TWI704151B - Erk抑制劑 - Google Patents
Erk抑制劑 Download PDFInfo
- Publication number
- TWI704151B TWI704151B TW104141368A TW104141368A TWI704151B TW I704151 B TWI704151 B TW I704151B TW 104141368 A TW104141368 A TW 104141368A TW 104141368 A TW104141368 A TW 104141368A TW I704151 B TWI704151 B TW I704151B
- Authority
- TW
- Taiwan
- Prior art keywords
- compound
- pharmaceutically acceptable
- acceptable salt
- cancer
- methyl
- Prior art date
Links
- 239000012824 ERK inhibitor Substances 0.000 title description 3
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 93
- 238000011282 treatment Methods 0.000 claims abstract description 39
- 201000011510 cancer Diseases 0.000 claims abstract description 29
- 230000000694 effects Effects 0.000 claims abstract description 18
- 108010007457 Extracellular Signal-Regulated MAP Kinases Proteins 0.000 claims abstract description 15
- 102000007665 Extracellular Signal-Regulated MAP Kinases Human genes 0.000 claims abstract description 15
- 150000001875 compounds Chemical class 0.000 claims description 217
- 150000003839 salts Chemical class 0.000 claims description 48
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 21
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 20
- 239000003814 drug Substances 0.000 claims description 20
- JNPRPMBJODOFEC-UHFFFAOYSA-N 6,6-dimethyl-2-[2-[(2-methylpyrazol-3-yl)amino]pyrimidin-4-yl]-5-(2-morpholin-4-ylethyl)thieno[2,3-c]pyrrol-4-one Chemical compound CC1(N(C(C2=C1SC(=C2)C1=NC(=NC=C1)NC1=CC=NN1C)=O)CCN1CCOCC1)C JNPRPMBJODOFEC-UHFFFAOYSA-N 0.000 claims description 19
- 206010009944 Colon cancer Diseases 0.000 claims description 19
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 19
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 17
- 238000004519 manufacturing process Methods 0.000 claims description 14
- 239000001257 hydrogen Substances 0.000 claims description 13
- 229910052739 hydrogen Inorganic materials 0.000 claims description 13
- 239000003795 chemical substances by application Substances 0.000 claims description 12
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 8
- 201000002528 pancreatic cancer Diseases 0.000 claims description 8
- 229960002633 ramucirumab Drugs 0.000 claims description 8
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 7
- 201000001441 melanoma Diseases 0.000 claims description 7
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 150000002431 hydrogen Chemical class 0.000 claims description 6
- 239000003085 diluting agent Substances 0.000 claims description 4
- UZWDCWONPYILKI-UHFFFAOYSA-N n-[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-(7-fluoro-2-methyl-3-propan-2-ylbenzimidazol-5-yl)pyrimidin-2-amine Chemical compound C1CN(CC)CCN1CC(C=N1)=CC=C1NC1=NC=C(F)C(C=2C=C3N(C(C)C)C(C)=NC3=C(F)C=2)=N1 UZWDCWONPYILKI-UHFFFAOYSA-N 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 3
- 239000000460 chlorine Substances 0.000 claims description 3
- 229910052801 chlorine Inorganic materials 0.000 claims description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 229910052731 fluorine Inorganic materials 0.000 claims description 3
- 239000011737 fluorine Substances 0.000 claims description 3
- 230000002401 inhibitory effect Effects 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- KHVSDEGLMIURSH-UHFFFAOYSA-N thieno[2,3-c]pyrrol-4-one Chemical class S1C=CC2=C1C=NC2=O KHVSDEGLMIURSH-UHFFFAOYSA-N 0.000 abstract 1
- 239000000203 mixture Substances 0.000 description 120
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 93
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 85
- 239000000243 solution Substances 0.000 description 78
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 72
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 66
- 230000002829 reductive effect Effects 0.000 description 61
- 235000019439 ethyl acetate Nutrition 0.000 description 46
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 44
- 239000011541 reaction mixture Substances 0.000 description 42
- 239000000706 filtrate Substances 0.000 description 41
- 238000002360 preparation method Methods 0.000 description 39
- 238000004458 analytical method Methods 0.000 description 35
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 33
- 238000006243 chemical reaction Methods 0.000 description 31
- 238000000034 method Methods 0.000 description 30
- 210000004027 cell Anatomy 0.000 description 29
- 239000007787 solid Substances 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 23
- 230000005764 inhibitory process Effects 0.000 description 23
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- -1 thieno[2,3-c]pyrrol-4-one compound Chemical class 0.000 description 20
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 19
- 239000012071 phase Substances 0.000 description 19
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 18
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 18
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 18
- 230000004614 tumor growth Effects 0.000 description 18
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 17
- 229910052757 nitrogen Inorganic materials 0.000 description 17
- 239000003112 inhibitor Substances 0.000 description 16
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 16
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 238000010898 silica gel chromatography Methods 0.000 description 15
- 238000012360 testing method Methods 0.000 description 15
- 229940124297 CDK 4/6 inhibitor Drugs 0.000 description 14
- 241000282414 Homo sapiens Species 0.000 description 14
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 13
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 13
- 229910052796 boron Inorganic materials 0.000 description 13
- 239000002904 solvent Substances 0.000 description 13
- 239000003981 vehicle Substances 0.000 description 13
- 241001465754 Metazoa Species 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 12
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 12
- 230000035772 mutation Effects 0.000 description 12
- 239000012312 sodium hydride Substances 0.000 description 12
- 229910000104 sodium hydride Inorganic materials 0.000 description 12
- 238000003828 vacuum filtration Methods 0.000 description 12
- 238000005481 NMR spectroscopy Methods 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- 238000003786 synthesis reaction Methods 0.000 description 11
- 108040008097 MAP kinase activity proteins Proteins 0.000 description 10
- 102000019149 MAP kinase activity proteins Human genes 0.000 description 10
- 101150040459 RAS gene Proteins 0.000 description 10
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 10
- 239000000284 extract Substances 0.000 description 10
- 230000037361 pathway Effects 0.000 description 10
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 10
- 102000016914 ras Proteins Human genes 0.000 description 10
- 238000001308 synthesis method Methods 0.000 description 10
- 238000002877 time resolved fluorescence resonance energy transfer Methods 0.000 description 10
- 239000012981 Hank's balanced salt solution Substances 0.000 description 9
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 9
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 9
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 9
- 101150076031 RAS1 gene Proteins 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000002585 base Substances 0.000 description 9
- 239000000872 buffer Substances 0.000 description 9
- 238000010828 elution Methods 0.000 description 9
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 9
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 9
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 9
- 229920006395 saturated elastomer Polymers 0.000 description 9
- 0 *CCN(C(*)(*)c1c2cc(-c3nc(Cl)ncc3*)[s]1)C2=O Chemical compound *CCN(C(*)(*)c1c2cc(-c3nc(Cl)ncc3*)[s]1)C2=O 0.000 description 8
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 8
- 239000000654 additive Substances 0.000 description 8
- 230000000996 additive effect Effects 0.000 description 8
- 239000003153 chemical reaction reagent Substances 0.000 description 8
- 235000019253 formic acid Nutrition 0.000 description 8
- 239000006166 lysate Substances 0.000 description 8
- 238000005259 measurement Methods 0.000 description 8
- 239000002480 mineral oil Substances 0.000 description 8
- 235000010446 mineral oil Nutrition 0.000 description 8
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 8
- 229910000027 potassium carbonate Inorganic materials 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 8
- 102100030708 GTPase KRas Human genes 0.000 description 7
- 101000584612 Homo sapiens GTPase KRas Proteins 0.000 description 7
- 230000004913 activation Effects 0.000 description 7
- 238000004440 column chromatography Methods 0.000 description 7
- 239000013078 crystal Substances 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- KBZMMIRFTBGDJE-UHFFFAOYSA-N 2-(2-chloropyrimidin-4-yl)-6,6-dimethyl-5-(2-morpholin-4-ylethyl)thieno[2,3-c]pyrrol-4-one Chemical compound ClC1=NC=CC(=N1)C1=CC2=C(C(N(C2=O)CCN2CCOCC2)(C)C)S1 KBZMMIRFTBGDJE-UHFFFAOYSA-N 0.000 description 6
- KMSGORGYCMNQCA-UHFFFAOYSA-N 2-bromo-6,6-dimethyl-5H-thieno[2,3-c]pyrrol-4-one Chemical compound BrC1=CC2=C(C(NC2=O)(C)C)S1 KMSGORGYCMNQCA-UHFFFAOYSA-N 0.000 description 6
- JESRNIJXVIFVOV-UHFFFAOYSA-N 2-methylpyrazol-3-amine Chemical compound CN1N=CC=C1N JESRNIJXVIFVOV-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 6
- 101000876610 Dictyostelium discoideum Extracellular signal-regulated kinase 2 Proteins 0.000 description 6
- 101001052493 Homo sapiens Mitogen-activated protein kinase 1 Proteins 0.000 description 6
- 102100024193 Mitogen-activated protein kinase 1 Human genes 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- 239000013530 defoamer Substances 0.000 description 6
- UQLDLKMNUJERMK-UHFFFAOYSA-L di(octadecanoyloxy)lead Chemical compound [Pb+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O UQLDLKMNUJERMK-UHFFFAOYSA-L 0.000 description 6
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 6
- 108010082117 matrigel Proteins 0.000 description 6
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 6
- 235000017557 sodium bicarbonate Nutrition 0.000 description 6
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 5
- 238000002965 ELISA Methods 0.000 description 5
- 102000043136 MAP kinase family Human genes 0.000 description 5
- 108091054455 MAP kinase family Proteins 0.000 description 5
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 5
- 210000001015 abdomen Anatomy 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 238000011717 athymic nude mouse Methods 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 5
- 229910000024 caesium carbonate Inorganic materials 0.000 description 5
- 230000008859 change Effects 0.000 description 5
- 229940125904 compound 1 Drugs 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- 238000003306 harvesting Methods 0.000 description 5
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 5
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 5
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 5
- 235000011056 potassium acetate Nutrition 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- 230000004580 weight loss Effects 0.000 description 5
- 101000984753 Homo sapiens Serine/threonine-protein kinase B-raf Proteins 0.000 description 4
- 102000046795 Mitogen-Activated Protein Kinase 3 Human genes 0.000 description 4
- 108700027649 Mitogen-Activated Protein Kinase 3 Proteins 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- 108091000080 Phosphotransferase Proteins 0.000 description 4
- 241000700159 Rattus Species 0.000 description 4
- 102100027103 Serine/threonine-protein kinase B-raf Human genes 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- 239000002246 antineoplastic agent Substances 0.000 description 4
- 239000011324 bead Substances 0.000 description 4
- 230000004663 cell proliferation Effects 0.000 description 4
- 230000002301 combined effect Effects 0.000 description 4
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 4
- 229940127089 cytotoxic agent Drugs 0.000 description 4
- 239000013024 dilution buffer Substances 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 125000004494 ethyl ester group Chemical group 0.000 description 4
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 4
- 239000012091 fetal bovine serum Substances 0.000 description 4
- 238000002513 implantation Methods 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 229940098779 methanesulfonic acid Drugs 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 4
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 4
- 239000002953 phosphate buffered saline Substances 0.000 description 4
- 102000020233 phosphotransferase Human genes 0.000 description 4
- 229920000136 polysorbate Polymers 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 230000011664 signaling Effects 0.000 description 4
- ZAIPVTWHZANEKU-UHFFFAOYSA-N tert-butyl 4-[[4-[6,6-dimethyl-5-(2-morpholin-4-ylethyl)-4-oxothieno[2,3-c]pyrrol-2-yl]-5-methylpyrimidin-2-yl]amino]-3-methoxypyrazole-1-carboxylate Chemical compound CC1(N(C(C2=C1SC(=C2)C1=NC(=NC=C1C)NC=1C(=NN(C=1)C(=O)OC(C)(C)C)OC)=O)CCN1CCOCC1)C ZAIPVTWHZANEKU-UHFFFAOYSA-N 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 3
- BTTNYQZNBZNDOR-UHFFFAOYSA-N 2,4-dichloropyrimidine Chemical compound ClC1=CC=NC(Cl)=N1 BTTNYQZNBZNDOR-UHFFFAOYSA-N 0.000 description 3
- WTTWSMJHJFNCQB-UHFFFAOYSA-N 2-(dibenzylamino)ethanol Chemical compound C=1C=CC=CC=1CN(CCO)CC1=CC=CC=C1 WTTWSMJHJFNCQB-UHFFFAOYSA-N 0.000 description 3
- RHENBAINPHPTMJ-UHFFFAOYSA-N 2-bromo-6,6-dimethyl-5-(2-morpholin-4-ylethyl)thieno[2,3-c]pyrrol-4-one Chemical compound BrC1=CC2=C(C(N(C2=O)CCN2CCOCC2)(C)C)S1 RHENBAINPHPTMJ-UHFFFAOYSA-N 0.000 description 3
- GPUIFFRSNPNTCI-UHFFFAOYSA-N 4-benzyl-2,2-difluoromorpholin-3-one Chemical compound C(C1=CC=CC=C1)N1C(C(OCC1)(F)F)=O GPUIFFRSNPNTCI-UHFFFAOYSA-N 0.000 description 3
- IZKKZGVEYKHLBN-UHFFFAOYSA-N 5-ethyl-4-nitro-1h-pyrazole Chemical compound CCC1=NNC=C1[N+]([O-])=O IZKKZGVEYKHLBN-UHFFFAOYSA-N 0.000 description 3
- LOWDJYPIYKKQAG-UHFFFAOYSA-N 6,6-dimethyl-5H-thieno[2,3-c]pyrrol-4-one Chemical compound CC1(NC(C2=C1SC=C2)=O)C LOWDJYPIYKKQAG-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 102100039788 GTPase NRas Human genes 0.000 description 3
- 101000744505 Homo sapiens GTPase NRas Proteins 0.000 description 3
- 229940124647 MEK inhibitor Drugs 0.000 description 3
- 208000035823 Non-specific autoimmune cerebellar ataxia without characteristic antibodies Diseases 0.000 description 3
- 238000002441 X-ray diffraction Methods 0.000 description 3
- GMQUEDBQYNHEEM-UHFFFAOYSA-N [B].[B].CC(C)(O)C(C)(C)O.CC(C)(O)C(C)(C)O Chemical compound [B].[B].CC(C)(O)C(C)(C)O.CC(C)(O)C(C)(C)O GMQUEDBQYNHEEM-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000001099 ammonium carbonate Substances 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 239000012351 deprotecting agent Substances 0.000 description 3
- ZOCHARZZJNPSEU-UHFFFAOYSA-N diboron Chemical compound B#B ZOCHARZZJNPSEU-UHFFFAOYSA-N 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 238000003821 enantio-separation Methods 0.000 description 3
- 230000002496 gastric effect Effects 0.000 description 3
- 238000003304 gavage Methods 0.000 description 3
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 3
- 229910052737 gold Inorganic materials 0.000 description 3
- 239000010931 gold Substances 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- TUKJJRKDXCTGHO-UHFFFAOYSA-N methyl 2-cyanothiophene-3-carboxylate Chemical compound COC(=O)C=1C=CSC=1C#N TUKJJRKDXCTGHO-UHFFFAOYSA-N 0.000 description 3
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 3
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 3
- 230000026731 phosphorylation Effects 0.000 description 3
- 238000006366 phosphorylation reaction Methods 0.000 description 3
- 238000000634 powder X-ray diffraction Methods 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 3
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 230000002195 synergetic effect Effects 0.000 description 3
- OCGSFJWGOBEIKR-UHFFFAOYSA-N tert-butyl 2-bromo-6,6-dimethyl-4-oxothieno[2,3-c]pyrrole-5-carboxylate Chemical compound BrC1=CC2=C(C(N(C2=O)C(=O)OC(C)(C)C)(C)C)S1 OCGSFJWGOBEIKR-UHFFFAOYSA-N 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- IBXMKLPFLZYRQZ-UHFFFAOYSA-N 1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1C=CC(=O)C=CC1=CC=CC=C1 IBXMKLPFLZYRQZ-UHFFFAOYSA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 2
- DQXNTSXKIUZJJS-UHFFFAOYSA-N 2,4-dichloro-5-methylpyrimidine Chemical compound CC1=CN=C(Cl)N=C1Cl DQXNTSXKIUZJJS-UHFFFAOYSA-N 0.000 description 2
- KLTDVRKEFQBKGF-UHFFFAOYSA-N 2-(2-bromo-6,6-dimethyl-4-oxothieno[2,3-c]pyrrol-5-yl)ethyl methanesulfonate Chemical compound CS(=O)(=O)OCCN1C(C2=C(C1=O)C=C(S2)Br)(C)C KLTDVRKEFQBKGF-UHFFFAOYSA-N 0.000 description 2
- GEGFMFLOPTVDJJ-UHFFFAOYSA-N 2-(2-chloropyrimidin-4-yl)-6,6-dimethyl-5H-thieno[2,3-c]pyrrol-4-one Chemical compound ClC1=NC=CC(=N1)C1=CC2=C(C(NC2=O)(C)C)S1 GEGFMFLOPTVDJJ-UHFFFAOYSA-N 0.000 description 2
- QREKBZMQAMHWHE-UHFFFAOYSA-N 2-(2-chloropyrimidin-4-yl)-6,6-dimethyl-5H-thieno[2,3-c]pyrrol-4-one hydrochloride Chemical compound Cl.CC1(C)NC(=O)c2cc(sc12)-c1ccnc(Cl)n1 QREKBZMQAMHWHE-UHFFFAOYSA-N 0.000 description 2
- KKFDCBRMNNSAAW-UHFFFAOYSA-N 2-(morpholin-4-yl)ethanol Chemical compound OCCN1CCOCC1 KKFDCBRMNNSAAW-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- JQWOMTXGSVQRCO-UHFFFAOYSA-N 2-bromo-5-(2-hydroxyethyl)-6,6-dimethylthieno[2,3-c]pyrrol-4-one Chemical compound BrC1=CC2=C(C(N(C2=O)CCO)(C)C)S1 JQWOMTXGSVQRCO-UHFFFAOYSA-N 0.000 description 2
- NVFKJZYEGQEVHQ-UHFFFAOYSA-N 2-bromo-6,6-dimethyl-5-[2-(5-oxa-8-azaspiro[2.6]nonan-8-yl)ethyl]thieno[2,3-c]pyrrol-4-one Chemical compound BrC1=CC2=C(C(N(C2=O)CCN2CCOCC3(CC3)C2)(C)C)S1 NVFKJZYEGQEVHQ-UHFFFAOYSA-N 0.000 description 2
- JBKINHFZTVLNEM-UHFFFAOYSA-N 2-bromoethoxy-tert-butyl-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OCCBr JBKINHFZTVLNEM-UHFFFAOYSA-N 0.000 description 2
- UGHOJWLZPSTTKB-UHFFFAOYSA-N 4-(2-bromoethyl)morpholine;hydrobromide Chemical compound Br.BrCCN1CCOCC1 UGHOJWLZPSTTKB-UHFFFAOYSA-N 0.000 description 2
- JLCGPSNHSULUGI-UHFFFAOYSA-N 4-(2-bromoethyl)morpholine;hydrochloride Chemical compound Cl.BrCCN1CCOCC1 JLCGPSNHSULUGI-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- AUYLLVHPLHWIJE-UHFFFAOYSA-N 4-benzyl-2,2-difluoromorpholine Chemical compound C(C1=CC=CC=C1)N1CC(OCC1)(F)F AUYLLVHPLHWIJE-UHFFFAOYSA-N 0.000 description 2
- YDJBUEGMBIPPMF-UHFFFAOYSA-N 5-[2-(2,2-difluoromorpholin-4-yl)ethyl]-6,6-dimethyl-2-[2-[(2-methylpyrazol-3-yl)amino]pyrimidin-4-yl]thieno[2,3-c]pyrrol-4-one Chemical compound FC1(CN(CCO1)CCN1C(C2=C(C1=O)C=C(S2)C1=NC(=NC=C1)NC1=CC=NN1C)(C)C)F YDJBUEGMBIPPMF-UHFFFAOYSA-N 0.000 description 2
- DVZKNNBUPFEFCJ-UHFFFAOYSA-N 6,6-dimethyl-2-[2-[(5-methyl-1H-pyrazol-4-yl)amino]pyrimidin-4-yl]-5-(2-morpholin-4-ylethyl)thieno[2,3-c]pyrrol-4-one Chemical compound CC1(N(C(C2=C1SC(=C2)C1=NC(=NC=C1)NC=1C=NNC=1C)=O)CCN1CCOCC1)C DVZKNNBUPFEFCJ-UHFFFAOYSA-N 0.000 description 2
- MDPWAXFQGBBQET-UHFFFAOYSA-N 6,6-dimethylthieno[2,3-c]furan-4-one Chemical compound CC1(OC(C2=C1SC=C2)=O)C MDPWAXFQGBBQET-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 102000008096 B7-H1 Antigen Human genes 0.000 description 2
- 108010074708 B7-H1 Antigen Proteins 0.000 description 2
- 108020004705 Codon Proteins 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- 229910016523 CuKa Inorganic materials 0.000 description 2
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 2
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102100029974 GTPase HRas Human genes 0.000 description 2
- 101000584633 Homo sapiens GTPase HRas Proteins 0.000 description 2
- 101000944909 Homo sapiens Ribosomal protein S6 kinase alpha-1 Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 206010069755 K-ras gene mutation Diseases 0.000 description 2
- HHCBMISMPSAZBF-UHFFFAOYSA-N LY3009120 Chemical compound CC1=NC2=NC(NC)=NC=C2C=C1C1=CC(NC(=O)NCCC(C)(C)C)=C(F)C=C1C HHCBMISMPSAZBF-UHFFFAOYSA-N 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- 101100519207 Mus musculus Pdcd1 gene Proteins 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 2
- 102100033536 Ribosomal protein S6 kinase alpha-1 Human genes 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 description 2
- 239000013504 Triton X-100 Substances 0.000 description 2
- 229920004890 Triton X-100 Polymers 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 238000003016 alphascreen Methods 0.000 description 2
- 235000012501 ammonium carbonate Nutrition 0.000 description 2
- 239000000908 ammonium hydroxide Substances 0.000 description 2
- 230000003042 antagnostic effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 239000013592 cell lysate Substances 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 238000002050 diffraction method Methods 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 2
- 230000007705 epithelial mesenchymal transition Effects 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 229940043355 kinase inhibitor Drugs 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- OFXIBQASCWPFRW-UHFFFAOYSA-N methyl 2-[2-(dibenzylamino)ethoxy]-2,2-difluoroacetate Chemical compound C(C1=CC=CC=C1)N(CCOC(C(=O)OC)(F)F)CC1=CC=CC=C1 OFXIBQASCWPFRW-UHFFFAOYSA-N 0.000 description 2
- ADKGOXMEPLSPNN-UHFFFAOYSA-N methyl 2-bromothiophene-3-carboxylate Chemical compound COC(=O)C=1C=CSC=1Br ADKGOXMEPLSPNN-UHFFFAOYSA-N 0.000 description 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920001296 polysiloxane Polymers 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- FGIUAXJPYTZDNR-UHFFFAOYSA-N potassium nitrate Chemical compound [K+].[O-][N+]([O-])=O FGIUAXJPYTZDNR-UHFFFAOYSA-N 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000012064 sodium phosphate buffer Substances 0.000 description 2
- 230000000707 stereoselective effect Effects 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 2
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- NRQHBNNTBIDSRK-YRNVUSSQSA-N (4e)-4-[(4-methoxyphenyl)methylidene]-2-methyl-1,3-oxazol-5-one Chemical compound C1=CC(OC)=CC=C1\C=C\1C(=O)OC(C)=N/1 NRQHBNNTBIDSRK-YRNVUSSQSA-N 0.000 description 1
- BNYCHCAYYYRJSH-UHFFFAOYSA-N 1h-pyrazole-5-carboxamide Chemical compound NC(=O)C1=CC=NN1 BNYCHCAYYYRJSH-UHFFFAOYSA-N 0.000 description 1
- VAFOWNAIGXFWLF-UHFFFAOYSA-N 2-(2-chloro-5-methylpyrimidin-4-yl)-6,6-dimethyl-5-(2-morpholin-4-ylethyl)thieno[2,3-c]pyrrol-4-one Chemical compound ClC1=NC=C(C(=N1)C1=CC2=C(C(N(C2=O)CCN2CCOCC2)(C)C)S1)C VAFOWNAIGXFWLF-UHFFFAOYSA-N 0.000 description 1
- XNIOWJUQPMKCIJ-UHFFFAOYSA-N 2-(benzylamino)ethanol Chemical compound OCCNCC1=CC=CC=C1 XNIOWJUQPMKCIJ-UHFFFAOYSA-N 0.000 description 1
- MYXLEIDXGNMPAD-UHFFFAOYSA-N 2-[2-(dibenzylamino)ethoxy]-2,2-difluoroacetic acid Chemical compound C(C1=CC=CC=C1)N(CCOC(C(=O)O)(F)F)CC1=CC=CC=C1 MYXLEIDXGNMPAD-UHFFFAOYSA-N 0.000 description 1
- CRUDMIDFICENNH-UHFFFAOYSA-N 2-[2-[(5-methoxy-1H-pyrazol-4-yl)amino]-5-methylpyrimidin-4-yl]-6,6-dimethyl-5-(2-morpholin-4-ylethyl)thieno[2,3-c]pyrrol-4-one hydrochloride Chemical compound Cl.COc1[nH]ncc1Nc1ncc(C)c(n1)-c1cc2C(=O)N(CCN3CCOCC3)C(C)(C)c2s1 CRUDMIDFICENNH-UHFFFAOYSA-N 0.000 description 1
- QHYMLNGXQQQFSM-UHFFFAOYSA-N 2-bromo-5-[2-[tert-butyl(dimethyl)silyl]oxyethyl]-6,6-dimethylthieno[2,3-c]pyrrol-4-one Chemical compound BrC1=CC2=C(C(N(C2=O)CCO[Si](C)(C)C(C)(C)C)(C)C)S1 QHYMLNGXQQQFSM-UHFFFAOYSA-N 0.000 description 1
- RVSXMPCELBYUSF-UHFFFAOYSA-N 2-bromothiophene-3-carboxylic acid Chemical compound OC(=O)C=1C=CSC=1Br RVSXMPCELBYUSF-UHFFFAOYSA-N 0.000 description 1
- OAWAZQITIZDJRB-UHFFFAOYSA-N 2-chloro-2,2-difluoroacetic acid Chemical compound OC(=O)C(F)(F)Cl OAWAZQITIZDJRB-UHFFFAOYSA-N 0.000 description 1
- WWXOWIKXBOZBIB-UHFFFAOYSA-N 2-cyclopropylpyrazol-3-amine Chemical compound NC1=CC=NN1C1CC1 WWXOWIKXBOZBIB-UHFFFAOYSA-N 0.000 description 1
- CUAWMPGXOJKBBM-UHFFFAOYSA-N 2-cyclopropylpyrazole-3-carboxylic acid Chemical compound OC(=O)C1=CC=NN1C1CC1 CUAWMPGXOJKBBM-UHFFFAOYSA-N 0.000 description 1
- TWBPWBPGNQWFSJ-UHFFFAOYSA-N 2-phenylaniline Chemical group NC1=CC=CC=C1C1=CC=CC=C1 TWBPWBPGNQWFSJ-UHFFFAOYSA-N 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 1
- CVMXEDZZSWLXPB-UHFFFAOYSA-N 4-(2-bromoethyl)morpholine Chemical compound BrCCN1CCOCC1 CVMXEDZZSWLXPB-UHFFFAOYSA-N 0.000 description 1
- NBJHDLKSWUDGJG-UHFFFAOYSA-N 4-(2-chloroethyl)morpholin-4-ium;chloride Chemical compound Cl.ClCCN1CCOCC1 NBJHDLKSWUDGJG-UHFFFAOYSA-N 0.000 description 1
- ZAPMTSHEXFEPSD-UHFFFAOYSA-N 4-(2-chloroethyl)morpholine Chemical compound ClCCN1CCOCC1 ZAPMTSHEXFEPSD-UHFFFAOYSA-N 0.000 description 1
- PKRKJRIVVOGXHM-UHFFFAOYSA-N 4-amino-1h-pyrazole-5-carbonitrile Chemical compound NC=1C=NNC=1C#N PKRKJRIVVOGXHM-UHFFFAOYSA-N 0.000 description 1
- OKVZAUTUZBGJMA-UHFFFAOYSA-N 5-[2-[cyclopropyl(methyl)amino]ethyl]-6,6-dimethyl-2-[2-[(2-methylpyrazol-3-yl)amino]pyrimidin-4-yl]thieno[2,3-c]pyrrol-4-one Chemical compound C1(CC1)N(CCN1C(C2=C(C1=O)C=C(S2)C1=NC(=NC=C1)NC1=CC=NN1C)(C)C)C OKVZAUTUZBGJMA-UHFFFAOYSA-N 0.000 description 1
- WPPUKXXCGOVFMP-UHFFFAOYSA-N 5-[2-[tert-butyl(dimethyl)silyl]oxyethyl]-2-(2-chloropyrimidin-4-yl)-6,6-dimethylthieno[2,3-c]pyrrol-4-one Chemical compound [Si](C)(C)(C(C)(C)C)OCCN1C(C2=C(C1=O)C=C(S2)C1=NC(=NC=C1)Cl)(C)C WPPUKXXCGOVFMP-UHFFFAOYSA-N 0.000 description 1
- NSBPIXZUFLIUGL-UHFFFAOYSA-N 5-[2-[tert-butyl(dimethyl)silyl]oxyethyl]-6,6-dimethyl-2-[2-[(2-methylpyrazol-3-yl)amino]pyrimidin-4-yl]thieno[2,3-c]pyrrol-4-one Chemical compound [Si](C)(C)(C(C)(C)C)OCCN1C(C2=C(C1=O)C=C(S2)C1=NC(=NC=C1)NC=1N(N=CC=1)C)(C)C NSBPIXZUFLIUGL-UHFFFAOYSA-N 0.000 description 1
- AMBUTUSRGNEOAG-UHFFFAOYSA-N 5-ethyl-1h-pyrazol-4-amine Chemical compound CCC1=NNC=C1N AMBUTUSRGNEOAG-UHFFFAOYSA-N 0.000 description 1
- CBNLNXLAIMQSTR-UHFFFAOYSA-N 5-ethyl-1h-pyrazole Chemical compound CCC1=CC=NN1 CBNLNXLAIMQSTR-UHFFFAOYSA-N 0.000 description 1
- GLNYWXCDHONDOZ-UHFFFAOYSA-N 5-methoxy-4-nitro-1h-pyrazole Chemical compound COC=1NN=CC=1[N+]([O-])=O GLNYWXCDHONDOZ-UHFFFAOYSA-N 0.000 description 1
- WMEWGPRHFFHUAV-UHFFFAOYSA-N 5-methyl-1h-pyrazol-4-amine Chemical compound CC1=NNC=C1N WMEWGPRHFFHUAV-UHFFFAOYSA-N 0.000 description 1
- SWIPIBOQWRVJQW-UHFFFAOYSA-N 5-oxa-8-azaspiro[2.6]nonane Chemical compound C1CC11COCCNC1 SWIPIBOQWRVJQW-UHFFFAOYSA-N 0.000 description 1
- MMOODUDIFAVCIO-UHFFFAOYSA-N 6,6-difluoro-1,4-oxazepane;hydrochloride Chemical compound Cl.FC1(F)CNCCOC1 MMOODUDIFAVCIO-UHFFFAOYSA-N 0.000 description 1
- YCERFLIXSISLBS-UHFFFAOYSA-N 6,6-dimethyl-2-[2-[(2-methylpyrazol-3-yl)amino]pyrimidin-4-yl]-5-(2-thiomorpholin-4-ylethyl)thieno[2,3-c]pyrrol-4-one Chemical compound CC1(N(C(C2=C1SC(=C2)C1=NC(=NC=C1)NC1=CC=NN1C)=O)CCN1CCSCC1)C YCERFLIXSISLBS-UHFFFAOYSA-N 0.000 description 1
- RZKOQFGRSNUEGB-UHFFFAOYSA-N 6,6-dimethyl-2-[2-[(2-methylpyrazol-3-yl)amino]pyrimidin-4-yl]-5-[2-(2-oxa-5-azabicyclo[4.1.0]heptan-5-yl)ethyl]thieno[2,3-c]pyrrol-4-one Chemical compound CC1(N(C(C2=C1SC(=C2)C1=NC(=NC=C1)NC1=CC=NN1C)=O)CCN1CCOC2CC12)C RZKOQFGRSNUEGB-UHFFFAOYSA-N 0.000 description 1
- RHXHGRAEPCAFML-UHFFFAOYSA-N 7-cyclopentyl-n,n-dimethyl-2-[(5-piperazin-1-ylpyridin-2-yl)amino]pyrrolo[2,3-d]pyrimidine-6-carboxamide Chemical compound N1=C2N(C3CCCC3)C(C(=O)N(C)C)=CC2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 RHXHGRAEPCAFML-UHFFFAOYSA-N 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 241000208340 Araliaceae Species 0.000 description 1
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CGZIPLXYJMFKND-UHFFFAOYSA-O CC(C)(C)OC(N(C(C)(C)c1c2cc(BOC(C)(C)C(C)(C)[OH2+])[s]1)C2=O)=O Chemical compound CC(C)(C)OC(N(C(C)(C)c1c2cc(BOC(C)(C)C(C)(C)[OH2+])[s]1)C2=O)=O CGZIPLXYJMFKND-UHFFFAOYSA-O 0.000 description 1
- PPPGTBSCWCSEHE-UHFFFAOYSA-N CC(C)(c1c2cc(-c3c(C)cnc(Nc4c[nH]nc4OC)n3)[s]1)N(CCN1CCOCC1)C2=O Chemical compound CC(C)(c1c2cc(-c3c(C)cnc(Nc4c[nH]nc4OC)n3)[s]1)N(CCN1CCOCC1)C2=O PPPGTBSCWCSEHE-UHFFFAOYSA-N 0.000 description 1
- ASNFFALLZKXOJQ-UHFFFAOYSA-N CC(C)(c1c2cc(-c3ccnc(Nc4ccn[n]4C)n3)[s]1)N(CCN(CCOC1)CC1(F)F)C2=O Chemical compound CC(C)(c1c2cc(-c3ccnc(Nc4ccn[n]4C)n3)[s]1)N(CCN(CCOC1)CC1(F)F)C2=O ASNFFALLZKXOJQ-UHFFFAOYSA-N 0.000 description 1
- XLZMWNWNBXSZKF-UHFFFAOYSA-N CC(C)N1CCOCC1 Chemical compound CC(C)N1CCOCC1 XLZMWNWNBXSZKF-UHFFFAOYSA-N 0.000 description 1
- RAUVLPTXTLUZNS-UHFFFAOYSA-N CC1=CC2=C(C=NC2=O)S1 Chemical compound CC1=CC2=C(C=NC2=O)S1 RAUVLPTXTLUZNS-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 108091008794 FGF receptors Proteins 0.000 description 1
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000712674 Homo sapiens TGF-beta receptor type-1 Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- IVRXNBXKWIJUQB-UHFFFAOYSA-N LY-2157299 Chemical compound CC1=CC=CC(C=2C(=C3CCCN3N=2)C=2C3=CC(=CC=C3N=CC=2)C(N)=O)=N1 IVRXNBXKWIJUQB-UHFFFAOYSA-N 0.000 description 1
- 102000001291 MAP Kinase Kinase Kinase Human genes 0.000 description 1
- 108060006687 MAP kinase kinase kinase Proteins 0.000 description 1
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 101100372766 Mus musculus Kdr gene Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 102000014736 Notch Human genes 0.000 description 1
- 108010070047 Notch Receptors Proteins 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 235000005035 Panax pseudoginseng ssp. pseudoginseng Nutrition 0.000 description 1
- 235000003140 Panax quinquefolius Nutrition 0.000 description 1
- 229940122907 Phosphatase inhibitor Drugs 0.000 description 1
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 238000011579 SCID mouse model Methods 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 102100033456 TGF-beta receptor type-1 Human genes 0.000 description 1
- 108091005735 TGF-beta receptors Proteins 0.000 description 1
- 102000016715 Transforming Growth Factor beta Receptors Human genes 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000011481 absorbance measurement Methods 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 238000000540 analysis of variance Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940124650 anti-cancer therapies Drugs 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 238000011319 anticancer therapy Methods 0.000 description 1
- 238000007080 aromatic substitution reaction Methods 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- 150000008038 benzoazepines Chemical class 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- ZYPBNISTOOQVDL-UHFFFAOYSA-N butyl-[2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane Chemical group CCCCPC1=CC=CC=C1C1=C(C(C)C)C=C(C(C)C)C=C1C(C)C ZYPBNISTOOQVDL-UHFFFAOYSA-N 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 230000007248 cellular mechanism Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 238000010568 chiral column chromatography Methods 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 229960002271 cobimetinib Drugs 0.000 description 1
- RESIMIUSNACMNW-BXRWSSRYSA-N cobimetinib fumarate Chemical compound OC(=O)\C=C\C(O)=O.C1C(O)([C@H]2NCCCC2)CN1C(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F.C1C(O)([C@H]2NCCCC2)CN1C(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F RESIMIUSNACMNW-BXRWSSRYSA-N 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125961 compound 24 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 230000006552 constitutive activation Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910000365 copper sulfate Inorganic materials 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- OCXGTPDKNBIOTF-UHFFFAOYSA-N dibromo(triphenyl)-$l^{5}-phosphane Chemical compound C=1C=CC=CC=1P(Br)(C=1C=CC=CC=1)(Br)C1=CC=CC=C1 OCXGTPDKNBIOTF-UHFFFAOYSA-N 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- REWLCYPYZCHYSS-UHFFFAOYSA-N ditert-butyl-[3,6-dimethoxy-2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane Chemical group COC1=CC=C(OC)C(C=2C(=CC(=CC=2C(C)C)C(C)C)C(C)C)=C1P(C(C)(C)C)C(C)(C)C REWLCYPYZCHYSS-UHFFFAOYSA-N 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- FCZCIXQGZOUIDN-UHFFFAOYSA-N ethyl 2-diethoxyphosphinothioyloxyacetate Chemical compound CCOC(=O)COP(=S)(OCC)OCC FCZCIXQGZOUIDN-UHFFFAOYSA-N 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 102000052178 fibroblast growth factor receptor activity proteins Human genes 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000012595 freezing medium Substances 0.000 description 1
- 229950000456 galunisertib Drugs 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 235000008434 ginseng Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- FRIJBUGBVQZNTB-UHFFFAOYSA-M magnesium;ethane;bromide Chemical compound [Mg+2].[Br-].[CH2-]C FRIJBUGBVQZNTB-UHFFFAOYSA-M 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- FBBDOOHMGLLEGJ-UHFFFAOYSA-N methane;hydrochloride Chemical compound C.Cl FBBDOOHMGLLEGJ-UHFFFAOYSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- VEBLEROFGPOMPB-UHFFFAOYSA-N n-methylcyclopropanamine Chemical compound CNC1CC1 VEBLEROFGPOMPB-UHFFFAOYSA-N 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 229960004390 palbociclib Drugs 0.000 description 1
- AHJRHEGDXFFMBM-UHFFFAOYSA-N palbociclib Chemical compound N1=C2N(C3CCCC3)C(=O)C(C(=O)C)=C(C)C2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 AHJRHEGDXFFMBM-UHFFFAOYSA-N 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000004323 potassium nitrate Substances 0.000 description 1
- 235000010333 potassium nitrate Nutrition 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000003223 protective agent Substances 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 108010077182 raf Kinases Proteins 0.000 description 1
- 102000009929 raf Kinases Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 229950003687 ribociclib Drugs 0.000 description 1
- 238000003118 sandwich ELISA Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- HELHAJAZNSDZJO-OLXYHTOASA-L sodium L-tartrate Chemical compound [Na+].[Na+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O HELHAJAZNSDZJO-OLXYHTOASA-L 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- CMZUMMUJMWNLFH-UHFFFAOYSA-N sodium metavanadate Chemical compound [Na+].[O-][V](=O)=O CMZUMMUJMWNLFH-UHFFFAOYSA-N 0.000 description 1
- 239000001433 sodium tartrate Substances 0.000 description 1
- 229960002167 sodium tartrate Drugs 0.000 description 1
- 235000011004 sodium tartrates Nutrition 0.000 description 1
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- ULMNBXMMAFNOPM-UHFFFAOYSA-N spiro[5H-thieno[2,3-c]pyrrole-6,1'-cyclopropane]-4-one Chemical compound C12(CC1)NC(C1=C2SC=C1)=O ULMNBXMMAFNOPM-UHFFFAOYSA-N 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000002626 targeted therapy Methods 0.000 description 1
- LGHAAAIUUYWURJ-UHFFFAOYSA-N tert-butyl 1h-pyrrole-2-carboxylate Chemical compound CC(C)(C)OC(=O)C1=CC=CN1 LGHAAAIUUYWURJ-UHFFFAOYSA-N 0.000 description 1
- QSAHWEDTMRXYOS-UHFFFAOYSA-N tert-butyl 2-(2-chloropyrimidin-4-yl)-6,6-dimethyl-4-oxothieno[2,3-c]pyrrole-5-carboxylate Chemical compound ClC1=NC=CC(=N1)C1=CC2=C(C(N(C2=O)C(=O)OC(C)(C)C)(C)C)S1 QSAHWEDTMRXYOS-UHFFFAOYSA-N 0.000 description 1
- HOOKYOCWRZOQME-UHFFFAOYSA-N tert-butyl 3-methoxy-4-nitropyrazole-1-carboxylate Chemical compound COC1=NN(C=C1[N+](=O)[O-])C(=O)OC(C)(C)C HOOKYOCWRZOQME-UHFFFAOYSA-N 0.000 description 1
- MEOUQOGOAHPOCE-UHFFFAOYSA-N tert-butyl 4-amino-3-methoxypyrazole-1-carboxylate Chemical compound NC=1C(=NN(C=1)C(=O)OC(C)(C)C)OC MEOUQOGOAHPOCE-UHFFFAOYSA-N 0.000 description 1
- PXHXZWFYSRVALR-UHFFFAOYSA-N tert-butyl N-(2-cyclopropylpyrazol-3-yl)carbamate Chemical compound C1(CC1)N1N=CC=C1NC(OC(C)(C)C)=O PXHXZWFYSRVALR-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 229960004066 trametinib Drugs 0.000 description 1
- LIRYPHYGHXZJBZ-UHFFFAOYSA-N trametinib Chemical compound CC(=O)NC1=CC=CC(N2C(N(C3CC3)C(=O)C3=C(NC=4C(=CC(I)=CC=4)F)N(C)C(=O)C(C)=C32)=O)=C1 LIRYPHYGHXZJBZ-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 238000013414 tumor xenograft model Methods 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Endocrinology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
本發明提供噻吩并[2,3-c]吡咯-4-酮化合物,其可抑制細胞外信號調控激酶(extracellular-signal-regulated kinase;ERK)之活性及可用於治療癌症。
Description
本發明係關於抑制細胞外信號調控激酶(ERK)之活性且可用於治療癌症之噻吩并[2,3-c]吡咯-4-酮化合物或其醫藥上可接受之鹽,及包含該等化合物之醫藥組合物。
ERK/MAPK路徑對於細胞增殖至關重要且通常觀察到在許多腫瘤中被活化。ERK1/2上游之RAS基因在若干種癌症(包括結腸直腸腫瘤、黑色素瘤、非小細胞肺癌以及乳房及胰臟腫瘤)中發生突變。在許多人類腫瘤中,高RAS活性伴隨有升高的ERK活性。多項研究亦已顯示ERK係RAS信號傳導之關鍵組份。該等觀察支持ERK1/2信號傳導路徑對研發廣譜人類腫瘤之抗癌療法之吸引力。
ERK抑制劑為業內已知;例如,參見WO2013130976。另外,其他胺基嘧啶化合物為業內已知;例如,參見WO 2010/022121。業內仍需要提供更具體而言用於治療癌症之替代性ERK抑制劑。因此,本發明提供可用於治療癌症之ERK1/2抑制劑。
本發明提供下式之化合物:

其中:
R1係

R2及R3獨立地係甲基或R2及R3可一起形成環丙基;R4係氫、甲基、氯、氟或三氟甲基;且R5係

或其醫藥上可接受之鹽。
本發明提供下式之化合物:

其中:
R1係

R2及R3獨立地係甲基或R2及R3可一起形成環丙基;R4係氫、甲基、氯、氟或三氟甲基;且R5係

或其醫藥上可接受之鹽。
本發明亦提供式I化合物之實施例,其中R2及R3係甲基。
本發明亦提供式I化合物之另一實施例,其中R4係氫。
本發明亦提供式I化合物之另一實施例,其中R1係
本發明亦提供式I化合物之另一實施例,其中R5係
較佳地,本發明提供化合物,其為6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮,或其醫藥上可接受之鹽。
作為具體實施例,本發明提供化合物,其為6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮。
本發明提供醫藥組合物,其包含6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮或其醫藥上可接受之鹽及醫藥上可接受之載劑、稀釋劑或賦形劑。本發明提供醫藥組合物,其包含6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮及醫藥上可接受之載劑、稀釋劑或賦形劑。
本發明提供治療癌症之方法,其包含向有需要之患者投與有效量之6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮或其醫藥上可接受之鹽。本發明提供治療癌症之方法,其包含向有需要之患者投與有效量之6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮。
本發明提供6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮或其醫藥上可接受之鹽,其用於療法中。本發明提供6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮或其醫藥上可接受之鹽,其用於治療癌症。本發明提供醫藥組合物用於治療癌症,該醫藥組合物包含6,6-二甲基-2-
{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮或其醫藥上可接受之鹽。
本發明亦提供6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮用於療法中。本發明提供6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮用於治療癌症。本發明提供醫藥組合物用於治療癌症,該醫藥組合物包含6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮。
本發明提供6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮或其醫藥上可接受之鹽之用途,其用於製造用來治療癌症之藥劑。本發明亦提供6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮之用途,其用於製造用來治療癌症之藥劑。
本發明提供呈結晶型之6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮。本發明亦提供呈結晶型之6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮,其特徵在於X射線粉末繞射圖案具有在19.3°出現之2θ±0.2°特徵峰與一或多個選自由以下組成之群之峰的組合:15.5°、17.1°、18.0°、20.2°、21.5°及22.1°。
另外,本發明提供如本文所述之方法及用途之較佳實施例,其中癌症選自由以下組成之群:黑色素瘤、結腸直腸癌、胰臟癌及非小細胞肺癌。較佳癌症係結腸直腸癌、胰臟癌及非小細胞肺癌。
本發明亦提供6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-
4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮或其醫藥上可接受之鹽,其與一或多種化學治療劑之組合同時、分別或依序投與用於治療癌症。用於此一組合之較佳化學治療劑係泛RAF抑制劑化合物,更具體而言1-(3,3-二甲基丁基)-3-(2-氟-4-甲基-5-(7-甲基-2-(甲基胺基)吡啶并[2,3-d]嘧啶-6-基)苯基)尿素);CDK4/6抑制劑化合物,更具體而言帕博西尼(palbociclib)、瑞博西尼(ribociclib)或[5-(4-乙基-六氫吡嗪-1-基甲基)-吡啶-2-基]-[5-氟-4-(7-氟-3-異丙基-2-甲基-3H-苯并咪唑-5-基)-嘧啶-2-基]-胺或其醫藥上可接受之鹽;或抗VEGFR2抗體,更具體而言雷莫蘆單抗(ramucirumab)。用於此一組合之其他較佳化學治療劑係TGF-β受體激酶抑制劑化合物,更具體而言蓋倫塞替(galunisertib)(參見WO 2004/048382);ALK-5激酶抑制劑,更具體而言EW-7197;MEK抑制劑化合物,更具體而言卡比替尼(cobimetinib)或曲美替尼(trametinib);或Notch抑制劑化合物,更具體而言4,4,4-三氟-N-[(1S)-2-[[(7S)-5-(2-羥基乙基)-6-側氧基-7H-吡啶并[2,3-d][3]苯并氮呯-7-基]胺基]-1-甲基-2-側氧基-乙基]丁醯胺(參見WO 2013/016081)。用於此一組合之其他較佳化學治療劑係PD-L1(程式化死亡配體1)抑制劑或PD-1(程式化死亡1)抑制劑。
本發明較佳含有具有以下取代基之式I化合物:
a)R1係 b)R2係甲基;c)R3係甲基;d)R4係氫;或
e)R5係
b)R2係甲基;c)R3係甲基;d)R4係氫;或
e)R5係
更佳地,本發明含有具有以下取代基組合之式I化合物:a)R2及R3係甲基;b)R2係甲基,R3係甲基,且R4係氫;
c)R1係 且R5係
且R5係 d)R2係甲基,R3係甲基,R4係氫,且R1係
d)R2係甲基,R3係甲基,R4係氫,且R1係 e)R2係甲基,R3係甲基,R4係氫,且R5係
e)R2係甲基,R3係甲基,R4係氫,且R5係 ;或f)R2係甲基,R3係甲基,R4係氫,且R1係
;或f)R2係甲基,R3係甲基,R4係氫,且R1係 ,且R5係
,且R5係

除非另外指示,否則如上文及在本發明描述通篇中所用之以下術語應理解為具有以下含義:「醫藥上可接受之載劑、稀釋劑或賦形劑」係業內公認用於將生物活性劑遞送至哺乳動物(例如人類)之介質。
「醫藥上可接受之鹽」或「一種醫藥上可接受之鹽」係指本發明化合物之相對無毒之無機及有機鹽。
「有效量」意指將使組織、系統、動物、哺乳動物或人類發出研究者、獸醫、醫師或其他臨床醫師正尋求之生物或醫學反應或期望治療效應的本發明化合物或其醫藥上可接受之鹽或含有本發明化合物或其醫藥上可接受之鹽之醫藥組合物的量。
術語「治療(treatment)」、「治療(treat)」、「治療(treating)」
及諸如此類意欲包括減緩或逆轉病症之進展。該等術語亦包括緩和、改善、減弱、消除或減輕病症或病況之一或多種症狀,即使實際上並不消除該病症或病況且即使自身並不減緩或逆轉該病症或病況之進展。
熟習此項技術者應理解,本發明化合物能夠形成鹽。本發明化合物含有鹼性雜環,且因此與多種無機及有機酸中之任一者反應形成醫藥上可接受之酸加成鹽。該等醫藥上可接受之酸加成鹽及其一般製備方法為業內所熟知。例如,參見P.Stahl等人,HANDBOOK OF PHARMACEUTICAL SALTS:PROPERTIES,SELECTION AND USE,(VCHA/Wiley-VCH,2008);S.M.Berge等人,「Pharmaceutical Salts」,Journal of Pharmaceutical Sciences,第66卷,第1期,1977年1月。
本發明化合物較佳調配成藉由多種途徑投與之醫藥組合物。較佳地,該等組合物用於口服投與。該等醫藥組合物及其製備方法為業內所熟知。例如,參見REMINGTON:THE SCIENCE AND PRACTICE OF PHARMACY(A.Gennaro等人編輯,第21版,Mack Publishing Co.,2005)。
本發明化合物通常在較寬劑量範圍內有效。舉例而言,每日劑量通常在約1mg至2000mg之每日範圍內。較佳地,該等劑量在50mg至1000mg之每日範圍內。更佳地,該等劑量在125mg至400mg之每日範圍內。在一些情況下,低於上述範圍之下限之劑量值可能超足量,而在其他情形下仍可使用更大劑量,且因此以上劑量範圍並不意欲以任何方式限制本發明之範疇。應理解,化合物之實際投與量將由內科醫師根據包括以下各項之相關情況確定:欲治療之病況、所選投與途徑、實際投與之化合物、個體患者之年齡、體重及反應以及患者症狀之嚴重程度。
熟習此項技術者將瞭解,本發明之某些化合物含有至少一個手性中心。本發明涵蓋所有個別鏡像異構體或非鏡像異構體以及該等化合物之鏡像異構體與非鏡像異構體之混合物(包括外消旋物)。含有至少一個手性中心之本發明化合物較佳係以單一鏡像異構物或非鏡像異構物形式存在。單一鏡像異構體或非鏡像異構體可自手性試劑開始或藉由立體選擇性或立體特異性合成技術來製備。另一選擇為,單一鏡像異構物或非鏡像異構物可藉由標準手性層析或結晶技術自混合物分離。
化合物名稱中「異構物1」之名稱表示當藉由手性層析分離一對鏡像異構物之混合物時,本發明之相應中間體或化合物係兩種溶析鏡像異構物中之第一者。化合物名稱中「異構物2」之名稱表示當藉由手性層析分離一對鏡像異構物之混合物時,本發明之相應中間體或化合物係兩種溶析鏡像異構物中之第二者。
本發明化合物可根據業內所熟知及瞭解之合成方法來製備。該等反應之步驟之適宜反應條件為業內所熟知且溶劑及共試劑之適當取代為熟習此項技術者所熟知。同樣,熟習此項技術者應瞭解,合成中間體可視需要藉由多種熟知技術來分離及/或純化,且通常可將各種中間體直接用於後續合成步驟中而經極少或不經純化。另外,熟習此項技術者應瞭解,在一些情況下,引入各部分之順序並不重要。製造本發明化合物所需步驟之具體順序取決於所合成之具體化合物、起始化合物及經取代部分之相對不穩定性,該等因素為熟習此項技術者所熟知。除非另外指示,否則所有取代基皆係如先前所定義,且所有試劑皆為業內所熟知及瞭解。
如本文所使用,以下術語具有所指示之含義:「ACN」係指乙腈;「DCM」係指二氯甲烷;「DMF」表示N,N-二甲基甲醯胺;「DMSO」係指二甲基亞碸;「DTT」係指二硫蘇糖醇;「EDTA」
係指乙二胺四乙酸;「EGTA」係指乙二醇四乙酸;「ELISA」係指酶聯免疫吸附分析;「EtOAc」係指乙酸乙酯;「EtOH」係指乙醇;「FBS」係指胎牛血清;「HBSS」係指漢克氏平衡鹽溶液(Hank’s Balanced Salt Solution);「IC50」係指半數最大抑制濃度;「IVTI」係指活體內靶抑制;「MS」係指質譜術;「MeOH」係指甲醇;「NMR」係指核磁共振;「PBST」係指含有Tween-20之磷酸鹽緩衝鹽水;「THF」係指四氫呋喃;「UVW」係指紫外波長,且「XRD」係指X射線繞射。
除非說明相反之情形,否則本文所說明之化合物係使用ACDLABS或Accelrys Draw 4.1來命名及編號。
本發明化合物可如以下方案中所圖解說明來合成,其中R1、R2、R3、R4及R5係如先前所定義。

方案1圖解說明式I化合物之一般合成。在熟知芳香族取代或偶合反應條件下,使化合物1與經適當取代之化合物2反應以提供式I化合物。更特定而言,在升高溫度下在適宜鹼(例如氫化鈉、異丙基氯化鎂、碳酸銫、碳酸鉀或第三丁醇鉀)存在下,使化合物1與化合物2反應。視情況,在適宜溶劑(例如1,4-二噁烷或第三丁基醇)中引入適宜配體劑(例如4,5-雙(二苯基膦基)-9,9-二甲基 或2-(二-第三丁基膦基)-2’,4’,6’-三異丙基-3,6-二甲氧基-1,1’-聯苯)及適宜觸媒(例如乙酸鈀(II)、參(二亞苄基丙酮)二鈀(0)或氯[2-(二-第三丁基膦基)-2',4',6'-
三異丙基-1,1'-聯苯][2-(2-胺基乙基)苯基)]鈀(II))亦可提供式I化合物。
或2-(二-第三丁基膦基)-2’,4’,6’-三異丙基-3,6-二甲氧基-1,1’-聯苯)及適宜觸媒(例如乙酸鈀(II)、參(二亞苄基丙酮)二鈀(0)或氯[2-(二-第三丁基膦基)-2',4',6'-
三異丙基-1,1'-聯苯][2-(2-胺基乙基)苯基)]鈀(II))亦可提供式I化合物。

方案2圖解說明當R5係3-甲氧基-1H-吡唑-4-基或3-環丙基-1H-吡唑-4-基時,式I化合物之合成。在如先前所述之熟知偶合反應條件下,使化合物1與化合物3(其係經諸如第三丁基氧基羰基等適宜氮保護基團適當取代之吡唑胺)反應以提供化合物4。當R5係3-甲氧基-1H-吡唑-4-基或3-環丙基-1H-吡唑-4-基時,在適宜溶劑(例如1,4-二噁烷或MeOH)中,使化合物4與適宜氮去保護劑(例如氯化氫或三氟乙酸)進一步反應以提供式I化合物。

方案3圖解說明式I化合物之替代性合成。在適宜鹼(例如氫化鈉)存在下在適宜溶劑(例如DMF)中,使化合物5與(2-溴乙氧基)-第三丁基二甲基矽烷反應以提供化合物6。然後在如先前所述之熟知偶合反應條件下,使化合物6與經適當取代之化合物2(R5-NH2)反應以提供化合物7。在適宜溶劑(例如THF及水之混合物)中,使化合物7與適宜去保護劑(例如乙酸)反應以提供化合物8。在適宜溶劑(例如DMF)中在適宜鹼(例如三乙胺)存在下,使化合物8與甲烷磺醯氯進一步反應以提供化合物9。在適宜溶劑(例如ACN)中,使化合物9與適宜胺反應以提供式I化合物。

方案4圖解說明合成化合物1之方法。在適宜溶劑(例如ACN)中,使化合物10與適宜溴化劑(例如N-溴琥珀醯亞胺)反應以提供化合物11。使化合物11與適宜鹼(例如氫化鈉或氫氧化鈉)及適宜N-烷基化劑(例如4-(2-氯乙基)嗎啉)反應以提供化合物12。在適宜溶劑(例如1,4-二噁烷)中在升高溫度下,使化合物12與雙(頻哪醇)二硼、適宜鹼(例如乙酸鉀)及適宜觸媒(例如(1,1’-雙(二苯基膦基)二茂鐵)氯化鈀(II))反應以提供化合物13。在適宜溶劑(例如1,4-二噁烷及水之混合物)中在升高溫度下,使化合物13與經適當取代之嘧啶化合物(例如2,4-二氯-5-甲基嘧啶)、適宜鹼(例如碳酸鉀)、適宜觸媒(例如四(三苯基膦)鈀)反應以提供化合物1。

方案5圖解說明合成化合物1之替代性方法。在適宜溶劑(例如ACN)中在適宜鹼(例如N,N-二異丙基乙胺)存在下,使化合物11與適宜氮保護劑(例如二碳酸二-第三丁基酯)反應以提供化合物14。在如先前所述之熟知偶合反應條件下,使化合物14與雙(頻哪醇)二硼反應以提供化合物15。在如先前所述之熟知偶合反應條件下,使化合物15與經適當取代之嘧啶化合物反應以提供化合物16。在適宜溶劑(例如
DCM或1,4-二噁烷)中,使用適宜去保護劑(例如三氟乙酸或氯化氫)對化合物16去保護以提供化合物17。在適宜溶劑(例如N-甲基吡咯啶酮)中在適宜鹼(例如氫化鈉)存在下,使化合物17與適宜烷基化劑(例如4-(2-溴乙基)嗎啉)反應以提供化合物1。

在20L 3頸燒瓶中,將3-噻吩羧酸(250g,1.95mol)於THF(9750mL)中之溶液冷卻至-70℃。向此溶液中緩慢添加正丁基鋰(2.5M於己烷中,1872mL,4.68mol),同時將溫度維持在-55℃以下。在-70℃下將反應混合物攪拌1小時。在-70℃下緩慢添加丙酮(187mL,2.55mol)。將反應混合物升溫至0℃且在0℃下攪拌3小時。在0℃下向所得溶液中添加4M HCl(1500mL)並將反應混合物升溫至室溫。將所得混合物攪拌過夜。經由矽藻土墊過濾反應混合物且用甲苯(3×500mL)洗滌墊。在減壓下濃縮濾液。在室溫下,將所得粗殘餘物溶解於甲苯(3750mL)及水(250mL)中且添加對甲苯磺酸(100.1g,0.526mol)。在100℃下將反應混合物回流16小時。將反應物冷卻至室溫且在減壓下在50℃下濃縮。將所得殘餘物溶解於水中並用EtOAc(2×10L)萃取。用飽和碳酸氫鈉水溶液及水洗滌有機層。經無水硫酸鈉乾燥有機層,過濾且在減壓下在50℃下濃縮濾液,以提供棕色黏性液體狀標題化合物200g(61%)。MS(m/z):169(M+1)。

用6,6-二甲基噻吩并[2,3-c]呋喃-4-酮(150g,0.891mol)於氫氧化銨(1000ml)中之溶液裝填5L高壓釜。在封閉環境中,使反應混合物小心地升溫至200℃之溫度且在200℃下攪拌4小時。4小時後,將反應混合物冷卻至室溫並釋放氨氣。用DCM(3×750mL)萃取反應混合物。用水(1×750mL)洗滌有機層,且經無水硫酸鈉乾燥,過濾並在減壓下在50℃下濃縮濾液,以獲得標題化合物100g(67%)。MS(m/z):168(M+1)。

用硫酸(2.5mL,45mmol)處理2-溴-3-噻吩甲酸(10.1g,49mmol)於MeOH(100mL)中之溶液。將反應物加熱至回流過夜。在減壓下濃縮混合物以去除有機溶劑且將所得混合物傾倒至冰冷水中。用EtOAc萃取冷溶液。用水、然後用飽和碳酸氫鈉水溶液洗滌合併之有機萃取物。經無水硫酸鈉乾燥有機溶液,過濾且在減壓下濃縮濾液,以獲得標題化合物10.77g(100%)。1H NMR(400.15MHz,DMSO-d 6)δ 7.65(d,J=6Hz,1H),7.34(d,J=6Hz,1H),3.78(s,3H)。

將2-溴噻吩-3-甲酸甲酯(28g,128mmol)及氰化銅(15g,167
mmol)於N-甲基吡咯啶酮(130mL)中之混合物加熱至120℃過夜。將反應物冷卻至室溫並用EtOAc稀釋。用飽和NaCl洗滌有機溶液,經無水硫酸鈉乾燥,過濾且在減壓下濃縮濾液。藉由矽膠管柱層析使用己烷中之25% EtOAc溶析來純化殘餘物,以獲得標題化合物15.2g(71%)。1H NMR(400.15MHz,DMSO-d 6)δ 8.10(d,J=5Hz,1H),7.58(d,J=5Hz,1H),3.86(s,3H)。

用乙基溴化鎂溶液(3M於二乙醚中,58mL,175mmol)處理2-氰基噻吩-3-甲酸甲酯(13.7g,79mmol)及四(異丙醇)鈦(24.8g,87.4mmol)於二乙醚(330mL)中之-70℃溶液。將反應混合物攪拌60分鐘。去除冷卻浴且經1小時將混合物至緩慢升溫至室溫。添加三氟化硼合乙醚(22.6mL,159mmol)且將混合物再攪拌1小時。用1N鹽酸(240mL)驟冷反應物且攪拌過夜。分離有機層且用額外醚反萃取水層。合併有機萃取物且用飽和碳酸氫鈉水溶液及飽和NaCl洗滌。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。藉由C18管柱上之HPLC(管柱:275g;移動相:A)水中之0.10%甲酸,B)ACN中之0.10%甲酸;梯度:5%-35% B;流速:80mL/min)純化殘餘物,以獲得標題化合物1.02g(35%)。1H NMR(400.15MHz,DMSO-d 6)δ 8.43(bs,1H),7.53(d,J=5Hz,1H),7.12(d,J=5Hz,1H),1.51(m,2H),1.38(m,2H)。

向含有6,6-二甲基-5H-噻吩并[2,3-c]吡咯-4-酮(835g,4.99mol)之20L燒瓶中添加ACN(10000mL),且將溶液冷卻至10℃。將N-溴琥珀醯亞胺(444.4g,2.49mol)分四等份添加至反應混合物中並在25℃下攪拌6小時。在減壓下濃縮反應混合物且將所得化合物於水中製成漿液並用EtOAc(3×4.1L)萃取。用水(3×4.1L)及飽和NaCl(4.1L)洗滌合併之有機萃取物,經無水硫酸鈉乾燥並過濾。將有機溶液與其他批次合併儲存。
使用與上述相同之方法,分別自650g及835g 6,6-二甲基-5H-噻吩并[2,3-c]吡咯-4-酮開始製備另外兩批次。合併來自所有三個運行之有機溶液且在減壓下在50℃下濃縮,以產生呈棕色黏性材料形式之2-溴-6,6-二甲基-5H-噻吩并[2,3-c]吡咯-4-酮。將所得產物於二乙醚/己烷(2:1 v/v)中製成漿液並過濾,以產生標題化合物1542g(45%)。MS(m/z):246/248(M+1/M+3)。
以下化合物基本上係藉由製備6之方法來製備。


經1小時,用4-嗎啉乙醇(32g,244mmol)於DCM(60mL)中之溶液逐滴處理二溴化三苯基膦(124g,293mmol)於DCM(2.44L)中之溶液,同時將反應溫度維持在25℃以下。在室溫下將混合物攪拌過夜。如上文自4-嗎啉乙醇(10g,76mmol)開始實施另一反應,適當地按比例增減試劑。合併反應混合物且藉由真空過濾收集固體,以獲得標題化合物76.7g(84%)。1H NMR(399.8MHz,DMSO-d 6)δ 4.05(m,2H),3.84(m,2H),3.78(t,J=7Hz,1H),3.67(t,J=7Hz,2H),3.56(m,2H),3.26(m,2H)。

用二碳酸二-第三丁基酯(35g,162mmol)處理2-溴-6,6-二甲基-5H-噻吩并[2,3-c]吡咯-4-酮(25g,102mmol)、4-二甲基胺基吡啶(1.25g,10mmol)及N,N-二異丙基乙胺(24mL,138mmol)於ACN(481mL)中之溶液。在室溫下將混合物攪拌過夜。在減壓下濃縮混合物。用己烷稀釋混合物,經由矽膠墊過濾混合物,且用己烷、然後用己烷中之20% DCM溶析墊。將濾液濃縮至乾燥,以獲得橙色油狀標題化合物36.5g(93%)。1H NMR(399.8MHz,CDCl3)δ 7.19(s,1H),1.74(s,6H),1.58(s,9H)。
用N,N-二異丙基乙胺(213mL,1219mmol)逐滴處理2-溴-6,6-二甲基-5H-噻吩并[2,3-c]吡咯-4-酮(200g,813mmol)、N,N-二甲基吡啶-4-胺(9.93g,81mmol)及二碳酸二-第三丁基酯(266g,1219mmol)於
ACN(2L)中之溶液。在室溫下將混合物攪拌4小時。將反應物加熱至30℃並保持2小時。將混合物冷卻至室溫且攪拌過夜。在減壓下濃縮混合物。用EtOAc稀釋混合物且用水(300mL)、然後用飽和NaCl(300mL)將所得有機溶液洗滌兩次。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。在矽膠墊上使用己烷中之0-20% EtOAc梯度溶析來純化殘餘物,以獲得標題化合物253g(90%)。MS(m/z):290/292(M-異丁烯+1/M-異丁烯+3)。1H NMR(399.8MHz,CDCl3)δ 7.19(s,1H),1.74(s,6H),1.58(s,9H)。

用氮將2-溴-6,6-二甲基-4-側氧基-噻吩并[2,3-c]吡咯-5-甲酸第三丁基酯(114g,329mmol)、雙(頻哪醇)二硼(125g,494mmol)及乙酸鉀(97g,988mmol)於1,4-二噁烷(1.6L)中之混合物脫氣10分鐘。添加(1,1’-雙(二苯基膦基)二茂鐵)氯化鈀(II)(5.38g,6.6mmol),且在90℃下將混合物加熱4小時。將反應物冷卻至室溫並經由CELITE®墊過濾。濃縮濾液,且然後用己烷中之10% EtOAc處理殘餘物。藉由真空過濾收集沈澱,以獲得標題化合物65.8g(40%)。MS(m/z):338(M-異丁烯+1)。

用氮將2-溴-6,6-二甲基-4-側氧基-噻吩并[2,3-c]吡咯-5-甲酸第三丁基酯(36g,104mmol)、雙(頻哪醇)二硼(59.8g,235mmol)及乙酸鉀(33.2g,338mmol)於1,4-二噁烷(520mL)中之混合物脫氣10分鐘。添加(1,1’-雙(二苯基膦基)二茂鐵)氯化鈀(II)(4.45g,5.5mmol)且將混合物加熱至90℃。在90℃下將混合物加熱2小時。將反應物冷卻至室溫且攪拌3小時。添加2,4-二氯嘧啶(22g,145mmol),然後添加碳酸鉀(20.4g,147mmol)於水(83mL)中之溶液。用氮將所得混合物脫氣10分鐘。添加四(三苯基膦)鈀(1.59g,1.38mmol),且將混合物加熱至90℃並保持2小時。將混合物冷卻至室溫並經由CELITE®墊過濾。用三份水及一份飽和NaCl洗滌濾液。在減壓下濃縮有機物。藉由矽膠管柱層析使用DCM中之0-25% EtOAc梯度溶析來純化殘餘物,以獲得標題化合物11g(59%)。MS(m/z):324(M-異丁烯+1)。
用氮將6,6-二甲基-4-側氧基-2-(4,4,5,5-四甲基-1,3,2-二氧雜硼烷-2-基)噻吩并[2,3-c]吡咯-5-甲酸第三丁基酯(11.7g,30mmol)、2,4-二氯嘧啶(13g,89mmol)、碳酸鉀(20.4g,147mmol)及水(50mL)於1,4-二噁烷(100mL)中之混合物脫氣10分鐘。添加四(三苯基膦)鈀(2.58g,2.2mmol),且將混合物加熱至87℃並保持1.5小時。將混合物冷卻至室溫。用EtOAc(1L)稀釋混合物且用水及飽和NaCl洗滌所得溶液。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。用己烷(200mL)中之30% EtOAc處理殘餘物且藉由真空過濾收集所得沈澱,以獲得標題化合物7.6g(67%)。MS(m/z):324(M-異丁烯+1)。

在室溫下,將2-(2-氯嘧啶-4-基)-6,6-二甲基-4-側氧基-噻吩并[2,3-c]吡咯-5-甲酸第三丁基酯(6.36g,16.7mmol)及三氟乙酸(25mL)於DCM(25mL)中之混合物攪拌2小時。在減壓下濃縮混合物且用DCM稀釋殘餘物。用飽和碳酸氫鈉水溶液分配混合物且自兩相乳液收集固體。用醚洗滌固體且在真空下在50℃下乾燥過夜,以獲得標題化合物4.65g(99%)。MS(m/z):280(M+1)。

在30℃下,將2-(2-氯嘧啶-4-基)-6,6-二甲基-4-側氧基-噻吩并[2,3-c]吡咯-5-甲酸第三丁基酯(66.7g,176mmol)及氯化氫(4M於1,4-二噁烷中,263mL,1054mmol)於1,4-二噁烷(585mL)中之溶液加熱5小時。去除加熱元件且在室溫下將混合物攪拌過夜。將己烷(800mL)緩慢添加至反應混合物中。將所得漿液攪拌10分鐘且藉由真空過濾收集固體。在真空下乾燥固體,以獲得標題化合物56g(100%)。MS(m/z):280(M+1)。
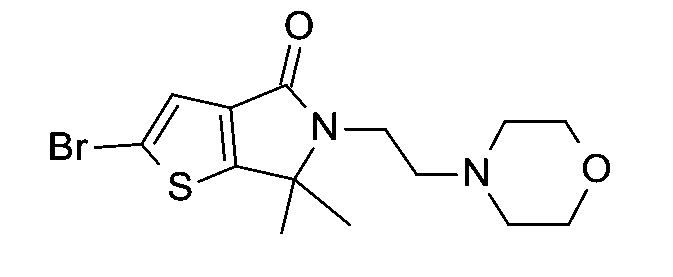
將氫氧化鈉(160g,4mol)添加至水(250mL)中且攪拌混合物直至產生澄清溶液。添加1,4-二噁烷(2L),然後添加2-溴-6,6-二甲基-5H-噻吩并[2,3-c]吡咯-4-酮(215g,874mmol)、四丁基碘化銨(300g,812mmol)及4-(2-氯乙基)嗎啉鹽酸鹽(300g,1564mmol)。在80℃下將混合物加熱1小時。將反應混合物冷卻至室溫。用水(2L)稀釋反應物且用EtOAc(3×2L)萃取混合物。經無水硫酸鈉乾燥合併之有機萃取物,過濾並在減壓下濃縮濾液。添加DCM(2L)及己烷(2L)且用飽和NaCl(2×1L)洗滌所得有機溶液。在減壓下將有機溶液濃縮至最小體積。過濾掉固體,以獲得標題化合物180g(57%)。MS(m/z):359/361(M+1/M+3)。

在0℃下,用2-溴-6,6-二甲基-5H-噻吩并[2,3-c]吡咯-4-酮(20g,81.3mmol)、然後用(2-溴乙氧基)-第三丁基二甲基矽烷(23.3g,97.5mmol)處理氫化鈉(60wt%於礦物油中,3.9g,97.5mmol)於DMF(203mL)中之懸浮液。在0℃下將反應物攪拌1小時。去除冰浴且將反應混合物攪拌過夜。用飽和氯化銨水溶液驟冷反應混合物並用EtOAc萃取。用飽和NaCl洗滌有機溶液。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。藉由矽膠管柱層析使用己烷中之20% EtOAc溶析
來純化殘餘物,以獲得標題化合物26g(79%)。MS(m/z):404/406(M+1/M+3)。
以下化合物基本上係藉由製備15之方法來製備。

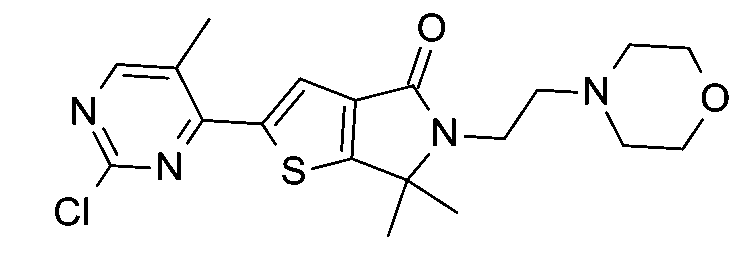
用氮將2-溴-6,6-二甲基-5-(2-嗎啉基乙基)噻吩并[2,3-c]吡咯-4-酮(5.76g,16mmol)、雙(頻哪醇)二硼(4.88g,19.2mmol)及乙酸鉀(4.86g,48mmol)於1,4-二噁烷(80mL)中之混合物脫氣20分鐘。添加(1,1’-雙(二苯基膦基)二茂鐵)氯化鈀(II)(668mg,0.80mmol)且在90℃下將混合物加熱過夜。將反應物冷卻至室溫,濃縮濾液,且然後用EtOAc處理殘餘物。藉由真空過濾收集沈澱。向固體(3.7g)中添加2,4-二氯-5-甲基嘧啶(1.5g,9.1mmol)、1,4-二噁烷(50mL)、碳酸鉀(3.8g,27mmol)及水(33mL)。用氮將所得混合物脫氣20分鐘。添加四(三苯基膦)鈀(790mg,0.68mmol)且將混合物加熱至90℃並保持2小時。將混合物冷卻至室溫並用EtOAc稀釋。用飽和NaCl洗滌有機溶液。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。藉由矽膠管柱層析使用EtOAc中之0-10% MeOH梯度溶析來純化殘餘物,以獲得標題化合物1.82g(19%)。MS(m/z):407(M+1)。
以下化合物基本上係藉由製備17之方法來製備。


用氮將2-溴-6,6-二甲基-5-(2-嗎啉基乙基)噻吩并[2,3-c]吡咯-4-酮
(200g,557mmol)、雙(頻哪醇)二硼(200g,788mmol)及乙酸鉀(200g,2038mmol)於1,4-二噁烷(1L)中之混合物脫氣15分鐘。添加(1,1’-雙(二苯基膦基)二茂鐵)氯化鈀(II)(20g,27mmol)且將混合物加熱至90℃。在90℃下將混合物加熱1小時。將反應物冷卻至50℃且添加碳酸鉀(250g,1809mmol)、2,4-二氯嘧啶(230g,1543mmol)及水(300mL)。在90℃下將混合物加熱1小時。將混合物冷卻至35℃且添加水(700mL)。用DCM(2L)萃取反應混合物。用DCM(500mL)反萃取水溶液。經無水硫酸鎂乾燥合併之有機溶液,過濾並在減壓下濃縮濾液。用己烷中之10% EtOAc(2L)稀釋殘餘物且攪拌1小時。傾析母液且用己烷(500mL)沖洗固體。將固體溶解於DCM(300mL)中且緩慢添加己烷(2L)。藉由真空過濾收集所得固體並乾燥,以獲得標題化合物150g(65%)。MS(m/z):393(M+1)。
使用冰水浴將2-(2-氯嘧啶-4-基)-6,6-二甲基-5H-噻吩并[2,3-c]吡咯-4-酮鹽酸鹽(10g,32mmol)及四丁基碘化銨(1.17g,3.16mmol)於N-甲基吡咯啶酮(211mL)中之混合物冷卻至0℃。逐份添加氫化鈉(60wt%於礦物油中,5.06g,126.5mmol)。在0℃下將混合物攪拌10分鐘,且然後添加4-(2-溴乙基)嗎啉氫溴酸鹽(13.9g,50.6mmol)。去除冰浴且將混合物攪拌4小時。用飽和氯化銨水溶液驟冷反應混合物且用水(1L)稀釋混合物。用乙酸異丙基酯(4×700mL)萃取混合物。經無水硫酸鈉乾燥合併之有機萃取物,過濾並在減壓下濃縮濾液。添加己烷中之20% EtOAc且將混合物攪拌1小時。藉由真空過濾收集固體並乾燥,以獲得標題化合物8.6g(69%)。MS(m/z):393(M+1)。
用氫化鈉(60wt%於礦物油中,129mg,3.2mmol)處理2-(2-氯嘧啶-4-基)-6,6-二甲基-5H-噻吩并[2,3-c]吡咯-4-酮(500mg,1.4mmol)
於DMF(14mL)中之溶液。將混合物攪拌10分鐘,且然後添加4-(2-溴乙基)嗎啉鹽酸鹽(412mg,1.8mmol)。在室溫下將反應混合物攪拌過夜。將混合物冷卻至0℃且添加4-(2-溴乙基)嗎啉鹽酸鹽(165mg,0.7mmol),然後添加氫化鈉(60wt%於礦物油中,14mg,0.4mmol)。去除冰浴且在室溫下將混合物攪拌過夜。添加氫化鈉(60wt%於礦物油中,14mg,0.4mmol)且在室溫下將所得混合物攪拌5小時。用水稀釋混合物並用EtOAc萃取。用5%氯化鋰水溶液洗滌有機萃取物。在減壓下濃縮有機溶液。藉由矽膠管柱層析使用DCM中之0-10% MeOH梯度溶析來純化殘餘物,以獲得標題化合物524mg(93%)。MS(m/z):393(M+1)。

用氮將5-[2-[第三丁基(二甲基)矽基]氧基乙基]-2-(2-氯嘧啶-4-基)-6,6-二甲基-噻吩并[2,3-c]吡咯-4-酮(6g,13.7mmol)、2-甲基吡唑-3-胺(1.60g,16.4mmol)、碳酸銫(8.92g,27.4mmol)、4,5-雙(二苯基膦基)-9,9-二甲基 (790mg,1.37mmol)及1,4-二噁烷(150mL)之混合物脫氣10分鐘。添加乙酸鈀(II)(610mg,2.74mmol)且在90℃下將混合物加熱2.5小時。將混合物冷卻至室溫且在室溫下將混合物攪拌過夜。用DCM中之10% MeOH稀釋反應混合物且將混合物攪拌15分鐘。經由CELITE®過濾混合物且用DCM中之10% MeOH洗滌固體。在減壓下濃縮濾液。藉由矽膠管柱層析使用DCM中之60%-100% EtOAc
梯度溶析來純化殘餘物,以獲得標題化合物5.73g(84%)。MS(m/z):499(M+1)。
(790mg,1.37mmol)及1,4-二噁烷(150mL)之混合物脫氣10分鐘。添加乙酸鈀(II)(610mg,2.74mmol)且在90℃下將混合物加熱2.5小時。將混合物冷卻至室溫且在室溫下將混合物攪拌過夜。用DCM中之10% MeOH稀釋反應混合物且將混合物攪拌15分鐘。經由CELITE®過濾混合物且用DCM中之10% MeOH洗滌固體。在減壓下濃縮濾液。藉由矽膠管柱層析使用DCM中之60%-100% EtOAc
梯度溶析來純化殘餘物,以獲得標題化合物5.73g(84%)。MS(m/z):499(M+1)。

用乙酸(120mL)及水(40mL)處理THF(40mL)中之2-溴-5-[2-[第三丁基(二甲基)矽基]氧基乙基]-6,6-二甲基-噻吩并[2,3-c]吡咯-4-酮(26g,64mmol)。在室溫下將混合物攪拌過夜。在減壓下濃縮反應混合物。用EtOAc稀釋殘餘物,且用飽和碳酸氫鈉水溶液、然後用飽和NaCl洗滌所得溶液。經無水硫酸鈉乾燥有機溶液,過濾並濃縮濾液,以獲得標題化合物18.97g(100%)。MS(m/z):290/292(M+1/M+3)。
以下化合物基本上係藉由製備26之方法來製備。


將2-溴-5-(2-羥基乙基)-6,6-二甲基-噻吩并[2,3-c]吡咯-4-酮(18.97g,65.4mmol)於DCM(300mL)中之溶液冷卻至0℃。用三乙胺(13.7mL,98.1mmol)及甲烷磺醯氯(8.24g,71.9mmol)處理混合物。在0
℃下將混合物攪拌2小時。用水及飽和NaCl洗滌溶液。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。藉由矽膠管柱層析使用EtOAc溶析來純化殘餘物,以獲得標題化合物23.8g(99%)。MS(m/z):368/370(M+1/M+3)。
以下化合物基本上係藉由製備28之方法來製備。
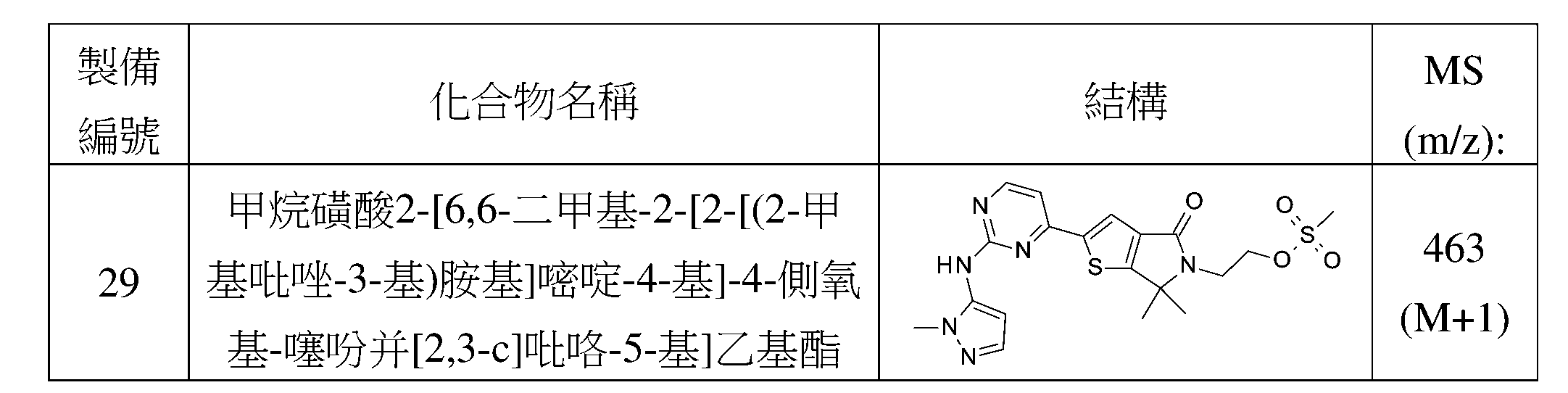

將3-乙基-1H-吡唑(1g,10.4mmol)溶解於硫酸(5mL)中且將混合物冷卻至-5℃。然後將硝酸鉀(1.16g,11.4mmol)逐份添加至混合物中。將混合物攪拌過夜,同時使其緩慢升溫至室溫。將混合物冷卻至0℃且用氫氧化銨緩慢驟冷,直至pH為約10。藉由真空過濾收集所得固體並用少量水洗滌。將濾液冷卻至0℃,且然後藉由真空過濾自濾液收集固體並用少量水洗滌。合併固體且溶解於DCM中。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。與二乙醚共蒸發一次,以獲得標題化合物1.34g(91%)。MS(m/z):140(M-1)。

用三乙胺(5.85mL,42mmol)處理5-甲氧基-4-硝基-1H-吡唑(3g,21mmol)、二碳酸二-第三丁基酯(6.9g,31.6mmol)及4-二甲基胺基吡啶(1.28g,10.5mmol)於DCM(300mL)中之懸浮液,且在室溫下將混合物攪拌過夜。在減壓下濃縮反應混合物。藉由矽膠管柱層析使用己烷中之1%-10% EtOAc梯度溶析來純化殘餘物,以獲得標題化合物3.48g(68%)。1H NMR(399.8MHz,DMSO-d 6)δ 9.10(s,1H),3.98(s,3H),1.56(s,9H)。

用3-乙基-4-硝基-1H-吡唑(300mg,2.13mmol)處理鈀(5wt%於碳上,450mg,0.21mmol)於EtOH(21mL)中之懸浮液。在氫氣氛下將所得混合物攪拌6.5小時。經由CELITE®過濾反應混合物且用額外EtOH沖洗固體。在減壓下濃縮濾液,以獲得標題化合物240g(99%)。1H NMR(400.1MHz,CD3CN)δ 7.02(s,1H),2.54(q,J=7Hz,3H),1.17(t,J=7Hz,9H)。
以下化合物基本上係藉由製備32之方法來製備。

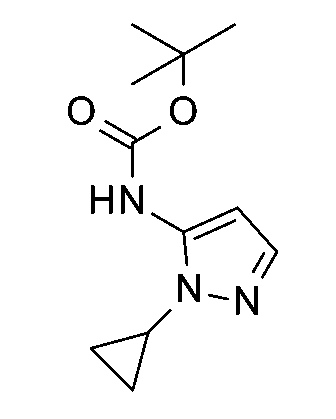
將2-環丙基吡唑-3-甲酸(4g,26mmol)於THF(35mL)中之溶液冷卻至0℃,且然後添加三乙胺(5.5mL,39mmol),然後添加二苯基磷醯疊氮化物(8.5mL,39mmol)。將混合物攪拌4小時,同時將反應溫度緩慢升溫至室溫。添加第三丁基醇(4.99mL)且在70℃下將混合物加熱18小時。在減壓下濃縮反應混合物。藉由矽膠管柱層析使用DCM中之0-20% MeOH梯度溶析來純化殘餘物,以獲得標題化合物5.39g(92%)。MS(m/z):224(M+1)。

用三氟乙酸(16mL,213mmol)處理N-(2-環丙基吡唑-3-基)胺基甲酸第三丁基酯(5.39g,24mmol)於DCM(12mL)中之溶液。在室溫下將溶液攪拌1小時。在減壓下濃縮反應混合物。將殘餘物溶解於DCM中且用飽和碳酸氫鈉水溶液處理直至水相之pH持續>7。分離各相並經無水硫酸鈉乾燥有機相。過濾混合物且在減壓下濃縮濾液。在減壓下濃縮水相。合併來自有機相及水相之殘餘物且藉由反相管柱層析(管柱:130g C18;移動相:A)水,B)ACN;梯度:0-20% B)純化。濃縮各部分且將殘餘物溶解於DCM中之25% MeOH中。過濾混合物且在減壓下濃縮濾液。將殘餘物溶解於EtOAc中且添加水。分離各層且用EtOAc(8×100mL)反萃取水層。在減壓下濃縮合併之有機萃
取物,以獲得標題化合物2.27g(76%)。1H NMR(400.1MHz,CD3CN)δ 7.02(d,J=2Hz,1H),5.33(d,J=2Hz,1H),4.28(bs,2H),3.10(m,1H),0.96(m,4H)。

用碳酸鉀(1.83g,13.2mmol)、然後用苄基溴(1.18mL,9.89mmol)處理2-(苄基胺基)乙醇(0.95mL,6.6mmol)於ACN(35mL)中之混合物。在80℃下將反應混合物加熱1.5小時。將反應物冷卻至室溫並過濾混合物。在減壓下濃縮濾液且藉由矽膠管柱層析使用己烷中之0-30% EtOAc梯度溶析來純化殘餘物,以獲得標題化合物1.7g(100%)。MS(m/z):242(M+1)。

在0℃下,用氫化鈉(60wt%於礦物油中,500mg,12.5mmol)處理2-(二苄基胺基)乙醇(1.5g,6.2mmol)及氯-2,2-二氟-乙酸鈉(950mg,6.19mmol)於THF(12mL)中之溶液。將反應混合物加熱至回流過夜。將氫化鈉(60wt%於礦物油中,120mg,3mmol)再添加至反應混合物自且繼續再加熱1小時。將反應物冷卻至室溫並用水稀釋。用二乙醚萃取混合物。分離各層且用6N鹽酸將水層調節至pH 6。用EtOAc萃取水溶液。合併所有有機溶液且經無水硫酸鈉乾燥。過濾混
合物並在減壓下濃縮濾液,以獲得標題化合物1.03g(49%)。MS(m/z):336(M+1)。

用(三甲基矽基)重氮甲烷(2M於己烷中,0.16mL,0.32mmol)逐滴處理2-[2-(二苄基胺基)乙氧基]-2,2-二氟-乙酸(100mg,0.298mmol)於甲苯(9mL)及MeOH(2mL)中之溶液。在室溫下將混合物攪拌15分鐘。用乙酸(0.1mL)驟冷反應物且在減壓下濃縮反應混合物,以獲得標題化合物102mg(98%)。MS(m/z):350(M+1)。

用EtOH(15mL)中之2-[2-(二苄基胺基)乙氧基]-2,2-二氟-乙酸甲酯(485mg,1.39mmol)處理鈀(10%於碳上,50mg,0.141mmol)於EtOH(15mL)中之懸浮液。在氫氣氛(氣球)下在室溫下將反應混合物攪拌過夜。經由CELITE®過濾反應混合物且在減壓下濃縮濾液,以獲得標題化合物294mg(93%)。MS(m/z):228(M+1)。

用硼二甲基硫複合物(2M於THF中,3.06mL,6.12mmol)處理4-
苄基-2,2-二氟-嗎啉-3-酮(290mg,1.28mmol)於THF(13mL)中之溶液。在55℃下將反應混合物加熱3.5小時,且然後去除熱並繼續攪拌過夜。將反應混合物加熱至55℃並再保持2小時。將反應混合物冷卻至室溫且藉由逐滴添加鹽酸(6N,3.06mL,18.4mmol)來驟冷。在100℃下將反應混合物加熱1小時。將混合物冷卻至室溫,並在減壓下濃縮。用水稀釋混合物且用2N氫氧化鈉將pH調節至12。用EtOAc萃取混合物。經無水硫酸鈉乾燥有機萃取物,過濾且在減壓下濃縮濾液,以獲得標題化合物120mg(44%)。MS(m/z):214(M+1)。

在80℃下,將甲烷磺酸2-(2-溴-6,6-二甲基-4-側氧基-噻吩并[2,3-c]吡咯-5-基)乙基酯(2.76g,6.9mmol)及5-氧雜-8-氮雜螺[2.6]壬烷(2.18,16.3mmol)於DMF(33mL)中之混合物加熱過夜。將混合物冷卻至室溫並用EtOAc稀釋。用飽和NaCl(3×30mL)洗滌有機溶液。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。藉由反相管柱層析(管柱:100g Gold C18;移動相:A)水中之0.1%甲酸,B)ACN中之0.1%甲酸;梯度:5% B保持5分鐘,5%-50% B經20分鐘;流速:53mL/分鐘)來純化殘餘物,以獲得標題化合物3.6g(85%)。MS(m/z):399/401(M+1/M+3)。
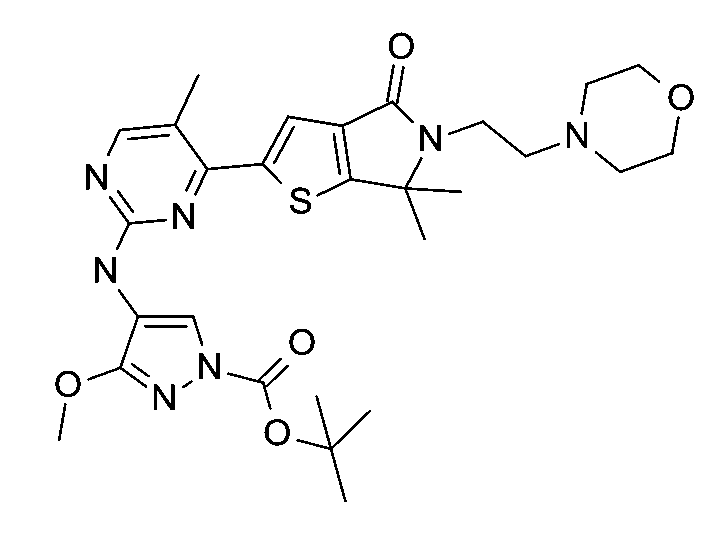
用氮將2-(2-氯-5-甲基-嘧啶-4-基)-6,6-二甲基-5-(2-嗎啉基乙基)噻吩并[2,3-c]吡咯-4-酮(250mg,0.61mmol)、4-胺基-3-甲氧基-吡唑-1-甲酸第三丁基酯(157mg,0.74mmol)、碳酸銫(300mg,0.92mmol)、4,5-雙(二苯基膦基)-9,9-二甲基 (71mg,0.12mmol)及1,4-二噁烷(6.4mL)之混合物脫氣15分鐘。添加乙酸鈀(II)(14mg,0.0614mmol)且在110℃下將混合物加熱過夜。將混合物冷卻至室溫並用EtOAc稀釋。用飽和NaCl洗滌有機溶液。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。藉由C18管柱上之HPLC(管柱:150g;移動相:A)水中之0.10%甲酸,B)ACN中之0.10%甲酸;梯度:10%-50% B;流速:60mL/min)來純化殘餘物,以獲得標題化合物101mg(28%)。MS(m/z):584(M+1)。
(71mg,0.12mmol)及1,4-二噁烷(6.4mL)之混合物脫氣15分鐘。添加乙酸鈀(II)(14mg,0.0614mmol)且在110℃下將混合物加熱過夜。將混合物冷卻至室溫並用EtOAc稀釋。用飽和NaCl洗滌有機溶液。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。藉由C18管柱上之HPLC(管柱:150g;移動相:A)水中之0.10%甲酸,B)ACN中之0.10%甲酸;梯度:10%-50% B;流速:60mL/min)來純化殘餘物,以獲得標題化合物101mg(28%)。MS(m/z):584(M+1)。
以下化合物基本上係藉由製備42之方法來製備。



將2-甲基吡唑-3-胺(75g,772mmol)緩慢添加至氫化鈉(60wt%於礦物油中,30g,750mmol)於N-甲基吡咯啶酮(500mL)中之懸浮液中。將所得混合物攪拌90分鐘。添加2-(2-氯嘧啶-4-基)-6,6-二甲基-5-(2-嗎啉基乙基)噻吩并[2,3-c]吡咯-4-酮(145g,369mmol)於N-甲基吡咯啶酮(200mL)中之溶液。將放熱反應冷卻至室溫且將反應物傾倒至水(3L)中。用濃鹽酸(200mL)將pH調節至約3。用DCM(4×2L)萃取混合物。使用5M氫氧化鈉中和水層。用DCM(2×2L)萃取此水溶液。合併該等有機萃取物且用水(2L)洗滌。經無水硫酸鈉乾燥有機物,過濾並在減壓下濃縮濾液。在矽膠塞(2kg)上相繼用DCM(2L)、DCM(2L)之2.5% EtOH、DCM(2L)中之5% EtOH、DCM(2L)中之
7.5% EtOH及最終DCM(10L)中之10% EtOH溶析來純化殘餘物。在減壓下濃縮適宜部分。添加EtOAc(1L)且在減壓下濃縮。添加EtOAc(1L)且在減壓下濃縮。添加EtOAc(500mL)及己烷(500mL)。藉由真空過濾收集固體且用己烷(500mL)洗滌固體。在真空下在50℃下乾燥固體,以獲得標題化合物65.7g(39%)。MS(m/z):454(M+1)。
用氮將2-(2-氯嘧啶-4-基)-6,6-二甲基-5-(2-嗎啉基乙基)噻吩并[2,3-c]吡咯-4-酮(20.8g,52.9mmol)、2-甲基吡唑-3-胺(5.7g,58.2mmol)、碳酸銫(37.9g,116.5mmol)、4,5-雙(二苯基膦基)-9,9-二甲基 (2.6g,4.5mmol)及1,4-二噁烷(529mL)之混合物脫氣10分鐘。添加參(二亞苄基丙酮)二鈀(0)(2.4g,2.6mmol)且將混合物加熱至85℃並保持4小時。將混合物冷卻至室溫並經由濾紙過濾混合物。在減壓下濃縮濾液。自8g2-(2-氯嘧啶-4-基)-6,6-二甲基-5-(2-嗎啉基乙基)噻吩并[2,3-c]吡咯-4-酮開始重複反應,且合併兩種殘餘物。藉由矽膠管柱層析(330g)使用(DCM中之10% EtOAc)中之5%-25% MeOH梯度溶析來純化殘餘物。彙集各部分且在減壓下濃縮。藉由矽膠管柱層析(330g)使用DCM中之10% EtOAc中之5%-25% MeOH梯度溶析再純化殘餘物。彙集各部分且在減壓下濃縮。將殘餘物溶解於DCM(400mL)中,且然後添加丙酮(1L)。在減壓下將混合物緩慢濃縮至約700mL。藉由真空過濾收集固體,以獲得標題化合物14.8g(48%)。MS(m/z):454(M+1)。
(2.6g,4.5mmol)及1,4-二噁烷(529mL)之混合物脫氣10分鐘。添加參(二亞苄基丙酮)二鈀(0)(2.4g,2.6mmol)且將混合物加熱至85℃並保持4小時。將混合物冷卻至室溫並經由濾紙過濾混合物。在減壓下濃縮濾液。自8g2-(2-氯嘧啶-4-基)-6,6-二甲基-5-(2-嗎啉基乙基)噻吩并[2,3-c]吡咯-4-酮開始重複反應,且合併兩種殘餘物。藉由矽膠管柱層析(330g)使用(DCM中之10% EtOAc)中之5%-25% MeOH梯度溶析來純化殘餘物。彙集各部分且在減壓下濃縮。藉由矽膠管柱層析(330g)使用DCM中之10% EtOAc中之5%-25% MeOH梯度溶析再純化殘餘物。彙集各部分且在減壓下濃縮。將殘餘物溶解於DCM(400mL)中,且然後添加丙酮(1L)。在減壓下將混合物緩慢濃縮至約700mL。藉由真空過濾收集固體,以獲得標題化合物14.8g(48%)。MS(m/z):454(M+1)。
用氮將2-(2-氯嘧啶-4-基)-6,6-二甲基-5-(2-嗎啉基乙基)噻吩并[2,3-c]吡咯-4-酮(250mg,0.64mmol)、2-甲基吡唑-3-胺(124mg,1.3mmol)、碳酸銫(622mg,1.9mmol)、4,5-雙(二苯基膦基)-9,9-二甲基 (55mg,0.095mmol)及1,4-二噁烷(6.4mL)之混合物脫氣15分
鐘。添加乙酸鈀(II)(14.3mg,0.0636mmol)且在90℃下將混合物加熱過夜。將混合物冷卻至室溫並經由濾紙過濾混合物。用DCM中之10% MeOH洗滌固體。在減壓下濃縮濾液。重複反應且合併兩種殘餘物。藉由C18管柱上之HPLC(30×75mm,5μm,xbridge ODB)使用水中之10mM碳酸銨(pH 10)中之9%-28% ACN 85mL/分鐘梯度溶析來純化殘餘物。彙集各部分且在減壓下濃縮以去除ACN。將水溶液凍乾,以獲得標題化合物100mg(18%)。MS(m/z):454(M+1)。
(55mg,0.095mmol)及1,4-二噁烷(6.4mL)之混合物脫氣15分
鐘。添加乙酸鈀(II)(14.3mg,0.0636mmol)且在90℃下將混合物加熱過夜。將混合物冷卻至室溫並經由濾紙過濾混合物。用DCM中之10% MeOH洗滌固體。在減壓下濃縮濾液。重複反應且合併兩種殘餘物。藉由C18管柱上之HPLC(30×75mm,5μm,xbridge ODB)使用水中之10mM碳酸銨(pH 10)中之9%-28% ACN 85mL/分鐘梯度溶析來純化殘餘物。彙集各部分且在減壓下濃縮以去除ACN。將水溶液凍乾,以獲得標題化合物100mg(18%)。MS(m/z):454(M+1)。
以下化合物基本上係藉由實例1之合成方法3來製備。




*:使用[(4,5-雙(二苯基膦基)-9,9-二甲基 )-2-(2'-胺基-1,1'-聯苯)]鈀(II)甲烷磺酸鹽作為配體。
)-2-(2'-胺基-1,1'-聯苯)]鈀(II)甲烷磺酸鹽作為配體。

在90℃下,將2-(2-氯嘧啶-4-基)-6,6-二甲基-5-(2-嗎啉基乙基)噻吩并[2,3-c]吡咯-4-酮(350mg,0.89mmol)、4-胺基-1H-吡唑-5-甲腈(116mg,1.07mmol)、碳酸鉀(320mg,2.32mmol)、2-(二-第三丁基膦基)-2’,4’,6’-三異丙基-3,6-二甲氧基-1,1’-聯苯(87mg,0.18mmol)、第三丁基醇(2.3mL)及乙酸(一滴)之溶液加熱2小時。將混合物冷卻至室溫。用DCM中之10% MeOH稀釋混合物並經由頂部含有少量矽膠之CELITE®管柱過濾溶液。用DCM中之10% MeOH洗滌管柱且在減壓下濃縮濾液。藉由反相管柱層析(管柱:C18,275g Gold;移動相:A)含有5% MeOH之水中之10mM碳酸氫銨,B)ACN;梯度:10% B保持5分鐘,梯度至40% B經25分鐘;流速:125mL/min)來純化殘餘物,以獲得標題化合物240mg(58%)。MS(m/z):465(M+1)。
以下化合物基本上係藉由實例20之方法來製備。


用氮將2-(2-氯嘧啶-4-基)-6,6-二甲基-5-(2-嗎啉基乙基)噻吩并[2,3-c]吡咯-4-酮(230mg,0.59mmol)、3-甲基-1H-吡唑-4-胺(142mg,0.73mmol)、氯[2-(二-第三丁基膦基)-2',4',6'-三異丙基-1,1'-聯苯][2-(2-胺基乙基)苯基)]鈀(II)(8mg,0.012mmol)、2-二-第三丁基膦基-2',4',6'-三異丙基聯苯(5mg,0.012mmol)及第三丁醇鈉(118mg,1.2mmol)之混合物吹掃三次。添加第三丁基醇(2mL),將反應密封且在室溫下將混合物攪拌1.5小時。用EtOAc處理反應混合物且將混合物攪拌過夜。在減壓下濃縮。將殘餘物溶解於EtOAc中且用飽和氯化銨水溶液洗滌有機溶液。用EtOAc將水層反萃取兩次。經無水硫酸鈉乾燥合併之有機萃取物,過濾並在減壓下濃縮濾液。藉由矽膠管柱層析使用DCM中之0-10% MeOH梯度溶析來純化殘餘物,以獲得標題化合物164mg(62%)。MS(m/z):454(M+1)。
以下化合物基本上係藉由實例22之方法來製備。


在60℃下,將甲烷磺酸2-[6,6-二甲基-2-[2-[(2-甲基吡唑-3-基)胺基]嘧啶-4-基]-4-側氧基-噻吩并[2,3-c]吡咯-5-基]乙基酯(260mg,0.56mmol)、硫嗎啉(116mg,1.12mmol)及三乙胺(0.18mL,0.38mmol)於ACN(2mL)中之混合物加熱過夜。將混合物冷卻至室溫並經由CELITE®過濾溶液。用DCM中之10% MeOH洗滌固體且在減壓下濃縮
濾液。藉由反相管柱層析(管柱:C18,275g Gold;移動相:A)水中之5% MeOH中之10mM氨,B)ACN;梯度:10% B保持5分鐘,梯度至10%-65% B經25min;流速:200mL/min)來純化殘餘物,以獲得標題化合物170mg(64%)。MS(m/z):470(M+1)。
以下化合物基本上係藉由實例25之方法來製備。



用EtOAc(5mL)中之4-苄基-2,2-二氟-嗎啉(95mg,0.45mmol)處理氫氧化鈀(20%於碳上,63mg,0.09mmol)於EtOAc(5mL)中之懸浮液。氫氣氛(氣球)下,在室溫下將反應混合物攪拌5小時。經由CELITE®過濾反應混合物且分離濾液。向濾液中添加三乙胺(0.11
mL,0.79mmol)及甲烷磺酸2-[6,6-二甲基-2-[2-[(2-甲基吡唑-3-基)胺基]嘧啶-4-基]-4-側氧基-噻吩并[2,3-c]吡咯-5-基]乙基酯(306mg,0.66mmol)。在80℃下將混合物加熱1小時。添加ACN(15mL)且在80℃下將混合物加熱2天。將反應混合物冷卻至室溫並在減壓下濃縮。藉由矽膠管柱層析使用EtOAc中之0-10% MeOH梯度溶析來純化殘餘物。在減壓下濃縮各部分。藉由矽膠管柱層析使用己烷中之50%-100% EtOAc梯度、然後使用EtOAc中之0-10% MeOH第二梯度溶析再純化殘餘物,以獲得標題化合物47mg(30%)。MS(m/z):490(M+1)。

用DCM(5mL)中之碳酸酯樹脂(3莫耳當量)處理6,6-二氟-1,4-氧氮雜環庚烷鹽酸鹽(200mg,0.15mmol)。使樹脂懸浮液旋轉1小時。藉由過濾去除固體且用對甲苯磺酸(250mg,1.45mmol)處理濾液。將所得混合物攪拌2小時,且然後在減壓下濃縮混合物。添加ACN(2mL)及甲烷磺酸2-[6,6-二甲基-2-[2-[(2-甲基吡唑-3-基)胺基]嘧啶-4-基]-4-側氧基-噻吩并[2,3-c]吡咯-5-基]乙基酯(200mg,0.43mmol)。用三乙胺(0.15mL,1.08mmol)處理所得溶液。密封反應容器且在80℃下將混合物加熱3小時。將反應混合物冷卻至室溫且濃縮。藉由反相管柱層析(管柱:50g C-18;移動相:A)水中之0.1% TFA,B)ACN中之0.1% TFA;梯度10%-80% B)來純化殘餘物。濃縮含有產物
之部分。將殘餘物溶解於DCM中且用飽和碳酸氫鈉水溶液洗滌。經無水硫酸鈉乾燥有機溶液,過濾並在減壓下濃縮濾液。藉由反相管柱層析(管柱:15g Gold C-18;移動相:A)含有10% MeOH之水中之10mM碳酸銨,B)ACN;梯度10%-80% B)純化殘餘物,以獲得標題化合物16mg(7%)。MS(m/z):504(M+1)。

在90℃下,將甲烷磺酸2-[6,6-二甲基-2-[2-[(2-甲基吡唑-3-基)胺基]嘧啶-4-基]-4-側氧基-噻吩并[2,3-c]吡咯-5-基]乙基酯(70mg,0.16mmol)及N-甲基環丙胺(77mg,1.08mmol)於DMF(1.7mL)中之溶液加熱過夜。將混合物冷卻至室溫。藉由反相管柱層析(管柱:100g Gold C-18;移動相:A)水中之0.1%甲酸,B)ACN中之0.1%甲酸;梯度:5% B保持5分鐘,梯度至65% B經25分鐘;流速:60mL/min)純化溶液,以獲得標題化合物70mg(37%)。MS(m/z):438(M+1)。
以下化合物基本上係藉由實例34之方法來製備。


在室溫下,將4-[[4-[6,6-二甲基-5-(2-嗎啉基乙基)-4-側氧基-噻吩并[2,3-c]吡咯-2-基]-5-甲基-嘧啶-2-基]胺基]-3-甲氧基-吡唑-1-甲酸第三丁基酯(101mg,0.173mmol)及氯化氫(4.0M於1,4-二噁烷中,2mL,8mmol)於MeOH(20mL)中之溶液攪拌12小時。將混合物濃縮至乾燥,以獲得標題化合物80mg(89%)。MS(m/z):484(M+1)。
以下化合物基本上係藉由實例36之方法來製備。

*:在反應後製備之游離鹼

藉由手性管柱層析(管柱:Lux Cellulose-4 21.2×250mm;移動相:40%異丙醇(0.2%異丙胺)/CO2);溶析時間:11分鐘)純化6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(2-氧雜-5-氮雜-二環[4.1.0]庚-5-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮(實例30),以獲得標題化合物24mg(34%)。MS(m/z):466(M+1)。
在於35kV及50mA下操作之配備有CuKa源(λ=1.54060 Å)及Vantec檢測器之Bruker D4 Endeavor X射線粉末繞射儀上獲得結晶固體之XRD圖案。在4°與40° 2θ之間掃描樣品,其中步長為0.009° 2θ且掃描速率為0.5秒/步,且使用0.6mm發散狹縫、5.28固定防散射狹縫及9.5mm檢測器狹縫。將乾燥粉末堆積於石英樣品架上並使用載玻片獲得光滑表面。在環境溫度及相對濕度下收集晶體形式繞射圖案。在晶體學領域中已眾所周知,對於任一給定晶體形式而言,繞射峰之相對強度可因由諸如晶體形態及習性等因素產生之較佳定向而變化。當存在較佳定向之影響時,峰強度會改變,但多晶型物之特徵峰位置不變。另外,在晶體學領域中亦眾所周知,對於任一給定晶體形式而言,峰角位置可略有變化。舉例而言,峰位置可因分析樣品時之溫度或濕度變化、樣品位移或存在或不存在內標而發生移位。在此情形下,±0.2 2θ之峰位置可變性將考慮到該等潛在變化,此並不妨礙所指示晶體形式之明確鑑別。晶體形式之確認可基於有區別之峰(通常
為更突出之峰,以° 2θ為單位)之任一獨特組合來實施。在8.853°及26.774° 2θ下基於NIST 675標準峰調節於環境溫度及相對濕度下收集之晶體形式繞射圖案。
6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮(結晶型1)
將6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮(103mg)添加至ACN(1mL)及水(1mL)之溶液中,且在70℃下將混合物加熱10分鐘。過濾並將溶液冷卻至室溫過夜。經5小時之時程將水(2mL)緩慢添加至溶液中。藉由真空過濾收集固體且用水洗滌。風乾固體,以獲得標題化合物94mg(92%)。
實例1結晶型1之所製備樣品之特徵在於XRD圖案(使用CuKa放射)具有如下表1中所述之繞射峰(2θ值),且具體而言具有19.3°峰與一或多個選自由以下組成之群之峰的組合:15.5°、17.1°、18.0°、20.2°、21.5°及22.1°;其中繞射角公差為0.2°。


若干證據線索指示,參與腫瘤起始,生長及進展之過程係藉由活化癌細胞中之一或多條信號傳導路徑來調介。促分裂原活化蛋白激酶(MAPK)路徑係細胞增殖及存活之關鍵調控劑。ERK係此路徑之下游成員且在傳輸來自活化受體酪胺酸激酶(RTK)(例如EGFR、FGFR、PDGFR、VEGFR等)之細胞外信號方面起關鍵作用。此路徑係由RAF、MEK及ERK(細胞外信號調控激酶)激酶組成之三層級激酶級聯,且此路徑之活化開始於小GTP酶RAS之活化。RAS之活化可招募絲胺酸/蘇胺酸激酶RAF及其活化。活化RAF隨後磷酸化並活化MEK1/2,MEK1/2進而磷酸化並活化ERK1/2。當活化時,ERK1/2使參與細胞增殖、生長、存活及EMT(上皮至間質轉變)之若干下游細胞質及核靶磷酸化。
RAS/MAPK路徑係最重要的細胞增殖路徑之一,且人們認為此路徑通常在約30%之所有人類癌症中被活化。組成型MAPK路徑活化可源自RAS、BRAF、MEK1中之活化性突變、腫瘤阻抑劑NF1之損失或由突變、擴增或配體介導之RTK活化介導之上游活化。已顯示,所有
三種RAS家族基因(KRAS、NRAS及HRAS)在若干癌症中發生體細胞突變,包括結腸直腸癌、黑色素瘤、肺癌及胰臟癌,此最通常歸因於密碼子12、13及61處之單一點突變。該等突變引起RAS之組成型活化,其伴隨有增加的ERK1/2活性及生長信號傳導。KRAS之密碼子12、13及61中之突變賦予抑制EGFR之化合物及單株抗體抗性。在30%之肺癌、90%之胰臟癌、10%之胃癌及50%之結腸直腸癌中發現KRAS突變。在約10%-25%之黑色素瘤中檢測到NRAS突變。另外,在約55%-60%之甲狀腺癌中已鑑別出RAS突變(HRAS、KRAS及NRAS)。BRAF中之體細胞點突變發生在約8%之人類腫瘤中,最通常發生在黑色素瘤(60%)、結腸直腸癌(10%)及甲狀腺癌(50%)中。在黑色素瘤中,所有BRAF突變似乎皆在激酶結構域內且單取代(T->A,V600E)佔據突變之80%。除了極少情況外,發現BRAF突變呈具有RAS突變之互斥模式,此表明該等遺傳變化活化共用下游效應物。
以下分析展示,本發明之實例性化合物係ERK1及ERK2激酶活性之抑制劑。以下分析之結果亦展示,本發明之實例性化合物抑制癌細胞中之ERK信號傳導。另外,實例1之化合物展示癌症之某些異種移植物腫瘤模型中之ERK路徑靶抑制。另外,在癌症之某些異種移植物腫瘤模型中,實例1之化合物抑制腫瘤生長。
此分析之目的係量測化合物抑制ERK1激酶活性之能力。使用TR-FRET分析來實施活體外ERK1激酶分析。藉由在384孔PROXIPLATETM(Perkin Elmer,編號GRN6260)中添加以下物質來起始反應(12.5μL):5μL ERK1酶(Invitrogen,編號PR5254B,最終濃度100ng/mL)加受質GFP-ATF2(Invitrogen,編號PV4445,最終濃度0.2μM)、5μL在激酶緩衝液(50mM Hepes pH 7.4、5mM MgCl2、0.1mM
EGTA、0.01% Triton X-100、1mM DTT)中製備之ATP溶液(Invitrogen,編號PV3227,最終濃度10μM)及2.5μL於DMSO溶液中之測試化合物(最終4%,v/v)。在室溫下將反應混合物培育60分鐘。藉由添加12.5μL於TR-FRET稀釋緩衝液(Invitrogen,編號PV3574)中之終止緩衝液(10mM EDTA、2nM Tb-抗pATF2(pThr71)抗體,Invitrogen,編號PV4448)來終止反應。在室溫下將板再培育60分鐘,且在ENVISION®(PerkinElmer)板讀取器上在激發波長340nm下讀取。藉由用GFP受體發射信號(在520nm下)除以Tb供體發射信號(在495nm下)來計算TR-FRET比率。使用化合物處理之孔相對於板上Max(DMSO對照)及Min(未添加酶)對照孔TR-FRET比率數據來計算抑制%{抑制%=100-[(測試化合物-中值Min)/(中值Max-中值Min)×100]}。使用1:3稀釋方案在10種濃度(20μM至0.001μM)下測試所有化合物。藉由使用ACTIVITYBASE® 7.3(ID Business Solutions Limited)將抑制%及10點濃度數據擬合至4參數非線性推理方程式(方程式205)導出Abs_IC50值。
在實質上如上文所述之此分析中測試在本發明範疇內之實例性化合物。此分析之結果展示,所有實例性化合物皆抑制ERK1激酶活性,且IC50值小於0.15μM。舉例而言,實例1之化合物之IC50值為4.86nM(±0.20,n=7)。
此分析之目的係量測化合物抑制ERK2激酶活性之能力。使用TR-FRET分析來實施活體外ERK2激酶分析。藉由在384孔PROXIPLATETM(Perkin Elmer,編號GRN6260)中添加以下物質來起始所有反應(12.5μL):5μL ERK2酶(Invitrogen,編號PV3595B,最終濃度50ng/mL)加受質GFP-ATF2(Invitrogen,編號PV4445,最終濃度0.2μM)、5μL在激酶緩衝液(50mM Hepes pH 7.4、5mM MgCl2、0.1
mM EGTA、0.01% Triton X-100、1mM DTT)中製備之ATP溶液(Invitrogen,編號PV3227,最終濃度10μM)及2.5μL於DMSO溶液中之測試化合物(最終4%,v/v)。在室溫下將反應物培育60分鐘。藉由添加12.5μL於TR-FRET稀釋緩衝液(Invitrogen,編號PV3574)中之終止緩衝液(10mM EDTA、2nM Tb-抗pATF2(pThr71)抗體,Invitrogen,編號PV4448)來終止反應。在室溫下將板再培育60分鐘,且在ENVISION®(PerkinElmer)板讀取器上在340nm之激發波長下讀取。藉由用GFP受體發射信號(在520nm下)除以Tb供體發射信號(在495nm下)來計算TR-FRET比率。使用化合物孔相對於板上Max(DMSO對照)及Min(未添加酶)對照孔TR-FRET比率數據來計算抑制%{抑制%=100-[(測試化合物-中值Min)/(中值Max-中值Min)×100]}。使用1:3稀釋方案在10種濃度(20μM至0.001μM)下測試所有化合物。藉由使用ACTIVITYBASE 7.3(ID Business Solutions Limited)將抑制%及10點濃度數據擬合至4參數非線性推理方程式(方程式205)導出Abs_IC50值。
在實質上如上文所述之此分析中測試在本發明範疇內之實例性化合物。此分析之結果展示,所有實例性化合物皆抑制ERK2激酶活性,且IC50值小於0.15μM。舉例而言,實例1之化合物之IC50值為5.24nM(±0.24,n=7)。
此分析之目的係量測化合物在活體外抑制癌細胞中之ERK信號傳導之能力。使用HCT116結腸直腸癌細胞系(ATCC,編號CCL-247)來實施pRSK1 Alphascreen分析。在T-150燒瓶中之含有5%胎牛血清(FBS)(Gibco,編號16000-044)之達爾伯克氏改良伊格爾培養基(Dulbecco's Modified Eagle's Medium,DMEM)(Hyclone,編號SH30022)生長培養基中以常規方式培養HCT116細胞,且在5% CO2培
育器在37℃下培育。當細胞鋪滿時收穫細胞並以1×10e7個細胞/mL冷凍於冷凍培養基中作為「分析備用冷凍細胞」且儲存在液氮中。為運行分析,將40,000個HCT116細胞/孔平鋪於96孔組織培養板中,且在37℃下在5% CO2培育器中培育過夜。以20μM最高濃度開始在10種濃度下測試化合物,且利用1:3稀釋方案(20μM至0.001μM),最終DMSO濃度為0.5%(v/v)。添加20μL無血清生長培養基中之化合物且在37℃下培育2小時。去除生長培養基,且將50μL含有1×halt蛋白酶及磷酸鹽抑制劑混合劑[Thermo,編號78441]之1×溶解緩衝液[Cell Signaling Technology,編號9803]添加至每孔中,並在室溫下在振盪器上培育10分鐘。將來自每孔之4μL細胞溶解物轉移至384孔分析板[Perkin Elmer,編號6006280]中之各別孔中,且添加5μL反應混合物[2000份1×分析緩衝液(Perkin Elmer,編號A1000)、1份生物素-RSK1抗體(Santa Cruz,編號sc-231-B-G)、4份pRSK1抗體(Abcam,編號ab32413)、35份受體珠粒(Perkin Elmer,編號6760617R)]。用箔板密封件(Beckman Coulter,編號538619)密封板並在室溫下培育2小時。將2μL供體珠粒[20份1×分析緩衝液、1份供體珠粒]添加至每孔中,且用透明板密封件(Applied Biosystems,編號4311971)密封板,並在室溫下在黑暗中培育2小時。藉由在ENVISION®(PerkinElmer)板讀取器中讀取板量測每孔之螢光強度。藉由使用ACTIVITYBASE® 7.3(ID Business Solutions Limited)將pRSK1抑制%[抑制%=100-[(測試化合物-中值Min)/(中值Max-中值Min)×100]及10點濃度數據擬合至4參數非線性推理方程式(Abase方程式205)導出Rel IC50值。
在實質上如上文所述之此分析中測試在本發明範疇內之實例性化合物。此分析之結果展示,所有實例性化合物皆抑制腫瘤細胞中之ERK受質(RSK)磷酸化,且IC50值小於3μM。舉例而言,實例1之化合物之IC50值為0.429μM(±0.173,n=8)。
此分析之目的係量測在動物模型中測試化合物抑制ERK1/2受質磷酸化之能力。向來自Harlan Laboratories之雌性無胸腺裸小鼠(22-25g)之在200μL 1:1漢克氏平衡鹽溶液(HBSS)+基質膠溶液中之右側腹區域中皮下植入5×10e6個HCT116結腸直腸癌細胞(ATCC,編號CCL-247)。自植入後第7天開始每週兩次量測腫瘤生長及體重。當腫瘤大小達到300-500mm3時,將動物隨機化且分成多個5動物組。向動物經口投用適宜劑量之於化合物特定媒劑中之化合物或單獨媒劑(媒劑:1% HEC/0.25% Tween 80/0.05%消泡劑),且在投用後以期望時間間隔收集腫瘤及血液。利用異氟烷麻醉加頸椎脫位殺死動物。快速冷凍腫瘤且儲存在-80℃下直至處理用於藉助ELISA分析來測定pRSK1含量。於EDTA管中收集血液且使血漿旋轉沈澱並冷凍於-80℃下96孔板中。使用標準方法測定化合物暴露。
在液氮中粉碎腫瘤,且在冷室(4℃)中在FastPrep-24TM細胞破碎機(MP Biomedical)中使用Matrix D珠粒(MP Biomedical,編號6913-500),使其溶解於含有1×halt蛋白酶及磷酸酶抑制劑混合劑(Thermo Scientific,編號0861281)、1mM苯基甲烷磺醯氟(PMSF)(Sigma,編號93482-50ML-F)及1μM偏釩酸鈉(Sigma,編號590088)之1×溶解緩衝液(MSD,編號R60TX-3)中。在4℃下在14000rpm下旋轉20分鐘後,將腫瘤溶解物轉移至新管中。使用Pierce BCA蛋白質分析套組(目錄編號23225,Thermo Scientific)測定腫瘤或細胞溶解物之蛋白質濃度。此套組含有三種主要組份-(1)BCA試劑A,其含有0.1M氫氧化鈉中之碳酸鈉、碳酸氫鈉、二辛可寧酸及酒石酸鈉,(2)BCA試劑B,其含有4%硫酸銅,及(3)白蛋白標準安瓿,其含有0.9%鹽水及0.05%疊氮化鈉中之2mg/mL。在96孔板中,在25μL一式兩份孔中針對20-2000μg/mL之濃度範圍添加牛血清白蛋白蛋白質標準物以產生
標準曲線。將於25μL 1×PBS中稀釋之細胞或腫瘤溶解物添加至一式兩份測試孔中。藉由將2%試劑B添加至試劑A中來製備工作BCA試劑(2mL B+98mL A),充分混合且將200μL添加至每一樣品或標準物中。充分混合,覆蓋板,且在37℃下培育30分鐘。將板冷卻至室溫且在板讀取器(來自Perkin Elmer之Envision板讀取器)上量測562nm處或其附近之吸光度。自所有其他個別標準物及未知(細胞或腫瘤溶解物)樣品複本之562nm量測減去空白標準複本之平均562nm吸光度量測。藉由繪製每一牛血清白蛋白標準物之平均空白校正之562nm量測對其濃度(μg/mL)來製備標準曲線。在Microsoft Excel中使用曲線擬合對數使用該標準曲線來確定每一未知樣品之蛋白質濃度。將剩餘腫瘤溶解物冷凍於-80℃下。藉由夾心ELISA使用冷凍-解凍一次之腫瘤溶解物來量測pRSK1表現。
在4℃下使用40ng RSK1山羊抗體(Santa Cruz,編號sc-231-G)將96孔板(Thermo,編號15042)包覆過夜,並在室溫下培育1小時且然後在4℃下過夜。用300μL PBST(含有0.05% Tween-20之1×磷酸鹽緩衝鹽水(PBS))將板洗滌三次,用100μL/孔封阻緩衝液(Thermo Scientific,編號37532)封阻且在室溫下培育2小時。用300μL PBST將板洗滌三次且將20μg腫瘤溶解物轉移至每孔中並在4℃下培育過夜。用300μL PBST將板洗滌三次,且在室溫下與100μL pRSK1(T359/S363)兔抗體(以1:1000稀釋於封阻緩衝液中)一起培育4小時。用300μL PBST將板洗滌三次,且與100μL HRP偶聯之抗兔二級抗體(GE Healthcare UK,編號NA934V,以1:10000稀釋於封阻緩衝液中)一起培育。在室溫下培育1小時。用300μL PBST將板洗滌三次,添加100μL SUPERSIGNAL® ELISA Femto最高靈敏度受質(Thermo,編號37075),且在振盪器上培育1分鐘。使用ENVISION®板讀取器測定發光信號。藉由將經單獨媒劑處理之動物之腫瘤溶解物視為100%來確
定每一腫瘤溶解物中之pRSK1含量。以一式兩份分析每一樣品且使用平均數進行計算。使用Excel及XL擬合來計算TED50。
在實質上如上文所述之此分析中測試在本發明範疇內之化合物。此分析之結果展示,在腫瘤異種移植物模型中實例1之化合物抑制RSK1磷酸化。舉例而言,實例1之化合物之TED50值為16mg/kg。
此分析之目的係量測因應測試化合物投與之腫瘤體積減小。在培養物中擴增人類結腸直腸癌細胞HCT116(ATCC,編號CCL-247),收穫且將200μL 1:1 HBSS+基質膠溶液中之5×10e6個細胞皮下注射至雌性無胸腺裸小鼠(22-25g,Harlan Laboratories)之右後側腹上。在培養物中擴增人類胰臟癌細胞MIA PACA-2(ATCC,編號CRL-1420)或人類非小細胞肺癌細胞CALU-6(ATCC,編號HTB-56)或人類結腸直腸癌細胞COLO-205(ATCC,編號CCL-222),收穫且將200μL 1:1 HBSS+基質膠溶液中之5×10e6個細胞皮下注射至雌性無胸腺裸小鼠(22-25g,Harlan Laboratories)之右後側腹上。自植入後第7天開始每週兩次量測腫瘤生長及體重。當腫瘤大小達到200-400mm3時,將動物隨機化且分成多個8至10動物組。製備適宜媒劑(媒劑:1% HEC/0.25% Tween 80/0.05%消泡劑)中之測試化合物且藉由經口胃管灌食投與達14至21天。藉由在療程期間每週一次實施之腫瘤體積量測來測定腫瘤反應。將體重視為毒性之一般量度。
在實質上如上文運行之此分析中測試在本發明範疇內之化合物。發現實例1之化合物具有如下表2中所提供之δ T/C%值。該等結果指示,在若干人類癌症異種移植物模型(包括HCT116、MIA PACA-2、CALU-6及COLO-205)中,實例1之化合物展示顯著抗腫瘤活性。

對腫瘤體積之分析係基於Log 10及SpatialPower共變數結構。
*:顯著(p<0.05)
NA:不適用
當治療組之終點腫瘤體積等於或大於基線腫瘤體積時,計算δ T/C%。該式係100*(T-T0)/(C-C0),其中T及C分別係治療組或對照組之平均終點腫瘤體積。T0及C0係彼等組之平均基線腫瘤體積。
當終點體積小於基線時,計算消退%。該式係100*(T-T0)/T0。其中T0係治療組之平均基線腫瘤體積。
對於HCT116、MIA PACA-2及CALU-6模型,分別使用第10天、第20天及第15天之所有組自基線之總平均值(隨機化)來計算T/C之變化%。
因腫瘤異質性所致,組合療法在某些類型之癌症治療中已成為有效療法所必需或用於克服所獲得之抗性。假設靶向療法之組合具有更有效地減緩或甚至終止癌症之潛能。在此背景下,測試實例1化合物與以下各項之組合之腫瘤生長抑制:泛RAF抑制劑化合物(參見WO 2013/134243,1-(3,3-二甲基丁基)-3-(2-氟-4-甲基-5-(7-甲基-2-(甲基胺基)吡啶并[2,3-d]嘧啶-6-基)苯基)尿素,下文稱為「泛RAF抑制劑化合物」)、CDK4/6抑制劑化合物(參見WO 2010/075074,[5-(4-乙基-六氫吡嗪-1-基甲基)-吡啶-2-基]-[5-氟-4-(7-氟-3-異丙基-2-甲基-3H-苯并咪唑-5-基)-嘧啶-2-基]-胺或其醫藥上可接受之鹽),下文稱為「CDK4/6抑制劑化合物」)或DC101(例如,參見Witte L.等人,Cancer Metastasis Rev.,17,155-161,1998,可在實驗中用作小鼠中抗VEGFR2 Ab之替代之針對小鼠VEGFR2之大鼠單株抗體,較佳雷莫蘆單抗(參見WO 2003/075840,亦稱為Cyramza®、IMC-1121b,CAS登記號947687-13-0))。更特定而言,在KRAS突變體結腸直腸癌異種移植物模型HCT116中測試實例1之化合物與泛RAF抑制劑化合物或CDK4/6抑制劑化合物之組合。同樣,在KRAS突變體非小細胞肺(NSCLC)異種移植物模型NCI-H441、A549及NCI-H2122中測試實例1之化合物與CDK4/6抑制劑化合物或DC101之組合。
在無胸腺裸大鼠中進行HCT116組合效能研究。在培養物中擴增人類結腸直腸癌細胞HCT116(ATCC,編號CCL-247),收穫且將200μL 1:1 HBSS+基質膠溶液中之5×10e6個細胞皮下注射至雌性NIH裸大鼠(120-145 gm,Taconic Farms)之右後側腹上。自植入後第7天開始每週兩次量測腫瘤生長及體重。當平均腫瘤大小達到200-300mm3時,將動物隨機化且分成多個5至7動物組。製備適宜媒劑(參見下文)中之測試化合物且藉由經口胃管灌食投與達21至28天。藉由在
療程期間每週一次實施之腫瘤體積量測來測定腫瘤反應。此研究中所用之媒劑係1% HEC(羥乙基纖維素)/0.25% Tween® 80/0.05%消泡劑。將實例1之化合物及泛RAF抑制劑化合物調配於1% HEC/0.25% Tween® 80/0.05%消泡劑中。將CDK4/6抑制劑化合物調配於25mM磷酸鈉緩衝液(pH 2)中之1% HEC中。以10mpk及20mpk QD投與實例1之化合物分別產生52%之單一藥劑活性及64%腫瘤生長抑制(表3)。相比之下,以10mpk及20mpk BID投與泛RAF抑制劑化合物分別產生29%及68%之單一藥劑活性。所有治療皆相對於媒劑對照在統計學上顯著(p<0.05),但10mpk BID之泛RAF抑制劑化合物除外。投與10mpk QD之實例1化合物與10mpk BID之泛RAF抑制劑化合物之組合產生94%腫瘤生長抑制(p<0.001),且組合結果係「協同」,如藉由Bliss獨立性方法所計算(表3)。此組合似乎可耐受,此乃因不存在顯著體重損失。投與10mpk QD之實例1化合物與20mpk BID之泛RAF抑制劑化合物之組合亦產生顯著(p<0.05)腫瘤生長抑制(95%),且組合結果係「加和的」,如藉由Bliss獨立性方法所計算(表3)。此組合似乎可耐受,此乃因不存在顯著體重損失。在相同研究中,投與10mpk QD之實例1化合物與20mpk QD之CDK4/6抑制劑化合物產生98%腫瘤生長抑制,而實例1化合物及CDK4/6抑制劑化合物之單一藥劑效能分別係52%及76%腫瘤生長抑制。該兩種藥劑之組合顯示統計學上顯著之「加和」結果,如藉由Bliss獨立性方法所計算(表3)。此組合似乎可耐受,此乃因不存在顯著體重損失。該等結果表明,實例1化合物與泛RAF抑制劑化合物或CDK4/6抑制劑化合物之組合可向患有KRAS突變體結腸直腸癌之患者提供更大益處。

對腫瘤體積之分析係基於Log 10及SpatialPower共變數結構。
*:顯著(p<0.05)
**藉由Bliss獨立性方法來測定兩種藥劑之組合之統計效應:首先,將重複量測模型擬合至log腫瘤體積對組、時間及組×時間。然後,使用對比陳述來測試每一時間點處之相互作用效應。根據腫瘤體積量表將該組合之預期加和反應(EAR)計算為EAR體積=V1 * V2/V0,其中V0、V1及V2分別係所估計之媒劑對照、單獨治療1及單獨治療2之平均腫瘤體積。若相互作用測試係顯著的(p<0.05),則在所測試劑量及時間表下,若所觀察到之組合體積小於EAR體積則宣佈組合效應為協同的,若所觀察到之組合體積大於EAR體積則宣佈組合效應為拮抗的,或否則係加和的。
NA:不適用
當治療組之終點腫瘤體積等於或大於基線腫瘤體積時,計算δ T/C%。該式係100*(T-T0)/(C-C0),其中T及C分別係治療組或對照組之平均終點腫瘤體積。T0及C0係彼等組之平均基線腫瘤體積。
在所有研究中持續28天投藥。
使用第11天之所有組自基線之總平均值(隨機化)來計算T/C之變化%。
亦在三種KRAS突變體NSCLC異種移植物模型(包括SCID小鼠中之A549(KRAS_G12S)以及無胸腺裸小鼠中之NCI-H441(KRAS_G12V)及NCI-H2122(KRAS_G12C))中測試組合效能。在培養物中擴增人類非小細胞肺癌細胞NCI-H441(ATCC,編號CRL-5807)及NCI-H2122(ATCC,編號CRL-5985),收穫且將200μL 1:1 HBSS+基質膠溶液中之5×10e6個細胞皮下注射至雌性無胸腺裸小鼠(20-22gm,Harlan Laboratories)之右後側腹上。在培養物中擴增人類非小細胞肺癌細胞A549(ATCC,編號CC1-185),收穫且將200μL 1:1 HBSS+基質膠溶液中之5×10e6個細胞皮下注射至雌性CB-17 SCID小鼠(18-20gm,Taconic Farms)之右後側腹上。對於所有細胞系,自植入後第7天開始每週兩次量測腫瘤生長及體重。當平均腫瘤大小達到200-300mm3時,將動物隨機化且分成多個5至7動物組。製備適宜媒劑(參見下文)中之測試化合物且藉由經口胃管灌食(實例1化合物及CDK4/6抑制劑化合物)或腹膜內(DC101)投與達21至28天。藉由在療程期間每週一次實施之腫瘤體積量測來測定腫瘤反應。該等研究中所用之媒劑係1% HEC/0.25% Tween® 80/0.05%消泡劑。將實例1之化合物調配於1% HEC/0.25% Tween® 80/0.05%消泡劑中,且將CDK4/6抑制劑化合物調配於25mM磷酸鈉緩衝液(pH 2)中之1% HEC中。以50mpk投與呈單一藥劑之實例1化合物在NCI-H2122及A549腫瘤中分別產生41%及91%腫瘤生長抑制;且在NCI-H441腫瘤中產生101%腫瘤生長抑制(即,
1%腫瘤消退)。以50mpk投與呈單一藥劑之CDK4/6抑制劑化合物在NCI-H2122、A549及NCI-H441模型中分別產生53%、51%及81%腫瘤生長抑制。同樣,實例1化合物(50mpk)與CDK4/6抑制劑化合物(50mpk)之組合在A549及NCI-H441腫瘤中分別產生151%(即51%消退)及147%(即47%消退)腫瘤生長抑制;且在NCI-H2122腫瘤中產生82%腫瘤生長抑制。所有三種腫瘤模型中之組合結果係「加和的」,如藉由Bliss獨立性方法所計算(表4)。一般而言,在該等研究中所有治療似乎可耐受,如藉由無顯著體重損失所指示。在相同NCI-H441異種移植物模型中,磷酸鹽緩衝液中之DC101亦作為單一藥劑或與實例1化合物組合經腹膜內以50mpk QD每週兩次(BIW)投與達28天。以20mpk BIW投與DC101產生102%腫瘤生長抑制(即2%消退)之單一藥劑活性。50mpk QD之實例1化合物與20mpk BIW之DC101之組合產生146%腫瘤生長抑制(即46%消退)。此組合結果係「加和的」,如藉由Bliss獨立性方法所計算(表4)。此組合似乎可耐受,此乃因不存在顯著體重損失。該等結果表明,實例1化合物與CDK4/6抑制劑化合物或抗VEGFR2抗體之組合可向具有KRAS突變之非小細胞肺癌患者提供更大益處。


+
腫瘤體積之分析係基於Log 10轉變及具有特殊檢定力共變數結構之重複量測ANOVA
*:顯著(p<0.05)
**藉由Bliss獨立性方法來測定兩種藥劑之組合之統計效應:首先,將重複量測模型擬合至log腫瘤體積對組、時間及組×時間。然後,使用對比陳述來測試每一時間點處之相互作用效應。根據腫瘤體積量表將該組合之預期加和反應(EAR)計算為EAR體積=V1 * V2/V0,其中V0、V1及V2分別係所估計之媒劑對照、單獨治療1及單獨治療2之平均腫瘤體積。若相互作用測試係顯著的(p<0.05),則在所測試劑量及時間表下,若所觀察到之組合體積小於EAR體積則宣佈組合效應為協同的,若所觀察到之組合體積大於EAR體積則宣佈組合效應為拮抗的,或否則係加和的。
NA:不適用
當治療組之終點腫瘤體積等於或大於基線腫瘤體積時,計算δ
T/C%。該式係100*(T-T0)/(C-C0),其中T及C分別係治療組或對照組之平均終點腫瘤體積。T0及C0係彼等組之平均基線腫瘤體積。
使用第11天(HCT116)、第25天(NCI-H441)、第22天(A549)、第18天(NCI-H2122)之基線(隨機化)來計算T/C之變化%。
在所有研究中持續28天投藥。
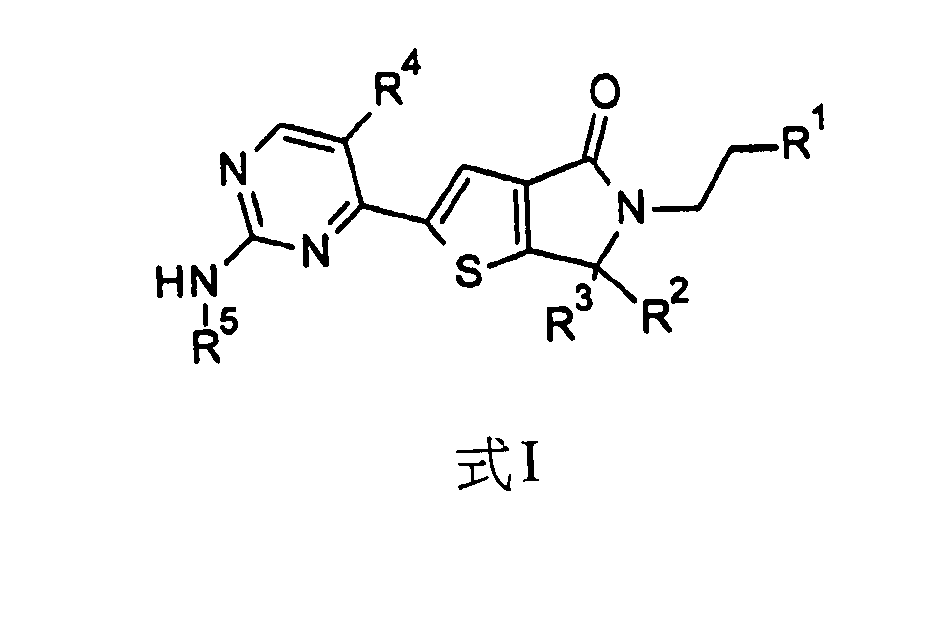
Claims (25)
- 一種下式之化合物或其醫藥上可接受之鹽



- 如請求項1之化合物或其醫藥上可接受之鹽,其中R2及R3獨立地係甲基。
- 如請求項2之化合物或其醫藥上可接受之鹽,其中R4係氫。
- 如請求項3之化合物或其醫藥上可接受之鹽,其中R1係

- 如請求項3之化合物或其醫藥上可接受之鹽,其中R5係
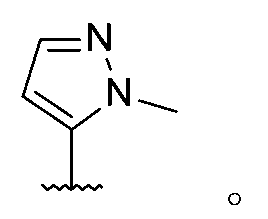
- 如請求項4之化合物或其醫藥上可接受之鹽,其中該化合物係6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮。
- 如請求項6之化合物或其醫藥上可接受之鹽,其係6,6-二甲基-2-{2-[(1-甲基-1H-吡唑-5-基)胺基]嘧啶-4-基}-5-[2-(嗎啉-4-基)乙基]-5,6-二氫-4H-噻吩并[2,3-c]吡咯-4-酮。
- 一種用於治療癌症之醫藥組合物,其包含如請求項1至7中任一項之化合物或其醫藥上可接受之鹽,及醫藥上可接受之載劑、稀釋劑或賦形劑。
- 一種如請求項1至7中任一項之化合物或其醫藥上可接受之鹽之用途,其用於製造抑制細胞外信號調控激酶(extracellular-signal-regulated kinase;ERK)活性之藥劑。
- 一種如請求項1至7中任一項之化合物或其醫藥上可接受之鹽之用途,其用於製造用來治療癌症之藥劑。
- 如請求項10之用途,其中該癌症選自由以下組成之群:黑色素瘤、結腸直腸癌、胰臟癌及非小細胞肺癌。
- 如請求項11之用途,其中該癌症係結腸直腸癌。
- 如請求項11之用途,其中該癌症係胰臟癌。
- 如請求項11之用途,其中該癌症係非小細胞肺癌。
- 一種包含(a)如請求項6之化合物或其醫藥上可接受之鹽及(b)雷莫蘆單抗(ramucirumab)之組合之用途,其用於製造治療非小細胞肺癌之藥劑,其中(a)及(b)係用於同時、分別或依序投與。
- 一種如請求項6之化合物或其醫藥上可接受之鹽之用途,其用於製造治療非小細胞肺癌之藥劑,其中該藥劑係用於與雷莫蘆單抗同時、分別或依序投與。
- 一種雷莫蘆單抗之用途,其用於製造治療非小細胞肺癌之藥劑,其中該藥劑係用於與如請求項6之化合物或其醫藥上可接受之鹽同時、分別或依序投與。
- 一種包含(a)如請求項6之化合物或其醫藥上可接受之鹽及(b)[5-(4-乙基-六氫吡嗪-1-基甲基)-吡啶-2-基]-[5-氟-4-(7-氟-3-異丙基-2-甲基-3H-苯并咪唑-5-基)-嘧啶-2-基]-胺或其醫藥上可接受之鹽之組合之用途,其用於製造治療非小細胞肺癌之藥劑,其中(a)及(b)係用於同時、分別或依序投與。
- 一種如請求項6之化合物或其醫藥上可接受之鹽之用途,其用於製造治療非小細胞肺癌之藥劑,其中該藥劑係用於與[5-(4-乙基-六氫吡嗪-1-基甲基)-吡啶-2-基]-[5-氟-4-(7-氟-3-異丙基-2-甲基-3H-苯并咪唑-5-基)-嘧啶-2-基]-胺或其醫藥上可接受之鹽同時、分別或依序投與。
- 一種[5-(4-乙基-六氫吡嗪-1-基甲基)-吡啶-2-基]-[5-氟-4-(7-氟-3-異丙基-2-甲基-3H-苯并咪唑-5-基)-嘧啶-2-基]-胺或其醫藥上可接受之鹽之用途,其用於製造治療非小細胞肺癌之藥劑,其中該藥劑係用於與如請求項6之化合物或其醫藥上可接受之鹽同時、分別或依序投與。
- 如請求項15至20中任一項之用途,其中該非小細胞肺癌為KRAS突變非小細胞肺癌。
- 一種包含(a)如請求項6之化合物或其醫藥上可接受之鹽及(b)[5-(4-乙基-六氫吡嗪-1-基甲基)-吡啶-2-基]-[5-氟-4-(7-氟-3-異丙基-2-甲基-3H-苯并咪唑-5-基)-嘧啶-2-基]-胺或其醫藥上可接受之鹽之組合之用途,其用於製造治療結腸直腸癌之藥劑,其中(a)及(b)係用於同時、分別或依序投與。
- 一種如請求項6之化合物或其醫藥上可接受之鹽之用途,其用於製造治療結腸直腸癌之藥劑,其中該藥劑係用於與[5-(4-乙基-六氫吡嗪-1-基甲基)-吡啶-2-基]-[5-氟-4-(7-氟-3-異丙基-2-甲基-3H-苯并咪唑-5-基)-嘧啶-2-基]-胺或其醫藥上可接受之鹽同時、分別或依序投與。
- 一種[5-(4-乙基-六氫吡嗪-1-基甲基)-吡啶-2-基]-[5-氟-4-(7-氟-3-異丙基-2-甲基-3H-苯并咪唑-5-基)-嘧啶-2-基]-胺或其醫藥上可接受之鹽之用途,其用於製造治療結腸直腸癌之藥劑,其中該藥劑係用於與如請求項6之化合物或其醫藥上可接受之鹽同時、分別或依序投與。
- 如請求項22至24中任一項之用途,其中該結腸直腸癌為KRAS突變結腸直腸癌。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462095185P | 2014-12-22 | 2014-12-22 | |
| US62/095,185 | 2014-12-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW201632530A TW201632530A (zh) | 2016-09-16 |
| TWI704151B true TWI704151B (zh) | 2020-09-11 |
Family
ID=55135525
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW104141368A TWI704151B (zh) | 2014-12-22 | 2015-12-09 | Erk抑制劑 |
Country Status (44)
Families Citing this family (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3363800A1 (en) | 2013-10-03 | 2018-08-22 | Kura Oncology, Inc. | Heterocyclic inhibitors of erk and methods of use |
| ES2700200T3 (es) | 2014-12-22 | 2019-02-14 | Lilly Co Eli | Inhibidores de ERK |
| TWI704151B (zh) * | 2014-12-22 | 2020-09-11 | 美商美國禮來大藥廠 | Erk抑制劑 |
| SG10201912959QA (en) | 2015-10-21 | 2020-02-27 | Otsuka Pharma Co Ltd | Benzolactam compounds as protein kinase inhibitors |
| US20200306254A1 (en) * | 2016-06-29 | 2020-10-01 | Eli Lilly And Company | Combination of erk1/2 inhibitor compound with gemcitabine or with gemcitabine and nab-paclitaxel for use in treatment of pancreatic cancer |
| WO2018081204A1 (en) * | 2016-10-26 | 2018-05-03 | Li George Y | DEUTERATED N-(5-((4-ETHYLPIPERAZIN-1-YL)METHYL) PYRIDIN-2-YL)-5-FLUORO-4-(4-FLUORO-1-ISOPROPYL-2-METHYL-1H-BENZO[d]IMIDAZOL-6-YL)PYRIMIDIN-2-AMINE |
| GB201706327D0 (en) | 2017-04-20 | 2017-06-07 | Otsuka Pharma Co Ltd | A pharmaceutical compound |
| US11691970B2 (en) | 2017-11-24 | 2023-07-04 | Janssen Pharmaceutica Nv | Pyrazolopyridinone compounds |
| CN112218634A (zh) * | 2018-04-09 | 2021-01-12 | G1治疗公司 | 具有驱动致癌突变的癌症的治疗 |
| KR102839394B1 (ko) * | 2018-05-22 | 2025-07-29 | 제이에스 이노메드 홀딩스 엘티디. | 키나아제 억제제로서 헤테로시클릭 화합물, 헤테로시클릭 화합물을 포함하는 조성물 및 그 사용 방법 |
| KR20210107069A (ko) | 2018-12-21 | 2021-08-31 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 컨쥬게이트와 키나아제 저해제의 조합 |
| WO2020197917A1 (en) | 2019-03-27 | 2020-10-01 | Eli Lilly And Company | Salts of 5,6-dihydro-4h-thieno[2,3-c]pyrrol-4-one compound as erk inhibitors |
| WO2020192750A1 (zh) * | 2019-03-28 | 2020-10-01 | 江苏恒瑞医药股份有限公司 | 噻吩并杂环类衍生物、其制备方法及其在医药上的应用 |
| TW202102505A (zh) * | 2019-03-29 | 2021-01-16 | 大陸商江蘇恆瑞醫藥股份有限公司 | 吡咯并雜環類衍生物、其製備方法及其在醫藥上的應用 |
| US20220168250A1 (en) * | 2019-05-16 | 2022-06-02 | Eli Lilly And Company | TRIPLE COMBINATION OF AN ERK1/2 INHIBITOR WITH A BRAF INHIBITOR AND AN EGFR INHIBITOR FOR USE IN THE TREATMENT OF BRAFv6ooE COLORECTAL CANCER |
| CN112457326B (zh) * | 2019-09-06 | 2022-02-15 | 上海凌达生物医药有限公司 | 一类芳香杂环并内酰胺类化合物、制备方法和用途 |
| PL4071152T3 (pl) | 2019-12-06 | 2025-08-11 | D3 Bio (Wuxi) Co., Ltd. | Związek spiro służący jako inhibitor erk i jego zastosowanie |
| CN110950876B (zh) * | 2019-12-10 | 2021-08-17 | 上海凌达生物医药有限公司 | 一类呋喃并内酰胺类化合物、制备方法和用途 |
| WO2021216777A1 (en) * | 2020-04-21 | 2021-10-28 | The Trustees Of The Stevens Institute Of Technology | Erk inhibitors for cancer therapy |
| CN114315837B (zh) * | 2020-09-29 | 2023-06-16 | 江苏恒瑞医药股份有限公司 | 一种erk抑制剂的结晶形式及其制备方法 |
| TW202221001A (zh) * | 2020-09-29 | 2022-06-01 | 大陸商江蘇恒瑞醫藥股份有限公司 | 吡咯並雜環類衍生物的晶型及其製備方法 |
| MX2024000084A (es) * | 2021-06-28 | 2024-01-18 | D3 Bio Wuxi Co Ltd | Compuesto de tiazololactama dimetil sustituido y uso de este. |
| US20240309018A1 (en) * | 2021-06-28 | 2024-09-19 | D3 Bio (Wuxi) Co., Ltd | Thiazole-lactam-spiroheterocyclic compounds and applications thereof |
| US20250144094A1 (en) * | 2021-11-15 | 2025-05-08 | Erasca, Inc. | Thiophene ulk1/2 inhibitors and their use thereof |
| WO2023137297A2 (en) * | 2022-01-11 | 2023-07-20 | Suvalent Therapeutics, Inc. | 5,6-dihydro-4h-thieno[2,3-c]pyrrole sumo inhibitors and uses thereof |
| US20240352029A1 (en) * | 2023-04-14 | 2024-10-24 | Prelude Therapeutics Incorporated | CDK Inhibitors And Their Use As Pharmaceuticals |
| TW202508595A (zh) | 2023-05-04 | 2025-03-01 | 美商銳新醫藥公司 | 用於ras相關疾病或病症之組合療法 |
| US20250049810A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025080946A2 (en) | 2023-10-12 | 2025-04-17 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025240847A1 (en) | 2024-05-17 | 2025-11-20 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025255438A1 (en) | 2024-06-07 | 2025-12-11 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025265060A1 (en) | 2024-06-21 | 2025-12-26 | Revolution Medicines, Inc. | Therapeutic compositions and methods for managing treatment-related effects |
| WO2026006747A1 (en) | 2024-06-28 | 2026-01-02 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026015790A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015825A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Use of ras inhibitor for treating pancreatic cancer |
| WO2026015796A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015801A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030064982A1 (en) * | 2000-09-15 | 2003-04-03 | Robert Davies | Pyrazole compounds useful as protein kinase inhibitors |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4023048A1 (de) * | 1990-07-20 | 1992-01-23 | Basf Ag | Dicarbonsaeureimide, verfahren zu ihrer herstellung und ihre verwendung als herbizide |
| ES2242771T5 (es) * | 2000-09-15 | 2011-10-14 | Vertex Pharmaceuticals Incorporated | Compuestos de pirazol útiles como inhibidores de proteína quinasas. |
| EP1916001B1 (en) | 2002-03-04 | 2011-05-25 | Imclone LLC | Human antibodies specific to KDR and uses thereof |
| EP1562938B1 (en) * | 2002-11-04 | 2007-08-29 | Vertex Pharmaceuticals Incorporated | Heteroaryl-pyrimidine derivatives as jak inhibitors |
| UA80571C2 (en) | 2002-11-22 | 2007-10-10 | Lilly Co Eli | Quinolinyl-pyrrolopyrazoles |
| US7803806B2 (en) * | 2005-11-03 | 2010-09-28 | Sgx Pharmaceuticals, Inc. | Pyrimidinyl-thiophene kinase modulators |
| US8697694B2 (en) | 2008-08-20 | 2014-04-15 | Merck Sharp & Dohme Corp. | Substituted pyridine and pyrimidine derivatives and their use in treating viral infections |
| CN102245600A (zh) * | 2008-10-08 | 2011-11-16 | 百时美施贵宝公司 | 吡咯烷酮黑色素浓集激素受体-1拮抗剂 |
| PA8852901A1 (es) * | 2008-12-22 | 2010-07-27 | Lilly Co Eli | Inhibidores de proteina cinasa |
| CA2809662C (en) * | 2010-09-01 | 2019-04-16 | Gilead Connecticut, Inc. | Pyridazinones, method of making, and method of use thereof |
| JO3148B1 (ar) | 2011-07-27 | 2017-09-20 | Lilly Co Eli | مركب مثبط لإشارات مسار notch |
| EP3321262B1 (en) | 2012-03-01 | 2021-01-13 | Array Biopharma, Inc. | Serine/threonine kinase inhibitors |
| AR090151A1 (es) | 2012-03-07 | 2014-10-22 | Lilly Co Eli | Compuestos inhibidores de raf |
| KR20150060687A (ko) * | 2012-09-28 | 2015-06-03 | 베링거 인겔하임 인터내셔날 게엠베하 | 이중 안지오포이에틴-2/Dll4 결합제 및 항-VEGF-R 제제를 포함하는 약제학적 조합물 |
| ES2700200T3 (es) * | 2014-12-22 | 2019-02-14 | Lilly Co Eli | Inhibidores de ERK |
| TWI704151B (zh) * | 2014-12-22 | 2020-09-11 | 美商美國禮來大藥廠 | Erk抑制劑 |
-
2015
- 2015-12-09 TW TW104141368A patent/TWI704151B/zh not_active IP Right Cessation
- 2015-12-10 JO JOP/2015/0310A patent/JO3596B1/ar active
- 2015-12-11 AR ARP150104045A patent/AR102977A1/es unknown
- 2015-12-16 ES ES15823869T patent/ES2738406T3/es active Active
- 2015-12-16 PT PT15823869T patent/PT3237423T/pt unknown
- 2015-12-16 HU HUE15823869A patent/HUE045933T2/hu unknown
- 2015-12-16 UA UAA201706046A patent/UA119686C2/uk unknown
- 2015-12-16 CN CN201580070315.6A patent/CN107108648B/zh not_active Expired - Fee Related
- 2015-12-16 MY MYPI2017702263A patent/MY178426A/en unknown
- 2015-12-16 MX MX2017008242A patent/MX371206B/es active IP Right Grant
- 2015-12-16 DK DK15823869.1T patent/DK3237423T3/da active
- 2015-12-16 PE PE2017001109A patent/PE20171043A1/es unknown
- 2015-12-16 CR CR20170182A patent/CR20170182A/es unknown
- 2015-12-16 CA CA2966559A patent/CA2966559C/en active Active
- 2015-12-16 LT LTEP15823869.1T patent/LT3237423T/lt unknown
- 2015-12-16 WO PCT/US2015/065940 patent/WO2016106029A1/en not_active Ceased
- 2015-12-16 EP EP15823869.1A patent/EP3237423B1/en active Active
- 2015-12-16 PL PL15823869T patent/PL3237423T3/pl unknown
- 2015-12-16 SG SG11201704521SA patent/SG11201704521SA/en unknown
- 2015-12-16 SI SI201530773T patent/SI3237423T1/sl unknown
- 2015-12-16 ME MEP-2019-201A patent/ME03490B/me unknown
- 2015-12-16 KR KR1020177016748A patent/KR101917972B1/ko not_active Expired - Fee Related
- 2015-12-16 JP JP2017533381A patent/JP6445701B2/ja not_active Expired - Fee Related
- 2015-12-16 MA MA41251A patent/MA41251B1/fr unknown
- 2015-12-16 RS RS20190906A patent/RS59015B1/sr unknown
- 2015-12-16 HR HRP20191268 patent/HRP20191268T1/hr unknown
- 2015-12-16 TR TR2019/09887T patent/TR201909887T4/tr unknown
- 2015-12-16 NZ NZ731531A patent/NZ731531A/en not_active IP Right Cessation
- 2015-12-16 EA EA201791133A patent/EA031659B1/ru not_active IP Right Cessation
- 2015-12-16 MD MDE20170231 patent/MD3237423T2/ro not_active IP Right Cessation
- 2015-12-16 TN TN2017000233A patent/TN2017000233A1/en unknown
- 2015-12-16 US US14/970,588 patent/US9469652B2/en not_active Ceased
- 2015-12-16 AU AU2015369983A patent/AU2015369983C1/en not_active Ceased
- 2015-12-16 BR BR112017011130A patent/BR112017011130A2/pt active Search and Examination
-
2016
- 2016-09-08 US US15/259,381 patent/US9526733B1/en active Active
-
2017
- 2017-04-20 ZA ZA2017/02786A patent/ZA201702786B/en unknown
- 2017-05-01 IL IL252065A patent/IL252065B/en active IP Right Grant
- 2017-05-15 SV SV2017005443A patent/SV2017005443A/es unknown
- 2017-05-16 DO DO2017000122A patent/DOP2017000122A/es unknown
- 2017-05-25 CO CONC2017/0005217A patent/CO2017005217A2/es unknown
- 2017-06-13 CL CL2017001514A patent/CL2017001514A1/es unknown
- 2017-06-19 GT GT201700134A patent/GT201700134A/es unknown
- 2017-06-20 PH PH12017501155A patent/PH12017501155B1/en unknown
- 2017-06-22 EC ECIEPI201738500A patent/ECSP17038500A/es unknown
-
2019
- 2019-05-22 US US16/419,141 patent/USRE48635E1/en active Active
- 2019-07-23 CY CY20191100777T patent/CY1121831T1/el unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030064982A1 (en) * | 2000-09-15 | 2003-04-03 | Robert Davies | Pyrazole compounds useful as protein kinase inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI704151B (zh) | Erk抑制劑 | |
| CN112805281B (zh) | Kras g12c抑制剂 | |
| CA2453881C (en) | 4-amino-6-phenyl-pyrrolo[2,3-d]pyrimidine derivatives | |
| JP6490215B2 (ja) | サイクリン依存性キナーゼ(cdk)阻害剤としての2−h−インダゾール誘導体およびその治療上の使用 | |
| JP6661048B2 (ja) | Cdk阻害剤としての2,4−二置換ピリミジン誘導体およびその薬学的に許容される塩、並びに、医薬品組成物 | |
| CN111601804B (zh) | 含氮杂芳类衍生物调节剂、其制备方法和应用 | |
| JP2023106404A (ja) | Tam阻害剤としてのピロロトリアジン化合物 | |
| CN105934432A (zh) | 2,4-二取代苯-1,5-二胺衍生物及其应用以及由其制备的药物组合物和药用组合物 | |
| CN107074874A (zh) | Erk抑制剂 | |
| KR20200115583A (ko) | Cdk4 및 cdk6 억제제로서의 2h-인다졸 유도체 및 그의 치료 용도 | |
| CN114763360A (zh) | 手性大环化合物作为蛋白激酶抑制剂及其用途 | |
| US7323469B2 (en) | 7H-pyrrolo[2,3-d]pyrimidine derivatives | |
| WO2020200154A1 (zh) | 一类噻吩并氮杂环类化合物、制备方法和用途 | |
| JP2014214138A (ja) | ヒドロキシル基を有するピペラジン誘導体及びその塩 | |
| HK1237340B (zh) | 作為erk抑制劑的噻吩並[2,3-c]吡咯-4-酮衍生物 | |
| HK1237340A1 (zh) | 作為erk抑制劑的噻吩並[2,3-c]吡咯-4-酮衍生物 | |
| OA19873A (en) | 2H-indazole derivatives as CDK4 and CDK6 inhibitors and therapeutic uses thereof. | |
| CN110642838A (zh) | Egfr抑制剂及其制备和应用 | |
| BR112018069612B1 (pt) | Compostos de pirrolotriazina, uso dos mesmos, composição farmacêutica que os compreende e métodos para inibir uma quinase tam, e quinase axl e mer in vitro |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | Annulment or lapse of patent due to non-payment of fees |