JP7335341B2 - Nlrp3モジュレーター - Google Patents
Nlrp3モジュレーター Download PDFInfo
- Publication number
- JP7335341B2 JP7335341B2 JP2021540533A JP2021540533A JP7335341B2 JP 7335341 B2 JP7335341 B2 JP 7335341B2 JP 2021540533 A JP2021540533 A JP 2021540533A JP 2021540533 A JP2021540533 A JP 2021540533A JP 7335341 B2 JP7335341 B2 JP 7335341B2
- Authority
- JP
- Japan
- Prior art keywords
- cancer
- independently
- mmol
- compound
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5386—1,4-Oxazines, e.g. morpholine spiro-condensed or forming part of bridged ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/08—Bridged systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Description
本出願は、2019年1月14日出願の米国仮出願番号第62/791,979号に対する優先権を主張するものであって、その内容の全てを参照により本明細書に組み込むものである。
本開示は、NLRP3シグナル伝達の増加が、対象(例えばヒト)における病理および/または症状および/または難治性の病状、疾患または障害(例えば、T細胞浸潤の少ないがん)の進行および/または治療に関与する、自然免疫活性の不足を正常化し得る病状、疾患または障害などの治療に有用なNLRP3を調節する(例えば、刺激または一部刺激する)、化学物質群(例えば、化合物またはその化合物の医薬的に許容される塩、および/または水和物、および/または共結晶、および/または合剤)を特徴とする。本開示はまた、上記化合物の組成物ならびに他のその使用および製造方法を特徴とする。

第1態様において、本発明は、とりわけ、式(I):

Wは、独立して、R6、-Y-R6、-Q-R6、および-Q-Y-R6から選択され;
Qは、独立して、NR5、CHR5、O、およびSから選択され;
Yは、独立して、C1-10アルキレン、C2-10アルケニレン、およびC2-10アルキニレンから選択され、それらは各々0~4個のRaで置換され、および/または各々適宜
(i)O;または
(ii)N(Rb)の1つが途中に含まれていてもよく;
X1は、独立して、NまたはCR1であり;
X2は、独立して、NまたはCR2であり;
X3は、独立して、NまたはCR3であり;
X4は、独立して、NまたはCR4であり;
ただし、X1、X3およびX4のうちの2個未満がNであり;
R1およびR3は、それぞれ独立して、H、ハロゲン、シアノ、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、およびC1-4ハロアルコキシから選択され;
R2およびR4は、それぞれ独立して、H、ハロゲン、シアノ、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、C1-4ハロアルコキシ、N(C1-4アルキル)2、および5個の環原子を含み、ここで1~4個の環原子がそれぞれ独立してN、NH、N(C1-4アルキル)、O、およびSから選択され、ヘテロアリール部分が0~3個のRdで置換された-(C0-3アルキレン)-ヘテロアリールから選択され;
R5は、独立して、HまたはC1-4アルキルであり;
R6は、独立して、5~12個の環原子を含む、二環または三環シクロアルキルまたはシクロアルケニルであり、ここでシクロアルキルまたはシクロアルケニルはスピロ環であり得るか、または-CR8R9-、-(CR8R9)2-、および-C(=O)-から選択される1個または2個の架橋リンカーを含有し得て、シクロアルキルまたはシクロアルケニル部分は0~4個のReで置換されており;
あるいは、R6は、独立して、5~12個の環原子を含み、ここで1~4個の環原子がそれぞれ独立して、N、N(Rb)、O、およびSから選択される、二環または三環ヘテロシクロアルキルであり、上記ヘテロシクロアルキルはスピロ環であり得るか、または-CR8R9-、-O-、-(CR8R9)2-、-OCR8R9-、および-CR8R9O-、および-C(=O)-から選択される架橋リンカーを含有し得て、ここでヘテロシクロアルキルは、0~4個のReで置換されており;
R7は、独立して、5個の環原子を含み、ここで1~4個の環原子がそれぞれ独立してN、NH、N(C1-4アルキル)、O、およびSから選択され、0~3個のRdで置換されたヘテロアリールであり;
R8およびR9は、それぞれ独立して、H、OH、ハロゲン、C1-4アルキルおよびC1-4アルコキシから選択され;
Raは、独立して、F、OH、シアノ、C1-4アルコキシ、C1-4ハロアルキル、C1-4ハロアルコキシ、および0~1個のRcで置換されたC1-4アルキルから選択され;
Rbは、独立して、H、0~1個のOHで置換されたC1-4アルキル、-C(O)(C1-4アルキル)、および-C(O)O(C1-4アルキル)から選択され;
Rcは、独立して、OH、CONH2およびC1-4アルコキシから選択され;
Rdは、独立して、ハロゲン、OH、シアノ、C1-4アルコキシ、C1-4アルコキシ、C1-4ハロアルキル、C1-4ハロアルコキシ、-C(O)O(C1-4アルキル)、NH2、N(C1-4アルキル)2、-C(O)NH2、-C(O)N(C1-4アルキル)2、C2-6アルケニル、C2-6アルキニル、および0~2個のRaで置換されたC1-6アルキルから選択され;および
Reは、独立して、オキソまたはRdである]
の化合物またはその立体異性体、互変異性体、または医薬的に許容される塩を提供する。
Qは、独立して、NH、N(C1-4アルキル)、CH2、およびOから選択され;
Yは、独立して、C1-10アルキレン、C2-6アルケニレン、およびC2-6アルキニレンから選択され、それらは各々0~4個のRaで置換されており;
R2は、独立して、H、ハロゲン、シアノ、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、およびC1-4ハロアルコキシから選択され;
R4は、独立して、H、ハロゲン、シアノ、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、C1-4ハロアルコキシ、N(C1-4アルキル)2、およびN、NH、O、およびSからそれぞれ独立して選択される1~2個の環原子を含む5員ヘテロアリールから選択され;
R6は、独立して、5~10個の環原子を含む二環または三環シクロアルキルまたはシクロアルケニルであり、ここでシクロアルキルまたはシクロアルケニルは、スピロ環であり得るか、または-CH2-および-CH2CH2-から選択される1個または2個の架橋リンカーを含有し得て、シクロアルキルまたはシクロアルケニル部分は0~4個のReで置換されており;
あるいは、R6は、独立して、5~10個の環原子を含み、ここで1~2個の環原子はそれぞれ独立して、N、N(Rb)、O、およびSから選択される二環ヘテロシクロアルキルであり、ここで二環式環は、スピロ環であり得るか、または-CH2-、-O-、-CH2CH2-、-OCH2-、および-CH2O-から選択される架橋リンカーを含有し得て、ここで上記ヘテロシクロアルキルは、0~4個のReで置換されており;
R7は、独立して、N、NH、O、およびSからそれぞれ独立して選択される1~2個の環原子を含む、5員ヘテロアリールであり;および
R8およびR9は、それぞれ独立して、HまたはC1-4アルキルである。
R7は、独立して、N、NHおよびSからそれぞれ独立して選択される1~2個の環原子を含む5員ヘテロアリールである。
R2は、独立して、H、ハロゲンおよびC1-4アルキルから選択され;
R4は、独立して、H、ハロゲン、シアノ、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、C1-4ハロアルコキシ、N(C1-4アルキル)2、およびピラゾリル、チエニルおよびイソチアゾリルから選択されるヘテロアリールから選択され;および
R7は、独立して、ピラゾリル、チエニルまたはイソチアゾリルである。

Wは、独立して、R6-NH-R6、または-NH-CH2-R6であり;
X2は、独立して、NまたはCR2であり;
R1、R2、R3およびR4は、それぞれ独立して、H、ハロゲンおよびC1-4アルキルから選択され;
R6は、独立して、5~10個の環原子を含む二環または三環シクロアルキルであり、ここでシクロアルキルはスピロ環であり得るか、または-CH2-および-CH2CH2-から選択される1個または2個の架橋リンカーを含有し得て、上記シクロアルキルは0~2個のReで置換されており;
あるいは、R6は、独立して、5~10個の環原子を含み、ここで1~2個の環原子はそれぞれ独立して、N、N(Rb)、およびOから選択される二環ヘテロシクロアルキルであり、ここで二環式環は、スピロ環であり得るか、または-CH2-、-O-、-CH2CH2-、-OCH2-、および-CH2O-から選択される架橋リンカーを含有し得て、上記ヘテロシクロアルキルは0~2個のReで置換されており;
Rbは、独立して、H、C1-4アルキル、および-C(O)O(C1-4アルキル)から選択され;および
Reは、独立して、オキソ、ハロゲン、シアノ、OH、CH2OH、C1-4アルコキシ、C1-4ハロアルキル、N(C1-4アルキル)2、-C(O)NH2、-C(O)O(C1-4アルキル)、C2-6アルケニル、および0~2個のC1-4アルコキシで置換されたC1-4アルキルから選択される]
の化合物またはその立体異性体、互変異性体、または医薬的に許容される塩を提供する。
Wは、独立して、-NH-R6、-NH-CH2-R6、

R6は、独立して、
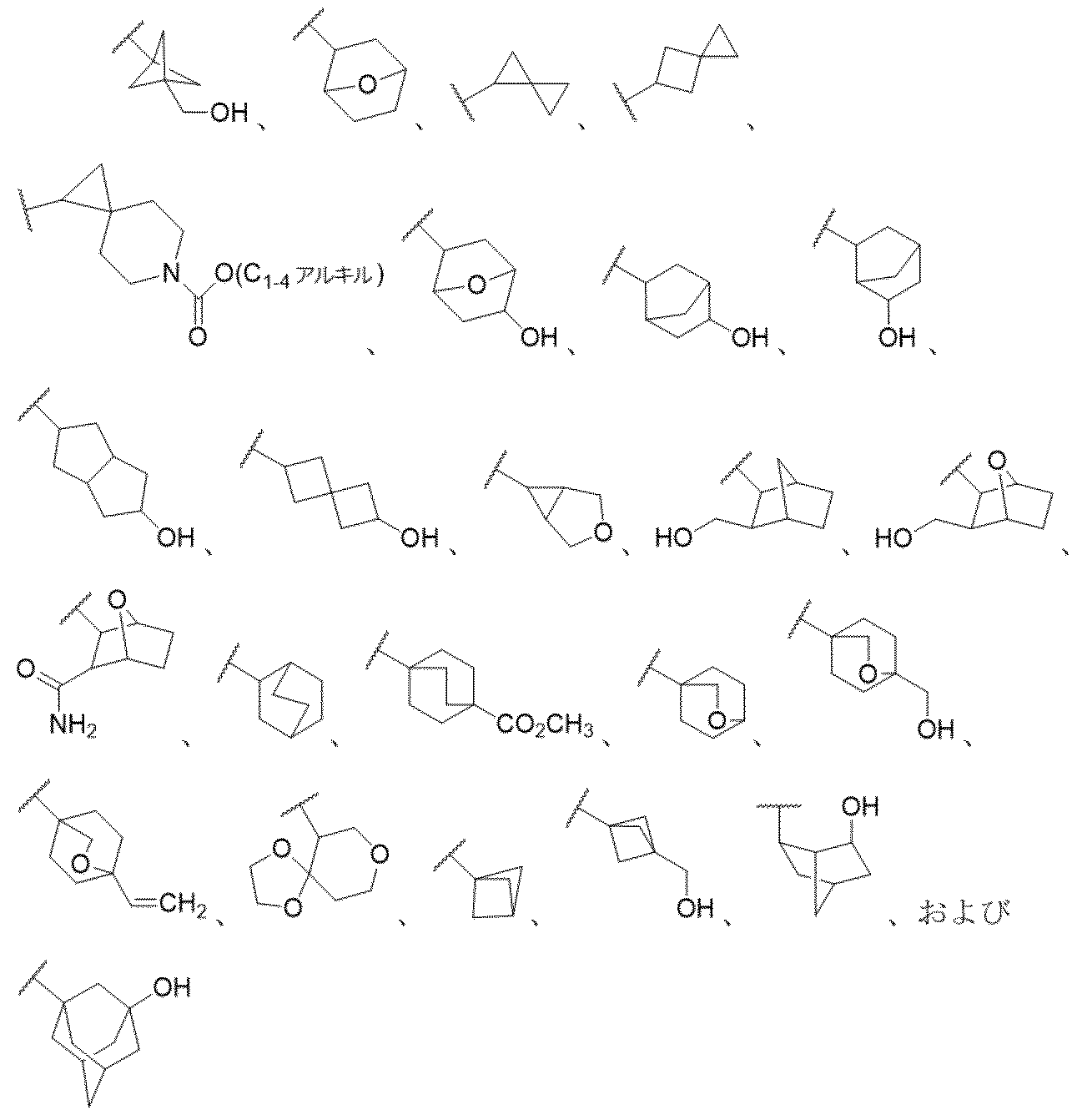
Wは、独立して、-NH-R6、

R6は、独立して、

Wは、独立して、-NH-R6、

R6は、独立して、


X2は、独立して、NまたはCR2であり;
R1、R2、R3およびR4は、それぞれ独立して、H、ハロゲンおよびC1-4アルキルから選択され;
R6は、独立して、5~10個の環原子を含む二環または三環シクロアルキルであり、ここでシクロアルキルはスピロ環であり得るか、または-CH2-および-CH2CH2-から選択される1個または2個の架橋リンカーを含有し得て、上記シクロアルキルは0~2個のReで置換されており;
あるいは、R6は、独立して、5~10個の環原子を含み、ここで1~2個の環原子はそれぞれ独立してNおよびOから選択される二環ヘテロシクロアルキルであり、この二環式環は、スピロ環であり得るか、または-CH2-、-O-、および-CH2CH2-から選択される架橋リンカーを含有し得て、上記ヘテロシクロアルキルは0~2個のReで置換されており;および
Reは、独立して、オキソ、OH、CH2OH、C1-4アルコキシ、および0~2個のC1-4アルコキシで置換されたC1-4アルキルから選択される]
の化合物またはその立体異性体、互変異性体、または医薬的に許容される塩を提供する。
X2は、独立して、NまたはCHであり;
R1、R3およびR4は、Hであり;および
R6は、独立して、

R6は、独立して、

ある態様において、本明細書に記載の化学物質(例えば、本明細書で一般的または特異的に記載されている化合物またはその医薬的に許容される塩またはそれを含む組成物)をNLRP3に接触させることを含む、NLRP3活性の調節方法(例えば、刺激、一部刺激、拮抗)が特徴付けられている。好ましい実施態様において、NLRP3活性の調節方法は、刺激および一部刺激することである。ある実施態様において、NLRP3活性の調節方法は刺激することである。ある実施態様において、NLRP3活性の調節方法は、一部刺激することである。その方法には、インビトロの方法(例えば、1つ以上のNLRP3を含む細胞(例えば、THP-1細胞)を含むサンプルに、上記化学物質を接触させること)が含まれる。また、その方法には、インビボの方法(例えば、NLRP3シグナル伝達の増加が、病理および/または症状および/または疾患の進行(例えばがん;例えば難治性がん)に関与する自然免疫活性の不足を正常化し得る疾患を有する対象(例えばヒト)に、化学物質を投与すること)も含まれる。
化学物質は、1つ以上の別のがん治療(例えば、手術、放射線療法、化学療法、毒素治療、免疫療法、凍結療法または遺伝子治療、またはその組み合わせ)と組み合わせて投与され得る(例えば、1つ以上(例えば、2、3、4、5、6つまたはそれ以上)の別の抗がん剤を投与することを含むがん治療)。以下に限らないが、例えば別の抗がん剤(化学療法剤)は、アルキル化剤(例えば、シスプラチン、カルボプラチン、メクロレタミン、シクロホスファミド、クロラムブシル、イホスファミドおよび/またはオキサリプラチン);代謝拮抗剤(例えば、アザチオプリンおよび/またはメルカプトプリン);テルペノイド(例えば、ビンカアルカロイド(例えば、ビンクリスチン、ビンブラスチン、ビノレルビンおよび/またはビンデシン)および/またはタキサン(タキソール、パクリタキセルおよび/またはドセタキセル));トポイソメラーゼ(例えば、I型トポイソメラーゼ(例えば、カンプトテシン、イリノテカンおよび/またはトポテカン)および/またはII型トポイソメラーゼ(アムサクリン、エトポシド、エトポシドリン酸塩および/またはテニポシド));細胞毒性抗生物質(例えば、アクチノマイシン、アントラサイクリン、ドキソルビシン、ダウノルビシン、バルルビシン、イダルビシン、エピルビシン、ブレオマイシン、プリカマイシンおよび/またはマイトマイシン);ホルモン(例えば、ホルモンアゴニストを放出する黄体形成ホルモン(例えば、ロイプロリド、ゴセレリン、トリプトレリン、ヒストレリン、ビカルタミド、フルタミドおよび/またはニルタミド));抗体(例えば、アブシキシマブ、アダリムマブ、アレムツズマブ、アトリズマブ、バシリキシマブ、ベリムマブ、ベバシズマブ、ブレンツキシマブ ベドチン、カナキヌマブ、セツキシマブ、セルトリズマブペゴル、ダクリズマブ、デノスマブ、エクリズマブ、エファリズマブ、ゲムツズマブ、ゴリムマブ、イブリツモマブチウキセタン、インフリキシマブ、イピリムマブ、ムロモナブ-CD3、ナタリズマブ、オファツムマブ、オマリズマブ、パリビズマブ、パニツムマブ、ラニビズマブ、リツキシマブ、トシリズマブ、トシツモマブおよび/またはトラスツズマブ);抗血管新生薬;サイトカイン; 血栓薬;増殖阻害剤;駆虫薬;およびCTLA-4、PD-1、PD-L1、PD-1-PD-L1、PD-1-PD-L2、T細胞免疫グロブリンおよびムチン3(TIM3またはHAVCR2)、ガレクチン9-TIM3、ホスファチジルセリン-TIM3、リンパ球活性化遺伝子3タンパク質(LAG3)、MHCクラスII-LAG3、4-1BB-4-1BBリガンド、OX40-OX40リガンド、GITR、GITRリガンド-GITR、CD27、CD70-CD27、TNFRSF25、TNFRSF25-TL1A、CD40L、CD40-CD40リガンド、HVEM-LIGHT-LTA、HVEM、HVEM-BTLA、HVEM-CD160、HVEM-LIGHT、HVEM-BTLA-CD160、CD80、CD80-PDL-1、PDL2-CD80、CD244、CD48-CD244、CD244、ICOS、ICOS-ICOSリガンド、B7-H3、B7-H4、VISTA、TMIGD2、HHLA2-TMIGD2、ブチロフィリン、例えばBTNL2、Siglecファミリー、TIGITおよびPVRファミリーメンバー、KIR、ILTおよびLIR、NKG2DおよびNKG2A、MICAおよびMICB、CD244、CD28、CD86-CD28、CD86-CTLA、CD80-CD28、ホスファチジルセリン、TIM3、ホスファチジルセリン-TIM3、SIRPA-CD47、VEGF、ニューロピリン、CD160、CD30、およびCD155(例えば、CTLA-4またはPD1またはPD-L1)から選択される免疫チェックポイント受容体を標的とする免疫チェックポイント阻害剤およびその他の免疫調節剤(例えば、インターロイキン-2(IL-2)、インドールアミン2,3-ジオキシゲナーゼ(IDO)、IL-10、トランスフォーミング増殖因子-β(TGFβ)、CD39、CD73、アデノシン-CD39-CD73、およびCXCR4-CXCL12)から選択される。
本明細書に記載の本開示の理解を進めるために、様々な他の用語が下記で定義される。一般に、本明細書で用いられる命名法および本明細書に記載の有機化学、医薬品化学、および薬理学における実験手順は、当業者に周知であり、一般に用いられる。特に断りが無い限り、本明細書で用いるあらゆる専門用語および科学用語は、一般に、本開示に関連する当業者により一般に理解されるものと同じ意味を持つ。

-CH2-)。
一部の実施態様において、化学物質(例えば、NLRP3を調節する(例えば、刺激するまたは一部刺激する)化合物、またはその医薬的に許容される塩、および/または水和物、および/または共結晶、および/または合剤)は、化学物質および1つ以上の医薬的に許容される賦形剤、本明細書に記載の1つ以上の別の治療剤を適宜含んでもよい医薬組成物として投与される。
一部の実施態様において、本明細書に記載の化学物質群またはその医薬組成物は、許容されるあらゆる投与経路により、その必要のある対象に投与され得る。許容される投与経路は、以下に限らないが、口腔内、皮膚、子宮頚管内、鼻腔内、気管内、腸内、硬膜外、間質内、腹腔内、動脈内、気管支内、滑液包内、脳内、大槽内、冠動脈内、真皮内、管内、十二指腸内、硬膜内、表皮内、食道内、胃内、歯肉内、回腸内、リンパ管内、髄内、髄膜内、筋肉内、卵巣内、腹腔内、前立腺内、肺内、腔内(intrasinal)、髄腔内、滑膜内、精巣内、くも膜下内、管内、腫瘍内、子宮内、血管内、静脈内、経鼻、経鼻胃、経口、非経口、経皮、経硬膜、直腸、呼吸器(吸入)、皮下、舌下、粘膜下、局所、経皮、経粘膜、経気管、尿管、尿道および腟内投与が挙げられる。特定の実施態様において、好ましい投与経路は、非経口投与(例えば、腫瘍内投与)である。特定の実施態様において、好ましい投与経路は、全身投与である。
投薬量は、患者の要望、治療する症状の重症度および用いられる特定の化合物によって変更されてもよい。特定の状況に対する適切な投薬量の決定は、医薬分野における当業者により決定され得る。1日の総投薬量は、1日を通して、分割し、少量ずつ投与されても、または連続的に投薬する手段により投与されてもよい。
前記投薬量は、1日あたりの基準量(例えば、単回用量、または2回以上の分割用量)または1日あたりではない基準量(例えば、隔日、2日毎、3日毎、週に1回、週に2回、2週に1回、1ヶ月に1回の用量)で投与され得る。
一部の実施態様において、NLRP3シグナル伝達の増加により、病状、疾患または障害(例えばがん)の病変および/または症状および/または進行に関与する自然免疫活性の不足を補正し得る、病状、疾患または障害(例えば、不十分な免疫応答に関連する病状、疾患または障害)を有する対象を治療する方法が提供される。
本明細書に記載のいずれかの方法において、対象はがんを有し得る。本明細書に記載のいずれかの方法の、一部の例において、哺乳動物は、がんを有すると確認されているか、またはがんを有すると診断されている。
本開示は、単剤療法計画ならびに併用療法計画の両方を意図する。
特定の実施態様において、免疫チェックポイント阻害剤は、ニボルマブ、ペムブロリズマブ、JS001、BGB-A317、INCSHR1210、TSR-042、GLS-010、STI-1110、MGD013、IBI308、BMS-936559、アテゾリズマブ、デュルバルマブ、アベルマブ、STI-1014、CX-072、KN035、LY3300054、CK-301、ウレルマブ、PF-05082566、MEDI6469、TRX518、バルリルマブ、BMS-986016、イピリムマブ、AGEN-1884、およびトレメリムマブから選択される。
これらの実施態様の特定の場合において、免疫チェックポイント阻害剤は、ウレルマブ、PF-05082566、MEDI6469、TRX518、バルリルマブ、CP-870893、ペムブロリズマブ(PD1)、ニボルマブ(PD1)、アテゾリズマブ(以前のMPDL3280A)(PDL1)、MEDI4736(PD-L1)、アベルマブ(PD-L1)、PDR001(PD1)、BMS-986016、MGA271、リリルマブ、IPH2201、エマクツズマブ、INCB024360、ガルニセルチブ、ウロクプルマブ、BKT140、バビツキシマブ、CC-90002、ベバシズマブ、およびMNRP1685Aから選択される。
一部の実施態様において、本明細書に記載の方法は、前記治療が必要な対象(例えば患者)を同定する工程(例えば、生検、内視鏡または当業者には既知の他の従来法による)をさらに含む。ある実施態様において、NLRP3タンパク質は、特定のがん種のバイオマーカーとして機能し得る。
当業者に理解され得るように、本明細書の式の化合物の合成方法は、当業者に明かである。例えば、本明細書に記載の化合物は、例えば、1つ以上の本明細書に記載の方法および/または例えば、US 2015/0056224に記載の方法を用いて合成され得る。本明細書に記載の化合物を合成する際に有用な、合成における化学変換および保護基の方法論(保護および脱保護)は当業者に既知であり、例えば、Larock, R.C., Comprehensive Organic Transformations, 2nd Edition, Wiley-VCH, New York, NY(1999); Wuts, P.G.M., Greene's Protective基 in Organic Synthesis, 5th Edition, Wiley(2014);L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994);およびL. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995)、およびその続編に記載されるものを含む。本発明の化合物の製造に用いられる出発物質は公知であり、既知の方法によって製造されるか、または市販品として入手可能である。当業者は、本明細書に記載の条件および試薬が、当分野において認められる代替物で置き換えられ得ることも認識している。例えば、多くの反応において、トリエチルアミンは、その他の塩基、例えば非求核性塩基(例えばジイソプロピルアミン、1,8-ジアザビシクロウンデカ-7-エン、2,6-ジ-tert-ブチルピリジン、またはテトラブチルホスファゼン)で置き換えられ得る。


スキーム1

ステップ2: この任意のステップにおいて、(ii)で示されたZ基は、異なる基、Xに変換されてもよい。このX基は所望により、最終生成物中に示されるR7基であってもよい。あるいは、X基は、後のステップでR7基に変換され得る基であってもよい。当業者は、ステップ2に選択される条件が、XおよびZ基の特徴に依存することを認識している。例えば、Z基がブロモ、およびX基がヘテロアリール環である場合、この変換は、触媒(例えばPdCl2(dppf))、および塩基(例えば炭酸セシウム)の存在下で、溶媒混合物(例えばジオキサンおよび水)中、適切なボロン酸またはボロン酸エステルとの反応により生じ得る。
ステップ3: ステップ3において、最終分子中のW基を導入するために、キナゾロン(iii)を、適当な試薬のセットと反応させる。例えば、所望のW基がアミンである場合、この変換は、(iii)を所望のアミン、試薬(例えばBOP)、および塩基(例えばDBU)と反応させることで生じ得る。所望のW基の特徴によって、この時点で追加の反応が実施され得る。例えば、導入した基がアルケンを含有し、所望のW基がジオールを含有する場合、この変換は、試薬(例えば四酸化オスミウム)および酸化剤(例えばNMO)との反応により得られうる。
ステップ4: この任意のステップにおいて、(iv)中のX基が、所望の最後の分子(v)中のR7基ではない場合、適切な条件下でR7基に変換され得る。例えば、所望のR7基が3-ピラゾリル、およびX基がテトラヒドロピラン基で保護されたピラゾールである場合、保護基は、酸および溶媒の適切な組合せ(例えばHClおよびメタノール、またはTFAおよびDCM)により除去され得る。
スキーム2

ステップ2: スキーム2のステップ2において、化合物(vii)は溶媒(例えばDCM)中、適当な酸化剤(例えばm-クロロ過安息香酸)と共に反応し、N-オキシド(viii)に変換される。
ステップ3: スキーム2のステップ3において、化合物(viii)は、適当な溶媒(例えばDCM)中、適当な活性化試薬(例えば塩化トシル)、およびアンモニア源(例えば塩化アンモニウムおよびトリエチルアミン)と共に反応し、アミン(ix)に変換される。
ステップ4: スキーム2のステップ4において、化合物(ix)のZ基は、化合物(x)中のR13基に変換される。R13基は、最終化合物中の所望のW基であり得る。あるいは、R13基は後の合成ステップでW基に変換され得る基であってもよい。当業者は、この変換を生じさせる方法がR13基およびZ基の性質によって変化することを理解している。例えば、Z基がクロロ、および所望のR13基がアミンである場合、化合物(ix)を溶媒(例えばDMSO)中、適当なアミンおよび塩基(例えばヒューニッヒ塩基)と共に適切な温度(例えば120℃)で加熱することでこの変換が生じ得る。あるいは、Z基がクロロ、および所望のR13基がエーテルである場合、化合物(ix)を溶媒(例えばNMP)中、適当なアルコールおよび塩基(例えばカリウムtert-ブトキシド)と共に適切な温度(例えば100℃)で加熱することでこの変換が生じ得る。あるいは、Z基がブロモ、および所望のR13基がアルキンである場合、化合物(ix)を適切な溶媒(例えばTHF)中、適当なアルキン、ヨウ化銅(I)、適当な塩基(例えばヒューニッヒ塩基)、および適切なパラジウム源(例えばテトラキス(トリフェニルホスフィン)パラジウム(0))と共に適切な温度(例えば70℃)で加熱することによってこの変換が生じ得る。あるいは、Z基がトリフレート、および所望のR13基が適宜置換されたアルキル基である場合、化合物(ix)を溶媒(例えばジオキサン)中、適当なアルキルボロン酸またはエステル、触媒(例えばPdCl2(dppf)-DCM錯体)、および塩基(例えば炭酸セシウム)と共に処理することでこのステップは完了し得る。
ステップ5: スキーム1のステップ5は、中間体(x)中のR12基を分子(xi)中のR7基に変換する任意の1ステップまたは一連のステップである。例えば、R12基がブロモ、および所望のR7基が芳香族またはヘテロ芳香族である場合、中間体(x)を混合溶媒(例えばジオキサンおよび水)中、適宜保護された芳香族またはヘテロ芳香族のボロン酸またはボロン酸エステル、触媒(例えばPdCl2(dppf)-DCM錯体)、および塩基(例えばリン酸三カリウム)と共に反応させることでこの変換が生じ得る。導入された基が保護基を有する場合、所望により適当な条件下で保護基の除去する、さらなる任意のステップが行われ得る。例えば、導入された基がテトラヒドロピラン保護基を含むピラゾールである場合、テトラヒドロピランは、溶媒(例えばジクロロメタン)中、酸(例えばトリフルオロ酢酸)と共に反応させることで除去され得る。あるいは、R12基がブロモ、および所望のR7基が芳香族またはヘテロ芳香族である場合、最初に中間体(x)を溶媒(例えばジオキサン)中、化合物(例えばPdCl2(dppf)-DCM錯体、ビス(ピナコラト)ジボロン)、試薬(例えば酢酸カリウム)、および触媒(例えばPdCl2(dppf)-DCM錯体)と共に反応させ、次いで得られたボロン酸エステルを適当な混合溶媒(例えばジオキサンおよび水)中、適当なアリールまたはヘテロアリールハライド、塩基(例えば炭酸ナトリウム)、および触媒(例えばテトラキス(トリフェニルホスフィン)パラジウム(0))と共に反応させることでこの変換が生じ得る。あるいは、R12基がブロモ、および所望のR7基が窒素原子で架橋されたヘテロ環である場合、このステップは、適当な溶媒(例えばDMSO)中、中間体(x)を銅源(例えばヨウ化銅(I))、適当なヘテロ環、塩基(例えば炭酸ナトリウム)、およびリガンド(例えばN,N'-ジメチルエタン-1,2-ジアミン)の存在下で共に反応させることで生じ得る。
ステップ6: スキーム2のステップ6は、中間体(xi)中のR13基を分子(xii)中のW基に変換する任意の1ステップまたは一連のステップである。例えば、R13基がBoc保護アミンを有し、所望のW基がアミドを有する場合、最初に適切な酸および溶媒の組合せ(例えば塩酸およびジオキサン)でBoc基を除去し、次いで溶媒(例えばDMF)中、適当なカルボン酸、カップリング剤(例えばT3P)、および塩基(例えばトリエチルアミン)と共に反応させることで所望のアミドを形成し、この変換が完了し得る。あるいは、R13基が不飽和基(例えばアルキン)を有し、所望のW基が完全飽和である場合、水素および適切な触媒(例えばパラジウム/炭素)と共に反応させることでこの変換が生じ得る。
ステップ7: スキーム2のステップ7は、中間体(xii)中のR11基を分子(xiii)中のR3基に変換するための任意の1ステップまたは一連のステップである。
ステップ8: スキーム2のステップ8は、中間体(xiii)中のR10基を分子(xiv)中のR2基に変換する任意の1ステップまたは一連のステップである。例えば、R10基がベンジルエーテルで保護されたアルコールを有し、所望のR2基が対応するアルコールである場合、適切な酸(例えば塩酸)と共に反応させることでこの変換が生じ得る。R10基がアルコールを有し、所望のR2基が同じ位置でアミンを有する場合、最初に中間体(xiii)を溶媒(例えばジクロロメタン)中、試薬(例えば塩化チオニル)と共に反応させ、次いで溶媒(例えばアセトニトリル)中、得られた塩化物をアミン(例えばエチルアミン)、ヨウ化ナトリウム、および塩基(例えば炭酸カリウム)と共に反応させることでこの変換が生じ得る。

ステップ2: スキーム3のステップ2において、化合物(xvi)は、溶媒(例えばエタノール)中、(xvi)を塩基(例えばナトリウムエトキシド)に高温(100℃)で曝露することで、塩基性触媒環化を介してキノリノール(xvii)に変換され得る。
ステップ3: スキーム3のステップ3は、中間体(xvii)中のR12基を分子(xviii)中のR7基に変換する、任意の1ステップまたは一連のステップである。例えば、R12がブロモであり、所望のR7基が芳香族またはヘテロ芳香族である場合、この変換は、中間体(xvii)を適宜保護された芳香族またはヘテロ芳香族のボロン酸またはボロン酸エステル、触媒(例えばPdCl2(dppf)-DCM錯体)、および塩基(例えばリン酸三カリウム)と混合溶媒(例えばジオキサンおよび水)中で反応させることで生じ得る。導入された基が保護基を有する場合、所望により保護基を適当な条件下で除去するために、さらに任意のステップが行われてもよい。例えば、導入された基がテトラヒドロピラン保護基を含むピラゾールである場合、テトラヒドロピランは、溶媒(例えばジクロロメタン)中、酸(例えばトリフルオロ酢酸)と共に反応させることで除去され得る。あるいは、R12がブロモであり、所望のR7基が芳香族またはヘテロ芳香族である場合、初めに中間体(xvii)を化合物(例えばビス(ピナコラト)ジボロン)、試薬(例えば酢酸カリウム)、および触媒(例えばPdCl2(dppf)-DCM錯体)と共に溶媒(例えばジオキサン)中で反応させ、次いで得られたボロン酸エステルを適当な混合溶媒(例えばジオキサンおよび水)中、適当なアリールまたはヘテロアリールハライド、塩基(例えば炭酸ナトリウム)、および触媒(例えばテトラキス(トリフェニルホスフィン)パラジウム(0))と共に反応させることでこの変換が生じ得る。あるいは、R12がブロモであり、所望のR7基が窒素原子を介したヘテロ環である場合、このステップは、中間体(xvii)を適当なヘテロ環と共に適当な溶媒(例えばDMSO)中、銅源(例えばヨウ化銅(I))、塩基(例えば炭酸ナトリウム)、およびリガンド(例えばN,N'-ジメチルエタン-1,2-ジアミン)の存在下で反応させることで生じ得る。
ステップ4: スキーム3のステップ4において、化合物(xviii)のアルコール基は、ハロゲン基またはスルホン酸エステル(例えばクロロ、ブロモ、またはトリフレート)に変換され得る。所望のZ基がクロロの場合、この変換は、溶媒(例えばトルエン)中、化合物(xviii)を試薬(例えば塩化ホスホリル)と共に反応させることで生じ得る。あるいは、所望のZ基がブロモである場合、この変換は、溶媒(例えばDMF)中、化合物(xviii)を試薬(例えば三臭化リン)と共に反応させることで生じ得る。あるいは、所望のZ基がトリフレートである場合、この変換は、溶媒(例えばジクロロメタン)中、化合物(xviii)を試薬(例えばトリフルオロメタンスルホニルクロリドおよび4-ジメチルアミノピリジン)、および塩基(例えばヒューニッヒ塩基)と共に反応させることで生じ得る。
ステップ5: スキーム3のステップ5において、化合物(xix)中のZ基は化合物(xx)中のR18基に変換される。R18基は最終化合物中の所望のW基であり得るか、あるいは、後の合成ステップでW基に変換され得る基であってもよい。当業者は、この変換を生じさせる方法がR18基およびZ基の性質によって変化することを理解している。例えば、Zがクロロであり、所望のR18基がアミンである場合、化合物(xix)を、溶媒(例えばDMSOまたはNMP)中、適当なアミンおよび塩基(例えばヒューニッヒ塩基)と共に適切な温度(例えば120℃)で加熱することで、この変換が生じ得る。あるいは、Z基がクロロであり、所望のR18基がエーテルである場合、化合物(xix)を、溶媒(例えばNMP)中、適当なアルコールおよび塩基(例えばカリウムtert-ブトキシド)と共に適切な温度(例えば100℃)で加熱することでこの変換が生じ得る。あるいは、Z基がブロモであり、所望のR18基がアルキンである場合、化合物(xix)を、適切な溶媒(例えばTHF)中、適当なアルキン、ヨウ化銅(I)、適当な塩基(例えばヒューニッヒ塩基)、および適切なパラジウム源(例えばテトラキス(トリフェニルホスフィン)パラジウム(0))と共に、適切な温度(例えば70℃)で加熱することでこの変換が生じ得る。あるいは、Z基がトリフレートであり、所望のR18基が適宜置換されていてもよいアルキル基である場合、このステップは、化合物(xix)を、溶媒(例えばジオキサン)中、適当なアルキルボロン酸またはエステル、触媒(例えばPdCl2(dppf)-DCM錯体)、および塩基(例えば炭酸セシウム)と共に加熱することで完了し得る。
ステップ6: スキーム3のステップ6は、中間体(xx)中のR18基を分子(xxi)中のW基に変換させる、任意の1ステップまたは一連のステップである。例えば、R18基がBoc保護アミンを有し、所望のW基がアミドを有する場合、この変換は、初めにBoc基を適切な組合せの酸および溶媒(例えば塩酸およびジオキサン)と反応させて除去し、次いで溶媒(例えばDMF)中、適当なカルボン酸、カップリング剤(例えばT3P)、および塩基(例えばトリエチルアミン)と共に反応させて所望のアミドを形成することで完了し得る。あるいは、R18基が不飽和基(例えばアルキン)を有し、所望のW基が完全飽和である場合、水素および適切な触媒(例えばパラジウム/炭素)と共に反応させることでこの変換が生じ得る。
ステップ7: スキーム3のステップ7は、中間体(xxi)中の置換基R11、R14、R16 、およびR17を分子(xxii)中の置換基R3、R1、R4、およびR2に変換する任意の1ステップまたは一連のステップである。
スキーム4(X2=CR2)

ステップ2: スキーム4のステップ2において、化合物(xxiv)は溶媒(例えばエタノール)中、(xxiv)を塩基(例えばナトリウムエトキシド)に高温(100℃)で曝露することで、塩基性触媒環化を介してキノリノール(xxv)に変換され得る。
ステップ3: スキーム4のステップ3は、中間体(xxv)中のR12基を分子(xxvi)中のR7基に変換する、任意の1ステップまたは一連のステップである。例えば、R12がブロモであり、所望のR7基が芳香族またはヘテロ芳香族である場合、化合物(xxv)を混合溶媒(例えばジオキサンおよび水)中、適宜保護された芳香族またはヘテロ芳香族のボロン酸またはボロン酸エステル、触媒(例えばPdCl2(dppf)-DCM錯体)、および塩基(例えばリン酸三カリウム)と共に反応させることでこの変換が生じ得る。導入された基が保護基を有する場合、所望により保護基を適当な条件下で除去するために、さらに任意のステップが行われてもよい。例えば、導入された基がテトラヒドロピラン保護基を含むピラゾールである場合、テトラヒドロピランは、溶媒(例えばジクロロメタン)中、酸(例えばトリフルオロ酢酸)と共に反応させることで除去され得る。あるいは、R12がブロモであり、所望のR7基が芳香族またはヘテロ芳香族である場合、初めに中間体(xxv)を溶媒(例えばジオキサン)中、化合物(例えばビス(ピナコラト)ジボロン)、試薬(例えば酢酸カリウム)、および触媒(例えばPdCl2(dppf)-DCM錯体)と共に反応させ、次いで得られたボロン酸エステルを適当な混合溶媒(例えばジオキサンおよび水)中、適当なアリールまたはヘテロアリールハライド、塩基(例えば炭酸ナトリウム)、および触媒(例えばテトラキス(トリフェニルホスフィン)パラジウム(0))と共に反応させることでこの変換が生じ得る。あるいは、R12がブロモであり、所望のR7基が窒素原子を介したヘテロ環である場合、このステップは、中間体(xxv)を適当なヘテロ環と共に適当な溶媒(例えばDMSO)中、銅源(例えばヨウ化銅(I))、塩基(例えば炭酸ナトリウム)、およびリガンド(例えばN,N'-ジメチルエタン-1,2-ジアミン)の存在下で反応させることで生じ得る。
ステップ4: スキーム4のステップ4において、化合物(xxvi)のアルコール基は、ハロゲン基またはスルホン酸エステル(例えばクロロ、ブロモ、またはトリフレート)に変換され得る。所望のZ基がクロロの場合、この変換は、溶媒(例えばトルエン)中、化合物(xxvi)を試薬(例えば塩化ホスホリル)と共に反応させることで生じ得る。あるいは、所望のZ基がブロモである場合、この変換は、化合物(xxvi)を溶媒(例えばDMF)中、試薬(例えば三臭化リン)と共に反応させることで生じ得る。あるいは、所望のZ基がトリフレートである場合、この変換は、溶媒(例えばジクロロメタン)中、化合物(xxvi)を試薬(例えばトリフルオロメタンスルホニルクロリドおよび4-ジメチルアミノピリジン)、および塩基(例えばヒューニッヒ塩基)と共に反応させることで生じ得る。
ステップ5: スキーム4のステップ5において、化合物(xxvii)中のZ基は、化合物(xxvii)中のR18基に変換される。R18基は最終化合物中の所望のW基であり得るか、あるいは、後の合成ステップでW基に変換され得る基であってもよい。当業者は、この変換を生じさせる方法がR18基およびZ基の性質によって変化することを理解している。例えば、Zがクロロであり、所望のR18基がアミンである場合、化合物(xxvii)を、溶媒(例えばDMSOまたはNMP)中、適当なアミンおよび塩基(例えばヒューニッヒ塩基)と共に適切な温度(例えば120℃)で加熱することで、この変換が生じ得る。あるいは、Z基がクロロであり、所望のR18基がエーテルである場合、化合物(xxvii)を、溶媒(例えばNMP)中、適当なアルコールおよび塩基(例えばカリウムtert-ブトキシド)と共に適切な温度(例えば100℃)で加熱することでこの変換が生じ得る。あるいは、Z基がブロモであり、所望のR18基がアルキンである場合、化合物(xxvii)を、適切な溶媒(例えばTHF)中、適当なアルキン、ヨウ化銅(I)、適当な塩基(例えばヒューニッヒ塩基)、および適切なパラジウム源(例えばテトラキス(トリフェニルホスフィン)パラジウム(0))と共に適切な温度(例えば70℃)で加熱することでこの変換が生じ得る。あるいは、Z基がトリフレートであり、所望のR18基が適宜置換されていてもよいアルキル基である場合、このステップは、化合物(xxvii)を溶媒(例えばジオキサン)中、適当なアルキルボロン酸またはエステル、触媒(例えばPdCl2(dppf)-DCM錯体)、および塩基(例えば炭酸セシウム)と共に加熱することで完了し得る。
ステップ6: スキーム4のステップ6は、中間体(xxviii)中のR18基を分子(xxix)中のW基に変換させる、任意の1ステップまたは一連のステップである。例えば、R18基がBoc保護アミンを有し、所望のW基がアミドを有する場合、初めにBoc基を適切な組合せの酸および溶媒(例えば塩酸およびジオキサン)と反応させて除去し、次いで溶媒(例えばDMF)中、適当なカルボン酸、カップリング剤(例えばT3P)、および塩基(例えばトリエチルアミン)と共に反応させることで所望のアミドを形成し、この変換が完了し得る。あるいは、R18基が不飽和基(例えばアルキン)を有し、所望のW基が完全飽和である場合、水素および適切な触媒(例えばパラジウム/炭素)と共に反応させることでこの変換が生じ得る。
ステップ7: スキーム4のステップ7は、中間体(xxix)の置換基X1(例えばC14)、X3(例えばC11)、X4(例えばC16)、およびR17を分子(xxx)中の置換基X1(例えばCR1)、X3(例えばCR3)、X4(例えばCR4)、およびR2に変換する任意の1ステップまたは一連のステップである。

ステップ2: スキーム5のステップ2は、化合物(xxxii)中のR12基を分子(xxxiii)中のR7基に変換する任意の1ステップまたは一連のステップである。例えば、R12がブロモであり、所望のR7基が芳香族またはヘテロ芳香族である場合、中間体(xxxii)を混合溶媒(例えばジオキサンおよび水)中、適宜保護された芳香族またはヘテロ芳香族のボロン酸またはボロン酸エステル、触媒(例えばPdCl2(dppf)-DCM錯体)、および塩基(例えばリン酸三カリウム)と共に反応させることでこの変換が生じ得る。導入された基が保護基を有する場合、所望により保護基を適当な条件下で除去するために、さらに任意のステップが行われてもよい。例えば、導入された基がテトラヒドロピラン保護基を含むピラゾールである場合、テトラヒドロピランは、酸(例えばトリフルオロ酢酸)と共に溶媒(例えばジクロロメタン)中で反応させることで除去され得る。あるいは、R12がブロモであり、所望のR7基が芳香族またはヘテロ芳香族である場合、初めに中間体(xxxii)を溶媒(例えばジオキサン)中、化合物(例えばビス(ピナコラト)ジボロン)、試薬(例えば酢酸カリウム)、および触媒(例えばPdCl2(dppf)-DCM錯体)と共に反応させ、次いで得られたボロン酸エステルを適当な混合溶媒(例えばジオキサンおよび水)中、適当なアリールまたはヘテロアリールハライドと共に、塩基(例えば炭酸ナトリウム)、および触媒(例えばテトラキス(トリフェニルホスフィン)パラジウム(0))と共に反応させることでこの変換が生じ得る。あるいは、R12がブロモであり、所望のR7基が窒素原子を介したヘテロ環である場合、このステップは、適当な溶媒(例えばDMSO)中、中間体(xxxii)を適当なヘテロ環と共に銅源(例えばヨウ化銅(I))、塩基(例えば炭酸ナトリウム)、およびリガンド(例えばN,N'-ジメチルエタン-1,2-ジアミン)の存在下で反応させることで生じ得る。
ステップ3: スキーム5のステップ3において、化合物(xxxiii)中のピリドンは化合物(xxxiv)中のR18基に変換される。R18基は最終化合物中の所望のW基であり得るか、あるいは、後の合成ステップでW基に変換され得る基であってもよい。当業者は、この変換を生じさせる方法がR18基の性質によって変化することを理解している。例えば、所望のR18基がアミンである場合、化合物(xxxiii)を溶媒(例えばDMF)中、適当なアミン、カップリング剤(例えばBOP)、および塩基(例えばDBU)と共に反応させることでこの変換が生じ得る。
ステップ4: スキーム5のステップ4は、中間体(xxxiv)中のR18基を分子(xxxv)中のW基に変換する任意の1ステップまたは一連のステップである。例えば、R18基がBoc保護アミンを有し、所望のW基がアミドを有する場合、初めにBoc基を適切な組合せの酸および溶媒(例えば塩酸およびジオキサン)と反応させて除去し、次いで溶媒(例えばDMF)中、適当なカルボン酸、カップリング剤(例えばT3P)、および塩基(例えばトリエチルアミン)と共に反応させて所望のアミドを形成することでこの変換は完了し得る。あるいは、R18基が不飽和基(例えばアルキン)を有し、所望のW基が完全飽和である場合、水素および適切な触媒(例えばパラジウム/炭素)と共に反応させることでこの変換が生じ得る。
ステップ5: スキーム5のステップ5は、中間体(xxxv)の置換基X1(例えばC14)、X3(例えばC11)、およびX4(例えばC16)を分子(xxxvi)の置換基X1(例えばC1)、X3(例えばC3)、およびX4(例えばC4)に変換する任意の1ステップまたは一連のステップである。当業者は、これらの様々なステップが最後の分子(xxxvi)の所望の基によって異なる順番で実施されてもよいことを理解している。例えば、一部の分子において、ステップ2で示したR12基からR7基への変換は、ステップ3で示したピリドンからR18基への変換の後で行われ得る。
PMA分化THP-1細胞におけるIL-1β生成の測定
THP-1細胞は、American Type Culture Collectionから購入し、サプライヤーの取扱説明書に従って継代培養を行った。実験前に、細胞をRPMI 1640(10%加熱不活性化FBS、ペニシリン(100ユニット/mL)およびストレプトマイシン(100μg/mL)含有)中で培養し、実験準備を行う前に対数期で保持した実験前に、THP-1をPMA(ホルボール12-ミリステート13-アセテート、10μg/mL)で24時間処理した。実験日に培養液を除去し、接触させる細胞をトリプシンで2分間処理し、細胞を次いで回収し、PBS(リン酸緩衝食塩水)で洗浄し、遠沈し、RPMI(2%加熱不活性化FBS含有)に1x106細胞/mLの濃度で再懸濁し、100μLを96ウェルプレートに播種した。化合物をジメチルスルホキシド(DMSO)に溶解し、所望の濃度(例えば100、30、10、3、1、0.3または0.1μM)になるまで培地に加えた。細胞を、化合物と共に4時間インキュベートした。細胞を含まない上清を回収し、IL-1βの生成をELISAにより評価した。ビークルのみのコントロールを各実験に対して同時に行った。最終的なDMSO濃度は1%であった。化合物は、PMA分化THP-1細胞において、用量に応じてIL-1β生成の増加を示した。
THP-1細胞は、American Type Culture Collectionから購入し、サプライヤーの取扱説明書に従って継代培養を行った。実験前に、細胞をRPMI 1640(10%加熱不活性化FBS、ペニシリン(100ユニット/mL)、ストレプトマイシン(100μg/mL)、HEPES(10mM)およびピルビン酸ナトリウム(1mM)含有)中で培養し、実験準備を行う前に対数期で保持した。実験前に、THP-1細胞をPMA(ホルボール12-ミリステート13-アセテート、20μg/mL)で終夜処理した。実験日に培養液を除去し、接触する細胞をトリプシンで2分間処理し、細胞を次いで回収し、PBS(リン酸緩衝食塩水)で洗浄し、遠心分離によりペレット状にし、384ウェルプレート中、RPMI(2%加熱不活性化FBS含有)に50,000細胞/ウェルの濃度で再懸濁した。細胞を含まない上清を回収し、IL-1βの生成をELISAにより評価した。化合物をジメチルスルホキシド(DMSO)に溶解し、所望の濃度(例えば100、30、10、3、1、0.3または0.1μM)になるまで培地に加えた。細胞を化合物と共に2時間インキュベートした。ビークルのみのコントロールを各実験に対して同時に行った。最終的なDMSO濃度は1%であった。化合物は、PMA分化THP-1細胞において、用量に応じてIL-1β生成の増加を示した。
化合物のDMSO溶液の段階希釈液を低容量384ウェルプレートに、ECHO 550 acoustic dispenser(Labcyte)を用いて100nL/ウェルで加え、アッセイ中の最終的な出発濃度が10μMになるようにした。
T175フラスコ中の、10%FBS含有RPMI(Gibco,11875)培地中の1x106細胞/mLの密度のTHP-1細胞を、分化させるために、最終濃度が50ng/mLのホルボール12-ミリステート13-アセテート(PMA、Sigma,P1585)で終夜37℃、5%CO2で処理した。dPBSでウェルを濯いだ翌日後、0.5%トリプシンを用いて細胞を回収した。2%FBS含有RPMI培地中、50μL/ウェルで50,000細胞にあたる、1x106細胞/mLの細胞溶液を調製した。マルチチャンネルピペットを用いて、384ウェル黒色透明底組織培養処理プレート(Greiner,781090)中の化合物希釈液に、細胞を播種した。該プレートを37℃、5%CO2のインキュベーターで2時間インキュベートした。
2時間インキュベート後、該細胞プレートを、遠心機を用いて5分間1200rpmで回転させた。Felix(CyBio)を用いて、上清(8μL)を384ウェルの低容量白色ProxiPlate(Perkin Elmer,6008230)に移した。ヒトIL-1βのhTRFキット(CISBIO,62HIL1BPEG)を用いて上清を分析した。該キット説明書に従ってIL-1β検量線を作成し、次いでキットの抗体をキット説明書の1:20ではなく、1:40で希釈した。混合した時点で、プレートに5μL/ウェルで抗体を加えた。このプレートを密封し、4℃で終夜インキュベートした。このプレートを次いでhTRFレーザーを用いてPerkin Elmer EnVision(665/615nm)で測定した。化合物は用量に応じて、IL-1β生成の増加を示した。
化合物のDMSO段階希釈液を低容量384ウェルプレートに、ECHO 550 acoustic dispenser(Labcyte)を用いて100nL/ウェルで、アッセイ中の最終的な出発濃度が10uMになるように加えた。
健常ドナーから得たヒト静脈全血を、1ng/mLのLPS(Invivogen,Cat#tlrl-eblps)で、95%空気/5%CO2の加湿インキュベーター中、37℃で4時間予め処理した。処理した血液を化合物のプレートに加え、さらに37℃で4時間インキュベートした。上清中のIL-1βを、AlphLISAキット(Cat#AL220)を用いてメーカーの説明書に従って測定した。化合物は、用量に応じてIL-1β生成の増加を示した。EC50は、前処理は行ったが化合物では処理していない血液をベースラインとして用いて決定した。
C57BL/6マウス由来の不死化マウスマクロファージを、Eicke Latz氏(University of Bonn/University of Massachusetts Worcester(MA))から得た。該細胞を、0.05%トリプシンを用いて回収し、PBSで洗浄した。細胞を2%FBS含有DMEM(Gibco,11965)の25uL中、30,000細胞/ウェルで播種し、37℃、5%CO2で10分間インキュベートした。LPS-EB(Invivogen、tlr-eblps)を、5uL/ウェルで最終濃度が200ng/mLになるまで加え、細胞を37℃、5%CO2で2時間インキュベートした。
化合物のDMSO段階希釈液を低容量384ウェルプレート中の細胞に、ECHO 550 acoustic dispenser(Labcyte)を用いて60nL/ウェルで、アッセイ中の最終的な出発濃度が50uMになるように加え、化合物と共に37℃、5%CO2でさらに2時間インキュベートした。
2時間インキュベート後、該細胞プレートを、遠心機を用いて5分間1200rpmで回転させた。Felix(CyBio)を用いて、上清(8μL)を384ウェルの低容量白色ProxiPlate(Perkin Elmer,6008230)に移した。ヒトIL-1βのhTRFキット(CISBIO,62MIL1BPEH)を用いて上清を分析した。該キット説明書に従ってIL-1β検量線を作成した(キットの抗体はキット説明書の1:20ではなく、1:40で希釈した)。混合した時点で、プレートに5μL/ウェルで抗体を加えた。このプレートを密封し、4℃で終夜インキュベートした。このプレートをhTRFレーザーを用いてPerkin Elmer EnVision(665/615nm)で測定した。次いでデータをIL-1βのpg/mL量に変換した。化合物は、用量に応じてIL-1β生成の増加を示した。
TLR7またはTLR8遺伝子およびNF-kB/AP1誘導性SEAP(分泌型胎盤アルカリホスファターゼ;Invivogen,San Diego(CA))レポーター遺伝子が共発現し、対数的に増殖するヒトHEK-Blue細胞を、384ウェルプレートの各ウェルに加え(15,000細胞/20μL/ウェル)、37℃、5%CO2で24時間静置した。翌日、試料化合物またはDMSOをacoustic liquid handling technologyを用いて各ウェルに分配(100nL/ウェル)し、細胞を続いて37℃、5%CO2で18時間インキュベートした。直前に調整したQUANTI-Blue試薬(メーカーの説明書に従って調製;Invivogen,San Diego(CA))をHEK-Blue TLR Nf-kB-SEAP細胞反応に加え、30分後にプレートリーダー機器(Envision)を用いて細胞のSEAP生成を測定した。あらゆるEC50値(半数効果濃度)は、独自のデータ分析ソフトウェアを用いて決定した。規格化EC50値とは、50μMの標準試料で処理した細胞から得た標準RLU(発光量)値を用いて、Yの最大値を100%と設定して定義される絶対値である。
分析HPLC/MSは以下の方法を用いて実施した。
方法A: カラム: Waters Acquity UPLC BEH C18、2.1x50mm、粒子径: 1.7μm; 移動相A: 5:95 アセトニトリル:水(0.1%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル:水(0.1%トリフルオロ酢酸含有); 温度: 50℃; グラジエント: 0~100%Bで3分かけて溶出し、次いで100%Bで0.75分間溶出; 流速: 1.0mL/分; 検出: UV(220nm)
方法B: カラム: Waters Acquity UPLC BEH C18、2.1x50mm、粒子径: 1.7μm; 移動相A: 5:95 アセトニトリル:水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル:水(10mM酢酸アンモニウム含有); 温度: 50℃; グラジエント: 0~100%Bで3分かけて溶出し、次いで100%Bで0.75分間溶出; 流速: 1.0mL/分; 検出: UV(220nm)
方法C: カラム: Waters XBridge C18、2.1mmx50mm、粒子: 1.7μm; 移動相A: 5:95 アセトニトリル:水(0.1%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル:水(0.1%トリフルオロ酢酸含有); 温度: 50℃; グラジエント: 0%B~100%Bで3分かけて溶出後、100%Bで0.50分間溶出; 流速: 1mL/分; 検出: MSおよびUV(220nm)
方法D: カラム: Waters Acquity UPLC BEH C18、2.1x50mm、粒子径: 1.7μm; 移動相A: 水(0.05%TFA含有); 移動相B: アセトニトリル(0.05%TFA含有); 温度: 50℃; グラジエント: 2~98%Bで1.0分かけて溶出し、次いで98%Bで0.50分間溶出; 流速: 0.80mL/分; 検出: UV(254nm)
方法E: ESI Pos/neg: カラム: Waters Acquity UPLC BEH C18、2.1x50mm、粒子径: 1.7μm; 移動相A: 95:5 水:アセトニトリル(10mM NH4OAc含有); 移動相B: 95:5 アセトニトリル:水(10mM NH4OAc含有); 温度: 50℃; グラジエント: 5~95%Bで1.0分かけて溶出し、次いで95%Bで0.50分間溶出; 流速: 0.80mL/分; 検出: UV(220nm)
化学シフトは、内部テトラメチルシラン(TMS)または重水素化NMR溶媒から推測されるTMSの位置から低磁場の百万分率(ppm)で表される。明らかな多重度は、シングレット-s、ダブレット-d、トリプレット-t、カルテット-q、またはマルチプレット-mと示した。ブロードなピークは、さらにbrと示した。積分値はおおよそである。ここで、積分強度、ピークの形状、化学シフトおよびカップリング定数は、溶媒、濃度、温度、pH、およびその他の要因によって変化し得ることに注意されたい。さらに、NMRスペクトル中の水または溶媒ピークと重複または交換されるピークは、信頼性のある積分強度を提供し得ない。一部のケースでは、不可視な重複ピーク、または形状および/または積分の変化が生じ得たNMRスペクトルは、水ピーク抑制を用いて得られうる。
1A. 2-アミノ-7-ブロモキナゾリン-4(3H)-オン
2-アミノ-4-ブロモ安息香酸メチル(485mg、2.1mmol)およびシアナミド(106mg、2.5mmol)/ジオキサン(5mL)の混合物に、4M HCl/ジオキサン(0.69mL、2.7mmol)を加えた。この応混合物を70℃で2時間加熱し、次いで温度を100℃に上げて1時間加熱した。得られた反応混合物を室温に冷却し、エーテルで希釈した。白色沈殿を濾過により回収し、エーテルで濯ぎ、続いて少量のエタノールおよび水で濯いだ。得られた固体を真空乾燥し、2-アミノ-7-ブロモキナゾリン-4(3H)-オン(429mg、85%収率)を得た。1HNMR(400MHz、DMSO)δ 8.33-8.04 (m, 2H), 7.88 (d, J=8.4Hz, 1H), 7.64 (d, J=1.6Hz, 1H), 7.51 (dd, J=8.4, 1.7Hz, 1H)
2-アミノ-7-ブロモキナゾリン-4(3H)-オン(2.86g、11.9mmol)、1-(テトラヒドロ-2H-ピラン-2-イル)-5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1Hピラゾール(3.97g、14.3mmol)および炭酸セシウム(11.6g、35.7mmol)/ジオキサン(120mL)および水(30mL)の混合物に、N2下でPdCl2(dppf)-CH2Cl2付加物(0.97g、1.2mmol)を加えた。この反応混合物を、還流冷却管を付けて110℃の加熱ブロックで加熱した。反応完了後、反応液を室温に冷却し、濃縮した。得られた残渣に1M NaOH(120mL)を加え、20分間撹拌した。この混合物を1M NaOHで濯いでセライト濾過した。塩基性水性母液をゆっくりと1Mクエン酸でpH 4に酸性化し、得られた薄黄褐色沈殿を水で濯いで濾過により回収した。固体を真空乾燥し、2-アミノ-7-(1-(テトラヒドロ-2Hピラン-2-イル)-1Hピラゾール-5-イル)キナゾリン-4(3H)-オン(2.28g、61.5%収率)を得た。1HNMR(400MHz、DMSO)δ 11.35-10.75 (m, 1H), 7.96 (br d, J=8.1Hz, 1H), 7.59 (s, 1H), 7.35 (s, 1H), 7.23 (br d, J=8.0Hz, 1H), 6.66-6.36 (m, 3H), 5.26 (br d, J=9.3Hz, 1H), 4.01 (br d, J=10.9Hz, 1H), 3.57 (br t, J=8.8Hz, 1H), 2.45-2.32 (m, 1H), 1.93 (br s, 1H), 1.78 (br d, J=12.5Hz, 1H), 1.65-1.48 (m, 3H)
7-オキサビシクロ[2.2.1]ヘプタ-5-エン-2-アミン・HCl(2.7461g、18.60mmol、Bioorg.Med.Chem.Lett. 9(7) 933-936(1999)に記載の通り製造したendoおよびexo異性体の混合物)/メタノール-塩化メチレン(50mL、1:1混合液)の懸濁液をBoc無水物(5.73mL、24.70mmol)および炭酸カリウム(3.3g、23.88mmol)で処理した。終夜撹拌後、得られた反応液を濾過し、濾液を濃縮した。この粗製生成物を、ISCOカラム(Iscoシリカゲル80g、溶出溶媒: 0~40%酢酸エチル/ヘキサン)で精製した。
第1溶出フラクションを蒸発し、rac-exo-tert-ブチル 7-オキサビシクロ[2.2.1]ヘプタ-5-エン-2-イルカルバメート(0.65g)を得た。1H NMR(400MHz、クロロホルム-d)δ ppm 6.38 (1 H, dd, J=5.94, 1.54 Hz), 6.29-6.35 (1 H, m), 4.99 (1 H, d, J=4.40 Hz), 4.89 (1 H, d, J=6.16 Hz), 4.74 (1 H, s), 3.75 (1 H, t, J=6.82 Hz), 1.83 (1 H, dd, J=11.88, 7.70 Hz), 1.45 (9 H, s), 1.36 (1 H, ddd, J=11.99, 4.51, 2.86 Hz)
第2溶出フラクションを蒸発し、rac-endo-tert-ブチル 7-オキサビシクロ[2.2.1]ヘプタ-5-エン-2-イルカルバメート(0.42g)を得た。1H NMR(400MHz、クロロホルム-d)δ ppm 6.44 (1 H, d, J=4.84 Hz), 6.23 (1 H, dd, J=5.72, 1.32 Hz), 4.91 (1 H, br. s.), 4.84 (1 H, dd, J=4.73, 1.21 Hz), 4.42 (1 H, d, J=4.18 Hz), 4.11 (1 H, br. s.), 2.18-2.29 (1 H, m), 1.34 (9 H, d, J=3.96 Hz), 0.79 (1 H, dd, J=11.77, 3.19 Hz)
丸底フラスコ(20mL)にexo-rac-tert-ブチル 7-オキサビシクロ[2.2.1]ヘプタ-5-エン-2-イルカルバメート(50mg、0.237mmol)およびPd/C(デグザタイプ、12.59mg、0.012mmol)/酢酸エチル(1mL)およびTHF(0.5mL)を加え、黒色懸濁液を得た。次いで水素をバルーンを介して加えた。反応完了後、得られた反応液を0.45μmの膜で濾過し、THFおよび酢酸エチルで濯いだ。溶媒を除去し、無色固体を得た。これをさらに精製せずにそのまま用いた。
バイアル(20mL)にrac-tert-ブチル((1R,2S,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)カルバメート(50mg、0.234mmol)/塩化メチレン(400μL)を加え、無色溶液を得た。4M HCl/ジオキサン(100μL、3.29mmol)の溶液を加え、終夜撹拌を続けた。得られた反応液を濃縮し、真空乾燥し、粗製生成物を得た。これをさらに精製せずにそのまま用いた。
バイアルに2-アミノ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キナゾリン-4(3H)-オン(48mg、0.154mmol)およびBOP(89mg、0.200mmol)/DMF(0.6mL)を加え、黄褐色懸濁液を得た。DBU(0.070mL、0.463mmol)を加え、最終的に橙色溶液を得た。数分後、rac-(1R,2S,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-アミン・HCl(34.6mg、0.231mmol)を加え、終夜撹拌を続けた。得られた反応液を10%塩化リチウム溶液および少量の飽和炭酸水素ナトリウム溶液で希釈し、薄黄褐色沈殿を得た。この固体を酢酸エチルに抽出した。有機層を硫酸マグネシウムで乾燥し、濾過し、濃縮し、粗製生成物を得た。この物質をエタノール(2mL)に溶解し、4M HCl/ジオキサン(0.19mL、0.77mmol)で処理した。約45分後、溶媒を濃縮し、得られた残渣を精製するためにメタノールに溶解した。この粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(0.05%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル: 水(0.05%トリフルオロ酢酸含有); グラジエント: 0%Bで3分間溶出し、0~40%Bで20分かけて溶出後、次いで100%Bで4分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSおよびUVシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、rac-N4-((1R,2S,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)-7-(1H-ピラゾール-5-イル)キナゾリン-2,4-ジアミン(19.3mg、0.044mmol、28%)をTFA塩として得た。HPLC RT: 0.98分; LCMS(M+H)+: 323.3(方法C)。1H-NMR(400MHz、DMSO)δ 9.00 (br d, J=5.7Hz, 1H), 8.48 (br d, J=8.5Hz, 1H), 8.38-7.96 (m, 2H), 7.88 (br s, 2H), 7.85 (br d, J=7.8Hz, 1H), 6.90 (d, J=2.0Hz, 1H), 4.68 (br t, J=4.6Hz, 1H), 4.54 (d, J=5.1Hz, 1H), 4.36-4.25 (m, 1H), 2.07-1.98 (m, 1H), 1.98-1.89 (m, 1H), 1.71-1.53 (m, 3H), 1.52-1.45 (m, 1H)(交換可能なプロトンの位置は明らかではない)
2-アミノ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キナゾリン-4-オール(25mg、0.080mmol、実施例1、ステップ2)および(2r,3aR,5r,6aS)-5-アミノオクタヒドロペンタレン-2-オール・HCl(35.7mg、0.201mmol)/DMF(0.5mL)の懸濁液に、DBU(0.061mL、0.401mmol)およびBOP(71.0mg、0.161mmol)を加えた。得られた反応液を終夜撹拌し、次いで水で希釈し、酢酸エチルで3回抽出した。この有機層を濃縮した。得られた残渣をメタノール(1mL)に溶解し、濃塩酸(0.05mL)を加えた。約1.5時間後、得られた反応液を濃縮し、塩化メチレンで共沸し、DMFに溶解し、シリンジフィルターを介して濾過した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(10mM酢酸アンモニウム含有); グラジエント: 1%Bで0分間溶出し、1~41%Bで20分かけて溶出後、次いで100%Bで4分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSおよびUVシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、(2r,3aR,5r,6aS)-5-((2-アミノ-7-(1H-ピラゾール-5-イル)キナゾリン-4-イル)アミノ)オクタヒドロペンタレン-2-オール(19.7mg、0.056mmol、62%)を得た。HPLC RT: 1.14分; LCMS(M+H)+: 351.3(方法C)。1H NMR(500MHz、DMSO-d6)δ 9.00 (br d, J=5.7Hz, 1H), 8.48 (br d, J=8.5Hz, 1H), 8.38-7.96 (m, 2H), 7.88 (br s, 2H), 7.85 (br d, J=7.8Hz, 1H), 6.90 (d, J=2.0Hz, 1H), 4.68 (br t, J=4.6Hz, 1H), 4.54 (d, J=5.1Hz, 1H), 4.36-4.25 (m, 1H), 2.07-1.98 (m, 1H), 1.98-1.89 (m, 1H), 1.71-1.53 (m, 3H), 1.52-1.45 (m, 1H)(交換可能な全てのプロトンは観測されていない)
2-アミノ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キナゾリン-4-オール(25mg、0.080mmol、実施例1、ステップ2)および6-アミノスピロ[3.3]ヘプタン-2-オール・HCl(36.0mg、0.220mmol)/DMF(0.5mL)の懸濁液に、DBU(0.061mL、0.401mmol)およびBOP(71.0mg、0.161mmol)を加えた。得られた反応液を終夜撹拌し、次いで水で希釈し、酢酸エチルで3回抽出した。この有機層を濃縮した。得られた残渣をメタノール(1mL)に溶解し、濃塩酸(0.05mL)を加えた。約45分後、得られた反応液を濃縮し、塩化メチレンで共沸し、DMFに溶解し、シリンジフィルターで濾過した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(10mM酢酸アンモニウム含有); グラジエント: 0%Bで0分間溶出し、0~40%Bで25分かけて溶出後、次いで100%Bで4分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、6-((2-アミノ-7-(1H-ピラゾール-5-イル)キナゾリン-4-イル)アミノ)スピロ[3.3]ヘプタン-2-オール(9.0mg、0.027mmol、32%)を得た。HPLC RT: 0.79分; LCMS(M+H)+: 337.0(方法C)。1H NMR(500MHz、DMSO-d6)δ 8.16-8.03 (m, 2H), 7.79-7.67 (m, 1H), 7.63 (br s, 1H), 7.54 (br d, J=6.7Hz, 1H), 7.25-7.14 (m, 1H), 6.80 (s, 1H), 6.42 (br d, J=2.1Hz, 1H), 4.67-4.52 (m, 1H), 4.06-3.93 (m, 1H), 2.42-2.34 (m, 2H), 2.33-2.24 (m, 1H), 2.23-2.17 (m, 1H), 2.16-2.07 (m, 2H), 1.89-1.79 (m, 2H)
9A. 4-(2-オキサ-5-アザビシクロ[2.2.1]ヘプタン-5-イル)-7-ブロモキナゾリン-2-アミン
バイアルに、2-アミノ-7-ブロモキナゾリン-4(3H)-オン(99mg、0.412mmol)、2-オキサ-5-アザビシクロ[2.2.1]ヘプタン・HCl(168mg、1.237mmol)、BOP(237mg、0.536mmol)、およびDBU(311μL、2.062mmol)/DMF(2062μL)を加えた。この反応液を終夜撹拌した。得られた反応液を酢酸エチルで希釈し、水で洗浄した。有機層を減圧濃縮し、所望の粗製生成物、4-(2-オキサ-5-アザビシクロ[2.2.1]ヘプタン-5-イル)-7-ブロモキナゾリン-2-アミン(132mg、0.411mmol、100%収率))を褐色の油状物として得た。HPLC RT: 0.55分; LCMS(M+H)+: 321(方法D)
ジオキサンおよび水を窒素で15分間脱気した。2ドラムバイアルに、4-(2-オキサ-5-アザビシクロ[2.2.1]ヘプタン-5-イル)-7-ブロモキナゾリン-2-アミン(132mg、0.411mmol)、3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール(120mg、0.616mmol)、PdCl2(dppf)-CH2Cl2付加物(33.6mg、0.041mmol)、ジオキサン(4110μL)およびリン酸三カリウム溶液(2M、616μL、1.233mmol)/水を加えた。この反応混合物を次いで95℃で3時間加熱した。得られた反応液を減圧濃縮し、DMFに溶解した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); グラジエント: 0%Bで0分間溶出後、0~30%Bで23分かけて溶出し、次いで100%Bで4分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、rac-4-(2-オキサ-5-アザビシクロ[2.2.1]ヘプタン-5-イル)-7-(1H-ピラゾール-3-イル)キナゾリン-2-アミン(30.2mg)をTFA塩として得た。HPLC RT: 0.96分; LCMS(M+H)+: 309.3(方法C)。1H NMR(500MHz、DMSO-d6)δ 8.14 (d, J=8.9Hz, 1H), 7.87 (br s, 2H), 7.79 (br d, J=7.3Hz, 1H), 6.88 (d, J=2.1Hz, 1H), 5.46 (s, 1H), 4.80 (s, 1H), 4.22 (br dd, J=11.6, 4.0Hz, 1H), 4.02-3.80 (m, 2H), 2.09-2.01 (m, 1H), 2.00-1.94 (m, 1H)(2つのプロトンが抑制された水ピークと重複している可能性が高く、消失している)

密封可能な反応バイアル中、2-アミノ-7-ブロモキナゾリン-4(1H)-オン(30mg、0.12mmol)およびBOP(71.9mg、0.16mmol)/無水DMF(0.8mL)の混合物に、室温で3-アミノ-1-アダマンタノール(62.7mg、0.38mmol)、続いて1,8-ジアザビシクロ-[5.4.0]ウンデカ-7-エン(0.1mL、0.66mmol)を加えた。得られた混合物を周囲温度で18時間撹拌し、EtOAcおよび水の間に分配した。層を分離し、水層をEtOAcで2回抽出した。この有機抽出物を元の有機層と合わせて減圧濃縮し、金色油状物を得た。これをさらに精製せずに、定量的な収率に基づいて次のステップにそのまま用いた。MS(ES): m/z=389/391 [M+H]+; Tr=0.65分
密封可能な反応バイアル中、室温で(1s,3r,5R,7S)-3-((2-アミノ-7-ブロモキナゾリン-4-イル)アミノ)アダマンタン-1-オール(0.12mmol)、3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン)-ピラゾール(53.4mg、0.28mmol)およびCs2CO3(122mg、0.38mmol)/ジオキサン(2.25mL)および水(0.3mL)の混合物を、アルゴンでおおよそ10分間スパージし、PdCl2(dppf)-CH2Cl2(20.4mg、0.025mmol)を加えた。上記バイアルを密封し、反応液を90℃で15.5時間加熱した。室温に冷却後、混合物をDMSOで希釈し、次いでシリンジフィルターで濾過し、EtOAcおよび水の間に分配した。層を分離し、水層を5%MeOH/CHCl3で抽出した。最初の有機抽出層を減圧濃縮し、それに第2有機抽出層を加え、次いで減圧濃縮し、暗褐色残渣を得た。これをDMFで希釈し、シリンジフィルターで濾過し、次いで分取HPLC/MSで精製し、表題化合物(10.7 mg; 22%収率)を得た。MS(ES): m/z=377 [M+H]+。tR=1.01分(方法C)。1H NMR(500MHz、DMSO-d6)δ 8.11 (d, J=8.5Hz, 1H), 7.73 (br s, 1H), 7.59 (d, J=1.5Hz, 1H), 7.50 (br d, J=7.9Hz, 1H), 6.94 (s, 1H), 6.79 (d, J=2.1Hz, 1H), 6.31 (br s, 2H), 3.61 (br s, 1H), 2.22-2.15 (m, 4H), 2.13-2.06 (m, 4H), 1.69-1.63 (m, 2H), 1.62-1.53 (m, 3H), 1.48-1.42 (m, 1H)(水ピークの抑制により、OHプロトンの積分値の出現が抑制されている)
14A. 7-ブロモ-4-クロロキノリン
7-ブロモキノリン-4-オール(2.5g、11.16mmol)/トルエン(20mL)の懸濁液に、POCl3(2.080mL、22.32mmol)を加えた。この反応液を100℃で加熱した。1.5時間後、得られた反応液を冷却し、次いで氷を加えた。得られた反応液を約30分間激しく撹拌し、次いで水を加えた。反応液をDCMで2回抽出した。この有機層を飽和NaHCO3水溶液および食塩水で洗浄し、次いで硫酸ナトリウムで乾燥し、濃縮した。LC/MSでは、一部の生成物が最初の水層に残っていることが示された。水層を撹拌し、飽和NaHCO3水溶液を慎重に加えた。沈殿した固体を濾過し、水で洗浄し、乾燥した。有機層の物質および濾過した固体を合わせて、高真空下で乾燥し、7-ブロモ-4-クロロキノリン(2.46g、10.14mmol、91%収率)を得た。1H NMR(400MHz、クロロホルム-d)δ 8.80 (d, J=4.7Hz, 1H), 8.33 (d, J=1.9Hz, 1H), 8.12 (d, J=9.0Hz, 1H), 7.75 (dd, J=9.0, 2.0Hz, 1H), 7.52 (d, J=4.8Hz, 1H)
7-ブロモ-4-クロロキノリン(2.0g、8.25mmol)/DCM(55.0mL)の溶液に、mCPBA(6.10g、24.74mmol)を加えた。この反応液を終夜撹拌し、次いで飽和チオ硫酸ナトリウム溶液でクエンチした。得られた反応液を0.5時間撹拌し、次いで飽和炭酸水素ナトリウム水溶液を加えた。この反応液を塩化メチレンで2回抽出した。この有機層を食塩水で洗浄し、硫酸ナトリウムで乾燥し、濃縮し、7-ブロモ-4-クロロキノリン1-オキシドを得た(2.16g、8.36mmol、定量的な収量)。1H NMR(400MHz、クロロホルム-d)δ 8.99 (d, J=1.9Hz, 1H), 8.43 (d, J=6.6Hz, 1H), 8.10 (d, J=9.0Hz, 1H), 7.86 (dd, J=9.0, 2.0Hz, 1H), 7.40 (d, J=6.6Hz, 1H)
丸底フラスコ(1)中、7-ブロモ-4-クロロキノリン1-オキシド(9400mg、36.4mmol)をDCM(150mL)に懸濁した。Ts-Cl(7626mg、40.0mmol)を加えた。この混合物を1時間撹拌した。別の丸底フラスコ(2)中、110℃のオーブンで終夜乾燥した塩化アンモニウム(9725mg、182mmol)をDCM(150mL)に懸濁した。トリエチルアミン(25.3mL、182mmol)を加え、混合物を0.5時間撹拌し、次いで丸底フラスコ(1)の内容物を丸底フラスコ(2)に加えた。得られた反応液を終夜撹拌し、次いで濾過し、濃縮した。残渣を熱したDCM(100mL)に溶解した。この溶液を室温に冷却し、固体を濾過した。濾過ケーキを-20℃のDCM(100mL)で洗浄した。濾過ケーキを水(50mL)に懸濁し、濾過した。この固体は所望の生成物、7-ブロモ-4-クロロキノリン-2-アミンである。塩化メチレンの濾液を濃縮し、水(100mL)に懸濁し、濾過した。濾過ケーキを-20℃のDCM(100mL)で洗浄し、さらに生成物を得た。固体を合わせて高真空下で乾燥し、7-ブロモ-4-クロロキノリン-2-アミン(6.52g、69.6%)を得た。1H NMR(400MHz、DMSO-d6)δ 7.79 (d, J=8.7Hz, 1H), 7.65 (d, J=1.9Hz, 1H), 7.39 (dd, J=8.8, 2.0Hz, 1H), 6.98 (s, 1H), 6.88 (s, 2H)
2つの耐圧バイアル(40mL)のそれぞれに、(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)ボロン酸(0.714g、3.64mmol)、7-ブロモ-4-クロロキノリン-2-アミン(0.750g、2.91mmol)、およびPdCl2(dppf)-DCM付加物(0.238g、0.291mmol)を加えた。バイアルを真空下に置き、窒素で3回充填した。ジオキサン(14.56mL)およびリン酸三カリウム水溶液(2M、4.37mL、8.74mmol)を各バイアルに加え、溶液を窒素バブリングし、次いで反応液を終夜100℃で加熱した。このバイアルを冷却し、EtOAcおよび水で希釈して合わせた。反応液をEtOAcで3回抽出し、次いで有機層を食塩水で洗浄し、硫酸ナトリウムで乾燥し、濃縮した。得られた残渣をISCO(80gカラム; 塩化メチレン/MeOH; グラジエント: 0~10%)で精製し、4-クロロ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キノリン-2-アミン(1.14g、59.5%収率)を得た。1H NMR(400MHz、クロロホルム-d)δ 8.11 (d, J=8.6Hz, 1H), 7.82 (d, J=1.4Hz, 1H), 7.65 (d, J=1.5Hz, 1H), 7.52 (dd, J=8.5, 1.7Hz, 1H), 6.90 (s, 1H), 6.45 (d, J=1.8Hz, 1H), 5.38-5.26 (m, 1H), 4.90 (br s, 1H), 4.22-4.09 (m, 2H), 3.65 (td, J=11.7, 2.3Hz, 1H), 2.68-2.51 (m, 1H), 2.14-1.51 (m, 5H)
(2r,3aR,5r,6aS)-5-アミノオクタヒドロペンタレン-2-オール・HCl(54.0mg、0.304mmol)および4-クロロ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キノリン-2-アミン(25mg、0.076mmol)/DMSO(0.5mL)の懸濁液に、ジイソプロピルエチルアミン(0.106mL、0.608mmol)を加えた。この反応液を120℃で加熱した。終夜撹拌後、さらに(2r,3aR,5r,6aS)-5-アミノオクタヒドロペンタレン-2-オール・HCl(54.0mg、0.304mmol)およびジイソプロピルエチルアミン(0.106mL、0.608mmol)を加えた。さらに1日撹拌後、冷却した反応液を水で希釈し、酢酸エチルで3回抽出した。この有機層を濃縮した。得られた残渣を次いでメタノール(1mL)に溶解し、濃HCl(0.05mL)を加えた。約2時間後、得られた反応液を濃縮し、塩化メチレンで共沸し、DMFに溶解し、シリンジフィルターを介して濾過した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(10mM酢酸アンモニウム含有); グラジエント: 2%Bで0分間溶出後、2~42%でB25分かけて溶出し、次いで100%Bで4分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、(2r,3aR,5r,6aS)-5-((2-アミノ-7-(1H-ピラゾール-5-イル)キノリン-4-イル)アミノ)オクタヒドロペンタレン-2-オール(5.8mg、0.0166mmol、21%)を得た。HPLC RT: 1.21分; LCMS(M+H)+: 350.3(方法C)。1H NMR(500MHz、DMSO-d6)δ 8.24 (br d, J=8.3Hz, 1H), 7.90-7.84 (m, 1H), 7.83-7.77 (m, 1H), 7.76-7.68 (m, 1H), 6.82 (d, J=1.9Hz, 1H), 5.89 (s, 1H), 4.12-4.00 (m, 1H), 3.92 (br t, J=5.9Hz, 1H), 2.10-2.00 (m, 2H), 1.95-1.85 (m, 2H), 1.82-1.71 (m, 2H), 1.30-1.15 (m, 2H)(側鎖の2つのプロトンピークがDMSOピークと重複している可能性が高く、消失している)
バイアルに4-クロロ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キノリン-2-アミン(50mg、0.152mmol)およびrac-(1R,2S,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-アミン・HCl(34.1mg、0.228mmol、実施例1参照、ステップ5)/NMP(0.8mL)を加え、橙色溶液を得た。ジイソプロピルエチルアミン(0.159mL、0.912mmol)を加え、得られた反応液を120℃の加熱ブロックで加熱した。約6時間後、得られた反応液を130℃に加熱し、終夜撹拌を続けた。冷却した反応を濃縮し、次いでメタノール(0.8mL)で希釈した。4M HCl/ジオキサン溶液(0.380mL、1.521mmol)を加えた。約1時間後、得られた反応液をメタノール(0.8mL)で希釈した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(10mM酢酸アンモニウム含有); グラジエント: 1%Bで0分間溶出後、1~41%Bで25分かけて溶出し、次いで100%Bで4分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、rac-N4-((1R,2S,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)-7-(1H-ピラゾール-5-イル)キノリン-2,4-ジアミン(25.9mg)を得た。HPLC RT: 1.01分; LCMS(M+H)+: 322.3(方法C)。1H NMR(500MHz、DMSO-d6)δ 8.14 (d, J=8.5Hz, 1H), 7.78 (s, 1H), 7.74 (br s, 1H), 7.56 (br d, J=7.6Hz, 1H), 6.79 (d, J=2.1Hz, 1H), 6.70-6.53 (m, 2H), 5.68 (s, 1H), 4.63 (t, J=4.6Hz, 1H), 4.51 (d, J=4.9Hz, 1H), 3.64-3.52 (m, 1H), 2.03 (dd, J=12.5, 7.6Hz, 1H), 1.86 (m, 1H), 1.69-1.53 (m, 3H), 1.53-1.40 (m, 1H)
4-クロロ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キノリン-2-アミン・HCl(30mg、0.082mmol)および(3-アミノビシクロ[1.1.1]ペンタン-1-イル)メタノール・HCl(42.0mg、0.281mmol)/DMSO(0.5mL)の溶液に、ジイソプロピルエチルアミン(0.143mL、0.821mmol)を加えた。この反応液を120℃で終夜加熱した。LCMSでは、反応中にTHP基が除去されたことが示された。反応液を一部濃縮し、メタノールで希釈し、シリンジフィルターを介して濾過した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(10mM酢酸アンモニウム含有); グラジエント:0%Bで0分間溶出し、0~28%Bで28分かけて溶出し、次いで100%Bで5分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、(3-((2-アミノ-7-(1H-ピラゾール-5-イル)キノリン-4-イル)アミノ)ビシクロ[1.1.1]ペンタン-1-イル)メタノール(6.2mg、0.019mmol、22%)を得た。HPLC RT: 0.83分; LCMS(M+H)+: 322.2(方法C)。1H NMR(500MHz、DMSO-d6)δ 8.06 (br d, J=8.9Hz, 1H), 7.89-7.81 (m, 1H), 7.79 (br s, 1H), 7.76-7.71 (m, 1H), 7.68-7.60 (m, 1H), 6.81 (s, 1H), 6.12 (s, 1H), 3.54 (s, 2H), 2.05 (s, 6H)
rac-((1R,2S,3R,4S)-3-アミノビシクロ[2.2.1]ヘプタン-2-イル)メタノール・HCl(108mg、0.608mmol)および4-クロロ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キノリン-2-アミン(50mg、0.152mmol)/DMSO(1mL)の懸濁液に、ジイソプロピルエチルアミン(0.212mL、1.217mmol)を加えた。この反応液を120℃で終夜加熱した。冷却した反応液を濃縮し、メタノール(1mL)に溶解した。少量の濃塩酸(0.05mL)を次いで加え、2時間撹拌を続けた。得られた反応液を次いで濃縮し、メタノールに溶解し、シリンジフィルターで濾過した。この粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(酢酸アンモニウム含有); グラジエント: 7%Bで0分間溶出後、7~47%Bで30分かけて溶出し、次いで100%Bで4分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはUVシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥した。この物質をさらに分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); グラジエント: 5%Bで0分間溶出後、5~45%Bで22分かけて溶出し、次いで100%Bで4分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、rac-((1R,2S,3R,4S)-3-((2-アミノ-7-(1H-ピラゾール-5-イル)キノリン-4-イル)アミノ)ビシクロ[2.2.1]ヘプタン-2-イル)メタノール(19.5mg、0.042mmol、28%)をTFA塩として得た。HPLC RT: 1.31分; LCMS(M+H)+: 350.0(方法C)。1H NMR(500MHz、DMSO-d6)δ 8.15-8.07 (m, 1H), 7.98-7.91 (m, 1H), 7.89-7.85 (m, 1H), 7.85-7.80 (m, 1H), 7.77 (br d, J=6.7Hz, 1H), 7.71-7.58 (m, 2H), 6.84 (d, J=1.8Hz, 1H), 5.88 (s, 1H), 3.58 (br t, J=7.3Hz, 1H), 3.51-3.42 (m, 1H), 2.40-2.28 (m, 2H), 2.03-1.94 (m, 1H), 1.89 (br d, J=10.1Hz, 1H), 1.65-1.50 (m, 2H), 1.36-1.17 (m, 2H), 1.14 (s, 1H)(1つのプロトンが抑制された水ピークと重複している可能性が高く、消失している)

24A. 2-アミノ-7-ブロモ-1,5-ナフチリジン-4-オール
アセトニトリル(2.261mL、43.3mmol)およびTHF(50mL)の撹拌溶液に、-78℃でn-ブチルリチウム(11.0M、ヘキサン溶液、1.967mL、21.64mmol)を加えた。-78℃で1時間撹拌後、3-アミノ-5-ブロモピコリン酸メチル(1.0g、4.33mmol)/THF(30mL)を滴下した。この反応液を-78℃で30分間撹拌し、次いで室温に加温した。3日後、反応混合物は、50%反応が完了した。次いで50℃で5時間および40℃で終夜撹拌することで85%反応が完了し、一部の非環状生成物と共に、3-(3-アミノ-5-ブロモピリジン-2-イル)-3-オキソプロパンニトリルが残った。得られた反応液を飽和NH4Cl水溶液(15mL)でクエンチし、減圧濾過し、固体を乾燥し、2-アミノ-7-ブロモ-1,5-ナフチリジン-4-オール(0.85g、82%収率)を黄褐色固体として得た。LCMS(M+H)+: 240.0
2-アミノ-7-ブロモ-1,5-ナフチリジン-4-オール(0.10g、0.417mmol)/原液POCl3(1.343mL、14.40mmol)の溶液を100℃で終夜撹拌した。この反応液を濃縮し、全てのPOCl3を除去し、MeOHで2回共沸し、7-ブロモ-4-クロロ-1,5-ナフチリジン-2-アミン(0.10g、93%収率)を得た。LCMS(M+H)+: 258.0
7-ブロモ-4-クロロ-1,5-ナフチリジン-2-アミン(0.025g、0.097mmol)および3-オキサ-8-アザビシクロ[3.2.1]オクタン塩酸塩(0.072g、0.484mmol)/NMP(0.5mL)の溶液に、DIEA(0.169mL、0.967mmol)を加えた。この反応混合物を150℃で終夜撹拌した。反応が完了後、続いてこの反応混合物とSuzuki反応を行った。LCMS(M+H)+: 335.1
4-(3-オキサ-8-アザビシクロ[3.2.1]オクタン-8-イル)-7-ブロモ-1,5-ナフチリジン-2-アミン(0.032g、0.095mmol)/ジオキサン(0.955mL)の溶液に、2Mリン酸三カリウム水溶液(0.143mL、0.286mmol)および5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール(0.046g、0.239mmol)を加えた。この混合物を窒素/真空で3回パージした。[1,1'ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)(3.49mg、4.77μmol)を加え、得られた溶液を窒素/真空でパージし、終夜100℃で撹拌した。ジオキサンを減圧除去し、粗製物をDMFに溶解し、濾過し、分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子径: 5μm; 移動相A: 5:95 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); グラジエント: 7%Bで0分間溶出後、7~47%Bで27分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製し、4-(3-オキサ-8-アザビシクロ[3.2.1]オクタン-8-イル)-7-(1H-ピラゾール-3-イル)-1,5-ナフチリジン-2-アミン(10.6mg、34%収率)を得た。
25A. 4-(アジドメチル)-1-ビニル-2-オキサビシクロ[2.2.2]オクタン
WO2013/003383に記載の方法で製造した、(1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル4-メチルベンゼンスルホネート(1.5g、4.65mmol)、アジ化ナトリウム(0.423g、6.51mmol)、およびヨウ化ナトリウム(0.139g、0.930mmol)/DMSO(5.82mL)の溶液を窒素下に加え、100℃で加熱した。2.5時間後、得られた反応液をNaN3(0.2g以上)およびNaI(0.1g以上)で処理し、さらに3時間加熱を続けた。反応液を周囲温度に冷却し、エーテルに注いだ。得られた混合物を水で3回、食塩水で1回洗浄し、乾燥し、減圧濃縮し、4-(アジドメチル)-1-ビニル-2-オキサビシクロ[2.2.2]オクタンを淡黄油状物として得た(25A、891mg、99%収率)。1HNMR(400MHz、CDCl3)δ 5.84 (dd, J=17.6, 10.9Hz, 1H), 5.17 (dd, J=17.6, 1.4Hz, 1H), 5.05 (dd, J=10.9, 1.4Hz, 1H), 3.78 (s, 2H), 3.12 (s, 2H), 1.87-1.98 (m, 2H), 1.67-1.79 (m, 4H), 1.60-1.65 (m, 2H)
4-(アジドメチル)-1-ビニル-2-オキサビシクロ[2.2.2]オクタン(25A、135mg、0.699mmol)/THF(4507μL)-水(150μL)の溶液をトリフェニルホスフィン(202mg、0.768mmol)で処理した。得られた溶液を50℃で加熱し、40分間撹拌し、周囲温度に冷却し、EtOAcに注いだ。この混合物を0.1M HCl水溶液で2回洗浄し、洗浄した水溶液を合わせて減圧濃縮した。EtOH、次いでエーテルから得られた残渣を濃縮し、(1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メタンアミン・HCl、0.5ジエチルエーテルをペースト状の灰白色固体として得た(25B、165mg、98%収率)。1H-NMR(400MHz、DMSO-d6)δ 5.79 (dd, J=17.5, 11.0Hz, 1H), 5.10 (dd, J=17.6, 1.8Hz, 1H), 4.96 (dd, J=10.9, 1.8Hz, 1H), 4.35-4.61 (m, 3H), 3.67 (s, 2H), 3.45 (q, J=7.0Hz, 2H), 2.60 (q, J=5.9Hz, 2H), 1.68-1.74 (m, 4H), 1.58-1.64 (m, 4H), 1.06 (t, J=7.0Hz, 2H)
(1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メタンアミン・HCl、0.5ジエチルエーテル(25B、126mg、0.524mmol)および7-ブロモ-4-クロロキノリン-2-アミン(90mg、0.349mmol)/NMP(699μL)の撹拌溶液を、DIEA(183μL、1.048mmol)で処理した。この溶液を95℃で5.5時間加熱し、次いで周囲温度に冷却した。反応液に50%HOAc水溶液を数滴加えてクエンチし、得られた懸濁溶液をDMFで希釈し、濾過し、分取HPLCで精製した。適当なフラクションを濃縮し、7-ブロモ-N4-((1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル)キノリン-2,4-ジアミン・TFA(25C、78mg、0.155mmol、44%収率)を白色粉末として得た。1H-NMR(400MHz、DMSO-d6)δ 12.38 (br. s, 1H), 8.24 (d, J=8.9Hz, 1H), 7.97 (t, J=6.2Hz, 1H), 7.73 (d, J=1.9Hz, 1H), 7.70 (br. s, 2H), 7.63 (dd, J=8.8, 1.9Hz, 1H), 5.94 (s, 1H), 5.79 (dd, J=17.5, 10.9Hz, 1H), 5.09 (dd, J=17.6, 1.9Hz, 1H), 4.96 (dd, J=10.9, 1.9Hz, 1H), 3.74 (s, 2H), 3.12 (d, J=6.2Hz, 2H), 1.62-1.76 (m, 8H)
7-ブロモ-N4-((1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル)キノリン-2,4-ジアミン・TFA(25C、58mg、0.115mmol)、3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール(56.0mg、0.289mmol)、PdCl2(dppf)-CH2Cl2付加物(9.43mg、0.012mmol)、および炭酸セシウム(113mg、0.346mmol)/脱気ジオキサン(1.2mL)の混合物を窒素下に置き、95℃で5時間加熱し、次いで周囲温度に冷却した。得られた反応液を濾過し、分取HPLC(Luna C18 カラム、MeOH-水-TFAグラジエント)で精製した。適当なフラクションを濃縮し、7-(1H-ピラゾール-3-イル)-N4-((1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル)キノリン-2,4-ジアミン・TFA(実施例25、35mg、0.072mmol、62%収率)を灰白色粉末として得た。

27A: (rac)-tert-ブチル1-((2-アミノ-7-ブロモキナゾリン-4-イル)アミノ)-6-アザスピロ[2.5]オクタン-6-カルボキシレート
密封可能な反応バイアル中、2-アミノ-7-ブロモキナゾリン-4(1H)-オン(30mg、0.125mmol)およびBOP(71.9mg、0.162mmol)/無水DMF(0.8mL)の混合物に、室温でtert-ブチル1-アミノ-6-アザスピロ[2.5]オクタン-6-カルボキシレート(56.6mg、0.250mmol)、続いて1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(0.1mL、0.663mmol)を加えた。得られた混合物を室温で終夜撹拌した。反応液を次いで酢酸エチルおよび水の間に分配した。層を分離し、水層を酢酸エチルで2回以上抽出した。合わせた有機層を減圧濃縮し、(rac)-tert-ブチル1-((2-アミノ-7-ブロモキナゾリン-4-イル)アミノ)-6-アザスピロ[2.5]オクタン-6-カルボキシレート(203mg)を金色油状物として得た。この物質は次のステップにそのまま用いた。
密封可能な反応バイアル中、tert-ブチル1-((2-アミノ-7-ブロモキナゾリン-4-イル)アミノ)-6-アザスピロ[2.5]オクタン-6-カルボキシレート(0.056g、0.125mmol)、3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン)-ピラゾール(0.053g、0.275mmol)およびCs2CO3(0.122g、0.375mmol)/ジオキサン(2.25mL)および水(0.3mL)の混合物を、室温下アルゴンで約5~10分間スパージし、PdCl2(dppf)-CH2Cl2付加物(0.020g、0.025mmol)を加え、このバイアルを密封し、得られた反応液を90℃で加熱した。終夜撹拌後、冷却した反応液を塩化メチレンおよび水の間に分配した。層を分離し、水層を塩化メチレンで2回以上、次いで5%メタノール/塩化メチレンで1回抽出した。抽出した有機層を合わせて減圧濃縮し、暗褐色残渣を得た。この物質の半分を精製(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(10mM酢酸アンモニウム含有); グラジエント: 14%Bで0分間溶出後、14~54%でB20分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)した。フラクションはMSおよびUVシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、(rac)-tert-ブチル1-((2-アミノ-7-(1H-ピラゾール-3-イル)キナゾリン-4-イル)アミノ)-6-アザスピロ[2.5]オクタン-6-カルボキシレート(12.5mg、0.028mmol)を得た。

29A. rac-tert-ブチル((1R,2R,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)カルバメート
丸底フラスコ(25mL)に、実施例1Cに記載の方法で製造したrac-tert-ブチル((1R,2R,4R)-7-オキサビシクロ[2.2.1]ヘプタ-5-エン-2-イル)カルバメート(116mg、0.549mmol)およびPd/C(デグザタイプ、29.2mg、0.027mmol)/THF(4mL)を加え、黒色懸濁液を得た。水素雰囲気をバルーンで導入した。約1時間後、得られた反応液をTHFで洗浄し、0.45μmフィルターで濾過した。洗浄液を合わせて濃縮し、rac-tert-ブチル((1R,2R,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)カルバメート(106.7mg)を無色の油状物として得た。この物質は精製せずに用いた。
バイアル(20mL)にrac-tert-ブチル((1R,2R,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)カルバメート(106mg、0.497mmol)/塩化メチレン(1mL)を加え、無色溶液を得た。HCl/ジオキサン溶液(4M、100μL、3.29mmol)を次いで加え、終夜撹拌を続けた。溶媒を次いで濃縮し、サンプルを真空乾燥し、rac-(1R,2R,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-アミン・HClを得た。この物質は精製せずに用いた。
バイアル(4mL)に2-アミノ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キナゾリン-4(3H)-オン(20mg、0.064mmol)およびBOP(36.9mg、0.084mmol)/DMF(0.5mL)を加え、懸濁液を得た。DBU(0.029mL、0.193mmol)を加え、均一な溶液とした。rac-(1R,2R,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-アミン・HCl(14.42mg、0.096mmol)を加え、この反応液を終夜撹拌した。次いで得られた反応液を水で希釈し、少量の飽和NaHCO3溶液および10%LiCl溶液で処理した。混合物をEtOAcで2回抽出した。合わせた有機層を次いで濃縮した。この物質をMeOH(1mL)に溶解し、HCl/ジオキサン溶液(4M、0.032mL、0.128mmol)で処理した。約0.5時間後、得られた反応液を濃縮した。得られた反応液をメタノールに溶解し、DIPEAで塩基性にした。この粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(酢酸アンモニウム含有); グラジエント: 0%Bで0分間溶出後、0~40%Bで20分かけて溶液し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSおよびUVシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、rac-N4-((1R,2R,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)-7-(1H-ピラゾール-5-イル)キナゾリン-2,4-ジアミン(18.5mg)を得た。
バイアル(20mL)に4-クロロ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キノリン-2-アミン(化合物14D、50mg、0.152mmol)およびrac-(1R,2R,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-アミン・HCl(化合物29B、59.2mg、0.395mmol)/NMP(1mL)を加え、橙色溶液を得た。ヒューニッヒ塩基(0.159mL、0.912mmol)を加え、得られた反応液を130℃で加熱した。終夜加熱後、冷却した反応液をメタノールで希釈し、RP-HPLC(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(酢酸アンモニウム含有); グラジエント: 0%Bで0分間溶出後、0~40%Bで20分かけて溶液し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSおよびUVシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、rac-N4-((1R,2R,4S)-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)-7-(1H-ピラゾール-5-イル)キノリン-2,4-ジアミン(38.9mg)を得た。

丸底フラスコ(20mL)にrac-tert-ブチル((1R,2S,4R)-7-オキサビシクロ[2.2.1]ヘプタ-5-エン-2-イル)カルバメート(1Cから得たexo-異性体、191mg、0.904mmol)/THF(1mL)を加え、無色溶液を得た。この反応液を氷浴で冷却し、ボラン-テトラヒドロフラン錯体(1M、THF溶液、2.260mL、2.260mmol)を滴下した。約15分後、反応系に針で通気口を作り、pH 7.2の緩衝液(1.4mL)で非常にゆっくりとクエンチした。添加終了後、過酸化水素(30%水溶液、0.646mL、6.33mmol)を加え、約20分間撹拌を続けた。得られた反応液を次いで飽和NaCl溶液で希釈し、EtOAcで回抽出した。合わせた有機層を氷水浴で冷却し、飽和チオ硫酸ナトリウム溶液を加えた。この混合物を氷水浴で約45分間すばやく撹拌した。次いで層を分離し、有機層を硫酸マグネシウムで乾燥し、濾過し、濃縮し、rac-tert-ブチル((1R,2S,4R,5S)-5-ヒドロキシ-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)カルバメートおよびrac-tert-ブチル((1R,2R,4S,6S)-6-ヒドロキシ-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)カルバメート(合計191mg)を得た。この物質を精製せずに次のステップで用いた。
バイアル(20mL)にrac-tert-ブチル((1R,2S,4R,5S)-5-ヒドロキシ-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)カルバメートおよびrac-tert-ブチル((1R,2R,4S,6S)-6-ヒドロキシ-7-オキサビシクロ[2.2.1]ヘプタン-2-イル)カルバメート(190mg、0.829mmol)/THF(2mL)を加え、無色溶液を得た。HCl/ジオキサン溶液(4M、0.829mL、3.31mmol)を加えた。約2時間後、さらにHCl/ジオキサンを加え、3日間撹拌を続けた。次いで得られた反応液を濃縮し、真空乾燥し、rac-(1R,2S,4R,5S)-5-アミノ-7-オキサビシクロ[2.2.1]ヘプタン-2-オールおよびrac-(1R,2S,4S,6R)-6-アミノ-7-オキサビシクロ[2.2.1]ヘプタン-2-オールをHCl塩として得た。この物質を精製せずに次のステップで用いた。
バイアル(8mL)に2-アミノ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キナゾリン-4(3H)-オン(50mg、0.161mmol)およびBOP(85mg、0.193mmol)/DMF(0.6mL)を加えた。DBU(0.097mL、0.642mmol)を加え、暗赤色/橙色溶液を得た。rac-(1R,2S,4R,5S)-5-アミノ-7-オキサビシクロ[2.2.1]ヘプタン-2-オール・HClおよびrac-(1R,2S,4S,6R)-6-アミノ-7-オキサビシクロ[2.2.1]ヘプタン-2-オール・HCl(39.9mg、0.241mmol)を加え、終夜撹拌を続けた。次いで得られた反応液を10%LiCl水溶液で希釈し、EtOAcで3回抽出した。合わせた有機層を次いで濃縮した。この残渣をEtOH(1mL)に溶解し、HCl/ジオキサン溶液(4M、0.201mL、0.803mmol)を加えた。LCMSで反応の完了が示された後、得られた反応液を濃縮し、精製するためにメタノールに再溶解した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(10mM酢酸アンモニウム含有); グラジエント: 0%Bで0分間溶出後、0~40%Bで25分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥した。この精製により一部が分離され、2つのサンプル、31A(18.9mg)および31B(8.5mg)を得た。31Bのサンプルは、LCMSおよび1H NMRにより1つの成分であると考えられる一方、31Aは31Bが多く含まれた不純物のままであった。

4-クロロ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キノリン-2-アミン(50mg、0.152mmol)/NMP(1mL)の溶液に、1,4,8-トリオキサスピロ[4.5]デカン-6-アミン(72.6mg、0.456mmol)およびDIEA(0.266mL、1.521mmol)を加えた。この反応混合物を130℃で3時間加熱した。化合物を部分的にRP-HPLC(Phen Luna 5μ, 30X100mmカラム、メタノール/水グラジエント+0.1%TFA)で精製した。得られた化合物をさらに精製した(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); グラジエント: 5%Bで0分間溶出後、5~45%Bで25分かけて溶出し、次いで100%Bで4分間溶出; 流速: 20mL/分; カラム温度: 25℃)。フラクションはMSおよびUVシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、7-(1H-ピラゾール-3-イル)-N4-(1,4,8-トリオキサスピロ[4.5]デカン-6-イル)キノリン-2,4-ジアミン(12.3mg)を得た。
スクリューバイアル(25mL)に(3-(アミノメチル)ビシクロ[1.1.1]ペンタン-1-イル)メタノール・HCl(24.55mg、150mmol)、2-アミノ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キナゾリン-4-オール(30mg、0.096mmol)、DBU(29.0μL、0.193mmol)、およびBOP(85mg、0.193mmol)/NMP(0.5mL)を加えた。この反応液を16時間撹拌し、TFA(500μL、6.49mmol)を加えた。3時間後、得られた反応液を濃縮し、高真空下で乾燥した。この反応混合物をDMF/酢酸(1:1、1mL)で希釈した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(酢酸アンモニウム含有); グラジエント: 0%Bで0分間溶出後、0~40%Bで20分かけて溶液し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSおよびUVシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、(3-(((2-アミノ-7-(1H-ピラゾール-5-イル)キナゾリン-4-イル)アミノ)メチル)ビシクロ[1.1.1]ペンタン-1-イル)メタノール・TFA(10.4mg、24%)を得た。

40A. 8-ヨードヘキサヒドロ-4,6-メタノシクロペンタ[d][1,3]オキサジン-2(1H)-オン
既知の8-ヨードヘキサヒドロ-4,6-メタノシクロペンタ[d][1,3]オキサジン-2(1H)-オン(220mg、0.902mmol、J.Org.Chem. 53 2665-2668 (1988), BOMC 12 (18) 4877-4884 (2004))を塩化メチレン(2.6mL)に溶解した。ヨウ素(275mg、1.08mmol)を加え、反応液を3時間撹拌した。得られた反応液を飽和チオ硫酸ナトリウム溶液でクエンチし、塩化メチレンで3回抽出した。有機抽出物を合わせて、水で洗浄し、硫酸ナトリウムで乾燥した。濾過および蒸発により、8-ヨードヘキサヒドロ-4,6-メタノシクロペンタ[d][1,3]オキサジン-2(1H)-オン(223mg、0.799mmol、88%収率)を得た。1H NMR(400MHz、クロロホルム-d)δ 5.23-5.16 (m, 1H), 3.86 (t, J=2.7Hz, 1H), 3.84-3.77 (m, 1H), 2.63 (br d, J=3.7Hz, 1H), 2.45-2.40 (m, 1H), 2.22-2.16 (m, 1H), 2.16-2.08 (m, 1H), 1.69 (dt, J=11.3, 1.5Hz, 1H), 1.46 (dt, J=13.5, 2.7Hz, 1H)
8-ヨードヘキサヒドロ-4,6-メタノシクロペンタ[d][1,3]オキサジン-2(1H)-オン(223mg、0.799mmol)をメタノール(6659μL)に溶解した。トリエチルアミン(200μL、1.438mmol)を加え、次いで得られた反応液を真空下に置き、窒素で3回充填した。約3時間後、Pd-C(25mg、0.012mmol)を加え、次いで水素バルーンを反応系に付けた。この反応系を真空下に置き、水素で3回充填した。終夜撹拌後、気体を窒素に交換した。得られた反応液を濾過し、メタノールで濯ぎ、濾液を濃縮した。得られた残渣を塩化メチレンおよび飽和炭酸水素ナトリウム溶液の間に分配した。水相を塩化メチレンで3回抽出した。蒸発により、ヘキサヒドロ-4,6-メタノシクロペンタ[d][1,3]オキサジン-2(1H)-オン(100mg、0.65mmol、82%)を得た。1H NMR(400MHz、クロロホルム-d)δ 6.93 (br s, 1H), 4.83-4.66 (m, 1H), 3.75 (dt, J=4.9, 2.4Hz, 1H), 2.40-2.31 (m, 1H), 2.26 (br s, 1H), 2.23-2.10 (m, 1H), 2.07-1.91 (m, 1H), 1.45 (s, 2H), 1.36-1.25 (m, 2H)
ヘキサヒドロ-4,6-メタノシクロペンタ[d][1,3]オキサジン-2(1H)-オン(100mg、0.653mmol)/エタノール(2332μL)の溶液を4M水酸化ナトリウム水溶液(1306μL、5.22mmol)で処理した。この反応液を80℃に加熱し、終夜撹拌した。得られた反応液を部分的に濃縮し、水で希釈し、塩化メチレンで3回抽出した。この有機層を硫酸ナトリウムで乾燥し、濾過し、濃縮し、(1S,2S,4R,6R)-6-アミノビシクロ[2.2.1]ヘプタン-2-オール(83mg)を得た。
2-アミノ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キナゾリン-4-オール(30mg、0.096mmol)および(2S,6S)-6-アミノビシクロ[2.2.1]ヘプタン-2-オール(24.51mg、0.193mmol)/DMF(0.5mL)の懸濁液に、DBU(0.044mL、0.289mmol)およびBOP(85mg、0.193mmol)を加えた。終夜撹拌後、得られた反応液を水で希釈し、酢酸エチルで3回抽出した。この有機層を濃縮し、得られた残渣をメタノール(1mL)および濃HCl(0.05mL)に溶解した。約45分後、得られた反応液を濃縮し、塩化メチレンで共沸し、DMFに溶解し、シリンジフィルターを介して濾過し、精製した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); グラジエント: 0%Bで3分間溶出後、0~40%Bで23分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、rac-(2S,6S)-6-((2-アミノ-7-(1H-ピラゾール-5-イル)キナゾリン-4-イル)アミノ)ビシクロ[2.2.1]ヘプタン-2-オール(13.1mg)を得た。

42A. rac-メチル(1R,2R,4R,5S)-5-ヒドロキシビシクロ[2.2.1]ヘプタン-2-カルボキシレートおよびrac-メチル(1S,2R,4R,6R)-6-ヒドロキシビシクロ[2.2.1]ヘプタン-2-カルボキシレート
丸底フラスコ(200mL)にrac-メチル(1R,2R,4R)-ビシクロ[2.2.1]ヘプタ-5-エン-2-カルボキシレート(2.81g、18.46mmol)/THF(40mL)を加え、溶液を得た。この反応液を0℃に冷却した。BH3-THF(1M、THF溶液、46.2mL、46.2mmol)を加えた。6時間後、水(1mL)をゆっくりと加え、30%過酸化水素(13.20mL、129mmol)を次いで滴下した。得られた反応液を酢酸エチル(100mL)および食塩水(25mL)の間に分配した。有機層を硫酸ナトリウムで乾燥し、濾過し、濃縮した。得られた粗製生成物を少量の塩化メチレンに溶解し、シリカゲルカートリッジ(220g)にロードし、溶出した(0%~100%酢酸エチル/ヘキサン)。蒸発により、rac-メチル(1R,2R,4R,5S)-5-ヒドロキシビシクロ[2.2.1]ヘプタン-2-カルボキシレートおよびrac-メチル(1S,2R,4R,6R)-6-ヒドロキシビシクロ[2.2.1]ヘプタン-2-カルボキシレート(2.23g)を得た。
丸底フラスコ(100mL)に、rac-メチル(1R,2R,4R,5S)-5-ヒドロキシビシクロ[2.2.1]ヘプタン-2-カルボキシレートおよびrac-メチル(1S,2R,4R,6R)-6-ヒドロキシビシクロ[2.2.1]ヘプタン-2-カルボキシレート(2.23g、13.10mmol)/アセトニトリル(60mL)の混合物を加えた。TBDPS-Cl(5.4mL、21.02mmol)、続いてDBU(5.92mL、39.3mmol)を加えた。反応完了後、酢酸エチル(50mL)およびHCl(1N、25mL)の間に分配した。有機層を硫酸ナトリウムで乾燥し、濾過し、濃縮した。得られた粗製生成物を少量の塩化メチレンに溶解し、シリカゲルカートリッジ(40g)にロードし、0%~70%酢酸エチル/ヘキサンのグラジエントで溶出した。蒸発により、rac-メチル(1R,2R,4R,5S)-5-((tert-ブチルジフェニルシリル)オキシ)ビシクロ[2.2.1]ヘプタン-2-カルボキシレートおよびrac-メチル(1S,2R,4R,6R)-6-((tert-ブチルジフェニルシリル)オキシ)ビシクロ[2.2.1]ヘプタン-2-カルボキシレート(4.19g)を得た。
反応バイアルにrac-メチル(1R,2R,4R,5S)-5-((tert-ブチルジフェニルシリル)オキシ)ビシクロ[2.2.1]ヘプタン-2-カルボキシレートおよびrac-メチル(1S,2R,4R,6R)-6-((tert-ブチルジフェニルシリル)オキシ)ビシクロ[2.2.1]ヘプタン-2-カルボキシレート(500mg、1.224mmol)/THF(5mL)およびメタノール(5mL)を加えた。水酸化ナトリウム溶液(1.63mL、4.89mmol、3N)を加えた。この反応液を室温で撹拌し、最終的に50℃および60℃で加熱した。反応が完了後、反応液を塩化メチレン(100mL)およびHCl(1N、50mL)の間に分配した。有機層を硫酸ナトリウムで乾燥し、濾過し、濃縮した。得られた粗製生成物を少量の塩化メチレンに溶解し、シリカゲルカートリッジ(80g)にロードし、グラジエント(0%~50%酢酸エチル/ヘキサン)をかけて溶出した。蒸発により、rac-(1S,2R,4R,6R)-6-((tert-ブチルジフェニルシリル)オキシ)ビシクロ[2.2.1]ヘプタン-2-カルボン酸(149mg)およびrac-(1R,2R,4R,5S)-5-((tert-ブチルジフェニルシリル)オキシ)ビシクロ[2.2.1]ヘプタン-2-カルボン酸(153mg)を得た。
丸底フラスコ(100mL)にrac-(1R,2R,4R,5S)-5-((tert-ブチルジフェニルシリル)オキシ)ビシクロ[2.2.1]ヘプタン-2-カルボン酸(640mg、1.622mmol)、トリエチルアミン(0.452mL、3.24mmol)、およびジフェニルホスホリルアジド(0.438mL、2.027mmol)/トルエン(30mL)を加えた。室温で1時間撹拌後、ベンジルアルコール(1.687mL、16.22mmol)を加えた。反応液を次いで100℃で16時間加熱した。反応が完了後、次いで濃縮し、真空乾燥させた。得られた粗製生成物を少量の塩化メチレンに溶解し、シリカゲルカートリッジ(80g)にロードし、グラジエント(0%~70%酢酸エチル/ヘキサン)をかけて溶出した。蒸発により、rac-ベンジル((1R,2R,4R,5S)-5-((tert-ブチルジフェニルシリル)オキシ)ビシクロ[2.2.1]ヘプタン-2-イル)カルバメート(811mg、84%)を得た。
反応バイアルにrac-ベンジル((1R,2R,4R,5S)-5-((tert-ブチルジフェニルシリル)オキシ)ビシクロ[2.2.1]ヘプタン-2-イル)カルバメート(683mg、1.367mmol)およびTBAF(1M、THF溶液、5.47mL、5.47mmol)/THF(20mL)を加えた。16時間後、得られた粗製生成物を塩化メチレンに溶解し、シリカゲルに吸着させて固体サンプルカートリッジ(25g)にチャージし、真空乾燥させた。サンプルを次いでシリカゲルカートリッジ(40g)に溶出させ、次いでグラジエントをかけて溶出した(0%~100%酢酸エチル/ヘキサン)。蒸発により、rac-ベンジル((1R,2R,4R,5S)-5-ヒドロキシビシクロ[2.2.1]ヘプタン-2-イル)カルバメート(350mg)を得た。
丸底フラスコ(100mL)にrac-ベンジル((1R,2R,4R,5S)-5-ヒドロキシビシクロ[2.2.1]ヘプタン-2-イル)カルバメート(175mg、0.670mmol)/メタノール(20mL)を加えた。Pd-C(71.3mg、0.067mmol)を加え、水素を導入した。16時間後、反応が完了後、真空および窒素で3回パージし、セライト濾過した。蒸発により、rac-(1R,2S,4R,5R)-5-アミノビシクロ[2.2.1]ヘプタン-2-オール(66mg)を得た。これをさらに精製せずにそのまま用いた。
丸底フラスコ(25mL)にrac-(1R,2S,4R,5R)-5-アミノビシクロ[2.2.1]ヘプタン-2-オール(61.3mg、0.482mmol)および2-アミノ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キナゾリン-4-オール(50mg、0.161mmol)/NMP(1mL)を加えた。DBU(72.6μL、0.482mmol)およびBOP(142mg、0.321mmol)を加え、反応液を16時間撹拌した。TFA(500μL、6.49mmol)を次いで加え、得られた反応液をさらに4時間撹拌した。得られた反応液をメタノールで希釈し、炭酸ナトリウムで中和した。サンプルを濾過し、濃縮した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(0.05%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル: 水(0.05%トリフルオロ酢酸含有); グラジエント: 0%Bで5分間溶出後、0~30%Bで25分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、rac-(1R,2R,4S,5S)-5-((2-アミノ-7-(1H-ピラゾール-5-イル)キナゾリン-4-イル)アミノ)ビシクロ[2.2.1]ヘプタン-2-オール(22mg)を得た。

42Cの2番目の酸、rac-(1S,2R,4R,6R)-6-((tert-ブチルジフェニルシリル)オキシ)ビシクロ[2.2.1]ヘプタン-2-カルボン酸を、実施例42に記載のように処理し、表題化合物を得た。

4-クロロ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キノリン-2-アミン・HCl(30mg、0.082mmol)およびヒューニッヒ塩基(0.057mL、0.329mmol)/DMSO(0.5mL)の溶液に、rac-(1S,2S,4R,6R)-6-アミノビシクロ[2.2.1]ヘプタン-2-オール(25mg、0.197mmol)を加えた。この反応液をはじめに110℃で加熱し、次いで120℃で加熱し、得られた反応液を部分的に濃縮し、冷却した。得られた残渣をメタノール(1mL)に溶解し、濃HCl(0.05mL)を加えた。約4時間後、この反応液を濃縮し、メタノールで共沸した。得られた反応液をDMFに溶解し、シリンジフィルターで濾過した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子径: 5μm; 移動相A: 5:95 アセトニトリル: 水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(10mM酢酸アンモニウム含有); グラジエント: 5%Bで0分間溶出後、5~45%Bで20分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、rac-(1S,2S,4R,6R)-6-((2-アミノ-7-(1H-ピラゾール-5-イル)キノリン-4-イル)アミノ)ビシクロ[2.2.1]ヘプタン-2-オール(4.1mg)を得た。
反応バイアルに7-ブロモ-4-クロロキノリン-2-アミン(40mg、0.155mmol)、(2-オキサビシクロ[2.2.2]オクタン-4-イル)メタノール(66.3mg、0.466mmol)、およびカリウムt-ブトキシド(1M、THF溶液、0.39mL、0.39mmol)/DMSO(0.78mL)を加えた。このバイアルを密封し、120℃で2時間加熱した。冷却した反応液を次いでRP-HPLC(グラジエント: メタノール/水+0.1%TFA)で精製した。生成物を含むフラクションを濃縮し、4-((2-オキサビシクロ[2.2.2]オクタン-4-イル)メトキシ)-7-ブロモキノリン-2-アミンを灰白色固体として得た。HPLC RT: 0.71分; LCMS(M+H)+: 365.1(方法C)。この物質(15mg、0.041mmol)を5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール(20.03mg、0.103mmol)/ジオキサン(脱気済み、0.41mL)と合わせた。炭酸セシウム(53.8mg、0.165mmol)およびPdCl2(dppf)-CH2Cl2付加物(6.74mg、8.26μmol)を加え、得られた反応液を窒素下に置いた。この反応液を次いで95℃で1時間加熱した。冷却した反応液を酢酸/水(1:1)数滴でクエンチし、DMF(2mL)で希釈した。サンプルを次いで分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子径: 5μm; 移動相A: 5:95 アセトニトリル: 水(酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(酢酸アンモニウム含有); グラジエント: 11%Bで0分間溶出後、11~51%Bで25分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、4-((2-オキサビシクロ[2.2.2]オクタン-4-イル)メトキシ)-7-(1H-ピラゾール-3-イル)キノリン-2-アミン(3.9mg、25%)を得た。
48A. (4-ヒドロキシシクロヘキサン-1,1-ジイル)ビス(メチレン)ビス(4-メチルベンゼンスルホネート)
既知の(4-オキソシクロヘキサン-1,1-ジイル)ビス(メチレン)ビス(4-メチルベンゼンスルホネート)(1.35g、2.89mmol、ACS Med.Chem.Lett. 5(5) 609-614 (2014))/塩化メチレン(6mL)の溶液を-40度に冷却し、DIBAL-H(3.47mL、3.47mmol、1M)/ヘキサンの溶液で処理した。30分間撹拌後、得られた反応液を-20℃まで加温した。さらにDIBAL-H(0.5mL)を加え、30分間撹拌を続けた。さらにDIBAL-H(0.5mL)を加え、15分間撹拌を続けた。得られた反応液を次いでロッシェル塩溶液でクエンチし、塩化メチレンで抽出した。乾燥、濾過および蒸発により、粗製生成物(4-ヒドロキシシクロヘキサン-1,1-ジイル)ビス(メチレン)ビス(4-メチルベンゼンスルホネート、1.25g)を得た。1H NMR(400MHz、DMSO-d6)δ 7.74 (dd, J=8.3, 6.8Hz, 4H), 7.54-7.43 (m, 4H), 4.49 (d, J=4.1Hz, 1H), 3.84 (s, 2H), 3.75 (s, 2H), 3.43-3.35 (m, 1H), 2.44 (d, J=1.8Hz, 6H), 1.44 (br dd, J=9.4, 5.9Hz, 4H), 1.16-1.02 (m, 4H)
(4-ヒドロキシシクロヘキサン-1,1-ジイル)ビス(メチレン)ビス(4-メチルベンゼンスルホネート)(1.1g、2.348mmol)/DME(47.0mL)の溶液を水素化ナトリウム(0.282g、7.04mmol、60%油状物に分散)で処理し、加熱還流した。1時間後、TLCは、反応が終了したことを示した。得られた反応液を~10℃に冷却し、塩化アンモニウム水溶液でクエンチした。得られた混合物を酢酸エチル/ヘキサン(1:1)、次いでジクロロメタンで抽出した。有機抽出物を合わせて食塩水で洗浄し、乾燥し、揮散させて淡黄色油状物を得た。これをフラッシュシリカゲルクロマトグラフィー(10~40%酢酸エチル/ヘキサン)で精製した。適当なフラクションを濃縮し、(2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル4-メチルベンゼンスルホネート(600mg、2.024mmol、86%収率)を無色油状物として得た。1H NMR(400MHz、DMSO-d6)δ 7.78 (d, J=8.3Hz, 2H), 7.54-7.45 (m, 2H), 3.70 (s, 2H), 3.66 (tt, J=3.8, 1.7Hz, 1H), 3.46 (t, J=1.3Hz, 2H), 2.43 (s, 3H), 1.88-1.76 (m, 2H), 1.62-1.32 (m, 6H)
(2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル4-メチルベンゼンスルホネート(590mg、1.991mmol)/DMSO(4977μL)の溶液を、アジ化ナトリウム(233mg、3.58mmol)およびヨウ化ナトリウム(59.7mg、0.398mmol)で処理した。得られた懸濁液を窒素下に置き、90℃で終夜加熱した。得られた反応液を室温に冷却し、エーテルに注いだ。この混合物を水で2回、食塩水で1回洗浄し、乾燥し、揮散させて4-(アジドメチル)-2-オキサビシクロ[2.2.2]オクタン(295mg、1.764mmol、89%収率)を無色の油状物として得た。1H NMR(400MHz、クロロホルム-d)δ 3.82 (t, J=4.0Hz, 1H), 3.70 (t, J=1.5Hz, 2H), 3.07 (s, 2H), 2.13-2.01 (m, 2H), 1.72-1.69 (m, 1H), 1.72-1.55 (m, 6H)
4-(アジドメチル)-2-オキサビシクロ[2.2.2]オクタン(300mg、1.794mmol)/エタノール(5980μL)の溶液を窒素下に置き、Pd-C(191mg、0.179mmol、10%)で処理した。この混合物を次いで真空にし、水素を加えた。HCl(493μL、1.974mmol、4M)/ジオキサンを加え、反応液を室温で終夜撹拌した。この反応液を塩化メチレンで希釈し、少量の硫酸マグネシウムで処理した。得られた反応液を軽く撹拌し、次いで濾過した。得られた濾液をクロロホルムで希釈し、水で洗浄した。水相(pH~2)を揮散させ、カラスを得た。これをEtOH/DCM(1:1)に溶解し、揮散させて黄褐色固体(300mg)を得た。1H NMR(400MHz、DMSO-d6)δ7.92 (br s, 3H), 3.69 (t, J=3.9Hz, 1H), 3.58 (s, 2H), 3.33 (s, 2H), 1.95-1.79 (m, 2H), 1.65-1.48 (m, 6H)
7-ブロモ-4-クロロキノリン-2-アミン(150mg、0.582mmol)および(2-オキサビシクロ[2.2.2]オクタン-4-イル)メタンアミン・HCl(155mg、0.874mmol)/NMP(1456μL)の溶液をDIPEA(407μL、2.330mmol)で処理した。この反応液を窒素下に置き、135℃で加熱した。5時間後、温度を125℃に下げ、終夜撹拌を続けた。得られた反応液を室温に冷却し、DMFおよび少量の酢酸で処理し、溶液を得た。分取HPLC(グラジエント: メタノール/水+0.1%TFA)で精製し、濃縮後、N4-((2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル)-7-ブロモキノリン-2,4-ジアミン・TFA(89mg、0.187mmol、32.1%収率)を灰白色固体として得た。1H NMR(400MHz、DMSO-d6)δ 12.70 (br s, 1H), 8.24 (d, J=8.9Hz, 1H), 7.95 (br t, J=6.0Hz, 1H), 7.83 (br s, 2H), 7.71 (d, J=1.7Hz, 1H), 7.61 (dd, J=8.8, 1.6Hz, 1H), 5.94 (s, 1H), 3.74-3.61 (m, 3H), 3.08 (br d, J=6.0Hz, 2H), 1.96-1.79 (m, 2H), 1.70-1.51 (m, 6H)
5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール(18.33mg、0.094mmol)、N4-((2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル)-7-ブロモキノリン-2,4-ジアミン・TFA(18mg、0.038mmol)、炭酸セシウム(43.1mg、0.132mmol)、およびPdCl2(dppf)-CH2Cl2付加物(4.63mg、5.67μmol)/脱気済みジオキサン(378μL)の懸濁液を窒素下に置き、95℃で5時間加熱した。この反応液を室温に冷却し、酢酸水溶液でクエンチし、DMFで2mLに希釈した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(酢酸アンモニウム含有); グラジエント: 3%Bで0分間溶出後、3~43%Bで20分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSおよびUVシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、N4-((2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル)-7-(1H-ピラゾール-3-イル)キノリン-2,4-ジアミン(9.5mg、72%)を得た。
チオフェン-3-イルボロン酸(7.25mg、0.057mmol)、N4-((2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル)-7-ブロモキノリン-2,4-ジアミン・TFA(18mg、0.038mmol、from 48E)、炭酸セシウム(43.1mg、0.132mmol)、およびPdCl2(dppf)-CH2Cl2付加物(4.63mg、5.67μmol)/脱気済みジオキサン(378μL)の懸濁液を窒素下に置き、95℃で2時間加熱した。得られた反応液を室温に冷却し、酢酸水溶液でクエンチし、DMFで2mLに希釈した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(10mM酢酸アンモニウム含有); グラジエント: 13%Bで0分間溶出後、13~53%Bで20分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSおよびUVシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、N4-((2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル)-7-(チオフェン-3-イル)キノリン-2,4-ジアミン(9.7mg、69%)を得た。
50A. メチル4-((トシルオキシ)メチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート
メチル4-(ヒドロキシメチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート(0.25g、1.261mmol)/ピリジン(1.261mL)の溶液をTs-Cl(0.245g、1.286mmol)で処理し、1日間撹拌した。さらにTsCl(0.2g)を加え、さらに1日間撹拌を続けた。この反応液を撹拌しながら10%水溶液HOAc(70mL)に移した。得られた沈殿を濾過し、水、次いで10%EtOAc/ヘキサンで濯ぎ、空気乾燥し、メチル4-((トシルオキシ)メチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート(320mg、0.908mmol、72.0%収率)を白色粉末として得た。1H NMR(400MHz、クロロホルム-d)δ 7.79 (d, J=8.3Hz, 2H), 7.37 (d, J=8.0Hz, 2H), 3.67 (s, 2H), 3.65 (s, 3H), 2.48 (s, 3H), 1.82-1.74 (m, 6H), 1.49-1.40 (m, 6H)
メチル4-((トシルオキシ)メチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート(310mg、0.880mmol)、アジ化ナトリウム(172mg、2.64mmol)、およびヨウ化ナトリウム(39.6mg、0.264mmol)/DMSO(1759μL)の混合物を窒素下に置き、100℃で加熱した。得られた反応液を4時間100℃で加熱し、次いで温度を90℃に下げた。翌日、この反応液を室温に冷却し、エーテルで希釈した。得られた混合物を水で3回および食塩水で1回洗浄し、乾燥し、揮散させ、メチル4-(アジドメチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート(190mg、0.851mmol、97%収率)を無色の油状物として得た。1H NMR(400MHz、クロロホルム-d)δ 3.67 (s, 3H), 3.06 (s, 2H), 1.88-1.77 (m, 6H), 1.52-1.43 (m, 6H)
メチル4-(アジドメチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート(0.19g、0.851mmol)/エタノール(5mL)の溶液を窒素下に置き、次いでパラジウム/炭素(0.181g、0.170mmol)を加えた。この混合物を次いでHCl(0.213mL、0.851mmol、4M)/ジオキサンで処理した。得られた反応液を水素で3時間処理した。得られた反応液を塩化メチレンで希釈し、少量の硫酸マグネシウムを加えて軽く撹拌し、次いで触媒を濾過により除去した。得られた溶液を減圧濃縮し、メチル4-(アミノメチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート・HCl、0.5エタノール(215mg、0.837mmol、98%収率)をろう状の白色固体として得た。
7-ブロモ-4-クロロキノリン-2-アミン(132mg、0.511mmol)、メチル4-(アミノメチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート・HCl、0.5エタノール(210mg、0.818mmol)、およびDIEA(357μL、2.045mmol)/N-メチル-2-ピロリジノン(2045μL)の混合物を100℃で1時間、120℃で2.5時間、最後に135℃で6日間加熱した。得られた反応液を次いで冷却し、酢酸水溶液に注いだ。得られた混合物を重炭酸ナトリウム水溶液でpH~7にした。この混合物を5%EtOH/CHCl3で2回抽出し、有機抽出物を合わせて乾燥し、揮散させ、RP-HPLC(グラジエント: メタノール/水+0.1%TFA)で精製した。適当なフラクションを濃縮し、メチル4-(((2-アミノ-7-ブロモキノリン-4-イル)アミノ)メチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート・TFA、エタノール(137mg、0.237mmol、46.3%収率)を黄褐色粉末として得た。1H NMR(400MHz、DMSO-d6)δ 12.29 (s, 1H), 8.26 (d, J=9.0Hz, 1H), 7.93 (br t, J=6.1Hz, 1H), 7.73 (d, J=2.0Hz, 1H), 7.65 (br s, 1H), 7.62 (dd, J=8.9, 2.0Hz, 1H), 5.95 (s, 1H), 3.57 (s, 2H), 3.08 (br d, J=6.2Hz, 1H), 1.78-1.66 (m, 4H), 1.57-1.46 (m, 4H)
3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール(15.10mg、0.078mmol)、メチル4-(((2-アミノ-7-ブロモキノリン-4-イル)アミノ)メチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート・TFA、エタノール(18mg、0.031mmol)、PdCl2(dppf)-CH2Cl2付加物(5.08mg、6.22μmol)、および炭酸セシウム(30.4mg、0.093mmol)/脱気済みジオキサン(311μL)の混合物を水(10μL)で処理し、窒素下に置いた。得られた混合物を95℃で加熱した。この反応液を次いで室温に冷却し、酢酸水溶液でpH~5にし、濾過した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(酢酸アンモニウム含有); グラジエント: 14%Bで0分間溶出後、14~54%でB20分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、メチル4-(((2-アミノ-7-(1H-ピラゾール-3-イル)キノリン-4-イル)アミノ)メチル)ビシクロ[2.2.2]オクタン-1-カルボキシレート(11.9mg、93%)を得た。
51A. (4-ヒドロキシ-4-ビニルシクロヘキサン-1,1-ジイル)ビス(メチレン)ビス(4-メチルベンゼンスルホネート)
ビニルマグネシウムブロマイド(16.85mL、16.85mmol、1M)/THFの溶液を-78℃に冷却した。これを20分かけて(4-オキソシクロヘキサン-1,1-ジイル)ビス(メチレン)ビス(4-メチルベンゼンスルホネート)(3.93g、8.42mmol)/THF(24.07mL)の溶液で処理した。得られた反応液を2時間撹拌し、次いで0℃に加温した。この反応液を酢酸水溶液でクエンチした。得られた混合物を軽く撹拌し、次いで酢酸エチル/ヘキサン(1:1)で抽出した。有機層を食塩水で洗浄し、乾燥し、減圧濃縮し、油状物を得た。この物質を、シリカゲル(エーテル/ヘキサン=50~90%)で精製した。適当なフラクションを濃縮し、(4-ヒドロキシ-4-ビニルシクロヘキサン-1,1-ジイル)ビス(メチレン)ビス(4-メチルベンゼンスルホネート)(3.21g、6.49mmol、77%収率)を無色ガラスとして得た。これを放置して固体とした。1H NMR(400MHz、DMSO-d6)δ 7.76 (dd, J=10.9, 8.3Hz, 4H), 7.52-7.45 (m, 4H), 5.79 (dd, J=17.3, 10.7Hz, 1H), 5.05 (dd, J=17.4, 1.9Hz, 1H), 4.90 (dd, J=10.7, 1.9Hz, 1H), 4.34 (s, 1H), 3.91 (s, 2H), 3.71 (s, 2H), 3.32 (s, 2H), 2.44 (s, 3H), 2.43 (s, 3H), 1.54-1.38 (m, 2H), 1.26-1.09 (m, 6H)
(4-ヒドロキシ-4-ビニルシクロヘキサン-1,1-ジイル)ビス(メチレン)ビス(4-メチルベンゼンスルホネート)(3.2g、6.47mmol)/DME(216mL)の溶液を水素化ナトリウム(0.518g、12.94mmol)で処理し、2時間加熱還流した。この反応液を室温に冷却し、塩化アンモニウム水溶液でクエンチした。得られた混合物を酢酸エチル/ヘキサン(1:1)で抽出し、有機層を食塩水で洗浄し、乾燥し、揮散させ、およびシリカゲル(酢酸エチル/ヘキサン=10~
30%)でクロマトグラフィーを行った。適当なフラクションを濃縮し、(1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル4-メチルベンゼンスルホネート(1.83g、5.68mmol、88%収率)を無色固体として得た。1H NMR(400MHz、クロロホルム-d)δ 7.76 (d, J=8.4Hz, 2H), 7.35 (d, J=7.9Hz, 2H), 5.78 (dd, J=17.6, 10.9Hz, 1H), 5.12 (dd, J=17.6, 1.4Hz, 1H), 5.01 (dd, J=10.9, 1.4Hz, 1H), 3.70 (s, 2H), 3.68 (t, J=1.4Hz, 2H), 2.45 (s, 3H), 1.91-1.80 (m, 2H), 1.75-1.60 (m, 4H), 1.53-1.45 (m, 2H)
(1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル4-メチルベンゼンスルホネート(1.5g、4.65mmol)、アジ化ナトリウム(0.423g、6.51mmol)、およびヨウ化ナトリウム(0.139g、0.930mmol)/DMSO(2.326mL)の溶液を窒素下に置き、100℃で2.5時間加熱した。温度を90℃に下げ、終夜撹拌を続けた。得られた反応液を冷却し、エーテルで希釈した。抽出層を水で3回、および食塩水で1回洗浄した。乾燥および蒸発後、黄色油状物を得た。この物質をフラッシュシリカゲルクロマトグラフィー(酢酸エチル/ヘキサン=10~30%)で精製した。適当なフラクションを濃縮し、4-(アジドメチル)-1-ビニル-2-オキサビシクロ[2.2.2]オクタン(560mg、2.90mmol、62.3%収率)を無色の油状物として得た。1H NMR(400MHz、クロロホルム-d)δ 5.81 (dd, J=17.6, 10.9Hz, 1H), 5.15 (dd, J=17.5, 1.4Hz, 1H), 5.03 (dd, J=10.9, 1.4Hz, 1H), 3.76 (t, J=1.5Hz, 2H), 3.09 (s, 2H), 1.98-1.85 (m, 2H), 1.78-1.65 (m, 4H), 1.64-1.55 (m, 2H)
4-(アジドメチル)-1-ビニル-2-オキサビシクロ[2.2.2]オクタン(510mg、2.64mmol)/THF(17mL)-水(0.57mL)の溶液をトリフェニルホスフィン(692mg、2.64mmol)で処理し、撹拌した。この反応液を室温で少しバブリングしておき、得られた反応液を次いで50℃で加熱し、~2時間撹拌した。冷却した反応液を酢酸エチル/ヘキサン(1:1)に注いだ。この混合物を0.1M HCl水溶液で3回洗浄し、合わせた洗浄液をロータリーエバポレーターで濃縮した。エタノール、次いで塩化メチレンを蒸発させ、白色固体(630mg)を得た。1H NMR(400MHz、DMSO-d6)δ 8.01 (br s, 3H), 5.79 (dd, J=17.5, 11.0Hz, 1H), 5.10 (dd, J=17.5, 1.8Hz, 1H), 4.96 (dd, J=10.9, 1.8Hz, 1H), 3.67 (s, 2H), 2.60 (q, J=5.9Hz, 2H), 1.76-1.66 (m, 4H), 1.65-1.53 (m, 4H)
7-ブロモ-4-クロロキノリン-2-アミン(506mg、1.964mmol)、(1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メタンアミン・HCl(600mg、2.95mmol)、およびヒューニッヒ塩基(1029μL、5.89mmol)/N-メチル-2-ピロリジノン(1964μL)の溶液を脱気し、窒素下に置いた。この反応液を120℃で3時間加熱した。次に、得られた反応液をLCMSが反応完了を示すまで135℃で加熱した。粗製物質をRP-HPLC(グラジエント: メタノール/水+0.1%TFA)で精製し、7-ブロモ-N4-((1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル)キノリン-2,4-ジアミン・TFA(700mg、1.394mmol、71.0%収率)を灰白色固体として得た。1H NMR(400MHz、DMSO-d6)δ 12.33 (s, 1H), 8.24 (d, J=9.0Hz, 1H), 7.97 (br t, J=6.4Hz, 1H), 7.74 (d, J=2.0Hz, 1H), 7.69 (br s, 1H), 7.63 (dd, J=8.9, 2.0Hz, 1H), 5.94 (s, 1H), 5.79 (dd, J=17.5, 10.9Hz, 1H), 5.09 (dd, J=17.5, 1.8Hz, 1H), 4.96 (dd, J=11.0, 1.8Hz, 1H), 3.75 (s, 2H), 3.12 (d, J=6.2Hz, 2H), 1.78-1.59 (m, 8H)
7-ブロモ-N4-((1-ビニル-2-オキサビシクロ[2.2.2]オクタン-4-イル)メチル)キノリン-2,4-ジアミン・TFA(350mg、0.697mmol)/アセトン(2903μL)-水(581μL)の溶液を4-メチルモルホリンN-オキシド水溶液(490mg、2.090mmol)、次いで2.5%OsO4/t-BuOH溶液(0.4mL)で処理した。この反応液を終夜撹拌した。得られた反応液を水(~25mL)で希釈し、10%エタノール-クロロホルム、次いで酢酸エチルで抽出した。第2抽出層は、第1抽出層と同様に高いUV活性を示していたため、水相を等体積の食塩水で希釈した。次いで酢酸エチルで2回抽出した。有機抽出物を合わせて、乾燥し、濃縮して油状固体を得た。これをエーテル/塩化メチレン(1:1)でトリチュレートした。得られた固体を濾過し、同じ溶液で濯ぎ、空気乾燥し、1-(4-(((2-アミノ-7-ブロモキノリン-4-イル)アミノ)メチル)-2-オキサビシクロ[2.2.2]オクタン-1-イル)エタン-1,2-ジオール(195mg、0.462mmol、66.3%収率)を粗い灰白色固体として得た。1H NMR(400MHz、DMSO-d6)δ 8.18 (d, J=8.9Hz, 1H), 7.65 (d, J=2.0Hz, 1H), 7.60-7.47 (m, 1H), 7.45 (br d, J=8.7Hz, 1H), 7.35-6.93 (m, 2H), 5.91 (s, 1H), 4.49 (d, J=4.4Hz, 1H), 4.36 (t, J=5.1Hz, 1H), 4.25 (br d, J=5.4Hz, 1H), 3.67 (s, 2H), 3.04 (br d, J=6.1Hz, 2H), 1.70-1.49 (m, 8H)
1-(4-(((2-アミノ-7-ブロモキノリン-4-イル)アミノ)メチル)-2-オキサビシクロ[2.2.2]オクタン-1-イル)エタン-1,2-ジオール(145mg、0.343mmol)/THF(2mL)の溶液を過ヨウ素酸ナトリウム(147mg、0.687mmol)/水(1.0mL)溶液で処理した。得られた混合物を軽くソニケーションし、次いで室温で2時間撹拌した。反応液を水(~20mL)で次いで希釈し、生成物が沈殿した。この生成物を濾過し、水で濯ぎ、空気乾燥して4-(((2-アミノ-7-ブロモキノリン-4-イル)アミノ)メチル)-2-オキサビシクロ[2.2.2]オクタン-1-カルボアルデヒド(66mg、0.169mmol、49.3%収率)を灰白色固体として得た。
4-(((2-アミノ-7-ブロモキノリン-4-イル)アミノ)メチル)-2-オキサビシクロ[2.2.2]オクタン-1-カルボアルデヒド(78mg、0.200mmol)/THF(1332μL)-水(666μL)の溶液を水素化ホウ素ナトリウム(15.12mg、0.400mmol)で処理し、室温で20分撹拌した。得られた反応液を水で希釈し、窒素気流下でTHFの大半を除去した。過剰な水素化ホウ素ナトリウムは酢酸数滴を加えてクエンチした。得られた反応液を軽く撹拌し、次いで炭酸ナトリウム水溶液で塩基性にした。得られた混合物をクロロホルムで2回抽出し、有機抽出物を合わせて、乾燥し、濃縮し、(4-(((2-アミノ-7-ブロモキノリン-4-イル)アミノ)メチル)-2-オキサビシクロ[2.2.2]オクタン-1-イル)メタノール(50mg、0.127mmol、63.8%収率)をガラスとして得た。1H NMR(400MHz、DMSO-d6)δ 7.95 (d, J=8.9Hz, 1H), 7.43 (d, J=2.0Hz, 1H), 7.15 (dd, J=8.8, 2.0Hz, 1H), 6.47 (br t, J=5.7Hz, 1H), 6.05 (s, 2H), 5.79 (s, 1H), 4.48 (t, J=6.0Hz, 1H), 3.69 (s, 2H), 3.16 (d, J=5.9Hz, 2H), 2.97 (d, J=6.0Hz, 2H), 1.69-1.56 (m, 8H)
チオフェン-3-イルボロン酸(9.78mg、0.076mmol)、(4-(((2-アミノ-7-ブロモキノリン-4-イル)アミノ)メチル)-2-オキサビシクロ[2.2.2]オクタン-1-イル)メタノール(20mg、0.051mmol)、PdCl2(dppf)-CH2Cl2付加物(4.16mg、5.10μmol)、および炭酸セシウム(49.8mg、0.153mmol)/脱気済みジオキサン(510μL)の混合物を窒素下に置き、95℃で4時間加熱した。得られた反応液を冷却し、酢酸水溶液でクエンチし、濾過し、DMFで2mLに希釈した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子径: 5μm; 移動相A: 5:95 アセトニトリル: 水(10mM酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(10mM酢酸アンモニウム含有); グラジエント: 6%Bで0分間溶出後、6~46%Bで23分間溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、(4-(((2-アミノ-7-(チオフェン-3-イル)キノリン-4-イル)アミノ)メチル)-2-オキサビシクロ[2.2.2]オクタン-1-イル)メタノール(12.8mg、64%)を得た。
52A. 3-(メトキシカルボニル)ビシクロ[1.1.1]ペンタン-1-カルボン酸
丸底フラスコ(200mL)にビシクロ[1.1.1]ペンタン-1,3-ジカルボン酸(2g、12.81mmol)/メタノール(100mL)を加えた。塩化チオニル(9.35mL、128mmol)をゆっくりと加えた。16時間後、得られた反応液を濃縮し、高真空下で乾燥させた。物質をTHF(30mL)で希釈し、水酸化ナトリウム溶液(12.81mL、12.81mmol、1M)を加えた。16時間後、pHを1N HClでpH 2に調節した。得られた反応混合物を水(50mL)で次いで希釈した。水相を酢酸エチル(2x100mL)で抽出した。合わせた有機層を硫酸ナトリウムで乾燥し、濾過し、濃縮し、3-(メトキシカルボニル)ビシクロ[1.1.1]ペンタン-1-カルボン酸(2.0g)を得た。
丸底フラスコ(25mL)に3-(メトキシカルボニル)ビシクロ[1.1.1]ペンタン-1-カルボン酸(250mg、1.469mmol)/エーテル(10mL)を加えた。塩化オキサリル(0.257mL、2.94mmol)をゆっくりと加えた。最後にDMF(0.02mL)を加えた。反応が完了後、濃縮し、高真空下で乾燥し、メチル3-(クロロカルボニル)ビシクロ[1.1.1]ペンタン-1-カルボキシレート(277mg、100%)を得た。1H NMR(400MHz、クロロホルム-d)δ 3.74 (s, 3H), 2.47 (s, 6H)
丸底フラスコ(50mL)にメチル3-(クロロカルボニル)ビシクロ[1.1.1]ペンタン-1-カルボキシレート(275mg、1.46mmol)/塩化メチレン(20mL)を加えた。アンモニアを溶液に30分間バブリングした。2時間撹拌後、得られた反応液を濾過し、濃縮し、メチル3-カルバモイルビシクロ[1.1.1]ペンタン-1-カルボキシレート(113mg、46%)を得た。1H NMR(400MHz、クロロホルム-d) δ 5.90-5.33 (m, 2H), 3.80-3.60 (m, 3H), 2.33 (s, 5H)
丸底フラスコ(100mL)にメチル3-カルバモイルビシクロ[1.1.1]ペンタン-1-カルボキシレート(133mg、0.786mmol)/THF(20mL)および塩化メチレン(20mL)を加えた。ボラン-ジメチルスルフィド錯体(5Mエーテル溶液、0.943mL、4.72mmol)を加えた。16時間撹拌後、Boc無水物(0.365mL、1.572mmol)および炭酸カリウム(435mg、3.14mmol)を加えた。70時間後、得られた反応液をメタノールでクエンチした。揮発性物質を除去し、得られた残渣を高真空下で乾燥した。粗製生成物を少量の塩化メチレンに溶解し、12gのシリカゲルカートリッジにチャージし、これを次いで溶出した(酢酸エチル/ヘキサン=0%~100%)。蒸発により、tert-ブチル((3-(ヒドロキシメチル)ビシクロ[1.1.1]ペンタン-1-イル)メチル)カルバメート(75mg、42%)を得た。1H NMR(400MHz、クロロホルム-d)δ 4.49 (br s, 1H), 3.63 (d, J=5.9Hz, 2H), 3.23 (br d, J=5.5Hz, 2H), 1.64 (s, 6H), 1.47 (s, 9H), 1.19 (t, J=6.0Hz, 1H)
丸底フラスコ(50mL)にtert-ブチル((3-(ヒドロキシメチル)ビシクロ[1.1.1]ペンタン-1-イル)メチル)カルバメート(75mg、0.330mmol)およびHCl(4Nジオキサン溶液、0.412mL、1.650mmol)/塩化メチレン(10mL)を加えた。この反応液を2時間撹拌し、揮発性物質を除去し、高真空下でサンプルを乾燥し、(3-(アミノメチル)ビシクロ[1.1.1]ペンタン-1-イル)メタノール・HCl(54mg、100%)を得た。
スクリューバイアル(5mL)に4-クロロ-7-(1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ピラゾール-5-イル)キノリン-2-アミン(25mg、0.076mmol)、(3-(アミノメチル)ビシクロ[1.1.1]ペンタン-1-イル)メタノール・HCl(24.89mg、0.152mmol)、およびヒューニッヒ塩基(80μL、0.456mmol)/NMP(1mL)を加えた。この反応液を120℃で16時間加熱した。冷却した反応液を濃縮し、真空乾燥させた。TFA(500μL、6.49mmol)を加え、撹拌を1時間続けた。得られた反応液を次いで濃縮し、高真空下で乾燥した。得られた反応混合物をDMF:酢酸(1:1、1mL)で希釈した。粗製物質を分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子径: 5μm; 移動相A: 5:95 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); 移動相B: 95:5 アセトニトリル: 水(0.1%トリフルオロ酢酸含有); グラジエント: 0%Bで0分間溶出後、0~40%Bで20分かけて溶液し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥した。この物質をさらに分取LC/MS(条件: カラム: XBridge C18、200mmx19mm、粒子: 5μm; 移動相A: 5:95 アセトニトリル: 水(酢酸アンモニウム含有); 移動相B: 95:5 アセトニトリル: 水(酢酸アンモニウム含有); グラジエント: 0%Bで0分溶液後、0~30%Bで25分かけて溶出し、次いで100%Bで0分間溶出; 流速: 20mL/分; カラム温度: 25℃)で精製した。フラクションはMSシグナルで判定して回収した。所望の生成物を含むフラクションを合わせて遠心エバポレーターで乾燥し、(3-(((2-アミノ-7-(1H-ピラゾール-5-イル)キノリン-4-イル)アミノ)メチル)ビシクロ[1.1.1]ペンタン-1-イル)メタノール(3.8mg、14%)を得た。













Claims (18)
- 式(I):
Wは、独立して、R6、-Y-R6、-Q-R6、および-Q-Y-R6から選択され;
Qは、独立して、NR5、CHR5、O、およびSから選択され;
Yは、独立して、C1-10アルキレン、C2-10アルケニレン、およびC2-10アルキニレンから選択され、それらは各々0~4個のRaで置換され、および/または各々適宜
(i)O;または
(ii)N(Rb)の1つが途中に含まれていてもよく;
X1は、独立して、NまたはCR1であり;
X2は、独立して、NまたはCR2であり;
X3は、独立して、NまたはCR3であり;
X4は、独立して、NまたはCR4であり;
ただし、X1、X3およびX4のうちの2個未満がNであり;
R1およびR3は、それぞれ独立して、H、ハロゲン、シアノ、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、およびC1-4ハロアルコキシから選択され;
R2およびR4は、それぞれ独立して、H、ハロゲン、シアノ、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、C1-4ハロアルコキシ、N(C1-4アルキル)2、および5個の環原子を含み、ここで1~4個の環原子がそれぞれ独立してN、NH、N(C1-4アルキル)、O、およびSから選択され、ヘテロアリール部分が0~3個のRdで置換された-(C0-3アルキレン)-ヘテロアリールから選択され;
R5は、独立して、HまたはC1-4アルキルであり;
R6は、独立して、5~12個の環原子を含む二環または三環シクロアルキルまたはシクロアルケニルであり、ここでシクロアルキルまたはシクロアルケニルはスピロ環であり得るか、または-CR8R9-、-(CR8R9)2-、および-C(=O)-から選択される1個または2個の架橋リンカーを含有し得て、シクロアルキルまたはシクロアルケニル部分は0~4個のReで置換されており;
あるいは、R6は、独立して、5~12個の環原子を含み、ここで1~4個の環原子がそれぞれ独立してN、N(Rb)、O、およびSから選択される二環または三環ヘテロシクロアルキルであり、上記ヘテロシクロアルキルはスピロ環であり得るか、または-CR8R9-、-O-、-(CR8R9)2-、-OCR8R9-、および-CR8R9O-、および-C(=O)-から選択される架橋リンカーを含有し得て、ここでヘテロシクロアルキルは、0~4個のReで置換されており;
R7は、独立して、5個の環原子を含み、ここで1~4個の環原子がそれぞれ独立してN、NH、N(C1-4アルキル)、O、およびSから選択され、0~3個のRdで置換されたヘテロアリールであり;
R8およびR9は、それぞれ独立して、H、OH、ハロゲン、C1-4アルキルおよびC1-4アルコキシから選択され;
Raは、独立して、F、OH、シアノ、C1-4アルコキシ、C1-4ハロアルキル、C1-4ハロアルコキシ、および0~1個のRcで置換されたC1-4アルキルから選択され;
Rbは、独立して、H、0~1個のOHで置換されたC1-4アルキル、-C(O)(C1-4アルキル)、および-C(O)O(C1-4アルキル)から選択され;
Rcは、独立して、OH、CONH2およびC1-4アルコキシから選択され;
Rdは、独立して、ハロゲン、OH、シアノ、C1-4アルコキシ、C1-4アルコキシ、C1-4ハロアルキル、C1-4ハロアルコキシ、-C(O)O(C1-4アルキル)、NH2、N(C1-4アルキル)2、-C(O)NH2、-C(O)N(C1-4アルキル)2、C2-6アルケニル、C2-6アルキニル、および0~2個のRaで置換されたC1-6アルキルから選択され;および
Reは、独立して、オキソまたはRdである]
の化合物またはその立体異性体、互変異性体、または医薬的に許容される塩。 - 式中、
Qが、それぞれ独立して、NH、N(C1-4アルキル)、CH2、およびOから選択され;
Yが、それぞれ独立して、C1-10アルキレン、C2-6アルケニレン、およびC2-6アルキニレンから選択され、それらは各々0~4個のRaで置換されており;
R2が、独立して、H、ハロゲン、シアノ、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、およびC1-4ハロアルコキシから選択され;
R4が、独立して、H、ハロゲン、シアノ、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、C1-4ハロアルコキシ、N(C1-4アルキル)2、およびN、NH、O、ならびにSからそれぞれ独立して選択される1~2個の環原子を含む5員ヘテロアリールから選択され;
R6が、独立して、5~10個の環原子を含む、二環または三環シクロアルキルまたはシクロアルケニルであり、ここでシクロアルキルまたはシクロアルケニルはスピロ環であり得るか、または-CH2-および-CH2CH2-から選択される1個または2個の架橋リンカーを含有し得て、シクロアルキルまたはシクロアルケニル部分は0~4個のReで置換されており;
あるいは、R6が、独立して、5~10個の環原子を含み、ここで1~2個の環原子はそれぞれ独立して、N、N(Rb)、O、およびSから選択される二環ヘテロシクロアルキルであり、ここで二環式環は、スピロ環であり得るか、または-CH2-、-O-、-CH2CH2-、-OCH2-、および-CH2O-から選択される架橋リンカーを含有し得て、ここで上記ヘテロシクロアルキルは、0~4個のReで置換されており;
R7が、独立して、N、NH、O、およびSからそれぞれ独立して選択される1~2個の環原子を含む、5員ヘテロアリールであり;および
R8およびR9が、それぞれ独立して、HまたはC1-4アルキルである、請求項1の化合物またはその立体異性体、互変異性体、または医薬的に許容される塩。 - 化合物が式(II):
Wは、独立して、R6-NH-R6、または-NH-CH2-R6であり;
X2は、独立して、NまたはCR2であり;
R1、R2、R3およびR4は、それぞれ独立して、H、ハロゲンおよびC1-4アルキルから選択され;
R6は、独立して、5~10個の環原子を含む二環または三環シクロアルキルであり、ここでシクロアルキルはスピロ環であり得るか、または-CH2-および-CH2CH2-から選択される1個または2個の架橋リンカーを含有し得て、上記シクロアルキルは0~2個のReで置換されており;
あるいは、R6は、独立して、5~10個の環原子を含み、ここで1~2個の環原子はそれぞれ独立してN、N(Rb)、およびOから選択される二環ヘテロシクロアルキルであり、ここで二環式環は、スピロ環であり得るか、または-CH2-、-O-、-CH2CH2-、-OCH2-、および-CH2O-から選択される架橋リンカーを含有し得て、上記ヘテロシクロアルキルは0~2個のReで置換されており;
Rbは、独立して、H、C1-4アルキル、および-C(O)O(C1-4アルキル)から選択され;および
Reは、独立して、オキソ、ハロゲン、シアノ、OH、CH2OH、C1-4アルコキシ、C1-4ハロアルキル、N(C1-4アルキル)2、-C(O)NH2、-C(O)O(C1-4アルキル)、C2-6アルケニル、および0~2個のC1-4アルコキシで置換されたC1-4アルキルから選択される]
である、請求項1または請求項2の化合物またはその立体異性体、互変異性体、または医薬的に許容される塩。 - 式中、
Wが、それぞれ独立して、-NH-R6、-NH-CH2-R6、
R6が、独立して
- 式中、
Wが、それぞれ独立して、-NH-R6、
R6が、独立して、
- 化合物が式(III):
X2は、独立して、NまたはCR2であり;
R1、R2、R3およびR4は、それぞれ独立して、H、ハロゲンおよびC1-4アルキルから選択され;
R6は、独立して、5~10個の環原子を含む二環または三環シクロアルキルであり、ここでシクロアルキルはスピロ環であり得るか、または-CH2-および-CH2CH2-から選択される1個または2個の架橋リンカーを含有し得て、上記シクロアルキルは0~2個のReで置換されており;
あるいは、R6は、独立して、5~10個の環原子を含み、ここで1~2個の環原子がそれぞれ独立してNおよびOから選択される二環ヘテロシクロアルキルであり、この二環式環は、スピロ環であり得るか、または-CH2-、-O-、および-CH2CH2-から選択される架橋リンカーを含有し得て、上記ヘテロシクロアルキルは0~2個のReで置換されており;および
Reは、独立して、オキソ、OH、CH2OH、C1-4アルコキシ、および0~2個のC1-4アルコキシで置換されたC1-4アルキルから選択される]
である、請求項1~4のいずれか一項の化合物またはその立体異性体、互変異性体、または医薬的に許容される塩。 - 式中、
X2が、独立して、NまたはCHであり;
R1、R3およびR4が、Hであり;および
R6が、独立して、
- 以下の化合物:
- 請求項1~8のいずれか一項に記載の化合物またはその医薬的に許容される塩、および1つ以上の医薬的に許容される賦形剤を含む、医薬組成物。
- 薬剤として用いるための、請求項1~8のいずれか一項に記載の化合物またはその医薬的に許容される塩、または請求項9に記載の医薬組成物。
- がんの治療に用いるための、請求項1~8のいずれか一項に記載の化合物またはその医薬的に許容される塩、または請求項9に記載の医薬組成物。
- がんが、急性骨髄性白血病、副腎皮質がん、カポジ肉腫、リンパ腫、肛門がん、虫垂癌、奇形腫/ラブドイド腫瘍、基底細胞がん、胆管がん、膀胱がん、骨がん、脳がん、乳がん、気管支腫瘍、カルチノイド腫瘍、心臓腫瘍、子宮頸がん、脊索腫、慢性リンパ性白血病、慢性骨髄増殖性腫瘍、結腸がん、結腸直腸癌、頭蓋咽頭腫、子宮内膜がん、上衣腫、食道がん、鼻腔神経芽細胞腫、ユーイング肉腫、眼腫瘍、卵管がん、胆嚢がん、消化管カルチノイド腫瘍、消化管間質腫瘍、胚細胞腫瘍、有毛細胞白血病、頭頸部がん、心臓腫瘍、肝臓がん、下咽頭がん、膵臓がん、腎臓がん、喉頭がん、慢性骨髄性白血病、口唇および口腔がん、肺がん、黒色腫、メルケル細胞がん、中皮腫、口唇がん、口唇部がん、骨肉腫、卵巣がん、陰茎がん、咽頭がん、前立腺がん、直腸がん、唾液腺がん、皮膚がん、小腸がん、軟部組織肉腫、精巣がん、喉のがん、甲状腺がん、尿道がん、子宮がん、腟がん、および外陰がんから選択される、請求項11に記載の化合物またはその医薬的に許容される塩、または医薬組成物。
- がんが、乳がん、結腸がん、直腸がん、結腸直腸がん、膵臓がん、および前立腺がんから選択される、請求項11に記載の化合物またはその医薬的に許容される塩、または医薬組成物。
- がんが、ホルモン受容体陽性乳がん、マイクロサテライト安定性結腸がんまたは直腸がん、膵臓がんおよび前立腺がんから選択される、請求項11に記載の化合物またはその医薬的に許容される塩、または医薬組成物。
- 化合物が、1つ以上の別のがん治療と組み合わせて投与される、請求項10~14のいずれか一項に記載の化合物またはその医薬的に許容される塩、または医薬組成物。
- 1つ以上の別のがん治療に、手術、放射線療法、化学療法、毒素治療、免疫療法、凍結療法または遺伝子治療、またはその組み合わせが含まれる、請求項15に記載の化合物またはその医薬的に許容される塩、または医薬組成物。
- 別のがん治療に、ニボルマブ、ペムブロリズマブ、PDR001、MEDI-0680、セミピリマブ、JS001、BGB-A317、INCSHR1210、TSR-042、GLS-010、AM-0001、STI-1110、AGEN2034、MGD013、IBI308、BMS-936559、アテゾリズマブ、デュルバルマブ、アベルマブ、STI-1014、CX-072、LY3300054、CK-301、ウレルマブ、PF-05082566、MEDI6469、TRX518、バルリルマブ、CP-870893、BMS-986016、MGA271、リリルマブ、IPH2201、エマクツズマブ、INCB024360、ガルニセルチブ、ウロクプルマブ、BKT140、バビツキシマブ、CC-90002、ベバシズマブ、MNRP1685A、イピリムマブ、MK-1308、AGEN-1884、およびトレメリムマブから選択される1つ以上の薬剤が含まれる、請求項15に記載の化合物またはその医薬的に許容される塩、または医薬組成物。
- 別のがん治療に、ニボルマブ、イピリムマブ、ペムブロリズマブ、アテゾリズマブ、デュルバルマブおよびアベルマブから選択される1つ以上の薬剤が含まれる、請求項15に記載の化合物またはその医薬的に許容される塩、または医薬組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962791979P | 2019-01-14 | 2019-01-14 | |
| US62/791,979 | 2019-01-14 | ||
| PCT/US2020/013264 WO2020150115A1 (en) | 2019-01-14 | 2020-01-13 | Nlrp3 modulators |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2022517109A JP2022517109A (ja) | 2022-03-04 |
| JPWO2020150115A5 JPWO2020150115A5 (ja) | 2022-09-05 |
| JP7335341B2 true JP7335341B2 (ja) | 2023-08-29 |
Family
ID=69469246
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021540533A Active JP7335341B2 (ja) | 2019-01-14 | 2020-01-13 | Nlrp3モジュレーター |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US12391675B2 (ja) |
| EP (1) | EP3911641A1 (ja) |
| JP (1) | JP7335341B2 (ja) |
| KR (1) | KR102865929B1 (ja) |
| CN (1) | CN113286786A (ja) |
| WO (1) | WO2020150115A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2022517112A (ja) * | 2019-01-14 | 2022-03-04 | イネイト・テューマー・イミュニティ・インコーポレイテッド | Nlrp3モジュレーター |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2022340126A1 (en) * | 2021-08-31 | 2024-04-11 | Japan Tobacco Inc. | 6-aminopyrazolopyrimidine compound and pharmaceutical use thereof |
| CN115282280A (zh) * | 2022-08-12 | 2022-11-04 | 中国科学技术大学 | TGF-β1信号抑制剂的新用途 |
| CN120752239A (zh) * | 2023-02-24 | 2025-10-03 | 日本烟草产业株式会社 | 经取代的吡唑并嘧啶化合物及其医药用途 |
| CN120731207A (zh) * | 2023-02-24 | 2025-09-30 | 日本烟草产业株式会社 | 6-烷氧基吡唑并嘧啶化合物及其药学用途 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017184746A1 (en) | 2016-04-19 | 2017-10-26 | Ifm Therapeutics, Inc | Nlrp3 modulators |
| WO2018152396A1 (en) | 2017-02-17 | 2018-08-23 | Innate Tumor Immunity, Inc. | Substituted imidazo-quinolines as nlrp3 modulators |
| JP2020527139A (ja) | 2017-07-14 | 2020-09-03 | イネイト・テューマー・イミュニティ・インコーポレイテッドInnate Tumor Immunity, Inc. | Nlrp3モジュレーター |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HRP20010551B1 (hr) | 1998-12-23 | 2013-05-31 | Pfizer Inc. | Humana monoklonalna antitijela za ctla-4 |
| CA2589418A1 (en) | 1999-08-24 | 2001-03-01 | Medarex, Inc. | Human ctla-4 antibodies and their uses |
| US7927613B2 (en) | 2002-02-15 | 2011-04-19 | University Of South Florida | Pharmaceutical co-crystal compositions |
| CN105330741B (zh) | 2005-07-01 | 2023-01-31 | E.R.施贵宝&圣斯有限责任公司 | 抗程序性死亡配体1(pd-l1)的人单克隆抗体 |
| KR101562580B1 (ko) | 2007-06-18 | 2015-10-22 | 머크 샤프 앤 도메 비.브이. | 사람 프로그램된 사멸 수용체 pd-1에 대한 항체 |
| BRPI0917592B1 (pt) | 2008-12-09 | 2021-08-17 | Genentech, Inc | Anticorpo anti-pd-l1, composição, artigos manufaturados e usos de uma composição |
| RS60033B1 (sr) | 2009-11-24 | 2020-04-30 | Medimmune Ltd | Ciljano vezujući agensi usmereni na b7-h1 |
| WO2012145493A1 (en) | 2011-04-20 | 2012-10-26 | Amplimmune, Inc. | Antibodies and other molecules that bind b7-h1 and pd-1 |
| US20140243302A1 (en) | 2011-06-27 | 2014-08-28 | Merck Sharp & Dohme Corporation | Bridged bicyclic compounds for the treatment of bacterial infections |
| EP2785375B1 (en) | 2011-11-28 | 2020-07-22 | Merck Patent GmbH | Anti-pd-l1 antibodies and uses thereof |
| CA3213528A1 (en) | 2012-05-15 | 2013-11-21 | Bristol-Myers Squibb Company | Cancer immunotherapy by disrupting pd-1/pd-l1 signaling |
| JP2015519375A (ja) | 2012-05-31 | 2015-07-09 | ソレント・セラピューティクス・インコーポレイテッドSorrento Therapeutics, Inc. | Pd−l1に結合する抗原結合蛋白質 |
| HRP20210122T1 (hr) | 2013-05-02 | 2021-04-16 | Anaptysbio, Inc. | Protutijela usmjerena protiv programirane smrti-1 (pd-1) |
| PT2996473T (pt) | 2013-05-18 | 2019-11-18 | Univ California | Composições e métodos para a ativação da sinalização dependente do ''estimulador de genes de interferão'' |
| US9676853B2 (en) | 2013-05-31 | 2017-06-13 | Sorrento Therapeutics, Inc. | Antigen binding proteins that bind PD-1 |
| WO2015035606A1 (en) | 2013-09-13 | 2015-03-19 | Beigene, Ltd. | Anti-pd1 antibodies and their use as therapeutics and diagnostics |
| SG10201804945WA (en) | 2013-12-12 | 2018-07-30 | Shanghai hengrui pharmaceutical co ltd | Pd-1 antibody, antigen-binding fragment thereof, and medical application thereof |
| TWI681969B (zh) | 2014-01-23 | 2020-01-11 | 美商再生元醫藥公司 | 針對pd-1的人類抗體 |
| JOP20200094A1 (ar) | 2014-01-24 | 2017-06-16 | Dana Farber Cancer Inst Inc | جزيئات جسم مضاد لـ pd-1 واستخداماتها |
| SG10201913297TA (en) | 2015-03-13 | 2020-02-27 | Cytomx Therapeutics Inc | Anti-pdl1 antibodies, activatable anti-pdl1 antibodies, and methods of use thereof |
| CN107849144B (zh) | 2015-05-29 | 2021-09-17 | 艾吉纳斯公司 | 抗-ctla-4抗体及其使用方法 |
| WO2017024465A1 (en) | 2015-08-10 | 2017-02-16 | Innovent Biologics (Suzhou) Co., Ltd. | Pd-1 antibodies |
| AR105654A1 (es) | 2015-08-24 | 2017-10-25 | Lilly Co Eli | Anticuerpos pd-l1 (ligando 1 de muerte celular programada) |
| HK1257840A1 (zh) | 2015-09-01 | 2019-11-01 | Agenus Inc. | 抗-pd-1抗体及其使用方法 |
| WO2017132827A1 (en) | 2016-02-02 | 2017-08-10 | Innovent Biologics (Suzhou) Co., Ltd. | Pd-1 antibodies |
| WO2017132825A1 (zh) | 2016-02-02 | 2017-08-10 | 华为技术有限公司 | 确定发射功率的方法、用户设备和基站 |
| CN113286787A (zh) * | 2019-01-14 | 2021-08-20 | 先天肿瘤免疫公司 | Nlrp3调节剂 |
| EP3911417B1 (en) * | 2019-01-14 | 2022-10-26 | Innate Tumor Immunity, Inc. | Heterocyclic nlrp3 modulators , for use in the treatment of cancer |
-
2020
- 2020-01-13 EP EP20703911.6A patent/EP3911641A1/en active Pending
- 2020-01-13 US US17/422,512 patent/US12391675B2/en active Active
- 2020-01-13 WO PCT/US2020/013264 patent/WO2020150115A1/en not_active Ceased
- 2020-01-13 KR KR1020217025495A patent/KR102865929B1/ko active Active
- 2020-01-13 JP JP2021540533A patent/JP7335341B2/ja active Active
- 2020-01-13 CN CN202080008819.6A patent/CN113286786A/zh active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017184746A1 (en) | 2016-04-19 | 2017-10-26 | Ifm Therapeutics, Inc | Nlrp3 modulators |
| WO2018152396A1 (en) | 2017-02-17 | 2018-08-23 | Innate Tumor Immunity, Inc. | Substituted imidazo-quinolines as nlrp3 modulators |
| JP2020527139A (ja) | 2017-07-14 | 2020-09-03 | イネイト・テューマー・イミュニティ・インコーポレイテッドInnate Tumor Immunity, Inc. | Nlrp3モジュレーター |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2022517112A (ja) * | 2019-01-14 | 2022-03-04 | イネイト・テューマー・イミュニティ・インコーポレイテッド | Nlrp3モジュレーター |
| JP7506080B2 (ja) | 2019-01-14 | 2024-06-25 | イネイト・テューマー・イミュニティ・インコーポレイテッド | Nlrp3モジュレーター |
Also Published As
| Publication number | Publication date |
|---|---|
| US12391675B2 (en) | 2025-08-19 |
| EP3911641A1 (en) | 2021-11-24 |
| KR20210116529A (ko) | 2021-09-27 |
| US20220089572A1 (en) | 2022-03-24 |
| KR102865929B1 (ko) | 2025-09-29 |
| CN113286786A (zh) | 2021-08-20 |
| WO2020150115A1 (en) | 2020-07-23 |
| JP2022517109A (ja) | 2022-03-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7335341B2 (ja) | Nlrp3モジュレーター | |
| CN110325534B (zh) | 作为nlrp3调节剂的取代的咪唑并喹啉 | |
| KR102768379B1 (ko) | 이미다조[4,5-c]퀴놀린 유래 NLRP3-조정제 | |
| JP7433291B2 (ja) | イミダゾ[4,5-c]キノリン誘導体のNLRP3モジュレーター | |
| JP7159282B2 (ja) | Nlrp3モジュレーター | |
| JP7506079B2 (ja) | がんの治療に用いるためのヘテロ環nlrp3モジュレーター | |
| JP7373571B2 (ja) | がん治療に用いるためのnlrp3モジュレーターとしての置換キナゾリン | |
| JP7506080B2 (ja) | Nlrp3モジュレーター | |
| JP7573622B2 (ja) | Nlrp3モジュレーター | |
| NZ757257B2 (en) | Substituted imidazo-quinolines as nlrp3 modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220826 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20220826 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230808 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20230810 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230817 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7335341 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |