CN1470506A - A kind of method of synthesizing docarbamine - Google Patents
A kind of method of synthesizing docarbamine Download PDFInfo
- Publication number
- CN1470506A CN1470506A CNA021361576A CN02136157A CN1470506A CN 1470506 A CN1470506 A CN 1470506A CN A021361576 A CNA021361576 A CN A021361576A CN 02136157 A CN02136157 A CN 02136157A CN 1470506 A CN1470506 A CN 1470506A
- Authority
- CN
- China
- Prior art keywords
- ethanoyl
- docarpamine
- methionyl
- homopiperony lamine
- preparing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
本发明提供一条合成多卡巴胺的新方法,采用胡椒乙胺或它的盐为起始原料与N-乙酰基-L-甲硫氨酸经混合缩合剂法缩合,得关键中间体N-(N-乙酰基-L-甲硫氨酰)胡椒乙胺再用三溴化硼等试剂脱亚甲基,最后用乙氧羰基保护两个羟基合成了多卡巴胺。本方法降低了成本,提高收率,纯度高且操作简便,适合于工业生产。The invention provides a new method for synthesizing docarbamine, which uses piperethylamine or its salt as the starting material to condense with N-acetyl-L-methionine through a mixed condensing agent method to obtain the key intermediate N-( N-acetyl-L-methionyl)piperethylamine was demethyleneized with reagents such as boron tribromide, and finally docarbamine was synthesized by protecting two hydroxyl groups with ethoxycarbonyl. The method reduces cost, improves yield, has high purity and is easy to operate, and is suitable for industrial production.
Description
Technical field
The present invention relates to medicine synthetic method improvement, the complete synthesis process modification of the medicine Docarpamine of more specifically saying so.
Background technologyDocarpamine is the oral prodrug of Dopamine HCL, and clinical in cardiostimulator, its structure is as follows:

Docarpamine be Dopamine HCL can be oral prodrug, clinical in cardiostimulator.Clinical renal failure, hypertension, chronic congestion heart trouble and the other diseases of being used for the treatment of of known Dopamine HCL, but owing to Dopamine HCL is difficult to absorb from digestion organs, so can only pass through drug administration by injection.And Dopamine HCL is by monoamine oxidase in the liver or other enzymes inactivation of degrading rapidly behind the intravenously administrable.Use Docarpamine to substitute Dopamine HCL and avoided above-mentioned shortcoming effectively.This medicine is by the exploitation of Japanese Tanabe company, and in 1994 in Japan's listing, the said firm reports the synthetic method of this compound in the United States Patent (USP) (US 4,228,183) of application in 1980.Route A:

Route A (I) generates mixed acid anhydride as active intermediate with N-ethanoyl-L-methionine(Met) and Vinyl chloroformate reaction earlier, and then with 3,4-diethoxy carbonyl Dopamine HCL tosic acid reactant salt generates final product, the separation and purification of recycle silicon glue chromatographic column.

In route A (II); earlier N-ethanoyl-L-methionine(Met) and N-hydroxy-succinamide are condensed into ester in the presence of DCC; react in DMF solution with dopamine hydrochloride again after the separation and purification and generate oily matter intermediate N (N-ethanoyl-L-methionyl) Dopamine HCL; with the pyridine is that the reaction of this intermediate of solvent and Vinyl chloroformate generates Docarpamine, still adopts the silica gel chromatography column separating purification.
After route A was attempted, we found to adopt the mixed anhydride method of article one route among the route A, and racemization is difficult to avoid all the time, and in the second route since directly the use dopamine hydrochloride, impurity is more in the product, brings inconvenience to aftertreatment.
Summary of the invention
Cost is low, technology is simple, product purity is high, product production is high, the method for suitable industrial synthesizing polycarbo amide to the present invention seeks to seek one.
It is that starting raw material and N-ethanoyl-L-methionine(Met) pass through the mixed condensing agent method that the present invention adopts homopiperony lamine or their salt; generate key intermediate; N-(N-ethanoyl-L-methionyl) homopiperony lamine (II); take off methylene radical with reagent such as alchlor, boron tribromide, boron trichlorides, again with two hydroxyls of ethoxycarbonyl protection.Synthesized Docarpamine, racemization has not taken place in the entire reaction course.
Reaction scheme is as follows:



The present invention adopts homopiperony lamine and N-ethanoyl-L-methionine(Met) condensation, and homopiperony lamine can be that free alkali can be the form of salt also, and wherein salt comprises hydrochloride, hydrobromate, oxalate, tosilate etc.Method of condensing is to adopt coupling condenser agent method, comprises HOBt-DCC, HOObt-DCC, HOSu-DCC.Solvent can use methylene dichloride, tetrahydrofuran (THF), and 1, the 4-dioxane, DMF, wherein with methylene dichloride and 1,4-dioxane the best.Temperature of reaction can be controlled between-15 ℃-15 ℃, with 5 ℃ of best results.When using piperethanamine salt to make reactant, should there be alkali to exist, can uses triethylamine or N-methylmorpholine.
In the reaction that removes methylene radical generation N-(N-ethanoyl-L-methionyl) Dopamine HCL, can use reagent such as alchlor, boron tribromide, boron trichloride.Solvent can use chloroform and methylene dichloride.Temperature of reaction can be controlled between-20 ℃-25 ℃, wherein with 0 ℃ of best results.
In the reaction of the synthesizing polycarbo amide with two hydroxyls of ethoxycarbonyl protection, solvent can use methylene dichloride, tetrahydrofuran (THF), 1,4-dioxane, pyridine.Alkali can use triethylamine or N-methylmorpholine.
The present invention compared with prior art has following beneficial effect:
1, to adopt homopiperony lamine be raw material in the present invention, and its price is lower than Dopamine HCL, thereby cost is reduced.
2, adopt homopiperony lamine elder generation and N-ethanoyl-L-methionine(Met) condensation; get key intermediate N-(N-ethanoyl-L-methionyl) homopiperony lamine; it has good stability; because the existing methylene radical protection of hydroxyl; therefore the side reaction in the condensation reaction obviously reduces, and racemization does not take place in the entire reaction course.
3, key intermediate is easy to crystallization purifying, so the impurity that enters next step reaction seldom, therefore purity is higher when synthetic final product Docarpamine.
4, final product can pass through the ordinary method purifying, has avoided separating with silica gel column chromatography, has shortened production process, is more suitable in industrial production.
Specific implementation method
Further the present invention is elaborated with the reaction example below, but the present invention is not limited.
Embodiment 1: synthetic N-ethanoyl-L-methionine(Met)
L-methionine(Met) 30.0g (0.20mol) is dissolved in 190ml water, transfers to PH10 with the 50%NaOH aqueous solution, and controlled temperature drips acetic anhydride at 5-10 ℃, keeps pH value between 9.5-10.5 with the NaOH aqueous solution simultaneously, drips in about 100 minutes.After continuing to stir 2h under this temperature, rise to room temperature gradually.Termination reaction.With the 6mol/L hydrochloric acid PH1 that neutralizes.Ethyl acetate extraction is told the organic layer anhydrous sodium sulfate drying, concentrates solid ether-acetone recrystallization.Get white crystal 32.6g.Yield: 90%.Mp 103-105 ℃ TLC:Rf=0.63 (propyl carbinol: acetone: Glacial acetic acid=10: 1: 1) [α]
D 20=-22.8; (C1, H
2O)
Embodiment 2: synthetic N-(N-ethanoyl-L-methionyl) homopiperony lamine
N-ethanoyl-L-methionine(Met) 5.0g (0.026mol), it is anhydrous 1 to add 80ml, and the dissolving of 4-dioxane adds 3.5gHOBt (0.026mol), and homopiperony lamine hydrochloride 5.275g (0.026mol), N-methylmorpholine 5.0ml.Be chilled to below 5 ℃ under stirring, add DCC/THF solution.Stirred stirring at room 3 hours, termination reaction 1 hour at 5 ℃.Remove by filter DCU, dense dried filtrate, resistates adds the 300ml ethyl acetate, uses sodium bicarbonate aqueous solution, and water, 0.5mol/L hydrochloric acid, water are washed one time successively, tell organic layer, and drying concentrates, and gets white solid 7.0g.Yield: 79% mp:168-170 ℃ TLC:Rf=5.9 (chloroform: acetone=7: 3) [α]
D 20=-25.6 (C3.2, CH
3OH) IR
V-max KBr(cm
-1) 3278,1633,1250 MS (EI): 338 (M+)
1H-NMR (CDCl
3, 400MHz): δ 1.98 (s 3H) δ 2.10 (s 3H) δ 1.81-2.10 (m 2H) δ 2.32-2.56 (m 2H) δ 2.75 (t2H) δ 3.38-3.56 (m 2H) δ 4.52 (q 1H) δ 5.92 (s 2H) δ 6.41 (m 2H) δ 6.60-6.80 (m 3H)
Embodiment 3: synthetic N-(N-ethanoyl-L-methionyl) homopiperony lamine
N-ethanoyl-L-methionine(Met) 5.0g (0.026mol) adds the dissolving of 80ml anhydrous tetrahydro furan, adds 3.5gHOBt (0.026mol), and homopiperony lamine 4.29g (0.026mol), is chilled to below 5 ℃ under stirring, and adds DCC/THF solution.At 5﹠amp; Ordm; C stirred 1 hour, stirring at room 3 hours, termination reaction.Remove by filter DCU, dense dried filtrate, resistates adds the 300ml ethyl acetate, uses sodium bicarbonate aqueous solution, and water, 0.5mol/L hydrochloric acid, water are washed one time successively, tell organic layer, and drying concentrates, and gets white solid 6.0g.The analytical results proof is same compound with embodiment 2.
Embodiment 4: synthetic N-(N-ethanoyl-L-methionyl) homopiperony lamine
N-ethanoyl-L-methionine(Met) 5.0g (0.026mol) adds the dissolving of 80ml anhydrous methylene chloride, adds 3.0gHOSu (0.026mol), and homopiperony lamine 4.29g (0.026mol), is chilled to below 5 ℃ under stirring, and adds DCC/CH
2Cl
2Solution.Stirred stirring at room 3 hours, termination reaction 1 hour at 5 ℃.Remove by filter DCU, dense dried filtrate, resistates adds the 300ml ethyl acetate, uses sodium bicarbonate aqueous solution, and water, 0.5mol/L hydrochloric acid, water are washed one time successively, tell organic layer, and drying concentrates, and gets white solid 6.8g.The analytical results proof is same compound with embodiment 2.
Embodiment 5: synthetic N-(N-ethanoyl-L-methionyl) Dopamine HCL
Compound (II) 2.0g (5.9mmol) is dissolved in the 60ml methylene dichloride, is cooled to 0 ℃, stirs to add BBr down
3/ CH
2Cl
2Solution [1.5g (6.0mmol)/40ml].Stir 5h at 0 ℃, rise to stir about 4h after the room temperature.Add 40ml methyl alcohol and stir after 20 minutes, remove solvent under reduced pressure, add the 100ml acetic acid ethyl dissolution.Wash twice with saturated sodium-chloride water solution, tell organic layer, the water layer ethyl acetate extraction merges organic layer, uses anhydrous sodium sulfate drying, concentrates, and gained caramel sample material is directly used in the next step.
Embodiment 6: synthetic N-(N-ethanoyl-L-methionyl) Dopamine HCL
Compound (II) 2.0g (5.9mmol) is dissolved in the 60ml methylene dichloride, is cooled to 4 ℃, stirs to add BCl down
3/ CH
2Cl
2Solution contains BCl
31.40g.Stir 5h at 4 ℃, rise to stir about 4h after the room temperature.Add 40ml methyl alcohol and stir after 20 minutes, remove solvent under reduced pressure, add the 100ml acetic acid ethyl dissolution.Wash twice with saturated sodium-chloride water solution, tell organic layer, the water layer ethyl acetate extraction merges organic layer, uses anhydrous sodium sulfate drying, concentrates, and products therefrom is identical with embodiment 5.
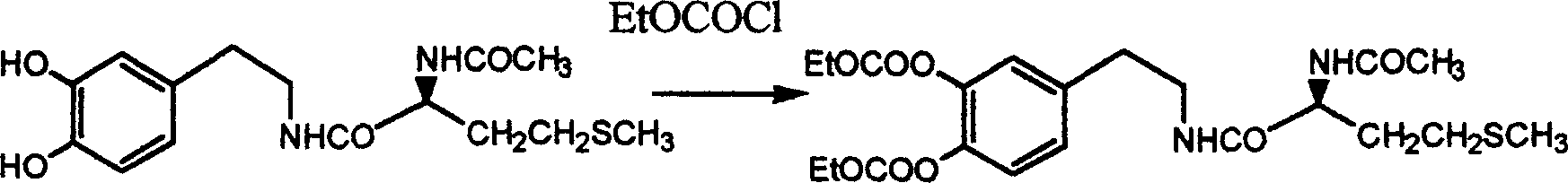
Embodiment 7: synthesizing polycarbo amide
It is anhydrous 1 that product adds 20ml in the example 5, and the dissolving of 4-dioxane adds the DMAP of catalytic amount, stirs to add the 3.0ml triethylamine down, adds the 1.0ml Vinyl chloroformate then, termination reaction after 30 minutes.Reaction solution is poured in the ice, after stirring melts the ice, used ethyl acetate extraction.Tell organic layer, use sodium bicarbonate aqueous solution successively, water, 0.5mol/L hydrochloric acid, washing is washed till neutrality at last.Tell the organic layer drying, solvent evaporated gets oily matter.Virahol-normal hexane crystallization gets white solid 2.0g two steps total recovery: 72%
Mp:103-105 ℃ of TLC:Rf=0.57 (chloroform: acetone=7: 3) [α]
D 20=-15.8 (C3.07, CH
3OH) IR
V-max KBr(cm
-1) 3286,3090,1755,1633,779
MS(EI):470(M+)
1H-NMR(CDCl
3,400MHz):δ1.35(t?6H)
δ1.80-2.15(m?2H)δ1.90(s?3H)δ2.05(s?3H)δ2.37-2.58(m?2H)
δ2.81(m?2H)δ3.35-3.68(m?2H)δ4.30(q?4H)δ4.51(q?1H)
δ6.38-6.56(m?2H)δ7.0-7.20(m?3H)
Ultimate analysis: theoretical value (%) C, 53.60; H, 6.43; N, 5.95; S, 6.81
Measured value (%) C, 53.72; H, 6.54; N, 5.94; S, 6.88
Claims (7)
1, a kind of is the method that starting raw material prepares Docarpamine by homopiperony lamine or their salt, is made up of following reaction scheme:
Homopiperony lamine or their salt and N-ethanoyl-L-methionine(Met) get key intermediate N-(N-ethanoyl-L-methionyl) homopiperony lamine through the condensation of mixed condensing agent method;
N-(N-ethanoyl-L-methionyl) homopiperony lamine is sloughed N-(N-ethanoyl-L-methionyl) Dopamine HCL that methylene radical gets open loop in the presence of boron tribromide;
 The N-of open loop (N-ethanoyl-L-methionyl) Dopamine HCL gets Docarpamine with two hydroxyls of ethoxycarbonyl protection.
The N-of open loop (N-ethanoyl-L-methionyl) Dopamine HCL gets Docarpamine with two hydroxyls of ethoxycarbonyl protection.
2, according to the described method for preparing Docarpamine of claim 1, the salt that it is characterized in that the homopiperony lamine described in the 1a is hydrochloride, hydrobromate, oxalate, tosilate.
3, according to the described method for preparing Docarpamine of claim 1, the mixed condensing agent described in the 1a comprises HOBt-DCC, HOObt-DCC, HOSu-DCC.
4, according to the described method for preparing Docarpamine of claim 1; the solvent of homopiperony lamine described in the 1a or their salt and N-ethanoyl-L-methionine(Met) condensation reaction is methylene dichloride, tetrahydrofuran (THF), 1; 4-dioxane, DMF, wherein with methylene dichloride, 1, the 4-dioxane is best.
5, according to the described method for preparing Docarpamine of claim 1, the N-described in the 1b (N-ethanoyl-L-methionyl) homopiperony lamine is that reagent removes methylene radical and gets N-(N-ethanoyl-L-methionyl) Dopamine HCL at alchlor, boron tribromide, boron trichloride.
6, according to the described method for preparing Docarpamine of claim 1, the solvent that N-described in the 1b (N-ethanoyl-L-methionyl) homopiperony lamine takes off methylene radical is chloroform, methylene dichloride, and temperature of reaction is at 10-35 ℃.
7, according to the described method for preparing Docarpamine of claim 1, the solvent of the protection of ethoxycarbonyl described in 1c hydroxyl reaction is methylene dichloride, tetrahydrofuran (THF), 1,4-dioxane, pyridine, and alkali uses triethylamine or N-methylmorpholine.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB021361576A CN1271049C (en) | 2002-07-23 | 2002-07-23 | Method for synthesizing polycarbo amide |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB021361576A CN1271049C (en) | 2002-07-23 | 2002-07-23 | Method for synthesizing polycarbo amide |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1470506A true CN1470506A (en) | 2004-01-28 |
| CN1271049C CN1271049C (en) | 2006-08-23 |
Family
ID=34146330
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB021361576A Expired - Fee Related CN1271049C (en) | 2002-07-23 | 2002-07-23 | Method for synthesizing polycarbo amide |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN1271049C (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102850189A (en) * | 2012-08-29 | 2013-01-02 | 中国林业科学研究院林产化学工业研究所 | Preparation method of high-purity saturated urushiol and its formal derivative |
| CN103408474A (en) * | 2013-08-28 | 2013-11-27 | 重庆紫光化工股份有限公司 | Efficient production method of high-purity N-acetyl-D, L-methionine |
| WO2014139161A1 (en) * | 2013-03-15 | 2014-09-18 | Techfields Pharma Co., Ltd. | Novel high penetration drugs and their compositions thereof for treatment of parkinson diseases |
| CN109053504A (en) * | 2018-08-16 | 2018-12-21 | 吉尔生化(上海)有限公司 | One kind (2S) -2-(acetylamino) -4-(methyl sulfinic acid acyl group) butyric acid synthetic method |
| JP2020040949A (en) * | 2017-12-28 | 2020-03-19 | テックフィールズ ファーマ カンパニー リミテッド | Novel high penetration drugs and compositions thereof for treatment of parkinson's disease |
-
2002
- 2002-07-23 CN CNB021361576A patent/CN1271049C/en not_active Expired - Fee Related
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102850189A (en) * | 2012-08-29 | 2013-01-02 | 中国林业科学研究院林产化学工业研究所 | Preparation method of high-purity saturated urushiol and its formal derivative |
| CN102850189B (en) * | 2012-08-29 | 2018-03-20 | 中国林业科学研究院林产化学工业研究所 | A kind of preparation method of high-purity saturation laccol and its formal derivatives |
| WO2014139161A1 (en) * | 2013-03-15 | 2014-09-18 | Techfields Pharma Co., Ltd. | Novel high penetration drugs and their compositions thereof for treatment of parkinson diseases |
| US9718766B2 (en) | 2013-03-15 | 2017-08-01 | Techfields Pharma Co., Ltd. | High penetration drugs and their compositions thereof for treatment of parkinson diseases |
| US11084813B2 (en) | 2013-03-15 | 2021-08-10 | Techfields Pharma Co., Ltd. | High penetration drugs and their compositions thereof for treatment of Parkinson diseases |
| US11685739B2 (en) | 2013-03-15 | 2023-06-27 | Techfields Pharma Co., Ltd. | High penetration drugs and their compositions thereof for treatment of Parkinson diseases |
| CN103408474A (en) * | 2013-08-28 | 2013-11-27 | 重庆紫光化工股份有限公司 | Efficient production method of high-purity N-acetyl-D, L-methionine |
| CN103408474B (en) * | 2013-08-28 | 2015-09-16 | 重庆紫光化工股份有限公司 | High purity N-acetyl-D, the high-efficiency method for producing of L-Methionine |
| JP2020040949A (en) * | 2017-12-28 | 2020-03-19 | テックフィールズ ファーマ カンパニー リミテッド | Novel high penetration drugs and compositions thereof for treatment of parkinson's disease |
| JP2022020671A (en) * | 2017-12-28 | 2022-02-01 | テックフィールズ ファーマ カンパニー リミテッド | Novel high penetration drugs and compositions thereof for treatment of parkinson's disease |
| CN109053504A (en) * | 2018-08-16 | 2018-12-21 | 吉尔生化(上海)有限公司 | One kind (2S) -2-(acetylamino) -4-(methyl sulfinic acid acyl group) butyric acid synthetic method |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1271049C (en) | 2006-08-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2018344099B2 (en) | A simple process for preparing avibactam | |
| CN110590635A (en) | Preparation method of levetiracetam and intermediate thereof | |
| CN1470506A (en) | A kind of method of synthesizing docarbamine | |
| CN112645833A (en) | Synthesis method of (S) -2, 6-diamino-5-oxohexanoic acid | |
| CN113372271A (en) | Preparation method of cisatracurium besylate | |
| CN102746207A (en) | Synthesis method of oxiracetam | |
| WO1994010185A1 (en) | Erythromycin derivative | |
| CN113773229B (en) | Alpha, beta-unsaturated amino acid derivative and DL-selenium-methyl seleno amino acid derivative, synthetic method and application thereof | |
| CN106146355A (en) | Leonurine and the preparation method of aspirin conjugate | |
| JP3533178B2 (en) | Method for producing high-purity mixed (meth) acrylic anhydride | |
| JP3207018B2 (en) | Method for producing benzylsuccinic acid derivative and intermediate for producing the same | |
| JP3207017B2 (en) | Method for producing benzylsuccinic acid derivative and intermediate for producing the same | |
| CN1986548B (en) | Industrial continuous preparing process of N-tert-butoxy carbonyl-5-aza-2-oxa-3-one-dicyclo-[2,2,1] heptane | |
| WO1998032736A1 (en) | Process for producing benzylsuccinic acid derivatives | |
| JP3193597B2 (en) | Method for producing glycine derivative | |
| CN111777554A (en) | Method for synthesizing cisatracurium besilate | |
| CN1314684C (en) | Synthesis method of saijiabin, its racemate and S-configuration, and preparation method of its amorphous powder | |
| JP4311889B2 (en) | Method for producing (meth) acrylic anhydride | |
| KR100483317B1 (en) | METHOD FOR THE PREPARATION OF α-PHENYL-α-PROPOXYBENZENEACETIC ACID 1-METHYL-4-PIPERIDINYL ESTER HYDROCHLORIDE | |
| JPH07267985A (en) | Production of tauroursodeoxycholic acid hydrate | |
| CN111662233B (en) | Method for synthesizing 4-chloro-1H-imidazole-2-carboxylic acid ethyl ester by one-step method | |
| CN118255707A (en) | Preparation method of gabapentin enroacalcet related substances | |
| JP4302974B2 (en) | Method for producing oxazole compound | |
| CN115572224A (en) | Synthesis method of (S) - (-) -3-cyclohexenecarboxylic acid | |
| CN1554654A (en) | Synthesis method of tiagabine and its racemate and S-configuration and preparation method of amorphous powder |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |