WO2013106646A2 - Compounds and methods for the inhibition of vcb e3 ubiquitin ligase - Google Patents
Compounds and methods for the inhibition of vcb e3 ubiquitin ligase Download PDFInfo
- Publication number
- WO2013106646A2 WO2013106646A2 PCT/US2013/021141 US2013021141W WO2013106646A2 WO 2013106646 A2 WO2013106646 A2 WO 2013106646A2 US 2013021141 W US2013021141 W US 2013021141W WO 2013106646 A2 WO2013106646 A2 WO 2013106646A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- group
- alkyl
- mmol
- compound according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *Cc1c[n](-c2ccccc2C(N(CCC2)[C@@]2C(NCc(cc2)ccc2Cl)=O)=O)nn1 Chemical compound *Cc1c[n](-c2ccccc2C(N(CCC2)[C@@]2C(NCc(cc2)ccc2Cl)=O)=O)nn1 0.000 description 17
- PUBUFLLZZJPLPP-ZMOSESBSSA-N CC(C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)N1c2ccccc2CC1 Chemical compound CC(C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)N1c2ccccc2CC1 PUBUFLLZZJPLPP-ZMOSESBSSA-N 0.000 description 2
- UTTGKBPJQAGBNL-XYPHTWIQSA-N C[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NC(C)(C)c(cc1)ccc1Cl)=O)=O)c1cc(C)n[o]1 Chemical compound C[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NC(C)(C)c(cc1)ccc1Cl)=O)=O)c1cc(C)n[o]1 UTTGKBPJQAGBNL-XYPHTWIQSA-N 0.000 description 2
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N C(C1)Cc2c1cccc2 Chemical compound C(C1)Cc2c1cccc2 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 1
- TVZWLAZOVACWKE-JLYBBMSXSA-N CC(C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)c1ccccc1 Chemical compound CC(C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)c1ccccc1 TVZWLAZOVACWKE-JLYBBMSXSA-N 0.000 description 1
- ZKFXPFPQKOTKSD-UHFFFAOYSA-N CC(C)(C)OC(CCOCCOCCOCCOc1cc(C(OC)=O)ccc1)=O Chemical compound CC(C)(C)OC(CCOCCOCCOCCOc1cc(C(OC)=O)ccc1)=O ZKFXPFPQKOTKSD-UHFFFAOYSA-N 0.000 description 1
- ZRVKACXYHYQNGM-CVEARBPZSA-N CC(C)(C)O[C@H](C1)CN[C@@H]1C(NCc(cc1)ccc1-c1cnc[o]1)=O Chemical compound CC(C)(C)O[C@H](C1)CN[C@@H]1C(NCc(cc1)ccc1-c1cnc[o]1)=O ZRVKACXYHYQNGM-CVEARBPZSA-N 0.000 description 1
- PKNJGCFWMVESEZ-DXPHZYLKSA-N CC(C)(C)[C@@H](C(N(CC(C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)NC(COCCOCCOCCOCCOCCCC[C@H](C1)[C@@H]([C@H](CC2)C(C)(CC3)[C@H]2O)[C@H]3c(cc2)c1cc2O)=O Chemical compound CC(C)(C)[C@@H](C(N(CC(C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)NC(COCCOCCOCCOCCOCCCC[C@H](C1)[C@@H]([C@H](CC2)C(C)(CC3)[C@H]2O)[C@H]3c(cc2)c1cc2O)=O PKNJGCFWMVESEZ-DXPHZYLKSA-N 0.000 description 1
- UPPDFYVGGFPFGN-JXFKEZNVSA-N CC(C)CN([C@@H](C)C(N(CCC1)[C@@H]1C(NCc(cc1)ccc1Cl)=O)=O)C(CNC(C)=O)=O Chemical compound CC(C)CN([C@@H](C)C(N(CCC1)[C@@H]1C(NCc(cc1)ccc1Cl)=O)=O)C(CNC(C)=O)=O UPPDFYVGGFPFGN-JXFKEZNVSA-N 0.000 description 1
- GGOYPZXHENWDBT-SQNIBIBYSA-N CC(C)[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)N Chemical compound CC(C)[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)N GGOYPZXHENWDBT-SQNIBIBYSA-N 0.000 description 1
- OUWPMPNUSXMZNO-ZZQHFIISSA-N CC(C)[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)NC(COCCOCCOCCOCCOCCCC[C@H](Cc1c2)[C@@H]([C@H](CC3)[C@](C)(CC4)[C@H]3O)[C@H]4c1ccc2O)=O Chemical compound CC(C)[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)NC(COCCOCCOCCOCCOCCCC[C@H](Cc1c2)[C@@H]([C@H](CC3)[C@](C)(CC4)[C@H]3O)[C@H]4c1ccc2O)=O OUWPMPNUSXMZNO-ZZQHFIISSA-N 0.000 description 1
- NXVYRNJMAKVCAP-PCDSSIDISA-N CC([C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)O)NC(C)=O)C1=[IH]=C1 Chemical compound CC([C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)O)NC(C)=O)C1=[IH]=C1 NXVYRNJMAKVCAP-PCDSSIDISA-N 0.000 description 1
- MOUBOVPEICVNHV-UEWDXFNNSA-N CC1=NOC(CC(N2[C@H](COCC=C)CCC2)=O)C1 Chemical compound CC1=NOC(CC(N2[C@H](COCC=C)CCC2)=O)C1 MOUBOVPEICVNHV-UEWDXFNNSA-N 0.000 description 1
- UPPABJWEIUNNIK-OWNMJYFTSA-N CC1NOC(CC(N(C[C@@H](C2)O)[C@@H]2C(N[C@@H](Cc(cc2)ccc2OC)c(cc2)ccc2Cl)=O)=O)=C1 Chemical compound CC1NOC(CC(N(C[C@@H](C2)O)[C@@H]2C(N[C@@H](Cc(cc2)ccc2OC)c(cc2)ccc2Cl)=O)=O)=C1 UPPABJWEIUNNIK-OWNMJYFTSA-N 0.000 description 1
- SJIHOWUBWIMPHG-PWSUYJOCSA-N CCOc1cc(C(N(C[C@@H](C2)O)[C@@H]2C(O)=O)=O)ccc1 Chemical compound CCOc1cc(C(N(C[C@@H](C2)O)[C@@H]2C(O)=O)=O)ccc1 SJIHOWUBWIMPHG-PWSUYJOCSA-N 0.000 description 1
- YMWLAHMAYWVXSG-IBGZPJMESA-N CN(C)c1cccc(C(N(CCC2)[C@@H]2C(NCc(cc2)ccc2Cl)=O)=O)c1 Chemical compound CN(C)c1cccc(C(N(CCC2)[C@@H]2C(NCc(cc2)ccc2Cl)=O)=O)c1 YMWLAHMAYWVXSG-IBGZPJMESA-N 0.000 description 1
- UOWCIIUJRDPPQQ-INIZCTEOSA-N COC(Cc1c[n](CC(N(CCC2)[C@@H]2C(NCc(cc2)ccc2Cl)=O)=O)nn1)=O Chemical compound COC(Cc1c[n](CC(N(CCC2)[C@@H]2C(NCc(cc2)ccc2Cl)=O)=O)nn1)=O UOWCIIUJRDPPQQ-INIZCTEOSA-N 0.000 description 1
- UHKGSFZMWLGESI-PHGOZILUSA-N C[C@@H](C(N(CC(C1)[O-3])[C@@H]1C1=[O]C(CCc2cnccc2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 Chemical compound C[C@@H](C(N(CC(C1)[O-3])[C@@H]1C1=[O]C(CCc2cnccc2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 UHKGSFZMWLGESI-PHGOZILUSA-N 0.000 description 1
- OTNZNNMYNQPLBH-CERFIVLJSA-N C[C@@H](C(N(CCC1)[C@@H]1C1=[O]C(CCc2cnccc2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 Chemical compound C[C@@H](C(N(CCC1)[C@@H]1C1=[O]C(CCc2cnccc2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 OTNZNNMYNQPLBH-CERFIVLJSA-N 0.000 description 1
- QZVUGBYQAOFQBK-AOVMDMDCSA-N C[C@@H](C(N(CCC1)[C@@H]1C1=[O]C(Cc2ccncc2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 Chemical compound C[C@@H](C(N(CCC1)[C@@H]1C1=[O]C(Cc2ccncc2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 QZVUGBYQAOFQBK-AOVMDMDCSA-N 0.000 description 1
- RDFJNIUYTKNUNQ-VJFRBFEXSA-N C[C@@H](C(N(CCC1)[C@@H]1C1=[O]C(Cc2nnn[nH]2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 Chemical compound C[C@@H](C(N(CCC1)[C@@H]1C1=[O]C(Cc2nnn[nH]2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 RDFJNIUYTKNUNQ-VJFRBFEXSA-N 0.000 description 1
- WMPDCKYVJGROGI-IPELMVKDSA-N C[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)NC(C1(C)COC1)=O Chemical compound C[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)NC(C1(C)COC1)=O WMPDCKYVJGROGI-IPELMVKDSA-N 0.000 description 1
- GGPWCNASPRRCQB-CKKISIDZSA-N C[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C1=[O]C(Cc2ccncc2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 Chemical compound C[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C1=[O]C(Cc2ccncc2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 GGPWCNASPRRCQB-CKKISIDZSA-N 0.000 description 1
- ZEUYYUDVUTXVSV-XXLBTEEXSA-N C[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C1=[O]C(Cc2nnn[nH]2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 Chemical compound C[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C1=[O]C(Cc2nnn[nH]2)(c(cc2)ccc2Cl)N1)=O)c1cc(C)n[o]1 ZEUYYUDVUTXVSV-XXLBTEEXSA-N 0.000 description 1
- KXJIVEJQOYZFRL-LSVBPWPTSA-N C[C@](CC1)([C@@H](CC2)[C@H]([C@H](CCCCOCCOCCO)Cc3c4)[C@H]1c3ccc4OCOC)[C@H]2OCOC Chemical compound C[C@](CC1)([C@@H](CC2)[C@H]([C@H](CCCCOCCOCCO)Cc3c4)[C@H]1c3ccc4OCOC)[C@H]2OCOC KXJIVEJQOYZFRL-LSVBPWPTSA-N 0.000 description 1
- FSUKHMUZGXDFSO-VPDMFFKESA-O C[C@](CC1)([C@@H](CC2)[C@H]([C@H](CCCCOCCOCCOCCOCCOCc3ccccc3)C3)[C@H]1c(cc1)c3cc1O)[C@H]2[OH2+] Chemical compound C[C@](CC1)([C@@H](CC2)[C@H]([C@H](CCCCOCCOCCOCCOCCOCc3ccccc3)C3)[C@H]1c(cc1)c3cc1O)[C@H]2[OH2+] FSUKHMUZGXDFSO-VPDMFFKESA-O 0.000 description 1
- HGBVWILTHJUMDU-FQEVSTJZSA-N Cc1c(-c2ccc(CNC([C@H](C=CC3)N3C(c3cccc(O)c3C)=O)=O)cc2)[s]cn1 Chemical compound Cc1c(-c2ccc(CNC([C@H](C=CC3)N3C(c3cccc(O)c3C)=O)=O)cc2)[s]cn1 HGBVWILTHJUMDU-FQEVSTJZSA-N 0.000 description 1
- JYVGFVLUGQKBPI-IBGZPJMESA-N Cc1c(-c2ccc(CNC([C@H](CCC3)N3c3nc(C#N)ccc3)=O)cc2)[s]cn1 Chemical compound Cc1c(-c2ccc(CNC([C@H](CCC3)N3c3nc(C#N)ccc3)=O)cc2)[s]cn1 JYVGFVLUGQKBPI-IBGZPJMESA-N 0.000 description 1
- WKFJBQPQAHKMDJ-SJORKVTESA-N Cc1c(-c2ccc(CNC([C@H](C[C@H](C3)O)N3C(c3ccc[s]3)=O)=O)cc2)[s]cn1 Chemical compound Cc1c(-c2ccc(CNC([C@H](C[C@H](C3)O)N3C(c3ccc[s]3)=O)=O)cc2)[s]cn1 WKFJBQPQAHKMDJ-SJORKVTESA-N 0.000 description 1
- BVUGHYGHSQJLRE-HNENSFHCSA-N Cc1ccc(CN/C(/O)=C(\CCC2)/N2C(c2cc(N)ccc2)=O)cc1 Chemical compound Cc1ccc(CN/C(/O)=C(\CCC2)/N2C(c2cc(N)ccc2)=O)cc1 BVUGHYGHSQJLRE-HNENSFHCSA-N 0.000 description 1
- KIFXSLYBMMOEJJ-OYKVQYDMSA-N Cc1ccc(CNC([C@H](CCC2)N2C(c(cc(cc2)N)c2Cl)=O)O)cc1 Chemical compound Cc1ccc(CNC([C@H](CCC2)N2C(c(cc(cc2)N)c2Cl)=O)O)cc1 KIFXSLYBMMOEJJ-OYKVQYDMSA-N 0.000 description 1
- RHXMDODFJMWJRJ-SFHVURJKSA-N Cc1ccc(CNC([C@H](CCC2)N2C(c2cc(N)ccc2)=O)=O)cc1 Chemical compound Cc1ccc(CNC([C@H](CCC2)N2C(c2cc(N)ccc2)=O)=O)cc1 RHXMDODFJMWJRJ-SFHVURJKSA-N 0.000 description 1
- QFIBOEWTAOFNIN-KRWDZBQOSA-N Cc1ccc(CNC([C@H](CCC2)N2C(c2ccnc(OC)c2)=O)=O)cc1 Chemical compound Cc1ccc(CNC([C@H](CCC2)N2C(c2ccnc(OC)c2)=O)=O)cc1 QFIBOEWTAOFNIN-KRWDZBQOSA-N 0.000 description 1
- IDQKZUGRIRWPQN-YLRAASNFSA-N Cc1n[o]c(C/C=C(\CCC2)/[C@@H]2C(NCc(cc2)ccc2Cl)=O)c1 Chemical compound Cc1n[o]c(C/C=C(\CCC2)/[C@@H]2C(NCc(cc2)ccc2Cl)=O)c1 IDQKZUGRIRWPQN-YLRAASNFSA-N 0.000 description 1
- QTMWSPIZWFMDBO-PXXRIBEUSA-N Cc1n[o]c(C/C=C(\C[C@@H](C2)O)/[C@@H]2C(NCc(cc2)ccc2Cl)=O)c1 Chemical compound Cc1n[o]c(C/C=C(\C[C@@H](C2)O)/[C@@H]2C(NCc(cc2)ccc2Cl)=O)c1 QTMWSPIZWFMDBO-PXXRIBEUSA-N 0.000 description 1
- YMZYDVLEMAVEIX-HNNXBMFYSA-N Cc1n[o]c(CC(N(CCC2)[C@@H]2C(NS(c(cc2)ccc2Cl)(=O)=O)=O)=O)c1 Chemical compound Cc1n[o]c(CC(N(CCC2)[C@@H]2C(NS(c(cc2)ccc2Cl)(=O)=O)=O)=O)c1 YMZYDVLEMAVEIX-HNNXBMFYSA-N 0.000 description 1
- ADIMRKFZWMXULO-NQIIRXRSSA-N Cc1n[o]c(CC(N(C[C@@H](C2)O)[C@@H]2C(NCc2cccc(-c3ccc[n]3C)c2)=O)=O)c1 Chemical compound Cc1n[o]c(CC(N(C[C@@H](C2)O)[C@@H]2C(NCc2cccc(-c3ccc[n]3C)c2)=O)=O)c1 ADIMRKFZWMXULO-NQIIRXRSSA-N 0.000 description 1
- PFKRWDRIUMNXQV-IRLDBZIGSA-N Cc1n[o]c(CC(N(C[C@@H](C2)O)[C@@H]2C(NCc2cccc(-c3ccccc3)c2)=O)=O)c1 Chemical compound Cc1n[o]c(CC(N(C[C@@H](C2)O)[C@@H]2C(NCc2cccc(-c3ccccc3)c2)=O)=O)c1 PFKRWDRIUMNXQV-IRLDBZIGSA-N 0.000 description 1
- NEMUHZSDDDMJDS-OCCSQVGLSA-N Cc1n[o]c(CC(N(C[C@@H](C2)O)[C@@H]2C(NCc2ccnc(Br)c2)=O)=O)c1 Chemical compound Cc1n[o]c(CC(N(C[C@@H](C2)O)[C@@H]2C(NCc2ccnc(Br)c2)=O)=O)c1 NEMUHZSDDDMJDS-OCCSQVGLSA-N 0.000 description 1
- ORXWQCANGZGPRO-DOMZBBRYSA-N Cc1n[o]c(CC(N(C[C@@H](C2)O)[C@@H]2C(NS(c(cc2)ccc2Cl)(=O)=O)=O)=O)c1 Chemical compound Cc1n[o]c(CC(N(C[C@@H](C2)O)[C@@H]2C(NS(c(cc2)ccc2Cl)(=O)=O)=O)=O)c1 ORXWQCANGZGPRO-DOMZBBRYSA-N 0.000 description 1
- QKKFLLUZXDZJSP-UXHICEINSA-N N#Cc1cc(C(N(C[C@@H](C2)O)[C@@H]2C(NCc(cc2)ccc2-c2cnc[o]2)=O)=O)ccc1 Chemical compound N#Cc1cc(C(N(C[C@@H](C2)O)[C@@H]2C(NCc(cc2)ccc2-c2cnc[o]2)=O)=O)ccc1 QKKFLLUZXDZJSP-UXHICEINSA-N 0.000 description 1
- KDGPGIRXRWYDQE-UHFFFAOYSA-N NCc(cc1)ccc1-c1cnc[o]1 Chemical compound NCc(cc1)ccc1-c1cnc[o]1 KDGPGIRXRWYDQE-UHFFFAOYSA-N 0.000 description 1
- SOPVQOQTQLADFQ-KRWDZBQOSA-N O=C([C@H](CCC1)N1C(c(cc1)ccc1Cl)=O)NCc(cc1)ccc1Cl Chemical compound O=C([C@H](CCC1)N1C(c(cc1)ccc1Cl)=O)NCc(cc1)ccc1Cl SOPVQOQTQLADFQ-KRWDZBQOSA-N 0.000 description 1
- PSGNTBOXLBGWJW-PKTZIBPZSA-N O[C@H](C[C@H]1C(NCc(cc2)ccc2Cl)=O)CN1C(c1cc(-c2cc(O)ccc2)ccc1)=O Chemical compound O[C@H](C[C@H]1C(NCc(cc2)ccc2Cl)=O)CN1C(c1cc(-c2cc(O)ccc2)ccc1)=O PSGNTBOXLBGWJW-PKTZIBPZSA-N 0.000 description 1
- ZUSLIQAIBLSPEZ-WUJWULDRSA-N [O-]C(C[C@H]1C(NCc(cc2)ccc2Cl)=O)CN1C(Cc1cnc[SH-]1)=O Chemical compound [O-]C(C[C@H]1C(NCc(cc2)ccc2Cl)=O)CN1C(Cc1cnc[SH-]1)=O ZUSLIQAIBLSPEZ-WUJWULDRSA-N 0.000 description 1
- UHOVQNZJYSORNB-UHFFFAOYSA-N c1ccccc1 Chemical compound c1ccccc1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- a pharmaceutically acceptable salt, enantiomer, diastereomer, solvate or polymorph thereof is a pharmaceutically acceptable salt, enantiomer, diastereomer, solvate or polymorph thereof.
- Preferred heteroaryl groups for R 2 include an optionally substituted quinoline (which may be attached to the pharmacophore or substituted on any carbon atom within the quinoline ring), an optionally substituted indole, an optionally substituted indolizine, an optionally substituted azaindolizine, an optionally substituted benzofuran, including an optionally substituted benzofuran, an optionally substituted isoxazole, an optionally substituted thiazole, an optionally substituted isothiazole, an optionally substituted thiophene, an optionally substituted pyridine (2-, 3, or 4-pyridine), an optionally substituted imidazole, an optionally substituted pyrrole, an optionally substituted diazole, an optionally substituted triazole, a tetrazole, an optionally substituted oximidazole, or a group according to the chemical structure:
- R 1 is a Cj-C 3 alkyl group, preferably H or CH 3 , more preferably H
- T is an optionally substituted -(CH 2 ) n - group, wherein each one of the methylene groups may be optionally substituted with one or two substituents, preferably selected from halogen, a C1-C3 alkyl group or the sidechain of an amino acid as otherwise described herein, preferably methyl, which may be optionally substituted; and n is 0 to 6, often 0, 1, 2, or 3, preferably 0 or 1.
- R HET is H, CN, N0 2 , halo (preferably CI or F), optionally substituted d-C 6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF 3 ), optionally substituted 0(Ci-C 6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C ⁇ C-R a where R a is H or a CrC6 alkyl group (preferably C]-C 3 alkyl);
- R 2 is an optionally substituted -NRj- an optionally substituted -NRi-X ⁇ ' -Aryl-HET or an optionally substituted -NR X ⁇ ' -HET- Aryl ,
- Aryl is phenyl which is optionally substituted with one or two substitutents, wherein said substituent(s) is preferably selected from -(CH 2 ) n OH, Ci-C 6 alkyl which itself is further optionally substituted with CN, halo (up to three halo groups), OH, -(CH 2 ) n O(C 1 -C6)alkyl, amine, mono- or di-(Ci-C 6 alkyl) amine wherein the alkyl group on the amine is optionally substituted with 1 or 2 hydroxyl groups or up to three halo (preferably F, CI) groups, or said Aryl group is substituted with -(CH 2 ) n OH, -(CH 2 ) n -0-(C 1 -C 6 )alkyl, -(CH 2 ) n -0-(CH 2 ) n - (d-C 6 )alkyl, -(CH 2 ) n -C(0)(
- R PRO is H, optionally substituted Ci-C 6 alkyl or an optionally substituted aryl (phenyl or napthyl), heteroaryl or heterocyclic group selected from the group consisting of oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, quinoline, (each preferably substituted with a Ci-C 3 alkyl group, preferably methyl or a halo group, preferably F or CI), benzofuran, indole, indolizine, azaindolizine;
- Y c is N or C-R YC , where R YC is H, OH, CN, N0 2 , halo (preferably CI or F), optionally substituted Ci-Ce alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF 3 ), optionally substituted 0(Ci-C 6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C ⁇ C-R a where R a is H or a Ci-C 6 alkyl group (preferably Ci-C 3 alkyl);
- Methods of stimulating erythropoiesis in a subject or patient including increasing the number of red blood cells and/or hematocrit of the patient, treating anemia, including chronic anemia and anemia associated with chronic kidney disease, dialysis, and cancer chemotherapy, ischemia, stroke and damage to cardiovascular tissue during cardiovascular ischemia as well as enhancing wound healing processes and preventing/reducing scarring associated with or secondary to the healing process represent additional aspects of the present invention.
- compositions of the present invention may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vaginally or via an implanted reservoir.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
- the compositions are administered orally, intraperitoneally or intravenously.
- Fmoc-Hyp(OtBu)OH (24.9 g, 60.8 mmol, 1 eq) was dissolved in DMF (300 mL) at room temperature.
- Sodium bicarbonate (12.8 g, 152 mmol, 2.5 eq) was added, followed by allyl bromide (25.3 mL, 300 mmol, 4.9 eq).
- the solution was fitted with an air condenser and heated to 50 °C for 20 hours. It was then cooled to room temperature, diluted with EtOAc, washed with aqueous 1 M HCl, saturated sodium bicarbonate, water and brine. The organic layer was dried with sodium sulfate, filtered and condensed.
- HPLC analysis was conducted on an XBridge CI 8 column (150mm x 30mm internal diameter, 5 ⁇ packing diameter) at ambient temperature.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Description
Compounds and Methods for the Inhibition of VCB E3 Ubiquitin Ligase
Related Applications and Grant Support
This application claims the benefit of priority of provisional application
US61/585,736, of identical title, filed January 12, 2011, the entire contents of which are incorporated by reference herein.
This invention was made with government support under grant nos AI084140, GM032136, BBSRC BB/G023123/1 and EC PIEF-GA-2010-275683 of the National Institutes of Health. The government has certain rights in the invention.
Field of the Invention
The present invention relates to novel compounds which find utility as modulators, especially inhibitors of the VCB E3 Ubiquitin Ligase and as bioactive agents for use as therapeutics for the stimulation of erythropoiesis in a patient or subject including inducement of EPO production in the patient or subject, for the treatment of chronic anemia and ischemia (limits brain injury during episodes of localized anemia, ischemia and/or stroke and damage to cardiovascular tissue during cardiovascular ischemia), as well as enhancing wound healing processes. Compounds according to the present invention also find use as standards and controls in bioassays, as intermediates in chemical synthesis and related applications, among others. Pharmaceutical compositions comprising effective amounts of compounds according to the present invention alone or in combination with an additional erythropoiesis stimulating agent such as EPO under the tradename procrit or epogen, or darbapoietin alfa under the tradename aranesp. Methods of stimulating erythropoiesis in a subject or patient, including increasing the number of red blood cells and/or hematocrit of the patient, treating anemia, including chronic anemia and anemia associated with chronic kidney disease, dialysis, and cancer chemotherapy, ischemia, stroke and damage to cardiovascular tissue during cardiovascular ischemia as well as enhancing wound healing processes and
preventing/reducing scarring secondary to healing represent additional aspects of the present invention. Local enhancement of angiogenesis and arteriogenesis through induction of VEGF including wound healing and reduction of stent occlusion remain additional aspects of the present invention.
Background of the Invention
E3 ubiquitin ligases (of which over 600 are known in humans)1 confer substrate specificity for ubiquitination and are more attractive therapeutic targets than general proteasome inhibitors3,4 due to their specificity for a small number of protein substrates. Unfortunately, the development of E3 ligase inhibitors has proven challenging, in part due to the fact that they must disrupt protein-protein interactions.5 These interactions are notoriously difficult to target using small molecules due to their large contact surfaces and the shallow grooves or flat interfaces involved. Conversely, most small molecule drugs bind enzymes or receptors in tight and well-defined pockets.6 Since the discovery of nutlins, the first small molecule E3 ligase inhibitors,7 additional compounds have been reported that target
Inhibitors of Apoptosis Proteins (IAPs),8'9 sCFMet30,10 and SCFcdc4;u however, the field remains underdeveloped.
One E3 ligase with exciting therapeutic potential is the von Hippel-Lindau (VHL) tumor suppressor, the substrate recognition subunit of the E3 ligase complex VCB, which also consists of elongins B and C, Cul2 and Rbxl.12 The primary substrate of VHL is
Hypoxia Inducible Factor l (HIF-la), a transcription factor that upregulates genes such as the pro-angiogenic growth factor VEGF and the red blood cell inducing cytokine
erythropoietin in response to low oxygen levels. While HIF-la is constitutively expressed, its intracellular levels are kept very low under normoxic conditions via its hydroxylation by prolyl hydroxylase domain (PHD) proteins and subsequent VHL-mediated ubiquitination (Figure 1). Small molecule inhibition of this pathway therefore would lead to increased endogenous erythropoietin production and could supplant the current use of recombinant erythropoietin to treat chronic anemia associated with chronic kidney disease and cancer chemotherapy.13 To this end, PHD inhibitors are under examination in clinical trials; however, a possible alternative would be the development of an inhibitor of the VHL/HIF-la interaction. Such an inhibitor may avoid the HIF-independent off target effects observed with PHD inhibitors.2
E3 ubiquitin ligases, which bind protein targets, leading to their ubiquitination and subsequent degradation, are highly desirable drug targets due to their exquisite substrate specificity. However, the development of small molecule inhibitors has proven
extraordinarily challenging as modulation of E3 ligase activities requires the targeting of
protein-protein interactions. Using rational design, we have generated the first small molecule inhibitors of Von Hippel Lindau (VHL), the substrate recognition subunit of the E3 ligase VCB, an important target in cancer, chronic anemia and ischemia.2 We have also obtained crystal structures of VHL with our most potent inhibitor, 15, confirming that the compound mimics the binding mode of the transcription factor HIF- la, a substrate of VHL. These results have the potential to guide future development of improved lead compounds as therapeutics for the treatment of chronic anemia and ischemia.
Anemia associated with chronic kidney disease, dialysis, and cancer chemotherapy is currently treated via injection of recombinant erythropoietin (Epogen). Given the need to inject Epogen and its high cost of manufacture, the present invention is directed to the development of a small molecule therapeutic that could induce endogenous EPO production.
Epogen sales for 2010 were $3.3 billion, and there is no small molecule competitor for that market. However, Fibrogen has a proline hydroxylase domain (PHD) protein inhibitor in Phil clinical trials that shows some promise in raising endogenous EPO levels in animal models of anemia. The VHL ligand of the present invention works via a different mechanism than Fibrogen' s PHD inhibitor by acting downstream of HIF hydroxylation. Moreover, since other proteins serve as PHD substrates (and there are three PHD family members), it is not clear what potential negative side effects may be associated with PHD inhibitors. To the best knowledge of the inventors, no attempt has been made to target the HIF:VHL interaction before. Our current best in class compound has a comparable affinity (1 μΜ) as the HIF peptide used in the FP competition assay.
From earlier biochemical and structural studies of a hydroxylated HIF peptide bound to VHL, it became clear that hydroxyproline played an important role in mediating this proteimprotein interaction. As a consequence of that work, the present inventors developed a hydroxylated HIF peptide:VHL fluorescence polarization (FP) binding assay with which they assayed >120 compounds possessing the central hydroxyproline residue flanked by non- peptidic moieties. Further to that research, the inventors now have co-crystal structures of VHL complexed with seven of the top compounds. Analysis of these ligand bound structures is driving the design/synthesis of the next generation of VHL ligands.
A second rationale for the present invention is the need for a small molecule E3 ligase ligand for the development of our Proteolysis Targeting Chimeric molecules (PROTACs). Our PROTAC technology brings targeted proteins to E3 ligases for ubiquitination and subsequent proteasomal degradation. In several proof-of-concept experiments, the present inventors demonstrated the utility of this approach using the short peptide sequence from HIF that binds VHL. In order to make a more 'drug-like' PROTAC, the inventors have replaced the HIF peptide with a 'small molecule' VHL ligand.
Objects of the Invention
It is an object of the invention to provide compounds which function as modulators, preferably inhibitors of VCB E3 Ubiquitin Ligase.
It is another object of the invention to provide pharmaceutical compositions based upon the above-described modulators, especially including inhibitors for therapeutic treatment of a patient or subject, preferably including a human patient or subject.
It is still another object of the invention to provide methods for stimulating erythropoiesis in a patient, including increasing EPO production, red blood cell formation and/or blood hematocrit of a patient or subject.
It is yet another aspect of the invention to provide methods for treating anemia, including chronic anemia, anemia associated with chronic kidney disease, dialysis, and cancer chemotherapy and ischemia in a patient or subject to limit neuronal (brain) injury and/or cardiovascular injury which may occur as a consequence of ischemia.
It is still another aspect of the invention to provide methods for enhancing wound healing in a patient or subject.
It is yet another object to provide methods for enhancing localized angiogenesis and arteriogenesis for reducing vascular and stent occlusions.
It is an additional object of the invention to provide compounds and compositions for use in a first medical application.
It is yet another aspect of the invention to provide compounds and pharmaceutical compositions for use in treating anemia, including chronic anemia, anemia associated with chronic kidney disease, dialysis, and cancer chemotherapy and ischemia in a patient or subject to limit neuronal (brain) injury and/or cardiovascular injury which may occur as a consequence of ischemia.
It is still another aspect of the invention to provide compounds and pharmaceutical compositions for use in enhancing wound healing in a patient or subject.
It is yet another object to provide compounds and pharmaceutical compositions for use in enhancing localized angiogenesis and arteriogenesis for reducing vascular and stent occlusions.
Any one or more of these and/or other objects of the invention may be readily gleaned from a routine scrutiny of the description of the invention which follows.
Brief Description of the Figures
Figure 1 shows (A) HIF- la accumulation leads to the transcriptional upregulation of genes involved in the hypoxic response, such as erythropoietin and VEGF. (B) Under normoxic conditions, HIF- la is hydroxylated, recognized by VHL, ubiquitinated and degraded by the proteasome, preventing transcriptional upregulation.
Figure 2. WaterLOGSY NMR spectroscopy shows binding of 3, but not L-Hyp or NAc-Hyp- Me to VHL.
Figure 3 shows a pictorial representation shows the key interactions between 15 and
VHL.
Figure 4 shows the 2.9 A co-crystal structure of 15 (lightest gray carbons) bound to VHL indicates that its binding mimics that of the HIF- la peptide (light gray carbons, pdb 1LM817)
Figure 5 shows the crystal structures of V54BC apo (A) and in complex with 15 (B). Electron density (2F0-FC) superimposed around Hyp binding site residues (sticks, yellow carbons) and conserved water molecules (red dots), and 15 (sticks, cyan carbons) are shown in blue and are contoured at 1.2σ. The protein surface is shown in green at 50% transparency.
Figures 6-12A and B show the activity of individual compounds according to the present invention in the described VHL polarization/displacement assay. Compounds according to the present invention are indicated with numerals at the top of each graph.
Control compound is presented in figure 15 B and served as minimum polarization
(maximum displacement) for comparison purposes. The percent inhibition as presented was determined by normalizing to maximum and minimum polarization, and graphed against the log [VL]. IC50 values were determined using Prism 5 for each replicate (n=9), where were then averaged to determine the average IC50 and the standard of error of the mean (SEM).
Figure 13 (along with Table 2- Affinity Table) shows numerous exemplary compounds according to the present invention.
Figure 14 shows numerous preferred compounds from Table 2 according to the present invention.
Figure 15 shows a further number of compounds according to the present invention and their activity. Most compounds are active below concentrations of 100 μΜ.
Figure 16 shows numerous preferred compounds from Figure 15 according to the present invention.
Figure 17 shows eight particularly preferred compounds from Figure 15 according to the present invention.
Figure 18 shows a representative bifunctional compound (protac) according to the present invention.
Brief Description of the Invention
In one aspect, the present invention relates to compounds according to the chemical structure:

Where R1 is an optionally substituted Ci-C6 alkyl group, an optionally substituted
-(CH2)nOH, an optionally substituted -(CH2)nSH, an optionally substituted (ΟΗ2)η-0-(^- C6)alkyl group, an optionally substituted (CH2)n-WCOCW-(Co-C6)alkyl group containing an epoxide moiety WCOCW where each W is independently H or a C1-C3 alkyl group, an optionally substituted -(CH2)nCOOH, an optionally substituted -(CH2)nC(0)-(C!-C6 alkyl), an optionally substituted -(CH2)nNHC(0)-Ri, an optionally substituted -(CH2)nC(0)-NR!R2, an optionally substituted -(CH2)nOC(0)-NRiR2, -(CH20)nH, an optionally substituted - (CH2)nOC(0)-(Ci-C6 alkyl), an optionally substituted -(CH2)nC(0)-0-(C1-C6 alkyl), an optionally substituted
-(CH20)nCOOH, an optionally substituted -(OCH2)nO-(C1-C6 alkyl), an optionally substituted -(CH20)nC(0)-(C]-C6 alkyl), an optionally substituted -(OCH2)nNHC(0)-Ri, an optionally substituted -(CH20)nC(0)-NRiR2, -(CH2CH20)„H, an optionally substituted
-(CH2CH20)„COOH, an optionally substituted -(OCH2CH2)nO-(Ci-C6 alkyl), an optionally substituted -(CH2CH20)nC(0)-(C1-C6 alkyl), an optionally substituted
-(OCH2CH2)nNHC(0)-R1, an optionally substituted -(CH2CH20)nC(0)-NRiR2,an optionally substituted -S02Rs, an optionally substituted S(0)Rs, N02, CN or halogen (F, CI, Br, I, preferably F or CI);
Ri and R2 are each independently H or a C\-Ce alkyl group which may be optionally substituted with one or two hydroxyl groups or up to three halogen groups (preferably fluorine);
Rs is a C C6 alkyl group, an optionally substituted aryl, heteroaryl or heterocycle group or a
 group,
X and X' are each independently C=0, 0=S, -S(0), S(0)2 , (preferably X and X' are both 0=0);
group,
X and X' are each independently C=0, 0=S, -S(0), S(0)2 , (preferably X and X' are both 0=0);
R2 is an optionally substituted -(CH2)n-(C=0)u(NR1)v(S02)walkyl group, an optionally substituted -(CH2)n-(C=0)u(NRi)v(SO2)wNRiNR2N group, an optionally substituted -(CH2)„-(C=0)u(NR1)v(S02)w-Aryl, an optionally substituted
-(CH2)n-(C=0)u(NR1)v(S02)w-Heteroaryl, an optionally substituted
-(CH2)n-(C=0)vNRi(S02)w-Heterocycle, an optionally substituted
-NR1-(CH2)n-C(0)u(NR1)v(S02)w-alkyl, an optionally substituted
-NR1-(CH2)n-C(0)u(NRI)v(S02)w- NR1NR2N, an optionally substituted
-NR1-(CH2)n-C(0)u(NR1)v(S02)w-NRiC(0)R1N, an optionally substituted
-NR1-(CH2)n-(C=0)u(NR1)v(S02)w-Aryl, an optionally substituted
-NR1-(CH2)n-(C=0)u(NRi)v(S02)w-Heteroaryl or an optionally substituted
-NR1-(CH2)n-(C=0)vNR](S02)w-Heterocycle, an optionally substituted
-Xs2 -alkyl group; an optionally substituted
-X1 2 - Aryl group; an optionally substituted
-X1*2 - Heteroaryl group; an optionally substituted
-X - Heterocycle group; an optionally substituted;
R3 is an optionally substituted alkyl, an optionally substituted
-(CH2)„-C(0)u(NR1)v(S02)w-alkyl, an optionally substituted
-(CH2)n-C(0)u(NR1)v(S02)w-NRiNR2N, an optionally substituted
-(CH2)n-C(0)u(NR1)v(S02)w-NRiC(0)R1N, an optionally substituted
-(CH2)n-C(0)u(NRi)v(S02)w-C(0)NRiR2, an optionally substituted
-(CH2)n-C(0)u(NRi)v(S02)w-Aryl, an optionally substituted
-(CH2)n-C(O)u(NR v(SO2)w-Heteroaryl, an optionally substituted
-(CH2)n-C(0)u(NRi)v(S02)w-Heterocycle, an optionally substituted
-NR1-(CH2)„-C(0)u(NR1)v(S02)w-alkyl, an optionally substituted
-NR1-(CH2)n-C(0)u(NR1)v(S02)w- NRiNR2N, an optionally substituted
-NR1-(CH2)n-C(0)u(NR1)v(S02)w- NR]C(0)R1N, an optionally substituted
-NRI-(CH2)n-C(0)u(NR1)v(S02)w-Aryl, an optionally substituted
-NR1-(CH2)n-C(0)u(NR1)v(S02)w-Heteroaryl, an optionally substituted
-NR1-(CH2)n-C(0)u(NR1)v(S02)w-Heterocycle, an optionally substituted
-O-(CH2)n-(C=0)u(NR1)v(SO2)w-alkyl, an optionally substituted
-0-(CH2)n-(C=0)u(NR1)v(SO2)w-NR1NR2N, an optionally substituted
-0-(CH2)n-(C=0)u(NRi)v(S02)w-NRiC(0)RiN, an optionally substituted
-0-(CH2)n-(C=0)u(NRi)v(S02)w-Aryl, an optionally substituted
-0-(CH2)n-(C=0)u(NRi)v(S02)w-Heteroaryl or an optionally substituted
-0-(CH2)n- (C=0)u(NRi)v(S02)w-Heterocycle;
-(CH2)n-(V)n'-(CH2)n-(V)n-alkyl group, an optionally substituted
-(CH2)n-(V)n'-(CH2)n-(V)n'-Aryl group, an optionally substituted
-(CH2)n-(V)n'-(CH2)n-(V)n-Heteroaryl group, an optionally substituted
-(CH2)n-(V)n'-(CH2)n-(V)n'-Heterocycle group, an optionally substituted
-(CH2)n-N(Ri group, an optionally substituted
-(CH2)n-N(R[
 group, an optionally substituted
group, an optionally substituted
-(CH2)n-N(Ri ')(C=0)m-(V)n-Heteroaryl group, an optionally substituted
-(CH2)n-N(Ri
 group, an optionally substituted
group, an optionally substituted
-XR3 - alkyl group; an optionally substituted
-XR3 - Aryl group; an optionally substituted
-XR3 - Heteroaryl group; an optionally substituted
-XR3 - Heterocycle group; an optionally substituted;
Where R[N and R2N are each independently H, Ci-C6 alkyl which is optionally substituted with one or two hydroxyl groups and up to three halogen groups or an optionally substituted
-(CH2)n-Aryl, -(CH2)n-Heteroaryl or -(CH2)n-Heterocycle group;
R1 and R,• are each independently H or a Ci-C3 alkyl group;
Ri is the same as above;
R1 and Rr are each independently H or a C1-C3 alkyl group;
X^' and X*3' are each independently an optionally substituted -CH2)n-, -CH2)n-
CH(XV)=CH(XV)- (cis or trans), -CH2)n-CH≡CH- , -(CH2CH20)n- or a C3-C6 cycloalkyl group, where Xv is H, a halo or a C1-C3 alkyl group which is optionally substituted;
Each m is independently 0, 1, 2, 3, 4, 5, 6;
Each m' is independently 0 or 1 ;
Each n is independently 0, 1, 2, 3, 4, 5, 6;
Each n' is indepdently 0 or 1 ;
Each u is independently 0 or 1 ;
Each v is independently 0 or 1 ;
Each w is independently 0 or 1 , or
A pharmaceutically acceptable salt, enantiomer, diastereomer, solvate or polymorph thereof.
In alternative aspects, the present invention relates to compounds according to the chemical structure:
R1

Wherein each of R1', R2 and R3 are the same as above and X is C=0, C=S, -S(O) group or a S(0)2 group, more preferably a C=0 group, or
A pharmaceutically acceptable salt, enantiomer, diastereomer, solvate or polymorph thereof.
In still further preferred aspects of the invention, compounds according to the present invention

Where R1 , R2 and R3 are the same as presented above, or
A pharmaceutically acceptable enantiomer, diastereomer, solvate or polymorph thereof.
In further preferred aspects of the invention, R1 is preferably a hydroxyl group or a group which may be metabolized to a hydroxyl or carboxylic group, preferably a hydroxyl group, such that the compound represents a prodrug form of an active compound. Exemplary preferred R1 groups include, for example, -(CH2)nOH, (CH2)n-0-(C1-C6)alkyl group, -
(CH2)nCOOH, -(CH20)nH, an optionally substituted -(CH2)nC(O)(C0-C6)alkyl, an optionally substituted -(CH2)nOC(0)-(CrC6)alkyl, or an optionally substituted -(CH2)nC(0)-0-(d- C6)alkyl, wherein n is 0 or 1. Most often, R1 is hydroxyl.
X and X', where present, are preferably a C=0, C=S, -S(O) group or a S(0)2 group, more preferably a C=0 group.
R 2' is preferably an optionally substituted -NR 1 -T-Aryl, an optionally substituted -NR^T-Heteroaryl group or an optionally substituted -NR'-T-Heterocycle, where R1 is a d- C3 alkyl group, preferably H or CH3, more preferably H and T is an optionally substituted -(CH2)n- group, wherein each one of the methylene groups within the alkylene chain may be optionally substituted with one or two substituents, preferably selected from halogen, a d-C3 alkyl group or a side chain of an amino acid as otherwise described herein, preferably one or two methyl groups, which may be optionally substituted; and n is 0 to 6, often 0, 1, 2 or 3, preferably 0 or 1. Alternatively, T may also be a -(CH20)n- group, a -(OCH2)n- group, a - (CH2CH20)n- group, a
-(OCH2CH2)n- group, all of which groups are optionally substituted.
Preferred Aryl groups for R2 include optionally substituted phenyl or naphthyl groups, preferably phenyl groups, wherein the phenyl group is optionally substituted with a halogen (preferably F or CI), an amine, monoalkyl- or dialkyl amine (preferably, dimethylamine), F, CI, OH, SH, COOH, Ci-C6 alkyl, preferably CH3, CF3, OMe, OCF3, N02, or CN group (each of which may be substituted in ortho-, meta- and/or para- positions of the phenyl ring, preferably para-), an optionally substituted phenyl group (the phenyl group itself is preferably substituted with at least one of F, CI, OH, SH, COOH, CH3, CF3, OMe, OCF3, N02, or CN group, which may be substituted in ortho-, meta- and/or para- positions of the phenyl ring, preferably para-), a naphthyl group, which may be optionally substituted, an optionally substituted heteroaryl, preferably an optionally substituted isoxazole including a
methylsubstituted isoxazole, an optionally substituted oxazole including a methylsubstituted oxazole, an optionally substituted thiazole including a methyl substituted thiazole, an optionally substituted isothiazole including a methyl substituted isothiazole, an optionally substituted pyrrole including a methylsubstituted pyrrole, an optionally substituted imidazole including a methylimidazole, an optionally substituted benzimidazole or
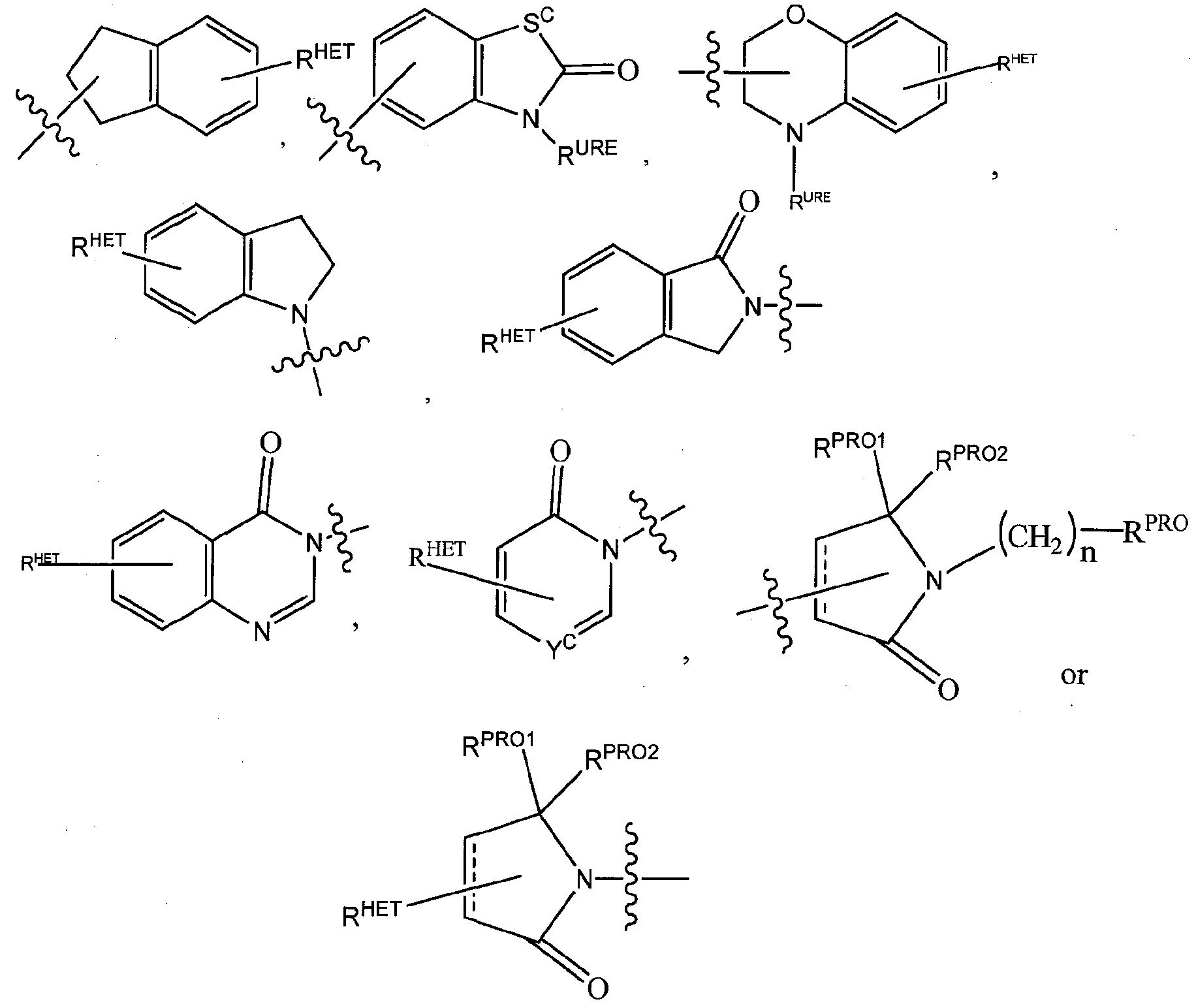
methoxybenzylimidazole, an optionally substituted oximidazole or methyloximidazole, an optionally substituted diazole group, including a methyldiazole group, an optionally substituted triazole group, including a methylsubstituted triazole group, an optionally substituted pyridine group, including a halo- (preferably, F) or methylsubstitutedpyridine group or an oxapyridine group (where the pyridine group is linked to the phenyl group by an oxygen), an optionally substituted furan, an optionally substituted benzofuran, an optionally substituted dihydrobenzofuran, an optionally substituted indole, indolizine or azaindolizine (2, 3, or 4-azaindolizine), an optionally substituted quinoline, an optionally substituted group according to the chemical structure:

Where Sc is CHR , NRURE, or O;
RHET is H, CN, N02, halo (preferably CI or F), optionally substituted Ci-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(^-ΰ6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where R¾ is H or a Ci-C6 alkyl group (preferably C1 -C3 alkyl);
Rss is H, CN, N02, halo (preferably F or CI), optionally substituted Ci-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups), optionally
substituted 0-(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to
three halo groups) or an optionally substituted -C(0)(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups);
RURE is H, a C]-C6 alkyl (preferably H or d-C3 alkyl) or a -C(0)(CrC6 alkyl) each of which groups is optionally substituted with one or two hydroxyl groups or up to three halogen, preferably fluorine groups, or an optionally substituted phenyl group, an optionally substituted heteroaryl, or an optionally substituted heterocycle, preferably for example piperidine, morpholine, pyrrolidine, tetrahydrofuran, among others);
RPR0 is H, optionally substituted C]-C6 alkyl or an optionally substituted aryl (phenyl or napthyl), heteroaryl or heterocyclic group selected from the group consisting of oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, quinoline, (each preferably substituted with a Ci-C3 alkyl group, preferably methyl or a halo group, preferably F or CI), benzofuran, indole, indolizine, azaindolizine;
RPR0I and RPR02 are each independently H, an optionally subsituted Ci-C3 alkyl group or together form a keto group; and
each n is independently 0, 1, 2, 3, 4, 5, or 6 (preferably 0 or 1), or
an optionally substituted heterocycle, preferably tetrahydrofuran, tetrahydrothiene, piperidine, piperazine or morpholine (each of which groups when substituted, are preferably substituted with a methyl or halo (F, Br, CI).
In certain preferred aspects,
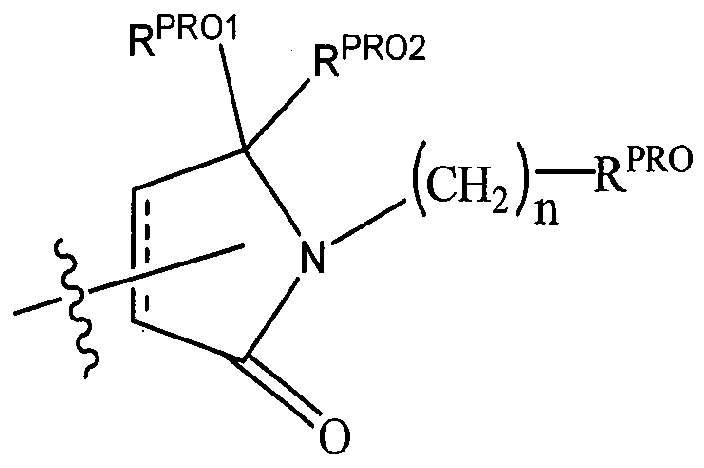

Preferred heteroaryl groups for R2 include an optionally substituted quinoline (which may be attached to the pharmacophore or substituted on any carbon atom within the quinoline ring), an optionally substituted indole, an optionally substituted indolizine, an optionally substituted azaindolizine, an optionally substituted benzofuran, including an optionally substituted benzofuran, an optionally substituted isoxazole, an optionally substituted thiazole, an optionally substituted isothiazole, an optionally substituted thiophene, an optionally substituted pyridine (2-, 3, or 4-pyridine), an optionally substituted imidazole, an optionally substituted pyrrole, an optionally substituted diazole, an optionally substituted triazole, a tetrazole, an optionally substituted oximidazole, or a group according to the chemical structure:

Where Sc is CHRSS, NRURE, or 0;
RHET is H, CN, N02, halo (preferably CI or F), optionally substituted d-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Ci-C6 alkyl group (preferably C1-C3 alkyl);
RSi> is H, CN, N02, halo (preferably F or CI), optionally substituted d-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups), optionally substituted 0-(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted -C(0)(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups);
RURE is H, a Ci-C6 alkyl (preferably H or CrC3 alkyl) or a -C(0)(C1-C6 alkyl), each of which groups is optionally substituted with one or two hydroxyl groups or up to three halogen, preferably fluorine groups, or an optionally substituted heterocycle, for example piperidine, morpholine, pyrrolidine, tetrahydrofuran, tetrahydrothiophene, piperidine, piperazine, each of which is optionally substituted, and
Yc is N or C-RYC, where RYC is H, OH, CN, N02, halo (preferably CI or F), optionally substituted Ci-C6 alkyl (preferably substituted with one or tvvo hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(C C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a C\-Ce alkyl group (preferably C1-C3 alkyl).
Preferred heterocycle groups for R2 include tetrahydroquinoline, piperidine, piperazine, pyrrollidine, morpholine, tetrahydrofuran, tetrahydrothiophene, oxane, thiane, each of which groups may be optionally substituted, or a group according to the chemical structure:

Where R is H, optionally substituted Ci-C^ alkyl or an optionally substituted aryl, heteroaryl or heterocyclic group;
R and R are each independently H, an optionally subsituted Q-C3 alkyl group or together form a keto group and
Each n is independently 0, 1 , 2, 3, 4, 5, or 6 (preferably 0 or 1).
Preferred R2 substituents for use in the present invention also include specifically (and without limitation to the specific compound disclosed) the R2 substituents which are found in the identified compounds disclosed herein (which includes the specific compounds which are disclosed in the present specification, and the figures which are attached hereto). Each of these R2 substituents may be used in conjunction with any number of R3 substituents which are also disclosed herein.
R3 is preferably an optionally substituted -T-Aryl, an optionally substituted
-T-Heteroaryl, an optionally substituted -T-Heterocycle, an optionally substituted
-NR'-T-Aryl, an optionally substituted -NR1 -T-Heteroaryl or an optionally substituted -NR^T-Heterocycle, where where R1 is a Cj-C3 alkyl group, preferably H or CH3, more preferably H, T is an optionally substituted -(CH2)n- group, wherein each one of the methylene groups may be optionally substituted with one or two substituents, preferably selected from halogen, a C1-C3 alkyl group or the sidechain of an amino acid as otherwise described herein, preferably methyl, which may be optionally substituted; and n is 0 to 6, often 0, 1, 2, or 3, preferably 0 or 1. Alternatively, T may also be a -(CH20)n- group, a -(OCH2)n- group, a -(CH2CH20)n- group, a -(OCH2CH2)„- group, each of which groups is optionally substituted.
Preferred aryl groups for R3 include optionally substituted phenyl or naphthyl groups (including tetrahydronaphthyl), preferably phenyl groups, wherein the phenyl or naphthyl group is optionally substituted with a halogen (preferably F or CI), an amine, monoalkyl- or dialkyl amine (preferably, dimethylamine), an amido group (preferably a -(CH2)m- NR!C(0)R2 group, where m, Ri and R2 are the same as above), a halo (often F, CI), OH, SH, CH3, CF3, OMe, OCF3, N02, CN or a S(0)2Rs group (Rs is a a Ci-C6 alkyl group, an optionally substituted aryl, heteroaryl or heterocycle group or a -(CH2)mNR1R2 group), each of which may be substituted in ortho-, meta- and/or para- positions of the phenyl ring, preferably para-), or an Aryl (preferably phenyl), Heteroaryl or Heterocycle. Preferably said substituent phenyl group is an optionally substituted phenyl group (i.e., the substituent phenyl
group itself is preferably substituted with at least one of F, CI, OH, SH, COOH, CH3, CF3, O e, OCF3, N02, or CN group, which may be substituted in ortho-, meta- and/or para- positions of the phenyl ring, preferably para-), a naphthyl group, which may be optionally substituted, an optionally substituted heteroaryl, including an optionally substituted isoxazole including a methylsubstituted isoxazole, an optionally substituted oxazole including a methylsubstituted oxazole, an optionally substituted thiazole including a methyl substituted thiazole, an optionally substituted pyrrole, including a methylsubstituted pyrrole, an optionally substituted imidazole including a methylimidazole, an optionally substituted benzylimidazole or methoxybenzylimidazole, an optionally substituted oximidazole or methyloximidazole, an optionally substituted diazole group, including a methyldiazole group, an optionally substituted triazole group, including a methylsubstituted triazole group, a tetrazole group, an optionally substituted pyridine group, including a halo- (preferably, F) or methylsubstitutedpyridine group or an optionally substituted oxapyridine group (where the pyridine group is linked to the phenyl group by an oxygen) or an optionally substituted heterocycle (tetrahydrofuran, terahydrothiophene, pyrrolidine, piperidine, morpholine, piperazine, oxane, thiane or tetrahydroquinoline).
Preferred Heteroaryl groups for R3 include an optionally substituted quinoline (which may be attached to the pharmacophore or substituted on any carbon atom within the quinoline ring), an optionally substituted indole (including dihydroindole), an optionally substituted indolizine, an optionally substituted azaindolizine (2, 3 or 4-azaindolizine) an optionally substituted benzimidazole, benzodiazole, benzoxofuran, an optionally substituted imidazole, an optionally substituted isoxazole, an optionally substituted oxazole (preferably methyl substituted), an optionally substituted diazole, an optionally substituted triazole, a tetrazole, an optionally substituted benzofuran, an optionally substituted thiophene, an optionally substituted thiazole (preferably methyl and/or thiol substituted), an optionally substituted isothiazole, an optionally substituted triazole (preferably a 1,2,3-triazole substituted with a methyl group, a triisopropylsilyl group, an optionally substituted -(CH2)m-0-Ci-C6 alkyl group or an optionally substituted -(CH2)m-C(0)-0-Ci-C6 alkyl group), an optionally substituted pyridine (2-, 3, or 4-pyridine) or a group according to the chemical structure:

Where Sc is CHRSS, NRURE, or O;
RHET is H, CN, N02, halo (preferably CI or F), optionally substituted d-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a CrC6 alkyl group (preferably C]-C3 alkyl);
Rss is H, CN, N02, halo (preferably F or CI), optionally substituted Ci-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups), optionally substituted 0-(C]-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted -C(0)(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups);
RURE is H, a Ci-Ce alkyl (preferably H or d-C3 alkyl) or a ~C(0)(C C6 alkyl), each of which groups is optionally substituted with one or two hydroxyl groups or up to three halogen, preferably fluorine groups, or an optionally substituted heterocycle, for example piperidine, morpholine, pyrrolidine, tetrahydrofurari, tetrahydrothiophene, piperidine, piperazine, each of which is optionally substituted, and
Yc is N or C-RYC, where RYC is H, OH, CN, N02, halo (preferably CI or F), optionally substituted Ci-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(Ci-C6 alkyl) (preferably substituted with one
or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Ci-C6 alkyl group (preferably Ci-C3 alkyl).
Preferred heterocycle groups for R include tetrahydroquinoline, piperidine, piperazine, pyrrolidine, morpholine, tetrahydrofuran, tetrahydrothiophene, oxane and thiane, each of which groups may be optionally substituted or a group according to the chemical structu

Preferably, a
 group,
group,
Where RPR0 is H, optionally substituted Ci-C6 alkyl or an optionally substituted aryl (phenyl or napthyl), heteroaryl or heterocyclic group selected from the group consisting of oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, quinoline, (each preferably substituted with a Ci-C3 alkyl group, preferably methyl or a halo group, preferably F or CI), benzofuran, indole, indolizine, azaindolizine;
RPR01 and RPR02 are each independently H, an optionally subsituted C[-C3 alkyl group or together form a keto group, and
Each n is 0, 1, 2, 3, 4, 5, or 6 (preferably 0 or 1).
Preferred R3 substituents for use in the present invention also include specifically (and without limitation to the specific compound disclosed) the R3 substituents which are found in the identified compounds disclosed herein (which includes the specific compounds which are disclosed in the present specification, and the figures which are attached hereto).
Each of these R3 substituents may be used in conjunction with any number of R2
substituents which are also disclosed in the present specification, especially including the R2' groups which are presented in the attached figures hereof.
In certain alternative preferred embodiments, R2 is an optionally substituted -NRj-
 an optionally substituted -NRi-X^'-Aryl-HET or an optionally substituted -NR X^'-HET- Aryl ,
an optionally substituted -NRi-X^'-Aryl-HET or an optionally substituted -NR X^'-HET- Aryl ,
Where R] is H or a C1-C3 alkyl group (preferably H);
X 2' is an optionally substituted -CH2)n- , -CH2)„-CH(XV)=CH(XV)- (cis or trans),
-CH2)n-CH≡CH- , -(CH2CH20)n- or a C3-C6 cycloalkyl group;
where Xv is H, a halo or a
 alkyl group which is optionally substituted with one or two hydroxyl groups or up to three halogen groups;
alkyl group which is optionally substituted with one or two hydroxyl groups or up to three halogen groups;
Alkyl is an optionally substituted CI-C10 alkyl (preferably a Ci-C6 alkyl) group (in certain preferred embodiments, the alkyl group is end-capped with a halo group, often a CI or Br); Aryl is an optionally substituted phenyl or naphthyl group (preferably, a phenyl group); and HET is an optionally substituted oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, benzofuran, indole, indolizine, azaindolizine, quinoline (when substituted, each preferably substituted with a C1-C3 alkyl group, preferably methyl or a halo group, preferably F or CI) or a group according to the chemical structure:
Where Sc is CHRSS, NRURE, or O;
RHET is H, CN, N02, halo (preferably CI or F), optionally substituted Q-Q alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Ci-C6 alkyl group (preferably CrC3 alkyl);
Rss is H, CN, N02, halo (preferably F or CI), optionally substituted Ci-Q alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups), optionally substituted 0-(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted -C(0)(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups);
RURE is H, a Ci-C6 alkyl (preferably H or C1-C3 alkyl) or a -C(0)(CrC6 alkyl), each of which groups is optionally substituted with one or two hydroxyl groups or up to three halogen,
preferably fluorine groups, or an optionally substituted heterocycle, for example piperidine, morpholine, pyrrolidine, tetrahydrofuran, tetrahydrothiophene, piperidine, piperazine, each of which is optionally substituted, and
Yc is N or C-RYC, where RYC is H, OH, CN, N02, halo (preferably CI or F), optionally substituted C]-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(C[-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Ci-C6 alkyl group (preferably Ci-C3 alkyl);
RPR0 is H, optionally substituted C[-C6 alkyl or an optionally substituted aryl (phenyl or napthyl), heteroaryl or heterocyclic group selected from the group consisting of oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, quinoline, (each preferably substituted with a Ci-C3 alkyl group, preferably methyl or a halo group, preferably F or CI), benzofuran, indole, indolizine, azaindolizine;
RPR01 and RPR02 are each independently H, an optionally subsituted C!-C3 alkyl group or together form a keto group, and
Each n is independently 0, 1, 2, 3, 4, 5, or 6 (preferably 0 or 1).
In certain alternative preferred embodiments of the present invention, R3 is an optionally substituted -(CH2)n-(V)n'-(CH2)n-(V)n'-RS3 group, an optionally substituted -(CH2)n-N(Ri (C=0)m>-(V)n>-RS3' group, an optionally substituted -XR3'-alkyl group, an optionally substituted -XR3 -Aryl group; an optionally substituted -XR3 -HET group, an optionally substituted -XR3 -Aryl-HET group or an optionally substituted -XR3'-HET-Aryl group,
Where RS3 is an optionally substituted alkyl group (Ci-Qo, preferably Q-C6 alkyl), an optionally substituted Aryl group or a HET group;
Rr is H or a C!-C3 alkyl group (preferably H);
V is O, S or NRr;
XR3' is -(CH2)n- , -(CH2CH20)n-, -CH2)„-CH(XV)=CH(XV)- (cis or trans), -CH2)„-CH≡CH- , or a C3-C6 cycloalkyl group, all optionally substituted;
where Xv is H, a halo or a Ci-C3 alkyl group which is optionally substituted with one or two hydroxyl groups or up to three halogen groups;
Alkyl is an optionally substituted Ci-Cio alkyl (preferably a Ci-C6 alkyl) group (in certain preferred embodiments, the alkyl group is end-capped with a halo group, often a CI or Br); Aryl is an optionally substituted phenyl or napthyl group (preferably, a phenyl group); and HET is an optionally substituted oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydroiuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, benzofuran, indole, indolizine, azaindolizine, quinoline (when substituted, each preferably substituted with a C!-C3 alkyl group, preferably methyl or a halo group, preferably F or CI), or a group according to the chemical structure:

Where Sc is CHR , NRURE, or O;
RHET is H, CN, N02, halo (preferably CI or F), optionally substituted Q-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to
three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Q-C6 alkyl group (preferably C1-C3 alkyl);
Rss is H, CN, N02, halo (preferably F or CI), optionally substituted Ci-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups), optionally substituted 0-(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted -C(0)(C!-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups);
RURE is H, a C,-C6 alkyl (preferably H or d-C3 alkyl) or a -C(O)(C0-C6 alkyl), each of which groups is optionally substituted with one or two hydroxyl groups or up to three halogen, preferably fluorine groups, or an optionally substituted heterocycle, for example piperidine, morpholine, pyrrolidine, tetrahydrofuran, tetrahydrothiophene, piperidine, piperazine, each of which is optionally substituted, and
Yc is N or C-RYC, where RYC is H, OH, CN, N02, halo (preferably CI or F), optionally substituted C!-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Ci-C6 alkyl group (preferably C1-C3 alkyl);
RPR0 is H, optionally substituted Ci-C6 alkyl or an optionally substituted aryl (phenyl or napthyl), heteroaryl or heterocyclic group selected from the group consisting of oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydirofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, quinoline, (each preferably substituted with a Q-C3 alkyl group, preferably methyl or a halo group, preferably F or CI), benzofuran, indole, indolizine, azaindolizine;
RPR01 and RPR02 are each independently H, an optionally subsituted C1-C3 alkyl group or together form a keto group, and
Each n is independently 0, 1, 2, 3, 4, 5, or 6 (preferably 0 or 1);
Each m' is 0 or 1 ; and
Each n' is 0 or 1.
In alternative embodiments, R3' is -(CH2)n-Aryl, -(CH2CH20)n-Aryl, -(CH2)n-HET or -(CH2CH20)n-HET;
Where Aryl is phenyl which is optionally substituted with one or two substitutents, wherein said substituent(s) is preferably selected from -(CH2)nOH, Ci-C6 alkyl which itself is further
optionally substituted with CN, halo (up to three halo groups), OH, -(CH2)nO(C1-C6)alkyl, amine, mono- or di-(Ci-C6 alkyl) amine wherein the alkyl group on the amine is optionally substituted with 1 or 2 hydroxyl groups or up to three halo (preferably F, CI) groups, or said Aryl group is substituted with -(CH2)nOH, -(CH2)n-0-(C1-C6)alkyl, -(CH2)n-0-(CH2)n- (d-C6)alkyl, -(CH2)n-C(0)(Co-C6) alkyl, -(CH2)n-C(O)O(C0-C6)alkyl, -(CH2)n-OC(O)(C0- C6)alkyl, amine, mono- or di-(C1-C6 alkyl) amine wherein the alkyl group on the amine is optionally substituted with 1 or 2 hydroxyl groups or up to three halo (preferably F, CI) groups, CN, N02, an optionally substituted -(CH2)n-(V)m'-CH2)n-(V)m-(C1-C6)alkyl group, a -(V)m-(CH2CH20)n-RPEG group where V is O, S or NRr, Rr is H or a d-C3 alkyl group (preferably H) and RPEG is H or a Ci-C6 alkyl group which is optionally substituted (including being optionally substituted with a carboxyl group), or said Aryl group is optionally substituted with a heterocycle, including a heteroaryl, selected from the group consisting of oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, quinoline, benzofuran, indole, indolizine, azaindolizine, (when substituted each preferably substituted with a Ci-C3 alkyl group, preferably methyl or a halo group, preferably F or CI), or a group according to the chemical
structure:

Where Sc is CHRS , NRURE, or O;
RHET is H, CN, N02, halo (preferably CI or F), optionally substituted d-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Ci-C alkyl group (preferably Ci-C3 alkyl);
Rss is H, CN, N02, halo (preferably F or CI), optionally substituted Ci-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups), optionally substituted 0-(C\-Ce alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted -C(0)(d-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups);
RURE is H, a Ci-C6 alkyl (preferably H or C C3 alkyl) or a -C(O)(C0-C6 alkyl), each of which groups is optionally substituted with one or two hydroxyl groups or up to three halogen, preferably fluorine groups, or an optionally substituted heterocycle, for example piperidine,
morpholine, pyrrolidine, tetrahydrofuran, tetrahydrothiophene, piperidine, piperazine, each of which is optionally substituted, and
Yc is N or C-RYC, where RYC is H, OH, CN, N02, halo (preferably CI or F), optionally substituted Ci-Ce alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Ci-C6 alkyl group (preferably Ci-C3 alkyl);
RPRO is H, optionally substituted Ci-C6 alkyl or an optionally substituted aryl (phenyl or napthyl), heteroaryl or heterocyclic group selected from the group consisting of oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, quinoline, (each preferably substituted with a Ci-C3 alkyl group, preferably methyl or a halo group, preferably F or CI), benzofuran, indole, indolizine, azaindolizine;
RPR01 and RPR02 are each independently H, an optionally subsituted C1-C3 alkyl group or together form a keto group;
HET is preferably oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene,
tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, quinoline, (each preferably substituted with a C1-C3 alkyl group, preferably methyl or a halo group, preferably F or CI), benzofuran, indole, indolizine, azaindolizine, or a group according to the chemical structure:

Where Sc is CHRSS, NR , or O;
RHET is H, CN, N02, halo (preferably CI or F), optionally substituted C C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a C]-C6 alkyl group (preferably Ci-C3 alkyl);
Rss is H, CN, N02, halo (preferably F or CI), optionally substituted Ci-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups), optionally substituted 0-(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted -C(0)(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups);
RURE is H, a Ci-C6 alkyl (preferably H or C,-C3 alkyl) or a -C(O)(C0-C6 alkyl), each of which groups is optionally substituted with one or two hydroxyl groups or up to three halogen, preferably fluorine groups, or an optionally substituted heterocycle, for example piperidine,
morpholine, pyrrolidine, tetrahydroftiran, tetrahydrothiophene, piperidine, piperazine, each of which is optionally substituted, and
Yc is N or C-RYC, where RYC is H, OH, CN, N02, halo (preferably CI or F), optionally substituted Ci-Ce alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Ci-C6 alkyl group (preferably Ci-C3 alkyl);
RPR0 is H, optionally substituted Ci-C6 alkyl or an optionally substituted aryl, heteroaryl or heterocyclic group;
RPR01 and RPR02 are each independently H, an optionally subsituted Ci-C3 alkyl group or together form a keto group,
Each m' is independently 0 or 1 , and
Each n is independently 0, 1, 2, 3, 4, 5, or 6 (preferably 0 or 1).
In still additional embodiments, preferred compounds include those according to the chemical structure:

Where Rr is OH or a group which is metabolized in a patient or subject to OH;
R2' is a -NH-CH2-Aryl-HET (preferably, a phenyl linked directly to a methyl substituted thiazole);
R3 is a -CHRCR3'-NH-C(0)-R3P1 group or a -CHRCR3'-R3P2 group;
Where RCR3 is a Q-Q alkyl group, preferably methyl, isopropyl or tert-butyl;
R3P1 is Ci-C3 alkyl (preferably methyl), an optionally substituted oxetane group (preferably methyl substituted, a -(CH2)„OCH3 group where n is 1 or 2 (preferably 2), or a
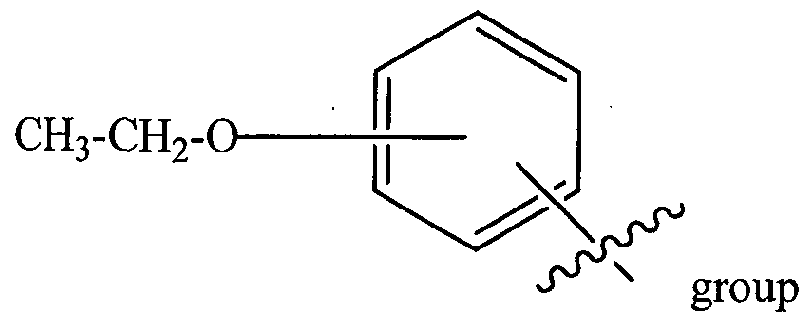 (the ethyl ether group is preferably meta- substituted on the phenyl moiety), a morpholino grop (linked to the carbonyl at the 2- or 3- position;
(the ethyl ether group is preferably meta- substituted on the phenyl moiety), a morpholino grop (linked to the carbonyl at the 2- or 3- position;
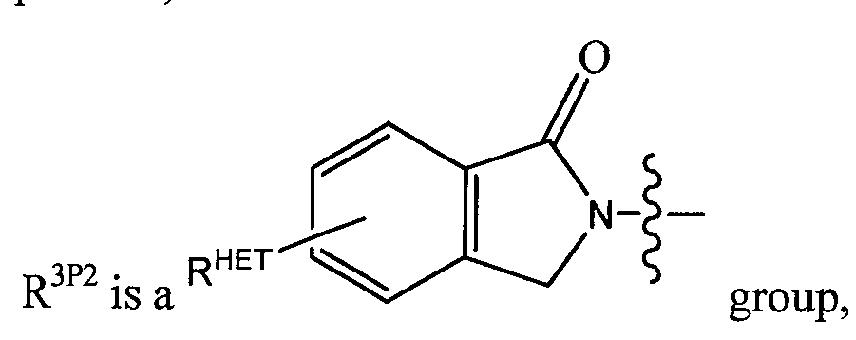
Where Aryl is phenyl;
HET is an optionally substituted thiazole or isothiazole; and
RHET is H or a halo group (preferably H),
Or a pharmaceutically acceptable salt, stereoisomer, solvate or polymorph thereof. Preferred compositions which pertain to this embodiment of the present application are presented in figure 17 hereof.
In another aspect, the compound according to the present invention is based upon an amino acid such as phenylanine as a portion (right hand) of the molecule according to the formula:

Where X is halogen, C1-C3 alkyl or an optionally substituted heterocycle; and
R1 and R2 are each independently H, Ci-C3 alkyl optionally substituted with one or two hydroxyl groups, or an optionally substituted phenyl group; and
n is 0, 1, 2, or 3, preferably 0 or 1, or
A pharmaceutically acceptable salt, enantiomer, diastereomer, solvate or polymorph thereof.
In another aspect, the present invention relates to pharmaceutical compositions comprising an effective amount of a compound as set forth hereinabove, in combination with a pharmaceutically acceptable carrier, additive or excipient.
In alternative aspects, the present invention relates to a method for enhancing erythropoiesis in a patient or subject in need, the method comprising administering to said patient or subject an effective amount of at least one compound as described hereinabove, optionally in combination with an additional erythropoiesis stimulating compound. The method according to the present invention may be used to increase the number of red blood cells (erythrocytes) and/or the hematocrit of the patient by virtue of the administration of effective amounts of at least one compound described herein.
Additional method aspects of the present invention relate to treating anemia, including chronic anemia or ischemia in a patient or subject in need, the method comprising administering to a patient in need an effective amount of at least one compound according to the present invention. The methods according to the present invention may be used to treat anemia, including chronic anemia such as anemia associate with chronic kidney disease, dialysis and chemotherapy and ischemia, including local ischemia, stroke and cardiovascular ischemia and limit the damage which occurs as a consequence of those disease states and/or conditions.
Additional method aspects of the present invention relate to enhancing wound healing and reducing scar tissue formation during wound healing by administering one or more compounds according to the present invention to a patient in need. Further methods include inducing local angiogenesis in a patient or subject in need by administering an effective amount of at least one compound of the present invention, optionally in combination with an additional erythropoiesis stimulating compound. Methods of stimulating erythropoiesis in a subject or patient, including increasing the number of red blood cells and/or hematocrit of the patient, treating anemia, including chronic anemia and anemia associated with chronic kidney disease, dialysis, and cancer chemotherapy, ischemia, stroke and damage to cardiovascular tissue during cardiovascular ischemia as well as enhancing wound healing processes and
preventing/reducing scarring associated with or secondary to the healing process represent additional aspects of the present invention.
Other methods of the present invention relate to the local enhancement of
angiogenesis through the induction of VEGF in a patient or subject using at least one compound according to the present invention, optionally in combination with an
erythropoiesis stimulating compound as otherwise described herein. An additional method of the present invention relates to the reduction and/or inhibition of occlusion in a surgically implanted stent in a patient or subject.
In addition, the present invention makes use of the compounds according to the present invention generically as protein modulators/degraders to provide complex
biiunctional compounds which include the present compounds to which is bonded a linker moiety through which is further bonded a protein binding moiety to provide a biiunctional compound which is capable of binding to and degrading specifically targeted proteins and modulating the activity of those targeted proteins. These biiunctional compounds, referred to as "VHL protac compounds", are useful for targeting VHL binding moieties according to the present invention to a specific protein in order to inactivate and/or otherwise modulate the protein by a protein degradation mechanisim. Thus, compounds according to the present invention also find use to create complex bifunctional compounds called VHL protacs for therapeutic use, including modulating protein activity In these compounds, each of the monofunctional VHL compounds described herein may be further modified on any appropriate position of the molecule by covalently linking a protein binding moiety (i.e., a moiety that selectively binds to a target protein and through that interaction, facilitates degradation of the bound protein by action of the VHL ligand) by covalently attaching a linker to the VHL compound and a protein binding moiety to the linker. The resulting bifunctional compound can be used to modulate levels of protein and/or protein activity as described above.
Detailed Description of the Invention
The following terms are used to describe the present invention. In instances where a term is not specifically defined herein, that term is given an art-recognized meaning by those of ordinary skill applying that term in context to its use in describing the present invention.
Where a range of values is provided, it is understood that each intervening value, to the tenth of the unit of the lower limit unless the context clearly dictates otherwise (such as in the case of a group containing a number of carbon atoms in which case each carbon atom number falling within the range is provided), between the upper and lower limit of that range and any other stated or intervening value in that stated range is encompassed within the invention. The upper and lower limits of these smaller ranges may independently be included in the smaller ranges is also encompassed within the invention, subject to any specifically excluded limit in the stated range. Where the stated range includes one or both of the limits, ranges excluding either both of those included limits are also included in the invention.
The term "compound", as used herein, unless otherwise indicated, refers to any specific chemical compound disclosed herein and includes tautomers, regioisomers, geometric isomers, and where applicable, stereoisomers, including optical isomers
(enantiomers) and other steroisomers (diastereomers) thereof, as well as pharmaceutically acceptable salts and derivatives (including prodrug forms) thereof where applicable, in context. Within its use in context, the term compound generally refers to a single compound, but also may include other compounds such as stereoisomers, regioisomers and/or optical isomers (including racemic mixtures) as well as specific enantiomers or enantiomerically enriched mixtures of disclosed compounds. The term also refers, in context to prodrug forms of compounds which have been modified to facilitate the administration and delivery of compounds to a site of activity. It is noted that in describing the present compounds, numerous substituents and variables associated with same, among others, are described. It is understood by those of ordinary skill that molecules which are described herein are stable compounds as generally described hereunder.

double bond and single bond are represented within the context of the compound shown.
The term "patient" or "subject" is used throughout the specification to describe an animal, preferably a human or a domesticated animal, to whom treatment, including prophylactic treatment, with the compositions according to the present invention is provided. For treatment of those infections, conditions or disease states which are specific for a specific animal such as a human patient, the term patient refers to that specific animal. In general, in
the present invention, the term patient refers to a human patient unless otherwise stated or implied from the context of the use of the term.
The term "effective" is used to describe an amount of a compound, composition or component which, when used within the context of its intended use, effects an intended result.
The term "VCB E3 Ubiquitin Ligase", "Hippel-Lindau E3 Ubiquitin Ligase" or "Ubiquitin Ligase" is used to describe the target enzyme(s) of compounds according to the present invention. VCB E3 is a multiprotein complex composed of pVHL, Elongin B, Elongin C, Cullin 2 and Rbxl proteins, that in combination with an E2 ubiquitin-conjugating enzyme causes the attachment of ubiquitin to a lysine on a target protein; the E3 ubiquitin ligase targets specific protein substrates for degradation by the proteasome. In general, the ubiquitin ligase is involved in polyubiquitination such that a second ubiquitin is attached to the first, a third is attached to the second, and so forth. Polyubiquitination marks proteins for degradation by the proteasome. However, there are some ubiquitination events that are limited to mono-ubiquitination, in which only a single ubiquitin is added by the ubiquitin ligase to a substrate molecule. Mono-ubiquitinated proteins are not targeted to the proteasome for degradation, but may instead be altered in their cellular location or function, for example, via binding other proteins that have domains capable of binding ubiquitin. Further
complicating matters, different lysines on ubiquitin can be targeted by an E3 to make chains. The most common lysine is Lys48 on the ubiquitin chain. This is the lysine used to make polyubiquitin, which is recognized by the proteasome. As a consequence of the complicated nature of ubiquitin ligase, finding modulators and in particular, inhibitors of the same is a difficult task.
The term "anemia" is used to describe a disease state or condition of a subject or patitent where the patient or subject does not have healthy red blood cells to carry adequate oxygen to tissues. There are many forms of anemia, each with its own cause. Anemia can be temporary or long term (chronic), and it can range from mild to severe. The use of EPO to treat anemia, including chronic anemia, as well as ischemia, and to enhance wound healing as well as enhancing the response of the brain to neuronal injury are additional aspects of this treatment.
The term "erythropoietin" or "EPO" is used to describe a glycoprotein hormone that controls erythropoi'esis, or red blood cell production. EPO is a cytokine for erythrocyte (red blood cell) precursors in the bone marrow. Also referred to as hematopoietin or hemopoietin, EPO is produced by interstitial fibroblasts in the kidney in close association with peritubular capillary and tubular epithelial cells. It is also produced in perisinusoidal cells in the liver. While liver production predominates in the fetal and perinatal period, renal production is predominant during adulthood. Erythropoietin is the hormone that regulates red blood cell production. It also has other known biological functions. For example, erythropoietin plays an important role in the brain's response to neuronal injury and functions to limit brain damage during ischemia. EPO is also involved in the wound healing process.
The term "angiogenesis" refers to the physiological process involving the growth of new blood vessels rom pre-existing vessels. Though there has been some debate over terminology, vasculogenesis is the term used for spontaneous blood-vessel formation, and intussusception is the term for the formation of new blood vessels by the splitting of exisiting ones. Angiogenesis is a normal and vital process in growth and development, as well as in wound healing and in granulation tissue. However, it is also a fundamental step in the transition of tumors from a dormant state to a malignant one.
The term "arteriogenesis" refers to an increase in the diameter of existing arterial vessels. Mechanically, arteriogenesis is linked to elevated pressure, which increases radial wall stress, and elevated flow, which increases endothelial surface stress. Endothelial cells have a receptor devoted to VEGF aptly named VEGF receptor- 1 that immediately signals rapid mitosis in the cells.
The term "pharmaceutically acceptable salt" is used throughout the specification to describe, where applicable, a salt form of one or more of the compounds described herein which are presented to increase the solubility of the compound in the gastic juices of the patient's gastrointestinal tract in order to promote dissolution and the bioavailability of the compounds. Pharmaceutically acceptable salts include those derived from pharmaceutically acceptable inorganic or organic bases and acids, where applicable. Suitable salts include those derived from alkali metals such as potassium and sodium, alkaline earth metals such as calcium, magnesium and ammonium salts, among numerous other acids and bases well known in the pharmaceutical art. Sodium and potassium salts are particularly preferred as
neutralization salts of the phosphates according to the present invention.
The term "pharmaceutically acceptable derivative" is used throughout the specification to describe any pharmaceutically acceptable prodrug form (such as an ester, amide other prodrug group) which, upon administration to a patient, provides directly or indirectly the present compound or an active metabolite of the present compound.
The term "independently" is used herein to indicate that the variable, which is independently applied, varies independently from application to application.
The term "hydrocarbyl" shall mean a compound which contains carbon and hydrogen and which may be fully saturated, partially unsaturated or aromatic and includes aryl groups, alkyl groups, alkenyl groups and alkynyl groups.
The term "alkyl" shall mean within its context a linear, branch-chained or cyclic fully saturated hydrocarbon radical or alkyl group, preferably a Cj-Cio, more preferably a Ci-Ce, alternatively a C1-C3 alkyl group, which may be optionally substituted. Examples of alkyl groups are methyl, ethyl, n-butyl, sec-butyl, n-hexyl, n-heptyl, n-octyl, n-nonyl, n-decyl, isopropyl, 2-methylpropyl, cyclopropyl, cyclopropylmethyl, cyclobutyl, cyclopentyl, cyclopentylethyl, cyclohexylethyl and cyclohexyl, among others. In certain preferred embodiments, compounds according to the present invention which may be used to covalently bind to dehalogenase enzymes. These compounds generally contain a side chain (often linked through a polyethylene glycol group) which terminates in an alkyl group which has a halogen substituent (often chlorine or bromine) on its distil end which results in covalent binding of the compound containing such a moiety to the protein. The term
"Alkenyl" refers to linear, branch-chained or cyclic C2-Ci0 (preferably C2-C6) hydrocarbon radicals containing at least one C=C bond. The term "Alkynyl" refers to linear, branch- chained or cyclic C2- 0 (preferably C2-C6) hydrocarbon radicals containing at least one C≡C bond. The term "alkylene" when used, refers to a -(CH2)n- group (n is an integer generally from 0-6), which may be optionally substituted. When substituted, the alkylene group preferably is substituted on one or more of the methylene groups with a CpC6 alkyl group (including a cyclopropyl group or a t-butyl group), more preferably a methyl group, but may also be substituted with one or more halo groups, preferably from 1 to 3 halo groups or one or two hydroxyl groups or 0-(Ci-C6 alkyl) groups. In certain embodiments, an alkylene group
may be substituted with a urethane or alkoxy group (or other group) which is further substituted with a polyethylene glycol chain (of from 1 to 10, preferably 1 to 6, often 1 to 4 ethylene glycol units) to which is substituted (preferably, but not exclusively on the distal end of the polyethylene glycol chain) an alkyl chain substituted with a single halogen group, preferably a chlorine group. In still other embodiments, the alkylene group may be substituted with an amino acid side chain such as group obtained from an amino acid (a natural or unnatural amino acid) such as, for example, alanine, β-alanine, arginine, asparagine, aspartic acid, cysteine, cystine, glutamic acid, glutamine, glycine, phenylalanine, histidine, isoleucine, lysine, leucine, methionine, proline, serine, threonine, valine, tryptophan or tyrosine.
The term "unsubstituted" shall mean substituted only with hydrogen atoms. A range of carbon atoms which includes C0 means that carbon is absent and is replaced with H. Thus, a range of carbon atoms which is Co-C6 includes carbons atoms of 1, 2, 3, 4, 5 and 6 and for Co, H stands in place of carbon. The term "substituted" or "optionally substituted" shall mean independently (i.e., where more than substituent occurs, each substituent is independent of another substituent), one or more substituents (independently, up to five substitutents, preferably up to three substituents, often 1 or 2 substituents on a moiety in a compound according to the present invention and may include substituents which themselves may be further substituted) at a carbon (or nitrogen) position anywhere on a molecule within context, and independently includes as substituents hydroxyl, thiol, carboxyl, cyano (C≡N), nitro (N02), halogen (preferably, 1, 2 or 3 halogens, especially on an alkyl, especially a methyl group such as a trifluoromethyl), an alkyl group (preferably, Ci-Qo , more preferably, Ci-C6), aryl (especially phenyl and substituted phenyl for example benzyl or benzoyl), alkoxy group (preferably, Ci-C6 alkyl or aryl, including phenyl and substituted phenyl), thioether (Ci-C6 alkyl or aryl), acyl (preferably, Q-C6 acyl), ester or thioester (preferably, Ci-C6 alkyl or aryl) including alkylene ester (such that attachment is on the alkylene group, rather than at the ester function which is preferably substituted with a Ci-C6 alkyl or aryl group), preferably, Ci-C6 alkyl or aryl, halogen (preferably, F or CI), amine (including a five- or six-membered cyclic alkylene amine, further including a Ci-C6 alkyl amine or a C]-C6 dialkyl amine which alkyl groups may be substituted with one or two hydroxyl groups) or an optionally substituted - N(Co-C6 alkyl)C(0)(0-C1-C6 alkyl) group (which may be optionally substituted with a polyethylene glycol chain to which is further bound an alkyl group containing a single halogen, preferably chlorine substituent), hydrazine, amido, which is preferably substituted with one or two -Q alkyl groups (including a carboxamide which is optionally substituted
with one or two Ci-C6 alkyl groups), alkanol (preferably, C\-C alkyl or aryl), or alkanoic acid (preferably, Ci-C6 alkyl or aryl). Substituents according to the present invention may include, for example
groups where each of R] and R2 is as otherwise described herein and R3 is H or a Ci-C6 alkyl group, preferably Ri, R2, R3 in this context is a Cj-C3 alkyl group (including an isopropyl or t-butyl group). Each of the above-described groups may be linked directly to the substituted moiety or alternatively, the substituent may be linked to the substituted moiety (preferably in the case of an aryl or heteraryl moiety) through an optionally substituted -(CH2)m- or alternatively an optionally substituted -(OCH2)m-, -(OCH2CH2)m- or - (CH2CH20)m- group, which may be substituted with any one or more of the above-described substituents. Alkylene groups -(CH2)m- or -(CH2)n- groups or other chains such as ethylene glycol chains, as identified above, may be substituted anywhere on the chain. Preferred substitutents on alkylene groups include halogen or C -Ce (preferably C]-C3) alkyl groups, which may be optionally substituted with one or two hydroxyl groups, one or two ether groups (O-C1-C6 groups), up to three halo groups (preferably F), an amino acid sidechain as otherwise described herein and optionally substituted amide (preferably carboxamide substituted as described above) or urethane groups (often with one or two C0-C6 alkyl substitutents, which group(s) may be further substituted). In certain embodiments, the alkylene group (often a single methylene group) is substituted with one or two optionally substituted Ci-C6 alkyl groups, preferably Ci-C4 alkyl group, most often methyl or O-methyl groups or an amino acid sidechain as otherwise disclosed herein. In the present invention, a moiety in a molecule may be optionally substituted with up to five substituents, preferably up to three substituents. Most often, in the present invention moieties which are substituted are substituted with one or two substituents.
The term "substituted" (each substituent being independent of another substituent) shall also mean within its context of use Q-C6 alkyl, C]-C6 alkoxy, halogen, amido, carboxamido, sulfone, including sulfonamide, keto, carboxy, C C6 ester (oxyester or carbonylester), -Ce keto, urethane -0-C(0)-NR1R2 or -N(Ri)-C(0)-0-R1, nitro, cyano and amine (especially including a Ci-C6 alkylene-NR1R2, a mono- or di- CpCe alkyl substituted amines which may be optionally substituted with one or two hydroxyl groups). Each of these groups contain unless otherwise indicated, within context, between 1 and 6 carbon atoms. In certain embodiments, preferred substituents will include for example, -NH-, -NHC(O)-, -0-,
=0, -(CH2)m- (here, m and n are in context, 1, 2, 3, 4, 5 or 6), -S-, -S(O)-, S02- or -NH-C(O)- NH-, -(CH2)„OH, -(CH2)„SH, -(CH2)nCOOH, Q-C6 alkyl, -(CH2)nO-(CI-C6 alkyl),
-(CH2)nC(0)-(C1-C6 alkyl), -(CH2)nOC(0)-(Ci-C6 alkyl), -(0η2)η0(Ο)Ο-(0ι-06 alkyl), -(CH2)nNHC(0)- !, -(CH2)nC(0)-NR1R2, -(OCH2)„OH, -(CH20)nCOOH, Q-Q alkyl, -(OCH2)„0-(C,-C6 alkyl), -(CH20)nC(0)-(C C6 alkyl), -(OCH2)„NHC(0)-Ru
-(CH20)„C(0)-NRiR2, -S(0)2-Rs, -S(0)-Rs (Rs is Cj-Q alkyl or a -(CH2)m-NR,R2 group), N02, CN or halogen (F, CI, Br, I, preferably F or CI), depending on the context of the use of the substituent. Rj and R2 are each, within context, H or a Q-C6 alkyl group (which may be optionally substituted with one or two hydroxyl groups or up to three halogen groups, preferably fluorine). The term "substituted" shall also mean, within the chemical context of the compound defined and substituent used, an optionally substituted aryl or heteroaryl group or an optionally substituted heterocyclic group as otherwise described herein. Alkylene groups may also be substituted as otherwise disclosed herein, preferably with optionally substituted C!-C6 alkyl groups (methyl, ethyl or hydroxymethyl or hydroxyethyl is preferred, thus providing a chiral center), an amido group as described hereinabove, or a urethane group 0-C(0)-NRiR2 group where R! and R2 are as otherwise described herein, although numerous other groups may also be used as substituents. Various optionally substituted moieties may be substituted indepencetnly with 3 or more substituents, preferably no more than 3 substituents and preferably with 1 or 2 substituents. It is noted that in instances where, in a compound at a particular position of the molecule substitution is required (principally, because of valency), but no substitution is indicated, then that substituent is construed or understood to be H, unless the context of the substitution suggests otherwise.
The term "aryl" or "aromatic", in context, refers to a substituted (as otherwise described herein) or unsubstituted monovalent aromatic radical having a single ring (e.g., benzene, phenyl, benzyl) or condensed rings (e.g., naphthyl, anthracenyl, phenanthrenyl, etc.) and can be bound to the compound according to the present invention at any available stable position on the ring(s) or as otherwise indicated in the chemical structure presented. Other examples of aryl groups, in context, may include heterocyclic aromatic ring systems
"heteroaryl" groups having one or more nitrogen, oxygen, or sulfur atoms in the ring
(moncyclic) such as imidazole, furyl, pyrrole, furanyl, thiene, thiazole, pyridine, pyrimidine, pyrazine, triazole, oxazole or fused ring systems such as indole, quinoline, indolizine, azaindolizine, benzofurazan, etc., among others, which may be optionally substituted as described above. Among the heteroaryl groups which may be mentioned include nitrogen-
containing heteroaryl groups such as pyrrole, pyridine, pyridone, pyridazine, pyrimidine, pyrazine, pyrazole, imidazole, triazole, triazine, tetrazole, indole, isoindole, indolizine, azaindolizine, purine, indazole, quinoline, dihydroquinoline, tetrahydroquinoline,
isoquinoline, dihydroisoquinoline, tetrahydroisoquinoline, quinolizine, phthalazine, naphthyridine, quinoxaline, quinazoline, cinnoline, pteridine, imidazopyridine,
imidazotriazine, pyrazinopyridazine, acridine, phenanthridine, carbazole, carbazoline, perimidine, phenanthroline, phenacene, oxadiazole, benzimidazole, pyrrolopyridine, pyrrolopyrimidine and pyridopyrimidine; sulfur-containing aromatic heterocycles such as thiophene and benzothiophene; oxygen-containing aromatic heterocycles such as furan, pyran, cyclopentapyran, benzofuran and isobenzofuran; and aromatic heterocycles comprising 2 or more hetero atoms selected from among nitrogen, sulfur and oxygen, such as thiazole, thiadizole, isothiazole, benzoxazole, benzothiazole, benzothiadiazole, phenothiazine, isoxazole, furazan, phenoxazine, pyrazoloxazole, imidazothiazole, thienofuran, furopyrrole, pyridoxazine, furopyridine, furopyrimidine, thienopyrimidine and oxazole, among others, all of which may be optionally substituted.
The term "heterocycle" refers to a cyclic group which contains at least one
heteroatom, i.e., 0, N or S, and may be aromatic (heteroaryl) or non-aromatic. Thus, the heteroaryl moieties are subsumed under the definition of heterocycle, depending on the context of its use. Exemplary heteroaryl groups are described hereinabove. Exemplary non- aromatic heterocyclic groups for use in the present invention include, for example,
pyrrolidinyl, pyrrolinyl, piperidinyl, piperazinyl, N-methylpiperazinyl, imidazolinyl, pyrazolidinyl, imidazolidinyl, morpholinyl, tetrahydropyranyl, azetidinyl, oxetanyl, oxathiolanyl, pyridone, 2-pyrrolidone, ethyleneurea, 1,3-dioxolane, 1,3-dioxane, 1,4-dioxane, phthalimide and succinimide, among others.
The term "amino acid sidechain" or "amino acid residue" shall mean, within context, a radical of a D- or L-amino acid sidechain (derived from an amino acid) which functions as a substituent on another group, often an alkylene (usually a methylene) group on R2 or R3 as otherwise described herein. Preferred amino acid sidechains for use in the present invention are derived from the sidechains of both natural and unnatural amino acids, preferably including, for example, alanine, β-alanine, arginine, asparagine, aspartic acid, cysteine, cystine, glutamic acid, glutamine, glycine, phenylalanine, histidine, isoleucine, lysine, leucine, methionine, proline, serine, threonine, valine, tryptophan or tyrosine, among others.
The terms "treat", "treating", and "treatment", etc., as used herein, refer to any action providing a benefit to a patient for which the present compounds may be administered, including stimulation of erythropoiesis in a patient or subject including inducement of EPO production in the patient or subject, for the treatment of chronic anemia and ischemia (which limits brain injury during episodes of localized anemia, ischemia and/or stroke and damage to cardiovascular tissue during cardiovascular ischemia), as well as enhancing wound healing processes. Methods of stimulating erythropoiesis in a subject or patient, including increasing the number of red blood cells and/or hematocrit of the patient, treating anemia, including chronic anemia and anemia associated with chronic kidney disease, dialysis, and cancer chemotherapy, ischemia, stroke and damage to cardiovascular tissue during cardiovascular ischemia as well as enhancing wound healing processes and preventing/reducing scarring secondary to healing represent additional treatment aspects of the present invention. Local enhancement of angiogenesis through induction of VEGF including wound healing and reduction of stent occlusion remain additional aspects of the present invention.
The term "coadministration" or "combination therapy" shall mean that at least two compounds or compositions are administered to the patient at the same time, such that effective amounts or concentrations of each of the two or more compounds may be found in the patient at a given point in time. Although compounds according to the present invention may be co-administered to a patient at the same time, the term embraces both administration of two or more agents at the same time or at different times, provided that effective concentrations of all coadministered compounds or compositions are found in the subject at a given time. In certain preferred aspects of the present invention, one or more of the present compounds described above, are coadministered in combination with at least one additional bioactive agent having erythropoiesis stimulating activity as otherwise described herein in order to enhance erythopoeisis, treat chronic anemia and ischemia (limit brain injury during episodes of localized anemia, ischemia and/or stroke and damage to cardiovascular tissue during cardiovascular ischemia), as well as enhancing wound healing processes. and stimulating angiogenesis and inhibiting or preventing occlusion in a surgically implanted stent . In particularly preferred aspects of the invention, the co-administration of compounds results in synergistic erythropoietic activity and/or therapy.
The term "additional erythropoisis stimulating agent" shall mean a traditional polypeptide such as EPO (procrit or epogen) or darbapoietin alfa (a synthetic form of erythropoietin).
Pharmaceutical compositions comprising combinations of an effective amount of at least one bifunctional compound according to the present invention, and one or more of the compounds otherwise described herein, all in effective amounts, in combination with a pharmaceutically effective amount of a carrier, additive or excipient, represents a further aspect of the present invention.
The compositions of the present invention may be formulated in a conventional manner using one or more pharmaceutically acceptable carriers and may also be administered in controlled-release formulations. Pharmaceutically acceptable carriers that may be used in these pharmaceutical compositions include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as prolamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene- polyoxypropylene-block polymers, polyethylene glycol and wool fat.
The compositions of the present invention may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vaginally or via an implanted reservoir. The term "parenteral" as used herein includes subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques. Preferably, the compositions are administered orally, intraperitoneally or intravenously.
Sterile injectable forms of the compositions of this invention may be aqueous or oleaginous suspension. These suspensions may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents. The sterile injectable preparation may also be a sterile injectable solution or suspension in a nontoxic parenterally-acceptable diluent or solvent, for example as a solution in 1, 3-butanediol.
Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution. In addition, sterile, fixed oils are
conventionally employed as a solvent or suspending medium. For this purpose, any bland fixed oil may be employed including synthetic mono- or di-glycerides. Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions. These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, such as Ph. Helv or similar alcohol.
The pharmaceutical compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, aqueous suspensions or solutions. In the case of tablets for oral use, carriers which are commonly used include lactose and corn starch. Lubricating agents, such as magnesium stearate, are also typically added. For oral administration in a capsule form, useful diluents include lactose and dried corn starch. When aqueous suspensions are required for oral use, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening, flavoring or coloring agents may also be added.
Alternatively, the pharmaceutical compositions of this invention may be administered in the form of suppositories for rectal administration. These can be prepared by mixing the agent with a suitable non-irritating excipient which is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug. Such materials include cocoa butter, beeswax and polyethylene glycols.
The pharmaceutical compositions of this invention may also be administered topically. Suitable topical formulations are readily prepared for each of these areas or organs. Topical application for the lower intestinal tract can be effected in a rectal suppository formulation (see above) or in a suitable enema formulation. Topically-acceptable transdermal patches may also be used.
For topical applications, the pharmaceutical compositions may be formulated in a suitable ointment containing the active component suspended or dissolved in one or more carriers. Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petrolatum, white petrolatum, propylene glycol,
polyoxyethylene, polyoxypropylene compound, emulsifying wax and water. In certain preferred aspects of the invention, the compounds may be coated onto a stent which is to be surgically implanted into a patient in order to inhibit or reduce the likelihood of occlusion occurring in the stent in the patient.
Alternatively, the pharmaceutical compositions can be formulated in a suitable lotion or cream containing the active components suspended or dissolved in one or more pharmaceutically acceptable carriers. Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2- octyldodecanol, benzyl alcohol and water.
For ophthalmic use, the pharmaceutical compositions may be formulated as micronized suspensions in isotonic, pH adjusted sterile saline, or, preferably, as solutions in isotonic, pH adjusted sterile saline, either with our without a preservative such as
benzylalkonium chloride. Alternatively, for ophthalmic uses, the pharmaceutical
compositions may be formulated in an ointment such as petrolatum.
The pharmaceutical compositions of this invention may also be administered by nasal aerosol or inhalation. Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance
bioavailability, fluorocarbons, and/or other conventional solubilizing or dispersing agents.
The amount of compound in a pharmaceutical composition of the instant invention that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host and disease treated, the particular mode of administration.
Preferably, the compositions should be formulated to contain between about 0.05 milligram to about 750 milligrams or more, more preferably about 1 milligram to about 600 milligrams, and even more preferably about 10 milligrams to about 500 milligrams of active ingredient, alone or in combination with at least one other compound according to the present invention or erythropoiesis stimulating agent (EPO, darbapoietin alfa) in order to inter alia enhance erythopoeisis, treat chronic anemia and ischemia (limits brain injury during episodes of localized anemia, ischemia and/or stroke and damage to cardiovascular tissue during
cardiovascular ischemia), as well as enhancing wound healing processes, and stimulating angiogenesis and inhibiting or preventing occlusion in a surgically implanted stent.
It should also be understood that a specific dosage and treatment regimen for any particular patient will depend upon a variety of factors, including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, rate of excretion, drug combination, and the judgment of the treating physician and the severity of the particular disease or condition being treated.
A patient or subject in need of therapy using compounds according to the present invention can be treated by administering to the patient (subject) an effective amount of the compound according to the present invention including pharmaceutically acceptable salts, solvates or polymorphs, thereof optionally in a pharmaceutically acceptable carrier or diluent, either alone, or in combination with other known erythopoiesis stimulating agents as otherwise identified herein.
These compounds can be administered by any appropriate route, for example, orally, parenterally, intravenously, intradermally, subcutaneously, or topically, including
transdermally, in liquid, cream, gel, or solid form, or by aerosol form.
The active compound is included in the pharmaceutically acceptable carrier or diluent in an amount sufficient to deliver to a patient a therapeutically effective amount for the desired indication, without causing serious toxic effects in the patient treated. A preferred dose of the active compound for all of the herein-mentioned conditions is in the range from about 10 ng/kg to 300 mg/kg, preferably 0.1 to 100 mg kg per day, more generally 0.5 to about 25 mg per kilogram body weight of the recipient/patient per day. A typical topical dosage will range from 0.01-5% wt/wt in a suitable carrier.
The compound is conveniently administered in any suitable unit dosage form, including but not limited to one containing less than lmg, 1 mg to 3000 mg, preferably 5 to 500 mg of active ingredient per unit dosage form. An oral dosage of about 25-250 mg is often convenient.
The active ingredient is preferably administered to achieve peak plasma concentrations of the active compound of about 0.00001-30 mM, preferably about 0.1-30 μΜ. This may be achieved, for example, by the intravenous injection of a solution or formulation of the active ingredient, optionally in saline, or an aqueous medium or administered as a bolus of the active ingredient. Oral administration is also appropriate to generate effective plasma concentrations of active agent.
The concentration of active compound in the drug composition will depend on absorption, distribution, inactivation, and excretion rates of the drug as well as other factors known to those of skill in the art. It is to be noted that dosage values will also vary with the severity of the condition to be alleviated. It is to be further understood that for any particular subject, specific dosage regimens should be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the
administration of the compositions, and that the concentration ranges set forth herein are exemplary only and are not intended to limit the scope or practice of the claimed composition. The active ingredient may be administered at once, or may be divided into a number of smaller doses to be administered at varying intervals of time.
Oral compositions will generally include an inert diluent or an edible carrier. They may be enclosed in gelatin capsules or compressed into tablets. For the purpose of oral therapeutic administration, the active compound or its prodrug derivative can be incorporated with excipients and used in the form of tablets, troches, or capsules. Pharmaceutically compatible binding agents, and/or adjuvant materials can be included as part of the
composition.
The tablets, pills, capsules, troches and the like can contain any of the following ingredients, or compounds of a similar nature: a binder such as microcrystalline cellulose, gum tragacanth or gelatin; an excipient such as starch or lactose, a dispersing agent such as alginic acid, Primogel, or corn starch; a lubricant such as magnesium stearate or Sterotes; a glidant such as colloidal silicon dioxide; a sweetening agent such as sucrose or saccharin; or a flavoring agent such as peppermint, methyl salicylate, or orange flavoring. When the dosage unit form is a capsule, it can contain, in addition to material of the above type, a liquid carrier such as a fatty oil. In addition, dosage unit forms can contain various other materials which modify the physical form of the dosage unit, for example, coatings of sugar, shellac, or
enteric agents.
The active compound or pharmaceutically acceptable salt thereof can be administered as a component of an elixir, suspension, syrup, wafer, chewing gum or the like. A syrup may contain, in addition to the active compounds, sucrose as a sweetening agent and certain preservatives, dyes and colorings and flavors.
The active compound or pharmaceutically acceptable salts thereof can also be mixed with other active materials that do not impair the desired action, or with materials that supplement the desired action, such as erythropoietin stimulating agents, including EPO and darbapoietin alfa, among others. In certain preferred aspects of the invention, one or more compounds according to the present invention are coadministered with another bioactive agent, such as an erythropoietin stimulating agent or a would healing agent, including an antibiotic, as otherwise described herein.
Solutions or suspensions used for parenteral, intradermal, subcutaneous, or topical application can include the following components: a sterile diluent such as water for injection, saline solution, fixed oils, polyethylene glycols, glycerine, propylene glycol or other synthetic solvents; antibacterial agents such as benzyl alcohol or methyl parabens; antioxidants such as ascorbic acid or sodium bisulfite; chelating agents such as ethylenediaminetetraacetic acid; buffers such as acetates, citrates or phosphates and agents for the adjustment of tonicity such as sodium chloride or dextrose. The parental preparation can be enclosed in ampoules, disposable syringes or multiple dose vials made of glass or plastic.
If administered intravenously, preferred carriers are physiological saline or phosphate buffered saline (PBS).
In one embodiment, the active compounds are prepared with carriers that will protect the compound against rapid elimination from the body, such as a controlled release
formulation, including implants and microencapsulated delivery systems. Biodegradable, biocompatible polymers can be used, such as ethylene vinyl acetate, polyanhydrides, polyglycolic acid, collagen, polyorthoesters, and polylactic acid. Methods for preparation of such formulations will be apparent to those skilled in the art.
Liposomal suspensions may also be pharmaceutically acceptable carriers. These may be prepared according to methods known to those skilled in the art, for example, as described in U.S. Pat. No. 4,522,811 (which is incorporated herein by reference in its entirety). For example, liposome formulations may be prepared by dissolving appropriate lipid(s) (such as stearoyl phosphatidyl ethanolamine, stearoyl phosphatidyl choline, arachadoyl phosphatidyl choline, and cholesterol) in an inorganic solvent that is then evaporated, leaving behind a thin film of dried lipid on the surface of the container. An aqueous solution of the active compound are then introduced into the container. The container is then swirled by hand to free lipid material from the sides of the container and to disperse lipid aggregates, thereby forming the liposomal suspension.
Detailed Synthetic Information
Generic scheme for the synthesis of all derivatives to be described here. Briefly, the compounds according to the present invention are synthesized pursuant to the general solution phase synthetic scheme (presented hereinbelow) and/or general scheme I, which is directed to phase synthesis of compounds according to the present invention. Initially a hydroxyl-protected carboxy substituted (and protected) pyrrolidine compound is reacted with a carboxylic acid containing reagent which introduces a carbonyl group at the amine of the pyrollidine ring to form an amide group. Alternatively, the pyrrolidine amine may be protected and the carboxylic acid moiety may be condensed with a nucleophilic group on a right hand fragment to provide an amide on the right hand portion of the pyrrolidine moiety. The left and right hand fragments to be condensed onto, respectively, the amine and carboxylic acid group of the pyrrolidine moiety are preferably prepared prior to condensing onto the pyrrolidine group, but other approaches may be taken to introduce groups onto the pyrrolidine group. Thus, a carboylic acid containing left hand fragment may be condensed onto the amine group of the pyrroline, thus forming an amide group with an R1 left hand fragment as depicted below. Onto the carboxyl group, any number of nucleophilic (preferably, amine containing) right hand fragments (pre-synthesized) may be condensed onto the carboxyl group to provide an amide group with an R2 right fragment as depicted below.
Formation of the pre-synthesized groups to condense onto the amine and/or the carboxyl moiety of the pyrrolidine proceeds in a facile manner. Virtually any compound can be synthesized readily using this approach. The solid phase synthetic method can also be used and employs similar methods used in the solution phase synthesis, the major difference being
that the hydroxyl group may be bound to a solid support as the other steps of the synthesis occur. The general synthetic methods are applicable to virtually all of the compounds of the present invention with facile modifications being made consistent with the state of chemical synthetic art as used directly or adapted from the specific teachings of the examples which follow.
Other synthetic methods and approaches for synthesizing compounds pursuant to the present invention are set forth in the further examples herein. Using the specific chemical syntheses which are presented in great detail herein, or alternative analogous methods which are presented in further detail hereinbelow, all of the compounds according to the present invention may be readily synthesized and characterized. Disclosed herein are methods for synthesizing the compounds which are presented herein, and in particular, the compounds which are set forth in Affinity Table II and the compounds which are presented in attached figure 15, hereof.
Scheme 1 Solution Phase Synthesis of Compounds According to the Present Invention
Route B
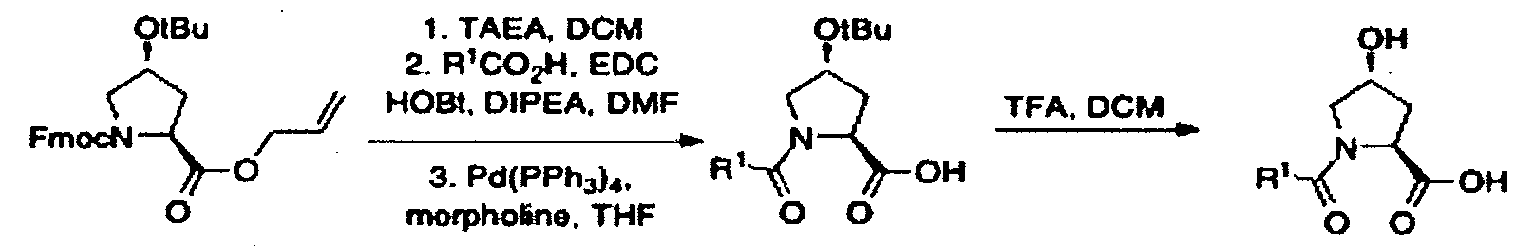
Hz R. EDC. HOB!. HJJNR. EDC, HOBt.
ROUt* A DIPEA DMF DIPEA, DMF

Scheme 2 Solid Phase Synthesis of Compounds According to the Present Invention
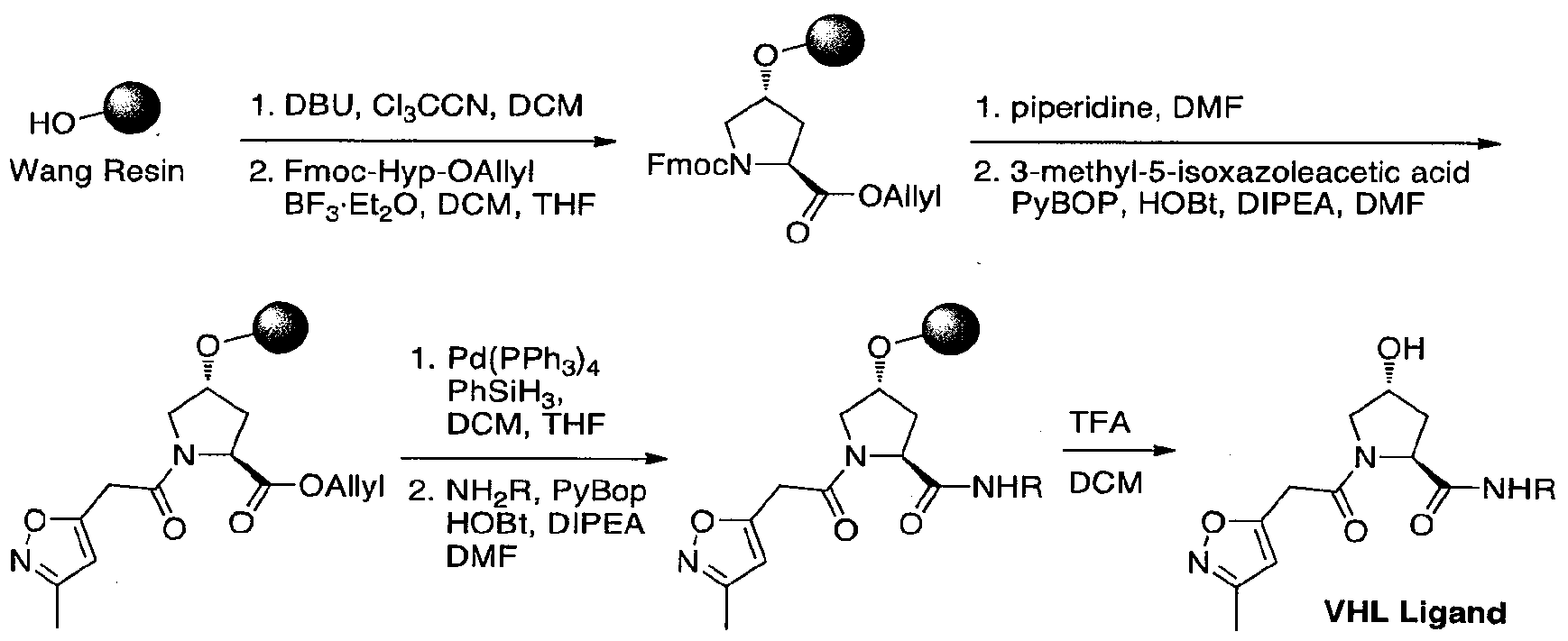
Examples
First Set
The inventors initially hypothesized that small molecule inhibitors of the VHL/HIF- la interaction could be rationally designed using hydroxyproline (Hyp) as a starting point, since residue Hyp564 on HIF- la makes key interactions with VHL14 and is crucial for HIF- la binding15. The inventors used the de-novo design software BOMB to guide the selection of plausible hydroxyproline analogs.16 1 and 2 were synthesized to test a promising design featuring an isoxazole moiety positioned to interact with a crystallographic water observed in the structure of VHL bound to the HIF peptide (549-582)14 and a benzyl group stacked along the side chain of Tyr98. Their ability to bind to VHL was measured by the competition of a fluorescent HIF- la peptide using fluorescence polarization (FP).17 Both were able to displace the fluorescent peptide albeit at high concentrations (Table 1 A). While the smaller 3 was unable to fully displace the fluorescent peptide, the observed binding to VHL through the use of WaterLOGSY and saturation transfer difference (STD) NMR. As no binding was observed with hydroxyproline alone, this suggested that the inventors identified a minimal
pharmacophore (see Figure 2).
Table 1A. Binding of Initial Ligands to VHL OH
1 117 10
2 120.1 7.1
3 CH3 > 250 N/A
'Average IC50 values were determined from three independent trials, each in triplicate.
Encouraged by these initial results, the inventors sought to increase the affinity of our VHL ligands by modifying the benzylamine moiety of 1 while maintaining the methyl- isoxazole fragment. In order to generate analogs rapidly, we developed a solid phase synthesis that involved the attachment of Fmoc-Hyp-OAllyl to Wang resin.18 Fmoc deprotection, coupling with 3-methyl-5-isoxazoleacetic acid followed by allyl ester deprotection and coupling with various amines and subsequent cleavage with trifluoroacetic acid led to the rapid generation of VHL ligands (Scheme 1).19,20 These ligands were then tested for their ability to bind VHL using the HIF peptide FP displacement assay.
Incorporation of various halogenated benzylamines showed that para substitution yielded the highest affinity and that there were only slight differences of affinity between chlorides and bromides, although the corresponding fluoride was less potent. We also found that substitution with electron withdrawing groups such as esters, nitro groups, nitriles, and ketones led to more potent ligands than substitution with the electron donating methoxy and t-butyl substituents. Molecular dynamics simulations suggested that Arg 107 is flexible and could accommodate bulkier groups at the para position. Therefore, we considered larger heterocyclic substituents at the para position of the benzylamine moiety and synthesized 15, which was found to bind with a 4.1 μΜ IC50 value (Table 2, below).
Fluorescence Polarization Assay
Ability of VHL ligands to compete for the HIFlo binding site on VCB was determined through a fluorescence polarization competition assay as described in the literature (Buckley et al. J ACS, 2012, 134, 4465-4468).
Table 2 Affinity Table- Most Compounds Showed Activity Within the Range of About 200 MicroMoles or Lower





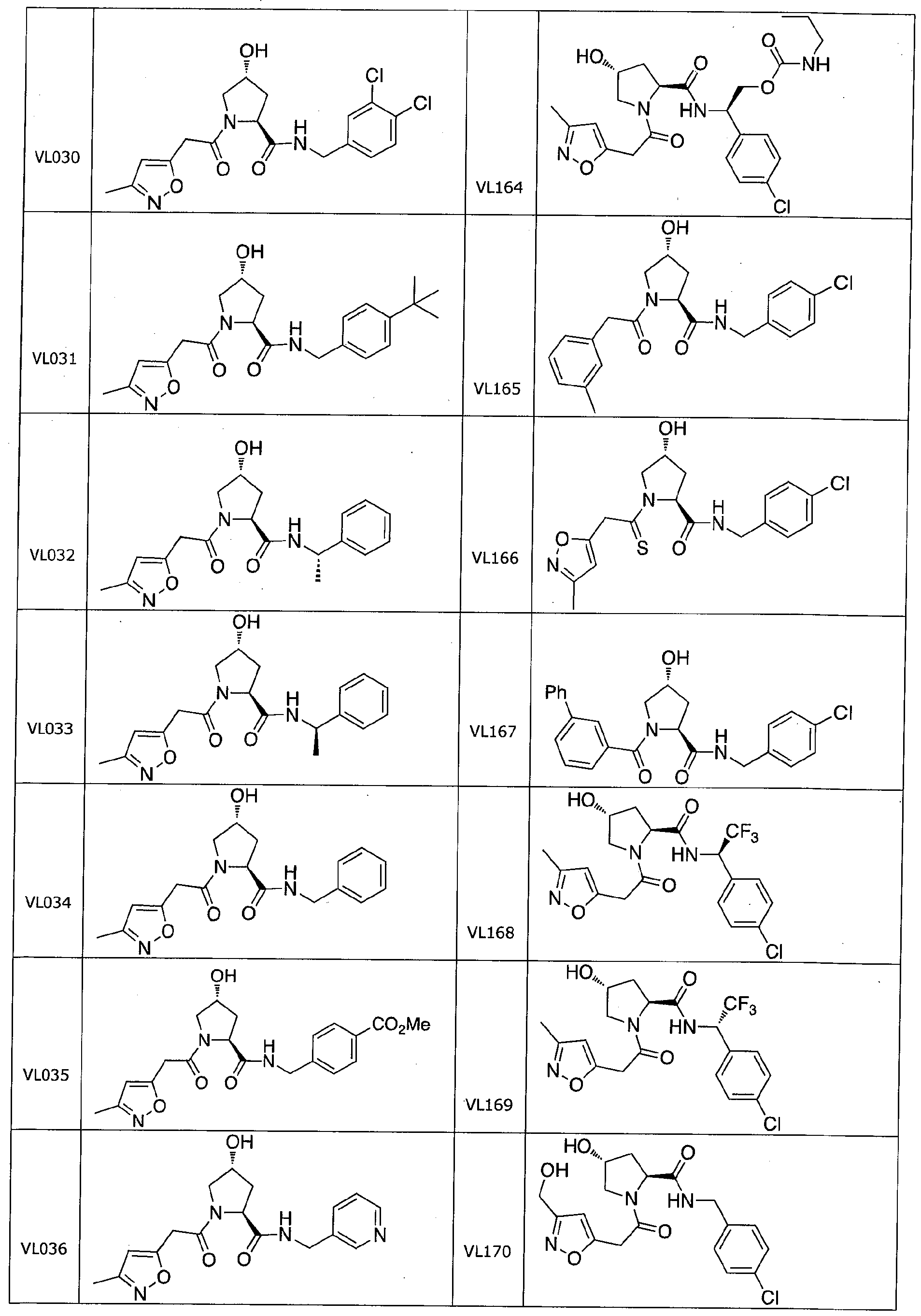










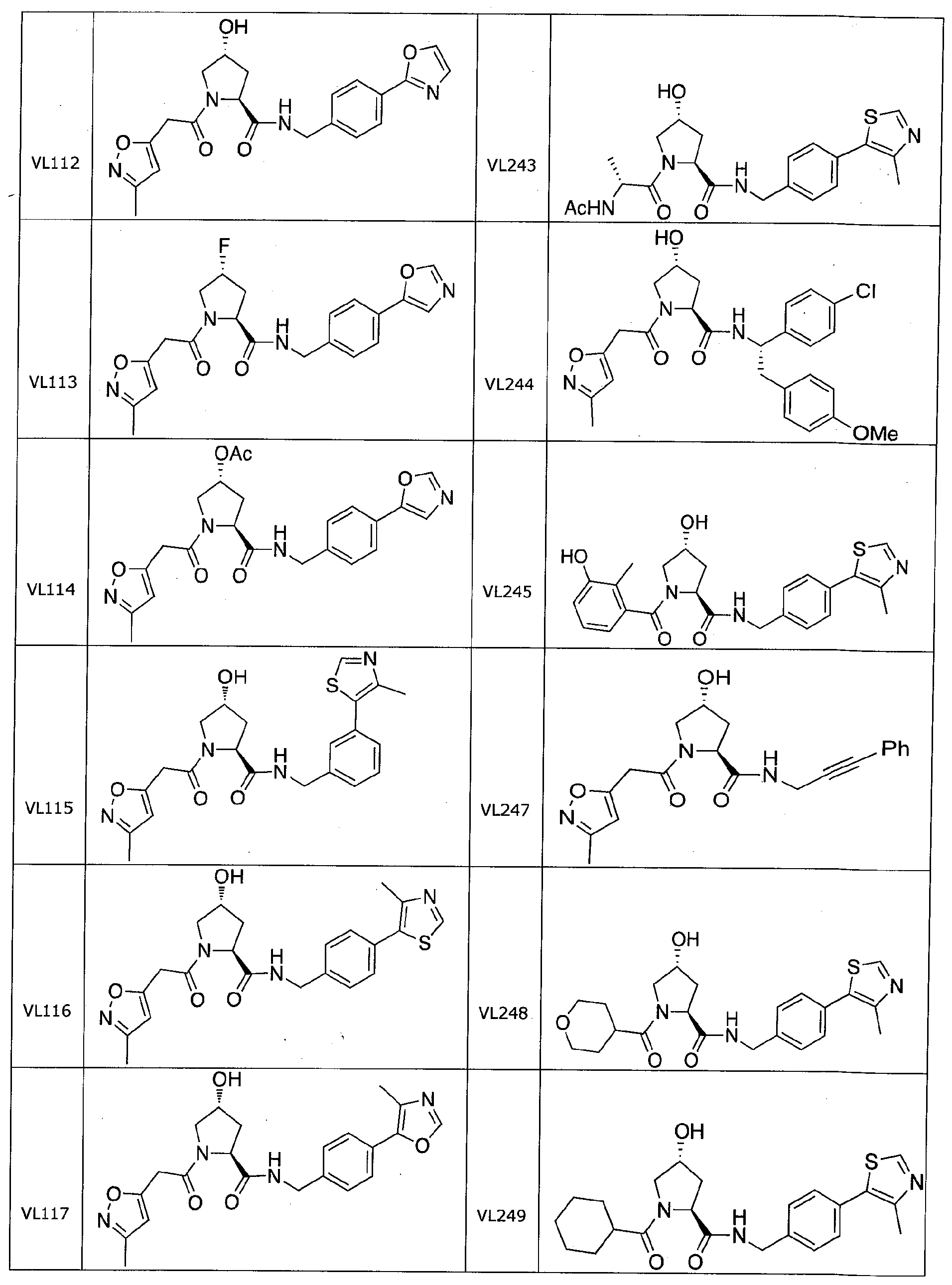



Synthetic Methods
General Chemistry
All reactions were performed in oven-dried or flame-dried glassware fitted with rubber septa under a positive pressure of nitrogen, unless otherwise noted. Air-and moisture-sensitive liquids were transferred via syringe or cannula. THF was distilled from
sodium/benzophenone. Dichloromethane was distilled from calcium hydride. Analytical thin layer chromatography (TLC) was performed using glass plates precoated with silica gel (0.25 mm). TLC plates were visualized by exposure to UV light (UV) or KMn04. Flash column chromatography was performed using silica gel 60 (230-400 mesh, Merck) with the indicated solvents.
!H and 13C spectra were recorded on Bruker Avance DPX-500 or Bruker Avance DPX- 400 NMR spectrometers. !H NMR spectra are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), integration, and coupling constant (J) in Hertz (Hz). Ή NMR chemical shifts are reported relative to CDC13
(7.26 ppm), d6- DMSO (2.50 ppm) and oVMeOD (3.31 ppm). 13C NMR was recorded relative to the central line of CDC13 (77.16 ppm), de-DMSO (39.52 ppm) and d4-MeOD (49.00 ppm). In most cases, only peaks of the major rotamer are reported. Mass spectra were obtained using a Perkin-Elmer API 150 EX spectrometer. MALDI-TOF analyses of purified samples were performed in a Voyager-DE-PRO 6268 (Applied Biosystems) using cyano-4- hydroxycinnamic acid matrices. Unless otherwise noted, HPLC was performed using a Dynamax SD200 solvent delivery system connected to a Dynamax UV-1 Absorbance Detector with a YMC-Pack ODS-AM preparative column (250 x 20 mm, 5 μπι particle size, 12 nm pore size). A linear gradient of MeCN in H20 from 20% to 100% MeCN, with constant 0.1% TF A was run over 40 minutes.
General Methods of Chemical Synthesis
The following eight (8) general chemical synthetic methods (Methods A through F and Solid Phase Synthesis A and B, described hereinbelow) are provided for synthesizing numerous compounds according to the present invention which are set forth in Table 2 Affinity Table above. Each method is presented with reference to a specific compound, the synthetic details of which are presented hereinabove. All of the compounds numbered may be synthesized relatively easily using the straight-forward methods which are set forth hereinbelow. In certain instances, more synthetic details are provided for certain preferred embodiments in order to present that information such that it may serve as a template for synthesizing a number of other compounds as otherwise disclosed herein.
A
 s an example, see the synthesis for compound VL133 of Table II, set forth below.
s an example, see the synthesis for compound VL133 of Table II, set forth below.

See the general synthesis for VLl 16 of Table II with protection of the hydroxyl group.
C

See the general synthesis for compound VL 156 of Table II, described below.
D

See the general synthesis for compound VL 217 of Table II, described below.
E
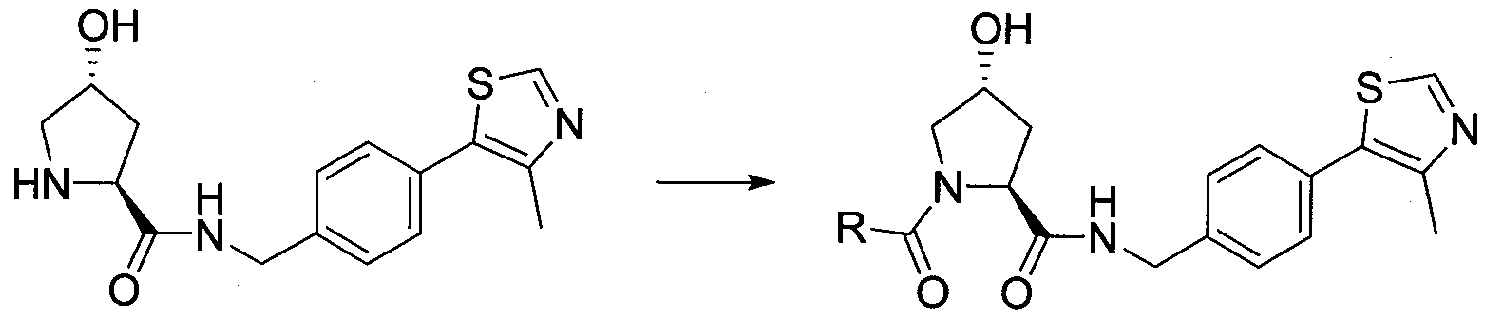

Method F subsumes methods C, D and E and is a general method which proceeds through commercially available amines.
Following the general synthetic methods set forth above and as previously described, the following compounds are synthesized by analogy.

(2S,4R)-l-((9H-fluoren-9-yl)methyl) 2-allyl 4-(tert-butoxy)pyrrolidine-l ,2-dicarboxylate (Fmoc-Hyp(OtBu)-OAUyl)

Fmoc-Hyp(OtBu)OH (24.9 g, 60.8 mmol, 1 eq) was dissolved in DMF (300 mL) at room temperature. Sodium bicarbonate (12.8 g, 152 mmol, 2.5 eq) was added, followed by allyl bromide (25.3 mL, 300 mmol, 4.9 eq). The solution was fitted with an air condenser and heated to 50 °C for 20 hours. It was then cooled to room temperature, diluted with EtOAc, washed with aqueous 1 M HCl, saturated sodium bicarbonate, water and brine. The organic layer was dried with sodium sulfate, filtered and condensed.15 Purification by column chromatography (15 to 33% EtOAc/hexanes) gave Fmoc-Hyp(OtBu)OAllyl (23.42 g, 52.1 mmol, 86%) as a faint yellow oil. !H NMR (500 MHz, CDC13) δ 7.76 (t, J= 6.3 Hz, 2H),
7.63 - 7.54 (m, 2H), 7.43 - 7.37 (m, 2H), 7.31 (t, J= 7.0 Hz, 2H), 5.99 - 5.79 (m, 1H), 5.39 - 5.18 (m, 2H), 4.66 (d, J= 5.6 Hz, 1H), 4.63 - 4.13 (m, 6H), 3.81 (ddd, J= 16.6, 10.7, 6.2 Hz, 1H), 3.48 - 3.33 (m, 1H), 2.31 - 2.18 (m, 1H), 2.18 - 2.08 (m, 1H), 1.21 (d, J= 11.6 Hz, 9H). 13C NMR (126 MHz, CDC13) (mixture of rotamers) δ 172.49, 155.01, 154.49, 144.31, 144.18, 144.06, 143.84, 141.44, 141.41, 141.36, 131.91, 131.74, 127.80, 127.76, 127.20, 127.16, 125.31, 125.28, 125.11, 120.08, 120.05, 118.93, 118.61, 74.29, 69.37, 68.48, 67.73, 65.86, 58.09, 57.79, 54.01, 53.52, 47.40, 47.28, 38.90, 37.87, 28.41, 28.37. MS (ESI) 450.5 (M+H).
(2S,4R)-l-((9H-fluoren-9-yl)methyI) 2-alIyI 4-hydroxypyrrolidine-l,2-dicarboxyIate (Fmoc-Hyp(OH)-OAHyl)

Fmoc-Hyp(OtBu)-OAllyl (23.42 g, 52.1 mmol) was dissolved in DCM (306 mL) at room temperature. TFA (54 mL, 15% vol/vol) was added and the solution was stirred for 13 hours. The solution was poured into water, neutralized by slow addition of saturated aqueous sodium bicarbonate and extracted twice with DCM and once with EtOAc. The combined organic layers were dried with sodium sulfate, filtered and condensed. Purification by column chromatography (30 to 80% EtOAc/hexanes) gave Fmoc-Hyp(OH)-OAllyl as a yellowish oil (16.7 g, 42.4 mmol, 81%). !H and 13C NMR spectra matched those reported in the
literature.16

Fmoc-Hyp(OWang)-OAllyl
 Wang Resin ( 12.1 g, 1.1 mmol/g loading, 13.3 mmol, 1 eq) was swelled with DCM (90 mL) in a glass reaction vessel and cooled to 4°C. Trichloroacetonitirle (20 mL, 200 mmol, 15 eq) was added, followed by the addition of DBU (3 mL, 20 mmol, 1.5 eq) in 3 portions over 3 minutes, manually shaking the reaction vessel in between additions. The reaction vessel was nutated at 4°C for 1 hour, then washed with DCM, DMSO, THF, then twice with DCM at room temperature.17 A solution of Fmoc- Hyp(OH)-OAllyl (26.15 g, 66.5 mmol, 5 eq) in DCM (40 mL) and THF (40 mL) was then added, and shaken for 30 minutes and then washed twice with DCM, thrice with DCM and then twice with MeOH followed by DCM. The initial DCM washes were condensed, and purified by column chromatography (33% to 80% EtOAc) to recover the Fmoc-Hyp(OH)- OAllyl starting material (21.51 g, 54.67 mmol, 82%). The resin was dried in air, then dried under vacuum to give 15.5 g of Fmoc-Hyp(OWang)-OAllyl. The loading of the resin was estimated to be 0.53 mmol/g based upon the increase in mass.
Wang Resin ( 12.1 g, 1.1 mmol/g loading, 13.3 mmol, 1 eq) was swelled with DCM (90 mL) in a glass reaction vessel and cooled to 4°C. Trichloroacetonitirle (20 mL, 200 mmol, 15 eq) was added, followed by the addition of DBU (3 mL, 20 mmol, 1.5 eq) in 3 portions over 3 minutes, manually shaking the reaction vessel in between additions. The reaction vessel was nutated at 4°C for 1 hour, then washed with DCM, DMSO, THF, then twice with DCM at room temperature.17 A solution of Fmoc- Hyp(OH)-OAllyl (26.15 g, 66.5 mmol, 5 eq) in DCM (40 mL) and THF (40 mL) was then added, and shaken for 30 minutes and then washed twice with DCM, thrice with DCM and then twice with MeOH followed by DCM. The initial DCM washes were condensed, and purified by column chromatography (33% to 80% EtOAc) to recover the Fmoc-Hyp(OH)- OAllyl starting material (21.51 g, 54.67 mmol, 82%). The resin was dried in air, then dried under vacuum to give 15.5 g of Fmoc-Hyp(OWang)-OAllyl. The loading of the resin was estimated to be 0.53 mmol/g based upon the increase in mass.

Solid Phase Synthesis General Method A
Fmoc-Hyp(OWang)-OAUyl resin (1 eq) was swelled DMF, then reacted with 20% piperidine in DMF for 30 minutes. The resin was then washed once with piperidine, and
reacted again with 20% piperidine for 30 minutes to ensure complete deprotection. The resin was then washed twice with DMF and once with MeOH followed by DCM. The resulting free amine was then coupled with 3-methyl-5-isoxazoleacetic acid (4 eq), PyBOP (4 eq) HOBt (4 eq) and DIPEA (7 eq) in DMF for 4 hours. The resin was then washed thrice with DMF and twice with MeOH followed by DCM. The resin was then swelled with freshly distilled DCM, and reacted with Pd(PPh3)4 (0.1 eq) and PhSiH3 (10 eq) for 30 minutes. The resin was then washed once with DCM, and reacted again with Pd(PPh3)4 (0.1 eq) and PhSiH3 (10 eq) in distilled DCM for 30 minutes, after which the resin was washed twice with DMF and once with MeOH followed by DCM. The resulting carboxylic acid was then coupled with the appropriate amine (or a salt of the appropriate amine), R H2 (4 eq) with PyBOP (4 eq), HOBt (4 eq) and DIPEA (7 eq for free amines, 8 eq for amine salts) in DMF for 4 hours. The resin was then washed 5 times with DMF, thrice with MeOH and 5 times with DCM. The resin was then reacted with 20% TFA in DCM for 2 hours. The reaction mixture was then drained and the resin was washed with DCM. Condensation under reduced pressure, and purification by column chromatography (1% to 10% 0.5M NH3 in MeOH/DCM or 1% to 10% MeOH in DCM) gave the desired VHL ligand.

VHL Ligand
Solid Phase Synthesis General Method B
Briefly, Fmoc-Hyp-(OWang)-OAllyl resin (1 eq) was swelled with freshly distilled DCM, and reacted with Pd(PPh3)4 (0.1 eq) and PhSiH3 (10 eq) for 30 minutes. The resin was
then washed once with DCM, and reacted again with Pd(PPh3)4 (0.1 eq) and PhSiH3 (10 eq) in distilled DCM for 30 minutes, after which the resin was washed twice with DMF and once with MeOH followed by DCM. The resulting carboxylic acid was then coupled with 4- chlorobenzylamine (4 eq), PyBOP (4 eq), HOBt (4 eq) and DIPEA (7 eq) in DMF for 4 hours. The resin was then reacted with 20% piperidine in DMF for 30 minutes. The resin was then washed once with DMF, and reacted again with 20% piperidine for 30 minutes to ensure complete deprotection. The resin was then coupled with the appropriate carboxylic acid (RC02H, 4 eq), PyBOP (4 eq), HOBt (4 eq) and DIPEA (7 eq) in DMF for 4 hours. The resin was then washed 4 times with DMF and twice with methanol followed by DCM. The resin was then reacted with 20% TFA in DCM for 2 hours. The reaction mixture was then drained and the resin was washed with DCM. Condensation under reduced pressure, and purification by column chromatography (1% to 10% 0.5M NH3 in MeOH DCM or 1% to 10% MeOH in DCM) gave the desired VHL ligand. Yields are based upon the loading of the resin, which was estimated based upon its change in mass.


Boc-Amb-OH (2.55 g, 10.16 mmol, 1 eq) was dissolved in DCM (68 mL) and cooled to 4 °C in an ice bath. EDC (2.34 g, 12.2 mmol, 1.2 eq), HOBt (1.65 g, 12.2 mmol, 1.2 eq) and DIPEA (6.2 mL, 35.6 mmol, 3.5 eq) were added. The solution was stirred for 30 minutes and
then Ν,Ο-Dimethylhydroxylamine hydrochloride (1.09 g, 11.2 mmol, 1.1 eq) was added. The solution warmed slowly to room temperature and after 21 hours was poured into brine, with a small amount of chloroform to break the resulting emulsion. After separation, the aqueous layer was extracted twice with EtOAc. The combined organic layer was dried over sodium sulfate, filtered and condensed. Purification by column chromatography (40 to 75%
EtOAc/hexanes) gave a colorless oil (2.45 g, 8.33 mmol, 82%). XH NMR (500 MHz, CDC13) 8 7.65 (d, J= 8.2 Hz, 2H), 7.31 (d, J= 8.1 Hz, 2H), 4.88 (s, 1H), 4.36 (d, J= 5.1 Hz, 2H), 3.55 (s, 3H), 3.35 (d, J= 4.7 Hz, 3H), 1.47 (s, 9H). 13C NMR (126 MHz, CDC13) δ 169.77, 156.04, 141.79, 133.21, 128.76, 127.03, 79.87, 61.20, 44.50, 33.88, 28.55. MS (ESI) 295.2 (M+H). tert- ut l 4-formylbenzyIcarbamate (Boc-Amb-H)

Boc-Amb-N(OMe)Me (2.45 g, 8.33 mmol, 1 eq) was dissolved in THF (83 mL) and cooled to -78 °C in a dry ice/acetone bath. Lithium aluminum hydride (0.41 g, 10.83 mmol, 1.3 eq) was added in 2 portions over 5 minutes. After 50 minutes, the suspension was warmed to 4 °C in an ice bath. After 3.5 hours, the reaction was deemed complete by TLC (mini workup in 10% potassium bisulfate and EtOAc, 50% EtOAc hexanes) and the reaction was quenched by the slow addition of 10% potassium bisulfate at 4 °C. The mixture was warmed to room temperature, and stirred for 30 minutes. Most of the THF was removed under reduced pressure and mixture was diluted with water and extracted thrice with EtOAc. The combined organic layer was washed once with brine, dried over sodium sulfate, filtered and condensed. Purification by column chromatography (40 to 50% EtOAc/hexanes) gave Boc-Amb-H as a white solid (1.66 g, 7.1 mmol, 85%). 1H NMR (500 MHz, CDC13) δ 9.96 (s, 1H), 7.81 (d, j = 8.2 Hz, 2H), 7.41 (d, J= 8.0 Hz, 2H), 5.12 (s, 1H), 4.37 (d, J= 5.6 Hz, 2H), 1.44 (s, 9H). 13C NMR (126 MHz, CDC13) 5 191.94, 156.03, 146.30, 135.62, 130.14, 127.78, 79.92, 44.44, 28.46. MS (ESI) 235.9 (M+H), 180.2 (M-tBu). tert-Butyl 4-(oxazol-5-yl)benzylcarbamate

Potassium carbonate (0.13 g, 0.94 mmol, 1.2 eq) and toluenesulfonylmethyl isocyanide (0.184 g, 0.94 mmol, 1.2 eq) were added to MeOH (7.8 mL) at room temperature. The round bottom was fitted with a reflux condenser and heated to 45 °C. After 15 minutes, Boc-Amb- H (0.1835 g, 0.78 mmol, 1 eq) was added and the mixture was heated to 75 °C for 3 hours and then cooled to room temperature. The MeOH was removed under reduced pressure and the crude material was resuspended in EtOAc and 1 :2 mixture of saturated sodium carbonate to water and separated. The aqueous layer was then extracted once with EtOAc. The combined organic layer was dried over sodium sulfate, filtered and condensed. Purification by column chromatography (20 to 35% EtOAc/hexanes) gave a white solid.Ή NMR (400 MHz, CDC13) δ 7.91 (s, 1H), 7.62 (d, J= 8.3 Hz, 2H), 7.35 (ob d, 2H), 7.34 (ob s, 1), 4.88 (s, 1H), 1.47 (s, 9H). 13C NMR (126 MHz, CDC13) 6 156.02, 151.40, 150.47, 139.78, 128.01 , 126.84, 124.67, 121.47, 79.68, 44.38, 28.47. MS (ESI) 275.5 (M+H).
(4-(Oxazol-5-yl)phenyl)methanamine trifluoroacetate salt

(2S,4R)-N-(3-chIorobenzyl)-4-hydroxy-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2- carboxamide (VL4)
 was synthesized according to General Method F as a white solid. 1H NMR (500MHz, MeOD):88.684 (1H, s); 7.33-7.23 (4H, m); 6.24 (1H, s); 4.56-4.53 (1H, t, J= 8 Hz); 4.51-4.50 (1H, m); 4.39-4.37 (2H, m); 3.96-3.92 (2H, m); 3.81- 3.3.78 (1H, dd, J= 9 Hz, 4 Hz); 3.64-3.62 (1H, m); 2.28-2.24 (4H, m); 2.09-2.04 (1H, m).13C NMR (125MHz, MeOD):8174.56, 168.67, 167.68, 161.58, 142.25, 135.35, 131.04, 128.43, 128.19, 126.76, 105.37, 70.86, 60.78, 56.96, 43.60, 39.33, 33.90, 11.21. MS (ESI) 378.2
was synthesized according to General Method F as a white solid. 1H NMR (500MHz, MeOD):88.684 (1H, s); 7.33-7.23 (4H, m); 6.24 (1H, s); 4.56-4.53 (1H, t, J= 8 Hz); 4.51-4.50 (1H, m); 4.39-4.37 (2H, m); 3.96-3.92 (2H, m); 3.81- 3.3.78 (1H, dd, J= 9 Hz, 4 Hz); 3.64-3.62 (1H, m); 2.28-2.24 (4H, m); 2.09-2.04 (1H, m).13C NMR (125MHz, MeOD):8174.56, 168.67, 167.68, 161.58, 142.25, 135.35, 131.04, 128.43, 128.19, 126.76, 105.37, 70.86, 60.78, 56.96, 43.60, 39.33, 33.90, 11.21. MS (ESI) 378.2
(M+H).
(2S,4R)-4-hydroxy-N-(4-hydroxyphenethyl)-l-(2-(3-methylisoxazoI-5- yl)acetyl)pyrrolidine-2-carboxamide (VL2) H

VL2 was synthesized according to General Method F. 1H NMR (500 MHz, MeOD) d 8.33 & 8.13 (due to the rotamers, both s, 1H), 7.02 (d, J= 8.3 Hz, 2H), 6.69 & 6.65 (due to the rotamers, both d, J= 8.3 Hz, 2H), 6.22 & 6.10 (due to the rotamers, both s, 1H), 4.43 (d, J= 7.7 Hz, 2H), 3.89 (d, J= 4.7 Hz, 2H), 3.74 (dd, J= 11.0, 4.3 Hz, 1H), 3.57 (d, J= 11.0 Hz, 1H), 3.42-3.36 (m, 2H), 2.73-2.63 (m, 2H), 2.25 (s, 3H), 2.16-2.12 (m, 1H), 1.96-1.91 (m, 1H). °C NMR (asterisk denotes the signals of the minor rotamer, 125 MHz, MeOD) d 174.2, 174.1, *173.9, *173.8, *169.0, 168.6, 167.6, *167.4, 161.6, * 161.5, * 157.0, 156.9, *131.2, 130.8, *116.2, 116.1, *105.6, 105.4, 70.7, *69.2, *61.0, *60.9, 60.7, 60.6, 56.9, *56.2, 42.4, 42.3, *42.0, *41.9, 41.5, 39.3, 35.5, *35.3, 33.9, *32.9, 11.2. MS (ESI) [M+H] 374.1,
[2M+Na] 769.6.
(2S,4R)-4-hydroxy-N-methyl-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2- carboxamide (VL26)

1H). 13C NMR (126 MHz, MeOD) δ 174.82, 168.66, 167.67, 161.58, 105.40, 70.79, 60.67, 56.91, 39.30, 33.89, 26.35, 11.20. MS (ESI) 291.1 (M+Na), 268.7 (M+H).
VL34

VL28

VL21

VL21 was synthesized from Fmoc-Hyp(OWang)-OAllyl (0.2 mmol) using Solid Phase Synthesis General Method A. It was isolated as a white solid (15.9 mg). 1H NMR (500 MHz, MeOD) δ 8.65 (s, 1H), 7.32 - 7.26 (m, 4H), 6.22 (s, 1H), 4.58 - 4.47 (m, 2H), 4.43 - 4.32 (m, 2H), 3.92 (d, J= 4.2 Hz, 2H), 3.80 (dd, J= 10.9, 4.3 Hz, 1H), 3.66 - 3.58 (m, 1H), 2.30 - 2.22 (m, 4H), 2.08 (dd, J= 8.3, 4.7 Hz, 1H). 13C NMR (126 MHz, MeOD) δ 174.48, 168.71, 167.66, 161.60, 138.67, 133.85, 129.99, 129.53, 105.37, 70.85, 60.80, 56.99, 43.45, 39.33, 33.95, 11.20. MS (ESI) 378.4 (M+H).
VL20
(0.15 mmol) using Solid Phase Synthesis General Method A. It was isolated as a white solid (9.9 mg). 1H NMR (400 MHz, MeOD) δ 8.64 (t, J= 5.6 Hz, 1H), 7.37 - 7.26 (m, 2H), 7.07 - 6.99 (m, 2H), 6.23 (s, 1H), 4.57 - 4.47 (m, 2H), 4.37 (dd, J= 8.4, 5.8 Hz, 2H), 3.93 (d, J = 3.0 Hz, 2H), 3.80 (dd, J= 11.0, 4.2 Hz, 1H), 3.66 - 3.58 (m, 1H), 2.28 - 2.22 (m, 4H), 2.08
(dd, 7= 8.3, 4.7 Hz, 1H). 1 C NMR (126 MHz, MeOD) δ 174.31, 168.69, 167.67, 163.44 (d, J= 243.5 Hz), 161.60, 135.78, 130.27 (d, J= 8.1 Hz), 116.09 (d, J= 21.6 Hz), 105.37, 70.85, 60.75, 56.99, 43.32, 39.33, 33.95, 1 1.20. MS (ESI) 362.3 (M+H).
VL29

(0.3 mmol) according to Solid Phase Synthesis General Method A. It was isolated as a light yellow solid (16.4 mg). 1H NMR (400 MHz, MeOD) δ 7.45 (dq, J= 9.0, 2.2 Hz, 2H), 7.23 (d, J= 8.5 Hz, 2H), 6.22 (s, 1H), 4.58 - 4.47 (m, 2H), 4.35 (dt, J= 18.9, 15.4 Hz, 2H), 3.92 (d, J = 2.6 Hz, 2H), 3.80 (dd, J= 10.9, 4.2 Hz, 1H), 3.63 (d, J= 11.0 Hz, 1H), 2.30 - 2.21 (m, 4H), 2.11 - 2.02 (m, 1H). 13C NMR (101 MHz, MeOD) δ 174.41, 168.71, 167.66, 161.61, 139.15, 132.55, 130.32, 121.77, 105.37, 70.85, 60.75, 56.99, 43.37, 39.33, 33.94, 11.22. MS (ESI) 424.1 (M+H).
VL31

VL47

(0.156 mmol) according to Solid Phase Synthesis General Method A. It was isolated as a white solid (9.1 mg). lH NMR (500 MHz, MeOD) δ 7.22 (dd, J= 8.4, 3.9 Hz, 2H), 6.86 (dd, J= 8.8, 2.2 Hz, 2H), 6.22 (s, 1H), 4.63 - 4.45 (m, 2H), 4.37 - 4.26 (m, 2H), 3.92 (d, J= 2.6 Hz, 2H), 3.83 - 3.70 (m, 4H), 3.61 (d, J= 11.2 Hz, 1H), 2.28 - 2.20 (m, 4H), 2.06 (ddd, J = 13.0, 8.1, 4.7 Hz, 1H). 13C NMR (126 MHz, MeOD) δ 174.13, 168.66, 167.68, 161.60, 160.39, 131.67, 129.76, 114.89, 105.37, 70.83, 60.74, 56.97, 55.67, 43.57, 39.34, 33.95, 11.21. MS (ESI) 374.5 (M+H).
VL35
OH

GAllyl (0.156 mmol) according to Solid Phase Synthesis General Method A. It was isolated as a white solid (14.1 mg). !H NMR (500 MHz, DMSO) δ 7.90 - 7.85 (m, 2H), 7.39 (d, J = 8.4 Hz, 2H), 6.23 (s, 1H), 5.17 (s, 1H), 4.37 (dd, J = 17.9, 10.4 Hz, 4H), 3.88 (s, 2H), 3.84 (s, 3H), 3.70 (dd, J= 10.5, 4.6 Hz, 1H), 3.47 (dd, J= 10.4, 2.5 Hz, 1H), 2.20 (d, J= 10.2 Hz, 3H), 2.11 - 2.03 (m, 1H), 1.92 (ddd, J= 12.5, 7.2, 4.9 Hz, 1H). 13C NMR (126 MHz, DMSO) 5 171.66, 166.66, 166.10, 165.66, 159.35, 145.22, 129.08, 127.99, 127.06, 103.94, 68.62, 58.76, 55.20, 52.02, 41.49, 38.17, 32.73, 10.95.MS (ESI) 402.6 (M+H).
VL48

(0.156 mmol) according to Solid Phase Synthesis General Method A. It was isolated as a white solid (11.4 mg). 1H NMR (400 MHz, MeOD) δ 8.28 - 8.05 (m, 2H), 7.55 (d, J= 8.8 Hz, 2H), 6.23 (s, 1H), 4.64 - 4.36 (m, 4H), 3.94 (d, J= 3.8 Hz, 2H), 3.81 (dd, J= 10.9, 4.2 Hz, 1H), 3.65 (dt, J= 11.0, 1.7 Hz, 1H), 2.34 - 2.21 (m, 4H), 2.09 (td, J= 8.5, 4.2 Hz, 1H) 13C MR (101 MHz, MeOD) δ 174.70, 168.79, 167.65, 161.63, 148.48, 147.72, 129.13, 124.56, 105.40, 70.88, 60.79, 57.03, 43.41, 39.32, 33.94, 11.20. MS (ESI) 389.3 (M+H), 411.4 (M+Na).
VL88

(0.156 mmol) according to Solid Phase Synthesis General Method A. It was isolated as a clear oil (8.0 mg). 1H NMR (500 MHz, MeOD) δ 8.77 (s, 1H), 7.58 (dd, J= 88.0, 8.1 Hz, 4H), 6.23 (d, J= 4.4 Hz, 1H), 4.61 - 4.33 (m, 4H), 3.93 (d, J= 9.7 Hz, 2H), 3.83 - 3.74 (m, 1H), 3.63 (dd, J= 10.4, 9.0 Hz, 1H), 2.33 - 2.27 (m, 1H), 2.26 (d, J= 3.7 Hz, 3H), 2.15 - 2.03 (m, 1H). 13C NMR (126 MHz, MeOD) δ 175.33, 168.76, 167.64, 161.61, 145.83, 133.39, 129.12, 119.74, 111.79, 105.28, 70.87, 70.87, 59.32, 57.02, 43.61, 38.79, 33.94, 11.21, 11.19. MS (ESI) 391.2 (M+Na).
VL95

(0.156 mmol) according to Solid Phase Synthesis General Method A. It was isolated as a white solid (23 mg). 1H NMR (400 MHz, MeOD) δ 8.71 (s, 1H), 7.96 - 7.91 (m, 2H), 7.43 (d, J= 8.5 Hz, 2H), 6.22 (s, 1H), 4.56 (t, J= 8.0 Hz, 1H), 4.49 (ddd, J= 18.6, 8.6, 4.1 Hz, 3H), 3.91 (s, 2H), 3.80 (dd, J= 10.9, 4.2 Hz, 1H), 3.65 - 3.58 (m, 1H), 2.58 (d, J= 1.6 Hz, 3H), 2.28 - 2.22 (m, 4H), 2.10 (dd, J= 8.3, 4.7 Hz, 1H). 13C NMR (101 MHz, MeOD) δ 200.08, 174.34, 168.43, 167.36, 161.39, 145.50, 136.96, 129.62, 128.28, 105.26, 70.65, 60.57, 56.83, 43.72, 39.14, 33.87, 26.73, 11.30. MS (ESI) 386.0 (M+H).
VL111

VL116 Right Hand Fragment (Representative Method B Synthesis)
HC!"H9N
 Pd{PtBti3!2
Pd{PtBti3!2
NBu4OH20
Cs¾C03. DMF

2-(trimethylsilyl)ethyl 4-bromobenzylcarbamate
TeocHN

4-Bromobenzylamine hydrochloride (354 mg, 1.59 mmol, 1 eq) was dissolved in DMF (6.4 mL) and water (2.1 mL) and stirred at room temperature. Triethylamine (0.33 mL, 2.39 mmol, 1.5 eq) and TeocOSu (454 mg, 1.75 mmol, 1.1 eq) were then added. After 12 hours, the mixture was diluted with EtOAc, washed with 1M HC1, saturated sodium
bicarbonate, water and brine. The organic layer was then dried over sodium sulfate, filtered, and concentrated under reduced pressure. Purification by column chromatography (10 to
20% EtOAc hexanes) gave a colorless oil (0.4158 g, 1.26 mmol, 79%). 1H NMR (500 MHz, CDC13) δ 7.48 - 7.43 (m, 2H), 7.17 (d, J = 8.1 Hz, 2H), 4.94 (s, 1H), 4.31 (d, J= 6.0 Hz, 2H), 4.23 - 4.15 (m, 2H), 1.04 - 0.93 (m, 2H), 0.04 (s, 9H). 13C NMR (126 MHz, CDCI3) δ 156.91, 137.95, 131.88, 129.32, 121.43, 63.53, 44.53, 17.92, -1.32. MS (ESI) 354.1 (M+H).
2-(trimethylsilyl)ethyl 4-(4-methylthiazol-5-yl)benzylcarbamate

- tr met y s y et y 4-bromobenzylcarbamate (132 mg, 0.4 mmol, 1 eq), 4- methylthiazole-5-carboxylic acid (114.5 mg, 0.8 mmol, 2 eq), tetrabutylammonium chloride hydrate (118 mg, 0.4 mmol, 1 eq), cesium carbonate (196 mg, 0.6 mmol, 1.5 eq) and
Pd(P(tBu)3)2 (40.8 mg, 0.08 mmol, 0.2 eq) were dissolved in DMF (4 mL).1 The reaction was heated to 170°C in a microwave reactor for 16 minutes. The mixture was then cooled to room temperature, diluted with EtOAc and washed thrice with brine, once with saturated sodium bicarbonate, water, and then brine. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. Purification by coulm chromatography (10 to 35% EtOAc/hexanes) gave a colorless oil (61.7 mg, 0.177 mmol, 44%). Ή NMR (400
MHz, CDCI3) δ 8.67 (s, 1H), 7.43 - 7.37 (m, 2H), 7.34 (d, J= 8.1 Hz, 2H), 5.09 (s, 1H), 4.39 (d, J= 6.0 Hz, 2H), 4.28 - 4.02 (m, 2H), 2.52 (s, 3H), 1.10 - 0.90 (m, 2H), 0.14 - -0.09 (m, 9H). 13C NMR (101 MHz, CDC13) δ 156.98, 150.42, 148.66, 138.76, 131.67, 131.18, 129.66, 127.89, 63.46, 44.71, 17.90, 16.18, -1.34. MS (ESI) 349.0 (M+H).
(4-(4-methylthiazol-5-yl)phenyl)methanamine

2-(trimethylsilyl)ethyl 4-(4-methylthiazol-5-yl)benzylcarbamate (51.8 mg, 0.149 mmol, 1 eq) was dissolvd in acetonittile (6 mL) at room temperature. A one molar solution of
tetrabutylammonium fluoride in THF (0.45 mL, 0.45 mmol, 3 eq) was added and the solution was stirred for 24 hours. The mixture was concentrated under reduced pressure. Purification by column chromatography (0.5 to 4% 0.5N N¾ (MeOH)/DCM) gave a light yellow oil
(27.2 mg, 0.133 mmol, 89%). Ή NMR (500 MHz, MeOD) δ 8.87 (s, 1H), 7.44 (s, 4H), 3.85 (s, 2H), 2.47 (s, 3H). I3C NMR (126 MHz, MeOD) δ 152.77, 149.07, 143.63, 133.42, 131.46, 130.49, 129.05, 46.23, 15.79. MS (ESI) 205.0 (M+H).
Alternate Route:
4-bromobenzonitrile (5.1 g, 28 mmol, 1 eq), 4-methylthiazole (5.56 g, 56 mmol, 2 eq) potassium acetate (5.5 g, 56 mmol, 2 eq), palladium (II) acetate (63 mg, 0.28 mmol, 1 mol %) were dissolved in dimethylacetamide and stirred under argon. (CITE JOC, 2009, 74, 1 179) The mixture was heated to 150 °C and stirred for 19 hours, then diluted with 500 mL EtOAc, and washed 4 times with 300 mL water. The first wash was then back extracted with 300 mL EtOAc, and then washed 4 times with 100 mL water. The combined organic layer was dried over sodium sulfate, filtered and concentrated under vacuum to give a beige solid (5.55 g, 27.7 mmol, 99%) that matched the reported spectral data.[S] The solid was then dissolved in MeOH (280 mL) and cooled to 4 °C. Cobalt chloride (9.9 g, 41.6 mmol, 1.5 eq) was added, followed by the slow, portionwise addition of sodium borohydride (5.2 g, 139 mmol, 5 eq), which was accompanied by vigorous bubbling. After 90 minutes, the reaction was quenched by the addition of water and ammonium hydroxide. The mixture was extracted 4 times with chloroform, and purified by column chromatography (10 to 30% 0.5M NH3 (MeOH)/DCM) to give a darker oil (4.12 g, 20.2 mmol, 73%»).
General Solution Phase Synthesis
(2»y,4R)-allyl 4-(feA,i-butoxy)pyrrolidine-2-carboxylate

(2S,4R)-l-((9H-fluoren-9-yl)methyl) 2-allyl 4-(tert-butoxy)pyrrolidine-l,2-dicarboxylate (7.0 g, 15.57 mmol, 1 eq) was dissolved in DCM (156 mL) and cooled to 4°C. Tris(2- aminoethyl)amine (5.8 mL, 38.9 mmol, 2.5 eq) was added, and the solution was stirred for 1 hour at 4°C and 4.5 hours at room temperature. The mixture was then mixed with silica gel (roughly 20 g), and concentrated under reduced pressure, and purified by column
chromatography (1 to 5% 0.5N NH3 (MeOH) DCM) to give an opaque oil (3.44 g, 15.1
mmol, 97%). *H NMR (400 MHz, MeOH) δ 6.03 - 5.88 (m, 1H), 5.25 (dq, J= 17.2, 1.8 Hz, 1H), 5.09 (dq, J = 10.5, 1.6 Hz, 1H), 4.33 - 4.23 (m, 1H), 4.06 (dt, J= 5.1, 1.6 Hz, 2H), 3.86 (t, J= 8.0 Hz, 1H), 3.18 (dd, J= 11.4, 5.7 Hz, 1H), 2.70 (dd, J= 11.4, 3.8 Hz, 1H), 2.00 (dd, = 8.0, 5.0 Hz, 2H), 1.19 (s, 9H). 13C NMR (101 MHz, MeOH) δ 175.89, 138.93, 114.88, 74.93, 72.93, 63.97, 59.77, 55.44, 40.18, 28.65. MS (ESI) 228.0 (M+H).
(2S,4R)-aIlyI 4-(tert-butoxy)-l-(2-(3-methylisoxazol-5-yl)acetyI)pyrroIidine-2- carboxylate

(2S,4R)-allyl 4-(tert-butoxy)pyrrolidine-2-carboxylate ( 0.148 g, 0.65 mmol, 1 eq) was dissolved in DMF (6.5 mL) and cooled to 4°C. 2-(3-methylisoxazol-5-yl)acetic acid (0.12 g, 0.85 mmol, 1.3 eq), EDC (0.163 g, 0.85 mmol, 1.3 eq), HOBt (0.123 g, 0.91 mmol, 1.4 eq), and DIPEA (0.283 mL, 1.63 mmol, 2.5 eq) were added, and the solution was allowed to warm slowly to room temperature. After 12 hours, the mixture was poured into brine and extracted four times with EtOAc. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. Purification by column chromatography (1 to 3% MeOH/DCM) gave a light yellow oil (0.2008 g, 0.573 mmol, 88%). ¾ NMR (500 MHz, CDCla) δ 6.17 (s, 1H), 5.95 - 5.85 (m, 1H), 5.29 (ddd, J= 13.8, 1 1.7, 1.3 Hz, 2H), 4.69 - 4.55 (m, 3H), 4.40 - 4.32 (m, 1H), 3.84 - 3.75 (m, 3H), 3.37 (dd, J= 10.0, 4.7 Hz, 1H), 2.27 (s, 3H), 2.15 (ddd, J= 18.5, 12.0, 5.9 Hz, 2H), 1.18 (s, 9H). °C NMR (126 MHz, CDC13) δ 171.87, 165.94, 165.63, 160.30, 131.84, 118.72, 104.04, 74.53, 69.55, 65.98, 57.96, 54.53, 37.31, 33.58, 28.35, 11.62. MS (ESI) 351.5 (M+H).
(2S,4R)-4-(tert-butoxy)-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylic acid

(2S,4R)-allyl 4-(tert-butoxy)-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylate (1.67 g, 4.77 mmol, 1 eq) was dissolved in THF (48 mL) at room temperature. Pd(PPb_3)4 (0.55 g, 0.48 mmol, 0.1 eq) and morpholine (4.2 mL, 48 mmol, 10 eq) were then added. After 35 minutes, the solution was concentrated under reduced pressure, redissolved in DCM, and washed four times with 1M HC1 (aq). The aqueous layer was then back extracted once with DCM. The combined organic layer was then dried over sodium sulfate, filtered and concentrated under reduced pressure. Purification by column chromatography (1 to 20% MeOH/DCM) gave a yellow solid (1.27 g, 4.1 mmol, 86%). H NMR (500 MHz, MeOH) δ 6.23 (s, 1H), 4.47 (t, J= 6.0 Hz, 2H), 3.94 - 3.80 (m, 3H), 3.48 (dd, J= 10.6, 3.8 Hz, 1H), 2.28 - 2.11 (m, 5H), 1.21 (s, 9H). 13C NMR (126 MHz, MeOH) δ 175.53, 168.41, 167.68, 161.59, 105.25, 75.57, 71.00, 59.36, 55.81, 38.49, 33.88, 28.48, 11.20. MS (ESI) 311.2 (M+H).
General Method B Representative Procedure (with hydroxyl group protection): VL116
(2S,4R)-4-(tert-butoxy)-l-(2-(3-methylisoxazol-5-yl)acetyl)-N-(4-(4-methylthiazol-5- yl)benz l)pyrrolidine-2-carboxamide

(2S,4R)-4-(tert-butoxy)-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylic acid (53.7 mg, 0.173 mmol, 1.3 eq), (4-(4-methylthiazol-5-yl)phenyl)methanamine (27.2 mg, 0.133 mmol, 1 eq), EDC (33.2 mg, 0.173 mmol, 1.3 eq), and HOBt (23.4 mg, 0.173 mmol, 1.3 eq) were dissolved in DMF (3.5 mL) at 4°C. DIPEA (0.07 mL, 0.4 mmol, 3 eq) was
added, and the solution was allowed to slowly warm to room tmeprature. After 19 hours, the mixture was poured into brine and extracted four times with EtOAc. The organic layer was dried with sodium sulfate, filtered and concentrated under reduced pressure. Purification by column chromatography (1 to 5% MeOH/DCM) gave a colorless oil (58.1 mg, 0.117 mmol,
88%). *H NMR (400 MHz, CDC13) δ 8.67 (s, 1H), 7.42 - 7.27 (m, 5H), 6.06 (s, 1H), 4.69 (dd, J= 8.4, 2.6 Hz, 1H), 4.59 - 4.35 (m, 3H), 3.82 - 3.71 (m, 3H), 3.34 (dd, J= 9.9, 6.3 Hz, 1H), 2.59 - 2.46 (m, 4H), 2.25 (s, 3H), 1.91 (dd, J= 8.2, 4.4 Hz, 1H), 1.25 - 1.14 (m, 9H). 13C NMR (101 MHz, CDC13) δ 170.70, 167.35, 165.30, 160.24, 150.42, 148.59, 138.09, 131.74, 131.05, 129.66, 127.85, 104.19, 74.48, 70.02, 59.12, 54.20, 43.25, 35.59, 33.49, 28.38, 16.19, 11.57. MS (ESI) 497.4 (M+H).
(2S,4R)-4-hydroxy-l-(2-(3-methylisoxazol-5-yl)acetyI)-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide (VL116)
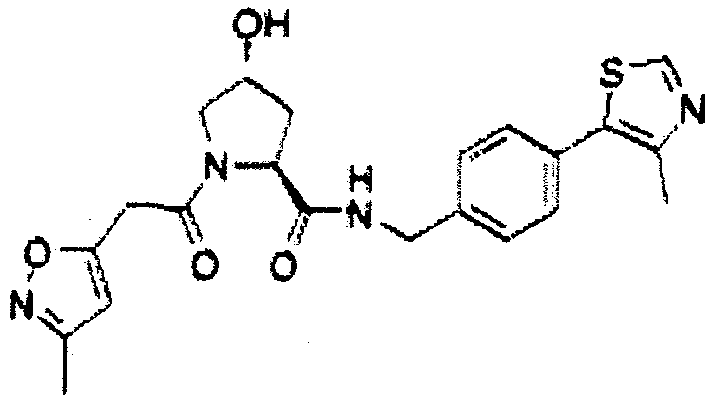
(2S,4R)-4-(tert-butoxy)-l-(2-(3-methylisoxazol-5-yl)acetyl)-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide (58.1 mg, 0.117 mmol) was dissolved in DCM (8 mL). TFA (2 mL, 20% vol/vol) was added and the solution was stirred for 12 hours at room temperature, after which it was concentrated under reduced pressure. Purification by column chromatography (1 to 10% 0.5N NH3 (MeOH)/DCM) gave a colorless oil (28.4 mg, 0.065 mmol, 56%). H NMR (400 MHz, MeOH) δ 8.87 (d, J= 2.1 Hz, 1H), 7.50 - 7.34 (m, 4H), 6.23 (s, 1H), 4.57 (t, J= 8.0 Hz, 1H), 4.54 - 4.38 (m, 3H), 3.93 (d, J= 2.4 Hz, 2H), 3.81 (dd, J= 10.9, 4.3 Hz, 1H), 3.63 (dd, J= 7.2, 5.5 Hz, 1H), 2.46 (d, J= 8.8 Hz, 3H), 2.33 - 2.20 (m, 4H), 2.10 (ddd, J= 13.1, 8.2, 4.7 Hz, 1H). 13C NMR (101 MHz, MeOH) δ 174.43, 168.71, 167.66, 161.58, 152.83, 149.04, 140.14, 133.39, 131.56, 130.43, 128.88, 105.39, 70.86, 60.78, 57.00, 43.65, 39.36, 33.96, 15.81, 11.22. MS (ESI) 441.3 (M+H).
General Method A Representative Procedure: VL133
(2S,4R)-4-hydroxy-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylic acid

(2S,4R)-4-(tert-butoxy)- 1 -(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylic acid (124.9 mg, 0.4 mmol,l eq) was dissolved in DCM (18 mL) at room temperature. TFA (2 mL, 10%) was added, and the solution was stirred for 12 hours. It was then concentrated under reduced pressure and purified by column chromatography (4 to 20% MeOH/DCM) to give a yellow oil (99.7 mg, 0.39 mmol, 98%). XH NMR (500 MHz, MeOD) δ 6.24 (s, 1H), 4.55 - 4.46 (m, 2H), 3.89 (d, J= 28.3 Hz, 2H), 3.77 (dd, J= 10.9, 4.3 Hz, 1H), 3.62 (d, J= 11.0 Hz, 1H), 2.36 - 2.22 (m, 4H), 2.10 (ddd, J= 13.1, 8.0, 4.8 Hz, 1H). 13C NMR (126 MHz, CDC13) δ 175.33, 168.51, 167.61, 161.61, 105.28, 70.86, 59.33, 56.60, 38.78, 33.85, 11.20. MS (ESI) 255.1 (M+H).
(2S,4R)-N-(4-(lH-pyrrol-3-yl)benzyl)-4-hydroxy-l-(2-(3-methylisoxazol-5- yl)acetyl)pyrrolidine-2-carboxamide (VL133)

(2S,4R)-4-hydroxy-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylic acid (52.6 mg, 0.207 mmol, 1.3 eq), (4-(lH-pyrrol-3-yl)phenyl)methanamine (27.3 mg, 0.159 mmol, 1 eq), EDC (39.7 mg, 0.207 mmol, 1.3 eq) and HOBt (28 mg, 0.207 mmol, 1.3 eq) were dissolved in DMF (4.1 mL) and cooled to 4°C. DIPEA (0.083 mL, 0.477 mmol, 3 eq) was added and the solution was allowed to slowly warm to room temperature. After 16 hours, the mixture was poured into half saturated sodium chloride (aqueous) and extracted 3 times with EtOAc. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. Purification by column chromatography (1 to 10% 0.5N NH3
(MeOH)/DCM) gave an off white solid (41.5 mg, 0.102 mmol, 64%). 1H NMR (400 MHz, DMSO) δ 8.40 (d, J= 6.0 Hz, 1H), 7.52 - 7.39 (m, 2H), 7.22 - 7.12 (m, 3H), 6.82 - 6.72 (m, 1H), 6.41 (d, J = 1.7 Hz, 1H), 6.24 (s, 1H), 5.17 (d, J= 3.9 Hz, 1H), 4.31 (ddd, J= 17.1 , 13.7, 6.4 Hz, 4H), 3.88 (s, 2H), 3.75 - 3.65 (m, 1H), 3.52 - 3.41 (m, 1H), 2.18 (d, J= 18.0 Hz, 3H), 2.12 - 1.99 (m, 1H), 1.94 - 1.85 (m, 1H). 13C NMR (101 MHz, DMSO) δ 171.36, 166.69, 165.54, 159.38, 135.66, 134.68, 127.20, 124.21, 123.00, 118.86, 114.71, 105.22, 103.99, 68.61, 58.76, 55.18, 41.63, 38.27, 32.78, 11.00. MS (ESI) 431.5 (M+Na).
For further reference see the following articles and the references cited therein:
(1) Buckley DL et al. J. Am. Chem. Soc 2012, 134, 4465-4468.
(2) Van Molle I et al. A Chemistry & Biology 2012, 19, 1300-1312
(3) Buckley, Ό Angew. Chem. Int. Ed., 2012, 51, 11463-11467
(4) Buckley, D. Let al. Angew. Chem. 2012, 124, 11630-11634.
Examples Second Set
VL50

VL50 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.156 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a white solid (29.8 mg, 0.084 mmol, 54%). 1H NMR (400MHz, CD3OD):87.34-7.27 (m, 4H); 5.43-5.35 (m, 4H); 3.81-3.78 (dd, J=8 Hz, 4 Hz, 1H); 3.61-3.57 (m, 1H); 2.65-2.61 (m, 2H); 2.57-2.51 (m, 2H); 2.28-2.21 (m, 1H); 2.08-2.02 (m, 1H). 13C NMR (100MHz, CD3OD):6 177.53, 174.74, 173.76, 138.75, 133.76, 129.96, 129.49, 70.71, 60.55, 56.47, 43.25, 39.33, 30.97, 30.64. MS (ESI) 354.2 (M+H).
VL52

VL52 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.156 mmol) according to the Solid Phase Synthesis General Method B. It was isolated as a white solid (7.7 mg, 0.021 mmol, 14%). Ή NMR (500MHz, CD3OD):57.41 (d, J= 2 Hz, IH); 7.30 (s, 4H); 6.35-6.34 (dd, J= 3 Hz, 2 Hz, IH); 6.26-6.25 (d, J= 3 Hz, IH); 4.49-4.32 (m, 4H); 3.82-3.73 (m, 3H); 3.65-2.62 (m, IH); 2.23-2.22 (m, IH); 2.09-2.06 (m, 1H).13C NMR (125MHz, CD3OD):6174.54, 170.58, 149.67, 143.24, 138.68, 133.84, 129.98, 129.53, 111.50, 108.98, 70.88, 60.75, 56.95, 43.31, 39.24, 35.58. MS (ESI) 365.2 (M+H), 385.3 (M+Na).
VL73

VL73 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.18 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a clear oil (38.9 mg, 0.099 mmol, 55%).1H NMR (500 MHz, CD3OD ) δ 7.51 - 6.99 (m, 8H), 4.72 (t, J = 8.2, IH), 4.55 - 4.33 (m, 3H), 3.60 (dd, J = 3.7, 11.3, IH), 3.19 (dd, J = 1.5, 11.3, IH), 2.36 - 2.25 (m, IH), 2.21 - 2.03 (m, 1H).13C NMR (126 MHz, CD3OD) δ 174.03, 169.66, 138.62, 137.31, 133.87, 132.12, 130.92, 130.48, 129.98, 129.56, 129.16, 128.53, 70.64, 60.38, 43.39, 39.25, 24.21. MS (ESI) 395.3 (M+H).
VL64

VL64 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.156 mmol) according to the Solid Phase Synthesis General Method B. It was isolated as a white solid (27.5 mg, 0.077 mmol, 49%).1H NMR (500 MHz, CD3OD) δ 7.66 - 7.59 (m, 2H), 7.54 - 7.22 (m, 7H), 4.75 (t, J = 8.6, 1H), 4.55 - 4.33 (m, 3H), 3.85 (dd, J = 3.0, 11.5, 1H), 3.43 (d, J = 11.5, 1H), 2.38 - 2.26 (m, 1H), 2.14 - 2.05 (m, 1H).13C NMR (126 MHz, CD3OD) δ 174.72, 172.78, 138.73, 137.14, 133.83, 131.74, 129.93, 129.55, 129.49, 128.56, 71.04, 60.85, 59.80, 43.34, 39.28. MS (ESI) 359.1 (M+H).
VL69

VL69 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.156 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a white solid (26.1 mg, 0.62 mmol, 40%). 1H NMR (500 MHz, DMSO) δ 7.30 (dt, J = 8.2, 25.1, 4H), 7.20 (dd, J = 1.7, 8.3, 1H), 7.13 (d, J = 1.7, 1H), 7.01 (d, J = 8.4, 1H), 4.98 (s, 1H), 4.56 (t, J = 8.6, 1H), 4.29 (d, J = 2.6, 2H), 3.76 (dd, J = 15.5, 30.6, 7H), 3.36 (d, J = 11.1, lH), 2.14 (dd, J = 7.7, 12.8, 1H), 1.96 - 1.83 (m, 1H). 13C NMR (126 MHz, DMSO) δ 171.91, 168.82, 150.39, 148.08, 138.70, 131.10, 128.68, 128.09, 121.00, 111.44, 110.75, 99.56, 68.90, 59.33, 59.30, 58.68, 55.57, 41.17, 38.01. MS (ESI) 418.8 (M+H).
VL70
OH

VL70 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.156 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a colorless oil (31.1mg, 0.083 mmol, 53%)}Έί NMR (500 MHz, CD3OD) δ 7.38 - 7.17 (m, 6H), 6.85 - 6.73 (m, 2H), 4.76 (t, J = 8.5, 1H), 4.53 - 4.31 (m, 3H), 3.85 (dd, J = 3.2, 1 1.6, 1H), 3.37 (d, J = 11.6, 1H), 2.50 - 2.24 (m, 1H), 2.08 (ddd, J = 4.3, 9.1 , 13.3, 1H).13C NMR (126 MHz, CD3OD) δ 174.72, 172.14, 145.18, 138.67, 133.87, 132.18, 129.97, 129.56, 128.80, 122.39, 1 18.87, 1 17.94, 71.01 , 60.29, 58.54, 43.40, 39.40. MS (ESI) 374.5 (M+H).
VL71

VL71 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.156 mmol) according to the Solid Phase Synthesis General Method B. It was isolated as a colorless oil (31.1 mg, 0.080 mmol, 51%). 1H NMR (500 MHz, CD3OD) δ 7.40 - 7.32 (m, 4H), 7.24 (t, J = 7.6, 1H), 7.09 (d, J = 7.9, 1H), 7.03 (d, J = 7.1, 1H), 4.74 (t, J = 8.2, 1H), 4.59 - 4.33 (m, 3H), 3.54 (d, J = 11.0, 1H), 3.20 (d, J = 1 1.2, 1H), 2.35 (dd, J = 8.7, 12.4, 1H), 2.26 (s, 3H), 2.14 (dd, J = 4.3, 9.3, 1H). 13C NMR (126 MHz, CD3OD) δ 174.37, 172.44, 140.91, 139.29, 138.69, 133.84, 129.93, 129.55, 128.37, 124.30, 121.45, 120.64, 70.73, 60.1 1, 43.36, 39.44, 13.89. MS (ESI) 388.1, 390.3 (M+H).
VL72
OH
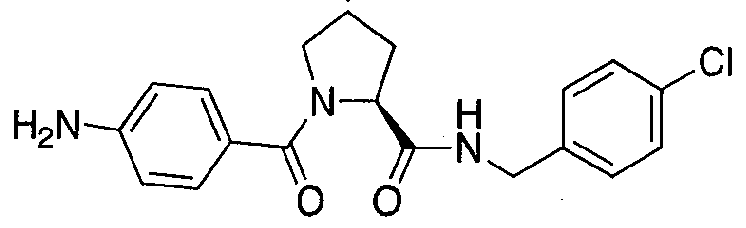
VL72 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.156 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a yellow oil (31.3 mg, 0.084 mmol, 54%). ¾ NMR (500 MHz, CD3OD) δ 7.48 (d, J = 8.3, 2H), 7.30 (s, 4H), 6.79 (d, J = 8.3, 2H), 4.79 - 4.69 (m, 1H), 4.53 - 4.29 (m, 3H), 3.95 - 3.83 (m, 1H), 3.54 (d, J = 1 1.4, 1H), 2.35 - 2.24 (m, 1H), 2.07 (ddd, J = 3.9, 10.1 , 13.5, 1H). I3C NMR (126 MHz, CD3OD) 5 175.03, 173.02, 149.95, 138.75, 133.80, 130.79, 129.91, 129.54, 126.23, 1 15.89, 71.16, 61.03, 60.12, 43.31, 39.16. MS (ESI) 375.0 (M+H).
VL74

VL74 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.18 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a white solid (36.3 mg, 0.092 mmol, 51%)}H NMR (500 MHz, CD3OD) δ 7.67 - 7.56 (m, 2H), 7.52 - 7.44 (m, 2H), 7.34 - 7.28 (m, 4H), 4.74 (dd, J = 7.7, 9.6, 1H), 4.55 - 4.30 (m, 3H), 3.85 (dd, J = 3.5, 1 1.4, 1H), 3.42 (d, J = 11.4, 1H), 2.37 - 2.28 (m, 1H), 2.15 - 2.05 (m, 1H). 13C NMR (126 MHz, CD3OD) δ 174.59, 171.54, 138.69, 137.75, 135.66, 133.84, 130.38, 129.92, 129.70, 129.55, 71.04, 60.92, 59.75, 43.34, 39.29. MS (ESI) 394.6 (M+H).
VL75
OH

VL75 was synthesized from Fmoc-Hyp(OWang)-OAUyl resin (0.18 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a white solid (25.0 mg, 0.066 mmol, 37%). 1H NMR (500 MHz, CD3OD) δ 7.64 - 6.87 (m, 8H), 4.73 (dd, J = 7.7, 9.6, 1H), 4.54 - 4.31 (m, 3H), 3.84 (dd, J = 3.5, 11.5, 1H), 3.42 (d, J = 11.4, lH), 2.33 (ddd, J = 1.6, 7.6, 13.0, 1H), 2.13 - 2.05 (m, 1H). 13C NMR (126 MHz, CD3OD) δ 174.64, 171.20, 163.83 (d, J = 246.5), 139.35, 138.72, 133.86, 131.60, 129.94, 129.56, 124.51, 118.51 (d, J=21.3), 115.56 (d, J=23.4), 71.02, 60.94, 59.70, 43.47, 39.31. MS (ESI) 377.4 (M+H).
VL76
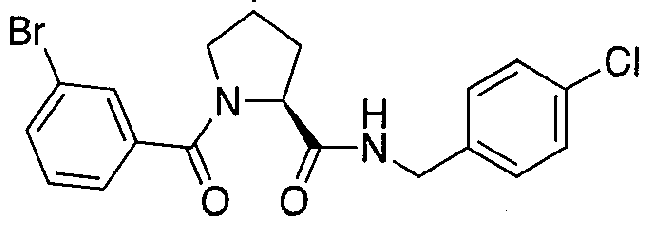
VL76 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.18 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a white solid (29.6 mg, 0.067 mmol, 38%). 1H NMR (400 MHz, CD3OD) δ 7.82 (s, 1H), 7.70 - 7.58 (m, 2H), 7.40 (t, J = 7.9, 1H), 7.36 - 7.18 (m, 4H), 4.73 (dd, J = 7.9, 9.4, 1H), 4.53 - 4.31 (m, 3H), 3.82 (dt, J = 5.2, 10.4, lH), 3.40 (d, J = 11.4, 1H), 2.33 (dd, J = 7.6, 13.2, 1H), 2.09 (ddd, J = 4.1, 9.7, 13.7, 1H). 13C NMR (101 MHz, CD3OD) δ 174.53, 170.95, 139.22, 138.68, 134.68, 133.85, 131.56, 131.44, 129.92, 129.56, 127.31, 123.35, 71.02, 60.90, 59.70, 43.33, 39.31. MS (ESI) 439.4 (M+H).
VL77
OH

VL77 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.18 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a white solid (31.0 mg, 0.081 mmol, 45%). JH NMR (500 MHz, DMSO) δ 8.07 (t, J = 1.4, 1H), 8.01 - 7.95 (m, 1H), 7.93 - 7.88 (m, 1H), 7.69 (t, J = 7.8, 1H), 7.40 - 7.23 (m, 4H), 4.56 (dd, J = 8.3, 16.4, 1H), 4.30 (dd, J = 8.1, 15.4, 3H), 3.79 (dd, J = 3.6, 11.0, 1H), 3.24 (d, J = 11.0, 1H), 2.23 - 2.15 (m, 1H), 1.92 (ddd, J = 4.2, 9.3, 13.2, 1H). 13C NMR (126 MHz, DMSO) 5 171.51, 167.29, 138.64, 137.27, 133.88, 132.29, 131.16, 131.08, 129.73, 128.68, 128.15, 118.25, 111.40, 68.82, 59.39, 59.36, 58.28, 38.19. MS (ESI) 383.8 (M+H).
VL79
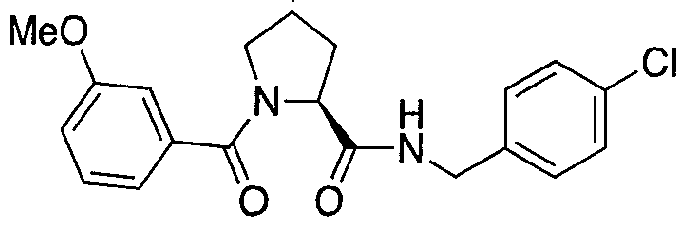
VL79 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.18 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a white solid (34.9 mg, 0.090 mmol, 50%). 1H NMR (500 MHz, CD3OD) δ 7.41 - 7.15 (m, 6H), 7.08 - 6.90 (m, 2H), 4.73 (dd, J = 7.7, 9.6, 1H), 4.54 - 4.31 (m, 3H), 3.87 - 3.74 (m, 4H), 3.43 (d, J = 11.5, 1H), 2.37 - 2.27 (m, 1H), 2.14 - 2.05 (m, 1H). 13C NMR (126 MHz, CD3OD) δ 174.72, 172.59, 161.07, 138.72, 138.40, 133.83, 130.67, 129.93, 129.56, 120.58, 117.52, 113.77, 71.01, 60.82, 59.80, 55.87, 43.33, 39.29. MS (ESI) 389.0 (M+H).
VL80
OH

VL80 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.18 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a white solid (41.2 mg, 0.1 10 mmol, 61%). *H NMR (500 MHz, CD3OD) δ 7.36 - 6.76 (m, 8H), 4.72 (dd, J = 7.8, 9.4, 1H), 4.53 - 4.31 (m, 3H), 3.82 (dd, J = 3.5, 11.6, 1H), 3.45 (d, J = 11.6, 1H), 2.34 - 2.27 (m, 1H), 2.14 - 2.03 (m, 1H). 13C NMR (126 MHz, CD3OD) δ 174.81, 172.77, 158.73, 138.74, 138.36, 133.83, 130.64, 129.93, 129.56, 119.39, 118.62, 115.26, 71.01, 60.81, 59.80, 43.46, 39.24. MS (ESI) 375.4 (M+H).
VL81

VL81 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.18 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a colorless oil (42.9 mg, 0.091 mmol, 50%). *H NMR (500 MHz, CD3OD) δ 7.77 - 7.27 (m, 6H), 7.04 (d, J = 8.3, 1H), 4.71 (t, J = 8.2, 1H), 4.56 - 4.30 (m, 3H), 3.59 (dd, J = 3.7, 11.2, 1H), 3.17 (d, J = 11.3, 1H), 2.37 - 2.25 (m, 1H), 2.19 - 2.09 (m, 1H). 13C NMR (126 MHz, CD3OD) δ 173.87, 169.41, 138.18, 137.15, 133.71, 130.60, 130.36, 130.00, 129.69, 129.56, 129.40, 120.48, 70.62, 69.41, 60.48, 43.53, 39.23. MS (ESI) 472.1 (M+H).
VL96

VL96 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.155 mmol) according to Solid Phase Synthesis General Method B. It was isolated as a light yellow oil (36.6 mg, 0.102 mmol, 66%). ¾ NMR (500 MHz, CD3OD) δ 8.81 (s, 1H), 8.66 (dd, J = 4.6, 1.5 Hz, 2H), 7.60 (dd, J = 4.5, 1.6 Hz, 2H), 7.32 - 7.25 (m, 4H), 4.70 (dd, J = 9.3, 7.9 Hz, 1H), 4.46 (dd, J = 15.3, 6.3 Hz, 1H), 4.39 (s, 1H), 4.33 (dd, J = 15.4, 5.5 Hz, 1H), 3.78 (dd, J = 11.4, 3.5 Hz, 1H), 3.27 (dt, J = 3.2, 1.6 Hz, 1H), 2.31 (dd, J = 13.2, 7.6 Hz, 1H), 2.08 (ddd, J = 13.5, 9.6, 4.2 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 174.32, 169.63, 150.65, 145.83, 138.68, 133.88, 129.96, 129.56, 123.32, 70.99, 60.88, 59.33, 43.49, 39.33. MS (ESI) 360.5 (M+H).
VL112
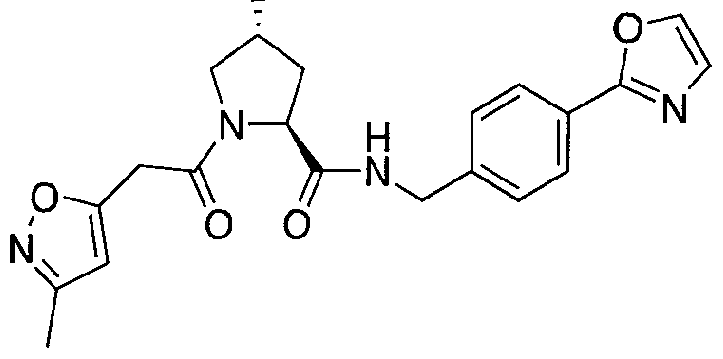
VL112 was synthesized from Fmoc-Hyp(OWang)-OAllyl resin (0.2 mmol) according to Solid Phase Synthesis General Method A. It was isolated as a cream colored solid (22.6 mg, 0.055 mmol, 28%). 1H NMR (500 MHz, CD3OD) δ 7.98 (d, J = 7.1 Hz, 3H), 7.46 (d, J = 8.0 Hz, 2H), 7.28 (s, 1H), 6.23 (s, 1H), 4.62 - 4.39 (m, 4H), 3.93 (d, J = 2.9 Hz, 2H), 3.81 (dd, J = 10.9, 4.1 Hz, 1H), 3.64 (d, J = 11.0 Hz, 1H), 2.33 - 2.17 (m, 4H), 2.15 - 2.04 (m, 1H). 13C NMR (126 MHz, CD3OD) δ 174.50, 168.72, 167.68, 163.41, 161.60, 142.96, 140.75, 129.45, 128.96, 127.53, 127.15, 105.36, 70.87, 60.78, 56.99, 43.72, 39.36, 33.95, 11.20. MS (ESI) 410.9 (M+H).
VL115

VLl 15 was synthesized according to General Method B. 1H NMR (500 MHz, CD3OD) δ 8.87 (s, 1H), 7.46 - 7.40 (m, 2H), 7.36 (dd, J= 8.8, 4.3 Hz, 2H), 6.20 (s, 1H), 4.55 (t, J= 8.0 Hz, 1H), 4.50 (d, J= 6.3 Hz, 1H), 4.48 - 4.42 (m, 2H), 3.92 (d, J= 4.5 Hz, 2H), 3.80 (dd, J = 10.9, 4.3 Hz, 1H), 3.62 (d, J= 11.0 Hz, 1H), 2.48 (d, J = 10.2 Hz, 3H), 2.31 - 2.21 (m, 4H), 2.08 (ddd, J= 13.0, 8.2, 4.7 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 174.48, 168.60, 167.70, 161.57, 152.92, 149.26, 140.81, 133.50, 133.09, 130.13, 129.24, 129.09, 128.34, 105.35, 70.86, 60.75, 56.95, 43.81, 39.38, 33.92, 15.87, 11.23. MS (ESI) 441.4 (M+H).
VL154

VLl 54 was synthesized according to General Method B. 1H NMR (500 MHz, CD3OD) δ 8.95 (s, 1H), 8.44 (t, J= 5.6, 1H), 7.84 (d, J= 8.2, 2H), 7.70 (d, J= 1.9, 1H), 7.35 (d, J = 8.2, 2H), 6.19 (s, 1H), 4.56 (t, J = 7.9, 1H), 4.51 (s, 1H), 4.43 (d, J= 5.7, 2H), 3.87 (s, 2H), 3.78 (dd, J= 10.9, 4.3, 1H), 3.58 (d, J= 10.8, 1H), 2.24 (obscured s, 4H), 2.16 - 2.07 (m, 1H); 13C NMR (126 MHz, CD3OD) δ 176.66, 170.98, 169.94, 164.15, 159.72, 157.62, 142.33, 136.89, 131.58, 130.33, 117.00, 108.05, 73.36, 63.27, 59.60, 46.75, 41.78, 36.80, 14.39; TLC: (EtOAC) R/=0.5; LRMS (ESI) 427.6 (M+H)+.
VL155

VL155 was synthesized according to General Method B. 1H NMR (500 MHz, CD3OD) δ 8.87 (d, J= 5.2, 1H), 8.54 (t, J= 5.7, 1H), 8.07 (s, 1H), 7.56 (d, J= 8.2, 2H), 7.36 (d, J= 8.2, 2H), 6.20 (s, 1H), 4.56 (t, J= 8.0, 1H), 4.51 (s, 1H), 4.42 (qd, J= 5.5, 15.5, 2H), 3.78 (dt, J = 9.2, 18.5, 1H), 3.60 (d, J= 11.1, 1H), 2.28 - 2.21 (m, 4H), 2.10 (ddd, J= 4.7, 8.0, 13.0, 1H); 13C NMR (126 MHz, CD3OD) δ 176.72, 171.03, 169.98, 164.12, 142.98, 142.96, 133.45, 132.34 131.86, 130.07, 108.02, 100.0, 73.37, 63.29, 59.60, 46.52, 41.81, 36.74, 14.27. TLC: (EtOAC) R/=0.5 ; LRMS (ESI) 427.4 (M+H)+.
VL118

VL118 was synthesized according to General Method A. lU NMR (500 MHz, CD3OD) δ 7.37 - 7.31 (m, 2H), 7.27 (d, J= 7.7 Hz, 1H), 7.22 (d, J= 7.5 Hz, 1H), 6.74 - 6.68 (m, 1H), 6.20 (s, 1H), 6.14 (dd, J = 3.5, 1.8 Hz, 1H), 6.10 - 6.05 (m, 1H), 4.55 (t, J= 8.0 Hz, 1H), 4.49 (s, 1H), 4.45 - 4.39 (m, 2H), 3.89 (t, J = 8.4 Hz, 1H), 3.79 (dd, J = 10.9, 4.3 Hz, 1H), 3.67 - 3.55 (m, 5H), 2.26 - 2.22 (m, 4H), 2.07 (ddd, J = 13.0, 8.1, 4.7 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 174.35, 168.57, 167.66, 161.57, 139.94, 135.44, 135.24, 129.58, 128.35, 128.22, 126.62, 124.93, 109.56, 108.54, 105.37, 70.84, 60.72, 56.91, 44.04, 39.36, 35.34, 33.88, 11.23. MS (ESI) 422.8 (M+H).
VL119

VL119 was synthesized according to General Method A. XH NMR (500 MHz, CD3OD) δ 7.38 - 7.30 (m, 4H), 6.76 - 6.67 (m, 1H), 6.23 (s, 1H), 6.10 (dd, J= 3.5, 1.8 Hz, 1H), 6.09 - 6.05 (m, 1H), 4.56 (t, J= 8.1 Hz, 1H), 4.51 (s, 1H), 4.47 - 4.39 (m, 2H), 3.93 (d, J= 3.0 Hz, 2H), 3.81 (dd, J= 10.9, 4.3 Hz, 1H), 3.63 (q, J= 5.8 Hz, 4H), 2,31 - 2.22 (m, 4H), 2.10 (ddd, J = 13.0, 8.1 , 4.7 Hz, 1H). 13C NMR (101 MHz, -1 :1 CD30D:CDC13) δ 172.63, 167.39, 166.19, 160.68, 136.80, 132.70, 129.06, 128.99, 127.75, 124.09, 108.75, 107.93, 104.62, 69.88, 59.64, 56.15, 43.36, 37.98, 35.19, 33.53, 11.37. MS (ESI) 423.6 (M+H).
VL131
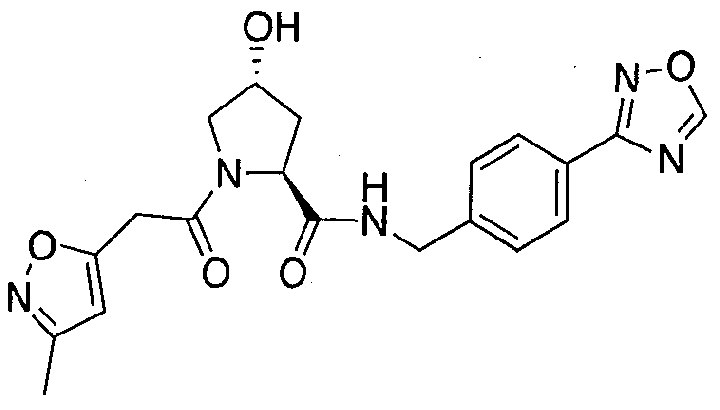
VL131 was synthesized according to General Method B. 1H NMR (400 MHz, CD3OD) δ 9.02 (d, J= 5.2 Hz, 1H), 8.02 (d, J= 8.4 Hz, 2H), 7.41 (d, J= 8.4 Hz, 2H), 6.17 (s, 1H), 4.52 - 4.38 (m, 4H), 3.84 (s, 2H), 3.76 (dd, J= 10.8, 4.3 Hz, 1H), 3.56 (d, J= 9.5 Hz, 1H), 2.30 - 2.18 (m, 4H), 2.14 (td, J= 8.1, 3.9 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 173.25, 167.86, 167.57, 166.44, 166.18, 160.85, 142.53, 128.33, 128.14, 125.56, 104.77, 70.04, 59.87, 56.31, 43.52, 38.30, 33.60, 11.39. MS (ESI) 413.3 (M+H).
VL138

VL138 was synthesized according to General Method B. 1H NMR (400 MHz, CD3OD) δ 7.42 (d, J= 8.2 Hz, 2H), 7.32 - 7.24 (m, 2H), 6.24 (s, 1H), 4.69 - 4.33 (m, 5H), 3.94 (d, J = 3.0 Hz, 1H), 3.82 (dd, J= 10.9, 4.3 Hz, 1H), 3.64 (d, J= 11.1 Hz, 1H), 2.38 (s, 3H), 2.31 - 2.24 (m, 4H), 2.23 (s, 3H), 2.10 (ddd, J= 13.1, 8.2, 4.7 Hz, 1H). 13C NMR (126 MHz, CD3OD) 8 174.41, 168.72, 167.67, 166.87, 161.59, 160.02, 139.46, 130.38, 129.37, 128.89, 117.72, 105.38, 70.87, 60.78, 57.01, 43.74, 39.37, 33.97, 11.38, 11.20, 10.66. MS (ESI) 438.6 (M+H).
VL139

VL139 was synthesized according to General Method B. 1H NMR (400 MHz, -1 :1 CD30D:CDC13) δ 7.89 (s, 2H), 7.49 (d, J = 8.1 Hz, 2H), 7.28 (d, J = 7.9 Hz, 2H), 6.20 (s, 1H), 4.54 (dd, J = 17.4, 9.5 Hz, 2H), 4.39 (d, J = 5.3 Hz, 2H), 3.93 - 3.46 (m, 4H), 2.32 - 2.16 (m, 4H), 2.16 - 2.05 (m, 1H). 13C NMR (126 MHz, -1 :1 CD3OD:CDCl3) δ 173.80, 168.23, 167.21, 161.29, 137.35, 132.66, 129.24, 128.77, 126.60, 126.48, 105.16, 70.53, 60.42, 56.73, 43.71, 39.00, 33.85, 11.30. MS (ESI) 410.0 (M+H).
VL152

VL152 was synthesized according to General Method B. !H NMR (400 MHz, CD3OD) δ 7.38 (d, J = 8.2 Hz, 2H), 7.25 (d, J = 8.2 Hz, 2H), 6.17 (d, J= 55.2 Hz, 1H), 4.65 - 4.30 (m, 4H), 4.05 - 3.72 (m, 3H), 3.64 (d, J = 11.1 Hz, 1H), 2.32 - 2.19 (m, 10H), 2.10 (ddd, J = 13.1 , 8.2, 4.7 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 174.37, 168.72, 167.67, 161.59, 143.18, 138.27, 132.85, 130.54, 129.12, 128.64, 105.40, 70.86, 60.78, 57.01 , 43.80, 39.38, 33.96, 11.21 , 11.07. MS (ESI) 438.5 (M+H).
VL158

VL158 was synthesized according to General Method B. Ή NMR (500 MHz, CD3OD) δ 8.03 (s, 1H), 7.62 (t, J= 8.7 Hz, 2H), 7.43 (d, J= 6.6 Hz, 1H), 7.33 (t, J= 6.5 Hz, 2H), 6.19 (s, 1H), 4.62 - 4.48 (m, 2H), 4.48 - 4.32 (m, 2H), 3.93 - 3.68 (m, 3H), 3.58 (s, 1H), 2.29 - 2.19 (m, 4H), 2.1 1 (ddd, J= 13.0, 8.0, 4.8 Hz, 1H). I3C NMR (126 MHz, CD3OD) δ 173.59, 168.01, 166.92, 161.11, 138.84, 135.95, 130.48, 129.06, 128.64, 125.89, 1 16.23, 105.04, 70.35, 60.24, 56.60, 43.56, 38.76, 33.78, 1 1.33. MS (ESI) 410.1 (M+H).
VL160

VL160 was synthesized according to General Method B. 1H NMR (500 MHz, CD3OD) δ 7.68 (d, J = 8.1 Hz, 2H), 7.63 (s, IH), 7.33 (d, J = 8.0 Hz, 2Η), 6.63 (s, IH), 6.19 (s, IH), 4.54 (t, J= 8.0 Hz, IH), 4.47 (s, IH), 4.38 (d, J= 4.6 Hz, 2H), 3.89 (d, J= 3.1 Hz, 2H), 3.78 (dd, J = 10.9, 4.3 Hz, IH), 3.60 (d, J = 11.1 Hz, IH), 2.28 - 2.14 (m, 4H), 2.06 (ddd, J = 13.0, 8.1, 4.7 Hz, IH). 13C NMR (126 MHz, CD3OD) δ 174.32, 168.69, 167.67, 161.61, 139.59, 132.31, 129.33, 128.86, 127.00, 126.87, 105.49, 105.38, 70.84, 60.78, 56.97, 43.80, 39.35, 33.95, 11.19. MS (ESI) 409.2 (M+H), 431.8 (M+Na).
(2S,4i?)-(9H-fluoren-9-yl)methyl 4-(teri-butoxy)-2-((4- chlorobenzyl)carbamoyl)pyrrolidine-l-carboxylate

Fmoc-Hyp(OtBu)-OH (1.23 g, 3 mmol, 1 eq) was dissolved in DCM (15 mL) and cooled to 4 °C. EDC (0.69g, 3.6 mmol, 1.2 eq) and HOBt (0.49 g, 3.6 mmol, 1.2 eq) were then added. After 20 minutes, 4-chlorobenzylamine (0.48 mL, 3.9 mmol, 1.3 eq) was added and the solution was allowed to warm slowly to room temperature. After 15 hours, the mixture was diluted with EtOAc and washed with 1M HC1, sodium bicarbonate, water and brine. The organic layer was dried with sodium sulfate, filtered and condensed. Purification by column chromatography (25 to 100% EtOAc/hexanes) gave a white foam (1.42 g, 2.66 mmol, 89%).
¾ NMR (400 MHz, CDC13) δ 7.77 (d, J= 7.2 Hz, 2Η), 7.57 (s, 2H), 7.40 (t, J= 7.3 Hz, 2Η), 7.31 (t, J = 7.3 Hz, 2H), 7.17 (dd, J = 27.2, 19.5 Hz, 4H), 4.58 - 3.94 (m, 7H), 3.60 (d, J = 6.7 Hz, 1H), 3.31 (d, J = 6.6 Hz, 1H), 2.50 (s, 1H), 1.96 (s, 1H), 1.28 - 1.10 (m, 9H). 13C NMR (101 MHz, CDCI3) δ 171.54, 156.13, 143.74, 141.32, 136.81, 133.02, 128.79, 128.71, 127.83, 127.12, 125.04, 120.07, 74.15, 69.63, 67.87, 59.17, 53.26, 47.10, 42.72, 36.34, 28.31. MS (ESI) 534.8 (M+H).
(25,4/?)-4-(te */-butoxy)-N-(4-chlorobenzyl)pyrrolidine-2-carboxamide

(2S',47?)-(9i/-fluoren-9-yl)methyl 4-(tert-butoxy)-2-((4-chlorobenzyl)carbamoyl)pyrrolidine- 1-carboxylate (0.5 g, 0.94 mmol, 1 eq) was dissolved in DCM (15 mL) and cooled to 4 °C. Tris(2-aminoethyl)amine (0.35 mL, 2.34 mmol, 2.5 eq) was added slowly, dropwise. After 30 minutes, the reaction was warmed to room temperature and stirred for an additional 14 hours. It was loaded directly onto a silica column, and purified by column chromatography (1 to 7% 0.5N methanolic ammonia/DCM) to give a white solid (0.2871 g, 0.92 mmol, 98%). *H NMR (400 MHz, CD3OD) δ 7.27 (dd, J= 20.1, 8.4 Hz, 4H), 4.35 (s, 2H), 4.22 (s, 1H), 3.84 (t, J- 8.0 Hz, 1H), 3.08 (dd, J = 11.4, 5.1 Hz, 1H), 2.76 (dd, J - 11.4, 2.8 Hz, 1H), 2.14 - 1.98 (m, 1H), 1.97 - 1.81 (m, 1H), 1.17 (s, 9H). 13C NMR (126 MHz, CD3OD) δ 176.48, 138.81, 133.83, 130.00, 129.52, 74.76, 73.37, 60.80, 55.61, 43.04, 40.76, 28.67.
General Method C: Representative Procedure: VL15


The white solid was dissolved in DCM (9 mL) at room temperature. TFA (1 mL) was added and the mixture was stirred for 12 hours and condensed. Purification by column
chromatography (1 to 20% 0.5 N methanolic ammonia/DCM) gave a white solid (39.8 mg, 0.11 mmol, 88% over 2 steps. 1H NMR (400 MHz, CD3OD) δ 8.73 (s, 1H), 7.47 (d, J = 16.9 Hz, 2H), 7.26 (s, 4H), 5.25 (dd, J= 37.5, 16.9 Hz, 2H), 4.56 (t, J= 7.9 Hz, 2H), 4.44 - 4.27 (m, 2H), 3.82 (dd, J= 10.8, 4.1 Hz, 1H), 3.63 (d, J= 10.8 Hz, 1H), 2.36 - 2.22 (m, 1H), 2.07 (ddd, J= 13.1, 8.3, 4.6 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 174.14, 166.34, 138.56, 138.20, 133.87, 129.97, 129.49, 124.55, 121.47, 70.94, 61.00, 55.75, 51.33, 43.35, 39.21. MS (ESI) 364.8 (M+H).
VL120

VL120 was synthesized according to General Method C. 1H NMR (500 MHz, CD3OD) δ 8.67 (d, J= 1.2 Hz, 1H), 7.36 (s, 1H), 7.29 (d, J= 9.2 Hz, 4H), 4.62 - 4.47 (m, 2H), 4.47 - 4.24 (m, 2H), 3.87 (ddd, J= 18.3, 15.1, 10.8 Hz, 3H), 3.66 (d, J= 11.0 Hz, 1H), 2.37 - 2.20 (m, 1H), 2.07 (ddd, J- 13.1, 8.4, 4.6 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 174.56, 169.45, 138.62, 135.15, 133.89, 129.98, 129.72, 129.52, 1 18.80, 70.89, 60.70, 56.84, 43.35, 39.46, 31.70. MS (ESI) 362.3 (M+H).
VL157
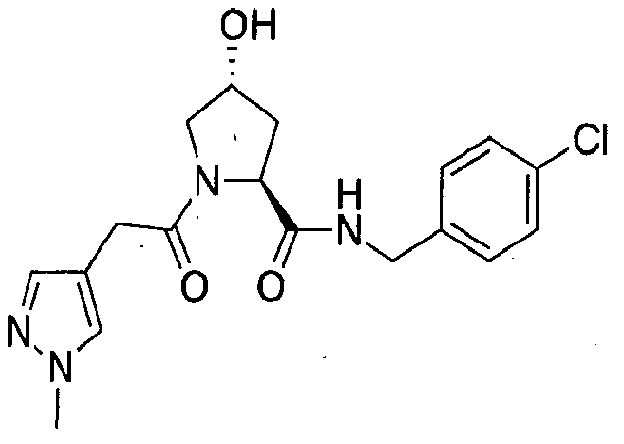
(2S,4R)-l-(2-azidoacetyl)-4-(ter -butoxy)-N-(4-chlorobenzyl)pyrrolidine-2-carboxamide

To a solution of azidoacetic acid (32 mg, 0.315 mmol) in CH2CI2-DMF (1.5 mL/1.5 mL)) at room temperature were added (25',4i?)-4-(terr-butoxy)-iV-(4-chlorobenzyl)pyrrolidine-2- carboxamide (93 mg, 0.300 mmol), DIPEA (0.19 mL, 1.080 mmol), and HOBt (48 mg, 0.360 mmol). The mixture was cooled to 0 °C, and then EDC (69 mg, 0.360 mmol) was added to the mixture at 0 °C. The reaction mixture was allowed to warm to rt, stirred at rt for 17 h, and cooled to 0 °C. The resulting mixture was quenched with H20 (5 mL) and extracted twice with ethylacetate. The combined extracts were washed with brine, dried over anhydrous Na2S04, filtered, and evaporated. The concentrate was purified by column chromatography (eluting with 5% ethyl acetate in hexane initially, grading to 40% ethyl acetate in hexane) on silica gel to afford the coupled product (110 mg, 93%). 1H NMR (400 MHz, CDC13) δ 7.31 (brs, 1H), 7.27 (d, J= 8.5 Hz, 2H), 7.18 (d, J= 8.5 Hz, 2H), 4.66 (dd, J= 8.4, 2.2 Hz, 1H), 4.58-4.52 (m, 1H), 4.41 (dd, J= 15.1, 6.1 Hz, 1H), 4.30 (dd, J= 15.1, 5.8 Hz, 1H), 3.87 (dd, J- 16.0, 16.0 Hz, 1H), 3.84 (dd, J- 16.0, 16.0 Hz, 1H), 3.61 (dd, J= 9.8, 7.0 Hz, 1H), 3.14 (dd, J= 9.8, 6.4 Hz, 1H), 2.50 (ddd, J= 12.6, 6.3, 2.3 Hz, 1H), 1.86 (dt, J- 12.6, 8.2 Hz, 1H), 1.19 (s, 9H). °C NMR, 100 MHz, CDC13) δ 170.3, 167.4, 162.5, 136.5, 133.1, 128.8, 128.7, 74.3, 69.9, 59.0, 2.7, 50.9, 42.8, 36.4, 35.1, 31.4, 28.2. MS (ESI) [M+H]+ 394.3.
(2S,4R)-4-(½rt-butoxy)-N-(4-chlorobenz l)-l-(2-(4-(methoxymethyl)-lH-l,2,3-triazol-l- yl)acetyl)pyrrolidine-2-carboxamide

To a solution of methyl propargyl ether (7 mg, 0.067 mmol) and (2^,4i?)-l-(2-azidoacetyl)-4- (te^butoxy)-N-(4-chlorobenzyl)pyrrolidine-2-carboxamide (25 mg, 0.0636 mmol) in t- B OH-H20 (1 : 1 , 1 mL) and THF (1 mL) at rt were added CuS04-5H20 (1.5 mg, 0.006 mmol) and sodium ascorbate (1.0 M in H20, 2 drops). The reaction mixture was stirred at rt for 19 h and evaporated. The residue was diluted with ¾0 (5 mL) and the mixture was extracted three times with ethyl acetate. The combined extracts were washed with brine, dried over Na2S0 , filtered, and concentrated. The crude residue was purified by flash
chromatography on silica gel to give the desired triazole (25 mg, 85%). *H NMR (500 MHz, CD3OD) d 7.91 (s, 1H), 7.30-7.22 (m, 4H), 5.43 (d, J= 16.8 Hz, 1H), 5.34 (d, J= 16.7 Hz, 1H), 4.54 (s, 2H), 4.53-4.49 (m, 2H), 4.37 (d, J = 15.4 Hz, 1H), 4.31 (d, 7= 15.4 Hz, 1H), 3.90 (dd, J= 10.3, 5.6 Hz, 1H), 3.49 (dd, J= 10.4, 4.3 Hz, 1H), 3.37 (s, 3H), 2.19-2.13 (m, 1 H), 2.11 -2.07 (m, 1 H), 1.22 (s, 9H). 13C NMR (asterisk denotes the signals of the minor rotamer, 125 MHz, CD3OD) d 174.1, * 173.4, * 166.9, 166.7, 145.7, * 138.6, 138.5, * 134.2, 133.8, *130.4, 129.9, *129.7, 129.5, 126.7, 75.6, *75.5, 71.1, *69.2, 66.3, *66.2, 60.8, *60.3, 58.4, *58.3, *55.5, 54.8, 52.6, +51.9, *43.7, 43.3, *41.1, 38.7, 28.5. MS (ESI) [M+H]+ 464.2.
VL141

To a stirred solution of the corresponding t-butyl ether (22 mg, 0.0475 mmol) in CH2C12 (1.5 niL) at 0 °C was added TFA (0.2 mL). The reaction mixture was stirred at rt for 12 h and concentrated. The residue was chromatographed (eluting with 100% CH2Cl2 initially, grading to 7% CH3OH in CH2C12) on silica gel to provide 5 (18.5 mg, 96%). *Η NMR (500 MHz, CD3OD/CDCI3 = 2:1) d 8.83 & 8.46 (due to the rotamers, both t, J= 5.7 Hz, 1H), 7.85 & 7.76 (due to the rotamers, both s, IH), 7.23 (d, J= 8.5 Hz, 2H), 7.18 (d, J= 8.5 Hz, 2H), 5.36 (d, J= 16.6 Hz, IH), 5.27 (d, J= 16.6 Hz, IH), 4.54 (s, 2H), 4.54-4.50 (m, 2H), 4.33 & 4.32 (due to the rotamers, both s, 2H), 3.77 (dd, J= 10.8, 4.2 Hz, IH), 3.60 (d, J= 10.8 Hz, IH), 3.37 & 3.36 (due to the rotamers, both s, 3H), 2.26-2.21 (m, IH), 2.09-2.04 (m, IH). 13C NMR (asterisk denotes the signals of the minor rotamer, 125 MHz, CD3OD/CDCI3 = 2:1) d •173.4, 173.3, *166.2, 165.9, 145.2, 137.5, *133.9, 133.5, 129.9, 129.4, 129.1, 126.0, *125.9, 70.4, *68.5, 66.0, *65.9, *60.4, 60.3, 58.4, *58.3, *56.1, 55.4, 52.2, *51.5, *43.2, 43.1, *41.1, 38.4. MS (ESI) [M+H 408.3. TLC (10% MeOH in CH2C12), Rf 0.48 (UV, CAM).
VL167

VL167 was synthesized according to General Method C. 1H NMR (500 MHz, CDC13) d 7.86 (s, IH), 7.71 (d, J= 9.5 Hz, IH), 7.61 (d, J= 7.4 Hz, 2H), 7.56 (d, J= 7.6 Hz, IH), 7.51 (d, J = 7.6 Hz, IH), 7.42 (dd, J= 7.4, 7.4 Hz, 2H), 7.38-7.30 (m, 2H), 7.30-7.26 (m, 3H), 4.66- 4.42 (m, 2H), 4.42-4.27 (m, 2H), 3.85 (dd, J= 11.4, 3.5 Hz, IH), 3.49 (d, J= 11.4 Hz, IH), 2.33-2.29 (m, IH), 2.15-2.09 (m, IH). 13C NMR (asterisk denotes the signals of the minor rotamer, 125 MHz, CDC13) d 172.5, 170.6, 162.7, 140.7, 139.3, 136.4, 135.6, 131.9, 128.2, *128.1, 128.0, 127.9, 127.6, *127.5, 126.8, 126.1, *126.0, 125.2, 125.1, 68.9, *67.4, *60.2, 58.7, 57.8, 41.5, *39.5, 37.2, 35.2, 30.0. MS (ESI) [M+H]+ 435.5.
VL216

VL216 was synthesized according to General Method C.


To a round bottom flask with stir bar was charged (2S,4 2)-l-(((9H-fluoren-9- yl)methoxy)carbonyl)-4-(teri-butoxy)pyrrolidine-2-carboxylic acid (0.587, 1.4 mmol 1.0 equiv)'EDC (380 mg, 2.0 mmol, 1.4 equiv), HOBt (310 mg, 2.0 mmol, 1.4 equiv) and (4- (oxazol-5-yl)phenyl)methanamine (250 mg, 1.4 mmol, 1.0 equiv). Upon stirring for 18 h the reaction was diluted with 25 ml DCM, and washed with citric acid (2X 50 mL), and saturated NaHC03 (2X 50 mL). The organic layer was dried with Na2S04, concentrated down then purified via silica gel chromatography (DCM to 2% MeOH (0,5 N NH3) to yield 515 mg (65% yield) of product as a viscous oil JH NMR (400 MHz, CDC13) δ 7.89 (s, 1H), 7.85 - 7.70 (m, 2H), 7.68 - 7.49 (m, 4H), 7.47 - 7.37 (m, 2H), 7.35 - 7.20 (m, 5H), 4.54 - 4.35 (m, 4H), 4.35 - 4.14 (m, 2H), 3.72 - 3.58 (m, 1H), 3.49 - 3.27 (m, 1H), 2.53 (s, 1H), 2.00 (dd, J = 8.1, 20.2, 1H), 1.25 (s, 9H); TLC: (9:1 DCM:MeOH (0.5 N NH3)) R^O.5; LRMS (ESI) 565.9(M+H)+.
(2S,4Jf)-l-(3-ethoxybenzoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide

To (25,4i?)-(9H-fluoren-9-yl)methyl 4-(te^butoxy)-2-((4-(oxazol-5-yl)benzyl)carbamoyl) pyrrolidine- 1-carboxylate (2,5 g, 3.61 mmol, 1.0 equiv) in 36 niL DCM was charged tris(2- aminoethyl)amine mol, 9.0 mmol, 2.5 equiv). D(400 upon stirring for 3 h the cloudy mixture was diluted with silica gel and concentrated down. The material was then dry loaded to a silica gel column and purified (DCM to 5% MeOH (0.5 N NH3) in DCM) to yield 820 mg (67% yield) as a white solid. 1H NMR (501 MHz, CDC13) δ 8.02 (s, 1H), 7.90 (s, 1H), 7.60 (d, J= 8.1, 2H), 7.33 - 7.29 (m, 3H), 4.44 (d, J= 6.1, 2H), 4.21 - 4.07 (m, 1H), 3.97 (dd, J= 7.2, 8.7, 1H), 2.87 (d, J= 11.6, 1H), 2.80 (dd, J = 4.3, 11.6, 1H), 2.17 (dd, J= 10.2, 12.4, 1H), 2.11 - 1.94 (m, 1H), 1.16 (s, 9H); 13C NMR (126 MHz, CDC13) δ 174.88, 151.27, 150.40, 139.33, 128.07, 126.79, 124.63, 121.43, 73.65, 72.30, 59.95, 54.97, 42.51, 39.27, 28.38; TLC: (9:1 DCM:MeOH (0.5 N NH3)) R/=0.42; LRMS (ESI) 344.2 (M+H)+.
!H NMR (400 MHz, CD3OD) δ 7.42 - 7.28 (m, 4H), 7.28 - 7.12 (m, 2H), 7.10 - 6.77 (m, 2H), 4.71 (dt, J= 30.7, 15.3 Hz, 1H), 4.58 - 4.30 (m, 3H), 4.18 - 3.92 (m, 2H), 3.87 - 3.77 (m, 1H), 3.44 (d, J= 11.4 Hz, 1H), 2.38 - 2.24 (m, 1H), 2.15 - 2.03 (m, 1H), 1.55 - 1.23 (m, 3H). 13C NMR (126 MHz, CD3OD) 8 174.72, 172.63, 160.34, 138.72, 138.35, 133.82, 130.65, 130.11, 129.92, 129.55, 120.47, 118.08, 114.27, 113.89, 71.01, 64.71, 60.81, 59.80, 43.33, 39.29, 15.09. MS (ESI) 403.2 (M+H).
General Method D: Representative Procedure:
VL217

(2S,4/.)-4-(tert-butoxy)-l-(3-ethoxybenzoyl)-N-(4-(oxazol-5-yl)benzyl)pyrrolidine-2- carboxamide

3-Ethoxybenzoic acid (13.3 mg, 0.08 mmol, 1 eq), EDC (16.9 mg, 0.088 mmol, 1.1 eq) and HOBt (11.9 mg, 0.88 mmol, 1.1 eq) were dissolved in DCM (0.8 mL) at room temperature. DIPEA (0.0279 mL, 0.16 mmol, 2 eq) was added, followed by (25',4/?)-4-(f^t-butoxy)-N-(4- (oxazol-5-yl)benzyl)pyrroli&ne-2-carboxarnide (33.0 mg, 0.096 mmol, 1.2 eq). The solution was stirred for 21 hours then diluted with EtOAc and washed with 10% citric acid, saturated sodium bicarbonate and brine. The organic layer was dried over sodium sulfate, filtered and condensed. Purification by column chromatography (1 to 5% MeOH/DCM) gave a colorless oil (36.1 mg, 0.073 mmol, 92%). ¾ NMR (400 MHz, CDC13) δ 7.90 (s, 1H), 7.61 (dd, J= 16.6, 6.9 Hz, 3H), 7.38 - 7.27 (m, 4H), 6.98 (dd, J= 16.0, 6.4 Hz, 3H), 4.92 (dd, J= 8.3, 4.7 Hz, 1H), 4.48 (d, J= 6.0 Hz, 2H), 4.43 - 4.31 (m, 1H), 4.03 (q, J= 7.0 Hz, 2H), 3.61 (dd, J = 10.9, 5.7 Hz, 1H), 3.31 (dd, J= 10.9, 4.4 Hz, 1H), 2.73 - 2.55 (m, 1H), 2.05 - 1.92 (m, 1H), 1.40 (t, J= 7.0 Hz, 3H), 1.13 (s, 9H). MS (ESI) 492.4 (M+H).
VL217

(25',4i?)-4-(tert-butoxy)- 1 -(3 -ethoxybenzoyl)-N-(4-(oxazol-5 -yl)benzyl)pyrrolidine-2- carboxamide (36.1 mg, 0.073 mmol, 1 eq) was dissolved in DCM (9 mL) at room temperature. TFA (1 mL) was added and the solution was stirred for 13 hours, then condensed. Purification by column chromatography (1 to 10% MeOH/DCM) gave a colorless oil (22.9 mg, 0.053 mmol, 72 %). 1H NMR (400 MHz, CD3OD) δ 8.24 (d, J= 12.0 Hz, 1H), 7.65 (dd, J= 28.0, 8.3 Hz, 2H), 7.47 (dd, J= 18.8, 10.6 Hz, 3H), 7.23 (ddd, J= 9.4, 4.6, 4.1 Hz, 3H), 7.09 - 6.87 (m, 2H), 4.75 (dd, J= 9.6, 7.7 Hz, 1H), 4.48 (dd, J= 49.7, 15.5 Hz, 3H), 4.06 (q, J = 7.0 Hz, 2H), 3.84 (dd, J = 11.5, 3.5 Hz, 1H), 3.44 (d, J = 11.5 Hz, 1H), 2.42 - 2.29 (m, 1H), 2.21 - 2.05 (m, 1H), 1.36 (dt, J = 24.0, 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 174.78, 172.66, 160.35, 153.14, 152.74, 140.85, 138.38, 130.66, 129.00, 127.71, 125.62, 121.77, 120.50, 118.08, 114.30, 71.02, 64.71, 60.85, 59.82, 43.72, 39.32,
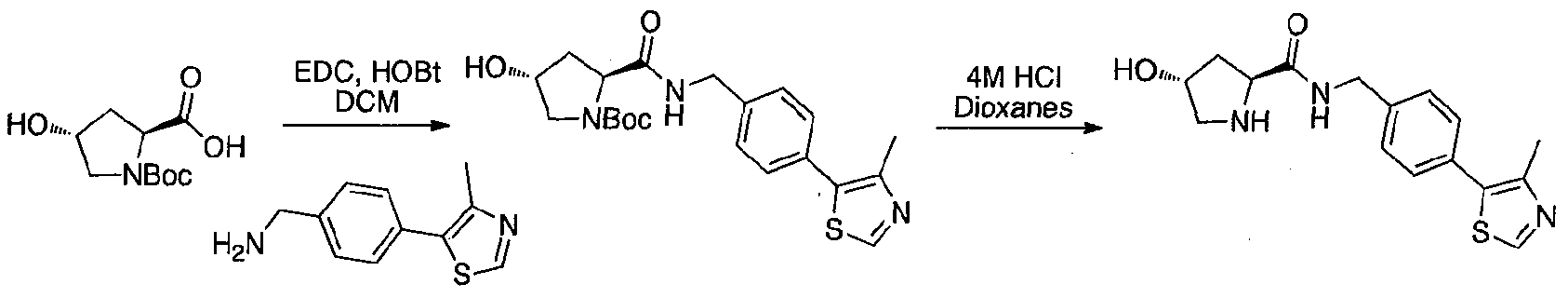
(3.S,4R)-tert-bvfiy\ 4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyI) pyrrolidine- 1-carboxylate

(25',4R)-l-(½rt-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid (366 mg, 1.58 mmol, 1 equiv.) was dissolved in 15 mL DMF and charged with EDC (380 mg, 2.0mmol 1.3 equiv,), and HOBt (310 mg, 2.0 mmol, 1.5 equiv) after 5 minutes of stirring (4-(4-
methylthiazol-5-yl)phenyl)methanamine (325 mg, 1.58 mmol, 1 equiv) was added. Upon stirring for 15 h the reaction was diluted with 25 mL EtOAc, and washed with 25 mL brine (2X), followed by 25 mL Sat. NaHC03 (2X). The organic layer was concentrated down to yield 650 mg (98 % yield) of the product as a yellow oil 1H NMR (400 MHz, CDC13) δ 8.67 (s, 1H), 7.43 - 7.29 (m, 4H), 4.49 (d, J= 16.7 Hz, 4H), 3.51 (dd, J= 1 1.0, 4.7 Hz, 2H), 2.61 - 2.45 (m, 4H), 2.03 (d, J = 7.4 Hz, 2H), 1.42 (s, 9H).TLC: (9:1 DCM:MeOH (0.5 N NH3)) Ry=0.20; MS (ESI) 417.5 (M+H)+.
(25',4R)-4-hydroxy-7V-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide

To (2iS',4i?)-tert-butyl 4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl) pyrrolidine- 1-carboxylate ( 650 mg, 1.40 mmol, 1 equiv) in a round bottom flask was charged 9 mL 4M HCL in dioxanes (36 mmol, 26 equiv). The reaction was left to stir for lh upon which time N2 was bubbled through for 1 h and the volatiles were removed by vacuum. The resulting viscous oil was washed dissolved in water and washed with 50 mL EtOAC. The aqueous layer was then basified to pH 12 with 1 M NaOH, and then extracted with 50 mL EtOAC (3X). The organic layer was dried and concentrated down to yield 250 mg (79% yield) of product as a brown viscous oil. *H NMR (501 MHz,CDCl3) δ 8.66 (s, 1H), 8.18 (t, J= 6.0, 1H), 7.38 (d, J= 8.1 , 2H), 7.30 (d, J= 8.1, 2H), 4.48 - 4.37 (m, 3H), 4.08 (t, J= 8.4, 1H),
3.02 (d, J= 13.3, 1H), 2.79 (dd, J= 3.2, 12.3, 1H), 2.51 (s, 3H), 2.33 (dd, J= 8.6, 13.9, 1H),
2.03 - 1.87 (m, 1H); 13C NMR (126 MHz, CDC13) δ 174.89, 150.35, 148.46, 138.30, 131.54, 130.95, 129.51 , 127.92, 72.90, 59.72, 55.35, 42.53, 39.98, 16.06; TLC: (9:1 DCM:MeOH) R^O.1 ; LRMS (ESI) 317.4 (M+H)+.


3-ethoxybenzoic acid (17 mg, 0.1 mmol, 1 equiv.) was dissolved in 1 mL 10:1 DCM:DMF and charged with EDC (25 mg, 0.13 mmol 1.3 equiv,), and HOBt (21 mg, 0.13 mmol, 1.3 equiv). After 5 minutes of stirring (2<S,,4i?)-4-hydroxy-7V-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide (31 mg, 0.095 mmol, 1 equiv) was added. Upon stirring for 18 h the reaction was diluted with 15 mL EtO Ac and washed with 25 mL 10% aqueous citric acid and 25 mL saturated NaHC03. The organic layer was dried with Na2S04 and concentrated by vacuum. The resultant oil was purified by silica gel chromatography (DCM to 9% MeOH (0.5 N NH3) in DCM) to yield 25 mg (56 % yield) of the product as a white solid. *H NMR (501 MHz, CD3OD) δ 8.87 (s, 1H), 7.51 - 7.42 (m, 4H), 7.37 (t, J= 8.1, 1H), 7.23 - 7.14 (m, 2H), 7.05 (dd, J= 2.2, 8.4, 1H), 4.79 (dd, J= 7.7, 9.5, 1H), 4.63 - 4.40 (m, 3H), 4.08 (q, J= 7.0, 2H), 3.86 (dt, J= 3.8, 7.6, 1H), 3.47 (d, J= 11.5, 1H), 2.47 (s, 3H) 2.36 (dd, J= 7.6, 13.2, 1H), 2.14 (ddd, J= 5.3, 10.2, 16.4, 1H), 1.41 (t, J= 7.0, 3H); 13C NMR (126 MHz, CD3OD) δ 174.74, 172.64, 160.34, 152.78, 149.05, 140.21, 138.40, 133.39, 131.55, 130.65, 130.44, 128.83, 120.49, 118.07, 114.32, 71.02, 64.71, 60.83, 59.81, 43.67,
39.30, 15.79, 15.06; TLC: (9:1 DCM:MeOH (0.5 N H3)) R/=0.25; LRMS (ESl)
466.1(M+H)+.
VL210

VL210 was synthesized according to General Method D. *H NMR (500 MHz, CD3OD) δ 8.23 (s, 1H), 7.82 (t, J = 1.7, 1H), 7.73 - 7.58 (m, 4H), 7.49 - 7.33 (m, 4H), 4.76 (dd, J - 7.6, 9.6, 1H), 4.59 - 4.32 (m, 3H), 3.84 (dd, J = 3.5, 11.4, 1H), 3.41 (d, J = 1 1.3, 1H), 2.43 - 2.30 (m, 1H), 2.18 - 2.07 (m, 1H); 13C NMR (126 MHz, CD3OD) δ 173.14, 169.54, 151.35, 139.41, 137.87, 133.28, 130.18, 130.01, 127.92, 127.61, 126.31, 125.93, 124.23, 121.93, 120.36, 69.62, 59.53, 58.30, 42.31, 37.95; LRMS (ESI) 471.5 (M+H)+.
VL224
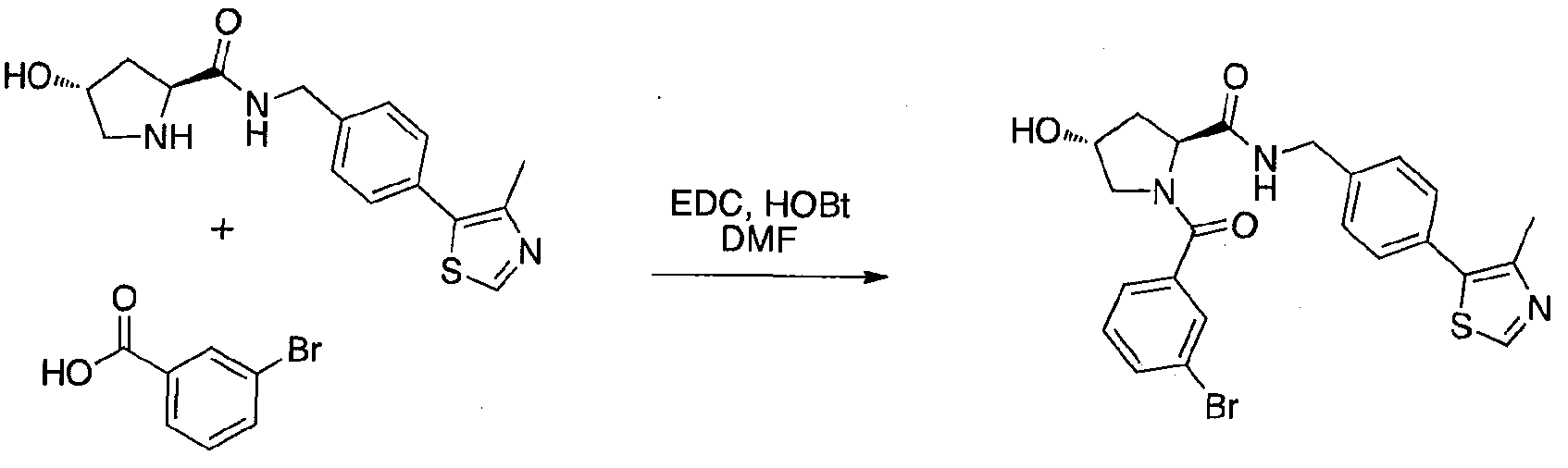
VL224 was synthesized according to General Method E. *H NMR (501 MHz, CD3OD) δ 8.87 (s, 1H), 7.83 (d, J= 1.5, 1H), 7.67 (d, J = 7.1, 1H), 7.61 (d, J= 6.7, 1H), 7.48 - 7.36 (m, 5H), 4.77 (t, J= 8.5, 1H), 4.60 - 4.39 (obscured m, 3H), 3.90 - 3.78 (m, 1H), 3.42 (d, J= 11.4, 1H), 2.47 (s, 3H), 2.41 - 2.30 (m, 1H), 2.19 - 2.06 (m, 1H); 13C NMR (126 MHz, CD3OD) 6 173.15, 169.55, 151.41, 147.64, 138.77, 137.85, 133.26, 132.78, 131.98, 130.16,
130.02, 129.05, 127.43, 125.91, 121.93, 69.64, 59.53, 58.32, 42.27, 37.94, 14.41; TLC: (9:1 DCM:MeOH (0.5 N NH3)) R =0.7; LRMS (ESI) 499.8(M+H)+.
VL215
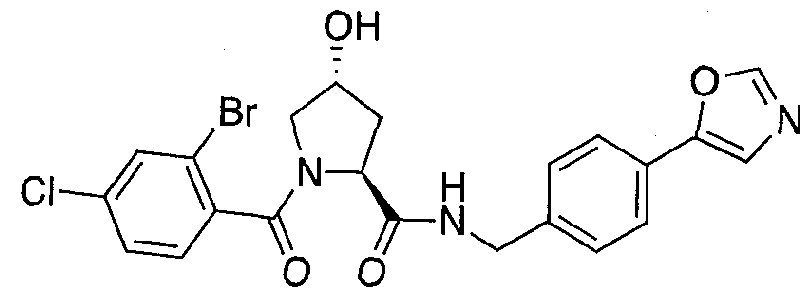
VL215 was synthesized according to General Method D. *H NMR (501 MHz, CD3OD) δ 8.24 (dd, J= 13.4, 7.1 Hz, 1H), 7.87 - 7.58 (m, 3H), 7.58 - 7.31 (m, 4H), 7.16 (s, 1H), 4.73 (d, J= 7.8 Hz, 1H), 4.63 - 4.50 (m, 1H), 4.49 - 4.29 (m, 2H), 3.80 (d, J = 10.5 Hz, 1H), 3.60 (s, 1H), 2.31 (s, 1H), 2.17 (s, 1H). 13C NMR (126 MHz, CD3OD) δ 179.37, 169.61, 153.12, 152.75, 140.72, 138.21, 137.16, 133.72, 130.38, 129.70, 129.40, 129.08, 127.76, 125.63, 121.80, 70.64, 60.46, 58.42, 43.80, 39.27. MS (ESI) 504.2 (M+H).
VL228

VL228 was synthesized according to General Method E. NMR (400 MHz, CD3OD) δ 8.88 (d, J= 9.3 Hz, 1H), 7.75 (d, J= 1.9 Hz, 1H), 7.48 (ddd, J= 13.9, 8.4, 4.1 Hz, 5H), 7.17 (d, J = 8.3 Hz, 1H), 4.74 (t, J= 8.2 Hz, 1H), 4.63 - 4.33 (m, 3H), 3.61 (dd, J= 11.3, 3.8 Hz, 1H), 3.18 (d, J= 1 1.3 Hz, 1H), 2.49 (d, J = 9.6 Hz, 3H), 2.33 (ddd, J = 7.8, 4.8, 2.1 Hz, 1H), 2.19 (ddd, J= 11.9, 7.8, 4.6 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 173.93, 169.45, 152.84, 149.07, 140.11, 138.21, 137.17, 133.73, 131.62, 130.53, 130.48, 130.39, 129.52, 129.41, 128.93, 70.65, 60.48, 58.39, 43.75, 39.26, 15.81. MS (ESI) 534.4 (M+H).
VL177

VL177 was synthesized according to General Method D. Ή NMR (500 MHz,
CDCI3/CD3OD) 8 7.89 (s, 1H); 7.84 (s, 1H); 7.80-7.75 (m, 2H); 7.69 (d, J= 7.8 Hz, 1H); 7.57-7.48 (m, 3H); 7.31 (d, J= 8.1 Hz, 2H); 4.76 (t, J= 8.3 Hz, 1H); 4.49-4.39 ( m, 3H); 3.72 (dd, J= 11.2, 3.5 Hz, 1H); 3.36 (d, J= 11.2 Hz, 1H); 2.93 (s, 1H); 2.31 (ddd, J= 13.2, 8.8, 4.4 Hz, 1H); 2.21 (dd, J= 13.5, 7.8 Hz, 1H). 13C NMR (125MHz, CDC13/CD30D) δ 171.8, 169.0, 151.5, 150.6, 138.9, 137.0, 133.9, 131.9, 131.3, 129.5, 128.1, 126.7, 124.7, 121.1, 117.9, 112.7, 69.9, 59.3, 58.5, 43.3, 37.4. TLC (10% MeOH in CH2C12), Rf 0.17 (UV, CAM), MS (ESI+): calculated for C23H21N404 [M+H]+ 417.2, found 417.1.
VL226

VL226 was synthesized according to General Method E. *H NMR (500 MHz,
CDC13/CD30D) δ 8.65 (s, 1H); 7.84 (s, 1H); 7.74 (dd, J= 13.3, 7.8 Hz, 2H); 7.53 (t, J= 7.8 Hz, 1H); 7.40-7.28 (m, 5H); 4.92 (t, J= 8.1 Hz, 1H); 4.53 (s, 1H); 4.48 (d, J= 5.9 Hz, 2H); 3.72 (dd, J= 11.3, 3.5 Hz, 1H); 3.52-3.44 (m, 1H); 2.85 (br s, 1H); 2.67-2.56 (m, 1H); 2.48 (s, 3H); 2.21 (dd, J= 13.5, 7.8 Hz, 1H). 13C NMR (125MHz, CDC13/CD30D) δ 170.8, 169.4, 150.5, 148.6, 138.0, 137.0, 134.1, 131.9, 131.4, 129.7, 129.6, 127.0, 118.0, 113.0, 70,4, 59.1, 58.6, 43.5, 36.8, 16.2. TLC (10% MeOH in CH2C12), Rf 0.32 (UV, CAM), MS (ESI+):
calculated for C24H23N403S [M+H]+ 447.2, found 447.0.
VL211
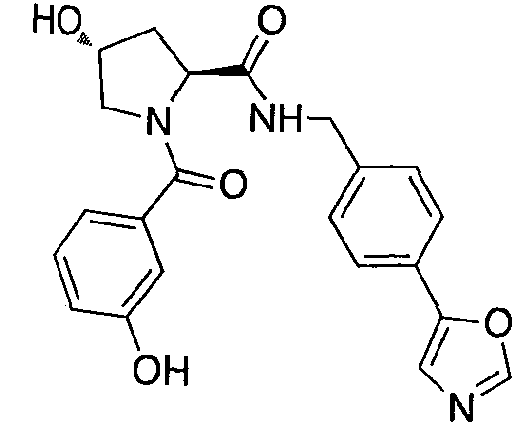
VL211 was synthesized according to General Method D. Ή NMR (500 MHz, CD3OD) δ 8.11 (s, 1H), 7.58 (dd, J= 2.3, 8.2, 2H), 7.42 - 7.30 (m, 3H), 7.16 (t, J= 7.9, 1H), 6.96 (d, J= 7.7, 1H), 6.92 (s, 1H), 6.80 (dd, J = 2.3, 8.1, 1H), 4.65 (t, J= 8.6, 1H), 4.47 - 4.26 (m, 3H), 3.71 (dt, J = 4.0, 8.0, 1H), 3.36 (d, J = 11.6, 1H), 2.32 - 2.16 (m, 1H), 2.08 - 1.94 (m, 1H); 13C NMR (126 MHz, CD3OD) δ 174.74, 172.77, 158.71, 153.14, 152.70, 140.83, 138.38, 130.62, 128.98, 127.69, 125.62, 121.75, 119.39, 118.61, 115.27, 71.01, 60.77, 59.79, 43.73, 39.25; TLC: (9:1 DCM:MeOH R^0.15; LRMS (ESI) 408.3 (M+H)+.
VL225

V1225 was synthesized according to General Method E. Ή NMR (501 MHz, CD3OD) δ 8.86 (s, 1H), 7.49 - 7.34 (m, 4H), 7.27 (t, J= 7.8, 1H), 7.11 - 7.00 (m, 2H), 6.93 - 6.84 (m, 1H), 4.77 (t, J= 8.5, 1H), 4.57- 4.38 (m, 3H), 3.84 (dd, J= 3.3, 11.5, 1H), 3.47 (d, J= 11.5, 1H), 2.46 (S, 3H) 2.34 (dd, J= 7.1, 12.5, 1H), 2.18 - 2.06 (m, 1H); 13C NMR (126 MHz, CD3OD) δ 173.35, 171.36, 157.31, 151.39, 147.62, 138.81, 136.95, 130.12, 129.21, 129.04, 127.62, 127.41, 117.97, 117.19, 1 13.84, 69.61, 59.37, 58.41, 42.24, 37.85, 14.37; TLC: (9:1
DCM:MeOH) R^0.3; LRMS (ESI) 437.0 (M+H)+.
VL178

VL178 was synthesized according to General Method D. 1H NMR (400 MHz, CD3OD) δ 8.84 (t, J= 5.9, 1H), 8.26 (d, J = 5.9, 1H), 7.73 (d, J= 8.3, 2H), 7.55 - 7.45 (m, 3H), 7.06 (t, J= 7.8, 1H), 6.86 - 6.76 (m, 1H), 6.68 (d, J= 7.3, 1H), 4.75 (t, J= 8.4, 1H), 4.66 - 4.35 (m, 3H), 3.55 (dd, J= 3.5, 11.6, 1H), 3.24 (d, J= 11.4, 1H), 2.41 - 2.26 (m, 1H), 2.21 - 2.10 (m, 4H); 13C NMR (126 MHz, CD3OD) δ 173.21 , 172.03, 151.32, 145.03, 139.46, 137.30, 130.95, 127.61, 126.53, 126.29, 124.22, 123.96, 120.35, 1 16.31, 1 15.97, 74.46, 69.35, 58.72, 42.46, 38.02, 23.61; LRMS (ESI) 420.4 (M+H)+.
VL229

(2S',4i?)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (24mg, 0.0756 mmol, 1 eq), 3-amino-2-methylbenzoic acid (13 mg, 0.083 mmol, 1.1 eq), EDC (16 mg, 0.083 mmol, 1.1 eq) and HOBt (11 mg, 0.083 mmol, 1.1 eq) were dissolved in DMF (0.76 mL) at room temperature. DIPEA (0.02 mL, 0.113 mmol, 1.5 eq) was added, and the solution was stirred for 17 hours. The solution was then partitioned between 1M NaOH and EtOAc, separated, and extracted twice more with EtOAc. The combined organic layer was dried over sodium sulfate, filtered and condensed. Purification by column chromatography (1 to 10 % 0.5N methanolic ammonia/DCM) gave a white solid (20.5 mg, 0.045 mmol, 60%). 1H NMR (501 MHz, CD3OD) δ 8.87 (t, J= 6.6 Hz, 1H), 7.45 (dt, J= 20.5, 7.7 Hz, 4H), 7.03 (t, J= 7.6 Hz, 1H), 6.78 (d, J= 7.8 Hz, 1H), 6.65 (s, 1H), 4.74 (t, J= 8.1 Hz, 1H), 4.67 - 4.34
(m, 3H), 3.53 (d, J= 11.3 Hz, 1H), 3.21 (d, J= 9.3 Hz, 1H), 2.48 (d, J= 3.1 Hz, 3H), 2.32 (d, J= 7.6 Hz, 1H), 2.14 (dd, J = 31.4, 20.5 Hz, 4H). 13C NMR (126 MHz, CD3OD) δ 174.56, 173.67, 152.89, 152.81, 149.04, 147.80, 140.23, 138.57, 133.44, 131.54, 130.46, 128.86, 127.83, 117.01, 116.36, 70.77, 69.66, 60.09, 43.70, 39.41, 15.81, 13.92. MS (ESI) 450.6 (M+H), 473.4 (M+Na).
For further reference see the following articles and the references cited therein:
(1) Buckley DL et al. J. Am. Chem. Soc 2012, 134, 4465-4468.
(2) Van Molle I et al. A Chemistry & Biology 2012, 19, 1300-1312
(3) Buckley, O Angew. Chem. Int. Ed., 2012, 51, 11463-11467
(4) Buckley, D. Let al Angew. Chem. 2012, 124, 11630-11634.
Examples- Compounds 165-266 of Affinity Table II
The following compounds were synthesized according to the stated General Method, were purified by standard chromatographic methods and had lE and 13C NMR and MS data consistent with the desired structure.
VL165:

VL165 was synthesized according to General Method B. VL168 & 169
The chiral RHS amine fragment was synthesized using the procedure from Surya Prakash, G.
K.; Mandal, M.; Olah, G. A. Angew. Chem. Int. Ed. 2001, 40, 589-690.


VL169 was synthesized according to General Method F VL174

VL174 General Method C
VL175: General Method C

VL190: General Method B


VL188: General Method B

VL189: General Method B
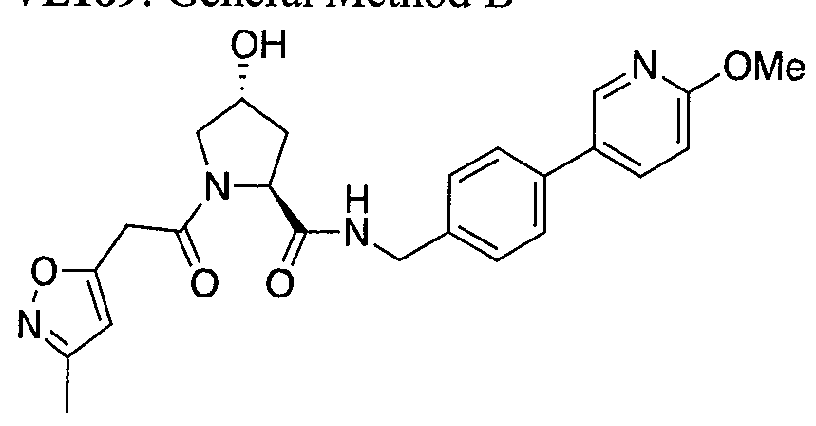
VL192-VL205: Solid Phase Synthesis General Method B


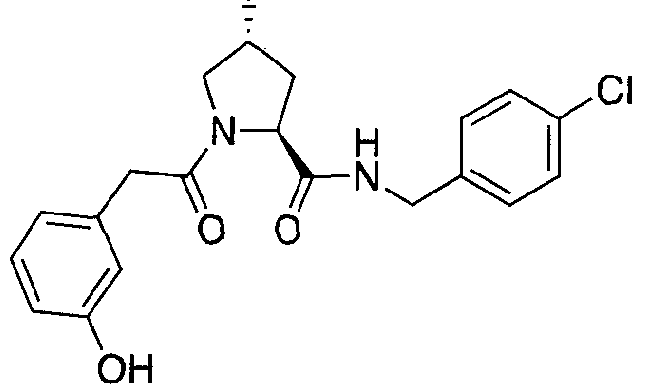
VL214 General Method C

VL218 General Method C

VL220 General Method C

VL221 General Method C

VL222 General Method C

VL256 General Method E

VL213: General Method C

VL223: General Method D

VL230: General Method D
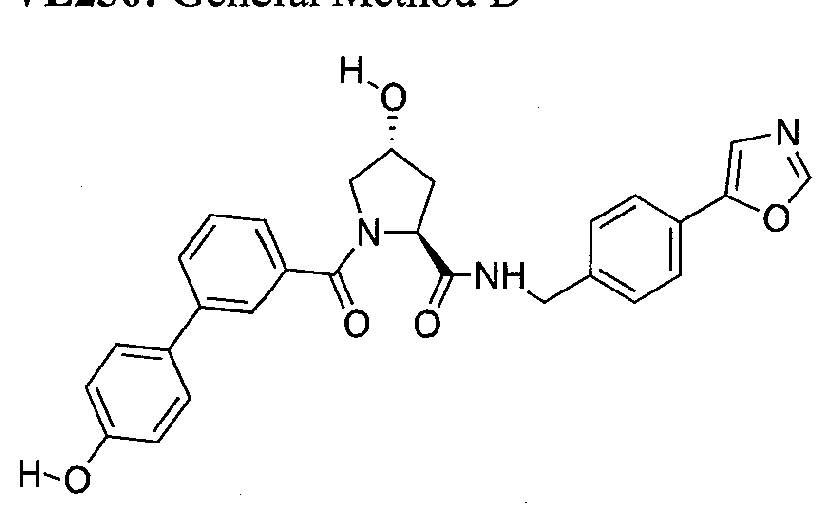
VL231: General Method E

VL238: General Method B

VL240: General Method E
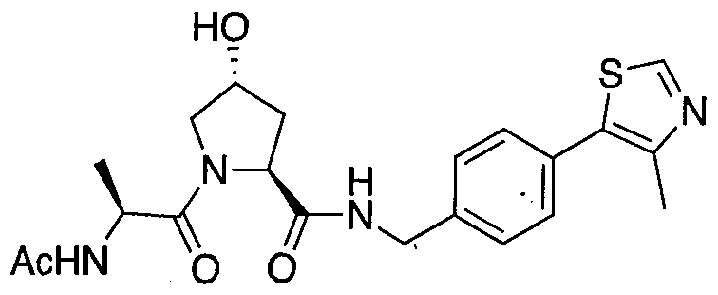
VL241 : General Method B

VL242: General Method E

VL243: General Method E

VL244: General Method B

3-hydroxy-2-methylbenzoic acid (26.3 mg, 0.173 mmol, 1.1 eq), EDC (33.2 mg) and HOBt (23.4 mg) were dissolved in DCM (0.8 mL) and DMF (0.1 mL) at 4 °C. After 10 minutes, (2S',4i?)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (0.5 mL of a 100 mg/mL solution in DCM) was added and the mixture was warmed to room temperature. After 21 hours, the mixture was diluted with 10 mL of half saturated sodium chloride and extracted thrice with 10 mL of EtOAc. The combined organic layer was dried over sodium sulfate, filtered and condensed. Purification by column chromatography (1 to 10%
MeOH/DCM) gave a white solid (29.2 mg, 0.0647, 1%). JH NMR (400 MHz, MeOD) δ
8.93 - 8.82 (m, 1H), 7.55 - 7.36 (m, 4H), 7.10 (t, J= 7.8 Hz, 1H), 6.83 (dd, J= 10.3, 7.9 Hz, 2H), 4.76 (t, J= 8.4 Hz, 1H), 4.66 - 4.38 (m, 3H), 3.56 (dd, J= 1 1.6, 3.6 Hz, 1H), 3.22 (d, J = 11.6 Hz, 1H), 2.54 - 2.47 (m, 3H), 2.41 - 2.31 (m, 1H), 2.19 (dt, J= 11.3, 10.9 Hz, 4H). I3C NMR (126 MHz, MeOD) δ 174.50, 173.09, 157.22, 152.89, 152.80, 149.01, 140.21 , 139.18, 133.42, 131.52, 130.44, 128.85, 127.94, 117.84, 116.42, 70.73, 69.62, 60.12, 43.69, 39.38, 15.79, 12.70. MS (ESI) 452.5 (M+H).
VL247: General Method C

VL248: General Method E
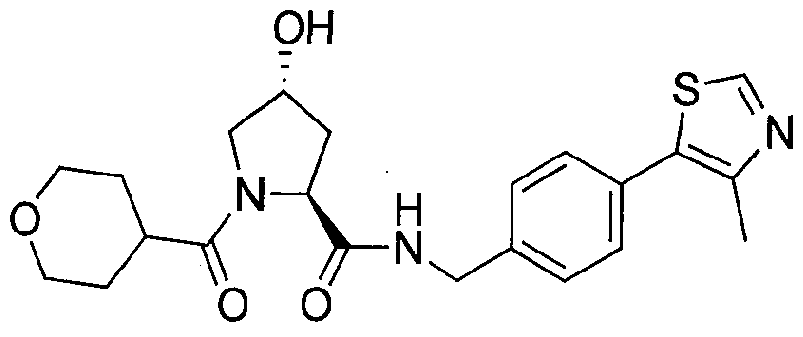
VL249: General Method E

VL250: General Method E

VL253: General Method C


VL257: General Method C

VL258: General Method C
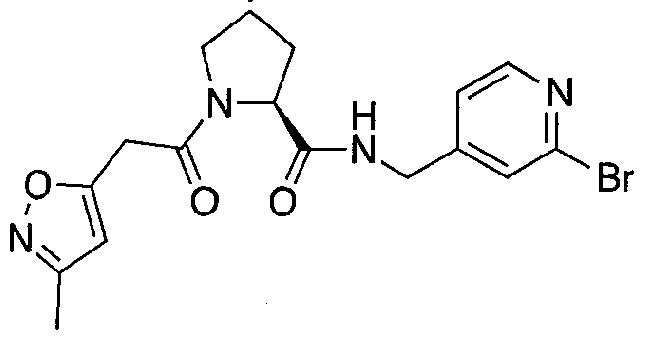
VL259: General Method C

VL260: General Method E
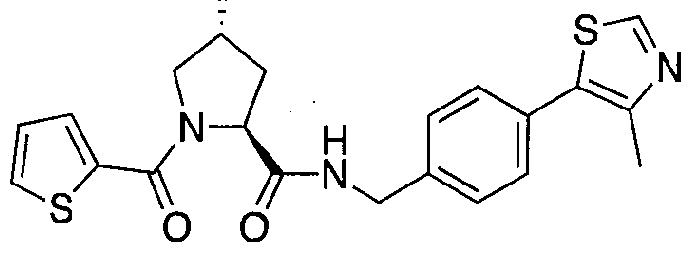
VL261 : General Method E
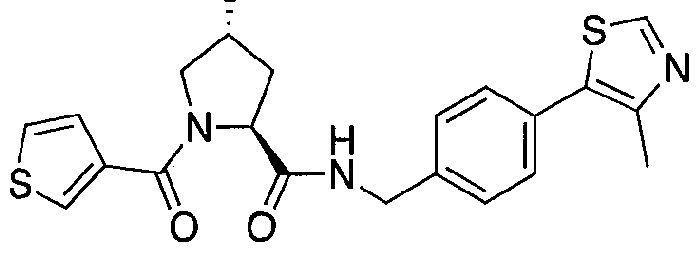
VL262: General Method E

VL263: General Method E

VL264: General Method E

VL265: General Method E

VL251
(2S',4i?)-l-((5)-2-((5^-2-acetamido-4-methylpentanamido)propanoyl)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide


Boc-Ala-OH (189 mg, 1.0- mmol) was dissolved in 10 mL DCM and charged with EDC (248 mg, 1.2 mmol), and HOBt (202 mg, 1.3 mmol) after 5 minutes of stirring (25',4i?)-4-hydroxy- N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carb-oxamide (317 mg, 1.0 mmol) was added. Upon stirring for 18 h the reaction was diluted with 10 mL DCM and washed with 10 mL 10% aqueous citric acid followed by 5 mL saturated NaHC03. The mixture was concentrated down and purified by silica gel chromatography (DCM/MeOH gradient) to yield 210 mg (43 % yield) of the product as a white solid. !H NMR (501 MHz, CDC13) δ
8.65 (s, 1H), 7.58 (s, 1H), 7.31 (d, J= 8.1, 2H), 7.26 (d, J= 8.0, 2H), 5.44 (d, J= 7.4, 1H),
4.66 (t, J= 7.6, 1H), 4.52 (s, 1H), 4.39 (m, 3H), 3.78 (d, J= 10.9, 1H), 3.59 (d, J= 7.0, 1H), 2.47 (s, 3H), 2.30 (s, 1H), 2.10 (s, 1H), 1.54 - 1.31 (m, 9H), 1.26 (d, J= 6.9, 3H); 13C NMR (126 MHz, CDC13) δ 173.1, 171.2, 155.5, 150.5, 148.2, 138.2, 130.7, 129.4, 127.6, 80.1, 70.1, 58.8, 55.2, 48.0, 42.3, 36.5, 28.3, 18.0, 16.0; LRMS (ESI) 489.4 (M+H)+.
VL251
(2S',4i?)-l-((5)-2-((5)-2-acetamido-4-methylpentanamido)propanoyl)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide

Boc-Ala-Hyp-benzyl thiazole (116 mg, 0.225 mmol) was dissolved in 1 mL DCM and charged with 2.3 mL 4M HCL in dioxanes. Upon stirring for one hour Nitrogen gas was sparged through the mixture for 15 minutes and the remaining volatiles removed by roto vap. The resultant foam was then dissolved in 5 mL 1 : 1 DCM: DMF and charged with EDC (56 mg, 0.29 mmol), HOBt (45 mg, 0.29 mmol), and Ac-Leu-OH (43 mg, 0.25 mmol) were added. After stirring for 5 minutes triethyl amine was added. Upon stirring for 18 h the
reaction was diluted with 10 n L EtOAc and washed with 10 niL 10% aqueous citric acid followed by 5 mL saturated NaHC03 The aqueous layer was then back extracted 2 X 10 mL DCM. The organic layers were combined and the mixture was concentrated down and purified by silica gel chromatography (DCM/MeOH gradient) to yield 35 mg (29 % yield) of the product as a white solid. *H NMR (501 MHz, CDC13) δ 8.68 (s, IH), 8.05 (s, IH), 7.70 (s, IH), 7.36. (d, J= 7.3, 2H), 7.28 (d, J= 8.6, 2H), 6.50 (s, IH), 4.85 - 4.73 (m, 2H), 4.68 (s, lH), 4.59 (s, IH), 4.40 (d, J = 32.4, 2H), 3.84 (d, J= 10.7, IH), 3.70 (d, J= 10.5, IH), 2.51 (s, 3H), 2.30 (s, IH), 2.21 (s, IH), 1.85 (s, 3H), 1.59 (s, IH), 1.50 (s, 2H), 1.34 (d, J = 6.7, 3H), 0.86 (t, J = 6.7, 6H); 13C NMR (126 MHz, CDC13) δ 172.1 , 172.0, 171.2, 170.8, 150.4, 148.4, 138.1 , 129.5, 129.4, 127.8, 1 10.0, 70.36, 58.9, 55.5, 51.8, 46.9, 43.1, 41.8, 36.9, 24.7, 23.2, 23.1, 21.8, 17.9, 16.0; LRMS (ESI) 545.1 (M+H)+.
VL252
(2>S',4i?)-l-((5}-2-((5)-2-amino-4-methylpentanamido)propanoyl)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide

Boc-Ala-Hyp-benzyl thiazole (1 16 mg, 0.225 mmol) was dissolved in 1 mL DCM and charged with 2.3 mL 4M HCL in dioxanes. Upon stirring for one hour Nitrogen gas was sparged through the mixture for 15 minutes and the remaining volatiles removed by roto vap. The resultant foam was then dissolved in 5 mL 1 :1 DCM: DMF and charged with EDC (56 mg, 0.29 mmol), HOBt (45 mg, 0.29 mmol), and Boc-Leu-OH (62 mg, 0.25 mmol) were added. After stirring for 5 minutes triethyl amine was added. Upon stirring for 18 h the reaction was diluted with 10 mL EtOAc and washed with 10 mL 10% aqueous citric acid followed by 5 mL saturated NaHC03. The organic layers were combined and the mixture was concentrated to yield 55 mg (40 % yield) of the product as a white solid. LRMS (ESI) 602.0 (M+H)+. Upon confirmation by mass spec the product was dissolved in 2 mL 1 : 1
DCM:MeOH and charged with 3 mL 4M HC1 in dioxanes. Upon stirring for 45 minutes the
reaction was quenched with 5 ml 0,5 N ammonia in methanol. The solvents were evaporated down and purified by silica gel chromatography (gradient of DCM/MeOH (0.5 N NH3) to yield 50 mg of pure product as a white solid. 1H NMR (501 MHz, CDC13) δ 8.25 (s, 1H), 6.91 (dd, J= 7.3, 18.0, 4H), 4.17 (d, J= 7.5, 2H), 4.06 (d, J= 22.4, 2H), 3.95 (d, J= 15.3, 1H), 3.56 - 3.42 (m, 2H), 3.24 (d, J= 7.8, 1H), 2.05 (s, 3H), 1.86 - 1.77 (m, 1H), 1.77 - 1.65 (m, 1H), 1.35 - 1.19 (m, 2H), 1.13 (s, 1H), 0.93 (d, J= 6.8, 3H), 0.50 (dd, J= 6.3, 9.8, 6H); 13C NMR (126 MHz, CDC13) δ 171.6, 171.5, 150.3, 147.7, 137.9, 131.4, 130.1, 129.0, 127.3, 109.9, 69.7, 58.6, 55.11, 46.9, 42.5, 36.7, 24.1, 22.4, 22.2, 16.2, 15.3; LRMS (ESI) 502.0 (M+H)+.
VL253: General Method C

VL254: General Method C

VL257: General Method C

VL258: General Method C

VL259: General Method C

VL260: General Method E

VL261: General Method E

VL262: General Method E

VL263: General Method E

VL264: General Method E

VL265: General Method E
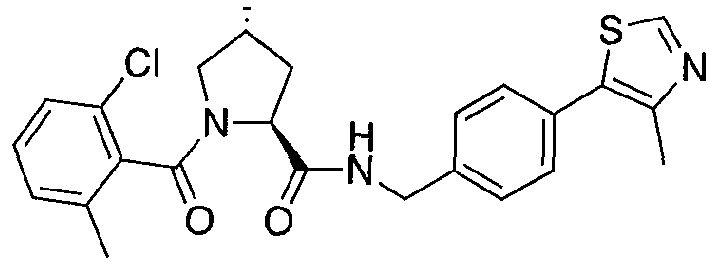
Examples (Compounds of Figure 15)
The following procedures were used to synthesize and/or characterize compounds according to the present invention as indicated
LCMS Method :
The analysis was conducted on an Acquity UPLC BEH CI 8 column (50mm x 2.1mm internal diameter 1.7μηι packing diameter) at 40°C.
The solvents employed were:
A = 0.1% v/v solution of formic acid in water.
B = 0.1 % v/v solution of formic acid in acetonitrile.
The gradient employed was as follows:
Time Flow Rate
% A % B
(minutes) (mL/min)
0 1 97 3
1.5 1 0 100
1.9 1 0 100
2.0 1 97 3
The UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
The following illustrates the mobile phases and gradients used when compounds underwent purification by mass-directed autopreparative HPLC.
Mass-Directed Autopreparative HPLC (Formic Acid Modifier)
The HPLC analysis was conducted on a Sunfire CI 8 column (150mm x 30mm internal diameter, 5μπι packing diameter) at ambient temperature.
The solvents employed were:
A = 0.1% v/v solution of formic acid in water.
B = 0.1% v/v solution of formic acid in acetonitrile.
Mass-Directed Autopreparative HPLC (Trifluoroacetic Acid Modifier)
The HPLC analysis was conducted on a Sunfire CI 8 column (150mm x 30mm internal diameter, 5μιη packing diameter) at ambient temperature.
The solvents employed were:
A = 0.1% v/v solution of trifluoroacetic acid in water.
B = 0.1% v/v solution of trifluoroacetic acid in acetonitrile.
Mass-Directed Autopreparative HPLC (Ammonium Bicarbonate Modifier)
The HPLC analysis was conducted on an XBridge CI 8 column (150mm x 30mm internal diameter, 5μηι packing diameter) at ambient temperature.
The solvents employed were:
A = 10 mM ammonium bicarbonate in water adjusted to pH 10 with ammonia solution. B = acetonitrile.
For each of the mass-directed autopreparative purifications, irrespective of the modifier used, the gradient employed was dependent upon the retention time of the particular compound undergoing purification as recorded in the analytical LCMS, and was as follows:
For compounds with an analytical LCMS retention time below 0.6 minutes the following gradient was used:

For compounds with an analytical LCMS retention time between 0.6 and 0.9 minutes the following gradient was used:
Time Flow Rate
% A % B
(minutes) (mL/min)
0 40 85 15
1 40 85 15
10 40 45 55
11 40 1 99
15 40 1 99
For compounds with an analytical LCMS retention time between 0.9 and 1.2 minutes the following gradient was used:

For compounds with an analytical LCMS retention time between 1.2 and 1.4 minutes the following gradient was used:

The UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
The chemical names were generated using ACD Name Pro version 6.02 from Advanced Chemistry Development, Inc.
Examples
(2S,4R)-l-(2-ethoxybenzoyl)-4-hydroxy-N-(4-(oxazoI-5-yl)benzyl)pyrroIidine-2- carboxamide

A solution of 2-ethoxybenzoic acid (commercially available from for example Aldrich) (29 mg, 0.17 mmol), (2S,4R)-4-hydroxy-N-(4-(oxazol-5-yl)benzyl)pyrrolidine-2-carboxamide (50 mg, 0.17 mmol) and DIPEA (0.061 mL, 0.35 mmol) in DMF (1 mL) was treated with HATU (80 mg, 0.21 mmol) and the mixture was stirred at ambient temperature for 2 hours. The product was then subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (38 mg, 0.087 mmol, 50% yield). LCMS RT= 0.72 min, ES+ve m/z 436 [M+H]+.
Using a method analogous to that for (2S,4R)-l-(2-ethoxybenzoyl)-4-hydroxy-N-(4-(oxazol- 5-yl)benzyl)pyrrolidine-2-carboxamide, the following compounds were prepared:
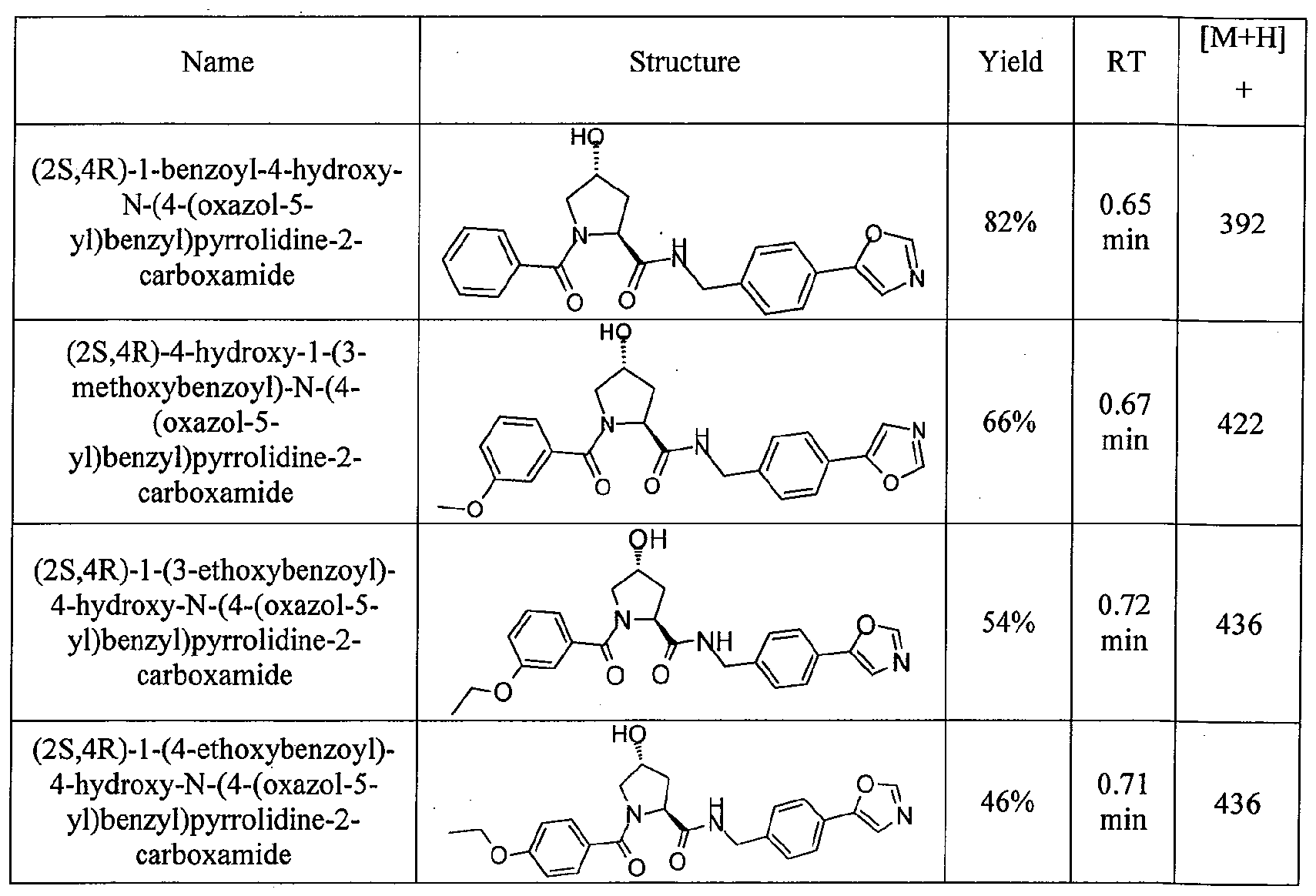

(S)-l-((2S,4R)-4-hydroxy-2-((4-(oxazol-5-yl)benzyl)carbamoyl)pyrrolidin-l-yl)-l- oxopropan-2-yl acetate and (2S,4R)-4-hydroxy-l-((S)-2-hydroxypropanoyl)-N-(4- (oxazol-5-yl)benzyl)pyrrolidine-2-carboxamide
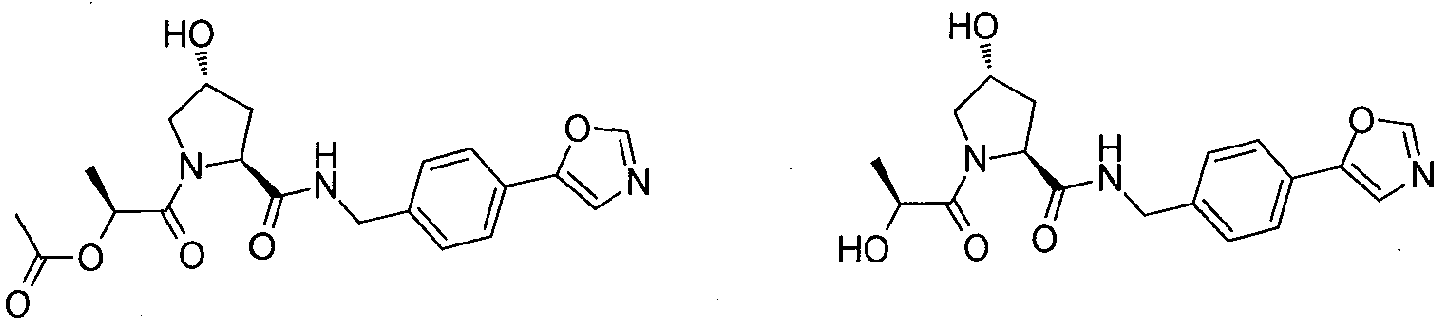
A stirred mixture of (2S,4R)-4-hydroxy-N-(4-(oxazol-5-yl)benzyl)pyrrolidine-2-carboxamide hydrochloride (60 mg, 0.1 mmol) and (S)-2-acetoxypropanoic acid (commercially available from for example Aldrich) (25 mg, 0.19 mmol) in DMF (1.2 mL) was treated with DIPEA (0.13 mL, 0.74 mmol) and then with HATU (85 mg, 0.22 mmol) and the mixture was stirred at ambient temperature for 30 minutes. Half of the reaction mixture was then subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford (S)-l-((2S,4R)-4-hydroxy-2-((4-(oxazol-5-yl)benzyl)carbamoyl)pyrrolidin-l-yl)-l- oxopropan-2-yl acetate (21 mg, 0.052 mmol, 28 % yield). LCMS RT= 0.58 min, ES+ve m/z 402 [M+H]+.
The remaining half of the reaction mixture was treated with ammonia (2M solution in methanol) (2 mL), sealed and allowed to stand for 1 day. The solution was then evaporated to dryness and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford (2S,4R)-4-hydroxy-l-((S)-2-hydroxypropanoyl)-N-(4- (oxazol-5-yl)benzyl)pyrrolidine-2-carboxamide (18 mg, 0.050 mmol, 27 % yield). LCMS RT= 0.53 min, ES+ve m/z 360 [M+H]+.
(2S,4R)-benzyl 4-hydroxy-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylate

An ice-cooled mixture of 2-(3-methylisoxazol-5-yl)acetic acid (commercially available from for example Aldrich) (0.91 g, 6.4 mmol) and (2S,4R)-benzyl 4-hydroxypyrrolidine-2- carboxylate, hydrochloride (1.67 g, 6.5 mmol) in DMF (9 mL) was treated with DIPEA (3.4 mL, 19 mmol) and then with HATU (2.56 g, 6.7 mmol) over 20 minutes. The mixture was stirred with cooling for 30 minutes and then overnight at ambient temperature. The mixture was then treated with saturated aqueous sodium bicarbonate (50 mL), extracted with dichloromethane (4 x 60 mL) and the combined organic phase was filtered through a hydrophobic frit and evaporated to dryness The product was purified by flash
chromatography (100 g cartridge) using a gradient elution from 0% to 15% methanol in
dichloromethane to afford the title compound (2.3 g, 6.7 mmol, quantitative). LCMS RT= 0.75 min, ES+ve m/z 345 [M+H]+.
(2S,4R)-N-((3,4-dihydro-2H-benzo[b][l,4]oxazin-2-yI)methyl)-4-hydroxy-l-(2-(3- methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxamide

A solution of (2S,4R)-4-hydroxy-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylic acid (90 mg, 0.35 mmol), (3,4-dihydro-2H-benzo[b][l,4]oxazin-2-yl)methanamine
(commercially available from for example Fluorochem) (58 mg, 0.35 mmol) and DIPEA (0.155 mL, 0.89 mmol) in DMF (2 mL) was treated with HATU (139 mg, 0.37 mmol) and stirred for 1 hour. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (84 mg, 0.21 mmol, 60 % yield) LCMS RT= 0.61 min, ES+ve m/z 401 [M+H]+.
(2S,4R)-N-(4-chlorobenzyl)-4-hydroxy-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2- carboxamide

A solution of (4-chlorophenyl)methanamine (commercially available from for example Aldrich) (0.021 mL, 0.17 mmol) and (2S,4R)-4-hydroxy-l-(2-(3-methylisoxazol-5- yl)acetyl)pyrrolidine-2-carboxylic acid (40 mg, 0.16 mmol) in DMF (1 mL) was treated with DIPEA (0.082 mL, 0.47 mmol) then with HATU (66 mg, 0.17 mmol) and the mixture was stirred at ambient temperature for 2 hours. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (24 mg, 0.064 mmol, 40 % yield). LCMS RT= 0.73 min, ES+ve m/z 378 [M+H]+.
(2S,4R)-4-hydroxy-l-(2-(3-methylisoxazol-5-yI)acetyl)-N-(4-(2-oxo-2,3- dihydrobenzo[d]oxazol-5-yl)benzyl)pyrrolidine-2-carboxamide

A mixture of (2S,4R)-4-hydroxy-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylic acid (60 mg, 0.24 mmol) and 5-(4-(aminomethyl)phenyl)benzo[d]oxazol-2(3H)-one, hydrochloride (65 mg, 0.24 mmol) in DMF (1.6 mL) was treated with DIPEA (0.124 mL, 0.71 mmol) and HATU (99 mg, 0.26 mmol) and the mixture was stirred for 30 minutes. The product was then subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (64 mg, 0.13 mmol, 57% yield). LCMS RT= 0.70 min, ES+ve m/z All [M+H]+.
(2S,4R)-N-(l-(benzofuran-2-yl)ethyl)-4-hydroxy-l-(2-(3-methylisoxazol-5- yl)acetyl)pyrrolidine-2-carboxamide
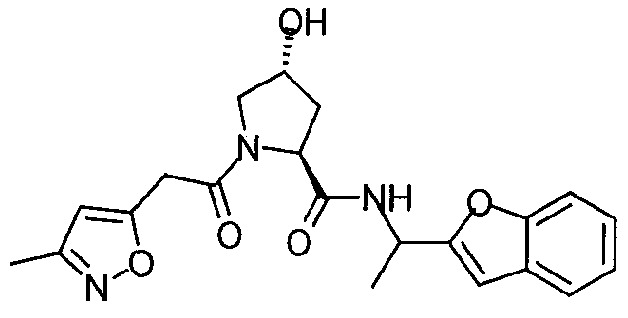
A stirred solution of (2S,4R)-4-hydroxy-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2- carboxylic acid (90 mg, 0.35 mmol), l-(benzofuran-2-yl)ethanamine (commercially available from for example Enamine) (57 mg, 0.35 mmol) and DIPEA (0.155 mL, 0.89 mmol) in DMF (2 mL) was treated with HATU (139 mg, 0.37 mmol). After 1 hour the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (91 mg, 0.21 mmol, 65 % yield) LCMS RT= 0.79 min, ES+ve m/z 398 [M+H]+.
(2S,4R)-N-([l,l'-biphenyl]-4-ylmethyl)-4-hydroxy-l-(2-(3-methylisoxazol-5- yl)acetyl)pyrrolidine-2-carboxamide

A stirred mixture of (2S,4R)-4-hydroxy-l-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2- carboxylic acid (30 mg, 0.12 mmol) and [l,l'-biphenyl]-4-ylmethanamine (commercially available from for example Aldrich) (22 mg, 0.12 mmol) in DMF (0.8 mL) was treated with
DIPEA (0.08 mL, 0.47 mmol) and then with HATU (49 mg, 0.13 mmol) and the mixture was stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (40 mg, 81 % yield). LCMS RT= 0.86 min, ES+ve m/z 420 [M+H]+.
Using a method analogous to that for (2S,4R)-N-([l,l '-biphenyl]-4-ylmethyl)-4-hydroxy-l - (2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxamide the following compounds were prepared:


(2S,4R)-l-((S)-2-acetamidopropanoyl)-4-hydroxy-N-phenethylpyrrolidine-2- carboxamide

A stirred mixture of (2S,4R)-l-((S)-2-acetamidopropanoyl)-4-hydroxypyrrolidine-2- carboxylic acid (24 mg, 0.10 mmol) and 2-phenylethanamine (commercially available from for example Aldrich) (0.012 mL, 0.10 mmol) in DMF (0.8 mL) was treated with DIPEA (0.07 mL, 0.39 mmol) and then with HATU (45 mg, 0.12 mmol), and the mixture was stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass- directed automated preparative HPLC (formic acid modifier) to afford the title compound (25 mg, 72% yield). LCMS RT= 0.56 min, ES+ve m/z 348 [M+H]+.
Using a method analogous to that for (2S,4R)-l-((S)-2-acetamidopropanoyl)-4-hydroxy-N- phenethylpyrrolidine-2-carboxamide the following compounds were prepared:
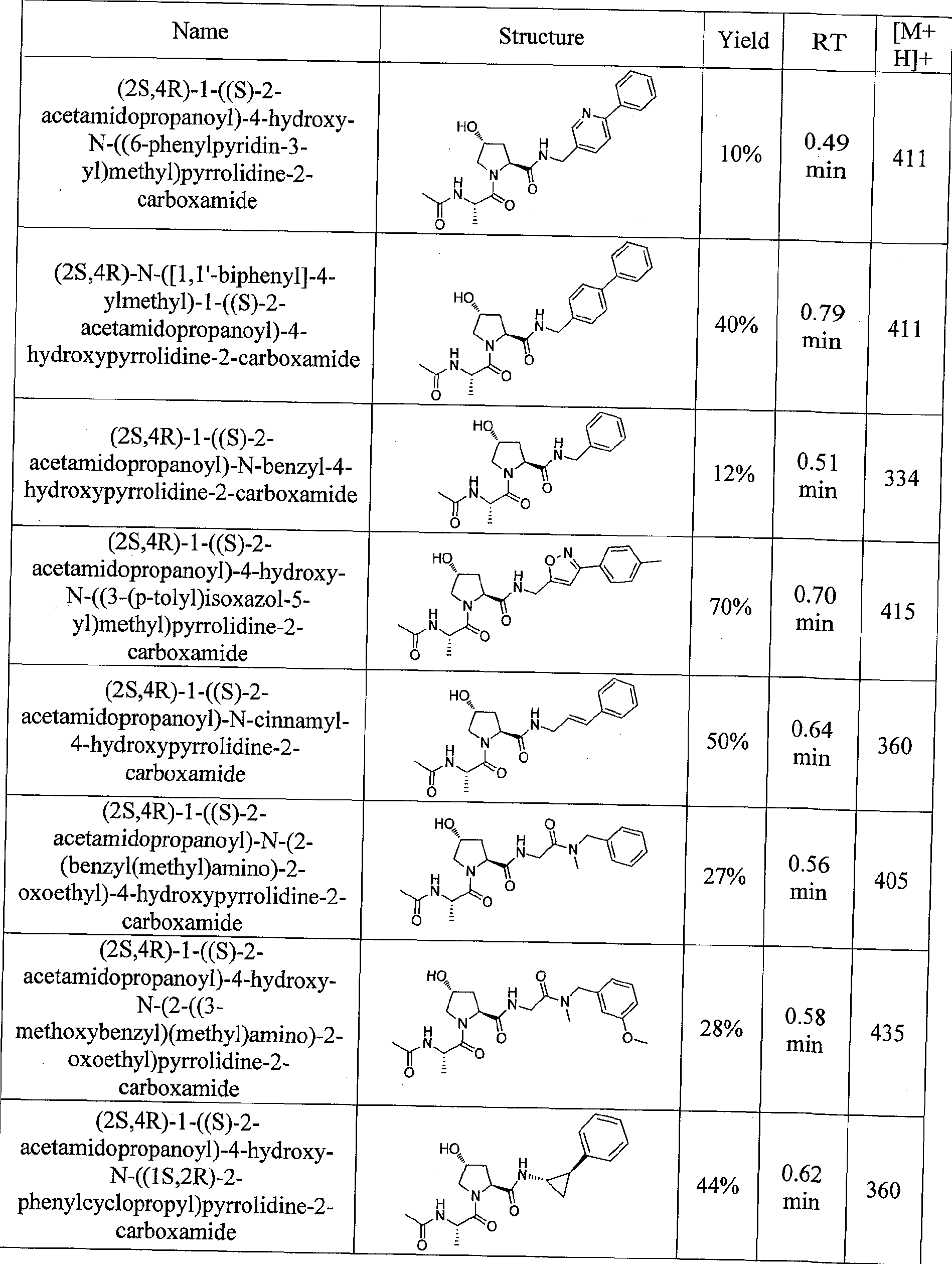

(S)-l-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrroIidin-l-yl)- l-oxopropan-2-yl acetate & (2S,4R)-l-acetyl-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrroIidine-2-carboxamide

An ice-cooled mixture of (2S,4R)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine- 2-carboxamide, hydrochloride (31 mg, 0.088 mmol) and (S)-2-acetoxypropanoic acid (commercially available from for example Aldrich) (8 μί, 0.09 mmol) in DMF (0.8 mL) was treated with DIPEA (0.074 mL, 0.42 mmol). HATU (34 mg, 0.088 mmol) was then added portion- wise over 10 minutes and the mixture was stirred at ambient temperature for 1 hour. The products were separated and purified by mass-directed automated preparative HPLC (formic acid modifier) to afford (S)-l-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidin-l-yl)-l-oxopropan-2-yl acetate (15 mg, 0.035 mmol, 41 % yield) LCMS RT= 0.64 min, ES+ve m/z 432 [M+H]+ and (2S,4R)-l-acetyl-4-hydroxy-N-(4- (4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (9 mg, 0.025 mmol, 30 % yield) LCMS RT= 0.57 min, ES+ve m/z 360 [M+H]+.
(2S,4R)-l-(2-(cyanomethyl)benzoyI)-4-hydroxy-N-(4-(4-methyIthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide

A stirred mixture of (2S,4R)-4-hydroxy-N-(4-(thiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (50 mg, 0.17 mmol) and 2-(cyanomethyl)benzoic acid (commercially available from for example Aldrich) (29 mg, 0.18 mmol) in DMF (0.7 mL) was treated with DIPEA (0.086 mL, 0.49 mmol) and then with HATU (69 mg, 0.18 mmol) and the mixture was stirred at ambient temperature for 10 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (31 mg, 0.067 mmol, 41 % yield). LCMS RT= 0.73 min, ES+ve m/z 461 [M+H]+.
Using a method analogous to that for (2S,4R)-l-(2-(cyanomethyl)benzoyl)-4-hydroxy-N-(4- (4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide the following compounds were prepared:




cyclopentyIpropanoyl)-4-
Mixture of
hydroxy-N-(4-(4- 0.82 methyIthiazol-5- diastereoisome 67% 442 min rs
yI)benzyI)pyrrolidine-2- carboxamide
OH
(2S,4R)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)- 1 - Mixture of
0.68 (2- diastereoisome 53% 459 morpho]inopropanoyl)pyrrolid min
rs
ine-2-carboxamide
Single
enantiomer,
stereochemistr
(2S,4R)-4-hydroxy-l- HO y unknown at
(indoline-2-carbonyl)-N-(4-(4- the undefined
methylthiazol-5- chiral centre, 0.70
64% 463 yi)benzyl)pyrrolidine-2- eluted first min
carboxamide during HPLC
purification
(formic acid
modifier)
Single
enantiomer,
(2S,4R)-l-(2-(4-chloro-lH- stereochemistr
OH y unknown at
pyrazol-l-yl)propanoyl)-4- hydroxy-N-(4-(4- the undefined
0.74 chiral centre, 39%
methylthiazol-5- 474 min eluted first
yl)benzyl)pyrrolidine-2- carboxamide during HPLC
purification
(formic acid
modifier)
(2S,4R)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)- 1 - Mixture of
(2-(l -oxoisoindolin-2- 0.73
diastereoisome 51% 505 yl)propanoyI)pyrrolidine-2- min
rs
carboxamide
Single
enantiomer,
(2S,4R)-l-(2-(3- stereochemistr
(difluoromethyl)-5-methyl- y unknown at
1 H-pyrazol- 1 -yl)propanoyl)- the undefined
hydroxy-N-(4-(4- 0.76
chiral centre, 31% 504 methylthiazol-5- min
eluted second
yl)benzyl)pyrrolidine-2- during HPLC
carboxamide purification
(formic acid
modifier)
(2S,4R)-4-hydroxy- 1 -(1 H- HQ
indole-2-carbonyl)-N-(4-(4- methylthiazol-5- 0.78
65% 461 yl)benzyl)pyrrolidine-2- min
carboxamide
 Single
Single
enantiomer,
9H stereochemistr
(2S,4R)-4-hydroxy-N-(4-(4- y unknown at
methylthiazol-5-yl)benzyl)- 1 - the undefined
0.78 (2- chiral centre, 44% 450 phenylpropanoyl)pyrrolidine- min
eluted first
2-carboxamide during HPLC
purification
(formic acid
modifier)
Single
enantiomer,
PH stereochemistr
(2S,4R)-4-hydroxy-N-(4-(4- y unknown at
methylthiazol-5-yl)benzyl)-l - the undefined
0.80 (2- chiral centre, 41% 450 min phenylpropanoyl)pyrrolidine- eluted second
2-carboxamide during HPLC
purification
(formic acid
modifier)
Single
enantiomer,
QH stereochemistr
(2S,4R)-4-hydroxy-N-(4-(4- y unknown at
methyIthiazol-5-yl)benzyl)- 1 - the undefined
0.62
(2-(2-oxopyridin- 1 (2H)- chiral centre, 33% 467 yl)propanoyl)pyrrolidine-2- min
eluted first
carboxamide during HPLC
purification
(formic acid
modifier)
Single
enantiomer,
stereochemistr
OH
(2S,4R)-4-hydroxy-l-(2- y unknown at
(indolin- 1 -yl)propanoyl)-N-(4- the undefined
(4-methylthiazol-5- chiral centre, 0.86
35% 491 yl)benzyl)pyrrolidine-2- min
eluted first
carboxamide during HPLC
purification
(formic acid
modifier)
Single
enantiomer,
stereochemistr
9H
(2S,4R)-4-hydroxy-l-(2- y unknown at
(indolin-1 -yl)propanoyl)-N-(4- the undefined
(4-methylthiazoI-5- 0.88
chiral centre, 40% 491 yl)benzyl)pyrrolidine-2- min
eluted second
carboxamide during HPLC
purification
(formic acid
modifier)
(2S,4R)-4-hydroxy-l-(2- HO
methyI-3- Mixture of
morpholinopropanoyl)-N-(4- 0.52
diastereoisome 57% 472 (4-methylthiazol-5 - min
rs
yl)benzyl)pyrrolidine-2- 0 o


3-(2-(2-(2-(3-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidine-l-carbonyl)phenoxy)ethoxy)ethoxy)ethoxy)propanoic acid

An ice-cooled mixture of (2S,4 )-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine- 2-carboxamide, hydrochloride (682 mg, 2.2 mmol), 3-((14,14-dimethyl-12-oxo-3,6,9,13- tetraoxapentadecyl)oxy)benzoic acid (778 mg, 2.0 mmol), DIPEA (1.36 mL, 7.8 mmol) in DMF (12 mL) was treated with HATU (817 mg, 2.2 mmol). The mixture was allowed to warm to ambient temperature and stirred for 30 minutes then treated with water (70 mL) and extracted with ethyl acetate (3 x 70 mL). The combined organic phase was washed with saturated aqueous sodium bicarbonate (100 mL), water (100 mL), brine (100 mL), dried over magnesium sulfate, filtered and evaporated to dryness. The crude product was dissolved in dichloromethane (6 mL) and treated with TFA (2.0 mL). After 1 hour, the reaction mixture was evaporated to dryness and the product was purified by flash chromatography (60 g CI 8 cartridge) using a gradient elution from 10 to 95% acetonitrile (+ 0.1% formic acid) in water (+ 0.1% formic acid) to afford the title compound (568 mg, 45% yield). LCMS RT= 0.73 min, ES+ve m/z 642 [M+H]+.
(2S,4R)-4-hydroxy-l-(3-(2-methoxyethoxy)benzoyl)-N-(4-(4-methylthiazoI-5- yl)benzyl)pyrrolidine-2-carboxamide

A mixture of (2S,4R)-4-hydroxy-l-(3-hydroxybenzoyl)-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide (55 mg, 0.13 mmol) and potassium carbonate (55 mg, 0.40 mmol) in DMF (0.8 mL) was treated with l-bromo-2-methoxyethane (commercially available from for example Aldrich) (0.024 mL, 0.25 mmol) and stirred at 50°C for 2.5 hours. Additional 1 -bromo-2-methoxyethane (0.024 mL, 0.25 mmol) was added and the mixture stirred at 50°C overnight. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (41 mg, 0.083 mmol, 66 % yield) LCMS RT= 0.74 min, ES+ve m/z 496 [M+H]+
(2S,4R)-4-hydroxy-l-(3-(2-(2-methoxyethoxy)ethoxy)benzoyl)-N-(4-(4-methylthiazol-5- yl)benzyI)pyrrolidine-2-carboxamide
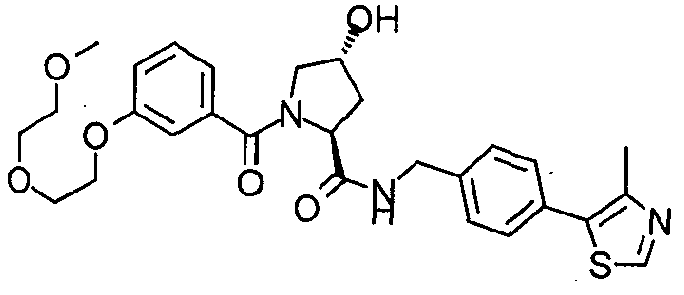
A mixture of (2S,4R)-4-hydroxy-l-(3-hydroxybenzoyl)-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide (55 mg, 0.13 mmol) and potassium carbonate (55 mg, 0.40 mmol) in DMF (0.8 mL) was treated with l-bromo-2-(2-methoxyethoxy)ethane
(commercially available from for example Aldrich) (0.034 mL, 0.25 mmol) and the reaction stirred at 50°C for 2.5 hours. Additional l-bromo-2-(2-methoxyethoxy)ethane (0.034 mL, 0.25 mmol) was added and the mixture stirred at 50°C overnight. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (40 mg, 0.074 mmol, 59 % yield) LCMS RT= 0.74 min, ES+ve m/z 540 [M+H]+.
(2S,4R)-l-((S)-2-((S)-2-acetamido-4-methylpentanamido)propanoyl)-4-hydroxy-N-(4-(4- methyIthiazoI-5-yl)benzyl)pyrrolidine-2-carboxamide

A stirred mixture of (2S,4R)-l-((S)-2-aminopropanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (50 mg, 0.12 mmol) and (S)-2- acetamido-4-methylpentanoic acid (commercially available from for example Aldrich) (22 mg, 0.12 mmol) in DMF (0.7 mL) was treated with DIPEA (0.062 mL, 0.35 mmol) and then with HATU (49 mg, 0.13 mmol) and the mixture was stirred at ambient temperature for 10 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (42 mg, 0.077 mmol, 66 % yield). LCMS RT= 0.68 min, ES+ve m/z 544 [M+H]+.
(2S,4R)-l-((S)-2-acetamido-3-methylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide
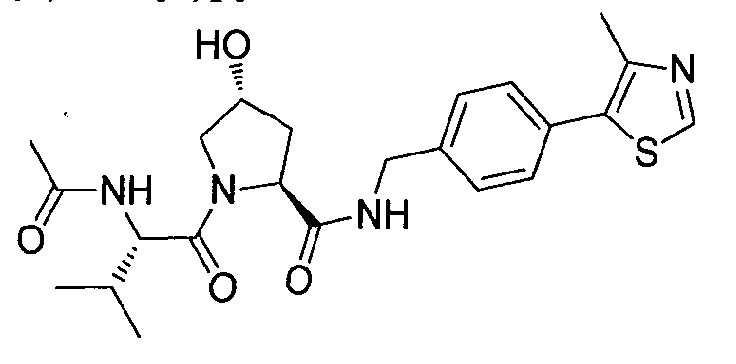
A solution of tert-butyl ((S)-l-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidin-l-yl)-3 -methyl- l-oxobutan-2-yl)carbamate (120 mg, 0.23 mmol) in dichloromethane (2 mL) and treated with 4 M hydrochloric acid in 1,4-dioxane (1 mL). The mixture was stirred at ambient temperature for 30 minutes and was then evaporated to dryness. The residue was dissolved in DMF (1 mL) and treated with triethylamine (0.08 mL, 0.58 mmol), followed by acetic anhydride (0.02 mL, 0.21 mmol) and the mixture was stirred at ambient temperature for 1 hour. The product was subjected to purification by mass- directed automated preparative HPLC (formic acid modifier) to afford the title compound (53 mg, 49% yield). LCMS RT= 0.65 min, ES+ve m/z 460 [M+H]+.
Using a method analogous to that for (2S,4R)-l-((S)-2-acetamido-3-methylbutanoyl)-4- hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, the following compounds were prepared:
Name Structure Yield RT [M+H]+

(2S,4R)-l-((S)-2-(3-ethoxy-N-methylbenzamido)propanoyl)-4-hydroxy-N-(4-(4- methylthiazoI-5-yl)benzyI)pyrrolidine-2-carboxamide

A mixture of (2S,4R)-4-hydroxy-l-((S)-2-(methylamino)propanoyl)-N-(4-(4-methylthiazol- 5-yl)benzyl)pyrrolidine-2-carboxamide (20 mg, 0.05 mmol), DIPEA (0.043 mL, 0.25 mmol) and 3-ethoxybenzoic acid (commercially available from for example Aldrich) (8 mg, 0.05
mmol) in DMF (1 mL) was treated with HATU (19 mg, 0.05 mmol) and the mixture was stirred for 20 minutes. The product was purified by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (14 mg, 0.025 mmol, 50 % yield). LCMS RT= 0.82 min, ES+ve m/z 551 [M+H]+.
(2S,4R)-4-hydroxy-l-((S)-2-(3-methoxypropanamido)propanoyl)-N-(4-(4-methylthiazol- 5-yl)benzyl)pyrrolidine-2-carboxamide

A solution of a mixture of (25',4i?)-l-((5)-2-aminopropanoyl)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (107 mg, 0.25 mmol), 3 -methoxypropionic acid (commercially available from for example Aldrich) (0.028 mL, 0.30 mmol) and DIPEA (0.2 mL, 1.1 mmol) in dry DMF (3 mL) was treated with HATU (115 mg, 0.30 mmol). The mixture was stirred at ambient temperature for 30 minutes. The mixture was loaded onto a methanol-preconditioned aminopropyl solid-phase extraction cartridge (2g), which was eluted with methanol (3 column volumes). The resulting eluant was evaporated to dryness and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (57 mg, 0.12 mmol, 48 % yield). LCMS RT= 0.62 min, ES+ve m/z 475 [M+H]+.
(2S,4R)-4-hydroxy-l-(2-(3-methoxypropanamido)-2-methylpropanoyI)- V-(4-(4- rboxamide

A solution of a mixture of (2.S',4i?)-l-(2-amino-2-methylpropanoyl)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (95 mg, 0.24 mmol), 3- methoxypropionic acid (0.028 mL, 0.30 mmol) and DIPEA (0.2 mL, 1.15 mmol) in dry DMF (3 mL) was treated with HATU (115 mg, 0.30 mmol) and the mixture was stirred at ambient temperature for 30 minutes The mixture was loaded onto a methanol-preconditioned aminopropyl solid-phase extraction cartridge (NH2) which was eluted with methanol (3 column volumes). The resulting eluant was evaporated to dryness and the product was
subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (45 mg, 0.09 mmol, 37 % yield). LCMS RT= 0.68 min, ES+ve m/z 489 [M+H]+.
(2S,4R)-4-hydroxy-l-((5)-2-(2-methoxyacetamido)-3-methylbutanoyl)-iV-(4-(4- methylthiazol-5-yl)benzyI)pyrroIidine-2-carboxamide

A mixture of (25',4i?)-l-((5)-2-amino-3-methylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (20 mg, 0.044 mmol), 2-methoxyacetic acid (3 nL, 0.039 mmol) and DIPEA (0.035 mL, 0.20 mmol) in DMF (1 mL) was treated with HATU (18 mg, 0.047 mmol) and stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (14 mg, 0.029 mmol, 73 % yield). LCMS RT=
0.70 min, ES+ve m/z 489 [M+H]+.
(2S,4 )-4-hydroxy-l-((S)-2-(2-methoxy^
methylthiazoI-5-yl)benzyl)pyrrolidine-2-carboxainide

A mixture of (25,,4 ?)-4-hydroxy- 1 -((5')-3-methyl-2-(methylamino)butanoyl)-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (19 mg, 0.041 mmol), 2-methoxyacetic acid (3 μί, 0.039 mmol) and DIPEA (0.035 mL, 0.20 mmol) in DMF (1 mL) was treated with HATU (18 mg, 0.047 mmol) and stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (16 mg, 0.032 mmol, 81 % yield). LCMS RT= 0.70 min, ES+ve m/z 503 [M+H]+.
W 201
169
(2S,4R)-l-((S)-2-(N,3-dimethyloxetane-3-carboxamido)propanoyl)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide

A mixture of (2S,4R)-4-hydroxy-l-((S)-2-(methylamino)propanoyl)-N-(4-(4-methylthiazol- 5-yl)benzyl)pyrrolidine-2-carboxamide (15 mg, 0.037 mmol), DIPEA (0.032 mL, 0.18 mmol) and 3-methyloxetane-3-carboxylic acid (commercially available from for example
Fluorochem) (3 μΐ,, 0.037 mmol) in DMF (1 mL) was treated with HATU (14 mg, 0.037 mmol) and the mixture was stirred for 1 hour. The product was purified by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (9 mg, 0.019 mmol, 51 % yield). LCMS RT= 0.62 min, ES+ve m/z 501 [M+H]+. loxetane-3-carboxamido)propanoyl)-N-(4-(4- e-2-carboxamide

A stirred mixture of (2S,4R)-l-((S)-2-aminopropanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide (21 mg, 0.043 mmol), DIPEA (0.043 mL, 0.25 mmol) and 3-methyloxetane-3-carboxylic acid (commercially available from for example
Fluorochem) (6 mg, 0.05 mmol) in DMF (2 mL) was treated with HATU (19 mg, 0.05 mmol) and stirred for 30 minutes. The product was purified by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (14 mg, 0.029 mmol, 60 % yield). LCMS RT= 0.59 min, ES+ve m/z 487 [M+H]+.
(2S,4R)-l-((S)-2-(3-ethoxybenzamido)propanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide

A stirred mixture of 3-ethoxybenzoic acid (commercially available from for example
Aldrich) (20 mg, 0.12 mmol) and (2S,4R)-l-((S)-2-aminopropanoyl)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (56 mg, 0.13 mmol) in
DMF (3.2 mL) was treated with DIPEA (0.063 mL, 0.36 mmol) and then with HATU (50 mg, 0.13 mmol) and the mixture was stirred at ambient temperature for 30 minutes. The product was then subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (36 mg, 0.067 mmol, 56 % yield). LCMS RT= 0.80 min, ES+ve m/z 537 [M+H]+.
(2S,4R)-N-((lH-indol-3-yI)methyl)-4-hydroxy-l-((S)-2-(l-oxoisoindolin-2- yl)propanoyl)pyrroIidine-2-carboxamide

A solution of (2S,4R)-4-hydroxy-l-((S)-2-(l-oxoisoindolin-2-yl)propanoyl)pyrrolidine-2- carboxylic acid (10 mg, 0.031 mmol), DIPEA (0.038 mL, 0.22 mmol) and (lH-indol-3- yl)methanamine (commercially available from for example Fluorochem) (6 mg, 0.041 mmol) in DMF (0.8 mL) was treated with HATU (15 mg, 0.039 mmol) and stirred for 1 hour. The product was purified by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (3.1 mg, 6.9 μηιοΐ, 22 % yield). LCMS RT= 0.75 min, ES+ve m/z 447 [M+H]+.
(2S,4R)-N-((R)-2,3-dihydrobenzofuran-3-yl)-4-hydroxy-l-((S)-2-(l-oxoisoindoIin-2- yl)propanoyl)pyrrolidine-2-carboxamide & (2S,4R)-N-((S)-2,3-dihydrobenzofuran-3-yl)- 2-carboxamide

A mixture of 2,3-dihydrobenzofuran-3-amine (commercially available from for example Chem-Impex International, Inc.) (13 mg, 0.094 mmol) and (2S,4R)-4-hydroxy-l-((S)-2-(l- oxoisoindolin-2-yl)propanoyl)pyrrolidine-2-carboxylic acid (25 mg, 0.079 mmol) in DMF (0.8 mL) was treated with DIPEA (0.055 mL, 0.31 mmol) and then HATU (33 mg, 0.086 mmol) and the mixture was stirred at ambient temperature for 30 minutes. The product mixture was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compounds: Isomer 1 (first-eluting) (12 mg, 0.027 mmol, 35 % yield). LCMS RT= 0.73 min, ES+ve m/z 436 [M+H]+. Isomer 2 (second-eluting) (13 mg, 0.030 mmol, 38% yield). LCMS RT= 0.74 min, ES+ve m/z 436 [M+H]+.
Using a method analogous to that for the two diastereoisomers of (2S,4R)-N-(2,3- dihydrobenzofuran-3 -yl)-4-hydroxy- 1 -((S)-2-( 1 -oxoisoindolin-2-yl)propanoyl)pyrrolidine-2- carboxamide, the following compounds were prepared:


(2S,4R)-4-hydroxy-N-(2-hydroxy-4-(4-methylthiazol-5-yl)benzyl)-l-((S)-3-methyl-2-(l- oxoisoindolin-2-yl)butanoyI)pyrrolidine-2-carboxamide

A mixture of (2S,4R)-4-hydroxy-N-(2-hya^oxy-4-(4-methylthiazol-5-yl)benzyl)pyrrolidine- 2-carboxamide, hydrochloride (125 mg, 0.34 mmol) and (S)-3-methyl-2-(l-oxoisoindolin-2- yl)butanoic acid (83 mg, 0.36 mmol) in DMF (1.6 mL) was treated with DIPEA (0.24 mL, 1.4 mmol) and HATU (140 mg, 0.37 mmol) and the mixture was stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (120 mg, 0.22 mmol, 65 % yield). LCMS RT= 0.81 min, ES+ve m/z 549 [M+H]+. ethylthiazol-5-yl)benzyl)-l-((R)-3-methyl-2-(l- 2-carboxamide

A mixture of (2S,4R)-4-hydroxy-N-(2-hydroxy-4-(4-methylthiazol-5-yl)benzyl)pyrrolidine- 2-carboxamide, hydrochloride (65 mg, 0.18 mmol) and (R)-3-methyl-2-(l-oxoisoindolin-2- yl)butanoic acid (43 mg, 0.19 mmol) in DMF (1.6 mL) was treated with DIPEA (0.123 mL, 0.70 mmol) and HATU (74 mg, 0.19 mmol) and the mixture was stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (64 mg, 0.12 mmol, 66 % yield). LCMS RT= 0.80 min, ES+ve m/z 549 [M+H]+.
,Λ Λ,, Α ί:
WO 2013/106646
173 tert-butyl ((S)-l-((2R,4S)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidin-l-yl)-l-oxopropan-2-yl)(methyl)carbamate

A stirred mixture of (S)-2-((tert-butoxycarbonyl)(methyl)amino)propanoic acid (1 15 mg, 0.57 mmol) and (2R,4S)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxarnide, hydrochloride (200 mg, 0.57 mmol) in DMF (0.7 mL) was treated with DIPEA (0.4 mL, 2.3 mmol) and then with HATU (215 mg, 0.57 mmol) and the mixture was stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (146 mg, 0.29 mmol, 51 % yield). LCMS RT= 0.84 min, ES+ve m/z 503 [M+H]\
(2S,4R)-4-hydroxy-l-((S)-2-(3-methoxy-N-methylpropanamido)propanoyl)-:
methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide

A stirred mixture of (2S,4R)-4-hydroxy-l-((S)-2-(methylamino)propanoyl)-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (50 mg, 0.12 mmol) and 3- methoxypropanoic acid (commercially available from for example Aldrich) (0.013 mL, 0.14 mmol) in DMF (0.8 mL) was treated with DIPEA (0.087 mL, 0.50 mmol) and then with HATU (52 mg, 0.14 mmol). The mixture was stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (43 mg, 71% yield). LCMS RT= 0.61 min, ES+ve m/z 489 [M+H]+.
(2S,4R)-4-hydroxy-l-((S)-2-((2-methoxyethyI)(methyl)amino)propanoyl)-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide

A stirred mixture of l-bromo-2-methoxyethane (commercially available from for example Aldrich) (0.013 mL, 0.14 mmol) and (2S,4R)-4-hydroxy-l-((S)-2-(memylamino)propanoyl N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (50 mg, 0.12 mmol) in DMF (0.8 mL) was treated with DIPEA (0.054 mL, 0.31 mmol) and the mixture was stirred at 85 °C for -18 hours. The reaction mixture was cooled and the product was subjected to purification by mass-directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title compound (44 mg, 77% yield). LCMS RT= 0.51 min, ES+ve m/z 461 [M+H]+.
(25,4J?)-4-hydroxy-l-((S)-4-(2-methoxyacetyl)morpholine-3-carbonyl)-N-(4-(4- methylthiazol-5-yl)benzyI)pyrrolidine-2-carboxamide

A mixture of (25',4if)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-l-((5)-morpholine-3- carbonyl)pyrrolidine-2-carboxamide, hydrochloride (19 mg, 0.041 mmol), 2-methoxyacetic acid (3 nL, 0.039 mmol) and DIPEA (0.035 mL, 0.20 mmol) in DMF (1 mL) was treated with HATU (18 mg, 0.047 mmol) and stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (13 mg, 0.026 mmol, 66 % yield). LCMS RT= 0.60 min, ES+ve m/z 503 [M+H]+.
(2S,4R)-N-(4-(2,4-dimethylthiazol-5-yl)benzyl)-4-hydroxy-l-(2-(3-methylisoxazoI-5- yl)acetyI)pyrrolidine-2-carboxamide

A stirred mixture of (2S,4R)-N-(4-(2,4-dimethylthiazol-5-yl)benzyl)-4-hydroxypyrrolidine-2- carboxamide (30 mg, 0.09 mmol) and 2-(3-methylisoxazol-5-yl)acetic acid (commercially available from for example Aldrich) (13 mg, 0.09 mmol) in DMF (0.8 mL) was treated with DIPEA (0.063 mL, 0.36 mmol) and then with HATU (41 mg, 0.11 mmol) and stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass- directed automated preparative HPLC (formic acid modifier) to afford the title compound (26 mg, 63% yield). LCMS RT= 0.66 min, ES+ve m/z 455 [M+H]+.
(2S,4R)-l-((S)-2-acetamidopropanoyl)-N-(4-(2,4-dimethylthiazol-5-yI)benzyl)-4-

A stirred mixture of (2S,4R)-N-(4-(2,4-dimethylthiazol-5-yl)benzyl)-4-hydroxypyrrolidine-2- carboxamide (30 mg, 0.09 mmol) and (S)-2-acetamidopropanoic acid (commercially available from for example Aldrich) (12 mg, 0.09 mmol) in DMF (0.8 mL) was treated with DIPEA (0.063 mL, 0.36 mmol) and then with HATU (41 mg, 0.11 mmol), and the mixture was stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (30 mg, 75% yield). LCMS RT= 0.58 min, ES+ve m/z 445 [M+H]+.
(2S,4R)-N-(4-bromobenzyl)-l-(3-ethoxybenzoyl)-4-hydroxypyrrolidine-2-carboxamide

An ice-cooled mixture of (2S,4R)-l-(3-ethoxybenzoyl)-4-hydroxypyrrolidine-2-carboxylic acid (73 mg, 0.26mmol) and (4-bromophenyl)methanamine, hydrochloride (commercially available from for example Aldrich) (58 mg, 0.26 mmol) in DMF (0.5 mL) was treated with a solution of DIPEA (0.145 mL, 0.83 mmol) in DMF (1 mL) and then with HATU (105 mg, 0.28 mmol) and stirred overnight at ambient temperature. The product was then subjected to
purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (62 mg, 53% yield). LCMS RT= 0.90 min, ES+ve m/z 447,449 [M+H]+.
(2S,4R)-N-([l, -biphenyl]-4-ylmethyl)-l-(3-ethoxybenzoyI)-4-hydroxypyrrolidine-2- carboxamide

A mixture of (2S,4R)-l-(3-ethoxybenzoyl)-4-hydroxypyrrolidine-2-carboxylic acid (30 mg, 0.11 mmol) and [l,l'-biphenyl]-4-ylmethanamine (commercially available from for example Aldrich) (20 mg, 0.11 mmol) in DMF (0.8 mL) was treated with DIPEA (0.075 mL, 0.43 mmol) and then with HATU (45 mg, 0.12 mmol) and stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (26 mg, 55% yield). LCMS RT= 0.98 min, ES+ve m/z 445 [M+H]+.
Using a method analogous to that for 2S,4R)-N-([l,l'-biphenyl]-4-ylmethyl)-l-(3- ethoxybenzoyl)-4-hydroxypyrrolidine-2-carboxamide, the following compounds were prepared:


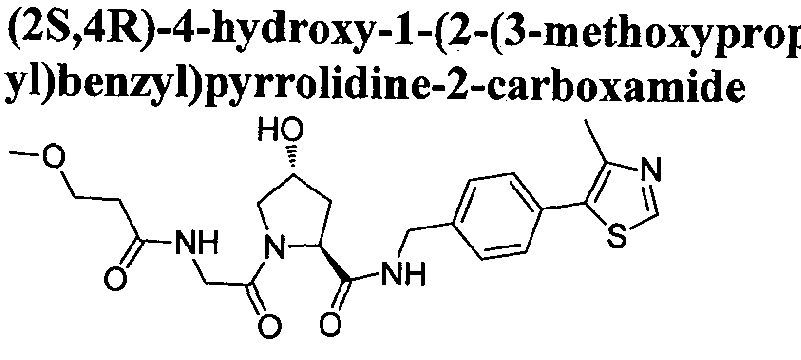
A mixture of (2S,4R)-l-(2-aminoacetyl)-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide (134 mg, 0.33 mmol) and 3-methoxypropanoic acid (commercially available from for example Aldrich) (37 mg, 0.36 mmol) in DMF (0.8 mL)
was treated with DIPEA (0.23 mL, 1.3 mmol) and then with HATU (136 mg, 0.36 mmol) and stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (36 mg, 24% yield). LCMS RT= 0.56 min, ES+ve m/z 461 [M+H]+.
(2S,4R)-l-((S)-3,3-dimethyl-2-(3-methyloxetane-3-carboxamido)butanoyl)-4-hydroxy-N- (4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide

A stirred mixture of (2S,4R)-l-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (20 mg, 0.04 mmol) and 3-methyloxetane-3-carboxylic acid (commercially available from for example
Chemgenx) (5 mg, 0.04 mmol) in DMF (0.6 mL) was treated with DIPEA (0.03 mL, 0.17 mmol) and then with HATU (20 mg, 0.05 mmol), and the mixture was stirred at ambient temperature for 1 hour. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to give the title compound (18 mg, 80% yield). LCMS RT= 0.76 min, ES+ve m/z 529 [M+H]+.
Using a method analogous to that for (2S,4R)-l-((S)-3,3-dimethyl-2-(3-methyloxetane-3- carboxamido)butanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, the following compounds were prepared:


carboxamide
(S)-N-((S)-l-((2S,4R)-4-hydroxy-2-((4-(4-methyIthiazol-5- yl)benzyl)carbamoyl)pyrroIidin-l-yl)-3,3-dimethyl-l-oxobutan-2-yl)morpholine-3- carboxamide, hydrochloride

A mixture of (2S,4R)-l-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (40 mg, 0.086 mmol) and (S)-4-(tert- butoxycarbonyl)morpholine-3-carboxylic acid (commercially available f om for example Astatech, Inc.) (20 mg, 0.086 mmol) in DMF (0.6 mL) was treated with DIPEA (0.06 mL, 0.35 mmol) and then with HATU (40 mg, 0.10 mmol) and stirred at ambient temperature for 1 hour. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to give the intermediate Boc-protected product. The intermediate was then dissolved in a mixture of dichloromethane (1 mL) and methanol (0.5 mL) and treated with 4M hydrochloric acid in 1,4-dioxane (0.4 mL, 1.6 mmol), After stirring at ambient temperature for 1 hour, the mixture was evaporated to dryness to afford the title compound (31 mg, 62% yield). LCMS RT= 0.60 min, ES+ve m/z 544 [M+H]+.
Using a method analogous to that for (S)-N-((S)-l-((2S,4R)-4-hydroxy-2-((4-(4- methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-l-yl)-3,3-dimethyl-l -oxobutan-2- yl)morpholine-3 -carboxamide, hydrochloride, the following compounds were re ared:


tert-butyl 4-((S)-l-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidin-l-yl)-l-oxopropan-2-yl)piperazine-l-carboxylate & tert-butyl 4-((R)-l-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazoI-5- yl)benzyl)carbamoyl)pyrrolidin-l-yl)-l-oxopropan-2-yl)piperazine-l-carboxylate

A stirred mixture of (2S,4R)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, hydrochloride (100 mg, 0.28 mmol) and 2-(4-(tert-butoxycarbonyl)-2- oxopiperazin-l-yl)propanoic acid (85 mg, 0.31 mmol) in DMF (0.8 mL) was treated with DIPEA (0.20 mL, 1.13 mmol) and then with HATU (129 mg, 0.34 mmol) and then stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass- directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title
compounds: Isomer 1 (first-eluting) (48 mg, 30 % yield). LCMS RT= 0.85 min, ES+ve m/z 572 [M+H]+. Isomer 2 (second-eluting) (51 mg, 32 % yield). LCMS RT= 0.86 min, ES+ve m/z 572 [M+H]+. (4-(4-
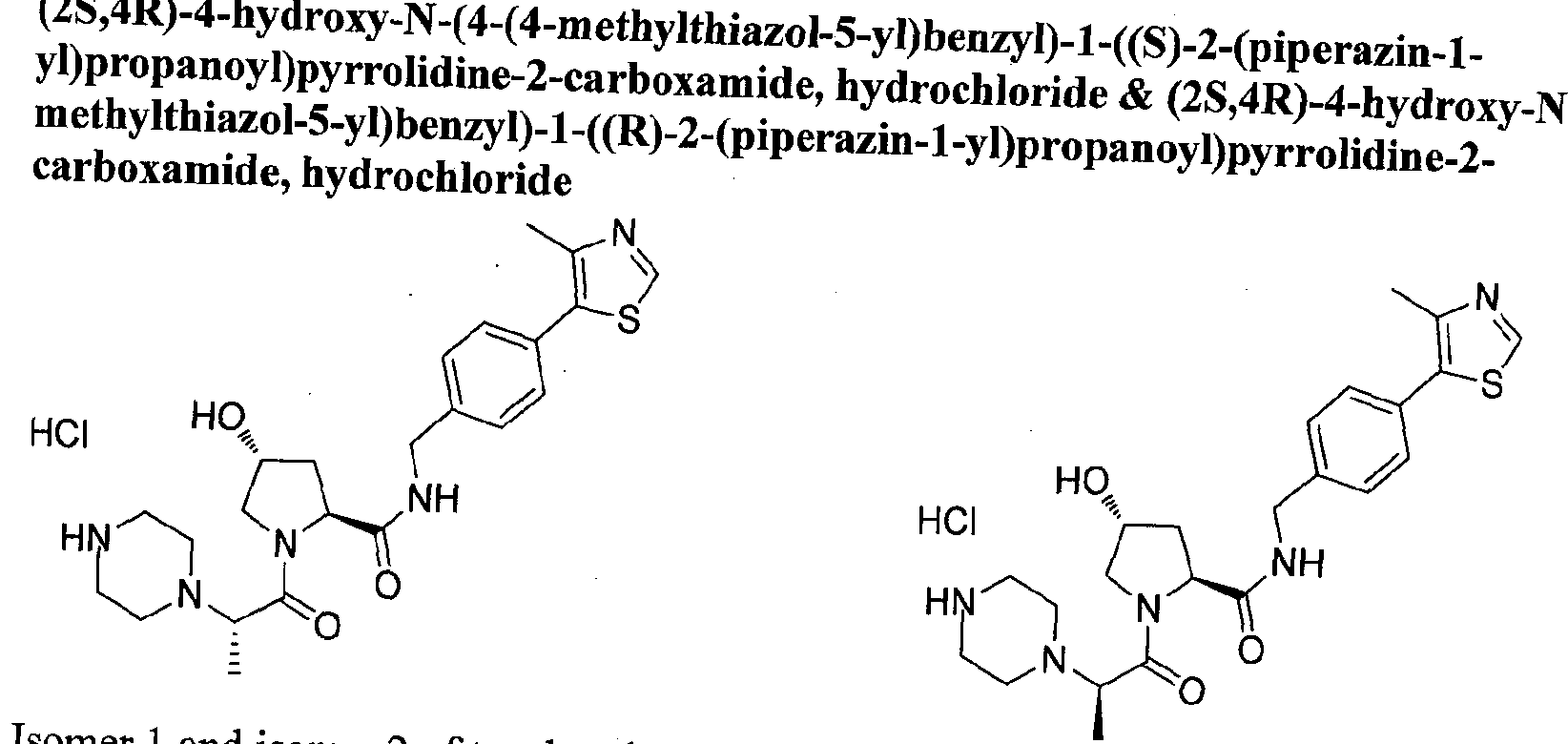
Isomer 1 and isomer 2 of tert-butyl 4-(l-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidin- 1 -yl)- 1 -oxopropan-2-yl)piperazine- 1 -carboxylate (48 mg, 0.08 mmol) were separately dissolved in a mixture of dichloromethane (0.3 mL) and methanol (0.1 mL) and treated with 4M hydrochloric acid in 1,4-dioxane (0.3 mL, 1.2 mmol) respectively. After stirring at ambient temperature for 1 hour, the reaction mixtures were evaporated to dryness to afford the title compounds as hydrochloride salts. Isomer 1 (42 mg, 99 % yield). LCMS RT= 0.62 min, ES+ve m/z 472 [M+H]+. Isomer 2 (42 mg, 99 % yield). LCMS RT= 0.60 min, ES+ve m/z All [M+H]+. utan-2-yl)-4-(2-

A mixture of l-bromo-2-methoxyethane (4 xL, 0.04 mmol), (S)-N-((S)-l-((2S,4R)-4- hydroxy-2-((4-(4-methylthiazol-5 -yl)benzyl)carbamoyl)pyrrolidin- 1 -yl)-3 ,3-dimethyl- 1 -
oxobutan-2-yl)morpholine-2-carboxamide, hydrochloride (20 mg, 0.04 mmol) and DIPEA (0.019 mL, 0.11 mmol) in DMF (0.5 mL) was stirred at 85 °C for 6 hours. The cooled product was subjected to purification by mass-directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title compound (9 mg, 41% yield). LCMS RT= 0.88 min, ES+ve m/z 602 [M+H]+.
Using a method analogous to that for (S)-N-((S)-l-((2S,4R)-4-hydroxy-2-((4-(4- methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin- 1 -yl)-3 ,3 -dimethyl- 1 -oxobutan-2-yl)-4-(2- methoxyethyl)morpholine-2-carboxamide the following compounds were prepared:


A mixture of (25,4i?)-l-((,S}-2-((5}-2-amino-4-methylpentanamido)propanoyl)-4-hydroxy-A^- (4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (102 mg, 0.19 mmol), DIPEA (0.165 mL, 0.95 mmol) and 4-methoxy-4-oxobutanoic acid (commercially available from for example Aldrich) (25 mg, 0.19 mmol) in DMF (2 mL) was treated with HATU (64 mg, 0.17 mmol) and the mixture was stirred at ambient temperature for 20 minutes. Brine (10 mL) was added and the product was extracted with ethyl acetate (20 mL). The organic phase was washed with brine (2 x 20 mL), dried using a hydrophobic frit and evaporated to dryness. The product was purified by chromatography on reverse phase silica using a gradient elution from 5% to 70% acetonitrile(+ 0.1% formic acid) in water(+ 0.1% formic acid) to afford the title compound (73 mg, 0.12 mmol, 63 % yield). LCMS RT= 0.74 min, ES+ve m/z 616 [M+H]+.
4-(3-((2S,4R)-4-Hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-l- carbonyl)phenoxy)butanoic acid (
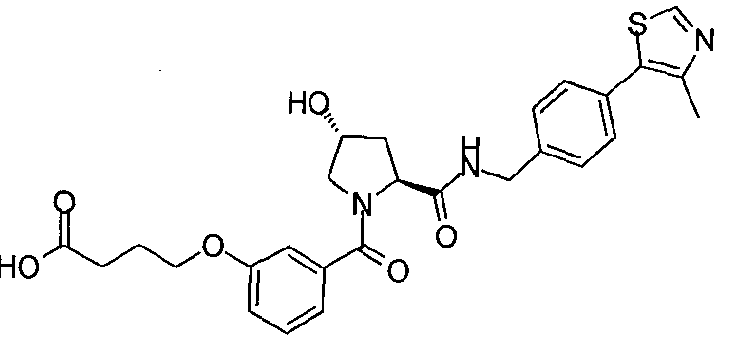
A solution of rt-butyl 4-(3-((25',4^)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidine-l-carbonyl)phenoxy)butanoate (130 mg, 0.22 mmol) in dichloromethane (3 mL) was treated with TFA (0.5 mL, 6.5 mmol) and stirred at ambient temperature for 5 hours. The solvent was evaporated to dryness and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (65 mg, 0.12 mmol, 55 % yield). LCMS RT= 0.70 min, ES+ve m/z 524 [M+H]+.
4-(((5)-l-(((S)-l-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazoI-5- yl)benzyl)carbamoyl)pyrrolidin-l-yl)-l-oxopropan-2-yl)amino)-4-methyl-l-oxopentan- 2-yI)amino)-4-oxobutanoic acid
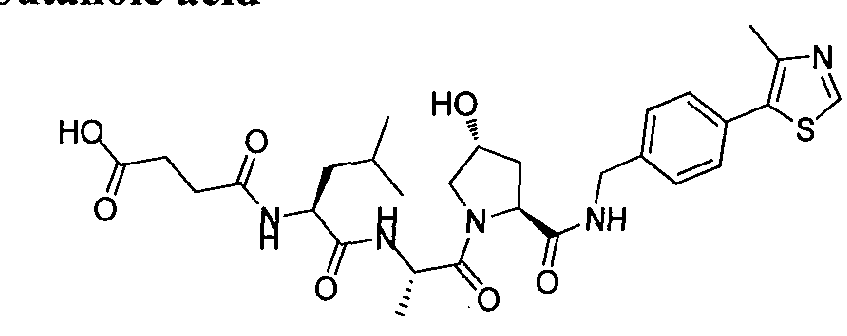
A solution of methyl 4-(((,¾-l-(((JS}-l-((25,4i?)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidin- 1 -yl)- 1 -oxopropan-2-yl)amino)-4-methyl- 1 -oxopentan-2- yl)amino)-4-oxobutanoate (73 mg, 0.12 mmol) in methanol (3 mL) was treated with aqueous sodium hydroxide (2 , 0.6 mL, 1.2 mmol) and the mixture was stirred at ambient temperature for 2 hours. The mixture was evaporated to dryness and the product was purified by chromatography on reverse phase silica using a gradient elution from 5% to 60% acetonitrile(+ 0.1% formic acid) in water(+ 0.1% formic acid) to afford the title compound
(53 mg, 0.088 mmol, 74 % yield). LCMS RT= 0.69 min, ES+ve m/z 602 [M+H]+.
(2S,4R)-4-hydroxy-l-((S)-2-((S)-2-(2-methoxyacetamido)-4- methylpentanamido)propanoyl)- V-(4-(4-methyIthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide

A mixture of (25',4i?)-l-((5)-2-((iS)-2-amino-4-methylpentanamido)propanoyl)-4-hydroxy-N- (4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (30 mg, 0.056 mmol), 2-methoxyacetic acid (commercially available from for example Aldrich) (4.3 uL, 0.056 mmol) and DIPEA (0.05 mL, 0.29 mmol) in DMF (1 mL) was treated with HATU (25 mg, 0.066 mmol) and stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (18 mg, 0.031 mmol, 56 % yield). LCMS RT= 0.73 min, ES+ve m/z 574 [M+H]+.
(25,4R)-4-hydroxy-l-((5)-2-((S)-2-(3-methoxypropanamido)-4- methylpentanamido)propanoyI)-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide

A mixture of (25',4i?)-l-((>S)-2-((S)-2-amino-4-methylpentanamido)propanoyl)-4-hydroxy-N- (4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (30 mg, 0.056 mmol), 3-methoxypropanoic acid (commercially available from for example Aldrich) (5.2 uL, 0.056 mmol) and DIPEA (0.05 mL, 0.29 mmol) in DMF (1 mL) and stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (20 mg, 0.034 mmol, 61 % yield). LCMS RT= 0.72 min, ES+ve m/z 588 [M+H]+.
(2S,4R)-l-(6-cyanopyridin-2-yI)-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide

A mixture of (2S,4R)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, hydrochloride (58 mg, 0.16 mmol) and 6-fluoropicolinonitrile (commercially available from for example Aldrich) (20 mg, 0.16 mmol) in DMSO (1 mL) was treated with DIPEA (0.10 mL, 0.57 mmol), sealed and heated in a Biotage "Initiator" microwave at 100°C for 60 minutes. The product was purified by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (23 mg, 0.055 mmol, 34 % yield). LCMS RT= 0.74 min, ES+ve m/z 420 [M+H]+.
Intermediates
4-(oxazol-5-yI)benzonitrile
 A mixture of 4-formylbenzonitrile (commercially available from for example Aldrich) (5.32 g, 41 mmol), l-((isocyanomethyl)sulfonyl)-4-methylbenzene (commercially available from for example Aldrich) (8.83 g, 45 mmol) and potassium carbonate (7.3 g, 53 mmol) in methanol (200 mL) was stirred at ambient temperature for 80 minutes. The mixture was then evaporated to dryness; the residue was treated with saturated aqueous sodium bicarbonate (100 mL) and extracted with dichloromethane (3 x 100 mL). The combined organics were washed with brine (75 mL), passed through a hydrophobic frit and then evaporated to dryness to afford the title compound (7.19 g, 42 mmol, quantitative). LCMS RT= 0.48 min, ES+ve m/z 171 [M+H]+.
A mixture of 4-formylbenzonitrile (commercially available from for example Aldrich) (5.32 g, 41 mmol), l-((isocyanomethyl)sulfonyl)-4-methylbenzene (commercially available from for example Aldrich) (8.83 g, 45 mmol) and potassium carbonate (7.3 g, 53 mmol) in methanol (200 mL) was stirred at ambient temperature for 80 minutes. The mixture was then evaporated to dryness; the residue was treated with saturated aqueous sodium bicarbonate (100 mL) and extracted with dichloromethane (3 x 100 mL). The combined organics were washed with brine (75 mL), passed through a hydrophobic frit and then evaporated to dryness to afford the title compound (7.19 g, 42 mmol, quantitative). LCMS RT= 0.48 min, ES+ve m/z 171 [M+H]+.
(4-(oxazol-5-yl)phenyl)methanamine
Under an atmosphere of nitrogen, an ice-cooled mixture of 4-(oxazol-5-yl)benzonitrile (900 mg, 5.29 mmol) and cobalt(II) chloride hexahydrate (commercially available from for example Aldrich) (1.8 g, 7.9 mmol) in methanol (50 mL) was treated portion- wise over 5 minutes with sodium borohydride (1 g, 26 mmol). The mixture was stirred for 30 minutes and then treated with water (50 mL) and concentrated aqueous ammonia (20 mL). The mixture was extracted with chloroform (3 x 150 mL), the combined organics were evaporated to dryness and the product was purified by chromatography on silica using a gradient elution from 0% to 30% methanol in dichloromethane (+ 0.1% triethylamine) to afford the title compound (580 mg, 3.3 mmol, 63 % yield). LCMS RT= 0.35 min, ES+ve m/z 175 [M+H]+. -2-((4-(oxazol-5-yI)benzyl)carbamoyl)pyrrolidine-l-

To a stirred solution of (2S,4R)-l-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid (0.66 g, 2.9 mmol) in dry DMF (20 mL) were added (4-(oxazol-5- yl)phenyl)methanamine (0.5 g, 2.87 mmol) and DIPEA (1 mL, 5.7 mmol). This solution was
then ice-cooled and HATU (1.09 g, 2.9 mmol) was added. The reaction mixture was stirred with cooling for an additional hour then treated with water (30 mL) and extracted with ethyl acetate (3x100 mL). The combined organic phase was washed with saturated aqueous sodium bicarbonate (60 mL), brine (60 mL), dried over magnesium sulfate, filtered and evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0% to 25% methanol in dichloromethane to afford the title compound (758 mg, 1.96 mmol, 68 % yield). LCMS RT= 0.73 min, ES+ve m/z 388 [M+H]+.
(2S,4R)-4-hydroxy-N-(4-(oxazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride

A solution of (2S,4R)-tert-butyl 4-hydroxy-2-((4-(oxazol-5-yl)benzyl)carbamoyl)pyrrolidine- 1-carboxylate (2.74 g, 7.1 mmol) in methanol (10 mL) and dichloromethane (15 mL) was treated with hydrochloric acid (4 M in 1,4-dioxane) (8.8 mL, 35 mmol) and the mixture was stirred at ambient temperature for 24 hours. The mixture was evaporated to dryness. The residue was suspended in methanol, filtered and dried under vacuum to afford the title compound (2.24 g, 6.9 mmol, 98 % yield). LCMS RT= 0.44 min, ES+ve m/z 288 [M+H]+. -tert-butyl 2-((4-bromobenzyl)carbamoyl)-4-hydroxypyrrolidine-l-carboxylate

An ice-cooled mixture of (2S,4R)-l-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid (commercially available from for example Aldrich) (7.95 g, 34 mmol) and (4- bromophenyl)methanamine (commercially available from for example Fluorochem) (6.4 g, 34 mmol) in DMF (200 mL) was treated with DIPEA (18 mL, 103 mmol) and then with HATU (14.4 g, 38 mmol) and the mixture was stirred at ambient temperature for 30 minutes. The mixture was treated with water (200 mL) and extracted with ethyl acetate (2 x 200 mL). The combined organic phase was washed with saturated aqueous sodium bicarbonate (2 x 300 mL), water (100 mL), brine (200 mL), dried over magnesium sulfate and evaporated to dryness. The product was purified by flash chromatography (750 g silica cartridge) using a
gradient elution from 0% to 10% methanol in dichloromethane to afford the title compound (12.9 g, 94% yield). LCMS RT= 0.87 min, ES+ve m/z 401 [M+H]+.
(2S,4R)-tert-butyl 4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine- 1-carboxylate

Under an atmosphere of nitrogen, a mixture of (2S,4R)-tert-butyl 2-((4- bromobenzyl)carbamoyl)-4-hydroxypyrrolidine-l-carboxylate (12.9 g, 32 mmol), 4- methylthiazole (commercially available from for example Aldrich) (5.9 mL, 65 mmol), palladium(II) acetate (commercially available from for example Aldrich) (0.145 g, 0.65 mmol) and potassium acetate (6.34 g, 65 mmol) in N-methyl-2-pyrrolidone (80 mL) was stirred at 120 °C for 18 hours. After cooling to ambient temperature, water (100 ml) was added and the product was extracted with ethyl acetate (4 x 300 mL). The combined organic phase was washed with brine (5 x 200 mL), dried over magnesium sulfate and evaporated to dryness. The product was purified by flash chromatography (750 g silica cartridge) using a gradient elution from 0% to 10% methanol in dichloromethane to afford the title compound (8.0 g, 59% yield). LCMS RT= 0.75 min, ES+ve m/z 418 [M+H]+.
(2S,4R)-4-hydroxy-N-(4-(4-methyIthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride

A solution of (2S,4R)-tert-butyl 4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidine-l-carboxylate (8 g, 19 mmol) in a mixture of methanol (30 mL) and dichloromethane (20 mL) was treated with 4M hydrochloric acid in 1,4-dioxane (8 mL, 32 mmol). The mixture was stirred at ambient temperature for 2 hours. The solvent was evaporated to dryness and the residue was triturated in dichloromethane, filtered and dried under vacuum to afford the title compound (6.7 g, 99 % yield). LCMS RT= 0.51 min, ES+ve w/z 318 [M+H .
-l-(3-ethoxybenzoyI)-4-hydroxypyrrolidine-2-carboxylic acid

3-Ethoxybenzoic acid (commercially available from for example Aldrich) (4 g, 24 mmol) was dissolved in thionyl chloride (24 mL, 329 mmol) and stirred at 60 °C for 1 hour and then at 50 °C for 18 hours. After cooling to ambient temperature the mixture was evaporated to dryness and the residue was treated with diethyl ether (5 mL). The mixture was then ice- cooled and treated with a solution of (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid, hydrochloride (commercially available from for example Aldrich) (4.44 g, 27 mmol) in 1M aqueous sodium hydroxide (27 mL, 27 mmol). The reaction was warmed to ambient temperature and stirred for 18 hours. The mixture was separated; the aqueous phase was washed with diethyl ether and then acidified with 2M aqueous hydrochloric acid. The product was extracted in diethyl ether (2 x 70 mL) and the combined ethereal phase was evaporated to dryness. The product was purified by flash chromatography (340 g CI 8 cartridge), using a gradient elution from 10 to 30% acetonitrile (+ 0.1% formic acid) in water (+ 0.1% formic acid) to afford the title compound (3.5 g, 52% yield). LCMS RT= 0.55 min, ES+ve m/z 280
[M+H]+.
-benzyl l-((S)-2-acetamidopropanoyl)-4-hydroxypyrrolidine-2-carboxylate

An ice-cooled mixture of (S)-2-acetamidopropanoic acid (commercially available from for example Aldrich) (2.80 g, 21 mmol) and (2S,4R)-benzyl 4-hydroxypyrrolidine-2-carboxylate, hydrochloride (commercially available from for example Aldrich) (5 g, 19 mmol) in DMF (5 mL) was treated with DIPEA (14 mL, 78 mmol), followed by HATU (8.11 g, 21 mmol) over 10 min. The mixture was warmed to ambient temperature and stirred for 1 hour then treated with saturated aqueous sodium bicarbonate (30 mL) and stirred for 5 min. The mixture was then extracted with ethyl acetate (3 x 100 mL) and the combined organic phase was washed with water (100 mL), brine (100 mL), dried over magnesium sulfate and evaporated to dryness. The product was purified by flash chromatography (330 g silica cartridge) using a
gradient elution from 0 to 10% methanol in dichloromethane to afford the title compound (2.0 g, 31% yield). LCMS RT= 0.63 min, ES+ve m/z 335 [M+H]+.
-l-((S)-2-acetamidopropanoyl)-4-hydroxypyrrolidine-2-carboxyIic acid

A solution of (2S,4R)-benzyl-l-((S)-2-acetamidopropanoyl)-4-hydroxypynOlidine-2- carboxylate (2 g, 6.0 mmol) in ethanol (10 mL) was added to a flask containing palladium on carbon (1.27 g, 1.2 mmol) (10%, Degussa type) under an atmosphere of nitrogen. The flask was filled with hydrogen and the solution was stirred at ambient temperature for 2 hours. The catalyst was removed by filtration through celite and the filtrate was evaporated under reduced pressure to afford the title compound (1.37 g, 94% yield). LCMS RT= 0.28 min, ES+ve m/z 244 [M+H]+.
(2S,4R)-4-hydroxy-l-(2-(3-methyIisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylic acid

Under an atmosphere of nitrogen, a solution of (2S,4R)-benzyl 4-hydroxy-l-(2-(3- methylisoxazol-5-yl)acetyl)pyrrolidine-2-carboxylate (2.3 g, 6.7 mmol) in ethanol (60 mL) was added to palladium on carbon (0.071 g, 0.67 mmol) (10 %, Degussa type) and then stirred under an atmosphere of hydrogen. After 2 hours, the mixture was filtered through celite. The filtrate was evaporated to dryness and the residue was triturated with cyclohexane and dried under vacuum to afford a white solid. The product was purified by mass-directed automated preparative HPLC (TFA modifier) to afford the title compound (650 mg, 2.6 mmol, 38 % yield). LCMS RT= 0.38 min, ES+ve m/z 255 [M+H]+.
(2S,4R)-methyI 4-hydroxy-l-((S)-2-(l-oxoisoindoIin-2-yl)propanoyl)pyrrolidine-2- carboxylate

(commercially available from for example Aldrich) (1.77 g, 9.8 mmol) and (S)-2-(l- oxoisoindolin-2-yl)propanoic acid (2 g, 9.8 mmol) in DMF (4 mL) was treated with DIPEA (5.11 mL, 29 mmol) and then with HATU (4.08 g, 10.7 mmol), and stirred at ambient temperature for 30 minutes. The mixture was treated with saturated aqueous sodium bicarbonate (100 mL) and extracted with ethyl acetate (2 x 200 mL). The combined organic phase was washed with water (100 mL), brine (100 mL), dried over magnesium sulfate and evaporated to dryness. The product was purified by flash chromatography (120 g CI 8 cartridge), using a gradient elution from 10% to 50% acetonitrile (+ 0.1% formic acid) in water (+ 0.1 % formic acid) to afford the title compound (1.0 g, 31 % yield). LCMS RT= 0.60 min, ES+ve m/z 333 [M+H]+.
(2S,4R)-4-hydroxy-l-((S)-2-(l-oxoisoindoIin-2-yl)propanoyl)pyrroIidine-2-carboxyIic acid
A solution of (2S,4R)-methyl 4-hydroxy-l-((S)-2-(l-oxoisoindolin-2- yl)propanoyl)pyrrolidine-2-carboxylate (1 g, 3.0 mmol) in methanol (2 mL) was treated with 2M aqueous sodium hydroxide (5 mL, 10 mmol) and the mixture was stirred at ambient temperature for 2 hours then acidified with 2M aqueous hydrochloric acid (6 mL). The mixture was then evaporated to about one half of the original volume and then ice-cooled. The resulting precipitate was filtered off and dried under vacuum to afford the title compound (615 mg, 64% yield). LCMS RT= 0.51 min, ES+ve m/z 319 [M+H]+.
Methyl 3-(4-(tert-butoxy)-4-oxobutoxy)beiizoate

A solution of methyl 3Thydroxybenzoate (commercially available from for example Aldrich) (1 g, 6.6 mmol) and K2C03 (1.82 g, 13.2 mmol) in DMF (10 mL) was treated with tert-butyl 4-bromobutanoate (commercially available from for example Aldrich) (2.2 g, 9.9 mmol) and the mixture was stirred at 60 °C for 16 hours. A further aliquot of K2C03 (1.82 g, 13.2 mmol) and tert-butyl 4-bromobutanoate (2.2 g, 9.9 mmol) were added and the mixture was heated at 60 °C for further 6 hours. The mixture was cooled to ambient temperature and partitioned
between ethyl acetate (50 mL) and water (50 mL). The organic phase was washed with brine (2 x 50 mL), dried (hydrophobic frit) and evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0% to 100% methyl tert-butyl ether in cyclohexane to afford the title compound (1.4 g, 4.8 mmol, 72 % yield). LCMS RT= 1.26 min, ES+ve m/z 312 [M+H]+.
3-(4-(rer/-butoxy)-4-oxobutoxy)benzoic acid

A mixture of methyl 3-(4-(tert-butoxy)-4-oxobutoxy)benzoate (1.4 g, 4.8 mmol) and aqueous sodium hydroxide (2M, 4.8 mL, 9.6 mmol) in methanol (10 mL) was stirred at ambient temperature for 5 hours. The methanol was removed under reduced pressure (no heat) and the aqueous phase was acidified to pH 3 with saturated aqueous citric acid. The product was extracted with ethyl acetate (60 mL) and the organic extract was washed with brine (20 mL), dried using a hydrophobic frit and evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0% to 25% methanol in
dichloromethane to afford the title compound (625 mg, 2.2 mmol, 47 % yield). LCMS RT= 1.06 min, ES+ve m/z 279 [M-H]\
aoxapentadecyl)oxy)benzoate

An ice-cooled mixture of tert-butyl 3-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)propanoate (commercially available from for example Aldrich) (2.0 g, 7.2 mmol), triphenylphosphine (2.3 g, 8.6 mmol) and methyl 3-hydroxybenzoate (commercially available from for example Aldrich) (1.2 g, 7.9 mmol) in THF (40 mL) was treated dropwise over 5 minutes with diisopropyl azodicarboxylate (1.68 mL, 8.6 mmol). The mixture was warmed to ambient temperature and stirred for 18 hours. The mixture was then evaporated to dryness and purified by flash column chromatography (100 g silica cartridge) using a gradient elution from 0 to 100% methyl tert-butyl ether in cyclohexane over 40 minutes to afford the title compound (2.53 g, 85% yield). LCMS RT= 1.14 min, ES+ve m/z 430 [M+NH4]+.
3-((14,14-dimethyl-12-oxo-3,6,9,13-tetraoxapentadecyl)oxy)benzoic acid (

A solution of methyl 3-((14,14-dimethyl-12-oxo-3,6,9,13-tetraoxapentadecyl)oxy)benzoate
(2.53 g, 4.9 mmol) in methanol (25 mL) was treated with 1M aqueous sodium hydroxide (0.3 g, 7.6 mmol) in water (7 mL), and the mixture was stirred at ambient temperature for 1 hour.
Acetic acid (0.45 mL, 7.9 mmol) was slowly added and the mixture was evaporated to dryness and purified by flash chromatography (100 g silica cartridge) using a gradient elution from 0% to 15% methanol in dichloromethane (+1% triethylamine) to afford the title compound (1.37 g, 70% yield). LCMS RT= 0.99 min, ES+ve m/z 399 [M+H]+.
tert-butyl ((S)-l-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyI)carbamoyI)pyrrolidiii-l-yl)-3-methyI-l-oxobutan-2-yl)carbamate

A stirred mixture of (2S,4R)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, hydrochloride (125 mg, 0.35 mmol) and (S)-2-((tert-butoxycarbonyl)amino)-3- methylbutanoic acid (commercially available from for example Aldrich) (77 mg, 0.35 mmol) in DMF (0.9 mL) was treated with DIPEA (0.22 mL, 1.3 mmol) and then with HATU (134 mg, 0.35 mmol) and the mixture was stirred at ambient temperature for 1 hour. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (120 mg, 72% yield). LCMS RT= 0.87 min, ES+ve w/z 517 [M+H]+.
Using a method analogous to that for tert-butyl-((S)-l-((2S,4R)-4-hydroxy-2-((4-(4- methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin- 1 -yl)-3 -methyl- 1 -oxobutan-2-yl)carbamate, the following compounds were prepared:
 (2S,4R)-l-(2-aminoacetyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide
(2S,4R)-l-(2-aminoacetyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide

A stirred mixture of (2S,4R)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, hydrochloride (100 mg, 0.28 mmol) and 2-((tert-butoxycarbonyl)amino)acetic acid (commercially available from for example Aldrich) (49 mg, 0.28 mmol) in DMF (3 mL) was treated with DIPEA (0.20 mL, 1.1 mmol) and then with HATU ( 18 mg, 0.31 mmol) and the mixture was stirred at ambient temperature for 30 minutes. Water (20 ml) was added and the product was extracted with ethyl acetate (3 x 20 mL). The combined organic phase was washed with saturated aqueous sodium bicarbonate (20 mL), water (20 mL), brine (20 mL) filtered through a hydrophobic frit and evaporated to dryness. The residue was then dissolved in dichloromethane (3 mL) and treated with TFA (1 mL, 13 mmol). After stirring at ambient temperature for 10 minutes, the reaction mixture was evaporated to dryness. The residue was dissolved in the minimum amount of methanol and then loaded onto a pre-conditioned (methanol) aminopropyl solid-phase extraction cartridge (5 g). The column was eluted with methanol (3 volumes) and the product-containing fractions were evaporated to dryness to afford the title compound (104 mg, 99 % yield). LCMS RT= 0.44 min, ES+ve m/z 375
[M+H]+.
(2S,4R)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-l-((S)-morphoIine-3- carbonyl)pyrrolidme-2-carboxamide

A solution of (S)-tert-butyl 3-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidine-l-carbonyl)morpholine-4-carboxylate (115 mg, 0.22 mmol) in dichloromethane (0.5 mL) was treated with TFA (0.5 mL) and the reaction mixture was stirred at ambient temperature for 1 hour. The mixture was evaporated to dryness and the residue was then dissolved in the minimum amount of a mixture of
methanol.'dichloromethane (1:1), and loaded onto a pre-conditioned (methanol) aminopropyl
solid-phase extraction cartridge (2 g). The column was eluted with methanol (3 volumes) and the product-containing fractions were evaporated under reduced pressure to afford the title compound (89 mg, 94% yield). LCMS RT= 0.47 min, ES+ve m/z 431 [M+H]+.
(2S,4R)-4-hydroxy-l-((S)-3-methyI-2-(methylamino)butanoyl)-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride
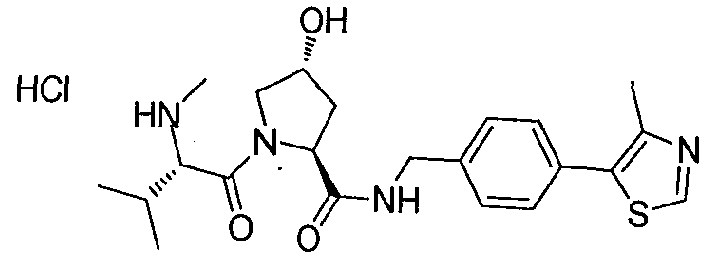
A mixture of (2S,4R)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, hydrochloride (100 mg, 0.28 mmol), (S)-2-((tert- butoxycarbonyl)(methyl)amino)-3-methylbutanoic acid (commercially available from for example Aldrich) (65 mg, 0.28 mmol) and DIPEA (0.247 mL, 1.41 mmol) in DMF (2 mL) was treated with HATU (118 mg, 0.31 mmol) and stirred at ambient temperature for 30 minutes. The Boc protected intermediate was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier). The purified intermediate was dissolved in methanol rdichloromethane (1 :1, 3 mL), treated with hydrochloric acid in 1,4-dioxane (4M, 3 mL, 12 mmol) and allowed to stand for 1 hour. The mixture was then evaporated to dryness to afford the title compound (107 mg, 0.23 mmol, 81 % yield). LCMS RT= 0.55 min, ES+ve m/z 431 [M+H]+.
(2S,4R)-l-((S)-2-amino-3-methylbutanoyl)-4-hydroxy- V-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride

A solution of rt-butyl ((5)-l-((25',4^)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidin-l-yl)-3-methyl-l-oxobutan-2-yl)carbamate (287 mg, 0.56 mmol) in THF (5 mL) and treated with 4M hydrochloric acid in 1,4-dioxan (10 mL) and stirred at ambient temperature for 2 hours. The mixture was evaporated to dryness to afford
the title compound (224 mg, 0.49 mmol, quantitative). LCMS RT= 0.55 min, ES+ve m/z 417
[M+H]+.
(2S,4R)-l-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methyIthiazol-S- yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride

A stirred mixture of (2S,4R)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, hydrochloride (70 mg, 0.20 mmol) and (S)-2-((tert-butoxycarbonyl)amino)-3,3- dimetbylbutanoic acid (commercially available from for example Fluka) (50 mg, 0.22 mmol) in DMF (1 mL) was treated with DIPEA (0.14 mL, 0.79 mmol) and then with HATU (90 mg, 0.24 mmol), and stirred at ambient temperature for 30 minutes. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to give the intermediate boc-protected product. The intermediate was then dissolved in a mixture of dichloromethane (0.5 mL) and methanol (0.1 mL) and treated with 4M hydrochloric acid in 1,4-dioxane (0.25 mL, 1 ,0 mmol), After stirring at ambient temperature for 1 hour, the reaction mixture was evaporated to dryness and the residue triturated to a solid with dichloromethane and dried under vacuum to afford the title compound (76 mg, 82 % yield). LCMS RT= 0.58 min, ES+ve m/z 431 [M+H]+.
(2»-»,4R)-l-((5)-2-((S)-2-ammo-4-metb.ylpentanamido)propanoyl)-4-hydroxy-N-(4-(4- methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride

HCI
A solution of a mixture of (25,,4 ?)-l-((5 -2-aminopropanoyl)-4-hydroxy-N-(4-(4- memylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (507 mg, 1.2 mmol), DIPEA (0.868 mL, 4.97 mmol) and (5)-2-((tert-butoxycarbonyl)amino)-4-methylpentanoic acid (commercially available from for example Aldrich) (230 mg, 0.99 mmol) in DMF (5 mL) was treated with HATU (416 mg, 1.1 mmol) and stirred at ambient temperature for 2
hours. Water (50 mL) was added and the product was extracted with ethyl acetate (50 mL). The organic phase was washed with brine (2 x 50 mL), dried using a hydrophobic frit and evaporated to dryness. The residue was dissolved in methanohdichloromethane (1:1, 15 mL), treated with hydrochloric acid in 1,4-dioxane (4M, 5 mL, 20 mmol) and stirred at ambient temperature for 3 hours. The mixture was evaporated to dryness and the residue was suspended in dichloromethane (10 mL), sonicated, filtered and dried under vacuum to afford the title compound (280 mg, 0.56 mmol, 56 % yield). LCMS RT= 0.55 min, ES+ve m/z 502 [M+H]+. lthiazol-5-yl)benzyl)carbamoyl)-4-

Under an atmosphere of nitrogen, a mixture of (2S,4R)-tert-butyl 2-((4- bromobenzyl)carbamoyl)-4-hydroxypyrrolidine-l-carboxylate (200 mg, 0.50 mmol), 2,4- dimethylthiazole (commercially available from for example Avocado) (113 mg, 1.0 mmol), palladium(II) acetate (commercially available from for example Aldrich) (2 mg, 10 μιηοΐ) and potassium acetate (98 mg, 1.0 mmol) in N-methyl-2-pyrrolidone (2 mL) was stirred at 120 °C for 18 hours. The cooled mixture was treated with water (25 ml) and the product was extracted with ethyl acetate (4 x 30 mL). The combined organic phase was washed with brine (5 x 20 mL), filtered through a hydrophobic frit and evaporated to dryness. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (142 mg, 66% yield). LCMS RT= 0.77 min, ES+ve m/z 432 [M+H]+.
(2S,4R)-N-(4-(2,4-dimethylthiazol-5-yl)benzyI)-4-hydroxypyrrolidine-2-carboxamide, hydrochloride

A solution of (2S,4R)-tert-butyl 2-((4-(2,4-dimethylthiazol-5-yl)benzyl)carbamoyl)-4- hydroxypyrrolidine-l-carboxylate (142 mg, 0.33 mmol) in a mixture of methanol (0.5 mL) and dichloromethane (1.5 mL) was treated with 4M hydrochloric acid in 1,4-dioxane (0.63
mL, 2.5 mmol) and stirred at ambient temperature for 2 hours. The solvent was evaporated to dryness and the residue was triturated to a solid with diethyl ether and dried under vacuum to afford the title compound (120 mg, 99% yield). LCMS RT= 0.49 min, ES+ve m/z 332
[ +H]+.
-3-methyl-2-(3-methyl-2,5-dioxo-2,5-dihydro-lH-pyrrol-l-yl)butanoic acid

A mixture of 3-methylfuran-2,5-dione (commercially available from for example Aldrich) (0.12 mL, 1.3 mmol) and (S)-2-amino-3-methylbutanoic acid (commercially available from for example Apollo Scientific) (150 mg, 1.3 mmol).in acetic acid (1 mL) was sealed and heated in a Biotage "Initiator" microwave at 120°C for 1 hour. The mixture was evaporated to dryness to afford the title compound (253 mg, 94% yield). LCMS RT= 0.75 min, ES+ve m/z 212 [M+H]+
benzyl 2-(3-oxomorpholino)propanoate

Under an atmosphere of nitrogen, an ice-cooled solution of morpholin-3-one (commercially available from for example Aldrich) (100 mg, 1.0 mmol) in DMF (2 mL) was treated with sodium hydride (60% w/w in mineral oil) (40 mg, 1.0 mmol). After 5 minutes, benzyl 2- bromopropanoate (commercially available from for example Aldrich) (240 mg, 1.0 mmol) was added and the mixture was stirred with cooling for 30 minutes and then at ambient temperature for a further 18 hours. The reaction mixture was cautiously treated with saturated aqueous sodium bicarbonate (10 mL) and then extracted with ethyl acetate (2 x 40 mL). The combined organic phase was washed with brine (25 mL), filtered through a hydrophobic frit, and evaporated to dryness. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to give the title compound (105 mg, 40% yield). LCMS RT= 0.80 min, ES+ve m/z 264 [M+H]+.
2-(3-oxomorpholino)propanoic acid
 Under an atmosphere of nitrogen, a solution of benzyl 2-(3-oxomorpholino)propanoate (90 mg, 0.34 mmol) in ethanol (3 mL) was added to a flask containing palladium on carbon (36 mg, 0.034 mmol) (10%, Degussa type). The flask was then filled with hydrogen and the mixture was stirred at ambient temperature for 1 hour. The catalyst was removed by filtration through celite and the filtrate was evaporated under reduced pressure to afford the title compound (55 mg, 93% yield). LCMS RT= 0.33 min, ES+ve m/z 174 [M+H]+.
Under an atmosphere of nitrogen, a solution of benzyl 2-(3-oxomorpholino)propanoate (90 mg, 0.34 mmol) in ethanol (3 mL) was added to a flask containing palladium on carbon (36 mg, 0.034 mmol) (10%, Degussa type). The flask was then filled with hydrogen and the mixture was stirred at ambient temperature for 1 hour. The catalyst was removed by filtration through celite and the filtrate was evaporated under reduced pressure to afford the title compound (55 mg, 93% yield). LCMS RT= 0.33 min, ES+ve m/z 174 [M+H]+.
pan-2-yl)-3-oxopiperazine-l-carboxylate

Under an atmosphere of nitrogen, an ice-cooled solution of tert-butyl 3-oxopiperazine-l- carboxylate (commercially available from for example Aldrich) (200 mg, 1.0 mmol) in DMF (4 mL) was treated with sodium hydride (60% w/w in mineral oil) (44 mg, 1.1 mmol). After 5 minutes, benzyl 2-bromopropanoate (commercially available from for example Aldrich) (255 mg, 1.05 mmol) was added. The mixture was stirred at 0 °C for 30 minutes and then at ambient temperature for a further 18 hours. The mixture was cautiously treated with saturated aqueous sodium bicarbonate (20 mL) and then extracted with ethyl acetate (2 x 40 mL). The combined organic phase was washed with brine (25 mL), filtered through a hydrophobic frit, and evaporated to dryness. The product was purified by flash column chromatography (20 g silica cartridge) using a gradient elution from 0% to 100% ethyl acetate in cyclohexane to afford the title compound (230 mg, 64% yield). LCMS RT= 1.05 min, ES+ve m/z 363
[M+H]+.
-(4-(tert-butoxycarbonyl)-2-oxopiperazin-l-yI)propani

Under an atmosphere of nitrogen, a solution of tert-butyl 4-(l-(benzyloxy)-l-oxopropan-2- yl)-3-oxopiperazine-l-carboxylate (230 mg, 0.64 mmol) in ethanol (3 mL) was added to a flask containing palladium on carbon (68 mg, 0.063 mmol) (10%, Degussa type). The flask was filled with hydrogen and the mixture was stirred at ambient temperature for 1 hour. The catalyst was removed by filtration through celite and the filtrate was evaporated under
reduced pressure to afford the title compound (171 mg, 99% yield). LCMS RT= ES+ve m/z 290 [M+H]+.
enzo [d] oxazol-5-yl)benzylcarbamate

Under an atmosphere of nitrogen, a mixture of (4-(((tert- butoxycarbonyl)amino)methyl)phenyl)boronic acid (commercially available from for example Aldrich) (387 mg, 1.54 mmol), 5-bromobenzo[d]oxazol-2(3H)-one (commercially available from for example Aldrich) (300 mg, 1.40 mmol) and sodium carbonate (446 mg, 4.21 mmol) in DMF (4 mL) was treated with dicholoro[l,l'- bis(diphenylphosphino)ferrocene]palladium(II) (commercially available from for example Aldrich) (72 mg, 0.098 mmol) then sealed and heated in a Biotage "Initiator" microwave at 110 °C for 1 hr. The cooled product mixture was treated with dichloromethane (50 mL) and water (10 mL). The mixture was separated and the organic fraction was evaporated to dryness. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (178 mg, 0.52 mmol, 37 % yield). LCMS RT= 1.03 min, ES+ve m/z 341 [M+H]+.
5-(4-(aminomethyl)phenyl)benzo[d]oxazol-2(3H)-one, hydrochloride

A solution of tert-butyl 4-(2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)benzylcarbamate (130 mg, 0.38 mmol) in THF (10 mL) was treated with hydrochloric acid (4M in 1,4-dioxan) (1 mL, 4 mmol) and the mixture was stirred at ambient temperature overnight. The mixture was treated with diethyl ether (40 mL); the resulting precipitate was filtered off and dried under vacuum to afford the title compound (87 mg, 0.31 mmol, 82 % yield). LCMS RT= 0.49 min, ES+ve m/z 241 [M+H]+.
(2S,4R)-4-hydroxy-l-(3-hydroxybenzoyl)-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-

-2-(l-oxoisoindolin-2-yl)propanoic acid

A mixture of phthalaldehyde (commercially available from for example Aldrich) (4 g, 30 mmol) and (S)-2-aminopropanoic acid (commercially available from for example Aldrich) (2.39 g, 27 mmol) in acetonitrile (150 mL) was heated at reflux for 5 hr then allowed to cool to ambient temperature and stood overnight. The resulting crystalline precipitate was filtered off, washed with acetonitrile and dried under vacuum to afford the title compound (4.46 g, 22 mmol, 73 % yield). LCMS RT= 0.59 min, ES+ve m/z 206 [M+H]+.
Using a method analogous to that for (S)-2-(l-oxoisoindolin-2-yl)propanoic acid, the following compounds were prepared:
Yiel [M+H]
Name Structure RT
d +
\
(S)-4-methoxy-2-(l-
0.62
oxoisoindolin-2- 46% 250
min
yl)butanoic acid
o o
(S)-2-(l -oxoisoindolin-2- 0.67
60% 220
yl)butanoic acid min
o o
(S)-2-(l -oxoisoindolin-2- -t 0.77
60% 234
yl)pentanoic acid Ογ min
O 0
(S)-2-cyclopropyI-2-(l-
0.69
oxoisoindolin-2-yl)acetic 54% 232
min
acid O 0
(S)-3-methyl-2-(l-
0.74
oxoisoindolin-2- 59% 234
min
yl)butanoic acid o o
(S)-3,3-dimethyI-2-(l-
0.82
oxoisoindolin-2- 69% 248
min
yl)butanoic acid
0 0
Ethyl 2-(5-methoxy-l-oxoisoindolin-2-yl)-3-methylbutanoate

Under an atmosphere of nitrogen, an ice-cooled solution of 5-methoxyisoindolin-l-one (commercially available from for example Chem Impex) (105 mg, 0.64 mmol) in DMF (2.5 mL) was treated with sodium hydride (60% w/w in mineral oil) (31 mg, 0.77 mmol). The mixture was warmed to ambient temperature, treated with ethyl 2-bromo-3-methylbutanoate (commercially available from for example Alfa Aesar) (135 mg, 0.64 mmol), stirred at ambient temperature for 2 hours, and then heated to 70 °C for a further 18 hours. The mixture was then ice-cooled and treated with additional sodium hydride (60% w/w in mineral oil) (31 mg, 0.77 mmol), followed by ethyl 2-bromo-3-methylbutanoate (135 mg, 0.64 mmol) and stirred at 70 °C for a further 24 hours. The cooled mixture was then cautiously treated with saturated aqueous ammonium chloride (20 mL) and the product was extracted with ethyl acetate (2 x 25 mL). The combined organic phase was washed with water (20 mL), brine (20 mL), filtered through a hydrophobic frit and evaporated to dryness. The product was purified by flash chromatography (20 g silica cartridge) using a gradient elution from 0% to 50% ethyl acetate in cyclohexane to afford the title compound (44 mg, 23% yield). LCMS RT= 1.01 min, ES+ve m/z 292 [M+H]+
Ethyl 2-(6-methoxy-l-oxoisoindolin-2-yI)-3-methylbutanoate
Under an atmosphere of nitrogen, an ice-cooled solution of 6-methoxyisoindolin-l-one (commercially available from for example Astatech) (105 mg, 0.64 mmol) in DMF (2.5 mL)
was treated with sodium hydride (60% w/w in mineral oil) (31 mg, 0.77 mmol) and the mixture was allowed to warm to ambient temperature. The mixture was then treated with ethyl 2-bromo-3-methylbutanoate (commercially available from for example Alfa Aesar) (135 mg, 0.64 mmol) and the mixture was stirred for 18 hours then cautiously treated with saturated aqueous ammonium chloride (20 mL). The product was extracted with ethyl acetate (2 x 25 mL) and the combined organic phase was washed with water (20 mL), brine (20 mL), filtered through a hydrophobic frit and evaporated to dryness. The product was purified by flash chromatography (20 g silica cartridge) using a gradient elution from 0 to 50% ethyl acetate in cyclohexane to afford the title compound (40 mg, 21% yield). LCMS RT= 1.03 min, ES+ve m/z 292 [M+H]+
-(6-methoxy-l-oxoisoindolin-2-yl)-3-methyIbutanoic acid

A solution of ethyl 2-(6-methoxy-l-oxoisoindolin-2-yl)-3-methylbutanoate (40 mg, 0.14 mmol) in ethanol (0.4 mL) was treated with 2M aqueous sodium hydroxide (0.20 mL, 0.41 mmol) and the mixture was stirred at ambient temperature for 2 hours. The reaction mixture was evaporated to dryness, treated with water (lOmL) and acidified to pH 3 using 2M aqueous hydrochloric acid. The product was extracted with ethyl acetate (2 x10 mL), and the combined organic phase was filtered through a hydrophobic frit and evaporated to dryness to afford the title compound (34 mg, 94% yield). LCMS RT= 0.80 min, ES+ve m/z 264 [M+H]+ -(5-methoxy-l-oxoisoindolin-2-yl)-3-methylbutanoic acid

A solution of ethyl 2-(5-methoxy-l-oxoisoindolin-2-yl)-3-methylbutanoate (40 mg, 0.14 mmol) in ethanol (0.4 mL) was treated with 2M aqueous sodium hydroxide (0.20 mL, 0.41 mmol) and the mixture was stirred at ambient temperature for 2 hours. The mixture was then evaporated to dryness; the residue was treated with water (10 mL) and acidified to pH 3 using 2M aqueous hydrochloric acid. The product was extracted with ethyl acetate (2 10 mL), and the combined organic phase was filtered through a hydrophobic frit and evaporated to
dryness to afford the title compound (33 mg, 93% yield). LCMS RT= 0.76 min, ES+ve m/z

Ethyl 2-(7-chloro-l-oxoisoindolin-2-yl)-3-methylbutanoate

Under a nitrogen atmosphere, an ice-cooled solution of 7-chloroisoindolin-l-one
(commercially available from for example JW Pharm) (150 mg, 0.90 mmol) in DMF (2.5 mL) was treated with sodium hydride (60% w/w in mineral oil) (50 mg, 1.25 mmol). The mixture was allowed to warm to ambient temperature, then treated with ethyl 2-bromo-3- methylbutanoate (commercially available from for example Alfa Aesar) (187 mg, 0.90 mmol) and stirred for 5 hours. The mixture was then ice-cooled, treated with additional sodium hydride (60% w/w in mineral oil) (50 mg, 1.25 mmol) and ethyl 2-bromo-3-methylbutanoate (187 mg, 0.90 mmol) and the mixture was stirred at ambient temperature for a further 24 hours. The mixture was then cautiously treated with saturated aqueous ammonium chloride (20 mL), and the product was extracted with ethyl acetate (2 x 25 mL). The combined organic phase was washed with water (20 mL), brine (20 mL), filtered through a hydrophobic frit and evaporated to dryness. The product was purified by flash chromatography (20 g silica cartridge) using a gradient elution from 0 to 50% ethyl acetate in cyclohexane to afford the title compound (40 mg, 15% yield). LCMS RT= 1.07 min, ES+ve m/z 296 [M+H]+.
2-(7-chloro-l -oxoisoindolin-2- l)-3-methylbutanoic acid

A solution of ethyl 2-(7-chloro-l-oxoisoindolin-2-yl)-3-methylbutanoate (40 mg, 0.14 mmol) in ethanol (0.4 mL) was treated with 2M aqueous sodium hydroxide (0.22 mL, 0.45 mmol) and the mixture was stirred at ambient temperature for 2 hours. The mixture was then evaporated to dryness, treated with water (10 mL) and then acidified to pH 3 using 2M aqueous hydrochloric acid. The product was extracted with ethyl acetate (2 x10 mL) and the combined organic phase was filtered through a hydrophobic frit and evaporated to dryness to afford the title compound (34 mg, 94% yield). LCMS RT= 0.82 min, ES+ve m/z 268 [M+H]+.
-Hydroxy-4-(4-methylthiazol-5-yl)benzonitrile

Under an atmosphere of nitrogen, a mixture of 4-bromo-2-hydroxybenzonitrile
(commercially available from for example Fluorochem) (15 g, 76 mmol), 4-methylthiazole (commercially available from for example Aldrich) (14 mL, 152 mmol), potassium acetate (14.9 g, 152 mmol) and palladium(II) acetate (0.34 g, 1.52 mmol) in l-methyl-2-pyrrolidone (125 mL) was heated at 110°C for 3 hours. The mixture was then cooled to 50°C, poured into water (300 mL) and extracted with ethyl acetate (3 x 350 mL). The combined organic fraction was filtered and the filtrate was then washed with brine (3 x 400 mL), filtered through a hydrophobic frit and evaporated to dryness. The residue was re-evaporated from toluene then from diethyl ether and then slurried in methanol to precipitate a yellow solid which was filtered off. The filtrate was evaporated to dryness and slurried in ice-cooled methanol to afford a second batch of yellow solid. The combined solid was dried under vacuum to afford the title compound (12 g, 56 mmol, 73 % yield). LCMS RT= 0.75 min, ES+ve m/z 217
[M+H]+. -(Aminomethyl)-5-(4-methylthiazol-5-yl)phenol

An ice-cooled solution of 2-hydroxy-4-(4-methylthiazol-5-yl)benzonitrile (12 g, 56 mmol) in THF (550 mL) was treated dropwise with lithium aluminium hydride (1M in THF, 140 mL, 140 mmol) over 5 minutes. The resulting mixture was then heated at 50°C for 30 minutes and additional lithium aluminium hydride (1M in THF, 20 mL, 20 mmol) was added. After a further 30 minutes the mixture was cooled in an ice bath and treated cautiously with water (14 mL), followed by aqueous sodium hydroxide (4M, 42 mL, 168 mmol) and finally water (14 mL). After standing for 3 days, the mixture was filtered and the filtered solid was washed with THF. The combined filtrate was evaporated to dryness and the residue was slurried in dichloromethane:methanol (4: 1) with Celite (about 20g) and filtered. The filtered solid was washed three times with dichloromethane/methanol (4:1) and the combined filtrate was evaporated to dryness. The product was purified by flash chromatography (330 g silica
cartridge) using a gradient elution from 0 to 15 % methanol in dichloromethane (+1 % triethylamine) to afford the title compound (6.2 g, 28 mmol, 51 % yield). LCMS RT= 0.41 min, ES+ve m/z 221 [M+H]+.
(2S,4R)-tert-Butyl 4-hydroxy-2-((2-hydroxy-4-(4-methylthiazol-5- yI)benzyl)carbamoyl)py rrolidine- 1 -carboxylate

An ice-cooled solution of 2-(aminomethyl)-5-(4-methylthiazol-5-yl)phenol (3.05 g, 13.8 mmol) and (2S,4R)-l-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid (2.94 mL, 13.8 mmol) in DMF (35 mL) was treated with DIPEA (7.25 mL, 42 mmol) followed by HATU (5.79 g, 15.2 mmol) and the mixture was stirred at ambient temperature for 1 hour. The mixture was treated with saturated aqueous sodium bicarbonate (50 mL) and extracted with ethyl acetate (3 x 80 mL). The combined organic phase was washed with brine (60 mL), filtered through a hydrophobic frit and evaporated to dryness. The product was purified by flash chromatography (330 g silica cartridge) using a gradient from 0 to 15% methanol in dichloromethane to afford the title product (4.8 g, 11 mmol, 80 % yield). LCMS RT= 0.76 min, ES+ve m/z 434 [M+H]+.
(2S,4R)-4-Hydroxy-N-(2-hydroxy-4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, hydrochloride

A solution of (2S,4R)-tert-butyl 4-hydroxy-2-((2-hydroxy-4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidine-l -carboxylate (4.8 g, 11 mmol) in
dichloromethane:methanol 20:1 (50 mL) was treated with hydrochloric acid (4M in 1,4- dioxane) (35 mL, 140 mmol) and the mixture was stirred overnight at ambient temperature. The mixture was then evaporated to dryness and the residual solid was suspended in dichloromethane and filtered. The filtered solid was washed with further dichloromethane and dried under vacuum to afford the title compound (4 g, 10.8 mmol, 98 % yield). LCMS RT= 0.46 min, ES+ve m/z 334 [M+H]+.
(2S,4R)-l-((5)-2-Aminopropanoyl)-4-hydroxy-A'-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride

A stirred mixture of (2>S',4i?)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, hydrochloride (100 mg, 0.28 mmol), (S)-2-((tert- butoxycarbonyl)amino)propanoic acid (commercially available from for example Aldrich) (64 mg, 0.34 mmol) and DIPEA (0.197 mL, 1.13 mmol) in dry DMF (3 mL) was treated with HATU (129 mg, 0.34 mmol and stirred at ambient temperature for 30 minutes. The mixture was then partitioned between ethyl acetate (30 mL) and water (30 mL) and the organic phase was washed with brine (30 mL), dried (hydrophobic frit) and evaporated to dryness. The residue was dissolved in methanol and added to a methanol-preconditioned aminopropyl solid-phase extraction cartridge (2g) eluting with methanol (3 column volumes). The resulting eluant was evaporated to dryness and the residue was dissolved in
dichloromethanermethanol (1 :1, 8 mL) and treated with hydrochloric acid, (4M in 1 ,4- dioxane) (1 mL, 4 mmol). The mixture was stirred at ambient temperature for 16 hours and then evaporated to dryness to afford the title compound (107 mg, 0.25 mmol, 89 % yield). LCMS RT= 0.51 min, ES+ve m/z 389 [M+H]+.
(25,4R)-l-(2-Amino-2-methylpropanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5- yI)benzyl)pyrrolidine-2-carboxamide

A solution of a mixture of (25',4i?)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine- 2-carboxamide, hydrochloride (100 mg, 0.28 mmol), 2-((tert-butoxycarbonyl)amino)-2- methylpropanoic acid (commercially available from for example Aldrich) (69 mg, 0.34 mmol) and DIPEA (0.197 mL, 1.13 mmol) in dry DMF (3 mL) was treated with HATU (129 mg, 0.34 mmol, stirred at ambient temperature for 30 minutes and then partitioned between ethyl acetate (30 mL) and water (30 mL). The organic phase was washed with brine (30 mL), dried (hydrophobic frit) and evaporated to dryness. The residue was dissolved in methanol
and added to a methanol-preconditioned aminopropyl solid-phase extraction cartridge (2g) eluting with methanol. The resulting eluant was evaporated to dryness and the residue was dissolved in dichloromethane methanol (1:1, 8 mL) and treated with hydrochloric acid, 4M in 1,4-dioxane (1 mL, 4 mmol). The reaction mixture was stirred at ambient temperature for 16 hours and then evaporated to dryness. The residue was suspended in dichloromethane (4 mL) and treated with TFA (1 mL) and the mixture was stirred at ambient temperature for 4 hours. The mixture was evaporated to dryness and the residue was dissolved in methanol and added to a methanol-preconditioned sulfonic acid solid-phase extraction cartridge (2g) and eluted with methanol (3 column volumes) and then with ammonia in methanol (2M, 3 column volumes). The product-containing fractions were evaporated to dryness to afford the title compound (95 mg, 0.24 mmol, 84 % yield). LCMS RT= 0.53 min, ES+ve m/z 403 [M+H]+.
(2S,4R)-4-Hydroxy-l-((S)-2-(methylamino)propanoyl)-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide

A stirred mixture of (2S,4R)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, hydrochloride (120 mg, 0.34 mmol) and (S)-2-((tert- butoxycarbonyl)(methyl)amino)propanoic acid (commercially available from for example Aldrich) (76 mg, 0.37 mmol) in DMF (2 mL) was treated with DIPEA (0.24 mL, 1.36 mmol) and then with HATU (155 mg, 0.41 mmol), and the mixture was stirred at ambient temperature for 30 minutes. The crude product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to give the intermediate Boc-protected product. The intermediate was suspended in dichloromethane (0.5 mL) and treated with TFA (0.5 mL). The mixture was stirred at ambient temperature for 1 hour and was then evaporated to dryness. The residue was dissolved in the minimum amount of a mixture of
methanohdichloromethane (1 :1), and then loaded onto a pre-conditioned (methanol) aminopropyl solid-phase extraction cartridge (5 g). The column was eluted with methanol (3 volumes) and the product-containing fractions were combined and evaporated to dryness to afford the title compound (103 mg, 75% yield). LCMS RT= 0.47 min, ES+ve m/z 403
[M+Hf.
(25,4J?)-4-Hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-l-((5)-morpholine-3- carbonyl)pyrrolidine-2-carboxamide, hydrochloride

A mixture of (2S,4R)-4-hydroxy-N-(4-(4-me hylthiazol-5-yl)benzyl)pyrrolidine-2- carboxamide, hydrochloride (100 mg, 0.28 mmol), (5)-4-(tert-butoxycarbonyl)morpholine-3- carboxylic acid (commercially available from for example Astatech) (65 mg, 0.28 mmol) and DIPEA (0.247 mL, 1.41 mmol) in DMF (2 mL) was treated with HATU (118 mg, 0.311 mmol) and stirred at ambient temperature for 30 minutes. The Boc protected intermediate was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier). The intermediate was dissolved in methanol:dichloromethane (1 :1, 3 mL), treated with hydrochloric acid in 1,4-dioxane (4M, 3 mL, 12 mmol) and allowed to stand for 1 hour. The mixture was evaporated to dryness to afford the title compound (110 mg, 0.24 mmol, 83 % yield). LCMS RT= 0.50 min, ES+ve m/z 431/432 [M+H]+.
Tert-butyl 4-(3-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5- yl)benzyl)carbamoyl)pyrrolidine-l-carbonyl)phenoxy)butanoate

A solution of a mixture of 3-(4-(tert-butoxy)-4-oxobutoxy)benzoic acid (95 mg, 0.34 mmol), (2S,4i?)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (100 mg, 0.28 mmol) and DIPEA (0.2 mL, 1.15 mmol) in dry DMF (3 mL) was treated with HATU (129 mg, 0.34 mmol) and the mixture was stirred at ambient temperature for 30 minutes. The mixture was added to an aminopropyl solid-phase extraction cartridge and eluted with methanol (3 column volumes). The resulting eluant was evaporated to dryness and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (130 mg, 0.22 mmol, 79 % yield). LCMS RT= 0.98 min, ES+ve m/z 580 [M+H]+.
Example- VHL Protac Which Binds Estrogen Receptor (Estrogen Protects)
Abbreviations:
DCM: dichloTomethane.
DIPEA: N,N-diisopropylethylamine.
DMF: N,N-dimethylformamide.
HATU: 2-(7-aza- 1 H-benzotriazole- 1 -yl)- 1 , 1 ,3,3 -tetramethyluronium hexafluorophosphate.
HPLC: high-performance liquid chromatography.
LCMS: liquid chromatography-mass spectrometry
Min: minutes.
RT: retention time.
tBu: tert-butoxide.
TFA: trifluoroacetic acid.
THF: tetrahydrofuran.
LCMS Method :
The analysis was conducted on an Acquity UPLC BEH CI 8 column (50mm x 2.1mm internal diameter 1.7μπι packing diameter) at 40°C.
The solvents employed were:
A = 0.1% v/v solution of formic acid in water.
B = 0.1% v/v solution of formic acid in acetonitrile.
The gradient employed was as follows:

The UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
The following illustrates the mobile phases and gradients used when compounds underwent purification by mass-directed autopreparative HPLC.
Mass-Directed Autopreparative HPLC (Formic Acid Modifier)
The HPLC analysis was conducted on a Sunfire CI 8 column (150mm x 30mm internal diameter, 5μπι packing diameter) at ambient temperature.
The solvents employed were:
A = 0.1% v/v solution of formic acid in water.
B = 0.1 % v/v solution of formic acid in acetonitrile.
Mass-Directed Autopreparative HPLC (Trifluoroacetic Acid Modifier)
The HPLC analysis was conducted on a Sunfire C18 column (150mm x 30mm internal diameter, 5μηι packing diameter) at ambient temperature.
The solvents employed were:
A = 0.1% v/v solution of trifluoroacetic acid in water.
B = 0.1% v/v solution of trifluoroacetic acid in acetonitrile.
Mass-Directed Autopreparative HPLC (Ammonium Bicarbonate Modifier)
The HPLC analysis was conducted on an XBridge C18 column (150mm x 30mm internal diameter, 5μιη packing diameter) at ambient temperature.
The solvents employed were:
A = 10 mM ammonium bicarbonate in water adjusted to pH 10 with ammonia solution. B = acetonitrile.
For each of the mass-directed autopreparative purifications, irrespective of the modifier used, the gradient employed was dependent upon the retention time of the particular compound undergoing purification as recorded in the analytical LCMS, and was as follows:
For compounds with an analytical LCMS retention time below 0.6 minutes the following gradient was used:

For compounds with an analytical LCMS retention time between 0.6 and 0.9 minutes the following gradient was used:

For compounds with an analytical LCMS retention time between 0.9 and 1.2 minutes the following gradient was used:
Time Flow Rate
% A % B
(minutes) (mL/min)
0 40 70 30
1 40 70 30
10 40 15 85
11 40 1 99
15 40 1 99
For compounds with an analytical LCMS retention time between 1.2 and 1.4 minutes the following gradient was used:

For compounds with an analytical LCMS retention time greater than 1.4 minutes (LCMS method A) or greater than 3.6 minutes (LCMS method B) the following gradient was used:

The UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
The chemical names were generated using ACD Name Pro version 6.02 from Advanced Chemistry Development, Inc.
8R,9S,13S,14S,17S)-3,17-bis(methoxymethoxy)-13-methyl-
7,8,9,11, 12, 13, 14,15,16, 17-decahydro-6H-cyclopenta[a]phenanthren-6-one

(8R.9S, 13S, 1 S, 17S)-3, 17-bis(methoxymethoxy)-13-methyl-
7,8,9, 11 , 12, 13, 14,15, 16,17-decahydro-6W-cyclopenta[a]phenanthren-6-one can be prepared according to the process described by Xiang-Rong Jiang, J. Walter Sowell,
Bao Ting Zhu, Steroids, 2006, 71, 334-342. (doi: 10.1016/j.steroids.2005. 1.008).
15-Bromo-1 -phenyl-2,5,8,11-tetraoxapentadecane

To a suspension of sodium hydride, 60 % w/w in mineral oil (0.250 g, 6.24 mmol) in DMF (2 mL) was added a solution of 2-(2-(2-(benzyloxy)ethoxy)ethoxy)ethanol (1 g, 4.16 mmol) (commercially available from for example Fluorochem) in DMF (2 mL) at 0 °C. After stirring for 25 minutes, 1,4-dibromobutane (commercially available from for example Aldrich) (4.04 g, 18.73 mmol) dissolved in DMF (2 mL) was added dropwise to the mixture. The reaction was stirred under an atmosphere of nitrogen for 2.5 hours. A further aliquot of sodium hydride, 60 % w/w in mineral oil (0.250 g, 6.24 mmol) was added and the reaction was stirred at 0 °C for 30 minutes. The reaction was warmed to room temperature and stirred for 30 minutes. A final aliquot of sodium hydride, 60 % w/w in mineral oil (0.250 g, 6.24 mmol) was added and the reaction stirred at room temperature for 2 hours then left standing over the weekend. The reaction mixture was filtered through celite and the solid washed with DCM. The filtrate was partitioned between DCM (30 mL) and water (30 mL). The organic extract was washed with brine (2 x 30 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 100% methyl tert-butyl ether in cyclohexane to afford the title compound (711 mg, 1.89 mmol, 46% yield). LCMS RT= 1.16 min, ES+ve m/z 375.2/377.1 [M+H]+. 15-Iodo-l-phenyl-2,5,8,ll-tetraoxapentadecane

A mixture of 15-bromo-l-phenyl-2,5,8,l 1-tetraoxapentadecane (711 mg, 1.894 mmol) and sodium iodide (568 mg, 3.79 mmol) in acetone (10 mL) was heated under reflux conditions for 4 hours. The reaction was cooled to room temperature. The mixture was filtered through celite and the solid washed with acetone. The solvent was removed under reduced pressure. The residue was dissolved in ethyl acetate (30 mL) and washed with water (30 mL) and brine (2 x 30 mL). The organic extract was dried using a hydrophobic frit and concentrated under reduced pressure to afford the title compound (759 mg, 1.797 mmol, 95% yield). LCMS RT= 1.23 min, ES+ve m/z 440.0 [M+NH4]+.
(7S,8R,9S,13S,14S,17S)-3,17-Bis(methoxymethoxy)-13-methyl-7-(l-phenyl-2,5,8,ll- tetraoxapentadecan-15-yl)-7,8,9,ll,12,13,14,15,16,17-decahydro-6H- cyclopenta [a] phenanthren-6-one

A solution of KOtBu in THF (1M, 1.282 mL, 1.282 mmol) was added to a cooled solution (0 °C) of (8R,9S, 13S,14S,17S)-3, 17-bis(methoxymethoxy)-l 3-methyl- 7,8,9,1 l,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-6-one (240 mg, 0.641 mmol) in anhydrous THF (2 mL). The reaction mixture was stirred at 0 °C for 45 minutes and then cooled to -78 °C. A solution of 15-iodo-l-phenyl-2,5,8,l 1-tetraoxapentadecane (789 mg, 1.868 mmol) in THF (1 mL) was added dropwise. The solution was stirred at -78 °C for 2 minutes, allowed to warm to 0 °C and stirred for 1.5 hours at that temperature. The reaction was partitioned between water (30 mL) and ethyl acetate (30 mL). The organic extract was dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 100% ethyl acetate in cyclohexane to afford the title compound (234 mg, 0.350 mmol, 55% yield). LCMS RT= 1.48 min, ES+ve m/z 669.3 [M+H]+, 686.4 [M+NLUf.
tetraoxapentadecan-15-yl)-7,8,9,ll,12,13,14,15,16,17-decahydro-6H- cyclopenta [a] phenanthren-6-one

A solution of aqueous HC1 (6M, 2.3 mL, 13.80 mmol) was added to a solution of
(7S,8R,9S, 13S, 14S, 175)-3 , 17-bis(methoxymethoxy)- 13-methyl-7-( 1 -phenyl-2,5,8, 11 - tetraoxapentadecan-15-yl)-7,8,9, 11 , 12, 13 , 14, 15, 16, 17-decahydro-6H- cyclopenta[a]phenanthren-6-one (234 mg, 0.350 mmol) in THF (2.3 mL). The reaction mixture was stirred at room temperature for 16 hours. Water (30 mL) was added and the product was extracted with ethyl acetate (50 mL). The organic extract was washed with brine (2 x 30 mL), dried using a hydrophobic frit and concentrated under reduced pressure to afford the title compound (200 mg, 0.344 mmol, 98% yield). LCMS RT= 1.07 min, ES+ve m/z 581.3 [M+H]+, 598.3 [M+NH4]+.
(7R,8R,9S,13S,14S,175)-13-Methyl-7-(l-phenyI-2,5,8,ll-tetraoxapentadecan-15-yl)- 7,8,9,ll512,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthrene-3,17-dioI

Triethylsilane (commercially available from for example Aldrich) (0.550 mL, 3.44 mmol) was added to a solution of (71S,8i?,91?,135,145,,171S)-3,17-dihydroxy-13-methyl-7-(l-phenyl- 2,5,8, 11 -tetraoxapentadecan- 15 -yl)-7,8,9, 11 , 12, 13 , 14, 15, 16, 17-decahydro-6H- cyclopenta[a]phenanthren-6-one (200 mg, 0.344 mmol) in TFA (2 mL, 26.0 mmol). The reaction was stirred at room temperature under an atmosphere of nitrogen for 16 hours. The mixture was partitioned between ethyl acetate (30 mL) and brine (30 mL). The organic extract was washed with brine (2 x 30 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The residue was dissolved in MeOH (5 mL) and treated with aqueous NaOH (2M, 5 mL, 10.00 mmol). The reaction mixture was stirred at room
temperature for 3 hours. The solvent was removed under reduced pressure. The residue was partitioned between ethyl acetate (30 mL) and a 10 % citric acid solution (30 mL). The organic extract was washed with brine (30 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by reverse phase
chromatography using a gradient elution from 5% to 95% acetonitrile (+ 0.1% formic acid) in water (+ 0.1% formic acid) to afford the title compound (150 mg, 0.265 mmol, 77% yield). LCMS RT= 1.18 min, ES+ve m/z 567.3 [M+H]+, 584.3 [M+NH4]+.
15-((7J?,8R,9*S',13S,14S,175)-3,17-Bis(methoxymethoxy)-13-methyl- 7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cycIopenta[a]phenanthren-7-yI)-l-phenyl- 2,5,8,11-tetraoxapentadecane

A vial was charged with (7R,8R,95,,131S,145,17,S)-13-methyl-7-(l-phenyl-2,5,8,l 1- tetraoxapentadecan- 15-yl)-7,8,9, 11 , 12, 13, 14, 15, 16,17-decahydro-6H- cyclopenta[a]phenanthrene-3,17-diol (150 mg, 0.265 mmol) and DIPEA (0.555 mL, 3.18 mmol) in THF (10 mL). The vial was sealed, the solution was cooled to 0 °C and
chloro(methoxy)methane (commercially available from for example Aldrich) (0.2 mL, 2.63 mmol) was added. The reaction mixture was warmed to room temperature, stirred for 1 hour and heated at 70 °C for 40 hours. The reaction was cooled to room temperature. The reaction was partitioned between ethyl acetate (100 mL) and water (100 mL). The organic extract was washed with brine (2 x 50 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 100% methyl tert-butyl ether in cyclohexane to afford the title compound
(122 mg, 0.186 mmol, 70% yield). LCMS RT= 1.60 min, ES+ve m/z 672.5 [M+NELt .
2-(2-(2-(4-((7if,8R,95,135,145,17S)-3,17-Bis(methoxymethoxy)-13-methyl-
7,8,9,ll»12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7- yl)butoxy)ethoxy)ethoxy)ethanol

A mixture of 15-((77?,8i?,95',131S,141S,,171S)-3,17-bis(methoxymethoxy)-13-methyl- 7,8,9, 11 , 12, 13, 14, 15 , 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)- 1 -phenyl-2,5,8,11- tetraoxapentadecane (115 mg, 0.176 mmol) and 10 % w/w palladium on carbon (100 mg, 0.094 mmol) in ethanol (4 mL) was stirred at room temperature under an atmosphere of hydrogen for 1.5 hours. The palladium on carbon was filtered through celite and the filtrate evaporated under reduced pressure. The residue was partitioned between ethyl acetate (15 mL) and brine (15 mL). The organic extract was dried using a hydrophobic frit and concentrated under reduced pressure to afford the title compound (81 mg, 0.143 mmol, 82% yield). LCMS RT= 1.36 min, ES+ve m/z 582.4

Tert-butyl 16-((7R,8R,9S,13S,14S,175)-3,17-bis(methoxymethoxy)-13-methyl- 7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3,6,9,12- tetraoxahexadecan- 1 -oate

Sodium hydride, 60 % w/w in mineral oil (10 mg, 0.250 mmol) was added to a cooled solution (0 °C) of 2-(2-(2-(4-((7i?,8i?,95,13,S,141S,175)-3,17-bis(methoxymethoxy)-13-methyl- 7,8,9,11, 12,13, 14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7- yl)butoxy)ethoxy)ethoxy)ethanol (81 mg, 0.143 mmol) in DMF (2 mL). The reaction was stirred at that temperature for 10 minutes and terf-butyl 2-bromoacetate (commercially available from for example Aldrich) (32 μL, 0.217 mmol) was added. The reaction was stirred at 0 °C for 1 hour and at room temperature for further 2 hours. The reaction mixture was partitioned between ethyl acetate (30 mL) and water (30 mL). The organic layer was washed with brine (30 mL), dried (hydrophobic frit) and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to
100% methyl tart-butyl ether in cyclohexane to afford the title compound (60 mg, 0.088 mmol, 62% yield). LCMS RT= 1.57 min, ES+ve m/z 696.5 [M+NH4]+.
16-((7R,8R,9^,13S,14S,17S)-3,17-Dihydroxy-13-methyl-7,8,9,ll,12,13,14,15,16,17- decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3,6,9,12-tetraoxahexadecan-l-oic acid

7¾rt-butyl 16-{fJR$R$St 13S, 14S, 175)-3 , 17-bis(methoxymethoxy)- 13 -methyl- 7,8,9, 11 , 12, 13, 14, 15 , 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3 ,6,9, 12- tetraoxahexadecan-l-oate (133 mg, 0.196 mmol) was dissolved in THF (1.5 mL) and treated with aqueous HC1 (6M, 1.5 mL, 9.00 mmol). The reaction mixture was stirred at room temperature for 8 hours. The reaction mixture was subjected directly to purification by mass- directed automated preparative HPLC (formic acid modifier) to afford the title compound (60 mg, 0.112 mmol, 57% yield). LCMS RT= 0.89 min, ES+ve m/z 535.3 [M+H]+, 552.3
[M+NH4]+.
(2S,4R)-l-((5)-19-((7R,8R,9S,13S,14S,17S)-3,17-Dihydroxy-13-methyl- 7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-2-isopropyI-4- oxo-6,9,12,15-tetraoxa-3-azanonadecan-l-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrroIidine-2-carboxamide

HATU (16 mg, 0.042 mmol) was added to a mixture of (2S,4J?>l-((.S)-2-amino-3- methylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (25 mg, 0.055 mmol), 16-((7i?,8i?,9S,135,145,17S)-3,17-dihydroxy-13-methyl- 7,8,9, 11 , 12, 13, 14, 15, 16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3,6,9, 12- tetraoxahexadecan-l-oic acid (15 mg, 0.028 mmol) and DIPEA (0.05 mL, 0.286 mmol) in
DMF (1 mL). The reaction was stirred at room temperature for 30 minutes. The reaction mixture was subjected directly to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (20 mg, 0.021 mmol, 76% yield). LCMS RT= 0.99 min, ES+ve m/z 933.3 [M+H]+.
(2S,4R)-l-((5)-2-(r^butyl)-19^
7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-4-oxo-
6,9,12,15-tetraoxa-3-azanonadecan-l-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrroIidine-2-carboxamide

HATU (22 mg, 0.058 mmol) was added to a mixture of (25',4i?)-l-((S)-2-amino-3,3- dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyn'olidine-2-carboxamide, hydrochloride (25 mg, 0.054 mmol), 16-((7J?,8i?,95,135,14S,175 -3,17-dihydroxy-13-methyl- 7,8,9, 11 , 12, 13 , 14, 15, 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3,6,9, 12- tetraoxahexadecan-l-oic acid (23 mg, 0.043 mmol) and DIPEA (0.040 mL, 0.229 mmol) in DMF (1 mL). The reaction was stirred at room temperature for 10 minutes. The reaction mixture was subjected directly to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (26 mg, 0.027 mmol, 64% yield). LCMS RT= 1.02 min, ES+ve m/z 947.8 [M+H]+. -(2-(4-Bromobutoxy)ethoxy)ethoxy)methyl)benzene

To a suspension of sodium hydride, 60 % w/w in mineral oil (0.92 g, 22.9 mmol) in DMF (5 mL) was added a solution of 2-(2-(benzyloxy)ethoxy)ethanol (commercially available from for example Aldrich) (2.74 mL, 15.29 mmol) in DMF (5 mL) at 0 °C. After stirring for 25 min, 1,4-dibromobutane (commercially available from for example Aldrich) (14.9 g, 68.8
mmol) dissolved in DMF (5 mL) was added dropwise to the mixture. The reaction was warmed to ambient temperature and stirred under an atmosphere of nitrogen for 2.5 hours. A further aliquot of sodium hydride, 60 % w/w in mineral oil (0.92 g, 22.9 mmol) was added and the reaction was stirred at 0 °C for 30 minutes and at ambient temperature for 30 minutes. A final aliquot of sodium hydride, 60 % w/w in mineral oil (0.92 g, 22.9 mmol) was added and the reaction stirred at ambient temperature for 2 hours then left standing overnight. The reaction mixture was filtered through celite and the solid washed with DCM. The filtrate was partitioned between DCM (30 mL) and water (30 mL). The organic extract was washed with brine (2 x 30 mL), dried using a hydrophobic frit, and concentrated under reduced pressure. The product was purified by chromatography on silica using a using a gradient elution from 0% to 80% methyl tert-butyl ether in cyclohexane to afford the title compound (3 g, 9.06 mmol, 59% yield). LCMS RT= 1.19 min, ES+ve m/z 331.2/333.2 [M+H]+. -(2-(4-Iodobutoxy)ethoxy)ethoxy)methyl)benzene

A mixture of ((2-(2-(4-bromobutoxy)ethoxy)ethoxy)methyl)benzene (3 g, 9.06 mmol) and sodium iodide (2.72 g, 18.11 mmol) in acetone (10 mL) was heated under reflux conditions for 3 hours. The reaction was cooled to room temperature. The mixture was filtered through celite and the solid washed with acetone. The solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate (30 mL) and washed with water (30 mL) and brine (2 x 30 mL). The organic extract was dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a using a gradient elution from 0% to 50% methyl tert-butyl ether in cyclohexane to afford the title compound (3.1 g, 8.2 mmol, 90% yield). LCMS RT= 1.25 min, ES+ve m/z 379.2 [M+H]+.
(7S,8R,9S,13S,14S,17S)-7-(4-(2-(2-(Benzyloxy)ethoxy)ethoxy)butyl)-3,17- bis(methoxymethoxy)-13-methyl-7,8,9,H,12,13,14,15,16,17-decahydro-6H- cyclopenta[a]phenanthren-6-one

A solution of KOtBu, in THF (1M, 3.2 mL, 3.2 mmol) was added to a cooled solution (0 °C)
of(8 ?,95,13S,14^,17^-3,17-bis(methoxymethoxy)-13-methyl-7,8,9,l l,12,13,14,15,16,17- decahydro-6H-cyclopenta[a]phenanthren-6-one (600 mg, 1.6 mmol) in anhydrous THF (6 mL). The reaction mixture was stirred at 0 °C for 45 minutes and then cooled to -78 °C. A solution of ((2-(2-(4-Iodobutoxy)ethoxy)ethoxy)methyl)benzene (910 mg, 2.4 mmol) in THF (3 mL) was added dropwise. The solution was stirred at -78 °C for 2 minutes then allowed to warm to 0 °C and stirred for 1.5 hours at that temperature. The reaction was partitioned between water (30 mL) and ethyl acetate (30 mL). The organic extract was separated, dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 50% ethyl acetate in cyclohexane to afford the title compound (450 mg, 0.72 mmol, 45% yield). LCMS RT= 1.49 min, ES+ve m/z 625.5 [M+H]+.
(7S,8R,9S,13^,14S,17S)-7-(4-(2-(2-(Benzyloxy)ethoxy)ethoxy)butyl)-3,17-dihydroxy-13- methyl-7,8,9,ll,12,13,14,15,16,17-decahydro-6^-cyclopenta[a]phenanthren-6-one

A solution of aqueous HC1 (6M, 4.6 mL, 27.6 mmol) was added to a solution of
(7S,SR,9S, 13S, 14S, 171S)-7-(4-(2-(2-(benzyloxy)ethoxy)ethoxy)butyl)-3, 17- bis(methoxymethoxy)- 13 -methyl-7,8,9, 11 , 12, 13 , 14, 15, 16, 17-decahydro-6H- cyclopenta[a]phenanthren-6-one (470 mg, 0.752 mmol) in THF (4.6 mL). The reaction mixture was stirred at room temperature for 18 hours. Water (30 mL) was added and the product was extracted with ethyl acetate (50 mL). The organic extract was washed with brine (2 x 30 mL), dried using a hydrophobic frit and concentrated under reduced pressure to afford the title compound (390 mg, 0.727 mmol, 97% yield). LCMS RT= 1.08 min, ES+ve m/z
537.2 [M+H]+, 554.2 [M+NH4]+.
(7R,8R,9S,135,145,17S)-7-(4-(2-(2-(BenzyIoxy)ethoxy)ethox )butyl)-13-methyl- 7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cycIopenta[a]phenanthrene-3,17-diol

Triethylsilane (commercially available from for example Aldrich) 1.161 mL, 7.27 mmol) was added to a solution of (7,S',8^,9S,,135,,141S',175)-7-(4-(2-(2-(benzyloxy)ethoxy)ethoxy)butyl)-
3 , 17-dihydroxy- 13 -methyl-7,8,9, 11 , 12, 13 , 14, 15, 16, 17-decahydro-6H- cyclopenta[a]phenanthren-6-one (390 mg, 0.727 mmol) in TFA (4.2 mL, 54.5 mmol). The reaction was stirred at room temperature under an atmosphere of nitrogen for 18 hours. The mixture was partitioned between ethyl acetate (50 mL) and brine (50 mL). The organic extract was washed with brine (2 x 30 mL), saturated sodium bicarbonate (30 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The residue was dissolved in MeOH (10 mL) and treated with aqueous NaOH (2M, 5 mL, 10.0 mmol). The reaction mixture was stirred at room temperature for 1 hour. The solvent was removed under reduced pressure. The residue was partitioned between ethyl acetate (30 mL) and aqueous HC1 solution (1M, 20 mL). The organic extract was washed with brine (20 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by reverse phase chromatography using a gradient elution from 5% to 95% acetonitrile (+ 0.1% formic acid) in water (+ 0.1% formic acid) to afford the title compound (270 mg, 0.517 mmol,
71% yield). LCMS RT= 1.18 min, ES+ve m/z 523.5 [M+H]+, 540.5 [M+NH4]+.
(7R,8i?,9S,13^,14S,17S)-7-(4-(2-(2-(Benzyloxy)ethoxy)ethoxy)butyl)-3,17- bis(methoxymethoxy)-13-methyl-7,8,9,ll,12,13,14,15,16,17-decahydro-6H- cyclopentaja] phenanthrene
Chloro(methoxy)methane (commercially available from for example Aldrich) (0.390 mL, 5.14 mmol) was added to a cooled (0 °C) solution of (7i?,8J?,9S i3S,14S,17S)-7-(4-(2-(2- (benzyloxy)ethoxy)ethoxy)butyl)- 13 -methyl-7,8,9, 11 , 12, 13 , 14, 15 , 16, 17-decahydro-6H- cyclopenta[a]phenanthrene-3,17-diol (270 mg, 0.517 mmol) and DIPEA (1.083 mL, 6.20
mmol) in THF (16 mL). The reaction mixture was warmed to room temperature, stirred for 1 hour and then heated at 70 °C for 40 hours. The reaction mixture was cooled to 0 °C, additional DIPEA (0.271 mL, 1.550 mmol) and chloro(methoxy)methane (0.098 mL, 1.291 mmol) was added. The reaction was heated to 70 °C and stirred for a further 24 hours. The reaction was cooled to room temperature, and was partitioned between ethyl acetate (100 mL) and water (100 mL). The organic extract was washed with brine (2 x 50 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 100% methyl ter/-butyl ether in cyclohexane to afford the title compound (220 mg, 0.36 mmol, 70% yield). LCMS RT= 1.62 min, ES+ve m/z 628.6 [M+NH4]+.
2-(2-(4-((7R,8i?,95,13 ,14S,17S)-3,17-Bis(methoxymethoxy)-13-methyl-
7,8,9,ll,12,13,14,15,16,l'7-decahydro-6Hr-cyclopenta[a]phenanthreii-7- yl)butoxy)ethoxy)ethanol
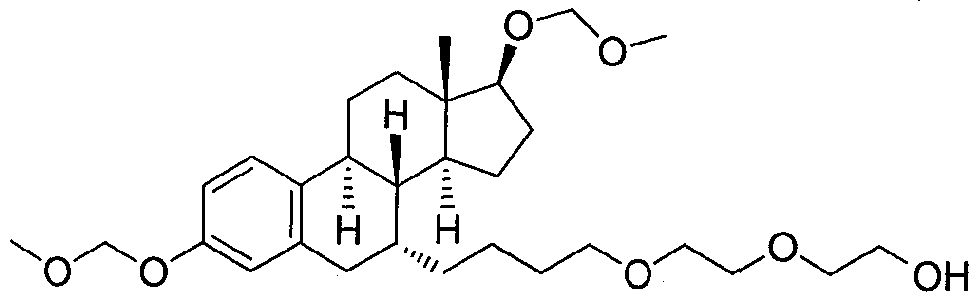
A mixture of (7i?,8i?,9,S,13^141S,175)-7-(4-(2-(2-(beiizyloxy)ethoxy)ethoxy)butyl)-3,17- bis(methoxymethoxy)- 13-methyl-7,8,9, 11 , 12, 13, 14, 15, 16,17-decahydro-6H- cyclopenta[a]phenanthrene (220 mg, 0.36 mmol) and 10 % w/w palladium on carbon (100 mg, 0.094 mmol) in ethanol (4 mL) was stirred at room temperature under an atmosphere of hydrogen for 1 hour. The palladium on carbon was filtered through celite, washed with ethanol (50 ml) and the filtrate was evaporated under reduced pressure to afford the title compound (186 mg, 0.357 mmol, 99% yield) LCMS RT= 1.36 min, ES+ve m/z 521.5
[M+H]+, 538.5 [M+NH4]+.
7¾/*-butyl 2-(2-(2-(4-((7J?,8R,9S,135,,14S,175)-3,17-is(methoxymethoxy)-13-methyl- 7,8,9,11 ,12,13,14,15,16,17-decahy dro-6H-cy clopenta [a]phenanthren-7- yl)butoxy)ethoxy)ethoxy)acetate
O
Sodium hydride, 60 % w/w mineral oil (25.0 mg, 0.625 mmol) was added to a cooled solution (0 °C) of 2-(2-(4-((7i?,8/?,95',135,145,175)-3,17-bis(methoxymethoxy)-13-methyl- 7,8,9, 11 , 12, 13 , 14, 15, 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7- yl)butoxy)ethoxy)ethanol (186 mg, 0.357 mmol) in DMF (4.5 mL). The reaction was stirred at 0 °C for 10 minutes and tert-butyl 2-bromoacetate (commercially available from for example Aldrich) (79 μΐ,, 0.536 mmol) was added. The reaction was stirred at 0 °C for 1 hour and at room temperature for a further 6 hours. The reaction was cooled to 0 °C and additional sodium hydride, 60 % w/w in mineral oil (15.72 mg, 0.393 mmol), followed by tert-butyl 2- bromoacetate (0.053 mL, 0.357 mmol) was added. The reaction was stirred at room temperature for a further 18 hours. The reaction mixture was partitioned between ethyl acetate (30 mL) and water (30 mL). The organic layer separated, washed with brine (30 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 100% methyl tert- butyl ether in cyclohexane to afford the title compound (90 mg, 0.142 mmol, 40% yield). LCMS RT= 1.56 min, ES+ve m/z 652.6 [M+NH4]+, 657.5 [M+Na]+.
2-(2-(2-(4-((7R,8R,9S,135,145,17S)-3,17-Dihydroxy-13-methyl- 7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7- yl)butoxy)ethoxy)ethoxy)acetic acid

7¾/ -butyl 2-(2-(2-(4-((7i?,8 i?,9S, 13S, 14S, 17S)-3, 17-bis(methoxymethoxy)-l 3-methyl- 7,8,9, 11 , 12, 13, 14, 15, 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7- yl)butoxy)ethoxy)ethoxy)acetate (80 mg, 0.126 mmol) was dissolved in THF (1 mL) and treated with aqueous HC1 (6M, 1 mL, 6.0 mmol). The reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was subjected directly to purification by mass- directed automated preparative HPLC (formic acid modifier) to afford the title compound (23 mg, 0.047 mmol, 37% yield). LCMS RT= 0.89 min, ES+ve m/z 491,4 [M+H]+.
(2S,4R)-l-((5 -16-((7R,8R,95',13 ,14S,17S)-3,17-Dihyclroxy-13-methyl-
7,8,9,1 l,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-2-isopropyl-4- oxo-6,9,12-trioxa-3-azahexadecan-l-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide

HATU (12 mg, 0.03 mmol) was added to a mixture of (25',4i?)-l-((5)-2-amino-3- methylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (23 mg, 0.05 mmol), 2-(2-(2-(4-((7i?,8i?,95,135,,14,S,175)-3,17-dihydroxy-13- methyl-7,8,9, 11 , 12, 13, 14, 15, 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7- yl)butoxy)ethoxy)ethoxy)acetic acid (10 mg, 0.02 mmol) and DIPEA (0.04 mL, 0.20 mmol) in DMF (0.8 mL). The reaction was stirred at room temperature for 30 min. The reaction mixture was subjected directly to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (15 mg, 0.017 mmol, 84% yield). LCMS RT= 0.98 min, ES+ve m/z 889.7 [M+H]+. -Bromo-l-phenyl-2,5,8,ll,14-pentaoxaoctadecane

To a suspension of sodium hydride, 60 % w/w in mineral oil (0.85 g, 21.3 mmol) in DMF (8 mL) was added a solution of 1 -phenyl -2,5, 8,1 l-tetraoxatridecan-13-ol (commercially available from for example TCI Europe Fine Chemicals) (4.0 g, 14.2 mmol) in DMF (8 mL) at 0 °C. After stirring for 25 minutes, 1 ,4-dibromobutane (commercially available from for example Aldrich) (7.62 mL, 63.8 mmol) dissolved in DMF (8 mL) was added dropwise to the mixture. The reaction was warmed to room temperature and stirred under an atmosphere of nitrogen for 30 minutes. A further aliquot of sodium hydride, 60 % w/w in mineral oil (0.85 g, 21.3 mmol) was added and the reaction was stirred at room temperature overnight. Another aliquot of sodium hydride, 60 % w/w in mineral oil (0.85 g, 21.3 mmol) was added and the reaction stirred at room temperature for 2 hours. A final aliquot of sodium hydride, 60 % w/w in mineral oil (0.43 g, 10.6 mmol) was added and the reaction stirred at room temperature for
1 hour. The reaction mixture was filtered through celite and the solid washed with DCM. The filtrate was partitioned between DCM (50 mL) and water (50 mL). The organic extract was washed with brine (2 x 50 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 85% methyl tert-butyl ether in cyclohexane to afford the title compound (3.93 g, 9.37 mmol, 63% yield). LCMS RT= 1.16 min, ES+ve m/z 419.3/421.2 [M+H]+. -Iodo-l-phenyl-2,5,8,ll»14-pentaoxaoctadecane

A mixture of 18-bromo-l-phenyl-2,5,8,l l,14-pentaoxaoctadecane (2.08 g, 4.91 mmol) and sodium iodide (1.47 g, 9.82 mmol) in acetone (10 mL) was heated under reflux conditions for 3 hours. The reaction was cooled to room temperature, filtered through celite and the solid was washed with acetone. The solvent was removed under reduced pressure. The residue was dissolved in ethyl acetate (30 mL), and washed with water (30 mL) and brine (2 x 30 mL). The organic extract was dried using a hydrophobic frit and concentrated under reduced pressure to afford the title compound (759 mg, 1.80 mmol, 95% yield).LCMS RT= 1.21 min, ES+ve m/z 467.0 [M+H]+, 484.0 [M+NH4]+.
(7 ,8R,9S,135,14S,17S)-3,17-Bis(methoxymethoxy)-13-methyl-7-(l-phenyl-2,5,8,ll,14- pentaoxaoctadecan-18-yl)-7,8,9,ll,12,13,14,15,16,17-decahydro-6H- cyclopenta [a] phenanthren-6-one
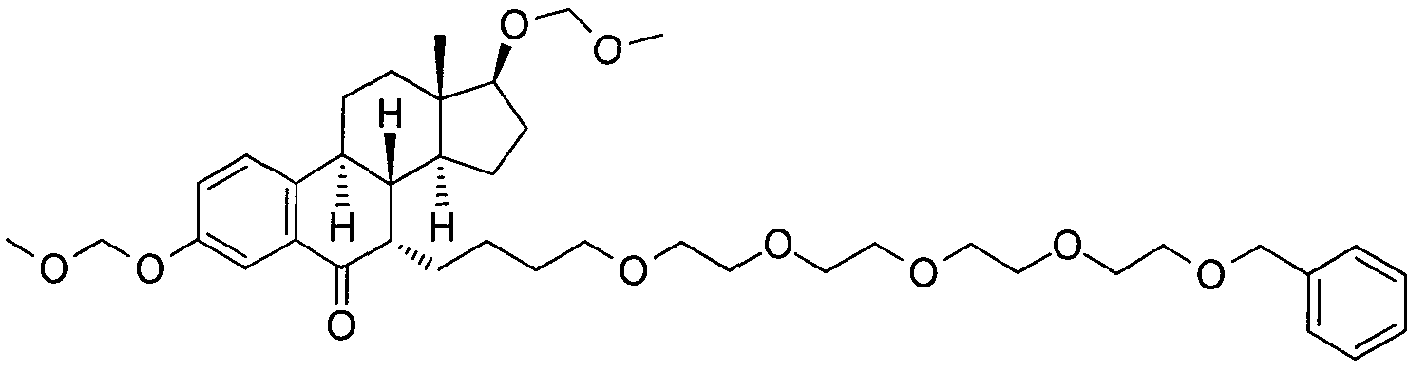
A solution of KOtBu in THF (1M¾5.34 mL, 5.34 mmol) was added to a cooled solution (0 °C) of (*R,9S, 13S, 14S, 17S)-3 , 17-bis(methoxymethoxy)- 13 -methyl-
7,8,9,11,12,13, 14,15, 16,17-decahydro-6H-cyclopenta[a]phenanthren-6-one (1 g, 2.67 mmol) in anhydrous THF (10 mL). The reaction mixture was stirred at 0 °C for 45 minutes and then cooled to -78 °C. 18-Iodo-l-phenyl-2,5,8,l 1,14-pentaoxaoctadecane (1.87 g, 4.01 mmol) in THF (5 mL) was added dropwise. The solution was stirred at -78 °C for 2 minutes, allowed to warm to 0 °C and stirred for 1.5 hours at that temperature. The reaction was partitioned
between water (50 mL) and ethyl acetate (2 x 50 mL). The organic extracts were dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 100% ethyl acetate in cyclohexane to afford the title compound (883 mg, 1.24 mmol, 46% yield). LCMS RT= 1.47 min, ES+ve m/z 713.5 [M+H]+. ,ll,14-

A solution of aqueous HC1 (6M, 9.2 mL, 55.2 mmol) was added to a solution of
(7S,8R,9S, 135, 145, 175)-3 , 17-bis(methoxymethoxy)- 13-methyl-7-( 1 -phenyl-2,5,8, 11,14- pentaoxaoctadecan- 18-yl)-7,8,9, 11,12,13,14,15,16,17-decahydro-6H- cyclopenta[a]phenanthren-6-one (883 mg, 1.24 mmol) in THF (9.2 mL). The reaction mixture was stirred at room temperature for 18 hours. Water (30 mL) was added and the product was extracted with ethyl acetate (50 mL). The organic extract was washed with brine (2 x 30mL), dried using a hydrophobic frit and concentrated under reduced pressure to afford the title compound (772 mg, 1.23 mmol, 99% yield). LCMS RT= 1.06 min, ES+ve m/z 625.3 [M+H]+, 642.3 [M+NH4]+.
(7i?,8R,9S,135,14S,175)-13-Methyl-7-(l-phenyl-2,5,8,ll,14-pentaoxaoctadecan-18-yI)- 7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthrene-3,17-diol

Triethylsilane (commercially available from for example Aldrich) (2.0 mL, 12.9 mmol) was added to a solution of (75,8R,95, 135, 145,175)-3,17-dihydroxy- 13 -methyl-7-(l -phenyl- 2,5,8, 11 , 14-pentaoxaoctadecan- 18-yl)-7,8,9, 11 , 12, 13 , 14, 15, 16, 17-decahydro-6H- cyclopenta[a]phenanthren-6-one (830 mg, 1.29 mmol) in TFA (8.5 mL, 110 mmol). The reaction was stirred at room temperature under an atmosphere of nitrogen for 16 hours. The
mixture was partitioned between ethyl acetate (50 mL) and brine (50 mL). The organic extract was washed with brine (2 x 50 mL), saturated sodium bicarbonate (50 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The residue was dissolved in MeOH (10 mL) and treated with aqueous NaOH (2M, 10 mL, 20.0 mmol). The reaction mixture was stirred at room temperature for 1 hour. The solvent was removed under reduced pressure. The residue was partitioned between ethyl acetate (40 mL) and 1M HC1 solution (20 mL). The organic extract was washed with brine (20 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by reverse phase chromatography using a gradient elution from 5% to 90% acetonitrile (+ 0.1% formic acid) in water (+ 0.1% formic acid) to afford the title compound (375 mg, 0.614 mmol, 47% yield). LCMS RT= 1.17 min, ES+ve m/z 611.5 [M+H]+, 628.6 [M+NH4]+.
18-((7R,8Ri95,13S,14S,l'7S)-3,17-Bis(methoxymethoxy)-13-methyl- 7,8,9,11)12,13,14,15,16,17-decahydro-6H-cycIopenta[a]phenanthren-7-yl)-l-phenyl- -pentaoxaoctadecane

Chloro(methoxy)methane (commercially available from for example Aldrich) (0.5 mL, 6.58 mmol) was added to a cooled (0 °C) solution of (7i?,8i?,95,,13,S',145,175)-13-methyl-7-(l- phenyl-2,5,8,l l,14-pentaoxaoctadecan-18-yl)-7,8,9,l l,12,13,14,15,16,17-decahydro-6H- cyclopenta[a]phenanthrene-3,17-diol (375 mg, 0.614 mmol) and DIPEA (1.5 mL, 8.59 mmol) in THF (20 mL). The reaction mixture was warmed to room temperature, stirred for 1 hour and heated at 70 °C for 72 hours. The reaction was cooled to room temperature. The reaction was partitioned between ethyl acetate (100 mL) and water (100 mL). The organic extract was washed with brine (2 x 50 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 100% methyl tert-butyl ether in cyclohexane to afford the title compound (357 mg, 0.51 mmol, 72% yield). LCMS RT= 1.60 min, ES+ve m/z 716.7 [Μ+Ν¾]+, 721.7 [M+Na]+.
16-((7R,8R,9S,13S,14S,17S)-3,17-Bis(methoxymethoxy)-13-methyl-
7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3,6,9,12- tetraoxahexadecan-l-ol

A mixture of ^-((V i^ i^^lS^H^iy^-S T-bisCmethoxymetho y)-^^^!- 7,8,9, 11 , 12, 13, 14, 15, 16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)- 1 -phenyl- 2,5,8,11,14-pentaoxaoctadecane (357 mg, 0.444 mmol) and 10 % w/w palladium on carbon (157 mg, 0.148 mmol) in ethanol (5 mL) was stirred at room temperature under an atmosphere of hydrogen for 1.5 hours. The palladium on carbon was filtered through celite and the filtrate evaporated under reduced pressure to afford the title compound (300 mg, 0.41 mmol, 93% yield) LCMS RT= 1.37 min, ES+ve m/z 609.6 [M+H]+, 631.6 [M+Na]+. 9,12,15-

Sodium hydride, 60 % w/w in mineral oil (30 mg, 0.75 mmol) was added to a cooled solution (0 °C) of 16-((7i?,8R,919,135,141S',175)-3,17-bis(methoxymethoxy)-13-methyl- 7,8,9, 11 , 12, 13 ,14, 15, 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3,6,9, 12- tetraoxahexadecan-l-ol (300 mg, 0.43 mmol) in DMF (5 mL). The reaction was stirred at that temperature for 10 minutes and tert-butyl 2-bromoacetate (0.095 mL, 0.643 mmol) was added. The reaction was stirred at 0 °C for 1 hour and then at room temperature for a further 18 hours. The reaction mixture was partitioned between ethyl acetate (30 mL) and water (30 mL). The water layer was extracted with additional ethyl acetate (2 x 30 mL), and the combined organic layers were washed with brine (2 x 30 mL), dried (hydrophobic frit) and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 100% methyl tert-butyl ether in cyclohexane to afford the title compound (177 mg, 0.245 mmol, 57% yield). LCMS RT= 1.58 min, ES+ve m/z 740.6 [Μ+Ν¾]+, 745.6 [M+Na]+.
19-((7i?,8R,95,13S,14S,175)-3,17-Dihydroxy-13-methyl-7,8,9,ll,12,13,14,15,16,17- decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3,6,9,12,15-pentaoxanonadecan-l-oic acid

Terr-butyl 19-((7R,SR,9S, 13S, 14S, 17S)-3, 17-bis(methoxymethoxy)- 13-methyl- 7,8,9, 11 , 12, 13 , 14, 15, 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3,6,9, 12, 15- pentaoxanonadecan-l-oate (177 mg, 0.189 mmol) was dissolved in THF (2 mL) and treated with aqueous HC1 (6M, 2 mL, 12.0 mmol). The reaction mixture was stirred at room temperature for 7 hours. The reaction mixture was subjected directly to purification by mass- directed automated preparative HPLC (formic acid modifier) to afford the title compound (64 mg, 0.1 1 1 mmol, 59% yield). LCMS RT= 0.92 min, ES+ve m/z 579.4 [M+H]+, 596.5
[M+NH4]+.
(25',4if)-l-(( )-22-((77?,8R,9 ,135,14 ,175)-3,17-Dihydroxy-13-methyl-
7,8,9,11, 12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-2-isopropyl-4- oxo-6,9,12,15,18-pentaoxa-3-azadocosan-l-oyl)-4-hydroxy-7V-(4-(4-methylthiazoI-5- yl)benzyl)pyrrolidine-2-carboxamide

HATU (16 mg, 0.04 mmol) was added to a mixture of (2S,4i?)-l-((,S 2-ammo-3- methylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (25 mg, 0.06 mmol), 19-((7i?,8i?,9,S,135,14>S,175}-3,17-dihydroxy-13-methyl- 7,8,9, 1 1 , 12, 13 , 14, 15, 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3 ,6,9,12,15- pentaoxanonadecan-l-oic acid (16 mg, 0.03 mmol) and DIPEA (0.048 mL, 0.28 mmol) in DMF (0.8 mL). The reaction was stirred at room temperature for 30 minutes. The reaction mixture was subjected directly to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (17.7 mg, 0.018 mmol, 65% yield).
LCMS RT= 0.99 min, ES+ve m z 97 '.4 [M+H]+.
(2S,4R)-l-((5)-2-(J^butyI)-22-((7^,8i?,95',13S,145,17>S)-3,17-dihydroxy-13-methyl- 7,8,9,ll,12,13,14,15,16,l'7-decahydro-6H-cyclopeiita[a]phenanthren-7-yl)-4-oxo- 6,9,12,15,18-pentaoxa-3-azadocosan-l-oyl)-4-hydroxy-A'-(4-(4-methylthiazol-5- yl)benzyl)pyrrolidine-2-carboxamide

HATU (16 mg, 0.04 mmol) was added to a mixture of (2S,4£)-l-((S)-2-amino-3,3- dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylmiazol-5-yl)benzyl)pyrrolidine-2-carboxamide hydrochloride (25 mg, 0.05 mmol), 19-((7^,8i?,95,,13S,,1419,175)-3,17-dihydroxy-13-methyl- 7,8,9,1 l,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-3,6,9,12,15- pentaoxanonadecan-l-oic acid (16 mg, 0.03 mmol) and DIPEA (0.048 mL, 0.28 mmol) in DMF (0.8 mL). The reaction was stirred at room temperature for 30 minutes. The reaction mixture was subjected directly to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (17.5 mg, 0.017 mmol, 63% yield). LCMS RT= 1.03 min, ES+ve m/z 991.4 [M+H]+.
(7S,8R,9 ,135,14S,175)-7-(5-(BenzyIoxy)pentyl)-3,17-dihydroxy-13-methyl- 7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthreii-6-one
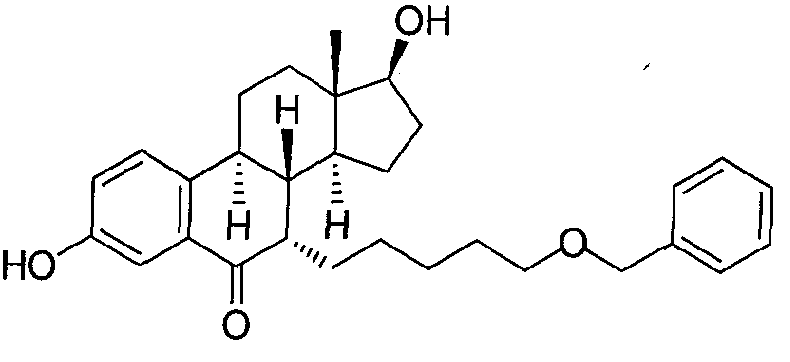
A solution of KOtBu in THF (1M, 4.81 mL, 4.81 mmol) was added to a cooled solution (0 °C) of (SR,9S, 13S, 1 AS, 175)-3, 17-bis(methoxymethoxy)-l 3-methyl- 7,8,9,11, 12,13, 14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-6-one (900 mg, 2.403 mmol) in anhydrous THF (10 mL). The reaction mixture was stirred at 0 °C for 45 minutes and then cooled to -78 0 °C. (((5-Iodopentyl)oxy)methyl)benzene (can be prepared following the procedure described in J. Chem. Soc, Perkin Trans. 1 1990, 129-132) (2.193 g, 7.21 mmol) in THF (0.5 mL) was added dropwise. The solution was stirred at -78 °C for 2 minutes and allowed to warm to room temperature and stirred for 1 hour at that temperature. The reaction was partitioned between water (70 mL) and ethyl acetate (70 mL). The organic
extract was dried using a hydrophobic frit and concentrated under reduced pressure. The intermediate was purified by chromatography on silica using a gradient elution from 0% to 50% ethyl acetate in cyclohexane. The residue was dissolved in THF (16 mL) and aqueous HC1 (6M, 16 mL, 96 mmol) was added. The reaction was stirred at room temperature for 16 hours. The reaction mixture was partitioned between ethyl acetate (20 mL) and water (20 mL). The organic extract was dried (hydrophobic frit) and concentrated under reduced pressure. The product was purified by reverse phase chromatography using a gradient elution from 5% to 85% acetonitrile (+ 0.1% formic acid) in water (+ 0.1% formic acid) to afford the title compound mg, 1.053 mmol, 44% yield). LCMS RT= 1.16 min, ES+ve m/z 463.4 [M+H]+.
(7 f,8J?,9S,13S,14S,17S)-7-(5-(Benzyloxy)pentyl)-13-methyl-7,8,9,ll,12,13,14,15,16,17- decahydro-6H-cyclopenta[a]phenanthrene-3,17-diol

Triethylsilane (1.681 mL, 10.53 mmol) was added to a solution of (7S,$R,9S,\ 35,14S.17S)-7- (5-(benzyloxy)pentyl)-3 , 17-dihydroxy- 13-methyl-7,8,9,l 1 , 12, 13 , 14, 15, 16, 17-decahydro-6H- cyclopenta[a]phenanthren-6-one (487 mg, 1.053 mmol) in TFA (6 mL, 78 mmol). The reaction was stirred at room temperature under an atmosphere of nitrogen for 16 hours. The mixture was partitioned between ethyl acetate (30 mL) and brine (30 mL). The organic extract was washed with brine (2 x 30 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The residue was dissolved in MeOH (4 mL) and treated with aqueous NaOH (2M, 4 mL, 8.00 mmol). The reaction mixture was stirred at room
temperature for 3 hours. The solvent was removed under reduced pressure. The residue was partitioned between ethyl acetate (30 mL) and water (30 mL). The organic extract was washed with brine (30 mL), dried (hydrophobic frit) and concentrated under reduced pressure. The product was purified by reverse phase chromatography using a gradient elution from 10% to 95% acetonitrile (+ 0.1% formic acid) in water (+ 0.1% formic acid) to afford the title compound (4\0 mg, 0.914 mmol, 87% yield). LCMS RT= 1.30 min, ES+ve m/z 449.1
[M+H]+.
(TR^Ji^Sjia^l^lT^-T-iS-CBenz lox ipent -S-lT-bisimethox metho yi-B-methyl- 7,8,9,11, 12,13,14,15,16,17-decahydro-6Hrcyclopenta[a]phenanthrene

Chloro(methoxy)methane (0.7 mL, 9.22 mmol) was added to solution of
(7i?,8i?,91S,13<S,14S,17¾-7-(5-(benzyloxy)pentyl)-13-methyl-7,8,9,l l, 12,13,14,15,16,17- decahydro-6H-cyclopenta[a]phenanthrene-3,17-diol (410 mg, 0.914 mmol) and DIPEA (2 mL, 11.45 mmol) in THF (8 mL). The reaction vessel was sealed, placed under an atmosphere of nitrogen and heated at 70 °C for 2 days. The reaction was cooled to room temperature. The reaction was partitioned between ethyl acetate (50 mL) and brine (50 mL). The organic extract was washed with brine (2 x 50 mL), dried using a hydrophobic frit and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 100% methyl tert-b tyl ether in cyclohexane to afford the title compound (41 '4 mg, 0.883 mmol, 97% yield). LCMS RT= 1.72 min, ES+ve m/z 554.5 [M+NH4]+.
5-((7R,8^,9S,135,14 ',175)-3,17-Bis(methoxymethoxy)-13-methyl- 7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yI)pentaii-l-ol

A mixture of (7R,$R,9S, 13S, 145", 175)-7-(5 -(benzyloxy)pentyl)-3 , 17-bis(methoxymethoxy 13-methyl-7,8,9,l l,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthrene (474 mg, 0.883 mmol) and 10% w/w palladium on carbon (100 mg, 0.094 mmol) in ethanol (5 mL) and methyl fert-butyl ether (2 mL) was stirred at room temperature under an atmosphere of hydrogen for 1.5 hours. The palladium was filtered through celite and the filtrate
concentrated under reduced pressure to afford the title compound (371 mg, 0.831 mmol, 94% yield). LCMS RT= 1.39 min, ES+ve m/z 447.5 [M+H]+ (weak ionisation).
S-rtTR-SR^^D^l^lTSJ-SjlV-Bisimetho metho i-lS-methyl- 7,8,9,ll,12,13,14,15,16,17-decahydro-6H-cycIopenta[a]phenanthren-7-yl)pentyl 4- methylbenzenesulfonate

4-Methylbenzene-l-sulfonyl chloride (400 rag, 2.098 mmol) was added to 5- ((1R$R,9S, 13S, 1 AS, 17S)-3, 17-bis(methoxymethoxy)- 13-methyl-7,8,9, 11,12, 13 , 14, 15, 16, 17- decahydro-6H-cyclopenta[a]phenanthren-7-yl)pentan-l-ol (371 mg, 0.831 mmol) in pyridine (5 mL). The reaction mixture was stirred at room temperature for 20 hours. The reaction mixture was partitioned between ethyl acetate (30 mL) and aqueous HC1 (2M, 30 mL). The organic extract was washed with sat Na2C03 (30 mL), brine (30 mL), dried (hydrophobic frit) and concentrated under reduced pressure. The product was purified by chromatography on silica using a gradient elution from 0% to 100% methyl tert-butyl ether in cyclohexane to afford the title compound (401 mg, 0.667 mmol, 80% yield). LCMS RT= 1.60 min, ES+ve m/z 623.4 [M+Na]+.
Tert-butyl 18-((7R,8R,9S,13S,14S,17S)-3,17-bis(methoxymethoxy)-13-methyl- 7,8,9,ll?12,13>14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)-13-inethyl- 4,7,10-trioxa-13-azaoctadecan-l-oate, formic acid salt

A microwave vial was charged with 5-((7i?,8i?,9iS',135',145',17S)-3,17-bis(methoxymethoxy)-
13- methyl-7,8 ,9, 1 1, 12,13, 14, 15,16,17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)pentyl 4-methylbenzenesulfonate (100 mg, 0.166 mmol), tert-butyl 5,8,1 l-trioxa-2-azatetradecan-
14- oate (can be prepared following the procedure described in WO2012054110A2) (145 mg, 0.499 mmol) and DIPEA (0.291 mL, 1.664 mmol) in THF (2 mL). The vial was sealed and placed under an atmosphere of nitrogen using a vacuum purge. The reaction was heated at 75 °C for 48 hours. The reaction was cooled to room temperature. The reaction mixture was subjected directly to purification by mass-directed automated preparative HPLC (formic acid
modifier) to afford the title compound (102 mg, 0.133 mmol, 80% yield). LCMS RT= 1.22 min, ES+ve m/z 720.6 [M+H]+. ,12,13,14,15,16,17- 0-trioxa-13-

7¾rt-butyl 1 X-((7RM,9S, 135, 1 S, 17S)-3 , 17-bis(methoxymethoxy)- 13 -methyl- 7,8,9, 11 , 12, 13 , 14, 15 , 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)- 13-methyl-4,7, 10- trioxa-13-azaoctadecan-l-oate, formic acid salt (100 mg, 0.131 mmol) was dissolved in THF (1 mL) and treated with aqueous HC1 (6M, 1 mL, 6.00 mmol). The reaction was stirred at room temperature for 6 hours. The reaction mixture was subjected directly to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (24 mg, 0.039 mmol, 30% yield). LCMS RT= 0.74 min, ES+ve m/z 576.5 [M+H]+.

dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide, hydrochloride (13 mg, 0.028 mmol), 18-((7i?,8#,9S, 135, 145,17S)-3,17-dihydroxy- 13 -methyl- 7,8,9, 11 , 12, 13 , 14, 15 , 16, 17-decahydro-6H-cyclopenta[a]phenanthren-7-yl)- 13 -methyl-4,7, 10- trioxa-13-azaoctadecan-l-oic acid, formic acid salt (12 mg, 0.019 mmol) and DIPEA (0.03 mL, 0.172 mmol) in DMF (0.8 mL). The reaction was stirred at room temperature for 10 minutes. The reaction mixture was subjected directly to two purifications by mass-directed
automated preparative HPLC (formic acid modifier followed by ammonium carbonate modifier) to afford the title compound (13 mg, 0.013 mmol, 68 % yield). LCMS RT= 0.84 min, ES+ve m/z 988.8 [M+H]+.
Claims
Claims:
1. A compound according to the chemical structure:

Where R1 is an optionally substituted Ci-C6 alkyl group, an optionally substituted
-(CH2)nOH, an optionally substituted -(CH2)nSH, an optionally substituted (CH2)n-0-(C1- C6)alkyl group, an optionally substituted (CH2)n-WCOCW-(Co-C6)alkyl group containing an epoxide moiety WCOCW where each W is independently H or a C]-C3 alkyl group, an optionally substituted -(CH2)„COOH, an optionally substituted -(CH2)nC(0)-(Ci-C6 alkyl), an optionally substituted -(CH2)nNHC(0)-Ri, an optionally substituted -(CH2)nC(0)-NR1R2, an optionally substituted
-(CH2)„OC(0)-NRiR2, -(CH20)nH, an optionally substituted -(CH2)nOC(0)-(C1-C6 alkyl), an optionally substituted -(CH2)nC(0)-0-(C1-C6 alkyl), an optionally substituted
-(CH20)nCOOH, an optionally substituted -(OCH2)nO-(Ci-C6 alkyl), an optionally substituted -(CH20)nC(0)-(Ci-C6 alkyl), an optionally substituted -(OCH2)nNHC(0)-Ri, an optionally substituted -(CH20)nC(0)-NRiR2, -(CH2CH20)„H, an optionally substituted
-(CH2CH20)„COOH, an optionally substituted -(OCH2CH2)nO-(C1-C6 alkyl), an optionally substituted -(CH2CH20)nC(0)-(C1-C6 alkyl), an optionally substituted
-(OCH2CH2)nNHC(0)-Ri, an optionally substituted -(CH2CH20)„C(0)-NRiR2,an optionally substituted -S02Rs, an optionally substituted S(0)Rs, N02, CN or halogen (F, CI, Br, I, preferably F or CI);
R\ and R2 are each independently H or a Ci-C6 alkyl group which may be optionally substituted with one or two hydroxyl groups or up to three halogen groups (preferably fluorine);
Rs is a C!-C6 alkyl group, an optionally substituted aryl, heteroaryl or heterocycle group or a -(C¾)raNRiR2 group,
X and X' are each independently C=0, C=S, -S(0), S(0)2 , (preferably X and X' are both C=0);
R2 is an optionally substituted -(CH2)n-(C=0)u(NRi)v(S02)w(Ci-C6)alkyl group, an optionally substituted -(CH2)n-(C=0)u(NRi)v(S0 )wNR1NR2N group, an optionally substituted -(CH2)n-(C=O)u(NRi)v(S02)w-Aryl, an optionally substituted
-(CH2)n-(C=0)u(NRi)v(S02)w-Heteroaryl, an optionally substituted
-(CH2)n-(C=0)vNRi(S02)w-Heterocycle, an optionally substituted
-NRI-(CH2)n-C(0)u(NRi)v(S02)w-CI-C6 alkyl, an optionally substituted
-NR1-(CH2)n-C(0)u(NR1)v(S02)w- NR1NR2N, an optionally substituted
-NR1-(CH2)n-C(0)u(NRI)v(S02)w-NRiC(0)R1N, an optionally substituted
-NR1-(CH2)n-(C=0)u(NRi)v(S02)w-Aryl, an optionally substituted
-NRI-(CH2)n-(C=0)u(NR1)v(S02)w-Heteroaryl or an optionally substituted
-NR1-(CH2)n-(C=0)vNR1(S02)w-Heterocycle, an optionally substituted
-X -C1-C6 alkyl group; an optionally substituted
-X1 2 - Aryl group; an optionally substituted
-X^2 - Heteroaryl group; an optionally substituted
n '
-X - Heterocycle group; an optionally substituted;
R3 is an optionally substituted Ci-C6 alkyl, an optionally substituted
-(CH2)n-C(0)u(NRi)v(S02)w-Ci-C6 alkyl, an optionally substituted
-(CH2)n-C(0)u(NRi)v(S02)w-NRiNR2N, an optionally substituted
-(CH2)n-C(0)u(NR1)v(S02)w-NR1C(0)R1N, an optionally substituted
-(CH2)n-C(0)u(NRI)v(S02)w-C(O)NR1R2, an optionally substituted
-(CH2)n-C(0)u(NRi)v(S02)w-Aryl, an optionally substituted
-(CH2)n-C(0)u(NRi)v(S02)w-Heteroaryl, an optionally substituted
-(CH2)n-C(0)u(NRi)v(S02)w-Heterocycle, an optionally substituted
-NR1-(CH2)n-C(0)u(NR1)v(S02)w-C1-C6 alkyl, an optionally substituted
- RI-(CH2)n-C(0)u(NR1)v(S02)w- NR!C(0)R1N, an optionally substituted
-NR1-(CH2)n-C(0)u(NR1)v(S02)w-Aryl, an optionally substituted
-NR1-(CH2)n-C(0)u(NR1)v(S02)w-Heteroaryl, an optionally substituted
-NR1-(CH2)n-C(0)u(NRi)v(S02)w-Heterocycle, an optionally substituted
-0-(CH2)n-(C=0)u(NRi)v(S02)w-Ci-C6 alkyl, an optionally substituted
-0-(CH2)n-(C=0)u(NR1)v(S02)w-NRiNR2N, an optionally substituted
-0-(CH2)n-(C=0)u(NRi)v(S02)w-NR1C(0)RIN, an optionally substituted -0-(CH2)n-(C=0)u(NRi)v(S02)w-Aryl, an optionally substituted
-0-(CH2)n-(C=0)u(NRi)v(S02)w-Heteroaryl or an optionally substituted
-0-(CH2)n- (C=0)u(NRi)v(S02)w-Heterocycle;
-(CH2)n-(V)n'-(CH2)n-(V)n'-Ci-C6 alkyl group, an optionally substituted
-(CH2)n-(V)n'-(CH2)n-(V)n'-Aryl group, an optionally substituted
-(CH2)n-(V)n-(CH2)n-(V)n-Heteroaryl group, an optionally substituted
-(CH2)n-(V)n'-(CH2)n-(V)n'-Heterocycle group, an optionally substituted
-(CH2)n-N(R1 (C=O)m-(V)n'-Ci-C6 alkyl group, an optionally substituted
-(CH2)n-N(Ri-)(C=0)m-(V)n-Aryl group, an optionally substituted
-(CH2)n-N(R1')(C=:0)m-(V)n'-Heteroaryl group, an optionally substituted
-(CH2)n-N(Ri >)(C=0)m>-(V)n'-Heterocycle group, an optionally substituted
-XR3 -Ci-C6 alkyl group; an optionally substituted
-XR3 - Aryl group; an optionally substituted
-XR3 - Heteroaryl group; an optionally substituted
-XR3 - Heterocycle group; an optionally substituted;
Where RJN and R2N are each independently H, C]-C6 alkyl which is optionally substituted with one or two hydroxyl groups and up to three halogen groups or an optionally substituted
-(CH2)n-Aryl, -(CH2)n-Heteroaryl or -(CH2)n-Heterocycle group;
V is 0, S or NRi;
Ri is the same as above;
R1 and Rp are each independently H or a C1-C3 alkyl group;
are each independently an optionally substituted -CH2)n-, -CH2)n- CH(XV)=CH(XV)- (cis or trans), -CH2)n-CH≡CH- , -(CH2CH20)n- or a C3-C6 cycloalkyl group, where Xv is H, a halo or a C1-C3 alkyl group which is optionally substituted;
Each m is independently 0, 1 , 2, 3, 4, 5, 6;
Each m' is independently 0 or 1 ;
Each n is independently 0, 1, 2, 3, 4, 5, 6;
Each n' is independently 0 or 1 ;
Each u is independently 0 or 1 ;
Each v is independently 0 or 1 ;
Each w is independently 0 or 1, or
A pharmaceutically acceptable salt, stereoisomer, solvate or polymorph thereof.
2. The compound according to claim 1 according to the chemical structure:

Wherein each of R1 , R2 and R3 are the same as in claim 1 above and X is C=0, C group or a S(0)2 group, or
A pharmaceutically acceptable salt, stereoisomer, solvate or polymorph thereof.
3. The compound according to claim 1 according to the chemical structure:

1 ' 2' 3 '
Where R' , R" and RJ are the same as presented above, or
A pharmaceutically acceptable enantiomer, diastereomer, solvate or polymorph thereof.
4. The compound according to any of claims 1-3 wherein R1 is a hydroxyl group or a group which may be metabolized to a hydroxyl or carboxylic group, such that the compound represents a prodrug form of an active compound.
5. The compound according to any of claims 1-4 wherein R1 is -(CH2)nOH, -(CH2)nSH -(CH2)n-0-(C i -C6)alkyl group, -(CH2)nCOOH, -(CH20)nH, an optionally substituted
-(CH2)nOC(0)-(C C6 alkyl), or an optionally substituted -(CH2)nC(0)-0-(Ci-C6 alkyl), wherein n is 0 or 1.
6. The compound according to any of claims 1-5 wherein X and X', where present, are a C=0, C=S, -S(O) group or a S(0)2 group.
7. The compound according to any of claims 1 -6 wherein X and X' are both C=0 groups.
8. The compound according to any of claims 1 -7 wherein R1 is OH.
9. The compound according to any of claims 1-8 wherein R2 is an optionally substituted -NR'-T-Aryl, an optionally substituted -NR'-T-Heteroaryl group or an optionally substituted -NR'-T-Heterocycle, where R1 is H or CH3, H; and
T is an optionally substituted -(CH2)n- group, wherein each one of the methylene groups may be optionally substituted with one or two substituents, which may be optionally substituted; and n is preferably 0, 1 , 2 or 3, preferably 0 or 1.
10. The compound according to claim 9 wherein said Aryl group is an optionally substituted phenyl or naphthyl group.
11. The compound according to claim 9 wherein said Aryl group is a phenyl group which is optionally substituted with a halogen group, an amine, monoalkyl- or dialkyl amine, OH, SH, CH3> CF3, OMe, OCF3, N02, or CN group, each of which may be substituted in the ortho-, meta- and/or para- positions of the phenyl ring.
12. The compound according to claim 10 wherein said Aryl group is a phenyl group which is optionally substituted with a heteroaryl group.
13. The compound according to claim 12 wherein said heteroaryl group is an optionally substituted isoxazole, an optionally substituted oxazole, an optionally substituted thiazole, an optionally substituted isothiazole, an optionally substituted pyrrole, an optionally substituted imidazole, an optionally substituted oximidazole, an optionally substituted diazole, an optionally substituted triazole group, an optionally substituted pyridine group or an optionallyl substituted oxapyridine group.
14. The compound according to claim 13 wherein said optionally substituted isoxazole is a methylsubstituted isoxazole, said optionally substituted oxazole is a methylsubstituted oxazole, said optionally substituted thiazole is a methyl substituted thiazole, said optionally substituted isothiazole is a methyl substituted isothiazole, said optionally substituted pyrrole is a methylsubstituted pyrrole, said optionally substituted imidazole is a
a methylimidazole, a benzylimidazole or a methoxybenzylimidazole, said optionally substituted oximidazole is a methyloximidazole, said optionally substituted diazole is a methyldiazole, said optionally substituted triazole group is a methylsubstituted triazole group, said optionally substituted pyridine group is a halosubstituted or methylsubstitutedpyridine and said oxapyridine group is linked to the phenyl group by an oxygen.
15. The compound according to claim 9 wherein said Aryl group is a phenyl group which is optionally substituted with a heterocycle selected from the group consisting of piperidine and morpholine.
16. The compound according to claim 9 wherein said Aryl group is a phenyl which is substituted with a methyl-substituted thiazole group.
17. The compound according to claim 9 wherein said Heteroaryl group is an optionally substituted quinoline, an optionally substituted indole, an optionally substituted azaindole, an optionally substituted isoxazole, an optionally substituted thiazole, an optionally substituted isothiazole, an optionally substituted benzofuran, an optionally substituted thiophene or an optionally substituted pyridine.
18. The compound according to claim 17 wherein said isoxazole is a methylsubstituted isoxazole.
19. The compound according to claim 17 wherein said thiophene is a methylsubstituted thiophene.
20. The compound according to claim 17 wherein said 2-, 3-, or 4-pyridine is
methylsubstituted.
21. The compound according to claim 17 wherein said thiazole is a methylsubstituted thiazole.
22. The compound according to claim 9 wherein said Heterocycle is selected from the group consisting of tetrahydroquinoline, tetrahydrofuran, tetrahydrothiophene, piperidine, piperazine, pyrrollidine, morpholine, oxane or thiane, each of which groups is optionally substituted.
23. The compound according to claim 10 wherein said Aryl group is a phenyl group which is optionally substituted with a halogen, an amine, monoalkylamine, dialkylamine, OH, SH, COOH, CH3, CF3, OMe, OCF3, N02, or CN group, an optionally substituted phenyl group, wherein said phenyl group is itself optionally substituted with at least one of F, CI, OH, SH, COOH, CH3, CF3, OMe, OCF3, N02, or CN group, a naphthyl group, which may be optionally substituted, or an optionally substituted heteroaryl or heterocycle group.
24. The compound according to claim 23 wherein said optionally substituted heteroaryl is an optionally substituted isoxazole, an optionally substituted oxazole, an optionally substituted thiazole, an optionally substituted pyrrole, an optionally substituted imidazole, an optionally substituted benzimidazole, an optionally substituted oximidazole an optionally substituted diazole group, including a methyldiazole group, an optionally substituted triazole group, including a methylsubstituted triazole group, an optionally substituted pyridine group, including a halo- (preferably, F) or methylsubstitutedpyridine group or an oxapyridine group, an optionally substituted furan, an optionally substituted benzofuran, an optionally substituted dihydrobenzofuran, an optionally substituted indole, an optionally substituted indolizine, an optionally substitutedazaindolizine, an optionally substituted quinoline, or an optionally substituted group according to the chemical structure:

Where Sc is CHR¾¾, NRUKfc, or O;
RHET is H, CN, N02, halo (preferably CI or F), optionally substituted Ci-C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups (e.g. CF3), optionally substituted 0(C]-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Q-C6 alkyl group (preferably Ci-C3 alkyl);
Rss is H, CN, N02, halo (preferably F or CI), optionally substituted C C6 alkyl (preferably substituted with one or two hydroxyl groups or up to three halo groups), optionally substituted 0-(C[-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups) or an optionally substituted -C(0)(Ci-C6 alkyl) (preferably substituted with one or two hydroxyl groups or up to three halo groups);
RURE is H, a d-C6 alkyl (preferably H or C C3 alkyl) or a -C(0)(Ci-C6 alkyl) each of which groups is optionally substituted with one or two hydroxyl groups or up to three halogen, preferably fluorine groups, or an optionally substituted phenyl group, an optionally substituted heteroaryl, or an optionally substituted heterocycle, preferably for example piperidine, morpholine, pyrrolidine, tetrahydrofuran);
RPRO is H, optionally substituted -C6 alkyl or an optionally substituted aryl group or an optionally substituted heteroaryl or heterocyclic group selected from the group consisting of oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine,
furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, quinoline, benzofuran, indole, indolizine, azaindolizine; RPR01 and RPR02 are each independently H, an optionally subsituted C!-C3 alkyl group or together form a keto group and
Each n is independently 0, 1, 2, 3, 4, 5, or 6,
Or a pharmaceutically acceptable salt, enantiomer, diastereomer, solvate or polymorph thereof.
25. The compound according to claim 23 wherein said optionally substituted heterocycle is selected from the group consisting of pyrollidine, furan, tetrahydrofuran, tetrahydrothiene, piperidine, piperazine and morpholine.
26. The compound according to any of claims 1-25 wherein R3' is an optionally substituted -T-Aryl, an optionally substituted, -T-Heteroaryl, an optionally substituted
-T-Heterocycle, an optionally substituted -NR1 -T-Aryl, an optionally substituted
-NR1 -T-Heteroaryl or an optionally substituted -NR1 -T-Heterocycle; and
T is a -(CH2)n group, -(CH20)n- group, a -(OCH2)n- group, a -(CH2CH20)n- group, or a -(OCH2CH2)n- group, each of which groups is optionally substituted with one or two substituents; and n is 0, 1, 2 or 3.
27. The compound according to claim 26 wherein said Aryl group is an optionally substituted phenyl or naphthyl group.
28. The compound according to claim 27 wherein said Aryl group is a phenyl group which is optionally substituted with a halogen (preferably F or CI), an amine, monoalkyl- or dialkyl amine, an amido group, OH, SH, CH3, CF3, OMe, OCF3, N02, ,CN or a S(0)2Rs group where Rs is a Ci-C6 alkyl group, a -(CH2)mNRiR2 group or an optionally substituted aryl, heteroaryl or heterocycle group, each of which may be substituted in ortho-, meta- and/or para- positions of the phenyl ring, or said phenyl group is optionally substituted with an aryl, heteroaryl or heterocycle, each of which groups is optionally substituted, where m is 0, 1, 2, 3, 4, 5 or 6 and Ri and R2 are each independently H or a Ci-C6 alkyl group which may be optionally substituted with one or two hydroxyl groups or up to three halogen groups.
29. The compound according to claim 28 wherein said Aryl group is a phenyl group which is optionally substituted with a heteroaryl group.
30. The compound according to claim 29 wherein said heteroaryl group is an optionally substituted isoxazole, an optionally substituted oxazole, an optionally substituted thiazole, an optionally substituted pyrrole, an optionally substituted imidazole, an optionally substituted oximidazole, an optionally substituted diazole is a methyldiazole, an optionally substituted triazole group, an optionally substituted pyridine group or an optionallyl substituted oxapyridine group.
31. The compound according to claim 30 wherein said optionally substituted isoxazole is a methylsubstituted isoxazole, said optionally substituted oxazole is a methylsubstituted oxazole, said optionally substituted thiazole a methyl substituted thiazole, said optionally substituted pyrrole is a methylsubstituted pyrrole, said optionally substituted imidazole is a a methylimidazole, a benzylimidazole or a methoxybenzylimidazole, said optionally substituted oximidazole is a methyloximidazole, said optionally substituted diazole is a methyldiazole, said optionally substituted triazole group is a methylsubstituted triazole group, said optionally substituted pyridine group is a halosubstituted or methylsubstitutedpyridine and said oxapyridine group is linked to the phenyl group by an oxygen.
32. The compound according to claim 28 wherein said Aryl group is a phenyl group which is optionally substituted with a heterocycle selected from the group consisting of piperidine and morpholine.
33. The compound according to claim 26 wherein said Heteroaryl group is an optionally substituted quinoline which may be attached to the pharmacophore or substituted on any carbon atom within the quinoline ring, an optionally substituted indole, an optionally substituted benzimidazole, an optionally substituted benzodiazole, an optionally substituted benzoxofuran, an optionally substituted imidazole, an optionally substituted isoxazole, an optionally substituted oxazole, an optionally substituted diazole, an optionally substituted benzofuran, an optionally substituted thiophene, an optionally substituted thiazole, an optionally substituted triazole, a triisopropylsilyl group, an optionally substituted -(CH2)m-0- Ci-C6 alkyl group, an optionally substituted -(CH2)m-C(0)-0-Ci-C6 alkyl group) or an optionally substituted 2-, 3, or 4-pyridine and m is 0, 1, 2, 3, 4, 5, or 6.
34. The compound according to claim 26 wherein said Heteroaryl group is a methylsubstituted oxazole or a triazole which is optionally substituted with a methyl group, a triisopropylsilyl group, an optionally substituted -(CH2)m-0-Ci-C6 alkyl group or an optionally substituted -(CH2)m-C(0)-0-Ci-C6 alkyl group).
35. The compound according to claim 26 wherein said Heterocycle is selected from the group consisting of tetrahydroquinoline, tetrahydrofuran, tetrahydrothiene, piperidine, piperazine, pyrrolidine and morpholine, each of which groups may be optionally substituted.
36. A compound according to the chemical structure:

Where R1' is OH or a group which can be metabolized in a patient or subject to OH;
R2' is an optionally substituted -NRi-X112 -alkyl group, an optionally substituted -NRi-X 2'-
Aryl group; an optionally substituted -NRi- Xs2 -HET group, an optionally substituted -NRi-
X^'-Aryl-HET group or an optionally substituted -NRj- X^'-HET-Aryl group,
is H or a C1-C3 alkyl group;
X 2' is an optionally substituted -CH2)n- , -CH2)n-CH(Xv)=CH(Xv)- (cis or trans),
-CH2)n-CH≡CH-, -(CH2CH20)n- or a C3-C6 cycloalkyl group;
R3' is an optionally substituted -(CH2)n-(V)n>-(CH2)n-(V)n'-RS3'group, an optionally substituted -(CH2)n-N(Rr)(C=0)m-(V)n-RS3' group, an optionally substituted -XR3'-alkyl group, an optionally substituted -XR3 -Aryl group; an optionally substituted -XR3 -HET group, an optionally substituted -XR3 -Aryl-HET group or an optionally substituted -XR3 -HET- Aryl group,
RS3 is an optionally substituted Cj-Cio alkyl group, an optionally substituted Aryl group or a HET group;
Ri' is H or a Q-C3 alkyl group;
V is O, S or NRp;
XR3' is an optionally substituted -CH )n- , -CH2)n-CH(Xv)=CH(Xv)- (cis or trans),
-CH2)„-CH≡CH-, -(CH2CH20)n- or a C3-C6 cycloalkyl group;
Xv is H, a halo or a Q-C3 alkyl group which is optionally substituted with one or two hydroxyl groups or up to three halogen groups;
Alkyl is an optionally substituted Ci-C10 alkyl group;
Aryl is an optionally substituted phenyl or naphthyl group; and
HET is an optionally substituted oxazole, isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahycirothiene, pyridine, piperidine, piperazine, morpholine, benzofbran, indole, indolizine, azaindolizine, quinoline or a group according to the chemical structure:

Where Sc is CHRSS, NRURE, or O;
RHET is H, CN, N02, halo, optionally substituted Q-C6 alkyl, optionally substituted 0(C!-C6 alkyl) or an optionally substituted acetylenic group -C≡C-Ra where Ra is H or a Ci-C6 alkyl group;
Rss is H, CN, N02, halo, optionally substituted Ci-C6 alkyl, optionally substituted 0-(C]-C6 alkyl) or an optionally substituted -C(0)(C1-C6 alkyl;
RURE is H, a Ci-C6 alkyl or a -C(0)(CrC6 alkyl), each of which groups is optionally substituted with one or two hydroxyl groups or up to three halogens, or an optionally substituted heterocycle; and
Yc is N or C-RYC, where RYC is H, OH, CN, N02, halo, optionally substituted Ci-C6 alkyl, optionally substituted 0(Ci-C6 alkyl) or an optionally substituted acetylenic group -C≡C-Ra where Ra is the same as above;
RP 0 is H, optionally substituted Ci-C6 alkyl or an optionally substituted aryl or an optionally substituted heteroaryl or heterocyclic group selected from the group consisting of oxazole,
isoxazole, thiazole, isothiazole, imidazole, diazole, oximidazole, pyrrole, pyrollidine, furan, dihydrofuran, tetrahydrofuran, thiene, dihydrothiene, tetrahydrothiene, pyridine, piperidine, piperazine, morpholine, quinoline, benzofuran, indole, indolizine, azaindolizine;
RPR01 and RPR02 are each independently H, an optionally subsituted C1-C3 alkyl group or together form a keto group,
m' is 0 or 1 ;
Each n is independently 0, 1 , 2, 3, 4, 5, or 6 and
Each n' is independently 0 or 1 , or a
pharmaceutically acceptable salt, stereoisomer, solvent or polymorph thereof.
37. A compound according to the chemical structure:

Where Rr is OH or a group which is metabolized in a patient or subject to OH;
R2' is a -NH-CH2-Aryl-HET;
R3' is a -CHRCR3'-NH-C(0)-R3P1 group or a -CHRCR3'-R3P2 group;
Where RCR3' is a C1-C4 alkyl group;
R3P1 is Q-C3 alkyl group, an optionally substituted oxetane group, a -(CH2)„OCH3 group
where n is 1 or 2, o
 CH3CH20- group is linked to phenyl in a meta or para position, or a morpholino group linked to the carbonyl at the 2- or 3-position;
CH3CH20- group is linked to phenyl in a meta or para position, or a morpholino group linked to the carbonyl at the 2- or 3-position;

Where Aryl is phenyl;
HET is an optionally substituted thiazole or isothiazole; and
RHET is H or a halo group,
Or a pharmaceutically acceptable salt, stereoisomer, solvate or polymorph thereof.
A compound according to claim 37 according to the chemical structure:
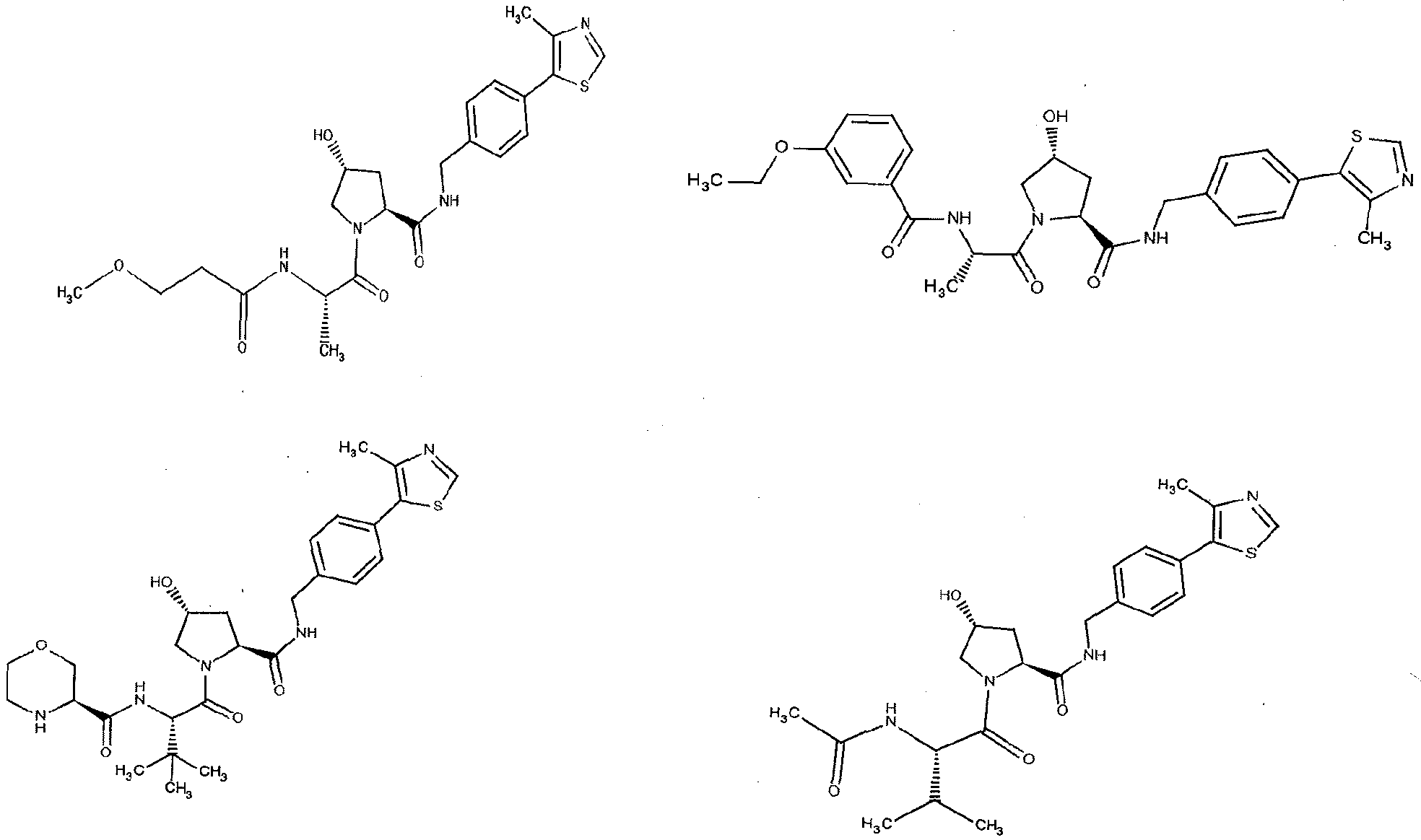


39. A compound of claim 1 as set forth in Affinity Table 2, figure 14, figure 15 or figure 16 hereof.
40. A compound according to claim 1 according to the chemical structure:




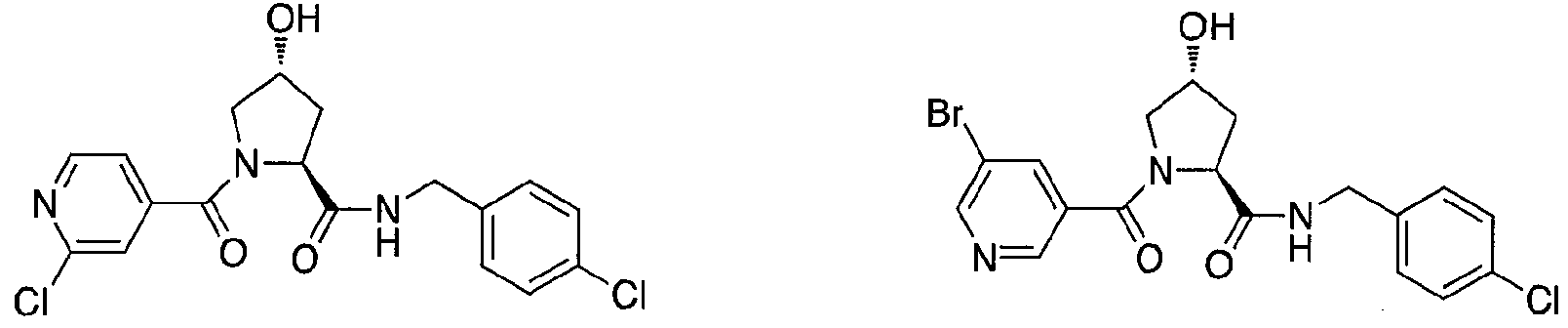















a pharmaceutically acceptable salt thereof.
A compound according to claim 1 according to
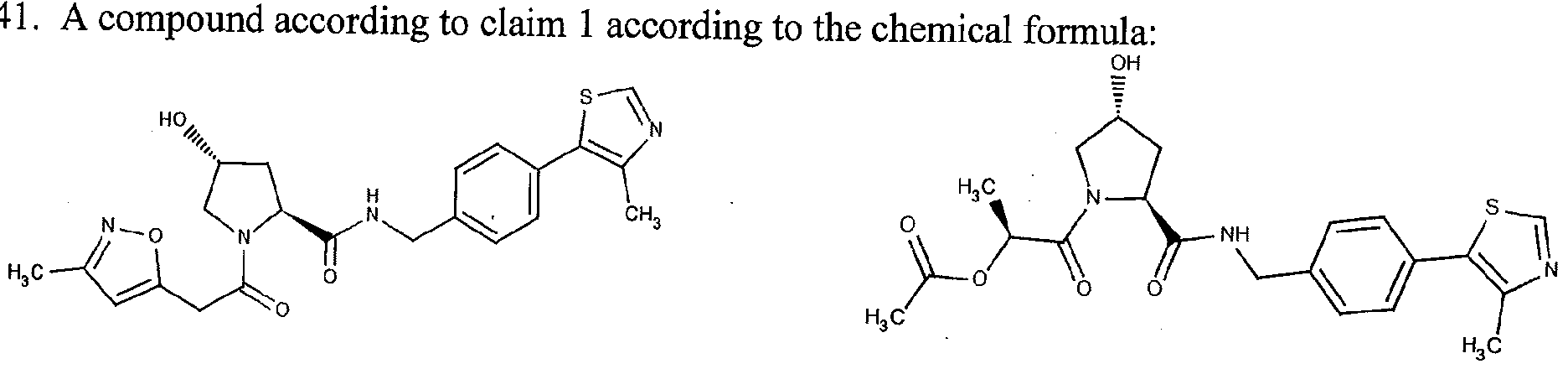
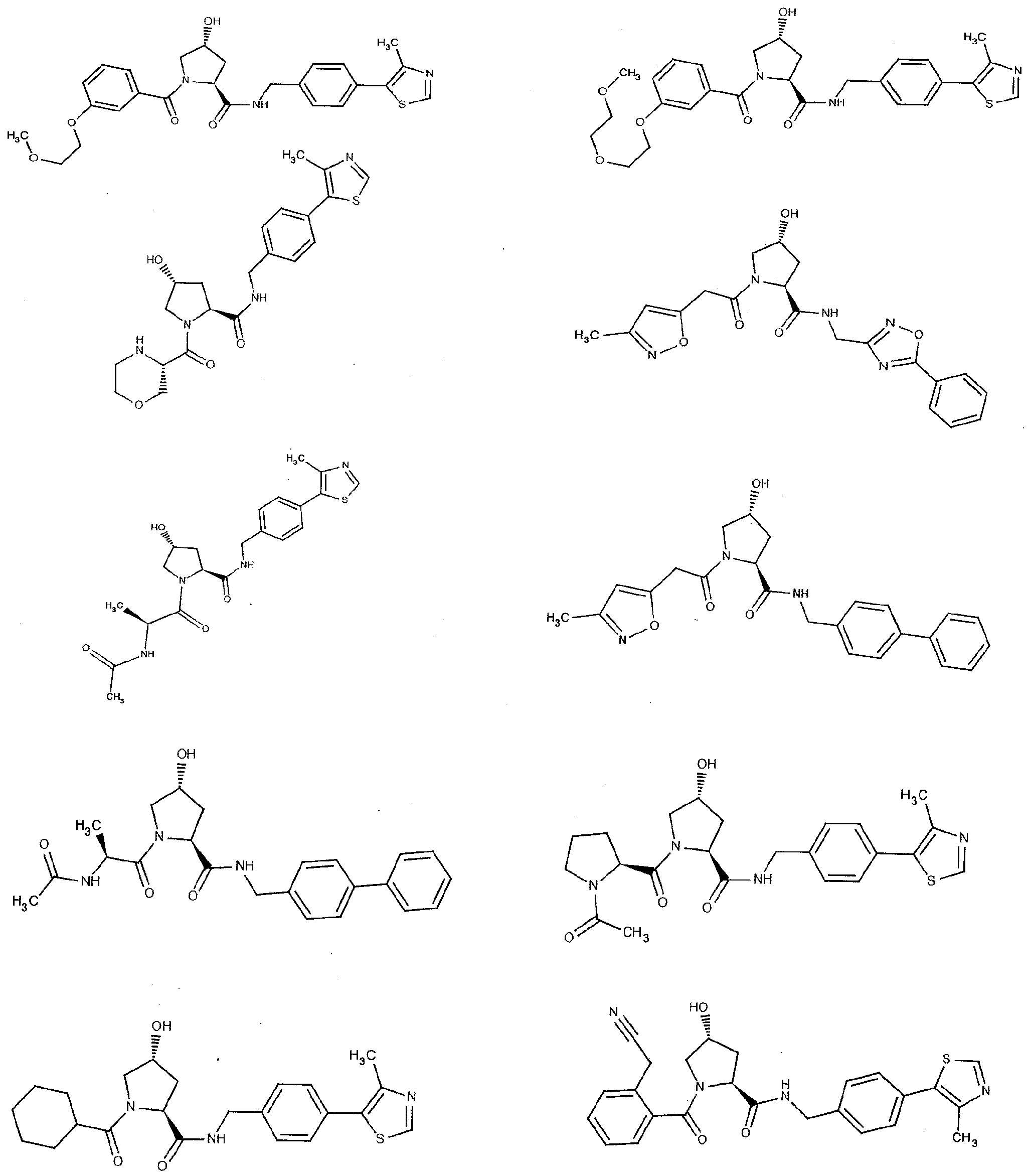

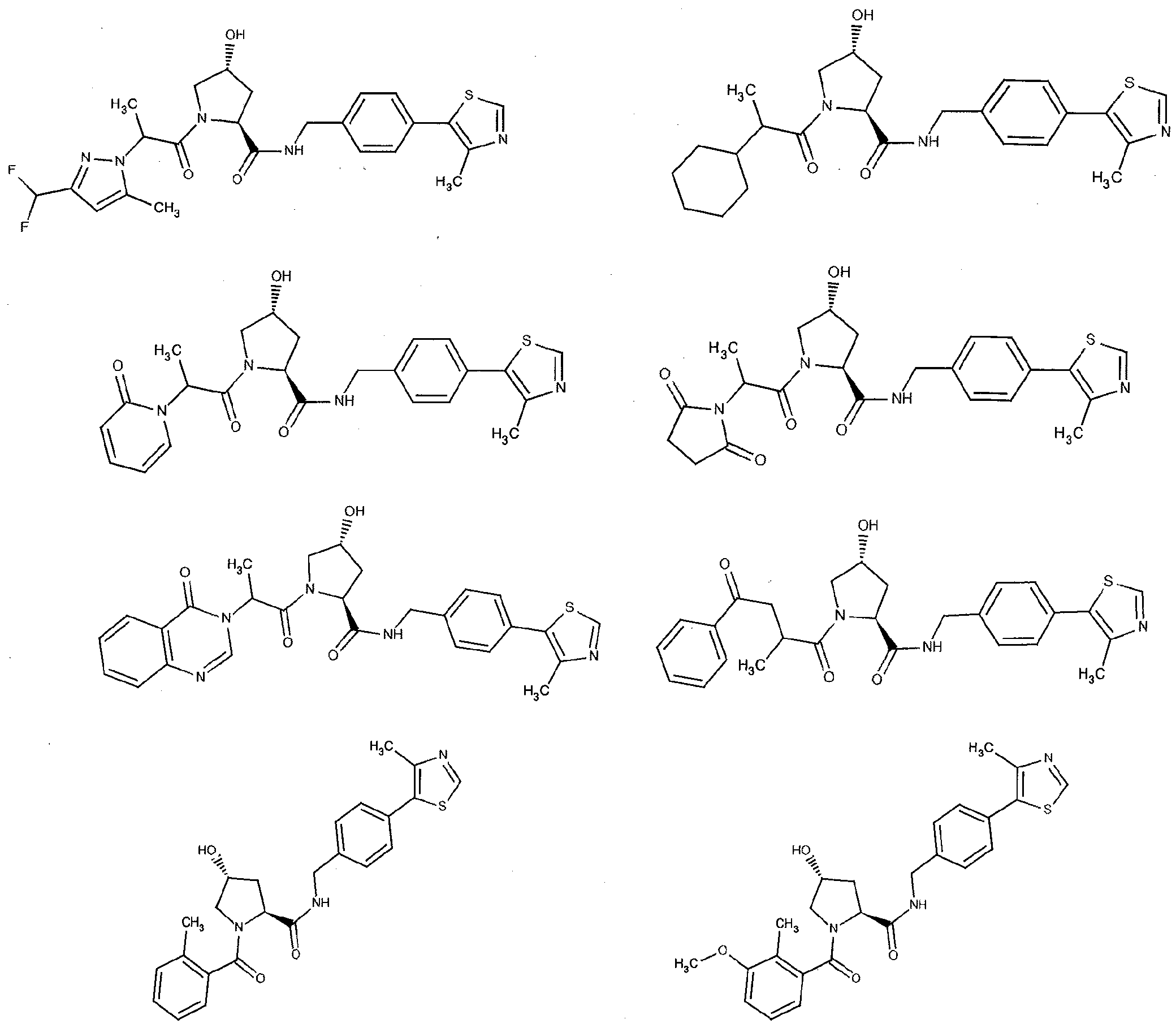




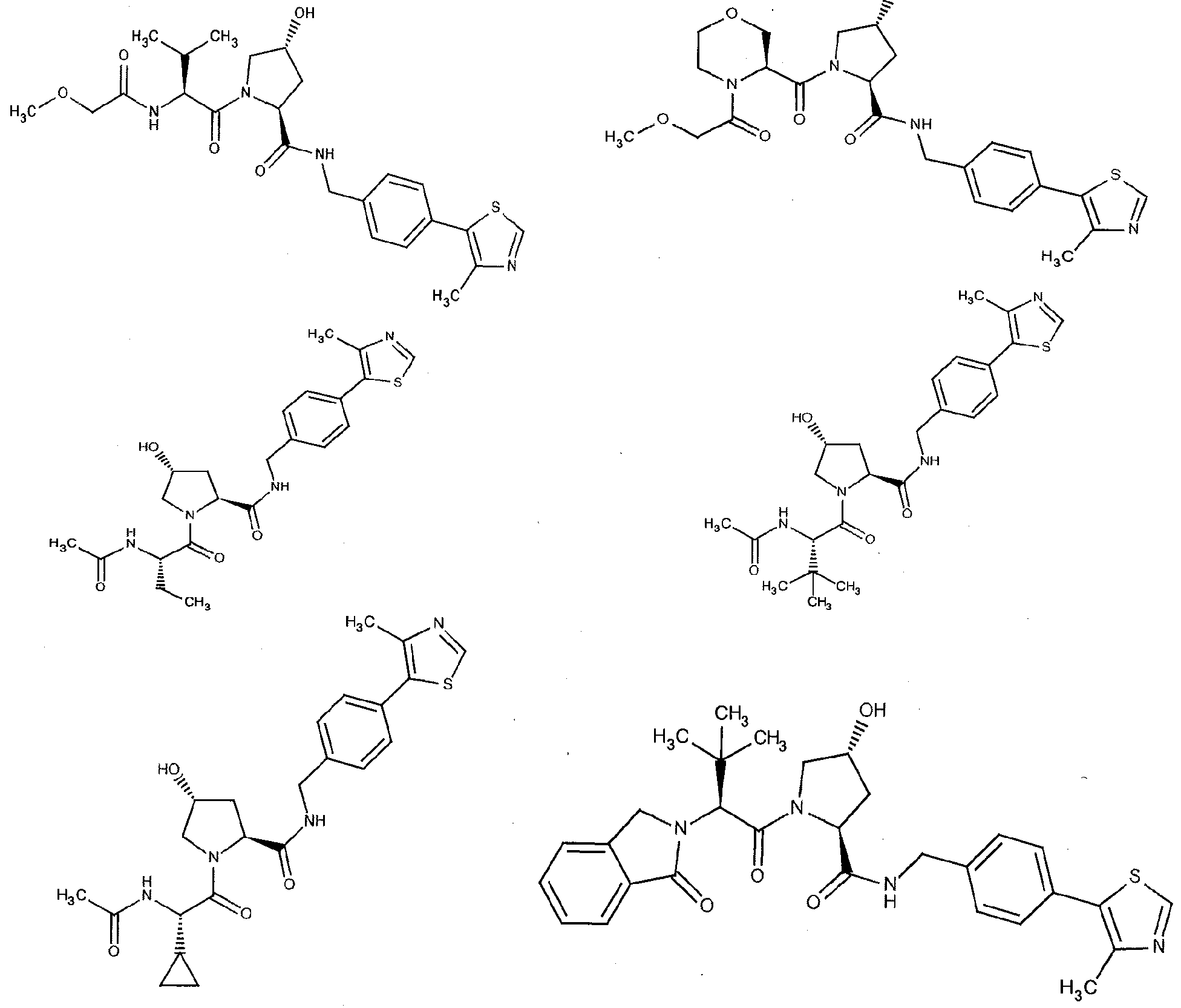
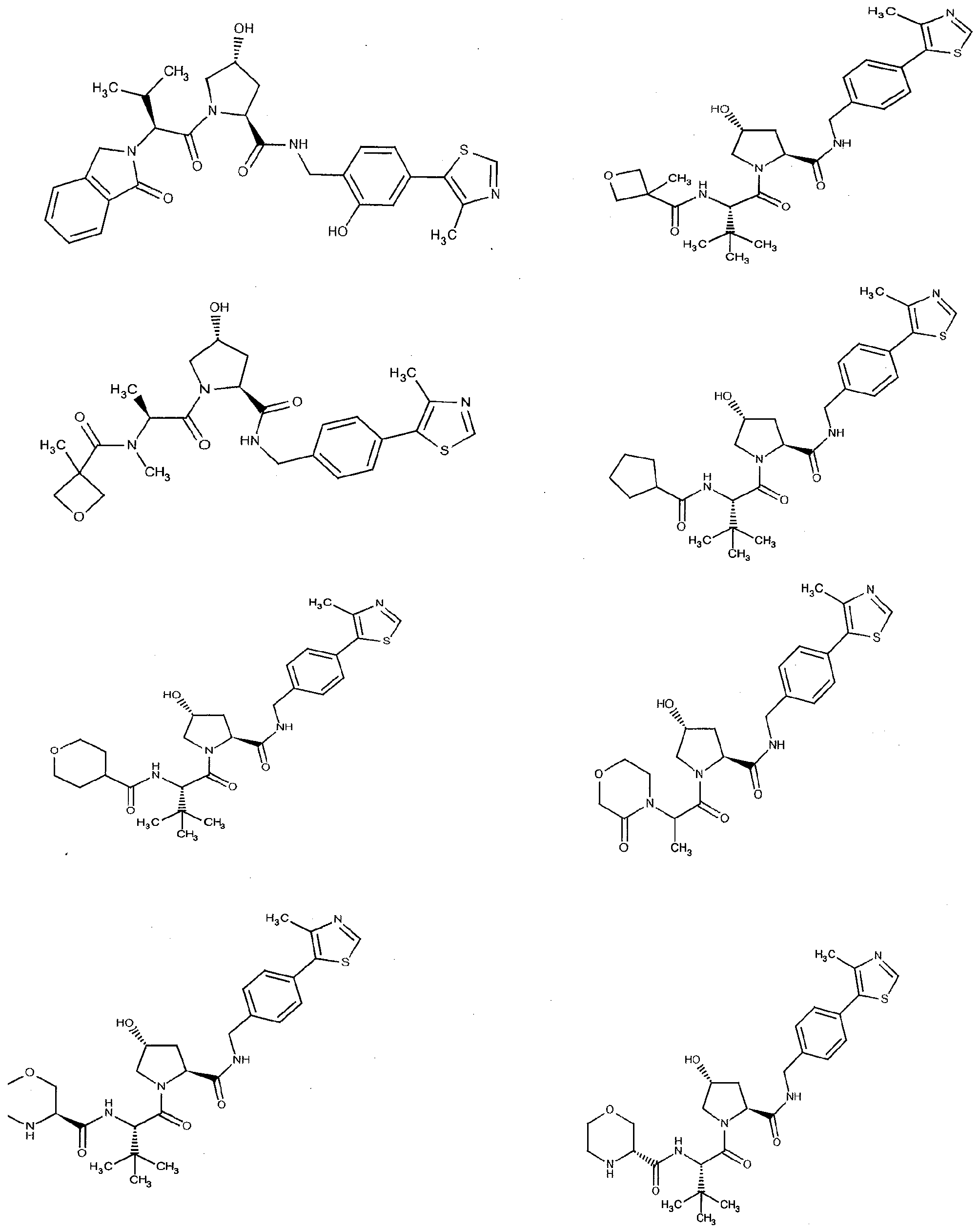

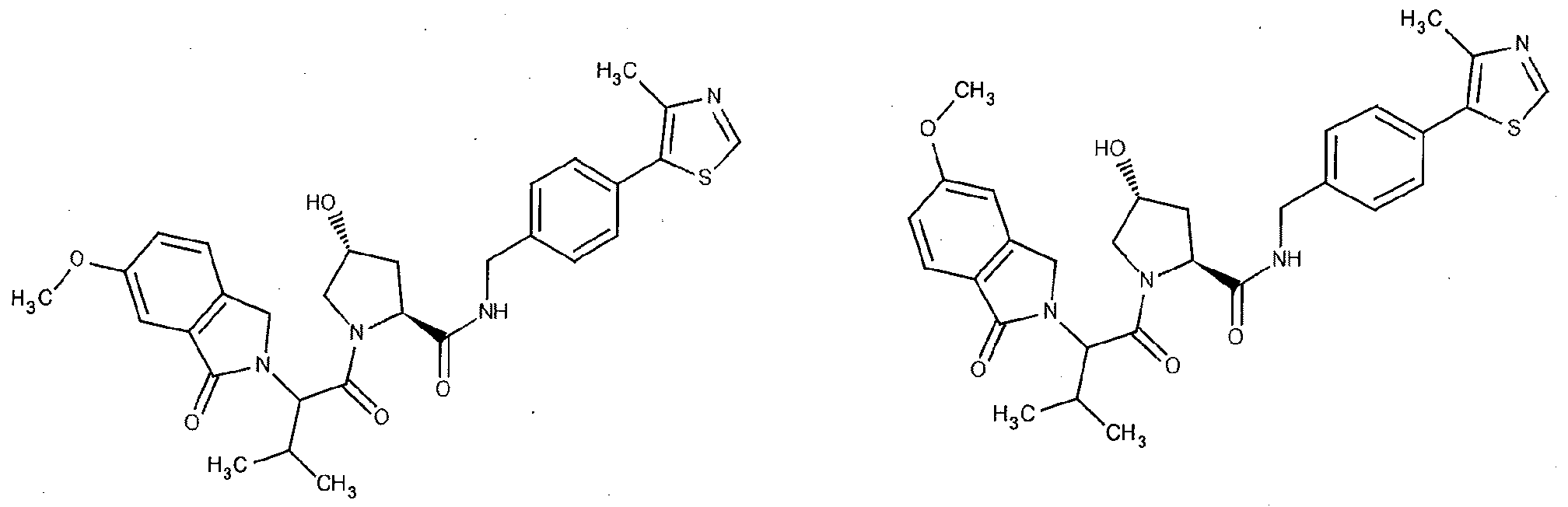

Or a pharmaceutically acceptable salt, enantiomer, diastereomer, solvate or polymorph thereof.
A compound according to claim 1 according to the chemical formula:

Or a pharmaceutically acceptable salt, enantiomer, diastereomer, solvate or polymorph thereof.
43. A compound according to claim 1 according to the chemical formula:
274


Or a pharmaceutically acceptable salt, enantiomer, diastereomer, solvate or polymorph thereof.
44. A compound according to claim 1 according to the chemical formula:

Where X is halogen, Q-C3 alkyl or an optionally substituted heterocycle; and
R1 and R2 are each independently H, Ci-C3 alkyl optionally substituted with one or two hydroxyl groups, or an optionally substituted phenyl group, or
A pharmaceutically acceptable salt, enantiomer, diastereomer, solvate or polymorph thereof.
45. A pharmaceutical composition comprising an effective amount of a compound according to any of claims 1-44 in combination with a pharmaceutically acceptable carrier, additive or excipient and optionally in further combination with a erythropoieses stimulating agent.
46. The composition according to claim 45 wherein said erythropoiesis stimulating agent is EPO or darbapoietin alfa.
47. A method of modulating VCB E3 Ubiquitin Ligase comprising exposing said ligase to a compound according to any of claims 1-44.
48. The method according to claim 47 wherein said modulation occurs in a patient or subject.
49. The method according to claim 47 or 48 wherein said compound is an inhibitor of said ligase.
50. A method of stimulating erythropoiesis in a subject or patient in need comprising administering to said patient an effective amount of a composition according to claim 45 or 46 to said patient.
51. The method according to claim 50 wherein said stimulating increases the red blood cells and/or the hematocrit of said patient or subject.
52. A method of treating anemia in a patient or subject comprising administering to said patient an effective amount of a composition according to claim 45 or 46 to said patient.
53. The method according to claim 52 wherein said anemia is chronic anemia or anemia associated with chronic kidney disease, dialysis and/or cancer chemotherapy.
54. A method of treating ischemia, stroke and/or damage to the cardiovascular system during ischemia in a patient in need comprising administering an effective amount of a composition according to claim 45 or 46 to said patient.
55. A method of enhancing wound healing in a patient in need comprising administering to said patient an effective amount of a composition according to claim 45 or 46 to said patient.
56. A method of reducing scarring secondary to wound healing in a patient in need comprising administering to said patient an effective amount of a composition according to claim 45 or 46 to said patient.
57. A method of enhancing angiogenesis and/or arteriogenesis in a patient in need comprising administering to said patient an effective amount of a composition according to claim 45 or 46.
58. The method according to claim 57 wherein said enhancing of angiogenesis occurs locally in said patient.
59. A method of reducing the likelihood of stent occlusion in a patient in need comprising administering to said patient an effective amount of a composition according to claim 45 or 46 to said patient.
60. Use of a compound according to any of claims 1-44 as a VHL ligand to prepare a bifunctional molecule which links said VHL ligand through a linking group to a protein binding moiety that selectively binds to a target protein and facilitates modulation of levels of said target protein by action of the VHL ligand moiety.
61. Use according to claim 60 wherein said linker is a polyethylene glycol having between 2 and 6 glycol units and the protein binding moiety is an estrogen binding moiety.
62. Use of a compound according to any of claims 1-44 in a first medical application.
63. Use of a compound according to any of claims 1-44 for modulating VCB E3 Ubiquitin Ligase.
64. Use according to claim 63wherein said modulation occurs in a patient or subject.
65. Use according to claim 63 or 64 wherein said compound is an inhibitor of said ligase.
66. Use of a composition according to claim 45 or 46 for stimulating erythropoiesis in a subject or patient in need.
67. Use according to claim 66 wherein said stimulating increases the red blood cells and/or the hematocrit of said patient or subject.
68. Use of a composition according to claim 45 or 46 for treating anemia in a patient or subject.
69. Use according to claim 68 wherein said anemia is chronic anemia or anemia associated with chronic kidney disease, dialysis and/or cancer chemotherapy.
70. Use of a composition according to claim 45 or 46 for treating ischemia, stroke and/or damage to the cardiovascular system during ischemia in a patient in need.
71. Use of a composition according to claim 45 or 46 for enhancing wound healing in a patient.
72. Use of a composition according to claim 45 or 46 for reducing scarring secondary to wound healing in a patient in need.
73. Use of a composition according to claim 45 or 46 for enhancing angiogenesis and/or arteriogenesis in a patient in need.
74. Use according to claim 73 wherein said enhancing of angiogenesis occurs locally in said patient.
75. Use of a composition according to claim 45 or 46 for reducing the likelihood of stent occlusion in a patient in need.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261585736P | 2012-01-12 | 2012-01-12 | |
| US61/585,736 | 2012-01-12 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2013106646A2 true WO2013106646A2 (en) | 2013-07-18 |
| WO2013106646A3 WO2013106646A3 (en) | 2013-09-06 |
Family
ID=48782089
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/021141 Ceased WO2013106646A2 (en) | 2012-01-12 | 2013-01-11 | Compounds and methods for the inhibition of vcb e3 ubiquitin ligase |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2013106646A2 (en) |
Cited By (100)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017185036A1 (en) * | 2016-04-22 | 2017-10-26 | Dana Farber Cancer Institute, Inc. | Bifunctional molecules for degradation of egfr and methods of use |
| WO2017201449A1 (en) | 2016-05-20 | 2017-11-23 | Genentech, Inc. | Protac antibody conjugates and methods of use |
| JP2018502097A (en) * | 2014-12-23 | 2018-01-25 | ダナ ファーバー キャンサー インスティテュート,インコーポレイテッド | Methods for inducing targeted proteolysis with bifunctional molecules |
| KR20180011759A (en) * | 2015-03-18 | 2018-02-02 | 아비나스 인코포레이티드 | Compounds and methods for improved degradation of targeted proteins |
| GB2554071A (en) * | 2016-09-14 | 2018-03-28 | Univ Dundee | Small molecules |
| JP2018509468A (en) * | 2015-03-13 | 2018-04-05 | ザ ユニバーシティ オブ ダンディーThe University Of Dundee | Derivatives of 1-[(cyclopentyl or 2-pyrrolidinyl) carbonylaminomethyl] -4- (1,3-thiazol-5-yl) benzene useful for the treatment of proliferative, autoimmune or inflammatory diseases |
| US9938264B2 (en) | 2015-11-02 | 2018-04-10 | Yale University | Proteolysis targeting chimera compounds and methods of preparing and using same |
| US9988376B2 (en) | 2013-07-03 | 2018-06-05 | Glaxosmithkline Intellectual Property Development Limited | Benzothiophene derivatives as estrogen receptor inhibitors |
| US9993514B2 (en) | 2013-07-03 | 2018-06-12 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US10071164B2 (en) | 2014-08-11 | 2018-09-11 | Yale University | Estrogen-related receptor alpha based protac compounds and associated methods of use |
| US10239888B2 (en) | 2016-09-29 | 2019-03-26 | Dana-Farber Cancer Institute, Inc. | Targeted protein degradation using a mutant E3 ubiquitin ligase |
| CN109678852A (en) * | 2018-12-20 | 2019-04-26 | 南京中医药大学 | 6- oxo hexahydropyrimidine derivative, preparation method and pharmacy application |
| WO2019133531A1 (en) | 2017-12-26 | 2019-07-04 | Kymera Therapeutics, Inc. | Irak degraders and uses thereof |
| US10464925B2 (en) | 2015-07-07 | 2019-11-05 | Dana-Farber Cancer Institute, Inc. | Methods to induce targeted protein degradation through bifunctional molecules |
| WO2020027225A1 (en) | 2018-07-31 | 2020-02-06 | ファイメクス株式会社 | Heterocyclic compound |
| US10584101B2 (en) | 2016-10-11 | 2020-03-10 | Arvinas, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| US10604506B2 (en) | 2017-01-26 | 2020-03-31 | Arvinas Operations, Inc. | Modulators of estrogen receptor proteolysis and associated methods of use |
| WO2020086858A1 (en) | 2018-10-24 | 2020-04-30 | Genentech, Inc. | Conjugated chemical inducers of degradation and methods of use |
| US10646575B2 (en) | 2016-05-10 | 2020-05-12 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| US10647698B2 (en) | 2016-12-01 | 2020-05-12 | Arvinas Operations, Inc. | Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degraders |
| US10660968B2 (en) | 2016-05-10 | 2020-05-26 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| WO2020113233A1 (en) | 2018-11-30 | 2020-06-04 | Kymera Therapeutics, Inc. | Irak degraders and uses thereof |
| US10723720B2 (en) | 2017-09-13 | 2020-07-28 | Amgen Inc. | Bisamide sarcomere activating compounds and uses therof |
| US10723717B2 (en) | 2016-12-23 | 2020-07-28 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of rapidly accelerated fibrosarcoma polypeptides |
| US10730862B2 (en) | 2012-01-12 | 2020-08-04 | Yale University | Compounds and methods for the enhanced degradation of targeted proteins and other polypeptides by an E3 ubiquitin ligase |
| US10772962B2 (en) | 2015-08-19 | 2020-09-15 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of bromodomain-containing proteins |
| US10787443B2 (en) | 2017-04-28 | 2020-09-29 | Zamboni Chem Solutions Inc. | RAF-degrading conjugate compounds |
| US10806737B2 (en) | 2016-12-23 | 2020-10-20 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of fetal liver kinase polypeptides |
| US10849982B2 (en) | 2016-05-10 | 2020-12-01 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
| US10849980B2 (en) | 2014-12-23 | 2020-12-01 | Dana-Farber Cancer Institute, Inc. | Methods to induce targeted protein degradation through bifunctional molecules |
| US10865202B2 (en) | 2016-09-15 | 2020-12-15 | Arvinas Operations, Inc. | Indole derivatives as estrogen receptor degraders |
| WO2020264499A1 (en) | 2019-06-28 | 2020-12-30 | Kymera Therapeutics, Inc. | Irak degraders and uses thereof |
| WO2021007286A1 (en) * | 2019-07-08 | 2021-01-14 | Board Of Regents, The University Of Texas System | Compositions and methods for cancer therapy |
| WO2021026349A1 (en) | 2019-08-08 | 2021-02-11 | Institute For Cancer Research D/B/A The Research Institute Of Fox Chase Cancer Center | Combination therapy for treatment of cancer |
| US10946017B2 (en) | 2015-06-05 | 2021-03-16 | Arvinas Operations, Inc. | Tank-binding kinase-1 PROTACs and associated methods of use |
| US10994015B2 (en) | 2016-12-23 | 2021-05-04 | Arvinas Operations, Inc. | EGFR proteolysis targeting chimeric molecules and associated methods of use |
| WO2021133920A1 (en) | 2019-12-23 | 2021-07-01 | Kymera Therapeutics, Inc. | Smarca degraders and uses thereof |
| US11065231B2 (en) | 2017-11-17 | 2021-07-20 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of interleukin-1 receptor- associated kinase 4 polypeptides |
| US11161841B2 (en) | 2018-04-04 | 2021-11-02 | Arvinas Operations, Inc. | Modulators of proteolysis and associated methods of use |
| US11173211B2 (en) | 2016-12-23 | 2021-11-16 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of rapidly accelerated Fibrosarcoma polypeptides |
| US11191741B2 (en) | 2016-12-24 | 2021-12-07 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of enhancer of zeste homolog 2 polypeptide |
| WO2021262731A2 (en) | 2020-06-23 | 2021-12-30 | Genentech, Inc. | Macrocyclic compounds and methods of use thereof |
| US11220515B2 (en) | 2018-01-26 | 2022-01-11 | Yale University | Imide-based modulators of proteolysis and associated methods of use |
| US11254672B2 (en) | 2017-09-04 | 2022-02-22 | C4 Therapeutics, Inc. | Dihydrobenzimidazolones for medical treatment |
| US11292792B2 (en) | 2018-07-06 | 2022-04-05 | Kymera Therapeutics, Inc. | Tricyclic CRBN ligands and uses thereof |
| WO2022104345A1 (en) * | 2020-11-11 | 2022-05-19 | Genentech, Inc. | 1-(2-(4-cyclopropyl-1h-1,2,3-triazol-1-yl)acetyl)-4-hydroxypyrrolidine-2-carboxa|mide derivatives as vhl inhibitors for the treatment of anemia |
| WO2022103411A1 (en) * | 2020-11-11 | 2022-05-19 | Genentech, Inc. | 1-(2-(4-cyclopropyl-1h-1,2,3-triazol-1-yl)acetyl)-4-hydroxy-n-(benzyl)pyrrolidin e-2-carboxamide derivatives as vhl inhibitors for the treatment of anemia and cancer |
| US11352351B2 (en) | 2015-01-20 | 2022-06-07 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| WO2022120355A1 (en) | 2020-12-02 | 2022-06-09 | Ikena Oncology, Inc. | Tead degraders and uses thereof |
| US11358948B2 (en) | 2017-09-22 | 2022-06-14 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| US11401256B2 (en) | 2017-09-04 | 2022-08-02 | C4 Therapeutics, Inc. | Dihydroquinolinones for medical treatment |
| WO2022174256A1 (en) * | 2021-02-12 | 2022-08-18 | The Scripps Research Institute | Small molecule activators of yap transcriptional activity for regenerative organ repair |
| US11427548B2 (en) | 2015-01-20 | 2022-08-30 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| US11459335B2 (en) | 2017-06-20 | 2022-10-04 | C4 Therapeutics, Inc. | N/O-linked Degrons and Degronimers for protein degradation |
| US11458123B2 (en) | 2016-11-01 | 2022-10-04 | Arvinas Operations, Inc. | Tau-protein targeting PROTACs and associated methods of use |
| US11485743B2 (en) | 2018-01-12 | 2022-11-01 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US11485750B1 (en) | 2019-04-05 | 2022-11-01 | Kymera Therapeutics, Inc. | STAT degraders and uses thereof |
| US11512080B2 (en) | 2018-01-12 | 2022-11-29 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| US11524949B2 (en) | 2017-11-16 | 2022-12-13 | C4 Therapeutics, Inc. | Degraders and Degrons for targeted protein degradation |
| US11584748B2 (en) | 2018-04-16 | 2023-02-21 | C4 Therapeutics, Inc. | Spirocyclic compounds |
| US11591332B2 (en) | 2019-12-17 | 2023-02-28 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11623932B2 (en) | 2017-09-22 | 2023-04-11 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US11623929B2 (en) | 2018-06-04 | 2023-04-11 | C4 Therapeutics, Inc. | Spirocyclic compounds |
| WO2023076161A1 (en) | 2021-10-25 | 2023-05-04 | Kymera Therapeutics, Inc. | Tyk2 degraders and uses thereof |
| US11685750B2 (en) | 2020-06-03 | 2023-06-27 | Kymera Therapeutics, Inc. | Crystalline forms of IRAK degraders |
| US11707452B2 (en) | 2018-08-20 | 2023-07-25 | Arvinas Operations, Inc. | Modulators of alpha-synuclein proteolysis and associated methods of use |
| US11707457B2 (en) | 2019-12-17 | 2023-07-25 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| CN116507617A (en) * | 2020-11-11 | 2023-07-28 | 基因泰克公司 | 1-(2-(4-Cyclopropyl-1H-1,2,3-triazol-1-yl)acetyl)-4-hydroxypyrrolidine-2-carboxamide as a VHL inhibitor for the treatment of anemia derivative |
| WO2023147328A1 (en) | 2022-01-26 | 2023-08-03 | Genentech, Inc. | Antibody-conjugated chemical inducers of degradation with hydolysable maleimide linkers and methods thereof |
| US11753397B2 (en) | 2018-03-26 | 2023-09-12 | C4 Therapeutics, Inc. | Cereblon binders for the degradation of ikaros |
| US11802131B2 (en) | 2017-09-04 | 2023-10-31 | C4 Therapeutics, Inc. | Glutarimides for medical treatment |
| JP2023551290A (en) * | 2020-11-30 | 2023-12-07 | ジェネンテック, インコーポレイテッド | Pyrrolidine derivatives and usage methods |
| US11883393B2 (en) | 2019-12-19 | 2024-01-30 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| US11912699B2 (en) | 2019-07-17 | 2024-02-27 | Arvinas Operations, Inc. | Tau-protein targeting compounds and associated |
| WO2024050016A1 (en) | 2022-08-31 | 2024-03-07 | Oerth Bio Llc | Compositions and methods for targeted inhibition and degradation of proteins in an insect cell |
| US11932624B2 (en) | 2020-03-19 | 2024-03-19 | Kymera Therapeutics, Inc. | MDM2 degraders and uses thereof |
| WO2024064358A1 (en) | 2022-09-23 | 2024-03-28 | Ifm Due, Inc. | Compounds and compositions for treating conditions associated with sting activity |
| US11957759B1 (en) | 2022-09-07 | 2024-04-16 | Arvinas Operations, Inc. | Rapidly accelerated fibrosarcoma (RAF) degrading compounds and associated methods of use |
| US11986532B2 (en) | 2021-04-16 | 2024-05-21 | Arvinas Operations, Inc. | Modulators of BCL6 proteolysis and associated methods of use |
| US12043612B2 (en) | 2020-05-09 | 2024-07-23 | Arvinas Operations, Inc. | Methods of manufacturing a bifunctional compound, ultrapure forms of the bifunctional compound, and dosage forms comprising the same |
| US12048747B2 (en) | 2016-05-10 | 2024-07-30 | C4 Therapeutics, Inc. | Substituted piperidine Degronimers for Target Protein degradation |
| US12091411B2 (en) | 2022-01-31 | 2024-09-17 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US12097261B2 (en) | 2021-05-07 | 2024-09-24 | Kymera Therapeutics, Inc. | CDK2 degraders and uses thereof |
| US12150995B2 (en) | 2020-12-30 | 2024-11-26 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US12162859B2 (en) | 2020-09-14 | 2024-12-10 | Arvinas Operations, Inc. | Crystalline and amorphous forms of a compound for the targeted degradation of estrogen receptor |
| US12171768B2 (en) | 2021-02-15 | 2024-12-24 | Kymera Therapeutics, Inc. | IRAK4 degraders and uses thereof |
| US12180193B2 (en) | 2020-08-28 | 2024-12-31 | Arvinas Operations, Inc. | Accelerating fibrosarcoma protein degrading compounds and associated methods of use |
| US12187744B2 (en) | 2021-10-29 | 2025-01-07 | Kymera Therapeutics, Inc. | IRAK4 degraders and synthesis thereof |
| US12208095B2 (en) | 2019-08-26 | 2025-01-28 | Arvinas Operations, Inc. | Methods of treating breast cancer with tetrahydronaphthalene derivatives as estrogen receptor degraders |
| US12239711B2 (en) | 2014-04-14 | 2025-03-04 | Arvinas Operations, Inc. | Cereblon ligands and bifunctional compounds comprising the same |
| WO2025049555A1 (en) | 2023-08-31 | 2025-03-06 | Oerth Bio Llc | Compositions and methods for targeted inhibition and degradation of proteins in an insect cell |
| US12310975B2 (en) | 2019-10-17 | 2025-05-27 | Arvinas Operations, Inc. | Modulators of BCL6 proteolysis and associated methods of use |
| US12441708B2 (en) | 2017-01-31 | 2025-10-14 | Arvinas Operations, Inc. | Cereblon ligands and bifunctional compounds comprising the same |
| US12448399B2 (en) | 2023-01-26 | 2025-10-21 | Arvinas Operations, Inc. | Cereblon-based KRAS degrading PROTACs and uses related thereto |
| US12454521B2 (en) | 2018-12-20 | 2025-10-28 | C4 Therapeutics, Inc. | Targeted protein degradation |
| US12454520B2 (en) | 2018-07-06 | 2025-10-28 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US12496301B2 (en) | 2023-12-08 | 2025-12-16 | Arvinas Operations, Inc. | Use of androgen receptor degrader for the treatment of spinal and bulbar muscular atrophy |
| US12521438B2 (en) | 2019-06-10 | 2026-01-13 | Kymera Therapeutics, Inc. | SMARCA degraders and uses thereof |
| US12528797B2 (en) | 2019-03-06 | 2026-01-20 | C4 Therapeutics, Inc. | Heterocyclic compounds for medical treatment |
| US12545659B2 (en) | 2020-06-29 | 2026-02-10 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12539292B2 (en) | 2018-04-01 | 2026-02-03 | Arvinas Operations, Inc. | BRM targeting compounds and associated methods of use |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2204082A1 (en) * | 1996-05-03 | 1997-11-03 | Michael William John Urquhart | Pharmaceutical compounds |
| WO2001054503A1 (en) * | 2000-01-28 | 2001-08-02 | Akkadix Corporation | Methods for killing nematodes and nematode eggs using 4-phenoxy-6-aminopyrimidine derivatives |
| WO2007022638A1 (en) * | 2005-08-26 | 2007-03-01 | Methylgene Inc. | Benzodiazepine and benzopiperazine analog inhibitors of histone deacetylase |
| WO2007106670A2 (en) * | 2006-03-03 | 2007-09-20 | Novartis Ag | N-formyl hydroxylamine compounds |
| WO2008011392A2 (en) * | 2006-07-20 | 2008-01-24 | Ligand Pharmacueticals Incorporated | Proline urea ccr1 antagonists for the treatment of autoimmune diseases or inflammation |
| JP5282091B2 (en) * | 2007-07-25 | 2013-09-04 | ブリストル−マイヤーズ スクイブ カンパニー | Triazine kinase inhibitor |
| US8629157B2 (en) * | 2009-01-05 | 2014-01-14 | Boehringer Ingelheim International Gmbh | Pyrrolidine compounds which modulate the CB2 receptor |
-
2013
- 2013-01-11 WO PCT/US2013/021141 patent/WO2013106646A2/en not_active Ceased
Cited By (188)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10730862B2 (en) | 2012-01-12 | 2020-08-04 | Yale University | Compounds and methods for the enhanced degradation of targeted proteins and other polypeptides by an E3 ubiquitin ligase |
| US9988376B2 (en) | 2013-07-03 | 2018-06-05 | Glaxosmithkline Intellectual Property Development Limited | Benzothiophene derivatives as estrogen receptor inhibitors |
| US9993514B2 (en) | 2013-07-03 | 2018-06-12 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US12239711B2 (en) | 2014-04-14 | 2025-03-04 | Arvinas Operations, Inc. | Cereblon ligands and bifunctional compounds comprising the same |
| US10071164B2 (en) | 2014-08-11 | 2018-09-11 | Yale University | Estrogen-related receptor alpha based protac compounds and associated methods of use |
| US10125114B2 (en) | 2014-12-23 | 2018-11-13 | Dana-Farber Cancer Institute, Inc. | Methods to induce targeted protein degradation through bifunctional molecules |
| US11059801B2 (en) | 2014-12-23 | 2021-07-13 | Dana-Farber Cancer Institute, Inc. | Methods to induce targeted protein degradation through bifunctional molecules |
| US11583586B2 (en) | 2014-12-23 | 2023-02-21 | Dana-Farber Cancer Institute, Inc. | Methods to induce targeted protein degradation through bifunctional molecules |
| JP2018502097A (en) * | 2014-12-23 | 2018-01-25 | ダナ ファーバー キャンサー インスティテュート,インコーポレイテッド | Methods for inducing targeted proteolysis with bifunctional molecules |
| US10849980B2 (en) | 2014-12-23 | 2020-12-01 | Dana-Farber Cancer Institute, Inc. | Methods to induce targeted protein degradation through bifunctional molecules |
| US10669253B2 (en) | 2014-12-23 | 2020-06-02 | Dana-Farber Cancer Institute, Inc. | Methods to induce targeted protein degradation through bifunctional molecules |
| US11427548B2 (en) | 2015-01-20 | 2022-08-30 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| US11352351B2 (en) | 2015-01-20 | 2022-06-07 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| US12312316B2 (en) | 2015-01-20 | 2025-05-27 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| JP2018509468A (en) * | 2015-03-13 | 2018-04-05 | ザ ユニバーシティ オブ ダンディーThe University Of Dundee | Derivatives of 1-[(cyclopentyl or 2-pyrrolidinyl) carbonylaminomethyl] -4- (1,3-thiazol-5-yl) benzene useful for the treatment of proliferative, autoimmune or inflammatory diseases |
| EP3268363B1 (en) * | 2015-03-13 | 2022-05-04 | The University of Dundee | Derivatives of 1-[(cyclopentyl or 2-pyrrolidinyl)carbonylaminomethyl]-4-(1,3-thiazol-5-yl) benzene which are useful for the treatment of proliferative, autoimmune or inflammatory diseases |
| US11179373B2 (en) | 2015-03-13 | 2021-11-23 | University Of Dundee | Derivatives of 1-[(cyclopentyl or 2-pyrrolidinyl)carbonylaminomethyl]-4-(l,3-thiazol-5-yl) benzene which are useful for the treatment of proliferative, autoimmune or inflammatory diseases |
| US10730870B2 (en) | 2015-03-18 | 2020-08-04 | Arvinas Operations, Inc. | Compounds and methods for the enhanced degradation of targeted proteins |
| JP2021073251A (en) * | 2015-03-18 | 2021-05-13 | アルビナス・オペレーションズ・インコーポレイテッドArvinas Operations, Inc. | Compound and method for decomposing and improving target protein |
| KR102616762B1 (en) * | 2015-03-18 | 2023-12-20 | 아비나스 오퍼레이션스, 인코포레이티드 | Compounds and methods for enhanced degradation of targeted proteins |
| KR20180011759A (en) * | 2015-03-18 | 2018-02-02 | 아비나스 인코포레이티드 | Compounds and methods for improved degradation of targeted proteins |
| JP2023109967A (en) * | 2015-03-18 | 2023-08-08 | アルビナス・オペレーションズ・インコーポレイテッド | Compound and method for decomposing and improving target protein |
| US11512083B2 (en) | 2015-03-18 | 2022-11-29 | Arvinas Operations, Inc. | Compounds and methods for the enhanced degradation of targeted proteins |
| JP7269731B2 (en) | 2015-03-18 | 2023-05-09 | アルビナス・オペレーションズ・インコーポレイテッド | Compounds and methods for enhancing target protein degradation |
| JP2018512449A (en) * | 2015-03-18 | 2018-05-17 | アルビナス インコーポレイテッド | Compounds and methods for improved degradation of target proteins |
| US12264157B2 (en) | 2015-03-18 | 2025-04-01 | Arvinas Operations, Inc. | Compounds and methods for the enhanced degradation of targeted proteins |
| US10946017B2 (en) | 2015-06-05 | 2021-03-16 | Arvinas Operations, Inc. | Tank-binding kinase-1 PROTACs and associated methods of use |
| US10464925B2 (en) | 2015-07-07 | 2019-11-05 | Dana-Farber Cancer Institute, Inc. | Methods to induce targeted protein degradation through bifunctional molecules |
| US10772962B2 (en) | 2015-08-19 | 2020-09-15 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of bromodomain-containing proteins |
| US11554171B2 (en) | 2015-08-19 | 2023-01-17 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of bromodomain-containing proteins |
| US12171831B2 (en) | 2015-08-19 | 2024-12-24 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of bromodomain- containing proteins |
| CN108366992A (en) * | 2015-11-02 | 2018-08-03 | 耶鲁大学 | Proteolysis targets chimera compound and its methods for making and using same |
| US9938264B2 (en) | 2015-11-02 | 2018-04-10 | Yale University | Proteolysis targeting chimera compounds and methods of preparing and using same |
| CN109475528A (en) * | 2016-04-22 | 2019-03-15 | 达纳-法伯癌症研究所股份有限公司 | Bifunctional molecules for EGFR degradation and methods of use |
| US11161842B2 (en) | 2016-04-22 | 2021-11-02 | Dana-Farber Cancer Institute, Inc. | Bifunctional molecules for degradation of EGFR and methods of use |
| JP2019519476A (en) * | 2016-04-22 | 2019-07-11 | ダナ−ファーバー キャンサー インスティテュート,インコーポレイテッド | Bifunctional molecule for degradation of EGFR and method of use |
| EP3892272A1 (en) * | 2016-04-22 | 2021-10-13 | Dana-Farber Cancer Institute, Inc. | Bifunctional molecules for degradation of egfr and methods of use |
| WO2017185036A1 (en) * | 2016-04-22 | 2017-10-26 | Dana Farber Cancer Institute, Inc. | Bifunctional molecules for degradation of egfr and methods of use |
| EP3445357A4 (en) * | 2016-04-22 | 2019-11-06 | Dana-Farber Cancer Institute, Inc. | BIFUNCTIONAL MOLECULES FOR EGFR DEGRADATION AND METHODS OF USE |
| CN109475528B (en) * | 2016-04-22 | 2022-01-11 | 达纳-法伯癌症研究所股份有限公司 | Bifunctional molecules for EGFR degradation and methods of use |
| US10450310B2 (en) | 2016-04-22 | 2019-10-22 | Dana-Farber Cancer Institute, Inc. | Bifunctional molecules for degradation of EGFR and methods of use |
| US10849982B2 (en) | 2016-05-10 | 2020-12-01 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
| US12076405B2 (en) | 2016-05-10 | 2024-09-03 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| US11992531B2 (en) | 2016-05-10 | 2024-05-28 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
| US12048747B2 (en) | 2016-05-10 | 2024-07-30 | C4 Therapeutics, Inc. | Substituted piperidine Degronimers for Target Protein degradation |
| US10905768B1 (en) | 2016-05-10 | 2021-02-02 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| US12048748B2 (en) | 2016-05-10 | 2024-07-30 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| US10660968B2 (en) | 2016-05-10 | 2020-05-26 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| US11185592B2 (en) | 2016-05-10 | 2021-11-30 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| US10646575B2 (en) | 2016-05-10 | 2020-05-12 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| WO2017201449A1 (en) | 2016-05-20 | 2017-11-23 | Genentech, Inc. | Protac antibody conjugates and methods of use |
| GB2554071A (en) * | 2016-09-14 | 2018-03-28 | Univ Dundee | Small molecules |
| US11234988B2 (en) | 2016-09-14 | 2022-02-01 | University Of Dundee | Fluorohydroxyproline derivatives useful in the preparation of proteolysis targeted chimeras |
| US10865202B2 (en) | 2016-09-15 | 2020-12-15 | Arvinas Operations, Inc. | Indole derivatives as estrogen receptor degraders |
| US11584743B2 (en) | 2016-09-15 | 2023-02-21 | Arvinas Operations, Inc. | Indole derivatives as estrogen receptor degraders |
| US10239888B2 (en) | 2016-09-29 | 2019-03-26 | Dana-Farber Cancer Institute, Inc. | Targeted protein degradation using a mutant E3 ubiquitin ligase |
| US10584101B2 (en) | 2016-10-11 | 2020-03-10 | Arvinas, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| US12077509B2 (en) | 2016-10-11 | 2024-09-03 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| CN111892577B (en) * | 2016-10-11 | 2023-07-07 | 阿尔维纳斯运营股份有限公司 | Compounds and methods for targeted degradation of androgen receptor |
| US10844021B2 (en) | 2016-10-11 | 2020-11-24 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| CN111892577A (en) * | 2016-10-11 | 2020-11-06 | 阿尔维纳斯运营股份有限公司 | Compounds and methods for targeted degradation of androgen receptors |
| US11952347B2 (en) | 2016-10-11 | 2024-04-09 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| US11236051B2 (en) | 2016-10-11 | 2022-02-01 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| US11964945B2 (en) | 2016-10-11 | 2024-04-23 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| US11458123B2 (en) | 2016-11-01 | 2022-10-04 | Arvinas Operations, Inc. | Tau-protein targeting PROTACs and associated methods of use |
| US10899742B1 (en) | 2016-12-01 | 2021-01-26 | Arvinas Operations, Inc. | Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degraders |
| US11597720B2 (en) | 2016-12-01 | 2023-03-07 | Arvinas Operations, Inc. | Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degraders |
| US10647698B2 (en) | 2016-12-01 | 2020-05-12 | Arvinas Operations, Inc. | Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degraders |
| US12172981B2 (en) | 2016-12-01 | 2024-12-24 | Arvinas Operations, Inc. | Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degraders |
| US11104666B2 (en) | 2016-12-01 | 2021-08-31 | Arvinas Operations, Inc. | Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degraders |
| US10994015B2 (en) | 2016-12-23 | 2021-05-04 | Arvinas Operations, Inc. | EGFR proteolysis targeting chimeric molecules and associated methods of use |
| US10723717B2 (en) | 2016-12-23 | 2020-07-28 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of rapidly accelerated fibrosarcoma polypeptides |
| US11986531B2 (en) | 2016-12-23 | 2024-05-21 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of rapidly accelerated fibrosarcoma polypeptides |
| US11173211B2 (en) | 2016-12-23 | 2021-11-16 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of rapidly accelerated Fibrosarcoma polypeptides |
| US10806737B2 (en) | 2016-12-23 | 2020-10-20 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of fetal liver kinase polypeptides |
| US11857519B2 (en) | 2016-12-24 | 2024-01-02 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of enhancer of zeste homolog 2 polypeptide |
| US11191741B2 (en) | 2016-12-24 | 2021-12-07 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of enhancer of zeste homolog 2 polypeptide |
| US11384063B2 (en) | 2017-01-26 | 2022-07-12 | Arvinas Operations, Inc. | Modulators of estrogen receptor proteolysis and associated methods of use |
| US10604506B2 (en) | 2017-01-26 | 2020-03-31 | Arvinas Operations, Inc. | Modulators of estrogen receptor proteolysis and associated methods of use |
| US12275716B2 (en) | 2017-01-26 | 2025-04-15 | Arvinas Operations, Inc. | Modulators of estrogen receptor proteolysis and associated methods of use |
| US12441708B2 (en) | 2017-01-31 | 2025-10-14 | Arvinas Operations, Inc. | Cereblon ligands and bifunctional compounds comprising the same |
| US10787443B2 (en) | 2017-04-28 | 2020-09-29 | Zamboni Chem Solutions Inc. | RAF-degrading conjugate compounds |
| US12180225B2 (en) | 2017-06-20 | 2024-12-31 | C4 Therapeutics, Inc. | N/O-linked degrons and degronimers for protein degradation |
| US11459335B2 (en) | 2017-06-20 | 2022-10-04 | C4 Therapeutics, Inc. | N/O-linked Degrons and Degronimers for protein degradation |
| US12441740B2 (en) | 2017-06-20 | 2025-10-14 | C4 Therapeutics, Inc. | N/O-linked degrons and degronimers for protein degradation |
| US11401256B2 (en) | 2017-09-04 | 2022-08-02 | C4 Therapeutics, Inc. | Dihydroquinolinones for medical treatment |
| US11802131B2 (en) | 2017-09-04 | 2023-10-31 | C4 Therapeutics, Inc. | Glutarimides for medical treatment |
| US11787802B2 (en) | 2017-09-04 | 2023-10-17 | C4 Therapeutics, Inc. | Dihydrobenzimidazolones for medical treatment |
| US12091397B2 (en) | 2017-09-04 | 2024-09-17 | C4 Therapeutics, Inc. | Dihydroquinolinones for medical treatment |
| US12365681B2 (en) | 2017-09-04 | 2025-07-22 | C4 Therapeutics, Inc. | Dihydrobenzimidazolones for medical treatment |
| US11254672B2 (en) | 2017-09-04 | 2022-02-22 | C4 Therapeutics, Inc. | Dihydrobenzimidazolones for medical treatment |
| US11254658B2 (en) | 2017-09-13 | 2022-02-22 | Amgen Inc. | Bisamide sarcomere activating compounds and uses thereof |
| US10899746B2 (en) | 2017-09-13 | 2021-01-26 | Amgen Inc. | Bisamide sarcomere activating compounds and uses thereof |
| US10723720B2 (en) | 2017-09-13 | 2020-07-28 | Amgen Inc. | Bisamide sarcomere activating compounds and uses therof |
| US11299479B1 (en) | 2017-09-13 | 2022-04-12 | Cytokinetics, Inc. | Bisamide sarcomere activating compounds and uses thereof |
| US11780826B2 (en) | 2017-09-13 | 2023-10-10 | Amgen Inc. | Bisamide sarcomere activating compounds and uses thereof |
| US11358948B2 (en) | 2017-09-22 | 2022-06-14 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| US11623932B2 (en) | 2017-09-22 | 2023-04-11 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US12540127B2 (en) | 2017-09-22 | 2026-02-03 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| US11524949B2 (en) | 2017-11-16 | 2022-12-13 | C4 Therapeutics, Inc. | Degraders and Degrons for targeted protein degradation |
| US12036209B2 (en) | 2017-11-17 | 2024-07-16 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of Interleukin-1 receptor-associated kinase 4 polypeptides |
| US11065231B2 (en) | 2017-11-17 | 2021-07-20 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of interleukin-1 receptor- associated kinase 4 polypeptides |
| WO2019133531A1 (en) | 2017-12-26 | 2019-07-04 | Kymera Therapeutics, Inc. | Irak degraders and uses thereof |
| US11723980B2 (en) | 2017-12-26 | 2023-08-15 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11318205B1 (en) | 2017-12-26 | 2022-05-03 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| EP4613773A2 (en) | 2017-12-26 | 2025-09-10 | Kymera Therapeutics, Inc. | Irak degraders and uses thereof |
| US10874743B2 (en) | 2017-12-26 | 2020-12-29 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US12168057B2 (en) | 2017-12-26 | 2024-12-17 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11932635B2 (en) | 2018-01-12 | 2024-03-19 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| US11485743B2 (en) | 2018-01-12 | 2022-11-01 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US12006329B2 (en) | 2018-01-12 | 2024-06-11 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US11512080B2 (en) | 2018-01-12 | 2022-11-29 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| US12516068B2 (en) | 2018-01-12 | 2026-01-06 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US11834460B2 (en) | 2018-01-26 | 2023-12-05 | Yale University | Imide-based modulators of proteolysis and associated methods of use |
| US11220515B2 (en) | 2018-01-26 | 2022-01-11 | Yale University | Imide-based modulators of proteolysis and associated methods of use |
| US11753397B2 (en) | 2018-03-26 | 2023-09-12 | C4 Therapeutics, Inc. | Cereblon binders for the degradation of ikaros |
| US11161841B2 (en) | 2018-04-04 | 2021-11-02 | Arvinas Operations, Inc. | Modulators of proteolysis and associated methods of use |
| US12227504B2 (en) | 2018-04-16 | 2025-02-18 | C4 Therepeutics, Inc. | Spirocyclic compounds |
| US11584748B2 (en) | 2018-04-16 | 2023-02-21 | C4 Therapeutics, Inc. | Spirocyclic compounds |
| US11623929B2 (en) | 2018-06-04 | 2023-04-11 | C4 Therapeutics, Inc. | Spirocyclic compounds |
| US11897882B2 (en) | 2018-07-06 | 2024-02-13 | Kymera Therapeutics, Inc. | Tricyclic crbn ligands and uses thereof |
| US11292792B2 (en) | 2018-07-06 | 2022-04-05 | Kymera Therapeutics, Inc. | Tricyclic CRBN ligands and uses thereof |
| US12454520B2 (en) | 2018-07-06 | 2025-10-28 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| WO2020027225A1 (en) | 2018-07-31 | 2020-02-06 | ファイメクス株式会社 | Heterocyclic compound |
| US12202829B2 (en) | 2018-07-31 | 2025-01-21 | Fimecs, Inc. | Heterocyclic compound |
| US11639354B2 (en) | 2018-07-31 | 2023-05-02 | Fimecs, Inc. | Heterocyclic compound |
| US11707452B2 (en) | 2018-08-20 | 2023-07-25 | Arvinas Operations, Inc. | Modulators of alpha-synuclein proteolysis and associated methods of use |
| WO2020086858A1 (en) | 2018-10-24 | 2020-04-30 | Genentech, Inc. | Conjugated chemical inducers of degradation and methods of use |
| US11352350B2 (en) | 2018-11-30 | 2022-06-07 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11117889B1 (en) | 2018-11-30 | 2021-09-14 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| WO2020113233A1 (en) | 2018-11-30 | 2020-06-04 | Kymera Therapeutics, Inc. | Irak degraders and uses thereof |
| US11807636B2 (en) | 2018-11-30 | 2023-11-07 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US12258341B2 (en) | 2018-11-30 | 2025-03-25 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| CN109678852B (en) * | 2018-12-20 | 2022-04-19 | 南京中医药大学 | 6-oxo-hexahydropyrimidine derivative, preparation method and pharmaceutical application thereof |
| CN109678852A (en) * | 2018-12-20 | 2019-04-26 | 南京中医药大学 | 6- oxo hexahydropyrimidine derivative, preparation method and pharmacy application |
| US12454521B2 (en) | 2018-12-20 | 2025-10-28 | C4 Therapeutics, Inc. | Targeted protein degradation |
| US12528797B2 (en) | 2019-03-06 | 2026-01-20 | C4 Therapeutics, Inc. | Heterocyclic compounds for medical treatment |
| US11746120B2 (en) | 2019-04-05 | 2023-09-05 | Kymera Therapeutics, Inc. | Stat degraders and uses thereof |
| US11485750B1 (en) | 2019-04-05 | 2022-11-01 | Kymera Therapeutics, Inc. | STAT degraders and uses thereof |
| US12077555B2 (en) | 2019-04-05 | 2024-09-03 | Kymera Therapeutics, Inc. | STAT degraders and uses thereof |
| US12521438B2 (en) | 2019-06-10 | 2026-01-13 | Kymera Therapeutics, Inc. | SMARCA degraders and uses thereof |
| WO2020264499A1 (en) | 2019-06-28 | 2020-12-30 | Kymera Therapeutics, Inc. | Irak degraders and uses thereof |
| WO2021007286A1 (en) * | 2019-07-08 | 2021-01-14 | Board Of Regents, The University Of Texas System | Compositions and methods for cancer therapy |
| US11912699B2 (en) | 2019-07-17 | 2024-02-27 | Arvinas Operations, Inc. | Tau-protein targeting compounds and associated |
| WO2021026349A1 (en) | 2019-08-08 | 2021-02-11 | Institute For Cancer Research D/B/A The Research Institute Of Fox Chase Cancer Center | Combination therapy for treatment of cancer |
| US12208095B2 (en) | 2019-08-26 | 2025-01-28 | Arvinas Operations, Inc. | Methods of treating breast cancer with tetrahydronaphthalene derivatives as estrogen receptor degraders |
| US12310975B2 (en) | 2019-10-17 | 2025-05-27 | Arvinas Operations, Inc. | Modulators of BCL6 proteolysis and associated methods of use |
| US11591332B2 (en) | 2019-12-17 | 2023-02-28 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11779578B2 (en) | 2019-12-17 | 2023-10-10 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US12539295B2 (en) | 2019-12-17 | 2026-02-03 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11707457B2 (en) | 2019-12-17 | 2023-07-25 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11883393B2 (en) | 2019-12-19 | 2024-01-30 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| US12427144B2 (en) | 2019-12-19 | 2025-09-30 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| WO2021133920A1 (en) | 2019-12-23 | 2021-07-01 | Kymera Therapeutics, Inc. | Smarca degraders and uses thereof |
| US11679109B2 (en) | 2019-12-23 | 2023-06-20 | Kymera Therapeutics, Inc. | SMARCA degraders and uses thereof |
| US12528785B2 (en) | 2020-03-19 | 2026-01-20 | Kymera Therapeutics, Inc. | MDM2 degraders and uses thereof |
| US11932624B2 (en) | 2020-03-19 | 2024-03-19 | Kymera Therapeutics, Inc. | MDM2 degraders and uses thereof |
| US12043612B2 (en) | 2020-05-09 | 2024-07-23 | Arvinas Operations, Inc. | Methods of manufacturing a bifunctional compound, ultrapure forms of the bifunctional compound, and dosage forms comprising the same |
| US11685750B2 (en) | 2020-06-03 | 2023-06-27 | Kymera Therapeutics, Inc. | Crystalline forms of IRAK degraders |
| US12187816B2 (en) | 2020-06-23 | 2025-01-07 | Genentech, Inc. | Macrocyclic compounds and methods of use thereof |
| WO2021262731A2 (en) | 2020-06-23 | 2021-12-30 | Genentech, Inc. | Macrocyclic compounds and methods of use thereof |
| US12545659B2 (en) | 2020-06-29 | 2026-02-10 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US12180193B2 (en) | 2020-08-28 | 2024-12-31 | Arvinas Operations, Inc. | Accelerating fibrosarcoma protein degrading compounds and associated methods of use |
| US12162859B2 (en) | 2020-09-14 | 2024-12-10 | Arvinas Operations, Inc. | Crystalline and amorphous forms of a compound for the targeted degradation of estrogen receptor |
| JP2023549187A (en) * | 2020-11-11 | 2023-11-22 | ジェネンテック, インコーポレイテッド | 1-(2-(4-cyclopropyl-1H-1,2,3-triazol-1-yl)acetyl)-4-hydroxy-N-(benzyl) as a VHL inhibitor for the treatment of anemia and cancer. ) Pyrrolidine E-2-carboxamide derivative |
| CN116783183A (en) * | 2020-11-11 | 2023-09-19 | 基因泰克公司 | 1-(2-(4-Cyclopropyl-1H-1,2,3-triazol-1-yl)acetyl)-4-hydroxy-N-(benzyl) as a VHL inhibitor for the treatment of anemia and cancer pyrrolidine-2-carboxamide derivatives |
| CN116507617A (en) * | 2020-11-11 | 2023-07-28 | 基因泰克公司 | 1-(2-(4-Cyclopropyl-1H-1,2,3-triazol-1-yl)acetyl)-4-hydroxypyrrolidine-2-carboxamide as a VHL inhibitor for the treatment of anemia derivative |
| WO2022104345A1 (en) * | 2020-11-11 | 2022-05-19 | Genentech, Inc. | 1-(2-(4-cyclopropyl-1h-1,2,3-triazol-1-yl)acetyl)-4-hydroxypyrrolidine-2-carboxa|mide derivatives as vhl inhibitors for the treatment of anemia |
| JP2025063287A (en) * | 2020-11-11 | 2025-04-15 | ジェネンテック, インコーポレイテッド | 1-(2-(4-cyclopropyl-1H-1,2,3-triazol-1-yl)acetyl)-4-hydroxy-N-(benzyl)pyrrolidine E-2-carboxamide derivatives as VHL inhibitors for the treatment of anemia and cancer |
| WO2022103411A1 (en) * | 2020-11-11 | 2022-05-19 | Genentech, Inc. | 1-(2-(4-cyclopropyl-1h-1,2,3-triazol-1-yl)acetyl)-4-hydroxy-n-(benzyl)pyrrolidin e-2-carboxamide derivatives as vhl inhibitors for the treatment of anemia and cancer |
| JP2023551290A (en) * | 2020-11-30 | 2023-12-07 | ジェネンテック, インコーポレイテッド | Pyrrolidine derivatives and usage methods |
| WO2022120355A1 (en) | 2020-12-02 | 2022-06-09 | Ikena Oncology, Inc. | Tead degraders and uses thereof |
| US12150995B2 (en) | 2020-12-30 | 2024-11-26 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| WO2022174256A1 (en) * | 2021-02-12 | 2022-08-18 | The Scripps Research Institute | Small molecule activators of yap transcriptional activity for regenerative organ repair |
| US12171768B2 (en) | 2021-02-15 | 2024-12-24 | Kymera Therapeutics, Inc. | IRAK4 degraders and uses thereof |
| US11986532B2 (en) | 2021-04-16 | 2024-05-21 | Arvinas Operations, Inc. | Modulators of BCL6 proteolysis and associated methods of use |
| US12097261B2 (en) | 2021-05-07 | 2024-09-24 | Kymera Therapeutics, Inc. | CDK2 degraders and uses thereof |
| WO2023076161A1 (en) | 2021-10-25 | 2023-05-04 | Kymera Therapeutics, Inc. | Tyk2 degraders and uses thereof |
| US12187744B2 (en) | 2021-10-29 | 2025-01-07 | Kymera Therapeutics, Inc. | IRAK4 degraders and synthesis thereof |
| WO2023147328A1 (en) | 2022-01-26 | 2023-08-03 | Genentech, Inc. | Antibody-conjugated chemical inducers of degradation with hydolysable maleimide linkers and methods thereof |
| US12091411B2 (en) | 2022-01-31 | 2024-09-17 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| WO2024050016A1 (en) | 2022-08-31 | 2024-03-07 | Oerth Bio Llc | Compositions and methods for targeted inhibition and degradation of proteins in an insect cell |
| US12156916B2 (en) | 2022-09-07 | 2024-12-03 | Arvinas Operations, Inc. | Rapid accelerated fibrosarcoma (RAF) degrading compounds and associated methods of use |
| US11957759B1 (en) | 2022-09-07 | 2024-04-16 | Arvinas Operations, Inc. | Rapidly accelerated fibrosarcoma (RAF) degrading compounds and associated methods of use |
| WO2024064358A1 (en) | 2022-09-23 | 2024-03-28 | Ifm Due, Inc. | Compounds and compositions for treating conditions associated with sting activity |
| US12448399B2 (en) | 2023-01-26 | 2025-10-21 | Arvinas Operations, Inc. | Cereblon-based KRAS degrading PROTACs and uses related thereto |
| WO2025049555A1 (en) | 2023-08-31 | 2025-03-06 | Oerth Bio Llc | Compositions and methods for targeted inhibition and degradation of proteins in an insect cell |
| US12496301B2 (en) | 2023-12-08 | 2025-12-16 | Arvinas Operations, Inc. | Use of androgen receptor degrader for the treatment of spinal and bulbar muscular atrophy |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2013106646A3 (en) | 2013-09-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2013106646A2 (en) | Compounds and methods for the inhibition of vcb e3 ubiquitin ligase | |
| AU2022205187B2 (en) | Compounds and methods for the enhanced degradation of targeted proteins and other polypeptides by an e3 ubiquitin ligase | |
| US20250223265A1 (en) | Compounds and methods for the targeted degradation of androgen receptor | |
| AU2016209349B2 (en) | Compounds and methods for the targeted degradation of the Androgen Receptor | |
| KR20200035435A (en) | Compounds and methods for targeted decomposition of androgen receptors | |
| EP3319944A1 (en) | Mdm2-based modulators of proteolysis and associated methods of use | |
| RU2781452C2 (en) | Compounds and methods for enhancing degradation of target proteins and other polypeptides, using e3 ubiquitin ligase | |
| HK40023141A (en) | Compounds & methods for the enhanced degradation of targeted proteins & other polypeptides by an e3 ubiquitin ligase | |
| RU2774863C2 (en) | Compounds and methods for targeted degradation of androgen receptor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13735940 Country of ref document: EP Kind code of ref document: A2 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13735940 Country of ref document: EP Kind code of ref document: A2 |