KR20080106919A - Improved Prodrugs of CC-1065 Analogs - Google Patents
Improved Prodrugs of CC-1065 Analogs Download PDFInfo
- Publication number
- KR20080106919A KR20080106919A KR1020087021749A KR20087021749A KR20080106919A KR 20080106919 A KR20080106919 A KR 20080106919A KR 1020087021749 A KR1020087021749 A KR 1020087021749A KR 20087021749 A KR20087021749 A KR 20087021749A KR 20080106919 A KR20080106919 A KR 20080106919A
- Authority
- KR
- South Korea
- Prior art keywords
- och
- alkyl
- prodrug
- formula
- linker
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 229940002612 prodrug Drugs 0.000 title claims abstract description 126
- 239000000651 prodrug Substances 0.000 title claims abstract description 126
- UOWVMDUEMSNCAV-WYENRQIDSA-N rachelmycin Chemical class C1([C@]23C[C@@H]2CN1C(=O)C=1NC=2C(OC)=C(O)C4=C(C=2C=1)CCN4C(=O)C1=CC=2C=4CCN(C=4C(O)=C(C=2N1)OC)C(N)=O)=CC(=O)C1=C3C(C)=CN1 UOWVMDUEMSNCAV-WYENRQIDSA-N 0.000 title claims description 62
- 239000003814 drug Substances 0.000 claims abstract description 64
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 27
- 125000006239 protecting group Chemical group 0.000 claims abstract description 22
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims abstract description 8
- BSCCSDNZEIHXOK-UHFFFAOYSA-N phenyl carbamate Chemical compound NC(=O)OC1=CC=CC=C1 BSCCSDNZEIHXOK-UHFFFAOYSA-N 0.000 claims abstract description 7
- 150000001875 compounds Chemical class 0.000 claims description 83
- -1 methoxy, hydroxyl Chemical group 0.000 claims description 76
- 239000011230 binding agent Substances 0.000 claims description 74
- 125000005647 linker group Chemical group 0.000 claims description 66
- 238000000034 method Methods 0.000 claims description 41
- 206010028980 Neoplasm Diseases 0.000 claims description 33
- 150000003839 salts Chemical class 0.000 claims description 33
- 201000011510 cancer Diseases 0.000 claims description 31
- 125000000217 alkyl group Chemical group 0.000 claims description 30
- 238000006243 chemical reaction Methods 0.000 claims description 27
- 125000006367 bivalent amino carbonyl group Chemical group [H]N([*:1])C([*:2])=O 0.000 claims description 24
- 238000011282 treatment Methods 0.000 claims description 23
- 150000003573 thiols Chemical class 0.000 claims description 19
- 229910052739 hydrogen Inorganic materials 0.000 claims description 14
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 claims description 13
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 claims description 13
- 125000006850 spacer group Chemical group 0.000 claims description 13
- 125000000623 heterocyclic group Chemical group 0.000 claims description 11
- 238000002360 preparation method Methods 0.000 claims description 11
- 230000003287 optical effect Effects 0.000 claims description 10
- 229920001223 polyethylene glycol Polymers 0.000 claims description 10
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 claims description 10
- 125000003118 aryl group Chemical group 0.000 claims description 9
- 238000004519 manufacturing process Methods 0.000 claims description 9
- 239000008194 pharmaceutical composition Substances 0.000 claims description 9
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 9
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 claims description 8
- 239000002202 Polyethylene glycol Substances 0.000 claims description 8
- 229910052799 carbon Inorganic materials 0.000 claims description 8
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 claims description 8
- 125000001072 heteroaryl group Chemical group 0.000 claims description 8
- 150000004677 hydrates Chemical class 0.000 claims description 8
- 230000008569 process Effects 0.000 claims description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 7
- 125000003368 amide group Chemical group 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 6
- 125000001424 substituent group Chemical group 0.000 claims description 6
- DGFLYYUXGFFPQW-LJQANCHMSA-N 3-[[(1s)-1-(chloromethyl)-3-[5-(3-sulfanylpropanoylamino)-1h-indole-2-carbonyl]-1,2-dihydrobenzo[e]indol-5-yl]oxycarbonylamino]benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC(NC(=O)OC=2C3=CC=CC=C3C=3[C@H](CCl)CN(C=3C=2)C(=O)C=2NC3=CC=C(NC(=O)CCS)C=C3C=2)=C1 DGFLYYUXGFFPQW-LJQANCHMSA-N 0.000 claims description 5
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 125000000524 functional group Chemical group 0.000 claims description 5
- 239000003102 growth factor Substances 0.000 claims description 5
- 150000004820 halides Chemical class 0.000 claims description 5
- 229940088597 hormone Drugs 0.000 claims description 5
- 239000005556 hormone Substances 0.000 claims description 5
- 239000001257 hydrogen Substances 0.000 claims description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 5
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 claims description 4
- 108010074338 Lymphokines Proteins 0.000 claims description 4
- 102000008072 Lymphokines Human genes 0.000 claims description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 4
- 239000012634 fragment Substances 0.000 claims description 4
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 4
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 4
- 125000003396 thiol group Chemical group [H]S* 0.000 claims description 4
- GGYGQZXBVWNQCA-HXUWFJFHSA-N 3-[[(1s)-1-(chloromethyl)-3-[5-[3-(methyldisulfanyl)propanoylamino]-1h-indole-2-carbonyl]-1,2-dihydrobenzo[e]indol-5-yl]oxycarbonylamino]benzenesulfonic acid Chemical compound C12=CC=CC=C2C([C@H](CCl)CN2C(=O)C=3NC4=CC=C(C=C4C=3)NC(=O)CCSSC)=C2C=C1OC(=O)NC1=CC=CC(S(O)(=O)=O)=C1 GGYGQZXBVWNQCA-HXUWFJFHSA-N 0.000 claims description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims description 3
- 102000014150 Interferons Human genes 0.000 claims description 3
- 108010050904 Interferons Proteins 0.000 claims description 3
- 125000003342 alkenyl group Chemical group 0.000 claims description 3
- 125000000304 alkynyl group Chemical group 0.000 claims description 3
- 125000004103 aminoalkyl group Chemical group 0.000 claims description 3
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 claims description 3
- 150000001768 cations Chemical class 0.000 claims description 3
- 230000001268 conjugating effect Effects 0.000 claims description 3
- 125000003107 substituted aryl group Chemical group 0.000 claims description 3
- 125000001302 tertiary amino group Chemical group 0.000 claims description 3
- 229940088594 vitamin Drugs 0.000 claims description 3
- 235000013343 vitamin Nutrition 0.000 claims description 3
- 239000011782 vitamin Substances 0.000 claims description 3
- 229930003231 vitamin Natural products 0.000 claims description 3
- MHKBMNACOMRIAW-UHFFFAOYSA-N 2,3-dinitrophenol Chemical compound OC1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O MHKBMNACOMRIAW-UHFFFAOYSA-N 0.000 claims description 2
- ZMCHBSMFKQYNKA-UHFFFAOYSA-N 2-aminobenzenesulfonic acid Chemical class NC1=CC=CC=C1S(O)(=O)=O ZMCHBSMFKQYNKA-UHFFFAOYSA-N 0.000 claims description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 claims description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 claims description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 2
- 150000001408 amides Chemical class 0.000 claims description 2
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 2
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims description 2
- 229940047124 interferons Drugs 0.000 claims description 2
- 125000005346 substituted cycloalkyl group Chemical group 0.000 claims description 2
- 125000000542 sulfonic acid group Chemical group 0.000 claims description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 claims description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 claims description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 claims 3
- 125000002947 alkylene group Chemical group 0.000 claims 1
- 125000004122 cyclic group Chemical group 0.000 claims 1
- 150000002431 hydrogen Chemical class 0.000 claims 1
- 230000003301 hydrolyzing effect Effects 0.000 claims 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims 1
- 229940079593 drug Drugs 0.000 abstract description 57
- 230000001472 cytotoxic effect Effects 0.000 abstract description 24
- 231100000433 cytotoxic Toxicity 0.000 abstract description 22
- 230000001225 therapeutic effect Effects 0.000 abstract description 11
- 229940127089 cytotoxic agent Drugs 0.000 abstract description 10
- 239000002254 cytotoxic agent Substances 0.000 abstract description 10
- 231100000599 cytotoxic agent Toxicity 0.000 abstract description 10
- 239000003153 chemical reaction reagent Substances 0.000 abstract description 7
- 150000003568 thioethers Chemical class 0.000 abstract description 7
- 230000027455 binding Effects 0.000 abstract description 5
- 150000002019 disulfides Chemical class 0.000 abstract description 4
- 239000003972 antineoplastic antibiotic Substances 0.000 abstract description 2
- UOWVMDUEMSNCAV-UHFFFAOYSA-N antibiotic cc 1065 Chemical compound N1C=2C(OC)=C(O)C=3N(C(N)=O)CCC=3C=2C=C1C(=O)N1CCC(C=2C=3)=C1C(O)=C(OC)C=2NC=3C(=O)N1CC2CC22C1=CC(=O)C1=C2C(C)=CN1 UOWVMDUEMSNCAV-UHFFFAOYSA-N 0.000 abstract 1
- 210000004027 cell Anatomy 0.000 description 144
- 239000000562 conjugate Substances 0.000 description 44
- 239000000203 mixture Substances 0.000 description 42
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 31
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 27
- 230000003013 cytotoxicity Effects 0.000 description 23
- 231100000135 cytotoxicity Toxicity 0.000 description 23
- 239000000243 solution Substances 0.000 description 23
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 22
- 238000000338 in vitro Methods 0.000 description 22
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 20
- 238000001727 in vivo Methods 0.000 description 20
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 18
- 201000010099 disease Diseases 0.000 description 17
- 238000005481 NMR spectroscopy Methods 0.000 description 15
- 239000007864 aqueous solution Substances 0.000 description 15
- 229910000104 sodium hydride Inorganic materials 0.000 description 14
- 239000000427 antigen Substances 0.000 description 12
- 108091007433 antigens Proteins 0.000 description 12
- 102000036639 antigens Human genes 0.000 description 12
- 210000001185 bone marrow Anatomy 0.000 description 12
- 208000009329 Graft vs Host Disease Diseases 0.000 description 10
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- 239000002253 acid Substances 0.000 description 10
- 230000021615 conjugation Effects 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- 208000024908 graft versus host disease Diseases 0.000 description 10
- 239000000546 pharmaceutical excipient Substances 0.000 description 10
- 108090000765 processed proteins & peptides Proteins 0.000 description 10
- 239000011780 sodium chloride Substances 0.000 description 10
- 230000015572 biosynthetic process Effects 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- TVEHPTDUSKNJAT-OAQYLSRUSA-N n-[2-[(1s)-1-(chloromethyl)-5-hydroxy-1,2-dihydrobenzo[e]indole-3-carbonyl]-1h-indol-5-yl]-5-[3-(methyldisulfanyl)propanoylamino]-1h-indole-2-carboxamide Chemical compound C1=C(O)C2=CC=CC=C2C([C@H](CCl)C2)=C1N2C(=O)C1=CC2=CC(NC(=O)C=3NC4=CC=C(C=C4C=3)NC(=O)CCSSC)=CC=C2N1 TVEHPTDUSKNJAT-OAQYLSRUSA-N 0.000 description 9
- 210000002966 serum Anatomy 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 9
- 239000002585 base Substances 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 238000004128 high performance liquid chromatography Methods 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- 238000003786 synthesis reaction Methods 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 7
- 235000011054 acetic acid Nutrition 0.000 description 7
- 125000003277 amino group Chemical group 0.000 description 7
- 230000037396 body weight Effects 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 239000003085 diluting agent Substances 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- ZMXDDKWLCZADIW-YYWVXINBSA-N N,N-dimethylformamide-d7 Chemical compound [2H]C(=O)N(C([2H])([2H])[2H])C([2H])([2H])[2H] ZMXDDKWLCZADIW-YYWVXINBSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000002246 antineoplastic agent Substances 0.000 description 6
- 210000002798 bone marrow cell Anatomy 0.000 description 6
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 6
- 230000002401 inhibitory effect Effects 0.000 description 6
- 230000002147 killing effect Effects 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- 210000004881 tumor cell Anatomy 0.000 description 6
- KTLUMOZOWZSJFC-UHFFFAOYSA-N 3-azatetracyclo[7.5.0.02,6.012,14]tetradeca-1,3,5,7,9,11,13-heptaene Chemical group C1=C2C=CN=C2C2=C3C=C3C=CC2=C1 KTLUMOZOWZSJFC-UHFFFAOYSA-N 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- 102000008100 Human Serum Albumin Human genes 0.000 description 5
- 108091006905 Human Serum Albumin Proteins 0.000 description 5
- 241001529936 Murinae Species 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- 210000001744 T-lymphocyte Anatomy 0.000 description 5
- 229940041181 antineoplastic drug Drugs 0.000 description 5
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 5
- 229940092714 benzenesulfonic acid Drugs 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 239000000499 gel Substances 0.000 description 5
- 230000012010 growth Effects 0.000 description 5
- 238000011534 incubation Methods 0.000 description 5
- 230000003389 potentiating effect Effects 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 102000004196 processed proteins & peptides Human genes 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- 230000001988 toxicity Effects 0.000 description 5
- 231100000419 toxicity Toxicity 0.000 description 5
- HJVJLAOFAYPFHL-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-pyridin-2-ylpropanedithioate Chemical compound C=1C=CC=NC=1C(C)C(=S)SN1C(=O)CCC1=O HJVJLAOFAYPFHL-UHFFFAOYSA-N 0.000 description 4
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 208000023275 Autoimmune disease Diseases 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 241000102542 Kara Species 0.000 description 4
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 4
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 4
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 4
- 244000180577 Sambucus australis Species 0.000 description 4
- 235000018734 Sambucus australis Nutrition 0.000 description 4
- 206010052779 Transplant rejections Diseases 0.000 description 4
- 239000012062 aqueous buffer Substances 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 4
- 230000000875 corresponding effect Effects 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 229940127121 immunoconjugate Drugs 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 210000004698 lymphocyte Anatomy 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 239000003607 modifier Substances 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- 230000008685 targeting Effects 0.000 description 4
- 229940124597 therapeutic agent Drugs 0.000 description 4
- 238000002054 transplantation Methods 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 206010006187 Breast cancer Diseases 0.000 description 3
- 208000026310 Breast neoplasm Diseases 0.000 description 3
- 0 CC(N(*)c1cc(*)c(*)c(*)c1*)=O Chemical compound CC(N(*)c1cc(*)c(*)c(*)c1*)=O 0.000 description 3
- 108090000371 Esterases Proteins 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 3
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 3
- 108091005804 Peptidases Proteins 0.000 description 3
- 102000035195 Peptidases Human genes 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 3
- 108010029180 Sialic Acid Binding Ig-like Lectin 3 Proteins 0.000 description 3
- 102000001555 Sialic Acid Binding Ig-like Lectin 3 Human genes 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 3
- 241000700605 Viruses Species 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 239000000611 antibody drug conjugate Substances 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 3
- 238000002512 chemotherapy Methods 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 208000029742 colonic neoplasm Diseases 0.000 description 3
- 230000003111 delayed effect Effects 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 239000000796 flavoring agent Substances 0.000 description 3
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 208000020816 lung neoplasm Diseases 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 201000001441 melanoma Diseases 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000002062 proliferating effect Effects 0.000 description 3
- 235000019833 protease Nutrition 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- ZCOSUVZGFDGWFV-UHFFFAOYSA-N pyrrolo[2,3-e]indole Chemical group C1=CC2=NC=CC2=C2N=CC=C21 ZCOSUVZGFDGWFV-UHFFFAOYSA-N 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 231100000057 systemic toxicity Toxicity 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- VRDGQQTWSGDXCU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-iodoacetate Chemical compound ICC(=O)ON1C(=O)CCC1=O VRDGQQTWSGDXCU-UHFFFAOYSA-N 0.000 description 2
- VQZYZXLBKBUOHE-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(pyridin-2-yldisulfanyl)butanoate Chemical compound C=1C=CC=NC=1SSC(C)CC(=O)ON1C(=O)CCC1=O VQZYZXLBKBUOHE-UHFFFAOYSA-N 0.000 description 2
- WGMMKWFUXPMTRW-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-[(2-bromoacetyl)amino]propanoate Chemical compound BrCC(=O)NCCC(=O)ON1C(=O)CCC1=O WGMMKWFUXPMTRW-UHFFFAOYSA-N 0.000 description 2
- JSHOVKSMJRQOGY-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(pyridin-2-yldisulfanyl)butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCSSC1=CC=CC=N1 JSHOVKSMJRQOGY-UHFFFAOYSA-N 0.000 description 2
- GTBCXYYVWHFQRS-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(pyridin-2-yldisulfanyl)pentanoate Chemical compound C=1C=CC=NC=1SSC(C)CCC(=O)ON1C(=O)CCC1=O GTBCXYYVWHFQRS-UHFFFAOYSA-N 0.000 description 2
- GKSPIZSKQWTXQG-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[1-(pyridin-2-yldisulfanyl)ethyl]benzoate Chemical compound C=1C=C(C(=O)ON2C(CCC2=O)=O)C=CC=1C(C)SSC1=CC=CC=N1 GKSPIZSKQWTXQG-UHFFFAOYSA-N 0.000 description 2
- QJGHJWKPZQIOSN-UHFFFAOYSA-N (4-aminophenyl)methanesulfonic acid Chemical compound NC1=CC=C(CS(O)(=O)=O)C=C1 QJGHJWKPZQIOSN-UHFFFAOYSA-N 0.000 description 2
- OFFSPAZVIVZPHU-UHFFFAOYSA-N 1-benzofuran-2-carboxylic acid Chemical class C1=CC=C2OC(C(=O)O)=CC2=C1 OFFSPAZVIVZPHU-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- WCOPHOIZTFAWAU-UHFFFAOYSA-N 3-aminobenzenesulfonic acid;n-ethyl-n-propan-2-ylpropan-2-amine Chemical compound CCN(C(C)C)C(C)C.NC1=CC=CC(S(O)(=O)=O)=C1 WCOPHOIZTFAWAU-UHFFFAOYSA-N 0.000 description 2
- QHSXWDVVFHXHHB-UHFFFAOYSA-N 3-nitro-2-[(3-nitropyridin-2-yl)disulfanyl]pyridine Chemical class [O-][N+](=O)C1=CC=CN=C1SSC1=NC=CC=C1[N+]([O-])=O QHSXWDVVFHXHHB-UHFFFAOYSA-N 0.000 description 2
- FSGFVDDKFPDQKT-UHFFFAOYSA-N 5-[3-(methyldisulfanyl)propanoylamino]-1h-indole-2-carboxylic acid Chemical compound CSSCCC(=O)NC1=CC=C2NC(C(O)=O)=CC2=C1 FSGFVDDKFPDQKT-UHFFFAOYSA-N 0.000 description 2
- VWLGQKLHWIVCCZ-UHFFFAOYSA-N 53992-33-9 Chemical compound OS(=O)(=O)CC1=CC=C([N+]([O-])=O)C=C1 VWLGQKLHWIVCCZ-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- 241000224489 Amoeba Species 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 208000035143 Bacterial infection Diseases 0.000 description 2
- 206010048396 Bone marrow transplant rejection Diseases 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 206010011831 Cytomegalovirus infection Diseases 0.000 description 2
- 230000004568 DNA-binding Effects 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- 108010007979 Glycocholic Acid Proteins 0.000 description 2
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 2
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 2
- 208000031886 HIV Infections Diseases 0.000 description 2
- 208000037357 HIV infectious disease Diseases 0.000 description 2
- 206010019315 Heart transplant rejection Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- HCUARRIEZVDMPT-UHFFFAOYSA-N Indole-2-carboxylic acid Chemical compound C1=CC=C2NC(C(=O)O)=CC2=C1 HCUARRIEZVDMPT-UHFFFAOYSA-N 0.000 description 2
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 206010023439 Kidney transplant rejection Diseases 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 206010024715 Liver transplant rejection Diseases 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 206010051604 Lung transplant rejection Diseases 0.000 description 2
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- 208000030852 Parasitic disease Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- 206010038389 Renal cancer Diseases 0.000 description 2
- 229920005654 Sephadex Polymers 0.000 description 2
- 239000012507 Sephadex™ Substances 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 102000011923 Thyrotropin Human genes 0.000 description 2
- 108010061174 Thyrotropin Proteins 0.000 description 2
- 102000004338 Transferrin Human genes 0.000 description 2
- 108090000901 Transferrin Proteins 0.000 description 2
- 208000036142 Viral infection Diseases 0.000 description 2
- 238000002679 ablation Methods 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 230000001464 adherent effect Effects 0.000 description 2
- BYRVKDUQDLJUBX-JJCDCTGGSA-N adozelesin Chemical compound C1=CC=C2OC(C(=O)NC=3C=C4C=C(NC4=CC=3)C(=O)N3C[C@H]4C[C@]44C5=C(C(C=C43)=O)NC=C5C)=CC2=C1 BYRVKDUQDLJUBX-JJCDCTGGSA-N 0.000 description 2
- 229950004955 adozelesin Drugs 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 230000000735 allogeneic effect Effects 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000003098 androgen Substances 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 229940049595 antibody-drug conjugate Drugs 0.000 description 2
- 239000012736 aqueous medium Substances 0.000 description 2
- 239000003125 aqueous solvent Substances 0.000 description 2
- 150000001504 aryl thiols Chemical class 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 208000022362 bacterial infectious disease Diseases 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 238000010322 bone marrow transplantation Methods 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 description 2
- 229930195731 calicheamicin Natural products 0.000 description 2
- BBZDXMBRAFTCAA-AREMUKBSSA-N carzelesin Chemical compound C1=2NC=C(C)C=2C([C@H](CCl)CN2C(=O)C=3NC4=CC=C(C=C4C=3)NC(=O)C3=CC4=CC=C(C=C4O3)N(CC)CC)=C2C=C1OC(=O)NC1=CC=CC=C1 BBZDXMBRAFTCAA-AREMUKBSSA-N 0.000 description 2
- 229950007509 carzelesin Drugs 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 239000003431 cross linking reagent Substances 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 208000037765 diseases and disorders Diseases 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 239000002158 endotoxin Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 210000003495 flagella Anatomy 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- RFDAIACWWDREDC-FRVQLJSFSA-N glycocholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 RFDAIACWWDREDC-FRVQLJSFSA-N 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 125000005179 haloacetyl group Chemical group 0.000 description 2
- 238000003306 harvesting Methods 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 2
- 238000013415 human tumor xenograft model Methods 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 2
- 229940051026 immunotoxin Drugs 0.000 description 2
- 239000002596 immunotoxin Substances 0.000 description 2
- 230000002637 immunotoxin Effects 0.000 description 2
- 231100000608 immunotoxin Toxicity 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 201000010982 kidney cancer Diseases 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- JJTUDXZGHPGLLC-UHFFFAOYSA-N lactide Chemical compound CC1OC(=O)C(C)OC1=O JJTUDXZGHPGLLC-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000006210 lotion Substances 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 206010025135 lupus erythematosus Diseases 0.000 description 2
- 230000001926 lymphatic effect Effects 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 239000007923 nasal drop Substances 0.000 description 2
- 229940100662 nasal drops Drugs 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- QCGPFSMQZNLBCA-UHFFFAOYSA-N s-(4-nitrophenyl) ethanethioate Chemical compound CC(=O)SC1=CC=C([N+]([O-])=O)C=C1 QCGPFSMQZNLBCA-UHFFFAOYSA-N 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 201000004409 schistosomiasis Diseases 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000008227 sterile water for injection Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 108700012359 toxins Proteins 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 239000012581 transferrin Substances 0.000 description 2
- 229940086542 triethylamine Drugs 0.000 description 2
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- 230000009385 viral infection Effects 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- PPODAMWXCOLZQP-DDWIOCJRSA-N (1S)-3-amino-1-(chloromethyl)-1,2-dihydrobenzo[e]indol-5-ol hydrochloride Chemical compound Cl.NN1C[C@@H](CCl)c2c1cc(O)c1ccccc21 PPODAMWXCOLZQP-DDWIOCJRSA-N 0.000 description 1
- JWDFQMWEFLOOED-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(pyridin-2-yldisulfanyl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCSSC1=CC=CC=N1 JWDFQMWEFLOOED-UHFFFAOYSA-N 0.000 description 1
- SKHUUKXTRZZCIO-UHFFFAOYSA-N (4-piperidin-1-ylpiperidin-1-yl) carbamate Chemical compound C1CN(OC(=O)N)CCC1N1CCCCC1 SKHUUKXTRZZCIO-UHFFFAOYSA-N 0.000 description 1
- IANQTJSKSUMEQM-UHFFFAOYSA-N 1-benzofuran Chemical group C1=CC=C2OC=CC2=C1 IANQTJSKSUMEQM-UHFFFAOYSA-N 0.000 description 1
- WMZYAQQKYLKWGI-UHFFFAOYSA-N 1-hydroxypyrrolidine-2,5-dione 4-(pyridin-2-yldisulfanyl)butanoic acid Chemical compound ON1C(=O)CCC1=O.OC(=O)CCCSSC1=CC=CC=N1 WMZYAQQKYLKWGI-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- LRQWVXZTIVYYHE-UHFFFAOYSA-N 2-(5-nitropyridin-2-yl)butanedithioic acid Chemical compound CCC(C1=NC=C(C=C1)[N+](=O)[O-])C(=S)S LRQWVXZTIVYYHE-UHFFFAOYSA-N 0.000 description 1
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- OWDQCSBZQVISPN-UHFFFAOYSA-N 2-[(2,5-dioxopyrrolidin-1-yl)amino]-4-(2-iodoacetyl)benzoic acid Chemical compound OC(=O)C1=CC=C(C(=O)CI)C=C1NN1C(=O)CCC1=O OWDQCSBZQVISPN-UHFFFAOYSA-N 0.000 description 1
- GHYWXMNHUWSLHA-UHFFFAOYSA-N 3-(methyldisulfanyl)propanoic acid Chemical compound CSSCCC(O)=O GHYWXMNHUWSLHA-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- LKNPLDRVWHXGKZ-UHFFFAOYSA-N 3-nitro-1h-pyridine-2-thione Chemical compound [O-][N+](=O)C1=CC=CN=C1S LKNPLDRVWHXGKZ-UHFFFAOYSA-N 0.000 description 1
- CSDQQAQKBAQLLE-UHFFFAOYSA-N 4-(4-chlorophenyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine Chemical compound C1=CC(Cl)=CC=C1C1C(C=CS2)=C2CCN1 CSDQQAQKBAQLLE-UHFFFAOYSA-N 0.000 description 1
- SLMVEZKWNOGJPD-UHFFFAOYSA-N 4-(pyridin-2-yldisulfanyl)butanoic acid Chemical compound OC(=O)CCCSSC1=CC=CC=N1 SLMVEZKWNOGJPD-UHFFFAOYSA-N 0.000 description 1
- VOLRSQPSJGXRNJ-UHFFFAOYSA-N 4-nitrobenzyl bromide Chemical compound [O-][N+](=O)C1=CC=C(CBr)C=C1 VOLRSQPSJGXRNJ-UHFFFAOYSA-N 0.000 description 1
- NKFIBMOQAPEKNZ-UHFFFAOYSA-N 5-amino-1h-indole-2-carboxylic acid Chemical compound NC1=CC=C2NC(C(O)=O)=CC2=C1 NKFIBMOQAPEKNZ-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 240000000662 Anethum graveolens Species 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 108010051152 Carboxylesterase Proteins 0.000 description 1
- 102000013392 Carboxylesterase Human genes 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 1
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 1
- 206010019851 Hepatotoxicity Diseases 0.000 description 1
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 1
- 108090001117 Insulin-Like Growth Factor II Proteins 0.000 description 1
- 102000048143 Insulin-Like Growth Factor II Human genes 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- XUMBMVFBXHLACL-UHFFFAOYSA-N Melanin Natural products O=C1C(=O)C(C2=CNC3=C(C(C(=O)C4=C32)=O)C)=C2C4=CNC2=C1C XUMBMVFBXHLACL-UHFFFAOYSA-N 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- BKAYIFDRRZZKNF-VIFPVBQESA-N N-acetylcarnosine Chemical compound CC(=O)NCCC(=O)N[C@H](C(O)=O)CC1=CN=CN1 BKAYIFDRRZZKNF-VIFPVBQESA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108010020346 Polyglutamic Acid Proteins 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000009052 Precursor T-Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 125000000066 S-methyl group Chemical group [H]C([H])([H])S* 0.000 description 1
- 239000012506 Sephacryl® Substances 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- 102000013275 Somatomedins Human genes 0.000 description 1
- 241000133426 Streptomyces zelensis Species 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 208000029052 T-cell acute lymphoblastic leukemia Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 241001061127 Thione Species 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000006747 Transforming Growth Factor alpha Human genes 0.000 description 1
- 101800004564 Transforming growth factor alpha Proteins 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 108010003533 Viral Envelope Proteins Proteins 0.000 description 1
- 108010067390 Viral Proteins Proteins 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- SWNGQBWPDLCTSG-UHFFFAOYSA-N [[1-(2,5-dioxopyrrolidin-1-yl)-2h-pyridin-3-yl]disulfanyl] propanoate Chemical group C1C(SSOC(=O)CC)=CC=CN1N1C(=O)CCC1=O SWNGQBWPDLCTSG-UHFFFAOYSA-N 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 201000011186 acute T cell leukemia Diseases 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910000102 alkali metal hydride Inorganic materials 0.000 description 1
- 150000008046 alkali metal hydrides Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 150000001356 alkyl thiols Chemical class 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229940030486 androgens Drugs 0.000 description 1
- 229940094957 androgens and estrogen Drugs 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 238000003491 array Methods 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 238000009395 breeding Methods 0.000 description 1
- 230000001488 breeding effect Effects 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- KDPAWGWELVVRCH-UHFFFAOYSA-M bromoacetate Chemical compound [O-]C(=O)CBr KDPAWGWELVVRCH-UHFFFAOYSA-M 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 150000001720 carbohydrates Chemical group 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 229940047120 colony stimulating factors Drugs 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000009849 deactivation Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 229940009976 deoxycholate Drugs 0.000 description 1
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- FSBVERYRVPGNGG-UHFFFAOYSA-N dimagnesium dioxido-bis[[oxido(oxo)silyl]oxy]silane hydrate Chemical compound O.[Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O FSBVERYRVPGNGG-UHFFFAOYSA-N 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 229910000397 disodium phosphate Inorganic materials 0.000 description 1
- 235000019800 disodium phosphate Nutrition 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- XBRDBODLCHKXHI-UHFFFAOYSA-N epolamine Chemical compound OCCN1CCCC1 XBRDBODLCHKXHI-UHFFFAOYSA-N 0.000 description 1
- 229950008932 epolamine Drugs 0.000 description 1
- 239000003687 estradiol congener Substances 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- DUYAAUVXQSMXQP-UHFFFAOYSA-N ethanethioic S-acid Chemical compound CC(S)=O DUYAAUVXQSMXQP-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- LIQODXNTTZAGID-OCBXBXKTSA-N etoposide phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 LIQODXNTTZAGID-OCBXBXKTSA-N 0.000 description 1
- 229960000752 etoposide phosphate Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 229940014144 folate Drugs 0.000 description 1
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 125000004970 halomethyl group Chemical group 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 231100001261 hazardous Toxicity 0.000 description 1
- 230000007686 hepatotoxicity Effects 0.000 description 1
- 231100000304 hepatotoxicity Toxicity 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- 102000056982 human CD33 Human genes 0.000 description 1
- 150000002440 hydroxy compounds Chemical class 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 125000002249 indol-2-yl group Chemical group [H]C1=C([H])C([H])=C2N([H])C([*])=C([H])C2=C1[H] 0.000 description 1
- 125000000814 indol-3-yl group Chemical group [H]C1=C([H])C([H])=C2N([H])C([H])=C([*])C2=C1[H] 0.000 description 1
- 125000004531 indol-5-yl group Chemical group [H]N1C([H])=C([H])C2=C([H])C(*)=C([H])C([H])=C12 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019792 magnesium silicate Nutrition 0.000 description 1
- 229910052919 magnesium silicate Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 210000002752 melanocyte Anatomy 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229960001047 methyl salicylate Drugs 0.000 description 1
- DVSDBMFJEQPWNO-UHFFFAOYSA-N methyllithium Chemical compound C[Li] DVSDBMFJEQPWNO-UHFFFAOYSA-N 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229940125645 monoclonal antibody drug Drugs 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 230000003071 parasitic effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 239000000813 peptide hormone Substances 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 125000006245 phosphate protecting group Chemical group 0.000 description 1
- 230000000886 photobiology Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 229920002643 polyglutamic acid Polymers 0.000 description 1
- 229920002503 polyoxyethylene-polyoxypropylene Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 231100000654 protein toxin Toxicity 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000005030 pyridylthio group Chemical group N1=C(C=CC=C1)S* 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000012217 radiopharmaceutical Substances 0.000 description 1
- 229940121896 radiopharmaceutical Drugs 0.000 description 1
- 230000002799 radiopharmaceutical effect Effects 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 239000003488 releasing hormone Substances 0.000 description 1
- 238000001226 reprecipitation Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 150000003902 salicylic acid esters Chemical class 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000003270 steroid hormone Substances 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229940014800 succinic anhydride Drugs 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- CNHYKKNIIGEXAY-UHFFFAOYSA-N thiolan-2-imine Chemical compound N=C1CCCS1 CNHYKKNIIGEXAY-UHFFFAOYSA-N 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 125000005413 thiopyridyl group Chemical group 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- KBMBVTRWEAAZEY-UHFFFAOYSA-N trisulfane Chemical compound SSS KBMBVTRWEAAZEY-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 238000002211 ultraviolet spectrum Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Images
















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/04—Amoebicides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/10—Anthelmintics
- A61P33/12—Schistosomicides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/58—[b]- or [c]-condensed
- C07D209/60—Naphtho [b] pyrroles; Hydrogenated naphtho [b] pyrroles
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Immunology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Tropical Medicine & Parasitology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Virology (AREA)
- Physical Education & Sports Medicine (AREA)
- Transplantation (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Indole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
본원에는 설폰산 함유 페닐 카바메이트를 함유하는 절단 가능한 보호 기를 갖는 항종양 항생제 CC-1065의 유사체의 프로드럭이 기재되어 있는데, 상기 보호 기는 프로드럭에 증강된 수 용해도를 부여해 주고, 본 발명의 프로드럭은 항체와 같은 세포 결합성 시약과 접합할 수 있는 잔기, 예를 들어 설파이드 또는 디설파이드를 갖고 있다. 상기 프로드럭 접합체의 치료적 용도가 또한 기재되어 있는데, 이러한 세포독성제의 프로드럭은 이들이 세포독성 프로드럭을 특정 세포 집단으로 전달시켜 표적화 방식으로 세포독성 약물로 효소적 전환되기 때문에 치료적 용도를 지니고 있다.Described herein is a prodrug of an analog of the antitumor antibiotic CC-1065 having a cleavable protecting group containing sulfonic acid containing phenyl carbamate, which gives the prodrug enhanced water solubility, Drugs have residues, such as sulfides or disulfides, that can be conjugated with cell binding reagents such as antibodies. Therapeutic uses of such prodrug conjugates are also described, prodrugs of such cytotoxic agents are intended for therapeutic use because they deliver cytotoxic prodrugs to specific cell populations and are enzymatically converted to cytotoxic drugs in a targeted manner. I have it.
Description
본 발명은 세포독성제의 신규한 프로드럭 (prodrug) 및 그의 치료적 용도에 관한 것이다. 보다 구체적으로 언급하면, 본 발명은 CC-1065의 유사체이고 세포 결합제와의 화학적 연결을 위한 잔기와 생체 내에서 절단되는 보호 기 둘 다를 포함하는, 세포독성제의 신규한 프로드럭에 관한 것이다. 이러한 프로드럭은 세포 결합제와 화학적으로 연결되어, 생체 내에서 활성화 및 방출될 수 있고 특정 세포 집단에 표적화 방식으로 전달될 수 있는 치료제를 제공할 수 있다.The present invention relates to novel prodrugs and therapeutic uses thereof of cytotoxic agents. More specifically, the present invention relates to novel prodrugs of cytotoxic agents, which are analogs of CC-1065 and contain both residues for chemical linkage with cell binding agents and protecting groups cleaved in vivo. Such prodrugs can be chemically linked with cell binding agents to provide therapeutic agents that can be activated and released in vivo and delivered in a targeted manner to a particular cell population.
모노클로날 항체-약물 접합체를 사용하여 종양 세포를 표적화하는 것에 관한 많은 보고서가 출판되었다 [참고: Sela et al, in Immunoconjugates, pp. 189-216 (C. Vogel, ed. 1987); Ghose et al, in Targeted Drugs, pp. 1-22 (E. Goldberg, ed. 1983); Diener et al, in Antibody Mediated Delivery Systems, pp. 1-23 (J. Rodwell, ed. 1988); Pietersz et al, in Antibody Mediated Delivery Systems, pp. 25-53 (J. Rodwell, ed. 1988); Bumol et al, in Antibody Mediated Delivery Systems, pp. 55-79 (J. Rodwell, ed. 1988); G.A. Pietersz & K.Krauer, 2 J. Drug Targeting, 183-215 (1994); R. V. J. Chari, 31 Adv. Drug Delivery Revs., 89-104 (1998); W.A. Blattler & R.V.J. Chari, in Anticancer Agents, Frontiers in Cancer Chemotherapy, 317-338, ACS Symposium Series 796; and I. Ojima et al eds, American Chemical Society 2001]. 메토트렉세이트, 다우노루비신, 독소루비신, 빈크리스틴, 빈블라스틴, 멜팔란, 미토마이신 C, 클로람부실, 칼리케아미신 및 마이탄시노이드 등의 세포독성 약물을 각종 뮤린 (murine) 모노클로날 항체에 접합시켜 왔다. 몇몇 경우에는, 약물 분자를 혈청 알부민 [참고: Garnett et al, 46 Cancer Res. 2407-2412 (1986); Ohkawa et al, 23 Cancer Immunol. Immunother. 81-86 (1986); Endo et al, 47 Cancer Res. 1076-1080 (1980)], 덱스트란 [참고: Hurwitz et al, 2 Appl. Biochem. 25-35 (1980); Manabi et al, 34 Biochem. Pharmacol. 289-291 (1985); Dillman et al, 46 Cancer Res. 4886-4891 (1986); and Shoval et al, 85 Proc. Natl. Acad. Sci. U.S.A. 8276-8280 (1988)], 또는 폴리글루탐산 [참고: Tsukada et al, 73 J. Natl. Canc. Inst. 721-729 (1984); Kato et al, 27 J. Med. Chem. 1602-1607 (1984); Tsukada et al, 52 Br. J. Cancer 111-116 (1985)]과 같은 중간 운반체 분자를 통하여 항체 분자에 접합시켰다.Many reports have been published on targeting tumor cells using monoclonal antibody-drug conjugates. Sela et al, in Immunoconjugates , pp. 189-216 (C. Vogel, ed. 1987); Ghose et al, in Targeted Drugs , pp. 1-22 (E. Goldberg, ed. 1983); Diener et al, in Antibody Mediated Delivery Systems , pp. 1-23 (J. Rodwell, ed. 1988); Pietersz et al, in Antibody Mediated Delivery Systems , pp. 25-53 (J. Rodwell, ed. 1988); Bumol et al, in Antibody Mediated Delivery Systems , pp. 55-79 (J. Rodwell, ed. 1988); GA Pietersz & K. Krarau, 2 J. Drug Targeting , 183-215 (1994); RVJ Chari, 31 Adv. Drug Delivery Revs., 89-104 (1998); WA Blattler & RVJ Chari, in Anticancer Agents , Frontiers in Cancer Chemotherapy , 317-338, ACS Symposium Series 796; and I. Ojima et al eds, American Chemical Society 2001]. Cytotoxic drugs such as methotrexate, daunorubicin, doxorubicin, vincristine, vinblastine, melphalan, mitomycin C, chlorambucil, calicheamicin, and maytansinoids can be applied to various murine monoclonal antibodies. It has been bonded. In some cases, the drug molecule is described as serum albumin [Ganett et al, 46 Cancer Res . 2407-2412 (1986); Ohkawa et al, 23 Cancer Immunol. Immunother . 81-86 (1986); Endo et al, 47 Cancer Res . 1076-1080 (1980)], dextran [Hurwitz et al, 2 Appl. Biochem . 25-35 (1980); Manabi et al, 34 Biochem. Pharmacol . 289-291 (1985); Dillman et al, 46 Cancer Res . 4886-4891 (1986); and Shoval et al, 85 Proc. Natl. Acad. Sci. USA 8276-8280 (1988)], or polyglutamic acid (Tsukada et al, 73 J. Natl. Canc. Inst. 721-729 (1984); Kato et al, 27 J. Med. Chem . 1602-1607 (1984); Tsukada et al, 52 Br. J. Cancer 111-116 (1985) to an antibody molecule via an intermediate carrier molecule.
이러한 면역 접합체를 제조하기 위한 광범위한 링커 어레이가 현재 입수 가능하며 이에는 절단 가능한 링커와 절단 가능하지 않은 링커 둘 다가 포함된다. 그러나 시험관내 세포독성 시험에서는, 항체-약물 접합체가 접합되지 않은 자유 약물과 동일한 세포독성 효력을 거의 발휘하지 않는 것으로 밝혀졌다. 이는 약물 분자가 접합된 항체로부터 방출되는 기전이 극히 비효율적이란 사실을 제안하고 있다. 면역독소 분야에 있어서의 초기 연구 결과는 모노클로날 항체와 촉매적 활성 단백질 독소 간의 디설파이드 브릿지를 통하여 형성된 접합체가 기타 링커를 함유하는 접합체보다 더 세포독성인 것으로 밝혀졌다 [참고: Lambert et al, 260 J. Biol. Chem. 12035-12041 (1985); Lambert et al, in Immunotoxins 175-209 (A. Frankel, ed. 1988); Ghetie et al, 48 Cancer Res. 2610-2617 (1988)]. 이러한 개선된 세포독성은 환원 글루타치온이 세포내에 고 농도로 존재하여 항체 분자와 독소 간의 디설파이드 결합을 효율적으로 절단시키는데 일조하였기 때문이다. 마이탄시노이드 (maytansinoid)와 칼리케아미신 (calicheamicin)은 모노클로날 항체를 디설파이드 결합을 통하여 연결시킨 고도의 세포독성 약물의 그 첫번째 예이다. 이들 약물의 항체 접합체는 시험관 내에서 강력한 효력을 보유하고 있고 마우스의 인간 종양 이종-이식편 모델에서 뛰어난 항종양 활성을 보유하고 있는 것으로 밝혀졌다 [참고: R. V. J. Chari et al., 52 Cancer Res., 127-131 (1992); C. Liu et al., 93, Proc. Natl. Acad. Sci., 8618-8623 (1996); L.M. Hinman et al., 53, Cancer Res., 3536-3542 (1993); and P.R. Hamann et al, 13, BioConjugate Chem., 40-46 (2002)].A wide range of linker arrays for making such immunoconjugates are currently available, including both cleavable and non-cleavable linkers. However, in vitro cytotoxicity tests have shown that antibody-drug conjugates hardly exert the same cytotoxic effects as unconjugated free drugs. This suggests that the mechanism by which drug molecules are released from conjugated antibodies is extremely inefficient. Initial research in the field of immunotoxins revealed that conjugates formed through disulfide bridges between monoclonal antibodies and catalytically active protein toxins are more cytotoxic than conjugates containing other linkers. Lambert et al, 260 J. Biol. Chem . 12035-12041 (1985); Lambert et al, in Immunotoxins 175-209 (A. Frankel, ed. 1988); Ghetie et al, 48 Cancer Res . 2610-2617 (1988). This improved cytotoxicity was due to the presence of high concentrations of reduced glutathione in the cells, which helped to efficiently cleave the disulfide bonds between the antibody molecule and the toxin. Maytansinoids and calicheamicin are the first examples of highly cytotoxic drugs that link monoclonal antibodies via disulfide bonds. The antibody conjugates of these drugs have been shown to have potent potency in vitro and to have excellent antitumor activity in the human tumor xenograft model of mice. RVJ Chari et al., 52 Cancer Res ., 127 -131 (1992); C. Liu et al., 93, Proc. Natl. Acad. Sci. 8618-8623 (1996); LM Hinman et al., 53, Cancer Res ., 3536-3542 (1993); and PR Hamann et al, 13, BioConjugate Chem ., 40-46 (2002).
이러한 세포독성 접합체를 제조하는 데에 있어서 관심을 끄는 후보 작용제가 CC-1065인데, 이는 스트렙토마이세스 젤렌시스 (Streptomyces zelensis) 배양액으로부터 분리시킨 강력한 항종양 항생제이다. CC-1065는 흔히 사용되고 있는 항암 약물, 예를 들어 독소루비신, 메토트렉세이트 및 빈크리스틴보다 시험관 내에서의 효력이 약 1000배 이상 더 강력하다 [참고: B.K. Bhuyan et al., Cancer Res., 42, 3532-3537 (1982)]. CC-1065의 구조 (화합물 1, 도 1A)가 x-선 결정학에 의해 결 정되었다 [참고: Martin, D. G. et al, 33 J. Antibiotics 902-903 (1980), and Chidester, C. G., et al, 103 J. Am. Chem. Soc. 7629-7635 (1981)]. CC-1065 분자는 아미드 결합에 의해 연결된 3개의 치환된 피롤로인돌 잔기로 이루어진다. "A" 소단위체는 분자 내에 단지 1개의 비대칭 탄소 만을 함유하는 시클로프로필 환을 갖고 있다. 이들 탄소의 상대적 입체 배치만을 x-선 데이터로부터 얻을 수 있지만, 절대적 입체 배치는 키랄 시약으로서 DNA를 사용함으로써 3b-R, 4a-S로서 추론되었다 [참고: Hurley, L. H. et al, 226 Science 843-844 (1984)]. CC-1065의 "B" 및 "C" 소단위체는 동일한 피롤로인돌 잔기이다.A candidate of interest in preparing such cytotoxic conjugates is CC-1065, which is a Streptomyces zelensis ) is a potent antitumor antibiotic isolated from culture. CC-1065 is about 1000 times more potent in vitro than commonly used anticancer drugs such as doxorubicin, methotrexate and vincristine [BK Bhuyan et al., Cancer Res ., 42, 3532-3537 (1982). The structure of CC-1065 (
CC-1065의 세포독성 효력은 그의 알킬화 활성과 그의 DNA-결합 활성 또는 DNA-삽입 활성과 상관이 있어 왔다. 이들 두 가지 활성은 해당 분자의 별개의 부분에 존재한다. 즉, 알킬화 활성은 시클로프로파피롤로인돌 (CPI) 소단위체 내에 함유되어 있고 DNA-결합 활성은 2개의 피롤로인돌 소단위체 내에 존재한다 (도 1A).The cytotoxic potency of CC-1065 has been correlated with its alkylation activity and its DNA-binding activity or DNA-insertion activity. These two activities are in separate parts of the molecule. That is, alkylation activity is contained in cyclopropyrroloindole (CPI) subunits and DNA-binding activity is present in two pyrroloindole subunits ( FIG. 1A ).
그러나, CC-1065가 세포독성제로서 흥미를 끄는 특정의 특징을 지니고 있긴 하지만, 이를 치료적으로 사용하기에는 한계가 있다. CC-1065를 마우스에게 투여하면 지연형 간독성이 유발되어 12.5 ㎍/kg을 1회 정맥내 투여한 후 50일째에는 사망에 이르게 되었다 [참고: V. L. Reynolds et al., J. Antibiotics, XXIX, 319-334 (1986)]. 이로 인해 지연형 독성을 유발시키지 않는 유사체를 개발하고자 하는 노력에 박차를 가하게 되었고, CC-1065를 모델로 한 보다 간단한 유사체의 합성이 보고되었다 [참고: M.A. Warpehoski et al., J. Med. Chem., 31, 590-603 (1988)]. 또 다른 일련의 유사체에서는, CPI 잔기를 시클로프로파벤즈인돌 (CBI) 잔기로 대체시켰다 [참고: D.L. Boger et al., J. Org. Chem., 55, 5823-5833, (1990), D.L. Boger et al., BioOrg. Med. Chem. Lett., 1, 115-120 (1991)]. 이들 화합물은 마우스 내에서 지연형 독성을 유발시키지 않으면서도 모 약물의 강력한 시험관내 효력을 유지하고 있다. CC-1065와 마찬가지로 이들 화합물은 공유 방식으로 DNA의 작은 홈 (minor groove)과 결합하여 세포 사멸을 유발시키는 알킬화제이다. 그러나, 가장 전도 유망한 유사체인 아도젤레신 (Adozelesin)과 카르젤레신 (Carzelesin)을 임상적으로 평가한 결과는 실망스러웠다 [참고: B.F. Foster et al., Investigational New Drugs, 13, 321-326 (1996); I. Wolff et al., Clin. Cancer Res., 2, 1717-1723 (1996)]. 이들 약물은 전신 독성이 높기 때문에 충분치 못한 치료 효과를 나타내었다.However, although CC-1065 has certain characteristics that are of interest as cytotoxic agents, there are limits to their therapeutic use. Administration of CC-1065 to mice induced delayed hepatotoxicity, which resulted in death 50 days after 12.5 μg / kg intravenous administration [VL Reynolds et al., J. Antibiotics , XXIX, 319- 334 (1986)]. This has spurred efforts to develop analogs that do not cause delayed toxicity, and the synthesis of simpler analogs modeled after CC-1065 has been reported. MA Warpehoski et al., J. Med. Chem. , 31, 590-603 (1988). In another series of analogues, CPI residues were replaced with cyclopropabenzindole (CBI) residues. See DL Boger et al., J. Org. Chem ., 55, 5823-5833, (1990), DL Boger et al., BioOrg. Med. Chem. Lett. , 1, 115-120 (1991). These compounds maintain the potent in vitro potency of the parent drug without causing delayed toxicity in mice. Like CC-1065, these compounds are alkylating agents that bind to the minor grooves of DNA in a covalent manner, causing cell death. However, the clinical evaluation of the most promising analogs Adozelesin and Carzelesin was disappointing [BF Foster et al., Investigational New Drugs , 13, 321-326 (1996). ); I. Wolff et al., Clin. Cancer Res. , 2, 1717-1723 (1996). These drugs exhibited insufficient therapeutic effects because of their high systemic toxicity.
CC-1065 유사체의 치료 효능은 종양 부위로의 표적화 전달을 통하여 생체내 분포를 변화시켜 비-표적화 조직에 대한 독성을 보다 저하시키고, 이로써 전신 독성을 저하시킴으로써 상당히 개선시킬 수 있다. 이러한 목표를 달성하기 위해, 종양 세포를 특이적으로 표적으로 하는 세포 결합제와 CC-1065의 유사체 및 유도체와의 접합체가 보고되었다 [참고: 미국 특허 제5,475,092호; 제5,585,499호; 제5,846,545호]. 이들 접합체는 전형적으로, 시험관 내에서 높은 표적-특이적 세포독성을 나타내고, 마우스의 인간 종양 이종-이식편 모델에서 뛰어난 항종양 활성을 나타내었다 [참고: R.V. J. Chari et al., Cancer Res., 55, 4079-4084 (1995)].The therapeutic efficacy of the CC-1065 analog can be significantly improved by altering the distribution in vivo through targeted delivery to the tumor site, thereby further reducing toxicity to non-targeted tissue, thereby lowering systemic toxicity. To achieve this goal, conjugates of cell binders that specifically target tumor cells with analogs and derivatives of CC-1065 have been reported. See US Pat. No. 5,475,092; No. 5,585,499; 5,846,545]. These conjugates typically exhibited high target-specific cytotoxicity in vitro and excellent antitumor activity in human tumor xenograft models of mice. See RVJ Chari et al., Cancer Res ., 55, 4079-4084 (1995)].
세포 결합제는 전형적으로 수성 매질에서만 가용성이고, 통상 수용액에 저장 된다. 따라서, 이들 유사체는 세포 결합제와의 효율적인 반응과 수용액 중에서의 후속 제형화를 허용하는 데에 충분한 수 용해도를 지니고 있어야 한다. 또한, 세포 결합제 접합체가 유용한 저장 수명을 가지기 위해서는, 이들 세포 결합제와 연결되는 CC-1065 유사체가 수용액 중에서 장기간 동안 안정적인 것이 중요하다.Cell binders are typically soluble only in aqueous media and are usually stored in aqueous solution. Therefore, these analogs should have sufficient water solubility to allow efficient reaction with the cell binder and subsequent formulation in aqueous solution. In addition, for cell binder conjugates to have a useful shelf life, it is important that CC-1065 analogs linked with these cell binders are stable for a long time in aqueous solution.
지금까지 보고된 CC-1065 유사체 [참고: 예를 들어, 도 1B 및 1C]는 거의 수불용성이다. CC-1065 유사체의 불용성으로 인해, 현재 세포 결합제와의 접합 반응은 극히 묽은 수용액에서 수행되어야만 한다. 따라서, 이들 프로드럭은 모 약물과 비교해서 수 용해도가 증강되어야 한다.The CC-1065 analogs reported so far [see, eg, FIGS. 1B and 1C ] are almost water insoluble. Due to the insolubility of the CC-1065 analogues, conjugation reactions with current cell binders must be performed in extremely dilute aqueous solutions. Therefore, these prodrugs should have enhanced water solubility compared to the parent drug.
또한, 지금까지 보고된 CC-1065 유사체는 다음 이유로 인해 수용액 중에서 다소 불안정하다. 약물의 seco-형태가 자발적으로 시클로프로필 형태로 전환된 다음, DNA가 존재하는 경우에는 이를 알킬화시킬 수도 있다. 그러나, 시클로프로필 형태와 물과의 경쟁적 반응으로 인해 시클로프로필 환의 개환 반응이 일어나 불활성인 히드록시 화합물이 수득된다. 따라서, 예를 들어 CC-1065 유사체의 프로드럭을 개발함으로써, 수용액 중에서의 그의 유용한 수명을 연장하기 위해 CC-1065 유사체의 반응성 부분을 보호시킬 필요가 있다.In addition, the CC-1065 analogs reported so far are somewhat unstable in aqueous solutions for the following reasons. The seco -form of the drug may be spontaneously converted to the cyclopropyl form and then alkylated if present. However, the competitive reaction of the cyclopropyl form with water results in the ring opening of the cyclopropyl ring resulting in an inert hydroxy compound. Thus, for example, by developing prodrugs of CC-1065 analogs, it is necessary to protect the reactive portion of the CC-1065 analogs in order to extend their useful life in aqueous solutions.
따라서, 수용액 중에서의 저장시 극히 안정적인 CC-1065 유사체의 프로드럭을 개발할 필요가 있다. 바람직하게는 이들 프로드럭이 생체 내에서 활성 약물로만 전환되어야 한다. 이러한 프로드럭이 일단 환자에게 주입되면, 이는 바람직하게 활성 약물로 효율적으로 전환되어야 한다.Thus, there is a need to develop prodrugs of CC-1065 analogs that are extremely stable upon storage in aqueous solution. Preferably these prodrugs should be converted only to the active drug in vivo. Once such a prodrug is injected into a patient, it should preferably be efficiently converted to the active drug.
카르젤레신은 아도젤레신 내의 페놀성 기를 페닐 카바메이트로서 보호시킨 프로드럭이다 [참고: L.H. Li et al., Cancer Res., 52, 4904-4913 (1992)]. 그러나, 이러한 프로드럭을 치료적으로 사용하기에는 너무 불안정하고, 모 약물과 비교해서 수 용해도 상의 증가를 전혀 제공하지도 못한다. 두 번째 예에서는, CC-1065 유사체의 페놀성 잔기를 당화시켜 프로드럭을 생성시켰다 [참고: 미국 특허 제5,646,298호]. 그러나 이러한 프로드럭은 생체 내에서 활성 약물로 전환되지 않았고 이를 세포독성 형태로 전환시키기 위해서는 세균성 공급원으로부터의 효소를 부가적으로 투여해야 한다.Carzelesin is a prodrug that protects phenolic groups in adozelesin as phenyl carbamate. LH Li et al., Cancer Res. , 52, 4904-4913 (1992). However, these prodrugs are too unstable for therapeutic use and provide no increase in water solubility compared to the parent drug. In the second example, the phenolic residues of the CC-1065 analog were glycosylated to produce prodrugs (US Pat. No. 5,646,298). However, these prodrugs have not been converted into active drugs in vivo and require additional administration of enzymes from bacterial sources to convert them into cytotoxic forms.
수용성 프로드럭으로 전환된 바 있지만 CC-1065와는 무관한 몇 가지 예의 항암 약물이 있다. 항암 약물 이리노테칸 (irinotecan)의 경우에는, 페놀성 기가 4-피페리디노-피페리디노 카바메이트에 의해 보호되어 있다. 이러한 보호 기가 해당 약물에 수용성을 부여하는 것으로 보고되었다. 또한, 상기 프로드럭은 인간의 생체 내에서 활성 약물로 용이하게 전환되는데, 이는 인간 혈청, 종양 조직 및 몇몇 기관에 천연적으로 존재하는 효소 카복실에스테라제에 의한 것으로 추정된다 [참고: A. Sparreboom, 4, Clin. Cancer Res., 2747-2754 (1998). L. P. Rivory et al., 52, Biochem Pharmacol., 1103-1111 (1996)].There are several examples of anticancer drugs that have been converted to water-soluble prodrugs but are not related to CC-1065. In the case of the anticancer drug irinotecan, the phenolic group is protected by 4-piperidino-piperidino carbamate. Such protecting groups have been reported to impart water solubility to the drug. In addition, the prodrugs are readily converted into active drugs in humans in vivo, presumably due to the enzyme carboxyl esterase that is naturally present in human serum, tumor tissue and some organs. See A. Sparreboom. , 4, Clin. Cancer Res ., 2747-2754 (1998). LP Rivory et al., 52, Biochem Pharmacol ., 1103-1111 (1996).
유사하게, 항암 약물 에토포시드 포스페이트는 아마도 내인성 알칼리성 포스파타제에 의한 가수분해를 통하여, 생체 내에서 활성 약물로 신속하게 전환되고 포스페이트 보호 기를 갖는 프로드럭의 한 예이다 [참고: S.Z. Fields et al., 1 Clin. Cancer Res., 105-111 (1995)].Similarly, the anticancer drug etoposide phosphate is an example of a prodrug with a phosphate protecting group that is rapidly converted into the active drug in vivo, possibly through hydrolysis by an endogenous alkaline phosphatase [SZ Fields et al., 1 Clin. Cancer Res ., 105-111 (1995).
최근에는 문헌 [참고: R.Y. Zhao et al. (US 6,756,397 B2)]에 해당 분자의 알킬화 부분의 페놀성 환 상에 카바메이트 또는 포스페이트 치환체를 포함하는, 제1 부류의 CC-1065 유사체의 수용성 프로드럭이 기재되었다. 본 발명자들은 이들 유형의 치환체를 함유하는 화합물의 용해도가 생리적 조건 하에서는 만족스럽지 않을 수도 있다는 사실을 밝혀내었다. 또한, 이들 화합물을 생물학적 활성 형태로 전환시키기 위해서는 포스파타제와 같은 특이적 작용제가 작용해야 한다. 따라서, 수용액 중에서의 용해도가 증가되고/되거나 생리적 조건 하에서도 용이하게 용해될 수 있는 CC-1065의 유사체가 일반적으로 필요하다. 부가적으로, 자신의 생물학적 활성은 유지하면서도 수용액 중에서 세포 결합제와의 접합을 촉진시키는 것이 고도로 요망된다. 또한, 독성 부작용을 저하시키기 위해서는, 주로 목적하는 치료 부위에서 바람직하게는 생리적 pH 하에서의 자발적 가수분해를 통하여 세포독성 약물로 전환되는 프로드럭 형태로 CC-1065 유사체를 제공하는 것이 유리할 것이다. 이들 이점들 모두는 본원에 기재된 본 발명에 의해 제공되며, 이는 당업자가 다음의 상세한 설명과 실시예로부터 명백히 알 수 있을 것이다.Recently, R.Y. Zhao et al. (US 6,756,397 B2) describe water-soluble prodrugs of the first class of CC-1065 analogues comprising carbamate or phosphate substituents on the phenolic ring of the alkylated portion of the molecule. We have found that the solubility of compounds containing these types of substituents may not be satisfactory under physiological conditions. In addition, specific agents such as phosphatase must act in order to convert these compounds into biologically active forms. Thus, there is a general need for analogs of CC-1065 that can increase solubility in aqueous solutions and / or readily dissolve even under physiological conditions. In addition, it is highly desirable to promote conjugation with cell binders in aqueous solution while maintaining their biological activity. In addition, to reduce toxic side effects, it would be advantageous to provide CC-1065 analogs in the form of prodrugs, which are converted primarily to cytotoxic drugs, preferably through spontaneous hydrolysis at desired site of treatment. All of these advantages are provided by the invention described herein, which will be apparent to one skilled in the art from the following detailed description and examples.
발명의 요약Summary of the Invention
본 발명의 목적은 수성 매질 중에서의 용해도를 증강시킨, CC-1065 유사체의 프로드럭을 제공하는 것이다. 이러한 목적과 기타 목적들은 해당 분자의 알킬화 부분의 페놀성 기를, 산성 수용액 중에서의 저장시 약물을 안정하게 만드는 작용기로 보호시킨 프로드럭을 제공함으로써 달성하였다. 또한, 이러한 보호 기는 보호되지 않은 유사체와 비교해서 약물에 대한 수 용해도를 증가시켜 준다. 보호 기는 생리적 pH 하의 생체 내에서 용이하게 절단되어 상응하는 활성 약물을 제공한다. 본원에 기재된 프로드럭에서는, 페놀성 치환체를 생리적 pH 하에서 전하를 보유하고 있는 설폰산 함유 페닐 카바메이트로서 보호함으로써, 수 용해도를 증강시켰다. 수 용해도를 추가로 증강시키기 위해서는, 임의의 폴리에틸렌 글리콜 스페이서를 디설파이드 기 등의 절단 가능한 연결과 인돌릴 소단위체 간의 링커 내로 도입할 수 있다. 이러한 스페이서의 도입은 약물의 효력을 변경시키지 않는다.It is an object of the present invention to provide prodrugs of CC-1065 analogs which have enhanced solubility in aqueous media. These and other objects have been achieved by providing prodrugs in which the phenolic group of the alkylated portion of the molecule is protected with a functional group that makes the drug stable upon storage in acidic aqueous solution. In addition, such protecting groups increase the water solubility of the drug compared to unprotected analogs. Protective groups are readily cleaved in vivo under physiological pH to provide the corresponding active drug. In the prodrugs described herein, the water solubility was enhanced by protecting the phenolic substituents as sulfonic acid containing phenyl carbamate, which retains charge under physiological pH. To further enhance water solubility, any polyethylene glycol spacer can be introduced into the linker between the cleavable linkage, such as a disulfide group, and the indolyl subunit. Introduction of such spacers does not alter the potency of the drug.
본 발명의 보다 구체적인 양태는 수 용해도와 안정성을 증강시켜 주는 보호 기를 갖고 있고 생리적 pH 하의 생체 내에서 절단될 수 있는 seco-시클로프로파벤즈인돌-함유 세포독성 약물의 유사체를 포함하는 프로드럭을 제공한다. 이러한 구체적 양태의 프로드럭은 제1 소단위체의 피롤 잔기의 2급 아미노 기에서 제2 소단위체의 C-2 카복실로의 아미드 결합에 의해 연결되는 제1 소단위체와 제2 소단위체를 갖고 있다. 이러한 제1 소단위체가 다음 화학식 I의 화합물로서 제시되고, 이는 다음 화학식 II 내지 XI의 화합물, 또는 그의 제약상 허용 가능한 염, 수화물 또는 수화 염, 또는 이들 화합물의 다형성 결정성 구조물 또는 그의 광학 이성체, 라세미체, 부분 입체이성체 또는 에난티오머 중에서 선택되는 제2 소단위체와 접합된다:More specific embodiments of the present invention provide prodrugs comprising analogs of seco -cyclopropagenzindole-containing cytotoxic drugs that have protecting groups that enhance water solubility and stability and can be cleaved in vivo under physiological pH. do. The prodrug of this specific embodiment has a first subunit and a second subunit linked by an amide bond from the secondary amino group of the pyrrole residue of the first subunit to the C-2 carboxyl of the second subunit. Such a first subunit is shown as a compound of Formula I, which is a compound of Formulas II to XI, or a pharmaceutically acceptable salt, hydrate or hydrate salt thereof, or a polymorphic crystalline structure of these compounds or an optical isomer thereof Conjugated with a second subunit selected from semi-, diastereomers or enantiomers:
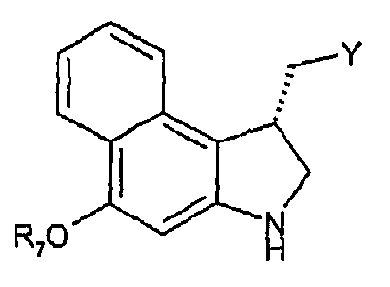





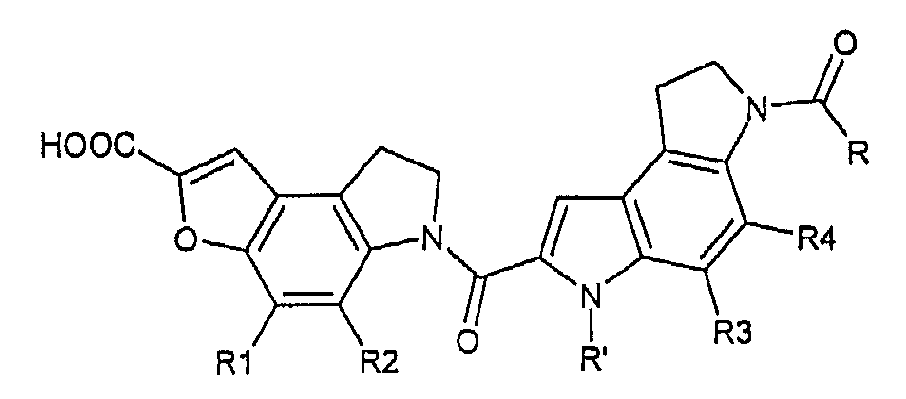




상기 식에서,Where
Y는 할라이드 (플루오라이드, 클로라이드, 브로마이드 또는 요오다이드), 메탄설포네이트, 파라-톨루엔설포네이트, 트리플루오로메탄 설포네이트, 모노 니트로 또는 디니트로 페놀레이트 중에서 선택된 이탈 기를 나타낸다. 바람직하게는, 이탈 기가 클로라이드 또는 브로마이드이다.Y represents a leaving group selected from halides (fluoride, chloride, bromide or iodide), methanesulfonate, para-toluenesulfonate, trifluoromethane sulfonate, mononitro or dinitrophenolate. Preferably, the leaving group is chloride or bromide.
R, R', R1-R6은 각각 독립적으로, 수소, C1-C3 선형 알킬, 메톡시, 히드록실, 1급 아미노, 2급 아미노, 3급 아미노 또는 아미도이거나, 또는 세포 결합제와 프로드럭과의 연결을 제공해 주는 링커이고, 이러한 연결은 바람직하게 설파이드 또는 디설파이드 결합을 통해서 이루어지는데, 단 R, R', R1-R6 중의 적어도 하나는 링커이다. 바람직하게는, R이 링커이다. 링커는 폴리에틸렌 글리콜 스페이서를 포함할 수 있다.R, R ', R 1 -R 6 are each independently hydrogen, C 1 -C 3 linear alkyl, methoxy, hydroxyl, primary amino, secondary amino, tertiary amino or amido, or a cell binder And a linker that provides a linkage to a prodrug, the linkage being preferably made via a sulfide or disulfide bond, provided that at least one of R, R ', R 1 -R 6 is a linker. Preferably, R is a linker. The linker may comprise a polyethylene glycol spacer.
R7은 생체 내에서 절단될 수 있고 시클로프로파벤즈인돌-함유 세포독성 약물의 수 용해도를 증강시킬 수 있는 보호 기이고, 설폰산 함유 페닐 카바메이트이다. R7은 다음 화학식 XII을 갖는다:R 7 is a protecting group that can be cleaved in vivo and can enhance the water solubility of cyclopropabenzindole-containing cytotoxic drugs and is sulfonic acid containing phenyl carbamate. R 7 has the formula

[상기 식에서, R9는 H 또는 C1-C4 분지 또는 선형 알킬이고; R10, R11, R12 또 는 R13은 동일하거나 상이하고, 이중 하나 이상이 -X-SO3 -M+ 기인데, M+는 H+ 또는 IA 족의 원자로부터 유래되는 양이온, 예를 들어 Na+, K+ 등이며, X는 직접 결합 또는 C1 내지 C4 선형 또는 분지 알킬, 알케닐 또는 알키닐, -O알킬-, -S알킬- 및 아미노알킬 중에서 선택된 스페이서이며, 기타 R10, R11, R12 또는 R13은 H, C1-C6 분지 또는 선형 알킬, -O알킬, -S알킬, 히드록실, 1급 아미노, 2급 아미노, 또는 아미도, 할라이드, 니트로, 아지도이다].[Wherein R 9 is H or C 1 -C 4 branched or linear alkyl; R 10, R 11, R 12 or R 13 are the same or different and are one or more double -X-SO 3 - groups inde + M, M + is a cation derived from the atoms of the H + or a Group IA, e. For example Na + , K +, etc., X is a direct bond or a spacer selected from C1 to C4 linear or branched alkyl, alkenyl or alkynyl, -Oalkyl-, -Salkyl- and aminoalkyl, and other R 10 , R 11 , R 12 or R 13 is H, C 1 -C 6 branched or linear alkyl, —Oalkyl, —Salkyl, hydroxyl, primary amino, secondary amino, or amido, halide, nitro, azido ].
바람직한 양태에 따르면, 본 발명의 프로드럭은 다음 화학식의 화합물, 또는 그의 제약상 허용 가능한 염, 수화물 또는 수화 염, 또는 이들 화합물의 다형성 결정성 구조물 또는 그의 광학 이성체, 라세미체, 부분 입체이성체 또는 에난티오머이다:According to a preferred embodiment, the prodrugs of the invention are compounds of the formula: or pharmaceutically acceptable salts, hydrates or hydrates thereof, or polymorphic crystalline structures of these compounds or optical isomers, racemates, diastereomers thereof or It is an enantiomer:
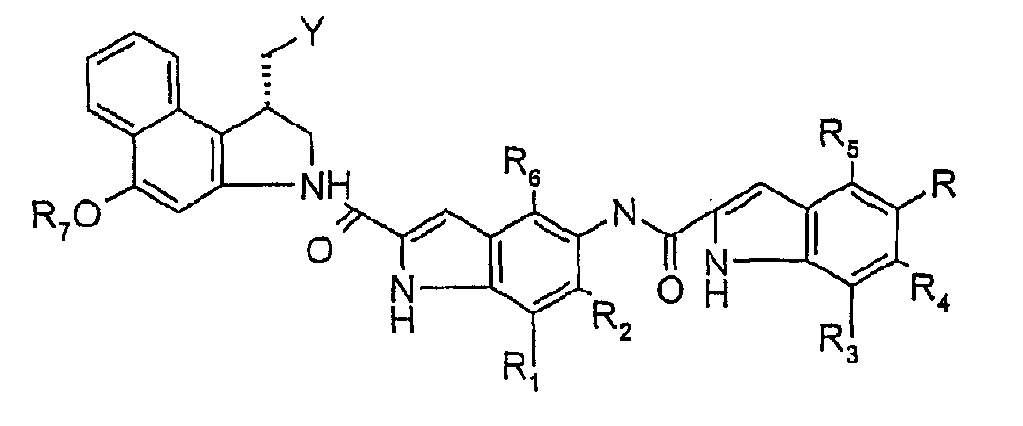
상기 식에서,Where
Y, R1, R2, R3, R4, R5, R6, R7 및 R은 상기 정의된 바와 같고, R은 본원에 정의된 바와 같은 링커이다. Y, R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 and R are as defined above and R is a linker as defined herein.
바람직하게, 상기 프로드럭은 다음으로 이루어진 군 중에서 선택된다:Preferably, the prodrug is selected from the group consisting of:
(S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐페닐)아미노카 보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-[(3-메틸디티오프로파노일)-아미노]-1H-인돌-2-카복스아미드;(S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylphenyl) aminocarbonyloxy] -3H-benz (e) indole- 3-yl} carbonyl] -1H-indol-5-yl] -5-[(3-methyldithiopropanoyl) -amino] -1H-indol-2-carboxamide;
(S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-[(3-머캅토프로파노일)-아미노]-1H-인돌-2-카복스아미드;(S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylphenyl) aminocarbonyloxy] -3H-benz (e) indole- 3-yl} carbonyl] -1H-indol-5-yl] -5-[(3-mercaptopropanoyl) -amino] -1H-indol-2-carboxamide;
(S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐메틸페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-{[3-(2-피리딜디티오)-프로파노일]-아미노}-1H-인돌-2-카복스아미드;(S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylmethylphenyl) aminocarbonyloxy] -3H-benz (e) indole- 3-yl} carbonyl] -1H-indol-5-yl] -5-{[3- (2-pyridyldithio) -propanoyl] -amino} -1H-indol-2-carboxamide;
(S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐메틸페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-[(3-머캅토프로파노일)-아미노]-1H-인돌-2-카복스아미드;(S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylmethylphenyl) aminocarbonyloxy] -3H-benz (e) indole- 3-yl} carbonyl] -1H-indol-5-yl] -5-[(3-mercaptopropanoyl) -amino] -1H-indol-2-carboxamide;
(S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐메틸페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-[(3-머캅토프로파노일)-아미노]-1H-인돌-2-카복스아미드; 또는(S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylmethylphenyl) aminocarbonyloxy] -3H-benz (e) indole- 3-yl} carbonyl] -1H-indol-5-yl] -5-[(3-mercaptopropanoyl) -amino] -1H-indol-2-carboxamide; or
그의 제약상 허용 가능한 염, 수화물 또는 수화 염, 또는 이들 화합물의 다형성 결정성 구조물 또는 그의 광학 이성체, 라세미체, 부분 입체이성체 또는 에난티오머.Pharmaceutically acceptable salts, hydrates or hydrated salts thereof, or polymorphic crystalline structures of these compounds or optical isomers, racemates, diastereomers or enantiomers thereof.
바람직한 또 다른 양태에 따르면, 본 발명의 프로드럭은 다음 화학식의 화합물, 또는 그의 제약상 허용 가능한 염, 수화물 또는 수화 염, 또는 이들 화합물의 다형성 결정성 구조물 또는 그의 광학 이성체, 라세미체, 부분 입체이성체 또는 에 난티오머이다:According to another preferred embodiment, the prodrugs of the present invention are compounds of the formula: or pharmaceutically acceptable salts, hydrates or hydrates thereof, or polymorphic crystalline structures of these compounds or optical isomers, racemates, diasteres thereof Isomers or ethiomers are:

상기식에서,In the above formula,
Y, R1, R2, R3, R4, R5, R6, R7 및 R은 상기 정의된 바와 같고, R은 본원에 정의된 바와 같은 링커이다. Y, R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 and R are as defined above and R is a linker as defined herein.
바람직하게, 상기 프로드럭은 다음으로 이루어진 군 중에서 선택된다:Preferably, the prodrug is selected from the group consisting of:
(S)-3-((1-(클로로메틸)-3-(5-(3-머캅토프로판아미도)-1H-인돌-2-카보닐)-2,3-디히드로-1H-벤조[e]인돌-5-일옥시)카보닐아미노)벤젠설폰산;(S) -3-((1- (chloromethyl) -3- (5- (3-mercaptopropaneamido) -1H-indole-2-carbonyl) -2,3-dihydro-1H-benzo [e] indol-5-yloxy) carbonylamino) benzenesulfonic acid;
(S)-3-((1-(클로로메틸)-3-(5-(3-(메틸디설파닐)프로판아미도)-1H-인돌-2-카보닐)-2,3-디히드로-1H-벤조[e]인돌-5-일옥시)카보닐아미노)벤젠설폰산; 또는(S) -3-((1- (chloromethyl) -3- (5- (3- (methyldisulfanyl) propaneamido) -1H-indole-2-carbonyl) -2,3-dihydro -1H-benzo [e] indol-5-yloxy) carbonylamino) benzenesulfonic acid; or
그의 제약상 허용 가능한 염, 수화물 또는 수화 염, 또는 이들 화합물의 다형성 결정성 구조물 또는 그의 광학 이성체, 라세미체, 부분 입체이성체 또는 에난티오머.Pharmaceutically acceptable salts, hydrates or hydrated salts thereof, or polymorphic crystalline structures of these compounds or optical isomers, racemates, diastereomers or enantiomers thereof.
본 발명의 프로드럭은 세포 결합제를 연결 기를 통하여 본 발명의 하나 이상의 프로드럭에 연결시킨 세포독성 접합체에 사용할 수 있다.Prodrugs of the invention can be used in cytotoxic conjugates in which cell binding agents are linked to one or more prodrugs of the invention via a linking group.
상기 연결 기는 세포 결합제의 티올, 설파이드 또는 디설파이드에 대해 반응성인 작용기와 연결된 상기 정의된 바와 같은 링커를 포함한다.The linking group includes a linker as defined above linked to a functional group reactive to thiol, sulfide or disulfide of the cell binder.
세포 결합제에는 항체 및 그의 단편, 인터페론, 림포카인, 비타민, 호르몬 및 성장 인자가 포함된다. 이러한 접합체를 함유하는 제약 조성물이 또한 제공된다.Cell binding agents include antibodies and fragments thereof, interferons, lymphokines, vitamins, hormones and growth factors. Pharmaceutical compositions containing such conjugates are also provided.
세포독성 접합체는 상기 제약 조성물의 유효량을 투여함으로써 특정 대상체를 치료하는 방법에 사용할 수 있다. 선택된 세포 결합제와 결합하는 세포 유형에 따라서, 많은 질병을 생체내, 생체외 또는 시험관 내에서 치료할 수 있다. 이러한 질병에는, 예를 들어 많은 종류의 암이 포함되는데, 이의 예로는 림프종, 백혈병, 폐암, 유방암, 결장암, 전립선암, 신장암, 췌장암 등이 있다.Cytotoxic conjugates can be used in methods of treating a particular subject by administering an effective amount of the pharmaceutical composition. Depending on the cell type that binds the selected cell binding agent, many diseases can be treated in vivo, ex vivo or in vitro. Such diseases include, for example, many types of cancers, including lymphomas, leukemias, lung cancers, breast cancers, colon cancers, prostate cancers, kidney cancers, pancreatic cancers, and the like.
따라서, 수용액 중에서의 용해도와 안정성이 개선되고, 활성화되었을 때 세포독성을 보유하고 있어 알킬화 약물을 생성시키며, 특이적 세포 결합제와의 접합을 통하여 특이적 세포 유형을 표적화하는 데에 있어서 유용한 CC-1065 유사체의 프로드럭이 제공된다.Thus, CC-1065 is useful for improving solubility and stability in aqueous solutions, retaining cytotoxicity when activated, producing alkylated drugs, and targeting specific cell types through conjugation with specific cell binding agents. Prodrugs of the analogs are provided.
도 1A는 CC-1065 및 그의 소단위체 A, B, 및 C의 구조를 도시한 것이다. 1A shows the structure of CC-1065 and its subunits A, B, and C. FIG.
도 1B 및 도 1C는 CC-1065의 2개의 공지된 유사체의 구조를 도시한 것이다. 1B and 1C show the structure of two known analogs of CC-1065.
도 1D는 중간체 DC1SMe의 제조를 도시한 것이다 [참고: 미국 특허 제6534660 B1호]. 1D depicts the preparation of intermediate DC1SMe (see US Pat. No. 6534660 B1).
도 2는 예시되는 본 발명의 CC-1065 유사체 및 프로드럭의 구조를 도시한 것이다. 2 illustrates the structure of the CC-1065 analogs and prodrugs of the present invention which are illustrated.
도 3은 예시되는 본 발명의 폴리에틸렌 글리콜-함유 프로드럭의 구조를 도시 한 것이다. Figure 3 illustrates the structure of the polyethylene glycol-containing prodrug of the invention illustrated.
도 4A 및 C는 (S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-[(3-메틸디티오프로파노일)-아미노]-1H-인돌-2-카복스아미드 (DC07SMe); (S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-[(3-머캅토프로파노일)-아미노]-1H-인돌-2-카복스아미드 (DC07); (S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐메틸페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-{[3-(2-피리딜디티오)-프로파노일]-아미노}-1H-인돌-2-카복스아미드 (DC08SPy); 및 (S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐메틸페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-[(3-머캅토프로파노일)-아미노]-1H-인돌-2-카복스아미드 (DC08); (S)-3-((1-(클로로메틸)-3-(5-(3-(메틸디설파닐)프로판아미도)-1H-인돌-2-카보닐)-2,3-디히드로-1H-벤조[e]인돌-5-일옥시)카보닐아미노)벤젠설폰산 (DC101SMe); (S)-3-((1-(클로로메틸)-3-(5-(3-머캅토프로판아미도)-1H-인돌-2-카보닐)-2,3-디히드로-1H-벤조[e]인돌-5-일옥시)카보닐아미노)벤젠설폰산(DC107)을 제조하기 위한 합성 도식이다. 4A and C show (S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylphenyl) aminocarbonyloxy] -3H-benz (e) indol-3-yl} carbonyl] -1H-indol-5-yl] -5-[(3-methyldithiopropanoyl) -amino] -1 H-indole-2-carboxamide (DC07SMe ); (S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylphenyl) aminocarbonyloxy] -3H-benz (e) indole- 3-yl} carbonyl] -1H-indol-5-yl] -5-[(3-mercaptopropanoyl) -amino] -1H-indol-2-carboxamide (DC07); (S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylmethylphenyl) aminocarbonyloxy] -3H-benz (e) indole- 3-yl} carbonyl] -1H-indol-5-yl] -5-{[3- (2-pyridyldithio) -propanoyl] -amino} -1H-indole-2-carboxamide ( DC08SPy); And (S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylmethylphenyl) aminocarbonyloxy] -3H-benz (e) indole -3-yl} carbonyl] -1 H-indol-5-yl] -5-[(3-mercaptopropanoyl) -amino] -1 H-indol-2-carboxamide (DC08); (S) -3-((1- (chloromethyl) -3- (5- (3- (methyldisulfanyl) propaneamido) -1H-indole-2-carbonyl) -2,3-dihydro -1H-benzo [e] indol-5-yloxy) carbonylamino) benzenesulfonic acid (DC101SMe); (S) -3-((1- (chloromethyl) -3- (5- (3-mercaptopropaneamido) -1H-indole-2-carbonyl) -2,3-dihydro-1H-benzo [e] Synthesis scheme for preparing indol-5-yloxy) carbonylamino) benzenesulfonic acid (DC107).
도 5는 생리적 pH 하에서 약물의 활성화/불활성화 공정을 나타낸 도식이다. 5 is a schematic of the activation / deactivation process of a drug under physiological pH.
도 6은 라모스 (Ramos) 및 HL60 세포에 대한 5일간 노출 후의 DC07-SMe 및 DC1-SMe의 시험관내 세포독성 데이터를 도시한 것이다. FIG. 6 shows in vitro cytotoxicity data of DC07-SMe and DC1-SMe after 5 days of exposure to Ramos and HL60 cells.
도 7은 라모스 및 HL60 세포에 대한 DC1-SPy 및 DC07-Spy의 시험관내 세포독 성 데이터를 도시한 것이다. 7 depicts in vitro cytotoxicity data of DC1-SPy and DC07-Spy for Ramos and HL60 cells.
도 8은 라모스 및 나말와 (Namalwa) 세포에 대한 DC08-Spy의 시험관내 세포독성 데이터를 도시한 것이다. 8 depicts in vitro cytotoxicity data of DC08-Spy against Ramos and Namalwa cells.
도 9는 HL-60 및 라모스 세포에 대한 DC101-SMe 및 DC107-SMe의 시험관내 세포독성 데이터를 도시한 것이다. 9 depicts in vitro cytotoxicity data of DC101-SMe and DC107-SMe against HL-60 and Ramos cells.
도 10은 카라 (KARA) 및 라모스 세포에 대한 MY9-6-DC07 (디설파이드 연결됨)의 시험관내 세포독성 데이터를 도시한 것이다. 10 depicts in vitro cytotoxicity data of MY9-6-DC07 (disulfide linked) for KARA and Ramos cells.
도 11은 COLO205 & A-375 세포에 대한 huC242-DC07 (티오에테르 연결됨)의 시험관내 세포독성 데이터를 도시한 것이다. FIG. 11 depicts in vitro cytotoxicity data of huC242-DC07 (thioether linked) for COLO205 & A-375 cells.
본 발명자들은 특정의 CC-1065 유사체의 안정성, 수 용해도 및 유용성이 이러한 유사체의 알킬화 잔기를 적합한 보호 기로 보호함으로써 증강된다는 사실을 밝혀내었다. 이로써 본 발명자들은 수 용해도가 증강되고 추가로 세포 결합제와 연결할 수 있는 CC-1065 유사체의 프로드럭을 제공하였는데, 이로 인해 이러한 CC-1065 유사체의 프로드럭의 치료 효능은 종양 부위로의 프로드럭의 표적화 전달을 통하여 생체내 분포도를 변화시켜 비-표적화 조직에 대한 독성을 보다 저하시키고, 이로써 전신 독성을 저하시킴으로써 개선된다. 생리적 pH는 실질적으로 상기 프로드럭을 그의 활성 약물 형태로 전환시키고, 세포 결합제에 대한 절단 가능한 링커를 갖는 양태에서는 CC-1065 유사체의 활성 약물 형태가 방출되므로 그의 세포독성 활성이 추가로 증강된다. 또 다른 한편, 세포 결합제에 대한 링커는 표적 세포 내부에서 먼저 절단되어 프로드럭을 방출시킬 수 있는데, 그 후에 이는 활성 약물로 내인성 전환된다.We have found that the stability, water solubility and utility of certain CC-1065 analogs are enhanced by protecting the alkylated moieties of such analogs with suitable protecting groups. This provided us with a prodrug of CC-1065 analogues that enhanced water solubility and could additionally be linked to cell binding agents, whereby the therapeutic efficacy of the prodrugs of such CC-1065 analogs was targeted to the tumor site. Delivery improves by changing the distribution in vivo to lower toxicity to non-targeted tissue, thereby lowering systemic toxicity. Physiological pH substantially converts the prodrug into its active drug form, and in embodiments with a cleavable linker for cell binders, the active drug form of the CC-1065 analog is released, thereby further enhancing its cytotoxic activity. On the other hand, the linker to the cell binding agent may be first cleaved inside the target cell to release the prodrug, after which it is endogenously converted to the active drug.
이러한 목표를 달성하기 위해, 본 발명자들은 (a) 수 용해도를 증강시키기 위해 페놀성 히드록실이 보호 기에 의해 보호되고 생리적 pH 하의 생체 내에서 절단되는 화학식 I의 제1 소단위체와, (b) 화학식 II 내지 XI 중의 하나로써 나타낸 구조를 갖고 있고 프로드럭을 세포 결합제에 접합시키기 위한 링커를 포함하는 제2 소단위체를 포함하는 seco-시클로프로파벤즈인돌 (CBI)-함유 세포독성 프로드럭인 CC-1065 유사체의 예시 프로드럭 (도 2-4B)을 합성하였다. 링커는 폴리에틸렌 글리콜 스페이서를 함유할 수 있다 (도 3). 프로드럭의 보호 기를 제거하면 모 약물의 고 세포독성을 보유하고 있는 활성 형태의 약물이 생성된다. 링커는 세포 결합제와의 접합을 위해 사용되는데, 바람직하게는 디설파이드 결합 또는 설파이드 (또는 본원에서 티오에테르로 지칭됨) 결합을 포함한다.To achieve this goal, the inventors have found that (a) the first subunit of formula (I) wherein phenolic hydroxyl is protected by a protecting group and cleaved in vivo under physiological pH to enhance water solubility, and (b) CC-, which is a seco -cyclopropagenzindole (CBI) -containing cytotoxic prodrug having a structure shown as one of II-XI and comprising a second subunit comprising a linker for conjugating the prodrug to the cell binder. Exemplary prodrugs of the 1065 analogs ( FIGS. 2-4B ) were synthesized. The linker may contain polyethylene glycol spacers ( FIG. 3 ). Removal of the prodrug's protecting group produces an active form of the drug that retains the high cytotoxicity of the parent drug. Linkers are used for conjugation with cell binders, preferably comprising disulfide bonds or sulfide (or thioethers) bonds herein.
디설파이드 결합과 같은 절단 가능한 연결을 이용하여 고도로 세포독성인 약물을 항체에 연결시키면 세포 내부에 완전히 활성인 약물이 방출된다는 사실과, 이러한 접합체가 항원 특이적 방식으로 세포독성이라는 사실이 기존에 밝혀진 바 있다 [참고: R. V. J. Chari et al, 52 Cancer Res. 127-131 (1992); R.V.J. Chari et al., 55 Cancer Res. 4079-4084 (1995); 및 미국 특허 제5,208,020호 및 제5,475,092호]. 본 발명에서, 본 발명자들은 CC-1065 유사체의 프로드럭의 합성, 이들을 모노클로날 항체와 접합시키는 과정, 및 이러한 접합체의 시험관내 세포독성 및 특이성을 평가하기 위한 과정을 서술하고 있다. 따라서, 본 발명은 부작용을 최소로 하면서도, 사멸 또는 용해시켜야만 병든 세포 또는 비정상 세포, 예를 들어 종양 세포, 바이러스 감염 세포, 미생물 감염 세포, 기생충 감염 세포, 자가면역 세포 (자가-항체를 생성시키는 세포), 활성화 세포 (이식편 거부 또는 이식편 대 숙주 질병에 관여한 세포), 또는 기타 모든 유형의 병든 세포 또는 비정상 세포를 제거시키고자 하는 치료제를 제조하는 데에 유용한 화합물을 제공한다.Linking highly cytotoxic drugs to antibodies using cleavable linkages, such as disulfide bonds, results in the release of fully active drugs inside the cell and that these conjugates are cytotoxic in an antigen-specific manner. [Reference: RVJ Chari et al, 52 Cancer Res. 127-131 (1992); R.V.J. Chari et al., 55 Cancer Res. 4079-4084 (1995); And US Pat. Nos. 5,208,020 and 5,475,092. In the present invention, we describe the synthesis of prodrugs of CC-1065 analogs, the conjugation of them with monoclonal antibodies, and the procedure for evaluating the in vitro cytotoxicity and specificity of such conjugates. Thus, the present invention is directed to killing or lysing diseased or abnormal cells, such as tumor cells, virus infected cells, microbial infected cells, parasitic infected cells, autoimmune cells (cells that produce auto-antibodies) with minimal side effects. ), Compounds that are useful for preparing a therapeutic agent intended to remove activated cells (cells involved in graft rejection or graft versus host disease), or any other type of diseased or abnormal cell.
따라서, 본 발명은 보호 기의 방출시 모 화합물 CC-1065의 높은 세포독성을 유지시켜 주는 세포 결합제와 화학적으로 연결될 수 있는 CC-1065의 프로드럭 유사체 및 유도체의 합성을 교시하고 있다. 추가로, 활성화시 이들 화합물이 세포 결합제와 연결되는 경우에 이는 세포 결합제와 결합하는 세포에 대해 세포독성이고 비-표적 세포에 대해서는 독성이 훨씬 덜하다.Thus, the present invention teaches the synthesis of prodrug analogs and derivatives of CC-1065 that can be chemically linked to cell binding agents that maintain high cytotoxicity of parent compound CC-1065 upon release of a protecting group. In addition, when these compounds are linked to cell binding agents upon activation, they are cytotoxic to cells that bind cell binding agents and much less toxic to non-target cells.
본 발명의 프로드럭Prodrug of the Invention
본 발명에 따르는 프로드럭은 해당 분자의 알킬화 부분의 페놀성 기를 보호시킨 CC-1065 유사체를 포함하고, 본 발명의 프로드럭은 이를 세포 결합제와 접합시킬 수 있는 링커를 추가로 포함한다. 상기 프로드럭은 아미드 결합을 통하여 연결되는 제1 및 제2 소단위체를 포함할 수 있다.Prodrugs according to the present invention comprise a CC-1065 analogue that protects the phenolic group of the alkylated portion of the molecule, and the prodrugs of the present invention further comprise a linker capable of conjugating it with a cell binder. The prodrug may comprise first and second subunits linked via amide bonds.
본 발명의 특정 양태에 따르면, CC-1065 유사체의 프로드럭은 그의 개방 할로메틸 형태의 seco-CBI (시클로프로파벤즈인돌 단위)인 제1 소단위체를 갖고 있는데, 이러한 제1 소단위체는 생체 내에서 절단될 수 있는 수용성 보호 기에 의해 보호되는 페놀성 히드록실을 갖는다. 본 발명의 특정 양태의 프로드럭의 제2 소단위체는 2-카복시-인돌 또는 2-카복시-벤조푸란 유도체이거나 또는 둘 다인 CC-1065의 B 소단위체 또는 B 소단위체와 C 소단위체 (도 1) 조합의 유사체를 포함하고, 이는 화학식 II 내지 XI로써 나타낸다. 천연 CC-1065 및 공개 발표된 유사체의 특성으로부터 확인할 수 있는 바와 같이 [참고: 예를 들어, Warpehoski et al, 31 J. Med. Chem. 590-603 (1988), Boger et al, 66 J. Org. Chem. 6654-6661 (2001)], B 및 C 소단위체는 기타 헤테로사이클 또는 치환된 헤테로사이클을 포함할 수도 있으므로, 화학식 II 내지 XI의 위치 R1 내지 R6에 상응하는, 인돌 또는 벤조푸란 환 상의 상이한 위치에 상이한 치환체를 수반할 수 있고 강력한 세포독성 활성을 여전히 보유할 수 있다.According to a particular embodiment of the invention, the prodrug of the CC-1065 analog has a first subunit which is seco- CBI (cyclopropabenzindole unit) in its open halomethyl form, which first subunit is in vivo It has a phenolic hydroxyl protected by a water-soluble protecting group that can be cleaved at The second subunit of a prodrug of certain embodiments of the present invention is a B-subunit or B subunit and a C subunit of CC-1065, which are either 2-carboxy-indole or 2-carboxy-benzofuran derivatives or both ( FIG. 1 ). Analogs of combinations, which are represented by Formulas II-XI. As can be seen from the properties of native CC-1065 and published published analogues, see, eg, Warpehoski et al, 31 J. Med. Chem. 590-603 (1988), Boger et al, 66 J. Org. Chem. 6654-6661 (2001)], B and C subunits may also contain other heterocycles or substituted heterocycles, and thus different on indole or benzofuran rings, corresponding to positions R 1 to R 6 of Formulas II to XI. It may involve different substituents at the position and still retain potent cytotoxic activity.
CC-1065 유사체의 프로드럭을 세포 결합제와 연결시키기 위해서는, 프로드럭이 먼저, 디설파이드 결합, 설파이드 (또는 본원에서 티오에테르로서 지칭됨) 결합, 산-불안정 기, 광-불안정 기, 펩티다제-불안정 기, 또는 에스테라제-불안정 기 등의 연결을 통하여 세포 결합제와 연결되도록 허용해주는 잔기를 포함해야만 한다. 프로드럭 유사체가, 예를 들어 디설파이드 결합, 티오에테르 결합, 산-불안정 기, 광-불안정 기, 펩티다제-불안정 기, 또는 에스테라제-불안정 기를 통하여 세포 결합제와 결합하는데 필요한 잔기를 함유하도록 프로드럭 유사체를 제조한다. 수용액 중에서의 용해도를 추가로 증강시키기 위해서는, 링커가 폴리에틸렌 글리콜 스페이서를 함유할 수 있다 (도 3).In order to link prodrugs of CC-1065 analogs with cell binders, the prodrugs must first be disulfide bonds, sulfide (or referred to herein as thioethers) bonds, acid-labile groups, photo-labile groups, peptidase- It should include residues that allow linkage with the cell binding agent through linkages such as labile groups, or esterase-labile groups. The prodrug analogs contain, for example, the residues needed to bind the cell binder via disulfide bonds, thioether bonds, acid-labile groups, photolabile groups, peptidase-labile groups, or esterase-labile groups. Prodrug analogs are prepared. To further enhance solubility in aqueous solution, the linker may contain polyethylene glycol spacers ( FIG. 3 ).
바람직하게는 디설파이드 연결을 사용하는데, 이는 표적화 세포의 환원성 환경으로 인해 디설파이드가 절단되고 프로드럭 (또는 세포 결합제로부터의 프로드럭의 절단과 보호 기의 가수분해의 상대적 순서에 따라서, 약물)이 방출되고, 이와 연관해서 세포독성이 증가하기 때문이다.Preferably a disulfide linkage is used, which is due to the reducing environment of the targeting cell to which the disulfide is cleaved and the prodrug (or drug, according to the relative order of cleavage of the prodrug from the cell binder and hydrolysis of the protecting group) is released. In this case, the cytotoxicity is increased.
보다 구체적으로 언급해서 본 발명의 특정 양태에 따르면, CC-1065 유사체의 프로드럭은 제1 소단위체의 피롤 잔기의 2급 아미노 기에서 화학식 II 내지 XI를 갖는 제2 소단위체의 C-2 카복시 기로의 아미드 결합을 통하여 공유적으로 연결시킨 제1 소단위체와 제2 소단위체를 포함한다.More specifically, in accordance with certain embodiments of the present invention, the prodrug of the CC-1065 analog is from the secondary amino group of the pyrrole moiety of the first subunit to the C-2 carboxy group of the second subunit having Formulas II to XI. It includes the first subunit and the second subunit covalently linked via an amide bond of.
화학식 II 내지 XI 내의 링커는 CC-1065 유사체의 프로드럭이 세포 결합제와 연결될 수 있도록 해준다. 이러한 링커는 폴리에틸렌 글리콜 스페이서를 함유할 수 있다. 이의 예에는 디설파이드 결합, 티오에테르 결합, 산-불안정 기, 광-불안정 기, 펩티다제-불안정 기, 또는 에스테라제-불안정 기를 통하여 연결시켜 줄 수 있는 잔기가 포함되고, 당해 분야에 널리 공지되어 있다 [참고: 예를 들어, 미국 특허 제5,846,545호; 이는 본원에 참고로 도입된다]. 바람직한 잔기는 디설파이드 결합, 예를 들어 티올 (DC1, DC07, DC08) 또는 디설파이드 (DC1-SMe, DC07-SMe, DC08SPy, 참고: 도 2-4B)를 통하여 연결시켜 줄 수 있는 잔기이다. 말단 이탈 기를 함유하는 혼합 디설파이드, 예를 들어 티오메틸 (DC1-SMe, DC07-SMe), DCO8SPy, 글루타치온, 알킬 티올, 피리딜티오, 아릴 티올, 니트로피리딜티오, 히드록시카보닐피리딜티오, (니트로)히드록시카보닐-피리딜티오 등을 사용할 수 있는데, 단 이러한 디설파이드는 프로드럭을 세포 결합제와 커플링시키기 위한 디설파이드-교환 반응을 진행할 수 있어야 한다. 링커는 임의로, 연결을 가능하게 해주는 부분의 반응성 기와 2-카복시-인돌 또는 2-카복시-벤조푸란 유도체 부분의 반응성 기 사이에 개입된 스페이서 영역을 추가로 포함할 수 있다.Linkers in Formulas II-XI allow prodrugs of the CC-1065 analogs to be linked to cell binders. Such linkers may contain polyethylene glycol spacers. Examples thereof include residues that can be linked through disulfide bonds, thioether bonds, acid-labile groups, photolabile groups, peptidase-labile groups, or esterase-labile groups, and are well known in the art. [See, eg, US Pat. No. 5,846,545; Which is incorporated herein by reference]. Preferred residues are residues which can be linked via disulfide bonds, for example thiols (DC1, DC07, DC08) or disulfides (DC1-SMe, DC07-SMe, DC08SPy, see FIGS. 2-4B ). Mixed disulfides containing terminal leaving groups such as thiomethyl (DC1-SMe, DC07-SMe), DCO8SPy, glutathione, alkyl thiols, pyridylthio, aryl thiols, nitropyridylthio, hydroxycarbonylpyridylthio, (Nitro) hydroxycarbonyl-pyridylthio and the like can be used provided that such disulfides are capable of undergoing a disulfide-exchange reaction for coupling the prodrug with the cell binder. The linker may optionally further comprise a spacer region interposed between the reactive group of the moiety that enables linkage and the reactive group of the 2-carboxy-indole or 2-carboxy-benzofuran derivative moiety.
바람직하게, 상기 링커는 다음 화학식을 갖는다:Preferably, the linker has the formula:
-G-D-(Z)p-S-Z'-G-D- (Z) p-S-Z '
상기식에서,In the above formula,
G는 단일 또는 이중 결합, -O-, -S- 또는 -NT-이고;G is a single or double bond, -O-, -S- or -NT-;
D는 단일 결합 또는 -E-, -E-NT-, -E-NT-F-, -E-O-, -E-O-F-, -E-NT-CO-, -E-NT-CO-F-, -E-CO-, -CO-E-, -E-CO-F, -E-S-, -E-S-F-, -E-NT-C-S-, -E-NT-CS-F-이며;D is a single bond or -E-, -E-NT-, -E-NT-F-, -EO-, -EOF-, -E-NT-CO-, -E-NT-CO-F-,- E-CO-, -CO-E-, -E-CO-F, -ES-, -ESF-, -E-NT-CS-, -E-NT-CS-F-;
T는 H 또는 C1-C6 분지 또는 선형 알킬이고;T is H or C 1 -C 6 branched or linear alkyl;
E 및 F는 동일하거나 상이하며, 선형 또는 분지 -(OCH2CH2)i알킬(OCH2CH2)j-, -알킬(OCH2CH2)i-알킬-, -(OCH2CH2)i-, -(OCH2CH2)i시클로알킬(OCH2CH2)j-, -(OCH2CH2)i헤테로사이클릭(OCH2CH2)j-, -(OCH2CH2)i아릴(OCH2CH2)j-, -(OCH2CH2)i헤테로아릴(OCH2CH2)j-, -알킬-(OCH2CH2)i알킬(OCH2CH2)j-, -알킬(OCH2CH2)i-, -알킬-(OCH2CH2)i시클로알킬(OCH2CH2)j-, -알킬(OCH2CH2)i헤테로사이클릭(OCH2CH2)j-, -알킬(OCH2CH2)i아릴(OCH2CH2)j-, -알킬(OCH2CH2)i헤테로아릴(OCH2CH2)j-, 시클로알킬-알킬-, -알킬-시클로알킬-, -헤테로사이클릭-알킬-, -알킬-헤테로사이클릭-, -알킬-아릴-, 아릴-알킬-, -알킬-헤테로아릴-, -헤테로아릴-알킬- 중에서 독립적으로 선택되고;E and F are the same or different and are linear or branched-(OCH 2 CH 2 ) i alkyl (OCH 2 CH 2 ) j- , -alkyl (OCH 2 CH 2 ) i -alkyl-,-(OCH 2 CH 2 ) i -,-(OCH 2 CH 2 ) i Cycloalkyl (OCH 2 CH 2 ) j -,-(OCH 2 CH 2 ) i Heterocyclic (OCH 2 CH 2 ) j -,-(OCH 2 CH 2 ) i Aryl (OCH 2 CH 2 ) j -,-(OCH 2 CH 2 ) i heteroaryl (OCH 2 CH 2 ) j- , -alkyl- (OCH 2 CH 2 ) i alkyl (OCH 2 CH 2 ) j -,- Alkyl (OCH 2 CH 2 ) i- , -alkyl- (OCH 2 CH 2 ) i Cycloalkyl (OCH 2 CH 2 ) j- , -alkyl (OCH 2 CH 2 ) i Heterocyclic (OCH 2 CH 2 ) j -, -Alkyl (OCH 2 CH 2 ) i aryl (OCH 2 CH 2 ) j- , -alkyl (OCH 2 CH 2 ) i heteroaryl (OCH 2 CH 2 ) j- , cycloalkyl-alkyl-, -alkyl- Independently selected from cycloalkyl-, -heterocyclic-alkyl-, -alkyl-heterocyclic-, -alkyl-aryl-, aryl-alkyl-, -alkyl-heteroaryl- and -heteroaryl-alkyl-;
i 및 j는 동일하거나 상이한 정수이며 0, 1 내지 2000 중에서 독립적으로 선택되고;i and j are the same or different integers and are independently selected from 0, 1 to 2000;
Z는 선형 또는 분지 -알킬-이고;Z is linear or branched-alkyl-;
p는 0 또는 1이며;p is 0 or 1;
Z'는 H, 티올 보호 기, 예를 들어 COR, R20 또는 SR2O인데, 여기서 R20은 H, 메틸, 알킬, 임의로 치환된 시클로알킬, 아릴, 헤테로아릴 또는 헤테로사이클릭을 나타낸다.Z 'is H, a thiol protecting group, for example COR, R 20 or SR 2O , wherein R 20 represents H, methyl, alkyl, optionally substituted cycloalkyl, aryl, heteroaryl or heterocyclic.
다음 양태 또는 그의 모든 조합이 바람직하다:The following aspects or all combinations thereof are preferred:
- G가 단일 결합 또는 -NT-이고;G is a single bond or -NT-;
- G가 -NT-이며;G is -NT-;
- D가 -CO-E-이고;D is -CO-E-;
- E가 선형 또는 분지 -알킬-이며;E is linear or branched-alkyl-;
- p가 0이고;p is 0;
- Z'가 H 또는 SR2O이며, R20이 알킬 또는 헤테로아릴을 나타낸다.Z 'is H or SR 2O and R 20 represents alkyl or heteroaryl.
링커의 바람직한 양태에는 (CH2)pNHCO(CH2)mSZ, (CH2)pNHCOC6H4(CH2)mSZ, (CH2)pO(CH2)mSZ, (CH2)pNHCO(CH2)m(OCH2CH2)nSZ, (CH2)pNHCOC6H4(CH2)m(OCH2CH2)nSZ, (CH2)pO(CH2)m(OCH2CH2)nSZ, (CH2)pNHCO(CH2)mCH(Me)SZ, (CH2)pNHCOC6H4(CH2)mCH(Me)SZ, (CH2)pO(CH2)mCH(Me)SZ, (CH2)pNHCO(CH2)m(OCH2CH2)nCH(Me)SZ, (CH2)pNHCOC6H4(CH2)m(OCH2CH2)nCH(Me)SZ, (CH2)pO(CH2)m(OCH2CH2)nCH(Me)SZ, (CH2)pNHCO(CH2)mC(Me)2SZ, (CH2)pNHCOC6H4(CH2)mC(Me)2SZ, (CH2)pO(CH2)mC(Me)2SZ, (CH2)pNHCO(CH2)m(OCH2CH2)nC(Me)2SZ, (CH2)pNHCOC6H4(CH2)m(OCH2CH2)nC(Me)2SZ, (CH2)pO(CH2)m(OCH2CH2)nC(Me2)SZ, (CH2)pOC6H4(CH2)mSZ, (CH2)pOC6H4(CH2)mCH(Me)SZ, (CH2)pOC6H4(CH2)m(OCH2CH2)nCHMeSZ, (CH2)pOC6H4(CH2)mCMe2SZ 또는 (CH2)pOC6H4(CH2)m(OCH2CH2)nC(Me)2SZ [여기서, Z는 H, 티올 보호 기, 예를 들어 Ac, R8 또는 SR8을 나타내고, R8은 메틸, 선형 알킬, 분지 알킬, 사이클릭 알킬, 단순 또는 치환된 아릴 또는 헤테로사이클릭을 나타내며, m은 0 내지 10의 정수를 나타내고, n은 0 내지 2000의 정수를 나타내며, p는 0 내지 10의 정수를 나타낸다]가 포함된다.Preferred embodiments of the linker include (CH 2 ) p NHCO (CH 2 ) m SZ, (CH 2 ) p NHCOC 6 H 4 (CH 2 ) m SZ, (CH 2 ) p O (CH 2 ) m SZ, (CH 2) ) p NHCO (CH 2 ) m (OCH 2 CH 2 ) n SZ, (CH 2 ) p NHCOC 6 H 4 (CH 2 ) m (OCH 2 CH 2 ) n SZ, (CH 2 ) p O (CH 2 ) m (OCH 2 CH 2 ) n SZ, (CH 2 ) p NHCO (CH 2 ) m CH (Me) SZ, (CH 2 ) p NHCOC 6 H 4 (CH 2 ) m CH (Me) SZ, (CH 2 ) p O (CH 2 ) m CH (Me) SZ, (CH 2 ) p NHCO (CH 2 ) m (OCH 2 CH 2 ) n CH (Me) SZ, (CH 2 ) p NHCOC 6 H 4 (CH 2 ) m (OCH 2 CH 2 ) n CH (Me) SZ, (CH 2 ) p O (CH 2 ) m (OCH 2 CH 2 ) n CH (Me) SZ, (CH 2 ) p NHCO (CH 2 ) m C (Me) 2 SZ, (CH 2 ) p NHCOC 6 H 4 (CH 2 ) m C (Me) 2 SZ, (CH 2 ) p O (CH 2 ) m C (Me) 2 SZ, (CH 2 ) p NHCO (CH 2 ) m (OCH 2 CH 2 ) n C (Me) 2 SZ, (CH 2 ) p NHCOC 6 H 4 (CH 2 ) m (OCH 2 CH 2 ) n C (Me) 2 SZ, ( CH 2 ) p O (CH 2 ) m (OCH 2 CH 2 ) n C (Me 2 ) SZ, (CH 2 ) p OC 6 H 4 (CH 2 ) m SZ, (CH 2 ) p OC 6 H 4 ( CH 2 ) m CH (Me) SZ, (CH 2 ) p OC 6 H 4 (CH 2 ) m (OCH 2 CH 2 ) n CHMeSZ, (CH 2 ) p OC 6 H 4 (CH 2 ) m CMe 2 SZ Or (CH 2 ) p OC 6 H 4 (CH 2 ) m (OC H 2 CH 2 ) n C (Me) 2 SZ [where Z represents H, a thiol protecting group such as Ac, R 8 or SR 8 and R 8 represents methyl, linear alkyl, branched alkyl, cyclic alkyl , A simple or substituted aryl or heterocyclic, m represents an integer from 0 to 10, n represents an integer from 0 to 2000, p represents an integer from 0 to 10.
R8로써 나타낸 선형 알킬의 예에는 메틸, 에틸, 프로필, 부틸, 펜틸 및 헥실이 포함된다. R8로써 나타낸 분지 알킬의 예에는 이소프로필, 이소부틸, 2급-부틸, 3급-부틸, 이소펜틸 및 1-에틸-프로필이 포함된다. R8로써 나타낸 사이클릭 알킬의 예에는 시클로프로필, 시클로부틸, 시클로펜틸 및 시클로헥실이 포함된다. R8로써 나타낸 단순 아릴의 예에는 페닐 및 나프틸이 포함된다. R8로써 나타낸 치환된 아릴의 예에는 알킬 기, 할로겐, 예를 들어 Cl, Br, F, 니트로 기, 아미노 기, 설폰산 기, 카복실산 기, 히드록시 기 및 알콕시 기로 치환된 페닐 또는 나프틸과 같은 아릴이 포함된다. R8로써 나타낸 헤테로사이클릭은 헤테로 원자가 O, N, 및 S 중에서 선택되는 화합물이고, 그의 예에는 푸릴, 피롤릴, 피리딜 (예: 2-치환된 피리미딘 기) 및 티오펜이 포함된다.Examples of linear alkyl represented by R 8 include methyl, ethyl, propyl, butyl, pentyl and hexyl. Examples of branched alkyl represented by R 8 include isopropyl, isobutyl, secondary-butyl, tert-butyl, isopentyl and 1-ethyl-propyl. Examples of cyclic alkyl represented by R 8 include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl. Examples of simple aryls represented as R 8 include phenyl and naphthyl. Examples of substituted aryls represented by R 8 include alkyl groups, halogens such as phenyl or naphthyl substituted with Cl, Br, F, nitro groups, amino groups, sulfonic acid groups, carboxylic acid groups, hydroxy groups and alkoxy groups; The same aryl is included. Heterocyclics represented by R 8 are compounds in which the hetero atom is selected from O, N, and S, examples of which include furyl, pyrrolyl, pyridyl (eg, 2-substituted pyrimidine groups) and thiophene.
링커의 가장 바람직한 양태에는 (CH2)pNHCO(CH2)2SH, (CH2)pNHCO(CH2)2C(Me)2SH, (CH2)pNHCO(CH2)2SSCH3, (CH2)pNHCO(CH2)2(OCH2CH2)nSH, (CH2)pNHCO(CH2)2(OCH2CH2)nC(Me)2SSCH3 및 (CH2)pNHCO(CH2)2(OCH2CH2)nSSMe가 포함된다.Most preferred embodiments of the linker include (CH 2 ) p NHCO (CH 2 ) 2 SH, (CH 2 ) p NHCO (CH 2 ) 2 C (Me) 2 SH, (CH 2 ) p NHCO (CH 2 ) 2 SSCH 3 , (CH 2 ) p NHCO (CH 2 ) 2 (OCH 2 CH 2 ) n SH, (CH 2 ) p NHCO (CH 2 ) 2 (OCH 2 CH 2 ) n C (Me) 2 SSCH 3 and (CH 2 ) p NHCO (CH 2 ) 2 (OCH 2 CH 2 ) n SSMe.
화학식 II 내지 XI 내에서, R, R', R1 내지 R6은 동일하거나 상이할 수 있고 독립적으로, 수소, C1-C3 선형 알킬, 메톡시, 히드록실, 1급 아미노, 2급 아미노, 3급 아미노 또는 아미도, 또는 링커를 나타내는데, 단 R, R', R1 내지 R6 중의 적어도 하나는 링커이다. 바람직하게는 R이 링커이다. 1급 아미노 기-함유 치환체의 예는 메틸 아미노, 에틸 아미노 및 이소프로필 아미노이다. 2급 아미노 기-함유 치환체의 예는 디메틸 아미노, 디에틸 아미노 및 에틸-프로필 아미노이다. 아미노 기의 예에는 N-메틸-아세트아미도, N-메틸-프로피온아미도, N-아세트아미도 및 N-프로피온아미도가 포함된다.Within Formulas II-XI, R, R ', R 1- R 6 may be the same or different and independently represent hydrogen, C 1 -C 3 linear alkyl, methoxy, hydroxyl, primary amino, secondary amino , Tertiary amino or amido, or a linker, provided that at least one of R, R ', R 1 to R 6 is a linker. Preferably R is a linker. Examples of primary amino group-containing substituents are methyl amino, ethyl amino and isopropyl amino. Examples of secondary amino group-containing substituents are dimethyl amino, diethyl amino and ethyl-propyl amino. Examples of amino groups include N-methyl-acetamido, N-methyl-propionamido, N-acetamido and N-propionamido.
화학식 II 내지 XI 내에서, R7은 seco-시클로프로파벤즈인돌-함유 세포독성 약물의 수 용해도를 증강시켜 주는 생체내-절단 가능한 보호 기이고, 이러한 기는 다음 화학식 XII의 설포산 함유 페닐 카바메이트 중에서 선택될 수 있다:Within Formulas II-XI, R 7 is an in vivo-cleavable protecting group that enhances the water solubility of seco -cyclopropagenzindole-containing cytotoxic drugs, such groups being sulfoic acid containing phenyl carbamate of Formula XII Can be chosen from:
화학식 XIIFormula XII

상기식에서,In the above formula,
R9는 H 또는 C1-C4 분지 또는 선형 알킬이고; R10, R11, R12 또는 R13은 동일하거나 상이하고, 이중 하나 이상이 -X-SO3 -M+ 기인데, M+는 H+ 또는 IA 족의 원자로부터 유래되는 양이온, 예를 들어 Na+, K+ 등이며, X는 직접 결합 또는 C1 내지 C4 선형 또는 분지 알킬, 알케닐 또는 알키닐, -O알킬-, -S알킬- 및 아미노알킬 중에서 선택된 스페이서이며, 기타 R10, R11, R12 또는 R13은 H, C1-C6 분지 또는 선형 알킬, -O알킬, -S알킬, 히드록실, 1급 아미노, 2급 아미노, 또는 아미도, 할라이드, 니트로, 아지도이다.R 9 is H or C 1 -C 4 branched or linear alkyl; R 10, R 11, R 12 or R 13 are the same or different and are one or more double -X-SO 3 - groups inde + M, M + is a cation, for, for example, derived from the atom of the H + or a Group IA Na + , K + and the like, X is a direct bond or a spacer selected from C1 to C4 linear or branched alkyl, alkenyl or alkynyl, -Oalkyl-, -Salkyl- and aminoalkyl, other R 10 , R 11 , R 12 or R 13 is H, C 1 -C 6 branched or linear alkyl, —Oalkyl, —Salkyl, hydroxyl, primary amino, secondary amino, or amido, halide, nitro, azido.
R9, R10, R11, R12 또는 R13으로써 나타낸 선형 알킬의 예에는 메틸, 에틸, 프로필, 부틸, 펜틸 및 헥실이 포함된다. R9, R10, R11, R12 또는 R13으로써 나타낸 분지 알킬의 예에는 이소프로필, 이소부틸, 2급-부틸, 3급-부틸, 이소펜틸 및 1-에틸-프로필이 포함된다. R9, R10, R11, R12 또는 R13으로써 나타낸 사이클릭 알킬의 예에는 시클로프로필, 시클로부틸, 시클로펜틸 및 시클로헥실이 포함된다.Examples of linear alkyl represented by R 9 , R 10 , R 11 , R 12 or R 13 include methyl, ethyl, propyl, butyl, pentyl and hexyl. Examples of branched alkyl represented by R 9 , R 10 , R 11 , R 12 or R 13 include isopropyl, isobutyl, secondary-butyl, tert-butyl, isopentyl and 1-ethyl-propyl. Examples of cyclic alkyl represented by R 9 , R 10 , R 11 , R 12 or R 13 include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
따라서, 설폰산 함유 페닐 카바메이트는 혈청 및 혈장에서 일어나는 생리적 pH 하에서의 가수분해에 의해 절단 가능한 것으로 밝혀졌다.Thus, sulfonic acid containing phenyl carbamate has been found to be cleavable by hydrolysis under physiological pH that occurs in serum and plasma.
본 발명의 CC-1065 유사체의 설파이드 또는 디설파이드-함유 및 머캅토-함유 프로드럭을 대상으로 하여, 항온 배양 조건하 시험관 내에서 불필요한 각종 세포주의 증식을 억제시킬 수 있는 능력에 관하여 평가할 수 있다. 예를 들어, 라모스 세포주 및 HL60과 같은 세포주가 이들 화합물의 세포독성을 평가하는 데에 용이하게 사용될 수 있다. 평가하고자 하는 세포를 상기 화합물에 24시간 동안 노출시킬 수 있고, 생존 세포 분획을 공지된 방법에 의해 직접 검정하여 측정하였다. 이어서, 이러한 검정 결과로부터 IC50 값을 계산할 수 있다.The sulfide or disulfide-containing and mercapto-containing prodrugs of the CC-1065 analogs of the present invention can be evaluated for their ability to inhibit the proliferation of unnecessary various cell lines in vitro under incubated conditions. For example, cell lines such as Ramos cell lines and HL60 can be readily used to assess the cytotoxicity of these compounds. The cells to be assessed can be exposed to the compound for 24 hours and the viable cell fractions measured by direct assay by known methods. The IC 50 value can then be calculated from these assay results.
본원에 사용된 바와 같은 표현 "세포 결합제와 연결 가능한"이란 해당 유도체를 세포 결합제에 결합시키는 데에 적합한 하나 이상의 링커 또는 그의 전구체를 포함하는 CC-1065 유사체 프로드럭을 지칭하고; 바람직한 링커는 티올, 설파이드 또는 디설파이드 결합, 또는 그의 전구체를 함유한다.As used herein, the expression “connectable with a cell binder” refers to a CC-1065 analog prodrug comprising one or more linkers or precursors thereof suitable for binding the derivative to the cell binder; Preferred linkers contain thiols, sulfide or disulfide bonds, or precursors thereof.
본원에 사용된 바와 같은 표현 "세포 결합제와 연결된"이란 적합한 링커 또는 그의 전구체를 통하여 세포 결합제와 결합된 하나 이상의 CC-1065 유사체 프로드럭을 포함하는 접합체 분자를 지칭하고; 바람직한 링커는 티올, 설파이드 또는 디설파이드 결합, 또는 그의 전구체를 함유한다.As used herein, the expression “linked to a cell binder” refers to a conjugate molecule comprising one or more CC-1065 analog prodrugs bound to the cell binder via a suitable linker or precursor thereof; Preferred linkers contain thiols, sulfide or disulfide bonds, or precursors thereof.
본원에서의 표현 "세포 결합제"에는 변형된 세포 결합제가 포함되기도 하는데, 이러한 세포 결합제는 반응성 작용기, 예를 들어 CC-1065 유사체 프로드럭의 링커의 머캅토, 설파이드 또는 디설파이드 결합에 대한 상기 세포 결합제의 반응성을 개선시켜 주는 변형제에 의해 변형된다. 이러한 변형제에는 N-설포석신이미딜-4-(5-니트로-2-피리딜디티오)부타노에이트 (SSNPB), 석신이미딜 4-[N-말레이미도메틸]시클로헥산-1-카복실레이트 (SMCC), 4-(2-피리딜디티오)부타노산 N-히드록시석신이미드 에스테르 (SPDB), 및 다음에 논의된 바와 같은 기타 작용제가 포함된다.The expression “cell binding agent” herein also includes modified cell binding agents, such cell binding agents for the binding of reactive cell groups such as the mercapto, sulfide or disulfide bonds of linkers of CC-1065 analogue prodrugs. Modified by modifiers that improve reactivity. Such modifiers include N-sulfosuccinimidyl-4- (5-nitro-2-pyridyldithio) butanoate (SSNPB), succinimidyl 4- [N-maleimidomethyl] cyclohexane-1- Carboxylate (SMCC), 4- (2-pyridyldithio) butanoic acid N-hydroxysuccinimide ester (SPDB), and other agents as discussed below.
본원에 사용된 바와 같은 용어 "유사체"는 특이적 화합물 또는 그의 부류의 화학적으로 변형된 형태를 포함하고 이러한 화합물 또는 부류의 특징적인 제약 및/또는 약리학적 활성을 보유하고 있는 화합물을 지칭한다.As used herein, the term “analogue” refers to a compound that includes a chemically modified form of a specific compound or class thereof and retains the characteristic pharmaceutical and / or pharmacological activity of that compound or class.
본원에 사용된 바와 같은 용어 "환자"는 본원에 기재된 한 가지 이상의 질병 및 질환을 앓고 있거나 또는 이러한 질병 및 질환에 걸릴 가능성이 있는 동물, 예를 들어 육종, 사업 또는 보호 목적으로 유용한 동물, 또는 바람직하게 인간 또는 어린이를 지칭한다.As used herein, the term “patient” refers to an animal suffering from, or likely to suffer from, one or more of the diseases and disorders described herein, eg, an animal useful for breeding, business, or protective purposes, or preferably Refers to human or child.
본원에 사용된 바와 같은 "치료적 유효량"은 본원에 기재된 질병 및 질환 증상을 예방, 저하, 제거, 치료 또는 억제하는 데에 유효한 본 발명의 화합물의 양을 지칭한다. 용어 "억제"는 본원에 기재된 질병 및 질환의 진행을 느리게 하거나, 차단시키거나, 저지시키거나 또는 중지시킬 수 있는 모든 과정을 지칭하지만, 반드시 모든 질병과 질환 증상을 완전히 없애는 것을 의미하지는 않으며, 이에는 예방적 처치가 포함된다.As used herein, “therapeutically effective amount” refers to an amount of a compound of the invention that is effective for preventing, decreasing, eliminating, treating or inhibiting the diseases and disease symptoms described herein. The term “inhibition” refers to any process that can slow, block, arrest or stop the progression of the diseases and conditions described herein, but does not necessarily mean the complete elimination of all diseases and disease symptoms, Prophylactic treatment is included.
본원에 사용된 바와 같은 용어 "제약상 허용 가능한"이란 신중한 의학적 판단 범위 내에서, 과도한 독성, 자극, 알레르기 반응 또는 기타 문제가 되는 합병증을 유발시키지 않으면서도 합리적인 유익/유해 비율에 알맞은 정도로 인간 및 동물의 조직과 접촉시키기에 적합한 화합물, 물질, 부형제, 조성물 또는 투여 형태를 지칭한다.As used herein, the term “pharmaceutically acceptable” means human and animal to the extent appropriate for reasonable benefit / hazardous ratios without causing excessive toxicity, irritation, allergic reactions or other problematic complications, within the scope of careful medical judgment. It refers to a compound, substance, excipient, composition or dosage form suitable for contact with tissues of.
본원에 사용된 바와 같은 "제약상 허용 가능한 염"은 모 화합물의 산 또는 염기 염을 만듬으로써 이를 변형시킨, 해당 화합물의 유도체를 지칭한다. 제약상 허용 가능한 염에는, 예를 들어 비독성 무기 또는 유기 산으로부터 형성된 모 화합물의 통상적인 비독성 염 또는 4급 암모늄 염이 포함된다. 예를 들어, 이러한 통상적인 비독성 염에는 무기 산, 예를 들어 염산, 브롬화수소산, 황산, 설팜산, 인산, 질산 등으로부터 유래된 염; 및 유기 산, 예를 들어 아세트산, 프로피온산, 석신산, 타르타르산, 시트르산, 메탄설폰산, 벤젠설폰산, 글루코론산, 글루탐산, 벤조산, 살리실산, 톨루엔설폰산, 옥살산, 푸마르산, 말레산, 락트산 등으로부터 제조된 염이 포함된다. 추가로 부가 염에는 암모늄 염, 예를 들어 트로메타민, 메글루민, 에폴라민 등, 금속 염, 예를 들어 나트륨, 칼륨, 칼슘, 아연 또는 마그네슘이 포함된다.As used herein, “pharmaceutically acceptable salts” refers to derivatives of such compounds, which are modified by making acid or base salts of the parent compound. Pharmaceutically acceptable salts include, for example, conventional nontoxic salts or quaternary ammonium salts of the parent compound formed from nontoxic inorganic or organic acids. For example, such conventional non-toxic salts include salts derived from inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, sulfamic acid, phosphoric acid, nitric acid, and the like; And organic acids such as acetic acid, propionic acid, succinic acid, tartaric acid, citric acid, methanesulfonic acid, benzenesulfonic acid, glucononic acid, glutamic acid, benzoic acid, salicylic acid, toluenesulfonic acid, oxalic acid, fumaric acid, maleic acid, lactic acid, and the like. Salts are included. Further addition salts include ammonium salts such as tromethamine, meglumine, epolamine and the like, metal salts such as sodium, potassium, calcium, zinc or magnesium.
본 발명의 제약상 허용 가능한 염은 염기성 또는 산성 잔기를 함유하는 모 화합물로부터 통상적인 화학적 방법에 의해 합성할 수 있다. 일반적으로, 이러한 염은 이들 화합물의 자유 산 또는 염기 형태를 물 또는 유기 용매, 또는 이들 둘의 혼합물 중에서 화학량적 양의 적당한 염기 또는 산과 반응시킴으로써 제조할 수 있다. 일반적으로, 비수성 매질, 예를 들어 에테르, 에틸 아세테이트, 에탄올, 이소프로판올 또는 아세토니트릴이 바람직하다. 적합한 염에 관한 목록은 본원에 참고로 도입된 다음 문헌을 참고할 수 있다 [참고: Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA, 1985, p. 1418].Pharmaceutically acceptable salts of the invention can be synthesized by conventional chemical methods from the parent compound containing a basic or acidic moiety. In general, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or an organic solvent, or a mixture of both. In general, non-aqueous media such as ether, ethyl acetate, ethanol, isopropanol or acetonitrile are preferred. For a list of suitable salts, see the following references incorporated herein by reference: Remington's Pharmaceutical Sciences , 17 th ed., Mack Publishing Company, Easton, PA, 1985, p. 1418].
추가 목적에 따르면, 본 발명은 또한, 본 발명의 화합물을 제조하는 방법에 관한 것이다.According to a further object, the present invention also relates to a process for preparing the compound of the present invention.
본 발명의 화합물 및 공정은 당업자에게 널리 공지된 수 많은 방식으로 제조할 수 있다. 본 발명의 화합물은, 예를 들어 다음에 기재된 방법을 적용 또는 적응시키거나, 또는 당업자가 잘 인지하는 바와 같은 변동을 줌으로써 합성할 수 있다. 적당한 변형과 치환은 용이하게 명백하고 널리 공지되어 있거나 당해 전문 분야에 관한 과학지로부터 용이하게 획득할 수 있다.The compounds and processes of the present invention can be prepared in a number of ways well known to those skilled in the art. The compounds of the present invention can be synthesized, for example, by applying or adapting the methods described below, or by making variations as is well recognized by those skilled in the art. Appropriate modifications and substitutions are readily apparent and well known or can be readily obtained from scientific journals of ordinary skill in the art.
특히 이러한 방법은 다음 문헌을 참고할 수 있다 [참고: R.C. Larock, Comprehensive Organic Transformations, Wiley-VCH Publishers, 1999].In particular, this method may be referred to the following literature [RC Larock, Comprehensive Organic Transformations , Wiley-VCH Publishers, 1999].
본 발명의 화합물이 하나 이상의 비대칭적으로 치환된 탄소 원자를 함유할 수 있고 광학 활성 또는 라세미 형태로 분리할 수 있다는 것을 인지해야 할 것이다. 따라서, 특이적 입체화학 또는 이성체 형태가 구체적으로 표시되지 않는 한은, 특정 구조의 모든 키랄, 부분 입체이성체, 라세미 형태 및 모든 기하 이성체 형태가 고려된다. 이러한 광학 활성 형태를 제조하고 분리하는 방법은 당해 분야에 널리 공지되어 있다. 예를 들어, 입체이성체의 혼합물은 표준 기술, 예를 들어 라세미 형태의 분해, 정상, 역상 및 키랄 크로마토그래피, 우선적 염 형성, 재결정화 등; 또는 키랄 출발 물질로부터의 키랄 합성 또는 표적 키랄 중심의 계획적인 합성에 의해 분리시킬 수 있다.It will be appreciated that the compounds of the present invention may contain one or more asymmetrically substituted carbon atoms and may be separated in optically active or racemic forms. Thus, unless specific stereochemical or isomeric forms are specifically indicated, all chiral, diastereomeric, racemic and all geometric isomeric forms of a particular structure are contemplated. Methods of making and isolating such optically active forms are well known in the art. For example, mixtures of stereoisomers can be prepared using standard techniques such as degradation of racemic forms, normal, reversed and chiral chromatography, preferential salt formation, recrystallization, and the like; Or by chiral synthesis from chiral starting materials or by deliberate synthesis of the target chiral center.
본 발명의 화합물은 각종 합성 경로에 의해 제조할 수 있다. 시약과 출발 물질은 시판되고 있거나, 또는 당업자가 널리 공지된 기술에 의해 용이하게 합성할 수 있다. 달리 표시되지 않는 한, 모든 치환체는 앞서 정의된 바와 같다.The compounds of the present invention can be prepared by various synthetic routes. Reagents and starting materials are commercially available or can be readily synthesized by those skilled in the art by well known techniques. Unless otherwise indicated, all substituents are as defined above.
후술되는 반응에서는, 반응성 작용기, 예를 들어 히드록시, 아미노, 이미노, 티오 또는 카복시 기를 보호시킬 필요가 있는데, 이들 작용기가 불필요하게 반응에 참여하는 것을 피하기 위해 최종 생성물 내에 보존되는 것이 요망된다. 통상적인 보호 기는 표준 실시에 따라서 사용할 수 있다 [참고: 예를 들어, T.W. Greene and P. G. M. Wuts in Protective Groups in Organic Chemistry, 3rd ed., John Wiley and Sons, 1999; J. F. W. McOmie in Protective Groups in Organic Chemistry, Plenum Press, 1973].In the reactions described below, it is necessary to protect reactive functional groups such as hydroxy, amino, imino, thio or carboxy groups, which are desired to be preserved in the final product to avoid unnecessary participation in the reaction. Conventional protecting groups can be used according to standard practice. See, eg, TW Greene and PGM Wuts in Protective Groups in Organic Chemistry , 3 rd ed., John Wiley and Sons, 1999; JFW McOmie in Protective Groups in Organic Chemistry , Plenum Press, 1973.
몇몇 반응은 염기의 존재하에 수행할 수도 있다. 이러한 반응에 사용될 염기의 종류에 관한 특별한 제한은 없고, 이러한 유형의 반응에 통상적으로 사용되고 있는 모든 염기가 마찬가지로 본원에 사용될 수 있는데, 단 분자의 기타 부분에 불리한 영향을 미치지 말아야 한다. 적합한 염기의 예에는 수산화나트륨, 탄산칼륨, 트리에틸아민, 알칼리 금속 수소화물, 예를 들어 수소화나트륨 및 수소화칼륨; 알킬리튬 화합물, 예를 들어 메틸리튬 및 부틸리튬; 및 알칼리 금속 알콕사이드, 예를 들어 나트륨 메톡사이드 및 나트륨 에톡사이드가 포함된다.Some reactions may be carried out in the presence of a base. There is no particular limitation as to the type of base to be used in this reaction, and any base commonly used in this type of reaction can likewise be used herein, provided that it does not adversely affect other parts of the molecule. Examples of suitable bases include sodium hydroxide, potassium carbonate, triethylamine, alkali metal hydrides such as sodium hydride and potassium hydride; Alkyllithium compounds such as methyllithium and butyllithium; And alkali metal alkoxides such as sodium methoxide and sodium ethoxide.
통상적으로는, 반응을 적합한 용매 중에서 수행할 수 있다. 각종 용매를 사용할 수 있는데, 단 이에 관여하는 시약이나 반응 자체에 불리한 영향을 미치지 말아야 한다. 적합한 용매의 예에는 방향족, 지방족 또는 지환족 탄화수소일 수 있는 탄화수소, 예를 들어 헥산, 시클로헥산, 벤젠, 톨루엔 및 크실렌; 아미드, 예를 들어 디메틸포름아미드; 알코올, 예를 들어 에탄올 및 메탄올; 및 에테르, 예를 들어 디에틸 에테르 및 테트라히드로푸란이 포함된다.Typically, the reaction can be carried out in a suitable solvent. Various solvents may be used provided that they do not adversely affect the reagents involved or the reaction itself. Examples of suitable solvents include hydrocarbons which may be aromatic, aliphatic or cycloaliphatic hydrocarbons, for example hexane, cyclohexane, benzene, toluene and xylene; Amides such as dimethylformamide; Alcohols such as ethanol and methanol; And ethers such as diethyl ether and tetrahydrofuran.
반응은 광범위한 온도 범위에 걸쳐 수행할 수 있다. 일반적으로, 본 발명자들은 해당 반응을 0 내지 150℃ (보다 바람직하게는 약 실온 내지 100℃) 하에서 수행하는 것이 편리하다는 것을 발견하였다. 반응에 소요되는 시간은 많은 요인들, 특히 반응 온도와 시약의 종류에 따라서 매우 다양할 수도 있다. 그러나, 반응을 상기 언급된 바람직한 조건 하에서 수행한다면, 3시간 내지 20시간이면 통상 족할 것이다.The reaction can be carried out over a wide temperature range. In general, the inventors have found it convenient to carry out the reaction under 0 to 150 ° C (more preferably about room temperature to 100 ° C). The time required for the reaction may vary greatly depending on many factors, especially the reaction temperature and the type of reagent. However, if the reaction is carried out under the above-mentioned preferred conditions, 3 to 20 hours will usually be sufficient.
이로써 제조된 화합물은 통상적인 수단에 의해 반응 혼합물로부터 회수할 수 있다. 예를 들어, 반응 혼합물로부터 용매를 증류 제거하거나, 또는 필요한 경우 반응 혼합물로부터 용매를 증류 제거한 후, 잔류물을 물에 주입한 다음 수-불혼화성 유기 용매로 추출하고, 이러한 추출물로부터 용매를 증류 제거함으로써 화합물을 회수할 수 있다. 부가적으로, 경우에 따라 널리 공지된 각종 기술, 예를 들어 재결정화, 재침전 또는 각종 크로마토그래피 기술, 특히 칼럼 크로마토그래피 또는 분취 박막 크로마토그래피에 의해 생성물을 추가로 정제할 수 있다.The compound thus prepared can be recovered from the reaction mixture by conventional means. For example, after distilling off the solvent from the reaction mixture, or distilling off the solvent from the reaction mixture, if necessary, the residue is poured into water and then extracted with a water-immiscible organic solvent, and the solvent is distilled off from this extract. The compound can be recovered by doing so. In addition, the product may be further purified, if desired, by various well-known techniques, for example recrystallization, reprecipitation or various chromatographic techniques, in particular column chromatography or preparative thin layer chromatography.
본 발명에 따르는 화학식의 화합물의 제조 방법이 본 발명의 추가의 목적이다.A further object of the invention is a process for the preparation of compounds of the formula according to the invention.
본 발명에 따르는 화학식의 화합물의 제조 방법은 CC-1065의 상응하는 유사체의 알킬화 부분의 페놀성 기를 설폰산 함유 카바메이트 작용기에 의해 보호시키는 단계를 포함한다. 이러한 반응은 다음 문헌에 기재된 바와 같이 OH 기를 카바메이트 작용기로 보호시키는 방법을 적용 또는 적응시킴으로써 수행할 수 있다 [참고: T.W. Greene and P. G. M. Wuts in Protective Groups in Organic Chemistry, 3rd ed., John Wiley and Sons, 1999 or J. F. W. McOmie in Protective Groups in Organic Chemistry, Plenum Press, 1973].The process for the preparation of the compounds of formula according to the invention comprises the step of protecting the phenolic groups of the alkylated portion of the corresponding analog of CC-1065 by sulfonic acid containing carbamate functional groups. This reaction can be carried out by applying or adapting the method of protecting an OH to a carbamate functional group, as described in the following described in reference:. TW Greene and PGM Wuts in Protective Groups in Organic Chemistry, 3 rd ed, John Wiley and Sons, 1999 or JFW McOmie in Protective Groups in Organic Chemistry , Plenum Press, 1973.
일반적으로, 상기 반응은 CDI/DMA와 다음 화학식의 상응하는 아미노페닐설포네이트 유도체를 이용하여 수행한다:In general, the reaction is carried out using CDI / DMA and the corresponding aminophenylsulfonate derivatives of the formula:

상기식에서,In the above formula,
R9, R10, R11, R12 및 R13은 화학식 XII에서 정의된 바와 같다.R 9 , R 10 , R 11 , R 12 and R 13 are as defined in formula (XII).
본 공정은 수득된 생성물을 분리시키는 추가의 단계를 포함할 수 있다.The process may comprise an additional step of separating the product obtained.
세포 결합제의 제조Preparation of Cell Binding Agents
본 발명의 프로드럭 화합물은 유효한 치료제로서 세포 결합제와의 접합체로서 사용할 수 있다. 세포 결합제는 현재 공지되어 있거나 공지될 어떠한 종류의 것일 수도 있고, 이에는 펩티드 및 비-펩티드가 포함된다. 일반적으로, 이들은 항체 (특히, 모노클로날 항체), 또는 하나 이상의 결합 부위를 함유하는 항체 단편, 림포카인, 호르몬, 성장 인자, 영양분-수송 분자 (예: 트랜스페린), 또는 기타 모든 세포 결합성 분자 또는 물질일 수 있다.The prodrug compounds of the invention can be used as conjugates with cell binding agents as effective therapeutic agents. Cell binding agents may be of any kind known or to be known now, including peptides and non-peptides. Generally, they are antibodies (especially monoclonal antibodies), or antibody fragments containing one or more binding sites, lymphokines, hormones, growth factors, nutrient-transporting molecules (eg transferrin), or any other cell binding agent. It may be a molecule or a substance.
사용될 수 있는 세포 결합제의 보다 구체적인 예에는 다음이 포함된다:More specific examples of cell binding agents that may be used include the following:
- 모노클로날 항체;Monoclonal antibodies;
- 단일 쇄 항체;Single chain antibodies;
- 항체의 단편, 예를 들어 Fab, Fab', F(ab')2 및 Fv [참고; Parham, 131 J. Immunol. 2895-2902 (1983); Spring et al, 113 J. Immunol. 470-478 (1974); Nisonoff et al, 89 Arch. Biochem. Biophys. 230-244 (1960)];Fragments of the antibodies, eg Fab, Fab ', F (ab') 2 and Fv [see; Parham, 131 J. Immunol. 2895-2902 (1983); Spring et al, 113 J. Immunol. 470-478 (1974); Nisonoff et al, 89 Arch. Biochem. Biophys. 230-244 (1960);
- 인터페론;Interferon;
- 펩티드;Peptides;
- 림포카인, 예를 들어 IL-2, IL-3, IL-4, IL-6;Lymphokines, for example IL-2, IL-3, IL-4, IL-6;
- 호르몬, 예를 들어 인슐린, TRH (티로트로핀 방출 호르몬), MSH (멜라닌 세포-자극 호르몬), 스테로이드 호르몬, 예를 들어 안드로겐 및 에스트로겐;Hormones such as insulin, TRH (tyrotropin releasing hormone), MSH (melanin cell-stimulating hormone), steroid hormones such as androgens and estrogens;
- 성장 인자 및 집락 자극 인자, 예를 들어 EGF, TGFα, 인슐린 유사 성장 인자 (IGF-I, IGF-II), G-CSF, M-CSF 및 GM-CSF [참고: Burgess, 5 Immunology Today 155-158 (1984)];Growth factors and colony stimulating factors such as EGF, TGFα, insulin-like growth factors (IGF-I, IGF-II), G-CSF, M-CSF and GM-CSF [Burgess, 5 Immunology Today 155- 158 (1984);
- 비타민, 예를 들어 엽산염; 및Vitamins, for example folate; And
- 트랜스페린 [참고: O'Keefe et al, 260 J. Biol. Chem. 932-937 (1985)].Transferrin [see O'Keefe et al, 260 J. Biol. Chem. 932-937 (1985).
모노클로날 항체 기술로 인해 극도로 선택적인 세포 결합제를 특이적 모노클로날 항체 형태로 생성할 수 있게 된다. 당해 분야에서 특히 널리 공지되어 있는 것은 마우스, 랫트, 햄스터 또는 기타 모든 포유류를 관심있는 항원, 예를 들어 본래의 표적 세포, 표적 세포로부터 분리한 항원, 완전 바이러스, 약독화 완전 바이러스, 및 바이러스성 단백질, 예를 들어 바이러스성 외피 단백질로 면역시킴으로써 생성된 모노클로날 항체를 창출시키는 기술이다.Monoclonal antibody technology allows the production of highly selective cell binding agents in the form of specific monoclonal antibodies. Particularly well known in the art are mice, rats, hamsters or any other mammals of interest, eg, original target cells, antigens isolated from target cells, complete viruses, attenuated complete viruses, and viral proteins. For example monoclonal antibodies produced by immunization with viral envelope proteins.
적당한 세포 결합제를 선별하는 것은 표적화시키고자 하는 특별한 세포 집단에 따라서 선택되지만, 일반적인 모노클로날 항체에서는 적당한 경우에 입수 가능한 것이 바람직하다.Selecting the appropriate cell binding agent is selected according to the particular cell population to be targeted, but in general monoclonal antibodies are preferably available where appropriate.
예를 들어, 모노클로날 항체 MY9는 CD33 항원과 특이적으로 결합하는 뮤린 IgG1 항체이고 [참고: J. D. Griffin et al 8 Leukemia Res., 521 (1984)], 이는 표적 세포가 급성 골수성 백혈병 (AML) 질환에서와 같이 CD33을 발현하는 경우에 사용할 수 있다. 유사하게, 모노클로날 항체 항-B4는 B 세포 상의 CD19 항원과 결합하는 뮤린 IgG1이고 [참고: Nadler et al, 131 J. Immunol. 244-250 (1983)], 이는 비호지킨 (non-Hodgkin's) 림프종 또는 만성 림프아구성 백혈병에서와 같이 표적 세포가 상기 항원을 발현하는 B 세포 또는 병든 세포인 경우에 사용할 수 있다.For example, the monoclonal antibody MY9 is a murine IgG 1 antibody that specifically binds to the CD33 antigen (JD Griffin et al 8 Leukemia Res., 521 (1984)), wherein the target cell is acute myeloid leukemia (AML). ) Can be used to express CD33 as in disease. Similarly, monoclonal antibody anti-B4 is a murine IgG 1 that binds to CD19 antigen on B cells and is described by Nadler et al, 131 J. Immunol. 244-250 (1983)], which can be used when the target cell is a B cell or diseased cell that expresses the antigen, such as in non-Hodgkin's lymphoma or chronic lymphocytic leukemia.
부가적으로, 골수 세포와 결합하는 GM-CSF를 급성 골수성 백혈병으로부터의 병든 세포에 대한 세포 결합제로서 사용할 수 있다. 활성화 T-세포와 결합하는 IL-2는 이식편 거부를 예방하고, 이식편 대 숙주 질병을 치료 및 예방하며, 급성 T-세포 백혈병을 치료하는 데에 사용할 수 있다. 멜라닌 세포와 결합하는 MSH는 흑색종을 치료하기 위해 사용할 수 있다.Additionally, GM-CSF which binds to bone marrow cells can be used as cell binding agent for diseased cells from acute myeloid leukemia. IL-2, which binds to activated T-cells, can be used to prevent graft rejection, to treat and prevent graft-versus-host disease, and to treat acute T-cell leukemia. MSH that binds to melanocytes can be used to treat melanoma.
프로드럭 접합체의 제조Preparation of Prodrug Conjugates
프로드럭과 세포 결합제의 접합체는 어떠한 기술을 이용해서도 형성시킬 수 있다. seco-CBI 유사체와 커플링된 인돌릴, 벤조푸라닐, 비스-인돌릴, 비스-벤조푸라닐, 인돌릴-벤조푸라닐 또는 벤조푸라닐-인돌릴 유도체는 자유 아미노 기를 함유하도록 제조한 다음, 산 불안정 링커 또는 광 불안정 링커를 통하여 항체 또는 기타 세포 결합제와 연결시킬 수 있다. 프로드럭 화합물은 적합한 서열을 지닌 펩티드와 축합시킨 다음, 이를 세포 결합제와 연결시켜 펩티다제 불안정 링커를 생성시킬 수 있다. 1급 히드록실 기를 함유하는 세포독성 화합물을 제조하고, 이를 석시닐화하고 세포 결합제에 연결시켜 접합체를 생성시키는데, 이는 세포내 에스테라제에 의해 절단되어 자유 프로드럭을 방출시킬 수 있다. 바람직하게는, PEG-함유 스페이서를 사용하거나 사용하지 않으면서, 자유 또는 보호된 티올 기를 함유하는 프로드럭 화합물을 합성한 다음, 하나 이상의 설파이드, 디설파이드 또는 티올-함유 프로드럭을 디설파이드 결합 또는 티오에테르 결합을 통하여 세포 결합제에 용이하게 공유적으로 연결시킨다.Conjugates of prodrugs and cell binders can be formed using any technique. Indolyl, benzofuranyl, bis-indolyl, bis-benzofuranyl, indolyl-benzofuranyl or benzofuranyl-indolyl derivatives coupled with seco- CBI analogues are prepared to contain free amino groups, The acid labile linker or light labile linker may be used to link the antibody or other cell binder. Prodrug compounds can be condensed with peptides with suitable sequences and then linked with cell binders to generate peptidase labile linkers. Cytotoxic compounds containing primary hydroxyl groups are prepared and succinylated and linked to cell binders to generate conjugates, which can be cleaved by intracellular esterases to release free prodrugs. Preferably, synthesis of prodrug compounds containing free or protected thiol groups, with or without PEG-containing spacers, is followed by one or more sulfide, disulfide or thiol-containing prodrugs to be disulfide bonds or thioether bonds. It is easily covalently linked to the cell binding agent through.
본 발명의 대표적인 접합체는 CC-1065 유사체의 프로드럭과 항체, 항체 단편, 표피 성장 인자 (EGF), 멜라닌 세포 자극 호르몬 (MSH), 갑상선 자극 호르몬 (TSH), 에스트로겐, 에스트로겐 유사체, 안드로겐 및 안드로겐 유사체와의 접합체이다.Representative conjugates of the invention include prodrugs and antibodies, antibody fragments, epidermal growth factor (EGF), melanocyte stimulating hormone (MSH), thyroid stimulating hormone (TSH), estrogens, estrogen analogs, androgens and androgen analogs of CC-1065 analogs. It is a conjugate with.
CC-1065 유사체의 프로드럭과 세포 결합제의 각종 접합체를 제조하는 것에 관한 대표적 예가 다음에 기재되어 있다.Representative examples of making various conjugates of prodrugs of the CC-1065 analog and cell binders are described below.
디설파이드 링커: 항 huMy-9-6은 대다수의 급성 골수성 백혈병 (AML) 증례를 포함하여, 인간 골수 세포 표면 상에서 발견되는 CD33 항원에 대항하여 유도된 뮤린 모노클로날 항체 My-9-6의 유전적 인간화 형태이다 [참고: E.J. Favaloro, K.F. Bradstock, A. Kabral, P. Grimsley & M. C. Berndt, Disease Markers, 5(4):215 (1987); M.G. Hoffee, D. Tavares, R.J. Lutz, Robert J., PCT Int. Appl. (2004) WO 20043344]. My-9-6은 접합체를 제조하는 데에 사용할 수 있다. 이 항체는 기존에 보고된 바와 같이 [참고: J. Carlsson, H. Drevin & R. Axen, Biochem. J., 173:723 (1978)] N-석신이미딜-3-피리딜디티오 프로피오네이트로 변형시켜 항체 분자당 평균 4개의 피리딜디티오 기를 도입시킨다. 이와 같이 변형시킨 항체를 티올 함유 프로드럭과 반응시켜 디설파이드 연결된 접합체를 생성시킨다. Disulfide linker : anti-huMy-9-6 is the genetic of the murine monoclonal antibody My-9-6 induced against the CD33 antigen found on the surface of human bone marrow cells, including the majority of acute myeloid leukemia (AML) cases Humanized forms [see: EJ Favaloro, KF Bradstock, A. Kabral, P. Grimsley & MC Berndt, Disease Markers , 5 (4): 215 (1987); MG Hoffee, D. Tavares, RJ Lutz, Robert J., PCT Int. Appl. (2004) WO 20043344. My-9-6 can be used to prepare conjugates. This antibody was previously reported as described in J. Carlsson, H. Drevin & R. Axen, Biochem. J. , 173: 723 (1978)] is modified with N-succinimidyl-3-pyridyldithio propionate to introduce an average of four pyridyldithio groups per antibody molecule. The antibody thus modified is reacted with a thiol containing prodrug to produce a disulfide linked conjugate.
티오에테르 링커: 본 발명의 티올 함유 유도체는 기존에 보고된 바와 같이 [참고: 미국 특허 제5,208,020호] 티오에테르 링커를 통하여 항체와 기타 세포 결합제에 연결시킬 수 있다. 이러한 항체 또는 기타 세포 결합제는 시판용 화합물, 예를 들어 N-석신이미딜 4-(말레이미도메틸)시클로헥산카복실레이트 (SMCC), N-석신이미딜-4-(N-말레이미도메틸)-시클로헥산-1-카복시-(6-아미도카프로에이트) [이는 SMCC의 "장쇄" 유사체 (LC-SMCC)이다]를 이용하여 변형시킬 수 있다. 이들 가교결합성 시약은 말레이미도-기재 잔기로부터 유래된 비-절단 가능한 링커를 형성한다.Thioether Linkers : Thiol-containing derivatives of the present invention can be linked to antibodies and other cell binders via thioether linkers as previously reported [US Pat. No. 5,208,020]. Such antibodies or other cell binding agents are commercially available compounds such as N-succinimidyl 4- (maleimidomethyl) cyclohexanecarboxylate (SMCC), N-succinimidyl-4- (N-maleimidomethyl) -cyclo Hexane-1-carboxy- (6-amidocaproate), which is a "long chain" analog of SMCC (LC-SMCC), can be modified. These crosslinkable reagents form non-cleavable linkers derived from maleimido-based residues.
할로아세틸-기재 잔기를 포함하는 가교결합성 시약에는 N-석신이미딜-4-(요오도아세틸)-아미노벤조에이트 (SIAB), N-석신이미딜 요오도아세테이트 (SIA), N-석신이미딜 브로모아세테이트 (SBA) 및 N-석신이미딜 3-(브로모-아세트아미도)프로피오네이트 (SBAP)가 포함된다. 이들 가교결합성 시약은 할로아세틸-기재 잔기로부터 유래된 비-절단 가능한 링커를 형성한다. 변형된 세포 결합제를 티올 함유 약물과 반응시켜 티오에테르-연결된 접합체를 제공할 수 있다.Crosslinking reagents comprising haloacetyl-based residues include N-succinimidyl-4- (iodoacetyl) -aminobenzoate (SIAB), N-succinimidyl iodoacetate (SIA), N-succinimi Dill bromoacetate (SBA) and N-succinimidyl 3- (bromo-acetamido) propionate (SBAP). These crosslinkable reagents form non-cleavable linkers derived from haloacetyl-based residues. Modified cell binders can be reacted with thiol containing drugs to provide thioether-linked conjugates.
산 불안정 링커: 본 발명의 아미노 기 함유 프로드럭은 기존에 보고된 바와 같이 [참고: W. A. Blattler et al, Biochemistry 24, 1517-1524 (1985); 미국 특허 제4,542,225호, 제4,569,789호, 제4,618,492호, 제4,764,368호] 산 불안정 링커를 통하여 항체 및 기타 세포 결합제에 연결시킬 수 있다. Acid labile linkers : The amino group containing prodrugs of the present invention are reported as previously described by WA Blattler et al, Biochemistry 24, 1517-1524 (1985); US Pat. Nos. 4,542,225, 4,569,789, 4,618,492, 4,764,368] can be linked to antibodies and other cell binding agents through acid labile linkers.
유사하게, 본 발명의 히드라지도 기 함유 프로드럭은 산 불안정 히드라존 링커를 통하여 항체의 탄수화물 부분과 기타 세포 결합제에 연결시킬 수 있다 [히드라존 링커의 예는 문헌 (B. C. Laguzza et al, J. Med. Chem., 32, 548-555 (1989); R. S. Greenfield et al, Cancer Res., 50, 6600-6607 (1990))을 참고할 수 있다].Similarly, the hydrazido group-containing prodrugs of the invention can be linked to the carbohydrate moiety and other cell binders of an antibody via an acid labile hydrazone linker. Examples of hydrazone linkers are described in BC Laguzza et al, J. Med. Chem., 32, 548-555 (1989); RS Greenfield et al, Cancer Res., 50, 6600-6607 (1990)).
광 불안정 링커: 본 발명의 아민 기 함유 프로드럭은 기존에 보고된 바와 같이 [참고: P. Senter et al, Photochemistry and Photobiology, 42, 231-237 (1985); 미국 특허 제4,625,014호] 광 불안정 링커를 통하여 항체 및 기타 세포 결합제에 연결시킬 수 있다. Light labile linkers : The amine group containing prodrugs of the present invention have been previously described as described in P. Senter et al, Photochemistry and Photobiology , 42, 231-237 (1985); US Patent No. 4,625,014] can be linked to antibodies and other cell binders via a light labile linker.
펩티다제 불안정 링커: 본 발명의 아민 기 함유 프로드럭은 또한, 펩티드 스페이서를 통하여 세포 결합제에 연결시킬 수 있다. 약물과 거대분자상 단백질 운반체 간의 짧은 펩티드 스페이서는 혈청에서는 안정적이지만, 세포내 펩티다제에 의해 용이하게 가수분해된다는 사실이 기존에 밝혀진 바 있다 [참고: A. Trouet et al, Proc. Natl. Acad. Sci., 79, 626-629 (1982)]. 아미노 기 함유 프로드럭은 1-에틸-3-(3-디메틸아미노프로필)카보디이미드-HCl (EDC-HCl) 등의 축합제를 사용하여 펩티드와 축합시켜 펩티드 유도체를 수득하고, 이를 세포 결합제에 연결시킬 수 있다. Peptidase labile linkers : The amine group containing prodrugs of the invention can also be linked to cell binders via peptide spacers. Short peptide spacers between drugs and macromolecular protein carriers have been previously found to be stable in serum but readily hydrolyzed by intracellular peptidase [see A. Trouet et al, Proc. Natl. Acad. Sci., 79, 626-629 (1982). The amino group containing prodrug is condensed with the peptide using a condensing agent such as 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide-HCl (EDC-HCl) to obtain a peptide derivative, which is added to the cell binder. Can be connected.
에스테라제 불안정 링커: 히드록시 알킬 기를 보유하고 있는 본 발명의 프로드럭은 석신산 무수물을 이용하여 석시닐화한 다음, 이를 세포 결합제에 연결시켜 접합체를 생성시킬 수 있는데, 이는 세포내 에스테라제에 의해 절단되어 자유 약물을 방출시킬 수 있다 [참고: 예를 들어, E. Aboud-Pirak et al, Biochem Pharmacol., 38, 641-648 (1989)]. Esterase labile linkers : The prodrugs of the present invention having hydroxy alkyl groups can be succinylated with succinic anhydride and then linked to cell binders to generate conjugates, which are incorporated into intracellular esterases. Can be cleaved to release free drug (see, eg, E. Aboud-Pirak et al, Biochem Pharmacol ., 38, 641-648 (1989)).
항체, 항체 단편, 단백질 또는 펩티드 호르몬, 단백질 또는 펩티드 성장 인자 및 기타 단백질의 CC-1065 유사체 접합체는 공지된 방법에 의해 동일한 방식으로 제조한다. 예를 들어, 펩티드 및 항체는 N-석신이미딜 3-(2-피리딜디티오)프로피오네이트, N-석신이미딜 4-(2-피리딜디티오)펜타노에이트 (SPP), 4-석신이미딜-옥시카보닐-α-메틸-α-(2-피리딜 디티오)-톨루엔 (SMPT), N-석신이미딜-3-(2-피리딜디티오) 부티레이트 (SDPB), 석신이미딜 피리딜-디티오프로피오네이트 (SPDP), 4-(2-피리딜디티오)부타노산 N-히드로석신이미드 에스테르 (SPDB), 석신이미딜 4-[N-말레이미도메틸]시클로헥산-1-카복실레이트 (SMCC), N-설포석신이미딜-3-(2-(5-니트로-피리딜디티오)부티레이트 (SSNPB), 2-이미노티올란 또는 S-아세틸석신산 무수물과 같은 가교 결합성 시약을 이용하여 공지된 방법에 의해 변형시킬 수 있다 [참고: Carlsson et al, 173 Biochem. J. 723-737 (1978); Blattler et al, 24 Biochem. 1517-1524 (1985); Lambert et al, 22 Biochem. 3913-3920 (1983); Klotz et al, 96 Arch. Biochem. Biophys. 605 (1962); and Liu et al, 18 Biochem. 690 (1979), Blakey and Thorpe, 1 Antibody, Immunoconjugates & Radiopharmaceuticals, 1-16 (1988), Worrell et al 1 Anti-Cancer Drug Design 179-184 (1986)]. 이어서, 이로써 유래된 자유 또는 보호된 티올 함유 세포 결합제를 디설파이드- 또는 티올-함유 CC-1065 프로드럭 유사체와 반응시켜 접합체를 생성시킨다. 이러한 접합체는 표준 칼럼 크로마토그래피, HPLC 또는 겔 여과에 의해 정제할 수 있다.CC-1065 analogue conjugates of antibodies, antibody fragments, protein or peptide hormones, protein or peptide growth factors and other proteins are prepared in the same manner by known methods. For example, peptides and antibodies include N-succinimidyl 3- (2-pyridyldithio) propionate, N-succinimidyl 4- (2-pyridyldithio) pentanoate (SPP), 4 -Succinimidyl-oxycarbonyl-α-methyl-α- (2-pyridyldithio) -toluene (SMPT), N-succinimidyl-3- (2-pyridyldithio) butyrate (SDPB), Succinimidyl pyridyl-dithiopropionate (SPDP), 4- (2-pyridyldithio) butanoic acid N-hydrosuccinimide ester (SPDB), succinimidyl 4- [N-maleimidomethyl] Cyclohexane-1-carboxylate (SMCC), N-sulfosuccinimidyl-3- (2- (5-nitro-pyridyldithio) butyrate (SSNPB), 2-iminothiolane or S-acetylsuccinic acid Crosslinking reagents, such as anhydrides, can be modified by known methods. See Carlsson et al, 173 Biochem. J. 723-737 (1978); Blattler et al, 24 Biochem . 1517-1524 (1985). Lambert et al, 22 Biochem . 3913-3920 (1983); Klotz et al, 96 Arch. Biochem. Biophy . s 605 (1962);. and Liu et al, 18 Biochem 690 (1979), Blakey and Thorpe, 1 Antibody, Immunoconjugates & Radiopharmaceuticals, 1-16 (1988), Worrell et al 1 Anti-Cancer Drug Design 179-184 (1986)] The free or protected thiol containing cell binder thus obtained is then reacted with a disulfide- or thiol-containing CC-1065 prodrug analog to generate a conjugate, which is prepared by standard column chromatography, HPLC or gel. It can be purified by filtration.
바람직하게, 모노클로날 항체- 또는 세포 결합제-CC-1065 프로드럭 유사체 접합체는 CC-1065 프로드럭 유사체를 전달할 수 있는, 상기 논의된 바와 같이 디설파이드 결합을 통하여 연결되는 것이다.Preferably, the monoclonal antibody- or cell binder-CC-1065 prodrug analog conjugate is one that is linked via disulfide bonds, as discussed above, capable of delivering a CC-1065 prodrug analog.
이러한 세포 결합성 접합체는 공지된 방법, 예를 들어 모노클로날 항체를 석신이미딜 피리딜-디티오프로피오네이트 (SPDP)로 변형시킴으로써 제조할 수 있다 [참고: Carlsson et al, 173 Biochem. J. 723-737 (1978)]. 이어서, 이로써 생성되는 티오피리딜 기는 티올 함유 CC-1065 프로드럭 유사체로 처리함으로써 전위시켜 디설파이드 연결된 접합체를 생성시킨다. 이러한 방법에 의한 접합은 미국 특허 제5,585,499호 (이는 본원에 참고로 도입된다)에 상세히 기재되어 있다.Such cell binding conjugates can be prepared by known methods, for example by modifying monoclonal antibodies with succinimidyl pyridyl-dithiopropionate (SPDP). Carlsson et al, 173 Biochem. J. 723-737 (1978). The resulting thiopyridyl group is then transposed by treatment with a thiol containing CC-1065 prodrug analog to produce a disulfide linked conjugate. Conjugation by this method is described in detail in US Pat. No. 5,585,499, which is incorporated herein by reference.
또 다른 한편, 아릴디티오-CC-1065 프로드럭 유사체의 경우에는, CC-1065 프로드럭 유사체의 아릴-티올을 앞서 항체 분자 내로 도입된 설프히드릴 기에 의해 직접 전위시킴으로써 세포 결합성 접합체를 형성시킨다. 디설파이드 브릿지를 통하여 연결된 1 내지 10개의 CC-1065 프로드럭 유사체를 함유하는 접합체는 어느 한 방법에 의해 용이하게 제조한다.On the other hand, in the case of aryldithio-CC-1065 prodrug analogues, cell-bonded conjugates are formed by directly transposing the aryl-thiol of the CC-1065 prodrug analogues with sulfhydryl groups previously introduced into the antibody molecule. . Conjugates containing 1 to 10 CC-1065 prodrug analogs linked through disulfide bridges are readily prepared by either method.
추가의 목적에 따르면, 본 발명은 또한, 본 발명의 접합체 분자 또는 상기 정의된 바와 같은 화학식 I의 화합물을 제약상 허용 가능한 담체와 함께 포함하는 제약 조성물에 관한 것이다.According to a further object, the present invention also relates to a pharmaceutical composition comprising a conjugate molecule of the invention or a compound of formula (I) as defined above together with a pharmaceutically acceptable carrier.
추가 목적에 따르면, 본 발명은 또한, 표적 세포 또는 표적 세포를 함유하는 조직을 본 발명에 따르는 유효량의 제약 조성물과 접촉시키는 것을 포함하는, 세포, 바람직하게는 선택된 세포 집단의 성장을 억제하거나 사멸시키는 방법에 관한 것이다.According to a further object, the present invention also provides for inhibiting or killing the growth of cells, preferably selected cell populations, comprising contacting the target cell or tissue containing the target cell with an effective amount of the pharmaceutical composition according to the invention. It is about a method.
선택된 세포 집단은 암성 및/또는 증식성 세포이다.The cell population of choice is cancerous and / or proliferative cells.
추가 목적에 따르면, 본 발명은 또한, 본 발명에 따르는 유효량의 제약 조성물을 이를 필요로 하는 환자에게 투여하는 것을 포함하는, 암을 치료하는 방법, 바람직하게는 암을 선택적으로 치료하는 방법에 관한 것이다.According to a further object, the present invention also relates to a method for treating cancer, preferably a method for selectively treating cancer, comprising administering to a patient in need thereof an effective amount of a pharmaceutical composition according to the invention. .
본 발명에 따르면, "암의 선택적 치료"는 정상 세포 및/또는 비-증식성 세포는 실질적으로 사멸시키지 않으면서 암성 세포 및/또는 증식성 세포를 사멸시키는 것을 지칭한다.According to the present invention, "selective treatment of cancer" refers to killing cancerous cells and / or proliferative cells without substantially killing normal and / or non-proliferative cells.
추가 목적에 따르면, 본 발명은 또한, 암 치료용 약물을 제조하기 위한, 본 발명의 접합체 분자 또는 상기 정의된 바와 같은 화학식 I의 화합물의 용도에 관한 것이다.According to a further object, the present invention also relates to the use of a conjugate molecule of the invention or a compound of formula (I) as defined above for the manufacture of a medicament for the treatment of cancer.
선택된 세포 집단의 성장을 억제시키는 방법은 시험관내, 생체내 또는 생체외에서 실시할 수 있다.Methods for inhibiting the growth of selected cell populations can be performed in vitro, in vivo or ex vivo.
시험관내 사용의 예에는 표적 항원을 발현하지 않는 목적하는 변이체를 제외한 모든 세포를 사멸시키거나 또는 바람직하지 못한 항원을 발현하는 변이체를 사멸시키기 위해 세포 배양물을 처리하는 것이 포함된다.Examples of in vitro use include treating cell culture to kill all cells except for the desired variant that does not express the target antigen or to kill variants that express undesirable antigens.
비-임상 시험관내 사용 조건은 당업자가 용이하게 결정할 수 있다.Non-clinical in vitro use conditions can be readily determined by one skilled in the art.
생체외 사용의 예에는 병든 세포 또는 악성 세포를 사멸시키기 위해 자기 유래의 골수를 동일한 환자에게 이식하기에 앞서 이러한 골수를 처치하는 것; 적격한 T 세포를 사멸시키고 이식편 대 숙주 질병 (GVHD)을 예방하기 위해 골수를 이식하기에 앞서 이를 처치하는 것이 포함된다.Examples of ex vivo use include treating such bone marrow prior to transplanting its own bone marrow into the same patient to kill the diseased or malignant cells; Treatment of prior bone marrow transplantation to kill qualified T cells and prevent graft-versus-host disease (GVHD).
암 치료 또는 자가면역 질환 치료에 있어서 자기 유래 이식에 앞서 골수로부터 종양 세포 또는 림프계 세포를 제거하거나, 또는 GVHD를 예방하기 위해 이식하기에 앞서 동종 골수 또는 조직으로부터 T 세포 및 기타 림프계 세포를 제거하기 위한 임상 생체외 처치는 다음과 같이 수행할 수 있다. 골수를 환자 또는 기타 개개인으로부터 수거한 다음, 본 발명의 세포독성제가 부가된 혈청을 함유하는 매질 내에서 약 37℃ 하에 약 30분 내지 약 48시간 동안 약 10 μM 내지 1 pM의 농도 범위로 항온 배양한다. 정확한 항온 배양 시간과 농도 조건 (=용량)은 당업자가 용이하게 결정할 수 있다. 항온 배양 후, 골수 세포를 혈청 함유 매질로 세척하고, 공지된 방법에 따라서 정맥내 주입함으로써 환자에게 돌려주었다. 골수를 수거한 시간과 처치한 세포를 재주입하는 시간 사이에 전신 방사선 조사 또는 절제 화학요법과 같은 기타 치료를 환자에게 제공하는 상황 하에서는, 상기와 같이 처치한 골수 세포를 표준 의학 장비를 이용하여 액체 질소에 동결 저장한다.To remove tumor cells or lymphoid cells from bone marrow prior to autologous transplantation in cancer treatment or autoimmune disease treatment, or to remove T cells and other lymphoid cells from allogeneic bone marrow or tissue prior to transplantation to prevent GVHD Clinical ex vivo treatment can be performed as follows. Bone marrow is harvested from patients or other individuals, and then incubated in a medium containing serum to which the cytotoxic agent of the invention is added at a concentration ranging from about 10 μM to 1 pM at about 37 ° C. for about 30 minutes to about 48 hours. do. The exact incubation time and concentration conditions (= dose) can be readily determined by one skilled in the art. After incubation, the bone marrow cells were returned to the patient by washing with serum containing medium and intravenously injecting according to known methods. Under the circumstances of providing the patient with other treatments, such as systemic irradiation or ablation chemotherapy, between the time of bone marrow harvest and the time of reinjecting the treated cells, the treated bone marrow cells may be removed using standard medical equipment. Store frozen in nitrogen.
임상 생체내 사용의 경우에는, 본 발명의 세포독성제를 용액으로서 공급하여 멸균성 및 내독소 수준에 관하여 시험하거나, 또는 동결건조된 고형물로서 공급하여 이를 주사용 멸균수에 재용해시킬 수 있다. 접합체 투여에 관한 적합한 프로토콜의 예는 다음과 같다: 접합체는 6주 동안 매주 정맥내 일시주입(bolus)으로 투여한다. 일시주입 용량은 인간 혈청 알부민을 부가할 수 있는 정상 식염수 50 내지 400 ml에 제공된다 (예를 들어, 0.5 내지 1 ml의 인간 혈청 알부민 농축 용액, 100 mg/ml). 투여량은 매주 약 50 ㎍ 내지 10 mg/체중 kg (정맥내 투여)일 것이다 (1회 주입당 10 ㎍ 내지 100 mg/kg). 처치한지 6주 후에 환자에게 제2 치료 과정을 실시할 수 있다. 투여 경로, 부형제, 희석제, 투여량, 시간 등과 관련한 구체적인 임상 프로토콜은 임상적 상황에 근거하여 당업자가 결정할 수 있다.For clinical in vivo use, the cytotoxic agents of the present invention may be supplied as a solution to test for sterile and endotoxin levels, or may be supplied as lyophilized solids and redissolved in sterile water for injection. Examples of suitable protocols for conjugate administration are as follows: The conjugate is administered by intravenous bolus weekly for 6 weeks. The bolus dose is given in 50 to 400 ml of normal saline to which human serum albumin can be added (e.g., 0.5 to 1 ml concentrated human serum albumin solution, 100 mg / ml). The dosage will be about 50 μg to 10 mg / kg body weight (intravenously) per week (10 μg to 100 mg / kg per injection). Six weeks after treatment, the patient may undergo a second course of treatment. Specific clinical protocols with respect to the route of administration, excipients, diluents, dosages, times, and the like can be determined by one skilled in the art based on clinical context.
선택된 세포 집단을 사멸시키는 생체내 또는 생체외 방법에 따라서 치료할 수 있는 의학적 질환의 예에는 모든 유형의 악성 종양, 예를 들어 폐암, 유방암, 결장암, 전립선암, 신장암, 췌장암, 난소암 및 림프계 기관 암; 흑색종; 자가 면역 질환, 예를 들어 전신성 루푸스, 류마티스성 관절염 및 다발성 경화증; 이식편 거부, 예를 들어 신장 이식체 거부, 간 이식체 거부, 폐 이식체 거부, 심장 이식체 거부 및 골수 이식체 거부; 이식편 대 숙주 질병; 바이러스성 감염, 예를 들어 CMV 감염, HIV 감염, AIDS 등; 세균성 감염; 및 기생충 감염, 예를 들어 편모충증, 아메바증, 주혈흡충증, 및 당업자에 의해 결정된 바와 같은 기타 질환이 포함된다.Examples of medical conditions that can be treated according to in vivo or ex vivo methods of killing selected cell populations include all types of malignant tumors, such as lung cancer, breast cancer, colon cancer, prostate cancer, kidney cancer, pancreatic cancer, ovarian cancer and lymphatic organs. cancer; Melanoma; Autoimmune diseases such as systemic lupus, rheumatoid arthritis and multiple sclerosis; Graft rejection, eg kidney transplant rejection, liver transplant rejection, lung transplant rejection, heart transplant rejection and bone marrow transplant rejection; Graft versus host disease; Viral infections such as CMV infection, HIV infection, AIDS, and the like; Bacterial infections; And parasitic infections such as flagella, amoeba, schistosomiasis, and other diseases as determined by one skilled in the art.
본원에 기재된 질병과 질환의 치료를 필요로 하는 대상체의 확인은 당업자의 능력과 지식 수준 내이다. 수의사 또는 당해 분야의 전문의는 임상 시험, 신체 검사, 의학/가족 병력 또는 생물학적 및 진단 시험을 사용함으로써 이러한 치료를 필요로 하는 대상체를 용이하게 확인할 수 있다.Identification of the subjects in need of treatment of the diseases and disorders described herein is within the skill and knowledge of those skilled in the art. Veterinarians or specialists in the art can readily identify subjects in need of such treatment by using clinical trials, physical examinations, medical / family history or biological and diagnostic tests.
치료적 유효량은 통상적인 기술을 사용하고 유사한 환경 하에 획득한 결과들을 관찰함으로써 담당 진단의에 의해 용이하게 결정될 수 있다. 치료적 유효량을 결정하는 데에 있어서, 수 많은 요인들이 담당 진단의에 의해 고려되는데, 이에는 대상체의 종; 그의 크기, 연령 및 일반적인 건강 상태; 관련된 구체적인 질병; 발병 정도 또는 질병 중증도; 개개 대상체의 반응; 투여된 특별한 화합물; 투여 방식; 투여된 제제의 생체내 이용 효율 특징; 선택된 용량 요법; 동시 투여되는 약물의 사용 여부; 및 기타 관련 상황이 포함되지만, 이에 제한되지 않는다.The therapeutically effective amount can be readily determined by the attending physician by using conventional techniques and observing the results obtained under similar circumstances. In determining the therapeutically effective amount, a number of factors are considered by the attending physician, including the species of the subject; Its size, age and general state of health; Related specific diseases; Prevalence or disease severity; The response of the individual subject; The particular compound administered; Mode of administration; Bioavailability characteristics of the administered agents; Selected dose regimen; Whether or not to use co-administered drugs; And other related situations, including but not limited to.
목적하는 생물학적 효과를 달성하는 데에 요구되는 화학식 I의 화합물 또는 접합체의 양은 수 많은 요인들, 예를 들어 이용된 화합물의 화학적 특징 (예: 소수성), 화합물의 효력, 질병 유형, 환자가 속하고 있는 종, 환자의 질병 상태, 투여 경로, 선택된 경로에 의한 화합물의 생체내 이용 효율; 투여하는데 필요한 투여 량, 전달 및 요법을 지시하는 모든 인자에 따라서 다양할 것이다.The amount of compound or conjugate of formula (I) required to achieve the desired biological effect depends on a number of factors, for example the chemical characteristics (eg hydrophobicity) of the compound used, the potency of the compound, the type of disease, Species present, disease state of the patient, route of administration, efficiency of bioavailability of the compound by the selected route; It will vary depending on all factors that dictate the dosage, delivery and therapy required to administer.
본원에 사용된 바와 같은"제약상 허용 가능한 부형제"에는 모든 담체, 희석제, 아주반트 (adjuvant) 또는 비히클, 예를 들어 방부제 또는 항산화제, 충진제, 붕해제, 습윤제, 유화제, 현탁화제, 용매, 분산 매질, 피복제, 항균제 및 항진균제, 등장성 및 흡수 지연제 등이 포함된다. 이러한 제약 활성 물질용 작용제 및 매질을 사용하는 것은 당해 분야에 널리 공지되어 있다. 통상적인 어떠한 매질이나 작용제도 활성 성분과 비화합성인 경우를 제외하고는, 이를 치료적 조성물에 사용하는 것이 고려된다. 보조 활성 성분을 적합한 치료적 배합물로서 해당 조성물 내로 혼입시킬 수도 있다.As used herein, “pharmaceutically acceptable excipients” include any carrier, diluent, adjuvant or vehicle, such as preservatives or antioxidants, fillers, disintegrants, wetting agents, emulsifiers, suspending agents, solvents, dispersions Media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like. The use of agents and media for such pharmaceutically active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, use thereof in the therapeutic compositions is contemplated. Supplementary active ingredients can also be incorporated into the compositions as suitable therapeutic combinations.
본 발명의 맥락에서, 본원에 사용된 바와 같은 "치료하는" 또는 "치료"는 이러한 용어가 적용되는 장애 또는 질환, 또는 이러한 장애 또는 질환의 한 가지 이상의 증상을 역전시키거나, 경감시키거나, 그의 진행을 억제시키거나 또는 예방하는 것을 의미한다.In the context of the present invention, “treating” or “treatment” as used herein refers to reversing, alleviating, or reducing the disorder or condition to which such term applies, or one or more symptoms of such disorder or disease. To inhibit or prevent progression.
일반적인 의미에서, 본 발명의 화합물은 비경구 투여의 경우에 0.1 내지 10% w/v 화합물을 함유하는 수성 생리적 완충 용액 중에 제공될 수 있다. 전형적인 용량 범위는 1일 1 ㎍/체중 kg 내지 0.1 g/체중 kg이고; 바람직한 용량 범위는 인간 어린이에게서 1일 0.01 mg/체중 kg 내지 10 mg/체중 kg 또는 등가 용량이다. 투여하고자 하는 약물의 바람직한 투여량은 질병 또는 장애의 진행 정도 및 유형, 특별한 환자의 전반적인 건강 상태, 선택된 화합물의 상대적 생물학적 효능, 화합물의 제형, 투여 경로 (정맥내, 근육내 등), 선택된 전달 경로에 의한 화합물의 약동학적 특성, 및 투여 속도 (일시주입 또는 지속적 주입) 및 투여 스케줄 (소정의 시간 동안의 반속 횟수)와 같은 변수에 좌우되는 것으로 예상된다.In a general sense, the compounds of the present invention may be provided in aqueous physiological buffer solutions containing 0.1 to 10% w / v compounds for parenteral administration. A typical dosage range is 1 μg / kg body weight to 0.1 g / kg body weight per day; Preferred dosage ranges are 0.01 mg / kg body weight to 10 mg / kg body weight or equivalent dose per day in human children. Preferred dosages of the drug to be administered are the extent and type of disease or disorder progressed, the general health of the particular patient, the relative biological efficacy of the selected compound, the formulation of the compound, the route of administration (intravenous, intramuscular, etc.), the chosen route of delivery. It is expected to depend on the pharmacokinetic properties of the compound by and variables such as the rate of administration (temporary or continuous infusion) and the schedule of administration (number of half-times for a given time).
본 발명의 화합물은 또한, 단위 투여 형태로 투여할 수 있는데, 용어 "단위 투여"는 환자에게 투여될 수 있고 용이하게 조작 및 패키징할 수 있어, 활성 화합물 그 자체를 포함하는 물리적 및 화학적으로 안정한 단위 용량으로서 존재하거나 또는 다음에 기재되는 바와 같은 제약상 허용 가능한 조성물로서 존재하는 단일 용량을 의미한다. 따라서, 전형적인 1일 총 용량 범위는 0.01 내지 100 mg/체중 kg이다. 일반적인 지침에 따르면, 인간의 경우에 단위 용량은 1일 1 mg 내지 3000 mg의 범위이다. 바람직하게는, 단위 용량 범위가 1 내지 500 mg으로, 이는 1일 1회 내지 6회 투여하고, 보다 더 바람직하게는 10 mg 내지 500 mg으로 1일 1회 투여한다. 본원에 제공된 화합물은 이를 하나 이상의 제약상 허용 가능한 부형제와 혼합함으로써 제약 조성물로 제형화할 수 있다. 이러한 단위 용량 조성물은 경구 투여용, 특히 정제, 단순 캅셀제 또는 연질 겔 캅셀제 형태; 또는 비내 투여용, 특히 산제, 비내 점적제 또는 에어로솔의 형태; 또는 피내 투여용, 예를 들어 전형적으로 연고, 크림, 로션, 겔 또는 스프레이 형태, 또는 경피 패치를 통하여 투여하도록 제조할 수 있다.The compounds of the present invention may also be administered in unit dosage form, wherein the term “unit dosage” may be administered to a patient and may be easily manipulated and packaged such that the physical and chemically stable units comprise the active compound itself. By a single dose is meant as a dose or as a pharmaceutically acceptable composition as described below. Thus, a typical daily total dose range is 0.01 to 100 mg / kg body weight. According to general guidelines, in humans the unit dose ranges from 1 mg to 3000 mg per day. Preferably, the unit dose ranges from 1 to 500 mg, which is administered once to six times daily, even more preferably from 10 mg to 500 mg once daily. The compounds provided herein can be formulated into pharmaceutical compositions by mixing them with one or more pharmaceutically acceptable excipients. Such unit dose compositions are for oral administration, in particular in the form of tablets, simple capsules or soft gel capsules; Or for intranasal administration, in particular in the form of powders, nasal drops or aerosols; Or for intradermal administration, eg, typically in the form of ointments, creams, lotions, gels or sprays, or via transdermal patches.
이러한 조성물은 단위 투여 형태로 투여하는 것이 편리할 수 있고, 이는 예를 들어, 다음 문헌에 기재된 바와 같이 제약 분야에 널리 공지된 방법에 의해 제조할 수 있다 [참고: Remington: The Science and Practice of Pharmacy, 20th ed.; Gennaro, A. R., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, 2000].Such compositions may be convenient to administer in unit dosage form, which may be prepared by methods well known in the art of pharmacy, for example, as described in Remington: The Science and Practice of Pharmacy. , 20 th ed .; Gennaro, AR, Ed .; Lippincott Williams & Wilkins: Philadelphia, PA, 2000].
바람직한 제형에는 본 발명의 화합물을 경우 또는 비경구 투여용으로 제형화시킨 제약 조성물이 포함된다.Preferred formulations include pharmaceutical compositions wherein the compounds of the invention are formulated for oral or parenteral administration.
경구 투여의 경우, 정제, 환제, 산제, 캅셀제, 트로키제 등은 다음 성분들 중의 한 가지 이상, 또는 유사한 종류의 화합물을 함유할 수 있다: 결합제, 예를 들어 미세 결정성 셀룰로스 또는 트라가칸드 검; 희석제, 예를 들어 전분 또는 락토스; 붕해제, 예를 들어 전분 및 셀룰로스 유도체; 윤활제, 예를 들어 마그네슘 스테아레이트; 활탁제, 예를 들어 콜로이드성 이산화규소; 감미제, 예를 들어 슈크로스 또는 삭카린; 또는 향미제, 예를 들어 페퍼민트 또는 메틸 살리실레이트. 캅셀제는 일반적으로 가소화제와 임의로 혼합된 젤라틴 블렌드로부터 만들어진 연질 캅셀제 또는 경질 캅셀제 형태 뿐만 아니라 전분 캅셀제 형태일 수 있다. 또한, 투여 단위 형태는 이러한 투여 단위의 물리적 형태를 변형시키는 기타 각종 물질, 예를 들어 당 피복제, 셸락 또는 장내 작용제를 함유할 수 있다. 기타 경구 투여 형태인 시럽 또는 엘릭서제는 감미제, 방부제, 염료, 착색제 및 향료를 함유할 수 있다. 또한, 활성 화합물을 신속 용해형, 변형 방출형 또는 지속 방출형 제제 및 제형 내로 혼입시킬 수 있는데, 이러한 지속 방출형 제형은 바람직하게 이봉 (bi-modal)이다. 바람직한 정제는 락토스, 옥수수 전분, 마그네슘 실리케이트, 크로스카멜로스 나트륨, 포비돈, 마그네슘 스테아레이트 또는 탈크를 어떠한 조합으로든 함유한다.For oral administration, tablets, pills, powders, capsules, troches and the like may contain one or more of the following ingredients, or compounds of a similar class: binders, for example microcrystalline cellulose or tragacanth gum ; Diluents such as starch or lactose; Disintegrants such as starch and cellulose derivatives; Lubricants such as magnesium stearate; Glidants such as colloidal silicon dioxide; Sweetening agents such as sucrose or saccharin; Or flavoring agents such as peppermint or methyl salicylate. Capsules may generally be in the form of soft or hard capsules made from gelatin blends optionally mixed with a plasticizer, as well as starch capsules. In addition, dosage unit forms may contain a variety of other materials that modify the physical form of such dosage units, such as sugar coatings, shellac or enteric agents. Syrups or elixirs in other oral dosage forms may contain sweetening, preservatives, dyes, colorants and flavorings. In addition, the active compounds may be incorporated into rapid dissolving, modified release or sustained release formulations and formulations, which are preferably bi-modal. Preferred tablets contain lactose, corn starch, magnesium silicate, croscarmellose sodium, povidone, magnesium stearate or talc in any combination.
비경구 투여용 액상 제제에는 멸균성 수성 또는 비수성 용제, 현탁제 및 에멀션이 포함된다. 액상 조성물은 결합제, 완충제, 방부제, 킬레이트제, 감미제, 향미제 및 착색제 등을 포함할 수도 있다. 비수성 용매에는 알코올, 프로필렌 글리콜, 폴리에틸렌 글리콜, 식물성 오일, 예를 들어 올리브유, 및 유기 에스테르, 예를 들어 에틸 올레에이트가 포함된다. 수성 담체에는 알코올과 물, 완충 매질 및 식염수와의 혼합물이 포함된다. 특히, 생적합성, 생분해성 락티드 중합체, 락티드/글리콜리드 공중합체, 또는 폴리옥시에틸렌-폴리옥시프로필렌 공중합체가 활성 화합물의 방출을 조절하는 데에 유용한 부형제일 수 있다. 정맥내 비히클에는 유체 및 영양분 보충제, 전해질 보충제, 예를 들어 링커 (Ringer) 덱스트로스에 의거한 보충제 등이 포함될 수 있다. 이들 활성 화합물에 대한 잠재적으로 유용한 기타 비경구 전달 시스템에는 에틸렌-비닐 아세테이트 공중합체 입자, 삼투압 펌프, 이식 가능한 주입 시스템 및 리포솜이 포함된다.Liquid preparations for parenteral administration include sterile aqueous or non-aqueous solvents, suspensions and emulsions. Liquid compositions may include binders, buffers, preservatives, chelating agents, sweetening agents, flavoring agents, coloring agents, and the like. Non-aqueous solvents include alcohols, propylene glycol, polyethylene glycols, vegetable oils such as olive oil, and organic esters such as ethyl oleate. Aqueous carriers include mixtures of alcohol with water, buffered media, and saline. In particular, biocompatible, biodegradable lactide polymers, lactide / glycolide copolymers, or polyoxyethylene-polyoxypropylene copolymers may be useful excipients in controlling the release of the active compound. Intravenous vehicles can include fluid and nutrient supplements, electrolyte supplements such as those based on Ringer dextrose, and the like. Other potentially useful parenteral delivery systems for these active compounds include ethylene-vinyl acetate copolymer particles, osmotic pumps, implantable infusion systems, and liposomes.
또 다른 투여 방식에는 흡입용 제형이 포함되는데, 이에는 건조 산제, 에어로솔 또는 점적제와 같은 수단이 포함된다. 이는, 예를 들어 폴리옥시에틸렌-9-라우릴 에테르, 글리코콜레이트 및 데옥시콜레이트를 함유하는 수성 용액, 또는 비내 점적제 형태로 투여하기 위한 오일성 용액, 또는 비내 투여될 겔일 수 있다. 볼내 투여용 제형에는, 예를 들어 로젠지 또는 향정이 포함되고, 이에는 향미 기재, 예를 들어 슈크로스 또는 아카시아 및 기타 부형제, 예를 들어 글리코콜레이트가 포함될 수도 있다. 직장 투여용으로 적합한 제형은 바람직하게, 고형에 의거한 담체, 예를 들어 코코아 버터와의 단위-용량 좌제로서 제시되고, 이에는 살리실레이트가 포함될 수 있다. 피부에 국소 적용하는데 적합한 제형은 바람직하게, 연고, 크림, 로션, 페이스트, 겔, 스프레이, 에어로솔 또는 오일 형태를 취한다. 사용될 수 있는 담체에는 석유 젤리, 라놀린, 폴리에틸렌 글리콜, 알코올 또는 그들의 조합물이 포함된다. 경피 투여용으로 적합한 제형은 별개의 패치로서 제시될 수 있고, 친지성 에멀션 또는 완충 수성 용액일 수 있거나 또는 중합체 또는 접착제에 용해 및/또는 분산될 수 있다.Another mode of administration includes inhalation formulations, including means such as dry powders, aerosols or drops. It may be, for example, an aqueous solution containing polyoxyethylene-9-lauryl ether, glycocholate and deoxycholate, or an oily solution for administration in the form of nasal drops, or a gel to be administered intranasally. Formulations for intranasal administration include, for example, lozenges or pastilles, which may include flavor bases such as sucrose or acacia and other excipients such as glycocholate. Formulations suitable for rectal administration are preferably presented as unit-dose suppositories with solid carriers, for example cocoa butter, which may include salicylates. Formulations suitable for topical application to the skin preferably take the form of ointments, creams, lotions, pastes, gels, sprays, aerosols or oils. Carriers that can be used include petroleum jelly, lanolin, polyethylene glycol, alcohols or combinations thereof. Formulations suitable for transdermal administration may be presented as separate patches, may be lipophilic emulsions or buffered aqueous solutions or may be dissolved and / or dispersed in a polymer or adhesive.
본 발명의 프로드럭과 세포 결합제 간의 접합체의 시험관내 세포독성In vitro Cytotoxicity of Conjugates Between Prodrugs and Cell Binding Agents of the Invention
본 발명의 프로드럭, 및 이들과 세포 결합제 간의 접합체의 세포독성은 보호 기를 절단하고 활성 약물로 전환시킨 후에 측정할 수 있다. 비부착성 세포주, 예를 들어 나말와 및 HL60에 대한 세포독성은 다음에 기재되는 바와 같이 세포 증식 곡선을 역-외삽함으로써 측정할 수 있다 [참고: Goldmacher et al, 135 J. Immunol. 3648-3651 (1985)]. 부착성 세포주, 예를 들어 A-375 및 SCaBER에 대한 이들 화합물의 세포독성은 다음에 기재되는 바와 같이 클론원성 검정에 의해 결정할 수 있다 [참고: Goldmacher et al, 102 J. Cell Biol. 1312-1319 (1986)].The cytotoxicity of the prodrugs of the invention, and the conjugates between them and the cell binder, can be measured after cleavage of the protecting group and conversion to the active drug. Cytotoxicity to non-adherent cell lines such as Namalwa and HL60 can be measured by de-extrapolating the cell proliferation curves as described below. Goldmacher et al, 135 J. Immunol . 3648-3651 (1985). Cytotoxicity of these compounds against adherent cell lines, such as A-375 and SCaBER, can be determined by clonality assays as described below. Goldmacher et al, 102 J. Cell Biol . 1312-1319 (1986).
선택된 세포 집단의 성장을 억제시키기 위한 치료제 및 방법Therapeutic Agents and Methods for Inhibiting Growth of Selected Cell Populations
본 발명은 또한, 다음을 포함하는 선택된 세포 집단의 성장을 억제시키기 위한 치료제를 제공한다.The present invention also provides a therapeutic for inhibiting the growth of a selected cell population comprising:
(a) 세포 결합제와 연결된 세포독성 량의 하나 이상의 상기 언급된 프로드럭; 및(a) at least one of the aforementioned prodrugs in a cytotoxic amount associated with a cell binding agent; And
(b) 제약상 허용 가능한 담체, 희석제 또는 부형제.(b) pharmaceutically acceptable carriers, diluents or excipients.
유사하게, 본 발명은 선택된 세포 집단으로부터의 세포를 함유하는 것으로 추정되는 조직 또는 세포 집단을, 세포 결합제와 연결된 하나 이상의 상기 언급된 프로드럭을 포함하는 세포독성 량의 세포독성제와 접촉시키는 것을 포함하는, 선택된 세포 집단의 성장을 억제시키는 방법을 제공한다.Similarly, the present invention includes contacting a tissue or cell population suspected of containing cells from a selected cell population with a cytotoxic amount of cytotoxic agent comprising one or more of the aforementioned prodrugs associated with a cell binding agent. To inhibit the growth of selected cell populations.
세포독성제는 상기 언급된 바와 같이 제조한다.Cytotoxic agents are prepared as mentioned above.
적합한 제약상 허용 가능한 담체, 희석제 및 부형제는 널리 공지되어 있고 임상적 상황에 근거하여 당업자가 결정할 수 있다.Suitable pharmaceutically acceptable carriers, diluents and excipients are well known and can be determined by one skilled in the art based on clinical circumstances.
적합한 담체, 희석제 및/또는 부형제의 예에는 (1) 약 1 mg/ml 내지 25 mg/ml 인간 혈청 알부민을 함유하는 둘벡코 (Dulbecco) 인산염 완충 식염수 (pH 약 7.4), (2) 0.9% 식염수 (0.9% w/v NaCl) 및 (3) 5% (w/v) 덱스트로스가 포함된다.Examples of suitable carriers, diluents and / or excipients include (1) Dulbecco phosphate buffered saline containing pH of about 1 mg / ml to 25 mg / ml human serum albumin (pH about 7.4), (2) 0.9% saline (0.9% w / v NaCl) and (3) 5% (w / v) dextrose.
선택된 세포 집단의 성장을 억제시키는 방법은 시험관내, 생체내 또는 생체외에서 실시할 수 있다.Methods for inhibiting the growth of selected cell populations can be performed in vitro, in vivo or ex vivo.
시험관내 사용의 예에는 표적 항원을 발현하지 않는 목적하는 변이체를 제외한 모든 세포를 사멸시키거나 또는 바람직하지 못한 항원을 발현하는 변이체를 사멸시키기 위해 세포 배양물을 처리하는 것이 포함된다.Examples of in vitro use include treating cell culture to kill all cells except for the desired variant that does not express the target antigen or to kill variants that express undesirable antigens.
비-임상 시험관내 사용 조건은 당업자가 용이하게 결정할 수 있다.Non-clinical in vitro use conditions can be readily determined by one skilled in the art.
생체외 사용의 예에는 병든 세포 또는 악성 세포를 사멸시키기 위해 자기 유래의 골수를 동일한 환자에게 이식하기에 앞서 이러한 골수를 처치하는 것; 적격한 T 세포를 사멸시키고 이식편 대 숙주 질병 (GVHD)을 예방하기 위해 골수를 이식하기에 앞서 이를 처치하는 것이 포함된다.Examples of ex vivo use include treating such bone marrow prior to transplanting its own bone marrow into the same patient to kill the diseased or malignant cells; Treatment of prior bone marrow transplantation to kill qualified T cells and prevent graft-versus-host disease (GVHD).
암 치료 또는 자가면역 질환 치료에 있어서 자기 유래 이식에 앞서 골수로부터 종양 세포 또는 림프계 세포를 제거하거나, 또는 GVHD를 예방하기 위해 이식하기에 앞서 동종 골수 또는 조직으로부터 T 세포 및 기타 림프계 세포를 제거하기 위한 임상 생체외 처치는 다음과 같이 수행할 수 있다. 골수를 환자 또는 기타 개개인으로부터 수거한 다음, 본 발명의 세포독성제가 부가된 혈청을 함유하는 매질 내에서 약 37℃ 하에 약 30분 내지 약 48시간 동안 약 10 μM 내지 1 pM의 농도 범위로 항온 배양한다. 정확한 항온 배양 시간과 농도 조건 (=용량)은 당업자가 용이하게 결정할 수 있다. 항온 배양 후, 골수 세포를 혈청 함유 매질로 세척하고, 공지된 방법에 따라서 정맥내 주입함으로써 환자에게 돌려주었다. 골수를 수거한 시간과 처치한 세포를 재주입하는 시간 사이에 전신 방사선 조사 또는 절제 화학요법과 같은 기타 치료를 환자에게 제공하는 상황 하에서는, 상기와 같이 처치한 골수 세포를 표준 의학 장비를 이용하여 액체 질소에 동결 저장한다.To remove tumor cells or lymphoid cells from bone marrow prior to autologous transplantation in cancer treatment or autoimmune disease treatment, or to remove T cells and other lymphoid cells from allogeneic bone marrow or tissue prior to transplantation to prevent GVHD Clinical ex vivo treatment can be performed as follows. Bone marrow is harvested from patients or other individuals, and then incubated in a medium containing serum to which the cytotoxic agent of the invention is added at a concentration ranging from about 10 μM to 1 pM at about 37 ° C. for about 30 minutes to about 48 hours. do. The exact incubation time and concentration conditions (= dose) can be readily determined by one skilled in the art. After incubation, the bone marrow cells were returned to the patient by washing with serum containing medium and intravenously injecting according to known methods. Under the circumstances of providing the patient with other treatments, such as systemic irradiation or ablation chemotherapy, between the time of bone marrow harvest and the time of reinjecting the treated cells, the treated bone marrow cells may be removed using standard medical equipment. Store frozen in nitrogen.
임상 생체내 사용의 경우에는, 본 발명의 세포독성제를 용액으로서 공급하여 멸균성 및 내독소 수준에 관하여 시험하거나, 또는 동결건조된 고형물로서 공급하여 이를 주사용 멸균수에 재용해시킬 수 있다. 접합체 투여에 관한 적합한 프로토콜의 예는 다음과 같다: 접합체는 6주 동안 매주 정맥내 일시 주입한다. 일시주입 용량은 인간 혈청 알부민을 부가할 수 있는 정상 식염수 50 내지 400 ml에 제공된다 (예를 들어, 0.5 내지 1 ml의 인간 혈청 알부민 농축 용액, 100 mg/ml). 투여량은 매주 약 50 ㎍ 내지 10 mg/체중 kg (정맥내 투여)일 것이다 (1회 주입당 10 ㎍ 내지 100 mg/kg). 처치한지 6주 후에 환자에게 제2 치료 과정을 실시한다. 투여 경로, 부형제, 희석제, 투여량, 시간 등과 관련한 구체적인 임상 프로토콜은 임상적 상황에 근거하여 당업자가 결정할 수 있다.For clinical in vivo use, the cytotoxic agents of the present invention may be supplied as a solution to test for sterile and endotoxin levels, or may be supplied as lyophilized solids and redissolved in sterile water for injection. Examples of suitable protocols for conjugate administration are as follows: The conjugate is injected intravenously, weekly, for 6 weeks. The bolus dose is given in 50 to 400 ml of normal saline to which human serum albumin can be added (e.g., 0.5 to 1 ml concentrated human serum albumin solution, 100 mg / ml). The dosage will be about 50 μg to 10 mg / kg body weight (intravenously) per week (10 μg to 100 mg / kg per injection). Six weeks after treatment, the patient undergoes a second course of treatment. Specific clinical protocols with respect to the route of administration, excipients, diluents, dosages, times, and the like can be determined by one skilled in the art based on clinical context.
선택된 세포 집단을 사멸시키는 생체내 또는 생체외 방법에 따라서 치료할 수 있는 의학적 질환의 예에는 모든 유형의 악성 종양, 예를 들어 폐암, 유방암, 결장암, 전립선암, 신장암, 췌장암, 난소암 및 림프계 기관 암; 흑색종; 자가 면역 질환, 예를 들어 전신성 루푸스, 류마티스성 관절염 및 다발성 경화증; 이식편 거부, 예를 들어 신장 이식체 거부, 간 이식체 거부, 폐 이식체 거부, 심장 이식체 거부 및 골수 이식체 거부; 이식편 대 숙주 질병; 바이러스성 감염, 예를 들어 CMV 감염, HIV 감염, AIDS 등; 세균성 감염; 및 기생충 감염, 예를 들어 편모충증, 아메바증, 주혈흡충증, 및 당업자에 의해 결정된 바와 같은 기타 질환이 포함된다.Examples of medical conditions that can be treated according to in vivo or ex vivo methods of killing selected cell populations include all types of malignant tumors, such as lung cancer, breast cancer, colon cancer, prostate cancer, kidney cancer, pancreatic cancer, ovarian cancer and lymphatic organs. cancer; Melanoma; Autoimmune diseases such as systemic lupus, rheumatoid arthritis and multiple sclerosis; Graft rejection, eg kidney transplant rejection, liver transplant rejection, lung transplant rejection, heart transplant rejection and bone marrow transplant rejection; Graft versus host disease; Viral infections such as CMV infection, HIV infection, AIDS, and the like; Bacterial infections; And parasitic infections such as flagella, amoeba, schistosomiasis, and other diseases as determined by one skilled in the art.
본 발명은 비-제한적 예를 참고로 하여 다음에 예시될 것이다. 달리 언급되지 않는 한, 모든 퍼센트, 비, 부 등은 중량을 기준으로 한 것이다.The invention will be illustrated next with reference to non-limiting examples. Unless stated otherwise, all percentages, ratios, parts, etc., are by weight.
재료 및 방법Materials and methods
융점은 전열 장치를 사용하여 측정하였고 정정하지 않았다. NMR 스펙트럼은 브루커 (Bruker) AVANCE400 (400 MHz) 분광계 상에서 기록하였다. 화학적 이동은 내부 표준으로서 TMS를 기준으로 하여 ppm으로 보고하였다. 질량 스펙트럼은 브루커 에스콰이어 (Bruker Esquire) 3000 시스템을 사용하여 획득하였다. 자외선 스펙트럼은 히타치 (Hitachi) U1200 분광광도계 상에서 기록하였다. HPLC는 벡크만 코울터 (Beckman Coulter) 시스템 GOLD 168 가변 파장 탐지기와 워터스 라디알팩 (Waters Radialpak) (역상 C-18 칼럼)이 장착된 벡크만 코울터 GOLD 125 시스템을 사용하여 수행하였다. 박층 크로마토그래피는 아날테크 (Analtech) GF 실리카 겔 TLC 판 상에서 수행하였다. 섬광 칼럼 크로마토그래피에 사용하기 위한 실리카 겔은 베이커 (Baker)로부터 공급되었다. 테트라히드로푸란은 나트륨 금속 상에서 증류시킴으로써 건조시켰다. 디메틸아세트아미드 및 디메틸포름아미드는 감압 하에 수소화칼슘 상에서 증류시킴으로써 건조시켰다. 사용된 기타 모든 용매는 시약 등급이거나 HPLC 등급이었다.Melting points were measured using a heat transfer device and were not corrected. NMR spectra were recorded on a Bruker AVANCE400 (400 MHz) spectrometer. Chemical shifts are reported in ppm based on TMS as an internal standard. Mass spectra were acquired using a Bruker Esquire 3000 system. Ultraviolet spectra were recorded on a Hitachi U1200 spectrophotometer. HPLC was performed using a Beckman Coulter GOLD 125 system equipped with a Beckman Coulter system GOLD 168 variable wavelength detector and a Waters Radialpak (reverse phase C-18 column). Thin layer chromatography was performed on an Analtech GF silica gel TLC plate. Silica gel for use in flash column chromatography was supplied by Baker. Tetrahydrofuran was dried by distillation over sodium metal. Dimethylacetamide and dimethylformamide were dried by distillation on calcium hydride under reduced pressure. All other solvents used were reagent grade or HPLC grade.
프로드럭 DC07 (7a), DC08 (8a) 및 DC107 (도 4)의 합성이 본원에 기재되어 있다. DC07 및 DC08 뿐만 아니라 DC107은 모 약물 DC1 및 DC101로부터 유래되는 반면, DC09 (9a)는 미국 특허 제6,756,397 B2호에 기재된 페길화 모 약물 DC5로부터 제조할 수 있다 (도 3). 프로드럭 DC07은 pH 5.5 이하의 수용액 중에서 극도로 안정적이고, 혈청 또는 혈장에서의 항온 배양에 의해 모 약물 DC1로 전환될 수 있다. 이들 약물은 또한, DC1과 비교해서 수 용해도가 증강되었다. DC07SMe를 마우스 혈청에서 항온 배양하면, 이는 모 약물 DC1SMe로 전환되었다.The synthesis of prodrugs DC07 ( 7a ), DC08 (8a) and DC107 ( FIG. 4 ) is described herein. DC07 and DC08 as well as DC107 are derived from parent drugs DC1 and DC101, while DC09 (9a) can be prepared from PEGylated parent drug DC5 described in US Pat. No. 6,756,397 B2 (FIG. 3). Prodrug DC07 is extremely stable in aqueous solutions up to pH 5.5 and can be converted to parent drug DC1 by incubation in serum or plasma. These drugs also enhanced water solubility compared to DC1. When incubated DC07SMe in mouse serum, it was converted to the parent drug DC1SMe.
인간 암 세포주 HL60, 나말와, A-375, COLO205 및 라모스는 공급원 [American Type Culture Collection (ATCC)]로부터 획득하였다. 카라 (Kara)는 인간 CD33 항원으로 안정적으로 형질감염시킨 뮤린 종양 세포주이다.Human cancer cell lines HL60, Namalwa, A-375, COLO205 and Ramos were obtained from the American Type Culture Collection (ATCC). Kara is a murine tumor cell line stably transfected with human CD33 antigen.
실시예 1Example 1
(S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐-페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-[(3-메틸디티오프로파노일)-아미노]-1H-인돌-2-카복스아미드 (DCO7-SMe, 7b)의 제조(S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonyl-phenyl) aminocarbonyloxy] -3H-benz (e) indole -3-yl} carbonyl] -1H-indol-5-yl] -5-[(3-methyldithiopropanoyl) -amino] -1 H-indole-2-carboxamide (DCO7-SMe, 7b Manufacturing
2.5 ml의 DMA 중의 11.6 mg (0.017 mmol)의 DC1SMe (1-[S]-(클로로메틸)-5-히드록시-3-{{5-[5-(3-메틸디티오프로피오닐)인돌-2-일-카보닐아미노]인돌-2-일}카보닐}-1,2-디히드로-3H-벤즈[e]인돌) 용액에 3.0 ㎕의 DIPEA를 가하였다. Ar 하에 3분 동안 교반시킨 후, 4.0 mg의 카보닐디이미다졸 (1.46 당량)을 가하였다. 혼합 물을 2시간 동안 교반시켰고, TLC는 모든 DC1SMe가 소모되었다는 것을 나타내었는데, 그 후에 신선하게 제조된 16 mg (3 당량)의 3-아미노벤젠설포네이트 디이소프로필에틸아민 염 (pH 5)을 가하였다. 혼합물을 Ar 하에 밤새 교반시켰고 HPLC는 반응이 완료되었다는 것을 나타내었다. 혼합물을 오일 펌프로 증발시키고, 100%의 0.01% 아세트산 내지 50%의 0.01% 아세트산 및 50% THF로 용출시킨 C-18 칼럼 (1.0 x 5 cm)을 이용하여 정제하였다. 분획을 모으고 증발시켜 11.8 mg의 표제 화합물 (DC07SMe)을 수득하였다; 1H NMR (DMF-d7) 11.85 (br, 2H), 11.79 (s, 1H), 11.70 (s, 1H), 10.53 (s, 1H), 10.27 (s, 1H), 10.03 (s, 1H), 8.50 (s, 1H), 8.41 (s, 1H), 8.21 (d, 1H, J = 1.7 Hz), 8.17 (d, 1H, J = 8.4 Hz), 8.12 (m, 1H), 7.46 (dd, 1H, J = 1.9, 8.8 Hz), 7.66 (m, 2H), 7.61 (d, 1H, J = 8.9 Hz), 7.58 (m, 2H), 7.48 (d, 1H, J = 1.4 Hz), 7.45 (dd, 1H, J = 1.9, 8.8 Hz), 7.35 (d, 1H, J = 1.6 Hz), 5.01 (t, 1H, J = 10.0 Hz), 4.84 (dd, 1H, J = 2.2, 10.9 Hz), 4.49 (m, 2H), 4.21 (dd, 1H, J = 3.2, 11.3 Hz), 4.10 (m, 2H), 3.12 (t, 2H, J = 7.0 Hz), 2.49 (s, 3H); 13C NMR 172.52, 170.33, 169.54, 162.94, 161.20, 160.41, 152.67, 147.94, 142.69, 139.11, 134.73, 134.56, 133.72, 133.37, 133.18, 132.16, 130.72, 128.76, 128.41, 128.34, 125.71, 125.61, 124.07, 123.16, 121.51, 120.05, 118.36, 113.54, 112.97, 112.92, 112.16, 111.61, 108.14, 107.56, 106.78, 104.02, 69.03, 67.95, 67.37, 55.84, 54.91, 48.13, 42.71, 37.05, 34.10, 23.12; MS m/z+ 831.14 (M + H)+, 883.0, 883.9, 884.9 (M+Na)+, 905.0, (M+K)+ 920.9, 922.9; MS m/z- 880.8, 881.8, 882.8, 883.8 (M-H)-.11.6 mg (0.017 mmol) of DC1SMe (1- [S]-(chloromethyl) -5-hydroxy-3-{{5- [5- (3-methyldithiopropionyl) indole- in 2.5 ml of DMA 3.0 μl of DIPEA was added to a 2-yl-carbonylamino] indol-2-yl} carbonyl} -1,2-dihydro-3H-benz [e] indole) solution. After stirring for 3 minutes under Ar, 4.0 mg of carbonyldiimidazole (1.46 equiv) were added. The mixture was stirred for 2 hours and TLC showed that all DC1SMe had been consumed, after which freshly prepared 16 mg (3 equiv) of 3-aminobenzenesulfonate diisopropylethylamine salt (pH 5) was added. Was added. The mixture was stirred under Ar overnight and HPLC showed the reaction was complete. The mixture was evaporated with an oil pump and purified using a C-18 column (1.0 × 5 cm) eluted with 100% 0.01% acetic acid to 50% 0.01% acetic acid and 50% THF. Fractions were combined and evaporated to give 11.8 mg of the title compound (DC07SMe); 1 H NMR (DMF-d7) 11.85 (br, 2H), 11.79 (s, 1H), 11.70 (s, 1H), 10.53 (s, 1H), 10.27 (s, 1H), 10.03 (s, 1H), 8.50 (s, 1H), 8.41 (s, 1H), 8.21 (d, 1H, J = 1.7 Hz), 8.17 (d, 1H, J = 8.4 Hz), 8.12 (m, 1H), 7.46 (dd, 1H , J = 1.9, 8.8 Hz), 7.66 (m, 2H), 7.61 (d, 1H, J = 8.9 Hz), 7.58 (m, 2H), 7.48 (d, 1H, J = 1.4 Hz), 7.45 (dd , 1H, J = 1.9, 8.8 Hz), 7.35 (d, 1H, J = 1.6 Hz), 5.01 (t, 1H, J = 10.0 Hz), 4.84 (dd, 1H, J = 2.2, 10.9 Hz), 4.49 (m, 2H), 4.21 (dd, 1H, J = 3.2, 11.3 Hz), 4.10 (m, 2H), 3.12 (t, 2H, J = 7.0 Hz), 2.49 (s, 3H); 13 C NMR 172.52, 170.33, 169.54, 162.94, 161.20, 160.41, 152.67, 147.94, 142.69, 139.11, 134.73, 134.56, 133.72, 133.37, 133.18, 132.16, 130.72, 128.76, 128.41, 128.125, 125.16, 125.16. , 121.51, 120.05, 118.36, 113.54, 112.97, 112.92, 112.16, 111.61, 108.14, 107.56, 106.78, 104.02, 69.03, 67.95, 67.37, 55.84, 54.91, 48.13, 42.71, 37.05, 34.10, 23.12; MS m / z + 831.14 (M + H) + , 883.0, 883.9, 884.9 (M + Na) + , 905.0, (M + K) + 920.9, 922.9; MS m / z - 880.8, 881.8, 882.8, 883.8 (MH) - .
실시예 2Example 2
(S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-[(3-머캅토프로파노일)-아미노]-1H-인돌-2-카복스아미드 (DCO7, 7a)의 제조(S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylphenyl) aminocarbonyloxy] -3H-benz (e) indole- Preparation of 3-yl} carbonyl] -1H-indol-5-yl] -5-[(3-mercaptopropanoyl) -amino] -1H-indol-2-carboxamide (DCO7, 7a)
2 ml의 H2O 중의 10 mg (0.104 mmol)의 TCEP 용액을, NaHCO3 분말을 부가하면서 pH 7.0으로 조정하였다. 이 용액에 3 ml의 DMA 중의 15 mg의 DC07SMe를 부가한 다음, 1.0 ml 20O mM NaH2PO4, pH 4.5 중의 5 mg의 DTT를 가하였다. 2시간 동안 교반시키고 MS가 DC07SMe 피크 (m/z 883 (M-1))가 사라졌다고 나타낸 후, 혼합물을 농축시키고, 60%의 10 mM NaH2PO4 및 40% DMA 내지 80% DMA로 40분 동안 용출시킨 분취 HPLC c-18 칼럼을 이용하여 정제하였다. 수집된 분획을 모으고, 농축시키며, DMA에 재용해시키고, 염을 여과시킨 다음, 다시 증발시켜 13 mg의 DC07을 수득하였다. MS m/z- 835.1 (M-H), 836.1, 837.1, 838.1.A 10 mg (0.104 mmol) TCEP solution in 2 ml H 2 O was adjusted to pH 7.0 with the addition of NaHCO 3 powder. To this solution was added 15 mg of DC07SMe in 3 ml of DMA followed by 5 mg of DTT in 1.0 ml 20 mM NaH 2
실시예 3Example 3
(S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐메틸페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-{[3-(2-피리딜디티오)-프로파노일]-아미노}-1H-인돌-2-카복스아미드 (DC08SPy)의 제조(S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylmethylphenyl) aminocarbonyloxy] -3H-benz (e) indole- 3-yl} carbonyl] -1H-indol-5-yl] -5-{[3- (2-pyridyldithio) -propanoyl] -amino} -1H-indole-2-carboxamide ( Manufacture of DC08SPy)
2 ml의 DMA 중의 21.0 mg의 (DC1SPy)를 10 마이크로리터의 디이소프로필에틸아민 (DIPEA)에 가하였다. Ar 하에 3분 동안 교반시킨 후, 8.0 mg의 카보닐 디이 미다졸 (CDI)을 가하고, 혼합물을 2시간 동안 교반시키며, C-18 칼럼 상에서 HPLC하여 검사한 결과, DC1SPy가 CDI와 완전히 반응하였다. 이 혼합물에 20 mg의 4-아미노벤질 설폰산 및 10 마이크로리터의 DIPEA를 가하였다. 밤새 교반시킨 후, 혼합물을 농축시키고, 10 mM NaH2PO4, pH 4.0 중의 30% DMA 내지 10 mM NaH2PO4, pH 4.0 중의 80% DMA로 용출시킨 C-18 칼럼 상에서 HPLC함으로써 정제하였다. 분획을 모으고, 농축시켜 20.2 mg의 표제 화합물을 수득하였다. 1H NMR (DMF-d7) 11.67 (d, 2H, J = 9.9 Hz), 10.58 (s, 1H), 10.23 (s, 1H), 9.81 (s, 1H), 8.86 (dddd, 1H, J = 0.8, 1.6, 2.5, 14.6), 8.41 (s, 1H), 8.27-8.20 (m, 4H), 8.12 (m, 1H), 7.95 (d, 1H, J = 8.2 Hz), 7.83 (m, 1H), 7.71 (dd, 1H, J = 2.0, 9.9 Hz), 7.60 - 7.51 (m, 4H), 7.47 - 7.40 (m, 4H), 7.30 (d, 1H, J = 1.6 Hz), 4.90 (t, 1H, J = 10.0 Hz), 4.75 (dd, 1H, J = 2.2, 10.9 Hz), 4.33 (m, 1H), 4.15 (dd, 1H, J = 3.2, 11.3 Hz), 4.11 (s, 2H), 3.97 (dd, 1H, J = 7.8, 11.0 Hz), 3.29 (t, 2H, J = 6.9 Hz), 2.76 (m, 3H); 13C NMR 172.35, 162.93, 161.20, 160.41, 155.77, 151.36, 147.94, 142.69, 140.14, 139.11, 134.73, 134.56, 133.82, 133.37, 132.97, 132.16, 131.49, 129.22, 128.82, 128.34, 125.01, 125.61, 124.17, 123.76, 121.81, 120.05, 119.32, 116.43, 113.31, 112.97, 112.92, 112.16, 111.61, 108.14, 107.56, 106.83, 101.78, 67.37, 56.37, 48.13, 42.71, 37.00, 34.10; MS m/z- 958.34 (M-1).21.0 mg (DC1SPy) in 2 ml of DMA was added to 10 microliters of diisopropylethylamine (DIPEA). After stirring for 3 minutes under Ar, 8.0 mg of carbonyl dimidazole (CDI) was added, the mixture was stirred for 2 hours, and examined by HPLC on a C-18 column, where DC1SPy reacted completely with CDI. To this mixture was added 20 mg of 4-aminobenzyl sulfonic acid and 10 microliters of DIPEA. After stirring overnight, the mixture was concentrated and purified by HPLC on a C-18 column eluted with 10 mM NaH 2
4-아미노벤질설폰산4-aminobenzylsulfonic acid
250 ml의 파르 (Par) 수소화용 병에 4-니트로벤질 설폰산 (6.80 g, 31.34 mmol), 10% Pd/C (0.4 g), CH3OH (150 ml)를 몇 방울의 물과 함께 채워 넣고 수소로 퍼징하였다. 반응 혼합물을 20 psi H2과 함께 밤새 진탕시켰다. 촉매를 셀라이트를 통하여 여과시킴으로써 제거하고 용매를 증발시켰다. 조 화합물을 3 x 50 ml의 물로 3회 공증발시키고 물/THF/EtAc/톨루엔 (1:10:10:100)으로 결정화하여 5.3 g (91%)의 표제 화합물을 수득하였다. 1H NMR (D2O), 7.28 (dd, 2H, J = 2.2, 4.3 Hz), 6.95 (dd, 2H, J = 2.2, 4.3 Hz), 4.10 (s, 2H); 13C NMR 134.30, 127.10, 121.04, 120.42, 59.14; MS m/z- 186.35 (M-H).Fill a 250 ml Par bottle with 4-nitrobenzyl sulfonic acid (6.80 g, 31.34 mmol), 10% Pd / C (0.4 g) and CH 3 OH (150 ml) with a few drops of water. Put and purged with hydrogen. The reaction mixture was shaken with 20 psi H 2 overnight. The catalyst was removed by filtration through celite and the solvent was evaporated. The crude compound was co-evaporated three times with 3 × 50 ml of water and crystallized with water / THF / EtAc / toluene (1: 10: 10: 100) to give 5.3 g (91%) of the title compound. 1 H NMR (D 2 O), 7.28 (dd, 2H, J = 2.2, 4.3 Hz), 6.95 (dd, 2H, J = 2.2, 4.3 Hz), 4.10 (s, 2H); 13 C NMR 134.30, 127.10, 121.04, 120.42, 59.14; MS m / z - 186.35 (MH).
4-니트로페닐티올 아세테이트4-nitrophenylthiol acetate
75 ml의 톨루엔 중의 5 ml (69.95 mmol)의 티올아세트산 및 12 ml의 트리에틸 아민 용액에 100 ml의 1:2 THF/톨루엔 중의 7.5 g (34.71 mmol)의 4-니트로벤질 브로마이드를 4시간 동안 적가하였다. 혼합물을 밤새 지속적으로 교반시키고, 증발시킨 다음, EtAc/톨루엔/헥산으로 결정화하여 7.1 g (96%)의 표제 화합물을 수득하였다. 1H NMR (CDCl3) 8.13 (ddd, 2H, J = 2.5, 4.5, 9.3 Hz), 7.44 (ddd, 2H, J = 2.5, 4.5, 9.3 Hz), 4.14 (s, 2H), 2.35 (S, 3H); 13C NMR 194.53, 145.73, 129.91, 124.07, 32.93, 30.50; MS M+ 234.34 (M + Na).To 5 ml (69.95 mmol) thiolacetic acid and 12 ml triethyl amine solution in 75 ml toluene was added dropwise 7.5 g (34.71 mmol) 4-nitrobenzyl bromide in 100 ml 1: 2 THF / toluene for 4 hours. It was. The mixture was continuously stirred overnight, evaporated and then crystallized with EtAc / toluene / hexanes to give 7.1 g (96%) of the title compound. 1 H NMR (CDCl 3) 8.13 (ddd, 2H, J = 2.5, 4.5, 9.3 Hz), 7.44 (ddd, 2H, J = 2.5, 4.5, 9.3 Hz), 4.14 (s, 2H), 2.35 (S, 3H ); 13 C NMR 194.53, 145.73, 129.91, 124.07, 32.93, 30.50; MS M + 234.34 (M + Na).
4-니트로벤질 설폰산4-nitrobenzyl sulfonic acid
7.0 g의 4-니트로페닐티올 아세테이트를 150 ml의 1:3 H2O2/HAc 용액에 5 분획으로 나누어 4시간 동안 가하고, 혼합물을 밤새 지속적으로 교반시켰다. 혼합물을 증발시키고, 3 x 50 ml 물로 3회 공증발시킨 다음, CH3OH/THF/톨루엔으로 결정화하여 6.82 g (95%)의 표제 화합물을 수득하였다. 1H NMR (D2O) 8.02 (ddd, 2H, J = 2.7, 4.6, 11.1 Hz), 7.20 (ddd, 2H, J = 2.6, 4.6, 11.1 Hz), 4.11 (s, 2H); 13C NMR 141.70, 139.30, 129.91, 122.07, 59.15; MS m/z- 216.21 (M-H).7.0 g of 4-nitrophenylthiol acetate was added to 150 ml of a 1: 3 H 2 O 2 / HAc solution in 5 portions for 4 hours, and the mixture was continuously stirred overnight. The mixture was evaporated and co-evaporated three times with 3 x 50 ml water and then crystallized with CH3OH / THF / toluene to give 6.82 g (95%) of the title compound. 1 H NMR (D 2 O) 8.02 (ddd, 2H, J = 2.7, 4.6, 11.1 Hz), 7.20 (ddd, 2H, J = 2.6, 4.6, 11.1 Hz), 4.11 (s, 2H); 13 C NMR 141.70, 139.30, 129.91, 122.07, 59.15; MS m / z - 216.21 (MH).
실시예 4Example 4
(S)-N-[2-{(1-클로로메틸)-1,2-디히드로-5-[(3-히드록시설포닐메틸페닐)아미노카보닐옥시]-3H-벤즈(e)인돌-3-일}카보닐]-1H-인돌-5-일]-5-[(3-머캅토프로파노일)-아미노]-1H-인돌-2-카복스아미드 (DC08)의 제조(S) -N- [2-{(1-chloromethyl) -1,2-dihydro-5-[(3-hydroxysulfonylmethylphenyl) aminocarbonyloxy] -3H-benz (e) indole- Preparation of 3-yl} carbonyl] -1H-indol-5-yl] -5-[(3-mercaptopropanoyl) -amino] -1 H-indole-2-carboxamide (DC08)
2 ml의 H2O 중의 10 mg (0.104 mmol)의 TCEP 용액을, NaHCO3 분말을 부가하면서 pH 7.0으로 조정하였다. 이 용액에 3 ml의 DMA 중의 15 mg의 DC08SPy를 부가한 다음, 1.0 ml 200 mM NaH2PO4, pH 4.5 중의 5 mg의 DTT를 가하였다. 2시간 교반시키고 MS가 DC08SPy 피크 (m/z 883 (M-1))가 사라졌다고 나타낸 후, 혼합물을 농축시키고, 60%의 10 mM NaH2PO4/40% DMA 내지 20%의 10 mM NaH2PO4/80% DMA로 40분 동안 용출시킨 분취 HPLC c-18 칼럼을 이용하여 정제하였다. 수집된 분획을 모으고, 건조 농축시키며, DMA에 재용해시키고, 염을 여과시킨 다음, 다시 증발시켜 13 mg의 DC08을 수득하였다. MS m/z- 849.1 (M-H), 850.1 , 851.1.A 10 mg (0.104 mmol) TCEP solution in 2 ml H 2 O was adjusted to pH 7.0 with the addition of NaHCO 3 powder. To this solution was added 15 mg of DC08SPy in 3 ml of DMA followed by 5 mg of DTT in 1.0 ml 200 mM NaH 2
실시예 5Example 5
(S)-N-(2-(1-((S) -N- (2- (1- ( 클로로메틸Chloromethyl )-5-히드록시-2,3-) -5-hydroxy-2,3- 디히드로Dehydro -1H--1H- 벤조(e)인돌Benzo (e) indole -3--3- 카보닐Carbonyl )-1H-인돌-5-일)-3-() -1H-indol-5-yl) -3- ( 메틸디설파닐Methyldisulfanyl )) 프로판아미드Propanamide ( ( DC101SMeDC101SMe )의 제조Manufacturing
7.0 ml의 DMA 중의 5-히드록시-3-아미노-1-[S]-(클로로메틸)-1,2-디히드로-3H-벤즈(e)인돌, 히드로클로라이드 염 (155 mg, 0.57 mmol) 및 5-(3-(메틸-디설파닐)프로판아미도)-1H-인돌-2-카복실산 (170 mg, 0.55 mmol) 용액에 EDC (300 mg, 1.56 mmol)를 Ar 하에 가하였다. 밤새 교반시킨 후, 혼합물을 건조 증발시키고, SiO2 크로마토그래피 (톨루엔 중의 20% 내지 40% THF)함으로써 정제하며, THF/톨루엔/헥산으로 결정화하여 218 mg (76%)의 DC101SMe를 수득하였다. 1H NMR (DMF-d7) 11.63 (s, 1H), 10.56 (s, 1H), 9.95 (s, 1H), 8.23 (s, 2H), 7.93 (d, 1H, J = 8.3 Hz), 7.56 (dd, 1H, J = 2.0, 8.1 Hz), 7.52 (s, 1H), 7.47 (dd, 1H, J = 1.9, 8.9 Hz), 7.41 (d, 1H, J = 1.7 Hz), 7.25 (d, 1H, J = 1.7 Hz), 4.88 (dd, 1H, J = 8.7, 11.0 Hz), 4.73 (dd, 1H, J = 2.3, 10.9 Hz), 4.31 (m, 1H), 4.12 (dd, 1H, J = 3.1, 11.0 Hz), 3.95 (dd, 1H, J = 8.4, 11.2 Hz), 3.21 (t, 2H, J = 7.1 Hz), 2.71 (t, 2H, J = 7.1 Hz), 2.45 (s, 3H); 13C NMR 169.54, 161.06, 155.36, 134.21, 133.39, 132.54, 131.05, 128.39, 127.99, 123.98, 123.74, 123.46, 123.31, 118.88, 115.99, 112.87, 112.31, 106.40, 101.33, 55.93, 48.10, 42.61, 36.92, 30.81, 25.34. MS m/z+ 548.2 (M+Na), 550.2 (M+2+Na).5-hydroxy-3-amino-1- [S]-(chloromethyl) -1,2-dihydro-3H-benz (e) indole, hydrochloride salt (7.0 mg, 0.57 mmol) in 7.0 ml DMA And EDC (300 mg, 1.56 mmol) was added under Ar to a solution of 5- (3- (methyl-disulfanyl) propaneamido) -1H-indole-2-carboxylic acid (170 mg, 0.55 mmol). After stirring overnight, the mixture was evaporated to dryness and purified by SiO 2 chromatography (20% to 40% THF in toluene) and crystallized with THF / toluene / hexanes to give 218 mg (76%) of DC 101 SMe. . 1 H NMR (DMF-d 7 ) 11.63 (s, 1H), 10.56 (s, 1H), 9.95 (s, 1H), 8.23 (s, 2H), 7.93 (d, 1H, J = 8.3 Hz), 7.56 (dd, 1H, J = 2.0, 8.1 Hz), 7.52 (s, 1H), 7.47 (dd, 1H, J = 1.9, 8.9 Hz), 7.41 (d, 1H, J = 1.7 Hz), 7.25 (d, 1H, J = 1.7 Hz), 4.88 (dd, 1H, J = 8.7, 11.0 Hz), 4.73 (dd, 1H, J = 2.3, 10.9 Hz), 4.31 (m, 1H), 4.12 (dd, 1H, J = 3.1, 11.0 Hz), 3.95 (dd, 1H, J = 8.4, 11.2 Hz), 3.21 (t, 2H, J = 7.1 Hz), 2.71 (t, 2H, J = 7.1 Hz), 2.45 (s, 3H ); 13 C NMR 169.54, 161.06, 155.36, 134.21, 133.39, 132.54, 131.05, 128.39, 127.99, 123.98, 123.74, 123.46, 123.31, 118.88, 115.99, 112.87, 112.31, 106.40, 101.33, 55.93, 48.93, 48.93, 48.93. , 25.34. MS m / z + 548.2 (M + Na), 550.2 (M + 2 + Na).
5-(3-(메틸디설파닐)프로판아미도)-1H-인돌-2-카복실산5- (3- (Methyldisulfanyl) propaneamido) -1 H-indole-2-carboxylic acid
50 ml의 DMA 중의 3-(메틸디설파닐)프로파노산 (1.18 g, 7.76 mmol) 용액에 히드록시석신이미드 (1.14 g, 9.90 mmol) 및 EDC (1.91 g, 9.96 mmol)를 아르곤 하에 가하였다. 혼합물을 밤새 교반시켰다. TLC (1:6 EtAc/헥산)에 의해 반응이 완료되었다는 것을 확인한 후, 혼합물을 추가의 정제 없이 다음 단계에 직접 사용하였다.To a solution of 3- (methyldisulfanyl) propanoic acid (1.18 g, 7.76 mmol) in 50 ml of DMA was added hydroxysuccinimide (1.14 g, 9.90 mmol) and EDC (1.91 g, 9.96 mmol) under argon. It was. The mixture was stirred overnight. After confirming that the reaction was complete by TLC (1: 6 EtAc / hexanes), the mixture was used directly in the next step without further purification.
0℃ 하에 50 ml의 1:4의 0.5 M Na2HPO4, pH 8.0/DMA 중의 5-아미노-1H-인돌-2-카복실산 (1.32 g, 7.49 mmol) 용액에 상기 제조된 NHS 에스테르 용액을 1시간 동안 적가하였다. 부가 후, 혼합물을 4℃ 하에 1시간 동안 지속적으로 교반시킨 다음, 실온 하에 1시간 동안 교반시켰다. 혼합물을 농축시키고, THF/톨루엔/아세트산 (1:4:02% 내지 1:2:0.02%)으로 용출시킨 SiO2 크로마토그래피 상에서 정제하여 표제 생성물을 수득하였다 (1.62 g, 70%). 1H NMR (DMF-d7) 12.80 (br, 0.8H), 11.66 (s, 1H), 9.96 (s, 1H), 8.17 (s, 1H), 7.44 (s, 2H), 7.14 (d, 1H, J = 2.0 Hz), 3.20 (t, 2H, J = 6.9 Hz), 2.72 (t, 2H, J = 7.0 Hz), 2.37 (s, 3H). 13C NMR 169.53, 163.45, 135.24, 133.38, 130.12, 127.99, 119.13, 113.10, 112.27, 108.04, 36.88, 30.60. MS m/z- 309.20 (M-1).To the solution of NHS ester prepared above was added dropwise to a solution of 5-amino-1H-indole-2-carboxylic acid (1.32 g, 7.49 mmol) in 50 ml 1: 4 0.5 M Na2HPO4, pH 8.0 / DMA at 0 ° C. for 1 hour. It was. After addition, the mixture was continuously stirred at 4 ° C. for 1 hour and then at room temperature for 1 hour. The mixture was concentrated and purified on SiO2 chromatography eluted with THF / toluene / acetic acid (1: 4: 02% to 1: 2: 0.02%) to give the title product (1.62 g, 70%). 1 H NMR (DMF-d7) 12.80 (br, 0.8H), 11.66 (s, 1H), 9.96 (s, 1H), 8.17 (s, 1H), 7.44 (s, 2H), 7.14 (d, 1H, J = 2.0 Hz), 3.20 (t, 2H, J = 6.9 Hz), 2.72 (t, 2H, J = 7.0 Hz), 2.37 (s, 3H). 13C NMR 169.53, 163.45, 135.24, 133.38, 130.12, 127.99, 119.13, 113.10, 112.27, 108.04, 36.88, 30.60. MS m / z - 309.20 (M-1).
실시예 6Example 6
(S)-3-((1-(클로로메틸)-3-(5-(3-(메틸디설파닐)프로판아미도)-1H-인돌-2-카보닐)-2,3-(S) -3-((1- (chloromethyl) -3- (5- (3- (methyldisulfanyl) propaneamido) -1H-indole-2-carbonyl) -2,3- 디히드로Dehydro -1H--1H- 벤조[e]인돌Benzo [e] indole -5--5- 일옥시Iloxy )) 카보닐아미노Carbonylamino )) 벤젠설폰산Benzene sulfonic acid ( ( DC107SMeDC107SMe ))
10 ml의 DMA 중의 DC101SMe ((S)-N-(2-(1-(클로로메틸)-5-히드록시-2,3-디히드로-1H-벤조[e]인돌-3-카보닐)-1H-인돌-5-일)-3-(메틸디설파닐)프로판아미드, 100 mg 0.19 mmol) 용액에 DIPEA (35 ㎕, 0.20 mmol)를 가하였다. Ar 하에 3분 동안 교반시킨 후, 32.0 mg (0.20 mmol)의 카보닐디이미다졸을 가하였다. 혼합물을 2시간 동안 교반시키면 TLC는 모든 DC101SMe가 소모되었다는 것을 나타냈으며, 이후 신선하게 제조된 3-아미노벤젠설포네이트 디이소프로필에틸아민 염 (65 mg, 0.21 mmol)을 가하였다. 혼합물을 Ar 하에 24시간 동안 교반시키면, HPLC는 반응이 완료되어 DC107SMe [(S)-3-((1-(클로로메틸)-3-(5-(3-(메틸디설파닐)프로판아미도)-1H-인돌-2-카보닐)-2,3-디히드로-1H-벤조[e]인돌-5-일옥시)카보닐아미노)벤젠설폰산]이 형성되었다는 것을 나타내었다. 이 혼합물을 오일 펌프를 이용하여 약 5 ml가 되도록 농축시키고, 추가의 정제없이 다음 단계에 직접 사용하였다.DC101SMe ((S) -N- (2- (1- (chloromethyl) -5-hydroxy-2,3-dihydro-1H-benzo [e] indole-3-carbonyl)-in 10 ml of DMA To a solution of 1H-indol-5-yl) -3- (methyldisulfanyl) propanamide, 100 mg 0.19 mmol) was added DIPEA (35 μl, 0.20 mmol). After stirring for 3 minutes under Ar, 32.0 mg (0.20 mmol) of carbonyldiimidazole were added. When the mixture was stirred for 2 hours, TLC showed that all DC101SMe were consumed, then freshly prepared 3-aminobenzenesulfonate diisopropylethylamine salt (65 mg, 0.21 mmol) was added. When the mixture was stirred under Ar for 24 hours, the HPLC completed the reaction and the reaction was complete with DC107SMe [(S) -3-((1- (chloromethyl) -3- (5- (3- (methyldisulfanyl) propaneamido ) -1H-indol-2-carbonyl) -2,3-dihydro-1H-benzo [e] indol-5-yloxy) carbonylamino) benzenesulfonic acid] was formed. This mixture was concentrated to about 5 ml using an oil pump and used directly in the next step without further purification.
실시예 7Example 7
(S)-3-((1-(클로로메틸)-3-(5-(3-머캅토프로판아미도)-1H-인돌-2-카보닐)-2,3-디(S) -3-((1- (chloromethyl) -3- (5- (3-mercaptopropaneamido) -1H-indole-2-carbonyl) -2,3-di 히He 드로-1H-Draw-1H- 벤조[e]인돌Benzo [e] indole -5--5- 일옥시Iloxy )) 카보닐아미노Carbonylamino )) 벤젠설폰산Benzene sulfonic acid ( ( DC107DC107 ))
상기 농축된 용액에 10 ml의 1:1 DMA/0.4 M NaH2PO4, pH 4.0 중의 60 mg (0.62 mmol)의 TCEP를 가하였다. Ar 하에 4시간 동안 교반시킨 후, 혼합물을 농축시키고, DMA 중의 90%의 0.01% 아세트산 내지 DMA 중의 40%의 0.01% 아세트산으로 용출시킨 C18 칼럼을 이용하여 정제하였다. 분획을 모으고 증발시켜 94 mg (73%, 2 단계)의 (S)-3-((1-(클로로메틸)-3-(5-(3-머캅토프로판아미도)-1H-인돌-2-카보닐)-2,3-디히드로-1H-벤조[e]인돌-5-일옥시)카보닐아미노)벤젠설폰산 (DC107)을 수 득하였다. 1H NMR (DMF-d7) 11.63 (s, 1H), 10.51 (s, 1H), 9.89 (s, 1H), 8.33 (s, 2H), 7.93 (d, 1H, J = 8.3 Hz), 7.58 (m, 2H), 7.52 (m, 2H), 7.47 (m, 2H), 7.41 (m, 1H), 7.38 (m, 1H), 7.25 (d, 1H, J = 1.7 Hz), 4.87 (dd, 1H, J = 8.7, 11.0 Hz), 4.74 (dd, 1H, J = 2.3, 10.9 Hz), 4.32 (m, 1H), 4.12 (dd, 1H, J = 3.0, 11.0 Hz), 3.93 (dd, 1H, J = 8.4, 11.2 Hz), 3.21 (t, 2H, J = 7.1 Hz), 2.70 (t, 2H, J = 7.1 Hz); 13C NMR 169.55, 161.30, 155.46, 150.21, 143.45, 134.22, 133.37, 132.54, 131.35, 130.31, 128.38, 127.95, 123.78, 123.72, 123.48, 123.33, 121.41, 118.88, 115.90, 112.88, 112.35, 106.44, 101.38, 55.95, 48.60, 42.67, 36.90, 25.01. MS m/z- 676.90 (M-H), ε340 nm= 19025 M-1cm-1.To the concentrated solution was added 60 mg (0.62 mmol) of TCEP in 10 ml 1: 1 DMA / 0.4 M NaH 2
모노클로날 항체와 DC07과의 접합. 인간 AML 세포 표면 상에 우선적으로 발현된 CD33 항원과 결합하는 huMy9-6 항체가 DC07과의 접합을 위해 선택되었다. Conjugation of Monoclonal Antibodies with DC07 . A huMy9-6 antibody that binds preferentially expressed CD33 antigen on human AML cell surface was selected for conjugation with DC07.
실시예 8Example 8
huMy9-6-SSNPB-DC07: 디설파이드 연결된 접합체의 제조huMy9-6-SSNPB-DC07: Preparation of Disulfide Linked Conjugates
제1 단계에서는, 항체를 변형제 N-설포석신이미딜 5-니트로-2-피리딜디티오부타노에이트 (SSNPB)와 반응시켜 니트로피리딜디티오 기를 도입하였다. 0.05 M 인산칼륨, 0.05 M 염화나트륨, 2 mM 에틸렌디아민테트라아세트산 (EDTA), 3% 디메틸아세트아미드 (DMA), pH 6.5를 함유하는 수성 완충액 중의 8.5 mg/mL 농도의 huMy9-6 항체 용액을 7.5배 몰 과량의 SSNPB 용액 (DMA 중의 20 mM 원액)으로 처리 하였다. 반응 혼합물을 실온 하에 90분 동안 교반시키고, 0.1 M NaH2PO4, 50 mM NaCl, pH 6.5를 함유하는 수성 완충액 내로 미리 평형시킨 바 있는 세파덱스 (Sephadex) G25 예비-패킹된 칼럼 상에서 정제하였다. 이와 같이 정제한 샘플은 98%의 생성물을 생성시켰다. 소 분취액의 변형된 항체를 디티오트레이톨로 처리하여 니트로-피리딜 디설파이드를 절단시키고, 방출된 니트로-피리딘-2-티온을 대상으로 하여 분광 광도계를 이용하여 검정하였다: 니트로-피리딘-2-티온의 경우, ε325nm = 10,964 M-1cm-1 및 ε280nm = 3,344 M-1cm-1이고, 항체의 경우에는 ε280nm = 206,460 M-1cm-1이다. 항체 분자당 평균 5.0개의 니트로-피리딜디설파이드 기가 연결되었다.In the first step, the antibody was reacted with the modifier N-sulfosuccinimidyl 5-nitro-2-pyridyldithiobutanoate (SSNPB) to introduce nitropyridyldithio groups. 7.5 times the huMy9-6 antibody solution at a concentration of 8.5 mg / mL in aqueous buffer containing 0.05 M potassium phosphate, 0.05 M sodium chloride, 2 mM ethylenediaminetetraacetic acid (EDTA), 3% dimethylacetamide (DMA), pH 6.5 Treated with molar excess of SSNPB solution (20 mM stock in DMA). The reaction mixture was stirred at room temperature for 90 minutes and purified on a Sephadex G25 pre-packed column that had previously been equilibrated into an aqueous buffer containing 0.1 M NaH 2 PO 4 , 50 mM NaCl, pH 6.5. The sample thus purified produced 98% of the product. Bovine aliquots of modified antibodies were treated with dithiothreitol to cleave nitro-pyridyl disulfides and assayed using a spectrophotometer on the released nitro-pyridine-2-thione: nitro-pyridine-2 - in the case of a thione, ε 325nm = 10,964 M -1 cm -1 and ε 280nm = 3,344 M -1 cm -1 and, in the case of the antibody, ε 280nm = 206,460 M -1 cm -1 . An average of 5.0 nitro-pyridyldisulfide groups were linked per antibody molecule.
변형된 항체는 pH 6.5 하의 상기 완충액 중에서 5% DMA를 사용하여 2.0 mg/mL가 되도록 희석시킨 다음, DMA 중의 DC07 용액으로 처리하였는데, 링커당 2.5개 약물로 약물 과량을 사용하였다. 접합 혼합물은 실온 하에 30분 동안 반응하였다. 반응 혼합물은, 20% DMA, 0.1 M NaH2PO4, 50 mM NaCl, pH 5.0에서 미리 평형시킨 바 있는 세파크릴 S300 겔 여과 칼럼을 통하여 통과시킴으로써 정제하였다. 단량체성 항체-DC07 접합체를 함유하는 분획을 모으고 10 mM 시트레이트, 135 mM NaCl, pH 5.0 내로 투석시켰다. 최종 접합체를 대상으로 하여 다음 흡광 계수를 이용하여 분광 광도계로 검정하였다: 약물의 경우, ε340nm = 38,000 M-1cm-1 및 ε280nm = 23,000 M-1cm-1이고, 항체의 경우에는 ε280nm = 206,460 M-1cm-1이다. 접합체는 항체당 3.5개의 약물을 함유하였다.Modified antibodies were diluted to 2.0 mg / mL with 5% DMA in the above buffer at pH 6.5 and then treated with a solution of DC07 in DMA, using drug excess at 2.5 drugs per linker. The conjugation mixture was reacted for 30 minutes at room temperature. The reaction mixture was purified by passing through a Sephacryl S300 gel filtration column previously equilibrated at 20% DMA, 0.1 M NaH 2 PO 4 , 50 mM NaCl, pH 5.0. Fractions containing monomeric antibody-DC07 conjugates were pooled and dialyzed into 10 mM citrate, 135 mM NaCl, pH 5.0. The final conjugates were assayed spectrophotometrically using the following absorbance coefficients: for drugs, ε 340 nm = 38,000 M -1 cm -1 and ε 280 nm = 23,000 M -1 cm -1 , and for antibodies ε 280 nm = 206,460 M −1 cm −1 . The conjugate contained 3.5 drugs per antibody.
실시예 9Example 9
huMy9-6-SMCC-DC07: 티오에테르 연결된 접합체의 제조huMy9-6-SMCC-DC07: Preparation of Thioether Linked Conjugates
제1 단계에서는, 항체를 변형제 석신이미딜 4-(N-말레이미도메틸)시클로헥산-1-카복실레이트 (SMCC)와 반응시켰다. 0.05 M 인산칼륨, 0.05 M 염화나트륨, 2 mM 에틸렌디아민테트라아세트산 (EDTA) 및 3% DMA, pH 6.5를 함유하는 수성 완충액 중의 8.5 mg/mL 농도의 huMy9-6 항체 용액을 실온 하에 90분 동안 8.5배 몰 과량의 SMCC 용액 (DMA 중의 20 mM)으로 처리하였다. 변형된 항체를, 0.1 M NaH2PO4, 50 mM NaCl, pH 6.5를 함유하는 수성 완충액 내로 미리 평형시킨 바 있는 세파덱스 G25 예비-패킹된 칼럼 상에서 정제하였다. 이와 같이 정제한 샘플을 대상으로 하여 분광 광도계로 검정하여 (ε280nm = 206,460 M-1cm-1) 항체의 농도를 계산하였다. 결합된 링커의 농도는 티올 함유 아미노산인 시스테인과의 반응에 이용 가능한 링커의 양을 간접적으로 측정해 주는 확립된 과정에 의해 결정하였다. 변형 후 항체당 6.0개 링커가 존재하는 것으로 결정되었다.In the first step, the antibody was reacted with the modifier succinimidyl 4- (N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC). A solution of huMy9-6 antibody at a concentration of 8.5 mg / mL in aqueous buffer containing 0.05 M potassium phosphate, 0.05 M sodium chloride, 2 mM ethylenediaminetetraacetic acid (EDTA) and 3% DMA, pH 6.5 was 8.5-fold for 90 minutes at room temperature. Treated with molar excess of SMCC solution (20 mM in DMA). Modified antibodies were purified on Sephadex G25 pre-packed columns that had previously been equilibrated into aqueous buffer containing 0.1 M NaH 2 PO 4 , 50 mM NaCl, pH 6.5. The sample thus purified was assayed with a spectrophotometer (ε 280 nm = 206,460 M -1 cm -1 ) to calculate the concentration of the antibody. The concentration of bound linker was determined by an established procedure that indirectly measured the amount of linker available for reaction with the thiol containing amino acid cysteine. After modification, it was determined that there were 6.0 linkers per antibody.
변형된 항체는 0.1 M NaH2PO4, 50 mM NaCl, 5% DMA, pH 6.5 중에서 2.0 mg/ml이 되도록 희석시키고, 0.20 mM DC07 (2.5 mol DC07/링커)로 처리하였다. 접합 반응을 실온 하에 80분 동안 진행시켰다. 이어서, 반응물을 5% 아세트산으로 산성화시키고, 20% DMA, 0.1 M NaH2PO4, 50 mM NaCl, pH 5.0에서 평형시킨 분석용 크기 배제 (SEC) 칼럼이 장착된 HPLC 시스템을 이용하여 분석용으로 제출하였는데, 이로써 반응되지 않은 DC07로부터 항체-약물 접합체를 격리시킬 수 있었다. 상기 칼럼으로부터의 용출액을 대상으로 하여 280 nM 및 340 nM 하에서의 흡광도를 모니터링하였다. 항체에 대한 DC07의 비를 분광 광도계로 측정한 결과 3.5인 것으로 결정되었다 (약물의 경우, ε340nm = 38,000 M-1cm-1 및 ε280nm = 23,000 M-1cm-1이고, 항체의 경우에는 ε280nm = 206,460 M-1cm-1이다).The modified antibody was diluted to 2.0 mg / ml in 0.1 M NaH 2 PO 4 , 50 mM NaCl, 5% DMA, pH 6.5 and treated with 0.20 mM DC07 (2.5 mol DC07 / linker). The conjugation reaction proceeded at room temperature for 80 minutes. The reaction was then acidified with 5% acetic acid and analyzed for analysis using an HPLC system equipped with an analytical size exclusion (SEC) column equilibrated at 20% DMA, 0.1 M NaH 2 PO 4 , 50 mM NaCl, pH 5.0. Submitted, it was possible to isolate the antibody-drug conjugate from unreacted DC07. Eluates from the column were monitored for absorbance under 280 nM and 340 nM. The ratio of DC07 to antibody was determined by spectrophotometer to be 3.5 (for drugs, ε 340 nm = 38,000 M -1 cm -1 and ε 280 nm = 23,000 M -1 cm -1 , and for antibodies ε 280nm = 206,460 M −1 cm −1 ).
SEC 분석으로부터 약물/Ab 비에 관한 계산:Calculation on Drug / Ab Ratio from SEC Analysis:
DC07 = A340/38000DC07 = A 340/38000
huMy9-6 = [(A280-(23000* A340))/38000]/206460 huMy9-6 = [(A 280 - ( 23000 * A 340)) / 38000] / 206460
약물/Ab = DC07/My9-6Drug / Ab = DC07 / My9-6
실시예 10Example 10
생물학적 결과Biological results
● 라모스 및 HL60/S 세포에 대한 DC07-SMe (도 2) 및 DC1-SMe (도 1D)의 세포독성은 시험관 내에서 5일간 노출시키면서 측정하였다. 그 결과가 도 6에 제시되었다. IC50이 다음에 제시되었다.Cytotoxicity of DC07-SMe (FIG. 2) and DC1-SMe (FIG. 1D) against Ramos and HL60 / S cells was measured with in vitro exposure for 5 days. The results are shown in FIG. IC50 is presented next.
● 라모스 및 HL60 세포에 대한 DC1-SPy 및 DC07-SPy의 세포독성을 측정하였다. 그 결과가 도 7에 제시되었다. IC50이 다음에 제시되었다.Cytotoxicity of DC1-SPy and DC07-SPy against Ramos and HL60 cells was measured. The results are shown in FIG. IC50 is presented next.
DC1-SPy: DC07-SPy:DC1-SPy: DC07-SPy:
세포주 IC50 (M) 세포주 IC50 (M)Cell Line IC 50 (M) Cell Line IC 50 (M)
라모스 7.5 x 10-12 라모스 2.8 x 10-11 Ramos 7.5 x 10 -12 Ramos 2.8 x 10 -11
HL60 7.5 x 10-12 HL60 2.0 x 10-11 HL60 7.5 x 10 -12 HL60 2.0 x 10 -11
● 나말와 및 라모스 세포에 대한 DC08-SSPy의 세포독성을 측정하였다. 그 결과가 도 8에 제시되었다. IC50이 다음에 제시되었다.Cytotoxicity of DC08-SSPy against Namalwa and Ramos cells was measured. The results are shown in FIG. IC50 is presented next.
● 항원 양성 카라 및 항원 음성 라모스 세포에 대한 MY9-6-DC07 (디설파이드 연결됨)의 세포독성을 측정하였다. 그 결과가 도 10에 제시되었고, IC50이 다음에 제시되었다.Cytotoxicity of MY9-6-DC07 (disulfide linked) against antigen positive Kara and antigen negative Ramos cells was measured. The results are shown in FIG. 10 and IC50 is shown next.
● 항원 양성 COLO 205 및 항원 음성 A-375 세포에 대한 huC242-DC07 (티오 에테르 연결됨)의 세포독성을 측정하였다. 그 결과가 도 11에 제시되었고, IC50이 다음에 제시되었다.Cytotoxicity of huC242-DC07 (thio ether linked) against antigen
특정의 특허 및 공개 공보가 본 명세서에 참고되었는데, 그의 교시는 전문이 본원에 참고로 도입되었다.Certain patents and publications are incorporated herein by reference, the teachings of which are incorporated herein by reference in their entirety.
본 발명이 그의 구체적 양태를 참고로 하여 상세히 기재되긴 하였지만, 본 발명의 요지 및 범위를 벗어나지 않고서도 각종 변화 및 변형이 이루어질 수 있다는 것이 당업자에게 명백할 것이다.Although the present invention has been described in detail with reference to specific embodiments thereof, it will be apparent to those skilled in the art that various changes and modifications may be made without departing from the spirit and scope of the invention.
Claims (34)



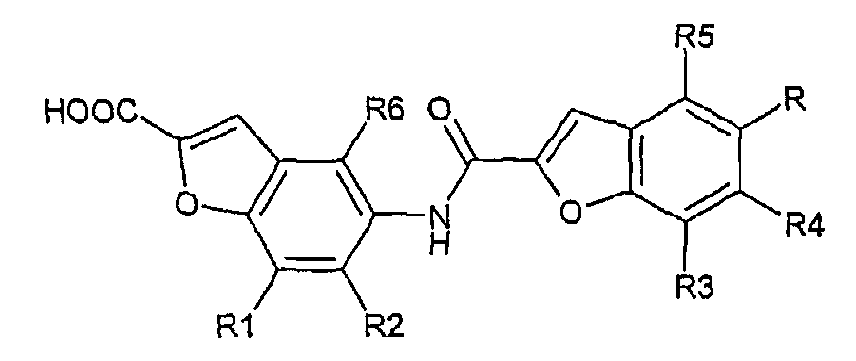
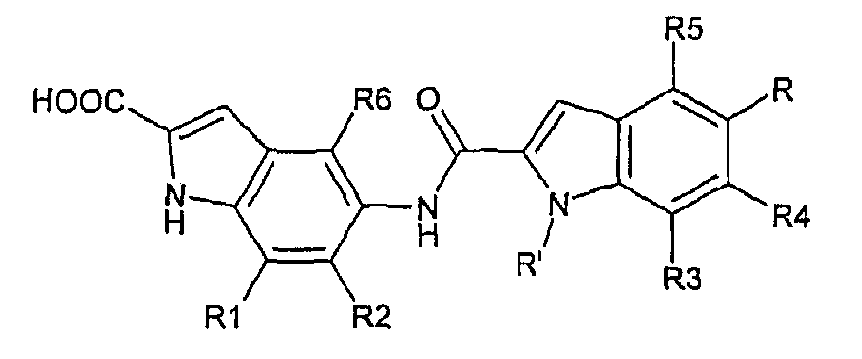





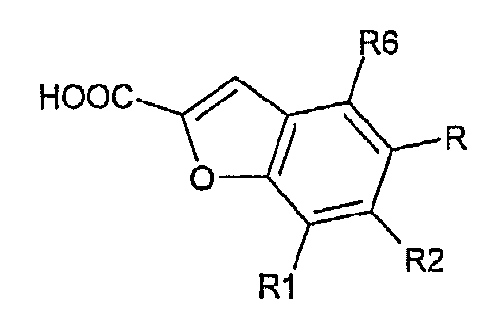

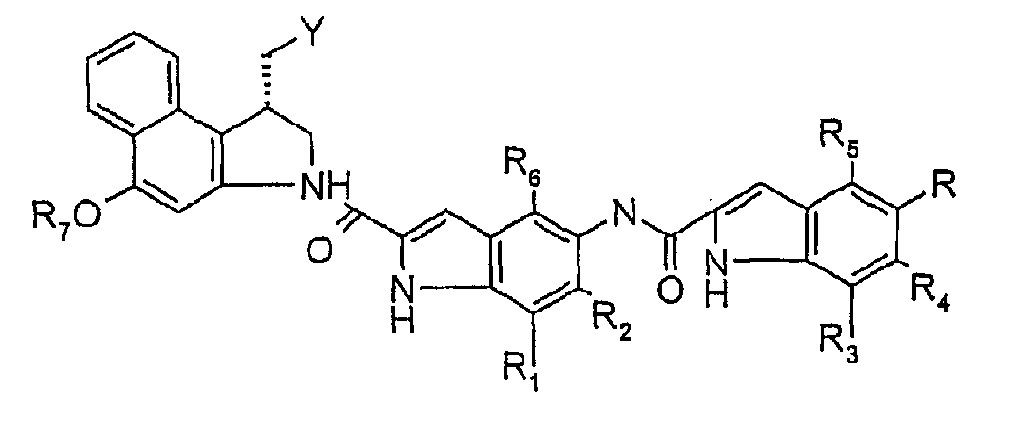
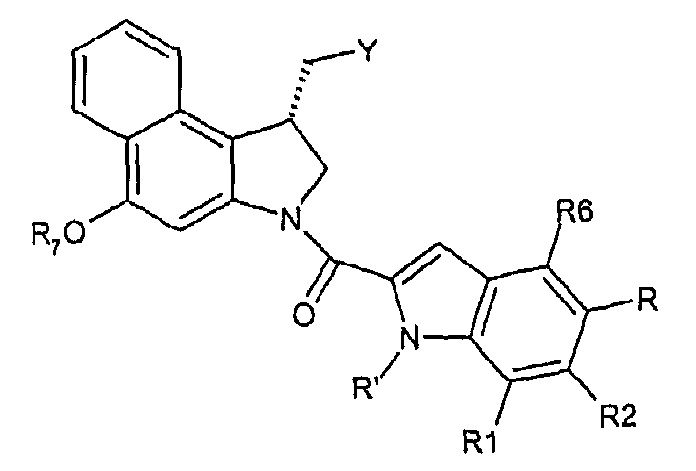

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06290379.4 | 2006-03-07 | ||
| EP06290379A EP1832577A1 (en) | 2006-03-07 | 2006-03-07 | Improved prodrugs of CC-1065 analogs |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20080106919A true KR20080106919A (en) | 2008-12-09 |
Family
ID=36679356
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020087021749A Withdrawn KR20080106919A (en) | 2006-03-07 | 2007-03-06 | Improved Prodrugs of CC-1065 Analogs |
Country Status (18)
| Country | Link |
|---|---|
| US (2) | US8012978B2 (en) |
| EP (2) | EP1832577A1 (en) |
| JP (1) | JP2009529030A (en) |
| KR (1) | KR20080106919A (en) |
| AR (1) | AR059767A1 (en) |
| AU (1) | AU2007222123A1 (en) |
| BR (1) | BRPI0708654A2 (en) |
| CA (1) | CA2642870A1 (en) |
| DO (1) | DOP2007000041A (en) |
| EA (1) | EA200870327A1 (en) |
| IL (1) | IL193673A0 (en) |
| MA (1) | MA30322B1 (en) |
| MX (1) | MX2008011431A (en) |
| NO (1) | NO20083910L (en) |
| PE (1) | PE20080691A1 (en) |
| TW (1) | TW200813028A (en) |
| UY (1) | UY30190A1 (en) |
| WO (1) | WO2007102069A1 (en) |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100056762A1 (en) | 2001-05-11 | 2010-03-04 | Old Lloyd J | Specific binding proteins and uses thereof |
| PT1392359E (en) | 2001-05-11 | 2010-01-27 | Ludwig Inst For Cancer Res Ltd | Specific binding proteins and uses thereof |
| US6756397B2 (en) * | 2002-04-05 | 2004-06-29 | Immunogen, Inc. | Prodrugs of CC-1065 analogs |
| WO2008010101A2 (en) | 2006-07-18 | 2008-01-24 | Sanofi-Aventis | Antagonist antibody against epha2 for the treatment of cancer |
| MX338185B (en) * | 2007-01-25 | 2016-04-05 | Dana Farber Cancer Inst Inc | Use of anti-egfr antibodies in treatment of egfr mutant mediated disease. |
| AU2008227123B2 (en) * | 2007-03-15 | 2014-03-27 | Ludwig Institute For Cancer Research Ltd. | Treatment method using EGFR antibodies and src inhibitors and related formulations |
| US9901567B2 (en) | 2007-08-01 | 2018-02-27 | Syntarga B.V. | Substituted CC-1065 analogs and their conjugates |
| JP5532486B2 (en) | 2007-08-14 | 2014-06-25 | ルードヴィッヒ インスティテュート フォー キャンサー リサーチ | Monoclonal antibody 175 targeting EGF receptor and derivatives and uses thereof |
| WO2009059309A2 (en) * | 2007-11-01 | 2009-05-07 | Panacea Pharmaceuticals, Inc. | Furin-cleavable peptide linkers for drug-ligand conjugates |
| US8889868B2 (en) | 2008-11-03 | 2014-11-18 | Syntarga Bv | CC-1065 analogs and their conjugates |
| FR2947269B1 (en) | 2009-06-29 | 2013-01-18 | Sanofi Aventis | NEW ANTICANCER COMPOUNDS |
| FR2949469A1 (en) | 2009-08-25 | 2011-03-04 | Sanofi Aventis | ANTICANCER DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION |
| US20110076232A1 (en) * | 2009-09-29 | 2011-03-31 | Ludwig Institute For Cancer Research | Specific binding proteins and uses thereof |
| TW201116297A (en) | 2009-10-02 | 2011-05-16 | Sanofi Aventis | Antibodies that specifically bind to the EphA2 receptor |
| AR078471A1 (en) | 2009-10-02 | 2011-11-09 | Sanofi Aventis | MAITANSINOID COMPOUNDS AND THE USE OF THESE TO PREPARE CONJUGATES WITH AN ANTIBODY WHICH ARE USED AS ANTI-BANKING AGENTS AND THE PREPARATION PROCEDURE OF THESE CONJUGATES |
| CN106749665B (en) | 2010-04-21 | 2021-03-26 | 辛塔佳有限公司 | Conjugates of CC-1065 analogs and bifunctional linkers |
| FR2963007B1 (en) | 2010-07-26 | 2013-04-05 | Sanofi Aventis | ANTICANCER DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION |
| WO2013148631A1 (en) * | 2012-03-30 | 2013-10-03 | The Scripps Research Institute | Cyclic prodrugs of duocarmycin analogs |
| WO2014009774A1 (en) | 2012-07-12 | 2014-01-16 | Hangzhou Dac Biotech Co., Ltd | Conjugates of cell binding molecules with cytotoxic agents |
| JP6133431B2 (en) | 2012-11-24 | 2017-05-24 | ハンジョウ ディーエーシー バイオテック シーオー.,エルティディ.Hangzhou Dac Biotech Co.,Ltd. | Use of hydrophilic conjugates and conjugation reactions between drug molecules and cell binding molecules |
| US9353150B2 (en) | 2012-12-04 | 2016-05-31 | Massachusetts Institute Of Technology | Substituted pyrazino[1′,2′:1 ,5]pyrrolo[2,3-b]-indole-1,4-diones for cancer treatment |
| PT2948184T (en) | 2014-01-10 | 2016-07-08 | Synthon Biopharmaceuticals Bv | Duocarmycin adcs showing improved in vivo antitumor activity |
| JP6224268B2 (en) | 2014-01-10 | 2017-11-01 | シントン・バイオファーマシューティカルズ・ビー.ブイ.Synthon Biopharmaceuticals B.V. | Duocarmycin ADC for use in the treatment of endometrial cancer |
| LT3092010T (en) | 2014-01-10 | 2018-10-25 | Synthon Biopharmaceuticals B.V. | Method for purifying cys-linked antibody-drug conjugates |
| US10464955B2 (en) | 2014-02-28 | 2019-11-05 | Hangzhou Dac Biotech Co., Ltd. | Charged linkers and their uses for conjugation |
| EP3319936B1 (en) | 2015-07-12 | 2025-12-17 | Hangzhou Dac Biotech Co., Ltd. | Bridge linkers for conjugation of cell-binding molecules |
| US9839687B2 (en) | 2015-07-15 | 2017-12-12 | Suzhou M-Conj Biotech Co., Ltd. | Acetylenedicarboxyl linkers and their uses in specific conjugation of a cell-binding molecule |
| US10918627B2 (en) | 2016-05-11 | 2021-02-16 | Massachusetts Institute Of Technology | Convergent and enantioselective total synthesis of Communesin analogs |
| TW201808336A (en) | 2016-05-11 | 2018-03-16 | 賽諾菲公司 | Treatment regimen using anti-MUC1 maytansinoid immunoconjugate antibody for the treatment of tumors |
| KR20220147721A (en) | 2016-11-14 | 2022-11-03 | 항저우 디에이씨 바이오테크 씨오, 엘티디 | Conjugation linkers, cell binding molecule-drug conjugates containing the likers, methods of making and uses such conjugates with the linkers |
| US11932650B2 (en) | 2017-05-11 | 2024-03-19 | Massachusetts Institute Of Technology | Potent agelastatin derivatives as modulators for cancer invasion and metastasis |
| US10640508B2 (en) | 2017-10-13 | 2020-05-05 | Massachusetts Institute Of Technology | Diazene directed modular synthesis of compounds with quaternary carbon centers |
| US11535634B2 (en) | 2019-06-05 | 2022-12-27 | Massachusetts Institute Of Technology | Compounds, conjugates, and compositions of epipolythiodiketopiperazines and polythiodiketopiperazines and uses thereof |
| WO2021191219A1 (en) * | 2020-03-23 | 2021-09-30 | Helmholtz-Zentrum für Infektionsforschung GmbH | N-phenyl-3-mercaptopropanamide derivatives as metallo-beta-lactamase inhibitors for the treatment of bacterial infections |
| WO2022182415A1 (en) | 2021-02-24 | 2022-09-01 | Massachusetts Institute Of Technology | Himastatin derivatives, and processes of preparation thereof, and uses thereof |
| AU2021362997A1 (en) | 2021-11-03 | 2024-05-16 | Hangzhou Dac Biotech Co., Ltd. | Specific conjugation of an antibody |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5278324A (en) * | 1990-08-28 | 1994-01-11 | Virginia Tech Intellectual Properties, Inc. | Water soluble derivatives of taxol |
| ES2149768T3 (en) * | 1992-03-25 | 2000-11-16 | Immunogen Inc | CONJUGATES OF BINDING AGENTS OF CELLS DERIVED FROM CC-1065. |
| US5646298A (en) | 1995-06-07 | 1997-07-08 | Procoron, Inc. | Cyclopropylindole prodrugs |
| US6756397B2 (en) * | 2002-04-05 | 2004-06-29 | Immunogen, Inc. | Prodrugs of CC-1065 analogs |
| US6534660B1 (en) | 2002-04-05 | 2003-03-18 | Immunogen, Inc. | CC-1065 analog synthesis |
-
2006
- 2006-03-07 EP EP06290379A patent/EP1832577A1/en not_active Withdrawn
-
2007
- 2007-03-05 PE PE2007000236A patent/PE20080691A1/en not_active Application Discontinuation
- 2007-03-06 AU AU2007222123A patent/AU2007222123A1/en not_active Abandoned
- 2007-03-06 WO PCT/IB2007/000521 patent/WO2007102069A1/en not_active Ceased
- 2007-03-06 TW TW096107668A patent/TW200813028A/en unknown
- 2007-03-06 UY UY30190A patent/UY30190A1/en not_active Application Discontinuation
- 2007-03-06 AR ARP070100932A patent/AR059767A1/en unknown
- 2007-03-06 CA CA002642870A patent/CA2642870A1/en not_active Abandoned
- 2007-03-06 JP JP2008557845A patent/JP2009529030A/en not_active Withdrawn
- 2007-03-06 DO DO2007000041A patent/DOP2007000041A/en unknown
- 2007-03-06 BR BRPI0708654-7A patent/BRPI0708654A2/en not_active Application Discontinuation
- 2007-03-06 MX MX2008011431A patent/MX2008011431A/en not_active Application Discontinuation
- 2007-03-06 EP EP07713102A patent/EP1993999A1/en not_active Withdrawn
- 2007-03-06 EA EA200870327A patent/EA200870327A1/en unknown
- 2007-03-06 KR KR1020087021749A patent/KR20080106919A/en not_active Withdrawn
-
2008
- 2008-08-25 IL IL193673A patent/IL193673A0/en unknown
- 2008-09-04 US US12/204,082 patent/US8012978B2/en not_active Expired - Fee Related
- 2008-09-12 NO NO20083910A patent/NO20083910L/en not_active Application Discontinuation
- 2008-09-29 MA MA31261A patent/MA30322B1/en unknown
-
2011
- 2011-07-15 US US13/184,255 patent/US20110280890A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| US20090028821A1 (en) | 2009-01-29 |
| BRPI0708654A2 (en) | 2011-06-07 |
| UY30190A1 (en) | 2008-10-31 |
| MA30322B1 (en) | 2009-04-01 |
| US20110280890A1 (en) | 2011-11-17 |
| EA200870327A1 (en) | 2009-02-27 |
| WO2007102069A1 (en) | 2007-09-13 |
| US8012978B2 (en) | 2011-09-06 |
| EP1832577A1 (en) | 2007-09-12 |
| IL193673A0 (en) | 2009-05-04 |
| EP1993999A1 (en) | 2008-11-26 |
| TW200813028A (en) | 2008-03-16 |
| PE20080691A1 (en) | 2008-06-04 |
| DOP2007000041A (en) | 2007-10-31 |
| JP2009529030A (en) | 2009-08-13 |
| NO20083910L (en) | 2008-12-01 |
| CA2642870A1 (en) | 2007-09-13 |
| AU2007222123A1 (en) | 2007-09-13 |
| MX2008011431A (en) | 2008-09-22 |
| AR059767A1 (en) | 2008-04-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8012978B2 (en) | Prodrugs of CC-A1065 analogs | |
| AU2003218062B2 (en) | Improved prodrugs of CC-1065 analogs | |
| TWI411609B (en) | Leptomycin derivatives | |
| HK1134240B (en) | Leptomycin derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0105 | International application |
Patent event date: 20080905 Patent event code: PA01051R01D Comment text: International Patent Application |
|
| PG1501 | Laying open of application | ||
| PC1203 | Withdrawal of no request for examination | ||
| WITN | Application deemed withdrawn, e.g. because no request for examination was filed or no examination fee was paid |