CN116178264A - 一种联苯吡菌胺的合成方法 - Google Patents
一种联苯吡菌胺的合成方法 Download PDFInfo
- Publication number
- CN116178264A CN116178264A CN202211678888.5A CN202211678888A CN116178264A CN 116178264 A CN116178264 A CN 116178264A CN 202211678888 A CN202211678888 A CN 202211678888A CN 116178264 A CN116178264 A CN 116178264A
- Authority
- CN
- China
- Prior art keywords
- dichlorophenyl
- solvent
- bixafen
- fluoronitrobenzene
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C201/00—Preparation of esters of nitric or nitrous acid or of compounds containing nitro or nitroso groups bound to a carbon skeleton
- C07C201/06—Preparation of nitro compounds
- C07C201/12—Preparation of nitro compounds by reactions not involving the formation of nitro groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/30—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of nitrogen-to-oxygen or nitrogen-to-nitrogen bonds
- C07C209/32—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of nitrogen-to-oxygen or nitrogen-to-nitrogen bonds by reduction of nitro groups
- C07C209/36—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of nitrogen-to-oxygen or nitrogen-to-nitrogen bonds by reduction of nitro groups by reduction of nitro groups bound to carbon atoms of six-membered aromatic rings in presence of hydrogen-containing gases and a catalyst
- C07C209/365—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of nitrogen-to-oxygen or nitrogen-to-nitrogen bonds by reduction of nitro groups by reduction of nitro groups bound to carbon atoms of six-membered aromatic rings in presence of hydrogen-containing gases and a catalyst by reduction with preservation of halogen-atoms in compounds containing nitro groups and halogen atoms bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F3/00—Compounds containing elements of Groups 2 or 12 of the Periodic Table
- C07F3/02—Magnesium compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F3/00—Compounds containing elements of Groups 2 or 12 of the Periodic Table
- C07F3/06—Zinc compounds
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本发明公开联苯吡菌胺的合成方法,包括步骤1:将镁粉和溶剂A混合,加入3,4‑二氯溴苯的溶剂A溶液反应,制得3,4‑二氯苯溴化镁金属化合物;步骤2:将3,4‑二氯苯溴化镁金属化合物用氯化锌进行金属转移化反应制得(3,4‑二氯苯基)氯化锌,将(3,4‑二氯苯基)氯化锌、2‑溴‑4‑氟硝基苯、催化剂和溶剂A混合反应,得到2‑(3,4‑二氯苯基)‑4‑氟硝基苯;步骤3:将2‑(3,4‑二氯苯基)‑4‑氟硝基苯、铁粉、氯化铵、溶剂B和水混合并加入冰醋酸反应,得到2‑(3,4‑二氯苯基)‑4‑氟苯胺;步骤4:将2‑(3,4‑二氯苯基)‑4‑氟苯胺和溶剂C混合,加入含1‑甲基‑3‑二氟甲基‑1H‑吡唑‑4‑甲酰氯的溶剂C溶液反应即得。本发明合成工艺操作简单,反应条件容易控制,各步骤产品收率高,纯度好,适合批量工业化生产。
Description
技术领域
本发明属于有机合成方法学领域,具体涉及一种联苯吡菌胺的合成方法。
背景技术
联苯吡菌胺是德国拜耳公司开发的吡唑酰胺类杀菌剂,属于线粒体呼吸链中琥珀酸辅酶Q还原酶抑制剂,主要用于谷物叶上叶斑病和叶锈病的防治,杀菌谱较广,可用于白粉病、锈病、霜霉病等多种病害的防治,尤其对大麦网斑病、苹果白粉病有很好的治疗和保护效果。
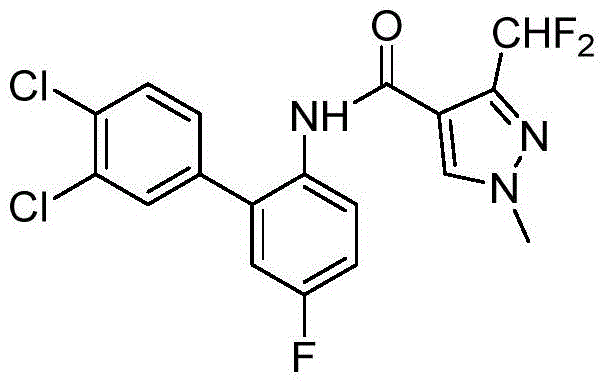
现有技术公开的关于联苯吡菌胺的合成方法主要有以下四种:
(1)US20060116414、WO2009106234、刘安昌等(新型杀菌剂氟唑菌酰胺和联苯吡菌胺的合成研究,现代农药,2016,15(1):16-18)公开了一种联苯吡菌胺的制备方法。以3,4-二氯溴苯为原料,经格氏反应得到3,4-二氯苯硼酸,然后与2-溴-4-氟苯胺偶联得到3',4'-二氯-5-氟-1,1'-联苯-2-胺,后与1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯缩合得目标产物,该路线的缺点是总收率50%以下,成本高,特别是3,4-二氯苯硼酸的制备,不仅存在收率低,而且溶剂难回收的问题。反应式如下:
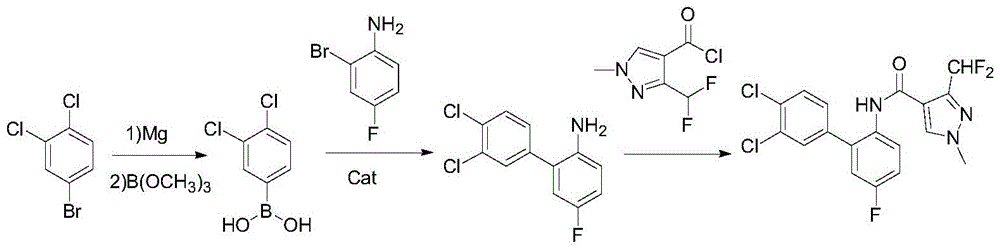
(2)Geralp P等(The Gomberg-Bachmann reaction for the arylation ofanilines with aryl diszotates,2012,18(37):11555-11559)、韩晓蕾等(联苯吡菌胺的合成,农药,2019,58(3):174-176)公开了一种联苯吡菌胺的制备方法。以3,4-二氯苯胺为原料先重氮化,再与对氟苯胺偶联的方法构造联苯胺中间体,再酰化生成目标产物,总收率分别为55.8%、62.87%。该路线原材料3,4-二氯苯胺价格较贵,采用重氮化反应有一定的安全隐患,且偶联反应转化率极低,反应过程有大量焦油状副产物。反应式如下:

(3)DE102006016462公开了一种联苯吡菌胺的制备方法。将2-溴-4-氟苯胺的氨基用乙酰基保护之后,和乙烯在钯催化剂的存在下发生heck偶联反应,然后与3,4-二氯噻吩砜反应成环,之后再氧化芳构化以及脱去乙酰基保护剂,生成3',4'-二氯-5-氟-2-联苯氨,最后酰化生成目标产物,总收率50.5%。该路线原料3,4-二氯噻吩砜在国内昂贵不易购买,易燃气体乙烯进行heck反应有一定的安全隐患,步骤较长,芳构化反应收率也不高,最高仅77%,导致总收率较前两条路线要低。反应式如下:

(4)US7750186,WO2006024388公开了一种联苯吡菌胺的制备方法。N-(2-溴-4-氟苯胺)乙酰胺在催化剂下,与2-甲基丁基-3-炔-2-醇反应制得N-[4-氟-2-(3-羟基-3-甲基丁基-1-炔-1-醇)苯基]乙酰胺,然后与甲醇钠反应,再与3,4-二氯噻吩-1,1-二氧化物反应制得N-(3',4'-二氯-5-氟联苯)2-基)乙酰胺,最后酰化生成目标产物。该路线对比路线三,此路线少了一步收率较低的芳构化但多了一步收率仍比较低的脱叔丁醇,总体收率比路线三略低(49.4%),工业化价值不大。反应式如下:
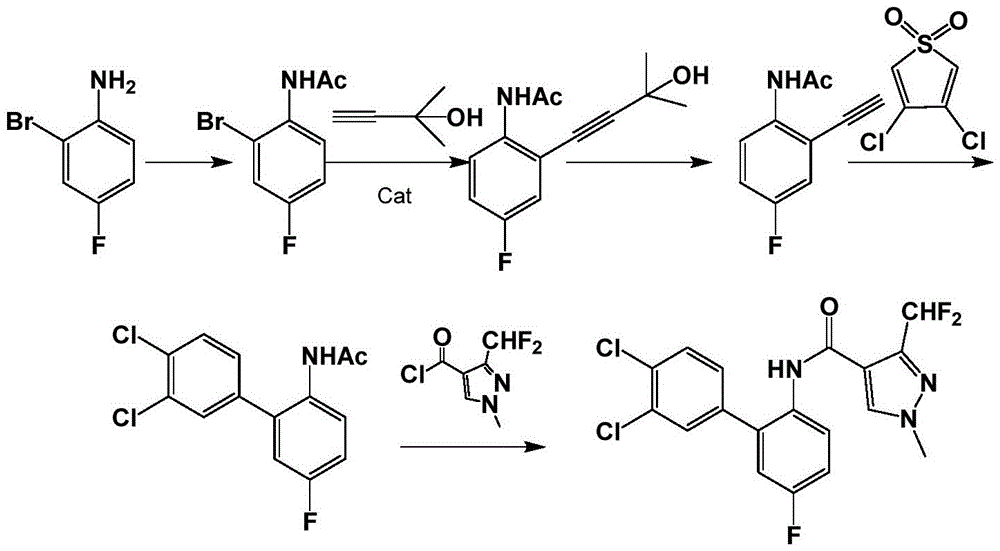
综上所述,现有技术中路线1使用了昂贵的钯催化剂,并且格氏反应制备3,4-二氯苯硼酸收率低,价格高,导致成本较高;路线2以3,4-二氯苯胺为原料,采用重氮化法存在原料价格昂贵,偶联反应转化率极低,反应过程有大量焦油状副产物等问题;路线3、4反应路线较长,原料3,4-二氯噻吩砜价格昂贵不易得,这就导致上述工艺均无法大规模应用。
发明内容
针对现有技术的不足,本发明的目的在于,提供一种路线短、收率高、成本低、适合批量工业化生产的联苯吡菌胺的合成方法。
为了实现上述目的,本发明采用如下技术方案予以实现:
一种联苯吡菌胺的合成方法,包括以下步骤:
步骤1:将镁粉和溶剂A混合,然后加入3,4-二氯溴苯的溶剂A溶液进行反应,制得3,4-二氯苯溴化镁金属化合物;
步骤2:将步骤1的3,4-二氯苯溴化镁金属化合物用氯化锌进行金属转移化反应制得(3,4-二氯苯基)氯化锌,接着将(3,4-二氯苯基)氯化锌、2-溴-4-氟硝基苯、催化剂和溶剂A混合进行反应,得到2-(3,4-二氯苯基)-4-氟硝基苯;
步骤3:将步骤2的2-(3,4-二氯苯基)-4-氟硝基苯、铁粉、氯化铵、溶剂B和水混合并加入冰醋酸反应,得到2-(3,4-二氯苯基)-4-氟苯胺;
步骤4:将步骤3的2-(3,4-二氯苯基)-4-氟苯胺和溶剂C混合,然后加入含1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的溶剂C溶液进行反应,得到联苯吡菌胺。
进一步的,步骤1的反应温度为40~65℃,反应时间为2h。
进一步的,所述溶剂A为四氢呋喃或甲基四氢呋喃;
其中,3,4-二氯溴苯和镁粉的摩尔比为1.0:1.0~3.0。
进一步的,步骤2的反应温度为25~30℃,反应时间为4h。
进一步的,所述催化剂为醋酸钯、双(三苯基膦)二氯化钯、四(三苯基膦)钯或Pd(dppf)Cl2;
其中,2-溴-4-氟硝基苯与催化剂的摩尔比为1.0:0.0001~0.05。
进一步的,步骤3的反应时间为5h;所述溶剂B为甲醇或乙醇;其中,2-(3,4-二氯苯基)-4-氟硝基苯、铁粉、氯化铵、冰醋酸的摩尔比为1.0:3.0~10.0:1.0~2.0:1.0~4.0。
进一步的,步骤4的反应时间为5h;所述溶剂C为甲苯、二甲苯、四氢呋喃、甲基四氢呋喃、二氯甲烷、二氯乙烷的一种或几种混合物;
其中,2-(3,4-二氯苯基)-4-氟苯胺、1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的摩尔比为1.0:1.0~2.0。
进一步的,3,4-二氯溴苯和镁的摩尔比为1.0:1.0~1.2;
2-溴-4-氟硝基苯、催化剂的摩尔比为1.0:0.0001~0.001;
2-(3,4-二氯苯基)-4-氟硝基苯、铁粉、氯化铵、醋酸的摩尔比为1.0:3.0~6.0:1.0~1.8:1.0~2.5;
2-(3,4-二氯苯基)-4-氟苯胺、1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的摩尔比为1.0:1.0~1.5。
进一步的,3,4-二氯溴苯和镁的摩尔比为1.0:1.1;
2-溴-4-氟硝基苯、催化剂的摩尔比为1.0:0.0005;
2-(3,4-二氯苯基)-4-氟硝基苯、铁粉、氯化铵、醋酸的摩尔比为1.0:5.0:1.5:2.0;
2-(3,4-二氯苯基)-4-氟苯胺、1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的摩尔比为1.0:1.2。
与现有技术相比,本发明的有益效果是:
本发明的整个合成工艺操作简单,反应条件容易控制,各步骤产品收率高,纯度好,适合批量工业化生产。步骤(1)中只做出格氏试剂中间体,不用做成3,4-二氯苯硼酸,反应步骤减少、溶剂可回收套用,收率较高,成本较低。步骤(2)中采用醋酸钯作为催化剂,催化剂用量少于千分之一、收率大于等于85%,成本更低。本发明的合成方法中,总收率达到75%,含量大于98%;溶剂可回收套用,生产成本低。
以下结合实施例来进一步说明本发明,其目的是能更好的理解本发明的内容所体现本发明的实质性特点,因此所举之例不应视为对本发明保护范围的限制。
本发明联苯吡菌胺的合成路线如下:
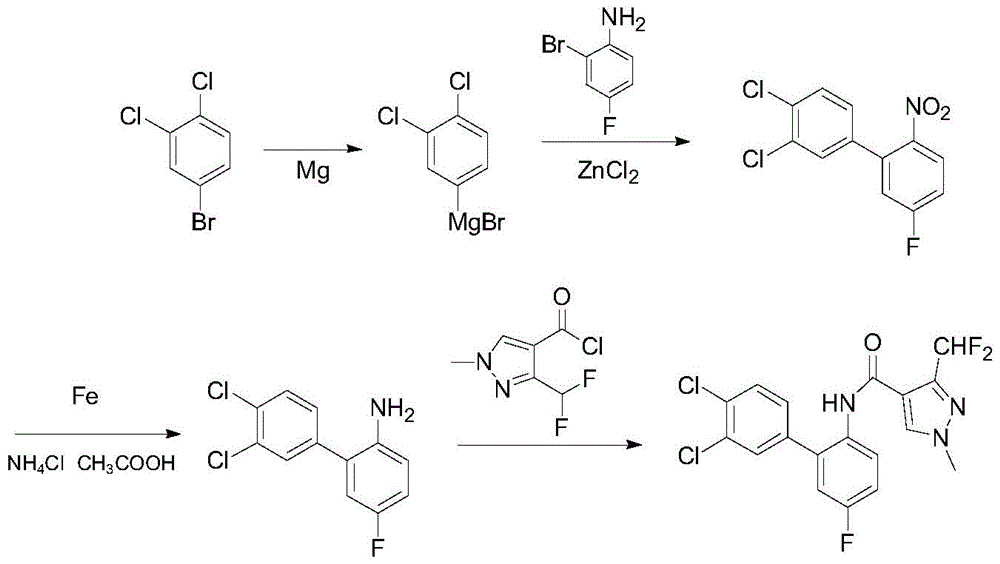
本发明的联苯吡菌胺的合成方法,包括以下步骤:
步骤1:氮气保护下,将镁粉、溶剂A,少许碘粒加入三口圆底烧瓶中,室温下缓慢滴加3,4-二氯溴苯的溶剂A溶液,反应引发后继续滴加3,4-二氯溴苯的溶剂A溶液,LC检测原料全部转化后,溶液冷却密封待用,制得3,4-二氯苯溴化镁金属化合物;
步骤2:将步骤1的3,4-二氯苯溴化镁金属化合物用氯化锌进行金属转移化反应制得(3,4-二氯苯基)氯化锌,氮气保护下,将2-溴-4-氟硝基苯、催化剂、溶剂A加入三口圆底烧瓶中,滴加(3,4-二氯苯基)氯化锌反应,然后用饱和氯化铵溶液洗涤,乙酸乙酯萃取,无水硫酸镁干燥后,减压蒸馏并重结晶得2-(3,4-二氯苯基)-4-氟硝基苯。
步骤3:将2-(3,4-二氯苯基)-4-氟硝基苯、还原铁粉、氯化铵、溶剂B和水加入三口圆底烧瓶中,升温至回流滴加冰醋酸,滴毕继续回流反应5h。冷却至室温后,过滤,浓缩后用二氯甲烷溶解,水洗至中性并干燥,浓缩得到2-(3,4-二氯苯基)-4-氟苯胺;
步骤4:将2-(3,4-二氯苯基)-4-氟苯胺、溶剂C加入三口圆底烧瓶中,搅拌并升温至回流,缓慢滴加含1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的溶剂C溶液,滴完后保温反应5h。冷却至室温后,分液并水洗,浓缩冷却得到联苯吡菌胺。
实施例1
本实施例给出制备联苯吡菌胺的合成方法,包括以下步骤:
1)氮气保护下,将5.2g(0.22mol)镁粉、20mL四氢呋喃,少许碘粒加入250mL三口圆底烧瓶中,室温下缓慢滴加45.2g(0.2mol)3,4-二氯溴苯的100mL四氢呋喃溶液,反应引发后继续滴加3,4-二氯溴苯的四氢呋喃溶液,滴毕升温至65℃保温反应2h,LC检测原料全部转化后,溶液冷却密封待用。
2)将上步制备的溶液用氯化锌(1M in THF,100ml,0.1mol),进行金属转移化反应。氮气保护下,将41.8g(0.19mol)2-溴-4-氟硝基苯、0.02g(0.05%mol)醋酸钯、四氢呋喃加入500mL三口圆底烧瓶中,滴加(3,4-二氯苯基)氯化锌,滴毕于25℃反应4h,然后用饱和氯化铵溶液200ml洗涤,乙酸乙酯(200ml*3)萃取,无水硫酸镁干燥后,减压蒸馏并重结晶得2-(3,4-二氯苯基)-4-氟硝基苯48.8g,纯度95.1%,收率85.4%。
1H NMR(δppm):7.15(ddd,2H),7.28(td,1H),7.44-7.46(m,1H),7.55(d,1H),8.06(dd,1H)。MS(m/z):285.9[M-H]-
3)将28.6g(0.1mol)2-(3,4-二氯苯基)-4-氟硝基苯、28g(0.5mol)还原铁粉、8g(0.15mol)氯化铵、100mL甲醇和20mL水加入250mL三口圆底烧瓶中,升温至回流滴加12g(0.2mol)冰醋酸,滴毕继续回流反应5h。冷却至室温后,过滤,浓缩后用二氯甲烷溶解,水洗至中性并干燥,浓缩得到2-(3,4-二氯苯基)-4-氟苯胺23.6g,纯度98.0%,收率92.1%。
1H NMR(δppm):3.59(s,2H),6.70(dd,1H),6.81(dd,1H),6.88(m,1H),7.28(m,1H),7.30~7.55(m,2H)。
4)将12.8g(0.05mol)2-(3,4-二氯苯基)-4-氟苯胺、40mL二甲苯加入250mL三口圆底烧瓶中,搅拌并升温至回流,缓慢滴加含11.7g(0.06mol)1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的40mL二甲苯溶液,滴完后保温反应5h。冷却至室温后,分液并水洗,浓缩冷却得到联苯吡菌胺20.7g,含量98.7%,收率96.2%,熔点141℃。
1H NMR(δppm):3.90(s,3H),6.76(t,1H),6.95-6.99(dd,1H),7.09-7.22(m,2H),7.46-7.52(m,2H),7.74(s,1H),7.89(s,1H),8.06-8.11(m,1H)。MS(m/z):413[M+]。
以3,4-二氯溴苯计,合成联苯吡菌胺的总收率为75.7%。
实施例2
1)氮气保护下,将14.4g(0.6mol)镁粉、20mL四氢呋喃,少许碘粒加入250mL三口圆底烧瓶中,室温下缓慢滴加45.2g(0.2mol)3,4-二氯溴苯的100mL四氢呋喃溶液,反应引发后继续滴加3,4-二氯溴苯的四氢呋喃溶液,滴毕升温至65℃保温反应2h,LC检测原料全部转化后,溶液冷却密封待用。
2)将上步制备的溶液用氯化锌(1M in THF,100ml,0.1mol),进行金属转移化反应。氮气保护下,将41.8g(0.19mol)2-溴-4-氟硝基苯、2.1g(5%mol)醋酸钯、四氢呋喃加入500mL三口圆底烧瓶中,滴加(3,4-二氯苯基)氯化锌,滴毕于30℃反应4h,然后用饱和氯化铵溶液200ml洗涤,乙酸乙酯(200ml*3)萃取,无水硫酸镁干燥后,减压蒸馏并重结晶得2-(3,4-二氯苯基)-4-氟硝基苯48.2g,纯度94.8%,收率88.6%。
3)将28.6g(0.1mol)2-(3,4-二氯苯基)-4-氟硝基苯、56g(1.0mol)还原铁粉、10.7g(0.2mol)氯化铵、100mL甲醇和20mL水加入250mL三口圆底烧瓶中,升温至回流滴加24g(0.4mol)冰醋酸,滴毕继续回流反应5h。冷却至室温后,过滤,浓缩后用二氯甲烷溶解,水洗至中性并干燥,浓缩得到2-(3,4-二氯苯基)-4-氟苯胺23.9g,纯度98.2%,收率93.4%。
4)将12.8g(0.05mol)2-(3,4-二氯苯基)-4-氟苯胺、40mL二甲苯加入250mL三口圆底烧瓶中,搅拌并升温至回流,缓慢滴加含19.5g(0.1mol)1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的40mL二甲苯溶液,滴完后保温反应5h。冷却至室温后,分液并水洗,浓缩冷却得到联苯吡菌胺19.9g,含量98.5%,收率95.9%。
以3,4-二氯溴苯计,合成联苯吡菌胺的总收率为79.4%。
实施例3:
1)氮气保护下,将4.8g(0.2mol)镁粉、20mL四氢呋喃,少许碘粒加入250mL三口圆底烧瓶中,室温下缓慢滴加45.2g(0.2mol)3,4-二氯溴苯的100mL四氢呋喃溶液,反应引发后继续滴加3,4-二氯溴苯的四氢呋喃溶液,滴毕升温至40℃保温反应2h,LC检测原料全部转化后,溶液冷却密封待用。
2)将上步制备的溶液用氯化锌(1M in THF,100ml,0.1mol),进行金属转移化反应。氮气保护下,将41.8g(0.19mol)2-溴-4-氟硝基苯、0.0004g(0.01%mol)醋酸钯、四氢呋喃加入500mL三口圆底烧瓶中,滴加(3,4-二氯苯基)氯化锌,滴毕于25℃反应4h,然后用饱和氯化铵溶液200ml洗涤,乙酸乙酯(200ml*3)萃取,无水硫酸镁干燥后,减压蒸馏并重结晶得2-(3,4-二氯苯基)-4-氟硝基苯38.9g,纯度96.8%,收率71.5%。
3)将28.6g(0.1mol)2-(3,4-二氯苯基)-4-氟硝基苯、16.8g(0.3mol)还原铁粉、5.3g(0.1mol)氯化铵、100mL甲醇和20mL水加入250mL三口圆底烧瓶中,升温至回流滴加6g(0.1mol)冰醋酸,滴毕继续回流反应5h。冷却至室温后,过滤,浓缩后用二氯甲烷溶解,水洗至中性并干燥,浓缩得到2-(3,4-二氯苯基)-4-氟苯胺21.6g,纯度97.0%,收率84.2%。
4)将12.8g(0.05mol)2-(3,4-二氯苯基)-4-氟苯胺、40mL二甲苯加入250mL三口圆底烧瓶中,搅拌并升温至回流,缓慢滴加含9.7g(0.05mol)1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的40mL二甲苯溶液,滴完后保温反应5h。冷却至室温后,分液并水洗,浓缩冷却得到联苯吡菌胺19.0g,含量98.3%,收率91.7%。
以3,4-二氯溴苯计,合成联苯吡菌胺的总收率为55.2%。
实施例4:
1)氮气保护下,将5.8g(0.24mol)镁粉、20mL四氢呋喃,少许碘粒加入250mL三口圆底烧瓶中,室温下缓慢滴加45.2g(0.2mol)3,4-二氯溴苯的100mL四氢呋喃溶液,反应引发后继续滴加3,4-二氯溴苯的四氢呋喃溶液,滴毕升温至40℃保温反应2h,LC检测原料全部转化后,溶液冷却密封待用。
2)将上步制备的溶液用氯化锌(1M in THF,100ml,0.1mol),进行金属转移化反应。氮气保护下,将41.8g(0.19mol)2-溴-4-氟硝基苯、0.004g(0.1%mol)醋酸钯、四氢呋喃加入500mL三口圆底烧瓶中,滴加(3,4-二氯苯基)氯化锌,滴毕于25℃反应4h,然后用饱和氯化铵溶液200ml洗涤,乙酸乙酯(200ml*3)萃取,无水硫酸镁干燥后,减压蒸馏并重结晶得2-(3,4-二氯苯基)-4-氟硝基苯47.2g,纯度97.3%,收率86.9%。
3)将28.6g(0.1mol)2-(3,4-二氯苯基)-4-氟硝基苯、33.6g(0.6mol)还原铁粉、9.6g(0.18mol)氯化铵、100mL甲醇和20mL水加入250mL三口圆底烧瓶中,升温至回流滴加15g(0.25mol)冰醋酸,滴毕继续回流反应5h。冷却至室温后,过滤,浓缩后用二氯甲烷溶解,水洗至中性并干燥,浓缩得到2-(3,4-二氯苯基)-4-氟苯胺23.1g,纯度98.5%,收率90.1%。
4)将12.8g(0.05mol)2-(3,4-二氯苯基)-4-氟苯胺、40mL二甲苯加入250mL三口圆底烧瓶中,搅拌并升温至回流,缓慢滴加含9.7g(0.05mol)1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的40mL二甲苯溶液,滴完后保温反应5h。冷却至室温后,分液并水洗,浓缩冷却得到联苯吡菌胺19.3g,含量99.0%,收率93.4%。
以3,4-二氯溴苯计,合成联苯吡菌胺的总收率为73.1%。
实施例5
该实施例与实施例1不同之处在于:步骤(2)使用的催化剂为四(三苯基膦)钯,合成联苯吡菌胺的总收率为80.6%。
实施例6:
该实施例与实施例1不同之处在于:步骤(2)使用的催化剂为双(三苯基膦)二氯化钯,合成联苯吡菌胺的总收率为81.2%。
实施例7:
该实施例与实施例1不同之处在于:步骤(2)使用的催化剂为Pd(dppf)Cl2,合成联苯吡菌胺的总收率为75.4%。
实施例8:
该实施例与实施例1的不同之处在于:步骤(4)中所用溶剂C不同。相应的收率如表1所示。
表1不同溶剂对反应的影响
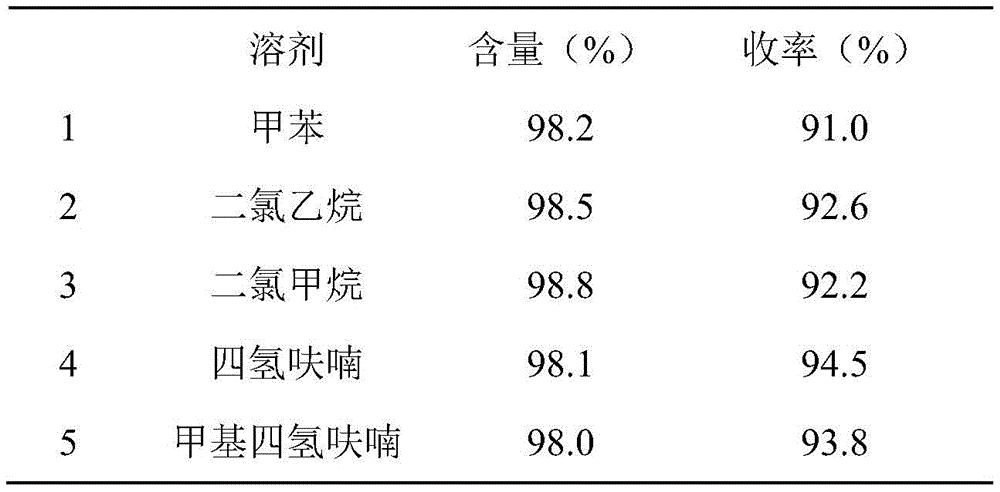
从表1可看出,步骤4中的溶剂C选用甲苯、四氢呋喃、甲基四氢呋喃、二氯甲烷或二氯乙烷对反应影响不大,收率均稳定在90%以上,其中,溶剂C选用四氢呋喃时,收率最高,为94.5%。
本发明的整个合成工艺操作简单,反应条件容易控制,各步骤产品收率高,纯度好,适合批量工业化生产。
以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
Claims (9)
1.一种联苯吡菌胺的合成方法,其特征在于,包括以下步骤:
步骤1:将镁粉和溶剂A混合,然后加入3,4-二氯溴苯的溶剂A溶液进行反应,制得3,4-二氯苯溴化镁金属化合物;
步骤2:将步骤1的3,4-二氯苯溴化镁金属化合物用氯化锌进行金属转移化反应制得(3,4-二氯苯基)氯化锌,接着将(3,4-二氯苯基)氯化锌、2-溴-4-氟硝基苯、催化剂和溶剂A混合进行反应,得到2-(3,4-二氯苯基)-4-氟硝基苯;
步骤3:将步骤2的2-(3,4-二氯苯基)-4-氟硝基苯、铁粉、氯化铵、溶剂B和水混合并加入冰醋酸反应,得到2-(3,4-二氯苯基)-4-氟苯胺;
步骤4:将步骤3的2-(3,4-二氯苯基)-4-氟苯胺和溶剂C混合,然后加入含1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的溶剂C溶液进行反应,得到联苯吡菌胺。
2.如权利要求1所述的联苯吡菌胺的合成方法,其特征在于,步骤1的反应温度为40~65℃,反应时间为2h。
3.如权利要求1所述的联苯吡菌胺的合成方法,其特征在于,所述溶剂A为四氢呋喃或甲基四氢呋喃;
其中,3,4-二氯溴苯和镁粉的摩尔比为1.0:1.0~3.0。
4.如权利要求1所述的联苯吡菌胺的合成方法,其特征在于,步骤2的反应温度为25~30℃,反应时间为4h。
5.如权利要求1所述的联苯吡菌胺的合成方法,其特征在于,所述催化剂为醋酸钯、双(三苯基膦)二氯化钯、四(三苯基膦)钯或Pd(dppf)Cl2;
其中,2-溴-4-氟硝基苯与催化剂的摩尔比为1.0:0.0001~0.05。
6.如权利要求1所述的联苯吡菌胺的合成方法,其特征在于,步骤3的反应时间为5h;
所述溶剂B为甲醇或乙醇;其中,2-(3,4-二氯苯基)-4-氟硝基苯、铁粉、氯化铵、冰醋酸的摩尔比为1.0:3.0~10.0:1.0~2.0:1.0~4.0。
7.如权利要求1所述的联苯吡菌胺的合成方法,其特征在于,步骤4的反应时间为5h;
所述溶剂C为甲苯、二甲苯、四氢呋喃、甲基四氢呋喃、二氯甲烷、二氯乙烷的一种或几种混合物;
其中,2-(3,4-二氯苯基)-4-氟苯胺、1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的摩尔比为1.0:1.0~2.0。
8.如权利要求1所述的联苯吡菌胺的合成方法,其特征在于,3,4-二氯溴苯和镁的摩尔比为1.0:1.0~1.2;
2-溴-4-氟硝基苯、催化剂的摩尔比为1.0:0.0001~0.001;
2-(3,4-二氯苯基)-4-氟硝基苯、铁粉、氯化铵、醋酸的摩尔比为1.0:3.0~6.0:1.0~1.8:1.0~2.5;
2-(3,4-二氯苯基)-4-氟苯胺、1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的摩尔比为1.0:1.0~1.5。
9.如权利要求1所述的联苯吡菌胺的合成方法,其特征在于,3,4-二氯溴苯和镁的摩尔比为1.0:1.1;
2-溴-4-氟硝基苯、催化剂的摩尔比为1.0:0.0005;
2-(3,4-二氯苯基)-4-氟硝基苯、铁粉、氯化铵、醋酸的摩尔比为1.0:5.0:1.5:2.0;
2-(3,4-二氯苯基)-4-氟苯胺、1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯的摩尔比为1.0:1.2。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202211678888.5A CN116178264A (zh) | 2022-12-26 | 2022-12-26 | 一种联苯吡菌胺的合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202211678888.5A CN116178264A (zh) | 2022-12-26 | 2022-12-26 | 一种联苯吡菌胺的合成方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN116178264A true CN116178264A (zh) | 2023-05-30 |
Family
ID=86439436
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202211678888.5A Pending CN116178264A (zh) | 2022-12-26 | 2022-12-26 | 一种联苯吡菌胺的合成方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN116178264A (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117886760A (zh) * | 2023-12-18 | 2024-04-16 | 爱斯特(成都)生物制药股份有限公司 | 3-(5-氟嘧啶-2-基)-2-甲氧基苯胺的合成方法 |
| CN119823043A (zh) * | 2024-12-18 | 2025-04-15 | 湖南化工研究院有限公司 | 利用气提和离子交换树脂脱酸制备联苯吡菌胺的方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007115685A1 (de) * | 2006-04-07 | 2007-10-18 | Bayer Cropscience Ag | Verfahren zum herstellen von biphenylaminen über vinylaniline |
| CN105294492A (zh) * | 2015-09-15 | 2016-02-03 | 联化科技(上海)有限公司 | 一种取代联苯的制备方法 |
-
2022
- 2022-12-26 CN CN202211678888.5A patent/CN116178264A/zh active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007115685A1 (de) * | 2006-04-07 | 2007-10-18 | Bayer Cropscience Ag | Verfahren zum herstellen von biphenylaminen über vinylaniline |
| CN105294492A (zh) * | 2015-09-15 | 2016-02-03 | 联化科技(上海)有限公司 | 一种取代联苯的制备方法 |
Non-Patent Citations (3)
| Title |
|---|
| A. S. VINOGRADOV 等: "Synthesis of Perfluorinated Biaryls by Reaction of Perfluoroarylzinc Compounds with Perfluoroarenes", 《SYNTHESIS OF PERFLUORINATED BIARYLS》, vol. 51, no. 10, 15 October 2015 (2015-10-15), pages 1419 * |
| 何敬文: "《药物合成反应》", 31 December 1995, 中国医药科技出版社, pages: 176 - 178 * |
| 杨光富: "《有机合成》", 31 August 2016, 华东理工大学出版社, pages: 51 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117886760A (zh) * | 2023-12-18 | 2024-04-16 | 爱斯特(成都)生物制药股份有限公司 | 3-(5-氟嘧啶-2-基)-2-甲氧基苯胺的合成方法 |
| CN119823043A (zh) * | 2024-12-18 | 2025-04-15 | 湖南化工研究院有限公司 | 利用气提和离子交换树脂脱酸制备联苯吡菌胺的方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN106478641B (zh) | 瑞博西尼中间体的合成方法 | |
| CN116178264A (zh) | 一种联苯吡菌胺的合成方法 | |
| CN103387541A (zh) | 一种取代吡唑醚类化合物的制备方法 | |
| TW201139400A (en) | Process for the synthesis of ketobenzofuran derivatives | |
| CN108033922B (zh) | 一种3-酰基喹喔啉酮衍生物的制备方法 | |
| CN105218448A (zh) | 一种1-甲基-3-二氟甲基-4-吡唑甲酸的合成方法 | |
| CN115974717A (zh) | 一种含七氟异丙基的双酰胺类化合物及其中间体化合物,其制备方法与用途 | |
| CN111662184A (zh) | 一种啶酰菌胺中间体2-(4’-氯苯基)苯胺的合成方法 | |
| CN107365262B (zh) | 一种5,5-二甲基-2-氰基环戊酮的制备方法 | |
| CN102666496B (zh) | 2-(2-正丁基-4-羟基-6-甲基-嘧啶-5-基)-n,n-二甲基乙酰胺的制备方法 | |
| CN110305018A (zh) | 一种3-溴-2-氟硝基苯的制备方法 | |
| CN115974727B (zh) | 一种联苯肼酯的合成方法 | |
| CN115232037B (zh) | 一种苯甲酰氨磺酰-n-取代苯甲酰胺化合物的制备方法 | |
| CN108373468B (zh) | 一种n-2-吡啶-5-嘧啶甲胺的制备方法 | |
| CN101723897B (zh) | 伊伐布雷定的合成方法 | |
| CN101407474B (zh) | 一种2-溴-4,5-二甲氧基苯丙腈的制备方法 | |
| CN119285548A (zh) | 一种氟唑菌酰胺的制备方法 | |
| CN118791471A (zh) | 一种贝舒地尔的制备方法 | |
| CN114478425B (zh) | 一种芳氧苯氧丙酸酯除草剂的合成方法 | |
| CN106243009B (zh) | 一种3-正丁胺基-4-溴-n-苯基马来酰亚胺的制备方法 | |
| CN115557823B (zh) | 一种合成酰胺类化合物的方法 | |
| TW201908296A (zh) | 嘧啶酮類化合物的製備方法 | |
| CN100463898C (zh) | 3,3’-二甲基-4,4’-二氨基二苯甲烷的制备方法 | |
| CN108794375A (zh) | 一种帕比司他中间体及其合成与应用 | |
| CN116178336A (zh) | 一种酸催化缩酮法合成n-芳基酮亚胺的方法及应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20230530 |