CN1470506A - 一种合成多卡巴胺的方法 - Google Patents
一种合成多卡巴胺的方法 Download PDFInfo
- Publication number
- CN1470506A CN1470506A CNA021361576A CN02136157A CN1470506A CN 1470506 A CN1470506 A CN 1470506A CN A021361576 A CNA021361576 A CN A021361576A CN 02136157 A CN02136157 A CN 02136157A CN 1470506 A CN1470506 A CN 1470506A
- Authority
- CN
- China
- Prior art keywords
- ethanoyl
- docarpamine
- methionyl
- homopiperony lamine
- preparing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000000034 method Methods 0.000 title claims abstract description 21
- 230000002194 synthesizing effect Effects 0.000 title abstract description 6
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 claims abstract description 10
- 150000003839 salts Chemical class 0.000 claims abstract description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 6
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 5
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 claims abstract description 5
- 239000003153 chemical reaction reagent Substances 0.000 claims abstract description 4
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 23
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 22
- 238000006243 chemical reaction Methods 0.000 claims description 20
- RRIRDPSOCUCGBV-UHFFFAOYSA-N methylenedioxyphenethylamine Chemical compound NCCC1=CC=C2OCOC2=C1 RRIRDPSOCUCGBV-UHFFFAOYSA-N 0.000 claims description 20
- CTENFNNZBMHDDG-UHFFFAOYSA-N Dopamine hydrochloride Chemical compound Cl.NCCC1=CC=C(O)C(O)=C1 CTENFNNZBMHDDG-UHFFFAOYSA-N 0.000 claims description 17
- ZLVMAMIPILWYHQ-INIZCTEOSA-N Docarpamine Chemical compound CCOC(=O)OC1=CC=C(CCNC(=O)[C@H](CCSC)NC(C)=O)C=C1OC(=O)OCC ZLVMAMIPILWYHQ-INIZCTEOSA-N 0.000 claims description 14
- 229950006045 docarpamine Drugs 0.000 claims description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 12
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 claims description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 12
- 229960004452 methionine Drugs 0.000 claims description 12
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 claims description 12
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 11
- 239000002904 solvent Substances 0.000 claims description 10
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 6
- HZVOZRGWRWCICA-UHFFFAOYSA-N methanediyl Chemical compound [CH2] HZVOZRGWRWCICA-UHFFFAOYSA-N 0.000 claims description 6
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 claims description 5
- 239000003513 alkali Substances 0.000 claims description 4
- -1 hydrobromate Chemical compound 0.000 claims description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 3
- 238000009833 condensation Methods 0.000 claims description 3
- 230000005494 condensation Effects 0.000 claims description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 3
- 239000002994 raw material Substances 0.000 claims description 3
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 claims description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 claims description 2
- 238000006482 condensation reaction Methods 0.000 claims description 2
- YYTWEEOFRNSTKS-UHFFFAOYSA-N n,n'-dicyclohexylmethanediimine;1-hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1.C1CCCCC1N=C=NC1CCCCC1 YYTWEEOFRNSTKS-UHFFFAOYSA-N 0.000 claims description 2
- 229950004288 tosilate Drugs 0.000 claims description 2
- 238000009776 industrial production Methods 0.000 abstract description 2
- XUYPXLNMDZIRQH-LURJTMIESA-N N-acetyl-L-methionine Chemical compound CSCC[C@@H](C(O)=O)NC(C)=O XUYPXLNMDZIRQH-LURJTMIESA-N 0.000 abstract 1
- 239000007858 starting material Substances 0.000 abstract 1
- 238000003756 stirring Methods 0.000 description 17
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 11
- 239000012044 organic layer Substances 0.000 description 10
- 239000000243 solution Substances 0.000 description 9
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- 238000001035 drying Methods 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 6
- 239000012141 concentrate Substances 0.000 description 6
- 150000001875 compounds Chemical class 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 235000002639 sodium chloride Nutrition 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 4
- 238000003810 ethyl acetate extraction Methods 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- PQLFROTZSIMBKR-UHFFFAOYSA-N ethenyl carbonochloridate Chemical compound ClC(=O)OC=C PQLFROTZSIMBKR-UHFFFAOYSA-N 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 230000006340 racemization Effects 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 229960001149 dopamine hydrochloride Drugs 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- NDYXFQODWGEGNU-UHFFFAOYSA-N 2-(1,3-benzodioxol-5-yl)ethanamine;hydron;chloride Chemical compound Cl.NCCC1=CC=C2OCOC2=C1 NDYXFQODWGEGNU-UHFFFAOYSA-N 0.000 description 1
- MIDXCONKKJTLDX-UHFFFAOYSA-N 3,5-dimethylcyclopentane-1,2-dione Chemical compound CC1CC(C)C(=O)C1=O MIDXCONKKJTLDX-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- FFEARJCKVFRZRR-UHFFFAOYSA-N L-Methionine Natural products CSCCC(N)C(O)=O FFEARJCKVFRZRR-UHFFFAOYSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- 229930195722 L-methionine Natural products 0.000 description 1
- 102000010909 Monoamine Oxidase Human genes 0.000 description 1
- 108010062431 Monoamine oxidase Proteins 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- 208000001647 Renal Insufficiency Diseases 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 235000013736 caramel Nutrition 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000027744 congestion Diseases 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- HWJHWSBFPPPIPD-UHFFFAOYSA-N ethoxyethane;propan-2-one Chemical compound CC(C)=O.CCOCC HWJHWSBFPPPIPD-UHFFFAOYSA-N 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000003292 glue Substances 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- HOGDNTQCSIKEEV-UHFFFAOYSA-N n'-hydroxybutanediamide Chemical compound NC(=O)CCC(=O)NO HOGDNTQCSIKEEV-UHFFFAOYSA-N 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
本发明提供一条合成多卡巴胺的新方法,采用胡椒乙胺或它的盐为起始原料与N-乙酰基-L-甲硫氨酸经混合缩合剂法缩合,得关键中间体N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺再用三溴化硼等试剂脱亚甲基,最后用乙氧羰基保护两个羟基合成了多卡巴胺。本方法降低了成本,提高收率,纯度高且操作简便,适合于工业生产。
Description
技术领域
本发明涉及药物合成的方法改进,更具体地说是药物多卡巴胺的全合成工艺改进。
背景技术多卡巴胺为多巴胺的口服的前药,临床用作心脏兴奋剂,其结构如下:
多卡巴胺为多巴胺的可口服的前药,临床用作心脏兴奋剂。已知多巴胺临床用于治疗肾衰竭、高血压、慢性充血性心脏病以及其他疾病,但是由于多巴胺难于从消化器官吸收,故只能通过注射给药。而且,静脉给药后多巴胺被肝脏中单胺氧化酶或其他酶迅速降解失活。使用多卡巴胺替代多巴胺有效地避免了上述缺点。该药由日本Tanabe公司开发,并于1994年在日本上市,该公司在1980年申请的美国专利(US 4,228,183)中对该化合物的合成方法进行了报道。路线A:
路线A(I)先用N-乙酰基-L-甲硫氨酸与氯甲酸乙酯反应生成混合酸酐作为活性中间体,然后再与3,4-二乙氧羰基多巴胺对甲苯磺酸盐反应,生成最终产物,再用硅胶色谱柱分离纯化。
在路线A(II)中,先把N-乙酰基-L-甲硫氨酸与N-羟基琥珀酰亚胺在DCC存在下缩合成酯,分离纯化后再与多巴胺盐酸盐在DMF溶液中反应生成油状物中间体N-(N-乙酰基-L-甲硫氨酰)多巴胺,以吡啶为溶剂该中间体与氯甲酸乙酯反应生成多卡巴胺,仍采用硅胶色谱柱分离纯化。
在对路线A进行尝试后,我们发现采用路线A中第一条路线的混合酸酐法,消旋始终难以避免,而在第二条路线中由于直接使用多巴胺盐酸盐,产物中杂质较多,给后处理造成不便。
发明内容
本发明目的是寻找一条成本低、工艺简单、产品纯度高、产品产量高、适合工业生产的合成多卡巴胺的方法。
本发明采用胡椒乙胺或它们的盐为起始原料与N-乙酰基-L-甲硫氨酸通过混合缩合剂法,生成关键中间体,N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺(II),用三溴化铝、三溴化硼、三氯化硼等试剂脱亚甲基,再用乙氧羰基保护两个羟基。合成了多卡巴胺,整个反应过程中未发生消旋。
反应路线如下:


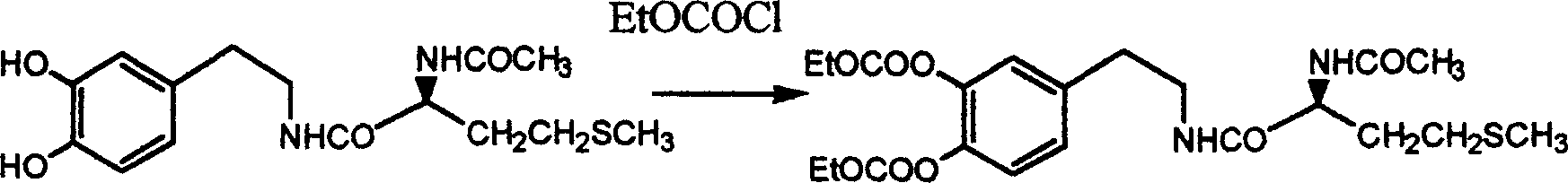
本发明采用胡椒乙胺和N-乙酰基-L-甲硫氨酸缩合,胡椒乙胺可以是游离碱也可以是盐的形式,其中盐包括盐酸盐,氢溴酸盐,草酸盐,对甲苯磺酸盐等。缩合方法是采用复合缩合剂法,包括HOBt-DCC,HOObt-DCC,HOSu-DCC。溶剂可以使用二氯甲烷,四氢呋喃,1,4-二氧六环,DMF,其中以二氯甲烷和1,4-二氧六环最佳。反应温度可控制在-15℃-15℃之间,以5℃效果最佳。使用胡椒乙胺盐作反应物时,应有碱存在,可使用三乙胺或N-甲基吗啉。
在脱除亚甲基生成N-(N-乙酰基-L-甲硫氨酰)多巴胺的反应中,可使用三溴化铝,三溴化硼,三氯化硼等试剂。溶剂可使用氯仿和二氯甲烷。反应温度可控制在-20℃-25℃之间,其中以0℃效果最佳。
在用乙氧羰基保护两个羟基而合成多卡巴胺的反应中,溶剂可使用二氯甲烷,四氢呋喃,1,4-二氧六环,吡啶。碱可使用三乙胺或N-甲基吗啉。
本发明与现有技术相比具有如下的有益效果:
1、本发明采用胡椒乙胺为原料,其价格低于多巴胺,从而使成本降低。
2、采用胡椒乙胺先与N-乙酰基-L-甲硫氨酸缩合,得关键中间体N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺,它有很好的稳定性,由于羟基已有亚甲基保护,因此缩合反应中的副反应明显减少,整个反应过程中未发生消旋。
3、关键中间体很容易结晶纯化,故进入下一步反应的杂质很少,因此在合成最终产物多卡巴胺时纯度较高。
4、最终产物可以通过常规方法纯化,避免了用硅胶层析分离,缩短了生产过程,更适合于工业生产。
具体实施方法
下面用反应实例进一步对本发明作详细说明,但对本发明不限制。
实施例1:合成N-乙酰基-L-甲硫氨酸
L-甲硫氨酸30.0g(0.20mol)溶于190ml水,用50%NaOH水溶液调至PH10,控制温度在5-10℃滴加醋酸酐,同时用NaOH水溶液维持PH值在9.5-10.5间,约100分钟滴加完。在该温度下继续搅拌2h后逐渐升至室温。终止反应。用6mol/L盐酸中和到PH1。乙酸乙酯萃取,分出有机层用无水硫酸钠干燥,浓缩,固体用乙醚-丙酮重结晶。得白色晶体32.6g。收率:90%。mp 103-105℃ TLC:Rf=0.63(正丁醇∶丙酮∶冰醋酸=10∶1∶1)[α]D 20=-22.8;(C1,H2O)
实施例2:合成N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺
N-乙酰基-L-甲硫氨酸5.0g(0.026mol),加80ml无水1,4-二氧六环溶解,加入3.5gHOBt(0.026mol),及胡椒乙胺盐酸盐5.275g(0.026mol),N-甲基吗啉5.0ml。搅拌下冷至5℃以下,加入DCC/THF溶液。在5℃搅拌1小时,室温搅拌3小时,终止反应。过滤除去DCU,浓干滤液,残余物加入300ml乙酸乙酯,用碳酸氢钠水溶液,水,0.5mol/L盐酸,水依次洗一遍,分出有机层,干燥,浓缩,得白色固体7.0g。收率:79% mp:168-170℃ TLC:Rf=5.9(氯仿∶丙酮=7∶3)[α]D 20=-25.6(C3.2,CH3OH)IRv-max KBr(cm-1)3278,1633,1250 MS(EI):338(M+)1H-NMR(CDCl3,400MHz):δ1.98(s 3H)δ 2.10(s 3H)δ1.81-2.10(m 2H)δ2.32-2.56(m 2H)δ2.75(t2H)δ3.38-3.56(m 2H)δ4.52(q 1H)δ5.92(s 2H)δ6.41(m 2H)δ6.60-6.80(m 3H)
实施例3:合成N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺
N-乙酰基-L-甲硫氨酸5.0g(0.026mol),加80ml无水四氢呋喃溶解,加入3.5gHOBt(0.026mol),及胡椒乙胺4.29g(0.026mol),搅拌下冷至5℃以下,加入DCC/THF溶液。在5º;C搅拌1小时,室温搅拌3小时,终止反应。过滤除去DCU,浓干滤液,残余物加入300ml乙酸乙酯,用碳酸氢钠水溶液,水,0.5mol/L盐酸,水依次洗一遍,分出有机层,干燥,浓缩,得白色固体6.0g。分析结果证明与实施例2为同一化合物。
实施例4:合成N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺
N-乙酰基-L-甲硫氨酸5.0g(0.026mol),加80ml无水二氯甲烷溶解,加入3.0gHOSu(0.026mol),及胡椒乙胺4.29g(0.026mol),搅拌下冷至5℃以下,加入DCC/CH2Cl2溶液。在5℃搅拌1小时,室温搅拌3小时,终止反应。过滤除去DCU,浓干滤液,残余物加入300ml乙酸乙酯,用碳酸氢钠水溶液,水,0.5mol/L盐酸,水依次洗一遍,分出有机层,干燥,浓缩,得白色固体6.8g。分析结果证明与实施例2为同一化合物。
实施例5:合成N-(N-乙酰基-L-甲硫氨酰)多巴胺
化合物(II)2.0g(5.9mmol)溶于60ml二氯甲烷,冷却到0℃,搅拌下加入BBr3/CH2Cl2溶液[1.5g(6.0mmol)/40ml]。在0℃搅拌5h,升至室温后搅拌约4h。加入40ml甲醇搅拌20分钟后,减压蒸除溶剂,加100ml乙酸乙酯溶解。用饱和氯化钠水溶液洗两次,分出有机层,水层用乙酸乙酯萃取,合并有机层,用无水硫酸钠干燥,浓缩,所得焦糖样物质直接用于下步反应。
实施例6:合成N-(N-乙酰基-L-甲硫氨酰)多巴胺
化合物(II)2.0g(5.9mmol)溶于60ml二氯甲烷,冷却到4℃,搅拌下加入BCl3/CH2Cl2溶液含BCl31.40g。在4℃搅拌5h,升至室温后搅拌约4h。加入40ml甲醇搅拌20分钟后,减压蒸除溶剂,加100ml乙酸乙酯溶解。用饱和氯化钠水溶液洗两次,分出有机层,水层用乙酸乙酯萃取,合并有机层,用无水硫酸钠干燥,浓缩,所得产物与实施例5相同。
实施例7:合成多卡巴胺
实例5中产物加20ml无水1,4-二氧六环溶解,加催化量的DMAP,搅拌下加入3.0ml三乙胺,然后加入1.0ml氯甲酸乙酯,30分钟后终止反应。将反应液倒入冰中,搅拌使冰融化后,用乙酸乙酯萃取。分出有机层,依次用碳酸氢钠水溶液,水,0.5mol/L盐酸,水洗,最后洗至中性。分出有机层干燥,蒸干溶剂得油状物。异丙醇-正己烷结晶,得白色固体2.0g两步总收率:72%
mp:103-105℃ TLC:Rf=0.57(氯仿∶丙酮=7∶3)[α]D 20=-15.8(C3.07,CH3OH)IRv-max KBr(cm-1)3286,3090,1755,1633,779
MS(EI):470(M+)1H-NMR(CDCl3,400MHz):δ1.35(t 6H)
δ1.80-2.15(m 2H)δ1.90(s 3H)δ2.05(s 3H)δ2.37-2.58(m 2H)
δ2.81(m 2H)δ3.35-3.68(m 2H)δ4.30(q 4H)δ4.51(q 1H)
δ6.38-6.56(m 2H)δ7.0-7.20(m 3H)
元素分析:理论值(%)C,53.60;H,6.43;N,5.95;S,6.81
实测值(%)C,53.72;H,6.54;N,5.94;S,6.88
Claims (7)
1、一种由胡椒乙胺或它们的盐为起始原料制备多卡巴胺的方法,由如下反应路线组成: 胡椒乙胺或它们的盐与N-乙酰基-L-甲硫氨酸经混合缩合剂法缩合,得关键中间体N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺;N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺在三溴化硼的存在下脱去亚甲基得开环的N-(N-乙酰基-L-甲硫氨酰)多巴胺; 开环的N-(N-乙酰基-L-甲硫氨酰)多巴胺用乙氧羰基保护两个羟基,得多卡巴胺。
开环的N-(N-乙酰基-L-甲硫氨酰)多巴胺用乙氧羰基保护两个羟基,得多卡巴胺。
2、根据权利要求1所述制备多卡巴胺的方法,其特征在于1a中所述的胡椒乙胺的盐为盐酸盐、氢溴酸盐、草酸盐、对甲苯磺酸盐。
3、根据权利要求1所述制备多卡巴胺的方法,1a中所述的混合缩合剂包括HOBt-DCC、HOObt-DCC、HOSu-DCC。
4、根据权利要求1所述制备多卡巴胺的方法,1a中所述的胡椒乙胺或它们的盐与N-乙酰基-L-甲硫氨酸缩合反应的溶剂为二氯甲烷、四氢呋喃、1,4-二氧六环、DMF,其中以二氯甲烷、1,4-二氧六环为最佳。
5、根据权利要求1所述制备多卡巴胺的方法,1b中所述的N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺在三溴化铝、三溴化硼、三氯化硼为试剂脱除亚甲基得N-(N-乙酰基-L-甲硫氨酰)多巴胺。
6、根据权利要求1所述制备多卡巴胺的方法,1b中所述N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺脱亚甲基的溶剂为氯仿、二氯甲烷,反应温度在10-35℃。
7、根据权利要求1所述制备多卡巴胺的方法,1c中所述乙氧羰基保护羟基反应的溶剂为二氯甲烷、四氢呋喃、1,4-二氧六环、吡啶,碱使用三乙胺或N-甲基吗啉。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB021361576A CN1271049C (zh) | 2002-07-23 | 2002-07-23 | 一种合成多卡巴胺的方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB021361576A CN1271049C (zh) | 2002-07-23 | 2002-07-23 | 一种合成多卡巴胺的方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1470506A true CN1470506A (zh) | 2004-01-28 |
| CN1271049C CN1271049C (zh) | 2006-08-23 |
Family
ID=34146330
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB021361576A Expired - Fee Related CN1271049C (zh) | 2002-07-23 | 2002-07-23 | 一种合成多卡巴胺的方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN1271049C (zh) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102850189A (zh) * | 2012-08-29 | 2013-01-02 | 中国林业科学研究院林产化学工业研究所 | 一种高纯度饱和漆酚及其缩甲醛衍生物的制备方法 |
| CN103408474A (zh) * | 2013-08-28 | 2013-11-27 | 重庆紫光化工股份有限公司 | 高纯度n-乙酰-d,l-蛋氨酸的高效生产方法 |
| WO2014139161A1 (en) * | 2013-03-15 | 2014-09-18 | Techfields Pharma Co., Ltd. | Novel high penetration drugs and their compositions thereof for treatment of parkinson diseases |
| CN109053504A (zh) * | 2018-08-16 | 2018-12-21 | 吉尔生化(上海)有限公司 | 一种(2s)-2-(乙酰氨基)-4-(甲基亚磺酸酰基)丁酸的合成方法 |
| JP2020040949A (ja) * | 2017-12-28 | 2020-03-19 | テックフィールズ ファーマ カンパニー リミテッド | パ−キンソン病治療用の新規高透過薬物及びその組成物 |
-
2002
- 2002-07-23 CN CNB021361576A patent/CN1271049C/zh not_active Expired - Fee Related
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102850189A (zh) * | 2012-08-29 | 2013-01-02 | 中国林业科学研究院林产化学工业研究所 | 一种高纯度饱和漆酚及其缩甲醛衍生物的制备方法 |
| CN102850189B (zh) * | 2012-08-29 | 2018-03-20 | 中国林业科学研究院林产化学工业研究所 | 一种高纯度饱和漆酚及其缩甲醛衍生物的制备方法 |
| WO2014139161A1 (en) * | 2013-03-15 | 2014-09-18 | Techfields Pharma Co., Ltd. | Novel high penetration drugs and their compositions thereof for treatment of parkinson diseases |
| US9718766B2 (en) | 2013-03-15 | 2017-08-01 | Techfields Pharma Co., Ltd. | High penetration drugs and their compositions thereof for treatment of parkinson diseases |
| US11084813B2 (en) | 2013-03-15 | 2021-08-10 | Techfields Pharma Co., Ltd. | High penetration drugs and their compositions thereof for treatment of Parkinson diseases |
| US11685739B2 (en) | 2013-03-15 | 2023-06-27 | Techfields Pharma Co., Ltd. | High penetration drugs and their compositions thereof for treatment of Parkinson diseases |
| CN103408474A (zh) * | 2013-08-28 | 2013-11-27 | 重庆紫光化工股份有限公司 | 高纯度n-乙酰-d,l-蛋氨酸的高效生产方法 |
| CN103408474B (zh) * | 2013-08-28 | 2015-09-16 | 重庆紫光化工股份有限公司 | 高纯度n-乙酰-d,l-蛋氨酸的高效生产方法 |
| JP2020040949A (ja) * | 2017-12-28 | 2020-03-19 | テックフィールズ ファーマ カンパニー リミテッド | パ−キンソン病治療用の新規高透過薬物及びその組成物 |
| JP2022020671A (ja) * | 2017-12-28 | 2022-02-01 | テックフィールズ ファーマ カンパニー リミテッド | パ-キンソン病治療用の新規高透過薬物及びその組成物 |
| CN109053504A (zh) * | 2018-08-16 | 2018-12-21 | 吉尔生化(上海)有限公司 | 一种(2s)-2-(乙酰氨基)-4-(甲基亚磺酸酰基)丁酸的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1271049C (zh) | 2006-08-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2018344099B2 (en) | A simple process for preparing avibactam | |
| CN110590635A (zh) | 左乙拉西坦及其中间体的制备方法 | |
| CN1470506A (zh) | 一种合成多卡巴胺的方法 | |
| CN112645833A (zh) | 一种(s)-2,6-二氨基-5-氧己酸的合成方法 | |
| CN113372271A (zh) | 一种顺苯磺酸阿曲库铵的制备方法 | |
| CN102746207A (zh) | 一种奥拉西坦的合成方法 | |
| WO1994010185A1 (en) | Erythromycin derivative | |
| CN113773229B (zh) | α,β-不饱和氨基酸衍生物及其DL-硒-甲基硒代氨基酸衍生物、合成方法和应用 | |
| CN106146355A (zh) | 益母草碱与阿司匹林缀合物的制备方法 | |
| JP3533178B2 (ja) | 高純度の混合無水(メタ)アクリル酸の製造方法 | |
| JP3207018B2 (ja) | ベンジルコハク酸誘導体の製造方法およびその製造中間体 | |
| JP3207017B2 (ja) | ベンジルコハク酸誘導体の製造方法およびその製造中間体 | |
| CN1986548B (zh) | N-叔丁氧羰基-5-氮杂-2-氧杂-3-酮-双环-[2,2,1]庚烷连续合成的工业化制备方法 | |
| WO1998032736A1 (fr) | Procede de production de derives d'acide benzylsuccinique | |
| JP3193597B2 (ja) | グリシン誘導体の製造方法 | |
| CN111777554A (zh) | 一种合成苯磺顺阿曲库铵的方法 | |
| US5280122A (en) | Resolution of 2-benzyl-4-piperidone-succinic acid | |
| JP4311889B2 (ja) | (メタ)アクリル酸無水物の製造方法 | |
| KR100483317B1 (ko) | α-페닐-α-프로폭시벤젠아세트산 1-메틸-4-피페리디닐에스테르 염산염의 제조방법 | |
| JP5704763B2 (ja) | トランス−4−アミノシクロペンタ−2−エン−1−カルボン酸誘導体の製造 | |
| JPH07267985A (ja) | タウロウルソデオキシコール酸水和物の製法 | |
| CN111662233B (zh) | 一种一步法合成4-氯-1h-咪唑-2-羧酸乙酯的方法 | |
| CN118255707A (zh) | 一种加巴喷丁恩那卡比有关物质的制备方法 | |
| JP4302974B2 (ja) | オキサゾール化合物の製造方法 | |
| CN115572224A (zh) | 一种(s)-(-)-3-环己烯甲酸的合成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |