C for 12 h. The resulting reaction mixture was filtered and the cake was washed with EtOAc (50 mL). The combined filtrate was concentrated to give a residue which was purified by flash silica gel chromatography (ISCO®; 20 g SepaFlash® Silica Flash Column, Eluent of 0~10% Ethyl acetate/Petroleum ether gradient, 20 mL/min) to give the product 3-chloro-6-(3,4-dimethylphenyl)-2- methoxy-pyridine (1.80 g, 5.96 mmol, 88.42 % yield) as a yellow solid. RT 0.645 min (method 1); m/z 247.9 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.77 (s, 1H), 7.76 - 7.72 (, J = 8.0 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.24 - 7.18 (J = 8.0 Hz, 1H), 4.13 (s, 3H), 2.35 (s, 3H), 2.31 (s, 3H) Preparation of Intermediate 25.2 6-(3,4-dimethylphenyl)-2-methoxy-3-((triisopropylsilyl)ethynyl)pyridine
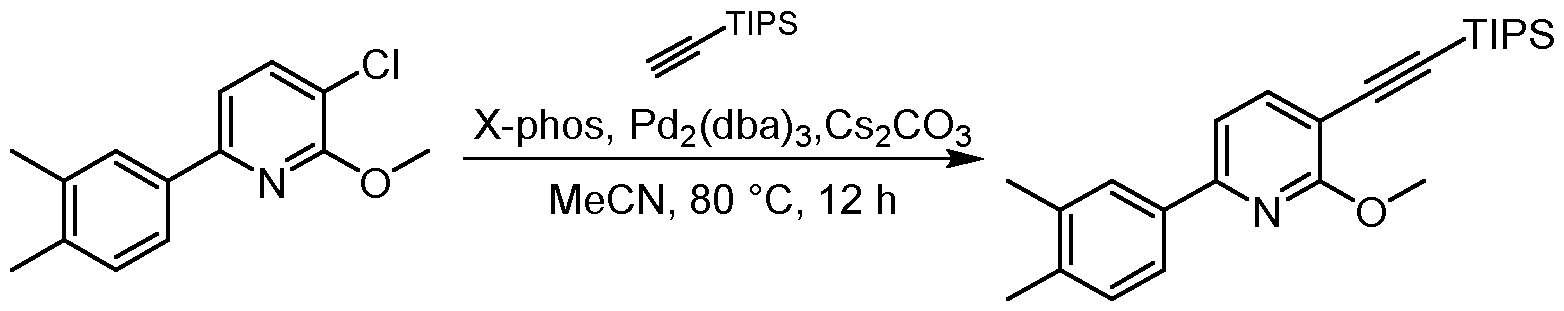 To a mixture of 3-chloro-6-(3,4-dimethylphenyl)-2-methoxy-pyridine (300 mg, 1.21 mmol), ethynyl(triisopropyl)silane (663 mg, 3.63 mmol), Cs2CO3 (1184 mg, 3.63 mmol), X-phos (173 mg, 0.363 mmol) in MeCN (7.5 mL) was added Pd2(dba)3 (111 mg, 0.121 mmol). The reaction mixture was degassed with N2 (3X), then stirred at 80 °C for 12 h, poured into water (50 mL) and extracted with EtOAc (50 mL, 3x). The combined organic layer was washed with brine (20 mL), dried over anhydrous Na2SO4, filtered concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO®; 20 g SepaFlash® Silica Flash Column, Eluent of 0~10% Ethyl acetate/Petroleum ether gradient, 20 mL/min)
and concentrated under vacuum to give the product 6-(3, 4-dimethylphenyl)-2-methoxy-3- ((triisopropylsilyl) ethynyl)pyridine (340 mg, 0.864 mmol, 71.32 % yield) as a yellow solid. RT 0.793 min (method 6); m/z 394.4 (M +H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.81 (s, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 8.0 Hz, 1H), 4.08 (s, 3H), 2.34 (s, 3H), 2.31 (s, 3H), 1.21-1.26 (m, 3H), 1.15 (s, 18H). Preparation of Intermediate 25.3 6-(3,4-dimethylphenyl)-3-ethynyl-2-methoxypyridine
To a mixture of 3-chloro-6-(3,4-dimethylphenyl)-2-methoxy-pyridine (300 mg, 1.21 mmol), ethynyl(triisopropyl)silane (663 mg, 3.63 mmol), Cs2CO3 (1184 mg, 3.63 mmol), X-phos (173 mg, 0.363 mmol) in MeCN (7.5 mL) was added Pd2(dba)3 (111 mg, 0.121 mmol). The reaction mixture was degassed with N2 (3X), then stirred at 80 °C for 12 h, poured into water (50 mL) and extracted with EtOAc (50 mL, 3x). The combined organic layer was washed with brine (20 mL), dried over anhydrous Na2SO4, filtered concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO®; 20 g SepaFlash® Silica Flash Column, Eluent of 0~10% Ethyl acetate/Petroleum ether gradient, 20 mL/min)
and concentrated under vacuum to give the product 6-(3, 4-dimethylphenyl)-2-methoxy-3- ((triisopropylsilyl) ethynyl)pyridine (340 mg, 0.864 mmol, 71.32 % yield) as a yellow solid. RT 0.793 min (method 6); m/z 394.4 (M +H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.81 (s, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 8.0 Hz, 1H), 4.08 (s, 3H), 2.34 (s, 3H), 2.31 (s, 3H), 1.21-1.26 (m, 3H), 1.15 (s, 18H). Preparation of Intermediate 25.3 6-(3,4-dimethylphenyl)-3-ethynyl-2-methoxypyridine
 To a solution of 6-(3,4-dimethylphenyl)-2-methoxy-3-((triisopropylsilyl)ethynyl)pyridine (340 mg, 0.864 mmol) in THF (3.5 mL) was added TBAF (271 mg, 1.04 mmol). The reaction mixture was stirred at 20°C for 4 h, then poured into water (50 mL) and extracted with EtOAc (20 mL; 3x). The combined organic layer was washed with brine (20 mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by preparative-TLC (UV, Rf=0.4, eluent: Ethyl acetate: Petroleum ether (1:5)) and concentrated under vacuum to give the product 6-(3,4-dimethylphenyl)-3-ethynyl-2-methoxy- pyridine (200 mg, 0.820 mmol, 94.94 % yield) as a yellow oil. RT 0.405 min (method 6); m/z 237.9 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.81 (s, 1H), 7.80 - 7.73 (m, 2H), 7.31 (d, J = 7.8 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 4.14 (s, 3H), 3.39 (s, 1H), 2.35 (s, 3H), 2.32 (s, 3H) Preparation of Example 25 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide
To a solution of 6-(3,4-dimethylphenyl)-2-methoxy-3-((triisopropylsilyl)ethynyl)pyridine (340 mg, 0.864 mmol) in THF (3.5 mL) was added TBAF (271 mg, 1.04 mmol). The reaction mixture was stirred at 20°C for 4 h, then poured into water (50 mL) and extracted with EtOAc (20 mL; 3x). The combined organic layer was washed with brine (20 mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by preparative-TLC (UV, Rf=0.4, eluent: Ethyl acetate: Petroleum ether (1:5)) and concentrated under vacuum to give the product 6-(3,4-dimethylphenyl)-3-ethynyl-2-methoxy- pyridine (200 mg, 0.820 mmol, 94.94 % yield) as a yellow oil. RT 0.405 min (method 6); m/z 237.9 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 7.81 (s, 1H), 7.80 - 7.73 (m, 2H), 7.31 (d, J = 7.8 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 4.14 (s, 3H), 3.39 (s, 1H), 2.35 (s, 3H), 2.32 (s, 3H) Preparation of Example 25 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide
 To solution of 3-azido-2,3-dihydrothiophene 1,1-dioxide (20 mg, 0.126 mmol), 6-(3,4- dimethylphenyl)-3-ethynyl-2-methoxypyridine (20 mg, 0.0843 mmol), CuSO4·5H2O (4.2 mg, 0.0169 mmol) in tert-butanol (0.5 mL) and water (0.5 mL) was added (+)-Sodium L-ascorbate (8.3 mg, 0.0421 mmol).
The reaction mixture was stirred at 70°C for 2 h, then filtered and the filtrate was concentrated. The crude product was purified by reversed-phase flash (ISCO®; 20 g Flash Column Welch Ultimate XB_C1820-40 m; 120 A, mobile phase: A: 0.1% formic acid in water, B: MeCN; B%: 40%-60%, 10 mL/min) and lyophilized directly to give the product 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3- triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide (0.93 mg, 0.00235 mmol, 1.86 % yield) as an off-white solid. LCMS: RT 0.553 min (method 1); m/z 397.0 (M+H)+ (ESI+); SFC: RT 1.866 min, 2.230 min (Method 8); 1H NMR (CDCl3, 400 MHz): 8.60 (d, J = 8.0 Hz, 1H), 8.19 (s, 1H), 7.86 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 6.7 Hz, 1H), 6.93 (d, J = 6.7 Hz, 1H), 6.26 - 6.17 (m, 1H), 4.19 (s, 3H), 3.99-3.93 (m, 1H), 3.57-3.52 (m, 1H), 2.36 (s, 3H), 2.33 (s, 3H). Preparation of Example 26 3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide
To solution of 3-azido-2,3-dihydrothiophene 1,1-dioxide (20 mg, 0.126 mmol), 6-(3,4- dimethylphenyl)-3-ethynyl-2-methoxypyridine (20 mg, 0.0843 mmol), CuSO4·5H2O (4.2 mg, 0.0169 mmol) in tert-butanol (0.5 mL) and water (0.5 mL) was added (+)-Sodium L-ascorbate (8.3 mg, 0.0421 mmol).
The reaction mixture was stirred at 70°C for 2 h, then filtered and the filtrate was concentrated. The crude product was purified by reversed-phase flash (ISCO®; 20 g Flash Column Welch Ultimate XB_C1820-40 m; 120 A, mobile phase: A: 0.1% formic acid in water, B: MeCN; B%: 40%-60%, 10 mL/min) and lyophilized directly to give the product 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3- triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide (0.93 mg, 0.00235 mmol, 1.86 % yield) as an off-white solid. LCMS: RT 0.553 min (method 1); m/z 397.0 (M+H)+ (ESI+); SFC: RT 1.866 min, 2.230 min (Method 8); 1H NMR (CDCl3, 400 MHz): 8.60 (d, J = 8.0 Hz, 1H), 8.19 (s, 1H), 7.86 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 6.7 Hz, 1H), 6.93 (d, J = 6.7 Hz, 1H), 6.26 - 6.17 (m, 1H), 4.19 (s, 3H), 3.99-3.93 (m, 1H), 3.57-3.52 (m, 1H), 2.36 (s, 3H), 2.33 (s, 3H). Preparation of Example 26 3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide
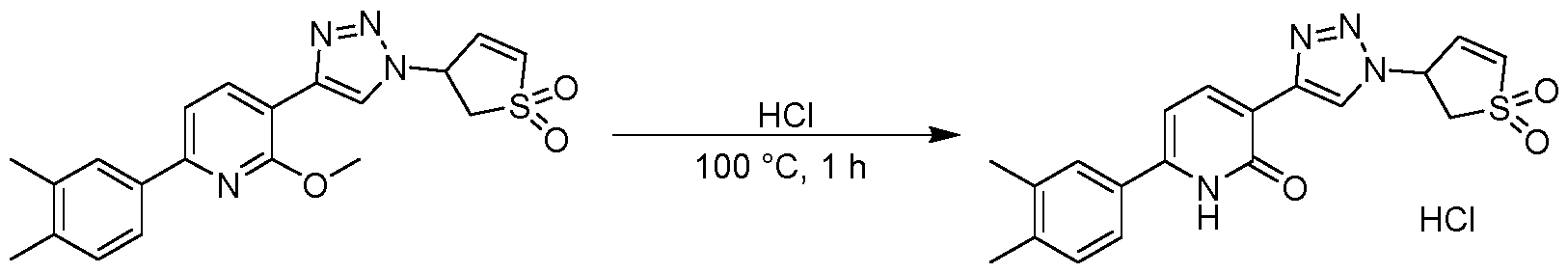 A solution of 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide (16 mg, 0.0404 mmol) in HCl (aq, 12 mol/L., 0.5 mL) was stirred at 100°C for 1 h. The resulting suspension was filtered,was washed with water (10 mL) and EtOH (10 mL), collected and dried under vacuum to give the product 3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3- triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide (4.6 mg, 0.0119 mmol, HCl salt, 29.54 % yield) as a yellow solid. LCMS: RT 0.692 min (method 4); m/z 383.0 (M+H)+ (ESI+); SFC: RT 1.247 min, 1.594 min (Method 8); 1H NMR (DMSO-d6, 400 MHz): 12.15 (br, 1H), 8.68 (s, 1H), 8.35 (d, J = 7.5 Hz, 1H), 7.61 (s, 1H), 7.60 - 7.50 (m, 2H), 7.33 - 7.19 (m, 2H), 6.75 (d, J = 7.5 Hz, 1H), 6.50 - 6.38 (m, 1H), 4.06 (dd, J = 8.6, 14.3 Hz, 1H), 3.63 (dd, J = 4.1, 14.3 Hz, 1H), 2.29 (s, 3H), 2.28 (s, 3H). Preparation of Example 25a&25b (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide and (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H- 1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide
A solution of 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide (16 mg, 0.0404 mmol) in HCl (aq, 12 mol/L., 0.5 mL) was stirred at 100°C for 1 h. The resulting suspension was filtered,was washed with water (10 mL) and EtOH (10 mL), collected and dried under vacuum to give the product 3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3- triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide (4.6 mg, 0.0119 mmol, HCl salt, 29.54 % yield) as a yellow solid. LCMS: RT 0.692 min (method 4); m/z 383.0 (M+H)+ (ESI+); SFC: RT 1.247 min, 1.594 min (Method 8); 1H NMR (DMSO-d6, 400 MHz): 12.15 (br, 1H), 8.68 (s, 1H), 8.35 (d, J = 7.5 Hz, 1H), 7.61 (s, 1H), 7.60 - 7.50 (m, 2H), 7.33 - 7.19 (m, 2H), 6.75 (d, J = 7.5 Hz, 1H), 6.50 - 6.38 (m, 1H), 4.06 (dd, J = 8.6, 14.3 Hz, 1H), 3.63 (dd, J = 4.1, 14.3 Hz, 1H), 2.29 (s, 3H), 2.28 (s, 3H). Preparation of Example 25a&25b (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide and (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H- 1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide
 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide (85 mg, 0.210 mmol) was separated by preparative SFC (column: Daicel Chiralpak IC 250x30mm I.D., 10 m particle size; Mobile Phase: A: Supercritical CO2, B: EtOH (NEU): 60% Phase B in Supercritical CO2, Gradient information:10 min, Flow Rate: 120 g/min) and lyophilized to give (R or S)- 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1- dioxide (30 mg, 0.0757 mmol, 36.0 % yield, ee%: 98.33% based on SFC method 8) as a light yellow solid and (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide (30 mg, 0.0757 mmol, 36.0 % yield, ee%: 100% based on SFC method 8) as a light yellow solid. Example 25a: LCMS: RT 0.562 min (method 1); m/z 397.1 (M+H)+ (ESI+); SFC: RT 1.853 min, ee%: 98.33% (Method 8); 1H NMR (CDCl3, 400 MHz): 8.61 (d, J = 8.0 Hz, 1H), 8.19 (s, 1H), 7.87 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 6.6 Hz, 1H), 6.93 (d, J = 6.6 Hz, 1H), 6.26 - 6.16 (m, 1H), 4.20 (s, 3H), 3.99-3.93 (m, 1H), 3.57-3.52 (m, 1H), 2.36 (s, 3H), 2.33 (s, 3H). Example 25b: LCMS: RT 0.563 min (method 1); m/z 397.1 (M+H)+ (ESI+); SFC: RT 2.225 min, ee%: 100% (SFC: Method 8); 1H NMR (CDCl3, 400 MHz): 8.61 (d, J = 8.0 Hz, 1H), 8.19 (s, 1H), 7.87 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 6.6 Hz, 1H), 6.93 (d, J =6.6 Hz, 1H), 6.25 - 6.18 (m, 1H), 4.20 (s, 3H), 3.99-3.93 (m, 1H), 3.57-3.52 (m, 1H), 2.36 (s, 3H), 2.33 (s, 3H). Preparation of Example 26a (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride
3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide (85 mg, 0.210 mmol) was separated by preparative SFC (column: Daicel Chiralpak IC 250x30mm I.D., 10 m particle size; Mobile Phase: A: Supercritical CO2, B: EtOH (NEU): 60% Phase B in Supercritical CO2, Gradient information:10 min, Flow Rate: 120 g/min) and lyophilized to give (R or S)- 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1- dioxide (30 mg, 0.0757 mmol, 36.0 % yield, ee%: 98.33% based on SFC method 8) as a light yellow solid and (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide (30 mg, 0.0757 mmol, 36.0 % yield, ee%: 100% based on SFC method 8) as a light yellow solid. Example 25a: LCMS: RT 0.562 min (method 1); m/z 397.1 (M+H)+ (ESI+); SFC: RT 1.853 min, ee%: 98.33% (Method 8); 1H NMR (CDCl3, 400 MHz): 8.61 (d, J = 8.0 Hz, 1H), 8.19 (s, 1H), 7.87 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 6.6 Hz, 1H), 6.93 (d, J = 6.6 Hz, 1H), 6.26 - 6.16 (m, 1H), 4.20 (s, 3H), 3.99-3.93 (m, 1H), 3.57-3.52 (m, 1H), 2.36 (s, 3H), 2.33 (s, 3H). Example 25b: LCMS: RT 0.563 min (method 1); m/z 397.1 (M+H)+ (ESI+); SFC: RT 2.225 min, ee%: 100% (SFC: Method 8); 1H NMR (CDCl3, 400 MHz): 8.61 (d, J = 8.0 Hz, 1H), 8.19 (s, 1H), 7.87 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 6.6 Hz, 1H), 6.93 (d, J =6.6 Hz, 1H), 6.25 - 6.18 (m, 1H), 4.20 (s, 3H), 3.99-3.93 (m, 1H), 3.57-3.52 (m, 1H), 2.36 (s, 3H), 2.33 (s, 3H). Preparation of Example 26a (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride
 A solution of (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide (15 mg, 0.0378 mmol) in conc. HCl (0.20 mL) was stirred at 100°C for 1 h. The reaction mixture was then filtered and the filter cake was washed with water (10 mL) and MeCN (10 mL). The cake was collected and dried under vacuum to give the product (R or S)-3-(4-(6-(3,4- dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide hydrochloride (7.1 mg, 0.0169 mmol, 44.61 % yield, ee%: 100% based on SFC method 8, HCl salt) as a yellow solid.
LCMS: RT 0.475 min (method 1); m/z 383.0 (M+H)+ (ESI+); SFC: RT 1.564 min, ee%: 100% (Method 8); 1H NMR (DMSO-d6, 400 MHz): 12.80 (br s, 1H), 8.69 (s, 1H), 8.36 (d, J = 7.4 Hz, 1H), 7.63 (s, 1H), 7.60 - 7.49 (m, 2H), 7.32 - 7.17 (m, 2H), 6.76 (d, J = 7.4 Hz, 1H), 6.44-6.40 (m, 1H), 4.10-4.04 (m, 1H), 3.66-3.61 (m, 1H), 2.29 (s, 3H), 2.28 (s, 3H). Preparation of Example 26b (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride
A solution of (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide (15 mg, 0.0378 mmol) in conc. HCl (0.20 mL) was stirred at 100°C for 1 h. The reaction mixture was then filtered and the filter cake was washed with water (10 mL) and MeCN (10 mL). The cake was collected and dried under vacuum to give the product (R or S)-3-(4-(6-(3,4- dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide hydrochloride (7.1 mg, 0.0169 mmol, 44.61 % yield, ee%: 100% based on SFC method 8, HCl salt) as a yellow solid.
LCMS: RT 0.475 min (method 1); m/z 383.0 (M+H)+ (ESI+); SFC: RT 1.564 min, ee%: 100% (Method 8); 1H NMR (DMSO-d6, 400 MHz): 12.80 (br s, 1H), 8.69 (s, 1H), 8.36 (d, J = 7.4 Hz, 1H), 7.63 (s, 1H), 7.60 - 7.49 (m, 2H), 7.32 - 7.17 (m, 2H), 6.76 (d, J = 7.4 Hz, 1H), 6.44-6.40 (m, 1H), 4.10-4.04 (m, 1H), 3.66-3.61 (m, 1H), 2.29 (s, 3H), 2.28 (s, 3H). Preparation of Example 26b (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride
 A solution of (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide (15 mg, 0.0378 mmol) in conc. HCl (0.20 mL) was stirred at 100°C for 1 h. The reaction mixture was then filtered and the filter cake was washed with water (10 mL) and EtOH (10 mL). The cake was collected and dried under vacuum to give the product (S or R)-3-(4-(6-(3,4- dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide hydrochloride (11 mg, 0.0252 mmol, 66.63 % yield, ee%: 100% based on SFC method 8, HCl salt) as a light yellow solid. LCMS: RT 0.466 min (method 1); m/z 383.1 (M+H)+ (ESI+); SFC: RT 1.222 min, ee%: 100% (Method 8); 1H NMR (DMSO-d6, 400 MHz): 12.15 (s, 1H), 8.68 (s, 1H), 8.35 (d, J = 7.4 Hz, 1H), 7.62 (s, 1H), 7.59 - 7.50 (m, 2H), 7.31 - 7.19 (m, 2H), 6.75 (d, J = 7.4 Hz, 1H), 6.47 - 6.37 (m, 1H), 4.09-4.03 (mm, 1H), 3.65-3.61 (m, 1H), 2.29 (s, 3H), 2.28 (s, 3H). Preparation of Intermediate 27.1 Bis(2,2,2-trifluoroethyl) ((methylsulfonyl)methyl)phosphonate
A solution of (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide (15 mg, 0.0378 mmol) in conc. HCl (0.20 mL) was stirred at 100°C for 1 h. The reaction mixture was then filtered and the filter cake was washed with water (10 mL) and EtOH (10 mL). The cake was collected and dried under vacuum to give the product (S or R)-3-(4-(6-(3,4- dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide hydrochloride (11 mg, 0.0252 mmol, 66.63 % yield, ee%: 100% based on SFC method 8, HCl salt) as a light yellow solid. LCMS: RT 0.466 min (method 1); m/z 383.1 (M+H)+ (ESI+); SFC: RT 1.222 min, ee%: 100% (Method 8); 1H NMR (DMSO-d6, 400 MHz): 12.15 (s, 1H), 8.68 (s, 1H), 8.35 (d, J = 7.4 Hz, 1H), 7.62 (s, 1H), 7.59 - 7.50 (m, 2H), 7.31 - 7.19 (m, 2H), 6.75 (d, J = 7.4 Hz, 1H), 6.47 - 6.37 (m, 1H), 4.09-4.03 (mm, 1H), 3.65-3.61 (m, 1H), 2.29 (s, 3H), 2.28 (s, 3H). Preparation of Intermediate 27.1 Bis(2,2,2-trifluoroethyl) ((methylsulfonyl)methyl)phosphonate
 At 0°C, to a solution of bis(2,2,2-trifluoroethyl) ((methylthio)methyl)phosphonate (2000 mg, 6.53 mmol) in DCM (20 mL) was added m-CPBA (3979 mg, 19.6 mmol, 85% purity). The mixture was stirred at 25°C for 1.5 h, then, poured into water (50 mL) and extracted with DCM (30 mL, 2x). The combined organic layer was washed with brine (80 mL, 2x), dried with anhydrous Na2SO4, filtered, concentrated
under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 40 g SepaFlash Silica Flash Column, Eluent of 0~30% Ethyl acetate/Petroleum ether gradient, 80mL/min) to give the product bis(2,2,2-trifluoroethyl) ((methylsulfonyl)methyl)phosphonate (1800 mg, 5.32 mmol, 81.48 % yield) as a white solid. 1H NMR (DMSO-d6, ,400 MHz) : 4.80 - 4.69 (m, 4H), 4.63 (s, 2H), 3.18 (s, 1H) Preparation of Intermediate 27.2 tert-butyl (S)-(4-(methylsulfonyl)but-3-en-2-yl)carbamate
At 0°C, to a solution of bis(2,2,2-trifluoroethyl) ((methylthio)methyl)phosphonate (2000 mg, 6.53 mmol) in DCM (20 mL) was added m-CPBA (3979 mg, 19.6 mmol, 85% purity). The mixture was stirred at 25°C for 1.5 h, then, poured into water (50 mL) and extracted with DCM (30 mL, 2x). The combined organic layer was washed with brine (80 mL, 2x), dried with anhydrous Na2SO4, filtered, concentrated
under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 40 g SepaFlash Silica Flash Column, Eluent of 0~30% Ethyl acetate/Petroleum ether gradient, 80mL/min) to give the product bis(2,2,2-trifluoroethyl) ((methylsulfonyl)methyl)phosphonate (1800 mg, 5.32 mmol, 81.48 % yield) as a white solid. 1H NMR (DMSO-d6, ,400 MHz) : 4.80 - 4.69 (m, 4H), 4.63 (s, 2H), 3.18 (s, 1H) Preparation of Intermediate 27.2 tert-butyl (S)-(4-(methylsulfonyl)but-3-en-2-yl)carbamate
 To a solution of bis(2,2,2-trifluoroethyl) ((methylsulfonyl)methyl)phosphonate (300 mg, 0.887 mmol) in THF (3 mL) was added K2CO3 (368 mg, 2.66 mmol) and tert-butyl (S)-(1-oxopropan-2- yl)carbamate (154 mg, 0.887 mmol). The mixture was stirred at 20 °C for 2 h then poured into water (20 mL) and extracted with EtOAc (30 mL, 2x). The combined organic layer was washed with brine (30 mL, 2x), dried over anhydrous Na2SO4, filtered, and concentrated under vacuum to give product tert-butyl (S)- (4-(methylsulfonyl)but-3-en-2-yl)carbamate (140 mg,0.519 mmol, 58.55 % yield) as coloress oil. RT 0.430 min (method 1); m/z 194.0 (M –tBu+H) + (ESI+).1H NMR (DMSO-d6, ,400 MHz): 6.86 (dd, J = 4.8, 15.2 Hz, 0.4H), 6.47 (d, J =15.2 Hz, 0.4H), 6.25 (d, J =11.2 Hz, 0.6 H), 6.19 (d, J =2.0, 11.2 Hz, 0.6 H), , 5.15-5.00 (m, 1H), 2.95 (s, 1.8H), 1.63-1.58 (m, 1.2H), 1.62-1.49 (m, 9H), 1.33-1.31 (m, 3 H). Preparation of Intermediate 27.3 (S)-4-(methylsulfonyl)but-3-en-2-amine
To a solution of bis(2,2,2-trifluoroethyl) ((methylsulfonyl)methyl)phosphonate (300 mg, 0.887 mmol) in THF (3 mL) was added K2CO3 (368 mg, 2.66 mmol) and tert-butyl (S)-(1-oxopropan-2- yl)carbamate (154 mg, 0.887 mmol). The mixture was stirred at 20 °C for 2 h then poured into water (20 mL) and extracted with EtOAc (30 mL, 2x). The combined organic layer was washed with brine (30 mL, 2x), dried over anhydrous Na2SO4, filtered, and concentrated under vacuum to give product tert-butyl (S)- (4-(methylsulfonyl)but-3-en-2-yl)carbamate (140 mg,0.519 mmol, 58.55 % yield) as coloress oil. RT 0.430 min (method 1); m/z 194.0 (M –tBu+H) + (ESI+).1H NMR (DMSO-d6, ,400 MHz): 6.86 (dd, J = 4.8, 15.2 Hz, 0.4H), 6.47 (d, J =15.2 Hz, 0.4H), 6.25 (d, J =11.2 Hz, 0.6 H), 6.19 (d, J =2.0, 11.2 Hz, 0.6 H), , 5.15-5.00 (m, 1H), 2.95 (s, 1.8H), 1.63-1.58 (m, 1.2H), 1.62-1.49 (m, 9H), 1.33-1.31 (m, 3 H). Preparation of Intermediate 27.3 (S)-4-(methylsulfonyl)but-3-en-2-amine
 To a solution of tert-butyl (S)-(4-(methylsulfonyl)but-3-en-2-yl)carbamate (125 mg, 0.464 mmol) in MeCN (2 mL) was added TsOH (96 mg, 0.557 mmol).The mixture was stirred at 50°C for 1 h and concentrated under vacuum to give the crude product (S)-4-(methylsulfonyl)but-3-en-2-amine (80 mg, 0.249 mmol, 53.66 % yield TsOH salt) as transparent oil. The crude product was used directly in the next step. Alternative preparation of Intermediate 7.2 2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide
To a solution of (3,4-dimethylphenyl)boronic acid (1000 mg, 6.67 mmol) and 5-bromo-2- (methoxycarbonyl)pyridine 1-oxide (1.20 eq, 1856 mg, 8.00 mmol) in 1,4-Dioxane (20 mL)and Water (2 mL) was added K2CO3 (2027 mg, 14.7 mmol)and Pd(dppf)Cl2 (544 mg, 0.667 mmol). The mixture was degassed with N2 (3x), stirred at 80°C for 1 h, then poured into water (20 mL) and extracted with EtOAc (60 mL, 2x). The combined organic layer was washed with brine (60 mL, 2x), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 40 g SepaFlash Silica Flash Column, Eluent of 0~10% Methyl alcohol/dichloromethane, 80mL/min) to give the product 2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide (1100 mg,3.62 mmol, 54.26 % yield) as a white solid. RT 0.497 min (method 1); m/z 244.1 (M+H) + (ESI+).1H NMR (DMSO-d6, ,400 MHz) : 9.11 (s, 1H), 8.30 (d, J = 8.2 Hz, 1H), 8.23 (d, J = 8.2 Hz, 1H)7.71 (s, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.31 (d, J = 7.6 Hz, 1H), 2.31 (s, 3H), 2.28 (s, 3H) Preparation of Example 27 (S,Z)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide
To a solution of tert-butyl (S)-(4-(methylsulfonyl)but-3-en-2-yl)carbamate (125 mg, 0.464 mmol) in MeCN (2 mL) was added TsOH (96 mg, 0.557 mmol).The mixture was stirred at 50°C for 1 h and concentrated under vacuum to give the crude product (S)-4-(methylsulfonyl)but-3-en-2-amine (80 mg, 0.249 mmol, 53.66 % yield TsOH salt) as transparent oil. The crude product was used directly in the next step. Alternative preparation of Intermediate 7.2 2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide
To a solution of (3,4-dimethylphenyl)boronic acid (1000 mg, 6.67 mmol) and 5-bromo-2- (methoxycarbonyl)pyridine 1-oxide (1.20 eq, 1856 mg, 8.00 mmol) in 1,4-Dioxane (20 mL)and Water (2 mL) was added K2CO3 (2027 mg, 14.7 mmol)and Pd(dppf)Cl2 (544 mg, 0.667 mmol). The mixture was degassed with N2 (3x), stirred at 80°C for 1 h, then poured into water (20 mL) and extracted with EtOAc (60 mL, 2x). The combined organic layer was washed with brine (60 mL, 2x), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 40 g SepaFlash Silica Flash Column, Eluent of 0~10% Methyl alcohol/dichloromethane, 80mL/min) to give the product 2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide (1100 mg,3.62 mmol, 54.26 % yield) as a white solid. RT 0.497 min (method 1); m/z 244.1 (M+H) + (ESI+).1H NMR (DMSO-d6, ,400 MHz) : 9.11 (s, 1H), 8.30 (d, J = 8.2 Hz, 1H), 8.23 (d, J = 8.2 Hz, 1H)7.71 (s, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.31 (d, J = 7.6 Hz, 1H), 2.31 (s, 3H), 2.28 (s, 3H) Preparation of Example 27 (S,Z)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide
 At 0°C, to a solution of 2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide (50 mg, 0.164 mmol) DIEA (0.088 mL, 0.493 mmol) and (S)-4-(methylsulfonyl)but-3-en-2-amine (80 mg, 0.249 mmol) in DMF (2 mL) was added T4P (77 mg, 0.214 mmol in EtOAc, wt%: 50%) .The reaction was stirred at 20 °C for 2 h, then poured into water (10 mL)and extracted with EtOAc (30 mL, 2x). The combined organic layer was washed with brine (30 mL, 2x), dried over anhydrous Na2SO4, filtered, and t concentrated under vacuum. The residue was purified by preparative-HPLC ( column: Phenomenex luna C18150*25mm* 10 m; mobile phase: A: 0.225% FA in water, B: MeCN; B%: 13%-43%,10 min) and lyophilized to give an impure product (~ 20 mg) which was purified by preparative-TLC(petroleum ether ethyl acetate=0:1,Rf=0.5)
and further purified by preparative-HPLC (column: Phenomenex luna C18150*25mm* 10 m;mobile phase: A: 0.225% TFA in water, B: MeCN; B%: 13%-43%,10min) to afford, after lyophilization, the product (S,Z)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide (2.5 mg, 0.00674 mmol, 4.10 % yield) as a white solid. LCMS: RT 0.497 min (method 1); m/z 375.1 (M+H)+ (ESI+); SFC: RT 1.652 min, 87.52% ee (method 9); 1H NMR (MeOD, 400 MHz) : 11.47 (d, J = 6.4 Hz, 1H,), 8.66 (s, 1H), 8.33 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.52 (s, 1H), 7.46 (d, J =8.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 6.47 (d, J = 10.8 Hz, 1H), 6.42 (dd, J = 6.4, 10.8 Hz, 1H), 5.74 - 5.62 (m, 1H), 3.23 (s, 3H), 2.36 (s, 3H), 2.34 (s, 3H), 1.47 (d, J = 6.8 Hz, 3H) Preparation of Intermediate 28.1 2-(4,5-dimethylthiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane
At 0°C, to a solution of 2-carboxy-5-(3,4-dimethylphenyl)pyridine 1-oxide (50 mg, 0.164 mmol) DIEA (0.088 mL, 0.493 mmol) and (S)-4-(methylsulfonyl)but-3-en-2-amine (80 mg, 0.249 mmol) in DMF (2 mL) was added T4P (77 mg, 0.214 mmol in EtOAc, wt%: 50%) .The reaction was stirred at 20 °C for 2 h, then poured into water (10 mL)and extracted with EtOAc (30 mL, 2x). The combined organic layer was washed with brine (30 mL, 2x), dried over anhydrous Na2SO4, filtered, and t concentrated under vacuum. The residue was purified by preparative-HPLC ( column: Phenomenex luna C18150*25mm* 10 m; mobile phase: A: 0.225% FA in water, B: MeCN; B%: 13%-43%,10 min) and lyophilized to give an impure product (~ 20 mg) which was purified by preparative-TLC(petroleum ether ethyl acetate=0:1,Rf=0.5)
and further purified by preparative-HPLC (column: Phenomenex luna C18150*25mm* 10 m;mobile phase: A: 0.225% TFA in water, B: MeCN; B%: 13%-43%,10min) to afford, after lyophilization, the product (S,Z)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide (2.5 mg, 0.00674 mmol, 4.10 % yield) as a white solid. LCMS: RT 0.497 min (method 1); m/z 375.1 (M+H)+ (ESI+); SFC: RT 1.652 min, 87.52% ee (method 9); 1H NMR (MeOD, 400 MHz) : 11.47 (d, J = 6.4 Hz, 1H,), 8.66 (s, 1H), 8.33 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.52 (s, 1H), 7.46 (d, J =8.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 6.47 (d, J = 10.8 Hz, 1H), 6.42 (dd, J = 6.4, 10.8 Hz, 1H), 5.74 - 5.62 (m, 1H), 3.23 (s, 3H), 2.36 (s, 3H), 2.34 (s, 3H), 1.47 (d, J = 6.8 Hz, 3H) Preparation of Intermediate 28.1 2-(4,5-dimethylthiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane
 To a solution of 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (292 mg, 1.57 mmol) and 5- bromo-2,3-dimethylthiophene (200 mg, 1.05 mmol) in THF (10 mL) was added n-BuLi (0.63 mL, 1.57 mmol, 2.5 M) at -70°C under N2. The mixture was stirred at -70°C for 1 h., thenpoured into NH4Cl (aq, sat., 5 ml) and extracted with EtOAc (5 ml; 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated to give the product 2-(4,5-dimethylthiophen-2-yl)-4,4,5,5-tetramethyl- 1,3,2-dioxaborolane (250 mg,1.05 mmol, crude) as a yellow oil which was directly used in the next step without further purification. 1H NMR (DMSO-d6, 400 MHz): 7.25 (s, 1H), 2.33 (s, 3H), 2.10 (s, 3H), 1.25 (s, 12H) Preparation of Intermediate 28.2 2-carboxy-5-(4,5-dimethylthiophen-2-yl)pyridine 1-oxide
To a solution of 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (292 mg, 1.57 mmol) and 5- bromo-2,3-dimethylthiophene (200 mg, 1.05 mmol) in THF (10 mL) was added n-BuLi (0.63 mL, 1.57 mmol, 2.5 M) at -70°C under N2. The mixture was stirred at -70°C for 1 h., thenpoured into NH4Cl (aq, sat., 5 ml) and extracted with EtOAc (5 ml; 3x). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated to give the product 2-(4,5-dimethylthiophen-2-yl)-4,4,5,5-tetramethyl- 1,3,2-dioxaborolane (250 mg,1.05 mmol, crude) as a yellow oil which was directly used in the next step without further purification. 1H NMR (DMSO-d6, 400 MHz): 7.25 (s, 1H), 2.33 (s, 3H), 2.10 (s, 3H), 1.25 (s, 12H) Preparation of Intermediate 28.2 2-carboxy-5-(4,5-dimethylthiophen-2-yl)pyridine 1-oxide
 A mixture of 5-bromo-2-(methoxycarbonyl)pyridine 1-oxide (200 mg, 0.862 mmol), 2-(4,5- dimethylthiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (246 mg, 1.03 mmol), K2CO3 (357 mg, 2.59 mmol) and Pd(PPh3)4 (100 mg, 0.0862 mmol) in 1,4-dioxane (2 mL) and water (0.2 mL) was stirred at 80°C under N2 atmosphere for 12 h. The resulting mixture was filtered and the filtrate was concentrated under vacuum. The residue was purified by reversed-phase flash chromatography (ISCO; 80 g Flash Column Welch Ultimate XB_C1820-40 m; 120 A, mobile phase: A: 0.1% HCl in water, B: MeCN, B%: 0%-40%, 80 mL/min) and lyophilized directly to give the product 2-carboxy-5-(4,5-dimethylthiophen-2- yl)pyridine 1-oxide (45 mg,0.0415 mmol, 4.82 % yield) as a yellow solid. LCMS: RT 0.510 min (method 1); m/z 250.0 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 9.04 (s, 1H), 8.22 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 7.73 (s, 1H), 2.39 (s, 3H), 2.15 (s, 3H) Preparation of Example 28 (R)-5-(4,5-dimethylthiophen-2-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide
A mixture of 5-bromo-2-(methoxycarbonyl)pyridine 1-oxide (200 mg, 0.862 mmol), 2-(4,5- dimethylthiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (246 mg, 1.03 mmol), K2CO3 (357 mg, 2.59 mmol) and Pd(PPh3)4 (100 mg, 0.0862 mmol) in 1,4-dioxane (2 mL) and water (0.2 mL) was stirred at 80°C under N2 atmosphere for 12 h. The resulting mixture was filtered and the filtrate was concentrated under vacuum. The residue was purified by reversed-phase flash chromatography (ISCO; 80 g Flash Column Welch Ultimate XB_C1820-40 m; 120 A, mobile phase: A: 0.1% HCl in water, B: MeCN, B%: 0%-40%, 80 mL/min) and lyophilized directly to give the product 2-carboxy-5-(4,5-dimethylthiophen-2- yl)pyridine 1-oxide (45 mg,0.0415 mmol, 4.82 % yield) as a yellow solid. LCMS: RT 0.510 min (method 1); m/z 250.0 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 9.04 (s, 1H), 8.22 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 7.73 (s, 1H), 2.39 (s, 3H), 2.15 (s, 3H) Preparation of Example 28 (R)-5-(4,5-dimethylthiophen-2-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide
 To a solution of 2-carboxy-5-(4,5-dimethylthiophen-2-yl)pyridine 1-oxide (30 mg, 0.120 mmol) and (R)-3-amino-2,3-dihydrothiophene 1,1-dioxide (26 mg, 0.120 mmol) in DMSO (1 mL) was added DIEA (0.064 mL, 0.361 mmol) and T4P (130 mg, 0.181 mmol in EtOAc, wt%: 50%). The mixture was stirred at 20°C for 1 h, then filtered and the filtrate was collected. The filtrate was diluted with H2O (5 mL) and extracted with EtOAc (5 mL; 2x). The combined organic layer was washed with brine (5 mL; 2x), dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in 2mL MeCN, the solution was purified by preparative HPLC (column: Waters Xbridge 150*25 mm* 5 µm; mobile phase: A: 10 mM aqueous solution of NH4HCO3, B: MeCN; B%: 30%-60%, 25 mL/min, 9 min) and the resulting product solution was lyophilized directly to give the product (R)-5-(4,5-dimethylthiophen-2-yl)-2-((1,1- dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (8.5 mg, 0.0225 mmol, 18.70 % yield) as a white solid. LCMS: RT 0.466 min (method 1); m/z 365.1 (M+H)+ (ESI+). SFC: RT 1.761 min; 90.14% ee (method 9).1H NMR (DMSO-d6, 400 MHz): 11.49 (d, J = 7.6 Hz, 1H), 8.72 (s, 1H), 8.17 (d, J = 8.4 Hz,
1H), 7.72 (d, J = 8.4 Hz, 1H), 7.64 (s, 1H), 7.31 - 7.26 (m, 1H), 7.03 - 6.98 (m, 1H), 5.43 - 5.32 (m, 1H), 3.79 - 3.70 (m, 1H), 3.39 - 3.34 (m, 1H), 2.37 (s, 3H), 2.13 (s, 3H); Preparation of Intermediate 29.1 2-carboxy-5-(5-(trifluoromethyl)thiophen-2-yl)pyridine 1-oxide
To a solution of 2-carboxy-5-(4,5-dimethylthiophen-2-yl)pyridine 1-oxide (30 mg, 0.120 mmol) and (R)-3-amino-2,3-dihydrothiophene 1,1-dioxide (26 mg, 0.120 mmol) in DMSO (1 mL) was added DIEA (0.064 mL, 0.361 mmol) and T4P (130 mg, 0.181 mmol in EtOAc, wt%: 50%). The mixture was stirred at 20°C for 1 h, then filtered and the filtrate was collected. The filtrate was diluted with H2O (5 mL) and extracted with EtOAc (5 mL; 2x). The combined organic layer was washed with brine (5 mL; 2x), dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in 2mL MeCN, the solution was purified by preparative HPLC (column: Waters Xbridge 150*25 mm* 5 µm; mobile phase: A: 10 mM aqueous solution of NH4HCO3, B: MeCN; B%: 30%-60%, 25 mL/min, 9 min) and the resulting product solution was lyophilized directly to give the product (R)-5-(4,5-dimethylthiophen-2-yl)-2-((1,1- dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (8.5 mg, 0.0225 mmol, 18.70 % yield) as a white solid. LCMS: RT 0.466 min (method 1); m/z 365.1 (M+H)+ (ESI+). SFC: RT 1.761 min; 90.14% ee (method 9).1H NMR (DMSO-d6, 400 MHz): 11.49 (d, J = 7.6 Hz, 1H), 8.72 (s, 1H), 8.17 (d, J = 8.4 Hz,
1H), 7.72 (d, J = 8.4 Hz, 1H), 7.64 (s, 1H), 7.31 - 7.26 (m, 1H), 7.03 - 6.98 (m, 1H), 5.43 - 5.32 (m, 1H), 3.79 - 3.70 (m, 1H), 3.39 - 3.34 (m, 1H), 2.37 (s, 3H), 2.13 (s, 3H); Preparation of Intermediate 29.1 2-carboxy-5-(5-(trifluoromethyl)thiophen-2-yl)pyridine 1-oxide
 A mixture of 5-bromo-2-(methoxycarbonyl)pyridine 1-oxide (200 mg, 0.862 mmol), (5- (trifluoromethyl)thiophen-2-yl)boronic acid (186 mg, 0.948 mmol), K2CO3 (357 mg, 2.59 mmol) and Pd(PPh3)4 (100 mg, 0.0862 mmol) in 1,4-dioxane (2 mL) and water (0.2 mL) was stirred at 80°C under N2 atmosphere for 12 h. The resulting mixture was filtered and the filtrated was concentrated under vacuum. The residue was purified by reversed-phase flash chromatography (ISCO; 40 g Flash Column Welch Ultimate XB_C1820-40 m; 120 A, mobile phase: A: 0.1% HCl in water, B: MeCN; B%: 0%-20%, 80 mL/min) and lyophilized directly to give the product 2-carboxy-5-(5-(trifluoromethyl)thiophen-2-yl)pyridine 1-oxide (40 mg,0.135 mmol, 15.67 % yield) as an off-white solid. LCMS: RT 0.511 min (method 1); m/z 289.9 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.54 (s, 1H), 7.81 - 7.71 (m, 2H), 7.51 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 8.0 Hz, 1H) Preparation of Example 29 (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen-2- yl)pyridine 1-oxide
A mixture of 5-bromo-2-(methoxycarbonyl)pyridine 1-oxide (200 mg, 0.862 mmol), (5- (trifluoromethyl)thiophen-2-yl)boronic acid (186 mg, 0.948 mmol), K2CO3 (357 mg, 2.59 mmol) and Pd(PPh3)4 (100 mg, 0.0862 mmol) in 1,4-dioxane (2 mL) and water (0.2 mL) was stirred at 80°C under N2 atmosphere for 12 h. The resulting mixture was filtered and the filtrated was concentrated under vacuum. The residue was purified by reversed-phase flash chromatography (ISCO; 40 g Flash Column Welch Ultimate XB_C1820-40 m; 120 A, mobile phase: A: 0.1% HCl in water, B: MeCN; B%: 0%-20%, 80 mL/min) and lyophilized directly to give the product 2-carboxy-5-(5-(trifluoromethyl)thiophen-2-yl)pyridine 1-oxide (40 mg,0.135 mmol, 15.67 % yield) as an off-white solid. LCMS: RT 0.511 min (method 1); m/z 289.9 (M+H)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.54 (s, 1H), 7.81 - 7.71 (m, 2H), 7.51 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 8.0 Hz, 1H) Preparation of Example 29 (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen-2- yl)pyridine 1-oxide
 At 0°C, to a solution of 2-carboxy-5-(5-(trifluoromethyl)thiophen-2-yl)pyridine 1-oxide (35 mg, 0.121 mmol) and (R)-3-amino-2,3-dihydrothiophene 1,1-dioxide hydrobromide (26 mg, 0.121 mmol) in DMF (0.2 mL) was added DIEA (0.065 mL, 0.363 mmol) and T4P (131 mg, 0.182 mmol in EtOAc, wt%: 50%).The mixture was stirred at 0°C for 1 h, then poured into water (5 mL) and extracted with ethyl acetate (5 mL; 2x). The combined organic layer was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was dissolved in 2mL MeCN, the solution was purified by preparative HPLC (column: Waters Xbridge 150*25 mm* 5 µm; mobile phase: A: 10 mM
aqueous solution of NH4HCO3, B: MeCN; B%: 32%-62%, 25 mL/min, 9 min) and the resulting product solution was lyophilized directly to give the product (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen-2-yl)pyridine 1-oxide (4.2 mg, 0.0103 mmol, 8.48 % yield) as a white solid. LCMS: RT 0.557 min (method 2); m/z 405.1 (M+H)+ (ESI+). SFC: RT 1.656 min; 92.37% ee.(method 10).1H NMR (DMSO-d6, 400 MHz): 11.41 (d, J = 7.6 Hz, 1H), 9.04 (s, 1H), 8.25 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 2.0 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.90 - 7.84 (J = 2.0 Hz, 1H), 7.30 - 7.20 (m, 1H), 7.02 - 7.01 (m, 1H), 5.45 - 5.34 (m, 1H), 3.82 - 3.71 (m, 1H), 3.42 - 3.35 (m, 1H) Preparation of Intermediate 30.1 5-bromo-2-chloro-4-phenoxypyrimidine
At 0°C, to a solution of 2-carboxy-5-(5-(trifluoromethyl)thiophen-2-yl)pyridine 1-oxide (35 mg, 0.121 mmol) and (R)-3-amino-2,3-dihydrothiophene 1,1-dioxide hydrobromide (26 mg, 0.121 mmol) in DMF (0.2 mL) was added DIEA (0.065 mL, 0.363 mmol) and T4P (131 mg, 0.182 mmol in EtOAc, wt%: 50%).The mixture was stirred at 0°C for 1 h, then poured into water (5 mL) and extracted with ethyl acetate (5 mL; 2x). The combined organic layer was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was dissolved in 2mL MeCN, the solution was purified by preparative HPLC (column: Waters Xbridge 150*25 mm* 5 µm; mobile phase: A: 10 mM
aqueous solution of NH4HCO3, B: MeCN; B%: 32%-62%, 25 mL/min, 9 min) and the resulting product solution was lyophilized directly to give the product (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen-2-yl)pyridine 1-oxide (4.2 mg, 0.0103 mmol, 8.48 % yield) as a white solid. LCMS: RT 0.557 min (method 2); m/z 405.1 (M+H)+ (ESI+). SFC: RT 1.656 min; 92.37% ee.(method 10).1H NMR (DMSO-d6, 400 MHz): 11.41 (d, J = 7.6 Hz, 1H), 9.04 (s, 1H), 8.25 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 2.0 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.90 - 7.84 (J = 2.0 Hz, 1H), 7.30 - 7.20 (m, 1H), 7.02 - 7.01 (m, 1H), 5.45 - 5.34 (m, 1H), 3.82 - 3.71 (m, 1H), 3.42 - 3.35 (m, 1H) Preparation of Intermediate 30.1 5-bromo-2-chloro-4-phenoxypyrimidine
 To a solution of 5-bromo-2,4-dichloropyrimidine (40.00 g, 176 mmol) and K2CO3 (60.65 g, 439 mmol) in DMF (400 mL) was added phenol (16.52 g, 176 mmol). The mixture was stirred at 100 °C for 12 h. The resulting reaction mixture was diluted with water (1 L) and then extracted with ethyl acetate (600 mL, 3x). The combined organic layer was washed with brine (1 L, 2x), separated, dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 330 g SepaFlash Silica Flash Column, Eluent of 0 to ~6% Ethyl acetate/Petroleum ether gradient @ 100 mL/min) and concentrated under vacuum to give the product 5-bromo-2-chloro-4- phenoxypyrimidine (35.00 g, 123 mmol, 69.83 % yield) as a white solid. RT 0.553 min (LCMS: method 7); m/z 286.9 (M+H+2)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 8.90 (s, 1H), 7.53-7.47 (m, 2H), 7.37-7.33 (m, 1H), 7.32-7.28 (m, 2H) Preparation of Intermediate 30.2 5-bromo-2-iodo-4-phenoxypyrimidine
To a solution of 5-bromo-2,4-dichloropyrimidine (40.00 g, 176 mmol) and K2CO3 (60.65 g, 439 mmol) in DMF (400 mL) was added phenol (16.52 g, 176 mmol). The mixture was stirred at 100 °C for 12 h. The resulting reaction mixture was diluted with water (1 L) and then extracted with ethyl acetate (600 mL, 3x). The combined organic layer was washed with brine (1 L, 2x), separated, dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 330 g SepaFlash Silica Flash Column, Eluent of 0 to ~6% Ethyl acetate/Petroleum ether gradient @ 100 mL/min) and concentrated under vacuum to give the product 5-bromo-2-chloro-4- phenoxypyrimidine (35.00 g, 123 mmol, 69.83 % yield) as a white solid. RT 0.553 min (LCMS: method 7); m/z 286.9 (M+H+2)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 8.90 (s, 1H), 7.53-7.47 (m, 2H), 7.37-7.33 (m, 1H), 7.32-7.28 (m, 2H) Preparation of Intermediate 30.2 5-bromo-2-iodo-4-phenoxypyrimidine
 To a solution of 5-bromo-2-chloro-4-phenoxypyrimidine (20.00 g, 70.0 mmol) in DCE (200 mL) at 0 °C was added HI (aq., 19 mL, 210 mmol, 11 mol/L) and NaI (23.10 g, 154 mmol). The mixture was heated to 80 °C, stirred for 12 h, cooled to room temperature, poured into NaHCO3 (aq., sat., 300 mL) and then,
extracted with ethyl acetate (400 mL, 3x). The combined organic layer was washed with brine (500 mL, 2x), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 120 g SepaFlash Silica Flash Column, Eluent of 0 to ~10% Ethyl acetate/Petroleum ether gradient @ 100mL/min) and concentrated under vacuum to give the product 5- bromo-2-iodo-4-phenoxypyrimidine (23.00 g, 61.0 mmol, 87.10 % yield) as a yellow solid. RT 0.566 min (LCMS: method 7); m/z 378.7 (M+H+2)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.68 (s, 1H), 7.54-7.45 (m, 2H), 7.37-7.31 (m, 1H), 7.31-7.24 (m, 2H) Preparation of Intermediate 30.3 (5-bromo-4-phenoxypyrimidin-2-yl)(cyclopropyl)methanone
To a solution of 5-bromo-2-chloro-4-phenoxypyrimidine (20.00 g, 70.0 mmol) in DCE (200 mL) at 0 °C was added HI (aq., 19 mL, 210 mmol, 11 mol/L) and NaI (23.10 g, 154 mmol). The mixture was heated to 80 °C, stirred for 12 h, cooled to room temperature, poured into NaHCO3 (aq., sat., 300 mL) and then,
extracted with ethyl acetate (400 mL, 3x). The combined organic layer was washed with brine (500 mL, 2x), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 120 g SepaFlash Silica Flash Column, Eluent of 0 to ~10% Ethyl acetate/Petroleum ether gradient @ 100mL/min) and concentrated under vacuum to give the product 5- bromo-2-iodo-4-phenoxypyrimidine (23.00 g, 61.0 mmol, 87.10 % yield) as a yellow solid. RT 0.566 min (LCMS: method 7); m/z 378.7 (M+H+2)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 8.68 (s, 1H), 7.54-7.45 (m, 2H), 7.37-7.31 (m, 1H), 7.31-7.24 (m, 2H) Preparation of Intermediate 30.3 (5-bromo-4-phenoxypyrimidin-2-yl)(cyclopropyl)methanone
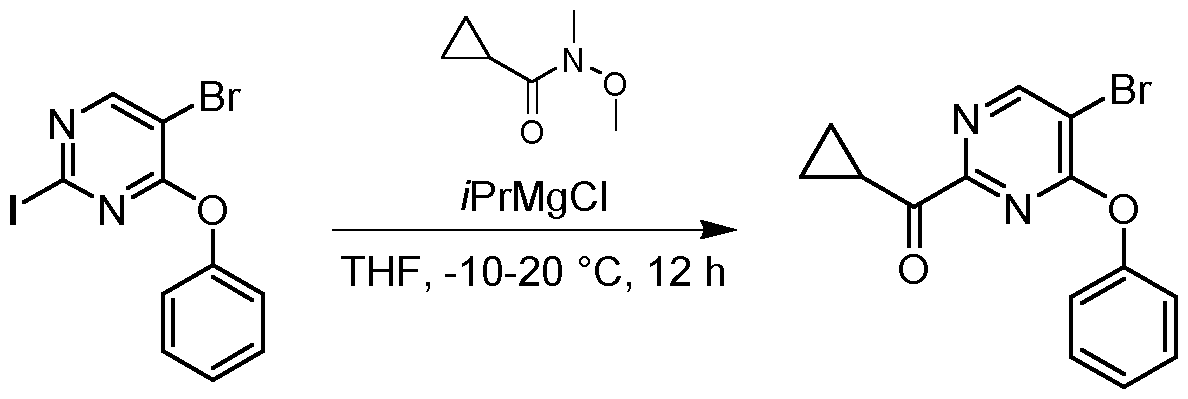 To a solution of 5-bromo-2-iodo-4-phenoxypyrimidine (12.5 g, 33.2 mmol) in THF (130 mL) was added iPrMgCl in THF (17 mL, 33.2 mmol) at -10 °C under N2 atmosphere. The mixture was stirred at - 10 °C for 1 h. Then, N-methoxy-N-methylcyclopropanecarboxamide (4.71 g, 36.5 mmol) was added and the mixture was stirred at 20 °C for 12 h under N2 atmosphere. The resulting reaction mixture was poured into NH4Cl (aq., sat., 100 mL) and extracted with ethyl acetate (50 ml, 3x). The combined organic layer was washed with brine (50 mL, 2x), dried over anhydrous Na2SO4 and concentrated under vacuum to give a residue, which was purified by flash silica gel chromatography (ISCO; 80 g SepaFlash Silica Flash Column, Eluent of 0 to ~7% Ethyl acetate/Petroleum ether gradient @ 60 mL/min) and concentrated under vacuum to give an impure product as a yellow solid which was further triturated in Petroleum ether (5 mL) at 25 °C for 30 min. The precipitate was filtered and dried under reduce pressure to give the product (5- bromo-4-phenoxypyrimidin-2-yl)(cyclopropyl)methanone (3.1 g, 9.71 mmol, 29.29 % yield) as a white solid. RT 0.553 min (LCMS: method 7; m/z 320.9 (M+H+2)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 9.15 (s, 1H), 7.55-7.44 (m, 2H), 7.38-7.27 (m, 3H), 3.10-2.81 (m, 1H), 1.10-1.03 (m, 2H), 1.02-0.90 (m, 2H) Preparation of Intermediate 30.4 5-bromo-2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine
A solution of (5-bromo-4-phenoxypyrimidin-2-yl)(cyclopropyl)methanone (3100 mg, 9.71 mmol) in DAST (32 mL, 9.71 mmol) was stirred at 30 °C for 48 h. The resulting mixture was cooled to room temperature, slowly poured into Na2SO3 (aq., sat., 50 mL) and extracted with ethyl acetate (50 ml, 3x). The combined organic layer was washed with brine (50 mL, 2x), dried over anhydrous Na2SO4 and concentrated under vacuum to give a residue, which was purified by flash silica gel chromatography (ISCO; 40 g SepaFlash Silica Flash Column, Eluent of 0 to ~5% Ethyl acetate/Petroleum ether gradient @ 60 mL/min) and concentrated under vacuum to give the product 5-bromo-2-(cyclopropyldifluoromethyl)- 4-phenoxypyrimidine (2600 mg, 7.62 mmol, 78.46 % yield) as a white solid. RT 0.613 min (LCMS: method 8); m/z 342.8 (M+H+2)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 9.08 (s, 1H), 7.53-7.45 (m, 2H), 7.36-7.26 (m, 3H), 1.68-1.47 (m, 1H), 0.64-0.49 (m, 4H); 19F NMR (DMSO-d6, 400 MHz): -102.75 (s, 2F) Preparation of Intermediate 30.5 Methyl 2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylate
To a solution of 5-bromo-2-iodo-4-phenoxypyrimidine (12.5 g, 33.2 mmol) in THF (130 mL) was added iPrMgCl in THF (17 mL, 33.2 mmol) at -10 °C under N2 atmosphere. The mixture was stirred at - 10 °C for 1 h. Then, N-methoxy-N-methylcyclopropanecarboxamide (4.71 g, 36.5 mmol) was added and the mixture was stirred at 20 °C for 12 h under N2 atmosphere. The resulting reaction mixture was poured into NH4Cl (aq., sat., 100 mL) and extracted with ethyl acetate (50 ml, 3x). The combined organic layer was washed with brine (50 mL, 2x), dried over anhydrous Na2SO4 and concentrated under vacuum to give a residue, which was purified by flash silica gel chromatography (ISCO; 80 g SepaFlash Silica Flash Column, Eluent of 0 to ~7% Ethyl acetate/Petroleum ether gradient @ 60 mL/min) and concentrated under vacuum to give an impure product as a yellow solid which was further triturated in Petroleum ether (5 mL) at 25 °C for 30 min. The precipitate was filtered and dried under reduce pressure to give the product (5- bromo-4-phenoxypyrimidin-2-yl)(cyclopropyl)methanone (3.1 g, 9.71 mmol, 29.29 % yield) as a white solid. RT 0.553 min (LCMS: method 7; m/z 320.9 (M+H+2)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 9.15 (s, 1H), 7.55-7.44 (m, 2H), 7.38-7.27 (m, 3H), 3.10-2.81 (m, 1H), 1.10-1.03 (m, 2H), 1.02-0.90 (m, 2H) Preparation of Intermediate 30.4 5-bromo-2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine
A solution of (5-bromo-4-phenoxypyrimidin-2-yl)(cyclopropyl)methanone (3100 mg, 9.71 mmol) in DAST (32 mL, 9.71 mmol) was stirred at 30 °C for 48 h. The resulting mixture was cooled to room temperature, slowly poured into Na2SO3 (aq., sat., 50 mL) and extracted with ethyl acetate (50 ml, 3x). The combined organic layer was washed with brine (50 mL, 2x), dried over anhydrous Na2SO4 and concentrated under vacuum to give a residue, which was purified by flash silica gel chromatography (ISCO; 40 g SepaFlash Silica Flash Column, Eluent of 0 to ~5% Ethyl acetate/Petroleum ether gradient @ 60 mL/min) and concentrated under vacuum to give the product 5-bromo-2-(cyclopropyldifluoromethyl)- 4-phenoxypyrimidine (2600 mg, 7.62 mmol, 78.46 % yield) as a white solid. RT 0.613 min (LCMS: method 8); m/z 342.8 (M+H+2)+ (ESI+); 1H NMR (DMSO-d6, 400 MHz): 9.08 (s, 1H), 7.53-7.45 (m, 2H), 7.36-7.26 (m, 3H), 1.68-1.47 (m, 1H), 0.64-0.49 (m, 4H); 19F NMR (DMSO-d6, 400 MHz): -102.75 (s, 2F) Preparation of Intermediate 30.5 Methyl 2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylate
 To a solution of 5-bromo-2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine (200 mg, 0.586 mmol) in methanol (4 mL) were added Pd(dppf)Cl2 (48 mg, 0.0586 mmol) and TEA (0.16 mL, 1.17 mmol). Then, the mixture was degassed with argon ( 3x) and stirred under CO atmosphere (50 psi) at 80 °C for 12 h. The resulting reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (ISCO; 20 g SepaFlash Silica Flash Column, Eluent of 0 to ~15% Ethyl acetate/Petroleum ether gradient @ 60 mL/min) and concentrated under vacuum to give the product methyl 2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylate (140 mg, 0.437 mmol, 74.56 % yield) as a white solid. RT 0.571 min (method 8); m/z 321.0 (M +H)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 9.22 (s, 1H), 7.51-7.45 (m, 2H), 7.34-7.30 (m, 1H), 7.27-7.25 (m, 2H), 3.92 (s, 3H), 1.65-1.53 (m, 1H), 0.61-0.52 (m, 4H).19F NMR (DMSO-d6, 400 MHz): -103.76 (s, 2F)
Preparation of Intermediate 30.6 2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylic acid
To a solution of 5-bromo-2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine (200 mg, 0.586 mmol) in methanol (4 mL) were added Pd(dppf)Cl2 (48 mg, 0.0586 mmol) and TEA (0.16 mL, 1.17 mmol). Then, the mixture was degassed with argon ( 3x) and stirred under CO atmosphere (50 psi) at 80 °C for 12 h. The resulting reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (ISCO; 20 g SepaFlash Silica Flash Column, Eluent of 0 to ~15% Ethyl acetate/Petroleum ether gradient @ 60 mL/min) and concentrated under vacuum to give the product methyl 2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylate (140 mg, 0.437 mmol, 74.56 % yield) as a white solid. RT 0.571 min (method 8); m/z 321.0 (M +H)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 9.22 (s, 1H), 7.51-7.45 (m, 2H), 7.34-7.30 (m, 1H), 7.27-7.25 (m, 2H), 3.92 (s, 3H), 1.65-1.53 (m, 1H), 0.61-0.52 (m, 4H).19F NMR (DMSO-d6, 400 MHz): -103.76 (s, 2F)
Preparation of Intermediate 30.6 2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylic acid
 To a solution of methyl 2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylate (110 mg, 0.343 mmol) in water (2 mL) and THF (2 mL) was added LiOH·H2O ( 72 mg, 1.72 mmol) at 0 °C. The mixture was stirred at 0 °C for 1 h and concentrated under reduced pressure. The residue was diluted with water (2 mL), the pH was adjusted to 3-4 by adding HCl (aq., 1N) and aqueous layer was extracted with ethyl acetate (5 mL; 3x). The combined organic layer was washed with brine (3 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to give the product 2- (cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylic acid (100 mg, 0.327 mmol, 95.07 % yield) as white solid. RT 0.503 min (method 8); m/z 307.0 (M+H)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 9.16 (s, 1H), 7.49-7.45 (m, 2H), 7.33-7.31 (m, 1H), 7.26-7.24 (m, 2H), 1.64-1.53 (m, 1H), 0.61-0.51 (m, 4H).19F NMR (DMSO-d6, 400 MHz): -103.67(s, 2F) Preparation of Intermediate 30.7 Tert-butyl (S,E)-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)carbamate and preparation of
To a solution of methyl 2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylate (110 mg, 0.343 mmol) in water (2 mL) and THF (2 mL) was added LiOH·H2O ( 72 mg, 1.72 mmol) at 0 °C. The mixture was stirred at 0 °C for 1 h and concentrated under reduced pressure. The residue was diluted with water (2 mL), the pH was adjusted to 3-4 by adding HCl (aq., 1N) and aqueous layer was extracted with ethyl acetate (5 mL; 3x). The combined organic layer was washed with brine (3 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to give the product 2- (cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylic acid (100 mg, 0.327 mmol, 95.07 % yield) as white solid. RT 0.503 min (method 8); m/z 307.0 (M+H)+ (ESI+).1H NMR (DMSO-d6, 400 MHz): 9.16 (s, 1H), 7.49-7.45 (m, 2H), 7.33-7.31 (m, 1H), 7.26-7.24 (m, 2H), 1.64-1.53 (m, 1H), 0.61-0.51 (m, 4H).19F NMR (DMSO-d6, 400 MHz): -103.67(s, 2F) Preparation of Intermediate 30.7 Tert-butyl (S,E)-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)carbamate and preparation of
 To a solution of tert-butyl (S)-(1-cyclopropyl-2-oxoethyl)carbamate (40 mg, 0.201 mmol) in THF (1 mL) were added diethyl (fluoro(methylsulfonyl)methyl)phosphonate (50 mg, 0.201 mmol) and K2CO3 (69 mg, 0.502 mmol). The reaction mixture was stirred at 80 °C for 3 h, then cooled to room temperature and filtered. The filtrate was purified by preparative-TLC (petroleum ether/ethyl acetate : 3/1) to give the product tert-butyl (S,Z)-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)carbamate (15 mg, 0.0511 mmol, 25.47 % yield) as a light yellow oil and the product tert-butyl (S,E)-(1-cyclopropyl-3-fluoro-3- (methylsulfonyl)allyl)carbamate (10 mg, 0.0341 mmol, 16.98 % yield) as a light yellow oil.
Intermediate 30.7: RT 0.494 min (LCMS: method 7; m/z 316.0 (M+Na)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 6.08 (dd, J = 8.8, 32.8 Hz, 1H), 4.91-4.70 (m, 1H), 3.99-3.76 (m, 1H), 3.05 (s, 3H), 1.45 (s, 9H), 1.05-0.94 (m, 1H), 0.66-0.55 (m, 2H), 0.47-0.33 (m, 2H).19F NMR (CDCl3, 400 MHz): -126.43 (s, 1F) Intermediate 30.8: 1H NMR (CDCl3, 400 MHz): 8.81 (dd, J = 10.4, 21.2 Hz, 1H), 4.95-4.65 (m, 1H), 4.43-4.28 (m, 1H), 3.23 (s, 3H), 1.43 (s, 9H), 0.99 - 0.88 (m, 1H), 0.58 - 0.42 (m, 4H).19F NMR (CDCl3, 400 MHz): -116.26 (s, 1F) Preparation of Intermediate 30.9 (S,E)-1-cyclopropyl-3-fluoro-3-(methylsulfonyl)prop-2-en-1-amine
To a solution of tert-butyl (S)-(1-cyclopropyl-2-oxoethyl)carbamate (40 mg, 0.201 mmol) in THF (1 mL) were added diethyl (fluoro(methylsulfonyl)methyl)phosphonate (50 mg, 0.201 mmol) and K2CO3 (69 mg, 0.502 mmol). The reaction mixture was stirred at 80 °C for 3 h, then cooled to room temperature and filtered. The filtrate was purified by preparative-TLC (petroleum ether/ethyl acetate : 3/1) to give the product tert-butyl (S,Z)-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)carbamate (15 mg, 0.0511 mmol, 25.47 % yield) as a light yellow oil and the product tert-butyl (S,E)-(1-cyclopropyl-3-fluoro-3- (methylsulfonyl)allyl)carbamate (10 mg, 0.0341 mmol, 16.98 % yield) as a light yellow oil.
Intermediate 30.7: RT 0.494 min (LCMS: method 7; m/z 316.0 (M+Na)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 6.08 (dd, J = 8.8, 32.8 Hz, 1H), 4.91-4.70 (m, 1H), 3.99-3.76 (m, 1H), 3.05 (s, 3H), 1.45 (s, 9H), 1.05-0.94 (m, 1H), 0.66-0.55 (m, 2H), 0.47-0.33 (m, 2H).19F NMR (CDCl3, 400 MHz): -126.43 (s, 1F) Intermediate 30.8: 1H NMR (CDCl3, 400 MHz): 8.81 (dd, J = 10.4, 21.2 Hz, 1H), 4.95-4.65 (m, 1H), 4.43-4.28 (m, 1H), 3.23 (s, 3H), 1.43 (s, 9H), 0.99 - 0.88 (m, 1H), 0.58 - 0.42 (m, 4H).19F NMR (CDCl3, 400 MHz): -116.26 (s, 1F) Preparation of Intermediate 30.9 (S,E)-1-cyclopropyl-3-fluoro-3-(methylsulfonyl)prop-2-en-1-amine
 A solution of tert-butyl (S,E)-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)carbamate (12 mg, 0.0409 mmol) in HCl in ethyl acetate (0.5 mL, 2M) was stirred at 25 °C for 1 h. The resulting mixture was concentrated under reduced pressure to give crude product (S,E)-1-cyclopropyl-3-fluoro-3- (methylsulfonyl)prop-2-en-1-amine hydrochloride (12 mg, HCl salt, crude) as a yellow solid. The crude was used directly in the next step. Preparation of Example 30 (E)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide
A solution of tert-butyl (S,E)-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)carbamate (12 mg, 0.0409 mmol) in HCl in ethyl acetate (0.5 mL, 2M) was stirred at 25 °C for 1 h. The resulting mixture was concentrated under reduced pressure to give crude product (S,E)-1-cyclopropyl-3-fluoro-3- (methylsulfonyl)prop-2-en-1-amine hydrochloride (12 mg, HCl salt, crude) as a yellow solid. The crude was used directly in the next step. Preparation of Example 30 (E)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide
 To a solution of 2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylic acid (5.0 mg, 0.0163 mmol), DIEA (17 uL, 0.0980 mmol) and (S,E)-1-cyclopropyl-3-fluoro-3-(methylsulfonyl)prop-2-en- 1-amine hydrochloride (5.2 mg, 0.0180 mmol, HCl salt) in DMF (1 mL) was added T4P in ethyl acetate (15 mg, 0.0212 mmol, purity: 50%). The mixture was stirred at 0 °C for 2 h and then filtered. The filtrate was purified by preparative HPLC (column: Waters xbridge 150*25mm 10 m; mobile phase: A: 10 mmol/L NH4HCO3 in water, B: MeCN; B%: 43%-73%, 10 min, @ 28 mL/min) and lyophilized directly to give the
product (E)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide (3.4 mg, 0.00700 mmol, 42.87 % yield) as a white solid. It is noted that in the process of preparation of Example 30, racemization of the stereogenic centre took place. It has been noted only at the final stage of the synthesis. However, it cannot be excluded that it took place already at an earlier stage, such as preparation of Intermediate 30.7. Thus, it cannot be excluded that intermediates 30.7, 30.8 and 30.9 are in fact not chiral. RT 0.687 min (LCMS: method 2; m/z 482.2 (M+H)+ (ESI+); RT 1.032 min (SFC: Method 6 showed 2 peaks, 15.83 ee%) ; 1H NMR (CDCl3, 400 MHz): 9.48 (s, 1H), 7.89 (d, J = 6.8 Hz, 1H), 7.55-7.46 (m, 2H), 7.41-7.34 (m, 1H), 7.25-7.19 (m, 2H), 6.22 (dd, J= 8.8, 32.4 Hz, 1H), 4.49-4.37 (m, 1H), 3.08 (s, 3H), 1.55-1.47 (m, 1H), 1.24-1.13 (m, 1H), 0.75-0.65 (m, 4H), 0.58-0.45 (m, 4H) Preparation of Intermediate 31.1 (S,Z)-1-cyclopropyl-3-fluoro-3-(methylsulfonyl)prop-2-en-1-amine
To a solution of 2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5-carboxylic acid (5.0 mg, 0.0163 mmol), DIEA (17 uL, 0.0980 mmol) and (S,E)-1-cyclopropyl-3-fluoro-3-(methylsulfonyl)prop-2-en- 1-amine hydrochloride (5.2 mg, 0.0180 mmol, HCl salt) in DMF (1 mL) was added T4P in ethyl acetate (15 mg, 0.0212 mmol, purity: 50%). The mixture was stirred at 0 °C for 2 h and then filtered. The filtrate was purified by preparative HPLC (column: Waters xbridge 150*25mm 10 m; mobile phase: A: 10 mmol/L NH4HCO3 in water, B: MeCN; B%: 43%-73%, 10 min, @ 28 mL/min) and lyophilized directly to give the
product (E)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide (3.4 mg, 0.00700 mmol, 42.87 % yield) as a white solid. It is noted that in the process of preparation of Example 30, racemization of the stereogenic centre took place. It has been noted only at the final stage of the synthesis. However, it cannot be excluded that it took place already at an earlier stage, such as preparation of Intermediate 30.7. Thus, it cannot be excluded that intermediates 30.7, 30.8 and 30.9 are in fact not chiral. RT 0.687 min (LCMS: method 2; m/z 482.2 (M+H)+ (ESI+); RT 1.032 min (SFC: Method 6 showed 2 peaks, 15.83 ee%) ; 1H NMR (CDCl3, 400 MHz): 9.48 (s, 1H), 7.89 (d, J = 6.8 Hz, 1H), 7.55-7.46 (m, 2H), 7.41-7.34 (m, 1H), 7.25-7.19 (m, 2H), 6.22 (dd, J= 8.8, 32.4 Hz, 1H), 4.49-4.37 (m, 1H), 3.08 (s, 3H), 1.55-1.47 (m, 1H), 1.24-1.13 (m, 1H), 0.75-0.65 (m, 4H), 0.58-0.45 (m, 4H) Preparation of Intermediate 31.1 (S,Z)-1-cyclopropyl-3-fluoro-3-(methylsulfonyl)prop-2-en-1-amine
 A solution of tert-butyl (S,Z)-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)carbamate (15 mg, 0.0511 mmol) in HCl in ethyl acetate (0.50 mL, 2 M) was stirred at 25 °C for 2 h. The mixture was concentrated to give crude product (S,Z)-1-cyclopropyl-3-fluoro-3-(methylsulfonyl)prop-2-en-1-amine (13 mg, HCl salt, crude) as a yellow solid. The crude was used directly in the next step. Preparation of Example 31 (Z)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide
A solution of tert-butyl (S,Z)-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)carbamate (15 mg, 0.0511 mmol) in HCl in ethyl acetate (0.50 mL, 2 M) was stirred at 25 °C for 2 h. The mixture was concentrated to give crude product (S,Z)-1-cyclopropyl-3-fluoro-3-(methylsulfonyl)prop-2-en-1-amine (13 mg, HCl salt, crude) as a yellow solid. The crude was used directly in the next step. Preparation of Example 31 (Z)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide
 To a solution of (S,Z)-1-cyclopropyl-3-fluoro-3-(methylsulfonyl)prop-2-en-1-amine hydrochloride (8.2 mg, 0.0359 mmol, HCl salt) in DMF (0.5 mL) was added 2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxylic acid (10 mg, 0.0327 mmol), DIEA (0.035 mL, 0.196 mmol) and then T4P in ethyl acetate ( 32 mg, 0.0424 mmol) at 0 °C. The mixture was stirred at 0 °C for 1 h and, then filtered.
The filtrate was collected and purified by preparative HPLC (column: Waters xbridge 150*25 mm 10 m; mobile phase: A: 10 mmol/L NH4HCO3 in water, B: MeCN; B%: 45%-75%, 10 min, @ 28 mL/min) and lyophilized directly to give an impure solid which was further purified by preparative TLC (Ethyl acetate/Petroleum ether =3/1) three times. After removal of the purification organic solvent, the residue was dissolved in MeCN (2mL) and water (80 mL) and the resulting solution was lyophilized to give (Z)-N- (1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5- carboxamide (2.12 mg, 0.00440 mmol, 13.49 % yield) as a white solid. It is noted that in the process of preparation of Example 31, racemization of the stereogenic centre took place. It has been noted only at the final stage of the synthesis. However, it cannot be excluded that it took place already at an earlier stage (see above). RT 0.704 min (LCMS: method 2; m/z 482.2 (M+H)+ (ESI+); RT 1.775 min (SFC: Method 11 showed 2 peaks, 1.79 ee%) ; 1H NMR (CDCl3, 400 MHz): 9.44 (s, 1H), 7.88 (d, J = 5.6 Hz, 1H), 7.57- 7.47 (m, 2H), 7.42-7.34 (m, 1H), 7.26-7.22 (m, 2H), 5.95 (dd, J = 10.4, 20.4 Hz, 1H), 4.88-4.72 (m, 1H), 3.32 (s, 3H), 1.55-1.45 (m, 1H), 1.19-1.08 (m, 1H), 0.73-0.68 (m, 2H), 0.68-0.60 (m, 3H), 0.58-0.48 (m, 3H) Preparation of Intermediate 32.1 Diethyl ((methylsulfinyl)methyl)phosphonate
To a solution of (S,Z)-1-cyclopropyl-3-fluoro-3-(methylsulfonyl)prop-2-en-1-amine hydrochloride (8.2 mg, 0.0359 mmol, HCl salt) in DMF (0.5 mL) was added 2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxylic acid (10 mg, 0.0327 mmol), DIEA (0.035 mL, 0.196 mmol) and then T4P in ethyl acetate ( 32 mg, 0.0424 mmol) at 0 °C. The mixture was stirred at 0 °C for 1 h and, then filtered.
The filtrate was collected and purified by preparative HPLC (column: Waters xbridge 150*25 mm 10 m; mobile phase: A: 10 mmol/L NH4HCO3 in water, B: MeCN; B%: 45%-75%, 10 min, @ 28 mL/min) and lyophilized directly to give an impure solid which was further purified by preparative TLC (Ethyl acetate/Petroleum ether =3/1) three times. After removal of the purification organic solvent, the residue was dissolved in MeCN (2mL) and water (80 mL) and the resulting solution was lyophilized to give (Z)-N- (1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidine-5- carboxamide (2.12 mg, 0.00440 mmol, 13.49 % yield) as a white solid. It is noted that in the process of preparation of Example 31, racemization of the stereogenic centre took place. It has been noted only at the final stage of the synthesis. However, it cannot be excluded that it took place already at an earlier stage (see above). RT 0.704 min (LCMS: method 2; m/z 482.2 (M+H)+ (ESI+); RT 1.775 min (SFC: Method 11 showed 2 peaks, 1.79 ee%) ; 1H NMR (CDCl3, 400 MHz): 9.44 (s, 1H), 7.88 (d, J = 5.6 Hz, 1H), 7.57- 7.47 (m, 2H), 7.42-7.34 (m, 1H), 7.26-7.22 (m, 2H), 5.95 (dd, J = 10.4, 20.4 Hz, 1H), 4.88-4.72 (m, 1H), 3.32 (s, 3H), 1.55-1.45 (m, 1H), 1.19-1.08 (m, 1H), 0.73-0.68 (m, 2H), 0.68-0.60 (m, 3H), 0.58-0.48 (m, 3H) Preparation of Intermediate 32.1 Diethyl ((methylsulfinyl)methyl)phosphonate
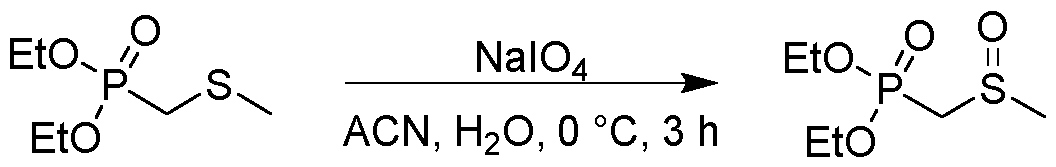 To a solution of diethyl ((methylthio)methyl)phosphonate (2000 mg, 10.1 mmol) in MeCN (20 mL) was added dropwise a solution of NaIO4 (4337 mg, 20.2 mmol) in Water (16 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 3 h, then diluted with water(20 mL) and extracted with ethyl acetate (30 mL;3x). The combined organic layer was washed with aq. Solution of Na2SO3 (sat., 30 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent DCM/MeOH gradient from 100/0 to ~90/~10 @ 60 mL/min) to give the product diethyl ((methylsulfinyl)methyl)phosphonate (870 mg, 4.06 mmol, 40.25 % yield) as colorless oil. MS: (method 1); m/z 215.1 (M+H)+ (ESI+).1H NMR (CDCl3, 400 MHz): 4.26-4.14 (m, 4H), 3.42-3.19 (m, 2H), 2.86 (s, 3H), 1.37 (t, J = 7.2 Hz, 6H) Preparation of Intermediate 32.2 Tert-butyl (((diethoxyphosphoryl)methyl)(methyl)(oxo)- 6-sulfaneylidene)carbamate
To a solution of diethyl ((methylthio)methyl)phosphonate (2000 mg, 10.1 mmol) in MeCN (20 mL) was added dropwise a solution of NaIO4 (4337 mg, 20.2 mmol) in Water (16 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 3 h, then diluted with water(20 mL) and extracted with ethyl acetate (30 mL;3x). The combined organic layer was washed with aq. Solution of Na2SO3 (sat., 30 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent DCM/MeOH gradient from 100/0 to ~90/~10 @ 60 mL/min) to give the product diethyl ((methylsulfinyl)methyl)phosphonate (870 mg, 4.06 mmol, 40.25 % yield) as colorless oil. MS: (method 1); m/z 215.1 (M+H)+ (ESI+).1H NMR (CDCl3, 400 MHz): 4.26-4.14 (m, 4H), 3.42-3.19 (m, 2H), 2.86 (s, 3H), 1.37 (t, J = 7.2 Hz, 6H) Preparation of Intermediate 32.2 Tert-butyl (((diethoxyphosphoryl)methyl)(methyl)(oxo)- 6-sulfaneylidene)carbamate

 To a solution of diethyl ((methylsulfinyl)methyl)phosphonate (840 mg, 3.92 mmol) in DCM (15 mL) was added tert-butyl carbamate (919 mg, 7.84 mmol), rhodium(II) acetate dimer powder (52 mg, 0.118 mmol) and MgO (627 mg, 15.7 mmol). The mixture was stirred at 20 °C under N2 for 20 min and PhI(OAc)2 (1895 mg, 5.88 mmol) was added. The mixture was stirred at 50 °C for 3 h under N2, then cooled to room temperature and filtered. The filtrate was purified by column chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent Petroleum ether/Ethyl acetate gradient from 60/40 to 0/100 @ 40 mL/min) to give the product tert-butyl (((diethoxyphosphoryl)methyl)(methyl)(oxo)- 6-sulfaneylidene)carbamate (1000 mg, 3.04 mmol, 77.43 % yield) as yellow oil. MS: (method 1); m/z 230.2(M+H-Boc)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 4.36 (t, J = 15.6 Hz, 1H), 4.31-4.19 (m, 4H), 3.95 (t, J = 15.6 Hz, 1H), 3.41 (s, 3H), 1.50 (s, 9H), 1.38 (t, J = 7.2 Hz, 6H) Preparation of Intermediate 32.3 Tert-
To a solution of diethyl ((methylsulfinyl)methyl)phosphonate (840 mg, 3.92 mmol) in DCM (15 mL) was added tert-butyl carbamate (919 mg, 7.84 mmol), rhodium(II) acetate dimer powder (52 mg, 0.118 mmol) and MgO (627 mg, 15.7 mmol). The mixture was stirred at 20 °C under N2 for 20 min and PhI(OAc)2 (1895 mg, 5.88 mmol) was added. The mixture was stirred at 50 °C for 3 h under N2, then cooled to room temperature and filtered. The filtrate was purified by column chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent Petroleum ether/Ethyl acetate gradient from 60/40 to 0/100 @ 40 mL/min) to give the product tert-butyl (((diethoxyphosphoryl)methyl)(methyl)(oxo)- 6-sulfaneylidene)carbamate (1000 mg, 3.04 mmol, 77.43 % yield) as yellow oil. MS: (method 1); m/z 230.2(M+H-Boc)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 4.36 (t, J = 15.6 Hz, 1H), 4.31-4.19 (m, 4H), 3.95 (t, J = 15.6 Hz, 1H), 3.41 (s, 3H), 1.50 (s, 9H), 1.38 (t, J = 7.2 Hz, 6H) Preparation of Intermediate 32.3 Tert-
 To a solution of tert-butyl (((diethoxyphosphoryl)methyl)(methyl)(oxo)- 6-sulfaneylidene)carbamate (400 mg, 1.21 mmol) in THF (8 mL) was added tert-butyl (S)-(1-cyclopropyl-2-oxoethyl)carbamate (290 mg, 1.46 mmol) and K2CO3 (420 mg, 3.04 mmol). The reaction mixture was stirred at 80 °C for 1 h, then cooled to room temperature, diluted with water (20 mL) and extracted with ethyl acetate (30 mL;3x). The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent Petroleum ether/Ethylacetate/ gradient from 90/10 to 70/30 @ 100 mL/min) to give the product Tert-butyl (E)-(3-(N- (tert-butoxycarbonyl)-S-methylsulfonimidoyl)-1-cyclopropylallyl)carbamate (130 mg, 0.347 mmol, 28.58 % yield) as yellow oil. It is noted that the starting material has likely racemized in the course of the reaction, as the product appears to be a mixture of four stereoisomers (as indicated by SFC). RT 0.601 min (LCMS: method 2; m/z 375.1 (M+H)+ (ESI+); RT 0.654 min, 0.747 min, 0.825 min, 1.021 min (SFC: Method 12 showed 4 peaks); 1H NMR (CDCl3, 400 MHz): 7.12-6.90 (m, 1H), 6.63-6.50 (m, 1H), 4.87-4.58 (m, 1H), 3.84-3.63 (m, 1H), 3.25 (m, 3H), 1.49 (s, 9H), 1.46 (m, 9H), 0.98-0.87 (m, 1H), 0.71-0.58 (m, 2H), 0.51-0.34 (m, 2H) Preparation of Intermediate 32.4 (E)-(3-amino-3-cyclopropylprop-1-en-1-yl)(imino)(methyl)- 6-sulfanone
A solution of tert-butyl (E)-(3-(N-(tert-butoxycarbonyl)-S-methylsulfonimidoyl)-1- cyclopropylallyl)carbamate (55 mg, 0.147 mmol) in HCl/EtOAc (2 M, 1.5 mL) was stirred at 25 °C for 2 h. The resulting mixture was concentrated under vacuum (30°C). The crude product was triturated with petroleum ether (2 mL at 20 °C for 10 min. Then, the mixture was filtered and the precipitate was dried under vacuum(30°C) to give the product (E)-(3-amino-3-cyclopropylprop-1-en-1-yl)(imino)(methyl)- 6- sulfanone (35 mg, crude) as a yellow solid which was used directly in the next step without any further purification. MS: method 1; m/z 175.2 (M+H)+ (ESI+); Preparation of Example 32 (E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide
To a solution of tert-butyl (((diethoxyphosphoryl)methyl)(methyl)(oxo)- 6-sulfaneylidene)carbamate (400 mg, 1.21 mmol) in THF (8 mL) was added tert-butyl (S)-(1-cyclopropyl-2-oxoethyl)carbamate (290 mg, 1.46 mmol) and K2CO3 (420 mg, 3.04 mmol). The reaction mixture was stirred at 80 °C for 1 h, then cooled to room temperature, diluted with water (20 mL) and extracted with ethyl acetate (30 mL;3x). The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography (ISCO; 12 g SepaFlash Silica Flash Column, Eluent Petroleum ether/Ethylacetate/ gradient from 90/10 to 70/30 @ 100 mL/min) to give the product Tert-butyl (E)-(3-(N- (tert-butoxycarbonyl)-S-methylsulfonimidoyl)-1-cyclopropylallyl)carbamate (130 mg, 0.347 mmol, 28.58 % yield) as yellow oil. It is noted that the starting material has likely racemized in the course of the reaction, as the product appears to be a mixture of four stereoisomers (as indicated by SFC). RT 0.601 min (LCMS: method 2; m/z 375.1 (M+H)+ (ESI+); RT 0.654 min, 0.747 min, 0.825 min, 1.021 min (SFC: Method 12 showed 4 peaks); 1H NMR (CDCl3, 400 MHz): 7.12-6.90 (m, 1H), 6.63-6.50 (m, 1H), 4.87-4.58 (m, 1H), 3.84-3.63 (m, 1H), 3.25 (m, 3H), 1.49 (s, 9H), 1.46 (m, 9H), 0.98-0.87 (m, 1H), 0.71-0.58 (m, 2H), 0.51-0.34 (m, 2H) Preparation of Intermediate 32.4 (E)-(3-amino-3-cyclopropylprop-1-en-1-yl)(imino)(methyl)- 6-sulfanone
A solution of tert-butyl (E)-(3-(N-(tert-butoxycarbonyl)-S-methylsulfonimidoyl)-1- cyclopropylallyl)carbamate (55 mg, 0.147 mmol) in HCl/EtOAc (2 M, 1.5 mL) was stirred at 25 °C for 2 h. The resulting mixture was concentrated under vacuum (30°C). The crude product was triturated with petroleum ether (2 mL at 20 °C for 10 min. Then, the mixture was filtered and the precipitate was dried under vacuum(30°C) to give the product (E)-(3-amino-3-cyclopropylprop-1-en-1-yl)(imino)(methyl)- 6- sulfanone (35 mg, crude) as a yellow solid which was used directly in the next step without any further purification. MS: method 1; m/z 175.2 (M+H)+ (ESI+); Preparation of Example 32 (E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide
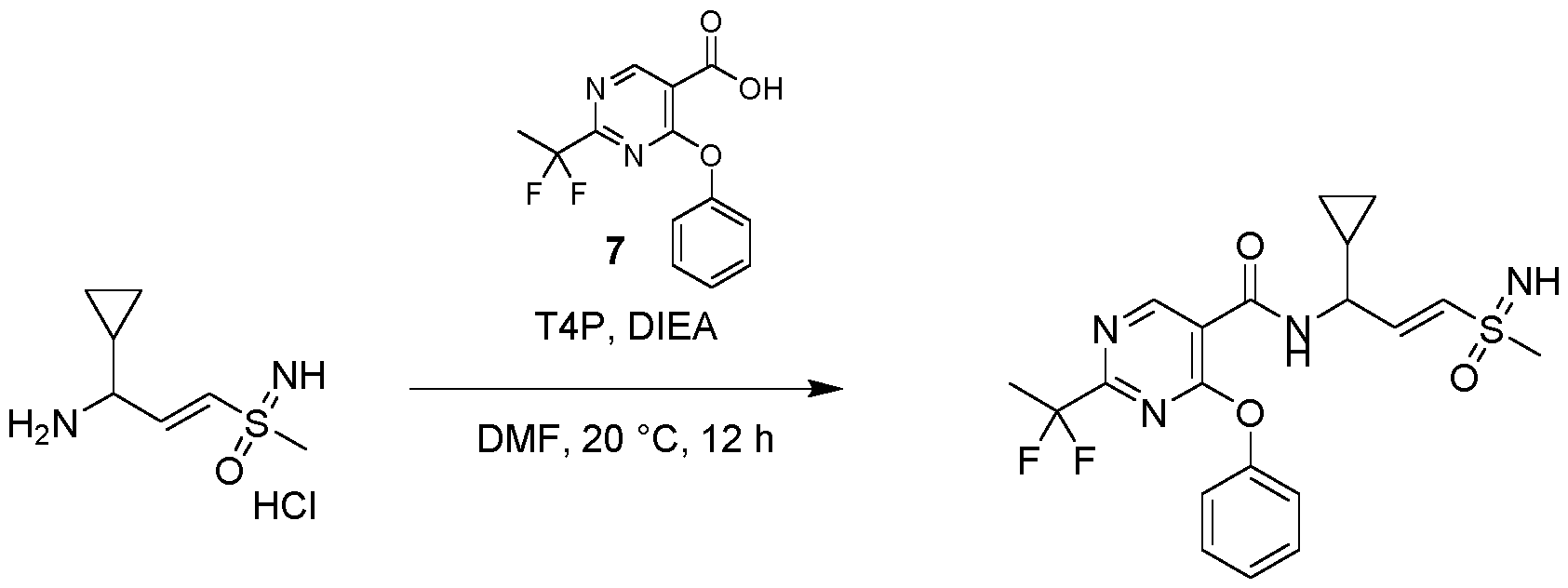 To a solution of (E)-(3-amino-3-cyclopropylprop-1-en-1-yl)(imino)(methyl)- 6-sulfanone hydrochloride (25 mg, 0.119 mmol), 2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxylic acid (25 mg, 0.0892 mmol) and DIEA (95 u, 0.535 mmol) in DMF (1 mL) was added T4P/ ethyl acetate (84 mg, 0.116 mmol, wt%: 50%) at 0 °C under N2. The mixture was stirred at 20 °C for 12 h, then diluted with water (3 mL) and extracted with ethyl acetate (5 mL, 3x). The organic layers were washed with brine, dried over Na2SO4, filtered and concentrated under vacuum. The residue was purified by Preparative TLC (ethyl acetate:100%),then, further purified by preparative HPLC (column: Unisil 3-100 C18 Ultra 150*50 mm*10 m; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 55%-85%, 10 min) and lyophilized directly to give the product (E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (6.0 mg, 0.0136 mmol, 15.27 % yield) as an off-white solid. The product is obtained as a mixture of four stereoisomers, as indicated by SFC.
RT 0.540 min (LCMS: method 8); m/z 437.2 (M +H)+ (ESI+). RT 1.333 min, 1.416 min, 1.529 min, 1.635 min (SFC: Method 13 showed 4 peaks); 1H NMR (CDCl3, 400 MHz): 9.48 (s, 1H), 7.78 (d, J = 6.8 Hz, 1H), 7.56-7.48 (m, 2H), 7.42-7.35 (m, 1H), 7.26-7.22 (m, 2H), 7.00-6.92 (m, 1H), 6.75-6.65 (m, 1H), 4.31-4.22 (m, 1H), 3.03 (s, 3H), 2.76-2.51 (brs, 1H), 1.86 (t, J = 18.4 Hz, 3H), 1.14-1.03 (m, 1H), 0.79- 0.71 (m, 1H), 0.71-0.62 (m, 1H), 0.55-0.44 (m, 2H).19F NMR (CDCl3, 400 MHz): -93.84 (s, 2F) Preparation of: Example 32a N-((S or R, E)-1-cyclopropyl-3-((R or S)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide Example 32b-((S or R, E)-1-cyclopropyl-3-((S or R)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide Example 32c N-((R or S, E)-1-cyclopropyl-3-((R or S)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide Example 32d N-((R or S, E)-1-cyclopropyl-3-((S or R)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide
To a solution of (E)-(3-amino-3-cyclopropylprop-1-en-1-yl)(imino)(methyl)- 6-sulfanone hydrochloride (25 mg, 0.119 mmol), 2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxylic acid (25 mg, 0.0892 mmol) and DIEA (95 u, 0.535 mmol) in DMF (1 mL) was added T4P/ ethyl acetate (84 mg, 0.116 mmol, wt%: 50%) at 0 °C under N2. The mixture was stirred at 20 °C for 12 h, then diluted with water (3 mL) and extracted with ethyl acetate (5 mL, 3x). The organic layers were washed with brine, dried over Na2SO4, filtered and concentrated under vacuum. The residue was purified by Preparative TLC (ethyl acetate:100%),then, further purified by preparative HPLC (column: Unisil 3-100 C18 Ultra 150*50 mm*10 m; mobile phase: A: 0.225% formic acid in water, B: MeCN; B%: 55%-85%, 10 min) and lyophilized directly to give the product (E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (6.0 mg, 0.0136 mmol, 15.27 % yield) as an off-white solid. The product is obtained as a mixture of four stereoisomers, as indicated by SFC.
RT 0.540 min (LCMS: method 8); m/z 437.2 (M +H)+ (ESI+). RT 1.333 min, 1.416 min, 1.529 min, 1.635 min (SFC: Method 13 showed 4 peaks); 1H NMR (CDCl3, 400 MHz): 9.48 (s, 1H), 7.78 (d, J = 6.8 Hz, 1H), 7.56-7.48 (m, 2H), 7.42-7.35 (m, 1H), 7.26-7.22 (m, 2H), 7.00-6.92 (m, 1H), 6.75-6.65 (m, 1H), 4.31-4.22 (m, 1H), 3.03 (s, 3H), 2.76-2.51 (brs, 1H), 1.86 (t, J = 18.4 Hz, 3H), 1.14-1.03 (m, 1H), 0.79- 0.71 (m, 1H), 0.71-0.62 (m, 1H), 0.55-0.44 (m, 2H).19F NMR (CDCl3, 400 MHz): -93.84 (s, 2F) Preparation of: Example 32a N-((S or R, E)-1-cyclopropyl-3-((R or S)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide Example 32b-((S or R, E)-1-cyclopropyl-3-((S or R)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide Example 32c N-((R or S, E)-1-cyclopropyl-3-((R or S)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide Example 32d N-((R or S, E)-1-cyclopropyl-3-((S or R)-S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide
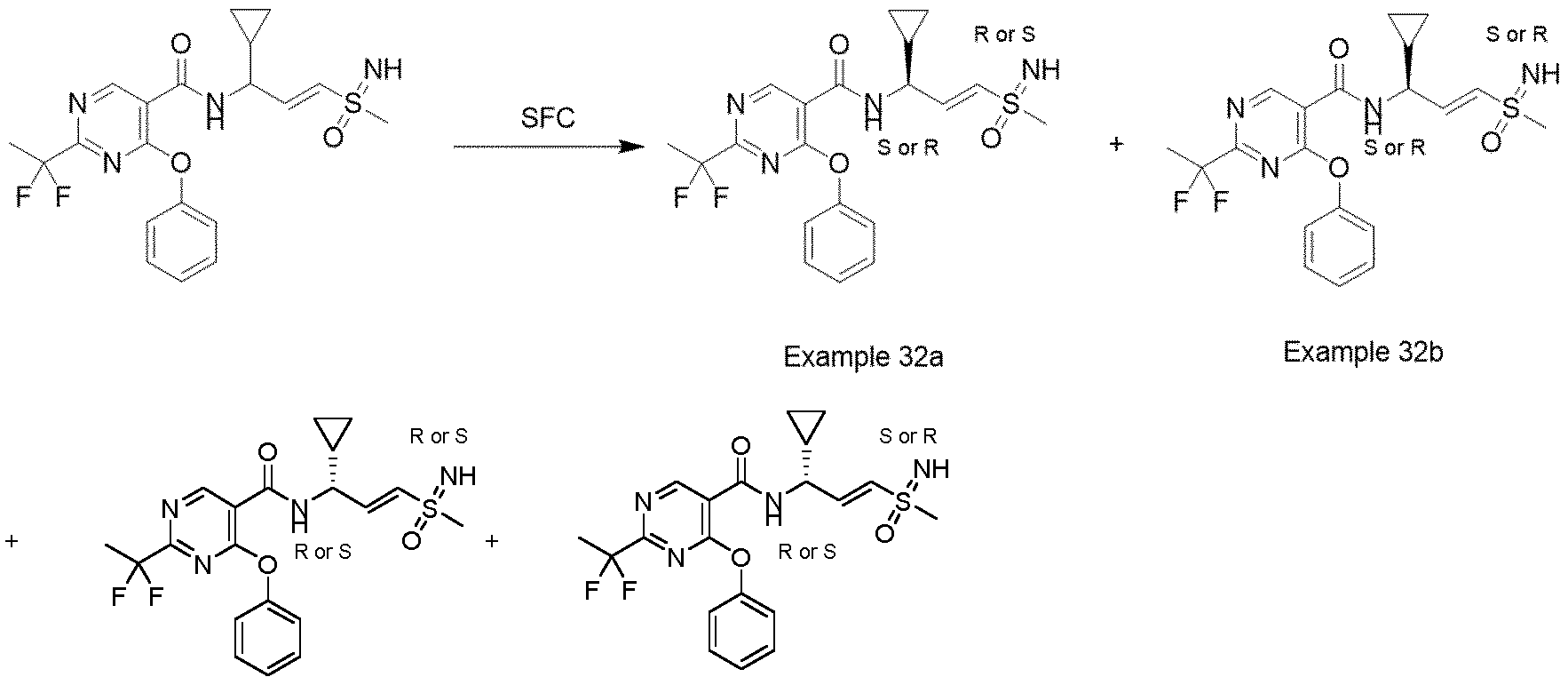 The (E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (350 mg, 0.802 mmol) was purified by preparative SFC (column: DAICEL CHIRALPAK IC(250mm*30 mm,10 µm); mobile phase: A for CO2, B for i-PrOH + ACN; Gradient elution:40% B in CO2, 4 mins)(SFC Method 6) to give two product solutions corresponding to peak 1 and peak 2. The solution containing peak 1 was concentrated under vacuum (30 °C) to give fraction A (SFC: RT: 1.323 min, 1.402 min (Method 13)). The solution containing peak 2 was concentrated under vacuum (30 °C) to give fraction B (SFC: RT: 1.528 min, 1.646 (Method 13)).
Fraction A (140 mg, 0.321 mmol) was further purified by preparative SFC (column: DAICEL CHIRALPAK IC(250 mm*30 mm,10 µm); mobile phase: A for CO2, B for i-PrOH/ACN(2:1); Gradient elution: 40% B in CO2, 4 mins) to afford 2 solutions which were concentrated separately under vacuum (30 °C) to give the respective products (it is noted that the stereochemistry has been assigned arbitrarily): Example 32a: N-((S or R,E)-1-cyclopropyl-3-((R or S)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (60 mg, 0.137 mmol, 99.39% purity,) as colorless gum. LCMS: RT 0.451 min (method 6); m/z 437.0 (M+H)+ (ESI+); SFC: RT: 1.325 min (Method 13); 100% ee ; 1H NMR (CDCl3, 400 MHz): 9.47 (s, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.58-7.47 (m, 2H), 7.44-7.34 (m, 1H), 7.26-7.23 (m, 2H), 6.97 (dd, J = 4.8, 15.2 Hz, 1H), 6.70 (dd, J = 1.6, 15.2 Hz, 1H), 4.37-4.14 (m, 1H), 3.04 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.15-1.05 (m, 1H), 0.80-0.64 (m, 2H), 0.57-0.44 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F). Example 32b: N-((S or R,E)-1-cyclopropyl-3-((S or R)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (51 mg, 0.115 mmol, 99.07% purity,) as colorless gum. LCMS: RT 0.452 min (method 6); m/z 437.1 (M+H)+ (ESI+); SFC: RT: 1.405 min (Method 13); 94.96% ee; 1H NMR (CDCl3, 400 MHz): 9.49 (s, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.57-7.48 (m, 2H), 7.46-7.35 (m, 1H), 7.28-7.23 (m, 2H), 6.97 (dd, J = 4.8, 15.2 Hz, 1H), 6.74 (dd, J = 1.6, 15.2 Hz, 1H), 4.37-4.14 (m, 1H), 3.04 (s, 3H), 1.87 (t, J = 18.4 Hz, 3H), 1.16-1.04 (m, 1H), 0.81-0.65 (m, 2H), 0.59-0.45 (m, 2H); 19F NMR (CDCl3, 400 MHz): 93.83 (s, 2F) Fraction B (140 mg, 0.321 mmol) was further purified by preparative SFC (column: DAICEL CHIRALPAK AD(250mm*30mm,10 m; mobile phase: A for CO2, B for i-PrOH; Gradient elution: 30% B in CO2, 2.6 mins) to afford 2 solutions which were concentrated separately under vacuum (30 °C) to give the respective products: Example 32c: N-((R or S,E)-1-cyclopropyl-3-((R or S)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (56 mg, 0.124 mmol, 97.57% purity,) as colorless gum. LCMS: RT 0.451 min (method 6); m/z 437.1(M+H)+ (ESI+); SFC : RT 1.533 min (Method 13); 96.16% ee; 1H NMR (CDCl3, 400 MHz): 9.47 (s, 1H), 7.78 (d, J = 7.2 Hz, 1H), 7.56-7.47 (m, 2H), 7.42-7.35 (m, 1H), 7.26-7.22 (m, 2H), 6.95 (dd, J = 4.8, 15.2 Hz, 1H), 6.72 (dd, J = 1.6, 15.2 Hz, 1H), 4.36-4.13 (m, 1H), 3.02 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.15-1.02 (m, 1H), 0.79-0.64 (m, 2H), 0.56-0.44 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F) Example 32d: N-((R or S,E)-1-cyclopropyl-3-((S or R)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (59 mg, 0.133 mmol, 98.74% purity,) as colorless gum. LCMS: RT 0.457 min (method 6); m/z 437.1 (M+H)+ (ESI+); SFC: RT: 1.646 min (Method 13); 97.49% ee; 1H NMR (CDCl3, 400 MHz): 9.49 (s, 1H), 7.81 (d, J = 7.2 Hz, 1H), 7.57-7.48 (m, 2H), 7.44-7.37 (m, 1H), 7.26-7.22 (m, 2H), 6.99 (dd, J = 4.8, 15.2 Hz, 1H), 6.72 (dd, J = 1.6, 15.2 Hz, 1H), 4.32-4.23 (m, 1H),
3.06 (s, 3H), 1.87 (t, J = 18.4 Hz, 3H), 1.15 - 1.05 (m, 1H), 0.82 - 0.65 (m, 2H), 0.58 - 0.46 (m, 2H) Preparation of Example 33 (E)-N-(1-cyclopropyl-3-(N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide
The (E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (350 mg, 0.802 mmol) was purified by preparative SFC (column: DAICEL CHIRALPAK IC(250mm*30 mm,10 µm); mobile phase: A for CO2, B for i-PrOH + ACN; Gradient elution:40% B in CO2, 4 mins)(SFC Method 6) to give two product solutions corresponding to peak 1 and peak 2. The solution containing peak 1 was concentrated under vacuum (30 °C) to give fraction A (SFC: RT: 1.323 min, 1.402 min (Method 13)). The solution containing peak 2 was concentrated under vacuum (30 °C) to give fraction B (SFC: RT: 1.528 min, 1.646 (Method 13)).
Fraction A (140 mg, 0.321 mmol) was further purified by preparative SFC (column: DAICEL CHIRALPAK IC(250 mm*30 mm,10 µm); mobile phase: A for CO2, B for i-PrOH/ACN(2:1); Gradient elution: 40% B in CO2, 4 mins) to afford 2 solutions which were concentrated separately under vacuum (30 °C) to give the respective products (it is noted that the stereochemistry has been assigned arbitrarily): Example 32a: N-((S or R,E)-1-cyclopropyl-3-((R or S)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (60 mg, 0.137 mmol, 99.39% purity,) as colorless gum. LCMS: RT 0.451 min (method 6); m/z 437.0 (M+H)+ (ESI+); SFC: RT: 1.325 min (Method 13); 100% ee ; 1H NMR (CDCl3, 400 MHz): 9.47 (s, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.58-7.47 (m, 2H), 7.44-7.34 (m, 1H), 7.26-7.23 (m, 2H), 6.97 (dd, J = 4.8, 15.2 Hz, 1H), 6.70 (dd, J = 1.6, 15.2 Hz, 1H), 4.37-4.14 (m, 1H), 3.04 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.15-1.05 (m, 1H), 0.80-0.64 (m, 2H), 0.57-0.44 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F). Example 32b: N-((S or R,E)-1-cyclopropyl-3-((S or R)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (51 mg, 0.115 mmol, 99.07% purity,) as colorless gum. LCMS: RT 0.452 min (method 6); m/z 437.1 (M+H)+ (ESI+); SFC: RT: 1.405 min (Method 13); 94.96% ee; 1H NMR (CDCl3, 400 MHz): 9.49 (s, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.57-7.48 (m, 2H), 7.46-7.35 (m, 1H), 7.28-7.23 (m, 2H), 6.97 (dd, J = 4.8, 15.2 Hz, 1H), 6.74 (dd, J = 1.6, 15.2 Hz, 1H), 4.37-4.14 (m, 1H), 3.04 (s, 3H), 1.87 (t, J = 18.4 Hz, 3H), 1.16-1.04 (m, 1H), 0.81-0.65 (m, 2H), 0.59-0.45 (m, 2H); 19F NMR (CDCl3, 400 MHz): 93.83 (s, 2F) Fraction B (140 mg, 0.321 mmol) was further purified by preparative SFC (column: DAICEL CHIRALPAK AD(250mm*30mm,10 m; mobile phase: A for CO2, B for i-PrOH; Gradient elution: 30% B in CO2, 2.6 mins) to afford 2 solutions which were concentrated separately under vacuum (30 °C) to give the respective products: Example 32c: N-((R or S,E)-1-cyclopropyl-3-((R or S)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (56 mg, 0.124 mmol, 97.57% purity,) as colorless gum. LCMS: RT 0.451 min (method 6); m/z 437.1(M+H)+ (ESI+); SFC : RT 1.533 min (Method 13); 96.16% ee; 1H NMR (CDCl3, 400 MHz): 9.47 (s, 1H), 7.78 (d, J = 7.2 Hz, 1H), 7.56-7.47 (m, 2H), 7.42-7.35 (m, 1H), 7.26-7.22 (m, 2H), 6.95 (dd, J = 4.8, 15.2 Hz, 1H), 6.72 (dd, J = 1.6, 15.2 Hz, 1H), 4.36-4.13 (m, 1H), 3.02 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.15-1.02 (m, 1H), 0.79-0.64 (m, 2H), 0.56-0.44 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F) Example 32d: N-((R or S,E)-1-cyclopropyl-3-((S or R)-S-methylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (59 mg, 0.133 mmol, 98.74% purity,) as colorless gum. LCMS: RT 0.457 min (method 6); m/z 437.1 (M+H)+ (ESI+); SFC: RT: 1.646 min (Method 13); 97.49% ee; 1H NMR (CDCl3, 400 MHz): 9.49 (s, 1H), 7.81 (d, J = 7.2 Hz, 1H), 7.57-7.48 (m, 2H), 7.44-7.37 (m, 1H), 7.26-7.22 (m, 2H), 6.99 (dd, J = 4.8, 15.2 Hz, 1H), 6.72 (dd, J = 1.6, 15.2 Hz, 1H), 4.32-4.23 (m, 1H),
3.06 (s, 3H), 1.87 (t, J = 18.4 Hz, 3H), 1.15 - 1.05 (m, 1H), 0.82 - 0.65 (m, 2H), 0.58 - 0.46 (m, 2H) Preparation of Example 33 (E)-N-(1-cyclopropyl-3-(N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide
 To a solution of (E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (10 mg, 0.0229 mmol) in DCM (0.30 mL) was added trimethyloxonium tetrafluoroborate (3.7 mg, 0.0252 mmol) in MeCN (0.04 mL) .The resulting mixture was stirred at 25 °C for 3 h, then quenched with aqueous NaHCO3 (sat.3 mL). The aqueous layer was extracted with DCM (3 mL; 3×). The combined organic layer was dried over Na2SO4, filtered and the filtrate was concentrated under vacuum (30 °C). The residue was purified by preparative HPLC (column: Waters xbridge 150*25mm 10 m; mobile phase: A: 10 mmol/L NH4HCO3 in water, B: MeCN; B%: 30%-60%, 9 min) and lyophilized directly to give the product (E)-N-(1-cyclopropyl-3-(N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)- 4-phenoxypyrimidine-5-carboxamide (1.5 mg, 0.00335 mmol, 14.63 % yield) as a white solid. RT 0.563 min (LCMS: method 9); m/z 451. (M +H)+ (ESI+). RT 0.796 min, 0.975 min, 1.133 min, 1.356 min (SFC: Method 14 showed 4 peaks); 1H NMR (CDCl3, 400 MHz): 9.48 (s, 1H), 7.78 (br d, J = 7.2 Hz, 1H), 7.57-7.44 (m, 2H), 7.42-7.36 (m, 1H), 7.28-7.22 (m, 2H), 6.97-6.82 (m, 1H), 6.40 (dd, J = 15.2, 1.6 Hz,, 1H), 4.38-4.22 (m, 1H), 2.99 (s1, , 1.7H, s2, 1.3 H), 2.72 (s1, 1.3H, s2, 1.7H), 1.86 (t, J = 18.4 Hz, 3H), 1.14-1.03 (m, 1H), 0.81-0.64 (m, 2H), 0.56-0.43 (m, 2H).19F NMR (CDCl3, 400 MHz): -93.84 (s, 2F) Preparation of: Example 33a N-((S or R, E)-1-cyclopropyl-3-((R or S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide Example 33b N-((S or R, E)-1-cyclopropyl-3-((S or R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide Example 33c
N-((R or S, E)-1-cyclopropyl-3-((R or S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide Example 33d N-((R or S, E)-1-cyclopropyl-3-((S or R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide
To a solution of (E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (10 mg, 0.0229 mmol) in DCM (0.30 mL) was added trimethyloxonium tetrafluoroborate (3.7 mg, 0.0252 mmol) in MeCN (0.04 mL) .The resulting mixture was stirred at 25 °C for 3 h, then quenched with aqueous NaHCO3 (sat.3 mL). The aqueous layer was extracted with DCM (3 mL; 3×). The combined organic layer was dried over Na2SO4, filtered and the filtrate was concentrated under vacuum (30 °C). The residue was purified by preparative HPLC (column: Waters xbridge 150*25mm 10 m; mobile phase: A: 10 mmol/L NH4HCO3 in water, B: MeCN; B%: 30%-60%, 9 min) and lyophilized directly to give the product (E)-N-(1-cyclopropyl-3-(N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)- 4-phenoxypyrimidine-5-carboxamide (1.5 mg, 0.00335 mmol, 14.63 % yield) as a white solid. RT 0.563 min (LCMS: method 9); m/z 451. (M +H)+ (ESI+). RT 0.796 min, 0.975 min, 1.133 min, 1.356 min (SFC: Method 14 showed 4 peaks); 1H NMR (CDCl3, 400 MHz): 9.48 (s, 1H), 7.78 (br d, J = 7.2 Hz, 1H), 7.57-7.44 (m, 2H), 7.42-7.36 (m, 1H), 7.28-7.22 (m, 2H), 6.97-6.82 (m, 1H), 6.40 (dd, J = 15.2, 1.6 Hz,, 1H), 4.38-4.22 (m, 1H), 2.99 (s1, , 1.7H, s2, 1.3 H), 2.72 (s1, 1.3H, s2, 1.7H), 1.86 (t, J = 18.4 Hz, 3H), 1.14-1.03 (m, 1H), 0.81-0.64 (m, 2H), 0.56-0.43 (m, 2H).19F NMR (CDCl3, 400 MHz): -93.84 (s, 2F) Preparation of: Example 33a N-((S or R, E)-1-cyclopropyl-3-((R or S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide Example 33b N-((S or R, E)-1-cyclopropyl-3-((S or R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide Example 33c
N-((R or S, E)-1-cyclopropyl-3-((R or S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide Example 33d N-((R or S, E)-1-cyclopropyl-3-((S or R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide
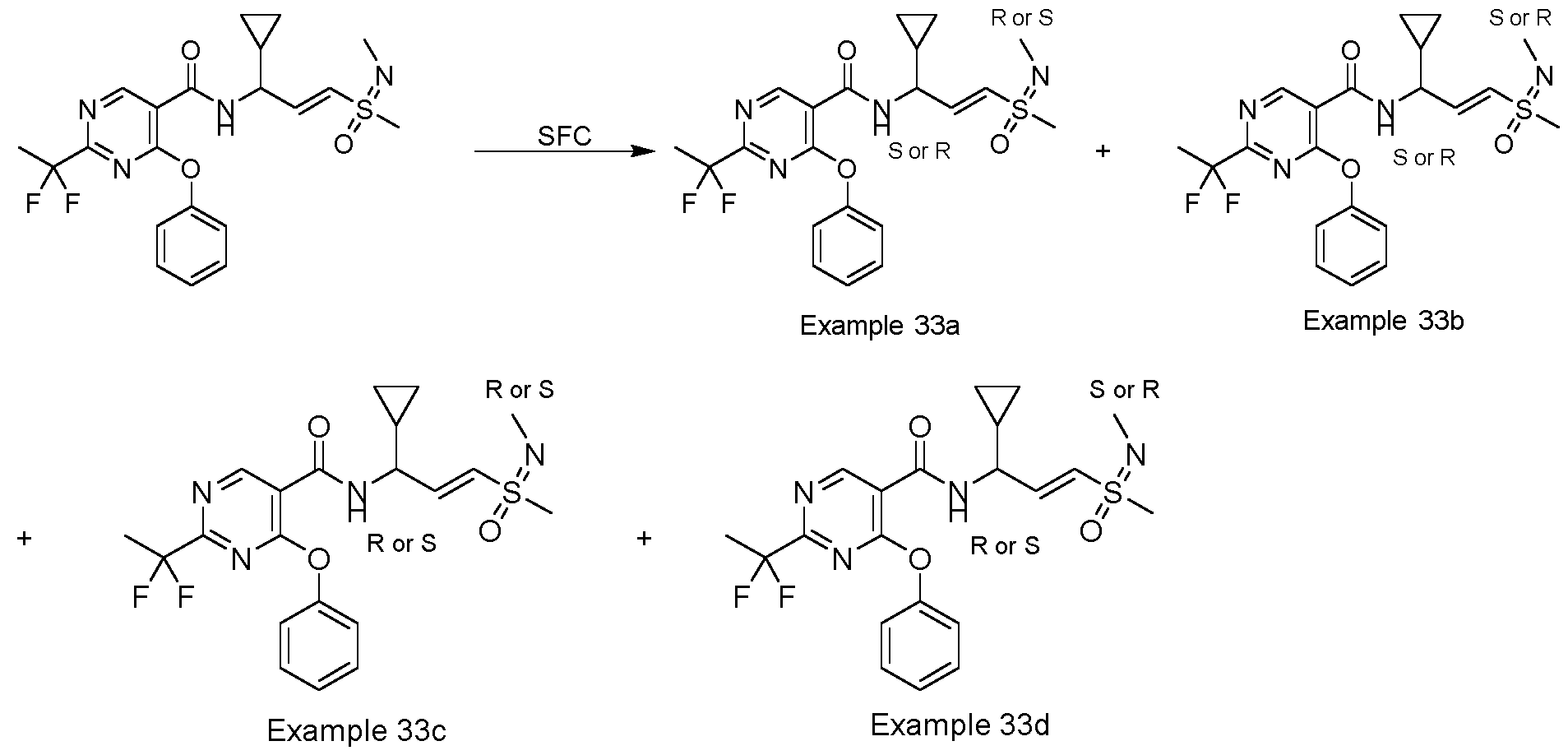 The (E)-N-(1-cyclopropyl-3-(N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (400 mg, 0.888 mmol) was purified by preparative SFC (column: DAICEL CHIRALCEL OX(250mm*30mm,10 m); mobile phase: A for CO2, B for i-PrOH; Gradient elution: 30% B in CO2, 10 mins) (SFC Method 43) to give three product solutions The solution from peak 1 was concentrated under vacuum (30 °C) to give Fraction A (SFC: RT 2.170 min, 2.368 min (Method 11)) containing 2 products. The solution from peak 2 was concentrated under vacuum (30 °C) to give Example 33a: N-((S or R,E)-1-cyclopropyl-3-((R or S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (85 mg, 0.184 mmol, 96.89% purity,) as colorless gum. LCMS: RT 0.570 min (method 10); m/z 451.1 (M+H)+ (ESI+); SFC: RT: 0.798 min (Method 14); 98.97% ee, 1H NMR (CDCl3, 400 MHz): 9.47 (s, 1H), 7.78 (d, J = 7.2 Hz, 1H), 7.57-7.49 (m, 2H), 7.41- 7.36 (m, 1H), 7.26-7.22 (m, 2H), 6.93 (dd, J = 4.8, 15.2 Hz, 1H), 6.42 (dd, J = 1.6, 15.2 Hz, 1H), 4.38-4.25 (m, 1H), 3.00 (s, 3H), 2.71 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.16 - 1.05 (m, 1H), 0.78 - 0.64 (m, 2H), 0.56 - 0.45 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F) The solution from peak 3 was concentrated under vacuum (30 °C) to give Example 33b: N-((S or R,E)-1-cyclopropyl-3-((S or R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (71 mg, 0.155 mmol, 98.73% purity) as a white solid.
LCMS: RT 0.570 min (method 10); m/z 451.1 (M+H)+ (ESI+); SFC: RT: 0.975 min (Method 14); 96.25% ee; 1H NMR (CDCl3, 400 MHz): 9.48 (s, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.54-7.48 (m, 2H), 7.42- 7.36 (m, 1H), 7.26-7.22 (m, 2H), 6.88 (dd, J = 5.2, 15.2 Hz, 1H), 6.41 (dd, J = 1.2, 15.2 Hz, 1H), 4.33-4.24 (m, 1H), 3.00 (s, 3H), 2.73 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.18 - 1.06 (m, 1H), 0.78 - 0.64 (m, 2H), 0.58 - 0.43 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F) Fraction A (SFC: RT 2.170 min, 2.368 min (Method 11)) (150 mg, 0.333 mmol) was further purified by preparative SFC (column: REGIS (s,s) WHELK-O1 (250mm*30mm,10 m); mobile phase: A for CO2, B for i-PrOH; Gradient elution: 35% B in CO2, 3.70 mins) to afford 2 solutions which were concentrated separately under vacuum (30 °C) to give: Example 33c: (SFC,) N-((R or S,E)-1-cyclopropyl-3-((R or S)-N,S-dimethylsulfonimidoyl)allyl)-2- (1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (58 mg, 0.129 mmol, 100% purity,) as a white solid.) LCMS: RT 0.567 min (method 10); m/z 451.1(M+H)+ (ESI+); SFC: RT: 1.127 min (Method 14); 94.68% ee; 1H NMR (CDCl3, 400 MHz): 9.48 (s, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.56-7.48 (m, 2H), 7.42- 7.35 (m, 1H), 7.26-7.23 (m, 2H), 6.88 (dd, J = 5.2, 15.2 Hz, 1H), 6.41 (d, J = 15.2 Hz, 1H), 4.34-4.23 (m, 1H), 2.99 (s, 3H), 2.73 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.17 - 1.05 (m, 1H), 0.78 - 0.65 (m, 2H), 0.57 - 0.44 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F) Example 33d: N-((R or S,E)-1-cyclopropyl-3-((S or R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (65 mg, 0.141 mmol, 98.53% purity) as a white solid. LCMS: RT 0.556 min (method 10); m/z 451.1 (M+H)+ (ESI+); SFC: RT: 1.334 min (Method 14), 97.42% ee; 1H NMR (CDCl3, 400 MHz): 9.47 (s, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.56-7.48 (m, 2H), 7.42- 7.35 (m, 1H), 7.26-7.21 (m, 2H), 6.93 (dd, J = 4.8, 15.2 Hz, 1H), 6.41 (d, J = 15.2 Hz, 1H), 4.36-4.27 (m, 1H), 3.00 (s, 3H), 2.73 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.17 - 1.04 (m, 1H), 0.80 - 0.63 (m, 2H), 0.57 - 0.45 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.84 (s, 2F) The following Table 1 provides an overview on the compounds described in the example section: Table 1 Example No. Structure Name of compound 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3- 1 dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide
6-(3,4-dimethylphenyl)-N-((1S or R,3 S)-1-imino-1- oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide o 6 x-( i3 d, o4 -- 2d ,i 3m -d e it hh yy dlp ro h -e 1n Hy -l) 1- lN 6--( th (1 io R ph o er n S -, 33 -y S l) ) -- 21 -- oim xo in -o 1- ,2 1 -- dihydropyridine-3-carboxamide 6-(3,4-dimethylphenyl)-N-((1S or R,3 R)-1-imino-1- oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide 6-(3,4-dimethylphenyl)-N-((1R or S,3 R)-1-imino-1- oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide 6-(3,4-dimethylphenyl)-N-(1-oxido-2,3- dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (R or S)-5-(3,4-dimethylphenyl)-2-((1,1-a dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide
(S or R)-5-(3,4-dimethylphenyl)-2-((1,1-b dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido- 2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)picolinamide 0 (R or S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)picolinamide 1 (S or R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)picolinamide 2 N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6- (piperidin-1-yl)-1,2-dihydropyridine-3-carboxamide 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-3 dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- sulfonamide 4 N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6- (prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxamide
dihyd 6 ro -( th 3, io 4 p-d hi em ne -t 3h -y yl lp )-h 2e -t n hy io l) x-N o-- 1(1 ,2 ,1 -d -d ih io yx di rd oo p- y2 r, i3 d- ine-3- carboxamide N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoro- 3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide 3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (R)-3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido- 2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)- 5-(4-(trifluoromethyl)phenyl)pyridine 1-oxide 4-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (R)-5-(2-(benzyloxy)phenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide ( y R l) ) c- a2 r- b(( a1 m,1 o- yd li )o -x 5i -d (5 o -- m2, e3 t- hd yih lt y hd io ro pt hh eio np -3 h -e yn l)- p3 y- ridine 1- oxide
2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)- 5-(spiro[3.3]hept-1-en-2-yl)pyridine 1-oxide 5-(2-chlorothiophen-3-yl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)- 1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1- dioxide 3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)- 1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1- dioxide hydrochloride (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-a methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide b ( mS e o th r o R x) y- p3 y-( r4 id -( in 6 -- 3(3 -y ,4 l)- -d 1i Hm -e 1t ,h 2y ,3 lp -h tr e ia n zy ol) l-- 12 -- yl)-2,3- dihydrothiophene 1,1-dioxide R or S N N N S O (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-a O hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- N OH dihydrothiophene 1,1-dioxide hydrochloride HCl (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-b hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride
b N-((S or R, E)-1-cyclopropyl-3-((S or R)-S- methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide c N-((R or S, E)-1-cyclopropyl-3-((R or S)-S- methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide d N-((R or S, E)-1-cyclopropyl-3-((S or R)-S- methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (E)-N-(1-cyclopropyl-3-(N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide a N-((S or R, E)-1-cyclopropyl-3-((R or S)-N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide b N di- m(( eS th o yr ls R u, lf E o) n- i1 m-c id y oc ylo l) p ar lo ly p l) y -l 2-3 -(- 1(( ,S 1- o dr ifl R uo )- rN o, eS th- yl)-4- phenoxypyrimidine-5-carboxamide
33c N-((R or S, E)-1-cyclopropyl-3-((R or S)-N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide 33d N-((R or S, E)-1-cyclopropyl-3-((S or R)-N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide The following Table 2 provides an overview on the compounds which were obtained following similar procedures as the ones described in the above example section or using procedures well known by someone skilled in the art. The skilled person is capable of obtaining any of the following compounds, by adapting and/or following the teaching provided herein for the synthesis of any one of the preceding compounds. Table 2 L + Example Chemical CMS data (RT + MH) number structure and SFC data (RT + Chemical name ee%) N-(1,1-dioxido-2,3- RT 0.337 min (LCMS dihydrothiophen-3-yl)-6- 34 Method 7), m/z 278.9 ethynyl-2-oxo-1,2- (M+H)+ (ESI+) dihydropyridine-3- carboxamide O O O N-(1,1-dioxido-2,3- S H O RT 0.366 min (LCMS dihydrothiophen-3-yl)-2- 35 N N H Method 7), m/z 324.1 oxo-6-(pyrrolidin-1-yl)- N (M+H)+ (ESI+) 1,2-dihydropyridine-3- carboxamide
59 N-(1,1-dioxido-2,3- RT 0.443 min (LCMS dihydrothiophen-3-yl)-6- Method 7), m/z 321.1 (3-methylbut-1-yn-1-yl)- (M+H)+ (ESI+) 2-oxo-1,2- dihydropyridine-3- carboxamide RT 0.465 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 335.0 dihydrothiophen-3-yl)-6- (M+H)+ (ESI+); RT 1.535 (3-methylbut-1-yn-1-yl)- min, 2.554 min 2-oxo-1,2- (SFCMethod 14); dihydropyridine-3- racemate carboxamide RT 0.540 min (LCMS Method 7), m/z 333.2 (R)-N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.692 dihydrothiophen-3-yl)-5- min (SFC Method 6); (spiro[3.3]heptan-2- ee: 88.20% yl)picolinamide RT 0.433 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 333.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.093 dihydrothiophen-3-yl)- min, 2.420 min SFC 1H-1,2,4-triazole-3- Method 24); racemate carboxamide RT 0.427 min (LCMS (R)-5-(3,4- Method 7), m/z 333.2 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 1.552 dioxido-2,3- min, (SFC Method 26); dihydrothiophen-3-yl)- ee: 90.09% 4H-1,2,4-triazole-3- carboxamide RT 0.443 min (LCMS (R)-2-((1,1-dioxido-2,3- Method 7), m/z 351.0 dihydrothiophen-3- (M+H)+ (ESI+); RT 2.239 yl)carbamoyl)-5-(5- min, (SFC Method 6); methylthiophen-2- ee: 84.88% yl)pyridine 1-oxide RT 0.455 min (LCMS (R)-5-(cyclohex-1-en-1- Method 7), m/z 335.1 yl)-2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.724 dihydrothiophen-3- min, ( SFC Method 17); yl)carbamoyl)pyridine 1- ee: 89.64% oxide RT 0.460 min (LCMS (R)-5-cyclohexyl-2-((1,1- Method 7), m/z 337.1 dioxido-2,3- (M+H)+ (ESI+); RT 1.449 dihydrothiophen-3- min, (SFC Method 7); yl)carbamoyl)pyridine 1- ee: 89.62% oxide
RT 0.364 min (LCMS Method 7), m/z 339.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.468 dihydrothiophen-3-yl)-2- min, 1.551 min, 1.641 oxo-6-(tetrahydro-2H- min, 1.733 min, pyran-3-yl)-1,2- (SFC Method 9); dihydropyridine-3- racemate carboxamide RT 0.402 min (LCMS Method 7), m/z 337.1 6-(3,4-dihydro-2H-pyran- (M+H)+ (ESI+); RT 1.718 6-yl)-N-(1,1-dioxido-2,3- min, 4.342 min, dihydrothiophen-3-yl)-2- (SFCMethod 18); oxo-1,2-dihydropyridine- racemate 3-carboxamide RT 0.394 min (LCMS Method 7), m/z 339.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT dihydrothiophen-3-yl)-2- 2.965 min, 3.441 min, oxo-6-(tetrahydro-2H- 9.181 min, 13.749 min, pyran-2-yl)-1,2- (SFC Method 32); dihydropyridine-3- racemate carboxamide 6-(3,6-dihydro-2H-pyran- RT 0.359 min (LCMS 4-yl)-N-(1,1-dioxido-2,3- Method 7), m/z 337.1 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); oxo-1,2-dihydropyridine- 3-carboxamide RT 0.321 min (LCMS (R)-N-(1,1-dioxido-2,3- Method 2), m/z 339.1 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 1.629 oxo-6-(tetrahydro-2H- min, (SFC Method 9 ); pyran-4-yl)-1,2- ee: 90.03% dihydropyridine-3- carboxamide O O S O 6-(5,6-dihydro-2H-pyran- HN N O RT 0.374 min (LCMS 3-yl)-N-(1,1-dioxido-2,3- H Method 7), m/z 337.0 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); oxo-1,2-dihydropyridine- 3-carboxamide RT 0.527 min (LCMS (R)-5-(3,4- Method 7), m/z 349.0 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 1.766 dioxido-2,3- min, (SFC Method 9); dihydrothiophen-3- ee: 91.02% yl)thiazole-2- carboxamide RT 0.471 min (LCMS Method 7), m/z 332.0 (R)-N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.437 dihydrothiophen-3-yl)-2- min, (SFC Method 6); oxo-6-phenyl-2H-pyran- ee: 93.30% 3-carboxamide
61 RT 0.411 min (LCMS Method 7), m/z 338.3 2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.393 dihydrothiophen-3- min, 1.914 min, (SFC yl)carbamoyl)-5- Method 13); (piperidin-1-yl)pyridine 1- racemate oxide RT 0.492 min (LCMS (R)-5-(3,4- Method 7), m/z 350.0 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 2.024 dioxido-2,3- min, (SFC Method 9), dihydrothiophen-3-yl)- ee: 90.65% 1,3,4-thiadiazole-2- carboxamide RT 0.476 min (LCMS (R)-5-(3,4- Method 7), m/z 332.2 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 2.718 dioxido-2,3- min, (SFC Method 26), dihydrothiophen-3-yl)- ee: 88.36% 1H-imidazole-2- carboxamide RT 0.405 min (LCMS (S)-N-(1,1-dioxido-2,3- Method 7), m/z 332.3 dihydrothiophen-3-yl)-1- (M+H)+ (ESI+); RT 1.445 methyl-2-(p-tolyl)-1H- min, (SFC Method 7); imidazole-4- ee: 84.59% carboxamide RT 0.395 min (LCMS (R)-N-(1,1-dioxido-2,3- Method 7), m/z 332.1 dihydrothiophen-3-yl)-1- (M+H)+ (ESI+); RT 1.633 methyl-2-(p-tolyl)-1H- min, (SFC Method 7); imidazole-4- ee: 83.87% carboxamide RT 0.404 min (LCMS 2-(3,4-dimethylphenyl)- Method 7), m/z 332.0 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.073 dihydrothiophen-3-yl)- min, 2.604 min, (SFC 1H-imidazole-4- Method 12); racemate carboxamide RT 0.468 min (LCMS (R)-5-(4-chlorophenyl)-2- Method 7), m/z 365.0 ((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.060 dihydrothiophen-3- min, (SFC Method 10); yl)carbamoyl)pyridine 1- ee: 88.14% oxide RT 0.439 min (LCMS (R)-N-(1,1-dioxido-2,3- Method 7), m/z 353.1 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 1.721 methoxy-6-(tetrahydro- min, (SFC Method 9); 2H-pyran-4- ee: 90.63% yl)nicotinamide
62 RT 0.442 min (LCMS 6-(3,4-dimethylphenyl)- Method 2), m/z 343.1 N-((3R)-1-oxido-2,3- (M+H)+ (ESI+); RT 1.407 dihydrothiophen-3-yl)-2- min, 2.758 min (SFC oxo-1,2-dihydropyridine- Method 33); racemate 3-carboxamide RT 0.480 min (LCMS 6-(3,4-dimethylphenyl)- Method 7), m/z 343.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); dihydrothiophen-3- yl)nicotinamide RT 0.605 min (LCMS Method 2), m/z 342.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.169 dihydrothiophen-3-yl)- min, 2.269 min (SFC 3',4'-dimethyl-[1,1'- Method 16); racemate biphenyl]-4-carboxamide RT 0.533 min (LCMS (R)-5-(3,4- Method 7), m/z 366.0 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 1.494 dioxido-2,3- min, (SFC Method 6) ee: dihydrothiophen-3-yl)-3- 93.06% fluorothiophene-2- carboxamide RT 0.511 min (LCMS 2-(3,4-dimethylphenyl)- Method 7), m/z 344.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); dihydrothiophen-3- yl)pyrimidine-5- carboxamide 5-(3,4-dimethylphenyl)- RT 0.471 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 344.1 dihydrothiophen-3- (M+H)+ (ESI+); yl)pyrimidine-2- carboxamide RT 0.486 min (LCMS Method 7), m/z 344.1 6-(3,4-dimethylphenyl)- (M+H)+ (ESI+); N-(1,1-dioxido-2,3- RT 1.624 min, 1.965 min, dihydrothiophen-3- (SFC Method 9); yl)pyridazine-3- racemate carboxamide RT 0.498 min (LCMS 2-((1,1-dioxido-2,3- Method 7), m/z 349.1 dihydrothiophen-3- (M+H)+ (ESI+); RT 0.797 yl)carbamoyl)-5- min, 1.892 min (SFC (spiro[3.3]heptan-2- Method 5) racemate yl)pyridine 1-oxide RT 0.478 min (LCMS 6-(3,4-dimethylphenyl)- Method 7), m/z 357.1 3-(6,6-dioxido-6-thia-2- (M+H)+ (ESI+); azaspiro[3.4]oct-7-en-2- yl)pyridin-2(1H)-one
63 RT 0.953 min (LCMS 3-((6-(3,4- Method 1), m/z 355.0 dimethylphenyl)-1H- (M+H)+ (ESI+); RT 1.614 indazol-3-yl)oxy)-2,3- min, 1.752 min (SFC dihydrothiophene 1,1- Method 15); racemate dioxide RT 0.914 min (LCMS 6-(3,4-dimethylphenyl)- Method 1), m/z 355.0 1-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.944 dihydrothiophen-3-yl)- min, 2.103 min (SFC 1,2-dihydro-3H-indazol- Method 16); racemate 3-one RT 0.542 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 354.1 2-(1,1-dioxido-2,3- (M+H)+ (ESI+); racemate dihydrothiophen-3- yl)isoindolin-1-one RT 0.465 min (LCMS (R)-N-(1,1-dioxido-2,3- Method 7), m/z 355.0 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 2.481 oxo-6-(phenylethynyl)- min (SFC Method 5); ee: 1,2-dihydropyridine-3- 90.45% carboxamide RT 0.446 min (LCMS Method 7), m/z 359.1 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 0.754 N-(1,1-dioxido-2,3- min, 1.727 min (SFC dihydrothiophen-3-yl)-4- Method 17); racemate hydroxypicolinamide RT 0.569 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 359.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); racemate dihydrothiophen-3-yl)-3- hydroxypicolinamide RT 0.423 min (LCMS 6-(3,4-dimethylphenyl)- Method 7), m/z 359.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.811 dihydrothiophen-3-yl)-4- min, 1.467 min (SFC oxo-1,4-dihydropyridine- Method 18); racemate 3-carboxamide RT 0.531 min (LCMS (R)-5-(2,4- Method 2), m/z 359.1 dimethylphenyl)-2-((1,1- (M+H)+ (ESI+); RT 2.003 dioxido-2,3- min (SFC Method 16); dihydrothiophen-3- ee: 88.35% yl)carbamoyl)pyridine 1- oxide
64 RT 0.491 min (LCMS (R)-5-(3,4- Method 7), m/z 399.0 dichlorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 2.171 dioxido-2,3- min (SFC Method 10); dihydrothiophen-3- ee: 96.65% yl)carbamoyl)pyridine 1- oxide RT 0.443 min (LCMS (R)-5-(3,4- Method 7), m/z 367.0 difluorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.661 dioxido-2,3- min (SFC Method 10); dihydrothiophen-3- ee: 87.27% yl)carbamoyl)pyridine 1- oxide RT 0.431 min (LCMS (R)-2-((1,1-dioxido-2,3- Method 7), m/z 361.1 dihydrothiophen-3- (M+H)+ (ESI+); RT 2.385 yl)carbamoyl)-5-(4- min (SFC Method 20); methoxyphenyl)pyridine ee: 91.51% 1-oxide RT 0.530 min (LCMS Method 7), m/z 361.2 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.569 N-(1,1-dioxido-2,3- min, 1.972 min (SFC dihydrothiophen-3-yl)-3- Method 9); racemate fluoropicolinamide RT 0.525 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 361.0 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); racemate dihydrothiophen-3-yl)-6- fluoropicolinamide RT 0.442 min (LCMS Method 7), m/z 357.1 6-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 0.731 N-(1-methyl-1-oxido-2,3- min, 1.074 min, 1.375 dihydrophosphol-3-yl)-2- min, 1.869 min (SFC oxo-1,2-dihydropyridine- Method 19); racemate 3-carboxamide RT 0.593 min (LCMS 3-amino-N-(1,1-dioxido- Method 2), m/z 357.2 2,3-dihydrothiophen-3- (M+H)+ (ESI+) yl)-3',4'-dimethyl-[1,1'- biphenyl]-4-carboxamide
65 RT 0.526 min (LCMS Method 7), m/z 357.2 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.333 N-(1,1-dioxido-2,3- min, 1.544 min (SFC dihydrothiophen-3-yl)-4- Method 9); racemate methylpicolinamide RT 0.528 min (LCMS Method 7), m/z 357.1 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.259 N-(1,1-dioxido-2,3- min, 1.462 min (SFC dihydrothiophen-3-yl)-6- Method 9); racemate methylpicolinamide RT 0.543 min (LCMS Method 7), m/z 357.1 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.435 N-(1,1-dioxido-2,3- min, 1.557 min, (SFC dihydrothiophen-3-yl)-3- Method 9); racemate methylpicolinamide RT 0.472 min (LCMS Method 7), m/z 357.3 2-amino-N-(1,1-dioxido- (M+H)+ (ESI+); RT 2.229 2,3-dihydrothiophen-3- min, 2.556 min, (SFC yl)-3',4'-dimethyl-[1,1'- Method 26); racemate biphenyl]-4-carboxamide RT 0.520 min (LCMS (S)-N-(1,1-dioxido-2,3- Method 7), m/z 389.0 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 1.915 (4- min, (SFC Method 26); (trifluoromethyl)phenyl)th ee: 84.50% iazole-4-carboxamide RT 0.521 min (LCMS (R)-N-(1,1-dioxido-2,3- Method 7), m/z 389.0 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 2.272 (4- min, (SFC Method 26); (trifluoromethyl)phenyl)th ee: 83.28% iazole-4-carboxamide RT 0.550 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 358.1 dihydrothiophen-3-yl)-3- (M+H)+ (ESI+); RT 2.303 hydroxy-3',4'-dimethyl- min, 2.711 min (SFC [1,1'-biphenyl]-4- Method 16); racemate carboxamide RT 0.538 min (LCMS Method 7), m/z 376.0 3-chloro-N-(1,1-dioxido- (M+H)+ (ESI+); RT 1.365 2,3-dihydrothiophen-3- min, 1.563 min (SFC yl)-3',4'-dimethyl-[1,1'- Method 9); racemate biphenyl]-4-carboxamide RT 0.525 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 360.2 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 1.136 fluoro-3',4'-dimethyl- min, 1.282 min (SFC [1,1'-biphenyl]-4- Method 9); racemate carboxamide
RT 0.532 min (LCMS Method 7), m/z 356.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.182 dihydrothiophen-3-yl)- min, 1.595 min (SFC 2,3',4'-trimethyl-[1,1'- Method 27); racemate biphenyl]-4-carboxamide RT 0.438 min (LCMS 2-(3,4-dimethylphenyl)- Method 7), m/z 360.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.857 dihydrothiophen-3-yl)-6- min, 1.109 min (SFC oxo-1,6- Method 28); racemate dihydropyrimidine-5- carboxamide RT 0.492 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 360.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.596 dihydrothiophen-3-yl)-3- min, 1.894 min (SFC oxo-3,4-dihydropyrazine- Method 25); racemate 2-carboxamide 2-amino-6-(3,4- H RT 0.540 min (LCMS dimethylphenyl)-N-(1,1-N N Method 7), m/z 358.2 dioxido-2,3- O NH2 (M+H)+ (ESI+); dihydrothiophen-3- yl)nicotinamide RT 0.493 min (LCMS (R)-2-((1,1-dioxido-2,3- Method 2), m/z 385.2 dihydrothiophen-3- (M+H)+ (ESI+); RT 2.133 yl)carbamoyl)-5-(2,4,6- min, (SFC Method 30); trifluorophenyl)pyridine ee: 87.55% 1-oxide 3-(3,4-dimethylphenyl)- RT 0.488 min (LCMS 6-((1,1-dioxido-2,3- Method 7), m/z 377.1 dihydrothiophen-3- (M+H)+ (ESI+); yl)carbamoyl)-2- fluoropyridine 1-oxide RT 0.492 min (LCMS 5-(3,4-dimethylphenyl)- Method 2), m/z 377.1 2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.538 dihydrothiophen-3- min, 1.772 min (SFC yl)carbamoyl)-4- Method 4); racemate fluoropyridine 1-oxide RT 0.556 min (LCMS 6-(3,4-dimethylphenyl)- Method 7), m/z 373.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); dihydrothiophen-3-yl)-2- methoxynicotinamide
67 RT 0.503 min (LCMS Method 7), m/z 373.1 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.892 N-(1,1-dioxido-2,3- min, 2.495 min (SFC dihydrothiophen-3-yl)-3- Method 9); racemate methoxypicolinamide RT 0.444 min (LCMS 6-(3,4-dimethylphenyl)- Method 7), m/z 373.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.370 dihydrothiophen-3-yl)-1- min, 1.462 min,(SFC methyl-4-oxo-1,4- Method 20); racemate dihydropyridine-3- carboxamide RT 0.514 min (LCMS 3-(3,4-dimethylphenyl)- Method 7), m/z 373.1 6-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.617 dihydrothiophen-3- min, 1.777 min,(SFC yl)carbamoyl)-2- Method 4); racemate methylpyridine 1-oxide RT 0.457 min (LCMS 3-(3,4-dimethylphenyl)- Method 7), m/z 375.1 6-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.610 dihydrothiophen-3- min, 0.799 min,(SFC yl)carbamoyl)-2- Method 21); racemate hydroxypyridine 1-oxide RT 0.441 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 375.1 2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.723 dihydrothiophen-3- min, 1.485 min,(SFC yl)carbamoyl)-4- Method 22); racemate hydroxypyridine 1-oxide RT 0.521 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 375.1 2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.128 dihydrothiophen-3- min, 2.774 min,(SFC yl)carbamoyl)-3- Method 23); racemate hydroxypyridine 1-oxide RT 0.508 min (LCMS (R)-5-chloro-N-(1,1- Method 7), m/z 392.1 dioxido-2,3- (M+H)+ (ESI+); RT 2.262 dihydrothiophen-3-yl)-2- min, (SFC Method 6); hydroxy-3',4'-dimethyl- ee: 89.35% [1,1'-biphenyl]-4- carboxamide RT 0.505 min (LCMS (S or R)-N-(1,1-dioxido- Method 7), m/z 376.0 2,3-dihydrothiophen-3- (M+H)+ (ESI+); RT 1.910 yl)-5-fluoro-2-hydroxy- min, (SFC Method 7); 3',4'-dimethyl-[1,1'- ee: 100.00% biphenyl]-4-carboxamide RT 0.501 min (LCMS (R or S)-N-(1,1-dioxido- Method 7), m/z 375.9 2,3-dihydrothiophen-3- (M+H)+ (ESI+); RT 2.430 yl)-5-fluoro-2-hydroxy- min, (SFC Method 7); 3',4'-dimethyl-[1,1'- ee: 100.00% biphenyl]-4-carboxamide
68 RT 0.552 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 372.1 dihydrothiophen-3-yl)-3- (M+H)+ (ESI+); RT 1.815 methoxy-3',4'-dimethyl- min, 2.733 min,(SFC [1,1'-biphenyl]-4- Method 24); racemate carboxamide RT 0.550 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 372.0 dihydrothiophen-3-yl)-3- (M+H)+ (ESI+); RT 1.387 methoxy-3',4'-dimethyl- min, 1.794 min, (SFC [1,1'-biphenyl]-4- Method 25); racemate carboxamide RT 0.521 min (LCMS 2-(3,4-dimethylphenyl)- Method 7), m/z 374.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.044 dihydrothiophen-3-yl)-4- min, 2.324 min ,(SFC methoxypyrimidine-5- Method 16); racemate carboxamide RT 0.498 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 374.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.069 dihydrothiophen-3-yl)-3- min, 2.300 min ,(SFC methoxypyrazine-2- Method 16); racemate carboxamide Cis or trans-6-(3,4- RT 0.433 min (LCMS dimethylphenyl)-N-(1- Method 7), m/z 372.2 (methylimino)-1-oxido- (M+H)+ (ESI+); RT 1.421 2,3-dihydro-1H-1 6- min, 1.461 min ,(SFC thiophen-3-yl)-2-oxo-1,2- Method 6); cis dihydropyridine-3- carboxamide Trans or cis-6-(3,4- RT 0.434 min (LCMS dimethylphenyl)-N-(1- Method 7), m/z 372.1 (methylimino)-1-oxido- (M+H)+ (ESI+); RT 1.566 2,3-dihydro-1H-1 6- min, 1.702 min ,(SFC thiophen-3-yl)-2-oxo-1,2- Method 6); trans dihydropyridine-3- carboxamide RT 0.432 min (LCMS (E)-6-(3,4- Method 7), m/z 370.1 dimethylphenyl)-3-(5-(2- (M+H)+ (ESI+); (methylsulfonyl)vinyl)- trans 1H-imidazol-2-yl)pyridin- 2-ol RT 0.497 min (LCMS (R)-5-(4- Method 7), m/z 371.1 cyclopropylphenyl)-2- (M+H)+ (ESI+); RT 2.234 ((1,1-dioxido-2,3- min, (SFC Method 10); dihydrothiophen-3- ee: 88.02% yl)carbamoyl)pyridine 1- oxide
RT 0.458 min (LCMS (R)-5-(benzofuran-5-yl)- Method 7), m/z 371.0 2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.713 dihydrothiophen-3- min, 1.045 min (SFC yl)carbamoyl)pyridine 1- Method 22) ee: 90.76% oxide RT 0.432 min (LCMS (R)-5-(2,3- Method 7), m/z 373.2 dihydrobenzofuran-5-yl)- (M+H)+ (ESI+); RT 0.747 2-((1,1-dioxido-2,3- min, 1.136 min (SFC dihydrothiophen-3- Method 22) ee: 88.80% yl)carbamoyl)pyridine 1- oxide 2-(6-(3,4- RT 0.619 min (LCMS dimethylphenyl)-2- Method 7, m/z 371.1 methoxypyridin-3-yl)-6- (M+H)+ (ESI+); thia-2-azaspiro[3.4]oct- 7-ene 6,6-dioxideO S O RT 0.519 min 7-(3,4-dimethylphenyl)- N O (LCMS Method 7), m/z 367.0 3-(1,1-dioxido-2,3- N (M+H)+ (ESI+); dihydrothiophen-3- yl)quinazolin-4(3H)-one 2-(3,4-dimethylphenyl)- RT 0.510 min (LCMS 6-(1,1-dioxido-2,3- Method 7), m/z 369.1 dihydrothiophen-3-yl)- (M+H)+ (ESI+); 7,8-dihydro-1,6- naphthyridin-5(6H)-one RT 0.543 min (LCMS Method 7), m/z 366.1 6-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.540 2-(1,1-dioxido-2,3- min, 2.136 min, (SFC dihydrothiophen-3- Method 9) racemate yl)isoquinolin-1(2H)-one O S O RT 0.555 min (LCMS 6-(3,4-dimethylphenyl)- N O Method 7), m/z 368.1 2-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.542 dihydrothiophen-3-yl)- min, 2.342 min, (SFC 3,4-dihydroisoquinolin- Method 9) racemate 1(2H)-one 7-(3,4-dimethylphenyl)- RT 0.495 min (LCMS 3-(1,1-dioxido-2,3- Method 7), m/z 368.0 dihydrothiophen-3- (M+H)+ (ESI+); yl)pyrido[2,3-d]pyrimidin- 4(3H)-one RT 0.399 min (LCMS 2-(6-(3,4- Method 7), m/z 370.2 dimethylphenyl)-2- (M+H)+ (ESI+); RT 1.890 hydroxypyridin-3-yl)-7- min, 2.129 min, (SFC thia-1,3- Method 9) racemate diazaspiro[4.4]nona-2,8- diene 7,7-dioxide
RT 0.392 min (LCMS (R)-2-(6-(3,4- Method 7), m/z 370.2 dimethylphenyl)-2- (M+H)+ (ESI+); RT 2.060 hydroxypyridin-3-yl)-7- min, (SFC Method 9) ee: thia-1,3- 100.00% diazaspiro[4.4]nona-2,8- diene 7,7-dioxide RT 0.396 min (LCMS (S)-2-(6-(3,4- Method 7), m/z 370.2 dimethylphenyl)-2- (M+H)+ (ESI+); RT 2.060 hydroxypyridin-3-yl)-7- min, (SFC Method 9) ee: thia-1,3- 97.03% diazaspiro[4.4]nona-2,8- diene 7,7-dioxide RT 0.506 min (LCMS (R,Z)-5-(3,4- Method 7), m/z 375.0 dimethylphenyl)-2-((4- (M+H)+ (ESI+); RT 1.540 (methylsulfonyl)but-3-en- min, (SFC Method 9) ee: 2-yl)carbamoyl)pyridine 86.68% 1-oxide RT 0.514 min (LCMS (R,E)-5-(3,4- Method 7), m/z 375.1 dimethylphenyl)-2-((4- (M+H)+ (ESI+); RT 1.806 (methylsulfonyl)but-3-en- min, (SFC Method 10) 2-yl)carbamoyl)pyridine ee: 97.79% 1-oxide RT 0.506 min (LCMS (S,E)-5-(3,4- Method 7), m/z 375.2 dimethylphenyl)-2-((4- (M+H)+ (ESI+); RT 1.534 (methylsulfonyl)but-3-en- min, (SFC Method 9) 2-yl)carbamoyl)pyridine ee: 90.33% 1-oxide 7-(5-(4- RT 0.522 min (LCMS (trifluoromethyl)phenyl)p Method 12, m/z 395.1 yridin-2-yl)-2-thia-7- (M+H)+ (ESI+); azaspiro[4.4]non-3-ene 2,2-dioxide RT 0.547 min (LCMS 7-(3,4-dimethylphenyl)- Method 7), m/z 385.3 3-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.728 dihydrothiophen-3-yl)-8- min, 1.956 min, (SFC fluoroquinazolin-4(3H)- Method 41 racemate one RT 0.609 min (LCMS 7-(6-(3,4- N S O Method 7), m/z 385.1 dimethylphenyl)-2- O (M+H)+ (ESI+); RT 2.117 methoxypyridin-3-yl)-2- N O min, 2.394 min, (SFC thia-7-azaspiro[4.4]non- Method 30 racemic 3-ene 2,2-dioxide RT 0.475 min (LCMS Method 7), m/z 388.1 (S)-4-(3-cyclopropyl-1H- (M+H)+ (ESI+); RT 1.882 pyrazol-1-yl)-N-(1,1- min, (SFC Method 12 dioxido-2,3- ee: 85.516% dihydrothiophen-3-yl)-2-
methoxy-6- methylbenzamide RT 0.469 min (LCMS (R)-4-(3-cyclopropyl-1H- Method 7), m/z 388.1 pyrazol-1-yl)-N-(1,1- (M+H)+ (ESI+); RT 2.246 dioxido-2,3- min, (SFC Method 13 dihydrothiophen-3-yl)-2- ee: 83.102% methoxy-6- methylbenzamide 7-(3,4-dimethylphenyl)- RT 0.502 min (LCMS 3-(1,1-dioxido-2,3- Method 7), m/z 384.1 dihydrothiophen-3- (M+H)+ (ESI+); yl)pyrido[2,3- d]pyrimidine-2,4(1H,3H)- dione O N 2-(6-(3,4- S O RT 0.454 m dimethylphenyl)-2- N in (LCMS Method 11), m/z methoxypyridin-3-yl)-7- H 384.3 (M+H)+ (ESI+); thia-1,3- diazaspiro[4.4]nona-2,8- diene 7,7-dioxide O N S O RT 0.632 min (LCMS (R)-2-(6-(3,4- Method 2), m/z 3 dimethylphenyl)-2- N 84.2 (M+H)+ (ESI+); RT 1 methoxypyridin-3-yl)-7- H .764 min, (SFC Method 10 thia-1,3- ee: 98.612% diazaspiro[4.4]nona-2,8- diene 7,7-dioxide O (S)-2-(6 S O RT 0.6 -(3,4- N 32 min (LCMS Me dimethylphenyl)-2- N thod 7), m/z 384.2 (M+ + + methoxypyridin-3-yl)-7- H H) (ESI); RT 1.340 min, (SFC Method 10 thia-1,3- ee: 100% diazaspiro[4.4]nona-2,8- diene 7,7-dioxide RT 0.437 min (LCMS (R)-7-(3,4- Method 6), m/z 382.1 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 1.761 dioxido-2,3- min, (SFC Method 9 ee: dihydrothiophen-3-yl)- 87.436% 1H-benzo[d]imidazole-4- carboxamide RT 0.459 min (LCMS N-(1-(cyanoimino)-1- Method 6), m/z 383.1 oxido-2,3-dihydro-1H- (M+H)+ (ESI+); RT 1.865 1 6-thiophen-3-yl)-6- min, 1.914 min, 2.129 (3,4-dimethylphenyl)-2- min, 2.169 min, (SFC oxo-1,2-dihydropyridine- Method 42 racemic 3-carboxamide RT 0.468 min (LCMS N-(1-(cyanoimino)-1- Method 7), m/z 383.1 oxido-4,5-dihydro-1H- (M+H)+ (ESI+); 1 6-thiophen-3-yl)-6- (3,4-dimethylphenyl)-2-
oxo-1,2-dihydropyridine- 3-carboxamide 3-(3,4-dimethylphenyl)- RT 0.487 min (LCMS 6-((1,1-dioxido-2,3- Method 7), m/z 389.0 dihydrothiophen-3- (M+H)+ (ESI+); yl)carbamoyl)-2- methoxypyridine 1-oxide 5-(3,4-dimethylphenyl)- RT 0.475 min (LCMS 2-((1,1-dioxido-2,3- Method 7), m/z 389.1 dihydrothiophen-3- (M+H)+ (ESI+); yl)carbamoyl)-4- methoxypyridine 1-oxide O O RT 0.441 min (LCMS 5-(3,4-dimethylphenyl)- S N O Method 7), m/z 389.1 2-((1,1-dioxido-2,3-N H (M+H)+ (ESI+); RT 2.036 dihydrothiophen-3- O min, 2.649 min, (SFC yl)carbamoyl)-3- Method 38); racemate methoxypyridine 1-oxide N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)-2- RT 0.433 min (LCMS oxo-6-(4- Method 7), m/z 406.0 (trifluoromethyl)piperidin- (M+H)+ (ESI+); 1-yl)-1,2- dihydropyridine-3- carboxamide RT 0.443 min (LCMS 2-((1,1-dioxido-2,3- Method 7), m/z 406.1 dihydrothiophen-3- (M+H)+ (ESI+); RT 1.109 yl)carbamoyl)-5-(4- min, 1.564 min, (SFC (trifluoromethyl)piperidin- Method 39); racemate 1-yl)pyridine 1-oxide RT 0.477 min (LCMS (R)-3-amino-2-((1,1- Method 7), m/z 420.0 dioxido-2,3- (M+H)+ (ESI+); RT 1.797 dihydrothiophen-3- min, 2.282min, (SFC yl)carbamoyl)-5-(5- Method 4); ee: 95.56% (trifluoromethyl)thiophen -3-yl)pyridine 1-oxide RT 0.482 min (LCMS (R)-3-amino-2-((1,1- Method 7), m/z 420.0 dioxido-2,3- (M+H)+ (ESI+); RT 1.874 dihydrothiophen-3- min 2.239 min, (SFC yl)carbamoyl)-5-(5- Method 6); ee: 93.88% (trifluoromethyl)thiophen -2-yl)pyridine 1-oxide RT 0.464 min (LCMS (R)-3-amino-2-((1,1- Method 7), m/z 419.9 dioxido-2,3- (M+H)+ (ESI+); RT 0.736 dihydrothiophen-3- min 1.667 min, (SFC yl)carbamoyl)-5-(4- Method 4); ee: 97.59% (trifluoromethyl)thiophen -2-yl)pyridine 1-oxide
RT 0.525 min (LCMS (R)-2-((1,1-dioxido-2,3- Method 7), m/z 413.2 dihydrothiophen-3- (M+H)+ (ESI+); RT 1.702 yl)carbamoyl)-5-(3- min, 1.780 min, (SFC methyl-4- Method 6); ee: 87.13% (trifluoromethyl)phenyl)p yridine 1-oxide RT 1.679 min (LCMS (S,Z)-N-(1-cyclopropyl-3- Method 10), m/z 401.1 (methylsulfonyl)allyl)-6- (M+H)+ (ESI+); RT 1.712 (3,4-dimethylphenyl)-2- min, 1.810 min, (SFC oxo-1,2-dihydropyridine- Method 11); ee: 81.26% 3-carboxamide RT 0.494 min (LCMS (S,E)-N-(1-cyclopropyl- Method 7), m/z 401.2 3-(methylsulfonyl)allyl)- (M+H)+ (ESI+); RT 1.939 6-(3,4-dimethylphenyl)- min, 2.107 min (SFC 2-oxo-1,2- Method 8); ee: 72.34% dihydropyridine-3- carboxamide RT 0.632 min (LCMS (S,Z)-2-((1- Method 9), m/z 401.1 cyclopropyl-3- (M+H)+ (ESI+); RT 2.270 (methylsulfonyl)allyl)carb min, 2.433 min (SFC amoyl)-5-(3,4- Method 8); ee: 50.83% dimethylphenyl)pyridine 1-oxide RT 0.505 min (LCMS (S,E)-2-((1-cyclopropyl- Method 8), m/z 401.2 3- (M+H)+ (ESI+); RT 2.232 (methylsulfonyl)allyl)carb min, 2.508 min (SFC amoyl)-5-(3,4- Method 8); ee: 52.57% dimethylphenyl)pyridine 1-oxide RT 0.522 min (LCMS (R)-5-(2-chloro-4- Method 7), m/z (trifluoromethyl)phenyl)- 433.1(M+H)+ (ESI+); RT 2-((1,1-dioxido-2,3- 1.118 min, RT 1.358 dihydrothiophen-3- min, (SFC Method 10); yl)carbamoyl)pyridine 1- ee: 88.36% oxide RT 0.503 min (LCMS O CF3 Method 7), m/z 6-(3,4-dimethylphenyl)- S O 413.1(M+H)+ (ESI+); 3-(1-((1,1-dioxido-2,3- HN N O RT H 1.457 min, 1.710min, dihydrothiophen-3- 1.941 min, 2.832 min yl)amino)-2,2,2- (SFC Method 14); trifluoroethyl)pyridin- racemate 2(1H)-one
74 7-(3,4-dimethylphenyl)- RT 0.487 min (LCMS 3-(1,1-dioxido-2,3- Method 8), m/z dihydrothiophen-3-yl)-8- 403.1(M+H)+ (ESI+); fluoro-2-hydroxy-2,3- dihydroquinazolin-4(1H)- one 8-(5-(4- RT 0.501 min (LCMS (trifluoromethyl)phenyl)p Method 7), m/z yridin-2-yl)-2-thia-8- 409.1(M+H)+ (ESI+); azaspiro[4.5]dec-3-ene 2,2-dioxide RT 0.548 min (LCMS (R)-5-(3,4- Method 7), m/z dimethylphenyl)-N-(1,1- 393.2(M+H)+ (ESI+); RT dioxido-2,3- 1.752 min, 2.032min, dihydrothiophen-3- (SFC Method 7); ee: yl)quinoline-8- 87.20% carboxamide RT 0.474 min (LCMS 2-(6-(3,4- Method 7), m/z dimethylphenyl)-2- 398.1(M+H)+ (ESI+); RT methoxypyridin-3-yl)-8- 1.599 min, 1.899min, thia-1,3- (SFC Method 26); diazaspiro[4.5]deca-2,6- racemate diene 8,8-dioxide 3-(6-(3,4- RT 0.516 min (LCMS dimethylphenyl)-2- Method 7), m/z hydroxypyridin-3-yl)-9- 398.3(M+H)+ (ESI+); thia-2,4- diazaspiro[5.5]undeca- 2,7-diene 9,9-dioxide RT 0.620 min (LCMS (R)-5-(3,4- Method 2), m/z 392.0 dimethylphenyl)-N-(1,1- (M-H)- (ESI-); RT 1.918 dioxido-2,3- min, RT 2.234 min, (SFC dihydrothiophen-3- Method 9); ee: 81.678% yl)quinazoline-8- carboxamide 3-(6-(3,4- RT 0.469 min (LCMS dimethylphenyl)-2- Method 7), m/z 412.1 methoxypyridin-3-yl)-9- (M+H)+ (ESI+) thia-2,4- diazaspiro[5.5]undeca- 2,7-diene 9,9-dioxide
RT 0.576 min (LCMS 3-(4-(6-cyclopentyl-2- Method 7), m/z 423.1 phenoxypyridin-3-yl)-1H- (M+H)+ (ESI+); RT 1.731 1,2,3-triazol-1-yl)-2,3- min, 1.872 min, (SFC dihydrothiophene 1,1- Method 30); racemate dioxide RT 0.429 min (LCMS 2-(2- Method 7), m/z 433.0 (cyclopropyldifluorometh (M+H)+ (ESI+); RT 1.964 yl)-4-phenoxypyrimidin- min, 2.272 min, (SFC 5-yl)-7-thia-1,3- Method 40); racemate diazaspiro[4.4]nona-2,8- diene 7,7-dioxide N S O (E)-2- N O RT (cyclopropyldifluorometh H 0.524 min (LCMS Method 7), m/z 433 yl)-5-(5-(2-N O .0 (M+H)+ (ESI+) (methylsulfonyl)vinyl)- 1H-imidazol-2-yl)-4- phenoxypyrimidine 3-(4-(6- RT 0.560 min (LCMS (isopropyl(methyl)amino) Method 7), m/z 426.1 -2-phenoxypyridin-3-yl)- (M+H)+ (ESI+) 1H-1,2,3-triazol-1-yl)- 2,3-dihydrothiophene 1,1-dioxide RT 0.538 min (LCMS (R)-5-(2- Method 7), m/z 436.1 (benzylamino)phenyl)-2- (M+H)+ (ESI+); RT 1.617 ((1,1-dioxido-2,3- min, (SFC Method 10); dihydrothiophen-3- ee: 88.98% yl)carbamoyl)pyridine 1- oxide
76 RT 0.496 min (LCMS 2-(6-(3,4- Method 7), m/z 446.2 dimethylphenyl)-2- (M+H)+ (ESI+); RT 2.234 phenoxypyridin-3-yl)-7- min, 2.464 min (SFC thia-1,3- Method 34); racemate diazaspiro[4.4]nona-2,8- diene 7,7-dioxide RT 0.498 min (LCMS (S)-2-(6-(3,4- Method 7), m/z 446.0 dimethylphenyl)-2- (M+H)+ (ESI+); RT 2.241 phenoxypyridin-3-yl)-7- min, 2.482 min (SFC thia-1,3- Method 34); ee: 61.74% diazaspiro[4.4]nona-2,8- diene 7,7-dioxide RT 0.521 min (LCMS (R)-5-(2-(benzyloxy)-3- Method 7), m/z 455.2 fluorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.261 dioxido-2,3- min, 1.439 min (SFC dihydrothiophen-3- Method 10); ee: 88.69% yl)carbamoyl)pyridine 1- oxide RT 0.517 min (LCMS (R)-5-(2-(benzyloxy)-4- Method 7), m/z 455.0 fluorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.569 dioxido-2,3- min, 1.675 min (SFC dihydrothiophen-3- Method 10); ee: 82.21% yl)carbamoyl)pyridine 1- oxide RT 0.508 min (LCMS (R)-5-(2-(benzyloxy)-5- Method 7), m/z 455.0 fluorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.543 dioxido-2,3- min, 1.735 min (SFC dihydrothiophen-3- Method 10); ee: 81.41% yl)carbamoyl)pyridine 1- oxide RT 0.509 min (LCMS (R)-5-(2-(benzyloxy)-6- Method 7), m/z 455.1 fluorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.451 dioxido-2,3- min, 1.608 min (SFC dihydrothiophen-3- Method 10); ee: 87.79% yl)carbamoyl)pyridine 1- oxide
77 RT 0.536 min (LCMS (R)-5-(2-(benzyloxy)-5- Method 7), m/z 451.2 methylphenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.720 dioxido-2,3- min, 1.826 min (SFC dihydrothiophen-3- Method 9); ee: 88.18% yl)carbamoyl)pyridine 1- oxide RT 0.526 min (LCMS (R)-5-(2-(benzyloxy)-3- Method 6), m/z 451.2 methylphenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.221 dioxido-2,3- min, 1.412 min (SFC dihydrothiophen-3- Method 10); ee: 87.70% yl)carbamoyl)pyridine 1- oxide RT 0.526 min (LCMS (R)-5-(2-(benzyloxy)-4- Method 7), m/z 451.3 methylphenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.708 dioxido-2,3- min, 1.837 min (SFC dihydrothiophen-3- Method 9); ee: 90.04% yl)carbamoyl)pyridine 1- oxide RT 0.635 min (LCMS (R)-5-(2-(benzyloxy)-6- Method 4), m/z 451.2 methylphenyl)-2-((1,1- (M+H)+ (ESI+); RT 2.072 dioxido-2,3- min, 2.213 min (SFC dihydrothiophen-3- Method 35); ee: 88.87% yl)carbamoyl)pyridine 1- oxide RT 0.510 min (LCMS (R)-2-((1,1-dioxido-2,3- Method 2), m/z 456.1 dihydrothiophen-3- (M+H)+ (ESI+); RT 1.654 yl)carbamoyl)-5-(5- min, 1.795 min (SFC fluoro-2-(pyridin-4- Method 35); ee: 88.87% ylmethoxy)phenyl)pyridin e 1-oxide 2-amino-3-(3,4- RT 0.461 min (LCMS dimethylphenyl)-6-((1,1- Method 7), m/z 374.3 dioxido-2,3- (M+H)+ (ESI+); RT 1.907 dihydrothiophen-3- min, 2.068 min (SFC yl)carbamoyl)pyridine 1- Method 36); racemate oxide 2,2,2- trifluoroacetate
(R)-3-((tert- RT 0.591 min (LCMS butoxycarbonyl)amino)- Method 7), m/z 520.1 2-((1,1-dioxido-2,3- 187 (M+H)+ (ESI+); RT 2.190 dihydrothiophen-3- min, 2.481 min (SFC yl)carbamoyl)-5-(5- Method 36); ee: 88.68% (trifluoromethyl)thiophen -2-yl)pyridine 1-oxide (R)-3-((tert- RT 0.577 min (LCMS butoxycarbonyl)amino)- Method 7), m/z 520.0 2-((1,1-dioxido-2,3- 188 (M+H)+ (ESI+); RT 1.298 dihydrothiophen-3- min, 1.895 min (SFC yl)carbamoyl)-5-(4- Method 4); ee: 86.19% (trifluoromethyl)thiophen -2-yl)pyridine 1-oxide (R)-3-((tert- RT 0.583min (LCMS butoxycarbonyl)amino)- Method 7), m/z 520.0 2-((1,1-dioxido-2,3- 189 (M+H)+ (ESI+); RT 1.569 dihydrothiophen-3- min, 2.148 min (SFC yl)carbamoyl)-5-(5- Method 37); ee: 94.65% (trifluoromethyl)thiophen -3-yl)pyridine 1-oxide Preparation of reference example 1 In the following, preparation of reference example 1 is described. This example is identical to example 87 reported in WO 2024/010782. Preparation of reference Intermediate 1.1 Ethyl 2-(1-ethoxyvinyl)-4-phenoxypyrimidine-5-carboxylate
The (E)-N-(1-cyclopropyl-3-(N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (400 mg, 0.888 mmol) was purified by preparative SFC (column: DAICEL CHIRALCEL OX(250mm*30mm,10 m); mobile phase: A for CO2, B for i-PrOH; Gradient elution: 30% B in CO2, 10 mins) (SFC Method 43) to give three product solutions The solution from peak 1 was concentrated under vacuum (30 °C) to give Fraction A (SFC: RT 2.170 min, 2.368 min (Method 11)) containing 2 products. The solution from peak 2 was concentrated under vacuum (30 °C) to give Example 33a: N-((S or R,E)-1-cyclopropyl-3-((R or S)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (85 mg, 0.184 mmol, 96.89% purity,) as colorless gum. LCMS: RT 0.570 min (method 10); m/z 451.1 (M+H)+ (ESI+); SFC: RT: 0.798 min (Method 14); 98.97% ee, 1H NMR (CDCl3, 400 MHz): 9.47 (s, 1H), 7.78 (d, J = 7.2 Hz, 1H), 7.57-7.49 (m, 2H), 7.41- 7.36 (m, 1H), 7.26-7.22 (m, 2H), 6.93 (dd, J = 4.8, 15.2 Hz, 1H), 6.42 (dd, J = 1.6, 15.2 Hz, 1H), 4.38-4.25 (m, 1H), 3.00 (s, 3H), 2.71 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.16 - 1.05 (m, 1H), 0.78 - 0.64 (m, 2H), 0.56 - 0.45 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F) The solution from peak 3 was concentrated under vacuum (30 °C) to give Example 33b: N-((S or R,E)-1-cyclopropyl-3-((S or R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (71 mg, 0.155 mmol, 98.73% purity) as a white solid.
LCMS: RT 0.570 min (method 10); m/z 451.1 (M+H)+ (ESI+); SFC: RT: 0.975 min (Method 14); 96.25% ee; 1H NMR (CDCl3, 400 MHz): 9.48 (s, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.54-7.48 (m, 2H), 7.42- 7.36 (m, 1H), 7.26-7.22 (m, 2H), 6.88 (dd, J = 5.2, 15.2 Hz, 1H), 6.41 (dd, J = 1.2, 15.2 Hz, 1H), 4.33-4.24 (m, 1H), 3.00 (s, 3H), 2.73 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.18 - 1.06 (m, 1H), 0.78 - 0.64 (m, 2H), 0.58 - 0.43 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F) Fraction A (SFC: RT 2.170 min, 2.368 min (Method 11)) (150 mg, 0.333 mmol) was further purified by preparative SFC (column: REGIS (s,s) WHELK-O1 (250mm*30mm,10 m); mobile phase: A for CO2, B for i-PrOH; Gradient elution: 35% B in CO2, 3.70 mins) to afford 2 solutions which were concentrated separately under vacuum (30 °C) to give: Example 33c: (SFC,) N-((R or S,E)-1-cyclopropyl-3-((R or S)-N,S-dimethylsulfonimidoyl)allyl)-2- (1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (58 mg, 0.129 mmol, 100% purity,) as a white solid.) LCMS: RT 0.567 min (method 10); m/z 451.1(M+H)+ (ESI+); SFC: RT: 1.127 min (Method 14); 94.68% ee; 1H NMR (CDCl3, 400 MHz): 9.48 (s, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.56-7.48 (m, 2H), 7.42- 7.35 (m, 1H), 7.26-7.23 (m, 2H), 6.88 (dd, J = 5.2, 15.2 Hz, 1H), 6.41 (d, J = 15.2 Hz, 1H), 4.34-4.23 (m, 1H), 2.99 (s, 3H), 2.73 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.17 - 1.05 (m, 1H), 0.78 - 0.65 (m, 2H), 0.57 - 0.44 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F) Example 33d: N-((R or S,E)-1-cyclopropyl-3-((S or R)-N,S-dimethylsulfonimidoyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (65 mg, 0.141 mmol, 98.53% purity) as a white solid. LCMS: RT 0.556 min (method 10); m/z 451.1 (M+H)+ (ESI+); SFC: RT: 1.334 min (Method 14), 97.42% ee; 1H NMR (CDCl3, 400 MHz): 9.47 (s, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.56-7.48 (m, 2H), 7.42- 7.35 (m, 1H), 7.26-7.21 (m, 2H), 6.93 (dd, J = 4.8, 15.2 Hz, 1H), 6.41 (d, J = 15.2 Hz, 1H), 4.36-4.27 (m, 1H), 3.00 (s, 3H), 2.73 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.17 - 1.04 (m, 1H), 0.80 - 0.63 (m, 2H), 0.57 - 0.45 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.84 (s, 2F) The following Table 1 provides an overview on the compounds described in the example section: Table 1 Example No. Structure Name of compound 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3- 1 dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide
6-(3,4-dimethylphenyl)-N-((1S or R,3 S)-1-imino-1- oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide o 6 x-( i3 d, o4 -- 2d ,i 3m -d e it hh yy dlp ro h -e 1n Hy -l) 1- lN 6--( th (1 io R ph o er n S -, 33 -y S l) ) -- 21 -- oim xo in -o 1- ,2 1 -- dihydropyridine-3-carboxamide 6-(3,4-dimethylphenyl)-N-((1S or R,3 R)-1-imino-1- oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide 6-(3,4-dimethylphenyl)-N-((1R or S,3 R)-1-imino-1- oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide 6-(3,4-dimethylphenyl)-N-(1-oxido-2,3- dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (R or S)-5-(3,4-dimethylphenyl)-2-((1,1-a dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide
(S or R)-5-(3,4-dimethylphenyl)-2-((1,1-b dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido- 2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)picolinamide 0 (R or S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)picolinamide 1 (S or R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)picolinamide 2 N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6- (piperidin-1-yl)-1,2-dihydropyridine-3-carboxamide 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-3 dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- sulfonamide 4 N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6- (prop-1-yn-1-yl)-1,2-dihydropyridine-3-carboxamide
dihyd 6 ro -( th 3, io 4 p-d hi em ne -t 3h -y yl lp )-h 2e -t n hy io l) x-N o-- 1(1 ,2 ,1 -d -d ih io yx di rd oo p- y2 r, i3 d- ine-3- carboxamide N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoro- 3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide 3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (R)-3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido- 2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)- 5-(4-(trifluoromethyl)phenyl)pyridine 1-oxide 4-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (R)-5-(2-(benzyloxy)phenyl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide ( y R l) ) c- a2 r- b(( a1 m,1 o- yd li )o -x 5i -d (5 o -- m2, e3 t- hd yih lt y hd io ro pt hh eio np -3 h -e yn l)- p3 y- ridine 1- oxide
2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)- 5-(spiro[3.3]hept-1-en-2-yl)pyridine 1-oxide 5-(2-chlorothiophen-3-yl)-2-((1,1-dioxido-2,3- dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)- 1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1- dioxide 3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)- 1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1- dioxide hydrochloride (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-a methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide b ( mS e o th r o R x) y- p3 y-( r4 id -( in 6 -- 3(3 -y ,4 l)- -d 1i Hm -e 1t ,h 2y ,3 lp -h tr e ia n zy ol) l-- 12 -- yl)-2,3- dihydrothiophene 1,1-dioxide R or S N N N S O (R or S)-3-(4-(6-(3,4-dimethylphenyl)-2-a O hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- N OH dihydrothiophene 1,1-dioxide hydrochloride HCl (S or R)-3-(4-(6-(3,4-dimethylphenyl)-2-b hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride
b N-((S or R, E)-1-cyclopropyl-3-((S or R)-S- methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide c N-((R or S, E)-1-cyclopropyl-3-((R or S)-S- methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide d N-((R or S, E)-1-cyclopropyl-3-((S or R)-S- methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide (E)-N-(1-cyclopropyl-3-(N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide a N-((S or R, E)-1-cyclopropyl-3-((R or S)-N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide b N di- m(( eS th o yr ls R u, lf E o) n- i1 m-c id y oc ylo l) p ar lo ly p l) y -l 2-3 -(- 1(( ,S 1- o dr ifl R uo )- rN o, eS th- yl)-4- phenoxypyrimidine-5-carboxamide
33c N-((R or S, E)-1-cyclopropyl-3-((R or S)-N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide 33d N-((R or S, E)-1-cyclopropyl-3-((S or R)-N,S- dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide The following Table 2 provides an overview on the compounds which were obtained following similar procedures as the ones described in the above example section or using procedures well known by someone skilled in the art. The skilled person is capable of obtaining any of the following compounds, by adapting and/or following the teaching provided herein for the synthesis of any one of the preceding compounds. Table 2 L + Example Chemical CMS data (RT + MH) number structure and SFC data (RT + Chemical name ee%) N-(1,1-dioxido-2,3- RT 0.337 min (LCMS dihydrothiophen-3-yl)-6- 34 Method 7), m/z 278.9 ethynyl-2-oxo-1,2- (M+H)+ (ESI+) dihydropyridine-3- carboxamide O O O N-(1,1-dioxido-2,3- S H O RT 0.366 min (LCMS dihydrothiophen-3-yl)-2- 35 N N H Method 7), m/z 324.1 oxo-6-(pyrrolidin-1-yl)- N (M+H)+ (ESI+) 1,2-dihydropyridine-3- carboxamide
59 N-(1,1-dioxido-2,3- RT 0.443 min (LCMS dihydrothiophen-3-yl)-6- Method 7), m/z 321.1 (3-methylbut-1-yn-1-yl)- (M+H)+ (ESI+) 2-oxo-1,2- dihydropyridine-3- carboxamide RT 0.465 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 335.0 dihydrothiophen-3-yl)-6- (M+H)+ (ESI+); RT 1.535 (3-methylbut-1-yn-1-yl)- min, 2.554 min 2-oxo-1,2- (SFCMethod 14); dihydropyridine-3- racemate carboxamide RT 0.540 min (LCMS Method 7), m/z 333.2 (R)-N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.692 dihydrothiophen-3-yl)-5- min (SFC Method 6); (spiro[3.3]heptan-2- ee: 88.20% yl)picolinamide RT 0.433 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 333.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.093 dihydrothiophen-3-yl)- min, 2.420 min SFC 1H-1,2,4-triazole-3- Method 24); racemate carboxamide RT 0.427 min (LCMS (R)-5-(3,4- Method 7), m/z 333.2 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 1.552 dioxido-2,3- min, (SFC Method 26); dihydrothiophen-3-yl)- ee: 90.09% 4H-1,2,4-triazole-3- carboxamide RT 0.443 min (LCMS (R)-2-((1,1-dioxido-2,3- Method 7), m/z 351.0 dihydrothiophen-3- (M+H)+ (ESI+); RT 2.239 yl)carbamoyl)-5-(5- min, (SFC Method 6); methylthiophen-2- ee: 84.88% yl)pyridine 1-oxide RT 0.455 min (LCMS (R)-5-(cyclohex-1-en-1- Method 7), m/z 335.1 yl)-2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.724 dihydrothiophen-3- min, ( SFC Method 17); yl)carbamoyl)pyridine 1- ee: 89.64% oxide RT 0.460 min (LCMS (R)-5-cyclohexyl-2-((1,1- Method 7), m/z 337.1 dioxido-2,3- (M+H)+ (ESI+); RT 1.449 dihydrothiophen-3- min, (SFC Method 7); yl)carbamoyl)pyridine 1- ee: 89.62% oxide
RT 0.364 min (LCMS Method 7), m/z 339.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.468 dihydrothiophen-3-yl)-2- min, 1.551 min, 1.641 oxo-6-(tetrahydro-2H- min, 1.733 min, pyran-3-yl)-1,2- (SFC Method 9); dihydropyridine-3- racemate carboxamide RT 0.402 min (LCMS Method 7), m/z 337.1 6-(3,4-dihydro-2H-pyran- (M+H)+ (ESI+); RT 1.718 6-yl)-N-(1,1-dioxido-2,3- min, 4.342 min, dihydrothiophen-3-yl)-2- (SFCMethod 18); oxo-1,2-dihydropyridine- racemate 3-carboxamide RT 0.394 min (LCMS Method 7), m/z 339.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT dihydrothiophen-3-yl)-2- 2.965 min, 3.441 min, oxo-6-(tetrahydro-2H- 9.181 min, 13.749 min, pyran-2-yl)-1,2- (SFC Method 32); dihydropyridine-3- racemate carboxamide 6-(3,6-dihydro-2H-pyran- RT 0.359 min (LCMS 4-yl)-N-(1,1-dioxido-2,3- Method 7), m/z 337.1 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); oxo-1,2-dihydropyridine- 3-carboxamide RT 0.321 min (LCMS (R)-N-(1,1-dioxido-2,3- Method 2), m/z 339.1 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 1.629 oxo-6-(tetrahydro-2H- min, (SFC Method 9 ); pyran-4-yl)-1,2- ee: 90.03% dihydropyridine-3- carboxamide O O S O 6-(5,6-dihydro-2H-pyran- HN N O RT 0.374 min (LCMS 3-yl)-N-(1,1-dioxido-2,3- H Method 7), m/z 337.0 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); oxo-1,2-dihydropyridine- 3-carboxamide RT 0.527 min (LCMS (R)-5-(3,4- Method 7), m/z 349.0 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 1.766 dioxido-2,3- min, (SFC Method 9); dihydrothiophen-3- ee: 91.02% yl)thiazole-2- carboxamide RT 0.471 min (LCMS Method 7), m/z 332.0 (R)-N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.437 dihydrothiophen-3-yl)-2- min, (SFC Method 6); oxo-6-phenyl-2H-pyran- ee: 93.30% 3-carboxamide
61 RT 0.411 min (LCMS Method 7), m/z 338.3 2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.393 dihydrothiophen-3- min, 1.914 min, (SFC yl)carbamoyl)-5- Method 13); (piperidin-1-yl)pyridine 1- racemate oxide RT 0.492 min (LCMS (R)-5-(3,4- Method 7), m/z 350.0 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 2.024 dioxido-2,3- min, (SFC Method 9), dihydrothiophen-3-yl)- ee: 90.65% 1,3,4-thiadiazole-2- carboxamide RT 0.476 min (LCMS (R)-5-(3,4- Method 7), m/z 332.2 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 2.718 dioxido-2,3- min, (SFC Method 26), dihydrothiophen-3-yl)- ee: 88.36% 1H-imidazole-2- carboxamide RT 0.405 min (LCMS (S)-N-(1,1-dioxido-2,3- Method 7), m/z 332.3 dihydrothiophen-3-yl)-1- (M+H)+ (ESI+); RT 1.445 methyl-2-(p-tolyl)-1H- min, (SFC Method 7); imidazole-4- ee: 84.59% carboxamide RT 0.395 min (LCMS (R)-N-(1,1-dioxido-2,3- Method 7), m/z 332.1 dihydrothiophen-3-yl)-1- (M+H)+ (ESI+); RT 1.633 methyl-2-(p-tolyl)-1H- min, (SFC Method 7); imidazole-4- ee: 83.87% carboxamide RT 0.404 min (LCMS 2-(3,4-dimethylphenyl)- Method 7), m/z 332.0 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.073 dihydrothiophen-3-yl)- min, 2.604 min, (SFC 1H-imidazole-4- Method 12); racemate carboxamide RT 0.468 min (LCMS (R)-5-(4-chlorophenyl)-2- Method 7), m/z 365.0 ((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.060 dihydrothiophen-3- min, (SFC Method 10); yl)carbamoyl)pyridine 1- ee: 88.14% oxide RT 0.439 min (LCMS (R)-N-(1,1-dioxido-2,3- Method 7), m/z 353.1 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 1.721 methoxy-6-(tetrahydro- min, (SFC Method 9); 2H-pyran-4- ee: 90.63% yl)nicotinamide
62 RT 0.442 min (LCMS 6-(3,4-dimethylphenyl)- Method 2), m/z 343.1 N-((3R)-1-oxido-2,3- (M+H)+ (ESI+); RT 1.407 dihydrothiophen-3-yl)-2- min, 2.758 min (SFC oxo-1,2-dihydropyridine- Method 33); racemate 3-carboxamide RT 0.480 min (LCMS 6-(3,4-dimethylphenyl)- Method 7), m/z 343.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); dihydrothiophen-3- yl)nicotinamide RT 0.605 min (LCMS Method 2), m/z 342.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.169 dihydrothiophen-3-yl)- min, 2.269 min (SFC 3',4'-dimethyl-[1,1'- Method 16); racemate biphenyl]-4-carboxamide RT 0.533 min (LCMS (R)-5-(3,4- Method 7), m/z 366.0 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 1.494 dioxido-2,3- min, (SFC Method 6) ee: dihydrothiophen-3-yl)-3- 93.06% fluorothiophene-2- carboxamide RT 0.511 min (LCMS 2-(3,4-dimethylphenyl)- Method 7), m/z 344.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); dihydrothiophen-3- yl)pyrimidine-5- carboxamide 5-(3,4-dimethylphenyl)- RT 0.471 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 344.1 dihydrothiophen-3- (M+H)+ (ESI+); yl)pyrimidine-2- carboxamide RT 0.486 min (LCMS Method 7), m/z 344.1 6-(3,4-dimethylphenyl)- (M+H)+ (ESI+); N-(1,1-dioxido-2,3- RT 1.624 min, 1.965 min, dihydrothiophen-3- (SFC Method 9); yl)pyridazine-3- racemate carboxamide RT 0.498 min (LCMS 2-((1,1-dioxido-2,3- Method 7), m/z 349.1 dihydrothiophen-3- (M+H)+ (ESI+); RT 0.797 yl)carbamoyl)-5- min, 1.892 min (SFC (spiro[3.3]heptan-2- Method 5) racemate yl)pyridine 1-oxide RT 0.478 min (LCMS 6-(3,4-dimethylphenyl)- Method 7), m/z 357.1 3-(6,6-dioxido-6-thia-2- (M+H)+ (ESI+); azaspiro[3.4]oct-7-en-2- yl)pyridin-2(1H)-one
63 RT 0.953 min (LCMS 3-((6-(3,4- Method 1), m/z 355.0 dimethylphenyl)-1H- (M+H)+ (ESI+); RT 1.614 indazol-3-yl)oxy)-2,3- min, 1.752 min (SFC dihydrothiophene 1,1- Method 15); racemate dioxide RT 0.914 min (LCMS 6-(3,4-dimethylphenyl)- Method 1), m/z 355.0 1-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.944 dihydrothiophen-3-yl)- min, 2.103 min (SFC 1,2-dihydro-3H-indazol- Method 16); racemate 3-one RT 0.542 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 354.1 2-(1,1-dioxido-2,3- (M+H)+ (ESI+); racemate dihydrothiophen-3- yl)isoindolin-1-one RT 0.465 min (LCMS (R)-N-(1,1-dioxido-2,3- Method 7), m/z 355.0 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 2.481 oxo-6-(phenylethynyl)- min (SFC Method 5); ee: 1,2-dihydropyridine-3- 90.45% carboxamide RT 0.446 min (LCMS Method 7), m/z 359.1 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 0.754 N-(1,1-dioxido-2,3- min, 1.727 min (SFC dihydrothiophen-3-yl)-4- Method 17); racemate hydroxypicolinamide RT 0.569 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 359.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); racemate dihydrothiophen-3-yl)-3- hydroxypicolinamide RT 0.423 min (LCMS 6-(3,4-dimethylphenyl)- Method 7), m/z 359.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.811 dihydrothiophen-3-yl)-4- min, 1.467 min (SFC oxo-1,4-dihydropyridine- Method 18); racemate 3-carboxamide RT 0.531 min (LCMS (R)-5-(2,4- Method 2), m/z 359.1 dimethylphenyl)-2-((1,1- (M+H)+ (ESI+); RT 2.003 dioxido-2,3- min (SFC Method 16); dihydrothiophen-3- ee: 88.35% yl)carbamoyl)pyridine 1- oxide
64 RT 0.491 min (LCMS (R)-5-(3,4- Method 7), m/z 399.0 dichlorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 2.171 dioxido-2,3- min (SFC Method 10); dihydrothiophen-3- ee: 96.65% yl)carbamoyl)pyridine 1- oxide RT 0.443 min (LCMS (R)-5-(3,4- Method 7), m/z 367.0 difluorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.661 dioxido-2,3- min (SFC Method 10); dihydrothiophen-3- ee: 87.27% yl)carbamoyl)pyridine 1- oxide RT 0.431 min (LCMS (R)-2-((1,1-dioxido-2,3- Method 7), m/z 361.1 dihydrothiophen-3- (M+H)+ (ESI+); RT 2.385 yl)carbamoyl)-5-(4- min (SFC Method 20); methoxyphenyl)pyridine ee: 91.51% 1-oxide RT 0.530 min (LCMS Method 7), m/z 361.2 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.569 N-(1,1-dioxido-2,3- min, 1.972 min (SFC dihydrothiophen-3-yl)-3- Method 9); racemate fluoropicolinamide RT 0.525 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 361.0 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); racemate dihydrothiophen-3-yl)-6- fluoropicolinamide RT 0.442 min (LCMS Method 7), m/z 357.1 6-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 0.731 N-(1-methyl-1-oxido-2,3- min, 1.074 min, 1.375 dihydrophosphol-3-yl)-2- min, 1.869 min (SFC oxo-1,2-dihydropyridine- Method 19); racemate 3-carboxamide RT 0.593 min (LCMS 3-amino-N-(1,1-dioxido- Method 2), m/z 357.2 2,3-dihydrothiophen-3- (M+H)+ (ESI+) yl)-3',4'-dimethyl-[1,1'- biphenyl]-4-carboxamide
65 RT 0.526 min (LCMS Method 7), m/z 357.2 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.333 N-(1,1-dioxido-2,3- min, 1.544 min (SFC dihydrothiophen-3-yl)-4- Method 9); racemate methylpicolinamide RT 0.528 min (LCMS Method 7), m/z 357.1 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.259 N-(1,1-dioxido-2,3- min, 1.462 min (SFC dihydrothiophen-3-yl)-6- Method 9); racemate methylpicolinamide RT 0.543 min (LCMS Method 7), m/z 357.1 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.435 N-(1,1-dioxido-2,3- min, 1.557 min, (SFC dihydrothiophen-3-yl)-3- Method 9); racemate methylpicolinamide RT 0.472 min (LCMS Method 7), m/z 357.3 2-amino-N-(1,1-dioxido- (M+H)+ (ESI+); RT 2.229 2,3-dihydrothiophen-3- min, 2.556 min, (SFC yl)-3',4'-dimethyl-[1,1'- Method 26); racemate biphenyl]-4-carboxamide RT 0.520 min (LCMS (S)-N-(1,1-dioxido-2,3- Method 7), m/z 389.0 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 1.915 (4- min, (SFC Method 26); (trifluoromethyl)phenyl)th ee: 84.50% iazole-4-carboxamide RT 0.521 min (LCMS (R)-N-(1,1-dioxido-2,3- Method 7), m/z 389.0 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 2.272 (4- min, (SFC Method 26); (trifluoromethyl)phenyl)th ee: 83.28% iazole-4-carboxamide RT 0.550 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 358.1 dihydrothiophen-3-yl)-3- (M+H)+ (ESI+); RT 2.303 hydroxy-3',4'-dimethyl- min, 2.711 min (SFC [1,1'-biphenyl]-4- Method 16); racemate carboxamide RT 0.538 min (LCMS Method 7), m/z 376.0 3-chloro-N-(1,1-dioxido- (M+H)+ (ESI+); RT 1.365 2,3-dihydrothiophen-3- min, 1.563 min (SFC yl)-3',4'-dimethyl-[1,1'- Method 9); racemate biphenyl]-4-carboxamide RT 0.525 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 360.2 dihydrothiophen-3-yl)-2- (M+H)+ (ESI+); RT 1.136 fluoro-3',4'-dimethyl- min, 1.282 min (SFC [1,1'-biphenyl]-4- Method 9); racemate carboxamide
RT 0.532 min (LCMS Method 7), m/z 356.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.182 dihydrothiophen-3-yl)- min, 1.595 min (SFC 2,3',4'-trimethyl-[1,1'- Method 27); racemate biphenyl]-4-carboxamide RT 0.438 min (LCMS 2-(3,4-dimethylphenyl)- Method 7), m/z 360.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.857 dihydrothiophen-3-yl)-6- min, 1.109 min (SFC oxo-1,6- Method 28); racemate dihydropyrimidine-5- carboxamide RT 0.492 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 360.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.596 dihydrothiophen-3-yl)-3- min, 1.894 min (SFC oxo-3,4-dihydropyrazine- Method 25); racemate 2-carboxamide 2-amino-6-(3,4- H RT 0.540 min (LCMS dimethylphenyl)-N-(1,1-N N Method 7), m/z 358.2 dioxido-2,3- O NH2 (M+H)+ (ESI+); dihydrothiophen-3- yl)nicotinamide RT 0.493 min (LCMS (R)-2-((1,1-dioxido-2,3- Method 2), m/z 385.2 dihydrothiophen-3- (M+H)+ (ESI+); RT 2.133 yl)carbamoyl)-5-(2,4,6- min, (SFC Method 30); trifluorophenyl)pyridine ee: 87.55% 1-oxide 3-(3,4-dimethylphenyl)- RT 0.488 min (LCMS 6-((1,1-dioxido-2,3- Method 7), m/z 377.1 dihydrothiophen-3- (M+H)+ (ESI+); yl)carbamoyl)-2- fluoropyridine 1-oxide RT 0.492 min (LCMS 5-(3,4-dimethylphenyl)- Method 2), m/z 377.1 2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.538 dihydrothiophen-3- min, 1.772 min (SFC yl)carbamoyl)-4- Method 4); racemate fluoropyridine 1-oxide RT 0.556 min (LCMS 6-(3,4-dimethylphenyl)- Method 7), m/z 373.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); dihydrothiophen-3-yl)-2- methoxynicotinamide
67 RT 0.503 min (LCMS Method 7), m/z 373.1 5-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.892 N-(1,1-dioxido-2,3- min, 2.495 min (SFC dihydrothiophen-3-yl)-3- Method 9); racemate methoxypicolinamide RT 0.444 min (LCMS 6-(3,4-dimethylphenyl)- Method 7), m/z 373.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.370 dihydrothiophen-3-yl)-1- min, 1.462 min,(SFC methyl-4-oxo-1,4- Method 20); racemate dihydropyridine-3- carboxamide RT 0.514 min (LCMS 3-(3,4-dimethylphenyl)- Method 7), m/z 373.1 6-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.617 dihydrothiophen-3- min, 1.777 min,(SFC yl)carbamoyl)-2- Method 4); racemate methylpyridine 1-oxide RT 0.457 min (LCMS 3-(3,4-dimethylphenyl)- Method 7), m/z 375.1 6-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.610 dihydrothiophen-3- min, 0.799 min,(SFC yl)carbamoyl)-2- Method 21); racemate hydroxypyridine 1-oxide RT 0.441 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 375.1 2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.723 dihydrothiophen-3- min, 1.485 min,(SFC yl)carbamoyl)-4- Method 22); racemate hydroxypyridine 1-oxide RT 0.521 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 375.1 2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.128 dihydrothiophen-3- min, 2.774 min,(SFC yl)carbamoyl)-3- Method 23); racemate hydroxypyridine 1-oxide RT 0.508 min (LCMS (R)-5-chloro-N-(1,1- Method 7), m/z 392.1 dioxido-2,3- (M+H)+ (ESI+); RT 2.262 dihydrothiophen-3-yl)-2- min, (SFC Method 6); hydroxy-3',4'-dimethyl- ee: 89.35% [1,1'-biphenyl]-4- carboxamide RT 0.505 min (LCMS (S or R)-N-(1,1-dioxido- Method 7), m/z 376.0 2,3-dihydrothiophen-3- (M+H)+ (ESI+); RT 1.910 yl)-5-fluoro-2-hydroxy- min, (SFC Method 7); 3',4'-dimethyl-[1,1'- ee: 100.00% biphenyl]-4-carboxamide RT 0.501 min (LCMS (R or S)-N-(1,1-dioxido- Method 7), m/z 375.9 2,3-dihydrothiophen-3- (M+H)+ (ESI+); RT 2.430 yl)-5-fluoro-2-hydroxy- min, (SFC Method 7); 3',4'-dimethyl-[1,1'- ee: 100.00% biphenyl]-4-carboxamide
68 RT 0.552 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 372.1 dihydrothiophen-3-yl)-3- (M+H)+ (ESI+); RT 1.815 methoxy-3',4'-dimethyl- min, 2.733 min,(SFC [1,1'-biphenyl]-4- Method 24); racemate carboxamide RT 0.550 min (LCMS N-(1,1-dioxido-2,3- Method 7), m/z 372.0 dihydrothiophen-3-yl)-3- (M+H)+ (ESI+); RT 1.387 methoxy-3',4'-dimethyl- min, 1.794 min, (SFC [1,1'-biphenyl]-4- Method 25); racemate carboxamide RT 0.521 min (LCMS 2-(3,4-dimethylphenyl)- Method 7), m/z 374.2 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.044 dihydrothiophen-3-yl)-4- min, 2.324 min ,(SFC methoxypyrimidine-5- Method 16); racemate carboxamide RT 0.498 min (LCMS 5-(3,4-dimethylphenyl)- Method 7), m/z 374.1 N-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 2.069 dihydrothiophen-3-yl)-3- min, 2.300 min ,(SFC methoxypyrazine-2- Method 16); racemate carboxamide Cis or trans-6-(3,4- RT 0.433 min (LCMS dimethylphenyl)-N-(1- Method 7), m/z 372.2 (methylimino)-1-oxido- (M+H)+ (ESI+); RT 1.421 2,3-dihydro-1H-1 6- min, 1.461 min ,(SFC thiophen-3-yl)-2-oxo-1,2- Method 6); cis dihydropyridine-3- carboxamide Trans or cis-6-(3,4- RT 0.434 min (LCMS dimethylphenyl)-N-(1- Method 7), m/z 372.1 (methylimino)-1-oxido- (M+H)+ (ESI+); RT 1.566 2,3-dihydro-1H-1 6- min, 1.702 min ,(SFC thiophen-3-yl)-2-oxo-1,2- Method 6); trans dihydropyridine-3- carboxamide RT 0.432 min (LCMS (E)-6-(3,4- Method 7), m/z 370.1 dimethylphenyl)-3-(5-(2- (M+H)+ (ESI+); (methylsulfonyl)vinyl)- trans 1H-imidazol-2-yl)pyridin- 2-ol RT 0.497 min (LCMS (R)-5-(4- Method 7), m/z 371.1 cyclopropylphenyl)-2- (M+H)+ (ESI+); RT 2.234 ((1,1-dioxido-2,3- min, (SFC Method 10); dihydrothiophen-3- ee: 88.02% yl)carbamoyl)pyridine 1- oxide
RT 0.458 min (LCMS (R)-5-(benzofuran-5-yl)- Method 7), m/z 371.0 2-((1,1-dioxido-2,3- (M+H)+ (ESI+); RT 0.713 dihydrothiophen-3- min, 1.045 min (SFC yl)carbamoyl)pyridine 1- Method 22) ee: 90.76% oxide RT 0.432 min (LCMS (R)-5-(2,3- Method 7), m/z 373.2 dihydrobenzofuran-5-yl)- (M+H)+ (ESI+); RT 0.747 2-((1,1-dioxido-2,3- min, 1.136 min (SFC dihydrothiophen-3- Method 22) ee: 88.80% yl)carbamoyl)pyridine 1- oxide 2-(6-(3,4- RT 0.619 min (LCMS dimethylphenyl)-2- Method 7, m/z 371.1 methoxypyridin-3-yl)-6- (M+H)+ (ESI+); thia-2-azaspiro[3.4]oct- 7-ene 6,6-dioxideO S O RT 0.519 min 7-(3,4-dimethylphenyl)- N O (LCMS Method 7), m/z 367.0 3-(1,1-dioxido-2,3- N (M+H)+ (ESI+); dihydrothiophen-3- yl)quinazolin-4(3H)-one 2-(3,4-dimethylphenyl)- RT 0.510 min (LCMS 6-(1,1-dioxido-2,3- Method 7), m/z 369.1 dihydrothiophen-3-yl)- (M+H)+ (ESI+); 7,8-dihydro-1,6- naphthyridin-5(6H)-one RT 0.543 min (LCMS Method 7), m/z 366.1 6-(3,4-dimethylphenyl)- (M+H)+ (ESI+); RT 1.540 2-(1,1-dioxido-2,3- min, 2.136 min, (SFC dihydrothiophen-3- Method 9) racemate yl)isoquinolin-1(2H)-one O S O RT 0.555 min (LCMS 6-(3,4-dimethylphenyl)- N O Method 7), m/z 368.1 2-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.542 dihydrothiophen-3-yl)- min, 2.342 min, (SFC 3,4-dihydroisoquinolin- Method 9) racemate 1(2H)-one 7-(3,4-dimethylphenyl)- RT 0.495 min (LCMS 3-(1,1-dioxido-2,3- Method 7), m/z 368.0 dihydrothiophen-3- (M+H)+ (ESI+); yl)pyrido[2,3-d]pyrimidin- 4(3H)-one RT 0.399 min (LCMS 2-(6-(3,4- Method 7), m/z 370.2 dimethylphenyl)-2- (M+H)+ (ESI+); RT 1.890 hydroxypyridin-3-yl)-7- min, 2.129 min, (SFC thia-1,3- Method 9) racemate diazaspiro[4.4]nona-2,8- diene 7,7-dioxide
RT 0.392 min (LCMS (R)-2-(6-(3,4- Method 7), m/z 370.2 dimethylphenyl)-2- (M+H)+ (ESI+); RT 2.060 hydroxypyridin-3-yl)-7- min, (SFC Method 9) ee: thia-1,3- 100.00% diazaspiro[4.4]nona-2,8- diene 7,7-dioxide RT 0.396 min (LCMS (S)-2-(6-(3,4- Method 7), m/z 370.2 dimethylphenyl)-2- (M+H)+ (ESI+); RT 2.060 hydroxypyridin-3-yl)-7- min, (SFC Method 9) ee: thia-1,3- 97.03% diazaspiro[4.4]nona-2,8- diene 7,7-dioxide RT 0.506 min (LCMS (R,Z)-5-(3,4- Method 7), m/z 375.0 dimethylphenyl)-2-((4- (M+H)+ (ESI+); RT 1.540 (methylsulfonyl)but-3-en- min, (SFC Method 9) ee: 2-yl)carbamoyl)pyridine 86.68% 1-oxide RT 0.514 min (LCMS (R,E)-5-(3,4- Method 7), m/z 375.1 dimethylphenyl)-2-((4- (M+H)+ (ESI+); RT 1.806 (methylsulfonyl)but-3-en- min, (SFC Method 10) 2-yl)carbamoyl)pyridine ee: 97.79% 1-oxide RT 0.506 min (LCMS (S,E)-5-(3,4- Method 7), m/z 375.2 dimethylphenyl)-2-((4- (M+H)+ (ESI+); RT 1.534 (methylsulfonyl)but-3-en- min, (SFC Method 9) 2-yl)carbamoyl)pyridine ee: 90.33% 1-oxide 7-(5-(4- RT 0.522 min (LCMS (trifluoromethyl)phenyl)p Method 12, m/z 395.1 yridin-2-yl)-2-thia-7- (M+H)+ (ESI+); azaspiro[4.4]non-3-ene 2,2-dioxide RT 0.547 min (LCMS 7-(3,4-dimethylphenyl)- Method 7), m/z 385.3 3-(1,1-dioxido-2,3- (M+H)+ (ESI+); RT 1.728 dihydrothiophen-3-yl)-8- min, 1.956 min, (SFC fluoroquinazolin-4(3H)- Method 41 racemate one RT 0.609 min (LCMS 7-(6-(3,4- N S O Method 7), m/z 385.1 dimethylphenyl)-2- O (M+H)+ (ESI+); RT 2.117 methoxypyridin-3-yl)-2- N O min, 2.394 min, (SFC thia-7-azaspiro[4.4]non- Method 30 racemic 3-ene 2,2-dioxide RT 0.475 min (LCMS Method 7), m/z 388.1 (S)-4-(3-cyclopropyl-1H- (M+H)+ (ESI+); RT 1.882 pyrazol-1-yl)-N-(1,1- min, (SFC Method 12 dioxido-2,3- ee: 85.516% dihydrothiophen-3-yl)-2-
methoxy-6- methylbenzamide RT 0.469 min (LCMS (R)-4-(3-cyclopropyl-1H- Method 7), m/z 388.1 pyrazol-1-yl)-N-(1,1- (M+H)+ (ESI+); RT 2.246 dioxido-2,3- min, (SFC Method 13 dihydrothiophen-3-yl)-2- ee: 83.102% methoxy-6- methylbenzamide 7-(3,4-dimethylphenyl)- RT 0.502 min (LCMS 3-(1,1-dioxido-2,3- Method 7), m/z 384.1 dihydrothiophen-3- (M+H)+ (ESI+); yl)pyrido[2,3- d]pyrimidine-2,4(1H,3H)- dione O N 2-(6-(3,4- S O RT 0.454 m dimethylphenyl)-2- N in (LCMS Method 11), m/z methoxypyridin-3-yl)-7- H 384.3 (M+H)+ (ESI+); thia-1,3- diazaspiro[4.4]nona-2,8- diene 7,7-dioxide O N S O RT 0.632 min (LCMS (R)-2-(6-(3,4- Method 2), m/z 3 dimethylphenyl)-2- N 84.2 (M+H)+ (ESI+); RT 1 methoxypyridin-3-yl)-7- H .764 min, (SFC Method 10 thia-1,3- ee: 98.612% diazaspiro[4.4]nona-2,8- diene 7,7-dioxide O (S)-2-(6 S O RT 0.6 -(3,4- N 32 min (LCMS Me dimethylphenyl)-2- N thod 7), m/z 384.2 (M+ + + methoxypyridin-3-yl)-7- H H) (ESI); RT 1.340 min, (SFC Method 10 thia-1,3- ee: 100% diazaspiro[4.4]nona-2,8- diene 7,7-dioxide RT 0.437 min (LCMS (R)-7-(3,4- Method 6), m/z 382.1 dimethylphenyl)-N-(1,1- (M+H)+ (ESI+); RT 1.761 dioxido-2,3- min, (SFC Method 9 ee: dihydrothiophen-3-yl)- 87.436% 1H-benzo[d]imidazole-4- carboxamide RT 0.459 min (LCMS N-(1-(cyanoimino)-1- Method 6), m/z 383.1 oxido-2,3-dihydro-1H- (M+H)+ (ESI+); RT 1.865 1 6-thiophen-3-yl)-6- min, 1.914 min, 2.129 (3,4-dimethylphenyl)-2- min, 2.169 min, (SFC oxo-1,2-dihydropyridine- Method 42 racemic 3-carboxamide RT 0.468 min (LCMS N-(1-(cyanoimino)-1- Method 7), m/z 383.1 oxido-4,5-dihydro-1H- (M+H)+ (ESI+); 1 6-thiophen-3-yl)-6- (3,4-dimethylphenyl)-2-
oxo-1,2-dihydropyridine- 3-carboxamide 3-(3,4-dimethylphenyl)- RT 0.487 min (LCMS 6-((1,1-dioxido-2,3- Method 7), m/z 389.0 dihydrothiophen-3- (M+H)+ (ESI+); yl)carbamoyl)-2- methoxypyridine 1-oxide 5-(3,4-dimethylphenyl)- RT 0.475 min (LCMS 2-((1,1-dioxido-2,3- Method 7), m/z 389.1 dihydrothiophen-3- (M+H)+ (ESI+); yl)carbamoyl)-4- methoxypyridine 1-oxide O O RT 0.441 min (LCMS 5-(3,4-dimethylphenyl)- S N O Method 7), m/z 389.1 2-((1,1-dioxido-2,3-N H (M+H)+ (ESI+); RT 2.036 dihydrothiophen-3- O min, 2.649 min, (SFC yl)carbamoyl)-3- Method 38); racemate methoxypyridine 1-oxide N-(1,1-dioxido-2,3- dihydrothiophen-3-yl)-2- RT 0.433 min (LCMS oxo-6-(4- Method 7), m/z 406.0 (trifluoromethyl)piperidin- (M+H)+ (ESI+); 1-yl)-1,2- dihydropyridine-3- carboxamide RT 0.443 min (LCMS 2-((1,1-dioxido-2,3- Method 7), m/z 406.1 dihydrothiophen-3- (M+H)+ (ESI+); RT 1.109 yl)carbamoyl)-5-(4- min, 1.564 min, (SFC (trifluoromethyl)piperidin- Method 39); racemate 1-yl)pyridine 1-oxide RT 0.477 min (LCMS (R)-3-amino-2-((1,1- Method 7), m/z 420.0 dioxido-2,3- (M+H)+ (ESI+); RT 1.797 dihydrothiophen-3- min, 2.282min, (SFC yl)carbamoyl)-5-(5- Method 4); ee: 95.56% (trifluoromethyl)thiophen -3-yl)pyridine 1-oxide RT 0.482 min (LCMS (R)-3-amino-2-((1,1- Method 7), m/z 420.0 dioxido-2,3- (M+H)+ (ESI+); RT 1.874 dihydrothiophen-3- min 2.239 min, (SFC yl)carbamoyl)-5-(5- Method 6); ee: 93.88% (trifluoromethyl)thiophen -2-yl)pyridine 1-oxide RT 0.464 min (LCMS (R)-3-amino-2-((1,1- Method 7), m/z 419.9 dioxido-2,3- (M+H)+ (ESI+); RT 0.736 dihydrothiophen-3- min 1.667 min, (SFC yl)carbamoyl)-5-(4- Method 4); ee: 97.59% (trifluoromethyl)thiophen -2-yl)pyridine 1-oxide
RT 0.525 min (LCMS (R)-2-((1,1-dioxido-2,3- Method 7), m/z 413.2 dihydrothiophen-3- (M+H)+ (ESI+); RT 1.702 yl)carbamoyl)-5-(3- min, 1.780 min, (SFC methyl-4- Method 6); ee: 87.13% (trifluoromethyl)phenyl)p yridine 1-oxide RT 1.679 min (LCMS (S,Z)-N-(1-cyclopropyl-3- Method 10), m/z 401.1 (methylsulfonyl)allyl)-6- (M+H)+ (ESI+); RT 1.712 (3,4-dimethylphenyl)-2- min, 1.810 min, (SFC oxo-1,2-dihydropyridine- Method 11); ee: 81.26% 3-carboxamide RT 0.494 min (LCMS (S,E)-N-(1-cyclopropyl- Method 7), m/z 401.2 3-(methylsulfonyl)allyl)- (M+H)+ (ESI+); RT 1.939 6-(3,4-dimethylphenyl)- min, 2.107 min (SFC 2-oxo-1,2- Method 8); ee: 72.34% dihydropyridine-3- carboxamide RT 0.632 min (LCMS (S,Z)-2-((1- Method 9), m/z 401.1 cyclopropyl-3- (M+H)+ (ESI+); RT 2.270 (methylsulfonyl)allyl)carb min, 2.433 min (SFC amoyl)-5-(3,4- Method 8); ee: 50.83% dimethylphenyl)pyridine 1-oxide RT 0.505 min (LCMS (S,E)-2-((1-cyclopropyl- Method 8), m/z 401.2 3- (M+H)+ (ESI+); RT 2.232 (methylsulfonyl)allyl)carb min, 2.508 min (SFC amoyl)-5-(3,4- Method 8); ee: 52.57% dimethylphenyl)pyridine 1-oxide RT 0.522 min (LCMS (R)-5-(2-chloro-4- Method 7), m/z (trifluoromethyl)phenyl)- 433.1(M+H)+ (ESI+); RT 2-((1,1-dioxido-2,3- 1.118 min, RT 1.358 dihydrothiophen-3- min, (SFC Method 10); yl)carbamoyl)pyridine 1- ee: 88.36% oxide RT 0.503 min (LCMS O CF3 Method 7), m/z 6-(3,4-dimethylphenyl)- S O 413.1(M+H)+ (ESI+); 3-(1-((1,1-dioxido-2,3- HN N O RT H 1.457 min, 1.710min, dihydrothiophen-3- 1.941 min, 2.832 min yl)amino)-2,2,2- (SFC Method 14); trifluoroethyl)pyridin- racemate 2(1H)-one
74 7-(3,4-dimethylphenyl)- RT 0.487 min (LCMS 3-(1,1-dioxido-2,3- Method 8), m/z dihydrothiophen-3-yl)-8- 403.1(M+H)+ (ESI+); fluoro-2-hydroxy-2,3- dihydroquinazolin-4(1H)- one 8-(5-(4- RT 0.501 min (LCMS (trifluoromethyl)phenyl)p Method 7), m/z yridin-2-yl)-2-thia-8- 409.1(M+H)+ (ESI+); azaspiro[4.5]dec-3-ene 2,2-dioxide RT 0.548 min (LCMS (R)-5-(3,4- Method 7), m/z dimethylphenyl)-N-(1,1- 393.2(M+H)+ (ESI+); RT dioxido-2,3- 1.752 min, 2.032min, dihydrothiophen-3- (SFC Method 7); ee: yl)quinoline-8- 87.20% carboxamide RT 0.474 min (LCMS 2-(6-(3,4- Method 7), m/z dimethylphenyl)-2- 398.1(M+H)+ (ESI+); RT methoxypyridin-3-yl)-8- 1.599 min, 1.899min, thia-1,3- (SFC Method 26); diazaspiro[4.5]deca-2,6- racemate diene 8,8-dioxide 3-(6-(3,4- RT 0.516 min (LCMS dimethylphenyl)-2- Method 7), m/z hydroxypyridin-3-yl)-9- 398.3(M+H)+ (ESI+); thia-2,4- diazaspiro[5.5]undeca- 2,7-diene 9,9-dioxide RT 0.620 min (LCMS (R)-5-(3,4- Method 2), m/z 392.0 dimethylphenyl)-N-(1,1- (M-H)- (ESI-); RT 1.918 dioxido-2,3- min, RT 2.234 min, (SFC dihydrothiophen-3- Method 9); ee: 81.678% yl)quinazoline-8- carboxamide 3-(6-(3,4- RT 0.469 min (LCMS dimethylphenyl)-2- Method 7), m/z 412.1 methoxypyridin-3-yl)-9- (M+H)+ (ESI+) thia-2,4- diazaspiro[5.5]undeca- 2,7-diene 9,9-dioxide
RT 0.576 min (LCMS 3-(4-(6-cyclopentyl-2- Method 7), m/z 423.1 phenoxypyridin-3-yl)-1H- (M+H)+ (ESI+); RT 1.731 1,2,3-triazol-1-yl)-2,3- min, 1.872 min, (SFC dihydrothiophene 1,1- Method 30); racemate dioxide RT 0.429 min (LCMS 2-(2- Method 7), m/z 433.0 (cyclopropyldifluorometh (M+H)+ (ESI+); RT 1.964 yl)-4-phenoxypyrimidin- min, 2.272 min, (SFC 5-yl)-7-thia-1,3- Method 40); racemate diazaspiro[4.4]nona-2,8- diene 7,7-dioxide N S O (E)-2- N O RT (cyclopropyldifluorometh H 0.524 min (LCMS Method 7), m/z 433 yl)-5-(5-(2-N O .0 (M+H)+ (ESI+) (methylsulfonyl)vinyl)- 1H-imidazol-2-yl)-4- phenoxypyrimidine 3-(4-(6- RT 0.560 min (LCMS (isopropyl(methyl)amino) Method 7), m/z 426.1 -2-phenoxypyridin-3-yl)- (M+H)+ (ESI+) 1H-1,2,3-triazol-1-yl)- 2,3-dihydrothiophene 1,1-dioxide RT 0.538 min (LCMS (R)-5-(2- Method 7), m/z 436.1 (benzylamino)phenyl)-2- (M+H)+ (ESI+); RT 1.617 ((1,1-dioxido-2,3- min, (SFC Method 10); dihydrothiophen-3- ee: 88.98% yl)carbamoyl)pyridine 1- oxide
76 RT 0.496 min (LCMS 2-(6-(3,4- Method 7), m/z 446.2 dimethylphenyl)-2- (M+H)+ (ESI+); RT 2.234 phenoxypyridin-3-yl)-7- min, 2.464 min (SFC thia-1,3- Method 34); racemate diazaspiro[4.4]nona-2,8- diene 7,7-dioxide RT 0.498 min (LCMS (S)-2-(6-(3,4- Method 7), m/z 446.0 dimethylphenyl)-2- (M+H)+ (ESI+); RT 2.241 phenoxypyridin-3-yl)-7- min, 2.482 min (SFC thia-1,3- Method 34); ee: 61.74% diazaspiro[4.4]nona-2,8- diene 7,7-dioxide RT 0.521 min (LCMS (R)-5-(2-(benzyloxy)-3- Method 7), m/z 455.2 fluorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.261 dioxido-2,3- min, 1.439 min (SFC dihydrothiophen-3- Method 10); ee: 88.69% yl)carbamoyl)pyridine 1- oxide RT 0.517 min (LCMS (R)-5-(2-(benzyloxy)-4- Method 7), m/z 455.0 fluorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.569 dioxido-2,3- min, 1.675 min (SFC dihydrothiophen-3- Method 10); ee: 82.21% yl)carbamoyl)pyridine 1- oxide RT 0.508 min (LCMS (R)-5-(2-(benzyloxy)-5- Method 7), m/z 455.0 fluorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.543 dioxido-2,3- min, 1.735 min (SFC dihydrothiophen-3- Method 10); ee: 81.41% yl)carbamoyl)pyridine 1- oxide RT 0.509 min (LCMS (R)-5-(2-(benzyloxy)-6- Method 7), m/z 455.1 fluorophenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.451 dioxido-2,3- min, 1.608 min (SFC dihydrothiophen-3- Method 10); ee: 87.79% yl)carbamoyl)pyridine 1- oxide
77 RT 0.536 min (LCMS (R)-5-(2-(benzyloxy)-5- Method 7), m/z 451.2 methylphenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.720 dioxido-2,3- min, 1.826 min (SFC dihydrothiophen-3- Method 9); ee: 88.18% yl)carbamoyl)pyridine 1- oxide RT 0.526 min (LCMS (R)-5-(2-(benzyloxy)-3- Method 6), m/z 451.2 methylphenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.221 dioxido-2,3- min, 1.412 min (SFC dihydrothiophen-3- Method 10); ee: 87.70% yl)carbamoyl)pyridine 1- oxide RT 0.526 min (LCMS (R)-5-(2-(benzyloxy)-4- Method 7), m/z 451.3 methylphenyl)-2-((1,1- (M+H)+ (ESI+); RT 1.708 dioxido-2,3- min, 1.837 min (SFC dihydrothiophen-3- Method 9); ee: 90.04% yl)carbamoyl)pyridine 1- oxide RT 0.635 min (LCMS (R)-5-(2-(benzyloxy)-6- Method 4), m/z 451.2 methylphenyl)-2-((1,1- (M+H)+ (ESI+); RT 2.072 dioxido-2,3- min, 2.213 min (SFC dihydrothiophen-3- Method 35); ee: 88.87% yl)carbamoyl)pyridine 1- oxide RT 0.510 min (LCMS (R)-2-((1,1-dioxido-2,3- Method 2), m/z 456.1 dihydrothiophen-3- (M+H)+ (ESI+); RT 1.654 yl)carbamoyl)-5-(5- min, 1.795 min (SFC fluoro-2-(pyridin-4- Method 35); ee: 88.87% ylmethoxy)phenyl)pyridin e 1-oxide 2-amino-3-(3,4- RT 0.461 min (LCMS dimethylphenyl)-6-((1,1- Method 7), m/z 374.3 dioxido-2,3- (M+H)+ (ESI+); RT 1.907 dihydrothiophen-3- min, 2.068 min (SFC yl)carbamoyl)pyridine 1- Method 36); racemate oxide 2,2,2- trifluoroacetate
(R)-3-((tert- RT 0.591 min (LCMS butoxycarbonyl)amino)- Method 7), m/z 520.1 2-((1,1-dioxido-2,3- 187 (M+H)+ (ESI+); RT 2.190 dihydrothiophen-3- min, 2.481 min (SFC yl)carbamoyl)-5-(5- Method 36); ee: 88.68% (trifluoromethyl)thiophen -2-yl)pyridine 1-oxide (R)-3-((tert- RT 0.577 min (LCMS butoxycarbonyl)amino)- Method 7), m/z 520.0 2-((1,1-dioxido-2,3- 188 (M+H)+ (ESI+); RT 1.298 dihydrothiophen-3- min, 1.895 min (SFC yl)carbamoyl)-5-(4- Method 4); ee: 86.19% (trifluoromethyl)thiophen -2-yl)pyridine 1-oxide (R)-3-((tert- RT 0.583min (LCMS butoxycarbonyl)amino)- Method 7), m/z 520.0 2-((1,1-dioxido-2,3- 189 (M+H)+ (ESI+); RT 1.569 dihydrothiophen-3- min, 2.148 min (SFC yl)carbamoyl)-5-(5- Method 37); ee: 94.65% (trifluoromethyl)thiophen -3-yl)pyridine 1-oxide Preparation of reference example 1 In the following, preparation of reference example 1 is described. This example is identical to example 87 reported in WO 2024/010782. Preparation of reference Intermediate 1.1 Ethyl 2-(1-ethoxyvinyl)-4-phenoxypyrimidine-5-carboxylate
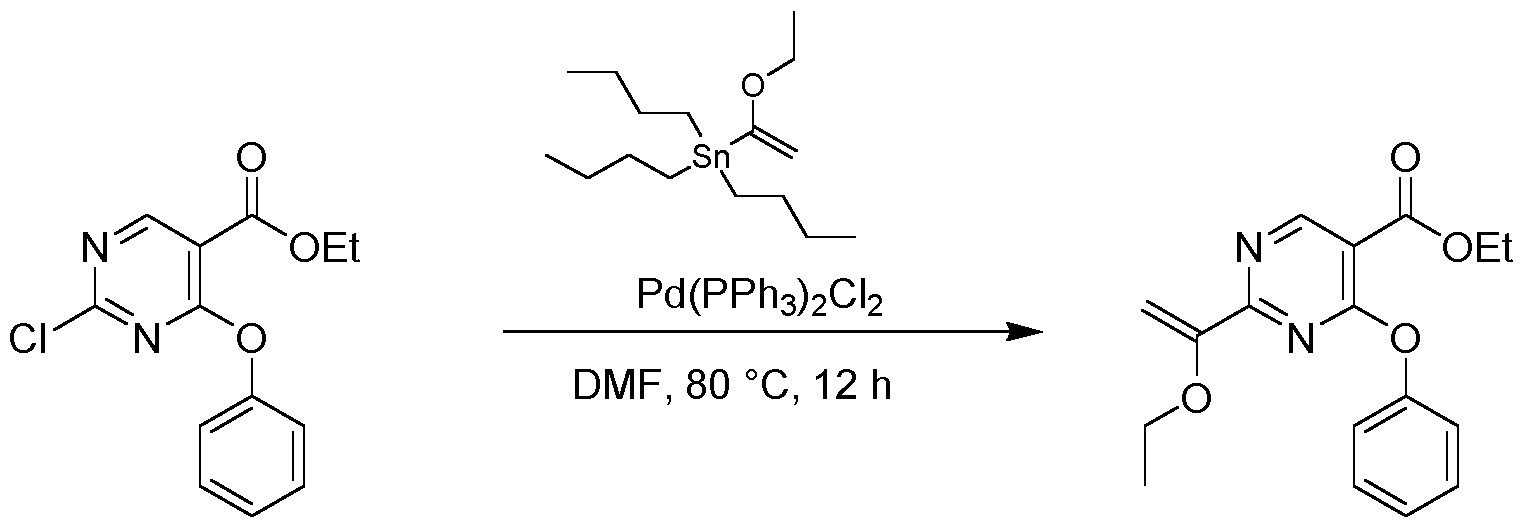 To a solution of ethyl 2-chloro-4-phenoxypyrimidine-5-carboxylate (3.00 g, 10.8 mmol) in DMF (30 mL) was added Pd(PPh3)2Cl2 (756 mg, 1.08 mmol) and tributyl(1-ethoxyvinyl)stannane (5.83 g, 16.1 mmol) under N2, and then stirred at 80 °C for 12 h under N2. The resulting mixture was poured into KF (aq., sat., 40 mL) and stirred for 30 min. The resulting mixture was filtered and the filtrate was extracted with EtOAc (40 mL; 2x). The combined organic layers were washed with brine (40 mL; 2x), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (ISCO; 40 g SepaFlash Silica Flash Column, Eluent of 0%-20% ethyl acetate
/petroleum ether; gradient @ 80 mL/min) and concentrated under vacuum to give the product ethyl 2-(1- ethoxyvinyl)-4-phenoxypyrimidine-5-carboxylate (2.70 g, 8.59 mmol, 79.79 % yield) as a brown oil. LCMS: RT 0.558 min (method 7); m/z 315.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 9.17 (s, 1H), 7.48-7.38 (m, 2H), 7.29-7.25 (m, 1H), 7.22-7.18 (m, 2H), 5.37 (d, J = 2.0 Hz, 1H), 4.53 (d, J = 2.0 Hz, 1H), 4.44 (q, J = 8.0 Hz, 2H), 3.94 (q, J = 8.0 Hz, 2H), 1.45-1.40 (m, 6H) Preparation of reference Intermediate 1.2 Ethyl 2-acetyl-4-phenoxypyrimidine-5-carboxylate
To a solution of ethyl 2-chloro-4-phenoxypyrimidine-5-carboxylate (3.00 g, 10.8 mmol) in DMF (30 mL) was added Pd(PPh3)2Cl2 (756 mg, 1.08 mmol) and tributyl(1-ethoxyvinyl)stannane (5.83 g, 16.1 mmol) under N2, and then stirred at 80 °C for 12 h under N2. The resulting mixture was poured into KF (aq., sat., 40 mL) and stirred for 30 min. The resulting mixture was filtered and the filtrate was extracted with EtOAc (40 mL; 2x). The combined organic layers were washed with brine (40 mL; 2x), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (ISCO; 40 g SepaFlash Silica Flash Column, Eluent of 0%-20% ethyl acetate
/petroleum ether; gradient @ 80 mL/min) and concentrated under vacuum to give the product ethyl 2-(1- ethoxyvinyl)-4-phenoxypyrimidine-5-carboxylate (2.70 g, 8.59 mmol, 79.79 % yield) as a brown oil. LCMS: RT 0.558 min (method 7); m/z 315.1 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 9.17 (s, 1H), 7.48-7.38 (m, 2H), 7.29-7.25 (m, 1H), 7.22-7.18 (m, 2H), 5.37 (d, J = 2.0 Hz, 1H), 4.53 (d, J = 2.0 Hz, 1H), 4.44 (q, J = 8.0 Hz, 2H), 3.94 (q, J = 8.0 Hz, 2H), 1.45-1.40 (m, 6H) Preparation of reference Intermediate 1.2 Ethyl 2-acetyl-4-phenoxypyrimidine-5-carboxylate
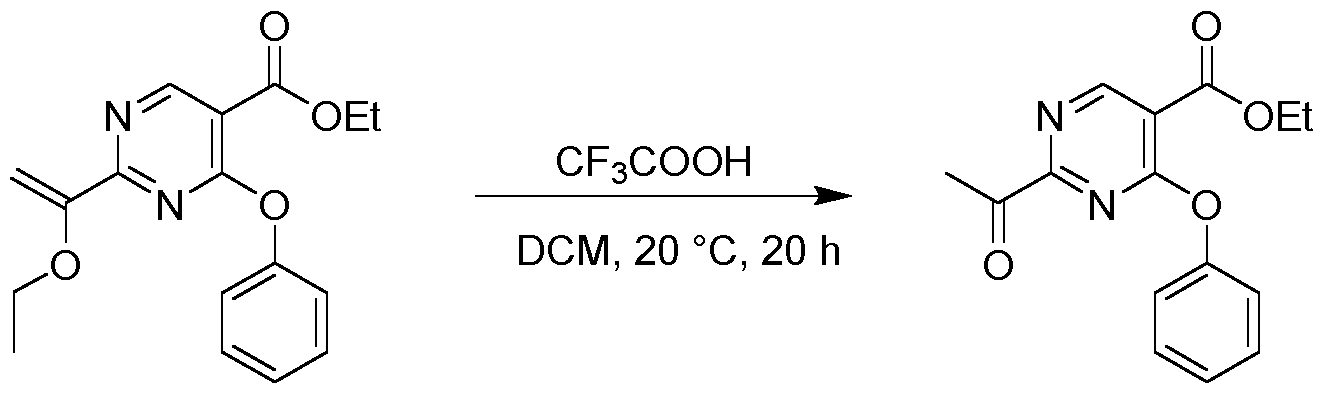 To a solution of ethyl 2-(1-ethoxyvinyl)-4-phenoxypyrimidine-5-carboxylate (2.7 g, 8.59 mmol) in DCM (50 mL) was added CF3COOH (2.0 mL, 35.4 mmol), and the mixture was stirred at 20 °C for 20 h. The mixture was diluted with DCM (100 mL) and washed with NaHCO3 (aq., sat, 50 mL; 2x). The organic layer was dried over anhydrous Na2SO4 and concentrated under vacuum to give the crude product ethyl 2-acetyl-4-phenoxypyrimidine-5-carboxylate (1600 mg, 5.59 mmol, 65.07 % yield) as a brown solid. The crude product was used directly without further purification. LCMS: RT 0.515 min (method 7); m/z 287.0 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 9.25 (s, 1H), 7.50-7.42 (m, 2H), 7.35-7.28 (m, 1H), 7.25 - 7.18 (m, 2H), 4.48 (q, J = 8.0 Hz, 2H), 2.47 (s, 3H), 1.44 (t, J = 8.0 Hz, 3H) Preparation of reference Intermediate 1.3 Ethyl 2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxylate
To a solution of ethyl 2-(1-ethoxyvinyl)-4-phenoxypyrimidine-5-carboxylate (2.7 g, 8.59 mmol) in DCM (50 mL) was added CF3COOH (2.0 mL, 35.4 mmol), and the mixture was stirred at 20 °C for 20 h. The mixture was diluted with DCM (100 mL) and washed with NaHCO3 (aq., sat, 50 mL; 2x). The organic layer was dried over anhydrous Na2SO4 and concentrated under vacuum to give the crude product ethyl 2-acetyl-4-phenoxypyrimidine-5-carboxylate (1600 mg, 5.59 mmol, 65.07 % yield) as a brown solid. The crude product was used directly without further purification. LCMS: RT 0.515 min (method 7); m/z 287.0 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 9.25 (s, 1H), 7.50-7.42 (m, 2H), 7.35-7.28 (m, 1H), 7.25 - 7.18 (m, 2H), 4.48 (q, J = 8.0 Hz, 2H), 2.47 (s, 3H), 1.44 (t, J = 8.0 Hz, 3H) Preparation of reference Intermediate 1.3 Ethyl 2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxylate
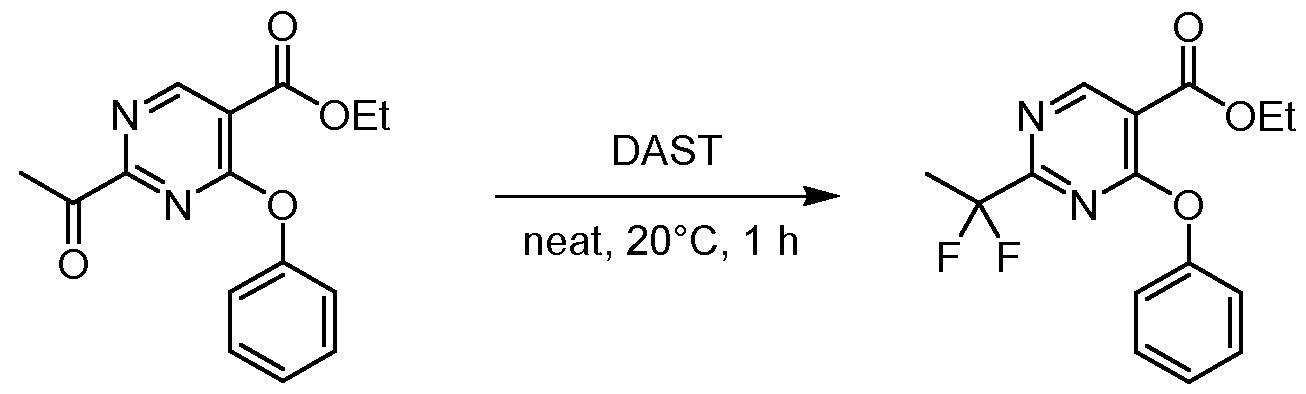 A solution of ethyl 2-acetyl-4-phenoxypyrimidine-5-carboxylate (800 mg, 2.79 mmol) in DAST (4.0 mL, 56.2 mmol) was stirred at 20 °C for 1 h. The resulting mixture was diluted with DCM (30 mL) and slowly poured into ice water (30 mL) over 5 min. The organic layer was washed with water (20 mL; 2x), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by column chromatography (ISCO; 20 g SepaFlash Silica Flash Column, Eluent of 5%~20% ethyl
acetate/petroleum ether @ 40 mL/min) to give the product ethyl 2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxylate (680 mg, 2.21 mmol, 78.94 % yield) as a brown solid. LCMS: RT 0.560 min (method 7); m/z 309.0 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 9.19 (s, 1H), 7.48-7.40 (m, 2H), 7.33-7.27 (m, 1H), 7.23-7.18 (m, 2H), 4.47 (q, J = 8.0 Hz, 2H), 1.84 (t, J = 18.4 Hz, 3H), 1.44 (t, J = 8.0 Hz, 3H); 19F NMR (CDCl3, 400 MHz): -93.95 (s, 2F) Preparation of reference Intermediate 1.4 2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxylic acid
A solution of ethyl 2-acetyl-4-phenoxypyrimidine-5-carboxylate (800 mg, 2.79 mmol) in DAST (4.0 mL, 56.2 mmol) was stirred at 20 °C for 1 h. The resulting mixture was diluted with DCM (30 mL) and slowly poured into ice water (30 mL) over 5 min. The organic layer was washed with water (20 mL; 2x), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by column chromatography (ISCO; 20 g SepaFlash Silica Flash Column, Eluent of 5%~20% ethyl
acetate/petroleum ether @ 40 mL/min) to give the product ethyl 2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxylate (680 mg, 2.21 mmol, 78.94 % yield) as a brown solid. LCMS: RT 0.560 min (method 7); m/z 309.0 (M+H)+ (ESI+); 1H NMR (CDCl3, 400 MHz): 9.19 (s, 1H), 7.48-7.40 (m, 2H), 7.33-7.27 (m, 1H), 7.23-7.18 (m, 2H), 4.47 (q, J = 8.0 Hz, 2H), 1.84 (t, J = 18.4 Hz, 3H), 1.44 (t, J = 8.0 Hz, 3H); 19F NMR (CDCl3, 400 MHz): -93.95 (s, 2F) Preparation of reference Intermediate 1.4 2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxylic acid
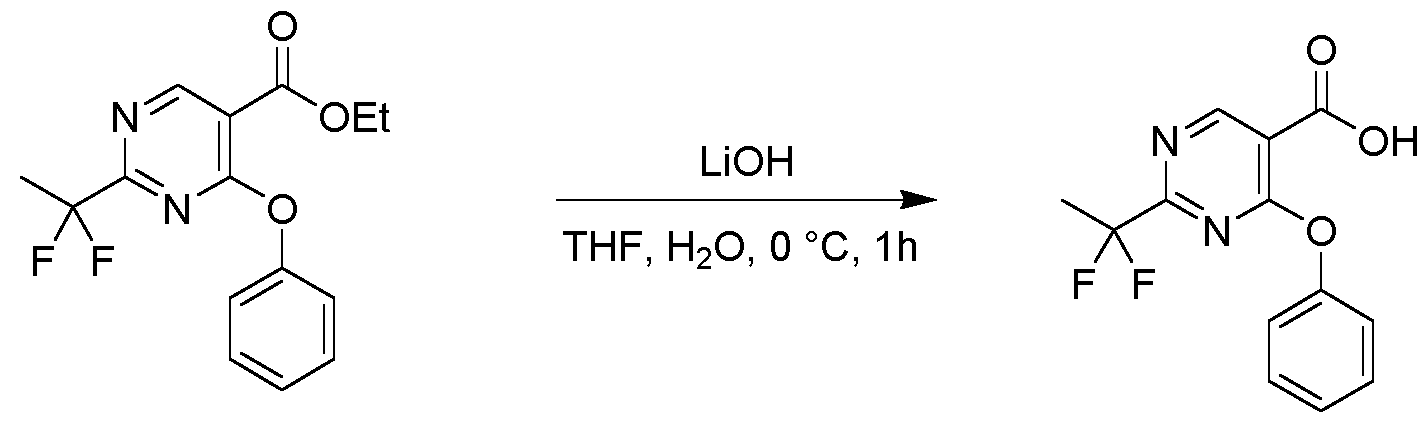 To a solution of ethyl 2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxylate (580 mg, 1.88 mmol) in THF (6 mL) and water (3 mL) at 0 °C was added LiOH.H2O (158 mg, 3.76 mmol), and the mixture was stirred at 0 °C for 1 h. The mixture was diluted with EtOAc (30 mL). The pH was adjusted to pH to 2-3 with HCl (aq., 1M) and the mixture was washed with water (20 mL; 2x). The combined organics layers were dried over anhydrous Na2SO4, filtered and concentrated under vacuum to give the product 2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxylic acid (410 mg,1.46 mmol, 77.77 % yield) as a brown solid. It was used directly in the next step without any further purification. LCMS: RT 0.476min (method 7); m/z 281.0 (M +H)+ (ESI+).;1H NMR (CDCl3, 400 MHz): 9.18 (s, 1H), 7.34-7.28 (m, 2H), 7.23-7.16 (m, 1H), 7.15-7.05 (m, 2H), 1.74 (t, J = 18.4 Hz, 3H); 19F NMR (CDCl3, 400 MHz): -93.86 (s, 2F) Preparation of reference Intermediate 1.5 Tert-butyl (S,E)-(1-cyclopropyl-3-(methylsulfonyl)allyl)carbamate
To a solution of ethyl 2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxylate (580 mg, 1.88 mmol) in THF (6 mL) and water (3 mL) at 0 °C was added LiOH.H2O (158 mg, 3.76 mmol), and the mixture was stirred at 0 °C for 1 h. The mixture was diluted with EtOAc (30 mL). The pH was adjusted to pH to 2-3 with HCl (aq., 1M) and the mixture was washed with water (20 mL; 2x). The combined organics layers were dried over anhydrous Na2SO4, filtered and concentrated under vacuum to give the product 2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxylic acid (410 mg,1.46 mmol, 77.77 % yield) as a brown solid. It was used directly in the next step without any further purification. LCMS: RT 0.476min (method 7); m/z 281.0 (M +H)+ (ESI+).;1H NMR (CDCl3, 400 MHz): 9.18 (s, 1H), 7.34-7.28 (m, 2H), 7.23-7.16 (m, 1H), 7.15-7.05 (m, 2H), 1.74 (t, J = 18.4 Hz, 3H); 19F NMR (CDCl3, 400 MHz): -93.86 (s, 2F) Preparation of reference Intermediate 1.5 Tert-butyl (S,E)-(1-cyclopropyl-3-(methylsulfonyl)allyl)carbamate
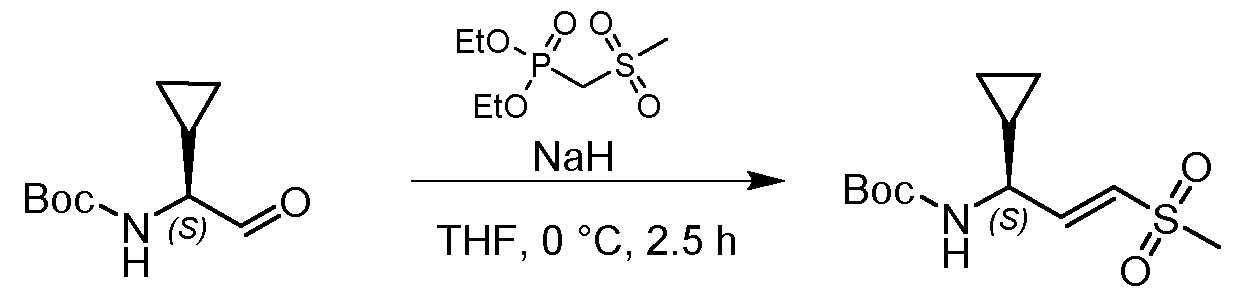 To a solution of diethyl ((methylsulfonyl)methyl)phosphonate (381 mg, 1.66 mmol) in THF (6 mL) was added NaH (66 mg, purity: 60%, 1.66 mmol,) at 0 °C and the mixture was stirred for 1 h. After 1h, tert-butyl N-[(1S)-1-cyclopropyl-2-oxo-ethyl]carbamate (300 mg, 1.51 mmol) was added and the mixture was stirred at 0 °C for 1.5 h. In parallel, another reaction was conducted with the same protocol but on different scale of diethyl ((methylsulfonyl)methyl)phosphonate (165 mg, 0.652 mmol). The combined
resulting mixture was poured into NH4Cl (aq., sat., 15 mL) and extracted with ethyl acetate (20 ml; 3x). The combined organic layers were washed with brine (30 mL), dried over anhydrous Na2SO4 and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 20 g SepaFlash Silica Flash Column, Eluent of 0 to ~15% ethyl acetate/petroleum ether gradient @ 60 mL/min) to give the product tert-butyl (S,E)-(1-cyclopropyl-3-(methylsulfonyl)allyl)carbamate (250 mg, 0.908 mmol, 42.07 % yield) as a white solid. LCMS: RT 0.452 min (method 7); m/z 298.1 (M+Na)+ (ESI+); SFC: RT 1.067 min (Method 44), ee:99.79%; 1H NMR (CDCl3, 400 MHz): 6.94 (dd, J = 4.8, 15.2 Hz, 1H), 6.53 (d, J =15.2 Hz, 1H), 4.77- 4.70 (m, 1H), 3.72 (br s, 1H), 2.96 (s, 3H), 1.46 (s, 9H), 0.96-0.84 (m, 1H), 0.73-0.56 (m, 2H), 0.51-0.42 (m, 1H), 0.40-0.31 (m, 1H) Preparation of reference Intermediate 1.6
To a solution of diethyl ((methylsulfonyl)methyl)phosphonate (381 mg, 1.66 mmol) in THF (6 mL) was added NaH (66 mg, purity: 60%, 1.66 mmol,) at 0 °C and the mixture was stirred for 1 h. After 1h, tert-butyl N-[(1S)-1-cyclopropyl-2-oxo-ethyl]carbamate (300 mg, 1.51 mmol) was added and the mixture was stirred at 0 °C for 1.5 h. In parallel, another reaction was conducted with the same protocol but on different scale of diethyl ((methylsulfonyl)methyl)phosphonate (165 mg, 0.652 mmol). The combined
resulting mixture was poured into NH4Cl (aq., sat., 15 mL) and extracted with ethyl acetate (20 ml; 3x). The combined organic layers were washed with brine (30 mL), dried over anhydrous Na2SO4 and concentrated under vacuum. The residue was purified by flash silica gel chromatography (ISCO; 20 g SepaFlash Silica Flash Column, Eluent of 0 to ~15% ethyl acetate/petroleum ether gradient @ 60 mL/min) to give the product tert-butyl (S,E)-(1-cyclopropyl-3-(methylsulfonyl)allyl)carbamate (250 mg, 0.908 mmol, 42.07 % yield) as a white solid. LCMS: RT 0.452 min (method 7); m/z 298.1 (M+Na)+ (ESI+); SFC: RT 1.067 min (Method 44), ee:99.79%; 1H NMR (CDCl3, 400 MHz): 6.94 (dd, J = 4.8, 15.2 Hz, 1H), 6.53 (d, J =15.2 Hz, 1H), 4.77- 4.70 (m, 1H), 3.72 (br s, 1H), 2.96 (s, 3H), 1.46 (s, 9H), 0.96-0.84 (m, 1H), 0.73-0.56 (m, 2H), 0.51-0.42 (m, 1H), 0.40-0.31 (m, 1H) Preparation of reference Intermediate 1.6
 A solution of tert-butyl (S,E)-(1-cyclopropyl-3-(methylsulfonyl)allyl)carbamate (230 mg, 0.835 mmol) in a solution of HCl in EtOAc (4.6 mL, 2M) was stirred at 20 °C for 2 h. The resulting mixture was concentrated under vacuum to give the crude product (S,E)-1-cyclopropyl-3-(methylsulfonyl)prop-2-en-1- amine hydrochloride (170 mg, 0.803 mmol, 96.14 % yield, HCl salt) as a white solid. The crude product was used directly in the next step without any further purification. 1H NMR (400 MHz, MeOD): 7.05-6.98 (m, 1H), 6.93-6.83 (m, 1H), 3.37-3.32 (m, 1H), 3.05 (s, 3H), 1.18-1.07 (m, 1H), 0.85-0.78 (m, 2H), 0.66-0.47 (m, 2H) Preparation of reference example 1 (S,E)-N-(1-cyclopropyl-3-(methylsulfonyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide
A solution of tert-butyl (S,E)-(1-cyclopropyl-3-(methylsulfonyl)allyl)carbamate (230 mg, 0.835 mmol) in a solution of HCl in EtOAc (4.6 mL, 2M) was stirred at 20 °C for 2 h. The resulting mixture was concentrated under vacuum to give the crude product (S,E)-1-cyclopropyl-3-(methylsulfonyl)prop-2-en-1- amine hydrochloride (170 mg, 0.803 mmol, 96.14 % yield, HCl salt) as a white solid. The crude product was used directly in the next step without any further purification. 1H NMR (400 MHz, MeOD): 7.05-6.98 (m, 1H), 6.93-6.83 (m, 1H), 3.37-3.32 (m, 1H), 3.05 (s, 3H), 1.18-1.07 (m, 1H), 0.85-0.78 (m, 2H), 0.66-0.47 (m, 2H) Preparation of reference example 1 (S,E)-N-(1-cyclopropyl-3-(methylsulfonyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide
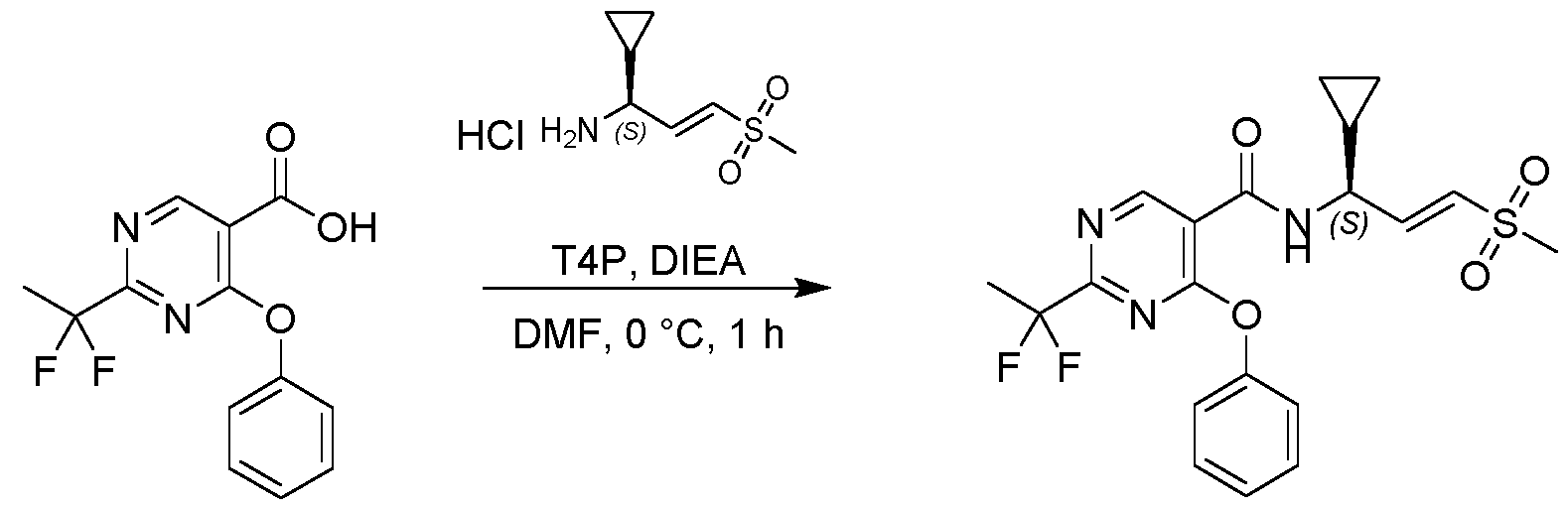 To a solution of (S,E)-1-cyclopropyl-3-(methylsulfonyl)prop-2-en-1-amine hydrochloride (68 mg, 0.321 mmol) and 2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxylic acid (90 mg, 0.321 mmol) in
DMF (2 mL) at 0°C was added DIEA (0.18 mL, 0.963 mmol) and T4P in EtOAc (694 mg, 50% purity, 0.963 mmol), and the mixture was stirred at 0 °C for 1 h. The resulting mixture was poured into ice-water (20 mL) and extracted with EtOAc (20 mL; 2x). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under vacuum. The residue was purified by preparative HPLC (column: Unisil 3-100 C18 Ultra 150*50mm*3 m;mobile phase: A: 0.225% FA in water, B: MeCN; B%: 38%-68%,10 min) and lyophilized directly to give the product (S,E)-N-(1-cyclopropyl-3-(methylsulfonyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (86.25 mg,0.197 mmol, 61.39 % yield) as a white solid. LCMS: RT 0.504min (method 7); m/z 438.1 (M +H)+ (ESI+). SFC: RT 1.270 min: (Method 13, 97.51% ee); 1H NMR (CDCl3, 400 MHz): 9.47 (s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.58-7.47 (m, 2H), 7.43- 7.35 (m, 1H), 7.27-7.20 (m, 2H), 7.03 (dd, J = 4.8, 15.2 Hz, 1H), 6.61 (d, J =15.2 Hz, 1H), 4.33-4.19 (m, 1H), 2.97 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.16-1.03 (m, 1H), 0.81-0.63 (m, 2H), 0.58 - 0.44 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F) Biological evaluation of the exemplary compounds Exemplary compounds of formula (I) were tested in selected biological and/or physicochemical assays one or more times. When tested more than once, data are reported as either average values or as median values, wherein the average value, also referred to as the arithmetic mean value, represents the sum of the values obtained divided by the number of times tested, and the median value represents the middle number of the group of values when ranked in ascending or descending order. If the number of values in the data set is odd, the median value is the middle value. If the number of values in the data set is even, the median is the arithmetic mean of the two middle values. The in vitro pharmacological, pharmacokinetic and physicochemical properties of the compounds can be determined according to the following assays and methods. WRN Protein Expression and Purification WRN (amino acid 517-1093, L1074F isoform) was purchased from CRELUX GmbH, Planegg, Germany. WRN was produced in a Sf21 expression system, purified, and stored and frozen in 50 mM HEPES / NaOH, 500 mM NaCl, 5% Glycerol and 0.5 mM TCEP, pH 7.5. Forked DNA Preparation The forked DNA was prepared by mixing the two DNA strands in a 1:1.8 ratio of OLIGOB-FLU (FAM(fluorescein)-GAACGA ACA CAT CGG GTA CGT TTT TTT TTT TTT TTT TTT TTT TTT TTT TT) (SEQ ID NO.: 1) to OLIGOA-BHQ1 (TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT CGT ACC CGA TGT GTT CGT TC-BHQ1) (SEQ ID NO.: 2) in DNA-annealing buffer (10 mM Tris/HCl pH 7.5, 50 mM NaCl & 1 mM EDTA, pH 8.0). The mixture is heated to 95 °C and cooled down to 35 °C at a rate of 5°C per minute in a Roche LightCycler® 96 System.
In vitro WRN Helicase assay A continuous, fluorescence-based helicase assay was set up to measure ATP-coupled unwinding of a forked DNA substrate by WRN (517-1093, L1074F isoform). Fluorescently labelled forked DNA was used as a DNA substrate, the DNA sequences were described by Sommers et al. 2019 (doi: 10.1371/journal.pone.0210525), with different fluorescent dyes. WRN mediated separation of the forked DNA leads to an increase in fluorescence. ssDNA complementary to the quenching strand, oligo-block (GAA CGA ACA CAT CGG GTA CG) (SEQ ID NO.: 3), is included in 10-fold excess to prevent reannealing of the forked DNA. Labelled DNA was purchased from IDT (Integrated DNA Technologies, Inc.) and ssDNA oligo-block from Microsynth AG. Reaction A typical reaction consists of 30 µL of 1 nM WRN in 25 mM Tris/HCl pH 7.5, 100 mM NaCl, 1 mM DTT, 0.01 % TWEEN-20, 0.025 mg/mL BSA, 0.1 mM forked DNA substrate, 1 mM Oligo-BLOCK and 0.5 mM Mg-ATP in a 384 well black plate (Thermo Scientific). To assess inhibition, IC50s were determined by pre-incubating compounds (10 mM DMSO stock solutions) with WRN in a 12-point custom dilution series to a final DMSO concentration of 1 %. DMSO only controls were included to represent no inhibition (high control) Compounds and WRN were pre-incubated for 2 or 6 hours at room temperature, before the addition of forked DNA, ssDNA oligo-block, and Mg-ATP to initiate the reaction. Fluorescence (Ex: 485 nm, Em: 535 nm, gain 66, 5 flashes per measurement) was measured on a Tecan Spark plate reader for 45 minutes, in 60 seconds intervals at 27.5°C. All points were measured in triplicate. Data Analysis Data analysis was performed using dotmatics (www.dotmatics.com). Enzymatic activity was assessed by the rates of DNA unwinding, measured by the rate of increase in fluorescence. The initial slopes were normalized to % activity, as follows: percentage of activity (Normalized) = (Sample Value / High Control Value (Average of DMSO only wells)) * 100%. The resulting dose response was fitted with the following equation: Y=Bottom + (Top-Bottom)/(1+(IC50/X)^HillSlope), where X values are concentrations and Y is the response (decreasing as X increases). The curve fitting was performed using the least squares regression method. Reported values are the mean of at least 2 independent replicates. The Absorbance IC50 value of compounds of Formula (I) in Examples 1 to 189 are provided in Table 3 below. In case there is a blank cell in the Table, this means that no data have been generated. Cellular Viability Assay The colon carcinoma cell lines HCT116 and SW48 (both microsatellite instability- high and WRN- inhibition sensitive cell lines) and SW620 (microsatellite stable and WRN-inhibition insensitive cell line) were obtained from ATCC.
HCT116 were cultured in growth medium composed of McCoy's 5A (Modified) Medium (ThermoFisher Scientific Cat# 16600082), 1x Penicillin-Streptomycin (ThermoFisher Scientific Cat# 15140163), and 10% fetal bovine serum (ThermoFisher Scientific Cat#A5256801). SW48 and SW620 were cultured in growth medium composed of Leibovitz's L-15 Medium (ThermoFisher Scientific Cat#11415056), 1x Penicillin-Streptomycin and 10% fetal bovine serum. HCT116, SW48 and SW620 were plated at 1500 cells/well, 2000 cells/well and 2000 cells/well, respectively, in 96-well black plates with clear flat bottom (Huberlab #.655983), in a volume of 200 µL per well. The outer wells of the plate were filled with DPBS (ThermoFisher Scientific Cat#14190250) to compensate evaporation mediated effects in the plate periphery. After 24 hours, the compounds were dispensed with the Tecan digital dispenser (D300e), starting at 20 µM for a 8-point dose curve at 1:4 dilution, in duplicates. The final concentration of DMSO was normalized to the highest compound concentration and a maximum of 0.1%.. After 4d of incubation, 145 µl of the growth medium were removed and 50 µL of Cell Titer-Glo (Promega Cat#G9243) were added per well. Plates were shaken for 2 minutes and after an incubation of 10 minutes, luminescence was read using a plate reader (Tecan Spark). For data analysis, the assay background signal which was determined in wells that contained medium only was subtracted from all the data points. Averaged values of the samples were normalized to DMSO treated control samples. The fitting of the logistic curve were performed using the following equation: Y=Bottom + (Top-Bottom)/(1+(IC50/X)^HillSlope), where X values are concentrations and Y is the response (decreasing as X increases). The curve fitting was performed using the least squares regression method. The IC50 value of compounds of Formula (I) in Examples 1 to 189 are provided in Table 3 below. In case there is a blank cell in the Table, this means that no data have been generated. Table 3 Example number IC50 in M determined in WRN helicase assay described under In vitro WRN Helicase assay following 6 hours incubation IC50 in M determined in WRN helicase assay described under In vitro WRN Helicase assay following 2 hours incubation IC50 in M determined in SW48 assay described under Cellular viability assay IC50 in M determined in HCT116 assay described under Cellular viability assay
IC50 in M determined in SW620 assay described under Cellular viability assay 0.970 5.3 3.6 0.928 0.469 0.373 2.85 0.351 4.9 1.3 1.9 5.0 6.6 0.608 1.1 3.39 10 10.9 21.8 5.1 36.1 1.25 1.6 0.877 1.5 0.117 0.254 0.081 0.112 4.63 5.82 >13.77 0.253 0.435 0.700 1.00 0.251 0.345 0.274 0.366 0.274 0.523 0.703 1.15 400
0.332 0.559 (67% inhibition at 100mM) a 400 b 1000 a 1000 0.143 (65% 0.07 (40% >20 >20b inhibition at inhibition at 100mM) 100mM) 0.135 0.169 6.76 >19.09 0.074 0.071 7.02 9.56 9.57 0.034 0.042 2.47 >15.93 1.5 2.8 <0.055 0.022 >20 400 6.006 9.55 >20 1.3 2.30 0.0545 0.153 >15.25a 1000 1.862 >12.88 >20b 400 1.405 5.85 >20c 4.85 0.0805 0.455 >20d 0.727 0.019 0.071 >20 1.35 0.0285 0.1195 5.358a 54.1 0.362 1.0895 5.225b 150 0.574 2.051 5.393c 0.4495 0.0095 0.043 5.3585d 0.8185 0.013 0.059 7.472 55.9 26.7 1.1 1.1 4.25 1.4 2.6 84.3 102
0.097 0.2075 0.142 0.27 0.149 0.277 23.7 1.3 1.95 6.5 6.1 24.2 9.4 8 2.6 4.05 12.2 25.8 26.4 400 400 80.6 0.164 0.2735 119.3 3.55 400 3.2 400 65.75 6.75 5.6 1 1.4 400 3.1
18 9.55 0.976 1.3 1000 3.35 1.8 3.1 0.097 0.138 0.173 0.319 2.8 2.35 0.25 0.63 1000 1.9 2.2 6.4 1.1 6.9 8.8 400 400 7.1 6.5 4 12.7 35.4 14.8 37.1 37.3 2 26.9 30.8
1.1 1.6 3 20.5 20.5 27.3 1.8 2.85 6.15 10.4 6.1 14.9 500.34 1.6 3.25 0.123 0.2335 4.7 3.7 6.05 5.5 6.6 9.8 26.8 10.2 0.104 0.2675 0.548 0.52 1.8 3.2 400 3.45 5.1 80.3 48.3 400 16 0.24167 0.122 3.322 3.1295 4.682
4.5 0.875 1.5 15.711 >20 3.3 5.55 >20 >20 2.25 4.05 >20 >20 1000 2.6 1000 23.8 42.4 39.3 400 5.6 36.1 4.8 18.3 15.2 1000 6.5 150 400 4.9 400 0.0555 0.14 5.304 6.5676 6.1632 0.019 0.024 2.896 5.241 8.0455 0.052 0.174 0.091 0.1945 0.105 0.263 0.034 0.094 2.81 7.2501 9.7306 0.128 0.464 0.6095 1.45
0.8335 1.5 16.3 1.25 400 1.2 2 9.2 1000 1000 2.6 3.75 200 8.8 8.554 8.353 37.1 6.945 9.5286 >20 400 6.9 4.644 6.838 0.2415 0.489 8.1 2.274 2.2626 3.7273 7.85 17.8 2.8 3.85 0.33 0.563 0.427 0.576 0.104 0.1485 0.703 1.2 3.8 0.247 0.443 2.5 3.55 0.351 0.8685 2.6 5 15 1000 5.9
Further assays Human plasma stability 1. Materials 1.1. Plasma Animal or human plasma were purchased from BioIVT or other qualified suppliers. 1.2. Control compounds: Propantheline is used as control compound in CD-1 mouse and human plasma. Enalapril, bisacodyl, and procaine are used as control compounds in SD rat, beagle dog, and cynomolgus monkey plasma, respectively. 2. Preparation of Working Solution Stock solution: 10 mM test compound and control compound (except propantheline) in DMSO; 10 mM propantheline in H2O. Working solution: 100 µM test compound and control compound (except propantheline) in 100% DMSO; 100 µM propantheline in H2O. 3. Assay Procedure 3.1. Plasma Preparation Frozen plasma was thawed under cold water for 10 to 20 minutes, and then centrifuged at 3220×g for 5 minutes. 3.2. Incubation Aliquots of 2.00 L test compound or control compounds working solutions were mixed with 98.0 L of blank plasma from animal and human in corresponding time points T0, T10, T30, T60, and T120 in duplicate, respectively. Incubate all sample plates in a water bath at 37.0 C. The final concentration of test compound and control compounds was 2.00 M in the incubation system. 3.3. Sample Processing and Extraction Procedure At each corresponding time point, all samples were extracted with a protein precipitation method by adding 500 µL of stop solution. All sample collection plates were then sealed and shaken for 10 minutes prior to centrifugation at 4000 rpm and 4°C for 20 minutes. And 150 µL/well of the resulting supernatant were transferred to corresponding bioanalysis plate prior to LC-MS/MS analysis. 4. Bioanalytical Analysis Concentrations of test compounds and positive controls in the samples are determined by using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. 5. Data Calculation In the determination of the in vitro elimination constant, ke, of test compound and control compounds, the peak area ratios (PAR) of analyte/internal standard were used to calculate the percentage
of remaining (%Remaining) with the following equation: %Remaining PAR of analyte tointernalstandardateach time point 100 PARof analyte tointernalstandardatT 0 C k t t C 0 e ,
To a solution of (S,E)-1-cyclopropyl-3-(methylsulfonyl)prop-2-en-1-amine hydrochloride (68 mg, 0.321 mmol) and 2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5-carboxylic acid (90 mg, 0.321 mmol) in
DMF (2 mL) at 0°C was added DIEA (0.18 mL, 0.963 mmol) and T4P in EtOAc (694 mg, 50% purity, 0.963 mmol), and the mixture was stirred at 0 °C for 1 h. The resulting mixture was poured into ice-water (20 mL) and extracted with EtOAc (20 mL; 2x). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under vacuum. The residue was purified by preparative HPLC (column: Unisil 3-100 C18 Ultra 150*50mm*3 m;mobile phase: A: 0.225% FA in water, B: MeCN; B%: 38%-68%,10 min) and lyophilized directly to give the product (S,E)-N-(1-cyclopropyl-3-(methylsulfonyl)allyl)-2-(1,1- difluoroethyl)-4-phenoxypyrimidine-5-carboxamide (86.25 mg,0.197 mmol, 61.39 % yield) as a white solid. LCMS: RT 0.504min (method 7); m/z 438.1 (M +H)+ (ESI+). SFC: RT 1.270 min: (Method 13, 97.51% ee); 1H NMR (CDCl3, 400 MHz): 9.47 (s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.58-7.47 (m, 2H), 7.43- 7.35 (m, 1H), 7.27-7.20 (m, 2H), 7.03 (dd, J = 4.8, 15.2 Hz, 1H), 6.61 (d, J =15.2 Hz, 1H), 4.33-4.19 (m, 1H), 2.97 (s, 3H), 1.86 (t, J = 18.4 Hz, 3H), 1.16-1.03 (m, 1H), 0.81-0.63 (m, 2H), 0.58 - 0.44 (m, 2H); 19F NMR (CDCl3, 400 MHz): -93.83 (s, 2F) Biological evaluation of the exemplary compounds Exemplary compounds of formula (I) were tested in selected biological and/or physicochemical assays one or more times. When tested more than once, data are reported as either average values or as median values, wherein the average value, also referred to as the arithmetic mean value, represents the sum of the values obtained divided by the number of times tested, and the median value represents the middle number of the group of values when ranked in ascending or descending order. If the number of values in the data set is odd, the median value is the middle value. If the number of values in the data set is even, the median is the arithmetic mean of the two middle values. The in vitro pharmacological, pharmacokinetic and physicochemical properties of the compounds can be determined according to the following assays and methods. WRN Protein Expression and Purification WRN (amino acid 517-1093, L1074F isoform) was purchased from CRELUX GmbH, Planegg, Germany. WRN was produced in a Sf21 expression system, purified, and stored and frozen in 50 mM HEPES / NaOH, 500 mM NaCl, 5% Glycerol and 0.5 mM TCEP, pH 7.5. Forked DNA Preparation The forked DNA was prepared by mixing the two DNA strands in a 1:1.8 ratio of OLIGOB-FLU (FAM(fluorescein)-GAACGA ACA CAT CGG GTA CGT TTT TTT TTT TTT TTT TTT TTT TTT TTT TT) (SEQ ID NO.: 1) to OLIGOA-BHQ1 (TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT CGT ACC CGA TGT GTT CGT TC-BHQ1) (SEQ ID NO.: 2) in DNA-annealing buffer (10 mM Tris/HCl pH 7.5, 50 mM NaCl & 1 mM EDTA, pH 8.0). The mixture is heated to 95 °C and cooled down to 35 °C at a rate of 5°C per minute in a Roche LightCycler® 96 System.
In vitro WRN Helicase assay A continuous, fluorescence-based helicase assay was set up to measure ATP-coupled unwinding of a forked DNA substrate by WRN (517-1093, L1074F isoform). Fluorescently labelled forked DNA was used as a DNA substrate, the DNA sequences were described by Sommers et al. 2019 (doi: 10.1371/journal.pone.0210525), with different fluorescent dyes. WRN mediated separation of the forked DNA leads to an increase in fluorescence. ssDNA complementary to the quenching strand, oligo-block (GAA CGA ACA CAT CGG GTA CG) (SEQ ID NO.: 3), is included in 10-fold excess to prevent reannealing of the forked DNA. Labelled DNA was purchased from IDT (Integrated DNA Technologies, Inc.) and ssDNA oligo-block from Microsynth AG. Reaction A typical reaction consists of 30 µL of 1 nM WRN in 25 mM Tris/HCl pH 7.5, 100 mM NaCl, 1 mM DTT, 0.01 % TWEEN-20, 0.025 mg/mL BSA, 0.1 mM forked DNA substrate, 1 mM Oligo-BLOCK and 0.5 mM Mg-ATP in a 384 well black plate (Thermo Scientific). To assess inhibition, IC50s were determined by pre-incubating compounds (10 mM DMSO stock solutions) with WRN in a 12-point custom dilution series to a final DMSO concentration of 1 %. DMSO only controls were included to represent no inhibition (high control) Compounds and WRN were pre-incubated for 2 or 6 hours at room temperature, before the addition of forked DNA, ssDNA oligo-block, and Mg-ATP to initiate the reaction. Fluorescence (Ex: 485 nm, Em: 535 nm, gain 66, 5 flashes per measurement) was measured on a Tecan Spark plate reader for 45 minutes, in 60 seconds intervals at 27.5°C. All points were measured in triplicate. Data Analysis Data analysis was performed using dotmatics (www.dotmatics.com). Enzymatic activity was assessed by the rates of DNA unwinding, measured by the rate of increase in fluorescence. The initial slopes were normalized to % activity, as follows: percentage of activity (Normalized) = (Sample Value / High Control Value (Average of DMSO only wells)) * 100%. The resulting dose response was fitted with the following equation: Y=Bottom + (Top-Bottom)/(1+(IC50/X)^HillSlope), where X values are concentrations and Y is the response (decreasing as X increases). The curve fitting was performed using the least squares regression method. Reported values are the mean of at least 2 independent replicates. The Absorbance IC50 value of compounds of Formula (I) in Examples 1 to 189 are provided in Table 3 below. In case there is a blank cell in the Table, this means that no data have been generated. Cellular Viability Assay The colon carcinoma cell lines HCT116 and SW48 (both microsatellite instability- high and WRN- inhibition sensitive cell lines) and SW620 (microsatellite stable and WRN-inhibition insensitive cell line) were obtained from ATCC.
HCT116 were cultured in growth medium composed of McCoy's 5A (Modified) Medium (ThermoFisher Scientific Cat# 16600082), 1x Penicillin-Streptomycin (ThermoFisher Scientific Cat# 15140163), and 10% fetal bovine serum (ThermoFisher Scientific Cat#A5256801). SW48 and SW620 were cultured in growth medium composed of Leibovitz's L-15 Medium (ThermoFisher Scientific Cat#11415056), 1x Penicillin-Streptomycin and 10% fetal bovine serum. HCT116, SW48 and SW620 were plated at 1500 cells/well, 2000 cells/well and 2000 cells/well, respectively, in 96-well black plates with clear flat bottom (Huberlab #.655983), in a volume of 200 µL per well. The outer wells of the plate were filled with DPBS (ThermoFisher Scientific Cat#14190250) to compensate evaporation mediated effects in the plate periphery. After 24 hours, the compounds were dispensed with the Tecan digital dispenser (D300e), starting at 20 µM for a 8-point dose curve at 1:4 dilution, in duplicates. The final concentration of DMSO was normalized to the highest compound concentration and a maximum of 0.1%.. After 4d of incubation, 145 µl of the growth medium were removed and 50 µL of Cell Titer-Glo (Promega Cat#G9243) were added per well. Plates were shaken for 2 minutes and after an incubation of 10 minutes, luminescence was read using a plate reader (Tecan Spark). For data analysis, the assay background signal which was determined in wells that contained medium only was subtracted from all the data points. Averaged values of the samples were normalized to DMSO treated control samples. The fitting of the logistic curve were performed using the following equation: Y=Bottom + (Top-Bottom)/(1+(IC50/X)^HillSlope), where X values are concentrations and Y is the response (decreasing as X increases). The curve fitting was performed using the least squares regression method. The IC50 value of compounds of Formula (I) in Examples 1 to 189 are provided in Table 3 below. In case there is a blank cell in the Table, this means that no data have been generated. Table 3 Example number IC50 in M determined in WRN helicase assay described under In vitro WRN Helicase assay following 6 hours incubation IC50 in M determined in WRN helicase assay described under In vitro WRN Helicase assay following 2 hours incubation IC50 in M determined in SW48 assay described under Cellular viability assay IC50 in M determined in HCT116 assay described under Cellular viability assay
IC50 in M determined in SW620 assay described under Cellular viability assay 0.970 5.3 3.6 0.928 0.469 0.373 2.85 0.351 4.9 1.3 1.9 5.0 6.6 0.608 1.1 3.39 10 10.9 21.8 5.1 36.1 1.25 1.6 0.877 1.5 0.117 0.254 0.081 0.112 4.63 5.82 >13.77 0.253 0.435 0.700 1.00 0.251 0.345 0.274 0.366 0.274 0.523 0.703 1.15 400
0.332 0.559 (67% inhibition at 100mM) a 400 b 1000 a 1000 0.143 (65% 0.07 (40% >20 >20b inhibition at inhibition at 100mM) 100mM) 0.135 0.169 6.76 >19.09 0.074 0.071 7.02 9.56 9.57 0.034 0.042 2.47 >15.93 1.5 2.8 <0.055 0.022 >20 400 6.006 9.55 >20 1.3 2.30 0.0545 0.153 >15.25a 1000 1.862 >12.88 >20b 400 1.405 5.85 >20c 4.85 0.0805 0.455 >20d 0.727 0.019 0.071 >20 1.35 0.0285 0.1195 5.358a 54.1 0.362 1.0895 5.225b 150 0.574 2.051 5.393c 0.4495 0.0095 0.043 5.3585d 0.8185 0.013 0.059 7.472 55.9 26.7 1.1 1.1 4.25 1.4 2.6 84.3 102
0.097 0.2075 0.142 0.27 0.149 0.277 23.7 1.3 1.95 6.5 6.1 24.2 9.4 8 2.6 4.05 12.2 25.8 26.4 400 400 80.6 0.164 0.2735 119.3 3.55 400 3.2 400 65.75 6.75 5.6 1 1.4 400 3.1
18 9.55 0.976 1.3 1000 3.35 1.8 3.1 0.097 0.138 0.173 0.319 2.8 2.35 0.25 0.63 1000 1.9 2.2 6.4 1.1 6.9 8.8 400 400 7.1 6.5 4 12.7 35.4 14.8 37.1 37.3 2 26.9 30.8
1.1 1.6 3 20.5 20.5 27.3 1.8 2.85 6.15 10.4 6.1 14.9 500.34 1.6 3.25 0.123 0.2335 4.7 3.7 6.05 5.5 6.6 9.8 26.8 10.2 0.104 0.2675 0.548 0.52 1.8 3.2 400 3.45 5.1 80.3 48.3 400 16 0.24167 0.122 3.322 3.1295 4.682
4.5 0.875 1.5 15.711 >20 3.3 5.55 >20 >20 2.25 4.05 >20 >20 1000 2.6 1000 23.8 42.4 39.3 400 5.6 36.1 4.8 18.3 15.2 1000 6.5 150 400 4.9 400 0.0555 0.14 5.304 6.5676 6.1632 0.019 0.024 2.896 5.241 8.0455 0.052 0.174 0.091 0.1945 0.105 0.263 0.034 0.094 2.81 7.2501 9.7306 0.128 0.464 0.6095 1.45
0.8335 1.5 16.3 1.25 400 1.2 2 9.2 1000 1000 2.6 3.75 200 8.8 8.554 8.353 37.1 6.945 9.5286 >20 400 6.9 4.644 6.838 0.2415 0.489 8.1 2.274 2.2626 3.7273 7.85 17.8 2.8 3.85 0.33 0.563 0.427 0.576 0.104 0.1485 0.703 1.2 3.8 0.247 0.443 2.5 3.55 0.351 0.8685 2.6 5 15 1000 5.9
Further assays Human plasma stability 1. Materials 1.1. Plasma Animal or human plasma were purchased from BioIVT or other qualified suppliers. 1.2. Control compounds: Propantheline is used as control compound in CD-1 mouse and human plasma. Enalapril, bisacodyl, and procaine are used as control compounds in SD rat, beagle dog, and cynomolgus monkey plasma, respectively. 2. Preparation of Working Solution Stock solution: 10 mM test compound and control compound (except propantheline) in DMSO; 10 mM propantheline in H2O. Working solution: 100 µM test compound and control compound (except propantheline) in 100% DMSO; 100 µM propantheline in H2O. 3. Assay Procedure 3.1. Plasma Preparation Frozen plasma was thawed under cold water for 10 to 20 minutes, and then centrifuged at 3220×g for 5 minutes. 3.2. Incubation Aliquots of 2.00 L test compound or control compounds working solutions were mixed with 98.0 L of blank plasma from animal and human in corresponding time points T0, T10, T30, T60, and T120 in duplicate, respectively. Incubate all sample plates in a water bath at 37.0 C. The final concentration of test compound and control compounds was 2.00 M in the incubation system. 3.3. Sample Processing and Extraction Procedure At each corresponding time point, all samples were extracted with a protein precipitation method by adding 500 µL of stop solution. All sample collection plates were then sealed and shaken for 10 minutes prior to centrifugation at 4000 rpm and 4°C for 20 minutes. And 150 µL/well of the resulting supernatant were transferred to corresponding bioanalysis plate prior to LC-MS/MS analysis. 4. Bioanalytical Analysis Concentrations of test compounds and positive controls in the samples are determined by using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. 5. Data Calculation In the determination of the in vitro elimination constant, ke, of test compound and control compounds, the peak area ratios (PAR) of analyte/internal standard were used to calculate the percentage
of remaining (%Remaining) with the following equation: %Remaining PAR of analyte tointernalstandardateach time point 100 PARof analyte tointernalstandardatT 0 C k t t C 0 e ,
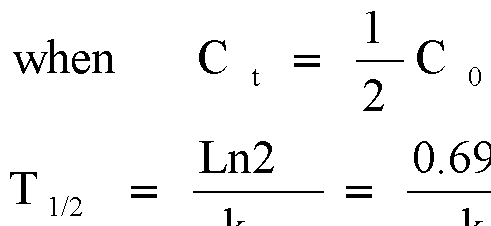 e e When the %Remaining value at the maximal incubation time, which was 120 min in this study, was higher than 75%, it is considered to be within the acceptable experimental variation, i.e., CV =25%. Therefore, a corresponding T1/2 value of >289.1 min is reported. 6. Reference [1] Li Di, Edward H. Kerns, Yan Hong, Hong Chen, International Journal of Pharmaceutics. Development and application of high throughput plasma stability assay for drug discovery 297 (2005) 110- 119. The T1/2 value is reported in table 4 for selected examples. As evidenced by the data in Table 4, Examples 32, 32d, 33 and 33d show an improved in vitro stability in human plasma in comparison to Reference Example 1 Table 4 The T1/2 value is reported for selected example compounds. Examples Human plasma T1/2 (mn) Example 32 159.5 Example 32d 123.9 Example 33 137.2 Example 33c 90 Reference Example 1 56.6 Kinetic solubility assay The Kinetic solubility assay employs the shake flask method followed by HPLC-UV analysis. For exemplary compounds, the kinetic solubility was measured according to the following protocol: 1. Samples were weighed and dissolved in 100% DMSO to make a stock solution of 10 mM. About 100 L of stock solution is needed to cover this assay.
2. Test compounds and controls (10 mM in DMSO, 10 L/tube) were added into the buffer (490 L/well) which were placed in a Mini-UniPrep filter. The buffer was prepared as the customer’s requirement. 3. The kinetic solubility samples were vortexed for 2 minutes. 4. The solubility solutions were shaken in an orbital shaker for 24 hr at room temperature . 5.200 L of each solubility solution were transferred into a 96-deep well for analysis when the samples were directly filtered by the syringeless filter device. 6. The test compound concentration of the filtrate were determined using HPLC-UV. 7. Three UV standard solutions were injected into HPLC from low to high concentrations, followed by testing of the K.S. supernatant. Testing samples were injected in duplicate. Bidirectional permeability in Caco2 The bidirectional permeability in Caco-2 cells assay was performed for the exemplary compounds of formula (I) according to the following protocol: 1. Caco-2 cells purchased from ATCC were seeded onto polyethylene membranes (PET) in 96- well BD insert plates at 1 x 105 cells/cm2, and refreshed medium every 4~5 days until to the 21st to 28th day for confluent cell monolayer formation. 2. The integrity of the monolayer was verified by performing Lucifer yellow rejection assay. 3. The quality of the monolayer was verified by measuring the unidirectional (A B) permeability of fenoterol/nadolol (low permeability marker), propranolol/metopronolol (high permeability marker) and bi-directional permeability of digoxin (a P-glycoprotein substrate marker) in duplicate wells. 4. Standard assay conditions for test compounds: test concentration: 2 M (DMSO 1%); replicates: n=2; directions: bi-directional transport including A B and B A; incubation time: single time point, 2hours; transport buffer: HBSS containing 10 mM HEPES, pH7.40±0.05; incubation condition: 37±1°C, 5% CO2, relatively saturated humidity. 5. Dosing solution were spiked and mixed with transport buffer and stop solution (containing an appropriate internal standard (IS)) as T0 sample. 6. At the end of incubation, sample solutions from both donor and receiver wells were mixed with stop solution immediately. 7. All samples including T0 samples, donor samples and receiver samples were analyzed using LC/MS/MS. Concentrations of test compound were expressed as peak area ratio of analytes versus
IS without a standard curve. Liver Microsomes Stability Assay 1. Materials 1.1 Liver microsomes Animal or human liver microsomes were purchased from Xenotech or Corning and stored in a freezer (lower than -60°C) before use. 1.2 -nicotinamide adenine dinucleotide phosphate reduced form, tetrasodium salt, Vendor: Chem-Impex International, Cat.No.00616 1.3 Control compounds: Testosterone, diclofenac and propafenone. 2. Preparation of Working Solution Stock Solution: 10 mM test compound in DMSO. Working solution: 100 µM test or control compounds in 100% acetonitrile (concentration of organic solvent: 1% (v/v) DMSO and 99% (v/v) acetonitrile) 3. Assay Procedure A total of two sample plates with 96-well format were prepared for incubation, labeled as 'Incubation' T60 and 'Incubation' NCF60. Empty 'Incubation' T60 and NCF60 plates were pre-warmed for 10 min minutes. Liver microsomes were diluted to 0.56 mg/mL in 100 mM phosphate buffer. Microsome working solutions (0.56 mg/mL) were transferred (445 uL) into pre-warmed 'Incubation' T60 and NCF60 plates, followed by incubation for 10 min at 37°C with constant shaking. Liver microsomes (54 µL) were transferred to a Blank60 plate, followed by the addition of 6 µL NAPDH cofactor and 180 µL stop solution (acetonitrile containing internal standards) into each well. An aliquot (5 µL) of compound working solution (100 M) was added into the 'incubation' plates (T60 and NCF60) containing microsomes and mixed 3 times thoroughly. For the 'Incubation' NCF60 plate, 50 uL of buffer was added and mixed 3 times thoroughly. The plates were incubated at 37°C for 60 min while shaking. samples were mixed once and 60 µL was transferred from the NCF60 incubation plate to the stop plate containing stop solution after the 60- min incubation. Stop solution (180 µL) and NAPDH cofactor (6 µL) were added to 'Quenching' plate T0. Plates were chilled to prevent evaporation. For the 'Incubation' T60 plate: mixed 3 times thoroughly, and immediately removed 54 µL mixture for the 0-min time point to stop plate ('Quenching' plate T0). NAPDH cofactor (44 µL) was added to the 'Incubation' T60 plate. The plate was incubated at 37°C for 60 min while shaking. At 5, 15,
30, 45, and 60 min, 180 µL stop solution was added to the 'Quenching' plates, samples were mixed once, and 60 µL was serially transferred from 'Incubation' T60 plate per time point. Consequently, for the wells containing the test or control compounds, the final concentration was 1 M for test compounds, testosterone, diclofenac and propafenone, 0.5 mg/mL for animal or human liver microsomes, 0.01% (v/v) for DMSO and 0.99% (v/v) for acetonitrile. All sampling plates were shaken for 10 min, then centrifuged at 3220 ×g for 20 minutes at 4°C. Supernatant (80 µL) was transferred into 240 µL HPLC water, and mixed by plate shaker for 10 min. Each bioanalysis plate was sealed and shaken for 10 minutes prior to LC-MS/MS analysis. 4. Bioanalytical Analysis Concentrations of test conpounds and positive controls, testosterone, diclofenac and propafenone in the samples were determined by using a liquid chromatography-tandem mass spectrometry (LC- MS/MS) method. 5. Data Calculation In the determination of the in vitro elimination constant, ke, of test compound and control compounds, the peak area ratios (PAR) of analyte/internal standard were used to calculate the percentage of remaining (%Remaining) with the following equation:
e e When the %Remaining value at the maximal incubation time, which was 120 min in this study, was higher than 75%, it is considered to be within the acceptable experimental variation, i.e., CV =25%. Therefore, a corresponding T1/2 value of >289.1 min is reported. 6. Reference [1] Li Di, Edward H. Kerns, Yan Hong, Hong Chen, International Journal of Pharmaceutics. Development and application of high throughput plasma stability assay for drug discovery 297 (2005) 110- 119. The T1/2 value is reported in table 4 for selected examples. As evidenced by the data in Table 4, Examples 32, 32d, 33 and 33d show an improved in vitro stability in human plasma in comparison to Reference Example 1 Table 4 The T1/2 value is reported for selected example compounds. Examples Human plasma T1/2 (mn) Example 32 159.5 Example 32d 123.9 Example 33 137.2 Example 33c 90 Reference Example 1 56.6 Kinetic solubility assay The Kinetic solubility assay employs the shake flask method followed by HPLC-UV analysis. For exemplary compounds, the kinetic solubility was measured according to the following protocol: 1. Samples were weighed and dissolved in 100% DMSO to make a stock solution of 10 mM. About 100 L of stock solution is needed to cover this assay.
2. Test compounds and controls (10 mM in DMSO, 10 L/tube) were added into the buffer (490 L/well) which were placed in a Mini-UniPrep filter. The buffer was prepared as the customer’s requirement. 3. The kinetic solubility samples were vortexed for 2 minutes. 4. The solubility solutions were shaken in an orbital shaker for 24 hr at room temperature . 5.200 L of each solubility solution were transferred into a 96-deep well for analysis when the samples were directly filtered by the syringeless filter device. 6. The test compound concentration of the filtrate were determined using HPLC-UV. 7. Three UV standard solutions were injected into HPLC from low to high concentrations, followed by testing of the K.S. supernatant. Testing samples were injected in duplicate. Bidirectional permeability in Caco2 The bidirectional permeability in Caco-2 cells assay was performed for the exemplary compounds of formula (I) according to the following protocol: 1. Caco-2 cells purchased from ATCC were seeded onto polyethylene membranes (PET) in 96- well BD insert plates at 1 x 105 cells/cm2, and refreshed medium every 4~5 days until to the 21st to 28th day for confluent cell monolayer formation. 2. The integrity of the monolayer was verified by performing Lucifer yellow rejection assay. 3. The quality of the monolayer was verified by measuring the unidirectional (A B) permeability of fenoterol/nadolol (low permeability marker), propranolol/metopronolol (high permeability marker) and bi-directional permeability of digoxin (a P-glycoprotein substrate marker) in duplicate wells. 4. Standard assay conditions for test compounds: test concentration: 2 M (DMSO 1%); replicates: n=2; directions: bi-directional transport including A B and B A; incubation time: single time point, 2hours; transport buffer: HBSS containing 10 mM HEPES, pH7.40±0.05; incubation condition: 37±1°C, 5% CO2, relatively saturated humidity. 5. Dosing solution were spiked and mixed with transport buffer and stop solution (containing an appropriate internal standard (IS)) as T0 sample. 6. At the end of incubation, sample solutions from both donor and receiver wells were mixed with stop solution immediately. 7. All samples including T0 samples, donor samples and receiver samples were analyzed using LC/MS/MS. Concentrations of test compound were expressed as peak area ratio of analytes versus
IS without a standard curve. Liver Microsomes Stability Assay 1. Materials 1.1 Liver microsomes Animal or human liver microsomes were purchased from Xenotech or Corning and stored in a freezer (lower than -60°C) before use. 1.2 -nicotinamide adenine dinucleotide phosphate reduced form, tetrasodium salt, Vendor: Chem-Impex International, Cat.No.00616 1.3 Control compounds: Testosterone, diclofenac and propafenone. 2. Preparation of Working Solution Stock Solution: 10 mM test compound in DMSO. Working solution: 100 µM test or control compounds in 100% acetonitrile (concentration of organic solvent: 1% (v/v) DMSO and 99% (v/v) acetonitrile) 3. Assay Procedure A total of two sample plates with 96-well format were prepared for incubation, labeled as 'Incubation' T60 and 'Incubation' NCF60. Empty 'Incubation' T60 and NCF60 plates were pre-warmed for 10 min minutes. Liver microsomes were diluted to 0.56 mg/mL in 100 mM phosphate buffer. Microsome working solutions (0.56 mg/mL) were transferred (445 uL) into pre-warmed 'Incubation' T60 and NCF60 plates, followed by incubation for 10 min at 37°C with constant shaking. Liver microsomes (54 µL) were transferred to a Blank60 plate, followed by the addition of 6 µL NAPDH cofactor and 180 µL stop solution (acetonitrile containing internal standards) into each well. An aliquot (5 µL) of compound working solution (100 M) was added into the 'incubation' plates (T60 and NCF60) containing microsomes and mixed 3 times thoroughly. For the 'Incubation' NCF60 plate, 50 uL of buffer was added and mixed 3 times thoroughly. The plates were incubated at 37°C for 60 min while shaking. samples were mixed once and 60 µL was transferred from the NCF60 incubation plate to the stop plate containing stop solution after the 60- min incubation. Stop solution (180 µL) and NAPDH cofactor (6 µL) were added to 'Quenching' plate T0. Plates were chilled to prevent evaporation. For the 'Incubation' T60 plate: mixed 3 times thoroughly, and immediately removed 54 µL mixture for the 0-min time point to stop plate ('Quenching' plate T0). NAPDH cofactor (44 µL) was added to the 'Incubation' T60 plate. The plate was incubated at 37°C for 60 min while shaking. At 5, 15,
30, 45, and 60 min, 180 µL stop solution was added to the 'Quenching' plates, samples were mixed once, and 60 µL was serially transferred from 'Incubation' T60 plate per time point. Consequently, for the wells containing the test or control compounds, the final concentration was 1 M for test compounds, testosterone, diclofenac and propafenone, 0.5 mg/mL for animal or human liver microsomes, 0.01% (v/v) for DMSO and 0.99% (v/v) for acetonitrile. All sampling plates were shaken for 10 min, then centrifuged at 3220 ×g for 20 minutes at 4°C. Supernatant (80 µL) was transferred into 240 µL HPLC water, and mixed by plate shaker for 10 min. Each bioanalysis plate was sealed and shaken for 10 minutes prior to LC-MS/MS analysis. 4. Bioanalytical Analysis Concentrations of test conpounds and positive controls, testosterone, diclofenac and propafenone in the samples were determined by using a liquid chromatography-tandem mass spectrometry (LC- MS/MS) method. 5. Data Calculation In the determination of the in vitro elimination constant, ke, of test compound and control compounds, the peak area ratios (PAR) of analyte/internal standard were used to calculate the percentage of remaining (%Remaining) with the following equation:
 CLint(mic) = 0.693 / T1/2 / mg microsome protein per mL CLint(liver) = CLint(mic) × mg microsomal protein/g liver weight × g liver weight/kg body weight According to the well-stirred model, hepatic intrinsic clearance and hepatic clearance can be calculated by the following formula. CL(liver) = (CLint(liver) × fu × Qh)/ (CLint(liver) × fu + Qh) The default value of fu (the fraction unbound in blood) is assumed as 1. The parameters in equations are listed in following table. Species Liver Weight Hepatic Blood Flow (Qh) (mL/min/kg) [1-2] Microsomal Protein
(g/kg Body Weight) [1-2] (mg/g liver weight) Mouse 88 90.0 Rat 40 55.2 Dog 32 30.9 45 Monkey 30 43.6 Human 20 20.7 When the %Remaining value at the maximal incubation time, which was 60 min in this study, was higher than 75%, it is considered to be within the acceptable experimental variation, i.e., CV =25%. Therefore, a corresponding T1/2 value of >145 min is reported. Consequently, the corresponding CLint(mic)value is reported as <9.6 L/min/mg protein. 6. References [1] Brian Davies and Tim Morris, Physiological Parameters in Laboratory Animals and Human. Pharmaceutical Research, Vol.10 No.7, 1993 [2] Journal of Pharmacology and Experimental Therapeutics, 1997, 283(1): 46-58 Hepatocyte Metabolic Stability 1. Materials 1.1 Hepatocyte Animal or human hepatocytes were purchased from BioreclamationIVT or RILD. 1.2 Control compounds: 7-Ethoxycoumarin and 7-Hydroxycoumarin 2. Preparation of Working Solution Stock Solution: 10 mM test compound and 30 mM control compound in DMSO. Working solution: 100 µM test compound or 300 µM control compounds in 100% acetonitrile (Concentration of organic solvent: 1% (v/v) DMSO and 99% (v/v) acetonitrile) 3. Assay Procedure Cryopreserved hepatocytes were thawed, isolated, and suspended in Williams’ Medium E, then diluted with pre-incubated Williams’ Medium E to a final concentration of 0.510×106 cells/mL. One hundred and ninety-eight (198) L of cells suspension (0.510×106 cells/mL) were added into appropriate wells. The incubation plate was pre-incubated in a 37.0 C incubator for about 10 minutes. Then 2 L of test compound and positive controls were added into plate except for the blank plate. Incubate all plates at 37.0°C in a 95.0% humidified incubator at 5.0% CO2 to start the reactions with constant shaking.
For the T0 plate, a corresponding quenching plate was prepared by adding 125 µL/well of acetonitrile containing 200 ng/mL tolbutamide and 200 ng/mL labetalol as internal standards (stop solution), and 25 µL/well of the incubation sample were transferred to this plate after shaking for 1 minute to ensure homogeneity. At each time-point, the corresponding plate was removed from the incubator, and 25 µL/well of the corresponding sample was transferred to its corresponding quenching plate containing 125 µL/well of stop solution. Medium control (MC) plates (T0-MC and T90-MC) were prepared by adding everything except for Williams’ Medium E at the corresponding time-points. The plates were then sealed and shaken for 10 minutes prior to centrifugation at 4000 rpm and 4°C for 20 minutes. 80 µL/well of the resulting supernatant were diluted with 240 µL/well of pure water and sealed and shaken for 10 minutes prior to LC-MS/MS analysis. 4. Bioanalytical Analysis Concentrations of test compounds and positive controls, 7-Ethoxycoumarin and 7- Hydroxycoumarin in the samples were determined by using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. 5. Data Calculation In the determination of the in vitro elimination constant, ke, of test compound and control compounds, the peak area ratios (PAR) of analyte/internal standard were used to calculate the percentage of remaining (%Remaining) with the following equation:
CLint(mic) = 0.693 / T1/2 / mg microsome protein per mL CLint(liver) = CLint(mic) × mg microsomal protein/g liver weight × g liver weight/kg body weight According to the well-stirred model, hepatic intrinsic clearance and hepatic clearance can be calculated by the following formula. CL(liver) = (CLint(liver) × fu × Qh)/ (CLint(liver) × fu + Qh) The default value of fu (the fraction unbound in blood) is assumed as 1. The parameters in equations are listed in following table. Species Liver Weight Hepatic Blood Flow (Qh) (mL/min/kg) [1-2] Microsomal Protein
(g/kg Body Weight) [1-2] (mg/g liver weight) Mouse 88 90.0 Rat 40 55.2 Dog 32 30.9 45 Monkey 30 43.6 Human 20 20.7 When the %Remaining value at the maximal incubation time, which was 60 min in this study, was higher than 75%, it is considered to be within the acceptable experimental variation, i.e., CV =25%. Therefore, a corresponding T1/2 value of >145 min is reported. Consequently, the corresponding CLint(mic)value is reported as <9.6 L/min/mg protein. 6. References [1] Brian Davies and Tim Morris, Physiological Parameters in Laboratory Animals and Human. Pharmaceutical Research, Vol.10 No.7, 1993 [2] Journal of Pharmacology and Experimental Therapeutics, 1997, 283(1): 46-58 Hepatocyte Metabolic Stability 1. Materials 1.1 Hepatocyte Animal or human hepatocytes were purchased from BioreclamationIVT or RILD. 1.2 Control compounds: 7-Ethoxycoumarin and 7-Hydroxycoumarin 2. Preparation of Working Solution Stock Solution: 10 mM test compound and 30 mM control compound in DMSO. Working solution: 100 µM test compound or 300 µM control compounds in 100% acetonitrile (Concentration of organic solvent: 1% (v/v) DMSO and 99% (v/v) acetonitrile) 3. Assay Procedure Cryopreserved hepatocytes were thawed, isolated, and suspended in Williams’ Medium E, then diluted with pre-incubated Williams’ Medium E to a final concentration of 0.510×106 cells/mL. One hundred and ninety-eight (198) L of cells suspension (0.510×106 cells/mL) were added into appropriate wells. The incubation plate was pre-incubated in a 37.0 C incubator for about 10 minutes. Then 2 L of test compound and positive controls were added into plate except for the blank plate. Incubate all plates at 37.0°C in a 95.0% humidified incubator at 5.0% CO2 to start the reactions with constant shaking.
For the T0 plate, a corresponding quenching plate was prepared by adding 125 µL/well of acetonitrile containing 200 ng/mL tolbutamide and 200 ng/mL labetalol as internal standards (stop solution), and 25 µL/well of the incubation sample were transferred to this plate after shaking for 1 minute to ensure homogeneity. At each time-point, the corresponding plate was removed from the incubator, and 25 µL/well of the corresponding sample was transferred to its corresponding quenching plate containing 125 µL/well of stop solution. Medium control (MC) plates (T0-MC and T90-MC) were prepared by adding everything except for Williams’ Medium E at the corresponding time-points. The plates were then sealed and shaken for 10 minutes prior to centrifugation at 4000 rpm and 4°C for 20 minutes. 80 µL/well of the resulting supernatant were diluted with 240 µL/well of pure water and sealed and shaken for 10 minutes prior to LC-MS/MS analysis. 4. Bioanalytical Analysis Concentrations of test compounds and positive controls, 7-Ethoxycoumarin and 7- Hydroxycoumarin in the samples were determined by using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. 5. Data Calculation In the determination of the in vitro elimination constant, ke, of test compound and control compounds, the peak area ratios (PAR) of analyte/internal standard were used to calculate the percentage of remaining (%Remaining) with the following equation:

 CLint (hep) = k / million cells per mL CLint (liver) = CLint (hep) × liver weight (g/kg body weight) × hepatocellularity According to the well-stirred model, hepatic intrinsic clearance and hepatic clearance can be calculated by the following formula. CL(liver) = (CLint(liver) × fu × Qh)/ (CLint(liver) × fu + Qh) The default value of fu (the fraction unbound in blood) is assumed as 1.
The parameters in equations are listed in following table. Liver Blo Hepatocellularity Species Liver Weight (g od Flow [2 /k -3 g ] Body Weight) (Qh) (mL/min/kg) [2-3]
CLint (hep) = k / million cells per mL CLint (liver) = CLint (hep) × liver weight (g/kg body weight) × hepatocellularity According to the well-stirred model, hepatic intrinsic clearance and hepatic clearance can be calculated by the following formula. CL(liver) = (CLint(liver) × fu × Qh)/ (CLint(liver) × fu + Qh) The default value of fu (the fraction unbound in blood) is assumed as 1.
The parameters in equations are listed in following table. Liver Blo Hepatocellularity Species Liver Weight (g od Flow [2 /k -3 g ] Body Weight) (Qh) (mL/min/kg) [2-3]
 Mouse 88 90.0 135×106 Rat 40 55.2 117×106 Dog 32 30.9 215×106 Monkey 30 43.6 120×106 Human 20 20.7 139×106 When the %Remaining value at the maximal incubation time, which was 90 min in this study, was higher than 75%, it is considered to be within the acceptable experimental variation, i.e., CV =25%. Therefore, a corresponding T1/2 value of >216.8 min is reported. Consequently, the corresponding CLint(hep) (µL/min/106 cells) is reported as <7.5. 6. References [1] Anna-Karin Sohlenius-Sternbeck. Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicology in Vitro, Vol.20 No.8, 2006 [2] Brian Davies and Tim Morris, Physiological Parameters in Laboratory Animals and Human. Pharmaceutical Research, Vol.10 No.7, 1993 [3] Obach R S, Baxter J G, Liston T E, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data [J]. Journal of Pharmacology and Experimental Therapeutics, 1997, 283(1): 46 Mouse PK Group Test Article No. of Sex Dose Dose Conc Dose Vehicle No. animals Route (mg/kg) (mg/mL) Volume 01 Example 32d 3 Female IV bolus 1 0.2 5 10%NMP+90% water 10%NMP+15%Kolliph 02 3 Female PO 10
Mouse 88 90.0 135×106 Rat 40 55.2 117×106 Dog 32 30.9 215×106 Monkey 30 43.6 120×106 Human 20 20.7 139×106 When the %Remaining value at the maximal incubation time, which was 90 min in this study, was higher than 75%, it is considered to be within the acceptable experimental variation, i.e., CV =25%. Therefore, a corresponding T1/2 value of >216.8 min is reported. Consequently, the corresponding CLint(hep) (µL/min/106 cells) is reported as <7.5. 6. References [1] Anna-Karin Sohlenius-Sternbeck. Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicology in Vitro, Vol.20 No.8, 2006 [2] Brian Davies and Tim Morris, Physiological Parameters in Laboratory Animals and Human. Pharmaceutical Research, Vol.10 No.7, 1993 [3] Obach R S, Baxter J G, Liston T E, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data [J]. Journal of Pharmacology and Experimental Therapeutics, 1997, 283(1): 46 Mouse PK Group Test Article No. of Sex Dose Dose Conc Dose Vehicle No. animals Route (mg/kg) (mg/mL) Volume 01 Example 32d 3 Female IV bolus 1 0.2 5 10%NMP+90% water 10%NMP+15%Kolliph 02 3 Female PO 10
 or EL
or EL
 +1%Tween20+74% (20%HP- -CD)
+1%Tween20+74% (20%HP- -CD)
 0.2 5 10%NMP+90% water 10%NMP+15%Kolliph
0.2 5 10%NMP+90% water 10%NMP+15%Kolliph
 or EL +1%Tween20+74% (20%HP- -CD) SAMPLE COLLECTION Group Dosage (mg/kg) Animal No. Matrix Sampling time point (hr) 01 1 M01, M02, Plasm 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M03 a post dose 02 10 M04, M05, Pl 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M06 asma post dose 03 1 M07, M08, 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M09 Plasma post dose 04 10 M10, M11, 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M12 Plasma post dose ANIMAL SPECIFICATIONS Strain: BALB/c nude Species: Mouse Vender: LC or other qualified vendors Gender: Female Age: 6-9 W Total Number of Animals: 6 Overnight Fast of Animals: True Comments: Fast for PO, fed for IV Food Returned: 4 hours post dose. Only for PO. Euthanasia: CO2 euthanasia Animal No. of Non-surgery: 6 Species Abbr: M
Table 5. Mouse PK parameters reported for selected examples PK parameters Example 32d Reference example 1 IV T1/2 [h], (1mpk) 0.88 0.25 IV CL [mL/min/kg], (1mpk) 37.9 48.1 AUC0-inf [ng.h/mL] (10 mpk) 4594 1794 Bioavailability [%], PO, 10mpk 104 52 As evidenced by the data in Table 5 example 32d shows a longer half-life (T1/2) and a reduced plasma clearance (CL) after iv application as well as improved exposure (AUC) and bioavailability after oral application in comparison to reference example 1.
or EL +1%Tween20+74% (20%HP- -CD) SAMPLE COLLECTION Group Dosage (mg/kg) Animal No. Matrix Sampling time point (hr) 01 1 M01, M02, Plasm 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M03 a post dose 02 10 M04, M05, Pl 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M06 asma post dose 03 1 M07, M08, 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M09 Plasma post dose 04 10 M10, M11, 8 points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr M12 Plasma post dose ANIMAL SPECIFICATIONS Strain: BALB/c nude Species: Mouse Vender: LC or other qualified vendors Gender: Female Age: 6-9 W Total Number of Animals: 6 Overnight Fast of Animals: True Comments: Fast for PO, fed for IV Food Returned: 4 hours post dose. Only for PO. Euthanasia: CO2 euthanasia Animal No. of Non-surgery: 6 Species Abbr: M
Table 5. Mouse PK parameters reported for selected examples PK parameters Example 32d Reference example 1 IV T1/2 [h], (1mpk) 0.88 0.25 IV CL [mL/min/kg], (1mpk) 37.9 48.1 AUC0-inf [ng.h/mL] (10 mpk) 4594 1794 Bioavailability [%], PO, 10mpk 104 52 As evidenced by the data in Table 5 example 32d shows a longer half-life (T1/2) and a reduced plasma clearance (CL) after iv application as well as improved exposure (AUC) and bioavailability after oral application in comparison to reference example 1.
Claims
New PCT international application based on US 63/587,354 and US 63/665,751 FoRx Therapeutics AG Vossius Ref.: AE3393 PCT BS Claims 1. A compound of formula
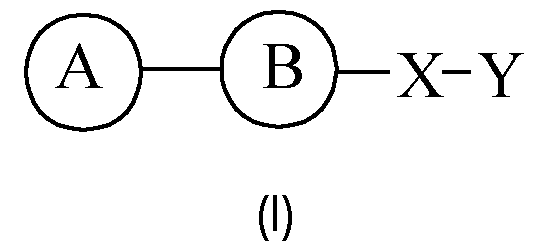 or a pharmaceutically acceptable salt thereof, wherein: A is selected from aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl, cycloalkenyl, C2 alkynyl, -N(C1-5 alkyl)(C1-5 alkyl) (such as -N(CH3)(CH(CH3)2)), C2-haloalkyl (such as -CF2CH3) and –(C1-2 haloalkylene)-cycloalkyl (such as -CF2-cyclopropyl), wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl, C1-6 haloalkyl, aryl (such as phenyl) or heteroaryl (such as thien-2-yl), preferably with C1-6 alkyl, C1-6 haloalkyl, or aryl (such as phenyl), more preferably with C1-6 alkyl or C1-6 haloalkyl, such as C1-6 alkyl; B is selected from arylene and heteroarylene, wherein said arylene and said hereroarylene are each optionally substituted with one or more R2; X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, -SO2N(C1-6 alkyl)- , -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, -CH(CF3)N(C1-6 haloalkyl)-, C2 alkenylene optionally substituted with one or more halogen, C1-6 alkyl or C1-6 haloalkyl, cycloalkylene optionally substituted with one or more C1-6 alkyl, -(heterocycloalkylene)-NH-, - (heterocycloalkylene)-N(C1-6 alkyl)-, heterocycloalkylene, heterocycloalkenylene and heteroarylene;
or a pharmaceutically acceptable salt thereof, wherein: A is selected from aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl, cycloalkenyl, C2 alkynyl, -N(C1-5 alkyl)(C1-5 alkyl) (such as -N(CH3)(CH(CH3)2)), C2-haloalkyl (such as -CF2CH3) and –(C1-2 haloalkylene)-cycloalkyl (such as -CF2-cyclopropyl), wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl, C1-6 haloalkyl, aryl (such as phenyl) or heteroaryl (such as thien-2-yl), preferably with C1-6 alkyl, C1-6 haloalkyl, or aryl (such as phenyl), more preferably with C1-6 alkyl or C1-6 haloalkyl, such as C1-6 alkyl; B is selected from arylene and heteroarylene, wherein said arylene and said hereroarylene are each optionally substituted with one or more R2; X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, -SO2N(C1-6 alkyl)- , -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, -CH(CF3)N(C1-6 haloalkyl)-, C2 alkenylene optionally substituted with one or more halogen, C1-6 alkyl or C1-6 haloalkyl, cycloalkylene optionally substituted with one or more C1-6 alkyl, -(heterocycloalkylene)-NH-, - (heterocycloalkylene)-N(C1-6 alkyl)-, heterocycloalkylene, heterocycloalkenylene and heteroarylene;
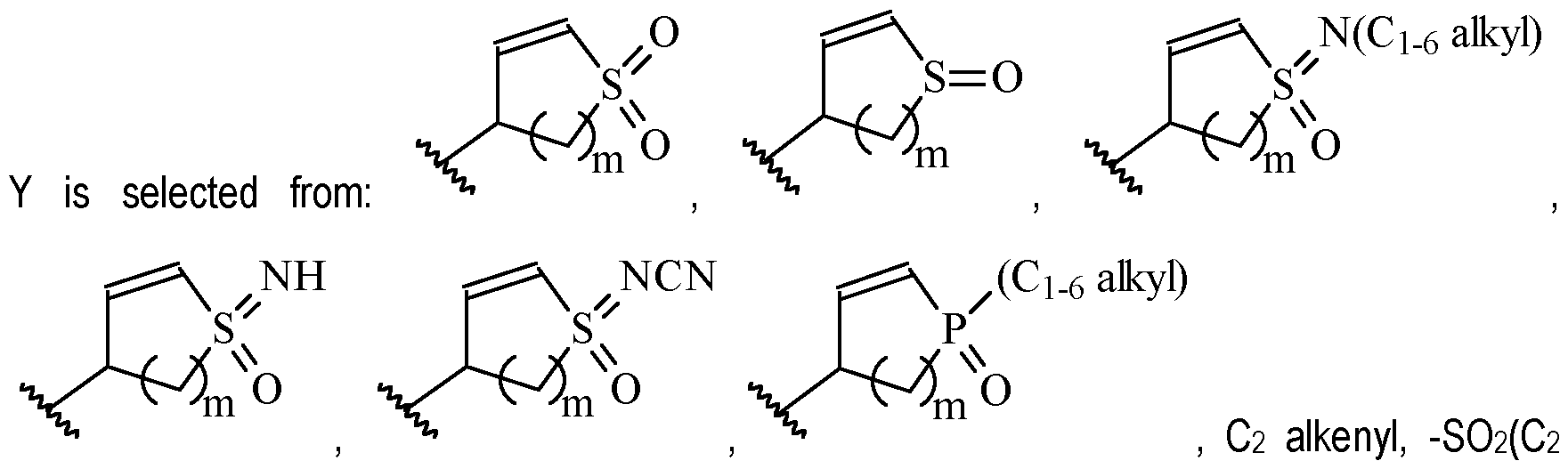 alkenyl), -CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein
said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6- membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, C1-4 haloalkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO3H, -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, -SO2- cycloalkyl, -SO(=NH)-cycloalkyl, -SO(=N-(C1-4 alkyl))-cycloalkyl, heterocycloalkyl, -SO2- heterocycloalkyl, -SO(=NH)-heterocycloalkyl, -SO(=N-(C1-4 alkyl))-heterocycloalkyl, aryl, -SO2- aryl, -SO(=NH)-aryl, -SO(=N-(C1-4 alkyl))-aryl, heteroaryl, -SO2-heteroaryl, -SO(=NH)-heteroaryl, -SO(=N-(C1-4 alkyl))-heteroaryl,-Hal, -CN and -CF3, and wherein said
alkenyl), -CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2, wherein
said -CH2- is optionally substituted with C1-4 alkyl, C1-4 haloalkyl, C3-8 cycloalkyl or 5 or 6- membered heterocyclyl, and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, C1-4 haloalkyl, -COOH, -COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO3H, -SO2(C1-4 alkyl), -SO(=NH)-(C1-4 alkyl), -SO(=N-(C1-4 alkyl))-(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, -SO2- cycloalkyl, -SO(=NH)-cycloalkyl, -SO(=N-(C1-4 alkyl))-cycloalkyl, heterocycloalkyl, -SO2- heterocycloalkyl, -SO(=NH)-heterocycloalkyl, -SO(=N-(C1-4 alkyl))-heterocycloalkyl, aryl, -SO2- aryl, -SO(=NH)-aryl, -SO(=N-(C1-4 alkyl))-aryl, heteroaryl, -SO2-heteroaryl, -SO(=NH)-heteroaryl, -SO(=N-(C1-4 alkyl))-heteroaryl,-Hal, -CN and -CF3, and wherein said
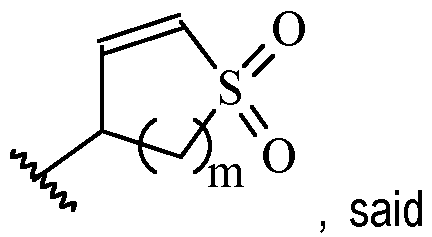
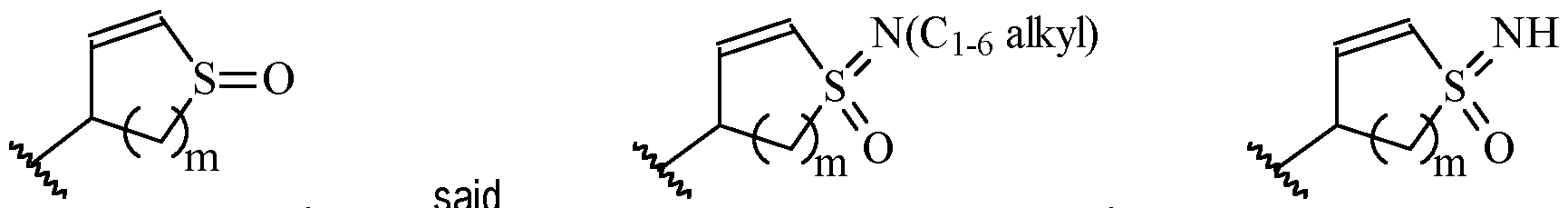 , , ,
, , ,
 are each optionally substituted by one or more C1-6 alkyl or Hal; each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3
alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl, -NH-(C0-3 alkylene)-carbocyclyl, -NH-(C0-3 alkylene)-heterocyclyl, -N(C1-5 alkyl)-(C0-3 alkylene)-carbocyclyl, -N(C1-5 alkyl)-(C0-3 alkylene)-heterocyclyl, -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said - O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -NH-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -N(C1-5 alkyl)-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl, the heterocyclyl moiety in said -NH-(C0-3 alkylene)-heterocyclyl, the heterocyclyl moiety in said - N(C1-5 alkyl)-(C0-3 alkylene)-heterocyclyl, and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, - NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), -CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)-CO(C1-4 alkyl); R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO- O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the
carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), - CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)- CO(C1-4 alkyl); or wherein A and Y are as defined hereinabove and B and X are combined to form a two-ring system according to formula:
are each optionally substituted by one or more C1-6 alkyl or Hal; each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3
alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl, -NH-(C0-3 alkylene)-carbocyclyl, -NH-(C0-3 alkylene)-heterocyclyl, -N(C1-5 alkyl)-(C0-3 alkylene)-carbocyclyl, -N(C1-5 alkyl)-(C0-3 alkylene)-heterocyclyl, -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said - O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -NH-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -N(C1-5 alkyl)-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl, the heterocyclyl moiety in said -NH-(C0-3 alkylene)-heterocyclyl, the heterocyclyl moiety in said - N(C1-5 alkyl)-(C0-3 alkylene)-heterocyclyl, and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, - NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), -CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)-CO(C1-4 alkyl); R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO- O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the
carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), - CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)- CO(C1-4 alkyl); or wherein A and Y are as defined hereinabove and B and X are combined to form a two-ring system according to formula:
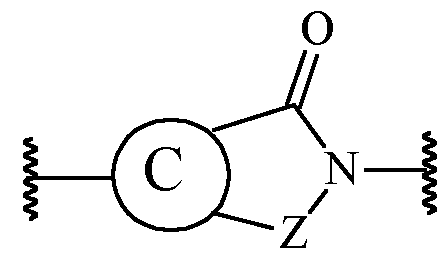 wherein C is aryl or heteroaryl ring, optionally substituted with one or more R2, Z is selected from C1-2 alkylene, -CH=CH-, -N=CH-, -N=C(C1-6 alkyl)-, -CH=N-, -C(C1-6 alkyl)=N-, -CO-, -SO-, -SO2- , -CONH-, -NHCO-, -CON(C1-6 alkyl)-, -N(C1-6 alkyl)CO-, -NH-, -N(C1-6 alkyl)- and -O-; or wherein A and B are as defined hereinabove, and X and Y form a two ring spiro system according to formula:
wherein C is aryl or heteroaryl ring, optionally substituted with one or more R2, Z is selected from C1-2 alkylene, -CH=CH-, -N=CH-, -N=C(C1-6 alkyl)-, -CH=N-, -C(C1-6 alkyl)=N-, -CO-, -SO-, -SO2- , -CONH-, -NHCO-, -CON(C1-6 alkyl)-, -N(C1-6 alkyl)CO-, -NH-, -N(C1-6 alkyl)- and -O-; or wherein A and B are as defined hereinabove, and X and Y form a two ring spiro system according to formula:
 wherein D is a nitrogen-containing heterocycloalkyl or heterocycloalkenyl, m is 1 or 2, and the
wherein D is a nitrogen-containing heterocycloalkyl or heterocycloalkenyl, m is 1 or 2, and the
 optionally substituted by one or more C1-6 alkyl or Hal.
optionally substituted by one or more C1-6 alkyl or Hal.
2. The compound of claim 1, wherein the compound is a compound of formula
 (I)
or a pharmaceutically acceptable salt thereof, wherein: A is selected from aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl, cycloalkenyl and C2 alkynyl, wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl or C1-6 haloalkyl; B is selected from arylene and heteroarylene, wherein said arylene and said hereroarylene are each optionally substituted with one or more R2; X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, -SO2N(C1-6 alkyl)- , -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, -CH(CF3)N(C1-6 haloalkyl)-, C2 alkenylene optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl, cycloalkylene optionally substituted with one or more C1-6 alkyl, -(heterocycloalkylene)-NH-, and - (heterocycloalkylene)-N(C1-6 alkyl)-;
(I)
or a pharmaceutically acceptable salt thereof, wherein: A is selected from aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl, cycloalkenyl and C2 alkynyl, wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl or C1-6 haloalkyl; B is selected from arylene and heteroarylene, wherein said arylene and said hereroarylene are each optionally substituted with one or more R2; X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, -SO2N(C1-6 alkyl)- , -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, -CH(CF3)N(C1-6 haloalkyl)-, C2 alkenylene optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl, cycloalkylene optionally substituted with one or more C1-6 alkyl, -(heterocycloalkylene)-NH-, and - (heterocycloalkylene)-N(C1-6 alkyl)-;
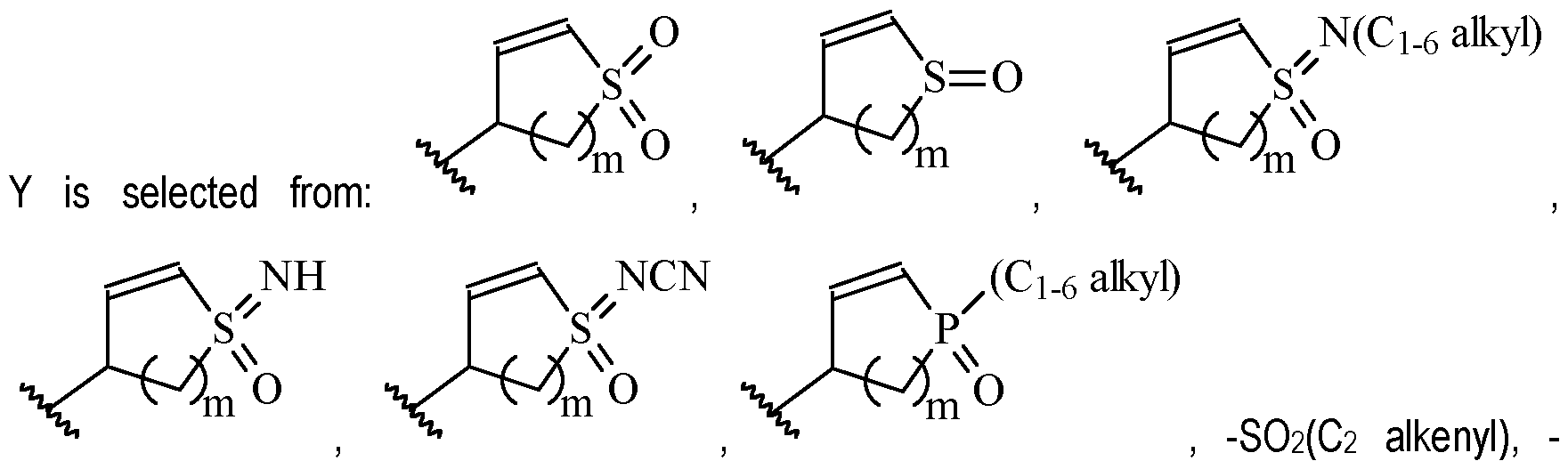 CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2 and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, - COO-(C1-4 alkyl), -CO-(C1-4 alkyl), -CONH-(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1- 4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -
CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2 and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, - COO-(C1-4 alkyl), -CO-(C1-4 alkyl), -CONH-(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1- 4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -
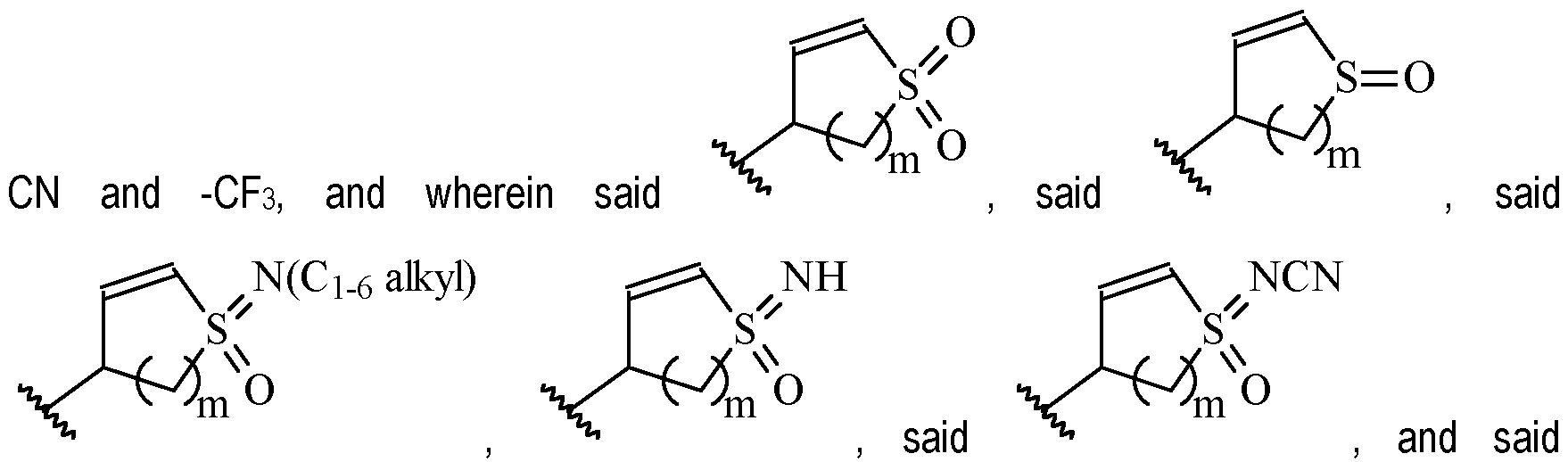
 are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F);
each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl, -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), - CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)- CO(C1-4 alkyl); and each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3
alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), - CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)- CO(C1-4 alkyl); or wherein A and Y are as defined hereinabove and B and X are combined to form a two-ring system according to formula:
are each optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F);
each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl, -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), - CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)- CO(C1-4 alkyl); and each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3
alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), - CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)- CO(C1-4 alkyl); or wherein A and Y are as defined hereinabove and B and X are combined to form a two-ring system according to formula:
 wherein C is aryl or heteroaryl ring, optionally substituted with one or more R2, Z is selected from C1-2 alkylene, -CH=CH-, -N=CH-, -N=C(C1-6 alkyl)-, -CH=N-, -C(C1-6 alkyl)=N-, -CO-, -SO-, -SO2- , -CONH-, -NHCO-, -CON(C1-6 alkyl)-, -N(C1-6 alkyl)CO-, -NH-, -N(C1-6 alkyl)- and -O-; or wherein A and B are as defined hereinabove, and X and Y form a two ring spiro system according to formula:
wherein C is aryl or heteroaryl ring, optionally substituted with one or more R2, Z is selected from C1-2 alkylene, -CH=CH-, -N=CH-, -N=C(C1-6 alkyl)-, -CH=N-, -C(C1-6 alkyl)=N-, -CO-, -SO-, -SO2- , -CONH-, -NHCO-, -CON(C1-6 alkyl)-, -N(C1-6 alkyl)CO-, -NH-, -N(C1-6 alkyl)- and -O-; or wherein A and B are as defined hereinabove, and X and Y form a two ring spiro system according to formula:
 wherein D is a nitrogen-containing heterocycloalkyl or heterocycloalkenyl, m is 1 or 2, and the
wherein D is a nitrogen-containing heterocycloalkyl or heterocycloalkenyl, m is 1 or 2, and the
 optionally substituted by one or more C1-6 alkyl or Hal.
optionally substituted by one or more C1-6 alkyl or Hal.
3. The compound of claim 1 or 2, wherein A is selected from aryl, heteroaryl, heterocycloalkyl, heterocycloalkenyl, cycloalkyl, cycloalkenyl and C2 alkynyl, wherein said aryl, said heteroaryl, said heterocycloalkyl, said heterocycloalkenyl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl or C1-6 haloalkyl; B is selected from arylene and heteroarylene, wherein said arylene and said hereroarylene are each optionally substituted with one or more R2; X is selected from -CONH-, -CON(C1-6 alkyl)-, -CON(C1-6 haloalkyl)-, -SO2NH-, -SO2N(C1-6 alkyl)- , -SO2N(C1-6 haloalkyl)-, -CH(CF3)NH-, -CH(CF3)N(C1-6 alkyl)-, -CH(CF3)N(C1-6 haloalkyl)-, C2 alkenylene optionally substituted with one or more C1-6 alkyl or C1-6 haloalkyl, cycloalkylene optionally substituted with one or more C1-6 alkyl, -(heterocycloalkylene)-NH-, and - (heterocycloalkylene)-N(C1-6 alkyl)-; ,
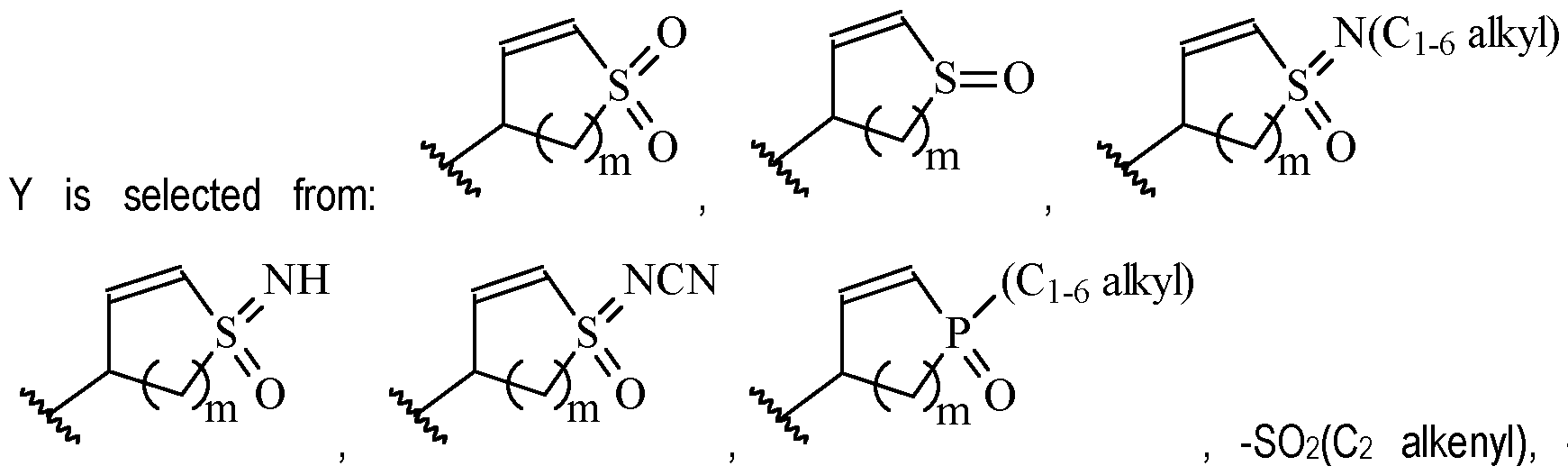 - CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2 and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, -- COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and - CF3, and wherein said
- CO(C2 alkenyl), -CH2(C2 alkenyl), and -(C1-2 alkylene)CN, wherein m is 1 or 2 and wherein said alkenyl is optionally substituted with one or more optional substituents selected from C1-4 alkyl, -- COO(C1-4 alkyl), -CO(C1-4 alkyl), -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -SO2(C1-4 alkyl), -SO2NH2, -SO2NH(C1-4 alkyl), -SO2N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)N(C1-4 alkyl)(C1-4 alkyl), -(C1-4 alkylene)(N-heterocycloalkyl), cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -Hal, -CN and - CF3, and wherein said
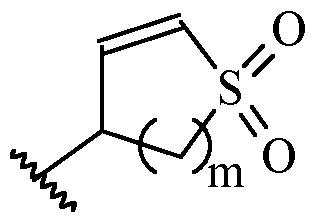 , said
, said
 , said
, , , and said
, said
, , , and said
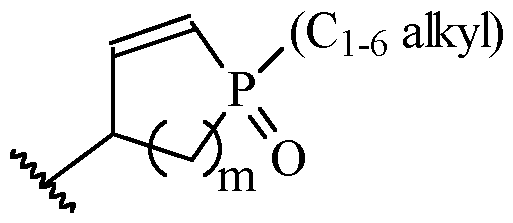 are each optionally substituted by one or more C1-6 alkyl or Hal; each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl, -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), - CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)- CO(C1-4 alkyl); and each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3
alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), - CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)- CO(C1-4 alkyl).
are each optionally substituted by one or more C1-6 alkyl or Hal; each R1 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl, -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), - CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)- CO(C1-4 alkyl); and each R2 is independently selected from C1-5 alkyl, C2-5 alkenyl, C2-5 alkynyl, -(C0-3 alkylene)-OH, -(C0-3 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-O(C1-5 alkylene)-OH, -(C0-3
alkylene)-O(C1-5 alkylene)-O(C1-5 alkyl), -(C0-3 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-S(C1-5 alkylene)-SH, -(C0-3 alkylene)-S(C1-5 alkylene)-S(C1-5 alkyl), -(C0-3 alkylene)-NH2, -(C0-3 alkylene)-NH(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-OH, -(C0-3 alkylene)-N(C1-5 alkyl)-OH, -(C0-3 alkylene)-NH-O(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-O(C1-5 alkyl), -(C0-3 alkylene)-halogen, -(C0-3 alkylene)-(C1-5 haloalkyl), -(C0-3 alkylene)-O-(C1-5 haloalkyl), -(C0-3 alkylene)-CN, -(C0-3 alkylene)-NO2, -(C0-3 alkylene)-CHO, -(C0-3 alkylene)-CO-(C1-5 alkyl), -(C0-3 alkylene)-COOH, -(C0-3 alkylene)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-(C1-5 alkyl), -(C0-3 alkylene)-CO-NH2, -(C0-3 alkylene)-CO-NH(C1-5 alkyl), -(C0-3 alkylene)-CO-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-(C1-5 alkyl), -(C0-3 alkylene)-NH-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-CO-O-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-NH-(C1-5 alkyl), -(C0-3 alkylene)-O-CO-N(C1-5 alkyl)-(C1-5 alkyl), -(C0-3 alkylene)-SO2-NH2, -(C0-3 alkylene)-SO2-NH(C1-5 alkyl), -(C0-3 alkylene)-SO2-N(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-NH-SO2-(C1-5 alkyl), -(C0-3 alkylene)-N(C1-5 alkyl)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO2-(C1-5 alkyl), -(C0-3 alkylene)-SO-(C1-5 alkyl), -(C0-3 alkylene)-Si(C1-5 alkyl)(C1-5 alkyl)(C1-5 alkyl), -(C0-3 alkylene)-SF5, -O-(C0-3 alkylene)-carbocyclyl, -O-(C0-3 alkylene)-heterocyclyl -(C0-3 alkylene)-carbocyclyl, and -(C0-3 alkylene)-heterocyclyl, wherein the carbocyclyl moiety in said -O-(C0-3 alkylene)-carbocyclyl, the carbocyclyl moiety in said -(C0-3 alkylene)-carbocyclyl, the heterocyclyl moiety in said -O-(C0-3 alkylene)-heterocyclyl and the heterocyclyl moiety in said -(C0-3 alkylene)-heterocyclyl are each optionally substituted with one or more groups independently selected from C1-4 alkyl, halogen, -CN, -NO2, -OH, -O-(C1-4 alkyl), -SH, -S-(C1-4 alkyl), -NH2, -NH(C1-4 alkyl), -N(C1-4 alkyl)(C1-4 alkyl), -COOH, -COO(C1-4 alkyl), - CONH2, -CONH(C1-4 alkyl), -CON(C1-4 alkyl)(C1-4 alkyl), -NHCO(C1-4 alkyl) and -N(C1-4 alkyl)- CO(C1-4 alkyl).
4. The compound of any one of claims 1 to 3, wherein the following provisions apply to formula (I): (e1) A is not selected from aryl, heteroaryl, cycloalkyl and cycloakenyl, wherein said aryl, said heteroaryl, said cycloalkyl and said cycloalkenyl are each optionally substituted with one or more R1; and/or (e2) B is not according to formula
 wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent
moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; and/or (e3) X is not -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and/or (e4) Y is not
wherein the left side of the depicted bivalent moiety is connected to A and the rights side of the depicted bivalent
moiety is connected to X, wherein RD1 is selected from hydrogen and C1-6 alkyl, and wherein the ring is optionally substituted with one or more groups selected from halogen, C1-6 alkyl, C1-6 haloalkyl, -OH, -O-(C1-6 alkyl), -NH-(C1-6 alkyl), -N(C1-6 alkyl)-(C1-6 alkyl) and -CN; and/or (e3) X is not -CONH- wherein the C atom in said -CONH- moiety is connected to B, and the N atom in said -CONH- moiety is connected to Y; and/or (e4) Y is not
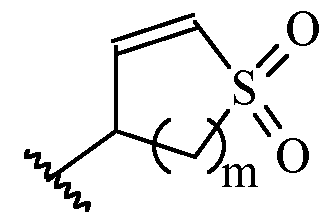 , wherein m is 1 or 2, and wherein said
, wherein m is 1 or 2, and wherein said
 is optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F).
is optionally substituted by one or more C1-6 alkyl (such as methyl) or Hal (such as F).
5. The compound of any one of claims 1 to 4, wherein A is selected from heterocycloalkyl, heterocycloalkenyl and C2 alkynyl, wherein said heterocycloalkyl and said heterocycloalkenyl are each optionally substituted with one or more R1, and wherein said C2 alkynyl is optionally substituted with C1-6 alkyl.
6. The compound of claim 5, wherein A are selected from:
 ,
,
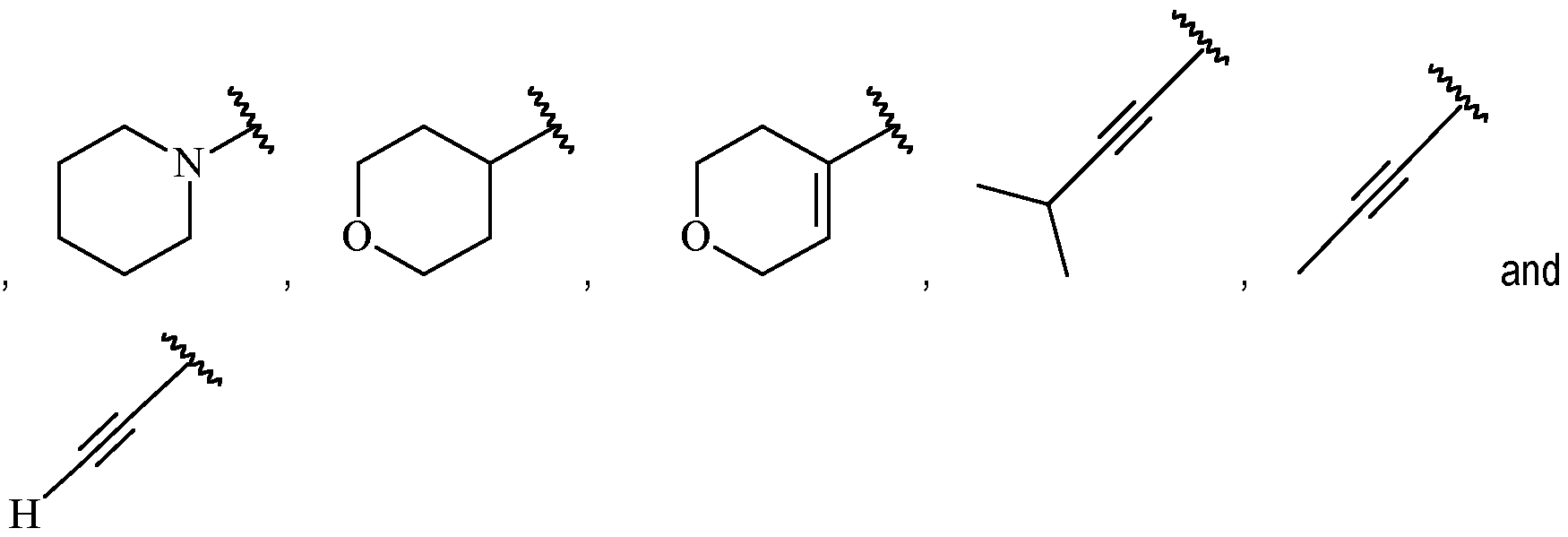 .
.
7. The compound of any one of claims 1 to 6, wherein B is arylene, optionally substituted with one or more R2.
8. The compound of claim 7, wherein
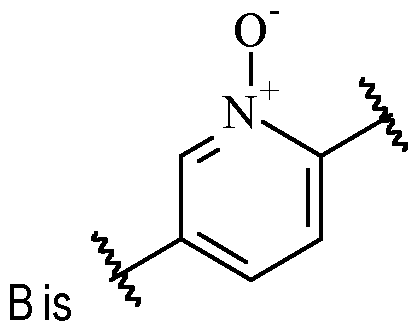 , optionally substituted with one or more R2.
, optionally substituted with one or more R2.
9. The compound of any one of claims 1 to 8, wherein X is -CONH-.
10. The compound of any one of claims 1 to 9, wherein Y is selected from:
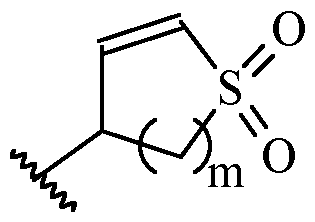 ,
,
 optionally substituted by one or more C1-6 alkyl or Hal.
optionally substituted by one or more C1-6 alkyl or Hal.
11. The compound of any one of claims 1 to 10, wherein
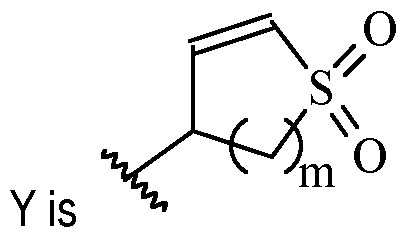 , wherein m is 1 or 2, wherein said
, wherein m is 1 or 2, wherein said
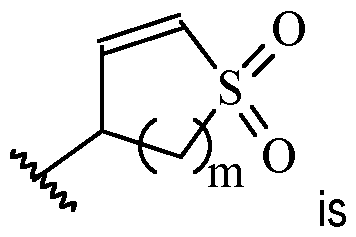 optionally substituted by one or more C1-6 alkyl or Hal.
optionally substituted by one or more C1-6 alkyl or Hal.
12. The compound of claim 10 or 11, wherein m is 1.
13. The compound of claim 1, selected from 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3 S)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide
6-(3,4-dimethylphenyl)-N-((1R or S,3 S)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3 R)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R or S,3 R)-1-imino-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2- oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-(1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-1l6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- sulfonamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3- carboxamide; and 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-thioxo-1,2-dihydropyridine-3- carboxamide, or its pharmaceutically acceptable salt.
14. The compound of claim 1, selected from: 6-(3,4-dimethylphenyl)-N-(1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3S or R)-1-imino-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R or S,3S or R)-1-imino-1-oxido-2,3-dihydro-1H-1 6-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1S or R,3R or S)-1-imino-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)- 2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-((1R or S,3R or S)-1-imino-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)-
2-oxo-1,2-dihydropyridine-3-carboxamide; 6-(3,4-dimethylphenyl)-N-(1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (R or S)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; (S or R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)picolinamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(piperidin-1-yl)-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- sulfonamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(prop-1-yn-1-yl)-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-thioxo-1,2-dihydropyridine-3- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; 3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-3-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-(trifluoromethyl)phenyl)pyridine 1- oxide; 4-amino-5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-5-(2-(benzyloxy)phenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-methylthiophen-3-yl)pyridine 1- oxide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(spiro[3.3]hept-1-en-2-yl)pyridine 1-oxide; 5-(2-chlorothiophen-3-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1-dioxide; 3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene
1,1-dioxide; (R)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide; (S)-3-(4-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide; (R)-3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride; (S)-3-(4-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide hydrochloride; (S,Z)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; (R)-5-(4,5-dimethylthiophen-2-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen-2- yl)pyridine 1-oxide; (E)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide ; (Z)-N-(1-cyclopropyl-3-fluoro-3-(methylsulfonyl)allyl)-2-(cyclopropyldifluoromethyl)-4- phenoxypyrimidine-5-carboxamide; (E)-N-(1-cyclopropyl-3-(S-methylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4-phenoxypyrimidine-5- carboxamide; (E)-N-(1-cyclopropyl-3-(N,S-dimethylsulfonimidoyl)allyl)-2-(1,1-difluoroethyl)-4- phenoxypyrimidine-5-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-ethynyl-2-oxo-1,2-dihydropyridine-3-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(pyrrolidin-1-yl)-1,2-dihydropyridine-3- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-(3-methylbut-1-yn-1-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-(3-methylbut-1-yn-1-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-5-(spiro[3.3]heptan-2-yl)picolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-1,2,4-triazole-3- carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4H-1,2,4-triazole-3- carboxamide;
(R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-methylthiophen-2-yl)pyridine 1- oxide; (R)-5-(cyclohex-1-en-1-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-cyclohexyl-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(tetrahydro-2H-pyran-3-yl)-1,2- dihydropyridine-3-carboxamide; 6-(3,4-dihydro-2H-pyran-6-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(tetrahydro-2H-pyran-2-yl)-1,2- dihydropyridine-3-carboxamide; 6-(3,6-dihydro-2H-pyran-4-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(tetrahydro-2H-pyran-4-yl)-1,2- dihydropyridine-3-carboxamide; 6-(5,6-dihydro-2H-pyran-3-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)thiazole-2-carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-phenyl-2H-pyran-3-carboxamide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(piperidin-1-yl)pyridine 1-oxide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1,3,4-thiadiazole-2- carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-imidazole-2- carboxamide; (S)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1-methyl-2-(p-tolyl)-1H-imidazole-4-carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1-methyl-2-(p-tolyl)-1H-imidazole-4-carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-imidazole-4-carboxamide; (R)-5-(4-chlorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxy-6-(tetrahydro-2H-pyran-4- yl)nicotinamide; 6-(3,4-dimethylphenyl)-N-((3R)-1-oxido-2,3-dihydrothiophen-3-yl)-2-oxo-1,2-dihydropyridine-3- carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)nicotinamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluorothiophene-2-
carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrimidine-5-carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrimidine-2-carboxamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyridazine-3-carboxamide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(spiro[3.3]heptan-2-yl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-3-(6,6-dioxido-6-thia-2-azaspiro[3.4]oct-7-en-2-yl)pyridin-2(1H)-one; 3-((6-(3,4-dimethylphenyl)-1H-indazol-3-yl)oxy)-2,3-dihydrothiophene 1,1-dioxide; 6-(3,4-dimethylphenyl)-1-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1,2-dihydro-3H-indazol-3-one; 5-(3,4-dimethylphenyl)-2-(1,1-dioxido-2,3-dihydrothiophen-3-yl)isoindolin-1-one; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(phenylethynyl)-1,2-dihydropyridine-3- carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-hydroxypicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-hydroxypicolinamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-oxo-1,4-dihydropyridine-3- carboxamide; (R)-5-(2,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-(3,4-dichlorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-(3,4-difluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-methoxyphenyl)pyridine 1-oxide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-fluoropicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-fluoropicolinamide; 6-(3,4-dimethylphenyl)-N-(1-methyl-1-oxido-2,3-dihydrophosphol-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 3-amino-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-methylpicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-methylpicolinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methylpicolinamide; 2-amino-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; (S)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-(4-(trifluoromethyl)phenyl)thiazole-4- carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-(4-(trifluoromethyl)phenyl)thiazole-4- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; 3-chloro-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide;
N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-fluoro-3',4'-dimethyl-[1,1'-biphenyl]-4-carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2,3',4'-trimethyl-[1,1'-biphenyl]-4-carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-6-oxo-1,6-dihydropyrimidine-5- carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-oxo-3,4-dihydropyrazine-2- carboxamide; 2-amino-6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)nicotinamide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(2,4,6-trifluorophenyl)pyridine 1- oxide; 3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-fluoropyridine 1- oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-4-fluoropyridine 1- oxide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxynicotinamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxypicolinamide; 6-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1-methyl-4-oxo-1,4- dihydropyridine-3-carboxamide; 3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-methylpyridine 1- oxide; 3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-hydroxypyridine 1- oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-4-hydroxypyridine 1- oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-3-hydroxypyridine 1- oxide; (R)-5-chloro-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; (S)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-5-fluoro-2-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; (R)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-5-fluoro-2-hydroxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxy-3',4'-dimethyl-[1,1'-biphenyl]-4- carboxamide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxy-3',4'-dimethyl-[1,1'-biphenyl]-4-
carboxamide; 2-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-4-methoxypyrimidine-5- carboxamide; 5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3-methoxypyrazine-2- carboxamide; 6-(3,4-dimethylphenyl)-N-(1-(methylimino)-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)-2-oxo-1,2- dihydropyridine-3-carboxamide; (E)-6-(3,4-dimethylphenyl)-3-(5-(2-(methylsulfonyl)vinyl)-1H-imidazol-2-yl)pyridin-2-ol; (R)-5-(4-cyclopropylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1- oxide; (R)-5-(benzofuran-5-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; (R)-5-(2,3-dihydrobenzofuran-5-yl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-oxide; 2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-6-thia-2-azaspiro[3.4]oct-7-ene 6,6-dioxide; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)quinazolin-4(3H)-one; 2-(3,4-dimethylphenyl)-6-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-7,8-dihydro-1,6-naphthyridin- 5(6H)-one; 6-(3,4-dimethylphenyl)-2-(1,1-dioxido-2,3-dihydrothiophen-3-yl)isoquinolin-1(2H)-one; 6-(3,4-dimethylphenyl)-2-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-3,4-dihydroisoquinolin-1(2H)- one; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrido[2,3-d]pyrimidin-4(3H)- one; 2-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7- dioxide; (R)-2-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (S)-2-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (R,Z)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; (R,E)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; (S,E)-5-(3,4-dimethylphenyl)-2-((4-(methylsulfonyl)but-3-en-2-yl)carbamoyl)pyridine 1-oxide; 7-(5-(4-(trifluoromethyl)phenyl)pyridin-2-yl)-2-thia-7-azaspiro[4.4]non-3-ene 2,2-dioxide; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-8-fluoroquinazolin-4(3H)-one; 7-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-2-thia-7-azaspiro[4.4]non-3-ene 2,2-dioxide;
(S)-4-(3-cyclopropyl-1H-pyrazol-1-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxy-6- methylbenzamide; (R)-4-(3-cyclopropyl-1H-pyrazol-1-yl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-methoxy-6- methylbenzamide; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)pyrido[2,3-d]pyrimidine- 2,4(1H,3H)-dione; 2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7- dioxide; (R)-2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (S)-2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (R)-7-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-1H-benzo[d]imidazole-4- carboxamide; N-(1-(cyanoimino)-1-oxido-2,3-dihydro-1H-16-thiophen-3-yl)-6-(3,4-dimethylphenyl)-2-oxo-1,2- dihydropyridine-3-carboxamide; N-(1-(cyanoimino)-1-oxido-4,5-dihydro-1H-16-thiophen-3-yl)-6-(3,4-dimethylphenyl)-2-oxo-1,2- dihydropyridine-3-carboxamide; 3-(3,4-dimethylphenyl)-6-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-2-methoxypyridine 1-oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-4-methoxypyridine 1-oxide; 5-(3,4-dimethylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-3-methoxypyridine 1-oxide; N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-2-oxo-6-(4-(trifluoromethyl)piperidin-1-yl)-1,2- dihydropyridine-3-carboxamide; 2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-(trifluoromethyl)piperidin-1-yl)pyridine 1-oxide; (R)-3-amino-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen- 3-yl)pyridine 1-oxide; (R)-3-amino-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-(trifluoromethyl)thiophen- 2-yl)pyridine 1-oxide; (R)-3-amino-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(4-(trifluoromethyl)thiophen- 2-yl)pyridine 1-oxide;
(R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(3-methyl-4- (trifluoromethyl)phenyl)pyridine 1-oxide; (S,Z)-N-(1-cyclopropyl-3-(methylsulfonyl)allyl)-6-(3,4-dimethylphenyl)-2-oxo-1,2-dihydropyridine- 3-carboxamide; (S,E)-N-(1-cyclopropyl-3-(methylsulfonyl)allyl)-6-(3,4-dimethylphenyl)-2-oxo-1,2-dihydropyridine- 3-carboxamide; (S,Z)-2-((1-cyclopropyl-3-(methylsulfonyl)allyl)carbamoyl)-5-(3,4-dimethylphenyl)pyridine 1- oxide; (S,E)-2-((1-cyclopropyl-3-(methylsulfonyl)allyl)carbamoyl)-5-(3,4-dimethylphenyl)pyridine 1- oxide; (R)-5-(2-chloro-4-(trifluoromethyl)phenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; 6-(3,4-dimethylphenyl)-3-(1-((1,1-dioxido-2,3-dihydrothiophen-3-yl)amino)-2,2,2- trifluoroethyl)pyridin-2(1H)-one; 7-(3,4-dimethylphenyl)-3-(1,1-dioxido-2,3-dihydrothiophen-3-yl)-8-fluoro-2-hydroxy-2,3- dihydroquinazolin-4(1H)-one; 8-(5-(4-(trifluoromethyl)phenyl)pyridin-2-yl)-2-thia-8-azaspiro[4.5]dec-3-ene 2,2-dioxide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)quinoline-8-carboxamide; 2-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-8-thia-1,3-diazaspiro[4.5]deca-2,6-diene 8,8- dioxide; 3-(6-(3,4-dimethylphenyl)-2-hydroxypyridin-3-yl)-9-thia-2,4-diazaspiro[5.5]undeca-2,7-diene 9,9- dioxide; (R)-5-(3,4-dimethylphenyl)-N-(1,1-dioxido-2,3-dihydrothiophen-3-yl)quinazoline-8-carboxamide; 3-(6-(3,4-dimethylphenyl)-2-methoxypyridin-3-yl)-9-thia-2,4-diazaspiro[5.5]undeca-2,7-diene 9,9-dioxide; 3-(4-(6-cyclopentyl-2-phenoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3-dihydrothiophene 1,1- dioxide; 2-(2-(cyclopropyldifluoromethyl)-4-phenoxypyrimidin-5-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8- diene 7,7-dioxide; (E)-2-(cyclopropyldifluoromethyl)-5-(5-(2-(methylsulfonyl)vinyl)-1H-imidazol-2-yl)-4- phenoxypyrimidine; 3-(4-(6-(isopropyl(methyl)amino)-2-phenoxypyridin-3-yl)-1H-1,2,3-triazol-1-yl)-2,3- dihydrothiophene 1,1-dioxide; (R)-5-(2-(benzylamino)phenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)pyridine 1-
oxide; 2-(6-(3,4-dimethylphenyl)-2-phenoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7- dioxide; (S)-2-(6-(3,4-dimethylphenyl)-2-phenoxypyridin-3-yl)-7-thia-1,3-diazaspiro[4.4]nona-2,8-diene 7,7-dioxide; (R)-5-(2-(benzyloxy)-3-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-4-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-5-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-6-fluorophenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-5-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-3-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-4-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-5-(2-(benzyloxy)-6-methylphenyl)-2-((1,1-dioxido-2,3-dihydrothiophen-3- yl)carbamoyl)pyridine 1-oxide; (R)-2-((1,1-dioxido-2,3-dihydrothiophen-3-yl)carbamoyl)-5-(5-fluoro-2-(pyridin-4- ylmethoxy)phenyl)pyridine 1-oxide; or a pharmaceutically acceptable salt thereof.
15. The compound of claim 1, wherein A and B are as defined in any one of claims 1 to 12, wherein X is -CONH-, wherein X is connected to B through its carbon atom, and wherein Y is a moiety according to formula:
 or a moiety according to formula:
wherein Ry1 is selected from H, C1-4 alkyl, C3-8 cycloalkyl, and 5 or 6-membered heterocyclyl, Ry2 is selected from -H, -F and -CN, and Ry3 is selected from C1-4 alkyl, C3-8 cycloalkyl, and 5 or 6- membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible.
or a moiety according to formula:
wherein Ry1 is selected from H, C1-4 alkyl, C3-8 cycloalkyl, and 5 or 6-membered heterocyclyl, Ry2 is selected from -H, -F and -CN, and Ry3 is selected from C1-4 alkyl, C3-8 cycloalkyl, and 5 or 6- membered heterocyclyl, and wherein the bonds drawn as indicate that both Z and E configurations of the double bond are possible.
16. The compound of claim 15, selected from

 .
.
17. A pharmaceutical composition comprising the compound of any one of claims 1 to 16, and a pharmaceutically acceptable excipient.
18. The compound of any one of claims 1 to 16 or the pharmaceutical composition of claim 17 for use as a medicament.
19. The compound of any one of claims 1 to 16 or the pharmaceutical composition of claim 17 for use in the treatment of cancer.
20. The compound for use or the pharmaceutical composition for use of claim 19, wherein the cancer is treatable by inhibition of WRN and/or the cancer is characterized by MSI-H and/or dMMR.
21. Use of the compound of any one of claims 1 to 16 in the manufacture of a medicament for treating cancer, preferably wherein the cancer is treatable by inhibition of WRN and/or the cancer is characterized by MSI-H and/or dMMR.
22. A method for treating a cancer in a subject in need thereof, the method comprising the step of administering to the subject the therapeutically effective amount of the compound of any one of claims 1 to 16, preferably wherein the cancer is treatable by inhibition of WRN and/or the cancer is characterized by MSI-H and/or dMMR.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2025/068557 WO2026003380A1 (en) | 2024-06-28 | 2025-06-30 | Wrn inhibitory compounds |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363587354P | 2023-10-02 | 2023-10-02 | |
| US63/587,354 | 2023-10-02 | ||
| US202463665751P | 2024-06-28 | 2024-06-28 | |
| US63/665,751 | 2024-06-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025073792A1 true WO2025073792A1 (en) | 2025-04-10 |
Family
ID=92973524
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/077789 Pending WO2025073792A1 (en) | 2023-10-02 | 2024-10-02 | Wrn inhibitory compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025073792A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025223559A1 (en) * | 2024-04-26 | 2025-10-30 | 上海济煜医药科技有限公司 | Aromatic amide compounds as wrn helicase inhibitors, preparation method therefor and use thereof |
| WO2025223549A1 (en) * | 2024-04-26 | 2025-10-30 | InventisBio Co., Ltd. | Compounds, preparation methods and uses thereof |
| WO2025261481A1 (en) * | 2024-06-21 | 2025-12-26 | 南京再明医药有限公司 | Five-membered heteroaromatic compound and use thereof |
| WO2025261486A1 (en) * | 2024-06-22 | 2025-12-26 | InventisBio Co., Ltd. | Compounds, preparation methods and uses thereof |
| WO2026003380A1 (en) * | 2024-06-28 | 2026-01-02 | Forx Therapeutics Ag | Wrn inhibitory compounds |
Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997022596A1 (en) | 1995-12-18 | 1997-06-26 | Zeneca Limited | Quinazoline derivatives |
| WO1997030035A1 (en) | 1996-02-13 | 1997-08-21 | Zeneca Limited | Quinazoline derivatives as vegf inhibitors |
| WO1997032856A1 (en) | 1996-03-05 | 1997-09-12 | Zeneca Limited | 4-anilinoquinazoline derivatives |
| WO1998013354A1 (en) | 1996-09-25 | 1998-04-02 | Zeneca Limited | Quinazoline derivatives and pharmaceutical compositions containing them |
| WO1999002166A1 (en) | 1997-07-08 | 1999-01-21 | Angiogene Pharmaceuticals Ltd. | Use of colchinol derivatives as vascular damaging agents |
| WO2000040529A1 (en) | 1999-01-07 | 2000-07-13 | Angiogene Pharmaceuticals Ltd. | Colchinol derivatives as vascular damaging agents |
| WO2000041669A2 (en) | 1999-01-15 | 2000-07-20 | Angiogene Pharmaceuticals Ltd. | Benzimidazole vascular damaging agents |
| WO2000047212A1 (en) | 1999-02-10 | 2000-08-17 | Astrazeneca Ab | Quinazoline derivatives as angiogenesis inhibitors |
| WO2001092224A1 (en) | 2000-05-31 | 2001-12-06 | Astrazeneca Ab | Indole derivatives with vascular damaging activity |
| WO2001094341A1 (en) | 2000-06-06 | 2001-12-13 | Astrazeneca Ab | Quinazoline derivatives for the treatment of tumours |
| WO2002004434A1 (en) | 2000-07-07 | 2002-01-17 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as vascular damaging agents |
| WO2002008213A1 (en) | 2000-07-07 | 2002-01-31 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as angiogenesis inhibitors |
| WO2022249060A1 (en) | 2021-05-26 | 2022-12-01 | Novartis Ag | Triazolo-pyrimidine analogues for treating diseases connected to the inhibiton of werner syndrome recq helicase (wrn) |
| WO2023062575A1 (en) | 2021-10-14 | 2023-04-20 | Ideaya Biosciences, Inc. | Cyclic vinyl sulfone compounds as wrn inhibitors |
| WO2024010782A1 (en) | 2022-07-06 | 2024-01-11 | Vividion Therapeutics, Inc. | Pharmaceutical compositions comprising wrn helicase inhibitors |
| WO2024010784A1 (en) | 2022-07-06 | 2024-01-11 | Vividion Therapeutics, Inc. | Pharmaceutical compositions comprising wrn helicase inhibitors |
| WO2024028169A1 (en) | 2022-08-01 | 2024-02-08 | Nerviano Medical Sciences S.R.L. | Novel specifically substituted thiophenolic compounds |
| WO2024079623A1 (en) | 2022-10-12 | 2024-04-18 | Novartis Ag | Tricyclic compounds and their uses |
| WO2024120378A2 (en) | 2022-12-05 | 2024-06-13 | Jiangsu Hansoh Pharmaceutical Group Co., Ltd. | Triazole compounds, preparation methods and medicinal uses thereof |
| WO2024222866A1 (en) * | 2023-04-27 | 2024-10-31 | Danatlas Pharmaceuticals Co., Ltd. | Cyclic derivatives, compositions and uses thereof |
-
2024
- 2024-10-02 WO PCT/EP2024/077789 patent/WO2025073792A1/en active Pending
Patent Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997022596A1 (en) | 1995-12-18 | 1997-06-26 | Zeneca Limited | Quinazoline derivatives |
| WO1997030035A1 (en) | 1996-02-13 | 1997-08-21 | Zeneca Limited | Quinazoline derivatives as vegf inhibitors |
| WO1997032856A1 (en) | 1996-03-05 | 1997-09-12 | Zeneca Limited | 4-anilinoquinazoline derivatives |
| WO1998013354A1 (en) | 1996-09-25 | 1998-04-02 | Zeneca Limited | Quinazoline derivatives and pharmaceutical compositions containing them |
| WO1999002166A1 (en) | 1997-07-08 | 1999-01-21 | Angiogene Pharmaceuticals Ltd. | Use of colchinol derivatives as vascular damaging agents |
| WO2000040529A1 (en) | 1999-01-07 | 2000-07-13 | Angiogene Pharmaceuticals Ltd. | Colchinol derivatives as vascular damaging agents |
| WO2000041669A2 (en) | 1999-01-15 | 2000-07-20 | Angiogene Pharmaceuticals Ltd. | Benzimidazole vascular damaging agents |
| WO2000047212A1 (en) | 1999-02-10 | 2000-08-17 | Astrazeneca Ab | Quinazoline derivatives as angiogenesis inhibitors |
| WO2001092224A1 (en) | 2000-05-31 | 2001-12-06 | Astrazeneca Ab | Indole derivatives with vascular damaging activity |
| WO2001094341A1 (en) | 2000-06-06 | 2001-12-13 | Astrazeneca Ab | Quinazoline derivatives for the treatment of tumours |
| WO2002004434A1 (en) | 2000-07-07 | 2002-01-17 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as vascular damaging agents |
| WO2002008213A1 (en) | 2000-07-07 | 2002-01-31 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as angiogenesis inhibitors |
| WO2022249060A1 (en) | 2021-05-26 | 2022-12-01 | Novartis Ag | Triazolo-pyrimidine analogues for treating diseases connected to the inhibiton of werner syndrome recq helicase (wrn) |
| WO2023062575A1 (en) | 2021-10-14 | 2023-04-20 | Ideaya Biosciences, Inc. | Cyclic vinyl sulfone compounds as wrn inhibitors |
| WO2024010782A1 (en) | 2022-07-06 | 2024-01-11 | Vividion Therapeutics, Inc. | Pharmaceutical compositions comprising wrn helicase inhibitors |
| WO2024010784A1 (en) | 2022-07-06 | 2024-01-11 | Vividion Therapeutics, Inc. | Pharmaceutical compositions comprising wrn helicase inhibitors |
| WO2024028169A1 (en) | 2022-08-01 | 2024-02-08 | Nerviano Medical Sciences S.R.L. | Novel specifically substituted thiophenolic compounds |
| WO2024079623A1 (en) | 2022-10-12 | 2024-04-18 | Novartis Ag | Tricyclic compounds and their uses |
| WO2024120378A2 (en) | 2022-12-05 | 2024-06-13 | Jiangsu Hansoh Pharmaceutical Group Co., Ltd. | Triazole compounds, preparation methods and medicinal uses thereof |
| WO2024222866A1 (en) * | 2023-04-27 | 2024-10-31 | Danatlas Pharmaceuticals Co., Ltd. | Cyclic derivatives, compositions and uses thereof |
Non-Patent Citations (23)
| Title |
|---|
| A. KOLACZEK ET AL., CHEMLK, vol. 68, 2014, pages 620 |
| ANNA-KARIN SOHLENIUS-STERNBECK: "Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements", TOXICOLOGY IN VITRO, vol. 20, no. 8, 2006 |
| ATZRODT J ET AL., BIOORG MED CHEM, vol. 20, no. 18, 2012, pages 5658 - 5667 |
| B. S. KADU, CATAL. SCI. TECHNOL., vol. 11, 2021, pages 1186 - 1221 |
| BAUDRIN ET AL., FRONT. ONCOL, 2018 |
| BRIAN DAVIESTIM MORRIS: "Physiological Parameters in Laboratory Animals and Human", PHARMACEUTICAL RESEARCH, vol. 10, no. 7, 1993 |
| DOBZHANSKY, T., GENETICS, vol. 31, 1946, pages 269 - 290 |
| DUDLEY, JONATHAN C. ET AL., CLINICAL CANCER RESEARCH, vol. 22, no. 4, 2016, pages 813 - 820 |
| E. MASSOLO ET AL., EUR. J. ORG. CHEM., 2020, pages 4641 |
| GHOSHAL ANIRBAN ET AL: "Structure Activity of [beta]-Amidomethyl Vinyl Sulfones as Covalent Inhibitors of Chikungunya nsP2 Cysteine Protease with Antialphavirus Activity", JOURNAL OF MEDICINAL CHEMISTRY, vol. 67, no. 18, 5 September 2024 (2024-09-05) - 5 September 2024 (2024-09-05), US, pages 16505 - 16532, XP093237111, ISSN: 0022-2623, DOI: 10.1021/acs.jmedchem.4c01346 * |
| HUANG ET AL., NATURE REVIEWS DRUG DISCOVERY, vol. 19, 2020, pages 23 - 38 |
| I. KANWAL ET AL., CATALYSTS, vol. 10, no. 4, 2020, pages 443 |
| J. MED. CHEM., vol. 47, 2004, pages 6658 - 6661 |
| LI DIEDWARD H. KERNSYAN HONGHONG CHEN: "Development and application of high throughput plasma stability assay for drug discovery", INTERNATIONAL JOURNAL OF PHARMACEUTICS, vol. 297, 2005, pages 110 - 119 |
| MODVIG A ET AL., J ORG CHEM, vol. 79, 2014, pages 5861 - 5868 |
| OBACH R SBAXTER J GLISTON T E ET AL.: "The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data [J", JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, vol. 283, no. 1, 1997, pages 46 - 58 |
| R. DOREL ET AL., ANGEW. CHEM. INT. ED, vol. 58, 2019, pages 17118 |
| R. N. SALVATOREET, TETRAHEDRON, 2001, pages 7785 |
| STERN ET AL., CRITICAL REVIEWS IN ONCOLOGY/HAEMATOLOGY, vol. 54, 2005, pages 1 1 - 29 |
| STN REGISTRY ET AL: "N-(2,3-dihydro-1,1-dioxido-3-thienyl)-1,2-dihydro-6-(4-methoxyphenyl)-2-oxo-3-pyridinecarboxamide", 12 March 2014 (2014-03-12), pages 1 - 1, XP093012912, Retrieved from the Internet <URL:www.stn.org> [retrieved on 20140312] * |
| VAN WIETMARSCHEN N ET AL., NATURE, vol. 586, 2020, pages 292 - 298 |
| WILLIAM JS ET AL., JOURNAL OF LABELLED COMPOUNDS AND RADIOPHARMACEUTICALS, vol. 53, no. 11-12, 2010, pages 635 - 644 |
| WU ET AL., AM J HUM GENETICS, vol. 65, 1999, pages 1291 - 1298 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025223559A1 (en) * | 2024-04-26 | 2025-10-30 | 上海济煜医药科技有限公司 | Aromatic amide compounds as wrn helicase inhibitors, preparation method therefor and use thereof |
| WO2025223549A1 (en) * | 2024-04-26 | 2025-10-30 | InventisBio Co., Ltd. | Compounds, preparation methods and uses thereof |
| WO2025261481A1 (en) * | 2024-06-21 | 2025-12-26 | 南京再明医药有限公司 | Five-membered heteroaromatic compound and use thereof |
| WO2025261486A1 (en) * | 2024-06-22 | 2025-12-26 | InventisBio Co., Ltd. | Compounds, preparation methods and uses thereof |
| WO2026003380A1 (en) * | 2024-06-28 | 2026-01-02 | Forx Therapeutics Ag | Wrn inhibitory compounds |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11680068B2 (en) | Compounds useful as RET inhibitors | |
| WO2025073792A1 (en) | Wrn inhibitory compounds | |
| EP2686318B1 (en) | Pyrrolopyridineamino derivatives as mps1 inhibitors | |
| WO2023057389A1 (en) | Parg inhibitory compounds | |
| RU2537945C2 (en) | Triazine, pyrimidine and pyridine analogues and use thereof as therapeutic agents and diagnostic samples | |
| WO2024074497A1 (en) | Parg inhibitory compound | |
| EP4413000A1 (en) | N,n-dimethyl-4-(7-(n-(1-methylcyclopropyl)sulfamoyl)-imidazo[1,5-a]pyridin-5-yl)piperazine-1-carboxamide derivatives and the corresponding pyrazolo[1,5-a]pyridine derivatives as parg inhibitors for the treatment of cancer | |
| CN118201896A (en) | PI 3K-alpha inhibitors and methods of use thereof | |
| TW202204351A (en) | Compounds having a macrocyclic structure and uses thereof | |
| WO2023175185A1 (en) | 2,4-dioxo-1,4-dihydroquinazoline derivatives as parg inhibitors for the treatment of cancer | |
| WO2023175184A1 (en) | 2,4-dioxo-1,4-dihydroquinazoline derivatives as parg inhibitors for the treatment of cancer | |
| WO2025133396A1 (en) | Novel bicyclo heteroaryl parg inhibitors | |
| WO2025133395A1 (en) | Bicyclic (hetero)arylene wrn inhibitory compounds | |
| WO2025093755A1 (en) | Novel parc inhibitors | |
| EP4688159A1 (en) | Parg inhibitory compounds | |
| WO2026003380A1 (en) | Wrn inhibitory compounds | |
| WO2025114480A1 (en) | Wrn inhibitory compounds | |
| WO2025191176A1 (en) | Wrn inhibitory compounds | |
| WO2026022150A1 (en) | Parg inhibitory compounds | |
| EP4598922A1 (en) | Parg inhibitory compound | |
| WO2025073870A1 (en) | Parg inhibitory compound | |
| WO2025046148A1 (en) | Novel parg inhibitors | |
| IL311851A (en) | Parg inhibitory compounds | |
| NZ787350A (en) | Heterocyclic compounds as ret kinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24783683 Country of ref document: EP Kind code of ref document: A1 |