WO2024089633A1 - Rna molecules encoding rsv-f and vaccines containing them - Google Patents
Rna molecules encoding rsv-f and vaccines containing them Download PDFInfo
- Publication number
- WO2024089633A1 WO2024089633A1 PCT/IB2023/060798 IB2023060798W WO2024089633A1 WO 2024089633 A1 WO2024089633 A1 WO 2024089633A1 IB 2023060798 W IB2023060798 W IB 2023060798W WO 2024089633 A1 WO2024089633 A1 WO 2024089633A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- aspects
- rna
- sequence
- rsv
- rna molecule
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/713—Double-stranded nucleic acids or oligonucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A61K39/155—Paramyxoviridae, e.g. parainfluenza virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/5123—Organic compounds, e.g. fats, sugars
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/88—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation using microencapsulation, e.g. using amphiphile liposome vesicle
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55555—Liposomes; Vesicles, e.g. nanoparticles; Spheres, e.g. nanospheres; Polymers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/31—Chemical structure of the backbone
- C12N2310/317—Chemical structure of the backbone with an inverted bond, e.g. a cap structure
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/33—Chemical structure of the base
- C12N2310/335—Modified T or U
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/18011—Paramyxoviridae
- C12N2760/18511—Pneumovirus, e.g. human respiratory syncytial virus
- C12N2760/18522—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/18011—Paramyxoviridae
- C12N2760/18511—Pneumovirus, e.g. human respiratory syncytial virus
- C12N2760/18534—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/18011—Paramyxoviridae
- C12N2760/18511—Pneumovirus, e.g. human respiratory syncytial virus
- C12N2760/18541—Use of virus, viral particle or viral elements as a vector
- C12N2760/18543—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/18011—Paramyxoviridae
- C12N2760/18511—Pneumovirus, e.g. human respiratory syncytial virus
- C12N2760/18571—Demonstrated in vivo effect
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/50—Vector systems having a special element relevant for transcription regulating RNA stability, not being an intron, e.g. poly A signal
Definitions
- RSV Respiratory syncytial virus
- BACKGROUND Respiratory syncytial virus is a respiratory virus that infects the lungs and breathing passages. RSV is the leading cause of serious viral lower respiratory tract illness in infants worldwide and an important cause of respiratory illness in the elderly.
- Two RSV protein subunit vaccines were approved in 2023, ABRYSVO (Pfizer) and AREXVY (GSK). However, no RNA vaccine has been approved for preventing RSV infection.
- RSV is a member of the Pneumoviridae family.
- RNA genome consists of a single-stranded, negative-sense RNA molecule that encodes 11 proteins, including nine structural proteins (three glycoproteins and six internal proteins) and two non-structural proteins.
- the structural proteins include three transmembrane surface glycoproteins: the attachment protein G, fusion protein F, and the small hydrophobic SH protein.
- the mature F glycoprotein has three general domains: ectodomain (ED), transmembrane domain (TM), and a cytoplasmic tail (CT). CT contains a single palmitoylated cysteine residue.
- the F glycoprotein of human RSV is initially translated from the mRNA as a single 574- amino acid polypeptide precursor (referred to “F0” or “F0 precursor”), which contains a signal peptide sequence (amino acids 1-25) at the N-terminus. Upon translation the signal peptide is removed by a signal peptidase in the endoplasmic reticulum.
- F0 574- amino acid polypeptide precursor
- the remaining portion of the F0 precursor may be further cleaved at two polybasic sites (a.a.109/110 and 136/137) by cellular proteases (in particular furin), removing a 27-amino acid intervening sequence designated pep27 (amino acids 110-136) and generating two linked fragments designated F1 (C-terminal portion; amino acids 137-574) and F2 (N-terminal portion; amino acids 26-109).
- F1 contains a hydrophobic fusion peptide at its N-terminus and two heptad-repeat regions (HRA and HRB). HRA is near the fusion peptide, and HRB is near the TM domain.
- the F1 and F2 fragments are linked together through two disulfide bonds. Either the uncleaved F0 protein without the signal peptide sequence or a F1-F2 heterodimer can form a RSV F protomer. Three such protomers assemble to form the final RSV F protein complex, which is a homotrimer of the three protomers.
- the F proteins of subtypes A and B are about 90 percent identical in amino acid sequence.
- An example sequence of the F0 precursor polypeptide for the A subtype is provided in SEQ ID NO: 1 (A2 strain; GenBank GI: 138251; Swiss Prot P03420), and for the B subtype is provided in SEQ ID NO: 2 (18537 strain; GenBank GI: 138250; Swiss Prot P13843).
- SEQ ID NO: 1 and SEQ ID NO: 2 are both 574 amino acid sequences.
- the signal peptide sequence for SEQ ID NO: 1 and SEQ ID NO: 2 has also been reported as amino acids 1-25 (GenBank and UniProt). In both sequences the TM domain is from approximately amino acids 530 to 550 but has alternatively been reported as 525-548.
- the cytoplasmic tail begins at either amino acid 548 or 550 and ends at amino acid 574, with the palmitoylated cysteine residue located at amino acid 550.
- RSV F protein is a primary antigen explored for RSV vaccines. The RSV F protein trimer mediates fusion between the virion membrane and the host cellular membrane and also promotes the formation of syncytia.
- Pre-fusion RSV F is recognized by monoclonal antibodies (mAbs) D25, AM22, and MPE8, without discrimination between oligomeric states.
- Pre-fusion F trimers are specifically recognized by mAb AM14 [Gilman MS, Moin SM, Mas V et al., PLoS Pathogens,11(7), 2015].
- pre-F pre-fusion state
- post-F post-fusion state
- the C-terminal coiled-coil of the pre-fusion molecule dissociates into its three constituent strands, which then wrap around the globular head and join three additional helices to form the post-fusion six helix bundle.
- a pre-fusion RSV F trimer is subjected to increasingly harsh chemical or physical conditions, such as elevated temperature, it undergoes structural changes.
- F-specific neutralizing antibodies presumably must bind the pre- fusion conformation of F on the virion, or potentially the extended intermediate, before the viral envelope fuses with a cellular membrane.
- the pre-fusion form of the F protein is considered the preferred conformation as the desired vaccine antigen [Ngwuta, J.O., Chen, M., Modjarrad, K., Joyce, M.G., Kanekiyo, M., Kumar, A., Yassine, H.M., Moin, S.M., Killikelly, A.M., Chuang, G.Y., Druz, A., Georgiev, I.S., Rundlet, E.J., Sastry, M., Stewart-Jones, G.B., Yang. Y., Zhang, B., Nason, M.C., Capella, C., Peeples, M., Ledgerwood, J.
- prefusion F as a vaccine antigen
- the neutralizing and protective antibodies function by interfering with virus entry, it is postulated that an F antigen that does not elicit pre- fusion specific antibodies is not expected to be as effective as an F antigen that elicits pre-fusion specific antibodies. Therefore, it is considered more desirable to utilize an F protein vaccine that contains a F protein immunogen in the pre-fusion form.
- Mutants of the RSV F protein have been provided to increase pre-fusion stability (see for example PCT application No WO2017/109629) and are promising vaccine candidates.
- RSV vaccines that incorporate F protein antigen have been under development.
- the present disclosure provides immunogenic compositions and methods for preventing, treating or ameliorating an infection, disease or condition in a subject comprising the administration of RNA molecules, e.g., immunogenic RNA polynucleotide encoding an amino acid sequence, e.g., an immunogenic antigen, comprising a Respiratory syncytial virus (RSV) protein, an immunogenic variant thereof, or an immunogenic fragment of the RSV protein or the immunogenic variant thereof, e.g., an antigenic peptide or protein.
- the immunogenic antigen comprises an epitope of a RSV protein for inducing an immune response against RSV, in the subject.
- RNA polynucleotide encoding an immunogenic antigen is administered to provide (following expression of the polynucleotide by appropriate target cells) antigen for induction, e.g., stimulation, priming, and/or expansion, of an immune response, e.g., antibodies and/or immune effector cells.
- an immune response e.g., antibodies and/or immune effector cells.
- the immune response to be induced according to the present disclosure is both B cell-mediated immune response, e.g., an antibody-mediated immune response as well as T-cell-mediated immune response.
- the immune response is an anti-RSV immune response.
- the immunogenic compositions described herein comprise RNA molecules comprising RNA (as the active principle) that may be translated into one or more proteins in a recipient’s cells.
- the RNA molecules may contain one or more structural elements optimized for maximal efficacy of the RNA with respect to stability and translational efficiency (5′ cap, 5′ UTR, subgenomic promoter, 3′ UTR, poly-A-tail). In one aspect, the RNA molecules contain all of these elements.
- the RNA molecules described herein may be complexed with lipids and/or proteins to generate RNA-particles (e.g., lipid nanoparticles (LNPs)) for administration. In one aspect, the RNA molecules described herein are complexed with lipids to generate RNA-lipid nanoparticles (e.g. RNA-LNPs) for administration.
- the RNA molecules described herein are complexed with proteins for administration. In one aspect, the RNA molecules described herein are complexed with lipids and proteins for administration. If a combination of different RNA molecules is used, the RNA molecules may be complexed together or complexed separately with lipids and/or proteins to generate RNA-particles for administration.
- the present disclosure provides for RNA molecules and RNA-LNPs that include at least one open reading frame (ORF) encoding a RSV antigen.
- the RSV antigen is a RSV polypeptide.
- the RSV polypeptide is RSV F protein. In some aspects, the RSV F protein is a full-length, truncated, fragment or variant thereof.
- the RSV F protein comprises at least one mutation.
- the present disclosure provides for RNA molecules and RNA-LNPs that include at least one ORF encoding an RSV polypeptide of Table 1.
- the RSV polypeptide comprises an amino acid sequence selected from SEQ ID NO: 1 to 6 or 71 to 74.
- the RSV polypeptide has, has at least, or has at most 90%, 91%, 92%, 93%, 94%, 95, 96%, 97%, 98% or 99% or higher identity to any of the amino acid sequences of Table 1, for example, any of SEQ ID NO: 1 to 6 or 71 to 74.
- the RSV polypeptide consists of any of the amino acid sequences of Table 1, for example, any of SEQ ID NO: 1 to 6 or 71 to 74.
- the present disclosure provides for RNA molecules and RNA-LNPs comprising at least one ORF transcribed from at least one DNA nucleic acid of Table 2.
- the RNA molecule is transcribed from a nucleic acid sequence selected from SEQ ID NO: 7 to 10 or 59 to 62.
- the RNA molecule comprises an ORF transcribed from a nucleic acid sequence that has, has at least, or has at most 90%, 91%, 92%, 93%, 94%, 95, 96%, 97%, 98% or 99% or higher identity to any of the nucleic acid sequences of Table 2, for example, any of SEQ ID NO: 7 to 10 or 59 to 62.
- the RNA molecule comprises an ORF transcribed from a nucleic acid sequence that consists of any of the nucleic acid sequences of Table 2, for example, any of SEQ ID NO: 7 to 10 or 59 to 62.
- RNA molecules and RNA-LNPs comprising at least one ORF comprising an RNA nucleic acid sequence of Table 3.
- the RNA molecule comprises a nucleic acid sequence selected from SEQ ID NO: 11 to 16 or 63 to 70.
- the RNA molecule comprises a nucleic acid sequence that has, has at least, or has at most 90%, 91%, 92%, 93%, 94%, 95, 96%, 97%, 98% or 99% identity to any of the nucleic acid sequences of Table 3, for example, any of SEQ ID NO: 11 to 16 or 63 to 70.
- the RNA molecule comprises a nucleic acid sequence that consists of any of the nucleic acid sequences of Table 3, for example, any of SEQ ID NO: 11 to 16 or 63 to 70.
- each uridine of any of SEQ ID NO: 11 to 16 is replaced by N1-methylpseudouridine ( ⁇ ) (e.g., modified RNA; modRNA).
- ⁇ N1-methylpseudouridine
- the present disclosure further provides for RNA molecules and RNA-LNPs that include a 5’ untranslated region (5’-UTR) and/or a 3’ untranslated region (3’-UTR).
- the RNA molecule includes a 5’ untranslated region (5’-UTR).
- the 5’ UTR comprises a sequence selected from any of SEQ ID NO: 17 to 19. In some aspects, the 5′ UTR comprises a sequence having at least 90%, 91%, 92%, 93%, 94%, 95, 96%, 97%, 98% or 99% or higher identity to any of SEQ ID NO: 17 to 19. In some aspects, the 5′ UTR comprises a sequence selected from any of SEQ ID NO: 17 to 19. In some aspects, the 5′ UTR comprises a sequence consisting of any of SEQ ID NO: 17 to 19. In some aspects, the RNA molecules and RNA-LNPs include a 3’ untranslated region (3’- UTR). In some aspects, the 3’ UTR comprises a sequence selected from any of SEQ ID NO: 20 to 25.
- the 3′ UTR comprises a sequence having at least 90%, 91%, 92%, 93%, 94%, 95, 96%, 97%, 98% or 99% or higher identity to any of SEQ ID NO: 20 to 25. In some aspects, the 3′ UTR comprises a sequence selected from any of SEQ ID NO: 20 to 25. In some aspects, the 3′ UTR comprises a sequence consisting of any of SEQ ID NO: 20 to 25.
- the present disclosure further provides for RNA molecules and RNA-LNPs that include a 5’ cap moiety. In some aspects, the 5′ cap moiety is (3′OMe) - m2 7,3′-O Gppp (m1 2′-O )ApG.
- RNA molecules and RNA-LNPs that include a 3’ poly-A tail.
- the poly-A tail comprises a sequence having SEQ ID NO: 26.
- the RNA molecule includes a 5’ UTR and 3’ UTR.
- the RNA molecule includes a 5’ cap, 5’ UTR, and 3’ UTR.
- the RNA molecule includes a 5’ cap, 5’ UTR, 3’ UTR, and poly-A tail.
- the RNA molecule includes a 5’ UTR, 3’ UTR, and poly-A tail.
- 1, 2, 3, or more of the foregoing elements can be excluded from the RNA molecule.
- each uridine of any of the 5′ UTR, 3′ UTR, and poly-A tail is replaced by N1-methylpseudouridine ( ⁇ ) (e.g., modified RNA; modRNA).
- the poly-A tail length may contain +1/-1 A.
- the uridine is N1-methylpseudouridine ( ⁇ ).
- the present disclosure provides for RNA molecules as described in Table 5.
- the RNA molecule comprises a 5′ UTR of SEQ ID NO: 18, a RSV ORF of SEQ ID NO: 11, a 3′ UTR of SEQ ID NO: 21 and/or a poly-A tail of SEQ ID NO: 26.
- the RNA molecule comprises a 5′ UTR of SEQ ID NO: 18, a RSV ORF of SEQ ID NO: 12, a 3′ UTR of SEQ ID NO: 21 and/or a poly-A tail of SEQ ID NO: 26.
- the RNA molecule comprises a 5′ UTR of SEQ ID NO: 18, a RSV ORF of SEQ ID NO: 63, a 3′ UTR of SEQ ID NO: 21 and/or a poly-A tail of SEQ ID NO: 26.
- the RNA molecule comprises a 5′ UTR of SEQ ID NO: 18, a RSV ORF of SEQ ID NO: 65, a 3′ UTR of SEQ ID NO: 21 and/or a poly-A tail of SEQ ID NO: 26.
- the RNA molecule comprises a 5′ UTR of SEQ ID NO: 18, a RSV ORF of SEQ ID NO: 67, a 3′ UTR of SEQ ID NO: 21 and/or a poly-A tail of SEQ ID NO: 26.
- the RNA molecule comprises a 5′ UTR of SEQ ID NO: 18, a RSV ORF of SEQ ID NO: 69, a 3′ UTR of SEQ ID NO: 21 and/or a poly-A tail of SEQ ID NO: 26.
- the RSV ORF further comprises a stop codon described herein.
- the poly-A tail length may contain +1/-1 A or +2/-2 A.
- each uridine of the RNA molecule is replaced by N1-methylpseudouridine ( ⁇ ) (e.g., modified RNA; modRNA).
- ⁇ N1-methylpseudouridine
- modRNA modified RNA
- the present disclosure further provides for RNA molecules that include at least one open reading frame that was generated from codon-optimized DNA.
- the open reading frame comprises a G/C content of at least, at most, exactly, or between (inclusive or exclusive) any two of 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%, 59%, 60%, 61%, 62%, 63%, 64%, 65%, 66%, 67%, 68%, 69%, 70%, 71%, 72%, 73%, 74%, or 75%, e.g., at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, is or is about 50% to 75%, or is or is or about 55% to 70%.
- the G/C content is or is about 58%, is or is about 66%, or is or is about 62%.
- the present disclosure further provides RNA molecules comprising stabilized RNA.
- the present disclosure further provides for RNA molecules that include RNA having at least one modified nucleotide (e.g., modified RNA; modRNA).
- the modified nucleotide is pseudouridine, N1-methylpseudouridine, N1-ethylpseudouridine, 2-thiouridine, 4′-thiouridine, 5- methylcytosine, 5-methyluridine, 2-thio-1-methyl-1-deaza-pseudouridine, 2-thio-1-methyl- pseudouridine, 2-thio-5-aza-uridine, 2-thio-dihydropseudouridine, 2-thio-dihydrouridine, 2-thio- pseudouridine, 4-methoxy-2-thio-pseudouridine, 4-methoxy-pseudouridine, 4-thio-1-methyl- pseudouridine, 4-thio-pseudouridine, 5-aza-uridine, dihydropseu
- the modified nucleotide is N1-methylpseudouridine ( ⁇ ). In some aspects, 1, 2, 3, 4, 5, or more of the foregoing modified nucleotides can be excluded from the RNA molecule.
- the present disclosure further provides for RNA molecules that are messenger-RNA (mRNA) or self-replicating RNA. In some aspects, the RNA is a mRNA.
- the present disclosure further provides for immunogenic compositions including the RNA molecules described herein.
- the RNA molecules may be formulated in, encapsulated in, complex with, bound to or adsorbed on a lipid nanoparticle (LNP) (e.g., RSV RNA-LNPs) in such immunogenic compositions.
- LNP lipid nanoparticle
- the lipid nanoparticle includes at least one of a cationic lipid, a polymer conjugated lipid (e.g., a PEGylated lipid), and at least one structural lipid (e.g., a neutral lipid and a steroid or steroid analog). In some aspects, 1, 2, 3, or more of the foregoing lipids can be excluded from the lipid nanoparticle. In some aspects, the lipid nanoparticle includes a cationic lipid. In some aspects, the cationic lipid is (4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC- 0315).
- the lipid nanoparticle includes a polymer conjugated lipid.
- the lipid nanoparticle includes a PEGylated lipid, also referred to as a PEG-lipid.
- the PEGylated lipid is PEG-modified phosphatidylethanolamine, PEG-modified phosphatidic acid, PEG-modified ceramides (e.g., PEG-CerC14 or PEG-CerC20), PEG-modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkylglycerols, 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide, glycol-lipids including PEG-c-DOMG, PEG-c-DMA, PEG-s-DMG, N-[(methoxy polyethylene glycol)2000)carbamoyl]-1,2-dimyristyloxlpropyl-3-amine (
- 1, 2, 3, 4, 5, or more of the foregoing PEGylated lipids can be excluded from the RNA molecule.
- the PEGylated lipid is 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC- 0159).
- the lipid nanoparticle includes at least one structural lipid, such as a neutral lipid.
- the neutral lipid is 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dipalmitoylphosphatidylcholine (DPPC), dioleoylphosphatidylglycerol (DOPG), dipalmitoylphosphatidylglycerol (DPPG), dioleoyl-phosphatidylethanolamine (DOPE), palmitoyloleoylphosphatidylcholine (POPC), palmitoyl-oleoyl-phosphatidylethanolamine (POPE), dioleoyl- phosphatidylethanolamine 4-(N-maleimidomethyl)-cyclohexane-1-carboxylate (DOPE- mal), dipalmitoylphosphatidylethanolamine (DPPE), dimyristoylphosphoethanolamine (DM)
- 1, 2, 3, 4, 5, or more of the foregoing structural lipids can be excluded from the RNA molecule.
- the neutral lipid is 1,2- distearoyl-sn-glycero-3-phosphocholine (DSPC).
- the lipid nanoparticle includes a second structural lipid, such as a steroid or steroid analog.
- the steroid or steroid analog is cholesterol.
- the lipid nanoparticle has a mean diameter of about 1 to about 500 nm, e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 1 nm, 10 nm, 20 nm, 30 nm, 40 nm, 50 nm, 60 nm, 70 nm, 80 nm, 90 nm, 100 nm, 110 nm, 120 nm, 130 nm, 140 nm, 150 nm, 160 nm, 170 nm, 180 nm, 190 nm, 200 nm, 210 nm, 220 nm, 230 nm, 240 nm, 250 nm, 260 nm, 270 nm, 280 nm, 290 nm, 300 nm, 310 nm, 320 nm, 330 nm, 340 nm, 350 nm, 360 nm, 370 nm,
- the RNA-LNP immunogenic composition is a liquid RNA-LNP composition comprising an RNA molecule/polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, encapsulated in LNPs with a lipid composition comprising a cationic lipid at a concentration of or of about 0.8 to 0.95 mg/mL (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 0.80, 0.81, 0.82, 0.83, 0.84, 0.85, 0.86, 0.87, 0.88, 0.89, 0.90, 0.91, 0.92, 0.93, 0.94, or 0.95 mg/mL), a PEGylated lipid at a concentration of or of about
- the liquid composition further comprises a buffer composition comprising a first buffer at a concentration of or of about 0.1 to 0.3 mg/mL (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 0.10, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, or 0.30 mg/mL), a second buffer at a concentration of or of about 1.25 to 1.4 mg/mL (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 1.25, 1.26, 1.27, 1.28, 1.29, 1.30, 1.31, 1.32, 1.33, 1.34, 1.35, 1.36, 1.37, 1.38, 1.39, or 1.40 mg/mL), and a stabilizing agent at a concentration of or of about 95 to 110 mg/mL (e.g.,
- the liquid RNA-LNP immunogenic composition comprises an RNA molecule/polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, encapsulated in LNPs with a lipid composition comprising ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2- hexyldecanoate) (ALC-0315) at a concentration of or of about 0.8 to 0.95 mg/m
- the liquid composition further comprises a Tris buffer composition comprising tromethamine at a concentration of or of about 0.1 to 0.3 mg/mL (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 0.10, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, or 0.30 mg/mL) and Tris hydrochloride (HCl) at a concentration of or of about 1.25 to 1.4 mg/mL (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 1.25, 1.26, 1.27, 1.28, 1.29, 1.30, 1.31, 1.32, 1.33, 1.34, 1.35, 1.36, 1.37, 1.38, 1.39, or 1.40 mg/mL), and sucrose at a concentration of or of about 95 to 110 mg/m
- the liquid RNA-LNP immunogenic composition comprises an RNA molecule/polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, encapsulated in a LNP, and further comprising of or of about 5 to 15 mM Tris buffer(e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 mM) and of or of about 200 to 400 mM sucrose (e.
- the RNA-LNP immunogenic composition is a lyophilized (reconstituted) RNA-LNP composition comprising an RNA molecule/polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, encapsulated in LNPs with a lipid composition comprising a cationic lipid at a concentration of or of about 0.8 to 0.95 mg/mL (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 0.80, 0.
- the lyophilized composition further comprises a first buffer at a concentration of or of about 0.01 and 0.15 mg/mL (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.10, 0.11, 0.12, 0.13, 0.14, or 0.15 mg/mL), a second buffer at a concentration of or of about 0.5 and 0.65 mg/mL (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 0.50, 0.51, 0.52, 0.53, 0.54, 0.55, 0.56, 0.57, 0.58, 0.59, 0.60, 0.61, 0.62, 0.63, 0.64, or 0.65 mg/mL), a stabilizing agent at a concentration of or of about 35 to 50 mg/mL (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 35, 36
- the lyophilized compositions are reconstituted in or in about 0.6 to 0.75 mL of the salt diluent (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 0.60, 0.61, 0.62, 0.63, 0.64, 0.65, 0.66, 0.67, 0.68, 0.69, 0.70, 0.71, 0.72, 0.73, 0.74, or 0.75 mL).
- Concentrations in the lyophilized RNA-LNP composition are determined post-reconstitution. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing elements can be excluded from the lyophilized RNA-LNP composition.
- a lyophilized (reconstituted) RNA-LNP composition comprises an RNA polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, encapsulated in LNPs with a lipid composition of ALC-0315 at a concentration of or of about 0.8 to 0.95 mg/mL (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 0.80, 0.81, 0.82, 0.83, 0.84, 0.85, 0.86, 0.87, 0.88, 0.89, 0.90, 0.91, 0.92, 0.93, 0.94, or 0.
- the lyophilized compositions are reconstituted in or in about 0.6 to 0.75 mL of sodium chloride (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 0.60, 0.61, 0.62, 0.63, 0.64, 0.65, 0.66, 0.67, 0.68, 0.69, 0.70, 0.71, 0.72, 0.73, 0.74, or 0.75 mL).
- Concentrations in the lyophilized RNA-LNP composition are determined post-reconstitution. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing elements can be excluded from the lyophilized RNA-LNP composition.
- 1, 2, 3, 4, 5, or more of the foregoing element concentrations can be excluded from the lyophilized RNA-LNP composition.
- the present disclosure provides for RNA molecules, RNA-LNPs and immunogenic compositions that may be administered to a subject at a dose per administration of at least, at most, exactly, or between (inclusive or exclusive) any two of 1 ⁇ g, 15 ⁇ g, 30 ⁇ g, 45 ⁇ g, 60 ⁇ g, 75 ⁇ g, 90 ⁇ g, 100 ⁇ g or higher of RSV RNA encapsulated in LNP.
- 1, 2, 3, 4, 5, or more of the foregoing concentrations of RSV RNA encapsulated in LNP can be excluded.
- RNA molecules, RNA-LNPs and immunogenic compositions that may be administered in a single dose.
- the present disclosure further provides for RNA molecules, RNA-LNPs and immunogenic compositions that may be administered twice (e.g., Day 0 and on or about Day 7, Day 0 and on or about Day 14, Day 0 and on or about Day 21, Day 0 and on or about Day 28, Day 0 and on or about Day 60, Day 0 and on or about Day 90, Day 0 and on or about Day 120, Day 0 and on or about Day 150, Day 0 and on or about Day 180, Day 0 and on or about 1 month later, Day 0 and on or about 2 months later, Day 0 and on or about 3 months later, Day 0 and on or about 6 months later, Day 0 and on or about 9 months later, Day 0 and on or about 12 months later, Day 0 and on or about 18 months later, Day 0 and on or about 2 years later, Day 0 and on or about 5 years later, or Day 0 and on or about
- the present disclosure further provides for RNA molecules, RNA-LNPs and immunogenic compositions that may be administered twice at Day 0 and on or about 2 months later.
- the present disclosure further provides for RNA molecules, RNA-LNPs and immunogenic compositions that may be administered twice at Day 0 and on or about 6 months later.
- the present disclosure further provides for RNA molecules, RNA-LNPs and immunogenic compositions that may be administered three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, or more times.
- periodic boosters at intervals of 1-5 years may be desirable to maintain protective levels of the antibodies.
- the present disclosure further provides for administration of at least one booster dose.
- the present disclosure provides for a method of inducing an immune response against RSV in a subject, including administering to the subject an effective amount of an RNA molecule, RNA- LNP and/or immunogenic composition described herein.
- the present disclosure further provides for the use of an RNA molecule, RNA-LNP and/or immunogenic composition described herein in the manufacture of a medicament for use in inducing an immune response against RSV in a subject.
- the present disclosure provides for a method of inducing an immune response against RSV in a subject, including administering to the subject an effective amount of an RNA molecule and/ or RNA-LNP that includes at least one open reading frame encoding a RSV polypeptide or immunogenic composition described herein.
- the present disclosure further provides for the use of an RNA molecule and/or RNA-LNP that includes at least one open reading frame encoding a RSV polypeptide or immunogenic composition described herein in the manufacture of a medicament for use in inducing an immune response against RSV in a subject.
- the present disclosure provides for a method of inducing an immune response against RSV in a subject, including administering to the subject an effective amount of an RNA molecule and/or RNA-LNP that includes at least one open reading frame encoding a polypeptide of a gene of interest or composition described herein.
- the present disclosure further provides for the use of an RNA molecule and/or RNA-LNP that includes at least one open reading frame encoding a polypeptide of a gene of interest or composition described herein in the manufacture of a medicament for use in inducing an immune response against RSV in a subject.
- the present disclosure provides for a method of preventing, treating, and/or ameliorating an infection, disease, or condition in a subject, including administering to a subject an effective amount of an RNA molecule, RNA-LNP and/or immunogenic composition described herein.
- the present disclosure further provides for the use of an RNA molecule, RNA-LNP and/or immunogenic composition described herein in the manufacture of a medicament for use in preventing, treating, and/or ameliorating an infection, disease, or condition in a subject.
- the infection, disease, or condition is associated with RSV .
- the infection, disease, or condition is acute lower respiratory infection (ALRI), including pneumonia and bronchitis.
- the infection, disease, or condition is acute lower respiratory infection (ALRI), including pneumonia and bronchitis.
- ARI acute lower respiratory infection
- the present disclosure provides for a method of preventing, treating, and/or ameliorating an infection, disease, or condition in a subject, including administering to a subject an effective amount of an RNA molecule and/or RNA-LNP that includes at least one open reading frame encoding a RSV polypeptide or immunogenic composition described herein.
- the present disclosure further provides for the use of an RNA molecule and/or RNA-LNP that includes at least one open reading frame encoding a RSV polypeptide or immunogenic composition described herein in the manufacture of a medicament for use in preventing, treating, and/or ameliorating an infection, disease, or condition in a subject.
- the infection, disease, or condition is associated with RSV .
- the infection, disease, or condition is acute lower respiratory infection (ALRI), including pneumonia and bronchitis.
- the infection, disease, or condition is acute lower respiratory infection (ALRI), including pneumonia and bronchitis.
- ARI acute lower respiratory infection
- the present disclosure further provides for a method of preventing, treating, and/or ameliorating an infection, disease, or condition in a subject, including administering to a subject an effective amount of RNA molecules and/or RNA-LNPs that include at least one open reading frame encoding a polypeptide of a gene of interest or immunogenic compositions described herein.
- RNA molecules and/or RNA-LNPs that include at least one open reading frame encoding a polypeptide of a gene of interest or immunogenic compositions described herein in the manufacture of a medicament for use in preventing, treating, and/or ameliorating an infection, disease, or condition in a subject.
- the infection, disease, or condition is associated with the gene of interest.
- the subject is at least, at most, exactly, or between (inclusive or exclusive) any two of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 months of age, or 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, or more years of age.
- the subject is, is at least, is at most, or is about less than 1 year of age, 1 year of age or older, 5 years of age or older, 10 years of age or older, 20 years of age or older, 30 years of age or older, 40 years of age or older, 50 years of age or older, 60 years of age or older, 70 years of age or older, or older. In some aspects, the subject the subject is or is about 50 years of age or older. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing age groups are not administered the RNA molecules and/or RNA- LNPs. In some aspects, the subject is immunocompetent. In some aspects, the subject is immunocompromised.
- the present disclosure provides for a method or use described herein, wherein the RNA molecule, RNA-LNP and/or immunogenic composition is administered as a vaccine.
- the present disclosure provides a method or use described herein, wherein the RNA molecule, RNA-LNP and/or immunogenic composition is administered by intradermal, intramuscular, or intranasal injection. It is contemplated that any aspect discussed in this specification may be implemented with respect to any method or composition of the disclosure, and vice versa. Furthermore, compositions of the disclosure may be used to achieve methods of the disclosure.
- FIG.1A to 1E show immunogenicity of modRNA-LNP formulations of RSV 847 in mice.
- Female BALB/c mice (10/group) were immunized intramuscularly at day 0 and 21 with RSV 847 constructs either as bivalent protein subunit (RSV 847A+B) or modRNA-LNP formulation either as monovalent (RSV 847A) or bivalent (RSV 847A+B) at indicated dose.
- RSV 847A+B bivalent protein subunit
- RSV 847A+B modRNA-LNP formulation either as monovalent (RSV 847A) or bivalent (RSV 847A+B) at indicated dose.
- serum was collected for RSV neutralizing assay and spleen for T-cell assays (ELISpot and Intracellular Cytokine Staining, ICS assays).
- FIG. 1B show neutralization assay results for RSV A and B expressed as 50% neutralizing titers (each symbol represents a titer from an individual animal. Bars represent geometric mean titer (GMT)).
- FIG.1C shows ELISpot assay results that measure the number of RSV A+B F-specific cells secreting IFN- ⁇ and expressed as spot forming cells (SFC) per million cells.
- FIG.1D and FIG.1E show ICS assay results that measured RSV A+B F-specific IFN- ⁇ -expressing cells within CD4+ and CD8+ T cells expressed as percentage of IFN- ⁇ + cells. Bars and errors bars depict median with interquartile range. NA: not analyzed.
- FIG.2 shows the immunogenicity of modRNA-LNP formulations encoding for different RSV A prefusion F (preF) designs in mice.
- Female BALB/c mice (10/group) were immunized intramuscularly at day 0 and 21 with modRNA-LNP formulations encoding RSV A prefusion F (preF) designs as depicted herein at 0.5 ⁇ g dose.
- preF prefusion F
- FIG.3A to 3F show the immunogenicity of modRNA-LNP and saRNA-LNP formulations of RSV prefusion F (preF) in mice.
- Female BALB/c mice (10/group) were immunized intramuscularly at day 0 and 21 with RSV preF constructs either as bivalent protein subunit (RSV preF A+B) or bivalent modRNA-LNP formulation or bivalent saRNA-LNP formulation at indicated dose.
- RSV preF A+B bivalent protein subunit
- 2W PD2 serum was collected for RSV neutralizing assay and on day 35 spleens were harvested for T-cell assays (Intracellular Cytokine Staining, ICS assay).
- Neutralization assay results are shown for RSV A and B expressed as 50% neutralizing titers at either 3W PD1 (FIG.3A and FIG.3B) or 2W PD2 (FIG.3C and FIG.3D). Each symbol represents a titer from an individual animal. Bars represent geometric mean titer (GMT).
- FIG.3E and FIG.3F show ICS assay results that measured RSV preF A+B F-specific IFN- ⁇ -expressing cells within CD4+ T cells and CD8+ T cells -. Bars and errors bars depict median with interquartile range. NT: not tested.
- FIG.4 schematically illustrates the wild-type (WT) RSV F protein (RSV WT) and variant RSV F protein constructs, where “SP” refers to a signal peptide sequence (amino acid residues 1-25 of each construct), “TM” refers to a transmembrane peptide sequence corresponding to the portion of the protein that spans the cell membrane, “CT” refers to a cytoplasmic tail peptide sequence corresponding to the portion of the protein that extends into the cell cytoplasm, and “ectodomain” refers to a peptide sequence corresponding to the portion of the protein that extends into the extracellular space wherein the ectodomain comprises amino acid residues 1-513 (without TM and CT, denoted by “ ⁇ TM & CT”).
- SP refers to a signal peptide sequence (amino acid residues 1-25 of each construct)
- TM refers to a transmembrane peptide sequence corresponding to the portion of the protein that spans the cell membrane
- RNA molecules e.g., RNA polynucleotide
- ORF open reading frame
- RSV antigen is a RSV polypeptide.
- RSV polypeptide is a RSV F polypeptide.
- RSV polypeptide comprises an amino acid sequence set forth in Table 1.
- the RNA molecules comprise an ORF transcribed from at least one DNA nucleic acid sequence of Table 2. In some aspects, the RNA molecules comprise an ORF comprising an RNA nucleic acid sequence of Table 3. In some aspects the RNA molecule comprises at least one of a 5’ cap, 5’ UTR, 3’ UTR and poly-A tail. In other aspects the RNA molecule comprises at least one of a 5’ cap, 3’ UTR and poly-A tail.
- the present disclosure provides for an RNA molecule comprising modified nucleotides (e.g., modified RNA; modRNA).
- the present disclosure provides for an immunogenic composition comprising any one of the RNA molecules encoding a RSV polypeptide described herein complexed with, encapsulated in, or formulated with one or more lipids, and forming lipid nanoparticles (RNA-LNPs).
- the present disclosure further provides for an immunogenic composition comprising any one of the RNA molecules comprising at least one RNA nucleic acid described herein complexed with, encapsulated in, or formulated with one or more lipids, and forming RNA-LNPs.
- the present disclosure further provides for a method of preventing, treating or ameliorating an infection, disease or condition (e.g., RSV infection-related Respiratory tract illness, including pneumonia and bronchitis) in a subject via administering to a subject an effective amount of an RNA molecule, RNA-LNP or an immunogenic composition described herein.
- an infection, disease or condition e.g., RSV infection-related Respiratory tract illness, including pneumonia and bronchitis
- the present disclosure further provides for the use of the RNA molecule, RNA-LNP and/or an immunogenic composition described herein as a vaccine.
- the present invention may be understood more readily by reference to the following detailed description of the embodiments of the invention and the Examples included herein. It is to be understood that this invention is not limited to specific methods of making that may of course vary.
- A, B, and/or C includes: A alone, B alone, C alone, a combination of A and B, a combination of A and C, a combination of B and C, or a combination of A, B, and C.
- “and/or” operates as an inclusive or.
- the phrase “essentially all” is defined as “at least 95%”; if essentially all members of a group have a certain property, then at least 95% of members of the group have that property.
- compositions and methods for their use may “comprise,” “consist essentially of,” or “consist of” any of the ingredients or steps disclosed throughout the specification.
- compositions and methods “consisting essentially of” any of the ingredients or steps disclosed limits the scope of the claim to the specified materials or steps which do not materially affect the basic and novel characteristic of the claimed disclosure.
- the words “consisting of” (and any form of consisting of, such as “consist of” and “consists of”) means including, and limited to, whatever follows the phrase “consisting of.” Thus, the phrase “consisting of” indicates that the listed elements are required or mandatory, and that no other elements may be present.
- inhibitors includes any measurable decrease (e.g., a 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99% decrease) or complete inhibition to achieve a desired result.
- the terms “improve,” “promote,” or “increase” or any variation of these terms includes any measurable increase (e.g., a 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99% increase) to achieve a desired result or production of a protein or molecule.
- the terms “reference,” “standard,” or “control” describe a value relative to which a comparison is performed. For example, an agent, subject, population, sample, or value of interest is compared with a reference, standard, or control agent, subject, population, sample, or value of interest.
- a reference, standard, or control may be tested and/or determined substantially simultaneously and/or with the testing or determination of interest for an agent, subject, population, sample, or value of interest and/or may be determined or characterized under comparable conditions or circumstances to the agent, subject, population, sample, or value of interest under assessment.
- isolated may refer to a nucleic acid or polypeptide that is substantially free of cellular material, bacterial material, viral material, or culture medium (when produced by recombinant DNA techniques) of their source of origin, or chemical precursors or other chemicals (when chemically synthesized).
- an isolated compound refers to one that may be administered to a subject as an isolated compound; in other words, the compound may not simply be considered “isolated” if it is adhered to a column or embedded in an agarose gel.
- an “isolated nucleic acid fragment” or “isolated peptide” is a nucleic acid or protein fragment that is not naturally occurring as a fragment and/or is not typically in the functional state and/or that is altered or removed from the natural state through human intervention.
- nucleic acid is a molecule comprising nucleic acid components and refers to DNA or RNA molecules.
- a nucleic acid molecule is a polymer comprising or consisting of nucleotide monomers, which are covalently linked to each other by phosphodiester-bonds of a sugar/phosphate-backbone. Nucleic acids may also encompass modified nucleic acid molecules, such as base-modified, sugar-modified or backbone-modified etc. DNA or RNA molecules.
- Nucleic acids may exist in a variety of forms such as: isolated segments and recombinant vectors of incorporated sequences or recombinant polynucleotides encoding polypeptides, such as antigens or one or both chains of an antibody, or a fragment, derivative, mutein, or variant thereof, polynucleotides sufficient for use as hybridization probes, PCR primers or sequencing primers for identifying, analyzing, mutating or amplifying a polynucleotide encoding a polypeptide, anti-sense nucleic acids for inhibiting expression of a polynucleotide, mRNA, saRNA, modRNA and complementary sequences of the foregoing described herein.
- Nucleic acids may encode an epitope to which antibodies may bind.
- epitope refers to a moiety that is specifically recognized by an immunoglobulin (e.g., antibody or receptor) binding component.
- an epitope is comprised of a plurality of chemical atoms or groups on an antigen.
- such chemical atoms or groups are surface-exposed when the antigen adopts a relevant three-dimensional conformation.
- such chemical atoms or groups are physically near to each other in space when the antigen adopts such a conformation.
- at least some such chemical atoms are groups are physically separated from one another when the antigen adopts an alternative conformation (e.g., is linearized).
- Nucleic acids may be single-stranded or double-stranded and may comprise RNA and/or DNA nucleotides and artificial variants thereof (e.g., peptide nucleic acids).
- a nucleic acid sequence may encode a polypeptide sequence with additional heterologous coding sequences, for example to allow for purification of the polypeptide, transport, secretion, post- translational modification, or for therapeutic benefits such as targeting or efficacy.
- a tag or other heterologous polypeptide may be added to the modified polypeptide-encoding sequence, wherein “heterologous” refers to a polypeptide that is not the same as the modified polypeptide.
- polynucleotide refers to a nucleic acid molecule that may be recombinant or has been isolated from total genomic nucleic acid. Included within the term “polynucleotide” are oligonucleotides (nucleic acids 100 residues or less in length), recombinant vectors, including, for example, plasmids, cosmids, phage, viruses, and the like. Polynucleotides include, in certain aspects, regulatory sequences, isolated substantially away from their naturally occurring genes or protein encoding sequences.
- Polynucleotides may be single-stranded (coding or antisense) or double-stranded, and may be RNA, DNA (genomic, cDNA, or synthetic), analogs thereof, or a combination thereof. Additional coding or non-coding sequences may, but need not, be present within a polynucleotide.
- polynucleotide variants having substantial identity to the sequences disclosed herein; those comprising equal to any one of, at least any one of, at most any one of, or between any two of 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% or higher sequence identity, compared to a polynucleotide sequence provided herein using the methods described herein (e.g., BLAST analysis using standard parameters).
- the isolated polynucleotide will comprise a nucleotide sequence encoding a polypeptide that has at least 90% identity to an amino acid sequence described herein, over the entire length of the sequence; or a nucleotide sequence complementary to said isolated polynucleotide. In some aspects, the isolated polynucleotide will comprise a nucleotide sequence encoding a polypeptide that has at least 95% identity to an amino acid sequence described herein, over the entire length of the sequence; or a nucleotide sequence complementary to said isolated polynucleotide.
- nucleic acid segments regardless of the length of the coding sequence itself, may be combined with other nucleic acid sequences, such as promoters, polyadenylation signals, additional restriction enzyme sites, multiple cloning sites, other coding segments, and the like, such that their overall length may vary considerably.
- the nucleic acids may be any length.
- nucleotides may be, for example, equal to any one of, at least any one of, at most any one of, or between any two of 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 75, 100, 125, 175, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 3000, 5000, 6000, 7000, 8000, 9000, 10000, 11000, 12000, 13000, 14000, 15000 or more nucleotides in length, and/or may comprise one or more additional sequences, for example, regulatory sequences, and/or be a part of a larger nucleic acid, for example, a vector.
- nucleic acid fragment of almost any length may be employed, with the total length being limited by the ease of preparation and use in the intended recombinant nucleic acid protocol.
- gene is used to refer to a nucleic acid that encodes a protein, polypeptide, or peptide (including any sequences required for proper transcription, post- translational modification, or localization).
- this term encompasses genomic sequences, expression cassettes, cDNA sequences, and smaller engineered nucleic acid segments that express, or may be adapted to express, proteins, polypeptides, domains, peptides, fusion proteins, and mutants.
- a nucleic acid encoding all or part of a polypeptide may contain a contiguous nucleic acid sequence encoding all or a portion of such a polypeptide. It also is contemplated that a particular polypeptide may be encoded by nucleic acids containing variations having slightly different nucleic acid sequences but, nonetheless, encode the same or substantially similar polypeptide.
- expression of a nucleic acid sequence refers to the generation of any gene product from the nucleic acid sequence.
- a gene product may be a transcript.
- a gene product may be a polypeptide.
- expression of a nucleic acid sequence involves one or more of the following: (1) production of an RNA template from a DNA sequence (e.g., by transcription); (2) processing of an RNA transcript (e.g., by splicing, editing, etc.); (3) translation of an RNA into a polypeptide or protein; and/or (4) post- translational modification of a polypeptide or protein.
- engineered refers to the aspect of having been manipulated by the hand of man.
- a polynucleotide is considered to be “engineered” when two or more sequences that are not linked together in that order in nature are manipulated by the hand of man to be directly linked to one another in the engineered polynucleotide and/or when a particular residue in a polynucleotide is non-naturally occurring and/or is caused through action of the hand of man to be linked with an entity or moiety with which it is not linked in nature.
- DNA means a nucleic acid molecule comprising nucleotides such as deoxy-adenosine-monophosphate, deoxy-thymidine-monophosphate, deoxy- guanosine-monophosphate and deoxy-cytidine-monophosphate monomers which are composed of a sugar moiety (deoxyribose), a base moiety and a phosphate moiety, and polymerize by a characteristic backbone structure.
- the backbone structure is, typically, formed by phosphodiester bonds between the sugar moiety of the nucleotide, e.g., deoxyribose, of a first and a phosphate moiety of a second, adjacent monomer.
- DNA sequence The specific order of the monomers, e.g., the order of the bases linked to the sugar/phosphate-backbone, is called the DNA sequence.
- DNA may be single stranded or double stranded. In the double stranded form, the nucleotides of the first strand typically hybridize with the nucleotides of the second strand, e.g. by A/T-base-pairing and G/C- base-pairing. DNA may contain all, or a majority of, deoxyribonucleotide residues.
- deoxyribonucleotide means a nucleotide lacking a hydroxyl group at the 2′ position of a ⁇ -D-ribofuranosyl group.
- DNA may encompass double stranded DNA, antisense DNA, single stranded DNA, isolated DNA, synthetic DNA, DNA that is recombinantly produced, and modified DNA.
- RNA means a nucleic acid molecule comprising nucleotides such as adenosine-monophosphate, uridine-monophosphate, guanosine-monophosphate and cytidine-monophosphate monomers which are connected to each other along a so-called backbone.
- the backbone is formed by phosphodiester bonds between the sugar, e.g., ribose, of a first and a phosphate moiety of a second, adjacent monomer.
- RNA may be obtainable by transcription of a DNA-sequence, e.g., inside a cell. In eukaryotic cells, transcription is typically performed inside the nucleus or the mitochondria. In vivo, transcription of DNA may result in premature RNA which is processed into messenger-RNA (mRNA). Processing of the premature RNA, e.g. in eukaryotic organisms, comprises various posttranscriptional modifications such as splicing, 5′ capping, polyadenylation, export from the nucleus or the mitochondria. Mature messenger RNA is processed and provides the nucleotide sequence that may be translated into an amino acid sequence of a peptide or protein.
- mRNA messenger-RNA
- a mature mRNA may comprise a 5′ cap, a 5′ UTR, an open reading frame, a 3′ UTR and a poly-A tail sequence.
- RNA may contain all, or a majority of, ribonucleotide residues.
- ribonucleotide means a nucleotide with a hydroxyl group at the 2′ position of a ⁇ -D-ribofuranosyl group.
- RNA may be messenger RNA (mRNA) that relates to a RNA transcript which encodes a peptide or protein.
- RNA generally contains a 5′ untranslated region (5′ UTR), a polypeptide coding region, and a 3′ untranslated region (3′ UTR).
- RNA may encompass double stranded RNA, antisense RNA, single stranded RNA, isolated RNA, synthetic RNA, RNA that is recombinantly produced, and modified RNA (modRNA).
- modified RNA modified RNA
- isolated RNA is defined as an RNA molecule that may be recombinant or has been isolated from total genomic nucleic acid.
- An isolated RNA molecule or protein may exist in substantially purified form, or may exist in a non-native environment such as, for example, a host cell.
- modified RNA refers to an RNA molecule having at least one addition, deletion, substitution, and/or alteration of one or more nucleotides as compared to naturally occurring RNA. Such alterations may refer to the addition of non-nucleotide material to internal RNA nucleotides, or to the 5′ and/or 3′ end(s) of RNA.
- such modRNA contains at least one modified nucleotide, such as an alteration to the base of the nucleotide.
- a modified nucleotide may replace one or more uridine and/or cytidine nucleotides.
- these replacements may occur for every instance of uridine and/or cytidine in the RNA sequence, or may occur for only select uridine and/or cytidine nucleotides.
- Such alterations to the standard nucleotides in RNA may include non-standard nucleotides, such as chemically synthesized nucleotides or deoxynucleotides.
- at least one uridine nucleotide may be replaced with N1-methylpseudouridine in an RNA sequence.
- Other such altered nucleotides are known to those of skill in the art.
- Such altered RNA molecules are considered analogs of naturally-occurring RNA.
- the RNA is produced by in vitro transcription using a DNA template, where DNA refers to a nucleic acid that contains deoxyribonucleotides.
- the RNA may be replicon RNA (replicon), in particular self-replicating RNA, or self-amplifying RNA (saRNA).
- replicon RNA
- saRNA self-amplifying RNA
- RNA may be used as a therapeutic modality to treat and/or prevent a number of conditions in mammals, including humans. Methods described herein comprise administration of the RNA described herein to a mammal, such as a human.
- RNA administered is in vitro transcribed RNA.
- RNA may be used to encode at least one antigen intended to generate an immune response in said mammal.
- Pathogenic antigens are peptide or protein antigens derived from a pathogen associated with infectious disease. In specific aspects, the pathogenic are peptide or protein antigens derived from RSV.
- Conditions and/or diseases that may be treated with RNA disclosed herein include, but are not limited to, those caused and/or impacted by viral infection. Such viruses include, but are not limited to, RSV.
- “Prevent” or “prevention,” as used herein when used in connection with the occurrence of a disease, disorder, and/or condition refers to reducing the risk of developing the disease, disorder and/or condition and/or to delaying onset of one or more characteristics or symptoms of the disease, disorder or condition. Prevention may be considered complete when onset of a disease, disorder, or condition has been delayed for a predefined period of time.
- risk of a disease, disorder, and/or condition refers to a likelihood that a particular individual will develop the disease, disorder, and/or condition.
- risk is expressed as a percentage.
- risk is, is at least, or is at most from 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90 up to 100%.
- risk is expressed as a risk relative to a risk associated with a reference sample or group of reference samples.
- a reference sample or group of reference samples have a known risk of a disease, disorder, condition and/or event.
- a reference sample or group of reference samples are from individuals comparable to a particular individual.
- risk may reflect one or more genetic attributes, e.g., which may predispose an individual toward development (or not) of a particular disease, disorder and/or condition.
- risk may reflect one or more epigenetic events or attributes and/or one or more lifestyle or environmental events or attributes.
- Susceptible to An individual who is “susceptible to” a disease, disorder, and/or condition is one who has a higher risk of developing the disease, disorder, and/or condition than does a member of the general public.
- an individual who is susceptible to a disease, disorder and/or condition may not have been diagnosed with the disease, disorder, and/or condition.
- an individual who is susceptible to a disease, disorder, and/or condition may exhibit symptoms of the disease, disorder, and/or condition. In some aspects, an individual who is susceptible to a disease, disorder, and/or condition may not exhibit symptoms of the disease, disorder, and/or condition. In some aspects, an individual who is susceptible to a disease, disorder, and/or condition will develop the disease, disorder, and/or condition. In some aspects, an individual who is susceptible to a disease, disorder, and/or condition will not develop the disease, disorder, and/or condition.
- the terms “protein,” “polypeptide,” or “peptide” are used herein as synonyms and refer to a polymer of amino acid monomers, e.g., a molecule comprising at least two amino acid residues.
- Polypeptides may include gene products, naturally occurring polypeptides, synthetic polypeptides, homologs, orthologs, paralogs, fragments and other equivalents, variants, and analogs of the foregoing. Polypeptides may be a single molecule or may be a multi-molecular complex such as a dimer, trimer or tetramer.
- a protein comprises one or more peptides or polypeptides, and may be folded into a 3-dimensional form, which may be required for the protein to exert its biological function.
- wild type or ”WT” or “native” refers to the endogenous version of a molecule that occurs naturally in an organism.
- wild type versions of a protein or polypeptide are employed, however, in other aspects of the disclosure, a modified protein or polypeptide is employed to generate an immune response.
- a “modified protein” or “modified polypeptide” or a “variant” refers to a protein or polypeptide whose chemical structure, particularly its amino acid sequence, is altered with respect to the wild type protein or polypeptide.
- a modified/variant protein or polypeptide has at least one modified activity or function (recognizing that proteins or polypeptides may have multiple activities or functions).
- a modified/variant protein or polypeptide may be altered with respect to one activity or function yet retain a wild type activity or function in other respects, such as immunogenicity.
- a protein is specifically mentioned herein, it is in general a reference to a native (wild type) or recombinant (modified) protein.
- the protein may be isolated directly from the organism of which it is native, produced by recombinant DNA/exogenous expression methods, produced by solid-phase peptide synthesis (SPPS), or other in vitro methods.
- SPPS solid-phase peptide synthesis
- fragment with reference to an amino acid sequence (peptide or protein), relates to a part of an amino acid sequence, e.g., a sequence which represents the amino acid sequence shortened at the N-terminus and/or C-terminus.
- a fragment shortened at the C- terminus (N-terminal fragment) is obtainable, e.g., by translation of a truncated open reading frame that lacks the 3′-end of the open reading frame.
- a fragment shortened at the N-terminus is obtainable, e.g., by translation of a truncated open reading frame that lacks the 5′-end of the open reading frame, as long as the truncated open reading frame comprises a start codon that serves to initiate translation.
- a fragment of an amino acid sequence comprises, e.g., at least 50 %, at least 60 %, at least 70 %, at least 80%, at least 90%, or at least 99% of the amino acid residues from an amino acid sequence.
- a fragment of a polypeptide, DNA nucleic acid or RNA nucleic acid sequence refers to a sequence having sequence identity of at least, at most, exactly, or between any two of 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% with a polypeptide, DNA nucleic acid or RNA nucleic acid sequence, from which it is derived.
- a fragment of a polypeptide, DNA nucleic acid or RNA nucleic acid sequence refers to a sequence having sequence identity of at least 70% with a polypeptide, DNA nucleic acid or RNA nucleic acid sequence, from which it is derived. In one aspect, a fragment of a polypeptide, DNA nucleic acid or RNA nucleic acid sequence refers to a sequence having sequence identity of at least 80% with a polypeptide, DNA nucleic acid or RNA nucleic acid sequence, from which it is derived.
- a fragment of a polypeptide, DNA nucleic acid or RNA nucleic acid sequence refers to a sequence having sequence identity of at least 85% with a polypeptide, DNA nucleic acid or RNA nucleic acid sequence, from which it is derived. In one aspect, a fragment of a polypeptide, DNA nucleic acid or RNA nucleic acid sequence refers to a sequence having sequence identity of at least 90% with a polypeptide, DNA nucleic acid or RNA nucleic acid sequence, from which it is derived.
- a fragment of a polypeptide, DNA nucleic acid or RNA nucleic acid sequence refers to a sequence having sequence identity of at least 95% with a polypeptide, DNA nucleic acid or RNA nucleic acid sequence, from which it is derived. In one aspect, a fragment of a polypeptide, DNA nucleic acid or RNA nucleic acid sequence refers to a sequence having sequence identity of at least 97% with a polypeptide, DNA nucleic acid or RNA nucleic acid sequence, from which it is derived.
- a fragment of a polypeptide, DNA nucleic acid or RNA nucleic acid sequence refers to a sequence having sequence identity of at least 99% with a polypeptide, DNA nucleic acid or RNA nucleic acid sequence, from which it is derived.
- the term “variant” refers to a molecule that shows significant structural identity with a reference molecule but differs structurally from the reference molecule, e.g., in the presence or absence or in the level of one or more chemical moieties as compared to the reference entity. In some aspects, a variant also differs functionally from its reference molecule.
- any biological or chemical reference molecule has certain characteristic structural elements.
- a variant by definition, is a distinct molecule that shares one or more such characteristic structural elements but differs in at least one aspect from the reference molecule.
- a variant polypeptide or nucleic acid may differ from a reference polypeptide or nucleic acid as a result of one or more differences in amino acid or nucleotide sequence and/or one or more differences in chemical moieties (e.g., carbohydrates, lipids, phosphate groups) that are covalently components of the polypeptide or nucleic acid (e.g., that are attached to the polypeptide or nucleic acid backbone).
- moieties e.g., carbohydrates, lipids, phosphate groups
- a variant polypeptide or nucleic acid shows an overall sequence identity with a reference polypeptide or nucleic acid that is at least, at most, exactly, or between any two of 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, or 99%.
- a variant polypeptide or nucleic acid does not share at least one characteristic sequence element with a reference polypeptide or nucleic acid.
- a reference polypeptide or nucleic acid has one or more biological activities.
- a variant polypeptide or nucleic acid shares one or more of the biological activities of the reference polypeptide or nucleic acid.
- a variant polypeptide or nucleic acid lacks one or more of the biological activities of the reference polypeptide or nucleic acid. In some aspects, a variant polypeptide or nucleic acid shows a reduced level of one or more biological activities as compared to the reference polypeptide or nucleic acid. In some aspects, a polypeptide or nucleic acid of interest is considered to be a “variant” of a reference polypeptide or nucleic acid if it has an amino acid or nucleotide sequence that is identical to that of the reference but for a small number of sequence alterations at particular positions.
- the variant polypeptide or nucleic acid sequence has at least one modification compared to the reference polypeptide or nucleic acid sequence, e.g., from 1 to about 20 modifications. In one aspect, the variant polypeptide or nucleic acid sequence has from 1 to about 10 modifications compared to the reference polypeptide or nucleic acid sequence. In one aspect, the variant polypeptide or nucleic acid sequence has from 1 to about 5 modifications compared to the reference polypeptide or nucleic acid sequence. In one aspect, the variant polypeptide or nucleic acid sequence has from 1 to about 4 modifications compared to the reference polypeptide or nucleic acid sequence.
- a variant polypeptide or nucleic acid comprises a very small number (e.g., fewer than about 5, about 4, about 3, about 2, or about 1) number of substituted, inserted, or deleted, functional residues (e.g., residues that participate in a particular biological activity) relative to the reference.
- a variant polypeptide or nucleic acid comprises about 10, about 9, about 8, about 7, about 6, about 5, about 4, about 3, about 2, or about 1 substituted residues as compared to a reference. In some aspects, a variant polypeptide or nucleic acid comprises fewer than about 25, about 20, about 19, about 18, about 17, about 16, about 15, about 14, about 13, about 10, about 9, about 8, about 7, about 6, and commonly fewer than about 5, about 4, about 3, or about 2 additions or deletions as compared to the reference. In some aspects, a variant polypeptide or nucleic acid comprises not more than about 5, about 4, about 3, about 2, or about 1 addition or deletion, and, in some aspects, comprises no additions or deletions, as compared to the reference.
- a reference polypeptide or nucleic acid is a “wild type” or “WT” or “native” sequence found in nature, including allelic variations.
- a wild type polypeptide or nucleic acid sequence has a sequence that has not been intentionally modified.
- variants of an amino acid sequence (peptide, protein, or polypeptide) comprise amino acid insertion variants, amino acid addition variants, amino acid deletion variants and/or amino acid substitution variants.
- “Variants” of a nucleotide sequence comprise nucleotide insertion variants, nucleotide addition variants, nucleotide deletion variants and/or nucleotide substitution variants.
- variant includes all mutants, splice variants, post-translationally modified variants, conformations, isoforms, allelic variants, species variants, and species homologs, in particular those which are naturally occurring.
- variant includes, in particular, fragments of an amino acid or nucleic acid sequence. Changes may be introduced by mutation into a nucleic acid, thereby leading to changes in the amino acid sequence of a polypeptide (e.g., an antigen or antibody or antibody derivative) that it encodes. Mutations may be introduced using any technique known in the art. In one aspect, one or more particular amino acid residues are changed using, for example, a site-directed mutagenesis protocol.
- one or more randomly selected residues are changed using, for example, a random mutagenesis protocol.
- a mutant polypeptide may be expressed and screened for a desired property. Mutations may be introduced into a nucleic acid without significantly altering the biological activity of a polypeptide that it encodes. For example, one may make nucleotide substitutions leading to amino acid substitutions at non-essential amino acid residues.
- one or more mutations may be introduced into a nucleic acid that selectively changes the biological activity of a polypeptide that it encodes. For example, the mutation may quantitatively or qualitatively change the biological activity. Examples of quantitative changes include increasing, reducing or eliminating the activity.
- sequence similarity indicates the percentage of amino acids that either are identical or that represent conservative amino acid substitutions.
- sequence identity indicates the percentage of amino acids that are identical between the sequences.
- sequence identity between two nucleic acid sequences indicates the percentage of nucleotides that are identical between the sequences.
- the terms “% identical,” “% identity,” or similar terms are intended to refer, in particular, to the percentage of nucleotides or amino acids which are identical in an optimal alignment between the sequences to be compared. Said percentage is purely statistical, and the differences between the two sequences may be but are not necessarily randomly distributed over the entire length of the sequences to be compared.
- Comparisons of two sequences are usually carried out by comparing the sequences, after optimal alignment, with respect to a segment or “window of comparison,” in order to identify local regions of corresponding sequences.
- the optimal alignment for a comparison may be carried out manually or with the aid of the local homology algorithm by Smith and Waterman, 1981, Ads App. Math.2, 482, with the aid of the local homology algorithm by Neddleman and Wunsch, 1970, J. Mol. Biol. 48, 443, with the aid of the similarity search algorithm by Pearson and Lipman, 1988, Proc. Natl Acad. Sci.
- percent identity of two sequences is determined using the BLASTN or BLASTP algorithm, as available on the United States National Center for Biotechnology Information (NCBI) website. Percentage identity is obtained by determining the number of identical positions at which the sequences to be compared correspond, dividing this number by the number of positions compared (e.g., the number of positions in the reference sequence) and multiplying this result by 100.
- the degree of similarity or identity is given for a region that is at least, at most, exactly, or between any two of about 50%, about 60%, about 70%, about 80%, about 90%, or about 100% of the entire length of the reference sequence.
- the degree of identity is given for at least, at most, exactly, or between any two of about 100, about 120, about 140, about 160, about 180, or about 200 nucleotides, in some aspects, continuous nucleotides.
- the degree of similarity or identity is given for the entire length of the reference sequence.
- Homologous amino acid sequences may exhibit at least, at most, exactly, or between any two of 40%, 50%, 60%, 70%, 80%, 90%, 95%, 98%, or 99% identity of the amino acid residues. In one aspect, homologous amino acid sequences exhibit at least 95% identity of the amino acid residues. In one aspect, homologous amino acid sequences exhibit at least 98% identity of the amino acid residues. In one aspect, homologous amino acid sequences exhibit at least 99% identity of the amino acid residues.
- a fragment or variant of an amino acid sequence may be a “functional fragment” or “functional variant.”
- the term “functional fragment” or “functional variant” of an amino acid sequence relates to any fragment or variant exhibiting one or more functional properties identical or similar to those of the amino acid sequence from which it is derived, e.g., it is functionally equivalent.
- one particular function is one or more immunogenic activities displayed by the amino acid sequence from which the fragment or variant is derived.
- the modifications in the amino acid sequence of the parent molecule or sequence do not significantly affect or alter the characteristics of the molecule or sequence.
- mutant of a wild-type RSV F protein, “mutant” of a RSV F protein, “RSV F protein mutant,” or “modified RSV F protein” refers to a polypeptide that displays introduced mutations relative to a wild-type F protein and is immunogenic against the wild-type F protein.
- the amino acid sequence which is derived from a particular amino acid sequence has an amino acid sequence that is identical, essentially identical, or homologous to that particular sequence or a fragment thereof.
- Amino acid sequences derived from a particular amino acid sequence may be variants of that particular sequence or a fragment thereof.
- the antigens suitable for use herein may be altered such that they vary in sequence from the naturally occurring or native sequences from which they were derived, while retaining the desirable activity of the native sequences.
- a vector refers to a nucleic acid molecule, such as an artificial nucleic acid molecule.
- a vector may be used to incorporate a nucleic acid sequence, such as a nucleic acid sequence comprising an open reading frame.
- Vectors include, but are not limited to, storage vectors, expression vectors, cloning vectors, transfer vectors.
- a vector may be an RNA vector or a DNA vector.
- the vector is a DNA molecule.
- the vector is a plasmid vector.
- the vector is a viral vector.
- an expression vector will contain a desired coding sequence and appropriate other sequences necessary for the expression of the operably linked coding sequence in a particular host organism (e.g., bacteria, yeast, plant, insect, or mammal) or in in vitro expression systems.
- Cloning vectors are generally used to engineer and amplify a certain desired fragment (typically a DNA fragment), and may lack functional sequences needed for expression of the desired fragment(s).
- compositions refers to an active agent, formulated together with one or more pharmaceutically acceptable carriers.
- Pharmaceutical compositions may be immunogenic compositions.
- active agent is present in unit dose amount appropriate for administration in a therapeutic regimen that shows a statistically significant probability of achieving a predetermined therapeutic effect when administered to a relevant population.
- pharmaceutical compositions may be specially formulated for parenteral administration, for example, by subcutaneous, intramuscular, intravenous or epidural injection as, for example, a sterile solution or suspension, or sustained-release formulation.
- vaccination refers to the administration of an immunogenic composition intended to generate an immune response, for example to a disease-associated (e.g., disease-causing) agent (e.g., a virus).
- a disease-associated agent e.g., a virus
- vaccination may be administered before, during, and/or after exposure to a disease-associated agent, and in certain aspects, before, during, and/or shortly after exposure to the agent.
- vaccination includes multiple administrations, appropriately spaced in time, of a vaccine composition.
- vaccination generates an immune response to an infectious agent.
- vaccination generates an immune response to a tumor; in some such aspects, vaccination is “personalized” in that it is partly or wholly directed to epitope(s) (e.g., which may be or include one or more neoepitopes) determined to be present in a particular individual’s tumors.
- An immune response refers to a humoral response, a cellular response, or both a humoral and cellular response in an organism.
- An immune response may be measured by assays that include, but are not limited to, assays measuring the presence or amount of antibodies that specifically recognize a protein or cell surface protein, assays measuring T-cell activation or proliferation, and/or assays that measure modulation in terms of activity or expression of one or more cytokines.
- the term “combination therapy” refers to those situations in which a subject is simultaneously exposed to two or more therapeutic regimens (e.g., two or more therapeutic agents).
- the two or more regimens may be administered simultaneously; in some aspects, such regimens may be administered sequentially (e.g., all “doses” of a first regimen are administered prior to administration of any doses of a second regimen); in some aspects, such agents are administered in overlapping dosing regimens.
- “administration” of combination therapy may involve administration of one or more agent(s) or modality(ies) to a subject receiving the other agent(s) or modality(ies) in the combination.
- combination therapy does not require that individual agents be administered together in a single composition (or even necessarily at the same time), although in some aspects, two or more agents, or active moieties thereof, may be administered together in a combination composition, or even in a combination compound (e.g., as part of a single chemical complex or covalent entity).
- dosing regimen may be used to refer to a set of unit doses (typically more than one) that are administered individually to a subject, typically separated by periods of time.
- a given therapeutic agent has a recommended dosing regimen, which may involve one or more doses.
- a dosing regimen comprises a plurality of doses each of which is separated in time from other doses.
- a dosing regimen comprises a plurality of doses and at least two different time periods separating individual doses. In some aspects, all doses within a dosing regimen are of the same unit dose amount. In some aspects, different doses within a dosing regimen are of different amounts. In some aspects, a dosing regimen comprises a first dose in a first dose amount, followed by one or more additional doses in a second dose amount different from the first dose amount. In some aspects, a dosing regimen comprises a first dose in a first dose amount, followed by one or more additional doses in a second dose amount same as the first dose amount.
- a dosing regimen is correlated with a desired or beneficial outcome when administered across a relevant population (e.g., is a therapeutic dosing regimen).
- RNA molecules e.g., RNA polynucleotides
- RSV respiratory syncytial virus
- the present disclosure further provides for an immunogenic composition comprising at least one RNA molecule encoding an RSV polypeptide complexed with, encapsulated in, or formulated with one or more lipids, and forming lipid nanoparticles (LNPs).
- the RSV polypeptide to be included in the immunogenic composition disclosed herein can be any RSV F protein in the prefusion conformation.
- prefusion conformation refers to a structural conformation adopted by an RSV F protein or mutant thereof that can be specifically bound by (i) antibody D25 or AM22 when the RSV F protein or mutant is in the form of a monomer or trimer, or (ii) by antibody AM14 when the RSV F protein mutant is in the form of a trimer.
- the prefusion trimer conformation is a subset of prefusion conformations.
- an RSV F protein or polypeptide or mutant thereof in prefusion conformation may be denoted as “RSV preF”.
- postfusion conformation refers to a structural conformation adopted by the RSV F protein that is not specifically bound by D25, AM22, or AM14.
- Native F protein adopts the postfusion conformation subsequent to the fusion of the virus envelope with the host cellular membrane.
- RSV F protein may also assume the postfusion conformation outside the context of a fusion event, for example, under stress conditions such as heat and low osmolality, when extracted from a membrane, when expressed as an ectodomain, or upon storage.
- the term “AM14” refers to an antibody described in WO 2008/147196 A2, which is hereby incorporated by reference in its entirety.
- the term “AM22” refers to an antibody described in WO 2011/043643 A1, which is hereby incorporated by reference in its entirety.
- the term “D25” refers to an antibody described in WO 2008/147196 A2, which is hereby incorporated herein by reference in its entirety.
- the RSV F protein is an RSV F protein of subtype A.
- the RSV F protein is an RSV F protein of subtype B.
- the terms “subtype” and “subgroup” are used interchangeably.
- strain refers to a specific isolate within each subtype or subgroup.
- the RSV F protein is a mutant of wild type RSV F protein.
- the RSV F protein is a mutant of wild type RSV F protein of subtype A.
- the RSV F protein is a mutant of wild type RSV F protein of subtype B.
- the mutants display introduced mutations in the amino acid sequence relative to the amino acid sequence of the corresponding wild-type RSV F protein and are immunogenic against the wild-type RSV F protein in the prefusion conformation or against a virus comprising the wild-type F protein.
- the amino acid mutations in the mutants include amino acid substitutions, deletions, or additions relative to a wild-type RSV F protein.
- the RSV F protein is an RSV protein mutant as described in WO2017/109629, which is hereby incorporated by reference in its entirety.
- the RSV F protein is a mutant of a wild-type RSV F protein, wherein the introduced amino acid mutations are mutation of a pair of amino acid residues in a wild-type RSV F protein to a pair of cysteines (”engineered disulfide mutation”).
- the introduced pair of cysteine residues allows for formation of a disulfide bond between the cysteine residues that stabilize the protein’s conformation or oligomeric state, such as the prefusion conformation.
- the RSV F protein mutants comprise amino acid mutations that are one or more cavity filling mutations.
- amino acids that may be replaced with the goal of cavity filling include small aliphatic (e.g. Gly, Ala, and Val) or small polar amino acids (e.g. Ser and Thr) and amino acids that are buried in the prefusion conformation, but exposed to solvent in the postfusion conformation.
- the RSV F protein mutant comprises a cavity filling mutation selected from the group consisting of: (1) substitution of S at positions 55, 62, 155, 190, or 290 with I, Y, L, H, or M; (2) substitution of T at position 54, 58, 189, 219, or 397 with I, Y, L, H, or M; (3) substitution of G at position 151 with A or H; (4) substitution of A at position 147 or 298 with I, L, H, or M; (5) substitution of V at position 164, 187, 192, 207, 220, 296, 300, or 495 with I, Y, H; and (6) substitution of R at position 106 with W.
- the RSV F protein mutant comprises at least one cavity filling mutation selected from the group consisting of: T54H, S190I, and V296I.
- the RSV F protein mutants comprise electrostatic mutations, which decrease ionic repulsion or increase ionic attraction between resides in a protein that are proximate to each other in the folded structure.
- the RSV F protein mutant includes an electrostatic substitution that reduces repulsive ionic interactions or increases attractive ionic interactions with acidic residues of Glu487 and Asp489 from another protomer of RSV F trimer.
- the RSV F protein mutant comprises an electrostatic mutation selected from the group consisting of: (1) substitution of E at position 82, 92, or 487 by D, F, Q, T, S, L, or H; (2) substitution of K at position 315, 394, or 399 by F, M, R, S, L, I, Q, or T; (3) substitution of D at position 392, 486, or 489 by H, S, N, T, or P; and (4) substitution of R at position 106 or 339 by F, Q, N, or W.
- the RSV F protein mutants comprise a combination of two or more different types of mutations selected from engineered disulfide mutations, cavity filling mutations, and electrostatic mutations.
- the RSV F protein mutants comprise a combination of mutations relative to the corresponding wild-type RSV F protein, wherein the combination of mutations is selected from the group consisting of: (1) combination of A103C, I148C, S190I, and D486S; (2) combination of T54H S55C L188C D486S; (3) combination of T54H, A103C, I148C, S190I, V296I, and D486S; (4) combination of T54H, S55C, L142C, L188C, V296I, and N371C; (5) combination of S55C, L188C, and D486S; (6) combination of T54H, S55C, L188C, and S190I; (7) combination of S55C, L188C, S190I, and D486S; (8) combination of T54H, S55C, L188C, S190I, and D486S; (9) combination of S155C, S190I, S290C, and D486S;
- the RSV F protein is of subtype A and comprises the mutations S155C, S190F, S290C, and V207L. In some embodiments, the RSV F protein is of subtype B and comprises the mutations S155C, S190F, S290C, and V207L. In some embodiments, the RSV F protein is of subtype A and comprises the mutations S155C, S190F, and S290C. In some embodiments, the RSV F protein is of subtype B and comprises the mutations S155C, S190F, and S290C. In some embodiments, the RSV F protein is of subtype A and comprises the mutations A103C, I148C, S190I, and D486S.
- the RSV F protein is of subtype B and comprises the mutations A103C, I148C, S190I, and D486S. In some embodiments, the RSV F protein is of subtype A and comprises the mutations T54H, A103C, I148C, S190I, and D486S. In some embodiments, the RSV F protein is of subtype B and comprises the mutations T54H, A103C, I148C, S190I, and D486S. In some embodiments, the RSV F protein is of subtype A and comprises the mutations T54H, S55C, L188C, and D486S.
- the RSV F protein is of subtype B and comprises the mutations T54H, S55C, L188C, and D486S.
- a person of ordinary skill in the art can easily compare amino acid positions between different native RSV F sequences to identify corresponding RSV F amino acid positions between different RSV strains and subtypes. For example, across nearly all identified native RSV F0 precursor proteins, the furin cleavage sites fall in the same amino acid positions.
- the conservation of native RSV F protein sequences across strains and subtypes allows use of a reference RSV F sequence for comparison of amino acids at particular positions in the RSV F protein.
- the RSV F protein amino acid positions are given with reference to the amino acid sequence of the full length native F precursor polypeptide of the RSV A2 strain; corresponding to GenInfo Identifier GI 138251 and Swiss Prot identifier P03420 (SEQ ID NO: 1).
- the RSV F protein is in the mature form of the RSV F protein, which comprises two separate polypeptide chains, namely the F1 polypeptide and F2 polypeptide.
- the F2 polypeptide is linked to the F1 polypeptide by one or two disulfide bonds to form a F2/F1 heterodimer.
- the RSV F mutants are in the form a single chain protein, wherein the F2 polypeptide is linked to the F1 polypeptide by a peptide bond or peptide linker.
- Any suitable peptide linkers for joining two polypeptide chains together may be used. Examples of such linkers include G, GG, GGG, GS, and SAIG linker sequences.
- the linker may also be the full length pep27 sequence or a fragment thereof, which full length pep27 sequence corresponds to amino acids at positions 110-136 of SEQ ID NO:1.
- the F1 polypeptide chain of the mutant may be of the same length as the full length F1 polypeptide of the corresponding wild-type RSV F protein; however, it may also have deletions, such as deletions of 1 up to 60 amino acid residues from the C-terminus of the full-length F1 polypeptide.
- a full-length F1 polypeptide of the RSV F mutants corresponds to amino acid positions 137-574 of the native RSV F0 precursor (SEQ ID NO: 1), and includes (from N- to C- terminus) an extracellular region (residues 137-524), a transmembrane domain (“TM”) (residues 525-550), and a cytoplasmic domain (“CT”) (residues 551-574).
- amino acid residues 514 onwards in a native F1 polypeptide sequence are optional sequences in a F1 polypeptide of the RSV F protein to be included in the immunogenic composition provided herein, and therefore may be absent from the F1 polypeptide of the mutant.
- the F1 polypeptide of the RSV F mutants lacks the entire cytoplasmic domain.
- the F1 polypeptide lacks the cytoplasmic domain and a portion of or all entire transmembrane domain.
- the mutant comprises a F1 polypeptide wherein the amino acid residues from position 510, 511, 512, 513, 514, 515, 520, 525, or 530 through 574 are absent.
- amino acids 514 through 574 can be absent.
- amino acid residues 514 through 574 are absent from the F1 polypeptide of the mutant.
- the F1 polypeptide of the RSV F mutants comprises or consists of amino acid residues 137-513 of a native F0 polypeptide sequence (SEQ ID NO: 1), such as the RSV 847A -Foldon polypeptide (SEQ ID NO: 74) or any of alternative F0 precursor sequence such as those disclosed in SEQ ID NOs: 1, 2, 4, 6, and 81- 270 of WO2017109629, which is hereby incorporated by reference in its entirety.
- the F1 polypeptide and F2 polypeptide of the RSV F protein mutants to which one or more mutations are introduced can be from any wild-type RSV F proteins known in the art or discovered in the future, including, without limitations, the F protein amino acid sequence of RSV subtype A, and subtype B strains, including A2 Ontario and wholesome Aires, or any other subtype.
- the RSV F mutant comprises a F1 and/or a F2 polypeptide from a RSV A virus, for example, a F1 and/or F2 polypeptide from a RSV F0 precursor protein set forth in any one of SEQ ID NOs: 1, 2, 4, 6, and 81-270 of WO2017109629, which sequences are hereby incorporated by reference in their entireties, to which one or more mutations are introduced.
- the RSV F mutant comprises a F1 and/or a F2 polypeptide from a RSV B virus, for example, a F1 and/or F2 polypeptide from a RSV F0 precursor protein set forth in any one of SEQ ID NOs:2, and 211- 263 of WO2017/109629, which sequences are hereby incorporated by reference in their entireties, to which one or more mutations are introduced.
- the RSV F mutant comprises a F1 and/or a F2 polypeptide from a RSV bovine virus, for example, a F1 and/or F2 polypeptide from a RSV F0 precursor protein set forth in any one of SEQ ID NOs:264-270 of WO2017109629, which sequences are hereby incorporated by reference in their entireties, to which one or more mutations are introduced.
- the term “F0 polypeptide” (F0) refers to the precursor polypeptide of the RSV F protein, which is composed of a signal polypeptide sequence, a F1 polypeptide sequence, a pep27 polypeptide sequence, and a F2 polypeptide sequence.
- F1 polypeptide refers to a polypeptide chain of a mature RSV F protein.
- Native F1 includes approximately residues 137-574 of the RSV F0 precursor and is composed of (from N- to C-terminus) an extracellular region (approximately residues 137-524), a transmembrane domain (“TM”) (approximately residues 525-550), and a cytoplasmic tail (“CT”) (approximately residues 551-574).
- F2 polypeptide refers to the polypeptide chain of a mature RSV F protein. Native F2 includes approximately residues 26-109 of the RSV F0 precursor.
- the term encompasses both native F2 polypeptides and F2 polypeptides including modifications (e.g., amino acid substitutions, insertions, or deletions) from the native sequence, for example, modifications designed to stabilize an RSV F protein mutant in a prefusion conformation or to enhance the immunogenicity of an RSV F protein mutant.
- modifications e.g., amino acid substitutions, insertions, or deletions
- the F2 polypeptide is linked to the F1 polypeptide by two disulfide bonds to form a F2-F1 heterodimer.
- foldon or “foldon domain” refers to an amino acid sequence that is capable of forming trimers.
- RNA molecule encodes an RSV F protein mutant as disclosed in WO2009/079796, WO2010/149745, WO2011/008974, WO2014/160463, WO2014/174018, WO2014/202570, WO2015/013551, WO2015/177312, WO2017/005848, WO2017/174564, WO2017/005844 and WO2018/109220.
- the RSV F proteins disclosed in these references are hereby incorporated by reference in their entirety.
- the term “respiratory syncytial virus” or “RSV” is not limited to any particular strain or variant.
- the RNA molecule comprises an open reading frame encoding a RSV antigen.
- the RSV antigen is a RSV polypeptide.
- the RSV polypeptide is a RSV glycoprotein or a fragment or a variant thereof.
- the RNA molecule encodes a RSV F protein.
- the RSV polypeptide is a full-length RSV polypeptide.
- the RSV polypeptide is a truncated RSV polypeptide. In some aspects, the RSV polypeptide is a variant of a RSV polypeptide. In some aspects, the RSV polypeptide is a fragment of a RSV polypeptide. In some aspects, the RSV polypeptide is a full-length RSV F protein. In some aspects, the RSV polypeptide is a truncated RSV F protein. In some aspects, the RSV polypeptide is a variant of a RSV F protein. In some aspects, the RSV polypeptide is a fragment of a RSV F protein. In some aspects, the RSV F protein comprises at least one mutation. In some aspects, the RSV F protein comprises at least two mutations.
- the RSV F protein comprises at least three mutations. In some aspects, the RSV F protein comprises at least four mutations. In some aspects, the RSV F protein comprises 4 mutations. In some aspects, the RSV F protein comprises at least five mutations. In some aspects, the RNA molecule encodes a RSV F protein as set forth in Table 1 (see Example 6). In some aspects, the RNA molecule encodes a RSV F protein comprising an amino acid sequence of any of SEQ ID NO: 1 to 6 and 71 to 74, or fragment or variant thereof.
- RSV F polypeptide may have at least, at most, exactly, or between any two of 70%, 71%, 72%, 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to any of the amino acid sequences of Table 1, for example, any of SEQ ID NO: 1 to 6 and 71 to 74.
- RSV F protein consists of any of the amino acid sequences of Table 1, for example, any of SEQ ID NO: 1 to 6 and 71 to 74.
- the RNA molecule sequence is transcribed from a DNA nucleic acid sequence (DNA polynucleotide) of Table 2 (see Example 6).
- the RNA molecule comprises an ORF transcribed from a nucleic acid sequence of any of SEQ ID NO: 7 to 10 and 59 to 62, or fragment or variant thereof.
- the RNA molecule comprises an ORF transcribed from a nucleic acid sequence that may have at least, at most, exactly, or between any two of 70%, 71%, 72%, 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to any of the nucleic acid sequences of Table 2, for example, any of SEQ ID NO: 7 to 10 and 59 to 62.
- the RNA molecule comprises an ORF transcribed from a nucleic acid sequence that consists of any of the nucleic acid sequences of Table 2, for example, any of SEQ ID NO:7 to 10 and 59 to 62.
- the RNA molecule comprises an ORF comprising an RNA nucleic acid sequence (RNA polynucleotide) of Table 3 (see Example 6).
- the RNA molecule comprises an ORF comprising a nucleic acid sequence of any of SEQ ID NO: 11 to 16 and 63 to 70, or fragment or variant thereof.
- the RNA molecule comprises an ORF comprising a nucleic acid sequence that may have at least, at most, exactly, or between any two of 70%, 71%, 72%, 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity to any of the RNA nucleic acid sequences of Table 3, for example, any of SEQ ID NO: 11 to 16 and 63 to 70.
- the RNA molecule comprises an ORF comprising a nucleic acid sequence that consists of any of the RNA nucleic acid sequences of Table 3, for example, any of SEQ ID NO: 11 to 16 and 63 to 70.
- the RNA molecule comprises stabilized RNA.
- the RNA molecule comprises a nucleic acid sequence having at least one uridine replaced by N1- methylpseudouridine.
- the RNA molecule comprises a sequence having all uridines replaced by N1-methylpseudouridine (designated as “ ⁇ ”).
- the RNA molecule comprises an ORF comprising a nucleic acid sequence of any of SEQ ID NO: 11 to 16 and 63 to 70, wherein all uridines have been replaced by N1-methylpseudouridine (designated as “ ⁇ ”).
- the RNA molecule comprises an open reading frame encoding a RSV F protein amino acid sequence that may be at least, at most, exactly, or between any two of 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any of the RSV F protein sequences of SEQ ID NO: 1 to 6 and 71 to 74 (Table 1) or other RSV prefusion F proteins described herein.
- the RNA molecule comprises an open reading frame encoding a RSV F protein amino acid sequence that consists of any of the RSV F protein sequences of SEQ ID NO: 1 to 6 and 71 to 74 (Table 1) or other RSV prefusion F protein described herein.
- the RNA molecule comprises an open reading frame transcribed from a DNA nucleic acid sequence that may be at least, at most, exactly, or between any two of 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any of the nucleic acid sequences of SEQ ID NO: 7 to 10 and 59 to 62 (Table 2) or other nucleic acid described herein.
- the RNA molecule comprises an open reading frame transcribed from a DNA nucleic acid sequence that consists of any of the nucleic acid sequences of SEQ ID NO: 7 to 10 and 59 to 62 (Table 2) or other nucleic acid described herein.
- the RNA molecule comprises an open reading frame comprising an RNA nucleic acid sequence that may be at least, at most, exactly, or between any two of 70%, 80%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to any of the nucleic acid sequences of SEQ ID NO: 11 to 16 and 63 to 70 (Table 3) or other nucleic acid described herein.
- the RNA molecule comprises an open reading frame comprising an RNA nucleic acid sequence that consists of any of the nucleic acid sequences of SEQ ID NO: 11 to 16 and 63 to 70 (Table 3) or other nucleic acid described herein.
- the RNA molecule comprises an ORF comprising a nucleic acid sequence of any of SEQ ID NO: 11 to 16 and 63 to 70 (Table 3), wherein all uridines have been replaced by N1-methylpseudouridine (designated as “ ⁇ ”).
- RNA MOLECULE In some aspects, the RNA molecule described herein is a coding RNA molecule.
- Coding RNA includes a functional RNA molecule that may be translated into a peptide or polypeptide.
- the coding RNA molecule includes at least one open reading frame (ORF) coding for at least one peptide or polypeptide.
- An open reading frame comprises a sequence of codons that is translatable into a peptide or protein.
- the coding RNA molecule may include one (monocistronic), two (bicistronic) or more (multicistronic) ORFs, which may be a sequence of codons that is translatable into a polypeptide or protein of interest.
- the coding RNA molecule may be a messenger RNA (mRNA) molecule, viral RNA molecule, or self-amplifying RNA molecule (saRNA, also referred to as a replicon).
- mRNA messenger RNA
- saRNA self-amplifying RNA molecule
- the RNA molecule is an mRNA.
- the RNA molecule of the present disclosure is an mRNA.
- the RNA molecule is modRNA.
- the RNA molecule is a saRNA.
- the saRNA molecule may be a coding RNA molecule.
- the RNA molecule may encode one polypeptide of interest or more, such as an antigen or more than one antigen, e.g., two, three, four, five, six, seven, eight, nine, ten or more polypeptides.
- one RNA molecule may also encode more than one polypeptide of interest, such as an antigen, e.g., a bicistronic, or tricistronic RNA molecule that encodes different or identical antigens.
- the sequence of the RNA molecule may be codon optimized or deoptimized for expression in a desired host, such as a human cell.
- a gene of interest (e.g., an antigen) described herein is encoded by a coding sequence which is codon-optimized and/or the guanosine/cytidine (G/C) content of which is increased compared to wild type coding sequence.
- G/C guanosine/cytidine
- one or more sequence regions of the coding sequence are codon-optimized and/or increased in the G/C content compared to the corresponding sequence regions of the wild type coding sequence.
- codon-optimization and/or increasing the G/C content does not change the sequence of the encoded amino acid sequence.
- coding regions are codon-optimized for optimal expression in a subject to be treated using an RNA polynucleotide described herein. Codon-optimization is based on the finding that the translation efficiency is also determined by a different frequency in the occurrence of tRNA molecules in cells.
- the sequence of RNA may be modified such that codons for which frequently occurring tRNA molecules are available are inserted in place of “rare codons.”
- G/C content of a coding region e.g., of a gene of interest sequence; open reading frame (ORF)
- ORF open reading frame
- the amino acid sequence encoded by the RNA is not modified compared to the amino acid sequence encoded by the wild type RNA. This modification of the RNA sequence is based on the fact that the sequence of any RNA region to be translated is important for efficient translation of that mRNA.
- Sequences having an increased G (guanosine)/C (cytidine) content are more stable than sequences having an increased A (adenosine)/U (uridine) content.
- the most favorable codons for the stability may be determined (so-called alternative codon usage).
- alternative codon usage the amino acid to be encoded by the RNA, there are various possibilities for modification of the RNA sequence, compared to its wild type sequence.
- G/C content of a coding region of an RNA described herein is increased by at least, at most, exactly, or between any two of 10%, 20%, 30%, 40%, 50%, 55%, or even more compared to the G/C content of a coding region of a wild type RNA.
- the coding region of the RSV RNA described herein comprises a G/C content of at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, or about 80%. In some aspects, the coding region of the RSV RNA described herein comprises a G/C content of about 50% to 75%, about 55% to 70%, about 50% to 60%, about 60% to 70%, about 70% to 80%, about 50% to 55%, about 55% to 60%, about 60% to 65%, about 65% to 70%, about 70% to 75%, or about 75% to 80%.
- the coding region of the RSV RNA described herein comprises a G/C content of about 50%, about 51%, about 52%, about 53%, about 54%, about 55%, about 56%, about 57%, about 58%, about 59%, about 60%, about 61%, about 62%, about 63%, about 64%, about 65%, about 66%, about 67%, about 68%, about 69%, about 70%, about 71%, about 72%, about 73%, about 74%, or about 75%.
- the coding region of the RSV RNA described herein comprises a G/C content of about 58%, about 66% or about 62%.
- the RNA molecule includes from about 20 to about 100,000 nucleotides (e.g., from 30 to 50, from 30 to 100, from 30 to 250, from 30 to 500, from 30 to 1,000, from 30 to 1,500, from 30 to 3,000, from 30 to 5,000, from 30 to 7,000, from 30 to 10,000, from 30 to 25,000, from 30 to 50,000, from 30 to 70,000, from 100 to 250, from 100 to 500, from 100 to 1,000, from 100 to 1,500, from 100 to 3,000, from 100 to 5,000, from 100 to 7,000, from 100 to 10,000, from 100 to 25,000, from 100 to 50,000, from 100 to 70,000, from 100 to 100,000, from 500 to 1,000, from 500 to 1,500, from 500 to 2,000, from 500 to 3,000, from 500 to 5,000, from 500 to 7,000, from 500 to 10,000, from 500 to 25,000, from 500 to 50,000, from 500 to 70,000, from 500 to 100,000, from 1,000 to 1,500, from 1,000, from 500 to 2,000, from 500 to 3,000, from 500 to 5,000, from 500
- the RNA molecule has at least, at most, exactly, or between any two of about 20, 40, 60, 80, 100, 120, 140, 160, 180, 200, 220, 240, 260, 280, 300, 320, 340, 360, 380, 400, 420, 440, 460, 480, 500, 520, 540, 560, 580, 600, 620, 640, 660, 680, 700, 720, 740, 760, 780, 800, 820, 840, 860, 880, 900, 920, 940, 960, 980, 1000, 1200, 1400, 1600, 1800, 2000, 2200, 2400, 2600, 2800, 3000, 3200, 3400, 3600, 3800, 4000, 4200, 4400, 4600, 4800, 5000, 5200, 5400, 5600, 5800, 6000, 6200, 6400, 6600, 6800, 7000, 7200, 7400, 7600, 7800,
- the RNA molecule includes at least 100 nucleotides.
- the RNA has a length between 100 and 15,000 nucleotides; between 7,000 and 16,000 nucleotides; between 8,000 and 15,000 nucleotides; between 9,000 and 12,500 nucleotides; between 11,000 and 15,000 nucleotides; between 13,000 and 16,000 nucleotides; between 7,000 and 25,000 nucleotides.
- the RNA molecule has at least, at most, exactly, or between any two of about 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1050, 1100, 1150, 1200, 1250, 1300, 1350, 1400, 1450, 1500, 1550, 1600, 1650, 1700, 1750, 1800, 1850, 1900, 1950, 2000, 2050, 2100, 2150, 2200, 2250, 2300, 2350, 2400, 2450, 2500, 2550, 2600, 2650, 2700, 2750, 2800, 2850, 2900, 2950, 3000, 3050, 3100, 3150, 3200, 3250, 3300, 3350, 3400, 3450, 3500, 3550, 3600, 3650, 3700, 3750, 3800, 3850, 3900, 3950, 4000, 4050, 4100, 4150, 4200,
- RNA molecules of the present disclosure may be prepared by any method know in the art, including chemical synthesis and in vitro methods, such as RNA in vitro transcription.
- the RNA of the present disclosure is prepared using in vitro transcription.
- the RNA molecule of the present disclosure is purified, e.g., such as by filtration that may occur via, e.g., ultrafiltration, diafiltration, or, e.g., tangential flow ultrafiltration/diafiltration.
- the RNA molecule of the present disclosure is lyophilized to be temperature stable.
- an RNA is or comprises messenger RNA (mRNA) that relates to an RNA transcript which encodes a polypeptide.
- mRNA messenger RNA
- an RNA disclosed herein comprises: a 5′ cap comprising a 5′ cap disclosed herein; a 5′ untranslated region comprising a cap proximal sequence (5′ UTR), a sequence encoding a protein (e.g. polypeptide) (e.g., a RSV prefusion F protein); a 3′ untranslated region (3′ UTR); and/or a polyadenylate (poly- A) sequence.
- an RNA disclosed herein comprises the following components in 5′ to 3′ orientation: a 5′ cap comprising a 5′ cap disclosed herein; a 5′ untranslated region comprising a cap proximal sequence (5′ UTR), a sequence encoding a protein (e.g.
- an RNA disclosed herein further comprises a signal peptide.
- signal peptides and amino acid and nucleic acid sequences encoding such peptides can be found in, e.g., WO2017/109629, the disclosure of which is incorporated by reference herein in its entirety.
- an RNA disclosed herein encodes an antigenic fusion protein.
- the encoded antigen or antigens may include two or more proteins (e.g., protein and/or protein fragment) joined together.
- an RNA disclosed herein encodes fusion proteins that comprise an antigen linked to a scaffold moieties.
- the RNA further encodes a linker located between at least one or each domain of the fusion protein.
- Non-limiting examples of such scaffold moieties and linkers can be found in, e.g., WO 2022/067010, the disclosure of which is incorporated by reference herein in its entirety.
- RNA molecules are not chemically modified and comprise the standard ribonucleotides consisting of adenosine, guanosine, cytosine and uridine.
- nucleotides and nucleosides of the present disclosure comprise standard nucleoside residues such as those present in transcribed RNA (e.g., A, G, C, and/or U).
- nucleotides and nucleosides of the present disclosure comprise standard deoxyribonucleosides such as those present in DNA (e.g., dA, dG, dC, and/or dT).
- the RNA molecules may comprise modified nucleobases which may be incorporated into modified nucleosides and nucleotides.
- the RNA molecule may include one or more modified nucleotides.
- the RNA molecule may include one or more modified nucleotides.
- Naturally occurring nucleotide modifications are known in the art.
- the RNA molecule may include a modified nucleotide.
- Non-limiting examples of modified nucleotides that may be included in the RNA molecule include pseudouridine, N1-methylpseudouridine, 5-methyluridine, 3-methyl-uridine, 5- methoxy-uridine, 5-aza-uridine, 6-aza-uridine, 2-thio-5-aza-uridine, 2-thio-uridine, 4-thio-uridine, 4-thio-pseudouridine, 2-thio-pseudouridine, 5-hydroxy-uridine, 5-aminoallyl-uridine, 5-halo- uridine (e.g., 5-iodo-uridine or 5-bromo-uridine), uridine 5-oxyacetic acid, uridine 5-oxyacetic acid methyl ester, 5-carboxymethyl-uridine, 1-carboxymethyl-pseudouridine, 5-carboxy hydroxymethyl-uridine, 5-carboxy hydroxy methyl-uridine methyl ester, 5- methoxycarbonylmethyl-uridine, 5-methoxycarbonyl
- RNA molecules disclosed herein can be excluded from the RNA molecules disclosed herein.
- Modifications that may be present in the RNA molecules further include, but are not limited to, e.g., the following: ms2io6A (2-methylthio-(N6-(cis-hydroxyisopentenyl)adenosine); ms2m6A (2-methylthio-N6-methyladenosine); ms2t6A 2-methylthio-N6-threonylcarbamoyladenosine; g6A (N6-glycinylcarbamoyladenosine); i6A (N6-isopentenyladenosine); m6A (N6-methyladenosine); t6A (N6-threonylcarbamoyladenosine); m′Am (1,2′-O-dimethyladenosine); m1A
- RNA molecules include a combination of at least two (e.g., 2, 3, 4, or more) of the aforementioned modified nucleobases. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing modifications can be excluded from the RNA molecules disclosed herein.
- modified nucleobases in RNA molecules comprise pseudouridine ( ⁇ ), 2-thiouridine (s2U), 4′-thiouridine, 5-methylcytosine, 2-thio-1-methyl-1-deaza-pseudouridine, 2- thio-1-methyl-pseudouridine, 2-thio-5-aza-uridine, 2-thio-dihydropseudouridine, 2-thio- dihydrouridine, 2-thio-pseudouridine, 4-methoxy-2-thio-pseudouridine, 4-methoxy- pseudouridine, 4-thio-1-methyl-pseudouridine, 4-thio-pseudouridine, 5-aza-uridine, dihydropseudouridine, 5-methyluridine, 5-methoxyuridine, 2′-O-methyl uridine, 1-methyl- pseudouridine (m1 ⁇ ), 1-ethyl-pseudouridine (e1 ⁇ ), 5-methoxy-uridine (mo5U),
- the RNA molecule includes a combination of at least two (e.g., 2, 3, 4, or more) of the aforementioned modified nucleobases, including but not limited to chemical modifications. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing modified nucleobases can be excluded from the RNA molecules disclosed herein.
- nucleobases and nucleosides having a modified cytosine include 5-aza- cytidine, 6-aza-cytidine, pseudoisocytidine, 3-methyl-cytidine (m3C), N4-acetyl-cytidine (ac4C), 5-formyl-cytidine (f5C), N4-methyl-cytidine (m4C), 5-methyl-cytidine (m5C), 5-halo-cytidine (e.g., 5-iodo-cytidine), 5-hydroxymethyl-cytidine (hm5C), 1-methyl-pseudoisocytidine, pyrrolo-cytidine, pyrrolo-pseudoisocytidine, 2-thio-cytidine (s2C), 2-thio-5-methyl-cytidine, 4-thio- pseudoisocytidine, 4-thio-1-methyl-pseudoisocytidine, 4-thi
- a modified nucleobase is a modified uridine.
- exemplary nucleobases and nucleosides having a modified uracil include pseudouridine ( ⁇ ), pyridin-4-one ribonucleoside, 5-aza-uridine, 6-aza-uridine, 2-thio-5-aza-uridine, 2-thio-uridine (s2U), 4-thio-uridine (s4U), 4- thio-pseudouridine, 2-thio-pseudouridine, 5-hydroxy-uridine (ho5U), 5-aminoallyl-uridine, 5-halo- uridine (e.g., 5-iodo-uridine or 5-bromo-uridine), 5-cyanouridine, 3-methyl-uridine (m3U), 5- methoxy-uridine (mo5U), uridine 5-oxyacetic acid (cmo
- modified nucleotides include any one of N1- methylpseudouridine and/or pseudouridine.
- the RNA molecule comprises nucleotides that are N1- methylpseudouridine modified.
- the RNA molecule comprises nucleotides that are pseudouridine modified.
- an RNA comprises a modified nucleoside in place of at least one uridine.
- an RNA comprises a modified nucleoside in place of each uridine.
- the RNA molecule comprises a sequence having at least one uridine replaced by N1- methylpseudouridine. In some aspects, the RNA molecule comprises a sequence having all uridines replaced by N1-methylpseudouridine. N1-methylpseudouridine is designated in sequences as “ ⁇ ”.
- uracil describes one of the nucleobases that may occur in the nucleic acid of RNA.
- uridine describes one of the nucleosides that may occur in RNA.
- RNA molecule comprises a nucleic acid sequence having at least one uridine replaced by N1-methylpseudouridine and/or pseudouridine.
- the RNA molecule comprises a nucleic acid sequence having at least, at most, exactly, or between (inclusive or exclusive) any two of 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24%, 25%, 26%, 27%, 28%, 29%, 30%, 31%, 32%, 33%, 34%, 35%, 36%, 37%, 38%, 39%, 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%, 59%, 60%, 61%, 62%, 63%, 64%, 65%, 66%, 67%, 68%, 69%, 70%, 71%, 72%, 73%, 74%, 75%, 76%
- the RNA molecule comprises a nucleic acid sequence having all uridines replaced by N1- methylpseudouridine and/or pseudouridine.
- a modified nucleobase is a modified adenine.
- nucleobases and nucleosides having a modified adenine include 2-amino-purine, 2, 6-diaminopurine, 2-amino- 6-halo-purine (e.g., 2-amino-6-chloro-purine), 6-halo-purine (e.g., 6-chloro-purine), 2-amino-6- methyl-purine, 8-azido-adenosine, 7-deaza-adenine, 7-deaza-8-aza-adenine, 7-deaza-2-amino- purine, 7-deaza-8-aza-2-amino-purine, 7-deaza-2,6-diaminopurine, 7-deaza-8-aza-2,6- diaminopurine, 1-methyl-adenosine (m1A), 2-methyl-adenine (m2A), N6-methyl -adenosine (m6A), 2-methylthio-N6-methyl-adenosine (m
- a modified nucleobase is a modified guanine.
- Exemplary nucleobases and nucleosides having a modified guanine include inosine (I), 1-methyl-inosine (m1I), wyosine (imG), methylwyosine (mimG), 4-demethyl-wyosine (imG-14), isowyosine (imG2), wybutosine (yW), peroxywybutosine (o2yW), hydroxywybutosine (OhyW), undermodified hydroxywybutosine (OhyW*), 7-deaza-guanosine, queuosine (Q), epoxyqueuosine (oQ), galactosyl-queuosine (galQ), mannosyl-queuosine (manQ), 7-
- RNA molecules are uniformly modified (e.g., fully modified, modified throughout the entire sequence) for a particular modification.
- the RNA molecules may be partially or fully (e.g., uniformly) modified along the entire length of the molecule.
- one or more or all or a given type of nucleotide e.g., purine and/or pyrimidine, or any one or more or all of A, G, U, C
- nucleotides X in a polynucleotide of the present disclosure are modified nucleotides, wherein X may be any one of nucleotides A, G, U, C, and/or any one of the combinations A+G, A+U, A+C, G+U, G+C, U+C, A+G+U, A+G+C, G+U+C and/or A+G+C.
- a polynucleotide can be uniformly modified with pseudouridine, meaning that all uridine residues in the RNA sequence are replaced with pseudouridine.
- a polynucleotide can be uniformly modified for any type of nucleoside residue present in the sequence by replacement with a modified residue such as those set forth above.
- the modified nucleotide can be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4, or more unique structures).
- RNA molecules may contain from or from about 1% to 100% modified nucleotides (either in relation to overall nucleotide content, or in relation to one or more types of nucleotide, e.g., any one or more of A, G, U and/or C) (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85,
- the RNA molecule may include phosphoramidate, phosphorothioate, and/or methylphosphonate linkages.
- the RNA molecules may include one or more structural and/or chemical modifications and/or alterations which impart useful properties to the polynucleotide including, in some aspects, reduced degradation in the cell or organism and/or lack of a substantial induction of the innate immune response of a cell into which the RNA molecule is introduced.
- a “structural” feature or modification is one in which two or more linked nucleotides are inserted, deleted, duplicated, inverted and/or randomized in an RNA molecule without significant chemical modification to the nucleotides themselves. Because chemical bonds will necessarily be broken and reformed to affect a structural modification, structural modifications are of a chemical nature and hence are chemical modifications. However, structural modifications will result in a different sequence of nucleotides. For example, the polynucleotide “ATCG” may be chemically modified to “AT-5meC-G”. The same polynucleotide may be structurally modified from “ATCG” to “ATCCCG”.
- a modified RNA molecule introduced to a cell or organism, exhibits reduced degradation in the cell or organism, respectively, relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides.
- a modified RNA molecule, introduced into a cell or organism may exhibit reduced immunogenicity in the cell or organism, respectively (e.g., a reduced innate response) relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides.
- the RNA molecule may include one or more modified nucleotides in addition to any 5′ cap structure.
- the RNA molecule does not include modified nucleotides, e.g., does not include modified nucleobases, and all of the nucleotides in the RNA molecule are conventional standard ribonucleotides A, U, G and C, with the exception of an optional 5′ cap that may include, for example, 7-methylguanosine, which is further described below.
- the RNA may include a 5′ cap comprising a 7’-methylguanosine, and the first 1, 2, or 35′ ribonucleotides may be methylated at the 2’ position of the ribose.
- the RNA molecule described herein includes a 5′ cap which generally “caps” the 5′ end of the RNA and stabilizes the RNA molecule.
- the 5′ cap moiety is a natural 5′ cap.
- a “natural 5′ cap” is defined as a cap that includes 7-methylguanosine connected to the 5′ end of an mRNA molecule through a 5′ to 5′ triphosphate linkage.
- a guanosine nucleoside included in a 5′ cap may be modified, for example, by methylation at one or more positions (e.g., at the 7-position) on a base (guanine), and/or by methylation at one or more positions of a ribose.
- a guanosine nucleoside included in a 5′ cap comprises a 3′O methylation at a ribose (3′OMeG). In some aspects, a guanosine nucleoside included in a 5′ cap comprises methylation at the 7- position of guanine (m7G). In some aspects, a guanosine nucleoside included in a 5′ cap comprises methylation at the 7-position of guanine and a 3′O methylation at a ribose (m7(3′OMeG)).
- the 5′ cap may be incorporated during RNA synthesis (e.g., co-transcriptional capping) or may be enzymatically engineered after RNA transcription (e.g., post-transcriptional capping).
- co-transcriptional capping with a cap disclosed herein improves the capping efficiency of an RNA compared to co-transcriptional capping with an appropriate reference comparator.
- improving capping efficiency may increase a translation efficiency and/or translation rate of an RNA, and/or increase expression of an encoded polypeptide.
- capping is performed after purification, e.g., tangential flow filtration, of the RNA molecule.
- an RNA described herein comprises a 5′ cap or a 5′ cap analog, e.g., a Cap 0, a Cap 1 or a Cap 2.
- a provided RNA does not have uncapped 5′- triphosphates.
- the 5′ end of the RNA is capped with a modified ribonucleotide.
- the 5′ cap moiety is a 5′ cap analog.
- an RNA may be capped with a 5′ cap analog.
- Cap structures include, but are not limited to, 7 mG(5′)ppp(5′)N 1 pN 2 p (Cap 0), 7 mG(5′)ppp(5′)N 1 m pNp (Cap 1), and 7 mG(5′)ppp(5′)N 1 m pN 2 m p (Cap 2).
- 1, 2, 3, 4, 5, or more of the foregoing cap structures can be excluded from the RNA molecules disclosed herein.
- an RNA described herein comprises a Cap 0.
- Cap 0 is a N7-methyl guanosine
- a Cap 0 structure comprises a guanosine nucleoside methylated at the 7-position of guanine (m7G).
- a Cap 0 structure is connected to an RNA via a 5′ to 5′-triphosphate linkage and is also referred to herein as m7G, m7Gppp, and/or m7G(5′)ppp(5′).
- ⁇ A 5′ cap may be methylated with the structure 7 mG(5′)ppp(5′)N1pN2p (Cap 0) or a derivative thereof, wherein N is the terminal 5′ nucleotide of the nucleic acid carrying the 5′ cap, typically the 5′-end of an mRNA.
- An exemplary enzymatic reaction for capping may include use of Vaccinia Virus Capping Enzyme (VCE) that includes mRNA triphosphatase, guanylyl- transferase and guanine-7-methytransferase, which catalyzes the construction of N7- monomethylated Cap 0 structures.
- VCE Vaccinia Virus Capping Enzyme
- Cap 0 structures play an important role in maintaining the stability and translational efficacy of the RNA molecule.
- the Cap 0 structure is essential for efficient translation of the mRNA that carries the cap.
- an RNA described herein comprises a Cap 1, e.g., as described herein.
- RNA molecules may be further modified on the 2′O position by a 2′-O- methyltransferase, which results in the generation of a Cap 1 structure (m7Gppp [m2′- ⁇ ] N), which may further increase translation efficacy.
- a Cap 1 structure comprises a guanosine nucleoside methylated at the 7-position of guanine (m7G) and a 2′O methylated first nucleotide in an RNA (2′OMeN1).
- a Cap 1 structure is connected to an RNA via a 5′- to 5′-triphosphate linkage and is also referred to herein as m7GpppN m , wherein N m denotes any nucleotide with a 2′O methylation, 7 mG(5′)ppp(5′)N1 m pNp, m7Gppp(2′OMeN1), and/or m7G(5′)ppp(5′)(2′OMeN 1 ).
- N 1 is chosen from A, C, G, or U.
- N 1 is A.
- N 1 is C.
- N 1 is G.
- N 1 is U.
- a m7G(5′)ppp(5′)(2′OMeN1) Cap 1 structure comprises a second nucleotide, N2, which is a cap proximal nucleotide at position 2 and is chosen from A, G, C, or U (m7G(5′)ppp(5′)(2′OMeN1)N2).
- N2 is A.
- N2 is C.
- N2 is G.
- N2 is U.
- a Cap 1 structure comprises a guanosine nucleoside methylated at the 7-position of guanine (m7G) and one or more additional modifications, e.g., methylation on a ribose, and a 2′O methylated first nucleotide in an RNA.
- a Cap 1 structure comprises a guanosine nucleoside methylated at the 7-position of guanine, a 3′O methylation at a ribose (m7(3′OMeG)), and a 2′O methylated first nucleotide in an RNA (2′OMeN1).
- a Cap 1 structure is connected to an RNA via a 5′- to 5′-triphosphate linkage and is also referred to herein as m7(3′OMeG)ppp(2′OMeN1) and/or m7(3′OMeG)(5′)ppp(5′)(2′OMeN1).
- N 1 is chosen from A, C, G, or U. In some aspects, N 1 is A. In some aspects, N 1 is C. In some aspects, N 1 is G. In some aspects, N 1 is U.
- a m7(3′OMeG)(5′)ppp(5′)(2′OMeN 1 ) Cap 1 structure comprises a second nucleotide, N 2 , which is a cap proximal nucleotide at position 2 and is chosen from A, G, C, or U (m7(3′OMeG)(5′)ppp(5′)(2′OmeN1)N2).
- N2 is A.
- N2 is C.
- N2 is G.
- N2 is U.
- 1, 2, 3, 4, 5, or more of the foregoing Cap 1 structures can be excluded from the RNA molecules disclosed herein.
- a second nucleotide in a Cap 1 structure may comprise one or more modifications, e.g., methylation.
- an RNA described herein comprises a Cap 2.
- a Cap 1 structure comprising a second nucleotide comprising a 2′O methylation is a Cap 2 structure.
- the RNA molecule may be enzymatically capped at the 5′ end using Vaccinia guanylyltransferase, guanosine triphosphate, and S-adenosyl-L-methionine to yield Cap 0 structure.
- An inverted 7-methylguanosine cap is added via a 5′ to 5′ triphosphate bridge.
- Cap 1 where, in addition to the Cap 0 structure, the 2′OH group is methylated on the penultimate nucleotide.
- S-adenosyl-L-methionine (SAM) is a cofactor utilized as a methyl transfer reagent.
- Non-limiting examples of 5′ cap structures are those which, among other things, have enhanced binding of cap-binding polypeptides, increased half-life, reduced susceptibility to 5′ endonucleases and/or reduced 5′ decapping, as compared to synthetic 5′ cap structures known in the art (or to a wild type, natural or physiological 5′ cap structure).
- recombinant Vaccinia Virus Capping Enzyme and recombinant 2′ O- methyltransferase enzyme may create a canonical 5′-5′-triphosphate linkage between the 5′- terminal nucleotide of an mRNA and a guanine cap nucleotide wherein the cap guanine includes an N7 methylation and the 5′-terminal nucleotide of the mRNA includes a 2′-O-methyl.
- Cap 1 structure Such a structure is termed the Cap 1 structure.
- This cap results in a higher translational-competency and cellular stability and a reduced activation of cellular pro-inflammatory cytokines, as compared, e.g., to other 5′ cap analog structures known in the art.
- a cap species may include one or more modified nucleosides and/or linker moieties.
- a cap may include a guanine nucleotide and a guanine (G) nucleotide methylated at the 7 position joined by a triphosphate linkage at their 5′ positions, e.g., m7G(5′)ppp(5′)G, commonly written as m7GpppG.
- G guanine nucleotide methylated at the 7 position joined by a triphosphate linkage at their 5′ positions, e.g., m7G(5′)ppp(5′)G, commonly written as m7GpppG.
- a cap species may also be an anti-reverse cap analog.
- a non- limiting list of possible cap species includes m7GpppG, m7Gpppm7G, m73′dGpppG, m27,O3′GpppG, m27,O3′GppppG, m27,O2′GppppG, m7Gpppm7G, m73′dGpppG, m27,O3′GpppG, m27,O3′GppppG, and m27,O2′GppppG.
- 1, 2, 3, 4, 5, or more of the foregoing cap species can be excluded from the RNA molecules disclosed herein.
- the 5′ terminal cap includes a cap analog
- a 5′ terminal cap may include a guanine analog.
- Exemplary guanine analogs include, but are not limited to, inosine, N1-methyl-guanosine, 2′-fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino- guanosine, LNA-guanosine, and 2-azido-guanosine.
- 1, 2, 3, 4, 5, or more of the foregoing guanine analogs can be excluded from the cap structures disclosed herein.
- the capping region may include a single cap or a series of nucleotides forming the cap.
- the capping region may be from 1 to 10, e.g., 2-9, 3-8, 4-7, 1-5, 5- 10, or at least 2, or 10 or fewer nucleotides in length. In this aspect, the capping region is at least, at most, exactly, or between (inclusive or exclusive) any two of 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides in length. In some aspects, the cap is absent. In some aspects, the first and second operational regions may range from 3 to 40, e.g., 5-30, 10-20, 15, or at least 4, or 30 or fewer nucleotides in length and may comprise, in addition to a Start and/or Stop codon, one or more signal and/or restriction sequences.
- the first and second operational regions are at least, at most, exactly, or between (inclusive or exclusive) any two of 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, or 40 nucleotides in length and may comprise, in addition to a Start and/or Stop codon, one or more signal and/or restriction sequences.
- 5′ cap structures include, but are not limited to, glyceryl, inverted deoxy abasic residue (moiety), 4′, 5′ methylene nucleotide, 1-(beta-D-erythrofuranosyl) nucleotide, 4′-thio nucleotide, carbocyclic nucleotide, 1,5-anhydrohexitol nucleotide, L- nucleotides, alpha-nucleotide, modified base nucleotide, threo-pentofuranosyl nucleotide, acyclic 3′,4′-seco nucleotide, acyclic 3,4-dihydroxybutyl nucleotide, acyclic 3,5 dihydroxypentyl nucleotide, 3′-3′-inverted nucleotide moiety, 3′-3′-inverted abasic moiety, 3′-2′-inverted nucleotide moiety, 3′-2′-inverted
- RNA molecules disclosed herein 1, 2, 3, 4, 5, or more of the foregoing 5′ cap structures can be excluded from the RNA molecules disclosed herein.
- the RNA molecule of the present disclosure comprises at least one 5′ cap structure. In some aspects, the RNA molecule of the present disclosure does not comprise a 5′ cap structure.
- the 5′ capping structure comprises a modified 5′ Cap 1 structure (m 7 G + m3′-5′-ppp- 5′-Am).
- the 5′ capping structure comprises is (3′OMe)-m2 7, 3′ -O Gppp(m1 2’-O )ApG (TriLink BioTechnologies). This molecule is identical to the natural RNA cap structure in that it starts with a guanosine methylated at N7, and is linked by a 5′ to 5′ triphosphate linkage to the first coded nucleotide of the transcribed RNA (in this case, an adenosine).
- This guanosine is also methylated at the 3′ hydroxyl of the ribose to mitigate possible reverse incorporation of the cap molecule.
- the 2’ hydroxyl of the ribose on the adenosine is methylated, conferring a Cap 1 structure.
- C. UNTRANSLATED REGIONS (UTRs) The 5′ UTR is a regulatory region situated at the 5′ end of a protein open reading frame that is transcribed into mRNA but not translated into an amino acid sequence and/or to the corresponding region in an RNA polynucleotide, such as an mRNA molecule.
- An untranslated region may be present 5′ (upstream) of an open reading frame (5′ UTR) and/or 3′ (downstream) of an open reading frame (3′ UTR).
- the UTR is derived from an mRNA that is naturally abundant in a specific tissue (e.g., lymphoid tissue), to which the mRNA expression is targeted.
- the UTR increases protein synthesis.
- the UTR may increase protein synthesis by increasing the time that the mRNA remains in translating polysomes (message stability) and/or the rate at which ribosomes initiate translation on the message (message translation efficiency). Accordingly, the UTR sequence may prolong protein synthesis in a tissue-specific manner.
- the regulatory features of a UTR can be incorporated into the RNAs of the present disclosure to, among other things, enhance the stability of the molecule.
- the specific features can also be incorporated to ensure controlled down-regulation of the transcript in case they are misdirected to undesired organs sites.
- a variety of 5′ UTR and the 3′ UTR sequences are known and available in the art. It should be understood that any UTR from any gene may be incorporated into the regions of the RNAs of the present disclosure.
- multiple wild-type UTRs of any known gene may be utilized. It is also within the scope of the present disclosure to provide artificial UTRs which are not variants of wild type regions.
- UTRs or portions thereof may be placed in the same orientation as in the transcript from which they were selected or may be altered in orientation and/or location. Hence a 5′ and/or 3′ UTR may be inverted, shortened, lengthened, and/or made with one or more other 5′ UTRs or 3′ UTRs.
- altered as it relates to a UTR sequence, means that the UTR has been changed in some way in relation to a reference sequence.
- 5′ UTRs and/or 3′ UTRs may be altered relative to a wild-type or native UTR by the change in orientation and/or location as taught above and/or may be altered by the inclusion of additional nucleotides, deletion of nucleotides, swapping, and/or transposition of nucleotides. Any of these changes produces an “altered” UTR (whether 5′ and/or 3′) including a variant UTR.
- a double, triple or quadruple UTR such as a 5′ and/or 3′ UTR may be used.
- a “double” UTR is one in which two copies of the same UTR are encoded either in series or substantially in series.
- a double beta-globin 3′ UTR may be used. It is also within the scope of the present disclosure to have patterned UTRs. As used herein “patterned UTRs” are those UTRs which reflect a repeating or alternating pattern, such as AB AB AB or AABBAABBAABB or ABCABCABC or variants thereof repeated once, twice, or more than 3 times. In these patterns, each letter, A, B, or C represent a different UTR at the nucleotide level.
- RNAs may encode polypeptides of interest belonging to a family of proteins that are expressed in a particular cell, tissue and/or at some time during development.
- the UTRs from any of these genes may be swapped for any other UTR of the same or different family of proteins to create a new RNA molecule.
- a “family of proteins” is used in the broadest sense to refer to a group of two or more polypeptides of interest which share at least one function, structure, feature, localization, origin, and/or expression pattern.
- the 5′ UTR and the 3′ UTR sequences are computationally derived.
- the 5′ UTR and the 3′ UTRs are derived from a naturally abundant mRNA in a tissue.
- the tissue may be, for example, liver, a stem cell and/or lymphoid tissue.
- the lymphoid tissue may include, for example, any one of a lymphocyte (e.g., a B-lymphocyte, a helper T- lymphocyte, a cytotoxic T-lymphocyte, a regulatory T-lymphocyte, and/or a natural killer cell), a macrophage, a monocyte, a dendritic cell, a neutrophil, an eosinophil and a reticulocyte.
- a lymphocyte e.g., a B-lymphocyte, a helper T- lymphocyte, a cytotoxic T-lymphocyte, a regulatory T-lymphocyte, and/or a natural killer cell
- a macrophage e.g., a monocyte, a dendritic cell, a neutrophil, an eosinophil and a reticulocyte.
- the 5′ UTR and the 3′ UTR are derived from an alphavirus.
- the 5′ UTR and the 3′ UTR are from a wild type
- an RNA disclosed herein comprises a 5′ UTR.
- a 5′ UTR if present, is located at the 5′ end and starts with the transcriptional start site upstream of the start codon of a protein encoding region.
- a 5′ UTR is downstream of the 5′ cap (if present), e.g. directly adjacent to the 5′ cap.
- the 5′ UTR may contain various regulatory elements, e.g., 5′ cap structure, stem- loop structure, and an internal ribosome entry site (IRES), which may play a role in the control of translation initiation.
- the 5′ UTR may harbor signatures like Kozak sequences, which are also involved in the process by which the ribosome initiates translation of many genes.5′ UTRs may also form secondary structures involved in elongation factor binding.
- a 5′ UTR disclosed herein comprises a cap proximal sequence, e.g., as disclosed herein.
- a cap proximal sequence comprises a sequence adjacent to a 5′ cap.
- a cap proximal sequence comprises nucleotides in positions +1, +2, +3, +4, and/or +5 of an RNA polynucleotide.
- a Cap structure comprises one or more polynucleotides of a cap proximal sequence.
- a Cap structure comprises an m7 Guanosine cap and nucleotide +1 (N1) of an RNA polynucleotide.
- a Cap structure comprises an m7 Guanosine cap and nucleotide +2 (N2) of an RNA polynucleotide.
- a Cap structure comprises an m7 Guanosine cap and nucleotides +1 and +2 (N 1 and N 2 ) of an RNA polynucleotide.
- one or more residues of a cap proximal sequence may be included in an RNA by virtue of having been included in a cap entity that (e.g., a Cap 1 structure, etc); alternatively, in some aspects, at least some of the residues in a cap proximal sequence may be enzymatically added (e.g., by a polymerase such as a T7 polymerase).
- a cap proximal sequence comprises N1 and/or N2 of a Cap structure, wherein N1 and N2 are any nucleotide, e.g., A, C, G or U.
- N1 is A.
- N 1 is C.
- N 1 is G.
- N 1 is U.
- N 2 is A. In some aspects, N 2 is C. In some aspects, N 2 is G. In some aspects, N 2 is U. In some aspects, a cap proximal sequence comprises N1 and N2 of a Cap structure and N3, N4 and N5, wherein N1 to N5 correspond to positions +1, +2, +3, +4, and/or +5 of an RNA polynucleotide. In some aspects, N1, N2, N3, N4, or N5 are any nucleotide, e.g., A, C, G or U.
- N1N2 comprises any one of the following: AA, AC, AG, AU, CA, CC, CG, CU, GA, GC, GG, GU, UA, UC, UG, or UU.
- N1N2 comprises AG and N3N4N5 comprises any one of the following: AAA, ACA, AGA, AUA, AAG, AGG, ACG, AUG, AAC, ACC, AGC, AUC, , AAU, ACU, AGU, AUU, CAA, CCA, CGA, CUA, CAG, CGG, CCG, CUG, CAC, CCC, CGC, CUC, , CAU, CCU, CGU, CUU, , GAA, GCA, GGA, GUA, , GAG, GGG, GCG, GUG, , GAC, GCC, GGC, GUC, , GAU, GCU, GGU, GUU, UAA, UCA, UGA, UUA, UCA, UGA,
- a cap proximal sequence comprises N1 and N2 of a Cap structure, and a sequence comprising: A3A4X5 (SEQ ID NO: 46; wherein X5 is A, G, C, or U), where N1 and N2 are each independently chosen from: A, C, G, or U.
- N1 is A and N2 is G.
- X 5 is chosen from A, C, G or U.
- X 5 is A.
- X 5 is C.
- X 5 is G.
- X 5 is U.
- a cap proximal sequence comprises N1 and N2 of a Cap structure, and a sequence comprising: C3A4X5 (SEQ ID NO: 47; wherein X5 is A, G, C, or U), where N1 and N2 are each independently chosen from: A, C, G, or U.
- N1 is A and N2 is G.
- X5 is chosen from A, C, G or U.
- X5 is A.
- X5 is C.
- X 5 is G.
- X 5 is U.
- a cap proximal sequence comprises N 1 and N 2 of a Cap structure, and a sequence comprising X 3 Y 4 X 5 (SEQ ID NO: 48; wherein X 3 or X 5 are each independently chosen from A, G, C, or U; and Y4 is not C).
- N1 and N2 are each independently chosen from: A, C, G, or U.
- N1 is A and N2 is G.
- X3 and X5 is each independently chosen from A, C, G or U.
- X3 and/or X5 is A.
- X 3 and/or X 5 is C.
- X 3 and/or X 5 is G.
- X 3 and/or X 5 is U.
- Y 4 is C. In other aspects, Y 4 is not C. In some aspects, Y 4 is A. In some aspects, Y 4 is G. In other aspects, Y4 is not G. In some aspects, Y4 is U.
- a cap proximal sequence comprises N1 and N2 of a Cap structure, and a sequence comprising A3C4A5 (SEQ ID NO: 49).
- N1 and N2 are each independently chosen from: A, C, G, or U. In some aspects, N1 is A and N2 is G.
- a cap proximal sequence comprises N 1 and N 2 of a Cap structure, and a sequence comprising A 3 U 4 G 5 (SEQ ID NO: 50).
- N 1 and N 2 are each independently chosen from: A, C, G, or U.
- N 1 is A and N 2 is G.
- 1, 2, 3, 4, 5, or more of the foregoing cap proximal sequences can be excluded from the 5′ UTR of the RNA molecules disclosed herein.
- a 5′ UTR is a heterologous UTR, e.g., is a UTR found in nature associated with a different ORF.
- a 5′ UTR is a synthetic UTR, e.g., does not occur in nature.
- Synthetic UTRs include UTRs that have been mutated or synthesized to improve their properties, e.g., to increase gene expression.
- the 5′ UTR is functionally linked to the ORF, e.g., associated with the ORF such that it may exert a function, e.g., increasing, enhancing, stabilizing, and/or prolonging protein production from an RNA molecule and/or increasing protein expression and/or total protein production from an RNA molecule, compared to a reference RNA molecule comprising a reference 5′ UTR or an RNA molecule lacking a 5′ UTR.
- exemplary 5′ UTRs include 5′ UTRs derived from Xenopus or human alpha globin or beta globin, human cytochrome b-245 a, hydroxysteroid (17b) dehydrogenase, Tobacco etch virus, the CMV immediate-early 1 (IE1) gene, TEV, HSP705′, c-Jun, or a homolog, fragment, or variant of any of the foregoing.
- IE1 immediate-early 1
- the 5′ UTR is a fragment, homolog or variant of a 5′ UTR of a TOP gene lacking the 5′ TOP motif (the oligopyrimidine tract), the 5′ UTR derived from ribosomal protein Large 32 (L32) gene, the 5′ UTR derived from the 5′ UTR of an hydroxysteroid (17p) dehydrogenase 4 gene (HSD17B4), or the 5′ UTR derived from the 5′ UTR of ATP5A1.
- 5′ UTRs are derived from SEQ ID NOs: 1-1363, SEQ ID NO: 1395, SEQ ID NO: 1421 and SEQ ID NO: 1422 of the patent application WO2013/143700, the disclosure of which is incorporated herein by reference in its entirety, or a sequence having at least, at most, exactly, or between (inclusive or exclusive) any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity with any of the foregoing sequences.
- the sequence GGGAUCCUACC may also be used.
- 1, 2, 3, 4, 5, or more of the foregoing 5′ UTR sequences may be excluded from the RNA molecules disclosed herein.
- the 5′ UTR comprises a sequence from the 5′ UTR region of a gene encoding RPSA, RPS2, RPS3, RPS3A, RPS4, RPS5, RPS6, RPS7, RPS8, RPS9, RPS10, RPS1 1 , RPS12, RPS13, RPS14, RPS15, RPS15A, RPS16, RPS17, RPS18, RPS19, RPS20, RPS21 , RPS23, RPS24, RPS25, RPS26, RPS27, RPS27A, RPS28, RPS29, RPS30, RPL3, RPL4, RPL5, RPL6, RPL7, RPL7A, RPL8, RPL9, RPL10, RPL10A, RPL11 , RPL12, RPL13, RPL13A, RPL14, RPL15, RPL17, RPL18, RPL18A, RPL19, RPL21 , RPL22, RPL23, RPL23A, RPL
- a DNA encoding a 5′ UTR disclosed herein comprises a sequence having at least, at most, exactly, or between (inclusive or exclusive) any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 17.
- the DNA encoding the 5′ UTR comprises a sequence of SEQ ID NO: 17.
- an RNA disclosed herein comprises a 5′ UTR comprising a sequence having at least, at most, exactly, or between any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to a 5’ UTR provided in any of SEQ ID NO: 18 or 19 in which the transcribed 5′ cap structure is underlined.
- the 5′ UTR comprises a sequence of any of SEQ ID NO: 18 or 19, in which the transcribed 5′ cap structure is underlined.
- a DNA encoding a 5′ UTR disclosed herein comprises a sequence having at least, at most, exactly, or between (inclusive or exclusive) any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 51.
- the DNA encoding the 5′ UTR comprises a sequence of SEQ ID NO: 51.
- an RNA disclosed herein comprises a 5′ UTR comprising a sequence having at least, at most, exactly, or between any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to a 5’ UTR provided in any of SEQ ID NO: 52 or 53.
- the 5′ UTR comprises a sequence of any of SEQ ID NO: 52 or 53, in which the transcribed 5′ cap structure is underlined.
- RNA disclosed herein comprises a 3′ UTR.
- a 3′ UTR if present, is situated downstream of a protein coding sequence open reading frame, e.g., downstream of the termination codon of a protein-encoding region.
- a 3′ UTR is typically the part of an mRNA which is located between the protein coding sequence and the poly-A tail of the mRNA.
- the 3′ UTR is upstream of the poly-A sequence (if present), e.g. directly adjacent to the poly-A sequence.
- the 3′ UTR may be involved in regulatory processes including transcript cleavage, stability and polyadenylation, translation, and mRNA localization. Natural or wild type 3′ UTRs comprise stretches of adenosines and uridines.
- AU rich elements can be separated into three classes: Class I AREs contain several dispersed copies of an AUUUA motif within U-rich regions. Class II AREs possess two or more overlapping UUAUUUA(U/A)(U/A) nonamers. Class III ARES do not contain an AUUUA motif. Most proteins binding to AREs are known to destabilize the molecule. Accordingly, introduction, removal and/or modification of 3′ UTR AREs can be used to modulate the stability of nucleic acids (e.g., RNA) of the disclosure.
- nucleic acids e.g., RNA
- one or more copies of an ARE can be introduced to make RNAs less stable and thereby curtail translation and decrease production of the resultant protein.
- AREs can be identified and removed and/or mutated to increase the intracellular stability and thus increase translation and production of the resultant protein.
- Transfection experiments can be conducted in relevant cell lines, using nucleic acids of the disclosure and protein production can be assayed at various time points post-transfection. For example, cells can be transfected with different ARE-engineering molecules and by using an ELISA kit to the relevant protein and assaying protein produced at 6 hour, 12 hour, 24 hour, 48 hour, and 7 days post-transfection.
- a 3′ UTR may have one or more AU-rich sequences removed. Alternatively the AU-rich sequences may remain in the 3′ UTR.
- a 3′ UTR may also comprise elements, which are not encoded in the template, from which an RNA is transcribed, but which are added after transcription during maturation, e.g. a poly-A tail.
- a 3′ UTR of the mRNA is not translated into an amino acid sequence.
- an RNA disclosed herein comprises a 3′ UTR comprising an F element and/or an I element.
- a 3′ UTR or a proximal sequence thereto comprises a restriction site.
- a restriction site is a BamHI site.
- a restriction site is a Xhol site.
- a 3′ UTR is a heterologous UTR, e.g., is a UTR found in nature associated with a different ORF.
- a 3′ UTR is a synthetic UTR, e.g., does not occur in nature.
- the 3′ UTR is functionally linked to the ORF, e.g., associated with the ORF such that it may exert a function, e.g., increasing, enhancing, stabilizing, and/or prolonging protein production from an RNA molecule and/or increasing protein expression and/or total protein production from an RNA molecule, compared to a reference RNA molecule comprising a reference 3′ UTR or an RNA molecule lacking a 3′ UTR.
- 1, 2, 3, 4, 5, or more of the foregoing 3′ UTR functions may be excluded.
- Exemplary 3′ UTRs include 3′ UTRs derived from an albumin gene, an a-globin gene, a ⁇ -globin gene, a ribosomal protein gene, a tyrosine hydroxylase gene, a lipoxygenase gene, and a collagen alpha gene, such as a collagen alpha 1 (1) gene, or from a homolog, fragment, or variant of a 3′ UTR of a gene comprising an albumin gene, an a-globin gene, a ⁇ - globin gene, a ribosomal protein gene, a tyrosine hydroxylase gene, a lipoxygenase gene, and/or a collagen alpha gene, such as a collagen alpha 1 (1) gene according to SEQ ID NOs: 1369-1390 of the patent application WO2013/143700, the disclosure of which is incorporated herein by reference in its entirety, or a sequence having at least, at most, exactly, or between (inclusive or exclusive) any two of 99%, 9
- the sequence UUUGAAUU is used. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing 3′ UTR sequences may be excluded from the RNA molecules disclosed herein.
- the 3′ UTR comprises a sequence of a transcript including NM_000661.4, NM_001024921 .2, NM_000967.3, NM_001033853.1 , NMJD00968.3, NM_000969.3, NM_001024662.1 , NM_000970.3, NM_000971 .3, NMJD00972.2, NM_000975.3, NM_001 199802.1 , NM_000976.3, NM__000977.3, NM_033251 .2, NMJ 01243130.1 , NM_001243131 , NM_000978.3, NM_000979.3, NM_001270490.1 , NMJD00980.3, NM_000981.3, NM_000982.3, NM_000983.3
- the 3′ UTR comprises a sequence from the 3′ UTR region of a gene encoding a ribosomal protein, e.g., ribosomal protein L9 (RPL9), ribosomal protein L3 (RPL3), ribosomal protein L4 (RPL4), ribosomal protein L5 (RPL5), ribosomal protein L6 (RPL6), ribosomal protein L7 (RPL7), ribosomal protein L7a (RPL7A), ribosomal protein L11 (RPL11 ), ribosomal protein L12 (RPL12), ribosomal protein L13 (RPL13), ribosomal protein L23 (RPL23), ribosomal protein L18 (RPL18), ribosomal protein L18a (RPL18A), ribosomal protein, ribosomal protein L9 (RPL9), ribosomal protein L3 (RPL3), ribosomal protein L4 (
- the 3′ UTR comprises a sequence from the 3′ UTR region of a gene encoding a ribosomal protein or from a gene comprising ubiquitin A-52 residue ribosomal protein fusion product 1 (UBA52), Finkel-Biskis-Reilly murine sarcoma virus (FBR-MuSV) ubiquitously expressed (FAU), ribosomal protein L22-like 1 (RPL22L1), ribosomal protein L39-like (RPL39L), ribosomal protein L10-like (RPL10L), ribosomal protein L36a-like (RPL36AL), ribosomal protein L3-like (RPL3L), ribosomal protein S27-like (RPS27L), ribosomal protein L26-like 1 (RPL26L1), ribosomal protein L7
- UAA52 ubiquitin A-52 residue ribosomal protein fusion product 1
- FBR-MuSV Finkel-Biskis
- 1, 2, 3, 4, 5, or more of the foregoing 3′ UTR sequences may be excluded from the RNA molecules disclosed herein.
- 5′ UTRs that are heterologous and/or synthetic may be used with any desired 3′ UTR sequence, and vice versa.
- a heterologous 5′ UTR may be used with a synthetic and/or heterologous 3′ UTR.
- a DNA encoding a 3′ UTR disclosed herein comprises a sequence having at least, at most, exactly, or between (inclusive or exclusive) any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 20.
- the DNA encoding the 3′ UTR comprises a sequence of SEQ ID NO: 20.
- an RNA disclosed herein comprises a 3′ UTR comprising a sequence having at least, at most, exactly, or between (inclusive or exclusive) any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to a 3’ UTR provided in any of SEQ ID NO: 21 or 22.
- the 3′ UTR comprises a sequence of any of SEQ ID NO: 21 or 22.
- the DNA encoding the 3′ UTR comprises a sequence of SEQ ID NO: 23.
- an RNA disclosed herein comprises a 3′ UTR comprising a sequence having at least, at most, exactly, or between any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to a 3’ UTR provided in any of SEQ ID NO: 24 or 25.
- the 3′ UTR comprises a sequence of any of SEQ ID NO: 24 or 25.
- the 5′ and 3′ UTRs may be operably linked to an open reading frame (ORF), which may be a sequence of codons that is capable of being translated into a polypeptide of interest.
- An open reading frame may be a sequence of several DNA or RNA nucleotide triplets, which may be translated into a peptide or protein.
- An ORF may begin with a start codon, e.g., a combination of three subsequent nucleotides coding usually for the amino acid methionine (ATG or AUG), at its 5’ end and a subsequent region, which usually exhibits a length which is a multiple of 3 nucleotides.
- An open reading frame may terminate with at least one stop codon, including but not limited to TAA, TAG, TGA or UAA, UAG or UGA, or any combination thereof.
- an open reading frame may terminate with one, two, three, four or more stop codons, including but not limited to TAATAA (SEQ ID NO: 27), TAATAG (SEQ ID NO: 28), TAATGA (SEQ ID NO: 29), TAGTGA (SEQ ID NO: 30), TAGTAA (SEQ ID NO: 31), TAGTAG (SEQ ID NO: 32), TGATGA (SEQ ID NO: 33), TGATAG (SEQ ID NO: 34), TGATAA (SEQ ID NO: 35) or UAAUAA (SEQ ID NO: 36), UAAUAG (SEQ ID NO: 37), UAAUGA (SEQ ID NO: 38), UAGUGA (SEQ ID NO: 39), UAGUAA (SEQ ID NO:40), UAGUAG (SEQ ID NO: 41), UG
- RNA molecule may include one (monocistronic), two (bicistronic) or more (multicistronic) open reading frames.
- the ORF encodes a non-structural viral gene.
- the ORF further includes one or more subgenomic promoters.
- the RNA molecule includes a subgenomic promoter operably linked to the ORF.
- a first RNA molecule does not include an ORF encoding any polypeptide of interest, whereas a second RNA molecule includes an ORF encoding a polypeptide of interest. In some aspects, the first RNA molecule does not include a subgenomic promoter.
- the present disclosure provides for an RNA molecule comprising at least one open reading frame encoding a respiratory syncytial virus (RSV) polypeptide. In some aspects, an RNA molecule comprising at least one open reading frame encoding a RSV F protein. In a preferred aspect, an RNA molecule comprising at least one open reading frame encoding a respiratory syncytial virus (RSV) prefusion F protein (preF) polypeptide.
- RSV respiratory syncytial virus
- preF prefusion F protein
- RNA molecules described herein may include a gene of interest.
- the gene of interest encodes a polypeptide of interest.
- polypeptides of interest include, e.g., biologics, antibodies, vaccines, therapeutic polypeptides or peptides, cell penetrating peptides, secreted polypeptides, plasma membrane polypeptides, cytoplasmic or cytoskeletal polypeptides, intracellular membrane bound polypeptides, nuclear polypeptides, polypeptides associated with human disease, targeting moieties, those polypeptides encoded by the human genome for which no therapeutic indication has been identified but which nonetheless have utility in areas of research and discovery, or combinations thereof.
- RNA molecules include a coding region for a gene of interest.
- a gene of interest is or comprises an antigenic polypeptide or an immunogenic variant or an immunogenic fragment thereof.
- an antigenic polypeptide comprises one epitope from an antigen.
- an antigenic polypeptide comprises a plurality of distinct epitopes from an antigen.
- an antigenic polypeptide comprising a plurality of distinct epitopes from an antigen is polyepitopic.
- an antigenic polypeptide comprises: an antigenic polypeptide from an allergen, a viral antigenic polypeptide, a bacterial antigenic polypeptide, a fungal antigenic polypeptide, a parasitic antigenic polypeptide, an antigenic polypeptide from an infectious agent, an antigenic polypeptide from a pathogen, a tumor antigenic polypeptide, or a self-antigenic polypeptide.
- 1, 2, 3, 4, 5, or more of the foregoing antigenic polypeptides may be excluded.
- antigen may refer to a substance, which is capable of being recognized by the immune system, e.g., by the adaptive immune system, and which is capable of eliciting an antigen-specific immune response, e.g., by formation of antibodies and/or antigen-specific T cells as part of an adaptive immune response.
- An antigen may be or may comprise a peptide or protein, which may be presented by the MHC to T cells.
- An antigen may be the product of translation of a provided nucleic acid molecule, e.g., an RNA molecule comprising at least one coding sequence as described herein.
- an antigen such as a peptide or a protein, comprising at least one epitope
- an RNA encoding a gene of interest e.g., an antigen
- the RNA is transiently expressed in cells of the subject.
- expression of a gene of interest, e.g., an antigen is at the cell surface.
- a gene of interest, e.g., an antigen is expressed and presented in the context of MHC.
- the RNA molecules include a coding region for a gene of interest, e.g., an antigen. In some aspects, the RNA molecules include a coding region for a gene of interest, e.g., an antigen, that is derived from a pathogen associated with an infectious disease. In some aspects, the RNA molecules include a coding region for a gene of interest, e.g., an antigen, that is derived from RSV. In some aspects, the RNA molecule encodes a RSV preF protein or a fragment or a variant thereof.
- an RNA polynucleotide described herein or a composition or medical preparation comprising the same comprises a nucleotide sequence disclosed herein. In some aspects, an RNA polynucleotide comprises a sequence having at least 80% identity to a nucleotide sequence disclosed herein. In some aspects, an RNA polynucleotide comprises a sequence encoding a polypeptide having at least 80% identity to a polypeptide sequence disclosed herein. In some aspects, an RNA polynucleotide described herein or a composition or medical preparation comprising the same is transcribed by a DNA template.
- a DNA template used to transcribe an RNA polynucleotide described herein comprises a sequence complementary to an RNA polynucleotide.
- a gene of interest described herein is encoded by an RNA polynucleotide described herein comprising a nucleotide sequence disclosed herein.
- an RNA polynucleotide encodes a polypeptide having at least 80% identity to a polypeptide sequence disclosed herein.
- a polypeptide described herein is encoded by an RNA polynucleotide transcribed by a DNA template comprising a sequence complementary to an RNA polynucleotide.
- the RNA molecule encodes a RSV preF protein comprising the sequence of any one of SEQ ID NOs: 1-6 and 71-74, or a fragment or variant thereof. In some aspects, the RNA molecule encodes a RSV preF protein synthesized from the nucleic acid sequence comprising any one of SEQ ID NOs: 7 to 10 and 59 to 62, or fragment or variant thereof.
- F. POLY-A TAIL In some aspects, RNA molecules disclosed herein comprise a poly-adenylate (poly-A) sequence, e.g., as described herein. In some aspects, a poly-A sequence is situated downstream of a 3′ UTR, e.g., adjacent to a 3′ UTR.
- a “poly-A tail” or “poly-A sequence” refers to a stretch of consecutive adenine residues, e.g., of up to or up to about 400 adenosine nucleotides, e.g., from or from about 20 to about 400, preferably from or from about 50 to about 400, more preferably from or from about 50 to about 300, even more preferably from or from about 50 to about 250, most preferably from or from about 60 to about 250 adenosine nucleotides, which may be attached to the 3′ end of the RNA molecule.
- Poly-A sequences are known to those of skill in the art and may follow the 3′ UTR in the RNA molecules described herein.
- the poly-A tail may increase the stability, half-life, and/or translational efficiency of the RNA molecule.
- 3′-end processing is a nuclear co-transcriptional process that promotes transport of mRNAs from the nucleus to the cytoplasm and affects the stability and the translation of mRNAs.
- this 3′ end occurs in a two-step reaction directed by the cleavage/polyadenylation machinery and depends on the presence of two sequence elements in mRNA precursors (pre-mRNAs); a hexanucleotide polyadenylation signal and a downstream G/U-rich sequence.
- pre-mRNAs mRNA precursors
- a hexanucleotide polyadenylation signal a downstream G/U-rich sequence.
- a first step pre-mRNAs are cleaved between these two elements to a free 3′ hydroxyl.
- the newly formed 3′ end is extended by polyadenylation or addition of a poly-A sequence.
- Polyadenylation refers to the addition of a poly-A sequence to an RNA molecule, e.g., to a premature mRNA.
- Polyadenylation may be induced by a so-called polyadenylation signal. This signal may be located within a stretch of nucleotides close to or at the 3′-end of an RNA molecule to be polyadenylated. A polyadenylation signal may also be comprised by the 3′ UTR of the artificial nucleic acid molecule.
- a polyadenylation signal typically comprises a hexamer consisting of adenine and uracil/thymine nucleotides, preferably the hexamer sequence AAUAAA, though other sequences, preferably hexamer sequences, are also conceivable. Polyadenylation typically occurs during processing of a pre-mRNA (also called premature-mRNA).
- RNA maturation comprises the step of polyadenylation.
- Poly-A tailing of in vitro transcribed mRNA can be achieved using various approaches including, but not limited to, cloning of a poly-T tract into the DNA template or by post-transcriptional addition using poly-A polymerase.
- the term may relate to polyadenylation of RNA as a cellular process or to polyadenylation carried out by enzymatic reaction in vitro with a suitable enzyme, such as E. coli poly-A polymerase, or by chemical synthesis.
- RNA molecules disclosed herein may have a poly-A sequence attached to the free 3′-end of the RNA by a template-independent RNA polymerase after transcription or a poly-A sequence encoded by DNA and transcribed by a template-dependent RNA polymerase.
- a poly-A sequence is attached during RNA transcription, e.g., during preparation of in vitro transcribed RNA, based on a DNA template comprising repeated dT nucleotides (deoxythymidylate) in the strand complementary to the coding strand.
- the DNA sequence encoding a poly-A sequence (coding strand) is referred to as poly-A cassette.
- the poly-A cassette present in the coding strand of DNA essentially consists of dA nucleotides, but is interrupted by a random sequence of the four nucleotides (dA, dC, dG, and dT).
- a random sequence may be at least, at most, exactly, or between (inclusive or exclusive) any two of 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, or 50 nucleotides in length.
- a poly-A cassette that essentially consists of dA nucleotides, but is interrupted by a random sequence having an equal distribution of the four nucleotides (dA, dC, dG, dT) and having a length of e.g., 5 to 50 nucleotides, shows, on a DNA level, constant propagation of plasmid DNA in E. coli and is still associated, on an RNA level, with the beneficial properties with respect to supporting RNA stability and translational efficiency.
- the poly-A sequence contained in an RNA polynucleotide described herein consists essentially of adenosine nucleotides, but is interrupted by a random sequence of the four nucleotides (A, C, G, U).
- a random sequence may be at least, at most, exactly, or between (inclusive or exclusive) any two of 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, or 50 nucleotides in length.
- the poly-A sequence may be located at any position within the 3′ UTR.
- no nucleotides other than adenosine nucleotides flank a poly-A sequence at its 3′-end, e.g., the poly-A sequence is not masked or followed at its 3′-end by a nucleotide other than adenosine.
- the poly-A sequence may be located at the 3′ terminus of the 3′ UTR, e.g., the 3′ UTR does not contain more than 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 nucleotides located 3′ of the poly-A sequence; more preferably the 3′ UTR does not contain further elements located 3′ to the poly-A sequence.
- poly-A sequence is located at the 3′ terminus of the RNA molecule, e.g., the artificial nucleic acid molecule does not contain more than 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 nucleotides located 3′ of the poly-A sequence.
- the poly-A sequence may be located at the 5′ terminus of the 3′ UTR, e.g., immediately 3′ of the ORF of the artificial nucleic acid molecule, or located within the 3′ UTR, e.g., flanked on the 5′ and on the 3′ side by other 3′ UTR elements.
- the poly-A sequence is flanked on the 3′ side by a poly- C sequence and/or a histone stem-loop sequence.
- the poly-A sequence can be flanked on the 5′ side by a 3′ UTR element derived from, e.g., a human albumin or globin gene.
- the RNA molecule may further include an endonuclease recognition site sequence immediately downstream of the poly-A tail sequence.
- the RNA molecule may further include a poly-A polymerase recognition sequence (e.g., a polyadenylation signal) (e.g., AAUAAA) near its 3′ end.
- the polyadenylation signal is located 3′ of the poly-A sequence comprised in the 3′ UTR.
- the poly-A sequence is separated from the polyadenylation signal by a nucleotide sequence comprising or consisting of at least, at most, exactly, or between (inclusive or exclusive) any two of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, or 150 nucleotides, wherein the nucleotide sequence does preferably not comprise more than 10, 9, 8, 7, 6, 5, 4, 3, or 2 consecutive adenine nucleotides.
- the nucleotide sequence that separates the poly-A sequence and the polyadenylation signal comprises from or from about 1 to about 200 nucleotides, e.g., from 10 to 90, from 20 to 85, from 30 to 80, from 40 to 80, from 50 to 75 or from 55 to 85 nucleotides, more preferably from 55 to 80 nucleotides, and the nucleotide sequence does not comprise more than 10, 9, 8, 7, 6, 5, 4, 3, or 2 consecutive adenine nucleotides.
- Such a consensus sequence may be recognized by most animal and bacterial cell-systems, for example, by the polyadenylation-factors, such as cleavage/polyadenylation specificity factor (CPSF) cooperating with CstF, PAP, PAB2, CFI and/or CFII.
- CPSF cleavage/polyadenylation specificity factor
- the polyadenylation signal (e.g., the consensus sequence NNUANA) is located less than or less than about 50 nucleotides, e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, or 50 nucleotides, downstream of the 3′-end of the 3′ UTR element as defined herein such that transcription of an RNA molecule will result in a premature-RNA containing the polyadenylation signal downstream of its 3′ UTR and subsequent attachment of a poly-A sequence to the premature-RNA.
- the polyadenylation signal e.g., the consensus sequence NNUANA
- a resulting RNA may comprise a 3′ UTR, which comprises at least one poly-A sequence, and wherein the 3′ UTR is followed by an additional poly-A sequence.
- the poly-A sequence may be of any length.
- the poly-A tail may be 5 to 300 nucleotides in length.
- the RNA molecule includes a poly-A tail that comprises, consists essentially of, or consists of a sequence of or of about 25 to about 400 adenosine nucleotides, a sequence of or of about 50 to about 400 adenosine nucleotides, a sequence of or of about 50 to about 300 adenosine nucleotides, a sequence of or of about 50 to about 250 adenosine nucleotides, a sequence of or of about 60 to about 250 adenosine nucleotides, or a sequence of or of about 40 to about 100 adenosine nucleotides.
- the poly-A tail comprises, consists essentially of, or consists of at least, at most, exactly, or between (inclusive or exclusive) any two of 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200, 205, 210, 215, 220, 225, 230, 235, 240, 245, 250, 255, 260, 265, 270, 275, 280, 285, 290, 295, 300, 305, 310, 315, 320, 325, 330, 335, 340, 345, 350, 355, 360, 365, 370, 375, 380, 385, 390, 395,
- “consists essentially of” means that most nucleotides in the poly-A sequence, typically at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% by number of nucleotides in the poly-A sequence are adenosine nucleotides, but permits remaining nucleotides to be nucleotides other than adenosine nucleotides, such as uridine, guanosine, and/or cytosine.
- RNA molecule includes a poly-A tail that includes a sequence of greater than 30 adenosine nucleotides. In some aspects, the RNA molecule includes a poly-A tail that includes or includes about 40 adenosine nucleotides. In some aspects, the RNA molecule includes a poly-A tail that includes or includes about 80 adenosine nucleotides.
- the 3′ poly-A tail has a stretch of at least 10 consecutive adenosine residues and at most 300 consecutive adenosine residues. In some specific aspects, the RNA molecule includes or includes about 40 consecutive adenosine residues. In some aspects, the RNA molecule includes or includes about 80 consecutive adenosine residues.
- Poly-A tails may play key regulatory roles in enhancing translation efficiency and regulating the efficiency of mRNA quality control and degradation. Short sequences or hyperpolyadenylation may signal for RNA degradation.
- a poly-A tail may be located within an RNA molecule or other nucleic acid molecule, such as, e.g., in a vector, for example, in a vector serving as template for the generation of an RNA, e.g., an mRNA, e.g., by transcription of the vector.
- the RNA molecule may not include a poly-A tail.
- a poly-A tail may be located within an RNA molecule or other nucleic acid molecule, such as, e.g., in a vector, for example, in a vector serving as template for the generation of an RNA, e.g. an mRNA, e.g., by transcription of the vector.
- the RNA molecule may not include a poly-A tail.
- a DNA encoding a poly-A tail disclosed herein comprises a sequence having at least, at most, exactly, or between (inclusive or exclusive) any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 26.
- the DNA encoding the poly-A tail comprises a sequence of SEQ ID NO: 26.
- an RNA disclosed herein comprises a poly-A tail comprising a sequence having at least, at most, exactly, or between (inclusive or exclusive) any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 26.
- the poly-A tail comprises a sequence of SEQ ID NO: 26.
- the poly-A tail comprises a sequence of SEQ ID NO: 26 +/- 2 adenosine (A) nucleotides.
- the poly-A tail comprises a sequence of SEQ ID NO: 26 +/- 1 adenosine (A) nucleotides.
- the poly-A tail comprises a sequence of SEQ ID NO: 26. In one aspect, the poly- A tail comprises a sequence of SEQ ID NO: 26 +/- 2 adenosine (A) nucleotides. In one aspect, the poly-A tail comprises a sequence of SEQ ID NO: 26 +/- 1 adenosine (A) nucleotides. In some aspects, the poly-A tail comprises a sequence of SEQ ID NO: 26. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing poly-A sequences may be excluded from the RNA molecules disclosed herein.
- RNA molecules additionally include a chain terminating nucleoside.
- a chain terminating nucleoside may include those nucleosides deoxygenated at the 2’ and/or 3′ positions of their sugar group.
- Such species may include 3′ deoxyadenosine (cordycepin), 3′ deoxyuridine, 3′ deoxycytosine, 3′ deoxyguanosine, 3′ deoxythymine, and 2',3′ dideoxynucleosides, such as 2',3′ dideoxyadenosine, 2',3′ dideoxyuridine, 2',3′ dideoxycytosine, 2',3′ dideoxyguanosine, and 2',3′ dideoxythymine.
- 1, 2, 3, 4, 5, or more of the foregoing chain terminating nucleosides may be excluded from the RNA molecules disclosed herein.
- incorporation of a chain terminating nucleotide into an mRNA may result in stabilization of the mRNA, as described, for example, in International Patent Publication No. WO 2013/103659.
- the RNA molecules additionally include a stem loop, such as a histone stem loop.
- a stem loop may include 2, 3, 4, 5, 6, 7, 8, or more nucleotide base pairs.
- a stem loop may include 4, 5, 6, 7, or 8 nucleotide base pairs.
- a stem loop may be located in any region of an mRNA.
- a stem loop may be located in, before, or after an untranslated region (a 5′ UTR or a 3′ UTR), a coding region, or a poly-A sequence or tail.
- a stem loop may affect one or more function(s) of an mRNA, such as initiation of translation, translation efficiency, and/or transcriptional termination.
- histone stem-loop sequences may be histone stem-loop sequences disclosed in WO 2012/019780, the disclosure of which is incorporated herein by reference in its entirety.
- Other non-limiting examples of histone stem loop structures and nucleic acid sequences encoding such structures can be found in, e.g., WO 2016/091391, the disclosure of which is incorporated by reference herein in its entirety.
- the combination of a poly-A sequence or polyadenylation signal and at least one histone stem-loop acts synergistically to increase the protein expression beyond the level observed with either of the individual elements.
- the synergistic effect of the combination of poly-A and at least one histone stem-loop does not depend on the order of the elements and/or the length of the poly-A sequence.
- the RNA does not comprise a histone downstream element (HDE).
- An HDE includes a purine-rich polynucleotide stretch of approximately 15 to 20 nucleotides 3′ of naturally occurring stem-loops, representing the binding site for the U7 snRNA, which is involved in processing of histone pre-mRNA into mature histone mRNA.
- the histone stem-loop is generally derived from histone genes, and includes an intramolecular base pairing of two neighbored partially or entirely reverse complementary sequences separated by a spacer, consisting of a short sequence, which forms the loop of the structure. The unpaired loop region is typically unable to base pair with either of the stem loop elements.
- the Stability of the stem-loop structure generally depends on the length, number of mismatches or bulges, and/or base composition of the paired region.
- wobble base pairing non-Watson-Crick base pairing
- the at least one histone stem-loop sequence comprises a length of 15 to 45 nucleotides.
- the RNA molecules include (e.g., within the 3′ UTR) a poly(C) sequence.
- the poly-C sequences has at least, at most, exactly, or between (inclusive or exclusive) any two of 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, or 200 cytidines. In some aspects, the poly-C sequences has or has about 30 cytidines.
- the RNA molecules include an internal ribosome entry site (IRES) sequence or IRES-motif. In some aspects, an IRES sequence separates ORFs, e.g., if the RNA encodes two or more peptides or proteins.
- RNA molecules may be a bi- or multicistronic nucleic acid molecule.
- the RNA does not comprise an intron.
- the RNA may instead or additionally include a microRNA binding site.
- RNA molecules including a combination of the elements disclosed herein can include, without limitation, in 5′-to-3′-direction, the following: ORF - poly-A sequence; ORF - IRES - ORF - poly-A sequence; ORF - 3′ UTR - poly-A sequence; ORF - poly-A sequence - 3′ UTR; ORF - 3′ UTR - poly-A sequence - poly(C) sequence - histone stem-loop; ORF - 3′ UTR - poly-A sequence - poly(C) sequence - poly-A sequence; ORF - 3′ UTR - poly-A sequence - histone stem-loop - poly-A sequence; 5′ UTR - ORF - 3′ UTR; 5′ UTR - ORF - poly-A sequence; 5′ UTR - ORF - poly-A sequence; 5′ UTR - ORF - poly-A sequence - poly(C) sequence - histone stem-loop; 5′ U
- RNA molecules disclosed herein may be an saRNA.
- Self-amplifying RNA refers to RNA with the ability to replicate itself.
- Self-amplifying RNA molecules may be produced by using replication elements derived from, e.g., alphaviruses, and substituting the structural viral polypeptides with a nucleotide sequence encoding a polypeptide of interest.
- a self- amplifying RNA molecule is typically a positive-strand molecule that may be directly translated after delivery to a cell, and this translation provides an RNA-dependent RNA polymerase that then produces both antisense and sense transcripts from the delivered RNA.
- the delivered RNA leads to the production of multiple daughter RNA molecules.
- These daughter RNA molecules, as well as collinear subgenomic transcripts, may be translated themselves to provide in situ expression of an encoded gene of interest, e.g., a viral antigen, and/or may be transcribed to provide further transcripts with the same sense as the delivered RNA that are translated to provide in situ expression of the antigen.
- the self-amplifying RNA includes at least one or more genes including any one of viral replicases, viral proteases, viral helicases and other nonstructural viral proteins, or combination thereof. In some aspects, 1, 2, 3, or more of the foregoing genes may be excluded from the self-amplifying RNA molecules disclosed herein.
- the self-amplifying RNA may also include 5′- and 3′-end tractive replication sequences, and optionally a heterologous sequence that encodes a desired amino acid sequence (e.g., an antigen of interest).
- a subgenomic promoter that directs expression of the heterologous sequence may be included in the self-amplifying RNA.
- the heterologous sequence e.g., an antigen of interest
- a self-amplifying RNA molecule described herein encodes (i) an RNA- dependent RNA polymerase that may transcribe RNA from the self-amplifying RNA molecule and (ii) a polypeptide of interest, e.g., a viral antigen.
- the polymerase may be an alphavirus replicase, e.g., including any one of alphavirus proteins nsP1, nsP2, nsP3, nsP4, or any combination thereof.
- 1, 2, 3, or more of the foregoing alphavirus proteins may be excluded from the RNA molecules disclosed herein.
- the self-amplifying RNA molecule may have two open reading frames.
- the first (5′) open reading frame may encode a replicase; the second (3′) open reading frame may encode a polypeptide comprising an antigen of interest.
- the RNA may have additional (e.g., downstream) open reading frames, e.g., to encode further antigens or to encode accessory polypeptides.
- the saRNA molecule further includes (1) an alphavirus 5′ replication recognition sequence, and (2) an alphavirus 3′ replication recognition sequence.
- the 5′ sequence of the self-amplifying RNA molecule is selected to ensure compatibility with the encoded replicase.
- the self-amplifying RNA molecule may encode a single polypeptide antigen or, optionally, two or more polypeptide antigens linked together in a way that each of the sequences retains its identity (e.g., linked in series) when expressed as an amino acid sequence.
- the polypeptides generated from the self-amplifying RNA may then be produced as a fusion polypeptide or engineered in such a manner to result in separate polypeptide or peptide sequences.
- the self-amplifying RNA described herein may encode one or more polypeptide antigens that include a range of epitopes.
- the self-amplifying RNA described herein may encode epitopes capable of eliciting either a helper T cell response or a cytotoxic T cell response or both.
- a self-amplifying RNA disclosed herein comprises a subgenomic promoter comprising a sequence having at least, at most, exactly, or between any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 54.
- the subgenomic promoter comprises a sequence of SEQ ID NO: 54.
- a self-amplifying RNA molecule described herein encodes (i) an RNA- dependent RNA polymerase that may transcribe RNA from the self-amplifying RNA molecule and (ii) a polypeptide of interest, e.g., a viral antigen.
- the polymerase may be an alphavirus replicase, e.g., including any one of alphavirus protein nsP1, nsP2, nsP3, nsP4, and any combination thereof.
- a self-amplifying RNA disclosed herein comprises an alphavirus replicase, e.g., including any one of alphavirus protein nsP1, nsP2, nsP3, nsP4, and any combination thereof, comprising a sequence having at least, at most, exactly, or between any two of 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to SEQ ID NO: 55-58, respectively.
- the alphavirus protein nsP1, nsP2, nsP3 and nsP4 each comprise a sequence of SEQ ID NO: 55-58, respectively.
- RNA TRANSCRIPTION In some aspects, the RNA disclosed herein is produced by in vitro transcription or chemical synthesis.
- transcription relates to a process, wherein the genetic code in a DNA sequence is transcribed into RNA. Subsequently, the RNA may be translated into peptide or protein.
- transcription comprises “in vitro transcription” or “IVT,” which refers to the process whereby transcription occurs in vitro in a non-cellular system to produce a synthetic RNA product for use in various applications, including, e.g., production of protein or polypeptides.
- ITT in vitro transcription
- the methodology for in vitro transcription of mRNA is well known in the art.
- Cloning vectors may be applied for the generation of transcripts. These cloning vectors are generally designated as transcription vectors and are according to the present invention encompassed by the term “vector.” According to specific aspects, the RNA used is in vitro transcribed RNA (IVT-RNA) and may be obtained by in vitro transcription of an appropriate DNA template.
- IVT-RNA in vitro transcribed RNA
- Template DNA can be prepared for in vitro transcription from a number of sources with appropriate techniques which are well known in the art including, but not limited to, plasmid DNA and polymerase chain reaction amplification (see Linpinsel, J.L and Conn, G.L., General protocols for preparation of plasmid DNA template, and Bowman, J.C., Azizi, B., Lenz, T.K., Ray, P., and Williams, L.D. in RNA in vitro transcription and RNA purification by denaturing PAGE in Recombinant and in vitro RNA syntheses, Methods 941 Conn G.L. (ed), New York, N.Y. Humana Press, 2012, each incorporated herein by reference).
- plasmid DNA and polymerase chain reaction amplification see Linpinsel, J.L and Conn, G.L., General protocols for preparation of plasmid DNA template, and Bowman, J.C., Azizi, B., Lenz, T.K., Ray, P.
- the promoter for controlling transcription may be any promoter for any RNA polymerase.
- RNA polymerases are the T7, T3, and SP6 RNA polymerases.
- the in vitro transcription according to the invention is controlled by a T7 or SP6 promoter.
- a DNA template for in vitro transcription may be obtained by cloning of a nucleic acid, in particular cDNA, and introducing it into an appropriate vector for in vitro transcription.
- the cDNA may be obtained by reverse transcription of RNA.
- Synthetic IVT RNA products may be translated in vitro or introduced directly into cells, where they may be translated.
- RNA refers to the process in the ribosomes of a cell by which a strand of mRNA directs the assembly of a sequence of amino acids to make a peptide or protein.
- synthetic RNA products include but are not limited to, e.g., mRNA molecules, saRNA molecules, antisense RNA molecules, shRNA molecules, long non-coding RNA molecules, ribozymes, aptamers, guide RNA molecules (e.g., for CRISPR), ribosomal RNA molecules, small nuclear RNA molecules, small nucleolar RNA molecules, and the like.
- 1, 2, 3, 4, 5, or more of the foregoing synthetic RNA products may be excluded.
- An IVT reaction typically utilizes a DNA template (e.g., a linear DNA template) as described and/or utilized herein, ribonucleotides (e.g., non-modified ribonucleotide triphosphates or modified ribonucleotide triphosphates), and an appropriate RNA polymerase.
- a DNA template e.g., a linear DNA template
- ribonucleotides e.g., non-modified ribonucleotide triphosphates or modified ribonucleotide triphosphates
- an appropriate RNA polymerase e.g., an mRNA is produced by in vitro transcription using a DNA template where DNA refers to a nucleic acid that contains deoxyribonucleotides.
- an RNA disclosed herein is in vitro transcribed RNA (IVT-RNA) and may be obtained by in vitro transcription of an appropriate DNA template.
- the promoter for controlling transcription may be any promoter
- a DNA template for in vitro transcription may be obtained by cloning of a nucleic acid, in particular cDNA, and introducing it into an appropriate vector for in vitro transcription.
- the cDNA may be obtained by reverse transcription of RNA.
- starting material for IVT may include linearized DNA template, nucleotides, Rnase inhibitor, pyrophosphatase, and/or a polymerase (e.g., a T7 RNA polymerase).
- the nucleotides may be manufactured in house, may be obtained from a supplier, or may be synthesized.
- the nucleotides may be, but are not limited to, those described herein including natural and unnatural (modified) nucleotides.
- RNA polymerases or variants may be used, including, but not limited to, a phage RNA polymerase, e.g., a T7 RNA polymerase, a T3 RNA polymerase, a SP6 RNA polymerase, and/or mutant polymerases such as, but not limited to, polymerases able to incorporate modified nucleic acids and/or modified nucleotides, including chemically modified nucleic acids and/or nucleotides.
- 1, 2, 3, 4, 5, or more of the foregoing RNA polymerases may be excluded from. Some embodiments exclude the use of Dnase.
- the IVT process is conducted in a bioreactor.
- the bioreactor may comprise a mixer.
- nucleotides may be added into the bioreactor throughout the IVT process.
- one or more post-IVT agents are added into the IVT mixture comprising RNA in the bioreactor after the IVT process.
- Exemplary post-IVT agents may include DNAse I configured to digest the linearized DNA template and/or proteinase K configured to digest DNAse I and T7 RNA polymerase.
- the post-IVT agents are incubated with the mixture in the bioreactor after IVT.
- the bioreactor may contain at least, at most, exactly, or between (inclusive or exclusive) any two of 60, 70, 80, 90, 100, 110, 120, 130, 140, 150 ,160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490, and 500 or more liters IVT mixture.
- the IVT mixture may have an RNA concentration that is or is not at least, at most, exactly, or between (inclusive or exclusive) any two of 3.3, 3.4, 3.5, 3.6, 3.7, 3.8, 3.9, 4.0, 4.1, 4.2, 4.3, 4.4, 4.5, 4.6, 4.7, 4.8, 4.9, 5.0, 5.1, 5.2, 5.3, 5.4, 5.5, 5.6, 5.7, 5.8, 5.9, 6.0, 7.0, 8.0, 9.0, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50, 60, 70, 80, 90, and 100 mg/mL or more RNA.
- the IVT mixture may include residual spermidine, residual DNA, residual proteins, peptides, HEPES, EDTA, ammonium sulfate, cations (e.g., Mg 2+ , Na + , Ca 2+ ), RNA fragments, residual nucleotides, free phosphates, or any combinations thereof. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing can be excluded from the IVT mixture.
- Isolation and/or purification of the nucleic acids described herein may include, but is not limited to, phenol/chloroform extraction and/or precipitation with either alcohol (ethanol, isopropanol) in the presence of monovalent cations or lithium chloride for nucleic acid clean-up, quality assurance and quality control.
- alcohol ethanol, isopropanol
- purification procedures include AGENCOURT® beads (Beckman Coulter Genomics, Danvers, MA), poly-T beads, LNATM oligo-T capture probes (EXIQON® Inc, Vedbaek, Denmark), HPLC based purification methods such as, but not limited to, strong anion exchange HPLC, weak anion exchange HPLC, reverse phase HPLC (RP-HPLC), and hydrophobic interaction HPLC (HIC-HPLC), size exclusion chromatography, and silica-based affinity chromatography and polyacrylamide gel electrophoresis.
- HPLC based purification methods such as, but not limited to, strong anion exchange HPLC, weak anion exchange HPLC, reverse phase HPLC (RP-HPLC), and hydrophobic interaction HPLC (HIC-HPLC), size exclusion chromatography, and silica-based affinity chromatography and polyacrylamide gel electrophoresis.
- Purification can be performed using a variety of commercially available kits including, but not limited to SV Total Isolation System (Promega) and In vitro Transcription Cleanup and Concentration Kit (Norgen Biotek). In some aspects, 1, 2, 3, 4, 5, or more of the foregoing purification may be excluded.
- the term “purified” when used in relation to a nucleic acid such as a “purified nucleic acid” refers to one that is separated from at least one contaminant.
- a “contaminant” is any substance that makes another unfit, impure or inferior.
- a purified nucleic acid e.g., DNA and RNA
- a purified nucleic acid is present in a form or setting different from that in which it is found in nature, or a form or setting different from that which existed prior to subjecting it to a treatment and/or purification method.
- at least a portion of the IVT mixture is filtered.
- the IVT mixture may be filtered via ultrafiltration and/or diafiltration to remove at least some impurities from the IVT mixture and/or to change buffer solution for the at least a portion of IVT mixture to produce a concentrated RNA solution as a retentate.
- both “ultrafiltration” and “diafiltration” refer to a membrane filtration process.
- Ultrafiltration typically uses membranes having pore sizes of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.001, 0.002, 0.003, 0.004, 0.005, 0.006, 0.007, 0.008, 0.009, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, and 0.1 ⁇ m.
- ultrafiltration membranes are typically classified by molecular weight cutoff (MWCO) rather than pore size.
- MWCO molecular weight cutoff
- the MWCO may be at least, at most, exactly, or between (inclusive or exclusive) any two of 30 kDa, 40 kDa, 50 kDa, 60 kDa, 70 kDa, 80 kDa, 90 kDa, 100 kDa, 110 kDa, 120 kDa, 130 kDa, 140 kDa, 150 kDa, 160 kDa, 170 kDa, 180 kDa, 190 kDa, 200 kDa, 210 kDa, 220 kDa, 230 kDa, 240 kDa, 250 kDa, 260 kDa, 270 kDa, 280 kDa, 290 kDa, 300 kDa, 310 kDa, 320 kDa, 330 kDa, 340 kDa, 350 kDa, 360 kDa, 370 kDa, 380
- ultrafiltration and diafiltration of the IVT mixture for purifying RNA may include (1) Direct Flow Filtration (DFF), also known as “dead-end” filtration, that applies a feed stream perpendicular to the membrane face and attempts to pass 100% of the fluid through the membrane, and/or (2) Tangential Flow Filtration (TFF), also known as crossflow filtration, where a feed stream passes parallel to the membrane face as one portion passes through the membrane (permeate) while the remainder (retentate) is retained and/or recirculated back to the feed tank.
- DFF Direct Flow Filtration
- TMF Tangential Flow Filtration
- the filtering of the IVT mixture is conducted via TFF comprising an ultrafiltration step, a first diafiltration step, and a second diafiltration step.
- the first diafiltration step is conducted in the presence of ammonium sulfate.
- the first diafiltration step may be configured to remove a majority of impurities from the IVT mixture.
- the second diafiltration step is conducted without ammonium sulfate.
- the second diafiltration step may be configured to transfer the RNA into a DS buffer formulation.
- a filtration membrane with an appropriate MWCO may be selected for ultrafiltration in the TFF process.
- the MWCO of a TFF membrane determines which solutes may pass through the membrane into the filtrate and which are retained in the retentate.
- the MWCO of a TFF membrane may be selected such that substantially all of the solutes of interest (e.g., desired synthesized RNA species) remain in the retentate, whereas undesired components (e.g., excess ribonucleotides, small nucleic acid fragments such as digested or hydrolyzed DNA template, peptide fragments such as digested proteins and/or other impurities) pass into the filtrate.
- the retentate comprising desired synthesized RNA species may be re-circulated to a feed reservoir to be re-filtered in additional cycles.
- a TFF membrane may have a MWCO of at least, at most, exactly, or between (inclusive or exclusive) any two of 30 kDa, 40 kDa, 50 kDa, 60 kDa, 70 kDa, 80 kDa, 90 kDa, or more. In some aspects, a TFF membrane may have a MWCO of at least, at most, exactly, or between (inclusive or exclusive) any two of 100 kDa, 150 kDa, 200 kDa, 250 kDa, 300 kDa, 350 kDa, 400 kDa, or more. In some aspects, a TFF membrane may have a MWCO of or of about 250-350 kDa.
- a TFF membrane e.g., a cellulose-based membrane
- a MWCO of or of about 30-300 kDa; 50-300 kDa, 100-300 kDa, or 200-300 kDa.
- Diafiltration may be performed either discontinuously, or alternatively, continuously.
- a diafiltration solution may be added to a sample feed reservoir at the same rate as filtrate is generated.
- the volume in the sample reservoir remains constant but small molecules (e.g., salts, solvents, etc.) that may freely permeate through a membrane are removed.
- each additional diafiltration volume (DV) reduces the solvent concentration further.
- discontinuous diafiltration a solution is first diluted and then concentrated back to the starting volume. This process is then repeated until the desired concentration of small molecules (e.g., salts, solvents, etc.) remaining in the reservoir is reached. Each additional diafiltration volume (DV) reduces the small molecule (e.g., solvent) concentration further.
- Continuous diafiltration typically requires a minimum volume for a given reduction of molecules to be filtered.
- Discontinuous diafiltration permits fast changes of the retentate condition, such as pH, salt content, and the like.
- the first diafiltration step is conducted with at least, at most, exactly, or between (inclusive or exclusive) any two of 2, 3, 4, 5, 6, 7, 8, 9, 10, or more diavolumes.
- the second diafiltration step is conducted with at least, at most, exactly, or between (inclusive or exclusive) any two of 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or more diavolumes.
- the first diafiltration step is conducted with 5 diavolumes
- second diafiltration step is conducted with 10 diavolumes.
- the IVT mixture is filtered at a rate of at least, at most, exactly, or between (inclusive or exclusive) any two of 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 500, 600, 700, 800, 900, or 1000 L/m 2 of filter area per hour, or more.
- the concentrated RNA solution may comprise at least, at most, exactly, or between (inclusive or exclusive) any two of 2.0, 2.1, 2.2, 2.3, 2.4, or 2.5 mg/mL single stranded RNA.
- the bioburden of the concentrated RNA solution via filtration to obtain an RNA product solution may also be reduced, in some aspects.
- the filtration for reducing bioburden may be conducted using one or more filters.
- the one or more filters may include a filter with a pore size that is or is not at least, at most, exactly, or between (inclusive or exclusive) any two of 0.2 ⁇ m, 0.45 ⁇ m, 0.65 ⁇ m, 0.8 ⁇ m, or any other pore size configured to remove bioburdens.
- reducing the bioburden may include draining a retentate tank containing retentate obtained from the ultrafiltration and/or diafiltration to obtain the retentate.
- Reducing the bioburden may include flushing a filtration system for ultrafiltration and/or diafiltration using a wash buffer solution to obtain a wash pool solution comprising residue RNA remaining in the filtration system.
- the retentate may be filtered to obtain a filtered retentate.
- the wash pool solution may be filtered using a first 0.2 ⁇ m filter to obtain a filtered wash pool solution.
- the retentate may be filtered using the first 0.2 ⁇ m filter or another 0.2 ⁇ m filter.
- the filtered wash pool solution and the filtered retentate may be combined to form a combined pool solution.
- the combined pool solution may be filtered using a second 0.2 ⁇ m filter to obtain a filtered combined pool solution, which is further filtered using a third 0.2 ⁇ m filter to produce an RNA product solution.
- a quality assurance and/or quality control check may be conducted using methods such as, but not limited to, gel electrophoresis, UV absorbance, and/or analytical HPLC.
- the nucleic acids may be sequenced by methods including, but not limited to reverse-transcriptase-PCR.
- the nucleic acid may be quantified using methods such as, but not limited to, ultraviolet visible spectroscopy (UV/Vis).
- a non-limiting example of a UV/Vis spectrometer is a NANODROP® spectrometer (ThermoFisher, Waltham, MA).
- the quantified nucleic acid may be analyzed in order to determine if the nucleic acid may be of proper size and/or to assess degradation.
- RNA ENCAPSULATION The RNA in an RNA product solution may be encapsulated, and the RNA solution may further comprise at least one encapsulating agent.
- the encapsulating agent comprises a lipid, a lipid nanoparticle (LNP), lipoplexes, polymeric particles, polyplexes, monolithic delivery systems, or a combination thereof. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing elements may be excluded as an encapsulating agent.
- the encapsulating agent is a lipid, and produced is lipid nanoparticle (LNP)- encapsulated RNA.
- a lipid may be a naturally occurring lipid or a synthetic lipid.
- a lipid is usually a biological substance.
- Biological lipids are well known in the art, and include for example, neutral fats, phospholipids, phosphoglycerides, steroids, terpenes, lysolipids, glycosphingolipids, glucolipids, sulphatides, lipids with ether and ester-linked fatty acids and polymerizable lipids, and combinations thereof.
- a lipid is a substance that is insoluble in water and extractable with an organic solvent. Compounds other than those specifically described herein are understood by one of skill in the art as lipids and are encompassed by the compositions and methods of the present disclosure.
- LNPs may be designed to protect RNA molecules (e.g., saRNA, mRNA) from extracellular Rnases and/or may be engineered for systemic delivery of the RNA to target cells.
- RNA molecules e.g., saRNA, mRNA
- such LNPs may be particularly useful to deliver RNA molecules (e.g., saRNA, mRNA) when RNA molecules are intravenously administered to a subject in need thereof.
- RNA molecules e.g., saRNA, mRNA
- RNA molecules e.g., saRNA, mRNA
- such LNPs may be particularly useful to deliver RNA molecules (e.g., saRNA, mRNA) when RNA molecules are intradermally administered to a subject in need thereof.
- such LNPs may be particularly useful to deliver RNA molecules (e.g., saRNA, mRNA) when RNA molecules are intranasally administered to a subject in need thereof.
- the RNA in the RNA product solution is at a concentration of ⁇ 1 mg/mL.
- the RNA is at a concentration of at least or at least about 0.05 mg/mL.
- the RNA is at a concentration of at least or at least about 0.5 mg/mL.
- the RNA is at a concentration of at least or at least about 1 mg/mL. In another aspect, the RNA concentration is from or from about 0.05 mg/mL to about 0.5 mg/mL. In another aspect, the RNA is at a concentration of at least 10 mg/mL. In another aspect, the RNA is at a concentration of at least 50 mg/mL.
- the RNA is or is not at a concentration of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 0.05 mg/mL, 0.5 mg/mL, 1 mg/mL, 10 mg/mL, 50 mg/mL, 75 mg/mL, 100 mg/mL, 150 mg/mL, 200 mg/mL, 250 mg/mL, 300 mg/mL, 400 mg/mL, or more.
- RNA product solution and a lipid preparation mixture or compositions thereof comprising at least one RNA encoding, e.g., an antigen (e.g., an RSV prefusion F protein) complexed with, encapsulated in, and/or formulated with one or more lipids, and forming lipid nanoparticles (LNPs), liposomes, lipoplexes and/or nanoliposomes.
- an antigen e.g., an RSV prefusion F protein
- LNPs lipid nanoparticles
- the composition comprises a lipid nanoparticle.
- a lipid nanoparticle or LNP refers to particles of any morphology generated when a cationic lipid and optionally one or more further lipids are combined, e.g., in an aqueous environment and/or in the presence of RNA.
- lipid nanoparticles are included in a formulation that may be used to deliver an active agent or therapeutic agent, such as a nucleic acid (e.g., mRNA) to a target site of interest (e.g., cell, tissue, organ, tumor, and the like).
- a nucleic acid e.g., mRNA
- the lipid nanoparticles of the present disclosure comprise a nucleic acid (e.g., mRNA).
- Such lipid nanoparticles typically comprise a cationic lipid and one or more excipients, e.g., one or more neutral lipids, charged lipids, steroids, polymer conjugated lipids, or combinations thereof.
- the LNPs comprise at least one cationic (e.g., ionizable) lipid, at least one neutral (e.g., non-cationic) lipid, at least one structural lipid (e.g., a steroid), and/or at least one polymer conjugated lipid (e.g., a polyethylene glycol (PEG)-modified lipid).
- a polyethylene glycol (PEG)-modified lipid e.g., 1, 2, 3, or more of the foregoing excipients may be excluded from the LNPs.
- the LNPs comprise 20-60 mol% cationic (e.g., ionizable) lipid(s).
- the LNPs may comprise 20-50 mol%, 20-40 mol%, 20-30 mol%, 30-60 mol%, 30-50 mol%, 30-40 mol%, 40-60 mol%, 40-50 mol%, or 50-60 mol% cationic (e.g., ionizable) lipid(s).
- the LNPs comprise or do not comprise at least, at most, exactly, or between (inclusive or exclusive) any two of 20 mol%, 30 mol%, 40 mol%, 50, or 60 mol% cationic (e.g., ionizable) lipid(s).
- the LNPs comprise 45 to 55 mole percent (mol%) cationic (e.g., ionizable) lipid(s).
- LNPs may comprise or not comprise at least, at most, exactly, or between (inclusive or exclusive) any two of 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, or 55 mol% cationic (e.g., ionizable) lipid(s).
- the LNPs comprise 5-25 mol% neutral (e.g., non-cationic) lipid(s).
- the LNPs may comprise 5-20 mol%, 5-15 mol%, 5-10 mol%, 10-25 mol%, 10-20 mol%, 10-25 mol%, 15-25 mol%, 15-20 mol%, or 20-25 mol% neutral (e.g., non-cationic) lipid(s).
- the LNPs are or are not at least, at most, exactly, or between (inclusive or exclusive) any two of 5 mol%, 10 mol%, 15 mol%, 20 mol%, or 25 mol% neutral (e.g., non- cationic) lipid(s).
- the LNPs comprise 5 to 15 mol% neutral (e.g., non-cationic) lipid(s).
- LNPs may comprise at least, at most, exactly, or between (inclusive or exclusive) any two of 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 mol% neutral (e.g., non-cationic) lipid(s).
- the LNPs comprise 25-55 mol% structural lipid(s) (e.g., a steroid).
- the LNPs may comprise 25-50 mol%, 25-45 mol%, 25-40 mol%, 25-35 mol%, 25-30 mol%, 30-55 mol%, 30-50 mol%, 30-45 mol%, 30-40 mol%, 30-35 mol%, 35-55 mol%, 35-50 mol%, 35-45 mol%, 35-40 mol%, 40-55 mol%, 40-50 mol%, 40-45 mol%, 45-55 mol%, 45-50 mol%, or 50-55 mol% structural lipid(s) (e.g., a steroid).
- structural lipid(s) e.g., a steroid
- the LNPs are or are not at least, at most, exactly, or between (inclusive or exclusive) any two of 25 mol%, 30 mol%, 35 mol%, 40 mol%, 45 mol%, 50 mol%, or 55 mol% structural lipid(s) (e.g., a steroid).
- the LNPs comprise 35 to 40 mol% structural lipid(s) (e.g., a steroid).
- LNPs may comprise at least, at most, exactly, or between (inclusive or exclusive) any two of 35, 36, 37, 38, 39, or 40 mol% structural lipid(s) (e.g., a steroid).
- the LNPs comprise 0.5-15 mol% polymer conjugated lipid(s) (e.g., a polyethylene glycol (PEG)-modified lipid).
- the lipid nanoparticle may comprise 0.5- 10 mol%, 0.5-5 mol%, 1-15 mol%, 1-10 mol%, 1-5 mol%, 2-15 mol%, 2-10 mol%, 2-5 mol%, 5- 15 mol%, 5-10 mol%, or 10-15 mol% polymer conjugated lipid(s) (e.g., a polyethylene glycol (PEG)-modified lipid).
- the lipid LNPs are or are not at least, at most, exactly, or between (inclusive or exclusive) any two of 0.5 mol%, 1 mol%, 2 mol%, 3 mol%, 4 mol%, 5 mol%, 6 mol%, 7 mol%, 8 mol%, 9 mol%, 10 mol%, 11 mol%, 12 mol%, 13 mol%, 14 mol%, or 15 mol% polymer conjugated lipid(s) (e.g., a polyethylene glycol (PEG)-modified lipid).
- the LNPs comprise 1 to 2 mol% polymer conjugated lipid(s) (e.g., a polyethylene glycol (PEG)- modified lipid).
- LNPs may comprise at least, at most, exactly, or between (inclusive or exclusive) any two of 1, 1.5, or 2 mol% polymer conjugated lipid(s) (e.g., a polyethylene glycol (PEG)-modified lipid).
- PEG polyethylene glycol
- the LNPs comprise 20-75 mol% cationic (e.g., ionizable) lipid(s) (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, and 75%), 0.5-25 mol% neutral (e.g., non-cationic) lipid(s) (e.g., at least, at most, exactly, or between (inclusive or exclusive) of 0.5%, 2.25%, 4%, 5.75%, 7.5%, 9.25%, 11%, 12.75%, 14.5%, 16.25%, 18%, 19.75%, 21.5%, 23.25%, and 25%), 5-55 mol% structural lipid(s) (e.g., a sterol) e.g., non-cationic) lipid(s) (e.g., at least, at most, exactly, or between (inclusive or exclusive) of 5%, 10%
- the molar lipid ratio is 50/10/38.5/1.5 (mol% cationic lipid/neutral lipid/structural lipid/polymer conjugated lipid), 60/7.5/31/1.5 (mol% cationic lipid/neutral lipid/structural lipid/polymer conjugated lipid), 57.5/7.5/31.5/3.5 (mol% cationic lipid/neutral lipid/structural lipid/polymer conjugated lipid), 57.2/7.1/34.3/1.4 (mol% cationic lipid/neutral lipid/structural lipid/polymer conjugated lipid), 40/15/40/5 (mol% cationic lipid/neutral lipid/structural lipid/polymer conjugated lipid), 50/10/35/4.5/0.5 (mol% cationic lipid/neutral lipid/structural lipid/polymer conjugated lipid), 50/10/35/5 (mol% cationic lipid/35/5 (mol% cationic lipid/cationic lipid/neutral lipid/structural lipid/polymer
- the active agent or therapeutic agent such as a nucleic acid (e.g., mRNA)
- a nucleic acid e.g., mRNA
- the active agent or therapeutic agent may be encapsulated in the lipid portion of the lipid nanoparticle and/or an aqueous space enveloped by some or all of the lipid portion of the lipid nanoparticle, thereby protecting it from enzymatic degradation or other undesirable effects induced by the mechanisms of the host organism or cells, e.g., an adverse immune response.
- the nucleic acid (e.g., mRNA) or a portion thereof may also be associated and complexed with the lipid nanoparticle.
- a lipid nanoparticle may comprise any lipid capable of forming a particle to which the nucleic acids are attached, and/or in which the one or more nucleic acids are encapsulated.
- provided RNA molecules e.g., saRNA, mRNA
- LNPs LNPs
- the lipid nanoparticles may or may not have a mean diameter of or of about 1 to 500 nm (e.g., at least, at most, exactly, or between (inclusive or exclusive) of 1, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490, or 500 nm).
- 1 to 500 nm e.g., at least, at most, exactly, or between (inclusive or exclusive) of 1, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220,
- the lipid nanoparticles have a mean diameter of or of from about 30 nm to about 150 nm, about 40 nm to about 150 nm, about 50 nm to about 150 nm, about 60 nm to about 130 nm, about 70 nm to about 110 nm, about 70 nm to about 100 nm, about 80 nm to about 100 nm, about 90 nm to about 100 nm, about 70 to about 90 nm, about 80 nm to about 90 nm, about 70 nm to about 80 nm, or at least, at most, exactly, or between (inclusive or exclusive) of 30 nm, 35 nm, 40 nm, 45 nm, 50 nm, 55 nm, 60 nm, 65 nm, 70 nm, 75 nm, 80 nm, 85 nm, 90 nm, 95 nm, 100 nm, 105 nm, 110 nm, 115
- mean diameter refers to the mean hydrodynamic diameter of particles as measured by dynamic laser light scattering (DLS) with data analysis using the so-called cumulant algorithm, which provides as results the so-called Z-average with the dimension of a length, and the polydispersity index (PI), which is dimensionless (Koppel, D., J. Chem. Phys.57, 1972, pp 4814- 4820, ISO 13321).
- PI polydispersity index
- “mean diameter,” “diameter,” or “size” for particles is used synonymously with the value of the Z-average.
- LNPs described herein may exhibit a polydispersity index less than or less than about 0.5, 0.4, 0.3, or 0.2 or less.
- the LNPs may or may not exhibit a polydispersity index of at least, at most, exactly, or between (inclusive or exclusive) of 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, 0.3, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.4, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, or 0.5.
- the polydispersity index is, in some aspects, calculated based on dynamic light scattering measurements by the so-called cumulant analysis referred to in the definition of “average diameter.” Under certain prerequisites, it may be taken as a measure of the size distribution of an ensemble of nanoparticles.
- an LNP of the disclosure comprises or does not comprise an N:P ratio of or of from about 2:1 to about 30:1, e.g., at least, at most, exactly, or between (inclusive or exclusive) of 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 11:1, 12:1, 13:1, 14:1, 15:1, 16:1, 17:1, 18:1, 19:1, 20:1, 21:1, 22:1, 23:1, 24:1, 25:1, 26:1, 27:1, 28:1, 29:1, or 30:1.
- an LNP of the disclosure comprises an N:P ratio of or of about 6:1.
- an LNP of the disclosure comprises an N:P ratio of or of about 3:1.
- an LNP of the disclosure comprises or does not comprise a wt/wt ratio of the cationic lipid component to the RNA of or of from about 5:1 to about 100:1, e.g., at least, at most, exactly, or between (inclusive or exclusive) of 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 11:1, 12:1, 13:1, 14:1, 15:1, 16:1, 17:1, 18:1, 19:1, 20:1, 21:1, 22:1, 23:1, 24:1, 25:1, 26:1, 27:1, 28:1, 29:1, 30:1, 31:1, 32:1, 33:1, 34:1, 35:1, 36:1, 37:1, 38:1, 39:1, 40:1, 41:1, 42:1, 43:1, 44:1, 45:1, 46:1, 47:1, 48:1, 49:1, 50:1, 51:1, 52:1, 53:1, 54:1, 55:1, 56:1, 57:1, 58:1, 59:1, 60:1, 61:1, 62:1, 63:1,
- an LNP of the disclosure comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of or of about 20:1. In some aspects, an LNP of the disclosure comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of or of about 10:1.
- nucleic acids e.g., RNA molecules
- LNPs are resistant in aqueous solution to degradation with a nuclease.
- LNPs are liver- targeting lipid nanoparticles. In some aspects, LNPs are cationic lipid nanoparticles comprising one or more cationic lipids (e.g., those described herein).
- cationic LNPs may comprise at least one cationic lipid, at least one polymer conjugated lipid, and at least one helper lipid (e.g., at least one neutral lipid).
- the RNA solution and lipid preparation mixture or compositions thereof may have at least, at most, exactly, between (inclusive or exclusive) of, or about 1%, 2%, 3%, 4% 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24%, 25%, 26%, 27%, 28%, 29%, 30%, 31%, 32%, 33%, 34%, 35%, 36%, 37%, 38%, 39%, 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%
- LNPs described herein can be generated using components, compositions, and methods as are generally known in the art, see, , e.g., PCT/US2016/052352; PCT/US2016/068300; PCT/US2017/037551; PCT/US2015/027400; PCT/US2016/047406; PCT/US2016000129; PCT/US2016/014280; PCT/US2016/014280; PCT/US2017/038426; PCT/US2014/027077; PCT/US2014/055394; PCT/US2016/52117; PCT/US2012/069610; PCT/US2017/027492; PCT/US2016/059575 and PCT/US2016/069491 all of which are incorporated by reference herein in their entirety.
- methods of preparing LNPs may involve obtaining a colloid from at least one cationic or cationically ionizable lipid or lipid-like material and/or at least one cationic polymer and mixing the colloid with nucleic acid to obtain nucleic acid particles.
- the term “colloid” as used herein relates to a type of homogeneous mixture in which dispersed particles do not settle out. The insoluble particles in the mixture are microscopic, with particle sizes between 1 and 1000 nanometers.
- the mixture may be termed a colloid or a colloidal suspension.
- colloids comprising at least one cationic or cationically ionizable lipid or lipid-like material and/or at least one cationic polymer
- methods are applicable herein that are conventionally used for preparing liposomal vesicles and are appropriately adapted.
- the most commonly used methods for preparing liposomal vesicles share the following fundamental stages: (i) lipids dissolution in organic solvents, (ii) drying of the resultant solution, and (iii) hydration of dried lipid (using various aqueous media).
- lipids are first dissolved in a suitable organic solvent and dried down to yield a thin film at the bottom of the flask.
- the obtained lipid film is hydrated using an appropriate aqueous medium to produce a liposomal dispersion.
- an additional downsizing step may be included.
- Reverse phase evaporation is an alternative method to film hydration for preparing liposomal vesicles that involves formation of a water-in-oil emulsion between an aqueous phase and an organic phase containing lipids. A brief sonication of this mixture is required for system homogenization. The removal of the organic phase under reduced pressure yields a milky gel that subsequently turns into a liposomal suspension.
- ethanol injection technique refers to a process in which an ethanol solution comprising lipids is rapidly injected into an aqueous solution through a needle. This action disperses the lipids throughout the solution and promotes lipid structure formation, for example, lipid vesicle formation such as liposome formation.
- RNA lipoplex particles described herein are obtainable by adding RNA to a colloidal liposome dispersion.
- colloidal liposome dispersion is, in some aspects, formed as follows: an ethanol solution comprising lipids, such as cationic lipids and additional lipids, is injected into an aqueous solution under stirring.
- the RNA lipoplex particles described herein are obtainable without a step of extrusion.
- the term “extruding” or “extrusion” refers to the creation of particles having a fixed, cross-sectional profile. In particular, it refers to the downsizing of a particle, whereby the particle is forced through filters with defined pores. Other methods for preparing a colloid having organic solvent free characteristics may also be used according to the present disclosure.
- LNP-encapsulated RNA may be produced by rapid mixing of an RNA solution described herein (e.g., the RNA product solution) and a lipid preparation described herein (comprising, e.g., at least one cationic lipid and optionally one or more other lipid components, in an organic solvent) under conditions such that a sudden change in solubility of lipid component(s) is triggered, which drives the lipids towards self-assembly in the form of LNPs.
- suitable buffering agents comprise tris, histidine, citrate, acetate, phosphate, and/or succinate. In some aspects, 1, 2, 3, or more of the foregoing buffering agents are excluded.
- the pH of a liquid formulation relates to the pKa of the encapsulating agent (e.g., cationic lipid).
- the pH of the acidifying buffer may be at least half a pH scale less than the pKa of the encapsulating agent (e.g., cationic lipid), and the pH of the final buffer may be at least half a pH scale greater than the pKa of the encapsulating agent (e.g., cationic lipid).
- properties of a cationic lipid are chosen such that nascent formation of particles occurs by association with an oppositely charged backbone of a nucleic acid (e.g., RNA).
- nucleic acids when present in the lipid nanoparticles, are resistant in aqueous solution to degradation with a nuclease.
- Lipid nanoparticles comprising nucleic acids and their method of preparation are disclosed in, e.g., U.S. Patent Publication Nos. 2004/0142025, 2007/0042031 and PCT Pub. Nos. WO 2013/016058 and WO 2013/086373, the full disclosures of which are herein incorporated by reference in their entirety for all purposes.
- each nucleic acid species is separately formulated as an individual LNP formulation.
- each individual LNP formulation will comprise one nucleic acid species.
- the individual LNP formulations may be present as separate entities, e.g., in separate containers.
- Such formulations are obtainable by providing each nucleic acid species separately (typically each in the form of a nucleic acid-containing solution) together with suitable cationic or cationically ionizable lipids or lipid-like materials and cationic polymers that allow the formation of LNPs.
- Respective particles will contain exclusively the specific nucleic acid species that is being provided when the particles are formed (individual particulate formulations).
- a composition such as a pharmaceutical composition comprises more than one individual LNP formulation.
- Respective pharmaceutical compositions are referred to as mixed LNP formulations.
- Mixed LNP formulations according to the invention are obtainable by forming, separately, individual LNP formulations, as described above, followed by a step of mixing of the individual LNP formulations. By the step of mixing, a formulation comprising a mixed population of nucleic acid-containing LNPs is obtainable. Individual LNP populations may be together in one container, comprising a mixed population of individual LNP formulations. Alternatively, it is possible that different nucleic acid species are formulated together as a combined LNP formulation.
- Such formulations are obtainable by providing a combined formulation (typically combined solution) of different RNA species together with suitable cationic or cationically ionizable lipids or lipid-like materials and cationic polymers that allow the formation of LNPs.
- a combined LNP formulation will typically comprise LNPs that comprise more than one RNA species.
- different RNA species are typically present together in a single particle.
- A. CATIONIC POLYMERIC MATERIALS Given their high degree of chemical flexibility, polymeric materials are commonly used for nanoparticle-based delivery. Typically, cationic materials are used to electrostatically condense the negatively charged nucleic acid into nanoparticles.
- a “polymeric material,” as used herein, is given its ordinary meaning, e.g., a molecular structure comprising one or more repeat units (monomers), connected by covalent bonds. In some aspects, such repeat units may all be identical; alternatively, in some cases, there may be more than one type of repeat unit present within the polymeric material.
- a polymeric material is biologically derived, e.g., a biopolymer such as a protein.
- additional moieties may also be present in the polymeric material, for example targeting moieties such as those described herein.
- a polymer (or polymeric moiety) utilized in accordance with the present disclosure may be a copolymer. Repeat units forming the copolymer may be arranged in any fashion.
- repeat units may be arranged in a random order; alternatively or additionally, in some aspects, repeat units may be arranged in an alternating order, or as a “block” copolymer, e.g., comprising one or more regions each comprising a first repeat unit (e.g., a first block), and one or more regions each comprising a second repeat unit (e.g., a second block), etc.
- Block copolymers may have two (a diblock copolymer), three (a triblock copolymer), or more numbers of distinct blocks.
- a polymeric material for use in accordance with the present disclosure is biocompatible. Biocompatible materials are those that typically do not result in significant cell death at moderate concentrations.
- a biocompatible material is biodegradable, e.g., is able to degrade, chemically and/or biologically, within a physiological environment, such as within the body.
- a polymeric material may be or comprise protamine or polyalkyleneimine, in particular protamine.
- protamine is often used to refer to any of various strongly basic proteins of relatively low molecular weight that are rich in arginine and are found associated especially with DNA in place of somatic histones in the sperm cells of various animals (as fish).
- protamine is often used to refer to proteins found in fish sperm that are strongly basic, are soluble in water, are not coagulated by heat, and yield chiefly arginine upon hydrolysis. In purified form, they are used in a long-acting formulation of insulin and to neutralize the anticoagulant effects of heparin.
- protamine as used herein is refers to a protamine amino acid sequence obtained or derived from natural or biological sources, including fragments thereof and/or multimeric forms of said amino acid sequence or fragment thereof, as well as (synthesized) polypeptides which are artificial and specifically designed for specific purposes and cannot be isolated from native or biological sources.
- a polyalkyleneimine comprises polyethylenimine and/or polypropylenimine.
- the polyalkyleneimine is polyethyleneimine (PEI).
- the polyalkyleneimine is a linear polyalkyleneimine, e.g., linear polyethyleneimine (PEI).
- Cationic materials e.g., polymeric materials, including polycationic polymers
- contemplated for use herein include those which are able to electrostatically bind nucleic acid.
- cationic polymeric materials contemplated for use herein include any cationic polymeric materials with which nucleic acid may be associated, e.g.
- particles described herein may comprise polymers other than cationic polymers, e.g., non-cationic polymeric materials and/or anionic polymeric materials. Collectively, anionic and neutral polymeric materials are referred to herein as non-cationic polymeric materials.
- lipid and “lipid-like material” are used herein to refer to molecules which comprise one or more hydrophobic moieties or groups and optionally also one or more hydrophilic moieties or groups.
- lipids and lipid-like materials may be cationic, anionic or neutral.
- Neutral lipids or lipid-like materials exist in an uncharged or neutral zwitterionic form at a selected pH.
- the term “lipid” refers to a group of organic compounds that are characterized by being insoluble in water but soluble in many organic solvents.
- lipids may be divided into eight categories: fatty acids and their derivatives (including tri-, di-, monoglycerides, and phospholipids), glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, polyketides, sterol lipids as well as sterol-containing metabolites such as cholesterol, and prenol lipids.
- fatty acids include, but are not limited to, fatty esters and fatty amides.
- glycerolipids include, but are not limited to, glycosylglycerols and glycerophospholipids (e.g., phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine).
- sphingolipids include, but are not limited to, ceramides phosphosphingolipids (e.g., sphingomyelins, phosphocholine), and glycosphingolipids (e.g., cerebrosides, gangliosides).
- sterol lipids include, but are not limited to, cholesterol and its derivatives and tocopherol and its derivatives.
- lipid-like material lipid-like compound
- lipid-like molecule relates to substances that structurally and/or functionally relate to lipids but may not be considered as lipids in a strict sense.
- the term includes compounds that are able to form amphiphilic layers as they are present in vesicles, multilamellar/unilamellar liposomes, or membranes in an aqueous environment and includes surfactants, or synthesized compounds with both hydrophilic and hydrophobic moieties.
- the term refers to molecules, which comprise hydrophilic and hydrophobic moieties with different structural organization, which may or may not be similar to that of lipids.
- the RNA solution and lipid preparation mixture or compositions thereof may comprise cationic lipids, neutral lipids, cholesterol, and/or polymer (e.g., polyethylene glycol) conjugated lipids which form lipid nanoparticles that encompass the RNA molecules.
- the LNP may comprise a cationic lipid and one or more excipients, e.g., one or more neutral lipids, charged lipids, steroids or steroid analogs (e.g., cholesterol), polymer conjugated lipids (e.g.
- the lipids are present in a composition in an amount that is effective to form a lipid nanoparticle and deliver a therapeutic agent, e.g., an RNA molecule, for treating a particular disease or condition of interest, e.g., those related to RSV.
- a therapeutic agent e.g., an RNA molecule
- the LNPs encompass, or encapsulate, the nucleic acid molecules.
- Cationic or cationically ionizable lipids or lipid-like materials refer to a lipid or lipid-like material capable of being positively charged and able to electrostatically bind nucleic acid.
- a “cationic lipid” or “cationic lipid-like material” refers to a lipid or lipid-like material having a net positive charge.
- Cationic lipids or lipid-like materials bind negatively charged nucleic acid by electrostatic interaction.
- cationic lipids possess a lipophilic moiety, such as a sterol, an acyl chain, a diacyl, or more acyl chains, and the head group of the lipid typically carries the positive charge.
- Exemplary cationic lipids include one or more amine group(s) which bear the positive charge.
- Cationic lipids may encapsulate negatively charged RNA.
- cationic lipids are ionizable such that they may exist in a positively charged or neutral form depending on pH. The ionization of the cationic lipid affects the surface charge of the lipid nanoparticle under different pH conditions. Without wishing to be bound by theory, this ionizable behavior is thought to enhance efficacy through helping with endosomal escape and reducing toxicity as compared with particles that remain cationic at physiological pH.
- a cationic lipid may comprise from or from about 10 mol % to about 100 mol %, about 20 mol % to about 100 mol %, about 30 mol % to about 100 mol %, about 40 mol % to about 100 mol %, or about 50 mol % to about 100 mol % of the total lipid present in the particle.
- a cationic lipid may or may not be at least, at most, exactly, or between (inclusive or exclusive) of 10 mol %, 20 mol %, 30 mol %, 40 mol %, 50 mol %, 60 mol %, 70 mol %, 80 mol %, 90 mol %, or 100 mol %, or any range or value derivable therein, of the total lipid present in the particle.
- cationic lipids include, but are not limited to: ((4- hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate), 1,2-dioleoyl-3- trimethylammonium propane (DOTAP), N,N-dimethyl-2,3-dioleyloxypropylamine (DODMA), 1,2- di-O-octadecenyl-3-trimethylammonium propane (DOTMA), 3-(N-( N′,N′-dimethylaminoethane)- carbamoyl)cholesterol (DC-Chol), dimethyldioctadecylammonium (DDAB), 1,2-dioleoyl-3- dimethylammonium-propane (DODAP), 1,2-diacyloxy-3-dimethylammonium propanes, 1,2- dialkyloxy-3-dimethylammonium propanes; dioct
- an ionizable cationic lipid of the disclosure comprises a compound of Formula (I): or a pharmaceutically acceptable salt, tautomer, prodrug, or stereoisomer thereof, wherein: R1 is a C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, or -R”M’R’; R2 and R3 are independently a H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, or -R*OR”, and/or R 2 and R 3 , together with the atom to which they are attached, form a heterocycle or carbocycle; R 4 is a C 3-6 carbocycle, -(CH 2 ) n Q, -(CH 2 ) n CHQR, -CHQR, -CQ(
- a subset of compounds of Formula (I) includes those in which when R4 is -(CH 2 ) n Q, -(CH 2 ) n CHQR, -CHQR, or -CQ(R) 2 , then (i) Q is not -N(R) 2 when n is 1, 2, 3, 4, or 5, or (ii) Q is not 5, 6, or 7-membered heterocycloalkyl when n is 1 or 2.
- R1 is a C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, or -R”M’R’;
- R2 and R3 are independently an H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, or -R*OR”, and/or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle;
- R 4 is a C 3-6 carbocycle, -(CH 2 ) n Q, -(CH 2 ) n CHQR, -CHQR, -CQ(R) 2 , or unsubstituted C 1-6 alkyl, where Q is a C 3-6 carbocycle, a 5- to 14-membered heteroaryl having one or more heteroatoms comprising N, O, or S, -OR, -O(CH2)nN(R)2,
- R1 is a C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, or -R”M’R’;
- R2 and R3 are independently an H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, or -R*OR”, and/or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle;
- R 4 is a C 3-6 carbocycle, -(CH 2 ) n Q, -(CH 2 ) n CHQR, -CHQR, -CQ(R) 2 , or unsubstituted C 1-6 alkyl, where Q is a C 3-6 carbocycle, a 5- to 14-membered heterocycle having one or more heteroatoms comprising N, O, or S, -OR, -O(CH2)nN(R)2,
- R1 is a C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, or -R”M’R’;
- R2 and R3 are independently an H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, or -R*OR”, and/or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle;
- R 4 is a C 3-6 carbocycle, -(CH 2 ) n Q, -(CH 2 ) n CHQR, -CHQR, -CQ(R) 2 , or unsubstituted C 1-6 alkyl, where Q is a C3-6 carbocycle, a 5- to 14-membered heteroaryl having one or more heteroatoms comprising N, O, or S, -OR, -O(CH2)nN(R)2,
- R 1 is a C 5-30 alkyl, C 5-20 alkenyl, -R*YR”, -YR”, or -R”M’R’;
- R 2 and R 3 are independently an H, C 2-14 alkyl, C 2-14 alkenyl, -R*YR”, -YR”, or -R*OR”, and/or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle;
- R4 is -(CH2)nQ or -(CH2)nCHQR, where Q is -N(R)2, and/or n is 3, 4, or 5;
- each R 5 is independently a C 1-3 alkyl, C 2-3 alkenyl, or H;
- each R 6 is independently a C 1-3 alkyl, C 2-3 alkenyl, or H;
- M and M’ are independently a -C(O)O-, -OC(O)-, -C(O)
- R 1 is a C 5-30 alkyl, C 5-20 alkenyl, -R*YR”, -YR”, or -R”M’R’;
- R 2 and R 3 are independently a C 1-14 alkyl, C 2-14 alkenyl, -R*YR”, -YR”, or -R*OR”, and/or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle;
- R4 is a -(CH2)nQ, -(CH2)nCHQR, -CHQR, or -CQ(R)2, where Q is -N(R)2, and/or n is 1, 2, 3, 4, or 5;
- each R5 is independently a C1-3 alkyl, C2-3 alkenyl, or H;
- each R 6 is independently a C 1-3 alkyl, C 2-3 alkenyl, or H;
- M and M' are independently a -C(O
- a subset of compounds of Formula (I) includes those of Formula (Ila), (lIb), (lIc), or (lIe): or a pharmaceutically acceptable salt, tautomer, prodrug, or stereoisomer thereof, wherein R4 is as described herein.
- a subset of compounds of Formula (I) includes those of Formula (IId): or a pharmaceutically acceptable salt, tautomer, prodrug, or stereoisomer thereof, wherein n is 2, 3, or 4; and m, R’, R”, and R2 through R6 are as described herein.
- each of R2 and R3 may be independently a C5-14 alkyl or C5-14 alkenyl.
- the ionizable cationic lipid comprises a compound having one of the following structures: or a pharmaceutically acceptable salt, tautomer, prodrug, or stereoisomer thereof, wherein: A is a 3 to 8-membered cycloalkyl or cycloalkylene ring; R 6 is, at each occurrence, independently H, OH or C1-C24 alkyl; and n is an integer ranging from 1 to 15.
- the ionizable cationic lipid comprises a compound having one of the following structures: or a pharmaceutically acceptable salt, tautomer, prodrug, or stereoisomer thereof, wherein y and z are each independently integers ranging from 1 to 12.
- one of L 1 or L 2 is -OCCO)-.
- the ionizable cationic lipid comprises a compound having one of the following structures: or a pharmaceutically acceptable salt, tautomer, prodrug, or stereoisomer thereof. In some of the foregoing aspects, the ionizable cationic lipid comprises a compound having one of the following structures: Or a pharmaceutically acceptable salt, tautomer, prodrug, or stereoisomer thereof.
- n is an integer ranging from 2 to 12, for example from 2 to 8 or from 2 to 4. For example, in some aspects, n is 3, 4, 5, or 6. In some aspects, n is 3. In some aspects, n is 4. In some aspects, n is 5. In some aspects, n is 6.
- y and z are each independently an integer ranging from 2 to 10.
- y and z are each independently an integer ranging from 4 to 9 or from 4 to 6.
- R 6 is H.
- R 6 is C 1 -C 24 alkyl.
- R 6 is OH.
- G is unsubstituted.
- G 3 is substituted.
- G 3 is linear C 1 -C 24 alkylene or linear C 1 - C24 alkenylene.
- R 1 or R 2 , or both, is C6-C24 alkenyl.
- R 1 and R 2 each, independently have the following structure: wherein: R 7a and R 7b are, at each occurrence, independently H or C 1 -C 12 alkyl; and a is an integer from 2 to 12, wherein R 7a , R 7b , and a are each selected such that R 1 and R 2 each independently comprise from 6 to 20 carbon atoms.
- a is an integer ranging from 5 to 9 or from 8 to 12.
- at least one occurrence of R 7a is H.
- R 7a is H at each occurrence.
- at least one occurrence of R 7b is C 1 -C 8 alkyl.
- C 1 -C 8 alkyl is methyl, ethyl, n-propyl, iso-propyl, n- butyl, iso-butyl, tert-butyl, n-hexyl, or n-octyl.
- R 1 or R 2 has one of the following:
- R 4 is methyl or ethyl.
- any aspect of the compounds set forth above, and any specific substituent and/or variable in the compounds set forth above, may be independently combined with other aspects and/or substituents and/or variables of compounds to form aspects of the inventions not specifically set forth above.
- substituents and/or variables may be listed for any particular substituent and/or variable in a particular embodiment and/or claim, it is understood that each individual substituent and/or variable may be deleted from the particular aspect and/or claim and that the remaining list of substituents and/or variables will be considered to be within the scope of the disclosure. It is understood that in the present description, combinations of substituents and/or variables of the depicted formulae are permissible only if such contributions result in stable compounds.
- the cationic lipid is In some embodiments, the cationic lipid is In some aspects, the lipid nanoparticles comprise one or more cationic lipids. In one aspect, the lipid nanoparticles comprise (4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2- hexyldecanoate) (ALC-0315), having the formula: Exemplary cationic lipids are disclosed in, e.g., U.S.10,166,298, the full disclosures of which are herein incorporated by reference in their entirety for all purposes. Representative cationic lipids include:
- RNA-LNPs comprise a cationic lipid, a RNA molecule as described herein and one or more of neutral lipids, steroids, pegylated lipids, or combinations thereof. If more than one cationic lipid is incorporated within the LNP, such percentages apply to the combined cationic lipids.
- the cationic lipid is present in the LNP in an amount such as at least, at most, exactly, or between (inclusive or exclusive) of, or about 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59 or 60 mole percent (mol%).
- two or more cationic lipids are incorporated within the LNP. If more than one cationic lipid is incorporated within the LNP, the foregoing percentages apply to the combined cationic lipids.
- the LNP comprises a combination or mixture of any the lipids described above. ii.
- the LNPs comprise a polymer conjugated lipid.
- polymer conjugated lipid refers to a molecule comprising both a lipid portion and a polymer portion.
- An example of a polymer conjugated lipid is a pegylated lipid (e.g., polyethylene glycol-lipid, PEG- lipid).
- the LNP comprises an additional, stabilizing lipid that is a pegylated lipid.
- pegylated lipid refers to a molecule comprising both a lipid portion and a polyethylene glycol portion.
- Pegylated lipids include, but are not limited to, PEG-modified phosphatidylethanolamine, PEG-modified phosphatidic acid, PEG-modified ceramides (e.g., PEG-CerC14 or PEG-CerC20), PEG-modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkylglycerols, 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide, and mixtures thereof.
- PEG-modified phosphatidylethanolamine PEG-modified phosphatidic acid
- PEG-modified ceramides e.g., PEG-CerC14 or PEG-CerC20
- PEG-modified dialkylamines e.g., PEG-modified diacylglycerols
- PEG-modified dialkylglycerols 2-[(polyethylene glycol)
- polyethylene glycol-lipids include PEG-c-DOMG, PEG-c-DMA, PEG-DSG, PEG-DPG, and PEG-s-DMG (1-(monomethoxy-polyethyleneglycol)-2,3- dimyristoylglycerol).
- the polyethylene glycol-lipid is N-[(methoxy polyethylene glycol)2000)carbamoyl]-1,2-dimyristyloxlpropyl-3-amine (PEG-c-DMA).
- the polyethylene glycol-lipid is PEG-2000-DMG.
- the polyethylene glycol-lipid is PEG- c-DOMG.
- the LNPs comprise a PEGylated diacylglycerol (PEG-DAG) such as 1-(monomethoxy-polyethyleneglycol)-2,3-dimyristoylglycerol (PEG-DMG), a PEGylated phosphatidylethanolamine (PEG-PE), a PEG succinate diacylglycerol (PEG-S-DAG) such as 4- O-(2′,3′-di(tetradecanoyloxy)propyl-1-O-((O-methoxy(polyethoxy)ethyl)butanedioate (PEG-S- DMG), a PEGylated ceramide (PEG-cer), or a PEG dialkoxypropylcarbamate such as co- methoxy(polyethoxy)ethyl-N-(2,3di(tetradecanoxy)propyl)carbamate or 2,3- di(tetrade
- PEG-lipids are disclosed in, e.g., U.S.9,737,619, the full disclosures of which is herein incorporated by reference in its entirety for all purposes. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing pegylated lipids may be excluded from the LNPs of the present disclosure.
- the composition comprises a pegylated lipid having the following structure: or a pharmaceutically acceptable salt, tautomer, or stereoisomer thereof, wherein: R 8 and R 9 are each independently a straight or branched, saturated or unsaturated alkyl chain containing from 10 to 30 carbon atoms, wherein the alkyl chain is optionally interrupted by one or more ester bonds; and w has a mean value ranging from 30 to 60. In some aspects, R 8 and R 9 are each independently straight, saturated alkyl chains containing from 12 to 16 carbon atoms. In some aspects, w has a mean value ranging from 43 to 53. In other aspects, the average w is or is about 45. In other different embodiments, the average w is or is about 49.
- the lipid nanoparticles comprise a polymer conjugated lipid.
- the lipid nanoparticle comprises 2-[(polyethylene glycol)-2000]-N,N- ditetradecylacetamide (ALC-0159), having the formula:
- the molar ratio of the cationic lipid to the pegylated lipid ranges from or from about 100:1 to about 20:1, e.g., 20:1, 25:1, 30:1, 35:1, 40:1, 45:1, 50:1, 55:1, 60:1, 65:1, 70:1, 75:1, 80:1, 85:1, 90:1, 95:1, or 100:1, or any range or value derivable therein.
- the PEG-lipid is or is not present in the LNP in an amount from or from about 1 to about 10 mole percent (mol %) (e.g., at least, at most, exactly, or between (inclusive or exclusive) of 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 mol %), relative to the total lipid content of the nanoparticle.
- the ratio of PEG in the lipid nanoparticle formulations may be increased or decreased and/or the carbon chain length of the PEG lipid may be modified to alter the pharmacokinetics and/or biodistribution of the lipid nanoparticle formulations.
- the LNP comprises one or more additional lipids or lipid-like materials that stabilize particles during their formation.
- Suitable stabilizing or structural lipids include non- cationic lipids, e.g., neutral lipids and anionic lipids.
- optimizing the formulation of LNPs by addition of other hydrophobic moieties, such as cholesterol and lipids, in addition to an ionizable/cationic lipid or lipid-like material may enhance particle stability and efficacy of nucleic acid delivery.
- an “anionic lipid” refers to any lipid that is negatively charged at a selected pH.
- neutral lipid refers to any one of a number of lipid species that exist in either an uncharged or neutral zwitterionic form at physiological pH.
- additional lipids comprise one of the following neutral lipid components: (1) a phospholipid, (2) cholesterol or a derivative thereof; or (3) a mixture of a phospholipid and cholesterol or a derivative thereof.
- Representative neutral lipids include phosphatidylcholines, phosphatidylethanolamines, phosphatidylglycerols, phosphatidic acids, phosphatidylserines, ceramides, sphingomyelins, dihydro-sphingomyelins, cephalins, and cerebrosides.
- Exemplary phospholipids include, for example, phosphatidylcholines, e.g., diacylphosphatidylcholines, such as distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dimyristoylphosphatidylcholine (DMPC), dipentadecanoylphosphatidylcholine, dilauroylphosphatidylcholine, dipalmitoylphosphatidylcholine (DPPC), dioleoylphosphatidylglycerol (DOPG), dipalmitoylphosphatidylglycerol (DPPG), diarachidoylphosphatidylcholine (DAPC), dibehenoylphosphatidylcholine (DBPC), ditricosanoylphosphatidylcholine (DTPC), dilignoceroylphatidylcholine (DLPC), palmitoyloleoy
- 1, 2, 3, 4, 5, or more of the foregoing neutral lipids may be excluded from the LNPs of the present disclosure.
- the neutral lipid is 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), having the formula:
- the LNPs comprise a neutral lipid, and the neutral lipid comprises one or more of DSPC, DPPC, DMPC, DOPC, POPC, DOPE, and/or SM.
- 1, 2, 3, 4, 5, or more of the foregoing neutral lipids may be excluded from the LNPs of the present disclosure.
- the LNPs further comprise a steroid or steroid analogue.
- a “steroid” is a compound comprising the following carbon skeleton:
- the steroid or steroid analogue is cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, ursolic acid, alpha- tocopherol, and mixtures thereof.
- 1, 2, 3, 4, 5, or more of the foregoing steroid or steroid analogues may be excluded from the LNPs of the present disclosure.
- the steroid or steroid analogue is cholesterol.
- cholesterol derivatives include, but are not limited to, cholestanol, cholestanone, cholestenone, coprostanol, cholesteryl-2′-hydroxyethyl ether, cholesteryl-4′-hydroxybutyl ether, tocopherol and derivatives thereof, and mixtures thereof.
- 1, 2, 3, 4, 5, or more of the foregoing cholesterol derivatives may be excluded from the LNPs of the present disclosure.
- the cholesterol has the formula: Without being bound by any theory, the amount of the at least one cationic lipid compared to the amount of the at least one additional lipid may affect important nucleic acid particle characteristics, such as charge, particle size, stability, tissue selectivity, and bioactivity of the nucleic acid.
- the molar ratio of the cationic lipid to the neutral lipid ranges from or from about 2:1 to about 8:1, or from or from about 10:0 to about 1:9, about 4:1 to about 1:2, or about 3:1 to about 1:1.
- the non-cationic lipid e.g., neutral lipid (e.g., one or more phospholipids and/or cholesterol)
- the non-cationic lipid e.g., neutral lipid (e.g., one or more phospholipids and/or cholesterol)
- VI. CHARACTERIZATION AND ANALYSIS OF RNA MOLECULE The RNA molecule described herein may be analyzed and characterized using various methods. Analysis may be performed before and/or after capping.
- analysis may be performed before and/or after poly-A capture-based affinity purification.
- analysis may be performed before and/or after additional purification steps, e.g., anion exchange chromatography and the like.
- RNA template quality may be determined using a Bioanalyzer chip-based electrophoresis system.
- RNA template purity is analyzed using analytical reverse phase HPLC.
- Capping efficiency may be analyzed using, e.g., total nuclease digestion followed by MS/MS quantitation of the dinucleotide cap species vs. uncapped GTP species.
- In vitro efficacy may be analyzed by, e.g., transfecting an RNA molecule into a human cell line.
- Protein expression of the polypeptide of interest may be quantified using methods such as ELISA and/or flow cytometry.
- Immunogenicity may be analyzed by, e.g., transfecting RNA molecules into cell lines that indicate innate immune stimulation, e.g., PBMCs.
- Cytokine induction may be analyzed using, e.g., methods such as ELISA to quantify a cytokine, e.g., Interferon- ⁇ .
- Biodistribution may be analyzed by, e.g., bioluminescence measurements. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing analytic methods may be excluded.
- an RNA polynucleotide disclosed herein is characterized in that, when assessed in an organism administered a composition or medical preparation comprising an RNA polynucleotide, elevated expression of a gene of interest (e.g., an antigen); increased duration of expression (e.g., prolonged expression) of a gene of interest (e.g., an antigen); elevated expression and increased duration of expression (e.g., prolonged expression) of a gene of interest (e.g., an antigen); decreased interaction with IFIT1 of an RNA polynucleotide; and/or increased translation of an RNA polynucleotide; is observed relative to an appropriate reference.
- a gene of interest e.g., an antigen
- increased duration of expression e.g., prolonged expression
- elevated expression and increased duration of expression e.g., prolonged expression
- decreased interaction with IFIT1 of an RNA polynucleotide e.g., an antigen
- a reference comprises an organism administered an otherwise similar RNA polynucleotide without a m7(3′OMeG)(5′)ppp(5′)(2′OMeAi)pG2 cap.
- a reference comprises an organism administered an otherwise similar RNA polynucleotide without a cap proximal sequence disclosed herein.
- a reference comprises an organism administered an otherwise similar RNA polynucleotide with a self-hybridizing sequence.
- elevated expression is determined at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, or at least 120 hours after administration of a composition or medical preparation comprising an RNA polynucleotide. In some aspects, elevated expression is determined at least 24 hours after administration of a composition or medical preparation comprising an RNA polynucleotide. In some aspects, elevated expression is determined at least 48 hours after administration of a composition or medical preparation comprising an RNA polynucleotide. In some aspects, elevated expression is determined at least 72 hours after administration of a composition or medical preparation comprising an RNA polynucleotide.
- elevated expression is determined at least 96 hours after administration of a composition or medical preparation comprising an RNA polynucleotide. In some aspects, elevated expression is determined at least 120 hours after administration of a composition or medical preparation comprising an RNA polynucleotide. In some aspects, elevated expression is determined at or at about 24-120 hours after administration of a composition or medical preparation comprising an RNA polynucleotide.
- elevated expression is determined at or at about 24-110 hours, 24-100 hours, 24- 90 hours, 24-80 hours, 24-70 hours, 24-60 hours, 24-50 hours, 24-40 hours, 24-30 hours, 30-120 hours, 40-120 hours, 50-120 hours, 60-120 hours, 70-120 hours, 80-120 hours, 90-120 hours, 100-120 hours, or 110-120 hours after administration of a composition or medical preparation comprising an RNA polynucleotide.
- expression of a gene of interest e.g., an antigen
- expression of a gene of interest is or is not elevated at least 2-fold to at least 10-fold.
- expression of a gene of interest e.g., an antigen is elevated at least 2-fold.
- expression of a gene of interest is elevated at least 3-fold. In some aspects, expression of a gene of interest (e.g., an antigen) is elevated at least 4-fold. In some aspects, expression of a gene of interest (e.g., an antigen) is elevated at least 6-fold. In some aspects, expression of a gene of interest (e.g., an antigen) is elevated at least 8-fold. In some aspects, expression of a gene of interest (e.g., an antigen) is elevated at least 10-fold. In some aspects, expression of a gene of interest (e.g., an antigen) is elevated or elevated about 2-fold to about 50-fold.
- expression of a gene of interest is elevated or elevated about 2-fold to about 45-fold, about 2-fold to about 40-fold, about 2-fold to about 30-fold, about 2-fold to about 25-fold, about 2-fold to about 20-fold, about 2-fold to about 15-fold, about 2-fold to about 10-fold, about 2-fold to about 8-fold, about 2-fold to about 5-fold, about 5-fold to about 50-fold, about 10-fold to about 50-fold, about 15-fold to about 50-fold, about 20-fold to about 50-fold, about 25-fold to about 50-fold, about 30-fold to about 50-fold, about 40- fold to about 50-fold, or about 45-fold to about 50-fold.
- a gene of interest e.g., an antigen
- expression of a gene of interest is or is not elevated at least, at most, exactly, or between (inclusive or exclusive) of 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 11-fold, 12-fold, 13- fold, 14-fold, 15-fold, 16-fold, 17-fold, 18-fold, 19-fold, 20-fold, 21-fold, 22-fold, 23-fold, 24-fold, 25-fold, 26-fold, 27-fold, 28-fold, 29-fold, 30-fold, 31-fold, 32-fold, 33-fold, 34-fold, 35-fold, 36- fold, 37-fold, 38-fold, 39-fold, 40-fold, 41-fold, 42-fold, 43-fold, 44-fold, 45-fold, 46-fold, 47-fold, 48-fold, 49-fold, or 50-fold, or any range or value derivable therein.
- elevated expression (e.g., increased duration of expression) of a gene of interest persists for at least, at most, exactly, or between (inclusive or exclusive) of 24 hours, 48 hours, 72 hours, 96 hours, or 120 hours after administration of a composition or a medical preparation comprising an RNA polynucleotide.
- elevated expression of a gene of interest persists for at least 24 hours after administration.
- elevated expression of a gene of interest persists for at least 48 hours after administration.
- elevated expression of a gene of interest (e.g., an antigen) persists for at least 72 hours after administration.
- elevated expression of a gene of interest persists for at least 96 hours after administration. In some aspects, elevated expression of a gene of interest (e.g., an antigen) persists for at least 120 hours after administration of a composition or medical preparation comprising an RNA polynucleotide. In some aspects, elevated expression of a gene of interest (e.g., an antigen) persists for or for about 24-120 hours after administration of a composition or medical preparation comprising an RNA polynucleotide.
- elevated expression persists for or for about 24-110 hours, 24-100 hours, 24-90 hours, 24-80 hours, 24-70 hours, 24-60 hours, 24-50 hours, 24-40 hours, 24-30 hours, 30-120 hours, 40-120 hours, 50-120 hours, 60-120 hours, 70-120 hours, 80- 120 hours, 90-120 hours, 100-120 hours, or 110-120 hours after administration of a composition or medical preparation comprising an RNA polynucleotide.
- elevated expression of a gene of interest persists or does not persist for at least, at most, exactly, or between (inclusive or exclusive) of 24 hours, 36 hours, 48 hours, 60 hours, 72 hours, 84 hours, 96 hours, 108 hours, or 120 hours, or any range or value derivable therein.
- a gene of interest e.g., an antigen
- the disclosure concerns evoking or inducing an immune response in a subject against an RSV protein, e.g., a wild type or variant RSV F protein.
- the immune response may protect against or treat a subject having, suspected of having, or at risk of developing an infection or related disease, particularly those related to RSV.
- the immunogenic compositions of the disclosure are to prevent RSV infections by inoculating or vaccination of a subject.
- the immunogenic compositions immunize the subject against RSV up to 1 year (e.g., for a single RSV season).
- the immunogenic compositions immunize the subject against RSV for up to 2 years.
- the immunogenic compositions immunize the subject against RSV for more than 2 years.
- the immunogenic compositions immunize the subject against RSV for more than 3 years.
- the immunogenic compositions immunize the subject against RSV for more than 4 years.
- the immunogenic compositions immunize the subject against RSV for 5-10 years. A.
- IMMUNOASSAYS The present disclosure includes the implementation of serological assays to evaluate whether and to what extent an immune response is induced or evoked by compositions of the disclosure.
- immunoassays encompassed by the present disclosure include, but are not limited to, those described in U.S. Patent 4,367,110 (double monoclonal antibody sandwich assay) and U.S. Patent 4,452,901 (western blot).
- Other assays include immunoprecipitation of labeled ligands and immunocytochemistry, both in vitro and in vivo.
- Immunoassays generally are binding assays.
- the immunoassays are the various types of enzyme linked immunosorbent assays (ELISAs) and radioimmunoassays (RIA) known in the art. Immunohistochemical detection using tissue sections is also particularly useful.
- ELISAs enzyme linked immunosorbent assays
- RIA radioimmunoassays
- Immunohistochemical detection using tissue sections is also particularly useful.
- antibodies or antigens are immobilized on a selected surface, such as a well in a polystyrene microtiter plate, dipstick, or column support. Then, a test composition suspected of containing the desired antigen or antibody, such as a clinical sample, is added to the wells. After binding and washing to remove non-specifically bound immune complexes, the bound antigen or antibody may be detected.
- Detection is generally achieved by the addition of another antibody, specific for the desired antigen or antibody, that is linked to a detectable label. This type of ELISA is known as a “sandwich ELISA.” Detection also may be achieved by the addition of a second antibody specific for the desired antigen, followed by the addition of a third antibody that has binding affinity for the second antibody, with the third antibody being linked to a detectable label. Competition ELISAs are also possible implementations in which test samples compete for binding with known amounts of labeled antigens or antibodies. The amount of reactive species in the unknown sample is determined by mixing the sample with the known labeled species before or during incubation with coated wells.
- ELISAs have certain features in common, such as coating, incubating or binding, washing to remove non-specifically bound species, and detecting the bound immune complexes.
- Antigen or antibodies may also be linked to a solid support, such as in the form of plate, beads, dipstick, membrane, or column matrix, and the sample to be analyzed is applied to the immobilized antigen or antibody. In coating a plate with either antigen or antibody, one will generally incubate the wells of the plate with a solution of the antigen or antibody, either overnight or for a specified period.
- the wells of the plate will then be washed to remove incompletely- adsorbed material. Any remaining available surfaces of the wells are then “coated” with a nonspecific protein that is antigenically neutral with regard to the test antisera. These include bovine serum albumin (BSA), casein, and solutions of milk powder. The coating allows for blocking of nonspecific adsorption sites on the immobilizing surface and thus reduces the background caused by nonspecific binding of antisera onto the surface.
- BSA bovine serum albumin
- casein casein
- solutions of milk powder solutions of milk powder.
- the coating allows for blocking of nonspecific adsorption sites on the immobilizing surface and thus reduces the background caused by nonspecific binding of antisera onto the surface.
- a method of detecting the presence of infections involves the steps of obtaining a sample suspected of being infected by one or more RSV strains, such as a sample taken from an individual, for example, from one’s blood, saliva, tissues, bone, muscle, cartilage, or skin.
- diagnostic assays utilizing the polypeptides, proteins, and/or peptides of the present disclosure may be carried out to detect the presence of RSV, and such assay techniques for determining such presence in a sample are well known to those skilled in the art and include methods such as radioimmunoassay, western blot analysis and ELISA assays.
- a method of diagnosing an infection wherein a sample suspected of being infected with RSV has added to it the polypeptide, protein, or peptide, in accordance with the present disclosure, and RSV is indicated by antibody binding to the polypeptides, proteins, and/or peptides, or polypeptides, proteins, and/or peptides binding to the antibodies in the sample.
- RNA molecules encoding RSV polypeptides, proteins, and/or peptides in accordance with the disclosure may be used for to treat, prevent, or reduce the severity of illness due to RSV infection (e.g., active or passive immunization) or for use as research tools.
- RNA molecules encoding RSV preF polypeptides, RNA-LNPs and compositions thereof, confer protective immunity to a subject.
- Protective immunity refers to a body’s ability to mount a specific immune response that protects the subject from developing a particular disease or condition that involves the agent against which there is an immune response.
- An immunogenically effective amount is capable of conferring protective immunity to the subject.
- the RNA molecules encoding RSV polypeptides, RNA- LNPs and compositions thereof of the present disclosure may be used to induce a balanced immune response against RSV comprising both cellular and humoral immunity, without many of the risks associated with attenuated virus vaccination.
- a “humoral” immune response refers to an immune response mediated by antibody molecules, including, e.g., secretory (IgA) or IgG molecules, while a “cellular” immune response is one mediated by T- lymphocytes (e.g., CD4+ helper and/or CD8+ T cells (e.g., CTLs) and/or other white blood cells.
- T- lymphocytes e.g., CD4+ helper and/or CD8+ T cells (e.g., CTLs) and/or other white blood cells.
- immune response or its equivalent “immunological response” refers to the development of a humoral (antibody mediated), cellular (mediated by antigen- specific T cells or their secretion products) or both humoral and cellular response directed against an antigen. Such a response may be an active response or a passive response.
- a cellular immune response is elicited by the presentation of polypeptide epitopes in association with Class I or Class II MHC molecules, to activate antigen-specific CD4 (+) T helper cells and/or CD8 (+) cytotoxic T cells.
- the response may also involve activation of monocytes, macrophages, NK cells, basophils, dendritic cells, astrocytes, microglia cells, eosinophils or other components of innate immunity.
- active immunity refers to any immunity conferred upon a subject from the production of antibodies in response to the presence of an of an antigen, e.g. an RSV F protein encoded by an RNA molecule of the present disclosure.
- passive immunity includes, but is not limited to, administration of activated immune effectors including cellular mediators or protein mediators (e.g., monoclonal and/or polyclonal antibodies) of an immune response.
- a monoclonal or polyclonal antibody composition may be used in passive immunization to treat, prevent, or reduce the severity of illness caused by infection by organisms that carry the antigen recognized by the antibody.
- An antibody composition may include antibodies that bind to a variety of antigens that may in turn be associated with various organisms.
- the antibody component may be a polyclonal antiserum.
- the antibody or antibodies are affinity purified from an animal or second subject that has been challenged with an antigen(s).
- an antibody mixture may be used, which is a mixture of monoclonal and/or polyclonal antibodies to antigens present in the same, related, or different microbes or organisms, such as viruses, including but not limited to RSV.
- Passive immunity may be imparted to a patient or subject by administering to the patient immunoglobulins (Ig) and/or other immune factors obtained from a donor or other non-patient source having a known immunoreactivity.
- an immunogenic composition of the present disclosure may be administered to a subject who then acts as a source or donor for globulin, produced in response to challenge with the immunogenic composition (“hyperimmune globulin”), that contains antibodies directed against a RSV or other organism.
- a subject thus treated would donate plasma from which hyperimmune globulin would then be obtained, via conventional plasma-fractionation methodology, and administered to another subject in order to impart resistance against or to treat RSV infection.
- epitopes and antigenic determinant are used interchangeably to refer to a site on an antigen to which B and/or T cells respond or recognize.
- B-cell epitopes may be formed both from contiguous amino acids or noncontiguous amino acids juxtaposed by tertiary folding of a protein. Epitopes formed from contiguous amino acids are typically retained on exposure to denaturing solvents whereas epitopes formed by tertiary folding are typically lost on treatment with denaturing solvents.
- An epitope typically includes at least 3, and more usually, at least 5 or 8-10 amino acids in a unique spatial conformation.
- Methods of determining spatial conformation of epitopes include, for example, x-ray crystallography and 2-dimensional nuclear magnetic resonance. See, e.g., Epitope Mapping Protocols (1996).
- Antibodies that recognize the same epitope may be identified in a simple immunoassay showing the ability of one antibody to block the binding of another antibody to a target antigen.
- T-cells recognize continuous epitopes of about nine amino acids for CD8 cells or about 13-15 amino acids for CD4 cells.
- T cells that recognize the epitope may be identified by in vitro assays that measure antigen-dependent proliferation, as determined by 3 H- thymidine incorporation by primed T cells in response to an epitope (Burke et al., 1994), by antigen-dependent killing (cytotoxic T lymphocyte assay, Tigges et al., 1996) or by cytokine secretion.
- the presence of a cell-mediated immunological response may be determined by proliferation assays (CD4 (+) T cells) or CTL (cytotoxic T lymphocyte) assays.
- the relative contributions of humoral and cellular responses to the protective or therapeutic effect of an immunogenic composition may be distinguished by separately isolating IgG and T-cells from an immunized syngeneic animal and measuring protective or therapeutic effect in a second subject.
- antibody or “immunoglobulin” are used interchangeably and refer to any of several classes of structurally related proteins that function as part of the immune response of an animal or recipient, which proteins include IgG, IgD, IgE, IgA, IgM and related proteins. Under normal physiological conditions antibodies are found in plasma and other body fluids and in the membrane of certain cells and are produced by lymphocytes of the type denoted B cells or their functional equivalent.
- RNA molecules and/or RNA-LNPs disclosed herein may be administered in a pharmaceutical composition or a medicament and may be administered in the form of any suitable pharmaceutical composition.
- a pharmaceutical composition is for therapeutic and/or prophylactic treatment.
- the disclosure relates to a composition for administration to a host.
- the host is a human.
- the host is a non-human.
- Formulations of the compositions described herein may be prepared by any method known or hereafter developed in the art of pharmacology.
- preparatory methods include the step of bringing an active ingredient (e.g., RNA molecules and/or RNA-LNPs) into association with an excipient and/or one or more other accessory ingredients, and then, if necessary and/or desirable, dividing, shaping and/or packaging the product into a desired single- or multi-dose unit.
- an active ingredient e.g., RNA molecules and/or RNA-LNPs
- a pharmaceutical composition or formulation in accordance with the present disclosure can be prepared, packaged, and/or sold in bulk, as a single unit dose, and/or as a plurality of single unit doses.
- Relative amounts of the active ingredient, the pharmaceutically acceptable excipient, and/or any additional ingredients in a composition in accordance with the disclosure will vary, depending upon the identity, size, and/or condition of the subject treated and further depending upon the route by which the composition is to be administered.
- the composition may comprise between 0.1% and 100%, e.g., between 0.5 and 50%, between 1-30%, between 5-80%, at least 80% (w/w), or at least, at most, exactly, or between (inclusive or exclusive) any two of 0.1%, 0.15%, 0.2%, 0.25%, 0.3%, 0.35%, 0.4%, 0.45%, 0.5%, 0.55%, 0.6%, 0.65%, 0.7%, 0.75%, 0.8%, 0.85%, 0.9%, 0.95%, 1%, 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 100% (w/w) active ingredient.
- 0.1% and 100% e.g., between 0.5 and 50%, between 1-30%, between 5-80%, at least 80% (w/w), or at least, at most, exactly, or between (inclusive or exclusive) any two of 0.1%, 0.15%, 0.2%, 0.25%, 0.3%,
- RNA molecules and/or RNA-LNPs disclosed herein may be administered in a pharmaceutical composition which may be formulated into preparations in solid, semi-solid, liquid, lyophilized, frozen, and/or gaseous forms.
- an RNA molecule and/or RNA-LNPs disclosed herein may be administered in a pharmaceutical composition which may comprise a pharmaceutically acceptable carrier and may optionally comprise one or more adjuvants, stabilizers, salts, buffers, preservatives, and optionally other therapeutic agents.
- a pharmaceutical composition disclosed herein comprises one or more pharmaceutically acceptable carriers, diluents and/or excipients.
- pharmaceutical compositions do not include an adjuvant (e.g., they are adjuvant free).
- excipient refers to a substance which may be present in a pharmaceutical composition of the present disclosure but is not an active ingredient.
- excipients include without limitation, carriers, diluents (e.g., solvents, dispersion media, and/or other liquid vehicles, dispersion or suspension aids), granulating and/or dispersing agents, surface active agents, isotonic agents, thickening and/or emulsifying agents, preservatives, binders, lubricants and/or oil, coloring, sweetening and/or flavoring agents, stabilizers, antioxidants, antimicrobial and/or antifungal agents, osmolality adjusting agents, pH adjusting agents, buffers, chelants, cryoprotectants, and/or bulking agents.
- diluents e.g., solvents, dispersion media, and/or other liquid vehicles, dispersion or suspension aids
- granulating and/or dispersing agents e.g., surface active agents, isotonic agents, thickening and/or emulsifying agents, preservatives, binders, lubricants and/
- carrier refers to a component which may be natural, synthetic, organic, or inorganic, in which the active component is combined in order to facilitate, enhance and/or enable administration of the pharmaceutical composition.
- a carrier as used herein may be one or more compatible solid or liquid fillers, diluents or encapsulating substances, which are suitable for administration to subject.
- Suitable carriers include, without limitation, sterile water, Ringer’s solution, Ringer’s lactate solution, sterile sodium chloride solution, isotonic saline, polyalkylene glycols, hydrogenated naphthalenes and, in particular, biocompatible lactide polymers, lactide/glycolide copolymers or polyoxyethylene/polyoxy-propylene copolymers.
- the pharmaceutical composition of the present disclosure includes sodium chloride.
- 1, 2, 3, 4, 5, or more of the foregoing carriers may be excluded from the pharmaceutical compositions disclosed herein.
- the term “diluent” relates a diluting and/or thinning agent.
- diluent includes any one or more of fluid, liquid or solid suspension and/or mixing media.
- suitable diluents for use in a pharmaceutical compositions of the present disclosure include, without limitation, ethanol, glycerol, saline, water, calcium or sodium carbonate, calcium phosphate, calcium hydrogen phosphate, sodium phosphate, lactose, sucrose, cellulose, microcrystalline cellulose, kaolin, mannitol, sorbitol, etc., and/or combinations thereof.
- 1, 2, 3, 4, 5, or more of the foregoing diluents may be excluded from the pharmaceutical compositions disclosed herein.
- suitable granulating and/or dispersing agents include, without limitation, starches, pregelatinized starches, or microcrystalline starch, alginic acid, guar gum, agar, poly(vinyl-pyrrolidone), (providone), cross-linked poly(vinyl-pyrrolidone) (crospovidone), cellulose, methylcellulose, carboxymethyl cellulose, cross-linked sodium carboxymethyl cellulose (croscarmellose), magnesium aluminum silicate (VEEGUM®), sodium lauryl sulfate, etc., and/or combinations thereof.
- 1, 2, 3, 4, 5, or more of the foregoing granulating and/or dispersing agents may be excluded from the pharmaceutical compositions disclosed herein.
- Suitable surface active agents for use in a pharmaceutical compositions of the present disclosure include, without limitation, natural emulsifiers (e.g., acacia, agar, alginic acid, sodium alginate, tragacanth, chondrux, cholesterol, xanthan, pectin, gelatin, egg yolk, casein, wool fat, cholesterol, wax, and lecithin), sorbitan fatty acid esters (e.g., polyoxyethylene sorbitan monooleate [TWEEN®80], sorbitan monopalmitate [SPAN®40], glyceryl monooleate, polyoxyethylene esters, polyethylene glycol fatty acid esters (e.g., CREMOPHOR®), polyoxyethylene ethers (e.g., polyoxyethylene lauryl ether [BRIJ®30]), PLUORINC®F 68, POLOXAMER®188, etc.
- natural emulsifiers e.g., acacia, a
- 1, 2, 3, 4, 5, or more of the foregoing surface active agents may be excluded from the pharmaceutical compositions disclosed herein.
- suitable preservatives for use in a pharmaceutical compositions of the present disclosure include, without limitation, benzalkonium chloride, chlorobutanol, paraben, thimerosal, vitamin A, vitamin C, vitamin E, beta-carotene, citric acid, ascorbic acid, butylated hydroxyanisole, ethylenediamine, sodium lauryl sulfate (SLS), sodium lauryl ether sulfate (SLES), etc., and combinations thereof.
- 1, 2, 3, 4, 5, or more of the foregoing preservatives may be excluded from the pharmaceutical compositions disclosed herein.
- suitable antimicrobial and/or antifungal agents for use in a pharmaceutical compositions of the present disclosure include, without limitation, benzalkonium chloride, benzethonium chloride, methyl paraben, ethyl paraben, propyl paraben, butyl paraben, benzoic acid, hydroxybenzoic acid, potassium or sodium benzoate, potassium or sodium sorbate, sodium propionate, sorbic acid, etc., and combinations thereof.
- 1, 2, 3, 4, 5, or more of the foregoing antimicrobial and/or antifungal agents may be excluded from the pharmaceutical compositions disclosed herein.
- Suitable binders for use in a pharmaceutical compositions of the present disclosure include, without limitation, starch, gelatin, sugars (e.g., sucrose, glucose, dextrose, dextrin, molasses, lactose, lactitol, mannitol), amino acids (e.g., glycine), natural and synthetic gums (e.g., acacia, sodium alginate), ethylcellulose, hydroxyethylcellulose, hydroxypropyl methylcellulose, etc., and combinations thereof. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing binders may be excluded from the pharmaceutical compositions disclosed herein.
- Suitable lubricants and/or oil for use in a pharmaceutical compositions of the present disclosure include, without limitation, magnesium stearate, calcium stearate, stearic acid, silica, talc, malt, hydrogenated vegetable oils, polyethylene glycol, sodium benzoate, sodium or magnesium lauryl sulfate, etc., and combinations thereof. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing lubricants and/or oils may be excluded from the pharmaceutical compositions disclosed herein.
- antioxidants for use in a pharmaceutical compositions of the present disclosure include, without limitation, alpha tocopherol, ascorbic acid, ascorbyl palmitate, benzyl alcohol, butylated hydroxyanisole, m-cresol, methionine, butylated hydroxytoluene, monothioglycerol, sodium or potassium metabisulfite, propionic acid, propyl gallate, sodium ascorbate, etc., and combinations thereof.
- 1, 2, 3, 4, 5, or more of the foregoing antioxidants may be excluded from the pharmaceutical compositions disclosed herein.
- Suitable osmolality adjusting agents, pH adjusting agents, and buffers for use in a pharmaceutical compositions of the present disclosure include, without limitation, sodium phosphate, sodium citrate, sodium succinate, histidine (or histidine-HCl), sodium malate, sodium carbonate, etc., and/or combinations thereof. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing osmolality adjusting agents may be excluded from the pharmaceutical compositions disclosed herein.
- suitable cryoprotectants for use in a pharmaceutical compositions of the present disclosure include, without limitation, mannitol, sucrose, trehalose, lactose, glycerol, dextrose, etc., and combinations thereof.
- cryoprotectants may be excluded from the pharmaceutical compositions disclosed herein.
- suitable bulking agents include, without limitation, sucrose, trehalose, mannitol, glycine, lactose, raffinose, and combinations thereof.
- 1, 2, 3, 4, 5, or more of the foregoing bulking agents may be excluded from the pharmaceutical compositions disclosed herein.
- Compositions can be formulated using one or more excipients (e.g., one or more carriers and/or diluents) to, e.g.: (1) increase stability; (2) increase cell transfection; (3) permit the sustained and/or delayed release (e.g., from a depot formulation); (4) alter the biodistribution (e.g., target to specific tissues and/or cell types); (5) increase the translation of encoded protein in vivo; and/or (6) alter the release profile of encoded protein (antigen) in vivo.
- excipients e.g., one or more carriers and/or diluents
- compositions comprises an RNA molecule comprising an open reading frame encoding an immunogenic polypeptide.
- immunogenic polypeptide comprises a RSV antigen.
- RSV antigen is a RSV F protein or a fragment or variant thereof.
- the composition comprises an RNA molecule comprising an open reading frame encoding a full-length RSV F protein.
- the encoded immunogenic polypeptide is a truncated RSV F protein.
- the encoded immunogenic polypeptide is a variant of a RSV F protein.
- the encoded immunogenic polypeptide is a fragment of a RSV F protein.
- a pharmaceutical composition comprises an RNA molecule (e.g., polynucleotide) disclosed herein formulated with a lipid-based delivery system.
- the composition includes a lipid-based delivery system (e.g., LNPs) (e.g., a lipid-based vaccine), which delivers a nucleic acid molecule to the interior of a cell, where it may then replicate, inhibit protein expression of interest, and/or express the encoded polypeptide of interest.
- the delivery system may have adjuvant effects which enhance the immunogenicity of an encoded antigen.
- the composition comprises at least one RNA molecule encoding a RSV polypeptide complexed with, encapsulated in, and/or formulated with one or more lipids, and forming lipid nanoparticles (LNPs), liposomes, lipoplexes and/or nanoliposomes.
- LNPs lipid nanoparticles
- the composition comprises a lipid nanoparticle.
- the present disclosure concerns compositions comprising one or more lipids associated with a nucleic acid or a polypeptide/peptide (e.g., RSV RNA-LNPs).
- the immunogenic composition including a lipid-based delivery system may further include one or more salts and/or one or more pharmaceutically acceptable surfactants, preservatives, carriers, diluents, and/or excipients, in some cases.
- the immunogenic composition including a lipid-based delivery system further includes a pharmaceutically acceptable vehicle.
- each of a buffer, stabilizing agent, and optionally a salt may be included in the immunogenic composition including a lipid-based delivery system.
- any one or more of a buffer, stabilizing agent, salt, surfactant, preservative, and excipient may be excluded from the immunogenic composition including a lipid-based delivery system.
- the immunogenic composition including a lipid-based delivery system further comprises a stabilizing agent.
- the stabilizing agent comprises sucrose, mannose, sorbitol, raffinose, trehalose, mannitol, inositol, sodium chloride, arginine, lactose, hydroxyethyl starch, dextran, polyvinylpyrolidone, glycine, or a combination thereof.
- the stabilizing agent is a disaccharide, or sugar.
- the stabilizing agent is sucrose.
- the stabilizing agent is trehalose.
- the stabilizing agent is a combination of sucrose and trehalose.
- the total concentration of the stabilizing agent(s) in the composition is or is about 5% to about 10% w/v.
- the total concentration of the stabilizing agent may or may not be equal to at least, at most, exactly, or between (inclusive or exclusive) of 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, or 20% w/v or any range or value derivable therein.
- the stabilizing agent concentration includes, but is not limited to, a concentration of or of about 10 mg/mL to about 400 mg/mL, about 100 mg/mL to about 200 mg/mL, about 100 mg/mL to about 150 mg/mL, about 100 mg/mL to about 140 mg/mL, about 100 mg/mL to about 130 mg/mL, about 100 mg/mL to about 120 mg/mL, about 100 mg/mL to about 110 mg/mL, or about 100 mg/mL to about 105 mg/mL.
- the concentration of the stabilizing agent is or is not equal to at least, at most, exactly, or between (inclusive or exclusive) of 10 mg/mL, 20 mg/mL, 50 mg/mL, 100 mg/mL, 101 mg/mL, 102 mg/mL, 103 mg/mL, 104 mg/mL, 105 mg/mL, 106 mg/mL, 107 mg/mL, 108 mg/mL, 109 mg/mL, 110 mg/mL, 150 mg/mL, 200 mg/mL, 300 mg/mL, 400 mg/mL, or more.
- the mass amount of the stabilizing agent and the mass amount of the RNA are in a specific ratio.
- the ratio of the mass amount of the stabilizing agent and the RNA is no greater than 5000. In another aspect, the ratio of the mass amount of the stabilizing agent and the RNA is no greater than 2000. In another aspect, the ratio of the mass amount of the stabilizing agent and the RNA is no greater than 1000. In another aspect, the ratio of the mass amount of the stabilizing agent and the RNA is no greater than 500. In another aspect, the ratio of the mass amount of the stabilizing agent and the RNA is no greater than 100. In another aspect, the ratio of the mass amount of the stabilizing agent and the pharmaceutical substance is no greater than 50. In another aspect, the ratio of the mass amount of the stabilizing agent and the RNA is no greater than 10.
- the ratio of the mass amount of the stabilizing agent and the RNA is no greater than 1. In another aspect, the ratio of the mass amount of the stabilizing agent and the RNA is no greater than 0.5. In another aspect, the ratio of the mass amount of the stabilizing agent and the RNA is no greater than 0.1. In another aspect, the stabilizing agent and RNA comprise a mass ratio of or of about 200 – 2000 of the stabilizing agent : 1 of the RNA. In some aspects, the immunogenic composition including a lipid-based delivery system further comprises a buffer.
- buffering agents include, but are not limited to, citrate buffer solutions, acetate buffer solutions, phosphate buffer solutions, ammonium chloride, calcium carbonate, calcium chloride, calcium citrate, calcium glubionate, calcium gluceptate, calcium gluconate, d-gluconic acid, calcium glycerophosphate, calcium lactate, calcium lactobionate, propanoic acid, calcium levulinate, pentanoic acid, dibasic calcium phosphate, phosphoric acid, tribasic calcium phosphate, calcium hydroxide phosphate, potassium acetate, potassium chloride, potassium gluconate, potassium mixtures, dibasic potassium phosphate, monobasic potassium phosphate, potassium phosphate mixtures, sodium acetate, sodium bicarbonate, sodium chloride, sodium citrate, sodium lactate, dibasic sodium phosphate, monobasic sodium phosphate, sodium phosphate mixtures, tromethamine, Tris hydrochloride (HCl), amino-sulfonate buffers (HC
- the buffer is a HEPES buffer, a Tris buffer, and/or a PBS buffer.
- the buffer is Tris buffer.
- the buffer is a HEPES buffer.
- the buffer is a PBS buffer.
- the buffer concentration may or may not be equal to at least, at most, exactly, or between (inclusive or exclusive) of 1 mM, 2 mM, 3 mM, 4 mM, 5 mM, 6 mM, 7 mM, 8 mM, 9 mM, 10 mM, 11 mM, 12 mM, 13 mM, 14 mM, 15 mM, 16 mM, 17 mM, 18 mM, 19 mM, or 20 mM, or any range or value derivable therein.
- the buffer may be at a neutral pH, pH 6.5 to 8.5, pH 7.0 to pH 8.0, or pH 7.2 to pH 7.6.
- the buffer may or may not be at least, at most, exactly, or between (inclusive or exclusive) of pH 6.5, 6.6, 6.7, 6.8, 6.9, 7, 7.1, 7.2, 7.3, 7.4, 7.5, 7.6, 7.7, 7.8, 7.9, 8, 8.1, 8.2, 8.3, 8.4, or 8.5, or any range or value derivable therein.
- the buffer is at pH 7.4.
- the immunogenic composition including a lipid-based delivery system may further comprise a salt.
- salts include but not limited to sodium salts and/or potassium salts.
- the salt is a sodium salt.
- the sodium salt is sodium chloride.
- the salt is a potassium salt.
- the potassium salt comprises potassium chloride.
- any one or more of the foregoing salts may be excluded from the immunogenic compositions disclosed herein.
- the concentration of the salts in the composition may be or be about 70 mM to about 140 mM.
- the salt concentration may or may not be equal to at least, at most, exactly, or between (inclusive or exclusive) of 50 mM, 60 mM, 70 mM, 80 mM, 90 mM, 100 mM, 120 mM, 130 mM, 140 mM, 150 mM, 160 mM, 170 mM, 180 mM, 190 mM, or 200 mM.
- the salt concentration includes, but is not limited to, a concentration of or of about 1 mg/mL to about 100 mg/mL, about 1 mg/mL to about 50 mg/mL, about 1 mg/mL to about 40 mg/mL, about 1 mg/mL to about 30 mg/mL, about 1 mg/mL to about 20 mg/mL, about 1 mg/mL to about 10 mg/mL, or about 1 mg/mL to about 15 mg/mL.
- the concentration of the salt is or is not equal to at least, at most, exactly, or between (inclusive or exclusive) of 1 mg/mL, 2 mg/mL, 3 mg/mL, 4 mg/mL, 5 mg/mL, 6 mg/mL, 7 mg/mL, 8 mg/mL, 9 mg/mL, 10 mg/mL, 11 mg/mL, 12 mg/mL, 13 mg/mL, 14 mg/mL, 15 mg/mL, 16 mg/mL, 17 mg/mL, 18 mg/mL, 19 mg/mL, 20 mg/mL, or more.
- the salt may be at a neutral pH, pH 6.5 to 8.5, pH 7.0 to pH 8.0, or pH 7.2 to pH 7.6.
- the salt may or may not be at a pH equal to at least, at most, exactly, or between (inclusive or exclusive) of 6.5, 6.6, 6.7, 6.8, 6.9, 7, 7.1, 7.2, 7.3, 7.4, 7.5, 7.6, 7.7, 7.8, 7.9, 8, 8.1, 8.2, 8.3, 8.4, or 8.5.
- the immunogenic composition including a lipid-based delivery system further comprises a surfactant, a preservative, any other excipient, or a combination thereof.
- any other excipient includes, but is not limited to, antioxidants, glutathione, EDTA, methionine, desferal, antioxidants, metal scavengers, and/or free radical scavengers.
- the surfactant, preservative, excipient or combination thereof is sterile water for injection (sWFI), bacteriostatic water for injection (BWFI), saline, dextrose solution, polysorbates, poloxamers, Triton, divalent cations, Ringer’s lactate, amino acids, sugars, polyols, polymers, and/or cyclodextrins.
- excipients may be excluded from the immunogenic compositions disclosed herein.
- excipients which refer to ingredients in the immunogenic compositions that are not active ingredients, include but are not limited to carriers, binders, diluents, lubricants, thickeners, surface active agents, preservatives, stabilizers, emulsifiers, buffers, flavoring agents, disintegrants, coatings, plasticizers, compression agents, wet granulation agents, and/or colorants.
- “pharmaceutically acceptable carrier” includes any and all aqueous solvents (e.g., water, alcoholic/aqueous solutions, saline solutions, parenteral vehicles, such as sodium chloride, Ringer’s dextrose, etc.), non-aqueous solvents (e.g., propylene glycol, polyethylene glycol, vegetable oil, and injectable organic esters, such as ethyl oleate), dispersion media, coatings, surfactants, antioxidants, preservatives (e.g., antibacterial and/or antifungal agents, anti-oxidants, chelating agents, and inert gases), isotonic agents, absorption delaying agents, salts, drugs, drug stabilizers, gels, binders, excipients, disintegration agents, lubricants, sweetening agents, flavoring agents, dyes, fluid and nutrient replenishers, such like materials and combinations thereof, as would be known to one of ordinary skill in the art.
- aqueous solvents e
- Diluents include but are not limited to ethanol, glycerol, water, sugars such as lactose, sucrose, mannitol, and sorbitol, and starches derived from wheat, corn rice, and potato; and celluloses such as microcrystalline cellulose.
- the amount of diluent in the composition may range from or from about 10% to about 90% by weight of the total composition, e.g., from or from about 25% to about 75%, about 30% to about 60% by weight, or about 12% to about 60%.
- Preservatives for use in the compositions disclosed herein include but are not limited to benzalkonium chloride, chlorobutanol, paraben and thimerosal.
- a pharmaceutical composition comprises an RSV RNA molecule encoding a RSV polypeptide as disclosed herein that is complexed with, encapsulated in, and/or formulated with one or more lipids to form RSV RNA-LNPs.
- the RSV RNA-LNP composition is a liquid. In some aspects, the RSV RNA-LNP composition is frozen. In some aspects, the RSV RNA-LNP composition is lyophilized. In some aspects, a RSV RNA-LNP composition comprises a RSV RNA polynucleotide molecule encoding a RSV polypeptide as disclosed herein, encapsulated in LNPs with a lipid composition of a cationic lipid, a PEGylated lipid (i.e. PEG- lipid), and one or more structural lipids (e.g., a neutral lipid).
- a lipid composition of a cationic lipid, a PEGylated lipid (i.e. PEG- lipid), and one or more structural lipids (e.g., a neutral lipid).
- a RSV RNA-LNP composition comprises a cationic lipid.
- the cationic lipid may comprise any one or more cationic lipids disclosed herein.
- the cationic lipid may comprise any one or more cationic lipids disclosed herein.
- the cationic lipid comprises ((4-hydroxybutyl)azanediyl)bis(hexane-6,1- diyl)bis(2-hexyldecanoate) (ALC-0315).
- the cationic lipid (e.g., ALC-0315) is or is not included in the composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, 0.3, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.4, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, 0.5, 0.51, 0.52, 0.53, 0.54, 0.55, 0.56, 0.57, 0.58, 0.59, 0.6, 0.61, 0.62, 0.63, 0.64, 0.65, 0.66, 0.67, 0.68
- the cationic lipid (e.g., ALC-0315) is or is not included in the composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 0.4, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, 0.5, 0.51, 0.52, 0.53, 0.54, 0.55, 0.56, 0.57, 0.58, 0.59, 0.6, 0.61, 0.62, 0.63, 0.64, 0.65, 0.66, 0.67, 0.68, 0.69, 0.7, 0.71, 0.72, 0.73, 0.74, 0.75, 0.76, 0.77, 0.78, 0.79, 0.8, 0.81, 0.82, 0.83, 0.84, 0.85, 0.86, 0.87, 0.88, 0.89, 0.9, 0.91, 0.92, 0.93, 0.94, 0.95, 0.96, 0.97, 0.98, 0.99, or 1 mg/mL.
- the cationic lipid (e.g., ALC- 0315) is included in the composition at a concentration of at least 0.4, at least 0.45, at least 0.5, at least 0.55, at least 0.6, at least 0.65, at least 0.7, at least 0.75, at least 0.8, at least 0.85, at least 0.9, at least 0.95, or at least 1 mg/mL.
- the cationic lipid (e.g., ALC-0315) is included in the composition at a concentration of between 0.4 and 0.5, between 0.5 and 0.6, between 0.6 and 0.7, between 0.7 and 0.8, between 0.8 and 0.9, or between 0.9 and 1.
- the cationic lipid (e.g., ALC-0315) is included in the composition at a concentration of between 0.4 and 0.45, between 0.45 and 0.5, between 0.5 and 0.55, between 0.55 and 0.6, between 0.6 and 0.65, between 0.65 and 0.7, between 0.7 and 0.75, between 0.75 and 0.8, between 0.8 and 0.85, between 0.85 and 0.9, between 0.9 and 0.95, or between 0.95 and 1 mg/mL.
- the cationic lipid (e.g., ALC-0315) is included in the composition at a concentration of 0.8 to 0.95 mg/mL.
- the cationic lipid (e.g., ALC-0315) is included in the composition at a concentration of or of about 0.8 to 0.9 mg/mL. In specific aspects, the cationic lipid (e.g., ALC-0315) is included in the composition at a concentration of or of about 0.85 to 0.9 mg/mL.
- the cationic lipid (e.g., ALC-0315) is or is not included in the composition at a concentration of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 0.8, 0.81, 0.82, 0.83, 0.84, 0.85, 0.86, 0.87, 0.88, 0.89, 0.9, 0.91, 0.92, 0.93, 0.94, or 0.95 mg/mL.
- the cationic lipid e.g., ALC-0315
- Concentrations for lyophilized compositions are determined post-reconstitution.
- a RSV RNA-LNP composition further comprises a PEGylated lipid (i.e., PEG-lipid).
- PEG-lipid i.e., PEG-lipid
- the PEGylated lipid may comprise any one or more PEGylated lipids disclosed herein.
- the PEGylated lipid comprises 2-[(polyethylene glycol)-2000]-N,N- ditetradecylacetamide (ALC-0159).
- the PEGylated lipid (e.g., ALC-0159) is or is not included in the composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, 0.3, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.4, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, 0.5, 0.51, 0.52, 0.53, 0.54, 0.55, 0.56, 0.57, 0.58, 0.59, 0.6, 0.61, 0.62, 0.63, 0.64, 0.65, 0.66, 0.67, 0.
- the PEGylated lipid (e.g., ALC-0159) is or is not included in the composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, 0.3, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.4, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, or 0.5 mg/mL.
- the PEGylated lipid (e.g., ALC-0159) is included in the composition at a concentration of at least 0.01, at least 0.05, at least 0.1, at least 0.15, at least 0.2, at least 0.25 mg/mL, at least 0.3 mg/mL, at least 0.35 mg/mL, at least 0.4 mg/mL, at least 0.45 mg/mL, or at least 0.5 mg/mL.
- the PEGylated lipid is included in the composition at a concentration of between 0.01 and 0.05, between 0.05 and 0.1, between 0.1 and 0.15, between 0.15 and 0.2, or between 0.2 and 0.25 mg/mL.
- the PEGylated lipid (e.g., ALC-0159) is included in the composition at a concentration of or of about 0.05 to 0.15 mg/mL. In specific aspects, the PEGylated lipid (e.g., ALC-0159) is included in the composition at a concentration of or of about 0.10 to 0.15 mg/mL. In specific aspects, the PEGylated lipid (e.g., ALC-0159) is or is not included in the composition at a concentration of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.11, 0.12, 0.13, 0.14, or 0.15 mg/mL.
- a RSV RNA-LNP composition further comprises one or more structural lipids.
- the one or more structural lipids may comprise any one or more structural lipids disclosed herein.
- the one or more structural lipids comprise a neutral lipid and a steroid or steroid analog.
- the one or more structural lipids comprise 1,2-distearoyl- sn-glycero-3-phosphocholine (DSPC) and cholesterol.
- the one or more structural lipids are or are not included in the composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, 0.3, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.4, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, 0.5, 0.51, 0.52, 0.53, 0.54, 0.55, 0.56, 0.57, 0.58, 0.59, 0.6, 0.61, 0.62, 0.63, 0.64, 0.65, 0.66, 0.67,
- the one or more structural lipids are or are not included in the composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, 0.3, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.4, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, or 0.5 mg/mL.
- the one or more structural lipids are included in the composition at a concentration of at least .05, at least 0.1, at least 0.15, at least 0.2, at least 0.25, at least 0.3, at least 0.35, at least 0.4, at least 0.45, at least 0.5, at least 0.55, at least 0.6, at least 0.65, at least 0.7, at least 0.75, at least 0.8, at least 0.85, at least 0.9, at least 0.95 or at least 1 mg/mL.
- the one or more structural lipids are included in the composition at a concentration of between 0.05 and 0.1, between 0.1 and 0.15, between 0.15 and 0.2, between 0.2 and 0.25, between 0.25 and 0.3, between 0.3 and 0.35, between 0.35 and 0.4, between 0.4 and 0.45, between 0.45 and 0.5, between 0.5 and 0.55, between 0.55 and 0.6, between 0.6 and 0.65, between 0.65 and 0.7, between 0.7 and 0.75, between 0.75 and 0.8, between 0.8 and 0.85, between 0.85 and 0.9, between 0.9 and 0.95, or between 0.95 and 1 mg/mL.
- the one or more structural lipids include DSPC, and the DSPC is included in the composition at a concentration of or of about 0.1 to 0.25 mg/mL. In specific aspects, the one or more structural lipids include DSPC, and the DSPC is included in the composition at a concentration of or of about 0.15 to 0.25 mg/mL.
- the one or more structural lipids include DSPC, and the DSPC is or is not included in the composition at a concentration of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, or 0.25 mg/mL.
- the DSPC is included in the composition at a concentration of or of about 0.19 mg/mL.
- the one or more structural lipids include cholesterol, and the cholesterol is included in the composition at a concentration of or of about 0.3 to 0.45 mg/mL.
- the one or more structural lipids include cholesterol, and the cholesterol is included in the composition at a concentration of or of about 0.3 to 0.4. In specific aspects, the one or more structural lipids include cholesterol, and the cholesterol is included in the composition at a concentration of or of about 0.35 to 0.45. In specific aspects, the one or more structural lipids include cholesterol, and the cholesterol is or is not included in the composition at a concentration of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 0.30, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.40, 0.41, 0.42, 0.43, 0.44, or 0.45 mg/mL.
- the cholesterol is included in the composition at a concentration of and/or of about 0.37 mg/mL. Concentrations for lyophilized compositions are determined post- reconstitution.
- the RSV RNA-LNP composition further comprises one or more buffers and stabilizing agents, and optionally, salt diluents.
- the RSV RNA-LNP composition comprises an cationic lipid, a PEGylated lipid, one or more structural lipids, one or more buffers, a stabilizing agent, and optionally, a salt diluent.
- 1, 2, 3, or more of the foregoing elements are excluded from the RSV RNA-LNP composition.
- a RSV RNA-LNP composition comprises one or more buffers.
- the one or more buffers may comprise any one or more buffering agents disclosed herein.
- the composition comprises a Tris buffer comprising at least a first buffer and a second buffer.
- the first buffer is tromethamine.
- the second buffer is Tris hydrochloride (HCl).
- the first buffer and second buffer of the Tris buffer are or are not included in the composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, 0.3, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.4, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, 0.5, 0.51, 0.52, 0.53, 0.54, 0.55, 0.56, 0.57, 0.58, 0.59, 0.6, 0.61, 0.62, 0.63, 0.64,
- the RSV RNA-LNP composition is a liquid composition comprising a Tris buffer.
- the Tris buffer comprises a first buffer.
- the first buffer is tromethamine.
- the first buffer e.g., tromethamine
- the first buffer is or is not included in the liquid composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, 0.3, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.4, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, or 0.5 mg/mL.
- the first buffer e.g., tromethamine
- the first buffer is included in the liquid composition at a concentration of at least 0.1, at least .05, at least 0.1, at least 0.15, at least 0.2, at least 0.25, at least 0.3, at least 0.35, at least 0.4, at least 0.45, at least 0.5, at least 0.55, at least 0.6, at least 0.65, at least 0.7, at least 0.75, at least 0.8, at least 0.85, at least 0.9, at least 0.95, or at least 1 mg/mL.
- the first buffer e.g., tromethamine
- the first buffer is included in the liquid composition at a concentration of between 0.05 and 0.15, between 0.15 and 0.25, between 0.25 and 0.35, between 0.35 and 0.45, between 0.45 and 0.55, between 0.55 and 0.65, between 0.65 and 0.75, between 0.75 and 0.85, or between 0.85 and 0.95.
- the first buffer e.g., tromethamine
- the first buffer is included in the liquid composition at a concentration of between 0.05 and 0.1, between 0.1 and 0.15, between 0.15 and 0.2, between 0.2 and 0.25, between 0.25 and 0.3, between 0.3 and 0.35, between 0.35 and 0.4, between 0.4 and 0.45, between 0.45 and 0.5, between 0.5 and 0.55, between 0.55 and 0.6, between 0.6 and 0.65, between 0.65 and 0.7, between 0.7 and 0.75, between 0.75 and 0.8, between 0.8 and 0.85, between 0.85 and 0.9, between 0.9 and 0.95, or between 0.95 and 1 mg/mL.
- the first buffer e.g., tromethamine
- the first buffer is included in the liquid composition at a concentration of or of about 0.1 to 0.3 mg/mL. In specific aspects, the first buffer (e.g., tromethamine) is included in the liquid composition at a concentration of or of about 0.15 to 0.25 mg/mL.
- the first buffer (e.g., tromethamine) is or is not included in the liquid composition at a concentration of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, or 0.3 mg/mL.
- the first buffer (e.g., tromethamine) is included in the liquid composition at a concentration of or of about 0.20 mg/mL.
- the RSV RNA-LNP composition is a liquid composition comprising a Tris buffer comprising a second buffer.
- the second buffer comprises Tris HCl.
- the second buffer (e.g., Tris HCl) is or is not included in the liquid composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 0.5, 0.55, 1, 1.01, 1.02, 1.03, 1.04, 1.05, 1.06, 1.07, 1.08, 1.09, 1.1, 1.11, 1.12, 1.13, 1.14, 1.15, 1.16, 1.17, 1.18, 1.19, 1.2, 1.21, 1.22, 1.23, 1.24, 1.25, 1.26, 1.27, 1.28, 1.29, 1.3, 1.31, 1.32, 1.33, 1.34, 1.35, 1.36, 1.37, 1.38, 1.39, 1.4, 1.41, 1.42, 1.43, 1.44, 1.45, 1.46, 1.47, 1.48, 1.49, or 1.5 mg/m
- the second buffer (e.g., Tris HCl) is included in the liquid composition at a concentration of at least 0.5, at least 0.55, at least 0.6, at least 0.65, at least 0.7, at least 0.75, at least 0.8, at least 0.85, at least 0.9, at least 0.95, at least 1, at least 1.05, at least 1.10, at least 1.15, at least 1.20, at least 1.25, at least 1.30, at least 1.35, at least 1.40, at least 1.45, or at least 1.50 mg/mL.
- Tris HCl Tris HCl
- the second buffer (e.g., Tris HCl) is included in the liquid composition at a concentration of between 0.5 and 0.6, between 0.6 and 0.7, between 0.7 and 0.8, between 0.8 and 0.9, between 0.9 and 1, between 1 and 1.10, between 1.10 and 1.20, between 1.20 and 1.30, between 1.30 and 1.40, or between 1.40 and 1.50 mg/mL.
- the second buffer e.g., Tris HCl
- the second buffer is included in the liquid composition at a concentration of or of about 1.25 to 1.40 mg/mL.
- the second buffer (e.g., Tris HCl) is included in the liquid composition at a concentration of or of about 1.30 to 1.40 mg/mL.
- the second buffer (e.g., Tris HCl) is or is not included in the liquid composition at a concentration of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 1.25, 1.26, 1.27, 1.28, 1.29, 1.30, 1.31, 1.32, 1.33, 1.34, or 1.35, 1.36, 1.37, 1.38, 1.39, or 1.40 mg/mL.
- the second buffer (e.g., Tris HCl) is included in the liquid composition at a concentration of or of about 1.32 mg/mL.
- the RSV RNA-LNP composition is a lyophilized composition comprising a Tris buffer.
- the Tris buffer comprises a first buffer.
- the first buffer is tromethamine.
- the first buffer (e.g., tromethamine) is or is not included in the lyophilized composition at a concentration, after reconstitution, of at least, at most, between (inclusive or exclusive) of, or exactly 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.2, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, 0.3, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.4, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, or 0.5 mg/mL.
- the first buffer e.g., tromethamine
- the first buffer is included in the lyophilized composition at a concentration, after reconstitution, of at least 0.01, of at least 0.05, of at least 0.1, of at least 0.15, of at least 0.2, of at least 0.25, of at least 0.3, of at least 0.35, of at least 0.4, of at least 0.45, or of at least 0.5 mg/mL.
- the first buffer (e.g., tromethamine (Tris base)) is included in the lyophilized composition at a concentration, after reconstitution, of between 0.01 and 0.05, between 0.05 and 0.1, between 0.1 and 0.15, between 0.15 and 0.2, between 0.2 and 0.25 mg/mL, between 0.25 and 0.3 mg/mL, between 0.3 and 0.35 mg/mL, between 0.35 and 0.4 mg/mL, between 0.4 and 0.45 mg/mL, or between 0.45 and 0.5 mg/mL.
- the first buffer (e.g., tromethamine) is included in the lyophilized composition at a concentration, after reconstitution, of or of about 0.01 and 0.15 mg/mL.
- the first buffer e.g., tromethamine
- the first buffer is included in the lyophilized composition at a concentration, after reconstitution, of or of about 0.01 and 0.10 mg/mL.
- the first buffer is included in the lyophilized composition at a concentration, after reconstitution, of or of about 0.05 and 0.15 mg/mL.
- the first buffer (e.g., tromethamine) is or is not included in the lyophilized composition at a concentration, after reconstitution, of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.11, 0.12, 0.13, 0.14, or 0.15 mg/mL.
- the first buffer e.g., tromethamine
- the first buffer is included in the lyophilized composition at a concentration, after reconstitution, of or of about 0.09 mg/mL.
- the RSV RNA-LNP composition is a lyophilized composition comprising a Tris buffer comprising a second buffer.
- the second buffer comprises Tris HCl.
- the second buffer (e.g., Tris HCl) is or is not included in the lyophilized composition at a concentration, after reconstitution, of at least, at most, between (inclusive or exclusive) of, or exactly 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5, 0.51, 0.52, 0.53, 0.54, 0.55, 0.56, 0.57, 0.58, 0.59, 0.6, 0.61, 0.62, 0.63, 0.64, 0.65, 0.66, 0.67, 0.68, 0.69, 0.7, 0.71, 0.72, 0.73, 0.74, 0.75, 0.76, 0.77, 0.78, 0.79, 0.8, 0.81, 0.82, 0.83, 0.84, 0.85, 0.86, 0.87, 0.88, 0.89, 0.9, 0.
- the second buffer (e.g., Tris HCl) is included in the lyophilized composition at a concentration, after reconstitution, of at least 0.1, at least 0.2, at least 0.3, at least 0.4, at least 0.5, at least 0.6, at least 0.7, at least 0.8, at least 0.9, or at least 1 mg/mL.
- the second buffer (e.g., Tris HCl) is included in the lyophilized composition at a concentration, after reconstitution, of between 0.1 and 0.2, between 0.2 and 0.3, between 0.3 and 0.4, between 0.4 and 0.5, between 0.5 and 0.6, between 0.6 and 0.7, between 0.7 and 0.8, between 0.8 and 0.9, or between 0.9 and 1 mg/mL.
- the second buffer (e.g., Tris HCl) is included in the lyophilized composition at a concentration, after reconstitution, of or of about 0.5 and 0.65 mg/mL.
- the second buffer e.g., Tris HCl
- the second buffer is included in the lyophilized composition at a concentration, after reconstitution, of or of about 0.5 and 0.6 mg/mL.
- the second buffer e.g., Tris HCl
- the second buffer is included in the lyophilized composition at a concentration, after reconstitution, of or of about 0.55 and 0.65 mg/mL.
- the second buffer (e.g., Tris HCl) is or is not included in the lyophilized composition at a concentration, after reconstitution, of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 0.5, 0.51, 0.52, 0.53, 0.54, 0.55, 0.56, 0.57, 0.58, 0.59, 0.6, 0.61, 0.62, 0.63, 0.64, or 0.65 mg/mL.
- the second buffer (e.g., Tris HCl) is included in the lyophilized composition at a concentration, after reconstitution, of or of about 0.57 mg/mL.
- a RSV RNA-LNP composition comprises a stabilizing agent.
- the stabilizing agent may comprise any one or more stabilizing agents disclosed herein.
- the stabilizing agent also functions as a cryoprotectant.
- the stabilizing agent comprises sucrose.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is or is not included in the composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85,
- the RSV RNA-LNP composition is a liquid composition
- the stabilizing agent e.g., sucrose
- the stabilizing agent is or is not included in the liquid composition at a concentration of at least, at most, between (inclusive or exclusive) of, or exactly 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, or 130 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent e.g., sucrose
- the stabilizing agent is included in the liquid composition at a concentration of at least 70, at least 75, at least 80, at least 85, at least 90, at least 95, at least 100, at least 105, at least 110, at least 115, at least 120, at least 125, or at least 130 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is included in the liquid composition at a concentration of between 70 and 80, between 80 and 90, between 90 and 100, between 100 and 110, between 110 and 120, or between 120 and 130 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is included in the liquid composition at a concentration of or of about 95 to 110 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is included in the liquid composition at a concentration of or of about 95 to 105 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is included in the liquid composition at a concentration of or of about 100 to 110 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is or is not included in the liquid composition at a concentration of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, or 110 mg/mL.
- the stabilizing agent e.g., sucrose
- the RSV RNA-LNP composition is a lyophilized composition
- the stabilizing agent e.g., sucrose
- the stabilizing agent is or is not included in the lyophilized composition at a concentration, after reconstitution, of at least, at most, between (inclusive or exclusive) of, or exactly 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, or 80 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is included in the lyophilized composition at a concentration, after reconstitution, of at least 20, at least 25, at least 30, at least 35, at least 40, at least 45, at least 50, at least 55, at least 60, at least 65, at least 70, at least 75, or at least 80 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is included in the lyophilized composition at a concentration, after reconstitution, of between 20 to 30, between 30 to 40, between 40 to 50, between 50 to 60, between 60 to 70, or between 70 to 80 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is included in the lyophilized composition at a concentration, after reconstitution, of or of about 35 to 50 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is included in the lyophilized composition at a concentration, after reconstitution, of or of about 35 to 45 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is included in the lyophilized composition at a concentration, after reconstitution, of or of about 40 to 50 mg/mL.
- the stabilizing agent e.g., sucrose
- the stabilizing agent is or is not included in the lyophilized composition at a concentration, after reconstitution, of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, or 50 mg/mL.
- the stabilizing agent e.g., sucrose
- lyophilized compositions are reconstituted in a suitable carrier and/or diluent.
- the carrier and/or diluent may comprise any one or more carriers and/or diluents disclosed herein.
- the carrier and/or diluent comprises a salt diluent, such as sodium chloride (NaCl) (e.g., saline, e.g., physiological or normal saline).
- the sodium chloride may comprise 0.9% sodium chloride for injection.
- the lyophilized compositions are or are not reconstituted in at least, at most, between (inclusive or exclusive) of, or exactly 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 0.10, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20, 0.21, 0.22, 0.23, 0.24, 0.25, 0.26, 0.27, 0.28, 0.29, 0.30, 0.31, 0.32, 0.33, 0.34, 0.35, 0.36, 0.37, 0.38, 0.39, 0.40, 0.41, 0.42, 0.43, 0.44, 0.45, 0.46, 0.47, 0.48, 0.49, 0.50, 0.51, 0.52, 0.53, 0.54, 0.55, 0.56, 0.57, 0.58, 0.59, 0.60, 0.61, 0.62, 0.63, 0.64, 0.65, 0.66, 0.67, 0.68, 0.69, 0.70, 0.71, 0.72, 0.73,
- the lyophilized compositions are reconstituted in at least 0.1, at least 0.2, at least 0.3, at least 0.4, at least 0.5, at least 0.6, at least 0.7, at least 0.8, at least 0.9, or at least 1 mL of sodium chloride. In specific aspects, the lyophilized compositions are reconstituted in or in about 0.6 to 0.75 mL of sodium chloride/saline. In specific aspects, the lyophilized compositions are reconstituted in or in about 0.65 to 0.75 mL of sodium chloride/saline.
- the lyophilized compositions are or are not reconstituted in or in at least, at most, exactly, between (inclusive or exclusive) any two of, or about 0.6, 0.61, 0.62, 0.63, 0.64, 0.65, 0.66, 0.67, 0.68, 0.69, 0.7, 0.71, 0.72, 0.73, 0,74, or 0.75 mL of sodium chloride/saline.
- the salt diluent e.g., NaCl
- the salt diluent is or is not included in the lyophilized composition at a concentration, after reconstitution, of at least, at most, between (inclusive or exclusive) of, or exactly 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 6.5, 7, 7.5, 8, 8.5, 9, 9.5, 10, 10.5, 11, 11.5, 12, 12.5, 13, 13.5, 14, 14.5, 15, 15.5, 16, 16.5, 17, 17.5, 18, 18.5, 19, 19.5, 20, 20.5, 21, 21.5, 22, 22.5, 23, 23.5, 24, 24.5, 25, 25.5, 26, 26.5, 27, 27.5, 28, 28.5, 29, 29.5, 30, 30.5, 31, 31.5, 32, 32.5, 33, 33.5, 34, 34.5, 35, 35.5, 36, 36.5, 37, 37.5, 38, 38.5, 39, 39.5, 40, 40.5, 41, 41.5, 42, 42.5, 43, 43.5, 44, 44.5, 45, 45.5, 46, 46.5
- the salt diluent e.g., NaCl
- the salt diluent is or is not included in the lyophilized composition at a concentration, after reconstitution, of in at least, at most, between (inclusive or exclusive) of, or exactly 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 6.5, 7, 7.5, 8, 8.5, 9, 9.5, 10, 10.5, 11, 11.5, 12, 12.5, 13, 13.5, 14, 14.5, 15, 15.5, 16, 16.5, 17, 17.5, 18, 18.5, 19, 19.5, or 20 mg/mL.
- the salt diluent e.g., NaCl
- the salt diluent is included in the lyophilized composition at a concentration, after reconstitution, of at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least 14, at least 15, or at least 20 mg/mL.
- the salt diluent e.g., NaCl
- the salt diluent is included in the lyophilized composition at a concentration, after reconstitution, of between or between about 5 and 15 mg/mL.
- the salt diluent e.g., NaCl
- the salt diluent is included in the lyophilized composition at a concentration, after reconstitution, of between or between about 5 and 10 mg/mL.
- the salt diluent e.g., NaCl
- the salt diluent is or is not included in the lyophilized composition at a concentration, after reconstitution, of or of at least, at most, exactly, between (inclusive or exclusive) any two of, or about 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 mg/mL.
- the salt diluent is included in the lyophilized composition at a concentration, after reconstitution, of or of about 9 mg/mL.
- the pH of the RSV RNA-LNP composition may or may not be at least, at most, exactly, or between (inclusive or exclusive) of pH 6.5, 6.6, 6.7, 6.8, 6.9, 7.0, 7.1, 7.2, 7.3, 7.4, 7.5, 7.6, 7.7, 7.8, 7.9, 8.0, 8.1, 8.2, 8.3, 8.4, or 8.5, or any range or value derivable therein.
- the RSV RNA-LNP composition is at a pH of at least 6.5, at least 7.0, at least 7.5, at least 8.0, or at least 8.5.
- the RSV RNA-LNP composition is at a pH between 6.0 and 7.5, between 6.5 and 7.5, between 7.0 and 8.0, between and 7.5 and 8.5. In specific aspects, the RSV RNA-LNP composition is between 7.0 and 8.0. In specific aspects, the RSV RNA-LNP composition is or is not at least, at most, exactly, between (inclusive or exclusive) any two of, or about pH 7.0, 7.1, 7.2, 7.3, 7.4, 7.5, 7.6, 7.7, 7.8, 7.9, or 8.0. In specific aspects, the RSV RNA-LNP composition is at or at about pH 7.4. In some aspects, sodium hydroxide buffer may be used for a buffer pH adjustment.
- a RSV RNA-LNP composition comprises a RSV RNA polynucleotide encoding a RSV polypeptide as disclosed herein, encapsulated in LNPs with a lipid composition of an cationic lipid at a concentration of or of about 0.8 to 0.95 mg/mL, a PEGylated lipid at a concentration of or of about 0.05 to 0.15 mg/mL, a first structural lipid at a concentration of or of about 0.1 to 0.25 mg/mL, and a second structural lipid at a concentration of or of about 0.3 to 0.45 mg/mL.
- one or more of the foregoing elements may be excluded from the RSV RNA-LNP composition.
- a RSV RNA-LNP composition comprises a RSV RNA polynucleotide encoding a RSV polypeptide as disclosed herein, encapsulated in LNPs with a lipid composition of ALC-0315 at a concentration of or of about 0.8 to 0.95 mg/mL, ALC-0159 at a concentration of or of about 0.05 to 0.15 mg/mL, DSPC at a concentration of or of about 0.1 to 0.25 mg/mL, and cholesterol at a concentration of about 0.3 to 0.45 mg/mL.
- one or more of the foregoing elements may be excluded from the RSV RNA-LNP composition.
- the RSV RNA-LNP composition is a liquid RSV RNA-LNP composition
- the liquid RSV RNA-LNP composition further comprises a buffer composition comprising a first buffer at a concentration of or of about 0.15 to 0.3 mg/mL, a second buffer at a concentration of or of about 1.25 to 1.4 mg/mL, and a stabilizing agent at a concentration of or of about 95 to 110 mg/mL.
- the RSV RNA-LNP composition is a liquid RSV RNA- LNP composition
- the liquid RSV RNA-LNP composition further comprises a Tris buffer composition comprising tromethamine at a concentration of or of about 0.1 to 0.3 mg/mL, Tris HCl at a concentration of or of about 1.25 to 1.4 mg/mL, and sucrose at a concentration of or of about 95 to 110 mg/mL.
- a Tris buffer composition comprising tromethamine at a concentration of or of about 0.1 to 0.3 mg/mL, Tris HCl at a concentration of or of about 1.25 to 1.4 mg/mL, and sucrose at a concentration of or of about 95 to 110 mg/mL.
- one or more of the foregoing elements may be excluded from the RSV RNA-LNP composition.
- a liquid RSV RNA-LNP composition comprises an cationic lipid at a concentration of or of about 0.8 to 0.95 mg/mL, a PEGylated lipid at a concentration of or of about 0.05 to 0.15 mg/mL, a first structural lipid at a concentration of or of about 0.1 to 0.25 mg/mL, a second structural lipid at a concentration of or of about 0.3 to 0.45 mg/mL, and further comprises a first buffer at a concentration of or of about 0.1 to 0.3 mg/mL, a second buffer at a concentration of or of about 1.25 to 1.4 mg/mL, and a stabilizing agent at a concentration of or of about 95 to 110 mg/mL.
- a liquid RSV RNA-LNP composition comprises ALC-0315 at a concentration of or of about 0.8 to 0.95 mg/mL, ALC-0159 at a concentration of or of about 0.05 to 0.15 mg/mL, DSPC at a concentration of or of about 0.1 to 0.25 mg/mL, cholesterol at a concentration of or of about 0.3 to 0.45 mg/mL, and further comprises a Tris buffer composition comprising tromethamine at a concentration of or of about 0.1 to 0.3 mg/mL, Tris HCl at a concentration of or of about 1.25 to 1.4 mg/mL, and sucrose at a concentration of or of about 95 to 110 mg/mL.
- the RSV RNA-LNP composition is a lyophilized RSV RNA-LNP composition, and the lyophilized RSV RNA-LNP composition further comprises (after reconstitution) a first buffer at a concentration of or of about 0.01 and 0.15 mg/mL, a second buffer at a concentration of or of about 0.5 and 0.65 mg/mL, a stabilizing agent at a concentration of or of about 35 to 50 mg/mL, and a salt diluent at a concentration of between or between about 5 and 15 mg/mL.
- a first buffer at a concentration of or of about 0.01 and 0.15 mg/mL
- a second buffer at a concentration of or of about 0.5 and 0.65 mg/mL
- a stabilizing agent at a concentration of or of about 35 to 50 mg/mL
- a salt diluent at a concentration of between or between about 5 and 15 mg/mL.
- the RSV RNA-LNP composition is a lyophilized RSV RNA-LNP composition
- the lyophilized RSV RNA-LNP composition further comprises (after reconstitution) a Tris buffer composition comprising tromethamine at a concentration of or of about 0.01 and 0.15 mg/mL, Tris HCl at a concentration of or of about 0.5 and 0.65 mg/mL, sucrose at a concentration of or of about 35 to 50 mg/mL, and sodium chloride (NaCl) at a concentration of or of about 5 to 15 mg/mL.
- Tris buffer composition comprising tromethamine at a concentration of or of about 0.01 and 0.15 mg/mL, Tris HCl at a concentration of or of about 0.5 and 0.65 mg/mL, sucrose at a concentration of or of about 35 to 50 mg/mL, and sodium chloride (NaCl) at a concentration of or of about 5 to 15 mg/mL.
- a lyophilized RSV RNA-LNP composition comprises (after reconstitution) a cationic lipid at a concentration of or of about 0.8 to 0.95 mg/mL, a PEGylated lipid at a concentration of or of about 0.05 to 0.15 mg/mL, a first structural lipid at a concentration of or of about 0.1 to 0.25 mg/mL, a second structural lipid at a concentration of or of about 0.3 to 0.45 mg/mL, and further comprises a first buffer at a concentration of or of about 0.01 and 0.15 mg/mL, a second buffer at a concentration of or of about 0.5 and 0.65 mg/mL, a stabilizing agent at a concentration of or of about 35 to 50 mg/mL, and a salt diluent at a concentration of or of about 5 to 15 mg/mL.
- a lyophilized RSV RNA-LNP composition comprises (after reconstitution) ALC-0315 at a concentration of or of about 0.8 to 0.95 mg/mL, ALC-0159 at a concentration of or of about 0.05 to 0.15 mg/mL, DSPC at a concentration of or of about 0.1 to 0.25 mg/mL, cholesterol at a concentration of or of about 0.3 to 0.45 mg/mL, and further comprises tromethamine at a concentration of or of about 0.01 and 0.15 mg/mL, Tris HCl at a concentration of or of about 0.5 and 0.65 mg/mL, sucrose at a concentration of or of about 35 to 50 mg/mL, and NaCl at a concentration of or of about 5 to 15 mg/
- the lyophilized compositions are reconstituted in 0.6 to 0.75 mL of NaCl (saline).
- NaCl saline
- one or more of the foregoing elements may be excluded from the RSV RNA-LNP composition. Concentrations in the lyophilized RSV RNA-LNP composition above are determined post- reconstitution.
- a RSV RNA-LNP composition (pre-lyophilization) comprises a cationic lipid at a concentration of or of about 1.0 to 3.0 mg/mL, a PEGylated lipid at a concentration of or of about 0.10 to 0.35 mg/mL, a first structural lipid at a concentration of or of about 0.4 to 0.55 mg/mL, a second structural lipid at a concentration of or of about 0.85 to 1.0 mg/mL, and further comprises a first buffer at a concentration of or of about 0.1 and 0.3 mg/mL, a second buffer at a concentration of or of about 1.25 and 1.40 mg/mL, a stabilizing agent at a concentration of or of about 95 to 110 mg/mL.
- a RSV RNA-LNP composition (pre-lyophilization) comprises ALC- 0315 at a concentration of or of about 1.0 to 3.0 mg/mL, ALC-0159 at a concentration of or of about 0.10 to 0.35 mg/mL, DSPC at a concentration of or of about 0.4 to 0.55 mg/mL, cholesterol at a concentration of or of about 0.85 to 1.0 mg/mL, and further comprises tromethamine at a concentration of or of about 0.1 and 0.3 mg/mL, Tris HCl at a concentration of or of about 1.25 and 1.40 mg/mL, sucrose at a concentration of or of about 95 to 110 mg/mL.
- a RSV RNA-LNP composition is a liquid RSV RNA-LNP composition comprising a RSV RNA polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, encapsulated in LNPs with a lipid composition of an cationic lipid at a concentration of or of about 0.8 to 0.95 mg/mL, a PEGylated lipid at a concentration of or of about 0.05 to 0.15 mg/mL, a first structural lipid at a concentration of or of about
- a liquid RSV RNA-LNP composition comprises a RSV RNA polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, and more preferably of or of about 0.06 mg/mL, encapsulated in LNPs with a lipid composition of ALC-0315 at a concentration of or of about 0.8 to 0.95 mg/mL, ALC-0159 at a concentration of or of about 0.05 to 0.15 mg/mL, DSPC at a concentration of or of about 0.1 to 0.25 mg/mL, and cholesterol at a concentration of or of about 0.3 to 0.45 mg/mL, and further comprising
- the RSV RNA-LNP composition is a lyophilized RSV RNA-LNP composition comprising a RSV RNA polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, encapsulated in LNPs with a lipid composition of a cationic lipid at a concentration of or of about 0.8 to 0.95 mg/mL, a PEGylated lipid at a concentration of or of about 0.05 to 0.15 mg/mL, a first structural lipid at a concentration of or of about 0.1 to 0.25 mg/mL, and a second structural lipid at a concentration of or of about
- one or more of the foregoing elements may be excluded from the RSV RNA-LNP composition.
- the lyophilized compositions are reconstituted in 0.6 to 0.75 mL of the salt diluent. Concentrations in the lyophilized RSV RNA-LNP composition are determined post-reconstitution.
- a lyophilized RSV RNA-LNP composition comprises a RSV RNA polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, and more preferably of or of about 0.06 mg/mL, encapsulated in LNPs with a lipid composition of ALC-0315 at a concentration of or of about 0.8 to 0.95 mg/mL, ALC-0159 at a concentration of or of about 0.05 to 0.15 mg/mL, DSPC at a concentration of or of about 0.1 to 0.25 mg/mL, and cholesterol at a concentration of or of about 0.3 to 0.45 mg/mL, and further comprising tromethamine at a concentration of or of about 0.01 and 0.15 mg/mL, Tris
- the lyophilized compositions are reconstituted in 0.6 to 0.75 mL of the NaCl diluent (saline). Concentrations in the lyophilized RSV RNA-LNP composition are determined post-reconstitution.
- a RSV RNA-LNP composition (pre-lyophilization) comprises a RSV RNA polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, encapsulated in LNPs with a lipid composition of a cationic lipid at a concentration of or of about 1.0 to 3.0 mg/mL, a PEGylated lipid at a concentration of or of about 0.10 to 0.35 mg/mL, a first structural lipid at a concentration of or of about 0.4 to 0.55 mg/mL, a second structural lipid at a concentration of or of about 0.85 to 1.0 mg/mL, and further comprises a first buffer at a concentration of or of about 0.1 and 0.3 mg/mL, a
- a RSV RNA-LNP composition (pre-lyophilization) comprises a RSV RNA polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, and more preferably 0.15 mg/mL, encapsulated in LNPs with a lipid composition of comprises ALC-0315 at a concentration of or of about 1.0 to 3.0 mg/mL, ALC-0159 at a concentration of or of about 0.10 to 0.35 mg/mL, DSPC at a concentration of or of about 0.4 to 0.55 mg/mL, cholesterol at a concentration of or of about 0.85 to 1.0 mg/mL, and
- the liquid RNA-LNP immunogenic composition comprises an RNA molecule/polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, encapsulated in a LNP, and further comprising or comprising about 5 to 15 mM Tris buffer and about 200 to 400 mM sucrose at a pH of or of about 7.0 to 8.0.
- the liquid RNA-LNP immunogenic composition comprises an RNA molecule/polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, and more preferably of or of about 0.06 mg/mL, encapsulated in a LNP, and further comprising or comprising about 10 mM Tris buffer and 300 mM sucrose at a pH of or of about 7.4.
- the RNA-LNP immunogenic composition (pre-lyophilized) comprises an RNA molecule/polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, encapsulated in a LNP, and further comprising or comprising about 5 to 15 mM Tris buffer and 200 to 400 mM sucrose at a pH of or of about 7.0 to 8.0, and reconstituted with 0.9% sodium chloride diluent.
- the RNA-LNP immunogenic composition (pre-lyophilized) comprises an RNA molecule/polynucleotide encoding a RSV polypeptide as disclosed herein at a concentration of at least, at most, exactly, or between (inclusive or exclusive) of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL, preferably of or of about 0.01 to 0.09 mg/mL, and more preferably 0.15 mg/mL, encapsulated in a LNP, and further comprising or comprising about 10 mM Tris buffer and 300 mM sucrose at a pH of or of about 7.4, and reconstituted with 0.9% sodium chloride diluent.
- a pharmaceutical composition described herein is an immunogenic composition for inducing an immune response.
- an immunogenic composition is a vaccine.
- the compositions described herein include at least one isolated nucleic acid or polypeptide molecule as described herein.
- the immunogenic compositions comprise nucleic acids, and the immunogenic compositions are nucleic acid vaccines.
- the immunogenic compositions comprise RNA (e.g., mRNA, saRNA), and vaccines are RNA vaccines.
- the immunogenic compositions comprise DNA, and vaccines are DNA vaccines.
- the immunogenic compositions comprise a polypeptide
- vaccines are polypeptide vaccines.
- Conditions and/or diseases that may be treated with the nucleic acid and/or peptide or polypeptide compositions include, but are not limited to, those caused and/or impacted by infection, cancer, rare diseases, and other diseases or conditions caused by overproduction, underproduction, and/or improper production of protein or nucleic acids.
- the composition is substantially free of one or more impurities or contaminants and, for instance, includes nucleic acid or polypeptide molecules that are equal to at least, at most, exactly, or between (inclusive or exclusive) of 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% pure; at least 98% pure, or at least 99% pure.
- the present disclosure includes methods for preventing, treating and/or ameliorating an infection, disease or condition in a subject, including administering to a subject an effective amount of an RNA molecule that includes at least one open reading frame encoding a polypeptide or composition described herein. As such, the disclosure contemplates vaccines for use in both active and passive immunization aspects.
- Immunogenic compositions may be prepared from RNA molecules encoding polypeptide(s), such as RSV preF polypeptides.
- immunogenic compositions are lyophilized for more ready formulation into a desired vehicle.
- the preparation of vaccines that contain nucleic acid and/or peptide or polypeptide as active ingredients is generally well understood in the art, as exemplified by U.S. Patents 4,608,251; 4,601,903; 4,599,231; 4,599,230; 4,596,792; and 4,578,770, all of which are incorporated herein by reference.
- such vaccines are prepared as injectables either as liquid solutions or suspensions; solid forms suitable for solution in or suspension in liquid prior to injection may also be prepared.
- the preparation may also be emulsified.
- the active immunogenic ingredient is often mixed with excipients that are pharmaceutically acceptable and compatible with the active ingredient. Suitable excipients are, for example, water, saline, dextrose, glycerol, ethanol, or the like and combinations thereof.
- the vaccine may contain amounts of auxiliary substances such as wetting or emulsifying agents, pH buffering agents, or adjuvants that enhance the effectiveness of the vaccines.
- vaccines are formulated with a combination of substances, as described in U.S.
- Patents 6,793,923 and 6,733,754 which are incorporated herein by reference.
- one or more of the foregoing elements may be excluded from a vaccine.
- Vaccines may be conventionally administered parenterally, by injection, for example, either subcutaneously or intramuscularly.
- Additional formulations which are suitable for other modes of administration include suppositories and, in some cases, oral formulations.
- binders and carriers may include, for example, polyalkylene glycols or triglycerides; such suppositories may be formed from mixtures containing the active ingredient in the range of or of about 0.5% to about 10%.
- suppositories may be formed from mixtures containing the active ingredient in the range of or of about 1% to about 2%.
- Oral formulations include such normally employed excipients as, for example, pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium carbonate and the like. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing excipients may be excluded from an oral formulation. These compositions take the form of solutions, suspensions, tablets, pills, capsules, sustained release formulations or powders and contain or contain about 10% to about 95% of active ingredient.
- the polypeptide-encoding nucleic acid constructs and polypeptides may be formulated into a vaccine as neutral or salt forms.
- “Pharmaceutically acceptable salt” includes both acid and base addition salts.
- “Pharmaceutically acceptable acid addition salt” refers to those salts which retain the biological effectiveness and properties of the free bases, which are not biologically or otherwise undesirable, and which are formed with inorganic acids such as, but are not limited to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, and organic acids such as, but not limited to, acetic acid, 2,2-dichloroacetic acid, adipic acid, alginic acid, ascorbic acid, aspartic acid, benzenesulfonic acid, benzoic acid, 4-acetamidobenzoic acid, camphoric acid, camphor-10-sulfonic acid, capric acid, caproic acid, caprylic acid, carbonic acid, cinnamic acid, citric acid, cyclamic acid, dodecylsulfuric acid, ethane-1,2-disulfonic acid
- “Pharmaceutically acceptable base addition salt” refers to those salts which retain the biological effectiveness and properties of the free acids, which are not biologically or otherwise undesirable. These salts are prepared from addition of an inorganic base or an organic base to the free acid. Salts derived from inorganic bases include, but are not limited to, the sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum salts and the like. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing inorganic bases may be excluded. Preferred inorganic salts are the ammonium, sodium, potassium, calcium, and magnesium salts.
- Salts derived from organic bases include, but are not limited to, salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as ammonia, isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, diethanolamine, ethanolamine, deanol, 2-dimethylaminoethanol, 2-diethylaminoethanol, dicyclohexylamine, lysine, arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine, benethamine, benzathine, ethylenediamine, glucosamine, methylglucamine, theobromine, triethanolamine, tromethamine, purines, piperazine, piperidine, N-ethylpiperidine, polyamine resins and the like.
- basic ion exchange resins such as
- Particularly preferred organic bases are isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexylamine, choline, and caffeine. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing organic bases may be excluded.
- the polypeptide-encoding nucleic acid constructs and polypeptides, or their pharmaceutically acceptable salts may contain one or more asymmetric centers and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)- or, as (D)- or (L)- for amino acids.
- the present disclosure is meant to include all such possible isomers, as well as their racemic and optically pure forms.
- Optically active (+) and (-), (R)- and (5)-, or (D)- and (L)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques, for example, chromatography and fractional crystallization.
- Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC).
- HPLC high pressure liquid chromatography
- a “stereoisomer” refers to a compound made up of the same atoms bonded by the same bonds but having different three- dimensional structures, which are not interchangeable.
- the present invention contemplates various stereoisomers and mixtures thereof and includes “enantiomers”, which refers to two stereoisomers whose molecules are non-superimposable mirror images of one another.
- a “tautomer” refers to a proton shift from one atom of a molecule to another atom of the same molecule. The present invention disclosure tautomers of any said compounds.
- Suitable protecting groups for hydroxy include trialkylsilyl or diarylalkylsilyl (for example, t- butyldimethylsilyl, t-butyldiphenylsilyl or trimethylsilyl), tetrahydropyranyl, benzyl, and the like.
- Suitable protecting groups for amino, amidino and guanidino include t-butoxycarbonyl, benzyloxycarbonyl, and the like.
- Suitable protecting groups for mercapto include -C(O)-R" (where R" is alkyl, aryl or arylalkyl), p-methoxybenzyl, trityl and the like.
- Suitable protecting groups for carboxylic acid include alkyl, aryl or arylalkyl esters.
- the protecting group may also be a polymer resin such as a Wang resin, Rink resin or a 2- chlorotrityl-chloride resin. In some aspects, 1, 2, 3, 4, 5, or more of the foregoing protecting groups may be excluded.
- Protecting groups may be added or removed in accordance with standard techniques, which are known to one skilled in the art (see, e.g., Green, T.W. and P.G.M. Wutz, Protective Groups in Organic Synthesis (1999), 3rd Ed., Wiley) and as described herein.
- prodrugs All prodrugs of compounds of this invention are included within the scope of the invention.
- vaccines are administered in a manner compatible with the dosage formulation, and in such amount as will be therapeutically effective and immunogenic.
- the quantity to be administered depends on the subject to be treated, including the capacity of the individual’s immune system to synthesize antibodies and the degree of protection desired. Precise amounts of active ingredient required to be administered depend on the judgment of the practitioner.
- suitable dosage ranges are of the order of several hundred micrograms of active ingredient per vaccination.
- Suitable regimes for initial administration and booster shots are also variable, but are typified by an initial administration followed by subsequent inoculations and/or other administrations.
- the manner of application may be varied widely. Any of the conventional methods for administration of a vaccine are applicable. These are believed to include oral application within a solid physiologically acceptable base or in a physiologically acceptable dispersion, parenterally, by injection and the like.
- the dosage of the vaccine will depend on the route of administration and will vary according to the size and health of the subject. In certain aspects, it will be desirable to have one administration of the vaccine. In some aspects, it will be desirable to have multiple administrations of the vaccine, e.g., 2, 3, 4, 5, 6, or more administrations.
- the vaccinations may be at 1, 2, 3, 4, 5, 6, 7, 8, to 5, 6, 7, 8, 9 ,10, 11, or 12 twelve week intervals, including all ranges there between. In some aspects, vaccinations may be at 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 month intervals, including all ranges there between. Periodic boosters at intervals of 1-5 years may be desirable to maintain protective levels of the antibodies.
- a pharmaceutically acceptable carrier may include the liquid or non-liquid basis of a composition. If a composition is provided in liquid form, the carrier may be water, such as pyrogen-free water; isotonic saline or buffered (aqueous) solutions, e.g. phosphate, citrate buffered solutions.
- Water or a buffer such as an aqueous buffer, may be used, containing a sodium salt, a calcium salt, and and/or a potassium salt.
- the sodium, calcium and/or potassium salts may occur in the form of their halogenides, e.g. chlorides, iodides, or bromides, in the form of their hydroxides, carbonates, hydrogen carbonates, or sulfates, etc.
- Examples of sodium salts include, but are not limited to, NaCI, Nal, NaBr, Na 2 CO 3 , NaHCO 3 , Na 2 SO 4 , Na 2 HPO 4 , Na 2 HPO 4 ⁇ 2 H 2 O
- examples of potassium salts include, but are not limited to, KCI, Kl, KBr, K 2 CO 3 , KHCO 3 , K 2 SO 4 , KH 2 PO 4
- examples of calcium salts include, but are not limited to, CaCl 2 , Cal 2 , CaBr2, CaCO3, CaSO4, Ca(OH)2.
- Examples of further carriers may include sugars, such as, for example, lactose, glucose, trehalose and sucrose; starches, such as, for example, com starch or potato starch; dextrose; cellulose and its derivatives, such as, for example, sodium carboxymethylcellulose, ethylcellulose, cellulose acetate; powdered tragacanth; malt; gelatin; tallow; solid glidants, such as, for example, stearic acid, magnesium stearate; calcium sulfate; vegetable oils, such as, for example, groundnut oil, cottonseed oil, sesame oil, olive oil, corn oil and oil from theobroma; polyols, such as, for example, polypropylene glycol, glycerol, sorbitol, mannitol and polyethylene glycol; alginic acid.
- sugars such as, for example, lactose, glucose, trehalose and sucrose
- starches such as, for example,
- Suitable adjuvants include all acceptable immunostimulatory compounds, such as cytokines, toxins, or synthetic compositions. A number of adjuvants may be used to enhance an antibody response. Adjuvants include, but are not limited to, oil-in-water emulsions, water-in-oil emulsions, mineral salts, polynucleotides, and natural substances.
- Specific adjuvants that may be used include Freund’s adjuvant, oil such as MONTANIDE® ISA51, IL1, IL2, IL3, IL4, IL5, IL6, IL7, IL8, IL9, IL10, IL12, alpha-interferon, PTNGg, GM-CSF, GMCSP, BCG, LT-a, aluminum salts, such as aluminum hydroxide or other aluminum compound, MDP compounds, such as thur- MDP and nor-MDP, CGP (MTP-PE), lipid A, monophosphoryl lipid A (MPL), lipopeptides (e.g., Pam3Cys).
- Freund’s adjuvant oil such as MONTANIDE® ISA51, IL1, IL2, IL3, IL4, IL5, IL6, IL7, IL8, IL9, IL10, IL12, alpha-interferon, PTNGg, GM-CSF, GMCSP, BCG, LT-a
- RIBI which contains three components extracted from bacteria, MPL, trehalose dimycolate (TDM), and cell wall skeleton (CWS) in a 2% squalene/Tween 80 emulsion. MHC antigens may even be used.
- Various methods of achieving adjuvant affect for the vaccine includes use of agents such as aluminum hydroxide or phosphate (alum), commonly used as about 0.05 to about 0.1% solution in phosphate buffered saline, admixture with synthetic polymers of sugars (CARBOPOL®) used as an about 0.25% solution, aggregation of the protein in the vaccine by heat treatment with temperatures ranging between about 70° to about 101°C for a 30-second to 2-minute period, respectively.
- agents such as aluminum hydroxide or phosphate (alum), commonly used as about 0.05 to about 0.1% solution in phosphate buffered saline, admixture with synthetic polymers of sugars (CARBOPOL®) used as an about 0.25% solution,
- Fab pepsin-treated antibodies to albumin
- mixture with bacterial cells e.g., C. parvum
- endotoxins or lipopolysaccharide components of Gram-negative bacteria emulsion in physiologically acceptable oil vehicles (e.g., mannide mono-oleate (Aracel A)); or emulsion with a 20% solution of a perfluorocarbon (FLUOSOL-DA®) used as a block substitute
- FLUOSOL-DA® perfluorocarbon
- 1, 2, 3, 4, 5, or more of the foregoing adjuvants may be excluded.
- BRM biologic response modifiers
- BRMs have been shown to upregulate T cell immunity or downregulate suppresser cell activity.
- BRMs include, but are not limited to, Cimetidine (CIM; 1200 mg/d) (Smith/Kline, PA); or low-dose Cyclophosphamide (CYP; 300 mg/m 2 ) (Johnson/ Mead, NJ) and cytokines such as ⁇ -interferon, IL-2, or IL-12 or genes encoding proteins involved in immune helper functions, such as B-7.
- C. COMBINATION THERAPY The compositions and related methods of the present disclosure, particularly administration of an RNA molecule encoding a RSV preF polypeptide, may also be used in combination with the administration of one or more other therapeutic agents.
- antiviral therapies such as acyclovir, valacyclovir, and famciclovir, or various combinations of antivirals.
- one or more therapies to treat one or more symptoms of RSV infection including, but not limited to, steroids including corticosteroids, anti-inflammatories including acetaminophen or ibuprofen, pain-relief agents, creams or lotions to relieve itching, cool compresses, or various combinations thereof.
- steroids including corticosteroids
- anti-inflammatories including acetaminophen or ibuprofen
- pain-relief agents creams or lotions to relieve itching, cool compresses, or various combinations thereof.
- 1, 2, 3, 4, 5, or more of the foregoing therapeutic agents may be excluded.
- Such combination therapy includes administration of a single pharmaceutical dosage formulation of a composition of the invention and one or more additional active agents, as well as administration of the composition of the invention and each active agent in its own separate pharmaceutical dosage formulation.
- a composition of the invention and the other active agent can be administered to the patient together in a single dosage composition such as an injection or tablet or capsule, or each agent administered in separate oral dosage formulations.
- the compounds of the invention and one or more additional active agents can be administered at essentially the same time, e.g., concurrently, or at separately staggered times, e.g., sequentially; combination therapy is understood to include all these regimens.
- a vaccine and/or therapy is used in conjunction with antiviral treatment.
- the vaccine and/or therapy may precede or follow treatment with another agent by intervals ranging from minutes to weeks.
- the other agents and/or vaccines are administered separately, one would generally ensure that a significant period of time did not expire between the time of each delivery, such that the agent and immunogenic composition would still be able to exert an advantageously combined effect on the subject.
- a pharmaceutical composition described herein may be administered intravenously, intranasally, subcutaneously, intradermally or intramuscularly.
- the RSV RNA molecules and/or RNA-LNP compositions are administered intramuscularly.
- the pharmaceutical composition is formulated for local administration or systemic administration. Systemic administration may include enteral administration, which involves absorption through the gastrointestinal tract, or parenteral administration.
- parenteral administration refers to the administration in any manner other than through the gastrointestinal tract, such as by intravenous injection.
- the pharmaceutical composition is formulated for intramuscular administration.
- the pharmaceutical composition is formulated for systemic administration, e.g., for intravenous administration. In some aspects, 1, 2, 3, or more of the foregoing administration routes may be excluded.
- Pharmaceutical compositions may be formulated into preparations in solid, semi-solid, liquid, lyophilized, frozen, and/or gaseous forms, such as tablets, capsules, powders, granules, ointments, solutions, suspensions, suppositories, injections, inhalants, gels, microspheres, and aerosols.
- compositions described herein are formulated so as to allow the active ingredients contained therein to be bioavailable upon administration of the composition to a patient.
- compositions that will be administered to a subject or patient take the form of one or more dosage units, where for example, a tablet may be a single dosage unit, and a container of a compound in aerosol form may hold a plurality of dosage units.
- the composition to be administered will, in any event, contain a therapeutically and/or prophylactically effective amount of a compound within the scope of this disclosure, or a pharmaceutically acceptable salt thereof, for treatment of a disease or condition of interest in accordance with the teachings described herein.
- a pharmaceutical composition within the scope of this disclosure may be in the form of a solid or liquid and may be frozen or lyophilized.
- the carrier(s) are particulate, so that the compositions are, for example, in tablet or powder form.
- the carrier(s) may be liquid, with the compositions being, for example, an oral syrup, injectable liquid, or an aerosol, which is useful in, for example, inhalatory administration.
- the pharmaceutical composition when intended for oral administration, is in either solid or liquid form, where semi-solid, semi-liquid, suspension, and gel forms are included within the forms considered herein as either solid or liquid.
- the pharmaceutical composition may be formulated into a powder, granule, compressed tablet, pill, capsule, chewing gum, wafer or the like form. Such a solid composition will typically contain one or more inert diluents or edible carriers.
- binders such as carboxymethylcellulose, ethyl cellulose, microcrystalline cellulose, gum tragacanth, or gelatin
- excipients such as starch, lactose, or dextrins
- disintegrating agents such as alginic acid, sodium alginate, PRIMOJEL®, corn starch and the like
- lubricants such as magnesium stearate or STEROTEX®
- glidants such as colloidal silicon dioxide
- sweetening agents such as sucrose or saccharin
- a flavoring agent such as peppermint, methyl salicylate, or orange flavoring
- a coloring agent such as peppermint, methyl salicylate, or orange flavoring
- the pharmaceutical composition When the pharmaceutical composition is in the form of a capsule, for example, a gelatin capsule, it may contain, in addition to materials of the above type, a liquid carrier such as polyethylene glycol or oil. In some aspects, 1, 2, 3, or more of the foregoing elements may be excluded from a solid composition.
- the pharmaceutical composition may be in the form of a liquid, for example, an elixir, syrup, solution, emulsion or suspension.
- the liquid may be for oral administration or for delivery by injection, as two examples.
- compositions when intended for oral administration, compositions contain, in addition to the present compounds, one or more of a sweetening agent, preservatives, dye/colorant, and flavor enhancer.
- a liquid pharmaceutical composition may include or exclude one or more of the following adjuvants: sterile diluents such as water for injection, saline solution, e.g., physiological saline, Ringer’s solution, isotonic sodium chloride, fixed oils such as synthetic mono or diglycerides which may serve as the solvent or suspending medium, polyethylene glycols, glycerin, propylene glycol or other solvents; antibacterial agents such as benzyl alcohol or methyl paraben; antioxidants such as ascorbic acid or sodium bisulfite; chelating agents such as ethylenediaminetetraacetic acid; buffers such as acetates, citrates, or phosphates; and agents for the adjustment of
- the parenteral preparation may be enclosed in ampoules, disposable syringes, or multiple dose vials made of glass or plastic.
- physiological saline is the adjuvant.
- an injectable pharmaceutical composition is sterile.
- a liquid pharmaceutical composition intended for either parenteral or oral administration should contain an amount of a compound such that a suitable dosage will be obtained.
- the pharmaceutical composition may include various materials, which modify the physical form of a solid or liquid dosage unit.
- the composition may include materials that form a coating shell around the active ingredients.
- the materials that form the coating shell are typically inert, and may be, for example, sugar, shellac, or other enteric coating agents.
- the pharmaceutical composition may include dosage units that can be administered as an aerosol.
- aerosol denote a variety of systems ranging from those of colloidal nature to systems consisting of pressurized packages. Delivery may be by a liquefied or compressed gas or by a suitable pump system that dispenses the active ingredients. Aerosols of compounds may be delivered in single phase, bi-phasic, or tri-phasic systems in order to deliver the active ingredient(s). Delivery of the aerosol includes the necessary container, activators, valves, subcontainers, and the like, which together may form a kit. One skilled in the art, without undue experimentation may determine preferred aerosols.
- the pharmaceutical compositions may be prepared by methodology well known in the pharmaceutical art.
- a pharmaceutical composition intended to be administered by injection may be prepared by combining the nucleic acid or polypeptide with sterile, distilled water or other carrier so as to form a solution.
- a surfactant may be added to facilitate the formation of a homogeneous solution or suspension.
- Surfactants are compounds that non-covalently interact with a compound consistent with the teachings herein so as to facilitate dissolution or homogeneous suspension of the compound in the aqueous delivery system.
- compositions according to the present disclosure are generally applied in a “therapeutically effective amount” or a “prophylactically effective amount” and in “a pharmaceutically acceptable preparation.”
- pharmaceutically acceptable refers to the non-toxicity of a material which does not interact with the action of the active component of the pharmaceutical composition.
- therapeutically effective amount and prophylactically effective amount refer to the amount which achieves a desired reaction or a desired effect alone or together with further doses.
- the desired reaction relates to inhibition of the course of the disease. This comprises slowing down the progress of the disease and, in particular, interrupting and/or reversing the progress of the disease.
- the desired reaction in a treatment of a disease may also be delay of the onset and/or a prevention of the onset of said disease or said condition.
- the compositions within the scope of the disclosure are administered in a therapeutically and/or prophylactically effective amount, which will vary depending upon a variety of factors including the activity of the specific therapeutic and/or prophylactic agent employed; the metabolic stability and length of action of the therapeutic and/or prophylactic agent; the individual parameters of the patient, including the age, body weight, general health, gender, and diet of the patient; the mode, time, and/or duration of administration; the rate of excretion; the drug combination; the severity of the particular disorder or condition; and the subject undergoing therapy.
- 1, 2, 3, 4, 5, or more of the factors may be excluded from determining a therapeutically and/or prophylactically effective amount. Accordingly, the doses administered of the compositions described herein may depend on various of such parameters. In the case that a reaction in a patient is insufficient with an initial dose, higher doses (or effectively higher doses achieved by a different, more localized route of administration) may be used.
- compositions may be administered at dosage levels sufficient to deliver 0.0001 ng/ ⁇ g/mg per kg to 100 ng/ ⁇ g/mg per kg, 0.001 ng/ ⁇ g/mg per kg to 0.05 ng/ ⁇ g/mg per kg, 0.005 ng/ ⁇ g/mg per kg to 0.05 ng/ ⁇ g/mg per kg, 0.001 ng/ ⁇ g/mg per kg to 0.005 ng/ ⁇ g/mg per kg, 0.05 ng/ ⁇ g/mg per kg to 0.5 ng/ ⁇ g/mg per kg, 0.01 ng/ ⁇ g/mg per kg to 50 ng/ ⁇ g/mg per kg, 0.1 ng/ ⁇ g/mg per kg to 40 ng/ ⁇ g/mg per kg, 0.5 ng/ ⁇ g/mg per kg to 30 ng/ ⁇ g/mg per kg, 0.01 ng/ ⁇ g/mg per kg to 10
- compositions may or may not be administered at dosage levels sufficient to deliver at least, at most, exactly, or between (inclusive or exclusive) any two of 0.0001, 0.0002, 0.0003, 0.0004, 0.0005, 0.0006, 0.0007, 0.0008, 0.0009, 0.001, 0.002, 0.003, 0.004, 0.005, 0.006, 0.007, 0.008, 0.009, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32,
- compositions may or may not be administered at a total dose of or at dosage levels sufficient to deliver a total dose of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.0001, 0.0002, 0.0003, 0.0004, 0.0005, 0.0006, 0.0007, 0.0008, 0.0009, 0.001, 0.002, 0.003, 0.004, 0.005, 0.006, 0.007, 0.008, 0.009, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46
- compositions may or may not be administered at a total dose of or at dosage levels sufficient to deliver a total dose of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.0001, 0.0002, 0.0003, 0.0004, 0.0005, 0.0006, 0.0007, 0.0008, 0.0009, 0.001, 0.002, 0.003, 0.004, 0.005, 0.006, 0.007, 0.008, 0.009, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46
- compositions may or may not be administered at dose levels of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.01, 0.15, 0.30, 0.45, 0.60, 0.75, or 0.90 mg/mL RSV RNA encapsulated in LNP.
- compositions e.g., RSV RNA-LNP compositions
- compositions may or may not be administered at a total dose of or at dosage levels sufficient to deliver a total dose of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.0001, 0.0002, 0.0003, 0.0004, 0.0005, 0.0006, 0.0007, 0.0008, 0.0009, 0.001, 0.002, 0.003, 0.004, 0.005, 0.006, 0.007, 0.008, 0.009, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55
- compositions may or may not be administered at dose levels of at least, at most, exactly, or between (inclusive or exclusive) any two of 1, 15, 30, 45, 60, 75, 90, 100 or higher ⁇ g/mL RSV RNA encapsulated in LNP.
- compositions e.g., RSV RNA-LNP compositions
- the desired dosage may be delivered multiple times a day (e.g., 1, 2, 3, 4, 5, or more times a day), every other day, every third day, every week, every two weeks, every three weeks, every four weeks, every 2 months, every three months, every 6 months, every year, etc.
- the desired dosage may be delivered using a single-dose administration.
- the desired dosage may be delivered using multiple administrations (e.g., two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, or more administrations). When multiple administrations are employed, split dosing regimens may be used.
- the time of administration between the initial administration of the composition and a subsequent administration of the composition may be, but is not limited to, 1 minute, 2 minutes, 3 minutes, 4 minutes, 5 minutes, 6 minutes, 7 minutes, 8 minutes, 9 minutes, 10 minutes, 15 minutes, 20 minutes 35 minutes, 40 minutes, 45 minutes, 50 minutes, 55 minutes, 1 hour, 2 hours, 3 hours, 4 hours, 5 hours, 6 hours, 7 hours, 8 hours, 9 hours, 10 hours, 11 hours, 12 hours, 13 hours, 14 hours, 15 hours, 16 hours, 17 hours, 18 hours, 19 hours, 20 hours, 21 hours, 22 hours, 23 hours, 1 day, 36 hours, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 10 days, 2 weeks, 3 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 1 year, 18 months, 2 years, 3 years, 4 years, 5 years, 6 years, 7 years, 8 years, 9 years, 10 years, 11 years, 12 years, 13 years
- compositions may be administered in a single dose.
- compositions e.g., RSV RNA-LNP compositions
- compositions e.g., RSV RNA-LNP compositions
- RSV RNA-LNP compositions may be administered three or four times.
- Periodic boosters at intervals of 1-5 years may be desirable to maintain protective levels of the antibodies.
- the term “booster” refers to an extra administration of a composition (e.g., a RSV RNA-LNP composition). A booster may be given after an earlier administration of the composition.
- compositions are or are not administered to a subject as a single dose of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.0001, 0.0002, 0.0003, 0.0004, 0.0005, 0.0006, 0.0007, 0.0008, 0.0009, 0.001, 0.002, 0.003, 0.004, 0.005, 0.006, 0.007, 0.008, 0.009, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58
- compositions are or are not administered the subject as a single dose of at least, at most, exactly, or between (inclusive or exclusive) any two of 1 ⁇ g, 15 ⁇ g, 30 ⁇ g, 45 ⁇ g, 60 ⁇ g, 75 ⁇ g, 90 ⁇ g, 100 ⁇ g or higher of RSV RNA encapsulated in LNP.
- compositions are or are not administered to a subject as two doses of at least, at most, exactly, or between (inclusive or exclusive) any two of 0.0001, 0.0002, 0.0003, 0.0004, 0.0005, 0.0006, 0.0007, 0.0008, 0.0009, 0.001, 0.002, 0.003, 0.004, 0.005, 0.006, 0.007, 0.008, 0.009, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58,
- compositions e.g., RSV RNA-LNP compositions
- compositions may or may not be administered twice (e.g., Day 0 and Day 28, Day 0 and Day 60, Day 0 and Day 180, Day 0 and 2 months later, Day 0 and 6 months later, Day 0 and one year later, etc.), with each administration at a total dose of or at dosage levels sufficient to deliver a total dose of at least, at most, exactly, or between (inclusive or exclusive) any two of 1 ⁇ g, 15 ⁇ g, 30 ⁇ g, 45 ⁇ g, 60 ⁇ g, 75 ⁇ g, 90 ⁇ g, 100 ⁇ g or higher RSV RNA encapsulated in LNP.
- twice e.g., Day 0 and Day 28, Day 0 and Day 60, Day 0 and Day 180, Day 0 and 2 months later, Day 0 and 6 months later, Day 0 and one year later, etc.
- each administration at a total dose of or at dosage levels sufficient to deliver a total dose of at least, at most, exactly, or between (inclusive or exclusive) any two of 1 ⁇
- compositions e.g., pharmaceutical compositions comprising RSV RNA molecules and/or RSV RNA-LNPs
- methods, kits and reagents for prevention and/or treatment of RSV in humans and other mammals may be used as therapeutic and/or prophylactic agents. They may be used in medicine to prevent and/or treat infectious disease.
- the RSV RNA compositions are used to provide prophylactic protection from acute lower respiratory infection (ALRI) of any genotype, strain, or isolate. It is envisioned that there may be situations where persons are at risk for infection with more than one strain of RSV.
- ALRI acute lower respiratory infection
- RSV RNA compositions are particularly amenable to combination vaccination approaches due to a number of factors including, but not limited to, speed of manufacture, ability to rapidly tailor vaccines to accommodate perceived geographical threat, and the like.
- the RSV RNA compositions e.g., RSV RNA-LNP compositions
- utilize the human body to produce the antigenic protein the RSV RNA compositions (e.g., RSV RNA-LNP compositions) are amenable to the production of larger, more complex antigenic proteins, allowing for proper folding, surface expression, antigen presentation, etc. in the human subject.
- a combination RSV RNA composition can be administered that includes RNA encoding at least one antigenic polypeptide protein (or antigenic portion thereof) of a first RSV and further includes RNA encoding at least one antigenic polypeptide protein (or antigenic portion thereof) of a second RSV.
- the RSV vaccines of the present disclosure may be used to prevent RSV (infection-associated illness, including pneumonia and bronchitis) and may be particularly useful for prevention and/or treatment of immunocompromised and elderly patients to prevent or to reduce the severity and/or duration of RSV infection.
- the RSV RNA compositions (e.g., RSV RNA-LNP compositions) of the disclosure are administered to a subject (e.g., a mammalian subject, such as a human subject), and the RNA polynucleotides are translated in vivo to produce an antigenic polypeptide.
- the RSV RNA compositions (e.g., RSV RNA-LNP compositions) may be induced for translation of a polypeptide (e.g., antigen or immunogen) in a cell, tissue or organism.
- a polypeptide e.g., antigen or immunogen
- the cell, tissue or organism is contacted with an effective amount of a RSV RNA composition (e.g., a RSV RNA-LNP composition) including an RNA molecule having at least one a translatable region encoding an antigenic polypeptide (e.g., a RSV antigen).
- a RSV RNA composition e.g., a RSV RNA-LNP composition
- the RSV RNA compositions of the disclosure may be used to prime immune effector cells, for example, to activate peripheral blood mononuclear cells (PBMCs) ex vivo, which are then infused (re-infused) into a subject.
- PBMCs peripheral blood mononuclear cells
- RNA-LNPs after administration of a RSV RNA molecule described herein, e.g., formulated as RNA-LNPs, at least a portion of the RNA is delivered to a target cell. In some aspects, at least a portion of the RNA is delivered to the cytosol of the target cell. In some aspects, the RNA is translated by the target cell to produce the polypeptide or protein it encodes. In some aspects, the target cell is a spleen cell. In some aspects, the target cell is an antigen presenting cell such as a professional antigen presenting cell in the spleen. In some aspects, the target cell is a dendritic cell and/or macrophage.
- RNA molecules such as RNA-LNPs described herein may be used for delivering RNA to such target cell. Accordingly, the present disclosure also relates to a method for delivering RNA to a target cell in a subject comprising the administration of the RNA-particles described herein to the subject.
- the RNA is delivered to the cytosol of the target cell.
- the RNA is translated by the target cell to produce the polypeptide or protein encoded by the RNA.
- Encoding refers to the inherent property of specific sequences of nucleotides in a polynucleotide, such as a gene, a cDNA, or an mRNA, to serve as templates for synthesis of other polymers and macromolecules in biological processes having either a defined sequence of nucleotides (e.g., rRNA, tRNA and mRNA) or a defined sequence of amino acids and the biological properties resulting therefrom.
- a gene encodes a protein if transcription and translation of mRNA corresponding to that gene produces the protein in a cell or other biological system.
- nucleic acid compositions described herein are characterized by (e.g., when administered to a subject) an induced and/or boosted immune response as a function of antigen production in the cell.
- Increased antigen production may be demonstrated by, e.g., increased cell transfection (the percentage of cells transfected with the RNA vaccine), increased protein translation from the polynucleotide, decreased nucleic acid degradation (as demonstrated, for example, by increased duration of protein translation from a modified polynucleotide), and/or altered antigen specific immune response of the host cell.
- the disclosure relates to a method of inducing an immune response against RSV in a subject. The method includes administering to the subject an effective amount of an RNA molecule, RNA-LNP and/or composition as described herein to produce an immune response against RSV.
- the disclosure relates to a method of vaccinating a subject.
- the method includes administering to the subject in need thereof an effective amount of an RNA molecule, RNA-LNP and/or composition described herein.
- the disclosure relates to a method of treating and/or preventing an infectious disease.
- the method includes administering to the subject an effective amount of an RNA molecule RNA-LNP and/or composition as described herein.
- the disclosure relates to a method of treating and/or preventing and/or reducing the severity of a RSV infection and/or illness caused by RSV .
- the method includes administering to the subject an effective amount of an RNA molecule, RNA-LNP and/or composition as described herein.
- the disclosure relates to a method of treating and/or preventing and/or reducing the severity of an infectious disease in a subject by, for example, inducing an immune response to an infectious disease in the subject.
- the method includes administering a priming composition that includes an effective amount of an RNA molecule, RNA- LNP and/or composition described herein, and administering a booster composition including an effective amount of an RNA molecule, RNA-LNP and/or composition.
- the composition elicits an immune response including an antibody response.
- the composition elicits an immune response including a T cell response and/or a B cell response.
- an immune response comprises a T cell response and a B cell response.
- the composition elicits a neutralizing immune response.
- a neutralizing immune response is an immune response that is a neutralizing antibody response and/or an effective neutralizing T cell response.
- a neutralizing antibody response produces a level of antibodies that meet or exceed a seroprotection threshold.
- the composition elicits an effective T cell response.
- An effective T cell response is a response which produces a baseline level of infectious disease-activated and/or infectious disease-specific T cells including CD8+ and CD4+ T helper type 1 cells.
- the effective T cells comprises a high proportion of CD8+ T cells and/or CD4+ T cells, relative to a baseline level (in a naive subject).
- these T cells are differentiated towards an early- differentiated memory phenotype with co-expression of CD27 and CD28.
- the disclosure relates to a method of treating and/or preventing and/or reducing the severity of a RSV infection and/or illness caused by RSV in a subject by, for example, inducing an immune response to RSV in the subject.
- the method includes administering a priming composition that includes an effective amount of an RNA molecule, RNA- LNP and/or composition described herein, and administering a booster composition including an effective amount of an RNA molecule RNA-LNP and/or composition as described herein.
- the composition elicits an immune response including an antibody response.
- the composition elicits an immune response including a T cell response and/or a B cell response.
- an immune response comprises a T cell response and a B cell response.
- the composition elicits a neutralizing immune response.
- a neutralizing immune response is an immune response that is a neutralizing antibody response and/or an effective neutralizing T cell response.
- a neutralizing antibody response produces a level of antibodies that meet or exceed a seroprotection threshold.
- the composition elicits an effective T cell response.
- An effective T cell response is a response which produces a baseline level of infectious disease-activated and/or infectious disease-specific T cells including CD8+ and CD4+ T helper type 1 cells.
- the effective T cells comprises a high proportion of CD8+ T cells and/or CD4+ T cells, relative to a baseline level (in a naive subject). In some embodiments these T cells are differentiated towards an early-differentiated memory phenotype with co-expression of CD27 and CD28.
- the methods disclosed herein may involve administering to the subject a RSV RNA-LNP composition comprising at least one RSV RNA molecule having an open reading frame encoding at least one RSV antigenic polypeptide, thereby inducing in the subject an immune response specific to RSV antigenic polypeptide, wherein anti-antigenic polypeptide antibody titer in the subject is increased following vaccination relative to anti-antigenic polypeptide antibody titer in a subject vaccinated with a prophylactically effective dose (e.g., a therapeutically effective dose that prevents infection with the virus at a clinically acceptable level) of a traditional vaccine against the RSV.
- a prophylactically effective dose e.g., a therapeutically effective dose that prevents infection with the virus at a clinically acceptable level
- An “anti-antigenic polypeptide antibody” is a serum antibody the binds specifically to the antigenic polypeptide.
- the anti-antigenic polypeptide antibody titer in the subject is or is not increased at least, at most, between (inclusive or exclusive) any two of, or exactly 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 log following administration of the RSV RNA-LNP composition relative to anti-antigenic polypeptide antibody titer in a subject administered a prophylactically effective dose of a traditional composition against RSV (e.g., the standard of care dose of a recombinant or purified RSV protein vaccine, a live attenuated or inactivated RSV vaccine, or a RSV VLP vaccine).
- a traditional composition against RSV e.g., the standard of care dose of a recombinant or purified RSV protein vaccine, a live attenuated or inactivated RSV vaccine, or a RSV VLP vaccine.
- the anti-antigenic polypeptide antibody titer in the subject is or is not increased at least, at most, between (inclusive or exclusive) any two of, or exactly 1-, 2-, 3-, 4-, 5-, 6-, 7-, 8-, 9-, 10-, 100-, or 1000-fold following administration of the RSV RNA-LNP composition relative to anti-antigenic polypeptide antibody titer in a subject administered a prophylactically effective dose of a traditional composition against RSV (e.g., the standard of care dose of a recombinant or purified RSV protein vaccine, a live attenuated or inactivated RSV vaccine, or a RSV VLP vaccine).
- a traditional composition against RSV e.g., the standard of care dose of a recombinant or purified RSV protein vaccine, a live attenuated or inactivated RSV vaccine, or a RSV VLP vaccine.
- an effective amount of a RSV RNA-LNP composition comprising at least one RSV RNA molecule having an open reading frame encoding at least one RSV antigenic polypeptide results in a 2-fold to 200-fold (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of a 2-, 3-, 4-, 5-, 6-, 7-, 8-, 9-, 10-, 20-, 30-, 40-, 50-, 60-, 70-, 80-, 90-, 100-, 110-, 120-, 130-, 140-, 150-, 160-, 170-, 180-, 190-, or 200-fold) increase in serum neutralizing antibodies against RSV , relative to a traditional composition against RSV (e.g., the standard of care dose of a recombinant or purified RSV protein vaccine, a live attenuated or inactivated RSV vaccine, or a RSV VLP vaccine).
- a traditional composition against RSV e.g., the standard of care dose of a
- an effective amount of a RSV RNA-LNP composition comprising at least one RSV RNA molecule having an open reading frame encoding at least one RSV antigenic polypeptide is a dose equivalent to an at least 2-fold reduction in a standard of care dose of a traditional composition against RSV .
- an effective amount of a RSV RNA-LNP composition may or may not be a dose equivalent to a 2-, 3-, 4-, 5-, 6-, 7-, 8-, 9-, 10-, 20-, 50-, 100-, 250-, 500-, or 1000-fold reduction in a standard of care dose of a traditional composition against RSV .
- the anti-RSV antigenic polypeptide antibody titer produced in a subject administered an effective amount of a RSV RNA-LNP composition is equivalent to an anti-RSV antigenic polypeptide antibody titer produced in a control subject administered the standard of care dose of a traditional composition against RSV .
- an effective amount of a RSV RNA-LNP composition is or is not a dose equivalent to a 2-fold to 1000-fold reduction (e.g., at least, at most, exactly, or between (inclusive or exclusive) any two of a 2-, 3 -,4 -,5 -,6-, 7-, 8-, 9-, 10-, 20-, 30-, 40-, 50-, 60-, 70-, 80-, 90-, 100-, 110-, 120-, 130-, 140-, 150-, 160-, 170-, 1280-, 190-, 200-, 210-, 220-, 230-, 240-, 250-, 260-, 270-, 280-, 290-, 300-, 310-, 320-, 330-, 340-, 350-, 360-, 370-, 380-, 390-, 400-, 410-, 420-, 430-, 440-, 450-, 4360-, 470-, 480-,
- an anti-RSV antigenic polypeptide antibody titer produced in the subject is equivalent to an anti-RSV antigenic polypeptide antibody titer produced in a control subject administered the standard of care dose of a traditional composition against RSV .
- a traditional composition against RSV refers to a composition other than the RNA molecules, RNA-LNPs and/or compositions described herein.
- a traditional composition includes, but is not limited, to live microorganism vaccines, killed microorganism vaccines, attenuated vaccines, subunit vaccines, protein antigen vaccines containing recombinant protein produced in a heterologous expression system or purified from large amounts of the pathogenic organism, DNA vaccines, virus-like particle (VLP) vaccines containing viral capsid proteins (e.g., pre- and/or post-fusion F proteins) but lacking viral genome, etc.
- a traditional vaccine is a vaccine that has achieved regulatory approval and/or is registered by a national drug regulatory body, for example the Food and Drug Administration (FDA) in the United States or the European Medicines Agency (EMA).
- FDA Food and Drug Administration
- EMA European Medicines Agency
- a “standard of care,” as provided herein, refers to a medical or psychological treatment guideline and can be general or specific. “Standard of care” specifies appropriate treatment based on scientific evidence and collaboration between medical professionals involved in the treatment of a given condition. It is the diagnostic and treatment process that a physician/ clinician should follow for a certain type of patient, illness or clinical circumstance.
- a “standard of care dose,” as provided herein, refers to the dose of a traditional composition against RSV that a physician/clinician or other medical professional would administer to a subject to treat and/or prevent RSV , or a RSV -related condition, while following the standard of care guideline for treating and/or preventing RSV , or a RSV -related condition.
- an RNA molecule, RNA-LNP and/or composition described herein e.g., a RSV RNA-LNP composition comprising at least one RSV RNA molecule having an open reading frame encoding at least one RSV antigenic polypeptide
- a RSV RNA-LNP composition comprising at least one RSV RNA molecule having an open reading frame encoding at least one RSV antigenic polypeptide
- the term antibody titer refers to the amount of antigen-specific antibody produces in a subject, e.g., a human subject.
- antibody titer is expressed as the inverse of the greatest dilution (in a serial dilution) that still gives a positive result.
- antibody titer is determined or measured by enzyme-linked immunosorbent assay (ELISA). In exemplary embodiments, antibody titer is determined or measured by neutralization assay, e.g., by microneutralization assay. In certain aspects, antibody titer measurement is expressed as a ratio, such as 1:40, 1:100, etc.
- ELISA enzyme-linked immunosorbent assay
- neutralization assay e.g., by microneutralization assay.
- antibody titer measurement is expressed as a ratio, such as 1:40, 1:100, etc.
- an efficacious an RNA molecule, RNA-LNP and/or composition described herein produces an antibody titer of greater than 1:10, greater that 1:100, greater than 1:400, greater than 1:1000, greater than 1:2000, greater than 1:3000, greater than 1:4000, greater than 1:5000, greater than 1:6000, greater than 1:7500, or greater than 1:10000.
- the antibody titer is produced or reached by 10 days following vaccination, by 20 days following vaccination, by 30 days following vaccination, by 40 days following vaccination, or by 50 or more days following vaccination.
- the titer is produced or reached following a single dose of vaccine administered to the subject.
- the titer is produced or reached following multiple doses, e.g., following a first and a second dose (e.g., a booster dose).
- the methods disclosed herein may involve administering to the subject a RSV RNA-LNP composition comprising at least one RSV RNA molecule having an open reading frame encoding at least one RSV antigenic polypeptide, thereby inducing in the subject an immune response specific to RSV antigenic polypeptide, wherein the immune response in the subject is equivalent to an immune response in a subject administered with a traditional composition against the RSV that is or is not at least, at most, in between (inclusive or exclusive) any two of, or exactly 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 34, 36, 38, 40, 42, 44, 46, 48, 50, 52, 54, 56, 58, 60, 62, 64, 66, 68, 70, 72, 74, 76, 78, 80, 82, 84, 86, 88, 90, 92, 94, 96, 98, or 100 times the dosage level relative to the RNA composition.
- the RNA molecule, RNA-LNP and/or composition is used as a vaccine.
- the RNA molecule, RNA-LNP and/or composition may be used in various therapeutic or prophylactic methods for preventing, treating or ameliorating of acute lower respiratory infection (ALRI), or a disorder related to respiratory illness, including pneumonia and bronchitis.
- the RNA molecule, RNA-LNP and/or composition may be used in various therapeutic or prophylactic methods for preventing, treating or ameliorating of acute lower respiratory infection (ALRI), including pneumonia and bronchitis.
- methods of the disclosure relate to prognosing, diagnosing, testing, monitoring, and/or treating a subject suspected of having had an infectious disease (e.g., RSV ), having an infectious disease (e.g., RSV ), at risk of having an infectious disease (e.g., RSV ), and/or having symptoms of an infectious disease (e.g., RSV ).
- the subject may have one or mor symptoms of an infectious disease (e.g., RSV ).
- the subject may be tested for one or more antigenic polypeptides or proteins (or antigenic portions thereof) from an infectious disease (e.g., RSV ) by the one or more diagnostic tests (e.g., PCR testing to detect RSV in skin lesions; Tzanck smear; IgM serologic testing; ELISA, glycoprotein-based ELISA, latex agglutination, and/or indirect fluorescent antibody for IgG detection; direct fluorescent antibody assay, viral culture, etc.).
- infectious disease e.g., RSV
- diagnostic tests e.g., PCR testing to detect RSV in skin lesions; Tzanck smear; IgM serologic testing; ELISA, glycoprotein-based ELISA, latex agglutination, and/or indirect fluorescent antibody for IgG detection; direct fluorescent antibody assay, viral culture, etc.
- the subject having had an infectious disease e.g., RSV
- having an infectious disease e.g., RSV
- at risk of having an infectious disease e.g., RSV
- symptoms of an infectious disease e.g., RSV
- a sample e.g., blood, saliva, tissues, bone, muscle, cartilage, and/or skin
- one or more antigenic polypeptides or proteins or antigenic portions thereof
- an infectious disease e.g., RSV
- diagnostic tests e.g., PCR testing to detect RSV ; Tzanck smear; IgM serologic testing; ELISA, glycoprotein-based ELISA, latex agglutination, and/or indirect fluorescent antibody for IgG detection; direct fluorescent antibody assay, viral culture, etc.
- RSV RNA compositions may be administered prophylactically to healthy subjects or early in infection during the incubation phase or during active infection after onset of symptoms.
- the subject is immunocompetent.
- the subject is immunocompromised.
- the RNA molecule, RNA-LNP and/or composition is administered in a single dose.
- a second, third or fourth dose may be given.
- the RNA molecule, RNA-LNP and/or composition is administered in multiple doses.
- the RNA molecule, RNA-LNP and/or composition is administered intramuscularly (IM) or intradermally (ID).
- IM intramuscularly
- ID intradermally
- the present disclosure further provides a kit comprising the RNA molecule, RNA-LNP, and/or composition.
- the RNA molecule, RNA-LNP and/or composition described herein is administered to a subject that is less than about 1 years old, or about 1 years old to about 10 years old, or about 10 years old to about 20 years old, or about 20 years old to about 50 years old, or about 60 years old to about 70 years old, or older.
- the subject is at least, at most, exactly, or between any two of less than 1 year of age, greater than 1 year of age, greater than 5 years of age, greater than 10 years of age, greater than 20 years of age, greater than 30 years of age, greater than 40 years of age, greater than 50 years of age, greater than 60 years of age, greater than 70 years of age, or older.
- the subject is greater than 50 years of age.
- the subject is at least, at most, exactly, or between (inclusive or exclusive) any two of about 1 year of age or older, about 5 years of age or older, about 10 years of age or older, about 20 years of age or older, about 30 years of age or older, about 40 years of age or older, about 50 years of age or older, about 60 years of age or older, about 70 years of age or older, or older. In some aspects, the subject may be about 50 years of age or older.
- the subject is at least, at most, exactly, or between (inclusive or exclusive) any two of 1 year of age or older, 5 years of age or older, 10 years of age or older, 20 years of age or older, 30 years of age or older, 40 years of age or older, 50 years of age or older, 60 years of age or older, 70 years of age or older, or older. In some aspects the subject may be 50 years of age or older.
- preF prefusion F
- modRNA modified RNA
- the RSV RNA-LNP vaccines may comprise RNA comprising a single-stranded, 5′-capped and polyadenylated modified RNA that is translated after entering the cell.
- the RNA comprises an open reading frame (ORF) that encodes variations of the RSV preF polypeptide.
- ORF open reading frame
- the RNA may comprise structural elements, such as untranslated regions (UTRs), optimized for high efficacy of the RNA.
- the RSV RNA-LNPs may comprise RNA as provided in Table 5 of Example 1 disclosed herein.
- the RSV RNA-LNPs may comprise RNA as provided in Tables 1 to 3 of Example 6 disclosed herein.
- the RNA may also comprise a substitution of 1-methyl-pseudouridine for uridine to decrease recognition of the vaccine RNA by innate immune sensors, such as toll-like receptors (TLRs) 7 and 8, resulting in decreased innate immune activation and increased protein translation.
- TLRs toll-like receptors
- the RNA molecules described herein are formulated/encapsulated into lipid nanoparticles (LNPs) to enable delivery of the RNA into host cells after, e.g., intramuscular (IM), intradermal (ID), or intranasal (IN) injection.
- IM intramuscular
- ID intradermal
- IN intranasal
- the LNP formulation may comprise two functional lipids, ALC- 0315 and ALC-0159, and two structural lipids, DSPC (1,2-distearoyl-sn-glycero-3- phosphocholine) and cholesterol. In some aspects, 1, 2, 3, or more of the forgoing lipids may be excluded from the LNP formulation.
- the potency of RNA vaccines is optimized by LNP encapsulation, which protects the RNA from degradation by extracellular RNases and facilitates delivery in the cell. After IM injection of RSV RNA-LNP vaccines, the LNPs are taken up by the cells, and the RNA is released into the cytosol. In the cytosol, the RNA is translated, and the encoded viral antigen is produced.
- the Examples herein demonstrate the RSV RNA-LNP vaccines of the present disclosure are immunogenic in mice and induce both humoral and cell mediated immune responses in mice.
- Clinical studies of the present disclosure evaluate the safety, tolerability, and immunogenicity of RSV RNA-LNP vaccines against RSV.
- the RSV RNA-LNPs vaccines may be indicated for active immunization for the prevention of acute lower respiratory infection (ALRI), including pneumonia and bronchitis caused by RSV for adults (e.g., ⁇ 45, ⁇ 50, ⁇ 55, ⁇ 60, ⁇ 70...etc. years of age or 50 through 69 years of age).
- ALRI acute lower respiratory infection
- RSV RNA-LNP vaccines may be administered in different dose level(s), dose formulation, number of doses and dosing schedules, as described herein, including but not limited to: - As a single-dose schedule or a two-dose schedule (e.g., Day 0 and on or about 2 months after or Day 0 and on or about 6 months after) - At different dose levels (e.g., of or of about 15 ⁇ g, 30 ⁇ g, 60 ⁇ g, 90 ⁇ g, 100 ⁇ g or higher per administration) - At different formulations (non-lyophilized and/or lyophilized) The RSV RNA-LNPs may be presented as a liquid or lyophilized formulation.
- RSV RNA-LNP vaccines may or may not be dosed in the range of or of about 15 ⁇ g, 30 ⁇ g, 60 ⁇ g, 90 ⁇ g, 100 ⁇ g or higher per dose with an injection volume of or of about 0.25 to 1 mL (e.g., of or of about 0.25, 0.5, 1 mL). Dilution with sterile 0.9% sodium chloride (normal saline) may be required.
- the objectives of RSV RNA-LNP clinical studies may include, but are not limited to: -To describe the safety and tolerability profile of RSV RNA-LNP vaccines administered at selected dose levels and schedules in participants.
- the efficacy (or effectiveness) against RSV of the RNA molecules encoding RSV polypeptides, RNA-LNPs and compositions thereof disclosed herein is or is not greater than 50% (e.g., at least, at most, exactly, or between any two of 50%, 60%, 70%, 80%, 90%, or more).
- Vaccine efficacy may be assessed using standard analyses (see, e.g., Weinberg et al, J Infect Dis.2010 Jun 1;201(11): 1607-10).
- vaccine efficacy may be measured by double-blind, randomized, clinical controlled trials such as those described herein.
- AR disease attack rate
- RR relative risk
- Efficacy (ARU ⁇ ARV)/ARU x 100
- Vaccine effectiveness is an assessment of how a vaccine (which may have already proven to have high vaccine efficacy) reduces disease in a population. This measure can assess the net balance of benefits and adverse effects of a vaccination program, not just the vaccine itself, under natural field conditions rather than in a controlled clinical trial. Vaccine effectiveness is proportional to vaccine efficacy (potency) but is also affected by how well target groups in the population are immunized, as well as by other non-vaccine-related factors that influence the real-world outcomes of hospitalizations, ambulatory visits, and/or costs. For example, a retrospective case control analysis may be used, in which the rates of vaccination among a set of infected cases and appropriate controls are compared.
- efficacy of the RSV polypeptides, RNA-LNPs and compositions thereof is at least 60% relative to unvaccinated control subjects.
- efficacy may be at least, at most, exactly, or between (inclusive or exclusive) any two of 65%, 70%, 75%, 80%, 85%, 95%, 98%, or 100% relative to unvaccinated control subjects.
- RNA constructs generated herein encode RSV F protein wild-type (WT) and RSV F protein variants/mutants (i.e. RSV pre-fusion F protein).
- Table 4 shows WT F proteins (WT F) and variant RSV preF proteins .
- RSV F proteins and description DNA sequences encoding RSV F proteins were prepared and utilized for in vitro transcription reactions to generate RNA. In vitro transcription of RNA is known in the art and is described herein. DNA templates were cloned into a plasmid vector with backbone sequence elements (T7 promoter, 5′ and 3′ UTR, poly-A tail) for improved RNA stability and translational efficiency.
- the DNA was purified, spectrophotometrically quantified and in vitro-transcribed by T7 RNA polymerase in the presence of a trinucleotide cap1 analogue ((m2 7,3′-O )Gppp(m 2’-O )ApG) (TriLink) and with N1-methylpseudouridine ( ⁇ ) replacing uridine (modified RNA (modRNA)).
- TriLink trinucleotide cap1 analogue
- ⁇ N1-methylpseudouridine
- modRNA modified RNA
- the RSV RNA was generated from codon-optimized (CO) DNA for stabilization and superior protein expression.
- Table 5 shows RNA constructs of the present disclosure, and corresponding sequences, comprising a 5’ UTR, an open reading frame encoding a respiratory syncytial virus (RSV) polypeptide, a 3’ UTR and a poly-A tail.
- RSV F modRNA constructs ’ * ’ 5 UTR sequence includes 5 cap sequence ** Poly-A tail length may contain +1/-1 A
- RSV F 847A saRNA construct comprising in order 5’cap-5’UTR-nsP1-nsP2-nsP3-nsP4- Subgenomic promoter-RSV [ORF]-3’UTR-polyA tail (encoding RSV F protein having SEQ ID NO: 4) 5’UTR 2 45 52 Table 7.
- RSV F 847B saRNA construct comprising in order 5’cap-5’UTR-nsP1-nsP2-nsP3-nsP4- Subgenomic promoter-RSV [ORF]-3’UTR-polyA tail (encoding RSV F protein having SEQ ID NO: 6)
- LNP formulation contains 2 functional lipids, ALC-0315 and ALC-0159, and 2 structural lipids DSPC (1,2distearoyl-sn-glycero-3-phosphocholine) and cholesterol.
- the physicochemical properties and the structures of the 4 lipids are shown in the Table 8 below.
- Lipid nanoparticles were prepared and tested according to the general procedures described in US Patent 9737619 (PCT Pub. No. WO2015/199952) and US Patent 10166298 (WO 2017/075531) and WO2020/146805, each of which is hereby incorporated by reference in its entirety.
- cationic lipid, DSPC, cholesterol and PEG-lipid were solubilized in ethanol at a molar ratio of about 47.5: 10: 40.7: 1.8.
- RSV preF expression This example serves to capture the in vitro expression (IVE) results generated for the respiratory syncytial virus (RSV) modRNA lipid nanoparticle (LNP) drug product described herein.
- the constructs encapsulated in the LNP encode for the full length RSV trimeric fusion glycoprotein (F-protein) from the A-strain and B-strain viruses, respectively.
- the LNP drug product was diluted in DPBS to a concentration of 80 ng/uL and serially diluted 8-pts with a dilution factor of 4. Then the 96-well culture plate was removed from the incubator and 50 uL of each step of the diluted LNP were added to duplicate wells of the 96-well culture plate to generate a titration curve ranging from 8,000 ng/well – 1.95 ng/well. The 96-well culture plate was placed back into the shaking incubator overnight. After incubation, 250 uL of cells are transferred to a 96-well u-bottom polystyrene plate and pelleted using a swinging bucket centrifuge (500 rcf, 5 min at RT).
- a swinging bucket centrifuge 500 rcf, 5 min at RT.
- the supernatant is removed and cells resuspended in 100 uL solution of Aqua405 live/dead stain.
- the plate is incubated 30 min at room temperature, protected from light. After incubation cells are washed with wash buffer and pelleted using centrifugation (500 rcf, 5 min at RT). The supernatant is removed and cells are resuspended in 100 uL fixation/permeabilization buffer and the plate is incubated 30 minutes at 2 -8° C, protected from light. Once incubation is complete the cells are pelleted using a swinging bucket centrifuge (500 rcf, 5 min at RT).
- the supernatant is removed and cells resuspended with 250 uL wash buffer, this is repeated for a total of 2 washes.
- the cells pelleted, supernatant removed, and resuspended in 50 uL of primary antibody solution.
- the plates are sealed and incubated for 45 minutes at 2 -8° C, protected from light. Once completed the cells are pelleted using a swinging bucket centrifuge (500 rcf, 5 min at RT). The supernatant is removed and cells resuspended with 250 uL wash buffer, this is repeated for a total of 2 washes.
- the cells are pelleted, supernatant removed, and resuspended in 50 uL of secondary antibody solution.
- the plates are sealed and incubated for 45 minutes at 2 -8° C, protected from light.
- the cells are pelleted using a swinging bucket centrifuge (500 rcf, 5 min at RT). The supernatant is removed and cells resuspended with 250 uL wash buffer, this is repeated for a total of 2 washes.
- cells are pelleted, supernatant removed, and resuspended in 200 uL of wash buffer and data acquired by flow cytometry.
- IVE In vitro Expression
- IVE for the modRNA LNP drug products was assessed by transfecting HEK293F cells with a dose titration curves and staining for antibodies; RSV mAb1 specific for the trimeric RSV F-protein and L4-6 specific for total RSV F-protein. These antibodies have been shown to recognize both the A-strain and B-strain RSV F-protein and were used in the assay with either permeabilizing or non-permeabilizing conditions to assess the total cell vs cell surface content of the RSV F-protein. The measured % positive cells (2,000 ng/well input) and EC50 of the dose response curves of the drug product lots are shown in Table 11. Table 11. Drug Product IVE Results Example 5.
- mice Female BALB/c mice were immunized with RSV prefusion F (847) in bivalent protein subunit version (RSV 847A + 847B) as described in WO2017/109629 or modRNA-LNP formulation described herein either as monovalent (RSV 847A) or bivalent (RSV 847A + 847B) or saRNA-LNP as bivalent formulation at different doses on day 0 and day 21. Immunogenicity was evaluated by measuring RSV neutralizing antibody response and RSV F-specific T-cell response.
- RSV microneutralization assay is a 3-day assay done using A549 cells (human alveolar basal epithelial cells) to measure functional antibodies in serum that neutralize RSV activity, preventing infection of a host cell monolayer.
- A549 cells (human alveolar basal epithelial cells; ATCC, cat # CCL-185) are seeded in 96-well tissue-culture treated plates at 2.5 x 10 4 cells per well and incubated for at least 20 hours to form a confluent monolayer.
- diluted virus (RSV A, M37; RSV B, B18537; 500 FFU/well) is added to 3-fold serial dilutions of heat inactivated test serum prepared in duplicate and incubated for 1 hour to allow antibodies to bind to the virus.
- the neutralization reaction is then transferred onto the prepared A549 cell monolayers and incubated for 2 hours. Additional media is supplemented onto the plates prior to an overnight incubation (at least 16 hours).
- a 50% neutralization titer is calculated as the last reciprocal serum dilution at which 50% of the virus is neutralized compared to wells containing virus only.
- a titer is reported as the geometric mean titer (GMT) of the two replicate titers of each sample.
- the assay titer range is 20 to 43,740. Any samples with a titer >43,740 are prediluted and repeated to extend the upper titer limit. Any samples below the lower limit of detection (LLOD) are reported at LLOD of 20.
- T-cell Response Measurement Vaccine-induced T-cell response to RSV F is assessed by ex vivo stimulation of splenocytes in the presence of RSV F (A+B) peptide pool to activate production of various cytokine such as IFN- ⁇ in antigen-specific T cells.
- the cytokines secreted outside the cells can be measured by ELISpot (expressed as spot forming cells, SFC per million cells) or cytokine secretion can be blocked within the cells to be measured by ICS (expressed as percentage of cytokine expressing CD4+ T cells and CD8+ T cells).
- the cytokine IFN- ⁇ secreted by activated T cells is captured by an anti- IFN- ⁇ antibody coated onto the polyvinylidene fluoride (PVDF) membrane of the well bottom on a microplate.
- the captured IFN- ⁇ is developed into a spot by another noncompeting biotinylated anti-IFN- ⁇ secondary antibody followed by an enzymatic color reaction using streptavidin-alkaline phosphatase (ALP) conjugate and the substrate solution, nitro-blue tetrazolium and 5-bromo-4- chloro-3'-indolyphosphate (BCIP/NBT-plus) that yield a dark purple precipitate or spot.
- ALP streptavidin-alkaline phosphatase
- BCIP/NBT-plus 5-bromo-4- chloro-3'-indolyphosphate
- T-cell IFN- ⁇ response is measured using Mabtech Mouse IFN- ⁇ ELISpot PLUS kit (ALP) and expressed as spot forming cells (SFC) per million cells.
- ICS staining can detect multiple cytokines, including IFN- ⁇ , produced in both CD4+ and CD8+ T cells following antigen peptide stimulation.
- Single cell suspensions of splenocytes (2 x 10 6 cells/well) were cultured ex vivo in cRPMI with media-DMSO (unstimulated) or specific peptide pool (15aa, 11aa overlap, 2 ⁇ g/mL/peptide) representing RSV F A+B for 5 hours at 37°C in the presence of anti-CD107a APC antibody and protein transport inhibitors, GolgiPlug and GolgiStop.
- splenocytes were incubated with fluorescently conjugated antibodies to the surface proteins CD19, CD3, CD4, CD8, CD44 (25 ⁇ 5 minutes at 18-25 °C) followed by fixation and permeabilization and staining for - IFN- ⁇ , TNF- ⁇ , IL-2 and CD40L/CD154 (25 ⁇ 5 minutes at 18-25 °C). After staining, the cells are washed and resuspended in FC buffer. Cells were acquired on LSR Fortessa and data analyzed by FlowJo (10.7.1). Results are background (media-DMSO) subtracted and shown as percentage of cytokine-expressing CD4+ T cells and CD8+ T cells.
- RSV A+B F-specific T-cell response induced by bivalent formulation is higher than that of monovalent formulation (RSV 847A), further supporting that a bivalent version of RSV 847 induces higher magnitude of T-cell response, similar to that of neutralizing response.
- study results demonstrate higher immunogenicity of modRNA-LNP formulation of RSV prefusion F constructs compared to that of the protein subunit vaccine in na ⁇ ve mice. Table 12. Immune response induced by protein subunit and modRNA-LNP formulations of RSV prefusion F in mice NA: not analyzed Next, the immunogenicity of modRNA-LNP encoding different RSV prefusion F (preF) antigen designs described herein was evaluated.
- mutant prefusion designs 847, 851 and 852 in full length version induce higher titers compared to DS-CAV1 and WT as well as 847A-Foldon, which is designed as an ectodomain with foldon (FIG.2).
- FOG.2 ectodomain with foldon
- saRNA-LNP The immunogenicity of saRNA-LNP was compared to modRNA-LNP and protein subunit using bivalent (A and B) formulations of RSVpreF.
- a and B bivalent formulations of RSVpreF.
- 3W PD1 3W PD1
- a dose-dependent effect was observed in the neutralizing response induced by both modRNA and saRNA vaccine against RSV A and B virus.
- the neutralizing response induced by saRNA at 0.02 ⁇ g dose was substantially higher than the modRNA vaccine in mice receiving 0.2 ⁇ g dose (FIG.3A and 3B), suggesting a dose-sparing response induced by saRNA.
- sequences may comprise any stop codon, including but not limited to the stop codons provided in the Tables below.
- Table 1 RSV Polypeptides A i id SLTLPSEINLCNVDIFNPKYDCKIMTSKTDVSSSVITSLGAIVSCYGK HNVNTGKSTTNIMITAIIIVIIVVLLSLIAIGLLLYCKAKNTPVTLSKDQL
- RNA molecule comprising at least one open reading frame encoding a respiratory syncytial virus (RSV) polypeptide.
- RSV respiratory syncytial virus
- RNA molecule of any one of paragraphs 1 to 5 wherein the RSV polypeptide comprises an amino acid of Table 1, including but not limited to any of SEQ ID NO: 1 to 6 or 71 to 74. 7.
- the RSV polypeptide comprises an amino acid sequence of any one of SEQ ID NOs: 1 to 6 or 71 to 74.
- RNA molecule of any one of paragraphs 1 to 8 wherein the open reading frame is transcribed from a nucleic acid sequence of Table 2, including by not limited to any of SEQ ID NO: 7 to 10 or 59 to 62. 10.
- the RNA molecule of any one of paragraphs 1 to 10 wherein the open reading frame comprises a nucleic acid sequence of Table 3, including but not limited to any of SEQ ID NO: 11 to 16 or 63 to 70. 12.
- each uridine is replaced by N1- methylpseudouridine ( ⁇ ).
- the RNA molecule of paragraph 15, wherein the 5’ UTR comprises a sequence selected from any of SEQ ID NO: 17 to 19. 17.
- the composition of paragraph 17, wherein the 3’ UTR comprises a sequence selected from any of SEQ ID NO: 20 to 25.
- the RNA molecule of any one of paragraphs 1 to 18, wherein the RNA molecule comprises a 5’ cap moiety.
- RNA molecule of paragraph 20 wherein the poly-A tail comprises a sequence having SEQ ID NO: 26, optionally comprising +/-1 or +/-2 adenosine (A).
- A adenosine
- 22 The RNA molecule of any one of paragraphs 1 to 21, wherein the RNA molecule comprises a 5’ UTR and 3’ UTR.
- 23 The RNA molecule of any one of paragraphs 1 to 22, wherein the RNA molecule comprises a 5’ cap, 5’ UTR, and 3’ UTR.
- 24 The RNA molecule of any one of paragraphs 1 to 23, wherein the RNA molecule comprises a 5’ cap, 5’ UTR, 3’ UTR, and poly-A tail. 25.
- RNA molecule of any one of paragraphs 1 to 26 wherein the open reading frame was generated from codon-optimized DNA.
- 28. The RNA molecule of any one of paragraphs 1 to 27, wherein the open reading frame comprises a G/C content of at least 55%, at least 60%, at least 65%, at least 70%, or at least 75%, of or of about 50% to 75% or 55% to 70%, or of or of about 58%, 66%, or 62%.
- 29. The RNA molecule of any one of paragraphs 1 to 28, wherein the encoded RSV polypeptide localizes in the cellular membrane, localizes in the Golgi and/or is anchored in the membrane and is secreted.
- RNA-LNP lipid nanoparticle
- composition of paragraph 36 or 37, wherein the lipid nanoparticle comprises a cationic lipid.
- the cationic lipid is (4- hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315).
- ALC-0315 The composition of any one of paragraphs 36 to 39, wherein the lipid nanoparticle comprises a PEGylated lipid.
- the PEGylatedlipid is PEG-modified phosphatidylethanolamine, PEG-modified phosphatidic acid, PEG-modified ceramides (e.g.
- PEG-CerC14 or PEG-CerC20 PEG-modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkylglycerols, 2-[(polyethylene glycol)-2000]-N,N- ditetradecylacetamide, glycol-lipids including PEG-c-DOMG, PEG-c-DMA, PEG-s-DMG,N- [(methoxy polyethylene glycol)2000)carbamyl]-1,2-dimyristyloxlpropyl-3-amine (PEG-c- DMA), and PEG-2000-DMG, PEGylated diacylglycerol (PEG-DAG) such as 1 - (monomethoxy-polyethyleneglycol)-2,3-dimyristoylglycerol (PEG-DMG), a PEGylated phosphatidylethanoloamine (PEG-PE), a PEG succinate diacylg
- composition of paragraph 43 wherein the neutral lipid is distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dipalmitoylphosphatidylcholine (DPPC), dioleoylphosphatidylglycerol (DOPG), dipalmitoylphosphatidylglycerol (DPPG), dioleoyl-phosphatidylethanolamine (DOPE), palmitoyloleoylphosphatidylcholine (POPC), palmitoyloleoyl-phosphatidylethanolamine (POPE) and dioleoyl-phosphatidylethanolamine 4-(N-maleimidomethyl)-cyclohexane- 1carboxylate (DOPE-mal), dipalmitoyl phosphatidyl ethanolamine (DPPE), dimyristoylphosphoethanolamine (DMPE), distearoyl- phosphatidylethanolamine (DSPE),
- composition of paragraph 43 or 44, wherein the neutral lipid is 1,2-distearoyl-sn- glycero-3-phosphocholine (DSPC).
- DSPC 1,2-distearoyl-sn- glycero-3-phosphocholine
- RNA is from: (i) RSV subtype A and (ii) RSV subtype B.
- RNA is from: (i) RSV subtype A and (ii) RSV subtype B.
- LNP lipid nanoparticle
- a cationic lipid at a concentration of or of about 0.8 to 0.95 mg/mL
- PEGylated lipid at a concentration of or of about 0.05 to 0.15 mg/mL
- a neutral lipid at a concentration of or of about 0.1 to 0.25 mg/mL
- steroid or steroid analog at a concentration of or of about 0.3 to 0.45 mg/mL.
- composition of any one of paragraphs 36 to 52 comprising an RNA molecule at a concentration of or of about 0.01 to 0.09 mg/mL formulated in a lipid nanoparticle (LNP) comprising (4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC- 0315) at a concentration of or of about 0.8 to 0.95 mg/mL, 2-[(polyethylene glycol)-2000]- N,N-ditetradecylacetamide (ALC-0159) at a concentration of or of about 0.05 to 0.15 mg/mL, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) at a concentration of or of about 0.1 to 0.25 mg/mL and cholesterol at a concentration of or of about 0.3 to 0.45 mg/mL.
- LNP lipid nanoparticle
- ALC- 0315 4-hydroxybutyl)azaned
- composition of any one of paragraphs 36 to 53 comprising an RNA molecule at a concentration of or of about 0.06 mg/mL formulated in a lipid nanoparticle (LNP).
- LNP lipid nanoparticle
- the composition of any one of paragraphs 36 to 54 further comprising at least one of a buffer, a stabilizing agent, salt, surfactant, preservative, excipient, and/or adjuvant.
- the composition of any one of paragraphs 36 to 55 further comprising at least a buffer and a stabilizing agent, and optionally, a salt diluent.
- the composition of paragraph 36 or 56 wherein the buffer is a Tris buffer. 58.
- Tris buffer comprises tromethamine and Tris hydrochloride (HCl).
- HCl Tris hydrochloride
- the tromethamine is at a concentration of or of about 0.1 to 0.3 mg/mL or of or of about 0.01 to 0.15 mg/mL.
- the Tris HCl is at a concentration of or of about 1.25 to 1.40 mg/mL or of or of about 0.5 to 0.65 mg/mL.
- the composition of any one of paragraphs 56 to 60, wherein the stabilizing agent is sucrose. 62.
- composition of paragraph 61 wherein the sucrose is at a concentration of or of about 95 to 110 mg/mL or of or of about 35 to 50 mg/mL.
- the salt diluent for reconstitution is sodium chloride.
- the sodium chloride is at a concentration of or of about 5 to 15 mg/mL.
- 65 The composition of any one of paragraphs 36 to 64, wherein the composition is a liquid or lyophilized. 66.
- composition of paragraph 65 comprising an RNA molecule at a concentration of or of about 0.01 to 0.09 mg/mL formulated in a lipid nanoparticle (LNP) comprising a cationic lipid at a concentration of or of about 0.8 to 0.95 mg/mL, a PEGylated lipid at a concentration of or of about 0.05 to 0.15 mg/mL, a neutral lipid at a concentration of or of about 0.1 to 0.25 mg/mL and a steroid or steroid analog at a concentration of or of about 0.3 to 0.45 mg/mL, and further comprising a Tris buffer comprising tromethamine at a concentration of or of about 0.1 to 0.3 mg/mL and Tris hydrochloride (HCl) at a concentration of or of about 1.25 to 1.40 mg/mL, and sucrose at a concentration of or of about 95 to 110 mg/mL, wherein the composition is a liquid composition.
- LNP lipid nanoparticle
- composition of paragraph 65 comprising an RNA molecule at a concentration of or of about 0.01 to 0.09 mg/mL formulated in a lipid nanoparticle (LNP) comprising a cationic lipid at a concentration of or of about 0.8 to 0.95 mg/mL, a PEGylated lipid at a concentration of or of about 0.05 to 0.15 mg/mL, a neutral lipid at a concentration of or of about 0.1 to 0.25 mg/mL and a steroid or steroid analog at a concentration of or of about 0.3 to 0.45 mg/mL, and further comprising a Tris buffer comprising tromethamine at a concentration of or of about 0.01 to 0.15 mg/mL and Tris hydrochloride (HCl) at a concentration of or of about 0.5 to 0.65 mg/mL, sucrose at a concentration of or of about 35 to 50 mg/mL.
- LNP lipid nanoparticle
- composition of paragraph 65 wherein the composition is reconstituted with sodium chloride at a concentration of or of about 5 to 15 mg/mL.
- composition of paragraph 65 wherein the composition is reconstituted with or with about 0.6 to 0.75 mL sodium chloride.
- 70 The composition of paragraph 65, further comprising or comprising about 5 to 15 mM Tris buffer, 200 to 400 mM sucrose at a pH of or of about 7.0 to 8.0, and optionally, 0.9% sodium chloride diluent to reconstitute.
- a method of inducing an immune response in a subject comprising administering to the subject an effective amount of the RNA molecule, RNA-LNP and/or composition of any one of paragraphs 1 to 70. 72.
- a method of preventing, treating or ameliorating an infection, disease or condition in a subject comprising administering to a subject an effective amount of the RNA molecule, RNA-LNP and/or composition of any one of paragraphs 1 to 70.
- the method of paragraph 72, wherein the infection, disease or condition is associated with RSV.
- the method of paragraph 72 or 73, wherein the infection, disease or condition is RSV infection-induced acute respiratory tract illness, including pneumonia and bronchitis.
- RNA-LNP Use of the RNA molecule, RNA-LNP and/or composition of any one of paragraphs 1 to 70 in the manufacture of a medicament for use in preventing, treating or ameliorating an infection, disease or condition in a subject.
- 77. The use of paragraph 76, wherein the infection, disease or condition is associated with RSV.
- 78. The use of paragraph 76 or 77, wherein the infection, disease or condition is RSV infection-related respiratory illness, including pneumonia and bronchitis. 79.
- any one of paragraphs 71 to 78 wherein the subject is less than about 1 year of age, about 1 year of age or older, about 5 years of age or older, about 10 years of age or older, about 20 years of age or older, about 30 years of age or older, about 40 years of age or older, about 50 years of age or older, about 60 years of age or older, about 70 years of age or older, or older.
- 80. The method or use of any one of paragraphs 71 to 79, wherein the subject the subject is about 50 years of age or older.
- RNA molecule or composition is administered as a vaccine.
- RNA molecule or composition is administered by intradermal or intramuscular injection.
- modRNA comprises nucleotides having SEQ ID NO: 13.
- the modRNA comprises nucleotides having SEQ ID NO: 14.
- the saRNA comprises nucleotides having SEQ ID NO: 15.
- RNA molecule of paragraph 83 wherein the modRNA comprises nucleotides having SEQ ID NO: 16.
- modRNA comprises nucleotides having SEQ ID NO: 16.
- any one of paragraphs 71 to 93 wherein the subject is administered a dose of at least or at least about 15 ⁇ g, 30 ⁇ g, 60 ⁇ g, 90 ⁇ g, 100 ⁇ g or higher RNA molecule and/or composition per administration.
- the subject is administered an injection with a volume of or of about 0.25 to 1 mL, including but not limited to, of or of about 0.25, 0.5, 1 mL. All of the methods disclosed and claimed herein can be made and executed without undue experimentation in light of the present disclosure.
- compositions and methods of this disclosure have been described in terms of certain aspects, it will be apparent to those of skill in the art that variations may be applied to the methods and in the steps or in the sequence of steps of the method described herein without departing from the concept, spirit and scope of the disclosure. More specifically, it will be apparent that certain agents which are both chemically and physiologically related may be substituted for the agents described herein while the same or similar results would be achieved. All such similar substitutes and modifications apparent to those skilled in the art are deemed to be within the spirit, scope and concept of the disclosure as defined by the appended claims.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Virology (AREA)
- Biochemistry (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Mycology (AREA)
- Immunology (AREA)
- Plant Pathology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Pulmonology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Oncology (AREA)
- Gastroenterology & Hepatology (AREA)
- Communicable Diseases (AREA)
- Nanotechnology (AREA)
- Optics & Photonics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Description

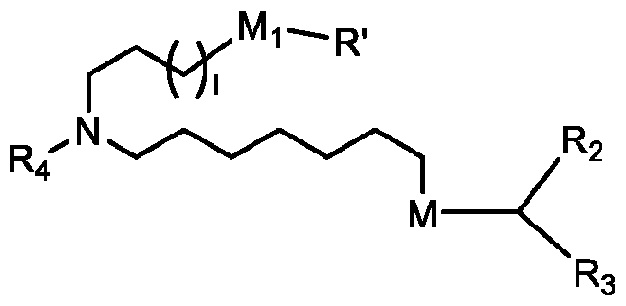



















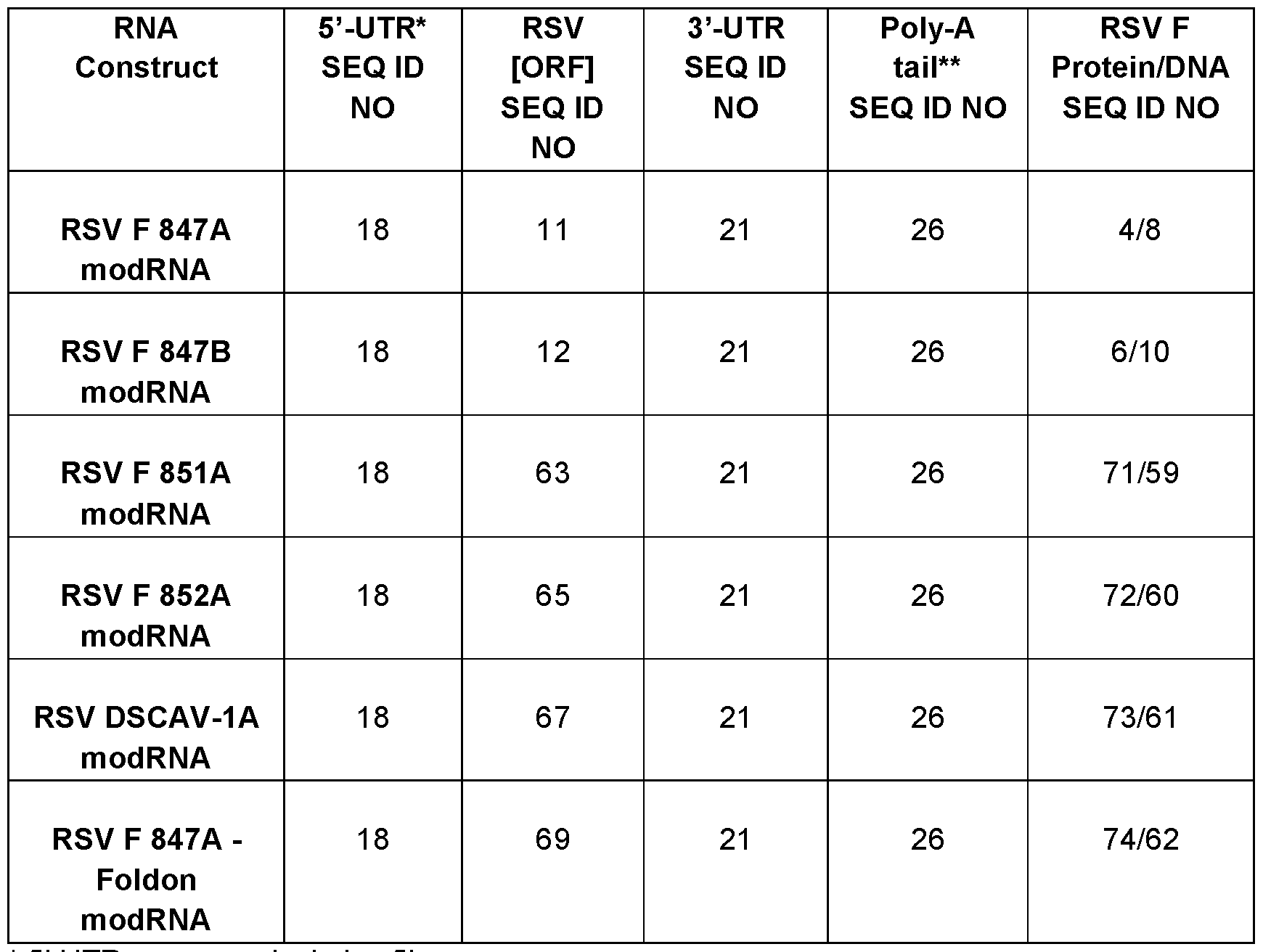


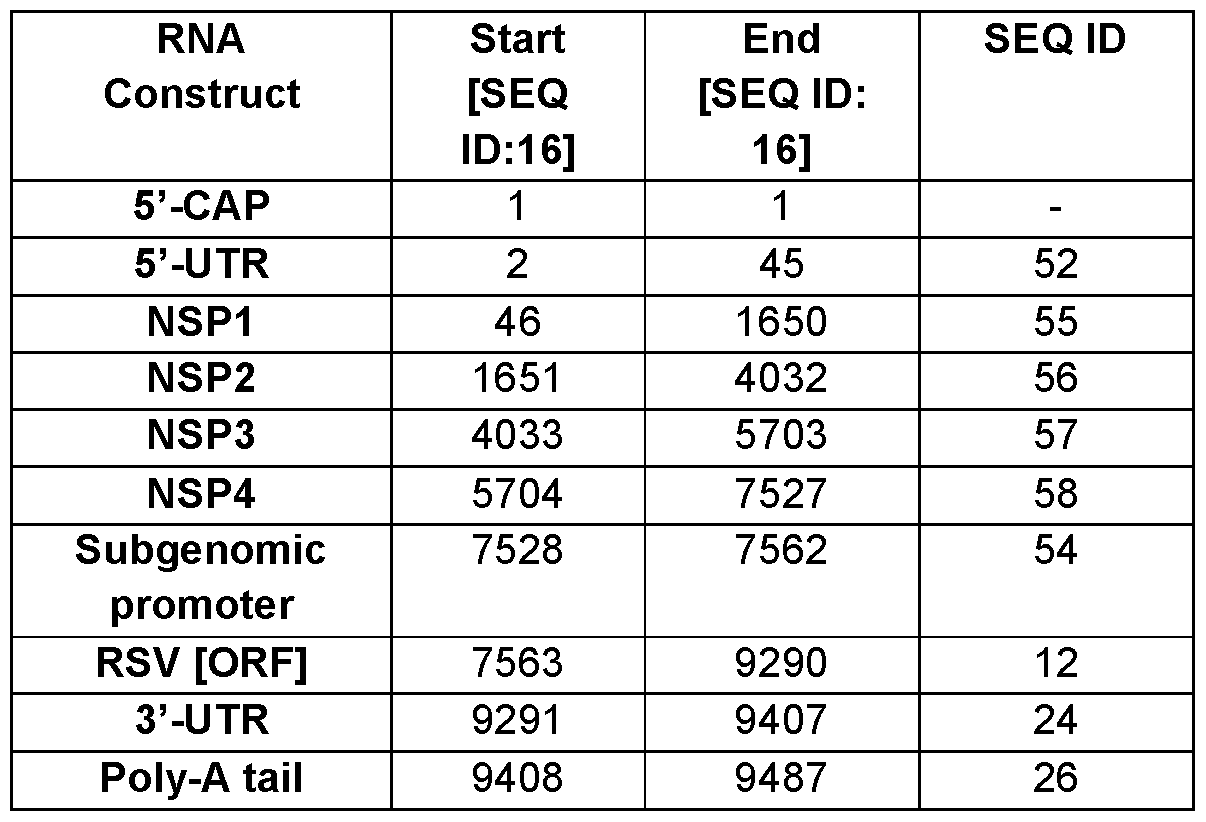

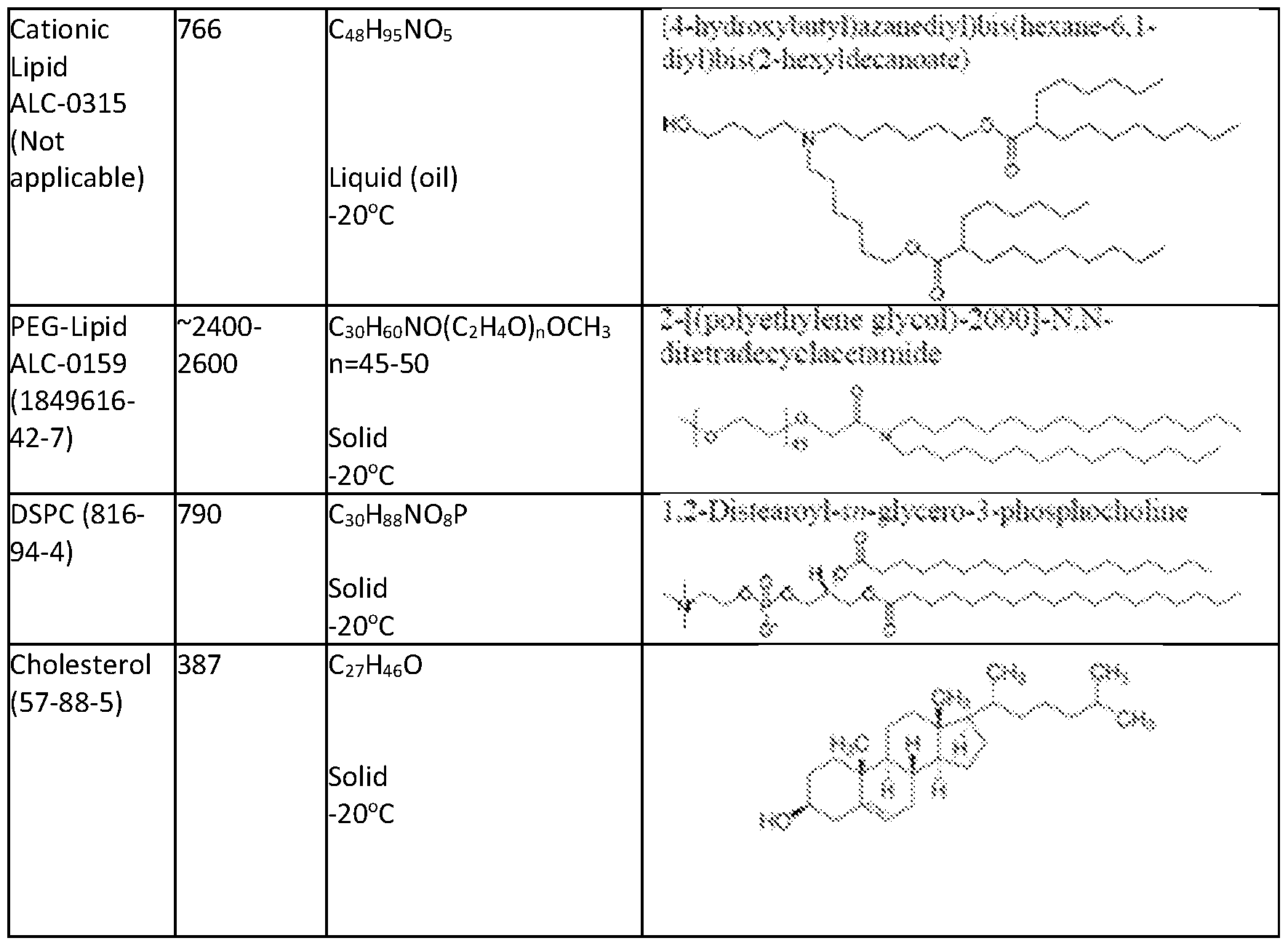



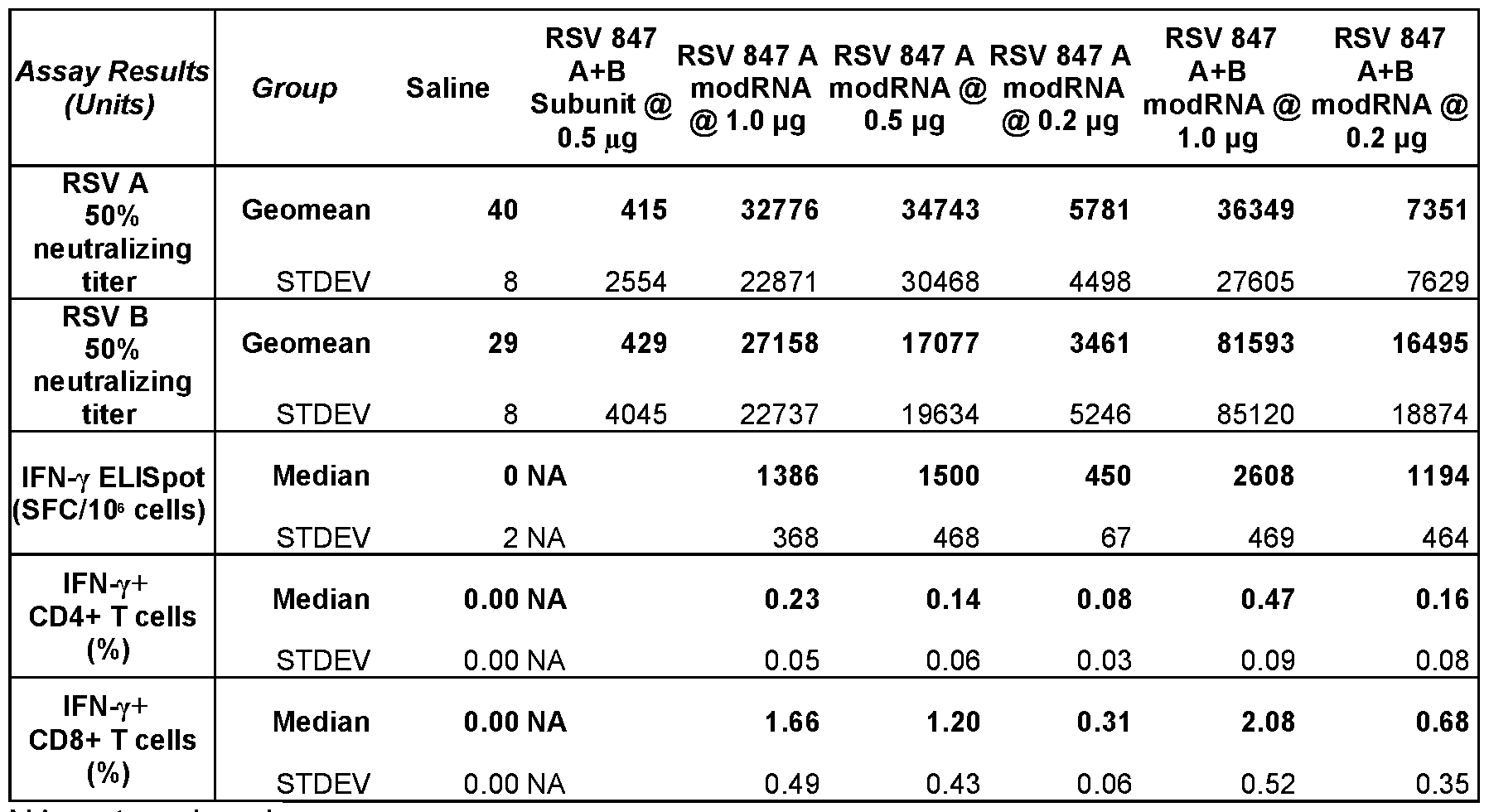




























Claims
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020257017392A KR20250096808A (en) | 2022-10-27 | 2023-10-26 | RNA molecule encoding RSV-F and vaccine comprising the same |
| EP23800587.0A EP4608440A1 (en) | 2022-10-27 | 2023-10-26 | Rna molecules encoding rsv-f and vaccines containing them |
| AU2023369585A AU2023369585A1 (en) | 2022-10-27 | 2023-10-26 | Rna molecules encoding rsv-f and vaccines containing them |
| PE2025000887A PE20251592A1 (en) | 2022-10-27 | 2023-10-26 | RNA MOLECULES THAT CODE RSV-F AND VACCINES THAT CONTAIN THEM |
| CN202380075365.8A CN120153078A (en) | 2022-10-27 | 2023-10-26 | RNA molecules encoding RSV-F and vaccines containing the same |
| IL320459A IL320459A (en) | 2022-10-27 | 2023-10-26 | RNA molecules encoding RSV-F and vaccines containing them |
| CONC2025/0005088A CO2025005088A2 (en) | 2022-10-27 | 2025-04-23 | RNA molecules encoding RSV-F and vaccines containing them |
| MX2025004821A MX2025004821A (en) | 2022-10-27 | 2025-04-24 | Rna molecules encoding rsv-f and vaccines containing them |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202263381238P | 2022-10-27 | 2022-10-27 | |
| US63/381,238 | 2022-10-27 | ||
| US202363585254P | 2023-09-26 | 2023-09-26 | |
| US63/585,254 | 2023-09-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024089633A1 true WO2024089633A1 (en) | 2024-05-02 |
Family
ID=88690013
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2023/060798 Ceased WO2024089633A1 (en) | 2022-10-27 | 2023-10-26 | Rna molecules encoding rsv-f and vaccines containing them |
Country Status (10)
| Country | Link |
|---|---|
| EP (1) | EP4608440A1 (en) |
| KR (1) | KR20250096808A (en) |
| CN (1) | CN120153078A (en) |
| AU (1) | AU2023369585A1 (en) |
| CO (1) | CO2025005088A2 (en) |
| IL (1) | IL320459A (en) |
| MX (1) | MX2025004821A (en) |
| PE (1) | PE20251592A1 (en) |
| TW (1) | TWI888970B (en) |
| WO (1) | WO2024089633A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025074292A2 (en) | 2023-10-06 | 2025-04-10 | Pfizer Inc. | Immunogenic compositions |
| WO2025186725A2 (en) | 2024-03-06 | 2025-09-12 | Pfizer Inc. | Improved lnp formulations and uses thereof |
Citations (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4367110A (en) | 1979-07-02 | 1983-01-04 | Toppan Printing Co. | Decorative laminate and a manufacturing method therefor |
| US4452901A (en) | 1980-03-20 | 1984-06-05 | Ciba-Geigy Corporation | Electrophoretically transferring electropherograms to nitrocellulose sheets for immuno-assays |
| US4578770A (en) | 1982-08-30 | 1986-03-25 | Musashi Engineering Kabushiki Kaisha | Method of discriminating sheet |
| US4596792A (en) | 1981-09-04 | 1986-06-24 | The Regents Of The University Of California | Safe vaccine for hepatitis containing polymerized serum albumin |
| US4599230A (en) | 1984-03-09 | 1986-07-08 | Scripps Clinic And Research Foundation | Synthetic hepatitis B virus vaccine including both T cell and B cell determinants |
| US4599231A (en) | 1984-03-09 | 1986-07-08 | Scripps Clinic And Research Foundation | Synthetic hepatitis B virus vaccine including both T cell and B cell determinants |
| US4601903A (en) | 1985-05-01 | 1986-07-22 | The United States Of America As Represented By The Department Of Health And Human Services | Vaccine against Neisseria meningitidis Group B serotype 2 invasive disease |
| US4608251A (en) | 1984-11-09 | 1986-08-26 | Pitman-Moore, Inc. | LHRH analogues useful in stimulating anti-LHRH antibodies and vaccines containing such analogues |
| US6733754B2 (en) | 1999-01-29 | 2004-05-11 | Pfizer, Inc. | Adjuvants for use in vaccines |
| US20040142025A1 (en) | 2002-06-28 | 2004-07-22 | Protiva Biotherapeutics Ltd. | Liposomal apparatus and manufacturing methods |
| US6793923B2 (en) | 2000-11-07 | 2004-09-21 | Immunovaccine Technologies, Inc. | Vaccines with enhanced immune response and methods for their preparation |
| US20070042031A1 (en) | 2005-07-27 | 2007-02-22 | Protiva Biotherapeutics, Inc. | Systems and methods for manufacturing liposomes |
| WO2008147196A2 (en) | 2007-06-01 | 2008-12-04 | Aimm Therapeutics B.V. | Rsv-specific binding molecules and means for producing them |
| WO2009079796A1 (en) | 2007-12-24 | 2009-07-02 | Id Biomedical Corporation Of Quebec | Recombinant rsv antigens |
| US20090226470A1 (en) | 2007-12-11 | 2009-09-10 | Mauro Vincent P | Compositions and methods related to mRNA translational enhancer elements |
| WO2010149745A1 (en) | 2009-06-24 | 2010-12-29 | Glaxosmithkline Biologicals S.A. | Recombinant rsv antigens |
| WO2011008974A2 (en) | 2009-07-15 | 2011-01-20 | Novartis Ag | Rsv f protein compositions and methods for making same |
| WO2011043643A1 (en) | 2009-10-06 | 2011-04-14 | Medimmune Ltd | Rsv-specific binding molecule |
| WO2012019780A1 (en) | 2010-08-13 | 2012-02-16 | Curevac Gmbh | Nucleic acid comprising or coding for a histone stem-loop and a poly(a) sequence or a polyadenylation signal for increasing the expression of an encoded protein |
| WO2013016058A1 (en) | 2011-07-22 | 2013-01-31 | Merck Sharp & Dohme Corp. | Novel bis-nitrogen containing cationic lipids for oligonucleotide delivery |
| WO2013078199A2 (en) | 2011-11-23 | 2013-05-30 | Children's Medical Center Corporation | Methods for enhanced in vivo delivery of synthetic, modified rnas |
| WO2013086373A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Lipids for the delivery of active agents |
| WO2013103659A1 (en) | 2012-01-04 | 2013-07-11 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | Stabilizing rna by incorporating chain-terminating nucleosides at the 3'-terminus |
| WO2013143700A2 (en) | 2012-03-27 | 2013-10-03 | Curevac Gmbh | Artificial nucleic acid molecules comprising a 5'top utr |
| WO2014160463A1 (en) | 2013-03-13 | 2014-10-02 | The United States Of America, As Represented By The Secretary, Department Of Health & Human Services | Prefusion rsv f proteins and their use |
| WO2014174018A1 (en) | 2013-04-25 | 2014-10-30 | Crucell Holland B.V. | Stabilized soluble prefusion rsv f polypeptides |
| WO2014202570A1 (en) | 2013-06-17 | 2014-12-24 | Crucell Holland B.V. | Stabilized soluble pre-fusion rsv f polypeptides |
| WO2015013551A1 (en) | 2013-07-25 | 2015-01-29 | Marshall Christopher Patrick | Conformationally stabilized rsv pre-fusion f proteins |
| WO2015177312A1 (en) | 2014-05-22 | 2015-11-26 | Glaxosmithkline Biologicals Sa | Rsvf trimerization domains |
| WO2015199952A1 (en) | 2014-06-25 | 2015-12-30 | Acuitas Therapeutics Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2016005324A1 (en) | 2014-07-11 | 2016-01-14 | Biontech Rna Pharmaceuticals Gmbh | Stabilization of poly(a) sequence encoding dna sequences |
| WO2016091391A1 (en) | 2014-12-12 | 2016-06-16 | Curevac Ag | Artificial nucleic acid molecules for improved protein expression |
| WO2017005848A1 (en) | 2015-07-07 | 2017-01-12 | Janssen Vaccines & Prevention B.V. | Stabilized soluble pre-fusion rsv f polypeptides |
| WO2017005844A1 (en) | 2015-07-07 | 2017-01-12 | Janssen Vaccines & Prevention B.V. | Vaccine against rsv |
| WO2017075531A1 (en) | 2015-10-28 | 2017-05-04 | Acuitas Therapeutics, Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017109629A1 (en) | 2015-12-23 | 2017-06-29 | Pfizer Inc. | Rsv f protein mutants |
| WO2017174564A1 (en) | 2016-04-05 | 2017-10-12 | Janssen Vaccines & Prevention B.V. | Vaccine against rsv |
| WO2018109220A2 (en) | 2016-12-16 | 2018-06-21 | Institute For Research In Biomedicine | Novel recombinant prefusion rsv f proteins and uses thereof |
| WO2020146805A1 (en) | 2019-01-11 | 2020-07-16 | Acuitas Therapeutics, Inc. | Lipids for lipid nanoparticle delivery of active agents |
| WO2021155243A1 (en) * | 2020-01-30 | 2021-08-05 | Modernatx, Inc. | Respiratory virus immunizing compositions |
| WO2022032154A2 (en) | 2020-08-06 | 2022-02-10 | Modernatx, Inc. | Compositions for the delivery of payload molecules to airway epithelium |
| WO2022067010A1 (en) | 2020-09-25 | 2022-03-31 | Modernatx, Inc. | Multi-proline-substituted coronavirus spike protein vaccines |
| WO2022101469A1 (en) * | 2020-11-16 | 2022-05-19 | BioNTech SE | Compositions and methods for stabilization of lipid nanoparticle mrna vaccines |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2018004917A (en) * | 2015-10-22 | 2019-04-01 | Modernatx Inc | Respiratory syncytial virus vaccine. |
-
2023
- 2023-10-26 CN CN202380075365.8A patent/CN120153078A/en active Pending
- 2023-10-26 PE PE2025000887A patent/PE20251592A1/en unknown
- 2023-10-26 IL IL320459A patent/IL320459A/en unknown
- 2023-10-26 AU AU2023369585A patent/AU2023369585A1/en active Pending
- 2023-10-26 WO PCT/IB2023/060798 patent/WO2024089633A1/en not_active Ceased
- 2023-10-26 KR KR1020257017392A patent/KR20250096808A/en active Pending
- 2023-10-26 TW TW112141022A patent/TWI888970B/en active
- 2023-10-26 EP EP23800587.0A patent/EP4608440A1/en active Pending
-
2025
- 2025-04-23 CO CONC2025/0005088A patent/CO2025005088A2/en unknown
- 2025-04-24 MX MX2025004821A patent/MX2025004821A/en unknown
Patent Citations (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4367110A (en) | 1979-07-02 | 1983-01-04 | Toppan Printing Co. | Decorative laminate and a manufacturing method therefor |
| US4452901A (en) | 1980-03-20 | 1984-06-05 | Ciba-Geigy Corporation | Electrophoretically transferring electropherograms to nitrocellulose sheets for immuno-assays |
| US4596792A (en) | 1981-09-04 | 1986-06-24 | The Regents Of The University Of California | Safe vaccine for hepatitis containing polymerized serum albumin |
| US4578770A (en) | 1982-08-30 | 1986-03-25 | Musashi Engineering Kabushiki Kaisha | Method of discriminating sheet |
| US4599230A (en) | 1984-03-09 | 1986-07-08 | Scripps Clinic And Research Foundation | Synthetic hepatitis B virus vaccine including both T cell and B cell determinants |
| US4599231A (en) | 1984-03-09 | 1986-07-08 | Scripps Clinic And Research Foundation | Synthetic hepatitis B virus vaccine including both T cell and B cell determinants |
| US4608251A (en) | 1984-11-09 | 1986-08-26 | Pitman-Moore, Inc. | LHRH analogues useful in stimulating anti-LHRH antibodies and vaccines containing such analogues |
| US4601903A (en) | 1985-05-01 | 1986-07-22 | The United States Of America As Represented By The Department Of Health And Human Services | Vaccine against Neisseria meningitidis Group B serotype 2 invasive disease |
| US6733754B2 (en) | 1999-01-29 | 2004-05-11 | Pfizer, Inc. | Adjuvants for use in vaccines |
| US6793923B2 (en) | 2000-11-07 | 2004-09-21 | Immunovaccine Technologies, Inc. | Vaccines with enhanced immune response and methods for their preparation |
| US20040142025A1 (en) | 2002-06-28 | 2004-07-22 | Protiva Biotherapeutics Ltd. | Liposomal apparatus and manufacturing methods |
| US20070042031A1 (en) | 2005-07-27 | 2007-02-22 | Protiva Biotherapeutics, Inc. | Systems and methods for manufacturing liposomes |
| WO2008147196A2 (en) | 2007-06-01 | 2008-12-04 | Aimm Therapeutics B.V. | Rsv-specific binding molecules and means for producing them |
| US20090226470A1 (en) | 2007-12-11 | 2009-09-10 | Mauro Vincent P | Compositions and methods related to mRNA translational enhancer elements |
| WO2009079796A1 (en) | 2007-12-24 | 2009-07-02 | Id Biomedical Corporation Of Quebec | Recombinant rsv antigens |
| WO2010149745A1 (en) | 2009-06-24 | 2010-12-29 | Glaxosmithkline Biologicals S.A. | Recombinant rsv antigens |
| WO2011008974A2 (en) | 2009-07-15 | 2011-01-20 | Novartis Ag | Rsv f protein compositions and methods for making same |
| WO2011043643A1 (en) | 2009-10-06 | 2011-04-14 | Medimmune Ltd | Rsv-specific binding molecule |
| WO2012019780A1 (en) | 2010-08-13 | 2012-02-16 | Curevac Gmbh | Nucleic acid comprising or coding for a histone stem-loop and a poly(a) sequence or a polyadenylation signal for increasing the expression of an encoded protein |
| WO2013016058A1 (en) | 2011-07-22 | 2013-01-31 | Merck Sharp & Dohme Corp. | Novel bis-nitrogen containing cationic lipids for oligonucleotide delivery |
| WO2013078199A2 (en) | 2011-11-23 | 2013-05-30 | Children's Medical Center Corporation | Methods for enhanced in vivo delivery of synthetic, modified rnas |
| WO2013086373A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Lipids for the delivery of active agents |
| WO2013103659A1 (en) | 2012-01-04 | 2013-07-11 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | Stabilizing rna by incorporating chain-terminating nucleosides at the 3'-terminus |
| WO2013143700A2 (en) | 2012-03-27 | 2013-10-03 | Curevac Gmbh | Artificial nucleic acid molecules comprising a 5'top utr |
| WO2014160463A1 (en) | 2013-03-13 | 2014-10-02 | The United States Of America, As Represented By The Secretary, Department Of Health & Human Services | Prefusion rsv f proteins and their use |
| WO2014174018A1 (en) | 2013-04-25 | 2014-10-30 | Crucell Holland B.V. | Stabilized soluble prefusion rsv f polypeptides |
| WO2014202570A1 (en) | 2013-06-17 | 2014-12-24 | Crucell Holland B.V. | Stabilized soluble pre-fusion rsv f polypeptides |
| WO2015013551A1 (en) | 2013-07-25 | 2015-01-29 | Marshall Christopher Patrick | Conformationally stabilized rsv pre-fusion f proteins |
| WO2015177312A1 (en) | 2014-05-22 | 2015-11-26 | Glaxosmithkline Biologicals Sa | Rsvf trimerization domains |
| WO2015199952A1 (en) | 2014-06-25 | 2015-12-30 | Acuitas Therapeutics Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US9737619B2 (en) | 2014-06-25 | 2017-08-22 | Acuitas Therapeutics, Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2016005324A1 (en) | 2014-07-11 | 2016-01-14 | Biontech Rna Pharmaceuticals Gmbh | Stabilization of poly(a) sequence encoding dna sequences |
| WO2016091391A1 (en) | 2014-12-12 | 2016-06-16 | Curevac Ag | Artificial nucleic acid molecules for improved protein expression |
| WO2017005844A1 (en) | 2015-07-07 | 2017-01-12 | Janssen Vaccines & Prevention B.V. | Vaccine against rsv |
| WO2017005848A1 (en) | 2015-07-07 | 2017-01-12 | Janssen Vaccines & Prevention B.V. | Stabilized soluble pre-fusion rsv f polypeptides |
| WO2017075531A1 (en) | 2015-10-28 | 2017-05-04 | Acuitas Therapeutics, Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US10166298B2 (en) | 2015-10-28 | 2019-01-01 | Acuitas Therapeutics, Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017109629A1 (en) | 2015-12-23 | 2017-06-29 | Pfizer Inc. | Rsv f protein mutants |
| WO2017174564A1 (en) | 2016-04-05 | 2017-10-12 | Janssen Vaccines & Prevention B.V. | Vaccine against rsv |
| WO2018109220A2 (en) | 2016-12-16 | 2018-06-21 | Institute For Research In Biomedicine | Novel recombinant prefusion rsv f proteins and uses thereof |
| WO2020146805A1 (en) | 2019-01-11 | 2020-07-16 | Acuitas Therapeutics, Inc. | Lipids for lipid nanoparticle delivery of active agents |
| WO2021155243A1 (en) * | 2020-01-30 | 2021-08-05 | Modernatx, Inc. | Respiratory virus immunizing compositions |
| WO2022032154A2 (en) | 2020-08-06 | 2022-02-10 | Modernatx, Inc. | Compositions for the delivery of payload molecules to airway epithelium |
| WO2022067010A1 (en) | 2020-09-25 | 2022-03-31 | Modernatx, Inc. | Multi-proline-substituted coronavirus spike protein vaccines |
| WO2022101469A1 (en) * | 2020-11-16 | 2022-05-19 | BioNTech SE | Compositions and methods for stabilization of lipid nanoparticle mrna vaccines |
Non-Patent Citations (24)
| Title |
|---|
| "GenBank", Database accession no. 138250 |
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING CO. |
| "Swiss Prot", Database accession no. P13843 |
| AMY S. ESPESETH ET AL: "Modified mRNA/lipid nanoparticle-based vaccines expressing respiratory syncytial virus F protein variants are immunogenic and protective in rodent models of RSV infection", NPJ VACCINES, vol. 5, no. 1, 16, 14 February 2020 (2020-02-14), XP055695604, DOI: 10.1038/s41541-020-0163-z * |
| BECKERT, B.MASQUIDA, B.: "Methods in Molecular Biology", vol. 703, 2010, HUMANA PRESS, article "Synthesis of RNA by In vitro Transcription in RNA" |
| BOWMAN, J.C.AZIZI, B.LENZ, T.K.RAY, P.WILLIAMS, L.D.: "Methods", vol. 941, 2012, HUMANA PRESS, article "RNA in vitro transcription and RNA purification by denaturing PAGE in Recombinant and in vitro RNA syntheses" |
| BRUNELLE, J.L.GREEN, R.: "In vitro transcription from plasmid or PCR-amplified DNA", METHODS IN ENZYMOLOGY, vol. 530, 2013, pages 101 - 114, XP009190475 |
| CHAIWATPONGSAKORN, S.EPAND, R.F.COLLINS, P.L.EPAND R.M.PEEPLES, M.E., J VIROL., vol. 85, no. 8, 2011, pages 3968 - 77 |
| DORMITZER, P.R.GRANDI, G.RAPPUOLI, R., NATURE REVIEWS MICROBIOL, vol. 10, 2012, pages 807 |
| GILMAN MSMOIN SMMAS V ET AL., PLOS PATHOGENS, vol. 11, no. 7, 2015 |
| GREEN, T.W.P.G.M. WUTZ: "Protective Groups in Organic Synthesis", 1999, WILEY |
| KAMAKAKA, R. T.KRAUS, W. L.: "In vitro Transcription", CURRENT PROTOCOLS IN CELL BIOLOGY, 2001 |
| KOPPEL, D., J. CHEM. PHYS., vol. 57, 1972, pages 4814 - 4820 |
| LINPINSEL, J.LCONN, G.L., GENERAL PROTOCOLS FOR PREPARATION OF PLASMID DNA TEMPLATE |
| LOSICK, R: "In vitro transcription", ANN REV BIOCHEM, vol. 41, 1972, pages 409 - 46, XP055712693, DOI: 10.1146/annurev.bi.41.070172.002205 |
| MCLELLAN JSCHEN MLEUNG S ET AL.: "Structure of RSV fusion glycoprotein trimer bound to a pre-fusion-specific neutralizing antibody", SCIENCE, vol. 340, 2013, pages 1113 - 1117 |
| NEDDLEMANWUNSCH, J. MOL. BIOL., vol. 48, 1970, pages 443 |
| NGWUTA, J.O.CHEN, M.MODJARRAD, K.JOYCE, M.G.KANEKIYO, M.KUMAR, A.YASSINE, H.M.MOIN, S.M.KILLIKELLY, A.M.CHUANG, G.Y., SCIENCE TRANSLAT. MED., vol. 14, no. 7, 2015, pages 309 |
| PEARSONLIPMAN, PROC. NATL ACAD. SCI. USA, vol. 88, 1988, pages 2444 |
| REMINGTON: "The Science and Practice of Pharmacy", 2005, LIPPINCOTT WILLIAMS & WILKINS |
| SCHOENMAKER LINDE ET AL: "mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability", INTERNATIONAL JOURNAL OF PHARMACEUTICS, vol. 601, 1 May 2021 (2021-05-01), NL, pages 120586, XP055828800, ISSN: 0378-5173, DOI: 10.1016/j.ijpharm.2021.120586 * |
| SMITHWATERMAN, ADS APP. MATH., vol. 2, 1981, pages 482 |
| WEINBERG ET AL., J INFECT DIS, vol. 201, no. 11, 1 June 2010 (2010-06-01), pages 1607 - 10 |
| YUNUS, A.S.JACKSON T.P.CRISAFI, K.BURIMSKI, I.KILGORE, N.R.ZOUMPLIS, D.ALLAWAY, G.P.WILD, CTSALZWEDEL, K., VIROLOGY, vol. 396, no. 2, 20 January 2010 (2010-01-20), pages 226 - 37 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025074292A2 (en) | 2023-10-06 | 2025-04-10 | Pfizer Inc. | Immunogenic compositions |
| WO2025186725A2 (en) | 2024-03-06 | 2025-09-12 | Pfizer Inc. | Improved lnp formulations and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CO2025005088A2 (en) | 2025-05-08 |
| AU2023369585A1 (en) | 2025-04-10 |
| KR20250096808A (en) | 2025-06-27 |
| MX2025004821A (en) | 2025-06-02 |
| TWI888970B (en) | 2025-07-01 |
| TW202424187A (en) | 2024-06-16 |
| IL320459A (en) | 2025-06-01 |
| CN120153078A (en) | 2025-06-13 |
| PE20251592A1 (en) | 2025-06-16 |
| EP4608440A1 (en) | 2025-09-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230233671A1 (en) | Rna molecules | |
| TWI888970B (en) | Rna molecules | |
| EP4448002A1 (en) | Polynucleotide compositions and uses thereof | |
| US20240252612A1 (en) | Immunogenic compositions and uses thereof | |
| US20250000960A1 (en) | Polynucleotide compositions and uses thereof | |
| EP4608441A1 (en) | Immunogenic compositions against influenza and rsv | |
| JP2024151316A (en) | RNA molecule | |
| WO2025126071A1 (en) | Rna molecules | |
| US20260021175A1 (en) | Immunogenic compositions and uses thereof | |
| JP2025166811A (en) | RNA molecules encoding RSV-F and vaccines containing the same | |
| WO2025126019A1 (en) | Rna molecules | |
| HK40122508A (en) | Rna molecules encoding rsv-f and vaccines containing them | |
| WO2025027492A1 (en) | Coronavirus antigen variants | |
| WO2026030275A1 (en) | Rna molecules | |
| EP4651890A1 (en) | Immunogenic compositions and methods of inducing an immune response against varicella zoster virus | |
| KR20240152232A (en) | Rna molecules | |
| WO2025186719A1 (en) | Immunogenic compositions and uses thereof | |
| WO2025186725A2 (en) | Improved lnp formulations and uses thereof | |
| CN118574637A (en) | RNA molecules | |
| HK40109231A (en) | Rna molecules | |
| JP2026504080A (en) | Immunogenic compositions and methods for inducing an immune response against varicella-zoster virus | |
| HK40125987A (en) | Immunogenic compositions and methods of inducing an immune response against varicella zoster virus | |
| WO2025202834A1 (en) | Polynucleotide compositions and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23800587 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P2025-00885 Country of ref document: AE Ref document number: AU2023369585 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 820053 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202590849 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 2023369585 Country of ref document: AU Date of ref document: 20231026 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112025006907 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2025523515 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 320459 Country of ref document: IL Ref document number: 2025523515 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2025/004821 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202517040292 Country of ref document: IN Ref document number: 202380075365.8 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 202517040292 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20257017392 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023800587 Country of ref document: EP Ref document number: 2025110755 Country of ref document: RU |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11202502014Q Country of ref document: SG |
|
| WWP | Wipo information: published in national office |
Ref document number: 11202502014Q Country of ref document: SG |
|
| ENP | Entry into the national phase |
Ref document number: 2023800587 Country of ref document: EP Effective date: 20250527 |
|
| WWP | Wipo information: published in national office |
Ref document number: MX/A/2025/004821 Country of ref document: MX |
|
| WWP | Wipo information: published in national office |
Ref document number: 202380075365.8 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020257017392 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2025110755 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2023800587 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 112025006907 Country of ref document: BR Kind code of ref document: A2 Effective date: 20250408 |