CN1052003C - 3-酰基-2-羟吲哚-1-甲酰胺类抗炎前药 - Google Patents
3-酰基-2-羟吲哚-1-甲酰胺类抗炎前药 Download PDFInfo
- Publication number
- CN1052003C CN1052003C CN94100697A CN94100697A CN1052003C CN 1052003 C CN1052003 C CN 1052003C CN 94100697 A CN94100697 A CN 94100697A CN 94100697 A CN94100697 A CN 94100697A CN 1052003 C CN1052003 C CN 1052003C
- Authority
- CN
- China
- Prior art keywords
- alkyl
- gram
- group
- compound
- milliliters
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000000651 prodrug Substances 0.000 title abstract description 18
- 229940002612 prodrug Drugs 0.000 title abstract description 18
- 230000003110 anti-inflammatory effect Effects 0.000 title abstract description 7
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 28
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 26
- 239000001257 hydrogen Substances 0.000 claims abstract description 16
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 10
- 150000002431 hydrogen Chemical class 0.000 claims abstract description 10
- 150000002367 halogens Chemical class 0.000 claims abstract description 9
- 150000001875 compounds Chemical class 0.000 claims description 129
- -1 4-piperidyl Chemical group 0.000 claims description 91
- HZVOZRGWRWCICA-UHFFFAOYSA-N methanediyl Chemical compound [CH2] HZVOZRGWRWCICA-UHFFFAOYSA-N 0.000 claims description 78
- 229910052760 oxygen Inorganic materials 0.000 claims description 54
- 239000001301 oxygen Substances 0.000 claims description 52
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 44
- 239000000460 chlorine Substances 0.000 claims description 42
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 34
- 229910052757 nitrogen Inorganic materials 0.000 claims description 28
- 229910052799 carbon Inorganic materials 0.000 claims description 27
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 18
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 17
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 8
- 229910052801 chlorine Inorganic materials 0.000 claims description 8
- 239000011737 fluorine Substances 0.000 claims description 8
- 229910052731 fluorine Inorganic materials 0.000 claims description 8
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 7
- 150000003053 piperidines Chemical class 0.000 claims description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 6
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 4
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 4
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 claims description 2
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 claims description 2
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 2
- 125000003342 alkenyl group Chemical group 0.000 claims description 2
- 125000001118 alkylidene group Chemical group 0.000 claims description 2
- 125000004122 cyclic group Chemical group 0.000 claims description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 claims description 2
- 125000005356 cycloalkylalkenyl group Chemical group 0.000 claims description 2
- 150000003951 lactams Chemical group 0.000 claims description 2
- UHHKSVZZTYJVEG-UHFFFAOYSA-N oxepane Chemical compound C1CCCOCC1 UHHKSVZZTYJVEG-UHFFFAOYSA-N 0.000 claims description 2
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 claims description 2
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 claims description 2
- 229910052717 sulfur Inorganic materials 0.000 claims description 2
- 150000003527 tetrahydropyrans Chemical class 0.000 claims description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims 5
- PCKPVGOLPKLUHR-UHFFFAOYSA-N OH-Indolxyl Natural products C1=CC=C2C(O)=CNC2=C1 PCKPVGOLPKLUHR-UHFFFAOYSA-N 0.000 abstract 1
- 230000001760 anti-analgesic effect Effects 0.000 abstract 1
- JYGFTBXVXVMTGB-UHFFFAOYSA-N indolin-2-one Chemical compound C1=CC=C2NC(=O)CC2=C1 JYGFTBXVXVMTGB-UHFFFAOYSA-N 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 193
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 186
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 130
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 129
- 239000007787 solid Substances 0.000 description 107
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 92
- 239000000243 solution Substances 0.000 description 81
- 239000000203 mixture Substances 0.000 description 79
- 238000000034 method Methods 0.000 description 77
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 75
- 238000001704 evaporation Methods 0.000 description 72
- 235000019439 ethyl acetate Nutrition 0.000 description 69
- 238000002360 preparation method Methods 0.000 description 67
- 238000004458 analytical method Methods 0.000 description 55
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 55
- 230000008020 evaporation Effects 0.000 description 49
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 48
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 46
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 44
- 239000000047 product Substances 0.000 description 44
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 43
- 239000000741 silica gel Substances 0.000 description 42
- 229910002027 silica gel Inorganic materials 0.000 description 42
- 229960001866 silicon dioxide Drugs 0.000 description 42
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 42
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 39
- 239000012043 crude product Substances 0.000 description 38
- 238000001035 drying Methods 0.000 description 36
- 238000003756 stirring Methods 0.000 description 34
- 238000007738 vacuum evaporation Methods 0.000 description 32
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 29
- 238000002425 crystallisation Methods 0.000 description 29
- 230000008025 crystallization Effects 0.000 description 29
- 238000001953 recrystallisation Methods 0.000 description 29
- 239000002904 solvent Substances 0.000 description 29
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 27
- 239000010410 layer Substances 0.000 description 27
- JPOXNPPZZKNXOV-UHFFFAOYSA-N bromochloromethane Chemical compound ClCBr JPOXNPPZZKNXOV-UHFFFAOYSA-N 0.000 description 26
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 25
- 238000001914 filtration Methods 0.000 description 25
- 239000002253 acid Substances 0.000 description 23
- 239000000706 filtrate Substances 0.000 description 23
- 235000009518 sodium iodide Nutrition 0.000 description 23
- 238000004611 spectroscopical analysis Methods 0.000 description 22
- 238000001816 cooling Methods 0.000 description 21
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 20
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 20
- 239000003480 eluent Substances 0.000 description 20
- 238000003818 flash chromatography Methods 0.000 description 19
- 239000000725 suspension Substances 0.000 description 19
- 238000005481 NMR spectroscopy Methods 0.000 description 18
- 238000000605 extraction Methods 0.000 description 18
- NPSSWQJHYLDCNV-UHFFFAOYSA-N prop-2-enoic acid;hydrochloride Chemical compound Cl.OC(=O)C=C NPSSWQJHYLDCNV-UHFFFAOYSA-N 0.000 description 18
- 238000005406 washing Methods 0.000 description 16
- 239000002585 base Substances 0.000 description 14
- 239000011541 reaction mixture Substances 0.000 description 14
- 150000001721 carbon Chemical group 0.000 description 13
- 239000003921 oil Substances 0.000 description 13
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 12
- 150000002148 esters Chemical class 0.000 description 12
- 238000009834 vaporization Methods 0.000 description 12
- 230000008016 vaporization Effects 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 10
- 229910052740 iodine Inorganic materials 0.000 description 10
- 239000011630 iodine Substances 0.000 description 10
- 239000007788 liquid Substances 0.000 description 10
- 238000004537 pulping Methods 0.000 description 10
- 150000003839 salts Chemical class 0.000 description 10
- 159000000000 sodium salts Chemical class 0.000 description 10
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 9
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 9
- 238000013019 agitation Methods 0.000 description 9
- 239000000463 material Substances 0.000 description 9
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 8
- 239000012141 concentrate Substances 0.000 description 8
- 238000000926 separation method Methods 0.000 description 8
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 8
- NYZBWOSRZJKQAI-UHFFFAOYSA-N Valerylglycine Chemical compound CCCCC(=O)NCC(O)=O NYZBWOSRZJKQAI-UHFFFAOYSA-N 0.000 description 7
- 235000019441 ethanol Nutrition 0.000 description 7
- 239000000284 extract Substances 0.000 description 7
- 238000010898 silica gel chromatography Methods 0.000 description 7
- 229910052938 sodium sulfate Inorganic materials 0.000 description 7
- 235000011152 sodium sulphate Nutrition 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 239000003643 water by type Substances 0.000 description 7
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 6
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 6
- 239000013078 crystal Substances 0.000 description 6
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 6
- 229910000474 mercury oxide Inorganic materials 0.000 description 6
- UKWHYYKOEPRTIC-UHFFFAOYSA-N mercury(ii) oxide Chemical compound [Hg]=O UKWHYYKOEPRTIC-UHFFFAOYSA-N 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- 238000001556 precipitation Methods 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 229960003328 benzoyl peroxide Drugs 0.000 description 5
- 238000004821 distillation Methods 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 235000014347 soups Nutrition 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 4
- QJXYINMYZCCIBW-UHFFFAOYSA-M CC([O-])=O.CC(O)=O.CC(O)=O.NC(N)=N.O.[Na+] Chemical compound CC([O-])=O.CC(O)=O.CC(O)=O.NC(N)=N.O.[Na+] QJXYINMYZCCIBW-UHFFFAOYSA-M 0.000 description 4
- YDLBXWIUWHVCCS-UHFFFAOYSA-N ICOC(C)=O.NC(=N)N.[O] Chemical compound ICOC(C)=O.NC(=N)N.[O] YDLBXWIUWHVCCS-UHFFFAOYSA-N 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 238000011097 chromatography purification Methods 0.000 description 4
- 238000005352 clarification Methods 0.000 description 4
- 238000000354 decomposition reaction Methods 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 4
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 4
- GSJFXBNYJCXDGI-UHFFFAOYSA-N methyl 2-hydroxyacetate Chemical class COC(=O)CO GSJFXBNYJCXDGI-UHFFFAOYSA-N 0.000 description 4
- GRJJQCWNZGRKAU-UHFFFAOYSA-N pyridin-1-ium;fluoride Chemical compound F.C1=CC=NC=C1 GRJJQCWNZGRKAU-UHFFFAOYSA-N 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- RINCXYDBBGOEEQ-UHFFFAOYSA-N succinic anhydride Chemical compound O=C1CCC(=O)O1 RINCXYDBBGOEEQ-UHFFFAOYSA-N 0.000 description 4
- 235000005074 zinc chloride Nutrition 0.000 description 4
- 239000011592 zinc chloride Substances 0.000 description 4
- HZDNNJABYXNPPV-UHFFFAOYSA-N (2-chloro-2-oxoethyl) acetate Chemical class CC(=O)OCC(Cl)=O HZDNNJABYXNPPV-UHFFFAOYSA-N 0.000 description 3
- ZLYYJUJDFKGVKB-OWOJBTEDSA-N (e)-but-2-enedioyl dichloride Chemical compound ClC(=O)\C=C\C(Cl)=O ZLYYJUJDFKGVKB-OWOJBTEDSA-N 0.000 description 3
- JJKWHOSQTYYFAE-UHFFFAOYSA-N 2-methoxyacetyl chloride Chemical compound COCC(Cl)=O JJKWHOSQTYYFAE-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- XQCKXOKBWVXUJH-UHFFFAOYSA-N CCOC([O])=O Chemical compound CCOC([O])=O XQCKXOKBWVXUJH-UHFFFAOYSA-N 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- 150000003862 amino acid derivatives Chemical class 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 125000001589 carboacyl group Chemical group 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- QOPVNWQGBQYBBP-UHFFFAOYSA-N chloroethyl chloroformate Chemical compound CC(Cl)OC(Cl)=O QOPVNWQGBQYBBP-UHFFFAOYSA-N 0.000 description 3
- 125000002603 chloroethyl group Chemical group [H]C([*])([H])C([H])([H])Cl 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 150000005690 diesters Chemical class 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- RZCCQWOCSZOHMQ-UHFFFAOYSA-N n,n-diethyl-2-hydroxyacetamide Chemical compound CCN(CC)C(=O)CO RZCCQWOCSZOHMQ-UHFFFAOYSA-N 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 229920000573 polyethylene Polymers 0.000 description 3
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 3
- 235000019345 sodium thiosulphate Nutrition 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- XMVQWNRDPAAMJB-UHFFFAOYSA-N (+)-13-Cyclopent-2-enyl-tridecansaeure Natural products OC(=O)CCCCCCCCCCCCC1CCC=C1 XMVQWNRDPAAMJB-UHFFFAOYSA-N 0.000 description 2
- XKBCNTPVQJGJPY-CMDGGOBGSA-N (e)-non-1-en-1-ol Chemical compound CCCCCCC\C=C\O XKBCNTPVQJGJPY-CMDGGOBGSA-N 0.000 description 2
- 125000001478 1-chloroethyl group Chemical group [H]C([H])([H])C([H])(Cl)* 0.000 description 2
- QGLVWTFUWVTDEQ-UHFFFAOYSA-N 2-chloro-3-methoxyphenol Chemical compound COC1=CC=CC(O)=C1Cl QGLVWTFUWVTDEQ-UHFFFAOYSA-N 0.000 description 2
- JNQVLKWNKVMFBN-UHFFFAOYSA-N 2-hydroxy-n,n-dimethylacetamide Chemical compound CN(C)C(=O)CO JNQVLKWNKVMFBN-UHFFFAOYSA-N 0.000 description 2
- XILORGWKMLOGKF-UHFFFAOYSA-N 4-(diethylamino)-4-oxobutanoic acid Chemical compound CCN(CC)C(=O)CCC(O)=O XILORGWKMLOGKF-UHFFFAOYSA-N 0.000 description 2
- MSHFRERJPWKJFX-UHFFFAOYSA-N 4-Methoxybenzyl alcohol Chemical compound COC1=CC=C(CO)C=C1 MSHFRERJPWKJFX-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Natural products CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- XMAIGJQOEDWYPQ-UHFFFAOYSA-N C(=O)(OCC1=CC=CC=C1)C(OC(=O)C1=CC=C(C(=O)Cl)C=C1)C1=CC=CC=C1 Chemical compound C(=O)(OCC1=CC=CC=C1)C(OC(=O)C1=CC=C(C(=O)Cl)C=C1)C1=CC=CC=C1 XMAIGJQOEDWYPQ-UHFFFAOYSA-N 0.000 description 2
- WHSXTOFGAIFRTI-UHFFFAOYSA-N C(=O)(OCC1=CC=CC=C1)C(OC(=O)CCC(=O)O)C1=CC=CC=C1 Chemical compound C(=O)(OCC1=CC=CC=C1)C(OC(=O)CCC(=O)O)C1=CC=CC=C1 WHSXTOFGAIFRTI-UHFFFAOYSA-N 0.000 description 2
- RWPZLLTWNXBVCO-UHFFFAOYSA-N ClCOC(C)=O.NC(=N)N.[O] Chemical compound ClCOC(C)=O.NC(=N)N.[O] RWPZLLTWNXBVCO-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 102000003820 Lipoxygenases Human genes 0.000 description 2
- 108090000128 Lipoxygenases Proteins 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 102000004005 Prostaglandin-endoperoxide synthases Human genes 0.000 description 2
- 108090000459 Prostaglandin-endoperoxide synthases Proteins 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 150000001266 acyl halides Chemical class 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- AOGYCOYQMAVAFD-UHFFFAOYSA-N chlorocarbonic acid Chemical class OC(Cl)=O AOGYCOYQMAVAFD-UHFFFAOYSA-N 0.000 description 2
- FSPIZMJSZQNNNL-UHFFFAOYSA-N chloromethyl 2-aminoacetate Chemical compound NCC(=O)OCCl FSPIZMJSZQNNNL-UHFFFAOYSA-N 0.000 description 2
- IXBDTGGFRAKVNX-UHFFFAOYSA-N chloromethyl 4-(diethylamino)-4-oxobutanoate Chemical compound CCN(CC)C(=O)CCC(=O)OCCl IXBDTGGFRAKVNX-UHFFFAOYSA-N 0.000 description 2
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 238000010908 decantation Methods 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- NTNZTEQNFHNYBC-UHFFFAOYSA-N ethyl 2-aminoacetate Chemical compound CCOC(=O)CN NTNZTEQNFHNYBC-UHFFFAOYSA-N 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- BWUDRSYMFIDDGB-UHFFFAOYSA-N iodomethyl 2-aminoacetate Chemical compound NCC(=O)OCI BWUDRSYMFIDDGB-UHFFFAOYSA-N 0.000 description 2
- QYXQRRLWXZBSGJ-UHFFFAOYSA-N iodomethyl 4-(diethylamino)-4-oxobutanoate Chemical compound CCN(CC)C(=O)CCC(=O)OCI QYXQRRLWXZBSGJ-UHFFFAOYSA-N 0.000 description 2
- KYXUBZSBLGVOQG-UHFFFAOYSA-N iodomethyl 4-(dimethylamino)-4-oxobutanoate Chemical compound CN(C)C(=O)CCC(=O)OCI KYXUBZSBLGVOQG-UHFFFAOYSA-N 0.000 description 2
- NDJPBTNFCNBLFW-UHFFFAOYSA-N iodomethyl 4-(dipropylamino)-4-oxobutanoate Chemical compound CCCN(CCC)C(=O)CCC(=O)OCI NDJPBTNFCNBLFW-UHFFFAOYSA-N 0.000 description 2
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- CFHIDWOYWUOIHU-UHFFFAOYSA-N oxomethyl Chemical compound O=[CH] CFHIDWOYWUOIHU-UHFFFAOYSA-N 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- RZWZRACFZGVKFM-UHFFFAOYSA-N propanoyl chloride Chemical compound CCC(Cl)=O RZWZRACFZGVKFM-UHFFFAOYSA-N 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- ATBIAJXSKNPHEI-UHFFFAOYSA-N pyridine-3-carbonyl chloride Chemical compound ClC(=O)C1=CC=CN=C1 ATBIAJXSKNPHEI-UHFFFAOYSA-N 0.000 description 2
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- WUWHFEHKUQVYLF-UHFFFAOYSA-M sodium;2-aminoacetate Chemical compound [Na+].NCC([O-])=O WUWHFEHKUQVYLF-UHFFFAOYSA-M 0.000 description 2
- UIUJIQZEACWQSV-UHFFFAOYSA-N succinic semialdehyde Chemical compound OC(=O)CCC=O UIUJIQZEACWQSV-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- TXTWXQXDMWILOF-UHFFFAOYSA-N (2-ethoxy-2-oxoethyl)azanium;chloride Chemical compound [Cl-].CCOC(=O)C[NH3+] TXTWXQXDMWILOF-UHFFFAOYSA-N 0.000 description 1
- QVHJQCGUWFKTSE-YFKPBYRVSA-N (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound OC(=O)[C@H](C)NC(=O)OC(C)(C)C QVHJQCGUWFKTSE-YFKPBYRVSA-N 0.000 description 1
- NILQLFBWTXNUOE-UHFFFAOYSA-N 1-aminocyclopentanecarboxylic acid Chemical compound OC(=O)C1(N)CCCC1 NILQLFBWTXNUOE-UHFFFAOYSA-N 0.000 description 1
- WADJDQQIPSKENN-UHFFFAOYSA-N 1-methoxy-4-methylbenzene Chemical class [CH2]C1=CC=C(OC)C=C1 WADJDQQIPSKENN-UHFFFAOYSA-N 0.000 description 1
- KPWDGTGXUYRARH-UHFFFAOYSA-N 2,2,2-trichloroethanol Chemical compound OCC(Cl)(Cl)Cl KPWDGTGXUYRARH-UHFFFAOYSA-N 0.000 description 1
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 1
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 1
- BWGOQNGZBLOTTI-UHFFFAOYSA-N 2-(4-chlorophenyl)-1-(6-fluoro-1,3-benzothiazol-2-yl)-4-hydroxy-3-(5-methylfuran-2-carbonyl)-2h-pyrrol-5-one Chemical compound O1C(C)=CC=C1C(=O)C1=C(O)C(=O)N(C=2SC3=CC(F)=CC=C3N=2)C1C1=CC=C(Cl)C=C1 BWGOQNGZBLOTTI-UHFFFAOYSA-N 0.000 description 1
- OXQGTIUCKGYOAA-UHFFFAOYSA-N 2-Ethylbutanoic acid Natural products CCC(CC)C(O)=O OXQGTIUCKGYOAA-UHFFFAOYSA-N 0.000 description 1
- CQQUWTMMFMJEFE-UHFFFAOYSA-N 2-chloro-n,n-diethylacetamide Chemical compound CCN(CC)C(=O)CCl CQQUWTMMFMJEFE-UHFFFAOYSA-N 0.000 description 1
- OFTKFKYVSBNYEC-UHFFFAOYSA-N 2-furoyl chloride Chemical compound ClC(=O)C1=CC=CO1 OFTKFKYVSBNYEC-UHFFFAOYSA-N 0.000 description 1
- RZNHSEZOLFEFGB-UHFFFAOYSA-N 2-methoxybenzoyl chloride Chemical compound COC1=CC=CC=C1C(Cl)=O RZNHSEZOLFEFGB-UHFFFAOYSA-N 0.000 description 1
- RLQZIECDMISZHS-UHFFFAOYSA-N 2-phenylcyclohexa-2,5-diene-1,4-dione Chemical class O=C1C=CC(=O)C(C=2C=CC=CC=2)=C1 RLQZIECDMISZHS-UHFFFAOYSA-N 0.000 description 1
- QISAUDWTBBNJIR-UHFFFAOYSA-N 2-phenylmethoxyacetyl chloride Chemical compound ClC(=O)COCC1=CC=CC=C1 QISAUDWTBBNJIR-UHFFFAOYSA-N 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- RUQIUASLAXJZIE-UHFFFAOYSA-N 3-methoxybenzoyl chloride Chemical compound COC1=CC=CC(C(Cl)=O)=C1 RUQIUASLAXJZIE-UHFFFAOYSA-N 0.000 description 1
- MGZORAUZXTVJSA-UHFFFAOYSA-N 4-(dimethylamino)-4-oxobutanoate;dimethylazanium Chemical compound C[NH2+]C.CN(C)C(=O)CCC([O-])=O MGZORAUZXTVJSA-UHFFFAOYSA-N 0.000 description 1
- WAIGPJMZARQZDX-UHFFFAOYSA-N 4-(dimethylamino)-4-oxobutanoic acid Chemical compound CN(C)C(=O)CCC(O)=O WAIGPJMZARQZDX-UHFFFAOYSA-N 0.000 description 1
- VHSXMWPTPBCNHB-UHFFFAOYSA-N 4-(dipropylamino)-4-oxobutanoic acid Chemical compound CCCN(CCC)C(=O)CCC(O)=O VHSXMWPTPBCNHB-UHFFFAOYSA-N 0.000 description 1
- WRSDJJXJDSEUSQ-UHFFFAOYSA-N 5-oxo-5-phenylmethoxypentanoic acid Chemical compound OC(=O)CCCC(=O)OCC1=CC=CC=C1 WRSDJJXJDSEUSQ-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- STDZLVHBSPRVBR-UHFFFAOYSA-N C(=O)=C1C(CCC1)C.[O].ClC Chemical compound C(=O)=C1C(CCC1)C.[O].ClC STDZLVHBSPRVBR-UHFFFAOYSA-N 0.000 description 1
- VMXZECKWBZVUET-UHFFFAOYSA-N CN(C)ClC=O Chemical compound CN(C)ClC=O VMXZECKWBZVUET-UHFFFAOYSA-N 0.000 description 1
- JAWWNCVQVOHGOB-UHFFFAOYSA-N CNC.CN(C)C(CCC(O)=O)=O.ClCCl Chemical compound CNC.CN(C)C(CCC(O)=O)=O.ClCCl JAWWNCVQVOHGOB-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- WRHYBDZCFBLCSH-UHFFFAOYSA-N ClC(C)C(C(=O)Cl)OC Chemical compound ClC(C)C(C(=O)Cl)OC WRHYBDZCFBLCSH-UHFFFAOYSA-N 0.000 description 1
- 229910020366 ClO 4 Inorganic materials 0.000 description 1
- 208000030453 Drug-Related Side Effects and Adverse reaction Diseases 0.000 description 1
- 206010053155 Epigastric discomfort Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 125000000174 L-prolyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])[C@@]1([H])C(*)=O 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 206010039361 Sacroiliitis Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- IWTYTFSSTWXZFU-QPJJXVBHSA-N [(e)-3-chloroprop-1-enyl]benzene Chemical compound ClC\C=C\C1=CC=CC=C1 IWTYTFSSTWXZFU-QPJJXVBHSA-N 0.000 description 1
- CDSUEGXTPYWXAB-UHFFFAOYSA-N acetic acid ethyl acetate Chemical compound CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.CCOC(C)=O CDSUEGXTPYWXAB-UHFFFAOYSA-N 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000005600 alkyl phosphonate group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- MXMOTZIXVICDSD-UHFFFAOYSA-N anisoyl chloride Chemical compound COC1=CC=C(C(Cl)=O)C=C1 MXMOTZIXVICDSD-UHFFFAOYSA-N 0.000 description 1
- 229910052728 basic metal Inorganic materials 0.000 description 1
- 150000003818 basic metals Chemical class 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 229960004217 benzyl alcohol Drugs 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- DVECBJCOGJRVPX-UHFFFAOYSA-N butyryl chloride Chemical compound CCCC(Cl)=O DVECBJCOGJRVPX-UHFFFAOYSA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- SKCNIGRBPJIUBQ-UHFFFAOYSA-N chloroform;ethyl acetate Chemical compound ClC(Cl)Cl.CCOC(C)=O SKCNIGRBPJIUBQ-UHFFFAOYSA-N 0.000 description 1
- CHHRXILMDYFSNF-UHFFFAOYSA-N chloromethyl 4-(dimethylamino)-4-oxobutanoate Chemical compound CN(C)C(=O)CCC(=O)OCCl CHHRXILMDYFSNF-UHFFFAOYSA-N 0.000 description 1
- LORSXHZWVXQHJH-UHFFFAOYSA-N chloromethyl 4-(dipropylamino)-4-oxobutanoate Chemical compound CCCN(CCC)C(=O)CCC(=O)OCCl LORSXHZWVXQHJH-UHFFFAOYSA-N 0.000 description 1
- BOXZXICVMMSYPE-UHFFFAOYSA-N chloromethyl benzoate Chemical compound ClCOC(=O)C1=CC=CC=C1 BOXZXICVMMSYPE-UHFFFAOYSA-N 0.000 description 1
- 230000006837 decompression Effects 0.000 description 1
- SXLDHZFJMXLFJU-UHFFFAOYSA-N diethyl cyclopropane-1,2-dicarboxylate Chemical compound CCOC(=O)C1CC1C(=O)OCC SXLDHZFJMXLFJU-UHFFFAOYSA-N 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- WEHWNAOGRSTTBQ-UHFFFAOYSA-N dipropylamine Chemical compound CCCNCCC WEHWNAOGRSTTBQ-UHFFFAOYSA-N 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 150000002084 enol ethers Chemical class 0.000 description 1
- 238000005837 enolization reaction Methods 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- IXZFDJXHLQQSGQ-UHFFFAOYSA-N ethyl 4-chloro-4-oxobutanoate Chemical compound CCOC(=O)CCC(Cl)=O IXZFDJXHLQQSGQ-UHFFFAOYSA-N 0.000 description 1
- 239000002024 ethyl acetate extract Substances 0.000 description 1
- 238000003810 ethyl acetate extraction Methods 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- FJBUQBPKURBOJF-UHFFFAOYSA-N formyl chloride;pyridine Chemical compound ClC=O.C1=CC=NC=C1 FJBUQBPKURBOJF-UHFFFAOYSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- 239000008274 jelly Substances 0.000 description 1
- 238000002386 leaching Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- RMIODHQZRUFFFF-UHFFFAOYSA-N methoxyacetic acid Chemical compound COCC(O)=O RMIODHQZRUFFFF-UHFFFAOYSA-N 0.000 description 1
- SRXOJMOGPYFZKC-UHFFFAOYSA-N methyl 4-chloro-4-oxobutanoate Chemical compound COC(=O)CCC(Cl)=O SRXOJMOGPYFZKC-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- ZPJZSEHCMJYUPI-UHFFFAOYSA-N methyl piperazine-1-carboxylate Chemical compound COC(=O)N1CCNCC1 ZPJZSEHCMJYUPI-UHFFFAOYSA-N 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- ZGEGCLOFRBLKSE-UHFFFAOYSA-N methylene hexane Natural products CCCCCC=C ZGEGCLOFRBLKSE-UHFFFAOYSA-N 0.000 description 1
- GRVDJDISBSALJP-UHFFFAOYSA-N methyloxidanyl Chemical group [O]C GRVDJDISBSALJP-UHFFFAOYSA-N 0.000 description 1
- AJFDBNQQDYLMJN-UHFFFAOYSA-N n,n-diethylacetamide Chemical compound CCN(CC)C(C)=O AJFDBNQQDYLMJN-UHFFFAOYSA-N 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- VIKNJXKGJWUCNN-XGXHKTLJSA-N norethisterone Chemical compound O=C1CC[C@@H]2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 VIKNJXKGJWUCNN-XGXHKTLJSA-N 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 150000005623 oxindoles Chemical class 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical compound [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 238000012453 sprague-dawley rat model Methods 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- QQWYQAQQADNEIC-RVDMUPIBSA-N tert-butyl [(z)-[cyano(phenyl)methylidene]amino] carbonate Chemical compound CC(C)(C)OC(=O)O\N=C(/C#N)C1=CC=CC=C1 QQWYQAQQADNEIC-RVDMUPIBSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 150000003866 tertiary ammonium salts Chemical class 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Indole Compounds (AREA)
Abstract
本发明公开了式(Ⅰ)的抗炎和止痛的羟吲哚前药,其中R10、R11、R12和R13为氢、烷基或卤素,R为亚甲基氧基链烷酰基、亚甲基氧基链烯酰基或链烯酰基。
Description
本发明涉及抗炎和止痛剂,具体地讲,涉及3-酰基-2-羟吲哚-1-甲酰胺类化合物的烯醇酯和醚前药,这是一类已知的非甾族抗炎剂。
羟吲哚类化合物作为抗炎剂的应用已被报道于美国专利3,634,453中,并且是由1-取代-2-羟吲哚-3-甲酰胺类化合物组成的。最近,一系列3-酰基-2-羟吲哚-1-甲酰胺类化合物被公开于美国专利4,556,672中,它们是环氧合酶(CO)和脂氧合酶(LO)抑制剂并且是有用的哺乳动物受治疗者的止痛剂和抗炎剂。某些3-酰基-2-羟吲哚-1-甲酰胺类前药被描述于公开的美国专利5,118,703中,其结构如式(A)所示: 其中X和Y各自为氢、氟或氯;R1为2-噻吩基或苄基;R为具有2-10个碳原子的链烷酰基、具有5-7个碳原子的环烷基羰基、具有7-10个碳原子的苯基链烷酰基、氯苯甲酰基、甲氧基苯甲酰基、噻吩甲酰基、所述烷氧基具有1-3个碳原子而所述链烷酰基具有3-5个碳原子的ω-烷氧羰基链烷酰基;具有2-10个碳原子的烷氧羰基;苯氧羰基;所述酰基具有1-4个碳原子而所述烷基具有2-4个碳原子的1-(酰氧基)烷基;所述烷氧基具有2-5个碳原子而所述烷基具有1-4个碳原子的1-(烷氧羰基氧基)烷基;具有1-3个碳原子的烷基;具有1-3个碳原子的烷基磺酰基;甲基苯基磺酰基或所述烷基各自具有1-3个碳原子的膦酸二烷基酯。美国专利5,118,703和美国专利4,556,672在这里引入作为参考文献。
其中X和Y各自为氢、氟或氯;R1为2-噻吩基或苄基;R为具有2-10个碳原子的链烷酰基、具有5-7个碳原子的环烷基羰基、具有7-10个碳原子的苯基链烷酰基、氯苯甲酰基、甲氧基苯甲酰基、噻吩甲酰基、所述烷氧基具有1-3个碳原子而所述链烷酰基具有3-5个碳原子的ω-烷氧羰基链烷酰基;具有2-10个碳原子的烷氧羰基;苯氧羰基;所述酰基具有1-4个碳原子而所述烷基具有2-4个碳原子的1-(酰氧基)烷基;所述烷氧基具有2-5个碳原子而所述烷基具有1-4个碳原子的1-(烷氧羰基氧基)烷基;具有1-3个碳原子的烷基;具有1-3个碳原子的烷基磺酰基;甲基苯基磺酰基或所述烷基各自具有1-3个碳原子的膦酸二烷基酯。美国专利5,118,703和美国专利4,556,672在这里引入作为参考文献。
本发明提供了式(I)的抗炎醚和酯前药, 其中R为式(II)-式(VI)基团,
其中R为式(II)-式(VI)基团,
 其中x为0或1;
其中x为0或1;
A为被最多达2个独立地选自C1-C7烷基或C3-C7环烷基的取代基任选取代的C1-C5亚烷基或C2-C6链烯基链;或者(CH2)nO(CH2)m,其中亚甲基可以被最多达2个独立地选自C1-C7烷基或C3-C7环烷基的取代基任选取代;或者被最多达2个C1-C3烷基任选取代的C3-C7环烷基或环烯基;或含有O、S或NR6连键的4-7元杂脂环基团;或被最多达2个独立地选自C1-C3烷基、C1-C3烷氧基、卤素或CF3的取代基任选取代的亚苯基;
B为C2-C6链烯基苯基、2、3或4-吡啶基、2、3或4-哌啶基、2或3-吡咯烷基、-OCH2CO2R1或-OCH2CONR2R3;
R1为H、C1-C8烷基、C3-C7环烷基、苯基(C1-C4)烷基、(CH2)pCO2R2、或(CH2)pCONR2R3、(CH2)pSi(CH3)2;或者R1可以与A形成任选被C1-C3烷基取代的5、6或7元内酯环;
R2和R3独立地为H、C1-C7烷基、C3-C7环烷基、苯基(C1-C4)烷基;或者R2和R3与所连的氮一起可以代表被最多达2个C1-C3烷基任选取代的吡咯烷、哌啶、吗啉或高哌啶基团;或者R2或R3可以与A形成被最多达2个C1-C3烷基任选取代的5、6或7元内酰胺环;
R4和R5独立地为H、C1-C7烷基、C3-C7环烷基、苯基(C1-C4)烷基、(CH2)pCO2R2、(CH2)pCONR2R3、(CH2)pNR7R8、(CH2)pOR6或(CH2)pSR6;或者R4与R5合在-起代表被最多达2个C1-C3烷基任选取代的C3-C7环烷基环;
R6为H、C1-C6烷基、(CH2)pCOOR2、被最多达2个C1-C6烷基任选取代的C3-C7环烷基、在苯环上被最多达2个独立地选自C1-C3烷基、C1-C3烷氧基、卤素或CF3的取代基任选取代的苯基(C1-C4)烷基、COR2、CONR2R3、或被最多达2个独立地选自C1-C3烷基、C1-C3烷氧基、卤素或CF3的取代基任选取代的苯基;或者R6与R4和所连的氧一起可代表被最多达2个C1-C3烷基任选取代的氧杂环丁烷、四氢呋喃、四氢吡喃或氧杂环庚烷;
R7和R8独立地为H、C1-C6烷基、C3-C7环烷基、苯基(C1-C4)烷基、COR2、COOR2;或R7和R8独立地为被最多达2个独立地选自C1-C6烷基、C3-C7环烷基、苯基(C1-C4)烷基、C3-C7支链烷基的取代基任选取代的C2-C7链烷酰基、C4-C8环烷酰基;或者R7和R8与所连的氮合在一起可以代表被最多达2个独立地选自C1-C6烷基、C3-C7环烷基、C3-C7支链烷基或氧代的取代基任选取代的吡咯烷、哌啶或高哌啶基团;
R9为H或甲基;
R10、R11、R12和R13独立地选自H、C1-C4烷基和卤素;
m和n独立地为0、1或2,其中m或n必须至少为1;以及
p为1-3。
优选的第一组化合物是其中R10和R11之一为5-氟而另一个为6-氯的式(I)化合物。
优选的第二组化合物是其中R10和R11之一为5-氟而另一个为6-氯且R为式(II),其中x为0,A为C2-C6链烯基链而R1为氢的式(I)化合物。在此组中特别优选的是其中A为几何构型为E构型的-CH=CH-而R12和R13为氢的化合物。在此组中还优选的是其中x为1,A为亚烷基而R1为苄基的化合物。
优选的第三组化合物是其中R10和R11之一为5-氟而另一个为6-氯,R为式(IV)而x为1的式(I)化合物。此组中特别优选的是其中R4、R9、R12和R13为氢,R5为氢、甲基或乙基且R6为氢、甲基、苄基或CH2COOR2的化合物。
优选的第四组化合物是其中R10和R11之一为5-氟而另一个为6-氯,x为1而R为式(V)的式(I)化合物。此组中优选的是其中R4、R9、R7、R8、R12和R13为氢,而R5为(CH2)pNR7R8、甲基或苄基的化合物。此组中还优选的是其中R7为COR2的化合物。
优选的第五组化合物是其中R10与R11之一为5-氟而另一个为6-氯,x为1,R为式(VI)且B为2-或3-吡咯烷的式(I)化合物。
本发明也包含治疗哺乳动物的炎症的方法,该方法包含给所说哺乳动物给药抗炎有效量的选自式(I)化合物的化合物。
本发明进一步包含治疗哺乳动物的疼痛的方法,该方法包含给所说哺乳动物给药止痛有效量的选自式(I)化合物的化合物。
本发明的烯醇醚和酯不是如母体化合物的烯醇酸,并且与所述母体化合物相比,显示出降低胃刺激的潜能。
术语“前药”是指这样一些药物前体化合物,它们在经给药和吸收后,通过某些新陈代谢过程在体内释放出该药物。
尽管所有通常的给药途径对于本发明化合物都是有用的,然而优选的给药途径是口服。经胃肠吸收后,本发明化合物在体内水解为其中R是氢的相应的式(I)化合物或其盐。因为本发明前药不是烯醇酸,因此胃肠道与酸性母体化合物的接触机会被减至最小。此外,由于胃肠道并发症已被认为是酸性非甾族抗炎药物的主要的不利反应[参见例如DelFavero,“Side Effects of Drugs Annual7”,Dukes和Elis,Eds.Excerpta Medica,Amsterdam,1983,p.104-115],所以本发明化合物(I)可能比母体烯醇化合物具有独特的优点。
在将3-酰基-2-羟吲哚-1-甲酰胺类化合物转化为式(I)化合物过程中,在3-位上的外向环双键上的取代基可以是顺式、反式或二者的混合物。因此,下式结构的化合物 或其混合物被描述为具有下式结构:
或其混合物被描述为具有下式结构: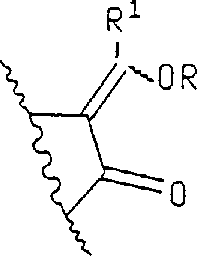 所有这些异构体均被认为是本发明的一部分。
所有这些异构体均被认为是本发明的一部分。
所需作为起始原料的3-酰基-2-羟吲哚-1-甲酰胺类化合物可以通过本领域的公知方法得到,参见例如,美国专利3,634,453和4,556,672。上面提到的其它起始试剂是可购到的,或由公知方法制备的,或者由在下面制备例部分所描述的方法制备得到。
本发明化合物的制备是易于完成的。适合的3-酰基-2-羟吲哚-1-甲酰胺的盐是在反应惰性溶剂中生成的,并经分离或不经分离就用于随后的与酰基卤或α-卤代烷基酯的反应中。这些反应条件不是关键性的;温度可以在约0℃至约50℃范围内变化,优选范围为约0℃至约20℃。反应时间依所选反应剂和温度而变化,但其范围在约8~90小时之间,优选时间为约20小时。
3-酰基-2-羟吲哚-1-甲酰胺的盐可以是碱金属盐、叔胺盐或季铵盐。碱金属包括锂、钠和钾。叔胺一般为低分子量脂族胺,例如三甲胺、三乙胺、三丁胺和混合胺,例如二异丙基乙胺、二乙基氨基吡啶;以及杂环胺例如吡啶和N-甲基吗啉。季铵化合物可以是对称的或混合的直链或支链烷基胺。钠盐、二异丙基乙胺盐、三乙胺盐和四丁基季铵盐是优选的。
酰基卤可以是酰基氯或酰基溴;而酰基氯是优选的。α-卤代酯可以是氯代、溴代或碘代酯,其中氯代和碘代酯是优选的。氯代酯优选与碘化钠一起使用,因此就地产生碘代酯。
本发明前药的生物利用率是通过该前药与母体化合物的比较进行测定的。
例如使禁食的雄性Sprague-Dawley大鼠口服3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺及所选前药,其剂量水平为3毫克当量3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺/千克作为在0.1%甲基纤维素中的溶液或悬浮液。每一药物制剂的给药体积保持在每1千克体重1毫升。口服后,血样是通过在服药后1、3和6小时向肝素化管中放入眶后
 (retroorbitat sinus)血并立即冷却而得到的。血浆贮存在-20℃下直至分析为止。
(retroorbitat sinus)血并立即冷却而得到的。血浆贮存在-20℃下直至分析为止。
给药3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺及前药后血浆中3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的浓度是通过高压液相色谱在360nm用紫外线检测进行测定的。对于3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的定量下限为0.2微克/毫升。
在口服给药3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺及每一前药后,对于3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的浓度-时间曲线下的面积[AUC(0-6小时)]是通过线性斜方形方法测定的。在每一前药给药后,对于3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的相对生物利用率是通过测定给药前药后3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的AUC(0-6)值与给药3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺后3-(羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的AUC(0-6)值的比率而进行评定的。
按照已知方法,例如大鼠足水肿试验、大鼠佐药诱导的关节炎试验或苯基苯醌诱导的小鼠扭曲试验,正如前面用于评价母体化合物的方法和描述于上述参考文献和其它文献中的方法进行式(1)前药的抗炎和止痛活性的评价,参见例如,C.A.Winter,“Progress inDrug Research”由E.Jucker编辑,Birkhauser Verlag,Basel,Vol.10,pp.139-192(1966).
以摩尔计,本发明前药一般以与衍生它们的已知的3-酰基-2-羟吲哚-1-甲酰胺类化合物相同的剂量水平和频率给药。然而,在需要用较高剂量抑制疼痛和炎症时,本发明化合物的非烯醇特性使其一般具有较高的耐受口服剂量。
本发明前药也是以与已知母体化合物相同的方式制成制剂的,并以相同的途径给药的,正如上面所述参考文献中所描述的那样。优选的给药途径是口服,这样便特别利用了本发明化合物的非烯醇特性。
本发明通过下列实施例加以说明,但并不限于这些实施例的特定细节。
实施例1
6-氯-5-氟-2,3-二氢-3-[(4-甲氧基苯甲酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1甲酰胺(5.08克,15.0毫摩尔)在CH2Cl2中制成浆液,并用三乙胺(1.67克,16.5毫摩尔)处理。将此黄色溶液在冰水浴中冷却并用4-甲氧基苯甲酰氯(12.8克,75.0毫摩尔)一次性处理。18小时后,过滤该反应混合物以除去黄色沉淀。滤液再用CH2Cl2稀释并用1N HCl(两次)和饱和NaHCO3/盐水混合物洗涤。用MgSO4干燥后,过滤,浓缩并用乙醇提取(两次),所得固体用EtOAc/己烷研制。收集固体,并将其与直接从反应混合物中分离出的固体合并,将全部固体用EtOAc/己烷(4/1)和一些丙酮重结晶,得到1.81克(26%)所期望的产物,为黄色晶体:mp 220-221℃;元素分析计算值:C22H14ClFN2O5S:C,55.88;H,2.98;N,5.92。实测值:C,56.04;H,2.82;N,5.88。
实施例2
6-氯-5-氟-2,3-二氢-3-[(肉桂酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了肉桂酰氯:mp 214-215℃;元素分析计算值:C23H14ClFN2O4S:C,58.91;H,3.01;N,5.97。实测值:C,58.52;H,2.89;N,5.91。
实施例3
6-氯-5-氟-2,3-二氢-3-[(3-甲氧基苯甲酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了3-甲氧基苯甲酰氯(4当量)并用氯仿作溶剂。产物用异丙醇重结晶:mp196-220℃。1H NMR谱表明样品中含有标题化合物的E和Z型两种几何异构体,比率为17∶83。元素分析计算值:C22H14ClFN2O5S:C,55.88;H,2.98;N,5.92。实测值:C,56.00;H,2.82;N,5.78。
实施例4
6-氯-5-氟-2,3-二氢-3-[(2-甲氧基苯甲酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了2-甲氧基苯甲酰氯,并用氯仿作溶剂。粗产物通过硅胶快速色谱法纯化(用95∶5 CHCl3/MeOH洗脱),然后通过用异丙醇重结晶。mp219-221℃。1H NMR谱表明样品中只含有标题化合物的E型几何异构体。元素分析计算值:C22H14ClFN2O5S:C,55.88;H,2.98;N,5.92。实测值:C,55.32;H,3.01;N,5.66。
实施例5
6-氯-5-氟-2,3-二氢-3-[烟酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了烟酰氯(2.5当量)和3.4当量三乙胺,并用氯仿作溶剂。粗产物通过快速色谱法纯化(用83∶17 CHCl3/MeOH作洗脱剂),然后用异丙醇重结晶:mp201-202.5°,元素分析计算值:C20H11ClFN3O4S:C,54.12;H,2.50;N,9.47。实测值:C,54.01;H,2.46;N,9.30。
实施例6
6-氯-5-氟-2,3-二氢-3-[异烟酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了异烟酰氯(1.1当量)和二异丙基乙基胺(2当量)。通过过滤反应混合物直接得到的产品用异丙醇重结晶:mp219.5-221°,元素分析计算值:C20H11ClFN3O4S:C,54.12;H,2.50;N,9.47。实测值:C,54.11;H,2.43;N,9.32。
实施例7
6-氯-5-氟-2,3-二氢-3-[吡啶甲酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了吡啶甲酰氯(1.1当量)和二异丙基乙基胺(2当量)。通过过滤反应混合物直接得到的产物用异丙醇重结晶:mp184-185°,元素分析计算值:C20H11ClFN3O4S:C,54.12;H,2.50;N,9.47。实测值:C,53.73;H,2.34;N,9.31。
实施例8
6-氯-5-氟-2,3-二氢-3-[3-(乙氧羰基)丙烯酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)3-(乙氧羰基)丙烯酰氯的制备用Lutz方法(J.Am.Chem.Soc.,1930,52,3430)。
b)该标题化合物的制备用实施例1的方法,所不同的是使用了3-(乙氧羰基)丙烯酰氯(2当量)和二异丙基乙基胺。粗产物通过快速色谱法纯化(用CHCl3洗脱,然后用98∶2 CHCl3/MeOH洗脱),然后经异丙醇重结晶:mp165.5-168℃。1H NMR谱表明该样品含有标题化合物的E和Z型两种几何异构体,比率为22∶78。元素分析计算值:C20H14ClFN2O6S:C,51.68;H,3.04;N,6.03。实测值:C,51.42;H,3.08;N,5.86。
实施例9
6-氯-5-氟-2,3-二氢-3-[3-(甲氧羰基)丙烯酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)3-(甲氧羰基)丙烯酰氯的制备是用Lutz方法(J.Am.Chem.Soc.,1930,52,3430)。
b)该标题化合物的制备用实施例1的方法,所不同的是使用了3-(甲氧羰基)丙烯酰氯(2当量)和二异丙基乙基胺。粗产物通过快速色谱法(用95∶5 CH2CL2/iPrOH洗脱)纯化,然后用异丙醇重结晶:mp183.5-186℃。1H NMR谱表明该样品含有标题化合物的E和Z型两种几何异构体,比率为34∶66。元素分析计算值:C19H12ClFN2O6S:C,50.62;H,2.68;N,6.21。实测值:C,50.56;H,2.84;N,6.04。
实施例10
6-氯-5-氟-2,3-二氢-3-[苄氧基乙酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了苄氧基乙酰氯(2当量)和二异丙基乙基胺(2当量)。粗产物用快速色谱法(用CHCl3洗脱)纯化,然后用异丙醇重结晶:mp 150-175℃。元素分析计算值:C23H16ClFN2O5S:C,56.74;H,3.31;N,5.75。实测值:C,58.41;H,3.17;N,5.00。
实施例11
6-氯-5-氟-2,3-二氢-3-[4-(苄氧羰基)苯甲酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了4-(苄氧羰基)苯甲酰氯(1.5当量)和二异丙基乙基胺(1.5当量)。粗产物通过与CHCl3研制加以纯化:mp 212-218℃。元素分析计算值:C29H18ClFN2O6S:C,60.37;H,3.14;N,4.85。实测值:C,60.94;H,3.00;N,4.27。
实施例12
6-氯-5-氟-2,3-二氢-3-[3-羧基丙烯酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)3-(2-三甲基甲硅烷基乙氧羰基)丙烯酰氯的制备用Lutz方法的变体(J.Am.Chem.Soc,1930,52,3430),其中剩余的富马酰氯和不需要的二酯是通过在真空下蒸馏除去的,在反应罐中剩下所期望的酰氯。
b)该标题化合物的制备用实施例1的方法,所不同的是使用了3-(2-三甲基甲硅烷基乙氧羰基)丙烯酰氯(1.3当量)和二异丙基乙基胺。粗产物用快速色谱法(用CH2Cl2、99∶1 CH2Cl2/MeOH及然后用98∶2 CH2Cl2/MeOH作洗脱剂)纯化,然后用乙腈重结晶。
c)在冰浴中在聚乙烯瓶中将氟化氢/吡啶配合物(500克)冷却。然后分批加入2-3克3-(2-三甲基甲硅烷基乙氧羰基)丙烯酰基衍生物(33.84克,63毫摩尔)。加毕,在冷却条件下将所得浆状物搅拌0.5小时。然后通过加入H2O使反应中止。在真空中干燥后,通过用热乙酸乙酯研制纯化产物。产量为19.8克。分析样品是通过用乙酸重结晶少量样品得到的:mp 229-232℃。1H NMR谱表明该样品只含有标题化合物的E型几何异构体。元素分析计算值:C18H10ClFN2O6S:C,49.50;H,2.31;N,6.41。实测值:C,49.23;H,2.23;N,6.37。
实施例13
6-氯-5-氟-2,3-二氢-3-[3-羧基苯甲酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)将间苯二酰二氯(40.6克,200毫摩尔)和2,2,2-三氯乙醇(30.9克,207毫摩尔)的甲苯溶液加热回流24小时。蒸发掉溶剂,剩余物质在真空下蒸馏。在0.2mmHg在125°~170℃之间蒸馏出所期望的产物以及一些不需要的双(2,2,2-三氯乙基)酯。经放置,该不需要的副产物作为固体分离出来。将所期望的产物,即3-(2,2,2-三氯乙氧羰基)苯甲酰氯(19.0克)滗析出,用于下一步骤中。
b)该标题化合物的制备用实施例1的方法,所不同的是使用了3-(2,2,2-三氯乙氧羰基)苯甲酰氯(2当量)和二异丙基乙基胺(2当量)。将通过过滤反应混合物直接得到的产物用CHCl3研制。
c)用锌粉(1.0克,16毫摩尔)处理3-(2,2,2-三氯乙氧羰基)苯甲酰基衍生物(1.0克,1.6毫摩尔)在乙酸(50毫升)中的溶液。将该混合物在油浴中温热至50℃,并保持18小时。在依然温热的同时,将混合物过滤(用乙酸洗涤),在冷却至室温后,将滤液倾入H2O(250毫升)中。通过过滤收集沉淀出的黄色固体,即6-氯-5-氟-2,3-二氢-3-[3-羧基苯甲酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺,并用乙酸重结晶:mp244-248℃。产量为130毫克。元素分析计算值:C19H14ClFN2O6S:C,50.40;H,3.12;N,6.19。实测值:C,51.21;H,2.96;N,5.99。
实施例14
6-氯-5-氟-2,3-二氢-3-[(丁氧羰基)丙烯酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)3-(丁氧羰基)丙烯酰氯的制备用Lutz方法(J.Am.Chem.Soc.,1930,52,3430)。
b)该标题化合物的制备用实施例1的方法,所不同的是使用了3-(丁氧羰基)丙烯酰氯(2当量)和二异丙基乙基胺。粗产物用快速色谱法(用CH2Cl2、95∶5 CH2Cl2/MeOH以及然后用90∶10 CH2Cl2/MeOH作洗脱剂)纯化,然后用乙腈重结晶:mp 196-197℃。1H NMR谱表明该样品只含有标题化合物的E型几何异构体。元素分析计算值:C22H18ClFN2O6S;C,53.61;H,3.68;N,5.68。实测值:C,53.53;H,3.55;N,5.78。
实施例15
6-氯-5-氟-2,3-二氢-3-[3-(辛氧羰基)丙烯酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)3-(辛氧羰基)丙烯酰氯的制备用Lutz方法(J.Am.Chem.Soc.,1930,52,3430)。
b)该标题化合物的制备用实施例1的方法,所不同的是使用了3-(辛氧羰基)丙烯酰氯(2当量)和二异丙基乙基胺。粗产物用快速色谱法(用99∶1 CH2Cl2/MeOH作洗脱剂)纯化,然后用乙腈重结晶:mp140-148℃。1H NMR谱表明该样品含有标题化合物的E和Z型两种几何异构体,比率为33∶67。元素分析计算值:C26H26ClFN2O6S:C,56.88;H,4.77;N,5.10。实测值:C,56.81;H,4.62;N,5.07。
实施例16
6-氯-5-氟-2,3-二氢-3-[3-(4-苯基丁氧羰基)丙烯酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)3-(4-苯基丁氧羰基)丙烯酰氯的制备用Lutz方法(J.AmChem.Soc.,1930,52,3430)。
b)该标题化合物的制备用实施例1的方法,所不同的是使用了3-(4-苯基丁氧羰基)丙烯酰氯(2当量)和二异丙基乙基胺。粗产物用快速色谱法(用98∶2 CH2Cl2/MeOH作洗脱剂)纯化,然后用乙腈重结晶。mp 159-161℃。1H NMR谱表明该样品含有标题化合物的E和Z型两种几何异构体,比率为27∶73。元素分析计算值:C28H22ClFN2O6S:C,59.10;H,3.90;N,4.92。实测值:C,58.89;H,3.71;N,4.72。
实施例17
6-氯-5-氟-2,3-二氢-3-[辛酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了辛酰氯(2当量),并用二异丙基乙基胺作为碱。粗产物用快速色谱法在硅胶(用CH2Cl2洗脱)上纯化,然后用乙腈重结晶:mp 149-150℃。1HNMR谱表明该样品只含有标题化合的Z型几何异构体。元素分析计算值:C22H22ClFN2O4S:C,56.83;H,4.77;N,6.03。实测值:C,56.85;H,4.49;N,6.03。
实施例18
6-氯-5-氟-2,3-二氢-3-[3-(1-甲基丙氧羰基)丙烯酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)3-(1-甲基丙氧羰基)丙烯酰氯的制备用Lutz方法(J.Am.Chem.Soc.,1930,52,3430)。
b)该标题化合物的制备用实施例1的方法,所不同的是使用了3-(1-甲基丙氧羰基)丙烯酰氯(1.2当量)和二异丙基乙基胺。粗产物通过快速色谱法(用CH2Cl2洗脱)纯化,然后用乙腈重结晶:mp180-186℃。1H NMR谱表明该样品含有标题化合物的E和Z型两种几何异构体,比率为60∶40。元素分析计算值:C22H18ClFN2O6S:C,53.61;H,3.68;N,5.68。实测值:C,53.60;H,3.57;N,5.67。
实施例19
6-氯-5-氟-2,3-二氢-3-[3-(甲氧羰基)丙酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了3-(甲氧羰基)丙酰氯(2当量)和二异丙基乙基胺。粗产物用快速色谱法(用96∶4 CH2Cl2/MeOH洗脱)纯化,然后用甲苯重结晶:mp179-183℃。1H NMR谱表明该样品含有标题化合物的E和Z型两种几何异构体,比率为31∶69。元素分析计算值:C22H12ClFN2O6S:C,54.27;H,2.48;N,5.75。实测值:C,53.80;H,2.18;N,5.71。
实施例20
6-氯-5-氟-2,3-二氢-3-[3-(乙氧羰基)丙酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用实施例1的方法,所不同的是使用了3-(乙氧羰基)丙酰氯(2当量)和二异丙基乙基胺(2.2当量)。粗产物用快速色谱法(用97.5∶2.5 CH2Cl2/MeOH洗脱)纯化,然后用CH2Cl2研制,并用乙腈重结晶:mp 182-183℃。1H NMR谱表明该样品只含有标题化合物的E型几何异构体。元素分析计算值:C20H16ClFN2O6S:C,51.45;H,3.45;N,6.00。实测值:C,51.38;H,3.27;N,5.97。
实施例21
6-氯-5-氟-2,3-二氢-3-[3-(N,N-二乙基甲酰氨基甲基)氧基羰基]丙酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)将琥珀酐(16.0克,160毫摩尔)和苄醇(17.1克,158毫摩尔)在甲苯(200毫升)中的溶液加热回流3.5小时。此时,在真空下除去溶剂,剩下3-(苄氧羰基)丙酸,为白色固体。
b)将3-(苄氧羰基)丙酸(15.0克,72毫摩尔)、2-氯-N,N-二乙基乙酰胺(11.9克,79.5毫摩尔)、三乙胺(11.2毫升,80.4毫摩尔)和碘化钠(1.1克,7.4毫摩尔)在乙酸乙酯(280毫升)中的溶液加热回流3小时。过滤所得混合物以除去三乙胺盐酸盐,将其用更多的乙酸乙酯洗涤。滤液用1N HCl溶液、饱和NaHCO3溶液和盐水洗涤。经干燥和过滤后,在真空下除去溶剂,剩下3-[(N,N-二乙基甲酰氨基甲基)氧基羰基]丙酸苄酯,为粘稠油状物(22.2克)。
c)向3-[(N,N-二乙基甲酰氨基甲基)氧基羰基]丙酸苄酯(22.2克,62.9毫摩尔)在乙醇(250毫升)中的溶液中加入10%钯/炭(1.0克)。在25℃下在氢气(3个大气压)气氛中将该混合物摇动18小时。通过用硅藻土过滤除去催化剂,蒸发溶剂剩下3-[(N,N-二乙基甲酰氨基甲基)氧基羰基]丙酸,为白色固体(10.47克)。
d)将3-[(N,N-二乙基甲酰氨基甲基)氧基羰基]丙酸(5.0克,21.6毫摩尔)和草酰氯(2.0毫升,23.5毫摩尔)在苯(100毫升)中的溶液加热回流1小时。在真空下除去溶剂,剩下3-[(N,N-二乙基甲酰氨基甲基)氧基羰基]丙酰氯,为油状物。
e)该标题化合物的制备用实施例1的方法,所不同的是使用了3-[(N,N-二乙基甲酰氨基甲基)氧基羰基]丙酰氯(2.2当量)和二异丙基乙基胺(2.2当量)。粗产物用快速色谱法(用CH2Cl2、然后用97.5∶2.5 CH2Cl2/MeOH洗脱)纯化,然后用1∶1乙醚/CH2Cl2研制,用甲苯重结晶:mp 165-167℃。1H NMR谱表明该样品只含有标题化合物的E型几何异构体。元素分析计算值:C24H23ClFN3O2S:C,52.22;H,4.20;N,7.61。实测值:C,52.17;H,4.06;N,7.50。
实施例22
5-氯-2,3-二氢-3-[3-羧基丙烯酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)3-(2-三甲基甲硅烷基乙氧羰基)丙烯酰氯的制备用Lutz方法变体(J.Am.Chem.Soc.,1930,52,3430)。其中剩下的富马酰氯和不需要的二酯是通过在真空下蒸馏除去的,剩下所期望的酰基氯留在反应罐中。
b)该标题化合物的制备用实施例1的方法,所不同的是使用了3-(2-三甲基甲硅烷基乙氧羰基)丙烯酰氯(1.3当量)、5-氯-2,3-二氢-3-[羟基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺和二异丙基乙基胺。粗产物用快速色谱法(用98∶2 CH2Cl2/EtOAc作洗脱剂)纯化,然后用乙腈重结晶。
c)在冰浴中在聚乙烯瓶中将氟化氢/吡啶配合物(20毫升)冷却。然后加入3-(2-三甲基甲硅烷基乙氧羰基)丙烯酰基衍生物(0.89克,1.71毫摩尔)。在冷却条件下将所得浆状物搅拌0.5小时。然后通过加入水使反应中止。过滤收集产物,用水洗涤,在真空中干燥。产量为0.60克,mp 198-200℃。1H NMR谱表明该样品只含有标题化合物的E型几何异构体。元素分析计算值:C18H11ClN2O6S:C,51.62;H,2.65;N,6.69。实测值:C,51.18;H,2.62;N,6.51。
实施例23
6-氯-5-氟-2,3-二氢-3-[3-羧基丙烯酰氧基-(4-氯-2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)3-(2-三甲基甲硅烷基乙氧羰基)丙烯酰氯的制备用Lutz方法变体(J.Am.Chem.Soc.,1930,52,3430),其中剩余的富马酰氯和不需要的二酯通过减压蒸馏除去,剩下所期望的酰基氯留在反应罐中。
b)该标题化合物的制备用实施例1的方法,所不同的是使用了3-(2-三甲基甲硅烷基乙氧羰基)丙烯酰氯(1.3当量)、6-氯-5-氟-2,3-二氢-3-[羟基-(4-氯-2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺及二异丙基乙基胺。粗产物用快速色谱法(用60∶40己烷/EtOAc作洗脱剂)纯化,然后用乙腈重结晶。
c)在冰浴中在聚乙烯瓶中将氟化氢/吡啶配合物(20毫升)冷却。然后加入3-(2-三甲基甲硅烷基乙氧羰基)丙烯酰基衍生物(0.82克,1.43毫摩尔)。在冷却条件下将所得浆状物搅拌0.5小时。然后通过加入H2O使反应中止。过滤收集产物,用H2O洗涤并于真空下干燥。产量为0.50克,mp 158℃。1H NMR谱表明该样品只含有标题化合物的E型几何异构体。元素分析计算值:C18H9Cl2FN2O6S:C,45.88;H,1.92;N,5.94。实测值:C,45.24;H,1.98;N,5.84。
实施例24
6-氯-5-氟-2,3-二氢-3-[乙氧羰基甲氧基乙酰氧基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)将二甘醇酸酐(12克,0.103摩尔)和乙醇(4.74克,0.103摩尔)在甲苯(100毫升)中的溶液加热回流16小时。将溶液冷却并蒸发,在减压下蒸馏残余物。在5托和109-135℃范围下收集产物二甘醇酸单乙酯(9.2克,55%)。
b)向硫酸氢四丁基铵(24.6克,0.0725摩尔)的水(250毫升)溶液中分批加入固体NaHCO3(12.2克,0.145摩尔),然后加入二甘醇酸单乙酯(11.75克,0.0725摩尔)溶液。将该混合物搅拌16小时,然后用二氯甲烷(3×500毫升)萃取。将合并的有机萃取液干燥,过滤并蒸发,剩下盐(21.1克,72%),该盐使用时无需进一步纯化。
c)在1.5小时内将二甘醇酸单乙酯的四丁基铵盐(16克,0.04摩尔)在二氯甲烷(125毫升)中的溶液加入到溴氯甲烷(200毫升)中。将所得溶液在室温下搅拌过夜,然后蒸发至干。将残余物在硅胶上进行色谱法分离,用乙酸乙酯/己烷40/60洗脱,合并含有产物的级分,并蒸发,得到二甘醇酸乙基二氯甲基酯(0.836克,10%),为无色油状物。NMR分析表明该物质纯度大约为70%,然而可直接用于下一步反应。
d)将由步骤c)得到的该氯甲基酯(0.836克,3.98毫摩尔)溶于丙酮(10毫升)中,然后加入碘化钠(1.8克,11.9毫摩尔),并将该混合物在室温下搅拌16小时。过滤该混合物,蒸发滤液至干,将其再溶于二氯甲烷(25毫升)中,然后用水(10毫升)、硫代硫酸钠溶液(10毫升)和饱和氯化钠溶液(10毫升)洗涤。将有机相分离出、干燥并蒸发,得到粗二甘醇酸乙基碘甲基酯(1克),该化合物直接用于最后步骤中。
e)在5分钟内向搅拌着的钠代-6-氯-5-氟2,3-二氢-3-[氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺(1.2克,3.3毫摩尔)在丙酮(30毫升)中的悬浮液中加入由步骤d)中得到的碘甲基酯在丙酮(10毫升)中的溶液。将该混合物在室温下搅拌16小时。所得悬浮液用丙酮稀释直到得到溶液为止,加入硅胶(2克),将该混合物蒸发至干。将所得无水硅胶浆状物加样到硅胶色谱柱上,然后用乙酸乙酯/己烷40/60洗脱。合并含有产物的级分,蒸发得到粗产物,该粗产物用乙醇重结晶得到标题化合物,为黄色晶状固体(136毫克,mp129-130℃)。
实施例25
6-氯-5-氟-2,3-二氢-3-[苄氧羰基甲氧基乙酰氧基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
该标题化合物的制备用与实施例24相同的方法,所不同的是用苄醇代替了步骤a)中的乙醇,得到二甘醇酸单苄基酯,用其进行其余的步骤。该标题化合物为黄色晶状固体,mp 122℃。
实施例26
6-氯-5-氟-2,3-二氢-3-[1-(苄氧羰基甲氧基乙酰氧基)乙氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)向二甘醇酸单苄基酯(7.87克,0.035毫摩尔)的二氯甲烷(75毫升)溶液中加入亚硫酰氯(42克,0.35摩尔),并将该混合物在室温下搅拌72小时。将该混合物蒸发至干,该粗酰基氯直接用于下一步骤中。
b)向由步骤a)得到的酰基氯(2.6克,0.011摩尔)和催化量的熔融的氯化锌的混合物中加入乙醛(0.96克,0.022摩尔),并将该混合物在室温下搅拌2小时。然后将该混合物蒸发至干,将残余物在硅胶上进行色谱法分离,用乙酸乙酯/己烷30/70洗脱。合并含有产物的级分,蒸发得到无色油状物(1.1克,35%)。
c)在5分钟内向搅拌着的钠代-6-氯-5-氟-2,3-二氧-3-[氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺(0.95克,2.62毫摩尔)在丙酮(20毫升)中的悬浮液中加入由步骤b)得到的氯乙基酯的丙酮(10毫升)溶液。然后加入碘化钠(0.13克,0.87毫摩尔),并将混合物加热回流7小时。将所得悬浮液冷却、用丙酮稀释至得到溶液为止,加入硅胶(2克),并将混合物蒸发至干。将所得的无水硅胶浆状物加样到硅胶色谱柱上,然后用乙酸乙酯/己烷40/60洗脱。合并含有产物的级分,蒸发得到粗产物。用乙醚重结晶得到标题化合物,为黄色晶状固体(20毫克,mp 89-91℃)。
实施例27
6-氯-5-氟-2,3-二氢-3-[苄氧羰基甲氧基乙酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
在5分钟内向冷却(0℃)的搅拌着的6-氯-5-氟-2,3-二氢-3-[氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺(1克,3毫摩尔)和三乙胺(0.33克,3.3毫摩尔)的二氯甲烷(10毫升)溶液中加入二甘醇酸单苄酯酰基氯(0.79克,3.3毫摩尔)(参见实施例26,步骤a))的二氯甲烷(10毫升)溶液。将该混合物在室温下搅拌2小时,然后将其吸附在硅胶上,并进行色谱法分离,用二氯甲烷/甲醇15/1洗脱。合并含有产物的级分并蒸发,用乙腈重结晶该粗产物,得到标题化合物,为黄色晶状固体(40毫克,mp 189-190℃)。
实施例28
6-氯-5-氟-2,3-二氢-3-[N,N-二乙基氨基甲酰基甲氧基乙酰氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)在10分钟内向搅拌着的二甘醇酸酐(1克,8.6毫摩尔)的甲苯(10毫升)溶液中加入二乙胺(0.628克,8.6毫摩尔)。产生温和的放热,将混合物在室温下搅拌16小时。蒸发溶剂得到产物,为无色粘稠油状物(1.7克,100%)。
b)向N,N-二乙基氨基甲酰基甲氧基乙酸(1.5克,7.3毫摩尔)的二氯甲烷(15毫升)溶液中加入亚硫酰氯(8.7克,73毫摩尔),将该混合物在室温下搅拌16小时。蒸发溶剂得到粗酰基氯,该其直接用于下一步骤中。
c)在5分钟内向搅拌着的6-氯-5-氟-2,3-二氢-3-[氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺(2.7克,8.2毫摩尔)和三乙胺(1克,9.8毫摩尔)的二氯甲烷(20毫升)溶液中加入N,N-二乙基氨基甲酰基甲氧基乙酰氯(1.7克,8.2毫摩尔)的二氯甲烷(10毫升)溶液。将该混合物在室温下搅拌72小时,然后将其吸附在硅胶上,并进行色谱法分离,用二氯甲烷/甲醇25/1洗脱。合并含有产物的级分并蒸发,粗产物用丙酮重结晶,得到标题化合物,为黄色晶状固体(126毫克,mp 205-208℃)。
实施例29
6-氯-5-氟-2,3-二氢-3-[(2-(1,1-二甲基乙氧羰基氨基)丙酰氧基)甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
将N-t-BOC丙氨酸的四丁基铵(TBA+)盐(50毫摩尔)(通过在乙醇中用TBA+氢氧化物滴定制备或通过在水中,用1当量TBA+HSO4 -和2当量NaHCO3处理并用含氯烃萃取制备)溶于BrCH2Cl(200毫升)中,并在暗处搅拌48小时。将浓缩,了的混合物溶于CHCl3中,用水洗涤,用MgSO4干燥,过滤并浓缩为油状物。将其进行色谱法分离(CH2Cl2为洗脱剂),得到6.25克(52%)N-t-BOC丙氨酸氯甲基酯,该化合物含有痕量的相应的溴甲基酯。
在丙酮(150毫升)中混合N-t-BOC丙氨酸氯甲基酯(6.20克,26.1毫摩尔)和碘化钠(19.5克,130毫摩尔),并在室温下搅拌18小时。然后过滤该混合物,并将滤液浓缩为油状物。将其溶于EtOAc中,并用10%硫代硫酸钠溶液和水洗涤。用MgSO4干燥,过滤并浓缩得到7.40克(86%)N-t-BOC丙氨酸碘甲基酯,为油状物。
在丙酮(75毫升)中混合N-t-BOC丙氨酸碘甲基酯(7.30克,22.2毫摩尔)和3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的钠盐(2.67克,7.4毫摩尔),并回流6小时。浓缩反应混合物,并将残余物预吸附在硅胶上。合并并浓缩适合的级分得到粗产物,将其重结晶(EtOAc/己烷)得到1.16克(29%)标题化合物:mp 165-170℃。元素分析计算值:C23H23ClFN3O7S:C,51.16;H,4.29;N,7.78。实测值:C,51.26;H,4.10;N,7.70。实施例30-40的制备用实施例29的方法。这些化合物的通式为 物理性质和“R”的定义列于下表中。
物理性质和“R”的定义列于下表中。

实施例41
通法
用两种方法将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的N-t-BOC保护的氨基酸衍生物转化为相应的去保护的化合物:
方法A:将N-t-BOC保护的氨基酸衍生物溶于冷却(冰-水浴)的TFA(0.1M)中,并将其在此温度下搅拌1小时。向该溶液中加入1当量p-TsOH·H2O,然后在真空下除去TFA。在用甲苯提取(2次)以进一步除去TFA后,残余固体用CH3OH/EtOAc重结晶以得到所期望的去保护的化合物。
方法B:将N-t-BOC保护的氨基酸衍生物溶于2/1二噁烷/EtOAc(0.1M)中,在保持温度低于10℃的同时,用HCl气体将此溶液饱和。3小时后,收集沉淀出的产物,用EtOAc洗涤,用EtOAc/EtOH重结晶得到所期望的去保护的化合物。
实施例42-45-5的制备用实施例41的方法A或B。

实施例46
6-氯-5-氟-2,3-二氨-3-基)[(1-(4-苯基甲氧羰基苯甲酰氧基)-1-乙氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
在真空中将氯化锌(524毫克)熔融,在冷却后,用4-(苄氧羰基)苯甲酰氯(21.48克,78.2毫摩尔)处理。30分钟后,在0℃下加入乙醛(3.44克,78毫摩尔)。在室温下将反应混合物搅拌30分钟,然后用CH2Cl2稀释。将混合物再搅拌30分钟,然后用水洗涤(3次)。经MgSO4干燥后,过滤,在真空中浓缩,得到绿/棕色固体,将其进行色谱法分离(用25∶75-CH2Cl2∶己烷洗脱),得到7.65克(31%)4-(苄氧羰基)苯甲酸(1-氯乙基)酯,为白色固体:mp 79-82℃。元素分析计算值C17H15ClO4:C,64.06;H,4.74;实测值C,63.88;H,4.44。在丙酮(40毫升)中将该氯乙基酯(4.00克,12.5毫摩尔)与3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的钠盐(4.51克,12.5毫摩尔)和碘化钠(644毫克,4.3毫摩尔)混合,并回流12小时。过滤反应混合物,将滤液在真空中浓缩。将该橙/棕色胶状物进行色谱法分离(用25∶75-EtOAc∶己烷洗脱,然后用1∶99-EtOAc∶CH2Cl2洗脱),得到1.947克(25%)橙色泡沫状物:元素分析计算值:C31H22ClFN2O7S·2/3 H2O:C,58.81;H,3.72;N,4.43.实测值:C,58.74;H,3.38;N,4.38。
实施例47标题化合物的制备用相同的反应顺序进行:
实施例49
3-[[[(5-苄氧基)戊二酰基]亚甲基]氧基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺
将114克(0.24摩尔)戊二酸苄基酯四丁基铵(A.R.English,D.Girard,V.J.Jasys,R.J.Martingano和M.S.Kellogg,J.Med.Chem.,1990,33,344)和600毫升溴氯甲烷的混合物在冰浴中在0℃下搅拌,并在16小时内将其缓慢温热至室温。在真空中除去过量的溴氯甲烷,将残余物溶于EtOAc中,依次用1N盐酸水溶液(2s 1升)和饱和碳酸氢钠水溶液(1×2升)洗涤,用硫酸钠干燥,蒸发得到35克油状物。在室温下将该油状物、55.2克(0.368摩尔)碘化钠和150毫升丙酮的混合物搅拌过夜。将残余物分配于EtOAc(500毫升)和水(500毫升)中。分离出有机层,用饱和硫代硫酸钠水溶液(2×500毫升)洗涤,用硫酸钠干燥,蒸发得到34.7克黄色油状物,将其用快速色谱法纯化,用EtOAc-己烷(4∶6)洗脱,得到10.7克(32%)[[5-(苄氧基)戊二酰基]氧基]甲基碘化物,为油状物。(Rf0.55,EtOAc-己烷(1∶1)).1H NMR(CDCl3)δ1.92-2.20(2H,m),2.41(2H,t,J=7),2.45(2H,t,J=7),5.16(s,2H),5.88(2H,s),7.30-7.40(5H,m).
将20.0克(0.059摩尔)3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺、590毫升(0.059摩尔)1N氢氧化钠水溶液和300毫升甲醇的混合物在真空中浓缩。残余物用乙醚充分洗涤,得到20.5克(95%)橙-黄色钠盐。
在室温下将10.1克(0.028摩尔)上述盐、10.0克(0.028摩尔)[[5-(苄氧基)戊二酰基]氧基]甲基碘化物和300毫升丙酮的悬浮液搅拌16小时。混合物变为均相,并将其在真空中浓缩,将残余物分配于350毫升EtOAc和350毫升饱和硫代硫酸钠水溶液中。分离出有机层,再用350毫升饱和硫代硫酸钠水溶液洗涤,用硫酸钠干燥,在真空下浓缩。通过快速色谱法纯化15.6克残余的黄色固体,用EtOAc-己烷(3∶7)洗脱,得到1.8克(11%)标题化合物,为黄色固体。(Rf0.3,EtOAc-己烷(1∶1))。1H NMR(CDCl3)δ1.90-2.06(2H,m),2.32-2.56(4H,m),5.12(2H,s),5.54(1H,bd s).5.72(2H,s),7.24-7.27(1H,m),7.28-7.41(5H,m),7.50-7.53(1H,m),7.75-7.78(2H,m),8.37-8.41(2H,m).
实施例50
6-氯-5-氟-3-[[(戊二酰基)亚甲基]氧基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺
在900毫克Pd(OH)2存在下,在45磅/英寸2条件下将1.75克3-[[[(5-苄氧基)戊二酰基]亚甲基]氧基-(2-噻吩基)亚甲基]6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的200毫升EtOAc溶液氢化2.5小时。再加入900毫升催化剂,然后数小时后再加入900毫克催化剂。然后继续氢化过夜。过滤出催化剂,并将滤液在真空中浓缩为2.2克橙色半固体。用快速色谱法(用甲醇-氯仿(2-98至10-90)作洗脱剂)纯化,收集含有Rf0.3(甲醇-氯仿1∶9)的物质的级分并进行浓缩。将残余物用乙醚研制得到66毫克(4%)标题化合物,为黄色固体,mp 184-186℃。1H NMR(DMSO-d6)δ1.62-1.80(2H,m),2.20(2H.t,J=7).2.40(2H,t,J=7),5.75(2H,s),7.30(1H,t,J=3),7.65(1H,d,J=2),7.76-7.85(2H,m),7.95(1H,s),8.05(1H,d,J=3),8.25(1H,d,J=7),8.32(1H,s).MS(m/e)337和339,295和297,211和213(碱)。
实施例51
6-氯-5-氟-2,3-二氢-3-[(2-呋喃甲酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
向搅拌着的(Z)-6-氯-5-氟-2,3-二氢-3-(羟基-2-噻吩基亚甲基)-2-氧代-1H-吲哚-1-甲酰胺的钠盐(1.4克,3.88毫摩尔)和碘化钠(20毫克,0.12毫摩尔)在30毫升二氯甲烷中的悬浮液中加入2-呋喃甲酰氯(380毫克,3.88毫摩尔)。然后将所得悬浮液在室温下搅拌20小时。过滤反应悬浮液并用二氯甲烷洗涤,以除去未反应的6-氯-5-氟-2,3-二氢-3-(羟基-2-噻吩基亚甲基)-2-氧代-1H-吲哚-1-甲酰胺的钠盐。真空蒸发滤液得到橙色固体,将其在硅胶柱(100克硅胶)上进行色谱法纯化,用二氯甲烷洗脱,得到粗的标题化合物,为固体。该固体经二氯甲烷和己烷结晶得到黄色固体,210毫克(12.6%):mp 215-216℃.元素分析计算值:C19H10ClFN2O5S:C,52.73;H,2.33;N,6.47.实测值:C,52.66;H,2.21;N,6.46。
实施例52
6-氯-5-氟-2,3-二氢-3-[(乙酰氧基乙酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
向搅拌的(Z)-6-氯-5-氟-2,3-二氢-3-(羟基-2-噻吩基亚甲基)-2-氧代-1H-吲哚-1-甲酰胺的钠盐(2.0克,5.6毫摩尔)和碘化钠(200毫克,1.33毫摩尔)在25毫升丙酮中的悬浮液中加入乙酰氧基乙酰氯(770毫克,5.6毫摩尔),并将该混和物加热至回流,并维持18小时。将该悬浮液冷却至室温并过滤以除去未反应的6-氯-5-氟-2,3-二氢-3-(羟基亚甲基)-2-氧代-1H-吲哚-1-甲酰胺的钠盐。将滤液在真空中蒸发至黄色固体,将该固体在硅胶柱(硅胶,75克)上进行色谱法纯化,用己烷和乙酸乙酯(9∶1)洗脱,得到粗的标题化合物,为固体,该化合物用乙腈结晶得到黄色固体,500毫克(20%)。mp 188-189℃.元素分析计算值:C18H12ClFN2O6S:C,49.27;H,2.76;N,6.38.实测值:C,49.14;H,2.54;N,6.17。
实施例53
6-氯-5-氟-2,3-二氢-3-[(甲氧基乙酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
用实施例52中所述的方法用甲氧基乙酰氯制备该标题化合物,所不同的是粗产物用色谱法(florasil)纯化,用乙酸乙酯洗脱。用乙腈结晶得到黄色固体,550毫克(24.2%):mp 199-200.5℃.元素分析计算值:C17H12ClFN2O5S:C,49.70;H,2.94;N,6.82.实测值:C,49.51,H,2.66;N,6.92。
实施例54
6-氯-5-氟-2,3-二氢-3-[反-(2-三甲基甲硅烷基乙氧羰基)-2-(环丁酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
向搅拌着的6-氯-5-氟-2,3-二氢-3-(羟基-2-噻吩基亚甲基)-2-氧代-1H-吲哚-1-甲酰胺(14.1克,0.0416毫摩尔)的120毫升二氯甲烷和二异丙基乙基胺(6.89克,0.053毫摩尔)的溶液中加入反-1-(2-三甲基甲硅烷基乙氧羰基)-2-环丁酰氯(14.0克,0.053摩尔)。在室温下搅拌18小时后,过滤所形成的固体,将滤液在真空中蒸发至黄色固体。该固体在硅胶柱上进行色谱法纯化,用己烷和乙酸乙酯(3∶1)洗脱得到粗固体,用丙酮结晶得到黄色固体,1.0克(4.3%):mp 183-184℃.元素分析计算值:C25H26ClFN2O6SSi:C,53.14;H,4.64;N,4.96.实测值:C,53.08;H,4.45;N,4.86。
实施例55
6-氯-5-氟-2,3-二氢-3-[反-1-(2-三甲基甲硅烷基乙氧羰基)-2-(环丙酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺Z型异构体
通过实施例54的方法,用适合的起始物质制备该标题化合物,所不同的是将其在硅胶垫板上分离,并用己烷和乙酸乙酯(4∶1)洗脱得到粗产物。用氯仿和己烷结晶得到黄色固体,140毫克(3.7%);mp 181-182℃.元素分析计算值:C24H24ClFN2O6SSi:C,52.31;H,4.39;N,5.08.实测值:C,52.32;H,4.36;N,5.12。
实施例56
6-氯-5-氟-2,3-二氢-3-[反-1-(2-三甲基甲硅烷基乙氧羰基)-2-(环丙酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺E型异构体
通过实施例54的方法用适合的起始物质制备该标题化合物。用氯仿和己烷结晶得到黄色固体,160毫克(4.2%);mp 175-176℃.元素分析计算值:C24H24ClFN2O6SSi:C,52.31;H,4.39;N,5.08.实测值:C,52.45;H,4.28;N,4.92。
实施例57
6-氯-5-氟-2,3-二氢-3-[(环戊酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
通过实施例54的方法用适合的起始物质制备该标题化合物,所不同的是硅胶色谱是用己烷和乙酸乙酯(7∶3)洗脱的,得到粗固体。用丙酮和己烷结晶得到黄色固体,260毫克(3.3%):mp 194℃(分解)。元素分析计算值:C20H16ClFN2O4S:C,55.24;H,3.71;N,6.44.实测值:C,55.31;H,3.47;N,6.37。
实施例58
6-氯-5-氟-2,3-二氢-3-[(2,2,3,3-四甲基环丙酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
通过实施例54的方法用适合的起始物质制备该标题化合物,所不同的是用硅胶垫板纯化,用己烷和乙酸乙酯(7∶3)洗脱得到粗固体。用丙酮结晶得到黄色固体,230毫克(2.5%):mp 202℃.元素分析计算值:C22H20ClFN2O4S:C,57.08;H,4.35;N,6.05.实测值:C,57.02;H,4.36;N,5.87。
实施例59
{1-[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]乙氧羰基氧基}乙酸甲酯
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(2.98克)和(1-碘代乙氧基)羰基氧基乙酸甲酯(2.18克)在丙酮中混合,并将该溶液在20℃下搅拌20小时。真空蒸发溶剂,并用乙醚萃取残余物。真空蒸发乙醚溶液,经放置得到部分结晶的残余油状物。滤出剩余油状物后,使残余固体在少量乙醚中成浆。并过滤得到495毫克黄色固体。通过将合并的乙醚可溶的残余物在硅胶上进行色谱法纯化(用带有2%丙酮的氯仿洗脱)得到另外295毫克可比质量的标题化合物。该产物通过NMR鉴定。实施例59-70的NMR数据列于实施例70后面的表中。
实施例60
{1-[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]乙氧羰基氧基}-N,N-二甲基乙酰胺
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(6.25克)和N,N-二甲基-(1-碘乙氧基)羰基氧基乙酰胺(5.55克)在60毫升丙酮中混合,并将该溶液在20℃下搅拌90小时。所存在的沉淀通过过滤分离并在真空中干燥。该固体在硅胶上进行色谱法纯化,用90∶10氯仿∶丙酮洗脱。将含有标题化合物的级分合并并蒸发。残余物用少量乙醚成浆并过滤得到820毫克黄色固体,mp 194.5-195.5℃。
实施例61
(1-[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]乙氧羰基氧基}-N,N-二乙基乙酰胺
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(13.85克)和N,N-二乙基-(1-碘代乙氧基)羰基氧基乙酰胺(13.6克)在100毫升丙酮中混合,并将该溶液在20℃下搅拌42小时。将不溶物质通过过滤分离,并将其用氯仿搅拌。滤掉剩余固体,并将该氯仿溶液真空蒸发。残余物在硅胶上进行色谱法纯化,用90∶10氯仿∶丙酮洗脱。将含有所需物质的级分合并并蒸发。将残余物用少量乙醚成浆,并过滤得到2.37克黄色固体,mp 172-174℃。
实施例62
N,N-二甲基琥珀酰胺酸{1-[1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]甲基}酯
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(6.65克)和N,N-二甲基琥珀酰胺酸碘甲基酯(3.27克)在100毫升丙酮中混合,并将该溶液在20℃下搅拌65小时。在真空中蒸发掉溶剂后,将残余物用氯仿萃取3次。合并氯仿萃取液,并在真空中蒸发。残余物在硅胶上进行色谱法纯化,用75∶25氯仿∶丙酮洗脱。将富含标题产物的级分合并并蒸发得到189毫克橙色油状物。将含有标题化合物的其他级分合并、蒸发并再于硅胶上进行色谱法纯化,用90∶10氯仿∶丙酮洗脱。将富含标题化合物的级分与189毫克上面提到的油状物合并,蒸发得到490毫克橙色固体。将该固体在少量乙醚中成浆,过滤得到180毫克橙色固体,mp 183-187℃。
实施例63
N,N-二乙基琥珀酰胺酸{[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]甲基}酯
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(4.36克)和N,N-二乙基琥珀酰胺酸碘甲基酯(2.36克)在30毫升丙酮中混合,并将该溶液在20℃下搅拌87小时。在真空中蒸发掉溶剂后,将残余物在硅胶上进行色谱法纯化,用90∶10氯仿∶丙酮洗脱。将含有标题产物的级分合并并浓缩为含有一些固体的油状物。向该油状固体中加入乙醚,滤出不溶物质。(该固体既含有TBA碘化物又含有C-烷基化产物)。蒸发乙醚滤液,将残余物在硅胶上进行色谱法纯化,用90∶10氯仿∶丙酮洗脱。合并富含标题产物的级分,真空蒸发至橙色油状物。将该油状物溶于乙醚中并令其放置过夜。过滤分离黄色固体沉淀,并在真空中干燥得到265毫克产物,mp 151-152.5℃。
实施例64
N,N-二丙基琥珀酰胺酸{[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]甲基}酯
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(6.07克)和N,N-二丙基琥珀酰胺酸碘甲基酯(2.6克)在50毫升丙酮中混合,并将该溶液在20℃下搅拌18小时。真空蒸发溶剂,并将残余物用乙醚搅拌。形成沉淀,并通过过滤除去。将乙醚溶液在真空中蒸发得到6.5克橙色油状物。将该油状物在硅胶上进行色谱法纯化,且95∶5氯仿∶丙酮洗脱。将富含标题产物的级分合并并真空浓缩。向残余物中加入乙醚,将溶液放置。滤出所形成的黄色沉淀得到210毫克产物。将含有标题产物的其它级分合并并蒸发,将残余物在硅胶上进行色谱法纯化,用50∶50乙酸乙酯∶己烷洗脱。将富含标题化合物的级分合并并浓缩至油状物,将其溶于乙醚中并放置18小时。滤出所形成的黄色沉淀得到另外220毫克产物,mp 131.5-133.5℃。
实施例65
N,N-六亚甲基琥珀酰胺酸{[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]甲基}酯
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(4.4克)和4-高哌啶子基-4-氧代丁酸碘甲基酯在30毫升丙酮中混合,并将该溶液在20℃下搅拌17小时。过滤该混合物,真空浓缩滤液。残余物在硅胶上进行色谱法纯化,用93∶7氯仿∶丙酮洗脱。将富含标题产物的级分合并并浓缩至橙色油状物。将该油状物在硅胶上进行色谱法纯化,用同样的洗脱剂洗脱。将富含标题产物的级分合并并蒸发。向残余物中加入乙醚,使该溶液放置。通过过滤分离所形成的黄色沉淀,得到250毫克物质,mp 149-151℃。
实施例66
N,N-二甲基氨基甲酰氧基乙酸{[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]甲基}酯
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(3.28克)和N,N-二甲基氨基羰基氧基乙酸碘甲基酯(1.18克)在15毫升丙酮中混合并将该溶液在20℃下搅拌17小时。过滤该混合物,真空浓缩滤液。残余物在硅胶上进行色谱法纯化,用95∶5氯仿∶丙酮洗脱。将富含标题产物的级分合并并浓缩至黄色油状物。向该油状物中加入乙醚,将该溶液放置。过滤分离所形成的黄色沉淀,干燥得到240毫克产物,mp185.5-187℃。
实施例67
N,N-二乙基氨基甲酰氧基乙酸{[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]甲基}酯
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(11.66克)和N,N-二乙基氨基羰基氧基乙酸碘甲基酯(6.36克)在100毫升丙酮中混合,并将该混合物在20℃下搅拌17小时。过滤后,真空浓缩滤液至半固体。加入乙醚,滤出不溶于乙醚的物质。将该乙醚溶液放置数小时。滤出所形成的白色沉淀(C-烷基化产物)。经进一步放置,该乙醚滤液沉积出黄色固体沉淀。过滤并干燥该黄色固体得到990毫克晶体,mp156.8-157.8℃。
实施例68
N-新戊酰基甘氨酸{[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]甲基}酯
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(2.80克)和N-新戊酰基甘氨酸碘甲基酯(1.45克)在20毫升丙酮中混合,并将该混合在20℃下搅拌17小时。真空蒸发溶剂后,将该黄橙色残余物用乙醚成浆并过滤。蒸发乙醚滤液,残余物用少量乙醚搅拌。过滤分离不溶性沉淀,并将其溶于乙酸乙酯中,以进行硅胶色谱法纯化,用乙酸乙酯洗脱。将含有标题产物的级分合并并蒸发至黄色固体。该固体用少量乙醚成浆,过滤得到140毫克产物,mp 200-204℃。
实施例69
N-(2-乙基丁酰基)甘氨酸{[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]甲基}酯
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(5.13克)和N-(2-乙基丁酰基)甘氨酸碘甲基酯(2.78克)在35毫升丙酮中混合,并将该混合物在20℃下搅拌17小时。真空蒸发溶剂后,将该橙色残余物用乙醚成浆,并过滤。蒸发乙醚滤液,将残余物用少量乙醚搅拌。过滤分离黄色沉淀(200毫克),将其溶于乙酸乙酯中以进行硅胶色谱法纯化,用乙酸乙酯洗脱。将含有标题产物的级分合并,蒸发得到40毫克黄色固体。通过在硅胶上将最初不溶于丙酮的级分进行色谱法纯化,用乙酸乙酯洗脱得到另外256毫克同样的标题产物。
实施例70
异丙基碳酸{1-[(1-氨基甲酰基-6-氯-5-氟-2-氧代-1,2-二氢吲哚-3-亚基)噻吩-2-基甲氧基]乙基}酯
将3-[羟基-(2-噻吩基)亚甲基]-6-氯-5-氟-2,3-二氢-2-氧代-1H-吲哚-1-甲酰胺的四丁基铵盐(13.62克)和异丙基碳酸(1-碘乙基)酯(Wan-Joo Kim等人,The Journal of Antibiotics,(1990),44,1086)(6.08克)在90毫升丙酮中混合,并将该混合物在20℃搅拌17小时。滤出不溶物,将其在氯仿中成浆,并过滤。将氯仿滤液与溶于丙酮的反应物级分合并,真空蒸发溶剂。残余物在硅胶上用色谱法纯化,用98∶2氯仿∶丙酮洗脱。将含有标题产物的级分合并,蒸发得到橙色油状物。在搅拌下加入乙醚,结晶出黄色固体。过滤并干燥得到1.10克产物,mp 183-184℃。
| 61 | Me | OCH2CONEt2 | 1.11(t,3H);2.28(t,3H);1.78(d,3H);3.16(q,2H);3.35(m,2H);4.66(ABq,2H);5.18(宽.s,1H);6.26(q,1H);7.23(dd,1H);7.58(dd,1H);7.70(dd,1H);7.82(d,1H);8.38(d,1H);8.42(宽.s1H). | |
| 62 | H | CH2CH2CONMe2 | 2.63(m,4H);2.90(s,3H);3.00(s,3H);5.16 (宽.s,1H);5.74(s,2H);7.26(dd,1H);7.54(dd,1H);7.73(dd,1H);7.78(d,1H);8.40(d,1H);8.45(宽.s,1H). | |
| 63 | H | CH2CH2CONEt2 | 1.04(t,3H);1.16(t,3H);2.62(m,4H);3.28(m,4H);5.22(宽.s,1H);5.70(s,2H);7.22(dd,1H);7.50(dd,1H);7.69(dd,1H);7.74(d,1H);8.34(d,1H);8.37(宽.s,1H) | |
| 64 | H | CH2CH2CONPr2 | 0.80(t,3H):0.88(t,3H);1.44(m,2H);1.56(m,2H);2.60(m,4H);3.16(q,4H);5.15(宽.s,1H);5.69(s,2H);7.20(dd,1H);7.46(dd,1H);7.68(dd,1H);7.74(d,1H);8.34(d,1H);8.38(宽.s,1H). | |
| 65 | H | (CH2)2CON(cH2)6 | 1.44-1.74(m,8H);2.60(m,4H);3.32-3.46(m,4H);5.22 (宽.s,1H);5.70(s,2H);7.20(dd,1H);7.47(dd,1H);7.68(dd,1H);7.73(d,1H);8.34(d,1H);8.36 (宽.s,1H). | |
| 66 | H | CH2OCONMe2 | 2.86(s,3H);2.90(s,3H);4.56(s,2H);5.14(宽.s,1H);5.74(s,2H);7.20(dd,1H);7.45(dd,1h);7.68(dd,1h);7.72(d,1H);8.33(宽.s,1H);8.34(d,1H). | |
| 67 | H | CH2OCONEt2 | 1.08(t,3H);1.11(t,3H);3.26(q,4H);4.59(s,2H);5.24(宽.s,1H);5.74(s,2H);7.20(dd,1H);7.44(dd,1H);7.68(dd,1H);7.72(d,1H);8.32(宽.s,1H);8.34(d,1H). | |
| 68 | H | CH2NHCOCMe3 | 1.20(s,9H);4.04(d,2H);5.15(宽.s,1H);5.75(s,2H);6.08(宽.s,1H);7.27(dd,1H);7.50(dd,1H);7.75(m,2H);8.40(宽.s,1H);8.42(d,1H). | |
| 69 | H | CH2NHCOCHEt2 | 0.88(t,6H);1.43-1.68(m,4H);1.05(m,1H);4.08(d,2H);5.18(宽.s,1H);5.77(s,2H);5.88(宽.s,1H);7.27(dd,1H);7.50(dd,1H);7.75(m,2H);8.39(宽.s,1H);8.41(d,1H). | |
| 70 | Me | OCHMe2 | 1.13(d,3H);1.35(d,3H);1.74(d,3H);4.74(m,1H);5.20(宽.s,1H);6.18(q,1H);7.24(dd,1H);7.56(dd,1H);7.72(dd,1H);7.80(d,1h);8.40(d,1H);8.45(宽.s,1H). |
实施例71
6-氯-5-氟-2,3-二氢-3-[1-(1-甲氧基乙酰氧基)乙氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)向甲氧基乙酰氯(10.0克,0.092摩尔)和催化量的熔融的氯化锌的混合物中加入乙醛(4.06克,0.092摩尔),将溶液冷却至-20℃。除掉冷却,在室温上搅拌1小时,在30托下蒸馏并收集50-80℃的馏分,得到油状物3.3克(22.9%)。
向搅拌着的钠-6-氯-5-氟-2,3-二氢-3-(羟基-2-噻吩基亚甲基)-2-氧代-1H-吲哚-1-甲酰胺(3.5克,9.7毫摩尔)和碘化钠(500毫克,3.3毫摩尔)在30毫升丙酮中的悬浮液中加入1-氯乙基甲氧基乙酰氯(1.8克,9.7毫摩尔)。将所得悬浮液回流7小时,冷却、真空蒸发得到橙色固体。该固体在硅胶柱(125毫升硅胶)上进行色谱法纯化,用己烷和乙酸乙酯(9∶1)洗脱得到粗产物。用乙腈结晶得到黄色固体,1.1克(25%):M.P.175-176℃。元素分析计算值C19H16ClFN2O6S:C,50.17;H,3.55;N,6.16。实测值:C,50.31;H,3.32;N,6.32
实施例72
6-氯-5-氟-2,3-二氢-3-[1-(吗啉氨基甲酰基)乙氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)向吗啉(8.7克,0.1摩尔)和三乙胺(10.1克,0.1摩尔)的二氯甲烷(120毫升)溶液中加入氯甲酸(1-氯乙基)酯(14.3克,0.1摩尔),加入的速度保持在使溶液不回流。搅拌10分钟后,将溶液倾入水(200毫升)中。分离出有机层,用硫酸镁干燥,蒸发得到浅棕色油状物17.8克(92%)。
向搅拌着的钠-6-氯-5-氟-2,3-二氢-3-(羟基-2-噻吩基亚甲基)-2-氧代-1H-吲哚-1-甲酰胺(10.0克,0.028摩尔)和碘化钠(300毫克,0.0017摩尔)在丙酮(45毫升)中的悬浮液中加入步骤a)中的吗啉氨基甲酰基酯(5.36克,0.028摩尔),并将混合物回流5.5小时。将悬浮液冷却,蒸发至固体,在硅胶柱(4.5厘米×42厘米)上进行色谱法纯化,用己烷和乙酸乙酯(7∶3)洗脱得到粗产物。用乙腈结晶得到黄色固体,2.37克(17.3%):M.P.124-126℃。元素分析计算值C21H19ClFN3O6S:C,50.86;H,3.86;N,8.47。实测值:C,50.91;H,3.78;N,8.54
实施例73
6-氯-5-氟-2,3-二氢-3-{1-(氨基-环戊酰氧基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺甲磺酸盐
a)1-N-t-BOC-氨基-1-环戊烷羧酸
向1-氨基-1-环戊烷羧酸(10.0克,0.0774摩尔)、三乙胺(11.75克,0.1161摩尔)、水(45毫升)和1,4-二噁烷(45毫升)的溶液中加入BOC-ON(20.9克,0.085摩尔),并在室温下搅拌7小时。将该混合物用乙酸乙酯萃取,乙酸乙酯层用水(50毫升)洗涤。水层用柠檬酸酸化至pH为3.5,并用乙酸乙酯(2×100毫升)萃取。乙酸乙酯层用硫酸镁干燥,蒸发得到油状产物14克(79%)。
b)1-N-t-BOC-氨基-1-氯甲基环戊酸酯
向在水(125毫升)中的硫酸氢四丁基铵(22.5克,0.066摩尔)中缓慢加入碳酸氢钠(11.1克,0.132摩尔)并搅拌15分钟。加入在氯仿(250毫升)中的a)中的化合物,并搅拌3小时。分离有机层并用硫酸镁干燥,蒸发为油状物。向该油状物中加入溴氯甲烷(225毫升),并在室温下将该溶液搅拌过夜。将混合物蒸发至粘稠油状物,将其用乙醚研制并过滤。将滤液蒸发得到油状粗产物,9.2克(50%)。
c)向搅拌着的6-氯-5-氟-2,3-二氢-3-(羟基-2-噻吩基亚甲基)-2-氧代-1H-吲哚-1-甲酰胺的钠盐(2.85克,7.92毫摩尔)和碘化钠(285毫克,1.9毫摩尔)在70毫升丙酮中的悬浮液中加入1-N-t-BOC-氨基-1-氯亚甲基环戊酸酯(2.2克,7.92毫摩尔)。将所得悬浮液回流7小时。将反应悬浮液冷却至室温并过滤除掉未反应的6-氯-5-氟-2,3-二氢-3-(羟基-2-噻吩基亚甲基)-2-氧代-1H-吲哚-1-甲酰胺的钠盐。滤液在真空中蒸发为橙色固体,将其在硅胶垫板(75克)上进行色谱法纯化,用己烷和乙酸乙酯(7∶3)洗脱得到固体,将其用乙腈结晶得到黄色固体,320毫克(7.1%):M.P.204℃(分解)。元素分析计算值C26H27ClFN3O7S:C,53.84;H,4.69;N,7.24。实测值:C,53.83;H,4.42;N.7.29
d)在室温下将步骤c)的1-N-t-BOC-氨基-1-环戊酰基衍生物(410毫克,0.7毫摩尔)在10毫升三氟乙酸中的溶液搅拌20分钟。在真空下蒸发该混合物得到油状固体,加入25毫升二氯甲烷和甲磺酸(67.9毫克,0.7毫摩尔),真空蒸发为黄色固体。将该黄色固体在25毫二氯甲烷中搅拌15分钟,过滤得到黄色固体,400毫克(99%):M.P.230℃(分解)。元素分析计算值C22H23ClFN3O3S2:C,45.87;H,4.02;N,7.30。实测值:C,45.63;H,3.95;N,7.19
实施例74
6-氯-5-氟-2,3-二氢-3-[乙酰氧基甲氧羰基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)向乙酰氧基乙酰氯(20克,0.146摩尔)和催化量的熔融氯化锌的混合物中加入多聚甲醛(6.2克,0.206摩尔);在蒸汽浴上将混合物加热3.5小时。冷却后,蒸馏反应物并收集72-82℃/1.2-1.9托的馏分,得到3.2克油状物(13%)。
b)用与实施例73所述相同的方式,使用步骤a)的中间体制备标题产物,所不同的是洗脱剂己烷和乙酸乙酯的比例为3∶1。用乙腈结晶得到黄色固体,900毫克(8.9%):M.P.176-177℃。元素分析计算值C19H14ClFN2O7S:C,48.68;H,3.01;N,5.98。实测值:C,48.60;H,3.02;N.6.00
实施例75
6-氯-5-氟-2,3-二氢-3-[苄基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)用与实施例74步骤a)相同的方式制备苯甲酸氯甲基酯。在74-76℃和2.0托下蒸馏反应产物得到粗产物,为油状物,16.42克(50%)。
b)使用步骤a)的中间体,用与实施例73所述相同的方式制备标题产物。用乙酸乙酯结晶得到黄色固体,130毫克(5.5%):M.P.202-203℃。元素分析计算值C22H14ClFN2O5S:C,55.88;H,2.98;N,5.92。实测值:C,55.69;H,2.69;N,5.86
实施例76
6-氯-5-氟-2,3-二氢-3-[1-乙酸-1-环戊基乙酰氧基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
1-(4-甲氧基苄氧羰基甲基)-1-环戊烷乙酸
a)将3,3-四亚甲基戊二酸酐(10.0克,5.95毫摩尔)、4-甲氧基苄基醇(8.2克,5.95毫摩尔)和吡啶(4.7克,5.95毫摩尔)的混合物进行混合,并在蒸汽浴上加热1.5小时。冷却,用6N HCl逐滴酸化,用乙醚和乙酸乙酯1/1(3×150毫升)萃取,用饱和碳酸氢钠溶液(3×150毫升)洗涤合并的有机层。用6N HCl酸化碱性水层,用乙酸乙酯和乙醚(200毫升)萃取,用硫酸镁干燥,真空蒸发得到粗油状物,11.6克(64%)。
b)用实施例73步骤b)中所述方法制备1-(4-甲氧基苄氧羰基甲基)氯甲氧羰基甲基环戊烷,蒸发得到粗产物,为油状物6.5克(48%),将其用于步骤c)中。
c)用与实施例73(c)中所述相同的方法制备标题化合物的4-甲氧基苄氧基衍生物。产量:300毫克(1.6%)为红色粘稠油状物。元素分析计算值C32H30ClFN2O8S:C,58.49;H,4.60;N,4.26实测值:C,58.89;H,4.32;N,4.33
d)将在15毫升三氟乙酸中的步骤c)的4-甲氧基苄基衍生物(400毫克,0.61毫摩尔)搅拌45分钟,真空蒸发得到黄色固体。加入60毫升二氯甲烷并蒸发。所得黄色固体用甲醇结晶得到产物600毫克(15%):M.P.187-188℃。元素分析计算值C24H22ClFN2O7S:C,53.68;H,4.13;N,5.22实测值:C,53.94;H,4.04;N,5.02
实施例77
6-氯-5-氟-2,3-二氢-3-[L-脯氨酰基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺甲磺酸盐
a)N-t-BOC-L-脯氨酸的氯甲基酯的制备用实施例73步骤b)中所述的方法。分离出来的粗产物为油状物,35.4克(83%)。
b)N-t-BOC-L-脯氨酰基衍生物的制备用与实施例73步骤c)中所述相同的方法。用乙醚和己烷结晶得到黄色固体,2.65克(9.36%):M.P.:168-169℃。元素分析计算值C25H26ClFN3O7S:C,52.96;H,4.62;N,7.41实测值:C,52.85;H,4.46;N,7.43
c)标题化合物的制备用与实施例73步骤d)中所述相同的方法。用2-丙醇结晶得到黄色固体,333毫克(34%):M.P.90-120℃,为无定形固体。元素分析计算值C20H17ClFN3O5SCH3SO3H1/4H2O:C,44.52;H,3.83;N,7.42实测值:C,44.35;H,3.92;N,7.23
实施例78
6-氯-5-氟-2,3-二氢-3-[1-羧酸-3-哌啶基羰基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺甲磺酸盐
a)N-t-BOC-哌啶-3-羧酸的制备用实施例73步骤a)中所述的方法。得到粗固体19克。用乙酸乙酯结晶得到白色固体,11.1克(48%):M.P.162-163℃。
b)1-N-t-BOC-哌啶-3-氯甲基酯的制备用与实施例73步骤b)中所述相同的方法。蒸发得到白色潮湿的固体13.5克(78%),并将其直接用于下一步骤。
c)用与实施例73所述相同的方法使用3-氯甲基酯-1-N-t-BOC-哌啶制备标题化合物的BOC衍生物。用乙醚和己烷结晶得到黄色固体,580毫克(2.5%):M.P.145-146℃。元素分析计算值C26H27ClFN3O7S:C,53.84;H,4.69;N,7.24实测值:C,53.82;H,4.57;N,7.16
d)标题化合物的制备用与实施例73步骤d)中所述相同的方法。用丙酮结晶得到黄色固体,280毫克(68.7%):M.P.184-185℃。元素分析计算值C22H23ClFN3O8S2:C,45.87;H,4.02;N,7.30实测值:C,45.75;H,3.85;N,7.17.CP-156,011.
实施例79
6-氯-5-氟-2,3-二氢-3-[1-羧基-3-乙基-3-甲基戊酰基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
标题化合物的制备用与实施例76相同的方法。用丙酮和己烷结晶得到黄色固体,380毫克(27%):M.P.124-125℃。元素分析计算值C23H22ClFN2O7S:C,52.62;H,4.22;N,5.34实测值:C,52.83;H,3.99;N,5.20
实施例80
6-氯-5-氟-2,3-二氢-3-[反-1-羧基-2-环己基羰基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
标题化合物的制备用与实施例76相同的方法。用丙酮结晶得到黄色固体,590毫克(28.3%):M.P.214-215℃。元素分析计算值C23H20ClFN2O7S:C,52.83;H,3.85;N,5.26实测值:C,53.17;H,3.72;N,5.10
实施例81
6-氯-5-氟-2,3-二氢-3-[反-1-羧基-2-环丙基羰基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺甲磺酸盐
a)标题化合物的1-N-t-BOC-环丙酰基衍生物的制备用与实施例73步骤c)中所述相同的方法。用乙醚结晶得到黄色固体,780毫克(4.4%):M.P.179-180℃。元素分析计算值C24H23ClFN3O7S:C,52.22;H,4.20;N,7.61实测值:C,52.30:H,4.03:N.7.67
b)标题化合物的制备用与实施例73中所述相同的方法。用甲醇结晶得到黄色固体,235毫克(78%):M.P.175-182℃,无定形固体。元素分析计算值C19H15ClFN3O5S·CH3SO3H·1/2H2O:C,43.13;H,3.62;N,7.54实测值:C,43.17;H,3.33;N,7.46
实施例82
6-氯-5-氟-2,3-二氢-3-[反-1-羧基-2-环丙基羰基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)向氢氧化钠(12.0克,0.3摩尔)和水(200毫升)的溶液中加入1,2-环丙烷二羧酸二乙酯(18.6克,0.10摩尔),并在80℃下搅拌5小时。在冷却(0-5℃)下蒸发溶液至固体。在冷却下缓慢滴加浓盐酸(50毫升),然后在蒸汽浴上加热,过滤掉不溶物,并在冰-水浴中冷却。收集所得到的晶体,将其溶于丙酮中并蒸发得到反-1,2-环丙烷二羧酸,6.2克(47.676):M.P.176-177℃。参考文献:JACS 4994-4999(1957)M.P.176-177℃。
b)将反-1,2-环丙烷二羧酸(3.0克,0.0231摩尔)和亚硫酰氯(20毫升)的混合物回流1.5小时,然后冷却并蒸发得到油状物。将该油状物溶于苯(20毫升)中并缓慢加入2-(三甲基甲硅烷基)乙醇(2.13克,0.018摩尔),然后在室温下搅拌过夜。将反应混合物过滤并蒸发得到粗产物,为浅棕色油状物,3.96克(42%)。
c)6-氯-5-氟-2,3-二氢-2-[反-1-(2-三甲基甲硅烷基氧基羰基)-2-(环丙酰基)氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺(Z)型异构体的制备用与实施例54中所述相同的方法,所不同的是将其在硅胶柱板上分离,用己烷和乙酸乙酯(4∶1)洗脱得到粗产物。用氯仿和己烷结晶得到黄色固体,140毫克(3.7%):M.P.181-182℃。元素分析计算值Q24H24ClFN2O6SSi:C,52.31;H,4.39;N,5.08实测值:C,52.32;H,4.36;N,5.12
d)E型异构体的分离用与步骤c)中所述相同的方法。用氯仿和己烷结晶得到黄色固体,160毫克(4.2%):M.P.175-176℃.CP-152,775。元素分析计算值C24H24ClFN2O6SSi:C,52.31;H,4.39;N,5.08实测值:C,52.45;H,4.28;N,4.92
e)将标题化合物的三甲基甲硅烷基氧基环丙基衍生物(800毫克,1.45毫摩尔)在氟化氢-吡啶配合物(5毫升)中的悬浮液在-20℃下搅拌30分钟,加入20毫升水并过滤粗固体。用乙醚结晶得到黄色固体,412毫克(61.7%):M.P.195℃(分解)。元素分析计算值C19H12ClFN2O6S·1/2H2O:C,49.63;H,2.85;N,6.09实测值:C,49.81;H,2.62:N,6.06。
实施例83
6-氯-5-氟-2,3-二氢-3-[1,3-二氧杂环己烷-5-羰基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)1,3-二氧杂环己烷-5-羧酸的合成用SynthesisCommunications 23(1974)中所述的方法。
b)其氯甲基酯的制备用与实施例73步骤c)中所述相同的方法。得到浅黄色油状物,4.3克(48%)。
c)其1,3-二氧杂环己烷衍生物的制备用与实施例73的制备相同的方法。用硅胶柱(42×5.5厘米)分离,用己烷和乙酸乙酯(7∶1)洗脱,真空蒸发得到黄色固体产物,150毫克(1.3%):M.P.199-200℃。元素分析计算值C20H16ClFN2O7S:C,49.75;H,3.34;N,5.80实测值:C,49.84;H,3.17;N,5.8
实施例84
6-氯-5-氟-2,3-二氢-3-[1-(4-甲氧基苄氧羰基)-5-(3-甲基)戊酰基甲氧基-(2-噻吩基)亚甲基]-2-氧代-1H-吲哚-1-甲酰胺
a)其氯甲基酯的制备用与实施例73步骤b)的合成中所述相同的方法。分离出的产物为油状物,9.92克(26%)。将其直接用于下一步骤中。
标题化合物的制备用与实施例76相同的方法。用乙醚结晶得到橙色固体,150毫克(7.6%);M.P.113-114℃。元素分析计算值C29H26ClFN2O3S:C,56.15;H,4.25;N,4.54实测值:C,56.32;H,4.13;N,4.56
制备例1
(1-氯乙氧基)羰基氧基乙酸甲酯
在搅拌下向用冰-水浴冷却的甘醇酸甲酯(13.5毫升)和N,N-二异丙基乙基胺(30.46毫升)的四氢呋喃(180毫升)溶液中逐滴加入25.0克氯甲酸(1-氯乙基)酯。撤掉0℃浴,并将该混合物再搅拌3小时。过滤以除掉不溶性的胺盐酸盐后,减压蒸发THF溶液。将残余物溶于乙酸乙酯与水的混合物中。分离乙酸乙酯层,用无水硫酸钠干燥,减压蒸发得到31.1克微红色油状物。
制备例2
(1-碘乙氧基)羰基氧基乙酸甲酯
将碘化钠(13.19克)、丙酮(35毫升)和(1-氯乙氧基)羰基氧基乙酸甲酯(8.65克)加热回流2.5小时。冷却后,过滤混合物,真空蒸发滤液至浅棕色油状物。将N-甲基吗啉(1.2毫升)和5.8毫升该浅棕色油状物溶于50毫升二氯甲烷中,并将混合物在室温下搅拌45分钟。过滤后,蒸发二氯甲烷溶液,向残余物中加入乙酸乙酯和水。分离乙酸乙酯相,用无水硫酸钠干燥,蒸发得到2.18克粗产物,该粗产物含有大约68%(1-碘乙氧基)羰基氧基乙酸甲酯和约32%(1-氯乙氧基)羰基氧基乙酸甲酯。该粗产物的使用无需进一步的纯化。
制备例3
N,N-二甲基-2-羟乙酰胺
向二甲基胺(25.52克)的150毫升甲醇溶液中加入14.8克甘醇酸甲酯,并将该混合物在20℃下放置18小时。减压蒸发过量的二甲基胺和甲醇,向残余物中加入乙醚以导致结晶。经过滤和干燥得到白色晶体(13.15克),M.P.44-46℃。
制备例4
N,N-二甲基-(1-氯乙氧基)羰基氧基乙酰胺
在搅拌下向用冰-水浴冷却的N,N-二甲基-2-羟乙酰胺(15.64克)和N,N-二异丙基乙基胺(26.4毫升)的四氢呋喃(100毫升)溶液中逐滴加入16.37毫升氯甲酸(1-氯乙基)酯。撤掉0℃浴,并将混合物再搅拌4小时。过滤除掉不溶性的胺盐酸盐后,减压蒸发THF溶液,剩下25.5克棕色固体。该粗产物在石油醚中成浆、过滤并真空干燥得到19.98克灰白色固体,M.P.59-60.8℃。
制备例5
N,N-二甲基-(1-碘乙氧基)羰基氧基乙酰胺
将碘化钠(17.16克)、丙酮(35毫升)和N,N-二甲基-(1-氯乙氧基)羰基氧基乙酰胺(12.0克)加热回流1.5小时。冷却后,过滤混合物,蒸发滤液。向残余物中加入乙酸乙酯,滤出不溶物。蒸发乙酸乙酯后,加入氯仿,并滤出不溶物。蒸发氯仿溶液得到13.0克微红色油状物。向残余物中加入乙醚,并将可溶于乙醚的部分从在乙醚中不溶的物质中滗析出来。蒸发乙醚溶液得到4.0克黄-橙色油状物。向该黄-橙色油状物中加入二氯甲烷(30毫升)和0.42毫升N-甲基吗啉,将混合物在室温下搅拌45分钟,然后真空浓缩。将残余物在乙酸乙酯和水中分配,分离乙酸乙酯层,用硫酸钠干燥,减压蒸发得到2.55克黄色固体,NMR表明其含有约66% N,N-二甲基-(1-碘乙氧基)羰基氧基乙酰胺和约33%N,N-二甲基-(1-氯乙氧基)羰基氧基乙酰胺。该粗物质使用时无需进一步纯化。
制备例6
N,N-二乙基-2-羟乙酰胺
在搅拌下向二乙胺(18.9毫升)和三乙胺(25.5毫升)的500毫升二氯甲烷的0℃溶液中逐滴加入25克乙酰氧基乙酰氯。撤掉冰浴,并将混合物温热至20℃,同时再搅拌2小时。过滤除掉胺盐酸盐后,减压蒸发二氯甲烷溶液得到29.1克粗的N,N-二乙基乙酰氧基乙酰胺,为黄色油状物。将黄色油状物溶于125毫升甲醇中,在搅拌下加入168毫升1N氢氧化钠。观察到稍微放热,在不进一步加热条件下将混合物搅拌2小时。减压除掉溶剂至1/4体积,剩下的水溶液用乙酸乙酯(200毫升)萃取。乙酸乙酯萃取液用无水硫酸钠干燥,减压蒸发得到14.0克黄色油状N,N-二乙基-2-羟乙酰胺。
制备例7
N,N-二乙基-(1-氯乙氧基)羰基氧基乙酰胺
在搅拌下向用冰-水浴冷却的N,N-二乙基-2-羟乙酰胺(19.25克)和N,N-二异丙基乙基胺(26.2毫升)的四氢呋喃(100毫升)溶液中逐滴加入15.8毫升氯甲酸(1-氯乙基)酯。撤去冰浴,将混合物再搅拌4小时。过滤以除去不溶性的胺盐酸盐后,减压蒸发THF溶液。将固体残余物溶于乙酸乙酯中,并用水洗涤该溶液,用无水硫酸钠干燥,蒸发至固体残余物。在乙醚中成浆后,将固体过滤并干燥得到18.4克灰白色晶体,M.P.76-78℃。
制备例8
N,N-二乙基-(1-碘乙氧基)羰基氧基乙酰胺
将碘化钠(15.14克)、丙酮(35毫升)和N,N-二乙基-(1-氯乙氧基)羰基氧基乙酰胺(12.0克)加热回流1.5小时。冷却后,过滤混合物,蒸发滤液。在搅拌下将乙酸乙酯和水加到残余物中。分离乙酸乙酯层,用无水硫酸钠干燥,真空蒸发得到13.6克微红色固体,nmr表明其含有大约58%N,N-二乙基-(1-碘乙氧基)羰基氧基乙酰胺和约42%N,N-二乙基-(1-氯乙氧基)羰基氧基乙酰胺。该物质在使用时,无需进一步纯化。
制备例9
N,N-二甲基琥珀酰胺酸二甲基铵
向预冷(0℃)的琥珀酐(10克)和四氢呋喃(75毫升)的混合物中缓慢地通入二甲胺气,鼓泡1小时,撤掉冷浴,将反应混合物再搅拌30分钟,然后减后蒸发得到17.57克澄清油状物。
制备例10
N,N-二甲基琥珀酰胺酸氯甲基酯
在搅拌下向50毫升二氯甲烷中加入10毫升水、31.4克硫酸氢四丁基铵、92.5毫升1N氢氧化钠以及溶于50毫升二氯甲烷中的17.57克N,N-二甲基琥珀酰胺酸二甲基铵。搅拌2.25小时后,分离二氯甲烷层,用无水硫酸钠干燥,真空蒸发得到26.8克油状物。将该油状物溶于100毫升溴氯甲烷中并在20℃下搅拌17小时。真空蒸发溴氯甲烷后,残余油状物用乙醚萃取数次。将乙醚萃取液合并并蒸发得到5.4克油状物。粗产物在硅胶上通过色谱法纯化,用70∶30氯仿-乙酸乙酯洗脱。将含有所需产物的级分合并并浓缩得到2.3克澄清油状物。
制备例11
N,N-二甲基琥珀酰胺酸碘甲基酯
将碘化钠(3.57克)加入到N,N-二甲基琥珀酰胺酸氯甲基酯(2.3克)的10毫升丙酮溶液中,将混合物在20℃下搅拌2小时。真空蒸发溶剂后,将残余物溶于二氯甲烷和水的混合物中。分离二氯甲烷层,用无水硫酸钠干燥,蒸发得到3.27克浅棕色油状物,其使用时无需进一步纯化。
制备例12
N,N-二乙基琥珀酰胺酸氯甲基酯
将硫酸氢四丁基铵(19.6克),N,N-二乙基琥珀酰胺酸(10.0克)、碳酸氢钠(4.85克)和1N氢氧化钠(58毫升)在搅拌下在400毫升二氯甲烷和80毫升水中混合。1.5小时后,进行相分离。二氯甲烷层用无水硫酸钠干燥,减压蒸发得到9.6克油状物。水层用另外的二氯甲烷萃取,在干燥并蒸发溶剂后又得到4.2克油状物。将一部分(4.2克)该油状四丁基铵盐溶于100毫升溴氯甲烷中并将溶液在20℃下搅拌40小时。真空蒸发溴氯甲烷后,残余物用乙醚反复萃取。将合并的乙醚萃取液蒸发至2.3克澄清油状物。该油状物用硅胶色谱法纯化,用80∶20氯仿-乙酸乙酯洗脱,得到1.62克油状物。
制备例13
N,N-二乙基琥珀酰胺酸碘甲基酯
将碘化钠(2.19克)和N,N-二乙基琥珀酰胺酸氯甲基酯(1.62克)与15毫升丙酮混合,将混合物在20℃下搅拌3小时。真空蒸发溶剂,使残余物在二氯甲烷和水之间分配。分离二氯甲烷层,用无水硫酸钠干燥,减压蒸发得到1.81克油状物,其使用时无需进一步纯化。
制备例14
N.N-二丙基琥珀酰胺酸二丙基铵
将琥珀酐(15.0克)加入到预冷(0℃)的二丙基胺(41毫升)的200毫升甲醇溶液中。撤掉冷浴。将混合物搅拌30分钟,然后蒸发溶剂得到42.8克浅黄色油状物。
制备例15
N,N-二丙基琥珀酰胺酸氯甲基酯
将含有N,N-二丙基琥珀酰胺酸二丙基铵(41.8克)、硫酸氢四丁基铵(48.05克)、二氯甲烷(250毫升)、水(80毫升)和1N氢氧化钠(142毫升)的混合物搅拌1.5小时,分离二氯甲烷层,用无水硫酸钠干燥,真空蒸发得到44.12克油状物。将20克该油状物溶于100毫升溴氯甲烷中,将溶液搅拌4.5小时。真空蒸发溴氯甲烷。残余物用乙醚萃取几次,合并乙醚萃取液,蒸发得到6.5克黄色油状物,其使用时无需进一步纯化。
制备例16
N,N-二丙基琥珀酰胺酸碘甲基酯
将碘化钠(7.8克)加入到N,N-二丙基琥珀酰胺酸氯甲基酯(6.5克)的30毫升丙酮溶液中,并将混合物搅拌2.5小时。真空蒸发溶剂后,将残余物溶于二氯甲烷和水的混合物中。分离二氯甲烷层,用无水硫酸钠干燥,蒸发得到8.8克橙色油状物,其使用时无需进一步纯化。
制备例17
4-高哌啶子基-4-氧代丁酸高哌啶鎓
将琥珀酐(15.0克)小心地加入到高哌啶(33.78毫升)的四氢呋喃(100毫升)溶液中,并将混合物搅拌30分钟。真空蒸发溶剂得到33.16克油状物。
制备例18
4-高哌啶子基-4-氧代丁酸氯甲基酯
将4-高哌啶子基-4-氧代丁酸高哌啶鎓(33.16克)、硫酸氢四丁基铵(49.74克)、二氯甲烷(200毫升)、水(50毫升)和1N氢氧化钠(146毫升)的混合物搅拌1.5小时。分离二氯甲烷相,用无水硫酸钠干燥,真空蒸发得到22.0克澄清油状物。将油状物溶于100毫升溴氯甲烷中并在20℃下搅拌17小时。真空蒸发溴氯甲烷后,将残余物用乙醚萃取。乙醚溶液用硫酸钠干燥,真空蒸发得到10.5克油状物。该油状物用硅胶色谱法纯化,用85∶15氯仿∶乙酸乙酯作洗脱剂。将含有所需氯甲基酯的级分合并并蒸发得到4.36克澄清油状物。
制备例19
4-高哌啶子基-4-氧代丁酸碘甲基酯
将碘化钠(5.28克)加入到4-高哌啶子基-4-氧代丁酸氯甲基酯(4.36克)的20毫升丙酮溶液中,并将混合物搅拌3小时。真空蒸发溶剂,将残余物分配于水和二氯甲烷之间。分离二氯甲烷层,用无水硫酸钠干燥,真空蒸发得到4.4克黄色油状物,其使用无需进一步纯化。
制备例20
N,N-二甲基氨基羰基氧基乙酸甲酯
在0℃下将吡啶(67毫升)、甘醇酸甲酯(12.85毫升)和二甲基氨基甲酰基氯(15.33毫升)混合,然后撤掉冷却浴,并将溶液在20℃下搅拌17小时,然后将反应混合物在65℃加热4小时,冷却,加入乙酸乙酯和水,通过加入1N HCl使混合物呈酸性。分离乙酸乙酯层,用盐水洗涤,用无水硫酸钠干燥,真空浓缩等到7.1克黄色油状物。用氯仿作洗脱剂在硅胶上对残余物进行色谱法纯化。将经测定含有所需产物的收集管中物进行合并,浓缩得到2.5克油状物。
制备例21
N,N-二甲基氨基羰基氧基乙酸钠盐
将上述酯(2.5克)溶于10毫升甲醇中,加入1N氢氧化钠(15.5毫升),并将溶液在20℃下搅拌1小时。减压蒸发反应混合物得到白色固体,其使用时无需进一步纯化。
制备例22
N,N-二甲基氨基羰基氧基乙酸氯甲基酯
将N,N-二甲基氨基羰基氧基乙酸钠盐(2.62克)与100毫升二氯甲烷、30毫升水、5.27克硫酸氢四丁基铵和1.3克碳酸氢钠混合,并搅拌1.5小时,分离二氯甲烷层后,用无水硫酸钠干燥,真空蒸发得到2.8克油状物。将该油状物溶于80毫升溴氯甲烷中并在20℃下搅拌18小时。真空蒸发二氯甲烷,并将残余物用乙醚萃取数次。合并萃取液并蒸发得到0.95克油状物,其使用时无需进一步纯化。
制备例23
N,N-二甲基氨基羰基氧基乙酸碘甲基酯
将碘化钠(1.46克)和N,N-二甲基氨基羰基氧基乙酸氯甲基酯(0.95克)与30毫升丙酮混合,并在20℃下搅拌19小时。真空蒸发丙酮得到1.18克油状物,其使用时无需进一步纯化。
制备例24
N,N-二乙基氨基羰基氧基乙酸甲酯
将吡啶(54毫升)、甘醇酸甲酯(10.3毫升)和二乙基氨基甲酰基氯(16.9毫升)混合,在65℃加热40小时,在85℃再加热24小时,另外在95℃加热24小时。冷却后,将反应混合物分配于乙酸乙酯和水之间。分离乙酸乙酯层,用1N HCl、水和盐水洗涤,用无水硫酸钠干燥,真空浓缩得到15.0克油状物,其使用时无需进一步纯化。
制备例25
N,N-二乙基氨基羰基氧基乙酸钠盐
将上述酯(15克)溶于100毫升甲醇中,加入1N氢氧化钠(79.3毫升),并将溶液在20℃下搅拌1.5小时。减压蒸发反应混合物得到白色固体,经P2O5干燥后为15.3克,其使用时无需进一步纯化。
制备例26
N,N-二乙基氨基羰基氧基乙酸碘甲基酯
将N,N-二乙基氨基羰基氧基乙酸钠盐(15.3克)与800毫升二氯甲烷、240毫升水、26.35克硫酸氢四丁基铵和6.52克碳酸氢钠混合,并搅拌1.5小时。分离二氯甲烷层,用无水硫酸钠干燥,真空蒸发,残余物用乙醚萃取数次。将萃取液合并,蒸发得到10.63克油状物。将该粗氯甲基酯与碘化钠(14.25克)混合于100毫升丙酮中并在20℃搅拌3.5小时。真空蒸发溶剂,残余物分配于氯仿和水之间。氯仿层用无水硫酸钠干燥并真空蒸发得到11.56克浅黄色油状物,其使用时无需进一步纯化。
制备例27
N-新戊酰基甘氨酸乙酯
向含有150毫升二氯甲烷的搅拌着的冰浴冷却的烧瓶中加入甘氨酸乙酯盐酯盐(25.0克)、新戊酰氯(20.9毫升)并缓慢加入62.4毫升N,N-二异丙基乙基胺。撤掉冰浴后,将反应溶液搅拌4小时。真空蒸发溶剂,将残余油状物分配于所加入的乙酸乙酯和水中。乙酸乙酯层用饱和碳酸氢钠水溶液和水的1∶1的混合物洗涤,用无水硫酸钠干燥,真空蒸发得到11.3克油状物。
制备例28
N-新戊酰基甘氨酸钠
向N-新戊酰基甘氨酸乙酯(11.3克)的50毫升甲醇溶液中加入69.2毫升1N氢氧化钠,并将溶液搅拌2小时。真空蒸发溶剂。加入新鲜的甲醇,并再蒸发得到12.45克灰白色固体。
制备例29
N-新戊酰基甘氨酸氯甲基酯
向N-新戊酰基甘氨酸钠(12.45克)和硫酸氢四丁基铵(21.8克)在100毫升二氯甲烷和50毫升水中的搅拌着的溶液中加入64.3毫升1N氢氧化钠,并将混合物搅拌3小时。分离二氯甲烷层,用硫酸钠干燥,真空蒸发得到20.9克油状物。将该油状物溶于50毫升溴氯甲烷中并在20℃下搅拌17小时。蒸发溴氯甲烷后,残余物用乙醚反复萃取。其空蒸发合并的萃取液得到2.2克油状物。用硅胶色谱法纯化,用氯仿-乙酸乙酯洗脱得到1.2克氯甲基酯。
制备例30
N-新戊酰基甘氨酸碘甲基酯
将碘化钠(1.71克)、N-新戊酰基甘氨酸氯甲基酯(1.2克)和丙酮(20毫升)的混合物在20℃下搅拌19小时。真空蒸发溶剂,将残余物分配于加入的氯仿和水之间。进行相分离,水层用氯仿反萃取。合并氯仿层,用无水硫酸钠干燥,真空蒸发得到1.45克油状物。该物质使用时无需进一步纯化。
制备例31
N-(2-乙基丁酰基)甘氨酸乙酯
向含有200毫升二氯甲烷的搅拌着的用冰浴冷却的烧瓶中加入甘氨酸乙酯盐酸盐(25.0克)、2-乙基丁酸酐(39.3毫升)并缓慢加入54.8毫升三乙胺。撤掉冰浴后,将反应溶液搅拌17小时。真空蒸发溶剂,将残余物分配于加入的乙酸乙酯和水中。乙酸乙酯层用饱和碳酸氢钠水溶液和水的1∶1混合物洗涤,用无水硫酸钠干燥,真空蒸发得到33.73克白色绒毛状固体。
制备例32
N-(2-乙基丁酰基)甘氨酸钠
向N-(2-乙基丁酰基)甘氨酸乙酯(33.73克)的100毫升甲醇溶液中加入167.6毫升1N氢氧化钠,并将溶液搅拌1.5小时。真空蒸发溶剂,然后加入新鲜甲醇,蒸发得到32.6克浅黄色油状物。
制备例33
N-(2-乙基丁酰基)甘氨酸氯甲基酯
向N-(2-乙基丁酰基)甘氨酸钠(32.6克)和硫酸氢四丁基铵(53.3克)在100毫升二氯甲烷和100毫升水中的搅拌着的溶液中加入157毫升1N氢氧化钠,并将混合物搅拌2.5小时。进行相分离,水层用400毫升二氯甲烷反萃取。合并有机层,用硫酸钠干燥,真空蒸发得到59.8克油状物。将该油状物溶于75毫升溴氯甲烷中并在20℃搅拌15小时。蒸发溴氯甲烷后,残余物用乙醚反复萃取。真空蒸发合并的萃取液得到7.84克油状物。用硅胶色谱法纯化、用氯仿-乙酸乙酯(3∶1)洗脱,得到2.33克氯甲基酯。
制备例34
N-(2-乙基丁酰基)甘氨酸碘甲基酯
将碘化钠(3.15克)、N-(2-乙基丁酰基)甘氨酸氯甲基酯(2.33克)和丙酮(20毫升)的混合物在20℃下搅拌17小时。真空蒸发溶剂,残余物分配于所加入的二氯甲烷和水中。进行相分离,水层用二氯甲烷反萃取两次。合并有机萃取液,用无水硫酸钠干燥,真空蒸发得到2.78克固体。该物质使用时无需进一步纯化。
制备例35
碳酸1-碘乙基异丙基酯
将氯甲酸(1-氯乙基)酯转化为碳酸1-碘乙基异丙基酯,所用方法见Wan-Joo Kim等人,The Journal of Antibiotics,(1991)44,pg.1086。
| 卤代烷基酯中间体 | ||||
| 制备例 | R1 | R2 | R3 | 1H NMR数据(CDCl2),δ |
| 1 | Me | OCH2CO2Me | Cl | 1.83(d,3H);3.77(s,3H);4.67(ABq,2H);6.40(q,1H). |
| 4 | Me | OCH2CONMe2 | Cl | 1.86 (d,3H);2.97(s,3H);2.99 (s,3H);2.99(s,3H);4.68 (d,1H);4.90(d,1H);6.45 (q,1H). |
| 7 | Me | OCH2CONEt2 | Cl | 1.10(t,3H);1.19(t,3H);1.81(d,3H);3.18(q,2H);3.37(q,2H);4.64(d,1H);4.86(d,1H);6.41(q,1H). |
| 10 | H | CH2CH2CONMe2 | Cl | 2.62-2.78(m,4H);2.96(s,3H);3.04(s,3H);5.72(s,2H). |
| 11 | H | CH2CH2CONMe2 | I | 2.67(s,4H);2.96(s,3H);3.03(s,3H);5.93(s,2H). |
| 12 | H | CH2CH2CONEt2 | Cl | 1.02(t,3H);1.14(t,3H);2.54-2.71(m,4H);3.21-3.38(m,4H);5.84(s,2H). |
| 13 | H | CH2CH2CONEt2 | I | 1.14(t,3H);1.24(t,3H);2.68-2.81(m,4H);3.32-3.50(m,4H);5.92(s,2H). |
| 15 | H | CH2CH2CONPr2 | Cl | 0.86(t,3H):0.94(t,3H);1.44-1.78(m,4H);2.60-2.84(m,4H);3.16-3.40(m,4H);5.72(s,2H). |
| 19 | H | (CH2)2CON(CH2)6 | I | 1.50-1.90(m,8H);2.65-2.80(m,4H);3.44-3.64(m,4H);5.93(s,2H). |
| 22 | H | CH2OCONMe2 | Cl | 2.92(s,3H);2.95(s,3H);4.63(s,2H);5.73(s,2H). |
| 23 | H | CH2OCONMe2 | I | 2.96(s,3h);2.99(s,3H);4.61(s,2H);5.95(s,2H). |
| 26 | H | CH2OCONEt2 | Cl | 1.12-1.23(m,6H);3.27-3.38(m,4H);4.70(s,2H);5.76(s,2H). |
| 29 | H | CH2NHCOCMe3 | Cl | 1.24(s,9H);4.12(d,2H);5.75(s,2H);6.15(宽.s,1H) |
| 30 | H | CH2NHCOCMe3 | I | 1.25(s,9H);4.06(d,2h);5.96(s,2H);6.15(宽s,1H). |
| 34 | H | CH2NHCOCHEt2 | I | 0.93(t,6H);1.44-1.75(m,4H);1.92-2.10(m,1H);4.10(ABq,2H);5.93(宽.s,1H);5.96(s,2H) |
Claims (17)
1、式(I)化合物:其中R为式(II)-式(VI)基团,
 其中x为0或1;
其中x为0或1;
A为被最多达2个独立地选自C1-C7烷基或C3-C7环烷基的取代基任选取代的C1-C5亚烷基或C2-C6链烯基链;或者(CH2)nO(CH2)m,其中亚甲基可以被最多达2个独立地选自C1-C7烷基或C3-C7环烷基的取代基任选取代;或者被最多达2个C1-C3烷基任选取代的C3-C7环烷基或环烯基;或含有O、S或NR6连键的4-7元杂脂环基团;或被最多达2个独立地选自C1-C3烷基、C1-C3烷氧基、卤素或CF3的取代基任选取代的亚苯基;
B为C2-C6链烯基苯基、2、3或4-吡啶基、2、3或4-哌啶基、2或3-吡咯烷基、-OCH2CO2R1或-OCH2CONR2R3;
R1为H、C1-C8烷基、C3-C7环烷基、苯基-(C1-C4)-烷基、(CH2)pCO2R2、或(CH2)pCONR2R3、(CH2)pSi(CH3)2;或者R1可以与A形成任选被C1-C3烷基取代的5、6或7元内酯环;
R2和R3独立地为H、C1-C7烷基、C3-C7环烷基、苯基-(C1-C4)-烷基;或者R2和R3与所连的氮一起可以代表被最多达2个C1-C3烷基任选取代的吡咯烷、哌啶、吗啉或高哌啶基团;或者R2或R3可以与A形成被最多达2个C1-C3烷基任选取代的5、6或7元内酰胺环;
R4和R5独立地为H、C1-C7烷基、C3-C7环烷基、苯基-(C1-C4)-烷基、(CH2)pCO2R2、(CH2)pCONR2R3、(CH2)pNR7R8、(CH2)pOR6或(CH2)pSR6;或者R4与R5合在一起代表被最多达2个C1-C3烷基任选取代的C3-C7环烷基环;
R6为H、C1-C6烷基、(CH2)pCOOR2、被最多达2个C1-C6烷基任选取代的C3-C7环烷基、在苯环上被最多达2个独立地选自C1-C3烷基、C1-C3烷氧基、卤素或CF3的取代基任选取代的苯基·(C1-C4)-烷基、COR2、CONR2R3、或被最多达2个独立地选自C1-C3烷基、C1-C3烷氧基、卤素或CF3的取代基任选取代的苯基;或者R6与R4和所连的氧一起可代表被最多达2个C1-C3烷基任选取代的氧杂环丁烷、四氢呋喃、四氢吡喃或氧杂环庚烷;
R7和R8独立地为H、C1-C6烷基、C3-C7环烷基、苯基-(C1-C4)-烷基、COR2、COOR2;或R7和R8独立地为被最多达2个独立地选自C1-C6烷基、C3-C7环烷基、苯基-(C1-C4)-烷基、C3-C7支链烷基的取代基任选取代的C2-C7链烷酰基、C4-C8环烷酰基;或者R7和R8与所连的氮合在一起可以代表被最多达2个独立地选自C1-C6烷基、C3-C7环烷基、C3-C7支链烷基或氧代的取代基任选取代的吡咯烷、哌啶或高哌啶基团;
R9为H或甲基;
R10、R11、R12和R13独立地选自H、C1-C4烷基和卤素;
m和n独立地为0、1或2,其中m或n必须至少为1;以及
p为1-3。
条件是:如果R为式II结构,且x是一个键,则A必须为非C2-C6亚烷基的基团。
2、权利要求1的化合物,其中R10和R11之一为5-氟,而另一个为6-氯。
3、权利要求2的化合物,其中R为下式基团,
4、权利要求3的化合物,其中x是0。
5、权利要求4的化合物,其中A是C2-C6链烯基链而R1为氢。
6、权利要求5的化合物,其中R12和R13为氢。
7、权利要求3的化合物,其中x为1。
8、权利要求7的化合物,其中A为C1-C5亚烷基。
9、权利要求8的化合物,其中R1为苄基。
10、权利要求2的化合物,其中R为下式基团, 且x是1。
且x是1。
11、权利要求10的化合物,其中R4、R9、R12及R13为氢,R5为氢、甲基或乙基,R6为氢、甲基、苄基或CH2COOR3。
12、权利要求2的化合物,其中R为下式基团, 且x是1。
且x是1。
13、权利要求12的化合物,其中R4、R9、R7、R8、R12和R13为氢,R5是(CH2)pNR7R8、甲基或苄基。
14、权利要求12的化合物,其中R7为COR2。
15、权利要求2的化合物,其中R为 且x为1。
且x为1。
16、权利要求15的化合物,其中B为2或3-吡咯烷。
17、权利要求2的化合物,其中R为下式基团,
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/009,188 US5270331A (en) | 1993-01-26 | 1993-01-26 | Prodrugs of antiinflammatory 3-acyl-2-oxindole-1-carboxamides |
| US009,188 | 1993-01-26 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1097740A CN1097740A (zh) | 1995-01-25 |
| CN1052003C true CN1052003C (zh) | 2000-05-03 |
Family
ID=21736103
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN94100697A Expired - Fee Related CN1052003C (zh) | 1993-01-26 | 1994-01-25 | 3-酰基-2-羟吲哚-1-甲酰胺类抗炎前药 |
Country Status (35)
| Country | Link |
|---|---|
| US (1) | US5270331A (zh) |
| EP (1) | EP0681580B1 (zh) |
| JP (1) | JP2703825B2 (zh) |
| KR (1) | KR0169827B1 (zh) |
| CN (1) | CN1052003C (zh) |
| AP (1) | AP443A (zh) |
| AT (1) | ATE155137T1 (zh) |
| AU (1) | AU678187B2 (zh) |
| BR (1) | BR9307768A (zh) |
| CA (1) | CA2152919C (zh) |
| CZ (1) | CZ181995A3 (zh) |
| DE (1) | DE69312093T2 (zh) |
| DK (1) | DK0681580T3 (zh) |
| EG (1) | EG20302A (zh) |
| ES (1) | ES2104187T3 (zh) |
| FI (1) | FI940365L (zh) |
| GR (1) | GR3024591T3 (zh) |
| HR (1) | HRP940034B1 (zh) |
| HU (1) | HUT69689A (zh) |
| IL (1) | IL108384A (zh) |
| MA (1) | MA23096A1 (zh) |
| MX (1) | MX9400677A (zh) |
| MY (1) | MY138629A (zh) |
| NO (1) | NO952949L (zh) |
| NZ (1) | NZ257278A (zh) |
| OA (1) | OA10563A (zh) |
| PL (1) | PL178857B1 (zh) |
| RU (1) | RU2124514C1 (zh) |
| SI (1) | SI9300567A (zh) |
| SK (1) | SK280929B6 (zh) |
| TW (1) | TW330935B (zh) |
| UA (1) | UA41348C2 (zh) |
| WO (1) | WO1994017061A1 (zh) |
| YU (1) | YU48796B (zh) |
| ZA (1) | ZA94463B (zh) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2097025T3 (es) * | 1993-02-09 | 1997-03-16 | Pfizer | Oxindol 1-(n-(alcoxicarbonil))carboxamidas y 1-(n-carboxamido)carboxamidas como agentes antiinflamatorios. |
| AU7284096A (en) * | 1995-10-09 | 1997-04-30 | Dieter Binder | Heterocyclically-substituted 1-indole carboxamides as cyclo-oxygenase-2 inhibitors |
| EP1058548B1 (en) * | 1998-03-06 | 2003-09-17 | Astrazeneca AB | The use of isatin and oxindole derivatives in the preparation of a medicament for use in the treatment of mycobacterial disease |
| EP0984012A3 (en) * | 1998-08-31 | 2001-01-10 | Pfizer Products Inc. | Nitric oxide releasing oxindole prodrugs with analgesic and anti-inflammatory properties |
| WO2003016259A2 (de) * | 2001-08-16 | 2003-02-27 | Pharmacon-Forschung Und Beratung Gmbh | Milchsäureelemente enthaltende verbindungen, deren herstellung sowie deren verwendung als pharmazeutische wirkstoffe |
| CN101628842A (zh) * | 2009-08-27 | 2010-01-20 | 中国人民解放军防化指挥工程学院 | 烷氧羰基甲氧基作为羧基保护基 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5118703A (en) * | 1988-10-18 | 1992-06-02 | Pfizer Inc. | Prodrugs of antiinflammatory 3-acyl-2-oxindole-1-carboxamides |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4556672A (en) * | 1984-03-19 | 1985-12-03 | Pfizer Inc. | 3-Substituted 2-oxindole-1-carboxamides as analgesic and anti-inflammatory agents |
| ES2075055T3 (es) * | 1988-10-18 | 1995-10-01 | Pfizer | Profarmacos de 3-acil-2-oxindol-1-carboxamidas antiinflamatorias. |
-
1993
- 1993-01-26 US US08/009,188 patent/US5270331A/en not_active Expired - Lifetime
- 1993-10-13 TW TW082108483A patent/TW330935B/zh active
- 1993-10-20 CA CA002152919A patent/CA2152919C/en not_active Expired - Fee Related
- 1993-10-20 RU RU95117091A patent/RU2124514C1/ru not_active IP Right Cessation
- 1993-10-20 DK DK93923879.6T patent/DK0681580T3/da active
- 1993-10-20 AT AT93923879T patent/ATE155137T1/de not_active IP Right Cessation
- 1993-10-20 SK SK912-95A patent/SK280929B6/sk unknown
- 1993-10-20 EP EP93923879A patent/EP0681580B1/en not_active Expired - Lifetime
- 1993-10-20 UA UA95073512A patent/UA41348C2/uk unknown
- 1993-10-20 PL PL93309989A patent/PL178857B1/pl not_active IP Right Cessation
- 1993-10-20 AU AU53598/94A patent/AU678187B2/en not_active Ceased
- 1993-10-20 NZ NZ257278A patent/NZ257278A/en unknown
- 1993-10-20 ES ES93923879T patent/ES2104187T3/es not_active Expired - Lifetime
- 1993-10-20 BR BR9307768A patent/BR9307768A/pt not_active Application Discontinuation
- 1993-10-20 DE DE69312093T patent/DE69312093T2/de not_active Expired - Fee Related
- 1993-10-20 JP JP6511478A patent/JP2703825B2/ja not_active Expired - Fee Related
- 1993-10-20 CZ CZ951819A patent/CZ181995A3/cs not_active IP Right Cessation
- 1993-10-20 WO PCT/US1993/009813 patent/WO1994017061A1/en active IP Right Grant
- 1993-10-21 AP APAP/P/1993/000583A patent/AP443A/en active
- 1993-10-28 SI SI9300567A patent/SI9300567A/sl not_active IP Right Cessation
-
1994
- 1994-01-16 EG EG2694A patent/EG20302A/xx active
- 1994-01-18 MY MYPI94000137A patent/MY138629A/en unknown
- 1994-01-20 IL IL10838494A patent/IL108384A/en not_active IP Right Cessation
- 1994-01-21 HR HR940034A patent/HRP940034B1/xx not_active IP Right Cessation
- 1994-01-24 ZA ZA94463A patent/ZA94463B/xx unknown
- 1994-01-25 HU HU9400209A patent/HUT69689A/hu unknown
- 1994-01-25 CN CN94100697A patent/CN1052003C/zh not_active Expired - Fee Related
- 1994-01-25 MX MX9400677A patent/MX9400677A/es not_active IP Right Cessation
- 1994-01-25 FI FI940365A patent/FI940365L/fi not_active Application Discontinuation
- 1994-01-25 YU YU2994A patent/YU48796B/sh unknown
- 1994-01-25 MA MA23402A patent/MA23096A1/fr unknown
-
1995
- 1995-07-25 OA OA60692A patent/OA10563A/en unknown
- 1995-07-25 NO NO952949A patent/NO952949L/no unknown
- 1995-07-26 KR KR1019950703074A patent/KR0169827B1/ko not_active IP Right Cessation
-
1997
- 1997-09-02 GR GR970402235T patent/GR3024591T3/el unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5118703A (en) * | 1988-10-18 | 1992-06-02 | Pfizer Inc. | Prodrugs of antiinflammatory 3-acyl-2-oxindole-1-carboxamides |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1038939C (zh) | 肽类化合物的制备方法 | |
| CN1032203C (zh) | 制备取代2-吡啶酮和吡啶-2-硫酮的方法 | |
| CN1036918C (zh) | 苯并吡喃衍生物及含有此衍生物的k+通道活化剂 | |
| CN1040986A (zh) | 环烷基取代的戊二酰胺衍生物的制备 | |
| CN1106017A (zh) | 核苷酸膦酸酯衍生物 | |
| CN1535968A (zh) | 四氢吡啶基或哌啶基杂环衍生物 | |
| CN1076928A (zh) | 1-[2-芳磺酰基氨基)-1-氧代乙基]哌啶衍生物及其制备和在治疗上的应用 | |
| CN1646531A (zh) | 作为髓过氧化物酶抑制剂的硫代黄嘌呤衍生物 | |
| CN1020098C (zh) | 硫代内酰胺-n-乙酸衍生物的生产 | |
| CN1038811A (zh) | 苯并吡喃衍生物及其制备方法 | |
| CN1075957A (zh) | 萘衍生物、其制备方法及其合成中间体 | |
| CN101039937A (zh) | 在呼吸系统疾病的治疗中用作β模拟剂的杂芳基化合物 | |
| CN1067053C (zh) | 单环β-内酰胺类抗生素衍生物的中间体的制备方法 | |
| CN1108657A (zh) | 雪花胺衍生物、其制备方法及其作为药物的用途 | |
| CN1278262A (zh) | 三环三唑并苯并氮杂衍生物、用于制备该衍生物的方法和抗变态反应剂 | |
| CN1103534A (zh) | 具有抗精神病作用的化合物 | |
| CN1054980A (zh) | 具有旋光活性的8-甲氧基喹诺酮羧酸衍生物,它们的制备方法以及它们的中间体 | |
| CN1069640C (zh) | 噁唑烷酮衍生物,其制备方法及含它们的药物组合物 | |
| CN1052003C (zh) | 3-酰基-2-羟吲哚-1-甲酰胺类抗炎前药 | |
| CN1047673A (zh) | 抗痉挛剂 | |
| CN1079745A (zh) | 新的9-氟-7-氧代-7H-吡啶并[1,2,3-d,e][1,4]苯并嗪-6-羧酸及其酯 | |
| CN1052482C (zh) | 喜树碱衍生物及其制备方法和抗肿瘤剂 | |
| CN1048706A (zh) | 3-取代乙烯基头孢菌素衍生物及其制备方法 | |
| CN1993367A (zh) | 稠合的嘧啶衍生物和黄嘌呤氧化酶抑制剂 | |
| CN1056847C (zh) | 7-酰基-3-(取代的氨基甲酰氧基)头孢烯化合物的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |