化合物
在一個方面,本公開提供式(I)化合物:
或其藥學上可接受的鹽,
其中,
X
1
、X
2
和X
3
各自獨立地為C或N,其限制條件為X
1
、X
2
和X
3
中的至少一個為N且X
1
、X
2
和X
3
中的至少一個為C;
短劃線“
”意指X
1
與X
2
之間的鍵及X
2
與X
3
之間的鍵可以是單鍵或雙鍵,其限制條件是X
1
與X
2
之間的鍵及X
2
與X
3
之間的鍵中的至少一個是單鍵;
R
1
不存在,為鹵素或C
1-6
烷基,其中所述C
1-6
烷基可以任選地經羥基、鹵素或氘單取代或獨立多取代;
各R
2
、R
3
和R
4
獨立地選自不存在、鹵素、羥基、氰基、C
1-6
烷基、C
1-6
烷氧基、-(CH
2
)
n
-Q,其可以任選地經以下各者單取代或獨立多取代:氘、羥基、氨基、氰基、鹵素、C
1-6
烷基、C
1-6
鹵烷基、(C≡N)-C
1-6
烷基、C
1-6
烷氧基、C
1-6
鹵烷氧基、C
3-8
環烷基、C
3-8
環烷氧基、3員至8員芳香基或3員至8員雜環基,
其中n為0、1或2,Q為3員至8員飽和或不飽和碳環基,或3員至8員飽和或不飽和雜環基;
環A為5員至12員芳香基、具有1到5個選自氧、硫和氮的環雜原子的5員至12員雜芳香基、具有0到5個選自氧、硫和氮的環雜原子的8員至10員雙環,其中環A不為苯基。
在一些實施例中,R
2
選自甲基、乙基、丙基、丁基、環丙基、環丁基、氧雜環丁烷基、環戊烷基、四氫呋喃基、環己烷基、四氫吡喃基、環庚烷基、呱啶基、苯基、吡啶基、吡啶酮基、氧雜環辛烷基、四氫吡喃基、二氫吡喃基、螺[3.3]庚烷基、螺[2.5]辛烷基、雙環[1.1.1]戊烷基、雙環[3.2.1]辛烷基、8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經以下各者單取代或獨立多取代:羥基、氰基、鹵素、C
1-6
烷基、C
1-6
鹵烷基、C
1-6
烷氧基、C
1-6
鹵烷氧基、C
3-8
環烷基、C
3-8
環烷氧基、3員至8員芳香基或3員至8員雜環基,其可以進一步任選地經以下各者單取代或獨立多取代:鹵素、氘、羥基、氨基、氰基、C
1-6
烷基、C
1-6
鹵烷基、C
1-6
烷氧基或C
1-6
鹵烷氧基。
在一些實施例中,R
2
選自:
,
其可以任選地經羥基、氰基、氟、氯、溴、甲基、乙基、甲氧基、二氟甲基、二氟甲氧基或三氟甲氧基單取代或獨立多取代。
在一些實施例中,R
2
為環己烷基或四氫吡喃基,其可以任選地經鹵素、C
1-6
烷基或C
1-6
烷氧基單取代或獨立多取代。
在一些實施例中,R
1
為甲基、乙基、正丙基、異丙基、正丁基、仲丁基、叔丁基或異丁基,其可以任選地經羥基、鹵素或氘單取代或獨立多取代。
在一些實施例中,R
1
為甲基、乙基、三氟甲基或三氘甲基。
在一些實施例中,環A為具有1個環雜原子氮的6員雜芳香基、具有2到3個選自氧、硫和氮的環雜原子的9員雙環,任選地,所述9員雙環為苯基或吡啶基稠合雙環,任選地,環A選自
。
在一些實施例中,每個R
3
和R
4
獨立地選自不存在、鹵素、羥基、氰基、C
1-6
烷基、CN-C
1-6
烷基、C
1-6
鹵烷基、C
1-6
烷氧基、C
1-6
鹵烷氧基、3員至8員飽和或不飽和雜環基,其中所述雜環基可以任選地進一步經C
1-3
烷基單取代或獨立多取代。
在一些實施例中,環A為
或
,每個R
3
和R
4
獨立地選自不存在、甲基、氰基、甲氧基、氯、氰基-甲基、吡唑基、惡唑基,其中所述吡唑基或惡唑基可以任選地進一步經C
1-3
烷基單取代或獨立多取代。
在一些實施例中,本文所提供的化合物具有式Ia的結構
和其藥學上可接受的鹽,其中R
1
、R
2
、R
3
、R
4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Ib的結構
和其藥學上可接受的鹽,其中R
1
、R
2
、R
3
、R
4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Ic的結構
和其藥學上可接受的鹽,其中R
1
、R
2
、R
3
、R
4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Id的結構
和其藥學上可接受的鹽,其中R
1
、R
2
、R
3
、R
4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Ie的結構
和其藥學上可接受的鹽,
其中,
X
1
和X
3
中的一個是N且另一個是C,短劃線“
”意指X
1
與N之間的鍵及N與X
3
之間的鍵可以是單鍵或雙鍵,其限制條件是X
1
與N之間的鍵和N與X
3
之間的鍵中的至少一個是單鍵;
R
1
為C
1-3
烷基,
R
2
為環戊基、環己烷基、四氫吡喃基或8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經鹵素或C
1-3
烷氧基單取代或獨立多取代,
Y
1
、Y
2
和Y
3
各自獨立地為C或N,其限制條件為Y
1
、Y
2
和Y
3
中的至少一個為N;
R
5
為鹵素或C
1-3
烷基,
R
6
為C
1-3
烷基。
在一些實施例中,式Ie的R
2
為未經取代的環戊基、環己烷基、四氫吡喃基或8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經鹵素或C
1-3
烷氧基單取代或獨立多取代。
在一些實施例中,式Ie的Y
3
為N且Y
1
和Y
2
的至少一個為N。
在一些實施例中,式Ie的R
5
為甲基。
式(I)的示範性化合物1到149闡述於下表1中。
應瞭解,為清楚起見而在單獨實施例的上下文中描述的本公開的某些特徵也可組合提供于單一實施例中。相反地,為簡潔起見描述於單個實施例的上下文中的本公開的各種特徵還可以分開地或以任何合適的子組合形式提供。
在本公開中的多處,描述了連接取代基。在結構明確需要鍵聯基團的情況下,關於所述基團所列的馬庫什變數(markush variable)應理解為鍵聯基團。例如,如果結構需要鍵聯基團並且所述變數的馬庫什群組定義列舉“烷基”,那麼應理解,所述“烷基”表示鍵聯亞烷基。
如本文所使用,當提到化學基團時,術語“經取代”意指所述化學基團有一個或多個氫原子被移除且經取代基置換。如本文所使用,術語“取代基”具有本領域中已知的普通含義並且是指共價連接至母基團或在適當時稠合至母基團的化學部分。如本文所用,術語“任選地經取代”或“任選地……經取代”意指化學基團可不具有取代基(即未經取代)或可具有一個或多個取代基(即經取代)。應理解,在給定原子處的取代受價數限制。
如本文所使用,術語“C
i-j
”指示碳原子數的範圍,其中i和j為整數,並且碳原子數的範圍包含端點(即i和j)和介於其間的每個整數點,並且其中j大於i。例如,C
1-6
指示一至六個碳原子的範圍,包含一個碳原子、兩個碳原子、三個碳原子、四個碳原子、五個碳原子及六個碳原子。在一些實施例中,術語“C
1-12
”指示1到12個,包含1到10個、1到8個、1到6個、1到5個、1到4個、1到3個或1到2個碳原子。
如本文所用,術語“烷基”無論作為另一術語的一部分或獨立地使用,都是指飽和或不飽和烴鏈,而後者可以進一步細分成具有至少一個雙鍵或三鍵的烴鏈(烯基或炔基)。在一些實施例中,烷基是指飽和烴鏈。以上提到的烴鏈可以是直鏈或分支鏈。術語“C
i-j
烷基”是指具有i至j個碳原子的烷基。飽和烷基的實例包含但不限於甲基、乙基、正丙基、異丙基、正丁基、叔丁基、異丁基、仲丁基;高級同系物,如2-甲基-1-丁基、正戊基、3-戊基、正己基、1,2,2-三甲基丁基等。不飽和烷基的實例包含但不限於乙烯基、正丙烯基、異丙烯基、正丁烯基、仲丁烯基、乙炔基、丙炔-1-基、丙炔-2-基等。“C
1-6
烷基”的實例包含但不限於甲基、乙基、丙基、異丙基、正丁基、異丁基及叔丁基。“C
1-3
烷基”的實例包含但不限於甲基、乙基、丙基及異丙基。
當“烷基”表示鍵聯亞烷基時,亞烷基的實例包含但不限於亞甲基、1,1-亞乙基、1,2-亞乙基、1,1-亞丙基、1,2-亞丙基、1,3-亞丙基、2,2-亞丙基、叔亞丁基等。
如本文所使用,術語“氨基”是指式“-NH
2
”的基團。
如本文所用,術語“氨甲醯基”是指氨基羰基(即NH
2
-C(=O)-)。
如本文所使用,術語“氰基”是指式“-C≡N”的基團。
如本文所使用,術語“鹵基”和“鹵素”是指氟基、氯基、溴基或碘基。
如本文所使用,術語“羥基”是指式“-OH”的基團。
如本文所用,術語“烷氧基”無論是作為另一術語的一部分或獨立地使用,都是指式-O-烷基的基團。
術語“C
i-j
烷氧基”意指烷氧基的烷基部分具有i至j個碳原子。烷氧基的實例包含但不限於甲氧基、乙氧基、丙氧基(例如正丙氧基和異丙氧基)、叔丁氧基等。“C
1-12
烷氧基”的實例為甲氧基、乙氧基和丙氧基。
如本文所用,術語“羥基C
1-12
烷基”是指式“-C
1-12
烷基-OH”的基團,其中所述基團的烷基部分具有1至12個碳原子,並且一個或多個羥基可鍵聯到烷基部分中的任何碳原子。在一些實施例中,“C
i-j
烷基-OH”具有一個羥基。“C
1-12
烷基-OH”的實例為羥甲基、1-羥乙基、2-羥乙基和1-羥異丙基。
如本文所用,術語“C
i-j
鹵烷基”是指經鹵素取代的(單取代或多取代的)C
i-j
烷基。“C
1-12
鹵烷基”的實例為氟甲基、二氟甲基、三氟甲基、氟乙基、二氟乙基、三氟乙基、氯乙基和溴異丙基。“二氟乙基”的實例為1,1-二氟乙基。“三氟乙基”的實例為2,2,2-三氟乙基和1,2,2-三氟乙基。
“C
i-j
鹵烷氧基”的實例為氟甲氧基、二氟甲氧基或三氟甲氧基。“三氟乙氧基”的實例為2,2,2-三氟乙氧基和1,2,2-三氟乙氧基。
如本文所使用,術語“芳香基”或“芳香族S”不管是作為另一術語的一部分還是獨立地使用,都是指在形成環的原子之間具有交替的雙鍵和單鍵的環系統。在本公開中,術語“芳香基”或“芳香族”也意圖包含假芳香族。術語“偽芳香族”是指並非絕對芳香族的環系統,但其借助於電子的離域而穩定且表現方式與芳環類似。芳香基或芳香族基團可具有單環或多環。芳基的實例包含但不限於苯基、萘基、四氫萘基、茚滿基等。
如本文所使用,如本文所使用的術語“雜芳基”是指含有至少一個選自O、S、N、P等的成環雜原子的芳基。雜芳基包含但不限於呋喃基、噻吩基、吡啶基(pyridinyl)、三嗪基、吡啶基(pyridyl)、吡咯基、惡唑基、噻唑基、咪唑基、吡唑基、異惡唑基、異噻唑基、吲哚嗪基、吲哚基、異吲哚基、吲哚啉基、1,2,3-惡二唑基、1,2,4-惡二唑基、1,2,4-惡二唑-5-酮、1,2,3-三唑基、1,3,4-噻二唑基、噠嗪基、嘧啶基、吡嗪基、喹唑啉基、異喹唑啉基,1,3,5-三嗪基、1H噻吩並[2,3-c]吡唑基、噻吩並[2,3-b]呋喃基、3H-吲哚基、苯並[b]呋喃基、苯並[b]噻吩基、1H-吲唑基、苯並咪唑基、四唑基、尿苷基和胞嘧啶基。
如本文所用,術語“碳環基”無論作為另一術語的一部分或獨立地使用,都是指任何環,包含單環或多環(例如具有2或3個稠環、橋環或螺環),其中所有環原子為碳且含有至少三個成環碳原子。如本文所用,術語“螺環”是指兩個環通過一個單一共同原子連接的環系統;術語“稠環”是指兩個環共用兩個相鄰原子的環系統;並且術語“橋環”是指兩個環共用三個或更多個原子的環系統。
在一些實施例中,碳環基可以含有3至12個成環碳原子(即3員至12員碳原子)、3至10個成環碳原子、3至9個成環碳原子或3至8個成環碳原子。碳環基可為飽和、部分不飽和或完全不飽和的。在一些實施例中,碳環基可以是飽和環狀烷基。在一些實施例中,碳環基可為在環系統中含有至少一個雙鍵的不飽和環狀烷基。在一些實施例中,不飽和碳環基可以含有一個或多個芳香環。在一些實施例中,飽和或不飽和碳環基的一個或多個成環-CH
2
-基團可經-C(O)-基團置換。
在一些實施例中,碳環基為單環烷基。在一些實施例中,碳環基為飽和單環烷基。飽和單環烷基的實例包含但不限於環丙基、環丁基、環戊基、環己基、環庚基、環辛基、環戊烯基、環己烯基等。
3員至8員“飽和或不飽和碳環基”為分別具有3至8個、3至6個或5至8個成環碳原子的飽和、部分不飽和或完全不飽和單環或多環環系統,其中一個或多個成環-CH
2
-基團可以任選地經-C(O)-基團置換。
“3員至8員飽和或不飽和碳環基”的實例為C
3-6
環烷基、環己基、環己烯基、環戊基、苯基、萘基和雙環[1.1.1]戊-1-基。“C
3-8
環烷基”的實例為環丙基、環丁基、環戊基、環己基、環庚基和環辛基。術語“C
3-8
環烷氧基”是指式“C
3-8
環烷基-O-”的基團。
如本文所用,術語“雜環基”是指碳環基,其中一個或多個(例如1、2或3個)環原子經雜原子置換,所述雜原子包含但不限於O、S、N、P等。在一些實施例中,雜環基是飽和雜環基。在一些實施例中,雜環基為在環系統中具有一個或多個雙鍵的不飽和雜環基。在一些實施例中,雜環基為部分不飽和雜環基。在一些實施例中,雜環基為完全不飽和雜環基。在一些實施例中,不飽和雜環基可含有一個或多個芳環。在一些實施例中,雜環基的一個或多個成環-CH
2
-基團可以任選地經
-C(O)- 、 -S- 、 -S(O)-
或
-S(O)2
- 基團
置換。在其中雜環基在其環系統中含有硫的一些實施例中,所述成環硫原子可任選地氧化以形成S-氧化物。在一些實施例中,雜環基經由其成環碳連接到化合物的其它部分。在一些實施例中,雜環基經由其成環氮連接到化合物的其它部分。
在一些實施例中,3員至8員飽和或不飽和單環或多環雜環基,其具有選自N、O或S的1、2或3個雜原子。
3員至8員“飽和或不飽和雜環基”為分別具有3到8個成環原子的飽和、部分不飽和或完全不飽和單環或多環(例如具有2或3個稠環、橋環或螺環),其中至少一個成環原子選自氮、硫或氧,除非另外規定,否則其可經由其成環碳或氮鍵聯到化合物的其它部分,其中飽和或不飽和雜環基的一個或多個成環-CH
2
-基團可經-C(O)-、-S-、-S(O)-或-S(O)
2
-基團置換,並且其中當雜環基在其環系統中含有硫時,所述環硫原子可以任選地氧化以形成S-氧化物。
示範性單環雜環基包含但不限於氧雜環丁烷基、1,1-二氧硫雜環丁烷基吡咯烷基、四氫呋喃基、四氫噻吩基、吡咯基、呋喃基、噻吩基、吡唑基、咪唑基、三唑基、惡唑基、噻唑基、呱啶基、呱啶基、呱嗪基、嗎啉基、吡啶基、吡嗪基、嘧啶基、噠嗪基、三嗪基、吡啶酮基、嘧啶酮基、吡嗪酮基、嘧啶酮基、噠嗪酮基、三嗪酮基等。
螺雜環基的實例包含但不限於螺吡喃基、螺惡嗪基等。稠合雜環基的實例包含但不限於苯基稠環或吡啶基稠環,如喹啉基、異喹啉基、喹喔啉基、喹嗪基、喹唑啉基、氮雜吲哚嗪基、喋啶基、色烯基、異色烯基、吲哚基、異吲哚基、吲哚嗪基、吲唑基、嘌呤基、苯並呋喃基、異苯並呋喃基、苯並咪唑基、苯並噻吩基、苯並噻唑基、哢唑基、吩嗪基、吩噻嗪基、啡啶基、咪唑並[1,2-a]吡啶基、[1,2,4]三唑並[4,3-a]吡啶基、[1,2,3]三唑並[4,3-a]吡啶基等。橋接雜環基的實例包含但不限於嗎啡烷基(morphanyl)、六亞甲基四氨基、8-氮雜-雙環[3.2.1]辛烷、1-氮雜-雙環[2.2.2]辛烷、1,4-二氮雜雙環[2.2.2]辛烷(DABCO)等。
除非另外說明,否則本公開的“化合物”意圖涵蓋所描繪的結構的所有立體異構體、幾何異構體和互變異構體。
術語“立體異構體”是指不對稱化合物(例如具有一個或多個不對稱取代的碳原子或“不對稱中心”的化合物)的各種立體異構構型(例如對映異構體、非對映異構體和外消旋體)中的任一種。含有不對稱中心的本公開化合物可以光學活性(對映異構體或非對映異構體)或光學非活性(外消旋)形式分離。術語“對映異構體”包含不互為可重疊的鏡像的立體異構體對。一對對映異構體的1:1混合物為“外消旋混合物”。術語“非對映異構體(diastereomer)”或“非對映異構體(diastereoisomer)”包含具有至少二個不對稱原子但不互為鏡像的立體異構體。含有一個或多個不對稱中心的某些化合物可產生對映異構體、非對映異構體或其它立體異構形式,其可根據卡恩-英格爾-普雷洛格(Cahn-Ingold-Prelog) R-S系統,在各不對稱中心處關於絕對構型定義為(R)-或(S)-。絕對構型未知的拆分的化合物可在不對稱中心處使用術語“或”指示。關於如何從外消旋混合物製備光學活性形式的方法是本領域中已知的,如通過HPLC拆分或立體選擇性合成。
術語“幾何異構體”或“順式和反式異構體”是指具有相同式但其官能團在三維空間中旋轉至不同取向的化合物。
術語“互變異構體”包含處於具有相同式和總電荷的化合物的異構質子化狀態的質子轉移互變異構體。質子轉移互變異構體的實例包含但不限於酮-烯醇對、醯胺-亞胺酸對、內醯胺-內醯亞胺對、烯胺-亞胺對,以及環狀形式,在環狀形式中,質子可以佔用雜環系統的兩個或更多個位置,例如1H-咪唑和3H-咪唑、1H-1,2,4-三唑、2H-1,2,4-三唑和4H-1,2,4-三唑、1H-異吲哚和2H-異吲哚,以及1H-吡唑和2H-吡唑。互變異構體可處於平衡狀態或通過適當取代而空間鎖定成一種形式。除非另外說明,否則通過名稱或結構標識為一種特定互變異構形式的本公開化合物意圖包含其它互變異構形式。
本公開的“化合物”還意圖涵蓋化合物中原子的所有同位素。原子的同位素包含具有相同原子數但質量數不同的原子。例如,除非另外規定,否則本公開“化合物”中的氫、碳、氮、氧、磷、硫、氟、氯、溴或碘意圖還包含其同位素,如但不限於
1
H、
2
H、
3
H、
11
C、
12
C、
13
C、
14
C、
14
N、
15
N、
16
O、
17
O、
18
O、
31
P、
32
P、
32
S、
33
S、
34
S、
36
S、
17
F、
19
F、
35
Cl、
37
Cl、
79
Br、
81
Br、
127
I和
131
I。在一些實施例中,氫包含氕、氘和氚。在一些實施例中,術語“經氘取代”或“氘取代的”用氘置換化學基團中的氫的另一種同功異型物(例如氕)。在一些實施例中,碳包含
12
C和
13
C。在一些實施例中,本公開的“化合物”僅涵蓋化合物中氫的同位素。在一些實施例中,本公開的“化合物”僅涵蓋呈天然豐度的原子的同位素。
還應理解,本公開的“化合物”可以呈溶劑化形式以及非溶劑化形式,例如水合形式、固體形式存在,並且本公開意圖涵蓋所有此類溶劑化形式和非溶劑化形式。
還應理解,本公開的“化合物”可以藥學上可接受的鹽形式存在。
如本文所用,術語“藥學上可接受”是指在合理醫學判斷的範圍內,適用於與人類和動物的組織接觸,而無過度毒性、刺激、過敏反應或其它問題或併發症,與合理的效益/風險比相稱的化合物、材料、組合物和/或劑型。在一些實施例中,藥學上可接受的化合物、材料、組合物和/或劑型是指被管理機構(如美國食品和藥物管理局(U.S. Food and Drug Administration)、中國食品和藥物管理局(China Food and Drug Administration)或歐洲藥物管理局(European Medicines Agency))批准或公認藥典(如美國藥典(U.S.Pharmacopoeia)、中國藥典(China Pharmacopoeia)或歐洲藥典(European Pharmacopoeia))中所列的可用於動物且特別是人類的那些化合物、材料、組合物和/或劑型。
如本文所用,“藥學上可接受的鹽”是指本公開化合物的衍生物,其中母體化合物通過將現有酸性部分(例如羧基等)或堿部分(例如胺、鹼金屬等)轉化為其鹽形式而修飾。在許多情況下,本公開化合物能夠憑藉氨基和/或羧基或其類似基團的存在而形成酸和/或堿鹽。藥學上可接受的鹽為保留母體化合物的生物有效性和特性,通常不會在生物學上或其它方面不合需要的酸和/或堿鹽。本公開化合物的適合的藥學上可接受的鹽包含例如酸加成鹽,其可以衍生自例如無機酸(例如鹽酸、氫溴酸、硫酸、硝酸、磷酸等)或有機酸(例如甲酸、乙酸、丙酸、乙醇酸、草酸、順丁烯二酸、丙二酸、琥珀酸、反丁烯二酸、酒石酸、苯均三酸、檸檬酸、乳酸、苯乙酸、苯甲酸、扁桃酸、甲烷磺酸、萘二磺酸、乙烷磺酸、甲苯磺酸、三氟乙酸、水楊酸、磺基水楊酸等)。在一些實施例中,本公開化合物的藥學上可接受的鹽為甲酸鹽。在一些實施例中,本公開化合物的藥學上可接受的鹽是TFA鹽。
本公開化合物的適合的藥學上可接受的鹽還包含例如堿加成鹽,其可以衍生自例如無機堿(例如週期表的第I列到第XII列的金屬(如鈣、鎂、鐵、銀、鋅、銅等)的鈉鹽、鉀鹽、銨鹽和氫氧化物、碳酸鹽、碳酸氫鹽)或有機堿(例如伯胺、仲胺和叔胺、包含天然存在的經取代胺的經取代胺、環胺、鹼性離子交換樹脂等)。某些有機胺包含但不限於異丙胺、苯乍生(benzathine)、膽酸鹽、二乙醇胺、二乙胺、賴氨酸、葡甲胺、呱嗪以及緩血酸胺。所屬領域的技術人員應理解,除了實例中所示以外,添加用於形成酸/堿加成鹽的酸或堿也是可能的。其它適合的鹽的列表可見于例如《雷明頓氏藥物科學(Remington's Pharmaceutical Sciences)》, 第20版, 馬克出版公司(Mack Publishing Company), 賓夕法尼亞州伊斯頓(Easton, Pa.), (1985);以及Stahl和Wermuth的《藥用鹽手冊:特性、選擇和使用(Handbook of Pharmaceutical Salts: Properties, Selection, and Use)》(德國魏因海姆的Wiley-VCH(Wiley-VCH, Weinheim, Germany), 2002)中。在一些實施例中,本公開化合物的適合的藥學上可接受的鹽為無機堿鹽。
本公開還包含本公開化合物的活性中間物、活性代謝物和前藥。如本文所用,“活性中間物”是指合成方法中的中間化合物,其展現與最終合成的化合物相同或基本上相同的生物活性。
如本文所用,“活性代謝物”是指通過在動物或人體內代謝或生物轉化產生的本公開化合物或其鹽或前藥的分解或最終產物,其展現與指定化合物相同或基本上相同的生物活性。此類代謝物可以由例如施用的化合物或鹽或前藥氧化、還原、水解、醯胺化、脫醯胺、酯化、去酯化、酶裂解等得到。
如本文所用,“前藥”是指當施用至動物或人類個體時釋放活性母體藥物的任何化合物或偶聯物。前藥可通過以一種方式修飾化合物中存在的官能團製備,所述方式使得修飾在常規操作中或在體內可由母體化合物裂解。前藥包含其中羥基、氨基、硫氫基或羧基鍵結到任何基團,使得當將其施用給哺乳動物個體時可分別裂解形成游離羥基、氨基、硫氫基或羧基的化合物。前藥的實例包含但不限於本公開化合物中醇和胺官能團的乙酸酯、甲酸酯和苯甲酸酯衍生物。前藥的製備和使用論述於THiguchi和V. Stella, “作為新穎遞送系統的前藥(Pro-drugs as Novel Delivery Systems)”, 《美國化學會會議論文集(A.C.S. Symposium Series)》第14卷,以及《藥物設計中的生物可逆載體(Bioreversible Carriers in Drug Design)》, Edward B.Roche編, 美國藥劑師協會和培格曼出版社(American Pharmaceutical Association and Pergamon Press), 1987,二者特此以全文引用的方式併入。
本文公開了可以選擇性抑制DNA-PK的新穎化合物或藥學上可接受的鹽。當與其它臨床上可用的DNA-PK抑制劑相比較時,前文公開的化合物或其藥學上可接受的鹽展現某些改進的特性,例如更高的BBB穿透(因此使其潛在地可用於治療已經轉移到CNS,特別是腦轉移和軟腦膜轉移的癌症)、更好的效力等。其還可具有相比已知DNA-PK調節劑有利的毒性概況和/或有利的代謝或藥物動力學概況。
因此,此類化合物或其藥學上可接受的鹽可尤其適用於治療癌症,尤其是具有腦部癌轉移的那些癌症。
合成方法
本文提供的化合物,包含其鹽、酯、水合物或溶劑化物或立體異構體的合成是在實例中的合成方案中說明。本文提供的化合物可使用任何已知的有機合成技術製備並且可以根據多種可能的合成途徑中的任一種合成,且因此,這些方案只是說明性的且不打算限制可用于製備本文提供的化合物的其它可能的方法。此外,所述方案中的步驟是為了更好地說明且在適當時可以改變。出於研究和可能提交給管理機構的目的,實例中的化合物實施例是在中國合成。
用於製備本公開化合物的反應可以在適合溶劑中進行,這些溶劑可以由有機合成領域的技術人員容易地選擇。適合溶劑可以在反應進行的溫度下,例如在從溶劑的冷凍溫度至溶劑的沸騰溫度範圍內的溫度下基本上不與起始物質(反應物)、中間物或產物反應。指定反應可以在一種溶劑或多於一種溶劑的混合物中進行。取決於特定反應步驟,用於特定反應步驟的適合溶劑可以由熟練技術人員選擇。
本公開化合物的製備可以涉及各種化學基團的保護和脫保護。對於保護和脫保護的需求,以及適當保護基的選擇可以由本領域技術人員容易地確定。保護基的化學性質可見於例如T. W. Greene和P. G. M. Wuts, 《有機合成中的保護基(Protective Groups in Organic Synthesis)》, 第3版,威利父子公司(Wiley & Sons, Inc.), 紐約(1999),其以全文引用的方式併入本文中。
可以根據本領域中已知的任何適合方法監測反應。例如,可以通過光譜手段,如核磁共振光譜(例如
1
H或
13
C)、紅外光譜、分光光度法(例如紫外-可見光)、質譜,或通過色譜法,如高壓液相色譜(HPLC)、液相色譜-質譜(LCMS)或薄層色譜(TLC)來監測產物形成。本領域的技術人員可以通過多種方法,包含高效液相色譜法(HPLC)(“製備型LC-MS純化:改善的化合物特異性方法優化(Preparative LC-MS Purification:Improved Compound Specific Method Optimization)”, Karl F.Blom, Brian Glass, Richard Sparks, Andrew P.Combs, 《組合化學雜誌(J.Combi.Chem.)》, 2004, 6(6), 874-883,以全文引用的方式併入本文中)和正相二氧化矽色譜法純化化合物。
如本文所使用的縮寫定義如下:“1×”或“×1”表示一次,“2×”或“×2”表示兩次,“3×”或“×3”表示三次,“4×”或“×4”表示四次,“5×”或“×5”表示兩次,“℃”表示攝氏度,“eq”或“eq.”表示當量,“g”表示克,“mg”表示毫克,“L”表示升,“mL”或“ml”表示毫升,“μL”表示微升,“N”表示正常,“M”表示摩爾,“mmol”表示毫摩爾,“min”表示分鐘,“h”或“hr”表示小時,“r.t.”或“rt”表示室溫,“atm”表示氛圍,“psi”表示每平方英寸磅數,“conc.”表示濃縮物,“sat”或“sat'd”表示飽和,“MS”或“Mass Spec”表示質譜,“ESI”表示電噴射電離質譜,“LCMS”表示液相色譜質譜法,“HPLC”表示高壓液相色譜,“RP”表示反相,“TLC”或“tlc”表示薄層色譜,“SM”表示起始物質,“NMR”表示核磁共振光譜,“
1
H”表示質子,“δ”表示△,“s”表示單重峰,“d”表示雙重峰,“t”表示三重峰,“q”表示四重峰,“m”表示多重峰,“br”表示廣譜,且“Hz”表示赫茲。“α”、“β”、“R”、“S”、“E”以及“Z”是本領域技術人員熟悉的立體化學名稱。
藥物組合物
本公開提供了包括至少一種本公開化合物的藥物組合物。在一些實施例中,所述藥物組合物包括多於一種本公開的化合物。在一些實施例中,所述藥物組合物包含一種或多種本公開的化合物和藥學上可接受的載劑。
藥學上可接受的載劑是本領域中的常規藥物載劑,其可以按藥學領域中眾所周知的方式製備。在一些實施例中,本公開的化合物可以與藥學上可接受的載劑混合以製備藥物組合物。
如本文所使用,術語“藥學上可接受的載劑”是指將本文提供的化合物從一個位置、體液、組織、器官(內部或外部)或身體的一部分載運或運輸至另一位置、體液、組織、器官或身體的一部分所涉及的藥學上可接受的材料、組合物或媒劑,如液體或固體填充劑、稀釋劑、賦形劑、溶劑或囊封材料。藥學上可接受的載劑可以是可用於接觸動物組織,而無過量毒性或不良作用的媒劑、稀釋劑、賦形劑或其它材料。示範性藥學上可接受的載劑包含糖、澱粉、纖維素、麥芽、黃芪膠、明膠、林格氏溶液(Ringer's solution)、海藻酸、等滲生理鹽水、緩衝劑等。可以用於本公開中的藥學上可接受的載劑包含本領域中一般已知的那些,如以引用的方式併入本文中的《雷氏藥學大全》, Mack Pub. Co., 新澤西州(New Jersey)(1991)中公開那些。
可以充當藥學上可接受的載劑的材料的一些實例包含:(1)糖,如乳糖、葡萄糖和蔗糖;(2)澱粉,如玉米澱粉和馬鈴薯澱粉;(3)纖維素和其衍生物,如羧甲基纖維素鈉、乙基纖維素和乙酸纖維素;(4)粉末狀黃芪膠;(5)麥芽;(6)明膠;(7)滑石;(8)賦形劑、如可哥脂和栓劑蠟;(9)油,如花生油、棉籽油、紅花油、芝麻油、橄欖油、玉米油和大豆油;(10)二醇,如丙二醇;(11)多元醇,如甘油、山梨糖醇、甘露醇和聚乙二醇;(12)酯,如油酸乙酯和月桂酸乙酯;(13)瓊脂;(14)緩衝劑,如氫氧化鎂和氫氧化鋁;(15)海藻酸;(16)無熱原質水;(17)等滲生理鹽水;(18)林格氏溶液;(19)醇,如乙醇和丙醇;(20)磷酸鹽緩衝溶液;以及(21)藥物配製物中採用的其它無毒可相容物質,如丙酮。
藥物組合物可視需要含有藥學上可接受的輔助物質以接近生理條件,如pH值調節劑和緩沖劑、毒性調節劑等,例如乙酸鈉、氯化鈉、氯化鉀、氯化鈣、乳酸鈉等。
藥物組合物的形式取決於多種標準,包含但不限於施用途徑、疾病程度或待施用的劑量。所述藥物組合物可以被配製成經口、經鼻、直腸、經皮、靜脈內或肌肉內施用。舉例來說,用於經鼻施用的劑型可以方便地配製為氣霧劑、溶液、滴劑、凝膠或乾粉;用於鼻內施用的劑型可以配製為流體配製物。根據希望的施用途徑,所述藥物組合物可以被配製成片劑、膠囊、丸劑、糖衣藥丸、散劑、顆粒劑、藥囊、扁囊劑、口含錠、懸浮液、乳液、溶液、糖漿、氣霧劑(呈固體形式或在液體介質中)、噴霧劑、油膏、糊漿、乳膏、洗劑、凝膠劑、貼片、吸入劑或栓劑形式。
藥物組合物可以被配製用於在通過採用本領域中已知的程式施用給患者之後,提供活性成分的快速、持續或延遲釋放。在一些實施例中,藥物組合物被配製成持續釋放形式。如本文所使用,術語“持續釋放形式”是指活性劑從藥物組合物釋放以使其變得在延長的時間段(延長釋放)內或在某一位置(控制釋放)可供個體,主要是個體的胃腸道生物吸收。在一些實施例中,所述延長的時間段可以是約1小時至24小時、2小時至12小時、3小時至8小時、4小時至6小時、1至2天或更長時間。在某些實施例中,所述延長的時間段是至少約4小時、至少約8小時、至少約12小時或至少約24小時。所述藥物組合物可以被配製成片劑形式。例如,活性劑的釋放速率不僅可以通過活性劑溶解於胃腸液中且隨後獨立於pH自片劑或丸劑擴散進行控制,而且還受片劑崩解和溶蝕的物理過程影響。在一些實施例中,“控制釋放的醫學應用(Medical Applications of Controlled Release)”, Langer和Wise(編), CRC Pres., 佛羅里達州波卡拉頓(Boca Raton, Florida)(1974);“控制性藥物生物利用率(Controlled Drug Bioavailability)”, 《藥品設計和性能(Drug Product Design and Performance)》, Smolen和Ball(編), Wiley, 紐約(1984);Ranger和Peppas, 1983, 《高分子科學-高分子化學評述(J Macromol.Sci.Rev. Macromol Chem.)》23:61中所公開的聚合材料可以用於持續釋放;還參見Levy等人, 1985, 《科學(Science)》228:190;During等人, 1989, 《神經病學年評(Ann. Neurol.)》25:351;Howard等人, 1989, 《神經外科雜誌(J. Neurosurg.)》71:105。以上參考文獻都以全文引用的方式併入本文中。
在某些實施例中,藥物組合物包括約0.0001mg到約100mg的本公開化合物(例如,約0.0001mg至約10mg、約0.001mg至約10mg、約0.01mg至約10mg、約0.1mg至約10mg、約0.1mg至約5mg、約0.1mg至約4mg、約0.1mg至約3mg、約0.1mg至約2mg、約0.1mg至約1mg、約0.1mg至約0.5mg、約1mg至約10mg、約1mg至約5mg、約5mg至約10mg、約5mg至約20mg、約5mg至約30mg、約5mg至約40mg、約5mg至約50mg、約10mg至約100mg、約20mg至約100mg、約30mg至約100mg、約40mg至約100mg、約50mg至約100mg)。每天每位個體的適合劑量可以是約0.1mg至約10mg,優選地是約0.1mg至約5mg、約5mg至約10mg、或約1mg至約5mg。
在某些實施例中,藥物組合物可按單位劑型配製,每種劑量含有約0.0001mg至約10mg、約0.001mg至約10mg、約0.01mg至約10mg、約0.1mg至約10mg、約0.1mg至約5mg、約0.1mg至約4mg、約0.1mg至約3mg、約0.1mg至約2mg、約0.1mg至約1mg、約0.1mg至約0.5mg、約1mg至約10mg、約5mg至約10mg、約5mg至約20mg、約5mg至約30mg、約5mg至約40mg、約5mg至約50mg、約10mg至約100mg、約20mg至約100mg、約30mg至約100mg、約40mg至約100mg、約50mg至約100mg的本公開化合物。術語“單位劑型”是指適合以單一劑量用於人類個體和其他哺乳動物的物理離散單元,每個單元含有與適合藥物載劑結合的經計算以產生所希望的治療作用的預定量的活性材料。
在一些實施例中,藥物組合物包括一種或多種本公開的化合物作為第一活性成分,並且還包含第二活性成分。第二活性成分可為所屬領域中已知的任何免疫調節劑或抗腫瘤劑,包含但不限於化學治療劑、免疫治療劑、細胞信號轉導抑制劑、細胞信號轉導抑制劑、烷化劑、拓撲異構酶抑制劑、有絲分裂抑制劑、抗激素劑等。此類免疫調節劑或抗腫瘤劑的實例為鉑類化學治療劑(例如順鉑(DDP)、卡鉑(CBP)、硫酸根-1,2-二氨基環己烷鉑(SHP)、奈達鉑(Nedaplatin)、奧沙利鉑(Oxaliplatin)(OXA)、樂鉑(Laboplatin))、多烯紫杉醇(Docetaxel)、太平洋紫杉醇(Paclitaxel)、多柔比星(Doxorubicin)、依託泊苷(Etoposide)、米托蒽醌(Mitoxantrone)、CTLA-4抑制劑、抗CTLA-4抗體、PD-1抑制劑、PD-L1抑制劑、抗PD-1/PD-L1抗體、CD39抑制劑、抗CD39抗體、CD73抑制劑、抗CD73抗體、CCR2抑制劑、抗CCR2抗體、EGFR抑制劑、CDK 4/6抑制劑、MELK抑制劑、OX40促效劑、抗雄激素抑制劑、IgG4同型抗體、酪氨酸激酶抑制劑、DNA甲基轉移酶抑制劑、Hsp90抑制劑、FGFR抑制劑、mTOR抑制劑、芳香酶抑制劑、VEGF抑制劑、LHRH拮抗劑、PI3K抑制劑、AKT抑制劑、極光激酶抑制劑、MEK抑制劑、HDAC抑制劑、BET抑制劑、PIK3CA抑制劑、蛋白酶體抑制劑、其他SERD、法呢基轉移酶抑制劑、VEGF-A抗體、ErbB3 (Her3)抗體、蛋白酶體抑制劑、蛋白激酶Cβ抑制劑、抗IGF-1R抗體、抗HER2抗體、SERM、IGF抑制劑、抗IgG抗體等。用於治療癌症或腫瘤的抗腫瘤劑的代表性實例可包含但不限於順鉑、卡鉑、SHP、奈達鉑、奧沙利鉑、樂鉑、多烯紫杉醇、太平洋紫杉醇、多柔比星、依託泊苷、米托蒽醌、長春新堿、長春堿、吉西他濱(gemcitabine)、環磷醯胺、苯丁酸氮芥(chlormabucil)、卡莫司汀(carmustine)、甲胺喋呤、氟尿嘧啶、放線菌素、表柔比星(epirubicin)、蒽環黴素、博萊黴素、絲裂黴素-C、伊立替康(irinotecan)、拓朴替康(topotecan)、替尼泊甙(teniposide)白介素、干擾素、曲美單抗(tremelimumab)、伊匹單抗(ipilimumab)、派立珠單抗(pembrolizumab)、納武單抗(nivolumab)、阿維魯單抗(avelumab)、度伐單抗(durvalumab)、阿特珠單抗(atezolizumab)、IPH 52、IPH 53、CPI-006、普洛利單抗(plozalizumab)、MLN1202、西妥昔單抗(cetuximab)、拉帕替尼(lapatinib)、埃羅替尼(erlotinib)、吉非替尼(gefitinib)、來那替尼(neratinib)、曲妥珠單抗(trastuzumab)、阿多-曲妥珠單抗恩他新(ado-trastuzumab emtansine)、帕妥珠單抗(pertuzumab)、MCLA-128、阿那曲唑(anastrazole)、雷諾昔酚(raloxifene)、G1T38、他莫昔芬(tamoxifen)、戈舍瑞林(goserelin)、恩雜魯胺(enzalutamide)、伏立諾他(vorinostat)、恩替諾特(entinostat)、舒尼替尼(sunitinib)、帕唑帕尼(pazopanib)、貝伐單抗(bevacizumab)、蘭比珠單抗(ranibizumab)、呱加他尼(pegaptanib)、西地尼布(cediranib)、達沙替尼(dasatinib)、GDC-0980、吉達昔布(gedatolisib)、阿普昔布(alpelisib)、BKM120、考帕昔布(copanlisib)、AZD8835、GDC-0941、塔瑟昔布(taselisib)、替西羅莫司(temsirolimus)、依維莫司(everolimus)、薩帕塞替(sapanisertib)、AZD5363、MK2206、帕尼單抗(panitumumab)、派立珠單抗(pembrolizumab)、索拉非尼(sorafenib)、帕博希布(palbociclib)、玻瑪西林(abemaciclib)、瑞博西林(ribociclib)、克唑替尼(crizotinib)、多韋替尼(dovitinib)、蘆可替尼(ruxolitinib)、阿紮胞苷(azacitidine)、CC-486、HSP90加利特皮(HSP90 ganetespib)、Debio 1347、伊達替尼(erdafitinib)、維瑟塞替(vitusertib)、阿立塞替(alisertib)、司美替尼(selumetinib)、G-5829、GSK525762、MLN9708、GDC-0810、AFP464、替吡法尼(tipifarnib)、塞利班單抗(seribantumab)、硼替佐米(bortezomib)、恩紮妥林(enzastaurin)、AVE1642、森吐珠單抗(xentuzumab)、達羅吐珠單抗(dalotuzumab)、AMG479等。
此類抗腫瘤劑的實例還可在V. T. Devita和S. Hellman(編)的《癌症原理和腫瘤學實踐(Cancer Principles and Practice of Oncology)》,第6版(2001年2月15日), 利平科特威廉姆斯和維爾金斯出版社(Lippincott Williams & Wilkins Publishers)中找到。本領域的普通技術人員還將能夠基於藥物的特定特徵和所涉及的癌症辨別哪些藥劑組合將是有用的。
根據本公開的這一方面,提供適用於治療癌症的組合,其包括如上文所定義的式(I)化合物或其藥學上可接受的鹽;以及上文所列之免疫調節劑或抗腫瘤劑中的任一種。
因此,在本公開的另一方面,提供一種與選自上文所列之免疫調節劑或化學治療劑的免疫調節劑或化學治療劑組合的式(I)化合物或其藥學上可接受的鹽。
本文中,在使用術語“組合”的情況下,應理解,這是指同時、分別或依序施用。在一些實施例中,“組合”是指同時施用。在本公開的另一方面,“組合”是指分開施用。在本公開的另一方面,“組合”是指依序施用。在依序或分別施用的情況下,施用第二組分的延遲不應使得失去所述組合的有益作用。
根據本公開的另一個方面,提供一種藥物組合物,其包括式(I)化合物或其藥學上可接受的鹽與選自以上列出的免疫調節劑或抗腫瘤劑的免疫調節劑或抗腫瘤劑的組合,以及藥學上可接受的稀釋劑或載劑。
根據本公開的另一個方面,提供一種藥物組合物,其包括式(I)化合物或其藥學上可接受的鹽與選自以上列出的一者的免疫調節劑或抗腫瘤劑的組合,以及藥學上可接受的稀釋劑或載劑,用於產生免疫調節或抗癌作用。
根據本公開的另一方面,提供一種藥物組合物,其包括式(I)化合物或其藥學上可接受的鹽與選自以上列出的一者的免疫調節劑或抗腫瘤劑的組合,以及藥學上可接受的稀釋劑或載劑,用於治療DNA-PK相關病症,例如NSCLC、RCC、前列腺癌或乳癌等。
根據本公開的另一方面,提供一種試劑盒,其包括式(I)化合物或其藥學上可接受的鹽與選自上文所列的一者的免疫調節劑或抗腫瘤劑的組合。
根據本公開的另一方面,提供一種試劑盒,其包含:
a)呈第一單位劑型的式(I)化合物或其藥學上可接受的鹽;
b)呈第二單位劑型的選自上文所列的一者的免疫調節劑或抗腫瘤劑;和
c)用於容納所述第一和第二劑型的容器。
除了用於治療藥物以外,式(I)化合物或其藥學上可接受的鹽還適用作體外和體內測試系統的開發和標準化中的藥理學工具,所述測試系統用於評估DNA-PK在如貓、狗、兔、猴、大鼠和小鼠的實驗動物中的活性或表達,作為探求新型治療劑的一部分。
在以上其它藥物組合物、過程、方法、用途和藥物製造特徵中,本文所述的本公開化合物的替代和優選實施例也適用。
治療方法
本公開提供一種治療DNA-PK相關病症的方法,其包括向個體施用有效量的一種或多種本公開的化合物、其藥學上可接受的鹽或藥物組合物。
本公開還提供一種治療DNA-PK相關病症的方法。在某些實施例中,所述方法包括向個體施用有效量的一種或多種本公開的化合物、其藥學上可接受的鹽或藥物組合物。
如本文所用,術語“DNA-PK相關病症”是指發作或發展或二者與DNA-PK的表達或活性有關的疾病。實例包含(但不限於)過度增生性病症(例如,癌症)。
在一些實施例中,DNA-PK相關病症為癌症,優選為過度表達DNA-PK的癌症。“過度表達DNA-PK的癌症”是與相同組織類型的非癌性細胞相比在癌症或腫瘤細胞中具有顯著較高水準的DNA-PK蛋白質的癌症。這種過度表達可能是由基因擴增或增加的轉錄或轉譯引起的。通過評估細胞上存在的DNA-PK蛋白質水準的增加(例如通過免疫組織化學分析;IHC),可在診斷或預後分析中測定DNA-PK過度表達。替代地或另外,可測量細胞中編碼DNA-PK的核酸的水準,例如通過螢光原位雜交(FISH;參見1998年10月公佈的WO98/45479)、DNA印跡法或聚合酶鏈反應(PCR)技術,如實時定量PCR(RT-PCR)(方法(Methods) 132: 73-80 (1990))。除了上述分析之外,所屬領域的技術人員可使用各種體內分析。例如,可將患者體內的細胞暴露於抗體,所述抗體任選地用可檢測標記(例如放射性同位素)標記,並且例如通過外部掃描放射性或通過分析從先前暴露于抗體的患者取得的活組織檢查,可評估抗體與患者細胞的結合。
確切地說,癌症包含不限於肺癌(例如非小細胞肺癌(NSCLC)、小細胞肺癌、肺腺癌、大細胞肺癌、鱗狀細胞肺癌)、腎細胞癌(RCC)、前列腺癌、乳癌、卵巢癌、子宮內膜癌、子宮頸癌、骨癌、子宮癌、結腸癌、白血病、成膠質細胞瘤、黑素瘤、軟骨肉瘤、腦癌、膽管癌、骨肉瘤、淋巴瘤、腺瘤、骨髓瘤、肝細胞癌、腎上腺皮質癌、胰腺癌、膀胱癌、肝癌、胃癌、結腸直腸癌、食道癌、睾丸癌、皮膚癌、腎癌、間皮瘤、成神經細胞瘤、甲狀腺癌、頭頸癌、食道癌、眼癌、鼻咽癌或口腔癌。在一些實施例中,癌症為NSCLC、RCC、前列腺癌或乳癌。除非另外規定,否則如本文中提到的癌症可處於任何分期。在一些實施例中,癌症為早期癌症。在一些實施例中,癌症為局部晚期癌症。在一些實施例中,癌症為局部晚期和/或轉移癌。在一些實施例中,癌症為侵入性癌症。在一些實施例中,癌症為對現有療法具有抗性的癌症。
如本文所使用,術語“治療(treatment/treat/treating)”是指逆轉、緩解如本文所描述的疾病或病症或其一種或多種症狀,延遲其發作或抑制其進展。在一些實施例中,治療可在已出現一或多種症狀後施用。在其它實施例中,治療可在症狀不存在的情況下施用。例如,治療可以在症狀發作之前施用給易感個體(例如根據症狀病史和/或根據遺傳學或其它易感因素)。也可以在症狀消退之後繼續治療,例如以預防或延遲其復發。
在一些實施例中,本文所提供的一種或多種化合物、其藥學上可接受的鹽或藥物組合物是通過腸胃外途徑或非腸胃外途徑施用。在一些實施例中,所述一種或多種化合物、其藥學上可接受的鹽、水合物、溶劑化物或立體異構體或藥物組合物是經口、經腸、經頰、經鼻、鼻內、經粘膜、經表皮、透皮、經皮、經眼、肺、經直腸、舌下、經陰道、經表面、皮下、靜脈內、肌肉內、動脈內、鞘內、囊內、眶內、心內、皮內、腹膜內、經氣管、表皮下、關節內、囊下、脊椎內、蛛網膜下或胸骨內施用。
本文提供的化合物可以呈純形式、與其它活性成分組合或呈本公開的藥物組合物形式施用。在一些實施例中,本文所提供的化合物可以與本領域中已知的一種或多種抗癌劑或消炎劑組合同時或依序向有需要的個體施用。這類組合中的個別化合物是在分開的或組合的藥物組合物中依序或同時施用。優選地,個別化合物將在組合的藥物組合物中同時施用。所屬領域的技術人員將瞭解已知治療劑的適當劑量。
在一些實施例中,施用是一天一次、一天兩次、一天三次或每兩天一次、每三天一次、每四天一次、每五天一次、每六天一次、一週一次進行。
如本文所提供的化合物或其藥學上可接受的鹽的治療有效量將取決於本領域中已知的各種因素,如體重、年齡、既往病史、當前藥物、個體的健康狀態和交叉反應、過敏、敏感和不良副作用的可能性,以及施用途徑和疾病發展程度。如由這些和其它情況或要求所指示,所屬領域的技術人員(例如醫生或獸醫)可按比例減少或增加劑量。
在一些實施例中,本文所提供的一種或多種化合物、其藥學上可接受的鹽或藥物組合物經口施用。對於經口施用,實現所希望的目標的任何劑量都是適當的。在一些實施例中,適合日劑量是在約0.001到100mg之間,優選地在0.1mg與5g之間,更優選地在5mg與1g之間,更優選地在10mg與500mg之間,並且施用是一天一次、一天兩次、一天三次、每天或一周3到5天進行。在一些實施例中,本文所提供的一種或多種化合物、其藥學上可接受的鹽或藥物組合物的劑量的範圍在每天約0.0001mg,優選地0.001mg、0.01mg、0.1mg、0.2mg、0.3mg、0.4mg、0.5mg、0.6mg、0.7mg、0.8mg、0.9mg、1mg、2mg、3mg、4mg、5mg、6mg、7mg、8mg、9mg、10mg之間。
化合物的用途
在某些實施例中,本公開提供本公開的化合物、其藥學上可接受的鹽或藥物組合物在製備用於治療DNA-PK相關病症的藥劑中的用途。在某些實施例中,DNA-PK相關病症包含癌症。
本公開中的化合物和其藥物組合物可用於預防或治療哺乳動物(尤其人類)中的DNA-PK相關病症(表達或活性)中的任一種的發作或發展。
在此類情形中,本公開還提供一種篩選適於用單獨的本公開的化合物或藥物組合物或其與其它成分(例如第二活性成分,例如抗炎劑或抗癌劑)的組合治療的患者的方法。所述方法包含對來自患者的組織樣品進行測序並且檢測患者中的DNA-PK的積聚。
實例
以下進一步闡明本公開的通用方法。本公開化合物可通過所屬領域中已知的方法製備。以下說明本公開的優選化合物的具體製備方法。然而,這些決不限制本公開化合物的製備方法。
合成實例
本文提供的化合物(包含其藥學上可接受的鹽)的合成是在實例中的合成方案中加以說明。本文提供的化合物可使用任何已知的有機合成技術製備並且可以根據多種可能的合成途徑中的任一種合成,且因此,這些方案只是說明性的且不打算限制可用于製備本文提供的化合物的其它可能的方法。此外,所述方案中的步驟是為了更好地說明且在適當時可以改變。出於研究和可能提交給管理機構的目的而合成實例中的化合物實施例。
用於製備本公開化合物的反應可在適合的溶劑中進行,所述溶劑可由有機合成領域的技術人員容易地選擇。適合的溶劑可在反應進行的溫度下,例如在溶劑的冷凍溫度至溶劑的沸騰溫度範圍內的溫度下基本上不與起始物質(反應物)、中間物或產物反應。指定反應可以在一種溶劑或多於一種溶劑的混合物中進行。取決於特定反應步驟,用於特定反應步驟的適合溶劑可以由熟練技術人員選擇。
本公開化合物的製備可涉及各種化學基團的保護和脫保護。對於保護和脫保護的需求,以及適當保護基的選擇可以由本領域技術人員容易地確定。保護基的化學性質可見於例如T. W. Greene和P. G. M. Wuts, 《有機合成中的保護基(Protective Groups in Organic Synthesis)》, 第3版, 威利父子公司(Wiley & Sons, Inc.), 紐約(1999),其以全文引用的方式併入本文中。
可以根據本領域中已知的任何適合方法監測反應。例如,可通過光譜手段,如核磁共振光譜(例如
1
H或
13
C)、紅外光譜(IR)、分光光度法(例如紫外-可見光)、質譜,或通過色譜法,如高壓液相色譜(HPLC)、液相色譜-質譜(LCMS)或薄層色譜(TLC)來監測產物形成。本領域的技術人員可以通過多種方法,包含高效液相色譜法(HPLC)(“製備型LC-MS純化:改善的化合物特異性方法優化(Preparative LC-MS Purification:Improved Compound Specific Method Optimization)”, Karl F.Blom, Brian Glass, Richard Sparks, Andrew P.Combs, 《組合化學雜誌(J.Combi.Chem.)》, 2004, 6(6), 874-883,以全文引用的方式併入本文中)和正相二氧化矽色譜法來純化化合物。
實例中的化合物的結構是通過核磁共振(NMR)或/和液相色譜-質譜(LC-MS)來表徵。NMR化學位移(
δ
)是以10
-6
(ppm)為單位給出。
1
H-NMR光譜是在Bruker AVANCE NMR(300 MHz或400 MHz)光譜儀上使用ICON-NMR(在TopSpin程式控制下),使用四甲基矽烷作為內標以二甲亞碸-
d6
(DMSO-
d6
)或CDCl
3
或CD
3
OD或D
2
O或丙酮_d
6
或CD
3
CN(來自伊諾凱(Innochem)或西格瑪-奧德里奇公司(Sigma-Aldrich)或劍橋同位素實驗室公司(Cambridge Isotope Lab., Inc.))記錄。
使用具有電噴霧源的Shimadzu 2020質譜儀在正離子和負離子模式下進行MS量測。
高效液相色譜(HPLC)測量是在Shimadzu LC-20AD系統或Shimadzu LC-20ADXR系統或Shimadzu LC-30AD系統上,使用勻漿填裝(Shim-pack) XR-ODS C18柱(3.0*50 mm,2.2 μm),或Ascentis Express C18柱(2.1*50 mm,2.7 μm),或Agilent Poroshell HPH-C18柱(3.0*50 mm,2.7 μm)進行。
使用國藥化學試劑北京有限公司(Sinopharm Chemical Reagent Beijing Co., Ltd.)和信諾化學(Xinnuo Chemical)矽膠板進行薄層色譜。用於薄層色譜(TLC)的矽膠板為175至225µm。通過TLC分離和純化產物所使用的矽膠板為1.0 mm。
純化色譜柱使用矽膠作為載體(100至200、200至300或300至400目,由乳山市上邦新材料有限公司(Rushanshi Shangbang Xincailiao Co., Ltd.)或乳山太陽乾燥劑有限公司(Rushan Taiyang Desiccant Co., Ltd. etc.)等製造),或Agela Technologies快速系統中的快速柱(反相C18柱20-45 μm,由艾傑爾科技公司(Agela Technologies)製造)。色譜柱的大小根據化合物的量進行調整。
本公開的已知起始物質可通過使用或根據所屬領域中已知的方法合成,或可購自阿法埃莎(Alfa Aesar)、梯希愛(TCI)、西格瑪-奧德里奇公司(Sigma-Aldrich)、書亞醫藥(Bepharm)、畢得醫藥科技(Bide pharmatech)、藥石(PharmaBlock)、Enamine、伊諾凱和傑達維醫藥科技(JW&Y PharmLab)等。
除非另外規定,否則反應全部在氬氣或氮氣氛圍下進行。氬氣或氮氛圍是指反應燒瓶被連接至體積為約1 L的氬氣或氮氣球。氫化通常是在壓力下進行。除非另外規定,否則實例中的反應溫度為環境溫度,其為10℃至30℃。通過TLC或/和LC-MS監測反應進展。用於反應的洗脫劑系統包含二氯甲烷-甲醇系統和石油醚-乙酸乙酯系統。溶劑的體積比根據化合物的不同極性進行調整。
用於純化化合物的柱色譜的洗脫系統以及TLC的洗脫劑系統包含二氯甲烷-甲醇系統和石油醚-乙酸乙酯系統。溶劑的體積比根據化合物的不同極性進行調整。可以添加少量鹼性或酸性試劑(0.1%至1%)(如甲酸、或乙酸、或TFA、或氨)進行調整。
在本文提供的化合物的合成中使用的化學試劑的縮寫列於下文:

製備1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例1)
步驟1. 5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶
在空氣氛圍下在0℃下將5-氯-1H-吡唑並[4,3-d]嘧啶(3.00 g,19.410 mmol,1.00當量)和NIS(7.86 g,34.936 mmol,1.80當量)於DMF(60.00 mL)中的混合物攪拌隔夜。用EtOAc(3×150 mL)萃取所得混合物。用鹽水(3×200 mL)洗滌合併的有機層。用Na
2
S
2
O
3
(3×200 mL)洗滌合併的有機層,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。通過矽膠柱色譜純化殘餘物,用PE/EtOAc(20:1)洗脫,得到呈黃色固體的5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶(2.4 g,44.09%)。LCMS: m/z (ESI), [M+H]
+
= 281.0。
步驟2. 5-氯-3-碘-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在0℃下將Cs
2
CO
3
(3.49 g,10.697 mmol,3當量)、CH
3
I(2.53 g,17.828 mmol,5.00當量)和5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶(1.00 g,3.566 mmol,1.00當量)於DMF(20.00 mL)中的混合物攪拌1 h。用水(100 mL)稀釋所得混合物並用EtOAc(3×80 mL)萃取。用鹽水(3×100 mL)洗滌合併的有機層,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。從EtOAc/PE(1:5 300 mL)再結晶粗產物,得到呈黃色固體的5-氯-3-碘-1-甲基-1H-吡唑並[4,3-d]嘧啶(850 mg,80.95%)。LCMS: m/z (ESI), [M+H]
+
= 295.0。
步驟3. 5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將含K
2
CO
3
(1210.85 mg,8.761 mmol,3當量)、Pd(dppf)Cl
2
CH
2
Cl
2
(476.98 mg,0.584 mmol,0.2當量)、2-(3,6-二氫-2H-吡喃-4-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(797.57 mg,3.797 mmol,1.3當量)和5-氯-3-碘-1-甲基-1H-吡唑並[4,3-d]嘧啶(860.00 mg,2.920 mmol,1.00當量)的二惡烷(15.00 mL)和H
2
O(3.00 mL)攪拌16個小時。用水(100 mL)稀釋所得混合物。用CH
2
Cl
2
/MeOH = 12:1(3×50 mL)萃取所得混合物。用鹽水(2×100 mL)洗滌合併的有機層,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(CH
2
Cl
2
/ MeOH = 12:1)來純化殘餘物,得到呈灰色固體的5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶(380 mg,51.90%)。LCMS: m/z (ESI), [M+H]
+
= 251.2。
步驟4. 3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs
2
CO
3
(2599.38 mg,7.978 mmol,2.50當量)、XantPhos(553.94 mg,0.957 mmol,0.30當量)、Pd(OAc)
2(
143.29 mg,0.638 mmol,0.20當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(567.40 mg,3.829 mmol,1.20當量)和5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶(800.00 mg,3.191 mmol,1.00當量)於二惡烷(20.00 mL)中的混合物攪拌隔夜。用水(200 mL)稀釋所得混合物。用CH
2
Cl
2
/MeOH = (12:1)(3×200 mL)萃取所得混合物,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。從EtOAc/PE(1:6 300 mL)再結晶粗產物,得到呈棕色固體的3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[4,3-d]嘧啶-5-胺(600 mg,51.88%)。LCMS: m/z (ESI), [M+H]
+
= 363.3。
步驟5. 1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(氧雜環己烷-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺.(實例1)
在氫氣氛圍下在室溫下將Pd/C(47.92 mg,0.450 mmol,1.36當量)和3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[4,3-d]嘧啶-5-胺(120 mg,0.331 mmol,1當量)於MeOH(200 mL)和THF(100 mL)中的混合物攪拌2小時。過濾所得混合物,用MeOH(3×100 mL)洗滌濾餅。減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內25 B至51 B)來純化粗產物(120 mg),得到呈白色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(氧雜環己烷-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(55 mg,45.12%)。LCMS: m/z (ESI), [M+H]
+
= 365.2。
1
H NMR (300 MHz, DMSO-
d6
) δ 1.95 (4H, t), 2.42 (3H, d), 3.14-3.30 (1H, m), 3.47 (2H, d), 3.93 (2H, d), 4.04 (3H, s), 7.71 (1H, t), 8.37 (1H, s), 8.84 (1H, s), 9.15 (1H, s), 9.34 (1H, s)
實例 2
製備1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例2)及3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺.(實例6)
步驟1. 3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺.(實例6)
在氮氣氛圍下在100℃下將Cs
2
CO
3
(682.34 mg,2.094 mmol,2.5當量)、XantPhos(96.94 mg,0.168 mmol,0.2當量)、Pd(OAc)
2
(37.61 mg,0.168 mmol,0.2當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(147.95 mg,1.005 mmol,1.2當量)和5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基吡唑並[4,3-d]嘧啶(210.00 mg,0.838 mmol,1.00當量)於二惡烷(6.00 mL)中的混合物攪拌隔夜。可通過LCMS來檢測所需產物。過濾所得混合物,用DCM(3×50 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至36 B)來純化粗產物(170 mg),得到呈黃色固體的3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(100 mg,57.65%)。LCMS: m/z (ESI), [M+H]
+
= 362.2。
1
H NMR (300 MHz, DMSO-
d6
) δ 2.30 (3H, d), 2.58 (2H, s), 3.83 (2H, t), 4.06 (3H, s), 4.25 (2H, d), 7.07-7.12 (1H, m), 7.43 (1H, d), 7.49 (1H, d), 7.84 (1H, t), 8.81 (2H, d), 9.14 (1H, s)
步驟2. 1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-3-(氧雜環己烷-4-基)吡唑並[4,3-d]嘧啶-5-胺.(實例2)
在氫氣氛圍下在40℃下將Pd/C(70.67 mg,0.664 mmol,3.00當量)和3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(80.00 mg,0.221 mmol,1.00當量)於MeOH(20.00 mL)中的混合物攪拌3小時。可通過LCMS來檢測所需產物。過濾所得混合物,用MeOH(3×10 mL)洗滌濾餅。減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內22 B至33 B;RT1:6.63)來純化粗產物(50 mg),得到呈黃色固體的1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-3-(氧雜環己烷-4-基)吡唑並[4,3-d]嘧啶-5-胺(20 mg,24.61%)。LCMS: m/z (ESI), [M+H]
+
= 364.2。
1
H NMR (300 MHz, DMSO-
d6
) δ 1.96 (4H, d), 2.28 (3H, d), 3.21 (1H, t), 3.47 (2H, d), 3.93 (2H, d), 4.03 (3H, s), 7.42 (1H, q), 7.49 (1H, d), 7.81 (1H, t), 8.69 (1H, s), 8.84 (1H, s), 9.09 (1H, s)
實例 3
製備7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-5H-吡咯並[3,2-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例3)
步驟1. 2-氯-7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-5H-吡咯並[3,2-d]嘧啶
在氮氣氛圍下在80℃下將2-氯-7-碘-5-甲基吡咯並[3,2-d]嘧啶(300.00 mg,1.022 mmol,1.00當量)、2-(3,6-二氫-2H-吡喃-4-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(279.16 mg,1.329 mmol,1.30當量)、Pd(dppf)Cl
2
(149.59 mg,0.204 mmol,0.2當量)和K
2
CO
3
(423.81 mg,3.067 mmol,3當量)於二惡烷(6.00 mL)和H
2
O(1.20 mL)中的混合物攪拌3小時。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:3)來純化殘餘物,得到呈棕色固體的2-氯-7-(3,6-二氫-2H-吡喃-4-基)-5-甲基吡咯並[3,2-d]嘧啶(196 mg,76.79%)。LCMS: m/z (ESI), [M+H]
+
= 250.2。
步驟2. 7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡咯並[3,2-d]嘧啶-2-胺
在氮氣氛圍下在100℃下將2-氯-7-(3,6-二氫-2H-吡喃-4-基)-5-甲基吡咯並[3,2-d]嘧啶(196.00 mg,0.785 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(139.56 mg,0.942 mmol,1.20當量)、Pd(AcO)
2
(35.25 mg,0.157 mmol,0.20當量)、XantPhos(136.25 mg,0.235 mmol,0.30當量)和Cs
2
CO
3
(639.37 mg,1.962 mmol,2.50當量)於二惡烷(3.00 mL)中的混合物攪拌3小時。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 10:1)來純化殘餘物,得到呈黃色固體的7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡咯並[3,2-d]嘧啶-2-胺(170 mg,59.93%)。LCMS: m/z (ESI), [M+H]
+
= 362.3。
步驟3. 7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-5H-吡咯並[3,2-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例3)
在氫氣氛圍下在室溫下將7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡咯並[3,2-d]嘧啶-2-胺(170.00 mg,0.470 mmol,1.00當量)和Pd/C (250.29 mg,2.352 mmol,5.00當量)於MeOH(20.00 mL)和THF(50.00 mL)中的混合物攪拌隔夜。過濾所得混合物,用MeOH(5×30 mL)洗滌濾餅。減壓濃縮濾液,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:25 mL/min;梯度:7 min內30 B至45 B;RT1:6.02)來純化粗產物(100 mg),得到呈白色固體的5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-7-(氧雜環己烷-4-基)吡咯並[3,2-d]嘧啶-2-胺(29 mg,16.96%)。LCMS: m/z (ESI), [M+H]
+
= 364.3。
1
H NMR (300 MHz, DMSO-d
6
) δ 1.76 (2H, d), 1.88 - 2.01 (2H, m), 2.46 (3H, d), 3.02 (1H, t), 3.47 (2H, td), 3.80 (3H, s), 3.88 - 4.05 (2H, m), 7.51 (1H, s), 7.61 - 7.75 (1H, m), 8.33 (2H, d), 8.74 (1H, s), 9.49 (1H, s)
實例 4
製備3-異丙基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例4)
步驟1. 5-氯-1-甲基-3-(丙-1-烯-2-基)-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將Pd(dppf)Cl
2
(124.24 mg,0.170 mmol,0.2當量)、K
2
CO
3
(293.33 mg,2.122 mmol,2.5當量)、4,4,5,5-四甲基-2-(丙-1-烯-2-基)-1,3,2-二氧雜環戊硼烷(213.99 mg,1.273 mmol,1.5當量)和5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(250.00 mg,0.849 mmol,1.00當量)於二惡烷(5.00 mL)和H
2
O(1.00 mL)中的混合物攪拌2小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:3)來純化殘餘物,得到呈粉色固體的5-氯-1-甲基-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶(130 mg,73.39%)。LCMS: m/z (ESI), [M+H]
+
= 209.6。
1
H NMR (300 MHz, DMSO-
d6
) δ 2.22 (3H, t), 4.18 (3H, s), 5.50 (1H, p), 6.43 (1H, d), 9.45 (1H, s)
步驟2. 1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(丙-1-烯-2-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs
2
CO
3
(468.47 mg,1.438 mmol,2.5當量)、XantPhos(66.56 mg,0.115 mmol,0.2當量)、Pd(AcO)
2
(25.82 mg,0.115 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(127.82 mg,0.863 mmol,1.50當量)和5-氯-1-甲基-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶(120.00 mg,0.575 mmol,1.00 當量)於二惡烷(4.00 mL)中的混合物攪拌3小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 15:1)來純化殘餘物,得到呈黃色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶-5-胺(90 mg,48.85%)。LCMS: m/z (ESI), [M+H]
+
= 321.3。
1
H NMR (300 MHz, DMSO-
d6
) δ 2.18 (3H, s), 2.45 (3H, d), 4.09 (3H, d), 5.33 (1H, d), 6.35 (1H, d), 7.70-7.78 (1H, m), 8.38 (1H, s), 8.96 (1H, s), 9.22 (1H, s), 9.37 (1H, s)
步驟3. 3-異丙基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例4)
在氫氣氛圍下在室溫下將Pd/C(89.69 mg,0.843 mmol,3當量)和1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶-5-胺(90.00 mg,0.281 mmol,1.00當量)於MeOH(10.00 mL)中的混合物攪拌隔夜。可通過LCMS來檢測所需產物。過濾所得混合物,用MeOH(4×50 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH
2
Cl
2
: MeOH 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:MeOH;流動速率:25 mL/min;梯度:7 min內58 B至70 B;RT1 5.57)來純化粗產物(80 mg),得到呈白色固體的3-異丙基-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(50 mg,54.66%)。LCMS: m/z (ESI), [M+H]
+
= 323.2。
1
H NMR (300 MHz, DMSO-
d6
) δ 1.39 (6H, d), 2.45 (3H, d), 3.18-3.34 (1H, m), 4.04 (3H, s), 7.69-7.75 (1H, m), 8.37 (1H, s), 8.79 (1H, s), 9.16 (1H, s), 9.44 (1H, s)
實例 5
製備3-環己基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例5)
步驟1. 5-氯-3-(環己-1-烯-1-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將含K
2
CO
3
(293.33 mg,2.122 mmol,2.50當量)、2-(環己-1-烯-1-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(265.01 mg,1.273 mmol,1.50當量)和5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(250.00 mg,0.849 mmol,1.00當量)的二惡烷(5.00 mL)和H
2
O(1.00 mL)攪拌2小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:2)來純化殘餘物,得到呈粉色固體的5-氯-3-(環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(150 mg,71.04%)。LCMS: m/z (ESI), [M+H]
+
= 249.3。
1
H NMR (300 MHz, DMSO-d
6
) δ 1.61 - 1.80 (4H, m), 2.29 (2H, s), 2.54 (2H, s), 4.14 (3H, s), 7.17 - 7.27 (1H, m), 9.40 (1H, s)
步驟2. 3-(環己-1-烯-1-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs
2
CO
3
(458.51 mg,1.407 mmol,2.5當量)、XantPhos(65.14 mg,0.113 mmol,0.2當量)、Pd(AcO)
2
(25.28 mg,0.113 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(125.11 mg,0.844 mmol,1.5當量)和5-氯-3-(環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(140.00 mg,0.563 mmol,1.00當量)於二惡烷(4.00 mL)中的混合物攪拌3小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 15:1)來純化殘餘物,得到呈黃色固體的3-(環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(140 mg,69.00%)。LCMS: m/z (ESI), [M+H]
+
= 361.3。
1
H NMR (300 MHz, DMSO-d
6
) δ 1.61 - 1.83 (4H, m), 2.17 - 2.33 (4H, m), 2.46 (3H, d), 4.06 (3H, s), 7.21 (1H, s), 7.73 (1H, s), 8.38 (1H, s), 8.88 (1H, s), 9.19 (1H, s), 9.46 (1H, s)
步驟3. 3-環己基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例5)
在氫氣氛圍下在室溫下將3-(環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(160.00 mg,0.444 mmol,1.00當量)和Pd/C(236.21 mg,2.220 mmol,5.00當量)於THF(40.00 mL)和MeOH (80.00 mL)中的溶液攪拌3天。通過過濾收集沉澱的固體並用MeOH(5×30 mL)洗滌。減壓濃縮所得混合物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內40 B至50 B;RT1:6.55)來純化粗產物(150 mg),得到呈白色固體的3-環己基-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(17.41 mg,10.82%)。LCMS: m/z (ESI), [M+H]
+
= 363.2。
1
H NMR (300 MHz, DMSO-d
6
) δ 1.33 (3H, d), 1.76 (5H, d), 2.00 (2H, d), 2.46 (3H, d), 2.88 - 3.05 (1H, m), 4.04 (3H, s), 7.72 (1H, t), 8.38 (1H, s), 8.78 (1H, s), 9.15 (1H, s), 9.51 (1H, s)
實例 8
製備3-((1r,4r)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例7)及3-((1s,4s)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例8)
步驟1. 5-氯-3-(4-甲氧基環己-1-烯-1-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將K
2
CO
3
(234.66 mg,1.698 mmol,2.5當量)、Pd(dppf)Cl
2
CH
2
Cl
2
(110.93 mg,0.136 mmol,0.2當量)、2-(4-甲氧基環己-1-烯-1-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(194.07 mg,0.815 mmol,1.2當量)和5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(200.00 mg,0.679 mmol,1.00當量)於二惡烷(5.00 mL)和H
2
O(1.00 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:2)來純化殘餘物,得到呈白色固體的5-氯-3-(4-甲氧基環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(175 mg,92.44%)。LCMS: m/z (ESI), [M+H]
+
= 279.3。
1
H NMR (300 MHz, CDCl
3
) δ 1.89 (2H, d), 2.11 (1H, m), 2.33 (1H, t), 2.66 (1H, m), 2.88 (1H, d), 3.43 (3H, s), 3.60 (1H, d), 4.12 (3H, s), 7.27 (1H, d), 8.89 (1H, s)
步驟2. 3-(4-甲氧基環己-1-烯-1-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs
2
CO
3
(496.78 mg,1.525 mmol,2.5當量)、XantPhos(70.58 mg,0.122 mmol,0.2當量)、Pd(AcO)
2
(27.39 mg,0.122 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(99.40 mg,0.671 mmol,1.1當量)和5-氯-3-(4-甲氧基環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(170.00 mg,0.610 mmol,1.00當量)於二惡烷(5.00 mL)中的混合物攪拌3小時。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物,得到呈黃色固體的3-(4-甲氧基環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(100 mg,41.99%)。LCMS: m/z (ESI), [M+H]
+
= 392.4。
1
H NMR (300 MHz, DMSO-
d6
) δ 1.67 (1H, d), 2.00 (1H, s), 2.27 (1H, m), 2.46 (3H, d), 2.72 (1H, d), 3.17 (2H, d), 3.33 (3H, s), 3.53 (1H, s), 4.07 (3H, s), 7.08 (1H, s), 7.73 (1H, s), 8.39 (1H, s), 8.91 (1H, s), 9.19 (1H, s), 9.43 (1H, s)
步驟3. 3-((1r,4r)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例7)及3-((1s,4s)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例8)
在氫氣氛圍下在室溫下將Pd/C(65.41 mg,0.615 mmol,3當量)和3-(4-甲氧基環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(80.00 mg,0.205 mmol,1.00當量)於MeOH(150.00 mL)和THF(80.00 mL)中的混合物攪拌隔夜。過濾所得混合物,用MeOH(3×50 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:25 mL/min;梯度:7 min內37 B至41 B;RT1:5.30/5.92)來純化粗產物(80 mg),得到呈白色固體的3-((1r,4r)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例7,60 mg,22.73%)。LCMS: m/z (ESI), [M+H]
+
= 393.3。
1
H NMR (400 MHz, DMSO-
d6
) δ 1.28 (2H, q), 1.83 (2H, q), 2.09 (4H, t), 2.43 (3H, d), 2.95 (1H, t), 3.30 (4H, m), 4.04 (3H, s), 7.73 (1H, s), 8.38 (1H, s), 8.81 (1H, s), 9.16 (1H, s), 9.51 (1H, s);
呈白色固體的3-((1s,4s)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例8,10 mg,12.19%)。LCMS: m/z (ESI), [M+H]
+
= 393.2。
1
H NMR (300 MHz, DMSO-
d6
) δ 1.57 (2H, t), 1.76 (2H, m), 1.86 (2H, m), 2.04 (2H, q), 2.43 (3H, d), 3.05 (1H, m), 3.20 (3H, s), 3.42 (1H, s), 4.04 (3H, s), 7.72 (1H, s), 8.37 (1H, s), 8.80 (1H, s), 9.14 (1H, s), 9.34 (1H, s)。
實例 9
製備3-(4,4-二氟環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例9)
步驟1. 5-氯-3-(4,4-二氟環己-1-烯-1-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(200.00 mg,0.679 mmol,1.00當量)、2-(4,4-二氟環己-1-烯-1-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(198.93 mg,0.815 mmol,1.20當量)、Pd(dppf)Cl
2
(99.39 mg,0.136 mmol,0.2當量)和K
2
CO
3
(234.66 mg,1.698 mmol,2.5當量)於二惡烷(3.00 mL)和H
2
O(0.60 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(PE/EtOAc 2:3)來純化殘餘物,得到呈黃色固體的5-氯-3-(4,4-二氟環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(100 mg,51.72%)。LCMS: m/z (ESI), [M+H]
+
= 285.3。
步驟2. 3-(4,4-二氟環己-1-烯-1-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將5-氯-3-(4,4-二氟環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(100.00 mg,0.351 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(62.45 mg,0.421 mmol,1.20當量)、Pd(OAc)
2
(15.77 mg,0.070 mmol,0.20當量)、XantPhos(60.97 mg,0.105 mmol,0.30當量)和Cs
2
CO
3
(286.12 mg,0.878 mmol,2.50當量)於二惡烷(2.50 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物,得到呈白色固體的3-(4,4-二氟環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(110 mg,79.00%)。LCMS: m/z (ESI), [M+H]
+
= 397.3。
步驟3. 3-(4,4-二氟環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例9)
在氫氣氛圍下在30℃下將3-(4,4-二氟環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(130.00 mg,0.328 mmol,1.00 當量)和Pd/C(174.50 mg,1.640 mmol,5.00當量)於MeOH(60.00 mL)和THF(30.00 mL)中的混合物攪拌5個小時。過濾所得混合物,用CH
2
Cl
2
(3×40 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內34 B至48 B;RT1:5.97)來純化粗產物(70 mg),得到呈粉色固體的3-(4,4-二氟環己基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(17 mg,13.01%)。LCMS: m/z (ESI), [M+H]
+
= 399.3。
1
H NMR (300 MHz, MeOD-d
4
) δ 1.80-2.22 (8H, m), 2.49 (3H, d), 3.22 (1H, s), 4.03 (3H, s), 7.58 (1H, t), 8.27 (1H, s), 8.98 (1H, s), 9.62 (1H, s)
實例 10
製備1-(甲基-d
3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例10)
步驟1. 5-氯-3-碘-1-(甲基-d
3
)-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在0℃下將Cs
2
CO
3
(1045.60 mg,3.209 mmol,3當量)、CD
3
I(775.31 mg,5.349 mmol,5當量)和5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶(300.00 mg,1.070 mmol,1.00當量)於DMF(6.00 mL)中的混合物攪拌1 h。可通過LCMS來檢測所需產物。用水(50 mL)稀釋所得混合物。用EtOAc(3×50 mL)萃取所得混合物。將合併的有機層用飽和鹽水(3×50 mL)和Na
2
S
2
O
3
(3×50 mL)洗滌且經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液,得到呈粉色固體的5-氯-3-碘-1-(甲基-d
3
)-1H-吡唑並[4,3-d]嘧啶(260 mg,81.70%)。LCMS: m/z (ESI), [M+H]
+
= 298.0。
1
H NMR (400 MHz, DMSO-
d6
) δ 9.45 (1H, s).
步驟2. 5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d
3
)-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將K
2
CO
3
(313.58 mg,2.269 mmol,2.5當量)、Pd(dppf)Cl
2
CH
2
Cl
2
(148.23 mg,0.182 mmol,0.2當量)、2-(3,6-二氫-2H-吡喃-4-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(228.79 mg,1.089 mmol,1.2當量)和5-氯-3-碘-1-(甲基-d
3
)-1H-吡唑並[4,3-d]嘧啶(270.00 mg,0.908 mmol,1.00當量)於二惡烷(5.00 mL)和水(1.00 mL)中的混合物攪拌2小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(己烷/EtOAc 1:2)來純化殘餘物,得到呈粉色固體的5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d
3
)-1H-吡唑並[4,3-d]嘧啶(100 mg,43.43%)。LCMS: m/z (ESI), [M+H]
+
= 254.2。
1
H NMR (400 MHz, DMSO-
d6
) δ 2.62 (2H, m), 3.86 (2H, t), 4.33 (2H, q), 7.18 (1H, t), 9.43 (1H, s)
步驟3. 3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d
3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs
2
CO
3
(385.28 mg,1.182 mmol,3當量)、XantPhos(45.61 mg,0.079 mmol,0.2當量)、Pd(OAc)
2
(17.70 mg,0.079 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(70.08 mg,0.473 mmol,1.20當量)和5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d
3
)-1H-吡唑並[4,3-d]嘧啶(100.00 mg,0.394 mmol,1.00當量)於二惡烷(3.00 mL)中的混合物攪拌2 h。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物,得到呈黃色固體的3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d
3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(100 mg,69.43%)。LCMS: m/z (ESI), [M+H]
+
= 366.3。
1
H NMR (400 MHz, DMSO-
d6
) δ 2.44 (2H, d), 2.61 (3H, s), 3.17 (1H, d), 3.85 (2H, t), 4.26 (2H, q), 7.72 (1H, s), 8.38 (1H, s), 8.94 (1H, s), 9.20 (1H, s), 9.37 (1H, s)。
步驟4. 1-(甲基-d
3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺.(實例10)
在氫氣氛圍下在35℃下將Pd/C(87.37 mg,0.821 mmol,3當量)和3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d
3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(100.00 mg,0.274 mmol,1.00當量)於MeOH (10.00 mL)和THF(10.00 mL)中的混合物攪拌2 h。可通過LCMS來檢測所需產物。過濾所得混合物,用MeOH(3×30 mL)和DCM(3×30 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內15 B至35 B;RT1:6.4)來純化粗產物(40 mg),得到呈白色固體的1-(甲基-d
3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(10 mg,9.85%)。LCMS: m/z (ESI), [M+H]
+
= 368.2。
1
H NMR (400 MHz, DMSO-
d6
) δ 1.94 (4H, d), 2.43 (3H, d), 3.24 (1H, d), 3.48 (2H, m), 3.93 (2H, m), 7.71 (1H, s), 8.37 (1H, s), 8.81 (1H, s), 9.15 (1H, s), 9.33 (1H, s).
實例 11/12
製備1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1s,4s)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例11)及1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1r,4r)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例12)
步驟1. 4-(5-氯-1-甲基-1H-吡唑並[4,3-d]嘧啶-3-基)環己-3-烯-1-醇
在室溫下向40 mL小瓶中添加5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(500.00 mg,1.698 mmol,1.00當量)和4-(4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷-2-基)環己-3-烯-1-醇(570.78 mg,2.547 mmol,1.50當量)、Pd(dppf)Cl
2
(248.47 mg,0.340 mmol,0.20當量)、K
2
CO
3
(938.64 mg,6.792 mmol,4當量)、二惡烷(10.00 mL)以及H
2
O(2.00 mL)。隨後在氮氣氛圍下在80℃下將混合物攪拌3h。真空濃縮所得混合物。通過製備型TLC(PE:EA = 1:4)來純化殘餘物,得到呈黃色固體的4-[5-氯-1-甲基吡唑並[4,3-d]嘧啶-3-基]環己-3-烯-1-醇(253 mg,56.29%)。LCMS: m/z (ESI), [M+H]
+
= 265.2。
步驟2. 5-氯-3-(4-(二氟甲氧基)環己-1-烯基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在50℃下向40 mL小瓶中添加4-[5-氯-1-甲基吡唑並[4,3-d]嘧啶-3-基]環己-3-烯-1-醇(253.00 mg,0.956 mmol,1.00當量)、CuI(63.71 mg,0.335 mmol,0.35當量)、MeCN(10.00 mL)且向混合物中逐滴添加2,2-二氟-2-(氟磺醯基)乙酸(510.61 mg,2.867 mmol,3.00當量)。隨後在空氣氛圍下在50℃下將混合物攪拌3h。用水(10 mL)稀釋所得混合物。用DCM(3×30 mL)萃取水層。用DCM(5 mL)稀釋所得混合物。過濾所得混合物,用DCM(10 mL*3)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(PE/EtOAc 1:1)來純化殘餘物,得到呈黃色固體的5-氯-3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基吡唑並[4,3-d]嘧啶(180 mg,47.87%)。LCMS: m/z (ESI), [M+H]
+
= 315.2。
步驟3. 3-(4-(二氟甲氧基)環己-1-烯基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在室溫下向40 mL小瓶中添加5-氯-3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基吡唑並[4,3-d]嘧啶(180.00 mg,0.572 mmol,1.00當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(101.69 mg,0.686 mmol,1.20當量)、Pd(OAc)
2
(38.52 mg,0.172 mmol,0.30當量)、XantPhos(99.28 mg,0.172 mmol,0.30 當量)、Cs
2
CO
3
(559.05 mg,1.716 mmol,3.00當量)以及二惡烷(10.00 mL)。隨後在氮氣氛圍下在70℃下將混合物攪拌3h。真空濃縮所得混合物。通過製備型TLC(DCM:MeOH 15:1)來純化殘餘物,得到呈黃色固體的3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(135 mg,55.35%)。LCMS: m/z (ESI), [M+H]
+
= 427.1。
1
H NMR (400 MHz, CDCl
3
) δ1.97 - 2.23 (3H, m), 2.55 (4H, s), 2.68 - 2.87 (2H, m), 2.98 (1H, d), 4.11 (3H, s), 4.54 - 4.62 (1H,m), 6.35 (1H, t), 7.00 (1H, s), 7.61 (1H, s), 8.29 (1H,s), 8.82 (1H, s), 9.90 (1H, s)
步驟4. 製備1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1s,4s)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例11)及1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1r,4r)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例12)
在空氣氛圍下向3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(135.00 mg,0.317 mmol,1.00當量)於MeOH(20 mL)中的攪拌混合物中添加Pd/C(168.45 mg,1.583 mmol,5.00當量)。在氫氣氛圍下在40℃下將所得混合物攪拌4h。過濾所得混合物,且用DCM(8×100 mL)洗滌濾餅。減壓濃縮濾液,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內34 B至54 B;RT1:5.93)來純化粗產物,得到固體。隨後,通過製備型HPLC利用以下條件(柱:CHIRALPAK IG,2*25cm,5um;移動相A:Hex:DCM=3:1,移動相B:EtOH;流動速率:20 mL/min;梯度:15 min內10 B至10 B;RT1:10;RT2:11)來純化產物,得到呈白色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1s,4s)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例11,15 mg,50.00%)和呈白色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1r,4r)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例12,5 mg,16.67%)。
(實例11) LCMS: m/z (ESI), [M+H]
+
= 429.3。
1
H NMR (300 MHz, DMSO-d
6
) δ1.87 (2H, d), 1.97 (4H, d), 2.09 (2H, d), 2.43 (3H, s), 3.07 (1H, d), 4.04 (3H, s), 4.37 (1H,s), 6.69 (1H, t), 7.70 (1H, s), 8.36 (1H, s), 8.79 (1H,s), 9.14 (1H, s), 9.33 (1H, s);
(實例12) LCMS: m/z (ESI), [M+H]
+
= 429.3。
1
H NMR (300 MHz, MeOD-d
4
) δ1.59-1.71 (2H, m), 1.95-2.09 (2H, m), 2.15-2.20 (4H, m), 2.55 (3H,s), 3.05-3.14 (1H, m), 4.09 (3H, s), 4.20-4.27 (1H, m), 6.71 (1H, t), 7.64 (1H, s), 8.31 (1H,s), 9.03 (1H, s), 9.85 (1H, s)。
實例 13
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例13)
步驟1. 6-氯-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在室溫下向1-(2,4-二氯嘧啶-5-基)乙酮(500.00 mg,2.618 mmol,1.00當量)和DIPEA(1353.26 mg,10.471 mmol,4.00當量)於THF(5.00 mL)中的攪拌混合物中分批添加氧雜環己烷-4-基肼(364.89 mg,3.141 mmol,1.20當量)。並且在氮氣氛圍下在室溫下攪拌混合物2 h。用EtOAc(3×50 mL)萃取所得混合物。用水(3×10 mL)洗滌合併的有機層,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(PE/EtOAc 2:1)來純化殘餘物,得到呈黃色固體的6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(350 mg,52.91%)。LCMS: m/z (ESI), [M+H]
+
= 253.2
步驟2. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例13)
在氮氣氛圍下在室溫下向6-氯-3-甲基-1-(氧雜環己烷-4-基)-1H-吡唑並[3,4-d]嘧啶(200 mg,0.791 mmol,1當量)、Cs
2
CO
3
(644.68 mg,1.979 mmol,2.5當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(140.72 mg,0.950 mmol,1.2當量)於二惡烷(20 mL)中的攪拌混合物中分批添加BrettPhos Pd G
3
(143.49 mg,0.158 mmol,0.2當量)。並且在氮氣氛圍下在100℃下攪拌混合物3 h。用EtOAc(3×50 mL)萃取所得混合物。用水(3×50 mL)洗滌合併的有機層,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:X select CSH OBD柱,30×150 mm 5 um;移動相A:水(0.05% TFA),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內18% B至29% B;t
R
:6.30 min)來純化粗產物,得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-(氧雜環己烷-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(74 mg,25.66%)。LCMS: m/z (ESI), [M+H]
+
= 365.3
1
H NMR (DMSO-d
6
, 300 MHz) δ 1.7-1.9 (2H, m), 2.0-2.2 (2H, m), 2.4-2.5 (6H, m), 3.4 (2H, td), 3.9-4.1 (2H, m), 4.5-4.7 (1H, m), 7.8 (1H, s), 8.6 (1H, s), 8.9 (1H, s), 9.3 (1H, s), 9.3 (1H, s)
實例 14
製備3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例14)
步驟1. 6-氯-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在0℃下將1-(2,4-二氯嘧啶-5-基)乙酮(250.00 mg,1.309 mmol,1.00當量)和氧雜環己烷-4-基肼(182.45 mg,1.571 mmol,1.20當量)、DIPEA(338.32 mg,2.618 mmol,2.00當量)於THF(10.00 mL)中的溶液攪拌2h。用EtOAc(20 mL×3)萃取所得混合物。用鹽水(10 mL×3)洗滌合併的有機層,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(PE/EtOAc 5:1)來純化殘餘物,得到呈白色固體的6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(230 mg,69.54%)。LCMS: m/z (ESI), [M+H]
+
= 253.2。
步驟2. 3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例14)
在氮氣氛圍下在100℃下將7-甲基咪唑[1,2-a]吡啶-6-胺(139.78 mg,0.950 mmol,1.50當量)和6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(160 mg,0.633 mmol,1.00當量)、BrettPhos Pd G
3
(57.40 mg,0.063 mmol,0.10當量)、Cs
2
CO
3
(412.59 mg,1.266 mmol,2.00當量)於二惡烷(5.00 mL)中的混合物攪拌3h。通過矽膠柱色譜純化殘餘物,用CH
2
Cl
2
/MeOH(10:1)洗脫,得到粗產物。通過製備型HPLC來純化粗產物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內17 B至37 B;RT1:6.75)來純化粗產物,得到呈灰白色固體的3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-胺(140 mg,60.84%)。LCMS: m/z (ESI), [M+H]
+
= 364.1,
1
H NMR (DMSO-d
6
, 300 MHz) δ1.79 (2H, d), 2.02-2.18 (2H, m), 2.23 (3H, d), 2.42 (3H, s), 3.44 (2H, dd), 3.94 (2H, dd), 4.62 (1H, dd), 7.42 (1H, s), 7.48 (1H, d), 7.84 (1H, s), 8.69 (1H, s), 8.85 (1H, s), 9.04 (1H, s)
實例 15
製備7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-7H-吡咯並[2,3-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例15)
步驟1. 2-氯-5-甲基-7-(四氫-2H-吡喃-4-基)-7H-吡咯並[2,3-d]嘧啶
在氮氣氛圍下在120℃下將2-氯-5-甲基-7H-吡咯並[2,3-d]嘧啶(400.00 mg,2.387 mmol,1.00當量)、4-溴惡烷(3.94 g,23.874 mmol,10.00當量)和K
2
CO
3
(824.62 mg,5.967 mmol,2.50當量)於DMSO(50.00 mL,12.922 mmol)中的混合物攪拌隔夜。用水(150 mL)稀釋所得混合物。用EtOAc(3×50 mL)萃取所得混合物。用鹽水(3×30 mL)洗滌合併的有機層,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(CH
2
Cl
2
/MeOH = 10:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內28 B至48 B;RT1:5.80)來純化粗產物(70 mg),得到呈黃色固體的2-氯-5-甲基-7-(氧雜環己烷-4-基)吡咯並[2,3-d]嘧啶(40 mg,6.66%)。
步驟2. 7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-7H-吡咯並[2,3-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例15)
在氮氣氛圍下在100℃下將2-氯-5-甲基-7-(氧雜環己烷-4-基)吡咯並[2,3-d]嘧啶(30.00 mg,0.119 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(21.19 mg,0.143 mmol,1.20當量)、Pd(AcO)
2
(5.35 mg,0.024 mmol,0.20當量)、XantPhos(20.69 mg,0.036 mmol,0.30當量)以及Cs
2
CO
3
(97.08 mg,0.298 mmol,2.50當量)於二惡烷(1.50 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至46 B;RT1:6.37)來純化粗產物(40 mg),得到呈白色固體的5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-7-(氧雜環己烷-4-基)吡咯並[2,3-d]嘧啶-2-胺(6 mg,13.85%)。LCMS: m/z (ESI), [M+H]
+
= 364.3。
1
H NMR (300 MHz, MeOD-d
4
) δ1.79-1.91 (2H, m), 1.95-2.14 (2H, m), 2.24 (3H, d), 2.43 (3H, d), 3.47 (2H, td), 3.93-4.05 (2H, m), 4.55-4.72 (1H, m), 7.18 (1H, d), 7.58-7.74 (1H, m), 8.37 (1H, s), 8.62 (2H, d), 9.32 (1H, s)
實例 16
製備N-(7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例16)
步驟1. N-(7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例16)
在氮氣氛圍下在100℃下將7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-胺(50.00 mg,0.297 mmol,1.00當量)、6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(74.95 mg,0.297 mmol,1.00當量)、Pd(AcO)
2
(13.32 mg,0.059 mmol,0.2當量)、XantPhos(51.48 mg,0.089 mmol,0.3當量)以及Cs
2
CO
3
(241.59 mg,0.741 mmol,2.5當量)於二惡烷(4.00 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:25 mL/min;梯度:7 min內30 B至40 B;RT1:6.62)來純化粗產物(90 mg),得到呈白色固體的N-[7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-胺(32.77mg,28.71%)。LCMS: m/z (ESI), [M+H]
+
= 385.2。
1
H NMR (300 MHz, DMSO-d
6
) δ1.84 (2H, d), 2.13 (2H, dd), 2.46 (3H, s), 3.39-3.57 (2H, m), 3.97 (2H, dd), 4.54-4.77 (1H, m), 8.23 (1H, s), 8.57 (1H, s), 8.93 (1H, s), 9.36 (2H, d)。
實例 17
製備N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例17)
步驟1. 6-氯-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在0℃下向6-氯-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶(340.00 mg,1.528 mmol,1.00當量)、氧雜環己烷-4-醇(624.11 mg,6.111 mmol,4.00當量)以及PPh
3
(1442.48 mg,5.500 mmol,3.60當量)於THF(2.50 mL)中的攪拌混合物中逐滴添加DIAD(1112.07 mg,5.500 mmol,3.60當量)。用EtOAc(3×20 mL)萃取所得混合物,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(CH
2
Cl
2
/MeOH = 10:1)來純化殘餘物,得到呈白色固體的6-氯-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶(370 mg,78.98%)。LCMS: m/z (ESI), [M+H]
+
= 307.3。
步驟2. N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例17)
在氮氣氛圍下在70℃下將6-氯-1-(氧雜環己烷-4-基)-3-(三氟甲基)吡唑並[3,4-d]嘧啶(70.00 mg,0.228 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(33.82 mg,0.228 mmol,1.00當量)、Pd(AcO)
2
(10.25 mg,0.046 mmol,0.20當量)、XantPhos(39.62 mg,0.068 mmol,0.30當量)以及Cs
2
CO
3
(185.93 mg,0.571 mmol,2.50當量)於二惡烷(3.00 mL)中的混合物攪拌3小時。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內30 B至50 B;RT1:6.67)來純化粗產物(90 mg),得到呈白色固體的N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-(氧雜環己烷-4-基)-3-(三氟甲基)吡唑並[3,4-d]嘧啶-6-胺(54.44 mg,53.86%)。LCMS: m/z (ESI), [M+H]
+
= 419.2。
1
H NMR (300 MHz, DMSO-d
6
) δ 1.77-2.01 (2H, m), 2.01-2.24 (2H, m), 2.39 (3H, d), 3.40-3.59 (2H, m), 3.98 (2H, d), 4.82 (1H, t), 7.78 (1H, d), 8.44 (1H, s), 9.13 (2H, d), 9.69 (1H, s)
實例 18
製備1-(4-甲氧基苯甲基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例18)
步驟1. 6-氯-1-(4-甲氧基苯甲基)-3-甲基-1H-吡唑並[3,4-d]嘧啶
在空氣氛圍下在0℃下向1-(2,4-二氯嘧啶-5-基)乙酮(640.00 mg,3.351 mmol,1.00當量)和DIPEA(433.04 mg,3.351 mmol,1.00當量)於THF中的攪拌混合物中逐滴添加[(4-甲氧基苯基)甲基]肼(764.93 mg,5.026 mmol,1.50當量)。在空氣氛圍下在室溫下攪拌所得混合物2h。減壓濃縮所得混合物。通過製備型TLC(PE:EA 1:1)來純化殘餘物,得到呈黃色固體的6-氯-1-[(4-甲氧基苯基)甲基]-3-甲基吡唑並[3,4-d]嘧啶(623 mg,64.40%)。LCMS: m/z (ESI), [M+H]
+
= 289.2。
1
H NMR (300 MHz, CDCl
3
) δ2.58 (3H, s), 3.77 (3H, s), 5.47 (2H, s), 6.82-6.87 (2H, m), 7.30-7.35 (2H, m), 8.90 (1H, s)
步驟2. 1-(4-甲氧基苯甲基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例18)
在空氣氛圍下在室溫下向6-氯-1-[(4-甲氧基苯基)甲基]-3-甲基吡唑並[3,4-d]嘧啶(100.00 mg,0.346 mmol,1.00當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(76.97 mg,0.519 mmol,1.50當量)於二惡烷(5 mL)中的攪拌混合物中逐滴添加Cs
2
CO
3
(338.53 mg,1.039 mmol,3.00當量)和XantPhos(40.08 mg,0.069 mmol,0.20當量)、Pd(AcO)
2
(15.55 mg,0.069 mmol,0.20當量)。在氮氣氛圍下在80℃下攪拌所得混合物2h。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 10:1)來純化殘餘物,得到黃色固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至46 B;RT1:7.07)來純化粗產物(70mg),得到呈白色固體的1-[(4-甲氧基苯基)甲基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(20 mg,28.57%)。LCMS: m/z (ESI), [M+H]
+
= 401.2。
1
H NMR (400 MHz, DMSO-d
6
) δ 2.38 (3H, s), 2.44 (3H, s), 3.71 (3H, s), 5.24 (2H, s), 6.85-6.88 (2H, m), 7.18 (2H, t), 7.77 (1H, s), 8.43 (1H, s), 8.95 (1H, s), 9.22 (2H, d)
實例 20
製備1-環己基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例20)
步驟1. 6-氯-1-環己基-3-甲基-1H-吡唑並[3,4-d]嘧啶
在空氣氛圍下在0℃下向1-(2,4-二氯嘧啶-5-基)乙酮(180.00 mg,0.942 mmol,1.00當量)和DIPEA(487.17 mg,3.769 mmol,4.00當量)於THF(10 mL)中的攪拌混合物中分批添加環己基鹽酸肼(184.56 mg,1.225 mmol,1.30當量)。在空氣氛圍下在25℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(PE/EA 1:2)來純化殘餘物,得到呈黃色固體的6-氯-1-環己基-3-甲基吡唑並[3,4-d]嘧啶(145 mg,61.37%)。LCMS: m/z (ESI), [M+H]
+
= 251.2。
1
H NMR (300 MHz, CDCl
3
) δ 1.42-1.48 (2H, m), 1.51-1.55 (2H, m), 1.99-2.08 (6H, m), 2.60 (3H, s), 4.66-4.76 (1H, m), 8.90 (1H, s)
步驟2. 1-環己基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例20)
在空氣氛圍下在室溫下向6-氯-1-環己基-3-甲基吡唑並[3,4-d]嘧啶(120.00 mg,0.479 mmol,1.00當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(106.37 mg,0.718 mmol,1.5當量)於二惡烷(6 mL)中的攪拌混合物中分批添加Cs
2
CO
3
(467.81 mg,1.436 mmol,3當量)和XantPhos (55.39 mg,0.096 mmol,0.2當量)、Pd(AcO)
2
(21.49 mg,0.096 mmol,0.2當量)。在氮氣氛圍下在80℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 10:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30*150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:% B;254;220 nm;RT1:6.50)來純化粗產物(80mg),得到呈白色固體的1-環己基-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(60 mg,34.59%)。LCMS: m/z (ESI), [M+H]
+
= 363.2.
1
H NMR (300 MHz, DMSO-d
6
) δ 1.23-1.38 (3H, m), 1.67 (1H, d), 1.81-1.97 (6H, m), 2.41-2.44 (6H, m), 4.36-4.43 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.15 (1H, s), 9.22 (1H, s).
實例 21
製備1-(4,4-二氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例21)
步驟1. 6-氯-1-(4,4-二氟環己基)-3-甲基-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在0℃下將DIPEA(338.32 mg,2.618 mmol,5當量)、(4,4-二氟環己基)肼(86.48 mg,0.576 mmol,1.10當量)以及1-(2,4-二氯嘧啶-5-基)乙酮(100.00 mg,0.524 mmol,1.00當量)於THF(5.00 mL)中的混合物攪拌3小時。減壓濃縮所得混合物。通過製備型TLC(己烷/EtOAc 1:1)來純化殘餘物,得到呈白色固體的6-氯-1-(4,4-二氟環己基)-3-甲基吡唑並[3,4-d]嘧啶(80 mg,53.30%)。LCMS: m/z (ESI), [M+H]
+
= 287.3。
1
H NMR (300 MHz, CDCl
3
) δ 2.03 (4H, d), 2.35 (4H, d), 2.60 (3H, s), 4.85 (1H, t), 8.92 (1H, s)。
步驟2. 1-(4,4-二氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例21)
在氮氣氛圍下在100℃下將Cs
2
CO
3
(227.28 mg,0.698 mmol,2.5當量)、XantPhos(32.29 mg,0.056 mmol,0.2當量)、Pd(AcO)
2
(12.53 mg,0.056 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(45.48 mg,0.307 mmol,1.1當量)以及6-氯-1-(4,4-二氟環己基)-3-甲基吡唑並[3,4-d]嘧啶(80.00 mg,0.279 mmol,1.00當量)於二惡烷(3.00 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內31 B至51 B;RT1:6.30)來純化粗產物(100 mg),得到呈白色固體的1-(4,4-二氟環己基)-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(80 mg,71.96%)。LCMS: m/z (ESI), [M+H]
+
= 399.3。
1
H NMR (300 MHz, DMSO-
d6
) δ 1.99 (3H, s), 2.15 (5H, s), 2.38 (3H, d), 2.45 (3H, s), 4.62 (1H, s), 7.75 (1H, d), 8.42 (1H, s), 8.93 (1H, s), 9.13 (1H, s), 9.22 (1H, s)。
實例 22
製備1-(4,4-二氟環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例22)
步驟1. 1-(4,4-二氟環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例22)
在氮氣氛圍下在80℃下將6-氯-1-(4,4-二氟環己基)-3-甲基吡唑並[3,4-d]嘧啶(67.00 mg,0.234 mmol,1.00當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(68.79 mg,0.467 mmol,2當量)、Pd(AcO)
2
(10.49 mg,0.047 mmol,0.20當量)、XantPhos(40.56 mg,0.070 mmol,0.30當量)以及Cs
2
CO
3
(190.35 mg,0.584 mmol,2.50當量)於二惡烷(5.00 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(10MMOL/L NH4HCO3+ 0.1%NH
3
.H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內38 B至50 B;RT1:5.63)來純化粗產物(50 mg),得到呈白色固體的1-(4,4-二氟環己基)-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(20 mg)。LCMS: m/z (ESI), [M+H]
+
= 398.2。
1
H NMR (300 MHz, DMSO-d
6
) δ 1.95 (3H, d), 2.15 (5H, d), 2.26 (3H, d), 2.44 (3H, s), 4.62 (1H, s), 7.44 (1H, s), 7.50 (1H, d), 7.86 (1H, t), 8.72 (1H, s), 8.88 (1H, s), 9.02 (1H, s)
實例 26
製備1-((1r,4r)-4-甲氧基環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例26)
步驟1. 1-(4-甲氧基苯甲基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在氮氣氛圍下在80℃下將6-氯-1-[(4-甲氧基苯基)甲基]-3-甲基吡唑並[3,4-d]嘧啶(200.00 mg,0.693 mmol,1.00當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(152.92 mg,1.039 mmol,1.50當量)、XantPhos(120.24 mg,0.208 mmol,0.30當量)、Pd(AcO)
2
(31.10 mg,0.139 mmol,0.20當量)以及Cs
2
CO
3
(564.21 mg,1.732 mmol,2.50當量)於二惡烷(3 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 12:1)來純化殘餘物,得到呈黃色固體的1-[(4-甲氧基苯基)甲基]-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(200mg,72.28%)。LCMS: m/z (ESI), [M+H]
+
= 400.3。
步驟2. 3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在空氣氛圍下在80℃下將1-[(4-甲氧基苯基)甲基]-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(190.00 mg,0.476 mmol,1.00當量)和TFA(70.00 mL,613.910 mmol,1981.34當量)的溶液攪拌2天。真空濃縮所得混合物。用DCM(10 mL)稀釋所得混合物。通過過濾用飽和NaHCO
3
(水溶液)將混合物調節到pH 8,並用DCM(2×3 mL)洗滌。收集沉澱的固體,得到3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(160 mg,80.00%)。LCMS: m/z (ESI), [M+H]
+
= 280.2
步驟3. 1-((1r,4r)-4-甲氧基環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例26)
在氮氣氛圍下在0℃下將3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(150.00 mg,0.537 mmol,1.00當量)、4-甲氧基環己-1-醇(174.79 mg,1.343 mmol,2.50當量)以及PPh
3
(422.58 mg,1.611 mmol,3.00當量)於THF(2.50 mL)中的混合物攪拌20 min,隨後添加DIAD(325.78 mg,1.611 mmol,3.00當量),且在70℃下攪拌混合物2小時。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/EtOAc =12:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(10MMOL/L NH
4
HCO
3
+ 0.1%NH
3
.H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:9 min內29 B至39 B;RT1:6.22,7.43)來純化粗產物(60.00 mg),得到呈白色固體的3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1-[(1r,4r)-4-甲氧基環己基]吡唑並[3,4-d]嘧啶-6-胺(16 mg,26.67%)。LCMS: m/z (ESI), [M+H
]+
= 392.2。
1
H NMR (300 MHz, MeOD-d
4
) δ 1.31-1.46 (2H, m), 2.00 (2H, s), 2.03-2.14 (2H, m), 2.21 (2H, d), 2.39 (3H, d), 2.50 (3H, s), 3.23 (1H, d), 3.38 (3H, s), 4.51 (1H, t), 7.46 (1H, s), 7.54 (1H, d), 7.80 (1H, s), 8.82 (2H, s)。
實例 27
製備1-((1s,4s)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例27)及1-((1r,4r)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例28)
步驟1. 4-氟環己-1-醇
在0℃下向4-氟環己-1-酮(1.00 g, 8.611 mmol,1.00當量)和MeOH (100.00 mL)的攪拌混合物中分批添加NaBH
4
(0.98 mg,0.026 mmol,3.0當量)。在室溫下攪拌所得混合物16 h。在0℃下用水淬滅反應物。減壓濃縮所得混合物。用水(50 mL)稀釋所得混合物。用CH
2
Cl
2
(3×50 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液,得到呈淡黃色固體的4-氟環己-1-醇(800 mg,78.63%)。
1
HNMR (300MHz, DMSO-d
6
) δ1.52 -1.56 (5H, m), 1.88 - 1.90 (3H, m), 3.54 - 3.57 (1H, m), 4.55 - 4.58(1H, m), 4.70 - 4.72(1H, m)
步驟2. 甲烷磺酸4-氟環己酯
在0℃下向4-氟環己-1-醇(400.00 mg,3.385 mmol,1.00當量)和TEA (1027.74 mg,10.156 mmol,3.00當量)於DCM(10.00 mL)中的攪拌混合物中逐滴添加甲烷磺醯氯(581.66 mg,5.078 mmol,1.5當量)。在室溫下攪拌所得混合物2小時。用水(50 mL)稀釋所得混合物。用CH
2
Cl
2
(3×50 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。減壓濃縮所得混合物,獲得呈淡黃色固體的甲烷磺酸4-氟環己酯(650 mg,97.84%)。粗產物不經進一步純化即直接用於下一步驟中。
1
HNMR (300MHz, DMSO-d
6
) δ 1.52 -1.90 (8H, m), 3.22 (3H, s), 4.60- 4.78(2H, m)
步驟3. 1-((1s,4s)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例27)及1-((1r,4r)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例28)
在氮氣氛圍下在110℃下將3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(150.00 mg,0.535 mmol,1.00當量)、甲烷磺酸4-氟環己酯(1050.18 mg,5.352 mmol,10.00當量)以及Cs
2
CO
3
(523.09 mg,1.605 mmol,3.00當量)於DMF(20.00 mL)中的混合物攪拌16 h。用水(20 mL)稀釋所得混合物。用EtOAc(3×20 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內39 B至59 B;RT1:6.4)來純化粗產物(100 mg),得到:
呈白色固體的1-((1s,4s)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例27,6.8 mg,3.34%)。LCMS: m/z (ESI), [M+H]
+
= 381.2。
1
HNMR (300MHz , DMSO-d
6
) δ 1.66 - 1.68 (1H, m), 1.75 (3H, d), 2.04 - 2.06(2H, m), 2.18 - 2.19 (2H, m), 2.38 (3H, d), 2.45 (3H, s), 4.50 - 4.53 (1H, m), 4.81 - 4.93 (1H, s), 7.73 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.15 (2H, d);
呈白色固體的1-((1r,4r)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例28,18.8 mg,9.23%)。LCMS: m/z (ESI), [M+H]
+
= 381.2。
1
HNMR (400MHz , DMSO-d
6
) δ 1.63 - 1.65(2H, m), 1.92 - 1.96 (4H, m), 2.00- 2.04 (2H, m), 2.14 (3H, s), 2.41 (3H, s), 4.47 - 4.49 (1H, m), 4.61 - 4.65 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.19 (2H, d)。
實例 42
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(2-氧雜螺[3.3]庚烷-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例42)
步驟1. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(2-氧雜螺[3.3]庚烷-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例42)
在N
2
氛圍下在0℃下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(70.00 mg,0.250 mmol,1.00當量)和2-氧雜螺[3.3]庚烷-6-醇(57.01 mg,0.499 mmol,2.00當量)以及PPh
3
(196.51 mg,0.749 mmol,3.00當量)於THF(10 mL)中的攪拌混合物中分批添加DIAD(151.50 mg,0.749 mmol,3.00當量)。在氮氣氛圍下在70℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(DCM/MeOH = 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(10MMOL/L NH
4
HCO
3
+0.1%NH
3
.H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至36 B;RT1:5.88)來純化粗產物(80 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[2-氧雜螺[3.3]庚烷-6-基]吡唑並[3,4-d]嘧啶-6-胺(40 mg,50.00%)。LCMS: m/z (ESI), [M+H]
+
= 337.3。
1
H NMR (300 MHz, DMSO-d
6
) δ 2.37 (3H, s), 2.44 (3H, s), 2.65-2.80 (4H, m), 4.45 (2H, s), 4.64 (2H, s), 4.84-4.89 (1H, m), 7.76 (1H, s), 8.42 (1H, s), 8.92 (1H, s), 9.13 (1H, s), 9.18 (1H, s)。
下表2中的以下化合物是通過實例42中所描述的類似方法合成。
實例 37
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[2.5]辛-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例37)
步驟1. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[2.5]辛-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例37)
在氮氣氛圍下在70℃下將TMAD(276.43 mg,1.605 mmol,3.00當量)、n-Bu
3
P(324.81 mg,1.605 mmol,3.00當量)、螺[2.5]辛-6-醇(202.61 mg,1.605 mmol,3.00當量)以及3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(150.00 mg,0.535 mmol,1.00當量)於THF(25.00 mL)中的混合物攪拌2h。用水(100 mL)稀釋所得混合物。用CH
2
Cl
2
(3×50 mL)萃取所得混合物,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。使粗產物從MeOH(20 mL)再結晶,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:MeOH;流動速率:25 mL/min;梯度:7 min內58 B至70 B)來純化粗產物(60 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[螺[2.5]辛-6-基]吡唑並[3,4-d]嘧啶-6-胺(30 mg,14.29%)。LCMS: m/z (ESI), [M+H]
+
= 389.3。
1
H NMR (400 MHz, DMSO-
d6
) δ 0.20 (2H, d), 0.33 (2H, d), 0.98 (2H, d), 1.84 (4H, m), 2.08 (2H, m), 2.39 (3H, d), 2.45 (3H, s), 4.45 (1H, d), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.16 (2H, d)
下表3中的以下化合物是通過實例37中所描述的類似方法合成。
實例 43
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[3.3] 庚烷-2-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例43)
步驟1. 甲烷磺酸螺[3.3]庚烷-2-基酯
在氮氣氛圍下在0℃下向螺[3.3]庚烷-2-醇(300.00 mg,2.674 mmol,1.00當量)和TEA(811.89 mg,8.023 mmol,3.00當量)於DCM(50.00 mL)中的攪拌混合物中逐滴添加甲烷磺醯氯(459.50 mg,4.012 mmol,1.50當量)。用水(20 mL)稀釋所得混合物。用CH
2
Cl
2
(3×20 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。這產生呈淡黃色油狀物的甲烷磺酸螺[3.3]庚烷-2-基酯(500 mg,98.26%)。
步驟2. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[3.3]庚烷-2-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例43)
在氮氣氛圍下在100℃下將3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(100.00 mg,0.357 mmol,1.00當量)、甲烷磺酸螺[3.3]庚烷-2-基酯(678.78 mg,3.568 mmol,10.00當量)以及Cs
2
CO
3
(348.73 mg,1.070 mmol,3.00當量)於DMF(20.00 mL)中的混合物攪拌16 h。用水(40 mL)稀釋所得混合物。用EtOAc(3×100 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na
2
SO
4
乾燥。在過濾後,減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05% NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內39 B至59 B;RT1:6.4)來純化粗產物,得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[螺[3.3]庚烷-2-基]吡唑並[3,4-d]嘧啶-6-胺(31.8mg,23.80%)。LCMS: m/z (ESI), [M+H]
+
= 375.3。
1
HNMR (400MHz , DMSO-d
6
) δ 1.81- 1.95 (2H, m), 1.89 - 1.91 (2H, m), 2.09 - 2.11 (2H, m), 2.42 (8H, d), 2.59 -2.63 (2H, m), 4.89 - 4.93(1H, m), 7.76 (1H, s), 8.42 (1H, s), 8.92 (1H, s), 9.17 (2H, d)
實例 44
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(氧雜環丁烷-3-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例44)
步驟1. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(氧雜環丁烷-3-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例44)
在空氣氛圍下在室溫下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(80.00 mg,0.285 mmol,1.00當量)和3-碘氧雜環丁烷(78.76 mg,0.428 mmol,1.50當量)於DMF(10 mL)中的攪拌混合物中添加K
2
CO
3
(118.34 mg,0.856 mmol,3.00當量)。在空氣氛圍下在80℃下攪拌所得混合物2h。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
: MEOH 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150 mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內12 B至32 B;RT1:6.62)來純化粗產物(100 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-(氧雜環丁烷-3-基)吡唑並[3,4-d]嘧啶-6-胺(實例44,40 mg,41.67%)。LCMS: m/z (ESI), [M+H]
+
= 337.2。
1
H NMR (300 MHz, DMSO-d
6
) δ 2.38 (3H, s), 2.49 (3H, s), 4.89-5.02 (4H, m), 5.72 (1H, t), 7.75 (1H, s), 8.42 (1H, s), 8.96 (1H, s), 9.15 (1H, s), 9.27 (1H, s)
下表4中的以下化合物是通過實例44中所描述的類似方法合成。
實例 59
製備1-異丙基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例59)
步驟1. 6-氯-1-異丙基-3-甲基-1H-吡唑並[3,4-d]嘧啶
在空氣氛圍下在0℃下向1-(2,4-二氯嘧啶-5-基)乙酮(150.00 mg,0.785 mmol,1.00當量)和DIPEA(54.13 mg,0.419 mmol,4.00當量)於THF中的攪拌混合物中逐滴添加異丙基肼(15.26 mg,0.136 mmol,1.30當量)。在空氣氛圍下在室溫下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(PE:EA 1:1)來純化殘餘物,得到呈黃色固體的6-氯-1-異丙基-3-甲基吡唑並[3,4-d]嘧啶(120 mg,72.53%)。LCMS: m/z (ESI), [M+H]
+
= 211.2。
1
H NMR (300 MHz, CDCl
3
) δ1.54 (6H, d), 2.61 (3H, s), 5.12 (1H, dd), 8.90 (1H, s)
步驟2. 1-異丙基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例59)
在空氣氛圍下在室溫下向6-氯-1-異丙基-3-甲基吡唑並[3,4-d]嘧啶(120.00 mg,0.570 mmol,1.00當量)和7-甲基咪唑[1,2-a]吡啶-6-胺(108.99 mg,0.740 mmol,1.30當量)於二惡烷(10 mL)中的攪拌混合物中添加XantPhos (65.92 mg,0.114 mmol,0.20當量)、Pd(AcO)
2
(25.58 mg,0.114 mmol,0.20當量)以及Cs
2
CO
3(
556.77 mg,1.709 mmol,3.00當量)。在氮氣氛圍下在60℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/ MeOH = 10:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;RT1:6.67)來純化粗產物(100 mg),得到呈白色固體的1-異丙基-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(62 mg,33.76%)。LCMS: m/z (ESI), [M+H]
+
= 323.4。
1
H NMR (300 MHz, DMSO-d
6
) δ 1.41 (6H, d), 2.40 (3H, d), 2.45 (3H, s), 4.73-4.82 (1H, m), 7.74 (1H, s), 8.41(1H, s), 8.92 (1H, s), 9.16 (1H, s), 9.19 (1H, s)
實例 60
製備1-異丙基-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例60)
步驟1. 1-異丙基-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例60)
在氮氣氛圍下在100℃下將6-氯-1-異丙基-3-甲基吡唑並[3,4-d]嘧啶(80.00 mg,0.380 mmol,1.00當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(67.07 mg,0.456 mmol,1.20當量)、Pd(AcO)
2
(17.05 mg,0.076 mmol,0.20當量)、XantPhos(65.92 mg,0.114 mmol,0.30當量)以及Cs
2
CO
3
(309.32 mg,0.949 mmol,2.50當量)於二惡烷(10.00 mL)中的混合物攪拌3小時。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 10:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內21 B至41 B;RT1:7.02)來純化粗產物(100 mg),得到呈白色固體的1-異丙基-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(55.77 mg,45.70%)。LCMS: m/z (ESI), [M+H]+ = 322.2。
1
H NMR (300 MHz, DMSO-d
6
) δ 1.38 (6H, d), 2.25 (3H, d), 2.44 (3H, s), 4.67-4.89 (1H, m), 7.47 (2H, dd), 7.87 (1H, t), 8.67 (1H, s), 8.86 (1H, s), 9.03 (1H, s)
實例 72
製備3-(3-甲基-6-((7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)丙腈(實例72)
步驟1. 3-(3-甲基-6-((7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)丙腈(實例72)
向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(70.00 mg,0.250 mmol,1.00當量)和3-溴-丙腈(66.92 mg,0.499 mmol,2.00當量)於DMF(10 mL)中的攪拌混合物中添加K
2
CO
3
(103.55 mg,0.749 mmol,3.00當量)。在空氣氛圍下在80℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(DCM: MeOH = 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內8 B至28 B;RT1:7.70)來純化粗產物,得到呈白色固體的3-[3-甲基-6-([7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]氨基)吡唑並[3,4-d]嘧啶-1-基]丙腈(40 mg,48.05%)。LCMS: m/z (ESI), [M+H]
+
= 334.3。
1
H NMR (300 MHz, DMSO-d
6
) δ2.39 (3H, s), 2.47(3H, s), 3.08(2H, t), 4.36(2H, t), 7.75(1H, s), 8.41(1H, s), 8.97(1H, s), 9.17 (1H, s), 9.29 (1H, s)。
下表5中的以下化合物是通過實例72中所描述的類似方法合成。
實例 73
製備4-(3-甲基-6-(7-甲基咪唑[1,2-a]吡啶-6-基氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)苯甲腈(實例73)
步驟1. 4-(3-甲基-6-(7-甲基咪唑[1,2-a]吡啶-6-基氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)苯甲腈(實例73)
在100℃下將7-甲基-N-[3-甲基-1H-吡唑並[3,4-d]嘧啶-6-基]-[1,2,4]三唑並[1,5-a]吡啶-6-胺(150.00 mg,0.535 mmol,1.00當量)、4-氟-苯甲腈(97.57 mg,0.806 mmol,1.50當量)、苯甲腈(97.22 mg,0.803 mmol,1.50當量)以及K
2
CO
3
(221.88 mg,1.605 mmol,3.0當量)於DMF(50.00 mL)中的混合物攪拌16 h。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH = 10:1)來純化殘餘物,得到呈淡黃色固體的4-[3-甲基-6-([7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]氨基)吡唑並[3,4-d]嘧啶-1-基]苯甲腈(40 mg,粗產物)。通過製備型HPLC利用以下條件(柱:XBridge Shield RP18 OBD柱,19*250mm,10um;移動相A:水(10MMOL/L NH
4
HCO
3
+0.1% NH
3
•H
2
O),移動相B:ACN;流動速率:20 mL/min;梯度:7 min內36 B至46 B;RT1:5.73)來純化粗產物(40 mg),得到呈白色固體的4-[3-甲基-6-([7-甲基咪唑[1,2-a]吡啶-6-基]氨基)吡唑並[3,4-d]嘧啶-1-基]苯甲腈(8.8mg,4.31%)。LCMS: m/z (ESI), [M+H]
+
= 381.3。
1
HNMR (300 MHz, CDCl
3
) δ 2.45 (3H, d), 2.62 (3H, s), 7.08 (1H, s), 7.54 (2H, s), 7.65 (1H, s), 7.75 (2H, d), 8.42 - 8.44 (2H, m), 8.85 (1H, s), 9.06 (1H, d)。
實例 89
製備6-甲氧基-4-甲基-N-[3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-基]吡啶-3-胺
步驟1. 6-甲氧基-4-甲基-N-[3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-基]吡啶-3-胺(實例89)
在氮氣氛圍下在80℃下將6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(80.00 mg,0.317 mmol,1.00當量)、6-甲氧基-4-甲基吡啶-3-胺(52.49 mg,0.380 mmol,1.20當量)、Pd(AcO)
2
(14.22 mg,0.063 mmol,0.20當量)、XantPhos(54.95 mg,0.095 mmol,0.30當量)以及Cs
2
CO
3
(257.87 mg,0.791 mmol,2.50當量)於二惡烷(2.50 mL)中的混合物攪拌2 h。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/ MeOH 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Shield RP18 OBD柱,30*150mm,5um;移動相A:水(0.05% NH
3
H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內25 B至55 B;254;220 nm; RT1:5.20)來純化粗產物(100 mg),得到呈白色固體的6-甲氧基-4-甲基-N-[3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-基]吡啶-3-胺(70 mg,62.39%)。LCMS: m/z (ESI), [M+H]
+
= 355.2。
1
H-NMR (300 MHz, DMSO-d
6
) δ 1.79 (2H, d), 1.99 - 2.20 (5H, m), 2.42 (3H, s), 3.43 - 3.56 (2H, m), 3.83 (3H, s), 3.96 (2H, dd), 4.58 (1H, t), 6.74 (1H, s), 8.11 (1H, s), 8.82 (1H, s), 8.98 (1H, s)。
實例 106
製備1-((1R,3r,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
步驟1. 1-((1R,3r,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在氮氣氛圍下在0℃下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(100.00 mg,0.357 mmol,1.00當量)、(1R,3S,5S)-8-氧雜雙環[3.2.1]辛-3-醇(137.18 mg,1.070 mmol,3.00當量)以及PPh
3
(280.72 mg,1.070 mmol,3當量)於THF(10.00 mL)中的攪拌混合物中逐滴添加DIAD(216.42 mg,1.070 mmol,3當量)。在氮氣氛圍下在70℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH 12:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內37 B至57 B)來純化粗產物,得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[(1R,3R,5S)-8-氧雜雙環[3.2.1]辛-3-基]吡唑並[3,4-d]嘧啶-6-胺(實例106,50 mg,50.00%)。LCMS: m/z (ESI), [M+H]
+
= 391.3。
1
H NMR (300 MHz, DMSO-d
6
) δ 1.74(4H, s), 2.22-2.45(10H, m), 4.35(2H, s), 4.63(1H, s), 7.75(1H, s), 8.41(1H, s), 8.93(1H, s), 9.17 (1H, s), 9.21(1H, s)。
實例 107
製備1-((1R,3s,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
步驟1. 1-((1R,3s,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在氮氣氛圍下在0℃下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(80.00 mg,0.285 mmol,1.00當量)、(1R,3R,5S)-8-氧雜雙環[3.2.1]辛-3-醇(109.75 mg,0.856 mmol,3.00當量)以及PPh
3
(224.58 mg,0.856 mmol,3.00當量)於THF(16.00 mL)中的攪拌混合物中逐滴添加DIAD(173.14 mg,0.856 mmol,3.00當量)。在氮氣氛圍下在70℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH 12:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH
3
•H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內37 B至50 B)來純化粗產物,得到呈白色固體的1-((1R,3s,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例107, 45mg,45.00%)。LCMS: m/z (ESI), [M+H]
+
= 391.4。
1
H NMR (300 MHz, DMSO-d
6
) δ 1.78-1.94(6H, m), 2.07-2.25(2H, m),2.42(3H.s), 2.45(3H,s), 4.44(2H, s), 4.88-4.96(1H, m), 7.73(1H, s), 8.41(1H, s), 8.92(1H, s), 9.18(1H, s), 9.29(1H, s)。
實例 108/109/110/111
1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108,異構體1)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例109,異構體2)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110,異構體3)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例111,異構體4)
步驟1. 製備1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(108/109的混合物)及製備1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(110/111的混合物)
在0℃下經10 min向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(220.00 mg,0.785 mmol,1.00當量)、PPh
3
(617.59 mg,2.355 mmol,3.00當量)以及3-甲氧基環丙-1-醇(273.52 mg,2.355 mmol,3.00當量)於THF(20.00 mL)中的攪拌混合物中逐滴添加含DIAD(476.13 mg,2.355 mmol,3.00當量)的THF(3 mL)。在氮氣氛圍下在70℃下攪拌所得混合物2 h。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH 10:1)來純化殘餘物,得到呈淡黃色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(150 mg)。通過製備型HPLC利用以下條件(柱:YMC-Actus Triart C18,30*250,5um;移動相A:水(0.05%NH
3
H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:8.5 min內34 B至46 B)來純化粗產物(150 mg),得到呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108/109的混合物,25 mg,16.67%)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
HNMR (400MHz , DMSO-d
6
) δ 1.81 - 1.85 (2H, m), 1.98 - 2.02 (2H, m), 2.09 - 2.11 (1H, m), 2.38 - 2.41 (4H, m), 2.45 (3H, s), 3.18 (3H, s), 3.83 - 3.85 (1H, m), 4.85 - 4.89 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.14 (2H, s), 及呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110/111的混合物,80 mg,53.33%)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
HNMR (400MHz, DMSO-d
6
) δ1.61 - 1.63 (1H, m), 1.82 - 1.98 (1H, m), 2.38 - 2.41 (4H, m), 2.39 (3H, s), 2.45 (3H, s), 3.18 (3H, s), 3.93 - 3.96 (1H, m), 5.00 - 5.06 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.15 (2H, d)。
步驟2. 製備1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108,異構體1)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例109,異構體2)
通過手性製備型HPLC利用以下條件(柱:CHIRALPAK AD-H,2.0cm I.D.*25cm L;移動相A:Hex(8mmol/L NH
3
.MeOH)--HPLC,移動相B:IPA--HPLC;流動速率:40 mL/min;梯度:18 min內20 B至20 B)來純化實例108/109的混合物(25 mg),得到呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108, 4.5mg,18.00%)(異構體1)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
HNMR (400MHz , DMSO-d
6
) δ
1.81 - 1.85 (2H, m), 1.98 - 2.01 (2H, m), 2.06 - 2.09 (1H, m), 2.38 - 2.43 (4H, m), 2.45 (3H, s), 3.18 (3H, s), 3.80 - 3.85 (1H, m), 4.82 - 4.87 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.14 (2H, s)及呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例109,3.8mg,12.00%)(異構體2)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
HNMR (400MHz , DMSO-d
6
) δ 1.81 - 1.83 (2H, m), 1.97 - 2.01 (2H, m), 2.10 - 2.12 (1H, m), 2.37 - 2.43 (4H, m), 2.45 (3H, s), 3.18 (3H, s), 3.81 - 3.85 (1H, m), 4.80 - 4.89 (1H, m), 7.73 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.14 (2H, s)。
步驟4. 1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110,異構體3)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例111,異構體4)
通過製備型HPLC利用以下條件(柱:CHIRALPAK AD-H,2.0cm I.D.*25cm L;移動相A:Hex(8mmol/L NH3.MeOH)--HPLC,移動相B:IPA--HPLC;流動速率:40 mL/min;梯度:12 min內30 B至30 B)來純化實例110/111的混合物(80 mg),得到呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110,39.1mg,48.87%)(異構體3)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
HNMR (400MHz, DMSO-d
6
) δ 1.67 - 1.69(1H, m), 1.93 - 1.99 (1H, m), 2.10 - 2.15 (4H, m), 2.39 (3H, s), 2.44 (3H, s), 3.16 (3H, s), 3.94 (1H, s), 5.00 - 5.08 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.16 (2H, d)及呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例111,35.2mg)(異構體4)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
HNMR (400MHz, DMSO-d
6
) δ 1.65 - 1.69 (1H, m), 1.93 - 1.97 (1H, m), 2.09 - 2.14 (4H, m), 2.39 (3H, d), 2.44 (3H, s), 3.15 (3H, s), 3.93 - 3.96 (1H, m), 5.00 - 5.08 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.16 (2H, d)。
實例 112/113/114/115.
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112,異構體1)/3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例113,異構體2)/3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114,異構體3)/3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例115,異構體4)
步驟1. 3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例112/113的混合物)和3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例114/115的混合物)
在0℃下攪拌3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(200.00 mg,0.714 mmol,1.00當量)、3-甲基氧雜環己烷-4-醇(248.65 mg,2.141 mmol,3.00當量)以及PPh
3
(561.45 mg,2.141 mmol,3.00當量)於THF(10.00 mL)中的混合物,且在氮氣氛圍下70℃下逐滴添加DIAD(432.85 mg,2.141 mmol,3.00當量),持續2小時。減壓濃縮所得混合物。通過製備型TLC(CH
2
Cl
2
/MeOH 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Shield RP18 OBD柱,30*150mm,5um;移動相A:水(0.05% NH
3
H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內18 B至48 B)來純化粗產物(120 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例112/113的混合物,30 mg,25.00%)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
HNMR (400MHz, DMSO-d
6
) δ 0.52 (3H, d), 1.74 (2H, d), 2.06 - 2.31 (1H, m), 2.36 (3H, d), 2.45 (3H, s), 3.07 (1H, t), 3.43 (1H, t), 3.82 - 4.00 (2H, m), 4.14 - 4.27 (1H, m), 7.69 - 7.76 (1H, m), 8.39 (1H, s), 8.91 (1H, s), 9.15 (2H, d)及呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[-3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例114/115的混合物,70mg,57.75%)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
HNMR (400MHz, DMSO-d
6
) δ 0.70 (3H, d), 1.74 - 1.91 (1H, m), 2.23 (1H, d), 2.37 (4H, d), 2.44 (3H, s), 3.44 - 3.65 (2H, m), 3.72 (1H, dd), 4.05 (1H, dt), 4.79 (1H, dt), 7.72 (1H, s), 8.39 (1H, s), 8.93 (1H, s), 9.17 (2H, s)
步驟2. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112,異構體1)和3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例113,異構體2)
通過製備型手性HPLC利用以下條件(柱:CHIRALPAK IE-3,4.6*50mm 3um;移動相A:Hex (0.1%DEA):EtOH = 50:50)來純化粗產物3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112/113的混合物,30.00 mg,0.079 mmol,1.00當量)),得到呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112,異構體1,12.22 mg,40.73%)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
HNMR (400MHz, DMSO-d
6
) δ 0.52 (3H, d), 1.74 (2H, d), 2.06 - 2.31 (1H, m), 2.36 (3H, d), 2.45 (3H, s), 3.07 (1H, t), 3.43 (1H, t), 3.82 - 4.00 (2H, m), 4.14 - 4.27 (1H, m), 7.69 - 7.76 (1H, m), 8.39 (1H, s), 8.91 (1H, s), 9.15 (2H, d)及呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例113,異構體2,8.37 mg,27.90%)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
HNMR (400MHz , DMSO-d
6
) δ 0.52 (3H, d), 1.74 (2H, d), 2.06 - 2.31 (1H, m), 2.36 (3H, d), 2.45 (3H, s), 3.07 (1H, t), 3.43 (1H, t), 3.82 - 4.00 (2H, m), 4.14 - 4.27 (1H, m), 7.69 - 7.76 (1H, m), 8.39 (1H, s), 8.91 (1H, s), 9.15 (2H, d)
步驟3. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114,異構體3)和3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例115,異構體4)
通過製備型手性HPLC利用以下條件(柱:CHIRALPAK IE-3,4.6*50mm 3um;移動相A:Hex (0.1%DEA):EtOH=50:50)來純化粗產物3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114/115的混合物,70.00 mg,0.185 mmol,1.00當量)),得到呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114,異構體3,32.98mg,47.11%)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
H NMR (300 MHz, DMSO-d
6
) δ 0.70 (3H, d), 1.74 - 1.91 (1H, m), 2.23 (1H, d), 2.37 (4H, d), 2.44 (3H, s), 3.44 - 3.65 (2H, m), 3.72 (1H, d), 4.05 (1H, t), 4.79 (1H, t), 7.72 (1H, s), 8.39 (1H, s), 8.93 (1H, s), 9.17 (2H, s), 及呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例115,異構體4,33.45 mg,41.39%)。LCMS: m/z (ESI), [M+H]
+
= 379.3。
1
H NMR (300 MHz, DMSO-d
6
) δ 0.70 (3H, d), 1.74 - 1.91 (1H, m), 2.23 (1H, d), 2.37 (4H, d), 2.44 (3H, s), 3.44 - 3.65 (2H, m), 3.72 (1H, d), 4.05 (1H, t), 4.79 (1H, t), 7.72 (1H, s), 8.39 (1H, s), 8.93 (1H, s), 9.17 (2H, s)
實例 116/117
製備1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例116,異構體1)及1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例117,異構體2)
步驟1.1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在0℃下向250 mL圓底燒瓶中添加含2,2-二甲基氧雜環己烷-4-醇(627.03 mg,4.816 mmol,3.00當量)和3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(450.00 mg,1.605 mmol,1.00當量)、PPh
3
(1263.26 mg,4.816 mmol,3.00當量)的THF(60.00 mL)。在N
2
下在0℃下將DIAD(973.90 mg,4.816 mmol,3.00當量)於THF(10 mL)中的溶液逐滴添加至以上溶液中,並在rt下攪拌3 min。在70℃下繼續攪拌反應混合物2h。減壓濃縮所得混合物。用DCM(20 mL)稀釋所得混合物且過濾,用DCM(2×5 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH
2
Cl
2
/MeOH 15:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:YMC-Actus Triart C18,30*250,5um;移動相A:水(0.05% NH
3
H
2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內37 B至57 B)來純化粗產物(250 mg),得到呈白色固體的1-[2,2-二甲基氧雜環己烷-4-基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(170mg,26.98%)。LCMS: m/z (ESI), [M+H]
+
= 393.2
步驟2. 1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例116,異構體1)/1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例117,異構體2)
通過手性製備型HPLC利用以下條件(柱:CHIRALPAK-AD-H-UL001,20*250mm,5 um;移動相A:Hex(8mmol/L NH
3
.MeOH)--HPLC,移動相B:IPA--HPLC;流動速率:20 mL/min;梯度:15 min內25 B至25 B;RT1:10.12;RT2:11.691)來純化粗產物(170 mg),得到rel-1-[(4R)-2,2-二甲基氧雜環己烷-4-基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(實例116,異構體1)(70mg,41.18%) LCMS: m/z (ESI), [M+H]
+
= 393.3。
1
H-NMR (400 MHz, DMSO-d
6
) δ 1.21 (6H, d), 1.80 (2H, dd), 1.89 (1H, t), 2.05 (1H, qd), 2.35 - 2.48 (6H, m), 3.66 - 3.81 (2H, m), 4.75 - 4.95 (1H, m), 7.74 (1H, d), 8.40 (1H, s), 8.92 (1H, s), 9.20 (2H, d); 及呈白色固體的rel-1-[(4R)-2,2-二甲基氧雜環己烷-4-基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(實例117,異構體2)(70mg,41.18%), LCMS: m/z (ESI), [M+H]
+
= 393.3。
1
H-NMR (400 MHz, DMSO-d
6
) δ1.21 (6H, d), 1.80 (2H, dd), 1.89 (1H, t), 2.05 (1H, qd), 2.35 - 2.48 (6H, m), 3.66 - 3.81 (2H, m), 4.75 - 4.95 (1H, m), 7.74 (1H, d), 8.40 (1H, s), 8.92 (1H, s), 9.20 (2H, d)。
生物實例
本文所公開的示範性化合物已經在以下一個或多個生物分析中表徵。
生物實例 1 :
酶分析
通過測量螢光標記的肽底物轉化成磷酸化產物的TRF-RET來測定化合物針對DNA-PK的抑制活性。所有分析都在黑色Greiner 384孔小體積培養板(Greiner)中進行,總反應體積為6 μL且最終DMSO濃度為0.5 %(v/v)。全長人類DNAPK蛋白、螢光素-P53 (Ser15)肽底物(螢光素-EPPLSQEAFADLWKK)以及LanthaScreen™ Tb-抗磷酸-p53 [pSer15]抗體試劑盒都是購自賽默飛世爾科技(Thermo Fisher Scientific)。最初,將DNA-PK蛋白質與化合物在室溫下在反應緩衝液(50 mM HEPES pH 7.5,0.01% Brij-35,10 mM MgCl2,1 mM EGTA,1 mM DTT,10 μg/ml小牛胸腺DNA)中培育30分鐘。然後通過添加ATP和螢光素-P53 (Ser15)肽底物引發反應。在60分鐘後通過添加含有20 mM EDTA、4 nM Tb抗磷酸-p53 [Serl5]抗體的6 μl終止緩衝液來淬滅激酶反應物(10 μΜ ATP,1.6 μΜ肽底物)。將反應物再培育一小時,且在Spark 20M(Tecan)上讀取培養板。分析資料,且使用利用XLfit的四倍參數邏輯擬合來計算化合物產生50%對相應激酶的抑制的濃度(IC
50
)。示範性化合物的DNA-PK抑制活性展示於下表5中。可以看出,本公開的化合物已展現出強效的DNA-PK抑制活性。
生物實例 2 :
代謝穩定性分析(大鼠肝細胞Clint)
使用台盼藍測定冷凍保存的肝細胞的存活力,並且用緩衝液將所述細胞濃縮液調節為每毫升106個細胞。將1 μM化合物(在乙腈中;0.01% DMSO)在96深孔培養板中與250 μL肝細胞(每毫升1百萬個細胞)一起培育。在不同時間點(0、0.5、5、15、30、45、60、80、100和120 min)通過添加3體積的冷卻乙腈到20 μL反應混合物並且在4℃下離心15 min來使反應停止。用純水將40 μL上清液稀釋到200 μL並使用LCMS/MS進行分析。
基於從初始濃度測定的化合物消失的消除半衰期(T
1/2
)來估計體外肝細胞清除率。計算每種化合物(測試或對照)與IS的峰面積比。藥物消除速率常數k(min-1)、T
1/2
(min)和體外固有清除率CL
int
(μL/min/E6)是根據以下等式計算:
k = -斜率
T
1/2
= 0.693/k
CL
int
= k/C
hep
其中C
hep
(細胞×μL
-1
)是培育系統中的細胞濃度。
資料展示如下表6中。
代謝穩定性分析(人類微粒體Clint)
將1 μM化合物在37℃下在含有1 mM NADPH溶液的250 μL緩衝液(100 mM磷酸鹽緩衝液,pH-7.4)中與1 mg/mL微粒體(具有20 mg/ml蛋白質錐體的混合HLM)一起培育。在新制96孔板中,在不同時間點0、0.5、5、10、15、20和30 min,用5體積的冷卻乙腈淬滅20 μL的培育混合物。將淬滅板在4000 rpm下離心15 min。用純水將40 μL上清液稀釋到200 μ L並使用LCMS/MS進行分析。
藥物在微粒體中的體外固有清除率CL
int
(µl/min/mg)是以與肝細胞中的CL
int
類似的方式計算的。資料也展示如下表6中。
MDCKII-MDR1-BCRP流出分析
跨越MDCKII-MDR1-BCRP細胞單層測量化合物在HBSS(25mM HEPES,pH 7.4)中的頂端到底外側(A-B)和底外側到頂端(B-A)轉運。在約37℃下進行培育120 min,其中使用5 μM地高辛作為陽性對照底物來確認測試系統的功能性。通過在培育期開始時定量供體隔室的培育介質中的底物濃度並且在培育期結束時定量供體隔室和受體隔室兩者中的底物濃度來測定5 μM化合物和對照化合物的轉運。資料用於計算表觀滲透率(Papp)。進行所有培育,重複三次,並且使用標記螢光黃確認細胞單層的完整性。
使用以下等式計算滲透率係數Pexact(cm/s):
Papp = (dCr/dt) x Vr / (A x C0)
Pexact = -(Vd×Vr)/(Vd+Vr)/A/t×ln(1-(Vd+Vr)×Cr/(Vd×Cd +Vr×Cr))
使用以下等式計算Pexact比:
Pexact或Papp比=Pexact或Papp(+抑制劑)/Pexact或Papp(-抑制劑)
使用以下等式計算流出比:
流出比 = Pexact或Papp(BA)/Pexact或Papp(AB)
其中dCr/dt為隨時間變化的接受體室中的化合物的累積濃度(µM/s);Vr為接受體室中的溶液體積(在頂端側為0.1 mL,在底外側為0.3 mL);A為用於轉運的表面積,即,單層面積為0.11 cm
2
;C0為供體室中的初始濃度(µM)。數據展示於下表6中。
生物實例 3 :
血腦屏障穿透分析
據信,Kp,uu是大腦和血漿中非結合藥物的濃度之間的關係,是CNS作用的預測的關鍵,並且應是針對藥物發現所測量和優化的主要參數(Di L等人,《藥物化學雜誌(Journal of Medicinal Chemistry)》[2013], 56:2-12)。
通過使用平衡透析方法用半滲透膜進行體外血漿和大腦結合分析。向血漿和稀釋的腦勻漿(1:4,其中DPBS pH7.4)摻入5 µM測試化合物(重複三次),並且在37℃下在緩慢旋轉的培養板中相對於相等體積的150 µL 100 mM PBS緩衝液(pH7.4)透析18小時。在培育結束時,取出來自接收器側的50 μL等分試樣和來自供體室的5 μL等分試樣。用45 μL空白血漿或腦勻漿進一步稀釋5 μL樣品。將配對樣品與緩衝液或空白血漿/腦勻漿進行基質匹配且混合2 min,並接著用150 μL低溫乙腈和100 ng/mL甲苯磺丁脲作為內標進行沉澱。在以4000 rpm離心20 min後,將上清液用0.1%甲酸水溶液稀釋並分析LC/MS/MS(API 4000, 美國應用生物系統公司(Applied Biosystems),福斯特城(Foster City))。通過緩衝液側回應與腦勻漿/血漿側回應的比率計算腦勻漿和稀釋血漿中測試化合物的未結合分數(fu),並且通過以下等式從勻漿和血漿中測量的fu計算未稀釋的血漿和組織中測試化合物的未結合分數(fu、pl和fu、br):fu,bl (fu,br) = (1/D) / [(1/fu -1) + 1/D)]。D是稀釋係數。
短口服吸收(SOA)模型是用於鑒別化合物的腦滲透的體內篩選模型。向從北京維通利華(Beijing Vital River)購買的六隻雄性Han Wistar大鼠經口給予10 mg/kg的含1%甲基纖維素的化合物。在給藥後0.5、1、2、4、7和16小時,從小腦延髓池收集腦部脊髓液(CSF)。將通過在收集的半小時內在約4℃下離心3,000 g來處理血漿的血漿樣品。將血漿樣品移到標記的試管且存儲在-80℃下直到分析為止。採集腦組織並在3×體積的100 mM磷酸鹽緩衝鹽水(pH7.4)中均質化。在LC/MS/MS分析之前,將所有樣品存儲在約-70℃。
通過摻入空白血漿、腦勻漿和範圍為0.5至500 ng/mL的人工CSF來製備標準品。通過添加3倍體積的含有低溫乙腈的內標(40 ng/mL地塞米松和40 ng/mL雙氯芬酸)沉澱均質化的腦組織以及血漿樣品,並且用100 μL含有低溫乙腈的內標沉澱10 μL CSF樣品。渦旋2分鐘並以14,000 rpm離心5 min後,通過LC/MS/MS(API 4000,美國應用生物系統公司,福斯特城)分析上清液。在血漿樣品分析中每個批次開始和結束時操作兩組標準曲線。對於腦和CSF樣品,分析一條標準曲線和測試樣品。
通過經口施用後齧齒動物中的AUC大腦/AUC血漿來測量表示為大腦/血液比率(K
p
)的總大腦水準。通過體外血漿和大腦結合分析測定生物基質中測試化合物的游離部分。通過以下等式計算Kp,uu:Kp,uu = AUC (大腦)/AUC (血漿)×(fu,大腦/fu.血漿)。數據展示於表7中。
儘管已經參照具體實施例(其中一部分是優選實施例)具體地顯示和描述本公開,但本領域的技術人員應理解,可以在不脫離如本文所公開的本公開的精神和範圍情況下,對其中的形式和細節作出各種變化。
 或其藥學上可接受的鹽,其中X1
、X2
、X3
、R1
、R2
、R3
、R4
和環A如本文所定義。
在另一方面,本公開提供由式(Ia)表示的化合物:
或其藥學上可接受的鹽,其中X1
、X2
、X3
、R1
、R2
、R3
、R4
和環A如本文所定義。
在另一方面,本公開提供由式(Ia)表示的化合物: 或其藥學上可接受的鹽,其中或其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
、n和環A如本文所定義。
在另一方面,本公開提供由式(Ib)表示的化合物:
或其藥學上可接受的鹽,其中或其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
、n和環A如本文所定義。
在另一方面,本公開提供由式(Ib)表示的化合物: 或其藥學上可接受的鹽,其中或其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
和環A如本文所定義。
在另一方面,本公開提供由式(Ic)表示的化合物:
或其藥學上可接受的鹽,其中或其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
和環A如本文所定義。
在另一方面,本公開提供由式(Ic)表示的化合物: 或其藥學上可接受的鹽,其中或其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
和環A如本文所定義。
在另一方面,本公開提供由式(Id)表示的化合物:
或其藥學上可接受的鹽,其中或其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
和環A如本文所定義。
在另一方面,本公開提供由式(Id)表示的化合物: 或其藥學上可接受的鹽,其中或其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
和環A如本文所定義。
在另一方面,本公開提供由式(Ie)表示的化合物:
或其藥學上可接受的鹽,其中或其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
和環A如本文所定義。
在另一方面,本公開提供由式(Ie)表示的化合物: 和其藥學上可接受的鹽,其中或其藥學上可接受的鹽,其中X1
、X3
、Y1
、Y2
、Y3
、R1
、R2
、R5
和R6
如本文所定義。
在另一方面,本公開提供一種藥物組合物,其包括一種或多種式(I)、式(Ia)、式(Ib)、式(Ic)、式(Id)、式(Ie)化合物或其藥學上可接受的鹽作為活性成分。
在另一方面,本公開進一步提供式(I)化合物或其藥學上可接受的鹽或一種或多種前述各者的藥物組合物,其用於抑制DNA-PK激酶。
在又一方面,本公開提供式(I)化合物或其藥學上可接受的鹽或一種或多種前述各者的藥物組合物的用途,其用於製造供抑制個體中的DNA-PK激酶用的藥劑。
在另一方面,本公開提供一種通過使用一種或多種式(I)化合物或其藥學上可接受的鹽或一種或多種前述各者的藥物組合物來抑制DNA-PK激酶的方法。
在另一方面,本公開提供一種通過使用式(I)化合物或其藥學上可接受的鹽或一種或多種前述各者的藥物組合物來治療DNA-PK相關病症(例如癌症)的方法。在另一方面,本公開提供式(I)化合物或其藥學上可接受的鹽,其與第二治療劑(優選地抗腫瘤劑)組合。
在另一方面,本公開提供式(I)化合物或其藥學上可接受的鹽與第二治療劑(優選地抗腫瘤劑)的組合用途。
和其藥學上可接受的鹽,其中或其藥學上可接受的鹽,其中X1
、X3
、Y1
、Y2
、Y3
、R1
、R2
、R5
和R6
如本文所定義。
在另一方面,本公開提供一種藥物組合物,其包括一種或多種式(I)、式(Ia)、式(Ib)、式(Ic)、式(Id)、式(Ie)化合物或其藥學上可接受的鹽作為活性成分。
在另一方面,本公開進一步提供式(I)化合物或其藥學上可接受的鹽或一種或多種前述各者的藥物組合物,其用於抑制DNA-PK激酶。
在又一方面,本公開提供式(I)化合物或其藥學上可接受的鹽或一種或多種前述各者的藥物組合物的用途,其用於製造供抑制個體中的DNA-PK激酶用的藥劑。
在另一方面,本公開提供一種通過使用一種或多種式(I)化合物或其藥學上可接受的鹽或一種或多種前述各者的藥物組合物來抑制DNA-PK激酶的方法。
在另一方面,本公開提供一種通過使用式(I)化合物或其藥學上可接受的鹽或一種或多種前述各者的藥物組合物來治療DNA-PK相關病症(例如癌症)的方法。在另一方面,本公開提供式(I)化合物或其藥學上可接受的鹽,其與第二治療劑(優選地抗腫瘤劑)組合。
在另一方面,本公開提供式(I)化合物或其藥學上可接受的鹽與第二治療劑(優選地抗腫瘤劑)的組合用途。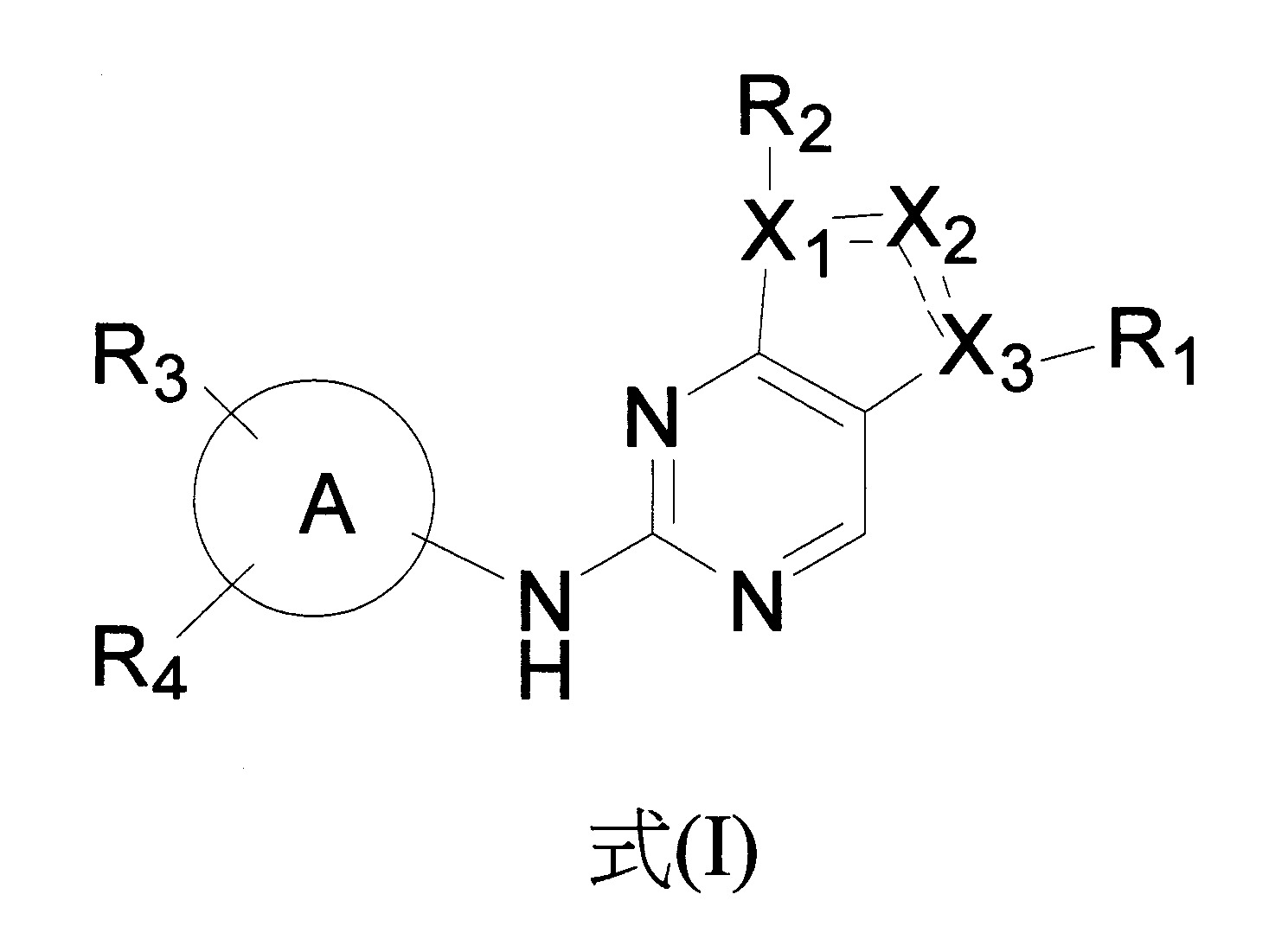 或其藥學上可接受的鹽,
其中,
X1
、X2
和X3
各自獨立地為C或N,其限制條件為X1
、X2
和X3
中的至少一個為N且X1
、X2
和X3
中的至少一個為C;
短劃線“
或其藥學上可接受的鹽,
其中,
X1
、X2
和X3
各自獨立地為C或N,其限制條件為X1
、X2
和X3
中的至少一個為N且X1
、X2
和X3
中的至少一個為C;
短劃線“ ”意指X1
與X2
之間的鍵及X2
與X3
之間的鍵可以是單鍵或雙鍵,其限制條件是X1
與X2
之間的鍵及X2
與X3
之間的鍵中的至少一個是單鍵;
R1
不存在,為鹵素或C1-6
烷基,其中所述C1-6
烷基可以任選地經羥基、鹵素或氘單取代或獨立多取代;
各R2
、R3
和R4
獨立地選自不存在、鹵素、羥基、氰基、C1-6
烷基、C1-6
烷氧基、-(CH2
)n
-Q,其可以任選地經以下各者單取代或獨立多取代:氘、羥基、氨基、氰基、鹵素、C1-6
烷基、C1-6
鹵烷基、(C≡N)-C1-6
烷基、C1-6
烷氧基、C1-6
鹵烷氧基、C3-8
環烷基、C3-8
環烷氧基、3員至8員芳香基或3員至8員雜環基,
其中n為0、1或2,Q為3員至8員飽和或不飽和碳環基,或3員至8員飽和或不飽和雜環基;
環A為5員至12員芳香基、具有1到5個選自氧、硫和氮的環雜原子的5員至12員雜芳香基、具有0到5個選自氧、硫和氮的環雜原子的8員至10員雙環,其中環A不為苯基。
在一些實施例中,R2
選自甲基、乙基、丙基、丁基、環丙基、環丁基、氧雜環丁烷基、環戊烷基、四氫呋喃基、環己烷基、四氫吡喃基、環庚烷基、呱啶基、苯基、吡啶基、吡啶酮基、氧雜環辛烷基、四氫吡喃基、二氫吡喃基、螺[3.3]庚烷基、螺[2.5]辛烷基、雙環[1.1.1]戊烷基、雙環[3.2.1]辛烷基、8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經以下各者單取代或獨立多取代:羥基、氰基、鹵素、C1-6
烷基、C1-6
鹵烷基、C1-6
烷氧基、C1-6
鹵烷氧基、C3-8
環烷基、C3-8
環烷氧基、3員至8員芳香基或3員至8員雜環基,其可以進一步任選地經以下各者單取代或獨立多取代:鹵素、氘、羥基、氨基、氰基、C1-6
烷基、C1-6
鹵烷基、C1-6
烷氧基或C1-6
鹵烷氧基。
在一些實施例中,R2
選自:
”意指X1
與X2
之間的鍵及X2
與X3
之間的鍵可以是單鍵或雙鍵,其限制條件是X1
與X2
之間的鍵及X2
與X3
之間的鍵中的至少一個是單鍵;
R1
不存在,為鹵素或C1-6
烷基,其中所述C1-6
烷基可以任選地經羥基、鹵素或氘單取代或獨立多取代;
各R2
、R3
和R4
獨立地選自不存在、鹵素、羥基、氰基、C1-6
烷基、C1-6
烷氧基、-(CH2
)n
-Q,其可以任選地經以下各者單取代或獨立多取代:氘、羥基、氨基、氰基、鹵素、C1-6
烷基、C1-6
鹵烷基、(C≡N)-C1-6
烷基、C1-6
烷氧基、C1-6
鹵烷氧基、C3-8
環烷基、C3-8
環烷氧基、3員至8員芳香基或3員至8員雜環基,
其中n為0、1或2,Q為3員至8員飽和或不飽和碳環基,或3員至8員飽和或不飽和雜環基;
環A為5員至12員芳香基、具有1到5個選自氧、硫和氮的環雜原子的5員至12員雜芳香基、具有0到5個選自氧、硫和氮的環雜原子的8員至10員雙環,其中環A不為苯基。
在一些實施例中,R2
選自甲基、乙基、丙基、丁基、環丙基、環丁基、氧雜環丁烷基、環戊烷基、四氫呋喃基、環己烷基、四氫吡喃基、環庚烷基、呱啶基、苯基、吡啶基、吡啶酮基、氧雜環辛烷基、四氫吡喃基、二氫吡喃基、螺[3.3]庚烷基、螺[2.5]辛烷基、雙環[1.1.1]戊烷基、雙環[3.2.1]辛烷基、8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經以下各者單取代或獨立多取代:羥基、氰基、鹵素、C1-6
烷基、C1-6
鹵烷基、C1-6
烷氧基、C1-6
鹵烷氧基、C3-8
環烷基、C3-8
環烷氧基、3員至8員芳香基或3員至8員雜環基,其可以進一步任選地經以下各者單取代或獨立多取代:鹵素、氘、羥基、氨基、氰基、C1-6
烷基、C1-6
鹵烷基、C1-6
烷氧基或C1-6
鹵烷氧基。
在一些實施例中,R2
選自:
 ,
其可以任選地經羥基、氰基、氟、氯、溴、甲基、乙基、甲氧基、二氟甲基、二氟甲氧基或三氟甲氧基單取代或獨立多取代。
在一些實施例中,R2
為環己烷基或四氫吡喃基,其可以任選地經鹵素、C1-6
烷基或C1-6
烷氧基單取代或獨立多取代。
在一些實施例中,R1
為甲基、乙基、正丙基、異丙基、正丁基、仲丁基、叔丁基或異丁基,其可以任選地經羥基、鹵素或氘單取代或獨立多取代。
在一些實施例中,R1
為甲基、乙基、三氟甲基或三氘甲基。
在一些實施例中,環A為具有1個環雜原子氮的6員雜芳香基、具有2到3個選自氧、硫和氮的環雜原子的9員雙環,任選地,所述9員雙環為苯基或吡啶基稠合雙環,任選地,環A選自
,
其可以任選地經羥基、氰基、氟、氯、溴、甲基、乙基、甲氧基、二氟甲基、二氟甲氧基或三氟甲氧基單取代或獨立多取代。
在一些實施例中,R2
為環己烷基或四氫吡喃基,其可以任選地經鹵素、C1-6
烷基或C1-6
烷氧基單取代或獨立多取代。
在一些實施例中,R1
為甲基、乙基、正丙基、異丙基、正丁基、仲丁基、叔丁基或異丁基,其可以任選地經羥基、鹵素或氘單取代或獨立多取代。
在一些實施例中,R1
為甲基、乙基、三氟甲基或三氘甲基。
在一些實施例中,環A為具有1個環雜原子氮的6員雜芳香基、具有2到3個選自氧、硫和氮的環雜原子的9員雙環,任選地,所述9員雙環為苯基或吡啶基稠合雙環,任選地,環A選自 。
在一些實施例中,每個R3
和R4
獨立地選自不存在、鹵素、羥基、氰基、C1-6
烷基、CN-C1-6
烷基、C1-6
鹵烷基、C1-6
烷氧基、C1-6
鹵烷氧基、3員至8員飽和或不飽和雜環基,其中所述雜環基可以任選地進一步經C1-3
烷基單取代或獨立多取代。
在一些實施例中,環A為
。
在一些實施例中,每個R3
和R4
獨立地選自不存在、鹵素、羥基、氰基、C1-6
烷基、CN-C1-6
烷基、C1-6
鹵烷基、C1-6
烷氧基、C1-6
鹵烷氧基、3員至8員飽和或不飽和雜環基,其中所述雜環基可以任選地進一步經C1-3
烷基單取代或獨立多取代。
在一些實施例中,環A為 或
或 ,每個R3
和R4
獨立地選自不存在、甲基、氰基、甲氧基、氯、氰基-甲基、吡唑基、惡唑基,其中所述吡唑基或惡唑基可以任選地進一步經C1-3
烷基單取代或獨立多取代。
在一些實施例中,本文所提供的化合物具有式Ia的結構
,每個R3
和R4
獨立地選自不存在、甲基、氰基、甲氧基、氯、氰基-甲基、吡唑基、惡唑基,其中所述吡唑基或惡唑基可以任選地進一步經C1-3
烷基單取代或獨立多取代。
在一些實施例中,本文所提供的化合物具有式Ia的結構 和其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Ib的結構
和其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Ib的結構 和其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Ic的結構
和其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Ic的結構 和其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Id的結構
和其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Id的結構 和其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Ie的結構
和其藥學上可接受的鹽,其中R1
、R2
、R3
、R4
、環A如本文所定義。
在一些實施例中,本文所提供的化合物具有式Ie的結構 和其藥學上可接受的鹽,
其中,
X1
和X3
中的一個是N且另一個是C,短劃線“
和其藥學上可接受的鹽,
其中,
X1
和X3
中的一個是N且另一個是C,短劃線“ ”意指X1
與N之間的鍵及N與X3
之間的鍵可以是單鍵或雙鍵,其限制條件是X1
與N之間的鍵和N與X3
之間的鍵中的至少一個是單鍵;
R1
為C1-3
烷基,
R2
為環戊基、環己烷基、四氫吡喃基或8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經鹵素或C1-3
烷氧基單取代或獨立多取代,
Y1
、Y2
和Y3
各自獨立地為C或N,其限制條件為Y1
、Y2
和Y3
中的至少一個為N;
R5
為鹵素或C1-3
烷基,
R6
為C1-3
烷基。
在一些實施例中,式Ie的R2
為未經取代的環戊基、環己烷基、四氫吡喃基或8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經鹵素或C1-3
烷氧基單取代或獨立多取代。
在一些實施例中,式Ie的Y3
為N且Y1
和Y2
的至少一個為N。
在一些實施例中,式Ie的R5
為甲基。
式(I)的示範性化合物1到149闡述於下表1中。
”意指X1
與N之間的鍵及N與X3
之間的鍵可以是單鍵或雙鍵,其限制條件是X1
與N之間的鍵和N與X3
之間的鍵中的至少一個是單鍵;
R1
為C1-3
烷基,
R2
為環戊基、環己烷基、四氫吡喃基或8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經鹵素或C1-3
烷氧基單取代或獨立多取代,
Y1
、Y2
和Y3
各自獨立地為C或N,其限制條件為Y1
、Y2
和Y3
中的至少一個為N;
R5
為鹵素或C1-3
烷基,
R6
為C1-3
烷基。
在一些實施例中,式Ie的R2
為未經取代的環戊基、環己烷基、四氫吡喃基或8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經鹵素或C1-3
烷氧基單取代或獨立多取代。
在一些實施例中,式Ie的Y3
為N且Y1
和Y2
的至少一個為N。
在一些實施例中,式Ie的R5
為甲基。
式(I)的示範性化合物1到149闡述於下表1中。





















 應瞭解,為清楚起見而在單獨實施例的上下文中描述的本公開的某些特徵也可組合提供于單一實施例中。相反地,為簡潔起見描述於單個實施例的上下文中的本公開的各種特徵還可以分開地或以任何合適的子組合形式提供。
在本公開中的多處,描述了連接取代基。在結構明確需要鍵聯基團的情況下,關於所述基團所列的馬庫什變數(markush variable)應理解為鍵聯基團。例如,如果結構需要鍵聯基團並且所述變數的馬庫什群組定義列舉“烷基”,那麼應理解,所述“烷基”表示鍵聯亞烷基。
如本文所使用,當提到化學基團時,術語“經取代”意指所述化學基團有一個或多個氫原子被移除且經取代基置換。如本文所使用,術語“取代基”具有本領域中已知的普通含義並且是指共價連接至母基團或在適當時稠合至母基團的化學部分。如本文所用,術語“任選地經取代”或“任選地……經取代”意指化學基團可不具有取代基(即未經取代)或可具有一個或多個取代基(即經取代)。應理解,在給定原子處的取代受價數限制。
如本文所使用,術語“Ci-j
”指示碳原子數的範圍,其中i和j為整數,並且碳原子數的範圍包含端點(即i和j)和介於其間的每個整數點,並且其中j大於i。例如,C1-6
指示一至六個碳原子的範圍,包含一個碳原子、兩個碳原子、三個碳原子、四個碳原子、五個碳原子及六個碳原子。在一些實施例中,術語“C1-12
”指示1到12個,包含1到10個、1到8個、1到6個、1到5個、1到4個、1到3個或1到2個碳原子。
如本文所用,術語“烷基”無論作為另一術語的一部分或獨立地使用,都是指飽和或不飽和烴鏈,而後者可以進一步細分成具有至少一個雙鍵或三鍵的烴鏈(烯基或炔基)。在一些實施例中,烷基是指飽和烴鏈。以上提到的烴鏈可以是直鏈或分支鏈。術語“Ci-j
烷基”是指具有i至j個碳原子的烷基。飽和烷基的實例包含但不限於甲基、乙基、正丙基、異丙基、正丁基、叔丁基、異丁基、仲丁基;高級同系物,如2-甲基-1-丁基、正戊基、3-戊基、正己基、1,2,2-三甲基丁基等。不飽和烷基的實例包含但不限於乙烯基、正丙烯基、異丙烯基、正丁烯基、仲丁烯基、乙炔基、丙炔-1-基、丙炔-2-基等。“C1-6
烷基”的實例包含但不限於甲基、乙基、丙基、異丙基、正丁基、異丁基及叔丁基。“C1-3
烷基”的實例包含但不限於甲基、乙基、丙基及異丙基。
當“烷基”表示鍵聯亞烷基時,亞烷基的實例包含但不限於亞甲基、1,1-亞乙基、1,2-亞乙基、1,1-亞丙基、1,2-亞丙基、1,3-亞丙基、2,2-亞丙基、叔亞丁基等。
如本文所使用,術語“氨基”是指式“-NH2
”的基團。
如本文所用,術語“氨甲醯基”是指氨基羰基(即NH2
-C(=O)-)。
如本文所使用,術語“氰基”是指式“-C≡N”的基團。
如本文所使用,術語“鹵基”和“鹵素”是指氟基、氯基、溴基或碘基。
如本文所使用,術語“羥基”是指式“-OH”的基團。
如本文所用,術語“烷氧基”無論是作為另一術語的一部分或獨立地使用,都是指式-O-烷基的基團。
術語“Ci-j
烷氧基”意指烷氧基的烷基部分具有i至j個碳原子。烷氧基的實例包含但不限於甲氧基、乙氧基、丙氧基(例如正丙氧基和異丙氧基)、叔丁氧基等。“C1-12
烷氧基”的實例為甲氧基、乙氧基和丙氧基。
如本文所用,術語“羥基C1-12
烷基”是指式“-C1-12
烷基-OH”的基團,其中所述基團的烷基部分具有1至12個碳原子,並且一個或多個羥基可鍵聯到烷基部分中的任何碳原子。在一些實施例中,“Ci-j
烷基-OH”具有一個羥基。“C1-12
烷基-OH”的實例為羥甲基、1-羥乙基、2-羥乙基和1-羥異丙基。
如本文所用,術語“Ci-j
鹵烷基”是指經鹵素取代的(單取代或多取代的)Ci-j
烷基。“C1-12
鹵烷基”的實例為氟甲基、二氟甲基、三氟甲基、氟乙基、二氟乙基、三氟乙基、氯乙基和溴異丙基。“二氟乙基”的實例為1,1-二氟乙基。“三氟乙基”的實例為2,2,2-三氟乙基和1,2,2-三氟乙基。
“Ci-j
鹵烷氧基”的實例為氟甲氧基、二氟甲氧基或三氟甲氧基。“三氟乙氧基”的實例為2,2,2-三氟乙氧基和1,2,2-三氟乙氧基。
如本文所使用,術語“芳香基”或“芳香族S”不管是作為另一術語的一部分還是獨立地使用,都是指在形成環的原子之間具有交替的雙鍵和單鍵的環系統。在本公開中,術語“芳香基”或“芳香族”也意圖包含假芳香族。術語“偽芳香族”是指並非絕對芳香族的環系統,但其借助於電子的離域而穩定且表現方式與芳環類似。芳香基或芳香族基團可具有單環或多環。芳基的實例包含但不限於苯基、萘基、四氫萘基、茚滿基等。
如本文所使用,如本文所使用的術語“雜芳基”是指含有至少一個選自O、S、N、P等的成環雜原子的芳基。雜芳基包含但不限於呋喃基、噻吩基、吡啶基(pyridinyl)、三嗪基、吡啶基(pyridyl)、吡咯基、惡唑基、噻唑基、咪唑基、吡唑基、異惡唑基、異噻唑基、吲哚嗪基、吲哚基、異吲哚基、吲哚啉基、1,2,3-惡二唑基、1,2,4-惡二唑基、1,2,4-惡二唑-5-酮、1,2,3-三唑基、1,3,4-噻二唑基、噠嗪基、嘧啶基、吡嗪基、喹唑啉基、異喹唑啉基,1,3,5-三嗪基、1H噻吩並[2,3-c]吡唑基、噻吩並[2,3-b]呋喃基、3H-吲哚基、苯並[b]呋喃基、苯並[b]噻吩基、1H-吲唑基、苯並咪唑基、四唑基、尿苷基和胞嘧啶基。
如本文所用,術語“碳環基”無論作為另一術語的一部分或獨立地使用,都是指任何環,包含單環或多環(例如具有2或3個稠環、橋環或螺環),其中所有環原子為碳且含有至少三個成環碳原子。如本文所用,術語“螺環”是指兩個環通過一個單一共同原子連接的環系統;術語“稠環”是指兩個環共用兩個相鄰原子的環系統;並且術語“橋環”是指兩個環共用三個或更多個原子的環系統。
在一些實施例中,碳環基可以含有3至12個成環碳原子(即3員至12員碳原子)、3至10個成環碳原子、3至9個成環碳原子或3至8個成環碳原子。碳環基可為飽和、部分不飽和或完全不飽和的。在一些實施例中,碳環基可以是飽和環狀烷基。在一些實施例中,碳環基可為在環系統中含有至少一個雙鍵的不飽和環狀烷基。在一些實施例中,不飽和碳環基可以含有一個或多個芳香環。在一些實施例中,飽和或不飽和碳環基的一個或多個成環-CH2
-基團可經-C(O)-基團置換。
在一些實施例中,碳環基為單環烷基。在一些實施例中,碳環基為飽和單環烷基。飽和單環烷基的實例包含但不限於環丙基、環丁基、環戊基、環己基、環庚基、環辛基、環戊烯基、環己烯基等。
3員至8員“飽和或不飽和碳環基”為分別具有3至8個、3至6個或5至8個成環碳原子的飽和、部分不飽和或完全不飽和單環或多環環系統,其中一個或多個成環-CH2
-基團可以任選地經-C(O)-基團置換。
“3員至8員飽和或不飽和碳環基”的實例為C3-6
環烷基、環己基、環己烯基、環戊基、苯基、萘基和雙環[1.1.1]戊-1-基。“C3-8
環烷基”的實例為環丙基、環丁基、環戊基、環己基、環庚基和環辛基。術語“C3-8
環烷氧基”是指式“C3-8
環烷基-O-”的基團。
如本文所用,術語“雜環基”是指碳環基,其中一個或多個(例如1、2或3個)環原子經雜原子置換,所述雜原子包含但不限於O、S、N、P等。在一些實施例中,雜環基是飽和雜環基。在一些實施例中,雜環基為在環系統中具有一個或多個雙鍵的不飽和雜環基。在一些實施例中,雜環基為部分不飽和雜環基。在一些實施例中,雜環基為完全不飽和雜環基。在一些實施例中,不飽和雜環基可含有一個或多個芳環。在一些實施例中,雜環基的一個或多個成環-CH2
-基團可以任選地經-C(O)- 、 -S- 、 -S(O)-
或-S(O)2
- 基團
置換。在其中雜環基在其環系統中含有硫的一些實施例中,所述成環硫原子可任選地氧化以形成S-氧化物。在一些實施例中,雜環基經由其成環碳連接到化合物的其它部分。在一些實施例中,雜環基經由其成環氮連接到化合物的其它部分。
在一些實施例中,3員至8員飽和或不飽和單環或多環雜環基,其具有選自N、O或S的1、2或3個雜原子。
3員至8員“飽和或不飽和雜環基”為分別具有3到8個成環原子的飽和、部分不飽和或完全不飽和單環或多環(例如具有2或3個稠環、橋環或螺環),其中至少一個成環原子選自氮、硫或氧,除非另外規定,否則其可經由其成環碳或氮鍵聯到化合物的其它部分,其中飽和或不飽和雜環基的一個或多個成環-CH2
-基團可經-C(O)-、-S-、-S(O)-或-S(O)2
-基團置換,並且其中當雜環基在其環系統中含有硫時,所述環硫原子可以任選地氧化以形成S-氧化物。
示範性單環雜環基包含但不限於氧雜環丁烷基、1,1-二氧硫雜環丁烷基吡咯烷基、四氫呋喃基、四氫噻吩基、吡咯基、呋喃基、噻吩基、吡唑基、咪唑基、三唑基、惡唑基、噻唑基、呱啶基、呱啶基、呱嗪基、嗎啉基、吡啶基、吡嗪基、嘧啶基、噠嗪基、三嗪基、吡啶酮基、嘧啶酮基、吡嗪酮基、嘧啶酮基、噠嗪酮基、三嗪酮基等。
螺雜環基的實例包含但不限於螺吡喃基、螺惡嗪基等。稠合雜環基的實例包含但不限於苯基稠環或吡啶基稠環,如喹啉基、異喹啉基、喹喔啉基、喹嗪基、喹唑啉基、氮雜吲哚嗪基、喋啶基、色烯基、異色烯基、吲哚基、異吲哚基、吲哚嗪基、吲唑基、嘌呤基、苯並呋喃基、異苯並呋喃基、苯並咪唑基、苯並噻吩基、苯並噻唑基、哢唑基、吩嗪基、吩噻嗪基、啡啶基、咪唑並[1,2-a]吡啶基、[1,2,4]三唑並[4,3-a]吡啶基、[1,2,3]三唑並[4,3-a]吡啶基等。橋接雜環基的實例包含但不限於嗎啡烷基(morphanyl)、六亞甲基四氨基、8-氮雜-雙環[3.2.1]辛烷、1-氮雜-雙環[2.2.2]辛烷、1,4-二氮雜雙環[2.2.2]辛烷(DABCO)等。
除非另外說明,否則本公開的“化合物”意圖涵蓋所描繪的結構的所有立體異構體、幾何異構體和互變異構體。
術語“立體異構體”是指不對稱化合物(例如具有一個或多個不對稱取代的碳原子或“不對稱中心”的化合物)的各種立體異構構型(例如對映異構體、非對映異構體和外消旋體)中的任一種。含有不對稱中心的本公開化合物可以光學活性(對映異構體或非對映異構體)或光學非活性(外消旋)形式分離。術語“對映異構體”包含不互為可重疊的鏡像的立體異構體對。一對對映異構體的1:1混合物為“外消旋混合物”。術語“非對映異構體(diastereomer)”或“非對映異構體(diastereoisomer)”包含具有至少二個不對稱原子但不互為鏡像的立體異構體。含有一個或多個不對稱中心的某些化合物可產生對映異構體、非對映異構體或其它立體異構形式,其可根據卡恩-英格爾-普雷洛格(Cahn-Ingold-Prelog) R-S系統,在各不對稱中心處關於絕對構型定義為(R)-或(S)-。絕對構型未知的拆分的化合物可在不對稱中心處使用術語“或”指示。關於如何從外消旋混合物製備光學活性形式的方法是本領域中已知的,如通過HPLC拆分或立體選擇性合成。
術語“幾何異構體”或“順式和反式異構體”是指具有相同式但其官能團在三維空間中旋轉至不同取向的化合物。
術語“互變異構體”包含處於具有相同式和總電荷的化合物的異構質子化狀態的質子轉移互變異構體。質子轉移互變異構體的實例包含但不限於酮-烯醇對、醯胺-亞胺酸對、內醯胺-內醯亞胺對、烯胺-亞胺對,以及環狀形式,在環狀形式中,質子可以佔用雜環系統的兩個或更多個位置,例如1H-咪唑和3H-咪唑、1H-1,2,4-三唑、2H-1,2,4-三唑和4H-1,2,4-三唑、1H-異吲哚和2H-異吲哚,以及1H-吡唑和2H-吡唑。互變異構體可處於平衡狀態或通過適當取代而空間鎖定成一種形式。除非另外說明,否則通過名稱或結構標識為一種特定互變異構形式的本公開化合物意圖包含其它互變異構形式。
本公開的“化合物”還意圖涵蓋化合物中原子的所有同位素。原子的同位素包含具有相同原子數但質量數不同的原子。例如,除非另外規定,否則本公開“化合物”中的氫、碳、氮、氧、磷、硫、氟、氯、溴或碘意圖還包含其同位素,如但不限於1
H、2
H、3
H、11
C、12
C、13
C、14
C、14
N、15
N、16
O、17
O、18
O、31
P、32
P、32
S、33
S、34
S、36
S、17
F、19
F、35
Cl、37
Cl、79
Br、81
Br、127
I和131
I。在一些實施例中,氫包含氕、氘和氚。在一些實施例中,術語“經氘取代”或“氘取代的”用氘置換化學基團中的氫的另一種同功異型物(例如氕)。在一些實施例中,碳包含12
C和13
C。在一些實施例中,本公開的“化合物”僅涵蓋化合物中氫的同位素。在一些實施例中,本公開的“化合物”僅涵蓋呈天然豐度的原子的同位素。
還應理解,本公開的“化合物”可以呈溶劑化形式以及非溶劑化形式,例如水合形式、固體形式存在,並且本公開意圖涵蓋所有此類溶劑化形式和非溶劑化形式。
還應理解,本公開的“化合物”可以藥學上可接受的鹽形式存在。
如本文所用,術語“藥學上可接受”是指在合理醫學判斷的範圍內,適用於與人類和動物的組織接觸,而無過度毒性、刺激、過敏反應或其它問題或併發症,與合理的效益/風險比相稱的化合物、材料、組合物和/或劑型。在一些實施例中,藥學上可接受的化合物、材料、組合物和/或劑型是指被管理機構(如美國食品和藥物管理局(U.S. Food and Drug Administration)、中國食品和藥物管理局(China Food and Drug Administration)或歐洲藥物管理局(European Medicines Agency))批准或公認藥典(如美國藥典(U.S.Pharmacopoeia)、中國藥典(China Pharmacopoeia)或歐洲藥典(European Pharmacopoeia))中所列的可用於動物且特別是人類的那些化合物、材料、組合物和/或劑型。
如本文所用,“藥學上可接受的鹽”是指本公開化合物的衍生物,其中母體化合物通過將現有酸性部分(例如羧基等)或堿部分(例如胺、鹼金屬等)轉化為其鹽形式而修飾。在許多情況下,本公開化合物能夠憑藉氨基和/或羧基或其類似基團的存在而形成酸和/或堿鹽。藥學上可接受的鹽為保留母體化合物的生物有效性和特性,通常不會在生物學上或其它方面不合需要的酸和/或堿鹽。本公開化合物的適合的藥學上可接受的鹽包含例如酸加成鹽,其可以衍生自例如無機酸(例如鹽酸、氫溴酸、硫酸、硝酸、磷酸等)或有機酸(例如甲酸、乙酸、丙酸、乙醇酸、草酸、順丁烯二酸、丙二酸、琥珀酸、反丁烯二酸、酒石酸、苯均三酸、檸檬酸、乳酸、苯乙酸、苯甲酸、扁桃酸、甲烷磺酸、萘二磺酸、乙烷磺酸、甲苯磺酸、三氟乙酸、水楊酸、磺基水楊酸等)。在一些實施例中,本公開化合物的藥學上可接受的鹽為甲酸鹽。在一些實施例中,本公開化合物的藥學上可接受的鹽是TFA鹽。
本公開化合物的適合的藥學上可接受的鹽還包含例如堿加成鹽,其可以衍生自例如無機堿(例如週期表的第I列到第XII列的金屬(如鈣、鎂、鐵、銀、鋅、銅等)的鈉鹽、鉀鹽、銨鹽和氫氧化物、碳酸鹽、碳酸氫鹽)或有機堿(例如伯胺、仲胺和叔胺、包含天然存在的經取代胺的經取代胺、環胺、鹼性離子交換樹脂等)。某些有機胺包含但不限於異丙胺、苯乍生(benzathine)、膽酸鹽、二乙醇胺、二乙胺、賴氨酸、葡甲胺、呱嗪以及緩血酸胺。所屬領域的技術人員應理解,除了實例中所示以外,添加用於形成酸/堿加成鹽的酸或堿也是可能的。其它適合的鹽的列表可見于例如《雷明頓氏藥物科學(Remington's Pharmaceutical Sciences)》, 第20版, 馬克出版公司(Mack Publishing Company), 賓夕法尼亞州伊斯頓(Easton, Pa.), (1985);以及Stahl和Wermuth的《藥用鹽手冊:特性、選擇和使用(Handbook of Pharmaceutical Salts: Properties, Selection, and Use)》(德國魏因海姆的Wiley-VCH(Wiley-VCH, Weinheim, Germany), 2002)中。在一些實施例中,本公開化合物的適合的藥學上可接受的鹽為無機堿鹽。
本公開還包含本公開化合物的活性中間物、活性代謝物和前藥。如本文所用,“活性中間物”是指合成方法中的中間化合物,其展現與最終合成的化合物相同或基本上相同的生物活性。
如本文所用,“活性代謝物”是指通過在動物或人體內代謝或生物轉化產生的本公開化合物或其鹽或前藥的分解或最終產物,其展現與指定化合物相同或基本上相同的生物活性。此類代謝物可以由例如施用的化合物或鹽或前藥氧化、還原、水解、醯胺化、脫醯胺、酯化、去酯化、酶裂解等得到。
如本文所用,“前藥”是指當施用至動物或人類個體時釋放活性母體藥物的任何化合物或偶聯物。前藥可通過以一種方式修飾化合物中存在的官能團製備,所述方式使得修飾在常規操作中或在體內可由母體化合物裂解。前藥包含其中羥基、氨基、硫氫基或羧基鍵結到任何基團,使得當將其施用給哺乳動物個體時可分別裂解形成游離羥基、氨基、硫氫基或羧基的化合物。前藥的實例包含但不限於本公開化合物中醇和胺官能團的乙酸酯、甲酸酯和苯甲酸酯衍生物。前藥的製備和使用論述於THiguchi和V. Stella, “作為新穎遞送系統的前藥(Pro-drugs as Novel Delivery Systems)”, 《美國化學會會議論文集(A.C.S. Symposium Series)》第14卷,以及《藥物設計中的生物可逆載體(Bioreversible Carriers in Drug Design)》, Edward B.Roche編, 美國藥劑師協會和培格曼出版社(American Pharmaceutical Association and Pergamon Press), 1987,二者特此以全文引用的方式併入。
本文公開了可以選擇性抑制DNA-PK的新穎化合物或藥學上可接受的鹽。當與其它臨床上可用的DNA-PK抑制劑相比較時,前文公開的化合物或其藥學上可接受的鹽展現某些改進的特性,例如更高的BBB穿透(因此使其潛在地可用於治療已經轉移到CNS,特別是腦轉移和軟腦膜轉移的癌症)、更好的效力等。其還可具有相比已知DNA-PK調節劑有利的毒性概況和/或有利的代謝或藥物動力學概況。
因此,此類化合物或其藥學上可接受的鹽可尤其適用於治療癌症,尤其是具有腦部癌轉移的那些癌症。合成方法
本文提供的化合物,包含其鹽、酯、水合物或溶劑化物或立體異構體的合成是在實例中的合成方案中說明。本文提供的化合物可使用任何已知的有機合成技術製備並且可以根據多種可能的合成途徑中的任一種合成,且因此,這些方案只是說明性的且不打算限制可用于製備本文提供的化合物的其它可能的方法。此外,所述方案中的步驟是為了更好地說明且在適當時可以改變。出於研究和可能提交給管理機構的目的,實例中的化合物實施例是在中國合成。
用於製備本公開化合物的反應可以在適合溶劑中進行,這些溶劑可以由有機合成領域的技術人員容易地選擇。適合溶劑可以在反應進行的溫度下,例如在從溶劑的冷凍溫度至溶劑的沸騰溫度範圍內的溫度下基本上不與起始物質(反應物)、中間物或產物反應。指定反應可以在一種溶劑或多於一種溶劑的混合物中進行。取決於特定反應步驟,用於特定反應步驟的適合溶劑可以由熟練技術人員選擇。
本公開化合物的製備可以涉及各種化學基團的保護和脫保護。對於保護和脫保護的需求,以及適當保護基的選擇可以由本領域技術人員容易地確定。保護基的化學性質可見於例如T. W. Greene和P. G. M. Wuts, 《有機合成中的保護基(Protective Groups in Organic Synthesis)》, 第3版,威利父子公司(Wiley & Sons, Inc.), 紐約(1999),其以全文引用的方式併入本文中。
可以根據本領域中已知的任何適合方法監測反應。例如,可以通過光譜手段,如核磁共振光譜(例如1
H或13
C)、紅外光譜、分光光度法(例如紫外-可見光)、質譜,或通過色譜法,如高壓液相色譜(HPLC)、液相色譜-質譜(LCMS)或薄層色譜(TLC)來監測產物形成。本領域的技術人員可以通過多種方法,包含高效液相色譜法(HPLC)(“製備型LC-MS純化:改善的化合物特異性方法優化(Preparative LC-MS Purification:Improved Compound Specific Method Optimization)”, Karl F.Blom, Brian Glass, Richard Sparks, Andrew P.Combs, 《組合化學雜誌(J.Combi.Chem.)》, 2004, 6(6), 874-883,以全文引用的方式併入本文中)和正相二氧化矽色譜法純化化合物。
如本文所使用的縮寫定義如下:“1×”或“×1”表示一次,“2×”或“×2”表示兩次,“3×”或“×3”表示三次,“4×”或“×4”表示四次,“5×”或“×5”表示兩次,“℃”表示攝氏度,“eq”或“eq.”表示當量,“g”表示克,“mg”表示毫克,“L”表示升,“mL”或“ml”表示毫升,“μL”表示微升,“N”表示正常,“M”表示摩爾,“mmol”表示毫摩爾,“min”表示分鐘,“h”或“hr”表示小時,“r.t.”或“rt”表示室溫,“atm”表示氛圍,“psi”表示每平方英寸磅數,“conc.”表示濃縮物,“sat”或“sat'd”表示飽和,“MS”或“Mass Spec”表示質譜,“ESI”表示電噴射電離質譜,“LCMS”表示液相色譜質譜法,“HPLC”表示高壓液相色譜,“RP”表示反相,“TLC”或“tlc”表示薄層色譜,“SM”表示起始物質,“NMR”表示核磁共振光譜,“1
H”表示質子,“δ”表示△,“s”表示單重峰,“d”表示雙重峰,“t”表示三重峰,“q”表示四重峰,“m”表示多重峰,“br”表示廣譜,且“Hz”表示赫茲。“α”、“β”、“R”、“S”、“E”以及“Z”是本領域技術人員熟悉的立體化學名稱。藥物組合物
本公開提供了包括至少一種本公開化合物的藥物組合物。在一些實施例中,所述藥物組合物包括多於一種本公開的化合物。在一些實施例中,所述藥物組合物包含一種或多種本公開的化合物和藥學上可接受的載劑。
藥學上可接受的載劑是本領域中的常規藥物載劑,其可以按藥學領域中眾所周知的方式製備。在一些實施例中,本公開的化合物可以與藥學上可接受的載劑混合以製備藥物組合物。
如本文所使用,術語“藥學上可接受的載劑”是指將本文提供的化合物從一個位置、體液、組織、器官(內部或外部)或身體的一部分載運或運輸至另一位置、體液、組織、器官或身體的一部分所涉及的藥學上可接受的材料、組合物或媒劑,如液體或固體填充劑、稀釋劑、賦形劑、溶劑或囊封材料。藥學上可接受的載劑可以是可用於接觸動物組織,而無過量毒性或不良作用的媒劑、稀釋劑、賦形劑或其它材料。示範性藥學上可接受的載劑包含糖、澱粉、纖維素、麥芽、黃芪膠、明膠、林格氏溶液(Ringer's solution)、海藻酸、等滲生理鹽水、緩衝劑等。可以用於本公開中的藥學上可接受的載劑包含本領域中一般已知的那些,如以引用的方式併入本文中的《雷氏藥學大全》, Mack Pub. Co., 新澤西州(New Jersey)(1991)中公開那些。
可以充當藥學上可接受的載劑的材料的一些實例包含:(1)糖,如乳糖、葡萄糖和蔗糖;(2)澱粉,如玉米澱粉和馬鈴薯澱粉;(3)纖維素和其衍生物,如羧甲基纖維素鈉、乙基纖維素和乙酸纖維素;(4)粉末狀黃芪膠;(5)麥芽;(6)明膠;(7)滑石;(8)賦形劑、如可哥脂和栓劑蠟;(9)油,如花生油、棉籽油、紅花油、芝麻油、橄欖油、玉米油和大豆油;(10)二醇,如丙二醇;(11)多元醇,如甘油、山梨糖醇、甘露醇和聚乙二醇;(12)酯,如油酸乙酯和月桂酸乙酯;(13)瓊脂;(14)緩衝劑,如氫氧化鎂和氫氧化鋁;(15)海藻酸;(16)無熱原質水;(17)等滲生理鹽水;(18)林格氏溶液;(19)醇,如乙醇和丙醇;(20)磷酸鹽緩衝溶液;以及(21)藥物配製物中採用的其它無毒可相容物質,如丙酮。
藥物組合物可視需要含有藥學上可接受的輔助物質以接近生理條件,如pH值調節劑和緩沖劑、毒性調節劑等,例如乙酸鈉、氯化鈉、氯化鉀、氯化鈣、乳酸鈉等。
藥物組合物的形式取決於多種標準,包含但不限於施用途徑、疾病程度或待施用的劑量。所述藥物組合物可以被配製成經口、經鼻、直腸、經皮、靜脈內或肌肉內施用。舉例來說,用於經鼻施用的劑型可以方便地配製為氣霧劑、溶液、滴劑、凝膠或乾粉;用於鼻內施用的劑型可以配製為流體配製物。根據希望的施用途徑,所述藥物組合物可以被配製成片劑、膠囊、丸劑、糖衣藥丸、散劑、顆粒劑、藥囊、扁囊劑、口含錠、懸浮液、乳液、溶液、糖漿、氣霧劑(呈固體形式或在液體介質中)、噴霧劑、油膏、糊漿、乳膏、洗劑、凝膠劑、貼片、吸入劑或栓劑形式。
藥物組合物可以被配製用於在通過採用本領域中已知的程式施用給患者之後,提供活性成分的快速、持續或延遲釋放。在一些實施例中,藥物組合物被配製成持續釋放形式。如本文所使用,術語“持續釋放形式”是指活性劑從藥物組合物釋放以使其變得在延長的時間段(延長釋放)內或在某一位置(控制釋放)可供個體,主要是個體的胃腸道生物吸收。在一些實施例中,所述延長的時間段可以是約1小時至24小時、2小時至12小時、3小時至8小時、4小時至6小時、1至2天或更長時間。在某些實施例中,所述延長的時間段是至少約4小時、至少約8小時、至少約12小時或至少約24小時。所述藥物組合物可以被配製成片劑形式。例如,活性劑的釋放速率不僅可以通過活性劑溶解於胃腸液中且隨後獨立於pH自片劑或丸劑擴散進行控制,而且還受片劑崩解和溶蝕的物理過程影響。在一些實施例中,“控制釋放的醫學應用(Medical Applications of Controlled Release)”, Langer和Wise(編), CRC Pres., 佛羅里達州波卡拉頓(Boca Raton, Florida)(1974);“控制性藥物生物利用率(Controlled Drug Bioavailability)”, 《藥品設計和性能(Drug Product Design and Performance)》, Smolen和Ball(編), Wiley, 紐約(1984);Ranger和Peppas, 1983, 《高分子科學-高分子化學評述(J Macromol.Sci.Rev. Macromol Chem.)》23:61中所公開的聚合材料可以用於持續釋放;還參見Levy等人, 1985, 《科學(Science)》228:190;During等人, 1989, 《神經病學年評(Ann. Neurol.)》25:351;Howard等人, 1989, 《神經外科雜誌(J. Neurosurg.)》71:105。以上參考文獻都以全文引用的方式併入本文中。
在某些實施例中,藥物組合物包括約0.0001mg到約100mg的本公開化合物(例如,約0.0001mg至約10mg、約0.001mg至約10mg、約0.01mg至約10mg、約0.1mg至約10mg、約0.1mg至約5mg、約0.1mg至約4mg、約0.1mg至約3mg、約0.1mg至約2mg、約0.1mg至約1mg、約0.1mg至約0.5mg、約1mg至約10mg、約1mg至約5mg、約5mg至約10mg、約5mg至約20mg、約5mg至約30mg、約5mg至約40mg、約5mg至約50mg、約10mg至約100mg、約20mg至約100mg、約30mg至約100mg、約40mg至約100mg、約50mg至約100mg)。每天每位個體的適合劑量可以是約0.1mg至約10mg,優選地是約0.1mg至約5mg、約5mg至約10mg、或約1mg至約5mg。
在某些實施例中,藥物組合物可按單位劑型配製,每種劑量含有約0.0001mg至約10mg、約0.001mg至約10mg、約0.01mg至約10mg、約0.1mg至約10mg、約0.1mg至約5mg、約0.1mg至約4mg、約0.1mg至約3mg、約0.1mg至約2mg、約0.1mg至約1mg、約0.1mg至約0.5mg、約1mg至約10mg、約5mg至約10mg、約5mg至約20mg、約5mg至約30mg、約5mg至約40mg、約5mg至約50mg、約10mg至約100mg、約20mg至約100mg、約30mg至約100mg、約40mg至約100mg、約50mg至約100mg的本公開化合物。術語“單位劑型”是指適合以單一劑量用於人類個體和其他哺乳動物的物理離散單元,每個單元含有與適合藥物載劑結合的經計算以產生所希望的治療作用的預定量的活性材料。
在一些實施例中,藥物組合物包括一種或多種本公開的化合物作為第一活性成分,並且還包含第二活性成分。第二活性成分可為所屬領域中已知的任何免疫調節劑或抗腫瘤劑,包含但不限於化學治療劑、免疫治療劑、細胞信號轉導抑制劑、細胞信號轉導抑制劑、烷化劑、拓撲異構酶抑制劑、有絲分裂抑制劑、抗激素劑等。此類免疫調節劑或抗腫瘤劑的實例為鉑類化學治療劑(例如順鉑(DDP)、卡鉑(CBP)、硫酸根-1,2-二氨基環己烷鉑(SHP)、奈達鉑(Nedaplatin)、奧沙利鉑(Oxaliplatin)(OXA)、樂鉑(Laboplatin))、多烯紫杉醇(Docetaxel)、太平洋紫杉醇(Paclitaxel)、多柔比星(Doxorubicin)、依託泊苷(Etoposide)、米托蒽醌(Mitoxantrone)、CTLA-4抑制劑、抗CTLA-4抗體、PD-1抑制劑、PD-L1抑制劑、抗PD-1/PD-L1抗體、CD39抑制劑、抗CD39抗體、CD73抑制劑、抗CD73抗體、CCR2抑制劑、抗CCR2抗體、EGFR抑制劑、CDK 4/6抑制劑、MELK抑制劑、OX40促效劑、抗雄激素抑制劑、IgG4同型抗體、酪氨酸激酶抑制劑、DNA甲基轉移酶抑制劑、Hsp90抑制劑、FGFR抑制劑、mTOR抑制劑、芳香酶抑制劑、VEGF抑制劑、LHRH拮抗劑、PI3K抑制劑、AKT抑制劑、極光激酶抑制劑、MEK抑制劑、HDAC抑制劑、BET抑制劑、PIK3CA抑制劑、蛋白酶體抑制劑、其他SERD、法呢基轉移酶抑制劑、VEGF-A抗體、ErbB3 (Her3)抗體、蛋白酶體抑制劑、蛋白激酶Cβ抑制劑、抗IGF-1R抗體、抗HER2抗體、SERM、IGF抑制劑、抗IgG抗體等。用於治療癌症或腫瘤的抗腫瘤劑的代表性實例可包含但不限於順鉑、卡鉑、SHP、奈達鉑、奧沙利鉑、樂鉑、多烯紫杉醇、太平洋紫杉醇、多柔比星、依託泊苷、米托蒽醌、長春新堿、長春堿、吉西他濱(gemcitabine)、環磷醯胺、苯丁酸氮芥(chlormabucil)、卡莫司汀(carmustine)、甲胺喋呤、氟尿嘧啶、放線菌素、表柔比星(epirubicin)、蒽環黴素、博萊黴素、絲裂黴素-C、伊立替康(irinotecan)、拓朴替康(topotecan)、替尼泊甙(teniposide)白介素、干擾素、曲美單抗(tremelimumab)、伊匹單抗(ipilimumab)、派立珠單抗(pembrolizumab)、納武單抗(nivolumab)、阿維魯單抗(avelumab)、度伐單抗(durvalumab)、阿特珠單抗(atezolizumab)、IPH 52、IPH 53、CPI-006、普洛利單抗(plozalizumab)、MLN1202、西妥昔單抗(cetuximab)、拉帕替尼(lapatinib)、埃羅替尼(erlotinib)、吉非替尼(gefitinib)、來那替尼(neratinib)、曲妥珠單抗(trastuzumab)、阿多-曲妥珠單抗恩他新(ado-trastuzumab emtansine)、帕妥珠單抗(pertuzumab)、MCLA-128、阿那曲唑(anastrazole)、雷諾昔酚(raloxifene)、G1T38、他莫昔芬(tamoxifen)、戈舍瑞林(goserelin)、恩雜魯胺(enzalutamide)、伏立諾他(vorinostat)、恩替諾特(entinostat)、舒尼替尼(sunitinib)、帕唑帕尼(pazopanib)、貝伐單抗(bevacizumab)、蘭比珠單抗(ranibizumab)、呱加他尼(pegaptanib)、西地尼布(cediranib)、達沙替尼(dasatinib)、GDC-0980、吉達昔布(gedatolisib)、阿普昔布(alpelisib)、BKM120、考帕昔布(copanlisib)、AZD8835、GDC-0941、塔瑟昔布(taselisib)、替西羅莫司(temsirolimus)、依維莫司(everolimus)、薩帕塞替(sapanisertib)、AZD5363、MK2206、帕尼單抗(panitumumab)、派立珠單抗(pembrolizumab)、索拉非尼(sorafenib)、帕博希布(palbociclib)、玻瑪西林(abemaciclib)、瑞博西林(ribociclib)、克唑替尼(crizotinib)、多韋替尼(dovitinib)、蘆可替尼(ruxolitinib)、阿紮胞苷(azacitidine)、CC-486、HSP90加利特皮(HSP90 ganetespib)、Debio 1347、伊達替尼(erdafitinib)、維瑟塞替(vitusertib)、阿立塞替(alisertib)、司美替尼(selumetinib)、G-5829、GSK525762、MLN9708、GDC-0810、AFP464、替吡法尼(tipifarnib)、塞利班單抗(seribantumab)、硼替佐米(bortezomib)、恩紮妥林(enzastaurin)、AVE1642、森吐珠單抗(xentuzumab)、達羅吐珠單抗(dalotuzumab)、AMG479等。
此類抗腫瘤劑的實例還可在V. T. Devita和S. Hellman(編)的《癌症原理和腫瘤學實踐(Cancer Principles and Practice of Oncology)》,第6版(2001年2月15日), 利平科特威廉姆斯和維爾金斯出版社(Lippincott Williams & Wilkins Publishers)中找到。本領域的普通技術人員還將能夠基於藥物的特定特徵和所涉及的癌症辨別哪些藥劑組合將是有用的。
根據本公開的這一方面,提供適用於治療癌症的組合,其包括如上文所定義的式(I)化合物或其藥學上可接受的鹽;以及上文所列之免疫調節劑或抗腫瘤劑中的任一種。
因此,在本公開的另一方面,提供一種與選自上文所列之免疫調節劑或化學治療劑的免疫調節劑或化學治療劑組合的式(I)化合物或其藥學上可接受的鹽。
本文中,在使用術語“組合”的情況下,應理解,這是指同時、分別或依序施用。在一些實施例中,“組合”是指同時施用。在本公開的另一方面,“組合”是指分開施用。在本公開的另一方面,“組合”是指依序施用。在依序或分別施用的情況下,施用第二組分的延遲不應使得失去所述組合的有益作用。
根據本公開的另一個方面,提供一種藥物組合物,其包括式(I)化合物或其藥學上可接受的鹽與選自以上列出的免疫調節劑或抗腫瘤劑的免疫調節劑或抗腫瘤劑的組合,以及藥學上可接受的稀釋劑或載劑。
根據本公開的另一個方面,提供一種藥物組合物,其包括式(I)化合物或其藥學上可接受的鹽與選自以上列出的一者的免疫調節劑或抗腫瘤劑的組合,以及藥學上可接受的稀釋劑或載劑,用於產生免疫調節或抗癌作用。
根據本公開的另一方面,提供一種藥物組合物,其包括式(I)化合物或其藥學上可接受的鹽與選自以上列出的一者的免疫調節劑或抗腫瘤劑的組合,以及藥學上可接受的稀釋劑或載劑,用於治療DNA-PK相關病症,例如NSCLC、RCC、前列腺癌或乳癌等。
根據本公開的另一方面,提供一種試劑盒,其包括式(I)化合物或其藥學上可接受的鹽與選自上文所列的一者的免疫調節劑或抗腫瘤劑的組合。
根據本公開的另一方面,提供一種試劑盒,其包含:
a)呈第一單位劑型的式(I)化合物或其藥學上可接受的鹽;
b)呈第二單位劑型的選自上文所列的一者的免疫調節劑或抗腫瘤劑;和
c)用於容納所述第一和第二劑型的容器。
除了用於治療藥物以外,式(I)化合物或其藥學上可接受的鹽還適用作體外和體內測試系統的開發和標準化中的藥理學工具,所述測試系統用於評估DNA-PK在如貓、狗、兔、猴、大鼠和小鼠的實驗動物中的活性或表達,作為探求新型治療劑的一部分。
在以上其它藥物組合物、過程、方法、用途和藥物製造特徵中,本文所述的本公開化合物的替代和優選實施例也適用。治療方法
本公開提供一種治療DNA-PK相關病症的方法,其包括向個體施用有效量的一種或多種本公開的化合物、其藥學上可接受的鹽或藥物組合物。
本公開還提供一種治療DNA-PK相關病症的方法。在某些實施例中,所述方法包括向個體施用有效量的一種或多種本公開的化合物、其藥學上可接受的鹽或藥物組合物。
如本文所用,術語“DNA-PK相關病症”是指發作或發展或二者與DNA-PK的表達或活性有關的疾病。實例包含(但不限於)過度增生性病症(例如,癌症)。
在一些實施例中,DNA-PK相關病症為癌症,優選為過度表達DNA-PK的癌症。“過度表達DNA-PK的癌症”是與相同組織類型的非癌性細胞相比在癌症或腫瘤細胞中具有顯著較高水準的DNA-PK蛋白質的癌症。這種過度表達可能是由基因擴增或增加的轉錄或轉譯引起的。通過評估細胞上存在的DNA-PK蛋白質水準的增加(例如通過免疫組織化學分析;IHC),可在診斷或預後分析中測定DNA-PK過度表達。替代地或另外,可測量細胞中編碼DNA-PK的核酸的水準,例如通過螢光原位雜交(FISH;參見1998年10月公佈的WO98/45479)、DNA印跡法或聚合酶鏈反應(PCR)技術,如實時定量PCR(RT-PCR)(方法(Methods) 132: 73-80 (1990))。除了上述分析之外,所屬領域的技術人員可使用各種體內分析。例如,可將患者體內的細胞暴露於抗體,所述抗體任選地用可檢測標記(例如放射性同位素)標記,並且例如通過外部掃描放射性或通過分析從先前暴露于抗體的患者取得的活組織檢查,可評估抗體與患者細胞的結合。
確切地說,癌症包含不限於肺癌(例如非小細胞肺癌(NSCLC)、小細胞肺癌、肺腺癌、大細胞肺癌、鱗狀細胞肺癌)、腎細胞癌(RCC)、前列腺癌、乳癌、卵巢癌、子宮內膜癌、子宮頸癌、骨癌、子宮癌、結腸癌、白血病、成膠質細胞瘤、黑素瘤、軟骨肉瘤、腦癌、膽管癌、骨肉瘤、淋巴瘤、腺瘤、骨髓瘤、肝細胞癌、腎上腺皮質癌、胰腺癌、膀胱癌、肝癌、胃癌、結腸直腸癌、食道癌、睾丸癌、皮膚癌、腎癌、間皮瘤、成神經細胞瘤、甲狀腺癌、頭頸癌、食道癌、眼癌、鼻咽癌或口腔癌。在一些實施例中,癌症為NSCLC、RCC、前列腺癌或乳癌。除非另外規定,否則如本文中提到的癌症可處於任何分期。在一些實施例中,癌症為早期癌症。在一些實施例中,癌症為局部晚期癌症。在一些實施例中,癌症為局部晚期和/或轉移癌。在一些實施例中,癌症為侵入性癌症。在一些實施例中,癌症為對現有療法具有抗性的癌症。
如本文所使用,術語“治療(treatment/treat/treating)”是指逆轉、緩解如本文所描述的疾病或病症或其一種或多種症狀,延遲其發作或抑制其進展。在一些實施例中,治療可在已出現一或多種症狀後施用。在其它實施例中,治療可在症狀不存在的情況下施用。例如,治療可以在症狀發作之前施用給易感個體(例如根據症狀病史和/或根據遺傳學或其它易感因素)。也可以在症狀消退之後繼續治療,例如以預防或延遲其復發。
在一些實施例中,本文所提供的一種或多種化合物、其藥學上可接受的鹽或藥物組合物是通過腸胃外途徑或非腸胃外途徑施用。在一些實施例中,所述一種或多種化合物、其藥學上可接受的鹽、水合物、溶劑化物或立體異構體或藥物組合物是經口、經腸、經頰、經鼻、鼻內、經粘膜、經表皮、透皮、經皮、經眼、肺、經直腸、舌下、經陰道、經表面、皮下、靜脈內、肌肉內、動脈內、鞘內、囊內、眶內、心內、皮內、腹膜內、經氣管、表皮下、關節內、囊下、脊椎內、蛛網膜下或胸骨內施用。
本文提供的化合物可以呈純形式、與其它活性成分組合或呈本公開的藥物組合物形式施用。在一些實施例中,本文所提供的化合物可以與本領域中已知的一種或多種抗癌劑或消炎劑組合同時或依序向有需要的個體施用。這類組合中的個別化合物是在分開的或組合的藥物組合物中依序或同時施用。優選地,個別化合物將在組合的藥物組合物中同時施用。所屬領域的技術人員將瞭解已知治療劑的適當劑量。
在一些實施例中,施用是一天一次、一天兩次、一天三次或每兩天一次、每三天一次、每四天一次、每五天一次、每六天一次、一週一次進行。
如本文所提供的化合物或其藥學上可接受的鹽的治療有效量將取決於本領域中已知的各種因素,如體重、年齡、既往病史、當前藥物、個體的健康狀態和交叉反應、過敏、敏感和不良副作用的可能性,以及施用途徑和疾病發展程度。如由這些和其它情況或要求所指示,所屬領域的技術人員(例如醫生或獸醫)可按比例減少或增加劑量。
在一些實施例中,本文所提供的一種或多種化合物、其藥學上可接受的鹽或藥物組合物經口施用。對於經口施用,實現所希望的目標的任何劑量都是適當的。在一些實施例中,適合日劑量是在約0.001到100mg之間,優選地在0.1mg與5g之間,更優選地在5mg與1g之間,更優選地在10mg與500mg之間,並且施用是一天一次、一天兩次、一天三次、每天或一周3到5天進行。在一些實施例中,本文所提供的一種或多種化合物、其藥學上可接受的鹽或藥物組合物的劑量的範圍在每天約0.0001mg,優選地0.001mg、0.01mg、0.1mg、0.2mg、0.3mg、0.4mg、0.5mg、0.6mg、0.7mg、0.8mg、0.9mg、1mg、2mg、3mg、4mg、5mg、6mg、7mg、8mg、9mg、10mg之間。化合物的用途
在某些實施例中,本公開提供本公開的化合物、其藥學上可接受的鹽或藥物組合物在製備用於治療DNA-PK相關病症的藥劑中的用途。在某些實施例中,DNA-PK相關病症包含癌症。
本公開中的化合物和其藥物組合物可用於預防或治療哺乳動物(尤其人類)中的DNA-PK相關病症(表達或活性)中的任一種的發作或發展。
在此類情形中,本公開還提供一種篩選適於用單獨的本公開的化合物或藥物組合物或其與其它成分(例如第二活性成分,例如抗炎劑或抗癌劑)的組合治療的患者的方法。所述方法包含對來自患者的組織樣品進行測序並且檢測患者中的DNA-PK的積聚。實例
以下進一步闡明本公開的通用方法。本公開化合物可通過所屬領域中已知的方法製備。以下說明本公開的優選化合物的具體製備方法。然而,這些決不限制本公開化合物的製備方法。合成實例
本文提供的化合物(包含其藥學上可接受的鹽)的合成是在實例中的合成方案中加以說明。本文提供的化合物可使用任何已知的有機合成技術製備並且可以根據多種可能的合成途徑中的任一種合成,且因此,這些方案只是說明性的且不打算限制可用于製備本文提供的化合物的其它可能的方法。此外,所述方案中的步驟是為了更好地說明且在適當時可以改變。出於研究和可能提交給管理機構的目的而合成實例中的化合物實施例。
用於製備本公開化合物的反應可在適合的溶劑中進行,所述溶劑可由有機合成領域的技術人員容易地選擇。適合的溶劑可在反應進行的溫度下,例如在溶劑的冷凍溫度至溶劑的沸騰溫度範圍內的溫度下基本上不與起始物質(反應物)、中間物或產物反應。指定反應可以在一種溶劑或多於一種溶劑的混合物中進行。取決於特定反應步驟,用於特定反應步驟的適合溶劑可以由熟練技術人員選擇。
本公開化合物的製備可涉及各種化學基團的保護和脫保護。對於保護和脫保護的需求,以及適當保護基的選擇可以由本領域技術人員容易地確定。保護基的化學性質可見於例如T. W. Greene和P. G. M. Wuts, 《有機合成中的保護基(Protective Groups in Organic Synthesis)》, 第3版, 威利父子公司(Wiley & Sons, Inc.), 紐約(1999),其以全文引用的方式併入本文中。
可以根據本領域中已知的任何適合方法監測反應。例如,可通過光譜手段,如核磁共振光譜(例如1
H或13
C)、紅外光譜(IR)、分光光度法(例如紫外-可見光)、質譜,或通過色譜法,如高壓液相色譜(HPLC)、液相色譜-質譜(LCMS)或薄層色譜(TLC)來監測產物形成。本領域的技術人員可以通過多種方法,包含高效液相色譜法(HPLC)(“製備型LC-MS純化:改善的化合物特異性方法優化(Preparative LC-MS Purification:Improved Compound Specific Method Optimization)”, Karl F.Blom, Brian Glass, Richard Sparks, Andrew P.Combs, 《組合化學雜誌(J.Combi.Chem.)》, 2004, 6(6), 874-883,以全文引用的方式併入本文中)和正相二氧化矽色譜法來純化化合物。
實例中的化合物的結構是通過核磁共振(NMR)或/和液相色譜-質譜(LC-MS)來表徵。NMR化學位移(δ
)是以10-6
(ppm)為單位給出。1
H-NMR光譜是在Bruker AVANCE NMR(300 MHz或400 MHz)光譜儀上使用ICON-NMR(在TopSpin程式控制下),使用四甲基矽烷作為內標以二甲亞碸-d6
(DMSO-d6
)或CDCl3
或CD3
OD或D2
O或丙酮_d6
或CD3
CN(來自伊諾凱(Innochem)或西格瑪-奧德里奇公司(Sigma-Aldrich)或劍橋同位素實驗室公司(Cambridge Isotope Lab., Inc.))記錄。
使用具有電噴霧源的Shimadzu 2020質譜儀在正離子和負離子模式下進行MS量測。
高效液相色譜(HPLC)測量是在Shimadzu LC-20AD系統或Shimadzu LC-20ADXR系統或Shimadzu LC-30AD系統上,使用勻漿填裝(Shim-pack) XR-ODS C18柱(3.0*50 mm,2.2 μm),或Ascentis Express C18柱(2.1*50 mm,2.7 μm),或Agilent Poroshell HPH-C18柱(3.0*50 mm,2.7 μm)進行。
使用國藥化學試劑北京有限公司(Sinopharm Chemical Reagent Beijing Co., Ltd.)和信諾化學(Xinnuo Chemical)矽膠板進行薄層色譜。用於薄層色譜(TLC)的矽膠板為175至225µm。通過TLC分離和純化產物所使用的矽膠板為1.0 mm。
純化色譜柱使用矽膠作為載體(100至200、200至300或300至400目,由乳山市上邦新材料有限公司(Rushanshi Shangbang Xincailiao Co., Ltd.)或乳山太陽乾燥劑有限公司(Rushan Taiyang Desiccant Co., Ltd. etc.)等製造),或Agela Technologies快速系統中的快速柱(反相C18柱20-45 μm,由艾傑爾科技公司(Agela Technologies)製造)。色譜柱的大小根據化合物的量進行調整。
本公開的已知起始物質可通過使用或根據所屬領域中已知的方法合成,或可購自阿法埃莎(Alfa Aesar)、梯希愛(TCI)、西格瑪-奧德里奇公司(Sigma-Aldrich)、書亞醫藥(Bepharm)、畢得醫藥科技(Bide pharmatech)、藥石(PharmaBlock)、Enamine、伊諾凱和傑達維醫藥科技(JW&Y PharmLab)等。
除非另外規定,否則反應全部在氬氣或氮氣氛圍下進行。氬氣或氮氛圍是指反應燒瓶被連接至體積為約1 L的氬氣或氮氣球。氫化通常是在壓力下進行。除非另外規定,否則實例中的反應溫度為環境溫度,其為10℃至30℃。通過TLC或/和LC-MS監測反應進展。用於反應的洗脫劑系統包含二氯甲烷-甲醇系統和石油醚-乙酸乙酯系統。溶劑的體積比根據化合物的不同極性進行調整。
用於純化化合物的柱色譜的洗脫系統以及TLC的洗脫劑系統包含二氯甲烷-甲醇系統和石油醚-乙酸乙酯系統。溶劑的體積比根據化合物的不同極性進行調整。可以添加少量鹼性或酸性試劑(0.1%至1%)(如甲酸、或乙酸、或TFA、或氨)進行調整。
在本文提供的化合物的合成中使用的化學試劑的縮寫列於下文:
應瞭解,為清楚起見而在單獨實施例的上下文中描述的本公開的某些特徵也可組合提供于單一實施例中。相反地,為簡潔起見描述於單個實施例的上下文中的本公開的各種特徵還可以分開地或以任何合適的子組合形式提供。
在本公開中的多處,描述了連接取代基。在結構明確需要鍵聯基團的情況下,關於所述基團所列的馬庫什變數(markush variable)應理解為鍵聯基團。例如,如果結構需要鍵聯基團並且所述變數的馬庫什群組定義列舉“烷基”,那麼應理解,所述“烷基”表示鍵聯亞烷基。
如本文所使用,當提到化學基團時,術語“經取代”意指所述化學基團有一個或多個氫原子被移除且經取代基置換。如本文所使用,術語“取代基”具有本領域中已知的普通含義並且是指共價連接至母基團或在適當時稠合至母基團的化學部分。如本文所用,術語“任選地經取代”或“任選地……經取代”意指化學基團可不具有取代基(即未經取代)或可具有一個或多個取代基(即經取代)。應理解,在給定原子處的取代受價數限制。
如本文所使用,術語“Ci-j
”指示碳原子數的範圍,其中i和j為整數,並且碳原子數的範圍包含端點(即i和j)和介於其間的每個整數點,並且其中j大於i。例如,C1-6
指示一至六個碳原子的範圍,包含一個碳原子、兩個碳原子、三個碳原子、四個碳原子、五個碳原子及六個碳原子。在一些實施例中,術語“C1-12
”指示1到12個,包含1到10個、1到8個、1到6個、1到5個、1到4個、1到3個或1到2個碳原子。
如本文所用,術語“烷基”無論作為另一術語的一部分或獨立地使用,都是指飽和或不飽和烴鏈,而後者可以進一步細分成具有至少一個雙鍵或三鍵的烴鏈(烯基或炔基)。在一些實施例中,烷基是指飽和烴鏈。以上提到的烴鏈可以是直鏈或分支鏈。術語“Ci-j
烷基”是指具有i至j個碳原子的烷基。飽和烷基的實例包含但不限於甲基、乙基、正丙基、異丙基、正丁基、叔丁基、異丁基、仲丁基;高級同系物,如2-甲基-1-丁基、正戊基、3-戊基、正己基、1,2,2-三甲基丁基等。不飽和烷基的實例包含但不限於乙烯基、正丙烯基、異丙烯基、正丁烯基、仲丁烯基、乙炔基、丙炔-1-基、丙炔-2-基等。“C1-6
烷基”的實例包含但不限於甲基、乙基、丙基、異丙基、正丁基、異丁基及叔丁基。“C1-3
烷基”的實例包含但不限於甲基、乙基、丙基及異丙基。
當“烷基”表示鍵聯亞烷基時,亞烷基的實例包含但不限於亞甲基、1,1-亞乙基、1,2-亞乙基、1,1-亞丙基、1,2-亞丙基、1,3-亞丙基、2,2-亞丙基、叔亞丁基等。
如本文所使用,術語“氨基”是指式“-NH2
”的基團。
如本文所用,術語“氨甲醯基”是指氨基羰基(即NH2
-C(=O)-)。
如本文所使用,術語“氰基”是指式“-C≡N”的基團。
如本文所使用,術語“鹵基”和“鹵素”是指氟基、氯基、溴基或碘基。
如本文所使用,術語“羥基”是指式“-OH”的基團。
如本文所用,術語“烷氧基”無論是作為另一術語的一部分或獨立地使用,都是指式-O-烷基的基團。
術語“Ci-j
烷氧基”意指烷氧基的烷基部分具有i至j個碳原子。烷氧基的實例包含但不限於甲氧基、乙氧基、丙氧基(例如正丙氧基和異丙氧基)、叔丁氧基等。“C1-12
烷氧基”的實例為甲氧基、乙氧基和丙氧基。
如本文所用,術語“羥基C1-12
烷基”是指式“-C1-12
烷基-OH”的基團,其中所述基團的烷基部分具有1至12個碳原子,並且一個或多個羥基可鍵聯到烷基部分中的任何碳原子。在一些實施例中,“Ci-j
烷基-OH”具有一個羥基。“C1-12
烷基-OH”的實例為羥甲基、1-羥乙基、2-羥乙基和1-羥異丙基。
如本文所用,術語“Ci-j
鹵烷基”是指經鹵素取代的(單取代或多取代的)Ci-j
烷基。“C1-12
鹵烷基”的實例為氟甲基、二氟甲基、三氟甲基、氟乙基、二氟乙基、三氟乙基、氯乙基和溴異丙基。“二氟乙基”的實例為1,1-二氟乙基。“三氟乙基”的實例為2,2,2-三氟乙基和1,2,2-三氟乙基。
“Ci-j
鹵烷氧基”的實例為氟甲氧基、二氟甲氧基或三氟甲氧基。“三氟乙氧基”的實例為2,2,2-三氟乙氧基和1,2,2-三氟乙氧基。
如本文所使用,術語“芳香基”或“芳香族S”不管是作為另一術語的一部分還是獨立地使用,都是指在形成環的原子之間具有交替的雙鍵和單鍵的環系統。在本公開中,術語“芳香基”或“芳香族”也意圖包含假芳香族。術語“偽芳香族”是指並非絕對芳香族的環系統,但其借助於電子的離域而穩定且表現方式與芳環類似。芳香基或芳香族基團可具有單環或多環。芳基的實例包含但不限於苯基、萘基、四氫萘基、茚滿基等。
如本文所使用,如本文所使用的術語“雜芳基”是指含有至少一個選自O、S、N、P等的成環雜原子的芳基。雜芳基包含但不限於呋喃基、噻吩基、吡啶基(pyridinyl)、三嗪基、吡啶基(pyridyl)、吡咯基、惡唑基、噻唑基、咪唑基、吡唑基、異惡唑基、異噻唑基、吲哚嗪基、吲哚基、異吲哚基、吲哚啉基、1,2,3-惡二唑基、1,2,4-惡二唑基、1,2,4-惡二唑-5-酮、1,2,3-三唑基、1,3,4-噻二唑基、噠嗪基、嘧啶基、吡嗪基、喹唑啉基、異喹唑啉基,1,3,5-三嗪基、1H噻吩並[2,3-c]吡唑基、噻吩並[2,3-b]呋喃基、3H-吲哚基、苯並[b]呋喃基、苯並[b]噻吩基、1H-吲唑基、苯並咪唑基、四唑基、尿苷基和胞嘧啶基。
如本文所用,術語“碳環基”無論作為另一術語的一部分或獨立地使用,都是指任何環,包含單環或多環(例如具有2或3個稠環、橋環或螺環),其中所有環原子為碳且含有至少三個成環碳原子。如本文所用,術語“螺環”是指兩個環通過一個單一共同原子連接的環系統;術語“稠環”是指兩個環共用兩個相鄰原子的環系統;並且術語“橋環”是指兩個環共用三個或更多個原子的環系統。
在一些實施例中,碳環基可以含有3至12個成環碳原子(即3員至12員碳原子)、3至10個成環碳原子、3至9個成環碳原子或3至8個成環碳原子。碳環基可為飽和、部分不飽和或完全不飽和的。在一些實施例中,碳環基可以是飽和環狀烷基。在一些實施例中,碳環基可為在環系統中含有至少一個雙鍵的不飽和環狀烷基。在一些實施例中,不飽和碳環基可以含有一個或多個芳香環。在一些實施例中,飽和或不飽和碳環基的一個或多個成環-CH2
-基團可經-C(O)-基團置換。
在一些實施例中,碳環基為單環烷基。在一些實施例中,碳環基為飽和單環烷基。飽和單環烷基的實例包含但不限於環丙基、環丁基、環戊基、環己基、環庚基、環辛基、環戊烯基、環己烯基等。
3員至8員“飽和或不飽和碳環基”為分別具有3至8個、3至6個或5至8個成環碳原子的飽和、部分不飽和或完全不飽和單環或多環環系統,其中一個或多個成環-CH2
-基團可以任選地經-C(O)-基團置換。
“3員至8員飽和或不飽和碳環基”的實例為C3-6
環烷基、環己基、環己烯基、環戊基、苯基、萘基和雙環[1.1.1]戊-1-基。“C3-8
環烷基”的實例為環丙基、環丁基、環戊基、環己基、環庚基和環辛基。術語“C3-8
環烷氧基”是指式“C3-8
環烷基-O-”的基團。
如本文所用,術語“雜環基”是指碳環基,其中一個或多個(例如1、2或3個)環原子經雜原子置換,所述雜原子包含但不限於O、S、N、P等。在一些實施例中,雜環基是飽和雜環基。在一些實施例中,雜環基為在環系統中具有一個或多個雙鍵的不飽和雜環基。在一些實施例中,雜環基為部分不飽和雜環基。在一些實施例中,雜環基為完全不飽和雜環基。在一些實施例中,不飽和雜環基可含有一個或多個芳環。在一些實施例中,雜環基的一個或多個成環-CH2
-基團可以任選地經-C(O)- 、 -S- 、 -S(O)-
或-S(O)2
- 基團
置換。在其中雜環基在其環系統中含有硫的一些實施例中,所述成環硫原子可任選地氧化以形成S-氧化物。在一些實施例中,雜環基經由其成環碳連接到化合物的其它部分。在一些實施例中,雜環基經由其成環氮連接到化合物的其它部分。
在一些實施例中,3員至8員飽和或不飽和單環或多環雜環基,其具有選自N、O或S的1、2或3個雜原子。
3員至8員“飽和或不飽和雜環基”為分別具有3到8個成環原子的飽和、部分不飽和或完全不飽和單環或多環(例如具有2或3個稠環、橋環或螺環),其中至少一個成環原子選自氮、硫或氧,除非另外規定,否則其可經由其成環碳或氮鍵聯到化合物的其它部分,其中飽和或不飽和雜環基的一個或多個成環-CH2
-基團可經-C(O)-、-S-、-S(O)-或-S(O)2
-基團置換,並且其中當雜環基在其環系統中含有硫時,所述環硫原子可以任選地氧化以形成S-氧化物。
示範性單環雜環基包含但不限於氧雜環丁烷基、1,1-二氧硫雜環丁烷基吡咯烷基、四氫呋喃基、四氫噻吩基、吡咯基、呋喃基、噻吩基、吡唑基、咪唑基、三唑基、惡唑基、噻唑基、呱啶基、呱啶基、呱嗪基、嗎啉基、吡啶基、吡嗪基、嘧啶基、噠嗪基、三嗪基、吡啶酮基、嘧啶酮基、吡嗪酮基、嘧啶酮基、噠嗪酮基、三嗪酮基等。
螺雜環基的實例包含但不限於螺吡喃基、螺惡嗪基等。稠合雜環基的實例包含但不限於苯基稠環或吡啶基稠環,如喹啉基、異喹啉基、喹喔啉基、喹嗪基、喹唑啉基、氮雜吲哚嗪基、喋啶基、色烯基、異色烯基、吲哚基、異吲哚基、吲哚嗪基、吲唑基、嘌呤基、苯並呋喃基、異苯並呋喃基、苯並咪唑基、苯並噻吩基、苯並噻唑基、哢唑基、吩嗪基、吩噻嗪基、啡啶基、咪唑並[1,2-a]吡啶基、[1,2,4]三唑並[4,3-a]吡啶基、[1,2,3]三唑並[4,3-a]吡啶基等。橋接雜環基的實例包含但不限於嗎啡烷基(morphanyl)、六亞甲基四氨基、8-氮雜-雙環[3.2.1]辛烷、1-氮雜-雙環[2.2.2]辛烷、1,4-二氮雜雙環[2.2.2]辛烷(DABCO)等。
除非另外說明,否則本公開的“化合物”意圖涵蓋所描繪的結構的所有立體異構體、幾何異構體和互變異構體。
術語“立體異構體”是指不對稱化合物(例如具有一個或多個不對稱取代的碳原子或“不對稱中心”的化合物)的各種立體異構構型(例如對映異構體、非對映異構體和外消旋體)中的任一種。含有不對稱中心的本公開化合物可以光學活性(對映異構體或非對映異構體)或光學非活性(外消旋)形式分離。術語“對映異構體”包含不互為可重疊的鏡像的立體異構體對。一對對映異構體的1:1混合物為“外消旋混合物”。術語“非對映異構體(diastereomer)”或“非對映異構體(diastereoisomer)”包含具有至少二個不對稱原子但不互為鏡像的立體異構體。含有一個或多個不對稱中心的某些化合物可產生對映異構體、非對映異構體或其它立體異構形式,其可根據卡恩-英格爾-普雷洛格(Cahn-Ingold-Prelog) R-S系統,在各不對稱中心處關於絕對構型定義為(R)-或(S)-。絕對構型未知的拆分的化合物可在不對稱中心處使用術語“或”指示。關於如何從外消旋混合物製備光學活性形式的方法是本領域中已知的,如通過HPLC拆分或立體選擇性合成。
術語“幾何異構體”或“順式和反式異構體”是指具有相同式但其官能團在三維空間中旋轉至不同取向的化合物。
術語“互變異構體”包含處於具有相同式和總電荷的化合物的異構質子化狀態的質子轉移互變異構體。質子轉移互變異構體的實例包含但不限於酮-烯醇對、醯胺-亞胺酸對、內醯胺-內醯亞胺對、烯胺-亞胺對,以及環狀形式,在環狀形式中,質子可以佔用雜環系統的兩個或更多個位置,例如1H-咪唑和3H-咪唑、1H-1,2,4-三唑、2H-1,2,4-三唑和4H-1,2,4-三唑、1H-異吲哚和2H-異吲哚,以及1H-吡唑和2H-吡唑。互變異構體可處於平衡狀態或通過適當取代而空間鎖定成一種形式。除非另外說明,否則通過名稱或結構標識為一種特定互變異構形式的本公開化合物意圖包含其它互變異構形式。
本公開的“化合物”還意圖涵蓋化合物中原子的所有同位素。原子的同位素包含具有相同原子數但質量數不同的原子。例如,除非另外規定,否則本公開“化合物”中的氫、碳、氮、氧、磷、硫、氟、氯、溴或碘意圖還包含其同位素,如但不限於1
H、2
H、3
H、11
C、12
C、13
C、14
C、14
N、15
N、16
O、17
O、18
O、31
P、32
P、32
S、33
S、34
S、36
S、17
F、19
F、35
Cl、37
Cl、79
Br、81
Br、127
I和131
I。在一些實施例中,氫包含氕、氘和氚。在一些實施例中,術語“經氘取代”或“氘取代的”用氘置換化學基團中的氫的另一種同功異型物(例如氕)。在一些實施例中,碳包含12
C和13
C。在一些實施例中,本公開的“化合物”僅涵蓋化合物中氫的同位素。在一些實施例中,本公開的“化合物”僅涵蓋呈天然豐度的原子的同位素。
還應理解,本公開的“化合物”可以呈溶劑化形式以及非溶劑化形式,例如水合形式、固體形式存在,並且本公開意圖涵蓋所有此類溶劑化形式和非溶劑化形式。
還應理解,本公開的“化合物”可以藥學上可接受的鹽形式存在。
如本文所用,術語“藥學上可接受”是指在合理醫學判斷的範圍內,適用於與人類和動物的組織接觸,而無過度毒性、刺激、過敏反應或其它問題或併發症,與合理的效益/風險比相稱的化合物、材料、組合物和/或劑型。在一些實施例中,藥學上可接受的化合物、材料、組合物和/或劑型是指被管理機構(如美國食品和藥物管理局(U.S. Food and Drug Administration)、中國食品和藥物管理局(China Food and Drug Administration)或歐洲藥物管理局(European Medicines Agency))批准或公認藥典(如美國藥典(U.S.Pharmacopoeia)、中國藥典(China Pharmacopoeia)或歐洲藥典(European Pharmacopoeia))中所列的可用於動物且特別是人類的那些化合物、材料、組合物和/或劑型。
如本文所用,“藥學上可接受的鹽”是指本公開化合物的衍生物,其中母體化合物通過將現有酸性部分(例如羧基等)或堿部分(例如胺、鹼金屬等)轉化為其鹽形式而修飾。在許多情況下,本公開化合物能夠憑藉氨基和/或羧基或其類似基團的存在而形成酸和/或堿鹽。藥學上可接受的鹽為保留母體化合物的生物有效性和特性,通常不會在生物學上或其它方面不合需要的酸和/或堿鹽。本公開化合物的適合的藥學上可接受的鹽包含例如酸加成鹽,其可以衍生自例如無機酸(例如鹽酸、氫溴酸、硫酸、硝酸、磷酸等)或有機酸(例如甲酸、乙酸、丙酸、乙醇酸、草酸、順丁烯二酸、丙二酸、琥珀酸、反丁烯二酸、酒石酸、苯均三酸、檸檬酸、乳酸、苯乙酸、苯甲酸、扁桃酸、甲烷磺酸、萘二磺酸、乙烷磺酸、甲苯磺酸、三氟乙酸、水楊酸、磺基水楊酸等)。在一些實施例中,本公開化合物的藥學上可接受的鹽為甲酸鹽。在一些實施例中,本公開化合物的藥學上可接受的鹽是TFA鹽。
本公開化合物的適合的藥學上可接受的鹽還包含例如堿加成鹽,其可以衍生自例如無機堿(例如週期表的第I列到第XII列的金屬(如鈣、鎂、鐵、銀、鋅、銅等)的鈉鹽、鉀鹽、銨鹽和氫氧化物、碳酸鹽、碳酸氫鹽)或有機堿(例如伯胺、仲胺和叔胺、包含天然存在的經取代胺的經取代胺、環胺、鹼性離子交換樹脂等)。某些有機胺包含但不限於異丙胺、苯乍生(benzathine)、膽酸鹽、二乙醇胺、二乙胺、賴氨酸、葡甲胺、呱嗪以及緩血酸胺。所屬領域的技術人員應理解,除了實例中所示以外,添加用於形成酸/堿加成鹽的酸或堿也是可能的。其它適合的鹽的列表可見于例如《雷明頓氏藥物科學(Remington's Pharmaceutical Sciences)》, 第20版, 馬克出版公司(Mack Publishing Company), 賓夕法尼亞州伊斯頓(Easton, Pa.), (1985);以及Stahl和Wermuth的《藥用鹽手冊:特性、選擇和使用(Handbook of Pharmaceutical Salts: Properties, Selection, and Use)》(德國魏因海姆的Wiley-VCH(Wiley-VCH, Weinheim, Germany), 2002)中。在一些實施例中,本公開化合物的適合的藥學上可接受的鹽為無機堿鹽。
本公開還包含本公開化合物的活性中間物、活性代謝物和前藥。如本文所用,“活性中間物”是指合成方法中的中間化合物,其展現與最終合成的化合物相同或基本上相同的生物活性。
如本文所用,“活性代謝物”是指通過在動物或人體內代謝或生物轉化產生的本公開化合物或其鹽或前藥的分解或最終產物,其展現與指定化合物相同或基本上相同的生物活性。此類代謝物可以由例如施用的化合物或鹽或前藥氧化、還原、水解、醯胺化、脫醯胺、酯化、去酯化、酶裂解等得到。
如本文所用,“前藥”是指當施用至動物或人類個體時釋放活性母體藥物的任何化合物或偶聯物。前藥可通過以一種方式修飾化合物中存在的官能團製備,所述方式使得修飾在常規操作中或在體內可由母體化合物裂解。前藥包含其中羥基、氨基、硫氫基或羧基鍵結到任何基團,使得當將其施用給哺乳動物個體時可分別裂解形成游離羥基、氨基、硫氫基或羧基的化合物。前藥的實例包含但不限於本公開化合物中醇和胺官能團的乙酸酯、甲酸酯和苯甲酸酯衍生物。前藥的製備和使用論述於THiguchi和V. Stella, “作為新穎遞送系統的前藥(Pro-drugs as Novel Delivery Systems)”, 《美國化學會會議論文集(A.C.S. Symposium Series)》第14卷,以及《藥物設計中的生物可逆載體(Bioreversible Carriers in Drug Design)》, Edward B.Roche編, 美國藥劑師協會和培格曼出版社(American Pharmaceutical Association and Pergamon Press), 1987,二者特此以全文引用的方式併入。
本文公開了可以選擇性抑制DNA-PK的新穎化合物或藥學上可接受的鹽。當與其它臨床上可用的DNA-PK抑制劑相比較時,前文公開的化合物或其藥學上可接受的鹽展現某些改進的特性,例如更高的BBB穿透(因此使其潛在地可用於治療已經轉移到CNS,特別是腦轉移和軟腦膜轉移的癌症)、更好的效力等。其還可具有相比已知DNA-PK調節劑有利的毒性概況和/或有利的代謝或藥物動力學概況。
因此,此類化合物或其藥學上可接受的鹽可尤其適用於治療癌症,尤其是具有腦部癌轉移的那些癌症。合成方法
本文提供的化合物,包含其鹽、酯、水合物或溶劑化物或立體異構體的合成是在實例中的合成方案中說明。本文提供的化合物可使用任何已知的有機合成技術製備並且可以根據多種可能的合成途徑中的任一種合成,且因此,這些方案只是說明性的且不打算限制可用于製備本文提供的化合物的其它可能的方法。此外,所述方案中的步驟是為了更好地說明且在適當時可以改變。出於研究和可能提交給管理機構的目的,實例中的化合物實施例是在中國合成。
用於製備本公開化合物的反應可以在適合溶劑中進行,這些溶劑可以由有機合成領域的技術人員容易地選擇。適合溶劑可以在反應進行的溫度下,例如在從溶劑的冷凍溫度至溶劑的沸騰溫度範圍內的溫度下基本上不與起始物質(反應物)、中間物或產物反應。指定反應可以在一種溶劑或多於一種溶劑的混合物中進行。取決於特定反應步驟,用於特定反應步驟的適合溶劑可以由熟練技術人員選擇。
本公開化合物的製備可以涉及各種化學基團的保護和脫保護。對於保護和脫保護的需求,以及適當保護基的選擇可以由本領域技術人員容易地確定。保護基的化學性質可見於例如T. W. Greene和P. G. M. Wuts, 《有機合成中的保護基(Protective Groups in Organic Synthesis)》, 第3版,威利父子公司(Wiley & Sons, Inc.), 紐約(1999),其以全文引用的方式併入本文中。
可以根據本領域中已知的任何適合方法監測反應。例如,可以通過光譜手段,如核磁共振光譜(例如1
H或13
C)、紅外光譜、分光光度法(例如紫外-可見光)、質譜,或通過色譜法,如高壓液相色譜(HPLC)、液相色譜-質譜(LCMS)或薄層色譜(TLC)來監測產物形成。本領域的技術人員可以通過多種方法,包含高效液相色譜法(HPLC)(“製備型LC-MS純化:改善的化合物特異性方法優化(Preparative LC-MS Purification:Improved Compound Specific Method Optimization)”, Karl F.Blom, Brian Glass, Richard Sparks, Andrew P.Combs, 《組合化學雜誌(J.Combi.Chem.)》, 2004, 6(6), 874-883,以全文引用的方式併入本文中)和正相二氧化矽色譜法純化化合物。
如本文所使用的縮寫定義如下:“1×”或“×1”表示一次,“2×”或“×2”表示兩次,“3×”或“×3”表示三次,“4×”或“×4”表示四次,“5×”或“×5”表示兩次,“℃”表示攝氏度,“eq”或“eq.”表示當量,“g”表示克,“mg”表示毫克,“L”表示升,“mL”或“ml”表示毫升,“μL”表示微升,“N”表示正常,“M”表示摩爾,“mmol”表示毫摩爾,“min”表示分鐘,“h”或“hr”表示小時,“r.t.”或“rt”表示室溫,“atm”表示氛圍,“psi”表示每平方英寸磅數,“conc.”表示濃縮物,“sat”或“sat'd”表示飽和,“MS”或“Mass Spec”表示質譜,“ESI”表示電噴射電離質譜,“LCMS”表示液相色譜質譜法,“HPLC”表示高壓液相色譜,“RP”表示反相,“TLC”或“tlc”表示薄層色譜,“SM”表示起始物質,“NMR”表示核磁共振光譜,“1
H”表示質子,“δ”表示△,“s”表示單重峰,“d”表示雙重峰,“t”表示三重峰,“q”表示四重峰,“m”表示多重峰,“br”表示廣譜,且“Hz”表示赫茲。“α”、“β”、“R”、“S”、“E”以及“Z”是本領域技術人員熟悉的立體化學名稱。藥物組合物
本公開提供了包括至少一種本公開化合物的藥物組合物。在一些實施例中,所述藥物組合物包括多於一種本公開的化合物。在一些實施例中,所述藥物組合物包含一種或多種本公開的化合物和藥學上可接受的載劑。
藥學上可接受的載劑是本領域中的常規藥物載劑,其可以按藥學領域中眾所周知的方式製備。在一些實施例中,本公開的化合物可以與藥學上可接受的載劑混合以製備藥物組合物。
如本文所使用,術語“藥學上可接受的載劑”是指將本文提供的化合物從一個位置、體液、組織、器官(內部或外部)或身體的一部分載運或運輸至另一位置、體液、組織、器官或身體的一部分所涉及的藥學上可接受的材料、組合物或媒劑,如液體或固體填充劑、稀釋劑、賦形劑、溶劑或囊封材料。藥學上可接受的載劑可以是可用於接觸動物組織,而無過量毒性或不良作用的媒劑、稀釋劑、賦形劑或其它材料。示範性藥學上可接受的載劑包含糖、澱粉、纖維素、麥芽、黃芪膠、明膠、林格氏溶液(Ringer's solution)、海藻酸、等滲生理鹽水、緩衝劑等。可以用於本公開中的藥學上可接受的載劑包含本領域中一般已知的那些,如以引用的方式併入本文中的《雷氏藥學大全》, Mack Pub. Co., 新澤西州(New Jersey)(1991)中公開那些。
可以充當藥學上可接受的載劑的材料的一些實例包含:(1)糖,如乳糖、葡萄糖和蔗糖;(2)澱粉,如玉米澱粉和馬鈴薯澱粉;(3)纖維素和其衍生物,如羧甲基纖維素鈉、乙基纖維素和乙酸纖維素;(4)粉末狀黃芪膠;(5)麥芽;(6)明膠;(7)滑石;(8)賦形劑、如可哥脂和栓劑蠟;(9)油,如花生油、棉籽油、紅花油、芝麻油、橄欖油、玉米油和大豆油;(10)二醇,如丙二醇;(11)多元醇,如甘油、山梨糖醇、甘露醇和聚乙二醇;(12)酯,如油酸乙酯和月桂酸乙酯;(13)瓊脂;(14)緩衝劑,如氫氧化鎂和氫氧化鋁;(15)海藻酸;(16)無熱原質水;(17)等滲生理鹽水;(18)林格氏溶液;(19)醇,如乙醇和丙醇;(20)磷酸鹽緩衝溶液;以及(21)藥物配製物中採用的其它無毒可相容物質,如丙酮。
藥物組合物可視需要含有藥學上可接受的輔助物質以接近生理條件,如pH值調節劑和緩沖劑、毒性調節劑等,例如乙酸鈉、氯化鈉、氯化鉀、氯化鈣、乳酸鈉等。
藥物組合物的形式取決於多種標準,包含但不限於施用途徑、疾病程度或待施用的劑量。所述藥物組合物可以被配製成經口、經鼻、直腸、經皮、靜脈內或肌肉內施用。舉例來說,用於經鼻施用的劑型可以方便地配製為氣霧劑、溶液、滴劑、凝膠或乾粉;用於鼻內施用的劑型可以配製為流體配製物。根據希望的施用途徑,所述藥物組合物可以被配製成片劑、膠囊、丸劑、糖衣藥丸、散劑、顆粒劑、藥囊、扁囊劑、口含錠、懸浮液、乳液、溶液、糖漿、氣霧劑(呈固體形式或在液體介質中)、噴霧劑、油膏、糊漿、乳膏、洗劑、凝膠劑、貼片、吸入劑或栓劑形式。
藥物組合物可以被配製用於在通過採用本領域中已知的程式施用給患者之後,提供活性成分的快速、持續或延遲釋放。在一些實施例中,藥物組合物被配製成持續釋放形式。如本文所使用,術語“持續釋放形式”是指活性劑從藥物組合物釋放以使其變得在延長的時間段(延長釋放)內或在某一位置(控制釋放)可供個體,主要是個體的胃腸道生物吸收。在一些實施例中,所述延長的時間段可以是約1小時至24小時、2小時至12小時、3小時至8小時、4小時至6小時、1至2天或更長時間。在某些實施例中,所述延長的時間段是至少約4小時、至少約8小時、至少約12小時或至少約24小時。所述藥物組合物可以被配製成片劑形式。例如,活性劑的釋放速率不僅可以通過活性劑溶解於胃腸液中且隨後獨立於pH自片劑或丸劑擴散進行控制,而且還受片劑崩解和溶蝕的物理過程影響。在一些實施例中,“控制釋放的醫學應用(Medical Applications of Controlled Release)”, Langer和Wise(編), CRC Pres., 佛羅里達州波卡拉頓(Boca Raton, Florida)(1974);“控制性藥物生物利用率(Controlled Drug Bioavailability)”, 《藥品設計和性能(Drug Product Design and Performance)》, Smolen和Ball(編), Wiley, 紐約(1984);Ranger和Peppas, 1983, 《高分子科學-高分子化學評述(J Macromol.Sci.Rev. Macromol Chem.)》23:61中所公開的聚合材料可以用於持續釋放;還參見Levy等人, 1985, 《科學(Science)》228:190;During等人, 1989, 《神經病學年評(Ann. Neurol.)》25:351;Howard等人, 1989, 《神經外科雜誌(J. Neurosurg.)》71:105。以上參考文獻都以全文引用的方式併入本文中。
在某些實施例中,藥物組合物包括約0.0001mg到約100mg的本公開化合物(例如,約0.0001mg至約10mg、約0.001mg至約10mg、約0.01mg至約10mg、約0.1mg至約10mg、約0.1mg至約5mg、約0.1mg至約4mg、約0.1mg至約3mg、約0.1mg至約2mg、約0.1mg至約1mg、約0.1mg至約0.5mg、約1mg至約10mg、約1mg至約5mg、約5mg至約10mg、約5mg至約20mg、約5mg至約30mg、約5mg至約40mg、約5mg至約50mg、約10mg至約100mg、約20mg至約100mg、約30mg至約100mg、約40mg至約100mg、約50mg至約100mg)。每天每位個體的適合劑量可以是約0.1mg至約10mg,優選地是約0.1mg至約5mg、約5mg至約10mg、或約1mg至約5mg。
在某些實施例中,藥物組合物可按單位劑型配製,每種劑量含有約0.0001mg至約10mg、約0.001mg至約10mg、約0.01mg至約10mg、約0.1mg至約10mg、約0.1mg至約5mg、約0.1mg至約4mg、約0.1mg至約3mg、約0.1mg至約2mg、約0.1mg至約1mg、約0.1mg至約0.5mg、約1mg至約10mg、約5mg至約10mg、約5mg至約20mg、約5mg至約30mg、約5mg至約40mg、約5mg至約50mg、約10mg至約100mg、約20mg至約100mg、約30mg至約100mg、約40mg至約100mg、約50mg至約100mg的本公開化合物。術語“單位劑型”是指適合以單一劑量用於人類個體和其他哺乳動物的物理離散單元,每個單元含有與適合藥物載劑結合的經計算以產生所希望的治療作用的預定量的活性材料。
在一些實施例中,藥物組合物包括一種或多種本公開的化合物作為第一活性成分,並且還包含第二活性成分。第二活性成分可為所屬領域中已知的任何免疫調節劑或抗腫瘤劑,包含但不限於化學治療劑、免疫治療劑、細胞信號轉導抑制劑、細胞信號轉導抑制劑、烷化劑、拓撲異構酶抑制劑、有絲分裂抑制劑、抗激素劑等。此類免疫調節劑或抗腫瘤劑的實例為鉑類化學治療劑(例如順鉑(DDP)、卡鉑(CBP)、硫酸根-1,2-二氨基環己烷鉑(SHP)、奈達鉑(Nedaplatin)、奧沙利鉑(Oxaliplatin)(OXA)、樂鉑(Laboplatin))、多烯紫杉醇(Docetaxel)、太平洋紫杉醇(Paclitaxel)、多柔比星(Doxorubicin)、依託泊苷(Etoposide)、米托蒽醌(Mitoxantrone)、CTLA-4抑制劑、抗CTLA-4抗體、PD-1抑制劑、PD-L1抑制劑、抗PD-1/PD-L1抗體、CD39抑制劑、抗CD39抗體、CD73抑制劑、抗CD73抗體、CCR2抑制劑、抗CCR2抗體、EGFR抑制劑、CDK 4/6抑制劑、MELK抑制劑、OX40促效劑、抗雄激素抑制劑、IgG4同型抗體、酪氨酸激酶抑制劑、DNA甲基轉移酶抑制劑、Hsp90抑制劑、FGFR抑制劑、mTOR抑制劑、芳香酶抑制劑、VEGF抑制劑、LHRH拮抗劑、PI3K抑制劑、AKT抑制劑、極光激酶抑制劑、MEK抑制劑、HDAC抑制劑、BET抑制劑、PIK3CA抑制劑、蛋白酶體抑制劑、其他SERD、法呢基轉移酶抑制劑、VEGF-A抗體、ErbB3 (Her3)抗體、蛋白酶體抑制劑、蛋白激酶Cβ抑制劑、抗IGF-1R抗體、抗HER2抗體、SERM、IGF抑制劑、抗IgG抗體等。用於治療癌症或腫瘤的抗腫瘤劑的代表性實例可包含但不限於順鉑、卡鉑、SHP、奈達鉑、奧沙利鉑、樂鉑、多烯紫杉醇、太平洋紫杉醇、多柔比星、依託泊苷、米托蒽醌、長春新堿、長春堿、吉西他濱(gemcitabine)、環磷醯胺、苯丁酸氮芥(chlormabucil)、卡莫司汀(carmustine)、甲胺喋呤、氟尿嘧啶、放線菌素、表柔比星(epirubicin)、蒽環黴素、博萊黴素、絲裂黴素-C、伊立替康(irinotecan)、拓朴替康(topotecan)、替尼泊甙(teniposide)白介素、干擾素、曲美單抗(tremelimumab)、伊匹單抗(ipilimumab)、派立珠單抗(pembrolizumab)、納武單抗(nivolumab)、阿維魯單抗(avelumab)、度伐單抗(durvalumab)、阿特珠單抗(atezolizumab)、IPH 52、IPH 53、CPI-006、普洛利單抗(plozalizumab)、MLN1202、西妥昔單抗(cetuximab)、拉帕替尼(lapatinib)、埃羅替尼(erlotinib)、吉非替尼(gefitinib)、來那替尼(neratinib)、曲妥珠單抗(trastuzumab)、阿多-曲妥珠單抗恩他新(ado-trastuzumab emtansine)、帕妥珠單抗(pertuzumab)、MCLA-128、阿那曲唑(anastrazole)、雷諾昔酚(raloxifene)、G1T38、他莫昔芬(tamoxifen)、戈舍瑞林(goserelin)、恩雜魯胺(enzalutamide)、伏立諾他(vorinostat)、恩替諾特(entinostat)、舒尼替尼(sunitinib)、帕唑帕尼(pazopanib)、貝伐單抗(bevacizumab)、蘭比珠單抗(ranibizumab)、呱加他尼(pegaptanib)、西地尼布(cediranib)、達沙替尼(dasatinib)、GDC-0980、吉達昔布(gedatolisib)、阿普昔布(alpelisib)、BKM120、考帕昔布(copanlisib)、AZD8835、GDC-0941、塔瑟昔布(taselisib)、替西羅莫司(temsirolimus)、依維莫司(everolimus)、薩帕塞替(sapanisertib)、AZD5363、MK2206、帕尼單抗(panitumumab)、派立珠單抗(pembrolizumab)、索拉非尼(sorafenib)、帕博希布(palbociclib)、玻瑪西林(abemaciclib)、瑞博西林(ribociclib)、克唑替尼(crizotinib)、多韋替尼(dovitinib)、蘆可替尼(ruxolitinib)、阿紮胞苷(azacitidine)、CC-486、HSP90加利特皮(HSP90 ganetespib)、Debio 1347、伊達替尼(erdafitinib)、維瑟塞替(vitusertib)、阿立塞替(alisertib)、司美替尼(selumetinib)、G-5829、GSK525762、MLN9708、GDC-0810、AFP464、替吡法尼(tipifarnib)、塞利班單抗(seribantumab)、硼替佐米(bortezomib)、恩紮妥林(enzastaurin)、AVE1642、森吐珠單抗(xentuzumab)、達羅吐珠單抗(dalotuzumab)、AMG479等。
此類抗腫瘤劑的實例還可在V. T. Devita和S. Hellman(編)的《癌症原理和腫瘤學實踐(Cancer Principles and Practice of Oncology)》,第6版(2001年2月15日), 利平科特威廉姆斯和維爾金斯出版社(Lippincott Williams & Wilkins Publishers)中找到。本領域的普通技術人員還將能夠基於藥物的特定特徵和所涉及的癌症辨別哪些藥劑組合將是有用的。
根據本公開的這一方面,提供適用於治療癌症的組合,其包括如上文所定義的式(I)化合物或其藥學上可接受的鹽;以及上文所列之免疫調節劑或抗腫瘤劑中的任一種。
因此,在本公開的另一方面,提供一種與選自上文所列之免疫調節劑或化學治療劑的免疫調節劑或化學治療劑組合的式(I)化合物或其藥學上可接受的鹽。
本文中,在使用術語“組合”的情況下,應理解,這是指同時、分別或依序施用。在一些實施例中,“組合”是指同時施用。在本公開的另一方面,“組合”是指分開施用。在本公開的另一方面,“組合”是指依序施用。在依序或分別施用的情況下,施用第二組分的延遲不應使得失去所述組合的有益作用。
根據本公開的另一個方面,提供一種藥物組合物,其包括式(I)化合物或其藥學上可接受的鹽與選自以上列出的免疫調節劑或抗腫瘤劑的免疫調節劑或抗腫瘤劑的組合,以及藥學上可接受的稀釋劑或載劑。
根據本公開的另一個方面,提供一種藥物組合物,其包括式(I)化合物或其藥學上可接受的鹽與選自以上列出的一者的免疫調節劑或抗腫瘤劑的組合,以及藥學上可接受的稀釋劑或載劑,用於產生免疫調節或抗癌作用。
根據本公開的另一方面,提供一種藥物組合物,其包括式(I)化合物或其藥學上可接受的鹽與選自以上列出的一者的免疫調節劑或抗腫瘤劑的組合,以及藥學上可接受的稀釋劑或載劑,用於治療DNA-PK相關病症,例如NSCLC、RCC、前列腺癌或乳癌等。
根據本公開的另一方面,提供一種試劑盒,其包括式(I)化合物或其藥學上可接受的鹽與選自上文所列的一者的免疫調節劑或抗腫瘤劑的組合。
根據本公開的另一方面,提供一種試劑盒,其包含:
a)呈第一單位劑型的式(I)化合物或其藥學上可接受的鹽;
b)呈第二單位劑型的選自上文所列的一者的免疫調節劑或抗腫瘤劑;和
c)用於容納所述第一和第二劑型的容器。
除了用於治療藥物以外,式(I)化合物或其藥學上可接受的鹽還適用作體外和體內測試系統的開發和標準化中的藥理學工具,所述測試系統用於評估DNA-PK在如貓、狗、兔、猴、大鼠和小鼠的實驗動物中的活性或表達,作為探求新型治療劑的一部分。
在以上其它藥物組合物、過程、方法、用途和藥物製造特徵中,本文所述的本公開化合物的替代和優選實施例也適用。治療方法
本公開提供一種治療DNA-PK相關病症的方法,其包括向個體施用有效量的一種或多種本公開的化合物、其藥學上可接受的鹽或藥物組合物。
本公開還提供一種治療DNA-PK相關病症的方法。在某些實施例中,所述方法包括向個體施用有效量的一種或多種本公開的化合物、其藥學上可接受的鹽或藥物組合物。
如本文所用,術語“DNA-PK相關病症”是指發作或發展或二者與DNA-PK的表達或活性有關的疾病。實例包含(但不限於)過度增生性病症(例如,癌症)。
在一些實施例中,DNA-PK相關病症為癌症,優選為過度表達DNA-PK的癌症。“過度表達DNA-PK的癌症”是與相同組織類型的非癌性細胞相比在癌症或腫瘤細胞中具有顯著較高水準的DNA-PK蛋白質的癌症。這種過度表達可能是由基因擴增或增加的轉錄或轉譯引起的。通過評估細胞上存在的DNA-PK蛋白質水準的增加(例如通過免疫組織化學分析;IHC),可在診斷或預後分析中測定DNA-PK過度表達。替代地或另外,可測量細胞中編碼DNA-PK的核酸的水準,例如通過螢光原位雜交(FISH;參見1998年10月公佈的WO98/45479)、DNA印跡法或聚合酶鏈反應(PCR)技術,如實時定量PCR(RT-PCR)(方法(Methods) 132: 73-80 (1990))。除了上述分析之外,所屬領域的技術人員可使用各種體內分析。例如,可將患者體內的細胞暴露於抗體,所述抗體任選地用可檢測標記(例如放射性同位素)標記,並且例如通過外部掃描放射性或通過分析從先前暴露于抗體的患者取得的活組織檢查,可評估抗體與患者細胞的結合。
確切地說,癌症包含不限於肺癌(例如非小細胞肺癌(NSCLC)、小細胞肺癌、肺腺癌、大細胞肺癌、鱗狀細胞肺癌)、腎細胞癌(RCC)、前列腺癌、乳癌、卵巢癌、子宮內膜癌、子宮頸癌、骨癌、子宮癌、結腸癌、白血病、成膠質細胞瘤、黑素瘤、軟骨肉瘤、腦癌、膽管癌、骨肉瘤、淋巴瘤、腺瘤、骨髓瘤、肝細胞癌、腎上腺皮質癌、胰腺癌、膀胱癌、肝癌、胃癌、結腸直腸癌、食道癌、睾丸癌、皮膚癌、腎癌、間皮瘤、成神經細胞瘤、甲狀腺癌、頭頸癌、食道癌、眼癌、鼻咽癌或口腔癌。在一些實施例中,癌症為NSCLC、RCC、前列腺癌或乳癌。除非另外規定,否則如本文中提到的癌症可處於任何分期。在一些實施例中,癌症為早期癌症。在一些實施例中,癌症為局部晚期癌症。在一些實施例中,癌症為局部晚期和/或轉移癌。在一些實施例中,癌症為侵入性癌症。在一些實施例中,癌症為對現有療法具有抗性的癌症。
如本文所使用,術語“治療(treatment/treat/treating)”是指逆轉、緩解如本文所描述的疾病或病症或其一種或多種症狀,延遲其發作或抑制其進展。在一些實施例中,治療可在已出現一或多種症狀後施用。在其它實施例中,治療可在症狀不存在的情況下施用。例如,治療可以在症狀發作之前施用給易感個體(例如根據症狀病史和/或根據遺傳學或其它易感因素)。也可以在症狀消退之後繼續治療,例如以預防或延遲其復發。
在一些實施例中,本文所提供的一種或多種化合物、其藥學上可接受的鹽或藥物組合物是通過腸胃外途徑或非腸胃外途徑施用。在一些實施例中,所述一種或多種化合物、其藥學上可接受的鹽、水合物、溶劑化物或立體異構體或藥物組合物是經口、經腸、經頰、經鼻、鼻內、經粘膜、經表皮、透皮、經皮、經眼、肺、經直腸、舌下、經陰道、經表面、皮下、靜脈內、肌肉內、動脈內、鞘內、囊內、眶內、心內、皮內、腹膜內、經氣管、表皮下、關節內、囊下、脊椎內、蛛網膜下或胸骨內施用。
本文提供的化合物可以呈純形式、與其它活性成分組合或呈本公開的藥物組合物形式施用。在一些實施例中,本文所提供的化合物可以與本領域中已知的一種或多種抗癌劑或消炎劑組合同時或依序向有需要的個體施用。這類組合中的個別化合物是在分開的或組合的藥物組合物中依序或同時施用。優選地,個別化合物將在組合的藥物組合物中同時施用。所屬領域的技術人員將瞭解已知治療劑的適當劑量。
在一些實施例中,施用是一天一次、一天兩次、一天三次或每兩天一次、每三天一次、每四天一次、每五天一次、每六天一次、一週一次進行。
如本文所提供的化合物或其藥學上可接受的鹽的治療有效量將取決於本領域中已知的各種因素,如體重、年齡、既往病史、當前藥物、個體的健康狀態和交叉反應、過敏、敏感和不良副作用的可能性,以及施用途徑和疾病發展程度。如由這些和其它情況或要求所指示,所屬領域的技術人員(例如醫生或獸醫)可按比例減少或增加劑量。
在一些實施例中,本文所提供的一種或多種化合物、其藥學上可接受的鹽或藥物組合物經口施用。對於經口施用,實現所希望的目標的任何劑量都是適當的。在一些實施例中,適合日劑量是在約0.001到100mg之間,優選地在0.1mg與5g之間,更優選地在5mg與1g之間,更優選地在10mg與500mg之間,並且施用是一天一次、一天兩次、一天三次、每天或一周3到5天進行。在一些實施例中,本文所提供的一種或多種化合物、其藥學上可接受的鹽或藥物組合物的劑量的範圍在每天約0.0001mg,優選地0.001mg、0.01mg、0.1mg、0.2mg、0.3mg、0.4mg、0.5mg、0.6mg、0.7mg、0.8mg、0.9mg、1mg、2mg、3mg、4mg、5mg、6mg、7mg、8mg、9mg、10mg之間。化合物的用途
在某些實施例中,本公開提供本公開的化合物、其藥學上可接受的鹽或藥物組合物在製備用於治療DNA-PK相關病症的藥劑中的用途。在某些實施例中,DNA-PK相關病症包含癌症。
本公開中的化合物和其藥物組合物可用於預防或治療哺乳動物(尤其人類)中的DNA-PK相關病症(表達或活性)中的任一種的發作或發展。
在此類情形中,本公開還提供一種篩選適於用單獨的本公開的化合物或藥物組合物或其與其它成分(例如第二活性成分,例如抗炎劑或抗癌劑)的組合治療的患者的方法。所述方法包含對來自患者的組織樣品進行測序並且檢測患者中的DNA-PK的積聚。實例
以下進一步闡明本公開的通用方法。本公開化合物可通過所屬領域中已知的方法製備。以下說明本公開的優選化合物的具體製備方法。然而,這些決不限制本公開化合物的製備方法。合成實例
本文提供的化合物(包含其藥學上可接受的鹽)的合成是在實例中的合成方案中加以說明。本文提供的化合物可使用任何已知的有機合成技術製備並且可以根據多種可能的合成途徑中的任一種合成,且因此,這些方案只是說明性的且不打算限制可用于製備本文提供的化合物的其它可能的方法。此外,所述方案中的步驟是為了更好地說明且在適當時可以改變。出於研究和可能提交給管理機構的目的而合成實例中的化合物實施例。
用於製備本公開化合物的反應可在適合的溶劑中進行,所述溶劑可由有機合成領域的技術人員容易地選擇。適合的溶劑可在反應進行的溫度下,例如在溶劑的冷凍溫度至溶劑的沸騰溫度範圍內的溫度下基本上不與起始物質(反應物)、中間物或產物反應。指定反應可以在一種溶劑或多於一種溶劑的混合物中進行。取決於特定反應步驟,用於特定反應步驟的適合溶劑可以由熟練技術人員選擇。
本公開化合物的製備可涉及各種化學基團的保護和脫保護。對於保護和脫保護的需求,以及適當保護基的選擇可以由本領域技術人員容易地確定。保護基的化學性質可見於例如T. W. Greene和P. G. M. Wuts, 《有機合成中的保護基(Protective Groups in Organic Synthesis)》, 第3版, 威利父子公司(Wiley & Sons, Inc.), 紐約(1999),其以全文引用的方式併入本文中。
可以根據本領域中已知的任何適合方法監測反應。例如,可通過光譜手段,如核磁共振光譜(例如1
H或13
C)、紅外光譜(IR)、分光光度法(例如紫外-可見光)、質譜,或通過色譜法,如高壓液相色譜(HPLC)、液相色譜-質譜(LCMS)或薄層色譜(TLC)來監測產物形成。本領域的技術人員可以通過多種方法,包含高效液相色譜法(HPLC)(“製備型LC-MS純化:改善的化合物特異性方法優化(Preparative LC-MS Purification:Improved Compound Specific Method Optimization)”, Karl F.Blom, Brian Glass, Richard Sparks, Andrew P.Combs, 《組合化學雜誌(J.Combi.Chem.)》, 2004, 6(6), 874-883,以全文引用的方式併入本文中)和正相二氧化矽色譜法來純化化合物。
實例中的化合物的結構是通過核磁共振(NMR)或/和液相色譜-質譜(LC-MS)來表徵。NMR化學位移(δ
)是以10-6
(ppm)為單位給出。1
H-NMR光譜是在Bruker AVANCE NMR(300 MHz或400 MHz)光譜儀上使用ICON-NMR(在TopSpin程式控制下),使用四甲基矽烷作為內標以二甲亞碸-d6
(DMSO-d6
)或CDCl3
或CD3
OD或D2
O或丙酮_d6
或CD3
CN(來自伊諾凱(Innochem)或西格瑪-奧德里奇公司(Sigma-Aldrich)或劍橋同位素實驗室公司(Cambridge Isotope Lab., Inc.))記錄。
使用具有電噴霧源的Shimadzu 2020質譜儀在正離子和負離子模式下進行MS量測。
高效液相色譜(HPLC)測量是在Shimadzu LC-20AD系統或Shimadzu LC-20ADXR系統或Shimadzu LC-30AD系統上,使用勻漿填裝(Shim-pack) XR-ODS C18柱(3.0*50 mm,2.2 μm),或Ascentis Express C18柱(2.1*50 mm,2.7 μm),或Agilent Poroshell HPH-C18柱(3.0*50 mm,2.7 μm)進行。
使用國藥化學試劑北京有限公司(Sinopharm Chemical Reagent Beijing Co., Ltd.)和信諾化學(Xinnuo Chemical)矽膠板進行薄層色譜。用於薄層色譜(TLC)的矽膠板為175至225µm。通過TLC分離和純化產物所使用的矽膠板為1.0 mm。
純化色譜柱使用矽膠作為載體(100至200、200至300或300至400目,由乳山市上邦新材料有限公司(Rushanshi Shangbang Xincailiao Co., Ltd.)或乳山太陽乾燥劑有限公司(Rushan Taiyang Desiccant Co., Ltd. etc.)等製造),或Agela Technologies快速系統中的快速柱(反相C18柱20-45 μm,由艾傑爾科技公司(Agela Technologies)製造)。色譜柱的大小根據化合物的量進行調整。
本公開的已知起始物質可通過使用或根據所屬領域中已知的方法合成,或可購自阿法埃莎(Alfa Aesar)、梯希愛(TCI)、西格瑪-奧德里奇公司(Sigma-Aldrich)、書亞醫藥(Bepharm)、畢得醫藥科技(Bide pharmatech)、藥石(PharmaBlock)、Enamine、伊諾凱和傑達維醫藥科技(JW&Y PharmLab)等。
除非另外規定,否則反應全部在氬氣或氮氣氛圍下進行。氬氣或氮氛圍是指反應燒瓶被連接至體積為約1 L的氬氣或氮氣球。氫化通常是在壓力下進行。除非另外規定,否則實例中的反應溫度為環境溫度,其為10℃至30℃。通過TLC或/和LC-MS監測反應進展。用於反應的洗脫劑系統包含二氯甲烷-甲醇系統和石油醚-乙酸乙酯系統。溶劑的體積比根據化合物的不同極性進行調整。
用於純化化合物的柱色譜的洗脫系統以及TLC的洗脫劑系統包含二氯甲烷-甲醇系統和石油醚-乙酸乙酯系統。溶劑的體積比根據化合物的不同極性進行調整。可以添加少量鹼性或酸性試劑(0.1%至1%)(如甲酸、或乙酸、或TFA、或氨)進行調整。
在本文提供的化合物的合成中使用的化學試劑的縮寫列於下文: 步驟1. 5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶
在空氣氛圍下在0℃下將5-氯-1H-吡唑並[4,3-d]嘧啶(3.00 g,19.410 mmol,1.00當量)和NIS(7.86 g,34.936 mmol,1.80當量)於DMF(60.00 mL)中的混合物攪拌隔夜。用EtOAc(3×150 mL)萃取所得混合物。用鹽水(3×200 mL)洗滌合併的有機層。用Na2
S2
O3
(3×200 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過矽膠柱色譜純化殘餘物,用PE/EtOAc(20:1)洗脫,得到呈黃色固體的5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶(2.4 g,44.09%)。LCMS: m/z (ESI), [M+H]+
= 281.0。
步驟2. 5-氯-3-碘-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在0℃下將Cs2
CO3
(3.49 g,10.697 mmol,3當量)、CH3
I(2.53 g,17.828 mmol,5.00當量)和5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶(1.00 g,3.566 mmol,1.00當量)於DMF(20.00 mL)中的混合物攪拌1 h。用水(100 mL)稀釋所得混合物並用EtOAc(3×80 mL)萃取。用鹽水(3×100 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。從EtOAc/PE(1:5 300 mL)再結晶粗產物,得到呈黃色固體的5-氯-3-碘-1-甲基-1H-吡唑並[4,3-d]嘧啶(850 mg,80.95%)。LCMS: m/z (ESI), [M+H]+
= 295.0。
步驟3. 5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將含K2
CO3
(1210.85 mg,8.761 mmol,3當量)、Pd(dppf)Cl2
CH2
Cl2
(476.98 mg,0.584 mmol,0.2當量)、2-(3,6-二氫-2H-吡喃-4-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(797.57 mg,3.797 mmol,1.3當量)和5-氯-3-碘-1-甲基-1H-吡唑並[4,3-d]嘧啶(860.00 mg,2.920 mmol,1.00當量)的二惡烷(15.00 mL)和H2
O(3.00 mL)攪拌16個小時。用水(100 mL)稀釋所得混合物。用CH2
Cl2
/MeOH = 12:1(3×50 mL)萃取所得混合物。用鹽水(2×100 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/ MeOH = 12:1)來純化殘餘物,得到呈灰色固體的5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶(380 mg,51.90%)。LCMS: m/z (ESI), [M+H]+
= 251.2。
步驟4. 3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs2
CO3
(2599.38 mg,7.978 mmol,2.50當量)、XantPhos(553.94 mg,0.957 mmol,0.30當量)、Pd(OAc)2(
143.29 mg,0.638 mmol,0.20當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(567.40 mg,3.829 mmol,1.20當量)和5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶(800.00 mg,3.191 mmol,1.00當量)於二惡烷(20.00 mL)中的混合物攪拌隔夜。用水(200 mL)稀釋所得混合物。用CH2
Cl2
/MeOH = (12:1)(3×200 mL)萃取所得混合物,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。從EtOAc/PE(1:6 300 mL)再結晶粗產物,得到呈棕色固體的3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[4,3-d]嘧啶-5-胺(600 mg,51.88%)。LCMS: m/z (ESI), [M+H]+
= 363.3。
步驟5. 1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(氧雜環己烷-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺.(實例1)
在氫氣氛圍下在室溫下將Pd/C(47.92 mg,0.450 mmol,1.36當量)和3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[4,3-d]嘧啶-5-胺(120 mg,0.331 mmol,1當量)於MeOH(200 mL)和THF(100 mL)中的混合物攪拌2小時。過濾所得混合物,用MeOH(3×100 mL)洗滌濾餅。減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內25 B至51 B)來純化粗產物(120 mg),得到呈白色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(氧雜環己烷-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(55 mg,45.12%)。LCMS: m/z (ESI), [M+H]+
= 365.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.95 (4H, t), 2.42 (3H, d), 3.14-3.30 (1H, m), 3.47 (2H, d), 3.93 (2H, d), 4.04 (3H, s), 7.71 (1H, t), 8.37 (1H, s), 8.84 (1H, s), 9.15 (1H, s), 9.34 (1H, s)實例 2
製備1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例2)及3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺.(實例6)
步驟1. 5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶
在空氣氛圍下在0℃下將5-氯-1H-吡唑並[4,3-d]嘧啶(3.00 g,19.410 mmol,1.00當量)和NIS(7.86 g,34.936 mmol,1.80當量)於DMF(60.00 mL)中的混合物攪拌隔夜。用EtOAc(3×150 mL)萃取所得混合物。用鹽水(3×200 mL)洗滌合併的有機層。用Na2
S2
O3
(3×200 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過矽膠柱色譜純化殘餘物,用PE/EtOAc(20:1)洗脫,得到呈黃色固體的5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶(2.4 g,44.09%)。LCMS: m/z (ESI), [M+H]+
= 281.0。
步驟2. 5-氯-3-碘-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在0℃下將Cs2
CO3
(3.49 g,10.697 mmol,3當量)、CH3
I(2.53 g,17.828 mmol,5.00當量)和5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶(1.00 g,3.566 mmol,1.00當量)於DMF(20.00 mL)中的混合物攪拌1 h。用水(100 mL)稀釋所得混合物並用EtOAc(3×80 mL)萃取。用鹽水(3×100 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。從EtOAc/PE(1:5 300 mL)再結晶粗產物,得到呈黃色固體的5-氯-3-碘-1-甲基-1H-吡唑並[4,3-d]嘧啶(850 mg,80.95%)。LCMS: m/z (ESI), [M+H]+
= 295.0。
步驟3. 5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將含K2
CO3
(1210.85 mg,8.761 mmol,3當量)、Pd(dppf)Cl2
CH2
Cl2
(476.98 mg,0.584 mmol,0.2當量)、2-(3,6-二氫-2H-吡喃-4-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(797.57 mg,3.797 mmol,1.3當量)和5-氯-3-碘-1-甲基-1H-吡唑並[4,3-d]嘧啶(860.00 mg,2.920 mmol,1.00當量)的二惡烷(15.00 mL)和H2
O(3.00 mL)攪拌16個小時。用水(100 mL)稀釋所得混合物。用CH2
Cl2
/MeOH = 12:1(3×50 mL)萃取所得混合物。用鹽水(2×100 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/ MeOH = 12:1)來純化殘餘物,得到呈灰色固體的5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶(380 mg,51.90%)。LCMS: m/z (ESI), [M+H]+
= 251.2。
步驟4. 3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs2
CO3
(2599.38 mg,7.978 mmol,2.50當量)、XantPhos(553.94 mg,0.957 mmol,0.30當量)、Pd(OAc)2(
143.29 mg,0.638 mmol,0.20當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(567.40 mg,3.829 mmol,1.20當量)和5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶(800.00 mg,3.191 mmol,1.00當量)於二惡烷(20.00 mL)中的混合物攪拌隔夜。用水(200 mL)稀釋所得混合物。用CH2
Cl2
/MeOH = (12:1)(3×200 mL)萃取所得混合物,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。從EtOAc/PE(1:6 300 mL)再結晶粗產物,得到呈棕色固體的3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[4,3-d]嘧啶-5-胺(600 mg,51.88%)。LCMS: m/z (ESI), [M+H]+
= 363.3。
步驟5. 1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(氧雜環己烷-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺.(實例1)
在氫氣氛圍下在室溫下將Pd/C(47.92 mg,0.450 mmol,1.36當量)和3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[4,3-d]嘧啶-5-胺(120 mg,0.331 mmol,1當量)於MeOH(200 mL)和THF(100 mL)中的混合物攪拌2小時。過濾所得混合物,用MeOH(3×100 mL)洗滌濾餅。減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內25 B至51 B)來純化粗產物(120 mg),得到呈白色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(氧雜環己烷-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(55 mg,45.12%)。LCMS: m/z (ESI), [M+H]+
= 365.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.95 (4H, t), 2.42 (3H, d), 3.14-3.30 (1H, m), 3.47 (2H, d), 3.93 (2H, d), 4.04 (3H, s), 7.71 (1H, t), 8.37 (1H, s), 8.84 (1H, s), 9.15 (1H, s), 9.34 (1H, s)實例 2
製備1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例2)及3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺.(實例6) 步驟1. 3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺.(實例6)
在氮氣氛圍下在100℃下將Cs2
CO3
(682.34 mg,2.094 mmol,2.5當量)、XantPhos(96.94 mg,0.168 mmol,0.2當量)、Pd(OAc)2
(37.61 mg,0.168 mmol,0.2當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(147.95 mg,1.005 mmol,1.2當量)和5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基吡唑並[4,3-d]嘧啶(210.00 mg,0.838 mmol,1.00當量)於二惡烷(6.00 mL)中的混合物攪拌隔夜。可通過LCMS來檢測所需產物。過濾所得混合物,用DCM(3×50 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至36 B)來純化粗產物(170 mg),得到呈黃色固體的3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(100 mg,57.65%)。LCMS: m/z (ESI), [M+H]+
= 362.2。1
H NMR (300 MHz, DMSO-d6
) δ 2.30 (3H, d), 2.58 (2H, s), 3.83 (2H, t), 4.06 (3H, s), 4.25 (2H, d), 7.07-7.12 (1H, m), 7.43 (1H, d), 7.49 (1H, d), 7.84 (1H, t), 8.81 (2H, d), 9.14 (1H, s)
步驟2. 1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-3-(氧雜環己烷-4-基)吡唑並[4,3-d]嘧啶-5-胺.(實例2)
在氫氣氛圍下在40℃下將Pd/C(70.67 mg,0.664 mmol,3.00當量)和3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(80.00 mg,0.221 mmol,1.00當量)於MeOH(20.00 mL)中的混合物攪拌3小時。可通過LCMS來檢測所需產物。過濾所得混合物,用MeOH(3×10 mL)洗滌濾餅。減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內22 B至33 B;RT1:6.63)來純化粗產物(50 mg),得到呈黃色固體的1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-3-(氧雜環己烷-4-基)吡唑並[4,3-d]嘧啶-5-胺(20 mg,24.61%)。LCMS: m/z (ESI), [M+H]+
= 364.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.96 (4H, d), 2.28 (3H, d), 3.21 (1H, t), 3.47 (2H, d), 3.93 (2H, d), 4.03 (3H, s), 7.42 (1H, q), 7.49 (1H, d), 7.81 (1H, t), 8.69 (1H, s), 8.84 (1H, s), 9.09 (1H, s)實例 3
製備7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-5H-吡咯並[3,2-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例3)
步驟1. 3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺.(實例6)
在氮氣氛圍下在100℃下將Cs2
CO3
(682.34 mg,2.094 mmol,2.5當量)、XantPhos(96.94 mg,0.168 mmol,0.2當量)、Pd(OAc)2
(37.61 mg,0.168 mmol,0.2當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(147.95 mg,1.005 mmol,1.2當量)和5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-甲基吡唑並[4,3-d]嘧啶(210.00 mg,0.838 mmol,1.00當量)於二惡烷(6.00 mL)中的混合物攪拌隔夜。可通過LCMS來檢測所需產物。過濾所得混合物,用DCM(3×50 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至36 B)來純化粗產物(170 mg),得到呈黃色固體的3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(100 mg,57.65%)。LCMS: m/z (ESI), [M+H]+
= 362.2。1
H NMR (300 MHz, DMSO-d6
) δ 2.30 (3H, d), 2.58 (2H, s), 3.83 (2H, t), 4.06 (3H, s), 4.25 (2H, d), 7.07-7.12 (1H, m), 7.43 (1H, d), 7.49 (1H, d), 7.84 (1H, t), 8.81 (2H, d), 9.14 (1H, s)
步驟2. 1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-3-(氧雜環己烷-4-基)吡唑並[4,3-d]嘧啶-5-胺.(實例2)
在氫氣氛圍下在40℃下將Pd/C(70.67 mg,0.664 mmol,3.00當量)和3-(3,6-二氫-2H-吡喃-4-基)-1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(80.00 mg,0.221 mmol,1.00當量)於MeOH(20.00 mL)中的混合物攪拌3小時。可通過LCMS來檢測所需產物。過濾所得混合物,用MeOH(3×10 mL)洗滌濾餅。減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內22 B至33 B;RT1:6.63)來純化粗產物(50 mg),得到呈黃色固體的1-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-3-(氧雜環己烷-4-基)吡唑並[4,3-d]嘧啶-5-胺(20 mg,24.61%)。LCMS: m/z (ESI), [M+H]+
= 364.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.96 (4H, d), 2.28 (3H, d), 3.21 (1H, t), 3.47 (2H, d), 3.93 (2H, d), 4.03 (3H, s), 7.42 (1H, q), 7.49 (1H, d), 7.81 (1H, t), 8.69 (1H, s), 8.84 (1H, s), 9.09 (1H, s)實例 3
製備7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-5H-吡咯並[3,2-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例3) 步驟1. 2-氯-7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-5H-吡咯並[3,2-d]嘧啶
在氮氣氛圍下在80℃下將2-氯-7-碘-5-甲基吡咯並[3,2-d]嘧啶(300.00 mg,1.022 mmol,1.00當量)、2-(3,6-二氫-2H-吡喃-4-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(279.16 mg,1.329 mmol,1.30當量)、Pd(dppf)Cl2
(149.59 mg,0.204 mmol,0.2當量)和K2
CO3
(423.81 mg,3.067 mmol,3當量)於二惡烷(6.00 mL)和H2
O(1.20 mL)中的混合物攪拌3小時。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:3)來純化殘餘物,得到呈棕色固體的2-氯-7-(3,6-二氫-2H-吡喃-4-基)-5-甲基吡咯並[3,2-d]嘧啶(196 mg,76.79%)。LCMS: m/z (ESI), [M+H]+
= 250.2。
步驟2. 7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡咯並[3,2-d]嘧啶-2-胺
在氮氣氛圍下在100℃下將2-氯-7-(3,6-二氫-2H-吡喃-4-基)-5-甲基吡咯並[3,2-d]嘧啶(196.00 mg,0.785 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(139.56 mg,0.942 mmol,1.20當量)、Pd(AcO)2
(35.25 mg,0.157 mmol,0.20當量)、XantPhos(136.25 mg,0.235 mmol,0.30當量)和Cs2
CO3
(639.37 mg,1.962 mmol,2.50當量)於二惡烷(3.00 mL)中的混合物攪拌3小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物,得到呈黃色固體的7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡咯並[3,2-d]嘧啶-2-胺(170 mg,59.93%)。LCMS: m/z (ESI), [M+H]+
= 362.3。
步驟3. 7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-5H-吡咯並[3,2-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例3)
在氫氣氛圍下在室溫下將7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡咯並[3,2-d]嘧啶-2-胺(170.00 mg,0.470 mmol,1.00當量)和Pd/C (250.29 mg,2.352 mmol,5.00當量)於MeOH(20.00 mL)和THF(50.00 mL)中的混合物攪拌隔夜。過濾所得混合物,用MeOH(5×30 mL)洗滌濾餅。減壓濃縮濾液,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:25 mL/min;梯度:7 min內30 B至45 B;RT1:6.02)來純化粗產物(100 mg),得到呈白色固體的5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-7-(氧雜環己烷-4-基)吡咯並[3,2-d]嘧啶-2-胺(29 mg,16.96%)。LCMS: m/z (ESI), [M+H]+
= 364.3。1
H NMR (300 MHz, DMSO-d6
) δ 1.76 (2H, d), 1.88 - 2.01 (2H, m), 2.46 (3H, d), 3.02 (1H, t), 3.47 (2H, td), 3.80 (3H, s), 3.88 - 4.05 (2H, m), 7.51 (1H, s), 7.61 - 7.75 (1H, m), 8.33 (2H, d), 8.74 (1H, s), 9.49 (1H, s)實例 4
製備3-異丙基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例4)
步驟1. 2-氯-7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-5H-吡咯並[3,2-d]嘧啶
在氮氣氛圍下在80℃下將2-氯-7-碘-5-甲基吡咯並[3,2-d]嘧啶(300.00 mg,1.022 mmol,1.00當量)、2-(3,6-二氫-2H-吡喃-4-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(279.16 mg,1.329 mmol,1.30當量)、Pd(dppf)Cl2
(149.59 mg,0.204 mmol,0.2當量)和K2
CO3
(423.81 mg,3.067 mmol,3當量)於二惡烷(6.00 mL)和H2
O(1.20 mL)中的混合物攪拌3小時。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:3)來純化殘餘物,得到呈棕色固體的2-氯-7-(3,6-二氫-2H-吡喃-4-基)-5-甲基吡咯並[3,2-d]嘧啶(196 mg,76.79%)。LCMS: m/z (ESI), [M+H]+
= 250.2。
步驟2. 7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡咯並[3,2-d]嘧啶-2-胺
在氮氣氛圍下在100℃下將2-氯-7-(3,6-二氫-2H-吡喃-4-基)-5-甲基吡咯並[3,2-d]嘧啶(196.00 mg,0.785 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(139.56 mg,0.942 mmol,1.20當量)、Pd(AcO)2
(35.25 mg,0.157 mmol,0.20當量)、XantPhos(136.25 mg,0.235 mmol,0.30當量)和Cs2
CO3
(639.37 mg,1.962 mmol,2.50當量)於二惡烷(3.00 mL)中的混合物攪拌3小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物,得到呈黃色固體的7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡咯並[3,2-d]嘧啶-2-胺(170 mg,59.93%)。LCMS: m/z (ESI), [M+H]+
= 362.3。
步驟3. 7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-5H-吡咯並[3,2-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例3)
在氫氣氛圍下在室溫下將7-(3,6-二氫-2H-吡喃-4-基)-5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡咯並[3,2-d]嘧啶-2-胺(170.00 mg,0.470 mmol,1.00當量)和Pd/C (250.29 mg,2.352 mmol,5.00當量)於MeOH(20.00 mL)和THF(50.00 mL)中的混合物攪拌隔夜。過濾所得混合物,用MeOH(5×30 mL)洗滌濾餅。減壓濃縮濾液,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:25 mL/min;梯度:7 min內30 B至45 B;RT1:6.02)來純化粗產物(100 mg),得到呈白色固體的5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-7-(氧雜環己烷-4-基)吡咯並[3,2-d]嘧啶-2-胺(29 mg,16.96%)。LCMS: m/z (ESI), [M+H]+
= 364.3。1
H NMR (300 MHz, DMSO-d6
) δ 1.76 (2H, d), 1.88 - 2.01 (2H, m), 2.46 (3H, d), 3.02 (1H, t), 3.47 (2H, td), 3.80 (3H, s), 3.88 - 4.05 (2H, m), 7.51 (1H, s), 7.61 - 7.75 (1H, m), 8.33 (2H, d), 8.74 (1H, s), 9.49 (1H, s)實例 4
製備3-異丙基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例4) 步驟1. 5-氯-1-甲基-3-(丙-1-烯-2-基)-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將Pd(dppf)Cl2
(124.24 mg,0.170 mmol,0.2當量)、K2
CO3
(293.33 mg,2.122 mmol,2.5當量)、4,4,5,5-四甲基-2-(丙-1-烯-2-基)-1,3,2-二氧雜環戊硼烷(213.99 mg,1.273 mmol,1.5當量)和5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(250.00 mg,0.849 mmol,1.00當量)於二惡烷(5.00 mL)和H2
O(1.00 mL)中的混合物攪拌2小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:3)來純化殘餘物,得到呈粉色固體的5-氯-1-甲基-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶(130 mg,73.39%)。LCMS: m/z (ESI), [M+H]+
= 209.6。1
H NMR (300 MHz, DMSO-d6
) δ 2.22 (3H, t), 4.18 (3H, s), 5.50 (1H, p), 6.43 (1H, d), 9.45 (1H, s)
步驟2. 1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(丙-1-烯-2-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs2
CO3
(468.47 mg,1.438 mmol,2.5當量)、XantPhos(66.56 mg,0.115 mmol,0.2當量)、Pd(AcO)2
(25.82 mg,0.115 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(127.82 mg,0.863 mmol,1.50當量)和5-氯-1-甲基-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶(120.00 mg,0.575 mmol,1.00 當量)於二惡烷(4.00 mL)中的混合物攪拌3小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 15:1)來純化殘餘物,得到呈黃色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶-5-胺(90 mg,48.85%)。LCMS: m/z (ESI), [M+H]+
= 321.3。1
H NMR (300 MHz, DMSO-d6
) δ 2.18 (3H, s), 2.45 (3H, d), 4.09 (3H, d), 5.33 (1H, d), 6.35 (1H, d), 7.70-7.78 (1H, m), 8.38 (1H, s), 8.96 (1H, s), 9.22 (1H, s), 9.37 (1H, s)
步驟3. 3-異丙基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例4)
在氫氣氛圍下在室溫下將Pd/C(89.69 mg,0.843 mmol,3當量)和1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶-5-胺(90.00 mg,0.281 mmol,1.00當量)於MeOH(10.00 mL)中的混合物攪拌隔夜。可通過LCMS來檢測所需產物。過濾所得混合物,用MeOH(4×50 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
: MeOH 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:MeOH;流動速率:25 mL/min;梯度:7 min內58 B至70 B;RT1 5.57)來純化粗產物(80 mg),得到呈白色固體的3-異丙基-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(50 mg,54.66%)。LCMS: m/z (ESI), [M+H]+
= 323.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.39 (6H, d), 2.45 (3H, d), 3.18-3.34 (1H, m), 4.04 (3H, s), 7.69-7.75 (1H, m), 8.37 (1H, s), 8.79 (1H, s), 9.16 (1H, s), 9.44 (1H, s)實例 5
製備3-環己基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例5)
步驟1. 5-氯-1-甲基-3-(丙-1-烯-2-基)-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將Pd(dppf)Cl2
(124.24 mg,0.170 mmol,0.2當量)、K2
CO3
(293.33 mg,2.122 mmol,2.5當量)、4,4,5,5-四甲基-2-(丙-1-烯-2-基)-1,3,2-二氧雜環戊硼烷(213.99 mg,1.273 mmol,1.5當量)和5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(250.00 mg,0.849 mmol,1.00當量)於二惡烷(5.00 mL)和H2
O(1.00 mL)中的混合物攪拌2小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:3)來純化殘餘物,得到呈粉色固體的5-氯-1-甲基-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶(130 mg,73.39%)。LCMS: m/z (ESI), [M+H]+
= 209.6。1
H NMR (300 MHz, DMSO-d6
) δ 2.22 (3H, t), 4.18 (3H, s), 5.50 (1H, p), 6.43 (1H, d), 9.45 (1H, s)
步驟2. 1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(丙-1-烯-2-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs2
CO3
(468.47 mg,1.438 mmol,2.5當量)、XantPhos(66.56 mg,0.115 mmol,0.2當量)、Pd(AcO)2
(25.82 mg,0.115 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(127.82 mg,0.863 mmol,1.50當量)和5-氯-1-甲基-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶(120.00 mg,0.575 mmol,1.00 當量)於二惡烷(4.00 mL)中的混合物攪拌3小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 15:1)來純化殘餘物,得到呈黃色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶-5-胺(90 mg,48.85%)。LCMS: m/z (ESI), [M+H]+
= 321.3。1
H NMR (300 MHz, DMSO-d6
) δ 2.18 (3H, s), 2.45 (3H, d), 4.09 (3H, d), 5.33 (1H, d), 6.35 (1H, d), 7.70-7.78 (1H, m), 8.38 (1H, s), 8.96 (1H, s), 9.22 (1H, s), 9.37 (1H, s)
步驟3. 3-異丙基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例4)
在氫氣氛圍下在室溫下將Pd/C(89.69 mg,0.843 mmol,3當量)和1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-(丙-1-烯-2-基)吡唑並[4,3-d]嘧啶-5-胺(90.00 mg,0.281 mmol,1.00當量)於MeOH(10.00 mL)中的混合物攪拌隔夜。可通過LCMS來檢測所需產物。過濾所得混合物,用MeOH(4×50 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
: MeOH 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:MeOH;流動速率:25 mL/min;梯度:7 min內58 B至70 B;RT1 5.57)來純化粗產物(80 mg),得到呈白色固體的3-異丙基-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(50 mg,54.66%)。LCMS: m/z (ESI), [M+H]+
= 323.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.39 (6H, d), 2.45 (3H, d), 3.18-3.34 (1H, m), 4.04 (3H, s), 7.69-7.75 (1H, m), 8.37 (1H, s), 8.79 (1H, s), 9.16 (1H, s), 9.44 (1H, s)實例 5
製備3-環己基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例5) 步驟1. 5-氯-3-(環己-1-烯-1-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將含K2
CO3
(293.33 mg,2.122 mmol,2.50當量)、2-(環己-1-烯-1-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(265.01 mg,1.273 mmol,1.50當量)和5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(250.00 mg,0.849 mmol,1.00當量)的二惡烷(5.00 mL)和H2
O(1.00 mL)攪拌2小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:2)來純化殘餘物,得到呈粉色固體的5-氯-3-(環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(150 mg,71.04%)。LCMS: m/z (ESI), [M+H]+
= 249.3。1
H NMR (300 MHz, DMSO-d6
) δ 1.61 - 1.80 (4H, m), 2.29 (2H, s), 2.54 (2H, s), 4.14 (3H, s), 7.17 - 7.27 (1H, m), 9.40 (1H, s)
步驟2. 3-(環己-1-烯-1-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs2
CO3
(458.51 mg,1.407 mmol,2.5當量)、XantPhos(65.14 mg,0.113 mmol,0.2當量)、Pd(AcO)2
(25.28 mg,0.113 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(125.11 mg,0.844 mmol,1.5當量)和5-氯-3-(環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(140.00 mg,0.563 mmol,1.00當量)於二惡烷(4.00 mL)中的混合物攪拌3小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 15:1)來純化殘餘物,得到呈黃色固體的3-(環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(140 mg,69.00%)。LCMS: m/z (ESI), [M+H]+
= 361.3。1
H NMR (300 MHz, DMSO-d6
) δ 1.61 - 1.83 (4H, m), 2.17 - 2.33 (4H, m), 2.46 (3H, d), 4.06 (3H, s), 7.21 (1H, s), 7.73 (1H, s), 8.38 (1H, s), 8.88 (1H, s), 9.19 (1H, s), 9.46 (1H, s)
步驟3. 3-環己基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例5)
在氫氣氛圍下在室溫下將3-(環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(160.00 mg,0.444 mmol,1.00當量)和Pd/C(236.21 mg,2.220 mmol,5.00當量)於THF(40.00 mL)和MeOH (80.00 mL)中的溶液攪拌3天。通過過濾收集沉澱的固體並用MeOH(5×30 mL)洗滌。減壓濃縮所得混合物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內40 B至50 B;RT1:6.55)來純化粗產物(150 mg),得到呈白色固體的3-環己基-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(17.41 mg,10.82%)。LCMS: m/z (ESI), [M+H]+
= 363.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.33 (3H, d), 1.76 (5H, d), 2.00 (2H, d), 2.46 (3H, d), 2.88 - 3.05 (1H, m), 4.04 (3H, s), 7.72 (1H, t), 8.38 (1H, s), 8.78 (1H, s), 9.15 (1H, s), 9.51 (1H, s)實例 8
製備3-((1r,4r)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例7)及3-((1s,4s)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例8)
步驟1. 5-氯-3-(環己-1-烯-1-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將含K2
CO3
(293.33 mg,2.122 mmol,2.50當量)、2-(環己-1-烯-1-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(265.01 mg,1.273 mmol,1.50當量)和5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(250.00 mg,0.849 mmol,1.00當量)的二惡烷(5.00 mL)和H2
O(1.00 mL)攪拌2小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:2)來純化殘餘物,得到呈粉色固體的5-氯-3-(環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(150 mg,71.04%)。LCMS: m/z (ESI), [M+H]+
= 249.3。1
H NMR (300 MHz, DMSO-d6
) δ 1.61 - 1.80 (4H, m), 2.29 (2H, s), 2.54 (2H, s), 4.14 (3H, s), 7.17 - 7.27 (1H, m), 9.40 (1H, s)
步驟2. 3-(環己-1-烯-1-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs2
CO3
(458.51 mg,1.407 mmol,2.5當量)、XantPhos(65.14 mg,0.113 mmol,0.2當量)、Pd(AcO)2
(25.28 mg,0.113 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(125.11 mg,0.844 mmol,1.5當量)和5-氯-3-(環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(140.00 mg,0.563 mmol,1.00當量)於二惡烷(4.00 mL)中的混合物攪拌3小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 15:1)來純化殘餘物,得到呈黃色固體的3-(環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(140 mg,69.00%)。LCMS: m/z (ESI), [M+H]+
= 361.3。1
H NMR (300 MHz, DMSO-d6
) δ 1.61 - 1.83 (4H, m), 2.17 - 2.33 (4H, m), 2.46 (3H, d), 4.06 (3H, s), 7.21 (1H, s), 7.73 (1H, s), 8.38 (1H, s), 8.88 (1H, s), 9.19 (1H, s), 9.46 (1H, s)
步驟3. 3-環己基-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例5)
在氫氣氛圍下在室溫下將3-(環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(160.00 mg,0.444 mmol,1.00當量)和Pd/C(236.21 mg,2.220 mmol,5.00當量)於THF(40.00 mL)和MeOH (80.00 mL)中的溶液攪拌3天。通過過濾收集沉澱的固體並用MeOH(5×30 mL)洗滌。減壓濃縮所得混合物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內40 B至50 B;RT1:6.55)來純化粗產物(150 mg),得到呈白色固體的3-環己基-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(17.41 mg,10.82%)。LCMS: m/z (ESI), [M+H]+
= 363.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.33 (3H, d), 1.76 (5H, d), 2.00 (2H, d), 2.46 (3H, d), 2.88 - 3.05 (1H, m), 4.04 (3H, s), 7.72 (1H, t), 8.38 (1H, s), 8.78 (1H, s), 9.15 (1H, s), 9.51 (1H, s)實例 8
製備3-((1r,4r)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例7)及3-((1s,4s)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例8) 步驟1. 5-氯-3-(4-甲氧基環己-1-烯-1-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將K2
CO3
(234.66 mg,1.698 mmol,2.5當量)、Pd(dppf)Cl2
CH2
Cl2
(110.93 mg,0.136 mmol,0.2當量)、2-(4-甲氧基環己-1-烯-1-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(194.07 mg,0.815 mmol,1.2當量)和5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(200.00 mg,0.679 mmol,1.00當量)於二惡烷(5.00 mL)和H2
O(1.00 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:2)來純化殘餘物,得到呈白色固體的5-氯-3-(4-甲氧基環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(175 mg,92.44%)。LCMS: m/z (ESI), [M+H]+
= 279.3。1
H NMR (300 MHz, CDCl 3
) δ 1.89 (2H, d), 2.11 (1H, m), 2.33 (1H, t), 2.66 (1H, m), 2.88 (1H, d), 3.43 (3H, s), 3.60 (1H, d), 4.12 (3H, s), 7.27 (1H, d), 8.89 (1H, s)
步驟2. 3-(4-甲氧基環己-1-烯-1-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs2
CO3
(496.78 mg,1.525 mmol,2.5當量)、XantPhos(70.58 mg,0.122 mmol,0.2當量)、Pd(AcO)2
(27.39 mg,0.122 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(99.40 mg,0.671 mmol,1.1當量)和5-氯-3-(4-甲氧基環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(170.00 mg,0.610 mmol,1.00當量)於二惡烷(5.00 mL)中的混合物攪拌3小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到呈黃色固體的3-(4-甲氧基環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(100 mg,41.99%)。LCMS: m/z (ESI), [M+H]+
= 392.4。1
H NMR (300 MHz, DMSO-d6
) δ 1.67 (1H, d), 2.00 (1H, s), 2.27 (1H, m), 2.46 (3H, d), 2.72 (1H, d), 3.17 (2H, d), 3.33 (3H, s), 3.53 (1H, s), 4.07 (3H, s), 7.08 (1H, s), 7.73 (1H, s), 8.39 (1H, s), 8.91 (1H, s), 9.19 (1H, s), 9.43 (1H, s)
步驟3. 3-((1r,4r)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例7)及3-((1s,4s)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例8)
在氫氣氛圍下在室溫下將Pd/C(65.41 mg,0.615 mmol,3當量)和3-(4-甲氧基環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(80.00 mg,0.205 mmol,1.00當量)於MeOH(150.00 mL)和THF(80.00 mL)中的混合物攪拌隔夜。過濾所得混合物,用MeOH(3×50 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:25 mL/min;梯度:7 min內37 B至41 B;RT1:5.30/5.92)來純化粗產物(80 mg),得到呈白色固體的3-((1r,4r)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例7,60 mg,22.73%)。LCMS: m/z (ESI), [M+H]+
= 393.3。1
H NMR (400 MHz, DMSO-d6
) δ 1.28 (2H, q), 1.83 (2H, q), 2.09 (4H, t), 2.43 (3H, d), 2.95 (1H, t), 3.30 (4H, m), 4.04 (3H, s), 7.73 (1H, s), 8.38 (1H, s), 8.81 (1H, s), 9.16 (1H, s), 9.51 (1H, s);
呈白色固體的3-((1s,4s)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例8,10 mg,12.19%)。LCMS: m/z (ESI), [M+H]+
= 393.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.57 (2H, t), 1.76 (2H, m), 1.86 (2H, m), 2.04 (2H, q), 2.43 (3H, d), 3.05 (1H, m), 3.20 (3H, s), 3.42 (1H, s), 4.04 (3H, s), 7.72 (1H, s), 8.37 (1H, s), 8.80 (1H, s), 9.14 (1H, s), 9.34 (1H, s)。實例 9
製備3-(4,4-二氟環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例9)
步驟1. 5-氯-3-(4-甲氧基環己-1-烯-1-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將K2
CO3
(234.66 mg,1.698 mmol,2.5當量)、Pd(dppf)Cl2
CH2
Cl2
(110.93 mg,0.136 mmol,0.2當量)、2-(4-甲氧基環己-1-烯-1-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(194.07 mg,0.815 mmol,1.2當量)和5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(200.00 mg,0.679 mmol,1.00當量)於二惡烷(5.00 mL)和H2
O(1.00 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(PE/EtOAc 1:2)來純化殘餘物,得到呈白色固體的5-氯-3-(4-甲氧基環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(175 mg,92.44%)。LCMS: m/z (ESI), [M+H]+
= 279.3。1
H NMR (300 MHz, CDCl 3
) δ 1.89 (2H, d), 2.11 (1H, m), 2.33 (1H, t), 2.66 (1H, m), 2.88 (1H, d), 3.43 (3H, s), 3.60 (1H, d), 4.12 (3H, s), 7.27 (1H, d), 8.89 (1H, s)
步驟2. 3-(4-甲氧基環己-1-烯-1-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs2
CO3
(496.78 mg,1.525 mmol,2.5當量)、XantPhos(70.58 mg,0.122 mmol,0.2當量)、Pd(AcO)2
(27.39 mg,0.122 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(99.40 mg,0.671 mmol,1.1當量)和5-氯-3-(4-甲氧基環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(170.00 mg,0.610 mmol,1.00當量)於二惡烷(5.00 mL)中的混合物攪拌3小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到呈黃色固體的3-(4-甲氧基環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(100 mg,41.99%)。LCMS: m/z (ESI), [M+H]+
= 392.4。1
H NMR (300 MHz, DMSO-d6
) δ 1.67 (1H, d), 2.00 (1H, s), 2.27 (1H, m), 2.46 (3H, d), 2.72 (1H, d), 3.17 (2H, d), 3.33 (3H, s), 3.53 (1H, s), 4.07 (3H, s), 7.08 (1H, s), 7.73 (1H, s), 8.39 (1H, s), 8.91 (1H, s), 9.19 (1H, s), 9.43 (1H, s)
步驟3. 3-((1r,4r)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例7)及3-((1s,4s)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例8)
在氫氣氛圍下在室溫下將Pd/C(65.41 mg,0.615 mmol,3當量)和3-(4-甲氧基環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(80.00 mg,0.205 mmol,1.00當量)於MeOH(150.00 mL)和THF(80.00 mL)中的混合物攪拌隔夜。過濾所得混合物,用MeOH(3×50 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:25 mL/min;梯度:7 min內37 B至41 B;RT1:5.30/5.92)來純化粗產物(80 mg),得到呈白色固體的3-((1r,4r)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例7,60 mg,22.73%)。LCMS: m/z (ESI), [M+H]+
= 393.3。1
H NMR (400 MHz, DMSO-d6
) δ 1.28 (2H, q), 1.83 (2H, q), 2.09 (4H, t), 2.43 (3H, d), 2.95 (1H, t), 3.30 (4H, m), 4.04 (3H, s), 7.73 (1H, s), 8.38 (1H, s), 8.81 (1H, s), 9.16 (1H, s), 9.51 (1H, s);
呈白色固體的3-((1s,4s)-4-甲氧基環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例8,10 mg,12.19%)。LCMS: m/z (ESI), [M+H]+
= 393.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.57 (2H, t), 1.76 (2H, m), 1.86 (2H, m), 2.04 (2H, q), 2.43 (3H, d), 3.05 (1H, m), 3.20 (3H, s), 3.42 (1H, s), 4.04 (3H, s), 7.72 (1H, s), 8.37 (1H, s), 8.80 (1H, s), 9.14 (1H, s), 9.34 (1H, s)。實例 9
製備3-(4,4-二氟環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例9) 步驟1. 5-氯-3-(4,4-二氟環己-1-烯-1-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(200.00 mg,0.679 mmol,1.00當量)、2-(4,4-二氟環己-1-烯-1-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(198.93 mg,0.815 mmol,1.20當量)、Pd(dppf)Cl2
(99.39 mg,0.136 mmol,0.2當量)和K2
CO3
(234.66 mg,1.698 mmol,2.5當量)於二惡烷(3.00 mL)和H2
O(0.60 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(PE/EtOAc 2:3)來純化殘餘物,得到呈黃色固體的5-氯-3-(4,4-二氟環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(100 mg,51.72%)。LCMS: m/z (ESI), [M+H]+
= 285.3。
步驟2. 3-(4,4-二氟環己-1-烯-1-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將5-氯-3-(4,4-二氟環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(100.00 mg,0.351 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(62.45 mg,0.421 mmol,1.20當量)、Pd(OAc)2
(15.77 mg,0.070 mmol,0.20當量)、XantPhos(60.97 mg,0.105 mmol,0.30當量)和Cs2
CO3
(286.12 mg,0.878 mmol,2.50當量)於二惡烷(2.50 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到呈白色固體的3-(4,4-二氟環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(110 mg,79.00%)。LCMS: m/z (ESI), [M+H]+
= 397.3。
步驟3. 3-(4,4-二氟環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例9)
在氫氣氛圍下在30℃下將3-(4,4-二氟環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(130.00 mg,0.328 mmol,1.00 當量)和Pd/C(174.50 mg,1.640 mmol,5.00當量)於MeOH(60.00 mL)和THF(30.00 mL)中的混合物攪拌5個小時。過濾所得混合物,用CH2
Cl2
(3×40 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內34 B至48 B;RT1:5.97)來純化粗產物(70 mg),得到呈粉色固體的3-(4,4-二氟環己基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(17 mg,13.01%)。LCMS: m/z (ESI), [M+H]+
= 399.3。1
H NMR (300 MHz, MeOD-d4
) δ 1.80-2.22 (8H, m), 2.49 (3H, d), 3.22 (1H, s), 4.03 (3H, s), 7.58 (1H, t), 8.27 (1H, s), 8.98 (1H, s), 9.62 (1H, s)實例 10
製備1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例10)
步驟1. 5-氯-3-(4,4-二氟環己-1-烯-1-基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(200.00 mg,0.679 mmol,1.00當量)、2-(4,4-二氟環己-1-烯-1-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(198.93 mg,0.815 mmol,1.20當量)、Pd(dppf)Cl2
(99.39 mg,0.136 mmol,0.2當量)和K2
CO3
(234.66 mg,1.698 mmol,2.5當量)於二惡烷(3.00 mL)和H2
O(0.60 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(PE/EtOAc 2:3)來純化殘餘物,得到呈黃色固體的5-氯-3-(4,4-二氟環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(100 mg,51.72%)。LCMS: m/z (ESI), [M+H]+
= 285.3。
步驟2. 3-(4,4-二氟環己-1-烯-1-基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將5-氯-3-(4,4-二氟環己-1-烯-1-基)-1-甲基吡唑並[4,3-d]嘧啶(100.00 mg,0.351 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(62.45 mg,0.421 mmol,1.20當量)、Pd(OAc)2
(15.77 mg,0.070 mmol,0.20當量)、XantPhos(60.97 mg,0.105 mmol,0.30當量)和Cs2
CO3
(286.12 mg,0.878 mmol,2.50當量)於二惡烷(2.50 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到呈白色固體的3-(4,4-二氟環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(110 mg,79.00%)。LCMS: m/z (ESI), [M+H]+
= 397.3。
步驟3. 3-(4,4-二氟環己基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例9)
在氫氣氛圍下在30℃下將3-(4,4-二氟環己-1-烯-1-基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(130.00 mg,0.328 mmol,1.00 當量)和Pd/C(174.50 mg,1.640 mmol,5.00當量)於MeOH(60.00 mL)和THF(30.00 mL)中的混合物攪拌5個小時。過濾所得混合物,用CH2
Cl2
(3×40 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內34 B至48 B;RT1:5.97)來純化粗產物(70 mg),得到呈粉色固體的3-(4,4-二氟環己基)-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(17 mg,13.01%)。LCMS: m/z (ESI), [M+H]+
= 399.3。1
H NMR (300 MHz, MeOD-d4
) δ 1.80-2.22 (8H, m), 2.49 (3H, d), 3.22 (1H, s), 4.03 (3H, s), 7.58 (1H, t), 8.27 (1H, s), 8.98 (1H, s), 9.62 (1H, s)實例 10
製備1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(實例10) 步驟1. 5-氯-3-碘-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在0℃下將Cs2
CO3
(1045.60 mg,3.209 mmol,3當量)、CD3
I(775.31 mg,5.349 mmol,5當量)和5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶(300.00 mg,1.070 mmol,1.00當量)於DMF(6.00 mL)中的混合物攪拌1 h。可通過LCMS來檢測所需產物。用水(50 mL)稀釋所得混合物。用EtOAc(3×50 mL)萃取所得混合物。將合併的有機層用飽和鹽水(3×50 mL)和Na2
S2
O3
(3×50 mL)洗滌且經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液,得到呈粉色固體的5-氯-3-碘-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶(260 mg,81.70%)。LCMS: m/z (ESI), [M+H]+
= 298.0。1
H NMR (400 MHz, DMSO-d6
) δ 9.45 (1H, s).
步驟2. 5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將K2
CO3
(313.58 mg,2.269 mmol,2.5當量)、Pd(dppf)Cl2
CH2
Cl2
(148.23 mg,0.182 mmol,0.2當量)、2-(3,6-二氫-2H-吡喃-4-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(228.79 mg,1.089 mmol,1.2當量)和5-氯-3-碘-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶(270.00 mg,0.908 mmol,1.00當量)於二惡烷(5.00 mL)和水(1.00 mL)中的混合物攪拌2小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(己烷/EtOAc 1:2)來純化殘餘物,得到呈粉色固體的5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶(100 mg,43.43%)。LCMS: m/z (ESI), [M+H]+
= 254.2。1
H NMR (400 MHz, DMSO-d6
) δ 2.62 (2H, m), 3.86 (2H, t), 4.33 (2H, q), 7.18 (1H, t), 9.43 (1H, s)
步驟3. 3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs2
CO3
(385.28 mg,1.182 mmol,3當量)、XantPhos(45.61 mg,0.079 mmol,0.2當量)、Pd(OAc)2
(17.70 mg,0.079 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(70.08 mg,0.473 mmol,1.20當量)和5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶(100.00 mg,0.394 mmol,1.00當量)於二惡烷(3.00 mL)中的混合物攪拌2 h。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到呈黃色固體的3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(100 mg,69.43%)。LCMS: m/z (ESI), [M+H]+
= 366.3。1
H NMR (400 MHz, DMSO-d6
) δ 2.44 (2H, d), 2.61 (3H, s), 3.17 (1H, d), 3.85 (2H, t), 4.26 (2H, q), 7.72 (1H, s), 8.38 (1H, s), 8.94 (1H, s), 9.20 (1H, s), 9.37 (1H, s)。
步驟4. 1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺.(實例10)
在氫氣氛圍下在35℃下將Pd/C(87.37 mg,0.821 mmol,3當量)和3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(100.00 mg,0.274 mmol,1.00當量)於MeOH (10.00 mL)和THF(10.00 mL)中的混合物攪拌2 h。可通過LCMS來檢測所需產物。過濾所得混合物,用MeOH(3×30 mL)和DCM(3×30 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內15 B至35 B;RT1:6.4)來純化粗產物(40 mg),得到呈白色固體的1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(10 mg,9.85%)。LCMS: m/z (ESI), [M+H]+
= 368.2。1
H NMR (400 MHz, DMSO-d6
) δ 1.94 (4H, d), 2.43 (3H, d), 3.24 (1H, d), 3.48 (2H, m), 3.93 (2H, m), 7.71 (1H, s), 8.37 (1H, s), 8.81 (1H, s), 9.15 (1H, s), 9.33 (1H, s).實例 11/12
製備1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1s,4s)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例11)及1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1r,4r)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例12)
步驟1. 5-氯-3-碘-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在0℃下將Cs2
CO3
(1045.60 mg,3.209 mmol,3當量)、CD3
I(775.31 mg,5.349 mmol,5當量)和5-氯-3-碘-1H-吡唑並[4,3-d]嘧啶(300.00 mg,1.070 mmol,1.00當量)於DMF(6.00 mL)中的混合物攪拌1 h。可通過LCMS來檢測所需產物。用水(50 mL)稀釋所得混合物。用EtOAc(3×50 mL)萃取所得混合物。將合併的有機層用飽和鹽水(3×50 mL)和Na2
S2
O3
(3×50 mL)洗滌且經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液,得到呈粉色固體的5-氯-3-碘-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶(260 mg,81.70%)。LCMS: m/z (ESI), [M+H]+
= 298.0。1
H NMR (400 MHz, DMSO-d6
) δ 9.45 (1H, s).
步驟2. 5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶
在氮氣氛圍下在80℃下將K2
CO3
(313.58 mg,2.269 mmol,2.5當量)、Pd(dppf)Cl2
CH2
Cl2
(148.23 mg,0.182 mmol,0.2當量)、2-(3,6-二氫-2H-吡喃-4-基)-4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷(228.79 mg,1.089 mmol,1.2當量)和5-氯-3-碘-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶(270.00 mg,0.908 mmol,1.00當量)於二惡烷(5.00 mL)和水(1.00 mL)中的混合物攪拌2小時。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(己烷/EtOAc 1:2)來純化殘餘物,得到呈粉色固體的5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶(100 mg,43.43%)。LCMS: m/z (ESI), [M+H]+
= 254.2。1
H NMR (400 MHz, DMSO-d6
) δ 2.62 (2H, m), 3.86 (2H, t), 4.33 (2H, q), 7.18 (1H, t), 9.43 (1H, s)
步驟3. 3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在氮氣氛圍下在100℃下將Cs2
CO3
(385.28 mg,1.182 mmol,3當量)、XantPhos(45.61 mg,0.079 mmol,0.2當量)、Pd(OAc)2
(17.70 mg,0.079 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(70.08 mg,0.473 mmol,1.20當量)和5-氯-3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-1H-吡唑並[4,3-d]嘧啶(100.00 mg,0.394 mmol,1.00當量)於二惡烷(3.00 mL)中的混合物攪拌2 h。可通過LCMS來檢測所需產物。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到呈黃色固體的3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(100 mg,69.43%)。LCMS: m/z (ESI), [M+H]+
= 366.3。1
H NMR (400 MHz, DMSO-d6
) δ 2.44 (2H, d), 2.61 (3H, s), 3.17 (1H, d), 3.85 (2H, t), 4.26 (2H, q), 7.72 (1H, s), 8.38 (1H, s), 8.94 (1H, s), 9.20 (1H, s), 9.37 (1H, s)。
步驟4. 1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺.(實例10)
在氫氣氛圍下在35℃下將Pd/C(87.37 mg,0.821 mmol,3當量)和3-(3,6-二氫-2H-吡喃-4-基)-1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(100.00 mg,0.274 mmol,1.00當量)於MeOH (10.00 mL)和THF(10.00 mL)中的混合物攪拌2 h。可通過LCMS來檢測所需產物。過濾所得混合物,用MeOH(3×30 mL)和DCM(3×30 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內15 B至35 B;RT1:6.4)來純化粗產物(40 mg),得到呈白色固體的1-(甲基-d3
)-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-(四氫-2H-吡喃-4-基)-1H-吡唑並[4,3-d]嘧啶-5-胺(10 mg,9.85%)。LCMS: m/z (ESI), [M+H]+
= 368.2。1
H NMR (400 MHz, DMSO-d6
) δ 1.94 (4H, d), 2.43 (3H, d), 3.24 (1H, d), 3.48 (2H, m), 3.93 (2H, m), 7.71 (1H, s), 8.37 (1H, s), 8.81 (1H, s), 9.15 (1H, s), 9.33 (1H, s).實例 11/12
製備1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1s,4s)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例11)及1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1r,4r)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例12) 步驟1. 4-(5-氯-1-甲基-1H-吡唑並[4,3-d]嘧啶-3-基)環己-3-烯-1-醇
在室溫下向40 mL小瓶中添加5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(500.00 mg,1.698 mmol,1.00當量)和4-(4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷-2-基)環己-3-烯-1-醇(570.78 mg,2.547 mmol,1.50當量)、Pd(dppf)Cl2
(248.47 mg,0.340 mmol,0.20當量)、K2
CO3
(938.64 mg,6.792 mmol,4當量)、二惡烷(10.00 mL)以及H2
O(2.00 mL)。隨後在氮氣氛圍下在80℃下將混合物攪拌3h。真空濃縮所得混合物。通過製備型TLC(PE:EA = 1:4)來純化殘餘物,得到呈黃色固體的4-[5-氯-1-甲基吡唑並[4,3-d]嘧啶-3-基]環己-3-烯-1-醇(253 mg,56.29%)。LCMS: m/z (ESI), [M+H]+
= 265.2。
步驟2. 5-氯-3-(4-(二氟甲氧基)環己-1-烯基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在50℃下向40 mL小瓶中添加4-[5-氯-1-甲基吡唑並[4,3-d]嘧啶-3-基]環己-3-烯-1-醇(253.00 mg,0.956 mmol,1.00當量)、CuI(63.71 mg,0.335 mmol,0.35當量)、MeCN(10.00 mL)且向混合物中逐滴添加2,2-二氟-2-(氟磺醯基)乙酸(510.61 mg,2.867 mmol,3.00當量)。隨後在空氣氛圍下在50℃下將混合物攪拌3h。用水(10 mL)稀釋所得混合物。用DCM(3×30 mL)萃取水層。用DCM(5 mL)稀釋所得混合物。過濾所得混合物,用DCM(10 mL*3)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(PE/EtOAc 1:1)來純化殘餘物,得到呈黃色固體的5-氯-3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基吡唑並[4,3-d]嘧啶(180 mg,47.87%)。LCMS: m/z (ESI), [M+H]+
= 315.2。
步驟3. 3-(4-(二氟甲氧基)環己-1-烯基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在室溫下向40 mL小瓶中添加5-氯-3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基吡唑並[4,3-d]嘧啶(180.00 mg,0.572 mmol,1.00當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(101.69 mg,0.686 mmol,1.20當量)、Pd(OAc)2
(38.52 mg,0.172 mmol,0.30當量)、XantPhos(99.28 mg,0.172 mmol,0.30 當量)、Cs2
CO3
(559.05 mg,1.716 mmol,3.00當量)以及二惡烷(10.00 mL)。隨後在氮氣氛圍下在70℃下將混合物攪拌3h。真空濃縮所得混合物。通過製備型TLC(DCM:MeOH 15:1)來純化殘餘物,得到呈黃色固體的3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(135 mg,55.35%)。LCMS: m/z (ESI), [M+H]+
= 427.1。1
H NMR (400 MHz, CDCl3
) δ1.97 - 2.23 (3H, m), 2.55 (4H, s), 2.68 - 2.87 (2H, m), 2.98 (1H, d), 4.11 (3H, s), 4.54 - 4.62 (1H,m), 6.35 (1H, t), 7.00 (1H, s), 7.61 (1H, s), 8.29 (1H,s), 8.82 (1H, s), 9.90 (1H, s)
步驟4. 製備1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1s,4s)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例11)及1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1r,4r)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例12)
在空氣氛圍下向3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(135.00 mg,0.317 mmol,1.00當量)於MeOH(20 mL)中的攪拌混合物中添加Pd/C(168.45 mg,1.583 mmol,5.00當量)。在氫氣氛圍下在40℃下將所得混合物攪拌4h。過濾所得混合物,且用DCM(8×100 mL)洗滌濾餅。減壓濃縮濾液,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內34 B至54 B;RT1:5.93)來純化粗產物,得到固體。隨後,通過製備型HPLC利用以下條件(柱:CHIRALPAK IG,2*25cm,5um;移動相A:Hex:DCM=3:1,移動相B:EtOH;流動速率:20 mL/min;梯度:15 min內10 B至10 B;RT1:10;RT2:11)來純化產物,得到呈白色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1s,4s)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例11,15 mg,50.00%)和呈白色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1r,4r)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例12,5 mg,16.67%)。
(實例11) LCMS: m/z (ESI), [M+H]+
= 429.3。1
H NMR (300 MHz, DMSO-d6
) δ1.87 (2H, d), 1.97 (4H, d), 2.09 (2H, d), 2.43 (3H, s), 3.07 (1H, d), 4.04 (3H, s), 4.37 (1H,s), 6.69 (1H, t), 7.70 (1H, s), 8.36 (1H, s), 8.79 (1H,s), 9.14 (1H, s), 9.33 (1H, s);
(實例12) LCMS: m/z (ESI), [M+H]+
= 429.3。1
H NMR (300 MHz, MeOD-d4
) δ1.59-1.71 (2H, m), 1.95-2.09 (2H, m), 2.15-2.20 (4H, m), 2.55 (3H,s), 3.05-3.14 (1H, m), 4.09 (3H, s), 4.20-4.27 (1H, m), 6.71 (1H, t), 7.64 (1H, s), 8.31 (1H,s), 9.03 (1H, s), 9.85 (1H, s)。實例 13
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例13)
步驟1. 4-(5-氯-1-甲基-1H-吡唑並[4,3-d]嘧啶-3-基)環己-3-烯-1-醇
在室溫下向40 mL小瓶中添加5-氯-3-碘-1-甲基吡唑並[4,3-d]嘧啶(500.00 mg,1.698 mmol,1.00當量)和4-(4,4,5,5-四甲基-1,3,2-二氧雜環戊硼烷-2-基)環己-3-烯-1-醇(570.78 mg,2.547 mmol,1.50當量)、Pd(dppf)Cl2
(248.47 mg,0.340 mmol,0.20當量)、K2
CO3
(938.64 mg,6.792 mmol,4當量)、二惡烷(10.00 mL)以及H2
O(2.00 mL)。隨後在氮氣氛圍下在80℃下將混合物攪拌3h。真空濃縮所得混合物。通過製備型TLC(PE:EA = 1:4)來純化殘餘物,得到呈黃色固體的4-[5-氯-1-甲基吡唑並[4,3-d]嘧啶-3-基]環己-3-烯-1-醇(253 mg,56.29%)。LCMS: m/z (ESI), [M+H]+
= 265.2。
步驟2. 5-氯-3-(4-(二氟甲氧基)環己-1-烯基)-1-甲基-1H-吡唑並[4,3-d]嘧啶
在50℃下向40 mL小瓶中添加4-[5-氯-1-甲基吡唑並[4,3-d]嘧啶-3-基]環己-3-烯-1-醇(253.00 mg,0.956 mmol,1.00當量)、CuI(63.71 mg,0.335 mmol,0.35當量)、MeCN(10.00 mL)且向混合物中逐滴添加2,2-二氟-2-(氟磺醯基)乙酸(510.61 mg,2.867 mmol,3.00當量)。隨後在空氣氛圍下在50℃下將混合物攪拌3h。用水(10 mL)稀釋所得混合物。用DCM(3×30 mL)萃取水層。用DCM(5 mL)稀釋所得混合物。過濾所得混合物,用DCM(10 mL*3)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(PE/EtOAc 1:1)來純化殘餘物,得到呈黃色固體的5-氯-3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基吡唑並[4,3-d]嘧啶(180 mg,47.87%)。LCMS: m/z (ESI), [M+H]+
= 315.2。
步驟3. 3-(4-(二氟甲氧基)環己-1-烯基)-1-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[4,3-d]嘧啶-5-胺
在室溫下向40 mL小瓶中添加5-氯-3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基吡唑並[4,3-d]嘧啶(180.00 mg,0.572 mmol,1.00當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(101.69 mg,0.686 mmol,1.20當量)、Pd(OAc)2
(38.52 mg,0.172 mmol,0.30當量)、XantPhos(99.28 mg,0.172 mmol,0.30 當量)、Cs2
CO3
(559.05 mg,1.716 mmol,3.00當量)以及二惡烷(10.00 mL)。隨後在氮氣氛圍下在70℃下將混合物攪拌3h。真空濃縮所得混合物。通過製備型TLC(DCM:MeOH 15:1)來純化殘餘物,得到呈黃色固體的3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(135 mg,55.35%)。LCMS: m/z (ESI), [M+H]+
= 427.1。1
H NMR (400 MHz, CDCl3
) δ1.97 - 2.23 (3H, m), 2.55 (4H, s), 2.68 - 2.87 (2H, m), 2.98 (1H, d), 4.11 (3H, s), 4.54 - 4.62 (1H,m), 6.35 (1H, t), 7.00 (1H, s), 7.61 (1H, s), 8.29 (1H,s), 8.82 (1H, s), 9.90 (1H, s)
步驟4. 製備1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1s,4s)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例11)及1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1r,4r)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例12)
在空氣氛圍下向3-[4-(二氟甲氧基)環己-1-烯-1-基]-1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[4,3-d]嘧啶-5-胺(135.00 mg,0.317 mmol,1.00當量)於MeOH(20 mL)中的攪拌混合物中添加Pd/C(168.45 mg,1.583 mmol,5.00當量)。在氫氣氛圍下在40℃下將所得混合物攪拌4h。過濾所得混合物,且用DCM(8×100 mL)洗滌濾餅。減壓濃縮濾液,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內34 B至54 B;RT1:5.93)來純化粗產物,得到固體。隨後,通過製備型HPLC利用以下條件(柱:CHIRALPAK IG,2*25cm,5um;移動相A:Hex:DCM=3:1,移動相B:EtOH;流動速率:20 mL/min;梯度:15 min內10 B至10 B;RT1:10;RT2:11)來純化產物,得到呈白色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1s,4s)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例11,15 mg,50.00%)和呈白色固體的1-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-[(1r,4r)-4-(二氟甲氧基)環己基]吡唑並[4,3-d]嘧啶-5-胺(實例12,5 mg,16.67%)。
(實例11) LCMS: m/z (ESI), [M+H]+
= 429.3。1
H NMR (300 MHz, DMSO-d6
) δ1.87 (2H, d), 1.97 (4H, d), 2.09 (2H, d), 2.43 (3H, s), 3.07 (1H, d), 4.04 (3H, s), 4.37 (1H,s), 6.69 (1H, t), 7.70 (1H, s), 8.36 (1H, s), 8.79 (1H,s), 9.14 (1H, s), 9.33 (1H, s);
(實例12) LCMS: m/z (ESI), [M+H]+
= 429.3。1
H NMR (300 MHz, MeOD-d4
) δ1.59-1.71 (2H, m), 1.95-2.09 (2H, m), 2.15-2.20 (4H, m), 2.55 (3H,s), 3.05-3.14 (1H, m), 4.09 (3H, s), 4.20-4.27 (1H, m), 6.71 (1H, t), 7.64 (1H, s), 8.31 (1H,s), 9.03 (1H, s), 9.85 (1H, s)。實例 13
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例13) 步驟1. 6-氯-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在室溫下向1-(2,4-二氯嘧啶-5-基)乙酮(500.00 mg,2.618 mmol,1.00當量)和DIPEA(1353.26 mg,10.471 mmol,4.00當量)於THF(5.00 mL)中的攪拌混合物中分批添加氧雜環己烷-4-基肼(364.89 mg,3.141 mmol,1.20當量)。並且在氮氣氛圍下在室溫下攪拌混合物2 h。用EtOAc(3×50 mL)萃取所得混合物。用水(3×10 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(PE/EtOAc 2:1)來純化殘餘物,得到呈黃色固體的6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(350 mg,52.91%)。LCMS: m/z (ESI), [M+H]+
= 253.2
步驟2. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例13)
在氮氣氛圍下在室溫下向6-氯-3-甲基-1-(氧雜環己烷-4-基)-1H-吡唑並[3,4-d]嘧啶(200 mg,0.791 mmol,1當量)、Cs2
CO3
(644.68 mg,1.979 mmol,2.5當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(140.72 mg,0.950 mmol,1.2當量)於二惡烷(20 mL)中的攪拌混合物中分批添加BrettPhos Pd G3
(143.49 mg,0.158 mmol,0.2當量)。並且在氮氣氛圍下在100℃下攪拌混合物3 h。用EtOAc(3×50 mL)萃取所得混合物。用水(3×50 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:X select CSH OBD柱,30×150 mm 5 um;移動相A:水(0.05% TFA),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內18% B至29% B;tR
:6.30 min)來純化粗產物,得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-(氧雜環己烷-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(74 mg,25.66%)。LCMS: m/z (ESI), [M+H]+
= 365.31
H NMR (DMSO-d6
, 300 MHz) δ 1.7-1.9 (2H, m), 2.0-2.2 (2H, m), 2.4-2.5 (6H, m), 3.4 (2H, td), 3.9-4.1 (2H, m), 4.5-4.7 (1H, m), 7.8 (1H, s), 8.6 (1H, s), 8.9 (1H, s), 9.3 (1H, s), 9.3 (1H, s)實例 14
製備3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例14)
步驟1. 6-氯-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在室溫下向1-(2,4-二氯嘧啶-5-基)乙酮(500.00 mg,2.618 mmol,1.00當量)和DIPEA(1353.26 mg,10.471 mmol,4.00當量)於THF(5.00 mL)中的攪拌混合物中分批添加氧雜環己烷-4-基肼(364.89 mg,3.141 mmol,1.20當量)。並且在氮氣氛圍下在室溫下攪拌混合物2 h。用EtOAc(3×50 mL)萃取所得混合物。用水(3×10 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(PE/EtOAc 2:1)來純化殘餘物,得到呈黃色固體的6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(350 mg,52.91%)。LCMS: m/z (ESI), [M+H]+
= 253.2
步驟2. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例13)
在氮氣氛圍下在室溫下向6-氯-3-甲基-1-(氧雜環己烷-4-基)-1H-吡唑並[3,4-d]嘧啶(200 mg,0.791 mmol,1當量)、Cs2
CO3
(644.68 mg,1.979 mmol,2.5當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(140.72 mg,0.950 mmol,1.2當量)於二惡烷(20 mL)中的攪拌混合物中分批添加BrettPhos Pd G3
(143.49 mg,0.158 mmol,0.2當量)。並且在氮氣氛圍下在100℃下攪拌混合物3 h。用EtOAc(3×50 mL)萃取所得混合物。用水(3×50 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:X select CSH OBD柱,30×150 mm 5 um;移動相A:水(0.05% TFA),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內18% B至29% B;tR
:6.30 min)來純化粗產物,得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-(氧雜環己烷-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(74 mg,25.66%)。LCMS: m/z (ESI), [M+H]+
= 365.31
H NMR (DMSO-d6
, 300 MHz) δ 1.7-1.9 (2H, m), 2.0-2.2 (2H, m), 2.4-2.5 (6H, m), 3.4 (2H, td), 3.9-4.1 (2H, m), 4.5-4.7 (1H, m), 7.8 (1H, s), 8.6 (1H, s), 8.9 (1H, s), 9.3 (1H, s), 9.3 (1H, s)實例 14
製備3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例14) 步驟1. 6-氯-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在0℃下將1-(2,4-二氯嘧啶-5-基)乙酮(250.00 mg,1.309 mmol,1.00當量)和氧雜環己烷-4-基肼(182.45 mg,1.571 mmol,1.20當量)、DIPEA(338.32 mg,2.618 mmol,2.00當量)於THF(10.00 mL)中的溶液攪拌2h。用EtOAc(20 mL×3)萃取所得混合物。用鹽水(10 mL×3)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(PE/EtOAc 5:1)來純化殘餘物,得到呈白色固體的6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(230 mg,69.54%)。LCMS: m/z (ESI), [M+H]+
= 253.2。
步驟2. 3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例14)
在氮氣氛圍下在100℃下將7-甲基咪唑[1,2-a]吡啶-6-胺(139.78 mg,0.950 mmol,1.50當量)和6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(160 mg,0.633 mmol,1.00當量)、BrettPhos Pd G3
(57.40 mg,0.063 mmol,0.10當量)、Cs2
CO3
(412.59 mg,1.266 mmol,2.00當量)於二惡烷(5.00 mL)中的混合物攪拌3h。通過矽膠柱色譜純化殘餘物,用CH2
Cl2
/MeOH(10:1)洗脫,得到粗產物。通過製備型HPLC來純化粗產物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內17 B至37 B;RT1:6.75)來純化粗產物,得到呈灰白色固體的3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-胺(140 mg,60.84%)。LCMS: m/z (ESI), [M+H]+
= 364.1,1
H NMR (DMSO-d6
, 300 MHz) δ1.79 (2H, d), 2.02-2.18 (2H, m), 2.23 (3H, d), 2.42 (3H, s), 3.44 (2H, dd), 3.94 (2H, dd), 4.62 (1H, dd), 7.42 (1H, s), 7.48 (1H, d), 7.84 (1H, s), 8.69 (1H, s), 8.85 (1H, s), 9.04 (1H, s)實例 15
製備7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-7H-吡咯並[2,3-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例15)
步驟1. 6-氯-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在0℃下將1-(2,4-二氯嘧啶-5-基)乙酮(250.00 mg,1.309 mmol,1.00當量)和氧雜環己烷-4-基肼(182.45 mg,1.571 mmol,1.20當量)、DIPEA(338.32 mg,2.618 mmol,2.00當量)於THF(10.00 mL)中的溶液攪拌2h。用EtOAc(20 mL×3)萃取所得混合物。用鹽水(10 mL×3)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(PE/EtOAc 5:1)來純化殘餘物,得到呈白色固體的6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(230 mg,69.54%)。LCMS: m/z (ESI), [M+H]+
= 253.2。
步驟2. 3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例14)
在氮氣氛圍下在100℃下將7-甲基咪唑[1,2-a]吡啶-6-胺(139.78 mg,0.950 mmol,1.50當量)和6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(160 mg,0.633 mmol,1.00當量)、BrettPhos Pd G3
(57.40 mg,0.063 mmol,0.10當量)、Cs2
CO3
(412.59 mg,1.266 mmol,2.00當量)於二惡烷(5.00 mL)中的混合物攪拌3h。通過矽膠柱色譜純化殘餘物,用CH2
Cl2
/MeOH(10:1)洗脫,得到粗產物。通過製備型HPLC來純化粗產物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內17 B至37 B;RT1:6.75)來純化粗產物,得到呈灰白色固體的3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-胺(140 mg,60.84%)。LCMS: m/z (ESI), [M+H]+
= 364.1,1
H NMR (DMSO-d6
, 300 MHz) δ1.79 (2H, d), 2.02-2.18 (2H, m), 2.23 (3H, d), 2.42 (3H, s), 3.44 (2H, dd), 3.94 (2H, dd), 4.62 (1H, dd), 7.42 (1H, s), 7.48 (1H, d), 7.84 (1H, s), 8.69 (1H, s), 8.85 (1H, s), 9.04 (1H, s)實例 15
製備7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-7H-吡咯並[2,3-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例15) 步驟1. 2-氯-5-甲基-7-(四氫-2H-吡喃-4-基)-7H-吡咯並[2,3-d]嘧啶
在氮氣氛圍下在120℃下將2-氯-5-甲基-7H-吡咯並[2,3-d]嘧啶(400.00 mg,2.387 mmol,1.00當量)、4-溴惡烷(3.94 g,23.874 mmol,10.00當量)和K2
CO3
(824.62 mg,5.967 mmol,2.50當量)於DMSO(50.00 mL,12.922 mmol)中的混合物攪拌隔夜。用水(150 mL)稀釋所得混合物。用EtOAc(3×50 mL)萃取所得混合物。用鹽水(3×30 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內28 B至48 B;RT1:5.80)來純化粗產物(70 mg),得到呈黃色固體的2-氯-5-甲基-7-(氧雜環己烷-4-基)吡咯並[2,3-d]嘧啶(40 mg,6.66%)。
步驟2. 7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-7H-吡咯並[2,3-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例15)
在氮氣氛圍下在100℃下將2-氯-5-甲基-7-(氧雜環己烷-4-基)吡咯並[2,3-d]嘧啶(30.00 mg,0.119 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(21.19 mg,0.143 mmol,1.20當量)、Pd(AcO)2
(5.35 mg,0.024 mmol,0.20當量)、XantPhos(20.69 mg,0.036 mmol,0.30當量)以及Cs2
CO3
(97.08 mg,0.298 mmol,2.50當量)於二惡烷(1.50 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至46 B;RT1:6.37)來純化粗產物(40 mg),得到呈白色固體的5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-7-(氧雜環己烷-4-基)吡咯並[2,3-d]嘧啶-2-胺(6 mg,13.85%)。LCMS: m/z (ESI), [M+H]+
= 364.3。1
H NMR (300 MHz, MeOD-d4
) δ1.79-1.91 (2H, m), 1.95-2.14 (2H, m), 2.24 (3H, d), 2.43 (3H, d), 3.47 (2H, td), 3.93-4.05 (2H, m), 4.55-4.72 (1H, m), 7.18 (1H, d), 7.58-7.74 (1H, m), 8.37 (1H, s), 8.62 (2H, d), 9.32 (1H, s)實例 16
製備N-(7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例16)
步驟1. 2-氯-5-甲基-7-(四氫-2H-吡喃-4-基)-7H-吡咯並[2,3-d]嘧啶
在氮氣氛圍下在120℃下將2-氯-5-甲基-7H-吡咯並[2,3-d]嘧啶(400.00 mg,2.387 mmol,1.00當量)、4-溴惡烷(3.94 g,23.874 mmol,10.00當量)和K2
CO3
(824.62 mg,5.967 mmol,2.50當量)於DMSO(50.00 mL,12.922 mmol)中的混合物攪拌隔夜。用水(150 mL)稀釋所得混合物。用EtOAc(3×50 mL)萃取所得混合物。用鹽水(3×30 mL)洗滌合併的有機層,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內28 B至48 B;RT1:5.80)來純化粗產物(70 mg),得到呈黃色固體的2-氯-5-甲基-7-(氧雜環己烷-4-基)吡咯並[2,3-d]嘧啶(40 mg,6.66%)。
步驟2. 7-甲基-N-(5-甲基-7-(四氫-2H-吡喃-4-基)-7H-吡咯並[2,3-d]嘧啶-2-基)-[1,2,4]三唑並[1,5-a]吡啶-6-胺(實例15)
在氮氣氛圍下在100℃下將2-氯-5-甲基-7-(氧雜環己烷-4-基)吡咯並[2,3-d]嘧啶(30.00 mg,0.119 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(21.19 mg,0.143 mmol,1.20當量)、Pd(AcO)2
(5.35 mg,0.024 mmol,0.20當量)、XantPhos(20.69 mg,0.036 mmol,0.30當量)以及Cs2
CO3
(97.08 mg,0.298 mmol,2.50當量)於二惡烷(1.50 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至46 B;RT1:6.37)來純化粗產物(40 mg),得到呈白色固體的5-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-7-(氧雜環己烷-4-基)吡咯並[2,3-d]嘧啶-2-胺(6 mg,13.85%)。LCMS: m/z (ESI), [M+H]+
= 364.3。1
H NMR (300 MHz, MeOD-d4
) δ1.79-1.91 (2H, m), 1.95-2.14 (2H, m), 2.24 (3H, d), 2.43 (3H, d), 3.47 (2H, td), 3.93-4.05 (2H, m), 4.55-4.72 (1H, m), 7.18 (1H, d), 7.58-7.74 (1H, m), 8.37 (1H, s), 8.62 (2H, d), 9.32 (1H, s)實例 16
製備N-(7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例16) 步驟1. N-(7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例16)
在氮氣氛圍下在100℃下將7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-胺(50.00 mg,0.297 mmol,1.00當量)、6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(74.95 mg,0.297 mmol,1.00當量)、Pd(AcO)2
(13.32 mg,0.059 mmol,0.2當量)、XantPhos(51.48 mg,0.089 mmol,0.3當量)以及Cs2
CO3
(241.59 mg,0.741 mmol,2.5當量)於二惡烷(4.00 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:25 mL/min;梯度:7 min內30 B至40 B;RT1:6.62)來純化粗產物(90 mg),得到呈白色固體的N-[7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-胺(32.77mg,28.71%)。LCMS: m/z (ESI), [M+H]+
= 385.2。1
H NMR (300 MHz, DMSO-d6
) δ1.84 (2H, d), 2.13 (2H, dd), 2.46 (3H, s), 3.39-3.57 (2H, m), 3.97 (2H, dd), 4.54-4.77 (1H, m), 8.23 (1H, s), 8.57 (1H, s), 8.93 (1H, s), 9.36 (2H, d)。實例 17
製備N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例17)
步驟1. N-(7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-基)-3-甲基-1-(四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例16)
在氮氣氛圍下在100℃下將7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-胺(50.00 mg,0.297 mmol,1.00當量)、6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(74.95 mg,0.297 mmol,1.00當量)、Pd(AcO)2
(13.32 mg,0.059 mmol,0.2當量)、XantPhos(51.48 mg,0.089 mmol,0.3當量)以及Cs2
CO3
(241.59 mg,0.741 mmol,2.5當量)於二惡烷(4.00 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:25 mL/min;梯度:7 min內30 B至40 B;RT1:6.62)來純化粗產物(90 mg),得到呈白色固體的N-[7-氯-[1,2,4]三唑並[1,5-a]吡啶-6-基]-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-胺(32.77mg,28.71%)。LCMS: m/z (ESI), [M+H]+
= 385.2。1
H NMR (300 MHz, DMSO-d6
) δ1.84 (2H, d), 2.13 (2H, dd), 2.46 (3H, s), 3.39-3.57 (2H, m), 3.97 (2H, dd), 4.54-4.77 (1H, m), 8.23 (1H, s), 8.57 (1H, s), 8.93 (1H, s), 9.36 (2H, d)。實例 17
製備N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例17) 步驟1. 6-氯-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在0℃下向6-氯-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶(340.00 mg,1.528 mmol,1.00當量)、氧雜環己烷-4-醇(624.11 mg,6.111 mmol,4.00當量)以及PPh3
(1442.48 mg,5.500 mmol,3.60當量)於THF(2.50 mL)中的攪拌混合物中逐滴添加DIAD(1112.07 mg,5.500 mmol,3.60當量)。用EtOAc(3×20 mL)萃取所得混合物,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物,得到呈白色固體的6-氯-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶(370 mg,78.98%)。LCMS: m/z (ESI), [M+H]+
= 307.3。
步驟2. N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例17)
在氮氣氛圍下在70℃下將6-氯-1-(氧雜環己烷-4-基)-3-(三氟甲基)吡唑並[3,4-d]嘧啶(70.00 mg,0.228 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(33.82 mg,0.228 mmol,1.00當量)、Pd(AcO)2
(10.25 mg,0.046 mmol,0.20當量)、XantPhos(39.62 mg,0.068 mmol,0.30當量)以及Cs2
CO3
(185.93 mg,0.571 mmol,2.50當量)於二惡烷(3.00 mL)中的混合物攪拌3小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內30 B至50 B;RT1:6.67)來純化粗產物(90 mg),得到呈白色固體的N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-(氧雜環己烷-4-基)-3-(三氟甲基)吡唑並[3,4-d]嘧啶-6-胺(54.44 mg,53.86%)。LCMS: m/z (ESI), [M+H]+
= 419.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.77-2.01 (2H, m), 2.01-2.24 (2H, m), 2.39 (3H, d), 3.40-3.59 (2H, m), 3.98 (2H, d), 4.82 (1H, t), 7.78 (1H, d), 8.44 (1H, s), 9.13 (2H, d), 9.69 (1H, s)實例 18
製備1-(4-甲氧基苯甲基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例18)
步驟1. 6-氯-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在0℃下向6-氯-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶(340.00 mg,1.528 mmol,1.00當量)、氧雜環己烷-4-醇(624.11 mg,6.111 mmol,4.00當量)以及PPh3
(1442.48 mg,5.500 mmol,3.60當量)於THF(2.50 mL)中的攪拌混合物中逐滴添加DIAD(1112.07 mg,5.500 mmol,3.60當量)。用EtOAc(3×20 mL)萃取所得混合物,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物,得到呈白色固體的6-氯-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶(370 mg,78.98%)。LCMS: m/z (ESI), [M+H]+
= 307.3。
步驟2. N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(四氫-2H-吡喃-4-基)-3-(三氟甲基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例17)
在氮氣氛圍下在70℃下將6-氯-1-(氧雜環己烷-4-基)-3-(三氟甲基)吡唑並[3,4-d]嘧啶(70.00 mg,0.228 mmol,1.00當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(33.82 mg,0.228 mmol,1.00當量)、Pd(AcO)2
(10.25 mg,0.046 mmol,0.20當量)、XantPhos(39.62 mg,0.068 mmol,0.30當量)以及Cs2
CO3
(185.93 mg,0.571 mmol,2.50當量)於二惡烷(3.00 mL)中的混合物攪拌3小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內30 B至50 B;RT1:6.67)來純化粗產物(90 mg),得到呈白色固體的N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-(氧雜環己烷-4-基)-3-(三氟甲基)吡唑並[3,4-d]嘧啶-6-胺(54.44 mg,53.86%)。LCMS: m/z (ESI), [M+H]+
= 419.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.77-2.01 (2H, m), 2.01-2.24 (2H, m), 2.39 (3H, d), 3.40-3.59 (2H, m), 3.98 (2H, d), 4.82 (1H, t), 7.78 (1H, d), 8.44 (1H, s), 9.13 (2H, d), 9.69 (1H, s)實例 18
製備1-(4-甲氧基苯甲基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例18) 步驟1. 6-氯-1-(4-甲氧基苯甲基)-3-甲基-1H-吡唑並[3,4-d]嘧啶
在空氣氛圍下在0℃下向1-(2,4-二氯嘧啶-5-基)乙酮(640.00 mg,3.351 mmol,1.00當量)和DIPEA(433.04 mg,3.351 mmol,1.00當量)於THF中的攪拌混合物中逐滴添加[(4-甲氧基苯基)甲基]肼(764.93 mg,5.026 mmol,1.50當量)。在空氣氛圍下在室溫下攪拌所得混合物2h。減壓濃縮所得混合物。通過製備型TLC(PE:EA 1:1)來純化殘餘物,得到呈黃色固體的6-氯-1-[(4-甲氧基苯基)甲基]-3-甲基吡唑並[3,4-d]嘧啶(623 mg,64.40%)。LCMS: m/z (ESI), [M+H]+
= 289.2。1
H NMR (300 MHz, CDCl3
) δ2.58 (3H, s), 3.77 (3H, s), 5.47 (2H, s), 6.82-6.87 (2H, m), 7.30-7.35 (2H, m), 8.90 (1H, s)
步驟2. 1-(4-甲氧基苯甲基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例18)
在空氣氛圍下在室溫下向6-氯-1-[(4-甲氧基苯基)甲基]-3-甲基吡唑並[3,4-d]嘧啶(100.00 mg,0.346 mmol,1.00當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(76.97 mg,0.519 mmol,1.50當量)於二惡烷(5 mL)中的攪拌混合物中逐滴添加Cs2
CO3
(338.53 mg,1.039 mmol,3.00當量)和XantPhos(40.08 mg,0.069 mmol,0.20當量)、Pd(AcO)2
(15.55 mg,0.069 mmol,0.20當量)。在氮氣氛圍下在80℃下攪拌所得混合物2h。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物,得到黃色固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至46 B;RT1:7.07)來純化粗產物(70mg),得到呈白色固體的1-[(4-甲氧基苯基)甲基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(20 mg,28.57%)。LCMS: m/z (ESI), [M+H]+
= 401.2。1
H NMR (400 MHz, DMSO-d6
) δ 2.38 (3H, s), 2.44 (3H, s), 3.71 (3H, s), 5.24 (2H, s), 6.85-6.88 (2H, m), 7.18 (2H, t), 7.77 (1H, s), 8.43 (1H, s), 8.95 (1H, s), 9.22 (2H, d)實例 20
製備1-環己基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例20)
步驟1. 6-氯-1-(4-甲氧基苯甲基)-3-甲基-1H-吡唑並[3,4-d]嘧啶
在空氣氛圍下在0℃下向1-(2,4-二氯嘧啶-5-基)乙酮(640.00 mg,3.351 mmol,1.00當量)和DIPEA(433.04 mg,3.351 mmol,1.00當量)於THF中的攪拌混合物中逐滴添加[(4-甲氧基苯基)甲基]肼(764.93 mg,5.026 mmol,1.50當量)。在空氣氛圍下在室溫下攪拌所得混合物2h。減壓濃縮所得混合物。通過製備型TLC(PE:EA 1:1)來純化殘餘物,得到呈黃色固體的6-氯-1-[(4-甲氧基苯基)甲基]-3-甲基吡唑並[3,4-d]嘧啶(623 mg,64.40%)。LCMS: m/z (ESI), [M+H]+
= 289.2。1
H NMR (300 MHz, CDCl3
) δ2.58 (3H, s), 3.77 (3H, s), 5.47 (2H, s), 6.82-6.87 (2H, m), 7.30-7.35 (2H, m), 8.90 (1H, s)
步驟2. 1-(4-甲氧基苯甲基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例18)
在空氣氛圍下在室溫下向6-氯-1-[(4-甲氧基苯基)甲基]-3-甲基吡唑並[3,4-d]嘧啶(100.00 mg,0.346 mmol,1.00當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(76.97 mg,0.519 mmol,1.50當量)於二惡烷(5 mL)中的攪拌混合物中逐滴添加Cs2
CO3
(338.53 mg,1.039 mmol,3.00當量)和XantPhos(40.08 mg,0.069 mmol,0.20當量)、Pd(AcO)2
(15.55 mg,0.069 mmol,0.20當量)。在氮氣氛圍下在80℃下攪拌所得混合物2h。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物,得到黃色固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至46 B;RT1:7.07)來純化粗產物(70mg),得到呈白色固體的1-[(4-甲氧基苯基)甲基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(20 mg,28.57%)。LCMS: m/z (ESI), [M+H]+
= 401.2。1
H NMR (400 MHz, DMSO-d6
) δ 2.38 (3H, s), 2.44 (3H, s), 3.71 (3H, s), 5.24 (2H, s), 6.85-6.88 (2H, m), 7.18 (2H, t), 7.77 (1H, s), 8.43 (1H, s), 8.95 (1H, s), 9.22 (2H, d)實例 20
製備1-環己基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例20) 步驟1. 6-氯-1-環己基-3-甲基-1H-吡唑並[3,4-d]嘧啶
在空氣氛圍下在0℃下向1-(2,4-二氯嘧啶-5-基)乙酮(180.00 mg,0.942 mmol,1.00當量)和DIPEA(487.17 mg,3.769 mmol,4.00當量)於THF(10 mL)中的攪拌混合物中分批添加環己基鹽酸肼(184.56 mg,1.225 mmol,1.30當量)。在空氣氛圍下在25℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(PE/EA 1:2)來純化殘餘物,得到呈黃色固體的6-氯-1-環己基-3-甲基吡唑並[3,4-d]嘧啶(145 mg,61.37%)。LCMS: m/z (ESI), [M+H]+
= 251.2。1
H NMR (300 MHz, CDCl3
) δ 1.42-1.48 (2H, m), 1.51-1.55 (2H, m), 1.99-2.08 (6H, m), 2.60 (3H, s), 4.66-4.76 (1H, m), 8.90 (1H, s)
步驟2. 1-環己基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例20)
在空氣氛圍下在室溫下向6-氯-1-環己基-3-甲基吡唑並[3,4-d]嘧啶(120.00 mg,0.479 mmol,1.00當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(106.37 mg,0.718 mmol,1.5當量)於二惡烷(6 mL)中的攪拌混合物中分批添加Cs2
CO3
(467.81 mg,1.436 mmol,3當量)和XantPhos (55.39 mg,0.096 mmol,0.2當量)、Pd(AcO)2
(21.49 mg,0.096 mmol,0.2當量)。在氮氣氛圍下在80℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30*150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:% B;254;220 nm;RT1:6.50)來純化粗產物(80mg),得到呈白色固體的1-環己基-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(60 mg,34.59%)。LCMS: m/z (ESI), [M+H]+
= 363.2.1
H NMR (300 MHz, DMSO-d6
) δ 1.23-1.38 (3H, m), 1.67 (1H, d), 1.81-1.97 (6H, m), 2.41-2.44 (6H, m), 4.36-4.43 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.15 (1H, s), 9.22 (1H, s).實例 21
製備1-(4,4-二氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例21)
步驟1. 6-氯-1-環己基-3-甲基-1H-吡唑並[3,4-d]嘧啶
在空氣氛圍下在0℃下向1-(2,4-二氯嘧啶-5-基)乙酮(180.00 mg,0.942 mmol,1.00當量)和DIPEA(487.17 mg,3.769 mmol,4.00當量)於THF(10 mL)中的攪拌混合物中分批添加環己基鹽酸肼(184.56 mg,1.225 mmol,1.30當量)。在空氣氛圍下在25℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(PE/EA 1:2)來純化殘餘物,得到呈黃色固體的6-氯-1-環己基-3-甲基吡唑並[3,4-d]嘧啶(145 mg,61.37%)。LCMS: m/z (ESI), [M+H]+
= 251.2。1
H NMR (300 MHz, CDCl3
) δ 1.42-1.48 (2H, m), 1.51-1.55 (2H, m), 1.99-2.08 (6H, m), 2.60 (3H, s), 4.66-4.76 (1H, m), 8.90 (1H, s)
步驟2. 1-環己基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例20)
在空氣氛圍下在室溫下向6-氯-1-環己基-3-甲基吡唑並[3,4-d]嘧啶(120.00 mg,0.479 mmol,1.00當量)和7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(106.37 mg,0.718 mmol,1.5當量)於二惡烷(6 mL)中的攪拌混合物中分批添加Cs2
CO3
(467.81 mg,1.436 mmol,3當量)和XantPhos (55.39 mg,0.096 mmol,0.2當量)、Pd(AcO)2
(21.49 mg,0.096 mmol,0.2當量)。在氮氣氛圍下在80℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30*150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:% B;254;220 nm;RT1:6.50)來純化粗產物(80mg),得到呈白色固體的1-環己基-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(60 mg,34.59%)。LCMS: m/z (ESI), [M+H]+
= 363.2.1
H NMR (300 MHz, DMSO-d6
) δ 1.23-1.38 (3H, m), 1.67 (1H, d), 1.81-1.97 (6H, m), 2.41-2.44 (6H, m), 4.36-4.43 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.15 (1H, s), 9.22 (1H, s).實例 21
製備1-(4,4-二氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例21) 步驟1. 6-氯-1-(4,4-二氟環己基)-3-甲基-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在0℃下將DIPEA(338.32 mg,2.618 mmol,5當量)、(4,4-二氟環己基)肼(86.48 mg,0.576 mmol,1.10當量)以及1-(2,4-二氯嘧啶-5-基)乙酮(100.00 mg,0.524 mmol,1.00當量)於THF(5.00 mL)中的混合物攪拌3小時。減壓濃縮所得混合物。通過製備型TLC(己烷/EtOAc 1:1)來純化殘餘物,得到呈白色固體的6-氯-1-(4,4-二氟環己基)-3-甲基吡唑並[3,4-d]嘧啶(80 mg,53.30%)。LCMS: m/z (ESI), [M+H]+
= 287.3。1
H NMR (300 MHz, CDCl 3
) δ 2.03 (4H, d), 2.35 (4H, d), 2.60 (3H, s), 4.85 (1H, t), 8.92 (1H, s)。
步驟2. 1-(4,4-二氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例21)
在氮氣氛圍下在100℃下將Cs2
CO3
(227.28 mg,0.698 mmol,2.5當量)、XantPhos(32.29 mg,0.056 mmol,0.2當量)、Pd(AcO)2
(12.53 mg,0.056 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(45.48 mg,0.307 mmol,1.1當量)以及6-氯-1-(4,4-二氟環己基)-3-甲基吡唑並[3,4-d]嘧啶(80.00 mg,0.279 mmol,1.00當量)於二惡烷(3.00 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內31 B至51 B;RT1:6.30)來純化粗產物(100 mg),得到呈白色固體的1-(4,4-二氟環己基)-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(80 mg,71.96%)。LCMS: m/z (ESI), [M+H]+
= 399.3。1
H NMR (300 MHz, DMSO-d6
) δ 1.99 (3H, s), 2.15 (5H, s), 2.38 (3H, d), 2.45 (3H, s), 4.62 (1H, s), 7.75 (1H, d), 8.42 (1H, s), 8.93 (1H, s), 9.13 (1H, s), 9.22 (1H, s)。實例 22
製備1-(4,4-二氟環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例22)
步驟1. 6-氯-1-(4,4-二氟環己基)-3-甲基-1H-吡唑並[3,4-d]嘧啶
在氮氣氛圍下在0℃下將DIPEA(338.32 mg,2.618 mmol,5當量)、(4,4-二氟環己基)肼(86.48 mg,0.576 mmol,1.10當量)以及1-(2,4-二氯嘧啶-5-基)乙酮(100.00 mg,0.524 mmol,1.00當量)於THF(5.00 mL)中的混合物攪拌3小時。減壓濃縮所得混合物。通過製備型TLC(己烷/EtOAc 1:1)來純化殘餘物,得到呈白色固體的6-氯-1-(4,4-二氟環己基)-3-甲基吡唑並[3,4-d]嘧啶(80 mg,53.30%)。LCMS: m/z (ESI), [M+H]+
= 287.3。1
H NMR (300 MHz, CDCl 3
) δ 2.03 (4H, d), 2.35 (4H, d), 2.60 (3H, s), 4.85 (1H, t), 8.92 (1H, s)。
步驟2. 1-(4,4-二氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例21)
在氮氣氛圍下在100℃下將Cs2
CO3
(227.28 mg,0.698 mmol,2.5當量)、XantPhos(32.29 mg,0.056 mmol,0.2當量)、Pd(AcO)2
(12.53 mg,0.056 mmol,0.2當量)、7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-胺(45.48 mg,0.307 mmol,1.1當量)以及6-氯-1-(4,4-二氟環己基)-3-甲基吡唑並[3,4-d]嘧啶(80.00 mg,0.279 mmol,1.00當量)於二惡烷(3.00 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內31 B至51 B;RT1:6.30)來純化粗產物(100 mg),得到呈白色固體的1-(4,4-二氟環己基)-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(80 mg,71.96%)。LCMS: m/z (ESI), [M+H]+
= 399.3。1
H NMR (300 MHz, DMSO-d6
) δ 1.99 (3H, s), 2.15 (5H, s), 2.38 (3H, d), 2.45 (3H, s), 4.62 (1H, s), 7.75 (1H, d), 8.42 (1H, s), 8.93 (1H, s), 9.13 (1H, s), 9.22 (1H, s)。實例 22
製備1-(4,4-二氟環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例22) 步驟1. 1-(4,4-二氟環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例22)
在氮氣氛圍下在80℃下將6-氯-1-(4,4-二氟環己基)-3-甲基吡唑並[3,4-d]嘧啶(67.00 mg,0.234 mmol,1.00當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(68.79 mg,0.467 mmol,2當量)、Pd(AcO)2
(10.49 mg,0.047 mmol,0.20當量)、XantPhos(40.56 mg,0.070 mmol,0.30當量)以及Cs2
CO3
(190.35 mg,0.584 mmol,2.50當量)於二惡烷(5.00 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(10MMOL/L NH4HCO3+ 0.1%NH3
.H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內38 B至50 B;RT1:5.63)來純化粗產物(50 mg),得到呈白色固體的1-(4,4-二氟環己基)-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(20 mg)。LCMS: m/z (ESI), [M+H]+
= 398.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.95 (3H, d), 2.15 (5H, d), 2.26 (3H, d), 2.44 (3H, s), 4.62 (1H, s), 7.44 (1H, s), 7.50 (1H, d), 7.86 (1H, t), 8.72 (1H, s), 8.88 (1H, s), 9.02 (1H, s)實例 26
製備1-((1r,4r)-4-甲氧基環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例26)
步驟1. 1-(4,4-二氟環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例22)
在氮氣氛圍下在80℃下將6-氯-1-(4,4-二氟環己基)-3-甲基吡唑並[3,4-d]嘧啶(67.00 mg,0.234 mmol,1.00當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(68.79 mg,0.467 mmol,2當量)、Pd(AcO)2
(10.49 mg,0.047 mmol,0.20當量)、XantPhos(40.56 mg,0.070 mmol,0.30當量)以及Cs2
CO3
(190.35 mg,0.584 mmol,2.50當量)於二惡烷(5.00 mL)中的混合物攪拌2小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(10MMOL/L NH4HCO3+ 0.1%NH3
.H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內38 B至50 B;RT1:5.63)來純化粗產物(50 mg),得到呈白色固體的1-(4,4-二氟環己基)-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(20 mg)。LCMS: m/z (ESI), [M+H]+
= 398.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.95 (3H, d), 2.15 (5H, d), 2.26 (3H, d), 2.44 (3H, s), 4.62 (1H, s), 7.44 (1H, s), 7.50 (1H, d), 7.86 (1H, t), 8.72 (1H, s), 8.88 (1H, s), 9.02 (1H, s)實例 26
製備1-((1r,4r)-4-甲氧基環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例26) 步驟1. 1-(4-甲氧基苯甲基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在氮氣氛圍下在80℃下將6-氯-1-[(4-甲氧基苯基)甲基]-3-甲基吡唑並[3,4-d]嘧啶(200.00 mg,0.693 mmol,1.00當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(152.92 mg,1.039 mmol,1.50當量)、XantPhos(120.24 mg,0.208 mmol,0.30當量)、Pd(AcO)2
(31.10 mg,0.139 mmol,0.20當量)以及Cs2
CO3
(564.21 mg,1.732 mmol,2.50當量)於二惡烷(3 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到呈黃色固體的1-[(4-甲氧基苯基)甲基]-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(200mg,72.28%)。LCMS: m/z (ESI), [M+H]+
= 400.3。
步驟2. 3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在空氣氛圍下在80℃下將1-[(4-甲氧基苯基)甲基]-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(190.00 mg,0.476 mmol,1.00當量)和TFA(70.00 mL,613.910 mmol,1981.34當量)的溶液攪拌2天。真空濃縮所得混合物。用DCM(10 mL)稀釋所得混合物。通過過濾用飽和NaHCO3
(水溶液)將混合物調節到pH 8,並用DCM(2×3 mL)洗滌。收集沉澱的固體,得到3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(160 mg,80.00%)。LCMS: m/z (ESI), [M+H]+
= 280.2
步驟3. 1-((1r,4r)-4-甲氧基環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例26)
在氮氣氛圍下在0℃下將3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(150.00 mg,0.537 mmol,1.00當量)、4-甲氧基環己-1-醇(174.79 mg,1.343 mmol,2.50當量)以及PPh3
(422.58 mg,1.611 mmol,3.00當量)於THF(2.50 mL)中的混合物攪拌20 min,隨後添加DIAD(325.78 mg,1.611 mmol,3.00當量),且在70℃下攪拌混合物2小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/EtOAc =12:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(10MMOL/L NH4
HCO3
+ 0.1%NH3
.H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:9 min內29 B至39 B;RT1:6.22,7.43)來純化粗產物(60.00 mg),得到呈白色固體的3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1-[(1r,4r)-4-甲氧基環己基]吡唑並[3,4-d]嘧啶-6-胺(16 mg,26.67%)。LCMS: m/z (ESI), [M+H]+
= 392.2。1
H NMR (300 MHz, MeOD-d4
) δ 1.31-1.46 (2H, m), 2.00 (2H, s), 2.03-2.14 (2H, m), 2.21 (2H, d), 2.39 (3H, d), 2.50 (3H, s), 3.23 (1H, d), 3.38 (3H, s), 4.51 (1H, t), 7.46 (1H, s), 7.54 (1H, d), 7.80 (1H, s), 8.82 (2H, s)。實例 27
製備1-((1s,4s)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例27)及1-((1r,4r)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例28)
步驟1. 1-(4-甲氧基苯甲基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在氮氣氛圍下在80℃下將6-氯-1-[(4-甲氧基苯基)甲基]-3-甲基吡唑並[3,4-d]嘧啶(200.00 mg,0.693 mmol,1.00當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(152.92 mg,1.039 mmol,1.50當量)、XantPhos(120.24 mg,0.208 mmol,0.30當量)、Pd(AcO)2
(31.10 mg,0.139 mmol,0.20當量)以及Cs2
CO3
(564.21 mg,1.732 mmol,2.50當量)於二惡烷(3 mL)中的混合物攪拌2小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 12:1)來純化殘餘物,得到呈黃色固體的1-[(4-甲氧基苯基)甲基]-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(200mg,72.28%)。LCMS: m/z (ESI), [M+H]+
= 400.3。
步驟2. 3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在空氣氛圍下在80℃下將1-[(4-甲氧基苯基)甲基]-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(190.00 mg,0.476 mmol,1.00當量)和TFA(70.00 mL,613.910 mmol,1981.34當量)的溶液攪拌2天。真空濃縮所得混合物。用DCM(10 mL)稀釋所得混合物。通過過濾用飽和NaHCO3
(水溶液)將混合物調節到pH 8,並用DCM(2×3 mL)洗滌。收集沉澱的固體,得到3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(160 mg,80.00%)。LCMS: m/z (ESI), [M+H]+
= 280.2
步驟3. 1-((1r,4r)-4-甲氧基環己基)-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例26)
在氮氣氛圍下在0℃下將3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(150.00 mg,0.537 mmol,1.00當量)、4-甲氧基環己-1-醇(174.79 mg,1.343 mmol,2.50當量)以及PPh3
(422.58 mg,1.611 mmol,3.00當量)於THF(2.50 mL)中的混合物攪拌20 min,隨後添加DIAD(325.78 mg,1.611 mmol,3.00當量),且在70℃下攪拌混合物2小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/EtOAc =12:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(10MMOL/L NH4
HCO3
+ 0.1%NH3
.H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:9 min內29 B至39 B;RT1:6.22,7.43)來純化粗產物(60.00 mg),得到呈白色固體的3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]-1-[(1r,4r)-4-甲氧基環己基]吡唑並[3,4-d]嘧啶-6-胺(16 mg,26.67%)。LCMS: m/z (ESI), [M+H]+
= 392.2。1
H NMR (300 MHz, MeOD-d4
) δ 1.31-1.46 (2H, m), 2.00 (2H, s), 2.03-2.14 (2H, m), 2.21 (2H, d), 2.39 (3H, d), 2.50 (3H, s), 3.23 (1H, d), 3.38 (3H, s), 4.51 (1H, t), 7.46 (1H, s), 7.54 (1H, d), 7.80 (1H, s), 8.82 (2H, s)。實例 27
製備1-((1s,4s)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例27)及1-((1r,4r)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例28) 步驟1. 4-氟環己-1-醇
在0℃下向4-氟環己-1-酮(1.00 g, 8.611 mmol,1.00當量)和MeOH (100.00 mL)的攪拌混合物中分批添加NaBH4
(0.98 mg,0.026 mmol,3.0當量)。在室溫下攪拌所得混合物16 h。在0℃下用水淬滅反應物。減壓濃縮所得混合物。用水(50 mL)稀釋所得混合物。用CH2
Cl2
(3×50 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液,得到呈淡黃色固體的4-氟環己-1-醇(800 mg,78.63%)。1
HNMR (300MHz, DMSO-d6
) δ1.52 -1.56 (5H, m), 1.88 - 1.90 (3H, m), 3.54 - 3.57 (1H, m), 4.55 - 4.58(1H, m), 4.70 - 4.72(1H, m)
步驟2. 甲烷磺酸4-氟環己酯
在0℃下向4-氟環己-1-醇(400.00 mg,3.385 mmol,1.00當量)和TEA (1027.74 mg,10.156 mmol,3.00當量)於DCM(10.00 mL)中的攪拌混合物中逐滴添加甲烷磺醯氯(581.66 mg,5.078 mmol,1.5當量)。在室溫下攪拌所得混合物2小時。用水(50 mL)稀釋所得混合物。用CH2
Cl2
(3×50 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。減壓濃縮所得混合物,獲得呈淡黃色固體的甲烷磺酸4-氟環己酯(650 mg,97.84%)。粗產物不經進一步純化即直接用於下一步驟中。1
HNMR (300MHz, DMSO-d6
) δ 1.52 -1.90 (8H, m), 3.22 (3H, s), 4.60- 4.78(2H, m)
步驟3. 1-((1s,4s)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例27)及1-((1r,4r)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例28)
在氮氣氛圍下在110℃下將3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(150.00 mg,0.535 mmol,1.00當量)、甲烷磺酸4-氟環己酯(1050.18 mg,5.352 mmol,10.00當量)以及Cs2
CO3
(523.09 mg,1.605 mmol,3.00當量)於DMF(20.00 mL)中的混合物攪拌16 h。用水(20 mL)稀釋所得混合物。用EtOAc(3×20 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內39 B至59 B;RT1:6.4)來純化粗產物(100 mg),得到:
呈白色固體的1-((1s,4s)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例27,6.8 mg,3.34%)。LCMS: m/z (ESI), [M+H]+
= 381.2。1
HNMR (300MHz , DMSO-d6
) δ 1.66 - 1.68 (1H, m), 1.75 (3H, d), 2.04 - 2.06(2H, m), 2.18 - 2.19 (2H, m), 2.38 (3H, d), 2.45 (3H, s), 4.50 - 4.53 (1H, m), 4.81 - 4.93 (1H, s), 7.73 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.15 (2H, d);
呈白色固體的1-((1r,4r)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例28,18.8 mg,9.23%)。LCMS: m/z (ESI), [M+H]+
= 381.2。1
HNMR (400MHz , DMSO-d6
) δ 1.63 - 1.65(2H, m), 1.92 - 1.96 (4H, m), 2.00- 2.04 (2H, m), 2.14 (3H, s), 2.41 (3H, s), 4.47 - 4.49 (1H, m), 4.61 - 4.65 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.19 (2H, d)。實例 42
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(2-氧雜螺[3.3]庚烷-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例42)
步驟1. 4-氟環己-1-醇
在0℃下向4-氟環己-1-酮(1.00 g, 8.611 mmol,1.00當量)和MeOH (100.00 mL)的攪拌混合物中分批添加NaBH4
(0.98 mg,0.026 mmol,3.0當量)。在室溫下攪拌所得混合物16 h。在0℃下用水淬滅反應物。減壓濃縮所得混合物。用水(50 mL)稀釋所得混合物。用CH2
Cl2
(3×50 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液,得到呈淡黃色固體的4-氟環己-1-醇(800 mg,78.63%)。1
HNMR (300MHz, DMSO-d6
) δ1.52 -1.56 (5H, m), 1.88 - 1.90 (3H, m), 3.54 - 3.57 (1H, m), 4.55 - 4.58(1H, m), 4.70 - 4.72(1H, m)
步驟2. 甲烷磺酸4-氟環己酯
在0℃下向4-氟環己-1-醇(400.00 mg,3.385 mmol,1.00當量)和TEA (1027.74 mg,10.156 mmol,3.00當量)於DCM(10.00 mL)中的攪拌混合物中逐滴添加甲烷磺醯氯(581.66 mg,5.078 mmol,1.5當量)。在室溫下攪拌所得混合物2小時。用水(50 mL)稀釋所得混合物。用CH2
Cl2
(3×50 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。減壓濃縮所得混合物,獲得呈淡黃色固體的甲烷磺酸4-氟環己酯(650 mg,97.84%)。粗產物不經進一步純化即直接用於下一步驟中。1
HNMR (300MHz, DMSO-d6
) δ 1.52 -1.90 (8H, m), 3.22 (3H, s), 4.60- 4.78(2H, m)
步驟3. 1-((1s,4s)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例27)及1-((1r,4r)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例28)
在氮氣氛圍下在110℃下將3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(150.00 mg,0.535 mmol,1.00當量)、甲烷磺酸4-氟環己酯(1050.18 mg,5.352 mmol,10.00當量)以及Cs2
CO3
(523.09 mg,1.605 mmol,3.00當量)於DMF(20.00 mL)中的混合物攪拌16 h。用水(20 mL)稀釋所得混合物。用EtOAc(3×20 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內39 B至59 B;RT1:6.4)來純化粗產物(100 mg),得到:
呈白色固體的1-((1s,4s)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例27,6.8 mg,3.34%)。LCMS: m/z (ESI), [M+H]+
= 381.2。1
HNMR (300MHz , DMSO-d6
) δ 1.66 - 1.68 (1H, m), 1.75 (3H, d), 2.04 - 2.06(2H, m), 2.18 - 2.19 (2H, m), 2.38 (3H, d), 2.45 (3H, s), 4.50 - 4.53 (1H, m), 4.81 - 4.93 (1H, s), 7.73 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.15 (2H, d);
呈白色固體的1-((1r,4r)-4-氟環己基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例28,18.8 mg,9.23%)。LCMS: m/z (ESI), [M+H]+
= 381.2。1
HNMR (400MHz , DMSO-d6
) δ 1.63 - 1.65(2H, m), 1.92 - 1.96 (4H, m), 2.00- 2.04 (2H, m), 2.14 (3H, s), 2.41 (3H, s), 4.47 - 4.49 (1H, m), 4.61 - 4.65 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.19 (2H, d)。實例 42
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(2-氧雜螺[3.3]庚烷-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例42) 步驟1. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(2-氧雜螺[3.3]庚烷-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例42)
在N2
氛圍下在0℃下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(70.00 mg,0.250 mmol,1.00當量)和2-氧雜螺[3.3]庚烷-6-醇(57.01 mg,0.499 mmol,2.00當量)以及PPh3
(196.51 mg,0.749 mmol,3.00當量)於THF(10 mL)中的攪拌混合物中分批添加DIAD(151.50 mg,0.749 mmol,3.00當量)。在氮氣氛圍下在70℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(DCM/MeOH = 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(10MMOL/L NH4
HCO3
+0.1%NH3
.H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至36 B;RT1:5.88)來純化粗產物(80 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[2-氧雜螺[3.3]庚烷-6-基]吡唑並[3,4-d]嘧啶-6-胺(40 mg,50.00%)。LCMS: m/z (ESI), [M+H]+
= 337.3。1
H NMR (300 MHz, DMSO-d6
) δ 2.37 (3H, s), 2.44 (3H, s), 2.65-2.80 (4H, m), 4.45 (2H, s), 4.64 (2H, s), 4.84-4.89 (1H, m), 7.76 (1H, s), 8.42 (1H, s), 8.92 (1H, s), 9.13 (1H, s), 9.18 (1H, s)。
下表2中的以下化合物是通過實例42中所描述的類似方法合成。
步驟1. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(2-氧雜螺[3.3]庚烷-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例42)
在N2
氛圍下在0℃下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(70.00 mg,0.250 mmol,1.00當量)和2-氧雜螺[3.3]庚烷-6-醇(57.01 mg,0.499 mmol,2.00當量)以及PPh3
(196.51 mg,0.749 mmol,3.00當量)於THF(10 mL)中的攪拌混合物中分批添加DIAD(151.50 mg,0.749 mmol,3.00當量)。在氮氣氛圍下在70℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(DCM/MeOH = 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(10MMOL/L NH4
HCO3
+0.1%NH3
.H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內26 B至36 B;RT1:5.88)來純化粗產物(80 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[2-氧雜螺[3.3]庚烷-6-基]吡唑並[3,4-d]嘧啶-6-胺(40 mg,50.00%)。LCMS: m/z (ESI), [M+H]+
= 337.3。1
H NMR (300 MHz, DMSO-d6
) δ 2.37 (3H, s), 2.44 (3H, s), 2.65-2.80 (4H, m), 4.45 (2H, s), 4.64 (2H, s), 4.84-4.89 (1H, m), 7.76 (1H, s), 8.42 (1H, s), 8.92 (1H, s), 9.13 (1H, s), 9.18 (1H, s)。
下表2中的以下化合物是通過實例42中所描述的類似方法合成。


 實例 37
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[2.5]辛-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例37)
實例 37
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[2.5]辛-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例37) 步驟1. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[2.5]辛-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例37)
在氮氣氛圍下在70℃下將TMAD(276.43 mg,1.605 mmol,3.00當量)、n-Bu3
P(324.81 mg,1.605 mmol,3.00當量)、螺[2.5]辛-6-醇(202.61 mg,1.605 mmol,3.00當量)以及3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(150.00 mg,0.535 mmol,1.00當量)於THF(25.00 mL)中的混合物攪拌2h。用水(100 mL)稀釋所得混合物。用CH2
Cl2
(3×50 mL)萃取所得混合物,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。使粗產物從MeOH(20 mL)再結晶,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:MeOH;流動速率:25 mL/min;梯度:7 min內58 B至70 B)來純化粗產物(60 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[螺[2.5]辛-6-基]吡唑並[3,4-d]嘧啶-6-胺(30 mg,14.29%)。LCMS: m/z (ESI), [M+H]+
= 389.3。1
H NMR (400 MHz, DMSO-d6
) δ 0.20 (2H, d), 0.33 (2H, d), 0.98 (2H, d), 1.84 (4H, m), 2.08 (2H, m), 2.39 (3H, d), 2.45 (3H, s), 4.45 (1H, d), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.16 (2H, d)
下表3中的以下化合物是通過實例37中所描述的類似方法合成。
步驟1. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[2.5]辛-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例37)
在氮氣氛圍下在70℃下將TMAD(276.43 mg,1.605 mmol,3.00當量)、n-Bu3
P(324.81 mg,1.605 mmol,3.00當量)、螺[2.5]辛-6-醇(202.61 mg,1.605 mmol,3.00當量)以及3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(150.00 mg,0.535 mmol,1.00當量)於THF(25.00 mL)中的混合物攪拌2h。用水(100 mL)稀釋所得混合物。用CH2
Cl2
(3×50 mL)萃取所得混合物,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。使粗產物從MeOH(20 mL)再結晶,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,19*250mm,5um;移動相A:水(0.05%NH3
•H2
O),移動相B:MeOH;流動速率:25 mL/min;梯度:7 min內58 B至70 B)來純化粗產物(60 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[螺[2.5]辛-6-基]吡唑並[3,4-d]嘧啶-6-胺(30 mg,14.29%)。LCMS: m/z (ESI), [M+H]+
= 389.3。1
H NMR (400 MHz, DMSO-d6
) δ 0.20 (2H, d), 0.33 (2H, d), 0.98 (2H, d), 1.84 (4H, m), 2.08 (2H, m), 2.39 (3H, d), 2.45 (3H, s), 4.45 (1H, d), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.16 (2H, d)
下表3中的以下化合物是通過實例37中所描述的類似方法合成。 實例 43
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[3.3] 庚烷-2-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例43)
實例 43
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[3.3] 庚烷-2-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例43) 步驟1. 甲烷磺酸螺[3.3]庚烷-2-基酯
在氮氣氛圍下在0℃下向螺[3.3]庚烷-2-醇(300.00 mg,2.674 mmol,1.00當量)和TEA(811.89 mg,8.023 mmol,3.00當量)於DCM(50.00 mL)中的攪拌混合物中逐滴添加甲烷磺醯氯(459.50 mg,4.012 mmol,1.50當量)。用水(20 mL)稀釋所得混合物。用CH2
Cl2
(3×20 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。這產生呈淡黃色油狀物的甲烷磺酸螺[3.3]庚烷-2-基酯(500 mg,98.26%)。
步驟2. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[3.3]庚烷-2-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例43)
在氮氣氛圍下在100℃下將3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(100.00 mg,0.357 mmol,1.00當量)、甲烷磺酸螺[3.3]庚烷-2-基酯(678.78 mg,3.568 mmol,10.00當量)以及Cs2
CO3
(348.73 mg,1.070 mmol,3.00當量)於DMF(20.00 mL)中的混合物攪拌16 h。用水(40 mL)稀釋所得混合物。用EtOAc(3×100 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05% NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內39 B至59 B;RT1:6.4)來純化粗產物,得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[螺[3.3]庚烷-2-基]吡唑並[3,4-d]嘧啶-6-胺(31.8mg,23.80%)。LCMS: m/z (ESI), [M+H]+
= 375.3。1
HNMR (400MHz , DMSO-d6
) δ 1.81- 1.95 (2H, m), 1.89 - 1.91 (2H, m), 2.09 - 2.11 (2H, m), 2.42 (8H, d), 2.59 -2.63 (2H, m), 4.89 - 4.93(1H, m), 7.76 (1H, s), 8.42 (1H, s), 8.92 (1H, s), 9.17 (2H, d)實例 44
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(氧雜環丁烷-3-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例44)
步驟1. 甲烷磺酸螺[3.3]庚烷-2-基酯
在氮氣氛圍下在0℃下向螺[3.3]庚烷-2-醇(300.00 mg,2.674 mmol,1.00當量)和TEA(811.89 mg,8.023 mmol,3.00當量)於DCM(50.00 mL)中的攪拌混合物中逐滴添加甲烷磺醯氯(459.50 mg,4.012 mmol,1.50當量)。用水(20 mL)稀釋所得混合物。用CH2
Cl2
(3×20 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。這產生呈淡黃色油狀物的甲烷磺酸螺[3.3]庚烷-2-基酯(500 mg,98.26%)。
步驟2. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(螺[3.3]庚烷-2-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例43)
在氮氣氛圍下在100℃下將3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(100.00 mg,0.357 mmol,1.00當量)、甲烷磺酸螺[3.3]庚烷-2-基酯(678.78 mg,3.568 mmol,10.00當量)以及Cs2
CO3
(348.73 mg,1.070 mmol,3.00當量)於DMF(20.00 mL)中的混合物攪拌16 h。用水(40 mL)稀釋所得混合物。用EtOAc(3×100 mL)萃取所得混合物。將合併的有機層用鹽水洗滌,經無水Na2
SO4
乾燥。在過濾後,減壓濃縮濾液。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05% NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內39 B至59 B;RT1:6.4)來純化粗產物,得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[螺[3.3]庚烷-2-基]吡唑並[3,4-d]嘧啶-6-胺(31.8mg,23.80%)。LCMS: m/z (ESI), [M+H]+
= 375.3。1
HNMR (400MHz , DMSO-d6
) δ 1.81- 1.95 (2H, m), 1.89 - 1.91 (2H, m), 2.09 - 2.11 (2H, m), 2.42 (8H, d), 2.59 -2.63 (2H, m), 4.89 - 4.93(1H, m), 7.76 (1H, s), 8.42 (1H, s), 8.92 (1H, s), 9.17 (2H, d)實例 44
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(氧雜環丁烷-3-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例44) 步驟1. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(氧雜環丁烷-3-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例44)
在空氣氛圍下在室溫下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(80.00 mg,0.285 mmol,1.00當量)和3-碘氧雜環丁烷(78.76 mg,0.428 mmol,1.50當量)於DMF(10 mL)中的攪拌混合物中添加K2
CO3
(118.34 mg,0.856 mmol,3.00當量)。在空氣氛圍下在80℃下攪拌所得混合物2h。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
: MEOH 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150 mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內12 B至32 B;RT1:6.62)來純化粗產物(100 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-(氧雜環丁烷-3-基)吡唑並[3,4-d]嘧啶-6-胺(實例44,40 mg,41.67%)。LCMS: m/z (ESI), [M+H]+
= 337.2。1
H NMR (300 MHz, DMSO-d6
) δ 2.38 (3H, s), 2.49 (3H, s), 4.89-5.02 (4H, m), 5.72 (1H, t), 7.75 (1H, s), 8.42 (1H, s), 8.96 (1H, s), 9.15 (1H, s), 9.27 (1H, s)
下表4中的以下化合物是通過實例44中所描述的類似方法合成。
步驟1. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(氧雜環丁烷-3-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例44)
在空氣氛圍下在室溫下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(80.00 mg,0.285 mmol,1.00當量)和3-碘氧雜環丁烷(78.76 mg,0.428 mmol,1.50當量)於DMF(10 mL)中的攪拌混合物中添加K2
CO3
(118.34 mg,0.856 mmol,3.00當量)。在空氣氛圍下在80℃下攪拌所得混合物2h。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
: MEOH 12:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150 mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內12 B至32 B;RT1:6.62)來純化粗產物(100 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-(氧雜環丁烷-3-基)吡唑並[3,4-d]嘧啶-6-胺(實例44,40 mg,41.67%)。LCMS: m/z (ESI), [M+H]+
= 337.2。1
H NMR (300 MHz, DMSO-d6
) δ 2.38 (3H, s), 2.49 (3H, s), 4.89-5.02 (4H, m), 5.72 (1H, t), 7.75 (1H, s), 8.42 (1H, s), 8.96 (1H, s), 9.15 (1H, s), 9.27 (1H, s)
下表4中的以下化合物是通過實例44中所描述的類似方法合成。

 實例 59
製備1-異丙基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例59)
實例 59
製備1-異丙基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例59) 步驟1. 6-氯-1-異丙基-3-甲基-1H-吡唑並[3,4-d]嘧啶
在空氣氛圍下在0℃下向1-(2,4-二氯嘧啶-5-基)乙酮(150.00 mg,0.785 mmol,1.00當量)和DIPEA(54.13 mg,0.419 mmol,4.00當量)於THF中的攪拌混合物中逐滴添加異丙基肼(15.26 mg,0.136 mmol,1.30當量)。在空氣氛圍下在室溫下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(PE:EA 1:1)來純化殘餘物,得到呈黃色固體的6-氯-1-異丙基-3-甲基吡唑並[3,4-d]嘧啶(120 mg,72.53%)。LCMS: m/z (ESI), [M+H]+
= 211.2。1
H NMR (300 MHz, CDCl3
) δ1.54 (6H, d), 2.61 (3H, s), 5.12 (1H, dd), 8.90 (1H, s)
步驟2. 1-異丙基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例59)
在空氣氛圍下在室溫下向6-氯-1-異丙基-3-甲基吡唑並[3,4-d]嘧啶(120.00 mg,0.570 mmol,1.00當量)和7-甲基咪唑[1,2-a]吡啶-6-胺(108.99 mg,0.740 mmol,1.30當量)於二惡烷(10 mL)中的攪拌混合物中添加XantPhos (65.92 mg,0.114 mmol,0.20當量)、Pd(AcO)2
(25.58 mg,0.114 mmol,0.20當量)以及Cs2
CO3(
556.77 mg,1.709 mmol,3.00當量)。在氮氣氛圍下在60℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/ MeOH = 10:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;RT1:6.67)來純化粗產物(100 mg),得到呈白色固體的1-異丙基-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(62 mg,33.76%)。LCMS: m/z (ESI), [M+H]+
= 323.4。1
H NMR (300 MHz, DMSO-d6
) δ 1.41 (6H, d), 2.40 (3H, d), 2.45 (3H, s), 4.73-4.82 (1H, m), 7.74 (1H, s), 8.41(1H, s), 8.92 (1H, s), 9.16 (1H, s), 9.19 (1H, s)實例 60
製備1-異丙基-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例60)
步驟1. 6-氯-1-異丙基-3-甲基-1H-吡唑並[3,4-d]嘧啶
在空氣氛圍下在0℃下向1-(2,4-二氯嘧啶-5-基)乙酮(150.00 mg,0.785 mmol,1.00當量)和DIPEA(54.13 mg,0.419 mmol,4.00當量)於THF中的攪拌混合物中逐滴添加異丙基肼(15.26 mg,0.136 mmol,1.30當量)。在空氣氛圍下在室溫下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(PE:EA 1:1)來純化殘餘物,得到呈黃色固體的6-氯-1-異丙基-3-甲基吡唑並[3,4-d]嘧啶(120 mg,72.53%)。LCMS: m/z (ESI), [M+H]+
= 211.2。1
H NMR (300 MHz, CDCl3
) δ1.54 (6H, d), 2.61 (3H, s), 5.12 (1H, dd), 8.90 (1H, s)
步驟2. 1-異丙基-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例59)
在空氣氛圍下在室溫下向6-氯-1-異丙基-3-甲基吡唑並[3,4-d]嘧啶(120.00 mg,0.570 mmol,1.00當量)和7-甲基咪唑[1,2-a]吡啶-6-胺(108.99 mg,0.740 mmol,1.30當量)於二惡烷(10 mL)中的攪拌混合物中添加XantPhos (65.92 mg,0.114 mmol,0.20當量)、Pd(AcO)2
(25.58 mg,0.114 mmol,0.20當量)以及Cs2
CO3(
556.77 mg,1.709 mmol,3.00當量)。在氮氣氛圍下在60℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/ MeOH = 10:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;RT1:6.67)來純化粗產物(100 mg),得到呈白色固體的1-異丙基-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(62 mg,33.76%)。LCMS: m/z (ESI), [M+H]+
= 323.4。1
H NMR (300 MHz, DMSO-d6
) δ 1.41 (6H, d), 2.40 (3H, d), 2.45 (3H, s), 4.73-4.82 (1H, m), 7.74 (1H, s), 8.41(1H, s), 8.92 (1H, s), 9.16 (1H, s), 9.19 (1H, s)實例 60
製備1-異丙基-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例60) 步驟1. 1-異丙基-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例60)
在氮氣氛圍下在100℃下將6-氯-1-異丙基-3-甲基吡唑並[3,4-d]嘧啶(80.00 mg,0.380 mmol,1.00當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(67.07 mg,0.456 mmol,1.20當量)、Pd(AcO)2
(17.05 mg,0.076 mmol,0.20當量)、XantPhos(65.92 mg,0.114 mmol,0.30當量)以及Cs2
CO3
(309.32 mg,0.949 mmol,2.50當量)於二惡烷(10.00 mL)中的混合物攪拌3小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內21 B至41 B;RT1:7.02)來純化粗產物(100 mg),得到呈白色固體的1-異丙基-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(55.77 mg,45.70%)。LCMS: m/z (ESI), [M+H]+ = 322.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.38 (6H, d), 2.25 (3H, d), 2.44 (3H, s), 4.67-4.89 (1H, m), 7.47 (2H, dd), 7.87 (1H, t), 8.67 (1H, s), 8.86 (1H, s), 9.03 (1H, s)實例 72
製備3-(3-甲基-6-((7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)丙腈(實例72)
步驟1. 1-異丙基-3-甲基-N-(7-甲基咪唑[1,2-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例60)
在氮氣氛圍下在100℃下將6-氯-1-異丙基-3-甲基吡唑並[3,4-d]嘧啶(80.00 mg,0.380 mmol,1.00當量)、7-甲基咪唑[1,2-a]吡啶-6-胺(67.07 mg,0.456 mmol,1.20當量)、Pd(AcO)2
(17.05 mg,0.076 mmol,0.20當量)、XantPhos(65.92 mg,0.114 mmol,0.30當量)以及Cs2
CO3
(309.32 mg,0.949 mmol,2.50當量)於二惡烷(10.00 mL)中的混合物攪拌3小時。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內21 B至41 B;RT1:7.02)來純化粗產物(100 mg),得到呈白色固體的1-異丙基-3-甲基-N-[7-甲基咪唑[1,2-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(55.77 mg,45.70%)。LCMS: m/z (ESI), [M+H]+ = 322.2。1
H NMR (300 MHz, DMSO-d6
) δ 1.38 (6H, d), 2.25 (3H, d), 2.44 (3H, s), 4.67-4.89 (1H, m), 7.47 (2H, dd), 7.87 (1H, t), 8.67 (1H, s), 8.86 (1H, s), 9.03 (1H, s)實例 72
製備3-(3-甲基-6-((7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)丙腈(實例72) 步驟1. 3-(3-甲基-6-((7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)丙腈(實例72)
向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(70.00 mg,0.250 mmol,1.00當量)和3-溴-丙腈(66.92 mg,0.499 mmol,2.00當量)於DMF(10 mL)中的攪拌混合物中添加K2
CO3
(103.55 mg,0.749 mmol,3.00當量)。在空氣氛圍下在80℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(DCM: MeOH = 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內8 B至28 B;RT1:7.70)來純化粗產物,得到呈白色固體的3-[3-甲基-6-([7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]氨基)吡唑並[3,4-d]嘧啶-1-基]丙腈(40 mg,48.05%)。LCMS: m/z (ESI), [M+H]+
= 334.3。1
H NMR (300 MHz, DMSO-d6
) δ2.39 (3H, s), 2.47(3H, s), 3.08(2H, t), 4.36(2H, t), 7.75(1H, s), 8.41(1H, s), 8.97(1H, s), 9.17 (1H, s), 9.29 (1H, s)。
下表5中的以下化合物是通過實例72中所描述的類似方法合成。
步驟1. 3-(3-甲基-6-((7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)丙腈(實例72)
向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(70.00 mg,0.250 mmol,1.00當量)和3-溴-丙腈(66.92 mg,0.499 mmol,2.00當量)於DMF(10 mL)中的攪拌混合物中添加K2
CO3
(103.55 mg,0.749 mmol,3.00當量)。在空氣氛圍下在80℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(DCM: MeOH = 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內8 B至28 B;RT1:7.70)來純化粗產物,得到呈白色固體的3-[3-甲基-6-([7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]氨基)吡唑並[3,4-d]嘧啶-1-基]丙腈(40 mg,48.05%)。LCMS: m/z (ESI), [M+H]+
= 334.3。1
H NMR (300 MHz, DMSO-d6
) δ2.39 (3H, s), 2.47(3H, s), 3.08(2H, t), 4.36(2H, t), 7.75(1H, s), 8.41(1H, s), 8.97(1H, s), 9.17 (1H, s), 9.29 (1H, s)。
下表5中的以下化合物是通過實例72中所描述的類似方法合成。 實例 73
製備4-(3-甲基-6-(7-甲基咪唑[1,2-a]吡啶-6-基氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)苯甲腈(實例73)
實例 73
製備4-(3-甲基-6-(7-甲基咪唑[1,2-a]吡啶-6-基氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)苯甲腈(實例73) 步驟1. 4-(3-甲基-6-(7-甲基咪唑[1,2-a]吡啶-6-基氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)苯甲腈(實例73)
在100℃下將7-甲基-N-[3-甲基-1H-吡唑並[3,4-d]嘧啶-6-基]-[1,2,4]三唑並[1,5-a]吡啶-6-胺(150.00 mg,0.535 mmol,1.00當量)、4-氟-苯甲腈(97.57 mg,0.806 mmol,1.50當量)、苯甲腈(97.22 mg,0.803 mmol,1.50當量)以及K2
CO3
(221.88 mg,1.605 mmol,3.0當量)於DMF(50.00 mL)中的混合物攪拌16 h。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物,得到呈淡黃色固體的4-[3-甲基-6-([7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]氨基)吡唑並[3,4-d]嘧啶-1-基]苯甲腈(40 mg,粗產物)。通過製備型HPLC利用以下條件(柱:XBridge Shield RP18 OBD柱,19*250mm,10um;移動相A:水(10MMOL/L NH4
HCO3
+0.1% NH3
•H2
O),移動相B:ACN;流動速率:20 mL/min;梯度:7 min內36 B至46 B;RT1:5.73)來純化粗產物(40 mg),得到呈白色固體的4-[3-甲基-6-([7-甲基咪唑[1,2-a]吡啶-6-基]氨基)吡唑並[3,4-d]嘧啶-1-基]苯甲腈(8.8mg,4.31%)。LCMS: m/z (ESI), [M+H]+
= 381.3。1
HNMR (300 MHz, CDCl3
) δ 2.45 (3H, d), 2.62 (3H, s), 7.08 (1H, s), 7.54 (2H, s), 7.65 (1H, s), 7.75 (2H, d), 8.42 - 8.44 (2H, m), 8.85 (1H, s), 9.06 (1H, d)。實例 89
製備6-甲氧基-4-甲基-N-[3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-基]吡啶-3-胺
步驟1. 4-(3-甲基-6-(7-甲基咪唑[1,2-a]吡啶-6-基氨基)-1H-吡唑並[3,4-d]嘧啶-1-基)苯甲腈(實例73)
在100℃下將7-甲基-N-[3-甲基-1H-吡唑並[3,4-d]嘧啶-6-基]-[1,2,4]三唑並[1,5-a]吡啶-6-胺(150.00 mg,0.535 mmol,1.00當量)、4-氟-苯甲腈(97.57 mg,0.806 mmol,1.50當量)、苯甲腈(97.22 mg,0.803 mmol,1.50當量)以及K2
CO3
(221.88 mg,1.605 mmol,3.0當量)於DMF(50.00 mL)中的混合物攪拌16 h。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH = 10:1)來純化殘餘物,得到呈淡黃色固體的4-[3-甲基-6-([7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]氨基)吡唑並[3,4-d]嘧啶-1-基]苯甲腈(40 mg,粗產物)。通過製備型HPLC利用以下條件(柱:XBridge Shield RP18 OBD柱,19*250mm,10um;移動相A:水(10MMOL/L NH4
HCO3
+0.1% NH3
•H2
O),移動相B:ACN;流動速率:20 mL/min;梯度:7 min內36 B至46 B;RT1:5.73)來純化粗產物(40 mg),得到呈白色固體的4-[3-甲基-6-([7-甲基咪唑[1,2-a]吡啶-6-基]氨基)吡唑並[3,4-d]嘧啶-1-基]苯甲腈(8.8mg,4.31%)。LCMS: m/z (ESI), [M+H]+
= 381.3。1
HNMR (300 MHz, CDCl3
) δ 2.45 (3H, d), 2.62 (3H, s), 7.08 (1H, s), 7.54 (2H, s), 7.65 (1H, s), 7.75 (2H, d), 8.42 - 8.44 (2H, m), 8.85 (1H, s), 9.06 (1H, d)。實例 89
製備6-甲氧基-4-甲基-N-[3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-基]吡啶-3-胺 步驟1. 6-甲氧基-4-甲基-N-[3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-基]吡啶-3-胺(實例89)
在氮氣氛圍下在80℃下將6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(80.00 mg,0.317 mmol,1.00當量)、6-甲氧基-4-甲基吡啶-3-胺(52.49 mg,0.380 mmol,1.20當量)、Pd(AcO)2
(14.22 mg,0.063 mmol,0.20當量)、XantPhos(54.95 mg,0.095 mmol,0.30當量)以及Cs2
CO3
(257.87 mg,0.791 mmol,2.50當量)於二惡烷(2.50 mL)中的混合物攪拌2 h。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/ MeOH 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Shield RP18 OBD柱,30*150mm,5um;移動相A:水(0.05% NH3
H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內25 B至55 B;254;220 nm; RT1:5.20)來純化粗產物(100 mg),得到呈白色固體的6-甲氧基-4-甲基-N-[3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-基]吡啶-3-胺(70 mg,62.39%)。LCMS: m/z (ESI), [M+H]+
= 355.2。1
H-NMR (300 MHz, DMSO-d6
) δ 1.79 (2H, d), 1.99 - 2.20 (5H, m), 2.42 (3H, s), 3.43 - 3.56 (2H, m), 3.83 (3H, s), 3.96 (2H, dd), 4.58 (1H, t), 6.74 (1H, s), 8.11 (1H, s), 8.82 (1H, s), 8.98 (1H, s)。實例 106
製備1-((1R,3r,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
步驟1. 6-甲氧基-4-甲基-N-[3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-基]吡啶-3-胺(實例89)
在氮氣氛圍下在80℃下將6-氯-3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶(80.00 mg,0.317 mmol,1.00當量)、6-甲氧基-4-甲基吡啶-3-胺(52.49 mg,0.380 mmol,1.20當量)、Pd(AcO)2
(14.22 mg,0.063 mmol,0.20當量)、XantPhos(54.95 mg,0.095 mmol,0.30當量)以及Cs2
CO3
(257.87 mg,0.791 mmol,2.50當量)於二惡烷(2.50 mL)中的混合物攪拌2 h。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/ MeOH 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Shield RP18 OBD柱,30*150mm,5um;移動相A:水(0.05% NH3
H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內25 B至55 B;254;220 nm; RT1:5.20)來純化粗產物(100 mg),得到呈白色固體的6-甲氧基-4-甲基-N-[3-甲基-1-(氧雜環己烷-4-基)吡唑並[3,4-d]嘧啶-6-基]吡啶-3-胺(70 mg,62.39%)。LCMS: m/z (ESI), [M+H]+
= 355.2。1
H-NMR (300 MHz, DMSO-d6
) δ 1.79 (2H, d), 1.99 - 2.20 (5H, m), 2.42 (3H, s), 3.43 - 3.56 (2H, m), 3.83 (3H, s), 3.96 (2H, dd), 4.58 (1H, t), 6.74 (1H, s), 8.11 (1H, s), 8.82 (1H, s), 8.98 (1H, s)。實例 106
製備1-((1R,3r,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺 步驟1. 1-((1R,3r,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在氮氣氛圍下在0℃下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(100.00 mg,0.357 mmol,1.00當量)、(1R,3S,5S)-8-氧雜雙環[3.2.1]辛-3-醇(137.18 mg,1.070 mmol,3.00當量)以及PPh3
(280.72 mg,1.070 mmol,3當量)於THF(10.00 mL)中的攪拌混合物中逐滴添加DIAD(216.42 mg,1.070 mmol,3當量)。在氮氣氛圍下在70℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH 12:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內37 B至57 B)來純化粗產物,得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[(1R,3R,5S)-8-氧雜雙環[3.2.1]辛-3-基]吡唑並[3,4-d]嘧啶-6-胺(實例106,50 mg,50.00%)。LCMS: m/z (ESI), [M+H]+
= 391.3。1
H NMR (300 MHz, DMSO-d6
) δ 1.74(4H, s), 2.22-2.45(10H, m), 4.35(2H, s), 4.63(1H, s), 7.75(1H, s), 8.41(1H, s), 8.93(1H, s), 9.17 (1H, s), 9.21(1H, s)。實例 107
製備1-((1R,3s,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
步驟1. 1-((1R,3r,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在氮氣氛圍下在0℃下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(100.00 mg,0.357 mmol,1.00當量)、(1R,3S,5S)-8-氧雜雙環[3.2.1]辛-3-醇(137.18 mg,1.070 mmol,3.00當量)以及PPh3
(280.72 mg,1.070 mmol,3當量)於THF(10.00 mL)中的攪拌混合物中逐滴添加DIAD(216.42 mg,1.070 mmol,3當量)。在氮氣氛圍下在70℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH 12:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內37 B至57 B)來純化粗產物,得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[(1R,3R,5S)-8-氧雜雙環[3.2.1]辛-3-基]吡唑並[3,4-d]嘧啶-6-胺(實例106,50 mg,50.00%)。LCMS: m/z (ESI), [M+H]+
= 391.3。1
H NMR (300 MHz, DMSO-d6
) δ 1.74(4H, s), 2.22-2.45(10H, m), 4.35(2H, s), 4.63(1H, s), 7.75(1H, s), 8.41(1H, s), 8.93(1H, s), 9.17 (1H, s), 9.21(1H, s)。實例 107
製備1-((1R,3s,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺 步驟1. 1-((1R,3s,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在氮氣氛圍下在0℃下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(80.00 mg,0.285 mmol,1.00當量)、(1R,3R,5S)-8-氧雜雙環[3.2.1]辛-3-醇(109.75 mg,0.856 mmol,3.00當量)以及PPh3
(224.58 mg,0.856 mmol,3.00當量)於THF(16.00 mL)中的攪拌混合物中逐滴添加DIAD(173.14 mg,0.856 mmol,3.00當量)。在氮氣氛圍下在70℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH 12:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內37 B至50 B)來純化粗產物,得到呈白色固體的1-((1R,3s,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例107, 45mg,45.00%)。LCMS: m/z (ESI), [M+H]+
= 391.4。1
H NMR (300 MHz, DMSO-d6
) δ 1.78-1.94(6H, m), 2.07-2.25(2H, m),2.42(3H.s), 2.45(3H,s), 4.44(2H, s), 4.88-4.96(1H, m), 7.73(1H, s), 8.41(1H, s), 8.92(1H, s), 9.18(1H, s), 9.29(1H, s)。實例 108/109/110/111
1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108,異構體1)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例109,異構體2)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110,異構體3)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例111,異構體4)
步驟1. 1-((1R,3s,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在氮氣氛圍下在0℃下向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(80.00 mg,0.285 mmol,1.00當量)、(1R,3R,5S)-8-氧雜雙環[3.2.1]辛-3-醇(109.75 mg,0.856 mmol,3.00當量)以及PPh3
(224.58 mg,0.856 mmol,3.00當量)於THF(16.00 mL)中的攪拌混合物中逐滴添加DIAD(173.14 mg,0.856 mmol,3.00當量)。在氮氣氛圍下在70℃下攪拌所得混合物2h。真空濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH 12:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Prep OBD C18柱,30×150mm 5um;移動相A:水(0.05%NH3
•H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內37 B至50 B)來純化粗產物,得到呈白色固體的1-((1R,3s,5S)-8-氧雜-雙環[3.2.1]辛-3-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例107, 45mg,45.00%)。LCMS: m/z (ESI), [M+H]+
= 391.4。1
H NMR (300 MHz, DMSO-d6
) δ 1.78-1.94(6H, m), 2.07-2.25(2H, m),2.42(3H.s), 2.45(3H,s), 4.44(2H, s), 4.88-4.96(1H, m), 7.73(1H, s), 8.41(1H, s), 8.92(1H, s), 9.18(1H, s), 9.29(1H, s)。實例 108/109/110/111
1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108,異構體1)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例109,異構體2)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110,異構體3)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例111,異構體4) 步驟1. 製備1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(108/109的混合物)及製備1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(110/111的混合物)
在0℃下經10 min向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(220.00 mg,0.785 mmol,1.00當量)、PPh3
(617.59 mg,2.355 mmol,3.00當量)以及3-甲氧基環丙-1-醇(273.52 mg,2.355 mmol,3.00當量)於THF(20.00 mL)中的攪拌混合物中逐滴添加含DIAD(476.13 mg,2.355 mmol,3.00當量)的THF(3 mL)。在氮氣氛圍下在70℃下攪拌所得混合物2 h。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH 10:1)來純化殘餘物,得到呈淡黃色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(150 mg)。通過製備型HPLC利用以下條件(柱:YMC-Actus Triart C18,30*250,5um;移動相A:水(0.05%NH3
H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:8.5 min內34 B至46 B)來純化粗產物(150 mg),得到呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108/109的混合物,25 mg,16.67%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz , DMSO-d6
) δ 1.81 - 1.85 (2H, m), 1.98 - 2.02 (2H, m), 2.09 - 2.11 (1H, m), 2.38 - 2.41 (4H, m), 2.45 (3H, s), 3.18 (3H, s), 3.83 - 3.85 (1H, m), 4.85 - 4.89 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.14 (2H, s), 及呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110/111的混合物,80 mg,53.33%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ1.61 - 1.63 (1H, m), 1.82 - 1.98 (1H, m), 2.38 - 2.41 (4H, m), 2.39 (3H, s), 2.45 (3H, s), 3.18 (3H, s), 3.93 - 3.96 (1H, m), 5.00 - 5.06 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.15 (2H, d)。
步驟2. 製備1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108,異構體1)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例109,異構體2)
通過手性製備型HPLC利用以下條件(柱:CHIRALPAK AD-H,2.0cm I.D.*25cm L;移動相A:Hex(8mmol/L NH3
.MeOH)--HPLC,移動相B:IPA--HPLC;流動速率:40 mL/min;梯度:18 min內20 B至20 B)來純化實例108/109的混合物(25 mg),得到呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108, 4.5mg,18.00%)(異構體1)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz , DMSO-d6
) δ
1.81 - 1.85 (2H, m), 1.98 - 2.01 (2H, m), 2.06 - 2.09 (1H, m), 2.38 - 2.43 (4H, m), 2.45 (3H, s), 3.18 (3H, s), 3.80 - 3.85 (1H, m), 4.82 - 4.87 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.14 (2H, s)及呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例109,3.8mg,12.00%)(異構體2)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz , DMSO-d6
) δ 1.81 - 1.83 (2H, m), 1.97 - 2.01 (2H, m), 2.10 - 2.12 (1H, m), 2.37 - 2.43 (4H, m), 2.45 (3H, s), 3.18 (3H, s), 3.81 - 3.85 (1H, m), 4.80 - 4.89 (1H, m), 7.73 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.14 (2H, s)。
步驟4. 1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110,異構體3)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例111,異構體4)
通過製備型HPLC利用以下條件(柱:CHIRALPAK AD-H,2.0cm I.D.*25cm L;移動相A:Hex(8mmol/L NH3.MeOH)--HPLC,移動相B:IPA--HPLC;流動速率:40 mL/min;梯度:12 min內30 B至30 B)來純化實例110/111的混合物(80 mg),得到呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110,39.1mg,48.87%)(異構體3)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ 1.67 - 1.69(1H, m), 1.93 - 1.99 (1H, m), 2.10 - 2.15 (4H, m), 2.39 (3H, s), 2.44 (3H, s), 3.16 (3H, s), 3.94 (1H, s), 5.00 - 5.08 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.16 (2H, d)及呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例111,35.2mg)(異構體4)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ 1.65 - 1.69 (1H, m), 1.93 - 1.97 (1H, m), 2.09 - 2.14 (4H, m), 2.39 (3H, d), 2.44 (3H, s), 3.15 (3H, s), 3.93 - 3.96 (1H, m), 5.00 - 5.08 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.16 (2H, d)。實例 112/113/114/115.
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112,異構體1)/3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例113,異構體2)/3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114,異構體3)/3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例115,異構體4)
步驟1. 製備1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(108/109的混合物)及製備1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(110/111的混合物)
在0℃下經10 min向3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(220.00 mg,0.785 mmol,1.00當量)、PPh3
(617.59 mg,2.355 mmol,3.00當量)以及3-甲氧基環丙-1-醇(273.52 mg,2.355 mmol,3.00當量)於THF(20.00 mL)中的攪拌混合物中逐滴添加含DIAD(476.13 mg,2.355 mmol,3.00當量)的THF(3 mL)。在氮氣氛圍下在70℃下攪拌所得混合物2 h。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH 10:1)來純化殘餘物,得到呈淡黃色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(150 mg)。通過製備型HPLC利用以下條件(柱:YMC-Actus Triart C18,30*250,5um;移動相A:水(0.05%NH3
H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:8.5 min內34 B至46 B)來純化粗產物(150 mg),得到呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108/109的混合物,25 mg,16.67%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz , DMSO-d6
) δ 1.81 - 1.85 (2H, m), 1.98 - 2.02 (2H, m), 2.09 - 2.11 (1H, m), 2.38 - 2.41 (4H, m), 2.45 (3H, s), 3.18 (3H, s), 3.83 - 3.85 (1H, m), 4.85 - 4.89 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.14 (2H, s), 及呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110/111的混合物,80 mg,53.33%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ1.61 - 1.63 (1H, m), 1.82 - 1.98 (1H, m), 2.38 - 2.41 (4H, m), 2.39 (3H, s), 2.45 (3H, s), 3.18 (3H, s), 3.93 - 3.96 (1H, m), 5.00 - 5.06 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.15 (2H, d)。
步驟2. 製備1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108,異構體1)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例109,異構體2)
通過手性製備型HPLC利用以下條件(柱:CHIRALPAK AD-H,2.0cm I.D.*25cm L;移動相A:Hex(8mmol/L NH3
.MeOH)--HPLC,移動相B:IPA--HPLC;流動速率:40 mL/min;梯度:18 min內20 B至20 B)來純化實例108/109的混合物(25 mg),得到呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例108, 4.5mg,18.00%)(異構體1)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz , DMSO-d6
) δ
1.81 - 1.85 (2H, m), 1.98 - 2.01 (2H, m), 2.06 - 2.09 (1H, m), 2.38 - 2.43 (4H, m), 2.45 (3H, s), 3.18 (3H, s), 3.80 - 3.85 (1H, m), 4.82 - 4.87 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.14 (2H, s)及呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例109,3.8mg,12.00%)(異構體2)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz , DMSO-d6
) δ 1.81 - 1.83 (2H, m), 1.97 - 2.01 (2H, m), 2.10 - 2.12 (1H, m), 2.37 - 2.43 (4H, m), 2.45 (3H, s), 3.18 (3H, s), 3.81 - 3.85 (1H, m), 4.80 - 4.89 (1H, m), 7.73 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.14 (2H, s)。
步驟4. 1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110,異構體3)/1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例111,異構體4)
通過製備型HPLC利用以下條件(柱:CHIRALPAK AD-H,2.0cm I.D.*25cm L;移動相A:Hex(8mmol/L NH3.MeOH)--HPLC,移動相B:IPA--HPLC;流動速率:40 mL/min;梯度:12 min內30 B至30 B)來純化實例110/111的混合物(80 mg),得到呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例110,39.1mg,48.87%)(異構體3)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ 1.67 - 1.69(1H, m), 1.93 - 1.99 (1H, m), 2.10 - 2.15 (4H, m), 2.39 (3H, s), 2.44 (3H, s), 3.16 (3H, s), 3.94 (1H, s), 5.00 - 5.08 (1H, m), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.16 (2H, d)及呈白色固體的1-(3-甲氧基環戊基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例111,35.2mg)(異構體4)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ 1.65 - 1.69 (1H, m), 1.93 - 1.97 (1H, m), 2.09 - 2.14 (4H, m), 2.39 (3H, d), 2.44 (3H, s), 3.15 (3H, s), 3.93 - 3.96 (1H, m), 5.00 - 5.08 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.16 (2H, d)。實例 112/113/114/115.
製備3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112,異構體1)/3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例113,異構體2)/3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114,異構體3)/3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例115,異構體4) 步驟1. 3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例112/113的混合物)和3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例114/115的混合物)
在0℃下攪拌3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(200.00 mg,0.714 mmol,1.00當量)、3-甲基氧雜環己烷-4-醇(248.65 mg,2.141 mmol,3.00當量)以及PPh3
(561.45 mg,2.141 mmol,3.00當量)於THF(10.00 mL)中的混合物,且在氮氣氛圍下70℃下逐滴添加DIAD(432.85 mg,2.141 mmol,3.00當量),持續2小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Shield RP18 OBD柱,30*150mm,5um;移動相A:水(0.05% NH3
H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內18 B至48 B)來純化粗產物(120 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例112/113的混合物,30 mg,25.00%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ 0.52 (3H, d), 1.74 (2H, d), 2.06 - 2.31 (1H, m), 2.36 (3H, d), 2.45 (3H, s), 3.07 (1H, t), 3.43 (1H, t), 3.82 - 4.00 (2H, m), 4.14 - 4.27 (1H, m), 7.69 - 7.76 (1H, m), 8.39 (1H, s), 8.91 (1H, s), 9.15 (2H, d)及呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[-3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例114/115的混合物,70mg,57.75%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ 0.70 (3H, d), 1.74 - 1.91 (1H, m), 2.23 (1H, d), 2.37 (4H, d), 2.44 (3H, s), 3.44 - 3.65 (2H, m), 3.72 (1H, dd), 4.05 (1H, dt), 4.79 (1H, dt), 7.72 (1H, s), 8.39 (1H, s), 8.93 (1H, s), 9.17 (2H, s)
步驟2. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112,異構體1)和3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例113,異構體2)
通過製備型手性HPLC利用以下條件(柱:CHIRALPAK IE-3,4.6*50mm 3um;移動相A:Hex (0.1%DEA):EtOH = 50:50)來純化粗產物3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112/113的混合物,30.00 mg,0.079 mmol,1.00當量)),得到呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112,異構體1,12.22 mg,40.73%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ 0.52 (3H, d), 1.74 (2H, d), 2.06 - 2.31 (1H, m), 2.36 (3H, d), 2.45 (3H, s), 3.07 (1H, t), 3.43 (1H, t), 3.82 - 4.00 (2H, m), 4.14 - 4.27 (1H, m), 7.69 - 7.76 (1H, m), 8.39 (1H, s), 8.91 (1H, s), 9.15 (2H, d)及呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例113,異構體2,8.37 mg,27.90%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz , DMSO-d6
) δ 0.52 (3H, d), 1.74 (2H, d), 2.06 - 2.31 (1H, m), 2.36 (3H, d), 2.45 (3H, s), 3.07 (1H, t), 3.43 (1H, t), 3.82 - 4.00 (2H, m), 4.14 - 4.27 (1H, m), 7.69 - 7.76 (1H, m), 8.39 (1H, s), 8.91 (1H, s), 9.15 (2H, d)
步驟3. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114,異構體3)和3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例115,異構體4)
通過製備型手性HPLC利用以下條件(柱:CHIRALPAK IE-3,4.6*50mm 3um;移動相A:Hex (0.1%DEA):EtOH=50:50)來純化粗產物3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114/115的混合物,70.00 mg,0.185 mmol,1.00當量)),得到呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114,異構體3,32.98mg,47.11%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
H NMR (300 MHz, DMSO-d6
) δ 0.70 (3H, d), 1.74 - 1.91 (1H, m), 2.23 (1H, d), 2.37 (4H, d), 2.44 (3H, s), 3.44 - 3.65 (2H, m), 3.72 (1H, d), 4.05 (1H, t), 4.79 (1H, t), 7.72 (1H, s), 8.39 (1H, s), 8.93 (1H, s), 9.17 (2H, s), 及呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例115,異構體4,33.45 mg,41.39%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
H NMR (300 MHz, DMSO-d6
) δ 0.70 (3H, d), 1.74 - 1.91 (1H, m), 2.23 (1H, d), 2.37 (4H, d), 2.44 (3H, s), 3.44 - 3.65 (2H, m), 3.72 (1H, d), 4.05 (1H, t), 4.79 (1H, t), 7.72 (1H, s), 8.39 (1H, s), 8.93 (1H, s), 9.17 (2H, s)實例 116/117
製備1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例116,異構體1)及1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例117,異構體2)
步驟1. 3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例112/113的混合物)和3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例114/115的混合物)
在0℃下攪拌3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(200.00 mg,0.714 mmol,1.00當量)、3-甲基氧雜環己烷-4-醇(248.65 mg,2.141 mmol,3.00當量)以及PPh3
(561.45 mg,2.141 mmol,3.00當量)於THF(10.00 mL)中的混合物,且在氮氣氛圍下70℃下逐滴添加DIAD(432.85 mg,2.141 mmol,3.00當量),持續2小時。減壓濃縮所得混合物。通過製備型TLC(CH2
Cl2
/MeOH 15:1)來純化殘餘物,得到粗固體。通過製備型HPLC利用以下條件(柱:XBridge Shield RP18 OBD柱,30*150mm,5um;移動相A:水(0.05% NH3
H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內18 B至48 B)來純化粗產物(120 mg),得到呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例112/113的混合物,30 mg,25.00%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ 0.52 (3H, d), 1.74 (2H, d), 2.06 - 2.31 (1H, m), 2.36 (3H, d), 2.45 (3H, s), 3.07 (1H, t), 3.43 (1H, t), 3.82 - 4.00 (2H, m), 4.14 - 4.27 (1H, m), 7.69 - 7.76 (1H, m), 8.39 (1H, s), 8.91 (1H, s), 9.15 (2H, d)及呈白色固體的3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1-[-3-甲基氧雜環己烷-4-基]吡唑並[3,4-d]嘧啶-6-胺(實例114/115的混合物,70mg,57.75%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ 0.70 (3H, d), 1.74 - 1.91 (1H, m), 2.23 (1H, d), 2.37 (4H, d), 2.44 (3H, s), 3.44 - 3.65 (2H, m), 3.72 (1H, dd), 4.05 (1H, dt), 4.79 (1H, dt), 7.72 (1H, s), 8.39 (1H, s), 8.93 (1H, s), 9.17 (2H, s)
步驟2. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112,異構體1)和3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例113,異構體2)
通過製備型手性HPLC利用以下條件(柱:CHIRALPAK IE-3,4.6*50mm 3um;移動相A:Hex (0.1%DEA):EtOH = 50:50)來純化粗產物3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112/113的混合物,30.00 mg,0.079 mmol,1.00當量)),得到呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例112,異構體1,12.22 mg,40.73%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz, DMSO-d6
) δ 0.52 (3H, d), 1.74 (2H, d), 2.06 - 2.31 (1H, m), 2.36 (3H, d), 2.45 (3H, s), 3.07 (1H, t), 3.43 (1H, t), 3.82 - 4.00 (2H, m), 4.14 - 4.27 (1H, m), 7.69 - 7.76 (1H, m), 8.39 (1H, s), 8.91 (1H, s), 9.15 (2H, d)及呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例113,異構體2,8.37 mg,27.90%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
HNMR (400MHz , DMSO-d6
) δ 0.52 (3H, d), 1.74 (2H, d), 2.06 - 2.31 (1H, m), 2.36 (3H, d), 2.45 (3H, s), 3.07 (1H, t), 3.43 (1H, t), 3.82 - 4.00 (2H, m), 4.14 - 4.27 (1H, m), 7.69 - 7.76 (1H, m), 8.39 (1H, s), 8.91 (1H, s), 9.15 (2H, d)
步驟3. 3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114,異構體3)和3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例115,異構體4)
通過製備型手性HPLC利用以下條件(柱:CHIRALPAK IE-3,4.6*50mm 3um;移動相A:Hex (0.1%DEA):EtOH=50:50)來純化粗產物3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114/115的混合物,70.00 mg,0.185 mmol,1.00當量)),得到呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例114,異構體3,32.98mg,47.11%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
H NMR (300 MHz, DMSO-d6
) δ 0.70 (3H, d), 1.74 - 1.91 (1H, m), 2.23 (1H, d), 2.37 (4H, d), 2.44 (3H, s), 3.44 - 3.65 (2H, m), 3.72 (1H, d), 4.05 (1H, t), 4.79 (1H, t), 7.72 (1H, s), 8.39 (1H, s), 8.93 (1H, s), 9.17 (2H, s), 及呈白色固體的3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1-(3-甲基四氫-2H-吡喃-4-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例115,異構體4,33.45 mg,41.39%)。LCMS: m/z (ESI), [M+H]+
= 379.3。1
H NMR (300 MHz, DMSO-d6
) δ 0.70 (3H, d), 1.74 - 1.91 (1H, m), 2.23 (1H, d), 2.37 (4H, d), 2.44 (3H, s), 3.44 - 3.65 (2H, m), 3.72 (1H, d), 4.05 (1H, t), 4.79 (1H, t), 7.72 (1H, s), 8.39 (1H, s), 8.93 (1H, s), 9.17 (2H, s)實例 116/117
製備1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例116,異構體1)及1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例117,異構體2) 步驟1.1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在0℃下向250 mL圓底燒瓶中添加含2,2-二甲基氧雜環己烷-4-醇(627.03 mg,4.816 mmol,3.00當量)和3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(450.00 mg,1.605 mmol,1.00當量)、PPh3
(1263.26 mg,4.816 mmol,3.00當量)的THF(60.00 mL)。在N2
下在0℃下將DIAD(973.90 mg,4.816 mmol,3.00當量)於THF(10 mL)中的溶液逐滴添加至以上溶液中,並在rt下攪拌3 min。在70℃下繼續攪拌反應混合物2h。減壓濃縮所得混合物。用DCM(20 mL)稀釋所得混合物且過濾,用DCM(2×5 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH 15:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:YMC-Actus Triart C18,30*250,5um;移動相A:水(0.05% NH3
H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內37 B至57 B)來純化粗產物(250 mg),得到呈白色固體的1-[2,2-二甲基氧雜環己烷-4-基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(170mg,26.98%)。LCMS: m/z (ESI), [M+H]+
= 393.2
步驟2. 1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例116,異構體1)/1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例117,異構體2)
通過手性製備型HPLC利用以下條件(柱:CHIRALPAK-AD-H-UL001,20*250mm,5 um;移動相A:Hex(8mmol/L NH3
.MeOH)--HPLC,移動相B:IPA--HPLC;流動速率:20 mL/min;梯度:15 min內25 B至25 B;RT1:10.12;RT2:11.691)來純化粗產物(170 mg),得到rel-1-[(4R)-2,2-二甲基氧雜環己烷-4-基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(實例116,異構體1)(70mg,41.18%) LCMS: m/z (ESI), [M+H]+
= 393.3。1
H-NMR (400 MHz, DMSO-d6
) δ 1.21 (6H, d), 1.80 (2H, dd), 1.89 (1H, t), 2.05 (1H, qd), 2.35 - 2.48 (6H, m), 3.66 - 3.81 (2H, m), 4.75 - 4.95 (1H, m), 7.74 (1H, d), 8.40 (1H, s), 8.92 (1H, s), 9.20 (2H, d); 及呈白色固體的rel-1-[(4R)-2,2-二甲基氧雜環己烷-4-基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(實例117,異構體2)(70mg,41.18%), LCMS: m/z (ESI), [M+H]+
= 393.3。1
H-NMR (400 MHz, DMSO-d6
) δ1.21 (6H, d), 1.80 (2H, dd), 1.89 (1H, t), 2.05 (1H, qd), 2.35 - 2.48 (6H, m), 3.66 - 3.81 (2H, m), 4.75 - 4.95 (1H, m), 7.74 (1H, d), 8.40 (1H, s), 8.92 (1H, s), 9.20 (2H, d)。生物實例
本文所公開的示範性化合物已經在以下一個或多個生物分析中表徵。生物實例 1 :
酶分析
通過測量螢光標記的肽底物轉化成磷酸化產物的TRF-RET來測定化合物針對DNA-PK的抑制活性。所有分析都在黑色Greiner 384孔小體積培養板(Greiner)中進行,總反應體積為6 μL且最終DMSO濃度為0.5 %(v/v)。全長人類DNAPK蛋白、螢光素-P53 (Ser15)肽底物(螢光素-EPPLSQEAFADLWKK)以及LanthaScreen™ Tb-抗磷酸-p53 [pSer15]抗體試劑盒都是購自賽默飛世爾科技(Thermo Fisher Scientific)。最初,將DNA-PK蛋白質與化合物在室溫下在反應緩衝液(50 mM HEPES pH 7.5,0.01% Brij-35,10 mM MgCl2,1 mM EGTA,1 mM DTT,10 μg/ml小牛胸腺DNA)中培育30分鐘。然後通過添加ATP和螢光素-P53 (Ser15)肽底物引發反應。在60分鐘後通過添加含有20 mM EDTA、4 nM Tb抗磷酸-p53 [Serl5]抗體的6 μl終止緩衝液來淬滅激酶反應物(10 μΜ ATP,1.6 μΜ肽底物)。將反應物再培育一小時,且在Spark 20M(Tecan)上讀取培養板。分析資料,且使用利用XLfit的四倍參數邏輯擬合來計算化合物產生50%對相應激酶的抑制的濃度(IC50
)。示範性化合物的DNA-PK抑制活性展示於下表5中。可以看出,本公開的化合物已展現出強效的DNA-PK抑制活性。
步驟1.1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺
在0℃下向250 mL圓底燒瓶中添加含2,2-二甲基氧雜環己烷-4-醇(627.03 mg,4.816 mmol,3.00當量)和3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]-1H-吡唑並[3,4-d]嘧啶-6-胺(450.00 mg,1.605 mmol,1.00當量)、PPh3
(1263.26 mg,4.816 mmol,3.00當量)的THF(60.00 mL)。在N2
下在0℃下將DIAD(973.90 mg,4.816 mmol,3.00當量)於THF(10 mL)中的溶液逐滴添加至以上溶液中,並在rt下攪拌3 min。在70℃下繼續攪拌反應混合物2h。減壓濃縮所得混合物。用DCM(20 mL)稀釋所得混合物且過濾,用DCM(2×5 mL)洗滌濾餅。減壓濃縮濾液。通過製備型TLC(CH2
Cl2
/MeOH 15:1)來純化殘餘物,得到粗產物。通過製備型HPLC利用以下條件(柱:YMC-Actus Triart C18,30*250,5um;移動相A:水(0.05% NH3
H2
O),移動相B:ACN;流動速率:60 mL/min;梯度:7 min內37 B至57 B)來純化粗產物(250 mg),得到呈白色固體的1-[2,2-二甲基氧雜環己烷-4-基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(170mg,26.98%)。LCMS: m/z (ESI), [M+H]+
= 393.2
步驟2. 1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例116,異構體1)/1-(2,2-二甲基四氫-2H-吡喃-4-基)-3-甲基-N-(7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基)-1H-吡唑並[3,4-d]嘧啶-6-胺(實例117,異構體2)
通過手性製備型HPLC利用以下條件(柱:CHIRALPAK-AD-H-UL001,20*250mm,5 um;移動相A:Hex(8mmol/L NH3
.MeOH)--HPLC,移動相B:IPA--HPLC;流動速率:20 mL/min;梯度:15 min內25 B至25 B;RT1:10.12;RT2:11.691)來純化粗產物(170 mg),得到rel-1-[(4R)-2,2-二甲基氧雜環己烷-4-基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(實例116,異構體1)(70mg,41.18%) LCMS: m/z (ESI), [M+H]+
= 393.3。1
H-NMR (400 MHz, DMSO-d6
) δ 1.21 (6H, d), 1.80 (2H, dd), 1.89 (1H, t), 2.05 (1H, qd), 2.35 - 2.48 (6H, m), 3.66 - 3.81 (2H, m), 4.75 - 4.95 (1H, m), 7.74 (1H, d), 8.40 (1H, s), 8.92 (1H, s), 9.20 (2H, d); 及呈白色固體的rel-1-[(4R)-2,2-二甲基氧雜環己烷-4-基]-3-甲基-N-[7-甲基-[1,2,4]三唑並[1,5-a]吡啶-6-基]吡唑並[3,4-d]嘧啶-6-胺(實例117,異構體2)(70mg,41.18%), LCMS: m/z (ESI), [M+H]+
= 393.3。1
H-NMR (400 MHz, DMSO-d6
) δ1.21 (6H, d), 1.80 (2H, dd), 1.89 (1H, t), 2.05 (1H, qd), 2.35 - 2.48 (6H, m), 3.66 - 3.81 (2H, m), 4.75 - 4.95 (1H, m), 7.74 (1H, d), 8.40 (1H, s), 8.92 (1H, s), 9.20 (2H, d)。生物實例
本文所公開的示範性化合物已經在以下一個或多個生物分析中表徵。生物實例 1 :
酶分析
通過測量螢光標記的肽底物轉化成磷酸化產物的TRF-RET來測定化合物針對DNA-PK的抑制活性。所有分析都在黑色Greiner 384孔小體積培養板(Greiner)中進行,總反應體積為6 μL且最終DMSO濃度為0.5 %(v/v)。全長人類DNAPK蛋白、螢光素-P53 (Ser15)肽底物(螢光素-EPPLSQEAFADLWKK)以及LanthaScreen™ Tb-抗磷酸-p53 [pSer15]抗體試劑盒都是購自賽默飛世爾科技(Thermo Fisher Scientific)。最初,將DNA-PK蛋白質與化合物在室溫下在反應緩衝液(50 mM HEPES pH 7.5,0.01% Brij-35,10 mM MgCl2,1 mM EGTA,1 mM DTT,10 μg/ml小牛胸腺DNA)中培育30分鐘。然後通過添加ATP和螢光素-P53 (Ser15)肽底物引發反應。在60分鐘後通過添加含有20 mM EDTA、4 nM Tb抗磷酸-p53 [Serl5]抗體的6 μl終止緩衝液來淬滅激酶反應物(10 μΜ ATP,1.6 μΜ肽底物)。將反應物再培育一小時,且在Spark 20M(Tecan)上讀取培養板。分析資料,且使用利用XLfit的四倍參數邏輯擬合來計算化合物產生50%對相應激酶的抑制的濃度(IC50
)。示範性化合物的DNA-PK抑制活性展示於下表5中。可以看出,本公開的化合物已展現出強效的DNA-PK抑制活性。 生物實例 2 :
代謝穩定性分析(大鼠肝細胞Clint)
使用台盼藍測定冷凍保存的肝細胞的存活力,並且用緩衝液將所述細胞濃縮液調節為每毫升106個細胞。將1 μM化合物(在乙腈中;0.01% DMSO)在96深孔培養板中與250 μL肝細胞(每毫升1百萬個細胞)一起培育。在不同時間點(0、0.5、5、15、30、45、60、80、100和120 min)通過添加3體積的冷卻乙腈到20 μL反應混合物並且在4℃下離心15 min來使反應停止。用純水將40 μL上清液稀釋到200 μL並使用LCMS/MS進行分析。
基於從初始濃度測定的化合物消失的消除半衰期(T1/2
)來估計體外肝細胞清除率。計算每種化合物(測試或對照)與IS的峰面積比。藥物消除速率常數k(min-1)、T1/2
(min)和體外固有清除率CLint
(μL/min/E6)是根據以下等式計算:
k = -斜率
T1/2
= 0.693/k
CLint
= k/Chep
其中Chep
(細胞×μL-1
)是培育系統中的細胞濃度。
資料展示如下表6中。
代謝穩定性分析(人類微粒體Clint)
將1 μM化合物在37℃下在含有1 mM NADPH溶液的250 μL緩衝液(100 mM磷酸鹽緩衝液,pH-7.4)中與1 mg/mL微粒體(具有20 mg/ml蛋白質錐體的混合HLM)一起培育。在新制96孔板中,在不同時間點0、0.5、5、10、15、20和30 min,用5體積的冷卻乙腈淬滅20 μL的培育混合物。將淬滅板在4000 rpm下離心15 min。用純水將40 μL上清液稀釋到200 μ L並使用LCMS/MS進行分析。
藥物在微粒體中的體外固有清除率CLint
(µl/min/mg)是以與肝細胞中的CLint
類似的方式計算的。資料也展示如下表6中。
MDCKII-MDR1-BCRP流出分析
跨越MDCKII-MDR1-BCRP細胞單層測量化合物在HBSS(25mM HEPES,pH 7.4)中的頂端到底外側(A-B)和底外側到頂端(B-A)轉運。在約37℃下進行培育120 min,其中使用5 μM地高辛作為陽性對照底物來確認測試系統的功能性。通過在培育期開始時定量供體隔室的培育介質中的底物濃度並且在培育期結束時定量供體隔室和受體隔室兩者中的底物濃度來測定5 μM化合物和對照化合物的轉運。資料用於計算表觀滲透率(Papp)。進行所有培育,重複三次,並且使用標記螢光黃確認細胞單層的完整性。
使用以下等式計算滲透率係數Pexact(cm/s):
Papp = (dCr/dt) x Vr / (A x C0)
Pexact = -(Vd×Vr)/(Vd+Vr)/A/t×ln(1-(Vd+Vr)×Cr/(Vd×Cd +Vr×Cr))
使用以下等式計算Pexact比:
Pexact或Papp比=Pexact或Papp(+抑制劑)/Pexact或Papp(-抑制劑)
使用以下等式計算流出比:
流出比 = Pexact或Papp(BA)/Pexact或Papp(AB)
其中dCr/dt為隨時間變化的接受體室中的化合物的累積濃度(µM/s);Vr為接受體室中的溶液體積(在頂端側為0.1 mL,在底外側為0.3 mL);A為用於轉運的表面積,即,單層面積為0.11 cm2
;C0為供體室中的初始濃度(µM)。數據展示於下表6中。
生物實例 2 :
代謝穩定性分析(大鼠肝細胞Clint)
使用台盼藍測定冷凍保存的肝細胞的存活力,並且用緩衝液將所述細胞濃縮液調節為每毫升106個細胞。將1 μM化合物(在乙腈中;0.01% DMSO)在96深孔培養板中與250 μL肝細胞(每毫升1百萬個細胞)一起培育。在不同時間點(0、0.5、5、15、30、45、60、80、100和120 min)通過添加3體積的冷卻乙腈到20 μL反應混合物並且在4℃下離心15 min來使反應停止。用純水將40 μL上清液稀釋到200 μL並使用LCMS/MS進行分析。
基於從初始濃度測定的化合物消失的消除半衰期(T1/2
)來估計體外肝細胞清除率。計算每種化合物(測試或對照)與IS的峰面積比。藥物消除速率常數k(min-1)、T1/2
(min)和體外固有清除率CLint
(μL/min/E6)是根據以下等式計算:
k = -斜率
T1/2
= 0.693/k
CLint
= k/Chep
其中Chep
(細胞×μL-1
)是培育系統中的細胞濃度。
資料展示如下表6中。
代謝穩定性分析(人類微粒體Clint)
將1 μM化合物在37℃下在含有1 mM NADPH溶液的250 μL緩衝液(100 mM磷酸鹽緩衝液,pH-7.4)中與1 mg/mL微粒體(具有20 mg/ml蛋白質錐體的混合HLM)一起培育。在新制96孔板中,在不同時間點0、0.5、5、10、15、20和30 min,用5體積的冷卻乙腈淬滅20 μL的培育混合物。將淬滅板在4000 rpm下離心15 min。用純水將40 μL上清液稀釋到200 μ L並使用LCMS/MS進行分析。
藥物在微粒體中的體外固有清除率CLint
(µl/min/mg)是以與肝細胞中的CLint
類似的方式計算的。資料也展示如下表6中。
MDCKII-MDR1-BCRP流出分析
跨越MDCKII-MDR1-BCRP細胞單層測量化合物在HBSS(25mM HEPES,pH 7.4)中的頂端到底外側(A-B)和底外側到頂端(B-A)轉運。在約37℃下進行培育120 min,其中使用5 μM地高辛作為陽性對照底物來確認測試系統的功能性。通過在培育期開始時定量供體隔室的培育介質中的底物濃度並且在培育期結束時定量供體隔室和受體隔室兩者中的底物濃度來測定5 μM化合物和對照化合物的轉運。資料用於計算表觀滲透率(Papp)。進行所有培育,重複三次,並且使用標記螢光黃確認細胞單層的完整性。
使用以下等式計算滲透率係數Pexact(cm/s):
Papp = (dCr/dt) x Vr / (A x C0)
Pexact = -(Vd×Vr)/(Vd+Vr)/A/t×ln(1-(Vd+Vr)×Cr/(Vd×Cd +Vr×Cr))
使用以下等式計算Pexact比:
Pexact或Papp比=Pexact或Papp(+抑制劑)/Pexact或Papp(-抑制劑)
使用以下等式計算流出比:
流出比 = Pexact或Papp(BA)/Pexact或Papp(AB)
其中dCr/dt為隨時間變化的接受體室中的化合物的累積濃度(µM/s);Vr為接受體室中的溶液體積(在頂端側為0.1 mL,在底外側為0.3 mL);A為用於轉運的表面積,即,單層面積為0.11 cm2
;C0為供體室中的初始濃度(µM)。數據展示於下表6中。
 生物實例 3 :
血腦屏障穿透分析
據信,Kp,uu是大腦和血漿中非結合藥物的濃度之間的關係,是CNS作用的預測的關鍵,並且應是針對藥物發現所測量和優化的主要參數(Di L等人,《藥物化學雜誌(Journal of Medicinal Chemistry)》[2013], 56:2-12)。
通過使用平衡透析方法用半滲透膜進行體外血漿和大腦結合分析。向血漿和稀釋的腦勻漿(1:4,其中DPBS pH7.4)摻入5 µM測試化合物(重複三次),並且在37℃下在緩慢旋轉的培養板中相對於相等體積的150 µL 100 mM PBS緩衝液(pH7.4)透析18小時。在培育結束時,取出來自接收器側的50 μL等分試樣和來自供體室的5 μL等分試樣。用45 μL空白血漿或腦勻漿進一步稀釋5 μL樣品。將配對樣品與緩衝液或空白血漿/腦勻漿進行基質匹配且混合2 min,並接著用150 μL低溫乙腈和100 ng/mL甲苯磺丁脲作為內標進行沉澱。在以4000 rpm離心20 min後,將上清液用0.1%甲酸水溶液稀釋並分析LC/MS/MS(API 4000, 美國應用生物系統公司(Applied Biosystems),福斯特城(Foster City))。通過緩衝液側回應與腦勻漿/血漿側回應的比率計算腦勻漿和稀釋血漿中測試化合物的未結合分數(fu),並且通過以下等式從勻漿和血漿中測量的fu計算未稀釋的血漿和組織中測試化合物的未結合分數(fu、pl和fu、br):fu,bl (fu,br) = (1/D) / [(1/fu -1) + 1/D)]。D是稀釋係數。
短口服吸收(SOA)模型是用於鑒別化合物的腦滲透的體內篩選模型。向從北京維通利華(Beijing Vital River)購買的六隻雄性Han Wistar大鼠經口給予10 mg/kg的含1%甲基纖維素的化合物。在給藥後0.5、1、2、4、7和16小時,從小腦延髓池收集腦部脊髓液(CSF)。將通過在收集的半小時內在約4℃下離心3,000 g來處理血漿的血漿樣品。將血漿樣品移到標記的試管且存儲在-80℃下直到分析為止。採集腦組織並在3×體積的100 mM磷酸鹽緩衝鹽水(pH7.4)中均質化。在LC/MS/MS分析之前,將所有樣品存儲在約-70℃。
通過摻入空白血漿、腦勻漿和範圍為0.5至500 ng/mL的人工CSF來製備標準品。通過添加3倍體積的含有低溫乙腈的內標(40 ng/mL地塞米松和40 ng/mL雙氯芬酸)沉澱均質化的腦組織以及血漿樣品,並且用100 μL含有低溫乙腈的內標沉澱10 μL CSF樣品。渦旋2分鐘並以14,000 rpm離心5 min後,通過LC/MS/MS(API 4000,美國應用生物系統公司,福斯特城)分析上清液。在血漿樣品分析中每個批次開始和結束時操作兩組標準曲線。對於腦和CSF樣品,分析一條標準曲線和測試樣品。
通過經口施用後齧齒動物中的AUC大腦/AUC血漿來測量表示為大腦/血液比率(Kp
)的總大腦水準。通過體外血漿和大腦結合分析測定生物基質中測試化合物的游離部分。通過以下等式計算Kp,uu:Kp,uu = AUC (大腦)/AUC (血漿)×(fu,大腦/fu.血漿)。數據展示於表7中。
生物實例 3 :
血腦屏障穿透分析
據信,Kp,uu是大腦和血漿中非結合藥物的濃度之間的關係,是CNS作用的預測的關鍵,並且應是針對藥物發現所測量和優化的主要參數(Di L等人,《藥物化學雜誌(Journal of Medicinal Chemistry)》[2013], 56:2-12)。
通過使用平衡透析方法用半滲透膜進行體外血漿和大腦結合分析。向血漿和稀釋的腦勻漿(1:4,其中DPBS pH7.4)摻入5 µM測試化合物(重複三次),並且在37℃下在緩慢旋轉的培養板中相對於相等體積的150 µL 100 mM PBS緩衝液(pH7.4)透析18小時。在培育結束時,取出來自接收器側的50 μL等分試樣和來自供體室的5 μL等分試樣。用45 μL空白血漿或腦勻漿進一步稀釋5 μL樣品。將配對樣品與緩衝液或空白血漿/腦勻漿進行基質匹配且混合2 min,並接著用150 μL低溫乙腈和100 ng/mL甲苯磺丁脲作為內標進行沉澱。在以4000 rpm離心20 min後,將上清液用0.1%甲酸水溶液稀釋並分析LC/MS/MS(API 4000, 美國應用生物系統公司(Applied Biosystems),福斯特城(Foster City))。通過緩衝液側回應與腦勻漿/血漿側回應的比率計算腦勻漿和稀釋血漿中測試化合物的未結合分數(fu),並且通過以下等式從勻漿和血漿中測量的fu計算未稀釋的血漿和組織中測試化合物的未結合分數(fu、pl和fu、br):fu,bl (fu,br) = (1/D) / [(1/fu -1) + 1/D)]。D是稀釋係數。
短口服吸收(SOA)模型是用於鑒別化合物的腦滲透的體內篩選模型。向從北京維通利華(Beijing Vital River)購買的六隻雄性Han Wistar大鼠經口給予10 mg/kg的含1%甲基纖維素的化合物。在給藥後0.5、1、2、4、7和16小時,從小腦延髓池收集腦部脊髓液(CSF)。將通過在收集的半小時內在約4℃下離心3,000 g來處理血漿的血漿樣品。將血漿樣品移到標記的試管且存儲在-80℃下直到分析為止。採集腦組織並在3×體積的100 mM磷酸鹽緩衝鹽水(pH7.4)中均質化。在LC/MS/MS分析之前,將所有樣品存儲在約-70℃。
通過摻入空白血漿、腦勻漿和範圍為0.5至500 ng/mL的人工CSF來製備標準品。通過添加3倍體積的含有低溫乙腈的內標(40 ng/mL地塞米松和40 ng/mL雙氯芬酸)沉澱均質化的腦組織以及血漿樣品,並且用100 μL含有低溫乙腈的內標沉澱10 μL CSF樣品。渦旋2分鐘並以14,000 rpm離心5 min後,通過LC/MS/MS(API 4000,美國應用生物系統公司,福斯特城)分析上清液。在血漿樣品分析中每個批次開始和結束時操作兩組標準曲線。對於腦和CSF樣品,分析一條標準曲線和測試樣品。
通過經口施用後齧齒動物中的AUC大腦/AUC血漿來測量表示為大腦/血液比率(Kp
)的總大腦水準。通過體外血漿和大腦結合分析測定生物基質中測試化合物的游離部分。通過以下等式計算Kp,uu:Kp,uu = AUC (大腦)/AUC (血漿)×(fu,大腦/fu.血漿)。數據展示於表7中。 儘管已經參照具體實施例(其中一部分是優選實施例)具體地顯示和描述本公開,但本領域的技術人員應理解,可以在不脫離如本文所公開的本公開的精神和範圍情況下,對其中的形式和細節作出各種變化。
儘管已經參照具體實施例(其中一部分是優選實施例)具體地顯示和描述本公開,但本領域的技術人員應理解,可以在不脫離如本文所公開的本公開的精神和範圍情況下,對其中的形式和細節作出各種變化。
 ”意指X1 與X2 之間的鍵及X2 與X3 之間的鍵可以是單鍵或雙鍵,其限制條件是X1 與X2 之間的鍵及X2 與X3 之間的鍵中的至少一個是單鍵; R1 不存在或為C1-6 烷基,其中該C1-6 烷基可以任選地經羥基、鹵素或氘單取代或獨立多取代; 各R2 、R3 和R4 獨立地選自不存在、鹵素、羥基、氰基、C1-6 烷基、C1-6 烷氧基、-(CH2 )n -Q,其可以任選地經以下各者單取代或獨立多取代:氘、羥基、氨基、氰基、鹵素、C1-6 烷基、C1-6 鹵烷基、(C≡N)-C1-6 烷基、C1-6 烷氧基、C1-6 鹵烷氧基、C3-8 環烷基、C3-8 環烷氧基、3員至8員芳香基或3員至8員雜環基, 其中n為0、1或2,Q為3員至8員飽和或不飽和碳環基,或3員至8員飽和或不飽和雜環基; 環A為5員至12員芳香基、具有1到5個選自氧、硫和氮的環雜原子的5員至12員雜芳香基、具有0到5個選自氧、硫和氮的環雜原子的8員至10員雙環,其中環A不為苯基。
”意指X1 與X2 之間的鍵及X2 與X3 之間的鍵可以是單鍵或雙鍵,其限制條件是X1 與X2 之間的鍵及X2 與X3 之間的鍵中的至少一個是單鍵; R1 不存在或為C1-6 烷基,其中該C1-6 烷基可以任選地經羥基、鹵素或氘單取代或獨立多取代; 各R2 、R3 和R4 獨立地選自不存在、鹵素、羥基、氰基、C1-6 烷基、C1-6 烷氧基、-(CH2 )n -Q,其可以任選地經以下各者單取代或獨立多取代:氘、羥基、氨基、氰基、鹵素、C1-6 烷基、C1-6 鹵烷基、(C≡N)-C1-6 烷基、C1-6 烷氧基、C1-6 鹵烷氧基、C3-8 環烷基、C3-8 環烷氧基、3員至8員芳香基或3員至8員雜環基, 其中n為0、1或2,Q為3員至8員飽和或不飽和碳環基,或3員至8員飽和或不飽和雜環基; 環A為5員至12員芳香基、具有1到5個選自氧、硫和氮的環雜原子的5員至12員雜芳香基、具有0到5個選自氧、硫和氮的環雜原子的8員至10員雙環,其中環A不為苯基。




 , 其可以任選地經羥基、氰基、氟、氯、溴、甲基、乙基、甲氧基、二氟甲基、二氟甲氧基或三氟甲氧基單取代或獨立多取代。
, 其可以任選地經羥基、氰基、氟、氯、溴、甲基、乙基、甲氧基、二氟甲基、二氟甲氧基或三氟甲氧基單取代或獨立多取代。

 ,每個R3和R4獨立地選自不存在、甲基、氰基、甲氧基、氯、氰基-甲基、吡唑基、惡唑基,其中該吡唑基或惡唑基可以任選地進一步經C1-3 烷基單取代或獨立多取代。
,每個R3和R4獨立地選自不存在、甲基、氰基、甲氧基、氯、氰基-甲基、吡唑基、惡唑基,其中該吡唑基或惡唑基可以任選地進一步經C1-3 烷基單取代或獨立多取代。
 ”意指X1 與N之間的鍵及N與X3 之間的鍵可以是單鍵或雙鍵,其限制條件是X1 與N之間的鍵和N與X3 之間的鍵中的至少一個是單鍵; R1 為C1-3 烷基, R2 為環戊基、環己烷基、四氫吡喃基或8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經鹵素或C1-3 烷氧基單取代或獨立多取代, Y1 、Y2 和Y3 各自獨立地為C或N,其限制條件為Y1 、Y2 和Y3 中的至少一個為N; R5 為鹵素或C1-3 烷基, R6 為C1-3 烷基。
”意指X1 與N之間的鍵及N與X3 之間的鍵可以是單鍵或雙鍵,其限制條件是X1 與N之間的鍵和N與X3 之間的鍵中的至少一個是單鍵; R1 為C1-3 烷基, R2 為環戊基、環己烷基、四氫吡喃基或8-氧雜雙環[3.2.1]辛-3-基,其可以任選地經鹵素或C1-3 烷氧基單取代或獨立多取代, Y1 、Y2 和Y3 各自獨立地為C或N,其限制條件為Y1 、Y2 和Y3 中的至少一個為N; R5 為鹵素或C1-3 烷基, R6 為C1-3 烷基。








