TWI867024B - 囊腫纖維化跨膜傳導調節蛋白之調節劑 - Google Patents
囊腫纖維化跨膜傳導調節蛋白之調節劑 Download PDFInfo
- Publication number
- TWI867024B TWI867024B TW109127317A TW109127317A TWI867024B TW I867024 B TWI867024 B TW I867024B TW 109127317 A TW109127317 A TW 109127317A TW 109127317 A TW109127317 A TW 109127317A TW I867024 B TWI867024 B TW I867024B
- Authority
- TW
- Taiwan
- Prior art keywords
- compound
- pharmaceutically acceptable
- acceptable salt
- mmol
- formula
- Prior art date
Links
- 108010079245 Cystic Fibrosis Transmembrane Conductance Regulator Proteins 0.000 title abstract description 70
- 102000012605 Cystic Fibrosis Transmembrane Conductance Regulator Human genes 0.000 title abstract description 5
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 63
- 201000003883 Cystic fibrosis Diseases 0.000 claims abstract description 46
- 150000001875 compounds Chemical class 0.000 claims description 421
- 150000003839 salts Chemical class 0.000 claims description 196
- 239000000203 mixture Substances 0.000 claims description 81
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 claims description 46
- 229910052805 deuterium Inorganic materials 0.000 claims description 30
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 28
- 229910052739 hydrogen Inorganic materials 0.000 claims description 26
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 23
- 239000001257 hydrogen Substances 0.000 claims description 22
- 125000000217 alkyl group Chemical group 0.000 claims description 21
- 239000003814 drug Substances 0.000 claims description 19
- 239000003937 drug carrier Substances 0.000 claims description 18
- 229910052736 halogen Inorganic materials 0.000 claims description 16
- 229940124597 therapeutic agent Drugs 0.000 claims description 15
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 14
- 238000002360 preparation method Methods 0.000 claims description 14
- 229910052710 silicon Inorganic materials 0.000 claims description 13
- 150000002367 halogens Chemical class 0.000 claims description 12
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 10
- 229910052799 carbon Inorganic materials 0.000 claims description 10
- 229910052732 germanium Inorganic materials 0.000 claims description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 10
- 125000004429 atom Chemical group 0.000 claims description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 8
- 150000002431 hydrogen Chemical group 0.000 claims description 8
- 150000001721 carbon Chemical group 0.000 claims description 7
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical group [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 claims description 7
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 6
- 125000001434 methanylylidene group Chemical group [H]C#[*] 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 6
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 claims description 6
- 229940079593 drug Drugs 0.000 claims description 4
- HTSGKJQDMSTCGS-UHFFFAOYSA-N 1,4-bis(4-chlorophenyl)-2-(4-methylphenyl)sulfonylbutane-1,4-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)C(C(=O)C=1C=CC(Cl)=CC=1)CC(=O)C1=CC=C(Cl)C=C1 HTSGKJQDMSTCGS-UHFFFAOYSA-N 0.000 claims 7
- 238000000034 method Methods 0.000 abstract description 77
- 230000008569 process Effects 0.000 abstract description 3
- 235000002639 sodium chloride Nutrition 0.000 description 173
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 116
- 239000000243 solution Substances 0.000 description 86
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 77
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 73
- 238000006243 chemical reaction Methods 0.000 description 68
- 229910001868 water Inorganic materials 0.000 description 64
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 62
- -1 Lactose Diethanolamine Chemical compound 0.000 description 62
- 102100023419 Cystic fibrosis transmembrane conductance regulator Human genes 0.000 description 61
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 53
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 52
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 48
- 239000012071 phase Substances 0.000 description 48
- 239000007787 solid Substances 0.000 description 47
- 235000019439 ethyl acetate Nutrition 0.000 description 41
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 40
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 40
- POOSGDOYLQNASK-UHFFFAOYSA-N tetracosane Chemical compound CCCCCCCCCCCCCCCCCCCCCCCC POOSGDOYLQNASK-UHFFFAOYSA-N 0.000 description 40
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 39
- 230000002829 reductive effect Effects 0.000 description 37
- DRSHXJFUUPIBHX-UHFFFAOYSA-N COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 Chemical compound COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 DRSHXJFUUPIBHX-UHFFFAOYSA-N 0.000 description 36
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 36
- 230000014759 maintenance of location Effects 0.000 description 36
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 34
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 32
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 32
- 239000003921 oil Substances 0.000 description 32
- 235000019198 oils Nutrition 0.000 description 32
- 238000007792 addition Methods 0.000 description 31
- 239000011541 reaction mixture Substances 0.000 description 31
- 230000035772 mutation Effects 0.000 description 30
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 29
- 239000010410 layer Substances 0.000 description 29
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 26
- 239000012267 brine Substances 0.000 description 25
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 25
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 24
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 22
- 210000004027 cell Anatomy 0.000 description 22
- 238000003756 stirring Methods 0.000 description 22
- 239000000725 suspension Substances 0.000 description 22
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 20
- 229910052757 nitrogen Inorganic materials 0.000 description 20
- 239000000047 product Substances 0.000 description 20
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 19
- 238000005481 NMR spectroscopy Methods 0.000 description 19
- 239000012065 filter cake Substances 0.000 description 19
- 229910052938 sodium sulfate Inorganic materials 0.000 description 19
- 235000011152 sodium sulphate Nutrition 0.000 description 19
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 17
- 230000032258 transport Effects 0.000 description 17
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 16
- 235000019253 formic acid Nutrition 0.000 description 16
- 239000012044 organic layer Substances 0.000 description 16
- 239000012074 organic phase Substances 0.000 description 16
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 15
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 14
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 14
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 14
- 239000008186 active pharmaceutical agent Substances 0.000 description 14
- 239000000706 filtrate Substances 0.000 description 14
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 14
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- 229910000027 potassium carbonate Inorganic materials 0.000 description 13
- 239000000523 sample Substances 0.000 description 13
- 108091006146 Channels Proteins 0.000 description 12
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 12
- 229910000077 silane Inorganic materials 0.000 description 12
- 125000001424 substituent group Chemical group 0.000 description 12
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 11
- 150000002148 esters Chemical class 0.000 description 11
- 229920006395 saturated elastomer Polymers 0.000 description 11
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 11
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 10
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- 230000015572 biosynthetic process Effects 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 238000001816 cooling Methods 0.000 description 10
- 239000013058 crude material Substances 0.000 description 10
- 238000010348 incorporation Methods 0.000 description 10
- 235000011181 potassium carbonates Nutrition 0.000 description 10
- 239000011734 sodium Substances 0.000 description 10
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 9
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 9
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 9
- 230000007547 defect Effects 0.000 description 9
- 230000002950 deficient Effects 0.000 description 9
- 201000010099 disease Diseases 0.000 description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 9
- 238000002347 injection Methods 0.000 description 9
- 239000007924 injection Substances 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 9
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 description 8
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 8
- 239000008346 aqueous phase Substances 0.000 description 8
- 238000003556 assay Methods 0.000 description 8
- 239000012043 crude product Substances 0.000 description 8
- 230000006870 function Effects 0.000 description 8
- 238000010438 heat treatment Methods 0.000 description 8
- 230000001404 mediated effect Effects 0.000 description 8
- 239000002245 particle Substances 0.000 description 8
- 238000010898 silica gel chromatography Methods 0.000 description 8
- 238000003786 synthesis reaction Methods 0.000 description 8
- 239000005909 Kieselgur Substances 0.000 description 7
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 7
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 239000007789 gas Substances 0.000 description 7
- 239000000499 gel Substances 0.000 description 7
- 239000012528 membrane Substances 0.000 description 7
- YKYONYBAUNKHLG-UHFFFAOYSA-N n-Propyl acetate Natural products CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 description 7
- 239000011570 nicotinamide Substances 0.000 description 7
- 239000012299 nitrogen atmosphere Substances 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 7
- 239000000741 silica gel Substances 0.000 description 7
- 229910002027 silica gel Inorganic materials 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 7
- 239000003643 water by type Substances 0.000 description 7
- ZXXMWYSGWPMYCL-ZETCQYMHSA-N (3S)-3-(3-hydroxypropyl)-5,5-dimethylpyrrolidin-2-one Chemical compound OCCC[C@@H]1C(NC(C1)(C)C)=O ZXXMWYSGWPMYCL-ZETCQYMHSA-N 0.000 description 6
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 6
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- OHCQJHSOBUTRHG-KGGHGJDLSA-N FORSKOLIN Chemical compound O=C([C@@]12O)C[C@](C)(C=C)O[C@]1(C)[C@@H](OC(=O)C)[C@@H](O)[C@@H]1[C@]2(C)[C@@H](O)CCC1(C)C OHCQJHSOBUTRHG-KGGHGJDLSA-N 0.000 description 6
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 6
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 150000001450 anions Chemical class 0.000 description 6
- 239000012230 colorless oil Substances 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 235000005152 nicotinamide Nutrition 0.000 description 6
- 229960003966 nicotinamide Drugs 0.000 description 6
- 239000000377 silicon dioxide Substances 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- JRHPOFJADXHYBR-HTQZYQBOSA-N (1r,2r)-1-n,2-n-dimethylcyclohexane-1,2-diamine Chemical compound CN[C@@H]1CCCC[C@H]1NC JRHPOFJADXHYBR-HTQZYQBOSA-N 0.000 description 5
- JDSZYMBVWCRVQB-UHFFFAOYSA-N 3-(2-methyl-2-nitropropyl)oxan-2-one Chemical compound CC(CC1C(OCCC1)=O)(C)[N+](=O)[O-] JDSZYMBVWCRVQB-UHFFFAOYSA-N 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- 108010067035 Pancrelipase Proteins 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 235000011089 carbon dioxide Nutrition 0.000 description 5
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 5
- 238000004440 column chromatography Methods 0.000 description 5
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 230000000155 isotopic effect Effects 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 235000019341 magnesium sulphate Nutrition 0.000 description 5
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- NJBGXPJITDKQPS-UHFFFAOYSA-N tert-butyl 5-oxo-1h-pyrazole-2-carboxylate Chemical compound CC(C)(C)OC(=O)N1C=CC(O)=N1 NJBGXPJITDKQPS-UHFFFAOYSA-N 0.000 description 5
- 239000003039 volatile agent Substances 0.000 description 5
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 4
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 4
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 4
- 229930182821 L-proline Natural products 0.000 description 4
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- 239000008367 deionised water Substances 0.000 description 4
- 229910021641 deionized water Inorganic materials 0.000 description 4
- 210000002919 epithelial cell Anatomy 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 150000002500 ions Chemical class 0.000 description 4
- PURKAOJPTOLRMP-UHFFFAOYSA-N ivacaftor Chemical compound C1=C(O)C(C(C)(C)C)=CC(C(C)(C)C)=C1NC(=O)C1=CNC2=CC=CC=C2C1=O PURKAOJPTOLRMP-UHFFFAOYSA-N 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 230000001717 pathogenic effect Effects 0.000 description 4
- 238000012545 processing Methods 0.000 description 4
- 229960002429 proline Drugs 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 4
- 230000003595 spectral effect Effects 0.000 description 4
- 238000001228 spectrum Methods 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- OGCJXRVPXCTPKO-UHFFFAOYSA-N (2,6-dichloro-5-trimethylsilylpyridin-3-yl)-imidazol-1-ylmethanone Chemical compound ClC1=NC(=C(C=C1C(=O)N1C=NC=C1)[Si](C)(C)C)Cl OGCJXRVPXCTPKO-UHFFFAOYSA-N 0.000 description 3
- DXLJNJLGCAFIKI-UHFFFAOYSA-N (2,6-dichloropyridin-3-yl)-trimethylsilane Chemical compound C[Si](C)(C)C1=CC=C(Cl)N=C1Cl DXLJNJLGCAFIKI-UHFFFAOYSA-N 0.000 description 3
- RSWYZAGKUWIIPR-UHFFFAOYSA-N 2,6-dichloro-5-trimethylsilylpyridine-3-carboxylic acid Chemical compound ClC1=NC(=C(C=C1C(=O)O)[Si](C)(C)C)Cl RSWYZAGKUWIIPR-UHFFFAOYSA-N 0.000 description 3
- NAIVUBHZTYECEQ-HNNXBMFYSA-N 2,6-dichloro-N-[6-[3-[(3S)-5,5-dimethylpyrrolidin-3-yl]propylamino]pyridin-2-yl]sulfonyl-5-trimethylsilylpyridine-3-carboxamide Chemical compound ClC1=NC(=C(C=C1C(=O)NS(=O)(=O)C1=NC(=CC=C1)NCCC[C@@H]1CNC(C1)(C)C)[Si](C)(C)C)Cl NAIVUBHZTYECEQ-HNNXBMFYSA-N 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- RTMVZYGLVYFSBP-UHFFFAOYSA-N 2-benzylsulfanyl-6-fluoropyridine Chemical compound FC1=CC=CC(SCC=2C=CC=CC=2)=N1 RTMVZYGLVYFSBP-UHFFFAOYSA-N 0.000 description 3
- YJPKYWZTVYMZQT-UHFFFAOYSA-N 3,3-dimethylbutyl-dimethyl-[2-(oxan-2-yl)pyrazol-3-yl]silane Chemical compound CC(CC[Si](C=1N(N=CC=1)C1OCCCC1)(C)C)(C)C YJPKYWZTVYMZQT-UHFFFAOYSA-N 0.000 description 3
- HKVBHYCAXBRNLY-QMMMGPOBSA-N 3-[(3S)-5,5-dimethylpyrrolidin-3-yl]propan-1-ol Chemical compound CC1(C[C@@H](CN1)CCCO)C HKVBHYCAXBRNLY-QMMMGPOBSA-N 0.000 description 3
- YGDKIQZYHCUZIC-UHFFFAOYSA-N 3-methylideneoxan-2-one Chemical compound C=C1CCCOC1=O YGDKIQZYHCUZIC-UHFFFAOYSA-N 0.000 description 3
- QXAUPUZBANXHAK-UHFFFAOYSA-N 5-oxo-1h-pyrazole-2-carboxylic acid Chemical compound OC(=O)N1C=CC(=O)N1 QXAUPUZBANXHAK-UHFFFAOYSA-N 0.000 description 3
- WYYFSNSPYJRQLU-UHFFFAOYSA-N 6-fluoropyridine-2-sulfonamide Chemical compound NS(=O)(=O)C1=CC=CC(F)=N1 WYYFSNSPYJRQLU-UHFFFAOYSA-N 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 3
- DSBILMGLKQKPCU-UHFFFAOYSA-N BrC1=NC(=C(C(=O)N)C=C1)F Chemical compound BrC1=NC(=C(C(=O)N)C=C1)F DSBILMGLKQKPCU-UHFFFAOYSA-N 0.000 description 3
- HKVBHYCAXBRNLY-UHFFFAOYSA-N CC1(C)CC(CCCO)CN1 Chemical compound CC1(C)CC(CCCO)CN1 HKVBHYCAXBRNLY-UHFFFAOYSA-N 0.000 description 3
- 101150029409 CFTR gene Proteins 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 3
- SUZLHDUTVMZSEV-UHFFFAOYSA-N Deoxycoleonol Natural products C12C(=O)CC(C)(C=C)OC2(C)C(OC(=O)C)C(O)C2C1(C)C(O)CCC2(C)C SUZLHDUTVMZSEV-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 239000007832 Na2SO4 Substances 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 229910000564 Raney nickel Inorganic materials 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 3
- ZDHTYMDPFVUNRA-UHFFFAOYSA-N [dimethyl-[1-(trifluoromethyl)cyclopropyl]silyl]methanol Chemical compound C[Si](C1(CC1)C(F)(F)F)(C)CO ZDHTYMDPFVUNRA-UHFFFAOYSA-N 0.000 description 3
- WIMWYKQJPBUJBJ-UHFFFAOYSA-N [dimethyl-[1-(trifluoromethyl)cyclopropyl]silyl]methyl acetate Chemical compound C(C)(=O)OC[Si](C1(CC1)C(F)(F)F)(C)C WIMWYKQJPBUJBJ-UHFFFAOYSA-N 0.000 description 3
- XPZOXIUZMQOHCD-UHFFFAOYSA-N [tert-butyl(dimethyl)silyl]methanol Chemical compound CC(C)(C)[Si](C)(C)CO XPZOXIUZMQOHCD-UHFFFAOYSA-N 0.000 description 3
- 230000001154 acute effect Effects 0.000 description 3
- 239000000908 ammonium hydroxide Substances 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 229940124630 bronchodilator Drugs 0.000 description 3
- 229910002092 carbon dioxide Inorganic materials 0.000 description 3
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- NRJXPOXYQRZZRG-UHFFFAOYSA-N chloromethyl-dimethyl-(3,3,3-trifluoroprop-1-en-2-yl)silane Chemical compound ClC[Si](C(=C)C(F)(F)F)(C)C NRJXPOXYQRZZRG-UHFFFAOYSA-N 0.000 description 3
- KYEBJAYLIDAMJQ-UHFFFAOYSA-N chloromethyl-dimethyl-[1-(trifluoromethyl)cyclopropyl]silane Chemical compound ClC[Si](C1(CC1)C(F)(F)F)(C)C KYEBJAYLIDAMJQ-UHFFFAOYSA-N 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- OHCQJHSOBUTRHG-UHFFFAOYSA-N colforsin Natural products OC12C(=O)CC(C)(C=C)OC1(C)C(OC(=O)C)C(O)C1C2(C)C(O)CCC1(C)C OHCQJHSOBUTRHG-UHFFFAOYSA-N 0.000 description 3
- 238000002648 combination therapy Methods 0.000 description 3
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 3
- 239000003172 expectorant agent Substances 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- 239000012530 fluid Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 239000012458 free base Substances 0.000 description 3
- 238000004108 freeze drying Methods 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 230000005445 isotope effect Effects 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 229940066491 mucolytics Drugs 0.000 description 3
- 235000015097 nutrients Nutrition 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 235000021317 phosphate Nutrition 0.000 description 3
- 235000015320 potassium carbonate Nutrition 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- 239000000376 reactant Substances 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- 230000028327 secretion Effects 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 229960000707 tobramycin Drugs 0.000 description 3
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- MENNSBVOTILFAX-UHFFFAOYSA-N trimethyl-[2-(1H-pyrazol-5-yloxy)ethyl]silane Chemical compound C[Si](CCOC1=NNC=C1)(C)C MENNSBVOTILFAX-UHFFFAOYSA-N 0.000 description 3
- PFGHCOGUUKXRLV-UHFFFAOYSA-N trimethyl-[3-(1H-pyrazol-5-yloxy)propyl]silane Chemical compound C=1(C=CNN=1)OCCC[Si](C)(C)C PFGHCOGUUKXRLV-UHFFFAOYSA-N 0.000 description 3
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 238000003828 vacuum filtration Methods 0.000 description 3
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 2
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical class OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 2
- MJUVRTYWUMPBTR-MRXNPFEDSA-N 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-n-[1-[(2r)-2,3-dihydroxypropyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)indol-5-yl]cyclopropane-1-carboxamide Chemical compound FC=1C=C2N(C[C@@H](O)CO)C(C(C)(CO)C)=CC2=CC=1NC(=O)C1(C=2C=C3OC(F)(F)OC3=CC=2)CC1 MJUVRTYWUMPBTR-MRXNPFEDSA-N 0.000 description 2
- IMZWSOSYNFVECD-UHFFFAOYSA-N 1-(oxan-2-yl)pyrazole Chemical compound O1CCCCC1N1N=CC=C1 IMZWSOSYNFVECD-UHFFFAOYSA-N 0.000 description 2
- LDDRFAQIRZNWPA-UHFFFAOYSA-N 3,3-dimethylbutyl-dimethyl-(1H-pyrazol-5-yl)silane Chemical compound C=1(C=CNN=1)[Si](CCC(C)(C)C)(C)C LDDRFAQIRZNWPA-UHFFFAOYSA-N 0.000 description 2
- ZXXMWYSGWPMYCL-UHFFFAOYSA-N 3-(3-hydroxypropyl)-5,5-dimethylpyrrolidin-2-one Chemical compound OCCCC1C(NC(C1)(C)C)=O ZXXMWYSGWPMYCL-UHFFFAOYSA-N 0.000 description 2
- RXOHDQQWGDAABG-UHFFFAOYSA-N 4-(3-hydroxypropyl)-2,2-dimethylpyrrolidine-1-carboxylic acid Chemical compound CC1(C)CC(CCCO)CN1C(O)=O RXOHDQQWGDAABG-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- OZJPLYNZGCXSJM-UHFFFAOYSA-N 5-valerolactone Chemical compound O=C1CCCCO1 OZJPLYNZGCXSJM-UHFFFAOYSA-N 0.000 description 2
- ACLUEOBQFRYTQS-UHFFFAOYSA-N 6-tert-butyl-2-(furan-2-carbonylamino)-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylic acid Chemical compound C1C(C(C)(C)C)CCC(C=2C(O)=O)=C1SC=2NC(=O)C1=CC=CO1 ACLUEOBQFRYTQS-UHFFFAOYSA-N 0.000 description 2
- 241001251200 Agelas Species 0.000 description 2
- 108700028369 Alleles Proteins 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- WZPBZJONDBGPKJ-UHFFFAOYSA-N Antibiotic SQ 26917 Natural products O=C1N(S(O)(=O)=O)C(C)C1NC(=O)C(=NOC(C)(C)C(O)=O)C1=CSC(N)=N1 WZPBZJONDBGPKJ-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- LBCNJYTUHZKYBT-KRWDZBQOSA-N BrC1=CC=C(C(=N1)N1C[C@H](CC1(C)C)CCCNC(=O)C1=C(C(=O)O)C=CC=C1)C(NS(=O)(=O)C1=NC(=CC=C1)F)=O Chemical compound BrC1=CC=C(C(=N1)N1C[C@H](CC1(C)C)CCCNC(=O)C1=C(C(=O)O)C=CC=C1)C(NS(=O)(=O)C1=NC(=CC=C1)F)=O LBCNJYTUHZKYBT-KRWDZBQOSA-N 0.000 description 2
- OKJNBNOYOOOKGK-AWEZNQCLSA-N BrC1=NC(=C(C(=O)N)C=C1)N1C(C[C@@H](C1)CCCN1C(C2=CC=CC=C2C1=O)=O)(C)C Chemical compound BrC1=NC(=C(C(=O)N)C=C1)N1C(C[C@@H](C1)CCCN1C(C2=CC=CC=C2C1=O)=O)(C)C OKJNBNOYOOOKGK-AWEZNQCLSA-N 0.000 description 2
- WIIDVSGDOCMTRV-JTQLQIEISA-N BrC1=NC(=C(C(=O)N)C=C1)N1C(C[C@@H](C1)CCCO)(C)C Chemical compound BrC1=NC(=C(C(=O)N)C=C1)N1C(C[C@@H](C1)CCCO)(C)C WIIDVSGDOCMTRV-JTQLQIEISA-N 0.000 description 2
- JDSZYMBVWCRVQB-ZETCQYMHSA-N CC(C[C@H]1C(OCCC1)=O)(C)[N+](=O)[O-] Chemical compound CC(C[C@H]1C(OCCC1)=O)(C)[N+](=O)[O-] JDSZYMBVWCRVQB-ZETCQYMHSA-N 0.000 description 2
- OHMJKMNGYYWCHB-HVMBLDELSA-N COC1=CC(=CC=C1\N=N\C1=CC=C2C(=CC(=C(N)C2=C1O)S(O)(=O)=O)S(O)(=O)=O)C1=CC=C(\N=N\C2=C(O)C3=C(N)C(=CC(=C3C=C2)S(O)(=O)=O)S(O)(=O)=O)C(OC)=C1 Chemical compound COC1=CC(=CC=C1\N=N\C1=CC=C2C(=CC(=C(N)C2=C1O)S(O)(=O)=O)S(O)(=O)=O)C1=CC=C(\N=N\C2=C(O)C3=C(N)C(=CC(=C3C=C2)S(O)(=O)=O)S(O)(=O)=O)C(OC)=C1 OHMJKMNGYYWCHB-HVMBLDELSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- XRHVZWWRFMCBAZ-UHFFFAOYSA-L Endothal-disodium Chemical compound [Na+].[Na+].C1CC2C(C([O-])=O)C(C(=O)[O-])C1O2 XRHVZWWRFMCBAZ-UHFFFAOYSA-L 0.000 description 2
- 206010064571 Gene mutation Diseases 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 235000019502 Orange oil Nutrition 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- 239000007868 Raney catalyst Substances 0.000 description 2
- 229910004298 SiO 2 Inorganic materials 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 241000534944 Thia Species 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- WZPBZJONDBGPKJ-VEHQQRBSSA-N aztreonam Chemical compound O=C1N(S([O-])(=O)=O)[C@@H](C)[C@@H]1NC(=O)C(=N/OC(C)(C)C(O)=O)\C1=CSC([NH3+])=N1 WZPBZJONDBGPKJ-VEHQQRBSSA-N 0.000 description 2
- 229960003644 aztreonam Drugs 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 238000010504 bond cleavage reaction Methods 0.000 description 2
- 239000000168 bronchodilator agent Substances 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 150000001975 deuterium Chemical group 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 2
- 229940043264 dodecyl sulfate Drugs 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000003792 electrolyte Substances 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- 238000000806 fluorine-19 nuclear magnetic resonance spectrum Methods 0.000 description 2
- 230000004907 flux Effects 0.000 description 2
- 238000004817 gas chromatography Methods 0.000 description 2
- 229940050410 gluconate Drugs 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 230000005283 ground state Effects 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 230000037427 ion transport Effects 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 210000003097 mucus Anatomy 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000010502 orange oil Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- 230000004783 oxidative metabolism Effects 0.000 description 2
- 229940045258 pancrelipase Drugs 0.000 description 2
- RLBIQVVOMOPOHC-UHFFFAOYSA-N parathion-methyl Chemical compound COP(=S)(OC)OC1=CC=C([N+]([O-])=O)C=C1 RLBIQVVOMOPOHC-UHFFFAOYSA-N 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 229920001992 poloxamer 407 Polymers 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229960003975 potassium Drugs 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 125000001453 quaternary ammonium group Chemical group 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 102200132108 rs80034486 Human genes 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 229940083542 sodium Drugs 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- CUJVOTFZEAXRSZ-UHFFFAOYSA-N tert-butyl 2,2-dimethyl-4-(3-methylsulfonyloxypropyl)pyrrolidine-1-carboxylate Chemical compound CC1(N(CC(C1)CCCOS(=O)(=O)C)C(=O)OC(C)(C)C)C CUJVOTFZEAXRSZ-UHFFFAOYSA-N 0.000 description 2
- ASBLCXVIWZIOFQ-UHFFFAOYSA-N tert-butyl 2,2-dimethyl-4-[3-[(6-sulfamoylpyridin-2-yl)amino]propyl]pyrrolidine-1-carboxylate Chemical compound CC1(N(CC(C1)CCCNC1=NC(=CC=C1)S(N)(=O)=O)C(=O)OC(C)(C)C)C ASBLCXVIWZIOFQ-UHFFFAOYSA-N 0.000 description 2
- XOFOBXWQPZWFIQ-UHFFFAOYSA-N tert-butyl 3-(2-trimethylsilylethoxy)pyrazole-1-carboxylate Chemical compound C[Si](CCOC1=NN(C=C1)C(=O)OC(C)(C)C)(C)C XOFOBXWQPZWFIQ-UHFFFAOYSA-N 0.000 description 2
- DVBPENKJOUBVQB-UHFFFAOYSA-N tert-butyl 3-(trimethylsilylmethoxy)pyrazole-1-carboxylate Chemical compound C[Si](C)(C)COC1=NN(C=C1)C(=O)OC(C)(C)C DVBPENKJOUBVQB-UHFFFAOYSA-N 0.000 description 2
- SJBIXEHEDPJIGQ-UHFFFAOYSA-N tert-butyl 3-[[tert-butyl(dimethyl)silyl]methoxy]pyrazole-1-carboxylate Chemical compound [Si](C)(C)(C(C)(C)C)COC1=NN(C=C1)C(=O)OC(C)(C)C SJBIXEHEDPJIGQ-UHFFFAOYSA-N 0.000 description 2
- GFCJGMPPLDUPEC-UHFFFAOYSA-N tert-butyl 4-(3-aminopropyl)-2,2-dimethylpyrrolidine-1-carboxylate Chemical compound NCCCC1CC(N(C1)C(=O)OC(C)(C)C)(C)C GFCJGMPPLDUPEC-UHFFFAOYSA-N 0.000 description 2
- UVDHPDPOUNSQER-UHFFFAOYSA-N tert-butyl-dimethyl-[2-(oxan-2-yl)pyrazol-3-yl]silane Chemical compound C(C)([Si](C)(C1=CC=NN1C1OCCCC1)C)(C)C UVDHPDPOUNSQER-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000003354 tissue distribution assay Methods 0.000 description 2
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical class O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- DVSZKTAMJJTWFG-SKCDLICFSA-N (2e,4e,6e,8e,10e,12e)-docosa-2,4,6,8,10,12-hexaenoic acid Chemical compound CCCCCCCCC\C=C\C=C\C=C\C=C\C=C\C=C\C(O)=O DVSZKTAMJJTWFG-SKCDLICFSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- YLQDXCAHIIESME-SNAWJCMRSA-N (3e)-3-(hydroxymethylidene)oxan-2-one Chemical compound O\C=C1/CCCOC1=O YLQDXCAHIIESME-SNAWJCMRSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- WXGDDUWFYSTFJV-TYYBGVCCSA-N (e)-but-2-enedioic acid;phosphoric acid Chemical compound OP(O)(O)=O.OC(=O)\C=C\C(O)=O WXGDDUWFYSTFJV-TYYBGVCCSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- XUXATUPUQBOACO-UHFFFAOYSA-N 1-[1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)indol-5-yl]cyclopropane-1-carboxamide Chemical compound FC=1C=C2N(CC(O)CO)C(C(C)(CO)C)=CC2=CC=1C1(C(N)=O)CC1 XUXATUPUQBOACO-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- BZMHKFKAXOWSBC-UHFFFAOYSA-N 2,2-dimethyl-4-(3-methylsulfonyloxypropyl)pyrrolidine-1-carboxylic acid Chemical compound CC1(C)CC(CCCOS(C)(=O)=O)CN1C(O)=O BZMHKFKAXOWSBC-UHFFFAOYSA-N 0.000 description 1
- AEQDJSLRWYMAQI-UHFFFAOYSA-N 2,3,9,10-tetramethoxy-6,8,13,13a-tetrahydro-5H-isoquinolino[2,1-b]isoquinoline Chemical compound C1CN2CC(C(=C(OC)C=C3)OC)=C3CC2C2=C1C=C(OC)C(OC)=C2 AEQDJSLRWYMAQI-UHFFFAOYSA-N 0.000 description 1
- XDAXHRXBCWWEHD-UHFFFAOYSA-N 2,3-dihydroxybutanedioic acid;2-hydroxynaphthalene-1-carboxylic acid;hydrochloride Chemical compound Cl.OC(=O)C(O)C(O)C(O)=O.C1=CC=C2C(C(=O)O)=C(O)C=CC2=C1 XDAXHRXBCWWEHD-UHFFFAOYSA-N 0.000 description 1
- ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 2,3-dimethylbutane Chemical group CC(C)C(C)C ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- FILKGCRCWDMBKA-UHFFFAOYSA-N 2,6-dichloropyridine Chemical compound ClC1=CC=CC(Cl)=N1 FILKGCRCWDMBKA-UHFFFAOYSA-N 0.000 description 1
- MBTGBRYMJKYYOE-UHFFFAOYSA-N 2,6-difluoropyridine Chemical compound FC1=CC=CC(F)=N1 MBTGBRYMJKYYOE-UHFFFAOYSA-N 0.000 description 1
- QKBKGNDTLQFSEU-UHFFFAOYSA-N 2-bromo-3,3,3-trifluoroprop-1-ene Chemical compound FC(F)(F)C(Br)=C QKBKGNDTLQFSEU-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- FGLBSLMDCBOPQK-UHFFFAOYSA-N 2-nitropropane Chemical compound CC(C)[N+]([O-])=O FGLBSLMDCBOPQK-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-M 3-carboxynaphthalen-2-olate Chemical compound C1=CC=C2C=C(C([O-])=O)C(O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-M 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 1
- YNUUCDHAZGAVEZ-UHFFFAOYSA-N 3-trimethylsilylpropan-1-ol Chemical compound C[Si](C)(C)CCCO YNUUCDHAZGAVEZ-UHFFFAOYSA-N 0.000 description 1
- GZJLLYHBALOKEX-UHFFFAOYSA-N 6-Ketone, O18-Me-Ussuriedine Natural products CC=CCC=CCC=CCC=CCC=CCC=CCCCC(O)=O GZJLLYHBALOKEX-UHFFFAOYSA-N 0.000 description 1
- CKSOKCDTRKHTPT-UHFFFAOYSA-N 6-bromo-2-fluoropyridine-3-carboxylic acid Chemical compound OC(=O)C1=CC=C(Br)N=C1F CKSOKCDTRKHTPT-UHFFFAOYSA-N 0.000 description 1
- ZYYNYUDHGGZBOB-UHFFFAOYSA-N 6-fluoropyridine-2-sulfonyl chloride Chemical compound FC1=CC=CC(S(Cl)(=O)=O)=N1 ZYYNYUDHGGZBOB-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 244000144730 Amygdalus persica Species 0.000 description 1
- 108091006515 Anion channels Proteins 0.000 description 1
- 102000037829 Anion channels Human genes 0.000 description 1
- 241001061264 Astragalus Species 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 239000012583 B-27 Supplement Substances 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- CZZCEEHPEMLQHF-KRWDZBQOSA-N BrC1=NC(=C(C(=O)NS(=O)(=O)C2=NC(=CC=C2)F)C=C1)N1C(C[C@@H](C1)CCCN1C(C2=CC=CC=C2C1=O)=O)(C)C Chemical compound BrC1=NC(=C(C(=O)NS(=O)(=O)C2=NC(=CC=C2)F)C=C1)N1C(C[C@@H](C1)CCCN1C(C2=CC=CC=C2C1=O)=O)(C)C CZZCEEHPEMLQHF-KRWDZBQOSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- VBFMZLLUSVKXSM-NSHDSACASA-N CS(=O)(=O)OCCC[C@@H]1CN(C(C1)(C)C)C1=NC(=CC=C1C(N)=O)Br Chemical compound CS(=O)(=O)OCCC[C@@H]1CN(C(C1)(C)C)C1=NC(=CC=C1C(N)=O)Br VBFMZLLUSVKXSM-NSHDSACASA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229940126062 Compound A Drugs 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 206010011224 Cough Diseases 0.000 description 1
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 1
- 208000000059 Dyspnea Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 102100021022 Gastrin Human genes 0.000 description 1
- 108010052343 Gastrins Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- 208000026350 Inborn Genetic disease Diseases 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- GSDSWSVVBLHKDQ-JTQLQIEISA-N Levofloxacin Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-JTQLQIEISA-N 0.000 description 1
- 206010024971 Lower respiratory tract infections Diseases 0.000 description 1
- 229940126560 MAPK inhibitor Drugs 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 108010021466 Mutant Proteins Proteins 0.000 description 1
- 102000008300 Mutant Proteins Human genes 0.000 description 1
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 1
- 239000012580 N-2 Supplement Substances 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- HTMSAZQUWJLSDE-UAIGNFCESA-N NCCN.OC(/C=C\C(O)=O)=O.Br Chemical compound NCCN.OC(/C=C\C(O)=O)=O.Br HTMSAZQUWJLSDE-UAIGNFCESA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- SUHOOTKUPISOBE-UHFFFAOYSA-N O-phosphoethanolamine Chemical compound NCCOP(O)(O)=O SUHOOTKUPISOBE-UHFFFAOYSA-N 0.000 description 1
- 208000035467 Pancreatic insufficiency Diseases 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- QSXMZJGGEWYVCN-UHFFFAOYSA-N Pirbuterol acetate Chemical compound CC(O)=O.CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=N1 QSXMZJGGEWYVCN-UHFFFAOYSA-N 0.000 description 1
- 206010035664 Pneumonia Diseases 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- HLCFGWHYROZGBI-JJKGCWMISA-M Potassium gluconate Chemical compound [K+].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O HLCFGWHYROZGBI-JJKGCWMISA-M 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 235000006040 Prunus persica var persica Nutrition 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 102000003673 Symporters Human genes 0.000 description 1
- 108090000088 Symporters Proteins 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- GKXVJHDEWHKBFH-UHFFFAOYSA-N [2-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=CC=C1CN GKXVJHDEWHKBFH-UHFFFAOYSA-N 0.000 description 1
- GPDHNZNLPKYHCN-DZOOLQPHSA-N [[(z)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-morpholin-4-ylmethylidene]-dimethylazanium;hexafluorophosphate Chemical compound F[P-](F)(F)(F)(F)F.CCOC(=O)C(\C#N)=N/OC(=[N+](C)C)N1CCOCC1 GPDHNZNLPKYHCN-DZOOLQPHSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- OMCWXFSQPKBREP-UHFFFAOYSA-N acetic acid;hydroiodide Chemical compound I.CC(O)=O OMCWXFSQPKBREP-UHFFFAOYSA-N 0.000 description 1
- WEVYAHXRMPXWCK-FIBGUPNXSA-N acetonitrile-d3 Chemical compound [2H]C([2H])([2H])C#N WEVYAHXRMPXWCK-FIBGUPNXSA-N 0.000 description 1
- AUALQMFGWLZREY-UHFFFAOYSA-N acetonitrile;methanol Chemical compound OC.CC#N AUALQMFGWLZREY-UHFFFAOYSA-N 0.000 description 1
- 229960004308 acetylcysteine Drugs 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 230000009056 active transport Effects 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000012574 advanced DMEM Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 150000008055 alkyl aryl sulfonates Chemical class 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- VMWZRHGIAVCFNS-UHFFFAOYSA-J aluminum;lithium;tetrahydroxide Chemical compound [Li+].[OH-].[OH-].[OH-].[OH-].[Al+3] VMWZRHGIAVCFNS-UHFFFAOYSA-J 0.000 description 1
- 229960004821 amikacin Drugs 0.000 description 1
- LKCWBDHBTVXHDL-RMDFUYIESA-N amikacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O LKCWBDHBTVXHDL-RMDFUYIESA-N 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000002924 anti-infective effect Effects 0.000 description 1
- 229960005475 antiinfective agent Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 235000006533 astragalus Nutrition 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 239000007640 basal medium Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- VZFLMGRPVHYDGH-UHFFFAOYSA-N benzenesulfonic acid 2-hydroxyethanesulfonic acid Chemical compound OCCS(O)(=O)=O.OS(=O)(=O)c1ccccc1 VZFLMGRPVHYDGH-UHFFFAOYSA-N 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- AEZRRMRHIVEUFV-UHFFFAOYSA-N benzoic acid;2-hydroxypropanoic acid Chemical compound CC(O)C(O)=O.OC(=O)C1=CC=CC=C1 AEZRRMRHIVEUFV-UHFFFAOYSA-N 0.000 description 1
- UENWRTRMUIOCKN-UHFFFAOYSA-N benzyl thiol Chemical compound SCC1=CC=CC=C1 UENWRTRMUIOCKN-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229960005069 calcium Drugs 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 239000004227 calcium gluconate Substances 0.000 description 1
- 235000013927 calcium gluconate Nutrition 0.000 description 1
- 229960004494 calcium gluconate Drugs 0.000 description 1
- NEEHYRZPVYRGPP-UHFFFAOYSA-L calcium;2,3,4,5,6-pentahydroxyhexanoate Chemical compound [Ca+2].OCC(O)C(O)C(O)C(O)C([O-])=O.OCC(O)C(O)C(O)C(O)C([O-])=O NEEHYRZPVYRGPP-UHFFFAOYSA-L 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- RBHJBMIOOPYDBQ-UHFFFAOYSA-N carbon dioxide;propan-2-one Chemical compound O=C=O.CC(C)=O RBHJBMIOOPYDBQ-UHFFFAOYSA-N 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000012159 carrier gas Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000011072 cell harvest Methods 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- LGEJHPHGNSBWOF-UHFFFAOYSA-N chloro-(3,3-dimethylbutyl)-dimethylsilane Chemical compound CC(C)(C)CC[Si](C)(C)Cl LGEJHPHGNSBWOF-UHFFFAOYSA-N 0.000 description 1
- ITKVLPYNJQOCPW-UHFFFAOYSA-N chloro-(chloromethyl)-dimethylsilane Chemical compound C[Si](C)(Cl)CCl ITKVLPYNJQOCPW-UHFFFAOYSA-N 0.000 description 1
- ZKNHDYVIBKUBQU-UHFFFAOYSA-N chloromethyl(trimethyl)germane Chemical compound C[Ge](C)(C)CCl ZKNHDYVIBKUBQU-UHFFFAOYSA-N 0.000 description 1
- OOCUOKHIVGWCTJ-UHFFFAOYSA-N chloromethyl(trimethyl)silane Chemical compound C[Si](C)(C)CCl OOCUOKHIVGWCTJ-UHFFFAOYSA-N 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 229940092125 creon Drugs 0.000 description 1
- 239000012045 crude solution Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 125000004431 deuterium atom Chemical group 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 239000012973 diazabicyclooctane Substances 0.000 description 1
- ACYGYJFTZSAZKR-UHFFFAOYSA-J dicalcium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Ca+2].[Ca+2].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O ACYGYJFTZSAZKR-UHFFFAOYSA-J 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 230000001079 digestive effect Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- GXGAKHNRMVGRPK-UHFFFAOYSA-N dimagnesium;dioxido-bis[[oxido(oxo)silyl]oxy]silane Chemical compound [Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O GXGAKHNRMVGRPK-UHFFFAOYSA-N 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 229910000397 disodium phosphate Inorganic materials 0.000 description 1
- 235000019800 disodium phosphate Nutrition 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 235000020669 docosahexaenoic acid Nutrition 0.000 description 1
- KAUVQQXNCKESLC-UHFFFAOYSA-N docosahexaenoic acid (DHA) Natural products COC(=O)C(C)NOCC1=CC=CC=C1 KAUVQQXNCKESLC-UHFFFAOYSA-N 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 108010067396 dornase alfa Proteins 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 229940009662 edetate Drugs 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- LJQKCYFTNDAAPC-UHFFFAOYSA-N ethanol;ethyl acetate Chemical compound CCO.CCOC(C)=O LJQKCYFTNDAAPC-UHFFFAOYSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- SRCZQMGIVIYBBJ-UHFFFAOYSA-N ethoxyethane;ethyl acetate Chemical compound CCOCC.CCOC(C)=O SRCZQMGIVIYBBJ-UHFFFAOYSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 231100000502 fertility decrease Toxicity 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 238000005187 foaming Methods 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 229960000308 fosfomycin Drugs 0.000 description 1
- YMDXZJFXQJVXBF-STHAYSLISA-N fosfomycin Chemical compound C[C@@H]1O[C@@H]1P(O)(O)=O YMDXZJFXQJVXBF-STHAYSLISA-N 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- DSLZVSRJTYRBFB-DUHBMQHGSA-N galactaric acid Chemical class OC(=O)[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O DSLZVSRJTYRBFB-DUHBMQHGSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 208000016361 genetic disease Diseases 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 150000002301 glucosamine derivatives Chemical class 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- DQKGOGJIOHUEGK-UHFFFAOYSA-M hydron;2-hydroxyethyl(trimethyl)azanium;carbonate Chemical compound OC([O-])=O.C[N+](C)(C)CCO DQKGOGJIOHUEGK-UHFFFAOYSA-M 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- XXSMGPRMXLTPCZ-UHFFFAOYSA-N hydroxychloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CCO)CC)=CC=NC2=C1 XXSMGPRMXLTPCZ-UHFFFAOYSA-N 0.000 description 1
- 229960004171 hydroxychloroquine Drugs 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000021267 infertility disease Diseases 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 229960003376 levofloxacin Drugs 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229960001078 lithium Drugs 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- OVEHNNQXLPJPPL-UHFFFAOYSA-N lithium;n-propan-2-ylpropan-2-amine Chemical compound [Li].CC(C)NC(C)C OVEHNNQXLPJPPL-UHFFFAOYSA-N 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- UFSKUSARDNFIRC-UHFFFAOYSA-N lumacaftor Chemical compound N1=C(C=2C=C(C=CC=2)C(O)=O)C(C)=CC=C1NC(=O)C1(C=2C=C3OC(F)(F)OC3=CC=2)CC1 UFSKUSARDNFIRC-UHFFFAOYSA-N 0.000 description 1
- 238000005710 macrocyclization reaction Methods 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000001755 magnesium gluconate Substances 0.000 description 1
- 235000015778 magnesium gluconate Nutrition 0.000 description 1
- 229960003035 magnesium gluconate Drugs 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940091250 magnesium supplement Drugs 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- IAKLPCRFBAZVRW-XRDLMGPZSA-L magnesium;(2r,3s,4r,5r)-2,3,4,5,6-pentahydroxyhexanoate;hydrate Chemical compound O.[Mg+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O IAKLPCRFBAZVRW-XRDLMGPZSA-L 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- FIXGUDASGASFSO-LURJTMIESA-N methyl (2S)-2,4-dimethyl-4-nitropentanoate Chemical compound C[C@H](C(=O)OC)CC(C)([N+](=O)[O-])C FIXGUDASGASFSO-LURJTMIESA-N 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 230000000510 mucolytic effect Effects 0.000 description 1
- PURKAOJPTOLRMP-ASMGOKTBSA-N n-[2-tert-butyl-4-[1,1,1,3,3,3-hexadeuterio-2-(trideuteriomethyl)propan-2-yl]-5-hydroxyphenyl]-4-oxo-1h-quinoline-3-carboxamide Chemical compound C1=C(O)C(C(C([2H])([2H])[2H])(C([2H])([2H])[2H])C([2H])([2H])[2H])=CC(C(C)(C)C)=C1NC(=O)C1=CNC2=CC=CC=C2C1=O PURKAOJPTOLRMP-ASMGOKTBSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 238000001208 nuclear magnetic resonance pulse sequence Methods 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000002220 organoid Anatomy 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- LVSJDHGRKAEGLX-UHFFFAOYSA-N oxolane;2,2,2-trifluoroacetic acid Chemical compound C1CCOC1.OC(=O)C(F)(F)F LVSJDHGRKAEGLX-UHFFFAOYSA-N 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 230000004203 pancreatic function Effects 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N phenylalanine group Chemical group N[C@@H](CC1=CC=CC=C1)C(=O)O COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000008055 phosphate buffer solution Substances 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 229960004994 pirbuterol acetate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-M pivalate Chemical compound CC(C)(C)C([O-])=O IUGYQRQAERSCNH-UHFFFAOYSA-M 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000004224 potassium gluconate Substances 0.000 description 1
- 235000013926 potassium gluconate Nutrition 0.000 description 1
- 229960003189 potassium gluconate Drugs 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 125000006308 propyl amino group Chemical group 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- 229940107568 pulmozyme Drugs 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- NYCVCXMSZNOGDH-UHFFFAOYSA-N pyrrolidine-1-carboxylic acid Chemical compound OC(=O)N1CCCC1 NYCVCXMSZNOGDH-UHFFFAOYSA-N 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 239000011435 rock Substances 0.000 description 1
- 102220002718 rs121908745 Human genes 0.000 description 1
- 102200132021 rs121909036 Human genes 0.000 description 1
- 102200132039 rs139304906 Human genes 0.000 description 1
- 102220020371 rs151020603 Human genes 0.000 description 1
- 102200132012 rs36210737 Human genes 0.000 description 1
- 102200128617 rs75961395 Human genes 0.000 description 1
- 102200132028 rs78194216 Human genes 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 229960004017 salmeterol Drugs 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 208000013220 shortness of breath Diseases 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 235000017550 sodium carbonate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000000176 sodium gluconate Substances 0.000 description 1
- 235000012207 sodium gluconate Nutrition 0.000 description 1
- 229940005574 sodium gluconate Drugs 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- SNOOUWRIMMFWNE-UHFFFAOYSA-M sodium;6-[(3,4,5-trimethoxybenzoyl)amino]hexanoate Chemical compound [Na+].COC1=CC(C(=O)NCCCCCC([O-])=O)=CC(OC)=C1OC SNOOUWRIMMFWNE-UHFFFAOYSA-M 0.000 description 1
- 239000007962 solid dispersion Substances 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 230000006103 sulfonylation Effects 0.000 description 1
- 238000005694 sulfonylation reaction Methods 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- UXQPRXPNOJXOQO-UHFFFAOYSA-N sulfuric acid;hydrobromide Chemical compound Br.OS(O)(=O)=O UXQPRXPNOJXOQO-UHFFFAOYSA-N 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 210000004233 talus Anatomy 0.000 description 1
- LRBQNJMCXXYXIU-NRMVVENXSA-N tannic acid Chemical class OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-NRMVVENXSA-N 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- ASBLCXVIWZIOFQ-AWEZNQCLSA-N tert-butyl (4S)-2,2-dimethyl-4-[3-[(6-sulfamoylpyridin-2-yl)amino]propyl]pyrrolidine-1-carboxylate Chemical compound CC1(N(C[C@H](C1)CCCNC1=NC(=CC=C1)S(N)(=O)=O)C(=O)OC(C)(C)C)C ASBLCXVIWZIOFQ-AWEZNQCLSA-N 0.000 description 1
- JRUPNCPAQXQNJI-UHFFFAOYSA-N tert-butyl 3-(3-trimethylsilylpropoxy)pyrazole-1-carboxylate Chemical compound C[Si](CCCOC1=NN(C=C1)C(=O)OC(C)(C)C)(C)C JRUPNCPAQXQNJI-UHFFFAOYSA-N 0.000 description 1
- IMTSOPLKKQBQEG-UHFFFAOYSA-N tert-butyl 3-[[dimethyl-[1-(trifluoromethyl)cyclopropyl]silyl]methoxy]pyrazole-1-carboxylate Chemical compound C[Si](C1(CC1)C(F)(F)F)(C)COC1=NN(C=C1)C(=O)OC(C)(C)C IMTSOPLKKQBQEG-UHFFFAOYSA-N 0.000 description 1
- VTLNXGOQNWRXNK-UHFFFAOYSA-N tert-butyl 4-(3-hydroxypropyl)-2,2-dimethylpyrrolidine-1-carboxylate Chemical compound OCCCC1CC(N(C1)C(=O)OC(C)(C)C)(C)C VTLNXGOQNWRXNK-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- BSJFXAFLSWDUPK-UHFFFAOYSA-N tert-butyl pyrazole-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1C=CC=N1 BSJFXAFLSWDUPK-UHFFFAOYSA-N 0.000 description 1
- PUBXADYSQDUPRU-UHFFFAOYSA-N tert-butyl-dimethyl-(1H-pyrazol-5-yl)silane Chemical compound [Si](C1=NNC=C1)(C(C)(C)C)(C)C PUBXADYSQDUPRU-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 229940054369 ultrase Drugs 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 238000002255 vaccination Methods 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 235000016804 zinc Nutrition 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/081—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te
- C07F7/0812—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/081—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te
- C07F7/0812—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring
- C07F7/0814—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring said ring is substituted at a C ring atom by Si
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/081—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te
- C07F7/0812—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring
- C07F7/0816—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring said ring comprising Si as a ring atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/30—Germanium compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
本揭示案提供囊腫纖維化跨膜傳導調節蛋白(CFTR)之調節劑、含有至少一種此類調節劑之醫藥組成物、使用此類調節劑及醫藥組成物治療囊腫纖維化之方法、及用於製備此類調節劑之製程。
Description
本揭示案提供囊腫纖維化跨膜傳導調節蛋白(CFTR)之調節劑、含有該等調節劑之醫藥組成物、使用此類調節劑及醫藥組成物治療囊腫纖維化之方法、及用於製備此類調節劑之製程。
囊腫纖維化(CF)為一種隱性遺傳疾病,全世界有約70,000名兒童及成人受到影響。儘管CF之治療有所進展,但仍無法治癒。
在患有CF之患者中,呼吸上皮細胞內源性表現之CFTR突變導致肺尖陰離子分泌減少,從而導致離子及液體運輸失衡。所引起之陰離子運輸減少導致肺中黏液蓄積增加,且伴隨微生物感染,最終導致CF患者死亡。除呼吸道疾病外,CF患者通常會出現胃腸道問題及胰臟功能不全,若不及時治療,將導致死亡。另外,大多數患有囊腫纖維化之男性為不育的,且患有囊腫纖維化之女性之生育力降低。
CFTR基因之序列分析揭示了多種致病性突變(Cutting, G. R.等人(1990) Nature 346:366-369;Dean, M.等人 (1990) Cell 61:863:870;及Kerem, B-S.等人(1989) Science 245:1073-1080;Kerem, B-S等人(1990) Proc. Natl. Acad. Sci. USA 87:8447-8451)。迄今為止,已經鑑別出超過2000種CF基因突變;目前,CFTR2資料庫僅包含關於該等經鑑別突變中之322個突變之信息,且有足夠證據將281個突變定義為致病原因。最普遍的致病性突變為CFTR胺基酸序列在位置508處之苯丙胺酸缺失,且通常稱為F508del突變。該突變在大多數囊腫纖維化病例中出現,且與嚴重疾病有關。
CFTR中殘基508之缺失阻止了初生蛋白正確折疊。這導致突變蛋白無法離開內質網(ER)並運送至質膜。因此,存在於膜中之用於陰離子運輸的CFTR通道數目遠少於在表現野生型CFTR(亦即無突變之CFTR)之細胞中觀測到之數目。除運送受損外,該突變會導致缺陷型通道閘控。總之,膜中通道數量減少及缺陷型閘控導致跨越上皮細胞之陰離子及流體運輸減少。(Quinton, P. M. (1990), FASEB J. 4: 2709-2727)。由於F508del突變而有缺陷之通道仍具有功能,儘管其功能低於野生型CFTR通道。(Dalemans等人(1991), Nature Lond. 354: 526-528;Pasyk及Foskett (1995), J. Cell. Biochem. 270: 12347-50)。除F508del外,亦可上調或下調導致缺陷型運輸、合成、及/或通道閘控之其他致病性CFTR突變,以改變陰離子分泌且改變疾病進程及/或嚴重性。
CFTR為在多種細胞類型(包括吸收性及分泌性上皮細胞)中表現之經cAMP/ATP介導之陰離子通道,其中該通道調節跨越膜之陰離子通量以及其他離子通道及蛋白之活性。在上皮細胞中,CFTR之正常功能對於維持電解質在整個身體(包括呼吸及消化組織)中之運輸為關鍵的。CFTR由編碼構成跨膜域銜接重複體之蛋白的1480個胺基酸組成,各跨膜域包含六個跨膜螺旋及一個核苷酸結合域。該兩個跨膜域藉由具有可調節通道活性及細胞運送之多個磷酸化位點之大極性調節(R)域連接。
氯離子運輸藉由存在於肺尖膜上之ENaC及CFTR及細菌細胞表面上表現之Na+
-K+
-ATP酶泵及Cl-通道之協同活性來發生。氯離子自管腔側繼發性主動運輸導致細胞內氯離子蓄積,然後可經由Cl-
通道被動地離開細胞,導致向量運輸。基側表面上之Na+
/2Cl-
/K+
共同運輸蛋白、Na+
-K+
-ATP酶泵及基側膜K+
通道及管腔側上之CFTR的排列協調管腔側上經由CFTR之氯離子分泌。因為水本身可能永遠不會主動運輸,所以水跨越上皮細胞之流動取決於由鈉及氯離子大流量所產生之微小經上皮滲透梯度。
最近已鑑別許多CFTR調節化合物。然而,仍需要可以治療囊腫纖維化及其他經CFTR介導疾病或減輕該等疾病之嚴重性且特別是該等疾病之更嚴重形式的化合物。
本發明之一個態樣提供了新穎化合物,包括式(1)、(2)、(3)及(4)化合物、其醫藥學上可接受之鹽及前述任一者之氘化衍生物,其中該等化合物各自含有至少一個矽原子或至少一個鍺原子。
例如,本文揭示了式(1)化合物及其醫藥學上可接受之鹽及氘化衍生物:(1)
其中:
X選自Si(R)3
、-(O)n
-(C1
-C8
烷基)、-(O)n
-(C3
-C10
環烷基),其中:
n為0或1,
各C1
-C8
烷基經0、1、2或3個選自鹵素、羥基、側氧基、C3
-C10
環烷基、C1
-C4
鹵烷基、及Si(R)3
基團之基團取代,
各C3
-C10
環烷基經0、1、2、3或4個選自鹵素、C1
-C4
鹵烷基、C1
-C4
烷基、及Si(R)3
基團之基團取代,且
各C1
-C8
烷基中之一個-CH2-
視情況經-Si(R)2
-置換;
Y選自氫及-Si(R)3
;
各Z獨立地選自-CH2-
及-Si(R)2
-;且
各R獨立地選自苯基及C1
-C6
烷基;且
其中各式(1)化合物含有至少一個Si原子。在一些實施例中,式(1)化合物選自化合物(1-1)-(1-11)及其醫藥學上可接受之鹽及氘化衍生物。
在一些實施例中,式(1)中之X選自Si(R)3
、、、、、、及,其中 各R獨立地選自苯基及C1
-C6
烷基。
另一個實施例提供式(2)化合物及其醫藥學上可接受之鹽及氘化衍生物:(2)
其中:
X選自Ge(R)3
、-(O)n
-(C1
-C8
烷基)、-(O)n
-(C3
-C10
環烷基),其中:
n為0或1,
各C1
-C8
烷基經0、1、2或3個選自鹵素、羥基、側氧基、C3
-C10
環烷基、C1
-C4
鹵烷基、及Ge(R)3
基團之基團取代,
各C3
-C10
環烷基經0、1、2、3或4個選自鹵素、C1
-C4
鹵烷基、C1
-C4
烷基、及Ge(R)3
基團之基團取代,且
各C1
-C8
烷基中之一個-CH2-
視情況經-Ge(R)2
-置換;
Y選自氫及-Ge(R)3
;
各Z獨立地選自-CH2-
及-Ge(R)2
-;且
各R獨立地選自苯基及C1
-C6
烷基;且
其中各式(2)化合物含有至少一個Ge原子。在一些實施例中,式(2)化合物選自化合物(2-1)及其醫藥學上可接受之鹽及氘化衍生物。
在一些實施例中,式(2)中之X選自Ge(R)3
、、、、、、及,其中
各R獨立地選自苯基及C1
-C6
烷基。
本發明之另一個實施例包括式(3)化合物及其醫藥學上可接受之鹽及氘化衍生物:(3),
其醫藥學上可接受之鹽及前述任一者之氘化衍生物,
其中:
- 式(3)之位置2處之碳原子經矽原子置換;
- 式(3)之位置6及7處之甲基中之至少一者經選自-Si(R)3
基團、-Si(R)2
(OR)基團、及-Si( R)(OR)2
基團之基團置換;
- 式(3)之位置3、5、及8處之亞甲基中之至少一者經選自>Si(R)2
基團及>Si(R)(OR)基團之基團置換;且/或
- 式(3)之位置4處之次甲基經選自≡Si(R)基團及≡Si(OR)基團之基團置換;且
其中各R可為相同或不同的,獨立地選自氫、苯基、及C1
-C6
烷基。
本發明之另一個實施例包括式(4)化合物及其醫藥學上可接受之鹽及氘化衍生物:(4),
其醫藥學上可接受之鹽及前述任一者之氘化衍生物,
其中:
- 式(4)之位置2處之碳原子經鍺原子取代;
- 式(4)之位置6及7處之甲基中之至少一者經選自-Ge(R)3
基團、-Ge(R)2
(OR)基團及-Ge( R)(OR)2
基團之基團置換;
- 式(4)之位置3、5、及8處之亞甲基中之至少一者經選自>Ge(R)2
基團及 >Ge(R)(OR)基團之基團置換;且/或
- 式(4)之位置4處之次甲基經選自≡Ge(R)基團及≡Ge(OR)基團之基團置換;且
其中各R可為相同或不同的,獨立地選自氫、苯基、及C1
-C6
烷基。
本發明之另一態樣提供醫藥組成物,該醫藥組成物包含至少一種選自本文揭示之新穎化合物、其醫藥學上可接受之鹽及前述任一者之氘化衍生物的化合物及至少一種醫藥學上可接受之載劑,該等組成物可進一步包括至少一種額外活性醫藥成分。因此,本發明之另一態樣提供治療經CFTR介導之疾病囊腫纖維化之方法,該等方法包含向有需要之受試者投與至少一種選自本文揭示之新穎化合物、其醫藥學上可接受之鹽及前述任一者之氘化衍生物之化合物及至少一種醫藥學上可接受之載劑,視情況作為包含至少一種額外組分之醫藥組成物之一部分。
在某些實施例中,本發明之醫藥組成物包含至少一種選自式(1)、(2)、(3)、及(4)、化合物(1-1)-(1-11)、化合物( 2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物。在一些實施例中,包含至少一種選自式(1)、(2)、(3)、及(4)、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物的組成物可視情況進一步包含(a)至少一種選自化合物II
及其化合物醫藥學上可接受之鹽及氘化衍生物之化合物;(b)至少一種選自化合物III
及其醫藥學上可接受之鹽及氘化衍生物之化合物,諸如化合物III-d
;及/或(c)至少一種選自化合物IV
及其醫藥學上可接受之鹽及氘化衍生物之化合物。
本發明之另一個態樣提供了治療經CFTR介導之疾病囊腫纖維化之方法,該等方法包含向有需要之患者投與至少一種選自以下之化合物,視情況作為包含至少一種額外組分之至少一種醫藥組成物之一部分:本文所揭示之新穎化合物、其醫藥學上可接受之鹽、及前述任一者之氘化衍生物、 (R
)-1-(2,2-二氟苯并[d][1,3]間二氧雜環戊烯-5-基)-N-(1-(2,3-二羥基丙基)-6-氟-2-(1-羥基-2-甲基丙烷-2-基)-1H-吲哚-5-基)環丙烷甲醯胺(化合物II
)、N-[2,4-雙(1,1-二甲基乙基)-5-羥基苯基]-1,4-二氫-4-側氧基喹啉-3-甲醯胺(化合物III
)或N-(2-(三級丁基)-5-羥基-4-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基)-4-側氧基-1,4-二氫喹啉-3-甲醯胺(化合物III-d
)、及3-(6-(1-(2,2-二氟苯并[d][1,3]間二氧雜環戊烯-5-基)環丙烷甲醯胺)-3-甲基吡啶-2-基)苯甲酸(化合物IV
)。在一些實施例中,化合物II
及/或化合物III
呈固體分散體之形式。
本申請案主張2019年8月14日申請之美國臨時申請案第62/886,511號之優先權,該申請案以引用方式整體併入本文。定義
如本文所用,「化合物II
」係指(R
)-1-(2,2-二氟苯并[d][1,3]間二氧雜環戊烯-5-基)-N-
(1-(2,3-二羥基丙基)-6-氟-2-(1-羥基-2-甲基丙烷-2-基)-1H-吲哚-5-基)環丙烷甲醯胺,其可經以下結構描繪: II
。
化合物II
可呈醫藥學上可接受之鹽之形式。化合物II以及製備及使用化合物II之方法揭示於WO 2010/053471、WO 2011/119984、及WO 2015/160787,該等專利各自以引用方式併入本文。
如貫穿本揭示案所用,「化合物III
」係指N-
(5-羥基-2,4-二-三級丁基
-苯基)-4-側氧基-1H-喹啉-3-甲醯胺,其經以下結構描繪: III
。
化合物III
亦可呈醫藥學上可接受之鹽之形式。化合物III
以及製備及使用化合物III
之方法揭示於WO 2006/002421、WO 2007/ 079139、及WO 2010/019239中,該等專利各自以引用方式併入本文中。
在一些實施例中,化合物III
之氘化衍生物(化合物III-d
)用於本文揭示之組成物及方法中。化合物III-d
之化學名稱為N-
(2-(三級丁基
)-5-羥基-4-(2-(甲基-d3)丙烷-2-基-1,1,1,3,3,3-d6)苯基)-4-側氧基-1,4-二氫喹啉-3-甲醯胺,如由以下結構描繪: III-d
。
化合物III-d
可呈醫藥學上可接受之鹽之形式。化合物III-d
以及製備及使用化合物III-d
之方法揭示於WO 2012/158885及WO 2014/078842中,該等專利以引用方式併入本文。
如本文所用,「化合物IV
」係指3-(6-(1-(2,2-二氟苯并[d][1,3]間二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸,其經以下化學結構描繪: IV
。
化合物IV
可呈醫藥學上可接受之鹽之形式。化合物IV
以及製備及使用化合物IV
之方法揭示於WO 2007/056341、WO 2009/073757、及WO 2009/076142中,該等專利以引用方式併入本文。
如本文所用,「-Si(R)3
基團」、「-Si(R)2
(OR)基團」、及「-Si(R)(OR)2
基團」係指具有三個取代基之單價基團,其中「-」符號表示自矽原子到化合物之連接點。
如本文所用,「>Si(R)2
基團」及「>Si(R)(OR)基團」係指具有兩個取代基之二價基團,其中「>」符號表示自矽原子到化合物之兩個連接點。
如本文所用,「≡Si(R)基團」及「≡Si(OR)基團」係指具有一個取代基之三價基團,且「≡」符號表示自矽原子到化合物之三個連接點。
如本文所用,「-Ge(R)3
基團」、「-Ge(R)2
(OR)基團」、及「-Ge(R)(OR)2
基團」係指具有三個取代基之單價基團,其中「-」符號表示自矽原子到化合物之連接點。
如本文所用,「>Ge(R)2
基團」及「>Ge(R)(OR)基團」係指具有兩個取代基之二價基團,其中「>」符號表示自矽原子到化合物之兩個連接點。
如本文所用,「≡Ge(R)基團」及「≡Ge(OR)基團」係指具有一個取代基之三價基團,且「≡」符號表示自矽原子到化合物之三個連接點。
如本文所用,「化合物(1-1)-(1-11)」係指化合物(1-1)、(1-2)、(1-3)、(1-4)、(1- 5)、(1-6)、(1-7)、(1-8)、(1-9)、(1-10)及(1-11)中之各者。
如本文所用,術語「烷基」係指含有碳原子(例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20個碳原子)之飽和支鏈或非支鏈脂族烴。烷基可經取代或未經取代。
如本文所用,術語「鹵烷基」係指經一或多個鹵素原子取代之烷基。
如本文所用,術語「烷氧基」係指共價鍵合至氧原子之烷基或環烷基。烷氧基可經取代或未經取代。
如本文所用,「環烷基」係指具有3至12個碳(例如3-10個碳)之環狀、雙環、三環或多環非芳族烴基。「環烷基」基團包括單環、雙環、三環、橋連環、稠合環及螺環,包括單螺環及雙螺環。環烷基之非限制性實例為環丙基、環丁基、環戊基、環己基、金剛烷基、降莰基、及雙螺[2.0.2.1]庚烷。環烷基可經取代或未經取代。
「經取代」表示「經取代」基團中至少一個氫經取代基取代。除非另有說明,否則「視情況經取代之」基團可在該基團之各可取代位置處具有合適取代基,且當任何給定結構中之多於一個位置可經選自指定基團之多於一個取代基取代時,在各位置處該取代基可以相同或不同。合適取代基為不消除未經取代化合物之CFTR調節活性的基團。
如本文所用,「氘化衍生物」係指相同化學結構,但其中一或多個氫原子經氘原子置換。
如本文所用,「CFTR」係指囊腫纖維化跨膜傳導調節蛋白。
如本文所用,術語「CFTR調節劑」係指增加CFTR活性之化合物。由CFTR調節劑引起之活性增加包括但不限於校正、增強、穩定及/或放大CFTR之化合物。
如本文所用,術語「 CFTR校正劑」係指促進CFTR加工及運送以增加細胞表面之CFTR量的化合物。本文揭示之新穎化合物為CFTR校正劑。
如本文所用,術語「CFTR增強劑」係指增加位於細胞表面之CFTR蛋白之通道活性,從而導致離子運輸增強之化合物。本文揭示之化合物III
及III-d
為CFTR增強劑之實例。應當理解,當本文提供選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物與其他指定CFTR調節劑之組合的描述時,結合該組合提及「化合物III
或III-d
」意指化合物III
或化合物III-d
(但並非二者)包括於該組合中。
如本文所用,術語「活性醫藥成分」或「治療劑」(「 API」)係指生物活性化合物。
術語「患者」及「受試者」可互換使用,且係指包括人類在內之動物。
術語「有效劑量」及「有效量」在本文中可互換使用,且係指化合物對所投與者產生期望效應(例如,改善CF或CF症狀或減輕CF或CF症狀之嚴重性)之量。有效劑量之確切量將取決於治療目的,且將由熟悉此項技藝者使用已知技術來確定(參見例如Lloyd (1999) The Art, Science and Technology of Pharmaceutical Compounding)。
如本文所用,術語「治療(treatment/treating)」等通常意指在受試者中改善CF或其症狀或減輕CF或其症狀之嚴重性。如本文所用,「治療」包括但不限於以下:受試者之生長增加、體重增量增加、肺黏液減少、胰臟及/或肝功能改善、胸部感染減少、及/或咳嗽或呼吸急促減少。根據此項技術已知之標準方法及技術,可以容易評估該等症狀中任一者之嚴重性的改善或減輕。
如本文所用,術語「與……組合」當提及兩種或更多種化合物、劑或額外活性醫藥成分時,意指在彼此之前、同時、或之後向患者投與該兩種或更多種化合物、劑或額外活性醫藥成分。
當與組成物或劑型之成分之劑量、量或重量百分比結合使用時,術語「約」及「大約」包括熟悉此項技藝者公認之指定劑量、量或重量百分比之值或劑量、量或重量百分比之範圍,以提供等效於由該指定劑量、量或重量百分比獲得之藥理學作用。術語「約」及「大約」可係指由熟悉此項技藝者確定之特定值的可接受誤差,其部分取決於如何量測或確定該等值。在一些實施例中,術語「約」及「大約」意指在給定值或範圍之20%、15%、10%、5%、4%、3%、2%、1%、或0.5%內。
如本文所用,術語「溶劑」係指產物至少部分可溶解於其中之任何液體(產物溶解度>1 g/l)。
如本文所用,術語「室溫」或「環境溫度」係指15℃至30℃。
應當理解,本發明之某些化合物可以作為單獨的立體異構物或鏡像異構物及/或彼等立體異構物或鏡像異構物之混合物存在。
即使僅描述了單個互變異構結構,但本文揭示之某些化合物可以作為互變異構物存在,且兩種互變異構形式均為預期的。例如,對化合物A之描述應理解為包括其互變異構物化合物B,且反之亦然,以及其混合物:
如本文所用,「最小功能(MF)突變」係指與最小CFTR功能相關之CFTR基因突變(幾乎無功能性CFTR蛋白),且包括例如與CFTR通道打開及關閉之能力嚴重缺陷相關之突變,這稱為缺陷型通道閘控或「閘控突變」;與CFTR細胞加工及其向細胞表面之傳遞中之嚴重缺陷相關之突變;與無(或最少)CFTR合成相關之突變;及與通道傳導之嚴重缺陷相關之突變。
如本文所用,術語「醫藥學上可接受之鹽」係指本揭示案之化合物之鹽形式,其中該鹽為無毒的。本揭示案之化合物之醫藥學上可接受之鹽包括衍生自合適無機及有機酸及鹼之彼等鹽。化合物之「遊離鹼」形式不含有離子鍵合之鹽。
關於本發明之一或多種化合物或式,片語「及其醫藥學上可接受之鹽及氘化衍生物」可與「及其醫藥學上可接受之鹽及前述任一者之氘化衍生物」互換使用。該等片語旨在涵蓋任一種參考化合物之醫藥學上可接受之鹽、任一種參考化合物之氘化衍生物、及彼等氘化衍生物之醫藥學上可接受之鹽。
此項技術普通技術人員將認識到,當揭示「一種化合物或其醫藥學上可接受之鹽」之量時,該化合物之醫藥學上可接受之鹽形式之量為等效於該化合物之該遊離鹼之濃度的量。注意,本文揭示之化合物或其醫藥學上可接受之鹽的量係基於其遊離鹼形式。
合適的醫藥學上可接受之鹽為例如S. M. Berge,等人 . J. Pharmaceutical Sciences
, 1977,66
, 1-19中所揭示之彼等者。例如,該文章之表1提供以下醫藥學上可接受之鹽:表 1:
| 乙酸鹽 | 碘化物 | 苄乙二胺青黴素 |
| 苯磺酸鹽 | 羥乙基磺酸鹽 | 氯普魯卡因 |
| 苯甲酸鹽 | 乳酸鹽 | 膽鹼 |
| 碳酸氫鹽 | 乳糖酸鹽 | 二乙醇胺 |
| 酒石酸氫鹽 | 蘋果酸鹽 | 乙二胺 |
| 溴化物 | 順丁烯二酸鹽 | 葡甲胺 |
| 依地酸鈣 | 苯乙醇酸鹽 | 普魯卡因 |
| 樟腦酸酯 | 甲磺酸酯 | 鋁 |
| 碳酸鹽 | 甲基溴化物 | 鈣 |
| 氯化物 | 甲硝酸鹽 | 鋰 |
| 檸檬酸鹽 | 甲硫酸鹽 | 鎂 |
| 二鹽酸鹽 | 黏酸鹽 | 鉀 |
| 依地酸鹽 | 萘磺酸鹽 | 鈉 |
| 乙二磺酸鹽 | 硝酸鹽 | 鋅 |
| 硫酸月桂酯鹽 | 雙羥萘酸鹽(思波酸鹽) | |
| 乙磺酸鹽 | 泛酸鹽 | |
| 反丁烯二酸鹽 | 磷酸鹽/磷酸氫鹽 | |
| 葡庚糖酸鹽 | 聚半乳醣醛酸鹽 | |
| 葡萄糖酸鹽 | 柳酸鹽 | |
| 麩胺酸鹽 | 硬脂酸鹽 | |
| 對羥乙醯胺基苯砷酸鹽(Glycollylarsanilate) | 亞乙酸鹽 | |
| 己基間苯二酸鹽 | 琥珀酸鹽 | |
| 海巴明(Hydrabamine) | 硫酸鹽 | |
| 氫溴酸鹽 | 鞣酸鹽 | |
| 鹽酸鹽 | 酒石酸鹽 | |
| 羥萘甲酸鹽 | 8-氯茶鹼鹽(Teociate) | |
| 三乙基碘 |
醫藥學上可接受之酸加成鹽之非限制性實例包括:以無機酸例如鹽酸、氫溴酸、磷酸、硫酸或過氯酸形成之鹽;以有機酸例如乙酸、草酸、順丁烯二酸、酒石酸、檸檬酸、琥珀酸或丙二酸形成之鹽;及藉由使用此項技術中所用之其他方法例如離子交換形成之鹽。醫藥學上可接受之鹽之非限制性實例包括己二酸鹽、藻酸鹽、抗壞血酸鹽、天冬胺酸鹽、苯磺酸鹽、苯甲酸鹽、硫酸氫鹽、硼酸鹽、丁酸鹽、樟腦酸鹽、樟腦磺酸鹽、檸檬酸鹽、環戊烷丙酸鹽、二葡萄糖酸鹽、十二烷基硫酸鹽、乙磺酸鹽、甲酸鹽、反丁烯二酸鹽、葡庚糖酸鹽、甘油磷酸鹽、葡萄糖酸鹽、半硫酸鹽、庚酸鹽、己酸鹽、氫碘酸鹽、2-羥基乙磺酸鹽、乳糖醛酸鹽、乳酸鹽、月桂酸鹽、月桂基硫酸鹽、蘋果酸鹽、順丁烯二酸鹽、丙二酸鹽、甲磺酸鹽、2-萘磺酸鹽、菸鹼酸鹽、硝酸鹽、油酸鹽、草酸鹽、棕櫚酸鹽、雙羥萘酸鹽、果膠酯酸鹽、過硫酸鹽、3-苯基丙酸鹽、磷酸鹽、苦味酸鹽、新戊酸鹽、丙酸鹽、硬脂酸鹽、琥珀酸鹽、硫酸鹽、酒石酸鹽、硫氰酸鹽、對甲苯磺酸鹽、十一烷酸鹽及戊酸鹽。衍生自適當鹼之醫藥學上可接受之鹽包括鹼金屬鹽、鹼土金屬鹽、銨鹽及N+
(C1-4
烷基)4
鹽。本揭示案亦設想本文揭示之化合物之任何鹼性含氮基團的四級銨化。鹼金屬鹽及鹼土金屬鹽之合適非限制性實例包括鈉、鋰、鉀、鈣及鎂。醫藥學上可接受之鹽之其他非限制性實例包括使用抗衡離子例如鹵離子、氫氧根、羧酸根、硫酸根、磷酸根、硝酸根、低級烷基磺酸根及芳基磺酸根形成之銨、四級銨及胺陽離子。醫藥學上可接受之鹽之其他合適非限制性實例包括苯磺酸鹽及葡糖胺鹽。治療方法
本文揭示之任何新穎化合物,例如式(1)、(2)、(3)及(4)化合物、化合物(1-1)-(1-11)、化合物(2- 1)、其醫藥學上可接受之鹽、及此類化合物及鹽之氘化衍生物可以充當CFTR調節劑,亦即其調節體內CFTR活性。具有CFTR編碼基因突變之個體可受益於接受CFTR調節劑。CFTR突變可能影響CFTR數量,亦即細胞表面處之CFTR通道數量,或者其可能影響CFTR功能,亦即各通道打開及運輸離子之功能性能力。影響CFTR數量之突變包括導致缺陷型合成(I類缺陷)之突變、導致缺陷型加工及運送(II類缺陷)之突變、導致CFTR合成減少(V類缺陷)之突變、及降低CFTR表面穩定性(VI類缺陷)之突變。影響CFTR功能之突變包括導致缺陷型閘控(III類缺陷)之突變及導致缺陷型傳導(IV類缺陷)之突變。一些CFTR突變表現出多種類別之特徵。CFTR基因中之某些突變會導致囊腫纖維化。
因此,在一些實施例中,本發明提供了治療患者之囊腫纖維化、減輕該囊腫纖維化之嚴重性或對症治療該囊腫纖維化之方法,該方法包括向該患者投與有效量的本文揭示之任何新穎化合物,例如式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、其醫藥學上可接受之鹽、及/或此類化合物及鹽之氘化衍生物,其單獨使用或與另一種活性成分(例如另一種CFTR調節劑)組合使用。在一些實施例中,患者俱有 F508del/最小功能(MF)基因型、F508del/F508del基因型(對於F508del突變為同型接合的)、F508del/閘控基因型、或 F508del/殘餘功能(RF)基因型。在一些實施例中,患者為異型接合的且具有一個F508del突變。在一些實施例中,患者對於N1303K突變為同型接合的。
在一些實施例中,患者為異型接合的,且在一個等位基因上具有F508del突變,且在另一個等位基因上具有選自表2之突變:表 2 : CFTR 突變
| 突變 | ||||||||
| Q2X | L218X | Q525X | R792X | E1104X | ||||
| S4X | Q220X | G542X | E822X | W1145X | ||||
| W19X | Y275X | G550X | W882X | R1158X | ||||
| G27X | C276X | Q552X | W846X | R1162X | ||||
| Q39X | Q290X | R553X | Y849X | S1196X | ||||
| W57X | G330X | E585X | R851X | W1204X | ||||
| E60X | W401X | G673X | Q890X | L1254X | ||||
| R75X | Q414X | Q685X | S912X | S1255X | ||||
| L88X | S434X | R709X | Y913X | W1282X | ||||
| E92X | S466X | K710X | Q1042X | Q1313X | ||||
| Q98X | S489X | Q715X | W1089X | Q1330X | ||||
| Y122X | Q493X | L732X | Y1092X | E1371X | ||||
| E193X | W496X | R764X | W1098X | Q1382X | ||||
| W216X | C524X | R785X | R1102X | Q1411X | ||||
| 185+1G→T | 711+5G→A | 1717-8G→A | 2622+1G→A | 3121-1G→A | ||||
| 296+1G→A | 712-1G→T | 1717-1G→A | 2790-1G→C | 3500-2A→G | ||||
| 296+1G→T | 1248+1G→A | 1811+1G→C | 3040G→C(G970R) | 3600+2insT | ||||
| 405+1G→A | 1249-1G→A | 1811+1.6kbA→G | 3850-1G→A | |||||
| 405+3A→C | 1341+1G→A | 1811+1643G→T | 3120G→A | 4005+1G→A | ||||
| 406-1G→A | 1525-2A→G | 1812-1G→A | 3120+1G→A | 4374+1G→T | ||||
| 621+1G→T | 1525-1G→A | 1898+1G→A | 3121-2A→G | |||||
| 711+1G→T | 1898+1G→C | |||||||
| 182delT | 1078delT | 1677delTA | 2711delT | 3737delA | ||||
| 306insA | 1119delA | 1782delA | 2732insA | 3791delC | ||||
| 306delTAGA | 1138insG | 1824delA | 2869insG | 3821delT | ||||
| 365-366insT | 1154insTC | 1833delT | 2896insAG | 3876delA | ||||
| 394delTT | 1161delC | 2043delG | 2942insT | 3878delG | ||||
| 442delA | 1213delT | 2143delT | 2957delT | 3905insT | ||||
| 444delA | 1259insA | 2183AA→G | 3007delG | 4016insT | ||||
| 457TAT→G | 1288insTA | 2184delA | 3028delA | 4021dupT | ||||
| 541delC | 1343delG | 2184insA | 3171delC | 4022insT | ||||
| 574delA | 1471delA | 2307insA | 3171insC | 4040delA | ||||
| 663delT | 1497delGG | 2347delG | 3271delGG | 4279insA | ||||
| 849delG | 1548delG | 2585delT | 3349insT | 4326delTC | ||||
| 935delA | 1609del CA | 2594delGT | 3659delC | |||||
| CFTRdele1 | CFTRdele16-17b | 1461ins4 | ||||||
| CFTRdele2 | CFTRdele17a,17b | 1924del7 | ||||||
| CFTRdele2,3 | CFTRdele17a-18 | 2055del9→A | ||||||
| CFTRdele2-4 | CFTRdele19 | 2105-2117del13insAGAAA | ||||||
| CFTRdele3-10,14b-16 | CFTRdele19-21 | 2372del8 | ||||||
| CFTRdele4-7 | CFTRdele21 | 2721del11 | ||||||
| CFTRdele4-11 | CFTRdele22-24 | 2991del32 | ||||||
| CFTR50kbdel | CFTRdele22,23 | 3667ins4 | ||||||
| CFTRdup6b-10 | 124del23bp | 4010del4 | ||||||
| CFTRdele11 | 602del14 | 4209TGTT→AA | ||||||
| CFTRdele13,14a | 852del22 | |||||||
| CFTRdele14b-17b | 991del5 | |||||||
| A46D | V520F | Y569D | N1303K | |||||
| G85E | A559T | L1065P | ||||||
| R347P | R560T | R1066C | ||||||
| L467P | R560S | L1077P | ||||||
| I507del | A561E | M1101K | ||||||
在一些實施例中,本揭示案亦係關於使用前述化合物之經同位素標記之化合物或其醫藥學上可接受之鹽的治療方法,其中此類化合物及鹽之式及變量各自且獨立地為如上所述或為上文所述之任何其他實施例,前提條件為其中一或多個原子經原子質量或質量數不同於通常天然存在之原子的原子質量或質量數之原子置換(經同位素標記)。可商購獲得且適用於本揭示案之同位素之實例包括氫、碳、氮、氧、磷、氟及氯之同位素,例如分別為2
H、3
H、13
C、14
C、15
N、18
O、17
O、31
P、32
P、35
S、18
F、及36
Cl。
經同位素標記之化合物及鹽可以多種有益方式使用。其可適合於藥物及/或各種類型之檢定,例如受質組織分佈檢定。例如,由於製備相對簡單且可偵測性優異,經氚(3
H)-及/或碳-14(14
C)標記之化合物特別可用於各種類型之檢定,例如受質組織分佈檢定。例如,經氘(2
H)標記之化合物為治療上可用的,與未經2
H標記之化合物相比具有潛在治療優勢。通常,由於下文所述之動力學同位素效應,與未經同位素標記之化合物及鹽相比,經氘(2
H)標記之化合物及鹽可以具有更高代謝穩定性。更高代謝穩定性直接導致可能需要的更長體內半衰期或更低劑量。經同位素標記之化合物及鹽通常可以藉由執行本文本之合成方案及相關描述、實例部分及製備部分中揭示之程序,用未經同位素標記之反應物置換易獲得之經同位素標記之反應物來製備。
在一些實施例中,經同位素標記之化合物及鹽為經氘(2
H)標記之化合物及鹽。在一些具體實施例中,經同位素標記之化合物及鹽為經氘(2
H)標記的,其中該等化合物及鹽中一或多個氫原子已經氘置換。在化學結構中,氘經表示為「D」。
當發現並開發治療劑時,熟悉此項技藝者試圖優化藥物動力學參數,同時保留期望的體外特性。可以合理地假設許多藥物動力學特性較差之化合物容易發生氧化代謝。
經氘(2
H)標記之化合物及鹽可以藉由主要動力學同位素效應來操控化合物之氧化代謝。主要動力學同位素效應為由同位素核之交換引起之化學反應的速率變化,這又由該同位素交換後形成共價鍵所必需之基態能量變化引起。較重同位素之交換通常會降低化學鍵之基態能量,且因此降低限速鍵斷裂。若沿著多產物反應之配位在鞍點區域內或附近發生鍵斷裂,則可顯著改變產物分佈比。解釋:若氘在不可交換位置處與碳原子鍵合,則速率差kM/
kD
= 2-7為典型的。有關進一步討論,請參見S. L. Harbeson及R. D. Tung,Deuterium In Drug Discovery and Development
, Ann. Rep. Med. Chem.2011
, 46, 403-417,該文獻以引用方式併入本文。
摻入到本揭示案之經同位素標記之化合物及鹽中之同位素(例如氘)的濃度可由同位素濃化因數定義。如本文所用,術語「同位素濃化因數」意指指定同位素之同位素豐度與自然豐度之間的比率。在一些實施例中,若本揭示案之化合物中之取代基經表示為氘,則該化合物對於各指定氘原子之同位素濃化因數為至少3500 (在各指定氘原子處摻入52.5%氘)、至少4000 (摻入60%氘)、至少4500 (摻入67.5%氘)、至少5000 (摻入75%氘)、至少5500 (摻入82.5%氘)、至少6000(摻入90%氘)、至少6333.3 (摻入95%氘)、至少6466.7 (摻入97%氘)、至少6600 (摻入99%氘)、或至少6633.3 (摻入99.5%氘)。組合療法
本文揭示之一個態樣提供使用本文揭示之任何新穎化合物,例如式(1)、(2)、(3)及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、其醫藥學上可接受之鹽、及此類化合物及鹽之氘化衍生物與至少一種額外活性醫藥成分(包括CFTR調節劑)之組合治療囊腫纖維化及其他經CFTR介導之疾病的方法。在一些實施例中,至少一種額外活性醫藥成分選自(a)化合物II
及其醫藥學上可接受之鹽;及(b)化合物III
或化合物III-d
及化合物III
或化合物III-d
之醫藥學上可接受之鹽。因此,在一些實施例中,本文提供之組合療法包含選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物、及至少一種選自化合物II
、(化合物III
或III-d)
、及其醫藥學上可接受之鹽之化合物。在一些實施例中,本文提供之組合療法包含至少一種選自式(1)、(2)、(3)及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物;及至少一種選自(化合物III
或III-d)
、化合物IV
、及/或其醫藥學上可接受之鹽之化合物。
在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、其醫藥學上可接受之鹽及氘化衍生物之化合物與至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物組合投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、其醫藥學上可接受之鹽及氘化衍生物之化合物與至少一種選自化合物III
及其醫藥學上可接受之鹽之化合物組合投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、其醫藥學上可接受之鹽及氘化衍生物之化合物與至少一種選自化合物III-d
及其醫藥學上可接受之鹽之化合物組合投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、其醫藥學上可接受之鹽及氘化衍生物之化合物與化合物II
或其醫藥學上可接受之鹽及至少一種選自化合物III
及其醫藥學上可接受之鹽之化合物組合投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、其醫藥學上可接受之鹽及氘化衍生物之化合物與至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物及至少一種選自化合物III-d
及其醫藥學上可接受之鹽之化合物組合投與。
式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、化合物II
、及化合物III
或III-d
、及其醫藥學上可接受之鹽及氘化衍生物各自可以獨立地每天一次、每天兩次或每天三次地投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物每天一次地投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物每天兩次地投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以及至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物每天一次地投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以及至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物每天兩次地投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以及至少一種選自化合物III
或III-d
及其醫藥學上可接受之鹽之化合物每天一次地投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以及至少一種選自化合物III
或III-d
及其醫藥學上可接受之鹽之化合物每天兩次地投與。
在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物、至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物、及至少一種選自化合物III
或III-d
及其醫藥學上可接受之鹽之化合物每天一次地投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物、至少一次選自化合物III
或III-d
及其醫藥學上可接受之鹽之化合物、及至少一種選自化合物IV
及其醫藥學上可接受之鹽之化合物每天一次地投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物、至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物、及至少一種選自化合物III
或III-d
及其醫藥學上可接受之鹽之化合物每天兩次地投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物、至少一種選自化合物III
或III-d
及其醫藥學上可接受之鹽之化合物、及至少一種選自化合物IV
及其醫藥學上可接受之鹽之化合物每天兩次地投與。
在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以及至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物每天一次地投與,且至少一種選自化合物III-d
及其醫藥學上可接受之鹽之化合物每天兩次地投與。在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以及至少一種選自化合物IV
及其醫藥學上可接受之鹽之化合物每天一次地投與,且至少一種選自化合物III-d
及其醫藥學上可接受之鹽之化合物每天兩次地投與。
式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、化合物II
、(III
或III-d
)、及其醫藥學上可接受之鹽及氘化衍生物可以單一醫藥組成物或單獨的醫藥組成物形式投與。此類醫藥組成物可以每天一次或每天多次,例如每天兩次地投與。如本文所用,片語給定量之API(例如化合物II
、(III
、III-d)
或其醫藥學上可接受之鹽)每天一次或每天兩次地投與意指按照劑量每天一次或兩次地投與該給定量。
在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以第一醫藥組成物投與;至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物以第二醫藥組成物投與;且至少一種選自化合物III
及其醫藥學上可接受之鹽之化合物以第三醫藥組成物投與。
在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以第一醫藥組成物投與;至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物以第二醫藥組成物投與;且至少一種選自化合物III-d
及其醫藥學上可接受之鹽之化合物以第三醫藥組成物投與。
在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以第一醫藥組成物投與;至少一種選自化合物III
或III-d
及其醫藥學上可接受之鹽之化合物以第二醫藥組成物投與;且至少一種選自化合物IV
及其醫藥學上可接受之鹽之化合物以第三醫藥組成物投與。
在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以第一醫藥組成物投與;至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物及至少一種選自化合物III
或III-d
及其醫藥學上可接受之鹽之化合物以第二醫藥組成物投與。在一些實施例中,第二醫藥組成物包含一半日劑量之該至少一種選自化合物III
、III-d
、及其醫藥學上可接受之鹽之化合物,且另一半之該至少一種選自化合物III
、III-d
、及其醫藥學上可接受之鹽之化合物以第三醫藥組成物投與。
在一些實施例中,至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物;至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物及至少一種選自化合物III
、III-d
及其醫藥學上可接受之鹽之化合物以第一醫藥組成物投與。在一些實施例中,第一醫藥組成物每天兩次向患者投與。在一些實施例中,第一醫藥組成物每天一次地投與。在一些實施例中,第一醫藥組成物每天一次地投與,且僅包含化合物III
之第二組成物每天一次地投與。
此項技術已知之任何合適醫藥組成物均可用於式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1 )、化合物II
、化合物III
、化合物III-d
、及其醫藥學上可接受之鹽及氘化衍生物。化合物II
及其醫藥學上可接受之鹽之一些示範性醫藥組成物可見於WO 2011/119984及WO 2014/014841,該等專利案以引用方式併入本文。化合物III
及其醫藥學上可接受之鹽之一些示範性醫藥組成物可見於WO 2007/134279、WO 2010/019239、WO 2011/019413、WO 2012/027731、及WO 2013/130669,且化合物III-d
及其醫藥學上可接受之鹽之一些示範性醫藥組成物可見於US 8,865,902、US 9,181,192、US 9,512,079、WO 2017/053455、及WO 2018/080591,該等專利案均以引用方式併入本文。化合物IV
及其醫藥學上可接受之鹽之一些示範性醫藥組成物可見於WO 2010/037066、WO 2011/127421、及WO 2014/071122,該等專利案均以引用方式併入本文。醫藥組成物
本發明之另一態樣提供了醫藥組成物,其包含至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物( 2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物。在一些實施例中,本發明提供醫藥組成物,其包含至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物與至少一種額外活性醫藥成分之組合。在一些實施例中,至少一種額外活性醫藥成分為CFTR調節劑。在一些實施例中,至少一種額外活性醫藥成分為CFTR校正劑。在一些實施例中,至少一種額外活性醫藥成分為CFTR增強劑。在一些實施例中,醫藥組成物包含至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以及至少兩種額外活性醫藥成分,其中一種為CFTR校正劑且一種為CFTR增效劑。
在一些實施例中,至少一種額外活性醫藥成分選自黏液分解劑、支氣管擴張劑、抗生素、抗感染劑、及抗炎劑。
在一些實施例中,額外治療劑為抗生素。本文可用之示範性抗生素包括妥布黴素(tobramycin)(包括妥布黴素吸入粉末(TIP))、阿奇黴素(azithromycin)、胺曲南(包括胺曲南之氣溶膠化形式)、阿米卡星(amikacin)(包括其脂質體調配物)、環丙沙星(包括適合於藉由吸入投與之調配物)、左氧氟沙星(包括其氣溶膠化調配物)、及兩種抗生素(例如磷黴素及妥布黴素)之組合。
在一些實施例中,額外劑為黏液分解劑。本文可用之示範性黏液分解劑包括Pulmozyme®。
在一些實施例中,額外劑為支氣管擴張劑。示範性支氣管擴張劑包括沙丁胺醇(albuterol)、硫酸間羥異丙腎上腺素(metaprotenerol sulfate)、乙酸吡布特羅(pirbuterol acetate)、沙美特羅(salmeterol)或硫酸特布他林(tetrabuline sulfate)。
在一些實施例中,該額外劑為抗炎劑,亦即可以減輕肺部炎症之劑。本文可用之示範性此類劑包括布洛芬(ibuprofen)、二十二碳六烯酸(DHA)、習多芬(sildenafil)、吸入麩胱甘肽(glutathione)、匹格列酮(pioglitazone)、羥氯奎(hydr氧基chloroquine)、或辛伐他汀(simavastatin)。
在一些實施例中,額外劑為營養劑。示範性營養劑包括胰脂肪酶(胰酶替代物),包括Pancrease®、Pancreacarb®、Ultrase®、或Creon®、Liprotomase® (先前為Trizytek®)、Aquadeks®、或麩胱甘肽吸入劑。在一個實施例中,額外營養劑為胰脂肪酶。
在某些實施例中,本發明提供一種醫藥組成物,其包含至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物以及至少一種醫藥學上可接受之載劑。
在一些實施例中,本發明提供一種醫藥組成物,其包含(a)至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1- 11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物,(b)至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物,及(c)至少一種醫藥學上可接受之載劑。
在一些實施例中,本揭示案提供一種醫藥組成物,其包含(a)至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1- 11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物,(b)至少一種選自化合物III
、III-d
、及其醫藥學上可接受之鹽之化合物,及(c)至少一種醫藥學上可接受之載劑。
在一些實施例中,本揭示案提供一種醫藥組成物,其包含(a)至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1- 11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物,(b)至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物,(c)至少一種選自化合物III
及其醫藥學上可接受之鹽之化合物,及(d)至少一種醫藥學上可接受之載劑。
在一些實施例中,本揭示案提供了醫藥組成物,其包含(a)至少一種選自式(1)、(2)、(3)、及(4)化合物、化合物(1-1)-(1- 11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物,(b)至少一種選自化合物II
及其醫藥學上可接受之鹽之化合物,(c)至少一種選自化合物III-d
及其醫藥學上可接受之鹽之化合物,及(d)至少一種醫藥學上可接受之載劑。
在一些實施例中,本揭示案提供一種醫藥組成物,其包含(a)至少一種選自式(1)、(2)、(3)及(4)化合物、化合物(1-1)-(1-11)、化合物(2-1)、及其醫藥學上可接受之鹽及氘化衍生物之化合物,(b)至少一種選自化合物III
或III-d
及其醫藥學上可接受之鹽之化合物,(c)至少一種選自化合物IV
及其醫藥學上可接受之鹽之化合物,及(d)至少一種醫藥學上可接受之載劑。
本文揭示之任何醫藥組成物可包含至少一種醫藥學上可接受之載劑。在一些實施例中,至少一種醫藥學上可接受之載劑選自醫藥學上可接受之媒劑及醫藥學上可接受之佐劑。在一些實施例中,至少一種醫藥學上可接受之選自醫藥學上可接受之填充劑、崩解劑、界面活性劑、黏合劑、潤滑劑。
本文所述之醫藥組成物可用於治療囊腫纖維化及其他經CFTR介導之疾病。
如上文所述,本文揭示之醫藥組成物可以視情況進一步包含至少一種醫藥學上可接受之載劑。該至少一種醫藥學上可接受之載劑可以選自佐劑及媒劑。如本文所用,至少一種醫藥學上可接受之載劑包括任何及所有溶劑、稀釋劑、其他液體媒劑、分散助劑、懸浮助劑、界面活性劑、等滲劑、增稠劑、乳化劑、防腐劑、固體黏合劑、及潤滑劑,以適合所需特定劑型。Remington:The Science and Practice of Pharmacy
, 第21版, 2005, ed. D.B. Troy, Lippincott Williams & Wilkins, Philadelphia, andEncyclopedia of Pharmaceutical Technology
, 編者J. Swarbrick及J. C. Boylan, 1988-1999, Marcel Dekker, New York揭示了用於調配醫藥組成物之各種載劑及其已知製備技術。除非任何常規載劑與本揭示案之化合物不相容,例如藉由產生任何不期望的生物學效應或以有害方式與醫藥組成物之任何其他組分相互作用,否則預期其用途處於本揭示案之範圍內。合適醫藥學上可接受之載劑之非限制性實例包括但不限於離子交換劑、氧化鋁、硬脂酸鋁、卵磷脂、血清蛋白(例如人血清白蛋白)、緩衝物質(例如磷酸鹽、甘胺酸、山梨酸及山梨酸鉀)、飽和植物脂肪酸、水、鹽、及電解質(例如硫酸魚精蛋白、磷酸氫二鈉、磷酸氫鉀、氯化鈉、及鋅鹽)之部分甘油酯混合物、膠體二氧化矽、三矽酸鎂、聚乙烯吡咯啶酮、聚丙烯酸酯、蠟、聚乙烯-聚氧丙烯嵌段聚合物、羊毛脂、糖(例如乳糖、葡萄糖及蔗糖)、澱粉(例如玉米澱粉及馬鈴薯澱粉)、纖維素及其衍生物(例如羧甲基纖維素鈉、乙基纖維素及乙酸纖維素)、黃芪膠粉、麥芽、明膠、滑石粉、賦形劑(例如可可脂及栓劑蠟)、油類(例如花生油、棉籽油、紅花子油、芝麻油、橄欖油、玉米油及大豆油)、二醇類(例如丙二醇及聚乙二醇)、酯類(例如油酸乙酯及月桂酸乙酯)、瓊脂、緩沖劑(例如氫氧化鎂及氫氧化鋁) 、海藻酸、無熱原水、等滲鹽水、林格氏溶液(Ringer's solution)、乙醇、磷酸鹽緩衝溶液、無毒相容性潤滑劑(諸如月桂硫酸鈉及硬脂酸鎂)、著色劑、脫模劑、塗佈劑、甜味劑、調味劑、加香劑、防腐劑、及抗氧化劑。
額外實施例包括:
1. 一種式(1)化合物:(1),
或其醫藥學上可接受之鹽或氘化衍生物,
其中:
X選自Si(R)3
、-(O)n
-(C1
-C8
烷基)、-(O)n
-(C3
-C10
環烷基),其中:
n為0或1,
各C1
-C8
烷基經0、1、2或3個選自鹵素、羥基、側氧基、C3
-C10
環烷基、C1
-C4
鹵烷基、及Si(R)3
基團之基團取代,
各C3
-C10
環烷基經0、1、2、3或4個選自鹵素、C1
-C4
鹵烷基、C1
-C4
烷基、及Si(R)3
基團之基團取代,且
各C1
-C8
烷基中之一個-CH2-
視情況經-Si(R)2
-置換;
Y選自氫及-Si(R)3
;
各Z獨立地選自-CH2-
及-Si(R)2
-;且
各R獨立地選自苯基及C1
-C6
烷基;且
其中該式(1)化合物含有至少一個Si原子。
2. 如實施例1之化合物,其中X選自Si(R)3
、、、、、、及。
3. 如實施例1或2中任一項之化合物,其中該化合物選自:
及其醫藥學上可接受之鹽及氘化衍生物。
4. 如實施例1至3中任一項之化合物,其中至少一個氫經氘置換。
5. 如實施例1至4中任一項之化合物,其中該化合物為醫藥學上可接受之鹽。
6. 一種式(2)化合物:(2),
或其醫藥學上可接受之鹽或氘化衍生物,
其中:
X選自Ge(R)3
、-(O)n
-(C1
-C8
烷基)、-(O)n
-(C3
-C10
環烷基),其中:
n為0或1,
各C1
-C8
烷基經0、1、2或3個選自鹵素、羥基、側氧基、C3
-C10
環烷基、C1
-C4
鹵烷基、及Ge(R)3
基團之基團取代,
各C3
-C10
環烷基經0、1、2、3或4個選自鹵素、C1
-C4
鹵烷基、C1
-C4
烷基、及Ge(R)3
基團之基團取代,且
各C1
-C8
烷基中之一個-CH2-
視情況經-Ge(R)2
-置換;
Y選自氫及-Ge(R)3
;
各Z獨立地選自-CH2-
及-Ge(R)2
-;且
各R獨立地選自苯基及C1
-C6
烷基;且
其中該式(2)化合物含有至少一個Ge原子。
7. 如實施例6之化合物,其中X選自Ge(R)3
、、、、、、及。
8. 如實施例6或7中任一項之化合物,其中該化合物選自:
及其醫藥學上可接受之鹽及氘化衍生物。
9. 如實施例6至8中任一項之化合物,其中至少一個氫經氘置換。
10. 如實施例6至9中任一項之化合物,其中該化合物為醫藥學上可接受之鹽。
11. 一種式(3)化合物:(3),
或其醫藥學上可接受之鹽或氘化衍生物,其中:
- 式(3)之位置2處之碳原子經矽原子置換;
- 式(3)之位置6及7處之甲基中之至少一者經選自-Si(R)3
基團、-Si(R)2
(OR)基團、及-Si( R)(OR)2
基團之基團置換;
- 式(3)之位置3、5、及8處之亞甲基中之至少一者經選自>Si(R)2
基團及>Si(R)(OR)基團之基團置換;且/或
- 式(3)之位置4處之次甲基經選自≡Si(R)基團及≡Si(OR)基團之基團置換;且
其中各R獨立地選自氫、苯基、及C1
-C6
烷基。
12. 一種式(4)化合物:(4),
或其醫藥學上可接受之鹽或氘化衍生物,其中:
- 式(4)之位置2處之碳原子經鍺原子取代;
- 式(4)之位置6及7處之甲基中之至少一者經選自-Ge(R)3
基團、-Ge(R)2
(OR)基團及-Ge( R)(OR)2
基團之基團置換;
- 式(4)之位置3、5、及8處之亞甲基中之至少一者經選自>Ge(R)2
基團及 >Ge(R)(OR)基團之基團置換;且/或
- 式(4)之位置4處之次甲基經選自≡Ge(R)基團及≡Ge(OR)基團之基團置換;且
其中各R獨立地選自氫、苯基、及C1
-C6
烷基。
13. 如實施例11或12之化合物,其中至少一個氫經氘置換。
14. 如實施例11至13中任一項之化合物,其中該化合物為醫藥學上可接受之鹽。
15. 一種醫藥組成物,其包含如實施例1-14中任一項之化合物及醫藥學上可接受之載劑。
16. 如實施例15之醫藥組成物,其進一步包含一或多種額外治療劑。
17. 如實施例16之醫藥組成物,其中該一或多種額外治療劑包含選自化合物II
、化合物III
、化合物III-d
、及其醫藥學上可接受之鹽的化合物。
18. 如實施例17之醫藥組成物,其中該組成物包含化合物II
及化合物III
。
19. 如實施例17之醫藥組成物,其中該組成物包含化合物II
及化合物III-d
。
20. 一種醫藥組成物,其包含:
(a) 至少一種選自如實施例1-14中任一項之化合物的化合物;
(b) 至少一種醫藥學上可接受之載劑;及
視情況以下一或多者:
(c) (i) 選自化合物II
之化合物:,
及其醫藥學上可接受之鹽及氘化衍生物;及
(ii) 選自化合物III
、化合物III-d
之化合物: III 、 III-d
及其醫藥學上可接受之鹽及氘化衍生物。
21. 一種治療囊腫纖維化之方法,其包含向有需要之患者投與如實施例1-14中任一項之化合物或如實施例15-20中任一項之醫藥組成物。
22. 如實施例21之方法,其進一步包含在該化合物或該醫藥組成物之前、同時、或之後向該患者投與一或多種額外治療劑。
23. 如實施例22之方法,其中該一或多種額外治療劑包含選自化合物II
、化合物III
、化合物III-d
、及其醫藥學上可接受之鹽之化合物。
24. 如實施例23之方法,其中該一或多種額外治療劑包含化合物II
及化合物III
。
25. 如實施例23之方法,其中該一或多種額外治療劑包含化合物II
及化合物III-d
。
26. 如實施例1-14中任一項之化合物或如實施例15-20中任一項之醫藥組成物,其用於治療囊腫纖維化。
27. 如實施例1-14中任一項之化合物或如實施例15-20中任一項之醫藥組成物,其用於製造用於治療囊腫纖維化之藥物。實例 一般實驗程序
| 化合物(1-1) | , |
| 化合物(1-2) | , |
| 化合物(1-3) | , |
| 化合物(1-4) | , |
| 化合物(1-5) | , |
| 化合物(1-6) | , |
| 化合物(1-7) | , |
| 化合物(1-8) | , |
| 化合物(1-9) | , |
| 化合物(1-10) | , |
| 化合物(1-11) | , |
| 化合物(2-1) | , |
化合物II
、III
、III-d
、及IV
可藉由此項技術中之任何合適方法來製備,例如PCT公開案第WO 2011/133751號、第WO 2011/133951號、第WO 2015/160787號/及美國專利第8,865,902號。縮寫列表
ACN:乙腈
Boc酸酐((BOC)2
O):二碳酸二三級
丁酯
CDI:羰基二咪唑
COMU:(1-氰基-2-乙氧基-2-側氧基伸乙基胺基氧基)二甲基胺基-N-嗎啉基六氟磷酸碳
DABCO:1,4-二氮雜雙環[2.2.2]辛烷
DBU:1,8-二氮雜雙環(5.4.0)十一碳-7-烯
DCM:二氯甲烷
DI水:去離子水
DIAD:偶氮二羧酸二異丙酯
DIEA (DIPEA;N,N-
二異丙基乙胺)
DMA:N,N-
二甲基乙醯胺
DMAP:4-二甲基胺基吡啶
DMF:N,N-
二甲基甲醯胺
DMSO:二甲基亞碸
EA:乙酸乙酯
EDC:1-乙基-3-(3-二甲基胺基丙基)碳二亞胺
Et2
O:乙醚
EtOAc:乙酸乙酯
EtOH:乙醇
HATU:1-[雙(二甲基胺基)亞甲基]-1H
-1,2,3-三唑并[4,5-b]吡啶鎓 3-氧化六氟磷酸鹽
HPLC:高效液相層析
HMPA:六甲基磷醯胺
IPA:異丙醇
LAH:氫化鋁鋰
LC:液相層析
LDA:二異丙胺鋰
MeCN:乙腈
MeOH:甲醇
MTBE:甲基三級丁基
醚
MeTHF或2-MeTHF:2-甲基四氫呋喃
NMP:N-
甲基-2-吡咯啶酮
NMM:N-
甲基嗎啉
Pd(dppf)Cl2:[1,1′-雙(二苯基膦基)二茂鐵]二氯鈀(II)
PTFE:聚四氟乙烯
Rpm:每分鐘轉數
rt:室溫
SFC:超臨界流體層析
TBS-Cl:三級丁基二甲基氯矽烷
TEA:三乙胺
TFA:三氟乙酸
THF:四氫呋喃
TMS:三甲基矽基
TMSCl:三甲基氯矽烷
TPPO-DIAD複合物:三苯基氧化膦與偶氮二羧酸二異丙酯之複合物
對-TsOH:對-甲苯磺酸
UPLC:超高效液相層析用於合成通用中間物之程序
除非另有說明,否則藉由商業來源獲得試劑及起始原料,且無需純化即使用。
在以分別為400及100 MHz之1
H及13
C共振頻率運行之Bruker Biospin DRX 400 MHz FTNMR譜儀上或在300 MHz NMR譜儀上採集質子及碳NMR譜。使用寬帶觀測(BBFO)探針以分別為0.1834及0.9083 Hz/Pt數字分辨率之20 Hz樣品旋轉採集一維質子及碳譜。使用標準、先前公佈之脈衝序列及常規處理參數在30℃下在溫度控制下採集所有質子及碳譜。
亦在分別以400 MHz及100 MHz運行且配備有5 mm多核Iprobe之Bruker AVNEO 400 MHz光譜儀上記錄NMR (1D & 2D)譜。
亦在Varian Mercury NMR儀器上使用45度脈衝角度、4800 Hz譜寬及28860採集點在300 MHz下記錄1
H之NMR譜。將FID零填充到32k點,且在傅立葉轉換前應用0.3Hz譜線增寬。使用30度脈衝角度、100 kHz譜寬及所採集之59202點在282 MHz處記錄19
F NMR譜。將FID零填充到64k點,且在傅立葉轉換前應用0.5 Hz譜線增寬。
亦在Bruker Avance III HD NMR儀器上使用30度脈衝角度、8000 Hz譜寬及128k採集點在400 MHz下記錄1
H之NMR譜。將FID零填充到256k點,且在傅立葉轉換前應用0.3Hz譜線增寬。使用30度脈衝角度、89286 Hz譜寬及所採集之128k點在377 MHz處記錄19
F NMR譜。將FID零填充到256k點,且在傅立葉轉換前應用0.3 Hz譜線增寬。
亦在配備有5mm QNP(H1/C13/F19/P31)探針(類型:250-SB, s#23055/0020)之Bruker AC 250MHz儀器上或在配備有ID PFG, 5 mm, 50-202/500 MHz探針(型號/零件號99337300)之Varian 500MHz儀器上記錄NMR譜。
藉由使用由Waters (pn: 186002350)製成之Acquity UPLC BEH C18
管柱(50 × 2.1 mm, 1.7 μm particle)及在3.0分鐘內自1-99%流動相B之雙重梯度運行來進行反相UPLC,確定化合物之最終純度。流動相A = H2
O (0.05% CF3
CO2
H)。流動相B = CH3
CN (0.035% CF3
CO2
H)。流速 = 1.2 mL/min,注入體積 = 1.5 μL,且管柱溫度 = 60℃。藉由對兩個UV跡線(220 nm、254 nm)之曲線下面積(AUC)取平均值來計算最終純度。低分辨率質譜經報告為使用配備有電噴霧電離(ESI)源之單四極桿質譜儀獲得之[M+1]+
種類,該電噴霧電離源能夠在整個偵測範圍內達成0.1 Da質量精度及1000最小分辨率(無分辨率單位)。使用對掌性氣相層析(GC)分析在Agilent 7890A/MSD 5975C上使用Restek Rt-βDEXcst (30 m x 0.25 mm x 0.25 µm_df)管柱以2.0 mL/min流速(H2
載氣)在220℃注入溫度及120℃烘箱溫度下15分鐘來確定(2S
)-2,4-二甲基-4-硝基-戊酸甲酯之光學純度。實例 1 : 3- 側氧基 -2,3- 二氫 -1H- 吡唑 -1- 甲酸三級
丁酯之合成
啟動50 L反應器,且將夾套設定在20℃,在150 rpm、回流冷凝器(10℃)及氮氣吹掃下攪拌。添加MeOH(2.860 L)及(E
)-3-甲氧基丙-2-烯酸甲酯(2.643 kg,22.76 mol),且將反應器加蓋。將反應物加熱至40℃內部溫度,且設定系統以將夾套溫度保持在40℃。在30 min內經由加料漏斗分批添加水合肼(1300 g 55%w/w,22.31 mol)。將反應物加熱至60℃達1 h。將反應混合物冷卻至20℃且分批添加三乙胺(2.483 kg,3.420 L,24.54 mol),保持反應溫度 < 30℃。分批添加Boc酸酐(二碳酸二-三級
丁酯) (4.967 kg,5.228 L,22.76 mol)於MeOH(2.860 L)中之溶液,保持溫度< 45℃。將反應混合物在20℃下攪拌16 h。將反應溶液部分濃縮,以去除MeOH,得到澄清淺琥珀色油狀物。將所得油狀物轉移到50 L反應器中,攪拌且添加水(7.150 L)及庚烷(7.150 L)。該等添加使少量產物沉澱。將水層排入乾淨容器中,且過濾界面及庚烷層,以分離出固體(產物)。將水層轉移回到反應器中,且將所收集之固體放回反應器中並將其與水層混合。將滴液漏斗添加至反應器中,且裝載乙酸(1.474 kg,1.396 L,24.54 mol),並逐滴添加。將該夾套設定為0℃,以吸收淬火放熱。添加完成後(pH=5),將反應混合物攪拌1 h。藉由過濾收集固體,且用水(7.150 L)洗滌,並再次用水(3.575 L)洗滌。將結晶固體轉移至20 L旋轉蒸發儀球罐中,並添加庚烷(7.150 L)。將混合物在45℃下漿化30 min,且蒸餾出1-2體積的溶劑。過濾旋轉蒸發燒瓶中之漿液,且將固體用庚烷(3.575 L)洗滌。在真空
(50℃,15 mbar)中進一步乾燥該固體,得到呈粗結晶固體之5-側氧基-1H-
吡唑-2-羧酸三級
丁酯(2921 g,71%)。1
HNMR (400 MHz, DMSO-d6
) δ 10.95 (s, 1H), 7.98 (d,J
= 2.9 Hz, 1H), 5.90 (d,J
= 2.9 Hz, 1H), 1.54 (s, 9H)。實例 2 : (14S)-8- 溴 -12,12- 二甲基 -2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(23),5(10),6,8,19,21- 己烯 -2,2,4- 三酮之合成 步驟 1 : 6- 溴 -2- 氟 - 吡啶 -3- 甲醯胺
向6-溴-2-氟-吡啶-3-羧酸(24.7 g,106.66 mmol)及Boc2
O(33 g,146.67 mmol)於2-MeTHF(250 mL)中之溶液中添加NMM(13.80 g,15 mL,136.44 mmol)。將混合物在室溫下攪拌30 min,且然後添加NH4
HCO3
(15 g,189.74 mmol)。將反應混合物在室溫下攪拌20小時。添加水(200 mL)及EtOAc(100 mL),且將混合物攪拌10 min。分離該兩個相。將有機層用飽和碳酸氫鈉、鹽水洗滌,經硫酸鈉乾燥且濃縮,得到呈淺色固體之6-溴-2-氟-吡啶-3-甲醯胺(23.5 g,96%)。ESI-MS m/z計算值217.9491,實測值219.3 (M+1)+
;保留時間:2.33分鐘(LC方法B)。步驟 2 : 3- 亞甲基四氫 -2H- 哌喃 -2- 酮
階段1
為5 L 3頸RB燒瓶裝備機械攪拌器、加熱套、加料漏斗、J-Kem溫度探針/控制器及氮氣入口/出口。在氮氣氛下,向容器中裝入氫化鈉(59.91 g 60%w/w,1.498 mol),然後裝入庚烷(1.5 L),這提供灰色懸浮液。開始攪拌,且在19℃下記錄鍋溫。然後,向容器中裝入經由注射器添加之乙醇(3.451 g,74.91 mmol),這導致氣體逸出。向加料漏斗中裝入四氫哌喃-2-酮(150 g,1.498 mol)及甲酸乙酯(111 g,1.50 mol)之澄清淡黃色溶液。在1小時內逐滴添加該溶液,這導致氣體放出且逐漸放熱至45℃。然後將所得濃稠白色懸浮液加熱至65℃達2小時,且然後使其冷卻至室溫。將混合物在室溫下繼續攪拌隔夜(約10小時)。在氮氣流下透過玻璃膠布氏漏斗(Buchner funnel) (中等孔隙率)真空過濾反應混合物。將濾餅用庚烷(2×250 mL)置換洗滌且吸拉幾分鐘。將稍微帶有庚烷之濕濾餅轉移到玻璃托盤,且在45℃真空烘箱中乾燥15小時,得到呈白色固體(205 g,1.36 mol,91%產率)之所要產物(E)-(2-側氧基四氫哌喃-3-亞基)甲醇鹽。1
HNMR (400 MHz, DMSO-d6
) δ 8.99 (s, 1H), 3.90- 3.83 (m, 2H), 2.09 (t, J = 6.3 Hz, 2H), 1.57 (qd, J = 6.4, 4.7 Hz, 2H)。
階段2
為5 L 3頸RB燒瓶裝備機械攪拌器、加熱套、加料漏斗、J-Kem溫度探針/控制器及氮氣入口/出口。在氮氣氛下向容器中裝入(E)-(2-側氧基四氫哌喃-3-亞基)甲醇鹽-Na鹽(205 g,1.366 mol)及四氫呋喃(1640 mL),得到白色懸浮液。開始攪拌,且在19℃下記錄鍋溫。然後向該容器中裝入作為固體一次性添加之多聚甲醛(136.6 g,4.549 mol)。將所得懸浮液加熱至63℃,且將該條件保持15小時。加熱後,反應混合物變得略微凝膠狀。將白色凝膠狀混合物在減壓下濃縮,以去除大部分四氫呋喃。將剩餘殘餘物在分液漏斗中分配於乙酸乙酯(1000 mL)、飽和氯化鈉(500 mL)及飽和碳酸氫鈉(500 mL)中。去除有機物,且將殘餘水層用乙酸乙酯(5×300mL)萃取。將經合併之有機物經硫酸鈉(500g)乾燥,且然後透過具有20 mm矽藻土層之玻璃膠布氏漏斗真空過濾。將濾餅用乙酸乙酯(250 mL)置換洗滌。將澄清濾液在減壓下濃縮,得到呈澄清淡黃色油狀物(135 g)之所要粗產物。藉由在1小時內以100%己烷至己烷中之60%乙酸乙酯之梯度溶離在Isco (1500g RediSep管柱)上進行矽膠管柱急速層析(液體負載)來純化該材料,收集450 mL流份。藉由在以3:1 Hex/EtOAc溶離之矽膠上進行TLC分析來偵測產物,且在紫外線下可視化。將產物流份合併且在減壓下濃縮,提供呈澄清無色油狀物(132 g,1.18mol,86%產率)之所要產物3-亞甲基四氫哌喃-2-酮。1
HNMR (400 MHz, DMSO-d6
) δ 6.18 (q, J = 1.9 Hz, 1H), 5.60 (q, J = 1.9 Hz, 1H), 4.40- 4.26 (m, 2H), 2.61 (ddt, J = 7.0, 6.3, 2.0 Hz, 2H), 1.90- 1.75 (m, 2H)。質子NMR指示約16重量%之殘留乙酸乙酯。經校正產率為:(100-16 = 84) 0.84 (132) = 110.9 g (72%產率)。步驟 3 : 3-(2- 甲基 -2- 硝基丙基 ) 四氫 -2H- 哌喃 -2- 酮
為5000 mL 3頸RB燒瓶裝備機械攪拌器、用作次級容器之冷卻浴、J-Kem溫度探針、加料漏斗及氮氣入口/出口。在氮氣氛下,向容器中裝入2-硝基丙烷(104.9 g,1.177 mol)。開始攪拌且在19℃下記錄鍋溫。然後向容器中裝入一次性添加之純淨的1,8-二氮雜雙環[5.4.0]十一碳-7-烯 (22.41 g,147.2 mmol),得到澄清淺黃色溶液。未觀測到放熱。向加料漏斗中裝入3-亞甲基四氫哌喃-2-酮(110 g,981.0 mmol)於乙腈(1100 mL)中之溶液,在1小時內逐滴添加該溶液,得到澄清淺黃色溶液,且逐漸放熱至24℃。將反應混合物在室溫下繼續攪拌3.5小時,且然後在減壓下濃縮。將剩餘殘留物溶解於二氯甲烷(1000 mL)中,且分配於500 mL 1莫耳檸檬酸溶液/飽和氯化鈉溶液之3:2混合物中。去除有機相,且將殘餘水相用二氯甲烷(300 mL)萃取。將經合併之有機相用飽和氯化鈉溶液(300 mL)洗滌,經硫酸鈉(250 g)乾燥,且然後透過玻璃膠布氏漏斗過濾。在減壓下濃縮濾液至約200 mL體積。將澄清淡藍色二氯甲烷溶液用甲基三級
丁基醚(1500 mL)稀釋,且將渾濁溶液在減壓下濃縮至約200 mL體積,從而提供懸浮液。將混合物再次用甲基三級
丁基醚(1500 mL)稀釋,且在減壓下濃縮至約250 mL體積。將所得懸浮液在室溫下靜置隔夜(約12小時)。藉由在玻璃膠布氏漏斗中真空過濾來收集固體,且將濾餅用冷的甲基三級
丁基醚(2×150 mL)置換洗滌,且然後吸拉30分鐘。將該材料在45℃真空烘箱中進一步乾燥5小時,以提供呈白色固體(160 g,0.795 mol,81%產率)之所要產物3-(2-甲基-2-硝基-丙基)四氫哌喃-2-酮。1
HNMR (400 MHz, DMSO-d6
) δ 4.34 (ddd, J = 11.1, 9.3, 4.3 Hz, 1H), 4.20 (dt, J = 11.1, 5.1 Hz, 1H), 2.75- 2.62 (m, 1H), 2.56 (dd, J = 14.9, 5.2 Hz, 1H), 2.01- 1.89 (m, 2H), 1.89- 1.67 (m, 2H), 1.55 (d, J = 6.0 Hz, 6H), 1.44 (dddd, J = 12.8, 11.5, 8.1, 6.6 Hz, 1H)。ESI-MS m/z計算值201.10011,實測值202.0 (M+1)+
;保留時間:0.97分鐘,呈灰白色固體。步驟 4 :外消旋 3-(2- 甲基 -2- 硝基丙基 ) 四氫 -2H- 哌喃 -2- 酮之對掌性分離
將外消旋3-(2-甲基-2-硝基丙基)四氫-2H-哌喃-2-酮以MeOH/ACN 70/30 v/v溶解至80 g/L +/- 8 g/L (目標80 +/- 2 g/L)且在作為固定相之Chiralpak AD 20 μm上使用MeOH/ACN 70/30 v/v作為流動相進行分離。(S
)-3-(2-甲基-2-硝基丙基)四氫-2H-哌喃-2-酮為峰2。步驟 5 : (S)- 3-(3- 羥丙基 )-5,5- 二甲基吡咯啶 -2- 酮
將雷氏鎳(Raney Nickel) 2400 (77重量%,2.8 kg)之懸浮液沉降2天。將靜置液體傾析到廢物中,且藉助於水(2.6 kg)將剩餘催化劑裝入反應器中,然後用N2
脫氣。在第二反應器中,將(S
)-3-(2-甲基-2-硝基丙基)四氫-2H-哌喃-2-酮(13.9 kg)及EtOH(170.8 kg)之混合物加熱至30℃,然後用N2
脫氣,然後將其轉移到含有雷氏鎳之反應器中。藉助於EtOH (29.8 kg)沖洗完成轉移。將該混合物用氮氣吹掃三次,且用氫氣吹掃三次。將反應器內容物加熱至60℃-65℃,且在H2
(4-8 psig)下攪拌,直到反應完成(18 h)。將混合物冷卻至15℃-20℃,然後用氮氣吹掃三次,然後透過以EtOH(3.2 kg)潤溼之矽藻土墊(3.0 kg)過濾。將反應器及矽藻土餅用EtOH (2×14.0 kg)洗滌。將濾液蒸餾至約25 L最終體積,然後將其加熱至45℃。然後裝入MTBE (269.4 kg),保持48℃-50℃,且然後在環境壓力、48℃-55℃下蒸餾至約30 L最終體積。依次添加另外兩份MTBE (269.4 kg,然後187.4 kg),然後濃縮至約30 L體積。
在40℃下用(S
)-3-(3-羥丙基)-5,5-二甲基吡咯啶-2-酮(70.1g)接種反應器內容物。在3.5 h時段內將經接種晶體漿液冷卻至15℃,然後在12℃-15℃之間攪拌16.5 h 30 min,然後過濾。然後將反應器及濾餅用冷(-2℃至-10℃) MTBE(2×10 kg)洗滌。將濾餅乾燥至恆重,得到呈白色結晶固體之(S
)-3-(3-羥丙基)-5,5-二甲基吡咯啶二-2-酮(10.4 kg;88%)。(S)-3-(3- 羥丙基 )-5,5- 二甲基吡咯啶 -2- 酮之再結晶
將(S
)-3-(3-羥丙基)-5,5-二甲基吡咯啶-2-酮(10.3 kg)及DCM(28.2 kg)之混合物攪拌且加熱至25℃達2 h,然後透過管線過濾器(45 um)將其轉移至另一反應器中。在21℃下將初始反應器用DCM (6.8 kg)沖洗10 min,然後透過管線過濾器將其轉移到反應器中。在25℃-30℃下向溶液中裝入MTBE (38.1 kg),然後在35℃-52℃、大氣壓下在2.5 h時段內蒸餾該混合物至約30 L最終體積。在45℃-50℃下向反應器中裝入約30 L. MTBE (38.2 kg)。將所得懸浮液在3.25 h時段內在49℃-55℃、大氣壓下蒸餾至約30 L最終體積。將反應器內容物在2.5 h時段內冷卻至21℃,且在20℃下攪拌16 h。過濾懸浮液。將反應器及濾餅用MTBE (7.7 kg,0℃)沖洗。將濾餅乾燥2天時段。產量:9.1 kg (88.3%)灰白色固體。步驟 6: (S
)-3-(5,5- 二甲基吡咯啶 -3- 基 ) 丙 -1- 醇
將LAH團粒(332.5 g,8.760 mol,1.50當量)在30℃-40℃下緩慢添加到反應器之2-MeTHF(10.00 L,10體積)中。然後將混合物加熱至75℃。在單獨反應器中製備(S
)-3-(3-羥丙基)-5,5-二甲基吡咯啶-2-酮(1,000 g,5.840 mol,1.00當量)及2-MeTHF (10.00 L,10體積)之混合物且將其加熱至65℃,然後在2 h內小心轉移至含有LAH混合物之反應器中。將混合物在70℃下攪拌,直至反應完成(18-24小時),然後冷卻至0-10℃。然後小心地添加水(400.0 mL,1 x LAH wt),同時保持混合物溫度< 30℃。然後添加15% NaOH水溶液(400.0 mL,1x LAH wt),然後添加水(400.0 mL,1x LAH wt),同時保持混合物溫度< 30℃。然後將所得混合物加熱至60℃且在該溫度下保持至少30分鐘。將混合物冷卻至20℃-30℃,然後添加矽藻土(200克,20重量%)。然後透過矽藻土墊過濾混合物。將反應器及濾餅用2-MeTHF(4.0 L,4.0體積)沖洗。在真空下濃縮濾液,得到呈澄清油狀物之(S
)-3-(5,5-二甲基吡咯啶-3-基)丙-1-醇(872 g;94.95%產率)。步驟 7 : (S
)-6- 溴 -2-(4-(3- 羥丙基 )-2,2- 二甲基吡咯啶 -1- 基 ) 菸鹼醯胺
攪拌(S
)-3-(5,5-二甲基吡咯啶-3-基)丙-1-醇(2325 g,14.8 mol)及6-溴-2-氟菸鹼醯胺(3400 g,15.5 mol)於2-甲基四氫呋喃(23 L)中之混合物,然後添加碳酸鉀(2650 g,19.2 mol)及去離子水(7 L)。將混合物在25℃下攪拌,直至反應完成(16 h)。去除水相,且將上層有機相用水(7 L)及2%氯化鈉水溶液(7 L)洗滌。將有機層在減壓下濃縮至約19L。藉由乙腈(2×20 L)之兩次連續添加及濃縮,接著蒸餾來自混合物中驅除2-甲基四氫呋喃。向剩餘溶液中添加乙腈(20 L),且將反應物升溫至85℃達2 h,且然後以10℃/h冷卻至25℃。將漿液冷卻至10℃且攪拌4 h,然後過濾。將濾餅用乙腈(2×3 L)沖洗兩次,然後將固體在真空下乾燥,得到呈結晶性白色固體之(S
)-6-溴-2-(4-(3-羥丙基)-2,2-二甲基吡咯啶-1-基)菸鹼醯胺(3850 g,73%產率)。步驟 8 : (S
)-6- 溴 -2-(4-(3-(1,3- 二側氧基異吲哚啉 -2- 基 ) 丙基 )-2,2- 二甲基吡咯啶 -1- 基 ) 菸鹼醯胺之合成
將(S
)-6-溴-2-(4-(3-羥丙基)-2,2-二甲基吡咯啶-1-基)菸鹼醯胺(2.65 kg,7.4 mol)、2-甲基四氫呋喃(16 L)及三乙胺(900 g,8.88 mol)之混合物在20℃下攪拌,然後在2 h內添加甲磺醯氯(933 g,8.14 mol)。將混合物在20℃下攪拌,直至反應完成(通常為16 h)。過濾所得混合物,且用三級
丁基甲基醚(2×4 L)沖洗濾餅。將經合併之濾液(含有甲磺酸(S
)-3-(1-(6-溴-3-胺甲醯基吡啶-2-基)-5,5-二甲基吡咯啶-3-基)丙酯)轉移至反應器中且用二甲基亞碸(16 L)稀釋。向混合物中添加鄰苯二甲醯亞胺(1198 g,8.14 mol)。攪拌混合物直至獲得溶液,然後添加碳酸鉀(1023 g,7.4 mol),且將混合物攪拌並加熱至70℃,直到反應完成(2 h)。將混合物冷卻至20℃,且用2-甲基四氫呋喃(16L)稀釋,接著添加去離子水(21 L)。分離各相,且將上層有機相用去離子水(10 L)及飽和氯化鈉水溶液(2×1 L)洗滌。將有機相用甲苯(16 L)稀釋,且在減壓下濃縮至約10 L體積。藉由過濾分離固體,且將濾餅用甲苯(2×2 L)沖洗。乾燥所得固體,得到呈灰白色固體之(S
)-6-溴-2-(4-(3-(1,3-二側氧基異吲哚啉-2-基)丙基)-2,2-二甲基吡咯啶-1-基)菸鹼醯胺(3393 g,6.99 mol,94%產率)。步驟 9 :磺醯化成 (S
)-6- 溴 -2-(4-(3-(1,3- 二側氧基異吲哚啉 -2- 基 ) 丙基 )-2,2- 二甲基吡咯啶 -1- 基 )-N
-((6- 氟吡啶 -2- 基 ) 磺醯基 ) 菸鹼醯胺
在0-5℃下將6-氟吡啶-2-磺醯氯(529 g,340 mL,2.71 mol)添加到(S
)-6-溴-2-(4-(3-(1,3-二側氧基異吲哚啉-2-基)丙基)-2,2-二甲基吡咯啶-1-基)菸鹼醯胺• 0.5 PhMe(1.20 kg,2.26 mol;91.2%效力)於2-MeTHF (6.56 L; 6體積當量)中之溶液中,然後添加2-甲基丁-2-酸鋰(t
-OAmLi;1.22 kg 40重量%,1.67 L 40重量%,5.19 mol;2.3當量),同時保持反應溫度在5℃-10℃之間。添加完成後,將反應溶液在0-10℃下攪拌,直至反應完成(HPLC顯示保留了< 1% AUC起始材料)。反應溶液無需任何進一步處理即可進行下一步驟。步驟 10 :鄰苯二甲醯亞胺開環成 (S
)-2-((3-(1-(6- 溴 -3-(((6- 氟吡啶 -2- 基 ) 磺醯基 ) 胺甲醯基 ) 吡啶 -2- 基 )- 5,5- 二甲基吡咯啶 -3- 基 ) 丙基 ) 胺甲醯基 ) 苯甲酸
使來自前一步驟之含有(S
)-6-溴-2-(4-(3-(1,3-二側氧基異吲哚啉-2-基)丙基)-2,2-二甲基吡咯啶-1-基)-N
-((6-氟吡啶-2-基)磺醯基)菸鹼醯胺之反應溶液冷卻,且當添加LiOH • H2
O (284 g,6.77 mol;3當量)於水(2.19 L;2體積當量)中之溶液時保持低於10℃。將兩相混合物在5℃-15℃下攪拌,直至反應完成(通常為2 h)。在保持反應溫度低於10℃時,在約1 h內逐滴添加2 M HCl (5.64 L,11.3 mol;5當量)。水相之pH約為2。分離各相,然後將有機相濃縮至最小體積,以去除大部分MeTHF (40 ℃/150-70托)。反應混合物無需任何進一步處理即可進行下一步驟。步驟 11 :鄰苯二甲醯亞胺脫保護為 (S
)-2-(4-(3- 胺基丙基 )-2,2- 二甲基吡咯啶 -1- 基 )-6- 溴 -N- ((6- 氟吡啶 -2- 基 ) 磺醯基 ) 菸鹼醯胺
將來自前一步驟之含有(S
)-2-((3-(1-(6-溴-3-(((6-氟吡啶-2-基)磺醯基)胺甲醯基)吡啶-2-基)-5,5-二甲基吡咯啶-3-基)丙基)胺甲醯基)苯甲酸之濃縮物用CH3
CN(6.56 L;6體積當量)及水(3.83 L;2體積當量)稀釋,然後添加草酸(508 g,5.64 mol;2.5當量),且將所得溶液在60℃下加熱,直至反應完成(通常至少4 h)。將溶液冷卻至0-10℃,然後逐滴添加K2
CO3
(2.18 kg,15.8 mol;7當量)於水(3.83 L;3.5體積當量)中之溶液,同時保持反應溫度低於10℃。藉由過濾收集固體。將濕濾餅用水(2 x 2.2 L;2體積當量)連續洗滌,且然後用i-
PrOH (2 x 600 mL;0.5體積當量)洗滌,抽吸風乾,且真空乾燥(50 ℃/30托),得到呈白色細粉之(S
)-2-(4-(3-胺基丙基)-2,2-二甲基吡咯啶-1-基)-6-溴-N-
((6-氟吡啶-2-基)磺醯基)菸鹼醯胺(959 g;對於3個步驟為83%;>98% AUC)。
1
HNMR (400 MHz, DMSO‐d6
) δ 8.09 (q, J = 7.9 Hz, 1H), 7.83 (dd, J = 7.5, 2.2 Hz, 1H), 7.67 (s, 3H), 7.39 (d, J = 7.6 Hz, 1H), 7.23 (dd, J = 8.2, 2.4 Hz, 1H), 6.58 (d, J = 7.6 Hz, 1H), 3.20 – 2.99 (m, 2H), 2.81 (td, J = 7.2, 4.7 Hz, 2H), 2.08 (dh, J = 15.3, 7.0 Hz, 1H), 1.84 (dd, J = 11.8, 5.7 Hz, 1H), 1.54 (q, J = 7.6 Hz, 2H), 1.48 (s, 3H), 1.47 (s, 3H), 1.37 (t, J = 11.9 Hz, 1H), 1.26 (ddd, J = 29.1, 13.8, 7.4 Hz, 2H)。步驟 12 :大環化成 (14S)-8- 溴 -12,12- 二甲基 -2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(23),5(10),6,8,19,21- 己烯 -2,2,4- 三酮
將(S
)-2-(4-(3-胺基丙基)-2,2-二甲基吡咯啶-1-基)-6-溴-N-
((6-氟吡啶-2-基)磺醯基)菸鹼醯胺(950 g,1.85 mol)及Na2
CO3
(392 g,3.69 mol;2當量)於DMSO (7.60 L;8體積當量)中之混合物加熱到85℃,直到反應完成 (約6 h)。將懸浮液冷卻至< 15℃且用MeTHF(19.0 L;20體積當量)稀釋。緩慢添加水(13.3 L),同時保持反應溫度< 15℃。在保持反應溫度< 15℃時,添加2 M HCl (4.62 L,9.24 mol;5當量) (pH約2)。分離各相,且將有機相用含有NaCl(190 g;2重量%)之水(9.50 L;10體積當量)洗滌兩次。將有機相濃縮至最小體積(45℃/180托)且用i-
PrOAc(2-3 x 500 mL)處理,以去除MeTHF。將濃縮物用i-
PrOAc (3.8 L;4體積當量)回填並在45℃下攪拌,直至發生結晶。將該懸浮液在攪拌下老化不超過30 min,然後將其冷卻至20℃。在20℃下老化至少2 h後,藉由過濾收集固體。將濾餅用1:1i-
PrOAc/MTBE (500-mL)洗滌,抽吸風乾,且真空乾燥(40℃-55℃/< 100托/通N2
),得到呈帶淡黃色之白色粉末之(14S
)-8-溴-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮• 0.8i-
PrOAc (830 g;對於i-
PrOAc溶劑合物校正為78%產率)。
藉由將濾液濃縮至約400 mL總體積來獲得第二收穫物。然後接種混合物且在15℃-20℃下老化。藉由過濾收集固體。將濾餅用1:1i-
PrOAc/MTBE (200 mL)及 MTBE (100 mL)連續洗滌,抽吸風乾,且真空乾燥(55℃/< 100托/通N2
),得到呈淺黃色固體之(14S
)-8-溴-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮• 0.67i-
PrOAc (113 g;11%經校正產率)。
1
HNMR (400 MHz, 氯仿‐d
) δ 9.13 (s, 1H), 7.66 (d,J
= 7.9 Hz, 1H), 7.54 (dd,J
= 8.4, 7.3 Hz, 1H), 7.43 (d,J
= 7.3 Hz, 1H), 6.79 (d,J
= 7.9 Hz, 1H), 6.56 (d,J
= 8.4 Hz, 1H), 4.99 (hept,J
= 6.3 Hz, 1H), 4.57 (d,J
= 8.8 Hz, 1H), 4.02 – 3.85 (m, 1H), 3.27 – 3.09 (m, 2H), 2.96 (t,J
= 10.2 Hz, 1H), 2.35 (p,J
= 9.5 Hz, 1H), 2.02 (s, 3H), 1.95 (dd,J
= 12.1, 6.7 Hz, 1H), 1.72 – 1.59 (m, 6H), 1.58 (s, 3H), 1.55 (s, 3H), 1.43 (d,J
= 40.1 Hz, 1H), 1.23 (d,J
= 6.3 Hz, 5H)。一般 UPLC/HPLC 分析方法
LC 方法 A
:使用由Waters (pn:186002350)製成之Acquity UPLC BEH C18
管柱(50×2.1 mm,1.7 μm顆粒)及在2.9分鐘內自1%-99%流動相B之雙重梯度運行來進行反相UPLC分析。流動相A = H2
O (0.05% CF3
CO2
H)。流動相B = CH3
CN (0.035% CF3
CO2
H)。流速 = 1.2 mL/min,注入體積 = 1.5 μL,且管柱溫度 = 60℃。
LC 方法 B
:Merckmillipore Chromolith SpeedROD C18
管柱(50 x 4.6 mm)及在6分鐘內自5%-100%流動相B之雙重梯度運行。流動相A = 水(0.1% CF3
CO2
H)。流動相B = 乙腈(0.1% CF3
CO2
H)。
LC 方法 C
:Merckmillipore Chromolith SpeedROD C18
管柱(50 x 4.6 mm)及在12分鐘內自5%-100%流動相B之雙重梯度運行。流動相A = 水(0.1% CF3
CO2
H)。流動相B = 乙腈(0.1% CF3
CO2
H)。
LC 方法 D
:由Waters (pn:186002349)製成之Acquity UPLC BEH C18
管柱(30×2.1 mm,1.7 μm顆粒)及在1.0分鐘內自1%-99%流動相B之雙重梯度運行。流動相A = H2
0 (0.05% CF3
CO2
H)。流動相B = CH3
CN (0.035% CF3
CO2
H)。流速 = 1.5 mL/min,注入體積 = 1.5 μL,且管柱溫度 = 60℃。
LC 方法 E :
Luna管柱C18
(2) 50 x 3 mm,3 µm。運行:2.5分鐘。流動相:含有0.1%甲酸之初始95%H2
O /含有0.1%甲酸之5% MeCN,在1.3 min內線性梯度至含有0.1%甲酸之95% MeCN,在含有0.1%甲酸之95% MeCN下保持1.2 min。溫度:45℃,流量:1.5 mL/min。
LC 方法 F :
SunFire管柱C18
75 x 4.6 mm 3.5 µm,運行:6分鐘。流動相條件:初始95%H2
O + 0.1%甲酸/ 5% MeCN + 0.1%甲酸,線性梯度至95% MeCN達4 min,在95% MeCN下保持2 min。T:45℃,流量:1.5 mL/min。
LC 方法 G :
使用由Waters (pn:186002350)製成之Acquity UPLC BEH C18
管柱(50×2.1 mm,1.7 μm顆粒)及在2.9分鐘內自30%-99%流動相B之雙重梯度運行來進行反相UPLC分析。流動相A = H2
0 (0.05% CF3
CO2
H)。流動相B = MeCN (0.035% CF3
CO2
H)。流速 = 1.2 mL/min,注入體積 = 1.5 μL,且管柱溫度 = 60℃。
LC 方法 H :
Water Cortex 2.7µ C18
(3.0 mm x 50 mm)管柱,溫度:55℃;流量:1.2 mL/min;流動相:具有0.1%三氟乙酸(TFA)之100%水,然後具有0.1%TFA酸之100%乙腈,在4 min內梯度為5%至100% B,且在100% B下停留0.5 min,在1.5 min內平衡至5% B。
LC 方法 I :
使用由Waters (pn:186002349)製成之Acquity UPLC BEH C18
管柱(30×2.1 mm,1.7 μm顆粒)及在1.0分鐘內自30%-99%流動相B之雙重梯度運行來進行反相UPLC。流動相A = H2
0 (0.05% CF3
CO2
H)。流動相B = CH3
CN (0.035% CF3
CO2
H)。流速 = 1.5 mL/min,注入體積 = 1.5 μL,且管柱溫度 = 60℃。
LC 方法 J
:SunFire C18
4.6 x 75mm,5 µM,6 min運行,50%-95% ACN/水 (0.1% FA改性劑),1.5 min平衡,在3 min內梯度,保持3 min。1.5 mL/min。
LC 方法 K
:SunFire C18
75 x 4.6mm 3.5 µm,運行:6 min。流動相條件:初始95% H2
O + 0.1%甲酸/5% CH3
CN + 0.1% FA,線性梯度至95% CH3
CN達4 min,在95% CH3
CN下保持2 min。T:45℃,流量:1.5 mL/min
LC 方法
L:Luna C18
3.0 x 50 mm 3.0 µM,溫度:45℃,流量:2.0 mL/min,運行時間:3分鐘。流動相:初始95% H2
O (0.1%甲酸)及5% CH3
CN (0.1%甲酸),線性梯度至95% CH3
CN (0.1%甲酸)達2.0 min,然後在95% CH3
CN (0.1%甲酸)下保持1.0 min。
LC 方法 M
:使用由Waters (pn:186002350)製成之Acquity UPLC BEH C18
管柱(50×2.1 mm,1.7 μm顆粒)及在5.0分鐘內自1%-99%流動相B之雙重梯度運行來進行反相UPLC分析。流動相A = 水(0.05%三氟乙酸)。流動相B = 乙腈(0.035%三氟乙酸)。流速 = 1.2 mL/min,注入體積 = 1.5 μL,且管柱溫度 = 60℃。
LC 方法 N
:使用由Waters (pn:186002349)製成之Acquity UPLC BEH C18
管柱(30×2.1 mm,1.7 μm顆粒)及在1.2分鐘內自1%-99%流動相B之雙重梯度運行來進行反相UPLC分析。流動相A = 水(0.05%三氟乙酸)。流動相B = 乙腈(0.035%三氟乙酸)。流速 = 1.5 mL/min,注入體積 = 1.5 μL,且管柱溫度 = 60℃實例 3 : (14S
)-8-[3-( 三級丁基 二甲基矽基 )-1H
- 吡唑 -1- 基 ]-12,12- 二甲基 -2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(23),5(10),6,8,19,21- 己烯 -2,2,4- 三酮,化合物 (1-5) 之製備 步驟 1 :三級
丁基 - 二甲基 -(2- 四氫哌喃 -2- 基吡唑 -3- 基 ) 矽烷
將1-四氫哌喃-2-基吡唑(5.065 g,33.28 mmol)溶解於THF (30 mL)中,且冷卻至-35℃。逐滴添加正
丁基鋰(16 mL於己烷中之2.5 M溶液,40.00 mmol),且將該溶液在-35℃下再攪拌1 h。逐滴添加三級
丁基-氯-二甲基-矽烷(5.1 g,33.84 mmol)於THF (7 mL)中之溶液,且使反應物升溫至室溫並攪拌3 h。此時,添加飽和氯化銨溶液,直到pH為約7且將混合物用乙醚萃取。分離有機物,將其用鹽水洗滌,經硫酸鎂乾燥且蒸發,得到呈橙色油狀物之三級
丁基-二甲基-(2-四氫哌喃-2-基吡唑-3-基)矽烷(8.41 g,95%)。1
HNMR (400 MHz, DMSO-d6
) δ 7.53 (d, J = 1.6 Hz, 1H), 6.43 (d, J = 1.7 Hz, 1H), 5.23 (dd, J = 10.1, 2.4 Hz, 1H), 3.96- 3.85 (m, 1H), 3.63- 3.48 (m, 1H), 2.43- 2.29 (m, 1H), 2.02- 1.92 (m, 1H), 1.84- 1.75 (m, 1H), 1.73- 1.56 (m, 1H), 1.56- 1.47 (m, 2H), 0.88 (s, 9H), 0.32 (s, 3H), 0.30 (s, 3H)。ESI-MS m/z計算值266.18143,實測值267.3 (M+1)+
;保留時間:0.79分鐘(LC方法D)。步驟 2 :三級
丁基 - 二甲基 -(1H
- 吡唑 -3- 基 ) 矽烷
將三級
丁基-二甲基-(2-四氫哌喃-2-基吡唑-3-基)矽烷(8.4 g,31.53 mmol)溶解於6M HCl水溶液(16 mL 96.00 mmol)、乙醇(8 mL)之混合物中且在50℃下加熱3 h。添加飽和NaHCO3
水溶液以淬滅酸,且將所得溶液用乙酸乙酯萃取兩次。將有機物合併,用鹽水洗滌,經硫酸鎂乾燥且蒸發。藉由用己烷中之0-100%乙酸乙酯溶離進行矽膠層析來純化粗材料,得到呈白色固體之三級丁基
-二甲基-(1H-
吡唑-3-基)矽烷(3.85 g,67%)。1
HNMR (400 MHz, DMSO-d6
) δ 12.77 (s, 1H), 7.52 (s, 1H), 6.40 (d, J = 1.6 Hz, 1H), 0.85 (s, 9H), 0.25 (s, 6H)。ESI-MS m/z計算值182.12393,實測值183.6 (M+1)+
;保留時間:0.57分鐘(LC方法D)。步驟 3 : (14S
)-8-[3-( 三級 丁基二甲基矽基 )-1H
- 吡唑 -1- 基 ]-12,12- 二甲基 -2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(23),5(10),6,8,19,21- 己烯 -2,2,4- 三酮,化合物 (1-5)
向(14S)
-8-溴-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(75 mg,0.1259 mmol)及三級
丁基-二甲基-(1H-
吡唑-3-基)矽烷(27 mg,0.1481 mmol)於乙酸丁酯(1 mL)及DMSO(0.3 mL)中之溶液中添加碳酸鉀(46 mg,0.3328 mmol)。將N2
鼓泡5 min。添加(1R
, 2R
)-N
1,N
2-二甲基環己烷-1,2-二胺(10.800 mg,12 μL,0.0759 mmol)及CuI (2.3 mg,0.0121 mmol),將管密封且將反應物在120℃下升溫5.5 h。將反應物冷卻至室溫,且經矽藻土過濾混合物,用MeTHF (10 mL)溶離。添加DMSO (0.5 mL),且在真空下去除揮發物。藉由使用12g C18 Agela柱、使用0.1%甲酸水溶液中之MeCN梯度溶離(對於2 CV為5%,然後在15 CV中為70%至100%)進行反相層析來純化粗混合物。凍乾後得到呈白色固體之(14S
)-8-[3-(三級丁基
二甲基矽基)-1H
-吡唑-1-基]-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(38 mg,49%)。1
HNMR (400 MHz, DMSO-d6
) δ 12.52 (br. s, 1H), 8.41 (d, J = 2.4 Hz, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.58 (t, J = 7.9 Hz, 1H), 7.16 (d, J = 8.3 Hz, 1H), 7.06 (d, J = 7.1 Hz, 1H), 6.98 (br. s., 1H), 6.71 (d, J = 8.6 Hz, 1H), 6.69 (d, J = 2.4 Hz, 1H), 4.02- 3.84 (m, 1H), 3.17 (br. s., 1H), 2.95 (d, J = 13.2 Hz, 1H), 2.82- 2.63 (m, 1H), 2.13 (br. s., 1H), 1.87 (dd, J = 11.4, 5.0 Hz, 1H), 1.82- 1.71 (m, 1H), 1.65- 1.55 (m, 6H), 1.53 (s, 3H), 1.32 (q, J = 11.7 Hz, 1H), 0.94 (s, 9H), 0.27 (d, J = 1.5 Hz, 6H)。ESI-MS m/z計算值595.2761,實測值596.3 (M+1)+
;保留時間:4.26分鐘(LC方法J)。實例 4 : (14S
)-8-{3-[( 三級 丁基二甲基矽基 ) 甲氧基 ]-1H
- 吡唑 -1- 基 }-12,12- 二甲基 -2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(23),5(10),6,8,19,21- 己烯 -2,2,4- 三酮,化合物 (1-2) 之製備 步驟 1 : [ 三級 丁基 ( 二甲基 ) 矽基 ] 甲醇
在氮氣下在經加熱槍乾燥之圓底燒瓶中,在0℃冰水浴中將正
丁基鋰(1 mL,1.600 mmol,己烷中之1.6 M溶液)與N,N,N',N'-
四甲基乙烷-1,2-二胺(200 µL,1.325 mmol)合併於無水THF (8 mL)中。然後藉由注射器在1分鐘內逐滴添加三級丁基
-二甲基-(三丁基錫烷基甲氧基)矽烷(500 mg,1.149 mmol)。將反應混合物在0℃下攪拌2分鐘,然後用乙酸(200 μL,3.517 mmol)淬滅。然後將反應混合物用飽和碳酸氫鈉水溶液(10 mL)、水(10 mL)及乙酸乙酯(15 mL)稀釋,且升溫至室溫。分離各層,且再藉由2×15 mL乙酸乙酯萃取水相。將經合併之有機物用鹽水洗滌,經硫酸鈉乾燥,且濃縮,得到無色油狀物。藉由矽膠層析(己烷中之1%-70%乙酸乙酯梯度,最初用己烷沖洗)純化該粗材料,得到呈白色固體之[三級
丁基(二甲基)矽基]甲醇(95 mg,57%)。1
HNMR (400 MHz, 氯仿-d) δ 3.46 (s, 2H), 0.90 (s, 9H), 0.00 (s, 6H)。(醇OH不可見)步驟 2 : 3-[[ 三級 丁基 ( 二甲基 ) 矽基 ] 甲氧基 ] 吡唑 -1- 羧酸三級
丁酯
將3-羥基吡唑-1-羧酸三級
丁酯(220 mg,1.194 mmol)、[三級
丁基(二甲基)矽基]甲醇(190 mg,1.299 mmol)及三苯膦(345 mg,1.315 mmol)合併於THF (2.5 mL)中且將其冷卻至0℃。逐滴添加DIAD (255 μL,1.317 mmol),且將反應混合物升溫至室溫達16小時。然後將反應混合物分配於30 mL 1M NaOH(水溶液)及乙酸乙酯(30 mL)之間。分離各層,且再用2×30 mL乙酸乙酯萃取水相。將經合併之有機物用鹽水洗滌,經硫酸鈉乾燥,且濃縮。藉由用己烷中之0-50%乙酸乙酯梯度溶離(開始時很淺,就在10%之前溶離之化合物)進行矽膠急速層析來純化所得粗材料,得到呈無色油狀物之3-[[三級
丁基(二甲基)矽基]甲氧基]吡唑-1-羧酸三級
丁酯(242 mg,65%)。1
HNMR (400 MHz, 氯仿-d) δ 7.81 (d, J = 2.7 Hz, 1H), 5.85 (d, J = 2.8 Hz, 1H), 4.06 (s, 2H), 1.61 (s, 9H), 0.94 (s, 9H), 0.06 (s, 6H)。ESI-MS m/z計算值312.18692,實測值313.3 (M+1)+
;保留時間:0.88分鐘(LC方法D)。步驟 3 :三級
丁基 - 二甲基 -(1H- 吡唑 -3- 基氧基甲基 ) 矽烷
在室溫下將3-[[三級
丁基(二甲基)矽基]甲氧基]吡唑-1-羧酸三級
丁酯(242 mg,0.7744 mmol)與TFA (750 μL,9.735 mmol)合併於DCM (2.5 mL),且攪拌15分鐘。然後將反應混合物在減壓下蒸發。將粗材料溶解於15 mL乙酸乙酯中,且用15 mL飽和碳酸氫鈉水溶液洗滌。將水層再用2×10 mL乙酸乙酯萃取,且將經合併之有機物用鹽水洗滌,經硫酸鈉乾燥,且濃縮,得到呈無色油狀物之三級
丁基-二甲基-(1H
-吡唑-3-基氧基甲基)矽烷(161 mg,98%)。1
HNMR (400 MHz, 氯仿-d) δ 7.35 (d, J = 2.4 Hz, 1H), 5.73 (d, J = 2.5 Hz, 1H), 3.92 (s, 2H), 0.95 (s, 9H), 0.08 (s, 6H) (NH不可見)。ESI-MS m/z計算值212.13449,實測值213.6 (M+1)+
;保留時間:0.66分鐘(LC方法D)。步驟 4 : (14S
)-8-{3-[( 三級 丁基二甲基矽基 ) 甲氧基 ]-1H
- 吡唑 -1- 基 }-12,12- 二甲基 -2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(23),5(10),6,8,19,21- 己烯 -2,2,4- 三酮,化合物 (1-2)
將碘化亞銅(7 mg,0.0368 mmol)添加到(14S)
-8-溴-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(230 mg,0.3861 mmol)、三級丁基
-二甲基-(1H-
吡唑-3-基氧基甲基)矽烷(100 mg,0.4709 mmol)、(1R
,2R
)-N
1,N
2-二甲基環己烷-1,2-二胺(31.500 mg,35 μL,0.2215 mmol)及碳酸鉀(120 mg,0.8683 mmol)於乙酸丁酯(3 mL)及二甲基亞碸(0.9 mL)中之脫氣溶液中。將反應混合物在120℃下升溫6小時。將反應物冷卻至室溫,且經矽藻土過濾混合物,用Me-THF (20 mL)溶離。將濾液用水(25 mL)及鹽水(3×25 mL)洗滌。將有機相經硫酸鈉乾燥,過濾且濃縮至乾燥。藉由正相層析純化(10 + 12 g SiO2
,以己烷中之20%至100%乙酸乙酯溶離)且藉由C18反相層法(30 g,以水中之50%至100%乙腈溶離)純化粗產物,且將其冷凍乾燥,得到呈白色固體之(14S
)-8-{3-[(三級
丁基二甲基矽基)甲氧基]-1H-
吡唑-1-基}-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(103 mg,43%)。1
HNMR (400 MHz, DMSO-d6
) δ 12.48 (br. s., 1H), 8.20 (d, J = 2.7 Hz, 1H), 7.81 (d, J = 8.3 Hz, 1H), 7.57 (t, J = 7.8 Hz, 1H), 7.05 (d, J = 7.1 Hz, 1H), 7.02- 6.86 (m, 2H), 6.71 (d, J = 8.6 Hz, 1H), 6.12 (d, J = 2.7 Hz, 1H), 4.03 (s, 2H), 3.93- 3.90 (m, 1H), 3.19- 3.13 (m, 1H), 2.95- 2.93 (m, 1H), 2.76- 2.65 (m, 1H), 2.18- 2.07 (m, 1H), 1.87- 1.83 (m, 1H), 1.76- 1.70 (m, 1H), 1.66- 1.45 (m, 9H), 1.37- 1.23 (m, 1H), 0.94 (s, 9H), 0.07 (s, 6H)。ESI-MS m/z計算值625.2867,實測值626.3 (M+1)+
;保留時間:5.49分鐘(LC方法K)。實例 5 : (14S
)-8-{3-[(3,3- 二甲基丁基 ) 二甲基矽基 ]-1H
- 吡唑 -1- 基 }-12,12- 二甲基 -2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(23),5(10),6,8,19,21- 己烯 -2,2,4- 三酮,化合物 (1-4) 之製備 步驟 1 : 3,3- 二甲基丁基 - 二甲基 -(2- 四氫哌喃 -2- 基吡唑 -3- 基 ) 矽烷
在6 min內在-35℃下在氮氣下自滴液漏斗向1-四氫哌喃-2-基吡唑(3 g,19.71 mmol)於無水四氫呋喃(25 mL)中之攪拌溶液中逐滴添加正
丁基鋰(己烷中之1.6 M) (15 mL,24.00 mmol)。在添加完成後,將所得溶液在-35℃下再攪拌1 h。然後,在5 min內自滴液漏斗中逐滴添加氯- (3,3-二甲基丁基)-二甲基-矽烷(3.90 g,21.82 mmol)於無水四氫呋喃(1 mL)中之溶液。添加結束後,將反應物在該溫度下再攪拌10分鐘,且然後去除浴,且使其升溫至室溫。然後再攪拌3 h後,添加飽和氯化銨溶液(30 mL),且用乙醚(3×30 mL)萃取混合物。將經合併之有機物用鹽水(20 mL)洗滌,經硫酸鈉乾燥,過濾且在減壓下濃縮。粗材料(淺橙色輕質油狀物) 3,3-二甲基丁基-二甲基-(2-四氫哌喃-2-基吡唑-3-基)矽烷(5.79 g,100%)無需進一步純化即用於下一步驟中。1
HNMR (400 MHz, 氯仿-d) δ 7.57 (d, J = 1.7 Hz, 1H), 6.39 (d, J = 1.7 Hz, 1H), 5.29 (dd, J = 9.8, 2.6 Hz, 1H), 4.08- 3.98 (m, 1H), 3.62 (td, J = 11.2, 2.6 Hz, 1H), 2.57- 2.42 (m, 1H), 2.13- 2.07 (m, 1H), 2.03- 1.96 (m, 1H), 1.77- 1.63 (m, 2H), 1.61- 1.56 (m, 1H), 1.19- 1.12 (m, 2H), 0.85 (s, 9H), 0.76- 0.70 (m, 2H), 0.30 (s, 6H)。ESI-MS m/z計算值294.21274,實測值295.2 (M+1)+
;保留時間:2.21分鐘(LC方法A)。步驟 2 : 3,3- 二甲基丁基 - 二甲基 -(1H- 吡唑 -3- 基 ) 矽烷
向3,3-二甲基丁基-二甲基-(2-四氫哌喃-2-基吡唑-3-基)矽烷(5.2 g,17.66 mmol)於乙醇(15 mL)中之攪拌溶液中添加鹽酸水溶液(11 mL 5.0 M,55.00 mmol)且在50℃下攪拌4 h。使反應物冷卻至環境溫度且緩慢添加飽和NaHCO3
溶液(劇烈CO2
氣體逸出)以淬滅酸,且將所得溶液用乙酸乙酯(3×30 mL)萃取。將經合併之有機物用鹽水(20 mL)洗滌,經硫酸鈉乾燥,過濾且在減壓下濃縮。在環境溫度下靜置後,粗材料(黏稠油狀物)變成褐色固體,其為3,3-二甲基丁基-二甲基-(1H-
吡唑-3-基)矽烷(3.64 g,98%)。其無需進一步純化即用於隨後反應。1
HNMR (400 MHz, 甲醇-d4
) δ 8.18 (d, J = 2.4 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 1.22- 1.15 (m, 2H), 0.87 (s, 9H), 0.86- 0.79 (m, 2H), 0.39 (s, 6H)。ESI-MS m/z計算值210.15523,實測值211.1 (M+1)+
;保留時間:1.66分鐘(LC方法A)。步驟 3 : (14S
)-8-{3-[(3,3- 二甲基丁基 ) 二甲基矽基 ]-1H
- 吡唑 -1- 基 }-12,12- 二甲基 -2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(23),5(10),6,8,19,21- 己烯 -2,2,4- 三酮,化合物 (1-4)
在密封管中,將(14S)-8-
溴-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(100 mg,0.1679 mmol)、3,3-二甲基丁基-二甲基-(1H-
吡唑-3-基)矽烷(39 mg,0.1854 mmol)、碘化亞銅(I) (3 mg,0.0158 mmol)、碳酸鉀(52 mg,0.3762 mmol)及(1R
, 2R
)-N
1,N
2-二甲基環己烷-1,2-二胺(12.600 mg,14 μL,0.0886 mmol)混合於乙酸丁酯(0.7 mL)及二甲基亞碸(0.2 mL)中。使用3個真空/氮氣回填循環對混合物進行脫氣。將反應物在120℃下攪拌5小時。將反應物冷卻至室溫,且經矽藻土過濾混合物,用MeTHF(10 mL)溶離。添加DMSO (0.5 mL),且在真空下去除揮發物。藉由使用12 g C18 Agela柱、使用0.1%甲酸水溶液中之MeCN梯度溶離(對於2 CV為5%,然後在15 CV中為50%至100%)進行反相層析來純化粗混合物。凍乾後,得到呈白色固體之(14S
)-8-{3-[(3,3-二甲基丁基)二甲基矽基]-1H
-吡唑-1-基}-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(33 mg,31%)。1
HNMR (400 MHz, DMSO-d6
) δ 12.55 (br. s., 1H), 8.40 (d, J = 2.4 Hz, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.15 (d, J = 8.1 Hz, 1H), 7.06 (d, J = 7.3 Hz, 1H), 6.99- 6.97 (m, 1H), 6.71 (d, J = 8.6 Hz, 1H), 6.68 (d, J = 2.4 Hz, 1H), 3.97- 3.88 (m, 1H), 3.20- 3.15 (m, 1H), 2.95 (d, J = 13.0 Hz, 1H), 2.74- 2.66 (m, 1H), 2.18- 2.07 (m, 1H), 1.89- 1.85 (m, 1H), 1.81- 1.71 (m, 1H), 1.67- 1.49 (m, 9H), 1.39- 1.27 (m, 1H), 1.25- 1.18 (m, 2H), 0.84 (s, 9H), 0.73- 0.66 (m, 2H), 0.27 (s, 6H)。ESI-MS m/z計算值623.3074,實測值624.3 (M+1)+
;保留時間:4.47分鐘。(LC方法J)。實例 6 : (14S
)-12,12- 二甲基 -8-{3-[( 三甲基矽基 ) 甲氧基 ]-1H
- 吡唑 -1- 基 }-2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5(10),6,8,19(23),20- 己烯 -2,2,4- 三酮 , 化合物 (1-8) 之製備 步驟 1 : 3-( 三甲基矽基甲氧基 ) 吡唑 -1- 羧酸三級
丁酯
向5 mL微波小瓶中裝入3-羥基吡唑-1-羧酸三級
丁酯(2 g,10.86 mmol)、氯甲基(三甲基)矽烷(1.6 mL,11.46 mmol)、碳酸鉀(3.00 g,21.71 mmol)及DMA (20 mL)。將反應物在120℃下加熱16小時。將反應物用水(50 mL)及乙酸乙酯(50 mL)稀釋。分離兩層後,將水層用乙酸乙酯(2×50 mL)萃取。將經合併之有機層用鹽水(3×50 mL)洗滌,經無水硫酸鈉乾燥且在真空下濃縮。將殘餘物溶解於THF (20 mL)中。將TEA (4.6 mL,33.00 mmol)及Boc2
O(4.75 g,21.764 mmol)添加到反應混合物中,隨後添加催化量之DMAP(157 mg,1.285 mmol)。將反應物在室溫下攪拌16小時。在真空下去除所有揮發物。藉由使用己烷中之0至10%乙醚進行矽膠層析來純化殘餘物,提供呈澄清油狀物之3-(三甲基矽基甲氧基)吡唑-1-羧酸三級
丁酯(2.146 g,72%)。1
HNMR (250 MHz, 氯仿-d) δ 7.82 (d, J = 2.9 Hz, 1H), 5.86 (d, J = 2.9 Hz, 1H), 3.98 (s, 2H), 1.61 (s, 9H), 0.11 (s, 9H)。ESI-MS m/z計算值270.14,實測值270.9 (M+1)+
;保留時間:6.62分鐘(LC方法C)。步驟 2 :三甲基 (1H- 吡唑 -3- 基氧基甲基 ) 矽烷
向3-(三甲基矽基甲氧基)吡唑-1-羧酸三級
丁酯(2.146 g,7.78 mmol)於DCM (24 mL)中之溶液中添加TFA (12 mL,155.76 mmol)。將反應物在室溫下攪拌2小時。在真空下去除所有揮發物,得到呈澄清油狀物之三甲基(1H-
吡唑-3-基氧基甲基)矽烷(三氟乙酸鹽) (3.03 g,100%)。1
HNMR (250 MHz, 氯仿-d) δ 7.72 (d, J = 2.9 Hz, 1H), 5.95 (d, J = 2.9 Hz, 1H), 3.92 (s, 2H), 0.17 (s, 9H)。ESI-MS m/z計算值170.0875,實測值171.3 (M+1)+
;保留時間:3.85分鐘(LC方法C)。步驟 3 : (14S
)-12,12- 二甲基 -8-{3-[( 三甲基矽基 ) 甲氧基 ]-1H
- 吡唑 -1- 基 }-2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5(10),6,8,19(23),20- 己烯 -2,2,4- 三酮,化合物 (1-8)
向三甲基(1H
-吡唑-3-基氧基甲基)矽烷(56 mg,0.3289 mmol)、(14S
)-8-溴-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(150 mg,0.2518 mmol)、L-脯胺酸(12 mg,0.1042 mmol)、及K2
CO3
(105 mg,0.7597 mmol)於DMSO (1 mL)中之溶液添加CuI (10 mg,0.0525 mmol)。將該溶液在真空/N2
下循環3次,然後密封且在110℃下加熱18 h。將反應物稀釋到10 mL水中,添加2 M HCl水溶液直至pH為酸性,且將反應物用DCM (3×20 mL)萃取。將經合併之有機層用鹽水(2 x 20 mL)洗滌,經Na2
SO4
乾燥,且在減壓下濃縮。使用己烷中之10%-50%丙酮溶離來進行管柱層析(二氧化矽,12 g),接著使用水中之0至100%乙腈(以0.1% TFA緩衝)進行反相HPLC,得到呈白色固體之(14S
)-12,12-二甲基-8-{3-[(三甲基矽基)甲氧基]-1H
-吡唑-1-基}-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(22),5(10),6,8,19(23),20-己烯-2,2,4-三酮(28.1 mg,19%)。1
HNMR (500 MHz, DMSO-d6
) δ 12.50 (s, 1H), 8.21 (d, J = 2.8 Hz, 1H), 7.82 (d, J = 8.2 Hz, 1H), 7.58 (dd, J = 8.5, 7.2 Hz, 1H), 7.06 (d, J = 7.3 Hz, 1H), 6.99 (d, J = 8.8 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.72 (dd, J = 8.5, 0.8 Hz, 1H), 6.13 (d, J = 2.7 Hz, 1H), 3.96 (s, 3H), 3.16 (s, 1H), 3.00 – 2.90 (m, 1H), 2.77 – 2.63 (m, 1H), 2.13 (s, 1H), 1.91 – 1.83 (m, 1H), 1.83 – 1.70 (m, 1H), 1.62 – 1.51 (m, 9H), 1.39 – 1.26 (m, 1H), 0.12 (s, 9H)。ESI-MS m/z計算值583.2397,實測值584.2 (M+1)+
;保留時間:3.33分鐘(LC方法H)。實例 7 : (14S
)-12,12- 二甲基 -8-{3-[2-( 三甲基矽基 ) 乙氧基 ]-1H
- 吡唑 -1- 基 }-2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5(10),6,8,19(23),20- 己烯 -2,2,4- 三酮,化合物 (1-7) 之製備 步驟 1 : 3-(2- 三甲基矽基乙氧基 ) 吡唑 -1- 羧酸三級
丁酯
在0℃下向3-羥基吡唑-1-羧酸三級
丁酯(5.01 g,27.20 mmol)及三苯膦(14.3 g,54.52 mmol)於無水THF (50 mL)中之溶液中添加DIAD (10.5 mL,54.21 mmol)。將反應物在室溫下攪拌隔夜。將反應物在真空下濃縮,以去除大部分THF。將殘餘物用乙酸乙酯(300 mL)稀釋,且用鹽水(100 mL)洗滌。將有機層經無水硫酸鈉乾燥且在真空下濃縮。將殘餘物懸浮於己烷溶液中之20%乙醚(400 mL)中。過濾出固體。在真空下濃縮濾液,且藉由使用己烷中之0至10%乙酸乙酯進行矽膠層析來純化,提供呈澄清油狀物之3-(2-三甲基矽基乙氧基)吡唑-1-羧酸三級
丁酯(4.559 g,59%)。ESI-MS m/z計算值284.1556,實測值285.1 (M+1)+
;保留時間:6.7分鐘(LC方法C)。步驟 2 :三甲基 -[2-(1H
- 吡唑 -3- 基氧基 ) 乙基 ] 矽烷
向3-(2-三甲基矽基乙氧基)吡唑-1-羧酸三級
丁酯(4.076 g,14.33 mmol)於THF (29 mL)及EtOH (58 mL)之溶劑混合物中之溶液中添加NaOH (14.5 mL 2M,29.00 mmol)。將反應物在室溫下攪拌3小時。將反應物用水(100 mL)稀釋,且用乙酸乙酯(3×100 mL)萃取。將經合併之有機層用鹽水(100 mL)洗滌,經無水硫酸鈉乾燥且在真空下濃縮,提供呈澄清油狀物之三甲基-[2-(1H
-吡唑-3-基氧基)乙基]矽烷(2.919 g,99%)。該產物無需純化即用於下一步驟。1
HNMR (250 MHz, 氯仿-d) δ 7.36 (d, J = 1.7 Hz, 1H), 5.72 (d, J = 1.8 Hz, 1H), 4.35 – 4.14 (m, 2H), 1.21 – 1.03 (m, 2H), 0.06 (s, 9H)。ESI-MS m/z計算值184.1032,實測值185.4 (M+1)+
;保留時間:4.44分鐘(LC方法C)。步驟 3 : (14S
)-12,12- 二甲基 -8-{3-[2-( 三甲基矽基 ) 乙氧基 ]-1H
- 吡唑 -1- 基 }-2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5(10),6,8,19(23),20- 己烯 -2,2,4- 三酮,化合物 (1-7)
向三甲基-[2-(1H
-吡唑-3-基氧基)乙基]矽烷(56 mg,0.3038 mmol)、(14S
)-8-溴-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(150 mg,0.2518 mmol)、L-脯胺酸(12 mg,0.1042 mmol)、及K2
CO3
(105 mg,0.7597 mmol)於DMSO(1 mL)中之溶液中添加CuI (15 mg,0.0788 mmol)。將該溶液在真空/N2
下循環3次,然後在110℃下加熱14 h。將反應物稀釋到10 mL水中,添加2 M HCl水溶液直至pH為酸性,然後將反應物用DCM (3×20 mL)萃取。將經合併之有機層用鹽水(2 x 20 mL)洗滌,經Na2
SO4
乾燥,且在減壓下濃縮。使用己烷中之10%-40%丙酮溶離來進行管柱層析(矽膠,12 g),接著使用水中之0-100%乙腈(以0.1%TFA緩衝)進行反相HPLC,得到呈白色固體之(14S
)-12,12-二甲基-8-{3-[2-(三甲基矽基)乙氧基]-1H
-吡唑-1-基}-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(22),5(10),6,8,19(23),20-己烯-2,2,4-三酮(53.1 mg,34%) (53.1 mg,34%)。1
HNMR (500 MHz, DMSO-d6
) δ 12.51 (s, 1H), 8.21 (d, J = 2.7 Hz, 1H), 7.82 (d, J = 8.3 Hz, 1H), 7.58 (dd, J = 8.5, 7.2 Hz, 1H), 7.06 (d, J = 7.2 Hz, 1H), 6.99 (d, J = 9.0 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.72 (d, J = 8.5 Hz, 1H), 6.09 (d, J = 2.8 Hz, 1H), 4.34 – 4.27 (m, 2H), 3.93 (d, J = 11.8 Hz, 1H), 3.16 (s, 1H), 3.01 – 2.91 (m, 1H), 2.71 (t, J = 10.4 Hz, 1H), 2.13 (s, 1H), 1.90 – 1.71 (m, 2H), 1.64 – 1.51 (m, 9H), 1.38 – 1.26 (m, 1H), 1.17 – 1.06 (m, 2H), 0.08 (s, 9H)。ESI-MS m/z計算值597.2554,實測值598.4 (M+1)+
;保留時間:3.42分鐘(LC方法H)。實例 8 : (14S
)-12,12- 二甲基 -8-{3-[3-( 三甲基矽基 ) 丙氧基 ]-1H
- 吡唑 -1- 基 }-2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5(10),6,8,19(23),20- 己烯 -2,2,4- 三酮,化合物 (1-6) 之製備 步驟 1 : 3-(3- 三甲基矽基丙氧基 ) 吡唑 -1- 羧酸三級
丁酯
在0℃下向3-羥基吡唑-1-羧酸三級
丁酯(2 g,10.86 mmol)、3-三甲基矽基丙-1-醇(1.9 mL,11.24 mmol)及三苯膦(5.7 g,21.73 mmol)於THF(20 mL)中之溶液中添加DIAD (3.2 mL,15.87 mmol)。在室溫下4 h後,在減壓下去除溶劑,且將反應物稀釋到EtOAc (100 mL)中。將有機層用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,且在減壓下濃縮。然後將粗物質懸浮於10:1己烷:Et2
O (v:v) (150 mL)且過濾。在減壓下自上清液中去除溶劑。使用己烷中之0-10% EtOAc溶離來進行管柱層析(二氧化矽,120g),得到呈澄清油狀物之3-(3-三甲基矽基丙氧基)吡唑-1-羧酸三級
丁酯(3.3g,53%)。1
HNMR (250 MHz, CDCl3
) δ 7.83 (d, J = 3.0 Hz, 1H), 5.86 (d, J = 3.0 Hz, 1H), 4.24 (t, J = 6.9 Hz, 2H), 1.84 – 1.67 (m, 2H), 1.62 (s, 9H), 0.71 – 0.46 (m, 2H), 0.01 (s, 9H)。ESI-MS m/z計算值298.1713,實測值299.3 (M+1)+
;保留時間:4.02分鐘(LC方法B)。步驟 2: 三甲基 -[3-(1H
- 吡唑 -3- 基氧基 ) 丙基 ] 矽烷
在室溫下向3-(3-三甲基矽基丙氧基)吡唑-1-羧酸三級
丁酯(3.1 g,5.67 mmol)於THF (25 mL)及EtOH(50 mL)中之溶液中添加NaOH水溶液(10.5 mL 2 M,21.00 mmol)。將反應物在室溫下攪拌4 h,然後稀釋到水(100 mL)中,且用EtOAc (3×100 mL)萃取。將有機部分用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且在減壓下濃縮。使用己烷中之10%-50% EtOAC溶離進行管柱層析(二氧化矽,120 g),得到呈澄清液體之三甲基-[3-(1H
-吡唑-3-基氧基)丙基]矽烷(877 mg,77%)。1
HNMR (250 MHz, CDCl3
) δ 7.36 (d, J = 2.4 Hz, 1H), 5.91 – 5.52 (m, 1H), 4.24 – 3.92 (m, 2H), 1.76 (m, 2H), 0.71 – 0.44 (m, 2H), 0.18 –-0.19 (m, 9H)。ESI-MS m/z計算值198.1188,發現199.3 (M+1)+
;保留時間:2.94分鐘(LC方法B)。步驟 3 : (14S
)-12,12- 二甲基 -8-{3-[3-( 三甲基矽基 ) 丙氧基 ]-1H
- 吡唑 -1- 基 }-2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5(10),6,8,19(23),20- 己烯 -2,2,4- 三酮,化合物 (1-6)
向三甲基-[3-(1H
-吡唑-3-基氧基)丙基]矽烷(50 mg,0.2521 mmol)、(14S)-8-
溴-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(150 mg,0.2518 mmol)、L-脯胺酸(7 mg,0.0608、mmol)、及K2
CO3
(106 mg,0.7670 mmol)於DMSO (2 mL)中之溶液中添加CuI (7 mg,0.0368 mmol)。將溶液用氮氣鼓泡30 min,然後在100℃下加熱24 h。將反應物稀釋到10 mL水中,且添加2 M HCl直至pH為酸性,且將反應物用DCM (3×20 mL)萃取。將經合併之有機層用鹽水(2 x 20 mL)洗滌,經Na2
SO4
乾燥,且在減壓下濃縮。使用己烷中之20%-50%丙酮溶離進行管柱層析(二氧化矽,12 g),接著使用水中之0至100%乙腈(以0.1%TFA緩衝)進行反相HPLC,得到呈白色固體之(14S
)-12,12-二甲基-8-{3-[3-(三甲基矽基)丙氧基]-1H
-吡唑-1-基}-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(22),5(10),6,8,19(23),20-己烯-2,2,4-三酮(12.4 mg,8%) (12.4 mg,8%)。1
HNMR (500 MHz, DMSO-d6
) δ 12.52 (s, 1H), 8.21 (d, J = 2.8 Hz, 1H), 7.82 (d, J = 8.3 Hz, 1H), 7.58 (dd, J = 8.5, 7.2 Hz, 1H), 7.06 (d, J = 7.1 Hz, 1H), 6.99 (d, J = 8.2 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.72 (d, J = 8.4 Hz, 1H), 6.11 (d, J = 2.7 Hz, 1H), 4.16 (t, J = 6.8 Hz, 2H), 3.93 (d, J = 11.8 Hz, 1H), 3.15 (s, 1H), 2.96 (d, J = 13.3 Hz, 1H), 2.77 – 2.65 (m, 1H), 2.11 (d, J = 23.4 Hz, 1H), 1.90 – 1.83 (m, 1H), 1.80 – 1.69 (m, 3H), 1.64 – 1.49 (m, 9H), 1.39 – 1.25 (m, 1H), 0.63 – 0.56 (m, 2H) 0.02 (s, 9H)。ESI-MS m/z計算值611.271,實測值612.5 (M+1)+;保留時間:3.55分鐘(LC方法H)。實例 9 : (14S
)-12,12- 二甲基 -8-{3-[( 三甲基甲鍺烷基 ) 甲氧基 ]-1H
- 吡唑 -1- 基 }-2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(23),5(10),6,8,19,21- 己烯 -2,2,4- 三酮,化合物 (2-1) 之製備 步驟 1 : 3-( 三甲基鍺烷基甲氧基 ) 吡唑 -1- 羧酸三級
丁酯
向75 mL燒瓶中裝入3-羥基吡唑-1-羧酸三級
丁酯(2.38 g,12.92 mmol)、氯甲基(三甲基)鍺烷(2.27 g,13.57 mmol)、碳酸鉀(3.4 g,24.60 mmol)及DMA( 25 mL)。將反應物在70℃油浴中加熱26小時。將反應混合物用NaHCO3
及乙酸乙酯稀釋。分離兩層。將有機層用水、鹽水洗滌,經無水硫酸鈉乾燥且濃縮,得到粗材料,藉由層析純化該粗材料,得到呈澄清油狀物之3-(三甲基鍺烷基甲氧基)吡唑-1-羧酸三級
丁酯(2.57g,62%)。1
HNMR (250 MHz, CDCl3
) δ 7.83 (d, J = 3.0 Hz, 1H), 5.85 (d, J = 3.1 Hz, 1H), 4.20 (s, 2H), 1.61 (s, 9H), 0.23 (s, 9H)。ESI-MS m/z計算值316.0842,實測值317.0 (M+1)+
;保留時間:3.33分鐘(LC方法B)。步驟 2 :三甲基 (1H
- 吡唑 -3- 基氧基甲基 ) 鍺烷
向3-(三甲基鍺烷基甲氧基)吡唑-1-羧酸三級
丁酯(2.56 g,7.97 mmol)於DCM(25 mL)中之溶液中添加TFA (12.5 mL)。將反應物在室溫下攪拌1小時。去除所有揮發物,且添加EtOAc。然後將所得混合物用飽和NaHCO3
水溶液洗滌一次,經硫酸鈉乾燥,過濾且濃縮,得到呈黃色油狀物之粗的三甲基(1H
-吡唑-3-基氧基甲基)鍺烷(1.7 g,94%),其直接用於下一步驟。1
HNMR (250 MHz, CDCl3
) δ 7.35 (d, J = 2.4 Hz, 1H), 5.73 (d, J = 2.5 Hz, 1H), 4.06 (s, 2H), 0.25 (s, 9H)。步驟 3 : (14S
)-12,12- 二甲基 -8-{3-[( 三甲基甲鍺烷基 ) 甲氧基 ]-1H
- 吡唑 -1- 基 }-2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(23),5(10),6,8,19,21- 己烯 -2,2,4- 三酮,化合物 (2-1)
將CuI (4.3 mg,0.0226 mmol)添加到(14S)-8-
溴-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(140 mg,0.2350 mmol)、三甲基(1H-
吡唑-3-基氧基甲基)鍺烷(61 mg,0.2839 mmol)、(1R
, 2R
)-N
1,N
2-二甲基環己烷-1,2-二胺( 19.800 mg,22 μL,0.1392 mmol)及碳酸鉀(86 mg,0.6223 mmol)於乙酸丁酯(1.9 mL)及DMSO(0.56 mL)之脫氣溶液中。將反應混合物在120℃下升溫6小時。將反應物冷卻至室溫,且經矽藻土過濾混合物,用MeTHF (20 mL)溶離。將濾液用水(25 mL)及鹽水(3×25 mL)洗滌。將有機相經硫酸鈉乾燥,過濾且濃縮至乾燥。藉由正相層析(10 + 12 g SiO2
,以己烷中之20%至100%乙酸乙酯溶離)且藉由C18反相層析(30 g,以水中之50%至100%乙腈溶離)純化粗產物,且將其冷凍乾燥,得到呈白色蓬鬆固體之(14S
)-12,12-二甲基-8-{3-[(三甲基甲鍺烷基)甲氧基]-1H
-吡唑-1-基}-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(67.3 mg,46%)。1
HNMR (400 MHz, DMSO-d6
) δ 12.48 (br. s., 1H), 8.20 (d, J = 2.7 Hz, 1H), 7.81 (d, J = 8.1 Hz, 1H), 7.57 (t, J = 7.9 Hz, 1H), 7.05 (d, J = 7.1 Hz, 1H), 7.02- 6.95 (m, 1H), 6.93 (d, J = 8.3 Hz, 1H), 6.71 (d, J = 8.6 Hz, 1H), 6.10 (d, J = 2.7 Hz, 1H), 4.22- 4.14 (m, 2H), 3.99- 3.85 (m, 1H), 3.22- 3.10 (m, 1H), 2.95 (d, J = 13.0 Hz, 1H), 2.78- 2.65 (m, 1H), 2.13 (br. s., 1H), 1.86 (dd, J = 11.6, 5.0 Hz, 1H), 1.82- 1.69 (m, 1H), 1.66- 1.53 (m, 6H), 1.51 (s, 3H), 1.38- 1.25 (m, 1H), 0.23 (s, 9H)。ESI-MS m/z計算值629.184,實測值630.2 (M+1)+;保留時間:3.75分鐘(LC方法J)。實施例 10 : (14S
)-8-[3-({ 二甲基 [1-( 三氟甲基 ) 環丙基 ] 矽基 } 甲氧基 )-1H
- 吡唑 -1- 基 ]-12,12- 二甲基 -2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5(10),6,8,19(23),20- 己烯 -2,2,4- 三酮,化合物 (1-1) 之製備 步驟 1 :氯甲基 - 二甲基 -[1-( 三氟甲基 ) 乙烯基 ] 矽烷
在圓底燒瓶中稱入2-溴-3,3,3-三氟丙-1-烯(3.28 g,18.186 mmol)及氯-(氯甲基)-二甲基矽烷(1.0604 g,1 mL,7.2627 mmol)。添加乙醚(80mL)。將經隔膜密封之燒瓶置於氮氣球下,且在乙醇/液體氮氣浴中冷卻。使用熱偶溫度計讀數來控制反應溫度。將浴保持在約-115℃下且攪拌15 min。然後經由注射器沿著反應燒瓶之內壁逐滴添加t-
BuLi (10.8 mL 1.7 M,18.360 mmol)。將溫度保持低於-110℃達15 min,然後在60 min內升溫至-60℃。在冷卻後期期間,將乾冰添加到冷卻乙醇中,以保持溫度低於-60℃。添加NH4
Cl (5 mL,飽和水溶液),然後添加水(10mL)。將混合物升溫至約0℃。分離各層。將乙醚層用鹽水洗滌,經無水MgSO4
乾燥,過濾並濃縮(>150 mm Hg,25℃浴溫),得到呈淡黃色油狀物之氯甲基-二甲基-[1-(三氟甲基)乙烯基]矽烷(1.4 g,76%)。1
HNMR (250 MHz, 氯仿-d) δ 6.47 (s, 1H), 5.95 (s, 1H), 2.91 (s, 2H), 0.35 (s, 6H)。步驟 2 :氯甲基 - 二甲基 -[1-( 三氟甲基 ) 環丙基 ] 矽烷
將氯甲基-二甲基-[1-(三氟甲基)乙烯基]矽烷(1.4 g,5.5260 mmol)溶解於THF (20 mL)中。添加甲基(二苯基)鋶鹽;四氟硼酸鹽(2.5 g,8.2433 mmol)。將混合物在乾冰丙酮 (約-75℃浴溫)中冷卻。逐滴添加LiHMDS (16mL 1M THF溶液,16.0 mmol)。然後將混合物緩慢升溫至室溫且攪拌15 h。將混合物加載到約80 g矽膠前置管柱,且使用80 g矽膠管柱、使用100%戊烷純化,提供呈淡黃色油狀物之粗產物氯甲基-二甲基-[1-(三氟甲基)環丙基]矽烷(720 mg,48%)。1
HNMR (250 MHz, 氯仿-d) δ 2.92 (s, 2H), 1.1-0.94 (m, 2H), 0.79 – 0.59 (m, 2H),0.27-0.03 (m, 6H)。步驟 3 :乙酸 [ 二甲基 -[1-( 三氟甲基 ) 環丙基 ] 矽基 ] 甲酯
在室溫下將氯甲基-二甲基-[1-(三氟甲基)環丙基]矽烷(610 mg,2.3927 mmol)溶解於DMF (5 mL)中。一次性添加NaOAc (196.28 mg,2.3927 mmol)。將混合物用隔膜密封,且在攪拌下置於100℃油浴中達20 h。然後將其冷卻至室溫,且分配於乙醚(50 mL)與水(50mL)之間。分離各層,且將醚層用水(30 mL×3)、鹽水(30 mL)洗滌,經無水MgSO4
乾燥且過濾。在>120 mm Hg (<26℃油浴)下濃縮濾液。淺棕色粗產物(含有乙醚)乙酸[二甲基-[1-(三氟甲基)環丙基]矽基]甲酯(1.18 g,99%)不經進一步純化即用於下一步驟中。1
HNMR (250 MHz, 氯仿-d) δ 3.86 (s, 2H), 2.06 (s, 3H), 1.07 – 0.95 (m, 2H), 0.77 – 0.59 (m,2H), 0.15 (s, 6H)。步驟 4 : [ 二甲基 -[1-( 三氟甲基 ) 環丙基 ] 矽基 ] 甲醇
將乙酸[二甲基-[1-(三氟甲基)環丙基]矽基]甲酯(1.18 g,2.3571 mmol)溶解於乙醚(15 mL)中,且在冰水浴中冷卻,在氮氣氛下攪拌。少量添加LAH (100 mg,0.1091 mL,2.6347 mmol)。無需外部冷卻,即攪拌混合物。2 h後,藉由移液管添加洛瑟耳鹽(Rochelle's salt) (10 mL飽和水溶液)。將混合物在室溫下攪拌30 min。分離各層且用更多乙醚(15 mL)萃取水層。將經合併之乙醚溶液經無水MgSO4
乾燥,過濾且濃縮,得到呈無色油狀物之粗的[二甲基-[1-(三氟甲基)環丙基]矽基]甲醇(800 mg,86%)。1
HNMR (250 MHz, 氯仿-d) δ 1.07 – 0.95 (m, 2H), 0.81 – 0.61 (m, 2H), 0.34 – 0.04 (m, 6H) (錯失的CH2
質子可能與殘餘乙醚峰重疊)。步驟 5 : 3-[[ 二甲基 -[1-( 三氟甲基 ) 環丙基 ] 矽基 ] 甲氧基 ] 吡唑 -1- 羧酸三級
丁酯
在室溫下將[二甲基-[1-(三氟甲基)環丙基]矽基]甲醇(800 mg,2.02 mmol)溶解於THF(10 mL)中。添加3-羥基吡唑-1-羧酸三級
丁酯(405 mg,2.20 mmol)、PPh3
(529 mg,2.02 mmol)及DIAD(0.39 mL,2.0176 mmol)。將混合物在室溫下攪拌30 min。HPLC分析顯示形成所要產物。將混合物在氮氣下再攪拌12 h。然後將其濃縮且藉由使用己烷中之0-15% EtOAc溶離進行矽膠層析(40g柱)純化殘餘物,得到澄清(帶有黃色)油狀物。產物3-[[二甲基-[1-(三氟甲基)環丙基]矽基]甲氧基]吡唑-1-羧酸三級
丁酯(380 mg,51%)在儲存於冰箱中之後固化成淡黃色固體。1
HNMR (250 MHz, 氯仿-d) δ 7.83 (d, J = 3.0 Hz, 1H), 5.85 (d, J = 3.0 Hz, 1H), 4.07 (s, 2H), 1.61 (s, 9H), 1.12 – 0.84 (m, 2H), 0.84 – 0.58 (m, 2H), 0.19 (s, 6H)。ESI-MS m/z計算值364.143,實測值365.5 (M+1)+
;保留時間:4.04分鐘(LC方法B)。步驟 6 :二甲基 -(1H
- 吡唑 -3- 基氧基甲基 )-[1-( 三氟甲基 ) 環丙基 ] 矽烷
在室溫下將3-[[二甲基-[1-(三氟甲基)環丙基]矽基]甲氧基]吡唑-1-羧酸三級
丁酯(550 mg,1.43 mmol)溶解於DCM (20 mL)中。一次性添加TFA (10 mL,129.80 mmol)。將混合物攪拌90 min。然後將其用DCM (20 mL)稀釋,且用NaHCO3
(10 mL,飽和水溶液)處理。分離各層,且將DCM層經無水Na2
SO4
乾燥,過濾且濃縮,得到呈無色油狀物之二甲基-(1H
-吡唑-3-基氧基甲基)-[1-(三氟甲基)環丙基]矽烷(390 mg,98%)。ESI-MS m/z計算值264.0906,實測值265.3 (M+1)+
;保留時間:3.16分鐘(LC方法B)。步驟 7 : (14S
)-8-[3-({ 二甲基 [1-( 三氟甲基 ) 環丙基 ] 矽基 } 甲氧基 )-1H
- 吡唑 -1- 基 ]-12,12- 二甲基 -2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5(10),6,8,19(23),20- 己烯 -2,2,4- 三酮,化合物 (1-1)
向二甲基-(1H
-吡唑-3-基氧基甲基)-[1-(三氟甲基)環丙基]矽烷(33 mg,0.12 mmol)、(14S)-8-
溴-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(23),5(10),6,8,19,21-己烯-2,2,4-三酮(55 mg,0.106 mmol)、L-脯胺酸(15 mg,0.1303 mmol)、及Na2
CO3
(70 mg,0.66 mmol)於DMSO (1 mL)中之溶液中添加CuI (12 mg,0.06 mmol)。將該溶液用真空/N2
吹掃三次,然後密封且在110℃下加熱18 h。將反應物稀釋到10 mL水中,添加3M HCl水溶液直至pH為酸性,且將產物用乙酸乙酯(2×50 mL)萃取。將經合併之有機層用鹽水(2 x 20 mL)洗滌,經Na2
SO4
乾燥,且在減壓下濃縮。藉由使用以0.1% TFA緩沖之水中之30%至80%乙腈進行製備型HPLC來純化殘餘物,得到:呈米色粉末之(14S
)-8-[3-({二甲基[1-(三氟甲基)環丙基]矽基}甲氧基)-1H
-吡唑-1-基]-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(22),5(10),6,8,19(23),20-己烯-2,2,4-三酮(6.4 mg,9%)。1
HNMR (400 MHz, DMSO-d6
) δ 12.49 (s, 1H), 8.21 (s, 1H), 7.82 (d, J = 8.2 Hz, 1H), 7.58 (s, 1H), 7.05 (d, J = 7.1 Hz, 1H), 6.98 (d, J = 8.6 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.71 (d, J = 8.5 Hz, 1H), 6.13 (s, 1H), 4.05 (s, 2H), 3.93 (s, 1H), 3.15 (s, 1H), 2.97 (s, 1H), 2.71 (s, 1H), 2.12 (s, 1H), 1.75 (s, 2H), 1.61-151 (m, 9H), 1.33 (s, 1H), 0.98 (s, 2H), 0.89 (s, 2H), 0.19 (s, 6H)。ESI-MS m/z計算值677.2427,實測值678.5 (M+1)+
;保留時間:3.83分鐘(LC方法H)。實施例 11 : (14S
)-8-[3-(2-{ 雙螺 [2.0.24.13] 庚 -7- 基 } 乙氧基 )-1H
- 吡唑 -1- 基 ]-12,12- 二甲基 -7-( 三甲基矽基 )-2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5,7,9,19(23),20- 己烯 -2,2,4- 三酮,化合物 (1-9) 步驟 1 : 2- 芐基硫烷基 -6- 氟 - 吡啶
在配備有頂置式攪拌器、溫度探針及加料漏斗之5 L三頸圓底燒瓶中,將2,6-二氟吡啶(200 g,1.738 mol)溶解於二甲基亞碸(2 L)中。添加碳酸銫(572.4 g,1.757 mol)。經由
加料漏斗逐滴添加苯甲硫醇(206 mL,1.755 mol)。在添加期間觀測到放熱。溫度升至約40℃。將反應物在室溫下攪拌隔夜。將反應物倒入水中,且用二氯甲烷萃取。將萃取物用水洗滌兩次,且經小矽膠塞過濾。將塞用二氯甲烷溶離,且將濾液在真空
中蒸發,得到呈桃色油狀物之2-芐基硫烷基-6-氟-吡啶(366 g,96%),其在真空下固化成大塊狀板。1
H NMR (400 MHz, 氯仿-d) δ 7.58 (q,J =
7.9 Hz, 1H), 7.48 - 7.41 (m, 2H), 7.36 - 7.25 (m, 3H), 7.06 (dd,J =
7.6, 2.1 Hz, 1H), 6.62 (dd,J =
7.9, 2.6 Hz, 1H), 4.43 (s, 2H)。步驟 2 : 6- 氟吡啶 -2- 磺醯胺
在配備有頂置式攪拌器及溫度探針之12 L三頸圓底燒瓶中,將2-芐基硫烷基-6-氟吡啶(303.2 g,1.383 mol)溶解於氯仿(2.0 L)中。添加水(1.5L),且將混合物在冰浴中冷卻至0℃,且劇烈攪拌。藉由插入穿過燒瓶第三頸之隔膜的巴斯德吸管(Pasteur pipet),將來自演講瓶之氯氣劇烈鼓泡到反應物中。迅速形成白色沉澱。在添加期間觀測到放熱。當溫度升至20℃時,停止添加氯。在添加更多氯氣前,使反應物再次冷卻。繼續加液直至反應物變成黃綠色,且在攪拌30分鐘後保持該狀態。此時,未觀測到進一步放熱。將反應物倒入40%亞硫酸氫鈉水溶液中。分離有機層,且將水層用另一部分氯仿萃取。合併有機層,經硫酸鎂乾燥,過濾,且在真空
中蒸發,得到淺黃色油狀物。在配備有頂置式攪拌器、溫度探針及加料漏斗之12 L三頸圓底燒瓶中,將該油溶解於二氯甲烷(1.5 L)中,且逐滴添加到氫氧化銨(1.5 L,40重量%,17.12 mol)。在添加前,將氫氧化銨溶液在冰浴中冷卻至0℃。調節添加速率,以使反應溫度保持低於10℃。將所得黃綠色溶液攪拌一小時,且倒入冰中。分離各層(有機層為深綠色),且用更多二氯甲烷萃取水層。丟棄有機層。將水層在冰浴中冷卻,且向水層中分批添加濃鹽酸水溶液直至pH為強酸性。隨著添加各批次,攪拌所得混合物。將所得水溶液用乙酸乙酯萃取兩次。將有機層合併,經硫酸鎂乾燥,過濾,且在真空
中蒸發,得到淺棕色固體。將固體與二氯甲烷(約500 mL)混合,且用磁力攪拌棒攪拌,直到大部分塊狀物破碎。添加約1.5 L戊烷,沉澱出許多淺棕色固體。短暫攪拌所得混合物,然後過濾。將濾餅用戊烷洗滌,且在真空
中乾燥,得到呈淺棕色固體之6-氟吡啶-2-磺醯胺(204.1 g,84%)。1
H NMR (300 MHz, 二甲基亞碸-d6
) δ 8.52 - 8.11 (m, 1H), 7.89 (dd,J =
7.8, 2.7 Hz, 1H), 7.67 (s, 2H), 7.57 - 7.44 (m, 1H)。步驟 3 : 3-(3- 羥丙基 )-5,5- 二甲基 - 吡咯啶 -2- 酮
為1000 mL 3頸圓底燒瓶裝備特氟龍攪拌棒、加熱套、J-Kem溫度探針/控制器及橡膠隔膜。向容器中裝入3-(2-甲基-2-硝基-丙基)四氫哌喃-2-酮(25 g,124.2 mmol)及乙醇(375 mL),提供了白色懸浮液。開始攪拌,且將懸浮液加熱至40℃達10 min,提供澄清的無色溶液。然後,為該容器裝備氣體分散管,且將溶液用氮氣脫氣15 min。然後在容器中裝入雷氏鎳(8.019 g 50重量%,68.31 mmol),且然後為該容器裝備隔膜。將該容器抽真空且置於氫氣氛下。將該過程重複三個循環。然後將容器置於1大氣壓之氫氣下,且將反應混合物逐漸加熱至60℃。將反應物在60℃下繼續攪拌24 h。冷卻至室溫後,為該容器裝備氣體分散管,且將反應混合物用氮氣脫氣15 min。透過具有20 mm矽藻土層之玻璃膠布氏漏斗真空過濾混合物。將濾餅用乙醇(2×100 mL)置換洗滌且吸拉直至略微乙醇潤濕,然後用水潤濕,且在水下丟棄所用雷氏鎳催化劑。將澄清淺琥珀色濾液在減壓下濃縮成澄清的黏稠淺琥珀色油狀物。用甲基三級
丁基醚(1500 mL)稀釋該油狀物,且將混濁溶液在減壓下濃縮至約150 mL體積,提供了懸浮液。再次用甲基三級
丁基醚(1500 mL)稀釋混合物,且在減壓下濃縮至約150 mL體積。將所得懸浮液在室溫下放置隔夜(約12 h)。藉由在玻璃膠布氏漏斗中真空過濾來收集固體,且將濾餅用冷的甲基三級
丁基醚(2×50 mL)置換洗滌,然後吸拉30分鐘。將該材料在45℃真空烘箱中進一步乾燥3 h,得到呈白色固體(1 9 g,0.111 mol,89%產率)之產物3-(3-羥丙基)-5,5-二甲基-吡咯啶酮-2-酮。1
H NMR (400 MHz, 二甲基亞碸-d6
) δ 7.63 (s, 1H), 3.38 (t,J =
6.5 Hz, 2H), 2.37 (tdd,J =
9.8, 8.5, 4.4 Hz, 1H), 2.02 (dd,J =
12.3, 8.6 Hz, 1H), 1.72 (tdd,J =
9.6, 7.5, 4.4 Hz, 1H), 1.52 - 1.32 (m, 3H), 1.28 - 1.03 (m, 7H)。步驟 4 : 3-(5,5- 二甲基吡咯啶 -3- 基 ) 丙 -1- 醇
為5 L 3頸圓底燒瓶裝備機械攪拌器、加熱套、加料漏斗、J-Kem溫度探針/控制器及氮氣入口/出口。在氮氣氛下,向該容器中裝入氫化鋁鋰團粒(19.39 g,510.9 mmol)。然後向容器中裝入四氫呋喃(500 mL,20 mL/g)。開始攪拌,且在20℃下記錄鍋溫。將混合物在室溫下攪拌0.5 h,以使團粒溶解。在24℃下記錄所得灰色懸浮液之鍋溫。向加料漏斗中裝入3-(3-羥丙基)-5,5-二甲基-吡咯啶-2-酮(25 g,146.0 mmol)於四氫呋喃(500 mL)中之溶液,且在90 min內逐滴添加澄清淡黃色溶液。需要輕微加熱以實現均勻性。在完成添加後,在24℃下記錄所得淺灰色懸浮液之鍋溫。然後將混合物加熱至65℃鍋溫,且將該條件保持72 h。此時反應混合物之分析表明仍殘留一些殘餘起始材料且產物形成無變化。隨後在此時停止反應。移去加熱套,且為該容器裝備冷卻浴。將該懸浮液在碎冰/水冷卻浴中至0℃,且然後藉由極其緩慢地逐滴添加水(19.93 mL),接著添加15重量%氫氧化鈉溶液(19.93 mL)且然後最後用水(59.79 mL)來淬滅。在5℃下記錄所得白色懸浮液之鍋溫。去除冷卻浴,且再次為容器裝備加熱套。將懸浮液升溫至60℃,且將該條件保持30 min。透過具有20 mm矽藻土層之玻璃膠布氏漏斗對溫熱懸浮液進行真空過濾。然後將濾餅用60℃四氫呋喃(2×250 mL)置換洗滌,且然後吸拉30 min。將澄清濾液在減壓下濃縮,以提供(23.5 g,0.149 mol,99%產率)呈澄清淺黃色黏性油狀物之所要產物3-(5,5-二甲基吡咯啶-3-基)丙烷-1-醇。1
H NMR (400 MHz, 二甲基亞碸-d6
) δ 3.37 (dt,J =
8.3, 6.4 Hz, 3H), 2.95 (dd,J =
10.6, 7.6 Hz, 1H), 2.40 (dd,J =
10.7, 7.7 Hz, 1H), 2.04 (dt,J =
16.1, 8.1 Hz, 1H), 1.69 (dd,J =
12.2, 8.2 Hz, 1H), 1.50 - 1.24 (m, 5H), 1.11 - 0.94 (m, 7H)。步驟 5 : 4-(3- 羥丙基 )-2,2- 二甲基吡咯啶 -1- 羧酸三級
丁酯
為1 L 3頸圓底燒瓶裝備機械攪拌器、冷卻浴、加料漏斗、J-Kem溫度探針及氮氣入口/出口。在氮氣氛下,向容器中裝入3-(5,5-二甲基吡咯啶-3-基)丙-1-醇(15 g,95.39 mmol)及二氯甲烷(225 mL,15 mL/g),提供了澄清淺黃色溶液。開始攪拌,且在19℃下記錄鍋溫。向冷卻浴中裝入碎冰/水且將鍋溫降低至0℃。向加料漏斗中裝入三乙胺(12.55 g,124.0 mmol),隨後在5 min內逐滴添加純淨物。未觀測到放熱。然後向加料漏斗中裝入溶解於二氯甲烷(225 mL)中之二碳酸二-三級
丁酯(22.89 g,104.9 mmol)。然後在30 min內逐滴添加澄清淡黃色溶液,這導致輕微氣體釋放。未觀測到放熱。去除冷卻浴,且將所得澄清淺黃色溶液升溫至室溫,且在室溫下繼續攪拌3 h。將反應混合物轉移至分液漏斗,且分配於水(75 mL)中。去除有機物,且將其用飽和氯化鈉溶液(75 mL)洗滌,經硫酸鈉(150g)乾燥,且然後透過玻璃膠布氏漏斗過濾。在減壓下濃縮濾液,提供(30g)呈澄清淺黃色油狀物之所要粗產物。藉由使用100%二氯甲烷至二氯甲烷中之10%甲醇溶離進行矽膠管柱急速層析(液體負載有二氯甲烷)來純化該材料,在60 min內收集50 mL流份。將所要產物流份合併且在減壓下濃縮,得到呈澄清淡黃色黏性油狀物之4-(3-羥丙基)-2,2-二甲基-吡咯啶-1-羧酸三級
丁酯(22 g,0.0855 mol,90%收率)。1
H NMR (400 MHz, DMSO-d6
) δ 4.38 (td,J =
5.2, 1.4 Hz, 1H), 3.54 (dt,J =
10.3, 6.7 Hz, 1H), 3.38 (td,J =
6.6, 3.5 Hz, 2H), 2.76 (q,J =
10.3 Hz, 1H), 2.07 (td,J =
11.6, 5.7 Hz, 1H), 1.87 (ddd,J =
16.7, 12.1, 6.0 Hz, 1H), 1.37 (dd,J =
14.2, 10.4 Hz, 17H), 1.24 (s, 3H)。步驟 6 : 2,2- 二甲基 -4-(3- 甲基磺醯氧基丙基 ) 吡咯啶 -1- 羧酸三級
丁酯
將4-(3-羥丙基)-2,2-二甲基吡咯啶-1-羧酸三級
丁酯(50.5 g,196.22 mmol)及三乙胺(39.711 g,54.698 mL,392.44 mmol)溶解於二氯甲烷(500 mL)中,且將所得溶液在冰水浴中冷卻30 min。在30 min時段內逐滴添加甲磺醯氯(24.725 g,16.706 mL,215.84 mmol),然後去除冰浴,且將混合物在室溫下攪拌一小時。然後將反應物用飽和碳酸氫鈉溶液(200 mL)淬滅。分離各相,且將有機相用飽和碳酸氫鈉(200 mL)及水(2×100 mL)萃取。丟棄水相,且將有機相經硫酸鈉乾燥,過濾且在真空
中濃縮,得到呈淡黃色油狀物之2,2-二甲基-4-(3-甲基磺醯氧基丙基)吡咯啶-1-羧酸三級
丁酯(64.2 g,93%)。ESI-MS m/z計算值335.1766,實測值336.4 (M+1)+
;保留時間:5.54分鐘(LC方法C)。步驟 7 : 4-(3- 胺基丙基 )-2,2- 二甲基吡咯啶 -1- 羧酸三級
丁酯
將2,2-二甲基-4-(3-甲基磺醯氧基丙基)吡咯啶-1-羧酸三級
丁酯(64.2 g,191.38 mmol)溶解於二噁烷(650 mL)中,且然後添加氫氧化銨(650 mL),且將所得混合物加熱至45℃達18 h。18 h後,將反應物冷卻至室溫。將溶液用1M氫氧化鈉(200 mL)稀釋,且然後用乙醚(3×650 mL)萃取。丟棄水相,將經合併之有機相用水(2×200 mL)萃取。丟棄水相,將有機相經硫酸鈉乾燥,過濾且在真空
中濃縮,得到呈淡黃色油狀物之4-(3-胺基丙基)-2,2-二甲基-吡咯啶-1-羧酸三級
丁酯(48.9g,95%)。ESI-MS m/z計算值256.2151,實測值257.3 (M+1)+
;保留時間:3.70分鐘(LC方法C)。步驟 8 : 2,2- 二甲基 -4-[3-[(6- 胺磺醯基 -2- 吡啶基 ) 胺基 ] 丙基 ] 吡咯啶 -1- 羧酸三級
丁酯
向於二甲基亞碸(75 mL)中之4-(3-胺基丙基)-2,2-二甲基吡咯啶-1-羧酸三級
丁酯(8.91 g,34.8 mmol)及6-氟吡啶-2-磺醯胺(6.13 g,34.8 mmol)中添加碳酸鉀(4.91 g,35.5 mmol),且將混合物在100℃下攪拌12 h,且然後將其冷卻至環境溫度,且再攪拌4 h (共16 h)。將反應混合物緩慢倒入水(200 mL)中之鹽酸(35 mL 1 M,35.00 mmol)中(有些起泡沫),且用乙酸乙酯(250 mL)稀釋。分離有機相,且用100 mL鹽水洗滌。將有機相經硫酸鎂乾燥,經矽藻土過濾,且在真空
中濃縮,得到深黃色油狀物。藉由用己烷中之0%-100%乙酸乙酯溶離進行矽膠層析來純化粗產物。收集純流份(9.0 g)及不純流份(3 g)。藉由使用己烷中之0%-100%乙酸乙酯溶離進行矽膠層析來純化不純流份,總共得到2,2-二甲基-4-[3-[(6-胺磺醯基-2-吡啶基)胺基]丙基]吡咯啶-1-羧酸三級
丁酯(10.0 g,69%)。1
H NMR (400 MHz, 二甲基亞碸-d6
) δ 7.52 (dd,J =
8.5, 7.2 Hz, 1H), 7.07 (s, 2H), 6.95 (dd,J =
7.2, 0.7 Hz, 2H), 6.61 (d,J =
8.5 Hz, 1H), 3.55 (q,J =
9.1 Hz, 1H), 3.32 - 3.24 (m, 2H), 2.79 (q,J =
10.0 Hz, 1H), 2.13 (d,J =
16.1 Hz, 1H), 1.96 - 1.82 (m, 1H), 1.51 (dt,J =
18.0, 9.3 Hz, 2H), 1.37 (dd,J =
12.9, 10.6 Hz, 15H), 1.24 (s, 3H)。ESI-MS m/z計算值412.21442,實測值413.1 (M+1)+
;保留時間:2.34分鐘(LC方法M)。步驟 9 : (4S
)-2,2- 二甲基 -4-[3-[(6- 胺磺醯基 -2- 吡啶基 ) 胺基 ] 丙基 ] 吡咯啶 -1- 羧酸三級
丁酯
藉由使用ChiralPak IG (250 x 21.2 mm管柱,5 μm粒徑)在11.0 min內使用40%甲醇/60%二氧化碳流動相在70 mL/min下(注入體積= 500 μL甲醇中之32 mg/mL溶液)進行SFC層析來對外消旋2,2-二甲基-4-[3-[(6-胺磺醯基-2-吡啶基)胺基]丙基]吡咯啶-1-羧酸三級
丁酯(7 g,16.97 mmol)進行對掌性分離,得到第一個溶離峰(4S
)-2,2-二甲基-4-[3-[(6-胺磺醯基-2-吡啶基)胺基]丙基]吡咯啶-1-羧酸三級
丁酯(3.4481 g,99%)。ESI-MS m/z計算值412.21442,實測值413.2 (M+1)+
;保留時間:0.63分鐘(LC方法N)。步驟 10 : (2,6- 二氯 -3- 吡啶基 )- 三甲基 - 矽烷
將2,6-二氯吡啶(4.01 g,27.10 mmol)溶解於THF(80 mL)且在乾冰/丙酮浴中冷卻。緩慢添加THF/庚烷/乙基苯中之LDA (15 mL 2 M,30.00 mmol),且將反應物在-75℃下攪拌1 h。此時,添加TMS-Cl (3.5 mL,27.58 mmol),且將反應混合物在-75℃下再攪拌1 h。用HCl水溶液(50 mL 1 M,50.00 mmol)淬滅反應物,且使其升溫至室溫。分離各層,且將水層進一步用乙醚萃取。將有機物合併,用鹽水洗滌,經硫酸鈉乾燥且蒸發。藉由使用己烷中之0-10%乙酸乙酯溶離進行矽膠層析來純化粗製液,得到呈澄清液體之(2,6-二氯-3-吡啶基)-三甲基-矽烷(3.933g,66%)。1
H NMR (400 MHz, DMSO-d6
) δ 7.93 (d,J =
7.7 Hz, 1H), 7.56 (d,J =
7.7 Hz, 1H), 0.36 (s, 9H)。ESI-MS m/z計算值219.00378,實測值220.1 (M+1)+
;保留時間:0.76分鐘(LC方法D)。步驟 11 : 2,6- 二氯 -5- 三甲基矽基 - 吡啶 -3- 羧酸
將(2,6-二氯-3-吡啶基)-三甲基矽烷(531 mg,2.41 mmol)溶解於THF(10 mL)中,且在乾冰:丙酮浴中冷卻至-75℃。緩慢添加THF/庚烷/乙基苯中之LDA (1.25 mL 2 M,2.50 mmol),且將反應混合物再攪拌1 h。使CO2
氣體鼓泡透過反應混合物1 min。使反應物升溫至室溫且攪拌15 min。將反應混合物用乙酸乙酯稀釋,且用1M HCl水溶液洗滌。分離有機物,將其用鹽水洗滌,經硫酸鈉乾燥且蒸發。將粗材料用己烷研磨,且藉由真空過濾收集所得固體。進一步乾燥固體,得到呈白色固體之2,6-二氯-5-三甲基矽基-吡啶-3-羧酸(353 mg,55%)。1
H NMR (400 MHz, DMSO-d6
) δ 13.96 (s, 1H), 8.21 (s, 1H), 0.38 (s, 9H)。ESI-MS m/z計算值262.99362,實測值264.1 (M+1)+
;保留時間:0.65分鐘(LC方法A)。步驟 12 : (2,6- 二氯 -5- 三甲基矽基 -3- 吡啶基 )- 咪唑 -1- 基 - 甲酮
將2,6-二氯-5-三甲基矽基-吡啶-3-羧酸(570 mg,2.158 mmol)及CDI (530 mg,3.269 mmol)溶解於THF(9 mL)中,且使其反應隔夜。將反應混合物在減壓下濃縮且在矽膠上使用0至60%乙酸乙酯純化所得殘餘物(所要產物在約40%乙酸乙酯下溶離),得到呈油狀物之(2,6-二氯-5-三甲基矽基-3-吡啶基)-咪唑-1-基-甲酮(430 mg,63%),其在靜置時緩慢結晶。1
H NMR (400 MHz, CDCl3
) δ 7.93 (s, 1H), 7.86 (s, 1H), 7.44 (s, 1H), 7.20 (d,J =
1.0 Hz, 1H), 0.48 - 0.34 (m, 9H)。ESI-MS m/z計算值313.0205,實測值314.0 (M+1)+
;保留時間:1.91分鐘(LC方法L)。步驟 13 : (4S
)-4-[3-[[6-[(2,6- 二氯 -5- 三甲基矽基 - 吡啶 -3- 羰基 ) 胺磺醯基 ]-2- 吡啶基 ] 胺基 ] 丙基 ]-2,2- 二甲基 - 吡咯啶 -1- 羧酸三級
丁酯
將DBU (1.2 mL,8.0243 mmol)添加到溶解於THF(72 mL)中之(2,6-二氯-5-三甲基矽基-3-吡啶基)-咪唑-1-基甲酮(1.2 g,3.8187 mmol)及(4S
)-2,2-二甲基-4-[3-[(6-胺磺醯基-2-吡啶基)胺基]丙基]吡咯啶-1-羧酸三級
丁酯(1.58 g,3.830 mmol)的溶液中,然後在室溫下攪拌18 h。將反應混合物在減壓下濃縮,且藉由使用水(甲酸含量為0.1%)中之5%至100%甲醇進行C18
柱反相層析來直接純化殘餘物,提供呈米色粉末之(4S
)-4-[3-[[6-[(2,6-二氯-5-三甲基矽基-吡啶-3-羰基)胺磺醯基]-2-吡啶基]胺基]丙基]-2,2-二甲基-吡咯啶-1-羧酸三級
丁酯(1.41 g,56%)。1
H NMR (400 MHz, DMSO-d6
) δ 12.80 (br. s., 1H), 7.93 (d,J =
2.7 Hz, 1H), 7.60 (t,J =
7.8 Hz, 1H), 7.28 - 7.10 (m, 2H), 6.73 (d,J =
8.6 Hz, 1H), 3.57 - 3.45 (m, 1H), 3.26 - 3.18 (m, 2H), 2.74 (t,J =
10.3 Hz, 1H), 2.01 (br. s., 1H), 1.87 - 1.71 (m, 1H), 1.47 (br. s., 2H), 1.40 - 1.27 (m, 15H), 1.20 (s, 3H), 0.38 (s, 9H)。ESI-MS m/z計算值657.1975,實測值558.2 (M-99)+
;保留時間:2.33分鐘(LC方法L)。步驟 14 : 2,6- 二氯 -N-[[6-[3-[(3S
)-5,5- 二甲基吡咯啶 -3- 基 ] 丙基胺基 ]-2- 吡啶基 ] 磺醯基 ]-5- 三甲基矽基 - 吡啶 -3- 甲醯胺
將(4S
)-4-[3-[[6-[(2,6-二氯-5-三甲基矽基-吡啶-3-羰基)胺磺醯基]-2-吡啶基]胺基]丙基]-2,2-二甲基-吡咯啶-1-羧酸三級
丁酯(1.41 g,2.1405 mmol)溶解於DCM (110 mL)中,且添加2,2,2-三氟乙酸(28 mL,365.89 mmol)。在室溫下攪拌40 min後,將溶劑在減壓下蒸發且將所得殘餘物置於高真空下達1 h。將產物溶解於在減壓下濃縮之DCM(150 mL)中,然後在高真空下乾燥,以提供6-二氯-N-[[6-[3-[(3S
)-5,5-二甲基吡咯啶-3-基]丙基胺基]-2-吡啶基]磺醯基]-5-三甲基矽基-吡啶-3-甲醯胺(三氟乙酸鹽)(2.2 g,131%)。藉由使用純水中之5%至100%甲醇進行反相層析來進一步純化粗產物,提供呈黃色粉末之2,6-二氯-N-[[6-[3-[(3S
)-5,5-二甲基吡咯啶-3-基]丙基胺基]-2-吡啶基]磺醯基]-5-三甲基矽基-吡啶-3-甲醯胺(920 mg,77%)。1
H NMR (400 MHz, 乙腈-d3
) δ 7.82 (s, 1H), 7.45 (t,J =
7.7 Hz, 1H), 7.07 (d,J =
7.1 Hz, 1H), 6.52 (d,J =
8.3 Hz, 1H), 5.63 (br. s., 1H), 3.97 - 3.79 (m, 1H), 3.58 (dd,J =
13.3, 6.5 Hz, 1H), 3.18 - 2.98 (m, 2H), 2.71 (br. s., 1H), 1.85 (dd,J =
12.5, 7.3 Hz, 1H), 1.64 - 1.33 (m, 8H), 1.24 (s, 3H), 0.35 (s, 9H)。ESI-MS m/z計算值557.145,實測值558.2 (M+1)+
;保留時間:1.51分鐘(LC方法L)。步驟 15 : (14S)-8- 氯 -12,12- 二甲基 -7-( 三甲基矽烷基 )-2λ6 - 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5,7,9,19(23),20- 己烯 -2,2,4- 三酮
在微波小瓶中在氮氣氛下,將2,6-二氯-N-[[6-[3-[(3S
)-5,5-二甲基吡咯啶-3-基]丙基胺基]-2-吡啶基]磺醯基]-5-三甲基矽基-吡啶-3-甲醯胺將(200 mg,0.3580 mmol)與DMSO(3.2 mL)中之K2
CO3
(50 mg,0.362 mmol)混合,將微波瓶蓋好,且然後在130℃油浴中加熱2 h。將反應混合物冷卻至室溫,然後直接注入C18
管柱,且使用水(含有0.1%甲酸)中之10%至100%甲醇梯度純化,得到呈淡黃色固體之(14S
)-8-氯-12,12-二甲基-7-(三甲基矽烷基)
-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(22),5,7,9,19(23),20-己烯-2,2,4-三酮(68 mg,36%)。1
H NMR (400 MHz, DMSO-d6
) δ 12.61 (br. s., 1H), 7.62 (s, 1H), 7.57 (t,J =
7.8 Hz, 1H), 7.06 (d,J =
7.3 Hz, 1H), 7.02 - 6.94 (m, 1H), 6.71 (d,J =
8.6 Hz, 1H), 3.95 - 3.75 (m, 1H), 3.15 - 3.06 (m, 1H), 2.95 (d,J =
13.2 Hz, 1H), 2.67 - 2.58 (m, 1H), 2.17 - 2.02 (m, 1H), 1.83 (dd,J =
11.6, 4.8 Hz, 1H), 1.78 - 1.69 (m, 1H), 1.63 - 1.49 (m, 6H), 1.44 (s, 3H), 1.36 - 1.24 (m, 1H), 0.32 (s, 9H)。ESI-MS m/z計算值521.1684,實測值522.2 (M+1)+
;保留時間:2.1分鐘(LC方法L)。步驟 16 : (14S
)-8-[3-(2-{ 雙螺 [2.0.24.13] 庚 -7- 基 } 乙氧基 )-1H
- 吡唑 -1- 基 ]-12,12- 二甲基 -7-( 三甲基矽基 )-2λ6
- 硫代 -3,9,11,18,23- 五氮雜四環 [17.3.1.111,14.05,10] 二十四烷 -1(22),5,7,9,19(23),20- 己烯 -2,2,4- 三酮,化合物 (1-9)
在微波小瓶中在氮氣(氣球)下,將碘化亞銅(10 mg,0.0525 mmol)添加到於DMSO (0.05 mL)中之反式N,N'
-二甲基環己烷-1,2-二胺(8 mg,0.0562 mmol)、(14S
)-8-氯-12,12-二甲基-7-(三甲基矽烷基)-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(22),5,7,9,19(23),20-己烯-2,2,4-三酮(10 mg,0.0192 mmol)、3-(2-雙螺[2.0.24.13]庚-7-基乙氧基)-1H-
吡唑(12 mg,0.0587 mmol)及碳酸鉀(9 mg,0.0651 mmol)。將小瓶加熱至130℃,過週末。將反應混合物冷卻至室溫,且將其與來自10 mg規模之另一反應運行之粗物質合併,且將其與DMSO + 乙腈混合,且在C18
管柱上藉由使用水(0.1%甲酸含量)中之10%至100%甲醇進行反相層析來純化,在凍乾後提供呈白色蓬鬆固體之(14S
)-8-[3-(2-{雙螺[2.0.24.13]庚-7-基}乙氧基)-1H
-吡唑-1-基]-12,12-二甲基-7-(三甲基矽基)-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(22),5,7,9,19(23),20-己烯-2,2,4-三酮(6.2 mg,46%)。1
H NMR (400 MHz, DMSO-d6
) δ 8.12 (br. s., 1H), 7.82 (s, 1H), 7.37 (t,J =
7.3 Hz, 1H), 6.88 (d,J =
6.6 Hz, 1H), 6.48 - 6.40 (m, 2H), 5.98 (br. s., 1H), 4.16 (t,J =
6.7 Hz, 2H), 3.99 - 3.85 (m, 1H), 3.55 - 3.40 (m, 1H), 3.15 - 2.98 (m, 1H), 2.95 - 2.82 (m, 1H), 2.13 - 2.00 (m, 1H), 1.81 (q,J =
6.6 Hz, 3H), 1.73 - 1.65 (m, 1H), 1.61 - 1.43 (m, 10H), 1.34 - 1.25 (m,J =
9.8 Hz, 1H), 0.84 - 0.80 (m, 4H), 0.68 - 0.62 (m, 2H), 0.53 - 0.46 (m, 2H), 0.22 (s, 9H)。ESI-MS m/z計算值689.318,實測值690.2 (M+1)+
;保留時間:5.69分鐘(LC方法F)。實例 12 : 26
-(3-(2-( 雙螺 [2.0.24
.13
] 庚 -7- 基 ) 乙氧基 )-1H- 吡唑 -1- 基 )-15
,15
,9,9- 四甲基 -5- 硫雜 -4,7- 二氮雜 -9- 矽雜 -2(2,3),6(2,6)- 二吡啶 -1(1,3)- 吡咯啶環癸烷 (cyclodecaphan)- 3- 酮 5,5- 二氧化物,化合物 (1-10) 之製備
(1-10)
以下流程顯示了化合物(1-10)之合成路線: 實例 13 : 26
-(3-(2-( 雙螺 [2.0.24
.13
] 庚 -7- 基 ) 乙氧基 )-1H- 吡唑 -1- 基 )-15,15,10,10- 四甲基 -5- 硫雜 -4,7- 二氮雜 -10- 矽雜 -2(2,3),6(2,6)- 二吡啶 -1(1,3)- 吡咯啶環癸烷 - 3- 酮 5,5- 二氧化物,化合物 (1-11) 之製備
(1-11)
以下流程顯示了化合物(1-11)之合成路線: 實例 14 :生物活性測定 溶液
基礎培養基(ADF+++)由Advanced DMEM/Ham’s F12、2 mM Glutamax、10 mM HEPES、1 µg/ml青黴素/鏈黴素組成。
腸道維持培養基(IEMM)由ADF+++、1x B27補充劑、1x N2補充劑、1.25 mMN-
乙醯半胱胺酸、10 mM菸鹼醯胺、50 ng/mL hEGF、10 nM胃泌素、1 µg/mL hR-脊椎蛋白-1、100 ng/mL hNoggiN、TGF-b 1型抑制劑A-83-01、100 µg/mL PrimociN、10 µM P38 MAPK抑制劑SB202190組成。
Bath 1緩衝液由1 mmol MgCl2
、160 mM NaCl、4.5 mM KCl、10 mM HEPES、10 mM葡萄糖、2 mM CaCl2
組成。
無氯緩衝液由1 mM葡萄糖酸鎂、2 mM葡萄糖酸鈣、4.5 mM葡萄糖酸鉀、160 mM葡萄糖酸鈉、10 mM HEPES及10 mM葡萄糖組成。
Bath1染料溶液由Bath 1緩衝液、0.04% Pluronic F127、20 µM甲基氧喏、30 µM CaCCinh-A01、30 µM芝加哥天藍組成。
無氯染料溶液由無氯緩衝液、0.04% Pluronic F127、20 µM甲基氧喏、30 µM CaCCinh-A01、30 µM芝加哥天藍組成。
無氯染料刺激溶液由無氯染料溶液、10 µM毛喉素、100 µM IBMX及300 nM化合物III組成。細胞培養
人腸上皮腸樣細胞獲自荷蘭烏得勒支之胡布勒發育生物學與乾細胞研究(Hubrecht Institute for Developmental Biology and Stem Cell Research, Utrecht, The Netherlands),且如先前所述在T燒瓶中擴增(Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, de Winter-de Groot KM, Brandsma AM, de Jong NWM, Bijvelds MJC, Scholte BJ, Nieuwenhuis EES, van den Brink S, Clevers H, van der Ent CK, Middendorp S及M Beekman JM. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013年7月;19(7):939-45.)。腸樣細胞收穫及接種
在細胞回收溶液中回收細胞,藉由在4℃下在650 rpm下離心5 min來收集細胞,將其重懸於TryPLE中,且在37℃下溫育5 min。然後藉由在4℃下在650 rpm下離心5 min來收集細胞,且將其重懸於含有10 µM ROCK抑制劑(RI)之IEMM中。使細胞懸浮液穿過40 µm細胞過濾器,且以1x106個細胞/mL重懸於含有10 µM RI之IEMM中。將細胞以5000個細胞/孔接種於多孔板中,且檢定之前在37℃、95%濕度及5% CO2
下溫育隔夜。膜電位染料檢定
在IEMM中將腸樣細胞與測試化合物在37℃、95%濕度及5% CO2
下溫育18-24小時。在化合物溫育後,使用FLIPR Tetra進行膜電位染料檢定,在急性添加10 µM毛喉素及300 nMN-
[2,4-雙 (
1,1-二甲基乙基)-5-羥基苯基]-1,4-二氫-4-側氧基喹啉-3-甲醯胺之後直接量測該測試化合物對經CFTR介導之氯離子運輸之效力及功效。簡言之,將細胞以Bath 1緩衝液洗滌5次。添加Bath 1染料溶液,且將細胞在室溫下溫育25 min。在染料溫育後,將細胞以無氯染料溶液洗滌3次。藉由添加無氯染料刺激溶液開始氯離子運輸,且讀取螢光信號達15 min。由對急性毛喉素及300 nMN-
[2,4-雙 (
1,1-二甲基乙基)-5-羥基苯基]-1,4-二氫-4-側氧基喹啉-3-甲醯胺刺激之螢光反應之AUC確定在各條件下經CFTR介導之氯離子運輸。然後將氯離子運輸表示為在使用1 µM (14S
)-8-[3-(2-{雙螺[2.0.2.1]庚-7-基}乙氧基)-1H
-吡唑-1-基]-12,12-二甲基-2λ6
-硫代-3,9,11,18,23-五氮雜四環[17.3.1.111,14.05,10]二十四烷-1(22),5,7,9,19(23),20-己烯-2,2,4-三酮、3 µM (R
)-1-(2,2-二氟苯并[d][1,3]間二氧雜環戊烯-5-基)-N-
(1-(2,3-二羥丙基)-6-氟-2-(1-羥基-2-甲基丙烷-2-基)-1H-
吲哚-5-基)環丙烷甲醯胺及300 nM急性N-
[2,4-雙(1,1-二甲基乙基)-5-羥基苯基]-1,4-二氫-4-側氧基喹啉-3-甲醯胺三重組合控制(%活性)處理後氯離子運輸之百分比。
表3表示使用本文所揭示之一或多種檢定生成之本發明的代表性化合物之CFTR調節活性。(最大活性:+++ 為 >60%;++ 為30%-60%;+ 為< 30%。EC50:+++ 為 < 1 µM;++ 為1-3 µM;+ 為 >3 µM;且ND為「未確定」。)表 3 :
其他實施例
| 化合物編號 | 結構 | EC50 (µM) | 最大活性(%) |
| (1-1) | +++ | +++ | |
| (2-1) | +++ | +++ | |
| (1-2) | +++ | +++ | |
| (1-3) | +++ | +++ | |
| (1-4) | +++ | +++ | |
| (1-5) | ND | ND | |
| (1-6) | +++ | +++ | |
| (1-7) | +++ | +++ | |
| (1-8) | +++ | +++ | |
| (1-9) | ND | ND | |
| (1-10) | ND | ND | |
| (1-11) | ND | ND |
前述論述僅揭示且描述了本揭示案之示範性實施例。熟悉此項技藝者由此類論述及圖式及發明申請專利範圍中將容易地認識到,在不背離如發明申請專利範圍所限定之本揭示案之精神及範圍的情況下,可在其中進行各種改變、修改及變化。
Claims (27)
- 一種式(1)化合物或其醫藥學上可接受之鹽或氘化衍生物,
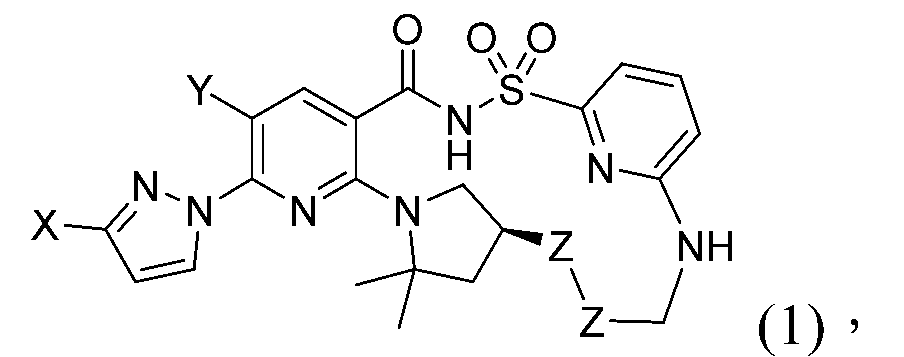
- 如請求項1之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中X選自Si(R)3、、
 、
、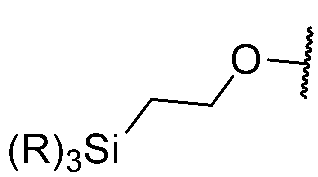 、
、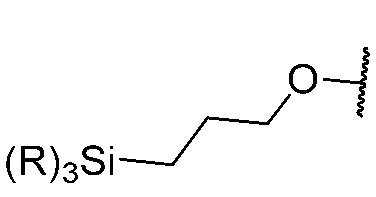 、
、

- 如請求項1之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中該化合物選自:
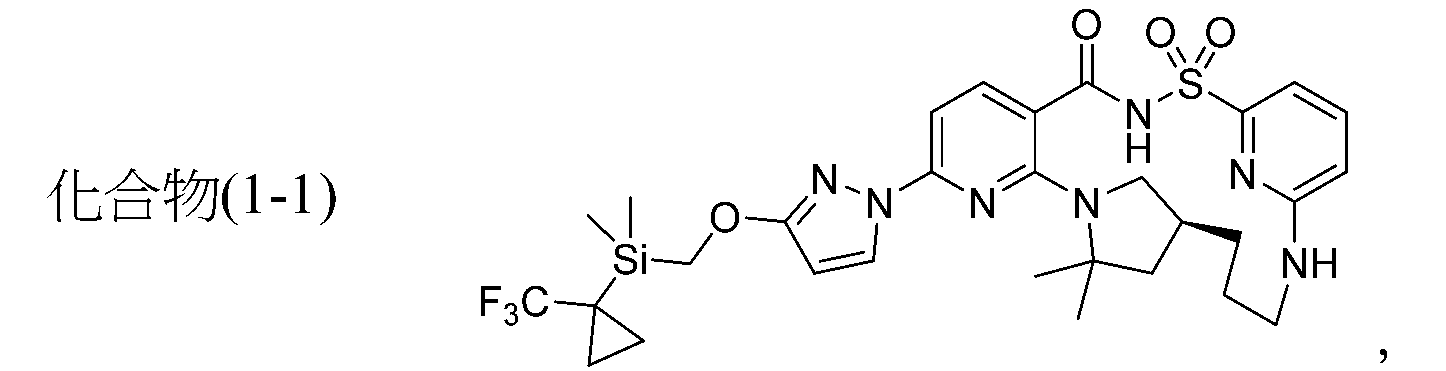
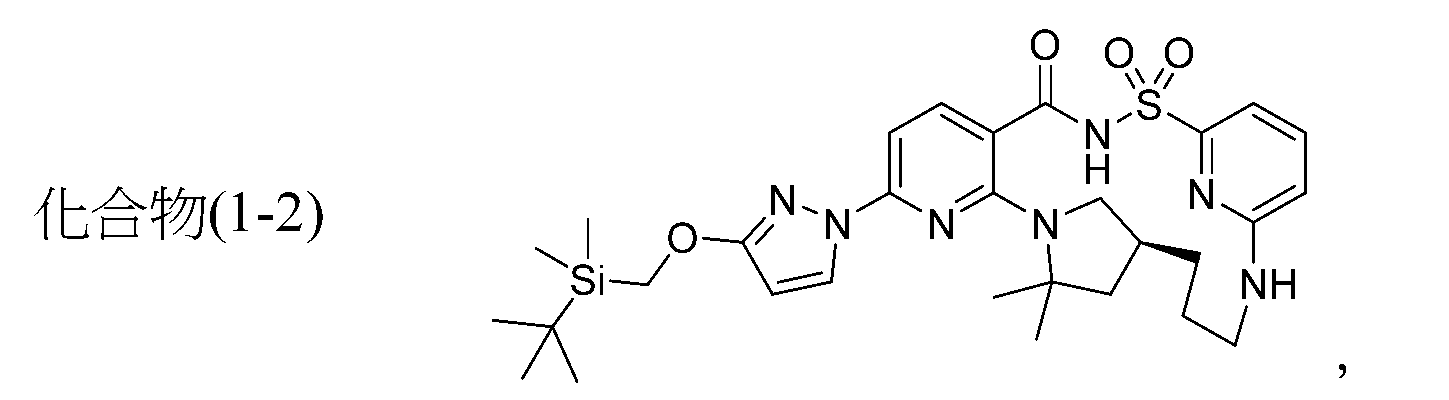
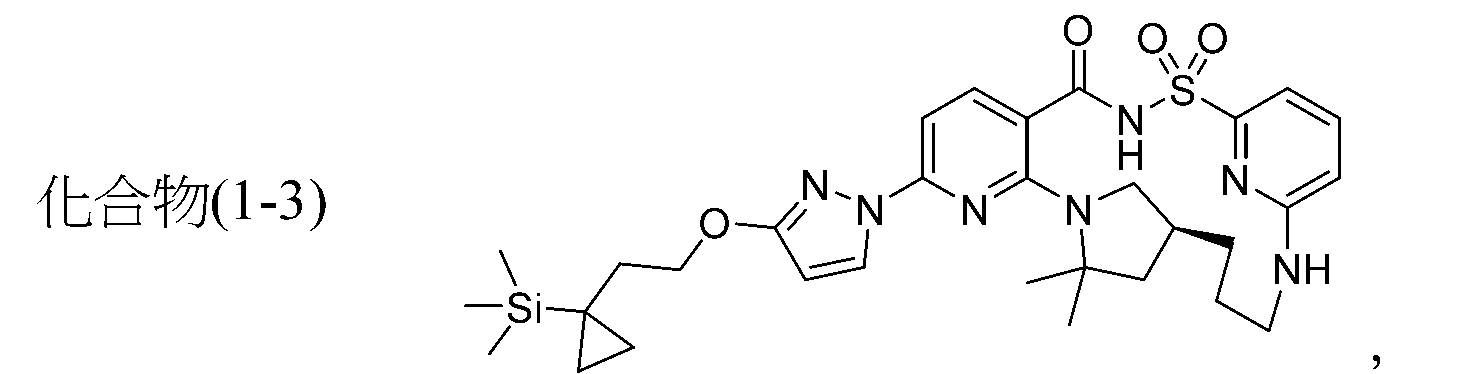
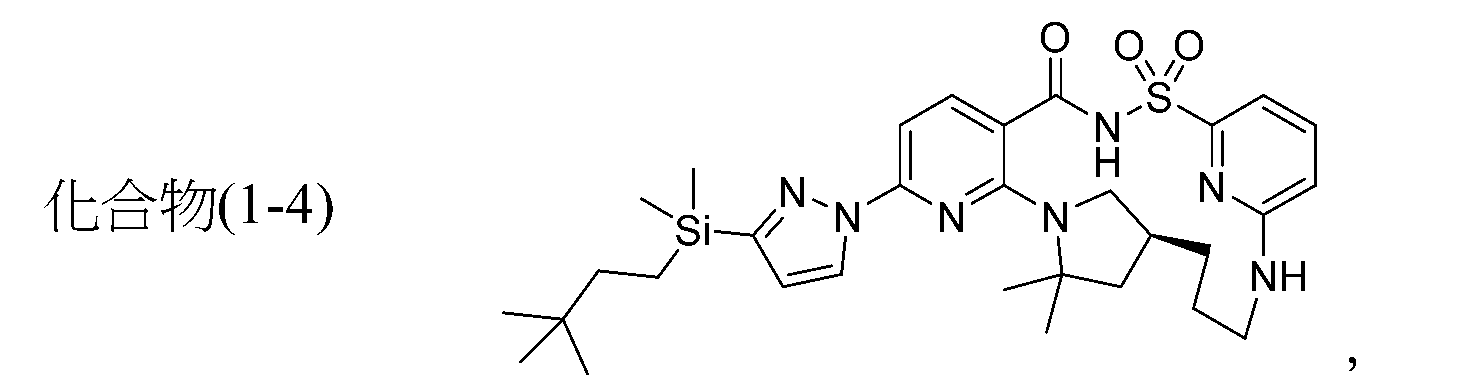

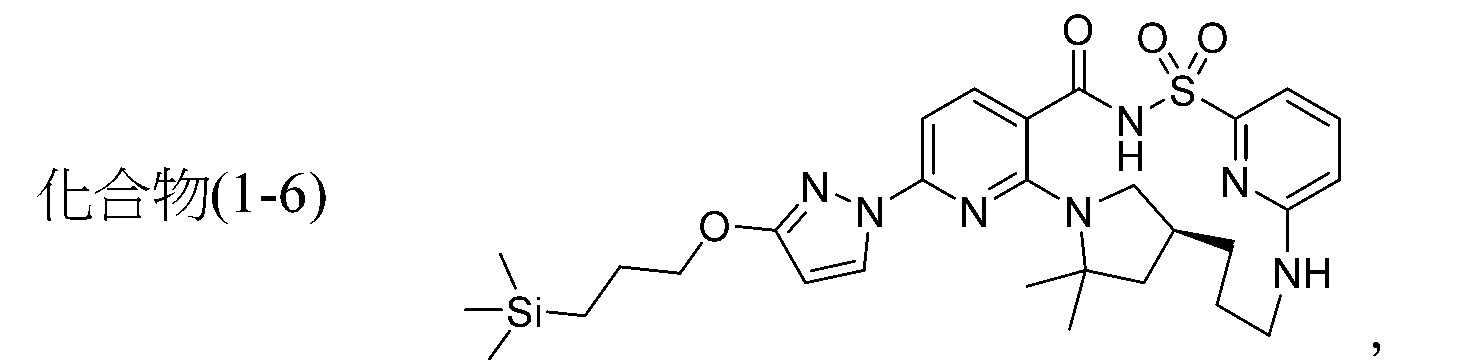

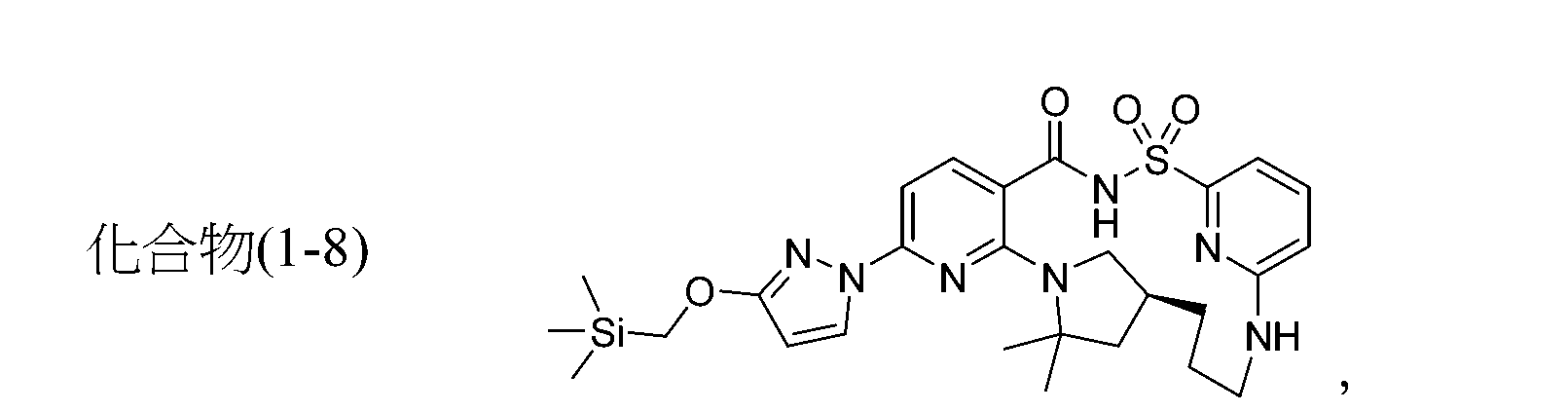


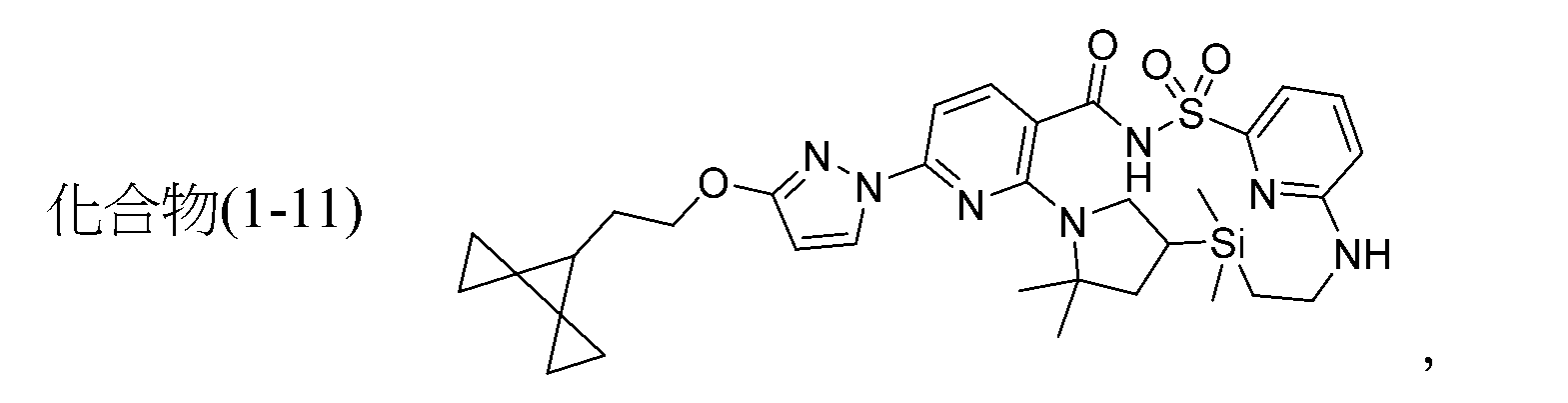
- 如請求項1之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中至少一個氫經氘置換。
- 如請求項1之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中該化合物為醫藥學上可接受之鹽。
- 一種式(2)化合物或其醫藥學上可接受之鹽或氘化衍生物,
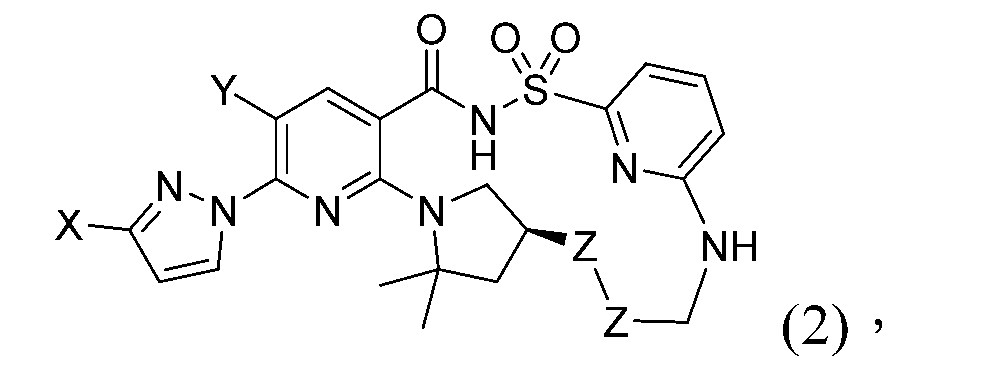
- 如請求項6之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中X選自Ge(R)3、、
 、
、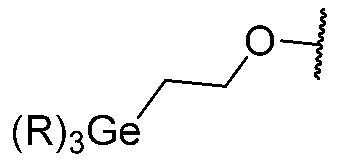 、
、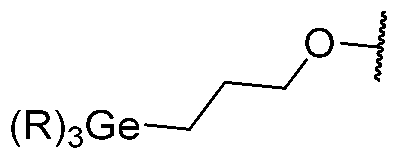

- 如請求項6之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中該化合物選自:

- 如請求項6之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中至少一個氫經氘置換。
- 如請求項6之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中該化合物為醫藥學上可接受之鹽。
- 一種式(3)化合物或其醫藥學上可接受之鹽或氘化衍生物,
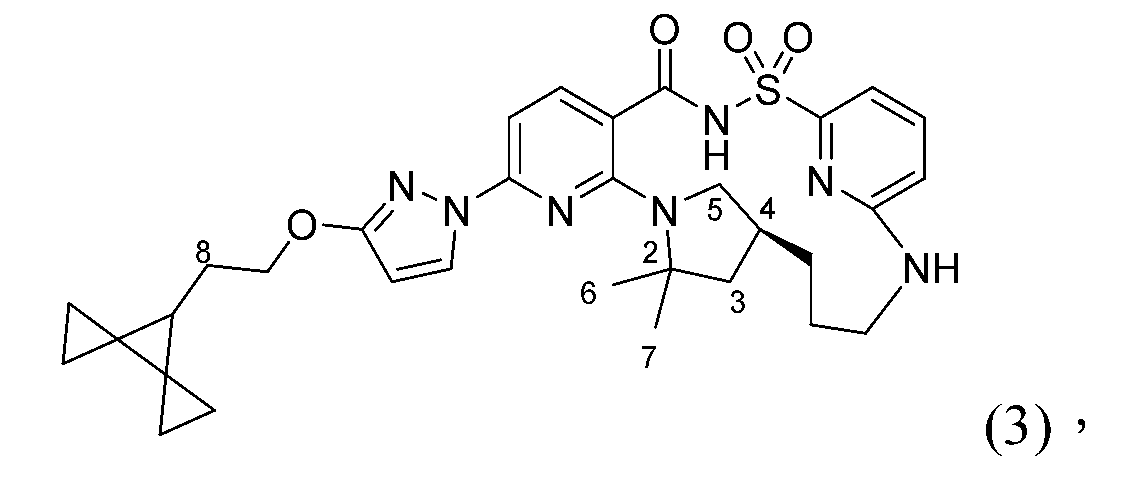
- 如請求項11之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中至少一個氫經氘置換。
- 如請求項11之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中該化合物為醫藥學上可接受之鹽。
- 一種式(4)化合物或其醫藥學上可接受之鹽或氘化衍生物,
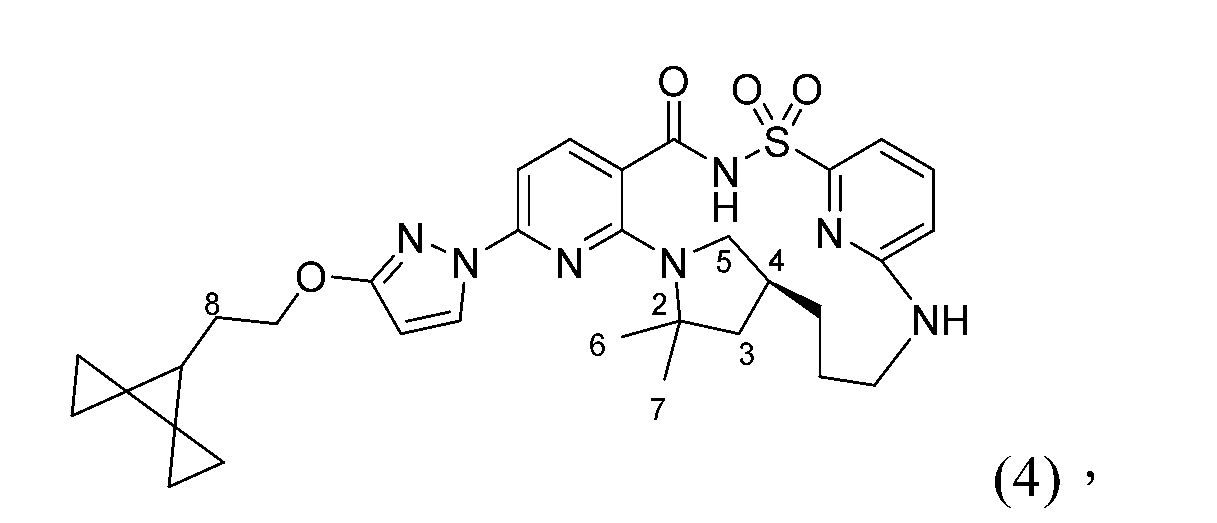
- 如請求項14之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中至少一個氫經氘置換。
- 如請求項14之化合物或其醫藥學上可接受之鹽或氘化衍生物,其中該化合物為醫藥學上可接受之鹽。
- 一種醫藥組成物,其包含如請求項1-16中任一項之化合物或其醫藥學上可接受之鹽或氘化衍生物及醫藥學上可接受之載劑。
- 如請求項17之醫藥組成物,其進一步包含一或多種額外治療劑。
- 如請求項18之醫藥組成物,其中該一或多種額外治療劑包含選自化合物II
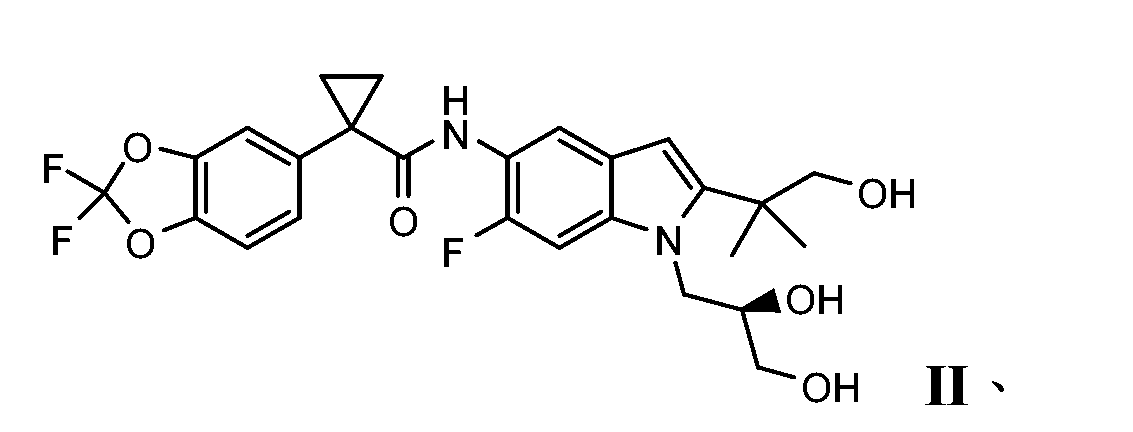
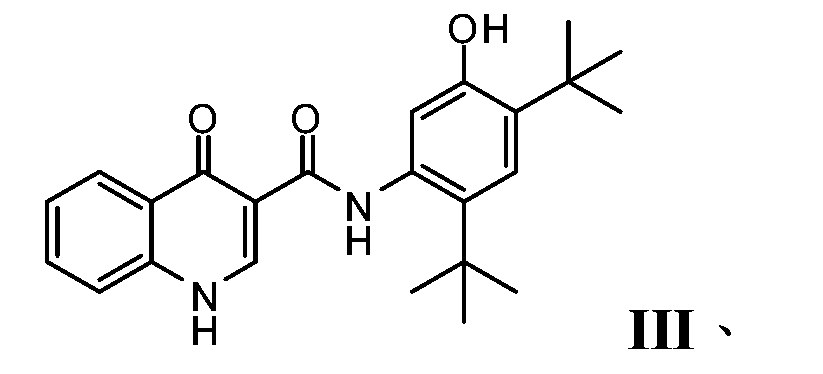

- 如請求項19之醫藥組成物,其中該組成物包含化合物II及化合物III。
- 如請求項19之醫藥組成物,其中該組成物包含化合物II及化合物III-d。
- 一種醫藥組成物,其包含:(a)至少一種選自如請求項1-16中任一項之化合物或其醫藥學上可接受之鹽或氘化衍生物的化合物;(b)至少一種醫藥學上可接受之載劑;及視情況以下一或多者:(c)(i)選自化合物II之化合物:

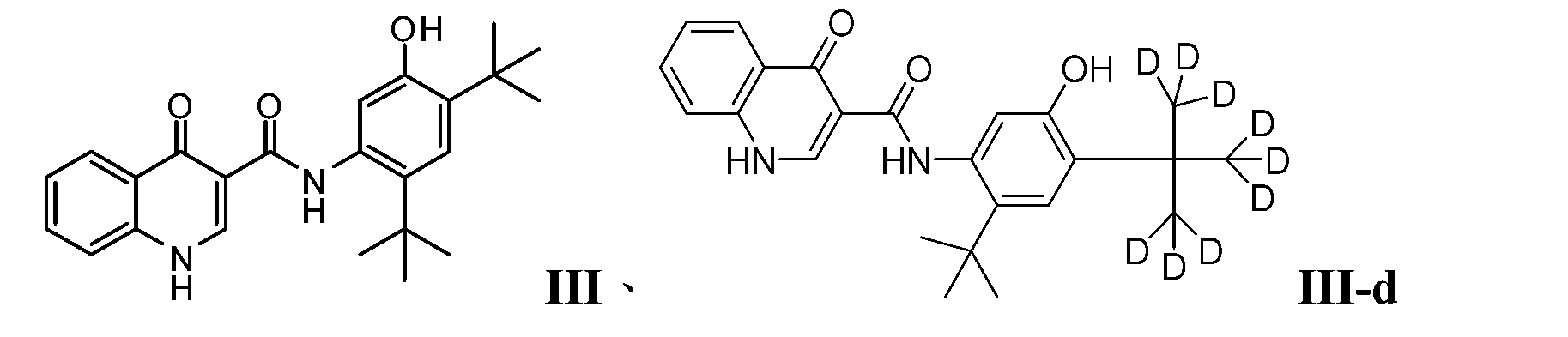
- 一種如請求項1-16中任一項之化合物或其醫藥學上可接受之鹽或氘化衍生物在製備用於治療囊性纖維化的藥物中的用途。
- 如請求項23之用途,其中該藥物經調配成與一種或多種額外治療劑一起投與。
- 如請求項24之用途,其中該一或多種額外治療劑包含選自化合物II


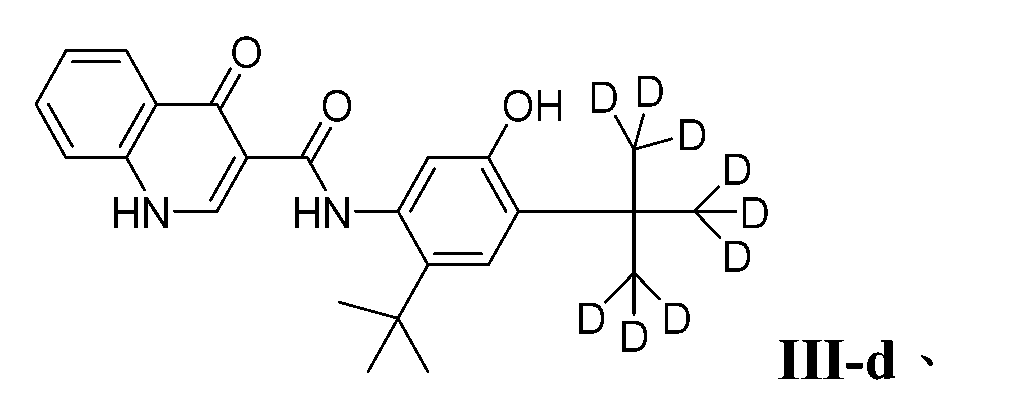
- 如請求項25之用途,其中該一或多種額外治療劑包含化合物II及化合物III。
- 如請求項25之用途,其中該一或多種額外治療劑包含化合物II及化合物III-d。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962886511P | 2019-08-14 | 2019-08-14 | |
| US62/886,511 | 2019-08-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW202115092A TW202115092A (zh) | 2021-04-16 |
| TWI867024B true TWI867024B (zh) | 2024-12-21 |
Family
ID=72291107
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW109127317A TWI867024B (zh) | 2019-08-14 | 2020-08-12 | 囊腫纖維化跨膜傳導調節蛋白之調節劑 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11591350B2 (zh) |
| EP (1) | EP4013763A1 (zh) |
| JP (1) | JP2022545633A (zh) |
| TW (1) | TWI867024B (zh) |
| WO (1) | WO2021030555A1 (zh) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2017004543A (es) | 2014-10-06 | 2017-10-04 | Vertex Pharma | Moduladores de regulador de conductancia transmembranal de fibrosis quística. |
| WO2018107100A1 (en) | 2016-12-09 | 2018-06-14 | Vertex Pharmaceuticals Incorporated | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| AU2018304168B2 (en) | 2017-07-17 | 2023-05-04 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| EP3720849A2 (en) | 2017-12-08 | 2020-10-14 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| ES2939775T3 (es) | 2018-02-15 | 2023-04-26 | Vertex Pharma | Macrociclos como moduladores del regulador de la conductancia transmembrana de la fibrosis quística, composiciones farmacéuticas de los mismos, su uso en el tratamiento de la fibrosis quística, y procesos para elaborarlos |
| AR118555A1 (es) | 2019-04-03 | 2021-10-20 | Vertex Pharma | Agentes moduladores del regulador de la conductancia transmembrana de la fibrosis quística |
| TWI899097B (zh) | 2019-08-14 | 2025-10-01 | 美商維泰克斯製藥公司 | 製備cftr調節劑之方法 |
| AU2020328028B2 (en) | 2019-08-14 | 2025-03-06 | Vertex Pharmaceuticals Incorporated | Crystalline forms of CFTR modulators |
| TWI867024B (zh) | 2019-08-14 | 2024-12-21 | 美商維泰克斯製藥公司 | 囊腫纖維化跨膜傳導調節蛋白之調節劑 |
| AU2020328568A1 (en) * | 2019-08-14 | 2022-03-03 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| CR20230120A (es) | 2020-08-07 | 2023-09-01 | Vertex Pharma | Moduladores del regulador de la conductancia transmembrana de la fibrosis quística |
| US12324802B2 (en) | 2020-11-18 | 2025-06-10 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| MX2023006770A (es) | 2020-12-10 | 2023-08-14 | Vertex Pharma | Metodos de tratamiento para fibrosis quistica. |
| AU2023218262A1 (en) | 2022-02-08 | 2024-08-22 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| GEAP202516620A (en) * | 2022-04-06 | 2025-04-10 | Vertex Pharma | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2024056791A1 (en) | 2022-09-15 | 2024-03-21 | Idorsia Pharmaceuticals Ltd | Combination of macrocyclic cftr modulators with cftr correctors and / or cftr potentiators |
| KR20250065909A (ko) | 2022-09-15 | 2025-05-13 | 이도르시아 파마슈티컬스 리미티드 | (3s,7s,10r,13r)-13-벤질-20-플루오로-7-이소부틸-n-(2-(3-메톡시-1,2,4-옥사디아졸-5-일)에틸)-6,9-디메틸-1,5,8,11-테트라옥소-10-(2,2,2-트리플루오로에틸)-1,2,3,4,5,6,7,8,9,10,11,12,13,14-테트라데카히드로-[1]옥사[4,7,10,14]테트라아자시클로헵타데시노[16,17-f]퀴놀린-3-카르복사미드의결정질 형태 |
| CA3267795A1 (en) | 2022-09-15 | 2024-03-21 | Idorsia Pharmaceuticals Ltd | MACROCYCLICAL CFTR MODULATORS |
| WO2025186214A1 (en) | 2024-03-05 | 2025-09-12 | Idorsia Pharmaceuticals Ltd | Macrocyclic cftr modulators |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201811766A (zh) * | 2016-08-29 | 2018-04-01 | 瑞士商諾華公司 | N-(吡啶-2-基)吡啶-磺醯胺衍生物及其用於疾病治療之用途 |
Family Cites Families (113)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2677682A (en) | 1951-08-24 | 1954-05-04 | American Cyanamid Co | Sulfonamido pteridines |
| ID18983A (id) | 1996-12-04 | 1998-05-28 | Lilly Co Eli | Pirazola sebagai inhibitor sekresi fosfolipase a2 non-pankreas pada manusia |
| SE0001899D0 (sv) | 2000-05-22 | 2000-05-22 | Pharmacia & Upjohn Ab | New compounds |
| CN1925854A (zh) | 2003-11-14 | 2007-03-07 | 沃泰克斯药物股份有限公司 | 可用作atp-结合弹夹转运蛋白调控剂的噻唑和噁唑 |
| WO2005075435A1 (en) | 2004-01-30 | 2005-08-18 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| RS56873B1 (sr) | 2004-06-24 | 2018-04-30 | Vertex Pharma | Modulatori atp-vezujućih kasetnih transportera |
| US8354427B2 (en) | 2004-06-24 | 2013-01-15 | Vertex Pharmaceutical Incorporated | Modulators of ATP-binding cassette transporters |
| MX2008002019A (es) | 2005-08-11 | 2008-04-16 | Vertex Pharma | Moduladores del regulador de conductancia transmembrana de fibrosis quistica. |
| WO2007053641A2 (en) | 2005-11-01 | 2007-05-10 | Mars, Incorporated | A-type procyanidins and inflammation |
| KR101561482B1 (ko) | 2005-11-08 | 2015-10-20 | 버텍스 파마슈티칼스 인코포레이티드 | Atp 결합 카세트 수송체의 헤테로사이클릭 조정제 |
| CA2635214A1 (en) | 2005-12-27 | 2007-07-05 | Vertex Pharmaceuticals Incorporated | Compounds useful in cftr assays and methods therewith |
| EP3219705B1 (en) | 2005-12-28 | 2020-03-11 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of the amorphous form of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| US7671221B2 (en) | 2005-12-28 | 2010-03-02 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-Binding Cassette transporters |
| US7691902B2 (en) | 2005-12-28 | 2010-04-06 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| PT2674428T (pt) | 2006-04-07 | 2016-07-14 | Vertex Pharma | Modeladores de transportadores de cassetes de ligação de atp |
| CA2652072A1 (en) | 2006-05-12 | 2007-11-22 | Vertex Pharmaceuticals Incorporated | Compositions of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| RU2009120976A (ru) | 2006-11-03 | 2010-12-10 | Вертекс Фармасьютикалз Инкорпорейтед (Us) | Производные азаиндола в качестве модуляторов cftr |
| MX2009013213A (es) | 2007-06-08 | 2010-03-30 | Abbott Lab | Indazoles 5-sustituidos 5-heteroarilo como inhibidores de cinasa. |
| CA2699292A1 (en) | 2007-09-14 | 2009-03-26 | Vertex Pharmaceuticals Incorporated | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| NZ585794A (en) | 2007-12-07 | 2012-05-25 | Vertex Pharma | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| HUE031913T2 (en) | 2007-12-07 | 2017-08-28 | Vertex Pharma | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| ES2647531T3 (es) | 2008-02-28 | 2017-12-22 | Vertex Pharmaceuticals Incorporated | Derivados de heteroarilo como moduladores de CFTR |
| US20100074949A1 (en) | 2008-08-13 | 2010-03-25 | William Rowe | Pharmaceutical composition and administration thereof |
| SMT202100077T1 (it) | 2008-08-13 | 2021-05-07 | Vertex Pharma | Composizione farmaceutica e sue somministrazioni |
| MX2011003249A (es) | 2008-09-29 | 2011-05-19 | Vertex Pharma | Unidades de dosificacion del acido 3-(6-(1-(2,2-difluorobenzo[d][1 ,3]dioxol-5-il)ciclopropancarboxamido)-3-metilpiridin-2-il)benzoi co. |
| JP5645833B2 (ja) | 2008-10-23 | 2014-12-24 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | 嚢胞性線維症膜コンダクタンス制御因子の修飾因子 |
| UA104876C2 (uk) | 2008-11-06 | 2014-03-25 | Вертекс Фармасьютікалз Інкорпорейтед | Модулятори atф-зв'язувальних касетних транспортерів |
| HRP20150288T1 (hr) | 2008-11-06 | 2015-04-24 | Vertex Pharmaceuticals Incorporated | Modulatori atp-vezujuä†ih kasetnih transportera |
| PT2408750E (pt) | 2009-03-20 | 2015-10-15 | Vertex Pharma | Processo para a produção de modeladores do regulador de condutância transmembranar da fibrose cística |
| WO2011050325A1 (en) | 2009-10-22 | 2011-04-28 | Vertex Pharmaceuticals Incorporated | Compositions for treatment of cystic fibrosis and other chronic diseases |
| AU2011227021A1 (en) | 2010-03-19 | 2012-10-18 | Vertex Pharmaceuticals Incorporated | Solid forms of N-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| EP3181561B1 (en) | 2010-03-25 | 2020-05-20 | Vertex Pharmaceuticals Incorporated | Synthetic intermediate of (r)-1(2,2 -difluorobenzo[d][1,3]dioxol-5yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2yl)-1h-indol-5yl)cyclopropanecarboxamide |
| ME02446B (me) | 2010-04-07 | 2016-09-20 | Vertex Pharma | Čvrste forme 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioksol-5-il)ciklopropankarboksiamido)-3-metilpiridin-2-il)benzoeve kiseline |
| LT3150198T (lt) | 2010-04-07 | 2021-12-10 | Vertex Pharmaceuticals Incorporated | 3-(6-(1-(2,2-difluorbenzo[d][1,3]dioksol-5-il) ciklopropankarboksamido)-3-metilpiridin-2-il)benzoinės rūgšties farmacinė kompozicija ir jos įvedimas |
| CA2796088C (en) | 2010-04-09 | 2018-02-06 | Ekso Bionics | Exoskeleton load handling system and method of use |
| EP3138563A1 (en) | 2010-04-22 | 2017-03-08 | Vertex Pharmaceuticals Inc. | Pharmaceutical compositions and administrations thereof |
| JP2013525371A (ja) | 2010-04-22 | 2013-06-20 | バーテックス ファーマシューティカルズ インコーポレイテッド | シクロアルキルカルボキサミド−インドール化合物の製造方法 |
| US8563593B2 (en) | 2010-06-08 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Formulations of (R)-1-(2,2-difluorobenzo[D] [1,3] dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| WO2012027247A2 (en) | 2010-08-23 | 2012-03-01 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition of (r)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxy propyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide and administration therof |
| RU2013113627A (ru) | 2010-08-27 | 2014-10-10 | Вертекс Фармасьютикалз Инкорпорейтед | Фармацевтическая композиция и ее введения |
| MX349159B (es) | 2011-05-18 | 2017-07-14 | Concert Pharmaceuticals Inc | Derivados deuterados de ivacaftor. |
| HUE047354T2 (hu) | 2011-05-18 | 2020-04-28 | Vertex Pharmaceuticals Europe Ltd | Ivacaftor deuterizált származékai |
| CN103946221B (zh) | 2011-09-16 | 2016-08-03 | 诺华股份有限公司 | 用于治疗囊性纤维化的杂环化合物 |
| MX2014005304A (es) | 2011-10-31 | 2015-03-20 | Xenon Pharmaceuticals Inc | Biaril eter sulfonamidas y su uso como agentes terapeuticos. |
| CA2852991C (en) | 2011-11-08 | 2019-12-31 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| CN108066306B (zh) | 2012-01-25 | 2021-09-07 | 沃泰克斯药物股份有限公司 | 药物制剂及其制备方法 |
| EP2819670A1 (en) | 2012-02-27 | 2015-01-07 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administration thereof |
| CA2869847A1 (en) | 2012-04-20 | 2013-10-24 | Vertex Pharmaceuticals Incorporated | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| GB201207406D0 (en) | 2012-04-27 | 2012-06-13 | Glaxo Group Ltd | Novel compounds |
| AR091112A1 (es) | 2012-05-22 | 2015-01-14 | Genentech Inc | Benzamidas n-sustituidas como inhibidores de los canales de sodio nav1.7 |
| WO2013185112A1 (en) | 2012-06-08 | 2013-12-12 | Vertex Pharmaceuticals Incorporated | Pharmaceuticl compositions for the treatment of cftr -mediated disorders |
| HK1209318A1 (zh) | 2012-07-16 | 2016-04-01 | Vertex Pharmaceuticals Incorporated | (r)-1-(2,2-二氟苯[d][1,3]二惡茂-5-基)-n-(1-(2,3-二羥基)-6-氟代-2-(1-羥基-2-甲基丙烷-2-基)-1氫-吲哚-5-基)環丙烷甲酰胺的藥劑學組合物和其應用 |
| SMT202500078T1 (it) | 2012-11-02 | 2025-03-12 | Vertex Pharma | Composizioni farmaceutiche per il trattamento di malattie mediate da cftr |
| WO2014078842A1 (en) | 2012-11-19 | 2014-05-22 | Concert Pharmaceuticals, Inc. | Deuterated cftr potentiators |
| WO2015051043A1 (en) | 2013-10-01 | 2015-04-09 | Amgen Inc. | Biaryl acyl-sulfonamide compounds as sodium channel inhibitors |
| NZ720958A (en) | 2013-11-12 | 2022-02-25 | Vertex Pharma | Process of preparing pharmaceutical compositions for the treatment of cftr mediated diseases |
| HUE039062T2 (hu) | 2014-04-15 | 2018-12-28 | Vertex Pharma | Gyógyszerészeti készítmények cisztás fibrózis transzmembrán konduktancia regulátor által mediált betegségek kezelésére |
| MX2017004543A (es) | 2014-10-06 | 2017-10-04 | Vertex Pharma | Moduladores de regulador de conductancia transmembranal de fibrosis quística. |
| US9701639B2 (en) | 2014-10-07 | 2017-07-11 | Vertex Pharmaceuticals Incorporated | Co-crystals of modulators of cystic fibrosis transmembrane conductance regulator |
| PT3221692T (pt) | 2014-11-18 | 2021-09-10 | Vertex Pharma | Processo de realização de testagem de alta produtividade por cromatografia líquida de alta eficiência |
| EP3277647A1 (en) | 2015-03-31 | 2018-02-07 | Vertex Pharmaceuticals (Europe) Limited | Deuterated vx-661 |
| JP6849686B2 (ja) | 2015-09-21 | 2021-03-24 | バーテックス ファーマシューティカルズ (ヨーロッパ) リミテッド | 重水素化されたcftr増強剤の投与 |
| EP3436432B1 (en) | 2016-03-30 | 2021-01-27 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| PL3436446T3 (pl) * | 2016-03-31 | 2023-09-11 | Vertex Pharmaceuticals Incorporated | Modulatory mukowiscydozowego przezbłonowego regulatora przewodnictwa |
| NZ746793A (en) | 2016-04-07 | 2022-10-28 | Proteostasis Therapeutics Inc | Silicone atoms containing ivacaftor analogues |
| CA3016303A1 (en) | 2016-04-26 | 2017-11-02 | Abbvie S.A.R.L. | Modulators of cystic fibrosis transmembrane conductance regulator protein |
| US10138227B2 (en) | 2016-06-03 | 2018-11-27 | Abbvie S.Á.R.L. | Heteroaryl substituted pyridines and methods of use |
| EP3472156B1 (en) | 2016-06-21 | 2023-06-07 | Proteostasis Therapeutics, Inc. | Compounds, compositions, and methods for increasing cftr activity |
| PT3519401T (pt) | 2016-09-30 | 2021-12-27 | Vertex Pharma | Modulador de regulador de condutância transmembranar de fibrose quística, composições farmacêuticas, métodos de tratamento e processo para fazer o modulador |
| US9981910B2 (en) | 2016-10-07 | 2018-05-29 | Abbvie S.Á.R.L. | Substituted pyrrolidines and methods of use |
| US10399940B2 (en) | 2016-10-07 | 2019-09-03 | Abbvie S.Á.R.L. | Substituted pyrrolidines and methods of use |
| WO2018081378A1 (en) | 2016-10-26 | 2018-05-03 | Proteostasis Therapeutics, Inc. | Compounds, compositions, and methods for modulating cftr |
| EP3532467A1 (en) | 2016-10-26 | 2019-09-04 | Proteostasis Therapeutics, Inc. | Pyridazine derivatives, compositions and methods for modulating cftr |
| WO2018081377A1 (en) | 2016-10-26 | 2018-05-03 | Proteostasis Therapeutics, Inc. | N-phenyl-2-(3-phenyl-6-oxo-1,6-dihydropyridazin-1-yl)acetamide derivatives for treating cystic fibrosis |
| CA3041819A1 (en) | 2016-10-27 | 2018-05-03 | Vertex Pharmaceuticals (Europe) Limited | Methods of treatment with deuterated cftr potentiators |
| CR20190292A (es) | 2016-11-18 | 2019-09-03 | Cystic Fibrosis Found Therapeutics Inc | Pirrolopirimidinas como potenciadores de cftr |
| WO2018107100A1 (en) | 2016-12-09 | 2018-06-14 | Vertex Pharmaceuticals Incorporated | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| EP3554506B1 (en) | 2016-12-16 | 2021-04-28 | Cystic Fibrosis Foundation | Bycyclic heteroaryl derivatives as cftr potentiators |
| WO2018116185A1 (en) | 2016-12-20 | 2018-06-28 | AbbVie S.à.r.l. | Deuterated cftr modulators and methods of use |
| EP3565815B1 (en) | 2017-01-07 | 2024-03-13 | Fochon Pharmaceuticals, Ltd. | Compounds as bcl-2-selective apoptosis-inducing agents |
| TW201831471A (zh) | 2017-02-24 | 2018-09-01 | 盧森堡商艾伯維公司 | 囊腫纖化症跨膜傳導調節蛋白的調節劑及其使用方法 |
| US20180280349A1 (en) | 2017-03-28 | 2018-10-04 | Vertex Pharmaceuticals Incorporated | Methods of treating cystic fibrosis in patients with residual function mutations |
| MA49061A (fr) | 2017-04-28 | 2021-04-21 | Proteostasis Therapeutics Inc | Dérivés 4-sulfonylaminocarbonylquinoline pour augmenter l'activité cftr |
| US11253509B2 (en) | 2017-06-08 | 2022-02-22 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| WO2019010092A1 (en) | 2017-07-01 | 2019-01-10 | Vertex Pharmaceuticals Incorporated | COMPOSITIONS AND METHODS FOR TREATING CYSTIC FIBROSIS |
| AU2018304168B2 (en) | 2017-07-17 | 2023-05-04 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US20200171015A1 (en) | 2017-07-17 | 2020-06-04 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| AR112467A1 (es) | 2017-08-02 | 2019-10-30 | Vertex Pharma | Procesos para preparar compuestos |
| US10988454B2 (en) | 2017-09-14 | 2021-04-27 | Abbvie Overseas S.À.R.L. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| CA3078230A1 (en) | 2017-10-06 | 2019-04-11 | Proteostasis Therapeutics, Inc. | Compounds, compositions and methods for increasing cftr activity |
| IL273831B2 (en) | 2017-10-19 | 2024-10-01 | Vertex Pharma | Crystalline forms and compositions of cftr modulators |
| US20210228489A1 (en) | 2017-12-04 | 2021-07-29 | Vertex Pharmaceuticals Incorporated | Compositions for treating cystic fibrosis |
| EP3720849A2 (en) | 2017-12-08 | 2020-10-14 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| TWI810243B (zh) | 2018-02-05 | 2023-08-01 | 美商維泰克斯製藥公司 | 用於治療囊腫纖化症之醫藥組合物 |
| ES2939775T3 (es) | 2018-02-15 | 2023-04-26 | Vertex Pharma | Macrociclos como moduladores del regulador de la conductancia transmembrana de la fibrosis quística, composiciones farmacéuticas de los mismos, su uso en el tratamiento de la fibrosis quística, y procesos para elaborarlos |
| WO2019191620A1 (en) | 2018-03-30 | 2019-10-03 | Vertex Pharmaceuticals Incorporated | Crystalline forms of modulators of cftr |
| US20210139514A1 (en) | 2018-04-05 | 2021-05-13 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2019200246A1 (en) | 2018-04-13 | 2019-10-17 | Alexander Russell Abela | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| MA54227B1 (fr) | 2018-11-14 | 2023-03-31 | Vertex Pharma | Méthodes de traitement de la fibrose kystique |
| CA3119656A1 (en) | 2018-12-21 | 2020-06-25 | Novartis Ag | Macrocyclic compounds and their use in the treatment of disease |
| WO2020161623A1 (en) | 2019-02-06 | 2020-08-13 | Novartis Ag | N-(pyridin-2-yl)pyridine-sulfonamide derivatives and their use in the treatment of disease |
| US20220160705A1 (en) | 2019-03-20 | 2022-05-26 | Cornell University | Methods for controlling prostaglandin-mediated biological processes |
| AR118555A1 (es) | 2019-04-03 | 2021-10-20 | Vertex Pharma | Agentes moduladores del regulador de la conductancia transmembrana de la fibrosis quística |
| WO2020214921A1 (en) | 2019-04-17 | 2020-10-22 | Vertex Pharmaceuticals Incorporated | Solid forms of modulators of cftr |
| WO2020242935A1 (en) | 2019-05-29 | 2020-12-03 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| AU2020328028B2 (en) | 2019-08-14 | 2025-03-06 | Vertex Pharmaceuticals Incorporated | Crystalline forms of CFTR modulators |
| TWI899097B (zh) | 2019-08-14 | 2025-10-01 | 美商維泰克斯製藥公司 | 製備cftr調節劑之方法 |
| TWI867024B (zh) | 2019-08-14 | 2024-12-21 | 美商維泰克斯製藥公司 | 囊腫纖維化跨膜傳導調節蛋白之調節劑 |
| AU2020328568A1 (en) | 2019-08-14 | 2022-03-03 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| EP4058434A1 (en) | 2019-11-12 | 2022-09-21 | Genzyme Corporation | 6-membered heteroarylaminosulfonamides for treating diseases and conditions mediated by deficient cftr activity |
| PH12022551139A1 (en) | 2019-11-12 | 2023-07-17 | Genzyme Corp | 5-membered heteroarylaminosulfonamides for treating conditions mediated by deficient cftr activity |
| CR20230120A (es) | 2020-08-07 | 2023-09-01 | Vertex Pharma | Moduladores del regulador de la conductancia transmembrana de la fibrosis quística |
| KR20230051216A (ko) | 2020-08-13 | 2023-04-17 | 버텍스 파마슈티칼스 인코포레이티드 | Cftr 조절제의 결정질 형태 |
-
2020
- 2020-08-12 TW TW109127317A patent/TWI867024B/zh active
- 2020-08-13 EP EP20764497.2A patent/EP4013763A1/en active Pending
- 2020-08-13 WO PCT/US2020/046120 patent/WO2021030555A1/en not_active Ceased
- 2020-08-13 US US16/992,448 patent/US11591350B2/en active Active
- 2020-08-13 JP JP2022508829A patent/JP2022545633A/ja active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201811766A (zh) * | 2016-08-29 | 2018-04-01 | 瑞士商諾華公司 | N-(吡啶-2-基)吡啶-磺醯胺衍生物及其用於疾病治療之用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202115092A (zh) | 2021-04-16 |
| US11591350B2 (en) | 2023-02-28 |
| US20210047350A1 (en) | 2021-02-18 |
| WO2021030555A1 (en) | 2021-02-18 |
| EP4013763A1 (en) | 2022-06-22 |
| JP2022545633A (ja) | 2022-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI867024B (zh) | 囊腫纖維化跨膜傳導調節蛋白之調節劑 | |
| CN113905785B (zh) | 囊性纤维化跨膜传导调节因子调节剂 | |
| TWI828358B (zh) | 囊性纖維化跨膜傳導調節因子之調節劑、醫藥組合物、治療方法及製造該調節劑之方法 | |
| JP7684279B2 (ja) | 嚢胞性線維症膜コンダクタンス制御因子モジュレーター | |
| EP3880197B9 (en) | Methods of treatment for cystic fibrosis | |
| TW202120517A (zh) | 製備cftr調節劑之方法 | |
| TW202229297A (zh) | 囊腫纖維化跨膜傳導調節蛋白之調節劑 | |
| CA3201793A1 (en) | Macrocycles containing a 1,3,4-oxadiazole ring for use as modulators of cystic fibrosis transmembrane conductance regulator | |
| IL301756A (en) | Modulators of cystic fibrosis transmembrane conductance regulator | |
| TW202229299A (zh) | 囊腫纖維化跨膜傳導調節蛋白之調節劑 | |
| TW202222306A (zh) | 囊腫纖維化跨膜傳導調節蛋白之調節劑 | |
| KR20230051216A (ko) | Cftr 조절제의 결정질 형태 | |
| CA3031073A1 (en) | N-(pyridin-2-yl)pyridine-sulfonamide derivatives and their use in the treatment of disease | |
| CN119731183A (zh) | 大环化合物作为cftr调节剂的固体形式和其制备 | |
| HK40096538A (zh) | 囊性纤维化的治疗方法 | |
| TW202515573A (zh) | 囊腫纖維化跨膜傳導調節蛋白之調節劑的固體形式 | |
| CN118974059A (zh) | 囊性纤维化跨膜传导调控因子的调节剂 | |
| EA046014B1 (ru) | Средства, модулирующие регулятор трансмембранной проводимости при муковисцидозе | |
| CN118696041A (zh) | 大环btk抑制剂 |