DE102014001468B4 - Heteroleptischer phenylbenzimidazol-komplex - Google Patents
Heteroleptischer phenylbenzimidazol-komplex Download PDFInfo
- Publication number
- DE102014001468B4 DE102014001468B4 DE102014001468.1A DE102014001468A DE102014001468B4 DE 102014001468 B4 DE102014001468 B4 DE 102014001468B4 DE 102014001468 A DE102014001468 A DE 102014001468A DE 102014001468 B4 DE102014001468 B4 DE 102014001468B4
- Authority
- DE
- Germany
- Prior art keywords
- mmol
- compound
- group
- tri
- substitution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table
- C07F15/0006—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table compounds of the platinum group
- C07F15/0033—Iridium compounds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/06—Luminescent, e.g. electroluminescent, chemiluminescent materials containing organic luminescent materials
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
- H10K85/341—Transition metal complexes, e.g. Ru(II)polypyridine complexes
- H10K85/342—Transition metal complexes, e.g. Ru(II)polypyridine complexes comprising iridium
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1029—Heterocyclic compounds characterised by ligands containing one nitrogen atom as the heteroatom
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1044—Heterocyclic compounds characterised by ligands containing two nitrogen atoms as heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/18—Metal complexes
- C09K2211/185—Metal complexes of the platinum group, i.e. Os, Ir, Pt, Ru, Rh or Pd
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K2101/00—Properties of the organic materials covered by group H10K85/00
- H10K2101/10—Triplet emission
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/11—OLEDs or polymer light-emitting diodes [PLED] characterised by the electroluminescent [EL] layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
- H10K85/321—Metal complexes comprising a group IIIA element, e.g. Tris (8-hydroxyquinoline) gallium [Gaq3]
- H10K85/324—Metal complexes comprising a group IIIA element, e.g. Tris (8-hydroxyquinoline) gallium [Gaq3] comprising aluminium, e.g. Alq3
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/615—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene
- H10K85/622—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene containing four rings, e.g. pyrene
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/657—Polycyclic condensed heteroaromatic hydrocarbons
- H10K85/6576—Polycyclic condensed heteroaromatic hydrocarbons comprising only sulfur in the heteroaromatic polycondensed ring system, e.g. benzothiophene
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Crystallography & Structural Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Physics & Mathematics (AREA)
- Optics & Photonics (AREA)
- Electroluminescent Light Sources (AREA)
Abstract
Verbindung mit der Formel

worin R2 Mono-, Di-, Tri-, Tetra-, Pentasubstitutionen oder keine Substitution darstellt,
worin R3, R4 und R5 jeweils Mono-, Di-, Tri-, Tetrasubstitutionen oder keine Substitution darstellen,
worin R9 Mono-, Di-, Trisubstitutionen oder keine Substitution darstellt,
worin R1, R2, R3, R4, R5 und R9 jeweils unabhängig aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Halogenid, Alkyl, Cycloalkyl, Heteroalkyl, Arylalkyl, Alkoxy, Aryloxy, Amino, Silyl, Alkenyl, Cycloalkenyl, Heteroalkenyl, Alkinyl, Aryl, Heteroaryl, Acyl, Carbonyl, Carbonsäuren, Ester, Nitril, Isonitril, Sulfanyl, Sulfinyl, Sulfonyl, Phosphino und Kombinationen davon, ausgewählt sind,
worin n 1 oder 2 ist,
worin keine Substitution bedeutet, dass der jeweilige Rest R2 bis R5 und R9 für alle verfügbaren Positionen Wasserstoff ist, und
worin die Verbindung ausgewählt ist aus der Gruppe, bestehend aus: 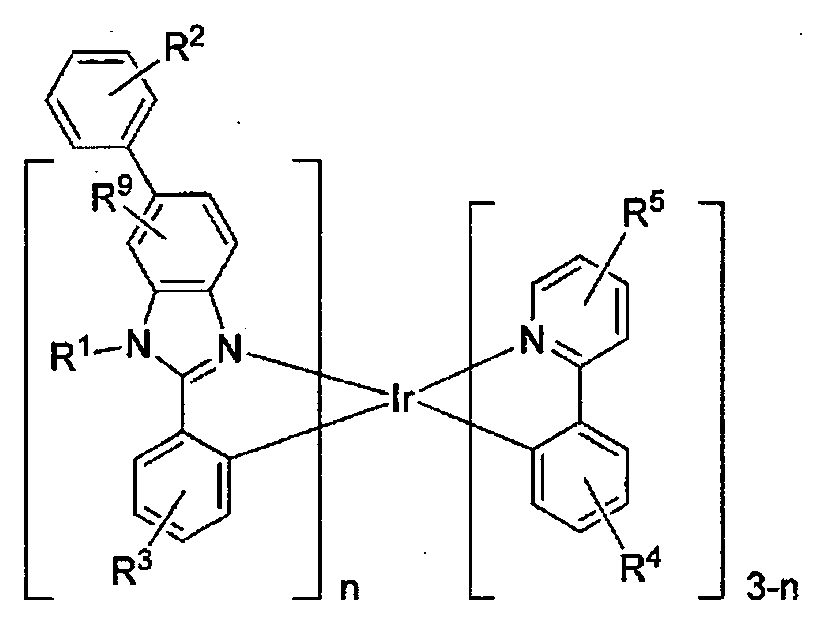
und

worin R2 Mono-, Di-, Tri-, Tetra-, Pentasubstitutionen oder keine Substitution darstellt,
worin R3, R4 und R5 jeweils Mono-, Di-, Tri-, Tetrasubstitutionen oder keine Substitution darstellen,
worin R9 Mono-, Di-, Trisubstitutionen oder keine Substitution darstellt,
worin R1, R2, R3, R4, R5 und R9 jeweils unabhängig aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Halogenid, Alkyl, Cycloalkyl, Heteroalkyl, Arylalkyl, Alkoxy, Aryloxy, Amino, Silyl, Alkenyl, Cycloalkenyl, Heteroalkenyl, Alkinyl, Aryl, Heteroaryl, Acyl, Carbonyl, Carbonsäuren, Ester, Nitril, Isonitril, Sulfanyl, Sulfinyl, Sulfonyl, Phosphino und Kombinationen davon, ausgewählt sind,
worin n 1 oder 2 ist,
worin keine Substitution bedeutet, dass der jeweilige Rest R2 bis R5 und R9 für alle verfügbaren Positionen Wasserstoff ist, und
worin die Verbindung ausgewählt ist aus der Gruppe, bestehend aus:
Description
- Die vorliegende Erfindung betrifft Verbindungen zur Verwendung als gelb/grün emittierende Materialien und Vorrichtungen, wie z.B. organische Licht-emittierende Dioden bzw. Leuchtdioden, welche diese als Teil einer organischen Schicht umfassen.
- Optoelektronische Vorrichtungen, die organische Materialien nutzen, werden aus einer Reihe von Gründen zunehmend wünschenswert. Viele der Materialien, die verwendet werden, um solche Vorrichtungen herzustellen, sind relativ billig, so dass organische optoelektronische Vorrichtungen ein Potenzial für Kostenvorteile gegenüber anorganischen Vorrichtungen aufweisen. Darüber hinaus können die inhärenten Eigenschaften von organischen Materialien, wie z.B. ihre Flexibilität, sie für bestimmte Anwendungen, wie z.B. die Fertigung auf einem flexiblen Substrat, gut geeignet machen. Beispiele für organische optoelektronische Vorrichtungen umfassen organische Licht-emittierende Vorrichtungen (OLEDs), organische Phototransistoren, organische Solarzellen und organische Photodetektoren. Für OLEDs können die organischen Materialien Leistungsvorteile gegenüber herkömmlichen Materialien haben. Beispielsweise könnte die Wellenlänge, bei der eine organische emittierende Schicht Licht emittiert, im Allgemeinen mit geeigneten Dotiermitteln einfach eingestellt werden.
- OLEDs nutzen dünne organische Filme, die Licht emittieren, wenn eine Spannung an die Vorrichtung angelegt wird. OLEDs werden zu einer zunehmend interessanten Technologie für den Einsatz in Anwendungen wie Flachbildschirmen, Beleuchtung und Hintergrundbeleuchtung. Einige OLED-Materialien und -Konfigurationen sind in den US-Patenten Nr.
5,844,363 ,6,303,238 und5,707,745 beschrieben, welche hier unter Bezugnahme in ihrer Gesamtheit einbezogen sind. - Eine Anwendung für phosphoreszierende emittierende Moleküle ist ein Farbdisplay bzw. eine Farbanzeige. Industriestandards für eine derartige Anzeige fordern Pixel, die angepasst sind, um bestimmte Farben, die als „gesättigte“ Farben bezeichnet werden, zu emittieren. Insbesondere fordern diese Standards gesättigte rote, grüne und blaue Pixel. Die Farbe kann unter Verwendung von CIE-Koordinaten, die in dem Fachgebiet gut bekannt sind, gemessen werden.
- Ein Beispiel für ein grün emittierendes Molekül ist Tris(2-phenylpyridin)iridium, das als Ir(ppy)3 bezeichnet wird und die folgende Struktur aufweist:

- In dieser und den hier später gezeigten Figuren ist die koordinative Bindung von Stickstoff an ein Metall (hier Ir) als eine Gerade gezeigt.
- Wie hier verwendet, umfasst der Begriff „organisch“ polymere Materialien sowie organische Materialien aus kleinen Molekülen, die verwendet werden können, um organische optoelektronische Vorrichtungen herzustellen. „Kleines Molekül“ bezieht sich auf jedwedes organische Material, das kein Polymer ist und „kleine Moleküle“ können tatsächlich ziemlich groß sein. Kleine Moleküle können in einigen Fällen Wiederholungseinheiten umfassen. Beispielsweise führt die Verwendung einer langkettigen Alkylgruppe als Substituent nicht dazu, dass ein Molekül nicht zu der Klasse eines „kleinen Moleküls“ gehört. Kleine Moleküle können auch in Polymere einbezogen werden, beispielsweise als Seitengruppe an einem Polymergrundgerüst oder als Teil des Grundgerüsts. Kleine Moleküle können auch als die Kerneinheit eines Dendrimers dienen, das aus einer Reihe von chemischen Hüllen besteht, welche um die Kerneinheit angeordnet sind. Die Kerneinheit eines Dendrimers kann eine kleine fluoreszierende oder phosphoreszierende Molekülemissionsquelle sein. Ein Dendrimer kann ein „kleines Molekül“ sein und es wird angenommen, dass alle Dendrimere, die gegenwärtig auf dem Gebiet von OLEDs verwendet werden, kleine Moleküle sind.
- Wie hier verwendet, bedeutet „oben“ am weitesten von dem Substrat entfernt, während „unten“ als dem Substrat am nächsten bedeutet. Wo eine erste Schicht als „angeordnet über“ einer zweiten Schicht beschrieben ist, ist die erste Schicht weiter von dem Substrat entfernt angeordnet. Es können weitere Schichten zwischen der ersten und der zweiten Schicht vorliegen, es sei denn, dass die erste Schicht als „in Kontakt mit“ der zweiten Schicht angegeben ist. Beispielsweise kann eine Kathode als „angeordnet über“ einer Anode beschrieben sein, auch wenn verschiedene organische Schichten dazwischen vorliegen.
- Wie hier verwendet, bedeutet „lösungsverarbeitbar“ in einem flüssigen Medium löslich, dispergierbar oder transportierbar in und/oder abscheidbar aus einem flüssigen Medium zu sein, und zwar entweder in Lösungs- oder Suspensionsform.
- Ein Ligand kann als „photoaktiv“ bezeichnet werden, wenn angenommen wird, dass der Ligand direkt zu den photoaktiven Eigenschaften eines emittierenden Materials beiträgt. Ein Ligand kann als „ergänzend“ bezeichnet werden, wenn angenommen wird, dass der Ligand nicht zu den photoaktiven Eigenschaften eines emittierenden Materials beiträgt, obwohl ein ergänzender Ligand die Eigenschaften eines photoaktiven Liganden verändern könnte.
- Wie hier verwendet und wie es allgemein vom Fachmann verstanden wird, ist ein erstes „Höchstes besetztes Molekülorbital“ (HOMO)- oder „Niedrigstes unbesetztes Molekülorbital“ (LUMO)-Energieniveau „größer als“ oder „höher als“ ein zweites HOMO- oder LUMO-Energieniveau, wenn das erste Energieniveau näher am Vakuumenergieniveau ist. Da lonisationspotenziale (IP) als negative Energie bezogen auf das Vakuumniveau gemessen werden, entspricht ein höheres HOMO-Energieniveau einem IP, das einen kleineren Absolutwert aufweist (ein IP, das weniger negativ ist). In ähnlicher Weise entspricht ein höheres LUMO-Energieniveau einer Elektronenaffinität (EA), die einen kleineren Absolutwert aufweist (eine EA, die weniger negativ ist). In einem herkömmlichen Energieniveaudiagramm, bei dem sich das Vakuumniveau oben befindet, ist das LUMO-Energieniveau eines Materials höher als das HOMO-Energieniveau desselben Materials. Ein „höheres“ HOMO- oder LUMO-Energieniveau erscheint näher an der Spitze eines solchen Diagramms als ein „niedrigeres“ HOMO- oder LUMO-Energieniveau.
- Wie hier verwendet und wie es allgemein vom Fachmann verstanden wird, ist eine erste Austrittsenergie „größer als“ oder „höher als“ eine zweite Austrittsenergie, wenn die erste Austrittsenergie einen höheren Absolutwert aufweist. Weil Austrittsenergien allgemein als negative Zahlen bezogen auf das Vakuumniveau gemessen werden, bedeutet dies, dass eine „höhere“ Austrittsenergie negativer ist. In einem herkömmlichen Energieniveaudiagramm, bei dem sich das Vakuumniveau oben befindet, ist eine „höhere“ Austrittsenergie weiter weg von dem Vakuumniveau in Abwärtsrichtung dargestellt. Daher gilt für die Definitionen von HOMO- und LUMO-Energieniveaus eine andere Konvention als für Austrittsenergien.
- Weitere Einzelheiten zu OLEDs und den vorstehend beschriebenen Definitionen sind in dem
US-Patent Nr. 7,279,704 angegeben, das hier unter Bezugnahme in dessen Gesamtheit aufgenommen ist. - Gemäß einer Ausführungsform wird eine Verbindung bereitgestellt, welche die Struktur der nachstehend gezeigten Formel I aufweist:

worin R2 Mono-, Di-, Tri-, Tetra-, Pentasubstitutionen oder keine Substitution darstellt,
worin R3, R4 und R5 jeweils Mono-, Di-, Tri-, Tetrasubstitutionen oder keine Substitution darstellen,
worin R9 Mono-, Di-, Trisubstitutionen oder keine Substitution darstellt,
worin R1, R2, R3, R4, R5 und R9 jeweils unabhängig aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Halogenid, Alkyl, Cycloalkyl, Heteroalkyl, Arylalkyl, Alkoxy, Aryloxy, Amino, Silyl, Alkenyl, Cycloalkenyl, Heteroalkenyl, Alkinyl, Aryl, Heteroaryl, Acyl, Carbonyl, Carbonsäuren, Ester, Nitril, Isonitril, Sulfanyl, Sulfinyl, Sulfonyl, Phosphino und Kombinationen davon, ausgewählt sind, worin n 1 oder 2 ist, und worin keine Substitution bedeutet, dass der jeweilige Rest R2 bis R5 und R9 für alle verfügbaren Positionen Wasserstoff ist, und worin die Verbindung ausgewählt ist aus der Gruppe, bestehend aus:
- Gemäß einer weiteren Ausführungsform wird auch eine erste Vorrichtung bereitgestellt, die eine erste organische Licht-emittierende Vorrichtung umfasst. Die erste Vorrichtung kann eine Anode, eine Kathode und eine organische Schicht, die zwischen der Anode und der Kathode angeordnet ist, umfassen. Die organische Schicht kann eine Verbindung der Formel I umfassen. Die erste Vorrichtung kann ein Konsumenten- bzw. Verbraucherprodukt, eine organische Licht-emittierende Vorrichtung und/oder eine Beleuchtungseinheit sein.
- Die vorliegende Offenbarung ist am Besten mittels der folgenden detaillierten Beschreibung verständlich, wenn diese im Zusammenhang mit den beigefügten Zeichnungen gelesen wird. Es wird betont, dass gemäß der üblichen Praxis die verschiedenen Merkmale der Zeichnungen nicht notwendigerweise maßstabsgetreu sind. Die Abmessungen der verschiedenen Merkmale sind aus Klarheitsgründen vielmehr willkürlich vergrößert oder verkleinert. In der Beschreibung und den Zeichnungen geben die gleichen Bezugszeichen die gleichen Merkmale an.
- Die
1 zeigt eine organische Licht-emittierende Vorrichtung. - Die
2 zeigt eine invertierte organische Licht-emittierende Vorrichtung, die keine separate Elektronentransportschicht aufweist. - Die
3 zeigt die Formel I, wie sie hier offenbart ist. - Im Allgemeinen umfasst eine OLED mindestens eine organische Schicht, die zwischen einer Anode und einer Kathode angeordnet und elektrisch damit verbunden ist. Wenn ein Strom angelegt wird, injiziert die Anode Löcher und die Kathode injiziert Elektronen in die organische(n) Schicht(en). Die injizierten Löcher und Elektronen wandern jeweils zur entgegengesetzt geladenen Elektrode. Wenn sich ein Elektron und ein Loch an demselben Molekül befinden, wird ein „Exciton“ gebildet, das ein lokalisiertes Elektron-Loch-Paar ist, das einen angeregten Energiezustand aufweist. Licht wird emittiert, wenn das Exciton durch einen Lichtemissionsmechanismus relaxiert. In einigen Fällen kann das Exciton an einem Excimer oder einem Exciplex lokalisiert sein. Nicht-strahlende Mechanismen, wie z.B. eine thermische Relaxation, können auch auftreten, sind aber im Allgemeinen nicht erwünscht.
- Die ersten OLEDs verwendeten emittierende Moleküle, die Licht von ihren Singulett-Zuständen („Fluoreszenz“) emittierten, wie es beispielsweise im
US-Patent Nr. 4,769,292 offenbart ist, das hier unter Bezugnahme in seiner Gesamtheit aufgenommen ist. Eine Fluoreszenzemission erfolgt im Allgemeinen in einem Zeitrahmen von weniger als 10 Nanosekunden. - In jüngerer Zeit sind OLEDs vorgestellt worden, die emittierende Materialien aufweisen, die Licht aus Triplettzuständen emittieren („Phosphoreszenz“). Baldo et al., „Highly Efficient Phosphorescent Emission from Organic Electroluminescent Devices", Nature, Band 395, 151-154, 1998, („Baldo-I") und Baldo et al., „Very high-efficiency green organic light-emitting devices based on electrophosphorescence", Appl. Phys. Lett., Band 75, Nr. 3, 4-6 (1999) („Baldo-II“), welche hier unter Bezugnahme in ihrer Gesamtheit aufgenommen sind. Eine Phosphoreszenz ist detaillierter in dem
US-Patent Nr. 7,279,704 in den Spalten 5-6 beschrieben, die hier unter Bezugnahme aufgenommen sind. - Die
1 zeigt eine organische Licht-emittierende Vorrichtung 100. Die Figuren sind nicht notwendigerweise maßstabsgetreu gezeichnet. Die Vorrichtung 100 kann ein Substrat 110, eine Anode 115, eine Lochinjektionsschicht 120, eine Lochtransportschicht 125, eine Elektronensperrschicht 130, eine emittierende Schicht 135, eine Lochsperrschicht 140, eine Elektronentransportschicht 145, eine Elektroneninjektionsschicht 150, eine Schutzschicht 155, eine Kathode 160 und eine Sperrschicht 170 umfassen. Die Kathode 160 ist eine Verbundkathode, die eine erste leitende Schicht 162 und eine zweite leitende Schicht 164 aufweist. Die Vorrichtung 100 kann durch aufeinander folgendes Abscheiden der beschriebenen Schichten hergestellt werden. Die Eigenschaften und Funktionen dieser verschiedenen Schichten sowie Beispielmaterialien sind detaillierter inUS 7,279,704 in den Spalten 6-10 beschrieben, die hier unter Bezugnahme aufgenommen sind. - Für jede dieser Schichten gibt es weitere Beispiele. Zum Beispiel ist eine flexible und transparente Substrat-Anode-Kombination im
US-Patent Nr. 5,844,363 offenbart, das unter Bezugnahme in dessen Gesamtheit aufgenommen ist. Ein Beispiel für eine p-dotierte Lochtransportschicht ist m-MTDATA, dotiert mit F4-TCNQ in einem molaren Verhältnis von 50:1, wie es in der US-Patentanmeldung mit der Veröffentlichungsnummer2003/0230980 offenbart ist, die unter Bezugnahme in ihrer Gesamtheit aufgenommen ist. Beispiele für emittierende Materialien und Wirtsmaterialien sind in dem US-Patent Nr.6,303,238 für Thompson et al. offenbart, das unter Bezugnahme in dessen Gesamtheit aufgenommen ist. Ein Beispiel für eine n-dotierte Elektronentransportschicht ist BPhen, das mit Li in einem molaren Verhältnis von 1:1 dotiert ist, wie es in der US-Patentanmeldung mit der Veröffentlichungsnummer2003/0230980 5,703,436 und5,707,745 , die hier unter Bezugnahme in ihrer Gesamtheit aufgenommen sind, offenbaren Beispiele von Kathoden, einschließlich Verbundkathoden, die eine dünne Schicht aus Metall, wie z.B. Mg:Ag, mit einer darüber liegenden transparenten elektrisch leitenden, durch Sputtern abgeschiedenen ITO-Schicht aufweisen. Die Theorie und Verwendung von Sperrschichten ist detaillierter im US-Patent Nr.6,097,147 und in der US-Patentanmeldung mit der Veröffentlichungsnummer2003/0230980 beschrieben, die unter Bezugnahme in ihrer Gesamtheit aufgenommen sind. Beispiele für Injektionsschichten sind in der US-Patentanmeldung mit der Veröffentlichungsnummer2004/0174116 angegeben, die unter Bezugnahme in ihrer Gesamtheit aufgenommen ist. Eine Beschreibung von Schutzschichten findet sich in der US-Patentanmeldung mit der Veröffentlichungsnummer2004/0174116 - Die
2 zeigt eine invertierte OLED 200. Die Vorrichtung umfasst ein Substrat 210, eine Kathode 215, eine emittierende Schicht 220, eine Lochtransportschicht 225 und eine Anode 230. Die Vorrichtung 200 kann durch aufeinander folgendes Abscheiden der beschriebenen Schichten hergestellt werden. Da in der häufigsten OLED-Konfiguration eine Kathode über der Anode angeordnet ist und bei der Vorrichtung 200 die Kathode 215 unter der Anode 230 angeordnet ist, kann die Vorrichtung 200 als eine „invertierte“ OLED bezeichnet werden. Materialien ähnlich zu jenen, die in Bezug auf die Vorrichtung 100 beschrieben sind, können in den entsprechenden Schichten der Vorrichtung 200 verwendet werden. Die2 zeigt ein Beispiel, wie einige Schichten aus der Struktur der Vorrichtung 100 weggelassen werden können. - Die einfache Schichtstruktur, die in den
1 und2 gezeigt ist, ist als nicht-beschränkendes Beispiel angegeben und es versteht sich, dass Ausführungsformen der Erfindung in Verbindung mit einer breiten Vielfalt von weiteren Strukturen verwendet werden können. Die beschriebenen spezifischen Materialien und Strukturen sind in ihrer Art beispielhaft und andere Materialien und Strukturen können verwendet werden. Funktionelle OLEDs können durch Kombinieren der beschriebenen verschiedenen Schichten auf verschiedene Weise erhalten werden oder Schichten können aufgrund der Gestaltung, der Leistung und von Kostenfaktoren vollständig weggelassen werden. Weitere Schichten, die nicht spezifisch beschrieben sind, können auch einbezogen werden. Materialien, die von denjenigen, die spezifisch beschrieben sind, verschieden sind, können verwendet werden. Obwohl viele der Beispiele, die hier angegeben sind, verschiedene Schichten so beschreiben, dass sie ein einziges Material enthalten, versteht es sich, dass Kombinationen von Materialien, wie z.B. ein Gemisch aus Wirtsmaterial und Dotiermittel, oder allgemeiner ein Gemisch, verwendet werden können. Die Schichten können auch verschiedene Unter- bzw. Teilschichten aufweisen. Die Bezeichnungen, die den verschiedenen Schichten hier gegeben werden, sind nicht streng begrenzend aufzufassen. Die Lochtransportschicht 225 transportiert zum Beispiel in der Vorrichtung 200 Löcher und injiziert Löcher in die emittierende Schicht 220 und kann als eine Lochtransportschicht oder eine Lochinjektionsschicht beschrieben werden. In einer Ausführungsform kann eine OLED so beschrieben sein, dass sie eine „organische Schicht“ aufweist, die zwischen einer Kathode und einer Anode angeordnet ist. Diese organische Schicht kann eine einzige Schicht umfassen oder kann ferner mehrere Schichten aus verschiedenen organischen Materialien umfassen, wie sie zum Beispiel mit Bezug auf die1 und2 beschrieben sind. - Strukturen und Materialien, die nicht spezifisch beschrieben sind, können auch verwendet werden, wie z.B. OLEDs, die polymere Materialien umfassen (PLEDs), wie sie im US-Patent Nr.
5,247,190 für Friend et al. offenbart sind, das unter Bezugnahme in dessen Gesamtheit aufgenommen ist. Als weiteres Beispiel können OLEDs, die eine einzige organische Schicht aufweisen, verwendet werden. OLEDs können gestapelt bzw. übereinander angeordnet werden, wie es zum Beispiel im US-Patent Nr.5,707,745 für Forrest et al. beschrieben ist, das unter Bezugnahme in dessen Gesamtheit aufgenommen ist. Die OLED-Struktur kann von der einfachen Schichtstruktur, wie sie in den1 und2 dargestellt ist, abweichen. Das Substrat kann zum Beispiel eine abgewinkelte reflektierende Oberfläche umfassen, um die Auskopplung zu verbessern, wie z.B. eine Mesastruktur, wie sie im US-Patent Nr.6,091,195 für Forrest et al. beschrieben ist, und/oder eine Vertiefungsstruktur wie sie im US-Patent Nr.5,834,893 für Bulovic et al. beschrieben ist, die unter Bezugnahme in ihrer Gesamtheit aufgenommen sind. - Sofern nichts anderes angegeben ist, kann jedwede der Schichten der verschiedenen Ausführungsformen durch jedwedes geeignete Verfahren abgeschieden werden. Bevorzugte Verfahren für die organischen Schichten umfassen thermisches Verdampfen, Tintenstrahl, wie es in den US-Patenten Nr.
6,013,982 und6,087,196 beschrieben ist, die unter Bezugnahme in ihrer Gesamtheit aufgenommen sind, organische Gasphasenabscheidung (OVPD), wie es im US-Patent Nr.6,337,102 für Forrest et al. beschrieben ist, das unter Bezugnahme in dessen Gesamtheit aufgenommen ist, und eine Abscheidung durch organisches Dampfstrahldrucken (OVJP), wie es in dem US-Patent Nr.7,431,968 beschrieben ist, das hier unter Bezugnahme in dessen Gesamtheit aufgenommen ist. Andere geeignete Abscheidungsverfahren umfassen Schleuderbeschichten und andere Verfahren auf Lösungsbasis. Verfahren auf Lösungsbasis werden bevorzugt in Stickstoff oder einer inerten Atmosphäre durchgeführt. Bevorzugte Verfahren für die anderen Schichten umfassen eine thermische Verdampfung. Bevorzugte Strukturierungsverfahren umfassen eine Abscheidung durch eine Maske, ein Kaltverschweißen, wie es in den US-Patenten Nr.6,294,398 und6,468,819 beschrieben ist, die unter Bezugnahme in ihrer Gesamtheit aufgenommen sind, und ein Strukturieren, das mit einigen der Abscheidungsverfahren wie Tintenstrahl und OVJD zusammenhängt. Andere Verfahren können auch verwendet werden. Die Materialien, die abgeschieden werden sollen, können modifiziert werden, um sie mit einem bestimmten Abscheidungsverfahren kompatibel zu machen. Beispielsweise können Substituenten wie Alkyl- und Arylgruppen, die verzweigt oder unverzweigt sind und die vorzugsweise mindestens 3 Kohlenstoffatome enthalten, in kleinen Molekülen verwendet werden, um ihre Fähigkeit zur Verarbeitung in Lösung zu verbessern. Es können Substituenten verwendet werden, die 20 Kohlenstoffatome oder mehr aufweisen, und ein bevorzugter Bereich sind 3-20 Kohlenstoffatome. Materialien mit asymmetrischen Strukturen können eine bessere Lösungsverarbeitbarkeit aufweisen als solche mit symmetrischen Strukturen, weil asymmetrische Materialien eine geringere Tendenz zur Rekristallisation aufweisen können. Dendrimer-Substituenten können verwendet werden, um die Fähigkeit der kleinen Moleküle, in Lösung verarbeitet zu werden, zu verbessern. - Vorrichtungen, die gemäß Ausführungsformen der vorliegenden Erfindung hergestellt werden, können gegebenenfalls ferner eine Sperrschicht umfassen. Ein Zweck der Sperrschicht ist es, die Elektroden und organischen Schichten vor einer schädigenden Einwirkung von schädlichen Spezies in der Umgebung, einschließlich Feuchtigkeit, Dampf und/oder Gasen, usw., zu schützen. Die Sperrschicht kann über, unter oder neben einem Substrat, einer Elektrode oder über jedweden anderen Teilen einer Vorrichtung, einschließlich einer Kante, abgeschieden sein. Die Sperrschicht kann eine einzelne Schicht oder mehrere Schichten umfassen. Die Sperrschicht kann durch verschiedene bekannte chemische Gasphasenabscheidungstechniken hergestellt werden und kann Zusammensetzungen umfassen, die eine einzelne Phase aufweisen, sowie Zusammensetzungen, die mehrere Phasen aufweisen. Jedwedes geeignete Material oder jedwede geeignete Kombination von Materialien kann für die Sperrschicht verwendet werden. Die Sperrschicht kann eine anorganische oder eine organische Verbindung oder beides enthalten. Die bevorzugte Sperrschicht umfasst ein Gemisch aus einem polymeren Material und einem nicht-polymeren Material, wie es im
US-Patent Nr. 7,968,146 , den PCT-Patentanmeldungen mit den Nummern PCT/US2007/023098 und PCT/US2009/042829 beschrieben ist, die hier unter Bezugnahme in ihrer Gesamtheit aufgenommen sind. Um als ein „Gemisch“ angesehen zu werden, sollten die genannten polymeren und nicht-polymeren Materialien, welche die Sperrschicht bilden, unter den gleichen Reaktionsbedingungen und/oder zur gleichen Zeit abgeschieden werden. Das Gewichtsverhältnis von polymerem zu nicht-polymerem Material kann im Bereich von 95:5 bis 5:95 liegen. Das polymere Material und das nicht-polymere Material können aus dem gleichen Ausgangsmaterial erzeugt werden. In einem Beispiel besteht das Gemisch aus einem polymeren Material und einem nicht-polymeren Material im Wesentlichen aus polymerem Silizium und anorganischem Silizium. - Vorrichtungen, die gemäß Ausführungsformen der Erfindung hergestellt werden, können in einer großen Vielzahl von Konsumgütern, einschließlich in Flachbildschirmen, Computermonitoren, medizinischen Monitoren, Fernsehgeräten, Werbetafeln, Leuchten bzw. Lampen für die Innen- oder Außenbeleuchtung und/oder Signalgebung, Head-up-Displays, vollständig transparenten Anzeigen bzw. Displays, flexiblen Anzeigen bzw. Displays, Laserdruckern, Telefonen, Mobiltelefonen, „personal digital assistants“ (PDAs), Laptop-Computer, Digitalkameras, Camcordern, Suchern, Mikroanzeigen bzw. -displays, Fahrzeugen, einer großen Wandfläche, einem Theater- oder Stadionbildschirm oder einem Schild eingebaut werden. Verschiedene Steuermechanismen können verwendet werden, um die Vorrichtungen, die gemäß der vorliegenden Erfindung hergestellt werden, zu steuern, einschließlich passiver Matrix und aktiver Matrix. Viele der Vorrichtungen sind für die Verwendung in einem für Menschen angenehmen Temperaturbereich, wie z.B. 18 °C bis 30°C, und mehr bevorzugt bei Raumtemperatur (20-25 °C), vorgesehen.
- Die Materialien und Strukturen, die hier beschrieben sind, können in Vorrichtungen verwendet werden, die von OLEDs verschieden sind. Zum Beispiel können die Materialien und Strukturen in anderen optoelektronischen Vorrichtungen, wie z.B. organischen Solarzellen und organischen Photodetektoren, verwendet werden. Allgemeiner können die Materialien und Strukturen in organischen Vorrichtungen, wie z.B. organischen Transistoren, verwendet werden.
- Die Begriffe Halo, Halogen, Alkyl, Cycloalkyl, Alkenyl, Alkinyl, Arylalkyl, heterocyclische Gruppe, Aryl, aromatische Gruppe und Heteroaryl sind in dem Fachgebiet bekannt und sind in
US 7,279,704 in den Spalten 31-32 definiert, die hier unter Bezugnahme aufgenommen sind. - Gemäß einer Ausführungsform werden heteroleptische Phenylbenzimidazol-Komplexe bereitgestellt, die in unerwarteter Weise als gelb und grün emittierende Materialien geeignet sind und die eine höhere Effizienz und niedrigere Steuerspannungen als erwartet aufweisen, wenn sie in OLED-Vorrichtungen einbezogen werden. Es wurde in unerwarteter Weise festgestellt, dass das Binden eines aromatischen Rings an den Benzimidazolteil von Formel I eine neue Klasse von gelb/grün phosphoreszierenden emittierenden Materialien erzeugt, die eine hohe Effizienz und eine lange Vorrichtungslebensdauer aufweisen. Dieses Ergebnis war vollkommen unerwartet, da das Hinzufügen von aromatischen Ringen entweder zu dem LUMO-Teil, bei dem es sich um den Benzimidazolteil der Formel I handelt, oder dem HOMO-Teil des Liganden die Konjugation erhöht, was typischerweise zu einer signifikanten Rotverschiebung der Emission führen würde.
- Die heteroleptischen Phenylbenzimidazol-Komplexe, die in vorteilhafter Weise in OLEDs verwendet werden können, weisen die Struktur der Formel I auf:
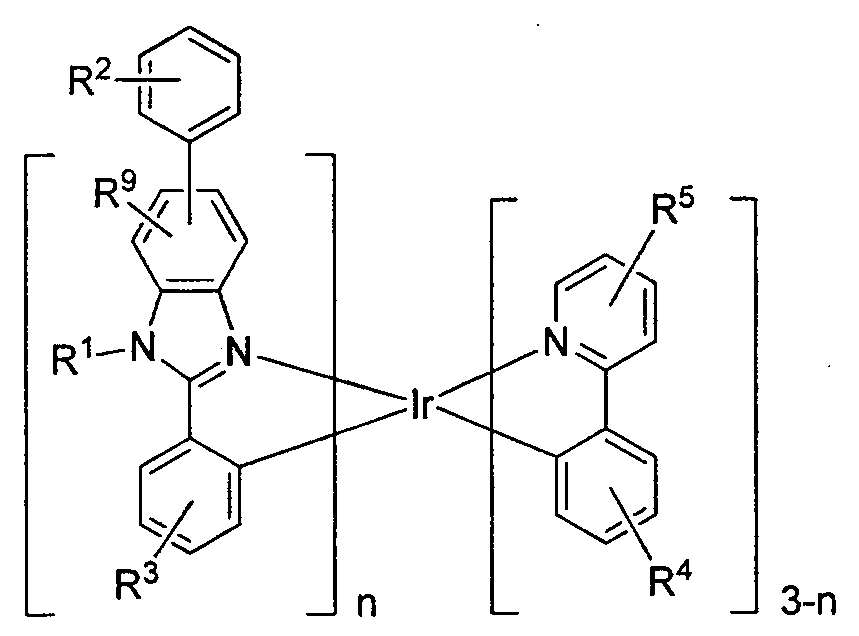
worin R2 Mono-, Di-, Tri-, Tetra-, Pentasubstitutionen oder keine Substitution darstellt,
worin R3, R4 und R5 jeweils Mono-, Di-, Tri-, Tetrasubstitutionen oder keine Substitution darstellen,
worin R9 Mono-, Di-, Trisubstitutionen oder keine Substitution darstellt,
worin R1, R2, R3, R4, R5 und R9 jeweils unabhängig aus der Gruppe bestehend aus Wasserstoff, Deuterium, Halogenid, Alkyl, Cycloalkyl, Heteroalkyl, Arylalkyl, Alkoxy, Aryloxy, Amino, Silyl, Alkenyl, Cycloalkenyl, Heteroalkenyl, Alkinyl, Aryl, Heteroaryl, Acyl, Carbonyl, Carbonsäuren, Ester, Nitril, Isonitril, Sulfanyl, Sulfinyl, Sulfonyl, Phosphino und Kombinationen davon, ausgewählt sind, worin n 1 oder 2 ist, worin keine Substitution bedeutet, dass der jeweilige Rest R2 bis R5 und R9 für alle verfügbaren Positionen Wasserstoff ist, und worin die Verbindung ausgewählt ist aus der Gruppe, bestehend aus:
- In einigen Ausführungsformen ist n = 1, während in einigen anderen Ausführungsformen n = 2 ist.
- Der gebundene aromatische Ring, der sich von dem Benzimidazolteil der Formel 1 erstreckt, kann an jedweder der vier verfügbaren Positionen des Benzolrings des Benzimidazolteils der Formel I gebunden sein. R9 kann an jedwede der verbleibenden Positionen des Benzolrings des Benzimidazolteils der Formel I gebunden sein.
- In einigen Ausführungsformen kann die Verbindung ein grün emittierendes Material sein. In einigen Ausführungsformen kann die Verbindung in einem Bereich von 520 bis 570 nm emittieren. In einigen Ausführungsformen kann die Verbindung in Bezug auf eine vergleichbare Verbindung ohne den gebundenen aromatischen Ring, der sich von dem Benzimidazolteil der Formel 1 erstreckt, blauverschoben sein.
- In einigen Ausführungsformen ist R1 aus der Gruppe, bestehend aus Alkyl, Cycloalkyl, Heteroalkyl, Arylalkyl, Alkoxy, Aryloxy, Alkenyl, Cycloalkenyl, Heteroalkenyl, Alkinyl, Aryl, Heteroaryl, Acyl, Carbonyl, Carbonsäuren, Ester, Nitril, Isonitril, Sulfanyl, Sulfinyl, Sulfonyl, Phosphino und Kombinationen davon, ausgewählt.
- In einigen Ausführungsformen sind R2, R3, R4, R5 und R9 jeweils unabhängig aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Alkyl, Cycloalkyl, Alkoxy, Aryloxy, Amino, Silyl, Aryl, Heteroaryl und Kombinationen davon, ausgewählt.
- In einigen Ausführungsformen kann R1 aus der Gruppe, bestehend aus Aryl, Heteroaryl, substituiertem Aryl und substituiertem Heteroaryl, ausgewählt sein.
- In einigen Ausführungsformen kann R1
- In einigen Ausführungsformen ist (a) R2 mono-, di-, tri-, tetra- oder pentasubstituiert, (b) R5 mono-, di-, tri- oder tetrasubstituiert oder (c) beides. In einigen Ausführungsformen ist R5 mono-, di-, tri- oder tetrasubstituiert. Wenn R5 mindestens monosubstituiert ist, kann R5 aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Alkyl, Cycloalkyl, Aryl und Kombinationen davon, ausgewählt sein.
- Wie hier verwendet bedeutet „substituiert“, dass ein Substituent, der von H verschieden ist, an den relevanten Kohlenstoff gebunden ist. Wenn R2 monosubstituiert ist, dann muss folglich ein R2 von H verschieden sein. Entsprechend müssen dann, wenn R3 disubstituiert ist, zwei von R3 von H verschieden sein. Entsprechend ist dann, wenn R2 unsubstituiert ist, R2 für alle verfügbaren Positionen Wasserstoff.
- In einigen Ausführungsformen ist R2 mono-, di-, tri-, tetra- oder pentasubstituiert. Wenn R2 mindestens monosubstituiert ist, kann R2 aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Alkyl, Cycloalkyl, Aryl und Kombinationen davon, ausgewählt sein.
- In einigen Ausführungsformen kann die Formel I die Strukturen der nachstehend gezeigten Formeln II aufweisen,

- Mindestens einer von R6 und R7 kann aus der Gruppe, bestehend aus Alkyl, Cycloalkyl, Aryl und Kombinationen davon, ausgewählt sein. Sowohl R6 als auch R7 kann aus der Gruppe, bestehend aus Alkyl, Cycloalkyl, Aryl und Kombinationen davon, ausgewählt sein.
- In einigen Ausführungsformen der Formel II ist R8 mindestens monosubstituiert und ist aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Alkyl, Cycloalkyl, Aryl und Kombinationen davon, ausgewählt.
- In einigen Ausführungsformen der Formel II ist (a) mindestens einer von R5 und R8 mono-, di-, tri- oder tetrasubstituiert, (b) R2 stellt Mono-, Di-, Tri-, Tetra-, Pentasubstitutionen dar oder (c) beides.
- In einigen Ausführungsformen sind die Reste R2, R3, R4, R5, R6, R7, R8 und R9 jeweils unabhängig aus der Gruppe bestehend aus Wasserstoff, Deuterium, Methyl, Ethyl, Propyl, 1-Methylethyl, Butyl, 1-Methylpropyl, 2-Methylpropyl, Pentyl, 1-Methylbutyl, 2-Methylbutyl, 3-Methylbutyl, 1,1-Dimethylpropyl, 1,2-Dimethylpropyl, 2,2-Dimethylpropyl, Cyclopentyl, Cyclohexyl, Phenyl, 2-Methylphenyl, 2,6-Dimethylphenyl, 2,4,6-Trimethylphenyl, 2-Isopropylphenyl, 2,6-Diisopropylphenyl, 2,4,6-Triisopropylphenyl, 2-Phenylphenyl, 2,6-Diphenylphenyl, 2,4,6-Triphenylphenyl und Kombinationen davon, ausgewählt.
und - Falls nichts anderes angegeben ist, sind alle Optionen für R1, R2, R3, R4, R5, R6, R7, R8 und R9, die auf die Formel I anwendbar sind, gleichermaßen auf die Formel II anwendbar, und alle Optionen für R1, R2, R3, R4, R5, R6, R7, R8 und R9, die auf die Formel II anwendbar sind, gleichermaßen auf die Formel I anwendbar. Entsprechend ist jedwede Kombination von R1, R2, R3, R4, R5, R6, R7, R8 und R9, die hier beschrieben ist, vorgesehen, und zwar ungeachtet davon, ob die Beschreibung eine Kombination von demjenigen ist, was als verschiedene Ausführungsformen bezeichnet ist. Dies gilt ungeachtet davon, wo die Optionen für R1, R2, R3, R4, R5, R6, R7, R8 und R9 angegeben sind.
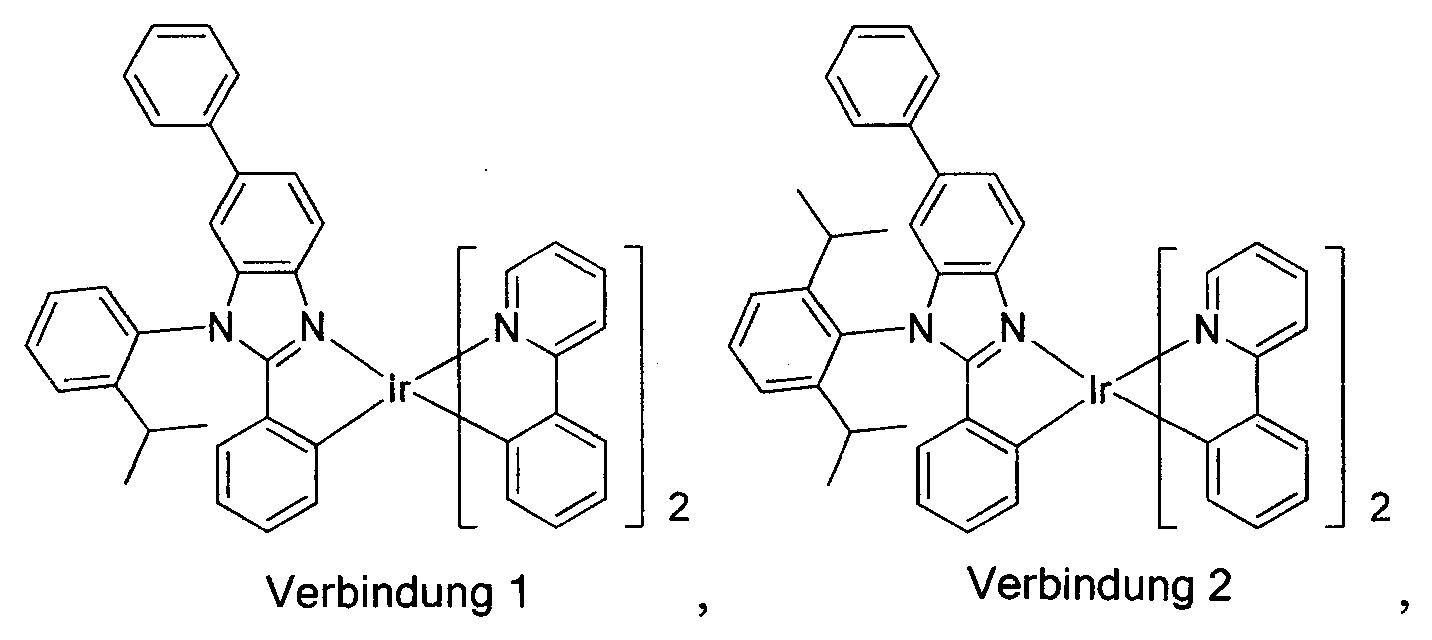
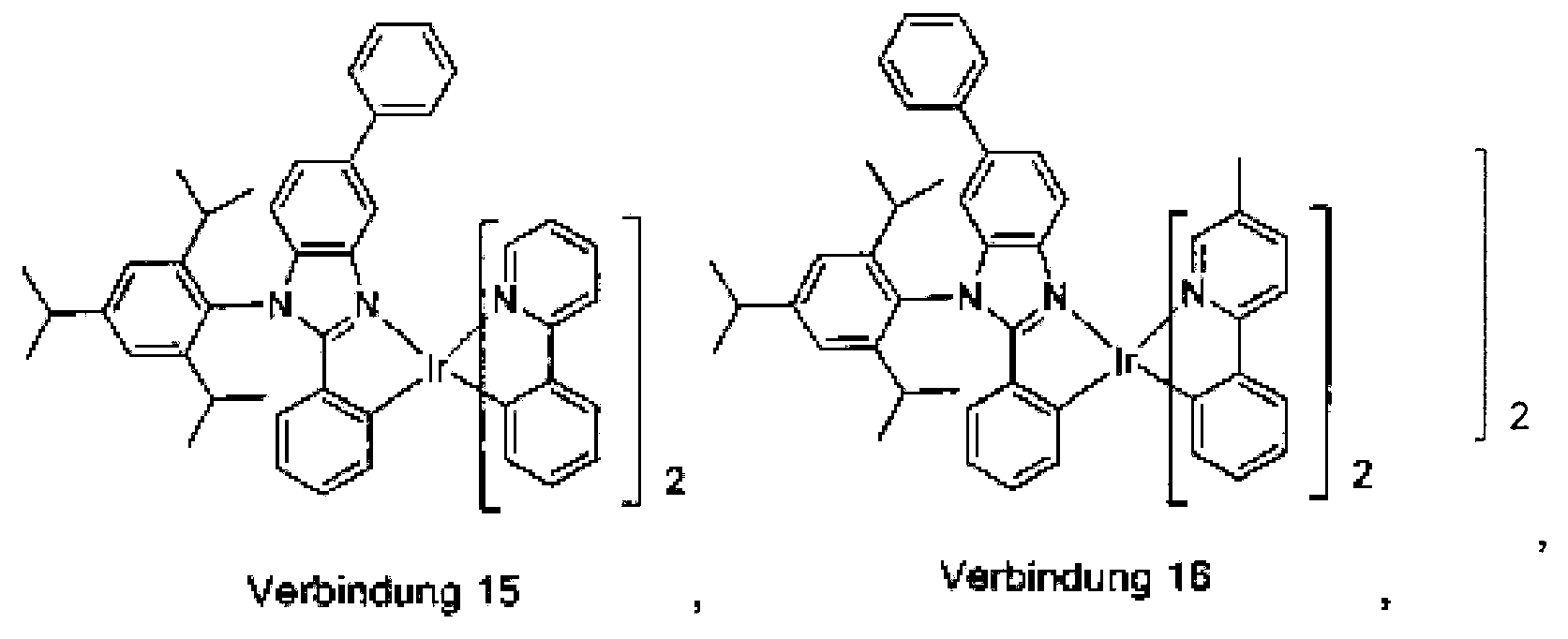
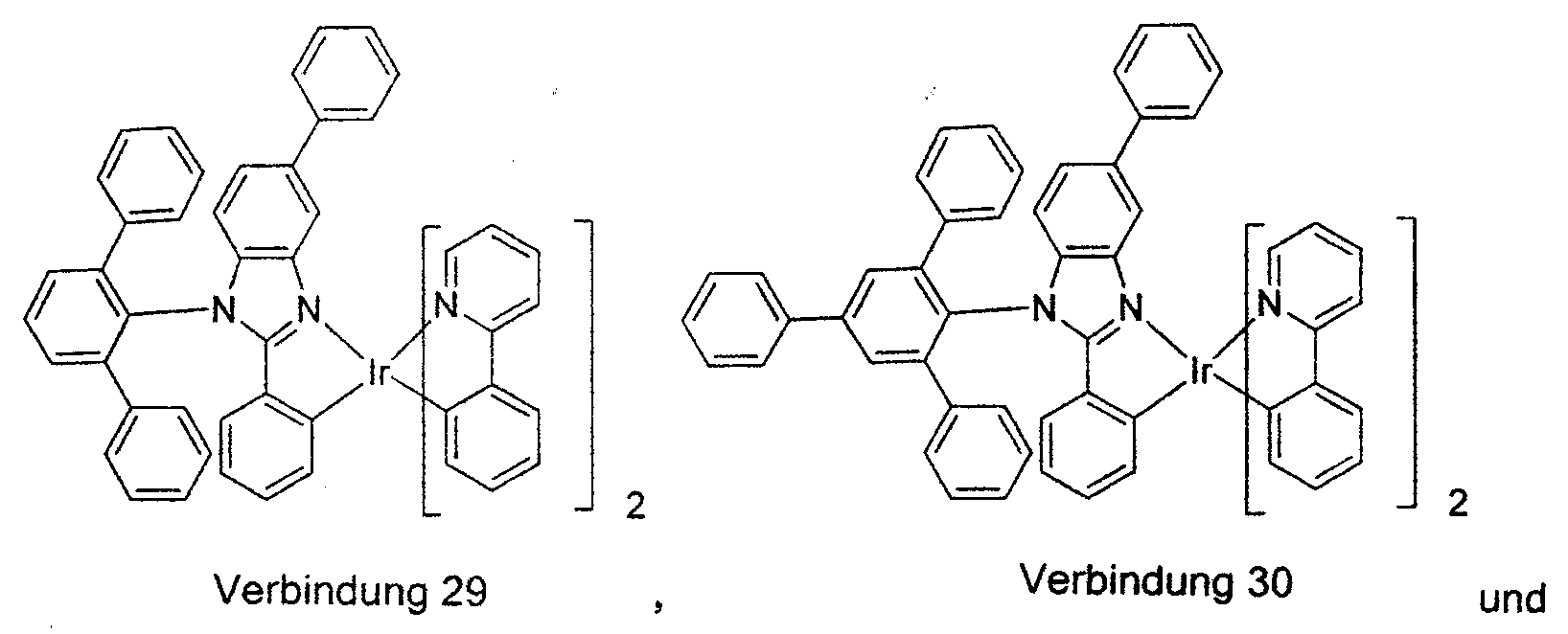
- In einigen Ausführungsformen kann der heteroleptische Phenylbenzimidazolkomplex aus der Gruppe, bestehend aus













- Gemäß eines anderen Aspekts der vorliegenden Offenbarung wird auch eine Vorrichtung bereitgestellt. Die Vorrichtung umfasst eine Anode, eine Kathode und eine organische Schicht, die zwischen der Anode und der Kathode angeordnet ist. Die organische Schicht kann eine Verbindung gemäß den Formeln I und II und deren Variationen umfassen, wie es beschrieben worden ist.
- Die Vorrichtung kann eines oder mehrere von einem Konsumgut bzw. Konsumenten- bzw. Verbraucherprodukt, einer organischen Licht-emittierenden Vorrichtung und einer Beleuchtungseinheit sein. Die organische Schicht kann eine emittierende Schicht sein und die Verbindung kann in einigen Ausführungsformen ein emittierendes Dotiermittel sein, während die Verbindung in anderen Ausführungsformen ein nicht-emittierendes Dotiermittel sein kann.
- Die organische Schicht kann auch eine Wirtsverbindung bzw. einen Wirt umfassen. Die Wirtsverbindung kann ein Triphenylen-enthaltendes Benzo-anelliertes Thiophen oder Benzo-anelliertes Furan sein. Jedweder Substituent in der Wirtsverbindung kann ein nichtanellierter Substituent sein, der unabhängig aus der Gruppe, bestehend aus CnH2n+1, OCnH2n+1, OAr1, N(CnH2n+1)2, N(Ar1)(Ar2), CH=CH-CnH2n+1, C≡CHCnH2n+1, Ar1, Ar1-Ar2, CnH2n-Ar1 oder keiner Substitution, ausgewählt ist. Bei den vorstehenden Substituenten kann n im Bereich von 1 bis 10 liegen und Ar1 und Ar2 können unabhängig aus der Gruppe, bestehend aus Benzol, Biphenyl, Naphthalin, Triphenylen, Carbazol und heteroaromatischen Analoga davon, ausgewählt sein.
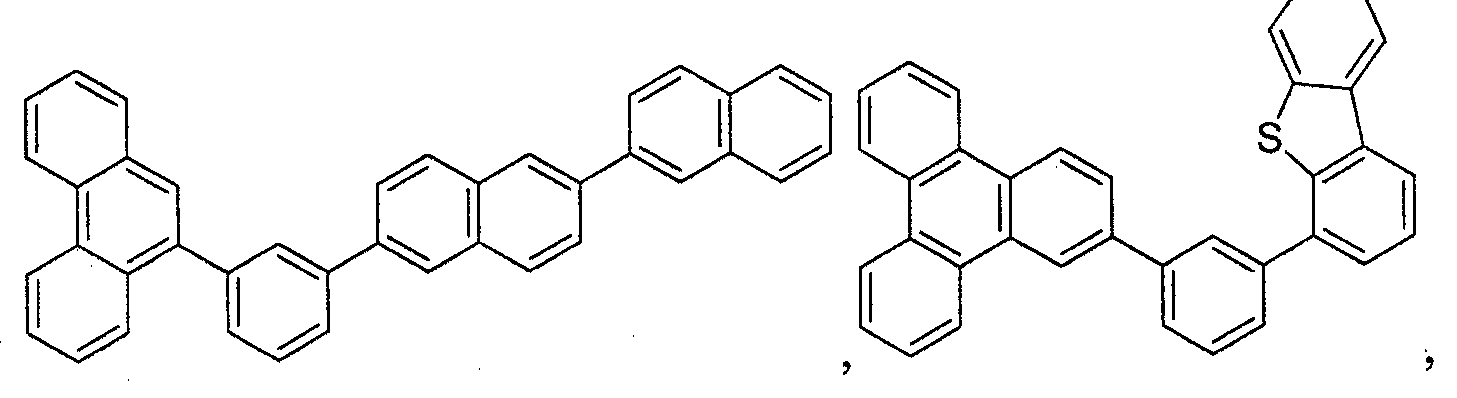
- Die Wirtsverbindung kann eine Verbindung sein, die aus der Gruppe, bestehend aus Carbazol, Dibenzothiophen, Dibenzofuran, Dibenzoselenophen, Azacarbazol, Azadibenzothiophen, Azadibenzofuran und Azadibenzoselenophen, ausgewählt ist. Die „Aza“-Bezeichnung in den vorstehend angegebenen Resten, d.h., Azadibenzofuran, Azadibenzothiophen, usw., bedeutet, dass eine oder mehrere der C-H-Gruppen in dem jeweiligen Rest durch ein Stickstoffatom ersetzt sein kann oder können, und beispielsweise umfasst ohne irgendeine Beschränkung Azatriphenylen sowohl Dibenzo[f,h]chinoxalin als auch Dibenzo[f,h]chinolin. Für den Fachmann sind in einfacher Weise andere Stickstoffanaloga der vorstehend beschriebenen Azaderivate offensichtlich und alle derartigen Analoga sollen von den hier angegebenen Begriffen umfasst sein. Die Wirtsverbindung kann einen Metallkomplex enthalten. Die Wirtsverbindung kann eine spezifische Verbindung sein, die aus der Gruppe, bestehend aus




- KOMBINATION MIT ANDEREN MATERIALIEN
- Die Materialien, die hier als nützlich für eine bestimmte Schicht in einer organischen Licht-emittierenden Vorrichtung beschrieben sind, können in Kombination mit vielen verschiedenen anderen Materialien, die in der Vorrichtung vorhanden sind, verwendet werden. Die hier offenbarten emittierenden Dotiermittel können zum Beispiel in Verbindung mit vielen verschiedenen Wirtsmaterialien, Transportschichten, Sperrschichten, Injektionsschichten, Elektroden und anderen Schichten, die vorhanden sein können, verwendet werden. Die beschriebenen oder nachstehend genannten Materialien sind nicht-beschränkende Beispiele von Materialien, die in einer Kombination mit den hier offenbarten Verbindungen nützlich sein können und der Fachmann kann einfach die Literatur konsultieren, um andere Materialien, die in einer Kombination nützlich sein können, zu bestimmen.
- HIL/HTL:
- Ein Lochinjektions/transportmaterial, das in der vorliegenden Erfindung verwendet wird, ist nicht besonders beschränkt und jedwede Verbindung kann verwendet werden, solange die Verbindung typischerweise als ein Lochinjektions/transportmaterial verwendet wird. Beispiele dieses Materials umfassen, sind aber nicht beschränkt auf: ein Phthalocyanin- oder Porphyrin-Derivat, ein aromatisches Aminderivat, ein Indolocarbazolderivat, ein Polymer, das Fluorkohlenwasserstoff enthält, ein Polymer mit Leitfähigkeitsdotiermitteln, ein leitendes Polymer, wie z.B. PEDOT/PSS, ein selbst-assemblierendes Monomer, das von Verbindungen wie z.B. Phosphonsäure- und Silanderivaten abgeleitet ist, ein Metalloxidderivat wie MoOx, eine p-Typ-halbleitende organische Verbindung, wie z.B. 1,4,5,8,9,12-Hexaazatriphenylen-hexacarbonitril, ein Metallkomplex und vernetzbare Verbindungen.
- Beispiele für aromatische Aminderivate, die in HIL oder HTL verwendet werden, umfassen, ohne darauf beschränkt zu sein, die folgenden allgemeinen Strukturen:

- Jeder von Ar1 bis Ar9 ist ausgewählt aus der Gruppe, bestehend aus aromatischen cyclischen Kohlenwasserstoffverbindungen, wie z.B. Benzol, Biphenyl, Triphenyl, Triphenylen, Naphthalin, Anthracen, Phenalen, Phenanthren, Fluoren, Pyren, Chrysen, Perylen, Azulen, der Gruppe, bestehend aus aromatischen heterocyclischen Verbindungen, wie z.B. Dibenzothiophen, Dibenzofuran, Dibenzoselenophen, Furan, Thiophen, Benzofuran, Benzothiophen, Benzoselenophen, Carbazol, Indolocarbazol, Pyridylindol, Pyrrolodipyridin, Pyrazol, Imidazol, Triazol, Oxazol, Thiazol, Oxadiazol, Oxatriazol, Dioxazol, Thiadiazol, Pyridin, Pyridazin, Pyrimidin, Pyrazin, Triazin, Oxazin, Oxathiazin, Oxadiazin, Indol, Benzimidazol, Indazol, Indoxazin, Benzoxazol, Benzisoxazol, Benzothiazol, Chinolin, Isochinolin, Cinnolin, Chinazolin, Chinoxalin, Naphthyridin, Phthalazin, Pteridin, Xanthen, Acridin, Phenazin, Phenothiazin, Phenoxazin, Benzofuropyridin, Furodipyridin, Benzothienopyridin, Thienodipyridin, Benzoselenophenopyridin, und Selenophenodipyridin, und der Gruppe, bestehend aus 2 bis 10 cyclischen Struktureinheiten, die Gruppen von demselben Typ oder verschiedenen Typen sind, die ausgewählt sind aus der aromatischen cyclischen Kohlenwasserstoffgruppe und der aromatischen heterocyclischen Gruppe und direkt oder über mindestens eines von einem Sauerstoffatom, Stickstoffatom, Schwefelatom, Siliziumatom, Phosphoratom, Boratom, einer Kettenstruktureinheit und einer aliphatischen cyclischen Gruppe aneinander gebunden sind. Jedes Ar ist ferner durch einen Substituenten substituiert, der aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Halogen, Alkyl, Cycloalkyl, Heteroalkyl, Arylalkyl, Alkoxy, Aryloxy, Amino, Silyl, Alkenyl, Cycloalkenyl, Heteroalkenyl, Alkinyl, Aryl, Heteroaryl, Acyl, Carbonyl, Carbonsäuren, Ester, Nitril, Isonitril, Sulfanyl, Sulfinyl, Sulfonyl, Phosphino, und Kombinationen davon, ausgewählt ist.
- In einem Aspekt sind Ar1 bis Ar9 unabhängig ausgewählt aus der Gruppe, bestehend aus:


- k ist eine ganze Zahl von 1 bis 20, X101 bis X108 sind C (einschließlich CH) oder N, Z101 ist NAr1, O oder S, Ar1 ist dieselbe Gruppe, wie es vorstehend definiert worden ist.
- Beispiele für Metallkomplexe, die in HIL oder HTL verwendet werden, umfassen, sind aber nicht beschränkt auf, die folgende allgemeine Formel:

- Met ist ein Metall, das ein Atomgewicht von mehr als 40 aufweisen kann, (Y101 - Y102) ist ein zweizähniger Ligand, Y101 und Y102 sind unabhängig ausgewählt aus C, N, O, P, und S, L101 ist ein ergänzender Ligand, k' ist eine ganze Zahl von 1 bis zur maximalen Anzahl von Liganden, die an das Metall gebunden sein können, und k' + k" ist die maximale Anzahl von Liganden, die an das Metall gebunden sein können.
- In einem Aspekt ist (Y101 - Y102) ein 2-Phenylpyridinderivat.
- In einem anderen Aspekt ist (Y101 - Y102) ein Carbenligand.
- In einem anderen Aspekt ist Met aus Ir, Pt, Os und Zn ausgewählt.
- In einem weiteren Aspekt weist der Metallkomplex ein kleinstes Oxidationspotential in Lösung gegen ein Fc+/Fc-Paar von weniger als etwa 0,6 V auf.
- Wirtsverbindung bzw. Wirtsmaterial:
- Die Licht-emittierende Schicht der organischen EL-Vorrichtung der vorliegenden Erfindung enthält vorzugsweise mindestens einen Metallkomplex als Licht-emittierendes Material, und sie kann ein Wirtsmaterial enthalten, bei dem der Metallkomplex als Dotiermittelmaterial verwendet wird. Beispiele für das Wirtsmaterial sind nicht speziell beschränkt und jedwede Metallkomplexe oder organischen Verbindungen können verwendet werden, solange die Triplettenergie des Wirts größer ist als diejenige des Dotiermittels. Während die nachstehende Tabelle die Wirtsmaterialien kategorisiert, die für die Vorrichtungen, welche verschiedene Farben emittieren, bevorzugt sind, kann jedwedes Wirtsmaterial mit jedwedem Dotiermittel verwendet werden, solange die Triplettkriterien erfüllt sind.
- Beispiele für Metallkomplexe, die als Wirtsverbindung bzw. Wirtsmaterial verwendet werden, weisen vorzugsweise die folgende allgemeine Formel auf:

- Met ist ein Metall, (Y103 - Y104) ist ein zweizähniger Ligand, Y103 und Y104 sind unabhängig ausgewählt aus C, N, O, P, und S, L101 ist ein weiterer Ligand, k' ist eine ganze Zahl von 1 bis zur maximalen Anzahl von Liganden, die an das Metall gebunden sein können, und k' + k" ist die maximale Anzahl von Liganden, die an das Metall gebunden sein können.
- In einem Aspekt sind die Metallkomplexe:

- (O-N) ist ein zweizähniger Ligand, der Metall aufweist, das an die Atome O und N koordiniert ist.
- In einem anderen Aspekt ist Met aus Ir und Pt ausgewählt.
- In einem weiteren Aspekt ist (Y103 - Y104) ein Carbenligand.
- Beispiele für organische Verbindungen, die als Wirtsverbindung verwendet werden, sind ausgewählt aus der Gruppe, bestehend aus aromatischen cyclischen Kohlenwasserstoffverbindungen, wie z.B. Benzol, Biphenyl, Triphenyl, Triphenylen, Naphthalin, Anthracen, Phenalen, Phenanthren, Fluoren, Pyren, Chrysen, Perylen, Azulen, der Gruppe, bestehend aus aromatischen heterocyclischen Verbindungen, wie z.B. Dibenzothiophen, Dibenzofuran, Dibenzoselenophen, Furan, Thiophen, Benzofuran, Benzothiophen, Benzoselenophen, Carbazol, Indolocarbazol, Pyridylindol, Pyrrolodipyridin, Pyrazol, Imidazol, Triazol, Oxazol, Thiazol, Oxadiazol, Oxatriazol, Dioxazol, Thiadiazol, Pyridin, Pyridazin, Pyrimidin, Pyrazin, Triazin, Oxazin, Oxathiazin, Oxadiazin, Indol, Benzimidazol, Indazol, Indoxazin, Benzoxazol, Benzisoxazol, Benzothiazol, Chinolin, Isochinolin, Cinnolin, Chinazolin, Chinoxalin, Naphthyridin, Phthalazin, Pteridin, Xanthen, Acridin, Phenazin, Phenothiazin, Phenoxazin, Benzofuropyridin, Furodipyridin, Benzothienopyridin, Thienodipyridin, Benzoselenophenopyridin, und Selenophenodipyridin, und der Gruppe, bestehend aus 2 bis 10 cyclischen Struktureinheiten, die Gruppen von demselben Typ oder verschiedenen Typen sind, die ausgewählt sind aus der aromatischen cyclischen Kohlenwasserstoffgruppe und der aromatischen heterocyclischen Gruppe und direkt oder über mindestens eines von einem Sauerstoffatom, Stickstoffatom, Schwefelatom, Siliziumatom, Phosphoratom, Boratom, einer Kettenstruktureinheit und einer aliphatischen cyclischen Gruppe aneinander gebunden sind. Jede Gruppe ist weiter durch einen Substituenten substituiert, der aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Halogen, Alkyl, Cycloalkyl, Heteroalkyl, Arylalkyl, Alkoxy, Aryloxy, Amino, Silyl, Alkenyl, Cycloalkenyl, Heteroalkenyl, Alkinyl, Aryl, Heteroaryl, Acyl, Carbonyl, Carbonsäuren, Ester, Nitril, Isonitril, Sulfanyl, Sulfinyl, Sulfonyl, Phosphino und Kombinationen davon, ausgewählt ist.
- In einem Aspekt enthält die Wirtsverbindung mindestens eine der folgenden Gruppen in dem Molekül:



- R101 bis R107 sind unabhängig ausgewählt aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Halogen, Alkyl, Cycloalkyl, Heteroalkyl, Arylalkyl, Alkoxy, Aryloxy, Amino, Silyl, Alkenyl, Cycloalkenyl, Heteroalkenyl, Alkinyl, Aryl, Heteroaryl, Acyl, Carbonyl, Carbonsäuren, Ester, Nitril, Isonitril, Sulfanyl, Sulfinyl, Sulfonyl, Phosphino und Kombinationen davon, wobei Aryl oder Heteroaryl entsprechend wie die vorstehend genannten Ar definiert ist.
- k ist eine ganze Zahl von 0 bis 20 oder 1 bis 20, k'" ist eine ganze Zahl von 0 bis 20.
- X101 bis X108 sind aus C (einschließlich CH) oder N ausgewählt.
- Z101 und Z102 sind aus NR101, O oder S ausgewählt.
- HBL:
- Eine Lochsperrschicht (HBL) kann verwendet werden, um die Anzahl von Löchern und/oder Excitonen, welche die emittierende Schicht verlassen, zu reduzieren. Die Anwesenheit einer solchen Sperrschicht in einer Vorrichtung kann verglichen mit einer ähnlichen Vorrichtung, der eine Sperrschicht fehlt, zu wesentlich höheren Effizienzen führen. Eine Sperrschicht kann auch verwendet werden, um eine Emission auf einen gewünschten Bereich einer OLED zu beschränken.
- In einem Aspekt enthält die in der HBL verwendete Verbindung dasselbe Molekül oder dieselben funktionellen Gruppen, die als Wirtsverbindung, wie vorstehend beschrieben, verwendet werden.
- In einem anderen Aspekt enthält die in der HBL verwendete Verbindung mindestens eine der folgenden Gruppen in dem Molekül:
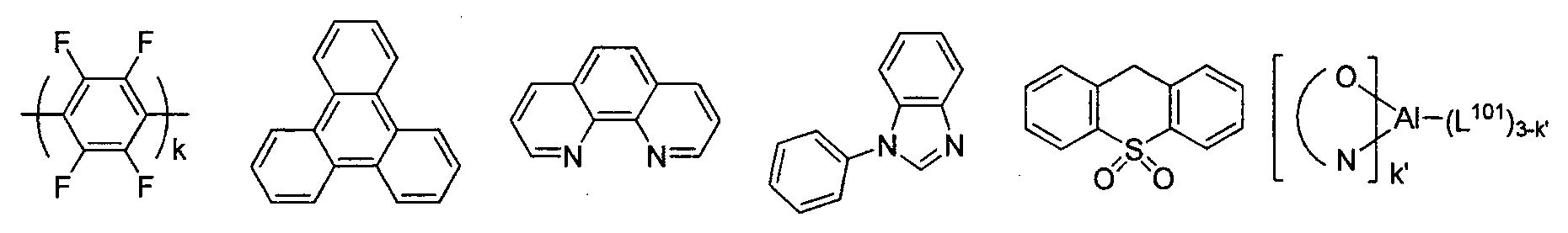
- k ist eine ganze Zahl von 0 bis 20, L101 ist ein weiterer Ligand, k' ist eine ganze Zahl von 1 bis 3.
- ETL:
- Die Elektronentransportschicht (ETL) kann ein Material umfassen, das Elektronen transportieren kann. Die Elektronentransportschicht kann intrinsisch (undotiert) oder dotiert sein. Das Dotieren kann verwendet werden, um die Leitfähigkeit zu erhöhen. Beispiele für das ETL-Material sind nicht besonders beschränkt und jedwede Metallkomplexe oder organischen Verbindungen können verwendet werden, solange sie typischerweise verwendet werden, um Elektronen zu transportieren.
- In einem Aspekt enthält die in der ETL verwendete Verbindung mindestens eine der folgenden Gruppen im Molekül:


- R101 ist ausgewählt aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Halogen, Alkyl, Cycloalkyl, Heteroalkyl, Arylalkyl, Alkoxy, Aryloxy, Amino, Silyl, Alkenyl, Cycloalkenyl, Heteroalkenyl, Alkinyl, Aryl, Heteroaryl, Acyl, Carbonyl, Carbonsäuren, Ester, Nitril, Isonitril, Sulfanyl, Sulfinyl, Sulfonyl, Phosphino und Kombinationen davon, wobei Aryl oder Heteroaryl entsprechend wie die vorstehend genannten Ar definiert ist.
- Ar1 bis Ar3 sind entsprechend wie die vorstehend genannten Ar definiert.
- k ist eine ganze Zahl von 1 bis 20.
- X101 bis X108 sind aus C (einschließlich CH) oder N ausgewählt.
- In einem anderen Aspekt enthalten die Metallkomplexe, die in der ETL verwendet werden, ohne darauf beschränkt zu sein, die folgenden allgemeinen Formeln:

- (O-N) oder (N-N) ist ein zweizähniger Ligand, der Metall aufweist, das an die Atome O, N oder N, N koordiniert ist, L101 ist ein weiterer Ligand, k' ist eine ganze Zahl von 1 bis zur maximalen Anzahl von Liganden, die an das Metall gebunden sein können.
- In jedweder der vorstehend genannten Verbindungen, die in jeder Schicht der OLED-Vorrichtung verwendet werden, können die Wasserstoffatome teilweise oder vollständig deuteriert sein. Daher umfasst jedweder spezifisch angegebene Substituent, wie z.B., ohne darauf beschränkt zu sein, Methyl, Phenyl, Pyridyl, usw., undeuterierte, teilweise deuterierte und vollständig deuterierte Versionen davon. Entsprechend umfassen Klassen von Substituenten wie z.B., ohne darauf beschränkt zu sein, Alkyl, Aryl, Cycloalkyl, Heteroaryl, usw., undeuterierte, teilweise deuterierte und vollständig deuterierte Versionen davon.
- Zusätzlich zu und/oder in Kombination mit den Materialien, die hier offenbart sind, können viele Lochinjektionsmaterialien, Lochtransportmaterialien, Wirtsmaterialien, Dotiermittelmaterialien, Exciton/Lochsperrschichtmaterialien, Elektronentransport- und Elektroneninjektionsmaterialien in einer OLED verwendet werden. Nicht-beschränkende Beispiele der Materialien, die in einer OLED in einer Kombination mit Materialien, die hier offenbart sind, verwendet werden können, sind in der nachstehenden Tabelle 1 angegeben. In der Tabelle 1 sind nicht-beschränkende Klassen von Materialien, nicht-beschränkende Beispiele von Verbindungen für jede Klasse und Dokumente, welche die Materialien offenbaren, angegeben. Tabelle 1
MATERIAL MATERIALBEISPIELE VERÖFFENTLICHUNGEN Lochinjektionsmaterialien Phthalocyanin- und Porphyrinverbindungen 
Appl. Phys. Lett. 69, 2160 (1996) „Starburst“-Triarylamine 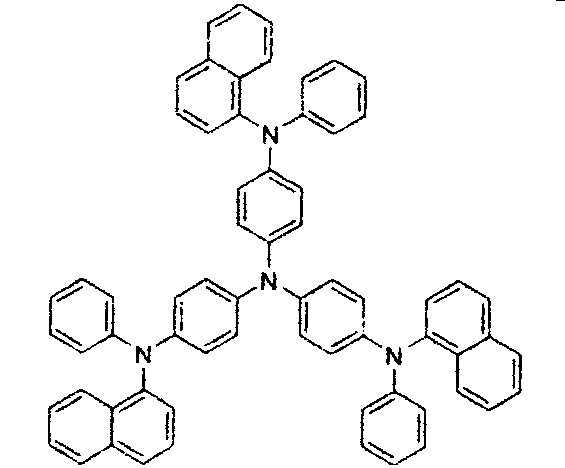
J. Lumin. 72-74, 985 (1997) CFx-Fluorkohlenwasserstoff-polymer 
Appl. Phys. Lett. 78, 673 (2001) Leitende Polymere (z.B. PEDOT:PSS, Polyanilin, Polythiophen) 
Synth. Met. 87, 171 (1997) WO2007002683 Phosphonsäure- und Silan-SAMs 
US20030162053 Triarylamin- oder Polythiophen-Polymere mit leitenden Dotiermitteln 
EP1725079A1 Organische Verbindungen mit leitenden anorganischen Verbindungen, wie z.B. Molybdän- und Wolframoxiden 
US20050123751 SID Symposium Digest, 37, 923 (2006)WO2009018009 n-Typ-halbleitende organische Komplexe 
US20020158242 Organometallische Metallkomplexe 
US20060240279 Vernetzbare Verbindungen 
US20080220265 Polymere und Copolymere auf Polythiophenbasis 
WO 2011075644 EP2350216 Lochtransportmaterialien Triarylamine (z.B. TPD, α-NPD) 
Appl. Phys. Lett. 51, 913 (1987) 
US5061569 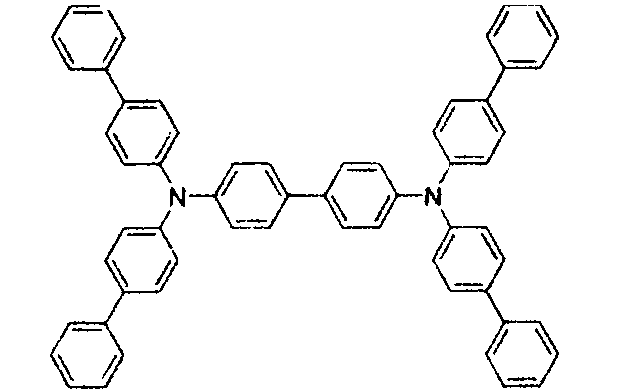
EP650955 
J. Mater. Chem. 3, 319 (1993) 
Appl. Phys. Lett. 90, 183503 (2007) 
Appl. Phys. Lett. 90, 183503 (2007) Triarylamin auf Spirofluorenkern 
Synth. Met. 91, 209 (1997) Arylamincarbazolverbindungen 
Adv. Mater. 6, 677 (1994), US20080124572 Triarylamin mit (Di)benzothiophen/(Di)benzofuran 
US20070278938 ,US20080106190 US20110163302 Indolocarbazole 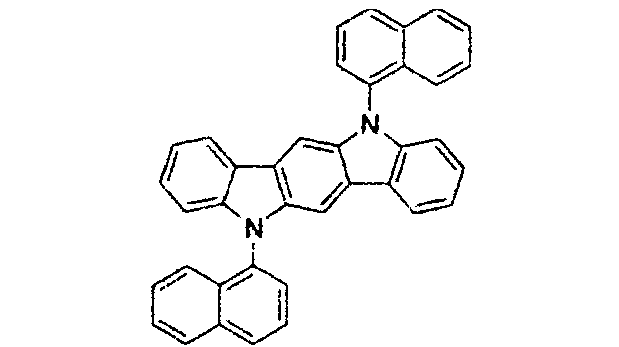
Synth. Met. 111, 421 (2000) Isoindolverbindungen 
Chem. Mater. 15, 3 Σ 48 (2003) Metall-Carben-Komplexe 
US20080018221 Phosphoreszierende OLED-Wirtsmaterialien Rote Wirtsmaterialien Arylcarbazole 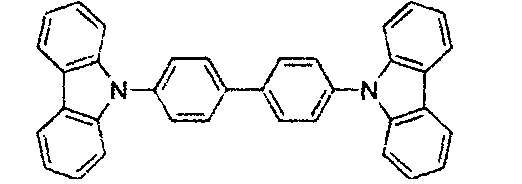
Appl. Phys. Lett. 78, 1622 (2001) Metall-8-hydroxychinolate (z.B. Alq3, BAlq) 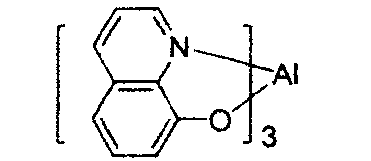
Nature 395, 151 (1998) 
US20060202194 
WO2005014551 
WO2006072002 Metall-Phenoxybenzothiazolverbindungen 
Appl. Phys. Lett. 90, 123509 (2007) Konjugierte Oligomere und Polymere (z.B. Polyfluoren) 
Org. Electron. 1, 15 (2000) Aromatische anellierte Ringe 
WO2009066779 ,WO2009066778 ,WO2009063833 ,US20090045731 ,US20090045730 ,WO2009008311 ,US20090008605 ,US20090009065 Zinkkomplexe 
WO2010056066 Verbindungen auf Chrysenbasis 
WO2011086863 Grüne Wirtsmaterialien Arylcarbazole 
Appl. Phys. Lett. 78, 1622 (2001) 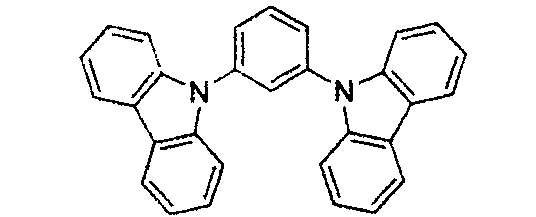
US20030175553 
WO2001039234 Aryltriphenylenverbindungen 
US20060280965 
US20060280965 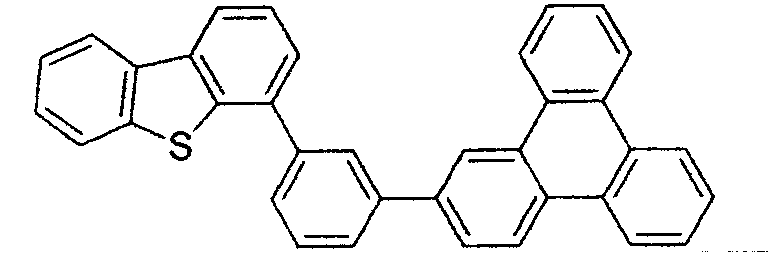
WO2009021126 Polyanellierte Heteroarylverbindungen 
US20090309488 US20090302743 US20100012931 Moleküle des Donor-Akzeptor-Typs 
WO2008056746 
WO2010107244 Azacarbazol/DBT/DBF 
JP2008074939 
US20100187984 Polymere (z.B. PVK) 
Appl. Phys. Lett. 77, 2280 (2000) Spirofluorenverbindungen 
WO2004093207 Metall-Phenoxybenzooxazolverbindungen 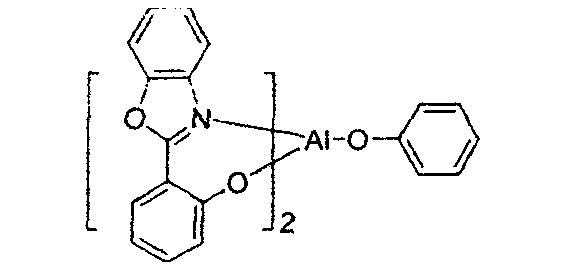
WO2005089025 
WO2006132173 
JP200511610 Spirofluoren-Carbazol-Verbindungen 
JP2007254297 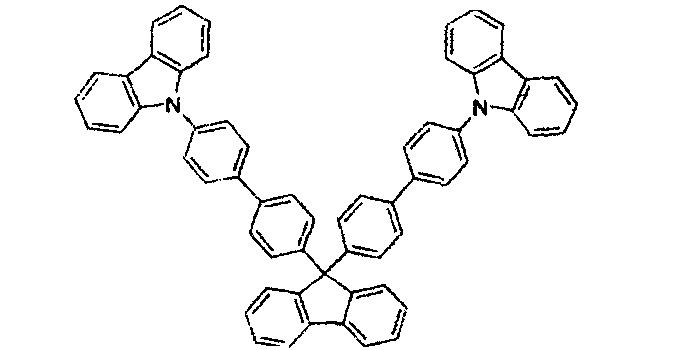
JP2007254297 Indolocarbazole 
WO2007063796 
WO2007063754 Elektronenmangel-Heterocyclen mit 5-gliedrigem Ring (z.B. Triazol, Oxadiazol) 
J. Appl. Phys. 90, 5048 (2001) 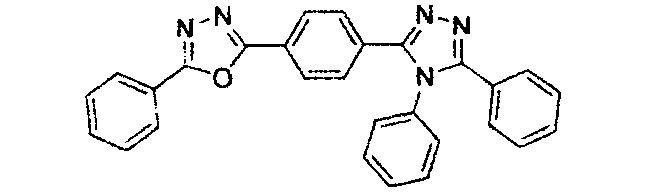
WO2004107822 Tetraphenylenkomplexe 
US20050112407 Metall-Phenoxypyridinverbindungen 
WO2005030900 Metallkoordinationskomplexe (z.B. Zn, AI mit N^N-Liganden) 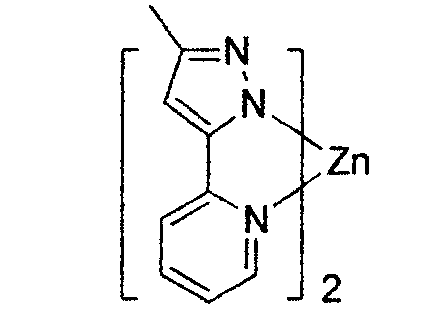
US20040137268 ,US20040137267 Blaue Wirtsverbindungen Arylcarbazole 
Appl. Phys. Lett, 82, 2422 (2003) 
US20070190359 Dibenzothiophen/Dibenzofuran-Carbazolverbindungen 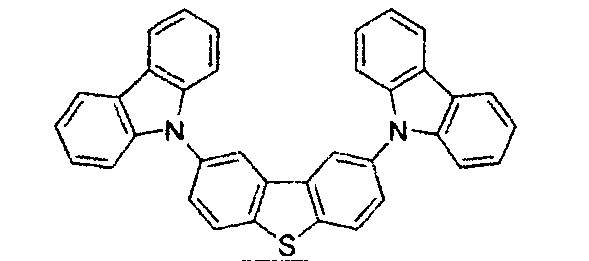
WO2006114966 ,US20090167162 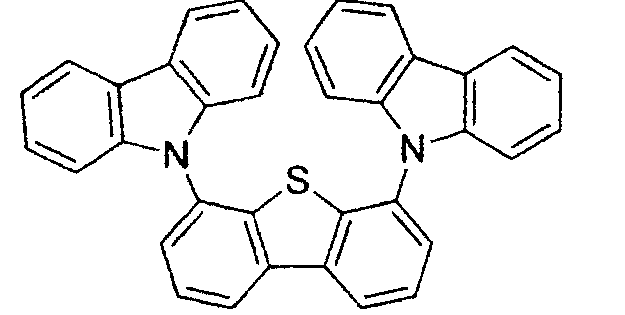
US20090167162 
WO2009086028 
US20090030202 ,US20090017330 
US20100084966 Siliziumarylverbindungen 
US20050238919 
WO2009003898 Silizium/Germanium-Arylverbindungen 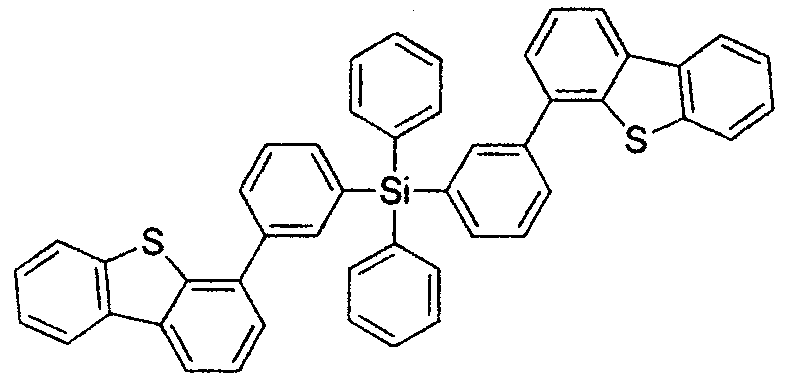
EP2034538A Arylbenzoylester 
WO2006100298 Durch nicht-konjugierte Gruppen verknüpftes Carbazol 
US20040115476 Azacarbazole 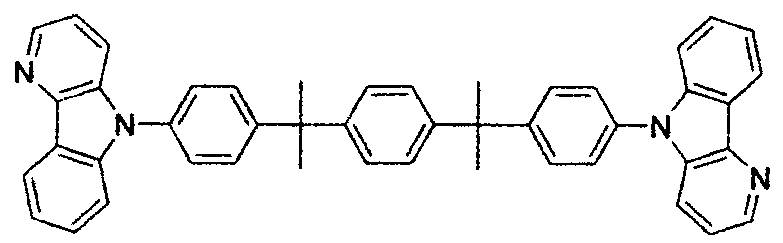
US20060121308 Organometallische Metallkomplexe mit hohem Triplett 
US7154114 Phosphoreszierende Dotiermittel Rote Dotiermittel Schwermetallporphyrine (z.B. PtOEP) 
Nature 395, 151 (1998) Organometallische Iridium(III)-Komplexe 
Appl. Phys. Lett. 78, 1622 (2001) 
US2006835469 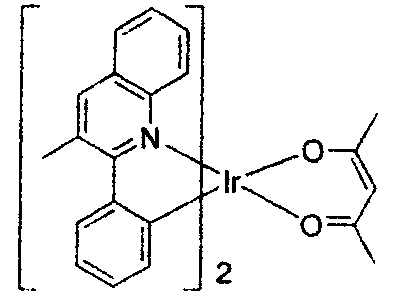
US2006835469 
US20060202194 
US20060202194 
US20070087321 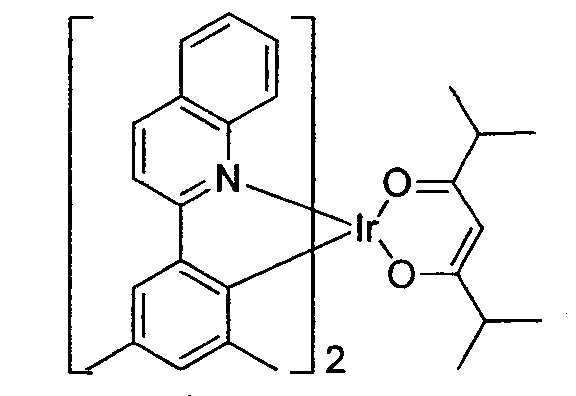
US20080261076 US20100090591 
US20070087321 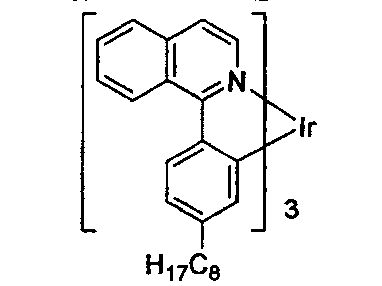
Adv. Mater. 19, 739 (2007) 
WO2009100991 
WO2008101842 
US7232618 Organometallische Platin(II)-Komplexe 
WO2003040257 
US20070103060 Osmium(III)-Komplexe 
Chem. Mater. 17,3532 (2005) Ruthenium(II)-Komplexe 
Adv. Mater. 17, 1059 (2005) Rhenium(I)-, (II)- und (III)-Komplexe 
US20050244673 Grüne Dotiermittel Organometallische Iridium(III)-Komplexe 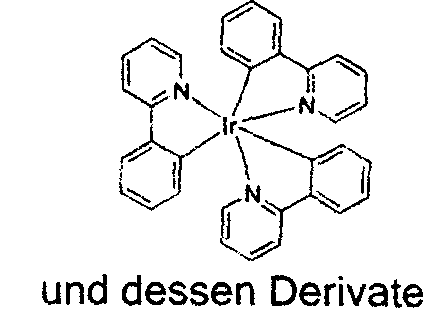
Inorg. Chem. 40, 1704 (2001) 
US20020034656 
US7332232 
US20090108737 
WO2010028151 
EP1841834B 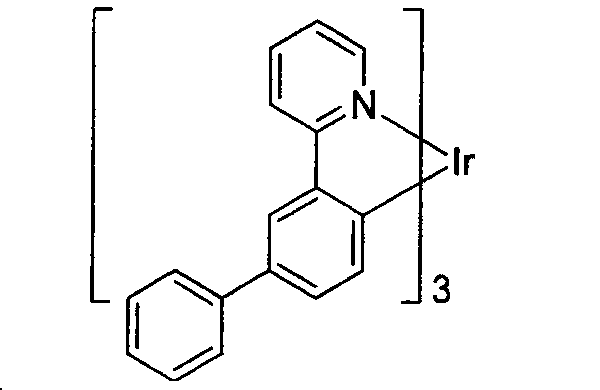
US20060127696 
US20090039776 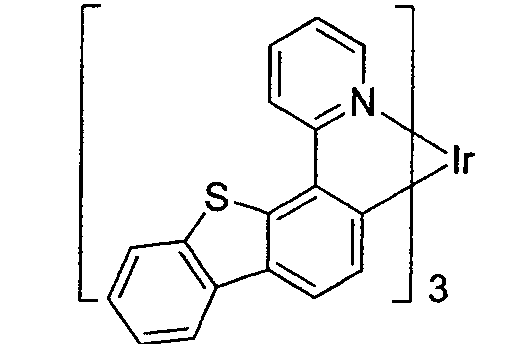
US6921915 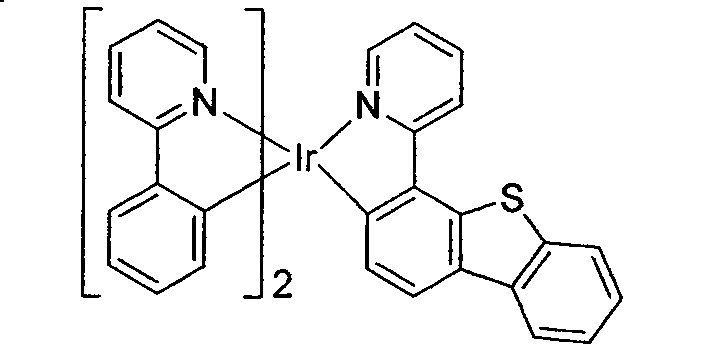
US20100244004 
US6687266 
Chem. Mater. 16, 2480 (2004) 
US20070190359 
US 20060008670 JP2007123392 
WO2010086089 ,WO2011044988 
Adv. Mater. 16, 2003 (2004) 
Angew. Chem. Int. Ed. 2006, 45, 7800 
WO2009050290 
US20090165846 
US20080015355 
US20010015432 
US20100295032 Monomer für polymere organometallische Metallverbindungen 
US7250226 ,US7396598 Organometallische Pt(II)-Komplexe, einschließlich mehrzähnige Liganden 
Appl. Phys. Lett. 86, 153505 (2005) 
Appl. Phys. Lett. 86, 153505 (2005) 
Chem. Lett. 34, 592 (2005) 
WO2002015645 
US20060263635 
US20060182992 US20070103060 Cu-Komplexe 
WO2009000673 
US20070111026 Goldkomplexe 
Chem. Commun. 2906 (2005) Rhenium(III)-Komplexe 
Inorg. Chem. 42, 1248 (2003) Osmium(II)-Komplexe 
US7279704 Deuterierte organometallische Komplexe 
US20030138657 Organometallische Komplexe mit zwei oder mehr Metallzentren 
US20030152802 
US7090928 Blaue Dotiermittel Organometallische Iridium(III)-Komplexe 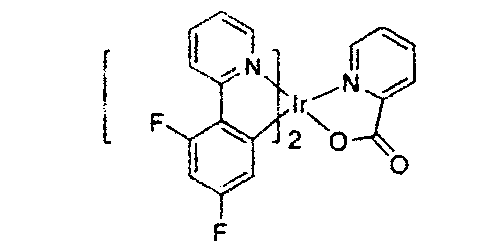
WO2002002714 
WO2006009024 
US20060251923 US20110057559 US20110204333 
US7393599 ,WO2006056418 ,US20050260441 , WO2005019373
US7534505 
WO2011051404 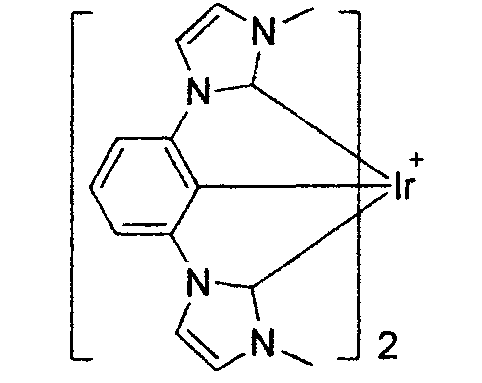
US7445855 
US20070190359 ,US20080297033 US20100148663 
US7338722 
US20020134984 
Angew. Chem. Int. Ed. 47, 1 (2008) 
Chem. Mater. 18, 5119 (2006) 
Inorg. Chem. 46, 4308 (2007) 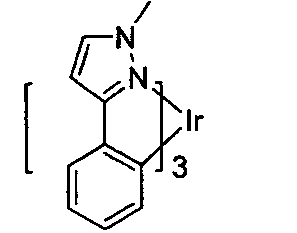
WO2005123873 
WO2005123873 
WO2007004380 
WO2006082742 Osmium(II)-Komplexe 
US7279704 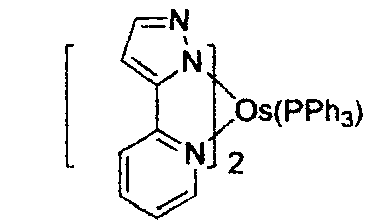
Organometallics 23, 3745 (2004) Goldkomplexe 
Appl. Phys. Lett.74,1361 (1999) Platin(II)-Komplexe 
WO2006098120 ,WO2006103874 Vierzähnige Pt-Komplexe mit mindestens einer Metall-Carben-Bindung 
US7655323 Exciton/Lochsperrschichtmaterialien Bathocuprinverbindungen (z.B. BCP, BPhen) 
Appl. Phys. Lett. 75, 4 (1999) 
Appl. Phys. Lett. 79, 449 (2001) Metall-8-hydroxychino-late (z.B. BAlq) 
Appl. Phys. Lett. 81, 162 (2002) Elektronenmangel-Heterocyclen mit 5-gliedrigem Ring, wie z.B. Triazol, Oxadiazol, Imidazol, Benzoimidazol 
Appl. Phys. Lett. 81, 162 (2002) Triphenylenverbindungen 
US20050025993 Fluorierte aromatische Verbindungen 
Appl. Phys. Lett. 79, 156 (2001) Phenothiazin-S-oxid 
WO2008132085 Silylierte fünfgliedrige Stickstoff-, Sauerstoff-, Schwefel- oder Phosphor-Dibenzoheterocyclen 
WO2010079051 Azacarbazole 
US20060121308 Elektronentransportmaterialien Anthracenbenzoimidazol-Verbindungen 
WO2003060956 
US20090179554 Azatriphenylenderivate 
US20090115316 Anthracen-Benzothiazolverbindungen 
Appl. Phys. Lett. 89, 063504 (2006) Metall-8-hydroxychino-late (z.B. Alq3, Zrq4) 
Appl. Phys. Lett. 51, 913 (1987) US7230107 Metallhydroxybenzochinolate 
Chem. Lett. 5, 905 (1993) Bathocuprinverbindungen, wie z.B. BCP, BPhen, usw. 
Appl. Phys. Lett. 91, 263503 (2007) 
Appl. Phys. Lett. 79, 449(2001) Elektronenmangel-Heterocyclen mit 5-gliedrigem Ring, (z.B. Triazol, Oxadiazol, Imidazol, Benzoimidazol) 
Appl. Phys. Lett. 74, 865 (1999) 
Appl. Phys. Lett. 55, 1489 (1989) 
Jpn. J. Apply. Phys. 32, L917 (1993) Silolverbindungen 
Org. Electron. 4, 113 (2003) Arylboranverbindungen 
J. Am. Chem. Soc. 120, 9714 (1998) Fluorierte aromatische Verbindungen 
J. Am. Chem. Soc. 122, 1832 (2000) Fullerene (z.B. C60) 
US20090101870 Triazinkomplexe 
US20040036077 Zn (N^N)-Komplexe 
US6528187 - EXPERIMENTELLES
- Vergleichsdaten - Peakemissionswellenlänge
- Bei phosphoreszierenden Iridiumkomplexen bestimmen die HOMO/LUMO-Niveaus häufig deren Emissionseigenschaften. Innerhalb der gleichen Ligandenfamilie kann das Einführen von Substitutionen zur Veränderung der HOMO/LUMO-Niveaus zu einer Blauverschiebung oder einer Rotverschiebung führen. Beispielsweise kann die Erweiterung der Konjugation des Liganden, wobei sich das LUMO am Liganden befindet, die Emission signifikant ins Rote verschieben. Beispielsweise emittiert Tris(2-phenylpyridin)iridium(III) bei etwa 517 nm in einer Dichlormethanlösung bei Raumtemperatur. Durch Substituieren des Pyridinrings mit einer Phenylgruppe an einem der Liganden weist die Verbindung Y unter den gleichen Bedingungen eine gelbe Emission mit einer Peakwellenlänge bei 578 nm auf. Die Phenylsubstitution verminderte das LUMO des Komplexes signifikant, was zu einer Rotverschiebung von 61 nm führt.

- Die US-Patentanmeldung mit der Veröffentlichungsnummer
20100141127 offenbart eine Vergleichsverbindung A (nachstehend) und beschreibt eine grüne Emission mit einer Peakwellenlänge von 518 nm in 2-Methyltetrahydrofuran bei Raumtemperatur.
- Das niedrigste unbesetzte Molekülorbital (LUMO) der Vergleichsverbindung A befindet sich an dem Benzimidazolteil des Liganden. Wenn eine Phenylsubstitution in den Benzolring eingeführt wird, wie es in der Verbindung 4 (vorstehend) der Fall ist, wurde erwartet, dass die Emission verglichen mit der Vergleichsverbindung A signifikant rotverschoben ist, da die Phenylsubstitution das LUMO des Komplexes vermindert. Dies würde verhindern, dass die Verbindung 4 als grün emittierendes Material geeignet ist.
- Eine Cyclovoltammetrie bestätigt, dass das LUMO für die Verbindung 4 tatsächlich niedriger war. Die Reduktionspotenziale der Vergleichsverbindung A und der Verbindung 4 wurden gemessen und betrugen -2,71 V bzw. -2,66 V gegen Fc/Fc+. Die Verbindung 4 zeigte jedoch in unerwarteter Weise eine grüne Emission mit einer Peakwellenlänge von 514 nm, was verglichen mit der Vergleichsverbindung A in Lösung bei Raumtemperatur etwa 4 nm blauverschoben ist. Daher zeigten die Verbindungen der Erfindung unerwartete photophysikalische Eigenschaften. Darüber hinaus wurde gefunden, dass Verbindungen der Erfindung eine höhere Effizienz und eine niedrigere Steuerspannung in OLED-Vorrichtungen zeigten.
- Synthese der Verbindung 1
- Herstellung von 3-Fluor-4-nitro-1,1'-biphenyl:

- Herstellung von N-(2-Isopropylphenyl)-4-nitro-[1,1'-biphenyl]-3-amin:

- Herstellung von N3-(2-Isopropylphenyl)-[1,1'-biphenyl]-3,4-diamin:

- Herstellung von 1-(2-lsopropylphenyl)-2,6-diphenyl-1H-benzo[d]imidazol:
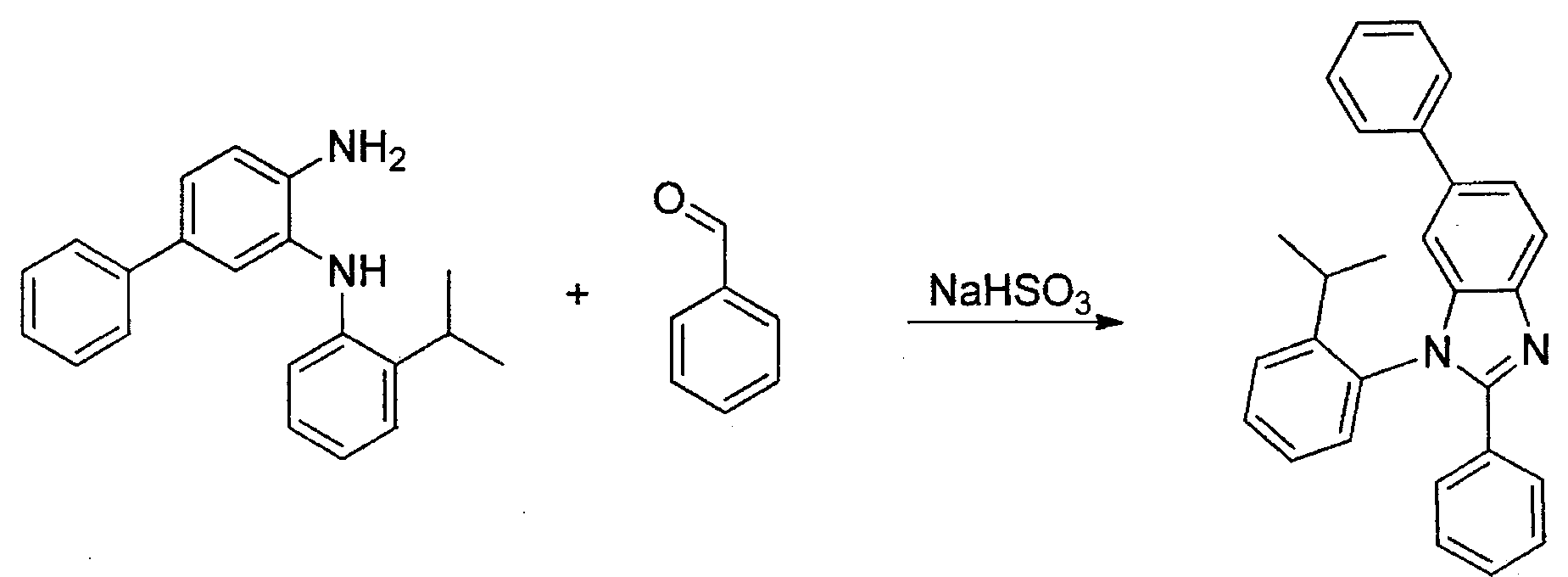
- Synthese der Verbindung 1:
-
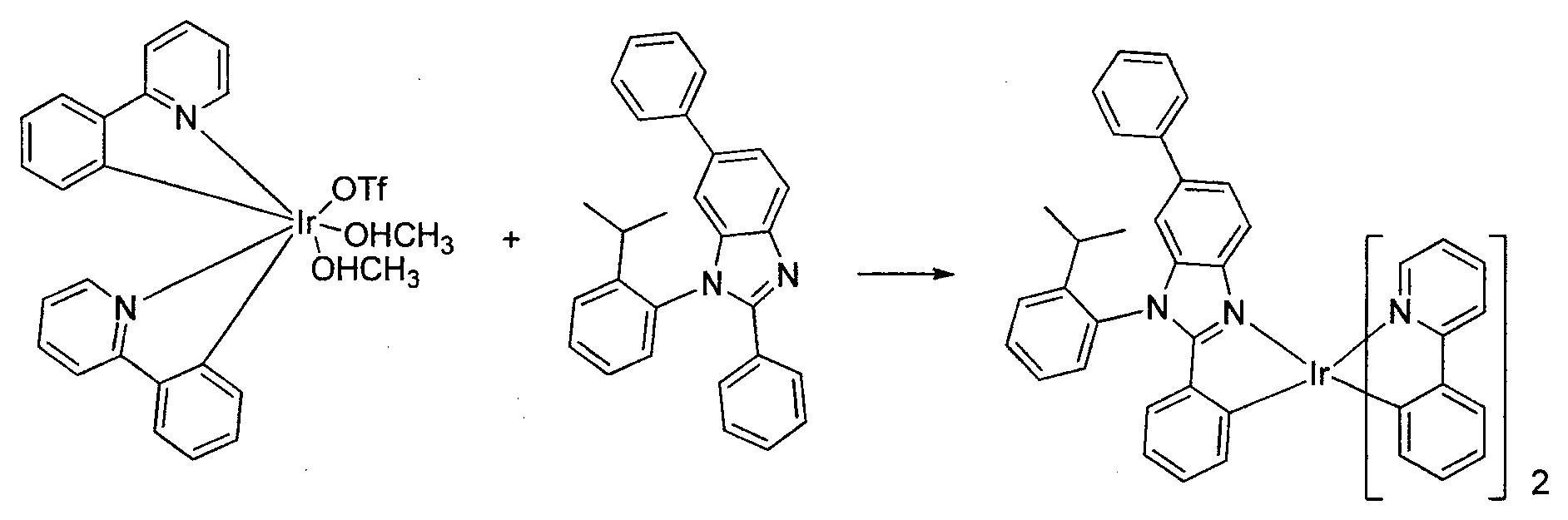
- Synthese der Verbindung 2
- Herstellung von N-(2,6-Diisopropylphenyl)-4-nitro-[1,1'-biphenyl]-3-amin:

- Herstellung von N3-(2,6-Diisopropylphenyl)-[1,1-biphenyl]-3,4-diamin

- Herstellung von 1-(2,6-Diisopropylphenyl)-2,6-diphenyl-1H-benzo[d]imidazol:
- Herstellung der Verbindung 2:
-

- Synthese der Verbindung 3
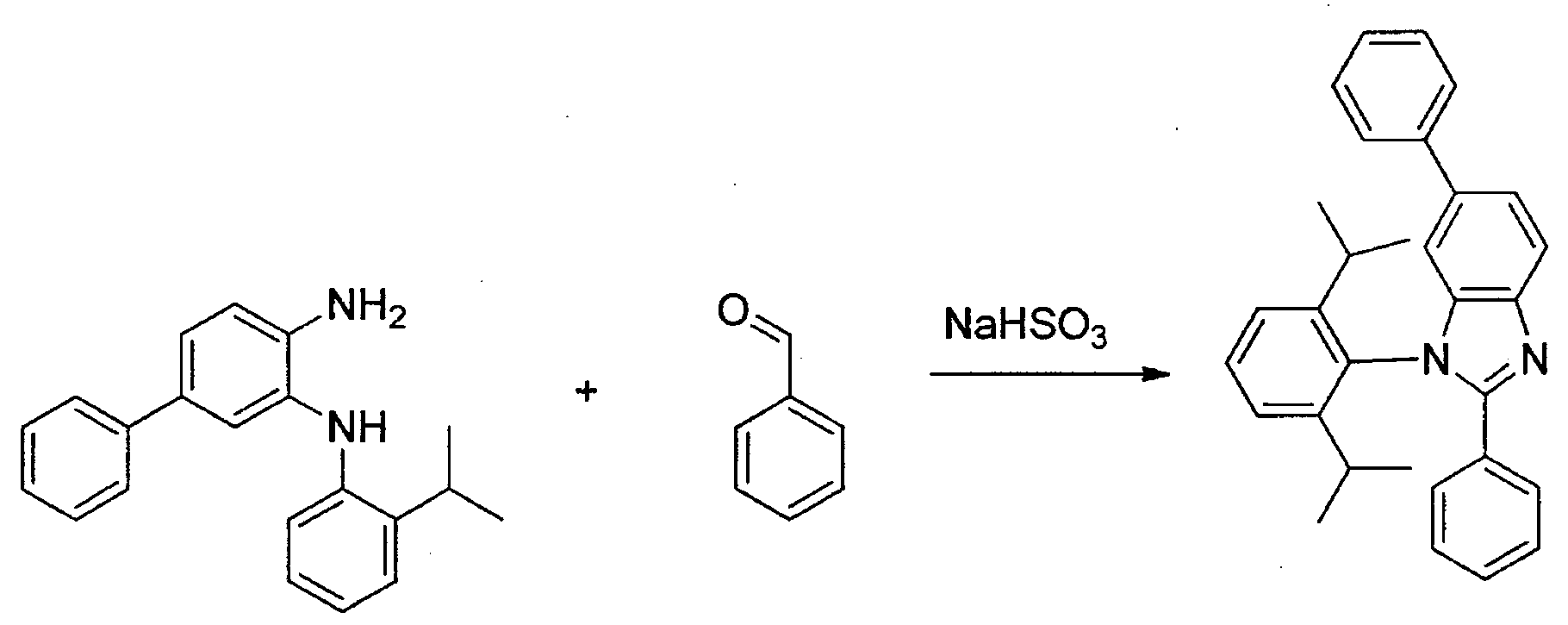
- Herstellung von 5-Fluor-2-methyl-4-nitro-1,1'-biphenyl:
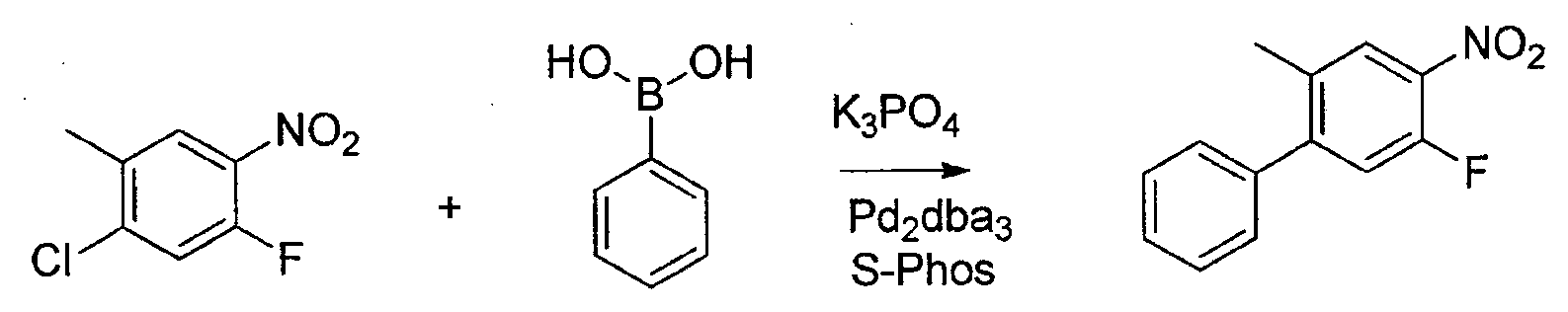
- Herstellung von N-(2-lsopropylphenyl)-6-methyl-4-nitro-[1,1'-biphenyl]-3-amin:

- Herstellung von N3-(2-lsopropylphenyl)-6-methyl-[1,1'-biphenyl]-3,4-diamin:
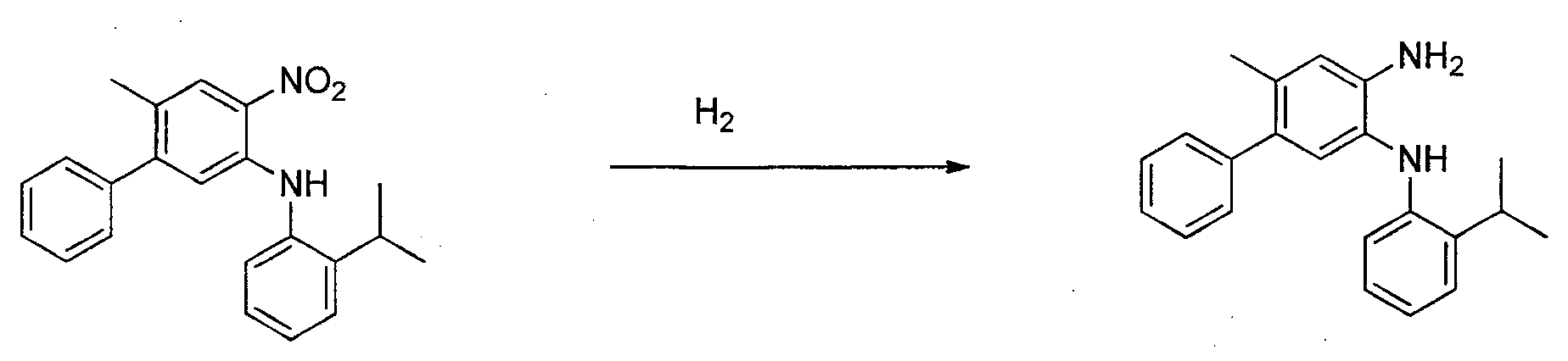
- Herstellung von 1-(2-Isopropylphenyl)-5-methyl-2,6-diphenyl-1H-benzo[d]imidazol:

- Synthese der Verbindung 3:
-
-

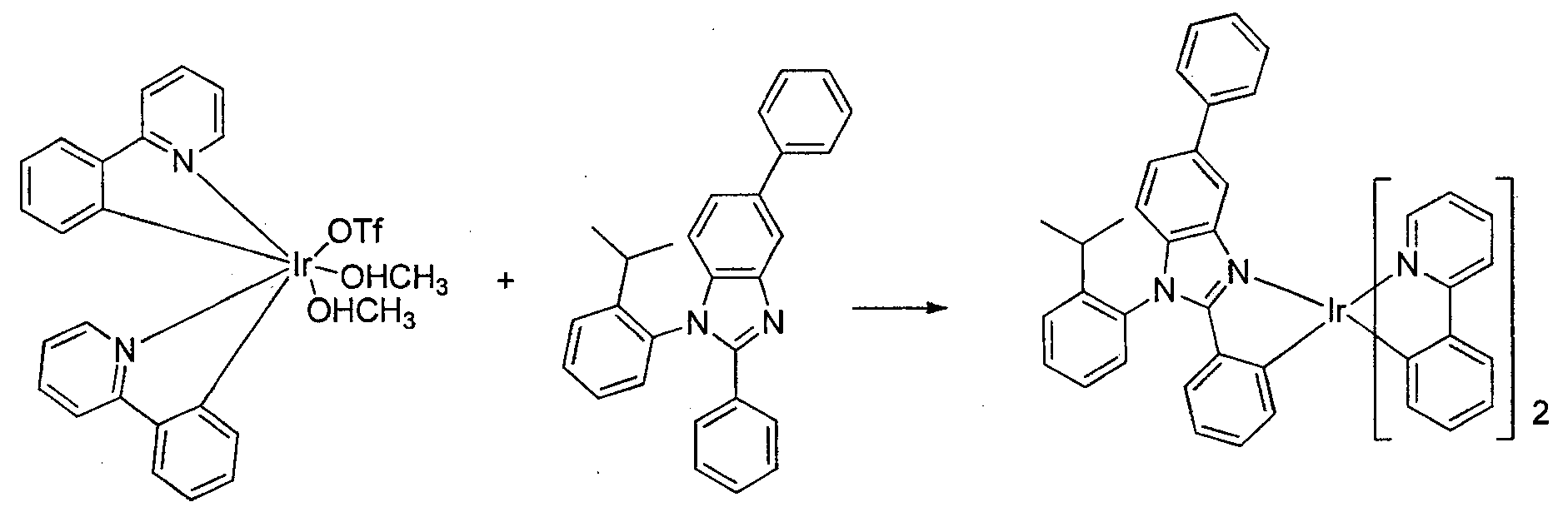
- Der Iridiumtriflatkomplex (2,96 g, 4,14 mmol) und 1-(2-Isopropylphenyl)-5-methyl-2,6-diphenyl-1 H-benzo[d]imidazol (5,0 g, 12,42 mmol) wurden einem 250 ml-Rundkolben zugesetzt. Ethanol (100 ml) wurde zugesetzt. Das Gemisch wurde 26 Stunden unter Rückfluss gehalten. Das Gemisch wurde durch Celite filtriert. Der Kuchen wurde mit Ethanol und Hexan gewaschen. Der Trichter wurde zu einem anderen Filtrationskolben überführt und das Produkt wurde mit DCM extrahiert. Das Filtrat wurde auf Celite® adsorbiert und chromatographiert (Silicagel). Eine Elution mit 1:1 DCM-Hexan ergab 1,72 Gramm (46 %) der Verbindung 3.
- Synthese der Verbindung 4
- Herstellung von N-(2-Isopropylphenyl)-3-nitro-[1,1'-biphenyl]-4-amin

- Herstellung von N4-(2-Isopropylphenyl)-[1,1'-biphenyl]-3,4-diamin:

- Herstellung von 1-(2-Isopropylphenyl)- 2,5-diphenyl-1H-benzo[d]imidazol:

- Synthese der Verbindung 4:
-
- Der Iridiumkomplex (2,450 g, 3,43 mmol) und 1-(2-Isopropylphenyl)- 2,5-diphenyl-1H-benzo[d]imidazol (4 g, 10,30 mmol) wurden 125 ml Ethanol zugesetzt und für 30 Minuten entgast. Das Reaktionsgemisch wurde für 24 Stunden unter Rückfluss erwärmt. Das Reaktionsgemisch, das eine gelbe Farbe aufwies, wurde dann auf Raumtemperatur abgekühlt und durch eine Celite®-Lage filtriert. Die Niederschläge wurden mit Ethanol und dann mit Hexanen gewaschen und schließlich wieder in DCM gelöst, so dass eine Lösung mit einer gelben Farbe erhalten wurde. Aus dem vereinigten Filtrat wurden Lösungsmittel unter vermindertem Druck entfernt, wobei eine Rohverbindung mit einer gelben Farbe erhalten wurde, die mittels Silicagel-Säulenchromatographie gereinigt wurde, wobei 0,4 g der Verbindung 4 erhalten wurden.
- Synthese der Verbindung 5
- Herstellung von N-(2,6-Diisopropylphenyl)-3-nitro-[1,1'-biphenyl]-4-amin:

- Herstellung von N4-(2,6-Diisopropylphenyl)-[1,1-biphenyl]-3,4-diamin:

- Herstellung von 1-(2,6-Diisopropylphenyl)-2,5-diphenyl-1H-benzo[d]imidazol:
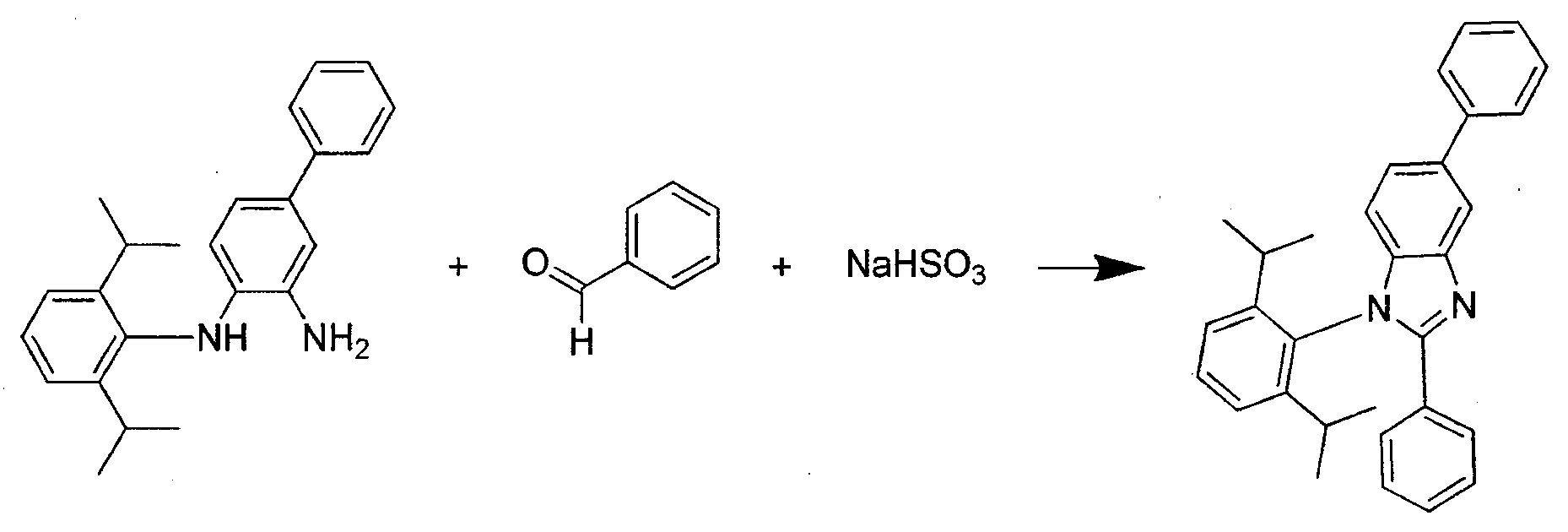
- Synthese der Verbindung 5:
-

- Der Iridiumtriflatkomplex (1,658 g, 2,322 mol) und 1-(2,6-Diisopropylphenyl)- 2,5-diphenyl-1H-benzo[d]imidazol (3 g, 6,97 mmol) wurden 80 ml Ethanol zugesetzt und für 30 Minuten entgast. Das Gemisch wurde für 24 Stunden unter Stickstoffgas unter Rückfluss gehalten. Das Reaktionsgemisch wurde durch eine Celite®-Lage filtriert. Die Niederschläge wurden in DCM wieder gelöst und organische Lösungsmittel wurden von dieser Lösung unter vermindertem Druck entfernt, wobei ein gelber Feststoff erhalten wurde. Der Feststoff wurde mittels Säulenchromatographie über Silicagel gereinigt, wobei 0,6 g der Verbindung 5 erhalten wurden.
- Synthese der Verbindung 6
- Herstellung von N-(2-Ethylphenyl)-3-nitro-[1,1'-biphenyl]-4-diamin:

- Herstellung von N4-(2-Ethylphenyl)-[1,1'-biphenyl]-3,4-diamin:

- Herstellung von 1-(2-Ethylphenyl)-2,5-diphenyl-1H-benzo[d]imidazol:

- Synthese der Verbindung 6:
-

- Synthese der Verbindung 7
- Herstellung von N-(2,6-Diethylphenyl)-3-nitro-[1,1'-biphenyl]-4-amin:

- Herstellung von N4-(2,6-Diethylphenyl)-[1,1'-biphenyl]-3,4-diamin:

- Herstellung von 1-(2,6-Diethylphenyl)-2,5-diphenyl-1H-benzo[d]imidazol:

- Synthese der Verbindung 7:
-
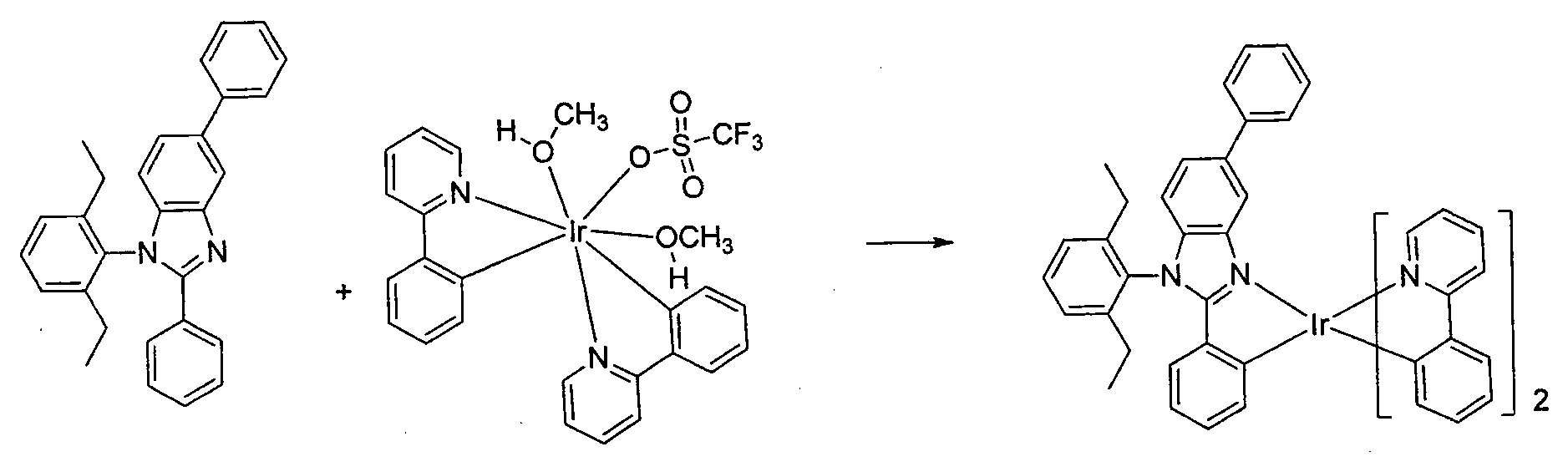
- Der Iridiumtriflatkomplex (2,78 g, 3,89 mmol) und 1-(2,6-Diethylphenyl)-2,5-diphenyl-1H-benzo[d]imidazol (4,7 g, 11,68 mmol) wurden einem 500 ml-Rundkolben zugesetzt. Ethanol wurde zugesetzt und das Gemisch wurde für 22 Stunden unter Rückfluss gerührt. Das Gemisch wurde durch Celite® filtriert und der Kuchen wurde zuerst mit Ethanol und Hexan gewaschen, dann wurde der Trichter zu einem zweiten Filterkolben überführt und das Produkt wurde mit DCM extrahiert. Das Filtrat wurde eingedampft. Das Produkt wurde auf einer Silicagelsäule chromatographiert, die mit 1:1 Hexan-DCM eluiert wurde. Es wurden 2,1 Gramm der Verbindung 7 als Produkt erhalten.
- Synthese der Verbindung 8
- Herstellung von 2-Fluor-3-nitro-1,1'-biphenyl:

- Herstellung von 3-Nitro-N-phenyl-[1, 1'-biphenyl]-2-amin:

- Herstellung von N2-Phenyl-[1,1'-biphenyl]-2,3-diamin:
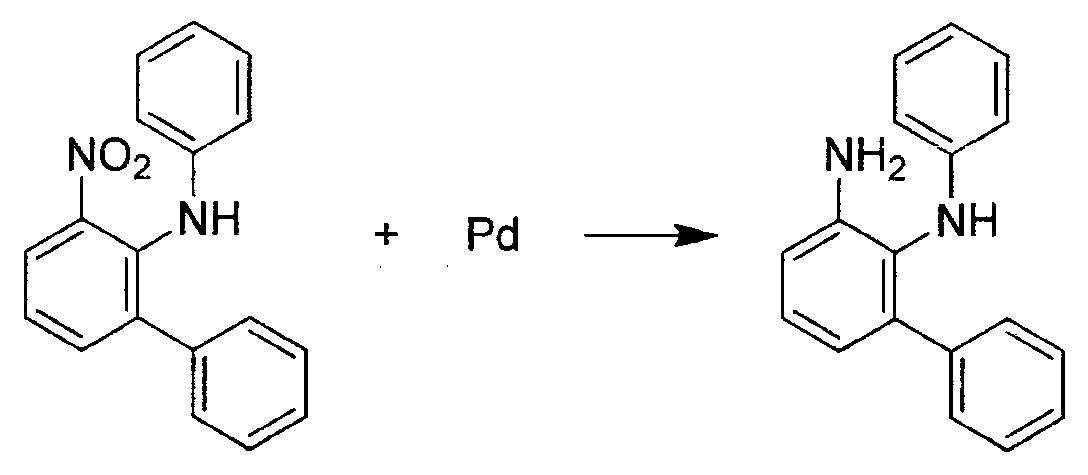
- Herstellung von 1,2,7-Triphenyl-1H-benzo[d]imidazol:

- Synthese der Verbindung 8:
-
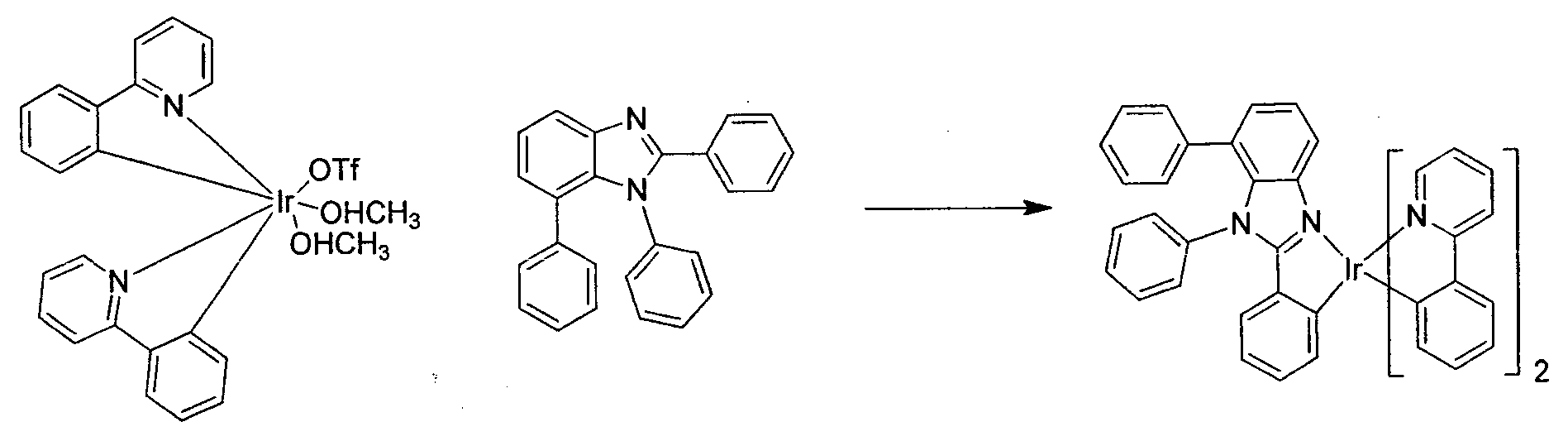
- Der Iridiumkomplex (1,717 g, 2,406 mmol) und 1,2,7-Triphenyl-1H-benzo[d]imidazol (2,5 g, 7,22 mmol) wurden 80 ml Ethanol zugesetzt und das Reaktionsgemisch wurde für 30 Minuten entgast. Es wurde für 48 Stunden unter einer inerten Atmosphäre unter Rückfluss gehalten. Das Reaktionsgemisch wurde durch eine Celite®-Lage filtriert. Die Niederschläge wurden gesammelt und mittels Säulenchromatographie gereinigt, wobei 0,29 g der Verbindung 8 erhalten wurden.
- Synthese der Verbindung 9
- Herstellung von N-(2-Isopropylphenyl)-3-nitro-[1,1'-biphenyl]-2-amin:

- Herstellung von N2-(2-Isopropylphenyl)-[1,1'-biphenyl]-2,3-diamin:

- Ein Gemisch von N-(2-Isopropylphenyl)-3-nitro-[1,1'-biphenyl]-2-amin (8,04 g, 24,19 mmol) und 10 % Palladium auf Aktivkohle (0,804 g, 7,55 mmol) wurde für 1 Stunde bei 50 psi hydriert. Das Reaktionsgemisch wurde durch eine Celite®-Lage filtriert. Organische Lösungsmittel wurden aus dem Filtrat unter vermindertem Druck entfernt und rohes N2-(2-Isopropy-Iphenyl)-[1,1'-biphenyl]-2,3-diamin-Material (6,42 g, 88 % Ausbeute) wurde für den nächsten Schritt ohne weitere Reinigung verwendet.
- Herstellung von 1-(2-Isopropylphenyl)-2,7-diphenyl-1H-benzo[d]imidazol:
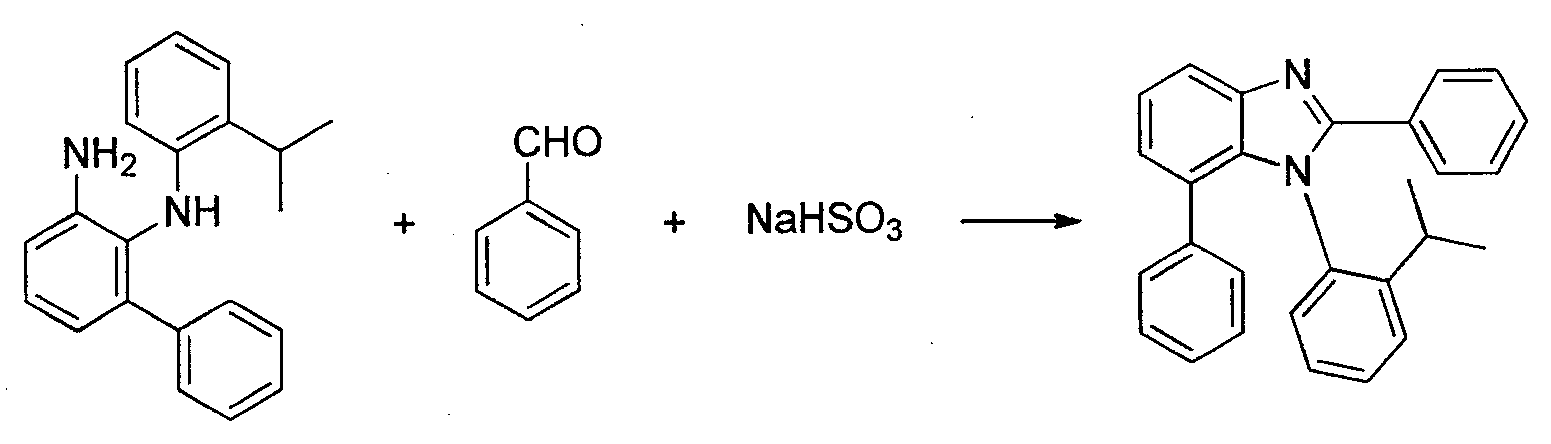
- Synthese der Verbindung 9 (nicht beansprucht)
-

- Der Iridiumkomplex (1,837 g, 2,57 mmol) und 1-(2-Isopropylphenyl)-2,7-diphenyl-1H-benzo[d]imidazol (3 g, 7,72 mmol) wurden 90 ml Ethanol zugesetzt und durch Einleiten von Stickstoffgas entgast. Das Gemisch wurde unter Stickstoffgas für 48 Stunden unter Rückfluss gehalten und dann durch eine Celite®-Lage filtriert. Die Niederschläge wurden gesammelt und mittels Säulenchromatographie unter Verwendung von 7:3 DCM/Hexane als Elutionsmittel gereinigt. Dieses Material wurde ferner sublimiert, wobei 0,5 g der Verbindung 9 erhalten wurden.
- Synthese der Verbindung 10
-
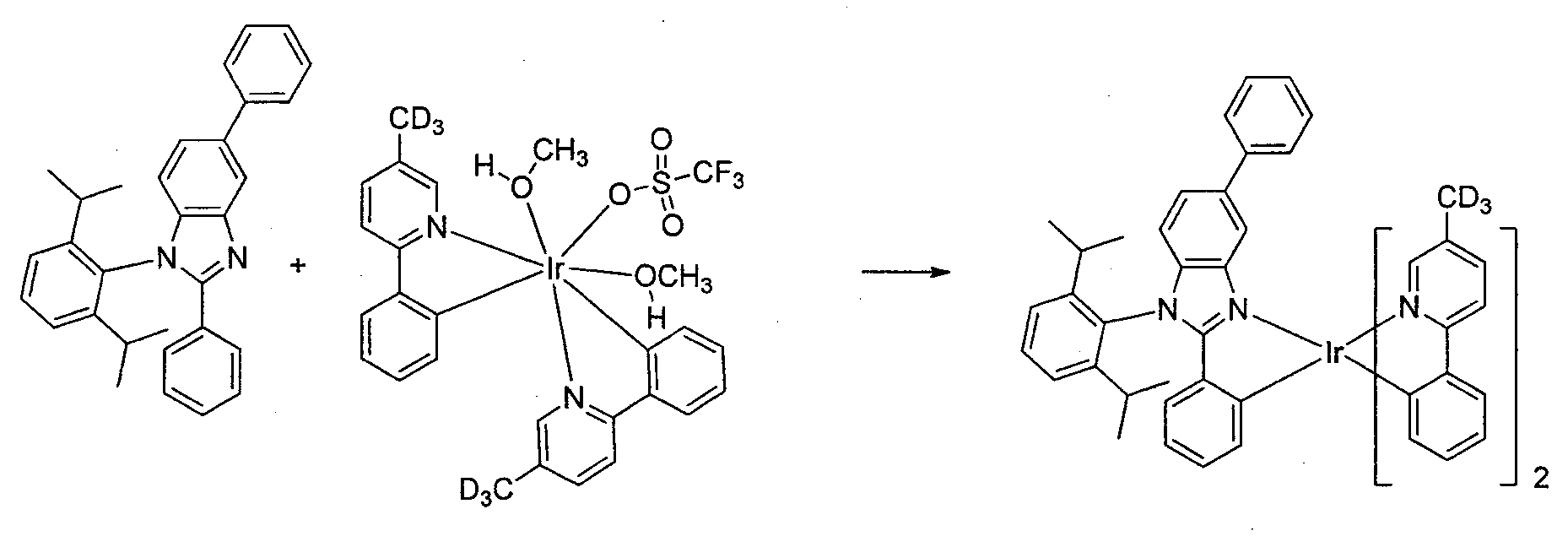
- 1-(2,6-Diisopropylphenyl)-2,5-diphenyl-1H-benzo[d]imidazol (3,1 g, 7,20 mmol) und der Iridiumkomplex (1,80 g, 2,40 mmol) wurden einem 250 ml-Rundkolben zugesetzt. Das Reaktionsgemisch wurde mit Ethanol (100 ml) verdünnt und dann für 26 Stunden unter Rückfluss gerührt. Die Suspension wurde durch Celite filtriert und der Kuchen wurde zuerst mit Ethanol und Hexan und dann mit DCM gewaschen. Das Produkt wurde chromatographiert (Silicagel). Die Verwendung einer mobilen Phase, die aus 1:1 DCM-Hexan bestand, ergab 1,2 Gramm (52 %) der Verbindung 10.
- Synthese der Verbindung 11
- Herstellung von d7-Isopropylzinkbromid:
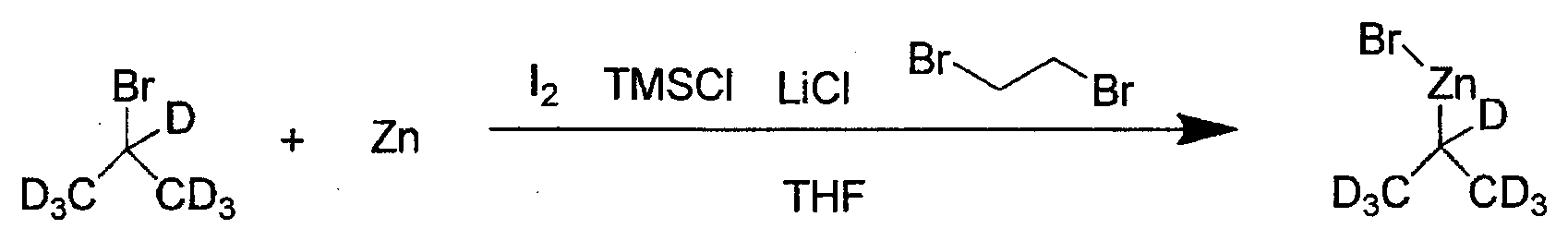
- Der Reaktionsbehälter wurde aus dem Ölbad entfernt und für 20 min auf Raumtemperatur abkühlen gelassen. Chlortrimethylsilan (0,769 ml, 0,769 mmol) und dann lod (0,098 g, 0,385 mmol), das in 0,5 ml THF gelöst war, wurden der Reaktion mittels einer Spritze durch eine Septumkappe zugesetzt. Der Reaktionsbehälter wurde wieder für 20 min in das auf 60 °C eingestellte Ölbad gestellt.
- Der Reaktionsbehälter wurde aus dem Ölbad entfernt und für 20 min auf Raumtemperatur abkühlen gelassen. d7-Isopropylbromid (10 g, 77 mmol) wurde mittels einer Spritze direkt durch die Septumkappe zugesetzt. Der Reaktionsbehälter wurde wieder über Nacht in das auf nun 50 °C eingestellte Ölbad gestellt.
- Die Reaktion wurde aus dem Ölbad entfernt und das Rühren wurde eingestellt. Das hergestellte Reagenz wurde direkt aus dem Kolben mit einer Spritze entnommen und als solches für die nächste Reaktion verwendet.
- Herstellung von 2-d7-Isopropylanilin:

- Herstellung von 4-Chlor-N-(2-d7-isopropylphenyl)-2-nitroanilin:
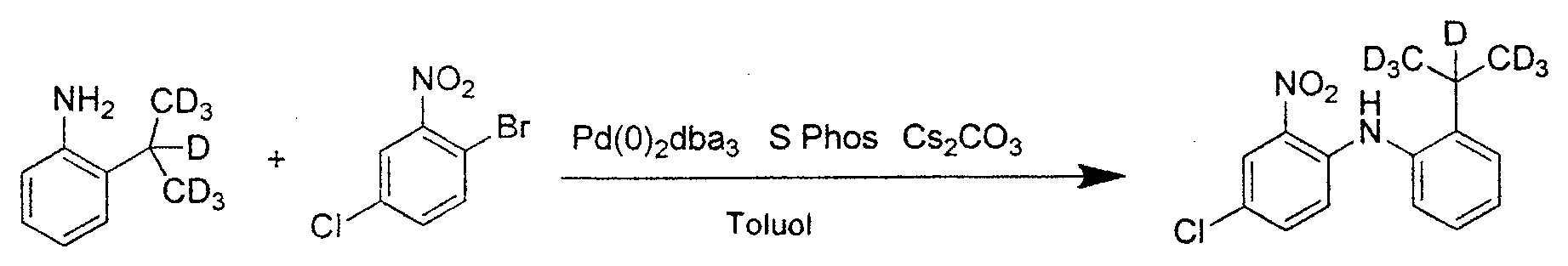
- Herstellung von 4-Chlor-N1-(2-d7-isopropylphenyl)-benzol-1,2-diamin:

- Nach drei Stunden wurde das Reaktionsgemisch durch Celite filtriert, um das Eisen zu entfernen, mit Wasser und dann Dichlormethan gewaschen. Die zwei Phasen wurden eingedampft, wobei ein feuchter, hellbrauner Feststoff erhalten wurde. Der Probe wurde Wasser zugesetzt und dann wurde Natriumcarbonat in Portionen unter heftigem Rühren zugesetzt, bis die Gasentwicklung endete und der wässrige Teil basisch war. Das Gemisch wurde in einen Scheidetrichter mit Ethylacetat überführt. Der wässrige Teil war eine graue Emulsion, so dass das Gemisch durch Celite® filtriert und gründlich mit Ethylacetat gewaschen wurde. Der wässrige Teil wurde abgetrennt und dann weiter zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Teile wurde zweimal mit Kochsalzlösung gewaschen, mit Natriumsulfat getrocknet, filtriert und unter Vakuum eingedampft, wobei 2,7 g eines beigen Feststoffs erhalten wurden, so dass das gewünschte 4-Chlor-N1-(2-d7-isopropylphenyl)-benzol-1,2-diamin-Produkt vollständig erhalten wurde (100 % Ausbeute).
- Herstellung von 5-Chlor-1-(2-d7-isopropylphenyl)-2-phenyl-1H-benzo[d]imidazol:
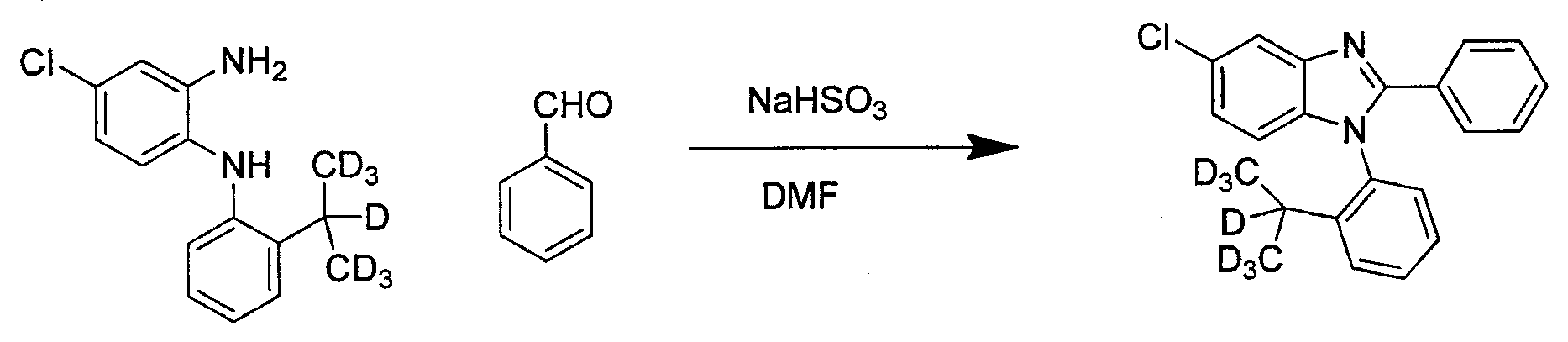
- Die rohe Probe wurde dann mittels einer Silicagel-Säulenchromatographie gereinigt. Die mobile Phase bestand aus einem 50/48/2 Hexan/Dichlormethan/Ethylacetat-Lösungsmittelsystem, das eine vollständige Trennung von Verunreinigungen ergab. Die gewünschten Fraktionen wurden vereinigt und eingedampft, wobei 3,25 g 5-Chlor-1-(2-d7-isopropylphenyl)-2-phenyl-1H-benzo[d]imidazol in der Form eines beigen Feststoffs mit einer Ausbeute von 91 % erhalten wurden.
- Herstellung von 1-(2-d7-Isopropylphenyl)-2,5-diphenyl-1H-benzo[d]imidazol:

- Ein GC/MS zeigte an, dass die Reaktion im Wesentlichen vollständig war. Das Reaktionsgemisch wurde in einen Scheidetrichter mit Ethylacetat und Wasser überführt. Es lag eine geringe Menge eines schwarzen unlöslichen Niederschlags vor, so dass eine geringe Menge Kochsalzlösung zugesetzt wurde, um die Phasentrennung zu unterstützen. Der wässrige Teil wurde dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Teile wurden einmal mit Kochsalzlösung gewaschen, mit Natriumsulfat getrocknet, filtriert und eingedampft, wobei 5 g eines braunen Öls erhalten wurden. Die Probe wurde chromatographiert (Silicagel) und mit einem 85/15 Hexan/Ethylacetat-Lösungsmittelsystem gereinigt, wobei eine vollständige Trennung des gewünschten Produkts von Verunreinigungen erreicht wurde. Die gewünschten Fraktionen wurden vereinigt und zu 3,24 g 1-(2-d7-Isopropylphenyl)-2,5-diphenyl-1H-benzo[d]imidazol in der Form eines weißen Feststoffs mit einer Ausbeute von 96,6 % eingedampft.
- Synthese der Verbindung 11
-

- Die rohe Probe wurde an 40 g Celite adsorbiert und auf einer Silicagelsäule chromatographiert, die mit einem 50/50 und dann 25/75 Hexan/Dichlormethan-Lösungsmittelsystem eluiert wurde. Dies ergab Fraktionen, die vereinigt wurden und mit einem Rotationsverdampfer eingedampft wurden, wobei 1,13 g der Verbindung 11 in der Form eines gelben Feststoffs erhalten wurden.
- Vorrichtungsbeispiele
- Alle Beispielvorrichtungen wurden durch eine thermische Verdampfung im Hochvakuum (< 10-7 Torr) hergestellt. Die Anodenelektrode war 1200 Å Indiumzinnoxid (ITO). Die Kathode bestand aus 10 Å LiF gefolgt von 1000 Å Al. Alle Vorrichtungen wurden unmittelbar nach der Herstellung in einem Stickstoff-Handschuhkasten (< 1 ppm H2O und O2) mit einer Glasabdeckung eingekapselt, die mit einem Epoxyharz versiegelt wurde, und ein Feuchtigkeitsabsorptionsmittel wurde in die Baugruppe einbezogen.
- Der organische Stapel der Vorrichtungsbeispiele bestand aufeinanderfolgend ausgehend von der ITO-Oberfläche aus 100 Å der Verbindung B als Lochinjektionsschicht (HIL), 300 Å 4,4'-Bis[N-(1-naphthyl)-N-phenylamino]biphenyl (α-NPD) als Lochtransportschicht (HTL), 300 Å der Verbindung der Formel I, die mit der Verbindung C als Wirtsmaterial dotiert war, mit 10 bis 15 Gew.-% der phosphoreszierenden Iridiumverbindung als emittierende Schicht (EML), 50 Å der Verbindung C als Blockierschicht (BL), 450 Å Alq (Tris-8-hydroxychi-nolinaluminium) als ETL. Das Vergleichsbeispiel mit der Verbindung A wurde entsprechend den Vorrichtungsbeispielen hergestellt, mit der Ausnahme, dass die Verbindung A als emittierende Verbindung in der EML verwendet wurde.
- Die Vorrichtungsergebnisse und -daten dieser Vorrichtungen sind in den Tabellen 2 und 3 zusammengefasst. Wie hier verwendet weisen NPD, Alq, die Verbindung A, die Verbindung B und die Verbindung C die folgenden Strukturen auf:


Beispielvorrichtungen HIL (100 Å) HTL (300 Å) EML (300 Å, Dotierung %) BL (50 Å) ETL (450 Å) Vergleichsbeispiel 1 Verbindung B NPD Verbindung C Verbindung A 10 % Verbindung C Alq Erfindungsgemäßes Beispiel 1 Verbindung B NPD Verbindung C Verbindung 1 10 % Verbindung C Alq Erfindungsgemäßes Beispiel 2 Verbindung B NPD Verbindung C Verbindung 2 10 % Verbindung C Alq Erfindungsgemäßes Beispiel 3 Verbindung B NPD Verbindung C Verbindung 3 10 % Verbindung C Alq Erfindungsgemäßes Beispiel 4 Verbindung B NPD Verbindung C Verbindung 4 10 % Verbindung C Alq Erfindungsgemäßes Beispiel 5 Verbindung B NPD Verbindung C Verbindung 5 15 % Verbindung C Alq Vorrichtungsbeispiele 1931 CIE bei 1000 cd/m2 x y λmax FWHM (nm) Spannung (V) LE (cd/A) EQE (%) PE (nm) (Im/W) Vergleichsbeispiel 1 0,331 0,618 520 72 6,3 51,8 14,5 25,9 Erfindungsgemä-ßes Beispiel 1 0,363 0,601 526 76 6,2 61,8 17,3 31,4 Erfindungsgemä-ßes Beispiel 2 0,358 0,604 524 74 6,3 56,2 15,8 27,9 Erfindungsgemä-ßes Beispiel 3 0,341 0,614 522 74 6,2 56,8 15,8 28,9 Erfindungsgemä-ßes Beispiel 4 0,333 0,619 520 70 6,0 57,9 16,1 30,2 Erfindungsgemä-ßes Beispiel 5 0,325 0,623 520 68 6,0 53,6 14,9 27,9 - Die Tabelle 3 fasst das Leistungsvermögen der Vorrichtungen zusammen. Die Steuerspannung (V), die Lichtausbeute (LE), die externe Quanteneffizienz (EQE) und die Leistungseffizienz (PE) wurden bei 1000 cd/m2 gemessen. Wie es in der Tabelle 2 gezeigt ist, erfordert jede erfindungsgemäße Verbindung die gleiche oder eine geringfügig niedrigere Spannung als das Vergleichsbeispiel. Zusätzlich zeigte jede erfindungsgemäße Verbindung eine hervorragende Effizienz in nahezu jeder Kategorie von LE, EQE und PE.
- Es sollte beachtet werden, dass die verschiedenen Ausführungsformen, die hier beschrieben sind, lediglich beispielhaft aufzufassen sind und den Bereich der Erfindung nicht beschränken sollen. Beispielsweise können viele der Materialien und Strukturen, die hier beschrieben sind, durch andere Materialien und Strukturen ersetzt werden, ohne vom Wesen der Erfindung abzuweichen. Die vorliegende Erfindung, wie sie beansprucht ist, kann daher Variationen der speziellen Beispiele und bevorzugten Ausführungsformen, die hier beschrieben sind, umfassen, wie es für den Fachmann geläufig ist. Es sollte beachtet werden, dass verschiedene Theorien dahingehend, warum die Erfindung ausgeführt werden kann, nicht beschränkend aufzufassen sind.
Claims (13)
- Verbindung mit der Formel


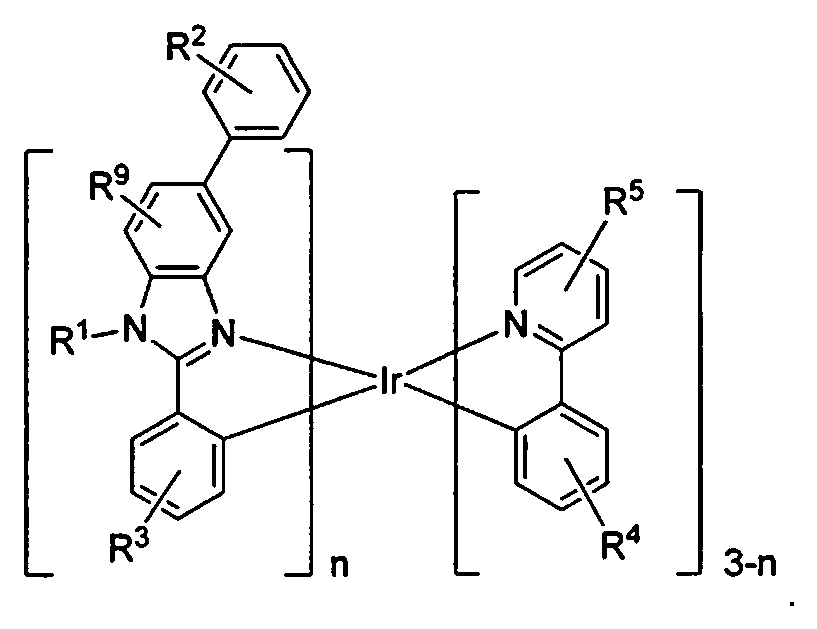
- Verbindung nach
Anspruch 1 , wobei R1 aus der Gruppe, bestehend aus Aryl, Heteroaryl, substituiertem Aryl und substituiertem Heteroaryl, ausgewählt ist. - Verbindung nach
Anspruch 1 , wobei (a) R2 mono-, di-, tri-, tetra- oder pentasubstituiert ist, (b) R5 mono-, di-, tri-, oder tetrasubstituiert ist, oder (c) beides. - Verbindung nach
Anspruch 1 , wobei R5 mono-, di-, tri-, oder tetrasubstituiert ist. - Verbindung nach
Anspruch 1 , wobei R2 mono-, di-, tri-, tetra- oder pentasubstituiert ist. - Verbindung nach
Anspruch 1 , wobei die Verbindung ausgewählt ist aus der Gruppe, bestehend aus: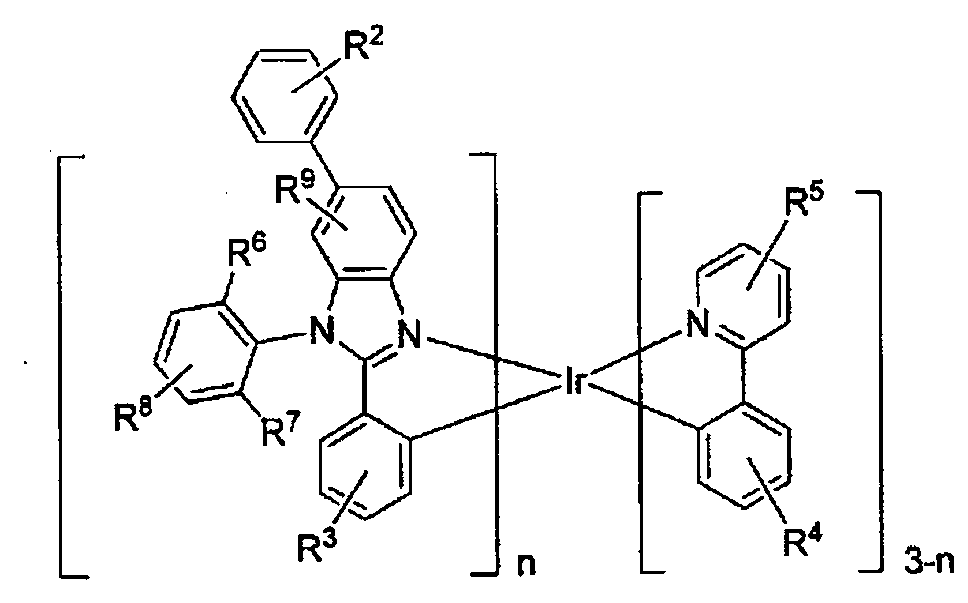

- Verbindung nach
Anspruch 6 , wobei mindestens einer von R6 und R7 aus der Gruppe bestehend aus Alkyl, Cycloalkyl, Aryl und Kombinationen davon ausgewählt ist. - Verbindung nach
Anspruch 6 , wobei R2, R3, R4, R5, R6, R7, R8 und R9 jeweils unabhängig aus der Gruppe bestehend aus Wasserstoff, Deuterium, Methyl, Ethyl, Propyl, 1-Methylethyl, Butyl, 1-Methylpropyl, 2-Methylpropyl, Pentyl, 1-Methylbutyl, 2-Methylbutyl, 3-Methylbutyl, 1,1-Dimethylpropyl, 1,2-Dimethylpropyl, 2,2-Dimethylpropyl, Cyclopentyl, Cyclohexyl, Phenyl, 2-Methylphenyl, 2,6-Dimethylphenyl, 2,4,6-Trimethylphenyl, 2-Isopropylphenyl, 2,6-Diisopropylphenyl, 2,4,6-Triisopropylphenyl, 2-Phenylphenyl, 2,6-Diphenylphenyl, 2,4,6-Triphenylphenyl, und Kombinationen davon, ausgewählt sind. - Verbindung nach
Anspruch 1 , wobei die Verbindung mit der Formel I ausgewählt ist aus der Gruppe, bestehend aus:

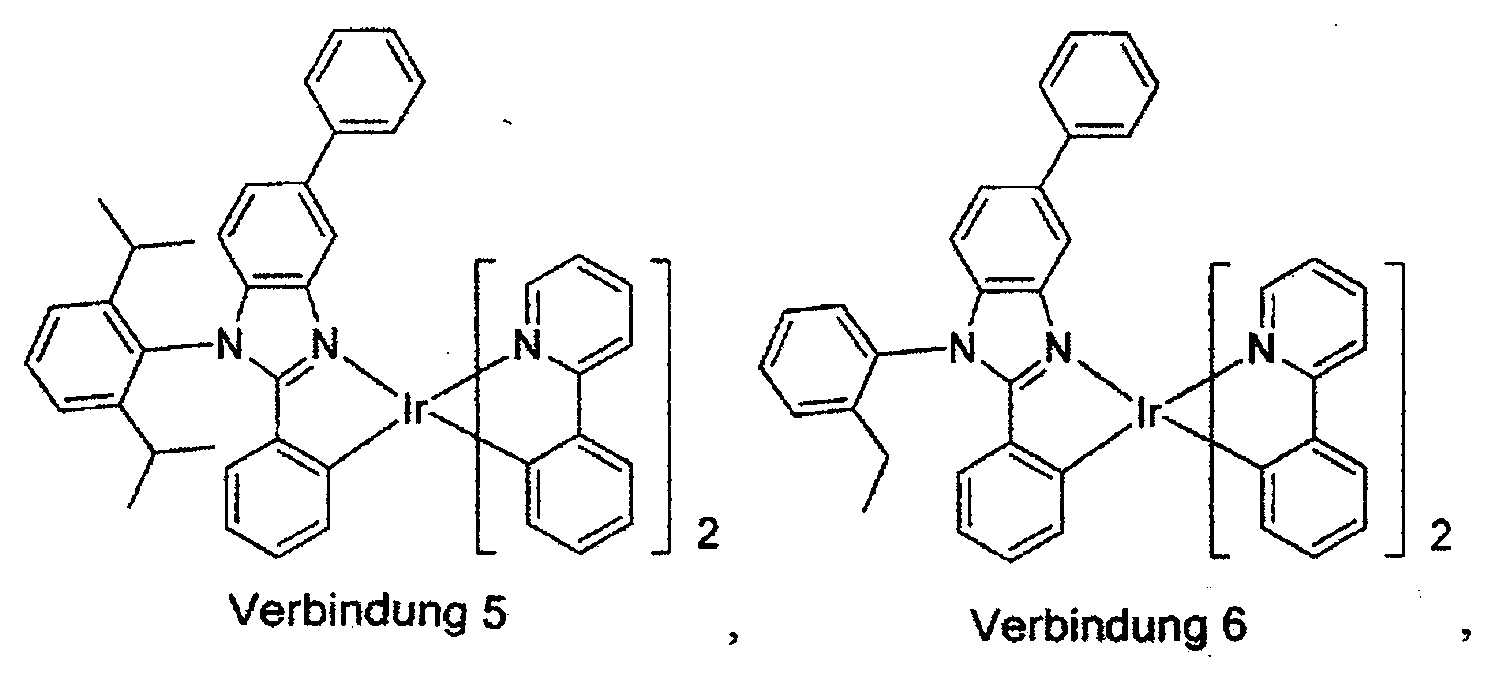

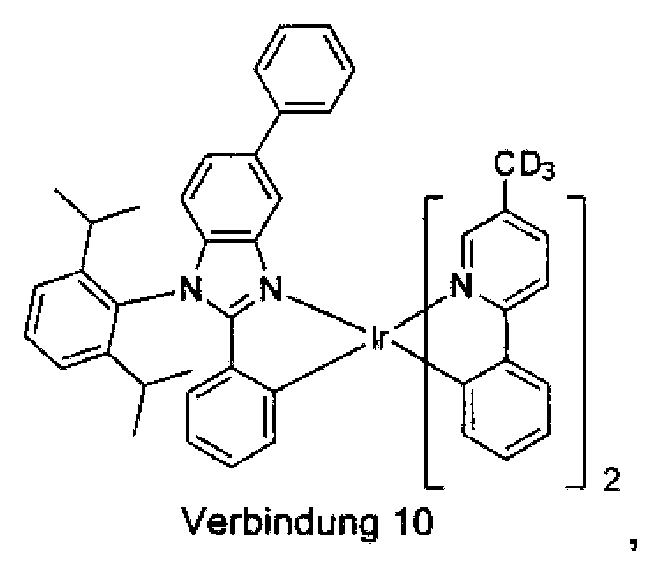







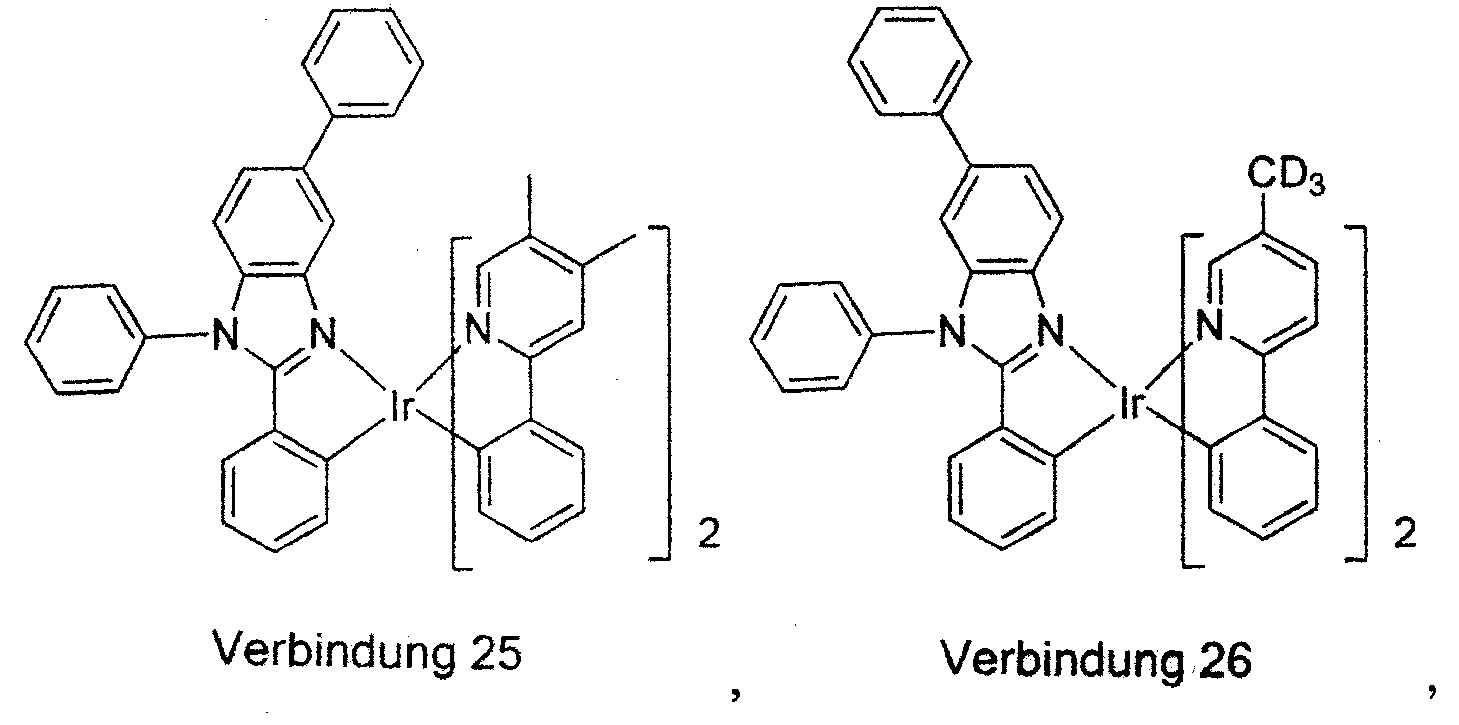

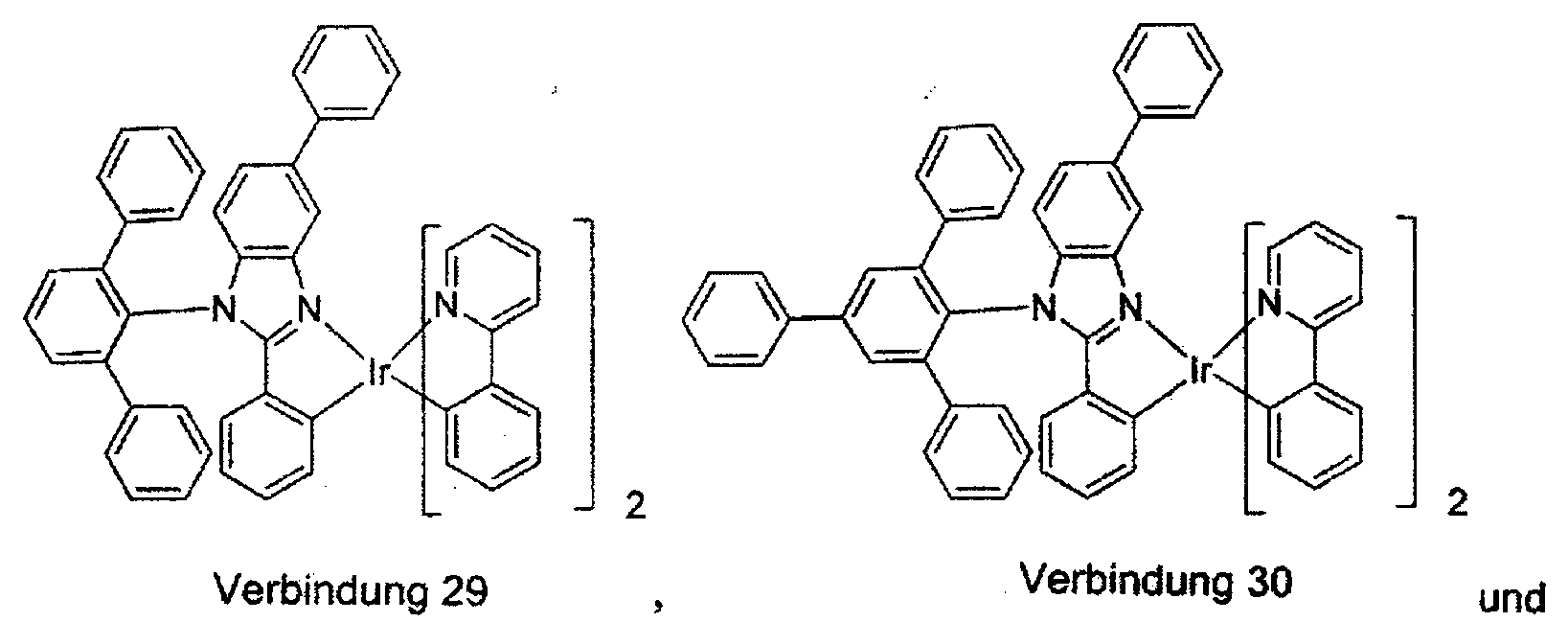

- Vorrichtung, die eine organische Licht-emittierende Vorrichtung umfasst, ferner umfassend: eine Anode, eine Kathode und eine organische Schicht, die zwischen der Anode und der Kathode angeordnet ist und die eine Verbindung gemäß einem der
Ansprüche 1 bis9 umfasst. - Vorrichtung nach
Anspruch 10 , wobei die Vorrichtung mindestens eines von einem Verbraucherprodukt, einer organischen Licht-emittierenden Vorrichtung und einer Beleuchtungseinheit ist. - Vorrichtung nach
Anspruch 10 , wobei die organische Schicht ferner eine Wirtsverbindung umfasst. - Vorrichtung nach
Anspruch 12 , wobei die Wirtsverbindung ein Triphenylenenthaltendes Benzo-anelliertes Thiophen oder Benzo-anelliertes Furan umfasst, wobei jedweder Substituent in der Wirtsverbindung ein nicht-anellierter Substituent ist, der unabhängig aus der Gruppe, bestehend aus CnH2n+1, OCnH2n+1, OAr1, N(CnH2n+1)2, N(Ar1)(Ar2), CH=CH-CnH2n+1, C=CCnH2n+1, Ar1, Ar1-Ar2, CnH2n-Ar1 oder keiner Substitution, ausgewählt ist, worin n 1 bis 10 ist, und worin Ar1 und Ar2 unabhängig aus der Gruppe, bestehend aus Benzol, Biphenyl, Naphthalin, Triphenylen, Carbazol und heteroaromatischen Analoga davon, ausgewählt sind.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/762,612 US10400163B2 (en) | 2013-02-08 | 2013-02-08 | Organic electroluminescent materials and devices |
| US13/762,612 | 2013-02-08 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE102014001468A1 DE102014001468A1 (de) | 2014-08-14 |
| DE102014001468B4 true DE102014001468B4 (de) | 2024-08-08 |
Family
ID=51226355
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE102014001468.1A Active DE102014001468B4 (de) | 2013-02-08 | 2014-02-05 | Heteroleptischer phenylbenzimidazol-komplex |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US10400163B2 (de) |
| KR (1) | KR102139384B1 (de) |
| CN (1) | CN103980321B (de) |
| DE (1) | DE102014001468B4 (de) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101880007B1 (ko) * | 2015-02-03 | 2018-07-18 | 스미또모 가가꾸 가부시키가이샤 | 조성물 및 그것을 사용한 발광 소자 |
| US11925102B2 (en) * | 2015-06-04 | 2024-03-05 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US20170324049A1 (en) | 2016-05-05 | 2017-11-09 | Universal Display Corporation | Organic Electroluminescent Materials and Devices |
| US11545636B2 (en) | 2016-12-15 | 2023-01-03 | Universal Display Corporation | Organic electroluminescent materials and devices |
| WO2018109621A1 (en) * | 2016-12-16 | 2018-06-21 | Semiconductor Energy Laboratory Co., Ltd. | Organometallic complex, light-emitting element, light-emitting device, electronic device, and lighting device |
| WO2019065388A1 (ja) * | 2017-09-29 | 2019-04-04 | 住友化学株式会社 | 組成物及びそれを用いた発光素子 |
| EP3798225B1 (de) | 2019-09-26 | 2022-11-02 | Samsung Electronics Co., Ltd. | Organometallische verbindung, organische lichtemittierende vorrichtung damit und elektronische vorrichtung mit der organischen lichtemittierenden vorrichtung |
| KR102824585B1 (ko) | 2019-11-28 | 2025-06-24 | 삼성전자주식회사 | 유기금속 화합물, 이를 포함한 유기 발광 소자 및 이를 포함한 진단용 조성물 |
Citations (156)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4769292A (en) | 1987-03-02 | 1988-09-06 | Eastman Kodak Company | Electroluminescent device with modified thin film luminescent zone |
| US5061569A (en) | 1990-07-26 | 1991-10-29 | Eastman Kodak Company | Electroluminescent device with organic electroluminescent medium |
| US5247190A (en) | 1989-04-20 | 1993-09-21 | Cambridge Research And Innovation Limited | Electroluminescent devices |
| EP0650955A1 (de) | 1993-11-01 | 1995-05-03 | Hodogaya Chemical Co., Ltd. | Aminverbindung und sie enthaltende Elektrolumineszenzvorrichtung |
| US5703436A (en) | 1994-12-13 | 1997-12-30 | The Trustees Of Princeton University | Transparent contacts for organic devices |
| US5707745A (en) | 1994-12-13 | 1998-01-13 | The Trustees Of Princeton University | Multicolor organic light emitting devices |
| US5834893A (en) | 1996-12-23 | 1998-11-10 | The Trustees Of Princeton University | High efficiency organic light emitting devices with light directing structures |
| US5844363A (en) | 1997-01-23 | 1998-12-01 | The Trustees Of Princeton Univ. | Vacuum deposited, non-polymeric flexible organic light emitting devices |
| US6013982A (en) | 1996-12-23 | 2000-01-11 | The Trustees Of Princeton University | Multicolor display devices |
| US6087196A (en) | 1998-01-30 | 2000-07-11 | The Trustees Of Princeton University | Fabrication of organic semiconductor devices using ink jet printing |
| US6091195A (en) | 1997-02-03 | 2000-07-18 | The Trustees Of Princeton University | Displays having mesa pixel configuration |
| US6097147A (en) | 1998-09-14 | 2000-08-01 | The Trustees Of Princeton University | Structure for high efficiency electroluminescent device |
| WO2001039234A2 (en) | 1999-11-24 | 2001-05-31 | The Trustees Of Princeton University | Organic light emitting diode having a blue phosphorescent molecule as an emitter |
| US20010015432A1 (en) | 2000-02-10 | 2001-08-23 | Tatsuya Igarashi | Light emitting device material comprising iridium complex and light emitting device using same material |
| US6294398B1 (en) | 1999-11-23 | 2001-09-25 | The Trustees Of Princeton University | Method for patterning devices |
| US6303238B1 (en) | 1997-12-01 | 2001-10-16 | The Trustees Of Princeton University | OLEDs doped with phosphorescent compounds |
| US6337102B1 (en) | 1997-11-17 | 2002-01-08 | The Trustees Of Princeton University | Low pressure vapor phase deposition of organic thin films |
| WO2002002714A2 (en) | 2000-06-30 | 2002-01-10 | E.I. Du Pont De Nemours And Company | Electroluminescent iridium compounds with fluorinated phenylpyridines, phenylpyrimidines, and phenylquinolines and devices made with such compounds |
| WO2002015645A1 (en) | 2000-08-11 | 2002-02-21 | The Trustees Of Princeton University | Organometallic compounds and emission-shifting organic electrophosphorescence |
| US20020034656A1 (en) | 1998-09-14 | 2002-03-21 | Thompson Mark E. | Organometallic complexes as phosphorescent emitters in organic LEDs |
| US20020134984A1 (en) | 2001-02-01 | 2002-09-26 | Fuji Photo Film Co., Ltd. | Transition metal complex and light-emitting device |
| US20020158242A1 (en) | 1999-12-31 | 2002-10-31 | Se-Hwan Son | Electronic device comprising organic compound having p-type semiconducting characteristics |
| US6528187B1 (en) | 1998-09-08 | 2003-03-04 | Fuji Photo Film Co., Ltd. | Material for luminescence element and luminescence element using the same |
| WO2003040257A1 (en) | 2001-11-07 | 2003-05-15 | E. I. Du Pont De Nemours And Company | Electroluminescent platinum compounds and devices made with such compounds |
| WO2003060956A2 (en) | 2002-01-18 | 2003-07-24 | Lg Chem, Ltd. | New material for transporting electrons and organic electroluminescent display using the same |
| US20030138657A1 (en) | 2000-12-07 | 2003-07-24 | Canon Kabushiki Kaisha | Deuterated semi-conducting organic compounds used for opto-electronic devices |
| US20030152802A1 (en) | 2001-06-19 | 2003-08-14 | Akira Tsuboyama | Metal coordination compound and organic liminescence device |
| US20030162053A1 (en) | 1996-06-25 | 2003-08-28 | Marks Tobin J. | Organic light - emitting diodes and methods for assembly and enhanced charge injection |
| US20030175553A1 (en) | 2001-12-28 | 2003-09-18 | Thompson Mark E. | White light emitting oleds from combined monomer and aggregate emission |
| US20030230980A1 (en) | 2002-06-18 | 2003-12-18 | Forrest Stephen R | Very low voltage, high efficiency phosphorescent oled in a p-i-n structure |
| US6687266B1 (en) | 2002-11-08 | 2004-02-03 | Universal Display Corporation | Organic light emitting materials and devices |
| US20040036077A1 (en) | 2002-08-22 | 2004-02-26 | Fuji Photo Film Co., Ltd. | Light emitting element |
| US20040115476A1 (en) | 2002-11-26 | 2004-06-17 | Tomohiro Oshiyama | Organic electroluminescent element, and display and illuminator |
| US20040137268A1 (en) | 2002-12-27 | 2004-07-15 | Fuji Photo Film Co., Ltd. | Organic electroluminescent device |
| US20040137267A1 (en) | 2002-12-27 | 2004-07-15 | Fuji Photo Film Co., Ltd. | Organic electroluminescent device |
| US20040174116A1 (en) | 2001-08-20 | 2004-09-09 | Lu Min-Hao Michael | Transparent electrodes |
| WO2004093207A2 (de) | 2003-04-15 | 2004-10-28 | Covion Organic Semiconductors Gmbh | Mischungen von organischen zur emission befähigten halbleitern und matrixmaterialien, deren verwendung und elektronikbauteile enthaltend diese mischungen |
| WO2004107822A1 (ja) | 2003-05-29 | 2004-12-09 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子 |
| JP2005011610A (ja) | 2003-06-18 | 2005-01-13 | Nippon Steel Chem Co Ltd | 有機電界発光素子 |
| US20050025993A1 (en) | 2003-07-25 | 2005-02-03 | Thompson Mark E. | Materials and structures for enhancing the performance of organic light emitting devices |
| WO2005014551A1 (ja) | 2003-08-07 | 2005-02-17 | Nippon Steel Chemical Co., Ltd. | 有機el材料用のアルミニウムキレート錯体 |
| WO2005030900A1 (ja) | 2003-09-25 | 2005-04-07 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子 |
| US20050112407A1 (en) | 2003-11-21 | 2005-05-26 | Fuji Photo Film Co., Ltd. | Organic electroluminescent device |
| US20050123751A1 (en) | 2003-08-25 | 2005-06-09 | Tetsuo Tsutsui | Electrode device for organic device, electronic device having electrode device for organic device, and method of forming electrode device for organic device |
| US6921915B2 (en) | 2001-03-08 | 2005-07-26 | Canon Kabushiki Kaisha | Metal coordination compound, luminescence device and display apparatus |
| WO2005089025A1 (ja) | 2004-03-15 | 2005-09-22 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子 |
| US20050238919A1 (en) | 2004-04-23 | 2005-10-27 | Fuji Photo Film Co., Ltd. | Organic electroluminescent device |
| US20050244673A1 (en) | 2002-08-27 | 2005-11-03 | Fujitsu Limited | Organometallic complex, organic EL element and organic EL display |
| US20050260441A1 (en) | 2004-05-18 | 2005-11-24 | Thompson Mark E | Luminescent compounds with carbene ligands |
| WO2005123873A1 (ja) | 2004-06-17 | 2005-12-29 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20060008670A1 (en) | 2004-07-06 | 2006-01-12 | Chun Lin | Organic light emitting materials and devices |
| WO2006009024A1 (ja) | 2004-07-23 | 2006-01-26 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| WO2006056418A2 (de) | 2004-11-25 | 2006-06-01 | Basf Aktiengesellschaft | Verwendung von übergangsmetall-carbenkomplexen in organischen licht-emittierenden dioden (oleds) |
| US20060121308A1 (en) | 2003-04-23 | 2006-06-08 | Eisaku Katoh | Material for organic electroluminescent device, organic electroluminescent device, illuminating device and display |
| US20060127696A1 (en) | 2002-08-24 | 2006-06-15 | Covion Organic Semiconductors Gmbh | Rhodium and iridium complexes |
| WO2006072002A2 (en) | 2004-12-30 | 2006-07-06 | E.I. Dupont De Nemours And Company | Organometallic complexes |
| WO2006082742A1 (ja) | 2005-02-04 | 2006-08-10 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US7090928B2 (en) | 2003-04-01 | 2006-08-15 | The University Of Southern California | Binuclear compounds |
| US20060182992A1 (en) | 2003-06-02 | 2006-08-17 | Kazumi Nii | Organic electroluminescent devices and metal complex compounds |
| US20060202194A1 (en) | 2005-03-08 | 2006-09-14 | Jeong Hyun C | Red phosphorescene compounds and organic electroluminescence device using the same |
| WO2006098120A1 (ja) | 2005-03-16 | 2006-09-21 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子 |
| WO2006100298A1 (de) | 2005-03-24 | 2006-09-28 | Basf Aktiengesellschaft | Verwendung von verbindungen, welche aromatische oder heteroaromatische über carbonyl-gruppen enthaltende gruppen verbundene ringe enthalten, als matrixmaterialien in organischen leuchtdioden |
| WO2006103874A1 (ja) | 2005-03-29 | 2006-10-05 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20060240279A1 (en) | 2005-04-21 | 2006-10-26 | Vadim Adamovich | Non-blocked phosphorescent OLEDs |
| WO2006114966A1 (ja) | 2005-04-18 | 2006-11-02 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20060251923A1 (en) | 2005-05-06 | 2006-11-09 | Chun Lin | Stability OLED materials and devices |
| EP1725079A1 (de) | 2004-03-11 | 2006-11-22 | Mitsubishi Chemical Corporation | Zusammensetzung für einen ladungstransportfilm und ionenzusammensetzungs-ladungstransportfilm und organische elektrolumineszenzeinrichtung damit und verfahren zur herstellung einer organischen elektrolumineszenzeinrichtung und verfahren zur herstellung eines ladungstransportfilms |
| US20060263635A1 (en) | 2005-05-06 | 2006-11-23 | Fuji Photo Film Co., Ltd. | Organic electroluminescent device |
| US20060280965A1 (en) | 2005-05-31 | 2006-12-14 | Raymond Kwong | Triphenylene hosts in phosphorescent light emitting diodes |
| WO2006132173A1 (ja) | 2005-06-07 | 2006-12-14 | Nippon Steel Chemical Co., Ltd. | 有機金属錯体及びこれを用いた有機電界発光素子 |
| US7154114B2 (en) | 2004-05-18 | 2006-12-26 | Universal Display Corporation | Cyclometallated iridium carbene complexes for use as hosts |
| WO2007002683A2 (en) | 2005-06-27 | 2007-01-04 | E. I. Du Pont De Nemours And Company | Electrically conductive polymer compositions |
| WO2007004380A1 (ja) | 2005-07-01 | 2007-01-11 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20070023098A1 (en) | 2003-10-22 | 2007-02-01 | Uster Technologies Ag | Holding element for a device for monitoring the quality on a mechanical weaving loom |
| US20070087321A1 (en) | 2003-09-09 | 2007-04-19 | Csaba Pribenszky | Post-thaw survival of chryopreserved biological material by hydrostatic pressure challenge |
| US20070103060A1 (en) | 2003-11-04 | 2007-05-10 | Takasago International Corporation | Platinum complex and light emitting device |
| JP2007123392A (ja) | 2005-10-26 | 2007-05-17 | Konica Minolta Holdings Inc | 有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20070111026A1 (en) | 2005-11-15 | 2007-05-17 | Deaton Joseph C | Oled devices with dinuclear copper compounds |
| WO2007063796A1 (ja) | 2005-12-01 | 2007-06-07 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子 |
| WO2007063754A1 (ja) | 2005-12-01 | 2007-06-07 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子用化合物及び有機電界発光素子 |
| US7230107B1 (en) | 2004-12-29 | 2007-06-12 | E. I. Du Pont De Nemours And Company | Metal quinoline complexes |
| US7232618B2 (en) | 2002-04-12 | 2007-06-19 | Canon Kabushiki Kaisha | Organic light-emitting device |
| US7250226B2 (en) | 2001-08-31 | 2007-07-31 | Nippon Hoso Kyokai | Phosphorescent compound, a phosphorescent composition and an organic light-emitting device |
| US20070190359A1 (en) | 2006-02-10 | 2007-08-16 | Knowles David B | Metal complexes of cyclometallated imidazo[1,2-ƒ]phenanthridine and diimidazo[1,2-a:1',2'-c]quinazoline ligands and isoelectronic and benzannulated analogs thereof |
| JP2007254297A (ja) | 2006-03-20 | 2007-10-04 | Nippon Steel Chem Co Ltd | 発光層化合物及び有機電界発光素子 |
| US7279704B2 (en) | 2004-05-18 | 2007-10-09 | The University Of Southern California | Complexes with tridentate ligands |
| US20070278938A1 (en) | 2006-04-26 | 2007-12-06 | Idemitsu Kosan Co., Ltd. | Aromatic amine derivative and electroluminescence device using the same |
| US20080015355A1 (en) | 2004-06-28 | 2008-01-17 | Thomas Schafer | Electroluminescent Metal Complexes With Triazoles And Benzotriazoles |
| US7332232B2 (en) | 2004-02-03 | 2008-02-19 | Universal Display Corporation | OLEDs utilizing multidentate ligand systems |
| US7338722B2 (en) | 2003-03-24 | 2008-03-04 | The University Of Southern California | Phenyl and fluorenyl substituted phenyl-pyrazole complexes of Ir |
| JP2008074939A (ja) | 2006-09-21 | 2008-04-03 | Konica Minolta Holdings Inc | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20080106190A1 (en) | 2006-08-23 | 2008-05-08 | Idemitsu Kosan Co., Ltd. | Aromatic amine derivatives and organic electroluminescent device using same |
| WO2008056746A1 (en) | 2006-11-09 | 2008-05-15 | Nippon Steel Chemical Co., Ltd. | Compound for organic electroluminescent device and organic electroluminescent device |
| US20080124572A1 (en) | 2006-11-24 | 2008-05-29 | Idemitsu Kosan Co., Ltd. | Aromatic amine derivative and organic electroluminescence device using the same |
| US7393599B2 (en) | 2004-05-18 | 2008-07-01 | The University Of Southern California | Luminescent compounds with carbene ligands |
| US7396598B2 (en) | 2001-06-20 | 2008-07-08 | Showa Denko K.K. | Light emitting material and organic light-emitting device |
| WO2008101842A1 (en) | 2007-02-23 | 2008-08-28 | Basf Se | Electroluminescent metal complexes with benzotriazoles |
| US20080220265A1 (en) | 2006-12-08 | 2008-09-11 | Universal Display Corporation | Cross-linkable Iridium Complexes and Organic Light-Emitting Devices Using the Same |
| US7431968B1 (en) | 2001-09-04 | 2008-10-07 | The Trustees Of Princeton University | Process and apparatus for organic vapor jet deposition |
| US20080261076A1 (en) | 2007-03-08 | 2008-10-23 | Universal Display Corporation | Phosphorescent materials |
| US7445855B2 (en) | 2004-05-18 | 2008-11-04 | The University Of Southern California | Cationic metal-carbene complexes |
| WO2008132085A1 (de) | 2007-04-26 | 2008-11-06 | Basf Se | Silane enthaltend phenothiazin-s-oxid oder phenothiazin-s,s-dioxid-gruppen und deren verwendung in oleds |
| US20080297033A1 (en) | 2006-02-10 | 2008-12-04 | Knowles David B | Blue phosphorescent imidazophenanthridine materials |
| WO2009000673A2 (en) | 2007-06-22 | 2008-12-31 | Basf Se | Light emitting cu(i) complexes |
| US20090009065A1 (en) | 2007-07-07 | 2009-01-08 | Idemitsu Kosan Co., Ltd. | Organic electroluminescence device and material for organic electroluminescence device |
| US20090008605A1 (en) | 2007-07-07 | 2009-01-08 | Idemitsu Kosan Co., Ltd. | Naphthalene derivative, material for organic electroluminescence device, and organic electroluminescence device using the same |
| WO2009003898A1 (de) | 2007-07-05 | 2009-01-08 | Basf Se | Organische leuchtdioden enthaltend carben-übergangsmetall-komplex-emitter und mindestens eine verbindung ausgewählt aus disilylcarbazolen; disilyldibenzofuranen, disilyldibenzothiophenen, disilyldibenzophospholen, disilyldibenzothiophen-s-oxiden und disilyldibenzothiophen-s,s-dioxiden |
| US20090017330A1 (en) | 2007-07-10 | 2009-01-15 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescence device and organic electroluminescence device utilizing the same |
| WO2009008311A1 (ja) | 2007-07-07 | 2009-01-15 | Idemitsu Kosan Co., Ltd. | クリセン誘導体及びそれを用いた有機エレクトロルミネッセンス素子 |
| US20090030202A1 (en) | 2007-07-10 | 2009-01-29 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescent element and organic electroluminescent element employing the same |
| WO2009018009A1 (en) | 2007-07-27 | 2009-02-05 | E. I. Du Pont De Nemours And Company | Aqueous dispersions of electrically conducting polymers containing inorganic nanoparticles |
| WO2009021126A2 (en) | 2007-08-08 | 2009-02-12 | Universal Display Corporation | Benzo-fused thiophene or benzo-fused furan compounds comprising a triphenylene group |
| US20090042829A1 (en) | 2007-08-06 | 2009-02-12 | Majed Matar | Nucleic Acid-Lipopolymer Compositions |
| US20090039776A1 (en) | 2007-08-09 | 2009-02-12 | Canon Kabushiki Kaisha | Organometallic complex and organic light-emitting element using same |
| US20090045731A1 (en) | 2007-07-07 | 2009-02-19 | Idemitsu Kosan Co., Ltd. | Organic electroluminescence device and material for organic electroluminescence device |
| US20090045730A1 (en) | 2007-07-07 | 2009-02-19 | Idemitsu Kosan Co., Ltd. | Organic electroluminescence device and material for organic electroluminescence device |
| EP2034538A1 (de) | 2006-06-02 | 2009-03-11 | Idemitsu Kosan Co., Ltd. | Material für ein organisches elektrolumineszenzelement und das material verwendendes organisches elektrolumineszenzelement |
| US20090101870A1 (en) | 2007-10-22 | 2009-04-23 | E. I. Du Pont De Nemours And Company | Electron transport bi-layers and devices made with such bi-layers |
| WO2009050290A1 (de) | 2007-10-17 | 2009-04-23 | Basf Se | Übergangsmetallkomplexe mit verbrückten carbenliganden und deren verwendung in oleds |
| US20090108737A1 (en) | 2006-12-08 | 2009-04-30 | Raymond Kwong | Light-emitting organometallic complexes |
| EP1841834B1 (de) | 2004-12-23 | 2009-05-06 | Ciba Holding Inc. | Elektrolumineszierende metallkomplexe mit nukleophilen carbenliganden |
| US20090115316A1 (en) | 2007-11-02 | 2009-05-07 | Shiying Zheng | Organic electroluminescent device having an azatriphenylene derivative |
| US7534505B2 (en) | 2004-05-18 | 2009-05-19 | The University Of Southern California | Organometallic compounds for use in electroluminescent devices |
| WO2009063833A1 (ja) | 2007-11-15 | 2009-05-22 | Idemitsu Kosan Co., Ltd. | ベンゾクリセン誘導体及びそれを用いた有機エレクトロルミネッセンス素子 |
| WO2009066779A1 (ja) | 2007-11-22 | 2009-05-28 | Idemitsu Kosan Co., Ltd. | 有機el素子 |
| WO2009066778A1 (ja) | 2007-11-22 | 2009-05-28 | Idemitsu Kosan Co., Ltd. | 有機el素子および有機el材料含有溶液 |
| US20090165846A1 (en) | 2005-09-07 | 2009-07-02 | Universitaet Braunschweig | Triplet emitter having condensed five-membered rings |
| US20090167162A1 (en) | 2007-12-28 | 2009-07-02 | Universal Display Corporation | Dibenzothiophene-containing materials in phosphorescent light emitting diodes |
| WO2009086028A2 (en) | 2007-12-28 | 2009-07-09 | Universal Display Corporation | Carbazole-containing materials in phosphorescent light emitting diodes |
| US20090179554A1 (en) | 2006-05-11 | 2009-07-16 | Hitoshi Kuma | Organic electroluminescent device |
| WO2009100991A1 (en) | 2008-02-12 | 2009-08-20 | Basf Se | Electroluminescent metal complexes with dibenzo[f,h]quinoxalines |
| US20090302743A1 (en) | 2008-06-05 | 2009-12-10 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescence device and organic electroluminescence device using the same |
| US20090309488A1 (en) | 2008-06-05 | 2009-12-17 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescence device and organic electroluminescence device using the same |
| US20100012931A1 (en) | 2008-06-05 | 2010-01-21 | Idemitsu Kosan Co., Ltd. | Polycyclic compounds and organic electroluminescence device employing the same |
| US7655323B2 (en) | 2004-05-18 | 2010-02-02 | The University Of Southern California | OLEDs utilizing macrocyclic ligand systems |
| WO2010028151A1 (en) | 2008-09-03 | 2010-03-11 | Universal Display Corporation | Phosphorescent materials |
| US20100084966A1 (en) | 2006-12-13 | 2010-04-08 | Konica Minolta Holdings, Inc. | Organic electroluminescent element, display device and lighting device |
| US20100090591A1 (en) | 2008-09-16 | 2010-04-15 | Universal Display Corporation | Phosphorescent materials |
| WO2010056066A1 (en) | 2008-11-13 | 2010-05-20 | Gracel Display Inc. | Novel organometallic compounds for electroluminescence and organic electroluminescent device using the same |
| US20100141127A1 (en) | 2008-11-11 | 2010-06-10 | Universal Display Corporation | Phosphorescent emitters |
| US20100148663A1 (en) | 2008-12-12 | 2010-06-17 | Universal Display Corporation | Blue Emitter with High Efficiency Based on Imidazo[1,2-f] Phenanthridine Iridium Complexes |
| WO2010079051A1 (de) | 2009-01-07 | 2010-07-15 | Basf Se | Silyl- und heteroatom-substituierte verbindungen ausgewählt aus carbazolen, dibenzofuranen, dibenzothiophenen und dibenzophospholen und ihre anwendung in der organischen elektronik |
| US20100187984A1 (en) | 2009-01-16 | 2010-07-29 | Universal Display Corporation | Materials with aza-dibenzothiophene or aza-dibenzofuran core for pholed |
| WO2010086089A1 (de) | 2009-02-02 | 2010-08-05 | Merck Patent Gmbh | Metallkomplexe |
| WO2010107244A2 (en) | 2009-03-20 | 2010-09-23 | Dow Advanced Display Materials, Ltd. | Novel organic electroluminescent compounds and organic electroluminescent device using the same |
| US20100244004A1 (en) | 2009-03-23 | 2010-09-30 | Universal Display Corporation | Heteroleptic iridium complex |
| US20100295032A1 (en) | 2009-05-20 | 2010-11-25 | Universal Display Corporation | Metal complexes with boron-nitrogen heterocycle containing ligands |
| US20110057559A1 (en) | 2007-12-28 | 2011-03-10 | Universal Display Corporation | Phosphorescent emitters and host materials with improved stability |
| WO2011044988A1 (de) | 2009-10-16 | 2011-04-21 | Merck Patent Gmbh | Metallkomplexe |
| WO2011051404A1 (de) | 2009-10-28 | 2011-05-05 | Basf Se | Heteroleptische carben-komplexe und deren verwendung in der organischen elektronik |
| WO2011075644A2 (en) | 2009-12-18 | 2011-06-23 | Plextronics, Inc. | Copolymers of 3,4-dialkoxythiophenes and methods for making and devices |
| US7968146B2 (en) | 2006-11-01 | 2011-06-28 | The Trustees Of Princeton University | Hybrid layers for use in coatings on electronic devices or other articles |
| US20110163302A1 (en) | 2008-06-30 | 2011-07-07 | Universal Display Corporation | Hole transport materials having a sulfur-containing group |
| WO2011086863A1 (ja) | 2010-01-15 | 2011-07-21 | 富士フイルム株式会社 | 有機電界発光素子 |
| EP2350216A1 (de) | 2008-10-27 | 2011-08-03 | Plextronics, Inc. | Ladungsinjektions- und transportschichten |
| US20110204333A1 (en) | 2010-02-25 | 2011-08-25 | Universal Display Corporation | Phosphorescent emitters |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0579151B1 (de) * | 1992-07-13 | 1996-10-09 | Eastman Kodak Company | Einen inneren Übergang aufweisende organisch elektrolumineszierende Vorrichtung mit einer neuen Zusammensetzung |
| US6835469B2 (en) | 2001-10-17 | 2004-12-28 | The University Of Southern California | Phosphorescent compounds and devices comprising the same |
| US6916554B2 (en) * | 2002-11-06 | 2005-07-12 | The University Of Southern California | Organic light emitting materials and devices |
| US7029765B2 (en) | 2003-04-22 | 2006-04-18 | Universal Display Corporation | Organic light emitting devices having reduced pixel shrinkage |
| DE10338550A1 (de) | 2003-08-19 | 2005-03-31 | Basf Ag | Übergangsmetallkomplexe mit Carbenliganden als Emitter für organische Licht-emittierende Dioden (OLEDs) |
| DE102007053771A1 (de) | 2007-11-12 | 2009-05-14 | Merck Patent Gmbh | Organische Elektrolumineszenzvorrichtungen |
| US9212197B2 (en) * | 2011-05-19 | 2015-12-15 | Universal Display Corporation | Phosphorescent heteroleptic phenylbenzimidazole dopants |
| EP2699027B1 (de) | 2011-08-02 | 2021-10-06 | Huawei Device Co., Ltd. | Verfahren, vorrichtung und endgerät zur erzeugung und verarbeitung von nachrichten |
-
2013
- 2013-02-08 US US13/762,612 patent/US10400163B2/en active Active
-
2014
- 2014-01-23 KR KR1020140008328A patent/KR102139384B1/ko active Active
- 2014-01-29 CN CN201410043782.7A patent/CN103980321B/zh active Active
- 2014-02-05 DE DE102014001468.1A patent/DE102014001468B4/de active Active
Patent Citations (158)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4769292A (en) | 1987-03-02 | 1988-09-06 | Eastman Kodak Company | Electroluminescent device with modified thin film luminescent zone |
| US5247190A (en) | 1989-04-20 | 1993-09-21 | Cambridge Research And Innovation Limited | Electroluminescent devices |
| US5061569A (en) | 1990-07-26 | 1991-10-29 | Eastman Kodak Company | Electroluminescent device with organic electroluminescent medium |
| EP0650955A1 (de) | 1993-11-01 | 1995-05-03 | Hodogaya Chemical Co., Ltd. | Aminverbindung und sie enthaltende Elektrolumineszenzvorrichtung |
| US5703436A (en) | 1994-12-13 | 1997-12-30 | The Trustees Of Princeton University | Transparent contacts for organic devices |
| US5707745A (en) | 1994-12-13 | 1998-01-13 | The Trustees Of Princeton University | Multicolor organic light emitting devices |
| US20030162053A1 (en) | 1996-06-25 | 2003-08-28 | Marks Tobin J. | Organic light - emitting diodes and methods for assembly and enhanced charge injection |
| US5834893A (en) | 1996-12-23 | 1998-11-10 | The Trustees Of Princeton University | High efficiency organic light emitting devices with light directing structures |
| US6013982A (en) | 1996-12-23 | 2000-01-11 | The Trustees Of Princeton University | Multicolor display devices |
| US5844363A (en) | 1997-01-23 | 1998-12-01 | The Trustees Of Princeton Univ. | Vacuum deposited, non-polymeric flexible organic light emitting devices |
| US6091195A (en) | 1997-02-03 | 2000-07-18 | The Trustees Of Princeton University | Displays having mesa pixel configuration |
| US6337102B1 (en) | 1997-11-17 | 2002-01-08 | The Trustees Of Princeton University | Low pressure vapor phase deposition of organic thin films |
| US6303238B1 (en) | 1997-12-01 | 2001-10-16 | The Trustees Of Princeton University | OLEDs doped with phosphorescent compounds |
| US6087196A (en) | 1998-01-30 | 2000-07-11 | The Trustees Of Princeton University | Fabrication of organic semiconductor devices using ink jet printing |
| US6528187B1 (en) | 1998-09-08 | 2003-03-04 | Fuji Photo Film Co., Ltd. | Material for luminescence element and luminescence element using the same |
| US6097147A (en) | 1998-09-14 | 2000-08-01 | The Trustees Of Princeton University | Structure for high efficiency electroluminescent device |
| US20020034656A1 (en) | 1998-09-14 | 2002-03-21 | Thompson Mark E. | Organometallic complexes as phosphorescent emitters in organic LEDs |
| US6294398B1 (en) | 1999-11-23 | 2001-09-25 | The Trustees Of Princeton University | Method for patterning devices |
| US6468819B1 (en) | 1999-11-23 | 2002-10-22 | The Trustees Of Princeton University | Method for patterning organic thin film devices using a die |
| WO2001039234A2 (en) | 1999-11-24 | 2001-05-31 | The Trustees Of Princeton University | Organic light emitting diode having a blue phosphorescent molecule as an emitter |
| US20020158242A1 (en) | 1999-12-31 | 2002-10-31 | Se-Hwan Son | Electronic device comprising organic compound having p-type semiconducting characteristics |
| US20010015432A1 (en) | 2000-02-10 | 2001-08-23 | Tatsuya Igarashi | Light emitting device material comprising iridium complex and light emitting device using same material |
| WO2002002714A2 (en) | 2000-06-30 | 2002-01-10 | E.I. Du Pont De Nemours And Company | Electroluminescent iridium compounds with fluorinated phenylpyridines, phenylpyrimidines, and phenylquinolines and devices made with such compounds |
| WO2002015645A1 (en) | 2000-08-11 | 2002-02-21 | The Trustees Of Princeton University | Organometallic compounds and emission-shifting organic electrophosphorescence |
| US20030138657A1 (en) | 2000-12-07 | 2003-07-24 | Canon Kabushiki Kaisha | Deuterated semi-conducting organic compounds used for opto-electronic devices |
| US20020134984A1 (en) | 2001-02-01 | 2002-09-26 | Fuji Photo Film Co., Ltd. | Transition metal complex and light-emitting device |
| US6921915B2 (en) | 2001-03-08 | 2005-07-26 | Canon Kabushiki Kaisha | Metal coordination compound, luminescence device and display apparatus |
| US20030152802A1 (en) | 2001-06-19 | 2003-08-14 | Akira Tsuboyama | Metal coordination compound and organic liminescence device |
| US7396598B2 (en) | 2001-06-20 | 2008-07-08 | Showa Denko K.K. | Light emitting material and organic light-emitting device |
| US20040174116A1 (en) | 2001-08-20 | 2004-09-09 | Lu Min-Hao Michael | Transparent electrodes |
| US7250226B2 (en) | 2001-08-31 | 2007-07-31 | Nippon Hoso Kyokai | Phosphorescent compound, a phosphorescent composition and an organic light-emitting device |
| US7431968B1 (en) | 2001-09-04 | 2008-10-07 | The Trustees Of Princeton University | Process and apparatus for organic vapor jet deposition |
| WO2003040257A1 (en) | 2001-11-07 | 2003-05-15 | E. I. Du Pont De Nemours And Company | Electroluminescent platinum compounds and devices made with such compounds |
| US20030175553A1 (en) | 2001-12-28 | 2003-09-18 | Thompson Mark E. | White light emitting oleds from combined monomer and aggregate emission |
| WO2003060956A2 (en) | 2002-01-18 | 2003-07-24 | Lg Chem, Ltd. | New material for transporting electrons and organic electroluminescent display using the same |
| US7232618B2 (en) | 2002-04-12 | 2007-06-19 | Canon Kabushiki Kaisha | Organic light-emitting device |
| US20030230980A1 (en) | 2002-06-18 | 2003-12-18 | Forrest Stephen R | Very low voltage, high efficiency phosphorescent oled in a p-i-n structure |
| US20040036077A1 (en) | 2002-08-22 | 2004-02-26 | Fuji Photo Film Co., Ltd. | Light emitting element |
| US20060127696A1 (en) | 2002-08-24 | 2006-06-15 | Covion Organic Semiconductors Gmbh | Rhodium and iridium complexes |
| US20050244673A1 (en) | 2002-08-27 | 2005-11-03 | Fujitsu Limited | Organometallic complex, organic EL element and organic EL display |
| US6687266B1 (en) | 2002-11-08 | 2004-02-03 | Universal Display Corporation | Organic light emitting materials and devices |
| US20040115476A1 (en) | 2002-11-26 | 2004-06-17 | Tomohiro Oshiyama | Organic electroluminescent element, and display and illuminator |
| US20040137267A1 (en) | 2002-12-27 | 2004-07-15 | Fuji Photo Film Co., Ltd. | Organic electroluminescent device |
| US20040137268A1 (en) | 2002-12-27 | 2004-07-15 | Fuji Photo Film Co., Ltd. | Organic electroluminescent device |
| US7338722B2 (en) | 2003-03-24 | 2008-03-04 | The University Of Southern California | Phenyl and fluorenyl substituted phenyl-pyrazole complexes of Ir |
| US7090928B2 (en) | 2003-04-01 | 2006-08-15 | The University Of Southern California | Binuclear compounds |
| WO2004093207A2 (de) | 2003-04-15 | 2004-10-28 | Covion Organic Semiconductors Gmbh | Mischungen von organischen zur emission befähigten halbleitern und matrixmaterialien, deren verwendung und elektronikbauteile enthaltend diese mischungen |
| US20060121308A1 (en) | 2003-04-23 | 2006-06-08 | Eisaku Katoh | Material for organic electroluminescent device, organic electroluminescent device, illuminating device and display |
| WO2004107822A1 (ja) | 2003-05-29 | 2004-12-09 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子 |
| US20060182992A1 (en) | 2003-06-02 | 2006-08-17 | Kazumi Nii | Organic electroluminescent devices and metal complex compounds |
| JP2005011610A (ja) | 2003-06-18 | 2005-01-13 | Nippon Steel Chem Co Ltd | 有機電界発光素子 |
| US20050025993A1 (en) | 2003-07-25 | 2005-02-03 | Thompson Mark E. | Materials and structures for enhancing the performance of organic light emitting devices |
| WO2005014551A1 (ja) | 2003-08-07 | 2005-02-17 | Nippon Steel Chemical Co., Ltd. | 有機el材料用のアルミニウムキレート錯体 |
| US20050123751A1 (en) | 2003-08-25 | 2005-06-09 | Tetsuo Tsutsui | Electrode device for organic device, electronic device having electrode device for organic device, and method of forming electrode device for organic device |
| US20070087321A1 (en) | 2003-09-09 | 2007-04-19 | Csaba Pribenszky | Post-thaw survival of chryopreserved biological material by hydrostatic pressure challenge |
| WO2005030900A1 (ja) | 2003-09-25 | 2005-04-07 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子 |
| US20070023098A1 (en) | 2003-10-22 | 2007-02-01 | Uster Technologies Ag | Holding element for a device for monitoring the quality on a mechanical weaving loom |
| US20070103060A1 (en) | 2003-11-04 | 2007-05-10 | Takasago International Corporation | Platinum complex and light emitting device |
| US20050112407A1 (en) | 2003-11-21 | 2005-05-26 | Fuji Photo Film Co., Ltd. | Organic electroluminescent device |
| US7332232B2 (en) | 2004-02-03 | 2008-02-19 | Universal Display Corporation | OLEDs utilizing multidentate ligand systems |
| EP1725079A1 (de) | 2004-03-11 | 2006-11-22 | Mitsubishi Chemical Corporation | Zusammensetzung für einen ladungstransportfilm und ionenzusammensetzungs-ladungstransportfilm und organische elektrolumineszenzeinrichtung damit und verfahren zur herstellung einer organischen elektrolumineszenzeinrichtung und verfahren zur herstellung eines ladungstransportfilms |
| WO2005089025A1 (ja) | 2004-03-15 | 2005-09-22 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子 |
| US20050238919A1 (en) | 2004-04-23 | 2005-10-27 | Fuji Photo Film Co., Ltd. | Organic electroluminescent device |
| US7655323B2 (en) | 2004-05-18 | 2010-02-02 | The University Of Southern California | OLEDs utilizing macrocyclic ligand systems |
| US7393599B2 (en) | 2004-05-18 | 2008-07-01 | The University Of Southern California | Luminescent compounds with carbene ligands |
| US20050260441A1 (en) | 2004-05-18 | 2005-11-24 | Thompson Mark E | Luminescent compounds with carbene ligands |
| US7445855B2 (en) | 2004-05-18 | 2008-11-04 | The University Of Southern California | Cationic metal-carbene complexes |
| US7279704B2 (en) | 2004-05-18 | 2007-10-09 | The University Of Southern California | Complexes with tridentate ligands |
| US7534505B2 (en) | 2004-05-18 | 2009-05-19 | The University Of Southern California | Organometallic compounds for use in electroluminescent devices |
| US7154114B2 (en) | 2004-05-18 | 2006-12-26 | Universal Display Corporation | Cyclometallated iridium carbene complexes for use as hosts |
| WO2005123873A1 (ja) | 2004-06-17 | 2005-12-29 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20080015355A1 (en) | 2004-06-28 | 2008-01-17 | Thomas Schafer | Electroluminescent Metal Complexes With Triazoles And Benzotriazoles |
| US20060008670A1 (en) | 2004-07-06 | 2006-01-12 | Chun Lin | Organic light emitting materials and devices |
| WO2006009024A1 (ja) | 2004-07-23 | 2006-01-26 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| WO2006056418A2 (de) | 2004-11-25 | 2006-06-01 | Basf Aktiengesellschaft | Verwendung von übergangsmetall-carbenkomplexen in organischen licht-emittierenden dioden (oleds) |
| US20080018221A1 (en) | 2004-11-25 | 2008-01-24 | Basf Aktiengesellschaft | Use Of Transition Metal Carbene Complexes In Organic Light-Emitting Diodes (Oleds) |
| EP1841834B1 (de) | 2004-12-23 | 2009-05-06 | Ciba Holding Inc. | Elektrolumineszierende metallkomplexe mit nukleophilen carbenliganden |
| US7230107B1 (en) | 2004-12-29 | 2007-06-12 | E. I. Du Pont De Nemours And Company | Metal quinoline complexes |
| WO2006072002A2 (en) | 2004-12-30 | 2006-07-06 | E.I. Dupont De Nemours And Company | Organometallic complexes |
| WO2006082742A1 (ja) | 2005-02-04 | 2006-08-10 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20060202194A1 (en) | 2005-03-08 | 2006-09-14 | Jeong Hyun C | Red phosphorescene compounds and organic electroluminescence device using the same |
| WO2006098120A1 (ja) | 2005-03-16 | 2006-09-21 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子 |
| WO2006100298A1 (de) | 2005-03-24 | 2006-09-28 | Basf Aktiengesellschaft | Verwendung von verbindungen, welche aromatische oder heteroaromatische über carbonyl-gruppen enthaltende gruppen verbundene ringe enthalten, als matrixmaterialien in organischen leuchtdioden |
| WO2006103874A1 (ja) | 2005-03-29 | 2006-10-05 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| WO2006114966A1 (ja) | 2005-04-18 | 2006-11-02 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20060240279A1 (en) | 2005-04-21 | 2006-10-26 | Vadim Adamovich | Non-blocked phosphorescent OLEDs |
| US20060263635A1 (en) | 2005-05-06 | 2006-11-23 | Fuji Photo Film Co., Ltd. | Organic electroluminescent device |
| US20060251923A1 (en) | 2005-05-06 | 2006-11-09 | Chun Lin | Stability OLED materials and devices |
| US20060280965A1 (en) | 2005-05-31 | 2006-12-14 | Raymond Kwong | Triphenylene hosts in phosphorescent light emitting diodes |
| WO2006132173A1 (ja) | 2005-06-07 | 2006-12-14 | Nippon Steel Chemical Co., Ltd. | 有機金属錯体及びこれを用いた有機電界発光素子 |
| WO2007002683A2 (en) | 2005-06-27 | 2007-01-04 | E. I. Du Pont De Nemours And Company | Electrically conductive polymer compositions |
| WO2007004380A1 (ja) | 2005-07-01 | 2007-01-11 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20090165846A1 (en) | 2005-09-07 | 2009-07-02 | Universitaet Braunschweig | Triplet emitter having condensed five-membered rings |
| JP2007123392A (ja) | 2005-10-26 | 2007-05-17 | Konica Minolta Holdings Inc | 有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US20070111026A1 (en) | 2005-11-15 | 2007-05-17 | Deaton Joseph C | Oled devices with dinuclear copper compounds |
| WO2007063754A1 (ja) | 2005-12-01 | 2007-06-07 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子用化合物及び有機電界発光素子 |
| WO2007063796A1 (ja) | 2005-12-01 | 2007-06-07 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子 |
| US20080297033A1 (en) | 2006-02-10 | 2008-12-04 | Knowles David B | Blue phosphorescent imidazophenanthridine materials |
| US20070190359A1 (en) | 2006-02-10 | 2007-08-16 | Knowles David B | Metal complexes of cyclometallated imidazo[1,2-ƒ]phenanthridine and diimidazo[1,2-a:1',2'-c]quinazoline ligands and isoelectronic and benzannulated analogs thereof |
| JP2007254297A (ja) | 2006-03-20 | 2007-10-04 | Nippon Steel Chem Co Ltd | 発光層化合物及び有機電界発光素子 |
| US20070278938A1 (en) | 2006-04-26 | 2007-12-06 | Idemitsu Kosan Co., Ltd. | Aromatic amine derivative and electroluminescence device using the same |
| US20090179554A1 (en) | 2006-05-11 | 2009-07-16 | Hitoshi Kuma | Organic electroluminescent device |
| EP2034538A1 (de) | 2006-06-02 | 2009-03-11 | Idemitsu Kosan Co., Ltd. | Material für ein organisches elektrolumineszenzelement und das material verwendendes organisches elektrolumineszenzelement |
| US20080106190A1 (en) | 2006-08-23 | 2008-05-08 | Idemitsu Kosan Co., Ltd. | Aromatic amine derivatives and organic electroluminescent device using same |
| JP2008074939A (ja) | 2006-09-21 | 2008-04-03 | Konica Minolta Holdings Inc | 有機エレクトロルミネッセンス素子材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US7968146B2 (en) | 2006-11-01 | 2011-06-28 | The Trustees Of Princeton University | Hybrid layers for use in coatings on electronic devices or other articles |
| WO2008056746A1 (en) | 2006-11-09 | 2008-05-15 | Nippon Steel Chemical Co., Ltd. | Compound for organic electroluminescent device and organic electroluminescent device |
| US20080124572A1 (en) | 2006-11-24 | 2008-05-29 | Idemitsu Kosan Co., Ltd. | Aromatic amine derivative and organic electroluminescence device using the same |
| US20090108737A1 (en) | 2006-12-08 | 2009-04-30 | Raymond Kwong | Light-emitting organometallic complexes |
| US20080220265A1 (en) | 2006-12-08 | 2008-09-11 | Universal Display Corporation | Cross-linkable Iridium Complexes and Organic Light-Emitting Devices Using the Same |
| US20100084966A1 (en) | 2006-12-13 | 2010-04-08 | Konica Minolta Holdings, Inc. | Organic electroluminescent element, display device and lighting device |
| WO2008101842A1 (en) | 2007-02-23 | 2008-08-28 | Basf Se | Electroluminescent metal complexes with benzotriazoles |
| US20080261076A1 (en) | 2007-03-08 | 2008-10-23 | Universal Display Corporation | Phosphorescent materials |
| WO2008132085A1 (de) | 2007-04-26 | 2008-11-06 | Basf Se | Silane enthaltend phenothiazin-s-oxid oder phenothiazin-s,s-dioxid-gruppen und deren verwendung in oleds |
| WO2009000673A2 (en) | 2007-06-22 | 2008-12-31 | Basf Se | Light emitting cu(i) complexes |
| WO2009003898A1 (de) | 2007-07-05 | 2009-01-08 | Basf Se | Organische leuchtdioden enthaltend carben-übergangsmetall-komplex-emitter und mindestens eine verbindung ausgewählt aus disilylcarbazolen; disilyldibenzofuranen, disilyldibenzothiophenen, disilyldibenzophospholen, disilyldibenzothiophen-s-oxiden und disilyldibenzothiophen-s,s-dioxiden |
| WO2009008311A1 (ja) | 2007-07-07 | 2009-01-15 | Idemitsu Kosan Co., Ltd. | クリセン誘導体及びそれを用いた有機エレクトロルミネッセンス素子 |
| US20090008605A1 (en) | 2007-07-07 | 2009-01-08 | Idemitsu Kosan Co., Ltd. | Naphthalene derivative, material for organic electroluminescence device, and organic electroluminescence device using the same |
| US20090045731A1 (en) | 2007-07-07 | 2009-02-19 | Idemitsu Kosan Co., Ltd. | Organic electroluminescence device and material for organic electroluminescence device |
| US20090045730A1 (en) | 2007-07-07 | 2009-02-19 | Idemitsu Kosan Co., Ltd. | Organic electroluminescence device and material for organic electroluminescence device |
| US20090009065A1 (en) | 2007-07-07 | 2009-01-08 | Idemitsu Kosan Co., Ltd. | Organic electroluminescence device and material for organic electroluminescence device |
| US20090030202A1 (en) | 2007-07-10 | 2009-01-29 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescent element and organic electroluminescent element employing the same |
| US20090017330A1 (en) | 2007-07-10 | 2009-01-15 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescence device and organic electroluminescence device utilizing the same |
| WO2009018009A1 (en) | 2007-07-27 | 2009-02-05 | E. I. Du Pont De Nemours And Company | Aqueous dispersions of electrically conducting polymers containing inorganic nanoparticles |
| US20090042829A1 (en) | 2007-08-06 | 2009-02-12 | Majed Matar | Nucleic Acid-Lipopolymer Compositions |
| WO2009021126A2 (en) | 2007-08-08 | 2009-02-12 | Universal Display Corporation | Benzo-fused thiophene or benzo-fused furan compounds comprising a triphenylene group |
| US20090039776A1 (en) | 2007-08-09 | 2009-02-12 | Canon Kabushiki Kaisha | Organometallic complex and organic light-emitting element using same |
| WO2009050290A1 (de) | 2007-10-17 | 2009-04-23 | Basf Se | Übergangsmetallkomplexe mit verbrückten carbenliganden und deren verwendung in oleds |
| US20090101870A1 (en) | 2007-10-22 | 2009-04-23 | E. I. Du Pont De Nemours And Company | Electron transport bi-layers and devices made with such bi-layers |
| US20090115316A1 (en) | 2007-11-02 | 2009-05-07 | Shiying Zheng | Organic electroluminescent device having an azatriphenylene derivative |
| WO2009063833A1 (ja) | 2007-11-15 | 2009-05-22 | Idemitsu Kosan Co., Ltd. | ベンゾクリセン誘導体及びそれを用いた有機エレクトロルミネッセンス素子 |
| WO2009066778A1 (ja) | 2007-11-22 | 2009-05-28 | Idemitsu Kosan Co., Ltd. | 有機el素子および有機el材料含有溶液 |
| WO2009066779A1 (ja) | 2007-11-22 | 2009-05-28 | Idemitsu Kosan Co., Ltd. | 有機el素子 |
| US20110057559A1 (en) | 2007-12-28 | 2011-03-10 | Universal Display Corporation | Phosphorescent emitters and host materials with improved stability |
| US20090167162A1 (en) | 2007-12-28 | 2009-07-02 | Universal Display Corporation | Dibenzothiophene-containing materials in phosphorescent light emitting diodes |
| WO2009086028A2 (en) | 2007-12-28 | 2009-07-09 | Universal Display Corporation | Carbazole-containing materials in phosphorescent light emitting diodes |
| WO2009100991A1 (en) | 2008-02-12 | 2009-08-20 | Basf Se | Electroluminescent metal complexes with dibenzo[f,h]quinoxalines |
| US20090302743A1 (en) | 2008-06-05 | 2009-12-10 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescence device and organic electroluminescence device using the same |
| US20090309488A1 (en) | 2008-06-05 | 2009-12-17 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescence device and organic electroluminescence device using the same |
| US20100012931A1 (en) | 2008-06-05 | 2010-01-21 | Idemitsu Kosan Co., Ltd. | Polycyclic compounds and organic electroluminescence device employing the same |
| US20110163302A1 (en) | 2008-06-30 | 2011-07-07 | Universal Display Corporation | Hole transport materials having a sulfur-containing group |
| WO2010028151A1 (en) | 2008-09-03 | 2010-03-11 | Universal Display Corporation | Phosphorescent materials |
| US20100090591A1 (en) | 2008-09-16 | 2010-04-15 | Universal Display Corporation | Phosphorescent materials |
| EP2350216A1 (de) | 2008-10-27 | 2011-08-03 | Plextronics, Inc. | Ladungsinjektions- und transportschichten |
| US20100141127A1 (en) | 2008-11-11 | 2010-06-10 | Universal Display Corporation | Phosphorescent emitters |
| WO2010056066A1 (en) | 2008-11-13 | 2010-05-20 | Gracel Display Inc. | Novel organometallic compounds for electroluminescence and organic electroluminescent device using the same |
| US20100148663A1 (en) | 2008-12-12 | 2010-06-17 | Universal Display Corporation | Blue Emitter with High Efficiency Based on Imidazo[1,2-f] Phenanthridine Iridium Complexes |
| WO2010079051A1 (de) | 2009-01-07 | 2010-07-15 | Basf Se | Silyl- und heteroatom-substituierte verbindungen ausgewählt aus carbazolen, dibenzofuranen, dibenzothiophenen und dibenzophospholen und ihre anwendung in der organischen elektronik |
| US20100187984A1 (en) | 2009-01-16 | 2010-07-29 | Universal Display Corporation | Materials with aza-dibenzothiophene or aza-dibenzofuran core for pholed |
| WO2010086089A1 (de) | 2009-02-02 | 2010-08-05 | Merck Patent Gmbh | Metallkomplexe |
| WO2010107244A2 (en) | 2009-03-20 | 2010-09-23 | Dow Advanced Display Materials, Ltd. | Novel organic electroluminescent compounds and organic electroluminescent device using the same |
| US20100244004A1 (en) | 2009-03-23 | 2010-09-30 | Universal Display Corporation | Heteroleptic iridium complex |
| US20100295032A1 (en) | 2009-05-20 | 2010-11-25 | Universal Display Corporation | Metal complexes with boron-nitrogen heterocycle containing ligands |
| WO2011044988A1 (de) | 2009-10-16 | 2011-04-21 | Merck Patent Gmbh | Metallkomplexe |
| WO2011051404A1 (de) | 2009-10-28 | 2011-05-05 | Basf Se | Heteroleptische carben-komplexe und deren verwendung in der organischen elektronik |
| WO2011075644A2 (en) | 2009-12-18 | 2011-06-23 | Plextronics, Inc. | Copolymers of 3,4-dialkoxythiophenes and methods for making and devices |
| WO2011086863A1 (ja) | 2010-01-15 | 2011-07-21 | 富士フイルム株式会社 | 有機電界発光素子 |
| US20110204333A1 (en) | 2010-02-25 | 2011-08-25 | Universal Display Corporation | Phosphorescent emitters |
Non-Patent Citations (1)
| Title |
|---|
| Baldo et al., „Highly Efficient Phosphorescent Emission from Organic Electroluminescent Devices", Nature, Band 395, 151-154, 1998, („Baldo-I") und Baldo et al., „Very high-efficiency green organic light-emitting devices based on electrophosphorescence", Appl. Phys. Lett., Band 75, Nr. 3, 4-6 (1999 |
Also Published As
| Publication number | Publication date |
|---|---|
| US10400163B2 (en) | 2019-09-03 |
| KR102139384B1 (ko) | 2020-07-30 |
| DE102014001468A1 (de) | 2014-08-14 |
| CN103980321B (zh) | 2018-10-26 |
| CN103980321A (zh) | 2014-08-13 |
| US20140225069A1 (en) | 2014-08-14 |
| KR20140101292A (ko) | 2014-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE102013019465B4 (de) | Organische elektrolumineszenzvorrichtung mit verzögerter fluoreszenz | |
| DE102012220691B4 (de) | Triphenylensilanwirte | |
| DE112011101498B4 (de) | Triphenylen-Benzofuran-/Benzothiophen-/Benzoselenophen-Verbindungen mit Substituenten, die sich zu fusionierten Ringen zusammenschließen sowie Vorrichtung diese umfassend | |
| DE112010005815B4 (de) | Bicarbazolverbindungen für OLEDs | |
| DE112011103404B4 (de) | Neuartige 3,9-verknüpfte Oligocarbazole und OLEDs, die diese neuartigen Oligocarbazole enthalten | |
| DE112011101447B4 (de) | Bicarbazol enthaltende Verbindungen für OLEDs | |
| DE112011101663B4 (de) | Azaborinverbindungen als Hostmaterialien und Dotiermittel für Pholeds | |
| DE112012002237B4 (de) | Host Materialien für OLEDS | |
| EP3205658B1 (de) | Organische elektrolumineszenzmaterialien und vorrichtungen | |
| EP2891659B1 (de) | Organische elektrolumineszente Materialien und Vorrichtungen | |
| EP2602302B1 (de) | Neuartige organische lichtemittierende materialien | |
| EP2718302B1 (de) | Heteroleptische iridium-carben-komplexe und lichtemittierende vorrichtung damit | |
| DE102014001468B4 (de) | Heteroleptischer phenylbenzimidazol-komplex | |
| DE102016201672A1 (de) | Organische elektrolumineszierende Materialien und Vorrichtungen | |
| EP2594572A1 (de) | Organische lichtemittierende Materialien | |
| DE102013208026A1 (de) | Asymmetrische Wirte mit Triarylsilanseitenketten | |
| DE112011101983T5 (de) | OLED mit verzögerter Fluoreszenz | |
| EP3001480B1 (de) | Organische elektrolumineszente materialien und vorrichtungen | |
| US9929361B2 (en) | Organic electroluminescent materials and devices | |
| DE102013214144B4 (de) | Diarylamino-substituierte Metallkomplexe | |
| DE102013003605B4 (de) | Sekundäre Lochtransportschicht mit Tricarbazolverbindungen | |
| EP3301099B1 (de) | Phosphoreszierende homoleptische tris-[deuterierte-2-(2-pyridinyl)phenyl]-iridium-komplexe und deren verwendung in lichtemitierenden gegenständen. | |
| EP2874192B1 (de) | Organische elektrolumineszente Materialien und Vorrichtungen | |
| DE102013020303B4 (de) | Lochtransportmaterialien mit verdrehten Arylgruppen | |
| DE102013200085B4 (de) | Hocheffiziente phosphoreszierende Materialien |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| R082 | Change of representative |
Representative=s name: MUELLER-BORE & PARTNER PATENTANWAELTE PARTG MB, DE Representative=s name: MAIWALD PATENTANWALTSGESELLSCHAFT MBH, DE Representative=s name: MAIWALD PATENTANWALTS- UND RECHTSANWALTSGESELL, DE |
|
| R082 | Change of representative |
Representative=s name: MAIWALD PATENTANWALTSGESELLSCHAFT MBH, DE Representative=s name: MAIWALD PATENTANWALTS- UND RECHTSANWALTSGESELL, DE |
|
| R012 | Request for examination validly filed | ||
| R016 | Response to examination communication | ||
| R016 | Response to examination communication | ||
| R018 | Grant decision by examination section/examining division | ||
| R020 | Patent grant now final |