JP2004509613A - 非同調的刺激pcr - Google Patents
非同調的刺激pcr Download PDFInfo
- Publication number
- JP2004509613A JP2004509613A JP2002502178A JP2002502178A JP2004509613A JP 2004509613 A JP2004509613 A JP 2004509613A JP 2002502178 A JP2002502178 A JP 2002502178A JP 2002502178 A JP2002502178 A JP 2002502178A JP 2004509613 A JP2004509613 A JP 2004509613A
- Authority
- JP
- Japan
- Prior art keywords
- primer
- target
- probe
- temperature
- strand
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000000523 sample Substances 0.000 claims abstract description 244
- 238000000137 annealing Methods 0.000 claims abstract description 97
- 230000000295 complement effect Effects 0.000 claims abstract description 68
- 150000007523 nucleic acids Chemical class 0.000 claims abstract description 67
- 102000039446 nucleic acids Human genes 0.000 claims abstract description 64
- 108020004707 nucleic acids Proteins 0.000 claims abstract description 64
- 238000002866 fluorescence resonance energy transfer Methods 0.000 claims abstract description 49
- 238000003199 nucleic acid amplification method Methods 0.000 claims abstract description 40
- 230000003321 amplification Effects 0.000 claims abstract description 39
- 238000000034 method Methods 0.000 claims description 133
- 108020004414 DNA Proteins 0.000 claims description 92
- 108091093088 Amplicon Proteins 0.000 claims description 68
- 238000009396 hybridization Methods 0.000 claims description 66
- 125000003729 nucleotide group Chemical group 0.000 claims description 64
- 239000002773 nucleotide Substances 0.000 claims description 60
- 239000000975 dye Substances 0.000 claims description 59
- 102000040430 polynucleotide Human genes 0.000 claims description 42
- 108091033319 polynucleotide Proteins 0.000 claims description 42
- 239000002157 polynucleotide Substances 0.000 claims description 42
- 102000053602 DNA Human genes 0.000 claims description 35
- 239000003153 chemical reaction reagent Substances 0.000 claims description 34
- -1 7-deaza-8-azaguanine Chemical compound 0.000 claims description 31
- 239000007850 fluorescent dye Substances 0.000 claims description 28
- 239000002299 complementary DNA Substances 0.000 claims description 24
- 239000000203 mixture Substances 0.000 claims description 20
- 230000008859 change Effects 0.000 claims description 19
- 239000001226 triphosphate Substances 0.000 claims description 19
- 235000000346 sugar Nutrition 0.000 claims description 16
- 238000004925 denaturation Methods 0.000 claims description 14
- 230000036425 denaturation Effects 0.000 claims description 14
- 230000008569 process Effects 0.000 claims description 14
- PEHVGBZKEYRQSX-UHFFFAOYSA-N 7-deaza-adenine Chemical compound NC1=NC=NC2=C1C=CN2 PEHVGBZKEYRQSX-UHFFFAOYSA-N 0.000 claims description 12
- 239000000178 monomer Substances 0.000 claims description 11
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical group O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 claims description 10
- 239000000872 buffer Substances 0.000 claims description 10
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical group NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 claims description 10
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical group O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 claims description 10
- 238000004519 manufacturing process Methods 0.000 claims description 10
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical group CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 claims description 10
- 239000011230 binding agent Substances 0.000 claims description 9
- 239000003607 modifier Substances 0.000 claims description 9
- PIINGYXNCHTJTF-UHFFFAOYSA-N 2-(2-azaniumylethylamino)acetate Chemical compound NCCNCC(O)=O PIINGYXNCHTJTF-UHFFFAOYSA-N 0.000 claims description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims description 6
- 230000003287 optical effect Effects 0.000 claims description 6
- 229930024421 Adenine Chemical group 0.000 claims description 5
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical group NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 claims description 5
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 5
- 239000004472 Lysine Substances 0.000 claims description 5
- 108020004682 Single-Stranded DNA Proteins 0.000 claims description 5
- 229960000643 adenine Drugs 0.000 claims description 5
- 229940104302 cytosine Drugs 0.000 claims description 5
- 229910052731 fluorine Inorganic materials 0.000 claims description 5
- 229940113082 thymine Drugs 0.000 claims description 5
- 229940035893 uracil Drugs 0.000 claims description 5
- PZOUSPYUWWUPPK-UHFFFAOYSA-N 4-methyl-1h-indole Chemical compound CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 claims description 4
- OIVLITBTBDPEFK-UHFFFAOYSA-N 5,6-dihydrouracil Chemical compound O=C1CCNC(=O)N1 OIVLITBTBDPEFK-UHFFFAOYSA-N 0.000 claims description 4
- 150000001413 amino acids Chemical group 0.000 claims description 4
- 125000003118 aryl group Chemical group 0.000 claims description 4
- 229910052799 carbon Inorganic materials 0.000 claims description 4
- 229910052801 chlorine Inorganic materials 0.000 claims description 4
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 claims description 4
- DRAVOWXCEBXPTN-UHFFFAOYSA-N isoguanine Chemical compound NC1=NC(=O)NC2=C1NC=N2 DRAVOWXCEBXPTN-UHFFFAOYSA-N 0.000 claims description 4
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 claims description 4
- LHCPRYRLDOSKHK-UHFFFAOYSA-N 7-deaza-8-aza-adenine Chemical compound NC1=NC=NC2=C1C=NN2 LHCPRYRLDOSKHK-UHFFFAOYSA-N 0.000 claims description 3
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 claims description 3
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 3
- 125000004432 carbon atom Chemical group C* 0.000 claims description 3
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 claims description 3
- 239000013612 plasmid Substances 0.000 claims description 3
- 239000001022 rhodamine dye Substances 0.000 claims description 3
- RGNOTKMIMZMNRX-XVFCMESISA-N 2-amino-1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-4-one Chemical compound NC1=NC(=O)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 RGNOTKMIMZMNRX-XVFCMESISA-N 0.000 claims description 2
- MPDKOGQMQLSNOF-GBNDHIKLSA-N 2-amino-5-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1h-pyrimidin-6-one Chemical compound O=C1NC(N)=NC=C1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 MPDKOGQMQLSNOF-GBNDHIKLSA-N 0.000 claims description 2
- MWBWWFOAEOYUST-UHFFFAOYSA-N 2-aminopurine Chemical compound NC1=NC=C2N=CNC2=N1 MWBWWFOAEOYUST-UHFFFAOYSA-N 0.000 claims description 2
- HCGYMSSYSAKGPK-UHFFFAOYSA-N 2-nitro-1h-indole Chemical compound C1=CC=C2NC([N+](=O)[O-])=CC2=C1 HCGYMSSYSAKGPK-UHFFFAOYSA-N 0.000 claims description 2
- FTBBGQKRYUTLMP-UHFFFAOYSA-N 2-nitro-1h-pyrrole Chemical compound [O-][N+](=O)C1=CC=CN1 FTBBGQKRYUTLMP-UHFFFAOYSA-N 0.000 claims description 2
- OGVOXGPIHFKUGM-UHFFFAOYSA-N 3H-imidazo[2,1-i]purine Chemical compound C12=NC=CN2C=NC2=C1NC=N2 OGVOXGPIHFKUGM-UHFFFAOYSA-N 0.000 claims description 2
- XXSIICQLPUAUDF-TURQNECASA-N 4-amino-1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-prop-1-ynylpyrimidin-2-one Chemical compound O=C1N=C(N)C(C#CC)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 XXSIICQLPUAUDF-TURQNECASA-N 0.000 claims description 2
- OVONXEQGWXGFJD-UHFFFAOYSA-N 4-sulfanylidene-1h-pyrimidin-2-one Chemical compound SC=1C=CNC(=O)N=1 OVONXEQGWXGFJD-UHFFFAOYSA-N 0.000 claims description 2
- NBAKTGXDIBVZOO-UHFFFAOYSA-N 5,6-dihydrothymine Chemical compound CC1CNC(=O)NC1=O NBAKTGXDIBVZOO-UHFFFAOYSA-N 0.000 claims description 2
- GSPMCUUYNASDHM-UHFFFAOYSA-N 5-methyl-4-sulfanylidene-1h-pyrimidin-2-one Chemical compound CC1=CNC(=O)N=C1S GSPMCUUYNASDHM-UHFFFAOYSA-N 0.000 claims description 2
- BXJHWYVXLGLDMZ-UHFFFAOYSA-N 6-O-methylguanine Chemical compound COC1=NC(N)=NC2=C1NC=N2 BXJHWYVXLGLDMZ-UHFFFAOYSA-N 0.000 claims description 2
- DZHQWVMWRUHHFF-GBNDHIKLSA-N 6-amino-5-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1h-pyrimidin-2-one Chemical compound NC1=NC(=O)NC=C1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 DZHQWVMWRUHHFF-GBNDHIKLSA-N 0.000 claims description 2
- CKOMXBHMKXXTNW-UHFFFAOYSA-N 6-methyladenine Chemical compound CNC1=NC=NC2=C1N=CN2 CKOMXBHMKXXTNW-UHFFFAOYSA-N 0.000 claims description 2
- LOSIULRWFAEMFL-UHFFFAOYSA-N 7-deazaguanine Chemical compound O=C1NC(N)=NC2=C1CC=N2 LOSIULRWFAEMFL-UHFFFAOYSA-N 0.000 claims description 2
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 claims description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 claims description 2
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 claims description 2
- 229930010555 Inosine Natural products 0.000 claims description 2
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 claims description 2
- 102100034434 Nebulin Human genes 0.000 claims description 2
- NWUTZAVMDAGNIG-UHFFFAOYSA-N O(4)-methylthymine Chemical compound COC=1NC(=O)N=CC=1C NWUTZAVMDAGNIG-UHFFFAOYSA-N 0.000 claims description 2
- 229930185560 Pseudouridine Natural products 0.000 claims description 2
- PTJWIQPHWPFNBW-UHFFFAOYSA-N Pseudouridine C Natural products OC1C(O)C(CO)OC1C1=CNC(=O)NC1=O PTJWIQPHWPFNBW-UHFFFAOYSA-N 0.000 claims description 2
- 235000003704 aspartic acid Nutrition 0.000 claims description 2
- WGDUUQDYDIIBKT-UHFFFAOYSA-N beta-Pseudouridine Natural products OC1OC(CN2C=CC(=O)NC2=O)C(O)C1O WGDUUQDYDIIBKT-UHFFFAOYSA-N 0.000 claims description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 claims description 2
- 235000013922 glutamic acid Nutrition 0.000 claims description 2
- 239000004220 glutamic acid Substances 0.000 claims description 2
- 229960003786 inosine Drugs 0.000 claims description 2
- 108010054130 nebulin Proteins 0.000 claims description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 2
- 229940096913 pseudoisocytidine Drugs 0.000 claims description 2
- PTJWIQPHWPFNBW-GBNDHIKLSA-N pseudouridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1C1=CNC(=O)NC1=O PTJWIQPHWPFNBW-GBNDHIKLSA-N 0.000 claims description 2
- 125000004219 purine nucleobase group Chemical group 0.000 claims description 2
- HBCQSNAFLVXVAY-UHFFFAOYSA-N pyrimidine-2-thiol Chemical compound SC1=NC=CC=N1 HBCQSNAFLVXVAY-UHFFFAOYSA-N 0.000 claims description 2
- BZWKPZBXAMTXNQ-UHFFFAOYSA-N sulfurocyanidic acid Chemical compound OS(=O)(=O)C#N BZWKPZBXAMTXNQ-UHFFFAOYSA-N 0.000 claims description 2
- XQTLDIFVVHJORV-UHFFFAOYSA-N tecnazene Chemical compound [O-][N+](=O)C1=C(Cl)C(Cl)=CC(Cl)=C1Cl XQTLDIFVVHJORV-UHFFFAOYSA-N 0.000 claims description 2
- 229960003087 tioguanine Drugs 0.000 claims description 2
- 229960005508 8-azaguanine Drugs 0.000 claims 2
- ANRHNWWPFJCPAZ-UHFFFAOYSA-M thionine Chemical compound [Cl-].C1=CC(N)=CC2=[S+]C3=CC(N)=CC=C3N=C21 ANRHNWWPFJCPAZ-UHFFFAOYSA-M 0.000 claims 2
- JRYMOPZHXMVHTA-DAGMQNCNSA-N 2-amino-7-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1h-pyrrolo[2,3-d]pyrimidin-4-one Chemical group C1=CC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O JRYMOPZHXMVHTA-DAGMQNCNSA-N 0.000 claims 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 claims 1
- UQDJGEHQDNVPGU-UHFFFAOYSA-N serine phosphoethanolamine Chemical compound [NH3+]CCOP([O-])(=O)OCC([NH3+])C([O-])=O UQDJGEHQDNVPGU-UHFFFAOYSA-N 0.000 claims 1
- 238000005382 thermal cycling Methods 0.000 abstract description 57
- 238000002372 labelling Methods 0.000 abstract description 19
- 238000002844 melting Methods 0.000 abstract description 17
- 230000008018 melting Effects 0.000 abstract description 17
- 230000000694 effects Effects 0.000 abstract description 13
- 238000012546 transfer Methods 0.000 abstract description 9
- 239000003298 DNA probe Substances 0.000 abstract description 3
- 108060002716 Exonuclease Proteins 0.000 abstract description 3
- 102000013165 exonuclease Human genes 0.000 abstract description 3
- 230000001404 mediated effect Effects 0.000 abstract description 2
- 239000013615 primer Substances 0.000 description 243
- 238000003752 polymerase chain reaction Methods 0.000 description 145
- 108091093037 Peptide nucleic acid Proteins 0.000 description 72
- 108091034117 Oligonucleotide Proteins 0.000 description 41
- 238000001514 detection method Methods 0.000 description 37
- 239000000047 product Substances 0.000 description 22
- 125000005647 linker group Chemical group 0.000 description 21
- 238000006243 chemical reaction Methods 0.000 description 20
- 239000002987 primer (paints) Substances 0.000 description 19
- 238000003556 assay Methods 0.000 description 16
- 101710163270 Nuclease Proteins 0.000 description 14
- 238000010791 quenching Methods 0.000 description 14
- 230000002441 reversible effect Effects 0.000 description 14
- 230000015572 biosynthetic process Effects 0.000 description 13
- 238000003776 cleavage reaction Methods 0.000 description 13
- 230000007017 scission Effects 0.000 description 13
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 12
- 238000003786 synthesis reaction Methods 0.000 description 12
- 238000004458 analytical method Methods 0.000 description 11
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 10
- 230000037230 mobility Effects 0.000 description 10
- 150000008300 phosphoramidites Chemical class 0.000 description 10
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 9
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 9
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 9
- 125000000524 functional group Chemical group 0.000 description 9
- 230000000171 quenching effect Effects 0.000 description 9
- 229910019142 PO4 Inorganic materials 0.000 description 8
- 238000007846 asymmetric PCR Methods 0.000 description 8
- 230000008901 benefit Effects 0.000 description 8
- 239000010452 phosphate Substances 0.000 description 8
- 108090000623 proteins and genes Proteins 0.000 description 8
- 239000007787 solid Substances 0.000 description 8
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 7
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 7
- 238000002474 experimental method Methods 0.000 description 7
- 238000005259 measurement Methods 0.000 description 7
- 150000004713 phosphodiesters Chemical class 0.000 description 7
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 7
- 238000011897 real-time detection Methods 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 239000011616 biotin Substances 0.000 description 6
- 229960002685 biotin Drugs 0.000 description 6
- 235000020958 biotin Nutrition 0.000 description 6
- 239000000499 gel Substances 0.000 description 6
- 239000000155 melt Substances 0.000 description 6
- 108020004999 messenger RNA Proteins 0.000 description 6
- 238000012544 monitoring process Methods 0.000 description 6
- 238000010647 peptide synthesis reaction Methods 0.000 description 6
- 238000000926 separation method Methods 0.000 description 6
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 5
- 238000010586 diagram Methods 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 239000002777 nucleoside Substances 0.000 description 5
- 239000002751 oligonucleotide probe Substances 0.000 description 5
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 5
- 230000000087 stabilizing effect Effects 0.000 description 5
- WCKQPPQRFNHPRJ-UHFFFAOYSA-N 4-[[4-(dimethylamino)phenyl]diazenyl]benzoic acid Chemical compound C1=CC(N(C)C)=CC=C1N=NC1=CC=C(C(O)=O)C=C1 WCKQPPQRFNHPRJ-UHFFFAOYSA-N 0.000 description 4
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 4
- BZTDTCNHAFUJOG-UHFFFAOYSA-N 6-carboxyfluorescein Chemical compound C12=CC=C(O)C=C2OC2=CC(O)=CC=C2C11OC(=O)C2=CC=C(C(=O)O)C=C21 BZTDTCNHAFUJOG-UHFFFAOYSA-N 0.000 description 4
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 4
- 108010085238 Actins Proteins 0.000 description 4
- 108091028043 Nucleic acid sequence Proteins 0.000 description 4
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 4
- 238000012408 PCR amplification Methods 0.000 description 4
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 238000003491 array Methods 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- BFMYDTVEBKDAKJ-UHFFFAOYSA-L disodium;(2',7'-dibromo-3',6'-dioxido-3-oxospiro[2-benzofuran-1,9'-xanthene]-4'-yl)mercury;hydrate Chemical compound O.[Na+].[Na+].O1C(=O)C2=CC=CC=C2C21C1=CC(Br)=C([O-])C([Hg])=C1OC1=C2C=C(Br)C([O-])=C1 BFMYDTVEBKDAKJ-UHFFFAOYSA-L 0.000 description 4
- 238000001962 electrophoresis Methods 0.000 description 4
- 230000002068 genetic effect Effects 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 238000010348 incorporation Methods 0.000 description 4
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 238000011002 quantification Methods 0.000 description 4
- 230000010076 replication Effects 0.000 description 4
- 230000035945 sensitivity Effects 0.000 description 4
- 238000012163 sequencing technique Methods 0.000 description 4
- 239000007790 solid phase Substances 0.000 description 4
- 238000010186 staining Methods 0.000 description 4
- 235000011178 triphosphate Nutrition 0.000 description 4
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 3
- 108700028369 Alleles Proteins 0.000 description 3
- 108020004635 Complementary DNA Proteins 0.000 description 3
- 239000003155 DNA primer Substances 0.000 description 3
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 3
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 150000001540 azides Chemical class 0.000 description 3
- 230000000875 corresponding effect Effects 0.000 description 3
- 230000001351 cycling effect Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 238000001502 gel electrophoresis Methods 0.000 description 3
- 230000014509 gene expression Effects 0.000 description 3
- 150000004820 halides Chemical class 0.000 description 3
- 230000000155 isotopic effect Effects 0.000 description 3
- 238000002493 microarray Methods 0.000 description 3
- 125000003835 nucleoside group Chemical group 0.000 description 3
- JAFCRLMYPPBVMU-UHFFFAOYSA-N o-pent-4-ynylhydroxylamine Chemical group NOCCCC#C JAFCRLMYPPBVMU-UHFFFAOYSA-N 0.000 description 3
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 3
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical group OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 description 3
- 229920002401 polyacrylamide Polymers 0.000 description 3
- 230000037452 priming Effects 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108700042226 ras Genes Proteins 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 230000001360 synchronised effect Effects 0.000 description 3
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 3
- QGKMIGUHVLGJBR-UHFFFAOYSA-M (4z)-1-(3-methylbutyl)-4-[[1-(3-methylbutyl)quinolin-1-ium-4-yl]methylidene]quinoline;iodide Chemical compound [I-].C12=CC=CC=C2N(CCC(C)C)C=CC1=CC1=CC=[N+](CCC(C)C)C2=CC=CC=C12 QGKMIGUHVLGJBR-UHFFFAOYSA-M 0.000 description 2
- 125000001917 2,4-dinitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C(=C1*)[N+]([O-])=O)[N+]([O-])=O 0.000 description 2
- 102000007469 Actins Human genes 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 238000001712 DNA sequencing Methods 0.000 description 2
- SHIBSTMRCDJXLN-UHFFFAOYSA-N Digoxigenin Natural products C1CC(C2C(C3(C)CCC(O)CC3CC2)CC2O)(O)C2(C)C1C1=CC(=O)OC1 SHIBSTMRCDJXLN-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 238000002944 PCR assay Methods 0.000 description 2
- 239000004952 Polyamide Substances 0.000 description 2
- 108091028664 Ribonucleotide Proteins 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 210000003050 axon Anatomy 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 2
- 239000004327 boric acid Substances 0.000 description 2
- 238000010804 cDNA synthesis Methods 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 238000004132 cross linking Methods 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 238000007405 data analysis Methods 0.000 description 2
- 238000013480 data collection Methods 0.000 description 2
- 239000005547 deoxyribonucleotide Substances 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- QONQRTHLHBTMGP-UHFFFAOYSA-N digitoxigenin Natural products CC12CCC(C3(CCC(O)CC3CC3)C)C3C11OC1CC2C1=CC(=O)OC1 QONQRTHLHBTMGP-UHFFFAOYSA-N 0.000 description 2
- SHIBSTMRCDJXLN-KCZCNTNESA-N digoxigenin Chemical compound C1([C@@H]2[C@@]3([C@@](CC2)(O)[C@H]2[C@@H]([C@@]4(C)CC[C@H](O)C[C@H]4CC2)C[C@H]3O)C)=CC(=O)OC1 SHIBSTMRCDJXLN-KCZCNTNESA-N 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 238000010195 expression analysis Methods 0.000 description 2
- 238000001917 fluorescence detection Methods 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 239000000138 intercalating agent Substances 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- 239000003068 molecular probe Substances 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- 150000003833 nucleoside derivatives Chemical class 0.000 description 2
- 238000002515 oligonucleotide synthesis Methods 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 2
- 229920002647 polyamide Polymers 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 238000003753 real-time PCR Methods 0.000 description 2
- 238000010223 real-time analysis Methods 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 239000002336 ribonucleotide Substances 0.000 description 2
- 125000002652 ribonucleotide group Chemical group 0.000 description 2
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- AVBGNFCMKJOFIN-UHFFFAOYSA-N triethylammonium acetate Chemical compound CC(O)=O.CCN(CC)CC AVBGNFCMKJOFIN-UHFFFAOYSA-N 0.000 description 2
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical class OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- 0 *N(*)P(O*)OI Chemical compound *N(*)P(O*)OI 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- YKBGVTZYEHREMT-KVQBGUIXSA-N 2'-deoxyguanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 YKBGVTZYEHREMT-KVQBGUIXSA-N 0.000 description 1
- 125000000143 2-carboxyethyl group Chemical group [H]OC(=O)C([H])([H])C([H])([H])* 0.000 description 1
- 125000001731 2-cyanoethyl group Chemical group [H]C([H])(*)C([H])([H])C#N 0.000 description 1
- ALNUOSXDMYFQNQ-UHFFFAOYSA-N 2-nitro-1,3-thiazole Chemical class [O-][N+](=O)C1=NC=CS1 ALNUOSXDMYFQNQ-UHFFFAOYSA-N 0.000 description 1
- OALHHIHQOFIMEF-UHFFFAOYSA-N 3',6'-dihydroxy-2',4',5',7'-tetraiodo-3h-spiro[2-benzofuran-1,9'-xanthene]-3-one Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC(I)=C(O)C(I)=C1OC1=C(I)C(O)=C(I)C=C21 OALHHIHQOFIMEF-UHFFFAOYSA-N 0.000 description 1
- CKTSBUTUHBMZGZ-ULQXZJNLSA-N 4-amino-1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-tritiopyrimidin-2-one Chemical compound O=C1N=C(N)C([3H])=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 CKTSBUTUHBMZGZ-ULQXZJNLSA-N 0.000 description 1
- 125000004042 4-aminobutyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])N([H])[H] 0.000 description 1
- LPXQRXLUHJKZIE-UHFFFAOYSA-N 8-azaguanine Chemical compound NC1=NC(O)=C2NN=NC2=N1 LPXQRXLUHJKZIE-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GNRLUBOJIGSVNT-UHFFFAOYSA-N Aminoethoxyacetic acid Chemical compound NCCOCC(O)=O GNRLUBOJIGSVNT-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 101100519158 Arabidopsis thaliana PCR2 gene Proteins 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 108020001019 DNA Primers Proteins 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- BVTJGGGYKAMDBN-UHFFFAOYSA-N Dioxetane Chemical class C1COO1 BVTJGGGYKAMDBN-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 229920001917 Ficoll Polymers 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 101000756632 Homo sapiens Actin, cytoplasmic 1 Proteins 0.000 description 1
- 101000739159 Homo sapiens Mammaglobin-A Proteins 0.000 description 1
- 208000026350 Inborn Genetic disease Diseases 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- 102100037273 Mammaglobin-A Human genes 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- OMIKXSTXQGXCJT-UHFFFAOYSA-N NP(O)O.NP(O)O.NP(O)O.N.N Chemical group NP(O)O.NP(O)O.NP(O)O.N.N OMIKXSTXQGXCJT-UHFFFAOYSA-N 0.000 description 1
- 206010067482 No adverse event Diseases 0.000 description 1
- 108010010677 Phosphodiesterase I Proteins 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 108091034057 RNA (poly(A)) Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 108010006785 Taq Polymerase Proteins 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 108020000999 Viral RNA Proteins 0.000 description 1
- WAWRKBQQBUDAMY-UHFFFAOYSA-N [6-(dimethylamino)-9-(2-methoxycarbonylphenyl)xanthen-3-ylidene]-dimethylazanium Chemical compound COC(=O)C1=CC=CC=C1C1=C2C=CC(=[N+](C)C)C=C2OC2=CC(N(C)C)=CC=C21 WAWRKBQQBUDAMY-UHFFFAOYSA-N 0.000 description 1
- WXIONIWNXBAHRU-UHFFFAOYSA-N [dimethylamino(triazolo[4,5-b]pyridin-3-yloxy)methylidene]-dimethylazanium Chemical compound C1=CN=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 WXIONIWNXBAHRU-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 125000005336 allyloxy group Chemical group 0.000 description 1
- 150000001408 amides Chemical group 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 238000005415 bioluminescence Methods 0.000 description 1
- 230000029918 bioluminescence Effects 0.000 description 1
- 239000007844 bleaching agent Substances 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- UDSAIICHUKSCKT-UHFFFAOYSA-N bromophenol blue Chemical compound C1=C(Br)C(O)=C(Br)C=C1C1(C=2C=C(Br)C(O)=C(Br)C=2)C2=CC=CC=C2S(=O)(=O)O1 UDSAIICHUKSCKT-UHFFFAOYSA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000005289 controlled pore glass Substances 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- SUYVUBYJARFZHO-RRKCRQDMSA-N dATP Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-RRKCRQDMSA-N 0.000 description 1
- SUYVUBYJARFZHO-UHFFFAOYSA-N dATP Natural products C1=NC=2C(N)=NC=NC=2N1C1CC(O)C(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-UHFFFAOYSA-N 0.000 description 1
- RGWHQCVHVJXOKC-SHYZEUOFSA-J dCTP(4-) Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)C1 RGWHQCVHVJXOKC-SHYZEUOFSA-J 0.000 description 1
- HAAZLUGHYHWQIW-KVQBGUIXSA-N dGTP Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 HAAZLUGHYHWQIW-KVQBGUIXSA-N 0.000 description 1
- NHVNXKFIZYSCEB-XLPZGREQSA-N dTTP Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C1 NHVNXKFIZYSCEB-XLPZGREQSA-N 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 239000003398 denaturant Substances 0.000 description 1
- 238000000326 densiometry Methods 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- 125000000664 diazo group Chemical group [N-]=[N+]=[*] 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- NAGJZTKCGNOGPW-UHFFFAOYSA-N dithiophosphoric acid Chemical class OP(O)(S)=S NAGJZTKCGNOGPW-UHFFFAOYSA-N 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 238000000295 emission spectrum Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000006862 enzymatic digestion Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 238000001506 fluorescence spectroscopy Methods 0.000 description 1
- 238000002189 fluorescence spectrum Methods 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 244000078673 foodborn pathogen Species 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 208000016361 genetic disease Diseases 0.000 description 1
- 238000003205 genotyping method Methods 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 125000005179 haloacetyl group Chemical group 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 238000010191 image analysis Methods 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000012678 infectious agent Substances 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical class NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 230000031700 light absorption Effects 0.000 description 1
- 239000012160 loading buffer Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- FDZZZRQASAIRJF-UHFFFAOYSA-M malachite green Chemical class [Cl-].C1=CC(N(C)C)=CC=C1C(C=1C=CC=CC=1)=C1C=CC(=[N+](C)C)C=C1 FDZZZRQASAIRJF-UHFFFAOYSA-M 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- YACKEPLHDIMKIO-UHFFFAOYSA-N methylphosphonic acid Chemical compound CP(O)(O)=O YACKEPLHDIMKIO-UHFFFAOYSA-N 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- HDCXQTPVTAIPNZ-UHFFFAOYSA-N n-({[4-(aminosulfonyl)phenyl]amino}carbonyl)-4-methylbenzenesulfonamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NC1=CC=C(S(N)(=O)=O)C=C1 HDCXQTPVTAIPNZ-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 150000004957 nitroimidazoles Chemical class 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000000538 pentafluorophenyl group Chemical group FC1=C(F)C(F)=C(*)C(F)=C1F 0.000 description 1
- 150000002972 pentoses Chemical class 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical group [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- SXADIBFZNXBEGI-UHFFFAOYSA-N phosphoramidous acid Chemical group NP(O)O SXADIBFZNXBEGI-UHFFFAOYSA-N 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 102000054765 polymorphisms of proteins Human genes 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000003252 repetitive effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 108020004418 ribosomal RNA Proteins 0.000 description 1
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 229940063675 spermine Drugs 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 239000005451 thionucleotide Substances 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical class [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 238000011282 treatment Methods 0.000 description 1
- 125000005208 trialkylammonium group Chemical group 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 1
- 125000005500 uronium group Chemical group 0.000 description 1
- 239000002023 wood Substances 0.000 description 1
- 125000001834 xanthenyl group Chemical class C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- NLIVDORGVGAOOJ-MAHBNPEESA-M xylene cyanol Chemical compound [Na+].C1=C(C)C(NCC)=CC=C1C(\C=1C(=CC(OS([O-])=O)=CC=1)OS([O-])=O)=C\1C=C(C)\C(=[NH+]/CC)\C=C/1 NLIVDORGVGAOOJ-MAHBNPEESA-M 0.000 description 1
Images
























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/686—Polymerase chain reaction [PCR]
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Analytical Chemistry (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Saccharide Compounds (AREA)
Abstract
Description
本願は、本明細書中で参考として援用される、2000年6月6日に出願された仮特許番号60/209883の優先権の利益を主張する。
【0002】
(I.発明の分野)
本発明は一般に、核酸ハイブリダイゼーションの分野、そしてより具体的には核酸増幅の方法に関する。
【0003】
(II.導入)
核酸増幅アッセイは、現代生物学において、遺伝性疾患の診断、ヒトの同定、微生物の同定、親子鑑定、ウイルス学およびDNA配列決定における多様な適用を有する、特定の標的配列の検出法の重要なクラスを含む。ポリメラーゼ連鎖反応(PCR)増幅法は、大きな感度および特異性によって、標的核酸配列の生成および検出を可能にする。PCR法は、クローニング、遺伝子発現の分析、DNA配列決定、遺伝子マッピング、薬物発見などに不可欠である(Gilliland(1990)Proc.Natl.Acad.Sci.、87:2725−2729;Bevan(1992)PCR Methods and Applications 1:222−228;Green(1991)PCR Methods and Applications 1:77−90;McPherson,M.J.、Quirke,P.およびTaylor,G.R.、PCR 2:A Practical Approach(1995)Oxford University Press、Oxford)。標的配列またはアンプリコンにハイブリダイズし得るオリゴヌクレオチドプローブを使用してPCR産物(アンプリコン)を検出するための方法は、Mullis、米国特許第4,683,195号および同第4,683,202号;EP第237,362号に記載される。
【0004】
従来のPCRにおいて、オリゴヌクレオチドプライマーは、目的の標的配列に隣接する相補的な標的鎖中の配列にアニーリングされ、そしてアニーリングされたプライマーは、標的配列の二本鎖(ds)コピーを生成するために、同時に伸長される。これらのプライマーは、ポリメラーゼ、好ましくは末端安定性ポリメラーゼによって伸長される(McPherson,M.編(1995)PCR 2:A Practical Approach、Oxford University Press、Oxfordで、IRL Press)。従来、PCRにおいて使用される2つのオリゴヌクレオチドプライマーの配列は、類似のアニーリングおよび伸長の効率を促進するために、等しいかまたは類似のTm値を有するように設計および選択される。
【0005】
非対称PCRは、標的配列由来のDNAの一本鎖コピーの生成のための用途を見出した(Gyllensten(1988)Proc.Natl.Acad.Sci USA、85:7652;McCabe,P.(1990)「Production of single−stranded DNA by asymmetric PCR」、PCR Protocols:A guide to Methods and Applications、Innis,M.編、Academic Press,Inc.、San Diego、76−83頁)。非等量の2つの増幅プライマーが使用される(例えば、低濃度プライマーおよび高濃度プライマーについて、それぞれ、1〜5pmolおよび50〜100pmol)。最初の20〜25サイクルの間、二本鎖DNAが指数関数的に生成され、そして、限定的なプライマーが涸渇した場合、一本鎖DNAが、残りの5〜10サイクルの間、直線的に蓄積する。PCRは最適状態には及ばない条件(すなわち、低濃度の1つのプライマー)で実行されなければならない点で、不利である。従って、増幅は、不十分であり得るか、または再現不能であり得る(Hopgood(1992)BioTechniques,13:82;Hunkapiller(1991)Current Opinion in Biotechnology、2:92)。一本鎖アンプリコンを生成する他のPCR法は、二本鎖アンプリコンの1本の鎖の酵素的消化、プライマー対の多重化セット、プライマーのネスティド(nested)セット、および逆増幅を含む。しかし、各方法は、扱いにくいか、または制限を有する(Higuchi(1989)Nucleic Acids Res.、17:5865;Sarkar(1989)Nucleic Acids Res.、16:5197;Stoflet(1988)Science、239:491;Bevan(1992)PCR Methods and Applications、1:22;Gyllensten,U.(1989)「Direct sequencing of in vitro amplified DNA」、PCR Technology:Principles and Applications for DNA Amplification、Erlich,H.編、Stockton Press、New York、50〜53頁)。
【0006】
(III.発明の要旨)
本発明は、核酸増幅のための新規熱サイクルプロトコールを含む、核酸増幅の方法に関する。進行(すなわち、増幅産物の生成)の検出は、標的配列の一本鎖形態に対して、検出可能なプローブをハイブリダイズさせることによって、容易化および改善され得る。一本鎖標的は、2つの段階、アニーリングプロトコールおよび伸長プロトコールにおける、中間体である。最初に、より高い融点のプライマーが標的の1本の鎖に選択的にアニーリングし、そして伸長され、二本鎖コピーおよび、未コピーの一本鎖標的を生じる。
【0007】
第一の局面において、本発明は、標的ポリヌクレオチドの相補的なポリヌクレオチド鎖を生成するための方法を含む。互いにハイブリダイズして塩基対形成構造体を形成し得る第一および第二の標的ポリヌクレオチド鎖を含む混合物が獲得され、この混合物は、標的配列、第一の標的ポリヌクレオチド鎖の第一の領域に相補的な第一のプライマー、および第二の標的ポリヌクレオチド鎖の第二の領域に相補的な第二のプライマーを含み、その結果、第一および第二の領域は、標的配列に隣接する。第一のプライマーは、第一の温度で伸長されて、第二のプライマーが第二の領域に実質的にハイブリダイズしないような条件下で第一の標的鎖にハイブリダイズする第一の相補鎖を含む第一の複合体を形成する。第二のプライマーは、第一の温度よりも低い第二の温度で伸長されて、第二の標的鎖にハイブリダイズする第二の相補鎖を含む第二の複合体を形成する。第二のプライマーを伸長させる前に、検出可能なプローブは、第二の標的鎖における相補的結合領域にハイブリダイズされ、そしてハイブリダイズしたプローブは、第二の標的鎖の測定として検出される。
【0008】
別の局面において、非同調的熱サイクルプロトコールは、以下の工程を包含する:
第一のアニーリング温度で、第一のプライマーを、変性した標的核酸の第一鎖にアニーリングさせる工程;
伸長温度または第一のアニーリング温度で、プライマー伸長試薬を用いて第一のプライマーを伸長させて、二本鎖の核酸を生成する工程であって、ここで、このプライマー伸長試薬は、ポリメラーゼ、ヌクレオチド5’−トリホスフェートおよび緩衝液を含む、工程;
プローブハイブリダイゼーション温度で、検出可能なプローブを、変性した標的核酸の第二鎖にアニーリングさせる工程;
第二のアニーリング温度で、第二のプライマーを、変性した標的核酸の第二鎖にアニーリングさせる工程であって、ここで、第二のアニーリング温度は、第一のアニーリング温度および伸長温度よりも低い、工程;
伸長温度で、プライマー伸長試薬を用いて第二のプライマーを伸長させて、二本鎖の核酸を生成する工程;ならびに
変性温度で、二本鎖標的核酸を第一鎖および第二鎖に変性させる工程。
【0009】
本発明の上記の方法によって、検出可能なプローブは、未コピーの一本鎖標的にアニーリングされる。このハイブリダイゼーション事象は、例えば、このプローブが標識のレポーター/クエンチャー(quencher)対を有する場合に、FRETによって検出される。このプローブは、DNAであり得、そしてポリメラーゼのヌクレアーゼ活性によって切断され得る。あるいは、このプローブは、切断不能であり得る。このプローブは、核酸アナログモノマー単位(例えば、2−アミノエチルグリシン)を含む核酸アナログまたはキメラであり得る。このプローブは、PNAまたはPNA/DNAキメラであり得る。PNA FRETプローブは、レポーターおよびクエンチャーの対に隣接する核酸塩基単位を有する、2−アミノエチルグリシンの配列から構成され得る。
【0010】
このプローブは、標的にハイブリダイズしたまま検出され得る。プローブの検出は、PCR(リアルタイム)の間の各サイクルで実施され得る。あるいは、プローブは、PCRの最後(例えば、2〜50サイクルの完了の後)、またはさらに、幾何級数的な増幅もしくは直線的な増幅の最後(終点)で検出または定量され得る。
【0011】
プローブの検出後、第一のプライマーよりも低いTmを有する第二のプライマーは、一本鎖の標的に選択的にアニーリングされ、そして、標的のコピーを作製するために伸長される。プローブ検出を伴う非同調的熱サイクル方法は、多サイクル反復され得、ここで、この混合物は、規定された時間にわたる規定された温度での変性、アニーリングおよびプライマー伸長の工程をもたらす温度変化を経る。
【0012】
非同調的熱サイクルプロトコールの1つの実施形態の間に、プローブは、増幅された核酸に特異的にハイブリダイズする。ハイブリダイズした場合、ポリメラーゼのヌクレアーゼ活性は、ヌクレオチド間切断によってプローブを分解し得、それによって、インタクトなプローブによって維持される分子内クエンチを除外する。このプローブは、増幅された標的核酸(アンプリコン)に特異的にハイブリダイズするように設計されることから、プローブの切断によって生じるPCR反応混合物由来の蛍光強度の増大は、増幅の進行(すなわち、標的配列の量および増幅の量)に相関し得る。
【0013】
一般に、サンプル中の標的核酸は、DNA、最も通常はゲノムDNAの配列である。しかし、本発明はまた、他の核酸(例えば、合成オリゴヌクレオチド、メッセンジャーRNA、リボソームRNA、ウイルスRNA、cDNA、またはクローニングされたDNA)を用いて実行され得る。適切な標的核酸サンプルは、本発明において使用するための一本鎖または二本鎖のDNAまたはRNAを含む。
【0014】
別の局面において、本発明は、標的ポリヌクレオチドの相補的ポリヌクレオチド鎖を生成するための方法を含む。互いにハイブリダイズして塩基対形成構造体を形成し得る第一および第二の標的ポリヌクレオチドを含む混合物が獲得され、この混合物は、標的配列、第一の標的ポリヌクレオチドの第一の領域に相補的な第一のプライマー、および第二の標的ポリヌクレオチドの第二の領域に相補的な第二のプライマーを含み、その結果、第一および第二の領域は、標的配列に隣接する。第一のプライマーは、第一の温度で伸長されて、第二のプライマーが第二の領域に実質的にハイブリダイズしないような条件下で第一の標的鎖にハイブリダイズする第一の相補鎖を含む第一の複合体を形成する。第二のプライマーは、第一の温度よりも低い第二の温度で伸長されて、第二の標的鎖にハイブリダイズする第二の相補鎖を含む第二の複合体を形成する。第一の複合体および第二の複合体は、変性され得る。第一のプライマー伸長、第二のプライマー伸長、および変性の工程は、1以上のサイクルで反復され得る。
【0015】
別の局面において、本発明は、過剰のssアンプリコンを生成するための非同調的熱サイクル方法を含み、この方法は、以下の工程を包含する:
第一のアニーリング温度で、第一のプライマーを、変性した標的核酸の第一鎖にアニーリングさせる工程;
伸長温度または第一のアニーリング温度で、プライマー伸長試薬を用いて第一のプライマーを伸長させて、二本鎖の核酸を生成する工程であって、ここで、このプライマー伸長試薬は、ポリメラーゼ、ヌクレオチド5’−トリホスフェートおよび緩衝液を含む、工程;
第二のアニーリング温度で、第二のプライマーを、変性した標的核酸の第二鎖にアニーリングさせる工程であって、ここで、第二のアニーリング温度は、第一のアニーリング温度および伸長温度よりも低い、工程;
伸長温度で、プライマー伸長試薬を用いて第二のプライマーを伸長させて、二本鎖の核酸を生成する工程;ならびに
変性温度で、二本鎖標的を第一鎖および第二鎖に変性させる工程。
【0016】
工程のサイクルは、2〜50サイクルまたはそれ以上反復されて、二本鎖(ds)アンプリコンを生成し得る。第二のプライマーのアニーリングおよび第二のプライマーの伸長の工程は、最後の1以上のサイクルを省略されて、過剰の一本鎖(ss)アンプリコンを生成し得る。
【0017】
別の局面において、本発明は、配列決定、すなわちハイブリダイゼーションによる配列決定(SBH)によってcDNAライブラリーを特徴付ける方法を含む。
【0018】
別の実施形態において、本発明は、サンプル核酸中の標的配列の存在または非存在を検出する非同調的熱サイクルプロトコールによってPCRアッセイを実行するために適切なキットに関する。このキットは、サンプル中の1以上の標的配列のリアルタイム検出または終点検出、あるいは定量を可能にし得る。1つの実施形態において、このキットは、約10〜30℃の異なる融点を有するプライマーを含む。このキットはまた、PCRまたは標的の測定および検出を実行するために選択される1以上のプローブ、ヌクレオチド、ポリメラーゼ、および他の試薬または組成物を含み得る。
【0019】
(V.詳細な説明)
ここで本発明の特定の実施形態(これらの例は、添付の図面において例示される)を詳細に言及る。本発明は、例示的な実施形態と組み合わせて記載されるが、これは、発明をこれらの実施形態に制限することを意図しないことが理解される。これに反して、本発明は、全ての代替案、改変および等価物を含むことが意図される。これらは、請求される本発明の範囲に含まれ得る。
【0020】
(V.1定義)
「核酸塩基」は、相補的な核酸塩基または核酸塩基アナログ(例えば、プリン、7−デアザプリンまたはピリミジン)との対合においてWatson−Crick水素結合を形成し得る窒素含有複素環部分を意味する。代表的な核酸塩基は、天然に存在する核酸塩基(アデニン、グアニン、シトシン、ウラシル、チミン)および天然に存在する核酸塩基のアナログ(例えば、7−デアザアデニン、7−デアザグアニン、7−デアザ−8−アザグアニン、7−デアザ−8−アザアデニン、イノシン、ネブラリン、ニトロピロール、ニトロインドール、2−アミノ−プリン、2,6−ジアミノ−プリン、ヒポキサンチン、プソイドウリジン、プソイドシチジン、プソイドイソシチジン、5−プロピニル−シチジン、イソシチジン、イソグアニン、7−デアザ−クアニン、2−チオーピリミジン、6−チオーグアニン、4−チオ−チミン、4−チオ−ウラシル、O6−メチル−グアニン、N6−メチル−アデニン、O4−メチル−チミン、5,6−ジヒドロチミン、5,6−ジヒドロウラシル、4−メチル−インドールおよびエテノアデニン)である(Fasman(1989)Practical Handbook of Biochemistry and Moleculuar Biology 385〜394頁,CRC Press,Boca Raton,F1)。
【0021】
「ヌクレオシド」は、リボース糖のC−1’炭素に結合した核酸塩基からなる化合物をいう。リボースは、置換されとも良いし、または置換されなくてもよい。置換されたリボース糖としては、以下が挙げられるが、これらに限定されない;1つ以上の炭素原子(例えば、2’炭素)が、1つ以上の同じかまたは異なるCl、F、−R、−OR、−NR2またはハロゲン基で置換されるリボース糖、ここで各Rは、独立してH、C1−C6アルキル、またはC5−C14アリールである。リボースの例としては、以下が挙げられる:リボース、2’−デオキシリボース、2’,3’−ジデオキシリボース、2’−ハロリボース、2’−フルオロリボース、2’−シクロリボースおよび2’−アルキルリボース(例えば、2’−O−メチル)、4’−α−アノマーヌクレオチド、1’−α−アノマーヌクレオチド、2’,4’−および3’,4’−結合ならびに他の「結合」または「LNA」、二環状糖改変(WO98/22489;WO98/39352;WO99/14226)。オリゴヌクレオチド内のLNA糖アナログは、以下の構造:
【0022】
【化3】
【0023】
リボースの2’位または3’位における改変としては、以下が挙げられる:水素、ヒドロキシ、メトキシ、エトキシ、アリルオキシ、イソプロポキシ、ブトキシ、イソブトキシ、メトキシエチル、アルコキシ、フェノキシ、アジド、アミノ、アルキルアミノ、フルオロ、クロロおよびブロモ。ヌクレオシドおよびヌクレオチドは、天然のD光学異性体形態およびL光学異性体形態を含む(Garbesi(1993)Nucl.Acids Res.21:4159−65;Fujimori(1990)J.Amer.Chem.Soc.112:7435;Urata,(1993)Nucleic Acids Symposium Ser.No.29:69−70)。核酸塩基がプリン(例えば、AまたはG)である場合、リボース糖は、核酸塩基のN9位に結合する。核酸塩基が、ピリミジン(例えば、C、TまたはU)の場合、ペントース糖は、核酸塩基のN1位に結合する(KornbergおよびBaker,(1992)DNA Replication,第2版,Freeman,San Francisco,CA)。
【0024】
「ヌクレオチド」は、モノマー単位としてまたは核酸内のヌクレオシドのリン酸エステルをいう。ヌクレオチドは、時々、リボース糖の構造的な特徴を特に指摘するために、「NTP」または「dNTP」および「ddNTP」として示される。「ヌクレオチド5’−トリホスフェート」は、5’位におけるトリホスフェートエステル基を有するヌクレオチドをいう。トリホスフェートエステル基は、種々の酸素に対する置換イオウ置換を含み得る(例えば、α−チオ−ヌクレオチド5’−トリホスフェート)。
【0025】
本明細書中で使用される場合、用語「ポリヌクレオチド」または「オリゴヌクレオチド」は、交換可能に使用され、そしてヌクレオチドモノマーの一本鎖ポリマーおよび二本鎖ポリマー(ヌクレオチド間リン酸ジエステル結合連結またはヌクレオチ間アナログおよび関連するカウンターイオン(例えば、H+、NH4 +、トリアルキルアンモニウム、Mg+、Na+、など)によって結合される2’−デオキシリボヌクレオチド(DNA)およびリボヌクレオチド(RNA)を含む)を意味する。ポリヌクレオチドは、完全にデオキシリボヌクレオチドから、完全にリボヌクレオチドからまたはそれらの化学的混合物を構成し得る。ポリヌクレオチドは、核酸塩基アナログおよび糖アナログから構成され得る。ポリヌクレオチドは、典型的に、少数の単量体単位(例えば、これらが、オリゴヌクレオチドとして当該分野で頻繁にいわれる場合、5〜40)から数千の単量体ヌクレオチド単位のサイズの範囲である。他に示さない限り、いつポリヌクレオチド配列が、示されようとも、これは、ヌクレオチドが、左から右の5’→3’順序であり、そして他に示さない限り、「A」は、デオキシアデノシンを示し、「C」は、デオキシシチジンを示し、「G」は、デオキシグアノシンを示し、そして「T」は、チミジンを示すことが理解される。
【0026】
「ヌクレオチド間アナログ」は、オリゴヌクレオチドのリン酸エステルアナログまたは非リン酸アナログを意味する。リン酸エステルアナログとしては、以下が挙げられる:(i)(C1−C4)アルキルホスホネート(例えば、メチルホスホネート);(ii)ホスホラミダイト:(iii)(C1−C6)アルキルまたは置換アルキルホスホトリエステル;(iv)ホスホロチオエート;および(v)ホスホロジチオエート。非リン酸アナログは、糖/リン酸部分が、アミド結合によって置換される場合、例えば、2−アミノエチルグリシン単位(通常、PNAとして称される)を含む(Nielsen(1991)Science 254:1497−1500)。
【0027】
「結合部位」とは、リンカーに共有的に結合するか、または共有的に結合し得る、部分または分子(例えば、色素、ヌクレオチドまたはPNA)における部位を意味する。
【0028】
「リンカー」とは、1つの部分または分子(例えば、色素をポリヌクレオチドに、または1つの色素と別の色素に共有結合する)を共有結合するケ有結合される原子の鎖をいう。
【0029】
「反応性結合基」は、別の分子と反応して、共有結合を形成し得る化学的に反応性の置換基または部分(例えば、求核剤または求電子剤)をいう。
【0030】
「複素環」とは、環系を伴う分子をいい、ここで、1つ以上の環原子は、ヘテロ原子(例えば、窒素、酸素およびイオウ(炭素に対して))である。
【0031】
「酵素学的に伸長可能な」は、以下のヌクレオチドをいう:(i)ポリメラーゼ酵素の作用を介してポリヌクレオチド鎖の末端に酵素学的に組み込まれ得るヌクレオチド、および(ii)さらにプライマー伸長を支持し得るヌクレオチド。酵素学的に伸長可能なヌクレオチドとしては、ヌクレオチド5’−トリホスフェート、すなわち、dNTPおよびNTPが挙げられる。
【0032】
「酵素学的に組み込み可能な」とは、ポリメラーゼ酵素の作用を介してポリヌクレオチド鎖の末端に酵素学的に組み込まれ得るヌクレオチドをいう。酵素学的に組み込み可能なヌクレオチドとしては、dNTP、NTPおよび2’,3’−ジデオキシ、ヌクレオチド5’−トリホスフェート、すなわち、ddNTPが挙げられる。
【0033】
「標的配列」とは、相補的なポリヌクレオチド(例えば、プライマーまたはプローブ)とのハイブリダイズの対象であるポリヌクレオチド配列を意味する。標的配列は、DNA、RNA、それらのアナログそしてそれらの組み合わせをから構成され得る。
【0034】
用語「プローブ」とは、標的核酸の配列との相補的な塩基対形成による二重鎖構造を形成するオリゴヌクレオチドを意味する。本発明において、プローブは、例えば、蛍光色素、または蛍光レポーター色素およびクエンチャーから構成される標識の組を用いて、検出可能に標識され得る。
【0035】
用語「標識」とは、分子に結合し得、そして:(i)検出可能なシグナルを提供するか;(ii)第2の標識と相互作用して、第2の標識(例えば、FRET)によって提供される検出可能なシグナルを改変するか;(iii)ハイブリダイゼーション(すなわち、二重鎖形成)を安定化するかまたは(iv)捕捉部分(すなわち、親和性、抗体/抗原、イオン性錯体化)を提供する任意の部分をいう。標識化は、公知の標識、結合、結合基、試薬、反応条件ならびに分析方法および精製方法を使用する、多くの公知の技術のうちのいずれかを使用して達成され得る。標識としては、蛍光、化学発光または生物発光によって検出可能なシグナルを生じる光放射化合物が挙げられる(Kricka,L.in Nonisotopic DNA Probe Techniques(1992),Academic Press,San Diego,3−28頁)。標識の別のクラスは、二重鎖のハイブリダイゼーションを増大、安定化、または影響を与えるのに役立つハイブリダイゼーション安定化部分である(例えば、インターカレーター、副溝結合剤および架橋官能基)(Blackburn,G.およびGait,M.編,「DNA and RNA structure」in Nucleic Acids in Chemistry and Biology,第2版,(1996)Oxford University Press,15−81頁)。標識のなお別のクラスは、特異的な捕捉基または非特異的な捕捉基によって分子の分離または固定化をもたらす(例えば、ビオチン、ジゴキシゲニンおよび他のハプテン)(Andrus,A.「Chemical methods for 5’non−isotopic labelling of PCR probes and primers」(1995) in PCR 2:A Practical Approach,Oxford University Press, Oxford,39−54頁)。
【0036】
用語「クエンチング」とは、機構に関係なく第2の部分(クエンチャー)によって起こされる第1の部分(レポーター色素)の蛍光の減少することをいう。
【0037】
本明細書中において使用される場合、「キメラ」は、1つ以上のヌクレオチドおよび1つ以上のヌクレオチドアナログ単位を含むオリゴヌクレオチドをいう。
【0038】
用語「アニーリング」および「ハイブリダイゼーション」は、交換可能に使用され、そして二重鎖構造または他の高次構造の形成を生じる別の核酸と1つの核酸の塩基対形成相互作用を意味する。第1の相互作用は、塩基特異的である(すなわち、Watson/CrickおよびHoogsteen型水素結合によるA/TおよびG/C)である。
【0039】
用語「終点分析」は、反応が、実質的に完全である場合、データ収集が生じる方法をいう。
【0040】
用語「リアルタイム分析」は、PCRの間の定期的なモニタリングをいう。特定の系(例えば、ABI 7700配列検出システム(Applied Biosystems,Foster City,CA))は、予め決定した時点または使用者が規定した時点で各熱サイクルの間モニタリングを実行する。FRETプローブを用いたPCRのリアルタイム分析は、サイクルからサイクルの蛍光色素の変化(好ましくは、任意の内部コントロールシグナルを差し引いて)を測定する。
【0041】
(V.2a.プライマーおよびプローブの合成)
オリゴヌクレオチドは、市販のホスホラミダイトヌクレオシド(Caruthers,米国特許第4,415,732号)、支持体(例えば、シリカ、制御された細孔ガラス(controlled−pore−glass)(Caruthers,米国特許第4,458,066号)およびポリスチレン(Andrus,米国特許第5,047,524号および同5,262,530号)ならびに自動合成機(Caruthers,米国特許第4,458,066号;Models 392,394,3948,3900 DNA/RNA Synthesizers,Applied Biosystems, Foster City, CA)を使用して、ホスホラミダイト方法(Caruthers,米国特許第4,973,679号;Beaucage(1992)Tetrahedron 48:2223−2311)によって固体支持体において通常合成される。
【0042】
(V.2b.プライマーおよびプローブ設計および選択)
非同調的熱サイクルプロトコールを実施するため、および伝統的かつ非同調的熱サイクルプロトコールを用いた比較実験のためのPCRプライマーおよびプローブは、Primer ExpressTM(Version 1.0,Applied Biosystems,CA)を使用して設計され得る。他のオリゴヌクレオチド選択および評価ソフトウェアプログラムは、報告または市販されている。標的核酸配列は、データベース(例えば、GenBank(http://www.ncbi.nlm. nih.gov/;Nuc.Acids Res.2000 Jan 1;28(1):15−8))のような遺伝子コードから入力されるかまたは読み込まれる。いくつかの実施形態において、標的に相補的なプライマーの結合部位の位置は、特定の部位において特定の長さのアンプリコンを増幅するために選択される。他の実施形態において、汎用なプライマー(すなわち、無作為にプライミングするプライマーまたは重複した冗長な塩基対を形成するヌクレオチドまたは乱交雑な塩基対を形成するヌクレオチドを用いてプライマーのセット)の使用における場合、プライマーの結合部位は、未知であり得る。
【0043】
加熱において、二重鎖は、融解し、そして濃色効果のシフトを起こす。特定のプライマーまたはプローブについてのTmは、集団の半数が、標的にハイブリダイズする温度である。Tmは、吸光度(例えば、260nm)対温度をプロットすることによって生じる特徴的な正弦波における変曲点として示される。ハイブリダイゼーション親和性は、プライマーの長さ、G+C含量、塩濃度、プライマーの化学的改変(例えば、2’−O−メチル(Stump(1999)Nucleic Acids Res.27:4642−48))、プライマーにおける標識、および安定化し得る試薬(例えば、インターカレーター)または不安定化し得る試薬(例えば、変性剤)、二重鎖形成によって影響を受ける。プライマーおよびプローブのTm値は、配列、長さ、G+C含量およびハイブリダイゼーション安定化改変を含むパラメーターのいくつかの組み合わせの選択によって設計され得て、特定の非同調的熱サイクルプロトコールにおける十分な増幅をもたらす特定のTm値を有する。
【0044】
非同調的PCR法において使用されるプライマーの配列および長さは、第2の、より低い温度で融解するプライマーが、標的にアニーリングしない第1のアニーリング温度で、第1の、より高い温度で融解するプライマーとの標的とするアニーリングが、起きるように選択される。プライマーの組または組のセットは、より高い温度で融解するプライマーとより低い温度で融解するプライマーとの間のTmにおける約10〜30℃の差異を確立するように選択される。例として、図2は、より高い温度で融解するプライマーの組は、約60〜70℃のTmを有するように設計され得、そしてより低い温度で融解するプライマーの組は、約45〜55℃のTmを有するように選択され得る。Tm値は、標準的な塩基対形成および最隣接のアルゴリズムを使用して推定され得る。典型的には、標的とのプライマーおよびプローブのアニーリングは、二重鎖の推定融解温度、10℃下までの温度で実施される(Ausubelら編「Preparation and Analysis of DNA」,および「The Polymerase Chain Reaction」in Current Protocols in Molecular Biology,(1993)John Wiley&Sons,New York)。
【0045】
プローブについてのTm値は、より短く、高親和性のプローブ(例えば、PNA FRETプローブ)を除いて、68〜70℃であり得る。これは、より低いTmを有し得る。プローブ配列は、標的ポリヌクレオチドに相補的であるように選択され、そして標的のプライマー結合部位間にある。プローブ配列は、第2の、より低いTmプライマーによって伸長される鎖に相補的であるように選択されるべきである。この鎖は、他の鎖をコピーするために第1の、より高いTmプライマーにアニーリングおよび伸長の後、実質的に一本鎖になる(図1)。プローブ配列は、レポーター色素標識およびクエンチャー標識の実行される近接を支持する非標的特異的な、自己相補的な配列を含むように設計され得る。自己相補的な配列は、プローブの5’末端および3’末端に位置し得る。このような「ヘアピン」配列は、分子間「ステム」領域および非塩基対形成「ループ」領域を有する。標的への結合において、レポーター色素およびクエンチャーは、空間的に分離され、そして蛍光は、増大する。
【0046】
プローブは、PCRの間、ポリメラーゼによって伸長不可能であるように設計される。PNA FRETプローブは、一般に、ポリメラーゼに対する基質ではない。DNAプローブは、3’ヒドロキシルにおいて、3’リン酸基または他の基で3’末端をブロックするかあるいは3’末端ヌクレオチドの核酸塩基をブロックすることによって伸長不可能にされ得る(Livak,米国特許第5,723,591号)。
【0047】
(V2c.核酸アナログ)
核酸アナログは、DNAおよびRNAの構造的なアナログであり、そして相補的な核酸配列にハイブリダイズするように設計される。ヌクレオチド間結合の改変を介して、本発明の糖および/または核酸塩基、核酸アナログは、以下の所望の性質のいずれかまたは全てを達成し得る:1)最適化されたハイブリダイゼーション特異性または親和性、2)ヌクレアーゼ耐性、3)化学的安定性、4)可溶性、5)膜透過性ならびに6)合成および精製の容易さまたは低費用。
【0048】
核酸アナログの有用かつアクセス可能なクラスは、ペプチド核酸(PNA)のファミリーであり、ここで、DNAまたはRNAの糖/リン酸骨格は、非環式、アキラルおよび中性のポリアミド結合に置換される。アミド骨格を介して連鎖に結合される核酸塩基を有する2−アミノエチルグリシンポリアミド連結は、PNAの実施形態として非常に研究されており、そして例外的なハイブリダイゼーション特異性および親和性を有することが示される(Buchardt,WO92/20702;Nielsen(1991)Science 254:1497−1500;Egholm(1993)Nature,365:566−68)。
【0049】
(V.2d PNA FRET プローブ)
PNAは、平行方向または逆平行方向のいずれかにおいてその標的相補鎖にハイブリダイズし得る。しかし、逆平行二重鎖(PNAのカルボキシル末端が、DNAの5’末端と整列化され、そしてPNAのアミノ末端が、DNAの3’末端と整列化される場合)は、典型的により安定である(Egholm(1993)Nature 365:566−68)。PNAプローブは、高い特異性および親和性を有する標的DNA配列に結合することが公知である(Coull,米国特許第6,110,676号)。レポーター部分およびクエンチャー部分を有する、本発明のPNA FRETプローブの例は、PNAが、標的配列に逆平行方向にアニーリングするように設計される。相補的な標的配列に結合されるPNAプローブは、一般に、PCRの間ポリメラーゼのヌクレアーゼ活性によって認められるほどには開裂しない、ハイブリダイズのみが、レポーター部分およびクエンチャー部分の十分な分離を起こして、クエンチングの減少によって蛍光の増大を生じる(図5)。
【0050】
PNAは、任意の規模で合成され得る。最も好都合なことに、PNAは、XALまたはPAL支持体を備えるExpedite Synthesizer(Applied Biosystems);あるいはMBHA支持体を備えるModel 433A Synthesizer(Applied Biosystems);あるいは他の自動化合成機においてFmoc/Bhoc、tBoc/Z、またはMMT保護基モノマーを使用して、2μモルの規模で合成される。PNA FRETプローブは、一般に、ペプチド合成に使用される多くの固体支持体において合成され得る。固相ペプチド合成の総説に関して、J.StewartおよびJ.Young,「Solid Phase Peptide Synthesis」,Pierce Chemical Co.Rockford,IL,1984;E.AthertonおよびR.C.Sheppard,「Solid phase peptide synthesis:A practical approach」,IRL Press,Oxford,1989;M.W.PenningtonおよびB.M.Dunn(編)「Methods in molecular biology,第35巻:Peptide synthesis protocols」,Humana Press,Totowa,NJ(1994),91頁;G.Grant(編),「Synthetic peptides」,W.H.Freeman&Co.,New York,NY,1992;G.B.Fields,Int.J.Peptide Protein Res.(1990)35:161;A.J.Smith in「techniques in protein chemistry III」,R.Angeletti(編),Academic Press,Orlando,FL,1992,219頁;G.B.Fields(編),「Methods in enzymology:第289巻」,Academic Press,New York,NY,1997;W.C.ChanおよびP.D.White,「Fmoc solid phase peptide synthesis:a practical approach,Oxford University Press,Oxford,UK,2000;P.Lloyd−WilliamsおよびF.Albericio(編),「Chemical approaches to the synthesis of peptides and proteins」, CRC Press, New York,NY 1997を参照のこと。
【0051】
合成が完了した後、粗PNAは、例えば、トリフルオロ酢酸を用いて、支持対から開裂され得、次いで、ジエチルエーテルを用いて沈殿され、そしてジエチルエーテルで2回洗浄される。PNAは、逆相HPLCによって精製され得、質量分析法によって分析され得、そして質量を使用して260nmの吸光度の補正することによって、定量化され得る。蛍光標識PNAプローブは、ハイブリダイゼーションアッセイにおける所望の性質を実証する(Hyldig−Nielsen,米国特許第5,985,563号;Coull,WO98/24933;Coull,WO99/22018;Gildea,WO99/21881;Coull,WO99/49293)。
【0052】
PNA−DNAキメラは、別個のPNA部分およびヌクレオチド部分を伴うオリゴマー分子である。これらは、実質的に任意の組換せまたは配列におけるPNAモノマーおよびヌクレオチドを共有結合することによって合成され得る。効率的かつ自動化された方法は、PNA−DNAキメラ合成のために開発されている(Vinayak(1997)Nucleosides&Nucleotides 16:1653−56;Uhlmann(1996)Angew.Chem.,Intl.編,Eng.35:2632−35;Uhlmann,EP 829542;Van der Laan(1997)Tetrahedron Lett.38:2249−52;Van der Laan(1998)Bioorg.Med.Chem.Lett.8:663−68)。PNA−DNAキメラは、PNAおよびDNAに見られる所望の性質(例えば、PNAのより良いハイブリダイゼーション性質およびDNAのような生物学的機能(DNA部分の3’OH末端を介するプライマー伸長を含む))を有するように設計される(Uhlmann(1998)Biol.Chem.379:1045−52)。
【0053】
PNAモノマー単位と標識の間のリンカーは、以下を含む:(i)共有結合;(ii)アルキリジイルスペーサー−(CH2)n−、ここでnは、1〜12である;(iii)エチレンオキシ−(CH2CH2O)n−、ここでnは、1〜12である;(iv)アリールジイル(C6−C20);または(v)1つ以上のアミノ酸。リジン側鎖、アスパラギン酸側鎖およびグルタミン酸側鎖は、PNA、FRETプローブにおける連鎖部位であり得る。リジンの側鎖のε−アミノ基は、標識(例えば、レポーター色素またはクエンチャー)の結合のための反応連結基であり得る。リンカーは、典型的には、PNAモノマー単位および固体支持体との縮合のための適合性のある保護基および反応性機能基を伴う対応するモノマー単位によってPNAのアミノ末端および/またはカルボキシル末端に結合される。例えば、「Oリンカー」(2−(2−アミノエトキシ)酢酸)の単位)は、任意のPNA骨格アミノ基または固体支持体のアミノ官能基のアミノ末端に結合し得る。
【0054】
(V.2e 標識化)
標識されたオリゴヌクレオチドは、当該分野において周知の方法(例えば、Hermanson,Bioconjugate Techniques,(1996)Academic Press,San Diego,CA.40−55頁,643−71を参照のこと)を使用して、適切な反応性標識と適切な溶媒中のオリゴヌクレオチドと適切な溶媒(両方とも可溶性である)中で反応させることによって形成され得る。粗標識化オリゴヌクレオチドは、任意の開始物質または所望でない副産物から精製され得、そして後の使用のために(好ましくは、低温で)乾燥貯蔵され得るか、または溶液中で貯蔵され得る。
【0055】
標識は、オリゴヌクレオチドまたはヌクレオチドに対する、1つの結合を介す共有結合性結合のための、1つの置換基の位置における反応性連結基(例えば、フルオレセインまたはローダミンの5−カルボキシまたは6−カルボキシ)の1つに反応性連結基を有し得る。一般に、この標識とオリゴヌクレオチドまたはヌクレオチドとを連結する結合は、(i)プライマー伸長を妨害すべきではなく、(ii)ポリメラーゼ活性を阻害すべきではなく、また(iii)色素標識の蛍光特性に悪影響(例えば、クエンチまたは漂白)を与えるべきではない。反応性連結基は、共有結合の形成が可能な部分であり、代表的にはオリゴヌクレオチド上の求核性基(例えば、アミンおよびチオール)との反応が可能な求電子性官能基である。反応性連結基の例としては、活性エステル、例えば、イソチオシアネート、スルホニルクロリド、スルホン酸エステル、シリルハライド,2,6−ジクロロトリアジニル、ホスホルアミダイト、マレイミド、ハロアセチル、エポキシド、ハロゲン化アルキル、ハロゲン化アリル、アルデヒド、ケトン、アシルアジド、無水物、およびヨードアセトアミドが挙げられる。活性エステルとしては、スクシンイミジル(NHS)、ヒドロキシベンゾトリアゾーリル(HOBt)およびペンタフルオロフェニルエステルが挙げられる。
【0056】
蛍光色素の1つの反応性連結基は、この蛍光色素のカルボキシル置換基のN−ヒドロキシスクシンイミジルエステル(NHS)である。この色素のNHSエステルは実施され得、単離され得、精製され得、そして/また特徴付けられ得、あるいはこれはインサイチュで形成され得、そしてオリゴヌクレオチドの求核性基と反応し得る。代表的には、この色素のカルボキシル形態は、以下の幾つかの組み合わせと反応することによって活性化されて、この色素のNHSエステルを与える:(1)カルボジイミド試薬、(例えば、ジシクロヘキシルカルボジイミド)、ジイソプロピルカルボジイミド、またはウロニウム(uronium)試薬、(例えば、TSTU(O−(N−スクシンイミジル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボレート、HBTU(O−ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオホスフェート)、またはHATU(O−(7−アザベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート));(2)アクチベーター(例えば、1−ヒドロキシベンゾトリアゾール(HOBt));および(3)N−ヒドロキシスクシンイミド。
【0057】
標識の別の反応性連結基は、蛍光色素、クエンチャー、小溝結合剤、および移動度改変因子(mobility modifier)のホスホルアミダイト形態である。ホスホルアミダイト色素試薬は、標識化されたオリゴヌクレオチドの自動合成機にとって特に有用である。このホスホルアミダイト試薬は、ヌクレオシド性(nucleosidic)であるか、または非ヌクレオシド性(non−nucleosidic)であり得る。非ヌクレオシド性形態のホスホルアミダイト色素試薬は、以下の一般式:
【0058】
【化4】
【0059】
上記の一般的なホスホルアミダイト色素試薬は、ヒドロキシル基(例えば、固体支持体に結合したオリゴヌクレオチドの5’末端のOH)と穏やかな酸活性化下で反応し、ヌクレオチド間亜リン酸基を形成し、次いでこの亜リン酸基は、ヌクレオチド間リン酸基へと酸化される。いくつかの例において、この色素は、ホスホルアミダイト試薬の合成の間、またはこれに続く分子(例えば、オリゴヌクレオチド)を標識するための使用の間のいずれかで、保護を必要とする官能基を含み得る。使用される保護基は、官能基の性質に依存し、そしてこれは当業者にとって明らかである(Greene,T.およびWuts,P.Protective Groups in Organic Synthesis、第2版、John Wiley&Sons,New York,1991)。この色素は、3’から5’への方向の合成結果として、オリゴヌクレオチドの5’末端に結合する。ヌクレオシド性および非ヌクレオシド性である、他のホスホルアミダイト色素試薬は、オリゴヌクレオチドの他の部位(例えば、3’末端、核酸塩基、ヌクレオチド間結合、糖)で標識を可能にする。核酸塩基、ヌクレオチド間結合、および糖部位での標識によって、蛍光色素での、内部標識および複数の標識が可能なる。
【0060】
ヌクレオチド5’−トリホスフェートは、本発明の特定の実施形態における使用のために標識され得る。ヌクレオチドの、糖部分または核酸塩基部分は、標識され得る。核酸塩基標識部位としては、プリン核酸塩基の8−C、7−デアザプリン核酸塩基の7−Cまたは8−C、およびピリミジン核酸塩基の5−位置が挙げられる。この標識されたヌクレオチドは、酵素的に取りこみ可能であり、かつ酵素的に伸長可能である。標識されたヌクレオチド5’−トリホスフェートは、以下の式:
【0061】
【化5】
【0062】
【化6】
【0063】
核酸塩基標識された、オリゴヌクレオチドプライマーまたはプローブは、以下の式を有し得る:
【0064】
【化7】
【0065】
【化8】
【0066】
種々の標識は、オリゴヌクレオチドプローブの3末端に共有結合的に結合され得る。標識を保有するか、または合成後反応によって標識され得る官能基を保有する、固体支持体は、オリゴヌクレオチド合成のための固体支持体として使用され得る(米国特許第5,141,813号;同第5,231,191号、同第5,401,837号;同第5,736,626号)。このアプローチによって、この標識またはこの官能基は、オリゴヌクレオチドの合成の間に存在する。切断および脱保護の間、この標識または官能基は、オリゴヌクレオチドに共有結合したままである。3’末端において標識されるオリゴヌクレオチドプローブは、以下の式を有し得る:
【0067】
【化9】
【0068】
1つの合成後化学標識方法において、オリゴヌクレオチドは以下のように標識される:NHS形態の6−カルボキシフルオレセインを、DMSOに溶解または懸濁し、そして、0.25M重炭酸/炭酸緩衝液中の5’−アミノへキシルオリゴヌクレオチドに対して、過剰(10〜20倍)に約pH9で、添加し、6時間反応させる(Fung、米国特許第4,757,141号)。色素標識されたオリゴヌクレオチド生成物は、緩衝液、例えば、0.1モル濃度の酢酸トリエチルアミン(TEAA)で溶出する、サイズ除去クロマトグラフィーカラムを通過させることによって未反応の色素から分離され得る。例えば、この粗標識化オリゴヌクレオチドを含むこの画分は、勾配溶離を使用する逆相HPLCによってさらに精製され得る。
本発明のオリゴヌクレオチドプライマーおよびプローブは、電気泳動移動の速度に影響を与える部分、すなわち移動度改変標識で標識され得る。移動度改変標識としては、以下が挙げられるが、これらに限定されない:ビオチン、蛍光色素、コレステロール、およびポリエチレンオキシ単位、―(CH2CH2O)n−(ここで、nは1〜100であり得る)(Grossman,米国特許第5,624,800号)。好ましくは、nは2〜20である。このポリエチレンオキシ単位は、リン酸基と共に散在され得る。蛍光標識プライマーを、別個で既知サイズであるポリエチレンオキシのさらなる標識で特異的に標識することによって、アンプリコンのサイズ(すなわちヌクレオチドの数)に実質的に依存しないアンプリコンの電気永動による分離を可能にする。すなわち、同じ長さのポリヌクレオチドは、分光学的に分離可能な色素標識の検出によって区別され得、そして移動度改変標識に基づいて分離され得る。色素標識および移動度改変標識の両方を保持するポリヌクレオチドは、単一標識したオリゴヌクレオチド成分または単一標識したヌクレオチド成分の、連結またはポリメラーゼ伸長、例えば、非同調的PCRによって、酵素的に形成され得る。
標識の1つのクラスは、蛍光、化学発光、および電気化学的発光によって、標識された伸長生成物および標識された増幅生成物の検出のためのシグナルを提供する(Kricka,L. Nonisotopic DNA Probe Techniques(1992),Academic Press,San Diego,pp.3−28)。化学発光標識としては、1,2−ジオキセタン化合物(米国特許第4,931,223号;Bronstein(1994)Anal.Biochemistry 219:169−81)が挙げられる。プローブ、プライマー、およびヌクレオチド5’−トリホスフェートの標識化のために有用な蛍光色素としては、フルオレセイン,ローダミン(米国特許第5,366,860号;同第5,936,087号;同第6,051,719号)、シアニン(Kubista,WO 97/45539)、および金属ポルフィリン錯体(Stanton,WO 88/04777)が挙げられる。
【0069】
蛍光レポーター色素としては、キサンテン化合物(例えば、フルオレセインIおよびローダミンII:
【0070】
【化10】
【0071】
IおよびIIの環の位置は、置換され得る。IIのアミノR基は、置換され得る。これらの置換基は、本発明のプライマー、プローブおよびヌクレオチドに対する共有結合を含む。IおよびIIの例としては、Xが、カルボキシル基、クロロ基、および他の基で置換されたフェニルである場合(米国特許第5,847,162号;同第6,025,505号;同第5,654,442号;同第5,188,934号;同第5,885,778号;同第6,008,379号;同第6,020,481号;同第5,936,087号)、およびXが水素である場合(Benson,米国特許第.6,051,719)が挙げられる。
【0072】
プローブ標識の別のクラスとしては、蛍光クエンチャーが挙げられる。クエンチャーの発光スペクトルは、蛍光色素の蛍光が、分子間の蛍光色素とオーバーラップし、その結果、蛍光共鳴エネルギー転移「FRET」現象によって減少またはクエンチされる(Clegg(1992)Meth.Enzymol.,211:353−388)。発蛍光団からクエンチャーへのエネルギー転移を許容する配置で、プローブに結合された、蛍光レポーター色素およびクエンチャーは、蛍光色素による蛍光の減少を生じ得る。このレポーターは、化学反応によって励起して化学発光を生成するか、または光吸収によって励起して蛍光を生成するかのいずれかである、蛍光化合物である。このクエンチャーは、レポーターと相互作用して、発光を変更し、通常はこのレポーターの発光効率を減少し得る。このクエンチ減少の効率は、レポーター分子とクエンチャー分子との間の距離に直接的に相関する(Yaron(1979) Analytical Biochemistry,95:228−35)。この自己クエンチ効果は、プローブのその相補体に対するハイブリダイゼーションの際、または蛍光レポーターおよびクエンチャーが分離してすぐのヌクレアーゼ切断の際に減少され得るか、または消失され得る(図5)。
特定のクエンチャーとしては、以下が挙げられるが、これらに限定されない:(i)以下からなる群より選択されるローダミン色素:テトラメチル−6−カルボキシローダミン(TAMRA)、テトラプロパノ−6−カルボキシローダミン(ROX)(Bergot,米国特許第5,366,860号):
【0073】
【化11】
【0074】
【化12】
【0075】
【化13】
標識の別のクラスは、特異的捕捉手段または非特異的捕捉手段による、標識化したアンプリコンの分離および固定を実施する役割を果たす(例えば、ビオチン;2,4−ジニトロフェニル(DNP);およびジゴキシゲニン(Andrus,A.「Chemical methods for 5’non−isotopic labelling of PCR probe and primers」(1995) PCR 2:A Practical Approach,Oxford University Press,Oxford,pp.39−54))。
【0076】
標識の別のクラスは、移動度改変因子(例えば、ポリエチレンオキシ(PEO)単位)である。このPEO標識は、荷電基(例えば、ホスホジエステル)が含まれ得、電荷を与え、そして電気泳動移動度(速度)を上昇する。このPEO標識は、非荷電であり得、そして電気泳動移動度またはクロマトグラフィー移動度を抑制するように作用する。このような改変因子は、例えば、蛍光検出による分析の間、増幅生成物の電気泳動速度に影響を与えるかまた標準化するように働き、分解能および分離を改善する(米国特許第5,470,705号)。
【0077】
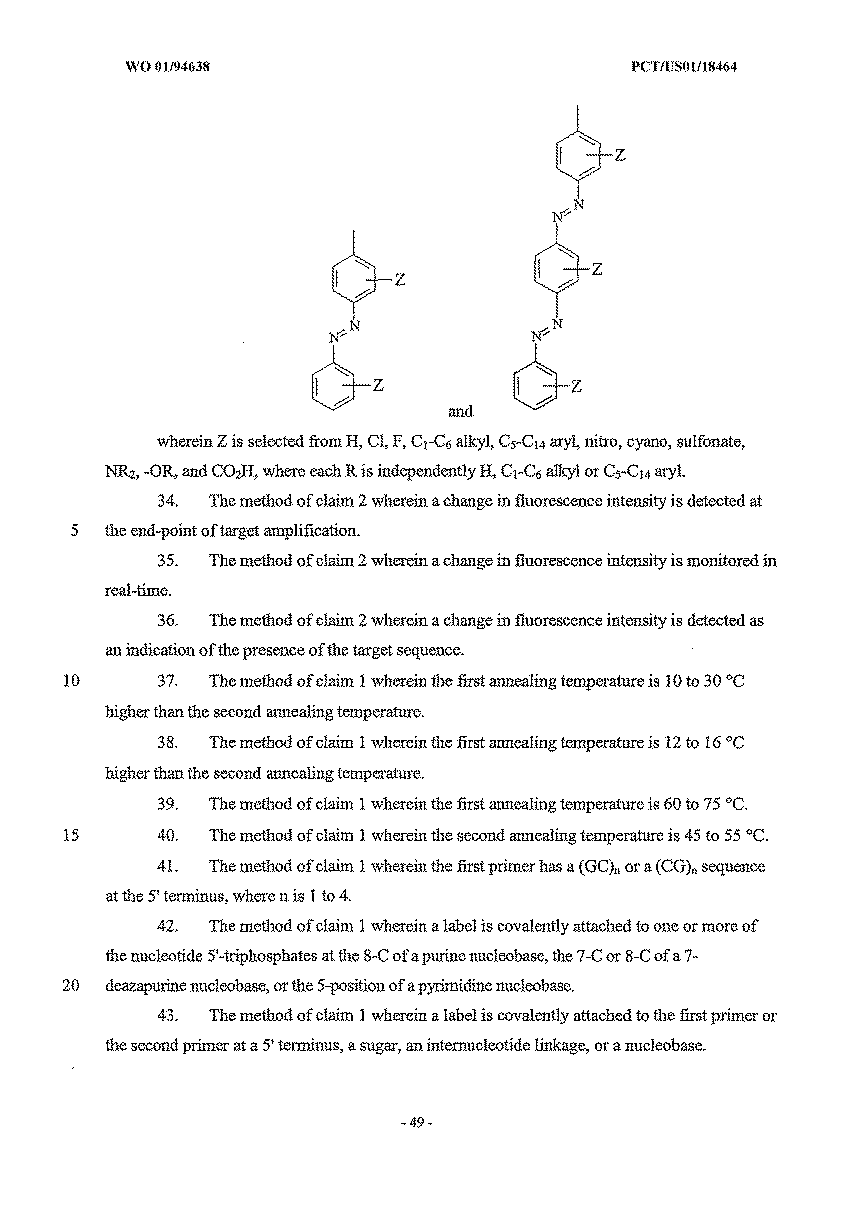
プローブ標識およびプライマー標識の別のクラスは、本明細書中でハイブリダイゼーションスタビライザーとして称され、これには以下が挙げられるが、これらに限定されない:小溝結合剤、インターカレーター、ポリカチオン(例えば、ポリリジンおよびスペルミン)、および架橋官能基。ハイブリダイゼーションスタビライザーは、プライマーと標的との、またはプローブと標的との塩基対合の安定性、すなわち親和性またはハイブリダイゼーションの速度を増加し得る(Corey(1995)J.Amer.Chem.Soc.117:9373−74)。ハイブリダイゼーションスタビライザーは、完全に相補的なオリゴヌクレオチド配列と標的配列との間のTmの大きな差違で例示される、塩基対形成の特異性を増加すように働き、ここで、得られる2重鎖は、1つ以上のワトソン/クリック塩基対形成のミスマッチを含む(Blackburn,G.およびGait,M.編「DNA and RNA structure」 Nucleic Acids in Chemistry and Biology,第2版(1996)Oxford University Press,pp. 15−81および337−46)。小溝結合剤としては、Hoechst 33258 Rajur、(1997)J.Org.Chem.62:523−29)、MGB1(Gong(1997)Biochem.and Biophys.Res.Comm.240:557−60)およびCDPI1−3(米国特許第.5,801,155;WO 96/32496)、例えば、CDPI3:
【0078】
【化14】
【0079】
(V.3 非同調的熱サイクルプロトコール)
本発明は、標的核酸のPCR増幅のための新規の非同調的熱サイクル方法を含む。標的は、プライマーの伸長および増幅が可能な任意のポリヌクレオチドであり得る。標的核酸としては、例えば、プラスミド、cDNA,アンプリコン、ゲノムDNA、制限消化DNA、および連結生成物が挙げられる。標的核酸は、様々な反復配列および単一塩基多型(SNP)を含む、多型であり得る。この方法は、異なるTm値のプライマーを使用する、多段階のアニーリングおよび伸長プロセスを使用する。このPCR増幅試薬は、プライマー伸長試薬(例えば、ポリメラーゼ)、ヌクレオチド5’−トリホスフェート、および緩衝液を含む。2つの有意な利点が、以下この方法のから認識され得る:(1)2本鎖標的よりもむしろ1本鎖標的の、PCR混合物中に存在するプローブでの標的化、および(2)1本鎖アンプリコンの過剰な生成または主要な生成。
【0080】
この本発明の熱サイクルプロトコールは、代表的には、規定された温度における、1連の時限工程を含む。一連の工程は、PCRプロセスが完了するまで、または所望の成果(例えば、特定のシグナル検出、またはデータの回収)が達成されるまで反復され得る。これらの工程の個々のパラメーターは、以下を含むPCR中の各事象を最適化するように選択される:(1)変性(1本鎖への二重鎖の熱融解);(2)アニーリング(標的に対するプライマーのハイブリダイゼーション);および(3)プライマー伸長(酵素的に伸長可能なヌクレオチドの取り込み)。いくつかのプロトコールにおいて、プローブハイブリダイゼーション工程を、このサイクルに取り込み得る。また、これらの事象のうちいくつかは、単一工程で実施され得る。例えば、1つ以上のプライマーのプローブハイブリダイゼーションおよびアニーリングは、同じ温度で起こり得る。プライマーのアニーリングおよび伸長は、単一の温度で起こり得る。
【0081】
これらの工程のパラメーター(例えば、順序、持続時間、および温度)は、成果を最適化するように選択され、そしてこれらのパラメーターは以下の様々な因子によって、おおきく左右される:プライマーおよびプローブのTm、(存在する場合)アンプリコンの長さ、標的の量および純度ならびに検出方法。低コピー数のゲノムDNA標的配列は、特定の工程の長い持続時間または高サイクル数を必要とし得る。
【0082】
本発明の方法における特定の実施形態は、ある変性温度における2つの鎖への二本鎖標的核酸の変性工程を含む。図1は、以下の工程を含む本発明の1つの実施形態に従う非同調的PCR熱サイクル法の概略図を示す:(i)二本鎖標的の変性工程、(ii)第1のプライマーのアニーリング工程、(iii)第1のプライマーの伸長工程、(iv)プローブハイブリダイゼーション、(v)第2のプライマーのアニーリング工程、および(vi)第2のプライマーの伸長工程。これらの温度および時間は、単なる例示である。他の実施形態において、この方法は、1本鎖標的核酸で開始し得る。
【0083】
第1の、より高い親和性のプライマーは、第1のアニーリング温度で、標的の一本鎖のその相補的な配列にアニーリングされる(図1のアニーリングI)。このより高い親和性のプライマーは、反応容器中の、第2の、より低い親和性のプライマーに比べより高いTmを有する。第1のアニーリング温度で、第2のプライマーは、第1のプライマーがその相補的な標的配列にアニールするより低い程度で、標的の他の鎖においてその相補的な配列にアニーリングする。なぜならば、第2のプライマー/標的の2重鎖は、第1のアニーリング温度で十分な安定性を有しないからである。この第1のプライマーは、ヌクレオチドの取り込み(すなわち、ポリメラーゼに媒介されるヌクレオチド5’―トリホスフェートの付加)によって、第1のアニーリング温度、すなわち伸長温度で伸長される(図1での伸長I)。この第1のアニーリング温度および伸長温度は単一の温度であり得、この温度において、第1のプライマーのアニーリングおよび伸長が、共通温度で起こり得る。本発明のこの実施形態のこの方法のこの段階において、この標的の一方のストランドは、2重鎖の一部分であり、そして他のストランドは1本鎖である。
【0084】
温度は、検出可能なプローブが、標的核酸の一本鎖形態にハイブリダイズするプローブハイブリダイゼーション温度(図1におけるハイブリダイゼーション)まで低められ得る。この検出可能なプローブは、ハイブリダイゼーションの際、またはポリメラーゼのヌクレアーゼ活性による切断の際に、蛍光において、例えばFRETによって、増加を示し得る。
【0085】
次いでこの温度は、第2アニーリング温度(図1におけるアニーリングII)に変えられるか、またはプローブハイブリダイゼーション温度に一定に保たれ、それによりこの第2プライマーは、標的のその相補鎖にアニーリングする。この第2アニーリング温度は、第1アニーリング温度よりも低く、そして第1プライマーの伸長温度よりも低い。第2プライマーは、第2アニーリング温度、またはより高い伸長温度で伸長する。伸長II温度は、伸長I温度と同じであっても異なっていても良い。サイクル中のこの時点で、標的の各鎖のコピーが作製される。図1は、非同調的熱サイクルプロトコールの1つの実施形態のサイクルを図示する。この工程の温度および時間は、単に例である。
【0086】
図1において示される本発明の実施形態において、第2サイクルおよび引く続くサイクルは、二本鎖標的の変性を伴って再び開始し、前述の他の工程が続く。このサイクルは、所望するだけの回数が繰り返され得るが、代表的に、検出可能なシグナルが明白化もしくは安定化されるまで、または十分な量のアンプリコンが産生されるまで繰り返される。代表的に、50サイクルが、アンプリコンを検出または産生するのに十分である。このサイクルにおける各工程の持続時間は、事象を完了させるために十分である(すなわち、変性、アニーリング、伸長、およびプローブハイブリダイゼーションが実質的に完了する)。
【0087】
非同調的熱サイクルプロトコールの代替的な実施形態は、検出可能なプローブもプローブハイブリダイゼーション工程も使用しない。この実施形態は、第1プライマーおよび第2プライマーの時間的に連続したアニーリング工程および伸長工程が、第1段階:変性、アニーリングI、伸長I、アニーリングIIおよび伸長II、続いて変性工程、アニーリングI工程および伸長I工程のみのサイクルの第2段階において行なわれる場合に有用であり得る。この第1段階は、2〜50サイクル行われ得、続いてプロトコールの後半部分として1〜25サイクルの第2段階が行われ得る。第2段階におけるアニーリングII工程および伸長II工程の省略は、第1プライマーに対する相補体のみのコピーまたは主にこの相補体のコピーを可能にする。従って、この生じたアンプリコンは、一本鎖核酸が優勢である。
【0088】
1つの実施形態において、第1プライマーと第2プライマーとの間のTmの差(ΔTm)は、より高い温度の第1アニーリング工程および第1伸長工程の間、より高いTmプライマーのみがアニーリングおよび伸長を受けるように、十分大きい。代表的に、アニーリング温度は、アニーリングおよび伸長されるべきプライマーのTmの0〜10℃下に設定される。第1アニーリング温度は、標的への第1プライマーのアニーリングを可能にし、そして標的への第2プライマーのアニーリングは実質的に行なわれないような任意の温度であり得る。第1プライマーについての伸長温度は、標的に対する第2プライマーの伸長を可能にし、そして標的への第2プライマーのアニーリングは実質的に行なわれないような任意の温度である。第1プライマーの伸長温度は、標的に対する第2プライマーの伸長を可能にする任意の温度である。第2プライマーについての伸長温度は、第2アニーリング温度と同じであっても異なっていても良い。より低いTmの第2プライマーのアニーリング工程および伸長工程の間、より高いTmの第1プライマーに対して相補的な、ほとんど全てまたは実質的に全ての標的配列は、図1において示されるように、伸長され、二重鎖として存在する。図2は、例示的なTm範囲(第1プライマーについて、例えばTm=60〜75℃、および第2プライマーについて、例えばTm=45〜55℃)を示す。
【0089】
1対よりも多くのプライマーは、本発明の非同調的熱サイクルプロトコールによって行われるPCR反応中に存在し得る。1対よりも多くのプライマーは、特定のアンプリコンを増幅し得る。1対よりも多くのプライマーが本発明のPCR反応物中に存在する場合、1つよりも多くのアンプリコンが生じ得る(すなわち、1つよりも多くの標的配列が増幅され得る)。特定のプライマー(例えば、より高温で融解する第1のプライマー、またはより低温で融解する第2のプライマー)は、1対よりも多くのプライマーを形成し得、そして1つより多くの標的配列を増幅し得る。例えば、より高温で融解するプライマーは、より低温で融解する1つのプライマーと100bpのアンプリコン、およびより低温で融解する異なるプライマーと200bpのアンプリコンを産生し得る。1つより多くのプローブが、本発明の非同調的熱サイクルプロトコールによって行われるPCR反応中に存在し得る。各プローブは、独特のの色素を有し得、そして特定の標的配列の相補鎖を検出するように(例えば、遺伝子の2つの対立遺伝子形態を検出するように)設計された配列を有し得る。
【0090】
PCR反応は、熱サイクルが可能である任意の囲いまたは部位中で行われ得る。容器としては、チューブ、フラスコ、ウェル、凹部(depression)、フリット(frit)、多孔性部位、および表面上のアドレス可能な位置(すなわち、アレイ)が挙げられる。
【0091】
(V.4 PNA FRETプローブを用いる非同調的PCRのモニタリング)
本発明の1つの実施形態において、レポーター色素およびクエンチャーで標識されたPNA FRETプローブは、ハイブリダイゼーションによって標的ポリヌクレオチドのリアルタイム増幅を検出およびモニタリングし得る。PNAプローブ(プライマー配列に対して内部のアンプリコン配列に相補的である)は、より高いTmのプライマーがアニーリングおよび伸長した後に、ssアンプリコンにハイブリダイズする。相補的な標的にハイブリダイズしたPNAプローブは、酵素(例えば、Taqポリメラーゼのエキソヌクレアーゼ活性)によって、PCR中に識別できるほどは切断されない。相補体に結合しない場合、このレポーター色素はクエンチされる。相補的配列にハイブリダイズした場合、このレポーター色素とクエンチャーとは、空間的に離れており、そして蛍光における増加が検出され得る。図5は、クエンチされた(離れた、ハイブリダイズされていない)状態およびクエンチされない(ハイブリダイズした)状態における、例示的な8〜18ntのPNA FRETプローブを示す。この蛍光強度の変化は、相補的なポリヌクレオチド(すなわち、アンプリコン)のハイブリダイゼーション(従って、存在および量)と相関し得る。このPNA FRETプローブは、逆に荷電したリンカー(例えば、カルボキシル化アミノ酸鎖(例えば、グルタミン酸およびアスパラギン酸)、ならびにアンモニウムアミノ酸側鎖(例えば、リジン)を取り込むことによって結合していない状態におけるクエンチングを最適化するように設計され得る。あるいは、このプローブの配列は、5’末端および3’末端で、分子内の塩基対形成の立体配置(すなわち、ヘアピン構造)をとる非標的相補的配列を含むように設計され得、この構造は蛍光色素部分とクエンチャー部分とを近位にする。
【0092】
(V.5 リアルタイム検出を用いる非同調的PCRの応用)
非同調的PCRプロトコールの1つの実施形態のリアルタイムモニタリングにおける工程は、高特異性条件下(すなわち、比較的高い温度)での検出可能なプローブのハイブリダイゼーションである。より高い特異性は、単一塩基対の区別を可能にする。このプローブは、切断性(例えば、DNA)でも良いし、または、非切断性(例えばPNAもしくはそのアナログ)でも良い。このプローブハイブリダイゼーション工程および検出工程は、任意の温度で実行され得、そしてより長い対応するプローブよりも内因的により特異的である非常に短いプローブの使用を可能にする。例示として、図13は、第2プライマーアニーリング工程の前に使用される、30〜37℃のプローブアニーリング工程を示す。このような低温のプローブアニーリング工程は、8ntおよび9ntほどの短いPNAプローブを使用して実行され得る(実施例5、図8)。
【0093】
PNA FRETプローブは、伝統的な熱サイクルプロトコールよりも非同調的熱サイクルPCRプロトコールの間に、より効果的にDNA標的に結合する。図9は、非同調的PCR(A−PCR)および伝統的な(通常)PCRの各々の間の、25〜30サイクルでの平均蛍光変化を示す。65℃でのアニーリング工程で、蛍光強度は、伝統的なPCRについて、A−PCRプロトコールについての、2倍のシグナル増加と比較してごくわずかに増加し、これは標準に対するPNAプローブのより少ない結合を示す。シグナルにおける、より大きな増加は、標的のより低いコピー数でのより高い検出感度(すなわち、より高いシグナル/ノイズ)を意味する。
【0094】
非同調的PCR方法の代替的な実施形態は、2つのうち1つのプライマーが、GもしくはC含有テイル、または「クランプ」(例えば5’(GC)− nまたは(CC)n(ここで、nは1〜4である)を有するように設計される、より少ないサイクルの伝統的な熱サイクルプロトコールを実行することである。あるいは、このテイルは、ポリGまたはポリCモチーフであり得る。このGCまたはCGテイルユニットは、あらゆる特定の標的配列に非相補的であるように設計される。このテイルは、「テイル付加していない」配列に比較してプライマーのTmを増加するのに役立つ。最初の数サイクルの間、両方のプライマーは標的に等しく良好にアニーリングし、単一アニーリング温度が、テイル付加していないプライマーのTmに等しいか、またはほとんど等しい、伝統的な熱サイクルプロトコールの間、比較的同調的な伸長を生じる。図14aは、アンプリコン中へのGCテイルの複製を伴う伝統的な熱サイクルプロトコールの2つのサイクルを示す。いくつかのサイクルの後、大多数のアンプリコンは5’末端でGCテイルを取り込み、そしてその相補体は3’末端でGCテイルを取り込む。次いでこのプライマーのGCテイルは、アンプリコンに対して相補的であり、そしてより高い温度(テイル付加していないプライマーはアニーリングしない温度)でアニーリングする。いくつかのサイクル(例えば、1〜5サイクルの伝統的なプロトコール)の後、熱サイクルプロトコールは、非同調的プロトコールに移行し、それによってssアンプリコンは、プライマーアニーリング工程と伸長工程との間で、プローブによって標的化され得るか、または過剰なssアンプリコンを産生するために標的化され得る。あるいは、非同調的プロトコールは、単独で使用され得る。GCテイルプライマー方法の1つの利点は、プライマーまたはアンプリコンの設計に存在し得る。
【0095】
非同調的PCRサイクルはまた、切断性DNA FRETプローブを用いるヌクレアーゼ切断アッセイにおいて有用性を有する。本発明の1つの実施形態は、5’−エキソヌクレアーゼ(TAQMAN(登録商標))増幅および検出プローブに対する改良を提供する(Holland(1991)Proc.Natl.Acad.Sci.、88:7276〜80;Livak、米国特許第5,538,848号;Gelfand、米国特許第5,210,015号および同第5,538,848号)。プライマー伸長を行い、そしてポリヌクレオチドを増幅するポリメラーゼはまた、「レポーター」色素および「クエンチャー」が結合し、そして配列が標的DNAに相補的である、標的にアニーリングしたプローブのホスホジエステル結合を切断することに役立つヌクレアーゼ活性を有し得る。切断は、検出のための、クエンチされていない標識化したフラグメントを放出し得る。プローブの切断は、レポーターとクエンチャーとの間の空隔間が、ハイブリダイゼーションの結果として調節されるFRETプローブを設計することによってハイブリダイゼーション事象の検出が達成されるいくつかのアッセイにおいて、必要ではない。(Morrison(1992)Nonisotopic DNA Probe Techniques、Kricka編、Academic Press,Inc.,San Diego、Calif.第13章;HellerおよびMorrison(1985)Rapid Detection and Indentification of Infectious Agents、Academic Press,Inc.,San Diego、CA、245〜256ページ)。この方法は、FRET、蛍光エネルギー転移(FET)、非放射性エネルギー転移、長期エネルギー転移、双極子結合エネルギー転移またはForsterエネルギー転移として文献中に種々に記載される、適切な蛍光標識が密接に近位にされた場合に生じる蛍光における変化に依存する。FRETプローブは、(標的へ結合していない場合、「ダーク(dark)」状態にする自己相補性「ヘアピン」配列を含み得、そしてハイブリダイゼーションアッセイにおける特異性を増大させ得る(Tyagi、米国特許第5,925,517号;同第6,037,130号;同第6,103,476号;同第6,150,097号)。エキソヌクレアーゼアッセイおよび他の定量的蛍光ベースのアレイを実行するシステムの例は、ABI PRISMTM7700、7200、および7900HT Sequence Detection Systems(Applied Biosystems)である。
【0096】
(V.6 終点検出を用いる非同調的PCRの応用)
非同調的PCR熱サイクルプロトコールによる感度性および特異性の増大の利点は、ヒト疾患診断、食物が原因となる病原体の検出、および微生物の検出についてのアッセイにおいて実現され得る。得られたアンプリコンは、PCRの終点で、電気泳動系(例えば、ABI PRISM 310、ABI PRISM 377、ABI PRISM 3100、およびABI PRISM 3700(Applied Biosystems))、または蛍光プレートリーダー、蛍光スキャナーもしくは画像化デバイスによって検出され得る。アンプリコンは、蛍光色素で標識化されたプライマーを用いてPCRによってか、またはインターカレーター色素染色(例えば、SYBR Green(Molecular Probes、Eugene、OR)によって検出され得る。
【0097】
PCRの終点分析は、熱サイクルおよび増幅が完了したときの蛍光色素シグナル測定を含む。結果は、PCR熱サイクルの最初から終わりまでの蛍光色素シグナル(好ましくは、あらゆる内部コントロールシグナル(を差し引く)の蛍光における変化(すなわち、蛍光強度ユニット)に関して報告される。
【0098】
本発明の非同調的PCR熱サイクルプロトコールは、標的DNAの対立遺伝子の区別のために有用である。各対立遺伝子に特異的なプローブは、閉じたチューブの均一なPCRアッセイにおいて、モニタリングされ得る。例えば、二対立遺伝子系において、2つのプローブは、各々異なる色素(例えば、FAMおよびTET)で、および各々の対立遺伝子形態について特異的な配列を伴って標識され得る(Livak(1995)Nature Genetics 9:341〜2;Livak(1999)「Allelic discrimination using fluorogenic probes and the 5’nuclease assay」Genetic Analysis:Biomolecular Engineering、Elsevier Press、14:143〜49)。プローブと標的との間のミスマッチは、このプローブがPNA FRETプローブであろうがまたはヌクレアーゼ切断性DNA FRETプローブであろうが、プローブハイブリダイゼーションの効率を大いに減少させる。従って、FAMまたはTETの蛍光シグナルにおける実質的な増加は、FAM特異的対立遺伝子またはTET特異的対立遺伝子のホモ接合性を示す。両方のシグナルにおける増加は、ヘテロ接合性を示す。
【0099】
本発明の非同調的PCR熱サイクルプロトコールはまた、遺伝子型決定および遺伝子発現分析について有用であり得る。FRETプローブを使用する遺伝子決定は、蛍光の測定がPCRが完了した後(終点)でなされることを要求する。実験のこれらの型は、ABI 7200または7700 Sequence Detection Systems(Applied Biosystems)に対して便利に行われる。このシステムは、約500〜650nmの完全な蛍光スペクトルを、PCR反応チューブにおいて直接測定する。このシステムソフトウェアは、遺伝的区別をするために蛍光データを自動的に処理する。
【0100】
(V.6.a cDNAライブラリースクリーニング、ハイブリダイゼーションによる均質配列決定(SHB))
非同調的PCRは、に対して特徴的なss cDNAアンプリコンを産生してcDNAライブラリーを特徴付けるために有用である。cDNAクローンは、寒天プレート上で、通常の実験室手順によって増殖され得、そして96または384ウェルプレートにおいて接種されて、マスター培養物を産生し得る。DNA精製は、対応する数のウェルを有する新しいプレート上の10〜20μl培養物を使用して、沸騰方法によって実行され得る。これらの手順は、自動化され得る(ABI 6700、Applied Biosystems、Foster City,CA)。次いで、このcDNA挿入物は、非同調的PCRによって、例えばプレート中の約100μlの量で、増幅され得る。このDNAは、必要であれば、<100bpのフラグメントに物理的に共有し得る。次いで、各PCR産物は、蒸留した、脱イオン水中で、例えば、2倍希釈され、そして32の同一のマイクロタイタープレート中でアリコートに分けられる。次いでこのPCR産物は、1つ以上の固有のPNA FRETプローブと混合され得る。各プローブは、アミノ末端で固有の色素(例えば、6FAM、TET、HEX、ROX)およびカルボキシル末端でクエンチャー(例えば、NTB、DABCYL)で標識される。次いで、蛍光は、蛍光マルチウェルプレートリーダー(例えば、CytoFluorII(Applied Biosystems)上で測定され得る。標準化した、適切な大きさにされた、98プローブの単一クローンに対するこの生じた蛍光強度は、ハイブリダイゼーションの表示であり、そして「ハイブリダイゼーションサイン」として規定される(Drmanac(1993)Science 260:1649〜52)。cDNAアンプリコンのハイブリダイゼーション部分の配列は、異なる既知の配列の多くのFRETプローブに対するハイブリダイゼーションに起因した、蛍光強度の逆重畳によって決定され得る(Dramanac(1994)BioTechniques 17:328〜9;Milosavljevic(1996)Genome Res.6:143〜141)。このシグナルの標準化は、特異的なプラスミド配列を標的化する内部コントロールプローブからのシグナルを越える個々の色素についてシグナルの比を使用することによって、実現され得る。ハイブリダイゼーションサインは、個々のクローン間またはcDNA配列間の配列類似性を割当てるために使用される。類似のハイブリダイゼーションサインを有するクローンは、1つの遺伝子提示クラスター中にグループ分けされる。クラスターは、特異的な全長cDNAまたは新規な遺伝子を、組織間または処置間のcDNAサインプロファイリングの差に基づいて同定するために有用である。
【0101】
図17は、PNA FRETプローブを使用する均一なSBHの概略図を示す。例示的な方法の工程は以下を包含する:(i)ss cDNAアンプリコンを作製するための非同調的PCRによって増幅されるcDNA;(ii)ss cDNAアンプリコンを整列させる;(iii)PNAプローブを、各ss cDNAアンプリコンにハイブリダイズする;(iv)蛍光検出によってハイブリダイゼーションサインを得る。この方法の利点としては、以下が挙げられる:(i)均一の条件;(ii)ハイスループット適用(すなわち、多くのサンプルを並行して処理すること)のための多重化;(iii)高いTmの短いPNAプローブを用いる、迅速なハイブリダイゼーション反応速度論、および(iv)より短いプローブのコストの利点。
【0102】
代表的な哺乳動物細胞は、10,000〜30,000の間の異なるmRNA配列を含む。これらの全てのmRNAが、cDNAライブラリーに等しく提示されているわけではない。少量のmRNA(約10コピー/細胞未満)は、全部のmRNAの約30%を構成し、従ってこの少量クラスに分けられる約11,000個の異なるmRNAが存在する(Wood(1984)Nature 312:330〜7)。所定のcDNAライブラリー中に存在する任意の珍しいcDNAクローンを得る少なくとも99%の確立を達成するために、1,000,000個までのクローンをスクリーニングしなければならない。図8は、8ntおよび9ntのPNA FRETプローブとの配列の特異性を用いた効率的な検出を示す。8nt PNA FRETプローブの完全ライブラリーは、48/2=32,000プローブからなり;ヒトゲノムにおける1,000,000よりも多いSNPを、cDNAライブラリースクリーニングによって検出するのに十分である。このライブラリーはまた、遺伝子発現モニタリングに適用可能である。
【0103】
cDNAスクリーニングに対するSBH法の利点は、cDNAライブラリー中の全ての遺伝子を一度に特徴付けする能力である。cDNAライブラリーを特徴付けするために1,000,000個のクローンが必要とされると仮定して、384ウェル形式の2604枚のプレートが、1,000,000個のPCR反応のために必要とされる。非同調的PCRは、ハイブリダイゼーションについて準備されている単一鎖アンプリコンの効率的な産生、およびアンプリコンの単離、変性、および精製の除外することによる顕著な利点を提供する。ss標的配列の産生には、しばしば、アレイ上のプローブに対する効率的なハイブリダイゼーションが必要とされる。
【0104】
(V.7 非同調的PCRによって産生されるssDNAについての応用)
非同調的PCRは、プライマー配列の選択に依存して、DNA標的の+鎖または−鎖のいずれかの増幅を可能にする。高いTmのプライマー相補鎖は、低いTmプライマー相補鎖に対して形成される。各非同調的サイクルには、2つのアニーリング工程および2つの伸長工程が含まれる。このプライマーは、プライマー長によって大部分もたらされる、顕著に異なるTm値を有する。Tmによって測定される親和性はまた、塩基含量(G+C含量)、配列およびハイブリダイゼーション安定化標識によって影響される。
【0105】
大多数の一本鎖DNAアンプリコンを生成するための方法は、一対の異なるTmのプライマーを用いて開発された。非同調的PCRは、指数関数的な増幅をもたらす多数のサイクル数について行なわれ、次いで、より高い融解プライマーによってハイブリダイゼーションおよび伸長のみを可能にするアニーリング温度および伸長温度で1つ以上のサイクルの熱サイクルが行なわれた(図20b)。これは、DNAアンプリコンの一方の鎖のみの直線的な増幅に役立つ、過剰または大多数のssDNAを生成する(図20a)。
【0106】
(V.8 キット)
本発明は、本発明の非同調的PCR方法に従った、標的核酸を増幅するための試薬を含むキットを含む。このキットは、第1プライマーおよび第2プライマーを含む。この第1プライマーおよび第2プライマーは、第1プライマーは標的にアニーリングし、そして伸長するが、第2プライマーはアニーリングも伸長もしない十分異なるTm差を有する。代表的に、ΔTmは、約10〜30℃である。第1プライマーまたは第2プライマーの1つは、標識化され得る。この標識は、蛍光色素、移動度改変因子、またはハイブリダイゼーション安定化部分であり得る。
【0107】
このキットはまた、検出可能なプローブ、ポリメラーゼ、およびヌクレオチドを含み得る。このプローブおよび/またはヌクレオチドは、蛍光標識され得る。このプローブは、蛍光部分およびクエンチャー部分で標識され得る。このプローブは、DNA、PNAまたは核酸アナログであり得る。
【0108】
このキットは、4つの異なるヌクレオチドのセットを含み得、これらは各々A、G、C、またはT核酸塩基を保有する。このセットは、核酸塩基、リンカー、および蛍光色素の組み合わせが、電気泳動条件下で分離するアンプリコンを生じる4つのヌクレオチドのセットを産生するように、設計され得る。
【0109】
(V.9 実施例)
本発明を記載してきたが、以下の実施例は、例示の目的で提供され、限定ではない。プライマー、プローブおよび標的配列について、DNAヌクレオチドは、大文字で記され、変異部位は下線および太字で記される。PNAプローブ配列は、小文字で記載される。他に記載しない限り、DNA配列の配向は、左が5’末端および右が3’末端である。PNA配列の配向は、左がアミノ末端および右がカルボキシル末端である。
【0110】
以下の実施例におけるPCRプライマーおよびプローブは、Primer ExpressTM(バージョン1.0、Applied Biosystems、CA)を使用して設計された。熱融解すなわちTmの値は、DNAプライマーおよびDNAプローブについて、基本式を使用する計算によって評価された:
Tm=81.5−16.6(log10[Na+]+0.41(%G+C)−(600/N)、
ここで、N=ヌクレオチド数でオリゴヌクレオチドの長さである(Bolton(1962)Proc.Natl.Acad.Sci.、48:1390;Sambrook,J.、Fritch,E,F.、Maniatis,T.編(1989)Molecular Cloning、A Laboratory Manual,第2版、第2巻、11.46頁、9.50〜9.51頁。基本式に対する精密化は、最近接効果および溶媒効果についてなされ得る。
【0111】
(実施例1)
(プライマーおよびPNA FRETプローブの融解温度Tmの決定)
PNA FRETプローブの融解温度(Tm)の測定を、Peltier温度コントローラーを備えるλ14分光光度計(Perkin−Elmer、Norwalk、CT)を使用して実行した。温度傾斜速度(temperature ramp rate)は、260nmで連続モニタリングしながら1℃/分であった。Tm値を、製造業者によって提供されるソフトウェアを使用して、A260 対 温度プロットの第1誘導体曲線の最大値を使用して計算した。Tmの決定を、10mMのリン酸ナトリウムおよび100mMの塩化ナトリウムを含む緩衝液において行った。各々のTmの測定の前に、種々のDNAテンプレートおよびPNAプローブの各々の鎖を、UV分光学を使用して定量し、そして最終濃度1μMの最終融解緩衝液になるように希釈した。この最終光学密度の範囲は、260nmで、0.2OD(光学密度単位)と0.8ODとの間であった。このサンプルを、90℃で5分間加熱することによって「前融解」し、そして融解プロファイルを実行する前に周囲温度までゆっくりと冷却した。あるいは、この前融解を、融解プロファイルを実行する前に、分光光度計で、高温までの迅速な傾斜(5℃/分)および出発温度まで温度を下降し戻す傾斜(2〜3℃/分)によって、実行した。
【0112】
(実施例2)
(ssDNAおよびdsDNAに対するPNA FRETプローブ結合反応速度論)(図6)
FRET PNAプローブのssDNAおよびdsDNAに対するハイブリダイゼーションの反応速度を測定した(図6)。プローブが標的に結合しない場合、または標的の存在下においてプローブのTmより低い場合、蛍光色素およびクエンチャーは、蛍光色素の実質的に完全なクエンチングが可能になる平均化された形態で存在する(図5)。プローブが標的にハイブリダイズする場合、蛍光色素およびクエンチャーは、空間的に分離されて、蛍光の増大をクエンチングの損失に依存して測定し得る。16nt PNA FRETプローブ(配列番号1)の蛍光強度の測定は、蛍光の基準を提供する。コントロール実験は、PNAプローブのみを含み、標的を含まない(図6上)。クエンチングは、温度の増大を通じて、実質的に完了する。プローブとds標的DNAの混合物を95℃に維持した。68nt(配列番号2)と74nt相補体(配列番号3)とをアニールすることによってdsDNAを形成して、3ntのオーバーハングを有する68bp二重鎖を形成した。次いで温度を60℃まで下げた(図6中)。
【0113】
蛍光を約5〜10秒間隔の時間の関数として10分間測定した(ABI 7700, Applied Biosystems,Foster City,CA)。蛍光強度は、約4倍増大し、いくらかのハイブリダイゼーションを示した。温度を約60℃まで下げた場合、両方のテンプレート鎖の存在下で、PNAの相補的テンプレート鎖に対する結合は、他の相補的DNA鎖によって競合(out−compete)されることが、蛍光がわずかに増大することからわかる(図6中)。PNAの結合が緩やかに置換されたことを示す、シグナルの緩やかな低下もまた注目される。最終的に、プローブおよびss標的DNA(配列番号2)の混合物を、95℃に維持し、次いで温度を60℃まで下げた(図6下)。16nt PNAプローブは、ssDNA標的に1分以内に結合することが、蛍光の8倍の増大からわかる(図6下)。しかし、サンプルプローブは、dsDNA標的により少なく結合する。ssDNAおよびdsDNAテンプレートの両方は、25、50、〜100nMの範囲である。濃度範囲を、指数関数的相からプラトー相までのPCR段階を模倣するよう選択した。この結果は、プローブ(例えば、PNA FRET 16nt)は、dsDNAより迅速にssDNAにハイブリダイズすることを示す(図3)。この結果はまた、dsテンプレートへのプローブ結合が、反応速度的および熱力学的の両方で支持されないことを例証する。
PNA 16nt:
【0114】
【化15】
(非同調的、伝統的、および非対称的なPCR熱サイクルプロトコールの比較)(図4aおよび4b)
非同調的な熱サイクルプロトコールを、伝統的な熱サイクルプロトコールと直接比較した。PCR反応を以下の条件を独立して変えることによって行った:(i)非同調的および伝統的(1回のアニーリングおよび1回の伸長工程)熱サイクルプロトコール;(ii)プライマーのTm;および(iii)プライマー濃度。他の条件は、一定に維持した。フォワードおよびリバースプライマーの3つの組み合せを用いて、標的DNAを増幅した。
【0115】
非同調的PCR(A−PCR)についてのサイクルを、図4aにおいて概説し、ここでプライマーを、これらのTm値が約15℃離れるように設計した。増幅サイクルの最初の半ばにおいて、高いTmプライマーは、標的にアニーリングして、次いで十分に伸長する。その後温度が下げて(例えば、52℃)、そしてこのサイクルの部分で蛍光を測定する。このサイクルの部分において、低温度プライマーは、標的配列に結合するが、実質的な範囲までプライマーを伸長し得ない。温度を上げて、第2のプライマーの伸長を完全にすることによって、このサイクルを完了する。
【0116】
同調的、伝統的および非対称的なPCR熱サイクルプロトコールからの結果を比較した(図4b)。2つのフォワードプライマーおよび2つのリバースプライマーを、4つの可能な組み合わせのうち3つ(66/52;66/61;60/61)において比較し、不同性Tmの対およびほぼ等しいTmの対を作った。非対称的PCRを、200nMおよび20nMのプライマー濃度で行った。アンプリコンおよび標的サイズは、68ntであった。フォワードプライマー(各プライマー対において、Tm=66℃および60℃)を、電気泳動可動性修飾因子として、6−カルボキシフルオレセイン(FAM)を用いて、5’標識した。FAM標識は、アンプリコンの電気泳動を遅延させて、変性分析ゲル条件において、鎖の分析を可能にする。標識化バンド(ゆっくり移動)および非標識化バンド(速く移動)の各レーンにおける分析は、種々の条件でのPCRの結果に由来する、二重鎖(FAM標識、ゆっくり移動、上のバンド)アンプリコンおよび一本鎖(非標識、速く移動、下のバンド)アンプリコンの存在を示す。電気泳動を15%ポリアクリルアミドで、変性条件下(電気泳動の間のゲル温度約55〜60℃、および7M尿素)において、SYBR GreenTMインターカレーター((Molecular Probes,Inc.,Eugene,OR)の存在下で行い、アンプリコンを染色および可視化した。
【0117】
図4bは、標的を増幅させたPCR産物のゲル電気泳動分析を示す。66℃および52℃のTmプライマーを用いた非同調的PCR(左から3番目のレーン)は、上のバンドが下のバンドに対して4:1の割合であることを、デンシトメトリー定量によって提示し、66℃および52℃のTmプライマーを用いた伝統的PCRよりもアンプリコンを多く生じることを示した。実際に、非同調的なプロトコールは、プライマーの全ての3つの組み合せを用いて、大量の産物を提供するが、ここで伝統的なプロトコール(真中のレーン)は、ほぼ等しいTmのプライマー対(61℃および60℃)に対してのみ有効であった。非同調的熱サイクルプロトコール(右レーン)は、全ての3つのプライマーの組み合せで相対的に、非能率的であった。従って、不同性のTmプライマーを用いて、プロトコールが、より高い温度でのみアニーリングして終了する場合、非同調的な熱サイクルプロトコールが効率的な増幅を行い、過剰のssアンプリコンの産生を可能にすることを、図4bは示す。
【0118】
【化16】
【0119】
PCR増幅反応物(50μl)は、以下を含んだ:DNAまたはRNA標的核酸、PCR緩衝液を含む2×Master mix(25μl)、dNTPs(dATP、dGTP、dCTP、dTTP)、およびMgCl2(Applied Biosystems),AmpliTaq Gold DNAポリメラーゼ、フォワードプライマーおよびリバースプライマー(各200〜900nM)、ならびにプローブ(200〜250nM)。
【0120】
(非対称的PCR)
非対称的PCRのための50μl混合物は、全ての反応成分を、伝統的および非同調的プロトコールにおけるものと同一量含んだ(ただし、特定のプライマーの量(25〜50pmol)が他方のプライマー(1.25〜2.5pmol)の20倍量であったことを除く)。非対称的PCRの熱サイクルプロトコールは、対称的、伝統的PCRプロトコールと同一であった(図4b下)。
【0121】
(変性PAGEおよび画像解析:)
PCR産物アンプリコン(0.5〜5μl)を最終濃度が1×ローディング緩衝液(45mM Trisベース、45mM ホウ酸、0.4mM EDTA、3%フィコール、0.02%ブロモフェノールブルー、0.02%キシレンシアノール)と混合して、95℃で10〜20分間変性した。サンプルを10〜15%変性PAGEゲル上にロードし、1×TBE(89mM Trisベース、89mMホウ酸、2mM EDTA、pH8.3)中で100〜160V、70℃で25〜60分間電気泳動した。ゲルを40〜120ml容量の1×TBE中で10〜30分間l×SYBR Green(Molecular Probes,Eugene,OR)を用いて染色することによって、伸長された産物を可視化した。ChemiImaging 2000ゲルドキュメンテーションシステムによって、画像を取り込んだ。SpotDensoプログラム(Alpha Innotech Corp.,CA)によって、ゲル上のバンド内のDNAの相対量を比較し、比率を算出し得る。
【0122】
(実施例4)
(短いPNA FRETプローブを用いた、ABI System上での、完全な一致および不一致の標的の増幅のリアルタイム検出)(図7aおよび7b) 非同調的PCR法を用いた高い特異性の達成を例証するため、以下の2つの異なる不一致を合成標的テンプレート中に導入した;許容性の弱いCT不一致および一般によく許容されるGT不一致(すなわち、対する識別が難しい)。PNA FRET 16ntプローブ(配列番号1)は、不一致とこれら間の数サイクルによる完全なテンプレートを容易に識別する(図7b)。蛍光を、各サイクルの間検出され、蛍光における対数的な変化(ΔRn)をサイクル数に対してプロットする。蛍光における変化(ΔRn)が閾値を越えるPCRプロトコールのサイクルは、CTとして示される。相対的に低いCT値は、アンプリコンの効率的な検出を示す。許容限界サイクルは、複製数量またはサンプル中の標的ポリヌクレオチド量に高い関連がある。図7bの完全一致実験は、プローブ/標的の検出を示し、ここで不一致標的実験は、CT閾値に達しなかった。従って、16nt PNA FRETプローブは、1つの塩基対不一致特異性を示した。同じ標的に対して相補的な14nt PNA FRETプローブ(配列番号8)を調製して、上記のような同じサイクルおよび同じプライマーセットで用いる。14nt PNA FRETプローブは、いずれの不一致標的でも、アンプリコンとさらに良好な認識を示した。いずれの不一致実験も、CT閾値に達せず、そしてΔRnは、増幅の後期のラウンドにおいてさえも、ほとんど明白でない(図7a)。
【0123】
リアルタイムPCRについて、因習的な熱サイクルプロトコールを、50℃で2分および95℃で10分で始め、次いで95℃で15秒および60℃で1分間を40サイクル進めた。リアルタイム非同調的PCRについて、各サイクルは、2回のアニーリングおよび伸長の工程(95℃で30秒、66〜69℃で30〜120秒、72℃で30〜60秒、52〜55℃で60〜120秒、および72℃で60秒を含む)を有する。全ての反応をABI 7700(Applied Biosystems,Foster City,CA)上で実施した。反応条件を、ABI 7700 Sequence Detectorに直接連結したPower Macintosh G3(Apple Computer,CA)上でプログラムした。データ分析をまた収集および分析ソフトウェア(Applied Biosystems)を備えたMacintoshコンピュータ上で実施した。
【0124】
【化17】
(短いPNA FRETプローブを用いた非同調的PCRによるリアルタイム検出)(図8)
蛍光の変化(ΔRn)とCT閾値の間のシヌソイド相関特異性から、特異性を例証した(図8)。ABI 7700上ならびに実施例3および4と同様の条件化でPCRを実施した。
【0125】
【化18】
(PNA FRETプローブを用いた非同調的PCRによるリアルタイム検出)(図10、11、12)
一連の3つのPNA FRETプローブ(15〜17nt、および合成ss68nt標的DNAに相補的)を、N末端(DNA上の5’末端に等しい)に、レポーター色素(F)としてカルボキシフルオレセインを用意し、C末端にクエンチャー(Q)としてdabcylを用意した。PNA FRETプローブをさらに、PNAオリゴマーとFの間の陰性荷電グルタミン酸部分に配置し、QとPNAオリゴマーの間に挿入されたさらなる陽性荷電リジンに配置した。反対の荷電を帯びたアミノ酸は、蛍光色素およびクエンチャーの近位を強いられ、従って、プローブが相補的配列(すなわち、標的核酸配列)にハイブリダイズしない場合、クエンチングの程度はより高くなる。ABI7700上ならびに実施例3および4と同様の条件でPCRを実施した。
【0126】
PNA FRETプローブを、非同調的熱サイクルプロトコールによる、合成DNA標的のリアルタイム検出に用いた。プライマーのTmは、14℃異なった。6つの異なる濃度の出発複製物:104、105、106、107、108および109を含む希釈によって標的サンプルを調製した。3つのプローブ(15nt、16nt、17nt)の各々を用いて、プローブをアニーリングすることによって各標的サンプル濃度を検出する(ここでプロトコール中のプローブをアニーリングする工程および蛍光(バックグラウンドから差し引かれ、標的へのプローブのハイブリダイゼーションにおけるFRETクエンチングの損失によって生じる)を測定する工程がある)。図10は、各プローブが効率的に、閾値の蛍光レベルを超えるアンプリコンを、標的の濃度の関数として検出することを示す。終点(40サイクル)においての蛍光の約20倍の増大(ΔRn)によって、各増幅を検出する。図11は、許容限界サイクルCTおよび出発複製数のプロットであり、標的サンプルと標準コントロールの間で高い相関係数を有する線形相関を示す。対照的に同じPNA FRETプローブおよびプライマーを用いて、伝統的な熱サイクルプロトコール(60)サイクルで同じ標的を増幅した。図12は、最も高い複製数標的サンプル(108および109)のみが、効率的な増幅および検出を提供することを示す。図12はまたCTと出発複製数の間に相関がないことを明らかにする。どの伝統的なプロトコール増幅も蛍光の約3倍を超える増大を示さなかった。
【0127】
【化19】
(3セットのプライマーおよびPNA FRETプローブを用いた、K−ras遺伝子標的上での非同調的PCRのリアルタイム検出)(図14b)
図14bは、3つの異なるプライマー対および16nt PNA FRETプローブを用いた、K−ras遺伝子を標的核酸とした、リアルタイムPCR検出アッセイを示す。ABI7700上で、図14aのサイクルに続いて40サイクルの非同調的熱サイクルプロトコールを用いて、アッセイを実施した。プライマー対は、以下を含む:(A)等価なTm(52℃)のフォワードおよびリバースプライマー、(B)5’(GC)4クランプフォワードプライマー(Tm 77.5℃)およびリバースプライマー(Tm 52℃)、ならびに(C)不同性Tmのフォワードおよびリバースプライマー(65℃および52℃)。全ての3つのプライマー対が、約16〜17のほぼ等価なCT値を伴う効率的な増幅を行ったことは、PCRの経過の間のΔRnのプロット(図14b)から見受けられ得る。GCクランプ対(B)は、蛍光強度に最も大きな増大を生じた。不同性のTmプライマー対(C)は、等しいTmプライマー対(A)より大きな蛍光強度の増大を提供し、このことは、小さい複製数標的サンプルに対して、より大きな感度を予測する。ABI7700上ならびに実施例3および4と同様の条件でPCRを実施した。
【0128】
【化20】
(ヌクレアーゼ切断アッセイのリアルタイム検出)(図15a、b、c)
非同調的および伝統的な熱サイクルプロトコールを、ABI 7700 System上で、切断DNA FRETプローブを用いて比較した。プローブ、プライマーおよび標的以外のPCR増幅反応物は、実施例2と同じ試薬を含む。標的核酸は、ゲノムDNAのβアクチン遺伝子内のアンプリコンであった。
【0129】
図15aは、伝統的PCRを実施した場合に、市販のアッセイ(Applied Biosystems,Foster City,CA)を用いた、ゲノムDNA中のヒトβアクチン遺伝子に対する等価なTmプライマーでのPCR検出の結果を示す。一連のゲノムDNA濃度(0.6pg〜50,000pgの範囲)を用いた。この範囲内において、連続的に5倍異なる濃度の8つの標的サンプルを用いた。伝統的PCRサイクルは、1回のアニーリング工程および1回の伸長工程を有する(図16下)。異なる長さおよび不同性のTm値を有するプライマーを非同調的な熱サイクルプロトコールを伴うアッセイに対して設計した(図16上)。図15bは、不同性Tmのプライマーを非同調的な熱サイクルプロトコールで用いた結果、およびβアクチン遺伝子に対する別の同じアッセイ中の8つの異なる濃度においての結果を示す。同じ切断可能なDNA FRETプローブ(配列番号23)を用いて、両方のプロトコールを実施した(図15c)。蛍光シグナル強度は顕著に増大し、そしてCT値は、伝統的プロトコール(図15a)と比較した非同調的プロトコール(図15b)について、かなり低かった。非同調的プロトコールによる検出制限は、1つの複製検出を可能にする。言い換えると、ヌクレアーゼ切断アッセイは、非同調的PCR方法によって顕著に増大される。非同調的PCR法はまた、特定の条件下で、より短い、切断DNA FRETプローブ(すなわち、低いTm)の使用を可能にし得る。
【0130】
【化21】
(5’標識化プライマーを用いたPCRの終点検出)(図20a)
A−PCRにおける増幅が、非同調的な様式で進むことを証明するために、フォワード(より高いTm)プライマーをビオチンで5’標識し、2つの産物の鎖が変性ポリアクリルアミドゲル電気泳動の間によく分離するようにした。この実施例の設計は、図20a下に概説した。最初の25サイクル、続く1サイクルの最初の半ばで非同調的PCRサイクルを実施し、ここで標識化プライマーのみがハイブリダイズして伸長する。2×ローディング色素(Novex,San Diego,CA)を添加し、95℃で20分間変性することによって、反応を直ちに停止した。増幅が本当に非同調的である場合、産物の鎖は、理論的に2:1の割合であるはずである。25回の十分なサイクルの後に止めた場合、割合は、1:1であったが、さらなる半サイクル後に1:0.67まで進んだ(図20a)。これは、実際に増幅が非同調的であり、好ましくは、より高い融解温度のプライマーが伸長し、そして過剰の一本鎖アンプリコンが産生されることを示す。25サイクルの非同調的熱サイクルプロトコールおよび高い温度での最後のアニーリングおよび伸長によって、PCRを実施した。PCR条件および分析は、実施例3の条件を用いた。
【0131】
【化22】
(非同調的PCRプロトコールによるssDNA増幅および標識)(図22) ssアンプリコンを相補的なアレイ、固相支持体結合プローブに対してハイブリダイズすることの有用性を調査した。2対のPCRプライマーを設計して伝統的PCRを非同調的PCRと、ガラススライドアレイ上にスポットされたプローブにハイブリダイズするアンプリコンの産生において比較した。フォワードプライマーの各対は、5’Cy5色素標識(Amersham Pharmacia Biotech,Piscataway,NJ)を有した。リバースプライマーは、非標識であった。21ntのフォワードプライマーおよび20ntのリバースプライマーの伝統的な対は、およそ等しく算出されるTm値を有した。25ntのフォワードプライマーおよび18ntのリバースプライマーの非対称的な対は、約15〜20℃の算出されたΔTmを有した。非同調的な対のフォワードプライマーは、伝統的な対のフォワードプライマーと相対的に、5’CGGC非標的相補テイルを有した。伝統的な熱サイクルプロトコールによってPCRを実施して、96bpのdsアンプリコンを生じ、非対称的な熱サイクルプロトコールによってPCRを実施して、100ntのssアンプリコンを生じた。各固定化プローブは、各アンプリコンに相補的な24ntの配列を有した。
【0132】
図22は、4つの異なる標的由来のCy色素5’標識化A−PCR(主にssDNA)および伝統的PCR(dsDNA)産物のガラススライドアレイへのハイブリダイゼーションを示す。4つの標的の例示的な列を、比較のため各アレイ位置上の四角で囲む。シグナルをコントロールハイブリダイゼーションによって規準化した。標識化A−PCR産物からの蛍光シグナルの平均化中央値(右)は、伝統的な熱サイクルプロトコールによって産生されたdsアンプリコンからのもの(左)に比べて、3〜4倍高かった。この結果は、ガラス表面に付着したアレイプローブが、ssDNAにより効果的にハイブリダイズしたことを示唆する。
【0133】
標的サンプルは、アレイプローブ特異的配列を含む。PCRをABI 7700 System上で実施した。PCR反応物は、総容積50μl中に以下を含む:10mM Tris−HCl、pH8.3、50mM KC1、2〜5mM MgCl2、0.01%ゼラチン、250μM各dNTP、0.5〜1μMフォワードプライマー、0.05〜0.1μMリバースプライマー、10μlの96nt合成標的DNA(1:1000希釈)、1〜5UのAmpliTaq Gold DNAポリメラーゼ(Applied Biosystems,Foster City,CA)。オリゴヌクレオチドのテンプレート依存的ライゲーションによって96nt合成標的DNAを調製した。非同調的PCRは、一連で実施される2つの熱サイクルプロトコールを含む。第1のプロトコールは、以下からなっていた:最初の95℃で10分間の変性に続いて、95℃で15秒、65℃で60秒(フォワードのプライミング)、52.5℃(50〜55℃)で60秒(リバースのプライミング)、72℃で60秒の15〜25サイクル、そして7分間の余分な伸長。一本鎖形態の色素標識化アンプリコンを生じるために直ちに続けられる第2のプロトコールは、95℃で30秒、67(66〜69)℃で90秒、および70℃で60秒の10〜80サイクルからなっていた。伝統的PCRは、実施例4におけるプロトコールによって実施された(50℃で2分そして95℃で10分、次いで95℃で15秒そして60℃で1分;または50℃で2分そして95℃で10分を40サイクル、次いで95℃で15秒、60℃で1分そして72℃で1分を40サイクル)。Microcon−100(Millipore,Medford,MA)上で3回洗浄してPCR産物を精製した。
【0134】
(マイクロアレイハイブリダイゼーション、洗浄、データ収集および分析)
合計64の異なる24nt DNAオリゴヌクレオチドプローブをガラススライド上にスポットした。各プローブの8つの複製をスライドごとにスポットした。ハイブリダイゼーション混合物(20〜30μl/スライド)は、以下を含む:4×SSC(生理食塩水−クエン酸ナトリウム)、0.3%SDS(ドデシル硫酸ナトリウム)、1μg/μl酵母tRNA、1μg/μlポリ(A)、および1〜2μlの50μl PCR産物。混合物を95℃で2〜4分間変性し、スライドに各20〜30μlを適用した。スライドをアレイチャンバー内に配置した。16〜20時間の50〜55℃の水浴中におけるハイブリダイゼーション後、マイクロアレイを50〜55℃の4×SSCおよび0.3%SDSで簡単に洗浄し、室温の1×SSCおよび0.3%SDSで1回、2分間洗浄し、続いて室温の0.06×SSCで2回、各2分間洗浄した。マイクロアレイをAxonスキャナーを用いて画像化し、画像をGenePix Pro 3.0ソフトウェア(Axon Instruments,Foster City,CA)で分析した。
【0135】
【化23】
【0136】
ここで十分に記載される本発明は、本発明の精神または範囲から逸脱することなしに、本発明に対して多くの変更および改変がなされ得ることが当業者によって、理解される。
【図面の簡単な説明】
【図1】
図1は、本発明の1つの実施形態に従う非同調的PCR熱サイクル方法についての模式図を示し、この方法は、以下の工程を包含する:
(i)二本鎖標的を変性させる工程、(ii)第一のプライマーをアニーリングさせる工程、(iii)第一のプライマーの伸長、(iv)プローブハイブリダイゼーション、(v)第二のプライマーのアニーリング、および(vi)第二のプライマーの伸長。温度および時間は、例示である。
【図2】
図2は、本発明の1つの実施形態に従う、標的核酸に対する、第二のプライマー(短い矢印)よりも高い温度での第一のプライマー(長い矢印)のハイブリダイゼーションについての模式図を示す。
【図3】
図3は、従来のPCRの間に(部分的に)二本鎖の標的に対してプライマーおよびプローブをハイブリダイズさせる工程(上)、ならびに本発明の1つの実施形態に従うプローブによる非同調的PCRの間に一本鎖標的に対してプライマーおよびFRETプローブをハイブリダイズさせる工程(下)の模式図を示す(F=レポーター色素、Q=クエンチャー)。
【図4a】
図4aは、特定の温度での連続的かつサイクル的な持続期間と共に、本発明の1つの実施形態に従う非同調的PCR(上)および従来のPCR(下)の熱サイクルプロトコールを示す。
【図4b】
図4bは、以下の3つのPCRプロトコール:非同調的、従来のおよび非対称的後の、変性条件(約55〜60℃、7M 尿素)下でのポリアクリルアミド(15%)ゲル電気泳動分析、およびアンプリコンのSYBR−Green染色(上)、ならびに順方向および逆方向のプライマーの3つの組み合わせを用いた標的DNAの増幅の模式図(下)を示す。順方向プライマーは、電気泳動移動度改変因子(例えば、ビオチンまたはFAM)で5’標識されている。
【図5】
図5は、グルタミン酸結合およびリジン結合によるレポーター色素(F)およびクエンチャー(Q)を含む、例示的なPNA FRETプローブを示す(上)。このプローブは、相補的な標的にハイブリダイズされる場合、レポーター色素のクエンチを引き起こす、少なくとも1つの立体構造で存在する(左下)。標的へのハイブリダイズに際して、クエンチは消失して、蛍光強度が増大する(右下)。
【図6】
図6は、16ntのPNA FRETプローブ(配列番号1)の、ABI 7700 Sequence Detection System(Applied Biosystems、Foster City、CA)による、ある時間にわたる蛍光強度の測定を示す:相補的DNAなし(上);相補的な68ntのDNA(配列番号2)および74ntのDNA(配列番号3)の二本鎖形態に対してハイブリダイズした(中);そして相補的な68ntのssDNA(配列番号2)に対してハイブリダイズした(下)。
【図7a】
図7aは、14ntのPNA FRETプローブ(配列番号8)が、相補的な標的に対して、完全一致、単一のG−Tミスマッチ、および単一のC−Tミスマッチでハイブリダイズする場合の、非同調的PCRの間にABI 7700で測定された蛍光の変化(ΔRn)を示す。
【図7b】
図7bは、16ntのPNA FRETプローブ(配列番号1)が、相補的な標的に対して、完全一致、単一のG−Tミスマッチ、および単一のC−Tミスマッチでハイブリダイズする場合の、非同調的PCRの間にABI 7700で測定された蛍光の変化(ΔRn)を示す。
【図8】
図8は、8ntのPNA FRETプローブ(配列番号11)および9ntのPNA FRETプローブ(配列番号12)を用いる、本発明の1つの実施形態に従う非同調的熱サイクルプロトコール(下)によって増幅される非同調的PCRの間に、ABI 7700で測定された蛍光の変化(ΔRn)を示す。
【図9】
図9は、例示的な、平均的な非同調的PCR熱サイクルおよび平均的な従来のPCR熱サイクルの過程の間の、16ntのPNA FRETプローブ(配列番号1)からの蛍光強度における2倍の増大を示す。各プロットは、合計40サイクルのうちの25〜30サイクルからの平均である。温度プロフィールが示される。
【図10】
図10は、本発明の1つの実施形態に従う非同調的PCR熱サイクルプロトコールの間、15nt(配列番号14)、16nt(配列番号1)および17nt(配列番号15)のPNA FRETプローブの各々が、6個のサンプル(68ntの合成ssDNA標的の104、105、106、107、108および109の開始コピー)を検出する場合に、PCRの間に、PNAプローブを使用してABI 7700でリアルタイム定量測定された蛍光の変化(ΔRn)を示す。
【図11】
図11は、本発明の1つの実施形態に従う非同調的PCR熱サイクルプロトコールの間の、検出可能な幾何級数的増幅の閾値サイクル(CT)と図10からの68ntの合成ssDNA標的の開始コピー数との間の線形の相関を示す。
【図12】
図12は、16ntのPNA FRETプローブ(配列番号1)、68ntの合成ssDNA標的の104〜109の開始コピー、および図10と同じ他の試薬を用いる従来のPCR熱サイクルプロトコールを使用する、PCRのリアルタイム定量についてのABI 7700からのデータの表示を示す。
【図13】
図13は、本発明の1つの実施形態に従う、低Tmの低温のハイブリダイゼーション温度(30〜37℃)、短いPNA FRETプローブを用いる非同調的PCR熱サイクルプロトコールについての模式図を示す。
【図14a】
図14aは、5’(GD)4クランプ(clamp)プライマーを用いるPCR熱サイクルプロトコールの最初の2サイクル、続く非同調的熱サイクルプロトコールの模式的例を示す。
【図14b】
図14bは、16ntのPNA FRETプローブ(配列番号16)が、以下:(A)等しいTmのプライマー、(B)5’(GC)4クランププライマー、および(C)別個のTmのプライマー、を用いる非同調的熱サイクルプロトコールの間に、K−ras遺伝子中のその完全一致の相補的な標的にハイブリダイズする場合に、PCRの間にABI 7700で測定された蛍光の変化(ΔRn)を示す。
【図15】
図15a(上)は、等しいTmのプライマーおよび8つの標的サンプル(5つの区切りによって異なる量のβ−アクチンゲノム標的dsDNAを含む)(左から右へ:50,000、10,000、2000、400、80、16、3、0.6pg)を用いる、従来のPCR熱サイクルプロトコールの間の、ABI 7700で測定される蛍光の変化(ΔRn)を示す。アンプリコンを、DNA FRETプローブ(配列番号23)を用いるヌクレアーゼ切断法によって検出した。
図15b(下)は、別個のTmのプライマーおよび8つの標的サンプル(図15aのβ−アクチンゲノム標的dsDNAの0.6〜50,000pg)(右から左へ)を用いる、非同調的PCR熱サイクルプロトコールの間の、ABI 7700で測定される蛍光の変化(ΔRn)を示す。アンプリコンを、DNA FRETプローブ(配列番号23)のヌクレアーゼ切断アッセイによって検出した。
【図15c】
図15cは、等しいTmのプライマーおよび従来のPCR熱サイクルプロトコール(上)、ならびに別個のTmの例示的なプライマーおよび例示的な非同調的PCR熱サイクルプロトコール(下)を使用する、DNA FRETプローブのヌクレアーゼ切断によるPCR検出のための模式図を示す。
【図16】
図16は、図15aの従来のPCR熱サイクルのための熱サイクルプロトコールおよび図15bで使用される例示的な非同調的PCR熱サイクルプロトコールを示す。
【図17】
図17は、ハイブリダイゼーションによる配列決定(SBH)による、PNA FRETプローブを用いるPCR cDNAクローンの均質な検出を示す。
【図18】
図18は、例示的な非同調的熱サイクルプロトコールによる、2つの別個のTmのプライマーを用いる指数関数的増幅、続いて、ハイブリダイゼーションおよび伸長が、より高いTmのプライマーのみがアニーリングおよび伸長するような十分に高い温度で実行される、多サイクルの高温プロトコールを含むPCRの方法についての模式図を示す。
【図19】
図19は、変性ポリアクリルアミドゲル電気泳動(PAGE)によるssアンプリコンおよびdsアンプリコンの検出ならびに定量と共に、実験的設計、および従来のPCRプロトコールと例示的な非同調的熱サイクルプロトコールとの間の比較を示す。
【図20a】
図20aは、非同調的PCR由来の産物の、変性条件(約55〜60℃、7M 尿素)下でのポリアクリルアミド(15%)ゲル電気泳動分析およびSYBR−Green染色を示す。二本鎖から分離される得られたssDNAは、濃度測定によって定量され、そして各レーンにおける上のバンドと下のバンドの比として示される。
【図20b】
図20bは、図20aの実験についての、非同調的PCR熱サイクルプロトコールを示す。
【図21】
図21は、例示的なFAMおよびDABCYLで標識されたPNA FRETプローブ構造:6−FAM−Glu−NH−PNA−C(O)−Lys−Lys−DABCYLの構造を示し、ここで、nは、2−アミノエチルグリシン単位の数である。
【図22】
図22は、従来の熱サイクルプロトコール(左)、および本発明の1つの実施形態に従う非同調的な熱サイクルプロトコール(右)によって生成される、5’標識されたPCR産物のハイブリダイゼーションの比較の、アレイ蛍光シグナル画像の結果を示す。
Claims (66)
- 核酸増幅の方法であって、該方法は、以下の工程:
第一のアニーリング温度で、第一プライマーを変性した標的核酸の第一鎖にアニーリングする工程;
伸長温度または該第一のアニーリング温度で、プライマー伸長試薬を用いて該第一プライマーを伸長させて、二本鎖核酸を生成する工程であり、ここで、該プライマー伸長試薬が、ポリメラーゼ、ヌクレオチド5’−トリホスフェート、および緩衝液を含む、工程;
プローブのハイブリダイゼーション温度で、検出可能なプローブを変性した標的核酸の第二鎖にアニーリングする工程;
第二のアニーリング温度で、第二プライマーを該変性した標的核酸の第二鎖にアニーリングする工程であり、ここで、該第二のアニーリング温度が、該第一のアニーリング温度および伸長温度よりも低い、工程;
該伸長温度で、プライマー伸長試薬を用いて該第二プライマーを伸長させて、二本鎖核酸を生成する工程;ならびに、
変性温度で、該二本鎖標的核酸を第一鎖と第二鎖とに変成させる工程、
を包含する、方法。 - 前記検出可能なプローブが、蛍光部分およびクエンチャー部分を含む、請求項1に記載の方法。
- 前記蛍光部分が、前記プローブの5’末端または3’末端に結合されており、かつ前記クエンチャー部分が、該プローブの5’末端または3’末端に結合されている、請求項2に記載の方法。
- 前記プローブが、前記第二プライマーの伸長前に検出される、請求項1に記載の方法。
- 前記工程が2〜50サイクル繰り返される、請求項1に記載の方法。
- 前記プローブが酵素学的に切断される、請求項1に記載の方法。
- 前記プローブが酵素学的に切断されない、請求項1に記載の方法。
- 前記標的核酸が、プラスミド、cDNA、アンプリコン、ゲノムDNA、制限消化物、およびライゲーション産物から選択される、請求項1に記載の方法。
- 前記標的核酸が単一のヌクレオチド多型を含む、請求項1に記載の方法。
- 前記第一プライマーおよび第二プライマーがDNAである、請求項1に記載の方法。
- 前記第一プライマーまたは第二プライマーがPNA/DNAキメラである、請求項1に記載の方法。
- 前記第一プライマーまたは第二プライマーが共有結合した蛍光色素を含む、請求項1に記載の方法。
- 前記第一プライマーまたは第二プライマーが共有結合した移動度改変因子を含む、請求項1に記載の方法。
- 前記第一プライマーまたは第二プライマーが共有結合した副溝結合剤を含む、請求項1に記載の方法。
- 前記プローブが、標的結合配列および2つの分子内塩基対形成配列を含む、請求項1に記載の方法。
- 前記プローブが、ヘアピン幹構造体およびループ構造体を形成する、請求項15に記載の方法。
- 前記分子内塩基対形成配列が前記プローブの5’末端および3’末端に位置している、請求項15に記載の方法。
- 前記プローブが、核酸塩基アナログ、2’−デオキシリボースアナログ、ヌクレオチド間アナログおよび光学異性体から選択される、1つ以上のヌクレオチドアナログを含む、請求項1に記載の方法。
- 請求項18に記載の方法であって、ここで、前記核酸塩基アナログが、7−デアザアデニン、7−デアザグアニン、7−デアザ−8−アザグアニン、7−デアザ−8−アザアデニン、イノシン、ネブラリン、ニトロピロール、ニトロインドール、2−アミノ−プリン、2,6−ジアミノ−プリン、ヒポキサンチン、プソイドウリジン、プソイドシチジン、プソイドイソシチジン、5−プロピニル−シチジン、イソシチジン、イソグアニン、7−デアザ−クアニン、2−チオ−ピリミジン、6−チオ−グアニン、4−チオ−チミン、4−チオ−ウラシル、O6−メチル−グアニン、N6−メチル−アデニン、O4−メチル−チミン、5,6−ジヒドロチミン、5,6−ジヒドロウラシル、4−メチル−インドール、およびエテノアデニンから選択される、方法。
- 請求項18に記載の方法であって、ここで、前記ヌクレオチドアナログは、2’−炭素原子において、Cl、F、−R、−ORまたは−NR2で置換されている2’−デオキシリボースアナログであり、ここで各Rは、独立して、−H、C1〜C6アルキルまたはC5〜C14アリールである、方法。
- 前記ヌクレオチドアナログがLNAである、請求項18に記載の方法。
- 前記ヌクレオチドアナログが、2’−デオキシリボースのL型光学異性体である、請求項18に記載の方法。
- 前記プローブが、1つ以上の2−アミノエチルグリシン(PNA)モノマー単位を含む、請求項1に記載の方法。
- 前記プローブがPNA/DNAキメラである、請求項23に記載の方法。
- 請求項1に記載の方法であって、ここで、前記プローブは、以下の構造:
Rは、蛍光部分であり;
L1およびL2は、リンカーであり;
Bは、核酸塩基であり;
Qは、クエンチャー部分であり;そして、
nは、5〜25の間の整数である、方法。 - L1またはL2が1つ以上のアミノ酸単位を含む、請求項25に記載の方法。
- L1およびL2が、独立して、アスパラギン酸、グルタミン酸およびリジンから選択される、請求項26に記載の方法。
- L1が1つ以上のアスパラギン酸単位またはグルタミン酸単位であり、L2が1つ以上のリジン単位である、請求項27に記載の方法。
- Bがウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、7−デアザグアノシン、7−デアザ−8−アザグアニン、および7−デアザ−8−アザアデニンから選択される、請求項25に記載の方法。
- 前記蛍光部分が、フルオレセイン色素、ローダミン色素またはシアニン色素である、請求項2に記載の方法。
- 前記クエンチャー部分が、ローダミン色素である、請求項2に記載の方法。
- 前記クエンチャー部分が、ニトロ置換シアニン色素である、請求項2に記載の方法。
- 請求項2に記載の方法であって、ここで、前記クエンチャー部分は、以下の構造:
ここで、Zは、H、Cl、F、C1〜C6アルキル、C5〜C14アリール、ニトロ、シアノ、スルホネート、NR2、−OR、およびCO2Hから選択され、ここで、各Rは、独立して、H、C1〜C6アルキルまたはC5〜C14アリールである、方法。 - 蛍光強度の変化が、標的増幅の終点において検出される、請求項2に記載の方法。
- 蛍光強度の変化が、リアルタイムでモニターされる、請求項2に記載の方法。
- 蛍光強度の変化が、標的配列の存在の指標として検出される、請求項2に記載の方法。
- 前記第一のアニーリング温度が、前記第二のアニーリング温度よりも10〜30℃高い、請求項1に記載の方法。
- 前記第一のアニーリング温度が、前記第二のアニーリング温度よりも12〜16℃高い、請求項1に記載の方法。
- 前記第一のアニーリング温度が、60〜75℃である、請求項1に記載の方法。
- 前記第二のアニーリング温度が、45〜55℃である、請求項1に記載の方法。
- 前記第一プライマーが、5’末端において(GC)n配列または(CG)n配列を有し、ここで、nが1〜4である、請求項1に記載の方法。
- 標識が、プリン核酸塩基の8−C、7−デアザプリン核酸塩基の7−Cもしくは8−C、またはピリミジン核酸塩基の5位において、1つ以上のヌクレオチド5’−トリホスフェートに共有結合している、請求項1に記載の方法。
- 標識が、5’末端、糖、ヌクレオチド間連鎖または核酸塩基において、前記第一プライマーまたは前記第二プライマーに共有結合している、請求項1に記載の方法。
- 標的ポリヌクレオチドの相補的なポリヌクレオチド鎖を生成するための方法であって、該方法は以下:
混合物を得る工程であり、該混合物は、相互にハイブリダイズして塩基対形成構造体を形成し得る、第一の標的ポリヌクレオチド鎖および第二の標的ポリヌクレオチド鎖を含み、該塩基対形成構造体は、標的配列、該第一の標的ポリヌクレオチド鎖の第一領域に相補的な第一プライマー、および該第二の標的ポリヌクレオチド鎖の第二領域に相補的な第二プライマーを含み、その結果、該第一領域および該第二領域が、該標的配列に隣接する、工程、
第一温度において該第一プライマーを伸長させて第一複合体を形成する工程であり、該第一複合体は、該第二プライマーが該第二領域に実質的にハイブリダイズしないような条件下で該第一標的鎖にハイブリダイズする、第一の相補鎖を含む、工程、ならびに、
該第一温度よりも低い第二温度において、該第二プライマーを伸長させて第二複合体を形成する工程であり、該第二複合体は、該第二標的鎖にハイブリダイズする第二の相補鎖を含む、工程、
を包含する方法であり、
ここで、該第二プライマーを伸長させる前に、検出可能なプローブが、該第二標的鎖の相補的な結合領域にハイブリダイズし、そして該ハイブリダイズしたプローブが、第二標的鎖の基準として検出される、方法。 - 核酸増幅の方法であって、該方法は、以下の工程:
第一のアニーリング温度で、第一プライマーを変性した標的核酸の第一鎖にアニーリングする工程;
伸長温度または該第一のアニーリング温度で、プライマー伸長試薬を用いて該第一プライマーを伸長させて、二本鎖核酸を生成する工程であり、ここで、該プライマー伸長試薬が、ポリメラーゼ、ヌクレオチド5’−トリホスフェート、および緩衝液を含む、工程;
第二のアニーリング温度で、第二プライマーを該変性した標的核酸の第二鎖にアニーリングする工程であり、ここで、該第二のアニーリング温度が、該第一のアニーリング温度および伸長温度よりも低い、工程;
該伸長温度で、プライマー伸長試薬を用いて該第二プライマーを伸長させて、二本鎖核酸を生成する工程;ならびに、
変性温度で、二本鎖標的を第一鎖と第二鎖とに変成させる工程、
を包含する、方法。 - 前記工程が2〜50サイクル繰り返される、請求項45に記載の方法。
- 前記第一プライマーの濃度が、前記第二プライマーの濃度よりも2〜10倍高い、請求項46に記載の方法。
- 請求項46に記載の方法であって、ここで、前記第二プライマーを変性した標的の第二鎖にアニーリングする工程、および前記第二プライマーを伸長させる工程が、最後の1〜25サイクル省略され、それによって、一本鎖DNAと二本鎖DNAとの混合物を生成する、方法。
- 請求項46に記載の方法であって、ここで、前記第二プライマーを変性した標的の第二鎖にアニーリングする工程、および前記第二プライマーを伸長させる工程が、最後の1〜10サイクル省略され、それによって、優勢なssDNAを生成する、方法。
- 前記標的がcDNAである、請求項45に記載の方法。
- 前記第一プライマーが蛍光色素で標識される、請求項45に記載の方法。
- 前記一本鎖DNAと二本鎖DNAとの生成混合物を、アレイ上に固定化された複数のプローブにハイブリダイズさせる工程をさらに包含する、請求項45に記載の方法。
- 前記プローブがFRETプローブである、請求項52に記載の方法。
- 標的ポリヌクレオチドの相補的なポリヌクレオチド鎖を生成するための方法であって、該方法は以下:
混合物を得る工程であり、該混合物は、相互にハイブリダイズして塩基対形成構造体を形成し得る、第一の標的ポリヌクレオチドおよび第二の標的ポリヌクレオチドを含み、該塩基対形成構造体は、標的配列、該第一の標的ポリヌクレオチドの第一領域に相補的な第一プライマー、および該第二の標的ポリヌクレオチドの第二領域に相補的な第二プライマーを含み、その結果、該第一領域および該第二領域が、該標的配列に隣接する、工程、
第一温度において該第一プライマーを伸長させて第一複合体を形成する工程であり、該第一複合体は、該第二プライマーが該第二領域に実質的にハイブリダイズしないような条件下で該第一標的鎖にハイブリダイズする、第一の相補鎖を含む、工程、ならびに、
該第一温度よりも低い第二温度において、該第二プライマーを伸長させて第二複合体を形成する工程であり、該第二複合体は、該第二標的鎖にハイブリダイズする第二の相補鎖を含む、工程、
を包含する、方法。 - 前記第二プライマーが伸長された後に、前記第一複合体および前記第二複合体を変性させる工程をさらに包含する、請求項54に記載の方法。
- 前記第一プライマーを伸長させる工程、前記第二プライマーを伸長させる工程、および前記変性させる工程を1サイクル以上繰り返す工程をさらに包含する、請求項55に記載の方法。
- 前記変性後に、前記第一温度で第一プライマーを伸長して、一本鎖形態の前記第二標的のポリヌクレオチドと二重鎖形態の前記第一複合体とを含む混合物を形成する、請求項55に記載の方法。
- 2つ以上のプライマーを含む標的ポリヌクレオチドを増幅するためのキットであって、ここで、第一プライマーおよび第二プライマーが、10〜30℃のTm差を有する、キット。
- 前記プライマーが蛍光色素で標識される、請求項58に記載のキット。
- ポリメラーゼをさらに含む、請求項58に記載のキット。
- 検出可能なプローブをさらに含む、請求項58に記載のキット。
- 前記検出可能なプローブがDNAであり、そして該プローブが蛍光部分およびクエンチャー部分を含む、請求項61に記載のキット。
- 前記検出可能なプローブがPNAであり、そして該プローブが蛍光部分およびクエンチャー部分を含む、請求項61に記載のキット。
- 前記プローブが、核酸塩基アナログ、2’−デオキシリボースアナログ、ヌクレオチド間アナログおよび光学異性体から選択される核酸アナログを含む、請求項61に記載のキット。
- 1つ以上の酵素学的に伸長可能なヌクレオチドをさらに含む、請求項58に記載のキット。
- ヌクレオチドが蛍光色素で標識される、請求項65に記載のキット。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US20988300P | 2000-06-06 | 2000-06-06 | |
| US09/875,211 US6887664B2 (en) | 2000-06-06 | 2001-06-05 | Asynchronous primed PCR |
| PCT/US2001/018464 WO2001094638A2 (en) | 2000-06-06 | 2001-06-06 | Asynchronous primed pcr |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004509613A true JP2004509613A (ja) | 2004-04-02 |
Family
ID=26904613
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002502178A Pending JP2004509613A (ja) | 2000-06-06 | 2001-06-06 | 非同調的刺激pcr |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US6887664B2 (ja) |
| EP (3) | EP2256213A1 (ja) |
| JP (1) | JP2004509613A (ja) |
| AT (1) | ATE448325T1 (ja) |
| AU (1) | AU6823501A (ja) |
| CA (1) | CA2412413A1 (ja) |
| DE (1) | DE60140487D1 (ja) |
| WO (1) | WO2001094638A2 (ja) |
Families Citing this family (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6887664B2 (en) * | 2000-06-06 | 2005-05-03 | Applera Corporation | Asynchronous primed PCR |
| CN100523216C (zh) * | 2001-03-02 | 2009-08-05 | 匹兹堡大学 | Pcr方法 |
| US20030175749A1 (en) | 2001-12-08 | 2003-09-18 | Jong-Yoon Chun | Annealing control primer and its uses |
| US7198897B2 (en) * | 2001-12-19 | 2007-04-03 | Brandeis University | Late-PCR |
| EP1942196B1 (en) | 2001-12-19 | 2012-05-30 | Brandeis University | Late-PCR |
| JP2003199568A (ja) * | 2002-01-10 | 2003-07-15 | Nichirei Corp | Dna増幅反応の効率向上方法 |
| EP2772552B1 (en) | 2002-10-23 | 2018-10-10 | University of Utah Research Foundation | Amplicon melting analysis with saturation dyes |
| JP4280519B2 (ja) | 2003-03-07 | 2009-06-17 | アークレイ株式会社 | Dna増幅法およびそのためのキット |
| JP2005000162A (ja) * | 2003-05-19 | 2005-01-06 | Canon Inc | 核酸のpcr増幅方法、pcrプライマー・セット、pcr増幅産物、ならびに、該増幅方法を利用する核酸の検出方法 |
| US20040235032A1 (en) * | 2003-05-19 | 2004-11-25 | Canon Kabushiki Kaisha | PCR amplification method, PCR primer set, PCR amplification product, and method for detection of nucleic acid using the amplification method |
| JP4322554B2 (ja) * | 2003-05-19 | 2009-09-02 | 株式会社ニチレイフーズ | Dna増幅反応の効率向上方法 |
| AU2003276765A1 (en) | 2003-11-10 | 2005-05-26 | Seegene, Inc. | Method for amplifying unknown dna sequence adjacent to known sequence |
| EP1735458B1 (en) * | 2004-01-28 | 2013-07-24 | 454 Life Sciences Corporation | Nucleic acid amplification with continuous flow emulsion |
| US7481997B1 (en) | 2004-02-12 | 2009-01-27 | Montana State University | Snow mountain virus genome sequence, virus-like particles and methods of use |
| US7387887B2 (en) * | 2004-04-20 | 2008-06-17 | University Of Utah Research Foundation | Nucleic acid melting analysis with saturation dyes |
| US9657347B2 (en) | 2004-04-20 | 2017-05-23 | University of Utah Research Foundation and BioFire Defense, LLC | Nucleic acid melting analysis with saturation dyes |
| WO2006112818A2 (en) | 2005-04-14 | 2006-10-26 | Applera Corporation | 3' modified oligonucleotides containing pseudoisocytosine nucleobase derivatives and applications thereof as primers or probes |
| EP1836319B1 (en) * | 2005-01-06 | 2012-09-19 | Life Technologies Corporation | Polypeptides having nucleic acid binding activity and methods for nucleic acid amplification |
| JP2008539730A (ja) * | 2005-05-02 | 2008-11-20 | ストラタジーン カリフォルニア | オリゴヌクレオチドのプローブ/プライマー組成物、及びポリヌクレオチドの検出方法 |
| EP1896617B1 (en) * | 2005-05-31 | 2013-01-02 | Life Technologies Corporation | Multiplex amplification of short nucleic acids |
| CN101495647B (zh) * | 2005-06-02 | 2014-12-17 | 阿德万德克斯公司 | 用于微生物分析的肽核酸探针 |
| US20070087360A1 (en) * | 2005-06-20 | 2007-04-19 | Boyd Victoria L | Methods and compositions for detecting nucleotides |
| US20070059713A1 (en) | 2005-09-09 | 2007-03-15 | Lee Jun E | SSB-DNA polymerase fusion proteins |
| US20070212704A1 (en) | 2005-10-03 | 2007-09-13 | Applera Corporation | Compositions, methods, and kits for amplifying nucleic acids |
| NZ571459A (en) | 2006-03-29 | 2011-09-30 | Merial Ltd | Vaccine against streptococcus equi |
| WO2007133703A2 (en) | 2006-05-10 | 2007-11-22 | Dxterity Diagnostics | Detection of nucleic acid targets using chemically reactive oligonucleotide probes |
| AU2007275762B2 (en) | 2006-07-17 | 2012-02-23 | Brandeis University | Specialized oligonucleotides and their use in nucleic acid amplification and detection |
| WO2008072933A1 (en) * | 2006-12-15 | 2008-06-19 | Panagene Inc. | Peptide nucleic acids conjugated with multi -amine linkers and nucleic acid detecting device using the same |
| KR101072900B1 (ko) * | 2007-12-04 | 2011-10-17 | 주식회사 파나진 | 고정화된 펩티드핵산 프로브를 사용한 표적핵산의 선택적 표지 및 검출 방법 |
| FI121379B (fi) * | 2008-01-17 | 2010-10-29 | Mobidiag Oy | Menetelmä yksijuosteisen DNA:n tuottamiseksi kaksivaiheisella symmetrisellä PCR:llä sekä menetelmään tarkoitettu reagenssipakkaus |
| GB0806041D0 (en) | 2008-04-03 | 2008-05-14 | Genomica S A | Method for detection of herpesvirus in test sample |
| US8993230B2 (en) * | 2008-12-04 | 2015-03-31 | Pacific Biosciences of Californ, Inc. | Asynchronous sequencing of biological polymers |
| AU2010232972B2 (en) | 2009-04-01 | 2016-10-27 | Dxterity Diagnostics Incorporated | Chemical ligation dependent probe amplification (CLPA) |
| US9334531B2 (en) * | 2010-12-17 | 2016-05-10 | Life Technologies Corporation | Nucleic acid amplification |
| US20110039304A1 (en) * | 2009-08-12 | 2011-02-17 | President And Fellows Of Harvard College | Methods to Generate Oligonucleotide Pools and Enrich Target Nucleic Acid Sequences |
| US9127309B2 (en) * | 2011-01-31 | 2015-09-08 | Qiagen Mansfield, Inc. | Methods of nucleic acid quantification and detection using anomalous migration |
| CA2836577C (en) | 2011-05-17 | 2019-06-25 | Dxterity Diagnostics Incorporated | Methods and compositions for detecting target nucleic acids |
| EP2722399A1 (en) | 2012-10-18 | 2014-04-23 | Roche Diagniostics GmbH | Method for preventing high molecular weight products during amplification |
| US10942184B2 (en) | 2012-10-23 | 2021-03-09 | Caris Science, Inc. | Aptamers and uses thereof |
| CN106879252B (zh) | 2014-06-10 | 2020-07-17 | 德克斯特里蒂诊断公司 | 用于收集和稳定生物样品的装置及方法 |
| EP3173488B1 (en) * | 2014-07-25 | 2020-09-09 | Panagene Inc. | Use of multiple target nucleic acid detection method using clamping probe and detection probe |
| JP6956012B2 (ja) * | 2015-05-18 | 2021-10-27 | サガ ダイアグノスティクス アーベー | 標的核酸及び多様体の検出 |
| ES3008759T3 (en) | 2016-08-02 | 2025-03-25 | Hoffmann La Roche | Helper oligonucleotide for improving efficiency of amplification and detection/quantitation of nucleic acids |
| CN108342489A (zh) * | 2017-01-23 | 2018-07-31 | 公安部物证鉴定中心 | 一种对男性个体进行y-snp分型的方法和系统 |
| US10513730B2 (en) * | 2017-11-22 | 2019-12-24 | Safeguard Biosystems Holdings Ltd. | Asymmetric PCR methods, primers and kits |
| KR101919896B1 (ko) * | 2018-05-03 | 2018-11-19 | 주식회사 유디피아 | 단일 형광 채널 유전자증폭기기를 이용한 다중 핵산 검출 방법 |
| EP3844303A4 (en) * | 2018-08-27 | 2022-06-01 | The Regents of The University of California | Reporter nucleic acids for type v crispr-mediated detection |
| KR20210026867A (ko) * | 2019-09-02 | 2021-03-10 | 주식회사 시선바이오머티리얼스 | 신규한 pna 올리고머, 이의 dna 메틸화 검출용도 및 이를 이용한 dna 메틸화 검출방법 |
| US20230040046A1 (en) * | 2020-01-10 | 2023-02-09 | Siemens Healthcare Diagnostics Inc. | Compositions, kits, and methods for performing rapid polymerase chain reactions |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5314809A (en) * | 1991-06-20 | 1994-05-24 | Hoffman-La Roche Inc. | Methods for nucleic acid amplification |
| EP0640828A1 (en) * | 1993-08-27 | 1995-03-01 | F. Hoffmann-La Roche AG | Monitoring multiple reactions simultaneously and analyzing same |
| US5972693A (en) * | 1995-10-24 | 1999-10-26 | Curagen Corporation | Apparatus for identifying, classifying, or quantifying DNA sequences in a sample without sequencing |
Family Cites Families (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4458066A (en) | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US4973679A (en) | 1981-03-27 | 1990-11-27 | University Patents, Inc. | Process for oligonucleo tide synthesis using phosphormidite intermediates |
| US4415732A (en) | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4757141A (en) | 1985-08-26 | 1988-07-12 | Applied Biosystems, Incorporated | Amino-derivatized phosphite and phosphate linking agents, phosphoramidite precursors, and useful conjugates thereof |
| CA1284931C (en) | 1986-03-13 | 1991-06-18 | Henry A. Erlich | Process for detecting specific nucleotide variations and genetic polymorphisms present in nucleic acids |
| US5151507A (en) | 1986-07-02 | 1992-09-29 | E. I. Du Pont De Nemours And Company | Alkynylamino-nucleotides |
| US4931223A (en) | 1986-07-24 | 1990-06-05 | Tropix, Inc. | Methods of using chemiluminescent 1,2-dioxetanes |
| DE3788356T2 (de) | 1986-12-15 | 1994-06-23 | British Tech Group Usa | Monomere phthalocyanin-reagenzien. |
| US5231191A (en) | 1987-12-24 | 1993-07-27 | Applied Biosystems, Inc. | Rhodamine phosphoramidite compounds |
| US5047524A (en) | 1988-12-21 | 1991-09-10 | Applied Biosystems, Inc. | Automated system for polynucleotide synthesis and purification |
| US5262530A (en) | 1988-12-21 | 1993-11-16 | Applied Biosystems, Inc. | Automated system for polynucleotide synthesis and purification |
| US5141813A (en) | 1989-08-28 | 1992-08-25 | Clontech Laboratories, Inc. | Multifunctional controlled pore glass reagent for solid phase oligonucleotide synthesis |
| US5366860A (en) | 1989-09-29 | 1994-11-22 | Applied Biosystems, Inc. | Spectrally resolvable rhodamine dyes for nucleic acid sequence determination |
| US5188934A (en) | 1989-11-14 | 1993-02-23 | Applied Biosystems, Inc. | 4,7-dichlorofluorescein dyes as molecular probes |
| US5654442A (en) | 1989-11-14 | 1997-08-05 | The Perkin-Elmer Corporation | 4,7-dichlorofluorescein dyes as molecular probes |
| US5210015A (en) | 1990-08-06 | 1993-05-11 | Hoffman-La Roche Inc. | Homogeneous assay system using the nuclease activity of a nucleic acid polymerase |
| DK51092D0 (da) | 1991-05-24 | 1992-04-15 | Ole Buchardt | Oligonucleotid-analoge betegnet pna, monomere synthoner og fremgangsmaade til fremstilling deraf samt anvendelser deraf |
| US5470705A (en) | 1992-04-03 | 1995-11-28 | Applied Biosystems, Inc. | Probe composition containing a binding domain and polymer chain and methods of use |
| WO1993020236A1 (en) | 1992-04-03 | 1993-10-14 | Applied Biosystems, Inc. | Probe composition and method |
| US5925517A (en) | 1993-11-12 | 1999-07-20 | The Public Health Research Institute Of The City Of New York, Inc. | Detectably labeled dual conformation oligonucleotide probes, assays and kits |
| US5538848A (en) | 1994-11-16 | 1996-07-23 | Applied Biosystems Division, Perkin-Elmer Corp. | Method for detecting nucleic acid amplification using self-quenching fluorescence probe |
| AU2522095A (en) | 1994-05-19 | 1995-12-18 | Dako A/S | Pna probes for detection of neisseria gonorrhoeae and chlamydia trachomatis |
| US5801155A (en) | 1995-04-03 | 1998-09-01 | Epoch Pharmaceuticals, Inc. | Covalently linked oligonucleotide minor grove binder conjugates |
| US5736626A (en) | 1996-01-29 | 1998-04-07 | The Perkin-Elmer Corporation | Solid support reagents for the direct synthesis of 3'-labeled polynucleotides |
| US6020481A (en) | 1996-04-01 | 2000-02-01 | The Perkin-Elmer Corporation | Asymmetric benzoxanthene dyes |
| JP3898228B2 (ja) | 1996-04-12 | 2007-03-28 | ザ パブリック ヘルス リサーチ インスティチュート オブ ザ シティー オブ ニューヨーク インク | 検出プローブ、キット及びアッセイ |
| US5847162A (en) | 1996-06-27 | 1998-12-08 | The Perkin Elmer Corporation | 4, 7-Dichlororhodamine dyes |
| SE506700C2 (sv) | 1996-05-31 | 1998-02-02 | Mikael Kubista | Sond och förfaranden för analys av nukleinsyra |
| US5821356A (en) | 1996-08-12 | 1998-10-13 | The Perkin Elmer Corporation | Propargylethoxyamino nucleotides |
| DE19637339A1 (de) | 1996-09-13 | 1998-03-19 | Hoechst Ag | Verfahren zur Amplifikation von Nukleinsäuren |
| DE69719220T2 (de) | 1996-11-18 | 2004-01-22 | Takeshi Imanishi | Neue nucleotidanaloga |
| US6110676A (en) | 1996-12-04 | 2000-08-29 | Boston Probes, Inc. | Methods for suppressing the binding of detectable probes to non-target sequences in hybridization assays |
| JP3756313B2 (ja) | 1997-03-07 | 2006-03-15 | 武 今西 | 新規ビシクロヌクレオシド及びオリゴヌクレオチド類縁体 |
| US5770716A (en) | 1997-04-10 | 1998-06-23 | The Perkin-Elmer Corporation | Substituted propargylethoxyamido nucleosides, oligonucleotides and methods for using same |
| CA2303299C (en) | 1997-09-12 | 2016-02-23 | Exiqon A/S | Oligonucleotide analogues |
| US6008379A (en) | 1997-10-01 | 1999-12-28 | The Perkin-Elmer Corporation | Aromatic-substituted xanthene dyes |
| WO1999022018A2 (en) | 1997-10-27 | 1999-05-06 | Boston Probes, Inc. | Methods, kits and compositions pertaining to pna molecular beacons |
| US6485901B1 (en) | 1997-10-27 | 2002-11-26 | Boston Probes, Inc. | Methods, kits and compositions pertaining to linear beacons |
| US5936087A (en) | 1997-11-25 | 1999-08-10 | The Perkin-Elmer Corporation | Dibenzorhodamine dyes |
| US6080868A (en) | 1998-01-23 | 2000-06-27 | The Perkin-Elmer Corporation | Nitro-substituted non-fluorescent asymmetric cyanine dye compounds |
| US6361942B1 (en) | 1998-03-24 | 2002-03-26 | Boston Probes, Inc. | Method, kits and compositions pertaining to detection complexes |
| DE69825142T2 (de) | 1998-05-01 | 2005-08-11 | F. Hoffmann-La Roche Ag | Vorrichtung zur gleichzeitigen Überwachung von Reaktionen, die in einer Vielzahl von Reaktionsgefässen stattfinden |
| US6117986A (en) | 1998-06-10 | 2000-09-12 | Intergen Company, L.P. | Pyrimidines linked to a quencher |
| US6037130A (en) | 1998-07-28 | 2000-03-14 | The Public Health Institute Of The City Of New York, Inc. | Wavelength-shifting probes and primers and their use in assays and kits |
| US6887664B2 (en) * | 2000-06-06 | 2005-05-03 | Applera Corporation | Asynchronous primed PCR |
-
2001
- 2001-06-05 US US09/875,211 patent/US6887664B2/en not_active Expired - Lifetime
- 2001-06-06 DE DE60140487T patent/DE60140487D1/de not_active Expired - Lifetime
- 2001-06-06 EP EP09014084A patent/EP2256213A1/en not_active Withdrawn
- 2001-06-06 AT AT01946152T patent/ATE448325T1/de not_active IP Right Cessation
- 2001-06-06 EP EP01946152A patent/EP1368489B1/en not_active Expired - Lifetime
- 2001-06-06 WO PCT/US2001/018464 patent/WO2001094638A2/en active IP Right Grant
- 2001-06-06 CA CA002412413A patent/CA2412413A1/en not_active Abandoned
- 2001-06-06 JP JP2002502178A patent/JP2004509613A/ja active Pending
- 2001-06-06 EP EP10184140A patent/EP2316975A1/en not_active Withdrawn
- 2001-06-06 AU AU6823501A patent/AU6823501A/xx active Pending
-
2004
- 2004-06-09 US US10/865,683 patent/US20050130178A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5314809A (en) * | 1991-06-20 | 1994-05-24 | Hoffman-La Roche Inc. | Methods for nucleic acid amplification |
| EP0640828A1 (en) * | 1993-08-27 | 1995-03-01 | F. Hoffmann-La Roche AG | Monitoring multiple reactions simultaneously and analyzing same |
| US5972693A (en) * | 1995-10-24 | 1999-10-26 | Curagen Corporation | Apparatus for identifying, classifying, or quantifying DNA sequences in a sample without sequencing |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2412413A1 (en) | 2001-12-13 |
| WO2001094638A3 (en) | 2003-07-10 |
| US20050130178A1 (en) | 2005-06-16 |
| ATE448325T1 (de) | 2009-11-15 |
| EP2316975A1 (en) | 2011-05-04 |
| EP1368489B1 (en) | 2009-11-11 |
| US20030207266A1 (en) | 2003-11-06 |
| EP1368489A2 (en) | 2003-12-10 |
| AU6823501A (en) | 2001-12-17 |
| WO2001094638A2 (en) | 2001-12-13 |
| DE60140487D1 (en) | 2009-12-24 |
| EP2256213A1 (en) | 2010-12-01 |
| WO2001094638A8 (en) | 2002-04-11 |
| US6887664B2 (en) | 2005-05-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2004509613A (ja) | 非同調的刺激pcr | |
| AU768240B2 (en) | Template-dependent ligation with PNA-DNA chimeric probes | |
| US6451588B1 (en) | Multipartite high-affinity nucleic acid probes | |
| EP1198594B1 (en) | Polymerase extension at 3' terminus of pna-dna chimera | |
| AU770217B2 (en) | Ligation assembly and detection of polynucleotides on solid-support | |
| JP4644370B2 (ja) | 標的ハイブリダイゼーション検出のための、プローブおよびクランプのバイナリー組成物ならびに方法 | |
| US6902900B2 (en) | Nucleic acid probes and methods to detect and/or quantify nucleic acid analytes | |
| US8268978B2 (en) | Modified oligonucleotides and applications thereof | |
| JP2004507248A (ja) | 核酸増幅に対する外部コントロールのための方法 | |
| CN102083846A (zh) | 包含2’-终止子核苷酸的核酸的合成和组合物 | |
| US20050176014A1 (en) | Fluorescent hybridization probes with reduced background | |
| US7408051B2 (en) | Modified oligonucleotides and applications thereof | |
| AU2001268235B2 (en) | Asynchronous primed PCR | |
| AU2001268235A1 (en) | Asynchronous primed PCR | |
| McGuire | Synthesis and studies of modified nucleotides and oligonucleotides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080528 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20090618 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110216 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20110512 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20110519 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20111027 |