JP2023525320A - B7‐h3抗体薬物複合体の単独使用又は併用のための方法 - Google Patents
B7‐h3抗体薬物複合体の単独使用又は併用のための方法 Download PDFInfo
- Publication number
- JP2023525320A JP2023525320A JP2022568560A JP2022568560A JP2023525320A JP 2023525320 A JP2023525320 A JP 2023525320A JP 2022568560 A JP2022568560 A JP 2022568560A JP 2022568560 A JP2022568560 A JP 2022568560A JP 2023525320 A JP2023525320 A JP 2023525320A
- Authority
- JP
- Japan
- Prior art keywords
- adc
- weeks
- dose
- once
- administration
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2827—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against B7 molecules, e.g. CD80, CD86
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6843—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a material from animals or humans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2818—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD28 or CD152
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
- A61K2039/507—Comprising a combination of two or more separate antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/54—Medicinal preparations containing antigens or antibodies characterised by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/94—Stability, e.g. half-life, pH, temperature or enzyme-resistance
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Zoology (AREA)
- Mycology (AREA)
- Endocrinology (AREA)
- Cell Biology (AREA)
- Microbiology (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Saccharide Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Steroid Compounds (AREA)
Abstract
Description
本出願は、米国特許出願第63/023,495号(2020年5月12日出願;係属中)、及び米国特許出願第63/180,795号(2021年4月28日出願;係属中)に対する優先権を主張するものであり、上記特許出願は、参照によりその全体が本出願に援用される。
本出願は、連邦規則法典第37巻第1.821節以下による配列表を含み、この配列表は、ASCIIフォーマットで電子出願されており、参照によりその全体が、あらゆる目的のために本出願に援用される。2021年5月5日に作成された上記配列表のASCIIコピーは、名称がMAC‐0112‐PC_SL.txtであり、サイズが49,512バイトである。
B7‐H3はB7‐CD28スーパーファミリーのメンバーであり、抗原提示細胞上で発現される。B7‐H3は、主要なヒト型が2つの細胞外タンデムIgV‐IgCドメイン(即ちIgV‐IgC‐IgV‐IgC)を含有する点において独特である(非特許文献1)。4免疫グロブリン細胞外ドメイン変異体(「4Ig‐B7‐H3」)は、最初はIgドメイン(IgV‐IgC、例えばNCBI配列NP_079516を参照)を2つだけ含むと考えられていたが、上記タンパク質のより一般的なヒト型であることが同定され、発見された(非特許文献2;例えばNCBI配列NP_001019907も参照)。B7‐H3 mRNA発現は、心臓、腎臓、精巣、肺、肝臓、膵臓、前立腺、結腸及び骨芽細胞において発見されている(非特許文献1)。タンパク質レベルでは、B7‐H3はヒトの肝臓、肺、膀胱、精巣、前立腺、乳房、膵臓及びリンパ器官に見られる(非特許文献3)。
B7‐H3は、多様な癌(例えば神経芽腫、胃癌、卵巣癌、非小細胞肺癌等、例えば非特許文献5を参照)及び培養された癌幹様細胞上で発現される。いくつかの独立した研究は、ヒト悪性腫瘍細胞がB7‐H3タンパク質の発現の著しい上昇を呈すること、及びこの著しい発現が、疾患の重篤度の上昇に関連すること(非特許文献6、7)を示しており、これはB7‐H3が腫瘍により、免疫回避経路として悪用されていることを示唆している(非特許文献3)。
免疫応答は、共刺激及び共阻害性リガンドと、「免疫チェックポイント」と呼ばれることが多い受容体とによって、厳密に制御される(非特許文献11、12)。これらの分子は、免疫応答を調節して感染症及び癌に対する保護を提供するポジティブ及びネガティブシグナルの平衡ネットワークを提供する。一部の癌細胞は、T細胞が持続的な抗原及び/又は炎症性シグナルにさらされるT細胞枯渇の状態を引き起こすことによって、免疫系から逃れることができる(非特許文献13)。T細胞枯渇に関与する2つの免疫チェックポイント分子、即ちプログラム細胞死‐1(Programmed Death-1:「PD‐1」)及びリンパ球活性化遺伝子3(Lymphocyte Activation Gene 3:LAG‐3)(非特許文献14)について、以下で更に詳細に説明する。
(A)B7‐H3‐ADC;及び
(B)PD‐1結合分子
を投与するステップを含む、上述のような方法の実施形態であって、上記方法は、B7‐H3‐ADCを上記被験者に、約0.5mg/kg~約5mg/kgの用量で3週間に1回投与するステップを含む、実施形態を提供する。
(A)B7‐H3‐ADC;及び
(B)PD‐1結合分子
を投与するステップを含む、上述のような方法の実施形態であって、上記方法は、B7‐H3‐ADCを上記被験者に、約0.5mg/kg~約5mg/kgの用量で4週間に1回投与するステップを含む、実施形態を提供する。
Ab‐(LM)m‐(D)n
で表される、上述のような方法の実施形態を提供し、ここで:
Abは、B7‐H3に結合し、かつ:
(i)その可変軽鎖(VL)ドメイン中に、CDRL1配列RASESIYSYLA(配列番号39)、CDRL2配列NTKTLPE(配列番号40)、及びCDRL3配列QHHYGTPPWT(配列番号41);並びに
(ii)その可変重鎖(VH)ドメイン中に、CDRH1配列SYGMS(配列番号42)、CDRH2配列TINSGGSNTYY PDSLKG(配列番号43)、及びCDRH3配列HDGGAMDY(配列番号44)
を含む、ヒト化B7‐H3抗体又はそのB7‐H3結合断片であり;
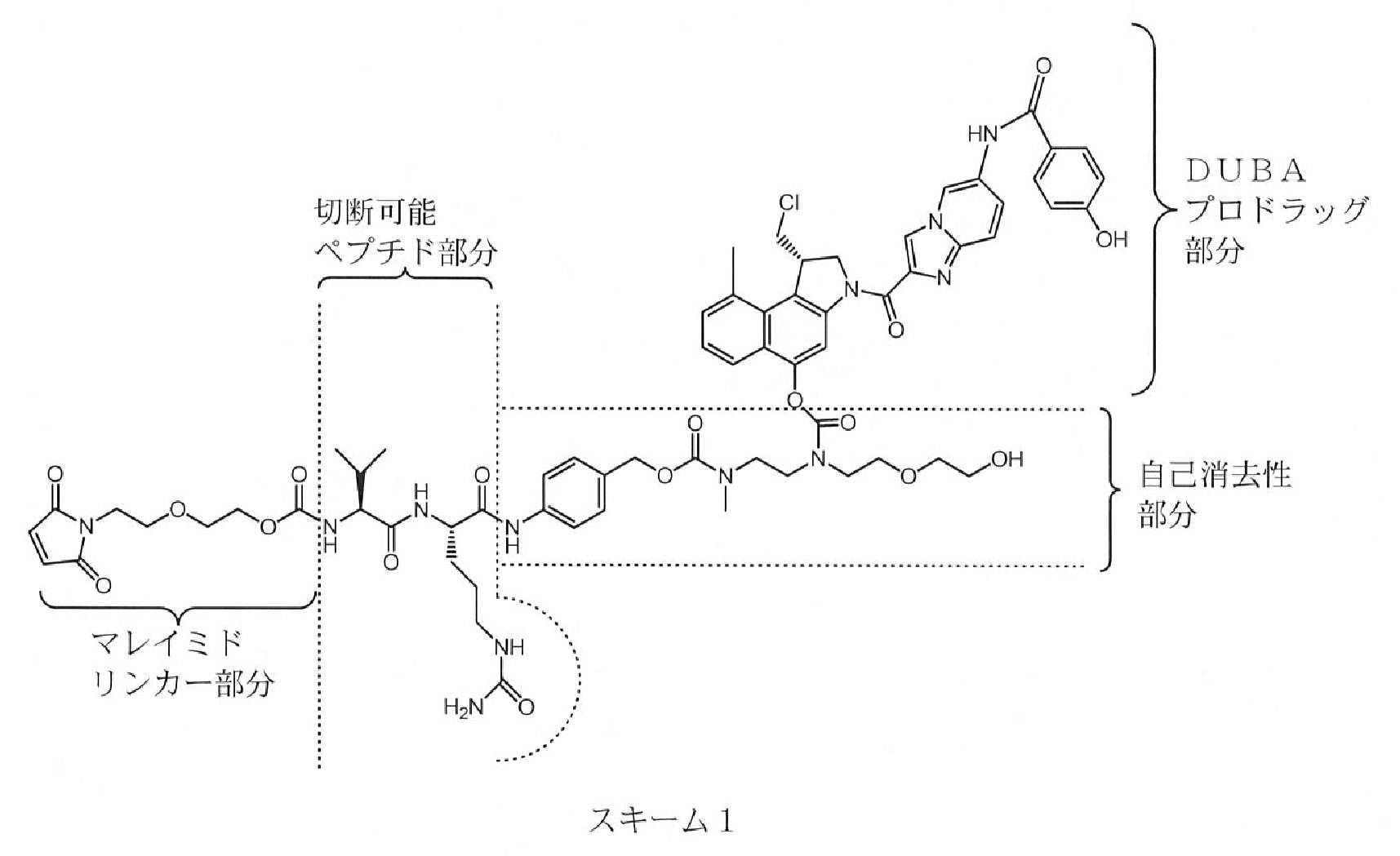
Dは、細胞傷害性デュオカルマイシン部分であり;
LMは、Ab及びDを共有結合させる少なくとも1つの結合又はリンカー分子(Linker Molecule)を含み;
mは0~nの整数であって、上記B7‐H3‐ADCの結合又はリンカー分子の数を表すが、LMが1つの結合である場合を除いてmは0ではなく;
nは1~10の整数であって、上記B7‐H3‐ADCに共有結合した上記細胞傷害性デュオカルマイシン部分の数を表す。
(I)配列番号17のアミノ酸配列を含むヒト化VLドメイン、及び
(II)配列番号18のアミノ酸配列を含むヒト化VHドメイン
を含む、上述のような方法の実施形態を提供する。
[V‐(W)k‐(X)1‐A]
で表され、従って上記B7‐H3‐ADCが以下の式:
Ab‐[V‐(W)k‐(X)1‐A]‐D
で表され、ここで:
Vは切断可能なリンカーであり;
(W)k‐(X)1‐Aは、l,(4+2n)消去によって自己消去する、細長い自己消去性スペーサ系であり;
W及びXはそれぞれl,(4+2n)電子カスケードスペーサであり、同一であるか又は異なっており;
Aは、式(Y)m(ここでYはl,(4+2n)電子カスケードスペーサである)のスペーサ基、又は式Uの基であり、環化除去スペーサであり;
k、1及びmは独立して、0~5(両端を含む)の整数であり;
nは0~10(両端を含む)の整数であり;
ただし:
Aが(Y)mである場合、k+l+m≧1であり;
k+l+m=lである場合、n>lであり;
AがUである場合、k+1≧1であり、
W、X及びYは独立して、以下の式:


Qは‐R5C=CR6‐、S、O、NR5、‐R5C=N‐又は‐N=CR5‐であり;
PはNR7、O又はSであり;
a、b及びcは独立して、0~5(両端を含む)の整数であり;
I、F及びGは独立して、式:

R1、R2、R3、R4、R5、R6、R7、R8及びR9は独立して、H、C1‐6アルキル、C3‐20ヘテロシクリル、C5‐20アリール、C1‐6アルコキシ、ヒドロキシ(OH)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)及びリン酸塩(OP(=O)(ORx)2)を表し、ここで:
Rx、Rx 1及びRx 2は独立して、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され;
上記置換基R1、R2、R3、R4、R5、R6、R7、R8又はR9のうちの2つ以上は任意に、互いに接続されて1つ以上の脂肪族又は芳香族環式構造を形成し;
Uは、式:

a、b及びcは独立して、0又は1の整数となるよう選択され;
ただしa+b+c=2又は3であり;
R1及び/又はR2は独立して、H、C1‐6アルキルを表し、上記アルキルは任意に、以下の基:ヒドロキシ(OH)、エーテル(ORx)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)、及びリン酸塩(OP(=O)(ORx)2)のうちの1つ以上によって置換され、ここでRx、Rx 1及びRx 2は、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され;
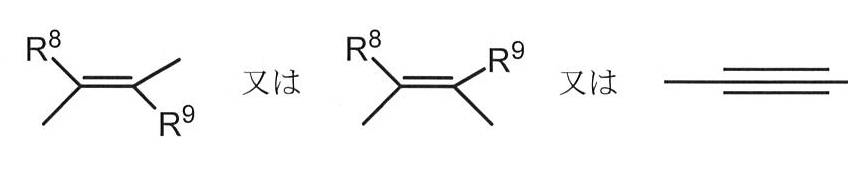
R3、R4、R5、R6、R7及びR8は独立して、H、C1‐6アルキル、C3‐20ヘテロシクリル、C5‐20アリール、C1‐6アルコキシ、ヒドロキシ(OH)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)、及びリン酸塩(OP(=O)(ORx)2)を表し、ここでRx、Rx 1及びRx 2は、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され、上記置換基R1、R2、R3、R4、R5、R6、R7、又はR8のうちの2つ以上は任意に、互いに接続されて1つ以上の脂肪族又は芳香族環式構造を形成する、上述のような方法の実施形態を提供する。
(1)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(2)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(3)p‐アミノシンナミルオキシカルボニル;
(4)p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(5)p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
(6)p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
(7)p‐アミノフェニルペンタジエニルオキシカルボニル;
(8)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
(9)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(10)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノフェニルペンタジエニルオキシカルボニル;
(11)p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(12)p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(13)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(14)p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(15)p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)‐カルボニル;
(16)p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(17)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジル;
(18)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル‐p‐アミノベンジル;
(19)p‐アミノシンナミル;
(20)p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジル;
(21)p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミル;
(22)p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミル;
(23)p‐アミノフェニルペンタジエニル;
(24)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノシンナミル;
(25)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノベンジル;又は
(26)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノフェニルペンタジエニルを含む、上述のような方法の実施形態を提供する。
上記VH CDR1はアミノ酸配列SYWMN(配列番号23)を含み;
上記VH CDR2はアミノ酸配列VIHPSDSETWLDQKFKD(配列番号24)を含み;
上記VH CDR3はアミノ酸配列EHYGTSPFAY(配列番号25)を含み、
上記抗体が、VL CDR1、VL CDR2、及びVL CDR3を含む可変軽鎖(VL)ドメインを含み:
上記VL CDR1はアミノ酸配列RASESVDNYGMSFMNW(配列番号26)を含み;
上記VL CDR2はアミノ酸配列AASNQGS(配列番号27)を含み;
上記VL CDR3はアミノ酸配列QQSKEVPYT(配列番号28)を含む、上述のような方法の実施形態を提供する。
(A)約0.5mg/ml~約5mg/mlのB7‐H3‐ADCを含む医薬組成物;及び
(B)説明資料
を含む医薬キットで提供され、上記説明資料は、B7‐H3‐ADCを含む上記医薬組成物を、PD‐1結合分子を含む医薬組成物と任意に組み合わせて投与することを指示する、上述のような方法の実施形態を提供する。
(I)配列番号17のアミノ酸配列を含むヒト化VLドメイン;及び
(II)配列番号18のアミノ酸配列を含むヒト化VHドメイン
を含む、上述のような方法の実施形態を提供する。
本発明の抗体は、当該免疫グロブリン分子の可変ドメインに位置する少なくとも1つの抗原認識部位によって、炭水化物、ポリヌクレオチド、脂質、ポリペプチド等の標的に特異的に結合できる、免疫グロブリン分子である。従って本発明のB7‐H3‐ADCは、B7‐H3に結合する抗体を含む。本明細書において使用される場合、用語「抗体(antibody及びantibodies)」は、モノクローナル抗体、多重特異性抗体、ヒト抗体、ヒト化抗体、合成抗体、キメラ抗体、ポリクローナル抗体、ラクダ化抗体、一本鎖Fv(scFv)、一本鎖抗体、Fab断片、F(ab’)断片、ジスルフィド結合二重特異性Fv(sdFv)、細胞内抗体、及び以上のうちのいずれかのエピトープ結合断片を包含する。特に、用語「抗体」は、免疫グロブリン分子及び免疫グロブリン分子の免疫学的に活性の断片、即ちエピトープ結合部位を含有する分子を含む。免疫グロブリン分子は、いずれのタイプ(例えばIgG、IgE、IgM、IgD、IgA及びIgY)、クラス(例えばIgG1、IgG2、IgG3、IgG4、IgA1及びIgA2)又はサブクラスのものとすることができる。抗体は、ポリペプチド又はタンパク質又は非タンパク質分子に「免疫特異的に結合(immunospecifically binding)」(又はこれらの分子に「免疫特異的な様式(immunospecific manner)」で結合)できる。というのは、このような分子上に特定のドメイン又は部分又は形態(「エピトープ(epitope)」)が存在するためである。エピトープ含有分子は、免疫学的活性を有することができ、これにより、動物における抗体産生応答を誘発する。このような分子を「抗原(antigen)」と呼ぶ。
IgG分子の可変ドメインは、エピトープと接触した残基を含有する複数の相補性決定領域(「CDR」)、及びフレームワークセグメント(「FR」)と呼ばれる非CDRセグメントからなり、上記フレームワークセグメントは一般に、CDRループの構造を維持してCDRの位置を決定することにより、このような接触を可能とする(ただし特定のフレームワーク残基も抗原に接触し得る)。従って、VL及びVHドメインは、構造n‐FR1‐CDR1‐FR2‐CDR2‐FR3‐CDR3‐FR4‐cを有する。CDRのアミノ酸配列は、ある抗体がある特定のエピトープに結合できるかどうかを決定する。抗体軽鎖と抗体重鎖との相互作用、及び特にこれらのVLドメインとVHドメインとの相互作用は、該抗体のエピトープ結合部位を形成する。
1.軽鎖の定常ドメイン
上述のように、抗体の各軽鎖は、可変ドメイン(「VL」)と定常ドメイン(「CL」)とを含有する。
RTVAAPSVFI FPPSDEQLKS GTASVVCLLN NFYPREAKVQ WKVDNALQSG
NSQESVTEQD SKDSTYSLSS TLTLSKADYE KHKVYACEVT HQGLSSPVTK
SFNRGEC
である。
QPKAAPSVTL FPPSSEELQA NKATLVCLIS DFYPGAVTVA WKADSSPVKA
GVETTPSKQS NNKYAASSYL SLTPEQWKSH RSYSCQVTHE GSTVEKTVAP
TECS
である。
上述のように、抗体の重鎖は、CH1、ヒンジドメイン、CH2及びCH3定常ドメインを含み得る。抗体の2つの重鎖のCH1ドメインは、抗体の軽鎖のCL定常ドメインと複合体化し、介在ヒンジドメインを介して重鎖CH2ドメインに付着する。
ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV
HTFPAVLQSS GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKRV
である。
ASTKGPSVFP LAPCSRSTSE STAALGCLVK DYFPEPVTVS WNSGALTSGV
HTFPAVLQSS GLYSLSSVVT VPSSSLGTKT YTCNVDHKPS NTKVDKRV
である。
231 240 250 260 270 280
APELLGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD
290 300 310 320 330
GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA
340 350 360 370 380
PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLTCLVK GFYPSDIAVE
390 400 410 420 430
WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE
440 447
ALHNHYTQKS LSLSPGX
であり、これはKabatに記載のEUインデックスによって番号付与されており、Xはリシン(K)であるか、又は不在である。
231 240 250 260 270 280
APEFLGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSQED PEVQFNWYVD
290 300 310 320 330
GVEVHNAKTK PREEQFNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKGLPS
340 350 360 370 380
SIEKTISKAK GQPREPQVYT LPPSQEEMTK NQVSLTCLVK GFYPSDIAVE
390 400 410 420 430
WESNGQPENN YKTTPPVLDS DGSFFLYSRL TVDKSRWQEG NVFSCSVMHE
440 447
ALHNHYTQKS LSLSLGX
であり、これはKabatに記載のEUインデックスによって番号付与されており、Xはリシン(K)であるか、又は不在である。
「mAb‐A」と称される例示的な抗B7‐H3抗体を、ヒトB7‐H3を発現する細胞、そのB7‐H3ポリペプチド又はペプチドエピトープを用いた免疫化によって生成されたハイブリドーマ細胞から単離した。抗体mAb‐Aはヒト化された。
マウス抗B7‐H3抗体mAb‐AのVLドメインのアミノ酸配列(配列番号15)を以下に示す(CDRL残基は下線を付して示されている):
DIQMTQSPAS LSVSVGETVT ITCRASESIY SYLAWYQQKQ GKSPQLLVYN
TKTLPEGVPS RFSGSGSGTQ FSLKINSLQP EDFGRYYCQH HYGTPPWTFG
GGTNLEIK
EVQQVESGGD LVKPGGSLKL SCAASGFTFS SYGMSWVRQT PDKRLEWVAT
INSGGSNTYY PDSLKGRFTI SRDNAKNTLY LQMRSLKSED TAMYYCARHD
GGAMDYWGQG TSVTVSS
抗B7‐H3抗体mAb‐Aの可変ドメインをヒト化して、ヒト化mAb‐A(「hmAb‐A」)を生成した。いくつかの例では、結合活性を最適化するため、及び/又は抗原性エピトープを除去するため、及び/又は潜在的に不安定なアミノ酸残基を除去するために、代替的なヒト化可変ドメインを生成してよい。
DIQMTQSPSS LSASVGDRVT ITCRASESIY SYLAWYQQKP GKAPKLLVYN
TKTLPEGVPS RFSGSGSGTD FTLTISSLQP EDFATYYCQH HYGTPPWTFG
QGTRLEIK
DIQMTQSPSS LSASVGDRVT ITCRASESIY SYLAWYQQKP GKAPKLLVYN
TKTLPEGVPS RFSGSGSGTD FTLTISSLQP EDFATYYCQH HYGTPPWTFG
QGTRLEIKRT VAAPSVFIFP PSDEQLKSGT ASVVCLLNNF YPREAKVQWK
VDNALQSGNS QESVTEQDSK DSTYSLSSTL TLSKADYEKH KVYACEVTHQ
GLSSPVTKSF NRGEC
EVQLVESGGG LVKPGGSLRL SCAASGFTFS SYGMSWVRQA PGKGLEWVAT
INSGGSNTYY PDSLKGRFTI SRDNAKNSLY LQMNSLRAED TAVYYCARHD
GGAMDYWGQG TTVTVSS
EVQLVESGGG LVKPGGSLRL SCAASGFTFS SYGMSWVRQA PGKGLEWVAT
INSGGSNTYY PDSLKGRFTI SRDNAKNSLY LQMNSLRAED TAVYYCARHD
GGAMDYWGQG TTVTVSSAST KGPSVFPLAP SSKSTSGGTA ALGCLVKDYF
PEPVTVSWNS GALTSGVHTF PAVLQSSGLY SLSSVVTVPS SSLGTQTYIC
NVNHKPSNTK VDKRVEPKSC DKTHTCPPCP APELLGGPSV FLFPPKPKDT
LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY
RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT
LPPSREEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS
DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LSLSPGK
本発明のFcドメイン含有分子(例えば抗体及びダイアボディ)のFcドメインは、完全なFcドメイン(例えば完全IgG Fcドメイン)、又は単にFcドメインの断片であってよい。任意に、本発明のFcドメイン含有分子のFcドメインは、C末端リシンアミノ酸残基を含まない。
APEFLGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSQED PEVQFNWYVD
GVEVHNAKTK PREEQFNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKGLPS
SIEKTISKAK GQPREPQVYT LPPSQEEMTK NQVSLTCLVK GFYPSDIAVE
WESNGQPENN YKTTPPVLDS DGSFFLYSRL TVDKSRWQEG NVFSCSVMHE
ALHNHYTQKS LSLSLGX
であり、Xはリシン(K)であるか、又は不在である。
本発明は、細胞傷害性薬物B7‐H3‐ADCに対して複合体化された上述の抗B7‐H3抗体hmAb‐Aに関する。上述のようなB7‐H3‐ADCは、特に癌の治療における抗B7‐H3療法の細胞傷害性を増強する。上述のように、本発明のB7‐H3‐ADCは、以下の式:
Ab‐(LM)m‐(D)n
で表され:
Abは、ヒト化可変重鎖(VH)ドメイン及びヒト化可変軽鎖(VL)ドメインを含むB7‐H3に結合する抗体、又はそのB7‐H3結合断片であり;
Dは、細胞傷害性デュオカルマイシン部分であり;
LMは、Ab及びDを共有結合させる結合又はリンカー分子であり;
mは0~nの整数であって、上記B7‐H3‐ADCの結合又はリンカー分子の数を表すが、LMが1つの結合である場合を除いてmは0ではなく;
nは1~10の整数であって、上記B7‐H3‐ADCに共有結合した上記細胞傷害性デュオカルマイシン部分の数を表す。
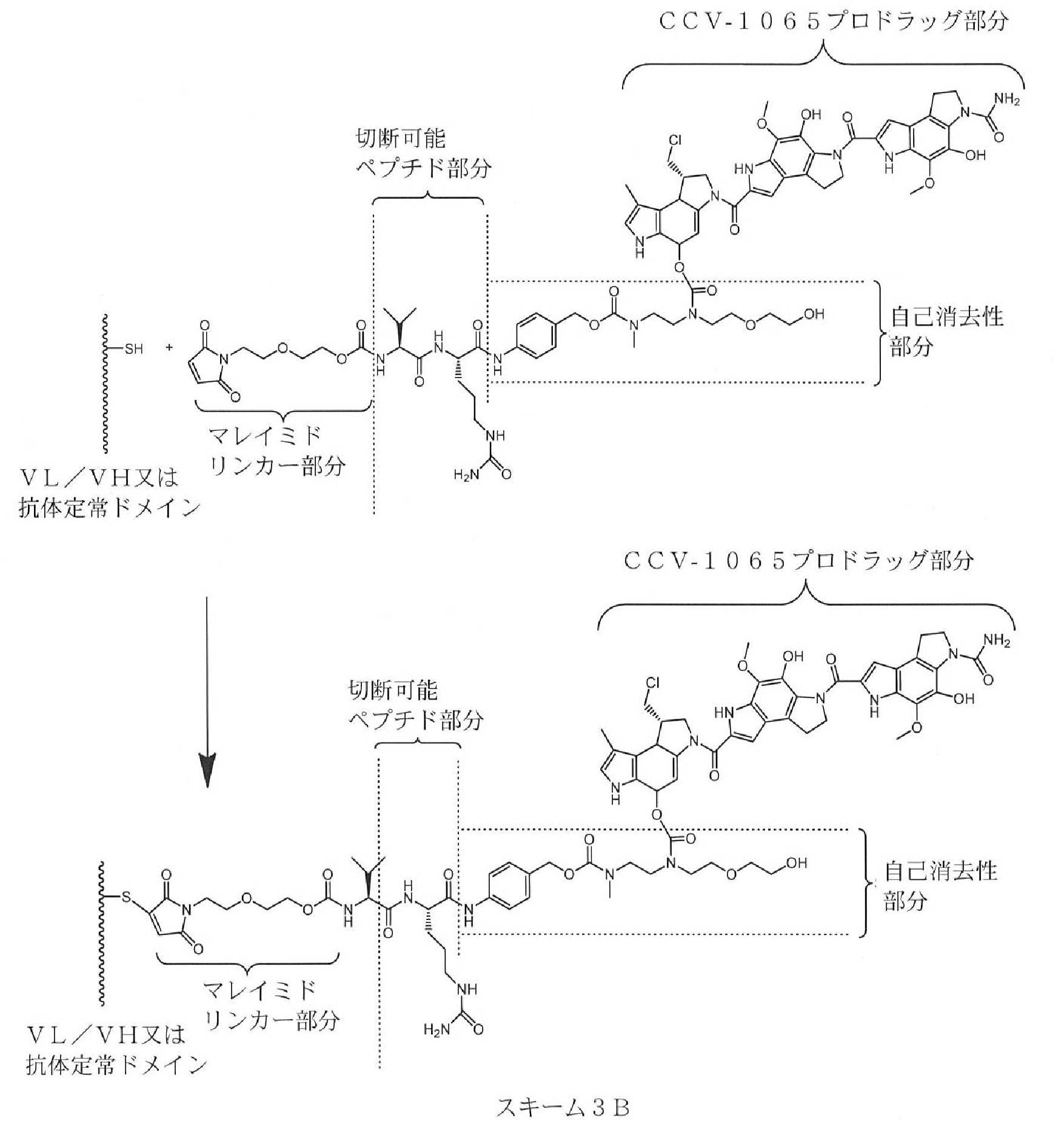
よって本発明は特に、LMがリンカー分子であり、かつ不在である(即ちm=0である)上述のようなB7‐H3‐ADCと、に2つ以上のリンカー分子LMを有し(即ちmが2~nの整数であり、nが2~10の整数であり)、各リンカー分子LMがB7‐H3‐ADCの細胞傷害性デュオカルマイシン部分D及びAbを共有結合させる、B7‐H3‐ADCとについて考察する。
Ab‐[V‐(W)k‐(X)1‐A]‐D
を有するB7‐H3‐ADCをもたらし、ここで:
Vは任意の切断可能部分であり;
(W)k‐(X)1‐Aは、l,(4+2n)消去によって自己消去する、細長い自己消去性スペーサ系であり;
W及びXはそれぞれl,(4+2n)電子カスケードスペーサであり、同一であるか又は異なっており;
Aは、式(Y)m(ここでYはl,(4+2n)電子カスケードスペーサである)のスペーサ基、又は式Uの基であり、環化除去スペーサであり;
k、1及びmは独立して、0~5(両端を含む)の整数であり;
nは0~10(両端を含む)の整数であり;
ただし:
Aが(Y)mである場合、k+l+m≧1であり;
k+l+m=lである場合、n>lであり;
AがUである場合、k+1≧1であり、
W、X及びYは独立して、以下の式:


Qは‐R5C=CR6‐、S、O、NR5、‐R5C=N‐又は‐N=CR5‐であり;
PはNR7、O又はSであり;
a、b及びcは独立して、0~5(両端を含む)の整数であり;
I、F及びGは独立して、式:

R1、R2、R3、R4、R5、R6、R7、R8及びR9は独立して、H、C1‐6アルキル、C3‐20ヘテロシクリル、C5‐20アリール、C1‐6アルコキシ、ヒドロキシ(OH)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)及びリン酸塩(OP(=O)(ORx)2)を表し、ここで:
Rx、Rx 1及びRx 2は独立して、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され;
上記置換基R1、R2、R3、R4、R5、R6、R7、R8又はR9のうちの2つ以上は任意に、互いに接続されて1つ以上の脂肪族又は芳香族環式構造を形成し;
Uは、式:

a、b及びcは独立して、0又は1の整数となるよう選択され;
ただしa+b+c=2又は3であり;
R1及び/又はR2は独立して、H、C1‐6アルキルを表し、上記アルキルは任意に、以下の基:ヒドロキシ(OH)、エーテル(ORx)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)、及びリン酸塩(OP(=O)(ORx)2)のうちの1つ以上によって置換され、ここでRx、Rx 1及びRx 2は、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され;
R3、R4、R5、R6、R7及びR8は独立して、H、C1‐6アルキル、C3‐20ヘテロシクリル、C5‐20アリール、C1‐6アルコキシ、ヒドロキシ(OH)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)、及びリン酸塩(OP(=O)(ORx)2)を表し、ここでRx、Rx 1及びRx 2は、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され、上記置換基R1、R2、R3、R4、R5、R6、R7、又はR8のうちの2つ以上は任意に、互いに接続されて1つ以上の脂肪族又は芳香族環式構造を形成する。
p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
p‐アミノシンナミルオキシカルボニル;
p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
p‐アミノ‐ベンジルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
p‐アミノフェニルペンタジエニルオキシカルボニル;
p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
p‐アミノフェニルペンタジエニルオキシカルボニル‐pアミノベンジルオキシカルボニル;
p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノフェニルペンタジエニルオキシカルボニル;
p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)‐カルボニル;
p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
p‐アミノベンジルオキシカルボニル‐p‐アミノベンジル;
p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル‐p‐アミノベンジル;
p‐アミノシンナミル;
p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジル;
p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミル;
p‐アミノ‐シンナミルオキシカルボニル‐p‐アミノシンナミル;
p‐アミノフェニルペンタジエニル;
p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノシンナミル;
p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノベンジル;及び
p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノフェニルペンタジエニルが挙げられる。
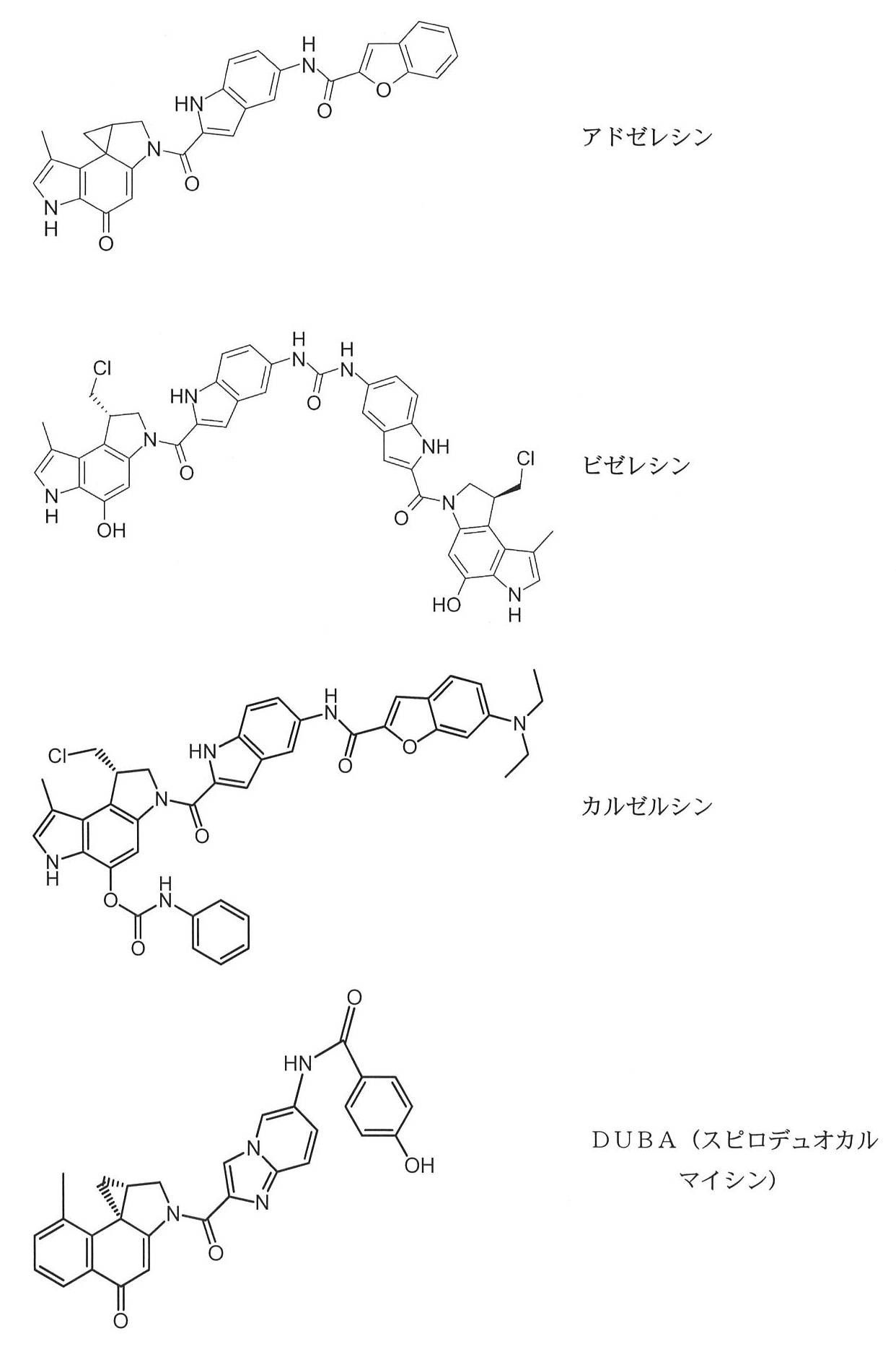
デュオカルマイシンは、初めはストレプトマイセス属の微生物から単離された一連の関連天然産物のメンバーであり、強力な抗腫瘍抗生物質である(Dokter, W. et al. (2014) “Preclinical Profile of the HER2‐Targeting ADC SYD983/SYD985: Introduction of a New Duocarmycin‐Based Linker‐Drug Platform,” Mol. Cancer Ther. 13(11):2618‐2629; Boger, D.L. et al. (1991). “Duocarmycins ‐ A New Class Of Sequence Selective DNA Minor Groove Alkylating Agents,” Chemtracts: Organic Chemistry 4 (5): 329‐349 (1991); Tercel et al. (2013) “The Cytotoxicity Of Duocarmycin Analogues Is Mediated Through Alkylation Of DNA, Not Aldehyde Dehydrogenase 1: A Comment,” Chem. Int. Ed. Engl. 52(21):5442‐5446; Boger, D.L. et al. (1995) “CC‐1065 And The Duocarmycins: Unraveling The Keys To A New Class Of Naturally Derived DNA Alkylating Agents,” Proc. Natl. Acad. Sci. (U.S.A.) 92(9):3642‐3649; Cacciari, B. et al. (2000) “CC‐1065 And The Duocarmycins: Recent Developments,” Expert Opinion on Therapeutic Patents 10(12):1853‐1871を参照)。



Rは脱離基であり;
R2、R2'、R3、R3'、R4、R4'、R12及びR19は独立して、H、OH、SH、NH2、N3、NO2、NO、CF3、CN、C(O)NH2、C(O)H、C(O)OH、ハロゲン、Ra、SRa、S(O)Ra、S(O)2Ra、S(O)ORa、S(O)2ORa、OS(O)Ra、OS(O)2Ra、OS(O)ORa、OS(O)2ORa、ORa、NHRa、N(Ra)Rb、+N(Ra)(Rb)Rc、P(O)(ORa)(ORb)、OP(O)(ORa)(ORb)、SiRaRbRc、C(O)Ra、C(O)ORa、C(O)N(Ra)Rb、OC(O)Ra、OC(O)ORa、OC(O)N(Ra)Rb、N(Ra)C(O)Rb、N(Ra)C(O)ORb及びN(Ra)C(O)N(Rb)Rcから選択され、ここでRa、Rb及びRcは独立して、H及び任意に置換されたC1‐3アルキル若しくはC1‐3ヘテロアルキルから選択され、又はR3+R3'及び/若しくはR4+R4'は独立して、=O、=S、=NOR18、=C(R18)R18'及び=NR18から選択され、R18及びR18'は独立して、H及び任意に置換されたC1‐3アルキルから選択され、R2、R2'、R3、R3'、R4、R4'及びR12のうちの2つ以上は、1つ以上の結合によって連結されて、1つ以上の任意に置換された炭素環及び/又は複素環を形成し;
X2はO、C(R14)(R14')及びNR14'から選択され、ここでR14及びR14'は、R7に関して定義されたものと同一の意味を有し、かつ独立して選択され、又はR14'及びR7'は不在であり、これによってR7'及びR14'を担持するよう指定された原子間に二重結合がもたらされ;
R5、R5'、R6、R6'、R7及びR7'は独立して、H、OH、SH、NH2、N3、NO2、NO、CF3、CN、C(O)NH2、C(O)H、C(O)OH、ハロゲン、Re、SRe、S(O)Re、S(O)2Re、S(O)ORe、S(O)2ORe、OS(O)Re、OS(O)2Re、OS(O)ORe、OS(O)2ORe、ORe、NHRe、N(Re)Rf、+N(Re)(Rf)Rg、P(O)(ORe)(ORf)、OP(O)(ORe)(ORf)、SiReRfRg、C(O)Re、C(O)ORe、C(O)N(Re)Rf、OC(O)Re、OC(O)ORe、OC(O)N(Re)Rf、N(Re)C(O)Rf、N(Re)C(O)ORf、N(Re)C(O)N(Rf)Rg及び水溶性基から選択され、ここで
Re、Rf及びRgは独立して、H及び任意に置換された(CH2CH2O)eeCH2CH2X13Re1、C1‐15アルキル、C1‐15ヘテロアルキル、C3‐15シクロアルキル、C1‐15ヘテロシクロアルキル、C5‐15アリール又はC1‐15ヘテロアリールから選択され、ここでeeは1~1000から選択され、X13はO、S及びNRf1から選択され、またRf1及びRe1は独立して、H及びC1‐3アルキルから選択され、Re、Rf、及び/又はRgの任意の置換基のうちの1つ以上は、任意に水溶性基であり、Re、Rf及びRgのうちの2つ以上は任意に、1つ以上の結合によって連結されて1つ以上の任意に置換された炭素環及び/又は複素環を形成し;
あるいはR5+R5'及び/若しくはR6+R6'及び/若しくはR7+R7'は独立して、=O、=S、=NORe3、=C(Re3)Re4及び=NRe3、Re3から選択され、またRe4は独立して、H及び任意に置換されたC1‐3アルキルから選択されるか、又はR5'+R6'及び/若しくはR6'+R7'及び/若しくはR7'+R14'は不在であり、これによってR5'+R6'及び/若しくはR6'+R7'及び/若しくはR7'+R14'それぞれを担持するよう指定された原子間に二重結合がもたらされ、R5、R5'、R6、R6'、R7、R7'、R14及びR14'のうちの2つ以上は任意に、1つ以上の結合によって連結されて1つ以上の任意に置換された炭素環及び/又は複素環を形成し;
X1はO、S及びNRから選択され、ここでRは、H及び任意に置換されたC1‐8アルキル又はC1‐8ヘテロアルキルから選択され、他のいずれの置換基と連結されず;
X3は、O、S、C(R15)R15'、‐C(R15)(R15')‐C(R15'')(R15''')‐、‐N(R15)‐N(R15')‐、‐C(R15)(R15')‐N(R15")‐、‐N(R15")‐C(R15)(R15')‐、‐C(R15)(R15')‐O‐、‐O‐C(R15)(R15')‐、‐C(R15)(R15')‐S‐、‐S‐C(R15)(R15')‐、‐C(R15)=C(R15')‐、=C(R15)‐C(R15')=、‐N=C(R15')‐、=N‐C(R15')=、‐C(R15)=N‐、=C(R15)‐N=、‐N=N‐、=N‐N=、CR15、N、NR15から選択され、又はDB1及びDB2では、‐X3‐は‐X3a及びX3b‐を表し、ここでX3aはX34に接続され、X34とX4との間には二重結合が存在し、またX3bはX11に接続され、ここでX3aは独立して、H及び任意に置換された(CH2CH2O)eeCH2CH2X13Re1、C1‐8アルキル又はC1‐8ヘテロアルキルから選択され、他のいずれの置換基と連結されず;
X4は、O、S、C(R16)R16'、NR16、N及びCR16から選択され;
X5は、O、S、C(R17)R17'、NOR17及びNR17から選択され、ここでR17及びR17'は独立して、H及び任意に置換されたC1‐8アルキル又はC1‐8ヘテロアルキルから選択され、他のいずれの置換基と連結されず;
X6は、CR11、CR11(R11')、N、NR11、O及びSから選択され;
X7は、CR8、CR8(R8')、N、NR8、O及びSから選択され;
X8は、CR9、CR9(R9')、N、NR9、O及びSから選択され;
X9は、CR10、CR10(R10')、N、NR10、O及びSから選択され;
X10は、CR20、CR20(R20')、N、NR20、O及びSから選択され;
X11は、C、CR21及びNから選択され、又はX11‐X3bは、CR21、CR21(R21')、N、NR21、O及びSから選択され;
X12は、C、CR22及びNから選択され;
X6*、X7*、X8*、X9*、X10*及びX11*は、それぞれX6、X7、X8、X9、X10及びX11に関して定義されたものと同一の意味を有し、また独立して選択され;
X34は、C、CR23及びNから選択され;
DB6及びDB7のX11*の環B原子は、環Aの環式原子に接続され、これによりDB6及びDB7の環A及び環Bは単一結合によって直接接続され;
ダッシュで描かれた二重結合は、指示されている結合が単一結合又は
任意に非局在化された非累積二重結合であってよいことを意味しており;
R8、R8'、R9、R9'、R10、R10'、R11、R11'、R15、R15'、R15"、R15"、R16、R16'、R20、R20'、R21、R21'、R22及びR23はそれぞれ独立して、H、OH、SH、NH2、N3、NO2、NO、CF3、CN、C(O)NH2、C(O)H、C(O)OH、ハロゲン、Rh、SRh、S(O)Rh、S(O)2Rh、S(O)ORh、S(O)2ORh、OS(O)Rh、OS(O)2Rh、OS(O)ORh、OS(O)2ORh、ORh、NHRh、N(Rh)Ri、+N(Rh)(Ri)Rj、P(O)(ORh)(ORi)、OP(O)(ORh)(ORi)、SiRhRiRj、C(O)Rh、C(O)ORh、C(O)N(Rh)Ri、OC(O)Rh、OC(O)ORh、OC(O)N(Rh)Ri、N(Rh)C(O)Ri、N(Rh)C(O)ORi、N(Rh)C(O)N(Ri)Rj及び水溶性基から選択され;ここで
Rh、Ri及びRjは独立して、H及び任意に置換された(CH2CH2O)eeCH2CH2X13Re1、C1‐15アルキル、C1‐15ヘテロアルキル、C3‐15シクロアルキル、C1‐15ヘテロシクロアルキル、C5‐15アリール、又はC1‐15ヘテロアリールから選択され、Rh、Ri及び/又はRjの任意の置換基のうちの1つ以上は任意に、水溶性基であり、Rh、Ri及びRjのうちの2つ以上は任意に、1つ以上の結合によって連結されて1つ以上の任意に置換された炭素環及び/又は複素環を形成し;
あるいは、R8+R8'及び/又はR9+R9'及び/又はR10+R10'及び/又はR11+R11'及び/又はR15+R15'及び/又はR15"+R15'"及び/又はR16+R16'及び/又はR20+R20'及び/又はR21+R21'は独立して、=O、=S、=NORh1、=C(Rhl)Rh2及び=NRhlから選択され、Rhl及びRh2は独立して、H及び及び任意に置換されたC1‐3アルキルから選択され、R8、R8'、R9、R9'、R10、R10'、R11、R11'、R15、R15'、R15"、R15'"、R16、R20、R20'、R21、R21'、R22及びR23のうちの2つ以上は任意に、1つ以上の結合によって連結されて1つ以上の任意に置換された炭素環及び/又は複素環を形成し;
R8b及びR9bは独立して選択され、またこれらが他のいずれの置換基と連結し得ないことを除いて、R8と同一の意味を有し;
R4及びR4'のうちの一方と、R16及びR16'のうちの一方とは任意に、1つ以上の結合によって連結されて1つ以上の任意に置換された炭素環及び/又は複素環を形成してよく;
R4及びR4'のうちの一方と、R16及びR16'のうちの一方とは任意に連結してよく;
R2、R2'、R3及びR3'のうちの1つと、R5及びR5'のうちの一方とは任意に、1つ以上の結合によって連結されて1つ以上の任意に置換された炭素環及び/又は複素環を形成してよく;
a及びbは独立して0~1から選択され;
DB部分は、DAI、DA2、DAI’又はDA2'部分を含まず;
DB1の環Bは複素環であり;
DB1のX3が‐X3a及びX3b‐を表し、かつ環Bが芳香族である場合、上記環B上の2つの近接した置換基は連結して、上記環Bに融合した、任意に置換された炭素環又は複素環を形成し;
DB2のX3が‐X3a及びX3b‐を表し、かつ環Bが芳香族である場合、上記環B上の2つの近接した置換基は連結して:上記環Bに融合した、任意に置換された複素環;上記環Bに融合した、任意に置換された非芳香族炭素環;又は上記環Bに融合し、かつヒドロキシ基、一次アミノ基又は二次アミノ基を含有する少なくとも1つの置換基が付着した、置換された芳香族炭素環を形成し、上記一次又は二次アミンは芳香環系中の環式原子でも、アミドの一部でもなく;
DB2の環Aが6員芳香環である場合、環B上の置換基は、環Bに融合した環を形成するように連結せず;
DB8の環A上の2つの近接した置換基は連結して、任意に置換された炭素環又は複素環を形成し、これは上記環Aに融合して二環式部分を形成し、これに環がこれ以上融合せず;
DB9の環Aは、上記環Aに融合したいずれの環と共に、少なくとも2つのヘテロ原子を含有する。








本発明のPD‐1結合分子は、二重特異性分子(例えば二重特異性抗体、二重特異性ダイアボディ等)、キメラ又はヒト化抗体、及び変異型Fc領域を有するこのような結合分子を含む。このようなPD‐1結合分子は、ヒトPD‐1(CD279)の連続した又は不連続の(例えば立体配座の)部分に結合する能力を示す。本発明のPD‐1結合分子は好ましくは、1つ以上の非ヒト種、特に霊長類種(及び特にカニクイザル等の霊長類種)のPD‐1分子への結合能力も示す。20アミノ酸残基シグナル配列及び268アミノ酸残基成熟タンパク質を含む、代表的なヒトPD‐1ポリペプチドは、NCBI配列NP_005009.2(配列番号22)によって提供されている。
ニボルマブのVHドメインのアミノ酸配列は、アミノ酸配列(配列番号36)(CDRH残基は下線を付して示されている):
QVQLVESGGG VVQPGRSLRL DCKASGITFS NSGMHWVRQA PGKGLEWVAV
IWYDGSKRYY ADSVKGRFTI SRDNSKNTLF LQMNSLRAED TAVYYCATND
DYWGQGTLVT VSS
を有する。
EIVLTQSPAT LSLSPGERAT LSCRASQSVS SYLAWYQQKP GQAPRLLIYD
ASNRATGIPA RFSGSGSGTD FTLTISSLEP EDFAVYYCQQ SSNWPRTFGQ
GTKVEIK
を有する。
ペムブロリズマブのVHドメインのアミノ酸配列は、アミノ酸配列(配列番号34)(CDRH残基は下線を付して示されている):
QVQLVQSGVE VKKPGASVKV SCKASGYTFT NYYMYWVRQA PGQGLEWMGG
INPSNGGTNF NEKFKNRVTL TTDSSTTTAY MELKSLQFDD TAVYYCARRD
YRFDMGFDYW GQGTTVTVSS
を有する。
EIVLTQSPAT LSLSPGERAT LSCRASKGVS TSGYSYLHWY QQKPGQAPRL
LIYLASYLES GVPARFSGSG SGTDFTLTIS SLEPEDFAVY YCQHSRDLPL
TFGGGTKVEIK
を有する。
特定の実施形態では、PD‐1結合分子は、hPD‐1 mAb‐AのVH及びVLドメインを含む。hPD‐1 mAb‐Aのアミノ酸配列は以下で提供されており、これはまた特許文献31、国際公開第2017/062619号、特許文献57、特許文献43でも開示されている。hPD‐1 mAb‐Aは、レチファンリマブ、MGA012、及びINCMGA‐00012(CAS登録番号2079108‐44‐2、IncyteとMacroGenics, Inc.との共同開発)としても公知である。hPD‐1 mAb‐Aのアミノ酸配列は以下に示されており、またWHO Drug Information 2019, Recommended INN: List 82, 33(1):611‐612で提供されている。


EIVLTQSPAT LSLSPGERAT LSCRASESVD NYGMSFMNWF QQKPGQPPKL
LIHAASNQGS GVPSRFSGSG SGTDFTLTIS SLEPEDFAVY FCQQSKEVPY
TFGGGTKVEI K
EIVLTQSPAT LSLSPGERAT LSCRASESVD NYGMSFMNWF QQKPGQPPKL
LIHAASNQGS GVPSRFSGSG SGTDFTLTIS SLEPEDFAVY FCQQSKEVPY
TFGGGTKVEI KRTVAAPSVF IFPPSDEQLK SGTASVVCLL NNFYPREAKV
QWKVDNALQS GNSQESVTEQ DSKDSTYSLS STLTLSKADY EKHKVYACEV
THQGLSSPVT KSFNRGEC
特定の実施形態では、PD‐1結合分子は、PD‐1及びLAG‐3に結合する二重特異性分子である。癌及び/又は病原体に関連する疾患の治療に使用するためのPD‐1×LAG‐3二重特異性分子は、特許文献51~56に記載されている。特定の実施形態では、二重特異性分子はPD‐1×LAG‐3二重特異性ダイアボディである。新規のPD‐1及びLAG‐3結合ドメインを有するPD‐1×LAG‐3二重特異性ダイアボディ、並びに例示的な活性は、特許文献57に記載されている。ある具体的実施形態では、上記ダイアボディは「PD‐1×LAG‐3 BD」である。PD‐1×LAG‐3 BDは、PD‐1に対して特異的な2つの結合部位、LAG‐3に対して特異的な2つの結合部位、Fc領域、及びシステイン含有E/Kコイルヘテロ二量体促進ドメインを有する、4鎖のFc領域含有ダイアボディである。PD‐1×LAG‐3 BDの一般的な構造が図1で提供されている。PD‐1×LAG‐3 BDは、PD‐1に結合するヒト化抗体のVL及びVHドメインと、LAG‐3に結合するヒト化抗体のVL及びVHドメインとを含む。よってPD‐1×LAG‐3 BDは、PD‐1のエピトープ及びLAG‐3のエピトープに特異的に結合できる。


本発明の結合分子(例えばB7‐H3‐ADC、hPD‐1 mAb A、及びPD‐1×LAG‐3 BD)は、組み換えによって作成でき、また組み換えタンパク質の製造に関して当該技術分野で公知のいずれの方法を用いて発現させることができる。例えば、このような結合分子のポリペプチド鎖をコードする核酸を構築し、発現ベクターに導入して、好適な宿主細胞で発現させることができる。上記結合分子は、細菌細胞(例えばE.coli細胞)又は真核細胞(例えばCHO、293E、COS、NS0細胞)中で組み換えによって製造してよい。更に上記結合分子は、Pichia又はSaccharomyces等の酵母細胞中で発現させることができる。
本発明のB7‐H3‐ADC及びPD‐1結合分子(例えばhPD‐1 mAb‐A及び/又はPD‐1×LAG‐3 BD)は、組成物として処方できる。本発明の組成物は、医薬組成物の製造に使用できるバルク薬物組成物(例えば純粋でない又は滅菌されていない組成物)、及び単位剤形の調製に使用できる医薬組成物(即ち被験者又は患者への投与に公的な組成物)を含む。このような組成物は、予防又は治療有効量の本発明のB7‐H3‐ADC、1つ以上のPD‐1結合分子、又はこれらの組み合わせと、1つ以上の薬学的に許容可能なキャリアとを含み、任意に1つ以上の追加の治療剤を更に含んでよい。上記医薬組成物は例えば、水溶液、又は薬学的に許容可能なキャリアを用いた再構成に特に適合された、若しくはこのようなキャリアを用いて再構成される、凍結乾燥粉末若しくは水非含有濃縮物として供給できる。
本発明はまた、1つ以上の医薬組成物と説明資料(例えば注意、添付文書、説明書等)とを内包する1つ以上のコンテナを含む、医薬パック又はキットを提供する。更に、疾患の治療に有用な1つ以上の他の予防又は治療剤も、医薬キットに含めることができる。このような医薬キットのコンテナは、1つ以上の気密バイアル、アンプル、サシェ等を含んでよく、これらには、これらが内包する活性剤の量が記されている。組成物が注入によって投与されるものである場合、コンテナは、滅菌された医薬グレードの溶液(例えば水、生理食塩水、緩衝液等)を内包した輸液ボトル、バッグであってよい。組成物が注射によって投与されるものである場合、医薬キットは、被験者(例えばヒト患者又は他の哺乳類)への投与のための医薬キットの構成要素の混合を容易にするために、注射用の滅菌水、生理食塩水、又は他の希釈剤のアンプルを内包してよい。特定の実施形態では、医薬パック又はキットは、B7‐H3‐ADC医薬組成物及び説明資料を含む。他の実施形態では、医薬パック又はキットは、B7‐H3‐ADC医薬組成物と、PD‐1結合分子組成物と、説明資料とを含む。
本発明のB7‐H3‐ADCは、任意に本発明のPD‐1結合分子と組み合わせて、特にB7‐H3を発現する癌である癌を含む様々な障害の治療又は予防に使用できる。従って本発明は、癌を治療する方法を提供し、上記方法は、本発明のB7‐H3‐ADCを、任意に本発明のPD‐1結合分子と組み合わせて、それを必要とする被験者に投与するステップを含む。本明細書中で使用される場合、用語「被験者(subject)」は、ヒト(即ちヒト患者)又は他の哺乳類を指す。それを必要とする被験者にこのような療法を投与するための例示的な投薬レジメンが、本明細書において提供される。
本発明の分子(例えばB7‐H3‐ADC及び/又はPD‐1結合分子)は、多様な方法で、被験者、例えばそれを必要とする被験者、例えばヒト患者に投与できる。多くの用途に関して、投与の経路は:静脈内注射又は注入(IV)、皮下注射(SC)、腹腔内(IP)、又は筋肉内注射のうちの1つである。関節内送達の使用も可能である。他の様式の非経口投与の使用も可能である。このような様式の例としては:動脈内、髄腔内、嚢内、眼窩内、心臓内、皮内、経気管、表皮下、関節内、被膜下、くも膜下、脊髄内、並びに硬膜外及び胸骨内注射が挙げられる。
本発明は部分的に、以下の非限定的な実施形態(E1~E119)に関する。
(A)B7‐H3‐ADC;及び
(B)PD‐1結合分子
を投与するステップを含む、癌を治療する方法であって、
上記方法は、上記B7‐H3‐ADCを上記被験者に、約0.5mg/kg~約5mg/kgの用量で3週間に1回投与するステップを含む、方法。
(A)B7‐H3‐ADC;及び
(B)PD‐1結合分子
を投与するステップを含む、癌を治療する方法であって、
上記方法は、上記B7‐H3‐ADCを上記被験者に、約0.5mg/kg~約5mg/kgの用量で4週間に1回投与するステップを含む、方法、
Ab‐(LM)m‐(D)n
で表され、ここで:
Abは、B7‐H3に結合し、かつ:
(i)その可変軽鎖(VL)ドメイン中に、CDRL1配列RASESIYSYLA(配列番号39)、CDRL2配列NTKTLPE(配列番号40)、及びCDRL3配列QHHYGTPPWT(配列番号41);並びに
(ii)その可変重鎖(VH)ドメイン中に、CDRH1配列SYGMS(配列番号42)、CDRH2配列TINSGGSNTYY PDSLKG(配列番号43)、及びCDRH3配列HDGGAMDY(配列番号44)
を含む、ヒト化B7‐H3抗体又はそのB7‐H3結合断片であり;
LMは、Ab及びDを共有結合させる少なくとも1つの結合又はリンカー分子(Linker Molecule)を含み;
mは0~nの整数であって、上記B7‐H3‐ADCの結合又はリンカー分子の数を表すが、LMが1つの結合である場合を除いてmは0ではなく;
nは1~10の整数であって、上記B7‐H3‐ADCに共有結合した上記細胞傷害性デュオカルマイシン部分の数を表す、E1~11のいずれか1つに記載の方法。
(I)配列番号17のアミノ酸配列を含むヒト化VLドメイン、及び
(II)配列番号18のアミノ酸配列を含むヒト化VHドメイン
を含む、E1~12のいずれか1つに記載の方法。
[V‐(W)k‐(X)1‐A]
で表され、従って上記B7‐H3‐ADCが以下の式:
Ab‐[V‐(W)k‐(X)1‐A]‐D
で表され、ここで:
Vは切断可能なリンカーであり;
(W)k‐(X)1‐Aは、l,(4+2n)消去によって自己消去する、細長い自己消去性スペーサ系であり;
W及びXはそれぞれl,(4+2n)電子カスケードスペーサであり、同一であるか又は異なっており;
Aは、式(Y)m(ここでYはl,(4+2n)電子カスケードスペーサである)のスペーサ基、又は式Uの基であり、環化除去スペーサであり;
k、1及びmは独立して、0~5(両端を含む)の整数であり;
nは0~10(両端を含む)の整数であり;
ただし:
Aが(Y)mである場合、k+l+m≧1であり;
k+l+m=lである場合、n>lであり;
AがUである場合、k+1≧1であり、
W、X及びYは独立して、以下の式:


Qは‐R5C=CR6‐、S、O、NR5、‐R5C=N‐又は‐N=CR5‐であり;
PはNR7、O又はSであり;
a、b及びcは独立して、0~5(両端を含む)の整数であり;
I、F及びGは独立して、式:

R1、R2、R3、R4、R5、R6、R7、R8及びR9は独立して、H、C1‐6アルキル、C3‐20ヘテロシクリル、C5‐20アリール、C1‐6アルコキシ、ヒドロキシ(OH)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)及びリン酸塩(OP(=O)(ORx)2)を表し、ここで:
Rx、Rx 1及びRx 2は独立して、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され;
上記置換基R1、R2、R3、R4、R5、R6、R7、R8又はR9のうちの2つ以上は任意に、互いに接続されて1つ以上の脂肪族又は芳香族環式構造を形成し;
Uは、式:
a、b及びcは独立して、0又は1の整数となるよう選択され;
ただしa+b+c=2又は3であり;
R1及び/又はR2は独立して、H、C1‐6アルキルを表し、上記アルキルは任意に、以下の基:ヒドロキシ(OH)、エーテル(ORx)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)、及びリン酸塩(OP(=O)(ORx)2)のうちの1つ以上によって置換され、ここでRx、Rx 1及びRx 2は、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され;
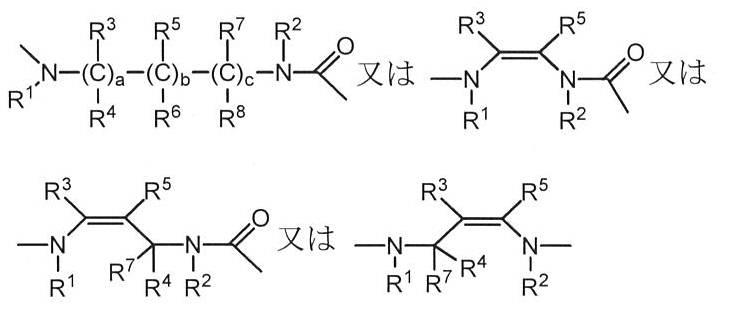
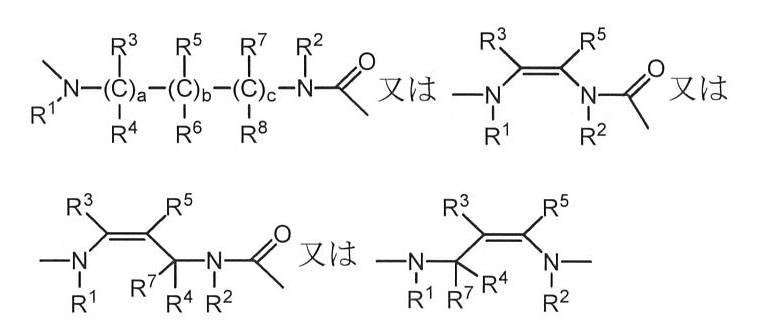
R3、R4、R5、R6、R7及びR8は独立して、H、C1‐6アルキル、C3‐20ヘテロシクリル、C5‐20アリール、C1‐6アルコキシ、ヒドロキシ(OH)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)、及びリン酸塩(OP(=O)(ORx)2)を表し、ここでRx、Rx 1及びRx 2は、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され、上記置換基R1、R2、R3、R4、R5、R6、R7、又はR8のうちの2つ以上は任意に、互いに接続されて1つ以上の脂肪族又は芳香族環式構造を形成する、E12~E20のいずれか1つに記載の方法。
(1)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(2)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(3)p‐アミノシンナミルオキシカルボニル;
(4)p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(5)p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
(6)p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
(7)p‐アミノフェニルペンタジエニルオキシカルボニル;
(8)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
(9)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(10)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノフェニルペンタジエニルオキシカルボニル;
(11)p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(12)p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(13)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(14)p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(15)p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)‐カルボニル;
(16)p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(17)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジル;
(18)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル‐p‐アミノベンジル;
(19)p‐アミノシンナミル;
(20)p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジル;
(21)p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミル;
(22)p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミル;
(23)p‐アミノフェニルペンタジエニル;
(24)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノシンナミル;
(25)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノベンジル;又は
(26)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノフェニルペンタジエニルを含む、E12~E21のいずれか1つに記載の方法。
上記VH CDR1はアミノ酸配列SYWMN(配列番号23)を含み;
上記VH CDR2はアミノ酸配列VIHPSDSETWLDQKFKD(配列番号24)を含み;
上記VH CDR3はアミノ酸配列EHYGTSPFAY(配列番号25)を含み、
上記抗体が、VL CDR1、VL CDR2、及びVL CDR3を含む可変軽鎖(VL)ドメインを含み:
上記VL CDR1はアミノ酸配列RASESVDNYGMSFMNW(配列番号26)を含み;
上記VL CDR2はアミノ酸配列AASNQGS(配列番号27)を含み;
上記VL CDR3はアミノ酸配列QQSKEVPYT(配列番号28)を含む、E10、E11、及びE41~E44のいずれか1つに記載の方法。
(A)約0.5mg/ml~約5mg/mlの上記B7‐H3‐ADCを含む医薬組成物;及び
(B)説明資料
を含む医薬キットで提供され、上記説明資料は、上記B7‐H3‐ADCを含む上記医薬組成物を、PD‐1結合分子を含む医薬組成物と任意に組み合わせて投与することを指示する、E1~E90のいずれか1つに記載の方法。
(I)配列番号17のアミノ酸配列を含むヒト化VLドメイン;及び
(II)配列番号18のアミノ酸配列を含むヒト化VHドメイン
を含む、E91又はE92に記載の方法。
PD‐1結合分子と組み合わされたB7‐H3‐ADCは、C57BL/6マウスにおいてインビボ抗腫瘍活性を示す
PD‐1結合分子と組み合わされた本発明のB7‐H3‐ADCの抗腫瘍活性を更に実証するために、任意に抗PD‐1抗体(RMP1‐14;BioXCell、米国ニューハンプシャー州レバノン)と組み合わされたB7‐H3‐ADCを、ヒトB7‐H3でトランスフェクトされたマウスMC38結腸直腸腫瘍細胞株(「MC38/hB7‐H3」)を用いて、C57BL/6同系マウスモデルにおいてインビボ毒性に関して評価した。簡潔に述べると、約5×105個の腫瘍細胞(1:1培地及びMATRIGEL(登録商標)中に懸濁)を、一群のC57BL/6マウス(Charles River Laboratories)に皮下移植した。15日目に腫瘍の体積がおよそ40~200mm3に達した時にマウスをランダム化し、B7‐H3‐ADC、抗PD‐1抗体、又は対照ビヒクルを腹腔内に投与した。これらの研究では、1用量のB7‐H3‐ADC(5mg/kg若しくは10mg/kg)又は対照ビヒクルを15日目に投与した。抗PD‐1抗体を、15、18、21、23、25、28、30、32、35、及び37日目に20mg/kg投与した。電子キャリパーを用いた直交測定によって、腫瘍を1週間に2回測定し、腫瘍体積を(長さ×幅×高さ)/2として算出した。(対照に対する)腫瘍体積を決定した(「T/C」)。処置済みの動物の腫瘍体積が研究期間中に≦5mm3まで減少するという発見は、完全奏功(Complete Response:「CR」)を意味するものと考えられた。部分奏功(Partial Response:「PR」)は、腫瘍が、研究中のいずれの時点において投薬日から50%以上減少している場合として定義された。抗腫瘍活性は、以下の米国立癌研究所(National Cancer Institute:NCI)の基準に従って評価された:T/C≦42%が抗腫瘍活性の最低レベルであり、>42%のT/C値は不活性である。T/C<10%は高活性とみなされる。
皮下移植されたMC38/hB7‐H3結腸直腸癌腫瘍細胞に対するこの研究の結果を表1及び図2に提示する。

PD‐1結合分子と組み合わされたB7‐H3‐ADCは、BALB/cマウスにおいてインビボ抗腫瘍活性を示す
PD‐1結合分子と組み合わされた本発明のB7‐H3‐ADCの抗腫瘍活性を更に実証するために、任意に抗PD‐1抗体(RMP1‐14;BioXCell、米国ニューハンプシャー州レバノン)と組み合わされたB7‐H3‐ADCを、ヒトB7‐H3でトランスフェクトされたマウスCT26結腸直腸腫瘍細胞株(「CT26/hB7‐H3」)を用いて、BALB/c同系マウスモデルにおいてインビボ毒性に関して評価した。簡潔に述べると、約5×105個の腫瘍細胞(1:1培地及びMATRIGEL(登録商標)中に懸濁)を、一群のBALB/cマウス(Charles River Laboratories)に皮下移植した。13日目に腫瘍の体積がおよそ40~100mm3に達した時にマウスをランダム化し、B7‐H3‐ADC、抗PD‐1抗体、又は対照ビヒクルを腹腔内に投与した。これらの研究では、1用量のB7‐H3‐ADC(10mg/kg)又は対照ビヒクルを13日目に投与した。抗PD‐1抗体を、13、16、19、22、26、29、33、及び36日目に20mg/kg投与した。電子キャリパーを用いた直交測定によって、腫瘍を1週間に2回測定し、腫瘍体積を(長さ×幅×高さ)/2として算出した。(対照に対する)腫瘍体積を決定した(「T/C」)。処置済みの動物の腫瘍体積が研究期間中に≦5mm3まで減少するという発見は、完全奏功(Complete Response:「CR」)を意味するものと考えられた。部分奏功(Partial Response:「PR」)は、腫瘍が、研究中のいずれの時点において投薬日から50%以上減少している場合として定義された。抗腫瘍活性は、以下の米国立癌研究所(National Cancer Institute:NCI)の基準に従って評価された:T/C≦42%が抗腫瘍活性の最低レベルであり、>42%のT/C値は不活性である。T/C<10%は高活性とみなされる。
皮下移植されたCT26/hB7‐H3結腸直腸癌腫瘍細胞に対するこの研究の結果を表2及び図3に提示する。

第I相試験
B7‐H3‐ADCに対する患者の忍容性を判断するために、第I相臨床研究を実施することになる。この研究は、用量漸増段階及びコホート拡大段階を含む。この研究は、各臨床施設の治験審査委員会によって承認され、全ての患者が書面によるインフォームドコンセントに署名する。


治療関連有害事象
Q3W用量漸増での23人の患者の治療後の所見を提供する。表6に示されているように、治療関連有害事象(treatment-related adverse event:TRAE)は22/23(91.7%)の患者で発生し、最も一般的なのは好中球減少症(n=6)、リンパ球減少症(n=3)、手掌・足底発赤知覚不全症候群(n=5)、及び斑状丘疹状皮疹(n=3)であった。グレード3以上のTRAEの割合は58.3%であった。3つの治療関連の重篤な有害事象が3人の患者で発生した:1)細菌性肺炎を併発した患者における肺臓炎;2)非感染性胃腸炎;及び3)慢性静脈不全患者におけるうっ血性皮膚炎。ベースラインまで回復したグレード4の好中球減少症の、1つの用量規定毒性(dose-limiting toxicity:「DLT」)が報告された。発熱性好中球減少症は観察されなかった。

図4は、0.5mg/kg、1mg/kg、2mg/kg、3mg/kg、又は4mg/kgでQ3WのB7‐H3‐ADC単剤療法を受けた、26人の応答評価可能な用量漸増及びコホート拡大患者間での、標的病変の縮小の割合を実証する、ウォーターフォールプロットを示す。患者は、用量漸増においては治療後6週間ごとに、コホート拡大においては治療後9週間ごとに撮像された。B7‐H3‐ADCを少なくとも1用量投与され、少なくとも1回のベースライン後腫瘍評価を受けた患者からのデータを示す。用量漸増コホートの患者は、非小細胞肺癌(NSCLC)、ブドウ膜黒色腫、黒色腫、前立腺癌、転移性去勢抵抗性前立腺癌(mCRPC)、小細胞肺癌(SCLC)、結腸直腸癌(CRC)、卵巣癌、腎細胞癌(RCC)、膵臓癌、肉腫、及び食道癌の患者を含んでいた。1人の転移性去勢抵抗性前立腺癌(mCRPC)患者は、B7‐H3‐ADCを5回投与され;最初の4回は2mg/kg投与され、5回目は1mg/kg投与された。このmCRPC患者では、標的病変がベースラインに比べておよそ30%縮小した。1人のNSCLC患者は、2mg/kgのB7‐H3‐ADCを6回投与され、標的病変がベースラインに比べて24%縮小した。図5は、Q3Wでの、2mg/kgのB7‐H3‐ADCの2回の投薬後の、この患者のコンピュータ断層撮影(CT)による肺の画像スキャンを示す。臨床研究者が指摘するように、前後方向の切片の(矢印で示されている)肺の病変は、およそ24%の縮小を示した。図4はまた、3mg/kgのB7‐H3‐ADCを投与された7人の応答評価可能なコホート拡大患者間での、標的病変の縮小の割合も示す。患者のうち2人はNSCLCに罹患しており、患者のうち4人はmCRPCに罹患している。患者はB7‐H3‐ADCを少なくとも1用量投与され、少なくとも1回のベースライン後腫瘍評価を受けた。この研究は進行中であり、データはまだ十分でない。
9人のmCRPC患者を、1mg/kg、2mg/kg、3mg/kg、又は4mg/kgのB7‐H3‐ADCを用いて、用量漸増のQ3Wで治療した。ベースライン、並びに6週目、12週目、及び19週目に血液試料を患者から採取し、PSAのレベルを試験した。PSAレベルは、通常の試験を用いて臨床現場で測定された。5人の患者(71%)で、PSAレベルはベースラインから50~95%の範囲で大幅に低下し、骨疾患が実質的に退縮した1人の患者が含まれていた。mCRPC患者、臨床応答、及びPSAの結果の概要を表7に示す。

免疫組織化学(IHC)を用いて、患者の前立腺腫瘍及び他の固形腫瘍からの生検でm,B7‐H3の発現を分析した。18人の患者が、B7‐H3発現に関して評価可能な組織試料を有していた。使用した具体的なアッセイは、Ventana Medical Systems, Inc.(「Ventana」;アリゾナ州ツーソン)のB7‐H3(SP206)IHCアッセイであった。腫瘍内の細胞の細胞膜、及び血管系における、B7‐H3の発現は、強度0~3+のHスコアで表される。各染色強度レベルの細胞の割合を算出し、以下の式を用いてHスコアを割り当てる:
Hスコア=[1×(%細胞1+)+2×(%細胞2+)+3×(%細胞3+)]
B7‐H3発現に関するHスコアの範囲は、腫瘍において82~279であり、スコア中央値は200であった。血管系におけるB7‐H3発現の範囲はゼロ~2+であり、スコア中央値は2+であった。
Claims (44)
- 抗B7‐H3抗体‐薬物コンジュゲート(B7‐H3‐ADC)の投与を必要とする被験者に前記B7‐H3‐ADCを投与するステップを含む、癌を治療する方法であって、
前記方法は、前記B7‐H3‐ADCを前記被験者に、約0.5mg/kg~約5mg/kgの用量で3週間に1回投与するステップを含む、方法。 - 前記B7‐H3‐ADCは以下の式:
Ab‐(LM)m‐(D)n
で表され、ここで:
Abは、B7‐H3に結合し、かつ:
(i)その可変軽鎖(VL)ドメイン中に、CDRL1配列RASESIYSYLA(配列番号39)、CDRL2配列NTKTLPE(配列番号40)、及びCDRL3配列QHHYGTPPWT(配列番号41);並びに
(ii)その可変重鎖(VH)ドメイン中に、CDRH1配列SYGMS(配列番号42)、CDRH2配列TINSGGSNTYY PDSLKG(配列番号43)、及びCDRH3配列HDGGAMDY(配列番号44)
を含む、ヒト化B7‐H3抗体又はそのB7‐H3結合断片であり;
Dは、細胞傷害性デュオカルマイシン部分であり;
LMは、Ab及びDを共有結合させる少なくとも1つの結合又はリンカー分子を含み;
mは0~nの整数であって、前記B7‐H3‐ADCの結合又はリンカー分子の数を表すが、LMが1つの結合である場合を除いてmは0ではなく;
nは1~10の整数であって、前記B7‐H3‐ADC分子に共有結合した前記細胞傷害性デュオカルマイシン部分の数を表す、請求項1に記載の方法。 - 前記Abは:
(i)配列番号17のアミノ酸配列を含むヒト化可変軽鎖(VL)ドメイン、及び
(ii)配列番号18のアミノ酸配列を含むヒト化可変重鎖(VH)ドメイン
を含む、請求項2に記載の方法。 - 前記AbはヒトIgGのFcドメインを更に含む、請求項2又は3に記載の方法。
- 前記ヒトIgGはヒトIgG1、IgG2、IgG3、又はIgG4である、請求項4に記載の方法。
- 前記Fcドメインは変異型Fcドメインであり、前記変異型Fcドメインは:
(a)FcγRに対する前記変異型Fcドメインの親和性を低減する1つ以上のアミノ酸修飾;及び/又は
(b)前記変異型Fcドメインの血清半減期を増大させる1つ以上のアミノ酸修飾
を含む、請求項4又は5に記載の方法。 - FcγRに対する前記変異型Fcドメインの親和性を低減する前記修飾は:L234A;L235A;又はL234A及びL235Aの置換を含み、前記番号付与はKabatに記載のEUインデックスのものである、請求項6に記載の方法。
- 前記変異型Fcドメインの血清半減期を増大させる前記修飾は:M252Y;M252Y及びS254T;M252Y及びT256E;M252Y、S254T及びT256E;又はK288D及びH435Kの置換を含み、前記番号付与はKabatに記載のEUインデックスのものである、請求項5又は6に記載の方法。
- 前記LMのうちの少なくとも1つはリンカー分子である、請求項2~8のいずれか1項に記載の方法。
- 前記LMリンカー分子はペプチドリンカーである、請求項2~9のいずれか1項に記載の方法。
- 前記ペプチドリンカーはバリン‐シトルリンジペプチドリンカーである、請求項10に記載の方法。
- 前記LMリンカー分子は、切断可能なリンカーとDとの間に自己消去性スペーサを更に含む、請求項2~11のいずれか1項に記載の方法。
- 前記自己消去性スペーサはパラ‐アミノベンジルオキシカルボニル部分を含む、請求項12に記載の方法。
- 前記リンカー分子は、前記切断可能なリンカーとAbとの間にマレイミドリンカー部分を更に含む、請求項2~13のいずれか1項に記載の方法。
- LMは以下の式:
[V‐(W)k‐(X)1‐A]
で表され、従って前記B7‐H3‐ADCは以下の式:
Ab‐[V‐(W)k‐(X)1‐A]‐D
で表され、ここで:
Vは切断可能なリンカーであり;
(W)k‐(X)1‐Aは、l,(4+2n)消去によって自己消去する、細長い自己消去性スペーサ系であり;
W及びXはそれぞれl,(4+2n)電子カスケードスペーサであり、同一であるか又は異なっており;
Aは、式(Y)m(ここでYはl,(4+2n)電子カスケードスペーサである)のスペーサ基、又は式Uの基であり、環化除去スペーサであり;
k、1及びmは独立して、0~5(両端を含む)の整数であり;
nは0~10(両端を含む)の整数であり;
ただし:
Aが(Y)mである場合、k+l+m≧1であり;
k+l+m=lである場合、n>lであり;
AがUである場合、k+1≧1であり、
W、X及びYは独立して、以下の式:
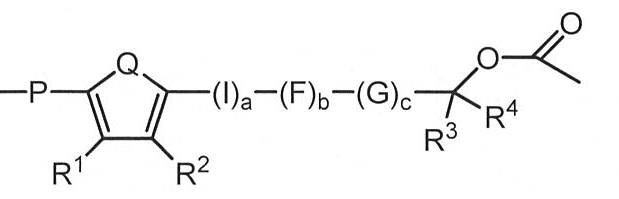

Qは‐R5C=CR6‐、S、O、NR5、‐R5C=N‐又は‐N=CR5‐であり;
PはNR7、O又はSであり;
a、b及びcは独立して、0~5(両端を含む)の整数であり;
I、F及びGは独立して、式:
R1、R2、R3、R4、R5、R6、R7、R8及びR9は独立して、H、C1‐6アルキル、C3‐20ヘテロシクリル、C5‐20アリール、C1‐6アルコキシ、ヒドロキシ(OH)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)及びリン酸塩(OP(=O)(ORx)2)を表し、ここで:
Rx、Rx 1及びRx 2は独立して、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され;
前記置換基R1、R2、R3、R4、R5、R6、R7、R8又はR9のうちの2つ以上は任意に、互いに接続されて1つ以上の脂肪族又は芳香族環式構造を形成し;
Uは、式:
a、b及びcは独立して、0又は1の整数となるよう選択され;
ただしa+b+c=2又は3であり;
R1及び/又はR2は独立して、H、C1‐6アルキルを表し、前記アルキルは任意に、以下の基:ヒドロキシ(OH)、エーテル(ORx)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)、及びリン酸塩(OP(=O)(ORx)2)のうちの1つ以上によって置換され、ここでRx、Rx 1及びRx 2は、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され;
R3、R4、R5、R6、R7及びR8は独立して、H、C1‐6アルキル、C3‐20ヘテロシクリル、C5‐20アリール、C1‐6アルコキシ、ヒドロキシ(OH)、アミノ(NH2)、モノ置換アミノ(NRxH)、ジ置換アミノ(NRx 1Rx 2)、ニトロ(NO2)、ハロゲン、CF3、CN、CONH2、SO2Me、CONHMe、環式C1‐5アルキルアミノ、イミダゾリル、C1‐6アルキルピペラジニル、モルホリノ、チオール(SH)、チオエーテル(SRx)、テトラゾール、カルボキシ(COOH)、カルボン酸塩(COORx)、スルホキシ(S(=O)2OH)、スルホン酸塩(S(=O)2ORx)、スルホニル(S(=O)2Rx)、スルフィキシ(S(=O)OH)、スルフィン酸塩(S(=O)ORx)、スルフィニル(S(=O)Rx)、ホスホノオキシ(OP(=O)(OH)2)、及びリン酸塩(OP(=O)(ORx)2)を表し、ここでRx、Rx 1及びRx 2は、C1‐6アルキル基、C3‐20ヘテロシクリル基又はC5‐20アリール基から選択され、前記置換基R1、R2、R3、R4、R5、R6、R7、又はR8のうちの2つ以上は任意に、互いに接続されて1つ以上の脂肪族又は芳香族環式構造を形成する、請求項1~14のいずれか1項に記載の方法。 - 前記LMリンカー分子は:
(1)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(2)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(3)p‐アミノシンナミルオキシカルボニル;
(4)p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(5)p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
(6)p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
(7)p‐アミノフェニルペンタジエニルオキシカルボニル;
(8)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル;
(9)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノベンジルオキシカルボニル;
(10)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノフェニルペンタジエニルオキシカルボニル;
(11)p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(12)p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(13)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(14)p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(15)p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)‐カルボニル;
(16)p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミルオキシカルボニル(メチルアミノ)エチル(メチルアミノ)カルボニル;
(17)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジル;
(18)p‐アミノベンジルオキシカルボニル‐p‐アミノベンジルオキシカルボニル‐p‐アミノベンジル;
(19)p‐アミノシンナミル;
(20)p‐アミノシンナミルオキシカルボニル‐p‐アミノベンジル;
(21)p‐アミノベンジルオキシカルボニル‐p‐アミノシンナミル;
(22)p‐アミノシンナミルオキシカルボニル‐p‐アミノシンナミル;
(23)p‐アミノフェニルペンタジエニル;
(24)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノシンナミル;
(25)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノベンジル;又は
(26)p‐アミノフェニルペンタジエニルオキシカルボニル‐p‐アミノフェニルペンタジエニルを含む、請求項15に記載の方法。 - 前記LMリンカー分子は、Abのポリペプチド鎖のアミノ酸の側鎖に対して複合体化されて、前記Abを前記細胞傷害性デュオカルマイシン部分Dの分子に結合させる、請求項2~16のいずれか1項に記載の方法。
- 前記細胞傷害性デュオカルマイシン部分Dは、デュオカルマイシンA、デュオカルマイシンB1、デュオカルマイシンB2、デュオカルマイシンC1、デュオカルマイシンC2、デュオカルマイシンD、デュオカルマイシンSA、CC‐1065、アドゼレシン、ビゼレシン、カルゼルシン(U‐80244)、セコ‐デュオカルマイシン、及びスピロ‐デュオカルマイシン(DUBA)からなる群から選択されるデュオカルマイシン細胞毒素を含む、請求項2~17のいずれか1項に記載の方法。
- 前記細胞傷害性デュオカルマイシン部分Dはセコ‐デュオカルマイシンを含む、請求項18に記載の方法。
- 前記LMリンカー分子は、還元型鎖間ジスルフィドを介して前記Abと共有結合する、請求項2~19のいずれか1項に記載の方法。
- 前記B7‐H3‐ADCは約3mg/kgの用量で投与される、請求項1~20のいずれか1項に記載の方法。
- 前記B7‐H3‐ADCは約3.5mg/kgの用量で投与される、請求項1~20のいずれか1項に記載の方法。
- 前記B7‐H3‐ADCは約4mg/kgの用量で投与される、請求項1~20のいずれか1項に記載の方法。
- 前記B7‐H3‐ADCは静脈内(IV)注入によって投与される、請求項1~23のいずれか1項に記載の方法。
- 前記IV注入は約60分の期間にわたる、請求項24に記載の方法。
- 前記B7‐H3‐ADCは、治療上有効な用量のPD‐1結合分子と組み合わせて投与される、請求項1~25のいずれか1項に記載の方法。
- 前記癌は:副腎癌;AIDS関連癌;胞巣状軟部肉腫;星細胞腫瘍;肛門癌;肛門管の扁平上皮癌(SCAC);膀胱癌;骨癌;脳及び脊髄癌;転移性脳腫瘍;B細胞癌;乳癌;HER2+乳癌;トリプルネガティブ乳癌(TNBC);頸動脈球腫瘍;子宮頸癌;軟骨肉腫;脊索腫;嫌色素性腎細胞癌;明細胞癌;大腸癌;結腸直腸癌;皮膚良性線維性組織球腫;線維形成性小円形細胞腫瘍;上衣腫;ユーイング腫瘍;骨外性粘液型軟骨肉腫;骨性線維形成不全症;線維性骨異形成症;胆嚢又は胆管癌;消化器癌;妊娠性絨毛性疾患;胚細胞腫瘍;頭頸部癌;神経膠芽腫;血液悪性腫瘍;肝細胞癌;膵島細胞腫;カポジ肉腫;腎臓癌;白血病;急性骨髄性白血病;脂肪肉腫/悪性リポソーム腫瘍;肝臓癌;リンパ腫;肺癌;非小細胞肺癌(NSCLC);髄芽腫;黒色腫;髄膜腫;中皮腫咽頭癌;多発性内分泌腫瘍;多発性骨髄腫;骨髄異形成症候群;神経芽細胞腫;神経内分泌腫瘍;卵巣癌;膵臓癌;乳頭状甲状腺癌;副甲状腺腫瘍;小児癌;末梢神経鞘腫瘍;褐色細胞腫;下垂体腫瘍;前立腺癌;転移性去勢抵抗性前立腺癌(mCRPC);後部ブドウ膜黒色腫;腎転移性癌;ラブドイド腫瘍;横紋筋肉腫;肉腫;皮膚癌;小児期の小円形青色細胞腫瘍;神経芽細胞腫;軟組織肉腫;扁平上皮細胞癌;頭頸部の扁平上皮細胞癌(SCCHN);胃癌;滑膜肉腫;精巣癌;胸腺癌;胸腺腫;甲状腺癌;甲状腺転移性癌;及び子宮癌からなる群から選択される、請求項1~26のいずれか1項に記載の方法。
- 前記癌は:副腎癌;肛門癌;SCAC;膀胱癌;乳癌;結腸直腸癌;消化器癌;神経膠芽腫;腎臓癌;肺癌;NSCLC;急性リンパ性白血病;急性骨髄性白血病;慢性リンパ球性白血病;慢性骨髄性白血病;有毛細胞白血病;バーキットリンパ腫;びまん性大細胞型B細胞リンパ腫;濾胞性リンパ腫;マントル細胞リンパ腫;辺縁帯リンパ腫;中皮腫咽頭癌;非ホジキンリンパ腫;小リンパ球性リンパ腫;多発性骨髄腫;黒色腫;卵巣癌;膵臓癌;後部ブドウ膜黒色腫;前立腺癌;mCRPC;皮膚癌;腎細胞癌;小児期の小円形青色細胞腫瘍;神経芽細胞腫;横紋筋肉腫;扁平上皮細胞癌;SCCHN;精巣癌;甲状腺癌;甲状腺転移性癌;及び子宮癌からなる群から選択される、請求項27に記載の方法。
- 前記癌は前立腺癌である、請求項27又は28に記載の方法。
- 前記前立腺癌はmCRPCである、請求項27~29のいずれか1項に記載の方法。
- 前記癌は肛門癌である、請求項27又は28に記載の方法。
- 前記肛門癌はSCACである、請求項27、28、及び31のいずれか1項に記載の方法。
- 前記癌は扁平上皮細胞癌である、請求項27又は28に記載の方法。
- 前記扁平上皮細胞癌はSCCHNである、請求項27、28、及び33のいずれか1項に記載の方法。
- 前記癌は乳癌である、請求項27又は28に記載の方法。
- 前記乳癌はTNBCである、請求項27、28、及び35のいずれか1項に記載の方法。
- 前記癌は黒色腫である、請求項27又は28に記載の方法。
- 前記黒色腫はブドウ膜黒色腫である、請求項27、28、及び37のいずれか1項のいずれか1項に記載の方法。
- 前記癌は肺癌である、請求項27又は28に記載の方法。
- 前記肺癌はNSCLCである、請求項27、28、及び39のいずれか1項に記載の方法。
- 治療又は予防有効量の、1つ以上の追加の治療剤又は化学療法剤を投与するステップを更に含む、請求項1~40のいずれか1項に記載の方法。
- 前記化学療法剤は白金系化学療法剤である、請求項41に記載の方法。
- 前記化学療法剤はタキサンである、請求項41に記載の方法。
- 前記B7‐H3‐ADCの投与を必要とする前記被験者はヒトである、請求項1~43のいずれか1項に記載の方法。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202063023495P | 2020-05-12 | 2020-05-12 | |
| US63/023,495 | 2020-05-12 | ||
| US202163180795P | 2021-04-28 | 2021-04-28 | |
| US63/180,795 | 2021-04-28 | ||
| PCT/US2021/031598 WO2021231309A1 (en) | 2020-05-12 | 2021-05-10 | Methods for the use of a b7-h3 antibody-drug conjugate alone or in combination |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2023525320A true JP2023525320A (ja) | 2023-06-15 |
| JPWO2021231309A5 JPWO2021231309A5 (ja) | 2024-05-20 |
Family
ID=78524901
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022568560A Pending JP2023525320A (ja) | 2020-05-12 | 2021-05-10 | B7‐h3抗体薬物複合体の単独使用又は併用のための方法 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20230226206A1 (ja) |
| EP (1) | EP4149549A4 (ja) |
| JP (1) | JP2023525320A (ja) |
| KR (1) | KR20230009397A (ja) |
| CN (1) | CN115484982A (ja) |
| AU (1) | AU2021273467A1 (ja) |
| BR (1) | BR112022022900A2 (ja) |
| CA (1) | CA3181535A1 (ja) |
| IL (1) | IL298105A (ja) |
| MX (1) | MX2022014068A (ja) |
| TW (1) | TW202207991A (ja) |
| WO (1) | WO2021231309A1 (ja) |
| ZA (1) | ZA202211455B (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7564958B2 (ja) * | 2020-12-18 | 2024-10-09 | シャンハイ フダン-チャンジャン バイオ-ファーマシューティカル カンパニー リミテッド | B7-h3を標的とする抗体薬物複合体、その製造方法と使用 |
| TW202515621A (zh) * | 2023-08-25 | 2025-04-16 | 美商宏觀基因有限公司 | B7—h3抗體藥物偶聯物 |
| WO2025167922A1 (zh) * | 2024-02-07 | 2025-08-14 | 上海翰森生物医药科技有限公司 | 抗体药物偶联物和抗egfr抗体的联合应用 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019520308A (ja) * | 2016-04-15 | 2019-07-18 | マクロジェニクス,インコーポレーテッド | 新規のb7‐h3結合分子、その抗体薬物コンジュゲート、及びその使用方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG10201604336VA (en) * | 2010-03-04 | 2016-07-28 | Macrogenics Inc | Antibodies Reactive With B7-H3, Immunologically Active Fragments Thereof And Uses Thereof |
| CN110305213B (zh) * | 2018-11-09 | 2023-03-10 | 泰州复旦张江药业有限公司 | 一种抗b7-h3抗体及其制备方法、其偶联物和应用 |
| KR20210063070A (ko) * | 2019-11-22 | 2021-06-01 | 주식회사 레고켐 바이오사이언스 | 항-b7-h3 항체를 포함하는 항체-약물 접합체 및 이의 용도 |
-
2021
- 2021-05-10 JP JP2022568560A patent/JP2023525320A/ja active Pending
- 2021-05-10 KR KR1020227040047A patent/KR20230009397A/ko active Pending
- 2021-05-10 CN CN202180031984.8A patent/CN115484982A/zh active Pending
- 2021-05-10 EP EP21805196.9A patent/EP4149549A4/en active Pending
- 2021-05-10 US US17/998,455 patent/US20230226206A1/en active Pending
- 2021-05-10 BR BR112022022900A patent/BR112022022900A2/pt unknown
- 2021-05-10 WO PCT/US2021/031598 patent/WO2021231309A1/en not_active Ceased
- 2021-05-10 MX MX2022014068A patent/MX2022014068A/es unknown
- 2021-05-10 IL IL298105A patent/IL298105A/en unknown
- 2021-05-10 AU AU2021273467A patent/AU2021273467A1/en active Pending
- 2021-05-10 CA CA3181535A patent/CA3181535A1/en active Pending
- 2021-05-11 TW TW110117006A patent/TW202207991A/zh unknown
-
2022
- 2022-10-19 ZA ZA2022/11455A patent/ZA202211455B/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019520308A (ja) * | 2016-04-15 | 2019-07-18 | マクロジェニクス,インコーポレーテッド | 新規のb7‐h3結合分子、その抗体薬物コンジュゲート、及びその使用方法 |
Non-Patent Citations (2)
| Title |
|---|
| CANCER RESEARCH, vol. Vol.78, Suppl.13, JPN6025026599, 2018, pages 820, ISSN: 0005628895 * |
| MOLECULAR CANCER THERAPEUTICS, vol. 19, no. 11, JPN6025026600, 2020, pages 2235 - 2244, ISSN: 0005628896 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CA3181535A1 (en) | 2021-11-18 |
| ZA202211455B (en) | 2023-11-29 |
| TW202207991A (zh) | 2022-03-01 |
| BR112022022900A2 (pt) | 2022-12-20 |
| EP4149549A1 (en) | 2023-03-22 |
| MX2022014068A (es) | 2022-12-07 |
| EP4149549A4 (en) | 2024-07-03 |
| CN115484982A (zh) | 2022-12-16 |
| IL298105A (en) | 2023-01-01 |
| AU2021273467A1 (en) | 2022-11-24 |
| US20230226206A1 (en) | 2023-07-20 |
| KR20230009397A (ko) | 2023-01-17 |
| WO2021231309A1 (en) | 2021-11-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12331132B2 (en) | Multispecific polypeptide constructs having constrained CD3 binding and related methods and uses | |
| US20230272084A1 (en) | B7-h3 directed antibody drug conjugates | |
| US20240101704A1 (en) | Multispecific polypeptide constructs having constrained cd3 binding and methods of using the same | |
| JP7745113B2 (ja) | 多重特異性抗体およびその使用 | |
| JP2023525320A (ja) | B7‐h3抗体薬物複合体の単独使用又は併用のための方法 | |
| US20250135022A1 (en) | Methods for the use of a b7-h3 antibody-drug conjugate in combination with a pd-1 x ctla-4 bispecific molecule | |
| CN118804928A (zh) | 用于与pd-1x ctla-4双特异性分子组合使用b7-h3抗体-药物缀合物的方法 | |
| AU2019251488B2 (en) | Multispecific polypeptide constructs having constrained CD3 binding and related methods and uses | |
| CN118215680A (zh) | 在治疗疾病中应用的多特异性抗体 | |
| EA042568B1 (ru) | Новые в7-н3 связывающие молекулы, содержащие их конъюгаты антитело-лекарственное средство и способы их применения |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240508 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20240508 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20250627 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20250924 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20251127 |