KR101410019B1 - 다가알코올과 염화수소의 반응에 의한 클로로히드린화합물의 제조방법 - Google Patents
다가알코올과 염화수소의 반응에 의한 클로로히드린화합물의 제조방법 Download PDFInfo
- Publication number
- KR101410019B1 KR101410019B1 KR1020070097675A KR20070097675A KR101410019B1 KR 101410019 B1 KR101410019 B1 KR 101410019B1 KR 1020070097675 A KR1020070097675 A KR 1020070097675A KR 20070097675 A KR20070097675 A KR 20070097675A KR 101410019 B1 KR101410019 B1 KR 101410019B1
- Authority
- KR
- South Korea
- Prior art keywords
- reactor
- reaction
- chlorohydrin
- feed
- distillation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 title claims abstract description 172
- 150000005846 sugar alcohols Polymers 0.000 title claims abstract description 34
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 title claims abstract description 31
- 229910000041 hydrogen chloride Inorganic materials 0.000 title claims abstract description 31
- 238000006243 chemical reaction Methods 0.000 title claims description 130
- 238000000034 method Methods 0.000 title claims description 71
- 230000008569 process Effects 0.000 title description 28
- -1 chlorohydrin compound Chemical class 0.000 title description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims abstract description 225
- 235000011187 glycerol Nutrition 0.000 claims abstract description 109
- 238000004821 distillation Methods 0.000 claims abstract description 94
- 238000004519 manufacturing process Methods 0.000 claims abstract description 64
- XENVCRGQTABGKY-ZHACJKMWSA-N chlorohydrin Chemical compound CC#CC#CC#CC#C\C=C\C(Cl)CO XENVCRGQTABGKY-ZHACJKMWSA-N 0.000 claims abstract description 45
- 239000003054 catalyst Substances 0.000 claims abstract description 33
- 239000000203 mixture Substances 0.000 claims abstract description 32
- 239000011541 reaction mixture Substances 0.000 claims abstract description 29
- 238000005660 chlorination reaction Methods 0.000 claims abstract description 14
- 150000007524 organic acids Chemical class 0.000 claims abstract description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 117
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 50
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 claims description 26
- SSZWWUDQMAHNAQ-UHFFFAOYSA-N 3-chloropropane-1,2-diol Chemical compound OCC(O)CCl SSZWWUDQMAHNAQ-UHFFFAOYSA-N 0.000 claims description 17
- 238000000926 separation method Methods 0.000 claims description 14
- 239000003377 acid catalyst Substances 0.000 claims description 13
- 150000002148 esters Chemical class 0.000 claims description 12
- 238000005457 optimization Methods 0.000 claims description 10
- 238000005292 vacuum distillation Methods 0.000 claims description 8
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 5
- 150000003839 salts Chemical class 0.000 claims description 5
- 238000004090 dissolution Methods 0.000 claims description 4
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 claims description 2
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 claims description 2
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 claims description 2
- DYPJJAAKPQKWTM-UHFFFAOYSA-N 2-chloropropane-1,3-diol Chemical compound OCC(Cl)CO DYPJJAAKPQKWTM-UHFFFAOYSA-N 0.000 claims description 2
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 2
- 229920000166 polytrimethylene carbonate Polymers 0.000 claims description 2
- 235000013772 propylene glycol Nutrition 0.000 claims description 2
- 150000001875 compounds Chemical class 0.000 claims 6
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims 1
- 150000008064 anhydrides Chemical class 0.000 claims 1
- 150000002763 monocarboxylic acids Chemical class 0.000 claims 1
- DEWLEGDTCGBNGU-UHFFFAOYSA-N 1,3-dichloropropan-2-ol Chemical compound ClCC(O)CCl DEWLEGDTCGBNGU-UHFFFAOYSA-N 0.000 abstract description 86
- 235000005985 organic acids Nutrition 0.000 abstract 1
- 235000011054 acetic acid Nutrition 0.000 description 38
- 239000000047 product Substances 0.000 description 31
- 239000007788 liquid Substances 0.000 description 27
- 239000002994 raw material Substances 0.000 description 16
- 239000000243 solution Substances 0.000 description 15
- 239000007791 liquid phase Substances 0.000 description 14
- 238000009835 boiling Methods 0.000 description 13
- 150000001735 carboxylic acids Chemical class 0.000 description 13
- 238000000066 reactive distillation Methods 0.000 description 13
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 230000014509 gene expression Effects 0.000 description 11
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 11
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 11
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 10
- 239000006227 byproduct Substances 0.000 description 10
- 239000000460 chlorine Substances 0.000 description 10
- 229910052801 chlorine Inorganic materials 0.000 description 10
- 238000010924 continuous production Methods 0.000 description 8
- 239000003960 organic solvent Substances 0.000 description 8
- OSDWBNJEKMUWAV-UHFFFAOYSA-N Allyl chloride Chemical compound ClCC=C OSDWBNJEKMUWAV-UHFFFAOYSA-N 0.000 description 7
- 239000003225 biodiesel Substances 0.000 description 7
- 239000003513 alkali Substances 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 238000011033 desalting Methods 0.000 description 5
- 230000003647 oxidation Effects 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- 238000007086 side reaction Methods 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 239000012808 vapor phase Substances 0.000 description 5
- ZXCYIJGIGSDJQQ-UHFFFAOYSA-N 2,3-dichloropropan-1-ol Chemical compound OCC(Cl)CCl ZXCYIJGIGSDJQQ-UHFFFAOYSA-N 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- XXROGKLTLUQVRX-UHFFFAOYSA-N allyl alcohol Chemical compound OCC=C XXROGKLTLUQVRX-UHFFFAOYSA-N 0.000 description 4
- 239000012295 chemical reaction liquid Substances 0.000 description 4
- 239000001279 citrus aurantifolia swingle expressed oil Substances 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 239000011259 mixed solution Substances 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000000376 reactant Substances 0.000 description 4
- 235000011121 sodium hydroxide Nutrition 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 238000007259 addition reaction Methods 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 230000001276 controlling effect Effects 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 150000004665 fatty acids Chemical class 0.000 description 3
- 238000004508 fractional distillation Methods 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- QWPPOHNGKGFGJK-UHFFFAOYSA-N hypochlorous acid Chemical compound ClO QWPPOHNGKGFGJK-UHFFFAOYSA-N 0.000 description 3
- 238000006386 neutralization reaction Methods 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 239000002699 waste material Substances 0.000 description 3
- 239000002351 wastewater Substances 0.000 description 3
- XEPXTKKIWBPAEG-UHFFFAOYSA-N 1,1-dichloropropan-1-ol Chemical compound CCC(O)(Cl)Cl XEPXTKKIWBPAEG-UHFFFAOYSA-N 0.000 description 2
- KNKRKFALVUDBJE-UHFFFAOYSA-N 1,2-dichloropropane Chemical compound CC(Cl)CCl KNKRKFALVUDBJE-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- BZHJMEDXRYGGRV-UHFFFAOYSA-N Vinyl chloride Chemical compound ClC=C BZHJMEDXRYGGRV-UHFFFAOYSA-N 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- XYAUIVRRMJYYHR-UHFFFAOYSA-N acetic acid;propane-1,2,3-triol Chemical compound CC(O)=O.OCC(O)CO XYAUIVRRMJYYHR-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 238000005886 esterification reaction Methods 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 239000013067 intermediate product Substances 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- BDJRBEYXGGNYIS-UHFFFAOYSA-N nonanedioic acid Chemical compound OC(=O)CCCCCCCC(O)=O BDJRBEYXGGNYIS-UHFFFAOYSA-N 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- HVAMZGADVCBITI-UHFFFAOYSA-M pent-4-enoate Chemical compound [O-]C(=O)CCC=C HVAMZGADVCBITI-UHFFFAOYSA-M 0.000 description 2
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 239000000344 soap Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 238000009834 vaporization Methods 0.000 description 2
- 230000008016 vaporization Effects 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 235000019871 vegetable fat Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 235000013311 vegetables Nutrition 0.000 description 2
- OWXJKYNZGFSVRC-NSCUHMNNSA-N (e)-1-chloroprop-1-ene Chemical compound C\C=C\Cl OWXJKYNZGFSVRC-NSCUHMNNSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- AVGQTJUPLKNPQP-UHFFFAOYSA-N 1,1,1-trichloropropane Chemical compound CCC(Cl)(Cl)Cl AVGQTJUPLKNPQP-UHFFFAOYSA-N 0.000 description 1
- ZAIDIVBQUMFXEC-UHFFFAOYSA-N 1,1-dichloroprop-1-ene Chemical compound CC=C(Cl)Cl ZAIDIVBQUMFXEC-UHFFFAOYSA-N 0.000 description 1
- CSVFWMMPUJDVKH-UHFFFAOYSA-N 1,1-dichloropropan-2-one Chemical compound CC(=O)C(Cl)Cl CSVFWMMPUJDVKH-UHFFFAOYSA-N 0.000 description 1
- IFDLXKQSUOWIBO-UHFFFAOYSA-N 1,3-dichloropropan-1-ol Chemical compound OC(Cl)CCCl IFDLXKQSUOWIBO-UHFFFAOYSA-N 0.000 description 1
- 229940035437 1,3-propanediol Drugs 0.000 description 1
- PSYQXTDKRKLJQC-UHFFFAOYSA-N 1-chloropropane-1,3-diol Chemical compound OCCC(O)Cl PSYQXTDKRKLJQC-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- PPQNQXQZIWHJRB-UHFFFAOYSA-N Methylcholanthrene Chemical compound C1=CC=C2C3=CC4=CC=C(C)C(CC5)=C4C5=C3C=CC2=C1 PPQNQXQZIWHJRB-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- 238000006137 acetoxylation reaction Methods 0.000 description 1
- 238000000184 acid digestion Methods 0.000 description 1
- 150000007933 aliphatic carboxylic acids Chemical class 0.000 description 1
- 239000012670 alkaline solution Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 238000010923 batch production Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 238000010961 commercial manufacture process Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000006837 decompression Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 238000006735 epoxidation reaction Methods 0.000 description 1
- 239000003822 epoxy resin Substances 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- ULYZAYCEDJDHCC-UHFFFAOYSA-N isopropyl chloride Chemical compound CC(C)Cl ULYZAYCEDJDHCC-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 229960003424 phenylacetic acid Drugs 0.000 description 1
- 239000003279 phenylacetic acid Substances 0.000 description 1
- 229920000647 polyepoxide Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- CSIGAEASXSGNKS-UHFFFAOYSA-N propane-1,1,3-triol Chemical compound OCCC(O)O CSIGAEASXSGNKS-UHFFFAOYSA-N 0.000 description 1
- ULWHHBHJGPPBCO-UHFFFAOYSA-N propane-1,1-diol Chemical compound CCC(O)O ULWHHBHJGPPBCO-UHFFFAOYSA-N 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 238000000197 pyrolysis Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 150000004072 triols Chemical class 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C31/00—Saturated compounds having hydroxy or O-metal groups bound to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/62—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by introduction of halogen; by substitution of halogen atoms by other halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/74—Separation; Purification; Use of additives, e.g. for stabilisation
- C07C29/76—Separation; Purification; Use of additives, e.g. for stabilisation by physical treatment
- C07C29/80—Separation; Purification; Use of additives, e.g. for stabilisation by physical treatment by distillation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C31/00—Saturated compounds having hydroxy or O-metal groups bound to acyclic carbon atoms
- C07C31/34—Halogenated alcohols
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
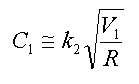


Claims (20)
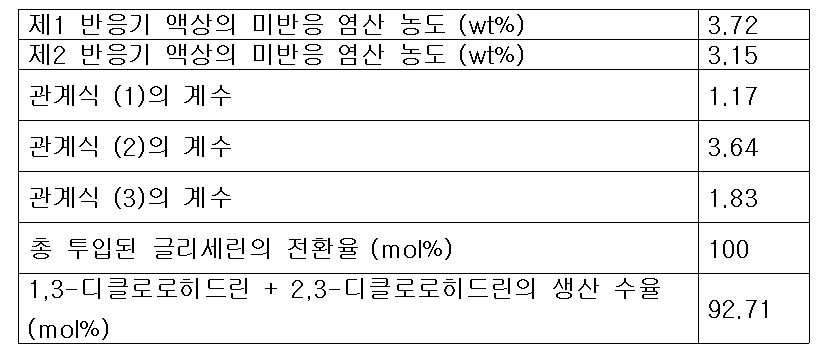
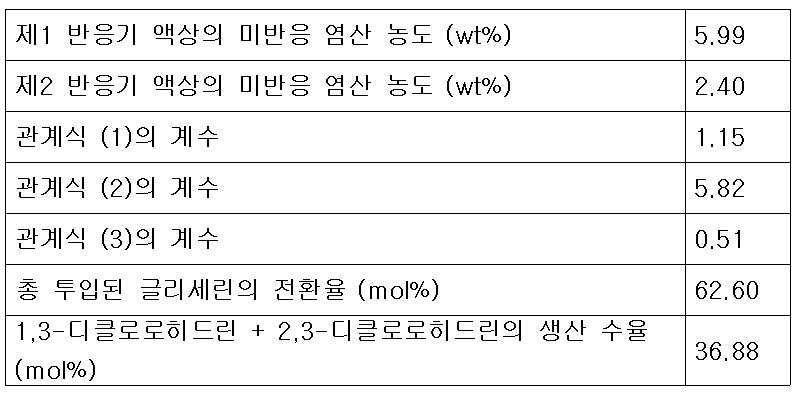
- 다가알코올, 염화수소 및 염소화 촉매인 유기산으로 이루어진 반응혼합물 피드가 제1반응기에 공급되어 상기 제1반응기로부터 염소화 반응에 의하여 클로로히드린이 제조되며;상기 제1반응기로부터 배출되는 클로로히드린 및 미반응된 반응혼합물을 포함하는 제1생성혼합물 피드와 추가의 다가알코올 피드가 상기 제2반응기로 공급되어 추가의 염소화반응에 의하여 클로로히드린이 제조되고;제2반응기로부터 배출되는 클로로히드린을 포함하는 제2생성혼합물 피드가 증류컬럼에 공급되어 클로로히드린을 포함하는 증류생산물이 증류컬럼 상부로 분리되며;상기 증류컬럼 하부로 배출되며 클로로히드린을 포함하는 증류잔류액 일부의 재순환 피드가 상기 제1반응기로 순환되는 것을 특징으로 하는 클로로히드린의 연속제조방법.
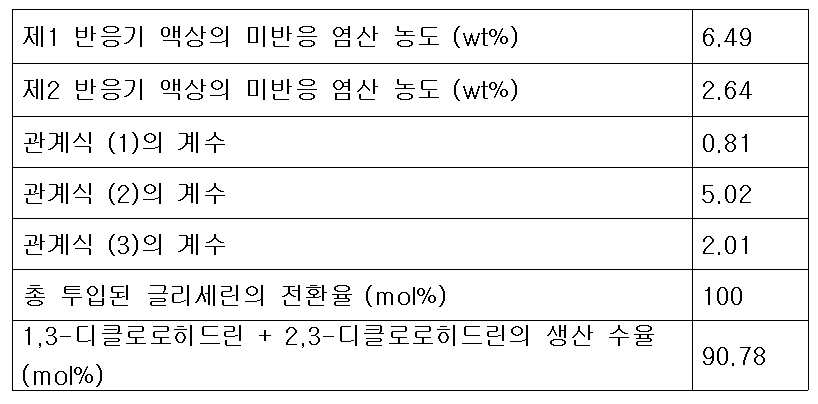
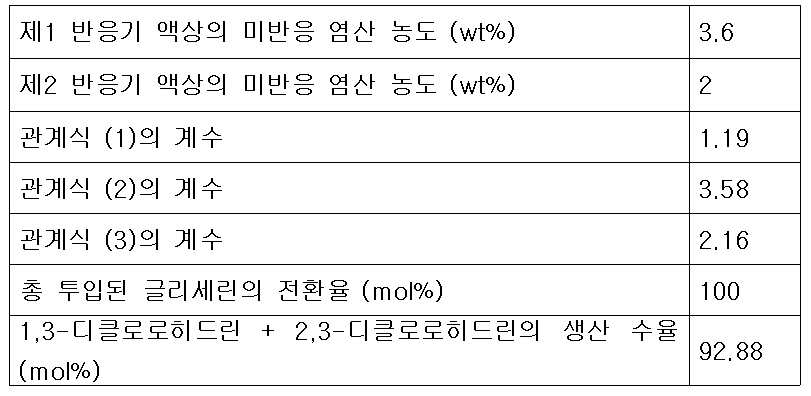
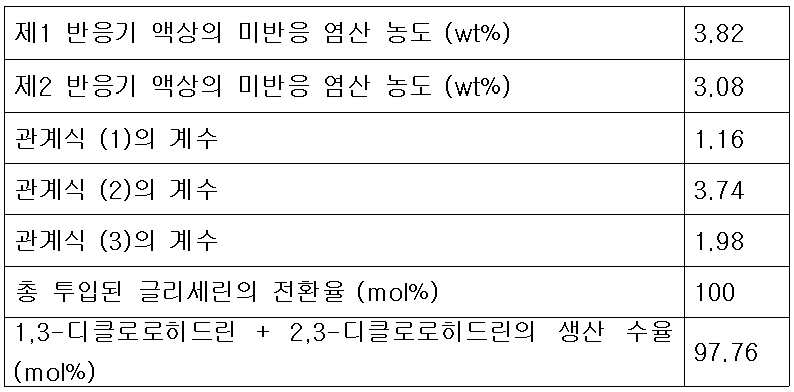
- 제 1항에 있어서,제1반응기 및 제2반응기에 투입되는 다가알코올 유량과 재순환 피드 유량의 비가 하기 식 (1)을 만족하는 것을 특징으로 하는 클로로히드린의 연속제조방법.(1)
 [상기 식 (1)에서 V1은 제1반응기의 유효 부피(ℓ)이고, G는 제1반응기 및 제2반응기에 투입되는 다가알코올의 총 투입 유량(kg/hr)이며, R은 증류컬럼에서 제1반응기로 순환되는 재순환 피드의 유량(kg/hr)이며, k1은 유량 최적화 상수로서 0.3 내지 5 kg/(ℓ-hr)의 값을 갖는다.]
[상기 식 (1)에서 V1은 제1반응기의 유효 부피(ℓ)이고, G는 제1반응기 및 제2반응기에 투입되는 다가알코올의 총 투입 유량(kg/hr)이며, R은 증류컬럼에서 제1반응기로 순환되는 재순환 피드의 유량(kg/hr)이며, k1은 유량 최적화 상수로서 0.3 내지 5 kg/(ℓ-hr)의 값을 갖는다.] - 제2항에 있어서,k1은 0.5 내지 1.5 kg/(ℓ-hr)인 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 1항에 있어서,제2반응기로 공급되는 제1생성혼합물 피드 중에 존재하는 미반응 염산의 농도가 하기 식 (2)를 만족하는 것을 특징으로 하는 클로로히드린의 연속제조방법.(2)
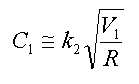 [상기 식 (2)에서 C1은 제1생성혼합물 피드 중에 존재하는 미반응 염산의 농 도(wt%)이고, V1은 제1반응기의 유효 부피(ℓ)이고, R은 증류컬럼에서 제1반응기로 순환되는 재순환 피드의 유량(kg/hr)이며, k2는 유량최적화 상수로서 3 내지 5의 값을 갖는다.]
[상기 식 (2)에서 C1은 제1생성혼합물 피드 중에 존재하는 미반응 염산의 농 도(wt%)이고, V1은 제1반응기의 유효 부피(ℓ)이고, R은 증류컬럼에서 제1반응기로 순환되는 재순환 피드의 유량(kg/hr)이며, k2는 유량최적화 상수로서 3 내지 5의 값을 갖는다.] - 제 1항에 있어서,제2반응기로 투입되는 추가의 다가알코올 피드 유량, 재순환 피드 유량 및 제1생성혼합물 중 미반응 염산의 농도가 하기 식 (3)을 만족하는 것을 특징으로 하는 클로로히드린의 연속제조방법.(3)
 [상기 식 (3)에서 G2는 제2반응기에 투입되는 다가알코올의 유량(kg/hr)이고, G1은 제1반응기의 반응혼합물 피드에 포함되는 다가알코올의 유량(kg/hr)이며, G는 제1반응기 및 제2반응기에 투입되는 다가알코올의 총 투입 유량(kg/hr)이고, R은 증류컬럼에서 제1반응기로 순환되는 재순환 피드의 유량(kg/hr)이며, C1은 제1생성혼합물 피드 중에 남아있는 미반응 염산의 농도(wt%)이고, k3는 유량 최적화 상수로서 0.7 내지 2.5 의 값을 갖는다.]
[상기 식 (3)에서 G2는 제2반응기에 투입되는 다가알코올의 유량(kg/hr)이고, G1은 제1반응기의 반응혼합물 피드에 포함되는 다가알코올의 유량(kg/hr)이며, G는 제1반응기 및 제2반응기에 투입되는 다가알코올의 총 투입 유량(kg/hr)이고, R은 증류컬럼에서 제1반응기로 순환되는 재순환 피드의 유량(kg/hr)이며, C1은 제1생성혼합물 피드 중에 남아있는 미반응 염산의 농도(wt%)이고, k3는 유량 최적화 상수로서 0.7 내지 2.5 의 값을 갖는다.] - 제 1항 내지 제 5항에서 선택되는 어느 한 항에 있어서,제1반응기로부터 배출되는 제1생성혼합물 피드 중 미반응된 반응혼합물은 다가알코올, 염화수소, 유기산 촉매 및 물을 포함하는 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 1항 내지 제 5항에서 선택되는 어느 한 항에 있어서,제1반응기로 순환되는 재순환 피드는 클로로히드린 및 미반응 다가알코올을 포함하는 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 1항 내지 제 5항에서 선택되는 어느 한 항에 있어서,다가알코올은 1,2-에탄디올, 1,2-프로판디올, 1,3-프로판디올, 3-클로로-1,2-프로판디올, 2-클로로-1,3-프로판디올 및 글리세린, 및 이들의 에스테르 화합물, 또는 이들의 혼합물인 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 8항에 있어서,다가알코올은 글리세린인 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 1항 내지 제 5항에서 선택되는 어느 한 항에 있어서,유기산은 모노카르복실산 또는 디카르복실산, 및 이들의 무수물, 이들의 염 또는 이들의 에스테르인 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 10항에 있어서,유기산은 아세트산인 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 1항 내지 제 5항에서 선택되는 어느 한 항에 있어서,염화수소는 무수물인 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 12항에 있어서,제1반응기의 반응온도는 70 내지 140℃인 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 12항에 있어서,제1반응기의 반응온도는 재순환 피드의 온도와 무수 염화수소의 용해열로부터 유지되는 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 14항에 있어서,무수 염화수소가 연속적으로 공급되어 가압하에 반응이 진행되는 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 1항 내지 제 5항에서 선택되는 어느 한 항에 있어서,증류컬럼 상부로부터 분리되는 증류생산물은 클로로히드린, 유기산 촉매 및 물을 포함하는 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 16항에 있어서,증류생산물 총 중량 기준으로 클로로히드린의 함량은 50 내지 90중량%인 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 제 16항에 있어서,상기 증류생산물은 감압증류에 의하여 분리되는 것을 특징으로 하는 클로로히드린의 연속제조방법.
- 삭제
- 제 16항에 따른 클로로히드린을 포함하는 증류생산물로부터 분리단계 없이 에피클로로히드린을 제조하는 방법.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020070097675A KR101410019B1 (ko) | 2007-09-28 | 2007-09-28 | 다가알코올과 염화수소의 반응에 의한 클로로히드린화합물의 제조방법 |
| PCT/KR2008/005474 WO2009041766A1 (en) | 2007-09-28 | 2008-09-17 | Process for preparing chlorohydrin by reaction of polyol with hydrochloric acid |
| CN2008801143217A CN101842339B (zh) | 2007-09-28 | 2008-09-17 | 通过多元醇与氢氯酸的反应制备氯代醇的方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020070097675A KR101410019B1 (ko) | 2007-09-28 | 2007-09-28 | 다가알코올과 염화수소의 반응에 의한 클로로히드린화합물의 제조방법 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20090032429A KR20090032429A (ko) | 2009-04-01 |
| KR101410019B1 true KR101410019B1 (ko) | 2014-06-26 |
Family
ID=40511628
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020070097675A Active KR101410019B1 (ko) | 2007-09-28 | 2007-09-28 | 다가알코올과 염화수소의 반응에 의한 클로로히드린화합물의 제조방법 |
Country Status (3)
| Country | Link |
|---|---|
| KR (1) | KR101410019B1 (ko) |
| CN (1) | CN101842339B (ko) |
| WO (1) | WO2009041766A1 (ko) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101705209B1 (ko) * | 2010-06-30 | 2017-02-09 | 롯데정밀화학 주식회사 | 클로로히드린류 조성물의 제조방법 및 그 방법에 의해 제조된 클로로히드린류 조성물을 사용하는 에피클로로히드린의 제조방법 |
| KR101705208B1 (ko) * | 2010-06-30 | 2017-02-09 | 롯데정밀화학 주식회사 | 클로로히드린류 조성물의 제조방법 및 그 방법에 의해 제조된 클로로히드린류 조성물을 사용하는 에피클로로히드린의 제조방법 |
| KR101705207B1 (ko) * | 2010-06-30 | 2017-02-09 | 롯데정밀화학 주식회사 | 클로로히드린류의 제조방법 및 그 방법에 의해 제조된 클로로히드린류를 사용하는 에피클로로히드린의 제조방법 |
| KR101705205B1 (ko) * | 2010-06-30 | 2017-02-09 | 롯데정밀화학 주식회사 | 클로로히드린류의 제조방법 및 그 방법에 의해 제조된 클로로히드린류를 사용하는 에피클로로히드린의 제조방법 |
| KR101705206B1 (ko) * | 2010-06-30 | 2017-02-09 | 롯데정밀화학 주식회사 | 클로로히드린류의 제조방법 및 그 방법에 의해 제조된 클로로히드린류를 사용하는 에피클로로히드린의 제조방법 |
| KR101705210B1 (ko) * | 2010-06-30 | 2017-02-09 | 롯데정밀화학 주식회사 | 클로로히드린류 조성물의 제조방법 및 그 방법에 의해 제조된 클로로히드린류 조성물을 사용하는 에피클로로히드린의 제조방법 |
| CN110922299A (zh) * | 2019-11-07 | 2020-03-27 | 无锡市银杏塑业科技有限公司 | 一种高含量2-氯乙醇的连续制备方法 |
| CN113234041B (zh) * | 2021-04-07 | 2023-03-10 | 江苏瑞恒新材料科技有限公司 | 一种环氧氯丙烷的制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4665240A (en) | 1986-05-27 | 1987-05-12 | Shell Oil Company | Process for the production of dichlorohydrin |
| WO2006020234A1 (en) | 2004-07-21 | 2006-02-23 | Dow Global Technologies Inc. | Conversion of a multihydroxylated-aliphatic hydrocarbon or ester thereof to a chlorohydrin |
| WO2006106154A1 (fr) | 2005-05-20 | 2006-10-12 | Solvay (Société Anonyme) | Procede continu de fabrication de chlorhydrines |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60258171A (ja) * | 1984-06-04 | 1985-12-20 | Showa Denko Kk | エピクロルヒドリンの製造方法 |
-
2007
- 2007-09-28 KR KR1020070097675A patent/KR101410019B1/ko active Active
-
2008
- 2008-09-17 CN CN2008801143217A patent/CN101842339B/zh not_active Expired - Fee Related
- 2008-09-17 WO PCT/KR2008/005474 patent/WO2009041766A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4665240A (en) | 1986-05-27 | 1987-05-12 | Shell Oil Company | Process for the production of dichlorohydrin |
| WO2006020234A1 (en) | 2004-07-21 | 2006-02-23 | Dow Global Technologies Inc. | Conversion of a multihydroxylated-aliphatic hydrocarbon or ester thereof to a chlorohydrin |
| WO2006106154A1 (fr) | 2005-05-20 | 2006-10-12 | Solvay (Société Anonyme) | Procede continu de fabrication de chlorhydrines |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009041766A1 (en) | 2009-04-02 |
| CN101842339B (zh) | 2013-08-28 |
| KR20090032429A (ko) | 2009-04-01 |
| CN101842339A (zh) | 2010-09-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101410019B1 (ko) | 다가알코올과 염화수소의 반응에 의한 클로로히드린화합물의 제조방법 | |
| TWI325417B (en) | Method of preparing dichloropropanols from glycerine | |
| TWI320036B (en) | Process for preparing a chlorohydrin starting from a polyhydroxylated aliphatic hydrocarbon | |
| JP6373554B2 (ja) | エポキシドの製造方法 | |
| US7906690B2 (en) | Batch, semi-continuous or continuous hydrochlorination of glycerin with reduced volatile chlorinated hydrocarbon by-products and chloracetone levels | |
| CN101208323A (zh) | 由多羟基脂肪烃和氯化剂制备环氧化物的方法 | |
| US9963436B2 (en) | Process for the manufacture of epoxy-monomers and epoxides | |
| EP2589585B1 (en) | Method for preparing chlorohydrins and method for preparing epichlorohydrin using chlorohydrins prepared thereby | |
| CN102040479B (zh) | 一种甘油与氯化氢自催化反应制备二氯丙醇的系统 | |
| TWI523834B (zh) | 多羥化脂族烴之氯化氫反應之方法 | |
| EP3844140B1 (en) | Method and apparatus to produce fatty acids from methyl esters throughout non-catalytic process | |
| JP3903513B2 (ja) | ジアセトキシブテンの製造方法 | |
| HK1096081B (en) | Method of preparing dichloropropanols from glycerine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0109 | Patent application |
Patent event code: PA01091R01D Comment text: Patent Application Patent event date: 20070928 |
|
| PG1501 | Laying open of application | ||
| A201 | Request for examination | ||
| PA0201 | Request for examination |
Patent event code: PA02012R01D Patent event date: 20120809 Comment text: Request for Examination of Application Patent event code: PA02011R01I Patent event date: 20070928 Comment text: Patent Application |
|
| E902 | Notification of reason for refusal | ||
| PE0902 | Notice of grounds for rejection |
Comment text: Notification of reason for refusal Patent event date: 20140221 Patent event code: PE09021S01D |
|
| E701 | Decision to grant or registration of patent right | ||
| PE0701 | Decision of registration |
Patent event code: PE07011S01D Comment text: Decision to Grant Registration Patent event date: 20140520 |
|
| PR0701 | Registration of establishment |
Comment text: Registration of Establishment Patent event date: 20140613 Patent event code: PR07011E01D |
|
| PR1002 | Payment of registration fee |
Payment date: 20140616 End annual number: 3 Start annual number: 1 |
|
| PG1601 | Publication of registration | ||
| FPAY | Annual fee payment |
Payment date: 20170526 Year of fee payment: 4 |
|
| PR1001 | Payment of annual fee |
Payment date: 20170526 Start annual number: 4 End annual number: 4 |
|
| FPAY | Annual fee payment |
Payment date: 20180403 Year of fee payment: 5 |
|
| PR1001 | Payment of annual fee |
Payment date: 20180403 Start annual number: 5 End annual number: 5 |
|
| FPAY | Annual fee payment |
Payment date: 20190307 Year of fee payment: 6 |
|
| PR1001 | Payment of annual fee |
Payment date: 20190307 Start annual number: 6 End annual number: 6 |
|
| PR1001 | Payment of annual fee |
Payment date: 20200330 Start annual number: 7 End annual number: 7 |
|
| PR1001 | Payment of annual fee |
Payment date: 20210326 Start annual number: 8 End annual number: 8 |
|
| PR1001 | Payment of annual fee |
Payment date: 20220323 Start annual number: 9 End annual number: 9 |
|
| PR1001 | Payment of annual fee |
Payment date: 20230329 Start annual number: 10 End annual number: 10 |
|
| PR1001 | Payment of annual fee |
Payment date: 20240325 Start annual number: 11 End annual number: 11 |
|
| PR1001 | Payment of annual fee |
Payment date: 20250325 Start annual number: 12 End annual number: 12 |