WO2025146626A1 - A method for the preparation of 1-(4-(5-(halomethyl)-1,2,4-oxadiazol-3-yl)phenyl)ethan-1-one - Google Patents
A method for the preparation of 1-(4-(5-(halomethyl)-1,2,4-oxadiazol-3-yl)phenyl)ethan-1-one Download PDFInfo
- Publication number
- WO2025146626A1 WO2025146626A1 PCT/IB2025/050009 IB2025050009W WO2025146626A1 WO 2025146626 A1 WO2025146626 A1 WO 2025146626A1 IB 2025050009 W IB2025050009 W IB 2025050009W WO 2025146626 A1 WO2025146626 A1 WO 2025146626A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- solvent
- iii
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
Definitions
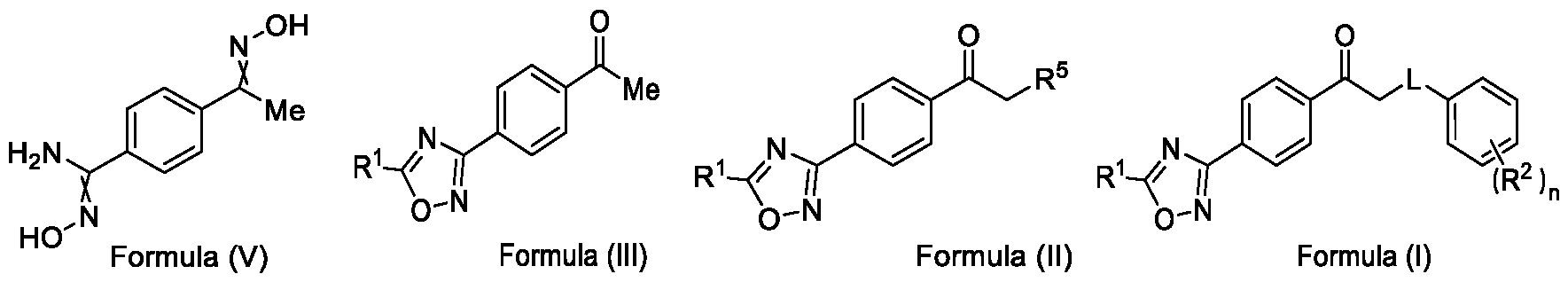
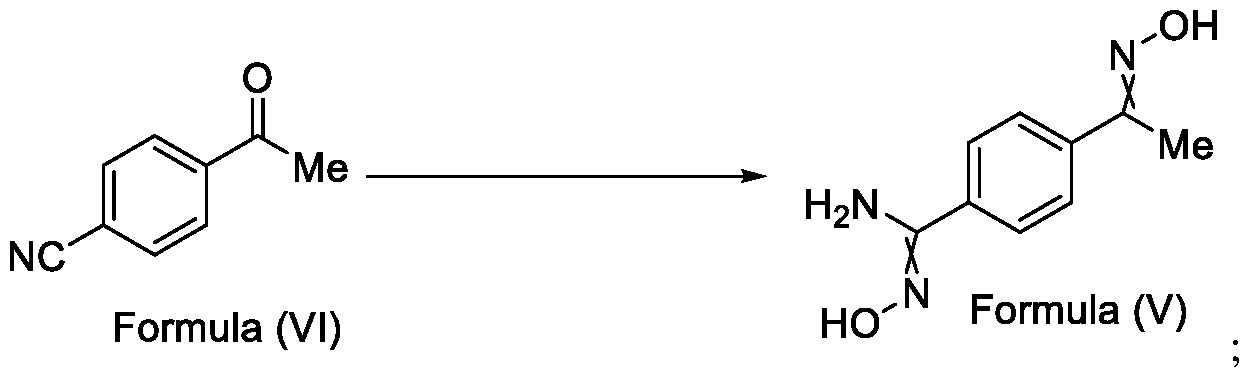
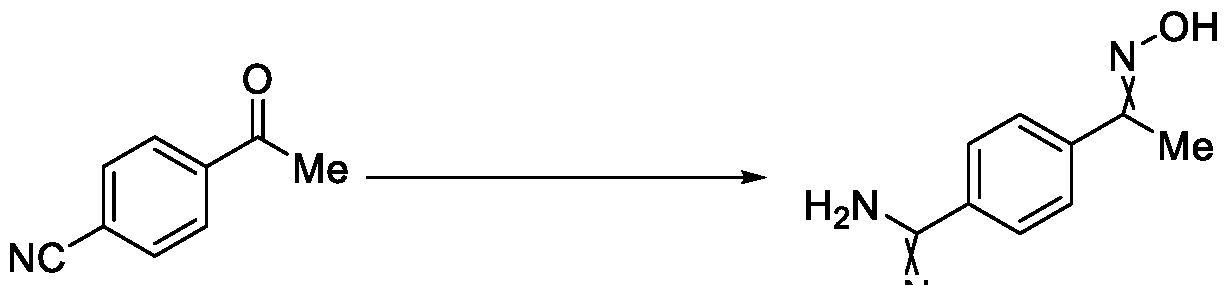
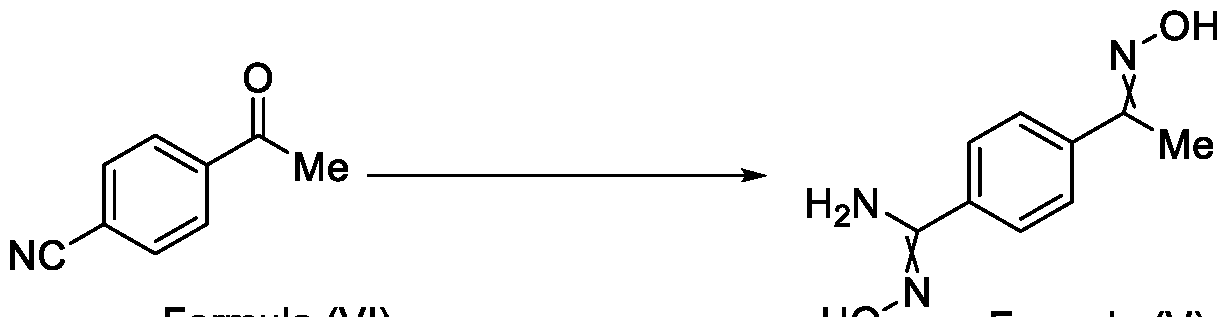
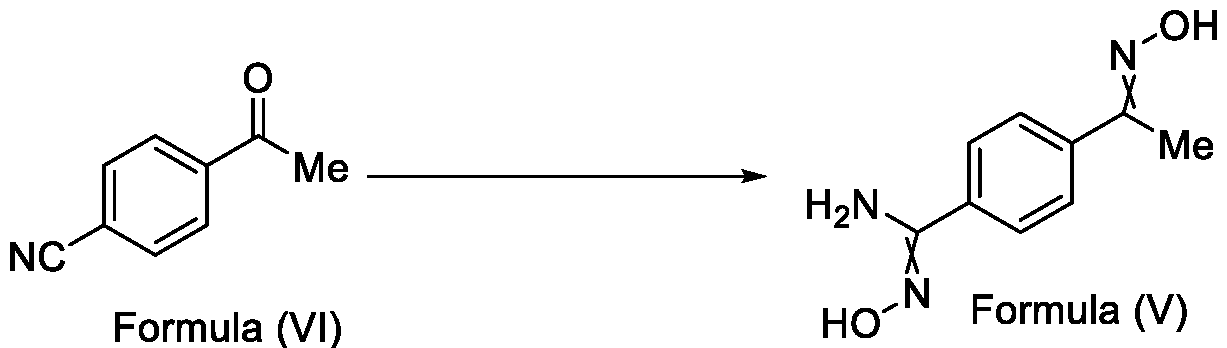
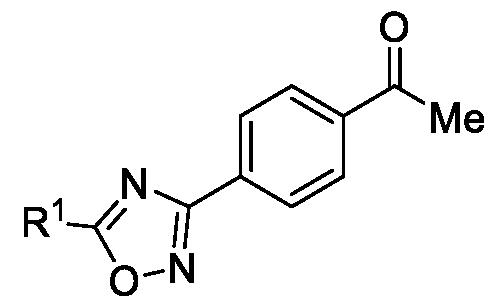
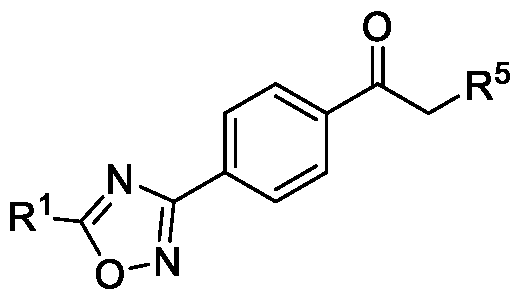
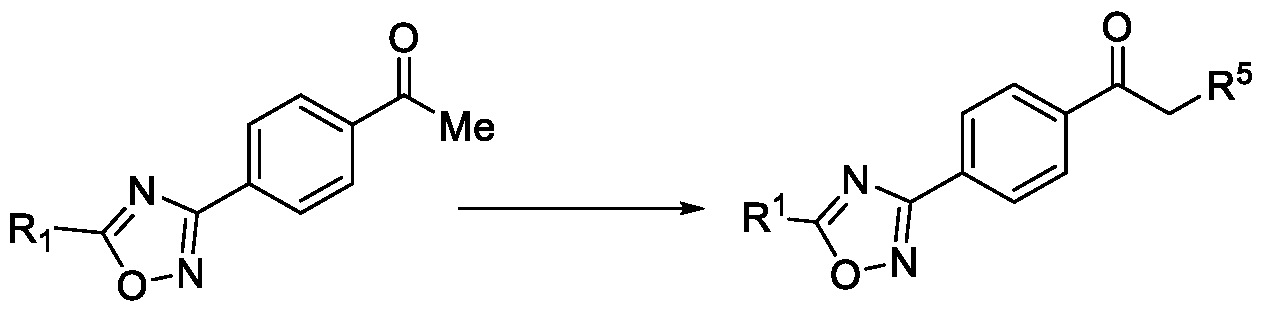
- the present invention relates to a method for the preparation of a compound (l-(4-(5-(halomethyl)- l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one) of Formula (III). Further the present invention also relates to a method for the preparation of a compound of Formula (I), an intermediate compound of Formula (II) and N-oxides or salts thereof, useful for combating phytopathogenic fungi.
- Oxadiazole based compounds are well known for their pesticidal activity.
- Various oxadiazoles have been disclosed in the literature.
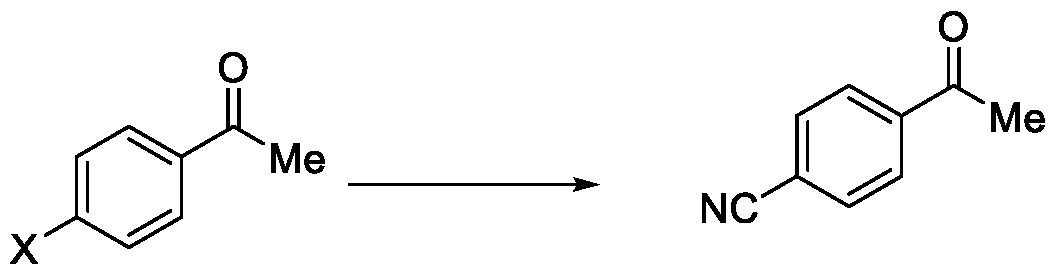
- WO2017118689 discloses a microbiocidal oxadiazole compound of Formula (I), as a fungicide, wherein l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one was used as an intermediate for its preparation.
- W02020208511 discloses the preparation of oxadiazole compounds bearing a carbonyl group, which includes protection of the carbonyl group of the nitrile reactant with ethylene glycol, followed by a reaction with aqueous hydroxylamine, furthermore by a cyclization with trifluoroacetic anhydride, and finally deprotection of the cyclic ether to obtain the oxadiazole compounds bearing a carbonyl group.
- the disclosed process is lengthy, involves too many reagents and purification steps at every stage, and is eventually less atom economic.
- the objective of the present invention is to provide a novel, cost effective and improved method for preparing the compound of Formula (I) and intermediate compounds of Formula (II) and Formula (III) and their N-oxides or salts thereof, in a reduced number of steps and with high isolated yields, suitable for commercial scale preparation.
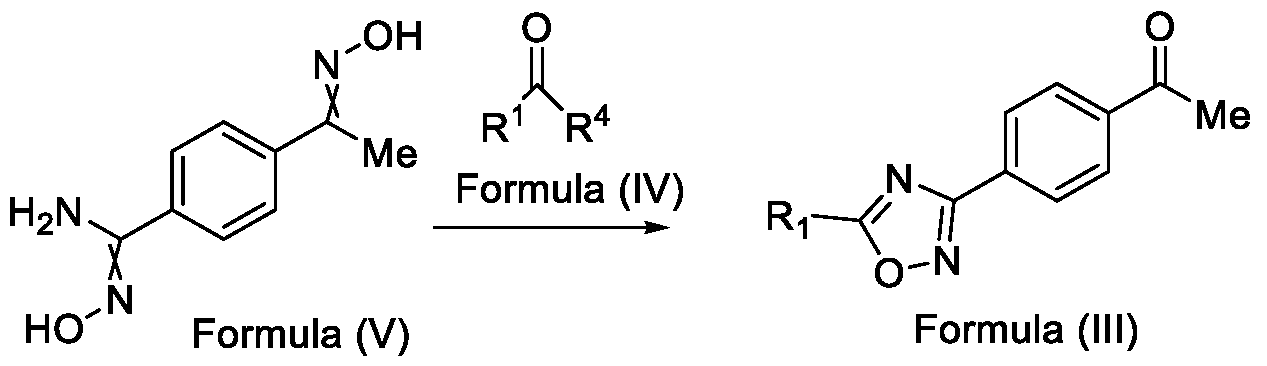
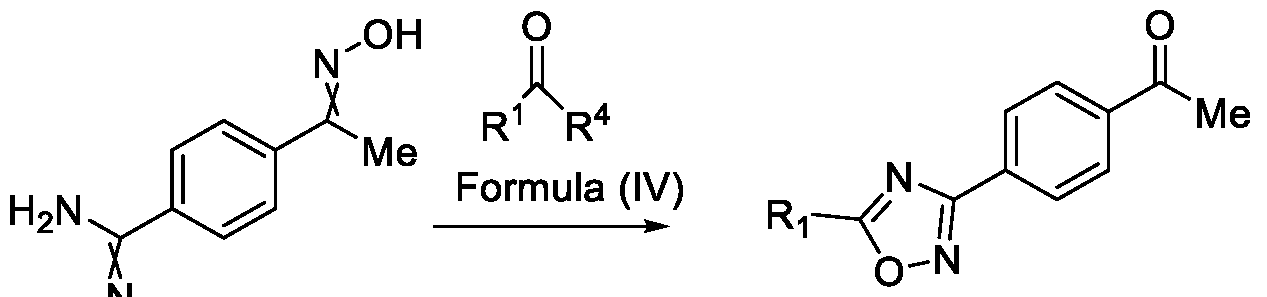
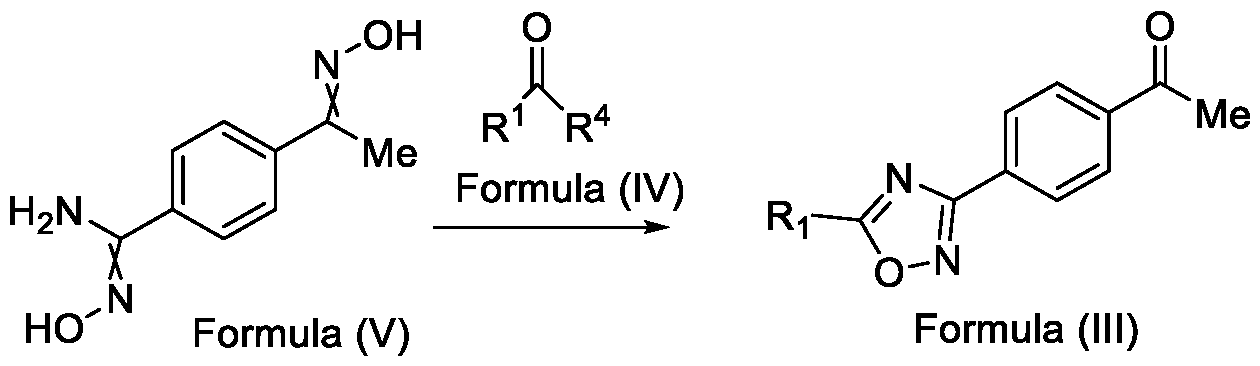
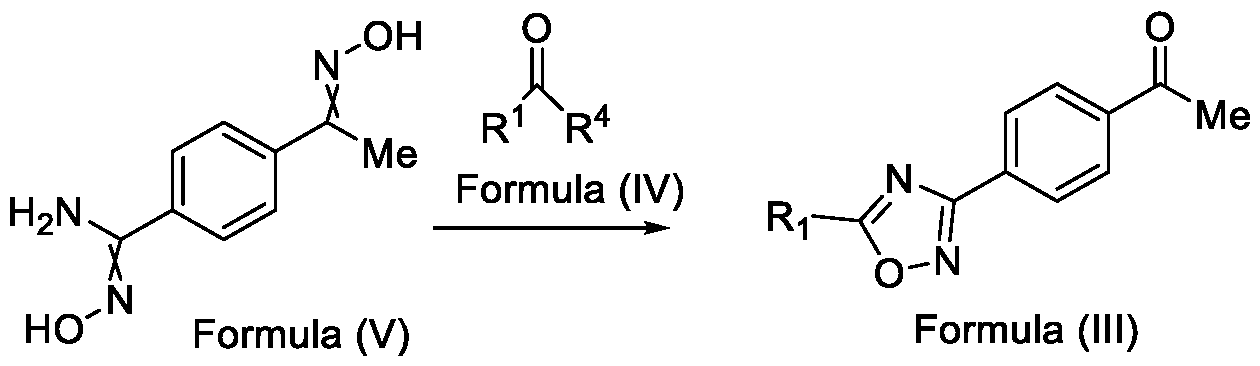
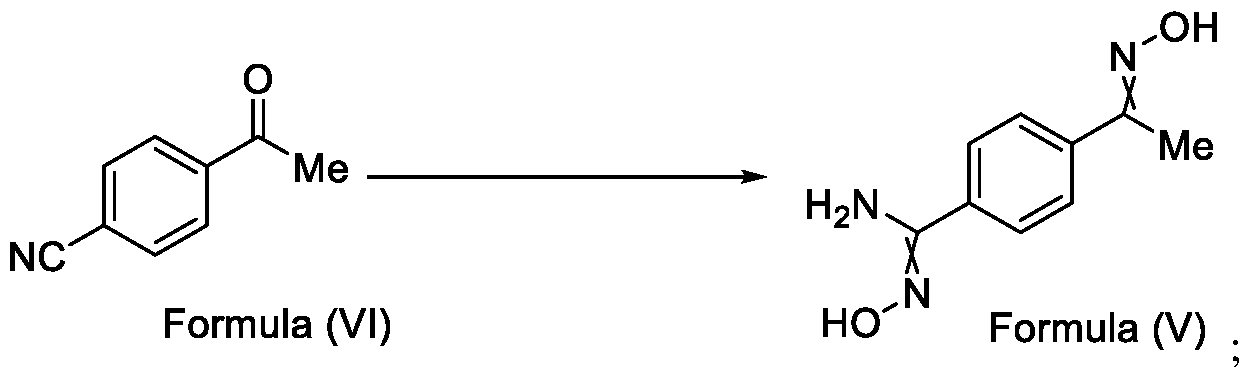
- the present invention provides a solution to this objective by offering a simple and economically amenable method for obtaining a compound of Formula (III). Further, the present invention also provides a method for the preparation of a compound of Formula (I), and intermediate compounds of Formula (II) or salts thereof, in high yields and a reduced number of reaction steps, wherein the said method comprises a novel and inventive step of simultaneous cyclization and de -protection of a compound of Formula (V) in a single step, by overcoming at least one of the shortcomings disclosed in the prior art.
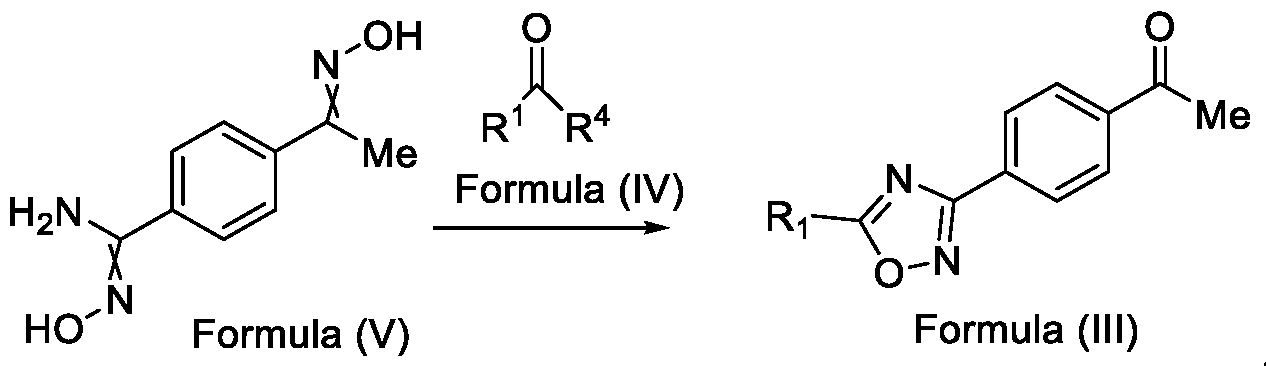
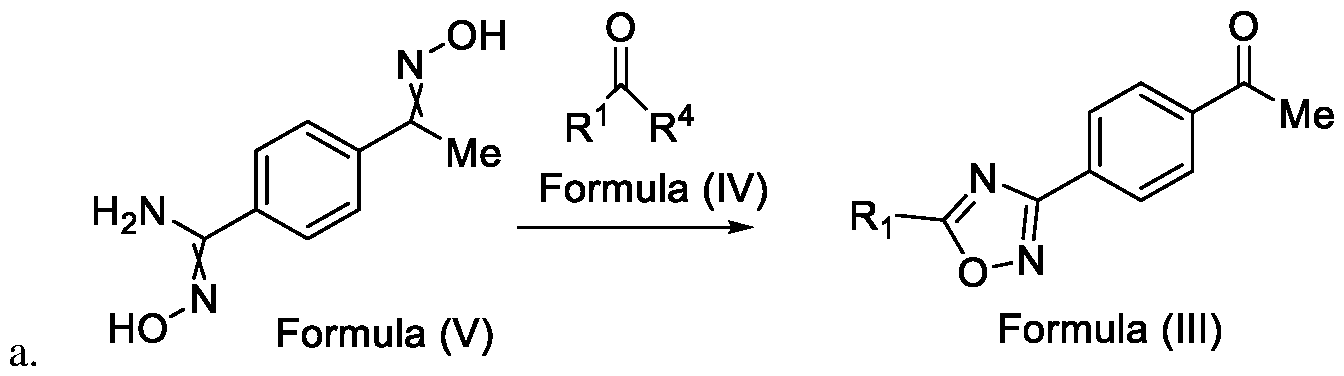
- the first aspect of the present invention provides a method for preparing a compound of Formula (III), or its N-oxides or salts thereof,
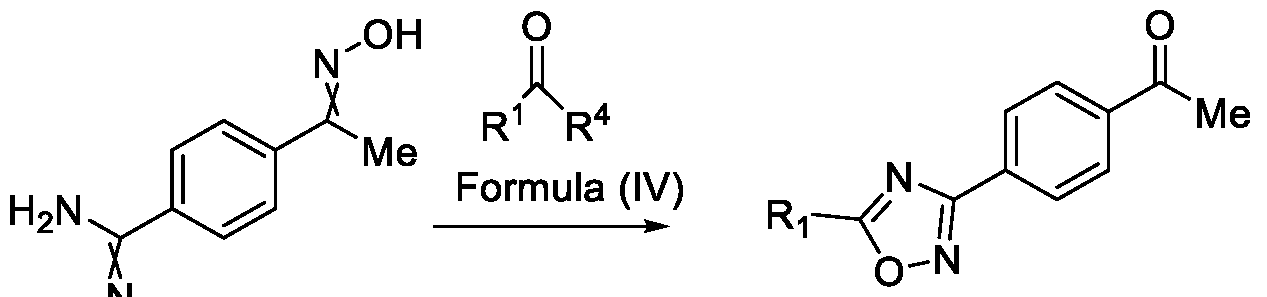
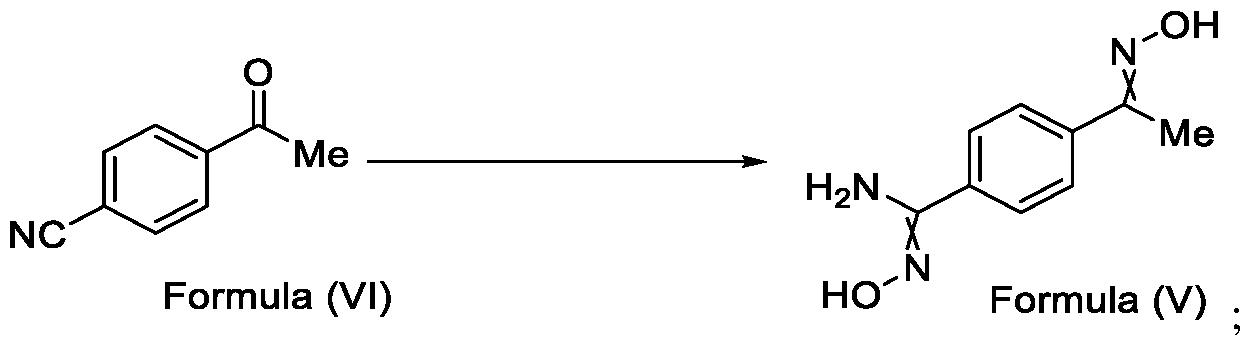
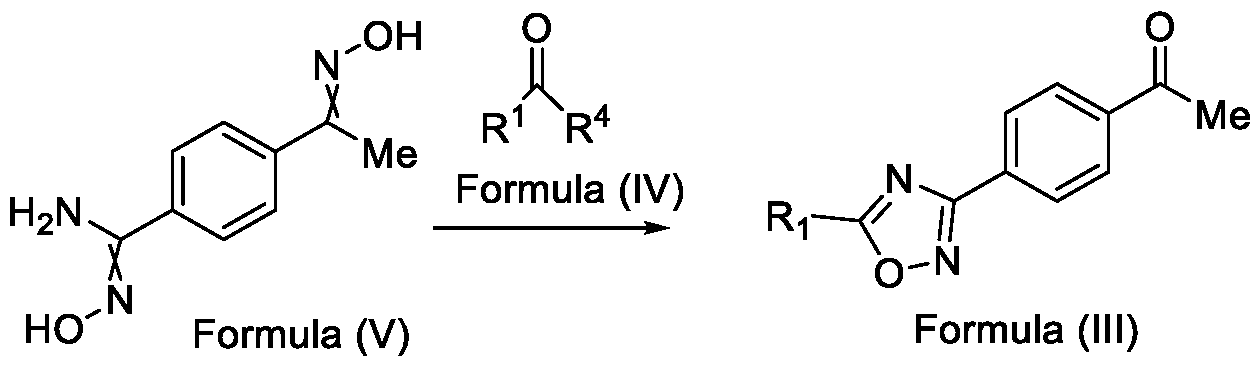
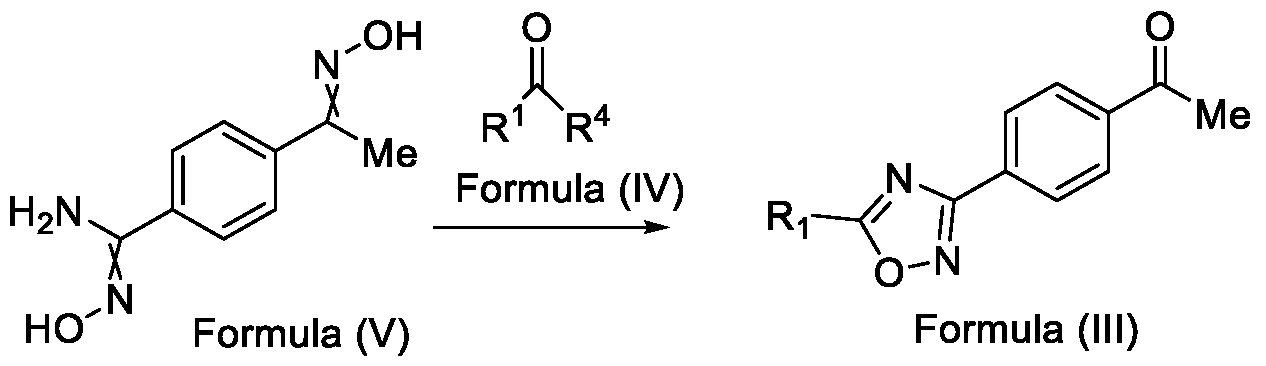
- Formula (III) wherein R 1 is selected from -CHF2, -CF2-CI or CF3, comprising the steps of: a. reacting a compound of Formula (VI) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V), and b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
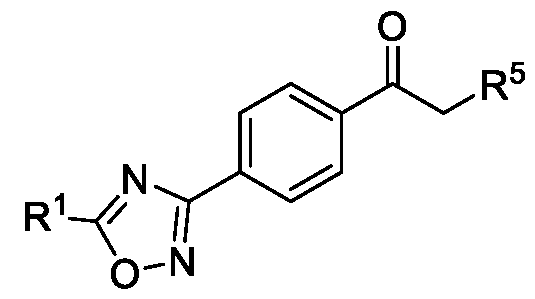
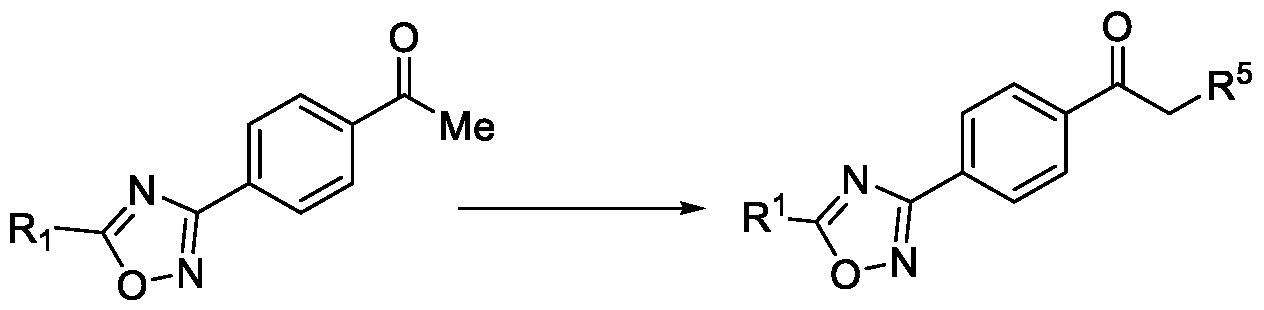
- the present invention provides a method for preparing a compound of Formula (II), or its N-oxides or salts thereof,
- R 1 is selected from -CHF2, -CF2-CI or CF3;
- R 5 is selected from fluoro, chloro, bromo, or iodo, comprising the steps of: a. reacting a compound of Formula (VI) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V), b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step, wherein R 4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; - and c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
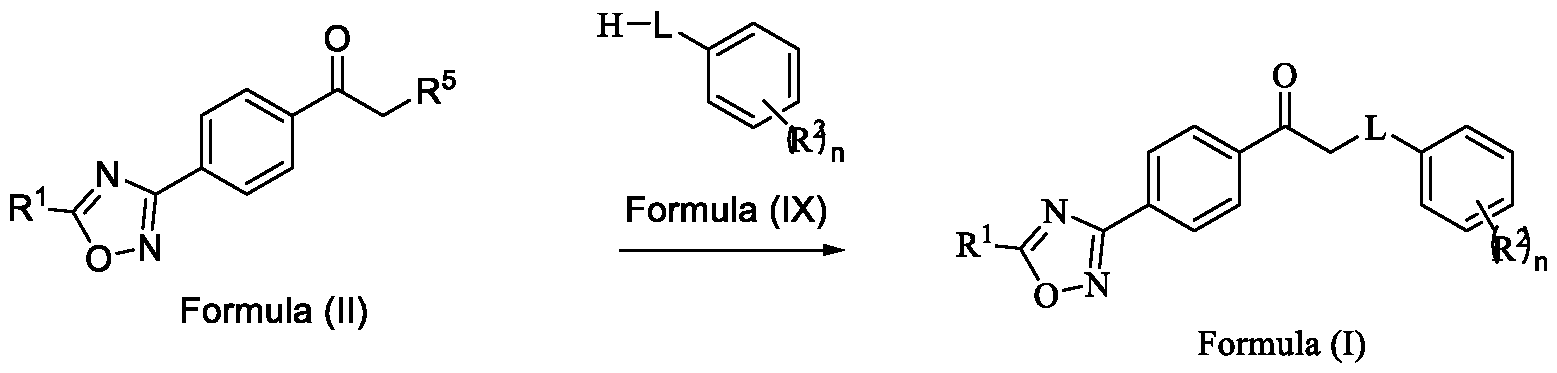
- the present invention provides a method for preparing a compound of Formula (I), or its N-oxides or salts thereof,
- R 1 is selected from -CHF2, -CF2-CI or CF3;
- L is selected from O, NR 3 or S;
- R 2 is selected from hydrogen, halogen, C i-Ce-alkyl, Ci-Ce-haloalkyl, Ci-Ce-alkoxy or C3-C6- cycloalkyl;
- R 3 is selected from hydrogen or C i-Ce-alkyl; and n is an integer selected from 1-2, comprising the steps of: a. reacting a compound of Formula (VI) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
- Formula (III) Formula (II) wherein R 5 is fluoro, chloro, bromo or iodo; and d. alkylating a compound of Formula (IX) with the compound of Formula (II) in the presence of a base, and optionally in the presence of a catalyst and a solvent, to obtain a compound of
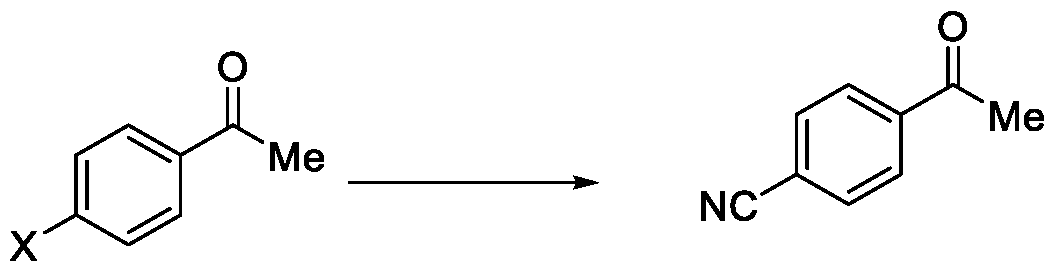
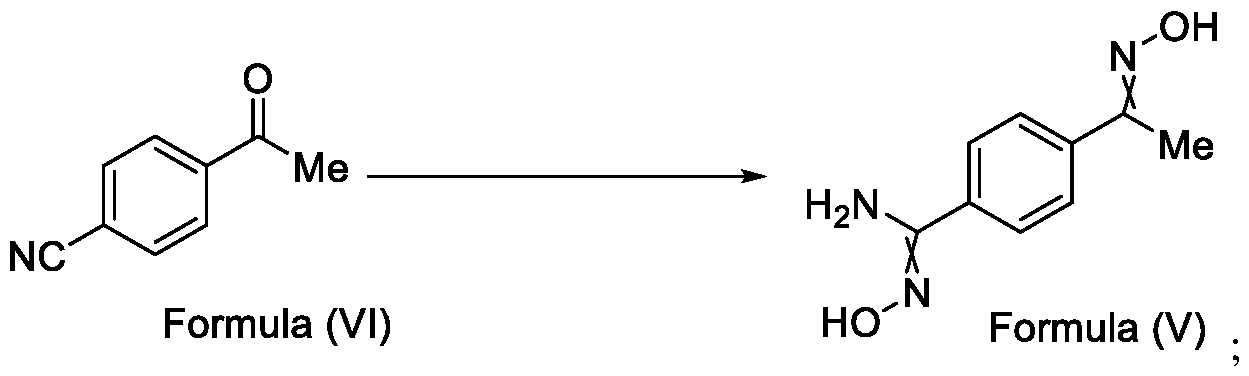
- the present invention provides a method for the synthesis of compounds of Formula (VI) and salts thereof, comprising the steps of: i. acetylating a compound of Formula (VIII) with acetyl chloride or acetic anhydride in the presence of a suitable catalyst and optionally in the presence of a solvent to obtain a compound of Formula (VII),
- R 4 is selected from halogen, Ci-Ce-alkoxy, OH or O-C(O)-R'.
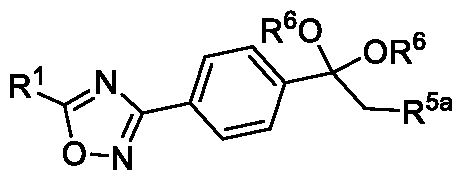
- the present invention further relates to an intermediate compound of formula (X) or formula (XI),
- the compounds of the present disclosure may be present either in pure form or as mixtures of different possible isomeric forms such as stereoisomers or constitutional isomers.
- the various stereoisomers include enantiomers, diastereomers, chiral isomers, atropisomers, conformers, rotamers, tautomers, optical isomers, polymorphs, and geometric isomers. Any desired mixtures of these isomers fall within the scope of the claims of the present disclosure.
- one stereoisomer may be more active and/or may exhibit beneficial effects when enriched relative to the other isomer(s) or when separated from the other isomer(s). Additionally, the person skilled in the art knows processes or methods or technology to separate, enrich, and/or to selectively prepare said isomers.
- the compounds of the present disclosure may be present in the form of N -oxides or salts.
- the compounds of the present invention may be an acid addition or base addition salt.
- the acid addition salt includes inorganic or organic acid preferably hydrochloric acid, trifluoroacetic acid, methanesulfonic acid, p-toluenesulfonic acid.
- the base addition salt includes inorganic or organic base preferably alkali metal or alkaline earth metal salt.
- halogen used in the present invention refers to fluoro, chloro, bromo or iodo.
- Ci-Ce alkyl used in the present invention refers to a linear or branched alkyl with 1 to 6 carbon atoms.
- Examples of Ci-Ce includes but not limited to methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, //-pentyl or //-hexyl and the like.
- Ci-Ce haloalkyl used in the present invention refers to a linear or a branched alkyl with 1 to 6 carbon atoms, which is substituted with one or more halogen. Examples includes but not limited to chloromethyl, dichloromethyl, trichloromethyl, trifluoromethyl, difluoromethyl, trifluoroethyl, perfluoroethyl and the like.
- Ci-Ce alkoxy used in the present invention refers to a linear or a branched alkoxy with 1 to 6 carbon atoms. Examples includes but not limited to methoxy, ethoxy, propoxy, isopropoxy, //-butoxy, t-butoxy and the like.
- C3-C6 cycloalkyl used in the present invention refers to a 3- to 6-membered saturated monocyclic carbocyclic ring. Examples includes but not limited to cyclopropyl, cyclobutyl, cyclopentyl and the like.
- Formula (VI) HO' N Formula (V) b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step, wherein R 4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; and c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
- R 3 is selected from hydrogen or C i-CT-alkyl; and n is an integer selected from 1-2, comprising the steps of: a. reacting the compound of Formula (VI) with hydroxylamine or its salt in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V), b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
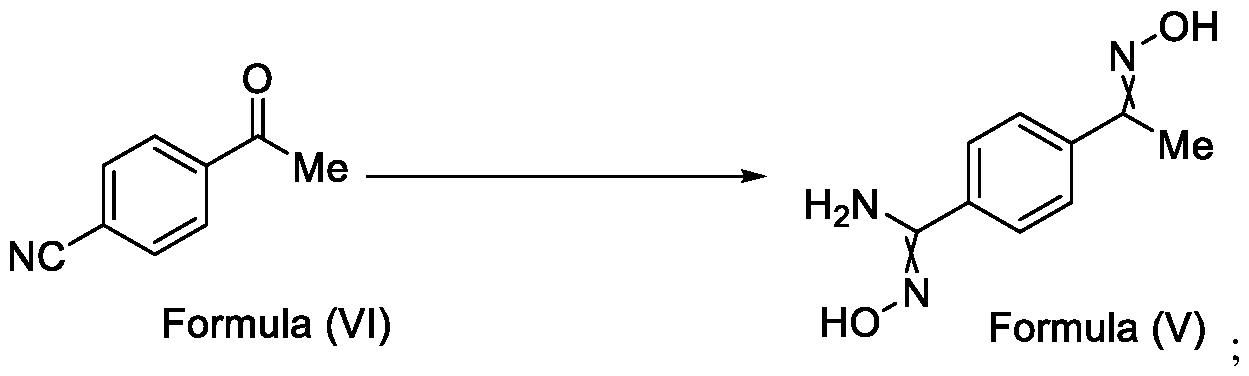
- Embodiment-004 The present invention still further provides a method for the synthesis of a compound of Formula (VI), comprising the steps of: i. acetylating a compound of Formula (VIII) with acetyl chloride or acetic anhydride in the presence of a suitable catalyst and optionally in the presence of a solvent to obtain a compound of Formula (VII),
- Embodiment-005 in yet another aspect, the present invention further relates to a method of preparing a compound of Formula (III), or its salts or N-oxides thereof, wherein the said method comprises the step (step-b) of cyclizing the dioxime compound of Formula (V) with a compound of Formula (IV) in a solvent and optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step, wherein, R 1 is selected from -CHF2, -CF2-CI or CF3 and
- R 4 is selected from halogen, OH, C i-Ce-alkoxy or O-C(O)-R
- Embodiment-006 In yet another aspect, the present invention provides a method for a compound of Formula (III), or its N-oxides or salts thereof,
- R 1 is selected from -CHF2, -CF2-CI or CF3, comprising the steps of:
- Formula (VIII) Formula (VII) . ii. reacting the compound of Formula (VII) with a cyanide source in a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (VI),
- Formula (VII) Formula (VI) a. reacting the compound of Formula (VI) obtained in step (A) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V), and b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step, wherein R 4 is selected from halogen, -OH, Ci-Ce-alkoxy or O-C(O)-R'.
- R 1 is selected from -CHF2, -CF2-CI or CF3;
- R 5 is selected from fluoro, chloro, bromo, or iodo, comprising the steps of:
- Embodiment-028 The methods of the present invention according to embodiments 001 , 002, 003, 004, 005, 006, 007, and 008, wherein the solvent can be selected from aliphatic or aromatic hydrocarbon, halogenated hydrocarbon, ethers, cyclic ethers, cyclic esters, cyclic carbonate ester, nitro based solvents, nitriles, amides, ketones, acids, alcohols, organosulfur, ionic liquids, water or mixture thereof.
- the solvent can be selected from aliphatic or aromatic hydrocarbon, halogenated hydrocarbon, ethers, cyclic ethers, cyclic esters, cyclic carbonate ester, nitro based solvents, nitriles, amides, ketones, acids, alcohols, organosulfur, ionic liquids, water or mixture thereof.
- Embodiment-030 The methods of the present invention according to embodiments 001 , 002, 003, 004, 005, 006, 007, and 008, wherein the base, if used, can be selected from an inorganic or organic base such as alkali metal hydrogen carbonate, alkali/alkaline earth metal carbonate, alkali/alkaline earth metal hydroxide, alkali metal phosphate, alkali metal hydride, alkali metal alkoxide, ethylamine, triethylamine, isopropylamine diisopropylamine, triisopropylamine, pyridine, picoline, piperidine, methylmorpholine, /V-mcthylpipcridinc N,N- (dimethylamino)pyridine (DMAP), lutidine, collidine, tetramethylammonium hydroxide, tetrabutylammonium hydroxide, choline hydroxide, l
- Embodiment-031 The methods of the present invention according to embodiments 001, 002, 003, 005, 006, 007, and 008, wherein the reagent hydroxylamine used in step-a can be either in an aqueous form or in a solid form such as a salt form.
- Embodiment-034 The methods of the present invention according to embodiment-031, wherein the reagent hydroxylamine is hydroxylamine hydrochloride salt.
- Hydroxylamine hydrochloride is soluble in polar organic solvents and is more stable to oxidation and is easy to handle on large quantity as compared to free hydroxylamine or aqueous hydroxylamine, and as a result, step-c of the present method was carried out using hydroxylamine hydrochloride.
- step-a was carried out in the presence of a base to neutralize the hydrochloride salt.
- Embodiment-035 The methods of the present invention according to embodiment-034, wherein the base can be selected from alkali metal hydrogen carbonate, alkaline earth metal hydrogen carbonate, alkali metal carbonate, alkaline earth metal carbonate, triethylamine, diisopropylamine, triisopropylamine, pyridine, or picoline.
- Embodiment-037 The methods of the present invention according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein step-a was performed in a suitable solvent selected from but not limited to alcohol such as ethanol, methanol, isopropanol, diethyl ether, /V,/V-di methyl f rm am ide, tetrahydrofuran or water or a mixture thereof.
- a suitable solvent selected from but not limited to alcohol such as ethanol, methanol, isopropanol, diethyl ether, /V,/V-di methyl f rm am ide, tetrahydrofuran or water or a mixture thereof.
- Embodiment-039 The methods of the present invention according to embodiments 001 , 002, 003, 005, 006, 007 and 008, wherein in step-a: i. the solvent is selected from ethanol, methanol, isopropanol, diethyl ether, N,N- dimethylformamide, tetrahydrofuran or water or a mixture thereof; and ii. the base if used, is selected from alkali metal hydrogen carbonate or alkaline earth metal hydrogen carbonate, alkali metal carbonate or alkaline earth metal carbonate, triethylamine, diisopropylamine, triisopropylamine, pyridine, or picoline.
- the solvent is selected from ethanol, methanol, isopropanol, diethyl ether, N,N- dimethylformamide, tetrahydrofuran or water or a mixture thereof
- the base if used is selected from alkali metal hydrogen carbonate or alkaline earth metal
- Embodiment-045 The methods of the present invention according to embodiments-041 and 042, wherein the acid used for the acidic treatment, can be selected from but not limited to sulphuric acid, hydrochloric acid, or hydrobromic acid.
- Embodiment-046 The methods of the present invention according to embodiment-045, wherein the acid is selected from hydrochloric acid and particularly from an aqueous hydrochloric acid.
- Embodiment-047 The step-b of the present methods according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein the solvent includes alcohol, tetrahydrofuran, 2- methyltetrahydrofuran, acetonitrile, or V,V-dimethylformamide or water.
- the solvent includes alcohol, tetrahydrofuran, 2- methyltetrahydrofuran, acetonitrile, or V,V-dimethylformamide or water.
- Embodiment-048 The methods of the present invention according to embodiment-047, wherein the solvent is selected from methanol, ethanol, isopropanol, tetrahydrofuran or water and particularly, it is selected from methanol, ethanol or tetrahydrofuran.
- the solvent is selected from methanol, ethanol, isopropanol, tetrahydrofuran or water and particularly, it is selected from methanol, ethanol or tetrahydrofuran.
- Embodiment-049 The methods of the present invention according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein in step-b: i. the solvent is selected from methanol, ethanol, isopropanol, tetrahydrofuran, 2- methyltetrahydrofuran, acetonitrile, V,V-dimethylformamide or water; ii. the base if used, is selected from alkali metal alkoxide or alkaline earth metal alkoxide; iii. the acid is selected from sulphuric acid, hydrochloric acid, or hydrobromic acid.
- the solvent is selected from methanol, ethanol, isopropanol, tetrahydrofuran, 2- methyltetrahydrofuran, acetonitrile, V,V-dimethylformamide or water
- the base if used is selected from alkali metal alkoxide or alkaline earth metal al
- Embodiment-050 The step-b of the present methods according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein the reaction temperature is between 0 °C-120 °C, and particularly, it is between 0 °C-40 °C during the cyclization and 70 °C -100 °C during the acidic treatment to obtain the compound of Formula (III).
- Embodiment-051 The step-c of the present methods according to embodiments 002, 003 and 007, 008, wherein the halogenating agent can be selected from but not limited to F2, CI2, Br2, 12, B in acetic acid, /V-bromosuccinimidc, copper bromide (CuB ). HBr, NaBr, NtBBr, phosphorus tribromide (PBi ;).
- phosphorus pentabromide a mixture of NJBBr and potassium peroxymonosulfate sulfate, Trimethylphenylammonium tribromide, N-chlorosuccinimide, phosphorus trichloride (PCh), phosphorus pentachloride (PCI5), sulfonyl chloride (SOCI2), sulfuryl chloride (SO2CI2), selenium oxychloride, benzyltrimethylammonium dichloroiodate, or trichloroisocyanuric acid.
- PCh phosphorus trichloride
- PCI5 phosphorus pentachloride
- SOCI2 sulfuryl chloride
- selenium oxychloride benzyltrimethylammonium dichloroiodate
- trichloroisocyanuric acid trichloroisocyanuric acid.
- Embodiment-052 The methods of the present invention according to embodiment-051, wherein the halogenating agent can be selected from B , /V-bromosuccinimidc, CI2, or N- chlorosuccinimide .
- Embodiment-054 The methods of the present invention according to embodiments-002, 003 and 007, 008, wherein the halogenating agent can be used in the presence of a catalyst selected from but not limited to p-toluenesulfonic acid, aluminium chloride (AlCh), monopotassium phosphate (K2HSO4), sulphuric acid, silica, amberlyst 15, or trimethylsilyl triflate.
- a catalyst selected from but not limited to p-toluenesulfonic acid, aluminium chloride (AlCh), monopotassium phosphate (K2HSO4), sulphuric acid, silica, amberlyst 15, or trimethylsilyl triflate.
- phosphorus pentabromide a mixture of NfhBr and potassium peroxymonosulfate sulfate, trimethylphenylammonium tribromide, N-chlorosuccinimide, phosphorus trichloride (PCI3), phosphorus pentachloride (PCI5), sulfonyl chloride (SOCI2), sulfuryl chloride (SO2CI2), selenium oxychloride, benzyltrimethylammonium dichloroiodate, or trichloroisocyanuric acid; ii.
- the solvent is selected from dichloromethane, dichloroethane (ethylene dichloride), chloroform, acetonitrile, diethyl ether, methyl tertiary-butyl ether, tetrahydrofuran, 2- methyl-tetrahydrofuran, ethyl acetate, alcoholic solvent selected from methanol, ethanol, or isopropanol, acetic acid or water or a mixture thereof; iii.
- the catalyst if used is selected from p-toluenesulfonic acid, aluminium chloride (AlCh), monopotassium phosphate (K2HSO4), sulphuric acid, silica, amberlyst 15, or trimethylsilyl triflate.
- Embodiment-058 The step-c of the present methods according to embodiments 002, 003 and 007, 008, wherein when the compound of formula (III) is subjected to halogenation using bromine or chlorine and in the presence of an alcoholic solvent such as methanol, ethanol or isopropanol followed by ketal de -protection in the presence of an acid to obtain a compound of formula (Ila), as shown below wherein,
- R 5a is selected from bromo or chloro
- R 6 is selected from methyl, ethyl, n-propyl or isopropyl, wherein the intermediate compound of formula (X) Formula (X) or f ormu ) a (XI)
- Formula (XI) is/are optionally isolated.
- Embodiment-059 The methods of the present invention according to embodiment-058, wherein the acid used for ketal deprotection is selected from hydrochloric acid.
- Embodiment-063 The step-d of the present methods according to embodiments 003 and 008, can be carried out with or without a solvent.
- Embodiment-090 The present invention further provides an intermediate compound of formula (XI),
- the isolation of the reaction product can be carried out by a technique which includes but is not limited to decantation, filtration, centrifugation, evaporation, liquid-liquid extraction, distillation, recrystallization, chromatography and the like or a combination thereof.
- Scheme 1 A process for preparing a compound of Formula (I) and its intermediate compounds of Formula (II) and Formula (III)
- reaction mixture was cooled to 25-30 °C and quenched by the addition of water (150 mL), and extracted with methyl tert-butyl ether (MTBE) (3 X 100 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate (NazSCH) and concentrated under reduced pressure to afford 4-acetylbenzonitrile (8.3 g, 78.99%) as an off-white solid.
- MTBE methyl tert-butyl ether
- Reagents 4-acetylbenzonitrile (300 g, 2046 mmol); hydroxylamine sulphate (402g, 2456 mmol); methanol (600 mL) and triethylamine (TEA) (371 mL, 2660 mmol).
- Step-b Preparation of l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one
- Method-2 Using Ethyl trifluoroacetate and NaOMe in MeOH To a stirred solution of /V'-hydroxy-4-(4 -(hydroxyimino) ethyl)benzimidamide (20 g, 104 mmol) in ethyl trifluoroacetate (36.8 g, 259 mmol), 30% sodium methoxide (NaOMe) in methanol (MeOH) (93.2 g, 518 mmol) was added in a dropwise manner at a temperature between 25 to 40 °C, and continued to stir further for 3 hours.
- reaction mixture was added into aqueous 5N hydrochloric acid (HC1) solution (200 mL) and further heated to a temperature between 70 to 80°C. The reaction mixture was then continued to stir for 6 hours at 80°C. During the reaction, the distillate was collected to recover ethyl trifluoroacetate and methyl trifluoroacetate formed in situ due to transesterification. After completion of the reaction, the reaction mixture was cooled to a temperature between 20 to25°C. The solid obtained was filtered.
- HC1 hydrochloric acid
- Step-c Preparation of 2-bromo-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l- one
- Method-1 To a stirred solution of l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (50 g, 195 mmol) in ethylene dichloride (EDC) (1.0 L), a solution of bromine (37.4 g, 234 mmol) in ethylene dichloride (500 mL) was added at a temperature between 40 to 45 °C in a drop wise manner. The reaction mixture was then stirred for 2 hours at a temperature between 40 to 45 °C. After the completion of the reaction, the reaction mixture was quenched by the addition of water (250 mL) and extracted with ethylene dichloride (2 x 250 mL).
- EDC ethylene dichloride
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
A method for the preparation of substituted 1-(4-(5-(halomethyl)-1,2,4-oxadiazol-3-yl)phenyl)ethan-1-one The present invention relates to a method for preparing a compound of Formula (I) and its intermediate compounds of Formula (II) and Formula (III), wherein R1, R2, L, R4, R5 and n are as defined in the description. The present method comprises a step of converting a dioxime compound of Formula (V) to a compound of Formula (III) in a single step via a simultaneous cyclization and deprotection of ketoxime to obtain the compound of Formula (III), which was then subjected to a subsequent halogenation and reaction with a compound of Formula (IX) to obtain the compound of Formula (I).
Description
Title: A method for the preparation of l-(4-(5-(halomethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one
Field of the Invention
The present invention relates to a method for the preparation of a compound (l-(4-(5-(halomethyl)- l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one) of Formula (III). Further the present invention also relates to a method for the preparation of a compound of Formula (I), an intermediate compound of Formula (II) and N-oxides or salts thereof, useful for combating phytopathogenic fungi.
Background and Prior art of the Invention
Oxadiazole based compounds are well known for their pesticidal activity. Various oxadiazoles have been disclosed in the literature. For example, WO2017118689 discloses a microbiocidal oxadiazole compound of Formula (I),
 as a fungicide, wherein l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one
as a fungicide, wherein l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one
 was used as an intermediate for its preparation.
was used as an intermediate for its preparation.
W02020208511 discloses the preparation of oxadiazole compounds bearing a carbonyl group, which includes protection of the carbonyl group of the nitrile reactant with ethylene glycol, followed by a reaction with aqueous hydroxylamine, furthermore by a cyclization with trifluoroacetic anhydride, and finally deprotection of the cyclic ether to obtain the oxadiazole compounds bearing a carbonyl group. The disclosed process is lengthy, involves too many reagents and purification steps at every stage, and is eventually less atom economic.
Hence, there is a need for a method for preparing such compounds which is short, high yielding and more atom economic, wherein both cyclization to oxadiazole, and deprotection of ketoxime takes place simultaneously in a single step.
Objective of the Invention
The objective of the present invention is to provide a novel, cost effective and improved method for preparing the compound of Formula (I) and intermediate compounds of Formula (II) and Formula (III)
and their N-oxides or salts thereof, in a reduced number of steps and with high isolated yields, suitable for commercial scale preparation.
The present invention provides a solution to this objective by offering a simple and economically amenable method for obtaining a compound of Formula (III). Further, the present invention also provides a method for the preparation of a compound of Formula (I), and intermediate compounds of Formula (II) or salts thereof, in high yields and a reduced number of reaction steps, wherein the said method comprises a novel and inventive step of simultaneous cyclization and de -protection of a compound of Formula (V) in a single step, by overcoming at least one of the shortcomings disclosed in the prior art.
Summary of the Invention
Accordingly, the first aspect of the present invention provides a method for preparing a compound of Formula (III), or its N-oxides or salts thereof,

Formula (III) wherein R1 is selected from -CHF2, -CF2-CI or CF3, comprising the steps of: a. reacting a compound of Formula (VI) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
 and b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
and b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
.N
HO Formula (V) Formula (III) wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'.
In a second aspect, the present invention provides a method for preparing a compound of Formula (II), or its N-oxides or salts thereof,
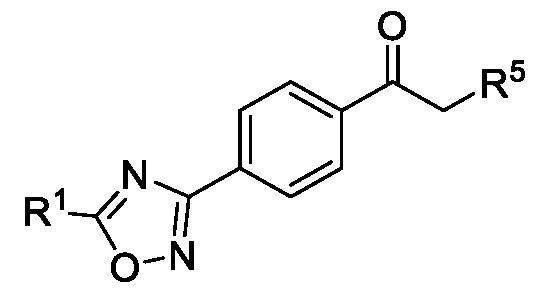
Formula (II) wherein,
R1 is selected from -CHF2, -CF2-CI or CF3; and
R5 is selected from fluoro, chloro, bromo, or iodo, comprising the steps of: a. reacting a compound of Formula (VI) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
 b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
 wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; - and
c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; - and
c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),

Formula (III) Formula (II)
In a third aspect, the present invention provides a method for preparing a compound of Formula (I), or its N-oxides or salts thereof,

Formula (I) wherein,
R1 is selected from -CHF2, -CF2-CI or CF3;
L is selected from O, NR3 or S;
R2 is selected from hydrogen, halogen, C i-Ce-alkyl, Ci-Ce-haloalkyl, Ci-Ce-alkoxy or C3-C6- cycloalkyl; and
R3 is selected from hydrogen or C i-Ce-alkyl; and n is an integer selected from 1-2, comprising the steps of: a. reacting a compound of Formula (VI) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
Formula (VI) HO"N Formula (V) b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
.N
HO Formula (V) Formula (III) wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
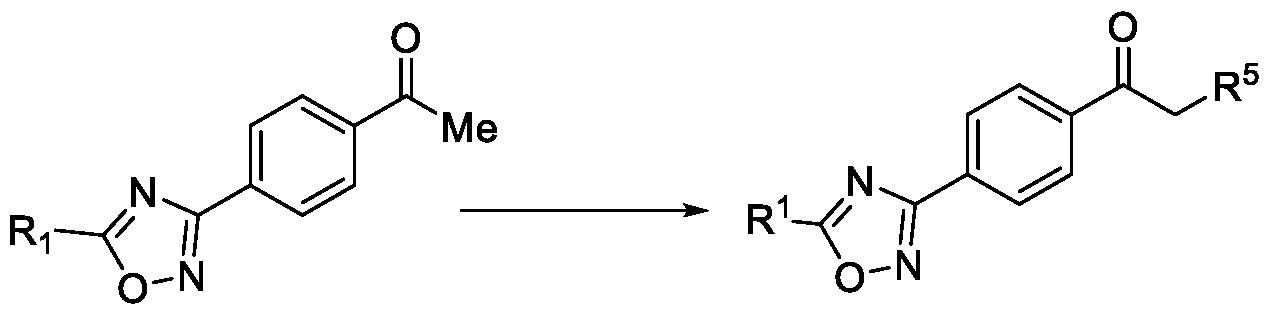
Formula (III) Formula (II) wherein R5 is fluoro, chloro, bromo or iodo; and d. alkylating a compound of Formula (IX) with the compound of Formula (II) in the presence of a base, and optionally in the presence of a catalyst and a solvent, to obtain a compound of
Formula (I),
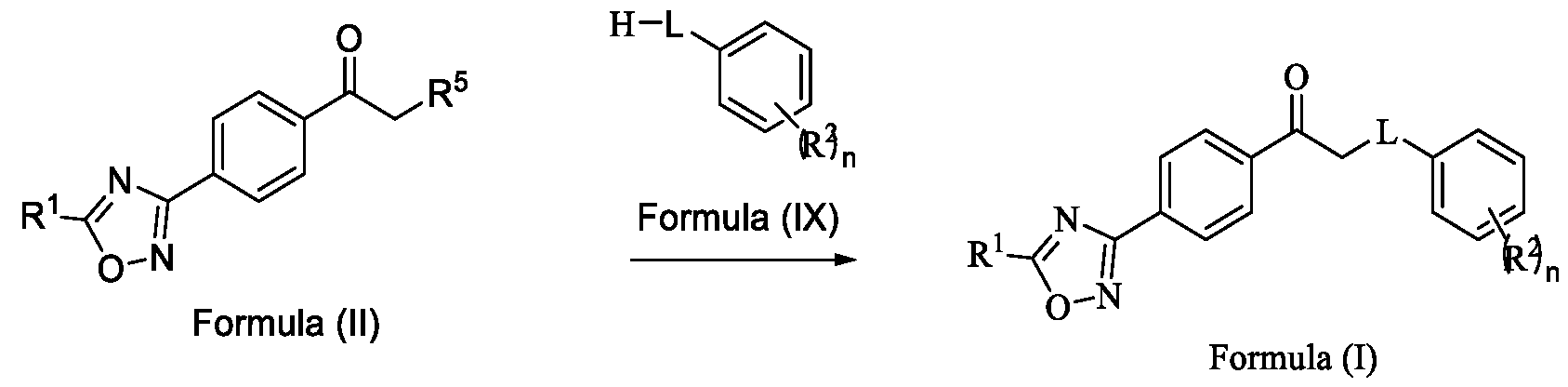
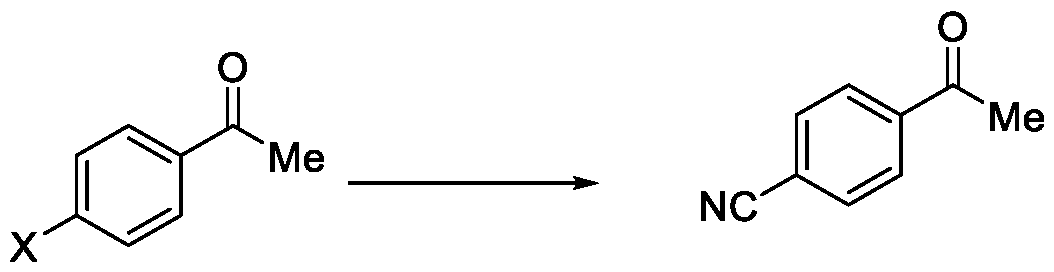
In a fourth aspect, the present invention provides a method for the synthesis of compounds of Formula (VI) and salts thereof, comprising the steps of: i. acetylating a compound of Formula (VIII) with acetyl chloride or acetic anhydride in the presence of a suitable catalyst and optionally in the presence of a solvent to obtain a compound of Formula (VII),
X = F, Cl, Br or I
Formula (VIII)
 and
ii. reacting the compound of Formula (VII) with a cyanide source in a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (VI),
and
ii. reacting the compound of Formula (VII) with a cyanide source in a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (VI),
Formula (VII) Formula <VI>
In yet another aspect, the present invention further relates to a method of preparing a compound of Formula (III), or its salts or N-oxides thereof, wherein the said method comprises the step (step-b) of cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
 wherein,
wherein,
R1 is selected from -CHF2, -CF2-CI or CF3; and
R4 is selected from halogen, Ci-Ce-alkoxy, OH or O-C(O)-R'.
In yet another aspect, the present invention further relates to an intermediate compound of formula (X) or formula (XI),
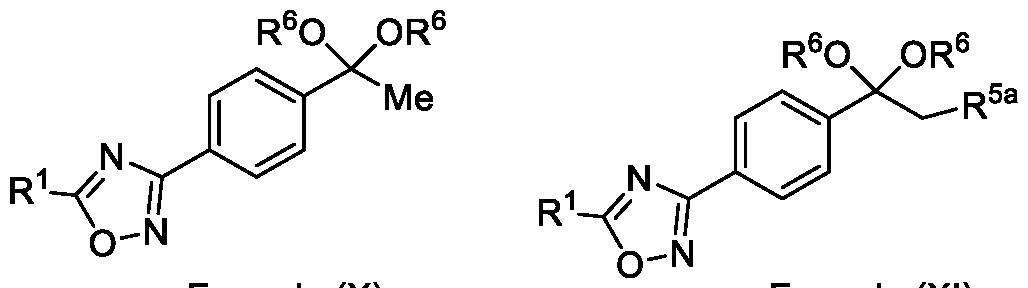
Formula (X) or Formula (XI) wherein R1, R6 and R5a as described in the description.
DETAILED DESCRIPTION OF THE INVENTION
As used herein, the terms “comprises”, “comprising”, “includes”, “including”, or any other variation thereof, are intended to cover a non-exclusive inclusion, subject to any limitation explicitly indicated.
For example, process or method that comprises a list of elements is not necessarily limited to only those elements but may include other elements not expressly listed or inherent to process or method.
Also, the indefinite articles “a” and “an” preceding an element or component of the present invention are intended to be non-restrictive regarding the number of instances (i.e. occurrences) of the element or component. Therefore “a” or “an” should be read to include one or at least one, and the singular word form of the element or component also includes the plural unless the number is obviously meant to be singular.
The compounds of the present disclosure may be present either in pure form or as mixtures of different possible isomeric forms such as stereoisomers or constitutional isomers. The various stereoisomers include enantiomers, diastereomers, chiral isomers, atropisomers, conformers, rotamers, tautomers, optical isomers, polymorphs, and geometric isomers. Any desired mixtures of these isomers fall within the scope of the claims of the present disclosure. One skilled in the art will appreciate that one stereoisomer may be more active and/or may exhibit beneficial effects when enriched relative to the other isomer(s) or when separated from the other isomer(s). Additionally, the person skilled in the art knows processes or methods or technology to separate, enrich, and/or to selectively prepare said isomers.
The compounds of the present disclosure may be present in the form of N -oxides or salts. The compounds of the present invention may be an acid addition or base addition salt. The acid addition salt includes inorganic or organic acid preferably hydrochloric acid, trifluoroacetic acid, methanesulfonic acid, p-toluenesulfonic acid. The base addition salt includes inorganic or organic base preferably alkali metal or alkaline earth metal salt.
Unless otherwise specifically mentioned, the term “halogen” used in the present invention refers to fluoro, chloro, bromo or iodo.
The term “Ci-Ce alkyl” used in the present invention refers to a linear or branched alkyl with 1 to 6 carbon atoms. Examples of Ci-Ce includes but not limited to methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, //-pentyl or //-hexyl and the like.
The term “Ci-Ce haloalkyl” used in the present invention refers to a linear or a branched alkyl with 1 to 6 carbon atoms, which is substituted with one or more halogen. Examples includes but not limited to chloromethyl, dichloromethyl, trichloromethyl, trifluoromethyl, difluoromethyl, trifluoroethyl, perfluoroethyl and the like.
The term “Ci-Ce alkoxy” used in the present invention refers to a linear or a branched alkoxy with 1 to 6 carbon atoms. Examples includes but not limited to methoxy, ethoxy, propoxy, isopropoxy, //-butoxy, t-butoxy and the like.
The term “C3-C6 cycloalkyl” used in the present invention refers to a 3- to 6-membered saturated monocyclic carbocyclic ring. Examples includes but not limited to cyclopropyl, cyclobutyl, cyclopentyl and the like.
In the context of the present invention, the term “atom economic” refers to maximizing the incorporation of material from the starting materials or reagents into the final product at the end of the chemical process.
In the context of the present invention, the term “optionally” when used in reference to any element, to intermediates, reagents or conditions, including any method step, e.g., the isolation of intermediates; is intended to mean that the subject element is isolated, or alternatively is not isolated from the reaction mixture and directly used for the subsequent chemical reaction. Similarly, this definition is applied in case for reagents or reaction conditions as well.
The specification herein and the various features and advantageous details thereof are explained with reference to the non-limiting examples in the description. Descriptions of well-known components and processing techniques are omitted so as to not unnecessarily obscure the embodiments herein. The examples used herein are intended merely to facilitate an understanding of ways in which the specification herein may be practiced and to further enable those of skilled in the art to practice the specification herein. Accordingly, the examples should not be construed as limiting the scope of the specification herein.
The description of the specific embodiments will so fully reveal the general nature of the embodiments herein that others can, by applying current knowledge, readily modify and/or adapt for various applications such specific embodiments without departing from the generic concept, and, therefore, such adaptations and modifications should and are intended to be comprehended within the meaning and range of equivalents of the disclosed embodiments. It is to be understood that the phraseology or terminology employed herein is for the purpose of description and not of limitation. Therefore, while the embodiments herein have been described in terms of preferred embodiments, those skilled in the art will recognize that the embodiments herein can be practiced with modification within the spirit and scope of the embodiments as described herein.
Any discussion of documents, acts, materials, devices, articles and the like that has been included in this specification is solely for the purpose of providing a context for the disclosure. It is not to be taken as an admission that any or all of these matters form a part of the prior art base or were common general knowledge in the field relevant to the disclosure as it existed anywhere before the priority date of this application.
It is well recognized that preparing a compound bearing a carbonyl moiety from a starting material containing a reactive carbonyl group, is tedious and may often lead to unwanted side products due to
competitive reactions among the reaction site and the reactive carbonyl group. Hence, such processes always require specific protection and de -protection of the reactive carbonyl group to eliminate such competitive reactions to minimise the side product formation. Therefore, there is a need for the development of a method for preparing the compound of Formula (III) and Formula (I), which does not require any specific protection and de -protection steps of the carbonyl group while not compromising on the yield of the final product.
Accordingly, the following embodiments describe the present invention in a detailed manner.
Embodiment-001 : The present invention provides a method for preparing a compound of Formula (III), or its N-oxides or salts thereof,
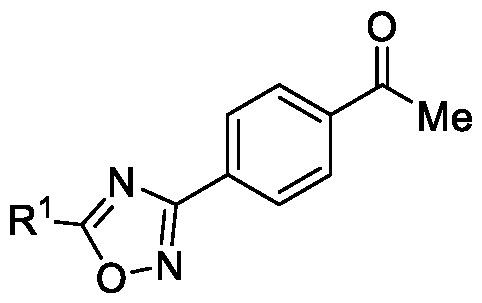
Formula (III) wherein R1 is selected from -CHF2, -CF2-CI or CF3, comprising the steps of: a. reacting the compound of Formula (VI) with hydroxylamine or its salt in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
Formula (VI) HO Formula (V) . and b. cyclizing the dioxime compound of Formula (V) by reaction with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
 wherein R4 is selected from halogen, OH, Ci-CT-alkoxy or O-C(O)-R'-.
Embodiment-002: The present invention also provides a method for preparing a compound of Formula (II), or its N-oxides or salts thereof,
wherein R4 is selected from halogen, OH, Ci-CT-alkoxy or O-C(O)-R'-.
Embodiment-002: The present invention also provides a method for preparing a compound of Formula (II), or its N-oxides or salts thereof,
Formula (II) wherein, R1 is selected from -CHF2, -CF2-CI or CF3; and
R5 is selected from fluoro, chloro, bromo, or iodo, comprising the steps of: a. reacting a compound of Formula (VI) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),

Formula (VI) HO'N Formula (V) b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
 wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; and c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; and c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),

Formula (III) Formula (II)
Embodiment-003: The present invention further relates to a method for preparing a compound of
Formula (I), or its N-oxides or salts thereof,

Formula (I) wherein,
R1 is selected from -CHF2, -CF2-CI or CF3;
L is selected from O, NR3 or S;
R2 is selected from hydrogen, halogen, C i-CT-alkyl, C i-CT-haloalkyl, Ci-CT-alkoxy or C3-C6- cycloalkyl;
R3 is selected from hydrogen or C i-CT-alkyl; and n is an integer selected from 1-2, comprising the steps of: a. reacting the compound of Formula (VI) with hydroxylamine or its salt in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
 b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,

HO' Formula (V) Formula (III) wherein R4 is selected from halogen, OH, Ci-CT-alkoxy or O-C(O)-R'; - c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
Formula (III) Formula (II) wherein R5 is fluoro, chloro, bromo or iodo; and d. alkylating a compound of Formula (IX) with the compound of Formula (II) in the presence of a base, and optionally in the presence of a catalyst and a solvent, to obtain a compound of Formula (I),

Embodiment-004: The present invention still further provides a method for the synthesis of a compound of Formula (VI), comprising the steps of: i. acetylating a compound of Formula (VIII) with acetyl chloride or acetic anhydride in the presence of a suitable catalyst and optionally in the presence of a solvent to obtain a compound of Formula (VII),

Formula (VIII) Formula (VII) . and ii. reacting the compound of Formula (VII) with a cyanide source in a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (VI),
Formula (VII) Formula (VI)
Embodiment-005: In yet another aspect, the present invention further relates to a method of preparing a compound of Formula (III), or its salts or N-oxides thereof, wherein the said method comprises the step (step-b) of cyclizing the dioxime compound of Formula (V) with a
compound of Formula (IV) in a solvent and optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
 wherein, R1 is selected from -CHF2, -CF2-CI or CF3 and
wherein, R1 is selected from -CHF2, -CF2-CI or CF3 and
R4 is selected from halogen, OH, C i-Ce-alkoxy or O-C(O)-R
Embodiment-006: In yet another aspect, the present invention provides a method for a compound of Formula (III), or its N-oxides or salts thereof,
Formula (III) wherein R1 is selected from -CHF2, -CF2-CI or CF3, comprising the steps of:
A. preparing a compound of formula (VI) by i. acetylating a compound of Formula (VIII) with acetyl chloride or acetic anhydride in the presence of a suitable catalyst and optionally in the presence of a solvent to obtain a compound of Formula (VII),

Formula (VIII) Formula (VII) . ii. reacting the compound of Formula (VII) with a cyanide source in a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (VI),
Formula (VII) Formula (VI)
a. reacting the compound of Formula (VI) obtained in step (A) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
 and b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
and b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
 wherein R4 is selected from halogen, -OH, Ci-Ce-alkoxy or O-C(O)-R'.
wherein R4 is selected from halogen, -OH, Ci-Ce-alkoxy or O-C(O)-R'.
Embodiment-007: In yet another aspect, the present invention also relates to a method for preparing a compound of Formula (II), or its N-oxides or salts thereof,
Formula (II) wherein,
R1 is selected from -CHF2, -CF2-CI or CF3; and
R5 is selected from fluoro, chloro, bromo, or iodo, comprising the steps of:
A. preparing a compound of formula (VI) by i. acetylating a compound of Formula (VIII) with acetyl chloride or acetic anhydride in the presence of a suitable catalyst and optionally in the presence of a solvent to obtain a compound of Formula (VII),

Formula (VIII) Formula (VII) ii. reacting the compound of Formula (VII) with a cyanide source in a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (VI),
Formula (VI)
Formula (VII) a. reacting the compound of Formula (VI) obtained in step (A) with hydroxylamine or its salt in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
 b. cyclizing the dioxime compound of Formula (V) with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
b. cyclizing the dioxime compound of Formula (V) with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
 wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; and c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; and c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),

Formula (III) Formula (II)
Embodiment-008: In yet another aspect, the present invention also relates to a method of preparing a compound of Formula (I), or its N-oxides or salts thereof,
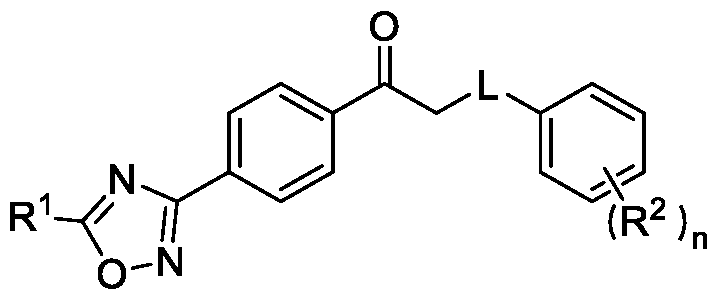
Formula (I) wherein,
R1 is selected from -CHF2, -CF2-CI or CF3;
L is selected from O, NR3 or S;
R2 is selected from hydrogen, halogen, C i-Ce-alkyl, Ci-Ce-haloalkyl, Ci-Ce-alkoxy or C3-C6- cycloalkyl; and
R3 is selected from hydrogen or C i-Ce-alkyl; and n is an integer selected from 1-2, comprising the steps of:
A. preparing a compound of formula (VI) by i. acetylating a compound of Formula (VIII) with acetyl chloride or acetic anhydride in the presence of a suitable catalyst and optionally in the presence of a solvent to obtain a compound of Formula (VII),
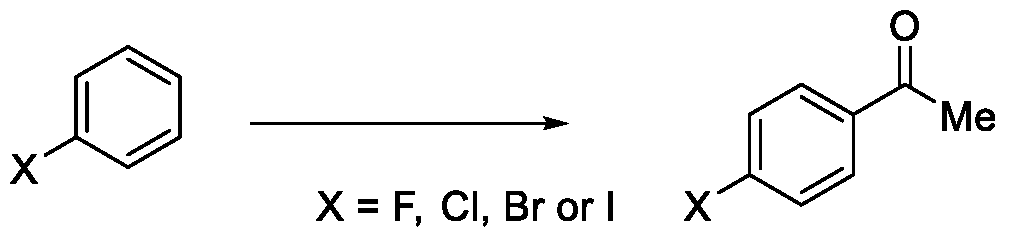 , ,
, ,
Formula (VIII) Formula (VII) ii. reacting the compound of Formula (VII) with a cyanide source in a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (VI),

Formula (VI)
Formula (VII) a. reacting the compound of Formula (VI) obtained in step (A) with hydroxylamine or its salt in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
 b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
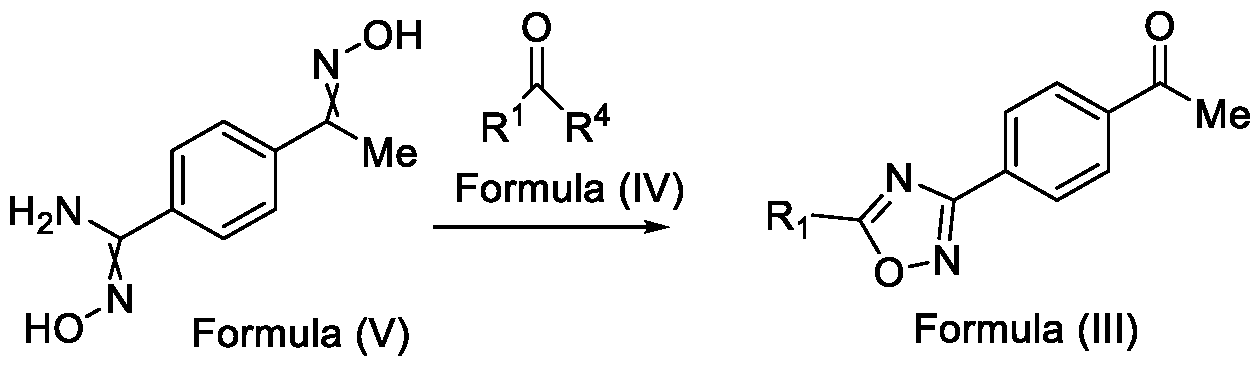 wherein R4 is selected from halogen, -OH, Ci-Ce-alkoxy or O-C(O)-R'; c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
wherein R4 is selected from halogen, -OH, Ci-Ce-alkoxy or O-C(O)-R'; c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
Formula (III) Formula (II) wherein R5 is fluoro, chloro, bromo or iodo; and d. alkylating a compound of Formula (IX) with the compound of Formula (II) in the presence of a base and, optionally in the presence of a catalyst and a solvent, to obtain a compound of Formula (I),
Embodiment-009: The methods of the present invention according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008 for preparing the compound of Formula (I), Formula (II), Formula (III), or their salts or N-oxides as described above, involve a novel and inventive step, wherein the dioxime intermediate of Formula (V) undergoes a simultaneous cyclization of the
/V-hydroxyimidamidc moiety and deprotection of ketoxime, to obtain the compound of Formula (III) in a single step.
Embodiment-010: The methods of the present invention according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008 for preparing the compound of Formula (I), Formula (II), Formula (III), their salts or N-oxides thereof as described above, can be obtained in fewer reaction steps by eliminating the reaction steps required for the separate protection and deprotection of the carbonyl group of the intermediates to obtain the final product. As a consequence, the present method(s) eliminate(s) the use of additional reactants, reagents and solvents required for performing said conversions and also, eventually avoid(s) the isolation or purification steps of the compounds formed during said conversions. Altogether, it enables the method(s) of the present invention for obtaining the compound of Formula (I) and its intermediate compounds of Formula (II) and Formula (III), in a simple, short, environmentally friendly and commercially viable method over the methods already known in the prior art.
Embodiment-011: Further, the method(s) of the present invention is more atom economic and provides the product and the key intermediates in a high isolated yield.
Embodiment-012: The methods of the present invention according to embodiments 001 , 002, 003 and embodiments 005, 006, 007, 008, wherein, if ethyl trifluoracetate (R1 = CF3 and R4 is COOEt in Formula (IV)) is used for the cyclization purpose to obtain the oxadiazole ring, then ethyl trifluoroacetate or its trans-esterified product i.e., methyl trifluoroacetate is obtained as a side-product, which can be recovered and re-used for the cyclization step and as a result, the present methods are significantly economic for commercial scale preparation, and also reduces the quantity of effluent generated during the process to minimize the environmental pollution.
Embodiment-013: The methods of the present invention according to embodiment 003 and embodiment 008, wherein for the compound of Formula (I), R1 may be selected from CF3 or CF2CI, preferably CF3; L may be selected from O, NH or S; R2 may be selected from hydrogen, halogen, C1-C3 alkyl or C1-C3 alkoxy; and n is 1-2.
Embodiment-014: The methods of the present invention according to embodiment-013, wherein for the compound of Formula (I), R1 is selected from CF3 or CF2CI; L is selected from O, NH or S; R2 is selected from hydrogen, fluoro, chloro, bromo, methyl or methoxy; and n is 1-2, particularly n = 1.
Embodiment-015: The methods of the present invention according to embodiment-014, wherein for the compound of Formula (I), R1 is CF3, L is O and n = 1, R2 is selected from fluoro (F), chloro (Cl), bromo (Br), methyl (Me) or methoxy (OMe); preferably 2-F, 3-F, 4-F, 2-C1, 3-C1, 4-C1, 2-Br, 3-Br, 4-Br, 2-Me, 3-Me, 4-Me, 2-OMe, 3-OMe, 4-OMe, 2,4-di-fluoro, or 3,4-di- fluoro.
Embodiment-016: The methods of the present invention according to embodiment-014, wherein for the compound of Formula (I), R1 is CF3, L is NH and n = 1-2, R2 is selected from fluoro, chloro, bromo, methyl or methoxy; preferably 2-F, 3-F, 4-F, 2-C1, 3-C1, 4-C1, 2-Br, 3-Br, 4-Br,
2-Me, 3-Me, 4-Me, 2-OMe, 3-OMe, 4-OMe, 2,4-di-fluoro, or 3,4-di-fluoro.
Embodiment-017: The methods of the present invention according to embodiment-014, wherein for the compound of Formula (I), R1 is CF3, L is S and n = 1, R2 is selected from fluoro, chloro, bromo, methyl or methoxy; preferably 2-F, 3-F, 4-F, 2-C1, 3-C1, 4-C1, 2-Br, 3-Br, 4-Br, 2-Me,
3-Me, 4-Me, 2-OMe, 3-OMe, 4-OMe, 2,4-di-fluoro, or 3,4-di-fluoro.
Embodiment-018: The methods of the present invention according to embodiments 002-003 and embodiments 007-008, wherein for the compound of Formula (II), R5 may be selected from chloro or bromo and R1 may be selected from CF3 or CF2CI, preferably CF3.
Embodiment-019: The methods of the present invention according to embodiments 001-003 and 005-008, wherein for the compound of Formula (III), R1 may be chosen from CF3 or CF2CI, preferably CF3.
Embodiment-020: The methods of the present invention according to embodiments 001, 002, 003 and 005, 006, 007, 008, wherein for the compound of Formula (IV), R1 may be selected from CF3 or CF2CI, preferably CF3, and R4 may be selected from Cl, Br, C1-C3 alkoxy or O-C(O)- CF3.
Embodiment-021: The methods of the present invention according to embodiment 020, wherein for the compound of Formula (IV), R1 is CF3 and R4 is OMe, OEt, or O-C(O)-CF3.
Embodiment-022: The methods of the present invention according to embodiment 021, wherein for the compound of Formula (IV), R1 is CF3 and R4 is OEt or O-C(O)-CF3.
Embodiment-023: The methods of the present invention according to embodiment 003 and embodiment 008, wherein for the compound of Formula (IX), L may be selected from O, NH or S; R2 may be selected from hydrogen, halogen, C1-C3 alkyl or C1-C3 alkoxy; and n is 1-2.
Embodiment-024: The methods of the present invention according to embodiment-023, wherein for the compound of Formula (IX), L is selected from O, NH or S; R2 is selected from hydrogen, fluoro, chloro, bromo, methyl or methoxy; and n is 1-2, preferably n = 1.
Embodiment-025: The methods of the present invention according to embodiment-024, wherein for the compound of Formula (IX), L is O and n = 1 , R2 is selected from fluoro, chloro or bromo, preferably 2-F, 4-F, 3-C1, 4-C1, 2-Br or 4-Br.
Embodiment-026: The methods of the present invention according to embodiment-024, wherein for the compound of Formula (IX), L is NH and n = 1-2, R2 is selected from hydrogen, fluoro, chloro, methyl or methoxy, preferably (when R2 is mono substitution) hydrogen, 2-Me, 4-Me, 2-F, 3-F or 4-F and (when R2 is di substitution) 2-Me & 4-C1, or 2,4- di-Fluoro.
Embodiment-027: The methods of the present invention according to embodiment-024, wherein for the compound of Formula (IX), L is S and n = 1, R2 is selected from hydrogen, fluoro, or chloro, preferably hydrogen, 2-F, 3-F, 2-C1 or 4-C1.
Embodiment-028: The methods of the present invention according to embodiments 001 , 002, 003, 004, 005, 006, 007, and 008, wherein the solvent can be selected from aliphatic or aromatic hydrocarbon, halogenated hydrocarbon, ethers, cyclic ethers, cyclic esters, cyclic carbonate ester, nitro based solvents, nitriles, amides, ketones, acids, alcohols, organosulfur, ionic liquids, water or mixture thereof.
Embodiment-029: The methods of the present invention according to embodiment-028, wherein the solvent can be selected from but not limited to acetonitrile, acetic acid, methanol, ethanol, iso-propanol, butanol, acetone, pentane, hexane, heptane, octane, nonane, decane, dodecane, cycloalkanes: cyclopropane, cyclobutane, cyclopentane, cyclohexane, cycloheptane, cyclooctane; dichloromethane, dichloroethane (ethylene dichloride), chloroform, ethyl acetate, iso-propyl acetate, toluene, xylene, mesitylene, benzene, nitro benzene, nitro methane, diethyl ether, diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, monoglyme, diglyme, methoxy-methane, methoxy-ethane ethoxy-ethane, dimethoxyethane, di-ethoxyethane, /V./V-dimcthylformaniidc, dimethyl sulfoxide, sulfolane, carbondisulfide, A-methyl-2-pyrrolidone, propylene carbonate, 1, 3-dimethyl-3, 4,5,6- tetrahydro-2(lH)-pyrimidinone, hexamethylphosphortriamide, 1 ,3-dimethyl-2- imidazolidinoneor of combinations thereof.
Embodiment-030: The methods of the present invention according to embodiments 001 , 002, 003, 004, 005, 006, 007, and 008, wherein the base, if used, can be selected from an inorganic or organic base such as alkali metal hydrogen carbonate, alkali/alkaline earth metal carbonate, alkali/alkaline earth metal hydroxide, alkali metal phosphate, alkali metal hydride, alkali metal alkoxide, ethylamine, triethylamine, isopropylamine diisopropylamine, triisopropylamine, pyridine, picoline, piperidine, methylmorpholine, /V-mcthylpipcridinc N,N- (dimethylamino)pyridine (DMAP), lutidine, collidine, tetramethylammonium hydroxide, tetrabutylammonium hydroxide, choline hydroxide, l,5,7-triazabicyclo[4.4.0]dec-5-ene, (TBD), 2,3,4,6,7,8,9,10-octahydropyrimidol[l,2-a]azepine (DBU) 1,5- diazabicyclo[4.3.0]non-5-ene (DBN), l,4-diazabicyclo[2.2.2]octane (DABCO), or triethylenediamine.
Embodiment-031: The methods of the present invention according to embodiments 001, 002, 003, 005, 006, 007, and 008, wherein the reagent hydroxylamine used in step-a can be either in an aqueous form or in a solid form such as a salt form.
Embodiment-032: The methods of the present invention according to embodiment-031, wherein the salt form can be selected from halide (HX), carboxylate, acetate, trifluoroacetate, sulfonate, trifluoromethanesulfonate, sulfate, preferably it is a hydrochloride salt.
Embodiment-033: The methods of the present invention according to embodiment-031, wherein the reagent hydroxylamine is 50% aqueous hydroxylamine.
Embodiment-034: The methods of the present invention according to embodiment-031, wherein the reagent hydroxylamine is hydroxylamine hydrochloride salt. Hydroxylamine hydrochloride is soluble in polar organic solvents and is more stable to oxidation and is easy to handle on large quantity as compared to free hydroxylamine or aqueous hydroxylamine, and as a result, step-c of the present method was carried out using hydroxylamine hydrochloride. When hydroxylamine hydrochloride was used, step-a, was carried out in the presence of a base to neutralize the hydrochloride salt.
Embodiment-035: The methods of the present invention according to embodiment-034, wherein the base can be selected from alkali metal hydrogen carbonate, alkaline earth metal hydrogen carbonate, alkali metal carbonate, alkaline earth metal carbonate, triethylamine, diisopropylamine, triisopropylamine, pyridine, or picoline.
Embodiment-036: The methods of the present invention according to embodiment-035, wherein the base is selected from sodium carbonate, sodium bicarbonate, potassium carbonate or triethylamine.
Embodiment-037: The methods of the present invention according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein step-a was performed in a suitable solvent selected from but not limited to alcohol such as ethanol, methanol, isopropanol, diethyl ether, /V,/V-di methyl f rm am ide, tetrahydrofuran or water or a mixture thereof.
Embodiment-038: The methods of the present invention according to embodiment-037, wherein the solvent can be selected from ethanol, methanol, isopropanol or water or a mixture thereof.
Embodiment-039: The methods of the present invention according to embodiments 001 , 002, 003, 005, 006, 007 and 008, wherein in step-a: i. the solvent is selected from ethanol, methanol, isopropanol, diethyl ether, N,N- dimethylformamide, tetrahydrofuran or water or a mixture thereof; and ii. the base if used, is selected from alkali metal hydrogen carbonate or alkaline earth metal hydrogen carbonate, alkali metal carbonate or alkaline earth metal carbonate, triethylamine, diisopropylamine, triisopropylamine, pyridine, or picoline.
Embodiment-040: The methods of the present invention according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein step-a was performed at a temperature ranging from 0 °C to 80 °C, and particularly from 0 °C to 60 °C.
Embodiment-041: The step-b of the present methods according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein the dioxime compound of Formula (V) was reacted initially with the compound of Formula (IV) without adding any base, depending on the reactivity of the reactants used. Once the reactants are consumed, the reaction mixture is
subjected to acidic treatment under heating condition to obtain the compound of Formula (III) in a single step.
Embodiment-042: The step-b of the present methods according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein the dioxime compound of Formula (V) was reacted initially with the compound of Formula (IV) in the presence of a base, depending on the reactivity of the reactants used. Once the reactants are consumed, the reaction mixture is subjected to acidic treatment under heating condition to obtain the compound of Formula (III) in a single step.
Embodiment-043: The methods of the present invention according to embodiment-042, wherein the base is selected from but not limited to an alkali metal alkoxide or alkaline earth metal alkoxide.
Embodiment-044: The methods of the present invention according to embodiment-043, wherein the base is selected from sodium ethoxide or sodium methoxide.
Embodiment-045: The methods of the present invention according to embodiments-041 and 042, wherein the acid used for the acidic treatment, can be selected from but not limited to sulphuric acid, hydrochloric acid, or hydrobromic acid.
Embodiment-046: The methods of the present invention according to embodiment-045, wherein the acid is selected from hydrochloric acid and particularly from an aqueous hydrochloric acid.
Embodiment-047: The step-b of the present methods according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein the solvent includes alcohol, tetrahydrofuran, 2- methyltetrahydrofuran, acetonitrile, or V,V-dimethylformamide or water.
Embodiment-048: The methods of the present invention according to embodiment-047, wherein the solvent is selected from methanol, ethanol, isopropanol, tetrahydrofuran or water and particularly, it is selected from methanol, ethanol or tetrahydrofuran.
Embodiment-049: The methods of the present invention according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein in step-b: i. the solvent is selected from methanol, ethanol, isopropanol, tetrahydrofuran, 2- methyltetrahydrofuran, acetonitrile, V,V-dimethylformamide or water; ii. the base if used, is selected from alkali metal alkoxide or alkaline earth metal alkoxide; iii. the acid is selected from sulphuric acid, hydrochloric acid, or hydrobromic acid.
Embodiment-050: The step-b of the present methods according to embodiments 001, 002, 003 and embodiments 005, 006, 007, 008, wherein the reaction temperature is between 0 °C-120 °C, and particularly, it is between 0 °C-40 °C during the cyclization and 70 °C -100 °C during the acidic treatment to obtain the compound of Formula (III).
Embodiment-051: The step-c of the present methods according to embodiments 002, 003 and 007, 008, wherein the halogenating agent can be selected from but not limited to F2, CI2, Br2, 12, B
in acetic acid, /V-bromosuccinimidc, copper bromide (CuB ). HBr, NaBr, NtBBr, phosphorus tribromide (PBi ;). phosphorus pentabromide (PBrs), a mixture of NJBBr and potassium peroxymonosulfate sulfate, Trimethylphenylammonium tribromide, N-chlorosuccinimide, phosphorus trichloride (PCh), phosphorus pentachloride (PCI5), sulfonyl chloride (SOCI2), sulfuryl chloride (SO2CI2), selenium oxychloride, benzyltrimethylammonium dichloroiodate, or trichloroisocyanuric acid.
Embodiment-052: The methods of the present invention according to embodiment-051, wherein the halogenating agent can be selected from B , /V-bromosuccinimidc, CI2, or N- chlorosuccinimide .
Embodiment-053: The methods of the present invention according to embodiment-052, wherein the halogenating agent is selected from bromine or chlorine and more particularly from bromine.
Embodiment-054: The methods of the present invention according to embodiments-002, 003 and 007, 008, wherein the halogenating agent can be used in the presence of a catalyst selected from but not limited to p-toluenesulfonic acid, aluminium chloride (AlCh), monopotassium phosphate (K2HSO4), sulphuric acid, silica, amberlyst 15, or trimethylsilyl triflate.
Embodiment-055: The step-c of the present methods according to embodiments 002, 003 and 007, 008, wherein the solvent can be selected from but not limited to dichloromethane, dichloroethane (ethylene dichloride), chloroform, acetonitrile, diethyl ether, methyl tertiary- butyl ether, tetrahydrofuran, 2-methyl-tetrahydrofuran, ethyl acetate, alcoholic solvent such as methanol, ethanol or isopropanol, acetic acid or water or a mixture thereof.
Embodiment-056: The methods of the present invention according to embodiment-055, wherein the solvent can be selected from dichloromethane, dichloroethane (ethylene dichloride), chloroform, alcoholic solvent selected from methanol, ethanol or isopropanol, or methyl tertiary-butyl ether.
Embodiment-057: The step-c of the present methods according to embodiments 002, 003 and 007, 008, wherein in step-c: i. the halogenating agent is selected from fluorine (F2), chlorine (CI2), bromine (B ). iodine (I2), bromine in acetic acid, N-bromosuccinimide, copper bromide (CuBr2), HBr, NaBr, NfhBr, phosphorus tribromide (PBn). phosphorus pentabromide (PBrs), a mixture of NfhBr and potassium peroxymonosulfate sulfate, trimethylphenylammonium tribromide, N-chlorosuccinimide, phosphorus trichloride (PCI3), phosphorus pentachloride (PCI5), sulfonyl chloride (SOCI2), sulfuryl chloride (SO2CI2), selenium oxychloride, benzyltrimethylammonium dichloroiodate, or trichloroisocyanuric acid; ii. the solvent is selected from dichloromethane, dichloroethane (ethylene dichloride), chloroform, acetonitrile, diethyl ether, methyl tertiary-butyl ether, tetrahydrofuran, 2-
methyl-tetrahydrofuran, ethyl acetate, alcoholic solvent selected from methanol, ethanol, or isopropanol, acetic acid or water or a mixture thereof; iii. the catalyst if used, is selected from p-toluenesulfonic acid, aluminium chloride (AlCh), monopotassium phosphate (K2HSO4), sulphuric acid, silica, amberlyst 15, or trimethylsilyl triflate.
Embodiment-058: The step-c of the present methods according to embodiments 002, 003 and 007, 008, wherein when the compound of formula (III) is subjected to halogenation using bromine or chlorine and in the presence of an alcoholic solvent such as methanol, ethanol or isopropanol followed by ketal de -protection in the presence of an acid to obtain a compound of formula (Ila), as shown below
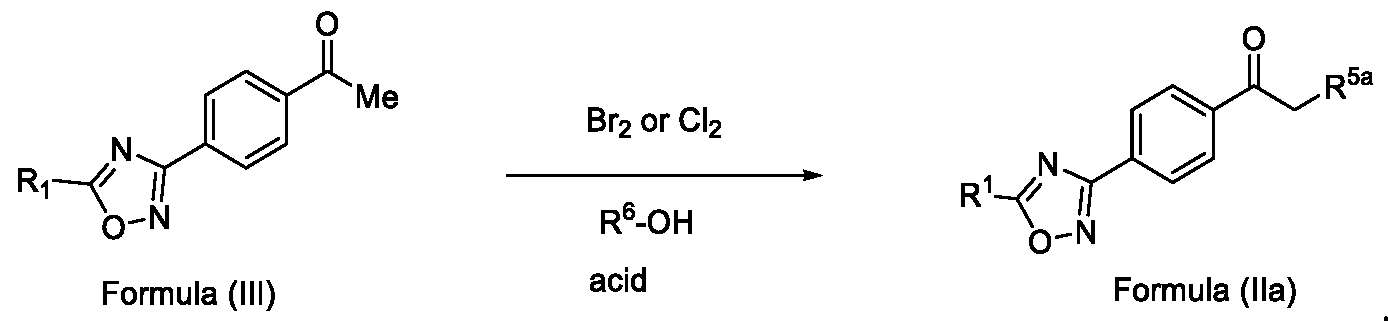 wherein,
wherein,
R1 is CHF2, -CF2-CI or CF3;
R5a is selected from bromo or chloro; and
R6 is selected from methyl, ethyl, n-propyl or isopropyl,
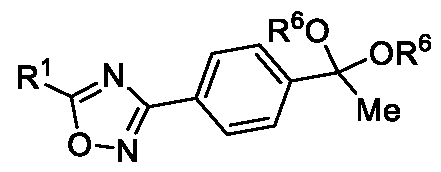 wherein the intermediate compound of formula (X) Formula (X) or formu)a (XI)
wherein the intermediate compound of formula (X) Formula (X) or formu)a (XI)
Formula (XI) is/are optionally isolated.
Embodiment-059: The methods of the present invention according to embodiment-058, wherein the acid used for ketal deprotection is selected from hydrochloric acid.
Embodiment-060: The step-c of the present methods according to embodiments 002, 003 and 007, 008, wherein the halogenation reaction can be performed at a temperature between 25 °C to 50 °C, particularly between 40 °C to 50 °C.
Embodiment-061: The step-d of the present methods according to embodiments 003 and 008, wherein the base used for the alkylation reaction can be selected from but not limited to sodium carbonate, sodium bicarbonate, potassium carbonate, potassium bicarbonate, 1,5,7-
triazabicyclo[4.4.0]dec-5-ene, pyridine, picoline or l,4-diazabicyclo[2.2.2]octane. Particularly, it can be selected from sodium carbonate, sodium bicarbonate, potassium carbonate or potassium bicarbonate.
Embodiment-062: The step-d of the present methods according to embodiments 003 and 008, wherein the alkylation can also be carried out in the presence of a catalyst such as sodium iodide, potassium iodide, l,4-diazabicyclo[2.2.2]octane (DABCO) or tetra alkylammonium iodide.
Embodiment-063: The step-d of the present methods according to embodiments 003 and 008, can be carried out with or without a solvent.
Embodiment-064: The step-d of the present methods according to embodiments 003 and 008, can be carried out under neat condition without using a solvent.
Embodiment-065: The step-d of the present methods according to embodiments 003 and 008, can be carried out in the presence of a solvent.
Embodiment-066: The step-d of the present methods according to embodiment-003, embodiment -008 and embodiment-065, wherein the solvent can be selected from but not limited to acetone, acetonitrile, diethyl ether, methyl ethyl ketone, methanol, ethanol, 1,4-dioxane, dichloromethane, toluene, cyclopentane, cyclohexane, /V,/V-dinicthylfornianiidc (DMF) or water or a mixture thereof; particularly the solvent is selected from acetone, acetonitrile, toluene, cyclohexane, ethanol or methanol.
Embodiment-067: The step-d of the present methods according to embodiments 003 and 008, wherein in the step-d: i. the base is selected from sodium carbonate, sodium bicarbonate, potassium carbonate, potassium bicarbonate, l,5,7-triazabicyclo[4.4.0]dec-5-ene, pyridine, picoline or 1,4- diazabicyclo [2.2.2] octane ; ii. the solvent if used, is selected from acetone, acetonitrile, diethyl ether, methyl ethyl ketone, methanol, ethanol, 1,4-dioxane, dichloromethane, dichlorethane, chlorobenzene, toluene, cyclopentane, cyclohexane, /V,/V-dinicthylfornianiidc (DMF) or water or a mixture thereof; and iii. the catalyst if used, is selected from sodium iodide, potassium iodide, 1,4- diazabicyclo[2.2.2]octane (DABCO) or tetra alkylammonium bromide, tetra alkylammonium iodide.
Embodiment-068: The step-d of the present methods according to embodiments 003 and 008, wherein the alkylation was carried out at a temperature ranging from 25 °C to 160 °C, particularly from 25 °C to 40°C.
Embodiment-069: The step-i of the present methods according to embodiment 004 and embodiments 006, 007, 008, wherein the catalyst can be selected from but not limited to
aluminium chloride (AlCh), iron chloride (FeCh), copper bromide (CuBr), or a metal triflate and preferably it is selected from aluminium chloride.
Embodiment-070: The step-i of the present methods according to embodiment 004 and embodiments 006, 007, 008, wherein the acetylation can be carried out in the presence or absence of a solvent.
Embodiment-071: The step-i of the present methods according to embodiment 004 and embodiments 006, 007, 008, wherein the acetylation can be performed in the absence of a solvent i.e., neat condition.
Embodiment-072: The step-i of the present methods according to embodiment 004 and embodiments 006, 007, 008, wherein the acetylation can be performed in the presence of a solvent.
Embodiment-073: The step-i of the present methods according to embodiment 004 and embodiments 006-008 and embodiment-072, wherein the solvent used for acetylation can be chosen from but not limited to dichloromethane, dichloroethane (ethylene dichloride), 1,4- dioxane, toluene or benzene.
Embodiment-074: The step-i of the present methods according to embodiment 004 and embodiments 006, 007, 008, wherein the reaction is carried out at a temperature ranging from 0 °C to 100 °C and particularly from 0 °C to 30 °C.
Embodiment-075: The step-ii of the present methods according to embodiment 004 and embodiments 006, 007, 008, wherein halo to cyano conversion was performed using the cyanide source selected from an alkali metal cyanide and the catalyst selected from a phase transfer catalyst, in a solvent at a temperature ranging from 25 °C to 150 °C.
Embodiment-076: The methods of the present invention according to embodiment-075, wherein the alkali metal cyanide is sodium cyanide or potassium cyanide.
Embodiment-077: The methods of the present invention according to embodiment-075, wherein the phase transfer catalyst is tetraalkylammonium salt, tetraalkylphosphonium salt or crown ether.
Embodiment-078: The methods of the present invention according to embodiment-077, wherein the phase transfer catalyst can be selected from tetraalkylammonium halide and particularly, tetrabutylammonium bromide (Bu4N+Br ) tetrabutylammonium chloride (BU4N+C1 ), tetraethylammonium chloride (Et4N+Cl ), tetraethylammonium bromide (Et4N+Br ). tetramethylammonium chloride (Me4N+Cl ).
Embodiment-079: The step-ii of the present methods according to embodiment- 075, wherein when X=F for the compound of Formula VII, then step-ii was carried out with alkali metal cyanide selected from sodium cyanide and a phase transfer catalyst selected from tetrabutylammonium bromide in a suitable solvent at a temperature ranging from 25 °C -150 °C, to obtain the desired compound of Formula (VI) in high yield, without using a transition
metal catalyst and/or a base or an additive/ligand such as alkyl substituted ethylenediamine. As a consequence, the present methods according to embodiment 004 and embodiments 006-008 are convenient, minimize operational difficulties and cost effective over known methods.
Embodiment-080: The step-ii of the present methods according to embodiment 004 and embodiments 006-008, can be performed in the presence of a metal catalyst, wherein the cyanide source may be selected from an alkali metal cyanide or alkali metal hexacyanoferrate (II) or its hydrates, preferably from sodium cyanide or potassium cyanide, sodium hexacyanoferrate (II) or potassium hexacyanoferrate (II) or their hydrates.
Embodiment-081: The step-ii of the present methods according to embodiment 080, wherein the metal catalyst may be selected from copper or copper (I) salt in the presence of a suitable ligand, wherein the copper (I) salt, can be selected from CuCl, CuBr or Cui, and the suitable ligand includes but not limited to dialkylethylenediamine, tetr alkylethylenediamine, 1,10- phenanthroline, or bipyridine. Particularly, the ligand may be chosen from 1,10-phenanthroline, bipyridine, bis(l,2-diphenylphosphino)ethane, triarylphosphine, triphenylphosphine, ethylenediamine, A'. ' Alimcthylcthylciicdiamiiic, N, N, N’, A' -tctiamcthylcthylcncdiaminc. An additive such as potassium iodide or tetraalkylammonium halide may also be used.
Embodiment-082: The step-ii of the present methods according to embodiment 080, wherein the metal catalyst may be selected from palladium source but not limited to Pd-nanoparticles, Pd on activated charcoal, 10% Pd/C or 5% Pd/C.
Embodiment-083: The cyanation reaction mentioned in step-ii of the present methods according to embodiment 004, embodiments 006 to 008 and embodiments 080 to 082, may be performed in the presence of one or more optional base selected from alkali metal carbonate or alkaline earth metal carbonate, organic amine, triethylamine or a mixture thereof, preferably the base is selected from sodium carbonate, potassium carbonate, triethylamine or a mixture thereof.
Embodiment-084: The step-ii of the present methods according to embodiment 004 and embodiments 006 to 008 and embodiment-080, wherein when X = Br for the compound of Formula (VII), it may be carried out with potassium ferrocyanide trihydrate (K4[Fe(CN)6]. 3H2O) in the presence of Cui, and a ligand such as N, V ’-dimethylethylenediamine in a suitable solvent; or it may be carried out with potassium ferrocyanide trihydrate (K4[Fe(CN)6].3H2O) in the presence of 10% Pd/C and a mixture of a base selected from sodium carbonate and triethylamine, in a suitable solvent.
Embodiment-085: The step-ii of the present methods according to embodiment 004, embodiments 006 to 008, embodiment-075, and embodiment-080, wherein the solvent can be selected from polar solvents but not limited to water, alcohol, acetonitrile, dimethylformamide, dimethylacetamide, N-methylpyrrolidinone, dimethylsulfoxide or a mixture thereof, and particularly, the solvent is dimethylformamide, N-methylpyrrolidinone, n-butanol or a mixture of n-butanol and A-methylpyrrolidinone.
Embodiment-086: The step-ii of the present methods according to embodiment 004, embodiments 006 to 008, embodiment-075, and embodiment-080, wherein the temperature can vary between the range of 25 °C to 150 °C and particularly, it is in the range of 25 °C to 40 °C during the addition of cyanide and then increased to a temperature in the range of 120 °C to 150 °C depending on the solvent used in step-ii.
Embodiment-087: The methods of the present invention according to embodiments 001 to 008, the reaction time is not critical and depends on the batch size, temperature, type of reaction, solvent and the used reagents and is usually between few minutes to few hours.
Embodiment-088: The present methods for preparing the compound of Formula (I) or its intermediate compound of Formula (II), Formula (III) or Formula (VI) as disclosed herein, wherein the steps can be carried out in a batch, semi-continuous or continuous reaction mode, specifically also under semi-continuous flow or continuous flow reaction conditions.
Embodiment-089: The present invention also provides an intermediate compound of formula (X),

Formula (X) •> wherein R1 is CHF2, -CF2-CI or CF3 and
R6 is selected from methyl, ethyl, n-propyl or isopropyl, preferably methyl.
Embodiment-090: The present invention further provides an intermediate compound of formula (XI),
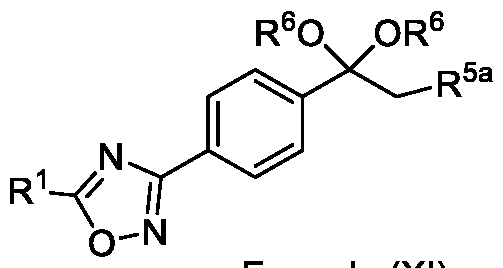
Formula (XI)
9
Wherein, R1 is CHF2, -CF2-CI or CF3;
R5a is selected from chloro or bromo: and
R6 is selected from methyl, ethyl, n-propyl or isopropyl, preferably methyl.
The person skilled in the art knows the best work-up of the reaction mixtures after the completion of the respective reactions. The work-up is usually carried out by isolation of the product, and optionally washing with solvent, and further optionally drying of the product if useful or required.
The isolation of the reaction product can be carried out by a technique which includes but is not limited to decantation, filtration, centrifugation, evaporation, liquid-liquid extraction, distillation, recrystallization, chromatography and the like or a combination thereof.
The reaction steps according to the invention are generally carried out under atmospheric pressure. Alternatively, however, it is also possible to work the method steps of the present method(s) under reduced pressure or higher pressure.
The invention is further illustrated by the following examples which are provided to be exemplary of the invention, and do not limit the scope of the invention. While the present invention has been described in terms of its specific embodiments, certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present invention.
Although the subject matter has been described in considerable detail with reference to certain examples and implementations thereof, other implementations are possible.
EXAMPLES
The disclosure will now be illustrated with the working examples, which is intended to illustrate the working of disclosure and not intended to take restrictively to imply any limitations on the scope of the present disclosure. Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood to one ordinary person skilled in the art to which this disclosure belongs. Although methods and materials similar or equivalent to those described herein can be used in the practice of the disclosed methods and compositions, the exemplary methods, devices and materials are described herein. It is to be understood that this disclosure is not limited to particular methods, and experimental conditions described, as such methods and conditions may apply.
The present invention for preparing a compound of Formula (I) and its intermediate compound of Formula (III) can be illustrated with the representative scheme 1 as provided below:
Scheme:
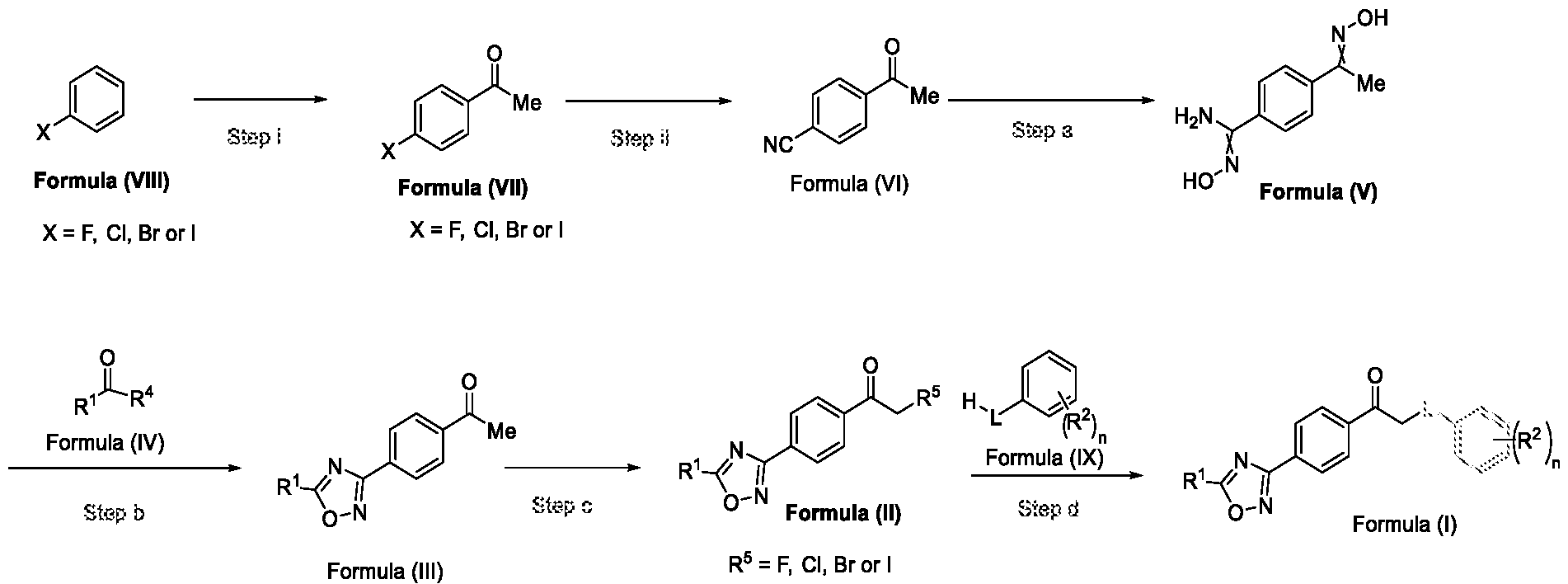
Scheme 1 : A process for preparing a compound of Formula (I) and its intermediate compounds of Formula (II) and Formula (III)
Experimental procedure
Step i: Synthesis of l-(4-fluorophenyl)ethan-l-one
Method-1:
To a solution of fluorobenzene (10 g, 104 mmol) in ethylene dichloride (EDC) (50 mL), aluminium chloride (16.32 g, 122 mmol) was added at 25 to 30 °C under nitrogen atmosphere. The reaction mass was cooled between 0 to 5 °C, and then acetyl chloride (8 mL, 112 mmol) was added in a dropwise manner. The reaction mixture was allowed to stir for 12 hours at 25 to 30 °C. After completion of the reaction, the reaction mixture was quenched by the addition of a saturated solution of sodium bicarbonate solution (250 mL). The reaction mixture was then extracted with ethylene dichloride (2 x 50 mL), the combined organic layer was washed with brine (50 mL) and concentrated under reduced pressure to afford l-(4-fluorophenyl)ethan-l-one (14.25 g, 99.14 % yield) as a colorless liquid.
'H-NMR (400 MHz, CDC13) 5 7.94-7.89 (m, 2H), 7.08-7.02 (m, 2H), 2.51 (s, 3H); GC-MS: m/z 138.1.
Method-2: Neat condition (in the absence of solvent)
To fluorobenzene (50 g, 510 mmol), aluminium chloride (76 g, 561 mmol) was added at a temperature between 0 andlO °C under nitrogen atmosphere. To this reaction mixture, acetyl chloride (61.3 g, 765 mmol) was added slowly in a drop wise manner. The reaction mixture was stirred at a temperature between 25 to 30°C for 3-5 h. After completion of the reaction, the reaction
mixture was quenched with ice cold water (800 mL) and stirred for 30 mins. The reaction mixture was then allowed to settle for 30 mins. The organic layer was separated and concentrated under reduced pressure to afford a crude material (66 g), which was then purified by distillation to obtain l-(4-fluorophenyl)ethan-l-one (58 g, 82.3 % yield) as a colorless liquid.
'H-NMR (400 MHz, CDC13) 5 7.94-7.89 (m, 2H), 7.08-7.02 (m, 2H), 2.51 (s, 3H); GC-MS: m/z 138.1.
Step i: Preparation of 4' -bromoacetophenone
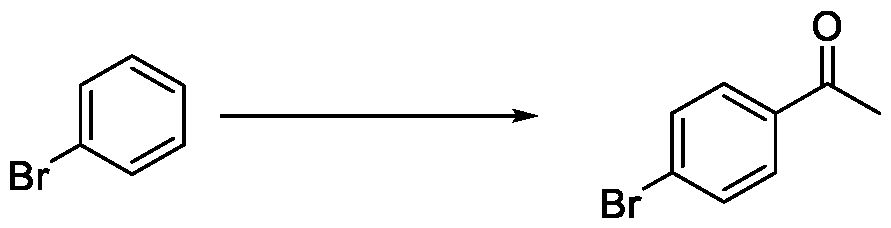
To a solution of bromobenzene (100 g, 637 mmol) in ethylene dichloride (EDC) (500 mL), aluminium chloride (102 g, 764 mmol) was added at a temperature between 25 to 30 °C under nitrogen atmosphere. The reaction mass was cooled to 0 to 5 °C and then acetyl chloride (54.5 mL, 764 mmol) was added in a dropwise manner. The reaction mass was allowed to stir for 12 hours at a temperature between 20 to 30°C. After completion of the reaction, the reaction mixture was quenched by pouring it into a saturated sodium bicarbonate solution (1000 mL). The reaction mixture was then extracted with ethylene dichloride (2 x 250 mL), the combined organic layers were washed with brine (500 mL) and concentrated under reduced pressure to afford 4'-bromoacetophenone (101.3 g, 79.91% yield) as a white solid.
‘H-NMR (400 MHz, DMSO-D6) 5 7.88-7.85 (m, 2H), 7.74-7.70 (m, 2H), 2.56 (s, 3H);
GC-MS: m/z 197.8
Step ii: Preparation of 4-acetylbenzonitrile
Method 1:
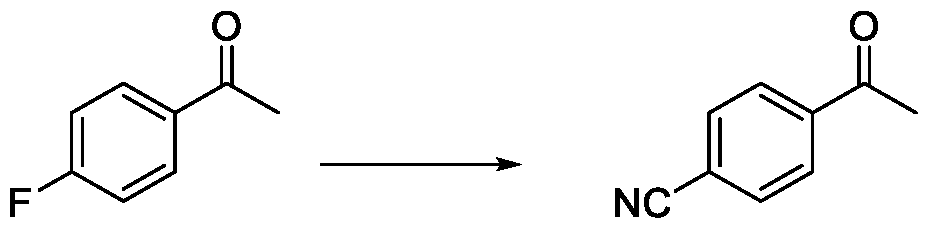
To a stirred suspension of l-(4-fluorophenyl) ethan-l-one (10 g, 72.4 mmol) in dimethylformamide (DMF) (50 mL), tetrabutylammonium bromide (TBAB), (2.3 g, 7.2 mmol) and sodium cyanide (7.1 g, 145 mmol) were added at a temperature between 25 to 30 °C under nitrogen atmosphere. The reaction mixture was heated at a temperature between 130 tol40 °C and stirred for 10 hours. After completion of the reaction, the reaction mixture was cooled to 25-30 °C and quenched by the addition of water (150 mL), and extracted with methyl tert-butyl ether (MTBE) (3 X 100 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate
(NazSCH) and concentrated under reduced pressure to afford 4-acetylbenzonitrile (8.3 g, 78.99%) as an off-white solid.
'H-NMR (400 MHz, DMS0-D6) 8.07 (d, J = 8.4 Hz, 2H), 7.98 (d, J = 8.4 Hz, 2H), 2.61 (s, 3H)
Method 2:
To a stirred suspension of l-(4-fluorophenyl) ethan-l-one (10 g, 72.4 mmol) in NMP (40 mL), Et4NCl (2.4 g, 14.48 mmol) and sodium cyanide (7.1 g, 145 mmol) was added at a temperature between 25 to 30 °C under nitrogen atmosphere. The reaction mixture was heated at 100 °C and stirred for 10 h. After completion of the reaction, the reaction mixture was cooled to 25 to 30 °C and quenched with water (50 mL) and then extracted with MTBE (3 X 80 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous NazSCL and concentrated under reduced pressure to afford 4-acetylbenzonitrile (8.5 g, 81.0%) as off-white solid.
'H-NMR (400 MHz, DMSO-D6) 8.07 (d, J = 8.4 Hz, 2H), 7.98 (d, J = 8.4 Hz, 2H), 2.61 (s, 3H)
Method-3
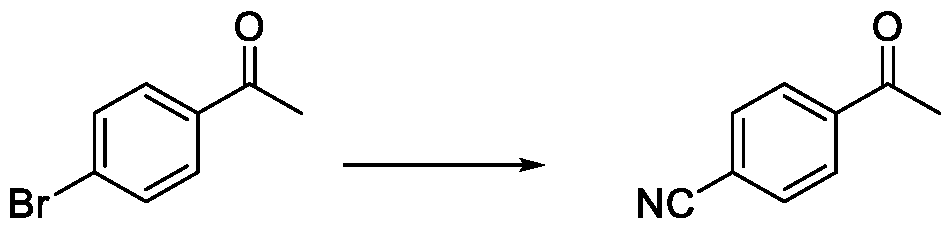
To a stirred suspension of l-(4-bromophenyl)ethan-l-one (10 g, 49.2 mmol) and /V-Mcthyl-2- pyrrolidone (NMP) (9.48 mL, 98 mmol) in //-butanol (60 mL), potassium ferrocyanide trihydrate (10.40 g, 24.62 mmol) and /V,/V'-dimcthylcthylcncdiaminc (5.30 mL, 49.2 mmol) were added at a temperature between 25 to 30°C. The reaction mixture was stirred for 5 min, then copper (I) iodide (Cui) (1.875 g, 9.85 mmol) was added at a temperature between 25 to30°C. The reaction mixture was heated at a temperature between 130 to 135 °C and stirred for 12 hours. After completion of the reaction, the reaction mixture was cooled to 25 °C and filtered through celite bed. The celite bed was washed with ethyl acetate (50 mL), collective filtrate was diluted with water (100 mL) and extracted with ethyl acetate (2 x 100 mL). The combined organic layers were washed with brine (2 x 100 mL), dried over anhydrous sodium sulfate (NazSCU), filtered and concentrated under reduced pressure to afford brown coloured solid material. This material was further stirred in cyclohexane (100 mL) for 1 hour at 25°C, filtered and dried under reduced pressure to afford 4-acetylbenzonitrile (5.4 g, 74.05 % yield) as an off-white sold.
‘H-NMR (400 MHz, DMSO-D6) 8.07 (d, J = 8.4 Hz, 2H), 7.98 (d, J = 8.4 Hz, 2H), 2.61 (s, 3H)
Method-4
To a stirred solution of l-(4-bromophenyl)ethan-l-one (10.0 g 50.2 mmol) in dimethylformamide (DMF) (50 mL), sodium carbonate (10.65 g, 100 mmol) and triethylamine (TEA) (14.01 mL, 100
mmol) were added at 25 to30°C. The reaction mixture was stirred for 5 min, followed by the addition of potassium ferrocyanide trihydrate (5.31 g, 12.56 mmol) and 10% Pd/C (50% wet, 2.67 g, 2.51 mmol) at 25 to 30°C. The reaction mixture was heated at 140 °C and stirred for 4 hours. After the completion of the reaction, the reaction mixture was cooled at 20-30°C, diluted with ethyl acetate (30 mL) and filtered through celite bed. The filtrate was washed with water (2 X 50 mL) and brine solution (50 mL). The organic phase was dried with anhydrous sodium sulfate (NazSCU), filtered, and concentrated under reduced pressure to obtain a crude product which was purified by silica gel chromatography with eluent ethyl acetate/n-hexane to afford an off white solid of 4- acetylbenzonitrile (7.0 g, 62.39% yield (assay basis)).
'H-NMR (400 MHz, DMSO-D6) 8.07 (d, J = 8.4 Hz, 2H), 7.98 (d, J = 8.4 Hz, 2H), 2.61 (s, 3H) .
Step-a: Preparation of N'-hydroxy-4-(l-(hydroxyimino)ethyl)benzimidamide
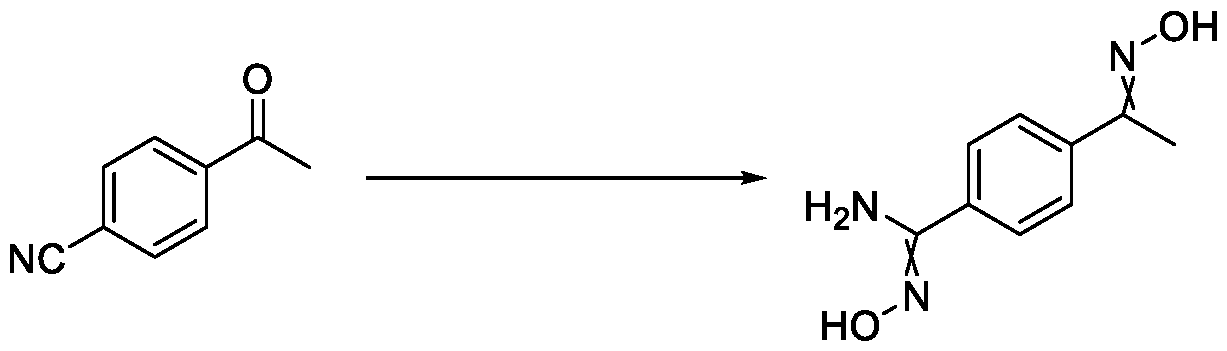
Method-1: Using EtOH/TEA
To a stirred solution of 4-acetylbenzonitrile (10.0 g, 68.9 mmol) in ethanol (100 mL), hydroxylamine hydrochloride (11.97 g, 172 mmol) and triethylamine (TEA) (17.43 g, 172 mmol) were added at 0 °C. The reaction mixture was stirred for 16 hours at a temperature between 50 to 60 °C. After the completion of the reaction, the reaction mixture was cooled to 0 °C and diluted with water (200 mL). The reaction mass was warmed to 25°C, stirred for 30 minutes and filtered. The wet cake was dried to afford A'-hydroxy-4-(l -(hydroxyimino) ethyl)benzimidamide as off white solid (11.8 g, 88.66 % yield).
‘H-NMR (400 MHz, DMSO-D6): 5 11.24 (s, 1H), 9.68 (s, 1H), 7.71-7.62 (m, 4H), 5.81 (bs, 2H), 2.15 (s, 3H); MS: m/z 193.95 [M+l]+.
Method-2: Using McOH/NaHCO;
To a stirred solution of 4-acetylbenzonitrile (100.0 g, 689 mmol) in methanol (500 ml), hydroxylamine hydrochloride (109.0 g, 1569 mmol) was added. The reaction mixture was stirred for 4 hours at a temperature between 50 to 60 °C. Then, the reaction temperature was cooled to 25 to 30°C followed by the addition of sodium bicarbonate (NaHCOs) (143 g, 1705 mmol). The stirring was continued further for 16 hours at a temperature between 25 to 30 °C. After completion of the reaction, the reaction mixture was concentrated under reduced pressure to recover methanol. To the resulting mass, demineralized (DM) water (200 mL) was added and stirred for 3 hours. The reaction
mass was filtered to obtain a wet cake, which was dried to afford /V'-hydroxy-4-( I -(hydroxyimino) ethyl)benzimidamide (125.0 g, 93.92 % yield).
‘H-NMR (400 MHz, DMS0-D6): 5 11.24 (s, 1H), 9.68 (s, 1H), 7.71-7.62 (m, 4H), 5.81 (bs, 2H), 2.15 (s, 3H); MS: m/z 193.95 [M+l]+.
Method-3: using hydroxylamine sulphate
Reaction is carried out according to method- 1
Reagents: 4-acetylbenzonitrile (300 g, 2046 mmol); hydroxylamine sulphate (402g, 2456 mmol); methanol (600 mL) and triethylamine (TEA) (371 mL, 2660 mmol).
Product: N’-hydroxy-4-(l -(hydroxyimino) ethyl)benzimidamide (376 g, 93.21 % yield) as off white solid.
‘H-NMR (400 MHz, DMSO-D6): 5 11.24 (s, 1H), 9.68 (s, 1H), 7.71-7.62 (m, 4H), 5.81 (bs, 2H), 2.15 (s, 3H); MS: m/z 193.95 [M+l]+.
Step-b: Preparation of l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one
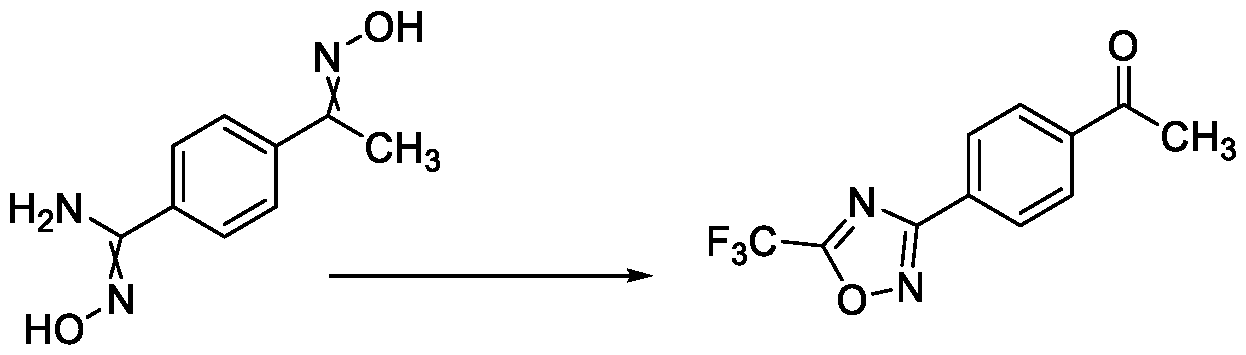
Method-1: Using trifluoroacetic anhydride
To a stirred solution of A'-hydroxy-4-(l-(hydroxyimino)ethyl)benzimidamide (15 g, 77 mmol) in tetrahydrofuran (180 mL), trifluoroacetic anhydride (20.79 g, 99 mmol) was added in a dropwise manner at 0 °C. The reaction mixture was warmed to 25 to 30°C and stirred for 16 hours. After the completion of the reaction, the reaction mixture was diluted with water (150 mL) and extracted with ethyl acetate (2 x 50 mL). The collective organic layer was concentrated under reduced pressure. To the residue obtained, water (100 mL) and concentrated hydrochloric acid (100 mL) were added at a temperature between 25 to 30 °C. The reaction mass was then heated to 90°C, stirred for 16 hours. The reaction mixture was cooled between 25 to 30°C, filtered and dried. The solid product was taken up in //-hexane (1.0 Lit), stirred for 30 min. and filtered. The wet cake was further dried to afford l-(4-(5- (trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (17.54 g, 83 % yield (Purity basis) as off white solid.
‘H-NMR (400 MHz, DMSO-D6): 5 8.18-8.12 (m, 4H), 2.63 (s, 3H); MS: m/z 255.10 [M-l]+.
Method-2: Using Ethyl trifluoroacetate and NaOMe in MeOH
To a stirred solution of /V'-hydroxy-4-(4 -(hydroxyimino) ethyl)benzimidamide (20 g, 104 mmol) in ethyl trifluoroacetate (36.8 g, 259 mmol), 30% sodium methoxide (NaOMe) in methanol (MeOH) (93.2 g, 518 mmol) was added in a dropwise manner at a temperature between 25 to 40 °C, and continued to stir further for 3 hours. After completion of the reaction, the reaction mixture was added into aqueous 5N hydrochloric acid (HC1) solution (200 mL) and further heated to a temperature between 70 to 80°C. The reaction mixture was then continued to stir for 6 hours at 80°C. During the reaction, the distillate was collected to recover ethyl trifluoroacetate and methyl trifluoroacetate formed in situ due to transesterification. After completion of the reaction, the reaction mixture was cooled to a temperature between 20 to25°C. The solid obtained was filtered. The wet cake was washed with water (100 mL) and dried to afford l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (23.5 g, 88.61 % yield) as off white solid.
‘H-NMR (400 MHz, DMSO-D6): 5 8.18-8.12 (m, 4H), 2.63 (s, 3H); MS: m/z 255.10 [M-l]+.
Step-c: Preparation of 2-bromo-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l- one

Method-1: To a stirred solution of l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (50 g, 195 mmol) in ethylene dichloride (EDC) (1.0 L), a solution of bromine (37.4 g, 234 mmol) in ethylene dichloride (500 mL) was added at a temperature between 40 to 45 °C in a drop wise manner. The reaction mixture was then stirred for 2 hours at a temperature between 40 to 45 °C. After the completion of the reaction, the reaction mixture was quenched by the addition of water (250 mL) and extracted with ethylene dichloride (2 x 250 mL). The combined organic layers were washed with brine (250 mL), dried over anhydrous sodium sulfate (NazSCL) and concentrated under reduced pressure. To the resulting residue, isopropyl alcohol (IP A) (100 mL) was added and stirred for 30 min. at a temperature between 0 tolO °C. The solid obtained was filtered and dried to afford 2-bromo-l-(4-(5- (trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (55.0 g, 84.10% yield) as off white solid. ‘H-NMR (400 MHz, DMSO-D6) 5 8.29-8.15 (m, 4H), 5.01 (s, 2H); MS: m/z 332.90 [M-l]+.
Step-d: Preparation of 2-(4-fluorophenoxy)-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3- yl)phenyl)ethan- 1-one
Method-1:

To a stirred solution of 4-fluorophenol (5.0 g, 44.6 mmol) in toluene (200 mL), potassium carbonate (15.41 g, 112 mmol) was added and stirred for 30 minutes at a temperature between 25 to30 °C. After that, 2-bromo-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (16.52 g, 49.3 mmol) was added into the reaction mixture and stirred for 10 hours at the same temperature. After the completion of the reaction, the reaction mixture was added into aqueous 2N hydrochloric acid (HC1) solution and extracted with ethyl acetate (2 x 160 mL). The combined organic layers were washed with brine (160 mL) and concentrated under reduced pressure. The resulting solid residue was charged into cyclohexane (48 mL) and stirred for 2 hours at room temperature. The solid obtained was filtered and dried to afford 2-(4-fluorophenoxy)- 1 -(4-(5-(trifluoromethyl)- 1 ,2,4-oxadiazol-3-yl)phenyl)ethan- 1 -one (15.6 g, 81.0% ) as an off-white solid.
'H-NMR (400 MHz, DMSO-D6) 5 8.24-8.20 (m, 4H), 7.13-7.08 (m, 2H), 7.03-7.00 (m, 2H), 5.62 (s, 2H); MS: m/z 364.75.1 [M-l]+.
Method-2: using catalytic amount of DABCO
To a solution of 4-fluorophenol (16.56 g, 145 mmol) in cyclohexane (250 mL), K2CO3 (26.5 g, 188 mmol) was added followed by DABCO (0.331 g, 2.89 mmol) at 25 to30°C. The reaction mixture was stirred for 10 min followed by the addition of 2-bromo-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3- yl)phenyl)ethan-l-one (50 g, 145 mmol) at 25 to 30°C. The reaction mixture was then stirred for 7 hr at 30°C. After the completion of the reaction, the reaction mixture was quenched with aqueous 2N HC1 solution (200 mL), stirred for 60 min at 25 to 30°C, filtered, washed with water (lOOmL) and dried to obtain 2-(4-fluorophenoxy)- 1 -(4-(5-(trifluoromethyl)- 1 ,2,4-oxadiazol-3-yl)phenyl)ethan- 1 -one (47 g, 80.0%) as off-white solid.
Method-3: Neat reaction
A mixture of 4-fluorophenol (0.662 g, 5.79 mmol), potassium carbonate (1.040 g, 7.53 mmol), 2-bromo- l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (2 g, 5.79 mmol) and DABCO (0.016 g, 0.145 mmol) was stirred for 2 hr at 70°C. After the completion of the reaction, the reaction mixture was diluted with MTBE (5 mL) and filtered. The filtrate was then quenched with 2N HC1 solution (20 mL). The organic layer was separated and concentrated to obtain 2-(4-fluorophenoxy)-l- (4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (1.7 g, 80.0%) as an off-white solid.
The following compounds were prepared by using the analogous procedure as described for example la and with appropriate phenols of Formula (IX):
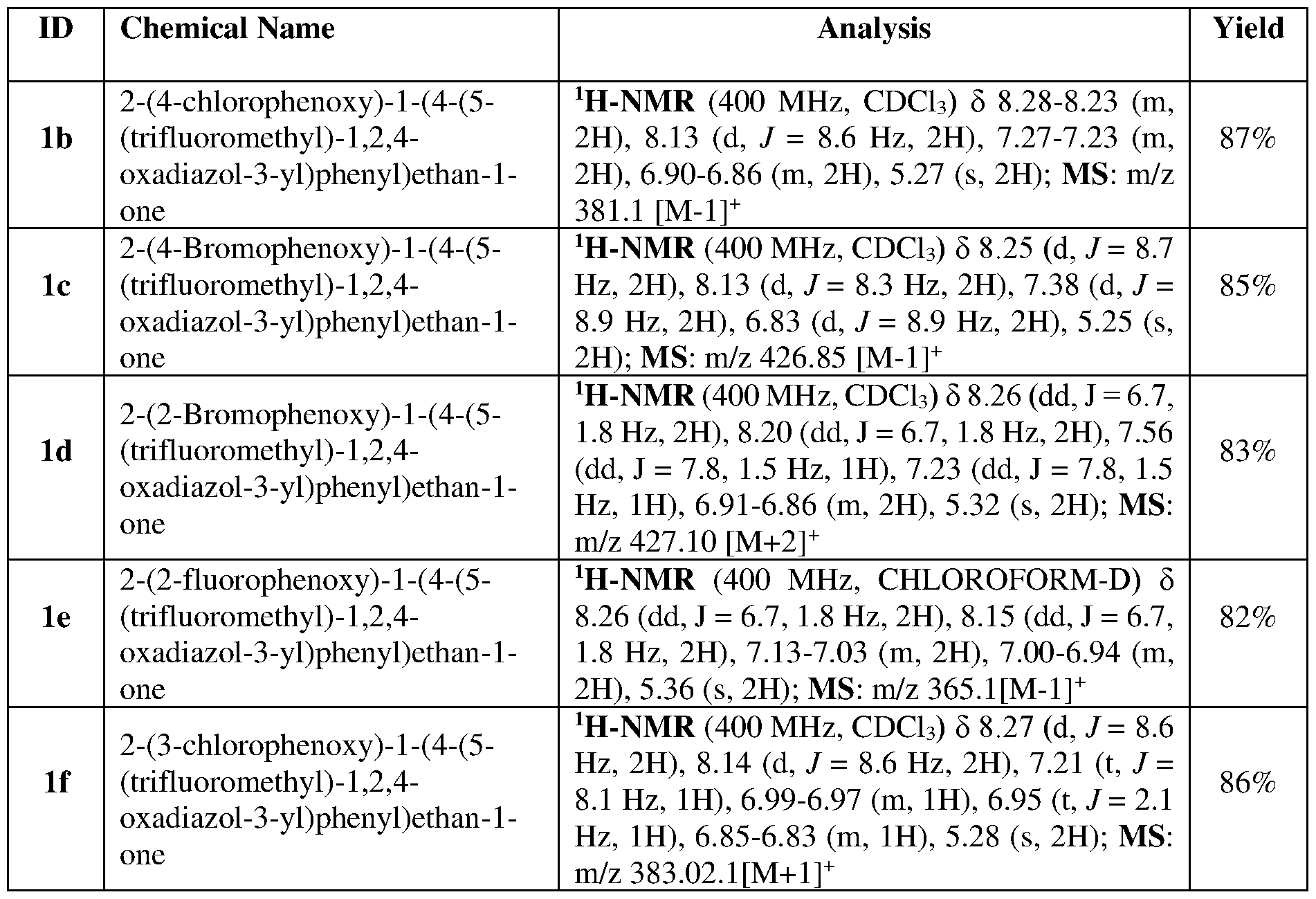
Step-d: Preparation of 2-((4-fluorophenyl)amino)-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3- yl)phenyl)ethan- 1-one (1g)

To a stirred solution of 4-fluoroaniline (3.65 g, 32.8 mmol) in ethanol (EtOH) (200 mL), sodium bicarbonate (2.507 g, 29.8 mmol) was added and stirred for 30 min at 25 to30°C. After that, 2-bromo- l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (10.00 g, 29.88 mmol) was added into the reaction mixture and stirred for 10 hours at 25 to30 °C. After completion of the reaction, the reaction mixture was added into aqueous 2N hydrochloric acid (HC1) solution and extracted with ethyl acetate (2 x 100 mL). The combined organic layers were washed with brine (100.0 mL) and concentrated under reduced pressure. The resulting solid residue was charged into cyclohexane (48.0 mL) and stirred for 2 hours at 25-30 °C. The solid obtained was filtered and dried to afford 2-(4-
fluorophenoxy)- l-(4-(5-(trifluoromethyl)-l, 2, 4-oxadiazol-3-yl)phenyl)ethan-l -one (1g, 10.1 g, 92.65 %) as an off-white solid.
'H-NMR (400 MHz, CDC13) 5 8.29 (dt, J= 8.5, 1.8 Hz, 2H), 8.16 (dt, J= 8.5, 1.8 Hz, 2H), 6.97-6.93 (m, 2H), 6.68-6.65 (m, 2H), 4.64 (s, 2H); MS: m/z 365.90 [M+l]+ The following compounds were prepared by using the analogous procedure as described in example 1g and with appropriate anilines of Formula (IX):

Step-d: Preparation of 2-((2-fluorophenyl)thio)-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3- yl)phenyl)ethan- 1-one (In)
 To a stirred solution of 2-fluorobenzenethiol (0.42 mg, 3.28 mmol) in toluene (20 mL), potassium carbonate (0.91 mg, 6.57 mmol) was added and stirred for 30 minutes at 25 to 30 °C. After that, 2- bromo-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (1.0 g, 2.98 mmol) was added into the reaction mixture and stirred for 10 hours at 25 to 30 °C. After the completion of the reaction, the reaction mixture was added into aqueous 2N hydrochloric acid (HC1) solution and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with brine (20.0 mL) and concentrated under reduced pressure. The resulting residue was charged into cyclohexane (5.0 mL) and stirred for 2 hours at 25 to30 °C. The solid obtained was filtered and dried to afford 2-((2- fluorophenyl)thio)-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (In, 0.8 g, 67.46%) as off-white solid.
To a stirred solution of 2-fluorobenzenethiol (0.42 mg, 3.28 mmol) in toluene (20 mL), potassium carbonate (0.91 mg, 6.57 mmol) was added and stirred for 30 minutes at 25 to 30 °C. After that, 2- bromo-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (1.0 g, 2.98 mmol) was added into the reaction mixture and stirred for 10 hours at 25 to 30 °C. After the completion of the reaction, the reaction mixture was added into aqueous 2N hydrochloric acid (HC1) solution and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with brine (20.0 mL) and concentrated under reduced pressure. The resulting residue was charged into cyclohexane (5.0 mL) and stirred for 2 hours at 25 to30 °C. The solid obtained was filtered and dried to afford 2-((2- fluorophenyl)thio)-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (In, 0.8 g, 67.46%) as off-white solid.
'H-NMR (400 MHz, CDC13) 5 8.23-8.21 (m, 2H), 8.07-8.05 (m, 2H), 7.42-7.38 (m, 1H), 7.32-7.27 (m, 1H), 7.11-7.05 (m, 2H), 4.28 (s, 2H);
MS: m/z 383.1 [M+l]+.
The following compounds were prepared by using the analogous procedure as described in example In and with appropriate benzenethiols of Formula (IX):
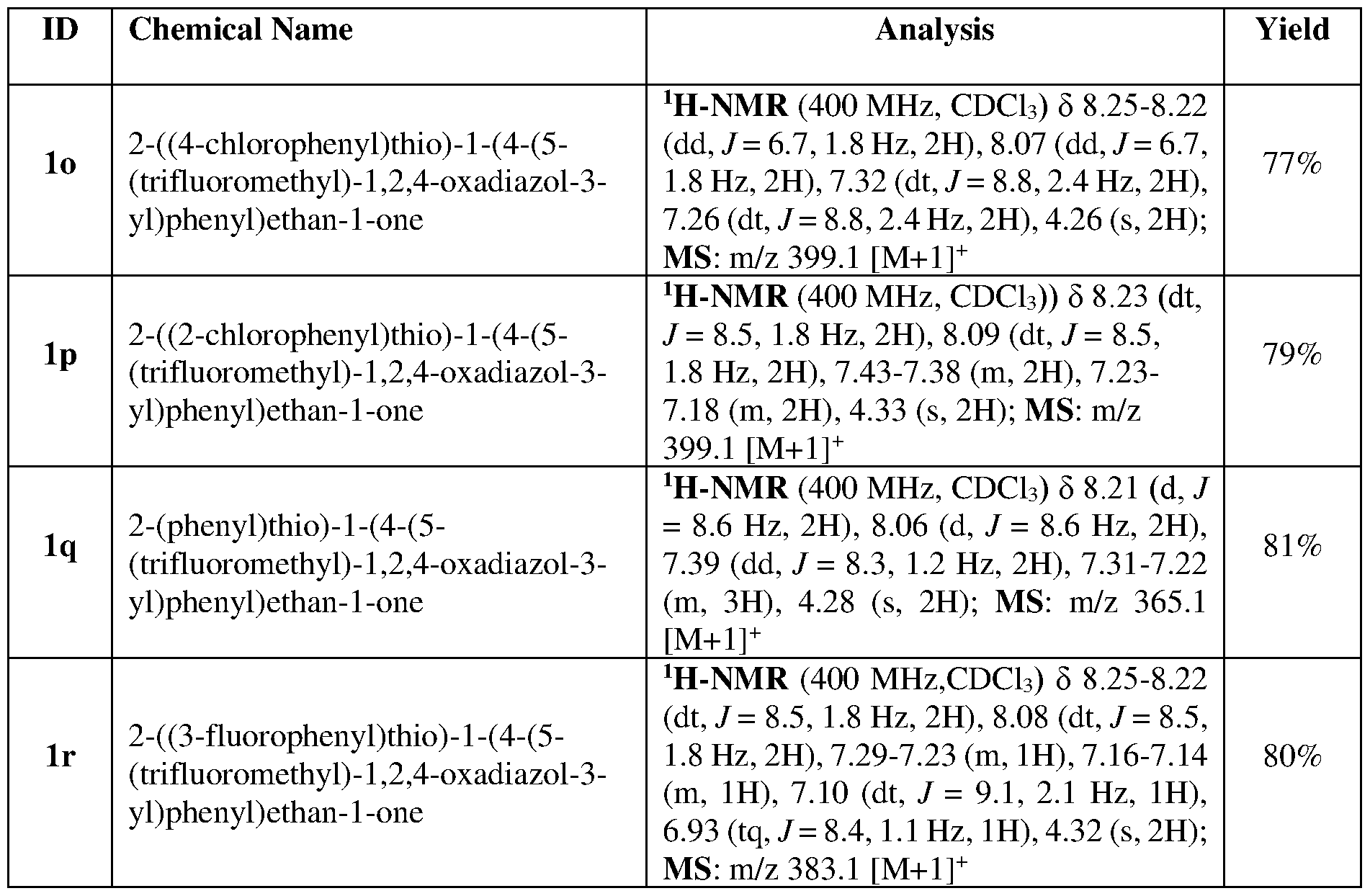
The following additional examples are provided to explain the formation of intermediate compound of formula (X) or formula (XI) during the halogenation of the compound of formula (III):
Synthesis of 3-(4-(2-bromo-l,l-dimethoxyethyl)phenyl)-5-(trifluoromethyl)-l,2,4-oxadiazole
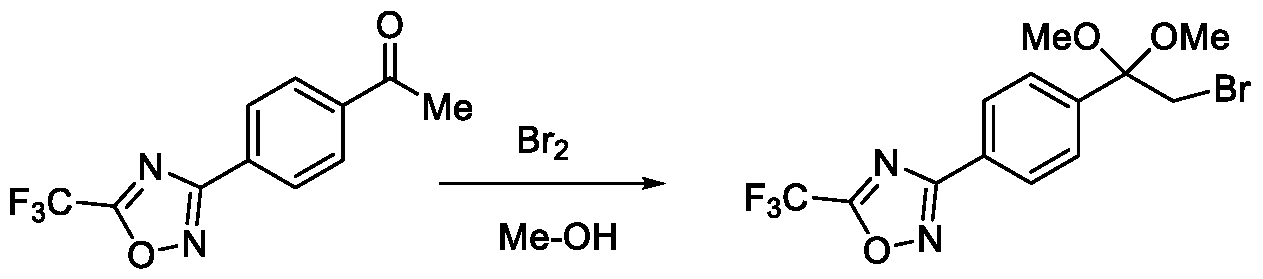
To a stirred solution of l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (20 g, 78 mmol) in methanol (300 mL), bromine (14.00 g, 86 mmol) was added at 25-35 °C in a drop wise manner. The reaction mixture was then stirred for 5 hours at 25-35°C. After completion of the reaction, the reaction mixture was quenched with saturated aqueous solution of sodium bicarbonate (20.08 g, 234 mmol) and then concentrated under reduced pressure to remove methanol solvent. After removing the methanol, the reaction mixture was extracted with ethyl acetate (2 x 250 mL) and the combined organic layer was concentrated under reduced pressure to afford 3-(4-(2-bromo-l,l-dimethoxyethyl)phenyl)-5- (trifluoromethyl)-l,2,4-oxadiazole (21.5g , 72% yield) as a pale yellow colour liquid.
'H-NMR (400 MHz, DMSO-D6) 5 8.09-8.07 (d, J = 8.0 Hz, 2H), 7.70-7.68 (d, J = 8.0 Hz, 2H), 3.85 (s, 2H), 3.13 (s, 6H); 19F-NMR (376.62 MHz, DMSO-D6) 5 -64.82.
Alternative method for the synthesis of 3-(4-(2-bromo-l,l-dimethoxyethyl)phenyl)-5- (trifluoromethyl)-l,2,4-oxadiazole
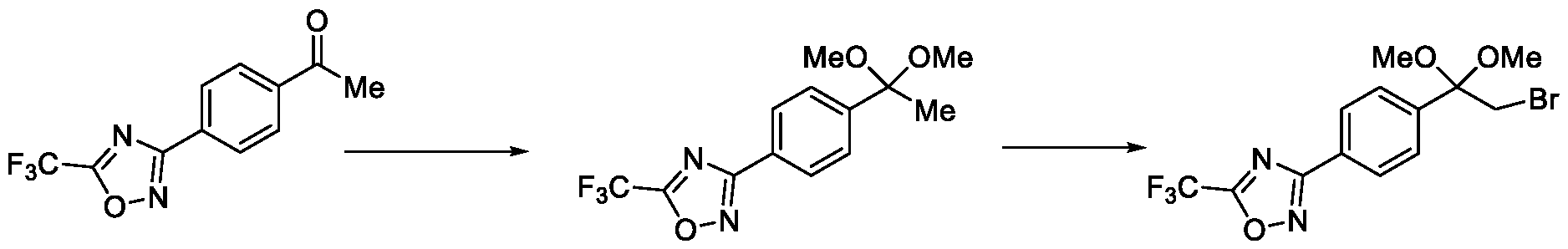
To a stirred solution of l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (20 g, 78 mmol) and trimethyl orthoformate (16.57 g, 156 mmol) in methanol (100 mL), catalytic amount of dioxane. HC1 (2mL) was added at 25-35 °C. The reaction mixture was stirred for 6 hours at 50-55°C. After completion of the reaction, the reaction mixture was quenched with saturated solution of sodium bicarbonate (pH = 7-8) and concentrated under reduced pressure. The obtained crude residue was extracted with ethyl acetate (2 x 150 mL) and the combined organic layer was concentrated under reduced pressure to afford 3-(4-(l,l-dimethoxyethyl)phenyl)-5-(trifluoromethyl)-l,2,4-oxadiazole (23.5g, 85.28% yield) as a white colour solid.
'H-NMR (400 MHz, CDC13) 5 8.09-8.07 (d, J = 8.0 Hz, 2H), 7.65-7.63 (d, J = 8.0 Hz, 2H), 3.19 (s, 6H), 1.54 (s, 3H); 19F-NMR (376.62 MHz, CDCh) 5 -65.49
To a stirred solution of 3-(4-(l,l-dimethoxyethyl)phenyl)-5-(trifluoromethyl)-l,2,4-oxadiazole (5 g, 15.76 mmol) in methanol (25 mL), bromine (2.57 g, 15.76 mmol) was added at 25-35 °C in a drop wise manner. The reaction mixture was then stirred for 10 hours at 25-50°C. After completion of the reaction, the reaction mixture was quenched with sat. NaHCCL solution and concentrated under vacuum. The obtained crude reaction mixture was then extracted with ethyl acetate (3 X 50 mL). The
combined organic layer was concentrated under reduced pressure to afford 3-(4-(2-bromo-l,l- dimethoxyethyl)phenyl)-5-(trifluoromethyl)-l,2,4-oxadiazole (7.2 g, 91% yield) as pale yellow colour liquid.
'H-NMR (400 MHz, DMSO-D6) 5 8.14-8.12 (d, J = 8.0 Hz, 2H), 7.68-7.66 (d, J = 8.0 Hz, 2H), 3.63 (s, 2H), 3.25 (s, 6H); 19F-NMR (376.62 MHz, DMSO-D6) 5 -65.43.
Synthesis of 2-bromo-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one from 3- (4-(l,l-dimethoxyethyl)phenyl)-5-(trifluoromethyl)-l,2,4-oxadiazole;
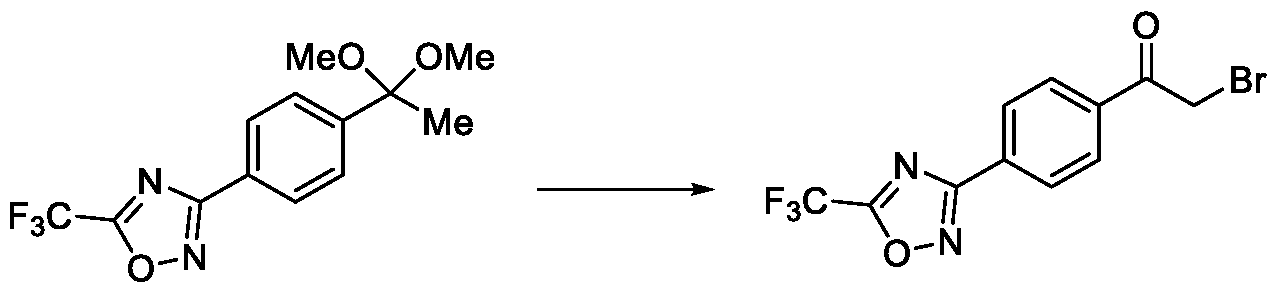
To a stirred solution of 3-(4-(l,l-dimethoxyethyl)phenyl)-5-(trifluoromethyl)-l,2,4-oxadiazole ( 2 g, 6.62 mmol) and catalytic amount of HC1 (4 mL) in methanol (20 mL), bromine (3.65 mL g, 6.94 mmol) was added at 25 °C in a drop wise manner. The reaction mixture was then stirred for 10 hours at 25 °C followed by 3 hours at 35°C. After completion of the reaction, the reaction mixture was diluted with water (20 mL) and concentrated on a rota evaporator to remove methanol. The obtained residue was filtered, washed with DM water (10 mL) and dried under reduced pressure to afford 2-bromo-l-(4-(5- (trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (1.7 g, 80% yield) as off white solid. 'H-NMR (400 MHz, CDCk) 5 8.29-8.11 (m, 4H), 4.48 (s, 2H); MS: m/z 332.90 [M-l]+.
Synthesis of 2-bromo-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one from 1- (4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one
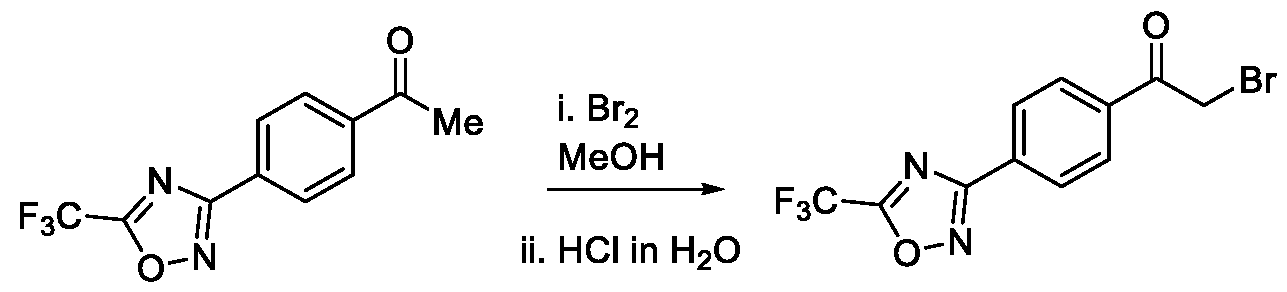
To a stirred solution of l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)phenyl)ethan-l-one (60 g, 234 mmol) in methanol (900 mL), bromine (37.4 g, 234 mmol) was charged at 25-35 °C in a drop wise manner. The reaction mixture was then stirred for 6 hours at 25-35°C. After completion of the reaction, the reaction mixture was concentrated under reduced pressure. To the crude residue, DM water (150 mL) was added followed by cone. HC1 (70 mL). The resulting mixture was stirred at 25-35°C for 3 hours. After completion of the reaction, the reaction mixture was filtered, washed with DM water (50 mL) and dried under reduced pressure to afford 2-bromo-l-(4-(5-(trifluoromethyl)-l,2,4-oxadiazol-3- yl)phenyl)ethan-l-one (75.0 g, 95% yield) as off white solid.
'H-NMR (400 MHz, CDC13) 5 8.29-8.11 (m, 4H), 4.48 (s, 2H); MS: m/z 332.90 [M-l]+.
Claims
1. A method for preparing a compound of Formula (III), or N-oxides or salts thereof,

Formula (III) wherein R1 is selected from -CHF2, -CF2-CI or CF3, said method comprising the steps of: a. reacting a compound of Formula (VI) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
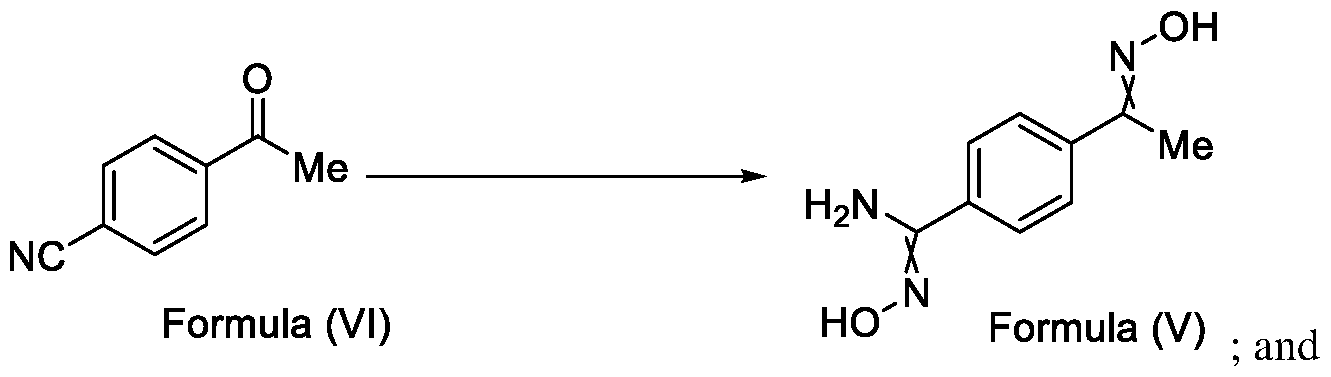 b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
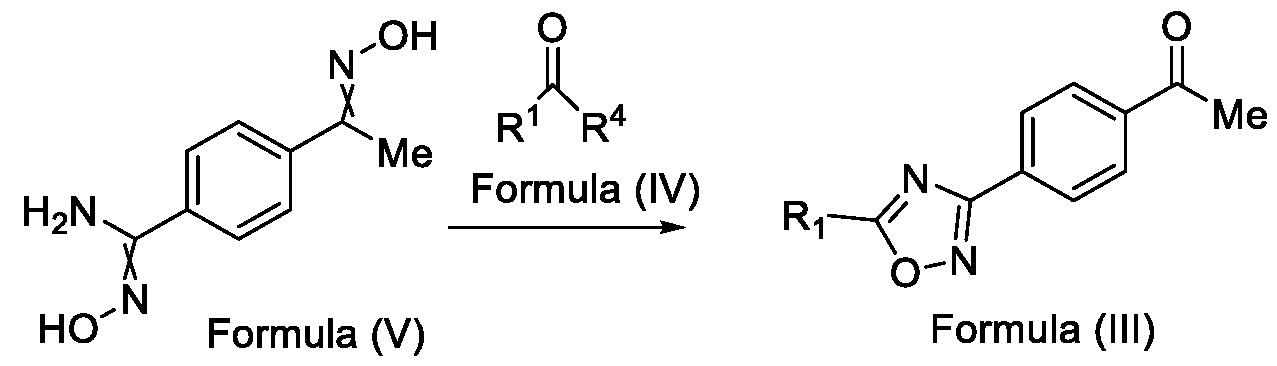 wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'.
wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'.
2. A method for preparing a compound of Formula (II), or N-oxides or salts thereof,
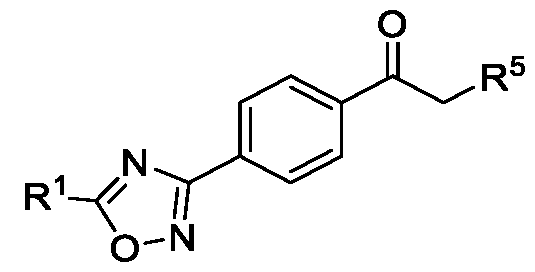
Formula (II) wherein,
R1 is selected from -CHF2, -CF2-CI or CF3; and
R5 is selected from fluoro, chloro, bromo, or iodo, said method comprising the step of a. reacting a compound of Formula (VI) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
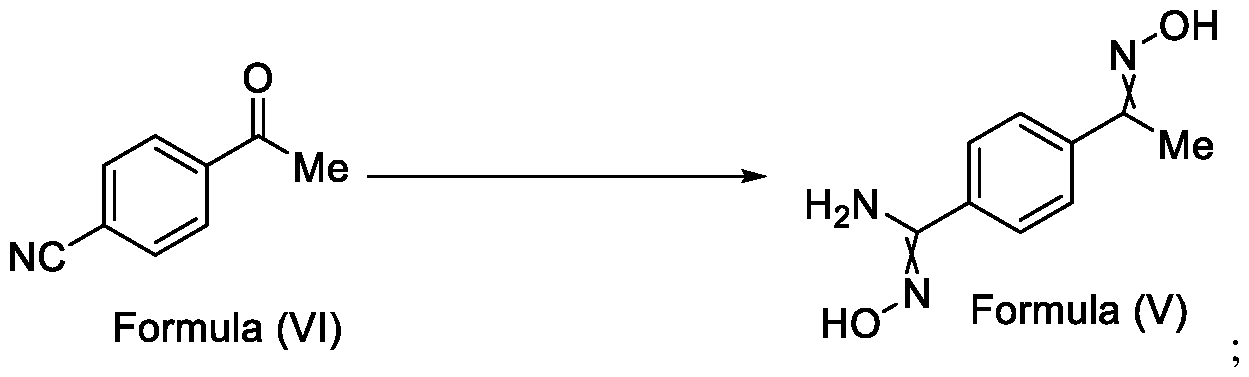 b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
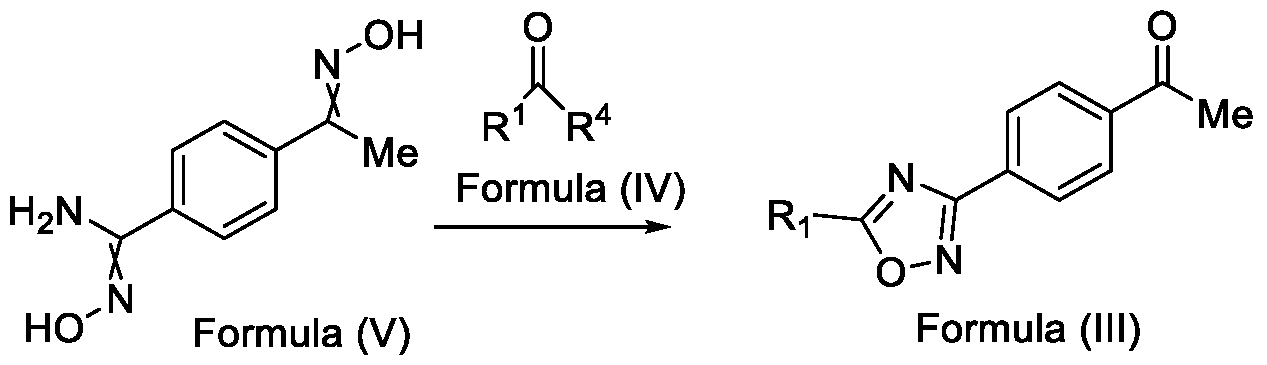 wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; and c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; and c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),

Formula (III) Formula (II)
3. A method for preparing the substituted l-(4-(5-(halomethyl)-l,2,4-oxadiazol-3- yl)phenyl)ethan-l-one of Formula (I), its N-oxides or salts thereof,
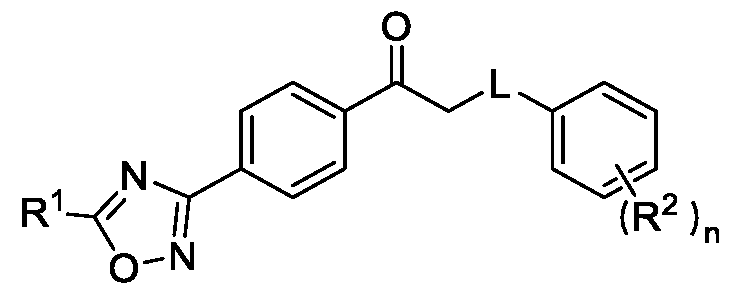
Formula (I) wherein,
R1 is selected from -CHF2, -CF2-CI or CF3;
L is selected from O, NR3 or S;
R2 is selected from hydrogen, halogen, C i-Ce-alkyl, Ci-Ce-haloalkyl, Ci-Ce-alkoxy or C Ce-cycloalkyl; and
R3 is selected from hydrogen or C i-Ce-alkyl; and n is an integer selected from 1-2, comprising the steps of: a. reacting a compound of Formula (VI) with hydroxylamine or its salts in a solvent, optionally in the presence of a base, to form a dioxime compound of Formula (V),
 b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
b. cyclizing the dioxime compound of Formula (V) by reacting it with a compound of Formula (IV) in a solvent, optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III) in a single-step,
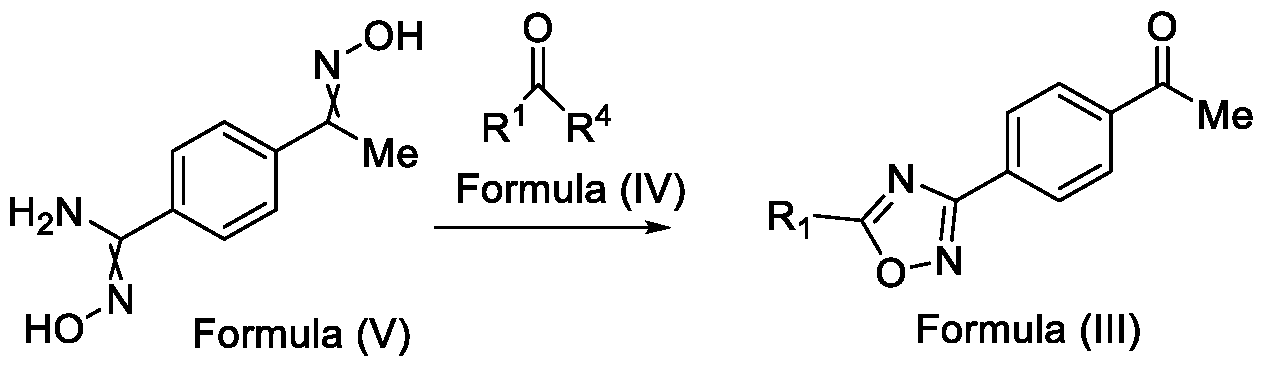 wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
wherein R4 is selected from halogen, OH, Ci-Ce-alkoxy or O-C(O)-R'; c. halogenating the compound of Formula (III) using a halogenating agent and a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (II),
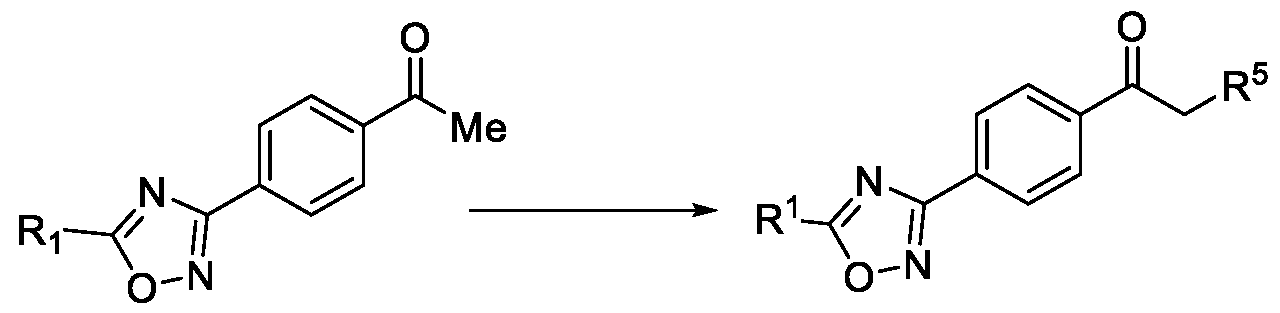
Formula (III) Formula (II) wherein R5 is fluoro, chloro, bromo or iodo; and d. alkylating a compound of Formula (IX) with the compound of Formula (II) in the presence of a base, and optionally in the presence of a catalyst and a solvent, to obtain a compound of Formula (I),
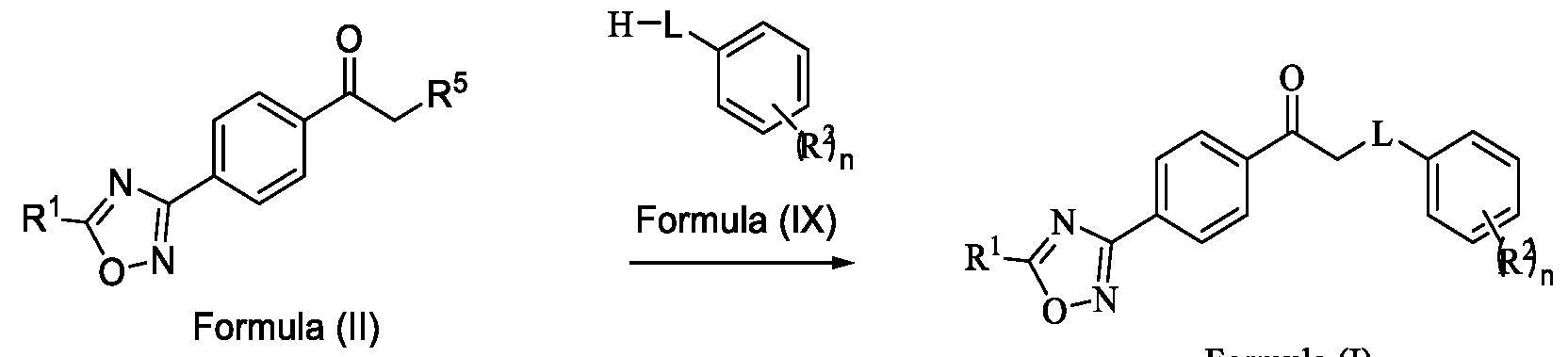
Formula (I)
4. A single-step method of preparing the compound of Formula (III), its salts or N-oxides thereof, wherein said method comprises the step (step-b) of cyclizing the dioxime compound of Formula (V) with a compound of Formula (IV) in a solvent and optionally in the presence of a base, followed by treatment with an acid, to obtain the compound of Formula (III),
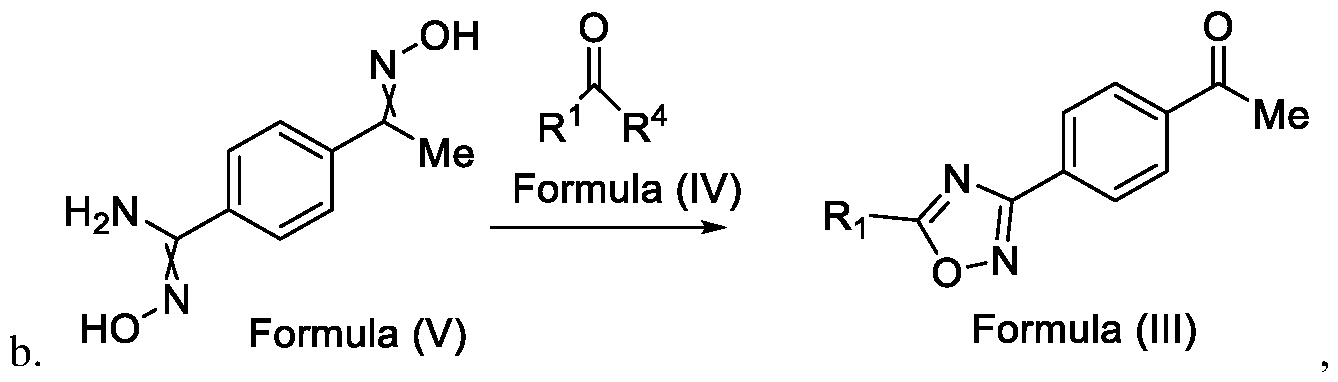 wherein,
wherein,
R1 is selected from -CHF2, -CF2-CI or CF3 and
R4 is selected from halogen, OH, C i-Ce-alkoxy or O-C(O)-R'.
5. The method as claimed in claim 1 or claim 4, wherein R1 is selected from CF3 or CF2CI.
6. The method as claimed in claim 2, wherein R1 is selected from CF3 or CF2CI; and R5 is selected from chloro or bromo.
7. The method as claimed in claim 3, wherein R1 is selected from CF3 or CF2CI; L is selected from O, NH or S; R2 is selected from hydrogen, fluoro, chloro, bromo, methyl or methoxy; R5 is selected from chloro or bromo; and n is 1-2.
8. The method as claimed in any one of the claims 1-4, wherein R4 is selected from Cl, Br, Ci- C3 alkoxy, OMe, OEt, or O-C(O)-CF3.
9. The method as claimed in any one of the claims 1 -4, wherein the reagent hydroxylamine in step-a is in an aqueous form or in a salt form.
10. The method as claimed in claim 9, wherein the salt form is selected from halide (HX), carboxylate, acetate, trifluoroacetate, sulfonate, trifluoromethanesulfonate, or sulfate.
11. The method as claimed in any one of the claims 1-4, wherein the solvent is selected from acetonitrile, acetic acid, methanol, ethanol, iso-propanol, butanol, acetone, pentane, hexane, heptane, octane, nonane, decane, dodecane, cyclobutane, cyclopentane, cyclohexane, cycloheptane, cyclooctane; dichloromethane, dichloroethane (ethylene dichloride),
chloroform, chlorobenzene, ethyl acetate, iso-propyl acetate, toluene, xylene, mesitylene, nitro benzene, nitro methane, diethyl ether, diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, monoglyme, diglyme, methoxymethane, methoxy-ethane, ethoxy-ethane, di-methoxy ethane, di-ethoxyethane, N,N- dimethylformamide, dimethyl sulfoxide, sulfolane, A-methyl-2-pyrrolidone, 1,3-dimethyl- 3 ,4,5 ,6-tetrahydro-2( 1 H)-pyrimidinone, hexamethylphosphortriamide, 1 ,3 -dimethyl-2- imidazolidinone or of combinations thereof.
12. The method as claimed in any one of the claims 1 -4, wherein the base is selected from alkali metal hydrogen carbonate, alkali or alkaline earth metal carbonate, alkali or alkaline earth metal hydroxide, alkali metal phosphate, alkali metal hydride, alkali metal alkoxide, ethylamine, triethylamine, isopropylamine diisopropylamine, triisopropylamine, pyridine, picoline, piperidine, methylmorpholine, /V-mcthylpipcridinc A,A-(dimethylamino)pyridine (DMAP), lutidine, collidine, tetramethylammonium hydroxide, tetrabutylammonium hydroxide, choline hydroxide, l,5,7-triazabicyclo[4.4.0]dec-5-ene, (TBD), 2,3,4,6,7,8,9,10- octahydropyrimidol[l,2-a]azepine (DBU) l,5-diazabicyclo[4.3.0]non-5-ene (DBN), 1,4- diazabicyclo[2.2.2]octane (DABCO), triethylenediamine or of combinations thereof.
13. The method as claimed in any one the claims 1-3, wherein in step-a i. the solvent is selected from ethanol, methanol, isopropanol, diethyl ether, N,N- dimethylformamide, tetrahydrofuran or water or a mixture thereof; and ii. the base if used, is selected from earth metal hydrogen carbonate, alkali metal carbonate or alkaline earth metal carbonate, triethylamine, diisopropylamine, or triisopropylamine, pyridine, or picoline.
14. The method as claimed in any one of the claims 1 -4, wherein in step-b i. the solvent is selected from methanol, ethanol, isopropanol, tetrahydrofuran, 2- methyltetrahydrofuran, acetonitrile, A,A-dimethylformamide or water; ii. the base if used, is selected from alkali metal alkoxide or alkaline earth metal alkoxide; iii. the acid is selected from sulphuric acid, hydrochloric acid, or hydrobromic acid.
15. The method as claimed in claim 2 or claim 3, wherein in step-c, i. the halogenating agent is selected from fluorine (F2), chlorine (CI2), bromine (B ). iodine (I2), Br2 in acetic acid, N-bromosuccinimide, copper bromide (CuBr2), HBr, NaBr, NFUBr, phosphorus tribromide (PBn). phosphorus pentabromide (PBrs), a mixture of NFUBr and potassium peroxymonosulfate sulfate, Trimethylphenylammonium tribromide, N-chlorosuccinimide, phosphorus trichloride (PCI3), phosphorus pentachloride (PCI5), sulfonyl chloride (SOCI2), sulfuryl chloride (SO2CI2), selenium oxychloride, benzyltrimethylammonium dichloroiodate, or trichloroisocyanuric acid;
ii. the solvent is selected from dichloromethane, dichloroethane (ethylene dichloride), chloroform, acetonitrile, diethyl ether, methyl tertiary-butyl ether, tetrahydrofuran, 2- methyl-tetrahydrofuran, ethyl acetate, alcoholic solvent selected from methanol, ethanol, or isopropanol, acetic acid or water or a mixture thereof; iii. the catalyst if used, is selected from -tolucncsul tonic acid, aluminium chloride (AlCh), monopotassium phosphate (K2HSO4), sulphuric acid, silica, amberlyst 15, or trimethylsilyl triflate.
16. The method as claimed in claim 2 or claim 3, wherein the compound of formula (III) is halogenated using halogenation reagents selected from bromine or chlorine in the presence of alcoholic solvent selected from methanol, ethanol or isopropanol, followed by ketal deprotection in the presence of an acid to obtain the compound of formula (Ila),
 wherein,
wherein,
R1 is CHF2, -CF2-CI or CF3;
R5a is selected from bromo or chloro; and
R6 is selected from methyl, ethyl, n-propyl or isopropyl,
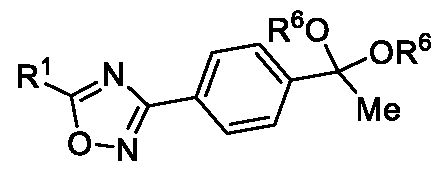 wherein the intermediate compound of formula (X) Formula (X) or formula (XI)
wherein the intermediate compound of formula (X) Formula (X) or formula (XI)
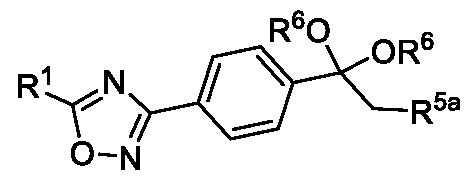
Formula (XI) is/are optionally isolated.
17. The method as claimed in claim 14, wherein the acid is selected from hydrochloric acid.
18. The method as claimed in claim 3, wherein in step-d, i. the base is selected from sodium carbonate, sodium bicarbonate, potassium carbonate, potassium bicarbonate, l,5,7-triazabicyclo[4.4.0]dec-5-ene, pyridine, picoline or 1,4- diazabicyclo [2.2.2] octane ; ii. the solvent if used, is selected from acetone, acetonitrile, diethyl ether, methyl ethyl ketone, methanol, ethanol, 1,4-dioxane, dichloromethane, dichloroethane (ethylene
dichloride), chlorobenzene, toluene, cyclopentane, cyclohexane, N,N- dimethylformamide (DMF) or water or a mixture thereof; and iii. the catalyst if used, is selected from sodium iodide, potassium iodide, 1,4- diazabicyclo[2.2.2]octane or tetra alkylammonium iodide.
19. The method as claimed in claim 1 or 2 or 3, wherein said method further comprises the following steps:
A. preparing a compound of formula (VI) by i. acetylating a compound of Formula (VIII) with acetyl chloride or acetic anhydride in the presence of a suitable catalyst and an optional solvent to obtain a compound of Formula (VII),
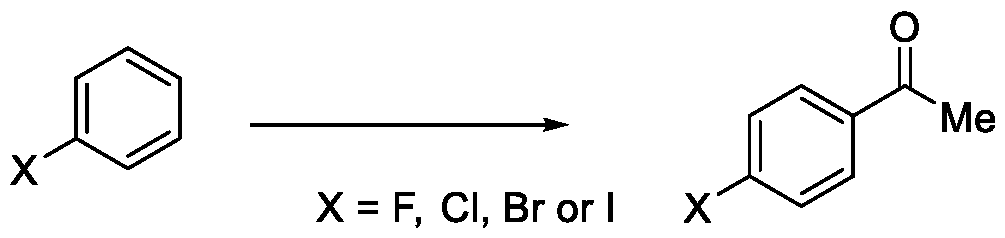
Formula (VIII) Formula (VII) . anj ii. reacting the compound of Formula (VII) with a cyanide source in a solvent, optionally in the presence of a catalyst, to obtain a compound of Formula (VI),
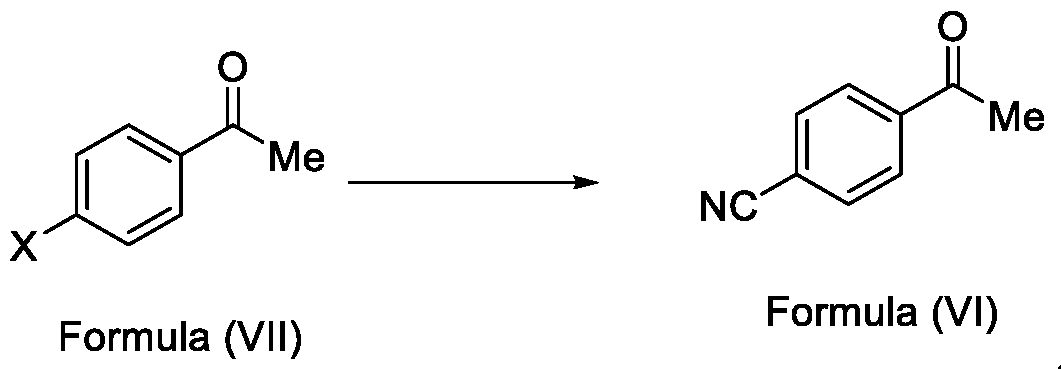
20. An intermediate compound of formula (X) or formula (XI);
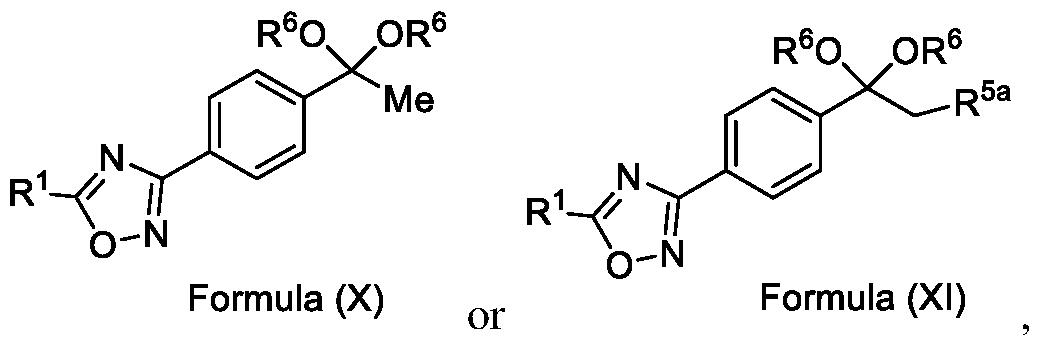 wherein R1 is CHF2, -CF2-CI or CF3;
wherein R1 is CHF2, -CF2-CI or CF3;
R5a is selected from bromo or chloro; and
R6 is selected from methyl, ethyl, n-propyl or isopropyl.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN202411000327 | 2024-01-03 | ||
| IN202411000327 | 2024-01-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025146626A1 true WO2025146626A1 (en) | 2025-07-10 |
Family
ID=96300136
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2025/050009 Pending WO2025146626A1 (en) | 2024-01-03 | 2025-01-02 | A method for the preparation of 1-(4-(5-(halomethyl)-1,2,4-oxadiazol-3-yl)phenyl)ethan-1-one |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202542121A (en) |
| WO (1) | WO2025146626A1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017220485A1 (en) * | 2016-06-21 | 2017-12-28 | Syngenta Participations Ag | Microbiocidal oxadiazole derivatives |
| WO2020208511A1 (en) * | 2019-04-08 | 2020-10-15 | Pi Industries Limited | Novel oxadiazole compounds for controlling or preventing phytopathogenic fungi |
| WO2022249074A1 (en) * | 2021-05-26 | 2022-12-01 | Pi Industries Ltd. | Fungicidal composition containing oxadiazole compounds |
-
2025
- 2025-01-02 WO PCT/IB2025/050009 patent/WO2025146626A1/en active Pending
- 2025-01-03 TW TW114100386A patent/TW202542121A/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017220485A1 (en) * | 2016-06-21 | 2017-12-28 | Syngenta Participations Ag | Microbiocidal oxadiazole derivatives |
| WO2020208511A1 (en) * | 2019-04-08 | 2020-10-15 | Pi Industries Limited | Novel oxadiazole compounds for controlling or preventing phytopathogenic fungi |
| WO2022249074A1 (en) * | 2021-05-26 | 2022-12-01 | Pi Industries Ltd. | Fungicidal composition containing oxadiazole compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202542121A (en) | 2025-11-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7553689B2 (en) | Method for producing phenylisoxazoline compounds | |
| JPH1045733A (en) | Method for producing 2- (4-alkoxy-3-cyanophenyl) thiazole derivative | |
| WO2024194896A1 (en) | A method for the preparation of anthranilic diamides | |
| DE102004041531A1 (en) | Process for the preparation of biphenylamines | |
| WO2025146626A1 (en) | A method for the preparation of 1-(4-(5-(halomethyl)-1,2,4-oxadiazol-3-yl)phenyl)ethan-1-one | |
| US20120029202A1 (en) | Process for the Preparation of 5-Substituted 3-Aryl-3-(trifluoromethyl)-3,4-dihydro-2H-pyrroles | |
| JP5140776B1 (en) | Process for producing 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid ester | |
| US5922916A (en) | Process to chloroketoamines using carbamates | |
| WO2025196653A1 (en) | A method for the preparation of substituted 1-(4-(5-(halomethyl)-1,2,4-oxadiazol-3-yl)phenyl)ethan-1-one | |
| US20040199002A1 (en) | Process for producing(2-nitrophenyl)acetonitrile derivative and intermediate therefor | |
| JP3006237B2 (en) | Preparation of aminopyrazole derivatives | |
| JP7547414B2 (en) | Method for preparing cis-alkoxy-substituted spirocyclic 1-H-pyrrolidine-2,4-dione derivatives | |
| RU2846415C2 (en) | Method of producing nucleocapsid inhibitor of hepatitis b virus | |
| KR101308227B1 (en) | Method for producing nicotinic acid derivative or salt thereof | |
| Nagano et al. | Synthesis of unsymmetrically and highly substituted thiophenes utilizing regioselective ring-expansion of gem-dichlorocyclopropyl ketones with Lawesson’s reagent | |
| US7601847B2 (en) | Preparation and purification of 4-(indazol-3-yl)phenols | |
| CN110914250B (en) | Novel process for synthesizing tilobaxib | |
| CN121039088A (en) | Methods for manufacturing 3-hydroxybiphenyl compounds and their derivatives | |
| JP2003171359A (en) | Method for producing (2-nitro-phenyl) acetonitrile derivative and synthetic intermediate thereof | |
| JP4356917B2 (en) | Process for producing bisaminomethyl-1,4-dithianes and intermediates thereof | |
| WO2003045934A1 (en) | Process for producing 5-substituted 2(5h)-furanone | |
| JP2004244362A (en) | Method for producing 1,2-disubstituted-1,4-dihydro-oxoquinoline derivative | |
| JP2008195697A (en) | Method for producing 3-alkyl-7,7,9,9-tetramethyl-1,3,8-triazaspiro[4,5]decane derivative | |
| JP2013010739A (en) | Method for producing isoxazoline compound | |
| JP2006008617A (en) | Process for producing 5-phthalanecarbonitrile compound, intermediate thereof and process for producing the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 25736470 Country of ref document: EP Kind code of ref document: A1 |