WO2025144938A1 - Systems for nucleic acid transfer - Google Patents
Systems for nucleic acid transfer Download PDFInfo
- Publication number
- WO2025144938A1 WO2025144938A1 PCT/US2024/061965 US2024061965W WO2025144938A1 WO 2025144938 A1 WO2025144938 A1 WO 2025144938A1 US 2024061965 W US2024061965 W US 2024061965W WO 2025144938 A1 WO2025144938 A1 WO 2025144938A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dna
- protein
- brambleberry
- mrna
- double membrane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0008—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition
- A61K48/0025—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the non-active part clearly interacts with the delivered nucleic acid
- A61K48/0041—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the non-active part clearly interacts with the delivered nucleic acid the non-active part being polymeric
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
- A61K48/0058—Nucleic acids adapted for tissue specific expression, e.g. having tissue specific promoters as part of a contruct
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/88—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation using microencapsulation, e.g. using amphiphile liposome vesicle
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/11—Antisense
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/14—Type of nucleic acid interfering nucleic acids [NA]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/14—Type of nucleic acid interfering nucleic acids [NA]
- C12N2310/141—MicroRNAs, miRNAs
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPR]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/10—Plasmid DNA
- C12N2800/106—Plasmid DNA for vertebrates
- C12N2800/107—Plasmid DNA for vertebrates for mammalian
Definitions
- the invention relates generally to systems for nucleic acid transfer.
- Systems for nucleic acid transfer are useful in the fields of gene therapy, vaccines, cell modification, and transfection.
- Nucleic acid transfer can include the introduction of nucleic acids into cells, such as mammalian cells.
- cells such as mammalian cells.
- an RNA or DNA molecule must be transferred not only into the cell, but also into the nucleus, where it can be expressed.
- Current non- viral nucleic acid transfer systems for introducing DNA into the nuclei of cells, such as non-dividing cells, include electroporation and lipid nanoparticles.
- Electroporation is an approach that facilitates a relatively efficient nucleic acid transfer into non-dividing cells, which is thought to be due to the creation of openings in both the plasma membrane and the nuclear envelope that allow DNA to pass directly into the nucleus.
- electroporation cannot be used on a large area and requires surgical intervention for transfer to internal organs. Further, the use of high voltage can damage nucleic acid and tissues.
- Lipid nanoparticles based on ionizable lipids deliver the RNA or DNA they carry into the cytoplasm of cells. It is contemplated that within minutes of contact with the cytoplasm, transfected DNA becomes sequestered into double membrane-enveloped compartments, which are derived from the endoplasmic reticulum (ER) and resemble a nuclear envelope topologically (Kobayashi et al. (2015) PROC. NATL. ACAD. SCL, 112(22): 7027-32).
- the present disclosure is based, in part, upon the discovery that double membrane fusogen proteins provide a solution to the topological problem of transferring DNA sequestered within a double membrane envelope to the nucleus.
- chromosomes become enveloped by a double membrane in structures known as karyomeres, which ultimately fuse to form the nucleus.
- zebrafish a protein that mediates this fusion is Brambleberry — so named, because in its absence, consolidation of karyomeres into a nucleus is arrested, and the unfused karyomeres resemble a brambleberry (Abrams et al.
- the disclosure relates, in part, to systems e.g., lipid-based delivery systems) and methods for the transfer of a non-native e.g., non-endogenous) DNA into a nucleus of a cell, such as a mammalian cell.
- the system or method comprises a non-native (e.g., non-endogenous) DNA and an RNA that encodes a double membrane fusogen protein, which, when present in the cell, mediates transfer of the non-native DNA into the nucleus of the cell.
- a plasmid with a CpG-free promoter when present in non-dividing cells is capable of persistently expressing a gene of interest, without being silenced, and without the loss of the plasmid, e.g. , by degradation.
- repositioning the TATA box in naturally occurring muscle-specific promoters results in improved expression.
- the disclosure also relates, in part, to engineered muscle-specific promoters, that can be used in combination with the systems described herein, or in other applications.
- the double membrane fusogen mediates the fusion of intracellular membranes, such as the fusion of a nuclear envelope of a cell with a double membrane-enveloped structure comprising the non-native DNA.
- the double membrane fusogen comprises a luminal domain, a transmembrane domain, a cytoplasmic domain, or a DNA-binding domain having at least 60%, at least 70%, at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to the corresponding domain present in SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, or 147.
- the at least one of features (a) - (c) does not naturally occur in the amino acid sequence of a wild-type double membrane fusogen having the greatest sequence identity to the encoded double membrane fusogen.
- the system can further comprises an immunosuppressive molecule or a pro-drug form thereof and/or an RNA encoding an immunosuppressive protein or a pro-drug form thereof.
- the immunosuppressive molecule can be a corticosteroid, a tyrosine kinase inhibitor, dexamethasone, tacrolimus, , fluticasone, fluticasone propionate, fluticasone furoate, clobetasol, betamethasone, halobetasol, amcinonide, desoximetasone, diflorasone, fluocinonide, mometasone, prednisone, prednicarbate, triamcinolone, triamcinolone acetonide, fluocinolone, alclometasone, desonide, cortisone, or hydrocortisone, or a pro-drug form of any of the foregoing.
- the immunosuppressive molecule or pro- drug form thereof can bind a human glucocorticoid receptor.
- the immunosuppressive molecule or pro-drug form thereof contains an ester bond.
- the immunosuppressive molecule or pro-drug form thereof is dexamethasone palmitate or fluticasone-furoate.
- the system comprises at least one immunosuppressive molecule or pro-drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, NLRP3 inflammasome inhibitors, MyD88 inhibitors, IRAK4 inhibitors, PKR inhibitors, PERK inhibitors, NFkB inhibitors, IKK inhibitors, JAK inhibitors, STAT inhibitors, GSK3 inhibitors, cGAS inhibitors, or STING inhibitors.
- immunosuppressive molecule or pro-drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, NLRP3 inflammasome inhibitors, MyD88 inhibitors, IRAK4 inhibitors, PKR inhibitors, PERK inhibitors, NFkB inhibitors, IKK inhibitors, JAK inhibitors, STAT inhibitors, GSK3 inhibitors, cGAS inhibitors, or STING inhibitors.
- the at least one immunosuppressive molecule or pro- drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, or NLRP3 inflammasome inhibitors.
- the immunosuppressive molecule or pro-drug form thereof is an NLRP3 inflammasome inhibitor.
- the NLRP3 inflammasome inhibitor can be oridonin, INF39, glyburide, 5-chloro-2-methoxy-N-[2-(4- sulfamoylphenyl)-ethyl]- benzamide (e.g. , 16673-34-0), JC124, FC11A-2, parthenolide, VX-740, VX-765, BAY 11- 7082, BHB, MCC950, MNS, CY-09, or Tranilast, OLT1177, or a pro-drug form of any of the foregoing.
- the galectin inhibitor can be GB 1107, galectin-3/galectin-8-IN-l, olitigaltin, selvigaltin, galectin-3/galectin-8-IN-2, galectin-3-IN-2, galectin-3-IN-3, galectin-3-IN-5, GB1490, GB1908, galectin-8-IN-l, galectin-8-IN-2, galectin-8N-IN-l, galectin-8N-IN-2, or thiodigalactoside, or a pro-drug form of any of the foregoing.
- the cathepsin inhibitor or cysteine protease inhibitor is cathepsin L-IN-2 (Z-Phe-Phe- FMK), disulfiram, belizatinib, cystatin B, cystatin C, E-64, E-64d, or a pro-drug form of any of the foregoing.
- the system comprises at least two different (e.g., at least three different) immunosuppressive molecules or pro-drug forms thereof.
- the immunosuppressive molecule or pro-drug form thereof has a LogP value greater than 3.0, 3.5, or 4.0.
- the system comprises an aqueous suspension of the immunosuppressive molecule or pro-drug form thereof.
- the aqueous suspension can include triamcinolone acetonide or betamethasone sodium phosphate.
- the system comprises at least one extracellular matrix (ECM) -modifying enzyme, for example, hyaluronidase or a fragment thereof.
- ECM extracellular matrix
- the cell is a non-dividing cell.
- the RNA encoding the double membrane fusogen comprises modified bases.
- the RNA encoding the double membrane fusogen is an mRNA.
- the RNA encoding the double membrane fusogen comprises one or more target sites for at least one micro RNA (miR).
- the DNA comprises modified CpG motifs.
- the DNA has fewer than 100 CpG motifs, the DNA is substantially free of unmodified CpG motifs, and/or the DNA is methylated at one or more CpG motifs.
- the DNA is substantially free of 6-methyladenine and/or 5 -methylcytosine.
- the DNA comprises at least one tissue- specific promoter.
- the tissue- specific promoter is expressed in non-dividing cells.
- the tissue- specific promoter is a muscle-specific promoter.
- the DNA comprises a promoter with fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs.
- the muscle-specific promoter comprises one or more transcription factor binding site core motifs selected from the group consisting of: a. 5’-CATTCC-3’, b. 5’-GGAATG-3’, c. 5’-CCWWWWWWGG-3’, d. 5’-CANNTG-‘3, e. 5 ’ -YTAAAAATA-3 ’ , and f. 5’-TATTTTTAR-3’, wherein N is any nucleotide, W is A or T, Y is T or C, and R is A or G.
- the TCTEX binder comprises a peptide with comprising an amino acid sequence at least five amino acids in length that is at least 80%, at least 85%, at least 90%, at least 95%, or 100% identical to a contiguous amino acid motif selected from the peptides GGFKLNIWDVGGQK (SEQ ID NO: 115), and GVSKTETSQVAPA (SEQ ID NO: 116).
- the disclosure relates to a composition
- a composition comprising the system described herein.
- the composition further comprises a pharmaceutically acceptable carrier.
- the disclosure relates to a pharmaceutical composition
- a pharmaceutical composition comprising an RNA encoding a double membrane fusogen, which, when delivered to a mammalian cell, mediates transfer of a non-native DNA into the nucleus of the mammalian cell and a pharmaceutically acceptable carrier.
- the system comprises the non- native DNA.
- the disclosure relates to nucleic acid comprising a muscle-specific promoter, wherein the muscle-specific promoter comprises fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs, and/or has at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to a nucleic acid sequence comprising SEQ ID NO: 7, 11, 13, 14, 19, 20, or 110 and a TATA box that is positioned within four nucleotides of the position of a TATA box present in SEQ ID NO: 7, 11, 13, 14, 19, 20, or 1 10 respectively.
- the muscle-specific promoter comprises one or more of the transcription factor binding site core motifs selected from the group consisting of: a. 5’-CATTCC-3’, b. 5’-GGAATG-3’, c. 5’-CCWWWWWWGG-3’, d. 5’-CANNTG-‘3, e. 5’-YTAAAAATA-3’, and f. 5’-TATTTTTAR-3’, wherein N is any nucleotide, W is A or T, Y is T or C, and R is A or G.
- the muscle-specific promoter has at least 80% identity to SEQ ID NO: 23 or 24, and wherein the muscle-specific promoter comprises fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs.
- the nucleic acid comprises a DNA polymerase III (Pol III) promoter, wherein the Pol III promoter comprises fewer than 3, 2, or 1 CpG motifs.
- Pol III DNA polymerase III
- the disclosure relates to a system, pharmaceutical composition, or nucleic acid as described herein, wherein the system, pharmaceutical composition, or nucleic acid comprises a DNA encoding a microRNA (miR), wherein the miR comprises: a. a 19-base guide sequence that is complementary to a target mRNA, b. an unpaired loop exactly 4 nucleotides in length, c. a 19-base sequence that is complementary to the 19-base guide sequence, which together with the guide sequence form a 19-base pair stem, and d.
- miR microRNA
- Poly III RNA Polymerase III
- the disclosure relates to a DNA encoding a microRNA (miR), wherein the miR comprises: a. a 19-base guide sequence that is complementary to a target mRNA, b. an unpaired loop exactly 4 nucleotides in length, c. a 19-base sequence that is complementary to the 19-base guide sequence, which together with the guide sequence form a 19-base pair stem, and d. a 3 ’ end that is a U, which is encoded by one of the U or T nucleotides of an RNA Polymerase III (Pol III) transcription termination signal comprising the sequence UUUUU, UUUUU, TTTTTT, or TTTTT.
- RNA Polymerase III Polymerase III
- the system, pharmaceutical composition, or nucleic acid includes an RNA Polymerase III promoter operably linked to the miR.
- the 3’ two nucleotides of the miR can be UU.
- the DNA encodes a microRNA (miR) operably linked to an Hl promoter that is at least 80% identical to nucleotides 1-99 of SEQ ID NO: 154.
- the miR targets a fucosylatransferase-8 (FUT8) gene sequence.
- FIGURES 1A-1B are drawings depicting the function of Brambleberry.
- zebrafish Dio rerio
- karyomeres containing zebrafish chromosomes associate (FIGURE 1A) and then fuse to form a nucleus (FIGURE IB).
- the fusion of karyomeres to form a nucleus is mediated by the Brambleberry (Bmb) protein.
- Brambleberry proteins can be used in nucleic acid transfer systems to mediate a process topologically similar to karyomere fusion in which double membrane- enveloped cellular structures containing exogenous DNA are fused with the nucleus of a cell to deposit the exogenous DNA into the nucleus.
- the double black lines represent lipid bilayers.
- the shaded gray area between the double black lines is topologically similar to the lumen of the endoplasmic reticulum (ER) and the region between the inner and outer membranes of the nuclear envelope.
- the white background represents cytoplasm.
- FIGURES 2A-2B provide depictions of the domain architecture of a Brambleberry protein.
- the signal peptide, luminal domain, transmembrane domains, and cytoplasmic domain of zebrafish (Danio rerio) Brambleberry (SEQ ID NO: 1) were predicted using TOPCONS (FIGURE 2A). These features are indicated on the full-length amino acid sequence of zebrafish Brambleberry (SEQ ID NO: 1) (FIGURE 2B).
- the predicted signal peptide for secretion into the endoplasmic reticulum (ER) is indicated in italics.
- the three predicted transmembrane domains are underlined.
- the predicted cytoplasmic domain is indicated in gray.
- the cytoplasmic domain contains a bipartite nuclear localization signal (NLS), i.e., a basic residue separated by a spacer of thirteen amino acids from three basic residues, where the basic residues are arginine (R) and/or lysine (K).
- the bipartite NLS is indicated in bold within the cytoplasmic domain.
- a canonical monopartite NLS consists of an amino acid sequence five amino acids in length where at least four out of five of the amino acids are basic residues.
- FIGURES 3A-3G depict monomeric and oligomeric structures of the luminal domain of zebrafish Brambleberry.
- the luminal domain of zebrafish (Danio rerio) Brambleberry (SEQ ID NO: 1) was predicted to form a folded domain of four short alpha helices at its N-terminus, whereas the majority of the luminal domain through its C-terminus formed an extended alpha helix (FIGURE 3A).
- An alternative predicted structure for the monomer of zebrafish Brambleberry has the extended helical domain folded back on itself (FIGURE 3B).
- FIGURES 5A-5B are line graphs of the results of experiments in which a Brambleberry mRNA and a DNA expressing a reporter gene were co-packaged into lipid nanoparticles (LNPs) that were used for nucleic acid transfer in differentiated C2C12 myotubes.
- LNPs lipid nanoparticles
- Gaussia luciferase plasmid DNA and zebrafish (Danio rerio) Brambleberry (Bmb) mRNA (or an irrelevant control mRNA) were co-packaged into LNPs and used to transfect differentiated C2C12 myotubes (FIGURE 5A).
- FIGURE 6 is a line graph of a titration experiment testing the dose-dependent effect of the amount of Brambleberry mRNA on the efficiency of nucleic acid transfer.
- Separate LNPs produced using a Nanoassemblr microfluidics device were generated containing NanoLuc plasmid DNA or zebrafish (Danio rerio) Brambleberry (Bmb) mRNA, in order to test the relationship between the amount of Bmb mRNA and nucleic acid transfer efficiency in differentiated C2C 12 myotubes.
- 50 ng plasmid DNA and the amount of Brambleberry indicated in the figure legend were the amounts of each nucleic acid used per well of a 96- well plate.
- NanoLuc luciferase activity was read in the supernatants on Days 1-9 post-nucleic acid transfer. RLU: relative light units.
- FIGURES 8A-8F are a series of bar and line graphs of experiments optimizing and characterizing a muscle-specific promoter.
- Adeno-associated virus (AAV) vectors expressing firefly luciferase were evaluated after intramuscular injection in mice.
- Desmin (DES) and Troponin- 1 (TNNI1) promoters were compared against a synthetic muscle- specific promoter (MSP) and a popular non-tissue specific promoter (CMV) 14 days post- injection (FIGURE 8A).
- MSP synthetic muscle- specific promoter
- CMV non-tissue specific promoter
- FIGURE 8A The effect of adding a TATA box to the MSP in two different orientations rotated 180° was evaluated 7 days post-injection (FIGURE 8B).
- the activity of the MSP with the TATA box was compared against the CASI promoter in a longitudinal experiment (FIGURE 8C). Time points with P ⁇ 0.05 significant differences are marked with an asterisk (*).
- FIG. 8D A version of the muscle-specific promoter with the TATA box lacking CpG motifs was synthesized, and 7 days post-injection luciferase was compared against the original version containing CpG motifs (FIGURE 8D). Plasmids expressing firefly luciferase from the CMV promoter or the MSP promoter were electroporated into the gastrocnemius muscles of mice, and luciferase expression was evaluated by in vivo imaging after 7 days (FIGURE 8E).
- the second plasmid had a CpG- firefly luciferase coding region and an Ori containing CpGs, and was not CpG-methylated by Sssl.
- the third plasmid had a CpG- firefly luciferase coding region and an Ori containing CpGs, and was CpG-methylated by Sssl.
- the fourth plasmid had a CpG- firefly luciferase coding region and an R6K Ori lacking CpGs, did not have CpG motifs, and was not CpG methylated.
- FIGURE 12 shows luciferase reporter gene expression in an experiment evaluating different attachment factors for promoting gene transfer by LNPs into differentiated C2C12 cells.
- the LNPs contained a plasmid DNA expressing NanoLuc luciferase from a muscle- specific promoter and a base-modified mRNA encoding Danio rerio Brambleberry at 1 :2 ratio by weight.
- the control used in this experiment was LNPs generated using the same components except for the lipid-conjugated attachment factor.
- the lipid-conjugated attachment factors evaluated were: ( 1) stearylated Ml 2 peptide with the amino acid sequence RRQPPRSISSHP (SEQ ID NO: 111) (StM12), (2) a first stearylated LAM1 peptide with the amino acid sequence YIGSR (SEQ ID NO: 112), (3) a second stearylated LAM1 peptide with the amino acid sequence RYVVLPR (SEQ ID NO: 113) that is thought to bind heparin sulfate, (4) a stearylated hyaluron-binding peptide with the amino acid sequence GAHWQFNALTVR (SEQ ID NO: 114), (5) an M12 peptide conjugated to a serine-octanoic acid as a second lipid in addition to being stearylated (SsoM12), (6) a DOPE-conjugated RGD 3 amino acid peptide (DOPE-RGD), a cholesterol-conju
- FIGURE 14 shows luciferase reporter gene expression in an experiment evaluating different lipid-conjugated intracellular transport ligands for promoting gene transfer by LNPs into differentiated C2C12 cells.
- the transport ligands evaluated were stearylated versions of peptides that interact with Tctex-1 (DYNLT-1), a dynein light chain protein.
- the two peptides are from Arl2 (GGFKLNIWDVGGQK) (SEQ ID NO: 115), and the C-terminus of rhodopsin D (GVSKTETSQVAPA) (SEQ ID NO: 116). These were conjugated to octyl- arginine (i.e.
- the controls were LNPs with no ligand for intracellular transport proteins (i.e., no dynein binder), and LNPs formulated with stearylated octyl-arginine (StR8). These were compared against identically -formulated LNPs containing stearylated octyl- arginine- Arl2 (StR8Arl2), or stearylated octyl-arginine-conjugated rhodopsin D C-terminus (StR8RhdCT).
- FIGURES 15A -C shows the results of three experiments where orthologs of Danio rerio Brambleberry were evaluated.
- DNA-mRNA LNPs where the mRNA encoded a Brambleberry protein, or negative control LNPs either with the mRNA but no DNA or with DNA but no mRNA, were evaluated for gene transfer efficiency in C2C12 myotubes that had been differentiated for 5 days in 2% horse serum.
- the Brambleberry mRNAs were all produced using N1 -methylpseudouridine (NlmpU) in place of uridine.
- NlmpU N1 -methylpseudouridine
- mRNA encoding Danio rerio Brambleberry (SEQ ID NOs: 1 -2) was compared against mRNAs encoding sheepshead pupfish (Cyprinodon variegatus) Brambleberry (SEQ ID NOs: 33-34) and Komodo dragon (Varanus komodoensis) Brambleberry (SEQ ID NOs: 29-30) (FIGURE 15 A).
- mRNA encoding Danio rerio Brambleberry also was compared against pigeon (Columba livia) Brambleberry (SEQ ID NOs: 27-28), sheepshead pupfish (Cyprinodon variegatus) Brambleberry (SEQ ID NOs: 33-34), and Hawaiian crow Brambleberry (Corvus hawaiiensis) (SEQ ID NOs: 119-120) (FIGURE 15B).
- FIGURE 15C shows the effect of including Brambleberry mRNA on gene transfer efficiency in C2C12 myotubes that had been differentiated for 10 days.
- the C2C12 cells were switched from media containing 20% FBS to media containing 2% horse serum.
- the relative light units (RLU) emitted by firefly luciferase indicates the relative efficiency of gene transfer.
- the conditions tested were a no LNP control, a DNA-only control, and DNA-mRNA LNPs where the mRNA encoded Danio rerio Brambleberry. Gene transfer efficiency was 472-fold higher for the DNA-mRNA LNPs containing Brambleberry mRNA than the DNA-only LNPs.
- FIGURE 17 is a graph showing average particle sizes determined by dynamic light scattering (DLS) of LNPs that were generated with different ionizable lipids and stored under different conditions.
- LNPs were generated using 56.9% ionizable lipid, 27.5% cholesterol, 11.2% DSPC, 1.5% DMG-PEG2000, 0.25% stearylated TCTEX-1 binder StR8RhdCT (SEQ ID NO: 1 18), and 4.3% DPG-galloyl.
- the ionizable lipids evaluated in this experiment included DLin-KC2-DMA (KC2), SM102, Lipid 29, and CL15F6.
- the resulting LNPs were sized by DLS after either being left unfiltered and stored at 4°C, filtered and 4°C, filtered and stored at -20°C, or filtered and stored at -80°C.
- FIGURES 18A-H depict the results of an experiment designed to assess the impact of different ionizable lipids on the efficiency of gene transfer by DNA-mRNA LNPs after cryopreservation.
- This experiment utilized the same preparations of LNPs as shown in FIGURE 17.
- the LNPs were generated using 27.5% cholesterol, 11.2% DSPC, 1.5% DMG- PEG2000, 0.25% stearylated TCTEX-1 binder StR8RhdCT (SEQ ID NO: 118), and 4.3% DPG-galloyl, and 56.9% of the ionizable lipids DLin-KC2-DMA (KC2), SM102, Lipid 29, or CL15F6.
- FIGURES 18A-D The chemical structures of the ionizable lipids used here, as well as the pKa of the ionizable head group, are shown in FIGURES 18E-H.
- FIGURE 19 is a graph of the results of an experiment designed to assess whether forms of polyethylene glycol (PEG) with shorter lipid anchors than DMG-PEG were optimal.
- PEG2000 with a cholesterol, stearic acid (SA), or C8 ceramide (C8C) anchor were compared against DMG-PEG2000.
- DNA-mRNA LNPs were generated using a mixture of lipids containing either IX (1.4% of total lipid by mass) or 2X (2.8% of total lipid by mass) of the PEGylated lipid. Unfiltered LNPs were evaluated as a quality control measure, to ensure the absence of signal loss due to unfilterable LNP aggregates.
- Filtered LNPs were evaluated after storage at 4°C or -80°C in 10-day differentiated C2C12 myotubes.
- the efficiency of gene transfer was assessed by measuring the firefly luciferase signal 4 days after adding the LNPs to 96- well plates containing the 10-day differentiated C2C12 myotubes.
- the firefly luciferase signal was quantified in terms of relative light units (RLU).
- FIGURE 20 is a graph of the results of an experiment designed to assess the impact of using lipid anchors of reduced size on DNA-mRNA LNP stability through cryopreservation and gene transfer efficiency.
- LNPs containing DNA were generated with and without Brambleberry mRNA, using the ionizable lipids DLin-KC2-DMA or CL15F6, and with either DMG-PEG2000 or C8C-PEG2000.
- the LNPs evaluated were either left unfiltered and stored at 4°C, filtered and then stored at 4°C, or filtered and then stored at - 80°C.
- the LNPs were evaluated for gene transfer efficiency in 10-day differentiated C2C12 myotubes.
- the amount of LNPs added to the cells was normalized to the amount of DNA, and the DNA-mRNA LNPs were generated using a 1:2 ratio of DNA to Brambleberry mRNA.
- Gene transfer efficiency was assessed by measuring firefly luciferase activity, which was quantified in terms of relative light units (RLU) 4 days after adding the LNPs to 10-day differentiated C2C12 myotubes.
- DNA-mRNA LNPs containing Brambleberry mRNA was 251-fold more efficient than the DNA-only LNPs, where both were made using DLin-KC2-DMA and DMG- PEG2000, 324-fold more efficient than the DNA-only LNPs in the context of LNPs made with CL15F6 and DMG-PEG2000 that were cryopreserved at -80°C, and 234-fold more efficient than the DNA-only LNPs in the context of LNPs made with CL15F6 and C8 ceramide (C8C)-PEG2000.
- FIGURES 21A-D show the results of a mouse experiment designed to validate the effect of including Brambleberry mRNA on the efficiency of gene transfer to a non-dividing cell type in vivo. This experiment was also designed to assess whether the 5’ cap Cap2 was non-inferior to Capl when present on the Brambleberry mRNA utilized to facilitate gene transfer. Mice received intramuscular injections of DNA-only control LNPs or DNA-mRNA LNPs where the mRNA encodes Danio rerio Brambleberry (SEQ ID NOs: 1-2), and the
- DNA is a circular DNA molecule containing only 14 CpG motifs per molecule that expresses firefly luciferase under a muscle-specific promoter (SEQ ID NO: 151).
- DNA and mRNA were encapsulated in LNPs at a ratio of 1 :2, and the amounts of LNPs administered to the mice by intramuscular injection in the quadriceps were normalized to 10 pg DNA per mouse.
- Mice were injected with the LNPs at week 0 and imaged on weeks 1, 2, 3, 4, 5, and 6 (FIGURE 21 A). The efficiency of gene transfer was quantified by reading the luminosity after weekly luciferin injection by IVIS (FIGURE 21B).
- the group injected with DNA-mRNA LNPs lacking chol-R8RhdCT was followed for 5 weeks and the luciferase expression was compared to the otherwise-identical group where the DNA-mRNA LNPs contained chol-R8RhdCT (FIGURE 21D).
- mice received intramuscular injections of DNA-only control LNPs or DNA-mRNA LNPs where the mRNA encodes Danio rerio Brambleberry (SEQ ID NOs: 1-2), and the DNA is a circular DNA molecule containing only 14 CpG motifs per molecule that expresses firefly luciferase under a muscle- specific promoter (SEQ ID NO: 151).
- DNA and mRNA were encapsulated in LNPs at a ratio of 1:2, and the amounts of LNPs administered to the mice by intramuscular injection in the quadriceps were normalized to 10 pig DNA per mouse.
- Mice were injected with the LNPs at week 0 and imaged on weeks 1, 2, 3, 4, 5, and 6 (FIGURE 22 A). The efficiency of gene transfer was quantified by reading the luminosity after weekly luciferin injection by IVIS (FIGURE 22B).
- FIGURE 24C shows the results of a similar knockdown experiment where the miR was expressed from either a wild-type U6 promoter, a wild-type Hl promoter, or versions of the Hl promoter where the CpG motifs were mutated to CA (version 1, vl) or TG (version 2, v2).
- FIGURES 25A-B are two bar graph charts showing the N-linked glycans attached to an eCD4-Ig protein made in cells with and without a miR that knocks down expression of Fut8.
- N-linked glycans were removed from eCD4-Ig protein by PNGase F, permethylated, and characterized by liquid chromatography-mass spectrometry (LC-MS).
- FIGURE 25A shows the N-linked glycan content of the eCD4-lg protein made in the parental cell line
- FIGURE 25B shows the N-linked glycan content of the eCD4-Ig protein made in the cell line expressing the miR that targets Fut8.
- FIGURE 26 shows the prevalence of an O-linked glycan attached to eCD4-Ig protein made with or without co-transfection with a plasmid expressing a miR that knocks down GALNT2.
- An eCD4-Ig protein was generated by transient transfection with an empty control plasmid or a plasmid expressing a miR that knocks down GALNT2 was characterized by mass spectrometry under the conditions described herein. The proportion of eCD4-Ig protein masses with and without an O-linked glycan were compared. 24% of the control eCD4-Ig protein was O-glycosylated, whereas 3.4% was O-glycosylated when made by co-transfection with the plasmid expressing the miR that knocks down GALNT2.
- FIGURE 27 shows a bar graph of a comparison of the efficiency of gene transfer by DNA-mRNA LNPs after intramuscular injection with and without hyaluronidase.
- Five nude mice per group received 25 pL intramuscular injections of LNPs containing 10 pg DNA and 20 pg Brambleberry mRNA in the quadriceps. Mice were imaged by IVIS after luciferin injection.
- Bovine hyaluronidase was added after thawing of cryopreserved LNPs such that its final concentration was 1 unit per pL after mixing with the thawed LNPs.
- the present disclosure is based, in part, on the discovery that double membrane fusogen proteins provide a solution to the topological problem of transferring DNA sequestered within a double membrane envelope to the nucleus.
- chromosomes become enveloped by a double membrane in structures known as karyomeres, which ultimately fuse to form the nucleus.
- the protein that mediates this fusion is Brambleberry — so named, because in its absence, consolidation of karyomeres into a nucleus is arrested, and the unfused karyomeres resemble a brambleberry (Abrams et al. (2012) supra) (FIGURE 1A).
- the disclosure relates, in part, to systems and methods for the transfer of a non-native DNA into a nucleus of a cell, such as a mammalian cell.
- the system or method comprises a non-native DNA and an RNA that encodes a double membrane fusogen protein, which, when present in the cell, mediates transfer of the non-native DNA into the nucleus of the cell.
- a plasmid with a CpG-free promoter in non- dividing cells is capable of persistently expressing a gene of interest, without being silenced, and without the loss of the plasmid, e.g., by degradation. Furthermore it has been discovered that repositioning the TATA box in naturally occurring muscle-specific promoters results in improved expression. Accordingly, the disclosure also relates, in part, to muscle-specific promoters that can be used in combination with the systems described herein, or in other applications.
- double membrane means a biological membrane consisting of two lipid bilayers separated by an aqueous phase.
- a well-known example of a double membrane is the nuclear envelope a eukaryotic cell.
- a nuclear envelope has two membrane layers, an inner bilayer and an outer bilayer.
- double membrane fusogen means any protein or group of proteins that mediate the fusion of a first double membrane with a second double membrane.
- Brambleberry means any protein that has a signal peptide for secretion into the ER, a luminal domain, and a transmembrane domain(s) and is capable of performing the same function as zebrafish (Danio rerio) Brambleberry fusing the nuclear envelope with another double membrane-enveloped structure, thereby introducing DNA contained within the double membrane-enveloped structure into the nucleus).
- Brambleberry proteins include any protein annotated by GenBank as a
- Brambleberry protein as well as protein with at least 70% (e.g., at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100%) sequence identity to a protein annotated by GenBank as a Brambleberry protein or other ortholog of a Brambleberry protein and which is capable of performing the same function as zebrafish Brambleberry (e.g.
- Exemplary Brambleberry proteins also include any chimeric protein that includes one or more portions of at least one Brambleberry protein and is capable of performing the same function as zebrafish Brambleberry as well as any split Brambleberry protein that is capable of performing the same function as zebrafish Brambleberry.
- split Brambleberry means two or more proteins capable of forming a protein complex, where the protein complex is capable of performing the same function as zebrafish Brambleberry, e.g., a first protein that includes the luminal and transmembrane domains of zebrafish Brambleberry plus a non-native dimerization domain, combined with a second protein that includes a matched non-native dimerization domain capable of dimerizing with that of the first protein and the DNA-binding domain of zebrafish Brambleberry.
- non-native or “non-endogenous”, as used herein, means not existing naturally in a place but coming from somewhere else.
- a non-native DNA or a non-endogenous DNA refers to a DNA existing, e.g., in a cell, where the DNA does not naturally occur in the cell.
- the term “functional fragment” of a protein refers to a fragment of a protein that retains, for example, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, or 100% of the protein activity of the corresponding full-length, naturally occurring protein. Protein activity may be assayed by any method known in the art.
- the functional fragment comprises at least 50, at least 75, at least 100, at least 125, or at least 150 consecutive amino acids present in the protein.
- the functional fragment comprises a truncation of about 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, or 50 amino acids as compared to the protein.
- non-coding RNA means an RNA molecule that mediates a function other than serving as a message for translation into protein, e.g., an shRNA, miR, ribozyme, or aptamer.
- denatured means DNA lacking canonical Watson-Crick base pairing.
- Denatured DNA can be generated by melting the DNA with increased temperatures, e.g., 95°C, optionally followed by rapid cooling.
- the present disclosure relates, in part, to a system (e.g. , a nanoparticle such as a lipid nanoparticle, a formulation, or a pharmaceutical formulation) for nucleic acid transfer in which an RNA molecule encoding a protein that mediates the entry of a DNA into the nucleus e.g. , a double membrane fusogen protein) is co-packaged with DNA, for example, DNA encoding a gene of interest such as a therapeutic gene.
- a system e.g. , a nanoparticle such as a lipid nanoparticle, a formulation, or a pharmaceutical formulation
- a system e.g. , a nanoparticle such as a lipid nanoparticle, a formulation, or a pharmaceutical formulation
- DNA for example, DNA encoding a gene of interest such as a therapeutic gene.
- the cytoplasmic domain of a double membrane fusogen protein can include a nuclear localization signal (NLS).
- NLS nuclear localization signal
- An exemplary NLS comprises or consists of an amino acid sequence five amino acids in length wherein at least four out of five of the amino acids are basic residues.
- Another exemplary NLS comprises or consists of an amino acid sequence comprising at least one basic residue separated by a spacer of ten to fifteen amino acids from three or more basic residues.
- the cytoplasmic domain can include from one to about 30 basic residues separated by about 10, 11, 12, 13, 14, or 15 amino acids from three, four, five, six, seven, eight, nine, ten, or more basic residues.
- the NLS does not naturally occur in the amino acid sequence of a naturally -occurring wild-type double membrane fusogen (e.g., Brambleberry protein) having the greatest sequence identity to the double membrane fusogen.
- the term “functional fragment” of a double membrane fusogen protein refers to fragment of a double membrane fusogen protein that retains, for example, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, or 100% of the fusogenic protein activity of the corresponding full-length, naturally occurring double membrane fusogen protein.
- Fusogenic protein activity may be assayed by any method known in the art, including, for example, by measuring one or more criteria indicative of membrane fusion activity, such as measuring expression of a reporter protein following co-transduction of a DNA encoding the reporter protein with an RNA encoding the double membrane fusogen protein fragment in a C2C12 myotube model, as described in Example 1.
- the functional fragment of a double membrane fusogen protein comprises an amino acid sequence having at least 80%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91 %, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to one or more of the signal peptide, the luminal domain, the transmembrane domain, and the cytoplasmic domain comprising a DNA-binding domain of SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, or 147.
- Fusogenic protein activity may be assayed by any method known in the art, including, for example, by measuring one or more criteria indicative of the membrane fusion activity, such as measuring expression of a reporter protein following co-transduction of a DNA encoding the reporter protein with an RNA encoding the double membrane fusogen protein fragment in a C2C12 myotube model, as described in Example 1.
- the variant of a double membrane fusogen protein comprises an amino acid sequence having at least 80%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to a double membrane fusogen protein sequence described herein.
- the variant of a double membrane fusogen protein comprises an amino acid substitution relative to a double membrane fusogen protein sequence provided herein.
- the variant of a double membrane fusogen protein comprises 1, up to 2, up to 3, up to 4, up to 5, up to 6, up to 7, up to 8, up to 9, or up to 10 amino acid substitutions relative to a double membrane fusogen protein sequence provided herein (e.g., SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, and 147).
- percent identity refers to the extent to which two sequences e.g., two polypeptides or two nucleic acids have the same respective amino acid or nucleotide at the same positions in an alignment.
- percent identity between a polypeptide sequence and a reference sequence is defined as the percentage of amino acid residues in the polypeptide sequence that are identical to the amino acid residues in the reference sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity.
- percent identity between a nucleic acid sequence and a reference sequence is defined as the percentage of nucleotides in the nucleic acid sequence that are identical to the nucleotides in the reference sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity. Alignment for purposes of determining percent sequence identity (e.g., nucleic acid sequence identity or amino acid sequence identity) can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST (Basic Local Alignment Search Tool), BLAST- 2, ALIGN, MEGALIGN (DNASTAR), CLUSTALW, CLUSTAL OMEGA, or MUSCLE software.
- BLAST Basic Local Alignment Search Tool
- BLAST- 2 ALIGN
- MEGALIGN MEGALIGN
- CLUSTALW CLUSTAL OMEGA
- MUSCLE software MUSCLE software
- the alignment algorithms above may take into account a scoring matrix to calculate an alignment score (see Chao et al., BIOMOLECULES (2022) 12(4): 546).
- the scoring matrix recommended by the BLAST algorithm is BLOSUM-62.
- the BLOSUM-62 scoring matrix assigns positive, zero, or negative scores between each pair or standard amino acid residues (see Henikoff and Henikoff, PROC. NATL. ACAD. SCI. USA (1992) 89, 10915-19 at FIG. 2).
- a positive score between two amino acid residues indicates that substitution of these amino acid residues for each other is conservative.
- the gene can be carried on a vector, e.g.. a recombinant plasmid or virus that is delivered into a cell.
- the vector can be circular and/or lack free 5’ and 3’ ends.
- the vector can carry a gene which does not naturally occur with the vector sequences flanking it.
- the gene can be operatively linked to regulatory components in a manner which permits transcription, translation, and/or expression of the gene in a target cell.
- the non-native gene can be derived from any organism. In certain embodiments, the non-native gene is derived from a human.
- the DNA comprises a coding region that is operably linked to a promoter.
- the promoter is a tissue-specific promoter.
- the tissue-specific promoter can be expressed in non-dividing cells, such as muscle cells.
- the DNA comprises a promoter with fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs.
- Muscle is an important target tissue for systems for nucleic acid transfer, but is comprised primarily of differentiated, non-dividing cells.
- fusogen proteins in nucleic acid delivery systems enables efficient non-viral systems for nucleic acid delivery to non-dividing cells including muscle.
- APCs transduced antigen-presenting cells
- the amount of the product of the DNA or interest presented by a transduced APC is thought to be far greater than the amount of product of the DNA an APC would be able to pick up from plasma or lymph.
- limiting expression of the product of the DNA in APCs may limit the potential for immune responses targeting the product of the DNA.
- Use of a muscle-specific promoter also has the safety advantage of reducing or preventing expression in other off-target cell types, in addition to APCs.
- the muscle specific promoters (MSP) of the disclosure may be engineered to improve expression of a gene (e.g., a gene present on a non- native DNA) that is operably linked to the MSP.
- a gene e.g., a gene present on a non- native DNA
- the system or method for transfer of a non-native DNA into a nucleus of a cell includes the non-native DNA and an RNA that encodes a membrane fusogen protein, which, when present in the cell, mediates transfer of the non- native DNA into the nucleus of the cell.
- the non-native DNA may include a muscle-specific promoter operably linked to a gene, which, when transferred to the nucleus of the cell, is expressed.
- TATA box e.g., TAT ATA
- TAT ATA a region of DNA that helps initiate transcription, at certain positions within a promoter
- promoters e.g., muscle-specific promoters, engineered to reduce the number of CpG motifs.
- a promoter e.g., a muscle-specific promoter
- a promoter is engineered to contain fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs.
- a promoter e.g., a muscle-specific promoter is engineered to contain no CpG motifs.
- MSP muscle-specific promoter
- enhancer created by random ligation of oligos containing muscle-specific transcription factor binding sites (Li et al. (1999) NAT. BIOTECHNOL., 17 (3): 241-245) and further including a TATA box at a position that improves expression, e.g., SEQ ID NO: 7.
- Other exemplary muscle-specific promoters comprising a TATA box positioned to improve expression include SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, and SEQ ID NO: 22 or a variant promoter having 1, 2, 3, 4, 5 ,6, 7, 8, 9, or 10 nucleotide substitutions as compared to any of the foregoing that is capable of expressing a non-native gene in a muscle tissue in an amount that is at least 80% (e.g., at least 85%, at least 90%, at least 95%) of the expression achievable using the promoter selected from SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, and SEQ ID NO: 22.
- the level of expression of a given promoter can he tested using a reporter gene in a muscle cell and/or tissue, e.g., as shown in the Examples herein.
- the regulatory sequences useful in the constructs provided herein may also contain an intron, desirably located between the promoter/enhancer sequence and the gene.
- One desirable intron sequence is derived from SV-40, and is a 100 bp mini-intron splice donor/splice acceptor referred to as SD-SA.
- the intron comprises the nucleotide sequence of SEQ ID NO: 10, or a codon-optimized or fragment thereof.
- Another suitable sequence includes the woodchuck hepatitis virus post-transcriptional element. (See, e.g., L. Wang and I. Verma (1999) PROC. NATL. ACAD. Set. USA, 96: 3906-3910).
- PolyA signals may be derived from many suitable species, including, without limitation SV-40, human and bovine.
- Enhancer sequences useful herein include the IRBP enhancer, immediate early cytomegalovirus enhancer, one derived from an immunoglobulin gene or SV40 enhancer, the cis-acting element identified in the mouse proximal promoter, etc.
- the DNA of the disclosure can comprise modified CpG motifs.
- the DNA can have fewer than 100 CpG motifs, the DNA can be substantially free of unmodified CpG motifs, and/or the DNA can be methylated at one or more CpG motifs.
- the DNA is substantially free of 6- methyladenine and/or 5 -methylcytosine.
- the DNA can include one or more genes encoding proteins for expression in a cell.
- the gene can encode a protein that can be used to correct or ameliorate gene deficiencies, which may include deficiencies in which normal genes are expressed at less than normal levels.
- the gene can encode an antibody (e.g., a monoclonal or a bispecific antibody), an scFv, a Fab, or an Fc region.
- the gene can encode a protein comprising at least one domain that binds a ligand and at least one effector domain.
- the nucleic acid transfer system allows for the transfer of the DNA to the nucleus where it is expressed.
- the DNA comprises a gene capable of expressing a secreted protein that is secreted from the cell.
- the HSA protein that has at least 80% (e.g., at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100%) sequence identity to HSA or a functional fragment thereof.
- a plasmid expressing an eCD4-Ig gene and/or a TPST2 gene lacks CpG motifs outside of the bacterial origin of replication (Ori).
- the CpG motifs of the Ori can be methylated prior to use as described herein.
- the plasmids can be generated and co-packaged with an mRNA encoding a double membrane fusogen protein (e.g., a Brambleberry-encoding mRNA) in LNPs as described herein.
- the mRNA can include modified uridine nucleotides.
- the LNPs can be formulated for injection in a pharmaceutically-acceptable carrier.
- An exemplary sequence of a plasmid for expressing eCD4-Ig under a CpG-free muscle-specific promoter is provided at SEQ ID NO: 35.
- the sequence of a similar plasmid for expressing TPST2 under a CpG-free muscle-specific promoter is provided at SEQ ID NO: 36.
- Versions of the plasmid for expressing eCD4-Ig that are further modified to express an shRNA for knocking down fucosyltransferase-8 (FUT8) by RNA interference (RNAi) (SEQ ID NO: 37), or a miR for knocking down FUT8 by RNAi (SEQ ID NO: 38), are provided.
- PCSK9 proprotein convertase subtilisin/kexin type 9
- sequences of monoclonal antibodies that bind PCSK9 are known in the art and can be used as coding sequences in the systems described herein.
- a PCSK9 antibody gene can be present on a plasmid.
- the plasmid sequence lacks CpG motifs outside of the bacterial origin of replication (Ori).
- the CpG motifs of the Ori can be methylated prior to use as described herein.
- the plasmids can be generated and co-packaged with an mRNA encoding a double membrane fusogen protein e.g., a Brambleberry-encoding mRNA) in LNPs as described herein.
- the mRNA can include modified uridine nucleotides.
- the LNPs can be formulated for injection in a pharmaceutically-acceptable carrier.
- the sequence of an exemplary plasmid for expressing evolocumab under a CpG- free muscle- specific promoter is provided at SEQ ID NO: 39.
- the plasmid is a bicistronic plasmid for co-expressing the heavy and light chains of evolocumab.
- GLP1R Glucagon-like Peptide- 1 Receptor
- an RNA polymerase III (pol III) promoter (e.g., an Hl pol III promoter) is operably linked to the miRNA.
- the RNA pol III promoter can be an Hl promoter.
- the Hl promoter has at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% identity to SEQ ID NO: 154.
- the pol III promoter lacks CpG sites.
- a plasmid expressing a non-coding RNA can be used as an off-switch for gene therapy.
- a non-coding RNA e.g., a miR
- a non-coding RNA that targets the therapeutic gene can be administered using the nucleic acid transfer systems and methods described herein.
- the therapeutic protein comprises a 3’ UTR sequence with one or more target sites for one or more miRs.
- an RNA polymerase III (pol III) promoter e.g., an Hl pol III promoter
- the RNA pol III promoter can be an Hl promoter.
- the Hl promoter has at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% identity to SEQ ID NO: 154.
- the pol III promoter lacks CpG sites.
- a plasmid expressing a miR (SEQ ID NO: 54) and a matched 3’ UTR sequence (SEQ ID NO: 55) with multiple target sites for that miR are provided.
- the miR is expressed from a version of the Hl RNA polymerase III (Pol III) promoter that has been modified to lack CpG sites.
- the plasmid can be methylated at CpG sites without the Hl promoter being methylated (and, thereby, potentially silenced), and the promoter does not provide recognition motifs for TLR9, thereby avoiding stimulation of the innate immune response.
- An exemplary miR contemplated herein includes the following features: a 19 base guide sequence, a loop of unpaired bases exactly 4 nucleotides in length, a 19 base sequence that is complementary to the guide sequence and forms a 19 base pair stem, and an RNA Polymerase III (Pol III) transcription termination signal (TTTTTT or UUUUUU) that overlaps with the 3’ end of the stem.
- TTTTTT or UUUUUU RNA Polymerase III transcription termination signal
- the 3’ end is a U.
- the 3’ end is a UU.
- RNA Polymerase III transcription termination signal
- an RNA Polymerase III promoter is operably linked to the miR.
- the non-coding RNA targets a gene involved in glycosylation.
- exemplary genes involved in glycosylation include Fut8 and GALNT2.
- the nucleic acid transfer system of the disclosure can include an immunosuppressive molecule or pro-drug form thereof, and/or an RNA encoding an immunosuppressive protein.
- the nucleic acid transfer systems of the disclosure can include an immunosuppressive non-coding RNA (e.g., an antisense oligonucleotide (ASO)) or a pro- drug form thereof that binds a nucleic acid sensing molecule such as C2mutl (as described above), a corticosteroid such as glucocorticoid, prednisone, prednisolone, triamcinolone, methylprednisolone, dexamethasone, dexamethasone palmitate, fluticasone, fluticasone propionate, fluticasone furoate, clobetasol, betamethasone, halobetasol, amcinonide, desoximetasone, diflorasone, mometasone
- immunosuppressive molecules include mRNAs encoding immunosuppressive molecules, such as indoleamine 2,3-dioxygenase-l (IDO1), NF kappa B inhibitor alpha (NFKBIA), inhibitor of kappa B subunit beta (IKBKB), TNF alpha induced protein 3 (TNFAIP3), interferon regulatory factor 4 (IRF4), interferon regulatory factor 8 (1RF8), cytotoxic T-lymphocyte-associated protein 4 (CTLA4), transforming growth factor beta-1 (TGFB1), interleukin 1 receptor type 2 (IL1R2), suppressor of cytokine signaling-1 (SOCS1), suppressor of cytokine signaling-2 (SOCS2), suppressor of cytokine signaling-3 (SOCS3), suppressor of cytokine signaling-4 (SOCS4), suppressor of cytokine signaling-5 (SOCS5), suppressor of cytokine signaling-6 (SOCS6), suppressor of cytokine signaling-7 (IDO
- mRNAs, siRNAs, and ASOs can be co-formulated with an mRNA encoding a double membrane fusogen protein (e.g. , Brambleberry mRNA) within an LNP, to reduce the potential for nucleic acid transfer to stimulate immune responses against the therapeutic gene.
- a double membrane fusogen protein e.g. , Brambleberry mRNA
- immunosuppressive molecules or pro-drug forms thereof including, e.g., corticosteroids (such as glucocorticoid), mTOR inhibitors, tyrosine kinase inhibitors, ASOs, and siRNAs, and/or mRNAs encoding immunosuppressive molecules, can be formulated with LNPs containing an mRNA encoding a double membrane fusogen protein (e.g., Brambleberry mRNA) and a DNA.
- Immunosuppressive molecules or pro-drug forms thereof can also include immunosuppressive molecule or pro-drug form thereof that bind human glucocorticoid receptor.
- the immunosuppressive molecule or pro-drug form thereof can contain an ester bond.
- the immunosuppressive molecule or prodrug form thereof is selected from the group consisting of: galectin inhibitors, such as GB1107, galectin- 3/galectin-8-IN-l, olitigaltin, selvigaltin, galectin-3/galectin-8-IN-2, galectin-3-IN-2, galectin-3-IN-3, galectin-3-IN-5, GB1490, GB1908, galectin-8-IN-l, galectin-8-IN-2, galectin-8N-IN-l, galectin-8N-IN-2, or thiodigalactoside, or pro-drug form of any of the foregoing; cysteine protease inhibitors, such as cathepsin inhibitors (such as cathepsin L-IN-2 (Z-Phe-Phe-FMK)), disulfiram, belizatinib, cystatin B, cystatin C, E-64, or E-64d or
- galectin inhibitors such
- the immunosuppressive molecule or pro-drug form thereof is selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, or NLRP3 inflammasome inhibitors. In certain embodiments, the immunosuppressive molecule or pro-drug form thereof is an NLRP3 inflammasome inhibitor.
- an LNP of the present disclosure comprises an ionizable lipid, a structural lipid, a PEGylated lipid (aka PEG lipid), and a phospholipid.
- an LNP comprises an ionizable lipid, a structural lipid, a PEGylated lipid (aka PEG lipid), and a zwitterionic amino acid lipid.
- an LNP further comprises a 5th lipid, besides any of the aforementioned lipid components.
- the LNP encapsulates one or more elements of the active agent of the present disclosure.
- an LNP further comprises a targeting moiety covalently or non-covalently bound to the outer surface of the LNP.
- the targeting moiety is a targeting moiety that binds to, or otherwise facilitates uptake by, cells of a particular organ system.
- the mol-% of the ionizable lipid may be from about 35 mol-% to about 55 mol-%. As a non-limiting example, the mol-% of the ionizable lipid may be from about 40 mol-% to about 50 mol-%.
- the mol-% of the phospholipid may be from about 1 mol-% to about 50 mol-%. In certain embodiments, the mol-% of the phospholipid may be from about 2 mol-% to about 45 mol-%. In certain embodiments, the mol-% of the phospholipid may be from about 3 mol-% to about 40 mol-%. In certain embodiments, the mol-% of the phospholipid may be from about 4 mol-% to about 35 mol-%. In certain embodiments, the mol-% of the phospholipid may be from about 5 mol-% to about 30 mol-%.
- the mol-% of the phospholipid may be from about 10 mol-% to about 20 mol- %. In some embodiments, the mol-% of the phospholipid may be from about 5 mol-% to about 20 mol-%.
- the mol-% of the structural lipid may be from about 10 mol-% to about 80 mol-%. In certain embodiments, the mol-% of the structural lipid may be from about 20 mol-% to about 70 mol-%. In certain embodiments, the mol-% of the structural lipid may be from about 30 mol-% to about 60 mol-%. In certain embodiments, the mol-% of the structural lipid may be from about 35 mol-% to about 55 mol-%. In some embodiments, the mol-% of the structural lipid may be from about 40 mol-% to about 50 mol-%.
- the mol-% of the PEG lipid may be from about 0.1 mol-% to about 10 mol-%. In certain embodiments, the mol-% of the PEG lipid may be from about 0.2 mol-% to about 5 mol-%. In certain embodiments, the mol-% of the PEG lipid may be from about 0.5 mol-% to about 3 mol-%. In certain embodiments, the mol-% of the PEG lipid may be from about 1 mol-% to about 2 mol-%. In certain embodiments, the mol-% of the PEG lipid may be about 1 .5 mol-%. In certain embodiments, the mol-% of the PEG lipid may be about 2.5 mol-%.
- Additional features of an ionizable lipid contemplated for use in the systems and methods herein include a pKa above 6.5, one or more branched acyl chains, one or more hydrolysable ester bonds, and few (e.g., less than 10, less than 9, less than 8, less than 7, less than 6, less than 5, less than 4, less than 3, less than 2, or no unsaturated bonds).
- Any lipid or combination of lipids that are known in the art can be used to produce a LNP.
- lipids used to produce LNPs are: DOTMA, DOSPA, DOTAP, DMRIE, DC-cholesterol, DOTAP-cholesterol, GAP-DMORIE-DPyPE, and GL67A-DOPE-DMPE- polyethylene glycol (PEG).
- cationic lipids are: 98N12-5, C12-200, DLin-KC2- DMA (KC2), DLin-MC3-DMA (MC3), XTC, MD1, SM102, Lipid 29, CL15F6, and 7C1.
- neutral lipids are: DPSC, DPPC, POPC, DOPE, and SM.
- PEG- modified lipids examples include PEG-DMG (l,2-Dimyristoyl-sn-glycero-3-methoxypolyethylene glycol), PEG-CerC14, and PEG-CerC20.
- Exemplary LNPs are described, for example, in U.S. Patent Nos. 8,058,069; 8,492,359; 8,822,668; 9,364,435; 9,504,651; 11,141,378; 9,404,127;
- peptides that can be conjugated to lipids or pegylated lipids that are useful in targeting muscle have been previously described, e.g., SEQ ID NOs: 103-105 (Tabebordbar et al. ,(2021), supra), SEQ ID NO: 56 (Ghosh and Barry (2005) J. VlROL., 79(21): 13667-13672), and SEQ ID NO: 57 (Schaffer et al. (2003) PROC. NATL. ACAD. Set. USA., 100:4435-4439, Jackson et al. (2020) MOL. THER. METHODS CLIN. DEV., 19: 496-506, and International Patent Application No. PCT/US2021/042200).
- Such peptides can be conjugated to lipids or pegylated lipids and used as attachment moieties to target the LNPs of the present invention to muscle.
- PEG500 to PEG5000 forms of PEG that may be suitable for use the systems and methods of the disclosure, depending on the circumstances, include PEG500 to PEG5000.
- PEG500, PEG1000, PEG1500, PEG2000, PEG2500, PEG3000, PEG3500, PEG4000, PEG4500, and PEG5000, or any range therein, can be used in accordance with the systems and methods herein.
- the present invention provides systems for nucleic acid transfer that promote nucleic acid transfer by electroporation.
- An isolated Brambleberry mRNA is injected intramuscularly, e.g., as a mixture in water, with DNA molecules encoding a therapeutic gene, and electrical pulses are applied to promote the entry of Brambleberry mRNA and the DNA vector into cells.
- Brambleberry allows DNA to enter the nucleus that otherwise would have entered the cell but lacked access to the nucleus.
- the present invention also provides systems for nucleic acid transfer that promote nucleic acid transfer that include dynein binders (e. ., a TCTEX-1 binder) and/or DNA binders. It is contemplated that the inclusion of a molecule containing both a DNA binder and a dynein binder may facilitate the intracellular trafficking of the DNA to a perinuclear localization that promotes gene transfer. Examples of dynein binders include small molecules and peptides. Dynein binders can be stearylated to facilitate their incorporation into LNPs.
- the dynein binder is a peptide selected from Arl2 (GGFKLNIWDVGGQK) (SEQ ID NO: 115) and the C-terminus of rhodopsin D (Rhd) (GVSKTETSQVAPA) (SEQ ID NO: 116).
- DNA binders include octyl- arginine.
- the dynein binder and the DNA binder are conjugated.
- Exemplary dynein-DNA binder conjugates include octyl-arginine conjugated to Arl2 (SEQ ID NO: 117) and octyl-arginine conjugated to Rhd (R8RhdCT, SEQ ID NO: 118).
- the nucleic acid transfer systems of the disclosure include at least one extracellular matrix (ECM)-modifying enzyme.
- ECM extracellular matrix
- the ECM-modifying enzyme is a hyaluronidase or a fragment thereof.
- the present disclosure also provides pharmaceutical compositions or formulations that include an active agent (e.g., a double membrane fusogen protein, a transgenic gene, a non-coding RNA, a system for DNA transfer) described herein.
- an active agent e.g., a double membrane fusogen protein, a transgenic gene, a non-coding RNA, a system for DNA transfer
- the pharmaceutical composition can be formulated for use in a variety of drug delivery systems.
- One or more pharmaceutically acceptable excipients or carriers can also be included in the composition for proper formulation. Suitable formulations for use in the present disclosure are found in Adeboye Adejare, Remington: The Science and Practice of Pharmacy (23rd ed. 2020).
- a pharmaceutical composition may contain formulation materials for modifying, maintaining or preserving, for example, the pH, osmolarity, viscosity, clarity, color, isotonicity, odor, sterility, stability, rate of dissolution or release, adsorption or penetration of the composition.
- suitable formulation materials include, but are not limited to, amino acids (such as glycine, glutamine, asparagine, arginine or lysine); antimicrobials; antioxidants (such as ascorbic acid, sodium sulfite or sodium hydrogen- sulfite); buffers (such as borate, bicarbonate, Tris-HCl, citrates, phosphates or other organic acids); bulking agents (such as mannitol or glycine); chelating agents (such as ethylenediamine tetraacetic acid (EDTA)); complexing agents (such as caffeine, polyvinylpyrrolidone, beta-cyclodextrin or hydroxypropyl-beta-cyclodextrin); fillers; monosaccharides; disaccharides; and other carbohydrates (such as glucose, mannose or dextrins); proteins (such as serum albumin, gelatin or immunoglobulins); coloring, flavoring and diluting agents; emulsifying agents
- amino acids
- compositions containing an active agent can be presented in a dosage unit form and can be prepared by any suitable method.
- a pharmaceutical composition should be formulated to be compatible with its intended route of administration. Examples of routes of administration are intravenous, intradermal, inhalation, transdermal, topical, transmucosal, intrathecal and rectal administration.
- Formulation components suitable for parenteral administration include a sterile diluent such as water for injection, saline solution, fixed oils, polyethylene glycols, glycerin, propylene glycol or other synthetic solvents; antibacterial agents such as benzyl alcohol or methyl parabens; antioxidants such as ascorbic acid or sodium bisulfite; chelating agents such as EDTA; buffers such as acetates, citrates or phosphates; and agents for the adjustment of tonicity such as sodium chloride or dextrose.
- a sterile diluent such as water for injection, saline solution, fixed oils, polyethylene glycols, glycerin, propylene glycol or other synthetic solvents
- antibacterial agents such as benzyl alcohol or methyl parabens
- antioxidants such as ascorbic acid or sodium bisulfite
- chelating agents such as EDTA
- buffers such as acetates, citrates or phosphates
- suitable carriers include physiological saline, bacteriostatic water, Cremophor ELTM (BASF, Parsippany, NJ) or phosphate buffered saline (PBS).
- the carrier should be stable under the conditions of manufacture and storage, and should be preserved against microorganisms.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyetheylene glycol), and suitable mixtures thereof.
- An intravenous drug delivery formulation of the present disclosure may be contained in a bag, a pen, or a syringe. In certain embodiments, the bag may be connected to a channel including a tube and/or a needle.
- the formulation is a liquid formulation.
- an aqueous formulation is prepared including the active agent (e.g., a double membrane fusogen protein, a transgenic gene, a non-coding RNA, a system for DNA transfer) in a pH-buffered solution.
- the pH of the liquid formulation may be set by addition of a pharmaceutically acceptable acid and/or base.
- the pharmaceutically acceptable acid may be hydrochloric acid.
- the base may be sodium hydroxide.
- a salt or buffer components may be added in an amount of 10 mM to 200 mM.
- the salts and/or buffers are pharmaceutically acceptable and are derived from various known acids (inorganic and organic) with “base forming” metals or amines.
- the buffer may be phosphate buffer.
- the buffer may be glycinate, carbonate, citrate buffers, in which case, sodium, potassium or ammonium ions can serve as counterion.
- Intravenous formulations can be diluted with 0.9% sodium chloride solution before administration.
- the diluted drug product for injection is isotonic and suitable for administration by intravenous infusion.
- the protein product of the present disclosure is formulated as a liquid formulation in either a USP / Ph Eur type I 50R vial closed with a rubber stopper and sealed with an aluminum crimp seal closure.
- the stopper may be made of elastomer complying with USP and Ph Eur.
- the liquid formulation may be diluted with 0.9% saline solution prior to use.
- Embodiment 8 The method of any one of embodiments 1-7, wherein the double membrane fusogen comprises a DNA-binding domain.
- Embodiment 15 The method of any one of embodiments 1-14, wherein the double membrane fusogen comprises a luminal domain, transmembrane domain, cytoplasmic domain, or DNA-binding domain that has least 60%, at least 70%, at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to the corresponding domain present in SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, or 147.
- Embodiment 17 The method of embodiment 16, wherein at least one of features (a) - (c) does not naturally occur in the amino acid sequence of a wild-type double membrane fusogen having the greatest sequence identity to the encoded double membrane fusogen.
- Embodiment 18 The method of any one of embodiments 1-17, wherein the double membrane fusogen is not a human protein.
- Embodiment 19 The method of any one of embodiments 1-18, wherein the double membrane fusogen is a Brambleberry protein, a protein involved in karyogamy, or a functional fragment, variant, or chimera of any of the foregoing.
- Embodiment 20 The method of any one of embodiments 1-19, wherein the double membrane fusogen is a Brambleberry protein or functional fragment or variant thereof.
- Embodiment 21 The method of any one of embodiments 19 or 20, wherein the Brambleberry protein or functional fragment or variant thereof is a zebrafish (Danio rerid) Brambleberry protein or functional fragment or variant thereof.
- the Brambleberry protein or functional fragment or variant thereof is a zebrafish (Danio rerid) Brambleberry protein or functional fragment or variant thereof.
- Embodiment 22 The method of any one of embodiments 19-21 wherein the Brambleberry protein is a split Brambleberry protein.
- Embodiment 23 The method of any one of embodiments 1-22, wherein the method further comprises an immunosuppressive molecule or a pro-drug form thereof, and/or an RNA encoding an immunosuppressive protein.
- Embodiment 24 The method of embodiment 23, wherein the immunosuppressive molecule or pro-drug form thereof is a corticosteroid or a tyrosine kinase inhibitor
- Embodiment 25 The method of embodiment 24, wherein the corticosteroid is a glucocorticoid.
- Embodiment 26 The method of any one of embodiments 23-25, wherein the immunosuppressive molecule or pro-drug form thereof binds human glucocorticoid receptor.
- Embodiment 27 The method of any one of embodiments 23-26, wherein the immunosuppressive molecule is dexamethasone, fluticasone, fluticasone propionate, fluticasone furoate, clobetasol, betamethasone, halobetasol, amcinonide, desoximetasone, diflorasone, fluocinonide, mometasone, prednisone, prednicarbate, triamcinolone, triamcinolone acetonide, fluocinolone, alclometasone, desonide, cortisone, or hydrocortisone, or a pro-drug form of any of the foregoing.
- the immunosuppressive molecule is dexamethasone, fluticasone, fluticasone propionate, fluticasone furoate, clobetasol, betamethasone, halobetasol, amcinonide, desoximetasone,
- Embodiment 28 The method of embodiment 23-27, wherein the immunosuppressive molecule is dexamethasone or tacrolimus.
- Embodiment 29 The method of any one of embodiments 23-28, wherein the immunosuppressive molecule or pro-drug form thereof contains an ester bond.
- Embodiment 31 The method of any one of embodiments 23-30, comprising at least one immunosuppressive molecule or pro-drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, NLRP3 inflammasome inhibitors, MyD88 inhibitors, IRAK4 inhibitors, PKR inhibitors, PERK inhibitors, NFkB inhibitors, IKK inhibitors, JAK inhibitors, STAT inhibitors, GSK3 inhibitors, cGAS inhibitors, or STING inhibitors.
- immunosuppressive molecule or pro-drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, NLRP3 inflammasome inhibitors, MyD88 inhibitors, IRAK4 inhibitors, PKR inhibitors, PERK inhibitors, NFkB inhibitors, IKK inhibitors, JAK inhibitors, STAT inhibitors, GSK3 inhibitors,
- Embodiment 32 The method of any one of embodiments 23-31, comprising at least one immunosuppressive molecule or pro-drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, or NLRP3 inflammasome inhibitors.
- Embodiment 33 The method of any one of embodiments 23-32, wherein the immunosuppressive molecule or pro-drug form thereof is an NLRP3 inflammasome inhibitor.
- Embodiment 34 The method of any one of embodiments 31-33, wherein the NLRP3 inflammasome inhibitor is oridonin, INF39, glyburide, 5-chloro-2-methoxy-N-[2-(4- sulfamoylphenyl)-ethyl]-benzamide (e.g., 16673-34-0), JC124, FC11A-2, parthenolide, VX- 740, VX-765, BAY 11-7082, BHB, MCC950, MNS, CY-09, or Tranilast, OLT1177, or a pro-drug form of any of the foregoing.
- the NLRP3 inflammasome inhibitor is oridonin, INF39, glyburide, 5-chloro-2-methoxy-N-[2-(4- sulfamoylphenyl)-ethyl]-benzamide (e.g., 16673-34-0), JC124, FC11A-2, part
- Embodiment 35 The method of any one of embodiments 31-32, wherein the galectin inhibitor is GB1107, galectin-3/galectin-8-IN-l, olitigaltin, selvigaltin, galectin- 3/galectin-8-IN-2, galectin-3-IN-2, galectin-3-IN-3, galectin-3-IN-5, GB1490, GB1908, galectin-8-IN-l, galectin- 8-IN-2, galectin-8N-IN-l, galectin-8N-IN-2, or thiodigalactoside, or a pro-drug form of any of the foregoing.
- the galectin inhibitor is GB1107, galectin-3/galectin-8-IN-l, olitigaltin, selvigaltin, galectin- 3/galectin-8-IN-2, galectin-3-IN-2, galectin-3-IN-3, galectin-3-IN-5, GB1490, GB
- Embodiment 36 The method of any one of embodiments 31-32, wherein the cathepsin inhibitor or cysteine protease inhibitor is cathepsin L-IN-2 (Z-Phe-Phe-FMK), Disulfiram, Belizatinib, Cystatin B, Cystatin C, E-64, E-64d, or a pro-drug form of any of the foregoing.
- the cathepsin inhibitor or cysteine protease inhibitor is cathepsin L-IN-2 (Z-Phe-Phe-FMK), Disulfiram, Belizatinib, Cystatin B, Cystatin C, E-64, E-64d, or a pro-drug form of any of the foregoing.
- Embodiment 37 The method of any one of embodiments 23-36, comprising at least two different immunosuppressive molecules or pro-drug forms thereof.
- Embodiment 38 The method of any one of embodiments 23-37, comprising at least three different immunosuppressive molecules or pro-drug forms thereof.
- Embodiment 39 The method of any one of embodiments 23-38, wherein the immunosuppressive molecule or pro-drug form thereof has a LogP value greater than 3.0, 3.5, or 4.0.
- Embodiment 40 The method of any one of embodiments 23-39, comprising an aqueous suspension of the immunosuppressive molecule or pro-drug form thereof.
- Embodiment 41 The method of embodiment 40, wherein the aqueous suspension comprises triamcinolone acetonide or betamethasone sodium phosphate.
- Embodiment 42 The method of any one of embodiments 1-41, comprising at least one extracellular matrix (ECM)-modifying enzyme.
- ECM extracellular matrix
- Embodiment 59 The method of any one of embodiments 1-58, wherein the DNA comprises a gene capable of expressing a secreted protein.
- Embodiment 62 The method of any one of embodiments 1-61, wherein the DNA comprises a gene encoding a protein comprising at least one domain that binds a ligand and at least one effector domain.
- CpG-methylation allows a CpG-containing bacterial origin of replication to be used in a system for nucleic acid transfer while minimizing recognition by TLR9.
- CpG-methylated plasmids also provide a means of gene regulation.
- CpG- methylation also has the utility of being a means of regulating or silencing genes or regions of the DNA that is introduced with a system for nucleic acid transfer.
- Example 10 Systems for gene transfer that include intracellular transport ligands
- Dynein binders were evaluated for their ability to promote gene transfer by promoting the trafficking of nucleic acids carried by LNPs into the cell.
- the dynein binders were dynein-binding peptides that interact with Tctex-1 (DYNLT-1), a dynein light chain protein.
- the two peptides are from Arl2 (GGFKLNIWDVGGQK) (SEQ ID NO: 115), and the C-terminus of rhodopsin D (Rhd) (GVSKTETSQVAPA) (SEQ ID NO: 1 16).
- octyl-arginine i.e., as in SEQ ID NOs: 117-118.
- the dynein binders were stearylated to facilitate their incorporation into the LNPs.
- Octyl-arginine was included as a DNA binder, which is cationic at intracellular pH (pH 7.4), and thus binds DNA through a charge interaction.
- the LNPs in this experiment carried DNA encoding NanoLuc luciferase under a muscle-specific promoter and mRNA encoding Danio rerio Brambleberry at a 1:2 ratio by weight.
- the controls included were LNPs formulated without any stearylated arginine or dynein binder, or with only stearylated octyl-arginine.
- the stearylated octyl- arginine-Arl2 peptide which is a dynein binder that binds Tctex-1 (DYNLT-1), increased gene transfer efficiency by 3.4-fold (FIGURE 14).
- the stearylated octyl-arginine-rhodopsin D peptide which is a dynein binder that binds Tctex-1 (DYNLT-1), increased gene transfer efficiency by 10-fold (FIGURE 14).
- transport ligands e.g., dynein binders targeting dynein proteins such as Tctex-1 (DYNLT-1)
- Tctex-1 Tctex-1
- the inclusion of a molecule containing both a DNA binder and a dynein binder may facilitate the intracellular trafficking of the DNA to a perinuclear localization that promotes gene transfer.
- Cyprinodon variegatus and Varanus komodoensis Brambleberry both were functional for facilitating gene transfer in mammalian cells.
- the activity of Danio rerio Brambleberry was compared against its orthologs from pigeon (Columba livia) (SEQ ID NOs: 27-28), sheepshead pupfish (Cyprinodon variegatus) (SEQ ID NOs: 33-34), and Hawaiian crow (Corvus hawaiiensis) (SEQ ID NOs: 119-120) (FIGURE 15B).
- the Brambleberry ortholog with the highest gene transfer efficiency was that from Cyprinodon variegatus and the second highest was that from Columba livia.
- Danio rerio Brambleberry was compared against Columba livia Brambleberry and a version of Columba livia Brambleberry with a non-native signal peptide (NNSP) (FIGURE 15C) for their ability to facilitate gene transfer in differentiated C2C12 myotubes.
- NSP non-native signal peptide
- This experiment demonstrated that Columba livia Brambleberry was functional with either the native or non-native signal peptide.
- this experiment demonstrates that Brambleberry orthologs from species other than Danio rerio can exhibit superior gene transfer efficiency to Danio rerio Brambleberry in mammalian cells.
- Brambleberry orthologs and mRNAs for expressing them are provided herein.
- the Brambleberry ortholog sequences provided are from zebrafish ⁇ Danio rerio) (SEQ ID NOs: 1-2), golden eagle Aquila chrysaetos chrysaetos) (SEQ ID NOs: 25-26), pigeon Columba livia) (SEQ ID NOs: 27-28), Komodo dragon ⁇ Varanus komodoensis) ) (SEQ ID NOs: 29-30), Goode’s thomscrub tortoise ⁇ Gopherus evgoodei) (SEQ ID NOs: 31-32), Sheepshead pupfish ⁇ Cyprinodon variegatus) (SEQ ID NOs: 33-34), Hawaiian crow ⁇ Corvus hawaiiensis) (SEQ ID NOs: 119-120),
- Example 12 Increasing the completeness of differentiation increases the relative impact of Brambleberry
- C2C12 myoblasts were differentiated into myotubes by culturing in media containing 2% horse serum instead of 20% FBS.
- DNA-mRNA LNPs where the DNA expressed firefly luciferase and the mRNA expressed Danio rerio Brambleberry were added to C2C12 myotube cultures 10 days post-differentiation.
- C2C12 myotube cultures 10 days post-differentiation rather than 5 days post-differentiation extended the impact of Brambleberry mRNA on gene transfer efficiency to 472-fold over a control with DNA-only LNPs without the Brambleberry mRNA (FIGURE 16).
- Minimizing the presence of dividing cells by culturing in differentiation media for 10 instead of 5 days increased the apparent impact of Brambleberry on the efficiency of gene transfer.
- Example 13 DNA-mRNA LNPs made with different ionizable lipids
- the ionizable lipid used in previous experiments had been DLin-KC2-DMA. Therefore, several different ionizable lipids were screened as potential alternatives to DLin- KC2-DMA.
- the lipids included were a DLin-KC2-DMA control, SMI 02, Lipid 29, and CL15F6. Due to the potential for freezing conditions to differently affect LNPs comprised of different lipids, LNPs made with the different lipids were evaluated after storage at 4°C, - 20°C, and -80°C. An unfiltered LNP control was included to monitor loss during filtration that can occur with excessive particle size or polydispersity.
- DNA-mRNA LNPs were formulated as follows. DNA and mRNA were included at a 1 :2 ratio by mass. The mRNA was made using N1 -methylpseudouridine rather than uridine. Lipids were premixed at ratios by mass of 56.9% ionizable lipid, 27.5% cholesterol, 11.2% DSPC, 1.5% DMG-PEG2000, 0.25% stearylated TCTEX-1 binder StR8RhdCT (SEQ ID NO: 118), and 4.3% DPG-galloyl.
- aqueous phase DNA + RNA + 0.2 M citrate buffer + water
- 800 pL of lipid phase in a Ignite Nanoassemblr instrument. 0.45 mL of the head and 0.05 mL of the tail were discarded. The middle portion of the mix ( ⁇ 2.7 mL) was used as the sample. Samples were immediately quenched with 0.5X TBS up to 14 mL and centrifuged in an Amicon 15 (30 kDa cutoff) centrifugal filter units at 4000 x g until the volume in the Amicon tubes was 500 mL (—50 minutes).
- the volume in the Amicon was again brought up to 14 mL with 0.5X TBS and the tubes were centrifuged at 4000 g until the volume in the Amicon tubes went down to 500 mL.
- the liquid was then transferred to an Amicon 4 (10 kDa) centrifugal filter units and centrifuged until about 100 uL volume remained.
- 100 ul of 0.5X TBS + 20% sucrose was added to the 100 ul of sample.
- Particle sizes were assessed by DLS for unfiltered material, filtered material stored at 4°C, filtered material stored at -20°C, and filtered material stored at -80°C (FIGURE 17).
- lipids evaluated included an ionizable head group with a pKa > 6.5.
- the new lipids evaluated (SM102, Lipid 29, and CL15F6), contained branched acyl chains and ester bonds hydrolysable by cellular esterases, but lacked unsaturated acyl chains (FIGURE 18E-H).
- features of an ionizable lipid including a pKa above 6.5, branched acyl chains, hydrolysable ester bonds, and a minimal or no unsaturated bonds may have contributed to the improved gene transfer efficiency in muscle cells observed with CL15F6.
- Example 14 DNA-mRNA LNPs made with PEG with short lipid anchors
- lipid anchors of reduced size relative to DMG may increase the kinetics with which the PEGylated lipid diffuses out of the LNP, thereby improving contact with cells and the efficiency of gene transfer.
- Forms of PEG2000 with a cholesterol, stearic acid (SA), or C8 ceramide (C8C) anchor were compared against DMG-PEG2000 (FIGURE 19).
- DNA-mRNA LNPs were generated using a mixture of lipids containing either 1 X ( 1 .4%) or 2X (2.8%) of the PEGylated lipid.
- Cholesterol was included at 27.5% for the IX PEG or 26.1% for the 2X PEG LNPs. In other words, the increased PEGylated lipid was subtracted from the amount of cholesterol added. The other lipid components were 56.9% CL15F6, 11.2% DSPC, and 3% DPG-galloyl.
- the mRNA was made with N 1 -methylpseudouridine instead of uridine.
- certain conditions were evaluated for gene transfer efficiency in 10-day differentiated C2C12 myotubes using firefly luciferase as a reporter gene.
- Use of PEGylated lipids with short lipid anchors substantially improved gene transfer efficiency over DMG-PEG2000.
- the two most efficient LNP preps had 1.4% C8C-PEG2000 or 2.8% cholesterol-PEG2000. Among these, 1 .4% C8C-PEG2000 exhibited superior gene transfer efficiency to 2.8% cholesterol-PEG2000. This experiment indicated that relatively short lipid anchors, e.g., C8C or cholesterol, can improve the efficiency of gene transfer by DNA-mRNA LNPs in muscle cells.
- LNPs containing DNA were generated with and without Brambleberry mRNA, using the ionizable lipids DLin-KC2-DMA or CL15F6, and with either DMG-PEG2000 or C8C-PEG2000.
- certain preparations of LNPs were evaluated for gene transfer efficiency in 10-day differentiated C2C12 myotubes.
- the amount of LNPs added to the cells was normalized to the amount of DNA, and the DNA-mRNA LNPs were generated using a 1 :2 ratio of DNA to Brambleberry mRNA.
- the LNPs were exchanged into TBS buffer containing 20% sucrose using centrifugal filter units.
- DNA-mRNA LNPs containing Brambleberry mRNA was 251- fold more efficient than the DNA-only LNPs, where both were made using DLin-KC2-DMA and DMG-PEG2000 (FIGURE 20).
- the presence of Brambleberry mRNA increased the efficiency of gene transfer by 324-fold.
- the presence of Brambleberry increased the efficiency of gene transfer by 234-fold.
- suppressing innate immune responses activated by DNA-mRNA LNPs may improve the expression of the Brambleherry mRNA, thereby improving the efficiency of gene transfer.
- coformulation e.g., coencapsulation
- immunosuppressive molecules affords a desirable means of limiting the potential for immune responses against the protein or proteins encoded by the DNA.
- Additional immunosuppressive molecules that can improve the efficiency of Brambleberry mRNA expression and the efficiency of gene transfer by DNA-mRNA LNPs include MyD88 inhibitors, TRAK4 inhibitors, PKR inhibitors, PERK inhibitors, NFkB inhibitors, IKK inhibitors, JAK inhibitors, STAT inhibitors, GSK3 inhibitors, cGAS inhibitors, and STING inhibitors.
- a miR gene under the control of a U6 Pol III promoter was described in Example 5.
- the sequence of a plasmid for expressing this miR is provided (SEQ ID NO: 54).
- This miR is matched to an optimized 3’ UTR, which contains 10 target sites for the miR, and which can be included in a transgene to enable its efficient knockdown by the miR through RNAi.
- the present example provides key data from the optimization of the miR included in SEQ ID NO: 54.
- the same miR construct was tested in C2C12 cells, either when firefly luciferase was expressed from a CMV promoter or when firefly luciferase was expressed from the muscle-specific promoter (MSP) (SEQ ID NO: 12).
- MSP muscle-specific promoter
- the miR and 3’ UTR target site afforded a system for efficient activation in muscle cells (FIGURE 24B).
- this particular miR design particularly in combination with a 3’ UTR with concatemeric target sites, provide a highly efficient means of knocking down a transgene by RNA, including in muscle cells.
- This miR construct may be particularly useful in the context of an off-switch, e.g., where a second plasmid expressing the miR is administered or otherwise activated in order to knock down the expression of a transgene by RNAi.
- H 1 promoter was modified in order to develop an RNA Polymerase III (Pol III) promoter lacking CpG motifs.
- the absence of CpG motifs in the Pol III promoter is useful to avoid including a ligand for TLR9, and to prevent silencing by promoter methylation.
- the Hl promoter was selected for modification due to its small size and paucity of CpG motifs.
- Two modified versions of the Hl promoter were generated, one where both CpG (i.e., CG) motifs were mutated to CA (version 1, vl), and the other where both CpG motifs were mutated to TG (version 2, v2).
- a miR construct was designed targeting the Fut8 mRNA that is expressed in CHO cells.
- This miR contained a 19 base guide sequence, a loop of unpaired bases exactly 4 nucleotides in length, a 19 base sequence that is complementary to the guide sequence and forms a 19 base pair stem, and an RNA Polymerase 111 (Pol III) transcription termination signal (TTTTTT) that overlaps by two nucleotides with the 3’ end of the stem.
- the target sequence in the Fut8 mRNA was SEQ ID NO: 150, which notably ends with a TT or UU motif.
- the guide sequence is SEQ ID NO: 151.
- the Fut8 knockdown cell line generated protein where just 5% of N-linked glycans were fucosylated.
- This substantial knockdown effect revealed the effective reduction in fucosylated N-linked glycans present on the eCD4-Ig protein made in the cell line expressing the miR relative to the parental cell line, thus demonstrating that this miR architecture is effective for knocking down Fut8.
- the miR operably linked to a CpG-free Hl promoter is provided as SEQ ID NO: 154.
- a miR construct was designed targeting the GALNT2 mRNA that is expressed in CHO cells.
- This miR contained a 19 base guide sequence, a loop of unpaired bases exactly 4 nucleotides in length, a 19 base sequence that is complementary to the guide sequence and forms a 19 base pair stem, and an RNA Polymerase III (Pol III) transcription termination signal (TTTTTT) that overlaps by two nucleotides with the 3’ end of the stem.
- the target sequence in the GALNT2 mRNA was SEQ ID NO: 155, which notably ends with a TT or UU motif.
- the whole miR operably linked to a U6 promoter is SEQ ID NO: 156.
- the presence or absence of an O-linked glycan on the eCD4-Ig protein generated was measured by mass spectrometry.
- the prevenance of an O-linked glycan was reduced from being present on 24% of the eCD4-Ig protein made in the control transfection to 3.4% of the protein made by co-transfection with the plasmid expressing the miR targeting GALNT2 (FIGURE 26).
- highly efficient knockdowns of a firefly luciferase gene containing an engineered 3’ UTR target site, Fut8, and GALNT2 were demonstrated using the miR architecture provided in the present example.
- DNA-mRNA LNPs can be coformulated with hyaluronidase to promote efficient gene transfer after intramuscular injection.
- the results of two studies were contrasted by comparing the area under the curve (AUC) of luminescence over the period from 2-4 weeks post- injection with the same amounts of identically-formulated DNA-mRNA LNPs with and without hyaluronidase.
- the LNPs administered in this example were made with 56.9% CL15F6, 27.2% cholesterol, 11.2% DSPC, 1.4% C8C-PEG2000, 0.32% of a cholesterol-conjugated TCTEX- 1 binder chol-R8RhdCT (SEQ ID NO: 118), and 3% DPG-galloyl by mass.
- DNA-mRNA LNPs were generated in which the DNA was a largely CpG-free DNA that expresses firefly luciferase from a muscle-specific promoter (SEQ ID NO: 149).
- the mRNA encoded Danio rerio Brambleberry DNA and mRNA were coencapsulated at a 1:2 ratio by mass.
- hyaluronidases provide a group of extracellular matrix (ECM) -modifying enzymes capable of improving the access of LNP to muscle cells after intramuscular injection.
- ECM extracellular matrix
- bovine hyaluronidase was used here, human hyaluronidase or a fragment thereof (e.g., Hylenex) could be used in a clinical setting.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Organic Chemistry (AREA)
- General Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Physics & Mathematics (AREA)
- Microbiology (AREA)
- Plant Pathology (AREA)
- Biophysics (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The disclosure relates generally to systems and methods for nucleic acid transfer. RNA encoding a double membrane fusogen protein facilitates the transfer of a DNA of interest into the nucleus of a cell where it can be expressed. Nucleic acid transfer can be used in connection with gene therapy, vaccine administration, cell modification, and the like. The methods and systems of nucleic acid transfer can be used to transfect non-dividing cells, such as muscle cells. The disclosure further relates to promoters for muscle-specific expression of a gene of interest.
Description
SYSTEMS FOR NUCLEIC ACID TRANSFER
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of and priority to U.S. Provisional Patent Application No. 63/614,723, filed December 26, 2023, which is incorporated by reference in its entirety herein.
FIELD OF THE INVENTION
[0002] The invention relates generally to systems for nucleic acid transfer. Systems for nucleic acid transfer are useful in the fields of gene therapy, vaccines, cell modification, and transfection.
BACKGROUND
[0003] Nucleic acid transfer can include the introduction of nucleic acids into cells, such as mammalian cells. For therapeutic applications such as gene therapy, an RNA or DNA molecule must be transferred not only into the cell, but also into the nucleus, where it can be expressed. Current non- viral nucleic acid transfer systems for introducing DNA into the nuclei of cells, such as non-dividing cells, include electroporation and lipid nanoparticles.
[0004] Electroporation is an approach that facilitates a relatively efficient nucleic acid transfer into non-dividing cells, which is thought to be due to the creation of openings in both the plasma membrane and the nuclear envelope that allow DNA to pass directly into the nucleus. However, electroporation cannot be used on a large area and requires surgical intervention for transfer to internal organs. Further, the use of high voltage can damage nucleic acid and tissues.
[0005] Lipid nanoparticles (LNPs) based on ionizable lipids deliver the RNA or DNA they carry into the cytoplasm of cells. It is contemplated that within minutes of contact with the cytoplasm, transfected DNA becomes sequestered into double membrane-enveloped compartments, which are derived from the endoplasmic reticulum (ER) and resemble a nuclear envelope topologically (Kobayashi et al. (2015) PROC. NATL. ACAD. SCL, 112(22): 7027-32). After the transfection of rapidly dividing cells (e.g., immortalized cell lines), it is thought that these compartments undergo dissolution during prometaphase, allowing transfected DNA to be incorporated into the nucleus and efficient transfection (Haraguchi et al. (2022) COMMUN. BIOL., 5(1): 78). However, incorporation of the sequestered DNA into
the nuclei of non-dividing cells typically is inefficient. Because DNA must be present in the nucleus to be expressed, inefficient transfer of the DNA to the nucleus results in low expression of the DNA.
[0006] Accordingly, there is a need in the art for methods and compositions that facilitate efficient nucleic acid transfer to the nuclei of cells, for example, to improve expression of a transgenic DNA of interest.
SUMMARY OF THE INVENTION
[0007] The present disclosure is based, in part, upon the discovery that double membrane fusogen proteins provide a solution to the topological problem of transferring DNA sequestered within a double membrane envelope to the nucleus. In early embryo development in animals with larger eggs than mammals, chromosomes become enveloped by a double membrane in structures known as karyomeres, which ultimately fuse to form the nucleus. In zebrafish (Danio rerio), a protein that mediates this fusion is Brambleberry — so named, because in its absence, consolidation of karyomeres into a nucleus is arrested, and the unfused karyomeres resemble a brambleberry (Abrams et al. (2012) CELL, 150 (3): 521-532) (FIGURE 1A). By contrast, in wild-type Danio rerio. the karyomeres fuse and form a nucleus (FIGURE IB) It has been discovered that transient expression of Brambleberry from an mRNA that is co-packaged with a plasmid DNA, e.g., in an LNP, mediates fusion of double membrane-enveloped vesicles sequestering DNA with the nucleus, thereby allowing successful nucleic acid transfer to non-dividing cells.
[0008] Accordingly, the disclosure relates, in part, to systems e.g., lipid-based delivery systems) and methods for the transfer of a non-native e.g., non-endogenous) DNA into a nucleus of a cell, such as a mammalian cell. The system or method comprises a non-native (e.g., non-endogenous) DNA and an RNA that encodes a double membrane fusogen protein, which, when present in the cell, mediates transfer of the non-native DNA into the nucleus of the cell.
[0009] In addition, it has been discovered that a plasmid with a CpG-free promoter when present in non-dividing cells is capable of persistently expressing a gene of interest, without being silenced, and without the loss of the plasmid, e.g. , by degradation. Furthermore, it has been discovered that repositioning the TATA box in naturally occurring muscle-specific promoters results in improved expression. Accordingly, the disclosure also relates, in part, to
engineered muscle-specific promoters, that can be used in combination with the systems described herein, or in other applications.
[0010] The discovery that a double membrane fusogen protein, such as Brambleberry, enables efficient nucleic acid transfer into non-dividing cells provides a non- viral system for nucleic acid transfer, which, when combined with a CpG-free muscle-specific promoter operably linked to a gene of interest, allows persistent long-term expression of the gene of interest in muscle.
[0011] Accordingly, in one aspect, the disclosure relates to a system for the transfer of non- native DNA into a nucleus of a cell, the system comprising the non-native DNA and RNA, wherein the RNA encodes a double membrane fusogen.
[0012] In certain embodiments, the double membrane fusogen mediates the fusion of intracellular membranes, such as the fusion of a nuclear envelope of a cell with a double membrane-enveloped structure comprising the non-native DNA.
[0013] The double membrane fusogen can include a signal peptide and a transmembrane domain. In certain embodiments, the double membrane fusogen comprises two or more (e.g. , three or more) transmembrane domains. In certain embodiments, the double membrane fusogen comprises a DNA-binding domain. In certain embodiments, the double membrane fusogen comprises a multimerization domain. In certain embodiments, the double membrane fusogen comprises an alpha helical domain at least 20, 30, 40, 50, 60, 70, 80, 90, or 100 amino acids in length. In certain embodiments, the double membrane fusogen comprises an alpha helical domain at least 50 amino acids in length. In certain embodiments, the double membrane fusogen comprises an alpha helical domain at least 60 amino acids in length. In certain embodiments, the double membrane fusogen comprises an alpha helical domain at least 80 amino acids in length. In certain embodiments, the double membrane fusogen comprises an alpha helical domain at least 100 amino acids in length.
[0014] In certain embodiments, the double membrane fusogen comprises a luminal domain, a transmembrane domain, a cytoplasmic domain, or a DNA-binding domain having at least 60%, at least 70%, at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to the corresponding domain present in SEQ ID NO: 1, 25,
27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, or 147.
In certain embodiments, the double membrane fusogen comprises a cytoplasmic domain, and the cytoplasmic domain comprises at least one of
(a) a nuclear localization signal (NLS),
(b) an amino acid sequence five amino acids in length wherein at least four out of five of the amino acids are basic residues, and
(c) an amino acid sequence comprising at least one basic residue separated by a spacer of ten to fifteen amino acids from three or more basic residues.
[0015] In certain embodiments, the at least one of features (a) - (c) does not naturally occur in the amino acid sequence of a wild-type double membrane fusogen having the greatest sequence identity to the encoded double membrane fusogen.
[0016] In certain embodiments, the double membrane fusogen is not a human protein.
[0017] In certain embodiments, the double membrane fusogen is a Brambleberry protein, a protein involved in karyogamy, or a functional fragment, variant, or chimera of any of the foregoing. In certain embodiments, the double membrane fusogen is a Brambleberry protein or functional fragment or variant thereof. For example, the Brambleberry protein or functional fragment or variant thereof can be a zebrafish (Danio rerio) Brambleberry protein or functional fragment or variant thereof. In certain embodiments, the Brambleberry protein is a split Brambleberry protein.
[0018] The system can further comprises an immunosuppressive molecule or a pro-drug form thereof and/or an RNA encoding an immunosuppressive protein or a pro-drug form thereof. For example, the immunosuppressive molecule can be a corticosteroid, a tyrosine kinase inhibitor, dexamethasone, tacrolimus, , fluticasone, fluticasone propionate, fluticasone furoate, clobetasol, betamethasone, halobetasol, amcinonide, desoximetasone, diflorasone, fluocinonide, mometasone, prednisone, prednicarbate, triamcinolone, triamcinolone acetonide, fluocinolone, alclometasone, desonide, cortisone, or hydrocortisone, or a pro-drug form of any of the foregoing. As another example, the immunosuppressive molecule or pro- drug form thereof can bind a human glucocorticoid receptor. In some embodiments, the immunosuppressive molecule or pro-drug form thereof contains an ester bond. In some
embodiments, the immunosuppressive molecule or pro-drug form thereof is dexamethasone palmitate or fluticasone-furoate.
[0019] In certain embodiments, the system comprises at least one immunosuppressive molecule or pro-drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, NLRP3 inflammasome inhibitors, MyD88 inhibitors, IRAK4 inhibitors, PKR inhibitors, PERK inhibitors, NFkB inhibitors, IKK inhibitors, JAK inhibitors, STAT inhibitors, GSK3 inhibitors, cGAS inhibitors, or STING inhibitors. In certain embodiments, the at least one immunosuppressive molecule or pro- drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, or NLRP3 inflammasome inhibitors.
[0020] In certain embodiments, the immunosuppressive molecule or pro-drug form thereof is an NLRP3 inflammasome inhibitor. For example, the NLRP3 inflammasome inhibitor can be oridonin, INF39, glyburide, 5-chloro-2-methoxy-N-[2-(4- sulfamoylphenyl)-ethyl]- benzamide (e.g. , 16673-34-0), JC124, FC11A-2, parthenolide, VX-740, VX-765, BAY 11- 7082, BHB, MCC950, MNS, CY-09, or Tranilast, OLT1177, or a pro-drug form of any of the foregoing.
[0021] The galectin inhibitor can be GB 1107, galectin-3/galectin-8-IN-l, olitigaltin, selvigaltin, galectin-3/galectin-8-IN-2, galectin-3-IN-2, galectin-3-IN-3, galectin-3-IN-5, GB1490, GB1908, galectin-8-IN-l, galectin-8-IN-2, galectin-8N-IN-l, galectin-8N-IN-2, or thiodigalactoside, or a pro-drug form of any of the foregoing.
[0022] The cathepsin inhibitor or cysteine protease inhibitor is cathepsin L-IN-2 (Z-Phe-Phe- FMK), disulfiram, belizatinib, cystatin B, cystatin C, E-64, E-64d, or a pro-drug form of any of the foregoing.
[0023] In certain embodiments, the system comprises at least two different (e.g., at least three different) immunosuppressive molecules or pro-drug forms thereof.
[0024] In certain embodiments, the immunosuppressive molecule or pro-drug form thereof has a LogP value greater than 3.0, 3.5, or 4.0.
[0025] In certain embodiments, the system comprises an aqueous suspension of the immunosuppressive molecule or pro-drug form thereof. The aqueous suspension can include triamcinolone acetonide or betamethasone sodium phosphate.
[0026] In certain embodiments, the system comprises at least one extracellular matrix (ECM) -modifying enzyme, for example, hyaluronidase or a fragment thereof.
[0027] In certain embodiments, the cell is a non-dividing cell.
[0028] In certain embodiments, the RNA encoding the double membrane fusogen comprises modified bases. In certain embodiments, the RNA encoding the double membrane fusogen is an mRNA. In certain embodiments, the RNA encoding the double membrane fusogen comprises one or more target sites for at least one micro RNA (miR). In certain embodiments, the DNA comprises modified CpG motifs. In certain embodiments, the DNA has fewer than 100 CpG motifs, the DNA is substantially free of unmodified CpG motifs, and/or the DNA is methylated at one or more CpG motifs. In certain embodiments, the DNA is substantially free of 6-methyladenine and/or 5 -methylcytosine. In certain embodiments, the DNA comprises at least one tissue- specific promoter. In certain embodiments, the tissue- specific promoter is expressed in non-dividing cells. In certain embodiments, the tissue- specific promoter is a muscle-specific promoter. In certain embodiments, the DNA comprises a promoter with fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs. In certain embodiments, the muscle-specific promoter comprises one or more transcription factor binding site core motifs selected from the group consisting of: a. 5’-CATTCC-3’, b. 5’-GGAATG-3’, c. 5’-CCWWWWWWGG-3’, d. 5’-CANNTG-‘3, e. 5 ’ -YTAAAAATA-3 ’ , and f. 5’-TATTTTTAR-3’, wherein N is any nucleotide, W is A or T, Y is T or C, and R is A or G.
[0029] In certain embodiments, the DNA lacks free 5’ and 3’ ends and/or is circular. In certain embodiments, the DNA is at least partially denatured. In certain embodiments, the DNA comprises a coding region that is operably linked to a promoter. In certain
embodiments, the DNA comprises a gene capable of expressing a secreted protein. In certain embodiments, the DNA comprises a gene encoding a protein containing an antibody Fc. In certain embodiments, the DNA comprises a gene encoding a protein comprising a monoclonal or bispecific antibody. In certain embodiments, the DNA comprises a gene encoding a protein comprising at least one domain that binds a ligand and at least one effector domain. In certain embodiments, the DNA comprises a gene encoding a protein that has at least 80% at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to human serum albumin (HSA). In certain embodiments, the DNA comprises a gene encoding an mRNA, wherein the mRNA comprises one or more target sites for a micro RNA (miR), short hairpin RNA (shRNA), or ribozyme. In certain embodiments, the DNA encodes at least one non-coding RNA. In certain embodiments, the at least one non- coding RNA comprises a micro RNA (miR), short hairpin RNA (shRNA), aptamer, or ribozyme. In certain embodiments, the at least one non-coding RNA targets an mRNA expressed by the DNA. In certain embodiments, the at least one non-coding RNA targets the mRNA of an immunomodulatory gene. In certain embodiments, the immunomodulatory gene is fucosyltransferase 8 (FUT8). In certain embodiments, the at least one non-coding RNA targets the mRNA of a human gene, and the DNA encodes a protein capable of functionally replacing a common form of that human gene.
[0030] In certain embodiments, the system comprises (e.g. , contains) one or more nanoparticles. In certain embodiments, the nanoparticles are lipid nanoparticles (LNPs). In certain embodiments, the LNPs comprise one or more ionizable lipids. In certain embodiments, the system comprises a polyplex. In certain embodiments, the nanoparticle comprises at least one attachment moiety. In certain embodiments, attachment moiety promotes attachment to muscle cells. In certain embodiments, the system comprises a lipid that is conjugated directly or indirectly to a glucose molecule. In certain embodiments, the system comprises a lipid that is conjugated to a glucose molecule, wherein the glucose molecule is conjugated to hydroquinone. In certain embodiments, the system comprises a lipid that is conjugated to a polyphenol. In certain embodiments, the system comprises a dynein binder. In certain embodiments, the system comprises at least one antisense oligonucleotide (ASO) or short-interfering RNA (siRNA). In certain embodiments, the system comprises a DNA binder that binds to DNA at pH 7.4.
[0031] In certain embodiments, the RNA encoding the double membrane fusogen comprises Cap2 or one or more internal ribosomal entry sites (IRES).
[0032] In certain embodiments, the system further comprises: a. a gene editing effector protein or an RNA encoding a gene editing effector protein, b. a transposase or an RNA encoding a transposase, c. an integrase or an RNA encoding an integrase, d. a recombinase or an RNA encoding a recombinase, or e. a reverse transcriptase or an RNA encoding a reverse transcriptase.
[0033] In certain embodiments, the system is a system for gene therapy.
[0034] In another aspect, the disclosure relates to a system for the transfer of non-native DNA into a nucleus of a cell, comprising the non-native DNA and a dynein binder. In certain embodiments, the dynein binder is or is conjugated to a DNA binder an RNA binder, a polymer, a peptide, a polypeptide, or a lipid. In certain embodiments, the DNA binder is cationic at pH 7.4. In certain embodiments, the dynein binder is a TCTEX binder. In certain embodiments, the dynein binder or TCTEX binder is a TCTEX-1 DYNLT-1) binder. In certain embodiments, the TCTEX binder comprises a peptide with comprising an amino acid sequence at least five amino acids in length that is at least 80%, at least 85%, at least 90%, at least 95%, or 100% identical to a contiguous amino acid motif selected from the peptides GGFKLNIWDVGGQK (SEQ ID NO: 115), and GVSKTETSQVAPA (SEQ ID NO: 116).
[0035] In another aspect, the disclosure relates to a composition comprising the system described herein. In certain embodiments, the composition further comprises a pharmaceutically acceptable carrier.
[0036] In another aspect, the disclosure relates to a pharmaceutical composition comprising an RNA encoding a double membrane fusogen, which, when delivered to a mammalian cell, mediates transfer of a non-native DNA into the nucleus of the mammalian cell and a pharmaceutically acceptable carrier. In certain embodiments, the system comprises the non- native DNA.
[0037] In another aspect, the disclosure relates to nucleic acid comprising a muscle-specific promoter, wherein the muscle-specific promoter comprises fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs, and/or has at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to a nucleic acid sequence comprising SEQ ID NO: 7, 11, 13, 14, 19, 20, or 110 and a TATA box that is positioned within four nucleotides of the position of a TATA box present in SEQ ID NO: 7, 11, 13, 14, 19, 20, or 1 10 respectively.
[0038] In certain embodiments, the muscle-specific promoter comprises one or more of the transcription factor binding site core motifs selected from the group consisting of: a. 5’-CATTCC-3’, b. 5’-GGAATG-3’, c. 5’-CCWWWWWWGG-3’, d. 5’-CANNTG-‘3, e. 5’-YTAAAAATA-3’, and f. 5’-TATTTTTAR-3’, wherein N is any nucleotide, W is A or T, Y is T or C, and R is A or G.
[0039] In certain embodiments, the muscle-specific promoter has at least 80% identity to SEQ ID NO: 23 or 24, and wherein the muscle-specific promoter comprises fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs.
[0040] In certain embodiments, the nucleic acid comprises a DNA polymerase III (Pol III) promoter, wherein the Pol III promoter comprises fewer than 3, 2, or 1 CpG motifs.
[0041] In another aspect, the disclosure relates to a lipid nanoparticle (LNP) comprising a lipid that is conjugated directly or indirectly to a glucose molecule. In certain embodiments, the lipid is covalently bonded to a glucose molecule. In certain embodiments, the glucose molecule is conjugated to hydroquinone. In certain embodiments, the lipid that is covalently bonded to a glucose molecule is cholesterol-undecanoate-glucose.
[0042] In another aspect, the disclosure relates to a lipid nanoparticle (LNP) comprising a lipid that is conjugated directly or indirectly to a polyphenol. In certain embodiments, the
lipid covalently bonded to a polyphenol. In certain embodiments, the polyphenol comprises galloyl or a galloyl group.
[0043] In another aspect, the disclosure relates to a system, pharmaceutical composition, or nucleic acid as described herein, wherein the system, pharmaceutical composition, or nucleic acid comprises a DNA encoding a microRNA (miR), wherein the miR comprises: a. a 19-base guide sequence that is complementary to a target mRNA, b. an unpaired loop exactly 4 nucleotides in length, c. a 19-base sequence that is complementary to the 19-base guide sequence, which together with the guide sequence form a 19-base pair stem, and d. a 3 ’ end that is a U, which is encoded by one of the U or T nucleotides of an RNA Polymerase III (Pol III) transcription termination signal comprising the sequence UUUUUU, UUUUU, TTTTTT, or TTTTT.
[0044] In another aspect, the disclosure relates to a DNA encoding a microRNA (miR), wherein the miR comprises: a. a 19-base guide sequence that is complementary to a target mRNA, b. an unpaired loop exactly 4 nucleotides in length, c. a 19-base sequence that is complementary to the 19-base guide sequence, which together with the guide sequence form a 19-base pair stem, and d. a 3 ’ end that is a U, which is encoded by one of the U or T nucleotides of an RNA Polymerase III (Pol III) transcription termination signal comprising the sequence UUUUUU, UUUUU, TTTTTT, or TTTTT.
[0045] In certain embodiments, the system, pharmaceutical composition, or nucleic acid includes an RNA Polymerase III promoter operably linked to the miR. The 3’ two nucleotides of the miR can be UU. In certain embodiments, the DNA encodes a microRNA (miR) operably linked to an Hl promoter that is at least 80% identical to nucleotides 1-99 of
SEQ ID NO: 154. In certain embodiments, the miR targets a fucosylatransferase-8 (FUT8) gene sequence.
[0046] In another aspect, the disclosure relates to a method of transfecting a non-native DNA into the nucleus of a nucleated mammalian cell. The method includes contacting the cell with the non-native DNA and an RNA encoding a double membrane fusogen, wherein, when the RNA is translated to produce the fusogen, the fusogen facilitates the transfer of the non- endogenous DNA into the nucleus of the cell. The method can include any one of the systems, compositions, pharmaceutical compositions, nucleic acids, LNPs, or DNA described herein.
[0047] In another aspect, the disclosure relates to an engineered, nucleated mammalian cell. The cell can include a nucleus, a non-native DNA, and an RNA encoding a double membrane fusogen, whereupon, when the RNA is transcribed into a fusogen, the fusogen facilitates transfer of the non-endogenous DNA into the nucleus of the cell. The cell can include any one of the systems, compositions, pharmaceutical compositions, nucleic acids, LNPs, or DNA described herein.
BRIEF DESCRIPTION OF THE DRAWINGS
[0048] FIGURES 1A-1B are drawings depicting the function of Brambleberry. During early zebrafish (Danio rerio) embryonic development, karyomeres containing zebrafish chromosomes associate (FIGURE 1A) and then fuse to form a nucleus (FIGURE IB). The fusion of karyomeres to form a nucleus is mediated by the Brambleberry (Bmb) protein. It has been discovered that Brambleberry proteins can be used in nucleic acid transfer systems to mediate a process topologically similar to karyomere fusion in which double membrane- enveloped cellular structures containing exogenous DNA are fused with the nucleus of a cell to deposit the exogenous DNA into the nucleus. The double black lines represent lipid bilayers. The shaded gray area between the double black lines is topologically similar to the lumen of the endoplasmic reticulum (ER) and the region between the inner and outer membranes of the nuclear envelope. The white background represents cytoplasm.
[0049] FIGURES 2A-2B provide depictions of the domain architecture of a Brambleberry protein. The signal peptide, luminal domain, transmembrane domains, and cytoplasmic domain of zebrafish (Danio rerio) Brambleberry (SEQ ID NO: 1) were predicted using TOPCONS (FIGURE 2A). These features are indicated on the full-length amino acid
sequence of zebrafish Brambleberry (SEQ ID NO: 1) (FIGURE 2B). The predicted signal peptide for secretion into the endoplasmic reticulum (ER) is indicated in italics. The three predicted transmembrane domains are underlined. Two of those transmembrane domains are separated by a single hydrophobic residue in zebrafish Brambleberry. The predicted cytoplasmic domain is indicated in gray. The cytoplasmic domain contains a bipartite nuclear localization signal (NLS), i.e., a basic residue separated by a spacer of thirteen amino acids from three basic residues, where the basic residues are arginine (R) and/or lysine (K). The bipartite NLS is indicated in bold within the cytoplasmic domain. (By contrast, a canonical monopartite NLS consists of an amino acid sequence five amino acids in length where at least four out of five of the amino acids are basic residues.)
[0050] FIGURES 3A-3G depict monomeric and oligomeric structures of the luminal domain of zebrafish Brambleberry. Present as a single copy (i.e., a monomer), the luminal domain of zebrafish (Danio rerio) Brambleberry (SEQ ID NO: 1) was predicted to form a folded domain of four short alpha helices at its N-terminus, whereas the majority of the luminal domain through its C-terminus formed an extended alpha helix (FIGURE 3A). An alternative predicted structure for the monomer of zebrafish Brambleberry has the extended helical domain folded back on itself (FIGURE 3B). Among the dimer structures predicted for zebrafish (Danio rerio) Brambleberry were parallel (FIGURE 3C) and anti-parallel (FIGURE 3D) homodimers. Thus, the transmembrane domains that are C-terminal to the luminal domains are held in close proximity by the oligomerized alpha helices. Side views are shown on the left of the figure, and views from the perspective looking toward the domain near the N-terminus are on the right. Trimers of zebrafish Brambleberry are predicted to form a three-helix bundle (FIGURE 3E), tetramers of zebrafish Brambleberry are predicted to form a four-helix bundle (FIGURE 3F), and pentamers of zebrafish Brambleberry are predicted to form a five-helix bundle (FIGURE 3G). Different polypeptides within the oligomer are shown with different shades of gray.
[0051] FIGURE 4 is a line graph of the results of an experiment showing that nucleic acid transfer is enhanced by co- transfection of an mRNA encoding Brambleberry. A time-course experiment was conducted, adding zebrafish (Danio rerio) Brambleberry (Bmb) mRNA at -3 hours, -1 hour, 0 hours, and +1 hour after adding Gaussia luciferase plasmid DNA. This experiment was conducted in differentiated C2C12 myotubes using Lipofectame 3000 as a transfection reagent. The graph shows the signal from Gaussia luciferase secreted into
supernatants for each condition on days 1-8 post-transfection. The amino acid sequence of zebrafish Brambleberry is provided as SEQ ID NO: 1, and the sequence of an mRNA encoding zebrafish Brambleberry is provided as SEQ ID NO: 2. RLU: relative light units.
[0052] FIGURES 5A-5B are line graphs of the results of experiments in which a Brambleberry mRNA and a DNA expressing a reporter gene were co-packaged into lipid nanoparticles (LNPs) that were used for nucleic acid transfer in differentiated C2C12 myotubes. Gaussia luciferase plasmid DNA and zebrafish (Danio rerio) Brambleberry (Bmb) mRNA (or an irrelevant control mRNA) were co-packaged into LNPs and used to transfect differentiated C2C12 myotubes (FIGURE 5A). NanoLuc luciferase DNA and Bmb mRNA, or DNA alone (0 ng Bmb mRNA) were co-packaged into LNPs on the Nanoassemblr microfluidics device, and used to transfect differentiated C2C12 myotubes (FIGURE 5B). Gaussia or NanoLuc luciferase were detected in supernatants harvested on days 1-5 post- nucleic acid transfer with LNPs. RLU: relative light units.
[0053] FIGURE 6 is a line graph of a titration experiment testing the dose-dependent effect of the amount of Brambleberry mRNA on the efficiency of nucleic acid transfer. Separate LNPs produced using a Nanoassemblr microfluidics device were generated containing NanoLuc plasmid DNA or zebrafish (Danio rerio) Brambleberry (Bmb) mRNA, in order to test the relationship between the amount of Bmb mRNA and nucleic acid transfer efficiency in differentiated C2C 12 myotubes. 50 ng plasmid DNA and the amount of Brambleberry indicated in the figure legend were the amounts of each nucleic acid used per well of a 96- well plate. NanoLuc luciferase activity was read in the supernatants on Days 1-9 post-nucleic acid transfer. RLU: relative light units.
[0054] FIGURE 7 is a line graph of an experiment assessing the impact of antisense oligonucleotides on nucleic acid transfer facilitated by Brambleberry mRNA. Separate LNPs produced using a Nanoassemblr microfluidics device were generated containing NanoLuc plasmid DNA, zebrafish (Danio rerio) Brambleberry (Bmb) mRNA, or an antisense oligonucleotide (ASO). The two ASOs evaluated here were C2mutl and A151. 50 ng plasmid DNA, 50 ng Brambleberry mRNA, and 50 ng ASO were the quantities of each used per well of a 96- well plate of differentiated C2C12 myotubes in this experiment. NanoLuc luciferase activity was read in the supernatants on Days 1 -9 post- nucleic acid transfer. As shown, the presence of the C2mutl ASO enhanced the efficiency of Brambleberry-mediated nucleic acid transfer by 3-to-5-fold. RLU: relative light units.
[0055] FIGURES 8A-8F are a series of bar and line graphs of experiments optimizing and characterizing a muscle-specific promoter. Adeno-associated virus (AAV) vectors expressing firefly luciferase were evaluated after intramuscular injection in mice. Desmin (DES) and Troponin- 1 (TNNI1) promoters were compared against a synthetic muscle- specific promoter (MSP) and a popular non-tissue specific promoter (CMV) 14 days post- injection (FIGURE 8A). The effect of adding a TATA box to the MSP in two different orientations rotated 180° was evaluated 7 days post-injection (FIGURE 8B). The activity of the MSP with the TATA box was compared against the CASI promoter in a longitudinal experiment (FIGURE 8C). Time points with P<0.05 significant differences are marked with an asterisk (*). A version of the muscle-specific promoter with the TATA box lacking CpG motifs was synthesized, and 7 days post-injection luciferase was compared against the original version containing CpG motifs (FIGURE 8D). Plasmids expressing firefly luciferase from the CMV promoter or the MSP promoter were electroporated into the gastrocnemius muscles of mice, and luciferase expression was evaluated by in vivo imaging after 7 days (FIGURE 8E). The human macrophage cell line THP-1 was transfected with plasmids expressing firefly luciferase driven by CMV, CASI, MSP, or DES promoters and then differentiated with PMA/ionomycin before reading luciferase activity (FIGURE 8F).
[0056] FIGURE 9 shows the persistence of gene expression from plasmid DNA (with and without ITRs), AAV DNA, and AAV vectors in mouse muscle. Firefly luciferase was used as a reporter gene, and its expression was quantified in terms of photons per second. Balb/C or immunodeficient nude mice received 20 pg CpG-i- plasmid DNA expressing firefly luciferase from a CpG- muscle-specific promoter (MSP) in their gastrocnemius muscle by electroporation (EP). AAV9 vectors containing an identical transgene were injected at 2.5x1011 vector genomes per kg (vg/kg). A group of nude mice received 6 pg of single- stranded vector DNA extracted from the same batch of AAV9, equal to 20 pg of plasmid on a molar basis.
[0057] FIGURE 10 is a DNA gel electrophoresis image showing CpG methylation. The restriction enzyme Hhal was used to discriminate between DNA that was methylated or not methylated at CpG sites. Hhal is blocked at methylated CpG motifs. Plasmid DNA that was either not methylated at CpG motifs, or methylated at CpG motifs by Sssl methyl transferase, was evaluated for size by agarose gel electrophoresis after treatment with no restriction enzymes, EcoRI alone, or EcoRI plus Hhal. EcoRI cuts the plasmid at a single site
linearizing the DNA, regardless of CpG methylation status. Two bands are seen for the unmethylated product digested with EcoRI plus Hhal, but only one band is seen (because Hhal was blocked by CpG methylation) for the CpG-methylated plasmid.
[0058] FIGURE 11 shows firefly luciferase activity in mice imaged after electroporation with plasmids with differences in CpG methylation. Four groups of n=5 Balb/C mice were electroporated with one of four plasmids expressing firefly luciferase from the CpG- free MSP. The first plasmid had a CpG-i- firefly luciferase coding region and a bacterial origin of replication (Ori) containing CpGs, and was not CpG-methylated by Sssl. The second plasmid had a CpG- firefly luciferase coding region and an Ori containing CpGs, and was not CpG-methylated by Sssl. The third plasmid had a CpG- firefly luciferase coding region and an Ori containing CpGs, and was CpG-methylated by Sssl. The fourth plasmid had a CpG- firefly luciferase coding region and an R6K Ori lacking CpGs, did not have CpG motifs, and was not CpG methylated.
[0059] FIGURE 12 shows luciferase reporter gene expression in an experiment evaluating different attachment factors for promoting gene transfer by LNPs into differentiated C2C12 cells. The LNPs contained a plasmid DNA expressing NanoLuc luciferase from a muscle- specific promoter and a base-modified mRNA encoding Danio rerio Brambleberry at 1 :2 ratio by weight. The control used in this experiment was LNPs generated using the same components except for the lipid-conjugated attachment factor. The lipid-conjugated attachment factors evaluated were: ( 1) stearylated Ml 2 peptide with the amino acid sequence RRQPPRSISSHP (SEQ ID NO: 111) (StM12), (2) a first stearylated LAM1 peptide with the amino acid sequence YIGSR (SEQ ID NO: 112), (3) a second stearylated LAM1 peptide with the amino acid sequence RYVVLPR (SEQ ID NO: 113) that is thought to bind heparin sulfate, (4) a stearylated hyaluron-binding peptide with the amino acid sequence GAHWQFNALTVR (SEQ ID NO: 114), (5) an M12 peptide conjugated to a serine-octanoic acid as a second lipid in addition to being stearylated (SsoM12), (6) a DOPE-conjugated RGD 3 amino acid peptide (DOPE-RGD), a cholesterol-conjugated glucose molecule in the form of cholesterol-undeconoate-glucose (chol-glu), and DPG-conjugated galloyl (DPG- GAL). Six replicate wells were included per condition. RLU means relative light units.
[0060] FIGURE 13 depicts the molecular structures of certain lipid-conjugated attachment factors. The molecular structure of DOPE-RGD is shown in FIGURE 13A, the molecular structure of cholesterol -undecanoate-glucose is shown in FIGURE 13B, and the molecular
structure of DPG-galloyl is shown in FIGURE 13C. The glucose moiety present in cholesterol-undecanoate-glucose is included within an arbutin moiety on this molecule.
[0061] FIGURE 14 shows luciferase reporter gene expression in an experiment evaluating different lipid-conjugated intracellular transport ligands for promoting gene transfer by LNPs into differentiated C2C12 cells. The transport ligands evaluated were stearylated versions of peptides that interact with Tctex-1 (DYNLT-1), a dynein light chain protein. The two peptides are from Arl2 (GGFKLNIWDVGGQK) (SEQ ID NO: 115), and the C-terminus of rhodopsin D (GVSKTETSQVAPA) (SEQ ID NO: 116). These were conjugated to octyl- arginine (i.e. , as in SEQ ID NOs: 117-118). The controls were LNPs with no ligand for intracellular transport proteins (i.e., no dynein binder), and LNPs formulated with stearylated octyl-arginine (StR8). These were compared against identically -formulated LNPs containing stearylated octyl- arginine- Arl2 (StR8Arl2), or stearylated octyl-arginine-conjugated rhodopsin D C-terminus (StR8RhdCT).
[0062] FIGURES 15A -C shows the results of three experiments where orthologs of Danio rerio Brambleberry were evaluated. DNA-mRNA LNPs where the mRNA encoded a Brambleberry protein, or negative control LNPs either with the mRNA but no DNA or with DNA but no mRNA, were evaluated for gene transfer efficiency in C2C12 myotubes that had been differentiated for 5 days in 2% horse serum. The Brambleberry mRNAs were all produced using N1 -methylpseudouridine (NlmpU) in place of uridine. mRNA encoding Danio rerio Brambleberry (SEQ ID NOs: 1 -2) was compared against mRNAs encoding sheepshead pupfish (Cyprinodon variegatus) Brambleberry (SEQ ID NOs: 33-34) and Komodo dragon (Varanus komodoensis) Brambleberry (SEQ ID NOs: 29-30) (FIGURE 15 A). mRNA encoding Danio rerio Brambleberry also was compared against pigeon (Columba livia) Brambleberry (SEQ ID NOs: 27-28), sheepshead pupfish (Cyprinodon variegatus) Brambleberry (SEQ ID NOs: 33-34), and Hawaiian crow Brambleberry (Corvus hawaiiensis) (SEQ ID NOs: 119-120) (FIGURE 15B). In a third experiment, mRNA encoding Danio rerio Brambleberry was compared against Columba livia Brambleberry and a version of Columba livia Brambleberry with a non-native signal peptide (NNSP) (FIGURE 15C). The reporter genes in all three experiments were NanoLuc luciferase. A version of NanoLuc luciferase with an optimized signal peptide was utilized in FIGURE 15C. RLU means relative light units.
[0063] FIGURE 16 shows the effect of including Brambleberry mRNA on gene transfer efficiency in C2C12 myotubes that had been differentiated for 10 days. Ten days prior to contacting cultured C2C12 myotubes with LNPs, the C2C12 cells were switched from media containing 20% FBS to media containing 2% horse serum. The DNA encoded firefly luciferase under a muscle-specific promoter. The relative light units (RLU) emitted by firefly luciferase indicates the relative efficiency of gene transfer. The conditions tested were a no LNP control, a DNA-only control, and DNA-mRNA LNPs where the mRNA encoded Danio rerio Brambleberry. Gene transfer efficiency was 472-fold higher for the DNA-mRNA LNPs containing Brambleberry mRNA than the DNA-only LNPs.
[0064] FIGURE 17 is a graph showing average particle sizes determined by dynamic light scattering (DLS) of LNPs that were generated with different ionizable lipids and stored under different conditions. LNPs were generated using 56.9% ionizable lipid, 27.5% cholesterol, 11.2% DSPC, 1.5% DMG-PEG2000, 0.25% stearylated TCTEX-1 binder StR8RhdCT (SEQ ID NO: 1 18), and 4.3% DPG-galloyl. The ionizable lipids evaluated in this experiment included DLin-KC2-DMA (KC2), SM102, Lipid 29, and CL15F6. The resulting LNPs were sized by DLS after either being left unfiltered and stored at 4°C, filtered and 4°C, filtered and stored at -20°C, or filtered and stored at -80°C.
[0065] FIGURES 18A-H depict the results of an experiment designed to assess the impact of different ionizable lipids on the efficiency of gene transfer by DNA-mRNA LNPs after cryopreservation. This experiment utilized the same preparations of LNPs as shown in FIGURE 17. The LNPs were generated using 27.5% cholesterol, 11.2% DSPC, 1.5% DMG- PEG2000, 0.25% stearylated TCTEX-1 binder StR8RhdCT (SEQ ID NO: 118), and 4.3% DPG-galloyl, and 56.9% of the ionizable lipids DLin-KC2-DMA (KC2), SM102, Lipid 29, or CL15F6. Firefly luciferase activity was read 4 days after adding the LNPs to C2C12 cells that had been differentiated in 2% horse serum for 10 days and cryopreserved at the temperature indicated. The results are shown in FIGURES 18A-D. The chemical structures of the ionizable lipids used here, as well as the pKa of the ionizable head group, are shown in FIGURES 18E-H.
[0066] FIGURE 19 is a graph of the results of an experiment designed to assess whether forms of polyethylene glycol (PEG) with shorter lipid anchors than DMG-PEG were optimal. Forms of PEG2000 with a cholesterol, stearic acid (SA), or C8 ceramide (C8C) anchor were compared against DMG-PEG2000. DNA-mRNA LNPs were generated using a mixture of
lipids containing either IX (1.4% of total lipid by mass) or 2X (2.8% of total lipid by mass) of the PEGylated lipid. Unfiltered LNPs were evaluated as a quality control measure, to ensure the absence of signal loss due to unfilterable LNP aggregates. Filtered LNPs were evaluated after storage at 4°C or -80°C in 10-day differentiated C2C12 myotubes. The efficiency of gene transfer was assessed by measuring the firefly luciferase signal 4 days after adding the LNPs to 96- well plates containing the 10-day differentiated C2C12 myotubes. The firefly luciferase signal was quantified in terms of relative light units (RLU).
[0067] FIGURE 20 is a graph of the results of an experiment designed to assess the impact of using lipid anchors of reduced size on DNA-mRNA LNP stability through cryopreservation and gene transfer efficiency. LNPs containing DNA were generated with and without Brambleberry mRNA, using the ionizable lipids DLin-KC2-DMA or CL15F6, and with either DMG-PEG2000 or C8C-PEG2000. The LNPs evaluated were either left unfiltered and stored at 4°C, filtered and then stored at 4°C, or filtered and then stored at - 80°C. The LNPs were evaluated for gene transfer efficiency in 10-day differentiated C2C12 myotubes. The amount of LNPs added to the cells was normalized to the amount of DNA, and the DNA-mRNA LNPs were generated using a 1:2 ratio of DNA to Brambleberry mRNA. Gene transfer efficiency was assessed by measuring firefly luciferase activity, which was quantified in terms of relative light units (RLU) 4 days after adding the LNPs to 10-day differentiated C2C12 myotubes. Among the conditions where the LNPs were cryopreserved at -80°C, DNA-mRNA LNPs containing Brambleberry mRNA was 251-fold more efficient than the DNA-only LNPs, where both were made using DLin-KC2-DMA and DMG- PEG2000, 324-fold more efficient than the DNA-only LNPs in the context of LNPs made with CL15F6 and DMG-PEG2000 that were cryopreserved at -80°C, and 234-fold more efficient than the DNA-only LNPs in the context of LNPs made with CL15F6 and C8 ceramide (C8C)-PEG2000.
[0068] FIGURES 21A-D show the results of a mouse experiment designed to validate the effect of including Brambleberry mRNA on the efficiency of gene transfer to a non-dividing cell type in vivo. This experiment was also designed to assess whether the 5’ cap Cap2 was non-inferior to Capl when present on the Brambleberry mRNA utilized to facilitate gene transfer. Mice received intramuscular injections of DNA-only control LNPs or DNA-mRNA LNPs where the mRNA encodes Danio rerio Brambleberry (SEQ ID NOs: 1-2), and the
DNA is a circular DNA molecule containing only 14 CpG motifs per molecule that expresses
firefly luciferase under a muscle-specific promoter (SEQ ID NO: 151). DNA and mRNA were encapsulated in LNPs at a ratio of 1 :2, and the amounts of LNPs administered to the mice by intramuscular injection in the quadriceps were normalized to 10 pg DNA per mouse. Mice were injected with the LNPs at week 0 and imaged on weeks 1, 2, 3, 4, 5, and 6 (FIGURE 21 A). The efficiency of gene transfer was quantified by reading the luminosity after weekly luciferin injection by IVIS (FIGURE 21B). The DNA-mRNA LNPs containing Brambleberry mRNA with either the Capl or Cap2 5’ cap structures were consistently more efficient at mediating gene transfer than the DNA-only control LNPs. To evaluate the significance of this difference, a 2-tailed t test was performed for the area under the curve (AUC) of luciferase signal that was detected over the course of the experiment (FIGURE 21C). Another group of mice received the same DNA-mRNA LNP formulation as the group that included Brambleberry mRNA with Capl, except that it did not include the cholesterol - conjugated TCTEX-1 (dynein) binder chol-R8RhdCT (SEQ ID NO: 118). The group injected with DNA-mRNA LNPs lacking chol-R8RhdCT was followed for 5 weeks and the luciferase expression was compared to the otherwise-identical group where the DNA-mRNA LNPs contained chol-R8RhdCT (FIGURE 21D).
[0069] FIGURES 22A-B show the results of a mouse experiment designed to validate the effect of including Brambleberry mRNA on the efficiency of gene transfer to a non-dividing cell type in vivo. This experiment was independent of that depicted in FIGURE 21 and was intended to demonstrate reproducibility of the effect of Brambleberry on the efficiency of gene transfer in non-dividing cells (e.g., muscle cells) in vivo. Mice received intramuscular injections of DNA-only control LNPs or DNA-mRNA LNPs where the mRNA encodes Danio rerio Brambleberry (SEQ ID NOs: 1-2), and the DNA is a circular DNA molecule containing only 14 CpG motifs per molecule that expresses firefly luciferase under a muscle- specific promoter (SEQ ID NO: 151). DNA and mRNA were encapsulated in LNPs at a ratio of 1:2, and the amounts of LNPs administered to the mice by intramuscular injection in the quadriceps were normalized to 10 pig DNA per mouse. Mice were injected with the LNPs at week 0 and imaged on weeks 1, 2, 3, 4, 5, and 6 (FIGURE 22 A). The efficiency of gene transfer was quantified by reading the luminosity after weekly luciferin injection by IVIS (FIGURE 22B).
[0070] FIGURES 23A-C are a series of graphs representing the results of mouse experiments where immunosuppressive molecules were coencapsulated within DNA-mRNA
LNPs. All of the LNPs evaluated in these experiments included an mRNA expressing Danio rerio Brambleberry, alongside a circular DNA molecule expressing firefly luciferase from a muscle-specific promoter. Mice were imaged at weekly time points after intramuscular injection of the DNA-mRNA LNPs in the quadriceps using an IVIS, and the luminescence of each injected quadricep was quantified after luciferin substrate injection. The first experiment tested whether gene transfer efficiency by DNA-mRNA LNPs was improved when dexamethasone-palmitate, INF39, or glyburide were coformulated with (/'.<?. , encapsulated in) the DNA-mRNA LNPs (FIGURE 23A). The second experiment tested whether gene transfer efficiency by DNA-mRNA LNPs was improved oridonin or glyburide were included, in addition to dexamethasone-palmitate, in the DNA-mRNA LNPs (FIGURE 23B). The third experiment compared gene transfer efficiency when dexamethasone-palmitate versus fluticasone-furoate were included in the DNA-mRNA LNPs (FIGURE 23C).
[0071] FIGURES 24A-C are a series of graphs showing RNAi knockdown experiments that utilize an optimized miR architecture. FIGURE 24A shows the extent of knockdown in an experiment conducted by 293T cell transfection using a firefly luciferase gene expressed from a CMV promoter as the target of the miR. The results are normalized to Renilla luciferase expression as a control for transfection efficiency. FIGURE 24B shows the results of a similar experiment, which was conducted in C2C12 muscle cells using a firefly luciferase reporter gene either under the control of a CMV promoter or the muscle-specific promoter (MSP) (SEQ ID NO: 12). FIGURE 24C shows the results of a similar knockdown experiment where the miR was expressed from either a wild-type U6 promoter, a wild-type Hl promoter, or versions of the Hl promoter where the CpG motifs were mutated to CA (version 1, vl) or TG (version 2, v2).
[0072] FIGURES 25A-B are two bar graph charts showing the N-linked glycans attached to an eCD4-Ig protein made in cells with and without a miR that knocks down expression of Fut8. N-linked glycans were removed from eCD4-Ig protein by PNGase F, permethylated, and characterized by liquid chromatography-mass spectrometry (LC-MS). FIGURE 25A shows the N-linked glycan content of the eCD4-lg protein made in the parental cell line, and FIGURE 25B shows the N-linked glycan content of the eCD4-Ig protein made in the cell line expressing the miR that targets Fut8.
[0073] FIGURE 26 shows the prevalence of an O-linked glycan attached to eCD4-Ig protein made with or without co-transfection with a plasmid expressing a miR that knocks down
GALNT2. An eCD4-Ig protein was generated by transient transfection with an empty control plasmid or a plasmid expressing a miR that knocks down GALNT2 was characterized by mass spectrometry under the conditions described herein. The proportion of eCD4-Ig protein masses with and without an O-linked glycan were compared. 24% of the control eCD4-Ig protein was O-glycosylated, whereas 3.4% was O-glycosylated when made by co-transfection with the plasmid expressing the miR that knocks down GALNT2.
[0074] FIGURE 27 shows a bar graph of a comparison of the efficiency of gene transfer by DNA-mRNA LNPs after intramuscular injection with and without hyaluronidase. Five nude mice per group received 25 pL intramuscular injections of LNPs containing 10 pg DNA and 20 pg Brambleberry mRNA in the quadriceps. Mice were imaged by IVIS after luciferin injection. Bovine hyaluronidase was added after thawing of cryopreserved LNPs such that its final concentration was 1 unit per pL after mixing with the thawed LNPs. Area under the curve (AUC) values were calculated for the period from weeks 2-4 post-injection. The use of hyaluronidase significantly improved the efficiency of gene transfer, in terms of the AUC of luciferase signal detected by IVIS (P=0.017).
DETAILED DESCRIPTION
[0075] The present disclosure is based, in part, on the discovery that double membrane fusogen proteins provide a solution to the topological problem of transferring DNA sequestered within a double membrane envelope to the nucleus. In early embryo development in animals with larger eggs than mammals, chromosomes become enveloped by a double membrane in structures known as karyomeres, which ultimately fuse to form the nucleus. The protein that mediates this fusion is Brambleberry — so named, because in its absence, consolidation of karyomeres into a nucleus is arrested, and the unfused karyomeres resemble a brambleberry (Abrams et al. (2012) supra) (FIGURE 1A). By contrast, in wild- type Danio rerio, the karyomeres fuse and form a nucleus (FIGURE IB). It has been discovered that transient expression of Brambleberry from an mRNA that is co-packaged with a plasmid DNA, e.g., in an LNP, mediates fusion of double membrane-enveloped vesicles sequestering DNA with the nucleus, thereby allowing successful nucleic acid transfer to non-dividing cells.
[0076] Accordingly, the disclosure relates, in part, to systems and methods for the transfer of a non-native DNA into a nucleus of a cell, such as a mammalian cell. The system or method comprises a non-native DNA and an RNA that encodes a double membrane fusogen protein, which, when present in the cell, mediates transfer of the non-native DNA into the nucleus of the cell.
[0077] In addition, it has been discovered that a plasmid with a CpG-free promoter in non- dividing cells is capable of persistently expressing a gene of interest, without being silenced, and without the loss of the plasmid, e.g., by degradation. Furthermore it has been discovered that repositioning the TATA box in naturally occurring muscle-specific promoters results in improved expression. Accordingly, the disclosure also relates, in part, to muscle-specific promoters that can be used in combination with the systems described herein, or in other applications.
[0078] The discovery that a double membrane fusogen protein, such as Brambleberry, enables efficient nucleic acid transfer into non-dividing cells provides a non- viral system for nucleic acid transfer, which, when combined with a CpG-free muscle-specific promoter operably linked to a gene of interest, allows persistent long-term expression of the gene of interest in muscle.
I. Definitions
[0079] The term “double membrane” means a biological membrane consisting of two lipid bilayers separated by an aqueous phase. A well-known example of a double membrane is the nuclear envelope a eukaryotic cell. A nuclear envelope has two membrane layers, an inner bilayer and an outer bilayer.
[0080] The term “double membrane fusogen” means any protein or group of proteins that mediate the fusion of a first double membrane with a second double membrane.
[0081] The term “Brambleberry”, as used herein, means any protein that has a signal peptide for secretion into the ER, a luminal domain, and a transmembrane domain(s) and is capable of performing the same function as zebrafish (Danio rerio) Brambleberry
 fusing the nuclear envelope with another double membrane-enveloped structure, thereby introducing DNA contained within the double membrane-enveloped structure into the nucleus).
fusing the nuclear envelope with another double membrane-enveloped structure, thereby introducing DNA contained within the double membrane-enveloped structure into the nucleus).
Exemplary Brambleberry proteins include any protein annotated by GenBank as a
Brambleberry protein as well as protein with at least 70% (e.g., at least 80%, at least 85%, at
least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100%) sequence identity to a protein annotated by GenBank as a Brambleberry protein or other ortholog of a Brambleberry protein and which is capable of performing the same function as zebrafish Brambleberry (e.g. , SEQ ID NOs: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, and 147). Exemplary Brambleberry proteins also include any chimeric protein that includes one or more portions of at least one Brambleberry protein and is capable of performing the same function as zebrafish Brambleberry as well as any split Brambleberry protein that is capable of performing the same function as zebrafish Brambleberry.
[0082] The term “split Brambleberry”, as used herein, means two or more proteins capable of forming a protein complex, where the protein complex is capable of performing the same function as zebrafish Brambleberry, e.g., a first protein that includes the luminal and transmembrane domains of zebrafish Brambleberry plus a non-native dimerization domain, combined with a second protein that includes a matched non-native dimerization domain capable of dimerizing with that of the first protein and the DNA-binding domain of zebrafish Brambleberry.
[0083] The term “non-native” or “non-endogenous”, as used herein, means not existing naturally in a place but coming from somewhere else. For example, a non-native DNA or a non-endogenous DNA refers to a DNA existing, e.g., in a cell, where the DNA does not naturally occur in the cell.
[0084] The term “signal peptide”, as used herein, means a peptide that confers a signal to a protein associated therewith for secretion into the endoplasmic reticulum (ER).
[0085] The term “functional fragment” of a protein refers to a fragment of a protein that retains, for example, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, or 100% of the protein activity of the corresponding full-length, naturally occurring protein. Protein activity may be assayed by any method known in the art. In certain embodiments, the functional fragment comprises at least 50, at least 75, at least 100, at least 125, or at least 150 consecutive amino acids present in the protein. In certain embodiments, the functional fragment comprises a truncation of about 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, or 50 amino acids as compared to the protein.
[0086] The term “non-coding RNA”, as used herein, means an RNA molecule that mediates a function other than serving as a message for translation into protein, e.g., an shRNA, miR, ribozyme, or aptamer.
[0087] The term “denatured,” used in reference to DNA, means DNA lacking canonical Watson-Crick base pairing. Denatured DNA can be generated by melting the DNA with increased temperatures, e.g., 95°C, optionally followed by rapid cooling.
[0088] The term “gene editing effector protein”, as used herein, means any protein component of a gene editing system that contributes to editing the genome of a cell, for example, a nuclease. In certain embodiments, the gene editing system is a nuclease-based genome editing system e.g., a Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) based genome editing system, a Transcription activator-like effector nuclease (TALEN) based genome editing system, a zinc-finger nuclease (ZFN) based genome editing system, a homing endonuclease (HE) based genome editing system, a prime editor (PE; reverse transcriptase coupled to an RNA-programmable nickase and a prime editing guide) based genome editing system, or a derivative of any of the foregoing).
[0089] The term “transduce”, as used herein, means to introduce DNA into a cell with any vector, regardless of whether the vector is a viral vector.
[0090] The term “effective amount”, as used herein, refers to the amount of an active agent e.g., a double membrane fusogen protein, a transgenic gene, a non-coding RNA, a system for DNA transfer) sufficient to effect beneficial or desired results. An effective amount can be administered in one or more administrations, applications or dosages and is not intended to be limited to a particular formulation or administration route.
[0091] As used herein, “treat”, “treating” and “treatment”, as used herein, mean the treatment of a disease in a subject, e.g., in a human. This includes: (a) inhibiting the disease, i.e. , arresting its development; and (b) relieving the disease, i.e., causing regression of the disease state. As used herein, the terms “subject” and “patient” refer to an organism to be treated by the methods and compositions described herein. Such organisms preferably include, but are not limited to, mammals (e.g. , murines, simians, equines, bovines, porcines, canines, felines, and the like), and more preferably includes humans.
[0092] The term “pro-drug” refers to a chemically modified, inactive drug that is converted into an active drug in the body of a subject through enzymatic or chemical reactions.
Prodrugs can be used to improve the properties of a drug, such as its pharmacokinetics, solubility, lipophilicity, permeability, and targeting.
[0093] The “LogP value” or “partition coefficient” refers to the quantification of a compound’s distribution between two immiscible liquids, typically octanol and water. The higher the LogP value the more lipophilic the molecule or compound.
[0094] The methods and compositions described herein can be used alone or in combination with other therapeutic agents and/or modalities. The term administered “in combination,” as used herein, is understood to mean that two (or more) different treatments are delivered to the subject during the course of the subject’s affliction with the disorder, such that the effects of the treatments on the patient overlap at a point in time. In certain embodiments, the delivery of one treatment is still occurring when the delivery of the second begins, so that there is overlap in terms of administration. This is sometimes referred to herein as “simultaneous” or “concurrent delivery.” In other embodiments, the delivery of one treatment ends before the delivery of the other treatment begins. In certain embodiments of either case, the treatment is more effective because of combined administration. For example, the second treatment is more effective, e.g. , an equivalent effect is seen with less of the second treatment, or the second treatment reduces symptoms to a greater extent, than would be seen if the second treatment were administered in the absence of the first treatment, or the analogous situation is seen with the first treatment. In certain embodiments, delivery is such that the reduction in a symptom, or other parameter related to the disorder is greater than what would be observed with one treatment delivered in the absence of the other. The effect of the two treatments can be partially additive, wholly additive, or greater than additive. The delivery can be such that an effect of the first treatment delivered is still detectable when the second is delivered.
[0095] Throughout the description, where compositions are described as having, including, or comprising specific components, or where processes and methods are described as having, including, or comprising specific steps, it is contemplated that, additionally, there are compositions of the present invention that consist essentially of, or consist of, the recited components, and that there are processes and methods according to the present invention that consist essentially of, or consist of, the recited processing steps.
[0096] In the application, where an element or component is said to be included in and/or selected from a list of recited elements or components, it should be understood that the element or component can be any one of the recited elements or components, or the element or component can be selected from a group consisting of two or more of the recited elements or components.
[0097] Further, it should be understood that elements and/or features of a composition or a method described herein can be combined in a variety of ways without departing from the spirit and scope of the present invention, whether explicit or implicit herein. For example, where reference is made to a particular compound, that compound can be used in various embodiments of compositions of the present invention and/or in methods of the present invention, unless otherwise understood from the context. In other words, within this application, embodiments have been described and depicted in a way that enables a clear and concise application to be written and drawn, but it is intended and will be appreciated that embodiments may be variously combined or separated without parting from the present teachings and invention(s). For example, it will be appreciated that all features described and depicted herein can be applicable to all aspects of the invention(s) described and depicted herein.
[0098] It should be understood that the expression “at least one of’ includes individually each of the recited objects after the expression and the various combinations of two or more of the recited objects unless otherwise understood from the context and use. The expression “and/or” in connection with three or more recited objects should be understood to have the same meaning unless otherwise understood from the context.
[0099] The use of the term “include,” “includes,” “including,” “have,” “has,” “having,” “contain,” “contains,” or “containing,” including grammatical equivalents thereof, should be understood generally as open-ended and non-limiting, for example, not excluding additional unrecited elements or steps, unless otherwise specifically stated or understood from the context.
[00100] Where the use of the term “about” is before a quantitative value, the present invention also includes the specific quantitative value itself, unless specifically stated otherwise. As used herein, the term “about” refers to a ±10% variation from the nominal value unless otherwise indicated or inferred.
[00101] It should be understood that the order of steps or order for performing certain actions is immaterial so long as the present invention remain operable. Moreover, two or more steps or actions may be conducted simultaneously.
[00102] The use of any and all examples, or exemplary language herein, for example, “such as” or “including,” is intended merely to illustrate better the present invention and does not pose a limitation on the scope of the invention unless claimed. No language in the specification should be construed as indicating any non-claimed element as essential to the practice of the present invention.
IL Systems for Nucleic Acid Transfer Including a Double Membrane Fusogen Protein
[00103] The present disclosure relates, in part, to a system (e.g. , a nanoparticle such as a lipid nanoparticle, a formulation, or a pharmaceutical formulation) for nucleic acid transfer in which an RNA molecule encoding a protein that mediates the entry of a DNA into the nucleus e.g. , a double membrane fusogen protein) is co-packaged with DNA, for example, DNA encoding a gene of interest such as a therapeutic gene. When delivered to a cell, e.g., by a lipid nanoparticle (LNP), the transient expression of the double membrane fusogen protein from the RNA molecule that is co-packaged with the DNA mediates fusion of the LNP and the nuclear membrane of a cell, thereby allowing successful transfer of the DNA to the cells. Such systems can also be used together with electroporation as the delivery method. Electroporation of an RNA encoding a double membrane fusogen protein and a DNA of interest (e.g., a therapeutic gene) allows DNA to enter the nucleus that otherwise would have entered the cell but lacked access to the nucleus.
A. Double Membrane Fusogen Protein and mRNA Encoding Same
[00104] The term “double membrane fusogen” means any protein or group of proteins that mediate the fusion of a first double membrane with a second double membrane. In the context of the instant disclosure, the double membrane fusogen protein functions to mediate fusion of biological membranes to allow for a DNA of interest, e.g., a DNA encoding a therapeutic protein, to enter the nucleus of a cell so that it can be expressed. Double membrane fusogen proteins can include one or more of the following domains: a signal peptide, a luminal domain, a transmembrane domain, and a cytoplasmic domain comprising a DNA-binding domain. Exemplary double membrane fusogen proteins include Brambleberry proteins (including, for example, split Brambleberry proteins), proteins involved in
karyogamy, or functional fragments, variants, or chimeras of any of the foregoing. In certain embodiments, the double membrane fusogen protein is not a human protein.
[00105] The cytoplasmic domain of a double membrane fusogen protein can include a nuclear localization signal (NLS). An exemplary NLS comprises or consists of an amino acid sequence five amino acids in length wherein at least four out of five of the amino acids are basic residues. Another exemplary NLS comprises or consists of an amino acid sequence comprising at least one basic residue separated by a spacer of ten to fifteen amino acids from three or more basic residues. For example, the cytoplasmic domain can include from one to about 30 basic residues separated by about 10, 11, 12, 13, 14, or 15 amino acids from three, four, five, six, seven, eight, nine, ten, or more basic residues. In certain embodiments, the NLS does not naturally occur in the amino acid sequence of a naturally -occurring wild-type double membrane fusogen (e.g., Brambleberry protein) having the greatest sequence identity to the double membrane fusogen.
[00106] In certain embodiments, the double membrane fusogen protein comprises an amino acid sequence having at least 80%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to any one of SEQ ID NOs: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, and 147. i. Double Membrane Fusogen Protein Fragments
[00107] As used herein, the term “functional fragment” of a double membrane fusogen protein refers to fragment of a double membrane fusogen protein that retains, for example, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, or 100% of the fusogenic protein activity of the corresponding full-length, naturally occurring double membrane fusogen protein. Fusogenic protein activity may be assayed by any method known in the art, including, for example, by measuring one or more criteria indicative of membrane fusion activity, such as measuring expression of a reporter protein following co-transduction of a DNA encoding the reporter protein with an RNA encoding the double membrane fusogen protein fragment in a C2C12 myotube model, as described in Example 1. In certain embodiments, the functional fragment comprises at least 50, at least 75, at least 100, at least 125, or at least 150 consecutive amino acids present in a
double membrane fusogen protein provided herein (e.g., one of SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, and 147). In certain embodiments, the functional fragment comprises a truncation of about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, or 50 amino acids as compared to a double membrane fusogen protein provided herein (e.g., one of SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, and 147).
[00108] In certain embodiments, the functional fragment of a double membrane fusogen protein includes one or more of the following domains: a signal peptide, a luminal domain, a transmembrane domain, a multimerization domain, and a cytoplasmic domain comprising a DNA-binding domain of SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, or 147. In certain embodiments, the functional fragment of a double membrane fusogen protein comprises an amino acid sequence having at least 80%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91 %, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to one or more of the signal peptide, the luminal domain, the transmembrane domain, and the cytoplasmic domain comprising a DNA-binding domain of SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, or 147. ii. Double Membrane Fusogen Protein Variants
[00109] As used herein, the term “variant” of a double membrane fusogen protein refers to variant of a double membrane fusogen protein or a functional fragment thereof that retains, for example, at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, or 100% of the fusogenic protein activity of the corresponding full-length, naturally occurring double membrane fusogen protein. Fusogenic protein activity may be assayed by any method known in the art, including, for example, by measuring one or more criteria indicative of the membrane fusion activity, such as measuring expression of a reporter protein following co-transduction of a DNA encoding the reporter protein with an RNA encoding the double membrane fusogen protein fragment in a C2C12 myotube model, as described in Example 1.
[00110] In certain embodiments, the variant of a double membrane fusogen protein comprises an amino acid sequence having at least 80%, at least 85%, at least 86%, at least
87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to a double membrane fusogen protein sequence described herein. In certain embodiments, the variant of a double membrane fusogen protein comprises an amino acid substitution relative to a double membrane fusogen protein sequence provided herein. For example, in certain embodiments, the variant of a double membrane fusogen protein comprises 1, up to 2, up to 3, up to 4, up to 5, up to 6, up to 7, up to 8, up to 9, or up to 10 amino acid substitutions relative to a double membrane fusogen protein sequence provided herein (e.g., SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, and 147).
[00111] In certain embodiments, the variant of a double membrane fusogen protein comprises a conservative substitution relative to a double membrane fusogen protein sequence provided herein. For example, in certain embodiments, the variant of a double membrane fusogen protein comprises 1 , up to 2, up to 3, up to 4, up to 5, up to 6, up to 7, up to 8, up to 9, or up to 10 conservative substitutions relative to a double membrane fusogen protein sequence provided herein. As used herein, the term “conservative substitution” refers to a substitution with a structurally similar amino acid. For example, conservative substitutions may include those within the following groups: Ser and Cys; Leu, IIe, and Vai; Glu and Asp; Lys, Arg, and His; Phe, Tyr, and Trp; and Gin, Asn, Glu, Asp. Conservative substitutions may also be defined by the BLAST (Basic Local Alignment Search Tool) algorithm, the BLOSUM substitution matrix (e.g., BLOSUM 62 matrix), or the PAM substitution^ matrix (e.g., the PAM 250 matrix). In certain embodiments, the double membrane fusogen protein comprises 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 conservative substitutions.
[00112] As used herein, the phrase “percent identity” and “% identity” refers to the extent to which two sequences e.g., two polypeptides or two nucleic acids have the same respective amino acid or nucleotide at the same positions in an alignment. As used herein, “percent identity” between a polypeptide sequence and a reference sequence is defined as the percentage of amino acid residues in the polypeptide sequence that are identical to the amino acid residues in the reference sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity. Similarly, “percent identity” between a nucleic acid sequence and a reference sequence is defined as the percentage of nucleotides in the nucleic acid sequence that are identical to the nucleotides in the reference
sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity. Alignment for purposes of determining percent sequence identity (e.g., nucleic acid sequence identity or amino acid sequence identity) can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST (Basic Local Alignment Search Tool), BLAST- 2, ALIGN, MEGALIGN (DNASTAR), CLUSTALW, CLUSTAL OMEGA, or MUSCLE software. For a discussion of basic issues in searching sequence databases see Altschul et al., (1994) NATURE GENETICS 6: 1 19-129, which is fully incorporated by reference herein. Those skilled in the art can determine appropriate parameters for aligning sequences, including any algorithms needed to achieve maximal alignment over the full length of the sequences being compared.
[00113] The alignment algorithms above may take into account a scoring matrix to calculate an alignment score (see Chao et al., BIOMOLECULES (2022) 12(4): 546). For example, for an amino acid sequence at least 85 amino acids in length, the scoring matrix recommended by the BLAST algorithm is BLOSUM-62. The BLOSUM-62 scoring matrix assigns positive, zero, or negative scores between each pair or standard amino acid residues (see Henikoff and Henikoff, PROC. NATL. ACAD. SCI. USA (1992) 89, 10915-19 at FIG. 2). A positive score between two amino acid residues indicates that substitution of these amino acid residues for each other is conservative. As used herein, “similarity” between a subject amino acid sequence and a reference amino acid sequence refers to the percentage of amino acid residues in the subject amino acid sequence that are identical or have a conservative substitution according to the BLOSUM-62 scoring matrix, relative to the amino acid residues in the reference sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum sequence alignment score. iii. Brambleberry Proteins
[00114] The domain architecture of Brambleherry provides information about structure- function relationships with respect to Brambleberry’s ability to mediate membrane fusion. A signal peptide exists at the N-terminus, consistent with a Type I transmembrane protein orientation (FIGURES 2A-B). The signal peptide is believed to mediate secretion of the protein into the endoplasmic reticulum (ER), and then is believed to be cleaved from the pre- protein, generating the mature Brambleberry protein. The existence of at least one transmembrane domain indicates that the protein is a membrane-bound protein. The region
between the signal peptide and transmembrane domain is a luminal domain, which would exist within the ER or cellular structures that are topologically similar to the ER. Three transmembrane domains are predicted by the membrane topology prediction program TOPCONS, as shown in FIGURES 2A-B. Two of the three predicted transmembrane regions are close in proximity to each other, e.g., separated by just one amino acid in zebrafish Brambleberry. At the C-terminus of Brambleberry is a cytoplasmic domain, which contains a DNA-binding domain (FIGURES 2A-B). It is contemplated that the proposed function of Brambleberry is to mediate the fusion of the nuclear envelope with other double membrane-enveloped structures, which, in the context of systems for nucleic acid transfer, contain DNA. The fusion of the nuclear envelope with the other double membrane- enveloped vesicles deposits DNA contained within those double membrane-enveloped vesicles into the nucleus. The transmembrane domains present in Brambleberry are thought to be membrane anchors when Brambleberry is in the nuclear envelope or in other double membrane-enveloped vesicles, and that have a role, e.g., including as a membrane anchor, in the fusion of the nuclear envelope with other double membrane-enveloped vesicles that contain DNA.
[00115] Predicted oligomerization structures of the Brambleberry luminal domain also are consistent with Brambleberry’s function as a membrane fusogen (i.e., a protein that is capable of mediating the fusion of membranes, and in the case of systems for nucleic acid transfer, the fusion of the nuclear envelope with other double membrane-enveloped structures containing DNA). Structures for oligomers of the zebrafish (Danio rerio) Brambleberry luminal domain were predicted using AlphaFold (FIGURE 3A-G). Monomers of Brambleberry luminal domains were predicted to have four short alpha helices at their N- terminus, whereas the majority of the protein through its C-terminus formed a single extended alpha helix (FIGURE 3A). An alternative structure for a monomer of the Brambleberry luminal domain formed a hairpin-like fold, in which the extended alpha helix folded back onto itself (FIGURE 3B). The zebrafish Brambleberry luminal domain was predicted to form alternative dimer structures, e.g., a parallel homodimer formed by the extended alpha helical domains (FIGURE 3C) or an anti-parallel homodimer formed from alpha helical domains with a hairpin- like fold (FIGURE 3D). It is contemplated that such parallel homodimer and antiparallel homodimer conformations may represent functional states of Brambleberry involved in fusing the nuclear envelope with other double membrane-
enveloped structures in the cell. The oligomerization of Brambleberry luminal domains in an extended conformation would bring together the transmembrane regions at the C-terminus of the luminal domains, potentially driving opposing membranes together and thus contributing to the mechanism of membrane fusion. The antiparallel homodimer conformation (FIGURE 3D) also may facilitate initial Brambleberry dimerization across opposing membranes. It is contemplated that the reorientation of Brambleberry luminal domains to the parallel orientation, and/or the dimerization of extended Brambleberry luminal domains in the parallel orientation, may be energetically-favorable conformational changes that drive opposing membranes together (e.g., the nuclear envelope and other double membrane-enveloped structures containing DNA) as part of a fusion mechanism that mediates the transfer of DNA into the nucleus.
[00116] Brambleberry proteins may tend to associate to form higher-order oligomers in the parallel orientation. AlphaFold predicts that three zebrafish Brambleberry luminal domains will tend to associate in a parallel orientation in a three-helix bundle (FIGURE 3C).
Likewise, AlphaFold predicts that four zebrafish Brambleberry luminal domains will tend to associate in a parallel orientation in a four-helix bundle (FIGURE 3D). AlphaFold predicts that five zebrafish Brambleberry luminal domains will tend to associate in a parallel orientation in a five-helix bundle (FIGURE 3E). Oligomers with few (e.g., two or three) participating Brambleberry proteins may generally represent intermediates that tend towards oligomers with additional participating Brambleberry proteins. The formation of parallel- oriented Brambleberry oligomers would drive the transmembrane regions close together, possibly as a mechanism involved in the fusion of the nuclear envelope with other double membrane-enveloped structures. Brambleberry monomers may unite with Brambleberry oligomers to drive the fusion of the membranes in which they are anchored, and/or Brambleberry oligomers may associate to form higher-order oligomers that drive the fusion of the membranes in which they are anchored.
[00117] Thus, in certain embodiments, a Brambleberry protein of the disclosure includes one or more of the following domains: a signal peptide, a luminal domain, a transmembrane domain, a multimerization domain, and a cytoplasmic domain comprising a DNA-binding domain. In certain embodiments, the Brambleberry protein comprises an alpha helical domain at least 20, 30, 40, 50, 60, 70, 80, 90, or 100 amino acids in length.
a. Split Brambleberry proteins
[00118] In some instances, a Brambleberry protein is a split Brambleberry protein, which includes two or more proteins or fragments thereof capable of forming a protein complex, where the protein complex is capable of performing the same function as zebrafish Brambleberry. For example, a split Brambleberry protein can include, e.g. , a first protein that includes (1) the luminal and transmembrane domains of a first Brambleberry protein (e.g., a Brambleberry protein domain from a first species) and a non-native dimerization domain, combined with (2) a second protein that includes the DNA-binding domain of second Brambleberry protein (e.g., a Brambleberry protein from a second species) and a second non- native dimerization domain capable of dimerizing with that of the first protein. The second non-native dimerization domain can be the same as or different that the first non-native dimerization domain, as long as it is capable of dimerizing with the first dimerization domain. It is contemplated that any combination of Brambleberry proteins or variants or fragments thereof can be combined to produce a split Brambleberry protein. For example, a first Brambleberry protein or fragment or variant thereof from a first species (or variant thereof) can contribute a luminal domain, a second Brambleberry protein or fragment or variant thereof from a second species (or variant thereof) can contribute a transmembrane domain, and a third Brambleberry protein or fragment or variant thereof from a third species (or variant thereof) can contribute a DNA-binding domain. b. Brambleberry proteins from various species and chimeras thereof
[00119] In one aspect, the disclosure provides systems for nucleic acid transfer that include Brambleberry proteins of species other than zebrafish, and functional fragments, variants, or chimeras thereof. For example, the Brambleberry protein of the Golden eagle (Aquila chrysaetos chrysaetos) is provided as SEQ ID NO: 25, and the sequence of an mRNA encoding the Brambleberry protein of Golden eagle is provided as SEQ ID NO: 26. The Brambleberry protein of the Passenger pigeon (Columba livia) is provided as SEQ ID NO: 27, and the sequence of an mRNA encoding the Brambleberry protein of the Passenger pigeon is provided as SEQ ID NO: 28. The Brambleberry protein of the Komodo dragon (Varanus komodoensis) is provided as SEQ ID NO: 29, and the sequence of an mRNA encoding the Brambleberry protein of the Komodo dragon is provided as SEQ ID NO: 30. The Brambleberry protein of Goode's thomscrub tortoise (Gopherus evgoodei) is provided as SEQ ID NO: 31, and the sequence of an mRNA encoding the Brambleberry protein of
Goode's thornscrub tortoise is provided as SEQ ID NO: 32. The Brambleberry protein of the Sheepshead pupfish {Cyprinodon variegatus) is provided as SEQ ID NO: 33, and the sequence of an mRNA encoding the Brambleberry protein of the Sheepshead pupfish is provided as SEQ ID NO: 34. The Brambleberry protein of the Hawaiian crow {Corvus hawaiiensis) is provided as SEQ ID NO: 119, and the sequence of an mRNA encoding the Brambleberry protein of the Hawaiian crow is provided as SEQ ID NO: 120. The Brambleberry protein of the swan Cygnus olor) is provided as SEQ ID NO: 121, and the sequence of an mRNA encoding the Brambleberry protein of the swan is provided as SEQ ID NO: 122. The Brambleberry protein of the bam owl {Tyto alba) is provided as SEQ ID NO: 123, and the sequence of an mRNA encoding the Brambleberry protein of the bam owl is provided as SEQ ID NO: 124. The Brambleberry protein of the hummingbird {Calypte anna) is provided as SEQ ID NO: 125, and the sequence of an mRNA encoding the Brambleberry protein of the hummingbird is provided as SEQ ID NO: 126. The Brambleberry protein of the crested ibis {Nipponia nippon) is provided as SEQ ID NO: 127, and the sequence of an mRNA encoding the Brambleberry protein of the crested ibis is provided as SEQ ID NO: 128. The Brambleberry protein of the bald eagle {Haliaeetus leucocephalus) is provided as SEQ ID NO: 129, and the sequence of an mRNA encoding the Brambleberry protein of the bald eagle is provided as SEQ ID NO: 130. The Brambleberry protein of the sparrow {Passer domesticus) is provided as SEQ ID NO: 131, and the sequence of an mRNA encoding the Brambleberry protein of the sparrow is provided as SEQ ID NO: 132. The Brambleberry protein of the swift {Apus apus) is provided as SEQ ID NO: 133, and the sequence of an mRNA encoding the Brambleberry protein of the swift is provided as SEQ ID NO: 134. The Brambleberry protein of the alligator {Alligator mississippiensis) is provided as SEQ ID NO: 135, and the sequence of an mRNA encoding the Brambleberry protein of the alligator is provided as SEQ ID NO: 136. The Brambleberry protein of the Sinaloan desert tortoise {Gopherus evgoodei) is provided as SEQ ID NO: 137, and the sequence of an mRNA encoding the Brambleberry protein of the Sinaloan desert tortoise is provided as SEQ ID NO: 138. The Brambleberry protein of the African clawed frog {Xenopus laevis) is provided as SEQ ID NO: 139, and the sequence of an mRNA encoding the Brambleberry protein of the African clawed frog is provided as SEQ ID NO: 140. The Brambleberry protein of the Nile tilapia {Oreochromis niloticus) is provided as SEQ ID NO: 141, and the sequence of an mRNA encoding the Brambleberry protein of the Nile tilapia is provided as SEQ ID NO:
142. The Brambleberry protein of the lamprey {Petromyzon marinus) is provided as SEQ ID
NO: 143, and the sequence of an mRNA encoding the Brambleberry protein of the lamprey is provided as SEQ ID NO: 144. The Brambleberry protein of the hydrothermal vent snail (Gigantopelta aegis) is provided as SEQ ID NO: 145, and the sequence of an mRNA encoding the Brambleberry protein of the hydrothermal vent snail is provided as SEQ ID NO: 146. The Brambleberry protein of the glass sponge (Oopsacas minuta) is provided as SEQ ID NO: 147, and the sequence of an mRNA encoding the Brambleberry protein of the glass sponge is provided as SEQ ID NO: 148.
[00120] These Brambleberry sequences are provided as examples, and are not intended to be limiting, as other Brambleberry proteins or functional fragments, variants, or chimeras thereof are expected to be functional in mediating nucleic acid transfer.
[00121] In certain embodiments, chimeric Brambleberry proteins are assembled from the proteins of one or more species. For example, the signal peptide, luminal domain, first transmembrane domain, second transmembrane domain, third transmembrane domain, membrane-proximal cytoplasmic domain, and DNA-binding domain can be swapped, individually or in combination, among different Brambleberry proteins. Thus, Brambleberry sequences from various species, including chimeras thereof, can be utilized in systems for nucleic acid transfer. iv. Inclusion of miR Target and De-targeting Sites in mRNA Encoding the Double Membrane Fusogen Protein and/or the RNA Transcript for the DNA of Interest
[00122] In one aspect, the disclosure provides one or more double membrane fusogen protein-encoding mRNAs that contain target sites for microRNAs (miRs). The inclusion of an miR target site in the mRNA transcript encoding a double membrane fusogen protein provides a means of de-targeting cell types that may, in certain instances, be undesirable to target (i.e. , are “off-target”). Off-target tissues and cell types may include myeloid lineage cells, e.g., antigen-presenting cells (APCs), and or liver cells (e.g., hepatocytes). Target sites for miRs can be engineered into the mRNA transcript, e.g., in multiple copies, in the 5’ UTR, and/or in the 3’ UTR. The de-targeting of APCs using one or more miR target sites is complementary with the use of a tissue specific promoter (e.g., a muscle-specific promoter) for preventing the expression of the therapeutic gene in APCs. Sequences of miR target sites for de-targeting APCs (e.g., myeloid lineage cells) include target sites for miR-126-3p (SEQ ID NO: 43), miR-142-3p (SEQ ID NO: 44), miR-142-5p (SEQ ID NO: 45), miR-146a-3p
(SEQ ID NO: 46), miR-146a-5p (SEQ ID NO: 47), and miR-146b-3p (SEQ ID NO: 48).
Such miR target sites, e.g., for de-targeting APCs, can be engineered into the fusogen protein-encoding mRNA and/or engineered into the transcript for the DNA of interest e.g., a therapeutic protein) delivered by the system for nucleic acid transfer of the present invention. The benefit of having such miR target sites in both the fusogen protein-encoding mRNA and the transcript for the DNA of interest (e.g., a therapeutic protein) is the intentional reduction of the efficiency of fusogen protein-mediated nucleic acid transfer in off-target cell populations such as APCs, combined with the reduction of the expression of the therapeutic protein in off-target cell populations that are transduced despite the reduced efficiency of fusogen protein-mediated nucleic acid transfer.
[00123] The liver can be de-targeted, e.g., by engineering target sites for miR-122 into the Brambleberry mRNA and/or the transcript for the therapeutic protein being delivered by this system for nucleic acid transfer. miR-122 is reported to be a liver- specific miR constituting 70% of the total miR content of hepatocytes (Jopling (2012) RNA BtOL., 9(2): 1 7-142). Target sites for miR-122-3p (SEQ ID NO: 50) and miR-122-5p (SEQ ID NO: 51) are provided. De-targeting transduction of the liver via the introduction of miR target sites into the fusogen protein-encoding mRNA and/or de-targeting expression of the therapeutic protein in the liver via the introduction of miR target sites into the transcript for the therapeutic protein, can be desirable when a tissue or organ other than liver, e.g., muscle, is the intended site for the production of the therapeutic protein.
[00124] Certain other miR target sites can be used to reduce the expression of the transgene in inflammatory and/or immunogenic contexts. For instance, the introduction of target sites for miRs that regulate inflammation and/or immunogenicity, e.g., miR-155-3p (SEQ ID NO: 52) and/or miR-155-5p (SEQ ID NO: 53), into the transcript for the therapeutic gene can be used to suppress its expression in inflammatory and/or immunogenic contexts, thereby reducing the potential for the therapeutic protein to be targeted by immune responses. v. Inclusion of a 5’ cap and/or an internal ribosome entry site (IRES)
[00125] The systems for nucleic acid transfer described herein can include an mRNA encoding a double membrane fusogen protein that comprises a 5 ’ cap and/or an internal ribosome entry site (IRES). 5’ caps comprise a guanine nucleotide connected to mRNA via a 5’ to 5’ triphosphate linkage. The guanosine is methylated on the 7 position after capping in
vivo by a methyltransferase (i.e., a 7-methylguanylate cap (m7G). Two 5’ caps suitable for use in the systems herein include m7G-ppp-Nm (Capl) or m7G-ppp-Nm-Nm (Cap2). Capl has a methylated 2'-hydroxy group on the first ribose sugar, while cap2 has methylated 2'- hydroxy groups on the first two ribose sugars.
[00126] An IRES sequence, or other suitable systems, may be used to produce more than one polypeptide from a single gene transcript. An IRES (or other suitable sequence) can be used to produce the double membrane fusogen protein and another protein within the same cell. Preferably, the IRES is located 3' to the gene in the mRNA.
B. Non-native DNA
[00127] In certain embodiments, a system for nucleic acid transfer comprises a non-native (also referred to herein as non-endogenous) DNA. The DNA can include one or more genes encoding proteins for expression in a cell. The gene can encode a protein that can be used to correct or ameliorate gene deficiencies, which may include deficiencies in which normal genes are expressed at less than normal levels. The gene can encode an antibody (e.g., a monoclonal or a bispecific antibody), an scFv, a Fab, or an Fc region. Following transfer of the DNA to the cell, the nucleic acid transfer system allows for the transfer of the DNA to the nucleus where it is expressed. In certain embodiments, the DNA comprises a gene capable of expressing a secreted protein that is secreted from the cell.
[00128] In certain embodiments, the DNA can include one or more genes encoding a non- coding RNA (such as an siRNA, tRNA, rRNA, microRNA (miR), short hairpin RNA (shRNA), aptamer, and ribozyme).
[00129] One or more non-coding RNAs can be expressed together with one or more proteins (e.g., two or more proteins, one or two non-coding RNA and one protein, etc.). For example, the DNA can comprise genes encoding an mRNA and a non-coding RNA, wherein the non- coding RNA targets the mRNA. In certain embodiments, the non-coding RNA targets the mRNA of an immunomodulatory gene, such as fucosyltransferase 8 (FUT8). In certain embodiments, the non-coding RNA targets the mRNA of a human gene, e.g., a mutated gene, and the DNA encodes a protein capable of functionally replacing a common form of the human gene.
[00130] In certain embodiments, the DNA is between about Ikb and about 1Mb. In certain embodiments, the DNA is between about Ikb and about 10kb. In certain embodiments, the
DNA is between about 10kb and about 100kb. In certain embodiments, the DNA is between about 100kb and about 1Mb. In certain embodiments, the DNA is 50kb. In certain embodiments, the DNA is 100kb. In certain embodiments, the DNA is 200kb. In certain embodiments, the DNA is 300kb. In certain embodiments, the DNA is 400kb. In certain embodiments, the DNA is 500kb. In certain embodiments, the DNA is 600kb. In certain embodiments, the DNA is 700kb. In certain embodiments, the DNA is 800kb. In certain embodiments, the DNA is 900kb. In certain embodiments, the DNA is IMb. i. Vectors and Regulatory Sequences
[00131] The gene can be carried on a vector, e.g.. a recombinant plasmid or virus that is delivered into a cell. The vector can be circular and/or lack free 5’ and 3’ ends. The vector can carry a gene which does not naturally occur with the vector sequences flanking it. The gene can be operatively linked to regulatory components in a manner which permits transcription, translation, and/or expression of the gene in a target cell. The non-native gene can be derived from any organism. In certain embodiments, the non-native gene is derived from a human.
[00132] The regulatory components can include conventional control elements which are operably linked to the transgene comprising a gene in a manner which permits its transcription, translation and/or expression in a cell transfected with the vector or infected with the virus produced as described herein. As used herein, “operably linked” sequences include both expression control sequences that are contiguous with the gene of interest and expression control sequences that act in trans or at a distance to control the gene of interest. Expression control sequences include appropriate transcription initiation, termination, promoter and enhancer sequences; efficient RNA processing signals such as splicing and polyadenylation (poly A) signals; sequences that stabilize cytoplasmic mRNA; sequences that enhance translation efficiency e.g. , Kozak consensus sequence); sequences that enhance protein stability; and when desired, sequences that enhance secretion of the encoded product. A great number of expression control sequences, including promoters, are known in the art and may be utilized. In certain embodiments, the DNA comprises a coding region that is operably linked to a promoter. In certain embodiments, the promoter is a tissue- specific promoter. The tissue-specific promoter can be expressed in non-dividing cells, such as muscle cells. In certain embodiments, the DNA comprises a promoter with fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs.
a. Muscle-Specific Promoters
[00133] Muscle is an important target tissue for systems for nucleic acid transfer, but is comprised primarily of differentiated, non-dividing cells. Thus, the discovery of the utility of fusogen proteins in nucleic acid delivery systems enables efficient non-viral systems for nucleic acid delivery to non-dividing cells including muscle.
[00134] One advantage of a muscle-specific promoter in a system for nucleic acid transfer to muscle cells is minimizing the expression of the transgenes in transduced antigen-presenting cells (APCs), such as macrophage and dendritic cells. The amount of the product of the DNA or interest presented by a transduced APC is thought to be far greater than the amount of product of the DNA an APC would be able to pick up from plasma or lymph. Thus, limiting expression of the product of the DNA in APCs may limit the potential for immune responses targeting the product of the DNA. Use of a muscle-specific promoter also has the safety advantage of reducing or preventing expression in other off-target cell types, in addition to APCs.
[00135] The muscle specific promoters (MSP) of the disclosure may be engineered to improve expression of a gene (e.g., a gene present on a non- native DNA) that is operably linked to the MSP. In certain embodiments, the system or method for transfer of a non-native DNA into a nucleus of a cell includes the non-native DNA and an RNA that encodes a membrane fusogen protein, which, when present in the cell, mediates transfer of the non- native DNA into the nucleus of the cell. The non-native DNA may include a muscle-specific promoter operably linked to a gene, which, when transferred to the nucleus of the cell, is expressed.
[00136] It has been discovered that an engineered promoter from which CpG sites have been removed (a CpG-free promoter) in non-dividing cells persistently expresses a gene of interest, potentially for the lifetime of a recipient, without being silenced, and without the loss of the plasmid (or other DNA lacking free 5’ and 3’ ends, such as circular DNAs produced in microbe-free systems) carrying the promoter and gene of interest (see, Example 7 herein). Accordingly, in certain embodiments, the systems of the present disclosure include a CPG- free promoter, such as a muscle-specific CPG-free promoter.
[00137] In addition, it has been discovered that the addition of a TATA box (e.g., TAT ATA), a region of DNA that helps initiate transcription, at certain positions within a
promoter, results in increased expression of a gene that is operably linked to the promoter (see Example 7). In addition, it has been discovered that the inclusion of certain transcription factor binding site core motifs results in increased expression of a gene that is operably linked to the promoter (see Example 7).
1. CpG Motifs
[00138] Provided herein are promoters, e.g., muscle-specific promoters, engineered to reduce the number of CpG motifs. In certain embodiments, a promoter (e.g., a muscle- specific promoter) is engineered to contain fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs. In certain embodiments, a promoter e.g., a muscle-specific promoter) is engineered to contain no CpG motifs.
[00139] In certain embodiments, the muscle-specific promoter engineered to reduce the number of CpG motifs comprises a nucleic acid sequence as set forth in SEQ ID NO: 10, SEQ ID NO: 1 1, SEQ ID NO: 12, SEQ ID NO: 17, SEQ ID NO: 18, SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, or SEQ ID NO: 24, or a variant promoter having 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotide substitutions as compared to any of the foregoing that is capable of expressing a non-native gene in a muscle tissue in an amount that is at least 80% (e.g., at least 85%, at least 90%, at least 95%) of the expression achievable using the promoter of SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 17, SEQ ID NO: 18, SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, or SEQ ID NO:24. The level of expression of a given promoter can be tested using a reporter gene in a muscle cell and/or tissue, e.g., as shown in the Examples herein.
2. TATA boxes
[00140] Provided herein are muscle-specific promoters derived from those known in the art (e.g., Spc5-12), and further comprising a TATA box (SEQ ID NOs: 13-14) positioned to improve expression, for example, as described in Example 7 herein. These sequences extended through a transcriptional start site (tss) are also provided (SEQ ID NOs: 15-16).
[00141] Also provided herein is a synthetic muscle-specific promoter (MSP) derived from an enhancer created by random ligation of oligos containing muscle-specific transcription factor binding sites (Li et al. (1999) NAT. BIOTECHNOL., 17 (3): 241-245) and further including a TATA box at a position that improves expression, e.g., SEQ ID NO: 7.
[00142] Other exemplary muscle-specific promoters comprising a TATA box positioned to improve expression include SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, and SEQ ID NO: 22 or a variant promoter having 1, 2, 3, 4, 5 ,6, 7, 8, 9, or 10 nucleotide substitutions as compared to any of the foregoing that is capable of expressing a non-native gene in a muscle tissue in an amount that is at least 80% (e.g., at least 85%, at least 90%, at least 95%) of the expression achievable using the promoter selected from SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, and SEQ ID NO: 22. The level of expression of a given promoter can he tested using a reporter gene in a muscle cell and/or tissue, e.g., as shown in the Examples herein.
3. Transcription Factor Binding Sites
[00143] Transcription factors with binding sites in these muscle-specific promoters include myocyte enhancer factor- 1 (MEFE), myocyte enhancer factor-2 (MEF2) paralogs, TEA domain transcription factor- 1 (TEAD1), and the serum response element (SRE). The consensus binding site for MEF1 is 5’-CANNTG-3’, e.g., 5’-CAGGTG-3’ or 5’-CACCTG- 3’, where N can be any nucleotide. The consensus binding site for MEF2 paralogs is 5’- YTAAAAATA-3’, e.g., 5’-CTAAAAATA-3’, where Y is C or T. The consensus binding site for TEAD1 is 5’-CATTCC-3’ or 5’-GGAATG-3’. The consensus SRE is 5’- CCWWWWWWGG-3’, e.g., 5’-CCAAATATGG-3’, where W is A or T. In certain embodiments, muscle-specific promoters include promoters containing the consensus binding sites for MEF1 , MEF2 paralogs, TEAD1, and/or the SRE. In certain embodiments, muscle- specific promoters include CpG-free promoters containing the consensus binding sites for MEF2 paralogs and/or TEADL The positions of MEF1, MEF2, TEAD1, and SRE consensus sites in a muscle-specific promoter with an optimally-positioned TATA box are indicated in SEQ ID NO: 110.
[00144] The regulatory sequences useful in the constructs provided herein may also contain an intron, desirably located between the promoter/enhancer sequence and the gene. One desirable intron sequence is derived from SV-40, and is a 100 bp mini-intron splice donor/splice acceptor referred to as SD-SA. In certain embodiments, the intron comprises the nucleotide sequence of SEQ ID NO: 10, or a codon-optimized or fragment thereof. Another suitable sequence includes the woodchuck hepatitis virus post-transcriptional element. (See, e.g., L. Wang and I. Verma (1999) PROC. NATL. ACAD. Set. USA, 96: 3906-3910). PolyA
signals may be derived from many suitable species, including, without limitation SV-40, human and bovine.
[00145] Another regulatory component that can be used in the methods described herein is an internal ribosome entry site (IRES). An IRES sequence, or other suitable systems, may be used to produce more than one polypeptide from a single gene transcript. An IRES (or other suitable sequence) is used to produce a protein that contains more than one polypeptide chain or to express two different proteins from or within the same cell. Preferably, the IRES is located 3’ to the gene in the vector.
[00146] Other regulatory sequences useful herein include enhancer sequences. Enhancer sequences useful herein include the IRBP enhancer, immediate early cytomegalovirus enhancer, one derived from an immunoglobulin gene or SV40 enhancer, the cis-acting element identified in the mouse proximal promoter, etc.
[00147] Selection of these and other common vector and regulatory elements are conventional and many such sequences are available. See, e.g., Sambrook et al., and references cited therein at, for example, pages 3.18-3.26 and 16.17-16.27 and Ausubel et al., Current Protocols in Molecular Biology, John Wiley & Sons, New York, 1989). It is understood that not all vectors and expression control sequences will function equally well to express all of the transgenes as described herein. However, one of skill in the art may make a selection among these, and other, expression control sequences to generate the DNA of the disclosure. ii. CpG Methylation
[00148] Toll-like receptor 9 (TLR) is an innate immune sensor for DNA, but does not efficiently recognize CpG-methylated DNA (Hemmi et al. (2000) NATURE, 408 (6813): 740- 745, and Rutz et al. (2004) EUR. J. IMMUNOL. 34 (9): 2541-2550). Thus, it is contemplated that CpG methylation of a double- stranded DNA (e.g., a vector such as a plasmid) used in a system for nucleic acid delivery provides a means of avoiding TLR9 signaling. However, CpG-methylation (for example, of a promoter) also may result in silencing of a gene of interest. Accordingly, in certain embodiments, a DNA that is included in a system for nucleic acid transfer can combine a promoter that lacks CpG motifs with a plasmid DNA that is CpG- methylated.
[00149] As described in Example 8 herein, it has been discovered that a plasmid having methylated CpG motifs in a bacterial origin of replication only, where the rest of the plasmid lacked CpG motifs, exhibited higher expression of the encoded protein as compared to plasmids lacking CpG motifs entirely. Therefore, CpG methylation in certain contexts, e.g., in a bacterial origin of replication, may be desirable in systems for nucleic acid transfer.
[00150] Accordingly, in certain embodiments, the DNA of the disclosure can comprise modified CpG motifs. For example, the DNA can have fewer than 100 CpG motifs, the DNA can be substantially free of unmodified CpG motifs, and/or the DNA can be methylated at one or more CpG motifs. In certain embodiments, the DNA is substantially free of 6- methyladenine and/or 5 -methylcytosine. iii. Genes
[00151] The DNA can include one or more genes encoding proteins for expression in a cell. The gene can encode a protein that can be used to correct or ameliorate gene deficiencies, which may include deficiencies in which normal genes are expressed at less than normal levels. The gene can encode an antibody (e.g., a monoclonal or a bispecific antibody), an scFv, a Fab, or an Fc region. The gene can encode a protein comprising at least one domain that binds a ligand and at least one effector domain. Following transfer of the DNA to the cell, the nucleic acid transfer system allows for the transfer of the DNA to the nucleus where it is expressed. In certain embodiments, the DNA comprises a gene capable of expressing a secreted protein that is secreted from the cell.
[00152] In certain embodiments, the DNA can include one or more genes encoding a non- coding RNA (such as an siRNA, tRNA, rRNA, microRNA (miR), short hairpin RNA (shRNA), aptamer, and ribozyme). One or more non-coding RNAs can be expressed together with one or more proteins (e.g., two or more proteins, one or two non-coding RNA and one protein, etc.). For example, the DNA can comprise genes encoding an mRNA and a non- coding RNA, wherein the non-coding RNA targets the mRNA. In certain embodiments, the non-coding RNA targets the mRNA of an immunomodulatory gene, such as fucosyltransferase 8 (FUT8). In certain embodiments, the non-coding RNA targets the mRNA of a human gene, e.g., a mutated gene, and the DNA encodes a protein capable of functionally replacing a common form of the human gene.
[00153] A nucleotide sequence of a gene that differs from the nucleotide sequence of a reference gene (e.g., a gene described herein) due to degeneracy in the genetic code are also within the scope of the disclosure. For example, a number of amino acids are designated by more than one triplet. Codons that specify the same amino acid, or synonyms (for example, CAU and CAC are synonyms for histidine) may result in “silent” mutations which do not affect the amino acid sequence of the protein. One skilled in the art will appreciate that these variations in one or more nucleotides (up to about 3-5% of the nucleotides) of the nucleic acids encoding a particular protein may exist among members of a given species due to natural allelic variation. Any and all such nucleotide variations are within the scope of this disclosure.
[00154] In certain embodiments, a gene present on the DNA described herein is expressed in a target cell, for example, a target cell that divides infrequently or does not divide. The target cell can be, for example, a cardiac cell (e.g. , a heart muscle cell), a neuron, a skeletal muscle cell, a corneal cell, a kidney cell, a platelet, a mature egg or sperm cell, a liver cell, etc. a. Reporter genes
[00155] In another embodiment, the DNA includes a sequence encoding a reporter gene, which upon expression produces a detectable signal. Such reporter sequences include, without limitation, DNA sequences encoding P-lactamase, P -galactosidase (LacZ), alkaline phosphatase, thymidine kinase, green fluorescent protein (GFP), red fluorescent protein (RFP), chloramphenicol acetyltransferase (CAT), luciferase, membrane bound proteins including, for example, CD2, CD4, CD8, the influenza hemagglutinin protein, and others well known in the art, to which high affinity antibodies directed thereto exist or can be produced by conventional means, and fusion proteins comprising a membrane bound protein appropriately fused to an antigen tag domain from, among others, hemagglutinin or Myc. These coding sequences, when associated with regulatory elements which drive their expression, provide signals detectable by conventional means, including enzymatic, radiographic, colorimetric, fluorescence or other spectrographic assays, fluorescent activating cell sorting assays and immunological assays, including enzyme linked immunosorbent assay (ELISA), radioimmunoassay (RIA) and immunohistochemistry. For example, where the marker sequence is the LacZ gene, the presence of the vector carrying the signal is detected by assays for beta-galactosidase activity. Where the transgene is green fluorescent protein or
luciferase, the vector carrying the signal may be measured visually by color or light production in a luminometer. b. Human Serum Albumin (HSA)
[00156] In another embodiment, the DNA includes a gene encoding a human serum albumin (HSA) protein or a fragment or variant thereof. Due to the interaction of HSA with the neonatal Fc receptor (FcRn) secreted proteins that are HSA fusion proteins can have extended half-lives in plasma. In this respect, HSA fusion proteins are analogous to proteins with Fc domains, which also can interact with FcRn to promote longevity in plasma. In certain embodiments, the HSA protein that has at least 80% (e.g., at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100%) sequence identity to HSA or a functional fragment thereof. c. eCD4-Ig and Tyrosylprotein Sulfotransferase-2 (TPST) Genes
[00157] In one embodiment, the DNA comprises one or more genes encoding eCD4-Ig (Gardner et al. (2015) NATURE, 519(7541): 87-91, PCT/US2019/023422) and/or tyrosylprotein sulfotransferase-2 (TPST2) (U.S. Patent No. 10,626,161). The eCD4-Ig gene and/or TPST2 gene can be expressed under a tissue specific promoter (e.g., a muscle-specific promoter), and/or a promoter engineered to have fewer CpG sites (e.g., a CpG free promoter) than a naturally occurring promoter. The eCD4-Ig gene and/or TPST2 gene can be expressed together with a non-coding RNA, for example, an shRNA or miR. The non-coding RNA (e.g., the shRNA or miR) can reduce the expression of an immunomodulatory gene such as fucosyltransferase-8 (FUT8).
[00158] In certain embodiments, a plasmid expressing an eCD4-Ig gene and/or a TPST2 gene lacks CpG motifs outside of the bacterial origin of replication (Ori). The CpG motifs of the Ori can be methylated prior to use as described herein. The plasmids can be generated and co-packaged with an mRNA encoding a double membrane fusogen protein (e.g., a Brambleberry-encoding mRNA) in LNPs as described herein. The mRNA can include modified uridine nucleotides. The LNPs can be formulated for injection in a pharmaceutically-acceptable carrier.
[00159] An exemplary sequence of a plasmid for expressing eCD4-Ig under a CpG-free muscle-specific promoter is provided at SEQ ID NO: 35. The sequence of a similar plasmid
for expressing TPST2 under a CpG-free muscle-specific promoter is provided at SEQ ID NO: 36. Versions of the plasmid for expressing eCD4-Ig that are further modified to express an shRNA for knocking down fucosyltransferase-8 (FUT8) by RNA interference (RNAi) (SEQ ID NO: 37), or a miR for knocking down FUT8 by RNAi (SEQ ID NO: 38), are provided. d. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Antibody Genes
[00160] Also provided herein is a system for nucleic acid transfer for expressing a monoclonal antibody that binds proprotein convertase subtilisin/kexin type 9 (PCSK9). The sequences of monoclonal antibodies that bind PCSK9 (e.g., evolocumab and alirocumab) are known in the art and can be used as coding sequences in the systems described herein.
PCSK9 antibodies can be expressed under a promoter, such as a tissue specific promoter e.g., a muscle-specific promoter), and/or a promoter engineered to have fewer CpG sites than a naturally occurring promoter. In certain embodiments, the promoter is a CpG free promoter. In certain embodiments, the promoter is a muscle-specific, CpG-free promoter.
[00161] A PCSK9 antibody gene can be present on a plasmid. In certain embodiments, the plasmid sequence lacks CpG motifs outside of the bacterial origin of replication (Ori). The CpG motifs of the Ori can be methylated prior to use as described herein. The plasmids can be generated and co-packaged with an mRNA encoding a double membrane fusogen protein e.g., a Brambleberry-encoding mRNA) in LNPs as described herein. The mRNA can include modified uridine nucleotides. The LNPs can be formulated for injection in a pharmaceutically-acceptable carrier.
[00162] The sequence of an exemplary plasmid for expressing evolocumab under a CpG- free muscle- specific promoter is provided at SEQ ID NO: 39. The plasmid is a bicistronic plasmid for co-expressing the heavy and light chains of evolocumab. e. Glucagon-like Peptide- 1 Receptor (GLP1R) Agonist Genes
[00163] In another exemplary embodiment, a system for nucleic acid transfer for expressing a glucagon-like peptide- 1 receptor (GLP1R) agonist is provided. Glucagon-like peptide- 1 (GLP-1) and variants thereof can be expressed as a secreted protein therapeutic. In certain embodiments, the DNA can include a sequence for a non-native signal peptide 5’ of the sequence encoding GLP-1 or a variant thereof. In certain embodiments, the GLP-1 gene or a variant thereof is expressed under a tissue specific promoter (e.g., a muscle-specific promoter), and/or a promoter engineered to have fewer CpG sites than a naturally occurring
promoter. In certain embodiments, the promoter is a CpG free promoter. In certain embodiments, the promoter is a muscle-specific, CpG-free promoter.
[00164] In certain embodiments, a plasmid expressing GLP-1 or a variant thereof lacks CpG motifs outside of the bacterial origin of replication (Ori). The CpG motifs of the Ori can be methylated prior to use as described herein. The sequence of a plasmid for expressing GLP-1 with a non-native signal peptide under a CpG-free muscle- specific promoter is provided at SEQ ID NO: 40. In other embodiments, the a plasmid expressing GLP-1 is completely CpG-free
 containing no CpG motifs), for example, due to use of a CpG-free R6K Ori (e.g., as described in U.S. Patent No. 7,244,609). Exemplary CpG-free plasmids are provided (SEQ ID NO: 41 and SEQ ID NO: 42).The plasmids can be generated and co- packaged with an mRNA encoding a double membrane fusogen protein (e.g., a Brambleberry-encoding mRNA) in LNPs as described herein. The mRNA can include modified uridine nucleotides. The LNPs can be formulated for injection in a pharmaceutically-acceptable carrier. f. Non-coding RNA
containing no CpG motifs), for example, due to use of a CpG-free R6K Ori (e.g., as described in U.S. Patent No. 7,244,609). Exemplary CpG-free plasmids are provided (SEQ ID NO: 41 and SEQ ID NO: 42).The plasmids can be generated and co- packaged with an mRNA encoding a double membrane fusogen protein (e.g., a Brambleberry-encoding mRNA) in LNPs as described herein. The mRNA can include modified uridine nucleotides. The LNPs can be formulated for injection in a pharmaceutically-acceptable carrier. f. Non-coding RNA
[00165] An mRNA encoding a double membrane fusogen protein (e.g., a Brambleberry mRNA) can be used to enhance nucleic acid transfer of a DNA that expresses an RNA, such as a non-coding RNA. In certain embodiments, the non-coding RNA is an siRNA, tRNA, rRNA, microRNA (miR), short hairpin RNA (shRNA), aptamer, or ribozyme. In certain embodiments, the non-coding RNA targets the mRNA of a human gene, e.g., a mutated gene, to prevent, lessen the severity of, or treat a disease (e.g., a genetic disease and/or a cancer).
[00166] In certain embodiments, an RNA polymerase III (pol III) promoter (e.g., an Hl pol III promoter) is operably linked to the miRNA. The RNA pol III promoter can be an Hl promoter. In certain embodiments, the Hl promoter has at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% identity to SEQ ID NO: 154. In certain embodiments, the pol III promoter lacks CpG sites.
[00167] One or more non-coding RNAs can be expressed together with one or more proteins (e.g., two or more proteins, one or two non-coding RNA and one protein, etc.). For example, a DNA can comprise genes encoding an mRNA and a non-coding RNA, wherein the non- coding RNA targets the mRNA. In certain embodiments, the non-coding RNA targets the
mRNA of an immunomodulatory gene, such as fucosyltransferase 8 (FUT8). In certain embodiments, the non-coding RNA targets the mRNA of a human gene, e.g., a mutated gene, and the DNA encodes a protein capable of functionally replacing a common form of the human gene.
[00168] A plasmid expressing a non-coding RNA (e.g., a miR) can be used as an off-switch for gene therapy. For example, to reduce or eliminate the amount of a therapeutic gene expressed in a subject who has received or is receiving a gene therapy, a non-coding RNA that targets the therapeutic gene can be administered using the nucleic acid transfer systems and methods described herein. To serve as an off-switch (to eliminate expression) or as a down-switch (to reduce expression), a plasmid expressing an non-coding RNA (e.g., a miR) can be co-delivered with an mRNA encoding a double membrane fusogen protein (e.g., Brambleberry mRNA), for example, in LNPs. The non-coding RNA can be delivered in an excess amount (e.g., an excess amount of material and/or in excess volume) to the original site of administration of the system for nucleic acid transfer for expressing the therapeutic protein to be switched off. The original site of injection can be located, e.g., by tattooing the site or using a standardized biometric approach for injection site selection and depth. Alternatively, if the target tissue is liver, intravenous injections can be used both to first administer the vector for expressing the therapeutic gene and then later to administer the miR-expression plasmid that serves as the off-switch.
[00169] In certain embodiments, the therapeutic protein comprises a 3’ UTR sequence with one or more target sites for one or more miRs. In certain embodiments, an RNA polymerase III (pol III) promoter (e.g., an Hl pol III promoter) is operably linked to the miRNA. The RNA pol III promoter can be an Hl promoter. In certain embodiments, the Hl promoter has at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% identity to SEQ ID NO: 154. In certain embodiments, the pol III promoter lacks CpG sites.
[00170] In an exemplary embodiment, a plasmid expressing a miR (SEQ ID NO: 54) and a matched 3’ UTR sequence (SEQ ID NO: 55) with multiple target sites for that miR are provided. The miR is expressed from a version of the Hl RNA polymerase III (Pol III) promoter that has been modified to lack CpG sites. Thus, the plasmid can be methylated at CpG sites without the Hl promoter being methylated (and, thereby, potentially silenced), and the promoter does not provide recognition motifs for TLR9, thereby avoiding stimulation of
the innate immune response. The miR-target pair (SEQ ID NO: 54 and SEQ ID NO: 55) was engineered to maximize the efficiency of knockdown of the gene of interest. To serve as an off-switch (or down-switch), the plasmid expressing the miR can be co-delivered with Brambleberry mRNA, e.g.. in LNPs, in an excess amount (in terms of both the amount of material and the volume) to the original site of administration of the system for nucleic acid transfer for expressing the therapeutic protein to be switched off.
[00171] An exemplary miR contemplated herein includes the following features: a 19 base guide sequence, a loop of unpaired bases exactly 4 nucleotides in length, a 19 base sequence that is complementary to the guide sequence and forms a 19 base pair stem, and an RNA Polymerase III (Pol III) transcription termination signal (TTTTTT or UUUUUU) that overlaps with the 3’ end of the stem. In certain embodiments, the 3’ end is a U. In certain embodiments, the 3’ end is a UU. Further contemplated herein is a DNA encoding a mR having 19 base guide sequence, a loop of unpaired bases exactly 4 nucleotides in length, a 19 base sequence that is complementary to the guide sequence and forms a 19 base pair stem, and an RNA Polymerase III (Pol III) transcription termination signal (TTTTTT) that overlaps with the 3’ end of the stem. In certain embodiments, an RNA Polymerase III promoter is operably linked to the miR.
[00172] In certain embodiments, the non-coding RNA targets a gene involved in glycosylation. Exemplary genes involved in glycosylation include Fut8 and GALNT2.
[00173] The nucleic acid transfer system of the disclosure can include an immunosuppressive molecule or pro-drug form thereof, and/or an RNA encoding an immunosuppressive protein. The nucleic acid transfer systems of the disclosure can include an immunosuppressive non-coding RNA (e.g., an antisense oligonucleotide (ASO)) or a pro- drug form thereof that binds a nucleic acid sensing molecule such as C2mutl (as described above), a corticosteroid such as glucocorticoid, prednisone, prednisolone, triamcinolone, methylprednisolone, dexamethasone, dexamethasone palmitate, fluticasone, fluticasone propionate, fluticasone furoate, clobetasol, betamethasone, halobetasol, amcinonide, desoximetasone, diflorasone, mometasone, prednicarbate, triamcinolone acetonide, fluocinolone, alclometasone, desonide, cortisone, hydrocortisone, and fluocinonide, or a pro- drug form thereof; mTOR inhibitors, such as rapamycin, sirolimus, and everolimus, or pro- drug forms thereof; a tyrosine kinase inhibitor such as deucravacitinib, baricitinib, tofacitinib, ruxolitinib, fedratinib, and fostamatinib, or pro-drug forms thereof; and a calcineurin inhibitor
such as tacrolimus, FK506 and cyclosporine A, or pro-drug forms thereof. Other non- limiting examples of immunosuppressive molecules, or pro-drug forms thereof include ASOs or siRNAs that target the transcripts encoding proteins involved in innate immune sensing, such as cGAS, TLR3, TLR7, TLR8, TLR9, RIG-I, MAVS, MDA5, and MYD88, or pro-drug forms thereof; ASOs or siRNAs, or pro-drug forms thereof, that target the transcripts encoding transcription factors involved in innate immune responses such as the transcripts of NFKB, IRF3, and IRF7. Other exemplary immunosuppressive molecules include mRNAs encoding immunosuppressive molecules, such as indoleamine 2,3-dioxygenase-l (IDO1), NF kappa B inhibitor alpha (NFKBIA), inhibitor of kappa B subunit beta (IKBKB), TNF alpha induced protein 3 (TNFAIP3), interferon regulatory factor 4 (IRF4), interferon regulatory factor 8 (1RF8), cytotoxic T-lymphocyte-associated protein 4 (CTLA4), transforming growth factor beta-1 (TGFB1), interleukin 1 receptor type 2 (IL1R2), suppressor of cytokine signaling-1 (SOCS1), suppressor of cytokine signaling-2 (SOCS2), suppressor of cytokine signaling-3 (SOCS3), suppressor of cytokine signaling-4 (SOCS4), suppressor of cytokine signaling-5 (SOCS5), suppressor of cytokine signaling-6 (SOCS6), suppressor of cytokine signaling-7 (SOCS7), ring finger protein 216 (RNF216), and CASP8 and FADD like apoptosis regulator (CFLAR), or pro-drug forms thereof.
[00174] mRNAs, siRNAs, and ASOs can be co-formulated with an mRNA encoding a double membrane fusogen protein (e.g. , Brambleberry mRNA) within an LNP, to reduce the potential for nucleic acid transfer to stimulate immune responses against the therapeutic gene. In certain embodiments, immunosuppressive molecules or pro-drug forms thereof, including, e.g., corticosteroids (such as glucocorticoid), mTOR inhibitors, tyrosine kinase inhibitors, ASOs, and siRNAs, and/or mRNAs encoding immunosuppressive molecules, can be formulated with LNPs containing an mRNA encoding a double membrane fusogen protein (e.g., Brambleberry mRNA) and a DNA. Immunosuppressive molecules or pro-drug forms thereof can also include immunosuppressive molecule or pro-drug form thereof that bind human glucocorticoid receptor. In certain embodiments, the immunosuppressive molecule or pro-drug form thereof can contain an ester bond.
[00175] In certain embodiments, the immunosuppressive molecule or prodrug form thereof is selected from the group consisting of: galectin inhibitors, such as GB1107, galectin- 3/galectin-8-IN-l, olitigaltin, selvigaltin, galectin-3/galectin-8-IN-2, galectin-3-IN-2, galectin-3-IN-3, galectin-3-IN-5, GB1490, GB1908, galectin-8-IN-l, galectin-8-IN-2,
galectin-8N-IN-l, galectin-8N-IN-2, or thiodigalactoside, or pro-drug form of any of the foregoing; cysteine protease inhibitors, such as cathepsin inhibitors (such as cathepsin L-IN-2 (Z-Phe-Phe-FMK)), disulfiram, belizatinib, cystatin B, cystatin C, E-64, or E-64d or a pro- drug form of any of the foregoing; NLRP3 inflammasome inhibitors, such as oridonin, INF39, glyburide, 5-chloro-2-methoxy-N-[2-(4- sulfamoylphenyl)-ethyl]-benzamide (e.g., 16673-34-0), JC124, FC11A-2, parthenolide, VX-740, VX-765, BAY 11-7082, BHB, MCC950, MNS, CY-09, Tranilast, OLT1177, or a pro-drug form thereof; MyD88 inhibitors, or pro-drug forms thereof; IRAK4 inhibitors, or pro-drug forms thereof; PKR inhibitors, or pro-drug forms thereof; PERK inhibitors, or pro-drug forms thereof; NFkB inhibitors, or pro- drug forms thereof; IKK inhibitors, or pro-drug forms thereof; JAK inhibitors, or pro-drug forms thereof; STAT inhibitors, or pro-drug forms thereof; GSK3 inhibitors, or pro-drug forms thereof; cGAS inhibitors, or pro-drug forms thereof; or STING inhibitors, or pro-drug forms thereof. In certain embodiments, the immunosuppressive molecule or pro-drug form thereof is selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, or NLRP3 inflammasome inhibitors. In certain embodiments, the immunosuppressive molecule or pro-drug form thereof is an NLRP3 inflammasome inhibitor.
[00176] In certain embodiments, the nucleic acid transfer systems of the disclosure include at least two, three, four, five, six, seven, eight, nine, or ten different immunosuppressive molecules or pro-drug forms thereof.
[00177] In certain embodiments, the nucleic acid transfer systems of the disclosure include an immunosuppressive molecule or pro-drug form thereof that has a LogP value greater than 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, or 5.0. In certain embodiments, the immunosuppressive molecule or pro-drug form thereof has a LogP value greater than 3.0, 3.5, or 4.0. In certain embodiments, the pro-drug has a higher LogP value than the drug itself, which can facilitate coencapsulation with DNA and mRNA in an LNP.
[00178] In certain embodiments, the nucleic acid transfer systems of the disclosure include an aqueous suspension of the immunosuppressive molecule or pro-drug form thereof. In certain embodiments, the aqueous suspension includes triamcinolone acetonide or betamethasone sodium phosphate.
[00179] The nucleic acid transfer systems of the disclosure can include a non-coding RNA that inhibits the binding of a cellular protein to DNA.
C. Lipid Nanoparticles
[00180] In order to transfer the RNA and the non-native DNA into the nucleus of a target cell, these components can be packaged within a suitable delivery system such as a lipid nanoparticle (LNP), (e.g., a vesicle, a micelle, a liposome, a solid lipid nanoparticle, a nanostructured lipid carrier, or other lipid-containing particle). In systems using LNPs for delivery, any suitable LNP known in the art can be used. An LNP refers to any particle having a diameter of less than 1000 nm, 500 nm, 250 nm, 200 nm, 150 nm, 100 nm, 75 nm, 50 nm, or 25 nm. Alternatively, a nanoparticle may range in size from 1-1000 nm, 1-500 nm, 1-250 nm, 25-200 nm, 25-100 nm, 35-75 nm, or 25-60 nm.
[00181] LNPs may be made from cationic, anionic, or neutral lipids. In certain embodiments, the LNP comprises one or more ionizable lipids. Neutral lipids, such as the fusogenic phospholipid DOPE or the membrane component cholesterol, may be included in LNPs as “helper lipids” to enhance transfection activity and nanoparticle stability.
Limitations of cationic lipids include low efficacy owing to poor stability and rapid clearance, as well as the generation of inflammatory or anti-inflammatory responses. LNPs may also comprise hydrophobic lipids, hydrophilic lipids, or both hydrophobic and hydrophilic lipids. In certain embodiments, the LNP comprises a polyplex.
[00182] In certain embodiments, an LNP of the present disclosure comprises an ionizable lipid, a structural lipid, a PEGylated lipid (aka PEG lipid), and a phospholipid. In certain embodiments, an LNP comprises an ionizable lipid, a structural lipid, a PEGylated lipid (aka PEG lipid), and a zwitterionic amino acid lipid. In certain embodiments, an LNP further comprises a 5th lipid, besides any of the aforementioned lipid components. In certain embodiments, the LNP encapsulates one or more elements of the active agent of the present disclosure. In certain embodiments, an LNP further comprises a targeting moiety covalently or non-covalently bound to the outer surface of the LNP. In certain embodiments, the targeting moiety is a targeting moiety that binds to, or otherwise facilitates uptake by, cells of a particular organ system.
[00183] In certain embodiments, the lipid nanoparticle compositions of the present disclosure are described according to the respective molar ratios of the component lipids in the formulation. As a non-limiting example, the mol-% of the ionizable lipid may be from about 10 mol-% to about 80 mol-%. As a non-limiting example, the mol-% of the ionizable
lipid may be from about 20 mol-% to about 70 mol-%. As a non-limiting example, the mol-% of the ionizable lipid may be from about 30 mol-% to about 60 mol-%. As a non-limiting example, the mol-% of the ionizable lipid may be from about 35 mol-% to about 55 mol-%. As a non-limiting example, the mol-% of the ionizable lipid may be from about 40 mol-% to about 50 mol-%.
[00184] In certain embodiments, the mol-% of the phospholipid may be from about 1 mol-% to about 50 mol-%. In certain embodiments, the mol-% of the phospholipid may be from about 2 mol-% to about 45 mol-%. In certain embodiments, the mol-% of the phospholipid may be from about 3 mol-% to about 40 mol-%. In certain embodiments, the mol-% of the phospholipid may be from about 4 mol-% to about 35 mol-%. In certain embodiments, the mol-% of the phospholipid may be from about 5 mol-% to about 30 mol-%. In certain embodiments, the mol-% of the phospholipid may be from about 10 mol-% to about 20 mol- %. In some embodiments, the mol-% of the phospholipid may be from about 5 mol-% to about 20 mol-%.
[00185] In certain embodiments, the mol-% of the structural lipid may be from about 10 mol-% to about 80 mol-%. In certain embodiments, the mol-% of the structural lipid may be from about 20 mol-% to about 70 mol-%. In certain embodiments, the mol-% of the structural lipid may be from about 30 mol-% to about 60 mol-%. In certain embodiments, the mol-% of the structural lipid may be from about 35 mol-% to about 55 mol-%. In some embodiments, the mol-% of the structural lipid may be from about 40 mol-% to about 50 mol-%.
[00186] In certain embodiments, the mol-% of the PEG lipid may be from about 0.1 mol-% to about 10 mol-%. In certain embodiments, the mol-% of the PEG lipid may be from about 0.2 mol-% to about 5 mol-%. In certain embodiments, the mol-% of the PEG lipid may be from about 0.5 mol-% to about 3 mol-%. In certain embodiments, the mol-% of the PEG lipid may be from about 1 mol-% to about 2 mol-%. In certain embodiments, the mol-% of the PEG lipid may be about 1 .5 mol-%. In certain embodiments, the mol-% of the PEG lipid may be about 2.5 mol-%.
[00187] Additional features of an ionizable lipid contemplated for use in the systems and methods herein include a pKa above 6.5, one or more branched acyl chains, one or more hydrolysable ester bonds, and few (e.g., less than 10, less than 9, less than 8, less than 7, less than 6, less than 5, less than 4, less than 3, less than 2, or no unsaturated bonds).
[00188] Any lipid or combination of lipids that are known in the art can be used to produce a LNP. Examples of lipids used to produce LNPs are: DOTMA, DOSPA, DOTAP, DMRIE, DC-cholesterol, DOTAP-cholesterol, GAP-DMORIE-DPyPE, and GL67A-DOPE-DMPE- polyethylene glycol (PEG). Examples of cationic lipids are: 98N12-5, C12-200, DLin-KC2- DMA (KC2), DLin-MC3-DMA (MC3), XTC, MD1, SM102, Lipid 29, CL15F6, and 7C1. Examples of neutral lipids are: DPSC, DPPC, POPC, DOPE, and SM. Examples of PEG- modified lipids are: PEG-DMG (l,2-Dimyristoyl-sn-glycero-3-methoxypolyethylene glycol), PEG-CerC14, and PEG-CerC20. Exemplary LNPs are described, for example, in U.S. Patent Nos. 8,058,069; 8,492,359; 8,822,668; 9,364,435; 9,504,651; 11,141,378; 9,404,127;
11,246,933; 11,382,979; 9,006,417; 9,404,127; 9,518,272; 11,135,312; 11,149,278; 11,241,493; 10,576,146; 10,485,884; 9,950,065; 11,318,195; 11,173,120; 10,808,242; 11,471,522; 10,703,789; 10,702,600; 10,577,403; 10,442,756; 10,266,485; 10,064,959; 9,868,692; 11,285,222; 10,799,602; 10,780,054; 9,669,089; 11,110,166; 11,141,476; 10,912,826; 11,478,552; 10,221,127; 10,166,298; 10, 106,490; 11,357,856; 11,453,639; 10,232,055; 10,006,007; 9,750,824; 9,371,511; 9,163,213; 9,012,219; 8,835,108; 8,748,089; 8,691,966; 8,278,036; 10,815,463; 10,233,148; 10,227,302; 9,580,711 ; 9,567,296;
11,015,204; 10,961,188; and 10,980,895; in U.S. Patent Application Nos. 20190240354; 20100130588; 20210087135; 20210128488; 20200121809; 20170119904; 20130108685; 20130195920; 20150005363; 20140308304; 20130053572; 20170210697; 20200206362; 20200164038; 20190314496; 20190351048; 20200345831; 20190083602; 20200246451; 202110361764; 20210401971; 20210128716; 20200046838; 20200283372; 20190022247; 20200163878; 20200172472; 2020297634; and 20200046830; and in International Patent Publication Nos. WO2016/118724; WO2016/118725; W02020/002598; W02020/097540; W02020/097548; W02009/086558; WO2011/036557; WO2021/204179; WO2019/232095; WO2021/077067; WO2019/152557; WO 2019/089828, or WO/2023/044343, each of which is incorporated by reference herein in their entirety. i. Attachment Moieties for Increasing Nucleic Acid Transfer to Desired Cell Types
[00189] In one aspect, the lipid nanoparticle including an RNA encoding a fusogen protein and a DNA of interest includes an attachment moiety that promotes attachment and/or internalization of the LNP. Lipids, pegylated lipids, or other lipophilic molecules such as cholesterol or dexamethasone conjugated to an attachment moiety can be included in LNPs generated using the methods described herein. For example, peptides including the three
amino acid motif Arg-Gly-Asp (RGD), which binds integrins can be used for increasing nucleic acid transfer to specific cell types, e.g., muscle. Lipid-conjugated RGD peptides are commercially available e.g., DOPE-RGD, Avanti Polar Lipids catalog number 870296, and DSPC-RGD Avanti Polar Lipids catalog number 870295). Extended peptides containing the RGD motif also have been described (Tabebordbar et al. (2021) CELL, 184(19): 4919-4938) (SEQ ID NOs: 56-102), and can be conjugated to lipids similarly as in DSPC-RGD and DOPE-RGD. Other peptides that can be conjugated to lipids or pegylated lipids that are useful in targeting muscle have been previously described, e.g., SEQ ID NOs: 103-105 (Tabebordbar et al. ,(2021), supra), SEQ ID NO: 56 (Ghosh and Barry (2005) J. VlROL., 79(21): 13667-13672), and SEQ ID NO: 57 (Schaffer et al. (2003) PROC. NATL. ACAD. Set. USA., 100:4435-4439, Jackson et al. (2020) MOL. THER. METHODS CLIN. DEV., 19: 496-506, and International Patent Application No. PCT/US2021/042200). Such peptides can be conjugated to lipids or pegylated lipids and used as attachment moieties to target the LNPs of the present invention to muscle.
[00190] Forms of polyethylene glycol (PEG) with shorter lipid anchors than DMG-PEG (PEGylated myristoyl) are contemplated to increase the kinetics with which the PEGylated lipid diffuses out of the LNP, thereby improving contact with cells and the efficiency of gene transfer. As shown in Example 14 herein, use of PEGylated lipids with short lipid anchors substantially improved gene transfer efficiency over DMG-PEG2000. PEGylated lipids that can be used with the systems and methods herein include C8C-PEG2000 and cholesterol- PEG2000. Thus, relatively short lipid anchors, e.g., C8C or cholesterol, can improve the efficiency of gene transfer by DNA-mRNA LNPs.
[00191] Forms of PEG that may be suitable for use the systems and methods of the disclosure, depending on the circumstances, include PEG500 to PEG5000. For example, PEG500, PEG1000, PEG1500, PEG2000, PEG2500, PEG3000, PEG3500, PEG4000, PEG4500, and PEG5000, or any range therein, can be used in accordance with the systems and methods herein.
[00192] Metabolites that interact with transporters or receptors on target cell membranes can be conjugated, directly or indirectly, to one or more lipids that are incorporated into the LNPs of the present invention in order to facilitate attachment, endocytosis, and/or gene transfer. For example, the molecule glucose can be conjugated via a linker to a lipid such as cholesterol (e.g., in cholesterol-undecanoate-glucose), and included in LNPs as an attachment
factor that promotes gene transfer. In certain embodiments, the glucose molecule can be conjugated to another compound, such as hydroquinone. An example of a glucose- hydroquinone conjugate is arbutin.
[00193] Polyphenols can be conjugated, directly or indirectly, to one or more lipids that are incorporated into the LNPs of the present invention in order to facilitate attachment, endocytosis, and/or gene transfer. For example, DPG-galloyl, a polyphenol linked to a lipid with two acyl chains can be included in the LNPs of the present invention to promote gene transfer through mechanisms, which, without the intention of being limited by a particular theory, promote attachment, endocytosis, and the efficiency of gene transfer.
D. Enhancement of electroporation efficiency with Brambleberry mRNA
[00194] In one aspect, the present invention provides systems for nucleic acid transfer that promote nucleic acid transfer by electroporation. An isolated Brambleberry mRNA is injected intramuscularly, e.g., as a mixture in water, with DNA molecules encoding a therapeutic gene, and electrical pulses are applied to promote the entry of Brambleberry mRNA and the DNA vector into cells. In the context of electroporation, Brambleberry allows DNA to enter the nucleus that otherwise would have entered the cell but lacked access to the nucleus.
E. Use of Dynein and/or DNA binders to promote gene transfer
[00195] The present invention also provides systems for nucleic acid transfer that promote nucleic acid transfer that include dynein binders (e. ., a TCTEX-1 binder) and/or DNA binders. It is contemplated that the inclusion of a molecule containing both a DNA binder and a dynein binder may facilitate the intracellular trafficking of the DNA to a perinuclear localization that promotes gene transfer. Examples of dynein binders include small molecules and peptides. Dynein binders can be stearylated to facilitate their incorporation into LNPs. In certain embodiments, the dynein binder is a peptide selected from Arl2 (GGFKLNIWDVGGQK) (SEQ ID NO: 115) and the C-terminus of rhodopsin D (Rhd) (GVSKTETSQVAPA) (SEQ ID NO: 116). Examples of DNA binders include octyl- arginine. In certain embodiments, the dynein binder and the DNA binder are conjugated. Exemplary dynein-DNA binder conjugates include octyl-arginine conjugated to Arl2 (SEQ ID NO: 117) and octyl-arginine conjugated to Rhd (R8RhdCT, SEQ ID NO: 118).
F. Extracellular matrix (ECM)-modifying enzymes
[00196] In certain embodiments, the nucleic acid transfer systems of the disclosure include at least one extracellular matrix (ECM)-modifying enzyme. In certain embodiments, the ECM-modifying enzyme is a hyaluronidase or a fragment thereof.
III. Pharmaceutical Compositions
[00197] The present disclosure also provides pharmaceutical compositions or formulations that include an active agent (e.g., a double membrane fusogen protein, a transgenic gene, a non-coding RNA, a system for DNA transfer) described herein. The pharmaceutical composition can be formulated for use in a variety of drug delivery systems. One or more pharmaceutically acceptable excipients or carriers can also be included in the composition for proper formulation. Suitable formulations for use in the present disclosure are found in Adeboye Adejare, Remington: The Science and Practice of Pharmacy (23rd ed. 2020).
[00198] In certain embodiments, a pharmaceutical composition may contain formulation materials for modifying, maintaining or preserving, for example, the pH, osmolarity, viscosity, clarity, color, isotonicity, odor, sterility, stability, rate of dissolution or release, adsorption or penetration of the composition. In such embodiments, suitable formulation materials include, but are not limited to, amino acids (such as glycine, glutamine, asparagine, arginine or lysine); antimicrobials; antioxidants (such as ascorbic acid, sodium sulfite or sodium hydrogen- sulfite); buffers (such as borate, bicarbonate, Tris-HCl, citrates, phosphates or other organic acids); bulking agents (such as mannitol or glycine); chelating agents (such as ethylenediamine tetraacetic acid (EDTA)); complexing agents (such as caffeine, polyvinylpyrrolidone, beta-cyclodextrin or hydroxypropyl-beta-cyclodextrin); fillers; monosaccharides; disaccharides; and other carbohydrates (such as glucose, mannose or dextrins); proteins (such as serum albumin, gelatin or immunoglobulins); coloring, flavoring and diluting agents; emulsifying agents; hydrophilic polymers (such as polyvinylpyrrolidone); low molecular weight polypeptides; salt-forming counterions (such as sodium); preservatives (such as benzalkonium chloride, benzoic acid, salicylic acid, thimerosal, phenethyl alcohol, methylparaben, propylparaben, chlorhexidine, sorbic acid or hydrogen peroxide); solvents (such as glycerin, propylene glycol or polyethylene glycol); sugar alcohols (such as mannitol or sorbitol); suspending agents; surfactants or wetting agents (such as pluronics, PEG, sorbitan esters, polysorbates such as polysorbate 20, polysorbate,
triton, tromethamine, lecithin, cholesterol, tyloxapal); stability enhancing agents (such as sucrose or sorbitol); tonicity enhancing agents (such as alkali metal halides, preferably sodium or potassium chloride, mannitol sorbitol); delivery vehicles; diluents; excipients and/or pharmaceutical adjuvants (see, Adeboye Adejare, Remington: The Science and Practice of Pharmacy (23rd ed. 2020)).
[00199] Pharmaceutical compositions containing an active agent (e.g. a double membrane fusogen protein, a transgenic gene, a non-coding RNA, a system for DNA transfer) can be presented in a dosage unit form and can be prepared by any suitable method. A pharmaceutical composition should be formulated to be compatible with its intended route of administration. Examples of routes of administration are intravenous, intradermal, inhalation, transdermal, topical, transmucosal, intrathecal and rectal administration. Formulation components suitable for parenteral administration include a sterile diluent such as water for injection, saline solution, fixed oils, polyethylene glycols, glycerin, propylene glycol or other synthetic solvents; antibacterial agents such as benzyl alcohol or methyl parabens; antioxidants such as ascorbic acid or sodium bisulfite; chelating agents such as EDTA; buffers such as acetates, citrates or phosphates; and agents for the adjustment of tonicity such as sodium chloride or dextrose.
[00200] For intravenous administration, suitable carriers include physiological saline, bacteriostatic water, Cremophor ELTM (BASF, Parsippany, NJ) or phosphate buffered saline (PBS). The carrier should be stable under the conditions of manufacture and storage, and should be preserved against microorganisms. The carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyetheylene glycol), and suitable mixtures thereof. An intravenous drug delivery formulation of the present disclosure may be contained in a bag, a pen, or a syringe. In certain embodiments, the bag may be connected to a channel including a tube and/or a needle.
[00201] In certain embodiments, the formulation is a liquid formulation. In certain embodiments, an aqueous formulation is prepared including the active agent (e.g., a double membrane fusogen protein, a transgenic gene, a non-coding RNA, a system for DNA transfer) in a pH-buffered solution. The pH of the liquid formulation may be set by addition of a pharmaceutically acceptable acid and/or base. In certain embodiments, the pharmaceutically acceptable acid may be hydrochloric acid. In certain embodiments, the
base may be sodium hydroxide. In certain embodiments, a salt or buffer components may be added in an amount of 10 mM to 200 mM. The salts and/or buffers are pharmaceutically acceptable and are derived from various known acids (inorganic and organic) with “base forming” metals or amines. In certain embodiments, the buffer may be phosphate buffer. In certain embodiments, the buffer may be glycinate, carbonate, citrate buffers, in which case, sodium, potassium or ammonium ions can serve as counterion. Intravenous formulations can be diluted with 0.9% sodium chloride solution before administration. In certain embodiments, the diluted drug product for injection is isotonic and suitable for administration by intravenous infusion.
[00202] In certain embodiments, the formulation is a lyophilized formulation including the active agent (e.g., a double membrane fusogen protein, a transgenic gene, a non-coding RNA, a system for DNA transfer) and a lyoprotectant. The lyoprotectant may be sugar, e.g., disaccharides. In certain embodiments, the lyoprotectant may be sucrose or maltose. The lyophilized formulation may also include one or more of a buffering agent, a surfactant, a bulking agent, and/or a preservative. The amount of sucrose or maltose useful for stabilization of the lyophilized drug product may be in a weight ratio of at least 1:2 protein to sucrose or maltose. In certain embodiments, the protein to sucrose or maltose weight ratio may be of from 1 :2 to 1 :5. Before lyophilization, the pH of the solution containing the protein of the present disclosure may be adjusted between 6 to 8. In certain embodiments, the pH range for the lyophilized drug product may be from 7 to 8. In certain embodiments, a “bulking agent” may be added. A “bulking agent” is a compound which adds mass to a lyophilized mixture and contributes to the physical structure of the lyophilized cake e.g., facilitates the production of an essentially uniform lyophilized cake which maintains an open pore structure). Illustrative bulking agents include mannitol, glycine, polyethylene glycol and sorbitol. The lyophilized formulations of the present disclosure may contain such bulking agents.
[00203] In certain embodiments, the lyophilized drug product may be constituted with an aqueous carrier. The aqueous carrier of interest herein is one which is pharmaceutically acceptable e.g. , safe and non-toxic for administration to a human) and is useful for the preparation of a liquid formulation, after lyophilization. Illustrative diluents include sterile water for injection (SWFI), bacteriostatic water for injection (BWFI), a pH buffered solution (e.g., phosphate-buffered saline), sterile saline solution, Ringer’s solution or dextrose
solution. In certain embodiments, the lyophilized protein product of the instant disclosure is constituted to about 4.5 mL water for injection and diluted with 0.9% saline solution (sodium chloride solution).
[00204] The pharmaceutical compositions may be sterilized by conventional sterilization techniques, or may be sterile filtered. The resulting aqueous solutions may be packaged for use as-is, or lyophilized, the lyophilized preparation being combined with a sterile aqueous carrier prior to administration. The pH of the preparations typically will be between 3 and 11 , more preferably between 5 and 9 or between 6 and 8, and most preferably between 7 and 8, such as 7 to 7.5. The resulting compositions in solid form may be packaged in multiple single dose units, each containing a fixed amount of the above-mentioned agent or agents. The composition in solid form can also be packaged in a container for a flexible quantity.
[00205] In certain embodiments, the present disclosure provides a formulation with an extended shelf life including the active agent (e.g., a double membrane fusogen protein, a transgenic gene, a non-coding RNA, a system for DNA transfer), in combination with mannitol, citric acid monohydrate, sodium citrate, disodium phosphate dihydrate, sodium dihydrogen phosphate dihydrate, sodium chloride, polysorbate 80, water, and sodium hydroxide.
[00206] A polyol, which acts as a tonicifier and may stabilize the antibody, may also be included in the formulation. The polyol is added to the formulation in an amount which may vary with respect to the desired isotonicity of the formulation. In certain embodiments, the aqueous formulation may be isotonic. The amount of polyol added may also be altered with respect to the molecular weight of the polyol. For example, a lower amount of a monosaccharide (e.g., mannitol) may be added, compared to a disaccharide (such as trehalose). In certain embodiments, the polyol which may be used in the formulation as a tonicity agent is mannitol. In certain embodiments, the mannitol concentration may be about 5 to about 20 mg/mL. In certain embodiments, the concentration of mannitol may be about 7.5 to about 15 mg/mL. In certain embodiments, the concentration of mannitol may be about 10 to about 14 mg/mL. In certain embodiments, the concentration of mannitol may be about 12 mg/mL. In certain embodiments, the polyol sorbitol may be included in the formulation.
[00207] A detergent or surfactant may also be added to the formulation. Exemplary detergents include nonionic detergents such as polysorbates (e. . , polysorbates 20, 80 etc.) or
poloxamers (e.g., poloxamer 188). The amount of detergent added is such that it reduces aggregation of the formulated antibody and/or minimizes the formation of particulates in the formulation and/or reduces adsorption. In certain embodiments, the formulation may include a surfactant which is a polysorbate. In certain embodiments, the formulation may contain the detergent polysorbate 80 or Tween 80. Tween 80 is a term used to describe polyoxyethylene (20) sorbitanmonooleate (see Fiedler, Lexikon der Hifsstoffe, Editio Cantor Verlag Aulendorf, 4th ed., 1996). In certain embodiments, the formulation may contain polysorbate 80 between about 0.1 mg/mL and about 10 mg/mL, or between about 0.5 mg/mL and about 5 mg/mL. In certain embodiments, about 0.1% polysorbate 80 may be added in the formulation.
[00208] In embodiments, the protein product of the present disclosure is formulated as a liquid formulation in either a USP / Ph Eur type I 50R vial closed with a rubber stopper and sealed with an aluminum crimp seal closure. The stopper may be made of elastomer complying with USP and Ph Eur. In certain embodiments, the liquid formulation may be diluted with 0.9% saline solution prior to use.
[00209] In certain embodiments, the liquid formulation of the disclosure may be prepared in combination with a sugar at stabilizing levels. In certain embodiments the liquid formulation may be prepared in an aqueous carrier. In certain embodiments, a stabilizer may be added in an amount no greater than that which may result in a viscosity undesirable or unsuitable for intravenous administration. In certain embodiments, the sugar may be di saccharides, e.g., sucrose. In certain embodiments, the liquid formulation may also include one or more of a buffering agent, a surfactant, and a preservative.
[00210] A preservative may be optionally added to the formulations herein to reduce bacterial action. The addition of a preservative may, for example, facilitate the production of a multi-use (multiple-dose) formulation.
[00211] In certain embodiments, a pharmaceutical composition may contain nanoparticles, e.g., polymeric nanoparticles, liposomes, or micelles (see Anselmo et al. (2016) BIOENG. TRANSL. MED. 1: 10-29).
[00212] In certain embodiments, a pharmaceutical composition may contain a sustained- or controlled-delivery formulation. Techniques for formulating sustained- or controlled- delivery means, such as liposome carriers, bio-erodible microparticles or porous beads and
depot injections, are also known to those skilled in the art. Sustained-release preparations may include, e.g. , porous polymeric microparticles or semipermeable polymer matrices in the form of shaped articles, e.g., films, or microcapsules. Sustained release matrices may include polyesters, hydrogels, polylactides, copolymers of L-glutamic acid and gamma ethyl-L- glutamate, poly (2-hydroxyethyl-inethacrylate), ethylene vinyl acetate, or poly-D(-)-3- hydroxybutyric acid. Sustained release compositions may also include liposomes that can be prepared by any of several methods known in the art.
[00213] In certain embodiments, a therapeutically effective amount of active component is in the range of 0. 1 mg/kg to 100 mg/kg, e.g., 1 mg/kg to 100 mg/kg, 1 mg/kg to 50 mg/kg, 1 mg/kg to 40 mg/kg, 1 mg/kg to 30 mg/kg, 1 mg/kg to 20 mg/kg, 1 mg/kg to 10 mg/kg, 1 mg/kg to 5 mg/kg, 50 mg/kg, 40 mg/kg, 30 mg/kg, 20 mg/kg, 10 mg/kg, 7.5 mg/kg, 5 mg/kg, or 2.5 mg/kg. The amount administered will depend on variables such as the type and extent of disease or indication to be treated, the overall health of the patient, the in vivo potency of the active component, the pharmaceutical formulation, and the route of administration. The initial dosage can be increased beyond the upper level in order to rapidly achieve the desired blood-level or tissue- level. Alternatively, the initial dosage can be smaller than the optimum, and the daily dosage may be progressively increased during the course of treatment. Human dosage can be optimized, e.g., in a conventional Phase I dose escalation study designed to run from 0.5 mg/kg to 30 mg/kg. Dosing frequency can vary, depending on factors such as route of administration, dosage amount, plasma half-life, and the disease being treated. Exemplary dosing frequencies are once per day, once per week, once every two weeks, once per month, once every two months, and once every three months. An exemplary route of administration is parenteral, e.g., intravenous infusion. In certain embodiments, the protein is administered subcutaneously. In certain embodiments, the protein or is lyophilized, and then reconstituted in buffered saline, at the time of administration. In certain embodiments, the protein is administered using a nebulizer.
[00214] The double membrane fusogen protein and non-native DNA and/or other components of a nucleic acid transfer system can be administered in one composition or in separate compositions. When administered in separate compositions, a double membrane fusogen protein can be administered before the non-native DNA and/or other components of the nucleic acid transfer system. For example, the double membrane fusogen protein can be
administered between about 30 minutes to about 12 hours before the non-native DNA and/or other components of the nucleic acid transfer system.
[00215] The systems for nucleic acid transfer can be co-formulated with immunosuppressive molecules or pro-drug forms thereof. For example, immunosuppressive molecules or pro- drug forms thereof can be incorporated into an LNP, or can be provided in an aqueous formulation buffer that contains LNPs. Immunosuppressive molecules or pro-drug forms thereof can include an ASO that binds a nucleic acid sensing molecule such as C2mutl (as described above), a corticosteroid such as glucocorticoid, prednisone, prednisolone, triamcinolone, methylprednisolone, dexamethasone, dexamethasone palmitate, fluticasone, fluticasone propionate, fluticasone furoate, clobetasol, betamethasone, halobetasol, amcinonide, desoximetasone, diflorasone, mometasone, prednicarbate, triamcinolone acetonide, fluocinolone, alclometasone, desonide, cortisone, hydrocortisone, and fluocinonide or a pro-drug form thereof; mTOR inhibitors, such as rapamycin, sirolimus, and everolimus; a tyrosine kinase inhibitor such as deucravacitinib, baricitinib, tofacitinib, ruxolitinib, fedratinib, and fostamatinib, or pro-drug forms thereof; and a calcineurin inhibitor such as tacrolimus, FK506 and cyclosporine A or pro-drug forms thereof. Other non-limiting examples of immunosuppressive molecules include ASOs or siRNAs that target the transcripts encoding proteins involved in innate immune sensing, such as cGAS, TLR3, TLR7, TLR8, TLR9, RIG-I, MAVS, MDA5, and MYD88, or pro-drug forms thereof; ASOs or siRNAs, or pro-drug forms thereof, that target the transcripts encoding transcription factors involved in innate immune responses such as the transcripts of NFKB, IRF3, and IRF7. Other exemplary immunosuppressive molecules include mRNAs encoding immunosuppressive molecules, such as indoleamine 2,3-dioxygenase-l (IDO1), NF kappa B inhibitor alpha (NFKBIA), inhibitor of kappa B subunit beta (IKBKB), TNF alpha induced protein 3 (TNFAIP3), interferon regulatory factor 4 (IRF4), interferon regulatory factor 8 (IRF8), cytotoxic T-lymphocyte- associated protein 4 (CTLA4), transforming growth factor beta-1 (TGFB1), interleukin 1 receptor type 2 (IL1R2), suppressor of cytokine signaling-1 (SOCS1), suppressor of cytokine signaling-2 (SOCS2), suppressor of cytokine signaling-3 (SOCS3), suppressor of cytokine signaling-4 (SOCS4), suppressor of cytokine signaling-5 (SOCS5), suppressor of cytokine signaling-6 (SOCS6), suppressor of cytokine signaling-7 (SOCS7), ring finger protein 216 (RNF216), and CASP8 and FADD like apoptosis regulator (CFLAR), or pro-drug forms thereof. mRNAs, siRNAs, and ASOs can be co-formulated with an
mRNA encoding a double membrane fusogen protein (e.g., Brambleberry mRNA) within an LNP, to reduce the potential for nucleic acid transfer to stimulate immune responses against the therapeutic gene. In certain embodiments, immunosuppressive molecules, or pro-drug forms thereof, including, e.g., corticosteroids (such as glucocorticoid), mTOR inhibitors, tyrosine kinase inhibitors, ASOs, and siRNAs, and/or mRNAs encoding immunosuppressive molecules, can be formulated with LNPs containing an mRNA encoding a double membrane fusogen protein (e.g., Brambleberry mRNA) and a DNA. Immunosuppressive molecules or pro-drug forms thereof can also include immunosuppressive molecule or pro-drug form thereof that bind human glucocorticoid receptor. In certain embodiments, the immunosuppressive molecule or pro-drug form thereof can contain an ester bond.
[00216] In certain embodiments, the immunosuppressive molecule or prodrug form thereof is selected from the group consisting of: galectin inhibitors, such as GB1107, galectin- 3/galectin-8-IN-l, olitigaltin, selvigaltin, galectin-3/galectin-8-IN-2, galectin-3-IN-2, galectin-3-IN-3, galectin-3-IN-5, GB1490, GB1908, galectin-8-IN-l , galectin-8-IN-2, galectin-8N-IN-l, galectin-8N-IN-2, or thiodigalactoside, or a pro-drug form of any of the foregoing; cysteine protease inhibitors, such as cathepsin inhibitors (e.g., L-IN-2 (Z-Phe-Phe- FMK)) or a pro-drug form thereof; NLRP3 inflammasome inhibitors, such as oridonin, INF39, glyburide, 5-chloro-2-methoxy-N-[2-(4- sulfamoylphenyl)-ethyl]-benzamide (e.g., 16673-34-0), JC124, FC11A-2, parthenolide, VX-740, VX-765, BAY 11-7082, BHB, MCC950, MNS, CY-09, Tranilast, or OLT1177, or a pro-drug form of any of the foregoing; MyD88 inhibitors, or pro-drug forms thereof; IRAK4 inhibitors, or pro-drug forms thereof; PKR inhibitors, or pro-drug forms thereof; PERK inhibitors, or pro-drug forms thereof; NFkB inhibitors, or pro-drug forms thereof; IKK inhibitors, or pro-drug forms thereof; JAK inhibitors, or pro-drug forms thereof; STAT inhibitors, or pro-drug forms thereof; GSK3 inhibitors, or pro-drug forms thereof; cGAS inhibitors, or pro-drug forms thereof; or STING inhibitors, or pro-drug forms thereof. In certain embodiments, the immunosuppressive molecule or pro-drug form thereof is selected from the group consisting of galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, and NLRP3 inflammasome inhibitors. In certain embodiments, the immunosuppressive molecule or pro-drug form thereof is an NLRP3 inflammasome inhibitor.
[00217] In certain embodiments, the nucleic acid transfer systems of the disclosure can include at least two, three, four, five, six, seven, eight, nine, or ten different immunosuppressive molecules or pro-drug forms thereof.
[00218] In certain embodiments, the nucleic acid transfer systems of the disclosure can include an immunosuppressive molecule or pro-drug form thereof that has a LogP value greater than 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, or 5.0. In certain embodiments, the immunosuppressive molecule or pro-drug form thereof has a LogP value greater than 3.0, 3.5, or 4.0.
[00219] In certain embodiments, a nucleic acid transfer systems of the disclosure comprises an aqueous suspension of the immunosuppressive molecule or pro-drug form thereof. In certain embodiments, the aqueous suspension comprises triamcinolone acetonide or betamethasone sodium phosphate.
[00220] It is further contemplated that extracellular matrix (ECM) -modifying enzymes such as hyaluronidase, or a fragment thereof, can be included in a system of the disclosure to improve the access of LNPs to muscle cells. In certain embodiments, the nucleic acid transfer systems of the disclosure comprises at least one extracellular matrix (ECM)-modifying enzyme. In certain embodiments, the ECM-modifying enzyme is a hyaluronidase or a fragment thereof. An exemplary hyaluronidase fragment is PH20 (Hylenex®).
IV. Therapeutic Uses
A. Gene Therapy
[00221] Described herein are various methods of preventing, treating, arresting progression of or ameliorating disease and disorders by administering a DNA encoding a protein of interest to prevent, treat, arrest the progression of, or ameliorate the disease or disorder.
Generally, the methods include administering to a subject, e.g., a mammalian subject, in need thereof, an effective amount of a composition comprising a nucleic acid transfer system as described herein, for example, carrying an mRNA encoding a double membrane fusogen protein (e.g., a Brambleberry protein), a DNA, and a pharmaceutically acceptable carrier. The DNA can encode a sequence which expresses a protein of interest in target cells of a subject. The protein may replace or supplement a protein that is missing or damaged in the cells of the subject.
[00222] In a certain aspect, the disclosure provides a method of treating a subject having a disease as described herein, comprising the step of administering to the subject a therapeutically effective amount of a nucleic acid transfer system as described herein, which may be in the range of 0.1 mg/kg to 100 mg/kg, e.g., 1 mg/kg to 100 mg/kg, 1 mg/kg to 50 mg/kg, 1 mg/kg to 40 mg/kg, 1 mg/kg to 30 mg/kg, 1 mg/kg to 20 mg/kg, 1 mg/kg to 10 mg/kg, 1 mg/kg to 5 mg/kg, 50 mg/kg, 40 mg/kg, 30 mg/kg, 20 mg/kg, 10 mg/kg, 7.5 mg/kg, 5 mg/kg, or 2.5 mg/kg. In certain embodiments, the pharmaceutical compositions of the disclosure comprise a pharmaceutically acceptable carrier.
[00223] In certain embodiments, a nucleic acid transfer system as described herein is capable of inducing at least 20%, 50%, 100%, 150%, 200%, 250%, 300%, 400%, 500%, 700%, 900%, 1,000%, 1,100%, 1,500%, or 2,000% higher expression of a protein of interest in a target cell as compared to the endogenous expression of the protein of interest in the target cell. Alternatively or in addition, expression of a nucleic acid transfer system as described herein in a target cell results in at least 20%, 50%, 100%, 150%, 200%, 250%, 300%, 400%, 500%, 700%, 900%, 1,000%, 1,100%, 1,500%, or 2,000% higher levels of activity of a protein of interest in the target cell as compared to endogenous levels of activity of the protein of interest in the target cell.
[00224] In certain embodiments, any of the treatment and/or prophylactic methods disclosed herein can be applied to a subject, e.g., a mammal, e.g., a human.
B. Gene Editing
[00225] The methods of the disclosure can be used to deliver a gene editing system (e.g., a CRISPR system) to a cell. For example, an mRNA encoding a double membrane fusogen protein, an mRNA encoding a gene editing nuclease, a guide RNA, and, optionally, a DNA template can be delivered to a cell according to the methods of the disclosure. In certain embodiments, the template comprises a coding sequence to be incorporated into the genome of the cell using the gene editing system. The double membrane fusogen protein encoded by the mRNA facilitates entry of the template into the nucleus of the cell, and the DNA template encodes a protein of interest that is inserted into the genome of a target cell via the gene editing system. In certain embodiments, the template is used as a repair template to correct a defect (e.g., deletion, mutation, and/or insertion) in the genome of a target cell (e.g., in a
protein coding sequence). In certain embodiments, the protein replaces or supplements a protein that is missing or damaged in the cells of the subject.
[00226] Gene editing systems that can be used in accordance with the methods herein include nuclease-based genome editing systems such as CRISPR systems, TALEN systems, ZFN systems, a homing endonuclease system, or a prime editing system. Such systems typically include a gene editing effector protein such as a nuclease. In a CRISPR system, nucleases can include CRISPR enzymes such as Casl, CaslB, Cas2, Cas3, Cas4, Cas5, Cas6, Cas7, Cas8, Cas9 (also known as Csnl and Csxl2), Cas10, Csyl, Csy2, Csy3, Csel, Cse2, Cscl, Csc2, Csa5, Csn2, Csm2, Csm3, Csm4, Csm5, Csm6, Cmrl, Cmr3, Cmr4, Cmr5, Cmr6, Csbl, Csb2, Csb3, Csxl7, Csxl4, Csx10, Csxl6, CsaX, Csx3, Csxl, Csxl5, Csfl, Csf2, Csf3, Csf4, Cpfl, Casl3a, Casl3b and Casl3d, homologs thereof, or modified versions thereof. These enzymes are known; for example, the amino acid sequence of .S'. pyogenes Cas9 protein may be found in the SwissProt database under accession number Q99ZW2. In certain embodiments, the unmodified CRISPR enzyme has DNA cleavage activity, such as Cas9, which may optionally be derived from S. pyogenes or S. pneumoniae . Additional exemplary CRISPR enzymes include MAD1, MAD2, MAD3, MAD4, MAD5, MAD6, MAD7, MAD8, MAD9 MAD 10, MAD11, or MAD 11 endonuclease (see, e.g., U.S. Patent No. 9,982,279). i. Generation of CAR-T Cells Using Gene Editing
[00227] The methods of the disclosure can be used to deliver a gene editing system (e.g., a CRISPR system) to a T cell. For example, an mRNA encoding a double membrane fusogen protein, an mRNA encoding a gene editing nuclease, a guide RNA, and a DNA template can be delivered to a cell according to the methods of the disclosure. In certain embodiments, the template encodes one or more chimeric T cell receptors that are incorporated into the genome of the T cell using the gene editing system. The double membrane fusogen protein encoded by the mRNA facilitates entry of the template into the nucleus of the T cell. Depending upon the circumstances, the chimeric receptor may be a chimeric T cell receptor that specifically binds CD 19 and/or CD20. In certain embodiments, the chimeric receptor is inserted into a T cell receptor alpha constant region (TRAC) sequence. An exemplary method for generating CAR-T cells using the nucleic acid transfer systems herein is discussed in Example 6.
C. Specific Embodiments of Methods of Use
[00228] Embodiment 1 : A method for the transfer of non-native DNA into a nucleus of a cell, the method comprising introducing the non-native DNA and RNA into a cell, wherein the RNA encodes a double membrane fusogen.
[00229] Embodiment 2: The method of embodiment 1, wherein the double membrane fusogen mediates the fusion of intracellular membranes.
[00230] Embodiment 3: The method of embodiment 2, wherein the double membrane fusogen mediates the fusion of a nuclear envelope of a cell with a double membrane- enveloped structure comprising the non-native DNA.
[00231] Embodiment 4: The method of any one of embodiments 1-3, wherein the double membrane fusogen comprises a signal peptide and a transmembrane domain.
[00232] Embodiment 5: The method of embodiment 4, wherein the double membrane fusogen comprises two or more transmembrane domains.
[00233] Embodiment 6: The method of embodiment 5, wherein the double membrane fusogen comprises three or more transmembrane domains.
[00234] Embodiment 7: The method of embodiment 6, wherein the double membrane fusogen comprises three or more transmembrane alpha helices.
[00235] Embodiment 8: The method of any one of embodiments 1-7, wherein the double membrane fusogen comprises a DNA-binding domain.
[00236] Embodiment 9: The method of any one of embodiments 1-8, wherein the double membrane fusogen comprises a multimerization domain.
[00237] Embodiment 10: The method of any one of embodiments 1-9, wherein double membrane fusogen comprises an alpha helical domain at least 20, 30, 40, 50, 60, 70, 80, 90, or 100 amino acids in length.
[00238] Embodiment 11 : The method of any one of embodiments 1-10, wherein double membrane fusogen comprises an alpha helical domain at least 50 amino acids in length.
[00239] Embodiment 12: The method of any one of embodiments 1-11, wherein double membrane fusogen comprises an alpha helical domain at least 60 amino acids in length.
[00240] Embodiment 13: The method of any one of embodiments 1-12, wherein double membrane fusogen comprises an alpha helical domain at least 80 amino acids in length.
[00241] Embodiment 14: The method of any one of embodiments 1-13, wherein double membrane fusogen comprises an alpha helical domain at least 100 amino acids in length.
[00242] Embodiment 15: The method of any one of embodiments 1-14, wherein the double membrane fusogen comprises a luminal domain, transmembrane domain, cytoplasmic domain, or DNA-binding domain that has least 60%, at least 70%, at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to the corresponding domain present in SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, or 147.
[00243] Embodiment 16: The method of any one of embodiments 1-15, wherein the double membrane fusogen comprises a cytoplasmic domain, and the cytoplasmic domain comprises at least one of (a) nuclear localization signal (NLS), (b) amino acid sequence five amino acids in length wherein at least four out of five of the amino acids are basic residues, and (c) an amino acid sequence comprising at least one basic residue separated by a spacer of ten to fifteen amino acids from three or more basic residues.
[00244] Embodiment 17: The method of embodiment 16, wherein at least one of features (a) - (c) does not naturally occur in the amino acid sequence of a wild-type double membrane fusogen having the greatest sequence identity to the encoded double membrane fusogen.
[00245] Embodiment 18: The method of any one of embodiments 1-17, wherein the double membrane fusogen is not a human protein.
[00246] Embodiment 19: The method of any one of embodiments 1-18, wherein the double membrane fusogen is a Brambleberry protein, a protein involved in karyogamy, or a functional fragment, variant, or chimera of any of the foregoing.
[00247] Embodiment 20: The method of any one of embodiments 1-19, wherein the double membrane fusogen is a Brambleberry protein or functional fragment or variant thereof.
[00248] Embodiment 21 : The method of any one of embodiments 19 or 20, wherein the Brambleberry protein or functional fragment or variant thereof is a zebrafish (Danio rerid) Brambleberry protein or functional fragment or variant thereof.
[00249] Embodiment 22: The method of any one of embodiments 19-21 wherein the Brambleberry protein is a split Brambleberry protein.
[00250] Embodiment 23: The method of any one of embodiments 1-22, wherein the method further comprises an immunosuppressive molecule or a pro-drug form thereof, and/or an RNA encoding an immunosuppressive protein.
[00251] Embodiment 24: The method of embodiment 23, wherein the immunosuppressive molecule or pro-drug form thereof is a corticosteroid or a tyrosine kinase inhibitor
[00252] Embodiment 25 : The method of embodiment 24, wherein the corticosteroid is a glucocorticoid.
[00253] Embodiment 26: The method of any one of embodiments 23-25, wherein the immunosuppressive molecule or pro-drug form thereof binds human glucocorticoid receptor.
[00254] Embodiment 27: The method of any one of embodiments 23-26, wherein the immunosuppressive molecule is dexamethasone, fluticasone, fluticasone propionate, fluticasone furoate, clobetasol, betamethasone, halobetasol, amcinonide, desoximetasone, diflorasone, fluocinonide, mometasone, prednisone, prednicarbate, triamcinolone, triamcinolone acetonide, fluocinolone, alclometasone, desonide, cortisone, or hydrocortisone, or a pro-drug form of any of the foregoing.
[00255] Embodiment 28: The method of embodiment 23-27, wherein the immunosuppressive molecule is dexamethasone or tacrolimus.
[00256] Embodiment 29: The method of any one of embodiments 23-28, wherein the immunosuppressive molecule or pro-drug form thereof contains an ester bond.
[00257] Embodiment 30: The method of any one of embodiments 23-29, wherein the immunosuppressive molecule or pro-drug form thereof is dexamethasone palmitate or fluticasone-furoate.
[00258] Embodiment 31 : The method of any one of embodiments 23-30, comprising at least one immunosuppressive molecule or pro-drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, NLRP3 inflammasome inhibitors, MyD88 inhibitors, IRAK4 inhibitors, PKR inhibitors, PERK inhibitors, NFkB inhibitors, IKK inhibitors, JAK inhibitors, STAT inhibitors, GSK3 inhibitors, cGAS inhibitors, or STING inhibitors.
[00259] Embodiment 32: The method of any one of embodiments 23-31, comprising at least one immunosuppressive molecule or pro-drug form thereof selected from the group
consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, or NLRP3 inflammasome inhibitors.
[00260] Embodiment 33: The method of any one of embodiments 23-32, wherein the immunosuppressive molecule or pro-drug form thereof is an NLRP3 inflammasome inhibitor.
[00261] Embodiment 34: The method of any one of embodiments 31-33, wherein the NLRP3 inflammasome inhibitor is oridonin, INF39, glyburide, 5-chloro-2-methoxy-N-[2-(4- sulfamoylphenyl)-ethyl]-benzamide (e.g., 16673-34-0), JC124, FC11A-2, parthenolide, VX- 740, VX-765, BAY 11-7082, BHB, MCC950, MNS, CY-09, or Tranilast, OLT1177, or a pro-drug form of any of the foregoing.
[00262] Embodiment 35: The method of any one of embodiments 31-32, wherein the galectin inhibitor is GB1107, galectin-3/galectin-8-IN-l, olitigaltin, selvigaltin, galectin- 3/galectin-8-IN-2, galectin-3-IN-2, galectin-3-IN-3, galectin-3-IN-5, GB1490, GB1908, galectin-8-IN-l, galectin- 8-IN-2, galectin-8N-IN-l, galectin-8N-IN-2, or thiodigalactoside, or a pro-drug form of any of the foregoing.
[00263] Embodiment 36: The method of any one of embodiments 31-32, wherein the cathepsin inhibitor or cysteine protease inhibitor is cathepsin L-IN-2 (Z-Phe-Phe-FMK), Disulfiram, Belizatinib, Cystatin B, Cystatin C, E-64, E-64d, or a pro-drug form of any of the foregoing.
[00264] Embodiment 37: The method of any one of embodiments 23-36, comprising at least two different immunosuppressive molecules or pro-drug forms thereof.
[00265] Embodiment 38: The method of any one of embodiments 23-37, comprising at least three different immunosuppressive molecules or pro-drug forms thereof.
[00266] Embodiment 39: The method of any one of embodiments 23-38, wherein the immunosuppressive molecule or pro-drug form thereof has a LogP value greater than 3.0, 3.5, or 4.0.
[00267] Embodiment 40: The method of any one of embodiments 23-39, comprising an aqueous suspension of the immunosuppressive molecule or pro-drug form thereof.
[00268] Embodiment 41 : The method of embodiment 40, wherein the aqueous suspension comprises triamcinolone acetonide or betamethasone sodium phosphate.
[00269] Embodiment 42: The method of any one of embodiments 1-41, comprising at least one extracellular matrix (ECM)-modifying enzyme.
[00270] Embodiment 43 : The method of embodiment 42, where in the ECM-modifying enzyme is a hyaluronidase or a fragment thereof.
[00271] Embodiment 44: The method of any one of embodiments 1-43, wherein the cell is a non-dividing cell.
[00272] Embodiment 45: The method of any one of embodiments 1-44, wherein the RNA encoding the double membrane fusogen comprises modified bases.
[00273] Embodiment 46: The method of any one of embodiments 1-45, wherein the RNA encoding the double membrane fusogen is an mRNA.
[00274] Embodiment 47: The method of any one of embodiments 1-46, wherein the RNA encoding the double membrane fusogen comprises one or more target sites for at least one micro RNA (miR).
[00275] Embodiment 48: The method of any one of embodiments 1-47, wherein the DNA comprises modified CpG motifs.
[00276] Embodiment 49: The method of embodiment 48, wherein the DNA has fewer than 100 CpG motifs per molecule, the DNA is substantially free of unmodified CpG motifs, and/or the DNA is methylated at one or more CpG motifs.
[00277] Embodiment 50: The method of embodiment 49, wherein the DNA is substantially free of 6-methyladenine and/or 5-methylcytosine.
[00278] Embodiment 51 : The method of any one of embodiments 1-50, wherein the DNA comprises at least one tissue-specific promoter.
[00279] Embodiment 52: The method of embodiment 51, wherein the tissue-specific promoter is expressed in non-dividing cells.
[00280] Embodiment 53: The method of embodiment 51 or 52, wherein the tissue-specific promoter is a muscle-specific promoter.
[00281] Embodiment 54: The method of any one of embodiments 1-53, wherein the DNA comprises a promoter with fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs.
[00282] Embodiment 55: The method of embodiments 53 or 54, wherein the muscle-specific promoter comprises one or more transcription factor binding site core motifs selected from the group consisting of: a. 5’-CATTCC-3’, b. 5’-GGAATG-3’, c. 5’-CCWWWWWWGG-3’, d. 5’-CANNTG-‘3, e. 5’-YTAAAAATA-3’, and f. 5’-TATTTTTAR-3’, wherein N is any nucleotide, W is A or T, Y is T or C, and R is A or G.
[00283] Embodiment 56: The method of any one of embodiments 1-55, wherein the DNA lacks free 5 ’ and 3 ’ ends and/or is circular.
[00284] Embodiment 57: The method of any one of embodiments 1-56, wherein the DNA is at least partially denatured.
[00285] Embodiment 58: The method of any one of embodiments 1-57, wherein the DNA comprises a coding region that is operably linked to a promoter.
[00286] Embodiment 59: The method of any one of embodiments 1-58, wherein the DNA comprises a gene capable of expressing a secreted protein.
[00287] Embodiment 60: The method of any one of embodiments 1-59, wherein the DNA comprises a gene encoding a protein containing an antibody Fc.
[00288] Embodiment 61 : The method of any one of embodiments 1-60 wherein the DNA comprises a gene encoding a protein comprising a monoclonal or bispecific antibody.
[00289] Embodiment 62: The method of any one of embodiments 1-61, wherein the DNA comprises a gene encoding a protein comprising at least one domain that binds a ligand and at least one effector domain.
[00290] Embodiment 63: The method of any one of embodiments 1-62, wherein the DNA comprises a gene encoding a protein that has at least 80% identity (e.g., at least 85%, at least
90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100%) to human serum albumin (HSA).
[00291] Embodiment 64: The method of any one of embodiments 1-63 wherein the DNA comprises a gene encoding an mRNA, wherein the mRNA comprises one or more target sites for a micro RNA (miR), short hairpin RNA (shRNA), or ribozyme.
[00292] Embodiment 65: The method of any one of embodiments 1-64, wherein the DNA encodes at least one non-coding RNA.
[00293] Embodiment 66: The method of embodiment 65, wherein the at least one non- coding RNA comprises a micro RNA (miR), short hairpin RNA (shRNA), aptamer, or ribozyme.
[00294] Embodiment 67 : The method of embodiment 65 or 66, wherein the at least one non- coding RNA targets an mRNA expressed by the DNA.
[00295] Embodiment 68: The method of any one of embodiments 65-67, wherein the at least one non-coding RNA targets the mRNA of an immunomodulatory gene.
[00296] Embodiment 69: The method of any one of embodiments 65-68, wherein the non- coding RNA targets the mRNA of the fucosyltransferase 8 (FUT8) gene.
[00297] Embodiment 70: The method of any one of embodiments 65-69, wherein the at least one non-coding RNA targets the mRNA of a human gene, and the DNA encodes a protein capable of functionally replacing a common form of that human gene.
[00298] Embodiment 71 : The method of any one of embodiments 1-70, comprising one or more nanoparticles.
[00299] Embodiment 72: The method of embodiment 71, wherein the nanoparticles are lipid nanoparticles (LNPs).
[00300] Embodiment 73: The method of any one of embodiments 1-72, comprising one or more ionizable lipids.
[00301] Embodiment 74: The method of any one of embodiments 1-73, comprising a polyplex.
[00302] Embodiment 75: The method of any one of embodiments 71-74, wherein the nanoparticle comprises at least one attachment moiety.
[00303] Embodiment 76: The method of embodiment 75, wherein the attachment moiety promotes attachment to muscle cells.
[00304] Embodiment 77: The method of any one of embodiments 71-77, further comprising a lipid that is conjugated directly or indirectly to a glucose molecule.
[00305] Embodiment 78: The method of embodiment 77, wherein the glucose molecule is conjugated to hydroquinone.
[00306] Embodiment 79: The method of any one of embodiments 71-77, comprising or further comprising a lipid that is conjugated to a polyphenol.
[00307] Embodiment 80: The method of any one of embodiments 1-79, further comprising a dynein binder.
[00308] Embodiment 81 : The method of any one of embodiments 1-80, further comprising at least one antisense oligonucleotide (ASO) or short-interfering RNA (siRNA).
[00309] Embodiment 82: The method of any one of embodiments 1-81, further comprising a DNA binder that binds to DNA at pH 7.4.
[00310] Embodiment 83: The method of any one of embodiments 1-82, wherein the RNA encoding the double membrane fusogen comprises Cap2 or one or more internal ribosomal entry site (IRES).
[00311] Embodiment 84: The method of any one of embodiments 1-83, further comprising: a. a gene editing effector protein or an RNA encoding a gene editing effector protein, b. a transposase or an RNA encoding a transposase, c. an integrase or an RNA encoding an integrase, d. a recombinase or an RNA encoding a recombinase, or e. a reverse transcriptase or an RNA encoding a reverse transcriptase.
[00312] Embodiment 85: The method of any one of embodiments 1-84, wherein the method is a method for gene therapy.
[00313] Embodiment 86: A method for the transfer of non-native DNA into a nucleus of a cell, comprising the non-native DNA and a dynein binder.
[00314] Embodiment 87: The method of any one of embodiments 80 or 86, wherein the dynein binder is or is conjugated to a DNA binder, an RNA binder, a polymer, a peptide, a polypeptide, or a lipid.
[00315] Embodiment 88: The method any one of embodiments 82 or 87, wherein the DNA binder is cationic at pH 7.4.
[00316] Embodiment 89: The method of any one of embodiments 80 or 86-88, wherein the dynein binder is a TCTEX binder.
[00317] Embodiment 90: The method of embodiment 89, wherein the dynein binder or TCTEX binder is a TCTEX- 1 (DYNLT-T) binder.
[00318] Embodiment 91 : The method of embodiment 89 or 90, wherein the TCTEX binder comprises a peptide with comprising an amino acid sequence at least five amino acids in length that is at least 80% (e.g., at least 90% or 100%) identical to a contiguous amino acid motif selected from the peptides: a. GGFKLNIWDVGGQK (SEQ ID NO: 115), and b. GVSKTETSQVAPA (SEQ ID NO: 116).
[00319] Embodiment 92: The method of embodiment 77, wherein the lipid that is covalently bonded to a glucose molecule is cholesterol-undecanoate-glucose.
[00320] Embodiment 93 : The method of embodiment 79, wherein the polyphenol comprises galloyl or a galloyl group.
[00321] Embodiment 94: The method of any one of embodiments 1-93, wherein the non- native DNA encodes a micro RNA (miR), wherein the miR comprises: a. a 19-base guide sequence that is complementary to a target mRNA, b. an unpaired loop exactly 4 nucleotides in length, c. a 19-base sequence that is complementary to the 19-base guide sequence, which together with the guide sequence form a 19-base pair stem, and
d. a 3 ’ end that is a U, which is encoded by an RNA Polymerase III (Pol III) UUUUUU or TTTTTT transcription termination sequence.
[00322] Embodiment 95: The method of embodiment 94, comprising an RNA Polymerase III promoter operably linked to the miR.
[00323] Embodiment 96: The method of embodiment 94 or 95, wherein the 3’ two nucleotides of the mIR are UU.
[00324] Embodiment 97 : The method of any preceding embodiment, comprising a DNA encoding a microRNA (miR) operably linked to an Hl promoter that is at least 80% identical to nucleotides 1-99 of SEQ ID NO: 154.
[00325] Embodiment 98: The method of any preceding embodiment, comprising a DNA encoding a microRNA (miR) operably linked to an Hl promoter that is at least 80% identical to nucleotides 1-99 of SEQ ID NO: 154.
[00326] Embodiment 99: comprising a DNA encoding a microRNA comprising a DNA encoding a microRNA (miR), wherein the miR targets a fucosylatransferase-8 (FUT8) gene sequence.
EXAMPLES
[00327] The following Examples are merely illustrative and are not intended to limit the scope or content of the invention in any way.
Example 1 — Use of Brambleberry to facilitate transfer of DNA into the nuclei of muscle cells
[00328] It was hypothesized that certain egg-laying animals may have a solution to the topological problem of transferring DNA sequestered within a double membrane envelope to the nucleus. In early embryo development in animals with larger eggs than mammals, chromosomes become enveloped by a double membrane in structures known as karyomeres, which ultimately fuse to form the nucleus. The protein that mediates this fusion is Brambleberry — so named, because in its absence, consolidation of karyomeres into a nucleus is arrested, and the unfused karyomeres resemble a brambleberry (Abrams el al. (2012), supra) (FIGURE 1A). By contrast, in wild-type Danio rerio. the karyomeres fuse and form a nucleus (FIGURE IB). It was hypothesized that transient expression of Brambleberry from an mRNA that is co-packaged with a plasmid DNA, e.g., in an LNP, would mediate
fusion of double membrane-enveloped vesicles sequestering DNA with the nucleus, thereby allowing successful nucleic acid transfer to non-dividing cells.
[00329] Differentiated C2C12 myotubes were used as a relevant tissue culture system to evaluate and optimize an LNP platform for co-delivery of an mRNA expressing Brambleberry and a plasmid DNA into non-dividing cells. C2C12 cells are mouse myoblasts, which can be efficiently differentiated into myotubes (i.e. , syncytial muscle fiber) in plates coated with ‘ECL Cell Attachment Matrix’ upon switching the culture media from 20% fetal bovine serum (FBS) to 2% horse serum plus insulin. Differentiation is easily observed by cell morphology. Differentiated C2C12 myotubes often twitch after adding fresh media containing insulin, demonstrating physiological similarity to muscle fibers. By 5 days post-differentiation, differentiated C2C12 myotubes are a tissue culture model for non- dividing muscle cells.
[00330] The differentiated C2C12 myotube model was used to assess the ability of an mRNA encoding zebrafish Danio rerio) Brambleberry to mediate the nucleic acid transfer to non-dividing cells. The amino acid sequence of zebrafish Brambleberry is provided as SEQ ID NO: 1, and the sequence of an mRNA encoding zebrafish Brambleberry is provided as SEQ ID NO: 2. Hypothesizing that the kinetics of early Brambleberry expression might be important, an experiment was designed to test whether adding Brambleberry mRNA 3 hours before, 1 hour before, 0 hours before, or 1 hour after the plasmid DNA could impact transfection efficiency (FIGURE 4). Lipofectamine 3000, which generates a polyplex or lipo-polyplex containing lipids, an anionic polymer, and RNA and/or DNA, was used as the transfection reagent in this experiment. Indeed, co-transfection with Brambleberry mRNA greatly enhanced transfection of differentiated C2C12 cells. Furthermore, a relationship was observed between the timing of when the Brambleberry mRNA (SEQ ID NO: 2) was added and transfection efficiency, with transfection efficiency being greatest when Bmb was added 3 hours in advance, second greatest when added 1 hour in advance, and least greatest when added 1 hour after the DNA. This experiment shows that Brambleberry can indeed facilitate efficient nuclear import of transfected DNA in non-dividing cells, and furthermore showed that its efficiency is sensitive to its expression kinetics.
[00331] Included in the present invention is a system for nucleic acid transfer in which an RNA molecule encoding a protein that mediates the entry of DNA into the nucleus (e.g. , a Brambleberry protein) is co-packaged with a DNA encoding a gene of interest (e.g. , a
therapeutic gene). A system for nucleic acid transfer based on LNPs containing a mixture of plasmid DNA and Brambleberry mRNA was evaluated. Here, mRNAs encoding a Brambleberry protein and a plasmid DNA encoding a gene of interest were both present as a mixture in the same LNPs. The LNPs in this experiment were made using the ionizable lipid DLin-KC2-DMA. By 5 days post-transfection, Gaussia luciferase expressed by the plasmid DNA and secreted into supernatants was 80-fold higher when the mRNA contained within the DNA-mRNA LNPs encoded zebrafish Brambleberry rather than an irrelevant mRNA control (FIGURE 5A). Supernatants were replaced every 24 hours, so the increase in Gaussia luciferase observed over time is due to the ongoing nuclear import of plasmid DNA facilitated by Brambleberry. Building on these early observations, the Nanoassemblr microfluidics device was used to co-package DNA and mRNA in DNA-mRNA LNPs made with the ionizable lipid DLin-KC2-DMA, and a secreted NanoLuc luciferase, which has a lower but more stable signal that makes it easier to measure than Gaussia luciferase. LNPs made with DLin-KC2-DMA by microfluidics were 2-fold more efficient than particles made using Lipofectamine 3000, when tested in parallel in the context of NanoLuc luciferase (data not shown). Brambleberry mRNA greatly enhanced nucleic acid transfer efficiency by DNA- mRNA LNPs made with DLin-KC2-DMA by microfluidics (FIGURE 5B). These experiments show that LNPs co-packaging Brambleberry mRNA and a plasmid DNA vector efficiently transfect non-dividing cells.
[00332] To test the hypothesis that providing more Brambleberry protein might improve nucleic acid transfer efficiency by increasing the amount of Brambleberry available early, a titration experiment was conducted in which separate NanoLuc DNA LNPs and Brambleberry mRNA LNPs made with DLin-KC2-DMA by microfluidics were added to differentiated C2C12 cells at the same time. Using separate LNPs for the mRNA and DNA facilitated the use of the same mRNA and DNA LNP batches across the entire experiment. Indeed, a clear relationship was observed, in which more Brambleberry mRNA increased nucleic acid transfer efficiency (FIGURE 6). With the highest amount of Brambleberry mRNA used (400 ng/well), a 176-fold increase in nucleic acid transfer efficiency was observed over the DNA-alone LNPs by Day 3. Brambleberry can facilitate the efficient import of plasmid DNA into the nucleus of non-dividing cells, but the efficiency is sensitive to the amount of Brambleberry expressed early. These experiments provided a rationale for developing a non- viral nucleic acid delivery system based on an mRNA encoding a double
membrane fusogen protein, such as Brambleberry mRNA, and plasmid DNA co-packaged in LNPs.
[00333] The impact of antisense oligonucleotides (AS Os) on Brambleberry-mediated nucleic acid transfer also was assessed. In one aspect, the invention includes the combination of Brambleberry mRNAs with additional molecules that promote nucleic acid transfer and/or reduce immunogenicity, such as ASOs, immunomodulatory molecules, or ASOs with immunomodulatory functions. It was hypothesized that inhibition of cyclic GMP-AMP synthase (cGAS) might promote Brambleberry-mediated nucleic acid transfer via at least one of two potential mechanisms. It is contemplated that inhibition of cGAS may promote nucleic acid transfer and/or reduce the immunogenicity of nucleic acid transfer by opposing cytosolic DNA sensing by cGAS. Also, it is contemplated that cGAS also may directly compete with Brambleberry for DNA binding. Therefore, the ability of an ASO that inhibits cGAS (C2mutl) (PCT/AU2022/050310, and Valentin et al. (2021) NUCLEIC ACIDS RES.
49(1 1): 6082-6099) to facilitate Brambleberry-mediated nucleic acid transfer was evaluated (FIGURE 7). Indeed, in an experiment where LNPs containing 50 ng NanoLuc-expressing plasmid DNA, LNPs containing 50 ng of Brambleberry mRNA, and LNPs containing 50 ng of the C2mutl ASO were added per well of 96-well plates of differentiated C2C12 myotubes, the presence of the C2mutl ASO enhanced the efficiency of Brambleberry-mediated nucleic acid transfer by 3-to-5-fold. Therefore, inhibition of cGAS and/or competition with cGAS may enhance Brambleberry-mediated nucleic acid transfer, and/or inhibiting innate immune signaling with an immunosuppressive molecule or pro-drug form thereof such as cGAS may enhance Brambleberry-mediated nucleic acid transfer. More broadly, this experiment suggests that inhibiting the binding of a cellular protein to DNA and/or providing an immunomodulatory molecule (e.g. , an immunosuppressive molecule or pro-drug form thereof), may facilitate Brambleberry-mediated nucleic acid transfer. Likewise, this experiment demonstrates the utility of combining an ASO and/or an immunomodulatory molecule with Brambleberry mRNA to facilitate nucleic acid transfer.
[00334] Brambleberry mRNAs were produced as follows. As a template, PCR product containing the T7 promoter containing the AGG initiation sequence, 5 ’ untranslated region (UTR) from tobacco etch virus (TEV), a coding sequence for Danio rerio Brambleberry that was codon-optimized for reduced Thymidine, a 3 ’ UTR from human hemoglobin alpha 1 (HBA1), and a 100 nucleotide polyA tail were used. In vitro transcription (IVT) was set up
according to the following protocol: An IVT 10x buffer was mixed to give 400 mM Tris-HCl (pH 8.0), 100 mM DTT, 21.2 mM Spermidine, 160 mM MgCh, and 150 mM HC1. Reactions were prepared by mixing NTPs, cap analogue, nuclease free water, template, 10x buffer, and finally enzymes, to give an initial reaction mixture of: 40 mM Tris-HCl (pH 8.0), 10 mM DTT, 2.12 mM Spermidine, 16 mM MgCl2, 15 mM HC1, CTP 6 mM, ATP 6 mM, GTP 4 mM, Nl-methyl-\|/TP 4 mM, CleanCap AG 10 mM, 25 ng/pL template DNA, 1 U/pL murine RNAse inhibitor, Inorganic Pyrophosphatase 0.002 U/pL, and T7 RNA polymerase 15 U/pL. Reactions were incubated for 2 hours at 37°C after which time additional NTPs and 10x buffer were added to give final post-feed concentration of 69.6 mM Tris-HCl (pH 8.0), 17.4 mM DTT, 3.7 mM Spermidine, 27.8 mM MgCl2, 26.1 mM HC1, CTP 9.2 mM, ATP 9.2 mM, GTP 6.2 mM, Nl-methyl-\|/TP 6.2 mM, CleanCap AG 7.4 mM, 18.4 ng/pL template DNA, 0.74 U/pL murine RNAse inhibitor, Inorganic Pyrophosphatase 0.0015 U/pL, and T7 RNA polymerase 11.1 U/pL. Reactions were incubated for an additional 2 hours at 37°C.
Following IVT, l/20th volume of TURBO DNAse (Thermo Fisher) was added and reactions were incubated 15 minutes at 37°C. Following template digestion, l/10th volume of 0.5M EDTA, and 1/10th volume of Proteinase K (NEB) were added and the mixture was incubated an additional 20 minutes at 37°C. At the end of this period 1 volume of water and 3/5,h volume of LiCl precipitation solution (Thermo) were added and incubated 15 minutes at - 20°C followed by maximum speed centrifugation (21,000 x g) at 4°C and 2 washes with cold ethanol. The resulting pellet was resuspended in THE RNA Storage solution (ThermoFisher).
[00335] Double- stranded RNA was removed as follows. Residual dsRNA was removed according to the method of Baiersdorfer (Baiersdorfer et al. (2019) THER. NUCLEIC ACIDS, 15: 26-35). Up to 500 pg of IVT product was reconstituted in 400 pL in 10 mM HEPES (pH 7.2), 0.1 mM EDTA, 125 mM NaCl, and 16% (v/v) ethanol and applied to a NucleoSpin Filters (Machery-Nagel) containing 100 mg cellulose (Sigma). Columns were rotated for 30 min and the Flow through was collected by centrifugation at 14,000 x g. The flow-through was applied to a second column and similarly rotated and collected. The flow-through from the second column was collected by centrifugation and the mRNA was recovered by LiCl precipitation.
[00336] 3’ triphosphates were removed as follows. Precipitated mRNA was resuspended in 200 pL Phosphatase Buffer (50 mM Tris HC1 pH 8.5, 0. 1 mM EDTA) to which were added 5 pL (100U) CIAP (Thermofisher), and incubated at 37°C for 1 hour.
[00337] The DNAs delivered using the systems for nucleic acid transfer described herein optimally lack free 5’ and 3’ ends. DNAs lacking free 5’ and 3’ ends can be circular DNAs (e.g., plasmids or circular products of microbe-free DNA synthesis), closed-end DNAs, or the like. The DNAs for nucleic acid transfer in the experiments described herein were plasmids generated in E. coli and modified by CpG methylation as described in more detail below.
However, microbe-free systems for generating circular or closed-end DNAs are known in the art as well. For instance, methods for generating circular DNAs from linear DNAs, such as products of polymerase chain reaction (PCR) or rolling circle amplification include incubation with a DNA ligase, a topoisomerase, or a mixture of enzymes, (e.g., as used in Gibson assembly, an exonuclease such as T5 exonuclease, a DNA polymerase, and a ligase such as Taq ligase). Methods based on rolling circle amplification can include, e.g., a Phi29 DNA polymerase, one or more restriction enzymes, and a DNA ligase, such as described by Grasemann et al. (2023) BtoRxtv, https://doi.org/10.1101/2023.06.26.546530. Methods for generating closed-end DNAs have been previously described (e.g., International Patent Publication No. PCT/US2019/014122). Thus, the DNAs that are delivered in the systems for nucleic acid transfer described in the present invention can be generated by a variety of techniques, including as a plasmid or plasmid-like circular DNA in a microbe such as E. coli, or manufactured in a microbe-free system.
[00338] LNPs were formulated on an Ignite NanoAssemblr (Precision Nanosystems) with nucleic acids at a concentration of 50 pg/mL in 50 mM citrate pH 4 for the aqueous phase. Lipids consisted of DLin-KC2-DMA, DSPC, Cholesterol, and PEG-DMA at a ratio of 50: 10:38.5:1.5 in ethanol at a concentration set to give an N:P ratio of 6 with a flow rate of 3:1 aqueous to ethanol. Mixed fractions were diluted with cold PBS, and buffer exchanged 3 times using an Amicon ultra 30 kDa ultrafiltration unit.
Example 2 — A system for nucleic acid transfer for expressing eCD4-Ig and knocking down FUT8
[00339] This example describes a system for nucleic acid transfer for expressing eCD4-Ig.
The sequence of a plasmid for expressing eCD4-Ig (Gardner et al. (2015), supra,
International Patent Application NO. PCT/US2019/023422) under a CpG-free muscle- specific promoter (SEQ ID NO: 35) can be used. The sequence of a similar plasmid for expressing tyrosylprotein sulfotransferase-2 (TPST2) (U.S. Patent No. 10,626,161) under a CpG-free muscle-specific promoter (SEQ ID NO: 36) can be used. Versions of the plasmid for expressing eCD4-Ig that are further modified to express an shRNA for knocking down fucosyltransferase-8 (FUT8) by RNA interference (RNAi) (SEQ ID NO: 37), or a miR for knocking down FUT8 by RNAi (SEQ ID NO: 38), are provided. The plasmid sequences provided herein lack CpG motifs outside of the bacterial origin of replication (Ori), which is methylated at CpG sites prior to use as described herein. These plasmids are generated and co-packaged with Brambleberry-encoding mRNAs containing modified uridine nucleotides in LNPs as described herein and formulated for injection in a pharmaceutically-acceptable carrier. It is expected that the DNA encoding for eCD4-Ig will be efficiently transferred to the nucleus of cells and expressed with minimal immune response due to the expression of the FUT8 non-coding RNA.
Example 3 — A system for nucleic acid transfer for expressing an anti-PCSK9 antibody
[00340] This example describes a system for nucleic acid transfer for expressing a monoclonal antibody that binds proprotein convertase subtilisin/kexin type 9 (PCSK9). The sequences of monoclonal antibodies that bind PCSK9 are known in the art (e.g., evolocumab and alirocumab), and methods for generating monoclonal antibodies are known in the art. The sequence of a plasmid for expressing evolocumab under a CpG-free muscle-specific promoter (SEQ ID NO: 39) can be used. This is a bicistronic plasmid for co-expressing the heavy and light chains of evolocumab. The plasmid sequences provided herein lack CpG motifs outside of the bacterial origin of replication (Ori), which is methylated at CpG sites prior to use as described herein. These plasmids are generated and co-packaged with Brambleberry-encoding mRNAs containing modified uridine nucleotides in LNPs as described herein and formulated for injection in a pharmaceutically-acceptable carrier. It is expected that the DNA encoding the PCSK9 antibody will be efficiently transferred to the nucleus of cells and expressed.
Example 4 — A system for nucleic acid transfer for expressing a GLP1R agonist
[00341] This example describes a system for nucleic acid transfer for expressing a glucagon- like peptide-1 receptor (GLP1R) agonist. Glucagon-like peptide-1 (GLP-1) and variants
thereof can be expressed as a secreted protein therapeutic, e.g., by introducing the sequence for a non-native signal peptide 5’ of a sequence coding for GLP-1. The sequence of a plasmid for expressing GLP- 1 with a non-native signal peptide under a CpG-free muscle- specific promoter (SEQ ID NO: 40) can be used. The plasmid sequences provided herein lack CpG motifs outside of the bacterial origin of replication (Ori), which is methylated at CpG sites prior to use as described herein. Sequences of plasmids completely CpG-free (i.e., containing no CpG motifs), in this case due to use of a CpG-free R6K Ori e.g., as described in U.S. Patent No. 7,244,609), are provided (SEQ ID NO: 41 and SEQ ID NO: 42). These plasmids are generated and co-packaged with Brambleberry-encoding mRNAs containing modified uridine nucleotides in LNPs as described herein and formulated for injection in a pharmaceutically-acceptable carrier. It is expected that the DNA encoding GLP-1 will be efficiently transferred to the nucleus of cells and expressed.
Example 5 — Brambleberry-mediated nucleic acid transfer of a DNA expressing a non- coding RNA
[00342] This example describes the use of a plasmid expressing a miR as an off-switch for gene therapy. The sequence of a plasmid expressing a miR (SEQ ID NO: 54) and a matched 3’ UTR sequence (SEQ ID NO: 55) with multiple target sites for that miR are provided. The miR is expressed from a version of the Hl RNA polymerase III (Pol III) promoter that has been modified to lack CpG sites. Thus, the plasmid can be methylated at CpG sites without the Hl promoter driving the miR being subject to CpG methylation, and also the promoter does not provide recognition motifs for TLR9. This miR-target pair was designed to maximize the efficiency of knockdown of the gene of interest. To serve as an off-switch (or down-switch), the plasmid expressing the miR is co-delivered with Brambleberry mRNA, e.g., in LNPs, in an excess amount (in terms of both the amount of material and the volume) to the original site of administration of the system for nucleic acid transfer for expressing the therapeutic protein that is now being switched off. The original site of injection can be located, e.g., by tattooing the site or using a standardized biometric approach for injection site selection and depth. Alternatively, if the target tissue is liver, intravenous injections can be used both to first administer the vector for expressing the therapeutic gene and then later to administer the miR-expression plasmid that serves as the off-switch. It is expected that the plasmid expressing the miR and matched 3 ’ UTR sequence will reduce or eliminate the amount of expression of the therapeutic protein that was previously delivered.
Example 6 — Generation of CAR-T cells using Brambleberry and CRISPR
[00343] This example describes a system for gene editing in unstimulated T cells by codelivery of mRNA encoding Brambleberry, mRNA encoding a CRISPR-Cas9 nuclease, a guide RNA, and a DNA template. The CRISPR -Cas9 mRNA is a commercially available capped 5-methoxyuridine containing mRNA (Trilink Catalog number L-7206). The TRAC guide RNA is specific to the human T cell receptor and is synthesized with 3 terminal phosphorthioate residues on each end (SEQ ID NO: 108) (Eyquem et cd. (2017) supra). The sequence of a 4183 bp DNA donor is provided (SEQ ID NO: 109) consisting of a 609 nucleotide TRAC left homology arm, a P2A cleavage site shortly after the start codon, followed by a signal peptide, two chimeric T cell receptors specific for CD 19 and CD20 separated by an F2A protease site, a polyadenylation site, and a 181 nucleotide TRAC right homology arm. All sequence between the right and left homology arms has been depleted of CpG motifs by codon optimization. The donor is provided as an insert within a minimal pUC57 plasmid that has been methylated with Sssl CpG methyltransferase. The mRNA encoding Brambleberry, mRNA encoding a CRISPR-Cas9 nuclease, guide RNA, and DNA template are coformulated into LNP. The LNP are incubated with primary T cells. It is expected that administration of the nucleic acid transfer system will allow for the introduction of the DNA template into the nucleus by Brambleberry so that it can serve as a template for CRISPR Cas9, and the chimeric T cell receptors will be introduced into the genome of the cell.
Example 7— -Generation of Muscle-Specific Promoters (MSP) for Nucleic Acid Transfer
[00344] Double membrane fusogen proteins are especially useful for enabling efficient nucleic acid transfer to non-dividing cells. Muscle is an important target tissue for systems for nucleic acid transfer, but is comprised primarily of differentiated, non-dividing cells. Thus, the discovery of the utility of double membrane fusogen proteins such as Brambleberry in nucleic acid delivery systems enables efficient non-viral systems for nucleic acid delivery to non-dividing cells including muscle.
[00345] One advantage of a muscle-specific promoter in a system for nucleic acid transfer to muscle cells is minimizing the expression of the transgenes in transduced antigen-presenting cells (APCs), such as macrophage and dendritic cells. The amount of the therapeutic gene
product presented by a transduced APC is thought to be far greater than the amount of therapeutic gene product an APC would be able to pick up from plasma or lymph. Thus, limiting expression of the therapeutic gene product in APCs may limit the potential for immune responses targeting the therapeutic gene product. Use of a muscle-specific promoter also has the safety advantage of preventing expression in other off-target cell types, in addition to APCs.
[00346] Ten (10) muscle-specific promoters were initially screened through in vitro in C2C12 differentiated mouse myotubes, and the 3 most promising promoter constructs were compared against each other and the CMV immediate-early promoter in the context of AAV vectors expressing firefly luciferase in mouse gastrocnemius muscle. A synthetic muscle- specific promoter (MSP) (SEQ ID NO: 3), which was derived from an enhancer created by random ligation of oligos containing muscle-specific transcription factor binding sites (Li et al. (1999), supra) performed better than the Desmin (DES) promoter (SEQ ID NO: 4) (Li et al. (1991) J. BIOL. CHEM. 266(10): 6562-6570, and Fuchs et al. (2020) MOL. THER. METHODS CLIN. DEV 16: 94-102), and the Troponin-1 (TNNI1) promoter (SEQ ID NO: 5), and was only about one order of magnitude less active than the CMV promoter (SEQ ID NO: 6) (FIGURE 8A). Adding a TATA box to the MSP was tested in two positions, offset by 5 bases (i.e., rotated 180° about the double helix). Inserting a TATA box at one position (SEQ ID NO: 7) but not the other (SEQ ID NO: 8) increased firefly luciferase activity in mouse muscle by 50-fold (FIGURE 8B). It is notable that the overlapping TATA box motif TATATA was utilized here. The AAV vector containing an MSP with the optimally- positioned TATA box (SEQ ID NO: 7) expressed about 5-fold more efficiently than CASI (SEQ ID NO: 9), which is a non-tissue-specific promoter favored by the AAV field (FIGURE 8C). Optimizing the position of a TATA box yielded a promoter that is comparable in strength, at least in mouse skeletal muscle, to the most popular non-tissue- specific promoters (i.e., CMV and CASI).
[00347] To eliminate the possibility of promoter silencing by CpG methylation in transduced cells, all of the CpG motifs were removed from the muscle-specific promoter (MSP). The sequence of the resulting CpG-free MSP is provided as SEQ ID NO: 10. The sequence of the CpG-free MSP with the optimally positioned TATA box is provided as SEQ ID NO: 11. The sequence of the CpG-free MSP with the optimally positioned TATA box through the transcriptional start site (tss) is provided as SEQ ID NO: 12. Thus, the novelty and utility of
this CpG-free muscle-specific promoter are, in part, that it is a muscle-specific promoter that cannot be silenced by promoter methylation. The version of the MSP lacking CpGs (SEQ ID NO: 11) expressed at least as efficiently, if not better, than the otherwise-identical MSP containing CpGs (SEQ ID NO: 7) after electroporation into mouse gastrocnemius muscle (FIGURE 8D). The MSP lacking CpGs (SEQ ID NO: 11) was then compared to the CMV promoter (SEQ ID NO: 6), and exhibited similar efficiency as the CMV promoter after electroporation into mouse muscle (FIGURE 8E). To confirm that the MSP lacked transcriptional activity in APCs, firefly luciferase expression constructs having the CMV, CASI, MSP, and Desmin (DES) promoters were transfected into the human macrophage cell line THP-1 (FIGURE 8F). Expression of the MSP in THP-1 cells was reduced by more than 300-fold in comparison to the CMV promoter. Interestingly, the DES promoter exhibited a relatively high level of activity in differentiated THP-1 macrophage cells, and is therefore not truly muscle-specific. This may be because the DES promoter contains a precise match for the consensus sequence of an interferon-stimulated response element (IS RE) (Honda et al. (2006) IUBMB LIFE, 58 (5-6): 290-295), which may be activated by transfection with CpG- containing DNA, and may be most active in the context of innate immune responses. Thus, a truly muscle-specific promoter was developed, which is inactive in human APCs, yet is more active than CASI and has comparable activity to the CMV promoter in skeletal muscle, and cannot be silenced by promoter methylation.
[00348] Sequences are provided derivatives of similar muscle-specific promoters known in the art (e.g., Spc5-12), but with the novel refinement described herein of an optimally- positioned TATA box (SEQ ID NOs: 13-14). These sequences extended through a transcriptional start site (tss) are also provided (SEQ ID NOs: 15-16). CpG-free versions of these muscle-specific promoters are also provided (SEQ ID NOs: 17-18), and the CpG-free versions with the optimal TATA box position described herein (SEQ ID NOs: 19-20). Sequences for these CpG-free muscle-specific promoters plus the optimally-positioned TATA boxes through a transcriptional start site are provided (SEQ ID NOs: 21-22). The systems for nucleic acid transfer described herein also can utilize, e.g., the muscle-specific promoter derived from the MHCK7 gene (SEQ ID NO: 23). Therefore, the sequence of a CpG-free version of the MHCK7 promoter is provided (SEQ ID NO: 24).
[00349] Transcription factors with binding sites in these muscle-specific promoters include myocyte enhancer factor-2 (MEF2) paralogs and TEA domain transcription factor- 1
(TEADI). The consensus binding site for MEF2 is 5’-(T/G)(C/T)TATTTTT-3’ or 5’- AAAAATA(A/G)(A/C)-3’, with the core motif 5’-TATTT-3’ or 5 ’AT AAA-3’ (Ma and Telese Common Integr Biol. 2015 Sep 23 ;8(6)). The consensus binding site for TEAD1 is 5’- (A/G)CATTC(C/T)(T/A)(C/G)-3’ or 5’-(G/C)(T/A)(A/G)GAATG(C/T)-3’, with the core motif 5’-CATTC-3’ or 5’-GAATG-3’. The present invention includes systems for gene therapy that include CpG-free promoters containing the consensus binding sites for MEF2 paralogs and/or TEAD1.
[00350] The expression of secreted protein therapeutics after electroporation of plasmid DNA into muscle has been reported to wane over time. For example, after electroporation into muscle in mice of plasmid DNAs encoding monoclonal antibodies against influenza virus HA protein, or plasmid DNAs encoding eCD4-Ig, plasma concentrations of these secreted protein therapeutics waned with half-lives of about 4-20 weeks (Andrews et al. (2017) MOL. THER. METHODS CLIN. DEV. 7: 74-82, and Xu et al. (2018) EBIOMEDICINE, 35: 97-105). Tt has been suggested that the waning kinetics of protein therapeutics expressed by plasmids electroporated into muscle is due to the loss of the plasmid over time. Without wishing to be bound by theory, it is contemplated that this waning may also be due, at least in part, to promoter silencing and immune responses. The development of a muscle-specific promoter that cannot be transcriptionally silenced by CpG methylation has allowed the investigation of mechanisms of DNA persistence in non-dividing muscle cells, without promoter silencing complicating the interpretation of expression kinetics.
[00351] In addition, it has been suggested that, within the field of gene therapy, adeno- associated virus (AAV) vectors are able to persist as episomes in non-dividing cells due to uncharacterized viral mechanisms that mediate retention in the nucleus. Relying in this conventional wisdom, it was hypothesized that the inverted terminal repeats (ITRs), which recombine with each other to circularize and concatemerize AAV vector genomes and are the only viral sequences present in AAV vector genomes, adopt a structure responsible for retention in the nucleus after recombination. Therefore, a recombined AAV ITR was cloned into a standard plasmid (pUC57), in order to recreate a covalently-closed circular DNA that is identical to that which is created upon AAV vector genome circularization. It was contemplated that the recombined ITR might allow a plasmid to persist in non-dividing cells, and thus allow an ITR-containing plasmid to be developed as a platform for non- viral gene therapy. Use of the optimized CpG-free MSP (SEQ ID NOs: 10-12) (FIGURE 8) allowed
for the assessment of the persistence of gene expression from plasmid DNA in muscle without the possibility of promoter silencing by CpG methylation being a confounding factor. The MSP was the only CpG-free DNA region in these plasmids, though, and the CpG motifs present throughout the remainder of the sequences would stimulate innate immune responses through TLR9. To prevent immune responses from complicating the question of persistence, this experiment was performed in immunodeficient nude mice. Thus, through the use of a CpG-free muscle-specific promoter and immunodeficient mice, the potential contribution of promoter silencing due to CpG methylation and immune responses was eliminated from complicating the interpretation of persistence experiments. Electroporation with the plasmids with and without a recombined ITR was compared against an AAV9 vector containing the identical firefly luciferase expression cassette driven by the MSP. As an additional control, single- stranded AAV vector genomes were extracted from that same batch of AAV9 vector, and electroporated into mouse muscle at the same molar amount as the plasmid DNAs. The plasmid DNA was also electroporated without the ITRs into immune-competent mice (Balb/C). This experiment produced a negative result, but not for the reason that was expected: the ITR did not appear to make any difference in persistence. Surprisingly, the plasmid without the recombined ITR also persisted in muscle with stable kinetics over the remaining lifetime of the mice (FIGURE 9). This experiment demonstrates that a plasmid lacking any features derived from AAV is capable of persisting in non-dividing skeletal muscle cells. The lack of persistence of the plasmid in immune-competent mice, therefore, appears to be fully attributable to immune responses. Thus, in the absence of promoter silencing and immune responses, a plasmid DNA is able to persist in skeletal muscle. This finding breaks with the conventional wisdom by implying that the only feature of AAV responsible for persistence as an episome is that its genome forms a circle, and that circle is not functionally distinct from a plasmid with respect to persistence in non-dividing cells.
[00352] Importantly, this experiment demonstrates that the engineered CpG-free MSP (SEQ ID NOs: 10-12) is persistently expressed in muscle. Consistent with design, it is not silenced over time. Thus, a CpG-free promoter, e.g. , a CpG-free muscle-specific promoter, is capable of stably expressing a gene of interest over the long-term.
[00353] Thus, the present invention is based, in part, on the discovery that a plasmid with a CpG-free promoter in non-dividing cells is a system for nucleic acid transfer that is capable of persistently expressing a gene of interest for the long term, potentially for the lifetime of
the recipient. The data presented herein show that a CpG-free muscle- specific promoter is capable of expressing a gene of interest for the long term, without being silenced, and apparently without the loss of the plasmid. This observation is not limited to plasmids, as other DNAs lacking free 5’ and 3’ ends, such as circular DNAs produced in microbe-free systems, would resist degradation similarly to a plasmid. The discovery that Brambleberry enables efficient nucleic acid transfer into non-dividing cells provides a non- viral system for nucleic acid transfer, which, when combined with a CpG-free muscle-specific promoter operably linked to the gene of interest, allows persistent long-term expression of the gene of interest in muscle.
[00354] To generate data on MSP activity by electroporation, mice received 20 pg DNA in a 20 pL volume of water by injection into the gastrocnemius muscle. Needletrodes were placed in the muscle on opposite sides of the injection site. BTX ECM 830 (Harvard Apparatus). A BTX ECM 830 (Harvard Apparatus) was used to administer 7 unipolar pulses having a square wave at 40V for 100 ms/pulse.
Example 8— -Systems for Nucleic acid transfer that include CpG-Methylated DNA
[00355] In one aspect, the invention includes the use of CpG-methylated DNA as part of a system for nucleic acid transfer. Toll-like receptor 9 (TLR) is an innate immune sensor for DNA, but does not efficiently recognize CpG-methylated DNA (Hemmi et al. (2000), supra and Rutz et al. (2004), supra). Thus, it is contemplated that CpG methylation of a double- stranded DNA (e.g., a plasmid) used in a system for nucleic acid delivery provides a means of avoiding TLR9 signaling. The systems for nucleic acid delivery described here are largely depleted of CpG motifs. However, the CpG-depleted pUC57 plasmid used in the nucleic acid delivery system here has a bacterial origin of replication that contains CpG motifs that cannot be removed. Others have utilized CpG-free bacterial origins of replication such as the R6K origin of replication (e.g., U.S. Patent No. 7,244,609), which requires particular host strains of E. coli for growth. The utility of a CpG-methylated standard origin of replication is partly that plasmids containing the standard (ColEl -derived pBR322) origin of replication grow to higher yields than those based on CpG-free origins of replication. For instance, the inventors observed that a largely CpG-free plasmid with a pBR322 origin of replication grows to approximately 5x the yield of the same plasmid containing a CpG-free R6K-derived origin of replication. CpG-methylation allows a CpG-containing bacterial origin of replication to be used in a system for nucleic acid transfer while minimizing recognition by TLR9.
[00356] CpG-methylated plasmids also provide a means of gene regulation. CpG- methylation also has the utility of being a means of regulating or silencing genes or regions of the DNA that is introduced with a system for nucleic acid transfer. CpG-methylation of a promoter provides a means of regulating the kinetics of its expression. The activity of a promoter that contains CpG motifs can be reduced by methylation prior to formulating the DNA within a system for nucleic acid delivery. To avoid silencing of the gene of interest, a DNA that is included in a system for nucleic acid transfer can combine a promoter that lacks CpG motifs with a plasmid DNA that is CpG-methylated.
[00357] CpG-methylated plasmid DNA was prepared as follows. CpG depleted plasmids were transformed into dcm-/dam- cells (NEB) in order to manufacture DNA that is free of 6- methyladenine and 5-methylcytosine. Single colonies were selected and grown in LB for preparation with Machery-Nagel Endotoxin Free prep as per the manufacturer’s instructions. 400 ug of freshly prepared plasmid were enzymatically methylated using Sssl methyltransferase (NEB). The reaction was performed in 300 u L NEB buffer 2 supplemented with 160 pM SAM and with 200 U of methyltransferase. After 2 hours the reaction volume was doubled with 300 L NEB2 with 160 pM SAM, but no additional enzyme. After 2 more hours, DNA was precipitated with isopropanol and resuspended in TE buffer. Complete methylation was confirmed by co-digestion with the restriction endonucleases Hhal (which is blocked by CpG methylation) and EcoRI, in comparison to digestion with EcoRI alone (FIGURE 10). Unmethylated DNA was used as a control. All control plasmid was digested into 2 large fragments by co-digestion, whereas methylated plasmid was linearized by EcoRI but protected from Hhal fragmentation. This restriction enzyme digest shows that the plasmid DNA was efficiently methylated using the methods described above.
[00358] CpG methylation may be advantageous for systems for nucleic acid transfer. An experiment was conducted comparing firefly luciferase expression from the CpG-free MSP after electroporation of plasmids with or without methylated CpGs into Balb/C mouse gastrocnemius muscle (FIGURE 11). Four plasmids were assessed. The plasmid backbones lacked CpG sites, except for in the bacterial origin of replication (Ori). The first plasmid had a firefly luciferase gene based on standard codon optimization for expression in mammalian cells where CpG motifs are present, an Ori containing CpGs, and was not CpG-methylated prior to administration. The second plasmid had a firefly luciferase gene lacking CpG motifs,
an Ori containing CpGs, and was not CpG-methylated prior to administration. The third plasmid had a firefly luciferase gene lacking CpG motifs, an Ori containing CpGs, and was CpG-methylated prior to administration. The fourth plasmid had a firefly luciferase gene lacking CpG motifs, an R6K Ori lacking CpGs, was devoid of CpG motifs, and was not CpG- methylated prior to administration. Interestingly, there was a trend towards the highest firefly luciferase expression in the group where the CpG motifs in the bacterial origin of replication, which were the only CpG motifs present in the plasmid, were methylated. Therefore, CpG methylation in certain contexts, e.g., in a bacterial origin of replication, may be a desirable in systems for nucleic acid transfer.
Example 9 — LNPs and other systems for gene transfer that include one or more attachment factors
[00359] Different attachment factors were evaluated for their ability to enhance gene transfer into muscle cells and thus synergize with Brambleberry mRNA expression to promote gene transfer. This experiment was performed in differentiated C2C12 muscle cells, 10 days post- differentiation. The control used in this experiment was LNPs generated using the same components except for the lipid-conjugated attachment factor. The lipid-conjugated attachment factors evaluated were: (1) stearylated M12 peptide with the amino acid sequence RRQPPRSISSHP (SEQ ID NO: 111) (StM12), (2) a first stearylated LAM1 peptide with the amino acid sequence YIGSR (SEQ ID NO: 112), (3) a second stearylated LAM1 peptide with the amino acid sequence RYVVLPR (SEQ ID NO: 113), (4) a stearylated hyaluron-binding peptide with the amino acid sequence GAHWQFNALTVR (SEQ ID NO: 114), (5) an M12 peptide conjugated to a serine-octanoic acid as a second lipid in addition to being stearylated (SsoM12), (6) a DOPE-conjugated RGD 3 amino acid peptide (DOPE-RGD), a cholesterol- conjugated glucose molecule in the form of cholesterol-undeconoate-glucose (chol-glu), and DPG-conjugated galloyl (DPG-GAL). In general, including a lipid-conjugated attachment factor indeed promoted gene transfer by the LNPs (FIGURE 12). The first stearylated LAM1 peptide increased gene transfer 3 -fold. The second stearylated LAM1 peptide, which is through to bind heparin sulfate, increased gene transfer almost 2-fold. The hyaluron-binding peptide increased gene transfer 3 -fold. Including the serine-octanoic acid as a second lipid in addition to stearylation of M12 increased gene transfer by approximately 50%. DOPE-RGD improved gene transfer by 37%. The glucose conjugate (cholesterol-undecanoate-glucose) improved gene transfer by 4-fold. Galloyl-conjugated DPG increased gene transfer efficiency
by the greatest extent (8.7-fold). The molecular structures of DOPE-RGD, cholesterol- undecanoate-glucose, and galloyl-DPG are provided (FIGURE 13). This experiment showed that attachment factors, e.g., attachment factors conjugated to lipids and included in the LNP, synergize with Brambleberry mRNA to promote gene transfer.
[00360] Lipid-conjugated metabolites such as lipid-conjugated glucose may be particularly useful for increasing the efficiency of LNPs and/or enhancing the efficiency of systems for gene transfer. Metabolites such as glucose have the advantage of being small, potentially allowing a high number of molecules of the metabolite to be included on the LNP. Also, their small size and presence in natural contexts reduces the potential of such metabolites to be targeted by an antibody response, thus improving the potential for re-dosing with LNPs containing the same attachment factor.
[00361] Polyphenol-conjugated lipids, such as galloyl-conjugated DPG, probably function as attachment factors to promote gene transfer. However, without the intention of being limited by a particular theory, polyphenols may have a second role in modulating the stability of the target cell membrane and/or the LNP. Thus, polyphenol-conjugated lipids may be useful in enhancing the efficiency of LNP-based systems in general, including synergizing with Brambleberry mRNA to improve the efficiency of systems for gene transfer.
Example 10 — Systems for gene transfer that include intracellular transport ligands
[00362] Dynein binders were evaluated for their ability to promote gene transfer by promoting the trafficking of nucleic acids carried by LNPs into the cell. The dynein binders were dynein-binding peptides that interact with Tctex-1 (DYNLT-1), a dynein light chain protein. The two peptides are from Arl2 (GGFKLNIWDVGGQK) (SEQ ID NO: 115), and the C-terminus of rhodopsin D (Rhd) (GVSKTETSQVAPA) (SEQ ID NO: 1 16). These were stearylated and conjugated to octyl-arginine (i.e., as in SEQ ID NOs: 117-118). The dynein binders were stearylated to facilitate their incorporation into the LNPs. Octyl-arginine was included as a DNA binder, which is cationic at intracellular pH (pH 7.4), and thus binds DNA through a charge interaction. The LNPs in this experiment carried DNA encoding NanoLuc luciferase under a muscle-specific promoter and mRNA encoding Danio rerio Brambleberry at a 1:2 ratio by weight. The controls included were LNPs formulated without any stearylated arginine or dynein binder, or with only stearylated octyl-arginine. The stearylated octyl- arginine-Arl2 peptide, which is a dynein binder that binds Tctex-1 (DYNLT-1), increased
gene transfer efficiency by 3.4-fold (FIGURE 14). The stearylated octyl-arginine-rhodopsin D peptide, which is a dynein binder that binds Tctex-1 (DYNLT-1), increased gene transfer efficiency by 10-fold (FIGURE 14). Thus, the inclusion of transport ligands, e.g., dynein binders targeting dynein proteins such as Tctex-1 (DYNLT-1), promote gene transfer efficiency and synergize with Brambleberry mRNA in doing so. It is contemplated that the inclusion of a molecule containing both a DNA binder and a dynein binder may facilitate the intracellular trafficking of the DNA to a perinuclear localization that promotes gene transfer.
Example 11 - Brambleberry Orthologs
[00363] Certain Brambleberry orthologs can exhibit superior gene transfer efficiency to zebrafish (Danio rerio) Brambleberry in mammalian cells. To evaluate whether orthologs of Danio rerio Brambleberry might exhibit superior gene transfer efficiency to Danio rerio Brambleberry in mammalian cells, we compared the efficiency with which the Brambleberry mRNAs from zebrafish (Danio rerio) (SEQ ID NOs: 1-2), sheepshead pupfish (Cyprinodon variegatus) (SEQ ID NOs: 33-34), and Komodo dragon (Varanus komodoensis) (SEQ ID NOs: 29-30) mediate gene transfer when co-encapsulated into LNPs with a luciferase DNA in 5-day differentiated C2C12 myotubes (FIGURE 15A). Indeed, Cyprinodon variegatus and Varanus komodoensis Brambleberry both were functional for facilitating gene transfer in mammalian cells. Next, the activity of Danio rerio Brambleberry was compared against its orthologs from pigeon (Columba livia) (SEQ ID NOs: 27-28), sheepshead pupfish (Cyprinodon variegatus) (SEQ ID NOs: 33-34), and Hawaiian crow (Corvus hawaiiensis) (SEQ ID NOs: 119-120) (FIGURE 15B). Among this set, the Brambleberry ortholog with the highest gene transfer efficiency was that from Cyprinodon variegatus and the second highest was that from Columba livia. In a third experiment, Danio rerio Brambleberry was compared against Columba livia Brambleberry and a version of Columba livia Brambleberry with a non-native signal peptide (NNSP) (FIGURE 15C) for their ability to facilitate gene transfer in differentiated C2C12 myotubes. This experiment demonstrated that Columba livia Brambleberry was functional with either the native or non-native signal peptide. Thus, this experiment demonstrates that Brambleberry orthologs from species other than Danio rerio can exhibit superior gene transfer efficiency to Danio rerio Brambleberry in mammalian cells.
[00364] Sequences of exemplary Brambleberry orthologs and mRNAs for expressing them are provided herein. The Brambleberry ortholog sequences provided are from zebrafish
{Danio rerio) (SEQ ID NOs: 1-2), golden eagle Aquila chrysaetos chrysaetos) (SEQ ID NOs: 25-26), pigeon Columba livia) (SEQ ID NOs: 27-28), Komodo dragon {Varanus komodoensis) ) (SEQ ID NOs: 29-30), Goode’s thomscrub tortoise {Gopherus evgoodei) (SEQ ID NOs: 31-32), Sheepshead pupfish {Cyprinodon variegatus) (SEQ ID NOs: 33-34), Hawaiian crow {Corvus hawaiiensis) (SEQ ID NOs: 119-120), swan {Cygnus olor) (SEQ ID NOs: 121-122), barn owl {Tyto alba) (SEQ ID NOs: 123-124), hummingbird {Calypte anna) (SEQ ID NOs: 125-126), crested ibis {Nipponia nippon) (SEQ ID NOs: 127-128), bald eagle {Haliaeetus leucocephalus) (SEQ ID NOs: 129-130), sparrow {Passer domesticus) (SEQ ID NOs: 131-132), swift {Apus apus) (SEQ ID NOs: 133-134), alligator {Alligator mississippiensis) (SEQ ID NOs: 135-136), Sinaloan desert tortoise {Gopherus evgoodei) (SEQ ID NOs: 137-138), African clawed frog {Xenopus laevis) (SEQ ID NOs: 139-140), Nile tilapia {Oreochromis niloticus) (SEQ ID NOs: 141-142), lamprey {Petromyzon marinus), (SEQ ID NOs: 143-144), hydrothermal vent snail {Gigantopelta aegis) (SEQ ID NOs: 145- 146), and glass sponge {Oopsacas minuta) (SEQ ID NOs: 147-148).
Example 12 - Increasing the completeness of differentiation increases the relative impact of Brambleberry
[00365] To minimize the potential for small numbers of undifferentiated C2C12 cells to be present in differentiated C2C12 cultures we extended the length of time after differentiation before use in gene transfer assays from 5 days to 10 days post-differentiation. C2C12 myoblasts were differentiated into myotubes by culturing in media containing 2% horse serum instead of 20% FBS. DNA-mRNA LNPs where the DNA expressed firefly luciferase and the mRNA expressed Danio rerio Brambleberry were added to C2C12 myotube cultures 10 days post-differentiation. Use of C2C12 myotube cultures 10 days post-differentiation rather than 5 days post-differentiation extended the impact of Brambleberry mRNA on gene transfer efficiency to 472-fold over a control with DNA-only LNPs without the Brambleberry mRNA (FIGURE 16). Minimizing the presence of dividing cells by culturing in differentiation media for 10 instead of 5 days increased the apparent impact of Brambleberry on the efficiency of gene transfer. These data confirm that Brambleberry facilitates gene transfer in non-dividing cells.
Example 13 - DNA-mRNA LNPs made with different ionizable lipids
[00366] The ionizable lipid used in previous experiments had been DLin-KC2-DMA. Therefore, several different ionizable lipids were screened as potential alternatives to DLin- KC2-DMA. The lipids included were a DLin-KC2-DMA control, SMI 02, Lipid 29, and CL15F6. Due to the potential for freezing conditions to differently affect LNPs comprised of different lipids, LNPs made with the different lipids were evaluated after storage at 4°C, - 20°C, and -80°C. An unfiltered LNP control was included to monitor loss during filtration that can occur with excessive particle size or polydispersity.
[00367] DNA-mRNA LNPs were formulated as follows. DNA and mRNA were included at a 1 :2 ratio by mass. The mRNA was made using N1 -methylpseudouridine rather than uridine. Lipids were premixed at ratios by mass of 56.9% ionizable lipid, 27.5% cholesterol, 11.2% DSPC, 1.5% DMG-PEG2000, 0.25% stearylated TCTEX-1 binder StR8RhdCT (SEQ ID NO: 118), and 4.3% DPG-galloyl. 2.4 mL of aqueous phase (DNA + RNA + 0.2 M citrate buffer + water) was mixed with 800 pL of lipid phase in a Ignite Nanoassemblr instrument. 0.45 mL of the head and 0.05 mL of the tail were discarded. The middle portion of the mix (~2.7 mL) was used as the sample. Samples were immediately quenched with 0.5X TBS up to 14 mL and centrifuged in an Amicon 15 (30 kDa cutoff) centrifugal filter units at 4000 x g until the volume in the Amicon tubes was 500 mL (—50 minutes). The volume in the Amicon was again brought up to 14 mL with 0.5X TBS and the tubes were centrifuged at 4000 g until the volume in the Amicon tubes went down to 500 mL. The liquid was then transferred to an Amicon 4 (10 kDa) centrifugal filter units and centrifuged until about 100 uL volume remained. 100 ul of 0.5X TBS + 20% sucrose was added to the 100 ul of sample. Particle sizes were assessed by DLS for unfiltered material, filtered material stored at 4°C, filtered material stored at -20°C, and filtered material stored at -80°C (FIGURE 17). The only condition where particle aggregation was observed was the CL15F6 LNPs stored at -20°C where the average particle size determined by DLS was 328 nm. However, under all of the other conditions tested, LNP sizes were in the desired range between 50 nm and 150 nm.
[00368] These batches of LNPs were tested for gene transfer efficiency in 10-day differentiated C2C12 myotube cultures. Among the screened lipids, CL15F6 improved Brambleberry-mediated gene transfer efficiency of C2C12 myotubes in tissue culture relative to DLin-KC2-DMA when stored at -4°C or -80°C (FIGURE 18A-D). The improvement for CL15F6 over DLin-KC2-DMA was approximately 3-fold. Based on the results of the
experiment, it is contemplated that CL15F6 is an ionizable lipid compatible with use in DNA-mRNA LNPs in muscle cells.
[00369] Features of the lipids evaluated included an ionizable head group with a pKa > 6.5. In contrast to DLin-KC2-DMA, the new lipids evaluated (SM102, Lipid 29, and CL15F6), contained branched acyl chains and ester bonds hydrolysable by cellular esterases, but lacked unsaturated acyl chains (FIGURE 18E-H). Without the intention of being limited by any particular theory, features of an ionizable lipid including a pKa above 6.5, branched acyl chains, hydrolysable ester bonds, and a minimal or no unsaturated bonds may have contributed to the improved gene transfer efficiency in muscle cells observed with CL15F6.
Example 14 - DNA-mRNA LNPs made with PEG with short lipid anchors
[00370] Forms of polyethylene glycol (PEG) with shorter lipid anchors than DMG-PEG were optimized and evaluated. Without the intention of being limited by any particular theory, lipid anchors of reduced size relative to DMG may increase the kinetics with which the PEGylated lipid diffuses out of the LNP, thereby improving contact with cells and the efficiency of gene transfer. Forms of PEG2000 with a cholesterol, stearic acid (SA), or C8 ceramide (C8C) anchor were compared against DMG-PEG2000 (FIGURE 19). DNA-mRNA LNPs were generated using a mixture of lipids containing either 1 X ( 1 .4%) or 2X (2.8%) of the PEGylated lipid. Cholesterol was included at 27.5% for the IX PEG or 26.1% for the 2X PEG LNPs. In other words, the increased PEGylated lipid was subtracted from the amount of cholesterol added. The other lipid components were 56.9% CL15F6, 11.2% DSPC, and 3% DPG-galloyl. The mRNA was made with N 1 -methylpseudouridine instead of uridine. Among the DNA-mRNA LNPs generated, certain conditions were evaluated for gene transfer efficiency in 10-day differentiated C2C12 myotubes using firefly luciferase as a reporter gene. Use of PEGylated lipids with short lipid anchors substantially improved gene transfer efficiency over DMG-PEG2000. The two most efficient LNP preps had 1.4% C8C-PEG2000 or 2.8% cholesterol-PEG2000. Among these, 1 .4% C8C-PEG2000 exhibited superior gene transfer efficiency to 2.8% cholesterol-PEG2000. This experiment indicated that relatively short lipid anchors, e.g., C8C or cholesterol, can improve the efficiency of gene transfer by DNA-mRNA LNPs in muscle cells.
[00371] LNPs containing DNA were generated with and without Brambleberry mRNA, using the ionizable lipids DLin-KC2-DMA or CL15F6, and with either DMG-PEG2000 or
C8C-PEG2000. Among these, certain preparations of LNPs were evaluated for gene transfer efficiency in 10-day differentiated C2C12 myotubes. The amount of LNPs added to the cells was normalized to the amount of DNA, and the DNA-mRNA LNPs were generated using a 1 :2 ratio of DNA to Brambleberry mRNA. The LNPs were exchanged into TBS buffer containing 20% sucrose using centrifugal filter units. Among the conditions where the LNPs were cryopreserved at -80°C, DNA-mRNA LNPs containing Brambleberry mRNA was 251- fold more efficient than the DNA-only LNPs, where both were made using DLin-KC2-DMA and DMG-PEG2000 (FIGURE 20). In the context of LNPs made with CL15F6 and DMG- PEG2000 cryopreserved at -80°C, the presence of Brambleberry mRNA increased the efficiency of gene transfer by 324-fold. For LNPs made with CL15F6 and C8C-PEG2000 that were cryopreserved at -80°C, the presence of Brambleberry increased the efficiency of gene transfer by 234-fold. These results clearly demonstrate that the inclusion of Brambleberry mRNA in LNPs alongside a DNA greatly enhances the efficiency of gene transfer into non-dividing cells. Furthermore, these results show that the enhancement of gene transfer afforded by Brambleberry mRNA extends to LNPs made with different lipids, e.g.. different ionizable lipids and/or different forms of PEG. Relatedly, these results show that forms of PEG with short lipid anchors e.g., C8C, which is a shorter lipid anchor than DMG) may facilitate gene transfer by DNA-mRNA LNPs into non-dividing cells such as muscle cells.
Example 15 - Use of Brambleberry mRNA to enhance gene transfer efficiency in vivo
[00372] The enhancement of gene transfer in muscle cells (a non-dividing cell type) afforded by Brambleberry mRNA was demonstrated in vivo. The DNA construct used in these experiments is a circular DNA that expresses a CpG-free Firefly luciferase gene from a CpG-free muscle-specific promoter (MSP) (SEQ ID NO: 12). The use of the muscle-specific promoter in this experiment ensured that the firefly luciferase signal that was detected after intramuscular injection owed to firefly luciferase expression by muscle cells, which are non- dividing. N1 -methylpsudouridine was used for the production of mRNA instead of uridine. DNA-mRNA LNPs were generated as described above using a mixture of lipids containing 56.9% CL15F6, 27.5% cholesterol, 11.2% DSPC, 3% DPG-galloyl, 0.32% of a cholesterol- conjugated TCTEX-1 binder chol-R8RhdCT (SEQ ID NO: 118), and 1.4% C8C-PEG2000. The circular DNA molecule and the mRNA were included at a ratio of 1 :2 by mass. LNPs
were assembled using a NanoAssemblr Ignite from Precision Nanosystems. The LNPs were exchanged into TBS buffer containing 20% sucrose using centrifugal filter units.
[00373] In addition to evaluating the enhancement of gene transfer efficiency afforded by Brambleberry mRNA in non-dividing cells, this experiment also was designed to evaluate whether different 5’ cap structures on the Brambleberry mRNA were favorable for the efficiency of gene transfer by DNA-mRNA LNPs. Brambleberry mRNA was synthesized containing either m7G-ppp-Nm (Capl) or m7G-ppp-Nm-Nm (Cap2) 5’ caps. The same identical Brambleberry sequence from Danio rerio (SEQ ID NOs: 1-2) was used in the context of Capl and Cap2 mRNAs.
[00374] LNPs containing a circular DNA encoding a firefly luciferase transgene under the muscle-specific promoter (SEQ ID NO: 12) and either no Brambleberry mRNA, Brambleberry mRNA with Capl, or Brambleberry mRNA with Cap2 were injected into the quadriceps of groups of n=5 female athymic nude mice in amounts normalized to 10 pg DNA per mouse on week 0. The mice were imaged using an IVIS bioluminescence imager on weeks 1, 2, 3, 4, 5, and 6 (FIGURE 21 ). The luciferase signal from each mouse at each time point was quantified, and the average luciferase signals of the groups of mice with no Brambleberry mRNA, Capl Brambleberry mRNA, and Cap2 Brambleberry mRNA were compared (FIGURE 21B). The luciferase signal in the groups that received the LNPs that included Brambleberry mRNA was consistently higher than the luciferase signal in the control group that received the LNPs without Brambleberry mRNA. At week 6, the group that received the LNPs containing Brambleberry mRNA with Capl had an average luciferase signal that was 11 -fold higher than that of the control group, and the group that received the LNPs containing Brambleberry mRNA with Cap2 had an average luciferase signal that was 10-fold higher than that of the control group. To evaluate the significance of this difference, a 2-tailed t test was performed for the area under the curve (AUC) of luciferase signal that was detected over the course of the experiment (FIGURE 21C). Brambleberry mRNA with Capl significantly improved the efficiency of gene transfer relative to the control DNA LNPs with no Brambleberry mRNA (P=0.01). Likewise, Brambleberry mRNA with Cap2 also significantly improved the efficiency of gene transfer relative to the control DNA LNPs (P=0.005). Thus, this experiment demonstrated the effect of Brambleberry mRNA on facilitating gene transfer in a non-dividing cell type in vivo. This experiment also demonstrated that Cap2 could be used on the Brambleberry mRNA without any significant
loss of gene transfer efficiency. Surprisingly, the kinetics of gene transfer appeared to be delayed by the use of Cap2 rather than Capl. These observations in mice validate in vivo the effect of Brambleberry mRNA that was observed in C2C12 myotube tissue culture.
[00375] Inclusion of the cholesterol-conjugated TCTEX-1 binder, chol-R8RhdCT (SEQ ID NO: 1 18), markedly improved gene transfer efficiency by DNA-mRNA LNPs in vivo (i.e., in mice). In parallel with the demonstration of the effect of Brambleberry in vivo using Brambleberry mRNA with either Capl or Cap2 (FIGURE 21A-B), another group of n=5 mice was included that received DNA-mRNA LNPs with Brambleberry mRNA with Capl but without the TCTEX-1 binder, chol-R8RhdCT (SEQ ID NO: 118). The comparison of the groups with and without chol-R8RhdCT demonstrate that the chol-R8RhdCT TCTEX-1 binder markedly improved the efficiency of gene transfer in muscle in vivo (FIGURE 21D). Thus, the inclusion of a dynein-binder markedly improved the efficiency of gene transfer in muscle cells in vivo. This particular dynein binder comprised a TCTEX-1 binder (SEQ ID NO: 1 16) conjugated to 8 arginine residues, which were the nucleic acid binder (e.g., an DNA binder and/or an RNA binder) component. Together, the 8-arginine nucleic acid binder and the TCTEX-1 binder used here are SEQ ID NO: 118. The 8-arginine motif was designed to interact with nucleic acid through a charge interaction in the cytoplasm (i.e., at neutral pH, pH 7.4). In this particular instance, the dynein binder, which is conjugated to a nucleic acid binder, also was conjugated to a lipid (e.g., cholesterol in FIGURE 21, and stearic acid in FIGURE 14). The dynein binder also can be conjugated to other types of molecules known in the art, such as polymers. This comparison demonstrated the marked improvement in the efficiency of gene transfer to muscle in vivo when a dynein binder is included in the system for gene transfer.
[00376] A follow-up experiment designed to evaluate the effect of Brambleberry mRNA on the efficiency of gene transfer was performed, this time with a lipidated attachment factor included on the LNP. DNA-mRNA LNPs were generated essentially as described above and included a Danio rerio Brambleberry mRNA with Ca l along with a firefly luciferase- encoding circular DNA under a muscle-specific promoter (SEQ ID NO: 12), and control DNA LNPs were generated with the same firefly luciferase-encoding circular DNA but no Brambleberry mRNA. Cholesterol-undecanoate-glucose (which has an arbutin moiety as the attachment factor) was included as 3.6% of the lipid mixture by mass. The other lipids included were CLF15F6 (48.3%), cholesterol (27.2%), DSPC (16.2%), C8C-PEG2000
(1.4%), 3% DPG-galloyl, 0.3% stearylated TCTEX-1 binder StR8RhdCT (SEQ ID NO: 118). The LNPs were exchanged into TBS buffer containing 20% sucrose using centrifugal filter units. DNA-mRNA LNPs or DNA LNPs were injected into the quadriceps of groups of n=5 female athymic nude mice in amounts normalized to 10 pg DNA per mouse on week 0. The mice were imaged using an IVIS bioluminescence imager on weeks 1, 2, 3, 4, 5, and 6 (FIGURE 22A). The luciferase signal from each mouse at each time point was quantified, and the average luciferase signals of the groups of mice with and without Brambleberry mRNA were compared (FIGURE 22B). At the week 6 time point, firefly luciferase expression was an average of 7.5 higher when Brambleberry mRNA was included in the LNPs than when the Brambleberry mRNA was omitted. This experiment confirmed the result that Brambleberry mRNA improves the efficiency of gene transfer in non-dividing cells (<?.g., muscle cells) in vivo.
Example 16 - Coformulation of DNA-mRNA LNPs with immunosuppressive molecules
[00377] Several mouse experiments were performed to assess whether coformulation (e.g., coencapsulation) of immunosuppressive molecules or pro-drug forms thereof with DNA- mRNA LNPs may improve the efficiency of gene transfer.
[00378] In particular, small molecule inhibitors (or pro-drug forms thereof) were selected that have LogP values consistent with efficient sorting into the LNP during encapsulation, e.g., having LogP values greater than 3.0, 3.5, or 4.0. For instance, a pro-drug form of the immunosuppressive molecule dexamethasone (dexamethasone-palmitate, which has a LogP value of 9.8) was evaluated due to its greater propensity to sort to the lipid phase during LNP formulation than dexamethasone itself, since dexamethasone itself has a LogP value of 1.83. Dexamethasone-palmitate is a pro-drug that contains an ester bond that is hydrolyzed by cellular esterases to release the immunosuppressive molecule dexamethasone. Thus, a modified form of an immunosuppressive molecule, where the modification increases the LogP value to greater than 3.0, 3.5, or 4.0, provides a generalizable strategy to facilitate the sorting of an immunosuppressive molecule into the LNP. Dexamethasone-palmitate provides an example of a pro-drug that is a modified form of an immunosuppressive molecule, where the pro-drug has a higher LogP value than the drug itself thus facilitating coencapsulation with DNA and mRNA in an LNP. In addition, the pro-drug exhibits extended release kinetics thus improving the amount of time it is present in cells that took up LNPs. Therefore, pro-drugs can have two desirable features for coformulation with LNPs. First, an increased
LogP value (e.g., at least 3.0, 3.5 or 4.0) facilitates coencapsulation in LNP. Second, the pro- drug from can have increased longevity in cells that have taken up LNPs relative to the longevity of the unmodified form of the molecule. Another feature exemplified by dexamethasone-palmitate is the presence of a chemical bond (e.g., an ester bond), which when cleaved by a cellular enzyme such as an esterase, releases the biologically-active or bioavailable form of the pro-drug. The example of dexamethasome-palmitate provided herein exemplifies these features.
[00379] In a first mouse experiment designed to evaluate the effect of including immunosuppressive molecules, DNA-mRNA LNPs were formulated with dexamethasone- palmitate, INF39, or glyburide. The LogP values of dexamethasone-palmitate, INF39, and glyburide are 9.8, 3.6, and 4.8, respectively. INF39 and glyburide inhibit the NLRP3 inflammasome, which was hypothesized to be activated by the endosomal escape mechanism through which LNPs deliver their contents to the cytoplasm. DNA-mRNA LNPs were generated in which the DNA was a largely CpG-free DNA that expresses firefly luciferase from a muscle-specific promoter (SEQ ID NO: 149). The mRNA encoded Danio rerio Brambleberry. DNA and mRNA were coencapsulated at a 1:2 ratio by mass. All of the LNPs, including the control LNPs, contained the same DNA and mRNA. The control LNPs were made using 56.9% CL15F6, 27.2% cholesterol, 11.2% DSPC, 1.4% C8C-PEG2000, 0.32% of a cholesterol-conjugated TCTEX-1 binder chol-R8RhdCT (SEQ ID NO: 118), and 3% DPG-galloyl by mass. For the DNA-mRNA LNPs that included dexamethasone-palmitate, INF39, or glyburide, 2.7% of the total lipids were subtracted from the cholesterol component and replaced with the immunosuppressive molecule or pro-drug form thereof. In other words, the cholesterol percentage was reduced from 27.2% to 24.5% and the immunosuppressive molecule or pro-drug form thereof was included as 2.7% of the total mass of lipids. The LNPs were exchanged into TBS buffer containing 20% sucrose using centrifugal filter units. 25 pL volumes containing an amount of DNA-mRNA LNPs containing 10 pg DNA and 20 pg mRNA were injected into the quadriceps muscles. Groups of n=5 nude immunodeficient female mice received either the control DNA-mRNA LNPs or DNA-mRNA LNPs coformulated with dexamethasone-palmitate, INF39, or glyburide. Dexamethasone-palmitate, INF39, and glyburide each markedly improved the efficiency of gene transfer by intramuscular injection of DNA-mRNA LNPs (FIGURE 23A). Thus, this experiment demonstrated that coformulation (e.g., coencapsulation) of an immunosuppressive molecule
(e.g., INF39 or glyburide) or pro-drug form thereof (e.g., dexamethasone-palmitate) improved the efficiency of gene transfer by DNA-mRNA LNPs.
[00380] A similar experiment was performed to confirm and extend the observation that coencapsulation of NLRP3 inflammasome inhibitors can facilitate gene transfer by DNA- mRNA LNPs. As described above, DNA-mRNA LNPs were generated in which the DNA was a largely CpG-free DNA that expresses firefly luciferase from a muscle-specific promoter (SEQ ID NO: 149). The mRNA encoded Danio rerio Brambleberry. DNA and mRNA were coencapsulated at a 1:2 ratio by mass. All of the LNPs, including the control LNPs, contained the same DNA and mRNA. The control LNPs were made using 56.9% CL15F6, 27.2% cholesterol, 11.2% DSPC, 1.4% C8C-PEG2000, 0.32% of a cholesteroL conjugated TCTEX-1 binder chol-R8RhdCT (SEQ ID NO: 118), and 3% DPG-galloyl by mass. For the DNA-mRNA LNPs that included an immunosuppressive molecule or pro-drug form thereof, 2.7% of the total lipids were subtracted from the cholesterol component and replaced with the immunosuppressive molecule or pro-drug form thereof. In this experiment, the NLRP3 inhibitor oridonin was compared against glyburide, when each were provided in combination with dexamethasone-palmitate. Dexamethasone-palmitate alone was included in one group as a control. Nude mice received 25 pL intramuscular injections of LNPs containing 10 pg DNA and 20 pg Brambleberry mRNA in the quadriceps. Luciferase activity was measured at weekly time points using an IVIS for 5 weeks (FIGURE 23B). Both oridonin and glyburide were observed to improve the efficiency of gene transfer and luciferase expression in comparison to dexamethasone-palmitate alone. Therefore, inclusion of inhibitors of the NLRP3 inflammasome can facilitate gene transfer by DNA-mRNA LNPs. Taken together, the improvement in the efficiency of gene transfer afforded by INF39, glyburide, and oridonin provide pharmacological validation of the approach of inhibiting the NRLP3 inflammasome as part of a strategy to facilitate gene transfer. Furthermore, a corticosteroid (e.g., a glucocorticoid such as dexamethasone-palmitate) in combination with an NLRP3 inflammasome inhibitor (e.g., INF39, glyburide, or oridonin), can promote the efficiency of gene transfer by DNA-mRNA LNPs.
[00381] NLRP3 inflammasome inhibitors known in the art include oridonin, INF39, glyburide, 5-chloro-2-methoxy-N-[2-(4- sulfamoylphenyl)-ethyl]-benzamide (e.g., 16673-34- 0), JC124, FC11A-2, parthenolide, VX-740, VX-765, BAY 11-7082, BHB, MCC950, MNS,
CY-09, Tranilast, and OLT1177. Such NLRP3 inhibitors, particularly those with LogP values of at least 3.0, 3.5, or 4.0, can be coencapuslated within LNPs to promote gene transfer.
[00382] The NLRP3 inflammasome may reside within an innate immune signaling pathway responsible, in part, for the immunogenicity of LNPs. The sensors upstream of the NLRP3 inflammasome in that innate immune pathway are thought to include galectins and cathepsins. Therefore, our results indicating that coformulation with immunosuppressive molecules that are inhibitors of the NLRP3 inflammasome improve the efficiency of gene transfer by DNA-mRNA LNPs also indicate that galectin inhibitors and/or cathepsin inhibitors can be used to improve the efficiency gene transfer by DNA-mRNA LNPs. Galectin inhibitors known in the art include GB1107, galectin-3/galectin-8-IN-l, olitigaltin, selvigaltin, galectin-3/galectin-8-IN-2, galectin- 3 -IN-2, galectin-3-IN-3, galectin-3-IN-5, GB1490, GB1908, galectin-8-IN-l, galectin-8-IN-2, galectin- 8N-IN-1, galectin-8N-IN-2, thiodigalactoside. Cathepsin inhibitors known in the art include L-IN-2 (Z-Phe-Phe-FMK). Cathepsins belong to the broader group of proteases known as cysteine proteases. Thus, broad cysteine protease inhibitors that inhibit cathepsins also can be used to inhibit cysteine protease activation upstream of NLRP3. Cysteine protease inhibitors known in the art include Disulfiram, Belizatinib, Cystatin B, Cystatin C, E-64 and E-64d.
[00383] To improve upon the increased gene transfer efficiency afforded by coencapsulation of dexamethasone-palmitate within DNA-mRNA LNPs, fluticasone-furoate was compared against dexamethasone-palmitate. DNA-mRNA LNPs were formulated as described above, including either 2.7% dexamethasone-palmitate or fluticasone-furoate. However, in this experiment, bovine hyaluronidase was included in the buffer with the LNPs at a concentration of 1 unit per pL. Without the intention of being limited by any particular theory, extracellular matrix (ECM)-modifying enzymes such as hyaluronidase, or a fragment thereof, can be included in order to improve the access of LNPs to muscle cells. The DNA- mRNA LNPs contained the same largely CpG-free DNA that expresses firefly luciferase from a muscle-specific promoter (SEQ ID NO: 149) and Brambleberry mRNA at a 1:2 ratio. The DNA-mRNA LNPs were cryopreserved at -80°C in 20% sucrose buffer. DNA-mRNA LNPs containing either 2.7% dexamethasone-palmitate or fluticasone-furoate were administered to nude mice by intramuscular injection in the quadriceps and the efficiency of gene transfer was quantitated by measuring luciferase at weekly time points for four weeks on an IVIS system (FIGURE 23C). By week 4, gene transfer appeared to be more efficient
when fluticasone-furoate was included in the DNA-mRNA LNP than when dexamethasone - palmitate was included. The LogP value of dexamethasone-palmitate is 9.8 and the LogP value of fluticasone-furoate is 4.8. Although the LogP value of fluticasone-furoate is lower than that of dexamethasone-palmitate, it is sufficiently high to facilitate efficient incorporation in the LNP. Without the intention of being limited by any particular theory, the improvement observed with fluticasone-furoate may owe in part to fluticasone-furoate being a biologically-active form of the molecule with a LogP value that facilitates coencapsulation in the LNPs and retention in cells that took up LNPs, without a requirement for hydrolysis by a cellular esterase to produce a biologically-active form of a molecule from a pro-drug.
[00384] The use of an aqueous suspension of an immunosuppressive molecule or pro-drug form thereof also provides a mechanism for retaining the immunosuppressive molecule at the site of injection. Such aqueous suspensions provide an alternative to, e.g., the attachment of a hydrophobic moiety such as palmitate to an immunosuppressive molecule. Aqueous suspensions of immunosuppressive molecules known in the art include triamcinolone acetonide (Kenalog) and betamethasone sodium phosphate. An immunosuppressive molecule provided as an aqueous suspension can be present in the aqueous phase of the formulation buffer containing DNA-mRNA LNPs.
[00385] Without the intention of being limited by any particular theory, suppressing innate immune responses activated by DNA-mRNA LNPs may improve the expression of the Brambleherry mRNA, thereby improving the efficiency of gene transfer. In addition to a direct role in the efficiency of gene transfer, coformulation (e.g., coencapsulation) with immunosuppressive molecules affords a desirable means of limiting the potential for immune responses against the protein or proteins encoded by the DNA.
[00386] Additional immunosuppressive molecules that can improve the efficiency of Brambleberry mRNA expression and the efficiency of gene transfer by DNA-mRNA LNPs include MyD88 inhibitors, TRAK4 inhibitors, PKR inhibitors, PERK inhibitors, NFkB inhibitors, IKK inhibitors, JAK inhibitors, STAT inhibitors, GSK3 inhibitors, cGAS inhibitors, and STING inhibitors.
Example 17 - Design, optimization, and use of a microRNA (miR)
[00387] A microRNA (miR) scaffold was designed and optimized for purposes including use in an off-switch and knockdown of cellular genes e.g., enzymes involved in
glycosylation) by RNA interference (RNAi). This miR was derived from a miR-451 scaffold. Advantages of the miR architecture used include that it bypasses Dicer and Drosha processing, improving RNAi knockdown efficiency. Salient features of the engineered miR include: a 19 base guide sequence, a loop of unpaired bases exactly 4 nucleotides in length, a 19 base sequence that is complementary to the guide sequence and forms a 19 base pair stem, and an RNA Polymerase III (Pol III) transcription termination signal (TTTTTT) that overlaps with the 3’ end of the stem. Thus, importantly, the Pol III transcription termination signal is used to make the 3’ end.
[00388] A miR gene under the control of a U6 Pol III promoter was described in Example 5. The sequence of a plasmid for expressing this miR is provided (SEQ ID NO: 54). This miR is matched to an optimized 3’ UTR, which contains 10 target sites for the miR, and which can be included in a transgene to enable its efficient knockdown by the miR through RNAi. The present example provides key data from the optimization of the miR included in SEQ ID NO: 54.
[00389] The ability of this miR to knock down expression of a firefly luciferase transgene containing the 3’ UTR of SEQ ID NO: 55 was evaluated in 293T cells. A plasmid expressing Renilla luciferase was included as a control for transfection efficiency, and data were normalized to Renilla luciferase expression. Firefly and Renilla luciferase were each expressed from a CMV promoter in this experiment. Co-transfection of 293T cells with a plasmid expressing the miR contained in SEQ ID NO: 55 knocked down the expression of firefly luciferase by 248-fold (FIGURE 24A). The same miR construct was tested in C2C12 cells, either when firefly luciferase was expressed from a CMV promoter or when firefly luciferase was expressed from the muscle-specific promoter (MSP) (SEQ ID NO: 12). The miR and 3’ UTR target site afforded a system for efficient activation in muscle cells (FIGURE 24B). Thus, this particular miR design, particularly in combination with a 3’ UTR with concatemeric target sites, provide a highly efficient means of knocking down a transgene by RNA, including in muscle cells. This miR construct may be particularly useful in the context of an off-switch, e.g., where a second plasmid expressing the miR is administered or otherwise activated in order to knock down the expression of a transgene by RNAi.
[00390] An H 1 promoter was modified in order to develop an RNA Polymerase III (Pol III) promoter lacking CpG motifs. The absence of CpG motifs in the Pol III promoter is useful to
avoid including a ligand for TLR9, and to prevent silencing by promoter methylation. The Hl promoter was selected for modification due to its small size and paucity of CpG motifs. Two modified versions of the Hl promoter were generated, one where both CpG (i.e., CG) motifs were mutated to CA (version 1, vl), and the other where both CpG motifs were mutated to TG (version 2, v2). These constructs were tested in a 293T transfection experiment similar to that described above, in which a firefly luciferase transgene with the 3’ UTR target site (SEQ ID NO: 55) and Renilla luciferase were each expressed from CMV promoters. The Renilla luciferase provided a transfection efficiency control, and data were normalized to Renilla luciferase expression. Version 1 (vl) of the Hl promoter, in which both CpG motifs were mutated to CA allowed expression of the miR to knock down expression of the firefly luciferase transgene to a comparable extent as was observed for the unmodified Hl promoter (FIGURE 24C). A control, which was the same miR with a U6 promoter, showed that the Hl promoter and the CpG-free version (v2) exhibited comparable efficiency to the U6 promoter. This Hl promoter where both CpG motifs were mutated to CA, operably linked to a miR capable of efficiently knocking down a transgene with target sites engineered into its 3’ UTR (SEQ ID NO: 55), is the promoter used in SEQ ID NO: 54. Thus, an example is provided of an efficient miR driven by a CpG-free Pol III promoter.
[00391] The same miR architecture was used to knock down enzymes involved in glycosylation. A miR construct was designed targeting the Fut8 mRNA that is expressed in CHO cells. This miR contained a 19 base guide sequence, a loop of unpaired bases exactly 4 nucleotides in length, a 19 base sequence that is complementary to the guide sequence and forms a 19 base pair stem, and an RNA Polymerase 111 (Pol III) transcription termination signal (TTTTTT) that overlaps by two nucleotides with the 3’ end of the stem. The target sequence in the Fut8 mRNA was SEQ ID NO: 150, which notably ends with a TT or UU motif. Thus, the guide sequence is SEQ ID NO: 151. The entire transcribed stem loop structure of the Fut8 miR is SEQ ID NO: 152. The promoter operably linked to a U6 promoter is provided at SEQ ID NO: 153. The miR of SEQ ID NO: 153 was used to generate a stable cell line expressing the miR. eCD4-Ig protein was generated by transient transfection of the parental control cell line (FIGURE 25A) or the cell line containing the miR (FIGURE 25B), and the N-linked glycans attached to the eCD4-Ig protein were characterized by liquid chromatography-mass spectrometry (LC-MS). Whereas the parental control cell line generated protein where 85% of the N-linked glycans were fucosylated, the Fut8 knockdown
cell line generated protein where just 5% of N-linked glycans were fucosylated. This substantial knockdown effect revealed the effective reduction in fucosylated N-linked glycans present on the eCD4-Ig protein made in the cell line expressing the miR relative to the parental cell line, thus demonstrating that this miR architecture is effective for knocking down Fut8. The miR operably linked to a CpG-free Hl promoter is provided as SEQ ID NO: 154.
[00392] The same miR architecture as used above also was demonstrated to be effective in knocking down expression of the gene GALNT2. A miR construct was designed targeting the GALNT2 mRNA that is expressed in CHO cells. This miR contained a 19 base guide sequence, a loop of unpaired bases exactly 4 nucleotides in length, a 19 base sequence that is complementary to the guide sequence and forms a 19 base pair stem, and an RNA Polymerase III (Pol III) transcription termination signal (TTTTTT) that overlaps by two nucleotides with the 3’ end of the stem. The target sequence in the GALNT2 mRNA was SEQ ID NO: 155, which notably ends with a TT or UU motif. The whole miR operably linked to a U6 promoter is SEQ ID NO: 156. The presence or absence of an O-linked glycan on the eCD4-Ig protein generated was measured by mass spectrometry. The prevenance of an O-linked glycan was reduced from being present on 24% of the eCD4-Ig protein made in the control transfection to 3.4% of the protein made by co-transfection with the plasmid expressing the miR targeting GALNT2 (FIGURE 26). Thus, highly efficient knockdowns of a firefly luciferase gene containing an engineered 3’ UTR target site, Fut8, and GALNT2 were demonstrated using the miR architecture provided in the present example.
Example 18 - Coformulation of DNA-mRNA LNPs with hyaluronidase
[00393] DNA-mRNA LNPs can be coformulated with hyaluronidase to promote efficient gene transfer after intramuscular injection. The results of two studies were contrasted by comparing the area under the curve (AUC) of luminescence over the period from 2-4 weeks post- injection with the same amounts of identically-formulated DNA-mRNA LNPs with and without hyaluronidase.
[00394] The LNPs administered in this example were made with 56.9% CL15F6, 27.2% cholesterol, 11.2% DSPC, 1.4% C8C-PEG2000, 0.32% of a cholesterol-conjugated TCTEX- 1 binder chol-R8RhdCT (SEQ ID NO: 118), and 3% DPG-galloyl by mass. DNA-mRNA LNPs were generated in which the DNA was a largely CpG-free DNA that expresses firefly
luciferase from a muscle-specific promoter (SEQ ID NO: 149). The mRNA encoded Danio rerio Brambleberry. DNA and mRNA were coencapsulated at a 1:2 ratio by mass. Five nude mice per group received 25 pL intramuscular injections of LNPs containing 10 pg DNA and 20 pg Brambleberry mRNA in the quadriceps. Mice were imaged by IVIS after luciferin injection. Bovine hyaluronidase was added after thawing of cryopreserved LNPs such that its final concentration was 1 unit per pL after mixing with the thawed LNPs. Area under the curve (AUC) values were calculated for the period from weeks 2-4 post-injection. The use of hyaluronidase significantly improved the efficiency of gene transfer, in terms of the AUC of luciferase signal detected by IVIS (P=0.017) (FIGURE 27). Without the intention of being limited by any particular theory, hyaluronidases provide a group of extracellular matrix (ECM) -modifying enzymes capable of improving the access of LNP to muscle cells after intramuscular injection. Whereas bovine hyaluronidase was used here, human hyaluronidase or a fragment thereof (e.g., Hylenex) could be used in a clinical setting.
INCORPORATION BY REFERENCE
[00395] The entire disclosure of each of the patent and scientific documents referred to herein is incorporated by reference for all purposes.
EQUIVALENTS
[00396] The invention may be embodied in other specific forms without departing from the spirit or essential characteristics thereof. The foregoing embodiments are therefore to be considered in all respects illustrative rather than limiting on the invention described herein. Scope of the invention is thus indicated by the appended claims rather than by the foregoing description, and all changes that come within the meaning and range of equivalency of the claims are intended to be embraced therein.
SEQUENCE LISTING:

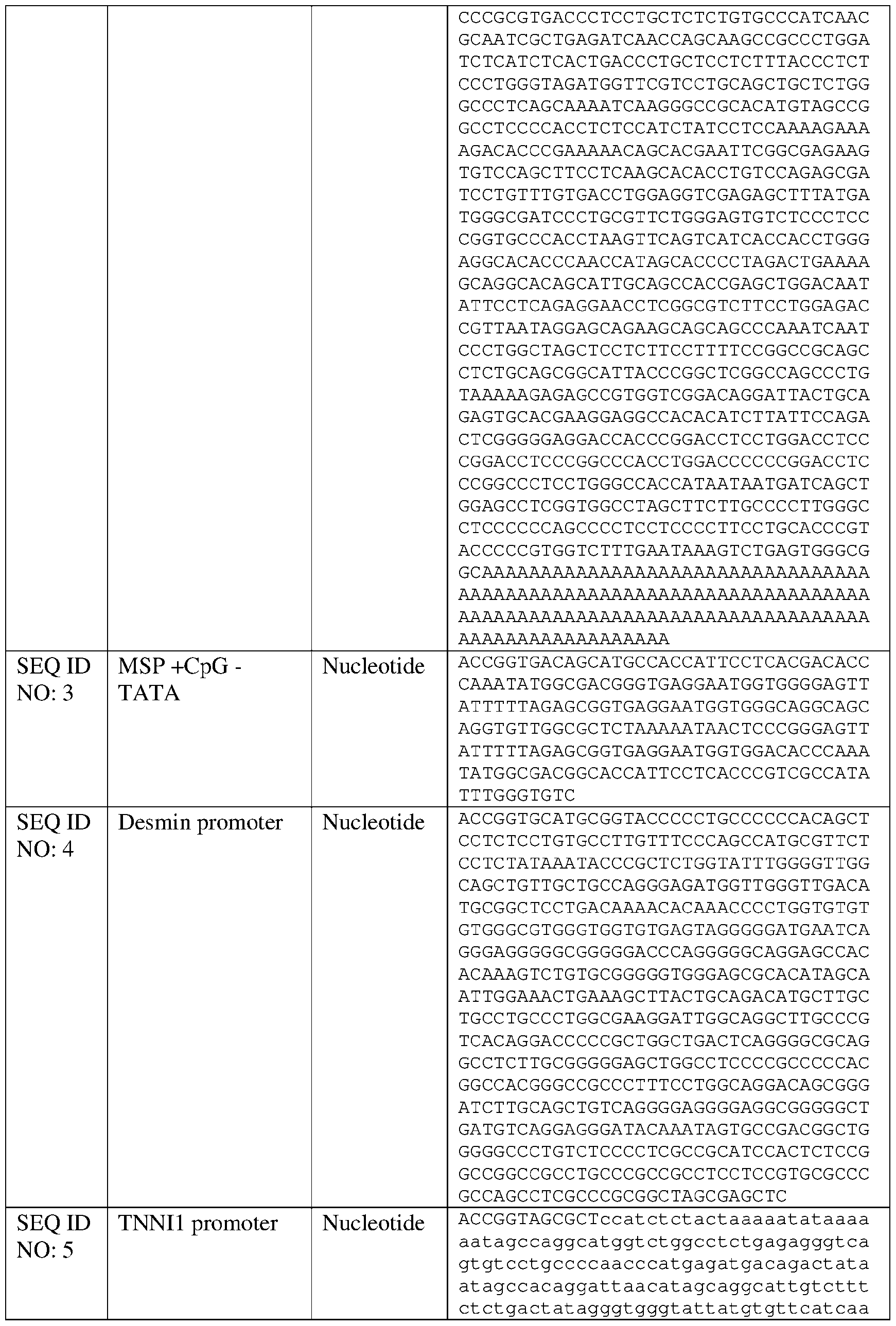
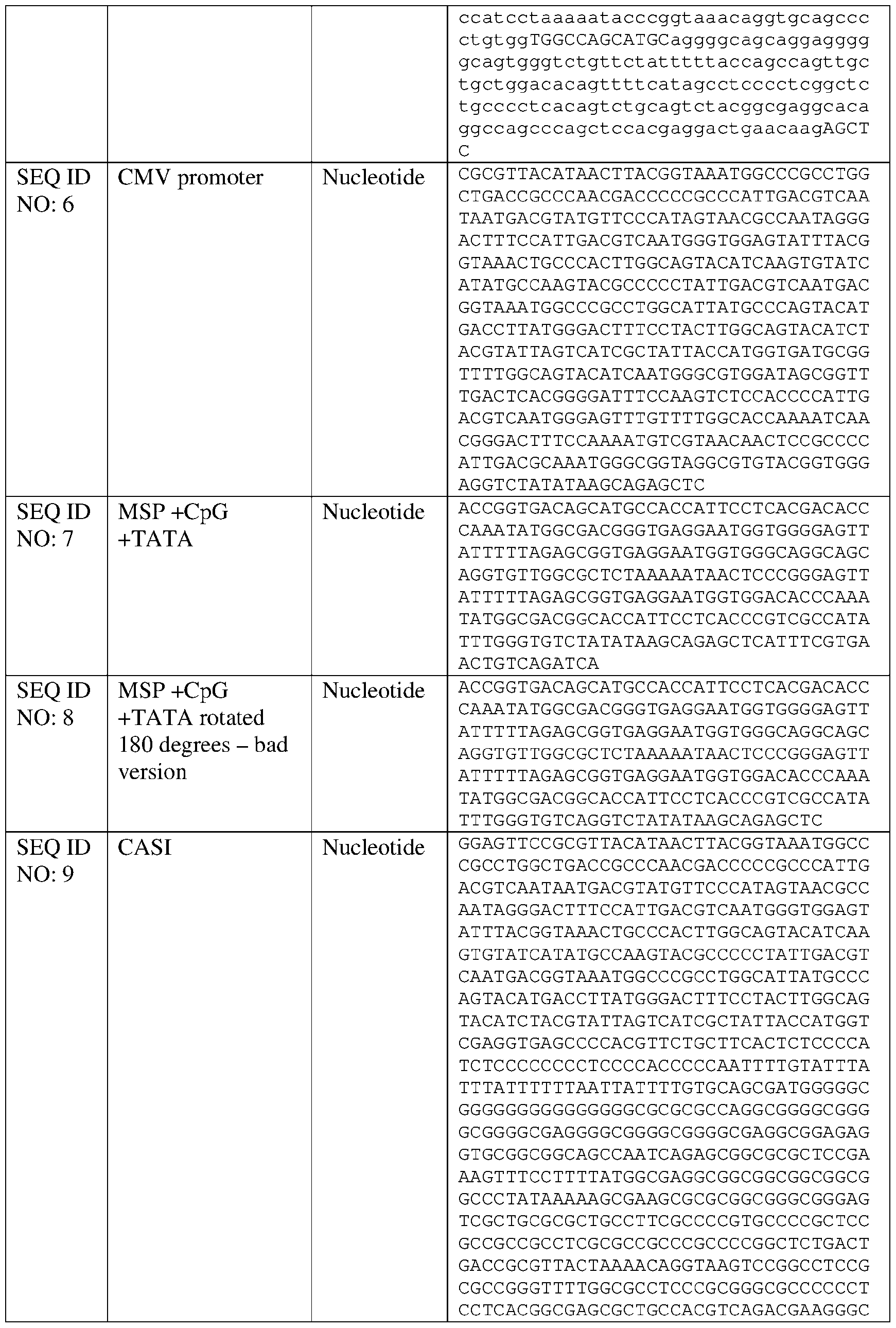
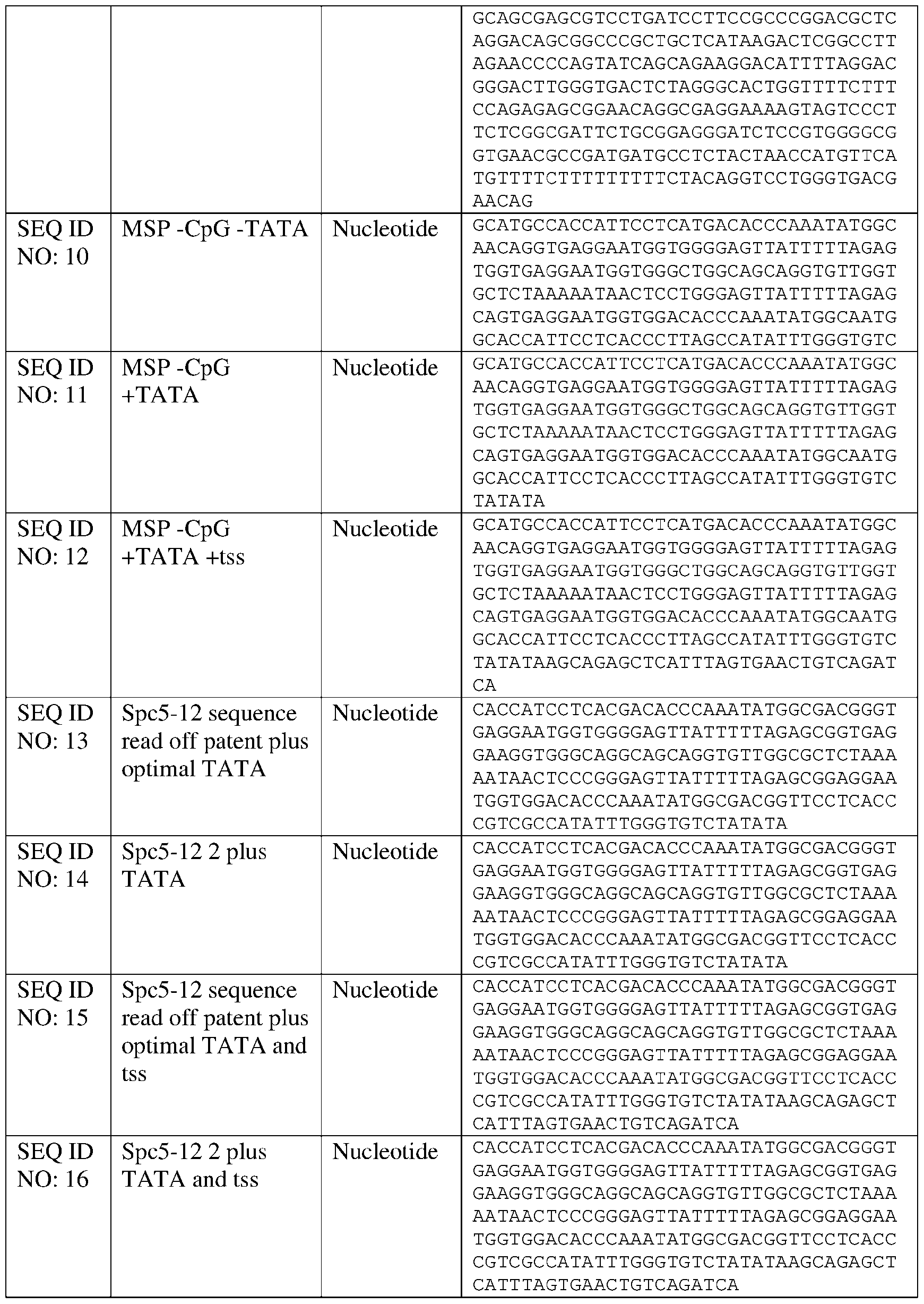
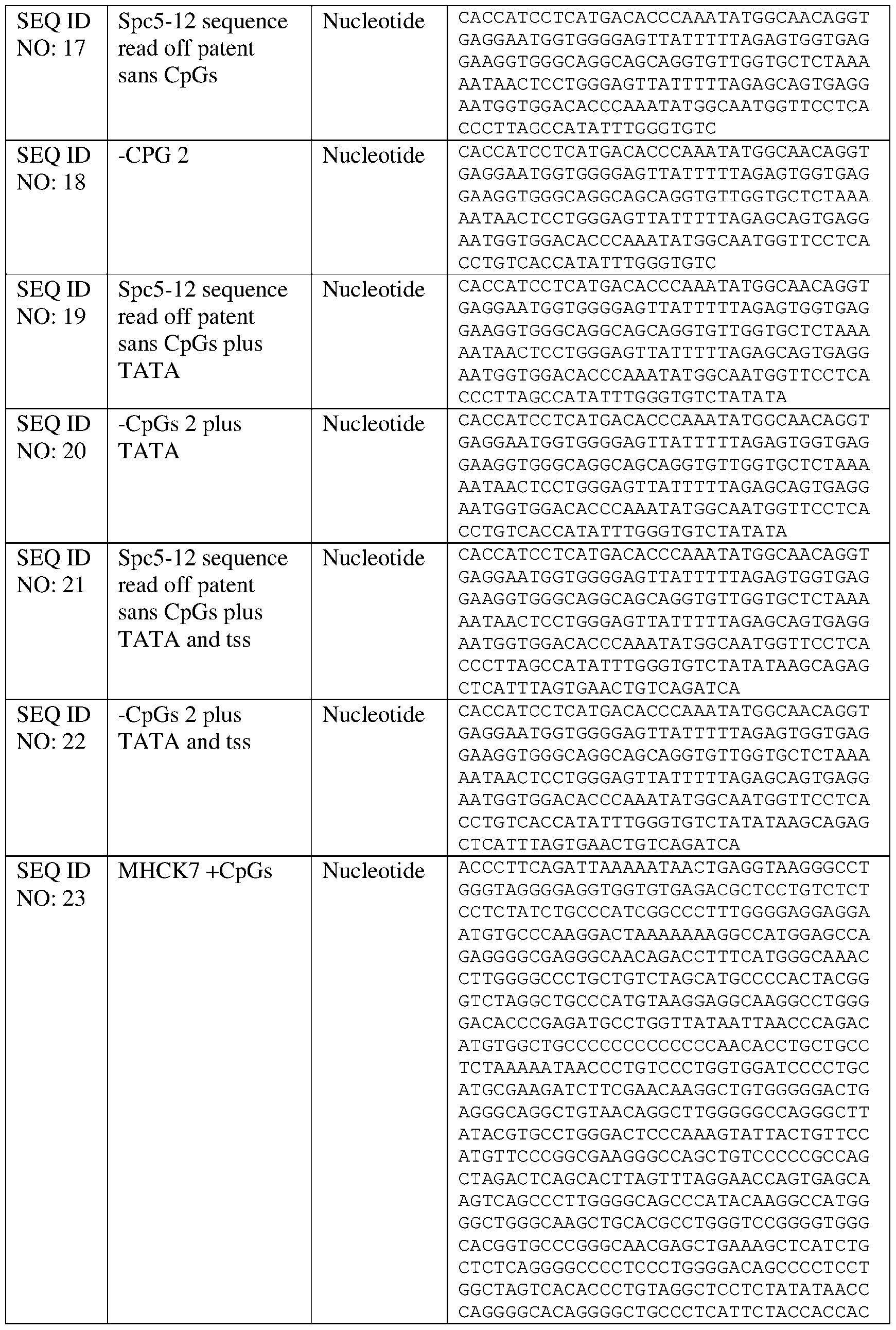



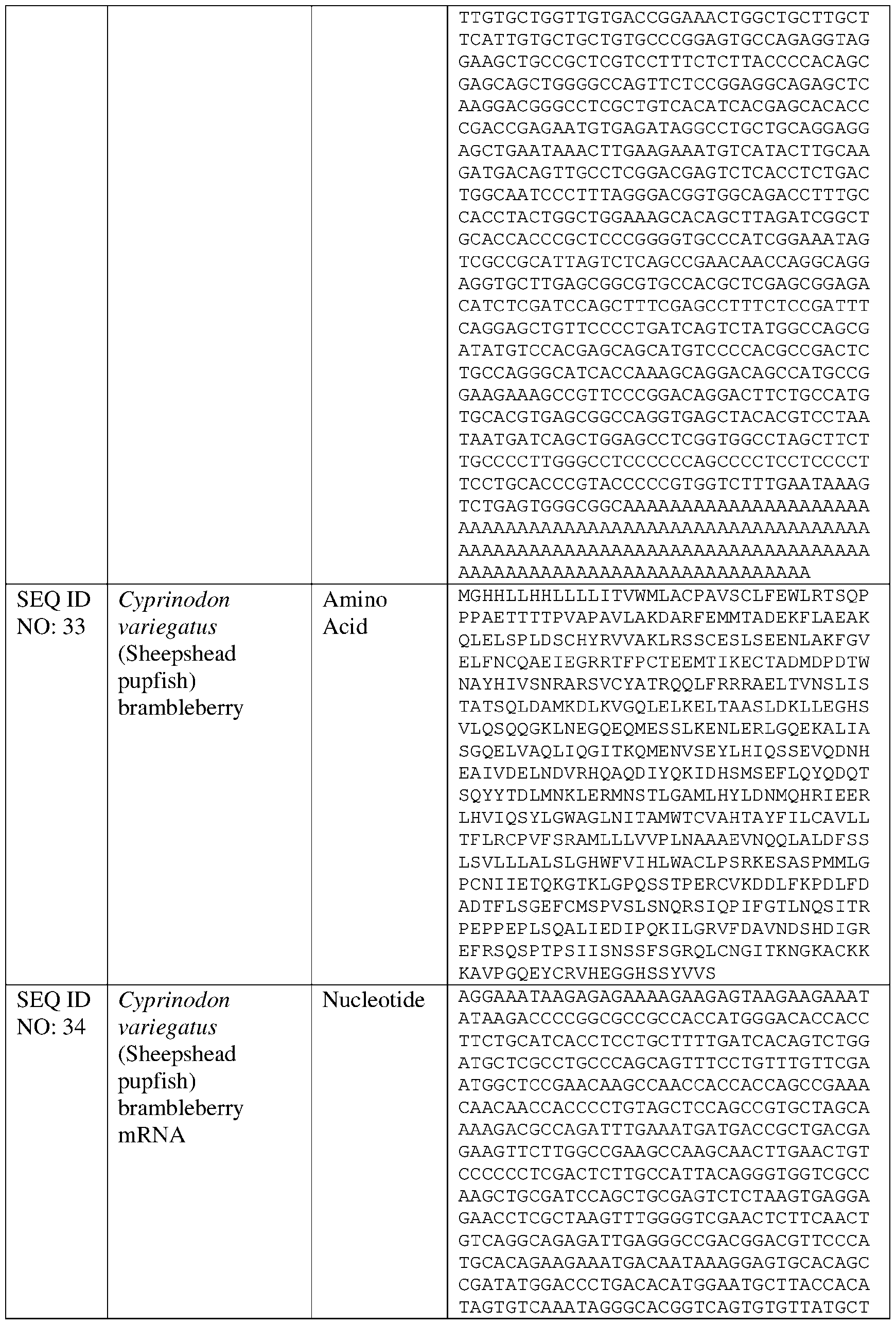



















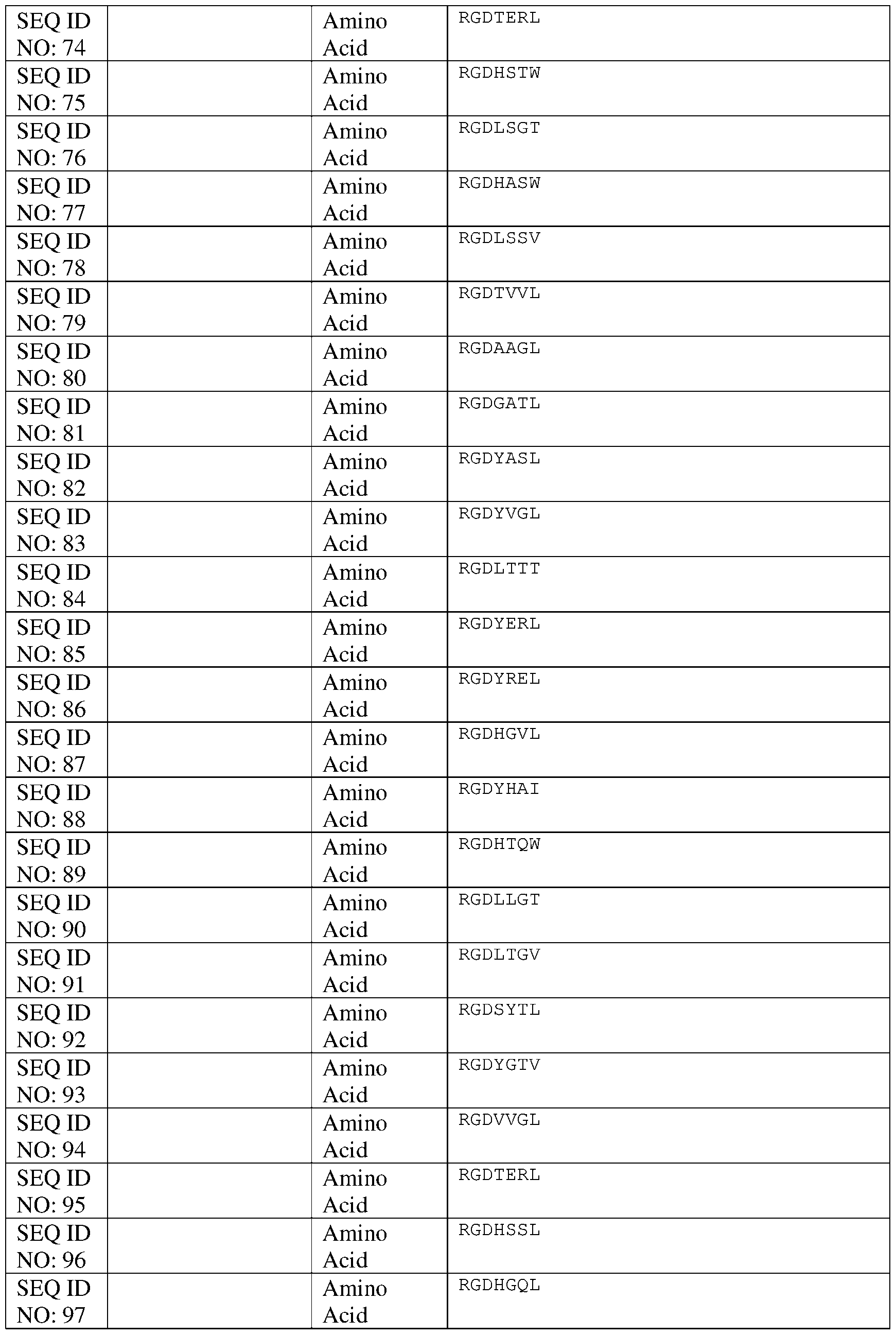

























Claims
1. A system for the transfer of non-native DNA into a nucleus of a cell, the system comprising the non-native DNA and RNA, wherein the RNA encodes a double membrane fusogen.
2. The system of claim 1 , wherein the double membrane fusogen mediates the fusion of intracellular membranes.
3. The system of claim 2, wherein the double membrane fusogen mediates the fusion of a nuclear envelope of a cell with a double membrane-enveloped structure comprising the non-native DNA.
4. The system of any one of claims 1-3, wherein the double membrane fusogen comprises a signal peptide and a transmembrane domain.
5. The system of claim 4, wherein the double membrane fusogen comprises two or more transmembrane domains.
6. The system of claim 5, wherein the double membrane fusogen comprises three or more transmembrane domains.
7. The system of claim 6, wherein the double membrane fusogen comprises three or more transmembrane alpha helices.
8. The system of any one of claims 1-7, wherein the double membrane fusogen comprises a DNA-binding domain.
9. The system of any one of claims 1-8, wherein the double membrane fusogen comprises a multimerization domain.
10. The system of any one of claims 1-9, wherein double membrane fusogen comprises an alpha helical domain at least 20, 30, 40, 50, 60, 70, 80, 90, or 100 amino acids in length.
11. The system of any one of claims 1-10, wherein the double membrane fusogen comprises an alpha helical domain at least 50 amino acids in length.
12. The system of any one of claims 1-11, wherein the double membrane fusogen comprises an alpha helical domain at least 60 amino acids in length.
13. The system of any one of claims 1-12, wherein the double membrane fusogen comprises an alpha helical domain at least 80 amino acids in length.
14. The system of any one of claims 1-13, wherein the double membrane fusogen comprises an alpha helical domain at least 100 amino acids in length.
15. The system of any one of claims 1-14, wherein the double membrane fusogen comprises a luminal domain, a transmembrane domain, a cytoplasmic domain, or a DNA- binding domain having at least 60%, at least 70%, at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, at least 99.5%, or 100% sequence identity to the corresponding domain present in SEQ ID NO: 1, 25, 27, 29, 31, 33, 119, 121, 123, 125, 127, 129, 131, 133, 135, 137, 139, 141, 143, 145, or 147.
16. The system of any one of claims 1-15, wherein the double membrane fusogen comprises a cytoplasmic domain, and the cytoplasmic domain comprises at least one of
(d) a nuclear localization signal (NLS),
(e) an amino acid sequence five amino acids in length wherein at least four out of five of the amino acids are basic residues, and
(f) an amino acid sequence comprising at least one basic residue separated by a spacer of ten to fifteen amino acids from three or more basic residues.
17. The system of claim 16, wherein the at least one of features (a) - (c) does not naturally occur in the amino acid sequence of a wild-type double membrane fusogen having the greatest sequence identity to the encoded double membrane fusogen.
18. The system of any one of claims 1-17, wherein the double membrane fusogen is not a human protein.
19. The system of any one of claims 1-18, wherein the double membrane fusogen is a Brambleberry protein, a protein involved in karyogamy, or a functional fragment, variant, or chimera of any of the foregoing.
20. The system of any one of claims 1-19, wherein the double membrane fusogen is a Brambleberry protein or functional fragment or variant thereof.
21. The system of any one of claims 19 or 20, wherein the Brambleberry protein or functional fragment or variant thereof is a zebrafish (Danio rerio) Brambleberry protein or functional fragment or variant thereof.
22. The system of any one of claims 19-21 wherein the Brambleberry protein is a split Brambleberry protein.
23. The system of any one of claims 1-22, wherein the system further comprises an immunosuppressive molecule or a pro-drug form thereof, and/or an RNA encoding an immunosuppressive protein or a pro-drug form thereof.
24. The system of claim 23, wherein the immunosuppressive molecule or pro-drug form thereof is a corticosteroid or a tyrosine kinase inhibitor.
25. The system of claim 24, wherein the corticosteroid is a glucocorticoid.
26. The system of any one of claims 23-25, wherein the immunosuppressive molecule or pro-drug form thereof binds a human glucocorticoid receptor.
27. The system of any one of claims 23-26, wherein the immunosuppressive molecule is dexamethasone, fluticasone, fluticasone propionate, fluticasone furoate, clobetasol, betamethasone, halobetasol, amcinonide, desoximetasone, diflorasone, fluocinonide, mometasone, prednisone, prednicarbate, triamcinolone, triamcinolone acetonide, fluocinolone, alclometasone, desonide, cortisone, or hydrocortisone, or a pro-drug form of any of the foregoing.
28. The system of claim 23-27, wherein the immunosuppressive molecule is dexamethasone or tacrolimus.
29. The system of any one of claims 23-28, wherein the immunosuppressive molecule or pro-drug form thereof contains an ester bond.
30. The system of any one of claims 23-29, wherein the immunosuppressive molecule or pro-drug form thereof is dexamethasone palmitate or fluticasone-furoate.
31. The system of any one of claims 23-30, comprising at least one immunosuppressive molecule or pro-drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, NLRP3 inflammasome inhibitors, MyD88 inhibitors, IRAK4 inhibitors, PKR inhibitors, PERK inhibitors, NFkB inhibitors, IKK inhibitors, JAK inhibitors, STAT inhibitors, GSK3 inhibitors, cGAS inhibitors, or STING inhibitors.
32. The system of any one of claims 23-31, comprising at least one immunosuppressive molecule or pro-drug form thereof selected from the group consisting of: galectin inhibitors, cathepsin inhibitors, cysteine protease inhibitors, or NLRP3 inflammasome inhibitors.
33. The system of any one of claims 23-32, wherein the immunosuppressive molecule or pro-drug form thereof is an NLRP3 inflammasome inhibitor.
34. The system of any one of claims 31-33, wherein the NLRP3 inflammasome inhibitor is oridonin, INF39, glyburide, 5-chloro-2-methoxy-N-[2-(4- sulfamoylphenyl)-ethyl]- benzamide (e.g. , 16673-34-0), JC124, FC11A-2, parthenolide, VX-740, VX-765, BAY 11- 7082, BHB, MCC950, MNS, CY-09, or Tranilast, OET1177, or a pro-drug form of any of the foregoing.
35. The system of any one of claims 31-32, wherein the galectin inhibitor is GB1107, galectin-3/galectin-8-IN- l, olitigaltin, selvigaltin, galectin-3/galectin-8-IN-2, galectin-3-IN-2, galectin-3-IN-3, galectin-3-IN-5, GB1490, GB1908, galectin-8-IN-l, galectin-8-IN-2, galectin-8N-IN-l, galectin-8N-IN-2, or thiodigalactoside, or a pro-drug form of any of the foregoing.
36. The system of any one of claims 31-32, wherein the cathepsin inhibitor or cysteine protease inhibitor is cathepsin E-IN-2 (Z-Phe-Phe-FMK), disulfiram, belizatinib, cystatin B, cystatin C, E-64, E-64d, or a pro-drug form of any of the foregoing.
37. The system of any one of claims 23-36, comprising at least two different immunosuppressive molecules or pro-drug forms thereof.
38. The system of any one of claims 23-37, comprising at least three different immunosuppressive molecules or pro-drug forms thereof.
39. The system of any one of claims 23-38, wherein the immunosuppressive molecule or pro-drug form thereof has a LogP value greater than 3.0, 3.5, or 4.0.
40. The system of any one of claims 23-39, comprising an aqueous suspension of the immunosuppressive molecule or pro-drug form thereof.
41. The system of claim 40, wherein the aqueous suspension comprises triamcinolone acetonide or betamethasone sodium phosphate.
42. The system of any one of claims 1-41 , comprising at least one extracellular matrix (ECM) -modifying enzyme.
43. The system of claim 42, where in the ECM-modifying enzyme is a hyaluronidase or a fragment thereof.
44. The system of any one of claims 1-43, wherein the cell is a non-dividing cell.
45. The system of any one of claims 1-44, wherein the RNA encoding the double membrane fusogen comprises modified bases.
46. The system of any one of claims 1-45, wherein the RNA encoding the double membrane fusogen is an mRNA.
47. The system of any one of claims 1-46, wherein the RNA encoding the double membrane fusogen comprises one or more target sites for at least one micro RNA (miR).
48. The system of any one of claims 1-47, wherein the DNA comprises modified CpG motifs.
49. The system of claim 48, wherein the DNA has fewer than 100 CpG motifs per molecule, the DNA is substantially free of unmodified CpG motifs, and/or the DNA is methylated at one or more CpG motifs.
50. The system of claim 49, wherein the DNA is substantially free of 6-methyladenine and/or 5 -methylcytosine.
51. The system of any one of claims 1-50, wherein the DNA comprises at least one tissue- specific promoter.
52. The system of claim 51, wherein the tissue-specific promoter is expressed in non- dividing cells.
53. The system of claim 51 or 52, wherein the tissue-specific promoter is a muscle- specific promoter.
54. The system of any one of claims 1-53, wherein the DNA comprises a promoter with fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs.
55. The system of claims 53 or 54, wherein the muscle-specific promoter comprises one or more transcription factor binding site core motifs selected from the group consisting of: a. 5’-CATTCC-3’, b. 5’-GGAATG-3’, c. 5’-CCWWWWWWGG-3’, d. 5’-CANNTG-‘3, e. 5’-YTAAAAATA-3’, and f. 5’-TATTTTTAR-3’, wherein N is any nucleotide, W is A or T, Y is T or C, and R is A or G.
56. The system of any one of claims 1-55, wherein the DNA lacks free 5’ and 3’ ends and/or is circular.
57. The system of any one of claims 1-56, wherein the DNA is at least partially denatured.
58. The system of any one of claims 1-57, wherein the DNA comprises a coding region that is operably linked to a promoter.
59. The system of any one of claims 1-58, wherein the DNA comprises a gene capable of expressing a secreted protein.
60. The system of any one of claims 1-59, wherein the DNA comprises a gene encoding a protein containing an antibody Fc.
61. The system of any one of claims 1-60 wherein the DNA comprises a gene encoding a protein comprising a monoclonal or bispecific antibody.
62. The system of any one of claims 1-61, wherein the DNA comprises a gene encoding a protein comprising at least one domain that binds a ligand and at least one effector domain.
63. The system of any one of claims 1-62, wherein the DNA comprises a gene encoding a protein that has at least 80% identity to human serum albumin (HSA).
64. The system of any one of claims 1-63 wherein the DNA comprises a gene encoding an mRNA, wherein the mRNA comprises one or more target sites for a micro RNA (miR), short hairpin RNA (shRNA), or ribozyme.
65. The system of any one of claims 1-64, wherein the DNA encodes at least one non- coding RNA.
66. The system of claim 65, wherein the at least one non-coding RNA comprises a micro RNA (miR), short hairpin RNA (shRNA), aptamer, or ribozyme.
67. The system of claim 65 or 66, wherein the at least one non-coding RNA targets an mRNA expressed by the DNA.
68. The system of any one of claims 65-67, wherein the at least one non-coding RNA targets the mRNA of an immunomodulatory gene.
69. The system of any one of claims 65-68, wherein the non-coding RNA targets the mRNA of the fucosyltransferase 8 (FUT8) gene.
70. The system of any one of claims 65-69, wherein the at least one non-coding RNA targets the mRNA of a human gene, and the DNA encodes a protein capable of functionally replacing a common form of that human gene.
71. The system of any one of claims 1-70, comprising one or more nanoparticles.
72. The system of any one of claims 1-70, wherein the system is a nanoparticle containing the non-native DNA and the RNA.
73. The system of claim 71 or claim 72, wherein the nanoparticles are lipid nanoparticles (LNPs).
74. The system of any one of claims 1-73, comprising one or more ionizable lipids.
75. The system of any one of claims 1-74, comprising a polyplex.
76. The system of any one of claims 71-75, wherein the nanoparticle comprises at least one attachment moiety.
77. The system of claim 76, wherein the attachment moiety promotes attachment to muscle cells.
78. The system of any one of claims 71-78, further comprising a lipid that is conjugated directly or indirectly to a glucose molecule.
79. The system of claim 78, wherein the glucose molecule is conjugated to hydroquinone.
80. The system of any one of claims 71-79, further comprising a lipid that is conjugated to a polyphenol.
81. The system of any one of claims 1-80, further comprising a dynein binder.
82. The system of any one of claims 1-81 , further comprising at least one antisense oligonucleotide (ASO) or short-interfering RNA (siRNA).
83. The system of any one of claims 1-82, further comprising a DNA binder that binds to DNA at pH 7.4.
84. The system of any one of claims 1-83, wherein the RNA encoding the double membrane fusogen comprises Cap2 or one or more internal ribosomal entry site (IRES).
85. The system of any one of claims 1-84, further comprising:
a. a gene editing effector protein or an RNA encoding a gene editing effector protein, b. a transposase or an RNA encoding a transposase, c. an integrase or an RNA encoding an integrase, d. a recombinase or an RNA encoding a recombinase, or e. a reverse transcriptase or an RNA encoding a reverse transcriptase.
86. The system of any one of claims 1-85, wherein the system is a system for gene therapy.
87. A system for the transfer of non-native DNA into a nucleus of a cell, comprising the non-native DNA and a dynein binder.
88. The system of any one of claims 81-86, wherein the dynein binder is or is conjugated to a DNA binder, an RNA binder, a polymer, a peptide, a polypeptide, or a lipid.
89. The system any one of claims 83-88, wherein the DNA binder is cationic at pH 7.4.
90. The system of any one of claims 81-89, wherein the dynein binder is a TCTEX binder.
91. The system of claim 90, wherein the dynein binder or TCTEX binder is a TCTEX- 1 (DYNLT-1) binder.
92. The system of claim 90 or 91 , wherein the TCTEX binder comprises a peptide with comprising an amino acid sequence at least five amino acids in length that is at least 80% identical to a contiguous amino acid motif selected from the peptides: a. GGFKLNIWDVGGQK (SEQ ID NO: 115), and b. GVSKTETSQVAPA (SEQ ID NO: 116).
93. A composition comprising the system of any one of claims 1-92.
94. The composition of claim 93, further comprising a pharmaceutically acceptable carrier.
95. A pharmaceutical composition comprising:
an RNA encoding a double membrane fusogen, which, when delivered to a mammalian cell, mediates transfer of a non-native DNA into the nucleus of the mammalian cell; and a pharmaceutically acceptable carrier.
96. The pharmaceutical composition of claim 95, further comprising the non-native DNA.
97. A nucleic acid comprising a muscle-specific promoter, wherein the muscle-specific promoter a. comprises fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs, and/or b. has at least 80% identity to a nucleic acid sequence comprising SEQ ID NO: 7, 11, 13, 14, 19, 20, or 110 and a TATA box that is positioned within four nucleotides of the position of a TATA box present in SEQ ID NO: 7, 11, 13, 14, 19, 20, or 110 respectively.
98. The nucleic acid of claim 97, wherein the muscle-specific promoter comprises one or more of the transcription factor binding site core motifs selected from the group consisting of: a. 5’-CATTCC-3’, b. 5’-GGAATG-3’, c. 5’-CCWWWWWWGG-3’, d. 5’-CANNTG-‘3, e. 5’-YTAAAAATA-3’, and f. 5’-TATTTTTAR-3’, wherein N is any nucleotide, W is A or T, Y is T or C, and R is A or G.
99. The nucleic acid of claim 97 or 98, wherein the muscle-specific promoter has at least 80% identity to SEQ ID NO: 23 or 24, and wherein the muscle-specific promoter comprises fewer than 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 CpG motifs.
100. The nucleic acid of any one of claims 97-99, further comprising a DNA polymerase III (Pol III) promoter, wherein the Pol III promoter comprises fewer than 3, 2, or 1 CpG motifs.
101. A lipid nanoparticle (LNP) comprising a lipid that is covalently bonded to a glucose molecule.
102. The LNP of claim 101, wherein the glucose molecule is conjugated to hydroquinone.
103. The system of any one of claims 78-92 or the LNP of claim 101, wherein the lipid that is covalently bonded to a glucose molecule is cholesterol-undecanoate-glucose.
104. A lipid nanoparticle (LNP) comprising a lipid that is covalently bonded to a polyphenol.
105. The system of any one of claims 80-92 or the LNP of claim 104, wherein the polyphenol comprises galloyl or a galloyl group.
106. The system of any one of claims 1-92, 103, or 105; the composition of claim 94; the pharmaceutical composition of any one of claims 95-96; the nucleic acid of claims 97-100; or the LNP of any one of claims 101, 102, or 104; comprising a DNA encoding a microRNA (miR), wherein the miR comprises: a. a 19-base guide sequence that is complementary to a target mRNA, b. an unpaired loop exactly 4 nucleotides in length, c. a 19-base sequence that is complementary to the 19-base guide sequence, which together with the guide sequence form a 19-base pair stem, and d. a 3’ end that is a U, which is encoded by an RNA Polymerase III (Pol III) UUUUUU, UUUUU, TTTTTT, or TTTTT transcription termination sequence.
107. A DNA encoding a microRNA (miR), wherein the miR comprises: a. a 19-base guide sequence that is complementary to a target mRNA, b. an unpaired loop exactly 4 nucleotides in length, c. a 19-base sequence that is complementary to the 19-base guide sequence, which together with the guide sequence form a 19-base pair stem, and
d. a 3 ’ end that is a U, which is encoded by one of the U or T nucleotides of an RNA Polymerase III (Pol III) transcription termination signal comprising the sequence UUUUUU, UUUUU, TTTTTT, or TTTTT.
108. The system of any one of claims 47-92, 103, or 105; the composition of claim 93 or 94; pharmaceutical composition of claim 95 or 96; the nucleic acid of claims 97-100; the LNP of any one of claims 101, 103, and 104; or the DNA of claim 107 comprising an RNA Polymerase III promoter operably linked to the miR.
109. The system of any one of claims 47-92, 103, or 105; the composition of claim 93 or 94; pharmaceutical composition of claim 95 or 96; the nucleic acid of claims 97-100; the LNP of any one of claims 101, 103, and 104; or the DNA of claim 107, wherein the 3’ two nucleotides of the mIR are UU.
110. The system, pharmaceutical composition, or nucleic acid of any one of claims 106- 109, comprising a DNA encoding a microRNA (miR) operably linked to an Hl promoter that is at least 80% identical to nucleotides 1-99 of SEQ ID NO: 154.
111. The system, pharmaceutical composition, or nucleic acid of claims 106-110, comprising a DNA encoding a microRNA (miR), wherein the miR targets a fucosylatransferase-8 (FUT8) gene sequence.
112. A method of transfecting a non-native DNA into the nucleus of a nucleated mammalian cell, the method comprising contacting the cell with the non-native DNA and an RNA encoding a double membrane fusogen, wherein, when the RNA is translated to produce the fusogen, the fusogen facilitates the transfer of the non-endogenous DNA into the nucleus of the cell.
113. The method of claim 112, wherein the method comprises the system of any one of claims 1-92, 103, or 105; the composition of claim 93 or 94; the pharmaceutical composition of claim 95 or 96; the nucleic acid of claims 97-100; the LNP of any one of claims 101, 103, and 104; or the DNA of claim 107.
114. An engineered, nucleated mammalian cell, the cell comprising a nucleus, a non-native DNA and an RNA encoding a double membrane fusogen, whereupon, when the RNA is
transcribed into a fusogen, the fusogen facilitates transfer of the non-endogenous DNA into the nucleus of the cell.
115. The engineered, nucleated mammalian cell of claim 114, wherein the cell comprises the system of any one of claims 1-92, 103, or 105; the composition of claim 93 or 94; the pharmaceutical composition of claim 95 or 96; the nucleic acid of claims 97-100; the LNP of any one of claims 101, 103, and 104; or the DNA of claim 107.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363614723P | 2023-12-26 | 2023-12-26 | |
| US63/614,723 | 2023-12-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025144938A1 true WO2025144938A1 (en) | 2025-07-03 |
Family
ID=94341323
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/061965 Pending WO2025144938A1 (en) | 2023-12-26 | 2024-12-26 | Systems for nucleic acid transfer |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025144938A1 (en) |
Citations (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7244609B2 (en) | 2001-03-09 | 2007-07-17 | Cayla | Synthetic genes and bacterial plasmids devoid of CpG |
| WO2009086558A1 (en) | 2008-01-02 | 2009-07-09 | Tekmira Pharmaceuticals Corporation | Improved compositions and methods for the delivery of nucleic acids |
| US20100130588A1 (en) | 2008-04-15 | 2010-05-27 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for nucleic acid delivery |
| WO2011036557A1 (en) | 2009-09-22 | 2011-03-31 | The University Of British Columbia | Compositions and methods for enhancing cellular uptake and intracellular delivery of lipid particles |
| US8278036B2 (en) | 2005-08-23 | 2012-10-02 | The Trustees Of The University Of Pennsylvania | RNA containing modified nucleosides and methods of use thereof |
| US20130053572A1 (en) | 2010-01-22 | 2013-02-28 | Steven L. Colletti | Novel Cationic Lipids for Oligonucleotide Delivery |
| US20130108685A1 (en) | 2010-04-28 | 2013-05-02 | Takeshi Kuboyama | Cationic lipid |
| US20130195920A1 (en) | 2011-12-07 | 2013-08-01 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20140308304A1 (en) | 2011-12-07 | 2014-10-16 | Alnylam Pharmaceuticals, Inc. | Lipids for the delivery of active agents |
| US20150005363A1 (en) | 2011-12-07 | 2015-01-01 | Alnylam Pharmaceuticals, Inc. | Branched Alkyl And Cycloalkyl Terminated Biodegradable Lipids For The Delivery Of Active Agents |
| US9006417B2 (en) | 2010-06-30 | 2015-04-14 | Protiva Biotherapeutics, Inc. | Non-liposomal systems for nucleic acid delivery |
| US9012219B2 (en) | 2005-08-23 | 2015-04-21 | The Trustees Of The University Of Pennsylvania | RNA preparations comprising purified modified RNA for reprogramming cells |
| WO2016118725A1 (en) | 2015-01-23 | 2016-07-28 | Moderna Therapeutics, Inc. | Lipid nanoparticle compositions |
| WO2016118724A1 (en) | 2015-01-21 | 2016-07-28 | Moderna Therapeutics, Inc. | Lipid nanoparticle compositions |
| US9504651B2 (en) | 2002-06-28 | 2016-11-29 | Protiva Biotherapeutics, Inc. | Lipid compositions for nucleic acid delivery |
| US9567296B2 (en) | 2013-11-18 | 2017-02-14 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US9580711B2 (en) | 2013-11-18 | 2017-02-28 | Arcturus Therapeutics, Inc. | Lipid particles with asymmetric cationic lipids for RNA delivery |
| US20170119904A1 (en) | 2015-10-28 | 2017-05-04 | Acuitas Therapeutics, Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US9669089B2 (en) | 2012-02-15 | 2017-06-06 | Curevac Ag | Nucleic acid comprising or coding for a histone stem-loop and a poly(A) sequence or a polyadenylation signal for increasing the expression of an encoded pathogenic antigen |
| US20170210697A1 (en) | 2015-09-17 | 2017-07-27 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| US9950065B2 (en) | 2013-09-26 | 2018-04-24 | Biontech Rna Pharmaceuticals Gmbh | Particles comprising a shell with RNA |
| US9982279B1 (en) | 2017-06-23 | 2018-05-29 | Inscripta, Inc. | Nucleic acid-guided nucleases |
| US10006007B2 (en) | 2009-12-07 | 2018-06-26 | The Trustees Of The University Of Pennsylvania | RNA preparations comprising purified modified RNA for reprogramming cells |
| US10064959B2 (en) | 2010-10-01 | 2018-09-04 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US10106490B2 (en) | 2014-06-25 | 2018-10-23 | Acuitas Therapeutics, Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US20190022247A1 (en) | 2015-12-30 | 2019-01-24 | Acuitas Therapeutics, Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US10221127B2 (en) | 2015-06-29 | 2019-03-05 | Acuitas Therapeutics, Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US10227302B2 (en) | 2017-02-09 | 2019-03-12 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US10233148B2 (en) | 2015-12-30 | 2019-03-19 | Arcturus Therapeutics, Inc. | Aromatic ionizable cationic lipid |
| US20190083602A1 (en) | 2015-12-22 | 2019-03-21 | Curevac Ag | Method for producing rna molecule compositions |
| WO2019089828A1 (en) | 2017-10-31 | 2019-05-09 | Acuitas Therapeutics, Inc. | Lamellar lipid nanoparticles |
| WO2019152557A1 (en) | 2018-01-30 | 2019-08-08 | Modernatx, Inc. | Compositions and methods for delivery of agents to immune cells |
| US20190240354A1 (en) | 2016-06-30 | 2019-08-08 | Arbutus Biopharma Corporation | Compositions and methods for delivering messenger rna |
| US20190314496A1 (en) | 2014-04-01 | 2019-10-17 | Curevac Ag | Polymeric carrier cargo complex for use as an immunostimulating agent or as an adjuvant |
| US20190351048A1 (en) | 2016-12-23 | 2019-11-21 | Curevac Ag | Mers coronavirus vaccine |
| US10485884B2 (en) | 2012-03-26 | 2019-11-26 | Biontech Rna Pharmaceuticals Gmbh | RNA formulation for immunotherapy |
| WO2019232095A1 (en) | 2018-05-30 | 2019-12-05 | Translate Bio, Inc. | Vitamin cationic lipids |
| WO2020002598A1 (en) | 2018-06-28 | 2020-01-02 | Curevac Ag | Bioreactor for rna in vitro transcription |
| US20200046830A1 (en) | 2017-03-30 | 2020-02-13 | The Government Of The United States, As Represented By The Secretary Of The Army | Nucleic acid vaccine composition comprising a lipid formulation, and method of increasing the potency of nucleic acid vaccines |
| US20200046838A1 (en) | 2017-04-13 | 2020-02-13 | Acuitas Therapeutics, Inc. | Lipids for delivery of active agents |
| US10577403B2 (en) | 2012-04-02 | 2020-03-03 | Modernatx, Inc. | Modified polynucleotides for the production of secreted proteins |
| US10626161B2 (en) | 2015-04-28 | 2020-04-21 | The Scripps Research Institute | Methods and compositions for protection against lentiviral infections |
| WO2020097540A1 (en) | 2018-11-09 | 2020-05-14 | Arbutus Biopharma Corporation | Lipid nanoparticle formulations |
| WO2020097548A1 (en) | 2018-11-09 | 2020-05-14 | Arbutus Biopharma Corporation | Lipid nanoparticle formulations |
| US20200163878A1 (en) | 2016-10-26 | 2020-05-28 | Curevac Ag | Lipid nanoparticle mrna vaccines |
| US20200164038A1 (en) | 2011-12-16 | 2020-05-28 | Modernatx, Inc. | Modified nucleoside, nucleotide, and nucleic acid compositions |
| US20200172472A1 (en) | 2017-08-17 | 2020-06-04 | Acuitas Therapeutics, Inc. | Lipids for use in lipid nanoparticle formulations |
| US20200206362A1 (en) | 2015-12-10 | 2020-07-02 | Moderna TX, Inc. | Compositions and methods for delivery of agents |
| US10702600B1 (en) | 2015-10-22 | 2020-07-07 | Modernatx, Inc. | Betacoronavirus mRNA vaccine |
| US20200246451A1 (en) | 2015-04-13 | 2020-08-06 | Curevac Real Estate Gmbh | Method for producing rna compositions |
| US20200283372A1 (en) | 2019-01-11 | 2020-09-10 | Acuitas Therapeutics, Inc. | Lipids for lipid nanoparticle delivery of active agents |
| US10780054B2 (en) | 2015-04-17 | 2020-09-22 | Curevac Real Estate Gmbh | Lyophilization of RNA |
| US10799602B2 (en) | 2013-08-21 | 2020-10-13 | Curevac Ag | Method for increasing expression of RNA-encoded proteins |
| US10808242B2 (en) | 2015-08-28 | 2020-10-20 | Biontech Rna Pharmaceuticals Gmbh | Method for reducing immunogenicity of RNA |
| US10815463B2 (en) | 2014-11-02 | 2020-10-27 | Arcturus Therapeutics, Inc. | Messenger UNA molecules and uses thereof |
| US20210087135A1 (en) | 2019-09-19 | 2021-03-25 | Modernatx, Inc. | Branched tail lipid compounds and compositions for intracellular delivery of therapeutic agents |
| US10961188B2 (en) | 2017-12-20 | 2021-03-30 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US10980895B2 (en) | 2016-12-21 | 2021-04-20 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| WO2021077067A1 (en) | 2019-10-18 | 2021-04-22 | The Trustees Of The University Of Pennsylvania | Lipid nanoparticles and formulations thereof for car mrna delivery |
| US20210128488A1 (en) | 2017-08-16 | 2021-05-06 | Acuitas Therapeutics, Inc. | Lipids for use in lipid nanoparticle formulations |
| US11015204B2 (en) | 2017-05-31 | 2021-05-25 | Arcturus Therapeutics, Inc. | Synthesis and structure of high potency RNA therapeutics |
| US11135312B2 (en) | 2001-06-05 | 2021-10-05 | Curevac Ag | Pharmaceutical composition containing a stabilised mRNA optimised for translation in its coding regions |
| WO2021204179A1 (en) | 2020-04-09 | 2021-10-14 | Suzhou Abogen Biosciences Co., Ltd. | Nucleic acid vaccines for coronavirus |
| US11149278B2 (en) | 2014-12-12 | 2021-10-19 | Curevac Ag | Artificial nucleic acid molecules for improved protein expression |
| US11173120B2 (en) | 2014-09-25 | 2021-11-16 | Biontech Rna Pharmaceuticals Gmbh | Stable formulations of lipids and liposomes |
| US11241493B2 (en) | 2020-02-04 | 2022-02-08 | Curevac Ag | Coronavirus vaccine |
| US11318195B2 (en) | 2016-07-15 | 2022-05-03 | BioNTech SE | Formulation for administration of RNA |
| WO2022165262A1 (en) * | 2021-01-28 | 2022-08-04 | The Broad Institute, Inc. | Compositions and methods for delivering cargo to a target cell |
| US11471522B2 (en) | 2017-03-24 | 2022-10-18 | BioNTech SE | Methods and compositions for stimulating immune response |
| US11478552B2 (en) | 2016-06-09 | 2022-10-25 | Curevac Ag | Hybrid carriers for nucleic acid cargo |
| US20230076395A1 (en) * | 2021-08-17 | 2023-03-09 | California Institute Of Technology | Cell-to-cell delivery of rna circuits |
| WO2023044343A1 (en) | 2021-09-14 | 2023-03-23 | Renagade Therapeutics Management Inc. | Acyclic lipids and methods of use thereof |
| WO2023147463A1 (en) * | 2022-01-27 | 2023-08-03 | Clemson University | Fusogenic peptide compositions and methods of cargo delivery |
| WO2023158487A1 (en) * | 2022-02-15 | 2023-08-24 | The Broad Institute, Inc. | Cell-type specific membrane fusion proteins |
-
2024
- 2024-12-26 WO PCT/US2024/061965 patent/WO2025144938A1/en active Pending
Patent Citations (106)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7244609B2 (en) | 2001-03-09 | 2007-07-17 | Cayla | Synthetic genes and bacterial plasmids devoid of CpG |
| US11135312B2 (en) | 2001-06-05 | 2021-10-05 | Curevac Ag | Pharmaceutical composition containing a stabilised mRNA optimised for translation in its coding regions |
| US9504651B2 (en) | 2002-06-28 | 2016-11-29 | Protiva Biotherapeutics, Inc. | Lipid compositions for nucleic acid delivery |
| US9163213B2 (en) | 2005-08-23 | 2015-10-20 | The Trustees Of The University Of Pennsylvania | RNA preparations comprising purified modified RNA for reprogramming cells |
| US8835108B2 (en) | 2005-08-23 | 2014-09-16 | The Trustees Of The University Of Pennsylvania | RNA containing modified nucleosides and methods of use thereof |
| US8278036B2 (en) | 2005-08-23 | 2012-10-02 | The Trustees Of The University Of Pennsylvania | RNA containing modified nucleosides and methods of use thereof |
| US10232055B2 (en) | 2005-08-23 | 2019-03-19 | The Trustees Of The University Of Pennsylvania | RNA containing modified nucleosides and methods of use thereof |
| US9750824B2 (en) | 2005-08-23 | 2017-09-05 | The Trustees Of The University Of Pennsylvania | RNA containing modified nucleosides and methods of use thereof |
| US9371511B2 (en) | 2005-08-23 | 2016-06-21 | The Trustees Of The University Of Pennsylvania | RNA preparations comprising purified modified RNA for reprogramming cells |
| US8691966B2 (en) | 2005-08-23 | 2014-04-08 | The Trustees Of The University Of Pennsylvania | RNA containing modified nucleosides and methods of use thereof |
| US8748089B2 (en) | 2005-08-23 | 2014-06-10 | The Trustees Of The University Of Pennsylvania | RNA containing modified nucleosides and methods of use thereof |
| US9012219B2 (en) | 2005-08-23 | 2015-04-21 | The Trustees Of The University Of Pennsylvania | RNA preparations comprising purified modified RNA for reprogramming cells |
| WO2009086558A1 (en) | 2008-01-02 | 2009-07-09 | Tekmira Pharmaceuticals Corporation | Improved compositions and methods for the delivery of nucleic acids |
| US8822668B2 (en) | 2008-04-15 | 2014-09-02 | Protiva Biotherapeutics, Inc. | Lipid formulations for nucleic acid delivery |
| US20100130588A1 (en) | 2008-04-15 | 2010-05-27 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for nucleic acid delivery |
| US11141378B2 (en) | 2008-04-15 | 2021-10-12 | Arbutus Biopharma Corporation | Lipid formulations for nucleic acid delivery |
| US9364435B2 (en) | 2008-04-15 | 2016-06-14 | Protiva Biotherapeutics, Inc. | Lipid formulations for nucleic acid delivery |
| US8492359B2 (en) | 2008-04-15 | 2013-07-23 | Protiva Biotherapeutics, Inc. | Lipid formulations for nucleic acid delivery |
| US8058069B2 (en) | 2008-04-15 | 2011-11-15 | Protiva Biotherapeutics, Inc. | Lipid formulations for nucleic acid delivery |
| WO2011036557A1 (en) | 2009-09-22 | 2011-03-31 | The University Of British Columbia | Compositions and methods for enhancing cellular uptake and intracellular delivery of lipid particles |
| US10006007B2 (en) | 2009-12-07 | 2018-06-26 | The Trustees Of The University Of Pennsylvania | RNA preparations comprising purified modified RNA for reprogramming cells |
| US20130053572A1 (en) | 2010-01-22 | 2013-02-28 | Steven L. Colletti | Novel Cationic Lipids for Oligonucleotide Delivery |
| US20130108685A1 (en) | 2010-04-28 | 2013-05-02 | Takeshi Kuboyama | Cationic lipid |
| US9404127B2 (en) | 2010-06-30 | 2016-08-02 | Protiva Biotherapeutics, Inc. | Non-liposomal systems for nucleic acid delivery |
| US9006417B2 (en) | 2010-06-30 | 2015-04-14 | Protiva Biotherapeutics, Inc. | Non-liposomal systems for nucleic acid delivery |
| US9518272B2 (en) | 2010-06-30 | 2016-12-13 | Protiva Biotherapeutics, Inc. | Non-liposomal systems for nucleic acid delivery |
| US10064959B2 (en) | 2010-10-01 | 2018-09-04 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US11246933B1 (en) | 2011-12-07 | 2022-02-15 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20140308304A1 (en) | 2011-12-07 | 2014-10-16 | Alnylam Pharmaceuticals, Inc. | Lipids for the delivery of active agents |
| US20130195920A1 (en) | 2011-12-07 | 2013-08-01 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US11382979B2 (en) | 2011-12-07 | 2022-07-12 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20150005363A1 (en) | 2011-12-07 | 2015-01-01 | Alnylam Pharmaceuticals, Inc. | Branched Alkyl And Cycloalkyl Terminated Biodegradable Lipids For The Delivery Of Active Agents |
| US20200164038A1 (en) | 2011-12-16 | 2020-05-28 | Modernatx, Inc. | Modified nucleoside, nucleotide, and nucleic acid compositions |
| US10912826B2 (en) | 2012-02-15 | 2021-02-09 | Curevac Ag | Nucleic acid comprising or coding for a histone stem-loop and a poly(A) sequence or a polyadenylation signal for increasing the expression of an encoded pathogenic antigen |
| US9669089B2 (en) | 2012-02-15 | 2017-06-06 | Curevac Ag | Nucleic acid comprising or coding for a histone stem-loop and a poly(A) sequence or a polyadenylation signal for increasing the expression of an encoded pathogenic antigen |
| US20210128716A1 (en) | 2012-02-15 | 2021-05-06 | Curevac Ag | Nucleic acid comprising or coding for a histone stem-loop and a poly(a) sequence or a polyadenylation signal for increasing the expression of an encoded pathogenic antigen |
| US20200345831A1 (en) | 2012-02-15 | 2020-11-05 | Curevac Ag | Nucleic acid comprising or coding for a histone stem-loop and a poly(a) sequence or a polyadenylation signal for increasing the expression of an encoded pathogenic antigen |
| US10485884B2 (en) | 2012-03-26 | 2019-11-26 | Biontech Rna Pharmaceuticals Gmbh | RNA formulation for immunotherapy |
| US10577403B2 (en) | 2012-04-02 | 2020-03-03 | Modernatx, Inc. | Modified polynucleotides for the production of secreted proteins |
| US10703789B2 (en) | 2012-04-02 | 2020-07-07 | Modernatx, Inc. | Modified polynucleotides for the production of secreted proteins |
| US10799602B2 (en) | 2013-08-21 | 2020-10-13 | Curevac Ag | Method for increasing expression of RNA-encoded proteins |
| US9950065B2 (en) | 2013-09-26 | 2018-04-24 | Biontech Rna Pharmaceuticals Gmbh | Particles comprising a shell with RNA |
| US10576146B2 (en) | 2013-09-26 | 2020-03-03 | Biontech Rna Pharmaceuticals Gmbh | Particles comprising a shell with RNA |
| US9580711B2 (en) | 2013-11-18 | 2017-02-28 | Arcturus Therapeutics, Inc. | Lipid particles with asymmetric cationic lipids for RNA delivery |
| US9567296B2 (en) | 2013-11-18 | 2017-02-14 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US11110166B2 (en) | 2014-04-01 | 2021-09-07 | Curevac Ag | Polymeric carrier cargo complex for use as an immunostimulating agent or as an adjuvant |
| US20190314496A1 (en) | 2014-04-01 | 2019-10-17 | Curevac Ag | Polymeric carrier cargo complex for use as an immunostimulating agent or as an adjuvant |
| US10106490B2 (en) | 2014-06-25 | 2018-10-23 | Acuitas Therapeutics, Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US11173120B2 (en) | 2014-09-25 | 2021-11-16 | Biontech Rna Pharmaceuticals Gmbh | Stable formulations of lipids and liposomes |
| US10815463B2 (en) | 2014-11-02 | 2020-10-27 | Arcturus Therapeutics, Inc. | Messenger UNA molecules and uses thereof |
| US11149278B2 (en) | 2014-12-12 | 2021-10-19 | Curevac Ag | Artificial nucleic acid molecules for improved protein expression |
| WO2016118724A1 (en) | 2015-01-21 | 2016-07-28 | Moderna Therapeutics, Inc. | Lipid nanoparticle compositions |
| WO2016118725A1 (en) | 2015-01-23 | 2016-07-28 | Moderna Therapeutics, Inc. | Lipid nanoparticle compositions |
| US20200246451A1 (en) | 2015-04-13 | 2020-08-06 | Curevac Real Estate Gmbh | Method for producing rna compositions |
| US10780054B2 (en) | 2015-04-17 | 2020-09-22 | Curevac Real Estate Gmbh | Lyophilization of RNA |
| US10626161B2 (en) | 2015-04-28 | 2020-04-21 | The Scripps Research Institute | Methods and compositions for protection against lentiviral infections |
| US10221127B2 (en) | 2015-06-29 | 2019-03-05 | Acuitas Therapeutics, Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US10808242B2 (en) | 2015-08-28 | 2020-10-20 | Biontech Rna Pharmaceuticals Gmbh | Method for reducing immunogenicity of RNA |
| US10266485B2 (en) | 2015-09-17 | 2019-04-23 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| US20170210697A1 (en) | 2015-09-17 | 2017-07-27 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| US9868692B2 (en) | 2015-09-17 | 2018-01-16 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| US10442756B2 (en) | 2015-09-17 | 2019-10-15 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| US10702600B1 (en) | 2015-10-22 | 2020-07-07 | Modernatx, Inc. | Betacoronavirus mRNA vaccine |
| US20170119904A1 (en) | 2015-10-28 | 2017-05-04 | Acuitas Therapeutics, Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US20200121809A1 (en) | 2015-10-28 | 2020-04-23 | Erikc A. HARWOOD | Lipid nanoparticle formulations |
| US10166298B2 (en) | 2015-10-28 | 2019-01-01 | Acuitas Therapeutics, Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US20200206362A1 (en) | 2015-12-10 | 2020-07-02 | Moderna TX, Inc. | Compositions and methods for delivery of agents |
| US11285222B2 (en) | 2015-12-10 | 2022-03-29 | Modernatx, Inc. | Compositions and methods for delivery of agents |
| US20190083602A1 (en) | 2015-12-22 | 2019-03-21 | Curevac Ag | Method for producing rna molecule compositions |
| US20190022247A1 (en) | 2015-12-30 | 2019-01-24 | Acuitas Therapeutics, Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| US10233148B2 (en) | 2015-12-30 | 2019-03-19 | Arcturus Therapeutics, Inc. | Aromatic ionizable cationic lipid |
| US11478552B2 (en) | 2016-06-09 | 2022-10-25 | Curevac Ag | Hybrid carriers for nucleic acid cargo |
| US20190240354A1 (en) | 2016-06-30 | 2019-08-08 | Arbutus Biopharma Corporation | Compositions and methods for delivering messenger rna |
| US11318195B2 (en) | 2016-07-15 | 2022-05-03 | BioNTech SE | Formulation for administration of RNA |
| US20200163878A1 (en) | 2016-10-26 | 2020-05-28 | Curevac Ag | Lipid nanoparticle mrna vaccines |
| US10980895B2 (en) | 2016-12-21 | 2021-04-20 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US20190351048A1 (en) | 2016-12-23 | 2019-11-21 | Curevac Ag | Mers coronavirus vaccine |
| US20210401971A1 (en) | 2016-12-23 | 2021-12-30 | Curevac Ag | Mers coronavirus vaccine |
| US11141476B2 (en) | 2016-12-23 | 2021-10-12 | Curevac Ag | MERS coronavirus vaccine |
| US10227302B2 (en) | 2017-02-09 | 2019-03-12 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US11471522B2 (en) | 2017-03-24 | 2022-10-18 | BioNTech SE | Methods and compositions for stimulating immune response |
| US20200046830A1 (en) | 2017-03-30 | 2020-02-13 | The Government Of The United States, As Represented By The Secretary Of The Army | Nucleic acid vaccine composition comprising a lipid formulation, and method of increasing the potency of nucleic acid vaccines |
| US11357856B2 (en) | 2017-04-13 | 2022-06-14 | Acuitas Therapeutics, Inc. | Lipids for delivery of active agents |
| US20200046838A1 (en) | 2017-04-13 | 2020-02-13 | Acuitas Therapeutics, Inc. | Lipids for delivery of active agents |
| US11015204B2 (en) | 2017-05-31 | 2021-05-25 | Arcturus Therapeutics, Inc. | Synthesis and structure of high potency RNA therapeutics |
| US9982279B1 (en) | 2017-06-23 | 2018-05-29 | Inscripta, Inc. | Nucleic acid-guided nucleases |
| US20210128488A1 (en) | 2017-08-16 | 2021-05-06 | Acuitas Therapeutics, Inc. | Lipids for use in lipid nanoparticle formulations |
| US20200172472A1 (en) | 2017-08-17 | 2020-06-04 | Acuitas Therapeutics, Inc. | Lipids for use in lipid nanoparticle formulations |
| WO2019089828A1 (en) | 2017-10-31 | 2019-05-09 | Acuitas Therapeutics, Inc. | Lamellar lipid nanoparticles |
| US10961188B2 (en) | 2017-12-20 | 2021-03-30 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| WO2019152557A1 (en) | 2018-01-30 | 2019-08-08 | Modernatx, Inc. | Compositions and methods for delivery of agents to immune cells |
| WO2019232095A1 (en) | 2018-05-30 | 2019-12-05 | Translate Bio, Inc. | Vitamin cationic lipids |
| WO2020002598A1 (en) | 2018-06-28 | 2020-01-02 | Curevac Ag | Bioreactor for rna in vitro transcription |
| WO2020097548A1 (en) | 2018-11-09 | 2020-05-14 | Arbutus Biopharma Corporation | Lipid nanoparticle formulations |
| WO2020097540A1 (en) | 2018-11-09 | 2020-05-14 | Arbutus Biopharma Corporation | Lipid nanoparticle formulations |
| US11453639B2 (en) | 2019-01-11 | 2022-09-27 | Acuitas Therapeutics, Inc. | Lipids for lipid nanoparticle delivery of active agents |
| US20200283372A1 (en) | 2019-01-11 | 2020-09-10 | Acuitas Therapeutics, Inc. | Lipids for lipid nanoparticle delivery of active agents |
| US20210087135A1 (en) | 2019-09-19 | 2021-03-25 | Modernatx, Inc. | Branched tail lipid compounds and compositions for intracellular delivery of therapeutic agents |
| WO2021077067A1 (en) | 2019-10-18 | 2021-04-22 | The Trustees Of The University Of Pennsylvania | Lipid nanoparticles and formulations thereof for car mrna delivery |
| US11241493B2 (en) | 2020-02-04 | 2022-02-08 | Curevac Ag | Coronavirus vaccine |
| WO2021204179A1 (en) | 2020-04-09 | 2021-10-14 | Suzhou Abogen Biosciences Co., Ltd. | Nucleic acid vaccines for coronavirus |
| WO2022165262A1 (en) * | 2021-01-28 | 2022-08-04 | The Broad Institute, Inc. | Compositions and methods for delivering cargo to a target cell |
| US20230076395A1 (en) * | 2021-08-17 | 2023-03-09 | California Institute Of Technology | Cell-to-cell delivery of rna circuits |
| WO2023044343A1 (en) | 2021-09-14 | 2023-03-23 | Renagade Therapeutics Management Inc. | Acyclic lipids and methods of use thereof |
| WO2023147463A1 (en) * | 2022-01-27 | 2023-08-03 | Clemson University | Fusogenic peptide compositions and methods of cargo delivery |
| WO2023158487A1 (en) * | 2022-02-15 | 2023-08-24 | The Broad Institute, Inc. | Cell-type specific membrane fusion proteins |
Non-Patent Citations (30)
| Title |
|---|
| ABRAMS ELLIOTT?W. ET AL: "Dynamic Assembly of Brambleberry Mediates Nuclear Envelope Fusion during Early Development", CELL, vol. 150, no. 3, 1 August 2012 (2012-08-01), Amsterdam NL, pages 521 - 532, XP093261964, ISSN: 0092-8674, Retrieved from the Internet <URL:https://pdf.sciencedirectassets.com/272196/1-s2.0-S0092867412X00163/1-s2.0-S0092867412008306/main.pdf?hash=34a73c88ee31da0e3b736419696c81b70f1bc60645d7a405303c89236e5b95ef&host=68042c943591013ac2b2430a89b270f6af2c76d8dfd086a07176afe7c76c2c61&pii=S0092867412008306&tid=spdf-1cb0a7ae-01de-420c-9586-fac> DOI: 10.1016/j.cell.2012.05.048 * |
| ABRAMS ET AL., CELL, vol. 150, no. 3, 2012, pages 521 - 532 |
| ADEBOYE ADEJARE, REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY, 2020 |
| ALTSCHUL, NATURE GENETICS, vol. 6, 1994, pages 119 - 129 |
| ANDREWS, MOL. THER. METHODS CLIN. DEV., vol. 7, 2017, pages 74 - 82 |
| ANSELMO ET AL., BIOENG. TRANSL. MED., vol. 1, 2016, pages 10 - 29 |
| AUSUBEL ET AL.: "Current Protocols in Molecular Biology", 1989, JOHN WILEY & SONS |
| BAIERSDORFER ET AL., THER. NUCLEIC ACIDS, vol. 15, 2019, pages 26 - 35 |
| CHAO ET AL., BIOMOLECULES, vol. 12, no. 4, 2022, pages 546 |
| FUCHS ET AL., MOL. THER. METHODS CLIN. DEV, vol. 16, 2020, pages 94 - 102 |
| GARDNER ET AL., NATURE, vol. 519, no. 7541, 2015, pages 87 - 91 |
| GHOSHBARRY, J. VIROL., vol. 79, no. 21, 2005, pages 13667 - 13672 |
| GRASEMANN ET AL., BIORXIV, 2023, Retrieved from the Internet <URL:https://doi.org/10.1101/2023.06.26.546530> |
| HARAGUCHI ET AL., COMMUN. BIOL, vol. 5, no. 1, 2022, pages 78 |
| HEMMI ET AL., NATURE, vol. 408, no. 6813, 2000, pages 740 - 745 |
| HENIKOFFHENIKOFF, PROC. NATL. ACAD. SCI. USA, vol. 89, 1992, pages 10915 - 19 |
| HONDA, IUBMB LIFE, vol. 58, no. 5-6, 2006, pages 290 - 295 |
| JACKSON ET AL., MOL. THER. METHODS CLIN. DEV., vol. 19, 2020, pages 496 - 506 |
| JOPLING, RNA BIOT,., vol. 9, no. 2, 2012, pages 137 - 142 |
| KOBAYASHI ET AL., PROC. NATL. ACAD. SCI., vol. 112, no. 22, 2015, pages 7027 - 32 |
| L. WANGI. VERMA, PROC. NATL. ACAD. SCI. USA, vol. 96, 1999, pages 3906 - 3910 |
| LI ET AL., J. BROL. CHEM., vol. 266, no. 10, 1991, pages 6562 - 6570 |
| LI ET AL., NAT. BIOTECHNOL., vol. 17, no. 3, 1999, pages 241 - 245 |
| MA, TELESE COMMUN INTEGR BIOL., vol. 8, no. 6, 23 September 2015 (2015-09-23) |
| RUTZ ET AL., EUR. J. IMMUNOL., vol. 34, no. 9, 2004, pages 2541 - 2550 |
| SCHAFFER ET AL., PROC. NATL. ACAD. SCI. USA., vol. 100, 2003, pages 4435 - 4439 |
| TABEBORDBAR ET AL., CELL, vol. 184, no. 19, 2021, pages 4919 - 4938 |
| VALENTIN ET AL., NUCLEIC ACIDS RES., vol. 49, no. 11, 2021, pages 6082 - 6099 |
| XU ET AL., EBIOMEDICINE, vol. 35, 2018, pages 97 - 105 |
| ZENG YE ET AL: "Fusogenic Coiled-Coil Peptides Enhance Lipid Nanoparticle-Mediated mRNA Delivery upon Intramyocardial Administration", ACS NANO, vol. 17, no. 23, 20 November 2023 (2023-11-20), US, pages 23466 - 23477, XP093261972, ISSN: 1936-0851, Retrieved from the Internet <URL:https://pubs.acs.org/doi/pdf/10.1021/acsnano.3c05341> DOI: 10.1021/acsnano.3c05341 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2024003220A (en) | Gene editing using modified closed-ended DNA (CEDNA) | |
| US20220280427A1 (en) | Lipid nanoparticle compositions comprising closed-ended dna and cleavable lipids and methods of use thereof | |
| US20250032642A1 (en) | Crispr-mediated transgene insertion in neonatal cells | |
| CN111868242A (en) | Closed-end DNA vectors obtainable from cell-free synthesis and methods for obtaining ceDNA vectors | |
| AU2017305404A1 (en) | Compositions and methods for treating CEP290 associated disease | |
| KR20210127935A (en) | Modification of Rep protein activity in closed-form DNA (ceDNA) production | |
| US20220133768A1 (en) | Crispr/rna-guided nuclease-related methods and compositions for treating rho-associated autosomal-dominant retinitis pigmentosa (adrp) | |
| JP2022506771A (en) | Modified Closed DNA (CEDNA) Containing Symmetrically Modified Inverted End Repeats | |
| TW202411426A (en) | Engineered class 2 type v crispr systems | |
| US20220288231A1 (en) | Methods and compositions for reducing gene or nucleic acid therapy-related immune responses | |
| TW202334194A (en) | Compositions and methods for expressing factor ix for hemophilia b therapy | |
| JP2022523806A (en) | Closed DNA (CEDNA) and immunomodulatory compounds | |
| TW202444910A (en) | Messenger rna encoding casx | |
| AU2023406273A1 (en) | Synthetic single stranded nucleic acid compositions and methods thereof | |
| WO2025144938A1 (en) | Systems for nucleic acid transfer | |
| WO2025085119A2 (en) | Synthetic partially single-stranded nucleic acid compositions and uses and methods therefor | |
| KR20250006975A (en) | Multicomponent system for site-specific genome modification | |
| US20240226334A1 (en) | Correction of duchenne muscular dystrophy mutations with all-in-one adeno-associated virus-delivered single-cut crispr | |
| WO2024249298A2 (en) | Modified closed-ended dna (cedna) vectors, compositions, and uses thereof | |
| WO2024249354A1 (en) | Methods for directing lipid nanoparticles in vivo | |
| Conley et al. | 17 Gene-Based Medicine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24841657 Country of ref document: EP Kind code of ref document: A1 |