WO2025027089A1 - Ionizable thiolipids and uses thereof - Google Patents
Ionizable thiolipids and uses thereof Download PDFInfo
- Publication number
- WO2025027089A1 WO2025027089A1 PCT/EP2024/071710 EP2024071710W WO2025027089A1 WO 2025027089 A1 WO2025027089 A1 WO 2025027089A1 EP 2024071710 W EP2024071710 W EP 2024071710W WO 2025027089 A1 WO2025027089 A1 WO 2025027089A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- lipid
- optionally substituted
- rna
- bnt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/04—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0043—Nose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/5123—Organic compounds, e.g. fats, sugars
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/30—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/31—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups having the sulfur atoms of the sulfonamide groups bound to acyclic carbon atoms
- C07C311/32—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups having the sulfur atoms of the sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/26—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C317/28—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to acyclic carbon atoms of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/24—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/25—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C335/00—Thioureas, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C335/04—Derivatives of thiourea
- C07C335/06—Derivatives of thiourea having nitrogen atoms of thiourea groups bound to acyclic carbon atoms
- C07C335/08—Derivatives of thiourea having nitrogen atoms of thiourea groups bound to acyclic carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/20—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carbonic acid, or sulfur or nitrogen analogues thereof
- C07D295/21—Radicals derived from sulfur analogues of carbonic acid
Definitions
- LNPs lipid nanoparticles
- cationic or ionizable lipids that are capable of forming complexes with nucleic acids, but have improved manufacturability and improved properties (e.g., improved transfection of nucleic acids and improved stability) relative to previous lipid formulations.
- the present disclosure provides cationic or ionizable lipid compounds comprising one or more sulfur-based moieties that avoid the problems associated with lipid compounds, while exhibiting improved properties (e.g., improved transfection) and ease of manufacture.
- the present disclosure provides a compound represented by formula I: or a pharmaceutically 1 2 L and L are each independently an optionally substituted C1-C30 aliphatic group; L 3 is a bond, optionally substituted C 1 -C 10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S; X 1 and X 2 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O) 2 N(R 1 )- , -N(R 1 )S(O) 2 , -S(O)-, -S(O) 2 -, -S(O)2C(R 1 ) 2 -, -OC(S)C(R 1 ) 2 -, -C(R 1 ) 2 C(S)O-, and –S-, wherein one or both of X 1 or X 2 is selected from
- the present disclosure provides a particle comprising a compound described herein and a nucleic acid.
- the present disclosure provides a composition (e.g., a pharmaceutical composition) comprising particles described herein.
- the present disclosure provides a method of treating a disease, disorder, or condition in a subject comprising administering to the subject a composition comprising particles described herein.
- the present disclosure provides a method of preparing a compound represented Formula IV: or a pharmaceutically accept able salt thereof, the method comprising: contacting a compound represented by Formula V with a compound represented b y one of Formulae VIa-VIc and a compound represented by one of Formulae VIIa-c in the presence of a reducing agent, wherein: each of L 4 and L 5 are each independently an optionally substituted C 1 -C 30 aliphatic group; L 6 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S; X 3 and X 4 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O) 2 N(R 40 )- , -N(R 40 )S(O) 2 , -S(O)-, -S(O) 2 -
- Figure 1 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- 51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59.
- Figure 2 is a bar graph illustrating zeta potential values of LNP formulations with BNT- 51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59.
- Figures 3A and 3B are a bar graph illustrating mRNA encapsulation efficiency ( Figure 3A) and agarose gel electropherogram (Figure 3B) of LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59.
- Figure 4 is a bar graph illustrating mRNA integrity (%) of the LNP formulations with BNT- 51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59.
- Figures 5A-5C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 on cell viability in C2C12 cells ( Figure 5A), HepG2 cells ( Figure 5B) and RAW cells (Figure 5C).
- Figures 6A-6C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 on transfection efficiency in C2C12 cells (Figure 6A), HepG2 cells (Figure 6B) and RAW cells (Figure 6C).
- Figure 7 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04.
- Figure 8 is a bar graph illustrating Zeta potential values of LNP formulations with BNT- sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04.
- Figures 9A and 9B are a bar graph illustrating mRNA encapsulation efficiency (Figure 9A) and agarose gel electropherogram (Figure 9B) of LNP formulations with BNT-sulfo- 01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04.
- Figure 10 is a bar graph mRNA integrity (%) of the LNP formulations with BNT-sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04.
- Figures 11A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04 on cell viability in C2C12 cells ( Figure 11A), HepG2 cells (Figure 11B), RAW cells (Figure 11C) and Hek293 cells ( Figure 11D).
- Figures 12A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 on transfection efficiency in C2C12 cells (Figure 12A), HepG2 cells ( Figure 12B), RAW cells (Figure 12C) and Hek293 cells (Figure 12D).
- Figures 13A and 13B are a bar graph illustrating the in vitro hemolytic effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 ( Figure 13A) and a bar graph illustrating the effect of LNP formulations on complement activation (Figure 13B).
- Figure 14 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- 51 and a helper lipid (DSPC or DOPE).
- Figure 15 is a bar graph illustrating zeta potential values of LNP formulations with BNT- 51 and a helper lipid (DSPC or DOPE).
- Figures 16A and 16B are a bar graph illustrating mRNA encapsulation efficiency (Figure 16A) and agarose gel electropherogram (Figure 16B) of LNP formulations with BNT-51 and a helper lipid.
- Figure 17 is a bar graph illustrating mRNA integrity (%) of the LNP formulations with BNT-51 and a helper lipid (DSPC or DOPE).
- Figures 18A-D are a bar graph illustrating the effect of the LNP formulations with BNT- 51 and a helper lipid on cell viability in C2C12 cells (Figure 18A), HepG2 cells (Figure 18B), RAW cells (Figure 18C) and Hek293 cells (Figure 18D).
- Figures 19A-D are a bar graph illustrating the effect of the LNP formulations with BNT- 51 and a helper lipid on transfection efficiency in C2C12 cells ( Figure 19A), HepG2 cells (Figure 19B), RAW cells (Figure 19C) and Hek293 cells (Figure 19D).
- Figure 20 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- 51 and a stealth lipid.
- Figure 21 is a bar graph illustrating zeta potential values of LNP formulations with BNT- 51 and a stealth lipid.
- Figures 22A-B are a set of bar graphs illustrating mRNA encapsulation efficiency (Figure 22A) and agarose gel electropherogram (Figure 22B) of LNP formulations with BNT-51 and a stealth lipid.
- Figure 23 is a bar graph illustrating mRNA integrity (%) of the LNP formulations with BNT-51 and a stealth lipid.
- Figures 24A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51 and a stealth lipid on cell viability C2C12 cells (Figure 24A), HepG2 cells (Figure 24B), RAW cells (Figure 24C) and Hek293 cells (Figure 24D).
- Figures 25A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51 and a stealth lipid on transfection efficiency in C2C12 cells ( Figure 25A), HepG2 cells (Figure 25B), RAW cells (Figure 25C) and Hek293 cells (Figure 25D).
- Figure 26 is a bar graph illustrating particle size and PDI of pre-formed LNPs with BNT51 and Ac-AEEA14-DMA subjected to at least three freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature.
- Figures 27A-C are a series of bar graphs illustrating characterization of BNT51 functionalized lipid nanoparticles prepared with Ac-AEEA14-DMA as stealth moiety and DSPE-PEG2k-Alfa lipid (Figure 27A); particle size and PDI of functionalized LNP1 and LNP2; ( Figure 27B); Agarose Gel Electrophoresis of controls, untreated functionalized LNP1 and LNP2 (upper row) and functionalized LNP1 and LNP2 treated with release solution (lower row) (Figure 27C); and particle size and PDI of functionalized LNP1 and LNP2 subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature, and after 2 weeks at 2-8°C and 25°C.
- Figures 28A-B are a series of bar graphs illustrating percentages of transfected cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs for both RNA (Thy1.1) ( Figure 28A) and DNA (Venus) ( Figure 28B), delivered using LNP1 or LNP2.
- Figure 29 is a bar graph illustrating particle size and PDI of preformed LNPs with BNT51 and Ac-AEEA14-VitE subjected to at least three freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature.
- Figures 30A-C are a series of bar graphs illustrating characterization of BNT51 functionalized lipid nanoparticles prepared with Ac-AEEA14-VitE as stealth moiety and DSPE-PEG2k-Alfa lipid (Figure 30A); particle size and PDI of functionalized LNP1 and LNP2 ( Figure 30B); Agarose Gel Electrophoresis of controls, untreated functionalized LNP1 and LNP2 (upper row) and functionalized LNP1 and LNP2 treated with release solution (lower row) ( Figure 30C); and particle size and PDI of functionalized LNP1 and LNP2, subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature, and after 2 weeks at 2-8°C and 25°C.
- Figures 31A-B are a series of bar graphs illustrating percentages of transfected cell (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs for both RNA (Thy1.1) ( Figure 31A) and DNA (Venus) ( Figure 31B), delivered using LNP1 or LNP2.
- Figures 32A-B are a set of bar graphs illustrating characterization of BNT51 functionalized lipid nanoparticles prepared with Ac-AEEA14-DMA as stealth moiety and DSPE-pAEEA14-Alfa lipid, including ( Figure 32A): particle size and PDI of preformed LNPs subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature; and ( Figure 32B): particle size and PDI of functionalized RNA/DNA-LNP2 at t0 and subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature.
- Figures 33A-B are a set of bar graphs illustrating characterization of BNT52 functionalized lipid nanoparticles prepared with Ac-AEEA14-DMA as stealth moiety and DSPE-pAEEA14-Alfa lipid
- Figure 33A particle size and PDI of preformed LNPs subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature
- Figure 33B Particle size and PDI of functionalized RNA/DNA-LNP2 at t0 and subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature.
- Figure 34 is a set of bar graphs illustrating use of thiolipids described herein for targeted transfection of T-cells.
- hPBMCs In vitro evaluation in hPBMCs. Comparison of three different LNPs containing three different ionizable lipids show the superior RNA and DNA transfection efficiency of thiolipids described herein which were formulated in LNP2 and LNP3. Depicted in the upper graph are the percentages of Thy1.1-expressing cell subtypes (CD4+ T cells, CD8+ T cells, CD19+ B cells) out of all single and alive cells (y ⁇ axes). Depicted in the lower graph are the percentages of Venus-Nanoplasmid expressing cells for CD4+ T cells and CD8 + T cells.
- Figures 35A and 35B illustrate the use of reported thiolipids for targeted transfection of T-cells. In vitro evaluation in hPBMCs. Comparison of two different LNPs containing two different ionizable lipids show high RNA and DNA transfection efficiency of reported thiolipids which were formulated in LNP1 and LNP2. Depicted in Figure 35A are the percentages of Thy1.1-expressing cell subtypes (CD4+ T cells, CD8+ T cells, CD19+ B cells, CD14+ Monocytes) out of all single and alive cells (y ⁇ axes). Depicted in Figure 35B are the percentages of Venus-Nanoplasmid expressing cells for CD4+ T cells and CD8 + T cells.
- Figures 36A and 36B are a set of bar charts illustrating characterization of the LNP formulations with BNT-72, including particle size and PDI ( Figure 36A) and zeta potential ( Figure 36B).
- Figures 37A and 37B are a set of bar graphs illustrating mRNA encapsulation efficiency (Figure 37A) and mRNA integrity (Figure 37B) of the of LNP formulations with BNT-72.
- Figures 38A-C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-72 on cell viability in C2C12 cells (Figure 38A), HepG2 cells (Figure 38B) and RAW cells (Figure 38C).
- Figures 39A-C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-72 on transfection efficiency in C2C12 cells (Figure 39A), HepG2 cells (Figure 39B) and RAW cells (Figure 39C).
- Figure 40 is a bar graph illustrating the size (nm) and polydispersity index (PDI) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM.
- Figure 41 is a bar graph illustrating Z-potential (mV) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM.
- Figure 42 is a bar graph illustrating RNA integrity (%) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM.
- Figure 43 is a bar graph illustrating osmolarity and pH of complexes comprising BNT- 76, BNT-90 or a benchmark lipid CM12_BM.
- Figures 44A-44D are a series of bar graphs illustrating transfection efficiency in C2C12 cells ( Figure 44A), HepG2 cells ( Figure 44B), RAW cells (Figure 44C) and Hek293 cells (Figure 44D).
- Figures 45A-45D are a series of bar graphs illustrating cell viability in C2C12 cells (Figure 45A), HepG2 cells (Figure 45B), RAW cells (Figure 45C) and Hek293 cells (Figure 45D).
- Figure 46 depicts the size and PDI of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C. The results of three measurements, as well as their average and error bars indicating 1x standard deviation are shown.
- Figure 47 depicts the percentages of transfected cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C.
- Figure 48 depicts the cell counts of alive cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs treated with aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, which were either applied freshly prepared or after one freeze/thaw cycle at -80°C.
- Figure 49 is a graph providing size and distribution (PDI) of provided LNPs comprising lipids reported herein.
- Figure 50 is a graph providing Z-potential of provided LNPs comprising lipids described herein.
- Figure 51 is a graph illustrating encapsulation efficiency of provided LNPs.
- Figure 52 is an agarose gel electrophoresis showing no free RNA for provided LNPs.
- Figures 53A-D are a series of graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage.
- Figures 54A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs.
- Figure 55 is a graph illustrating size and distribution (PDI) for provided LNPs comprising lipid compounds described herein.
- Figure 56 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein.
- Figure 57 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein.
- Figure 58 is an agarose gel electrophoresis image showing no free RNA for all provided LNPs.
- Figures 59A-D are a series of graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage.
- Figures 60A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs.
- Figure 61 is a graph illustrating size and distribution (PDI) of LNPs comprising lipid compounds described herein.
- Figure 62 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein.
- Figure 63 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein.
- Figure 64 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs.
- Figures 65A-D are a series graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage.
- Figures 66A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs.
- Figure 67 is a graph illustrating size and distribution (PDI) of LNPs comprising lipid compounds described herein.
- Figure 68 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein.
- Figure 69 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein.
- Figure 70 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs.
- Figures 71A-D are a series graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage.
- Figures 72A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs.
- Figure 73 is a graph illustrating size and distribution (PDI) of LNPs comprising lipid compounds described herein.
- Figure 74 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein.
- Figure 75 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein.
- Figure 76 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs.
- Figures 77A-B are a series graphs illustrating cytotoxicity in provided cell lines HepG2 and Hek as measured by viability percentage.
- Figures 78A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs.
- the present disclosure provides, among other things, the surprising discovery of particular cationic or ionizable lipids comprising at least one sulfur-containing moiety.
- Such cationic or ionizable lipids exhibit improved properties (e.g., improved transfection to cells) relative to previous lipids.
- thiolipids of the present disclosure exhibit improvements over previous lipids that lack a sulfur-containing moiety, including, for example, improved transfection of cells with RNA.
- Particles prepared using thiolipid compounds described herein exhibit narrow size distribution (i.e., are substantially uniform in size), high encapsulation efficiency, low cytotoxicity, and improved biodistribution and efficacy relative to particles comprising previous lipid compounds.
- Compounds and Definitions Compounds of this disclosure include those described generally above and are further illustrated by the classes, subclasses, and species disclosed herein. As used herein, the following definitions shall apply unless otherwise indicated.
- the chemical elements are identified in accordance with the Periodic Table of Elements, CAS version, Handbook of Chemistry and Physics, 75 th Ed.
- the term “approximately” or “about” may encompass a range of values that are within (i.e., ⁇ ) 25%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or less of the referred value.
- Administering typically refers to the administration of a composition to a subject to achieve delivery of an agent that is, or is included in, a composition to a target site or a site to be treated.
- administration may be ocular, oral, parenteral, topical, etc.
- administration may be bronchial (e.g., by bronchial instillation), buccal, dermal (which may be or comprise, for example, one or more of topical to the dermis, intradermal, interdermal, transdermal, etc.), enteral, intra-arterial, intradermal, intragastric, intramedullary, intramuscular, intranasal, intraperitoneal, intrathecal, intravenous, intraventricular, within a specific organ (e.g., intrahepatic), mucosal, nasal, oral, rectal, subcutaneous, sublingual, topical, tracheal (e.g., by intratracheal instillation), vaginal, vitreal, etc.
- bronchial e.g., by bronchial instillation
- buccal which may be or comprise, for example, one or more of topical to the dermis, intradermal, interdermal, transdermal, etc.
- enteral intra-arterial, intradermal, intragas
- administration may be parenteral. In some embodiments, administration may be oral. In some particular embodiments, administration may be intravenous. In some particular embodiments, administration may be subcutaneous. In some embodiments, administration may involve only a single dose. In some embodiments, administration may involve application of a fixed number of doses. In some embodiments, administration may involve dosing that is intermittent (e.g., a plurality of doses separated in time) and/or periodic (e.g., individual doses separated by a common period of time) dosing. In some embodiments, administration may involve continuous dosing (e.g., perfusion) for at least a selected period of time. In some embodiments, administration may comprise a prime-and-boost protocol.
- a prime-and-boost protocol can include administration of a first dose of a pharmaceutical composition (e.g., an immunogenic composition, e.g., a vaccine) followed by, after an interval of time, administration of a second or subsequent dose of a pharmaceutical composition (e.g., an immunogenic composition, e.g., a vaccine).
- a prime-and-boost protocol can result in an increased immune response in a patient.
- Aliphatic refers to a straight-chain (i.e., unbranched) or branched, substituted or unsubstituted hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation, or a monocyclic hydrocarbon or bicyclic hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic (also referred to herein as “cycloaliphatic”), that has a single point or more than one points of attachment to the rest of the molecule.
- aliphatic groups contain 1-12 aliphatic carbon atoms. In some embodiments, aliphatic groups contain 1-6 aliphatic carbon atoms (e.g., C 1-6 ).
- aliphatic groups contain 1-5 aliphatic carbon atoms (e.g., C 1-5 ). In other embodiments, aliphatic groups contain 1-4 aliphatic carbon atoms (e.g., C1-4). In still other embodiments, aliphatic groups contain 1-3 aliphatic carbon atoms (e.g., C1-3), and in yet other embodiments, aliphatic groups contain 1-2 aliphatic carbon atoms (e.g., C 1-2 ). Suitable aliphatic groups include, but are not limited to, linear or branched, substituted or unsubstituted alkyl, alkenyl, or alkynyl groups and hybrids thereof.
- a preferred aliphatic group is C 1-6 alkyl.
- Alkyl The term “alkyl”, used alone or as part of a larger moiety, refers to a saturated, optionally substituted straight or branched chain hydrocarbon group having (unless otherwise specified) 1-12, 1-10, 1-8, 1-6, 1-4, 1-3, or 1-2 carbon atoms (e.g., C 1-12 , C 1-10 , C 1-8 , C 1-6 , C 1-4 , C 1-3 , or C 1-2 ).
- Exemplary alkyl groups include methyl, ethyl, propyl, butyl, pentyl, hexyl, and heptyl.
- Alkylene refers to a bivalent alkyl group. In some embodiments, “alkylene” is a bivalent straight or branched alkyl group. In some embodiments, an "alkylene chain" is a polymethylene group, i.e., -(CH2)n-, wherein n is a positive integer, e.g., from 1 to 6, from 1 to 4, from 1 to 3, from 1 to 2, or from 2 to 3.
- An optionally substituted alkylene chain is a polymethylene group in which one or more methylene hydrogen atoms is optionally replaced with a substituent. Suitable substituents include those described below for a substituted aliphatic group and also include those described in the specification herein.
- two substituents of the alkylene group may be taken together to form a ring system.
- two substituents can be taken together to form a 3- to 7-membered ring.
- the substituents can be on the same or different atoms.
- the suffix “-ene” or “-enyl” when appended to certain groups herein are intended to refer to a bifunctional moiety of said group.
- Alkenyl refers to an optionally substituted straight or branched chain or cyclic hydrocarbon group having at least one double bond and having (unless otherwise specified) 2-12, 2-10, 2-8, 2-6, 2-4, or 2-3 carbon atoms(e.g., C 2-12 , C 2-10 , C 2-8 , C 2-6 , C 2-4 , or C 2-3 ).
- alkenyl groups include ethenyl, propenyl, butenyl, pentenyl, hexenyl, and heptenyl.
- cycloalkenyl refers to an optionally substituted non-aromatic monocyclic or multicyclic ring system containing at least one carbon-carbon double bond and having about 3 to about 10 carbon atoms.
- exemplary monocyclic cycloalkenyl rings include cyclopentenyl, cyclohexenyl, and cycloheptenyl.
- Alkynyl refers to an optionally substituted straight or branched chain hydrocarbon group having at least one triple bond and having (unless otherwise specified) 2-12, 2-10, 2-8, 2-6, 2-4, or 2-3 carbon atoms (e.g., C 2-12 , C 2-10 , C 2-8 , C 2-6 , C 2-4 , or C 2-3 ).
- exemplary alkynyl groups include ethynyl, propynyl, butynyl, pentynyl, hexynyl, and heptynyl.
- Aryl refers to monocyclic and bicyclic ring systems having a total of six to fourteen ring members (e.g., C 6 -C 14 ), wherein at least one ring in the system is aromatic and wherein each ring in the system contains three to seven ring members. In some embodiments, an “aryl” group contains between six and twelve total ring members (e.g., C6-C12). The term “aryl” may be used interchangeably with the term “aryl ring”. In certain embodiments, “aryl” refers to an aromatic ring system which includes, but not limited to, phenyl, biphenyl, naphthyl, anthracyl and the like, which may bear one or more substituents.
- aryl groups are hydrocarbons.
- an “aryl” ring system is an aromatic ring (e.g., phenyl) that is fused to a non-aromatic ring (e.g., cycloalkyl). Examples of aryl rings include that are fused include .
- Two events or entities are “associated” with one another, as that term is used herein, if the presence, level and/or form of one is correlated with that of the other.
- a particular entity e.g., polypeptide, genetic signature, metabolite, microbe, etc
- a particular disease, disorder, or condition if its presence, level and/or form correlates with incidence of and/or susceptibility to the disease, disorder, or condition (e.g., across a relevant population).
- two or more entities are physically “associated” with one another if they interact, directly or indirectly, so that they are and/or remain in physical proximity with one another.
- bicyclic ring or “bicyclic ring system” refers to any bicyclic ring system, i.e., carbocyclic or heterocyclic, saturated or having one or more units of unsaturation, having one or more atoms in common between the two rings of the ring system.
- the term includes any permissible ring fusion, such as ortho-fused or spirocyclic.
- heterocyclic is a subset of “bicyclic” that requires that one or more heteroatoms are present in one or both rings of the bicycle. Such heteroatoms may be present at ring junctions and are optionally substituted, and may be selected from nitrogen (including N-oxides), oxygen, sulfur (including oxidized forms such as sulfones and sulfonates), phosphorus (including oxidized forms such as phosphates), boron, etc.
- a bicyclic group has 7-12 ring members and 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- bridged bicyclic refers to any bicyclic ring system, i.e., carbocyclic or heterocyclic, saturated or partially unsaturated, having at least one bridge.
- a “bridge” is an unbranched chain of atoms or an atom or a valence bond connecting two bridgeheads, where a “bridgehead” is any skeletal atom of the ring system which is bonded to three or more skeletal atoms (excluding hydrogen).
- a bridged bicyclic group has 7-12 ring members and 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- bridged bicyclic groups are well known in the art and include those groups set forth below where each group is attached to the rest of the molecule at any substitutable carbon or nitrogen atom. Unless otherwise specified, a bridged bicyclic group is optionally substituted with one or more substituents as set forth for aliphatic groups. Additionally or alternatively, any substitutable nitrogen of a bridged bicyclic group is optionally substituted.
- Exemplary bicyclic rings include:
- biological sample typically refers to a sample obtained or derived from a biological source (e.g., a tissue or organism or cell culture) of interest, as described herein.
- a source of interest comprises an organism, such as an animal or human.
- a biological sample is or comprises biological tissue or fluid.
- a biological sample may be or comprise bone marrow; blood; blood cells; ascites; tissue or fine needle biopsy samples; cell-containing body fluids; free floating nucleic acids; sputum; saliva; urine; cerebrospinal fluid, peritoneal fluid; pleural fluid; feces; lymph; gynecological fluids; skin swabs; vaginal swabs; oral swabs; nasal swabs; washings or lavages such as a ductal lavages or broncheoalveolar lavages; aspirates; scrapings; bone marrow specimens; tissue biopsy specimens; surgical specimens; feces, other body fluids, secretions, and/or excretions; and/or cells therefrom, etc.
- a biological sample is or comprises cells obtained from an individual.
- obtained cells are or include cells from an individual from whom the sample is obtained.
- a sample is a “primary sample” obtained directly from a source of interest by any appropriate means.
- a primary biological sample is obtained by methods selected from the group consisting of biopsy (e.g., fine needle aspiration or tissue biopsy), surgery, collection of body fluid (e.g., blood, lymph, feces etc.), etc.
- sample refers to a preparation that is obtained by processing (e.g., by removing one or more components of and/or by adding one or more agents to) a primary sample. For example, filtering using a semi-permeable membrane.
- a “processed sample” may comprise, for example, nucleic acids or proteins extracted from a sample or obtained by subjecting a primary sample to techniques such as amplification or reverse transcription of mRNA, isolation and/or purification of certain components, etc.
- Carrier refers to a diluent, adjuvant, excipient, or vehicle with which a composition is administered.
- carriers can include sterile liquids, such as, for example, water and oils, including oils of petroleum, animal, vegetable or synthetic origin, such as, for example, peanut oil, soybean oil, mineral oil, sesame oil and the like. In some embodiments, carriers are or include one or more solid components.
- Combination therapy refers to those situations in which a subject is simultaneously exposed to two or more therapeutic regimens (e.g., two or more therapeutic agents or modality(ies)).
- the two or more regimens may be administered simultaneously; in some embodiments, such regimens may be administered sequentially (e.g., all “doses” of a first regimen are administered prior to administration of any doses of a second regimen); in some embodiments, such agents are administered in overlapping dosing regimens.
- “administration” of combination therapy may involve administration of one or more agent(s) or modality(ies) to a subject receiving the other agent(s) or modality(ies) in the combination.
- combination therapy does not require that individual agents be administered together in a single composition (or even necessarily at the same time), although in some embodiments, two or more agents, or active moieties thereof, may be administered together in a combination composition, or even in a combination compound (e.g., as part of a single chemical complex or covalent entity).
- the term “comparable” refers to two or more agents, entities, situations, sets of conditions, etc., that may not be identical to one another but that are sufficiently similar to permit comparison therebetween so that one skilled in the art will appreciate that conclusions may reasonably be drawn based on differences or similarities observed.
- comparable sets of conditions, circumstances, individuals, or populations are characterized by a plurality of substantially identical features and one or a small number of varied features.
- Those of ordinary skill in the art will understand, in context, what degree of identity is required in any given circumstance for two or more such agents, entities, situations, sets of conditions, etc. to be considered comparable.
- sets of circumstances, individuals, or populations are comparable to one another when characterized by a sufficient number and type of substantially identical features to warrant a reasonable conclusion that differences in results obtained or phenomena observed under or with different sets of circumstances, individuals, or populations are caused by or indicative of the variation in those features that are varied.
- composition may be used to refer to a discrete physical entity that comprises one or more specified components.
- a composition may be of any form – e.g., gas, gel, liquid, solid, etc.
- Cycloaliphatic As used herein, the term “cycloaliphatic” refers to a monocyclic C 3-8 hydrocarbon or a bicyclic C 6-10 hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic, that has a single point or more than one points of attachment to the rest of the molecule.
- Cycloalkyl refers to an optionally substituted saturated ring monocyclic or polycyclic system of about 3 to about 10 ring carbon atoms.
- Exemplary monocyclic cycloalkyl rings include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl.
- DNA refers to a polymeric molecule of nucleotides that are typically double-stranded and comprise adenine, cytosine, guanine and thymine, and a deoxyribose sugar backbone structure as specified in the definition “Nucleic Acid/Polynucleotide.” In some embodiments, DNA is linear DNA, plasmid DNA, minicircle DNA, nanoplasmid DNA, doggybone DNA, or a transposon.
- Deoxyribonucleotide As used herein, the term “deoxyribonucleotide” refers to unmodified and modified deoxyribonucleotides.
- unmodified deoxyribonucleotides include the purine bases adenine (A) and guanine (G), and the pyrimidine bases cytosine (C) and thymine (T).
- Modified deoxyribonucleotides may include one or more modifications including, but not limited to, for example, (a) end modifications, e.g., 5' end modifications (e.g., phosphorylation, dephosphorylation, conjugation, inverted linkages, etc.), 3' end modifications (e.g., conjugation, inverted linkages, etc.), (b) base modifications, e.g.
- Dosage form or unit dosage form may be used to refer to a physically discrete unit of an active agent (e.g., a therapeutic or diagnostic agent) for administration to a subject. Typically, each such unit contains a predetermined quantity of active agent.
- an active agent e.g., a therapeutic or diagnostic agent
- such quantity is a unit dosage amount (or a whole fraction thereof) appropriate for administration in accordance with a dosing regimen that has been determined to correlate with a desired or beneficial outcome when administered to a relevant population (i.e., with a therapeutic dosing regimen).
- Dosing regimen or therapeutic regimen Those skilled in the art will appreciate that the terms “dosing regimen” and “therapeutic regimen” may be used to refer to a set of unit doses (typically more than one) that are administered individually to a subject, typically separated by periods of time. In some embodiments, a given therapeutic agent has a recommended dosing regimen, which may involve one or more doses.
- a dosing regimen comprises a plurality of doses each of which is separated in time from other doses. In some embodiments, individual doses are separated from one another by a time period of the same length; in some embodiments, a dosing regimen comprises a plurality of doses and at least two different time periods separating individual doses. In some embodiments, all doses within a dosing regimen are of the same unit dose amount. In some embodiments, different doses within a dosing regimen are of different amounts. In some embodiments, a dosing regimen comprises a first dose in a first dose amount, followed by one or more additional doses in a second dose amount different from the first dose amount.
- a dosing regimen comprises a first dose in a first dose amount, followed by one or more additional doses in a second dose amount same as the first dose amount.
- a dosing regimen is correlated with a desired or beneficial outcome when administered across a relevant population (i.e., is a therapeutic dosing regimen).
- Effective Amount refers to the amount of a compound sufficient to effect beneficial or desired results (e.g., a therapeutic, ameliorative, inhibitory, or preventative result). An effective amount can be administered in one or more administrations, applications, or dosages and is not intended to be limited to a particular formulation or administration route.
- Excipient refers to a non-therapeutic agent that may be included in a pharmaceutical composition, for example, to provide or contribute to a desired consistency or stabilizing effect.
- suitable pharmaceutical excipients include, for example, starch, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, sodium stearate, glycerol monostearate, talc, sodium chloride, dried skim milk, glycerol, propylene, glycol, water, ethanol and the like.
- Halogen The term “halogen” or “halo” means F, Cl, Br, or I.
- heteroaliphatic or “heteroaliphatic group”, as used herein, denotes an optionally substituted hydrocarbon moiety having, in addition to carbon atoms, from one to five heteroatoms, that may be straight–chain (i.e., unbranched), branched, or cyclic (“heterocyclic”) and may be completely saturated or may contain one or more units of unsaturation, but which is not aromatic.
- heteroatom refers to nitrogen, oxygen, or sulfur, and includes any oxidized form of nitrogen or sulfur, and any quaternized form of a basic nitrogen.
- nitrogen also includes a substituted nitrogen.
- heteroaliphatic groups contain 1–10 carbon atoms wherein 1–3 carbon atoms are optionally and independently replaced with heteroatoms selected from oxygen, nitrogen, and sulfur. In some embodiments, heteroaliphatic groups contain 1–4 carbon atoms, wherein 1–2 carbon atoms are optionally and independently replaced with heteroatoms selected from oxygen, nitrogen, and sulfur. In yet other embodiments, heteroaliphatic groups contain 1–3 carbon atoms, wherein 1 carbon atom is optionally and independently replaced with a heteroatom selected from oxygen, nitrogen, and sulfur. Suitable heteroaliphatic groups include, but are not limited to, linear or branched, heteroalkyl, heteroalkenyl, and heteroalkynyl groups.
- a 1- to 10 atom heteroaliphatic group includes the following exemplary groups: -O-CH 3 , -CH 2 -O-CH 3 , -O-CH 2 -CH 2 -O-CH 2 -CH 2 -O-CH 3 , and the like.
- Heteroaryl and “heteroar—”, used alone or as part of a larger moiety, e.g., “heteroaralkyl”, or “heteroaralkoxy”, refer to monocyclic or bicyclic ring groups having 5 to 10 ring atoms (e.g., 5- to 6-membered monocyclic heteroaryl or 9- to 10-membered bicyclic heteroaryl); having 6, 10, or 14 ⁇ -electrons shared in a cyclic array; and having, in addition to carbon atoms, from one to five heteroatoms.
- Heteroaryl groups include, without limitation, thienyl, furanyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, indolizinyl, purinyl, naphthyridinyl, pteridinyl, imidazo[1,2-a]pyrimidinyl, imidazo[1,2-a]pyridyl, imidazo[4,5-b]pyridyl, imidazo[4,5- c]pyridyl, pyrrolopyridyl, pyrrolopyrazinyl, thienopyrimidinyl, triazolopyridyl, and benzoisox
- heteroaryl and “heteroar—”, as used herein, also include groups in which a heteroaromatic ring is fused to one or more aryl, cycloaliphatic, or heterocyclyl rings, where the radical or point of attachment is on the heteroaromatic ring (i.e., a bicyclic heteroaryl ring having 1 to 3 heteroatoms).
- Nonlimiting examples include indolyl, isoindolyl, benzothienyl, benzofuranyl, dibenzofuranyl, indazolyl, benzimidazolyl, benzotriazolyl, benzothiazolyl, benzothiadiazolyl, benzoxazolyl, quinolyl, isoquinolyl, cinnolinyl, phthalazinyl, quinazolinyl, quinoxalinyl, 4H–quinolizinyl, carbazolyl, acridinyl, phenazinyl, phenothiazinyl, phenoxazinyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, pyrido[2,3–b]–1,4–oxazin–3(4H)–one, 4H-thieno[3,2-b]pyrrole, and benzoisoxazolyl.
- heteroaryl may be used interchangeably with the terms “heteroaryl ring”, “heteroaryl group”, or “heteroaromatic”, any of which terms include rings that are optionally substituted.
- Heteroatom refers to nitrogen, oxygen, or sulfur, and includes any oxidized form of nitrogen or sulfur, and any quaternized form of a basic nitrogen.
- Heterocycle As used herein, the terms “heterocycle”, “heterocyclyl”, “heterocyclic radical”, and “heterocyclic ring” are used interchangeably and refer to a stable 3- to 8- membered monocyclic, a 6- to 10-membered bicyclic, or a 10- to 16-membered polycyclic heterocyclic moiety that is either saturated or partially unsaturated, and having, in addition to carbon atoms, one or more, such as one to four, heteroatoms, as defined above.
- nitrogen includes a substituted nitrogen.
- the nitrogen may be N (as in 3,4-dihydro-2H-pyrrolyl), NH (as in pyrrolidinyl), or NR + (as in N-substituted pyrrolidinyl).
- a heterocyclic ring can be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure and any of the ring atoms can be optionally substituted.
- saturated or partially unsaturated heterocyclic radicals include, without limitation, azetidinyl, oxetanyl, tetrahydrofuranyl, tetrahydrothienyl, pyrrolidinyl, piperidinyl, decahydroquinolinyl, oxazolidinyl, piperazinyl, dioxanyl, dioxolanyl, diazepinyl, oxazepinyl, thiazepinyl, morpholinyl, and thiamorpholinyl.
- a heterocyclyl group may be mono-, bi-, tri-, or polycyclic, preferably mono-, bi-, or tricyclic, more preferably mono- or bicyclic.
- a bicyclic heterocyclic ring also includes groups in which the heterocyclic ring is fused to one or more aryl rings.
- Exemplary bicyclic heterocyclic groups include indolinyl, isoindolinyl, benzodioxolyl, 1,3- dihydroisobenzofuranyl, 2,3-dihydrobenzofuranyl, and tetrahydroquinolinyl.
- a bicyclic heterocyclic ring can also be a spirocyclic ring system (e.g., 7- to 11-membered spirocyclic fused heterocyclic ring having, in addition to carbon atoms, one or more heteroatoms as defined above (e.g., one, two, three or four heteroatoms)).
- a bicyclic heterocyclic ring can also be a bridged ring system (e.g., 7- to 11-membered bridged heterocyclic ring having one, two, or three bridging atoms.
- Nanoparticle refers to a discrete entity of small size, e.g., typically having a longest dimension that is shorter than about 1000 nanometers (nm) and often is shorter than 500 nm, or even 100 nm or less. In many embodiments, a nanoparticle may be characterized by a longest dimension between about 1 nm and about 100 nm, or between about 1 ⁇ m and about 500 nm, or between about 1 nm and 1000 nm.
- a population of microparticles is characterized by an average size (e.g., longest dimension) that is below about 1000 nm, about 500 nm, about 100 nm, about 50 nm, about 40 nm, about 30 nm, about 20 nm, or about 10 nm and often above about 1 nm.
- a microparticle may be substantially spherical (e.g., so that its longest dimension may be its diameter).
- a nanoparticle has a diameter of less than 100 nm as defined by the National Institutes of Health.
- nanoparticles are micelles in that they comprise an enclosed compartment, separated from the bulk solution by a micellar membrane, typically comprised of amphiphilic entities which surround and enclose a space or compartment (e.g., to define a lumen).
- a micellar membrane is comprised of at least one polymer, such as for example a biocompatible and/or biodegradable polymer.
- Nucleic acid/ Polynucleotide As used herein, the term “nucleic acid” refers to a polymer of at least 10 nucleotides or more.
- a nucleic acid is or comprises DNA.
- a nucleic acid is or comprises RNA.
- a nucleic acid is or comprises a mixture of DNA and RNA. In some embodiments, a nucleic acid is or comprises peptide nucleic acid (PNA). In some embodiments, a nucleic acid is or comprises a single stranded nucleic acid. In some embodiments, a nucleic acid is or comprises a double-stranded nucleic acid. In some embodiments, a nucleic acid comprises both single and double-stranded portions. In some embodiments, a nucleic acid comprises a backbone that comprises one or more phosphodiester linkages. In some embodiments, a nucleic acid comprises a backbone that comprises both phosphodiester and non-phosphodiester linkages.
- PNA peptide nucleic acid
- a nucleic acid may comprise a backbone that comprises one or more phosphorothioate, phosphorodithioate, phosphoramide, phosphite-borane complexes, or 5'-N-phosphoramidite linkages and/or one or more peptide bonds, e.g., as in a “peptide nucleic acid”.
- a nucleic acid comprises one or more, or all, natural residues (e.g., adenine, cytosine, deoxyadenosine, deoxycytidine, deoxyguanosine, deoxythymidine, guanine, thymine, uracil).
- a nucleic acid comprises on or more, or all, non-natural residues.
- a non-natural residue comprises a nucleoside analog (e.g., 2-aminoadenosine, 2- thiothymidine, inosine, pyrrolo-pyrimidine, 3 -methyl adenosine, 5-methylcytidine, C-5 propynyl-cytidine, C-5 propynyl-uridine, 2-aminoadenosine, C5-bromouridine, C5- fluorouridine, C5-iodouridine, C5-propynyl-uridine, C5 -propynyl-cytidine, C5- methylcytidine, 2-aminoadenosine, 7-deazaadenosine, 7-deazaguanosine, 8- oxoadenosine, 8-oxoguanosine, 6-O-methylguanine
- a non-natural residue comprises one or more modified sugars (e.g., 2'-fluororibose, ribose, 2'- deoxyribose, arabinose, and hexose) as compared to those in natural residues.
- a nucleic acid has a nucleotide sequence that encodes a functional gene product such as an RNA or polypeptide.
- a nucleic acid has a nucleotide sequence that comprises one or more introns.
- a nucleic acid may be prepared by isolation from a natural source, enzymatic synthesis (e.g., by polymerization based on a complementary template, e.g., in vivo or in vitro, reproduction in a recombinant cell or system, or chemical synthesis.
- enzymatic synthesis e.g., by polymerization based on a complementary template, e.g., in vivo or in vitro, reproduction in a recombinant cell or system, or chemical synthesis.
- a nucleic acid is at least 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 600, 700, 800, 900, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 5000, 5500, 6000, 6500, 7000, 7500, 8000, 8500, 9000, 9500, 10,000, 10,500, 11,000, 11,500, 12,000, 12,500, 13,000, 13,500, 14,000, 14,500, 15,000, 15,500, 16,000, 16,500, 17,000, 17,500, 18,000, 18,500, 19,000, 19,500, or 20,000 or more residues or nucleotides long.
- Nucleic acid particle can be used to deliver nucleic acid to a target site of interest (e.g., cell, tissue, organ, and the like).
- a nucleic acid particle may be formed from at least one cationic or cationically ionizable lipid or lipid-like material, at least one cationic polymer such as protamine, or a mixture thereof and nucleic acid.
- Nucleic acid particles include lipid nanoparticle (LNP)-based and lipoplex (LPX)-based formulations.
- Nucleotide As used herein, the term “nucleotide” refers to its art-recognized meaning.
- a certain number of nucleotides refers to the number of nucleotides on a single strand, e.g., of a polynucleotide.
- parenteral administration and “administered parenterally” as used herein have their art-understood meaning referring to modes of administration other than enteral and topical administration, usually by injection, and include, without limitation, intravenous, intramuscular, intra-arterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intraarticulare, subcapsular, subarachnoid, intraspinal, and intrasternal injection and infusion.
- Partially unsaturated As used herein, the term “partially unsaturated” refers to a ring moiety that includes at least one double or triple bond between ring atoms.
- a patient or subject refers to any organism to which a provided composition is or may be administered, e.g., for experimental, diagnostic, prophylactic, cosmetic, and/or therapeutic purposes. Typical patients or subjects include animals (e.g., mammals such as mice, rats, rabbits, non-human primates, and/or humans). In some embodiments, a patient is a human. In some embodiments, a patient or a subject is suffering from or susceptible to one or more disorders or conditions.
- a patient or subject displays one or more symptoms of a disorder or condition.
- a patient or subject has been diagnosed with one or more disorders or conditions.
- a patient or a subject is receiving or has received certain therapy to diagnose and/or to treat a disease, disorder, or condition.
- Pharmaceutical composition refers to an active agent, formulated together with one or more pharmaceutically acceptable carriers.
- the active agent is present in unit dose amount appropriate for administration in a therapeutic or dosing regimen that shows a statistically significant probability of achieving a predetermined therapeutic effect when administered to a relevant population.
- compositions may be specially formulated for administration in solid or liquid form, including those adapted for the following: oral administration, for example, drenches (aqueous or non-aqueous solutions or suspensions), tablets, e.g., those targeted for buccal, sublingual, and systemic absorption, boluses, powders, granules, pastes for application to the tongue; parenteral administration, for example, by subcutaneous, intramuscular, intravenous or epidural injection as, for example, a sterile solution or suspension, or sustained-release formulation; topical application, for example, as a cream, ointment, or a controlled-release patch or spray applied to the skin, lungs, or oral cavity; intravaginally or intrarectally, for example, as a pessary, cream, or foam; sublingually; ocularly; transdermally; or nasally, pulmonary, and to other mucosal surfaces.
- oral administration for example, drenches (aqueous or non-aqueous solutions or suspension
- pharmaceutically acceptable refers to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- Pharmaceutically acceptable salt refers to salts of such compounds that are appropriate for use in pharmaceutical contexts, i.e., salts which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio.
- salts are well known in the art.
- S. M. Berge, et al. describes pharmaceutically acceptable salts in detail in J. Pharmaceutical Sciences, 66: 1-19 (1977).
- examples of pharmaceutically acceptable, nontoxic acid addition salts are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange.
- salts include adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2–hydroxy– ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2–naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pec
- Salts derived from appropriate bases include alkali metal, alkaline earth metal, ammonium and N + (C 1–4 alkyl) 4 salts.
- Representative alkali or alkaline earth metal salts include sodium, lithium, potassium, calcium, magnesium, and the like.
- Further pharmaceutically acceptable salts include, when appropriate, nontoxic ammonium, quaternary ammonium, and amine cations formed using counterions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, loweralkyl sulfonate and aryl sulfonate.
- Physiological conditions as used herein, has its art-understood meaning referencing conditions under which cells or organisms live and/or reproduce.
- physiological conditions are those conditions present within the body of a human or non-human animal, especially those conditions present at and/or within a surgical site.
- Physiological conditions typically include, e.g., a temperature range of 20 - 40°C, atmospheric pressure of 1, pH of 6-8, glucose concentration of 1-20 mM, oxygen concentration at atmospheric levels, and gravity as it is encountered on earth.
- conditions in a laboratory are manipulated and/or maintained at physiologic conditions.
- physiological conditions are encountered in an organism.
- polycyclic refers to a saturated or unsaturated ring system having two or more rings (for example, heterocyclyl rings, heteroaryl rings, cycloalkyl rings, or aryl rings), having between 7 and 20 atoms, in which one or more carbon atoms are common to two adjacent rings.
- a polycyclic ring system refers to a saturated or unsaturated ring system having three or more rings (for example, heterocyclyl rings, heteroaryl rings, cycloalkyl rings, or aryl rings), having between 14 and 20 atoms, in which one or more carbon atoms are common to two adjacent rings.
- polycyclic ring system may be fused (i.e., bicyclic or tricyclic), spirocyclic, or a combination thereof.
- An example polycyclic ring is a steroid.
- Polypeptide The term “polypeptide” or “peptide”, as used herein, typically has its art- recognized meaning of a polymer of at least three amino acids or more.
- polypeptide is intended to be sufficiently general as to encompass not only polypeptides having a complete sequence recited herein, but also to encompass polypeptides that represent functional, biologically active, or characteristic fragments, portions or domains (e.g., fragments, portions, or domains retaining at least one activity) of such complete polypeptides.
- polypeptides may contain L-amino acids, D-amino acids, or both and/or may contain any of a variety of amino acid modifications or analogs known in the art. Useful modifications include, e.g., terminal acetylation, amidation, methylation, etc.
- polypeptides may comprise natural amino acids, non-natural amino acids, synthetic amino acids, and combinations thereof (e.g., may be or comprise peptidomimetics).
- Reference As used herein describes a standard or control relative to which a comparison is performed. For example, in some embodiments, an agent, animal, individual, population, sample, sequence or value of interest is compared with a reference or control agent, animal, individual, population, sample, sequence or value. In some embodiments, a reference or control is tested and/or determined substantially simultaneously with the testing or determination of interest. In some embodiments, a reference or control is a historical reference or control, optionally embodied in a tangible medium.
- Ribonucleotide encompasses unmodified ribonucleotides and modified ribonucleotides.
- unmodified ribonucleotides include the purine bases adenine (A) and guanine (G), and the pyrimidine bases cytosine (C) and uracil (U).
- Modified ribonucleotides may include one or more modifications including, but not limited to, for example, (a) end modifications, e.g., 5' end modifications (e.g., phosphorylation, dephosphorylation, conjugation, inverted linkages, etc.), 3' end modifications (e.g., conjugation, inverted linkages, etc.), (b) base modifications, e.g. , replacement with modified bases, stabilizing bases, destabilizing bases, or bases that base pair with an expanded repertoire of partners, or conjugated bases, (c) sugar modifications (e.g., at the 2' position or 4' position) or replacement of the sugar, and (d) internucleoside linkage modifications, including modification or replacement of the phosphodiester linkages.
- end modifications e.g., 5' end modifications (e.g., phosphorylation, dephosphorylation, conjugation, inverted linkages, etc.), 3' end modifications (e.g., conjugation, inverted linkages, etc.)
- base modifications
- RNA Ribonucleic acid
- an RNA refers to a polymer of ribonucleotides.
- an RNA is single stranded.
- an RNA is double stranded.
- an RNA comprises both single and double stranded portions.
- an RNA can comprise a backbone structure as described in the definition of “Nucleic acid / Polynucleotide” above.
- RNA can be a regulatory RNA (e.g., siRNA, microRNA, etc.), or a messenger RNA (mRNA).
- mRNA messenger RNA
- an RNA typically comprises at its 3’ end a poly(A) region.
- an RNA typically comprises at its 5’ end an art-recognized cap structure, e.g., for recognizing and attachment of a mRNA to a ribosome to initiate translation.
- an RNA is a synthetic RNA.
- Synthetic RNAs include RNAs that are synthesized in vitro (e.g., by enzymatic synthesis methods and/or by chemical synthesis methods). Substituted or optionally substituted: As described herein, compounds of the invention may contain “optionally substituted” moieties. In general, the term “substituted,” whether preceded by the term “optionally” or not, means that one or more hydrogens of the designated moiety are replaced with a suitable substituent. “Substituted” applies to one or more hydrogens that are either explicit or implicit from the structure (e.g., refers to at least ; and refers to at least , , or ).
- an “optionally substituted” group may have a suitable substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position.
- Combinations of substituents envisioned by this invention are preferably those that result in the formation of stable or chemically feasible compounds.
- stable refers to compounds that are not substantially altered when subjected to conditions to allow for their production, detection, and, in certain embodiments, their recovery, purification, and use for one or more of the purposes provided herein.
- Groups described as being “substituted” preferably have between 1 and 4 substituents, more preferably 1 or 2 substituents.
- Groups described as being “optionally substituted” may be unsubstituted or be “substituted” as described above.
- Suitable monovalent substituents on R° are independently halogen, — (CH 2 ) 0–2 R l , –(haloR l ), –(CH 2 ) 0–2 OH, –(CH 2 ) 0–2 OR l , –(CH 2 ) 0–2 CH(OR l ) 2 , -O(haloR l ), –CN, – N 3 , –(CH 2 ) 0–2 C(O)R l , –(CH 2 ) 0 –2C(O)OH, –(CH2)0–2C(O)OR l , –(CH2)0–2SR l , –(CH2)0–2SH, – (CH 2 ) 0–2 NH 2 , –(CH 2 ) 0–2 NHR l , –(CH 2
- Suitable divalent substituents that are bound to vicinal substitutable carbons of an “optionally substituted” group include: –O(CR * 2 ) 2–3 O–, wherein each independent occurrence of R * is selected from hydrogen, C1–6 aliphatic which may be substituted as defined below, or an unsubstituted 5–6–membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Suitable substituents on the aliphatic group of R * include halogen, –R l , -(haloR l ), -OH, – OR l , –O(haloR l ), –CN, –C(O)OH, –C(O)OR l , –NH 2 , –NHR l , –NR l 2 , or –NO 2 , wherein each R l is unsubstituted or where preceded by “halo” is substituted only with one or more halogens, and is independently C1–4 aliphatic, –CH2Ph, –O(CH2)0–1Ph, or a 3- to 6- membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Suitable substituents on a substitutable nitrogen of an “optionally substituted” group include –R ⁇ , –NR ⁇ 2 , –C(O)R ⁇ , –C(O)OR ⁇ , –C(O)C(O)R ⁇ , – C(O)CH 2 C(O)R ⁇ , -S(O) 2 R ⁇ , -S(O) 2 NR ⁇ 2 , –C(S)NR ⁇ 2 , –C(NH)NR ⁇ 2 , or –N(R ⁇ )S(O) 2 R ⁇ ; wherein each R ⁇ is independently hydrogen, C1–6 aliphatic which may be substituted as defined below, unsubstituted –oPh, or an unsubstituted 3- to 6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or, notwithstanding the definition above, two independent
- Suitable substituents on the aliphatic group of R ⁇ are independently halogen, – R l , -(haloR l ), –OH, –OR l , –O(haloR l ), –CN, –C(O)OH, –C(O)OR l , –NH2, –NHR l , –NR l 2, or -NO 2 , wherein each R l is unsubstituted or where preceded by “halo” is substituted only with one or more halogens, and is independently C 1–4 aliphatic, –CH 2 Ph, –O(CH 2 ) 0–1 Ph, or a 3- to 6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Small molecule means a low molecular weight organic and/or inorganic compound.
- a “small molecule” is a molecule that is less than about 5 kilodaltons (kD) in size.
- a small molecule is less than about 4 kD, 3 kD, about 2 kD, or about 1 kD.
- the small molecule is less than about 800 daltons (D), about 600 D, about 500 D, about 400 D, about 300 D, about 200 D, or about 100 D.
- a small molecule is less than about 2000 g/mol, less than about 1500 g/mol, less than about 1000 g/mol, less than about 800 g/mol, or less than about 500 g/mol. In some embodiments, a small molecule is not a polymer. In some embodiments, a small molecule does not include a polymeric moiety. In some embodiments, a small molecule is not and/or does not comprise a protein or polypeptide (e.g., is not an oligopeptide or peptide). In some embodiments, a small molecule is not and/or does not comprise a polynucleotide (e.g., is not an oligonucleotide).
- a small molecule is not and/or does not comprise a polysaccharide; for example, in some embodiments, a small molecule is not a glycoprotein, proteoglycan, glycolipid, etc.). In some embodiments, a small molecule is not a lipid. In some embodiments, a small molecule is a modulating agent (e.g., is an inhibiting agent or an activating agent). In some embodiments, a small molecule is biologically active. In some embodiments, a small molecule is detectable (e.g., comprises at least one detectable moiety). In some embodiments, a small molecule is a therapeutic agent.
- such a small molecule may be utilized in accordance with the present disclosure in the form of an individual enantiomer, diastereomer or geometric isomer, or may be in the form of a mixture of stereoisomers; in some embodiments, such a small molecule may be utilized in accordance with the present disclosure in a racemic mixture form.
- certain small molecule compounds have structures that can exist in one or more tautomeric forms.
- such a small molecule may be utilized in accordance with the present disclosure in the form of an individual tautomer, or in a form that interconverts between tautomeric forms.
- small molecule compounds have structures that permit isotopic substitution (e.g., 2 H or 3 H for H; 11 C, 13 C or 14 C for 12 C; 13 N or 15 N for 14 N; 17 O or 18 O for 16 O; 36 Cl for 35 Cl or 37 Cl; 18 F for 19 F; 131 I for 127 I; etc.).
- such a small molecule may be utilized in accordance with the present disclosure in one or more isotopically modified forms, or mixtures thereof.
- reference to a particular small molecule compound may relate to a specific form of that compound.
- a particular small molecule compound may be provided and/or utilized in a salt form (e.g., in an acid-addition or base-addition salt form, depending on the compound); in some such embodiments, the salt form may be a pharmaceutically acceptable salt form.
- a small molecule compound is one that exists or is found in nature
- that compound may be provided and/or utilized in accordance in the present disclosure in a form different from that in which it exists or is found in nature.
- a preparation of a particular small molecule compound that contains an absolute or relative amount of the compound, or of a particular form thereof, that is different from the absolute or relative (with respect to another component of the preparation including, for example, another form of the compound) amount of the compound or form that is present in a reference preparation of interest is distinct from the compound as it exists in the reference preparation or source.
- a preparation of a single stereoisomer of a small molecule compound may be considered to be a different form of the compound than a racemic mixture of the compound; a particular salt of a small molecule compound may be considered to be a different form from another salt form of the compound; a preparation that contains only a form of the compound that contains one conformational isomer ((Z) or (E)) of a double bond may be considered to be a different form of the compound from one that contains the other conformational isomer ((E) or (Z)) of the double bond; a preparation in which one or more atoms is a different isotope than is present in a reference preparation may be considered to be a different form; etc.
- treat refers to a point of attachment between two atoms. Additionally or alternatively, the symbol refers to a point of attachment ring in a spirocyclic manner.
- Treat refers to any method used to partially or completely alleviate, ameliorate, relieve, inhibit, prevent, delay onset of, reduce severity of, and/or reduce incidence of one or more symptoms or features of a disease, disorder, and/or condition. Treatment may be administered to a subject who does not exhibit signs of a disease, disorder, and/or condition.
- treatment may be administered to a subject who exhibits only early signs of the disease, disorder, and/or condition, for example, for the purpose of decreasing the risk of developing pathology associated with the disease, disorder, and/or condition.
- Ionizable Thiolipid Compounds The present disclosure provides, among other things, cationic or ionizable thiolipid compounds useful for forming particles comprising nucleic acids.
- the present disclosure provides a compound represented by formula I: or a pharmaceutically ac ceptable salt thereof, wherein: L 1 and L 2 are each independently an optionally substituted C1-C30 aliphatic group; L 3 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S; X 1 and X 2 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O) 2 N(R 1 )- , -N(R 1 )S(O) 2 , -S(O)-, -S(O) 2 -, -S(O) 2 C(R 1 ) 2 -, -OC(S)C(R 1 ) 2 -, -C(R 1 ) 2 C(S)O-, and —S-,
- L 1 and L 2 are each independently an optionally substituted C1-C30 aliphatic group.
- L 1 is optionally substituted C 1 -C 30 aliphatic.
- L 1 is C 1 -C 10 aliphatic.
- L 1 is optionally substituted C 1 -C 30 alkylene.
- L 1 is optionally substituted C 1 -C 30 alkenylene.
- L 1 is C1-C10 alkylene.
- L 1 is –(CH2)1-10-.
- L 2 is optionally substituted C1-C30 aliphatic.
- L 2 is C1-C10 aliphatic.
- L 2 is optionally substituted C1-C30 alkylene. In some embodiments, L 2 is optionally substituted C1-C30 alkenylene. In some embodiments, L 2 is C 1 -C 10 alkylene. In some embodiments, L 2 is –(CH 2 ) 1-10 -. In some embodiments, L 1 is –(CH 2 ) 6 -, and L 2 is –(CH 2 ) 8 -. In some embodiments, L 1 and L 2 are each C 1 -C 30 alkylene. In some embodiments, L 1 and L 2 are each –(CH 2 ) 6-12 -. In some embodiments, L 1 and L 2 are each –(CH2)6-10-.
- L 1 and L 2 are each –(CH2)6-. In some embodiments, L 1 and L 2 are each –(CH2)7-. In some embodiments, L 1 and L 2 are each –(CH2)8-. In some embodiments, L 1 and L 2 are each –(CH2)9-. In some embodiments, L 1 and L 2 are the same. In some embodiments, L 1 and L 2 are different.
- X 1 and X 2 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O)2N(R 1 )-, -N(R 1 )S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R 1 )2-, -OC(S)C(R 1 )2-, - C(R 1 )2C(S)O-, and –S-, wherein one or both of X 1 or X 2 is selected from -S(O)2N(R 1 )-, - N(R 1 )S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R 1 )2-, -OC(S)C(R 1 )2-, -C(R 1 )2C(S)O-, and –S-.
- one of X 1 and X 2 is a bond, -OC(O)-, -C(O)O-, -S(O) 2 N(R 1 )-, - N(R 1 )S(O) 2 , -S(O)-, -S(O) 2 -, -S(O) 2 C(R 1 ) 2 -, -OC(S)C(R 1 ) 2 -, -C(R 1 ) 2 C(S)O-, or –S-, and the other of X 1 and X 2 is -S(O)2N(R 1 )-, -N(R 1 )S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R 1 )2-, - OC(S)C(R 1 )2-, -C(R 1 )2C(S)O-, or –S-.
- X 1 and X 2 are each independently selected from a -S(O) 2 N(R 1 )-, -N(R 1 )S(O) 2 , -S(O)-, -S(O) 2 -, -S(O) 2 C(R 1 ) 2 - , -OC(S)C(R 1 ) 2 -, -C(R 1 ) 2 C(S)O-, and –S-.
- one of X 1 and X 2 is a bond, -OC(O)-, or -C(O)O-, and the other of X 1 and X 2 is -S(O) 2 N(R 1 )-, -N(R 1 )S(O) 2 , - S(O)-, -S(O)2-, -S(O)2C(R 1 )2-, -OC(S)C(R 1 )2-, -C(R 1 )2C(S)O-, or –S-.
- X 1 is a bond, -OC(O)-, -C(O)O-, -S(O)2N(R 1 )-, -N(R 1 )S(O)2, -S(O)-, -S(O)2- , -S(O)2C(R 1 )2-, -OC(S)C(R 1 )2-, -C(R 1 )2C(S)O-, or –S-.
- X 1 is - S(O) 2 N(R 1 )-, -N(R 1 )S(O) 2 , -S(O)-, -S(O) 2 -, -S(O) 2 C(R 1 ) 2 -, -OC(S)C(R 1 ) 2 -, -C(R 1 ) 2 C(S)O-, or –S-.
- X 1 is a bond, -OC(O)-, or -C(O)O-.
- X 2 is a bond, -OC(O)-, -C(O)O-, -S(O) 2 N(R 1 )-, -N(R 1 )S(O) 2 , -S(O)-, -S(O) 2 -, -S(O) 2 C(R 1 ) 2 - , -OC(S)C(R 1 )2-, -C(R 1 )2C(S)O-, or –S-.
- X 2 is -S(O)2N(R 1 )-, - N(R 1 )S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R 1 )2-, -OC(S)C(R 1 )2-, -C(R 1 )2C(S)O-, or –S-.
- X 2 is a bond, -OC(O)-, or -C(O)O-.
- X 2 is a bond.
- X 2 is -OC(O)-.
- X 2 is -C(O)O-.
- X 1 and X 2 are each independently selected from -S(O) 2 N(R 1 )-, - N(R 1 )S(O) 2 , and -S(O) 2 -.
- X 1 is -S(O) 2 N(R 1 )-.
- X 1 is -S(O)2N(H)-.
- X 1 is -S(O)2N(R 1 )-, where R 1 is C1-C10 aliphatic.
- X 1 is -N(R 1 )S(O)2.
- X 1 is -N(H)S(O)2.
- X 1 is -N(R 1 )S(O)2, where R 1 is C1-C10 aliphatic. In some embodiments, X 1 is -S(O)2-. In some embodiments, X 2 is -S(O)2N(R 1 )-. In some embodiments, X 2 is -S(O) 2 N(H)-. In some embodiments, X 2 is -S(O) 2 N(R 1 )-, where R 1 is C 1 -C 10 aliphatic. In some embodiments, X 2 is -N(R 1 )S(O) 2 . In some embodiments, X 2 is -N(H)S(O) 2 .
- X 2 is -N(R 1 )S(O) 2 , where R 1 is C 1 -C 10 aliphatic. In some embodiments, X 2 is -S(O)2-. In some embodiments, X 1 is -S(O)2N(R 1 )-, where R 1 is C1-C10 aliphatic, and X 2 is –C(O)O- . In some embodiments, X 1 and X 2 are each -S(O)2N(R 1 )-, where each R 1 is independently R 1 is C 1 -C 10 aliphatic.
- X 1 and X 2 are each - S(O) 2 N(R 1 )-, where each R 1 is –(CH 2 ) 3-10 -CH 3 . In some embodiments, X 1 and X 2 are each -S(O)2N(R 1 )-, where each R 1 is –(CH2)3-CH3. In some embodiments, X 1 and X 2 are each -S(O)2N(R 1 )-, where each R 1 is –(CH2)4-CH3.
- X 1 and X 2 are each - S(O)2N(R 1 )-, where each R 1 is –(CH2)5-CH3. In some embodiments, X 1 and X 2 are each -S(O) 2 N(R 1 )-, where each R 1 is –(CH 2 ) 6 -CH 3 . In some embodiments, X 1 and X 2 are each -S(O)2N(R 1 )-, where each R 1 is –(CH2)7-CH3. In some embodiments, X 1 and X 2 are the same. In some embodiments, X 1 and X 2 are different.
- T 1 and T 2 are each independently an optionally substituted C 3 -C 30 aliphatic.
- T 1 is C3-C30 aliphatic.
- T 1 is optionally substituted C3-C30 alkyl.
- T 1 is optionally substituted C3- C20 alkyl.
- T 1 is optionally substituted C5-C20 alkyl.
- T 1 is optionally substituted straight-chained C 3 -C 20 alkyl.
- T 1 is optionally substituted branched C 3 -C 20 alkyl.
- T 1 is optionally substituted C 3 -C 20 alkenyl.
- T 1 is optionally substituted C10-C20 alkenyl.
- T 2 is C3-C30 aliphatic. In some embodiments, T 2 is optionally substituted C3-C30 alkyl. In some embodiments, T 2 is optionally substituted C3-C20 alkyl. In some embodiments, T 2 is optionally substituted C5- C 20 alkyl. In some embodiments, T 2 is optionally substituted straight chained C 3 -C 20 alkyl. In some embodiments, T 2 is optionally substituted branched C 3 -C 20 alkyl. In some embodiments, T 2 is optionally substituted C 3 -C 20 alkenyl.
- T 1 is optionally substituted C10-C20 alkenyl.
- T 1 and T 2 are the same. In some embodiments, T 1 and T 2 are different. In some embodiments, T 1 and T 2 are each independently selected from: , , , , In some embodiments, moiety –L 1 -X 1 -T 1 is selected from the group consisting of: In some embodiments, moiety –L 2 -X 2 -T 2 is selected from the group consisting of: In some embodiments, moiety –L 1 -X 1 -T 1 and moiety –L 2 -X 2 -T 2 are each independently selected from: As described herein, L 3 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S.
- L 3 is a bond. In some embodiments L 3 is optionally substituted C 1 -C 10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S. In some embodiments, L 3 is optionally substituted C 1 -C 10 aliphatic. In some embodiments, L 3 is optionally substituted C1-C10 alkylene. In some embodiments, L 3 is optionally substituted C1-C10 alkenylene. In some embodiments, L 3 is optionally substituted alkynylene. In some embodiments, L 3 is C1-C6 alkylene. In some embodiments, L 3 is –(CH2)1-6-.
- L 3 is –(CH2)2-4-. In some embodiments, L 3 is –(CH 2 ) 2 -. In some embodiments, L 3 is –(CH 2 ) 3 -. In some embodiments, L 3 is –(CH 2 ) 4 -. In some embodiments, L 3 is –(CH 2 ) 5 -. In some embodiments, L 3 is , , or . In some embodiments, L 3 is optionally substituted 2- to 10-membered heteroaliphatic comprising 1 to 4 heteroatoms selected from N, O, and S. In some embodiments, L 3 is optionally substituted 2- to 10-membered heteroaliphatic comprising 1 to 4 heteroatoms selected from N, O, and S.
- L 3 is optionally substituted 2- to 8- membered heteroaliphatic comprising 1 to 4 heteroatoms selected from N, O, and S. In some embodiments, L 3 is optionally substituted 2- to 6-membered heteroaliphatic comprising 1 to 2 heteroatoms selected from N, O, and S. In some embodiments, L 3 is optionally substituted 5-membered heteroaliphatic comprising 1 or 2 heteroatoms selected from N, O, and S.
- L 3 is: , , or As described herein, G is -N(R 2 )C(S)N(R 2 )2, -OH, -N(R 2 )2, , -N + (R 3 )3, -N(R 5 )C(O)R 3 , - N(R 5 )S(O)2R 3 , -N(R 5 )C(O)N(R 3 )2, -CH(N-R 2 ), -R 4 , or -S(O)2R 3 .
- G is -N(R 2 )C(S)N(R 2 )2, -OH, -N(R 2 )2, -N(R 5 )C(O)R 3 , -N(R 5 )S(O)2R 3 , -N(R 5 )C(O)N(R 3 )2, - CH(N-R 2 ), -R 4 , or -S(O)2R 3 .
- G is -N(R 2 )C(S)N(R 2 ) 2 .
- G is - N(H)C(S)N(R 2 ) 2 .
- G is -N(H)C(S)N(H)(R 2 ) . In some embodiments, G is -N(CH 3 )C(S)N(R 2 ) 2 . In some embodiments, G is –N(OH)C(S)N(R 2 ) 2 . In some embodiments, G is -N(H)C(S)N(R 2 )2, where each R 2 is selected from optionally substituted C1-C6 aliphatic and OH. In some embodiments, G is -N(H)C(S)N(OH)(R 2 ), where R 2 is optionally substituted C1-C6 aliphatic.
- G is - N(H)C(S)N(R 2 ) 2 , where each R 2 is selected from optionally substituted C 1 -C 6 aliphatic. In some embodiments, G is -N(H)C(S)N(R 2 ) 2 , where each R 2 is methyl, ethyl, propyl, or butyl. In some embodiments, G is -N(H)C(S)N(CH 3 ) 2 . In some embodiments, G is – N(H)C(S)N(CH3)(OCH3). In some embodiments, G is –N(H)C(S)N(CH3)(OH).
- G is –N(OH)C(S)N(CH3)2. In some embodiments, G is -N(H)C(S)N(R 2 )2, where two instances of R 2 come together with the atoms to which they are attached to form an optionally substituted 4- to 12- membered heterocycle ring or an optionally substituted 4- to 12-membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N, O, and S. In some embodiments, G is -N(H)C(S)N(R 2 )2, where two instances of R 2 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring comprising 1 to 4 heteroatoms selected from N, O, and S.
- G is - N(H)C(S)N(R 2 )2, where two instances of R 2 come together with the atoms to which they are attached to form an optionally substituted azetidine, pyrrolidine, piperidine, piperazine, or azepane ring.
- G is - N(R 2 )C(S)N(H)(R 2 ) , where two instances of R 2 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring comprising 1 to 4 heteroatoms selected from N, O, and S.
- G is: In some embodimen ts, G is OH.
- G is –N(R 2 )2. In some embodiments, G is –N(R 2 )2, wherein each R 2 is independently H or optionally substituted C1-C6 aliphatic. In some embodiments, G is –N(R 2 ) 2 , wherein each R 2 is optionally substituted C 1 -C 6 aliphatic. In some embodiments, G is –N(R 2 ) 2 , wherein each R 2 is independently methyl, ethyl, propyl, butyl, pentyl, or hexyl. In some embodiments, G is –N(CH 3 ) 2 . In some embodiments, G is –N + (R 3 )2.
- G is -N(R 5 )S(O)2R 3 . In some embodiments, G is –N(H)S(O)2R 3 . In some embodiments, G is –N(H)S(O) 2 -C 1 -C 6 aliphatic. In some embodiments, G is – N(H)S(O) 2 -CH 3 . In some embodiments, G is -N(R 5 )C(O)N(R 3 ) 2 . In some embodiments, G is – N(H)C(O)N(R 3 )2. In some embodiments, G is –N(H)C(O)N(H)(R 3 ).
- G is — N(H)C(O)N(H)(CH3). G is –N(H)C(O)N(CH3)2. In some embodiments, G is -CH(N-R 2 ). In some embodiments, G is –CH(N-C1-C6 aliphatic). In some embodiments, G is –CH(N-CH 3 ). In some embodiments, G is . In some embodiments, G is R 4 , and R 4 is optionally substituted 4- to 12-membered heterocycle. In some embodiments, G is optionally substituted 4- to 6-membered monocyclic heterocycle. In some embodiments, G is optionally substituted azetidine, pyrrolidine, piperidine, piperazine, or azepane.
- G is 6- to 12- membered bicyclic heterocycle comprising 1 to 4 heteroatoms selected from N, O, and S.
- G is 4- to 12-membered heterocycle comprising 1 to 4 heteroatoms selected from N, O, and S, and optionally substituted with -(CH 2 ) 0–4 N(R°) 2 or –(CH2)0–4OR°, and where R° is hydrogen or C1–C6 aliphatic.
- G is R 4 and R 4 is optionally substituted 5- to 12-membered heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S.
- G is optionally substituted 5- to 6-membered monocyclic heteroaryl comprising 1 to 3 heteroatoms selected from N, O, and S. In some embodiments, G is optionally substituted pyrrole, imidazole, pyrazole, thiazole, oxazole, or furan. In some embodiments, G is 6- to 12-membered bicyclic heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S. In some embodiments, G is 5- to 12-membered heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S, and optionally substituted with –(CH2)0–4SR ⁇ , and where R° is hydrogen or C1–C6 aliphatic.
- G is In some embodiments, G is R 4 , where R 4 is C 6 -C 12 aryl substituted with one or more of –(CH 2 ) 0-6 -OH or –(CH 2 ) 0-6 -N(R 5 ) 2. In some embodiments, G is phenyl substituted one or more of –(CH 2 ) 0-6 -OH or –(CH 2 ) 0-6 -N(R 5 )2. In some embodiments, G is R 4 , where R 4 C3-C12 cycloaliphatic substituted one or more of oxo, –(CH2)0-6-OH or –(CH2)0-6-N(R 5 )2.
- G is C3-C6 cycloaliphatic substituted with one or more of oxo, –(CH 2 ) 0-6 -OH, or –(CH 2 ) 0-6 –N(R 5 ) 2 .
- G is cyclobutyl, cyclopentyl, or cyclohexyl optionally substituted with one or more of oxo, -OH, or –N(R 5 )2.
- G is -S(O)2R 3 .
- G is -S(O)2-C1-C6 aliphatic.
- G is selected from: and .
- a moiety –L 3 -G can be a cationic or ionizable group, such that particular atoms, e.g., nitrogen atoms, in the moiety –L 3 -G can have a positive charge at either neutral pH or at physiological pH.
- a head group further comprises a suitable counterion (e.g., a halogen atom, such as Cl-, Br-, I-, F-, and the like).
- a moiety –L 3 -G is selected from:
- each R 2 is, independently, at each instance, selected from the group consisting of H, optionally substituted C1-C6 aliphatic and OR 3 ; or two instances of R 2 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring or an optionally substituted 4- to 12- membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N, O, and S.
- each R 2 is, independently, at each instance, selected from the group consisting of H, optionally substituted C1-C6 aliphatic, and OR 3 .
- R 2 two instances come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring or an optionally substituted 4- to 12-membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N, O, and S.
- each R 3 is, independently, at each instance, selected from the group consisting of H and optionally substituted C1-C10 aliphatic.
- R 4 is optionally substituted 4- to 12-membered heterocycle comprising 1 to 4 heteroatoms selected from N, O, and S, optionally substituted 4- to 12 membered heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S, C6-C12 aryl substituted with one or more of –(CH 2 ) 0-6 –OH or –(CH 2 ) 0-6 –N(R 5 ) 2 , or C 3 -C 12 cycloaliphatic substituted with one or more of oxo, –(CH 2 ) 0-6 –OH, or –(CH 2 ) 0-6 –N(R 5 ) 2 .
- each R 5 is independently selected from H and optionally substituted C1-C6 aliphatic.
- a compound described herein is a compound represented by Formula IIa: or a pharmaceutically acceptable salt thereof, wherein G, L 1 , L 2 , and L 3 are as described in classes and subclasses herein.
- a compound described herein is a compound represented by Formula IIb: or a pharmaceuti cally acceptable salt thereof, wherein G, L , L 2 , and L 3 are as described in classes and subclasses herein.
- a compound described herein is a compound represented by Formula IIc: or a pharmaceutically acceptable salt thereof, wherein G, L 1 , L 2 , and L 3 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIIa: or a pharmaceut ically acceptable salt thereof, wherein L 1 , L 2 , X 1 , X 2 , T 1 , and T 2 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIIb: or a pharmaceutically acceptable salt thereof, wherein L 1 , L 2 , X 1 , X 2 , R 1 , T 1 , and T 2 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIIc: or a pharmac eutically acceptable salt thereof, wherein L , L , X , X , T 1 , and T 2 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIId: or a pharmaceutic ally acceptable salt thereof, wherein L , L , X , X , T 1 , and T 2 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIId-1: or a pharmaceutica lly acceptable salt thereof, wherein L , L , X , X 2 , T 1 , and T 2 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIId-2: or a pharmaceut ically acceptable salt thereof, wherein L , L , X 1 , X 2 , T 1 , and T 2 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIIe: or a pharmaceutically acceptable salt thereof, wherein L 1 , L 2 , R 1 , T 1 , and T 2 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIIf: or a pharmaceutically acceptable salt thereof, wherein L 1 , L 2 , T 1 , and T 2 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIIg: or a pharma ceutically acceptable salt thereof, wherein L , L , X , X , T , and T 2 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIIg-1: or a pharm aceutically acceptable salt thereof, wherein L 1 , L 2 , R 1 , T 1 , and T 2 are as described in classes and subclasses herein.
- a compound described herein is represented by Formula IIIh-1:
- a compound described herein is represented by Formula IIIh-2: o r a pharmaceutically acceptable salt thereof, wherein X is S, S(O) , or S(O) 2 , and n’ is 1-6.
- a compound described herein is represented by Formula IIIh-3: or a pharmaceutically acceptable salt thereof, wherein X 50 is –S-, -S(O)-, or -S(O)2-, and n’ is 1-6.
- a compound described herein is represented by Formula IIIh-4:
- n’ is 1-6 and R 100 is optionally substituted C 1 -C 16 aliphatic.
- a compound described herein is represented by Formula IIIh-5: or a pharmaceutically acceptable salt thereof, wherein n’ is 1-6.
- a compound described herein is selected from Table 1: Table 1
- provided compounds are provided and/or utilized in a salt form (e.g., a pharmaceutically acceptable salt form). Reference to a compound provided herein is understood to include reference to salts thereof, unless otherwise indicated.
- thiolipid compounds of the present disclosure can be prepared according to General Schemes 1 , 2, and 3, depicted in Figures 79, 80, and 81 , respectively.
- a tail-linker moiety is any bivalent linker, such as an aliphatic or heteroaliphatic group; a tail-end is a hydrophobic group, e.g., an aliphatic group, a head group is a polar or cationic or ionizable head group, a tail junction is a biodegradable group, such as an ester, or a sulfur-containing moiety (e.g., thioether, sulfonyl, or sulfonamide), and a headtail junction is a central atom or functional group connecting a tail or tails to the head group (e.g., a tertiary amine group).
- a tail-end is a hydrophobic group, e.g., an aliphatic group
- a head group is a polar or cationic or ionizable head group
- a tail junction is a biodegradable group, such as an ester, or a sulfur-containing moiety (
- the present disclosure provides a method of preparing a compound represented by Formula IV: or a pharmaceutically acceptable salt thereof, the method comprising: contacting a compound represented by Formula V with a compound represented by one of Formulae Vla-VIc and a compound represented by one of Formulae Vlla-Vllc in the presence of a reducing agent, wherein: each of L 4 and L 5 are each independently an optionally substituted C1-C30 aliphatic group; L 6 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S;
- X 3 and X 4 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O) 2 N(R 40 )- , -N(R 40 )S(O) 2 , -S(O)-, -S(O) 2 -, -S(O) 2 C(R 40 ) 2 -, -OC(S)C(R 40 ) 2 -, -C(R 40 ) 2 C(S)0-, or -S-, wherein one or both of X 3 or X 4 is selected from -S(O) 2 N(R 40 )-, -N(R 40 )S(O) 2 , -8(0)-, - S(O) 2 -, -S(O) 2 C(R 40 ) 2 -, -OC(S)C(R 40 ) 2 -, -C(R 40 ) 2 C(S)0-, or -S-; each R 40
- G 1 is -N(R 6 )C(S)N(R 6 ) 2 , -OH, -N(R 6 ) 2 , -N(R 9 )C(O)R 7 , -N(R 9 )S(O) 2 R 7 , -N(R 9 )C(O)N(R 7 ) 2 , -CH(N-R 7 ), or-R 8 ; each G 2 is independently O or N 2 ; each G 3 is independently halogen (e.g., Cl, Br, or I), -OTs, or OTf; each R 6 is, independently, at each instance, selected from the group consisting of H, optionally substituted C 1 -C 6 aliphatic or OR 7 ; or two instances of R 6 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring or an optionally substituted 4- to 12-membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N,
- a reducing agent is NaBH 3 CN or NaBH(OCOCH 3 ) 3 .
- a reducing agent is NaBH 3 CN.
- a reducing agent is NaBH(OCOCH 3 ) 3 .
- a method of preparing a compound described herein further comprises preparing a compound represented by Formula VIa or the compound represented by Formula VIIa, wherein G 1 is O, by contacting a co compound represented by Formula IX with an oxidi
- an oxidizing agent is DMSO, PCC, or DMP.
- an oxidizing agent is DMSO
- the method further comprises contacting the compound represented by Formula VIII or IX and DMSO with a sulfur trioxide pyridine complex (SO3•pyridine).
- a method of preparing a compound described herein further comprises preparing the compound represented by Formula VIII or IX: by contacting a compound represented by Formula X or a compound represented by Formula XI with a compound represented by Formula XII or a compound represented by Formula XIII: in the presence of H 2 O 2 and SOCI 2 ; and wherein each V 1 is halogen.
- particles of the present disclosure comprise a compound described herein (e.g., a compound of one or more of formulae l-lllh-5), a nucleic acid (such as RNA (e.g., mRNA), DNA or mixtures thereof), and one or more of a polymer- conjugated lipid, a helper lipid, and a steroid.
- particles of the present disclosure comprise a compound described herein (e.g., a compound of one or more of formulae l-lllh-5), a nucleic acid (such as RNA (e.g., mRNA), DNA or mixtures thereof), a helper lipid, and a steroid.
- particles of the present disclosure comprise a compound described herein (e.g., a compound of one or more of formulae l-lllh-5), a nucleic acid (such as RNA (e.g., mRNA), DNA or mixtures thereof), a polymer-conjugated lipid, a helper lipid, and a steroid.
- a compound described herein e.g., a compound of one or more of formulae l-lllh-5
- a nucleic acid such as RNA (e.g., mRNA), DNA or mixtures thereof
- a polymer-conjugated lipid such as a helper lipid, and a steroid.
- Thiolipid compounds described herein comprise a cationic or cationically ionizable moiety that is capable of forming electrostatic interactions between the positively charged moiety and negatively charged nucleic acids, resulting in particle formation.
- particles described herein comprise more than one type of nucleic acid molecules, where the molecular parameters of the nucleic acid molecules may be similar or different from each other, like with respect to molar mass or fundamental structural elements such as molecular architecture, capping, coding regions or other features.
- a nucleic acid particle described herein is a nanoparticle.
- nanoparticle refers to a particle having an average diameter suitable for parenteral administration and is less than 1000 nm in diameter.
- a composition comprising nanoparticles can have an average nanoparticle size (e.g., mean diameter) of about 30 nm to about 150 nm, about 40 nm to about 150 nm, about 50 nm to about 150 nm, about 60 nm to about 130 nm, about 70 nm to about 110 nm, about 70 nm to about 100 nm, about 70 to about 90 nm, or about 70 nm to about 80 nm.
- a composition comprising nanoparticles can have an average nanoparticle size (e.g., mean diameter) of about 50 nm to about 100 nm.
- a composition comprising nanoparticles can have an average nanoparticle size (e.g., mean diameter) of about 50 nm to about 150 nm. In some embodiments, a composition comprising nanoparticles can have an average nanoparticle size (e.g., mean diameter) of about 60 nm to about 120 nm.
- a composition comprising nanoparticles can have an average nanoparticle size (e.g., mean diameter) of about 30 nm, 35 nm, 40 nm, 45 nm, 50 nm, 55 nm, 60 nm, 65 nm, 70 nm, 75 nm, 80 nm, 85 nm, 90 nm, 95 nm, 100 nm, 105 nm, 110 nm, 115 nm, 120 nm, 125 nm, 130 nm, 135 nm, 140 nm, 145 nm, or 150 nm.
- average nanoparticle size e.g., mean diameter
- a composition comprising nucleic acid particles (e.g., ribonucleic acid particles or deoxyribonucleic acid particles) described herein may exhibit a polydispersity index less than about 0.5, less than about 0.4, less than about 0.3, or about 0.2 or less of said nanoparticles.
- a composition comprising nucleic acid particles (e.g., ribonucleic acid particles or deoxyribonucleic acid particles) described herein can exhibit a polydispersity index in a range of about 0.1 to about 0.3 or about 0.2 to about 0.3.
- Nucleic acid particles e.g., ribonucleic acid particles or deoxyribonucleic acid particles
- N/P ratio is the molar ratio of cationic (nitrogen) groups (the “N” in N/P) in the cationic polymer to the anionic (phosphate) groups (the “P” in N/P) in RNA.
- N cationic
- a cationic group is one that is either in cationic form (e.g., N + ), or one that is ionizable to become cationic.
- nucleic acid particle e.g., a ribonucleic acid particle
- N/P ratio greater than or equal to 4.
- nucleic acid particle e.g., a ribonucleic acid particle
- nucleic acid particle e.g., a ribonucleic acid particle
- N/P ratio that is about 4 to about 16.
- nucleic acid particle e.g., a ribonucleic acid particle
- nucleic acid particle e.g., a ribonucleic acid particle
- a nucleic acid particle described herein has an N/P ratio that is about 4 to about 12. In some embodiments, a nucleic acid particle (e.g., a ribonucleic acid particle) described herein has an N/P ratio that is about 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, or 16. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is about 6. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is about 12. Compounds described herein are also referred to as “ionizable” or “cationic” lipids.
- Such lipids are intended to mean compounds that, in some embodiments, are capable of becoming cationic (i.e., becoming positively charged) at physiological pH.
- the term “average diameter” or “mean diameter” refers to the mean hydrodynamic diameter of particles as measured by dynamic laser light scattering (DLS) with data analysis using the so-called cumulant algorithm, which provides as results the so-called Z-average with the dimension of a length, and the polydispersity index (PDI), which is dimensionless (Koppel, D., J. Chem. Phys.57, 1972, pp 4814-4820, ISO 13321).
- PDI polydispersity index
- average diameter,” “mean diameter,” “diameter,” or “size” for particles is used synonymously with this value of the Z-average.
- the “polydispersity index” is preferably calculated based on dynamic light scattering measurements by the so-called cumulant analysis as mentioned in the definition of the “average diameter.” Under certain prerequisites, it can be taken as a measure of the size distribution of an ensemble of ribonucleic acid nanoparticles (e.g., ribonucleic acid nanoparticles).
- ribonucleic acid nanoparticles e.g., ribonucleic acid nanoparticles.
- Different types of nucleic acid particles have been described previously to be suitable for delivery of nucleic acid in particulate form (e.g. Kaczmarek, J. C. et al., 2017, Genome Medicine 9, 60).
- nanoparticle encapsulation of nucleic acid physically protects nucleic acid from degradation and, depending on the specific chemistry, can aid in cellular uptake and endosomal escape.
- Some embodiments described herein relate to compositions, methods and uses involving more than one, e.g., 2, 3, 4, 5, 6 or even more nucleic acid species.
- the nucleic acid species may be RNA and/or DNA.
- the particles described herein may contain one species of RNA (e.g., one species of mRNA) and one species of DNA.
- each nucleic acid species is separately formulated as an individual nucleic acid particle formulation.
- each individual nucleic acid particle formulation will comprise one nucleic acid species.
- the individual nucleic acid particle formulations may be present as separate entities, e.g., in separate containers. Such formulations are obtainable by providing each nucleic acid species separately (typically each in the form of a nucleic acid-containing solution) together with a particle-forming agent, thereby allowing the formation of particles. Respective particles will contain exclusively the specific nucleic acid species that is being provided when the particles are formed (individual particulate formulations).
- a composition such as a pharmaceutical composition comprises more than one individual nucleic acid particle formulation.
- Respective pharmaceutical compositions are referred to as “mixed particulate formulations.”
- Mixed particulate formulations according to the present disclosure are obtainable by forming, separately, individual nucleic acid particle formulations, as described above, followed by a step of mixing of the individual nucleic acid particle formulations. By the step of mixing, a formulation comprising a mixed population of nucleic acid-containing particles is obtainable. Individual nucleic acid particle populations may be together in one container, comprising a mixed population of individual nucleic acid particle formulations.
- nucleic acid species are formulated together as a “combined particulate formulation.”
- Such formulations are obtainable by providing a combined formulation (typically combined solution) of different nucleic acid species together with a particle-forming agent, thereby allowing the formation of particles.
- a “combined particulate formulation” will typically comprise particles that comprise more than one nucleic acid species.
- nucleic acids when present in provided nucleic acid particles are resistant in aqueous solution to degradation with a nuclease.
- nucleic acid particles are lipid nanoparticles.
- lipid nanoparticles are cationic lipid nanoparticles comprising one or more cationic lipids (e.g., ones described herein), a nucleic acid (e.g., RNA and/or DNA), a polymer-conjugated lipid, a helper lipid, and a sterol.
- Lipid nanoparticles have proven useful for the delivery of nucleic acid cargo to tissue of interest. LNPs are used, for example, in certain commercial vaccines for treatment of COVID-19.
- LNPs of the present disclosure comprise i) a cationic lipid (e.g., a thiolipid compound described herein, such as a compound of any one of formulae I- IIIh-5); ii) a helper lipid; iii) a polymer-conjugated lipid (e.g., a polyethylene glycol bound lipid “a PEG lipid”); and iv) a steroid.
- a cationic lipid e.g., a thiolipid compound described herein, such as a compound of any one of formulae I- IIIh-5
- helper lipid e.g., a helper lipid
- a polymer-conjugated lipid e.g., a polyethylene glycol bound lipid “a PEG lipid”
- a steroid e.g., a steroid.
- LNPs of the present disclosure may comprise i) a cationic lipid (e.g., a thiolipid compound described herein, such as a compound of any one of formulae I-IIIh-5); ii) a helper lipid; and iii) a steroid.
- LNPs described herein can further comprise additional additives, as described herein.
- LNPs of the present disclosure can be useful in a variety of contexts. For example, LNPs comprising a nucleic acid described herein are useful for delivery of said nucleic acid into the cell of a subject. In some embodiments, LNPs comprising a nucleic acid described herein are useful for causing increased expression of a protein in a subject.
- LNPs comprising a nucleic acid described herein are useful for causing a pharmacological effect induced by expression of a protein in a subject.
- Lipid nanoparticles described herein are characterized by molar percentage (mol%) of components in the lipid nanoparticle. A mol% used in reference to a lipid component of a lipid nanoparticle is relative to the total other lipid components in the lipid nanoparticle.
- Helper lipids As described herein, LNPs of the present disclosure comprise a helper lipid. In some embodiments, a helper lipid is a phospholipid.
- a helper lipid is or comprises 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dipalmitoyl-sn- glycero-3-phosphocholine (DPPC), 1,2-dimyristoyl-sn-glycero-3- phosphocholine (DMPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-sn- glycero-3-phosphocholine (DOPC), phosphatidyl ethanol amines such as 1,2-dioleoyl- sn-glycero-3-phosphoethanolamine (DOPE), sphingomyelins (SM), 1,2 ⁇ diacylglyceryl-3- O-4'-(N,N,N-trimethyl)-homoserine (DGTS), ceramides, cholesterol, steroids, such as sterols and their derivative
- a helper lipid is or comprises phosphatidylcholines, phosphatidylethanolamines, phosphatidylglycerols, phosphatidic acids, phosphatidylserines or sphingomyelin.
- a helper lipid is or comprises diacylphosphatidylcholines, such as distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dimyristoylphosphatidylcholine (DMPC), dipentadecanoylphosphatidylcholine, dilauroylphosphatidylcholine, dipalmitoylphosphatidylcholine (DPPC), diarachidoylphosphatidylcholine (DAPC), dibehenoylphosphatidylcholine (DBPC), ditricosanoylphosphatidylcholine (DTPC), dilignoceroylphatidylcholine (DLPC), palmitoyloleoyl-phosphatidylcholine (POPC), 1,2- di-O-octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), 1-olelphosphatid
- a helper lipid is selected from the group consisting of DSPC, DOPC, DMPC, DPPC, POPC, DOPE, DOPG, DPPG, POPE, DPPE, DMPE, DSPE, and SM.
- the neutral lipid is selected from the group consisting of DSPC, DPPC, DMPC, DOPC, POPC, DOPE and SM.
- the neutral lipid is DSPC.
- Helper lipids may be synthetic or naturally derived.
- a lipid nanoparticle comprises about 5 to about 15 mol% of a phospholipid. In some embodiments, a lipid nanoparticle comprises about 8 to about 12 mol% of a phospholipid. In some embodiments, a lipid nanoparticle comprises about 10 mol% of a phospholipid. In some embodiments, a lipid nanoparticle comprises about 5 to about 15 mol% of DSPC.
- a lipid nanoparticle comprises about 8 to about 12 mol% of DSPC. In some embodiments, a lipid nanoparticle comprises about 10 mol% of DSPC.
- LNPs of the present disclosure comprise a polymer-conjugated lipid. In some embodiments, a polymer conjugated lipid is a lipid conjugated to polyethylene glycol (PEG-lipid).
- a PEG lipid is selected from pegylated diacylglycerol (PEG-DAG) such as l-(monomethoxy-polyethyleneglycol)- 2,3- dimyristoylglycerol (PEG-DMG) (e.g., 1,2-dimyristoyl-rac-glycero-3- methoxypolyethylene glycol-2000 (PEG2000-DMG)), a pegylated phosphatidylethanoloamine (PEG-PE), a PEG succinate diacylglycerol (PEG-S-DAG) such as 4-O-(2',3'-di(tetradecanoyloxy)propyl-1-O-( ⁇ - methoxy(polyethoxy)ethyl)butanedioate (PEG-S-DMG), 1,2-distearoyl-sn-glycero-3- phosphoethanolamine-N-[amino(polyethylene)-DMG
- a PEG-lipid is PEG2000-DMG: In some embodiments, a PEG-lipid is DMG-PEG. In some embodiments, a PEG-lipid is provided in WO2021/026358, WO 2017/075531, or WO 2018/081480, each of which is incorporated by reference in its entirety.
- a polymer-conjugated lipid is 2-[(polyethylene glycol)-2000]-N,N- ditetradecylacetamide (ALC-0159). In some embodiments, a polymer-conjugated lipid is: or a pharmaceutically acceptable salt thereof, where n’ is an integer from about 45 to about 50.
- a polymer conjugated lipid is C16 PEG2000:
- the PEG- lipid has the following structure: wherein n in the formula above is from 30 to 60, such as about 50.
- the PEG-conjugated lipid is PEG 2000 -C-DMA which preferably refers to 3-N-[( ⁇ -methoxy poly(ethylene glycol)2000)carbamoyl]-1,2-dimyristyloxy-propylamine (MPEG-(2 kDa)-C-DMA) or methoxy-polyethylene glycol-2,3- bis(tetradecyloxy)propylcarbamate (2000).
- a PEG-lipid is selected from PEG-DAG, PEG-PE, PEG-S-DAG, PEG2000-DMG, PEG-S-DMG, PEG-cer, a PEG dialkyoxypropylcarbamate (e.g., w- methoxy(polyethoxy)ethyl-N-(2,3-di(tetradecanoxy)propyl)carbamate or 2,3- di(tetradecanoxy)propyl-N-(w methoxy(polyethoxy)ethyl)carbamate), ALC-0159, and combinations thereof.
- a PEG-lipid is ALC-0159 or PEG2000- DMG.
- a PEG-lipid is ALC-0159. In some embodiments, a PEG- lipid is PEG2000-DMG. In some embodiments, a PEG-lipid is PEG-DAG. In some embodiments, a PEG-lipid is PEG-PE. In some embodiments, a PEG-lipid is PEG-S- DAG. In some embodiments, a PEG-lipid is PEG-cer. In some embodiments, a PEG- lipid is a PEG dialkyoxypropylcarbamate.
- a PEG group that is part of a PEG-lipid has, on average in a composition comprising one or more PEG-lipid molecules, a weight average molecular weight (M w ) of about 2000 g/mol.
- a polymer-conjugated lipid is a polysarcosine-conjugated lipid, also referred to herein as sarcosinylated lipid or pSar-lipid.
- the term "sarcosinylated lipid” refers to a molecule comprising both a lipid portion and a polysarcosine (poly(N- methylglycine) portion.
- a polymer-conjugated lipid is a polyoxazoline (POX)-conjugated and/or polyoxazine (POZ)-conjugated lipid, also referred to herein as a conjugate of a POX and/or POZ polymer and one or more hydrophobic chains or as oxazolinylated and/or oxazinylated lipid or POX- and/or POZ-lipid.
- POX polyoxazoline
- POZ polyoxazine
- oxazinylated lipid or “POZ-lipid” refers to a molecule comprising both a lipid portion and a polyoxazine portion.
- oxazolinylated/oxazinylated lipid or “POX/POZ-lipid” or “POXZ-lipid” refers to a molecule comprising both a lipid portion and a portion of a copolymer of polyoxazoline and polyoxazine.
- an LNP described herein may comprise a sarcosinylated lipid.
- the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein comprise a sarcosinylated lipid and are substantially free of a pegylated lipid (or do not contain a pegylated lipid).
- the nucleic acid compositions (such as DNA or RNA compositions) described herein comprise a cationic/cationically ionizable lipid as described herein and a sarcosinylated lipid (pSAR-conjugated lipid).
- the nucleic acid compositions may further comprise a neutral lipid (e.g., a phospholipid, cholesterol or a derivative thereof) or a combination of neutral lipids (e.g., a phospholipid, and cholesterol or a derivative thereof).
- the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein comprise a cationic/cationically ionizable lipid as described herein, a sarcosinylated lipid, a neutral lipid (e.g., a phospholipid), and cholesterol or a derivative thereof.
- the phospholipid is DSPC.
- nucleic acid compositions such as DNA or RNA compositions, especially mRNA compositions
- said compositions are substantially free of a pegylated lipid (or do not contain a pegylated lipid).
- the sarcosinylated lipid comprises between 2 and 200 sarcosine units, such as between 5 and 100 sarcosine units, between 10 and 50 sarcosine units, between 15 and 40 sarcosine units, e.g., about 23 sarcosine units.
- the sarcosinylated lipid comprises the structure of the following general formula (XVII): wherein s is the number of sarcosine units.
- the sarcosinylated lipid comprises the structure of the following general formula (XVIII): wherein one of R 21 and R 22 comprises a hydrophobic group and the other is H, a hydrophilic group or a functional group optionally comprising a targeting moiety; and x is the number of sarcosine units.
- an LNP herein may comprise an oxazolinylated and/or/oxazinylated lipid.
- the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein comprise an oxazolinylated and/or/oxazinylated lipid and are substantially free of a pegylated lipid (or do not contain a pegylated lipid).
- a polymer-conjugated lipid comprises monomers of 2-(2-(2- aminoethoxy)ethoxy)acetic acid.
- a polymer-conjugated lipid comprises monomers of 2-(2-(2-aminoethoxy)ethoxy)acetic acid.
- the polymer of the polymer-conjugated lipid is or comprises poly-2-(2-(2- aminoethoxy)ethoxy)acetic acid (pAEEA) or poly-2-(2-(2- methylaminoethoxy)ethoxy)acetic acid (pMAEEA), or a derivative thereof, as defined herein.
- the polymer comprises the following general formula: wherein X 11 and X 12 taken together are optionally substituted amide, optionally substituted thioamide or ester; Y is -CH 2 -, -(CH 2 ) 2 -, or -(CH 2 ) 3 -; z is 2 to 24; and n is 1 to 100.
- X 11 when X 11 is -C(O)- then X 12 is -NR 10 -; (ii) when X 11 is -NR 10 - then X 12 is -C(O)-; (iii) when X 11 is -C(S)- then X 12 is -NR 1 -; (iv) when X 11 is -NR 1 - then X 12 is -C(S)-; (v) when X 11 is -C(O)- then X 12 is -O-; or (vi) when X 11 is -O- then X 12 is - C(O)-; wherein R 10 is hydrogen or C 1-8 alkyl.
- X 11 is -C(O)- and X 12 is -NR 10 -, wherein R 10 is hydrogen or C1-8 alkyl. In some embodiments, X 11 is -C(O)- and X 12 is -NR 10 -, wherein R 10 is hydrogen or methyl. In some embodiments, X 11 is -C(O)- and X 12 is -NR 10 -, wherein R 10 is hydrogen. In some embodiments, Y is -CH2- or -(CH2)2- . In some embodiments, Y is -CH2-. In some embodiments, the polymer comprises the following general formula: wherein R 10 is hydrogen or C1-8 alkyl; z is 2 to 24; and n is 1 to 100.
- z is 2 to 10. In some embodiments, z is 2 to 5. In some embodiments, z is 2. In some embodiments of the above formulas, R 10 is hydrogen or methyl. In some embodiments, R 10 is hydrogen. In some embodiments, a polymer conjugated lipid comprises “n” monomers of the following structure: In some embodiments of the above formulas, n is 5 to 50. In some embodiments, n is 5 to 25. In some embodiments, n is 7 to 14. In some embodiments, n is 10 to 25. In some embodiments, n is 14 to 17. In some embodiments, n is 8 or 14. In some embodiments, a polymer conjugated lipid comprises monomers of the following structure: . In some embodiments, a polymer conjugated lipid is selected from the table below:
- a polymer-conjugated lipid is about 0.5 to about 5 mol% relative to total lipids in the LNP.
- an LNP comprises about 1.0 to about 2.5 mol% of a polymer-conjugated lipid.
- an LNP comprises about 1.5 to about 2.0 mol% of a polymer-conjugated lipid.
- an LNP comprises about 1.5 to about 1.8 mol% of a polymer-conjugated lipid.
- a molar ratio of total cationic lipid to total polymer-conjugated lipid is from about 100:1 to about 20:1.
- a molar ratio of total cationic lipid to total polymer-conjugated lipid is from about 50:1 to about 20:1. In some embodiments, a molar ratio of total cationic lipid to total polymer-conjugated lipid is from about 40:1 to about 20:1. In some embodiments, a molar ratio of total cationic lipid to total polymer-conjugated lipid (e.g., PEG-conjugated lipid) is from about 35:1 to about 25:1.
- LNPs of the present disclosure may further comprise a compound of Formula (A1): L-X10-P-X20-B (A1) wherein P comprises a polymer; L comprises a hydrophobic moiety (e.g., lipid) attached to a first end of the polymer; B comprises a binding moiety attached to a second end of the polymer; X10 is absent or a first linking moiety; and X20 is absent or a second linking moiety.
- X10 comprises a carbonyl group.
- X20 comprises the reaction product of a thiol or cysteine reactive group, e.g., a maleimide group, with a thiol or cysteine group of a compound comprising the binding moiety.
- L comprises a lipid as described above.
- L comprises DSPE (distearoylphosphatidyl- ethanolamine), DPPE (dipalmitoylphosphatidylethanolamine), DOPE (dioleoylphosphatidylethanolamine), and POPE (palmitoyloleylphosphatidyl- ethanolamine) which may be linked to P by an amide group.
- P comprises a polymer as described above.
- P comprises a polymer selected from the group consisting of poly(ethylene glycol) (PEG), polysarcosine (pSar) (poly(N-methylglycine), polyoxazoline (POX), polyoxazine (POZ), poly-2-(2-(2-aminoethoxy)-ethoxy)acetic acid (pAEEA), and poly-2-(2-(2-methylamino-ethoxy)ethoxy)acetic acid (pMAEEA) (including derivatives thereof).
- P comprises poly(ethylene glycol) (PEG); e.g., PEG as described above.
- the binding moiety B comprises a moiety (e.g., an antibody or antigen-binding fragment thereof) binding to a cell surface antigen (e.g., a receptor, antigen or marker displayed on the surface of a target cell, such as a T cell, a B cell or a cancer cell).
- a cell surface antigen e.g., a receptor, antigen or marker displayed on the surface of a target cell, such as a T cell, a B cell or a cancer cell.
- the binding moiety B comprises or is a polypeptide (e.g., such as an epitope tag), e.g., an ALFA-tag, as described herein.
- the binding moiety B comprises or is an ALFA ⁇ tag comprises or consisting of a sequence selected from the group consisting of SEQ.ID.NO.1, SEQ.ID.NO.2, SEQ.ID.NO.3, and SEQ.ID.NO.4.
- the compound of formula (A1) may be a peptide-conjugated lipid.
- the compound of formula (A1) may be DSPE-PEG2K-alfa.
- the LNPs of the present disclosure may further comprise a peptide-conjugated lipid, which is optionally DSPE-PEG2K-alfa.
- LNPs comprising a compound of formula (A1), wherein the binding moiety B comprises or is an ALFA ⁇ tag (as described herein) can be further functionalized by binding to the ALFA ⁇ tag a “docking compound” comprising (i) an NbAlfa VHH domain comprising the CDR1 sequence SEQ.ID.NO.5, the CDR2 sequence SEQ.ID.NO.6, and the CDR3 sequence SEQ.ID.NO. 7; and (ii) an anti-CD3 binding VHH comprising the CDR1 sequence SEQ.ID.NO. 8, the CDR2 sequence SEQ.ID.NO. 9, and the CDR3 sequence SEQ.ID.NO.10.
- a “docking compound” comprising (i) an NbAlfa VHH domain comprising the CDR1 sequence SEQ.ID.NO.5, the CDR2 sequence SEQ.ID.NO.6, and the CDR3 sequence SEQ.ID.NO. 7; and (ii) an anti-CD3 binding VHH comprising the CDR
- a nucleic acid particle further comprises a steroid.
- a steroid is a sterol.
- a sterol is ⁇ -sitosterol, stigmasterol, cholesterol, cholecalciferol, ergocalciferol, calcipotriol, botulin, lupeol, ursolic acid, oleanolic acid, cycloartenol, lanosterol, or ⁇ -tocopherol.
- a sterol is ⁇ -sitosterol.
- a sterol is stigmasterol.
- a sterol is cholesterol.
- a sterol is cholecalciferol. In some embodiments, a sterol is ergocalciferol. In some embodiments, a sterol is calcipotriol. In some embodiments, a sterol is botulin. In some embodiments, a sterol is lupeol. In some embodiments, a sterol is ursolic acid. In some embodiments, a sterol is oleanolic acid. In some embodiments, a sterol is cycloartol. In some embodiments, a sterol is lanosterol. In some embodiments, a sterol is ⁇ -tocopherol.
- a lipid nanoparticle comprises about 30 to about 50 mol% of a steroid. In some embodiments, a lipid nanoparticle comprises about 35 to about 45 mol% of a steroid. In some embodiments, a lipid nanoparticle comprises about 38 to about 40 mol% of a steroid. In some embodiments, a lipid nanoparticle comprises about 38.5 mol% of a steroid. In some embodiments, a lipid nanoparticle comprises about 40 mol% of a steroid. In some embodiments, a lipid nanoparticle comprises about 30 to about 50 mol% of cholesterol. In some embodiments, a lipid nanoparticle comprises about 35 to about 45 mol% of cholesterol.
- a lipid nanoparticle comprises about 38 to about 41 mol% of cholesterol. In some embodiments, a lipid nanoparticle comprises about 38.5 mol% of cholesterol. In some embodiments, a lipid nanoparticle comprises about 40.7 mol% of cholesterol. In some embodiments, a lipid nanoparticle comprises: about 40 to about 50 mol% of a thiolipid compound; about 30 to about 50 mol% of a steroid; about 5 to about 15 mol% of a helper lipid; and about 1 to about 5 mol% of a polymer conjugated lipid.
- a lipid nanoparticle comprises: about 45 to about 50 mol% of a thiolipid compound; about 35 to about 45 mol% of a steroid; about 5 to about 15 mol% of a helper lipid; and about 1 to about 5 mol% of a polymer conjugated lipid.
- a lipid nanoparticle comprises: about 47.5 mol% of a thiolipid compound; about 40.5 mol% of a steroid; about 10 mol% of a helper lipid; and about 2 mol% of a polymer conjugated lipid.
- Lipids and lipid nanoparticles comprising nucleic acids and their method of preparation are known in the art, including, e.g., as described in U.S. Patent Nos. 8,569,256, 5,965,542 and U.S. Patent Publication Nos.
- cationic lipids, helper lipids, and steroids are solubilized in ethanol at a pre- determined weight or molar ratios/percentages (e.g., ones described herein).
- lipid nanoparticles are prepared at a total lipid to RNA or DNA molar ratio of approximately 10:1 to 30:1.
- RNA or DNA can be diluted to 0.2 mg/mL in acetate buffer.
- a colloidal lipid dispersion comprising nucleic acids e.g., RNA or DNA
- an ethanol solution comprising lipids, such as thiolipids described herein, helper lipids, steroids, and polymer-conjugated lipids is injected into an aqueous solution comprising nucleic acids (e.g., RNA or DNA).
- lipid and nucleic acid solutions can be mixed at room temperature by pumping each solution (e.g., a lipid solution comprising a thiolipid compound described herein, a helper lipid, steroids, and any other additives) at controlled flow rates into a mixing unit, for example, using piston pumps.
- the flow rates of a lipid solution and a nucleic acid solution into a mixing unit are maintained at a ratio of 1:3.
- nucleic acid-lipid particles are formed as the ethanolic lipid solution is diluted with aqueous RNAs. The lipid solubility is decreased, while cationic lipids bearing a positive charge interact with the negatively charged nucleic acid.
- a solution comprising nucleic acid (e.g., RNA)-encapsulated lipid nanoparticles can be processed by one or more of concentration adjustment, buffer exchange, formulation, and/or filtration.
- a composition or complex described herein further comprises a pharmaceutically acceptable surfactant.
- a pharmaceutically acceptable surfactant is selected from a polysorbate (e.g., polysorbate 20 (Tween20), polysorbate 40 (Tween40), polysorbate 60 (Tween60), and polysorbate 80 (Tween80)), poloxamers, and an amphiphilic group comprising a moiety selected from polyalkylene glycols (e.g., polyethylene glycol), poly(2-oxazoline), poly(2-methyl-2-oxazoline), polysarcosine, polyvinylpyrrolidone, and poly[N-(2-hydroxypropyl)methacrylamide, wherein the moiety is bound to one or more C12-C20 aliphatic groups.
- a polysorbate e.g., polysorbate 20 (Tween20), polysorbate 40 (Tween40), polysorbate 60 (Tween60), and polysorbate 80 (Tween80)
- poloxamers e.g., poloxamers, and an amphiphilic
- a particle described herein comprises one or more oligosaccharide compositions and a nucleic acid.
- a nucleic acid is RNA.
- an RNA amenable to technologies described herein is a single- stranded RNA.
- an RNA as disclosed herein is a linear RNA.
- a single-stranded RNA is a non-coding RNA in that its nucleotide sequence does not include an open reading frame (or complement thereof).
- a single-stranded RNA has a nucleotide sequence that encodes (or is the complement of a sequence that encodes) a polypeptide or a plurality of polypeptides (e.g., epitopes) of the present disclosure.
- an RNA is or comprises an siRNA, a miRNA, or other non-coding RNA.
- a relevant RNA includes at least one open reading frame (ORF) (e.g., is an mRNA); in some embodiments, a relevant RNA includes a single ORF; in some embodiments, a relevant RNA includes more than one ORF.
- ORF open reading frame
- an RNA comprises an ORF, e.g., encoding a polypeptide of interest or encoding a plurality of polypeptides of interest.
- an RNA produced in accordance with technologies provided herein comprises a plurality of ORFs (e.g., encoding a plurality of polypeptides).
- an RNA produced in accordance with technologies herein comprises a single ORF that encodes a plurality of polypeptides.
- polypeptides are or comprise antigens or epitopes thereof (e.g., relevant antigens).
- an ORF for use in accordance with the present disclosure encodes a polypeptide that includes a signal sequence, e.g., that is functional in mammalian cells, such as an intrinsic signal sequence or a heterologous signal sequence.
- a signal sequence directs secretion of an encoded polypeptide
- a signal sequence directs transport of an encoded polypeptide into a defined cellular compartment, preferably the cell surface, the endoplasmic reticulum (ER) or the endosomal-lysosomal compartment.
- an ORF encodes a polypeptide that includes a multimerization element (e.g., an intrinsic or heterologous multimerization element).
- an ORF that encodes a surface polypeptide includes a multimerization element.
- an ORF encodes a polypeptide that includes a transmembrane element or domain.
- an ORF is codon-optimized for expression in a cells of a particular host, e.g., a mammalian host, e.g., a human.
- an RNA includes unmodified uridine residues; an RNA that includes only unmodified uridine residues may be referred to as a “uRNA”.
- an RNA includes one or more modified uridine residues; in some embodiments, such an RNA (e.g., an RNA including entirely modified uridine residues) is referred to as a “modRNA”.
- an RNA may be a self-amplifying RNA (saRNA).
- an RNA may be a trans-amplifying RNA (taRNA) (see, for example, WO2017/162461).
- a relevant RNA includes a polypeptide-encoding portion or a plurality of polypeptide-encoding portions.
- such a portion or portions may encode a polypeptide or polypeptides that is or comprises a biologically active polypeptide or portion thereof (e.g., an enzyme or cytokine or therapeutic protein such as a replacement protein or antibody or portion thereof).
- a portion or portions may encode a polypeptide or polypeptides that is or comprises an antigen (or an epitope thereof), a cytokine, an enzyme, etc.
- an encoded polypeptide or polypeptides may be or include one or more neoantigens or neoepitopes associated with a tumor.
- an encoded polypeptide or polypeptides may be or include one or more antigens (or epitopes thereof) of an infectious agent (e.g., a bacterium, fungus, virus, etc.).
- an encoded polypeptide may be a variant of a wild type polypeptide.
- a single-stranded RNA e.g., mRNA
- may comprise a secretion signal-encoding region e.g., a secretion signal-encoding region that allows an encoded target entity or entities to be secreted upon translation by cells.
- such a secretion signal-encoding region may be or comprise a non-human secretion signal.
- such a secretion signal-encoding region may be or comprise a human secretion signal.
- a single-stranded RNA e.g., mRNA
- may comprise at least one non-coding element e.g., to enhance RNA stability and/or translation efficiency.
- non-coding elements include but are not limited to a 3’ untranslated region (UTR), a 5’ UTR, a cap structure (e.g., in some embodiments, an enzymatically-added cap; in some embodiments, a co-transcriptional cap), a poly adenine (polyA) tail (e.g., that, in some embodiments, may be or comprise 100 A residues or more, and/or in some embodiments may include one or more “interrupting” [i.e., non-A] sequence elements), and any combinations thereof.
- UTR untranslated region
- 5 UTR
- a cap structure e.g., in some embodiments, an enzymatically-added cap; in some embodiments, a co-transcriptional cap
- polyA tail e.g., that, in some embodiments, may be or comprise 100 A residues or more, and/or in some embodiments may include one or more “interrupting” [i.e., non-A] sequence elements
- non-coding elements may be found, for example, in WO2011015347, WO2017053297, US 10519189, US 10494399, WO2007024708, WO2007036366, WO2017060314, WO2016005324, WO2005038030, WO2017036889, WO2017162266, and WO2017162461, each of which is incorporated herein by referenced in its entirety.
- RNA pharmaceutical compositions e.g., immunogenic compositions or vaccines
- uRNA mRNA
- modRNA nucleoside-modified mRNA
- saRNA self-amplifying mRNA
- trans-amplifying RNAs RNAs.
- a non-modified uridine platform may include, for example, one or more of intrinsic adjuvant effect, good tolerability and safety, and strong antibody and T cell responses.
- modified uridine (e.g., pseudouridine) platform may include reduced adjuvant effect, blunted immune innate immune sensor activating capacity and thus augmented antigen expression, good tolerability and safety, and strong antibody and CD4-T cell responses.
- self-amplifying platform may include, for example, long duration of polypeptide (e.g., protein) expression, good tolerability and safety, higher likelihood for efficacy with very low vaccine dose.
- polypeptide e.g., protein
- a self-amplifying platform (e.g., saRNA) comprises a nucleic acid molecule, encoding both a replicase (e.g., a viral replicase) and a gene of interest, wherein the nucleic acid molecule is capable of being replicated by said replicase in cis (cis-replication system).
- a trans-amplifying platform (e.g., taRNA) comprises two nucleic acid molecules, wherein one nucleic acid molecule encodes a replicase (e.g., a viral replicase) and the other nucleic acid molecule is capable of being replicated (e.g., a replicon) by said replicase in trans (trans-replication system).
- a self/trans-amplifying platform (e.g., RNA) comprises a plurality of nucleic acid molecules, wherein said nucleic acids encode a plurality of replicases and/or replicons.
- a trans-replication system comprises the presence of both nucleic acid molecules in a single host cell.
- a nucleic acid encoding a replicase (e.g., a viral replicase) is not capable of self-replication in a target cell and/or target organism.
- a nucleic acid encoding a replicase (e.g., a viral replicase) lacks at least one conserved sequence element important for (-) strand synthesis based on a (+) strand template and/or for (+) strand synthesis based on a (-) strand template.
- a self-amplifying RNA comprises a 5’-cap; in some trans- replication systems, at least an RNA encoding a replicase is capped. Without wishing to be bound by any one theory, it has been found that a 5’-cap can be important for high level expression of a gene of interest in trans.
- a self/trans-amplifying platform does not require propagation of virus particles (e.g., is not associated with undesired virus-particle formation). In some embodiments, a self/trans-amplifying platform is not capable of forming virus particles.
- an RNA may comprise an Internal Ribosomal Entry Site (IRES) element. In some embodiments, an RNA does not comprise an IRES site; in particular, in some embodiments, an saRNA does not comprise an IRES site. In some such embodiments, translation of a gene of interest and/or replicase is not driven by an IRES element. In some embodiments, an IRES element is substituted by a 5’-cap.
- a complex described herein comprises modRNA, saRNA, taRNA, or uRNA.
- a complex comprises modRNA.
- a complex comprises saRNA.
- a complex comprises taRNA.
- a complex comprises uRNA.
- a disease, disorder, or condition is an infectious disease, cancer, an autoimmune disease, or a rare disease.
- an infectious disease is caused by or associated with a viral pathogen.
- a viral pathogen is of a family selected from poxviridae, rhabdoviridae, filoviridae, paramyxoviridae, hepadnaviridae, coronaviridae, caliciviridae, picornaviridae, reoviridae, retroviridae, and orthomyxoviridae.
- an infectious disease is caused by or associated with a virus selected from SARS-CoV-2, influenza, Crimean-Congo Hemorrhagic Fever (CCHF), Ebola virus, Lassa virus, Marburg virus, HIV, Nipah virus, and MERS-CoV.
- an infectious disease is caused by or associated with a bacterial pathogen.
- a bacterial pathogen is of a species selected from Actinomyces israelii, bacillus antracis, Bacteroides fragilis, Bordetella pertussis, Borrelia burgdorferi, Borrelia garinii, Borrelia afzelii, Borrelia recurrentis, Brucella abortus, Brucella canis, Brucella melitensis, Brucella suis, Campolobacter jejuni, Chlamydia pneumoniae, Chlamydia trachomatis, Chlamydophila psittaci, Clostridium botulinum, Clostridium difficile, Clostridium perfringens, Clostridium tetani, Corynebacterium idphteriae, Ehrlichia canis, Ehrlichia chaffeensis, Enterococcus faecalis, Enterococcus faecium, Escherichia
- an infectious disease is caused by or associated with a parasite.
- a parasite is of a family selected from Plasmodium, Leishmania, Cryptosporidium, Entamoeba, Trypanosomas, Schistosomes, Ascaris, Echinococcus and Taeniidae.
- a disease, disorder, or condition is a cancer.
- a cancer is selected from bladder cancer, breast cancer, colorectal cancer, kidney cancer, lung cancer, lymphoma, melanoma, oral/oropharyngeal cancer, pancreatic cancer, prostate cancer, thyroid cancer, and uterine cancer.
- a disease, disorder, or condition is a genetic disorder.
- a genetic disorder is associated with a gain-of-function mutation or a loss- of-function mutation.
- a disease, disorder, or condition is an autoimmune disease.
- an autoimmune disease is selected from Addison disease, celiac disease, rheumatoid arthritis, lupus, inflammatory bowel disease, dermatomyositis, multiple sclerosis, diabetes, Guillain-Barre syndrome, chronic inflammatory demyelinating polyneuropathy, psoriasis, pernicious anemia, graves’ disease, Hashimoto’s thyroiditis, myasthenia gravis, and vasculitis Sjörgen syndrome.
- a disease, disorder, or condition is a rare disease.
- a rare disease refers to a life-threatening or chronically debilitating diseases which are of such low prevalence (e.g., fewer than 1/2000 people) that special combined efforts are needed to address them.
- the present disclosure provides complexes that can selectively target particular systems within a body.
- targeting a particular system refers to causing increased expression of RNA derived from cargo in the complex in the desired system.
- complexes described herein can selectively target the lungs, liver, spleen, heart, brain, lymph nodes, bladder, kidneys, and pancreas.
- a complex “selectively targets” an organ when a single target expresses mRNA in an amount that is 65% or greater than expression in other organs post administration (e.g., 65% or more of mRNA throughout the body is expressed from a single organ, with the remaining 35% distributed between one or more different organs).
- a complex described herein selectively targets the lungs.
- a complex described herein selectively targets the liver.
- a complex described herein selectively targets the spleen.
- a complex described herein selectively targets the heart.
- a particle that is incorporated into a composition e.g., a pharmaceutical composition or a pharmaceutical formulation, as referred to herein
- a composition comprising particles described herein is administered as a monotherapy.
- a composition comprising particles described herein is administered as part of a combination therapy.
- a concentration of total RNA (e.g., a total concentration of all of the one or more RNA molecules) in a composition described herein is of about 0.01 mg/mL to about 0.5 mg/mL, or about 0.05 mg/mL to about 0.1 mg/mL.
- compositions may additionally comprise a pharmaceutically acceptable excipient, which, as used herein, includes any and all solvents, dispersion media, diluents, or other liquid vehicles, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired.
- a pharmaceutically acceptable excipient includes any and all solvents, dispersion media, diluents, or other liquid vehicles, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired.
- Gennaro (Lippincott, Williams & Wilkins, Baltimore, MD, 2006; incorporated herein by reference in its entirety) discloses various excipients used in formulating pharmaceutical compositions and known techniques for the preparation thereof. Except insofar as any conventional excipient medium is incompatible with a substance or its derivatives, such as by producing any undesirable biological effect or otherwise interacting in a deleterious manner with any other component(s) of the pharmaceutical composition, its use is contemplated to be within the scope of this disclosure.
- an excipient is approved for use in humans and for veterinary use.
- an excipient is approved by the United States Food and Drug Administration.
- an excipient is pharmaceutical grade.
- an excipient meets the standards of the United States Pharmacopoeia (USP), the European Pharmacopoeia (EP), the British Pharmacopoeia, and/or the International Pharmacopoeia.
- Pharmaceutically acceptable excipients used in the manufacture of pharmaceutical compositions include, but are not limited to, inert diluents, dispersing and/or granulating agents, surface active agents and/or emulsifiers, disintegrating agents, binding agents, preservatives, buffering agents, lubricating agents, and/or oils. Such excipients may optionally be included in pharmaceutical formulations.
- Excipients such as cocoa butter and suppository waxes, coloring agents, coating agents, sweetening, flavoring, and/or perfuming agents can be present in the composition, according to the judgment of the formulator.
- General considerations in the formulation and/or manufacture of pharmaceutical agents may be found, for example, in Remington: The Science and Practice of Phar ma cy 21st ed., Lippincott Williams & Wilkins, 2005 (incorporated herein by reference in its entirety).
- compositions provided herein may be formulated with one or more pharmaceutically acceptable carriers or diluents as well as any other known adjuvants and excipients in accordance with conventional techniques such as those disclosed in Remington: The Science and Practice of Phar ma cy 21st ed., Lippincott Williams & Wilkins, 2005 (incorporated herein by reference in its entirety).
- Pharmaceutical complexes and compositions described herein can be administered by appropriate methods known in the art. As will be appreciated by a skilled artisan, the route and/or mode of administration may depend on a number of factors, including, e.g., but not limited to stability and/or pharmacokinetics and/or pharmacodynamics of pharmaceutical compositions described herein.
- compositions described herein are formulated for parenteral administration, which includes modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intraarticular, subcapsular, subarachnoid, intraspinal, epidural and intrasternal injection and infusion.
- parenteral administration which includes modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intraarticular, subcapsular, subarachnoid, intraspinal, epidural and intrasternal injection and infusion.
- pharmaceutical compositions described herein are formulated for intravenous administration.
- pharmaceutically acceptable carriers that may be useful for intravenous administration include sterile aqueous solutions or dispersions and sterile powders for preparation of sterile injectable solutions or dispersions.
- pharmaceutical compositions described herein are formulated for subcutaneous (s.c) administration.
- pharmaceutical compositions described herein are formulated for intramuscular (i.m) administration.
- Therapeutic compositions typically must be sterile and stable under the conditions of manufacture and storage.
- the composition can be formulated as a solution, dispersion, powder (e.g., lyophilized powder), microemulsion, lipid nanoparticles, or other ordered structure suitable to high drug concentration.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyethylene glycol, and the like), and suitable mixtures thereof.
- the proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants.
- isotonic agents for example, sugars, polyalcohols such as mannitol, sorbitol, or sodium chloride in the composition.
- prolonged absorption of the injectable compositions can be brought about by including in the composition an agent that delays absorption, for example, monostearate salts and gelatin.
- Sterile injectable solutions can be prepared by incorporating the active compound in the required amount in an appropriate solvent with one or a combination of ingredients enumerated above, as required, followed by sterilization microfiltration.
- dispersions are prepared by incorporating the active compound into a sterile vehicle that contains a basic dispersion medium and the required other ingredients from those enumerated above.
- aqueous and nonaqueous carriers which may be employed in the pharmaceutical compositions described herein include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate.
- Proper fluidity can be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
- These compositions may also contain adjuvants such as preservatives, wetting agents, emulsifying agents and dispersing agents. Prevention of the presence of microorganisms may be ensured both by sterilization procedures, and by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid, and the like. It may also be desirable to include isotonic agents, such as sugars, sodium chloride, and the like into pharmaceutical compositions described herein.
- compositions described herein may be prepared by any method known or hereafter developed in the art of pharmacology. In general, such preparatory methods include the step of bringing active ingredient(s) into association with a diluent or another excipient and/or one or more other accessory ingredients, and then, if necessary and/or desirable, shaping and/or packaging the product into a desired single- or multi-dose unit.
- a pharmaceutical composition in accordance with the present disclosure may be prepared, packaged, and/or sold in bulk, as a single unit dose, and/or as a plurality of single unit doses.
- a "unit dose" is discrete amount of the pharmaceutical composition comprising a predetermined amount of at least one RNA product produced using a system and/or method described herein.
- an active agent that may be included in a pharmaceutical composition described herein is or comprises a therapeutic agent administered in a combination therapy described herein.
- Pharmaceutical compositions described herein can be administered in combination therapy, i.e., combined with other agents.
- such therapeutic agents may include agents leading to depletion or functional inactivation of regulatory T cells.
- a combination therapy can include a provided pharmaceutical composition with at least one immune checkpoint inhibitor.
- pharmaceutical composition described herein may be administered in conjunction with radiotherapy and/or autologous peripheral stem cell or bone marrow transplantation.
- a pharmaceutical composition described herein can be frozen to allow long-term storage.
- Embodiment 1 A compound represented by formula I: or a pharmaceutically acceptable salt thereof, wherein: L 1 and L 2 are each independently an optionally substituted C 1 -C 30 aliphatic group; L 3 is a bond, optionally substituted C 1 -C 10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S; X 1 and X 2 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O) 2 N(R 1 )- , -N(R 1 )S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R 1 )2-, -OC(S)C(R 1 )2-, -C(R 1 )2C(S)O
- Embodiment 2 The compound of Embodiment 1, wherein L 1 and L 2 are each C 1 - C 30 alkylene.
- Embodiment 3. The compound of Embodiments 1 or 2, wherein L 1 and L 2 are each independently –(CH 2 ) 6-12 -.
- Embodiment 4. The compound of any one of Embodiments 1-3, wherein L 1 and L 2 are the same.
- Embodiment 5. The compound of any one of Embodiments 1-3, wherein L 1 and L 2 are different.
- X 1 and X 2 are each independently selected from a -S(O) 2 N(R 1 )-, -N(R 1 )S(O) 2 , -S(O)-, -S(O) 2 -, - S(O) 2 C(R 1 ) 2 -, -OC(S)C(R 1 ) 2 -, -C(R 1 ) 2 C(S)O-, and –S-.
- Embodiment 9 The compound of any one of Embodiments 1-5, wherein X 1 is - S(O)2N(R 1 )-, where R 1 is C1-C10 aliphatic, and X 2 is –C(O)O-.
- Embodiment 9 The compound of any one of Embodiments 1-5, wherein X 1 and X 2 are each -S(O) 2 N(R 1 )-, where each R 1 is independently R 1 is C 1 -C 10 aliphatic.
- Embodiment 10 The compound of any one of Embodiments 1-5, wherein X 1 and X 2 are the same.
- Embodiment 11 The compound of any one of Embodiments 1-5, wherein X 1 and X 2 are different.
- Embodiment 15 The compound of any one of the preceding Embodiments, wherein a moiety –L 2 -X 2 -T 2 is selected from the group consisting of: Embodiment 16.
- the compound of any one of the preceding Embodiments, wherein a moiety –L 1 -X 1 -T 1 and moiety –L 2 -X 2 -T 2 are each independently selected from: Embodiment 17.
- Embodiment 19 The compound of any one of Embodiments 1-17, wherein G is - N(R 2 )C(S)N(R 2 )2 or -N(R 5 )S(O)2R 3 .
- Embodiment 19 The compound of any one of the preceding Embodiments, wherein G is -N(R 2 )C(S)N(R 2 ) 2 .
- Embodiment 20 The compound of any one of the preceding Embodiments, wherein G is -N(H)C(S)N(R 2 )2, where each R 2 is selected from optionally substituted C 1 - C 6 aliphatic and OH.
- Embodiment 21 The compound of any one of the preceding Embodiments, wherein G is –OH.
- Embodiment 22 The compound of any one of the preceding Embodiments, wherein G is optionally substituted 4- to 12-membered heterocycle.
- Embodiment 23 The compound of any one of the preceding Embodiments, wherein G is selected from: wherein moiety –L 3 -G is selected from:
- Embodiment 25 The compound of any one of the preceding Embodiments, wherein -L 3 -G is selected from: Embodiment 26.
- Embodiment 27 The compound of any one of the preceding Embodiments, wherein the compound is represented by Formula IIb: or a pharmaceutically acceptable salt thereof.
- Embodiment 28 The compound of any one of the preceding Embodiments, wherein the compound is represented by Formula IIc: or a pharmaceutically acceptable salt thereof.
- Embodiment 29 The compound of any one of the preceding Embodiments, wherein the compound is represented by Formula IIc: or a pharmaceutically acceptable salt thereof.
- Embodiment 30 The compound of any one of the preceding Embodiments, wherein the compound is represented by Formula IIIb: or a pharmaceutica lly acceptable salt thereof.
- Embodiment 31 The compound of any one of the preceding Embodiments, wherein the compound is represented by Formula IIIc: or a pharmaceuticall y acceptable salt thereof.
- Embodiment 32 The compound of any one of the preceding Embodiments, wherein the compound is represented by Formula IIId: or a pharmaceutically acceptable salt thereof.
- Embodiment 33 The compound of any one of the preceding Embodiments, wherein the compound is represented by Formula IIIa: or a pharmaceutically acceptable salt thereof.
- Embodiment 34 The compound of any one of the preceding Embodiments, wherein the compound is represented by Formula IIIf: or a pharmaceutic ally acceptable salt thereof.
- Embodiment 35 The compound of any one of the preceding Embodiments, wherein the compound is represented by Formula IIIg: or a pharmaceutic ally acceptable salt thereof.
- Embodiment 36 The compound of any one of the preceding Embodiments, wherein the compound is selected from Table 1.
- Embodiment 37 A particle comprising a compound of any one of Embodiments 1- 36, and a nucleic acid.
- Embodiment 38 The compound of any one of the preceding Embodiments, wherein the compound is selected from Table 1.
- Embodiment 37 wherein the nucleic acid is RNA, DNA, or mixtures thereof.
- Embodiment 39 The particle of Embodiment 38, the RNA is mRNA.
- Embodiment 40 The particle of Embodiment 39, wherein the RNA is modRNA, circRNA, saRNA, taRNA, or uRNA.
- Embodiment 41 The particle of Embodiment 37, wherein the DNA is linear DNA, plasmid DNA, minicircle DNA, nanoplasmid DNA, doggybone DNA, or a transposon.
- Embodiment 42 The particle of Embodiment 37, wherein the nucleic acid is RNA, DNA, or mixtures thereof.
- Embodiment 39 The particle of Embodiment 38, the RNA is mRNA.
- Embodiment 40 The particle of Embodiment 39, wherein the RNA is modRNA, circRNA, saRNA, taRNA, or uRNA.
- Embodiment 41 The
- Embodiment 43 The particle of Embodiment 42, wherein the helper lipid is a phospholipid.
- the particle of Embodiment 43, wherein the phospholipid is selected from: phosphatidylcholines, phosphatidylethanolamines, phosphatidylglycerols, phosphatidic acids, phosphatidylserines and sphingomyelins, more preferably selected from the group consisting of distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dimyristoylphosphatidylcholine (DMPC), dipentadecanoylphosphatidylcholine, dilauroylphosphatidylcholine, dipalmitoylphosphatidylcholine (DPPC), diarachidoylphosphatidylcholine (DAPC), dibehenoylphosphatidylcholine (DBPC), ditricosanoylphosphatidylcholine (DTPC), dilignoceroylphatidylcholine (DLPC),
- Embodiment 45 The particle of any one of Embodiments 42-44, wherein the polymer-conjugated lipid is selected from the group consisting of a poly(ethylene glycol) (PEG)-conjugated lipid, a poly(sarcosine) (pSar)-conjugated lipid, a poly(aminoethoxy ethoxy acetic acid) (pAEEA)-conjugated lipid; and a poly(2-methylaminoethoxy ethoxy acetic acid) (pMAEEA)-conjugated lipid.
- PEG poly(ethylene glycol)
- pSar poly(sarcosine)
- pAEEA poly(aminoethoxy ethoxy acetic acid)
- pMAEEA poly(2-methylaminoethoxy ethoxy acetic acid)
- the polymer-conjugated lipid is a PEG-lipid selected from PEG-DAG, PEG-PE, PEG-S-DAG, PEG2000-DMG, PEG-cer, a PEG dialkyoxypropylcarbamate, and combinations thereof.
- Embodiments 42-46 wherein the sterol is selected from ⁇ -sitosterol, stigmasterol, cholesterol, cholecalciferol, ergocalciferol, calcipotriol, botulin, lupeol, ursolic acid, oleanolic acid, cycloartenol, lanosterol, or ⁇ - tocopherol.
- Embodiment 48 The particle of any one of Embodiments 42-47, wherein the particle is characterized by an N/P ratio that is about 4 to about 16.
- Embodiment 49 Embodiment 49.
- a method of increasing or causing increased expression of RNA in a target in a subject comprising administering to the subject a composition comprising particles of any one of Embodiments 42-48.
- Embodiment 50 The method of Embodiment 49, wherein the target is selected from the lungs, liver, spleen, heart, brain, lymph nodes, bladder, kidneys, and pancreas.
- Embodiment 51 A method of treating a disease, disorder, or condition in a subject comprising administering to the subject a composition comprising the particles of any one of Embodiments 42-48.
- Embodiment 52 A method of increasing or causing increased expression of RNA in a target in a subject, the method comprising administering to the subject a composition comprising particles of any one of Embodiments 42-48.
- Embodiment 51 wherein the disease, disorder, or condition is an infectious disease, cancer, a genetic disorder, an autoimmune disease, or a rare disease.
- Embodiment 53 The method of any one of Embodiments 48-52, wherein the particle is administered parenterally or intranasally.
- Embodiment 54 The method of Embodiment 53, wherein the particle is administered intramuscularly, subcutaneously, intradermally, or intravenously.
- Embodiment 55 A particle of any one of Embodiments 42-48 for use as a medicament.
- Embodiment 56 is
- the disease or disorder is an infectious disease, cancer, a genetic disorder, an autoimmune disease, or a rare disease.
- Reference to an Alfa-Tag or Alfa peptide refers to a peptide of sequence SEQ.ID.NO.1
- Reference to NbAlfa refers to a VHH domain comprising the CDR1 sequence SEQ.ID.NO. 5, the CDR2 sequence SEQ.ID.NO. 6, and the CDR3 sequence SEQ.ID.NO. 7.
- Reference to aCD3-VHH refers to an anti-CD3 binding VHH comprising the CDR1 sequence SEQ.ID.NO. 8, the CDR2 sequence SEQ.ID.NO. 9, and the CDR3 sequence SEQ.ID.NO. 10.
- the present example provides a method preparing tail-end-fragments with tail-linkers via unoxidized junction residue “-S-“ having an oxidation state of -2. ( Figure 79)
- Example compounds BNT-21, BNT-46, and BNT-20 were prepared according to the methods provided above.
- the reaction mixture was evaporated to an oil/solid mixture, then taken up in an emulsion of either n-hexane or diethyl ether (150mL) and 10% aqueous NaH 2 PO 4 (150 mL) dependent on the solubility of the product.
- the organic phase was washed with brine (150 mL), then dried over anhydrous Na2SO4.
- the crude product was purified by flash chromatography on silica gel using ethyl acetate/n-hexane. The product fractions were identified using ELSD and UV detectors of the effluent. Evaporation of the product fractions resulted in the thiolipid alcohol as colorless oil representing the precursor for the tail group aldehydes.
- reaction mixture may be temporarily cooled by an ice bath. After continuous stirring for more than 2 hours at room temperature the reaction is found to be complete by TLC (hexane, ethyl acetate).
- TLC hexane, ethyl acetate
- the reaction mixture was transferred into 10% aqueous NaH 2 PO 4 (200 mL) and diethyl ether (200mL). After the phase separation occurred, the turbid organic phase was removed. The turbid aqueous phase was washed twice successively with 100mL diethyl ether each.
- the organic phases are combined and passed through a silica 60-plug (height: 30 mm, diameter 55mm) to remove the turbidity resulting from remaining SO 3 - complex.
- the filtrate was concentrated to a clear oil that may be chromatographed on silica or used directly for the formation of thiolipids using the following multicomponent reaction.
- the salt is removed by filtration, the filtrate evaporated to a pale yellow or white oil which may be taken up in hexane prior to subjection to chromatography on silica.
- the latter is performed using a gradient of hexane/1% triethyl amine in chloroform: 100/0-0/100(v/v); 1% triethyl amine in chloroform/methanol 100/0- 93/7(v/v).
- the product is detected in the effluent with an ELSD.
- the product fractions were pooled and evaporated to a colorless oil.
- the reaction mixture was poured into ice-water (50 mL). From the resulting emulsion, the product was extracted with chloroform (30 mL). The dark extract was treated with active carbon and subsequently dried with sodium sulfate. After filtration through Celite 545, the filtrate was evaporated to a black solid and a yellow oil. Only the latter was soluble in n- hexane/EtOAc: 9/1, v/v where it can be purified with the same solvent mixture.
- Methanesulfonic anhydride (14.0 g, 80.4 mmol, 1.5 equiv.) was added in portions, the reaction mixture was warmed and stirred for 2h at rt. The progress of the reaction was monitored with TLC by using solvent mixture (n-hexane/EtOAc; 4:1). The reaction mixture was neutralized with (200 ml) NaHCO 3 , the organic layer was separated, and the aq. Layer was further extracted with (2 X 100 ml) DCM. The combined organic phase was washed with (2 X 100 ml) brine, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was used in the next step without any further purification.
- reaction mixture was neutralized with 2 M HCl at 0°C, the mixture was diluted with (100 ml) diethyl ether, washed with (2 X 100 ml) H2O, and (2 X 100 ml) brine. The organic layer was collected, dried over Na 2 SO 4 , and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO 2 ; n-hexane).
- reaction mixture was neutralized with (10 ml) NaHCO 3 at 0°C, the mixture was diluted (100 ml) EtOAc, washed with (2 X 50 ml) H 2 O, then with (2 X 50 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane/EtOAc; 1/1).
- 7- octene-1-sulfonyl chloride can be prepared in high yield from 7-bromo-1-octene with sodium sulfite and treatment of the resultant acid with phosphorylchloride.
- 7-octene-1-sulfonyl chloride (CAS:923279-52-1) was added to a solution of octyl hexyl amine (CAS:82223-69-6) in THF and DEA:
- the terminal alkene (compound 3) was converted to an aldehyde by ozonolysis to provide a compound 4.
- General procedure for the synthesis of sulfonamides with terminal aldehydes was performed analogously as reported in WO2021250102.
- the aldehyde compound (e.g., compound 4 in the scheme above) was reacted with the primary amine bearing the head group in presence of Na[HB(OAc)3], Na2SO4, in DCM at RT to obtain the lipid with either a functional head group or a protected precursor of the final lipid.
- Compounds described herein can be prepared according to the scheme below:
- BNT51, 4-amino-butanol (CAS: 13325-10-5) was reacted with aldehydes of compound 4.
- the remaining head groups in the scheme above can be prepared using the reagents below, which are commercially available: CAS: 127346-48-9: N-t-Butyloxycarbonyl-1,3-diaminopropane hydrochloride CAS: 75178-96-0: tert-Butyl 3-aminopropylcarbamate CAS: 68076-36-8: tert-Butyl 4-aminobutylcarbamate CAS: 33545-98-1: N-t-Butyloxycarbonyl-1,4-diaminobutane hydrochloride
- the BOC-derivatized lipids can be deprotected using TFA.
- CAS: 16420-13-6 Dimethylthiocarbamoyl chloride, commercially available.
- CAS: 2241238-65-1 1-azeticinecabothioyl chloride, prepared according to the methods described in WO2018140730 CAS: 19009-42-8: 1-Pyrrolidinecarbothioyl chloride, commercially available.
- CAS: 16420-13-6 Dimethylthiocarbamoyl chloride, commercial available.
- CAS: 2241238-65-1 1-azeticinecabothioyl chloride, prepared according to the methods described in WO2018140730.
- CAS: 19009-42-8 1-Pyrrolidinecarbothioyl chloride, commercially available.
- Thiolipid compounds of the present disclosure can be prepared according to analogous methods known to those of skill in the art.
- Example 2 Manufacturability and in vitro investigation of LNP formulations consisting of novel ionizable lipids, BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59
- LNPs lipid nanoparticles
- thiolipid compounds e.g., BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT- 59
- Lipids with the thiourea head group led to formulations with smaller sizes compared to lipids with hydroxyl head group (BNT-51 from tail group 1 and BNT-56 from tail group 2).
- thiourea heads with the hydroxyl groups showed the lowest sizes in their corresponding groups.
- Tail group 1 showed smaller sizes compared to their tail group 2 analogs indicating that the branching position and the length of the branches effect the hydrodynamic diameters.
- the zeta potential values of the formulations have been investigated by electrophoretic light scattering (Figure 2).
- the formulations showed neutral-to-negative zeta potential values.
- the mRNA encapsulation efficiency of the formulations has been investigated by Ribogreen assay (Figure 3A). All formulations except BNT-56, showed very high encapsulation efficiency ( ⁇ 87%). No free mRNA has been seen as shown by AGE ( Figure 3B). mRNA integrity of the formulations has been determined by capillary electrophoresis, fragment analyzer ( Figure 4). As seen in Figure 4, negligible amount of mRNA fragmentation has been observed for all formulations.
- Example 3 Manufacturability and in vitro investigation of LNP formulations consisting of ionizable lipids, BNT-sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT- sulfo-04.
- the present example describes manufacturability and in vitro biological effects of the LNPs consisting of the following lipids: BNT-sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04.
- BNT-51 and BNT-52 have also been formulated for the present example.
- Example LNPs have been prepared by microfluidic mixing of the lipid mixture in ethanol and RNA solution in aqueous, acidic buffer.
- cytotoxicity and transfection efficiency of the formulations have been tested at three different mRNA concentrations (12.5, 25 and 50 ng) on four different cell lines (C2C12, HepG2, RAW 264.7 and Hek 293) ( Figures 11A-D and Figures 12A-D)
- the formulations did not show significant cytotoxicity at the tested conditions.
- Formulations were effectively able to transfect the cell lines that have been tested ( Figures 12A-D).
- BNT-51 has showed the highest in vitro transfection efficiency at all concentrations on all tested cell lines.
- BNT-sulfo-04 has showed the lowest transfection efficiency most likely due to decreased potency caused by the short tailing.
- BNT-52 showed slightly better expression than BNT-sulfo-01.
- Example 4 In vitro investigation of the effect of helper lipid on the LNP formulations consisting of novel ionizable lipid, BNT-51
- the present example investigates the manufacturability and in vitro biological effects of the LNPs consisting of BNT-51 with two different helper lipids (DSPC or DOPE).
- LNPs with the same composition but different helper lipids have been prepared, characterized, and tested in vitro.
- LNPs have been prepared by microfluidic mixing of the lipid mixture in ethanol and RNA solution in aqueous, acidic buffer. The details of the formulations have been given in the table below.
- the sizes of the LNPs were analyzed DLS ( Figure 14).
- the hydrodynamic diameter of the DSPC having formulation was found to be higher than DOPE having BNT-51 formulation (106 and 72 nm, respectively).
- BNT-51_DOPE has displayed higher PDI (0.28) than BNT-51_DSPC (0.13).
- the zeta potential values of the formulations have been investigated by electrophoretic light scattering ( Figure 15).
- the helper lipid did not significantly affect the zeta potential values.
- the formulations showed neutral zeta potential.
- the mRNA encapsulation efficiency of the formulations has been investigated by Ribogreen assay ( Figure 16A). Both formulations, regardless of the helper lipid, showed very high encapsulation efficiency ( ⁇ 90%).
- Example 5 In vitro investigation of the effect of stealth lipid and the stealth lipid anchor on the LNP formulations consisting of novel ionizable lipid, BNT-51
- the present example reports manufacturability and in vitro biological effects of the LNPs consisting of the same novel ionizable lipid (BNT-51), varying stealth lipids (PEG-DMG, VE-(AEEA)14_AC_2 or DMG-(AEEA)14_AC_2)
- LNPs with the same composition but different stealth lipids have been prepared, characterized, and tested in vitro.
- LNPs have been prepared by microfluidic mixing of the lipid mixture in ethanol and RNA solution in aqueous, acidic buffer. The details of the formulations have been given in the table below.
- the sizes of the LNPs have been investigated by DLS ( Figure 20).
- Well-defined formulations with diameters of ⁇ 100 nm and PDI of ⁇ 0.2 (narrow polydispersity) were able to be obtained.
- the hydrodynamic diameters and PDIs of the formulations are substantially similar.
- the zeta potential values of the formulations have been investigated by electrophoretic light scattering ( Figure 21). All formulations have showed very similar, neutral-to-slightly negative, zeta potential values.
- the mRNA encapsulation efficiency of the formulations was assessed by Ribogreen assay (Figure 22A).
- BNT-51_VitE-AEEA has showed negligible amount of free mRNA (8.5%, semi-quantified by ImageJ analysis).
- mRNA integrity of the formulations has been determined by capillary electrophoresis, fragment analyzer ( Figure 23). As seen in the graph, no mRNA fragmentation has bene observed for the formulations.
- the cytotoxicity and transfection efficiency of the formulations have been tested at three different mRNA concentrations (12.5, 25 and 50 ng) on four different cell lines (C2C12, HepG2, RAW 264.7, HEK-293) ( Figures 24A-D and Figure 25A-D) The formulations have not showed any cytotoxicity at the tested conditions.
- BNT-51_PEG-DMG and BNT_51_DMG-AEEA formulation have showed slightly higher transfection efficiency than BNT-51 formulation with Vitamin E lipid anchor (BNT-51_VitE-AEEA).
- Example 6 Preparation and characterization of RNA/DNA-lipid nanoparticles (BNT51/Ac-AEEA14-DMA/DSPE-PEG2k-Alfa-Tag lipid) Manufacturing of pre-formed lipid nanoparticles (BNT51/Ac-AEEA14-DMA/DSPE- PEG2k-Alfa-Tag lipid) by ethanol injection
- Alfa ⁇ tagged pre-formed lipid nanoparticles were prepared using a lipid mix consisting of BNT51, DSPC, cholesterol, Ac-AEEA14-DMA and DSPE ⁇ PEG2k ⁇ Alfa peptide (a DSPE- PEG2K polymer functionalized with an Alfa peptide, having a sequence of SEQ.ID.NO.
- the organic solvent in the obtained raw colloid nanoparticles was removed by dialysis against 5mM AcOH using Slide-A-Lyzer dialysis cassettes of 10K molecular weight cut- off (MCWO) (Thermo Fisher Scientific, Waltham, MA, USA).
- MCWO molecular weight cut- off
- the nanoparticles were concentrated to 2X times through centrifugation at 3000rpm x 10 min at 4°C, using Amico Ultra-15 of 30K molecular weight cut-off (MCWO) (Merck, Darmstadt, Germany). After concentration, the nanoparticles were diluted to a sucrose concentration of 10%.
- Frozen stability of pre-formed lipid (BNT51/Ac-AEEA14-DMA/DSPE-PEG2k-Alfa-Tag lipid) nanoparticles
- the frozen stability of the pre-formed lipid nanoparticles was assessed by cycling the formulations from ⁇ 20 °C and -80 °C (overnight) to +25 °C (2 h) for at least three times.
- the particle size and polydispersity index of the formulations were measured for freeze ⁇ thaw samples.
- the formulations between thaw and freeze cycles were mixed by gentle inversions before the next freezing cycle.
- RNA/DNA-lipid particles were prepared by an aqueous-aqueous protocol (LNP2), as described herein.
- Reporter Thy1.1 RNA and reporter Venus DNA were used for characterization experiments.
- RNA (Thy1.1) in aqueous buffer (HEPES 10 mM, EDTA 0.1 mM, pH 7.0) and DNA (Venus) in water were mixed with pre-formed lipid nanoparticles composed of BNT51, DSPC, cholesterol, Ac-AEEA14-DMA and DSPE ⁇ PEG2k ⁇ alfa peptide in a molar ratio of 47.5:10:40.5:1.8:0.2, respectively, in aqueous solution (5 mM acetic acid, 10% sucrose), in a volume ratio of 1:1.
- RNA/DNA-lipid particles were then functionalized with an aCD3-VHH ligand at ligand/cargo ratio of 0.48 w/w, and further diluted with 60 mM HEPES pH 6.0, 20% (w/v) sucrose to a final nucleic acid concentration of 0.1 mg/mL (final buffer composition: 22 mM HEPES, 9% sucrose, pH ⁇ 5).
- RNA/DNA-lipid particles were prepared at N/P ratio of 6:1 and filtered through a 0.22 ⁇ m polyethersulfone (PES) filter.
- PES polyethersulfone
- Manufacturing of functionalized RNA/DNA-lipid particles by aqueous-organic protocol (LNP1) RNA/DNA-lipid particles were alternatively prepared by an aqueous-ethanol mixing protocol (LNP1), as described herein.
- Reporter Thy1.1 RNA and reporter Venus DNA were also used for characterization experiments.
- RNA (Thy1.1) in aqueous buffer condition (HEPES 10 mM, EDTA 0.1 mM, pH 7.0) and DNA (Venus) in water were mixed with ethanolic lipid mix comprising BNT51, DSPC, cholesterol, Ac-AEEA14-DMA and DSPE ⁇ PEG2k ⁇ alfa peptide in a molar ratio of 47.5:10:40.5:1.8:0.2, respectively, at 14.4 mM total lipid concentration, in a volume ratio of 3 parts nucleic acids and 1 part lipid mix.
- the mixing was achieved using a standard syringe pump based set-up equipped with in-line dilution, with a total flow of 90 ml/min (45 ml/min for nucleic acid phase, 15 ml/min for lipid phase, 30 ml/min for in-line dilution) using T mixing element (0.5 mm T mixer).
- T mixing element 0.5 mm T mixer.
- the organic solvent in the obtained raw colloid nanoparticles was removed by dialysis against 20 mM HEPES pH 5.5 using Slide-A-Lyzer dialysis cassettes G2 of 10K molecular weight cut-off (MCWO) (Thermo Fisher Scientific, Waltham, MA, USA).
- RNA/DNA-lipid particles were then functionalized with an aCD3-VHH ligand at ligand/cargo ratio of 0.48 w/w, and further diluted to a final nucleic acid concentration of 0.1 mg/mL and storage matrix of 20 mM HEPES pH 5.5, 10% (w/v) sucrose.
- the RNA/DNA-lipid particles were prepared at N/P ratio of 6:1 and filtered through a 0.22 ⁇ m polyethersulfone (PES) filter. Characterization of the Formulations Particle size was determined by dynamic light scattering using a DynaPro Plate Reader II (Wyatt, Dernbach, Germany).
- RNA/DNA-LNP1 and RNA/DNA-LNP2 were calculated from the cumulant analysis using Dynamics 7.8.1.3 software.
- samples were diluted 1:10 in water and analysis was performed in triplicates. pH was measured on 150 ⁇ L sample after calibration of the device with pH 4, 7, and 10 standards. Successful cargos incorporation was verified via Agarose Gel Electrophoresis. Size and PDI was determined for functionalized sterile filtered RNA/DNA-LNP1 and RNA/DNA-LNP2 (prepared as described above) stored at final nucleic concentration of 0.1 mg/mL via DLS measurement.
- RNA/DNA-LNP1 and RNA/DNA-LNP2 can be successfully manufactured with Ac- AEEA14-DMA stealth moiety, resulting in particles of size ⁇ 100 nm and a PDI ⁇ 0.3 (Figure 27A).
- Successful cargo incorporation was verified via Agarose gel electrophoresis: no RNA or DNA bands are present in the samples, indicating that the formulation did not have any free cargo.
- all the respective RNA and DNA bands were clearly visible when the samples were treated with a release solution (controls), confirming the successful encapsulation of all the cargos (Figure 27B).
- RNA/DNA-lipid nanoparticles BNT51/Ac-AEEA14-DMA/Alfa- Tag lipid
- freeze thaw studies were conducted by cycling the formulations from ⁇ 20°C and -80 °C (overnight) to +25 °C (2 h) for at least three times. The particle size and polydispersity index of the formulations were measured for freeze ⁇ thaw samples. The formulations between thaw and freeze cycles were mixed by gentle inversions before the next freezing cycle.
- FIG. 29A shows the percentages of transfected cell (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs (Transfection, y-axes) per tested formulation condition (as indicated) for both RNA (Thy1.1) and DNA (Venus).
- Example 7 Manufacturing of RNA/DNA lipid nanoparticles (BNT51/Ac-AEEA14- VitE/ DSPE-PEG2k-Alfa-Tag lipid) Manufacturing of pre-formed lipid nanoparticles (BNT51/Ac-AEEA14-VitE/DSPE- PEG2k-Alfa-Tag lipid) by ethanol injection
- a lipid mix consisting of BNT51, DSPC, cholesterol, Ac-AEEA14-VitE and DSPE ⁇ PEG2k ⁇ peptide in a molar ratio of 47.5:10:40.5:1.8:0.2 was dissolved in organic solvent (ethanol) at 20 mM total lipid concentration and mixed with an aqueous phase (5mM AcOH)
- the organic solvent in the obtained raw colloid nanoparticles was removed by dialysis against 5mM AcOH using Slide-A-Lyzer dialysis cassettes of 10K molecular weight cut-off (MCWO) (Thermo Fisher Scientific, Waltham, MA, USA).
- MCWO molecular weight cut-off
- the nanoparticles were concentrated to 2X times through centrifugation at 3000rpm x 10 min at 4°C, using Amico Ultra-15 of 30K molecular weight cut-off (MCWO) (Merck, Darmstadt, Germany). After up-concentration, the nanoparticles were diluted to a sucrose concentration of 10%.
- Frozen stability of pre-formed lipid (BNT51/Ac-AEEA14-VitE/DSPE-PEG2k-Alfa-Tag lipid) nanoparticles
- the frozen stability of the pre-formed lipid nanoparticles has been investigated by cycling the formulations from ⁇ 20 °C and -80 °C (overnight) to +25 °C (2 h) for at least three times.
- the particle size and polydispersity index of the formulations were measured for freeze ⁇ thaw samples.
- the formulations between thaw and freeze cycles were mixed by gentle inversions before the next freezing cycle.
- RNA/DNA-lipid particles were prepared by an aqueous-aqueous protocol (LNP2), as described herein.
- RNA (Thy1.1) in aqueous buffer conditions HPES 10 mM, EDTA 0.1 mM, pH 7.0
- DNA Venus
- pre-formed lipid nanoparticles composed of BNT51, DSPC, cholesterol, Ac-AEEA14-VitE and DSPE ⁇ PEG2k ⁇ alfa peptide in a molar ratio of 47.5:10:40.5:1.8:0.2, respectively, in aqueous solution (5 mM acetic acid, 10% sucrose), in a volume ratio of 1:1.
- RNA/DNA-lipid particles were then functionalized with an aCD3-VHH ligand at ligand/cargo ratio of 0.48 w/w, and further diluted with 60 mM HEPES pH 6.0, 20% (w/v) sucrose to a final nucleic acid concentration of 0.1 mg/mL (final buffer composition: 22 mM HEPES, 9% sucrose, pH ⁇ 5).
- RNA/DNA-lipid particles were prepared at N/P ratio of 6:1 and filtered through a 0.22 ⁇ m polyethersulfone (PES) filter. Manufacturing of functionalized RNA/DNA-lipid particles by aqueous-organic protocol RNA/DNA-lipid particles were prepared by an aqueous-ethanol mixing protocol (LNP1), as described herein.
- LNP1 aqueous-ethanol mixing protocol
- RNA (Thy1.1) in aqueous buffer conditions HPES 10 mM, EDTA 0.1 mM, pH 7.0
- DNA (Venus) in water were mixed with ethanolic lipid mix comprising BNT51, DSPC, cholesterol, Ac-AEEA14-VitE and DSPE ⁇ PEG2k ⁇ alfa peptide in a molar ratio of 47.5:10:40.5:1.8:0.2, respectively, at 14.4 mM total lipid concentration, in a volume ratio of 3 parts nucleic acids and 1 part lipid mix.
- the mixing was achieved using a standard syringe pump-based set-up equipped with in-line dilution, with a total flow of 90 ml/min (45 ml/min for nucleic acid phase, 15 ml/min for lipid phase, 30 ml/min for in-line dilution) using T mixing element (0.5 mm T mixer).
- T mixing element 0.5 mm T mixer.
- the organic solvent in the obtained raw colloid nanoparticles was removed by dialysis against 20 mM HEPES pH 5.5 using Slide-A-Lyzer dialysis cassettes G2 of 10K molecular weight cut-off (MCWO) (Thermo Fisher Scientific, Waltham, MA, USA).
- RNA/DNA-lipid particles were then functionalized with an aCD3-VHH ligand at ligand/cargo ratio of 0.48 w/w, and further diluted to a final nucleic acid concentration of 0.1 mg/mL and storage matrix of 20 mM HEPES pH 5.5, 10% (w/v) sucrose.
- the RNA/DNA-lipid particles were prepared at N/P ratio of 6:1 and filtered through a 0.22 ⁇ m polyethersulfone (PES) filter. Characterization of the formulations Particle size was determined by dynamic light scattering using a DynaPro Plate Reader II (Wyatt, Dernbach, Germany).
- RNA/DNA-LNP1 and RNA/DNA-LNP2 were calculated from the cumulant analysis using Dynamics 7.8.1.3 software.
- samples were diluted 1:10 in water and analysis was performed in triplicates. pH was measured on 150 ⁇ L sample after calibration of the device with pH 4, 7, and 10 standards. Successful cargos incorporation was verified via Agarose Gel Electrophoresis. Size and PDI was determined for functionalized sterile filtered RNA/DNA-LNP1 and RNA/DNA-LNP2 stored at final nucleic concentration of 0.1 mg/mL via DLS measurement.
- RNA/DNA-LNP1 and RNA/DNA-LNP2 can be successfully manufactured with Ac-AEEA14-VitE stealth moiety, resulting in particles of size ⁇ 100 nm and a PDI ⁇ 0.3 (Figure 30A).
- Successful cargo incorporation was verified via Agarose gel electrophoresis: no RNA or DNA bands are present in the samples, indicating that the formulation did not have any free cargo.
- all the respective RNA and DNA bands were clearly visible when the samples were treated with a release solution (controls), confirming the successful encapsulation of all the cargos (Figure 30B).
- RNA/DNA-lipid nanoparticles BNT51/Ac-AEEA14-VitE/DSPE- PEG2k-Alfa-Tag lipid
- freeze thaw studies were conducted by cycling the formulations from ⁇ 20°C and -80°C (overnight) to +25°C (2 h) for at least three times. The particle size and polydispersity index of the formulations were measured for freeze ⁇ thaw samples. The formulations between thaw and freeze cycles were mixed by gentle inversions before the next freezing cycle.
- FIGS 31A-B show the percentages of transfected cell (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs (Transfection, y-axes) per tested formulation condition (as indicated) for both RNA (Thy1.1) and DNA (Venus).
- Example 8 Manufacturing of RNA/DNA lipid nanoparticles (BNT51/Ac-AEEA14- DMA/ DSPE-pAEEA14-Alfa-Tag lipid) Manufacturing of pre-formed lipid nanoparticles (BNT51/Ac-AEEA14-DMA/DSPE- pAEEA14-Alfa-Tag lipid) by ethanol injection
- a lipid mix consisting of BNT51, DSPC, cholesterol, Ac-AEEA14-DMA and DSPE ⁇ pAEEA14 ⁇ alfa peptide in a molar ratio of 47.5:10:40.5:1.8:0.2 was dissolved in organic solvent (ethanol) at 20 mM total lipid concentration and mixed with an aqueous phase (5mM AcOH) in a 3:1
- the organic solvent in the obtained raw colloid nanoparticles was removed by dialysis against 5mM AcOH using Slide-A-Lyzer dialysis cassettes of 10K molecular weight cut-off (MCWO) (Thermo Fisher Scientific, Waltham, MA, USA).
- MCWO molecular weight cut-off
- the nanoparticles were concentrated to 2X times through centrifugation at 3000rpm x 10 min at 4°C, using Amico Ultra-15 of 30K molecular weight cut-off (MCWO) (Merck, Darmstadt, Germany). After concentration, the nanoparticles were diluted to a sucrose concentration of 10%.
- Frozen stability of pre-formed lipid (BNT51/Ac-AEEA14-DMA/DSPE-pAEEA14-Alfa-Tag lipid) nanoparticles
- the frozen stability of the pre-formed lipid nanoparticles has been investigated by cycling the formulations from ⁇ 20°C and -80°C (overnight) to +25°C (2 h) for at least two times.
- the particle size and polydispersity index of the formulations were measured for freeze ⁇ thaw samples.
- the formulations between thaw and freeze cycles were mixed by gentle inversions before the next freezing cycle.
- RNA/DNA-lipid particles were prepared by an aqueous-aqueous protocol (LNP2), as described herein.
- RNA in aqueous buffer conditions of HEPES 10 mM, EDTA 0.1 mM, pH 7.0 and DNA in water were mixed with pre-formed lipid nanoparticles composed of BNT51, DSPC, cholesterol, Ac-AEEA14-DMA and DSPE ⁇ pAEEA14 ⁇ alfa peptide in a molar ratio of 47.5:10:40.5:1.8:0.2, respectively, in aqueous solution of 5 mM acetic acid, 10% sucrose, in a volume ratio of 1:1.
- RNA/DNA-lipid particles were then functionalized with an aCD3-VHH ligand at ligand/cargo ratio of 0.48 w/w, and further diluted to a final nucleic acid concentration of 0.1 mg/mL and storage matrix 22 mM HEPES pH 5.5, 10% (w/v) sucrose.
- the RNA/DNA-lipid particles were prepared at N/P ratio of 12:1 and filtered through a 0.22 ⁇ m polyethersulfone (PES) filter.
- PES polyethersulfone
- the particle size and polydispersity index of the formulations were measured for freeze ⁇ thaw samples.
- the formulations between thaw and freeze cycles were mixed by gentle inversions before the next freezing cycle.
- Size and PDI was determined for functionalized sterile filtered RNA/DNA-LNP2 stored at final nucleic acid concentration of 0.1 mg/mL via DLS measurement. It was observed that aCD3 ⁇ VHH-functionalized LNP2 can be successfully manufactured with Ac- AEEA14-DMA stealth moiety and DSPE-pAEEA14-Alfa lipid, resulting in particles of size ⁇ 100 nm and a PDI ⁇ 0.3 ( Figure 32B). Successful cargo incorporation was verified via Agarose gel electrophoresis.
- Example 9 Manufacturing of RNA/DNA lipid nanoparticles (BNT52/Ac-AEEA14-DMA/ DSPE-pAEEA14-Alfa-Tag lipid) Manufacturing of pre-formed lipid nanoparticles (BNT52/Ac-AEEA14-DMA/DSPE- pAEEA14-Alfa-Tag lipid) by ethanol injection
- a lipid mix consisting of BNT52, DSPC, cholesterol, Ac-AEEA14-DMA and DSPE ⁇ pAEEA14 ⁇ alfa peptide in a molar ratio of 47.5:10:40.5:1.8:0.2 was dissolved in organic solvent (ethanol) at 20 mM total lipid concentration and mixed with an aqueous phase (5mM AcOH) in a 3:1 volume ratio (aqueous : organic), using a standard
- the organic solvent in the obtained raw colloid nanoparticles was removed by dialysis against 5mM AcOH using Slide-A-Lyzer dialysis cassettes of 10K molecular weight cut-off (MCWO) (Thermo Fisher Scientific, Waltham, MA, USA).
- MCWO molecular weight cut-off
- the nanoparticles were concentrated to 2X times through centrifugation at 3000rpm x 10 min at 4°C, using Amico Ultra-15 of 30K molecular weight cut-off (MCWO) (Merck, Darmstadt, Germany). After concentration, the nanoparticles were diluted to a sucrose concentration of 10%.
- Frozen stability of pre-formed lipid nanoparticles (BNT52/Ac-AEEA14-DMA/DSPE- pAEEA14-Alfa-Tag lipid)
- the frozen stability of the pre-formed lipid nanoparticles has been investigated by cycling the formulations from ⁇ 20°C and -80°C (overnight) to +25°C (2 h) for at least two times.
- the particle size and polydispersity index of the formulations were measured for freeze ⁇ thaw samples.
- the formulations between thaw and freeze cycles were mixed by gentle inversions before the next freezing cycle.
- RNA/DNA-lipid particles were prepared by an aqueous-aqueous protocol (LNP2), as described herein.
- RNA in aqueous buffer condition HPES 10 mM, EDTA 0.1 mM, pH 7.0
- DNA in water were mixed with pre-formed lipid nanoparticles composed of BNT52, DSPC, cholesterol, Ac-AEEA14-DMA and DSPE ⁇ pAEEA14 ⁇ alfa peptide in a molar ratio of 47.5:10:40.5:1.8:0.2, respectively, in aqueous solution of 5 mM acetic acid, 10% sucrose, in a volume ratio of 1:1.
- RNA/DNA-lipid particles were then functionalized with an aCD3-VHH ligand at ligand/cargo ratio of 0.48 w/w, and further diluted to a final nucleic acid concentration of 0.1 mg/mL and storage matrix 22 mM HEPES pH 5.5, 10% (w/v) sucrose.
- the RNA/DNA-lipid particles were prepared at N/P ratio of 12:1 and filtered through a 0.22 ⁇ m polyethersulfone (PES) filter.
- PES polyethersulfone
- the particle size and polydispersity index of the formulations were measured for freeze ⁇ thaw samples.
- the formulations between thaw and freeze cycles were mixed by gentle inversions before the next freezing cycle.
- Size and PDI were determined for functionalized sterile filtered RNA/DNA-LNP2 stored at final nucleic acid concentration of 0.1 mg/mL via DLS measurement. It was observed that aCD3 ⁇ VHH-functionalized LNP2 can be successfully manufactured with Ac- AEEA14-DMA stealth moiety and DSPE-pAEEA14-Alfa lipid, resulting in particles of size ⁇ 100 nm and a PDI ⁇ 0.3 ( Figure 33B). Successful cargo incorporation was verified via Agarose gel electrophoresis.
- Example 10 Use of thiolipids for target transfection of T-cells
- the present example assessed the suitability of LNPs containing thiolipids described herein for cell-specific targeting by introducing a targeting system specific for primary T- cells.
- the present example compared thiolipids BNT51 and BNT52 with the known ionizable lipid DODMA, formulated in functionalized LNPs.
- the composition of the LNPs, which were produced at an N/P ratio of 6, can be found in the following table.
- RNA and DNA delivery a cargo mixture containing both Thy1.1 RNA and Venus nanoplasmid DNA was formulated into an LNP with the above lipid mixture, using methods known to those of skill in the art.
- Tag-lipid DSPE-PEG2k-ALFA containing LNPs were incubated with an anti-CD3 specific VHH_NbAlfa construct, which binds to the AlfaTag (having a sequence SEQ.ID.NO.1) presented on the LNP surface.
- the resulting particles showed acceptable particle properties (by DLS: diameter ⁇ 200nm, PDI ⁇ 0.5) and negligible amounts of free RNA/DNA as evaluated by Agarose gel electrophoresis.
- the particles thus obtained were tested in a PBMC assay.
- PBMCs were analyzed for cell type-specific Thy1.1-RNA and Venus-Nanoplasmid DNA transfection by flow cytometry.
- the LNPs containing thiolipids described herein showed greater RNA and DNA transfection in T-cells in comparison to DODMA containing LNPs.
- Figures 33A-B are bar graphs illustrating use of thiolipids described herein for targeted transfection of T-cells. In vitro evaluation in hPBMCs. Comparison of three different LNPs containing three different ionizable lipids show the superior RNA and DNA transfection efficiency of thiolipids described herein which were formulated in LNP2 and LNP3.
- Example 11 Use of additional thiolipid compounds for targeted transfection of T-cells The present example assesses lipid BNT52 and lipid BNT76 formulated in functionalized LNPs. The composition of the LNPs, which were produced at an N/P ratio of 12, can be found in the following table.
- RNA and DNA delivery a cargo mixture containing both Thy1.1 RNA and Venus nanoplasmid DNA was formulated into an LNP with the above lipid mixture, using methods of the art.
- Tag-lipid DSPE-PEG2k-ALFA containing LNPs were incubated with an anti-CD3 specific VHH_NbAlfa construct, which binds to the AlfaTag presented on the LNP surface.
- the resulting particles showed acceptable particle properties (by DLS: diameter ⁇ 200nm, PDI ⁇ 0.5) and negligible amounts of free RNA/DNA as evaluated by Agarose gel electrophoresis. The particles thus obtained were tested in a PBMC assay.
- Figures 44A and 44B show cell type-specific RNA-transfection (Thy1.1-RNA expression) and DNA- transfection (Venus-Nanoplasmid DNA expression), analyzed via flow cytometry.
- the RNA and DNA transfection observed in B-cells and Monocytes is low for both LNPs.
- the LNPs containing the novel lipids show high RNA and DNA transfection in T-cells. Both tested novel lipids are potent candidates for targeted transfection of T-cells.
- Figures 35A and 35B illustrate the use of reported thiolipids for targeted transfection of T-cells. In vitro evaluation in hPBMCs.
- FIG. 44A Depicted in Figure 44A are the percentages of Thy1.1-expressing cell subtypes (CD4+ T cells, CD8+ T cells, CD19+ B cells, CD14+ Monocytes) out of all single and alive cells (y ⁇ axes). Depicted in Figure 44B are the percentages of Venus-Nanoplasmid expressing cells for CD4+ T cells and CD8 + T cells.
- Example 12 Manufacturability and in vitro investigation of LNP formulations consisting of novel ionizable lipid, BNT-72
- the present example assessed the manufacturability and in vitro biological effects of the LNP formulations consisting of provided lipids (with sulfone-branched tail and hydroxyl head), BNT-72.
- BNT-72 has been formulated against a benchmark (BM) formulation and LNPs have been characterized and tested in vitro.
- BM benchmark
- the particles have been prepared at three different N/P ratios.
- LNPs have been prepared by microfluidic mixing of the lipid mixture in ethanol and RNA solution in aqueous, acidic buffer. The details of the formulations have been given in the table below.
- the sizes of the LNPs was assessed by DLS (Figure 36A).
- the hydrodynamic diameter of the benchmark formulation was found to be 70 nm as expected.
- the size of BNT-72 formulation has changed with the N/P ratio.
- N/P 4 and 8 formulations with diameter of 148 nm and 191 nm has been obtained, respectively.
- the size of the BNT- 72 formulation has significantly decreased (114 nm) PDI values was found to be ⁇ 0.2 indicating a narrow size distribution.
- the zeta potential values of the formulations was assessed by electrophoretic light scattering (Figure 36B). Formulations have showed neutral zeta potential.
- the mRNA encapsulation efficiency of the formulations was assessed by Ribogreen assay ( Figure 37A).
- BNT-72 formulation has showed high accessibility at N/P of 4 and 8.
- mRNA integrity of the formulations was assessed by capillary electrophoresis, fragment analyzer ( Figure 37B). In general, no fragmentation has been observed for the formulations.
- the cytotoxicity and transfection efficiency of the formulations was assessed at three different mRNA concentrations (12.5, 25 and 50 ng) on three different cell lines (C2C12, HepG2, RAW 264) ( Figures 38A-C). In general, the formulations did not show significant cytotoxicity at the tested conditions. Formulations were effectively able to transfect the cell lines that have been tested ( Figures 39A-C). The transfection ability of the BNT-72 formulation has increased significantly with the increase in N/P ratio.
- Example 13 Comparative Analysis of Tail Groups
- BNT-76 thiolipid
- BNT-90 traditional lipid
- Figure 40 is a bar graph illustrating the size (nm) and polydispersity index (PDI) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM.
- Figure 41 is a bar graph illustrating Z-potential (mV) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM.
- Figure 42 is a bar graph illustrating RNA integrity (%) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM.
- Figure 43 is a bar graph illustrating osmolarity and pH of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM.
- Figures 44A-44D are a series of bar graphs illustrating transfection efficiency in C2C12 cells (Figure 44A), HepG2 cells (Figure 44B), RAW cells (Figure 44C) and Hek293 cells ( Figure 44D).
- Figures 45A-45D are a series of bar graphs illustrating cell viability in C2C12 cells (Figure 45A), HepG2 cells ( Figure 45B), RAW cells (Figure 45C) and Hek293 cells ( Figure 45D).
- Example 14 Synthesis of Example Thiolipids Synthesis of BL-199 and BL-200
- Method B A 50 mL round bottom flask was charged with sodium 6-hydroxyhexane-1-sulfonate bromide (6.7 g, 1 Eq, 22 mmol) and POCl3 (33 g, 20 mL, 10 Eq, 0.22 mol). The resulting white suspension was heated to 110 °C and stirred for 30h whereafter the heating was stopped, and the reaction was stirred over the weekend at rt. The reaction was stopped and concentrated under reduced pressure. The crude was dissolved in 15 mL DCM, filtered and stripped 3x with 20 mL DCM to give 6-chlorohexane-1-sulfonyl chloride (4.7 g, 21 mmol, 98 %) which was used without further purification.
- 6-chloro-N-decylhexane-1-sulfonamide (6) Method A: A solution of decan-1-amine (280 mg, .8 Eq, 1.78 mmol), triethylamine (451 mg, 621 ⁇ L, 2 Eq, 4.45 mmol) in DCM (5.0 mL) was cooled to 0°C, next 6-chlorohexane- 1-sulfonyl chloride (488 mg, 1 Eq, 2.23 mmol) in 3 ml DCM was slowly added. The mixture was stirred overnight at room temperature.
- 6-chloro-N-decyl-N-octylhexane-1-sulfonamide (7) A solution of N-octyldecan-1-amine (2.0 g, 1 Eq, 7.4 mmol), triethylamine (2.3 g, 3.1 mL, 3 Eq, 22 mmol) in DCM (70 mL) was cooled to 0°C, next 6-chlorohexane-1-sulfonyl chloride (2.2 g, 1.33 Eq, 9.9 mmol) in 8 ml DCM was slowly added and stirred overnight. The reaction was concentrated under reduced pressure to give the crude product.
- heptadecan-9-yl 8-((2-hydroxyethyl)amino)octanoate (391 mg, 1 Eq, 885 ⁇ mol) and 6-chloro-N-decyl-N-octylhexane-1-sulfonamide (400 mg, 1 Eq, 885 ⁇ mol) were mixed in CPME (4.0 mL) / MeCN (4.0 mL) and potassium iodide (294 mg, 2 Eq, 1.77 mmol) and potassium carbonate (489 mg, 4 Eq, 3.54 mmol) were added. The mixture was heated at 90 °C overnight.
- the reaction mixture was allowed to cool to rt, filtered through a pad of Celite and evaporated in vacuo.
- the crude material was dissolved in a minimal amount of heptane/EtOAc and purified on silica column to afford heptadecan-9-yl 8-((6-(N-decyl-N-octylsulfamoyl)hexyl)(2- hydroxyethyl)amino)octanoate (254 mg, 296 ⁇ mol, 33.5 %).
- thionyl chloride (77.8 g, 47.7 mL, 10 Eq, 654 mmol) was added dropwise, and the mixture was stirred at reflux for 3 days. The mixture was evaporated to semi-dryness, coevaporated twice with toluene. Water was added and the product was extracted with EtOAc (2x), washed with brine, dried and evaporated to dryness to afford ethyl 7- (chlorosulfonyl)heptanoate (24.2 g, 66 mmol).
- reaction mixture was evaporated in vacuo, the residue was coated on hydromatrix and purified using silica column chromatography using 5%-40%-100% EtOAc in Heptane to afford a mixture of 7-chloro-N,N- dioctylheptane-1-sulfonamide en 7-bromo-N,N-dioctylheptane-1-sulfonamide (300 mg, 6.2-6.8 mmol) which was used without further purification.
- 9-chlorononane-1-sulfonyl chloride (21) 9-hydroxynonane-1-sulfonate.NaBr salt (6.18 g, 1 Eq, 17.7 mmol) was suspended in POCl3 (30 g, 18 mL, 11 Eq, 0.19 mol) and the reaction was stirred at (initially) 110°C. After 70h, the reaction was concentrated and co-evaporated with toluene and DCM to yield the crude 9-chlorononane-1-sulfonyl chloride. Cl:Br ratio 1:0.35. Product was used without further purification.
- N-butyl-9-chloro-N-hexylnonane-1-sulfonamide (22)
- N-butylhexan-1-amine (2.09 g, 0.75 Eq, 13.3 mmol)
- triethylamine (5.37 g, 7.38 mL, 3 Eq, 53.1 mmol)
- DCM 32 mL
- 9-chlorononane-1- sulfonyl chloride/bromide (4.62 g, 1 Eq, ⁇ 17.7 mmol) (crude mixture) in DCM (22 mL) was slowly added and the reaction was stirred at rt for 4 days.
- reaction mixture was concentrated in vacuo, re-suspended in DCM/MeOH, coated on hydromatrix and purified by FCC using 1-25% EtOAc in heptane to obtain a mixture of Cl/Br N-butyl-9-halogen- N-hexylnonane-1-sulfonamide (1.5 gram), ratio Cl/Br 1:1 according to NMR.
- N,N-dioctyl-7-oxoheptane-1-sulfonamide 400 mg, 2.5 Eq, 958 ⁇ mol
- 4-aminobutan-1-ol 34.1 mg, 1 Eq, 383 ⁇ mol
- sodium triacetoxyborohydride 203 mg, 2.5 Eq, 958 ⁇ mol
- tert-butyl (4-(bis(7-(N,N-dioctylsulfamoyl)heptyl)amino)butyl)carbamate (27) N,N-dioctyl-7-oxoheptane-1-sulfonamide (600 mg, 1Eq, 1.26 mmol) and tert-butyl (4- aminobutyl)carbamate (238 mg, 1 Eq, 1.26 mmol) were dissolved in DCM (5.06 mL), after 3 min sodium triacetoxyborohydride (670 mg, 2.5 Eq, 3.16 mmol) was added and the mixture was stirred overnight at rt.
- Method B A 50 mL round bottom flask was charged with sodium 6-hydroxyhexane-1-sulfonate bromide (6.7 g, 1 Eq, 22 mmol) and POCl3 (33 g, 20 mL, 10 Eq, 0.22 mol). The resulting white suspension was heated to 110 °C and stirred for 30 h whereafter the heating was stopped and the reaction was stirred over the weekend at rt. The reaction was stopped and concentrated under reduced pressure. The crude was dissolved in 15 mL DCM, filtered and stripped 3x with 20 mL DCM to give 6-chlorohexane-1-sulfonyl chloride (4.7 g, 21 mmol, 98 %) which was used without further purification.
- 6-chloro-N-hexyl-N-octylhexane-1-sulfonamide To a solution of N-hexyloctan-1-amine (1.5 g, 1.0 Eq, 6.8 mmol), triethylamine (1.4 g, 1.9 mL, 2 Eq, 14 mmol) in DCM (34 mL) was added 6-chlorohexane-1-sulfonyl chloride (1.5 g, 1 Eq, 6.8 mmol). The resulting mixture was stirred overnight. The reaction was diluted with 25 mL DCM, washed with water (3x 25 ml), brine, dried over Na2SO4, filtered and concentrated in vacuo to afford the crude product.
- tert-butyl (4-(bis(6-(N-hexyl-N-octylsulfamoyl)hexyl)amino)butyl)carbamate To a solution of 6-chloro-N-hexyl-N-octylsulfamoyl)hexyl)amino)butyl)carbamate to a solution of 6-chloro-N-hexyl-N-octylsulfamoyl)hexyl)amino)butyl)carbamate (100 mg, 102 ⁇ L, 1 Eq, 531 ⁇ mol) in a mixture CPME (2.66 mL) MeCN (2.66 mL) was added KI (353 mg, 4 Eq, 2.12 mmol), K2CO3 (367 mg, 5 Eq, 2.66 mmol) and this was heated to 90 °C and stirred for 48 hours.
- Method B A 50 mL round bottom flask was charged with sodium 6-hydroxyhexane-1-sulfonate bromide (6.7 g, 1 Eq, 22 mmol) and POCl3 (33 g, 20 mL, 10 Eq, 0.22 mol). The resulting white suspension was heated to 110 °C and stirred for 30 h whereafter the heating was stopped, and the reaction was stirred over the weekend at rt. The reaction was stopped and concentrated under reduced pressure. The crude was dissolved in 15 mL DCM, filtered and stripped 3x with 20 mL DCM to give 6-chlorohexane-1-sulfonyl chloride (4.7 g, 21 mmol, 98 %) which was used without further purification.
- Example 15 Preparation of Functionalized LNPs by a fluid-path system Ethanolic solutions containing lipids in the following ratio were prepared: cationically ionizable lipid (46.84 mol-%) (including heptadecan-9-yl 8- ⁇ (2-hydroxyethyl)[6-oxo-6- (undecyloxy)hexyl]amino ⁇ -octanoate) (SM-102) used as a control lipid), cholesterol (36.33 mol-%), DSPC (14.33 mol-%), C16-Ceramide-PEG2K (1.8 mol-%) and the targeting compound DSPE-PEG2K-ALFA tag (SEQ.ID.NO.
- cationically ionizable lipid 46.84 mol-%) (including heptadecan-9-yl 8- ⁇ (2-hydroxyethyl)[6-oxo-6- (undecyloxy)hexyl]amino ⁇ -
- syringe a mixture of one or several nucleic acids (total nucleic acid concentration 0.15 g/L) and an acidifier (50mM) in water was loaded (typically, this comprised at least one reporter DNA and one reporter RNA). Both syringes were loaded into a microfluidic device (Precision Nanosystems Nanoassemblr Ignite®) for mixing.
- the product fraction was loaded into a dialysis cassette (Slide-A-LyzerTM, 10,000 MWCO) and dialyzed against the formulation buffer. To balance volume changes during dialysis, the product is up-concentrated by centrifugal filtration; cryoprotectant (trehalose, to a final concentration of 10% (w/w)) and formulation buffer (HEPES 20Mm; pH 6) are added to achieve the desired final concentration.
- cryoprotectant trehalose, to a final concentration of 10% (w/w)
- formulation buffer HEPES 20Mm; pH 6
- the obtained ALFA-tagged LNPs (‘parent LNP’) are subsequently mixed with the desired amount of a docking compound (bispecific construct comprising an anti-ALFA Tag VHH and aCD3 VHH) construct and formulation buffer and incubated for 1 h at room temperature.
- Figure 46 depicts the size and PDI of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C. The results of three measurements, as well as their average and error bars indicating 1x standard deviation are shown.
- Figure 47 depicts the percentages of transfected cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C.
- Figure 48 depicts the cell counts of alive cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs treated with aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, which were either applied freshly prepared or after one freeze/thaw cycle at -80°C.
- Example 16 Effect of Oxidation State of the Sulfur Tail on Transfection Efficiency Synthesis of BNT-71 2-Butyloctyl methanesulfonate
- 2-butyloctyl methanesulfonate A solution of 2-butyloc tan-1-ol (10.0 g, 53.6 mmol, 1.00 equiv.), and triethylamine (16.2 g, 160 mmol, 3.00 equiv.) in 100 ml DCM was stirred under argon at 0°C.
- Methanesulfonic anhydride (14.0 g, 80.4 mmol, 1.5 equiv.) was added in portions, the reaction mixture was warmed and stirred for 2 h at rt.
- the progress of the reaction was monitored with TLC by using solvent mixture (n-hexane/EtOAc; 4:1).
- the reaction mixture was neutralized with (200 ml) NaHCO3, the organic layer was separated, and the aq. Layer was further extracted with (2 X 100 ml) DCM.
- the combined organic phase was washed with (2 X 100 ml) brine, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was used in the next step without any further purification.
- reaction mixture was neutralized with 2 M HCl at 0°C, the mixture was diluted with (100 ml) diethyl ether, washed with (2 X 100 ml) H 2 O, and (2 X 100 ml) brine. The organic layer was collected, dried over Na 2 SO 4 , and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO 2 ; n-hexane) to afford a colorless oil compound.
- reaction mixture was quenched with 100 ml 5% citric acid, exactrated with (3 X 100 ml) DCM, and (2 X 100 ml) brine.
- the organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure.
- the product was purified with column chromatography (SiO2; n-hexane/EtOAc; 9:1) to afford a colorless oil compound.
- the product was purified with column chromatography (SiO 2 ; from n-hexane/EtOAc/TEA; 4/1/1% to n-hexane/EtOAc/TEA; 1/1/1% for BNT-23, -71 & BL-213) to afford the title compound as a colorless oil.
- reaction mixture was neutralized with (10 ml) NaHCO3 at 0°C, the mixture was diluted (100 ml) EtOAc, washed with (2 X 50 ml) H2O, then with (2 X 50 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure.
- the product was purified with column chromatography (SiO 2 ; n-hexane/EtOAc; 1/1).
- the product was purified with column chromatography (SiO2; from n-hexane/EtOAc/TEA; 1/1/1%to EtOAc/TEA; 99%/1% for BNT-102, -106 & - 114).
- Example 17 LNP Formulation Production Lipid nanoparticles of the present example were prepared by mixing an aqueous phase (0.15 mg/mL mRNA diluted in 0.1 M citrate buffer, pH 4.0) and an organic phase (a lipid mixture of ionizable cationic lipid:DSPC:cholesterol:stealth lipid, with the components dissolved in ethanol at a molar fraction of 47.5:40.5: 10: 2 %, respectively, having an N/P of 6 and a total concentration of 17.05 mM) at a 3:1 volume ratio and at a rate of 12 mL/min, using a microfluidic instrument (NanoAssemblr® Benchtop, Precision NanoSystems, Vancouver, Canada).
- the mixture was dialyzed against 1x DPBS (GIBCO, pH 7.4) for 3 h in a Slide-A-Lyser 10K MWCO dialysis cassette (Thermo Fisher Scientific, Waltham, MA, USA.) Residual ethanol was regularly controlled by osmolality measurements.
- the physicochemical characterization size, polydispersity, zeta potential, RNA accessibility and total RNA concentration was performed on the day of preparation. After complete characterization, formulations were stored at 4°C for not more than 1 day. Lipid nanoparticles were diluted in PBS to the desired RNA concentration prior to in vitro testing.
- Particle Size Measurement Particle size analysis was performed by dynamic light scattering using a DynaPro Plate Reader II (Wyatt, Dernbach, Germany). LNP formulations were diluted to 0.005 mg/mL in 1X PBS and the l20 ⁇ L of diluted sample were measured in triplicate in a 96- well plate. From the measurements, size (Zaverage), and polydispersity indices (PdI) were calculated from the cumulant analysis using Dynamics 7.8.1.3 software. Potentiometric Measurement of pH The pH value of the sample solution was tested potentiometrically with a calibrated pH meter (Ph. Eur. 2.2.3, seven compact pH-meters equipped with a micro-electrode; Mettler-Toledo).
- pH-meter was calibrated in the expected pH-range prior measurement. 500 ⁇ L liposome sample were dispensed into a 1.5 mL Eppendorf tube. The pH-meter micro-electrode was then immersed into the liposome sample to initiate the pH measurement. Agarose Gel Electrophoresis Agarose gel electrophoresis was performed to evaluate free RNA.
- the gel was poured by using 1 g agarose dissolved in 100 mL of lx TAE Buffer pH 7.4 (Tris-acetate-EDTA) (Rotiphorese® 50X TAE, Carl Roth, Düsseldorf, Germany), 1 mL of 5% Sodium hypochlorite, and 10 pL of GelRed Nucleic Acid Gel Stain (Biotium, Hayward, CA, USA).
- the gel could set for at least 25 min at room temperature.
- the gel was then placed in a gel electrophoresis tank and lx TAE running buffer (pH 7.4) was used. Before loading, the samples were incubated at 40 °C with or without 2% of Triton X- 100, for total and free RNA, respectively.
- the gel was run at 80 V for 40 minutes. Gel images were taken on a Chemidoc XRS imaging system (Bio-Rad, Berkeley, CA, USA). Results The present example evaluates the effect of the different oxidation states of S- tails on the transfection efficiency of the ionizable lipids for engineering mRNA nanoparticles using the LNP technology comprising BNT-71 or BNT-72 or BNT-74 as an ionizable lipid. Therefore, LNPs were prepared by mixing the RNA aqueous phase and the lipid organic phase using a microfluidic instrument. These ionizable lipids, the helper lipid DSPC or DOPE, cholesterol and Peg-DMG.
- Figure 50 presents the RNA accessibility of the formulations. All LNPs including BMs have encapsulation efficiency of more than 80% as shown in Figure 51. Agarose gel electrophoresis shows no free RNA for all LNPs as shown in Figure 52. No cytotoxicity was observed for all formulations using 12.5, 25, 50 ng in all different cell lines, for instance, C2C12, HepG2, Raw cells and Hek cells as shown in Figures 53A-D.
- Example 18 Evaluation of E, Z Configuration on Head Group for Lipids Having Tail Comprising SO 2 Moiety
- the present example evaluates the effect of the E and Z configurations of the unsaturated head groups on the transfection efficiency of the ionizable lipids for engineering mRNA nanoparticles using the LNP technology comprising BNT-72 or BNT- 76 or BL-195 as an ionizable lipid.
- the LNPs of the present example were characterized by the same methods provided in Example 17.
- LNPs were prepared by mixing the RNA aqueous phase and the lipid organic phase using a microfluidic instrument.
- the physicochemical properties and biological performance of the ionizable cationic lipids in LNPs were compared with provided benchmarks BM_1 and BM_2. A summary of all the formulations used is provided in the table below.
- the size and distribution (PDI) of LNPs comprising provided lipids is shown in Figure 55.
- the size and PDI of BM formulations are also included.
- Figure 56 shows the Z-potential of various novel lipids containing LNPs measured in 0.1XPBS at pH 7.
- the results of BM formulations are also included. As can be seen, neutral surface charge was obtained for all LNPs including BMs as shown in Figure 56.
- the accessibility of RNA and the amount of free RNA in the LNP formulations was measured by Ribogreen Assay and agarose gel electrophoresis methods, respectively.
- Figure 57 presents the RNA accessibility of the formulations.
- Figure 58 is an agarose gel electrophoresis image showing no free RNA for all provided LNPs. No cytotoxicity was observed for all formulations using 12.5, 25, 50 ng in all different cell lines, for instance, C2C12, HepG2, Raw cells and Hek cells as shown in Figures 59A-D.
- the performance of LNPs comprising BL-195 (Z-configuration) and BNT-76 (E- configuration) is provided in different cell lines, as shown in Figures 60A-D.
- Previous examples showed that sulfone tails exhibited improved properties when formulated with DOPE rather than with DSPC. The present example was repeated to determine whether a Z configuration for a head group is better than E configuration when combined with a sulfone tail.
- LNPs were prepared by mixing the RNA aqueous phase and the lipid organic phase using a microfluidic instrument.
- Two LNPs were formulated with acetate buffer to determine the effect of acetate buffer in the size of particle formation.
- the physicochemical properties and biological performance of these different 3 ionizable lipids containing formulations were compared with benchmarks BM_1 and BM_2. A summary of all the formulations used in this study is given in the table below.
- the size and distribution (PDI) of LNPs comprising provided lipid compounds described herein (based on saturated and unsaturated head groups with both configurations with same sulfone tail, in addition to different of mRNA buffers) are shown in Figure 61.
- the size and PDI of BM formulations are also included.
- Figure 62 shows the Z-potential of various novel lipids containing LNPs measured in 0.1XPBS at pH 7.
- the results of benchmark (BM) formulations are also included.
- the accessibility of RNA and the amount of free RNA in the LNP formulations was measured by Ribogreen Assay and Agarose gel electrophoresis methods, respectively.
- Figure 63 presents the RNA accessibility of the formulations.
- Figure 64 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs. No cytotoxicity was observed for all formulations using 12.5, 25, 50 ng in all different cell lines, for instance, C2C12, HepG2, Raw cells and Hek cells as shown in Figures 65A-D. The performance of BL-195 (Z-configuration) and BNT-76 (E-configuration) in different cell lines and in both buffers were assessed in vitro as shown in Figures 66A-D. The particle size of different sulfone containing tails formulated with acetate buffer and citrate buffer was assessed. In the present example, LNPs were prepared by mixing the RNA aqueous phase and the lipid organic phase using a microfluidic instrument.
- the ionizable lipids, the helper lipid DSPC or DOPE, cholesterol and PEG-DMG were prepared at respective molar fractions 47.5:40.5: 10:2 and N/P ratio of 6.
- the size and distribution (PDI) of provided ionizable lipids in LNPs is shown in Figure 67.
- the size and PDI of BM formulations are also included.
- Figure 68 shows the Z-potential of various novel lipids containing LNPs measured in 0.1XPBS at pH 7.
- the results of BM formulations are also included.
- Figure 69 presents the RNA accessibility of the formulations.
- Figure 70 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs. No cytotoxicity was observed for all formulations using 12.5, 25, 50 ng in all different cell lines, for instance, C2C12, HepG2, Raw cells and Hek cells as shown in Figures 71A-D.
- the transfection performance of BL-195 (Z-configuration) and BNT-76 (E-configuration) in different cell lines and in both buffers provided cells in vitro is shown in Figures 72A- D.
- Example 19 Evaluation of E, Z Configuration on Head Group for Lipids Having Tail Comprising S or NSO 2 Procedure for Preparing BL-207, -214 & -215
- a solution of N,N-dioctyl-7-oxoheptane-1-sulfonamide (2.00 equiv.), NH2-R (1.00 equiv.) in 5 ml DCM was stirred under argon for 30 min. at rt., then NaBH(OAc)3 (4.00 equiv.) was added to the reaction mixture under argon and the reaction mixture was stirred for extra 12 h at rt.
- the reaction was diluted with (50 ml) DCM, washed with (3 X 50 ml) NaHCO 3 , (2 X 50 ml) brine.
- the organic layer was collected, dried over Na 2 SO 4 , and the solvent was evaporated under reduced pressure.
- the product was purified with column chromatography (SiO2; from n-hexane/EtOAc/TEA;1/1/1% to EtOAc/TEA; 99%/1% for BL-207, -214 & BL-215).
- BL-207, BL-214, and BL-215 can be prepared according to the following scheme:
- the present example evaluates the effect of the E and Z configurations of the unsaturated head groups on the transfection efficiency of other sulfur containing tails in provided ionizable lipids for use in preparing LNPs.
- the present example evaluated LNPs comprising BNT-23, BL-207 (saturated head), BL-213, BL-214 or BL-215 as the ionizable cationic lipid.
- the present LNPs were evaluated using the same methods provided in Example 17.
- LNPs were prepared by mixing the RNA aqueous phase and the lipid organic phase using a microfluidic instrument.
- LNPs had an N/P ratio of 6. Furthermore, the physicochemical properties and biological performance of these different 5 ionizable lipids containing formulations were compared with benchmarks BM_1 and BM_2. A summary of the formulations used in this study are provided in the table below.
- the size and distribution (PDI) of provided lipids contained in LNPs (based on saturated and unsaturated head groups with both configurations with same thioether and sulfonamide tails with both helper lipids) are shown in Figure 73.
- Figure 74 shows the Z-potential of various novel lipids containing LNPs measured in 0.1XPBS at pH 7. The results of BM formulations are also included. The accessibility of RNA and the amount of free RNA in the LNP formulations was measured by Ribogreen Assay and agarose gel electrophoresis methods, respectively.
- Figure 75 presents the RNA accessibility of the formulations.
- Figure 76 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs. No cytotoxicity was observed for all formulations using 12.5, 25, 50 ng in both cell lines HepG2 and Hek cells as shown in Figures 77A-B.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Dermatology (AREA)
- Optics & Photonics (AREA)
- Communicable Diseases (AREA)
- Otolaryngology (AREA)
- Oncology (AREA)
- Nanotechnology (AREA)
- Physics & Mathematics (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The present disclosure provides a compound of formula (I): (I), or a pharmaceutically acceptable salt thereof, that is useful for forming particles (e.g., lipid nanoparticles) for delivery of nucleic acids. The present disclosure further provides particle compositions comprising the compound of formula I, as well as uses thereof.
Description
Ionizable Thiolipids and Uses Thereof DESCRIPTION Background Particles for delivery of nucleic acids have been the subject of much recent work. Certain particles, such as lipid nanoparticles (LNPs) are particularly useful for the transport of therapies such as nucleic acid therapies to cells. See Tenchov, et al., ACS Nano, 2021, 15, 1116982-17015. LNPs comprise, among other things, cationic or ionizable lipids that, through electrostatic interaction, form stable complexes that encapsulate nucleic acids and thereby facilitate delivery into the cell. Summary There remains a need for cationic or ionizable lipids that are capable of forming complexes with nucleic acids, but have improved manufacturability and improved properties (e.g., improved transfection of nucleic acids and improved stability) relative to previous lipid formulations. The present disclosure provides cationic or ionizable lipid compounds comprising one or more sulfur-based moieties that avoid the problems associated with lipid compounds, while exhibiting improved properties (e.g., improved transfection) and ease of manufacture. In some embodiments, the present disclosure provides a compound represented by formula I: or a pharmaceutically 1 2
 L and L are each independently an optionally substituted C1-C30 aliphatic group; L3 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S; X1 and X2 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O)2N(R1)- , -N(R1)S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R1)2-, -OC(S)C(R1)2-, -C(R1)2C(S)O-, and –S-, wherein one or both of X1 or X2 is selected from -S(O)2N(R1)-, -N(R1)S(O)2, -S(O)-, - S(O)2-, -S(O)2C(R1)2-, -OC(S)C(R1)2-, -C(R1)2C(S)O-, and –S-; each R1 is, independently, at each instance, optionally substituted C1-C20 aliphatic or H; T1 and T2 are each independently an optionally substituted C3-C30 aliphatic;
G is -N(R2)C(S)N(R2)2, -OH, -N(R2)2, -N+(R3)3, -N(R5)C(O)R3, -N(R5)S(O)2R3, - N(R5)C(O)N(R3)2, -CH(N-R2), -R4, or -S(O)2R3; each R2 is, independently, at each instance, selected from the group consisting of H, optionally substituted C1-C6 aliphatic and OR3; or two instances of R2 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring or an optionally substituted 4- to 12-membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N, O, and S; each R3 is, independently, at each instance, selected from the group consisting of H and optionally substituted C1-C10 aliphatic; and R4 is optionally substituted 4- to 12-membered heterocycle comprising 1 to 4 heteroatoms selected from N, O, and S, optionally substituted 4- to 12 membered heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S, C6-C12 aryl substituted with one or more of –(CH2)0-6–OH or –(CH2)0-6–N(R5)2, or C3-C12 cycloaliphatic substituted with one or more of oxo, –(CH2)0-6–OH, or –(CH2)0-6–N(R5)2; each R5 is independently selected from H and optionally substituted C1-C6 aliphatic. In some embodiments, the present disclosure provides a particle comprising a compound described herein and a nucleic acid. In some embodiments, the present disclosure provides a composition (e.g., a pharmaceutical composition) comprising particles described herein. In some embodiments, the present disclosure provides a method of treating a disease, disorder, or condition in a subject comprising administering to the subject a composition comprising particles described herein. In some embodiments, the present disclosure provides a method of preparing a compound represented Formula IV: or a pharmaceutically accept
L and L are each independently an optionally substituted C1-C30 aliphatic group; L3 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S; X1 and X2 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O)2N(R1)- , -N(R1)S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R1)2-, -OC(S)C(R1)2-, -C(R1)2C(S)O-, and –S-, wherein one or both of X1 or X2 is selected from -S(O)2N(R1)-, -N(R1)S(O)2, -S(O)-, - S(O)2-, -S(O)2C(R1)2-, -OC(S)C(R1)2-, -C(R1)2C(S)O-, and –S-; each R1 is, independently, at each instance, optionally substituted C1-C20 aliphatic or H; T1 and T2 are each independently an optionally substituted C3-C30 aliphatic;
G is -N(R2)C(S)N(R2)2, -OH, -N(R2)2, -N+(R3)3, -N(R5)C(O)R3, -N(R5)S(O)2R3, - N(R5)C(O)N(R3)2, -CH(N-R2), -R4, or -S(O)2R3; each R2 is, independently, at each instance, selected from the group consisting of H, optionally substituted C1-C6 aliphatic and OR3; or two instances of R2 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring or an optionally substituted 4- to 12-membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N, O, and S; each R3 is, independently, at each instance, selected from the group consisting of H and optionally substituted C1-C10 aliphatic; and R4 is optionally substituted 4- to 12-membered heterocycle comprising 1 to 4 heteroatoms selected from N, O, and S, optionally substituted 4- to 12 membered heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S, C6-C12 aryl substituted with one or more of –(CH2)0-6–OH or –(CH2)0-6–N(R5)2, or C3-C12 cycloaliphatic substituted with one or more of oxo, –(CH2)0-6–OH, or –(CH2)0-6–N(R5)2; each R5 is independently selected from H and optionally substituted C1-C6 aliphatic. In some embodiments, the present disclosure provides a particle comprising a compound described herein and a nucleic acid. In some embodiments, the present disclosure provides a composition (e.g., a pharmaceutical composition) comprising particles described herein. In some embodiments, the present disclosure provides a method of treating a disease, disorder, or condition in a subject comprising administering to the subject a composition comprising particles described herein. In some embodiments, the present disclosure provides a method of preparing a compound represented Formula IV: or a pharmaceutically accept
 able salt thereof, the method comprising: contacting a compound represented by Formula V with a compound represented b
able salt thereof, the method comprising: contacting a compound represented by Formula V with a compound represented b
 y one of Formulae VIa-VIc
y one of Formulae VIa-VIc
 and a compound represented by one of Formulae VIIa-c
and a compound represented by one of Formulae VIIa-c
 in the presence of a reducing agent, wherein: each of L4 and L5 are each independently an optionally substituted C1-C30 aliphatic group; L6 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S; X3 and X4 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O)2N(R40)- , -N(R40)S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R40)2-, -OC(S)C(R40)2-, -C(R40)2C(S)O-, or –S-, wherein one or both of X3 or X4 is selected from -S(O)2N(R40)-, -N(R40)S(O)2, -S(O)-, - S(O)2-, -S(O)2C(R40)2-, -OC(S)C(R40)2-, -C(R40)2C(S)O-, or –S-; each R40 is, independently, at each instance, optionally substituted C1-C20 aliphatic or H; T3 and T4 are each independently an optionally substituted C3-C20 aliphatic; G1 is -N(R6)C(S)N(R6)2, -OH, -N(R6)2, -N(R9)C(O)R7, -N(R9)S(O)2R7, -N(R9)C(O)N(R7)2, -CH(N-R7),–R8, or -S(O)2R6; each G2 is independently O or N2; each G3 is independently halogen (e.g., Cl, Br, or I), -OTs, or OTf; each R6 is, independently, at each instance, selected from the group consisting of H, optionally substituted C1-C6 aliphatic and OR7; or two instances of R6 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring or an optionally substituted 4- to 12-membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N, O, and S; each R7 is, independently, at each instance, selected from the group consisting of H and optionally substituted C1-C6 aliphatic; R8 is optionally substituted 4- to 12-membered heterocycle comprising 1 to 4 heteroatoms selected from N, O, and S, optionally substituted 4- to 12 membered
heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S, C6-C12 aryl substituted with one or more of –(CH2)0-6–OH or –(CH2)0-6–N(R9)2, or C3-C12 cycloaliphatic substituted with one or more of oxo, –(CH2)0-6–OH, or –(CH2)0-6–N(R9)2; and each R9 is independently selected from H and optionally substituted C1-C6 aliphatic. Brief Description of the Drawing Figure 1 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- 51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59. Figure 2 is a bar graph illustrating zeta potential values of LNP formulations with BNT- 51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59. Figures 3A and 3B are a bar graph illustrating mRNA encapsulation efficiency (Figure 3A) and agarose gel electropherogram (Figure 3B) of LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59. Figure 4 is a bar graph illustrating mRNA integrity (%) of the LNP formulations with BNT- 51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59. Figures 5A-5C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 on cell viability in C2C12 cells (Figure 5A), HepG2 cells (Figure 5B) and RAW cells (Figure 5C). Figures 6A-6C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 on transfection efficiency in C2C12 cells (Figure 6A), HepG2 cells (Figure 6B) and RAW cells (Figure 6C). Figure 7 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04. Figure 8 is a bar graph illustrating Zeta potential values of LNP formulations with BNT- sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04. Figures 9A and 9B are a bar graph illustrating mRNA encapsulation efficiency (Figure 9A) and agarose gel electropherogram (Figure 9B) of LNP formulations with BNT-sulfo- 01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04. Figure 10 is a bar graph mRNA integrity (%) of the LNP formulations with BNT-sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04. Figures 11A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04 on cell viability in C2C12 cells (Figure 11A), HepG2 cells (Figure 11B), RAW cells (Figure 11C) and Hek293 cells (Figure 11D).
Figures 12A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 on transfection efficiency in C2C12 cells (Figure 12A), HepG2 cells (Figure 12B), RAW cells (Figure 12C) and Hek293 cells (Figure 12D). Figures 13A and 13B are a bar graph illustrating the in vitro hemolytic effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 (Figure 13A) and a bar graph illustrating the effect of LNP formulations on complement activation (Figure 13B). Figure 14 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- 51 and a helper lipid (DSPC or DOPE). Figure 15 is a bar graph illustrating zeta potential values of LNP formulations with BNT- 51 and a helper lipid (DSPC or DOPE). Figures 16A and 16B are a bar graph illustrating mRNA encapsulation efficiency (Figure 16A) and agarose gel electropherogram (Figure 16B) of LNP formulations with BNT-51 and a helper lipid. Figure 17 is a bar graph illustrating mRNA integrity (%) of the LNP formulations with BNT-51 and a helper lipid (DSPC or DOPE). Figures 18A-D are a bar graph illustrating the effect of the LNP formulations with BNT- 51 and a helper lipid on cell viability in C2C12 cells (Figure 18A), HepG2 cells (Figure 18B), RAW cells (Figure 18C) and Hek293 cells (Figure 18D). Figures 19A-D are a bar graph illustrating the effect of the LNP formulations with BNT- 51 and a helper lipid on transfection efficiency in C2C12 cells (Figure 19A), HepG2 cells (Figure 19B), RAW cells (Figure 19C) and Hek293 cells (Figure 19D). Figure 20 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- 51 and a stealth lipid. Figure 21 is a bar graph illustrating zeta potential values of LNP formulations with BNT- 51 and a stealth lipid. Figures 22A-B are a set of bar graphs illustrating mRNA encapsulation efficiency (Figure 22A) and agarose gel electropherogram (Figure 22B) of LNP formulations with BNT-51 and a stealth lipid. Figure 23 is a bar graph illustrating mRNA integrity (%) of the LNP formulations with BNT-51 and a stealth lipid. Figures 24A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51 and a stealth lipid on cell viability C2C12 cells (Figure 24A), HepG2 cells (Figure 24B), RAW cells (Figure 24C) and Hek293 cells (Figure 24D).
Figures 25A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51 and a stealth lipid on transfection efficiency in C2C12 cells (Figure 25A), HepG2 cells (Figure 25B), RAW cells (Figure 25C) and Hek293 cells (Figure 25D). Figure 26 is a bar graph illustrating particle size and PDI of pre-formed LNPs with BNT51 and Ac-AEEA14-DMA subjected to at least three freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature. Figures 27A-C are a series of bar graphs illustrating characterization of BNT51 functionalized lipid nanoparticles prepared with Ac-AEEA14-DMA as stealth moiety and DSPE-PEG2k-Alfa lipid (Figure 27A); particle size and PDI of functionalized LNP1 and LNP2; (Figure 27B); Agarose Gel Electrophoresis of controls, untreated functionalized LNP1 and LNP2 (upper row) and functionalized LNP1 and LNP2 treated with release solution (lower row) (Figure 27C); and particle size and PDI of functionalized LNP1 and LNP2 subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature, and after 2 weeks at 2-8°C and 25°C. Figures 28A-B are a series of bar graphs illustrating percentages of transfected cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs for both RNA (Thy1.1) (Figure 28A) and DNA (Venus) (Figure 28B), delivered using LNP1 or LNP2. Figure 29 is a bar graph illustrating particle size and PDI of preformed LNPs with BNT51 and Ac-AEEA14-VitE subjected to at least three freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature. Figures 30A-C are a series of bar graphs illustrating characterization of BNT51 functionalized lipid nanoparticles prepared with Ac-AEEA14-VitE as stealth moiety and DSPE-PEG2k-Alfa lipid (Figure 30A); particle size and PDI of functionalized LNP1 and LNP2 (Figure 30B); Agarose Gel Electrophoresis of controls, untreated functionalized LNP1 and LNP2 (upper row) and functionalized LNP1 and LNP2 treated with release solution (lower row) (Figure 30C); and particle size and PDI of functionalized LNP1 and LNP2, subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature, and after 2 weeks at 2-8°C and 25°C. Figures 31A-B are a series of bar graphs illustrating percentages of transfected cell (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs for both RNA (Thy1.1) (Figure 31A) and DNA (Venus) (Figure 31B), delivered using LNP1 or LNP2. Figures 32A-B are a set of bar graphs illustrating characterization of BNT51 functionalized lipid nanoparticles prepared with Ac-AEEA14-DMA as stealth moiety and
DSPE-pAEEA14-Alfa lipid, including (Figure 32A): particle size and PDI of preformed LNPs subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature; and (Figure 32B): particle size and PDI of functionalized RNA/DNA-LNP2 at t0 and subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature. Figures 33A-B are a set of bar graphs illustrating characterization of BNT52 functionalized lipid nanoparticles prepared with Ac-AEEA14-DMA as stealth moiety and DSPE-pAEEA14-Alfa lipid including (Figure 33A): particle size and PDI of preformed LNPs subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature; and (Figure 33B): Particle size and PDI of functionalized RNA/DNA-LNP2 at t0 and subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature. Figure 34 is a set of bar graphs illustrating use of thiolipids described herein for targeted transfection of T-cells. In vitro evaluation in hPBMCs. Comparison of three different LNPs containing three different ionizable lipids show the superior RNA and DNA transfection efficiency of thiolipids described herein which were formulated in LNP2 and LNP3. Depicted in the upper graph are the percentages of Thy1.1-expressing cell subtypes (CD4+ T cells, CD8+ T cells, CD19+ B cells) out of all single and alive cells (y‐axes). Depicted in the lower graph are the percentages of Venus-Nanoplasmid expressing cells for CD4+ T cells and CD8 + T cells. Figures 35A and 35B illustrate the use of reported thiolipids for targeted transfection of T-cells. In vitro evaluation in hPBMCs. Comparison of two different LNPs containing two different ionizable lipids show high RNA and DNA transfection efficiency of reported thiolipids which were formulated in LNP1 and LNP2. Depicted in Figure 35A are the percentages of Thy1.1-expressing cell subtypes (CD4+ T cells, CD8+ T cells, CD19+ B cells, CD14+ Monocytes) out of all single and alive cells (y‐axes). Depicted in Figure 35B are the percentages of Venus-Nanoplasmid expressing cells for CD4+ T cells and CD8 + T cells. Figures 36A and 36B are a set of bar charts illustrating characterization of the LNP formulations with BNT-72, including particle size and PDI (Figure 36A) and zeta potential (Figure 36B). Figures 37A and 37B are a set of bar graphs illustrating mRNA encapsulation efficiency (Figure 37A) and mRNA integrity (Figure 37B) of the of LNP formulations with BNT-72.
Figures 38A-C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-72 on cell viability in C2C12 cells (Figure 38A), HepG2 cells (Figure 38B) and RAW cells (Figure 38C). Figures 39A-C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-72 on transfection efficiency in C2C12 cells (Figure 39A), HepG2 cells (Figure 39B) and RAW cells (Figure 39C). Figure 40 is a bar graph illustrating the size (nm) and polydispersity index (PDI) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM. Figure 41 is a bar graph illustrating Z-potential (mV) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM. Figure 42 is a bar graph illustrating RNA integrity (%) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM. Figure 43 is a bar graph illustrating osmolarity and pH of complexes comprising BNT- 76, BNT-90 or a benchmark lipid CM12_BM. Figures 44A-44D are a series of bar graphs illustrating transfection efficiency in C2C12 cells (Figure 44A), HepG2 cells (Figure 44B), RAW cells (Figure 44C) and Hek293 cells (Figure 44D). Figures 45A-45D are a series of bar graphs illustrating cell viability in C2C12 cells (Figure 45A), HepG2 cells (Figure 45B), RAW cells (Figure 45C) and Hek293 cells (Figure 45D). Figure 46 depicts the size and PDI of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C. The results of three measurements, as well as their average and error bars indicating 1x standard deviation are shown. Figure 47 depicts the percentages of transfected cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C. Figure 48 depicts the cell counts of alive cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs treated with aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, which were either applied freshly prepared or after one freeze/thaw cycle at -80°C. Figure 49 is a graph providing size and distribution (PDI) of provided LNPs comprising lipids reported herein.
Figure 50 is a graph providing Z-potential of provided LNPs comprising lipids described herein. Figure 51 is a graph illustrating encapsulation efficiency of provided LNPs. Figure 52 is an agarose gel electrophoresis showing no free RNA for provided LNPs. Figures 53A-D are a series of graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage. Figures 54A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs. Figure 55 is a graph illustrating size and distribution (PDI) for provided LNPs comprising lipid compounds described herein. Figure 56 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein. Figure 57 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein. Figure 58 is an agarose gel electrophoresis image showing no free RNA for all provided LNPs. Figures 59A-D are a series of graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage. Figures 60A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs. Figure 61 is a graph illustrating size and distribution (PDI) of LNPs comprising lipid compounds described herein. Figure 62 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein. Figure 63 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein. Figure 64 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs. Figures 65A-D are a series graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage. Figures 66A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs. Figure 67 is a graph illustrating size and distribution (PDI) of LNPs comprising lipid compounds described herein.
Figure 68 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein. Figure 69 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein. Figure 70 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs. Figures 71A-D are a series graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage. Figures 72A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs. Figure 73 is a graph illustrating size and distribution (PDI) of LNPs comprising lipid compounds described herein. Figure 74 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein. Figure 75 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein. Figure 76 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs. Figures 77A-B are a series graphs illustrating cytotoxicity in provided cell lines HepG2 and Hek as measured by viability percentage. Figures 78A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs. Detailed Description of Certain Embodiments The present disclosure provides, among other things, the surprising discovery of particular cationic or ionizable lipids comprising at least one sulfur-containing moiety. Such cationic or ionizable lipids exhibit improved properties (e.g., improved transfection to cells) relative to previous lipids. For example, as illustrated in the Examples provided herein, thiolipids of the present disclosure exhibit improvements over previous lipids that lack a sulfur-containing moiety, including, for example, improved transfection of cells with RNA. Particles prepared using thiolipid compounds described herein exhibit narrow size distribution (i.e., are substantially uniform in size), high encapsulation efficiency, low cytotoxicity, and improved biodistribution and efficacy relative to particles comprising previous lipid compounds.
Compounds and Definitions Compounds of this disclosure include those described generally above and are further illustrated by the classes, subclasses, and species disclosed herein. As used herein, the following definitions shall apply unless otherwise indicated. For purposes of this disclosure, the chemical elements are identified in accordance with the Periodic Table of Elements, CAS version, Handbook of Chemistry and Physics, 75th Ed. Additionally, general principles of organic chemistry are described in “Organic Chemistry”, Thomas Sorrell, University Science Books, Sausalito: 1999, and “March’s Advanced Organic Chemistry”, 5th Ed., Ed.: Smith, M.B. and March, J., John Wiley & Sons, New York: 2001, the entire contents of which are hereby incorporated by reference. Unless otherwise stated, structures depicted herein are meant to include all stereoisomeric (e.g., enantiomeric or diastereomeric) forms of the structure, as well as all geometric or conformational isomeric forms of the structure. For example, the R and S configurations of each stereocenter are contemplated as part of the disclosure. Therefore, single stereochemical isomers, as well as enantiomeric, diastereomeric, and geometric (or conformational) mixtures of provided compounds are within the scope of the disclosure. For example, in some cases, Table 1 shows one or more stereoisomers of a compound, and unless otherwise indicated, represents each stereoisomer alone and/or as a mixture. Unless otherwise stated, all tautomeric forms of provided compounds are within the scope of the disclosure. Unless otherwise indicated, structures depicted herein are meant to include compounds that differ only in the presence of one or more isotopically enriched atoms. For example, compounds having the present structures including replacement of hydrogen by deuterium or tritium, or replacement of a carbon by 13C- or 14C-enriched carbon are within the scope of this disclosure. About or approximately: As used herein, the term "approximately" or "about," as applied to one or more values of interest, refers to a value that is similar to a stated reference value. In general, those skilled in the art, familiar within the context, will appreciate the relevant degree of variance encompassed by "about" or "approximately" in that context. For example, in some embodiments, the term "approximately" or "about" may encompass a range of values that are within (i.e., ±) 25%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or less of the referred value. Administering: As used herein, the term "administering" or "administration" typically refers to the administration of a composition to a subject to achieve delivery of an agent
that is, or is included in, a composition to a target site or a site to be treated. Those of ordinary skill in the art will be aware of a variety of routes that may, in appropriate circumstances, be utilized for administration to a subject, for example a human. For example, in some embodiments, administration may be ocular, oral, parenteral, topical, etc. In some particular embodiments, administration may be bronchial (e.g., by bronchial instillation), buccal, dermal (which may be or comprise, for example, one or more of topical to the dermis, intradermal, interdermal, transdermal, etc.), enteral, intra-arterial, intradermal, intragastric, intramedullary, intramuscular, intranasal, intraperitoneal, intrathecal, intravenous, intraventricular, within a specific organ (e.g., intrahepatic), mucosal, nasal, oral, rectal, subcutaneous, sublingual, topical, tracheal (e.g., by intratracheal instillation), vaginal, vitreal, etc. In some embodiments, administration may be parenteral. In some embodiments, administration may be oral. In some particular embodiments, administration may be intravenous. In some particular embodiments, administration may be subcutaneous. In some embodiments, administration may involve only a single dose. In some embodiments, administration may involve application of a fixed number of doses. In some embodiments, administration may involve dosing that is intermittent (e.g., a plurality of doses separated in time) and/or periodic (e.g., individual doses separated by a common period of time) dosing. In some embodiments, administration may involve continuous dosing (e.g., perfusion) for at least a selected period of time. In some embodiments, administration may comprise a prime-and-boost protocol. A prime-and-boost protocol can include administration of a first dose of a pharmaceutical composition (e.g., an immunogenic composition, e.g., a vaccine) followed by, after an interval of time, administration of a second or subsequent dose of a pharmaceutical composition (e.g., an immunogenic composition, e.g., a vaccine). In the case of an immunogenic composition, a prime-and-boost protocol can result in an increased immune response in a patient. Aliphatic: The term “aliphatic” refers to a straight-chain (i.e., unbranched) or branched, substituted or unsubstituted hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation, or a monocyclic hydrocarbon or bicyclic hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic (also referred to herein as “cycloaliphatic”), that has a single point or more than one points of attachment to the rest of the molecule. Unless otherwise specified, aliphatic groups contain 1-12 aliphatic carbon atoms. In some embodiments, aliphatic groups contain 1-6 aliphatic carbon atoms (e.g., C1-6). In some embodiments, aliphatic groups contain 1-5 aliphatic carbon atoms (e.g., C1-5). In
other embodiments, aliphatic groups contain 1-4 aliphatic carbon atoms (e.g., C1-4). In still other embodiments, aliphatic groups contain 1-3 aliphatic carbon atoms (e.g., C1-3), and in yet other embodiments, aliphatic groups contain 1-2 aliphatic carbon atoms (e.g., C1-2). Suitable aliphatic groups include, but are not limited to, linear or branched, substituted or unsubstituted alkyl, alkenyl, or alkynyl groups and hybrids thereof. A preferred aliphatic group is C1-6 alkyl. Alkyl: The term “alkyl”, used alone or as part of a larger moiety, refers to a saturated, optionally substituted straight or branched chain hydrocarbon group having (unless otherwise specified) 1-12, 1-10, 1-8, 1-6, 1-4, 1-3, or 1-2 carbon atoms (e.g., C1-12, C1-10, C1-8, C1-6, C1-4, C1-3, or C1-2). Exemplary alkyl groups include methyl, ethyl, propyl, butyl, pentyl, hexyl, and heptyl. Alkylene: The term “alkylene” is refers to a bivalent alkyl group. In some embodiments, “alkylene” is a bivalent straight or branched alkyl group. In some embodiments, an "alkylene chain" is a polymethylene group, i.e., -(CH2)n-, wherein n is a positive integer, e.g., from 1 to 6, from 1 to 4, from 1 to 3, from 1 to 2, or from 2 to 3. An optionally substituted alkylene chain is a polymethylene group in which one or more methylene hydrogen atoms is optionally replaced with a substituent. Suitable substituents include those described below for a substituted aliphatic group and also include those described in the specification herein. It will be appreciated that two substituents of the alkylene group may be taken together to form a ring system. In certain embodiments, two substituents can be taken together to form a 3- to 7-membered ring. The substituents can be on the same or different atoms. The suffix “-ene” or “-enyl” when appended to certain groups herein are intended to refer to a bifunctional moiety of said group. For example, “-ene” or “-enyl”, when appended to “cyclopropyl” becomes “cyclopropylene” or “cyclopropylenyl” and is intended to refer to a bifunctional cyclopropyl group, e.g., . Alkenyl: The term “alkenyl”, used alone or as part of a larger moiety, refers to an optionally substituted straight or branched chain or cyclic hydrocarbon group having at least one double bond and having (unless otherwise specified) 2-12, 2-10, 2-8, 2-6, 2-4, or 2-3 carbon atoms(e.g., C2-12, C2-10, C2-8, C2-6, C2-4, or C2-3). Exemplary alkenyl groups include ethenyl, propenyl, butenyl, pentenyl, hexenyl, and heptenyl. The term “cycloalkenyl” refers to an optionally substituted non-aromatic monocyclic or multicyclic ring system containing at least one carbon-carbon double bond and having about 3 to
about 10 carbon atoms. Exemplary monocyclic cycloalkenyl rings include cyclopentenyl, cyclohexenyl, and cycloheptenyl. Alkynyl: The term “alkynyl”, used alone or as part of a larger moiety, refers to an optionally substituted straight or branched chain hydrocarbon group having at least one triple bond and having (unless otherwise specified) 2-12, 2-10, 2-8, 2-6, 2-4, or 2-3 carbon atoms (e.g., C2-12, C2-10, C2-8, C2-6, C2-4, or C2-3). Exemplary alkynyl groups include ethynyl, propynyl, butynyl, pentynyl, hexynyl, and heptynyl. Aryl: The term “aryl” refers to monocyclic and bicyclic ring systems having a total of six to fourteen ring members (e.g., C6-C14), wherein at least one ring in the system is aromatic and wherein each ring in the system contains three to seven ring members. In some embodiments, an “aryl” group contains between six and twelve total ring members (e.g., C6-C12). The term “aryl” may be used interchangeably with the term “aryl ring”. In certain embodiments, “aryl” refers to an aromatic ring system which includes, but not limited to, phenyl, biphenyl, naphthyl, anthracyl and the like, which may bear one or more substituents. Unless otherwise specified, “aryl” groups are hydrocarbons. In some embodiments, an “aryl” ring system is an aromatic ring (e.g., phenyl) that is fused to a non-aromatic ring (e.g., cycloalkyl). Examples of aryl rings include that are fused include .
in the presence of a reducing agent, wherein: each of L4 and L5 are each independently an optionally substituted C1-C30 aliphatic group; L6 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S; X3 and X4 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O)2N(R40)- , -N(R40)S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R40)2-, -OC(S)C(R40)2-, -C(R40)2C(S)O-, or –S-, wherein one or both of X3 or X4 is selected from -S(O)2N(R40)-, -N(R40)S(O)2, -S(O)-, - S(O)2-, -S(O)2C(R40)2-, -OC(S)C(R40)2-, -C(R40)2C(S)O-, or –S-; each R40 is, independently, at each instance, optionally substituted C1-C20 aliphatic or H; T3 and T4 are each independently an optionally substituted C3-C20 aliphatic; G1 is -N(R6)C(S)N(R6)2, -OH, -N(R6)2, -N(R9)C(O)R7, -N(R9)S(O)2R7, -N(R9)C(O)N(R7)2, -CH(N-R7),–R8, or -S(O)2R6; each G2 is independently O or N2; each G3 is independently halogen (e.g., Cl, Br, or I), -OTs, or OTf; each R6 is, independently, at each instance, selected from the group consisting of H, optionally substituted C1-C6 aliphatic and OR7; or two instances of R6 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring or an optionally substituted 4- to 12-membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N, O, and S; each R7 is, independently, at each instance, selected from the group consisting of H and optionally substituted C1-C6 aliphatic; R8 is optionally substituted 4- to 12-membered heterocycle comprising 1 to 4 heteroatoms selected from N, O, and S, optionally substituted 4- to 12 membered
heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S, C6-C12 aryl substituted with one or more of –(CH2)0-6–OH or –(CH2)0-6–N(R9)2, or C3-C12 cycloaliphatic substituted with one or more of oxo, –(CH2)0-6–OH, or –(CH2)0-6–N(R9)2; and each R9 is independently selected from H and optionally substituted C1-C6 aliphatic. Brief Description of the Drawing Figure 1 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- 51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59. Figure 2 is a bar graph illustrating zeta potential values of LNP formulations with BNT- 51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59. Figures 3A and 3B are a bar graph illustrating mRNA encapsulation efficiency (Figure 3A) and agarose gel electropherogram (Figure 3B) of LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59. Figure 4 is a bar graph illustrating mRNA integrity (%) of the LNP formulations with BNT- 51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59. Figures 5A-5C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 on cell viability in C2C12 cells (Figure 5A), HepG2 cells (Figure 5B) and RAW cells (Figure 5C). Figures 6A-6C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 on transfection efficiency in C2C12 cells (Figure 6A), HepG2 cells (Figure 6B) and RAW cells (Figure 6C). Figure 7 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04. Figure 8 is a bar graph illustrating Zeta potential values of LNP formulations with BNT- sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04. Figures 9A and 9B are a bar graph illustrating mRNA encapsulation efficiency (Figure 9A) and agarose gel electropherogram (Figure 9B) of LNP formulations with BNT-sulfo- 01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04. Figure 10 is a bar graph mRNA integrity (%) of the LNP formulations with BNT-sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04. Figures 11A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-sulfo-01, BNT-sulfo-02, BNT-sulfo-03, or BNT-sulfo-04 on cell viability in C2C12 cells (Figure 11A), HepG2 cells (Figure 11B), RAW cells (Figure 11C) and Hek293 cells (Figure 11D).
Figures 12A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 on transfection efficiency in C2C12 cells (Figure 12A), HepG2 cells (Figure 12B), RAW cells (Figure 12C) and Hek293 cells (Figure 12D). Figures 13A and 13B are a bar graph illustrating the in vitro hemolytic effect of the LNP formulations with BNT-51, BNT-52, BNT-54, BNT-56, BNT-57 or BNT-59 (Figure 13A) and a bar graph illustrating the effect of LNP formulations on complement activation (Figure 13B). Figure 14 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- 51 and a helper lipid (DSPC or DOPE). Figure 15 is a bar graph illustrating zeta potential values of LNP formulations with BNT- 51 and a helper lipid (DSPC or DOPE). Figures 16A and 16B are a bar graph illustrating mRNA encapsulation efficiency (Figure 16A) and agarose gel electropherogram (Figure 16B) of LNP formulations with BNT-51 and a helper lipid. Figure 17 is a bar graph illustrating mRNA integrity (%) of the LNP formulations with BNT-51 and a helper lipid (DSPC or DOPE). Figures 18A-D are a bar graph illustrating the effect of the LNP formulations with BNT- 51 and a helper lipid on cell viability in C2C12 cells (Figure 18A), HepG2 cells (Figure 18B), RAW cells (Figure 18C) and Hek293 cells (Figure 18D). Figures 19A-D are a bar graph illustrating the effect of the LNP formulations with BNT- 51 and a helper lipid on transfection efficiency in C2C12 cells (Figure 19A), HepG2 cells (Figure 19B), RAW cells (Figure 19C) and Hek293 cells (Figure 19D). Figure 20 is a bar graph illustrating particle size and PDI of LNP formulations with BNT- 51 and a stealth lipid. Figure 21 is a bar graph illustrating zeta potential values of LNP formulations with BNT- 51 and a stealth lipid. Figures 22A-B are a set of bar graphs illustrating mRNA encapsulation efficiency (Figure 22A) and agarose gel electropherogram (Figure 22B) of LNP formulations with BNT-51 and a stealth lipid. Figure 23 is a bar graph illustrating mRNA integrity (%) of the LNP formulations with BNT-51 and a stealth lipid. Figures 24A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51 and a stealth lipid on cell viability C2C12 cells (Figure 24A), HepG2 cells (Figure 24B), RAW cells (Figure 24C) and Hek293 cells (Figure 24D).
Figures 25A-D are a series of bar graphs illustrating the effect of the LNP formulations with BNT-51 and a stealth lipid on transfection efficiency in C2C12 cells (Figure 25A), HepG2 cells (Figure 25B), RAW cells (Figure 25C) and Hek293 cells (Figure 25D). Figure 26 is a bar graph illustrating particle size and PDI of pre-formed LNPs with BNT51 and Ac-AEEA14-DMA subjected to at least three freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature. Figures 27A-C are a series of bar graphs illustrating characterization of BNT51 functionalized lipid nanoparticles prepared with Ac-AEEA14-DMA as stealth moiety and DSPE-PEG2k-Alfa lipid (Figure 27A); particle size and PDI of functionalized LNP1 and LNP2; (Figure 27B); Agarose Gel Electrophoresis of controls, untreated functionalized LNP1 and LNP2 (upper row) and functionalized LNP1 and LNP2 treated with release solution (lower row) (Figure 27C); and particle size and PDI of functionalized LNP1 and LNP2 subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature, and after 2 weeks at 2-8°C and 25°C. Figures 28A-B are a series of bar graphs illustrating percentages of transfected cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs for both RNA (Thy1.1) (Figure 28A) and DNA (Venus) (Figure 28B), delivered using LNP1 or LNP2. Figure 29 is a bar graph illustrating particle size and PDI of preformed LNPs with BNT51 and Ac-AEEA14-VitE subjected to at least three freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature. Figures 30A-C are a series of bar graphs illustrating characterization of BNT51 functionalized lipid nanoparticles prepared with Ac-AEEA14-VitE as stealth moiety and DSPE-PEG2k-Alfa lipid (Figure 30A); particle size and PDI of functionalized LNP1 and LNP2 (Figure 30B); Agarose Gel Electrophoresis of controls, untreated functionalized LNP1 and LNP2 (upper row) and functionalized LNP1 and LNP2 treated with release solution (lower row) (Figure 30C); and particle size and PDI of functionalized LNP1 and LNP2, subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature, and after 2 weeks at 2-8°C and 25°C. Figures 31A-B are a series of bar graphs illustrating percentages of transfected cell (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs for both RNA (Thy1.1) (Figure 31A) and DNA (Venus) (Figure 31B), delivered using LNP1 or LNP2. Figures 32A-B are a set of bar graphs illustrating characterization of BNT51 functionalized lipid nanoparticles prepared with Ac-AEEA14-DMA as stealth moiety and
DSPE-pAEEA14-Alfa lipid, including (Figure 32A): particle size and PDI of preformed LNPs subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature; and (Figure 32B): particle size and PDI of functionalized RNA/DNA-LNP2 at t0 and subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature. Figures 33A-B are a set of bar graphs illustrating characterization of BNT52 functionalized lipid nanoparticles prepared with Ac-AEEA14-DMA as stealth moiety and DSPE-pAEEA14-Alfa lipid including (Figure 33A): particle size and PDI of preformed LNPs subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature; and (Figure 33B): Particle size and PDI of functionalized RNA/DNA-LNP2 at t0 and subjected to at least two freeze thaw cycles from -20°C to room temperature and from -80°C to room temperature. Figure 34 is a set of bar graphs illustrating use of thiolipids described herein for targeted transfection of T-cells. In vitro evaluation in hPBMCs. Comparison of three different LNPs containing three different ionizable lipids show the superior RNA and DNA transfection efficiency of thiolipids described herein which were formulated in LNP2 and LNP3. Depicted in the upper graph are the percentages of Thy1.1-expressing cell subtypes (CD4+ T cells, CD8+ T cells, CD19+ B cells) out of all single and alive cells (y‐axes). Depicted in the lower graph are the percentages of Venus-Nanoplasmid expressing cells for CD4+ T cells and CD8 + T cells. Figures 35A and 35B illustrate the use of reported thiolipids for targeted transfection of T-cells. In vitro evaluation in hPBMCs. Comparison of two different LNPs containing two different ionizable lipids show high RNA and DNA transfection efficiency of reported thiolipids which were formulated in LNP1 and LNP2. Depicted in Figure 35A are the percentages of Thy1.1-expressing cell subtypes (CD4+ T cells, CD8+ T cells, CD19+ B cells, CD14+ Monocytes) out of all single and alive cells (y‐axes). Depicted in Figure 35B are the percentages of Venus-Nanoplasmid expressing cells for CD4+ T cells and CD8 + T cells. Figures 36A and 36B are a set of bar charts illustrating characterization of the LNP formulations with BNT-72, including particle size and PDI (Figure 36A) and zeta potential (Figure 36B). Figures 37A and 37B are a set of bar graphs illustrating mRNA encapsulation efficiency (Figure 37A) and mRNA integrity (Figure 37B) of the of LNP formulations with BNT-72.
Figures 38A-C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-72 on cell viability in C2C12 cells (Figure 38A), HepG2 cells (Figure 38B) and RAW cells (Figure 38C). Figures 39A-C are a series of bar graphs illustrating the effect of the LNP formulations with BNT-72 on transfection efficiency in C2C12 cells (Figure 39A), HepG2 cells (Figure 39B) and RAW cells (Figure 39C). Figure 40 is a bar graph illustrating the size (nm) and polydispersity index (PDI) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM. Figure 41 is a bar graph illustrating Z-potential (mV) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM. Figure 42 is a bar graph illustrating RNA integrity (%) of complexes comprising BNT-76, BNT-90 or a benchmark lipid CM12_BM. Figure 43 is a bar graph illustrating osmolarity and pH of complexes comprising BNT- 76, BNT-90 or a benchmark lipid CM12_BM. Figures 44A-44D are a series of bar graphs illustrating transfection efficiency in C2C12 cells (Figure 44A), HepG2 cells (Figure 44B), RAW cells (Figure 44C) and Hek293 cells (Figure 44D). Figures 45A-45D are a series of bar graphs illustrating cell viability in C2C12 cells (Figure 45A), HepG2 cells (Figure 45B), RAW cells (Figure 45C) and Hek293 cells (Figure 45D). Figure 46 depicts the size and PDI of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C. The results of three measurements, as well as their average and error bars indicating 1x standard deviation are shown. Figure 47 depicts the percentages of transfected cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C. Figure 48 depicts the cell counts of alive cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs treated with aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, which were either applied freshly prepared or after one freeze/thaw cycle at -80°C. Figure 49 is a graph providing size and distribution (PDI) of provided LNPs comprising lipids reported herein.
Figure 50 is a graph providing Z-potential of provided LNPs comprising lipids described herein. Figure 51 is a graph illustrating encapsulation efficiency of provided LNPs. Figure 52 is an agarose gel electrophoresis showing no free RNA for provided LNPs. Figures 53A-D are a series of graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage. Figures 54A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs. Figure 55 is a graph illustrating size and distribution (PDI) for provided LNPs comprising lipid compounds described herein. Figure 56 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein. Figure 57 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein. Figure 58 is an agarose gel electrophoresis image showing no free RNA for all provided LNPs. Figures 59A-D are a series of graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage. Figures 60A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs. Figure 61 is a graph illustrating size and distribution (PDI) of LNPs comprising lipid compounds described herein. Figure 62 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein. Figure 63 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein. Figure 64 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs. Figures 65A-D are a series graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage. Figures 66A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs. Figure 67 is a graph illustrating size and distribution (PDI) of LNPs comprising lipid compounds described herein.
Figure 68 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein. Figure 69 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein. Figure 70 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs. Figures 71A-D are a series graphs illustrating cytotoxicity in provided cell lines C2C12, HepG2, Raw, and Hek as measured by viability percentage. Figures 72A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs. Figure 73 is a graph illustrating size and distribution (PDI) of LNPs comprising lipid compounds described herein. Figure 74 is a graph illustrating Z-potential of provided LNPs comprising lipids described herein. Figure 75 is a graph illustrating encapsulation efficiency of provided LNPs comprising lipids described herein. Figure 76 is an image of an agarose gel electrophoresis showing no free RNA for provided LNPs. Figures 77A-B are a series graphs illustrating cytotoxicity in provided cell lines HepG2 and Hek as measured by viability percentage. Figures 78A-D are a series of graphs illustrating transfection efficiency of provided LNPs as measured by RLUs. Detailed Description of Certain Embodiments The present disclosure provides, among other things, the surprising discovery of particular cationic or ionizable lipids comprising at least one sulfur-containing moiety. Such cationic or ionizable lipids exhibit improved properties (e.g., improved transfection to cells) relative to previous lipids. For example, as illustrated in the Examples provided herein, thiolipids of the present disclosure exhibit improvements over previous lipids that lack a sulfur-containing moiety, including, for example, improved transfection of cells with RNA. Particles prepared using thiolipid compounds described herein exhibit narrow size distribution (i.e., are substantially uniform in size), high encapsulation efficiency, low cytotoxicity, and improved biodistribution and efficacy relative to particles comprising previous lipid compounds.
Compounds and Definitions Compounds of this disclosure include those described generally above and are further illustrated by the classes, subclasses, and species disclosed herein. As used herein, the following definitions shall apply unless otherwise indicated. For purposes of this disclosure, the chemical elements are identified in accordance with the Periodic Table of Elements, CAS version, Handbook of Chemistry and Physics, 75th Ed. Additionally, general principles of organic chemistry are described in “Organic Chemistry”, Thomas Sorrell, University Science Books, Sausalito: 1999, and “March’s Advanced Organic Chemistry”, 5th Ed., Ed.: Smith, M.B. and March, J., John Wiley & Sons, New York: 2001, the entire contents of which are hereby incorporated by reference. Unless otherwise stated, structures depicted herein are meant to include all stereoisomeric (e.g., enantiomeric or diastereomeric) forms of the structure, as well as all geometric or conformational isomeric forms of the structure. For example, the R and S configurations of each stereocenter are contemplated as part of the disclosure. Therefore, single stereochemical isomers, as well as enantiomeric, diastereomeric, and geometric (or conformational) mixtures of provided compounds are within the scope of the disclosure. For example, in some cases, Table 1 shows one or more stereoisomers of a compound, and unless otherwise indicated, represents each stereoisomer alone and/or as a mixture. Unless otherwise stated, all tautomeric forms of provided compounds are within the scope of the disclosure. Unless otherwise indicated, structures depicted herein are meant to include compounds that differ only in the presence of one or more isotopically enriched atoms. For example, compounds having the present structures including replacement of hydrogen by deuterium or tritium, or replacement of a carbon by 13C- or 14C-enriched carbon are within the scope of this disclosure. About or approximately: As used herein, the term "approximately" or "about," as applied to one or more values of interest, refers to a value that is similar to a stated reference value. In general, those skilled in the art, familiar within the context, will appreciate the relevant degree of variance encompassed by "about" or "approximately" in that context. For example, in some embodiments, the term "approximately" or "about" may encompass a range of values that are within (i.e., ±) 25%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or less of the referred value. Administering: As used herein, the term "administering" or "administration" typically refers to the administration of a composition to a subject to achieve delivery of an agent
that is, or is included in, a composition to a target site or a site to be treated. Those of ordinary skill in the art will be aware of a variety of routes that may, in appropriate circumstances, be utilized for administration to a subject, for example a human. For example, in some embodiments, administration may be ocular, oral, parenteral, topical, etc. In some particular embodiments, administration may be bronchial (e.g., by bronchial instillation), buccal, dermal (which may be or comprise, for example, one or more of topical to the dermis, intradermal, interdermal, transdermal, etc.), enteral, intra-arterial, intradermal, intragastric, intramedullary, intramuscular, intranasal, intraperitoneal, intrathecal, intravenous, intraventricular, within a specific organ (e.g., intrahepatic), mucosal, nasal, oral, rectal, subcutaneous, sublingual, topical, tracheal (e.g., by intratracheal instillation), vaginal, vitreal, etc. In some embodiments, administration may be parenteral. In some embodiments, administration may be oral. In some particular embodiments, administration may be intravenous. In some particular embodiments, administration may be subcutaneous. In some embodiments, administration may involve only a single dose. In some embodiments, administration may involve application of a fixed number of doses. In some embodiments, administration may involve dosing that is intermittent (e.g., a plurality of doses separated in time) and/or periodic (e.g., individual doses separated by a common period of time) dosing. In some embodiments, administration may involve continuous dosing (e.g., perfusion) for at least a selected period of time. In some embodiments, administration may comprise a prime-and-boost protocol. A prime-and-boost protocol can include administration of a first dose of a pharmaceutical composition (e.g., an immunogenic composition, e.g., a vaccine) followed by, after an interval of time, administration of a second or subsequent dose of a pharmaceutical composition (e.g., an immunogenic composition, e.g., a vaccine). In the case of an immunogenic composition, a prime-and-boost protocol can result in an increased immune response in a patient. Aliphatic: The term “aliphatic” refers to a straight-chain (i.e., unbranched) or branched, substituted or unsubstituted hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation, or a monocyclic hydrocarbon or bicyclic hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic (also referred to herein as “cycloaliphatic”), that has a single point or more than one points of attachment to the rest of the molecule. Unless otherwise specified, aliphatic groups contain 1-12 aliphatic carbon atoms. In some embodiments, aliphatic groups contain 1-6 aliphatic carbon atoms (e.g., C1-6). In some embodiments, aliphatic groups contain 1-5 aliphatic carbon atoms (e.g., C1-5). In
other embodiments, aliphatic groups contain 1-4 aliphatic carbon atoms (e.g., C1-4). In still other embodiments, aliphatic groups contain 1-3 aliphatic carbon atoms (e.g., C1-3), and in yet other embodiments, aliphatic groups contain 1-2 aliphatic carbon atoms (e.g., C1-2). Suitable aliphatic groups include, but are not limited to, linear or branched, substituted or unsubstituted alkyl, alkenyl, or alkynyl groups and hybrids thereof. A preferred aliphatic group is C1-6 alkyl. Alkyl: The term “alkyl”, used alone or as part of a larger moiety, refers to a saturated, optionally substituted straight or branched chain hydrocarbon group having (unless otherwise specified) 1-12, 1-10, 1-8, 1-6, 1-4, 1-3, or 1-2 carbon atoms (e.g., C1-12, C1-10, C1-8, C1-6, C1-4, C1-3, or C1-2). Exemplary alkyl groups include methyl, ethyl, propyl, butyl, pentyl, hexyl, and heptyl. Alkylene: The term “alkylene” is refers to a bivalent alkyl group. In some embodiments, “alkylene” is a bivalent straight or branched alkyl group. In some embodiments, an "alkylene chain" is a polymethylene group, i.e., -(CH2)n-, wherein n is a positive integer, e.g., from 1 to 6, from 1 to 4, from 1 to 3, from 1 to 2, or from 2 to 3. An optionally substituted alkylene chain is a polymethylene group in which one or more methylene hydrogen atoms is optionally replaced with a substituent. Suitable substituents include those described below for a substituted aliphatic group and also include those described in the specification herein. It will be appreciated that two substituents of the alkylene group may be taken together to form a ring system. In certain embodiments, two substituents can be taken together to form a 3- to 7-membered ring. The substituents can be on the same or different atoms. The suffix “-ene” or “-enyl” when appended to certain groups herein are intended to refer to a bifunctional moiety of said group. For example, “-ene” or “-enyl”, when appended to “cyclopropyl” becomes “cyclopropylene” or “cyclopropylenyl” and is intended to refer to a bifunctional cyclopropyl group, e.g., . Alkenyl: The term “alkenyl”, used alone or as part of a larger moiety, refers to an optionally substituted straight or branched chain or cyclic hydrocarbon group having at least one double bond and having (unless otherwise specified) 2-12, 2-10, 2-8, 2-6, 2-4, or 2-3 carbon atoms(e.g., C2-12, C2-10, C2-8, C2-6, C2-4, or C2-3). Exemplary alkenyl groups include ethenyl, propenyl, butenyl, pentenyl, hexenyl, and heptenyl. The term “cycloalkenyl” refers to an optionally substituted non-aromatic monocyclic or multicyclic ring system containing at least one carbon-carbon double bond and having about 3 to
about 10 carbon atoms. Exemplary monocyclic cycloalkenyl rings include cyclopentenyl, cyclohexenyl, and cycloheptenyl. Alkynyl: The term “alkynyl”, used alone or as part of a larger moiety, refers to an optionally substituted straight or branched chain hydrocarbon group having at least one triple bond and having (unless otherwise specified) 2-12, 2-10, 2-8, 2-6, 2-4, or 2-3 carbon atoms (e.g., C2-12, C2-10, C2-8, C2-6, C2-4, or C2-3). Exemplary alkynyl groups include ethynyl, propynyl, butynyl, pentynyl, hexynyl, and heptynyl. Aryl: The term “aryl” refers to monocyclic and bicyclic ring systems having a total of six to fourteen ring members (e.g., C6-C14), wherein at least one ring in the system is aromatic and wherein each ring in the system contains three to seven ring members. In some embodiments, an “aryl” group contains between six and twelve total ring members (e.g., C6-C12). The term “aryl” may be used interchangeably with the term “aryl ring”. In certain embodiments, “aryl” refers to an aromatic ring system which includes, but not limited to, phenyl, biphenyl, naphthyl, anthracyl and the like, which may bear one or more substituents. Unless otherwise specified, “aryl” groups are hydrocarbons. In some embodiments, an “aryl” ring system is an aromatic ring (e.g., phenyl) that is fused to a non-aromatic ring (e.g., cycloalkyl). Examples of aryl rings include that are fused include .
 Associated: Two events or entities are “associated” with one another, as that term is used herein, if the presence, level and/or form of one is correlated with that of the other. For example, a particular entity (e.g., polypeptide, genetic signature, metabolite, microbe, etc) is considered to be associated with a particular disease, disorder, or condition, if its presence, level and/or form correlates with incidence of and/or susceptibility to the disease, disorder, or condition (e.g., across a relevant population). In some embodiments, two or more entities are physically “associated” with one another if they interact, directly or indirectly, so that they are and/or remain in physical proximity with one another. In some embodiments, two or more entities that are physically associated with one another are covalently linked to one another; in some embodiments, two or more entities that are physically associated with one another are not covalently linked to one another but are non-covalently associated, for example by means of hydrogen bonds, van der Waals interaction, hydrophobic interactions, magnetism, and combinations thereof. Bicyclic: The term “bicyclic ring” or “bicyclic ring system” refers to any bicyclic ring system, i.e., carbocyclic or heterocyclic, saturated or having one or more units of
unsaturation, having one or more atoms in common between the two rings of the ring system. Thus, the term includes any permissible ring fusion, such as ortho-fused or spirocyclic. As used herein, the term “heterobicyclic” is a subset of “bicyclic” that requires that one or more heteroatoms are present in one or both rings of the bicycle. Such heteroatoms may be present at ring junctions and are optionally substituted, and may be selected from nitrogen (including N-oxides), oxygen, sulfur (including oxidized forms such as sulfones and sulfonates), phosphorus (including oxidized forms such as phosphates), boron, etc. In some embodiments, a bicyclic group has 7-12 ring members and 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur. As used herein, the term “bridged bicyclic” refers to any bicyclic ring system, i.e., carbocyclic or heterocyclic, saturated or partially unsaturated, having at least one bridge. As defined by IUPAC, a “bridge” is an unbranched chain of atoms or an atom or a valence bond connecting two bridgeheads, where a “bridgehead” is any skeletal atom of the ring system which is bonded to three or more skeletal atoms (excluding hydrogen). In some embodiments, a bridged bicyclic group has 7-12 ring members and 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur. Such bridged bicyclic groups are well known in the art and include those groups set forth below where each group is attached to the rest of the molecule at any substitutable carbon or nitrogen atom. Unless otherwise specified, a bridged bicyclic group is optionally substituted with one or more substituents as set forth for aliphatic groups. Additionally or alternatively, any substitutable nitrogen of a bridged bicyclic group is optionally substituted. Exemplary bicyclic rings include:
Associated: Two events or entities are “associated” with one another, as that term is used herein, if the presence, level and/or form of one is correlated with that of the other. For example, a particular entity (e.g., polypeptide, genetic signature, metabolite, microbe, etc) is considered to be associated with a particular disease, disorder, or condition, if its presence, level and/or form correlates with incidence of and/or susceptibility to the disease, disorder, or condition (e.g., across a relevant population). In some embodiments, two or more entities are physically “associated” with one another if they interact, directly or indirectly, so that they are and/or remain in physical proximity with one another. In some embodiments, two or more entities that are physically associated with one another are covalently linked to one another; in some embodiments, two or more entities that are physically associated with one another are not covalently linked to one another but are non-covalently associated, for example by means of hydrogen bonds, van der Waals interaction, hydrophobic interactions, magnetism, and combinations thereof. Bicyclic: The term “bicyclic ring” or “bicyclic ring system” refers to any bicyclic ring system, i.e., carbocyclic or heterocyclic, saturated or having one or more units of
unsaturation, having one or more atoms in common between the two rings of the ring system. Thus, the term includes any permissible ring fusion, such as ortho-fused or spirocyclic. As used herein, the term “heterobicyclic” is a subset of “bicyclic” that requires that one or more heteroatoms are present in one or both rings of the bicycle. Such heteroatoms may be present at ring junctions and are optionally substituted, and may be selected from nitrogen (including N-oxides), oxygen, sulfur (including oxidized forms such as sulfones and sulfonates), phosphorus (including oxidized forms such as phosphates), boron, etc. In some embodiments, a bicyclic group has 7-12 ring members and 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur. As used herein, the term “bridged bicyclic” refers to any bicyclic ring system, i.e., carbocyclic or heterocyclic, saturated or partially unsaturated, having at least one bridge. As defined by IUPAC, a “bridge” is an unbranched chain of atoms or an atom or a valence bond connecting two bridgeheads, where a “bridgehead” is any skeletal atom of the ring system which is bonded to three or more skeletal atoms (excluding hydrogen). In some embodiments, a bridged bicyclic group has 7-12 ring members and 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur. Such bridged bicyclic groups are well known in the art and include those groups set forth below where each group is attached to the rest of the molecule at any substitutable carbon or nitrogen atom. Unless otherwise specified, a bridged bicyclic group is optionally substituted with one or more substituents as set forth for aliphatic groups. Additionally or alternatively, any substitutable nitrogen of a bridged bicyclic group is optionally substituted. Exemplary bicyclic rings include:




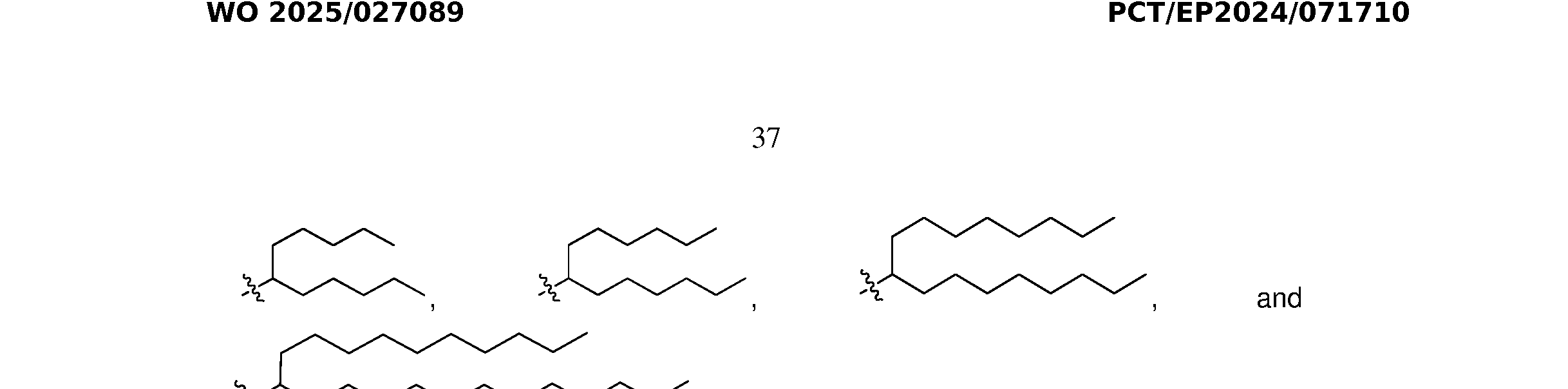












































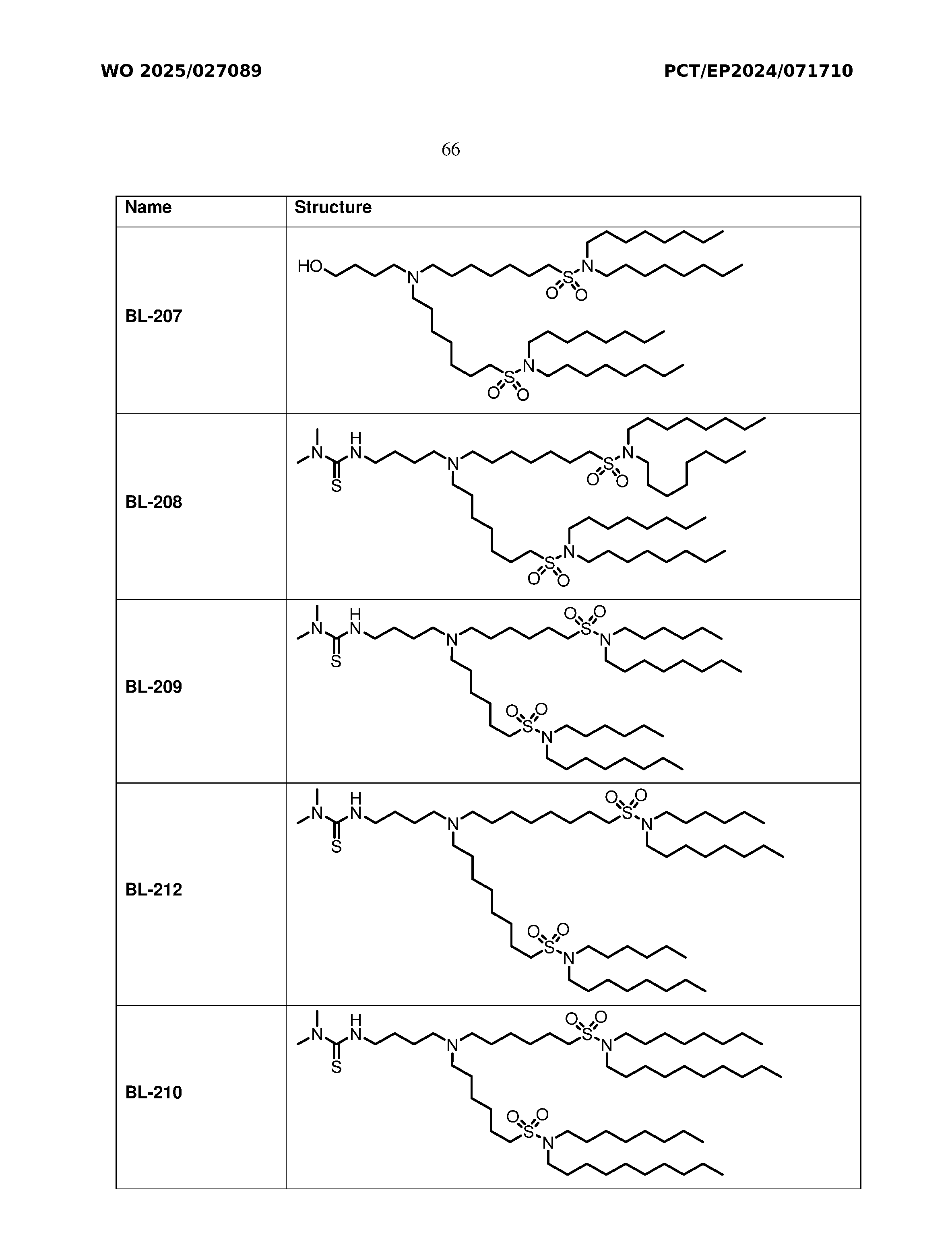
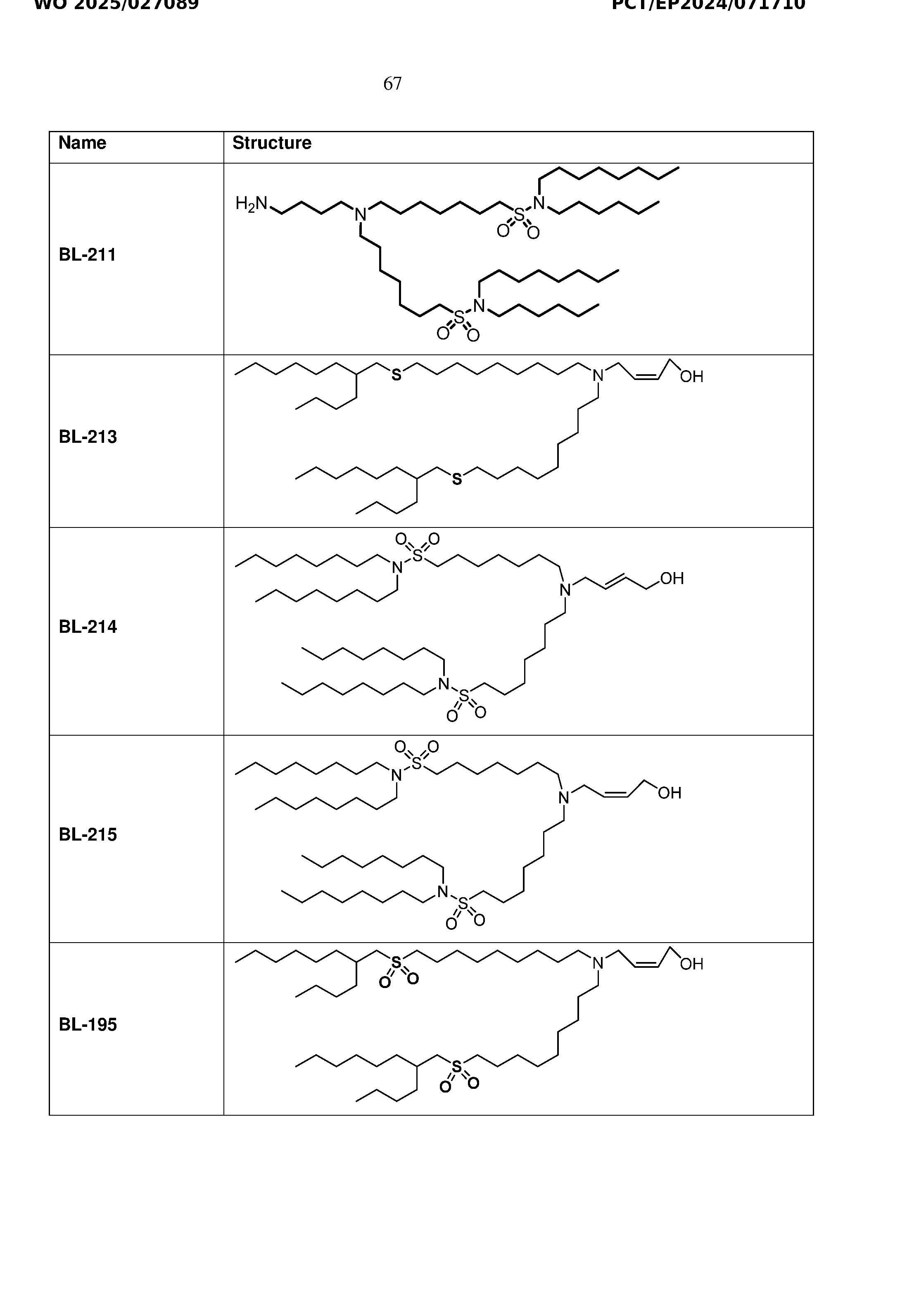
Methods of Preparing Thiolipids
Compounds described herein are particularly useful over previous lipid compounds due, at least in part, to the ease of preparation. In particular, diverse fragments can be coupled through sulfur linkages in order to provide a wide array of thiolipid compounds that are useful for the preparation of nucleic acid particles. For example, thiolipid compounds of the present disclosure can be prepared according to General Schemes 1 , 2, and 3, depicted in Figures 79, 80, and 81 , respectively.
where a tail-linker moiety is any bivalent linker, such as an aliphatic or heteroaliphatic group; a tail-end is a hydrophobic group, e.g., an aliphatic group, a head group is a polar or cationic or ionizable head group, a tail junction is a biodegradable group, such as an ester, or a sulfur-containing moiety (e.g., thioether, sulfonyl, or sulfonamide), and a headtail junction is a central atom or functional group connecting a tail or tails to the head group (e.g., a tertiary amine group).
As described herein, in some embodiments, the present disclosure provides a method of preparing a compound represented by Formula IV:
 or a pharmaceutically acceptable salt thereof, the method comprising: contacting a compound represented by Formula V
or a pharmaceutically acceptable salt thereof, the method comprising: contacting a compound represented by Formula V
 with a compound represented by one of Formulae Vla-VIc
with a compound represented by one of Formulae Vla-VIc
 and a compound represented by one of Formulae Vlla-Vllc
and a compound represented by one of Formulae Vlla-Vllc
 in the presence of a reducing agent, wherein: each of L4 and L5 are each independently an optionally substituted C1-C30 aliphatic group; L6 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S;
in the presence of a reducing agent, wherein: each of L4 and L5 are each independently an optionally substituted C1-C30 aliphatic group; L6 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S;
X3 and X4 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O)2N(R40)- , -N(R40)S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R40)2-, -OC(S)C(R40)2-, -C(R40)2C(S)0-, or -S-, wherein one or both of X3 or X4 is selected from -S(O)2N(R40)-, -N(R40)S(O)2, -8(0)-, - S(O)2-, -S(O)2C(R40)2-, -OC(S)C(R40)2-, -C(R40)2C(S)0-, or -S-; each R40 is, independently, at each instance, optionally substituted Ci-C2o aliphatic or H; T3 and T4 are each independently an optionally substituted Cs-C2o aliphatic;
G1 is -N(R6)C(S)N(R6)2, -OH, -N(R6)2, -N(R9)C(O)R7, -N(R9)S(O)2R7, -N(R9)C(O)N(R7)2, -CH(N-R7), or-R8; each G2 is independently O or N2;
each G3 is independently halogen (e.g., Cl, Br, or I), -OTs, or OTf; each R6 is, independently, at each instance, selected from the group consisting of H, optionally substituted C1-C6 aliphatic or OR7; or two instances of R6 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring or an optionally substituted 4- to 12-membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N, O, and S; each R7 is, independently, at each instance, selected from the group consisting of H and optionally substituted C1-C6 aliphatic; R8 is optionally substituted 4- to 12-membered heterocycle comprising 1 to 4 heteroatoms selected from N, O, and S, optionally substituted 4- to 12 membered heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S, C6-C12 aryl substituted with one or more of –(CH2)0-6–OH or –(CH2)0-6–N(R9)2, or C3-C12 cycloaliphatic substituted with one or more of oxo, –(CH2)0-6–OH, or –(CH2)0-6–N(R9)2; and each R9 is independently selected from H and optionally substituted C1-C6 aliphatic. In some embodiments, a reducing agent is NaBH3CN or NaBH(OCOCH3)3. In some embodiments, a reducing agent is NaBH3CN. In some embodiments, a reducing agent is NaBH(OCOCH3)3. In some embodiments, a method of preparing a compound described herein further comprises preparing a compound represented by Formula VIa or the compound represented by Formula VIIa, wherein G1 is O, by contacting a co compound represented by
 Formula IX with an oxidi In some em
Formula IX with an oxidi In some em
 bodiments, an oxidizing agent is DMSO, PCC, or DMP. In some embodiments, an oxidizing agent is DMSO, and the method further comprises contacting the compound represented by Formula VIII or IX and DMSO with a sulfur trioxide pyridine complex (SO3•pyridine).
In some embodiments, a method of preparing a compound described herein further comprises preparing the compound represented by Formula VIII or IX:
bodiments, an oxidizing agent is DMSO, PCC, or DMP. In some embodiments, an oxidizing agent is DMSO, and the method further comprises contacting the compound represented by Formula VIII or IX and DMSO with a sulfur trioxide pyridine complex (SO3•pyridine).
In some embodiments, a method of preparing a compound described herein further comprises preparing the compound represented by Formula VIII or IX:
 by contacting a compound represented by Formula X or a compound represented by Formula XI
by contacting a compound represented by Formula X or a compound represented by Formula XI
 with a compound represented by Formula XII or a compound represented by Formula XIII:
with a compound represented by Formula XII or a compound represented by Formula XIII:
 in the presence of H2O2 and SOCI2; and wherein each V1 is halogen.
in the presence of H2O2 and SOCI2; and wherein each V1 is halogen.
Particles for Nucleic Acid Delivery
In some embodiments, particles of the present disclosure comprise a compound described herein (e.g., a compound of one or more of formulae l-lllh-5), a nucleic acid (such as RNA (e.g., mRNA), DNA or mixtures thereof), and one or more of a polymer- conjugated lipid, a helper lipid, and a steroid. In some embodiments, particles of the present disclosure comprise a compound described herein (e.g., a compound of one or more of formulae l-lllh-5), a nucleic acid (such as RNA (e.g., mRNA), DNA or mixtures thereof), a helper lipid, and a steroid. In some embodiments, particles of the present disclosure comprise a compound described herein (e.g., a compound of one or more of formulae l-lllh-5), a nucleic acid (such as RNA (e.g., mRNA), DNA or mixtures thereof), a polymer-conjugated lipid, a helper lipid, and a steroid.
Thiolipid compounds described herein comprise a cationic or cationically ionizable moiety that is capable of forming electrostatic interactions between the positively charged moiety and negatively charged nucleic acids, resulting in particle formation.
In some embodiments, particles described herein (e.g., nucleic acid particles, e.g., ribonucleic acid particles or deoxyribonucleic acid particles) comprise more than one type of nucleic acid molecules, where the molecular parameters of the nucleic acid molecules may be similar or different from each other, like with respect to molar mass or
fundamental structural elements such as molecular architecture, capping, coding regions or other features. In some embodiments, a nucleic acid particle described herein is a nanoparticle. As used in the present disclosure, “nanoparticle” refers to a particle having an average diameter suitable for parenteral administration and is less than 1000 nm in diameter. In some embodiments, a composition comprising nanoparticles can have an average nanoparticle size (e.g., mean diameter) of about 30 nm to about 150 nm, about 40 nm to about 150 nm, about 50 nm to about 150 nm, about 60 nm to about 130 nm, about 70 nm to about 110 nm, about 70 nm to about 100 nm, about 70 to about 90 nm, or about 70 nm to about 80 nm. In some embodiments, a composition comprising nanoparticles can have an average nanoparticle size (e.g., mean diameter) of about 50 nm to about 100 nm. In some embodiments, a composition comprising nanoparticles can have an average nanoparticle size (e.g., mean diameter) of about 50 nm to about 150 nm. In some embodiments, a composition comprising nanoparticles can have an average nanoparticle size (e.g., mean diameter) of about 60 nm to about 120 nm. In some embodiments, a composition comprising nanoparticles can have an average nanoparticle size (e.g., mean diameter) of about 30 nm, 35 nm, 40 nm, 45 nm, 50 nm, 55 nm, 60 nm, 65 nm, 70 nm, 75 nm, 80 nm, 85 nm, 90 nm, 95 nm, 100 nm, 105 nm, 110 nm, 115 nm, 120 nm, 125 nm, 130 nm, 135 nm, 140 nm, 145 nm, or 150 nm. A composition comprising nucleic acid particles (e.g., ribonucleic acid particles or deoxyribonucleic acid particles) described herein may exhibit a polydispersity index less than about 0.5, less than about 0.4, less than about 0.3, or about 0.2 or less of said nanoparticles. By way of example, a composition comprising nucleic acid particles (e.g., ribonucleic acid particles or deoxyribonucleic acid particles) described herein can exhibit a polydispersity index in a range of about 0.1 to about 0.3 or about 0.2 to about 0.3. Nucleic acid particles (e.g., ribonucleic acid particles or deoxyribonucleic acid particles) described herein can be characterized by an “N/P ratio,” which is the molar ratio of cationic (nitrogen) groups (the “N” in N/P) in the cationic polymer to the anionic (phosphate) groups (the “P” in N/P) in RNA. It is understood that a cationic group is one that is either in cationic form (e.g., N+), or one that is ionizable to become cationic. Use of a single number in an N/P ratio (e.g., an N/P ratio of about 5) is intended to refer to that number over 1, e.g., an N/P ratio of about 4 is intended to mean about 4:1. In some embodiments, a nucleic acid particle (e.g., a ribonucleic acid particle) described herein has an N/P ratio greater than or equal to 4. In some embodiments, a nucleic acid particle (e.g., a ribonucleic acid particle) described herein has an N/P ratio that is about 4 to
about 16. In some embodiments, a nucleic acid particle (e.g., a ribonucleic acid particle) described herein has an N/P ratio that is about 6 to about 12. In some embodiments, a nucleic acid particle described herein has an N/P ratio that is about 4 to about 12. In some embodiments, a nucleic acid particle (e.g., a ribonucleic acid particle) described herein has an N/P ratio that is about 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, or 16. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is about 6. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is about 12. Compounds described herein are also referred to as “ionizable” or “cationic” lipids. Such lipids are intended to mean compounds that, in some embodiments, are capable of becoming cationic (i.e., becoming positively charged) at physiological pH. The term “average diameter” or “mean diameter” refers to the mean hydrodynamic diameter of particles as measured by dynamic laser light scattering (DLS) with data analysis using the so-called cumulant algorithm, which provides as results the so-called Z-average with the dimension of a length, and the polydispersity index (PDI), which is dimensionless (Koppel, D., J. Chem. Phys.57, 1972, pp 4814-4820, ISO 13321). Here “average diameter,” “mean diameter,” “diameter,” or “size” for particles is used synonymously with this value of the Z-average. The “polydispersity index” is preferably calculated based on dynamic light scattering measurements by the so-called cumulant analysis as mentioned in the definition of the “average diameter.” Under certain prerequisites, it can be taken as a measure of the size distribution of an ensemble of ribonucleic acid nanoparticles (e.g., ribonucleic acid nanoparticles). Different types of nucleic acid particles have been described previously to be suitable for delivery of nucleic acid in particulate form (e.g. Kaczmarek, J. C. et al., 2017, Genome Medicine 9, 60). For non-viral nucleic acid delivery vehicles, nanoparticle encapsulation of nucleic acid physically protects nucleic acid from degradation and, depending on the specific chemistry, can aid in cellular uptake and endosomal escape. Some embodiments described herein relate to compositions, methods and uses involving more than one, e.g., 2, 3, 4, 5, 6 or even more nucleic acid species. The nucleic acid species may be RNA and/or DNA. For example, the particles described herein may contain one species of RNA (e.g., one species of mRNA) and one species of DNA. In a nucleic acid particle composition, it is possible that each nucleic acid species is separately formulated as an individual nucleic acid particle formulation. In that case, each individual nucleic acid particle formulation will comprise one nucleic acid species. The
individual nucleic acid particle formulations may be present as separate entities, e.g., in separate containers. Such formulations are obtainable by providing each nucleic acid species separately (typically each in the form of a nucleic acid-containing solution) together with a particle-forming agent, thereby allowing the formation of particles. Respective particles will contain exclusively the specific nucleic acid species that is being provided when the particles are formed (individual particulate formulations). In some embodiments, a composition such as a pharmaceutical composition comprises more than one individual nucleic acid particle formulation. Respective pharmaceutical compositions are referred to as “mixed particulate formulations.” Mixed particulate formulations according to the present disclosure are obtainable by forming, separately, individual nucleic acid particle formulations, as described above, followed by a step of mixing of the individual nucleic acid particle formulations. By the step of mixing, a formulation comprising a mixed population of nucleic acid-containing particles is obtainable. Individual nucleic acid particle populations may be together in one container, comprising a mixed population of individual nucleic acid particle formulations. Alternatively, it is possible that different nucleic acid species are formulated together as a “combined particulate formulation.” Such formulations are obtainable by providing a combined formulation (typically combined solution) of different nucleic acid species together with a particle-forming agent, thereby allowing the formation of particles. As opposed to a “mixed particulate formulation,” a “combined particulate formulation” will typically comprise particles that comprise more than one nucleic acid species. In a combined particulate composition different nucleic acid species are typically present together in a single particle. In certain embodiments, nucleic acids, when present in provided nucleic acid particles are resistant in aqueous solution to degradation with a nuclease. Lipid Nanoparticles In some embodiments, nucleic acid particles are lipid nanoparticles. In some embodiments, lipid nanoparticles are cationic lipid nanoparticles comprising one or more cationic lipids (e.g., ones described herein), a nucleic acid (e.g., RNA and/or DNA), a polymer-conjugated lipid, a helper lipid, and a sterol. Lipid nanoparticles (LNPs) have proven useful for the delivery of nucleic acid cargo to tissue of interest. LNPs are used, for example, in certain commercial vaccines for treatment of COVID-19. In some embodiments, LNPs of the present disclosure comprise i) a cationic lipid (e.g., a thiolipid compound described herein, such as a compound of any one of formulae I-
IIIh-5); ii) a helper lipid; iii) a polymer-conjugated lipid (e.g., a polyethylene glycol bound lipid “a PEG lipid”); and iv) a steroid. In some embodiments, LNPs of the present disclosure may comprise i) a cationic lipid (e.g., a thiolipid compound described herein, such as a compound of any one of formulae I-IIIh-5); ii) a helper lipid; and iii) a steroid. In some embodiments, LNPs described herein can further comprise additional additives, as described herein. LNPs of the present disclosure can be useful in a variety of contexts. For example, LNPs comprising a nucleic acid described herein are useful for delivery of said nucleic acid into the cell of a subject. In some embodiments, LNPs comprising a nucleic acid described herein are useful for causing increased expression of a protein in a subject. In some embodiments, LNPs comprising a nucleic acid described herein are useful for causing a pharmacological effect induced by expression of a protein in a subject. Lipid nanoparticles described herein are characterized by molar percentage (mol%) of components in the lipid nanoparticle. A mol% used in reference to a lipid component of a lipid nanoparticle is relative to the total other lipid components in the lipid nanoparticle. (i) Helper lipids As described herein, LNPs of the present disclosure comprise a helper lipid. In some embodiments, a helper lipid is a phospholipid. In some embodiments, a helper lipid is or comprises 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dipalmitoyl-sn- glycero-3-phosphocholine (DPPC), 1,2-dimyristoyl-sn-glycero-3- phosphocholine (DMPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-sn- glycero-3-phosphocholine (DOPC), phosphatidyl ethanol amines such as 1,2-dioleoyl- sn-glycero-3-phosphoethanolamine (DOPE), sphingomyelins (SM), 1,2‐diacylglyceryl-3- O-4'-(N,N,N-trimethyl)-homoserine (DGTS), ceramides, cholesterol, steroids, such as sterols and their derivatives. In some embodiments, a helper lipid is or comprises phosphatidylcholines, phosphatidylethanolamines, phosphatidylglycerols, phosphatidic acids, phosphatidylserines or sphingomyelin. In some embodiments, a helper lipid is or comprises diacylphosphatidylcholines, such as distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dimyristoylphosphatidylcholine (DMPC), dipentadecanoylphosphatidylcholine, dilauroylphosphatidylcholine, dipalmitoylphosphatidylcholine (DPPC), diarachidoylphosphatidylcholine (DAPC), dibehenoylphosphatidylcholine (DBPC), ditricosanoylphosphatidylcholine (DTPC), dilignoceroylphatidylcholine (DLPC), palmitoyloleoyl-phosphatidylcholine (POPC), 1,2- di-O-octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), 1-oleoyl-2-
cholesterylhemisuccinoyl-sn-glycero-3-phosphocholine (OChemsPC), 1-hexadecyl-sn- glycero-3-phosphocholine (C16 Lyso PC) and phosphatidylethanolamines, including, for example diacylphosphatidylethanolamines, such as dioleoylphosphatidylethanolamine (DOPE), distearoyl-phosphatidylethanolamine (DSPE), dipalmitoyl- phosphatidylethanolamine (DPPE), dimyristoyl-phosphatidylethanolamine (DMPE), dilauroyl-phosphatidylethanolamine (DLPE), diphytanoyl-phosphatidylethanolamine (DPyPE), 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine (DOPG), 1,2- dipalmitoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DPPG), 1-palmitoyl-2-oleoyl-sn- glycero-3-phosphoethanolamine (POPE), N-palmitoyl-D-erythro- sphingosylphosphorylcholine (SM). In some embodiments, a helper lipid is selected from the group consisting of DSPC, DOPC, DMPC, DPPC, POPC, DOPE, DOPG, DPPG, POPE, DPPE, DMPE, DSPE, and SM. In some embodiments, the neutral lipid is selected from the group consisting of DSPC, DPPC, DMPC, DOPC, POPC, DOPE and SM. In some embodiments, the neutral lipid is DSPC. Helper lipids may be synthetic or naturally derived. Other helper lipids suitable for use in a lipid nanoparticle are described in WO2021/026358, WO 2017/075531, and WO 2018/081480, the entire contents of each of which are incorporated herein by reference in their entirety. In some embodiments, a lipid nanoparticle comprises about 5 to about 15 mol% of a phospholipid. In some embodiments, a lipid nanoparticle comprises about 8 to about 12 mol% of a phospholipid. In some embodiments, a lipid nanoparticle comprises about 10 mol% of a phospholipid. In some embodiments, a lipid nanoparticle comprises about 5 to about 15 mol% of DSPC. In some embodiments, a lipid nanoparticle comprises about 8 to about 12 mol% of DSPC. In some embodiments, a lipid nanoparticle comprises about 10 mol% of DSPC. (ii) Polymer-conjugated lipids As described herein, LNPs of the present disclosure comprise a polymer-conjugated lipid. In some embodiments, a polymer conjugated lipid is a lipid conjugated to polyethylene glycol (PEG-lipid). In some embodiments, a PEG lipid is selected from pegylated diacylglycerol (PEG-DAG) such as l-(monomethoxy-polyethyleneglycol)- 2,3- dimyristoylglycerol (PEG-DMG) (e.g., 1,2-dimyristoyl-rac-glycero-3- methoxypolyethylene glycol-2000 (PEG2000-DMG)), a pegylated phosphatidylethanoloamine (PEG-PE), a PEG succinate diacylglycerol (PEG-S-DAG) such as 4-O-(2',3'-di(tetradecanoyloxy)propyl-1-O-(ω-
methoxy(polyethoxy)ethyl)butanedioate (PEG-S-DMG), 1,2-distearoyl-sn-glycero-3- phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG2000 amine), a pegylated ceramide (PEG-cer), or a PEG dialkoxypropylcarbamate such as ω-m ethoxy (polyethoxy)ethyl-N-(2,3-di(tetradecanoxy)propyl)carbamate, 2,3- di(tetradecanoxy)propyl-1-Ν-(ω methoxy(polyethoxy)ethyl)carbamate, and N-palmitoyl- sphingosine-1-{succinyl[methoxy(polyethylene glycol)2000]} (C16-PEG2000 ceramide or C16Cer-PEG2K). In some embodiments, a PEG-lipid is PEG2000-DMG:
 In some embodiments, a PEG-lipid is DMG-PEG. In some embodiments, a PEG-lipid is provided in WO2021/026358, WO 2017/075531, or WO 2018/081480, each of which is incorporated by reference in its entirety. In some embodiments, a polymer-conjugated lipid is 2-[(polyethylene glycol)-2000]-N,N- ditetradecylacetamide (ALC-0159). In some embodiments, a polymer-conjugated lipid is:
In some embodiments, a PEG-lipid is DMG-PEG. In some embodiments, a PEG-lipid is provided in WO2021/026358, WO 2017/075531, or WO 2018/081480, each of which is incorporated by reference in its entirety. In some embodiments, a polymer-conjugated lipid is 2-[(polyethylene glycol)-2000]-N,N- ditetradecylacetamide (ALC-0159). In some embodiments, a polymer-conjugated lipid is:
 or a pharmaceutically acceptable salt thereof, where n’ is an integer from about 45 to about 50. In some embodiments, a polymer conjugated lipid is C16 PEG2000:
or a pharmaceutically acceptable salt thereof, where n’ is an integer from about 45 to about 50. In some embodiments, a polymer conjugated lipid is C16 PEG2000:
 In some embodiments, the PEG- lipid has the following structure: wherein n in the formula above is from 30 to 60, such as about 50. In one embodiment, the PEG-conjugated lipid (pegylated lipid) is PEG2000-C-DMA which preferably refers to 3-N-[(ω-methoxy poly(ethylene glycol)2000)carbamoyl]-1,2-dimyristyloxy-propylamine
(MPEG-(2 kDa)-C-DMA) or methoxy-polyethylene glycol-2,3- bis(tetradecyloxy)propylcarbamate (2000). In some embodiments, a PEG-lipid is selected from PEG-DAG, PEG-PE, PEG-S-DAG, PEG2000-DMG, PEG-S-DMG, PEG-cer, a PEG dialkyoxypropylcarbamate (e.g., w- methoxy(polyethoxy)ethyl-N-(2,3-di(tetradecanoxy)propyl)carbamate or 2,3- di(tetradecanoxy)propyl-N-(w methoxy(polyethoxy)ethyl)carbamate), ALC-0159, and combinations thereof. In some embodiments, a PEG-lipid is ALC-0159 or PEG2000- DMG. In some embodiments, a PEG-lipid is ALC-0159. In some embodiments, a PEG- lipid is PEG2000-DMG. In some embodiments, a PEG-lipid is PEG-DAG. In some embodiments, a PEG-lipid is PEG-PE. In some embodiments, a PEG-lipid is PEG-S- DAG. In some embodiments, a PEG-lipid is PEG-cer. In some embodiments, a PEG- lipid is a PEG dialkyoxypropylcarbamate. In some embodiments, a PEG group that is part of a PEG-lipid has, on average in a composition comprising one or more PEG-lipid molecules, a weight average molecular weight (Mw) of about 2000 g/mol. In some embodiments, a polymer-conjugated lipid is a polysarcosine-conjugated lipid, also referred to herein as sarcosinylated lipid or pSar-lipid. The term "sarcosinylated lipid" refers to a molecule comprising both a lipid portion and a polysarcosine (poly(N- methylglycine) portion. In some embodiments, a polymer-conjugated lipid is a polyoxazoline (POX)-conjugated and/or polyoxazine (POZ)-conjugated lipid, also referred to herein as a conjugate of a POX and/or POZ polymer and one or more hydrophobic chains or as oxazolinylated and/or oxazinylated lipid or POX- and/or POZ-lipid. The term "oxazolinylated lipid" or "POX-lipid" refers to a molecule comprising both a lipid portion and a polyoxazoline portion. The term "oxazinylated lipid" or "POZ-lipid" refers to a molecule comprising both a lipid portion and a polyoxazine portion. The term "oxazolinylated/oxazinylated lipid" or "POX/POZ-lipid" or "POXZ-lipid" refers to a molecule comprising both a lipid portion and a portion of a copolymer of polyoxazoline and polyoxazine. In some embodiments, an LNP described herein may comprise a sarcosinylated lipid. In some embodiments, the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein comprise a sarcosinylated lipid and are substantially free of a pegylated lipid (or do not contain a pegylated lipid). In some embodiments, the nucleic acid compositions (such as DNA or RNA compositions) described herein comprise a cationic/cationically ionizable lipid as described herein and a sarcosinylated lipid (pSAR-conjugated lipid). In some
embodiments, the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein may further comprise a neutral lipid (e.g., a phospholipid, cholesterol or a derivative thereof) or a combination of neutral lipids (e.g., a phospholipid, and cholesterol or a derivative thereof). In some embodiments, the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein comprise a cationic/cationically ionizable lipid as described herein, a sarcosinylated lipid, a neutral lipid (e.g., a phospholipid), and cholesterol or a derivative thereof. In some embodiments, the phospholipid is DSPC. In some embodiments of the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein which comprise a sarcosinylated lipid, said compositions are substantially free of a pegylated lipid (or do not contain a pegylated lipid). In some embodiments, the sarcosinylated lipid comprises between 2 and 200 sarcosine units, such as between 5 and 100 sarcosine units, between 10 and 50 sarcosine units, between 15 and 40 sarcosine units, e.g., about 23 sarcosine units. In some embodiments, the sarcosinylated lipid comprises the structure of the following general formula (XVII):
In some embodiments, the PEG- lipid has the following structure: wherein n in the formula above is from 30 to 60, such as about 50. In one embodiment, the PEG-conjugated lipid (pegylated lipid) is PEG2000-C-DMA which preferably refers to 3-N-[(ω-methoxy poly(ethylene glycol)2000)carbamoyl]-1,2-dimyristyloxy-propylamine
(MPEG-(2 kDa)-C-DMA) or methoxy-polyethylene glycol-2,3- bis(tetradecyloxy)propylcarbamate (2000). In some embodiments, a PEG-lipid is selected from PEG-DAG, PEG-PE, PEG-S-DAG, PEG2000-DMG, PEG-S-DMG, PEG-cer, a PEG dialkyoxypropylcarbamate (e.g., w- methoxy(polyethoxy)ethyl-N-(2,3-di(tetradecanoxy)propyl)carbamate or 2,3- di(tetradecanoxy)propyl-N-(w methoxy(polyethoxy)ethyl)carbamate), ALC-0159, and combinations thereof. In some embodiments, a PEG-lipid is ALC-0159 or PEG2000- DMG. In some embodiments, a PEG-lipid is ALC-0159. In some embodiments, a PEG- lipid is PEG2000-DMG. In some embodiments, a PEG-lipid is PEG-DAG. In some embodiments, a PEG-lipid is PEG-PE. In some embodiments, a PEG-lipid is PEG-S- DAG. In some embodiments, a PEG-lipid is PEG-cer. In some embodiments, a PEG- lipid is a PEG dialkyoxypropylcarbamate. In some embodiments, a PEG group that is part of a PEG-lipid has, on average in a composition comprising one or more PEG-lipid molecules, a weight average molecular weight (Mw) of about 2000 g/mol. In some embodiments, a polymer-conjugated lipid is a polysarcosine-conjugated lipid, also referred to herein as sarcosinylated lipid or pSar-lipid. The term "sarcosinylated lipid" refers to a molecule comprising both a lipid portion and a polysarcosine (poly(N- methylglycine) portion. In some embodiments, a polymer-conjugated lipid is a polyoxazoline (POX)-conjugated and/or polyoxazine (POZ)-conjugated lipid, also referred to herein as a conjugate of a POX and/or POZ polymer and one or more hydrophobic chains or as oxazolinylated and/or oxazinylated lipid or POX- and/or POZ-lipid. The term "oxazolinylated lipid" or "POX-lipid" refers to a molecule comprising both a lipid portion and a polyoxazoline portion. The term "oxazinylated lipid" or "POZ-lipid" refers to a molecule comprising both a lipid portion and a polyoxazine portion. The term "oxazolinylated/oxazinylated lipid" or "POX/POZ-lipid" or "POXZ-lipid" refers to a molecule comprising both a lipid portion and a portion of a copolymer of polyoxazoline and polyoxazine. In some embodiments, an LNP described herein may comprise a sarcosinylated lipid. In some embodiments, the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein comprise a sarcosinylated lipid and are substantially free of a pegylated lipid (or do not contain a pegylated lipid). In some embodiments, the nucleic acid compositions (such as DNA or RNA compositions) described herein comprise a cationic/cationically ionizable lipid as described herein and a sarcosinylated lipid (pSAR-conjugated lipid). In some
embodiments, the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein may further comprise a neutral lipid (e.g., a phospholipid, cholesterol or a derivative thereof) or a combination of neutral lipids (e.g., a phospholipid, and cholesterol or a derivative thereof). In some embodiments, the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein comprise a cationic/cationically ionizable lipid as described herein, a sarcosinylated lipid, a neutral lipid (e.g., a phospholipid), and cholesterol or a derivative thereof. In some embodiments, the phospholipid is DSPC. In some embodiments of the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein which comprise a sarcosinylated lipid, said compositions are substantially free of a pegylated lipid (or do not contain a pegylated lipid). In some embodiments, the sarcosinylated lipid comprises between 2 and 200 sarcosine units, such as between 5 and 100 sarcosine units, between 10 and 50 sarcosine units, between 15 and 40 sarcosine units, e.g., about 23 sarcosine units. In some embodiments, the sarcosinylated lipid comprises the structure of the following general formula (XVII):
 wherein s is the number of sarcosine units. In some embodiments, the sarcosinylated lipid comprises the structure of the following general formula (XVIII):
wherein s is the number of sarcosine units. In some embodiments, the sarcosinylated lipid comprises the structure of the following general formula (XVIII):
 wherein one of R21 and R22 comprises a hydrophobic group and the other is H, a hydrophilic group or a functional group optionally comprising a targeting moiety; and x is the number of sarcosine units. In some embodiments, an LNP herein may comprise an oxazolinylated and/or/oxazinylated lipid. In some embodiments, the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein comprise
an oxazolinylated and/or/oxazinylated lipid and are substantially free of a pegylated lipid (or do not contain a pegylated lipid). In some embodiments, a polymer-conjugated lipid comprises monomers of 2-(2-(2- aminoethoxy)ethoxy)acetic acid. In some embodiments, a polymer-conjugated lipid comprises monomers of 2-(2-(2-aminoethoxy)ethoxy)acetic acid. In some embodiments, the polymer of the polymer-conjugated lipid is or comprises poly-2-(2-(2- aminoethoxy)ethoxy)acetic acid (pAEEA) or poly-2-(2-(2- methylaminoethoxy)ethoxy)acetic acid (pMAEEA), or a derivative thereof, as defined herein. In some embodiments, the polymer comprises the following general formula:
wherein one of R21 and R22 comprises a hydrophobic group and the other is H, a hydrophilic group or a functional group optionally comprising a targeting moiety; and x is the number of sarcosine units. In some embodiments, an LNP herein may comprise an oxazolinylated and/or/oxazinylated lipid. In some embodiments, the nucleic acid compositions (such as DNA or RNA compositions, especially mRNA compositions) described herein comprise
an oxazolinylated and/or/oxazinylated lipid and are substantially free of a pegylated lipid (or do not contain a pegylated lipid). In some embodiments, a polymer-conjugated lipid comprises monomers of 2-(2-(2- aminoethoxy)ethoxy)acetic acid. In some embodiments, a polymer-conjugated lipid comprises monomers of 2-(2-(2-aminoethoxy)ethoxy)acetic acid. In some embodiments, the polymer of the polymer-conjugated lipid is or comprises poly-2-(2-(2- aminoethoxy)ethoxy)acetic acid (pAEEA) or poly-2-(2-(2- methylaminoethoxy)ethoxy)acetic acid (pMAEEA), or a derivative thereof, as defined herein. In some embodiments, the polymer comprises the following general formula:
 wherein X11 and X12 taken together are optionally substituted amide, optionally substituted thioamide or ester; Y is -CH2-, -(CH2)2-, or -(CH2)3-; z is 2 to 24; and n is 1 to 100. In some embodiments, (i) when X11 is -C(O)- then X12 is -NR10-; (ii) when X11 is -NR10- then X12 is -C(O)-; (iii) when X11 is -C(S)- then X12 is -NR1-; (iv) when X11 is -NR1- then X12 is -C(S)-; (v) when X11 is -C(O)- then X12 is -O-; or (vi) when X11 is -O- then X12 is - C(O)-; wherein R10 is hydrogen or C1-8 alkyl. In some embodiments, X11 is -C(O)- and X12 is -NR10-, wherein R10 is hydrogen or C1-8 alkyl. In some embodiments, X11 is -C(O)- and X12 is -NR10-, wherein R10 is hydrogen or methyl. In some embodiments, X11 is -C(O)- and X12 is -NR10-, wherein R10 is hydrogen. In some embodiments, Y is -CH2- or -(CH2)2- . In some embodiments, Y is -CH2-. In some embodiments, the polymer comprises the following general formula: wherein R10 is hydrogen or C1-8 alkyl; z is 2 to 24; and n is 1 to 100. In some embodiments of the above formulas, z is 2 to 10. In some embodiments, z is 2 to 5. In some embodiments, z is 2. In some embodiments of the above formulas, R10 is hydrogen or methyl. In some embodiments, R10 is hydrogen.
In some embodiments, a polymer conjugated lipid comprises “n” monomers of the following structure:
wherein X11 and X12 taken together are optionally substituted amide, optionally substituted thioamide or ester; Y is -CH2-, -(CH2)2-, or -(CH2)3-; z is 2 to 24; and n is 1 to 100. In some embodiments, (i) when X11 is -C(O)- then X12 is -NR10-; (ii) when X11 is -NR10- then X12 is -C(O)-; (iii) when X11 is -C(S)- then X12 is -NR1-; (iv) when X11 is -NR1- then X12 is -C(S)-; (v) when X11 is -C(O)- then X12 is -O-; or (vi) when X11 is -O- then X12 is - C(O)-; wherein R10 is hydrogen or C1-8 alkyl. In some embodiments, X11 is -C(O)- and X12 is -NR10-, wherein R10 is hydrogen or C1-8 alkyl. In some embodiments, X11 is -C(O)- and X12 is -NR10-, wherein R10 is hydrogen or methyl. In some embodiments, X11 is -C(O)- and X12 is -NR10-, wherein R10 is hydrogen. In some embodiments, Y is -CH2- or -(CH2)2- . In some embodiments, Y is -CH2-. In some embodiments, the polymer comprises the following general formula: wherein R10 is hydrogen or C1-8 alkyl; z is 2 to 24; and n is 1 to 100. In some embodiments of the above formulas, z is 2 to 10. In some embodiments, z is 2 to 5. In some embodiments, z is 2. In some embodiments of the above formulas, R10 is hydrogen or methyl. In some embodiments, R10 is hydrogen.
In some embodiments, a polymer conjugated lipid comprises “n” monomers of the following structure:
 In some embodiments of the above formulas, n is 5 to 50. In some embodiments, n is 5 to 25. In some embodiments, n is 7 to 14. In some embodiments, n is 10 to 25. In some embodiments, n is 14 to 17. In some embodiments, n is 8 or 14. In some embodiments, a polymer conjugated lipid comprises monomers of the following structure: .
In some embodiments of the above formulas, n is 5 to 50. In some embodiments, n is 5 to 25. In some embodiments, n is 7 to 14. In some embodiments, n is 10 to 25. In some embodiments, n is 14 to 17. In some embodiments, n is 8 or 14. In some embodiments, a polymer conjugated lipid comprises monomers of the following structure: .
 In some embodiments, a polymer conjugated lipid is selected from the table below:
In some embodiments, a polymer conjugated lipid is selected from the table below:


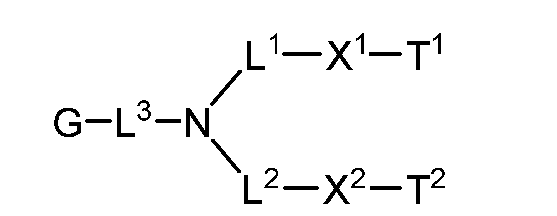










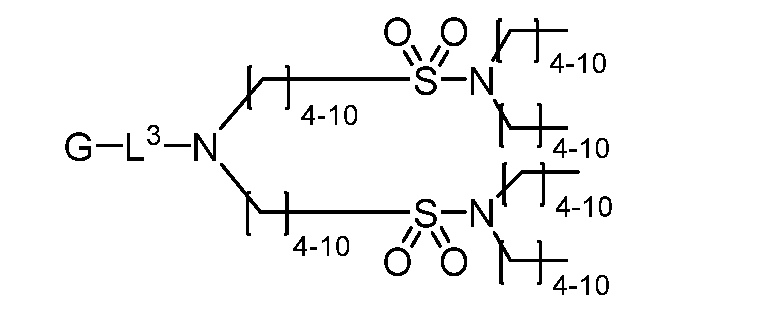







Example 1: Preparation and characterization of sulfonamide lipids
Synthesis of Fragments:
The present example provides a method preparing tail-end-fragments with tail-linkers via unoxidized junction residue “-S-“ having an oxidation state of -2. (Figure 79)



















Synthesis of BL-205 and BL-206
 9-hydroxynonane-1-sulfonate.NaBr (20) 9-bromononan-1-ol (9.6 g, 1 Eq, 43 mmol) was suspended in water (0.11 L), sodium sulfite (6.5 g, 1.2 Eq, 52 mmol) was added and the mixture was stirred overnight at 95°C. The mixture was cooled down, washed with MTBE, and the water layer was evaporated to dryness to afford sodium 9-hydroxynonane-1-sulfonate bromide.NaBr (18.3 g, 52.4 mmol). Product was used without further purification. 9-chlorononane-1-sulfonyl chloride (21) 9-hydroxynonane-1-sulfonate.NaBr salt (6.18 g, 1 Eq, 17.7 mmol) was suspended in POCl3 (30 g, 18 mL, 11 Eq, 0.19 mol) and the reaction was stirred at (initially) 110°C. After 70h, the reaction was concentrated and co-evaporated with toluene and DCM to yield the crude 9-chlorononane-1-sulfonyl chloride. Cl:Br ratio 1:0.35. Product was used without further purification. N-butyl-9-chloro-N-hexylnonane-1-sulfonamide (22) A solution of N-butylhexan-1-amine (2.09 g, 0.75 Eq, 13.3 mmol), triethylamine (5.37 g, 7.38 mL, 3 Eq, 53.1 mmol) in DCM (32 mL) was cooled to 0°C, next 9-chlorononane-1- sulfonyl chloride/bromide (4.62 g, 1 Eq, ~17.7 mmol) (crude mixture) in DCM (22 mL) was slowly added and the reaction was stirred at rt for 4 days. The reaction mixture was concentrated in vacuo, re-suspended in DCM/MeOH, coated on hydromatrix and purified
by FCC using 1-25% EtOAc in heptane to obtain a mixture of Cl/Br N-butyl-9-halogen- N-hexylnonane-1-sulfonamide (1.5 gram), ratio Cl/Br 1:1 according to NMR. BL-205 2-aminoethan-1-ol (42.1 mg, 1 Eq, 689 μmol), N-butyl-9-chloro-N-hexylnonane-1- sulfonamide (500 mg, 1.9 Eq, 1.31 mmol), K2CO3 (95.2 mg, 1 Eq, 689 μmol) and KI (114 mg, 1 Eq, 689 μmol) were suspended in a mixture of MeCN (3.27 mL) and CPME (3.27 mL) in a sealed tube and heated to 120 °C overnight. The mixture was evaporated, taken up in EtOAc/Water and extracted using EtOAc (2x), washed with brine, dried (Na2SO4) and evaporated to dryness. Purification by FCC using 0-100% EtOAc in heptane afforded 9,9'-((2- hydroxyethyl)azanediyl)bis(N-butyl-N-hexylnonane-1-sulfonamide) (244 mg, 324 μmol, 47.1 %). tert-butyl (4-(bis(9-(N-butyl-N-hexylsulfamoyl)nonyl)amino)butyl)carbamate (23) To a solution of N-butyl-9-chloro-N-hexylnonane-1-sulfonamide (1.14 g, 3 Eq, 2.98 mmol) and tert-butyl (4-aminobutyl)carbamate (187 mg, 190 μL, 1 Eq, 993 μmol) in a mixture CPME (4.97 mL) MeCN (4.97 mL) was added KI (660 mg, 4 Eq, 3.97 mmol), K2CO3 (686 mg, 5 Eq, 4.97 mmol) and this was heated to 90 °C and stirred overnight. The reaction was stopped, filtered over celite washed 2x with heptane and concentrated under reduced pressure to give crude product which was coated on hydromatrix and purified using FCC using 5-45% EtOAc:EtOH (3:1) in heptane to afford tert-butyl (4- (bis(9-(N-butyl-N-hexylsulfamoyl)nonyl)amino)butyl)carbamate (550 mg, 625 μmol, 63.0 %). 9,9'-((4-aminobutyl)azanediyl)bis(N-butyl-N-hexylnonane-1-sulfonamide) hydrochloride (24) To a solution of tert-butyl (4-(bis(9-(N-butyl-N- hexylsulfamoyl)nonyl)amino)butyl)carbamate (550 mg, 1 Eq, 625 μmol) in DCM (1.0 mL) was added HCl (456 mg, 3.13 mL, 4.0 molar, 20 Eq, 12.5 mmol) and this was stirred for 2 h and subsequently evaporated in vacuo to give 9,9'-((4-aminobutyl)azanediyl)bis(N- butyl-N-hexylnonane-1-sulfonamide) hydrochloride (495 mg, 607 μmol, 97.0 %). Product was used without further purification. BL-206
To a solution of 9,9'-((4-aminobutyl)azanediyl)bis(N-butyl-N-hexylnonane-1- sulfonamide) hydrochloride (350 mg, 1 Eq, 429 μmol) and Et3N (130 mg, 179 μL, 3 Eq, 1.29 mmol) in DCM (5.0 mL) was added dropwise at 0 °C thiophosgene (69.6 mg, 46.4 μL, 85% Wt, 1.2 Eq, 515 μmol) and the reaction mixture was allowed to stir at rt for 5 h whereafter, extra thiophosgene (29.6 mg, 19.7 μL, 0.6 Eq, 257 μmol) was added and the reaction was stirred for 3 h. The reaction was cooled to 0°C and a solution of dimethylamine in THF (193 mg, 2.15 mL, 2.0 molar, 10 Eq, 4.29 mmol) was added. The mixture was heated to 40 °C and stirred for 1 hour. The solvent was evaporated in vacuo and the crude product was coated on hydromatrix. Purification by FCC using 5-50% EtOAc:EtOH (3:1) in heptane afforded 9,9'-((4-(3,3- dimethylthioureido)butyl)azanediyl)bis(N-butyl-N-hexylnonane-1-sulfonamide) (340 mg, 392 μmol, 80.1 %). Synthesis of BL-207 and BL-208
9-hydroxynonane-1-sulfonate.NaBr (20) 9-bromononan-1-ol (9.6 g, 1 Eq, 43 mmol) was suspended in water (0.11 L), sodium sulfite (6.5 g, 1.2 Eq, 52 mmol) was added and the mixture was stirred overnight at 95°C. The mixture was cooled down, washed with MTBE, and the water layer was evaporated to dryness to afford sodium 9-hydroxynonane-1-sulfonate bromide.NaBr (18.3 g, 52.4 mmol). Product was used without further purification. 9-chlorononane-1-sulfonyl chloride (21) 9-hydroxynonane-1-sulfonate.NaBr salt (6.18 g, 1 Eq, 17.7 mmol) was suspended in POCl3 (30 g, 18 mL, 11 Eq, 0.19 mol) and the reaction was stirred at (initially) 110°C. After 70h, the reaction was concentrated and co-evaporated with toluene and DCM to yield the crude 9-chlorononane-1-sulfonyl chloride. Cl:Br ratio 1:0.35. Product was used without further purification. N-butyl-9-chloro-N-hexylnonane-1-sulfonamide (22) A solution of N-butylhexan-1-amine (2.09 g, 0.75 Eq, 13.3 mmol), triethylamine (5.37 g, 7.38 mL, 3 Eq, 53.1 mmol) in DCM (32 mL) was cooled to 0°C, next 9-chlorononane-1- sulfonyl chloride/bromide (4.62 g, 1 Eq, ~17.7 mmol) (crude mixture) in DCM (22 mL) was slowly added and the reaction was stirred at rt for 4 days. The reaction mixture was concentrated in vacuo, re-suspended in DCM/MeOH, coated on hydromatrix and purified
by FCC using 1-25% EtOAc in heptane to obtain a mixture of Cl/Br N-butyl-9-halogen- N-hexylnonane-1-sulfonamide (1.5 gram), ratio Cl/Br 1:1 according to NMR. BL-205 2-aminoethan-1-ol (42.1 mg, 1 Eq, 689 μmol), N-butyl-9-chloro-N-hexylnonane-1- sulfonamide (500 mg, 1.9 Eq, 1.31 mmol), K2CO3 (95.2 mg, 1 Eq, 689 μmol) and KI (114 mg, 1 Eq, 689 μmol) were suspended in a mixture of MeCN (3.27 mL) and CPME (3.27 mL) in a sealed tube and heated to 120 °C overnight. The mixture was evaporated, taken up in EtOAc/Water and extracted using EtOAc (2x), washed with brine, dried (Na2SO4) and evaporated to dryness. Purification by FCC using 0-100% EtOAc in heptane afforded 9,9'-((2- hydroxyethyl)azanediyl)bis(N-butyl-N-hexylnonane-1-sulfonamide) (244 mg, 324 μmol, 47.1 %). tert-butyl (4-(bis(9-(N-butyl-N-hexylsulfamoyl)nonyl)amino)butyl)carbamate (23) To a solution of N-butyl-9-chloro-N-hexylnonane-1-sulfonamide (1.14 g, 3 Eq, 2.98 mmol) and tert-butyl (4-aminobutyl)carbamate (187 mg, 190 μL, 1 Eq, 993 μmol) in a mixture CPME (4.97 mL) MeCN (4.97 mL) was added KI (660 mg, 4 Eq, 3.97 mmol), K2CO3 (686 mg, 5 Eq, 4.97 mmol) and this was heated to 90 °C and stirred overnight. The reaction was stopped, filtered over celite washed 2x with heptane and concentrated under reduced pressure to give crude product which was coated on hydromatrix and purified using FCC using 5-45% EtOAc:EtOH (3:1) in heptane to afford tert-butyl (4- (bis(9-(N-butyl-N-hexylsulfamoyl)nonyl)amino)butyl)carbamate (550 mg, 625 μmol, 63.0 %). 9,9'-((4-aminobutyl)azanediyl)bis(N-butyl-N-hexylnonane-1-sulfonamide) hydrochloride (24) To a solution of tert-butyl (4-(bis(9-(N-butyl-N- hexylsulfamoyl)nonyl)amino)butyl)carbamate (550 mg, 1 Eq, 625 μmol) in DCM (1.0 mL) was added HCl (456 mg, 3.13 mL, 4.0 molar, 20 Eq, 12.5 mmol) and this was stirred for 2 h and subsequently evaporated in vacuo to give 9,9'-((4-aminobutyl)azanediyl)bis(N- butyl-N-hexylnonane-1-sulfonamide) hydrochloride (495 mg, 607 μmol, 97.0 %). Product was used without further purification. BL-206
To a solution of 9,9'-((4-aminobutyl)azanediyl)bis(N-butyl-N-hexylnonane-1- sulfonamide) hydrochloride (350 mg, 1 Eq, 429 μmol) and Et3N (130 mg, 179 μL, 3 Eq, 1.29 mmol) in DCM (5.0 mL) was added dropwise at 0 °C thiophosgene (69.6 mg, 46.4 μL, 85% Wt, 1.2 Eq, 515 μmol) and the reaction mixture was allowed to stir at rt for 5 h whereafter, extra thiophosgene (29.6 mg, 19.7 μL, 0.6 Eq, 257 μmol) was added and the reaction was stirred for 3 h. The reaction was cooled to 0°C and a solution of dimethylamine in THF (193 mg, 2.15 mL, 2.0 molar, 10 Eq, 4.29 mmol) was added. The mixture was heated to 40 °C and stirred for 1 hour. The solvent was evaporated in vacuo and the crude product was coated on hydromatrix. Purification by FCC using 5-50% EtOAc:EtOH (3:1) in heptane afforded 9,9'-((4-(3,3- dimethylthioureido)butyl)azanediyl)bis(N-butyl-N-hexylnonane-1-sulfonamide) (340 mg, 392 μmol, 80.1 %). Synthesis of BL-207 and BL-208
 Ethyl 7-(N,N-dioctylsulfamoyl)heptanoate (25) To a solution of dioctylamine (3.437 g, 4.30 mL, 3 Eq, 14.23 mmol) in DCM (13.0 mL) at 0 °C, was added dropwise a cold (cooled to 0 °C) solution of ethyl 7-
(chlorosulfonyl)heptanoate (1400 mg, 87% Wt, 1 Eq, 4.744 mmol) in DCM (10 mL). The mixture was stirred for 20 min. The reaction mixture was coated on hydromatrix and purified by FCC. N,N-dioctyl-7-oxoheptane-1-sulfonamide (26) Ethyl 7-(N,N-dioctylsulfamoyl)heptanoate (1100 mg, 1 Eq, 2.382 mmol) was stripped twice with toluene, dissolved in Toluene (25 mL) under an Argon atmosphere, and cooled to -78 °C. Next a solution of DIBAL-H 1M in Toluene (372.7 mg, 2.620 mL, 1.0 molar, 1.1 Eq, 2.620 mmol) in Toluene (5.0 mL) was added dropwise while keeping the temp - 78 °C. The mixture was left stirring for 1 hour, the reaction was quenched using Methanol (1.5 mL) at -78°C and the mixture was stirred for 20 min. Next, water and 2M NaOH were added and the crude product was extracted with EtOAc affording N,N-dioctyl-7- oxoheptane-1-sulfonamide (1.35 g, 2.8 mmol, 120 %) which was used without further purification. BL-207 In a 20 mL screw capped vial, N,N-dioctyl-7-oxoheptane-1-sulfonamide (400 mg, 2.5 Eq, 958 μmol) and 4-aminobutan-1-ol (34.1 mg, 1 Eq, 383 μmol) were dissolved in DCM (3.83 mL). Next sodium triacetoxyborohydride (203 mg, 2.5 Eq, 958 μmol) was added and the mixture was stirred for 30 min. Mixture was quenched with sat. NaHCO3 and the crude product was extracted with DCM (2x), the combined DCM layers were washed with brine, dried and evaporated to dryness. The crude product was coated on hydromatrix and purified by FCC using 0-50% EtOAc:EtOH (3:1) in heptane affording 7,7'-((4-hydroxybutyl)azanediyl)bis(N,N-dioctylheptane-1-sulfonamide) (198 mg, 222 μmol, 57.9 %). tert-butyl (4-(bis(7-(N,N-dioctylsulfamoyl)heptyl)amino)butyl)carbamate (27) N,N-dioctyl-7-oxoheptane-1-sulfonamide (600 mg, 1Eq, 1.26 mmol) and tert-butyl (4- aminobutyl)carbamate (238 mg, 1 Eq, 1.26 mmol) were dissolved in DCM (5.06 mL), after 3 min sodium triacetoxyborohydride (670 mg, 2.5 Eq, 3.16 mmol) was added and the mixture was stirred overnight at rt. The solvent was evaporated in vacuo and the crude product was purified FCC using 0-25% EtOAc in heptane to afford tert-butyl (4- (bis(7-(N,N-dioctylsulfamoyl) heptyl)amino)butyl)carbamate (440 mg, 444 μmol, 35.1 %, CAD purity: 98.4%). 7,7'-((4-aminobutyl)azanediyl)bis(N,N-dioctylheptane-1-sulfonamide) hydrochloride (28) To a solution of tert-butyl (4-(bis(7-(N,N-dioctylsulfamoyl)heptyl)amino)butyl)carbamate (440 mg, 1 Eq, 444 μmol) in DCM (2.22 mL) was added 4M HCl in Dioxane (162 mg, 1.11 mL, 4.0 molar, 10 Eq, 4.44 mmol) and this was stirred overnight. The solvent was
evaporated in vacuo to give 7,7'-((4-aminobutyl)azanediyl)bis(N,N-dioctylheptane-1- sulfonamide) hydrochloride (418 mg, 450 μmol, 102 %). Product used without further purification. BL-208 To a solution of 7,7'-((4-aminobutyl)azanediyl)bis(N,N-dioctylheptane-1-sulfonamide) hydrochloride (412 mg, 1 Eq, 444 μmol) and Et3N (135 mg, 186 μL, 3 Eq, 1.33 mmol) in DCM (6.0 mL) was added dropwise at 0 °C thiophosgene (72.1 mg, 48.0 μL, 85% Wt, 1.2 Eq, 533 μmol) and the reaction mixture was allowed to stir at rt for 4 h. The reaction was cooled to 0°C and a solution of dimethylamine in THF (20.0 mg, 222 μL, 2.0 molar, 1 Eq, 444 μmol) was added after which the mixture was stirred overnight at rt. The reaction concentrated in vacuo and the crude product was coated on hydromatrix. Purification by FCC using 20-70% EtOAc:EtOH (3:1) in heptane afforded 7,7'-((4-(3,3- dimethylthioureido)butyl)azanediyl)bis(N,N-dioctylheptane-1-sulfonamide) (281 mg, 287 μmol, 64.7 %). Synthesis of BL-209
Ethyl 7-(N,N-dioctylsulfamoyl)heptanoate (25) To a solution of dioctylamine (3.437 g, 4.30 mL, 3 Eq, 14.23 mmol) in DCM (13.0 mL) at 0 °C, was added dropwise a cold (cooled to 0 °C) solution of ethyl 7-
(chlorosulfonyl)heptanoate (1400 mg, 87% Wt, 1 Eq, 4.744 mmol) in DCM (10 mL). The mixture was stirred for 20 min. The reaction mixture was coated on hydromatrix and purified by FCC. N,N-dioctyl-7-oxoheptane-1-sulfonamide (26) Ethyl 7-(N,N-dioctylsulfamoyl)heptanoate (1100 mg, 1 Eq, 2.382 mmol) was stripped twice with toluene, dissolved in Toluene (25 mL) under an Argon atmosphere, and cooled to -78 °C. Next a solution of DIBAL-H 1M in Toluene (372.7 mg, 2.620 mL, 1.0 molar, 1.1 Eq, 2.620 mmol) in Toluene (5.0 mL) was added dropwise while keeping the temp - 78 °C. The mixture was left stirring for 1 hour, the reaction was quenched using Methanol (1.5 mL) at -78°C and the mixture was stirred for 20 min. Next, water and 2M NaOH were added and the crude product was extracted with EtOAc affording N,N-dioctyl-7- oxoheptane-1-sulfonamide (1.35 g, 2.8 mmol, 120 %) which was used without further purification. BL-207 In a 20 mL screw capped vial, N,N-dioctyl-7-oxoheptane-1-sulfonamide (400 mg, 2.5 Eq, 958 μmol) and 4-aminobutan-1-ol (34.1 mg, 1 Eq, 383 μmol) were dissolved in DCM (3.83 mL). Next sodium triacetoxyborohydride (203 mg, 2.5 Eq, 958 μmol) was added and the mixture was stirred for 30 min. Mixture was quenched with sat. NaHCO3 and the crude product was extracted with DCM (2x), the combined DCM layers were washed with brine, dried and evaporated to dryness. The crude product was coated on hydromatrix and purified by FCC using 0-50% EtOAc:EtOH (3:1) in heptane affording 7,7'-((4-hydroxybutyl)azanediyl)bis(N,N-dioctylheptane-1-sulfonamide) (198 mg, 222 μmol, 57.9 %). tert-butyl (4-(bis(7-(N,N-dioctylsulfamoyl)heptyl)amino)butyl)carbamate (27) N,N-dioctyl-7-oxoheptane-1-sulfonamide (600 mg, 1Eq, 1.26 mmol) and tert-butyl (4- aminobutyl)carbamate (238 mg, 1 Eq, 1.26 mmol) were dissolved in DCM (5.06 mL), after 3 min sodium triacetoxyborohydride (670 mg, 2.5 Eq, 3.16 mmol) was added and the mixture was stirred overnight at rt. The solvent was evaporated in vacuo and the crude product was purified FCC using 0-25% EtOAc in heptane to afford tert-butyl (4- (bis(7-(N,N-dioctylsulfamoyl) heptyl)amino)butyl)carbamate (440 mg, 444 μmol, 35.1 %, CAD purity: 98.4%). 7,7'-((4-aminobutyl)azanediyl)bis(N,N-dioctylheptane-1-sulfonamide) hydrochloride (28) To a solution of tert-butyl (4-(bis(7-(N,N-dioctylsulfamoyl)heptyl)amino)butyl)carbamate (440 mg, 1 Eq, 444 μmol) in DCM (2.22 mL) was added 4M HCl in Dioxane (162 mg, 1.11 mL, 4.0 molar, 10 Eq, 4.44 mmol) and this was stirred overnight. The solvent was
evaporated in vacuo to give 7,7'-((4-aminobutyl)azanediyl)bis(N,N-dioctylheptane-1- sulfonamide) hydrochloride (418 mg, 450 μmol, 102 %). Product used without further purification. BL-208 To a solution of 7,7'-((4-aminobutyl)azanediyl)bis(N,N-dioctylheptane-1-sulfonamide) hydrochloride (412 mg, 1 Eq, 444 μmol) and Et3N (135 mg, 186 μL, 3 Eq, 1.33 mmol) in DCM (6.0 mL) was added dropwise at 0 °C thiophosgene (72.1 mg, 48.0 μL, 85% Wt, 1.2 Eq, 533 μmol) and the reaction mixture was allowed to stir at rt for 4 h. The reaction was cooled to 0°C and a solution of dimethylamine in THF (20.0 mg, 222 μL, 2.0 molar, 1 Eq, 444 μmol) was added after which the mixture was stirred overnight at rt. The reaction concentrated in vacuo and the crude product was coated on hydromatrix. Purification by FCC using 20-70% EtOAc:EtOH (3:1) in heptane afforded 7,7'-((4-(3,3- dimethylthioureido)butyl)azanediyl)bis(N,N-dioctylheptane-1-sulfonamide) (281 mg, 287 μmol, 64.7 %). Synthesis of BL-209
 Sodium 6-hydroxyhexane-1-sulfonate.Sodiumbromide 6-bromohexan-1-ol (2.0 g, 1 Eq, 11 mmol) was dissolved in Water (10 mL) and EtOH (10 mL), next sodium sulfite (2.1 g, 1.5 Eq, 17 mmol) was added and the mixture was heated to 100 °C and stirred overnight. The mixture was evaporated and co-evaporated with 2x
20 mL toluene , 2x 20 mL EtOH and 2x 20 mL MeCN to give the crude sodium 6- hydroxyhexane-1-sulfonate.Sodiumbromide (4.1 g, 11 mmol, 100 %). 6-chlorohexane-1-sulfonyl chloride Method A: To a solution of sodium 6-hydroxyhexane-1-sulfonate bromide (3.85 g, 1 Eq, 12.5 mmol) in DCM (40 mL) and DMF (0.1 mL) was added thionyl chloride (5.97 g, 3.66 mL, 4 Eq, 50.1 mmol) and this was heated to reflux and stirred for 4 hours. hereafter the reaction was stopped by concentrating it under reduced pressure to give crude product. This product was dissolved in DCM, filtered, stripped 3x with DCM and used as crude in the next reaction. Method B: A 50 mL round bottom flask was charged with sodium 6-hydroxyhexane-1-sulfonate bromide (6.7 g, 1 Eq, 22 mmol) and POCl3 (33 g, 20 mL, 10 Eq, 0.22 mol). The resulting white suspension was heated to 110 °C and stirred for 30 h whereafter the heating was stopped and the reaction was stirred over the weekend at rt. The reaction was stopped and concentrated under reduced pressure. The crude was dissolved in 15 mL DCM, filtered and stripped 3x with 20 mL DCM to give 6-chlorohexane-1-sulfonyl chloride (4.7 g, 21 mmol, 98 %) which was used without further purification. 6-chloro-N-hexyl-N-octylhexane-1-sulfonamide To a solution of N-hexyloctan-1-amine (1.5 g, 1.0 Eq, 6.8 mmol), triethylamine (1.4 g, 1.9 mL, 2 Eq, 14 mmol) in DCM (34 mL) was added 6-chlorohexane-1-sulfonyl chloride (1.5 g, 1 Eq, 6.8 mmol). The resulting mixture was stirred overnight. The reaction was diluted with 25 mL DCM, washed with water (3x 25 ml), brine, dried over Na2SO4, filtered and concentrated in vacuo to afford the crude product. Purification by FCC using 0-20% EtOAc in heptane afforded 6-chloro-N-hexyl-N-octylhexane-1-sulfonamide (110 mg, 278 μmol, 4.1 %). tert-butyl (4-(bis(6-(N-hexyl-N-octylsulfamoyl)hexyl)amino)butyl)carbamate To a solution of 6-chloro-N-hexyl-N-octylhexane-1-sulfonamide (631 mg, 3 Eq, 1.59 mmol) and tert-butyl (4-aminobutyl)carbamate (100 mg, 102 μL, 1 Eq, 531 μmol) in a mixture CPME (2.66 mL) MeCN (2.66 mL) was added KI (353 mg, 4 Eq, 2.12 mmol), K2CO3 (367 mg, 5 Eq, 2.66 mmol) and this was heated to 90 °C and stirred for 48 hours. The mixture was concentrated in vacuo, and purified by FCC using 5-50% EtOAc:EtOH (3:1) in heptane to afford tert-butyl (4-(bis(6-(N-hexyl-N- octylsulfamoyl)hexyl)amino)butyl)carbamate (300 mg, 331 μmol, 62.2 %).
6,6'-((4-aminobutyl)azanediyl)bis(N-hexyl-N-octylhexane-1-sulfonamide) hydrochloride To a solution of tert-butyl (4-(bis(6-(N-hexyl-N- octylsulfamoyl)hexyl)amino)butyl)carbamate (610 mg, 1 Eq, 672 μmol) in DCM (4.0 mL) was added 4M HCl in Dioxane (245 mg, 1.68 mL, 4.0 molar, 10 Eq, 6.72 mmol) and this was stirred overnight, and concentrated in vacuo to give the crude 6,6'-((4- aminobutyl)azanediyl)bis(N-hexyl-N-octylhexane-1-sulfonamide) hydrochloride (507 mg, 601 μmol, 89.4 %) which was used without further purification. BL-209 To a solution of 6,6'-((4-aminobutyl)azanediyl)bis(N-hexyl-N-octylhexane-1- sulfonamide) hydrochloride (507 mg, 1 Eq, 601 μmol) and TEA (182 mg, 251 μL, 3 Eq, 1.80 mmol) in DCM (5.0 mL) was added dropwise at 0 °C thiophosgene (97.5 mg, 65.0 μL, 85% Wt, 1.2 Eq, 721 μmol) and the reaction mixture was stirred overnight at rt. The reaction was cooled to 0°C and to this a solution of dimethylamine in THF (271 mg, 3.00 mL, 2.0 molar, 10 Eq, 6.01 mmol) was added and stirred for 3.5 hours. The reaction mixture was concentrated in vacuo to give crude product. Purification by FCC using 5- 75% EtOAc:EtOH (3:1) in heptane provided 6,6'-((4-(3,3- dimethylthioureido)butyl)azanediyl)bis(N-hexyl-N-octylhexane-1-sulfonamide) (99 mg, 0.10 mmol, 17 %). Synthesis of BL-210
Sodium 6-hydroxyhexane-1-sulfonate.Sodiumbromide 6-bromohexan-1-ol (2.0 g, 1 Eq, 11 mmol) was dissolved in Water (10 mL) and EtOH (10 mL), next sodium sulfite (2.1 g, 1.5 Eq, 17 mmol) was added and the mixture was heated to 100 °C and stirred overnight. The mixture was evaporated and co-evaporated with 2x
20 mL toluene , 2x 20 mL EtOH and 2x 20 mL MeCN to give the crude sodium 6- hydroxyhexane-1-sulfonate.Sodiumbromide (4.1 g, 11 mmol, 100 %). 6-chlorohexane-1-sulfonyl chloride Method A: To a solution of sodium 6-hydroxyhexane-1-sulfonate bromide (3.85 g, 1 Eq, 12.5 mmol) in DCM (40 mL) and DMF (0.1 mL) was added thionyl chloride (5.97 g, 3.66 mL, 4 Eq, 50.1 mmol) and this was heated to reflux and stirred for 4 hours. hereafter the reaction was stopped by concentrating it under reduced pressure to give crude product. This product was dissolved in DCM, filtered, stripped 3x with DCM and used as crude in the next reaction. Method B: A 50 mL round bottom flask was charged with sodium 6-hydroxyhexane-1-sulfonate bromide (6.7 g, 1 Eq, 22 mmol) and POCl3 (33 g, 20 mL, 10 Eq, 0.22 mol). The resulting white suspension was heated to 110 °C and stirred for 30 h whereafter the heating was stopped and the reaction was stirred over the weekend at rt. The reaction was stopped and concentrated under reduced pressure. The crude was dissolved in 15 mL DCM, filtered and stripped 3x with 20 mL DCM to give 6-chlorohexane-1-sulfonyl chloride (4.7 g, 21 mmol, 98 %) which was used without further purification. 6-chloro-N-hexyl-N-octylhexane-1-sulfonamide To a solution of N-hexyloctan-1-amine (1.5 g, 1.0 Eq, 6.8 mmol), triethylamine (1.4 g, 1.9 mL, 2 Eq, 14 mmol) in DCM (34 mL) was added 6-chlorohexane-1-sulfonyl chloride (1.5 g, 1 Eq, 6.8 mmol). The resulting mixture was stirred overnight. The reaction was diluted with 25 mL DCM, washed with water (3x 25 ml), brine, dried over Na2SO4, filtered and concentrated in vacuo to afford the crude product. Purification by FCC using 0-20% EtOAc in heptane afforded 6-chloro-N-hexyl-N-octylhexane-1-sulfonamide (110 mg, 278 μmol, 4.1 %). tert-butyl (4-(bis(6-(N-hexyl-N-octylsulfamoyl)hexyl)amino)butyl)carbamate To a solution of 6-chloro-N-hexyl-N-octylhexane-1-sulfonamide (631 mg, 3 Eq, 1.59 mmol) and tert-butyl (4-aminobutyl)carbamate (100 mg, 102 μL, 1 Eq, 531 μmol) in a mixture CPME (2.66 mL) MeCN (2.66 mL) was added KI (353 mg, 4 Eq, 2.12 mmol), K2CO3 (367 mg, 5 Eq, 2.66 mmol) and this was heated to 90 °C and stirred for 48 hours. The mixture was concentrated in vacuo, and purified by FCC using 5-50% EtOAc:EtOH (3:1) in heptane to afford tert-butyl (4-(bis(6-(N-hexyl-N- octylsulfamoyl)hexyl)amino)butyl)carbamate (300 mg, 331 μmol, 62.2 %).
6,6'-((4-aminobutyl)azanediyl)bis(N-hexyl-N-octylhexane-1-sulfonamide) hydrochloride To a solution of tert-butyl (4-(bis(6-(N-hexyl-N- octylsulfamoyl)hexyl)amino)butyl)carbamate (610 mg, 1 Eq, 672 μmol) in DCM (4.0 mL) was added 4M HCl in Dioxane (245 mg, 1.68 mL, 4.0 molar, 10 Eq, 6.72 mmol) and this was stirred overnight, and concentrated in vacuo to give the crude 6,6'-((4- aminobutyl)azanediyl)bis(N-hexyl-N-octylhexane-1-sulfonamide) hydrochloride (507 mg, 601 μmol, 89.4 %) which was used without further purification. BL-209 To a solution of 6,6'-((4-aminobutyl)azanediyl)bis(N-hexyl-N-octylhexane-1- sulfonamide) hydrochloride (507 mg, 1 Eq, 601 μmol) and TEA (182 mg, 251 μL, 3 Eq, 1.80 mmol) in DCM (5.0 mL) was added dropwise at 0 °C thiophosgene (97.5 mg, 65.0 μL, 85% Wt, 1.2 Eq, 721 μmol) and the reaction mixture was stirred overnight at rt. The reaction was cooled to 0°C and to this a solution of dimethylamine in THF (271 mg, 3.00 mL, 2.0 molar, 10 Eq, 6.01 mmol) was added and stirred for 3.5 hours. The reaction mixture was concentrated in vacuo to give crude product. Purification by FCC using 5- 75% EtOAc:EtOH (3:1) in heptane provided 6,6'-((4-(3,3- dimethylthioureido)butyl)azanediyl)bis(N-hexyl-N-octylhexane-1-sulfonamide) (99 mg, 0.10 mmol, 17 %). Synthesis of BL-210
 6-hydroxyhexane-1-sulfonate bromide 6-bromohexan-1-ol (2.0 g, 1 Eq, 11 mmol) was dissolved in Water (10 mL) and EtOH (10 mL), next sodium sulfite (2.1 g, 1.5 Eq, 17 mmol) was added and the mixture was heated to 100 °C and stirred overnight. The mixture was evaporated and co-evaporated with 2x 20 mL toluene, 2x 20 mL EtOH and 2x 20 mL MeCN to give the crude 6-hydroxyhexane- 1-sulfonic acid (4.1 g, 11 mmol, 100 %). 6-chlorohexane-1-sulfonyl chloride Method A: To a solution of sodium 6-hydroxyhexane-1-sulfonate bromide (3.85 g, 1 Eq, 12.5 mmol) in DCM (40 mL) and DMF (0.1 mL) was added thionyl chloride (5.97 g, 3.66 mL, 4 Eq, 50.1 mmol) and this was heated to reflux and stirred for 4 hours. hereafter the reaction was stopped by concentrating it under reduced pressure to give crude product. This was dissolved in DCM, filtered, stripped 3x with DCM and used as crude in the next reaction. Method B: A 50 mL round bottom flask was charged with sodium 6-hydroxyhexane-1-sulfonate bromide (6.7 g, 1 Eq, 22 mmol) and POCl3 (33 g, 20 mL, 10 Eq, 0.22 mol). The resulting white suspension was heated to 110 °C and stirred for 30 h whereafter the heating was stopped, and the reaction was stirred over the weekend at rt. The reaction was stopped and concentrated under reduced pressure. The crude was dissolved in 15 mL DCM, filtered and stripped 3x with 20 mL DCM to give 6-chlorohexane-1-sulfonyl chloride (4.7 g, 21 mmol, 98 %) which was used without further purification. 6-chloro-N-decyl-N-octylhexane-1-sulfonamide A solution of N-octyldecan-1-amine (2.0 g, 1 Eq, 7.4 mmol), triethylamine (2.3 g, 3.1 mL, 3 Eq, 22 mmol) in DCM (70 mL) was cooled to 0°C, next 6-chlorohexane-1-sulfonyl chloride (2.2 g, 1.33 Eq, 9.9 mmol) in 8 ml DCM was slowly added and stirred overnight. The reaction was concentrated under reduced pressure to give the crude product. This was coated on hydromatrix and purified using FCC heptane/EtOAc 0=>25% fraction containing product were combined and concentrated under reduced pressure to give 6- chloro-N-decyl-N-octylhexane-1-sulfonamide (657 mg, 1.45 mmol, 20 %). Tert-butyl (4-(bis(6-(N-decyl-N-octylsulfamoyl)hexyl)amino)butyl)carbamate To a solution of 6-chloro-N-decyl-N-octylhexane-1-sulfonamide (681 mg, 2.7 Eq, 1.51 mmol) and tert-butyl (4-aminobutyl)carbamate (105 mg, 1 Eq, 558 μmol) in a mixture CPME (2.79 mL) MeCN (2.79 mL) was added KI (370 mg, 4 Eq, 2.23 mmol), K2CO3 (385 mg, 5 Eq, 2.79 mmol) and this was heated to 90 °C and stirred overnight. The reaction was concentrated in vacuo, and purified by FCC using 5-50% EtOAc:EtOH (3:1)
in heptane to afford tertbutyl (4-((6-(N-decyl-N- octylsulfamoyl)hexyl)amino)butyl)carbamate (115 mg, 190 μmol, 34.1 %). 6,6'-((4-aminobutyl)azanediyl)bis(N-decyl-N-octylhexane-1-sulfonamide) hydrochloride To a solution of tert-butyl (4-(bis(6-(N-decyl-N- octylsulfamoyl)hexyl)amino)butyl)carbamate (265 mg, 1 Eq, 260 μmol) in DCM (3.0 mL) was added 4M HCl in Dioxane (190 mg, 1.30 mL, 4.0 molar, 20 Eq, 5.20 mmol) and this was stirred overnight, and concentrated in vacuo to give the crude 6,6'-((4- aminobutyl)azanediyl)bis(N-decyl-N-octylhexane-1-sulfonamide) hydrochloride (230 mg, 241 μmol, 92.6 %), which was used without further purification. BL-210 To a solution of 6,6'-((4-aminobutyl)azanediyl)bis(N-decyl-N-octylhexane-1- sulfonamide) hydrochloride (230 mg, 1 Eq, 241 μmol) and TEA (73.0 mg, 101 μL, 3 Eq, 722 μmol) in DCM (5.0 mL) was added dropwise at 0 °C thiophosgene (39.0 mg, 26.0 μL, 85% Wt, 1.2 Eq, 289 μmol) and the mixture was stirred overnight at rt. To the reaction was added dimethylamine in THF (108 mg, 1.20 mL, 2.0 molar, 10 Eq, 2.41 mmol) and stirred for 1.5 hour. The reaction mixture was concentrated in vacuo to give crude product. Purification by FCC using 5-75% EtOAc:EtOH (3:1) in heptane afforded 6,6'- ((4-(3,3-dimethylthioureido)butyl)azanediyl)bis(N-decyl-N-octylhexane-1-sulfonamide) (149.8 mg, 139.9 μmol, 58.2 %). Example 15: Preparation of Functionalized LNPs by a fluid-path system Ethanolic solutions containing lipids in the following ratio were prepared: cationically ionizable lipid (46.84 mol-%) (including heptadecan-9-yl 8-{(2-hydroxyethyl)[6-oxo-6- (undecyloxy)hexyl]amino}-octanoate) (SM-102) used as a control lipid), cholesterol (36.33 mol-%), DSPC (14.33 mol-%), C16-Ceramide-PEG2K (1.8 mol-%) and the targeting compound DSPE-PEG2K-ALFA tag (SEQ.ID.NO. 2; 0.2 mol%), and were diluted with additional ethanol to obtain a volume that allows a 3:1 volume ratio of aqueous nucleic phase and ethanolic lipid phase, and were loaded into a syringe. In a second syringe, a mixture of one or several nucleic acids (total nucleic acid concentration 0.15 g/L) and an acidifier (50mM) in water was loaded (typically, this comprised at least one reporter DNA and one reporter RNA). Both syringes were loaded into a microfluidic device (Precision Nanosystems Nanoassemblr Ignite®) for mixing. The product fraction was loaded into a dialysis cassette (Slide-A-Lyzer™, 10,000 MWCO) and dialyzed against the formulation buffer. To balance volume changes during dialysis, the product
is up-concentrated by centrifugal filtration; cryoprotectant (trehalose, to a final concentration of 10% (w/w)) and formulation buffer (HEPES 20Mm; pH 6) are added to achieve the desired final concentration. The obtained ALFA-tagged LNPs (‘parent LNP’) are subsequently mixed with the desired amount of a docking compound (bispecific construct comprising an anti-ALFA Tag VHH and aCD3 VHH) construct and formulation buffer and incubated for 1 h at room temperature. Figure 46 depicts the size and PDI of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C. The results of three measurements, as well as their average and error bars indicating 1x standard deviation are shown. Figure 47 depicts the percentages of transfected cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C. Figure 48 depicts the cell counts of alive cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs treated with aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, which were either applied freshly prepared or after one freeze/thaw cycle at -80°C. Example 16: Effect of Oxidation State of the Sulfur Tail on Transfection Efficiency Synthesis of BNT-71 2-Butyloctyl methanesulfonate A solution of 2-butyloc
6-hydroxyhexane-1-sulfonate bromide 6-bromohexan-1-ol (2.0 g, 1 Eq, 11 mmol) was dissolved in Water (10 mL) and EtOH (10 mL), next sodium sulfite (2.1 g, 1.5 Eq, 17 mmol) was added and the mixture was heated to 100 °C and stirred overnight. The mixture was evaporated and co-evaporated with 2x 20 mL toluene, 2x 20 mL EtOH and 2x 20 mL MeCN to give the crude 6-hydroxyhexane- 1-sulfonic acid (4.1 g, 11 mmol, 100 %). 6-chlorohexane-1-sulfonyl chloride Method A: To a solution of sodium 6-hydroxyhexane-1-sulfonate bromide (3.85 g, 1 Eq, 12.5 mmol) in DCM (40 mL) and DMF (0.1 mL) was added thionyl chloride (5.97 g, 3.66 mL, 4 Eq, 50.1 mmol) and this was heated to reflux and stirred for 4 hours. hereafter the reaction was stopped by concentrating it under reduced pressure to give crude product. This was dissolved in DCM, filtered, stripped 3x with DCM and used as crude in the next reaction. Method B: A 50 mL round bottom flask was charged with sodium 6-hydroxyhexane-1-sulfonate bromide (6.7 g, 1 Eq, 22 mmol) and POCl3 (33 g, 20 mL, 10 Eq, 0.22 mol). The resulting white suspension was heated to 110 °C and stirred for 30 h whereafter the heating was stopped, and the reaction was stirred over the weekend at rt. The reaction was stopped and concentrated under reduced pressure. The crude was dissolved in 15 mL DCM, filtered and stripped 3x with 20 mL DCM to give 6-chlorohexane-1-sulfonyl chloride (4.7 g, 21 mmol, 98 %) which was used without further purification. 6-chloro-N-decyl-N-octylhexane-1-sulfonamide A solution of N-octyldecan-1-amine (2.0 g, 1 Eq, 7.4 mmol), triethylamine (2.3 g, 3.1 mL, 3 Eq, 22 mmol) in DCM (70 mL) was cooled to 0°C, next 6-chlorohexane-1-sulfonyl chloride (2.2 g, 1.33 Eq, 9.9 mmol) in 8 ml DCM was slowly added and stirred overnight. The reaction was concentrated under reduced pressure to give the crude product. This was coated on hydromatrix and purified using FCC heptane/EtOAc 0=>25% fraction containing product were combined and concentrated under reduced pressure to give 6- chloro-N-decyl-N-octylhexane-1-sulfonamide (657 mg, 1.45 mmol, 20 %). Tert-butyl (4-(bis(6-(N-decyl-N-octylsulfamoyl)hexyl)amino)butyl)carbamate To a solution of 6-chloro-N-decyl-N-octylhexane-1-sulfonamide (681 mg, 2.7 Eq, 1.51 mmol) and tert-butyl (4-aminobutyl)carbamate (105 mg, 1 Eq, 558 μmol) in a mixture CPME (2.79 mL) MeCN (2.79 mL) was added KI (370 mg, 4 Eq, 2.23 mmol), K2CO3 (385 mg, 5 Eq, 2.79 mmol) and this was heated to 90 °C and stirred overnight. The reaction was concentrated in vacuo, and purified by FCC using 5-50% EtOAc:EtOH (3:1)
in heptane to afford tertbutyl (4-((6-(N-decyl-N- octylsulfamoyl)hexyl)amino)butyl)carbamate (115 mg, 190 μmol, 34.1 %). 6,6'-((4-aminobutyl)azanediyl)bis(N-decyl-N-octylhexane-1-sulfonamide) hydrochloride To a solution of tert-butyl (4-(bis(6-(N-decyl-N- octylsulfamoyl)hexyl)amino)butyl)carbamate (265 mg, 1 Eq, 260 μmol) in DCM (3.0 mL) was added 4M HCl in Dioxane (190 mg, 1.30 mL, 4.0 molar, 20 Eq, 5.20 mmol) and this was stirred overnight, and concentrated in vacuo to give the crude 6,6'-((4- aminobutyl)azanediyl)bis(N-decyl-N-octylhexane-1-sulfonamide) hydrochloride (230 mg, 241 μmol, 92.6 %), which was used without further purification. BL-210 To a solution of 6,6'-((4-aminobutyl)azanediyl)bis(N-decyl-N-octylhexane-1- sulfonamide) hydrochloride (230 mg, 1 Eq, 241 μmol) and TEA (73.0 mg, 101 μL, 3 Eq, 722 μmol) in DCM (5.0 mL) was added dropwise at 0 °C thiophosgene (39.0 mg, 26.0 μL, 85% Wt, 1.2 Eq, 289 μmol) and the mixture was stirred overnight at rt. To the reaction was added dimethylamine in THF (108 mg, 1.20 mL, 2.0 molar, 10 Eq, 2.41 mmol) and stirred for 1.5 hour. The reaction mixture was concentrated in vacuo to give crude product. Purification by FCC using 5-75% EtOAc:EtOH (3:1) in heptane afforded 6,6'- ((4-(3,3-dimethylthioureido)butyl)azanediyl)bis(N-decyl-N-octylhexane-1-sulfonamide) (149.8 mg, 139.9 μmol, 58.2 %). Example 15: Preparation of Functionalized LNPs by a fluid-path system Ethanolic solutions containing lipids in the following ratio were prepared: cationically ionizable lipid (46.84 mol-%) (including heptadecan-9-yl 8-{(2-hydroxyethyl)[6-oxo-6- (undecyloxy)hexyl]amino}-octanoate) (SM-102) used as a control lipid), cholesterol (36.33 mol-%), DSPC (14.33 mol-%), C16-Ceramide-PEG2K (1.8 mol-%) and the targeting compound DSPE-PEG2K-ALFA tag (SEQ.ID.NO. 2; 0.2 mol%), and were diluted with additional ethanol to obtain a volume that allows a 3:1 volume ratio of aqueous nucleic phase and ethanolic lipid phase, and were loaded into a syringe. In a second syringe, a mixture of one or several nucleic acids (total nucleic acid concentration 0.15 g/L) and an acidifier (50mM) in water was loaded (typically, this comprised at least one reporter DNA and one reporter RNA). Both syringes were loaded into a microfluidic device (Precision Nanosystems Nanoassemblr Ignite®) for mixing. The product fraction was loaded into a dialysis cassette (Slide-A-Lyzer™, 10,000 MWCO) and dialyzed against the formulation buffer. To balance volume changes during dialysis, the product
is up-concentrated by centrifugal filtration; cryoprotectant (trehalose, to a final concentration of 10% (w/w)) and formulation buffer (HEPES 20Mm; pH 6) are added to achieve the desired final concentration. The obtained ALFA-tagged LNPs (‘parent LNP’) are subsequently mixed with the desired amount of a docking compound (bispecific construct comprising an anti-ALFA Tag VHH and aCD3 VHH) construct and formulation buffer and incubated for 1 h at room temperature. Figure 46 depicts the size and PDI of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C. The results of three measurements, as well as their average and error bars indicating 1x standard deviation are shown. Figure 47 depicts the percentages of transfected cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs of aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, freshly prepared as well as after one freeze/thaw cycle at -80°C. Figure 48 depicts the cell counts of alive cells (CD14+ Monocytes, CD19+ B cells, CD4+ T cells or CD8+ T cells) within all transfected PBMCs treated with aCD3-functionalized LNPs prepared with different ionizable lipids according to the present example, which were either applied freshly prepared or after one freeze/thaw cycle at -80°C. Example 16: Effect of Oxidation State of the Sulfur Tail on Transfection Efficiency Synthesis of BNT-71 2-Butyloctyl methanesulfonate A solution of 2-butyloc
 tan-1-ol (10.0 g, 53.6 mmol, 1.00 equiv.), and triethylamine (16.2 g, 160 mmol, 3.00 equiv.) in 100 ml DCM was stirred under argon at 0°C. Methanesulfonic anhydride (14.0 g, 80.4 mmol, 1.5 equiv.) was added in portions, the reaction mixture was warmed and stirred for 2 h at rt. The progress of the reaction was monitored with TLC by using solvent mixture (n-hexane/EtOAc; 4:1). The reaction mixture was neutralized with (200 ml) NaHCO3, the organic layer was separated, and the aq. Layer was further extracted with (2 X 100 ml) DCM. The combined organic phase was washed with (2 X 100 ml) brine, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was used in the next step without any further purification.
2-Butyloctyl ethanethioacetate A solution of 2-Butyloctyl methanesulfonate (5.00 g, 20.5 mmol, 1.00 equiv.) in 10 ml DMF was stirred under argon at rt., potassium thioacetate (7.00 g, 61.5 mmol, 3.00 equiv.) was added in portions, the reaction mixture was stirred under argon at 85 °C for 12 h. The resulting reaction mixture was subsequently diluted with (200 ml) EtOAc and washed with (100 ml) 1 M HCL, (100 ml) NaHCO3, (100 ml) H2O, and (2 X 100 ml) brine. The organic phase was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane/EtOAc = 95/5). 2-butyloctane-1-thiol A solution of NaOH (2.00 g, 50.0 mmol, 2.00 equiv.) in 2.5 ml H2O was added dropwise to a solution of 2-Butyloctyl ethanethioacetate (5.00 g, 24.7 mmol, 1.00 equiv.) in 20 ml EtOH. The reaction mixture was stirred at 40°C for 2 h. The progress of the reaction was monitored with TLC by using solvent mixture (n-hexane). The reaction mixture was neutralized with 2 M HCl at 0°C, the mixture was diluted with (100 ml) diethyl ether, washed with (2 X 100 ml) H2O, and (2 X 100 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane) to afford a colorless oil compound. 9-((2-Butyloctyl)thio)nonan-1-ol In a 3-ne
tan-1-ol (10.0 g, 53.6 mmol, 1.00 equiv.), and triethylamine (16.2 g, 160 mmol, 3.00 equiv.) in 100 ml DCM was stirred under argon at 0°C. Methanesulfonic anhydride (14.0 g, 80.4 mmol, 1.5 equiv.) was added in portions, the reaction mixture was warmed and stirred for 2 h at rt. The progress of the reaction was monitored with TLC by using solvent mixture (n-hexane/EtOAc; 4:1). The reaction mixture was neutralized with (200 ml) NaHCO3, the organic layer was separated, and the aq. Layer was further extracted with (2 X 100 ml) DCM. The combined organic phase was washed with (2 X 100 ml) brine, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was used in the next step without any further purification.
2-Butyloctyl ethanethioacetate A solution of 2-Butyloctyl methanesulfonate (5.00 g, 20.5 mmol, 1.00 equiv.) in 10 ml DMF was stirred under argon at rt., potassium thioacetate (7.00 g, 61.5 mmol, 3.00 equiv.) was added in portions, the reaction mixture was stirred under argon at 85 °C for 12 h. The resulting reaction mixture was subsequently diluted with (200 ml) EtOAc and washed with (100 ml) 1 M HCL, (100 ml) NaHCO3, (100 ml) H2O, and (2 X 100 ml) brine. The organic phase was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane/EtOAc = 95/5). 2-butyloctane-1-thiol A solution of NaOH (2.00 g, 50.0 mmol, 2.00 equiv.) in 2.5 ml H2O was added dropwise to a solution of 2-Butyloctyl ethanethioacetate (5.00 g, 24.7 mmol, 1.00 equiv.) in 20 ml EtOH. The reaction mixture was stirred at 40°C for 2 h. The progress of the reaction was monitored with TLC by using solvent mixture (n-hexane). The reaction mixture was neutralized with 2 M HCl at 0°C, the mixture was diluted with (100 ml) diethyl ether, washed with (2 X 100 ml) H2O, and (2 X 100 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane) to afford a colorless oil compound. 9-((2-Butyloctyl)thio)nonan-1-ol In a 3-ne
 ck round bottom flask, 60 ml MeOH was added under argon, NaH 60% (780 mg, 19.7 mmol, 1.00 equiv.) was added in portions under argon at 0°C for 10 min, then 2-butyloctane-1-thiol (4.00 g, 19.7 mmol, 1.00 equiv.) was added under argon at rt., and the reaction mixture was stirred for 10 min, after that 9-bromonona-1-ol (4.40 g, 19.7 mmol, 1.00 equiv.) was added under argon, the reaction mixture was stirred for 3 h at
65°C. The solvent was evaporated under reduced pressure, dilute the mixture was (200 ml) n-hexane, NaH was filtered, the filtrate was collected, washed (2 X 100 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane/EtOAc; 4/1) to afford a colorless oil for the title compound. 9-((2-butyloctyl)thio)nonanal To a solution of 9-((2-Butyloctyl)thio)nonan-1-ol (2.00 g, 5.80 mmol, 1.00 equiv.), DMSO (2.00 ml, 29.0 mmol, 5.00 equiv.), and DIEA (7.00 ml, 40.0 mmol, 7.00 equiv.) in 20 ml DCM, pyridine sulfur trioxide (4.60 g, 29.0 mmol) was added under N2 and the reaction mixture was stirred for 3 h at 0°C. The reaction mixture was quenched with 100 ml 5% citric acid, exactrated with (3 X 100 ml) DCM, and (2 X 100 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane/EtOAc; 9:1) to afford a colorless oil compound. Synthesis of BNT-71, BNT-23 & BL-213 A solution of 9-((2-butyloctyl)thio)nonanal (2.00 equiv.), NH2-R (1.00 equiv.) in 5 ml DCM was stirred under argon for 30 min at rt., then NaBH(OAc)3 (4.00 equiv.) was added to the reaction mixture under argon and the reaction mixture was stirred for extra 12 h at rt. The reaction was diluted with (50 ml) DCM, washed with (3 X 50 ml) NaHCO3, (2 X 50 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; from n-hexane/EtOAc/TEA; 4/1/1% to n-hexane/EtOAc/TEA; 1/1/1% for BNT-23, -71 & BL-213) to afford the title compound as a colorless oil.
BNT-71: 1H-NMR (400 MHz, CDCL3): δ = 0.89 – 0.94 (m, 12H), 1.24 – 1.41 (m, 52 H) 1.46 – 1.63 (m, 10 H), 1.66 – 1.72 (m, 2H), 2.40 – 2.44 (m, 4H), 2.74 – 2.51 (m, 8 H), 2.65 (t, J = 8 Hz, 2H), 3.82 (t, J = 8 Hz, 2H), 4.70 (br s, 1H, OH). BNT-231H-NMR (400 MHz, CDCL3): δ = 0.85 – 0.93 (m, 12H), 1.27 – 1.46 (m, 58H), 1.51 – 1.63 (m, 4H), 2.47 – 2.51 (m, 6H), 3.00 (d, J = 4Hz, 2H), 4.16 (d, J = 4 Hz, 4H), 5.05 (br s, 1H, OH), 5.76 (ddd, J = 4, 12, 16 Hz, 2H) BL-2131H-NMR (400 MHz, CDCL3): δ = 0.89 – 0.94 (m, 12H), 1.27 – 1.42 (m, 54H), 1.46 – 1.62 (m, 10H), 2.44 – 2.51 (m, 10H), 3.08 (dd, J = 4, 8 Hz, 2H), 4.17 (dd, J = 4, 8 Hz, 2H), 5.05 (br s, OH), 5.64 – 5.70 (m, 1H), 5.87 – 5.92 (m, 1H) Scheme of BNT-23, BNT-71 & BL-213
ck round bottom flask, 60 ml MeOH was added under argon, NaH 60% (780 mg, 19.7 mmol, 1.00 equiv.) was added in portions under argon at 0°C for 10 min, then 2-butyloctane-1-thiol (4.00 g, 19.7 mmol, 1.00 equiv.) was added under argon at rt., and the reaction mixture was stirred for 10 min, after that 9-bromonona-1-ol (4.40 g, 19.7 mmol, 1.00 equiv.) was added under argon, the reaction mixture was stirred for 3 h at
65°C. The solvent was evaporated under reduced pressure, dilute the mixture was (200 ml) n-hexane, NaH was filtered, the filtrate was collected, washed (2 X 100 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane/EtOAc; 4/1) to afford a colorless oil for the title compound. 9-((2-butyloctyl)thio)nonanal To a solution of 9-((2-Butyloctyl)thio)nonan-1-ol (2.00 g, 5.80 mmol, 1.00 equiv.), DMSO (2.00 ml, 29.0 mmol, 5.00 equiv.), and DIEA (7.00 ml, 40.0 mmol, 7.00 equiv.) in 20 ml DCM, pyridine sulfur trioxide (4.60 g, 29.0 mmol) was added under N2 and the reaction mixture was stirred for 3 h at 0°C. The reaction mixture was quenched with 100 ml 5% citric acid, exactrated with (3 X 100 ml) DCM, and (2 X 100 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane/EtOAc; 9:1) to afford a colorless oil compound. Synthesis of BNT-71, BNT-23 & BL-213 A solution of 9-((2-butyloctyl)thio)nonanal (2.00 equiv.), NH2-R (1.00 equiv.) in 5 ml DCM was stirred under argon for 30 min at rt., then NaBH(OAc)3 (4.00 equiv.) was added to the reaction mixture under argon and the reaction mixture was stirred for extra 12 h at rt. The reaction was diluted with (50 ml) DCM, washed with (3 X 50 ml) NaHCO3, (2 X 50 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; from n-hexane/EtOAc/TEA; 4/1/1% to n-hexane/EtOAc/TEA; 1/1/1% for BNT-23, -71 & BL-213) to afford the title compound as a colorless oil.
BNT-71: 1H-NMR (400 MHz, CDCL3): δ = 0.89 – 0.94 (m, 12H), 1.24 – 1.41 (m, 52 H) 1.46 – 1.63 (m, 10 H), 1.66 – 1.72 (m, 2H), 2.40 – 2.44 (m, 4H), 2.74 – 2.51 (m, 8 H), 2.65 (t, J = 8 Hz, 2H), 3.82 (t, J = 8 Hz, 2H), 4.70 (br s, 1H, OH). BNT-231H-NMR (400 MHz, CDCL3): δ = 0.85 – 0.93 (m, 12H), 1.27 – 1.46 (m, 58H), 1.51 – 1.63 (m, 4H), 2.47 – 2.51 (m, 6H), 3.00 (d, J = 4Hz, 2H), 4.16 (d, J = 4 Hz, 4H), 5.05 (br s, 1H, OH), 5.76 (ddd, J = 4, 12, 16 Hz, 2H) BL-2131H-NMR (400 MHz, CDCL3): δ = 0.89 – 0.94 (m, 12H), 1.27 – 1.42 (m, 54H), 1.46 – 1.62 (m, 10H), 2.44 – 2.51 (m, 10H), 3.08 (dd, J = 4, 8 Hz, 2H), 4.17 (dd, J = 4, 8 Hz, 2H), 5.05 (br s, OH), 5.64 – 5.70 (m, 1H), 5.87 – 5.92 (m, 1H) Scheme of BNT-23, BNT-71 & BL-213
 Synthesis of BNT-74 9-((2-butyloctyl)sulfinyl)nonanal A solution of 9-((2-butyioctayl)sulfinyl)nonan-1-ol (4.15 g, 11.5 mmol, 1.00 equiv.) in 10 ml glacial AcOH was stirred at rt. H2O230% (2.00 ml, 92.0 mmol, 8.00 equiv.) was added dropwise to the reaction and the reaction mixture was stirred for 2 h at rt. The reaction mixture was neutralized with (10 ml) NaHCO3 at 0°C, the mixture was diluted (100 ml) EtOAc, washed with (2 X 50 ml) H2O, then with (2 X 50 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane/EtOAc; 1/1). BNT-74 A solution of 9-((2-butyloctyl)sulfinyl)nonanal (2.00 equiv.), 3-aminopropan-1-ol (1.00 equiv.) in 5 ml DCM was stirred under argon for 30 min . at rt., then NaBH(OAc)3 (4.00 equiv.) was added to the reaction mixture under argon and the reaction mixture was stirred for extra 12 h at rt. The reaction was diluted with (50 ml) DCM, washed with (3 X 50 ml) NaHCO3, (2 X 50 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; from n-hexane/EtOAc/TEA; 1/1/1% to /EtOAc/TEA; 99 %/1%) - 1H-NMR (400 MHz, CDCL3): δ = 0.89 – 0.94 (m, 12H), 1.27 – 1.37 (m, 42 H) 1.44 – 1.52 (m, 12 H), 1.67 – 1.73 (m, 4H), 1.75 – 1.82 (m, 6H), 1.92 – 2.00 (m, 2H), 2.38 – 2.51 (m, 6H) 2.59 – 2.74 (m, 6H), 3.82 (t, J = 4 Hz, 2H), 4.44 (br s, 1H, OH). Reaction Scheme of BNT-74
Synthesis of BNT-74 9-((2-butyloctyl)sulfinyl)nonanal A solution of 9-((2-butyioctayl)sulfinyl)nonan-1-ol (4.15 g, 11.5 mmol, 1.00 equiv.) in 10 ml glacial AcOH was stirred at rt. H2O230% (2.00 ml, 92.0 mmol, 8.00 equiv.) was added dropwise to the reaction and the reaction mixture was stirred for 2 h at rt. The reaction mixture was neutralized with (10 ml) NaHCO3 at 0°C, the mixture was diluted (100 ml) EtOAc, washed with (2 X 50 ml) H2O, then with (2 X 50 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; n-hexane/EtOAc; 1/1). BNT-74 A solution of 9-((2-butyloctyl)sulfinyl)nonanal (2.00 equiv.), 3-aminopropan-1-ol (1.00 equiv.) in 5 ml DCM was stirred under argon for 30 min . at rt., then NaBH(OAc)3 (4.00 equiv.) was added to the reaction mixture under argon and the reaction mixture was stirred for extra 12 h at rt. The reaction was diluted with (50 ml) DCM, washed with (3 X 50 ml) NaHCO3, (2 X 50 ml) brine. The organic layer was collected, dried over Na2SO4, and the solvent was evaporated under reduced pressure. The product was purified with column chromatography (SiO2; from n-hexane/EtOAc/TEA; 1/1/1% to /EtOAc/TEA; 99 %/1%) - 1H-NMR (400 MHz, CDCL3): δ = 0.89 – 0.94 (m, 12H), 1.27 – 1.37 (m, 42 H) 1.44 – 1.52 (m, 12 H), 1.67 – 1.73 (m, 4H), 1.75 – 1.82 (m, 6H), 1.92 – 2.00 (m, 2H), 2.38 – 2.51 (m, 6H) 2.59 – 2.74 (m, 6H), 3.82 (t, J = 4 Hz, 2H), 4.44 (br s, 1H, OH). Reaction Scheme of BNT-74











Claims
CLAIMS 1. A compound represented by formula I: or a pharmaceutically acce
 ptable salt thereof, wherein: L1 and L2 are each independently an optionally substituted C1-C30 aliphatic group; L3 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S; X1 and X2 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O)2N(R1)- , -N(R1)S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R1)2-, -OC(S)C(R1)2-, -C(R1)2C(S)O-, and –S-, wherein one or both of X1 or X2 is selected from -S(O)2N(R1)-, -N(R1)S(O)2, -S(O)-, - S(O)2-, -S(O)2C(R1)2-, -OC(S)C(R1)2-, -C(R1)2C(S)O-, and –S-; each R1 is, independently, at each instance, optionally substituted C1-C20 aliphatic or H; T1 and T2 are each independently an optionally substituted C3-C30 aliphatic; G is -N(R2)C(S)N(R2)2, -OH, -N(R2)2, -N+(R3)3, -N(R5)C(O)R3, -N(R5)S(O)2R3, - N(R5)C(O)N(R3)2, -CH(N-R2), -R4, or -S(O)2R3; each R2 is, independently, at each instance, selected from the group consisting of H, optionally substituted C1-C6 aliphatic and OR3; or two instances of R2 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring or an optionally substituted 4- to 12-membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N, O, and S; each R3 is, independently, at each instance, selected from the group consisting of H and optionally substituted C1-C10 aliphatic; and R4 is optionally substituted 4- to 12-membered heterocycle comprising 1 to 4 heteroatoms selected from N, O, and S, optionally substituted 4- to 12 membered heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S, C6-C12 aryl substituted with one or more of –(CH2)0-6–OH or –(CH2)0-6–N(R5)2, or C3-C12 cycloaliphatic substituted with one or more of oxo, –(CH2)0-6–OH, or –(CH2)0-6–N(R5)2; each R5 is independently selected from H and optionally substituted C1-C6 aliphatic.
ptable salt thereof, wherein: L1 and L2 are each independently an optionally substituted C1-C30 aliphatic group; L3 is a bond, optionally substituted C1-C10 aliphatic group, or optionally substituted 2- to 10-membered heteroaliphatic group comprising 1 to 4 heteroatoms selected from N, O, and S; X1 and X2 are each independently selected from a bond, -OC(O)-, -C(O)O-, -S(O)2N(R1)- , -N(R1)S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R1)2-, -OC(S)C(R1)2-, -C(R1)2C(S)O-, and –S-, wherein one or both of X1 or X2 is selected from -S(O)2N(R1)-, -N(R1)S(O)2, -S(O)-, - S(O)2-, -S(O)2C(R1)2-, -OC(S)C(R1)2-, -C(R1)2C(S)O-, and –S-; each R1 is, independently, at each instance, optionally substituted C1-C20 aliphatic or H; T1 and T2 are each independently an optionally substituted C3-C30 aliphatic; G is -N(R2)C(S)N(R2)2, -OH, -N(R2)2, -N+(R3)3, -N(R5)C(O)R3, -N(R5)S(O)2R3, - N(R5)C(O)N(R3)2, -CH(N-R2), -R4, or -S(O)2R3; each R2 is, independently, at each instance, selected from the group consisting of H, optionally substituted C1-C6 aliphatic and OR3; or two instances of R2 come together with the atoms to which they are attached to form an optionally substituted 4- to 12-membered heterocycle ring or an optionally substituted 4- to 12-membered heteroaryl ring comprising 1 to 4 heteroatoms selected from N, O, and S; each R3 is, independently, at each instance, selected from the group consisting of H and optionally substituted C1-C10 aliphatic; and R4 is optionally substituted 4- to 12-membered heterocycle comprising 1 to 4 heteroatoms selected from N, O, and S, optionally substituted 4- to 12 membered heteroaryl comprising 1 to 4 heteroatoms selected from N, O, and S, C6-C12 aryl substituted with one or more of –(CH2)0-6–OH or –(CH2)0-6–N(R5)2, or C3-C12 cycloaliphatic substituted with one or more of oxo, –(CH2)0-6–OH, or –(CH2)0-6–N(R5)2; each R5 is independently selected from H and optionally substituted C1-C6 aliphatic.
2. The compound of claim 1, wherein L1 and L2 are each C1-C30 alkylene.
3. The compound of claims 1 or 2, wherein L1 and L2 are each independently – (CH2)6-10-.
4. The compound of any one of claims 1-3, wherein L1 and L2 are the same.
5. The compound of any one of claims 1-3, wherein L1 and L2 are different.
6. The compound of any one of claims 1-5, wherein X1 and X2 are each independently selected from a -S(O)2N(R1)-, -N(R1)S(O)2, -S(O)-, -S(O)2-, -S(O)2C(R1)2- , -OC(S)C(R1)2-, -C(R1)2C(S)O-, and –S-.
7. The compound of any one of claims 1-5, wherein one of X1 and X2 is a bond, - OC(O)-, or -C(O)O-, and the other of X1 and X2 is -S(O)2N(R1)-, -N(R1)S(O)2, -S(O)-, - S(O)2-, -S(O)2C(R1)2-, -OC(S)C(R1)2-, -C(R1)2C(S)O-, or –S-.
8. The compound of any one of claims 1-5, wherein X1 is -S(O)2N(R1)-, where R1 is C1-C10 aliphatic, and X2 is –C(O)O-.
9. The compound of any one of claims 1-5, wherein X1 and X2 are each -S(O)2N(R1)- , where each R1 is independently R1 is C1-C10 aliphatic.
10. The compound of any one of claims 1-5, wherein X1 and X2 are the same.
11. The compound of any one of claims 1-5, wherein X1 and X2 are different.
12. The compound of any one of claims 1-11, wherein T1 and T2 are each independently selected from optionally substituted C3-C20 alkyl.
13. The compound of any one of claims 1-11, wherein T1 and T2 are each independently selected from:


14. The compound of claim 1, wherein a moiety –L1-X1-T1 is selected from the group consisting of:

15. The compound of claim 1 or claim 14, wherein a moiety –L2-X2-T2 is selected from the group consisting of:

16. The compound of claim 1, wherein a moiety –L1-X1-T1 and moiety –L2-X2-T2 are each independently selected from:


17. The compound of any one of claims 1-16, wherein L3 is optionally substituted C1- C10 aliphatic.
18. The compound of any one of claims 1-17, wherein G is -N(R2)C(S)N(R2)2 or - N(R5)S(O)2R3.
19. The compound of claim 18, wherein G is -N(R2)C(S)N(R2)2.
20. The compound of claim 19, wherein G is -N(H)C(S)N(R2)2, where each R2 is selected from optionally substituted C1-C6 aliphatic and OH.
21. The compound of any one of claims 1-17, wherein G is –OH.
22. The compound of any one of claims 1-17, wherein G is optionally substituted 4- to 12-membered heterocycle.
23. The compound of any one of claims 1-17, wherein G is selected from:

24. The compound of claim 1, wherein moiety –L3-G is selected from:

25. The compound of claim 1, wherein L G is selected from:

26. The compound of claim 1, wherein the compound is represented by Formula IIa: or a pharmaceutically
 acceptable salt thereof.
acceptable salt thereof.
27. The compound of claim 1, wherein the compound is represented by Formula IIb: or a pharmaceutical
 ly acceptable salt thereof.
ly acceptable salt thereof.
28. The compound of claim 1, wherein the compound is represented by Formula IIc: or a pharmaceutica
 lly acceptable salt thereof.
lly acceptable salt thereof.
29. The compound of claim 1, wherein the compound is represented by Formula IIIa: IIIa or a pharmaceutically acceptable salt thereof.
30. The compound of claim 1, wherein the compound is represented by Formula IIIb: IIIb or a pharmaceutically acceptable salt thereof.
31. The compound of claim 1, wherein the compound is represented by Formula IIIc: IIIc or a pharmaceutically acceptable salt thereof.
32. The compound of claim 1, wherein the compound is represented by Formula IIId: IIId or a pharmaceutically acceptable salt thereof.
33. The compound of claim 1, wherein the compound is represented by Formula IIIe:
or a pharmace
 utically acceptable salt thereof.
utically acceptable salt thereof.
34. The compound of claim 1, wherein the compound is represented by Formula IIIf: or a pharmaceuti
 cally acceptable salt thereof.
cally acceptable salt thereof.
35. The compound of claim 1, wherein the compound is represented by Formula IIIg: or a pharmaceu
 tically acceptable salt thereof.
tically acceptable salt thereof.
36. The compound of claim 1, wherein the compound is selected from Table 1.
37. A particle comprising a compound of any one of claims 1-36, and a nucleic acid.
38. The particle of claim 37, wherein the nucleic acid is RNA, DNA, or mixtures thereof.
39. The particle of claim 38, wherein the RNA is mRNA.
40. The particle of claim 39, wherein the RNA is modRNA, circRNA, saRNA, taRNA, or uRNA.
41. The particle of claim 37, wherein the DNA is linear DNA, plasmid DNA, minicircle DNA, nanoplasmid DNA, doggybone DNA, or a transposon.
42. The particle of any one of claims 37-41, wherein the particle further comprises one or more of a helper lipid, a polymer-conjugated lipid, or a sterol.
43. The particle of claim 42, wherein the helper lipid is a phospholipid.
44. The particle of claim 43, wherein the phospholipid is selected from: phosphatidylcholines, phosphatidylethanolamines, phosphatidylglycerols, phosphatidic acids, phosphatidylserines and sphingomyelins, more preferably selected from the group consisting of distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dimyristoylphosphatidylcholine (DMPC), dipentadecanoylphosphatidylcholine, dilauroylphosphatidylcholine, dipalmitoylphosphatidylcholine (DPPC), diarachidoylphosphatidylcholine (DAPC), dibehenoylphosphatidylcholine (DBPC), ditricosanoylphosphatidylcholine (DTPC), dilignoceroylphatidylcholine (DLPC), palmitoyloleoyl-phosphatidylcholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3- phosphocholine (18:0 Diether PC), 1-oleoyl-2-cholesterylhemisuccinoyl-sn-glycero-3- phosphocholine (OChemsPC), 1-hexadecyl-sn-glycero-3-phosphocholine (C16 Lyso PC), dioleoylphosphatidylethanolamine (DOPE), distearoyl-phosphatidylethanolamine (DSPE), dipalmitoyl-phosphatidylethanolamine (DPPE), dimyristoyl- phosphatidylethanolamine (DMPE), dilauroyl-phosphatidylethanolamine (DLPE), diphytanoyl-phosphatidylethanolamine (DPyPE), and combinations thereof.
45. The particle of any one of claims 42-44, wherein the polymer-conjugated lipid is selected from the group consisting of a poly(ethylene glycol) (PEG)-conjugated lipid, a poly(sarcosine) (pSar)-conjugated lipid, a poly(aminoethoxy ethoxy acetic acid) (pAEEA)-conjugated lipid; and a poly(2-methylaminoethoxy ethoxy acetic acid) (pMAEEA)-conjugated lipid.
46. The particle of any one of claims 42-45, wherein the polymer-conjugated lipid is a PEG-lipid selected from PEG-DAG, PEG-PE, PEG-S-DAG, PEG2000-DMG, PEG-cer, a PEG dialkyoxypropylcarbamate, and combinations thereof.
47. The particle of any one of claims 42-46, wherein the sterol is selected from β- sitosterol, stigmasterol, cholesterol, cholecalciferol, ergocalciferol, calcipotriol, botulin, lupeol, ursolic acid, oleanolic acid, cycloartenol, lanosterol, or α-tocopherol.
48. The particle of any one of claims 42-47, wherein the particle is characterized by an N/P ratio that is about 4 to about 16.
49. A method of increasing or causing increased expression of RNA in a target in a subject, the method comprising administering to the subject a composition comprising particles of any one of claims 42-48.
50. The method of claim 49, wherein the target is selected from the lungs, liver, spleen, heart, brain, lymph nodes, bladder, kidneys, and pancreas.
51. A method of treating a disease, disorder, or condition in a subject comprising administering to the subject a composition comprising the particles of any one of claims 42-48.
52. The method of claim 51, wherein the disease, disorder, or condition is an infectious disease, cancer, a genetic disorder, an autoimmune disease, or a rare disease.
53. The method of any one of claims 48-52, wherein the particle is administered parenterally or intranasally.
54. The method of claim 53, wherein the particle is administered intramuscularly, subcutaneously, intradermally, or intravenously.
55. A particle of any one of claims 42-48 for use as a medicament.
56. A particle of any one of claims 42-48 for use in the treatment and/or prevention of a disease or disorder, wherein the disease or disorder is an infectious disease, cancer, a genetic disorder, an autoimmune disease, or a rare disease.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2024315222A AU2024315222A1 (en) | 2023-08-01 | 2024-07-31 | Ionizable thiolipids and uses thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EPPCT/EP2023/071270 | 2023-08-01 | ||
| PCT/EP2023/071270 WO2025026545A1 (en) | 2023-08-01 | 2023-08-01 | Ionizable thioplipids and uses thereof |
| US202463639459P | 2024-04-26 | 2024-04-26 | |
| US63/639,459 | 2024-04-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025027089A1 true WO2025027089A1 (en) | 2025-02-06 |
Family
ID=92708884
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/071710 Pending WO2025027089A1 (en) | 2023-08-01 | 2024-07-31 | Ionizable thiolipids and uses thereof |
Country Status (3)
| Country | Link |
|---|---|
| AU (1) | AU2024315222A1 (en) |
| TW (1) | TW202519509A (en) |
| WO (1) | WO2025027089A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025176732A1 (en) | 2024-02-20 | 2025-08-28 | BioNTech SE | Compositions and methods |
| WO2025228975A1 (en) | 2024-04-29 | 2025-11-06 | BioNTech SE | Particles, compositions and methods |
Citations (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999039741A2 (en) | 1998-02-03 | 1999-08-12 | Inex Pharmaceuticals Corporation | Systemic delivery of serum stable plasmid lipid particles for cancer therapy |
| US5965542A (en) | 1997-03-18 | 1999-10-12 | Inex Pharmaceuticals Corp. | Use of temperature to control the size of cationic liposome/plasmid DNA complexes |
| US6009076A (en) | 1995-06-15 | 1999-12-28 | Fujitsu Limited | Repeating installation |
| WO2001007548A1 (en) | 1999-07-26 | 2001-02-01 | The Procter & Gamble Company | Cationic charge boosting systems |
| US20040142025A1 (en) | 2002-06-28 | 2004-07-22 | Protiva Biotherapeutics Ltd. | Liposomal apparatus and manufacturing methods |
| US20050017054A1 (en) | 2003-07-23 | 2005-01-27 | Tom Iverson | Flyback transformer wire attach method to printed circuit board |
| US20050064595A1 (en) | 2003-07-16 | 2005-03-24 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering RNA |
| WO2005038030A1 (en) | 2003-10-14 | 2005-04-28 | Johannes Gutenberg-Universität Mainz, Vertreten Durch Den Präsidenten | Recombinant vaccines and use thereof |
| US20050118253A1 (en) | 1998-02-03 | 2005-06-02 | Protiva Biotherapeutics, Inc. | Systemic delivery of serum stable plasmid lipid particles for cancer therapy |
| US20050175682A1 (en) | 2003-09-15 | 2005-08-11 | Protiva Biotherapeutics, Inc. | Polyethyleneglycol-modified lipid compounds and uses thereof |
| US20060008910A1 (en) | 2004-06-07 | 2006-01-12 | Protiva Biotherapeuties, Inc. | Lipid encapsulated interfering RNA |
| US20060083780A1 (en) | 2004-06-07 | 2006-04-20 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods of use |
| US20070042031A1 (en) | 2005-07-27 | 2007-02-22 | Protiva Biotherapeutics, Inc. | Systems and methods for manufacturing liposomes |
| WO2007024708A2 (en) | 2005-08-23 | 2007-03-01 | The Trustees Of The University Of Pennsylvania | Rna containing modified nucleosides and methods of use thereof |
| WO2007036366A2 (en) | 2005-09-28 | 2007-04-05 | Johannes Gutenberg-Universität Mainz, Vertreten Durch Den Präsidenten | Modification of rna, producing an increased transcript stability and translation efficiency |
| US20100130588A1 (en) | 2008-04-15 | 2010-05-27 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for nucleic acid delivery |
| WO2011015347A1 (en) | 2009-08-05 | 2011-02-10 | Biontech Ag | Vaccine composition comprising 5'-cap modified rna |
| US20110076335A1 (en) | 2009-07-01 | 2011-03-31 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for delivery of therapeutic agents to solid tumors |
| US20110117125A1 (en) | 2008-01-02 | 2011-05-19 | Tekmira Pharmaceuticals Corporation | Compositions and methods for the delivery of nucleic acids |
| WO2011141705A1 (en) | 2010-05-12 | 2011-11-17 | Protiva Biotherapeutics, Inc. | Novel cationic lipids and methods of use thereof |
| US20110311583A1 (en) | 2008-11-10 | 2011-12-22 | Alnylam Pharmaceuticals, Inc. | Novel lipids and compositions for the delivery of therapeutics |
| US20120027803A1 (en) | 2010-06-03 | 2012-02-02 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20120172411A1 (en) | 2010-09-17 | 2012-07-05 | Protiva Biotherapeutics, Inc. | Novel trialkyl cationic lipids and methods of use thereof |
| US20120295832A1 (en) | 2011-05-17 | 2012-11-22 | Arrowhead Research Corporation | Novel Lipids and Compositions for Intracellular Delivery of Biologically Active Compounds |
| US20130017223A1 (en) | 2009-12-18 | 2013-01-17 | The University Of British Columbia | Methods and compositions for delivery of nucleic acids |
| US20130022649A1 (en) | 2009-12-01 | 2013-01-24 | Protiva Biotherapeutics, Inc. | Snalp formulations containing antioxidants |
| WO2013016058A1 (en) | 2011-07-22 | 2013-01-31 | Merck Sharp & Dohme Corp. | Novel bis-nitrogen containing cationic lipids for oligonucleotide delivery |
| US20130086373A1 (en) | 2011-09-29 | 2013-04-04 | Apple Inc. | Customized content for electronic devices |
| WO2013086373A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Lipids for the delivery of active agents |
| WO2013086322A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Branched alkyl and cycloalkyl terminated biodegradable lipids for the delivery of active agents |
| US20130195920A1 (en) | 2011-12-07 | 2013-08-01 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20130245107A1 (en) | 2011-12-16 | 2013-09-19 | modeRNA Therapeutics | Dlin-mc3-dma lipid nanoparticle delivery of modified polynucleotides |
| US8569256B2 (en) | 2009-07-01 | 2013-10-29 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods for the delivery of therapeutic agents |
| US20130323269A1 (en) | 2010-07-30 | 2013-12-05 | Muthiah Manoharan | Methods and compositions for delivery of active agents |
| US20130338210A1 (en) | 2009-12-07 | 2013-12-19 | Alnylam Pharmaceuticals, Inc. | Compositions for nucleic acid delivery |
| WO2014008334A1 (en) | 2012-07-06 | 2014-01-09 | Alnylam Pharmaceuticals, Inc. | Stable non-aggregating nucleic acid lipid particle formulations |
| US20140200257A1 (en) | 2011-01-11 | 2014-07-17 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| US20150203446A1 (en) | 2011-09-27 | 2015-07-23 | Takeda Pharmaceutical Company Limited | Di-aliphatic substituted pegylated lipids |
| WO2015199952A1 (en) | 2014-06-25 | 2015-12-30 | Acuitas Therapeutics Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2016005324A1 (en) | 2014-07-11 | 2016-01-14 | Biontech Rna Pharmaceuticals Gmbh | Stabilization of poly(a) sequence encoding dna sequences |
| WO2016005318A1 (en) * | 2014-07-08 | 2016-01-14 | Incella Gmbh | Synthesis and use of amino lipids |
| WO2017004143A1 (en) | 2015-06-29 | 2017-01-05 | Acuitas Therapeutics Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017036889A1 (en) | 2015-08-28 | 2017-03-09 | Biontech Rna Pharmaceuticals Gmbh | Method for reducing immunogenicity of rna |
| WO2017053297A1 (en) | 2015-09-21 | 2017-03-30 | Trilink Biotechnologies, Inc. | Compositions and methods for synthesizing 5'-capped rnas |
| WO2017060314A2 (en) | 2015-10-07 | 2017-04-13 | Biontech Rna Pharmaceuticals Gmbh | 3' utr sequences for stabilization of rna |
| WO2017075531A1 (en) | 2015-10-28 | 2017-05-04 | Acuitas Therapeutics, Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017162266A1 (en) | 2016-03-21 | 2017-09-28 | Biontech Rna Pharmaceuticals Gmbh | Rna replicon for versatile and efficient gene expression |
| WO2017162461A1 (en) | 2016-03-21 | 2017-09-28 | Biontech Rna Pharmaceuticals Gmbh | Trans-replicating rna |
| WO2018081480A1 (en) | 2016-10-26 | 2018-05-03 | Acuitas Therapeutics, Inc. | Lipid nanoparticle formulations |
| WO2018140730A1 (en) | 2017-01-27 | 2018-08-02 | Signalrx Pharmaceuticals, Inc. | Thienopyranones and furanopyranones as kinase, bromodomain, and checkpoint inhibitors |
| WO2019027055A1 (en) * | 2017-08-04 | 2019-02-07 | 協和発酵キリン株式会社 | Nucleic acid-containing lipid nanoparticles |
| WO2021026358A1 (en) | 2019-08-07 | 2021-02-11 | Moderna TX, Inc. | Compositions and methods for enhanced delivery of agents |
| WO2021250102A1 (en) | 2020-06-10 | 2021-12-16 | Janssen Pharmaceutica Nv | Macrocyclic 2-amino-3-fluoro-but-3-enamides as inhibitors of mcl-1 |
| CN114989027A (en) * | 2022-08-03 | 2022-09-02 | 深圳市瑞吉生物科技有限公司 | Cationic lipid compounds and compositions for delivery of nucleic acids and uses |
| WO2023029928A1 (en) * | 2021-09-01 | 2023-03-09 | 深圳虹信生物科技有限公司 | Amino lipid and application thereof |
-
2024
- 2024-07-31 WO PCT/EP2024/071710 patent/WO2025027089A1/en active Pending
- 2024-07-31 TW TW113128599A patent/TW202519509A/en unknown
- 2024-07-31 AU AU2024315222A patent/AU2024315222A1/en active Pending
Patent Citations (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6009076A (en) | 1995-06-15 | 1999-12-28 | Fujitsu Limited | Repeating installation |
| US5965542A (en) | 1997-03-18 | 1999-10-12 | Inex Pharmaceuticals Corp. | Use of temperature to control the size of cationic liposome/plasmid DNA complexes |
| US20050118253A1 (en) | 1998-02-03 | 2005-06-02 | Protiva Biotherapeutics, Inc. | Systemic delivery of serum stable plasmid lipid particles for cancer therapy |
| WO1999039741A2 (en) | 1998-02-03 | 1999-08-12 | Inex Pharmaceuticals Corporation | Systemic delivery of serum stable plasmid lipid particles for cancer therapy |
| WO2001007548A1 (en) | 1999-07-26 | 2001-02-01 | The Procter & Gamble Company | Cationic charge boosting systems |
| US20040142025A1 (en) | 2002-06-28 | 2004-07-22 | Protiva Biotherapeutics Ltd. | Liposomal apparatus and manufacturing methods |
| US20110216622A1 (en) | 2002-06-28 | 2011-09-08 | Protiva Biotherapeutics, Inc. | Liposomal apparatus and manufacturing method |
| US20050064595A1 (en) | 2003-07-16 | 2005-03-24 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering RNA |
| US20120058188A1 (en) | 2003-07-16 | 2012-03-08 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering rna |
| US20060240093A1 (en) | 2003-07-16 | 2006-10-26 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering rna |
| US20050017054A1 (en) | 2003-07-23 | 2005-01-27 | Tom Iverson | Flyback transformer wire attach method to printed circuit board |
| US20050175682A1 (en) | 2003-09-15 | 2005-08-11 | Protiva Biotherapeutics, Inc. | Polyethyleneglycol-modified lipid compounds and uses thereof |
| US20110091525A1 (en) | 2003-09-15 | 2011-04-21 | Protiva Biotherapeutics, Inc. | Polyethyleneglycol-modified lipid compounds and uses thereof |
| WO2005038030A1 (en) | 2003-10-14 | 2005-04-28 | Johannes Gutenberg-Universität Mainz, Vertreten Durch Den Präsidenten | Recombinant vaccines and use thereof |
| US20110060032A1 (en) | 2004-06-07 | 2011-03-10 | Protiva Biotherapeutics, Inc. | Lipid encapsulating interfering rna |
| US20060008910A1 (en) | 2004-06-07 | 2006-01-12 | Protiva Biotherapeuties, Inc. | Lipid encapsulated interfering RNA |
| US20060083780A1 (en) | 2004-06-07 | 2006-04-20 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods of use |
| US20110262527A1 (en) | 2004-06-07 | 2011-10-27 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods of use |
| US20070042031A1 (en) | 2005-07-27 | 2007-02-22 | Protiva Biotherapeutics, Inc. | Systems and methods for manufacturing liposomes |
| WO2007024708A2 (en) | 2005-08-23 | 2007-03-01 | The Trustees Of The University Of Pennsylvania | Rna containing modified nucleosides and methods of use thereof |
| WO2007036366A2 (en) | 2005-09-28 | 2007-04-05 | Johannes Gutenberg-Universität Mainz, Vertreten Durch Den Präsidenten | Modification of rna, producing an increased transcript stability and translation efficiency |
| US20110117125A1 (en) | 2008-01-02 | 2011-05-19 | Tekmira Pharmaceuticals Corporation | Compositions and methods for the delivery of nucleic acids |
| US20120183581A1 (en) | 2008-04-15 | 2012-07-19 | Protiva Biotherapeutics, Inc | Novel lipid formulations for nucleic acid delivery |
| US20100130588A1 (en) | 2008-04-15 | 2010-05-27 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for nucleic acid delivery |
| US20110311583A1 (en) | 2008-11-10 | 2011-12-22 | Alnylam Pharmaceuticals, Inc. | Novel lipids and compositions for the delivery of therapeutics |
| US20110311582A1 (en) | 2008-11-10 | 2011-12-22 | Muthiah Manoharan | Novel lipids and compositions for the delivery of therapeutics |
| US20160199485A1 (en) | 2008-11-10 | 2016-07-14 | Tekmira Pharmaceuticals Corporation | Novel lipids and compositions for the delivery of therapeutics |
| US20150265708A1 (en) | 2008-11-10 | 2015-09-24 | Tekmira Pharmaceuticals Corporation | Novel lipids and compositions for the delivery of therapeutics |
| US8569256B2 (en) | 2009-07-01 | 2013-10-29 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods for the delivery of therapeutic agents |
| US20110076335A1 (en) | 2009-07-01 | 2011-03-31 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for delivery of therapeutic agents to solid tumors |
| WO2011015347A1 (en) | 2009-08-05 | 2011-02-10 | Biontech Ag | Vaccine composition comprising 5'-cap modified rna |
| US20130022649A1 (en) | 2009-12-01 | 2013-01-24 | Protiva Biotherapeutics, Inc. | Snalp formulations containing antioxidants |
| US20130338210A1 (en) | 2009-12-07 | 2013-12-19 | Alnylam Pharmaceuticals, Inc. | Compositions for nucleic acid delivery |
| US20130017223A1 (en) | 2009-12-18 | 2013-01-17 | The University Of British Columbia | Methods and compositions for delivery of nucleic acids |
| US20130123338A1 (en) | 2010-05-12 | 2013-05-16 | Protiva Biotherapeutics, Inc. | Novel cationic lipids and methods of use thereof |
| WO2011141705A1 (en) | 2010-05-12 | 2011-11-17 | Protiva Biotherapeutics, Inc. | Novel cationic lipids and methods of use thereof |
| US20160009637A1 (en) | 2010-06-03 | 2016-01-14 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20120027803A1 (en) | 2010-06-03 | 2012-02-02 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20130323269A1 (en) | 2010-07-30 | 2013-12-05 | Muthiah Manoharan | Methods and compositions for delivery of active agents |
| US20120172411A1 (en) | 2010-09-17 | 2012-07-05 | Protiva Biotherapeutics, Inc. | Novel trialkyl cationic lipids and methods of use thereof |
| US20140200257A1 (en) | 2011-01-11 | 2014-07-17 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| US20120295832A1 (en) | 2011-05-17 | 2012-11-22 | Arrowhead Research Corporation | Novel Lipids and Compositions for Intracellular Delivery of Biologically Active Compounds |
| WO2013016058A1 (en) | 2011-07-22 | 2013-01-31 | Merck Sharp & Dohme Corp. | Novel bis-nitrogen containing cationic lipids for oligonucleotide delivery |
| US20150203446A1 (en) | 2011-09-27 | 2015-07-23 | Takeda Pharmaceutical Company Limited | Di-aliphatic substituted pegylated lipids |
| US20130086373A1 (en) | 2011-09-29 | 2013-04-04 | Apple Inc. | Customized content for electronic devices |
| US20140308304A1 (en) | 2011-12-07 | 2014-10-16 | Alnylam Pharmaceuticals, Inc. | Lipids for the delivery of active agents |
| WO2013086373A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Lipids for the delivery of active agents |
| US20150005363A1 (en) | 2011-12-07 | 2015-01-01 | Alnylam Pharmaceuticals, Inc. | Branched Alkyl And Cycloalkyl Terminated Biodegradable Lipids For The Delivery Of Active Agents |
| US20150273068A1 (en) | 2011-12-07 | 2015-10-01 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20130195920A1 (en) | 2011-12-07 | 2013-08-01 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| WO2013086322A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Branched alkyl and cycloalkyl terminated biodegradable lipids for the delivery of active agents |
| US20130245107A1 (en) | 2011-12-16 | 2013-09-19 | modeRNA Therapeutics | Dlin-mc3-dma lipid nanoparticle delivery of modified polynucleotides |
| WO2014008334A1 (en) | 2012-07-06 | 2014-01-09 | Alnylam Pharmaceuticals, Inc. | Stable non-aggregating nucleic acid lipid particle formulations |
| WO2015199952A1 (en) | 2014-06-25 | 2015-12-30 | Acuitas Therapeutics Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2016005318A1 (en) * | 2014-07-08 | 2016-01-14 | Incella Gmbh | Synthesis and use of amino lipids |
| WO2016005324A1 (en) | 2014-07-11 | 2016-01-14 | Biontech Rna Pharmaceuticals Gmbh | Stabilization of poly(a) sequence encoding dna sequences |
| WO2017004143A1 (en) | 2015-06-29 | 2017-01-05 | Acuitas Therapeutics Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017036889A1 (en) | 2015-08-28 | 2017-03-09 | Biontech Rna Pharmaceuticals Gmbh | Method for reducing immunogenicity of rna |
| WO2017053297A1 (en) | 2015-09-21 | 2017-03-30 | Trilink Biotechnologies, Inc. | Compositions and methods for synthesizing 5'-capped rnas |
| US10519189B2 (en) | 2015-09-21 | 2019-12-31 | Trilink Biotechnologies, Inc. | Compositions and methods for synthesizing 5′-Capped RNAs |
| US10494399B2 (en) | 2015-09-21 | 2019-12-03 | Trilink Biotechnologies, Inc. | Compositions and methods for synthesizing 5′-Capped RNAS |
| WO2017060314A2 (en) | 2015-10-07 | 2017-04-13 | Biontech Rna Pharmaceuticals Gmbh | 3' utr sequences for stabilization of rna |
| WO2017075531A1 (en) | 2015-10-28 | 2017-05-04 | Acuitas Therapeutics, Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017162266A1 (en) | 2016-03-21 | 2017-09-28 | Biontech Rna Pharmaceuticals Gmbh | Rna replicon for versatile and efficient gene expression |
| WO2017162461A1 (en) | 2016-03-21 | 2017-09-28 | Biontech Rna Pharmaceuticals Gmbh | Trans-replicating rna |
| WO2018081480A1 (en) | 2016-10-26 | 2018-05-03 | Acuitas Therapeutics, Inc. | Lipid nanoparticle formulations |
| WO2018140730A1 (en) | 2017-01-27 | 2018-08-02 | Signalrx Pharmaceuticals, Inc. | Thienopyranones and furanopyranones as kinase, bromodomain, and checkpoint inhibitors |
| WO2019027055A1 (en) * | 2017-08-04 | 2019-02-07 | 協和発酵キリン株式会社 | Nucleic acid-containing lipid nanoparticles |
| WO2021026358A1 (en) | 2019-08-07 | 2021-02-11 | Moderna TX, Inc. | Compositions and methods for enhanced delivery of agents |
| US20220296517A1 (en) * | 2019-08-07 | 2022-09-22 | Moderna TX, Inc. | Compositions and methods for enhanced delivery of agents |
| WO2021250102A1 (en) | 2020-06-10 | 2021-12-16 | Janssen Pharmaceutica Nv | Macrocyclic 2-amino-3-fluoro-but-3-enamides as inhibitors of mcl-1 |
| WO2023029928A1 (en) * | 2021-09-01 | 2023-03-09 | 深圳虹信生物科技有限公司 | Amino lipid and application thereof |
| CN114989027A (en) * | 2022-08-03 | 2022-09-02 | 深圳市瑞吉生物科技有限公司 | Cationic lipid compounds and compositions for delivery of nucleic acids and uses |
Non-Patent Citations (13)
| Title |
|---|
| "Remington: The Science and Practice of Pharmacy", 2005, LIPPINCOTT WILLIAMS & WILKINS |
| "Remington's The Science and Practice of PharmaCy", 2006, LIPPINCOTT, WILLIAMS & WILKINS |
| ANDERSON ET AL.: "The Practice of Medicinal Chemistry", 1996, ACADEMIC PRESS |
| KACZMAREK, J. C. ET AL., GENOME MEDICINE, vol. 9, 2017, pages 60 |
| KIRSCHBERGTHORSTEN A. ET AL., BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 24, no. 923279-52-1, 2014, pages 969 - 972 |
| KOPPEL, D., J. CHEM. PHYS., vol. 57, 1972, pages 4814 - 4820 |
| P. GOULD, INTERNATIONAL J. OF PHARMACEUTICS, vol. 33, 1986, pages 201 - 217 |
| P. STAHL ET AL.: "Handbook of Pharmaceutical Salts", 2002, WILEY-VCH |
| S. BERGE ET AL., JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 66, no. 1, 1977, pages 1 - 19 |
| S. M. BERGE ET AL.: "describes pharmaceutically acceptable salts in detail", J. PHARMACEUTICAL SCIENCES, vol. 66, 1977, pages 1 - 19 |
| SMITH, M.B.MARCH, J.: "March's Advanced Organic Chemistry", 2001, JOHN WILEY & SONS |
| TENCHOV ET AL., ACS NANO, vol. 15, no. 11, 2021, pages 16982 - 17015 |
| THOMAS SORRELL: "Organic Chemistry", 1999, UNIVERSITY SCIENCE |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025176732A1 (en) | 2024-02-20 | 2025-08-28 | BioNTech SE | Compositions and methods |
| WO2025228975A1 (en) | 2024-04-29 | 2025-11-06 | BioNTech SE | Particles, compositions and methods |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202519509A (en) | 2025-05-16 |
| AU2024315222A1 (en) | 2026-02-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7596343B2 (en) | Compounds and compositions for intracellular delivery of therapeutic agents - Patents.com | |
| WO2025027089A1 (en) | Ionizable thiolipids and uses thereof | |
| AU2021409638A1 (en) | Nanomaterials comprising carbonates | |
| CN112424214A (en) | Cationic lipids comprising steroidal moieties | |
| AU2019277355A1 (en) | Phosphoester cationic lipids | |
| US20240335541A1 (en) | Nanomaterials comprising diamines | |
| JP2019508371A (en) | Compounds and compositions for intracellular delivery of drugs | |
| AU2022210317A1 (en) | Nanomaterials comprising an ionizable lipid | |
| WO2023121964A1 (en) | Nanomaterials comprising disulfides | |
| AU2022418511A1 (en) | Ionizable amine lipids and lipid nanoparticles | |
| US20250057960A1 (en) | Oligosaccharide complexes and uses | |
| AU2021409377A1 (en) | Nanomaterials comprising acetals | |
| EP4402149A1 (en) | Disulfide oligosaccharide compounds and complexes | |
| WO2025026545A1 (en) | Ionizable thioplipids and uses thereof | |
| WO2023232747A1 (en) | Complexes for delivery of nucleic acids | |
| WO2025134062A2 (en) | Ionizable lipids | |
| WO2025262672A1 (en) | Lipid compositions for nucleic acid delivery | |
| WO2025134066A1 (en) | Ionizable lipids | |
| EP4285933A1 (en) | Oligosaccharide complexes and uses | |
| EP4286004A1 (en) | Disulfide oligosaccharide compounds and complexes | |
| EP4186528A1 (en) | Oligosaccharide complexes and uses | |
| WO2026033380A1 (en) | Process for preparing oligosaccharide complexes | |
| EP4169579A1 (en) | Disulfide oligosaccharide compounds and complexes | |
| WO2025124711A1 (en) | Glycolipid compositions | |
| HK40096911A (en) | Lipid compounds and lipid nanoparticle compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24767995 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AU2024315222 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 326176 Country of ref document: IL |