WO2025010245A1 - Fused pyrazole derivatives as usp1 inhibitors - Google Patents
Fused pyrazole derivatives as usp1 inhibitors Download PDFInfo
- Publication number
- WO2025010245A1 WO2025010245A1 PCT/US2024/036492 US2024036492W WO2025010245A1 WO 2025010245 A1 WO2025010245 A1 WO 2025010245A1 US 2024036492 W US2024036492 W US 2024036492W WO 2025010245 A1 WO2025010245 A1 WO 2025010245A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- cancer
- independently
- ring
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/18—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/18—Bridged systems
Definitions
- Deubiquitinases are a class of enzymes that act on ubiquitinated substrates to catalyze the removal of ubiquitin moieties.
- the human genome contains approximately 100 genes that encode DUBs.
- Human DUBs are classified into five different families (Nijman, S.M. et al. (2005) Cell 123, 773-86, Nalepa, G. et al. (2006) Nat Rev Drug Discov 5, 596-613).
- USP1 ubiquitin specific protease 1 belongs to the USP subfamily of DUBs.
- the USP1 gene encodes a 785 amino acid protein having a conserved USP domain amino-terminal Cys box motif and a carboxy-terminal His box motif (Nijman, S.M. et al. (2005) Mol Cell 17, 331-9).
- UAF1 a WD40 repeat-containing protein
- USP1 gene transcription is regulated in a cell cycle-dependent manner.
- USP1 mRNA levels of USP1 remain low during G1 phase and reach a peak during S phase (Nijman, S.M. et al. (2005) Mol Cell 17, 331-9). The expression of USP1 is also regulated at the protein level by proteasomal degradation (Cataldo F, Mol Cell Biol (2013), 33(12):2485–2496). [0004] USP1 is a nuclear protein and localizes to chromatin where it is specifically associated with Fanconi anemia protein FANCD2. USP1 regulates several important steps in the DNA damage response pathway, including the Fanconi anemia (FA) pathway and the process of translesion synthesis (TLS).
- FA Fanconi anemia
- TLS translesion synthesis
- USP1 deubiquitinates monoubiquitinated FANCD2, which plays an important role in DNA damage repair (Nijman, S.M. et al. (2005) Mol Cell 17, 331-9, Guervilly, J.H. et al. (2011) Hum Mol Genet). While DNA-dependent mono- ubiquitination of FANCD2 facilitates DNA repair, it is deubiquitinated by USP1 to block the DNA-repairing response. USP1 is also critical for the deubiquitination of monoubiquitinated PCNA and thus negatively regulates PCNA-mediated TLS during DNA repair (Huang TT. et al. Nat Cell Biol (2006), 8(4):339–347). The expression of USP1 is significantly increased in several cancers (Das DS.
- ring Y is C 3 -C 8 cycloalkyl, C 5 -C 10 cycloalkeny , wherein the asterisk marks the point of attachment to rin ring Z is 3- to 15-membered heterocycloalkyl or 5- to 14-membered heteroaryl;
- X 1 , X 2 and X 3 are independently N or C;
- Y 1 to Y 4 are independently N or C, wherein 0-2 of Y 1 to Y 4 are N;
- Y 5 and Y 6 are independently N, C, O or S;
- each R X is independently C1-C6 alkyl, C1-C6 alkoxy or C3-C8 cycloalkyl;
- R Y is halo, C 1 -C 6 alkyl, C 1 -C 6 alkoxy or C 1 -C 6 alkylamino;
- each R Z is independently hydrogen, halo, C1-
- R 4 and R 4 ⁇ are not hydrogen or alkyl when: (a) each of R 2 , R 2 ⁇ , R 3 , R 3 ⁇ is hydrogen, (b) ring Y is phenyl, thiophenyl, thiazolyl, 6-membered heteroaryl or cubanyl, and (c) ring Z is imidazolyl; or R 4 and R 3 are taken together to form a C 1 -C 3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; subscript p is 1, 2 or 3; subscript q is 2, 3 or 4; subscript m is 0 or
- compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and at least one pharmaceutically acceptable excipient, such as a pharmaceutically acceptable carrier or diluent.
- pharmaceutically acceptable excipient such as a pharmaceutically acceptable carrier or diluent.
- Compounds of formula (I) are useful for the inhibition of USP1, and the therapeutic use of such compounds is also provided.
- DETAILED DESCRIPTION [0012] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as is commonly understood by one of skill in art to which the subject matter herein belongs. As used in the specification and the appended claims, unless specified to the contrary, the following terms have the meaning indicated to facilitate the understanding of the present disclosure.
- alkyl refers to saturated aliphatic hydrocarbon chains, including C1-C10 straight or C1-C10 branched alkyl chains, (e.g., C 1 -C 6 straight or branched alkyl chains).
- alkyl include but are not limited to methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, isopentyl or neopentyl and the like.
- alkylene refers to an alkyl group, as defined above, linking at least two other groups (i.e., a divalent alkyl radical).
- the two moieties linked to the alkylene group can be linked to the same carbon atom or different carbon atoms of the alkylene group.
- the alkylene group preferably has 1-6 carbon atoms and, more preferably, 1- 3 carbon atoms.
- halo and “halogen,” alone or in combination with other terms, are used interchangeably to refer to fluorine, chlorine, bromine or iodine.
- haloalkyl refers to alkyl substituted with one or more halogen atoms, wherein the alkyl groups are as defined above.
- haloalkyl include but are not limited to fluoromethyl, difluoromethyl, chloromethyl, trifluoromethyl, 2,2,2-trifluoroethyl and the like.
- alkoxy alone or in combination with other terms, refers to the group alkyl-O- or -O-alkyl, where the alkyl groups are as defined above.
- alkoxy examples include but are not limited to methoxy, ethoxy, n-propoxy, n-butoxy, t-butoxy and the like.
- alkoxyalkyl alone or in combination with other terms, refers to the “alkoxy” as defined above linked to the rest of the molecule via an alkyl moiety.
- alkoxyalkyl include but not limited to -CH 2 -OCH 3 , -C 2 H 4 -OCH 3 , -CH2-OCH2CH3, -CH2-OC3H7, -C2H4-OCH2CH3 and the like.
- amino and “amine,” alone or in combination with other terms, refer to a primary amine (–NH 2 ), a secondary amine (wherein ‘N’ is substituted with two substituents other than hydrogen) or a tertiary amine (wherein ‘N’ is substituted with three substituents other than hydrogen).
- alkylamino alone or in combination with other terms, refers to an amino group substituted with one or more alkyl groups, wherein the alkyl group and amino group are as defined above.
- alkylamino groups include but are not limited to -NHCH3, -NHCH2CH3, -N(CH3)2, -N(CH3)(CH2CH3) and the like.
- alkylaminoalkyl alone or in combination with other terms, refers to an alkylamino group as defined above linked to the rest of the molecule via an alkyl moiety.
- alkylaminoalkyl include but are not limited to -CH 2 -NHCH 3 , -C2H4-NHCH3, -CH2-NHCH2CH3, -CH2-N(CH3)2, -CH2-N(CH3)(CH2CH3) and the like.
- cycloalkyl refers to a C 3 -C 10 saturated cyclic hydrocarbon ring.
- a cycloalkyl may be monocyclic, typically containing from 3 to 7 carbon ring atoms. Examples of monocyclic cycloalkyls include but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and the like.
- a cycloalkyl may alternatively be polycyclic, containing more than one ring. Examples of polycyclic cycloalkyls include bridged, fused and spirocyclic cycloalkyls and the like.
- cycloalkenyl refers to a partially unsaturated C5-C12 cyclic hydrocarbon ring with at least one C-C double bond.
- a cycloalkenyl may be monocyclic, typically containing from 5 to 10 carbon ring atoms. Examples of monocyclic cycloalkenyls include but are not limited to, cyclopentenyl, cyclohexenyl, cyclohepentyl, 1,3-cyclohexadienyl and the like.
- a cycloalkenyl may alternatively be a polycyclic moiety containing more than one ring.
- polycyclic cycloalkenyls include, but are not limited to, bridged, fused and spirocyclic ring systems such as bicyclo[2.2.1]hept-2-enyl, bicyclo[4.4.0]dec-1-enyl and the like.
- aryl alone or in combination with other terms, refers to an unsubstituted or substituted monocyclic, bicyclic or polycyclic aromatic hydrocarbon ring system of about 6 to 14 carbon atoms.
- aryl include, but are not limited to phenyl, naphthyl, anthryl, and biphenyl.
- An aryl group may be unsubstituted or substituted with one or more suitable groups.
- arylalkyl refers to an aryl group as defined above linked to the rest of the molecule via an alkyl moiety.
- arylalkyl include but are not limited to (phenyl)alkyl-, (naphthyl)alkyl-, (anthryl)alkyl- and the like.
- heteroatom refers to a sulfur, nitrogen or oxygen atom.
- heterocycloalkyl refers to a non-aromatic, saturated or partially saturated monocyclic or polycyclic ring system of 3 to 15 members having at least one heteroatom or heterogroup selected from O, N, S, S(O), S(O) 2 and NH, with the remaining ring atoms being independently selected from carbon, oxygen, nitrogen and sulfur.
- a monocyclic heterocycloalkyl typically contains 4 to 7 ring atoms.
- heterocycloalkyl examples include, but are not limited to azetidinyl, oxetanyl, imidazolidinyl, pyrrolidinyl, oxazolidinyl, thiazolidinyl, pyrazolidinyl, tetrahydrofuranyl, piperidinyl, piperazinyl, tetrahydropyranyl, morpholinyl, oxopiperazinyl, oxopiperidinyl, tetrahydrothiophenyl, dihydropyranyl, indolinyl, azepanyl and N-oxides thereof.
- heterocycloalkyl can be unsubstituted or substituted with one or more suitable groups.
- heteroaryl alone or in combination with other terms, refers to an aromatic unsaturated ring system containing a total of 5 to 14 ring atoms. At least one of the ring atoms is a heteroatom (oxygen, nitrogen, or sulfur), with the remaining ring atoms/groups being independently selected from carbon, oxygen, nitrogen and sulfur.
- a heteroaryl may be a single-ring (monocyclic) or polycyclic ring system.
- heteroaryl examples include but are not limited to pyridyl, indolyl, benzimidazolyl, benzothiazolyl, pyrrolyl, pyrazolyl, imidazolyl, pyrimidinyl, pyrazinyl, pyridazinyl and the like.
- heteroarylalkyl alone or in combination with other terms, refers to the heteroaryl as defined above is linked to the rest of the molecule via an alkyl moiety.
- heteroarylalkyl examples include but are not limited to (pyridine)alkyl-, (indole)alkyl-, (benzimidazole)alkyl-, (pyrrole)alkyl-, (pyrazole)alkyl-, (imidazole)alkyl-, (pyrimidine)- alkyl-, (pyrazine)alkyl-, (pyridazine)alkyl- and the like.
- the term “optionally substituted” or “substituted” or “optionally substituted with suitable groups” refers to replacement of one or more hydrogen radicals in a given structure with a radical of a substituent such as halo, alkyl, alkenyl, alkynyl, aryl, heteroaryl, heterocycloalkyl, thiol, alkylthio, arylthio, alkylthioalkyl, arylthioalkyl, alkylsulfonyl, alkylsulfonylalkyl, arylsulfonylalkyl, alkoxy, aryloxy, arylalkoxy, aminocarbonyl, alkylaminocarbonyl, arylaminocarbonyl, alkoxycarbonyl, aryloxycarbonyl, haloalkyl (e.g., trifluoromethyl), amino, cyano, nitro, alkylamin
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- the terms “treat”, “treating” and “treatment” refer to the alleviation or abrogation of a disease and/or its attendant symptoms.
- the terms “prevent”, “preventing” and “prevention” refer to preventing the onset of a disease and/or its attendant symptoms or barring a subject from acquiring a disease.
- “prevent”, “preventing” and “prevention” also include delaying the onset of a disease and/or its attendant symptoms and reducing a subject's risk of acquiring a disease.
- the term “therapeutically effective amount” refers to the amount of the compound being administered sufficient to prevent development of or alleviate to some extent one or more of the symptoms of the disease or disorder being treated.
- “Pharmaceutically acceptable” refers to substances that are generally safe, non-toxic and neither biologically undesirable nor otherwise undesirable, including substances which are acceptable for human pharmaceutical use and veterinary use.
- a pharmaceutically acceptable excipient for example, is compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- the phrase “pharmaceutically acceptable excipient” refers to a pharmaceutically acceptable material that aids the administration of an active agent to a subject.
- excipients include, but are not limited to, liquid or solid fillers, diluents, solvents, encapsulating materials, disintegrants, lubricants, glidants, sweeteners, flavors, and colors. See, e.g., Remington: The Science and Practice of Pharmacy, 23 rd ed.; Academic Press: Cambridge, Mass., 2020; Handbook of Pharmaceutical Excipients, 9th ed.; Sheskey et al, Eds.; Pharmaceutical Press 2020; Handbook of Pharmaceutical Additives, 3rd ed.; Ash and Ash Eds.; Gower Publishing Company: 2007; Pharmaceutical Preformulation and Formulation, 2nd ed.; Gibson Ed.; CRC Press LLC: Boca Raton, Fla., 2009.
- pharmaceutically acceptable salt(s) refers to a parent compound which is modified by converting an existing acid or base moiety to its salt form.
- examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts of the present disclosure include conventional non-toxic salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- the pharmaceutically acceptable salts of the present disclosure can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods.
- such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, non-aqueous media like ether, ethyl acetate, alcohols or acetonitrile (ACN) are preferred.
- non-aqueous media like ether, ethyl acetate, alcohols or acetonitrile (ACN) are preferred.
- stereoisomers refers to any enantiomers, diastereoisomers, or geometric isomers of the compounds of Formula (I), (IA), (IB), (IC), (ID), (IE), (IF), (IG) and (IH), wherever they are chiral or when they bear one or more double bonds.
- Individual stereoisomers of compounds can be prepared synthetically from commercially available starting materials which contain chiral centers or by preparation of mixtures of enantiomeric products followed by separation, e.g., via conversion to a mixture of diastereomers followed by separation or recrystallization, chromatographic techniques (including direct separation of enantiomers on chiral chromatographic columns), or any other appropriate method known in the art.
- Starting compounds of particular stereochemistry are either commercially available or can be made and resolved by techniques known in the art.
- the compounds of the present disclosure may exist as geometric isomers.
- the present disclosure includes all cis and trans, syn and anti,
- the present disclosure provides fused pyrazole derivatives of formula (I), which are useful for the inhibition of USP1.
- the present disclosure further provides pharmaceutical compositions comprising the compounds of formula (I), and their derivatives as therapeutic agents. It will be apparent to those skilled in the art that various modifications and variations can be made to the compounds, compositions, and methods described herein without departing from the scope or spirit of various embodiments disclosed herein. For instance, features illustrated or described as part of one embodiment can be applied to another embodiment to yield a still further embodiment. Thus, it is intended that the present application includes such modifications and variations and their equivalents. Other objects, features, and aspects of the present application are disclosed in the following detailed description.
- fused pyrazole derivatives of formula (I), and pharmaceuticall wherein: ; ring Y is C3-C8 cycloalkyl, C5-C10 cycloalkeny , wherein the asterisk marks the point of attachment to rin ring Z is 3- to 15-membered heterocycloalkyl or 5- to 14-membered heteroaryl; X 1 , X 2 and X 3 are independently N or C; Y 1 to Y 4 are independently N or C, wherein 0-2 of Y 1 to Y 4 are N; Y 5 and Y 6 are independently N, C, O or S; each R X is independently C1-C6 alkyl, C1-C6 alkoxy or C3-C8 cycloalkyl; R Y is halo, C1-C6 alkyl, C1-C6 alkoxy or C1-C6 alkylamino; each R Z is independently hydrogen, halo, C 1 halo, C 1 hale
- R 4 and R 4 ⁇ are not hydrogen or alkyl when: (a) each of R 2 , R 2 ⁇ , R 3 , R 3 ⁇ is hydrogen, (b) ring Y is phenyl, thiophenyl, thiazolyl, 6-membered heteroaryl or cubanyl, and (c) ring Z is imidazolyl; or R 4 and R 3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; subscript p is 1, 2 or 3; subscript q is 2, 3 or 4; subscript m is 0 or
- the present application provides compounds of formula (IA), or pharmaceutically acceptable salts thereof or stereoisomers thereof.
- compounds of formula (IA) and pharmaceutically acceptable salts thereof and stereoisomers thereof are provided, wherein: ring Y is , wherein the asterisk marks the point of attachment to ring Z; ring Z is imidazolyl or pyrazolyl; X 1 and X 2 are N; Y 1 to Y 4 are independently N or C, wherein 1-2 of Y 1 to Y 4 are N; each R X is independently C 1 -C 6 alkoxy or C 3 -C 8 cycloalkyl; each R Z is independently C1-C6 alkyl or halo(C1-C6)alkyl; G is -C(R 4 R 4 ⁇ )-; R 1 is hydrogen, C1-C6 alkyl, or halo(C1-C6)alkyl; R 2 ⁇ and R 3 ⁇ are
- ring Y is pyridin-2-yl, pyridin-3-yl, or pyrazin-2-yl
- ring Z is imidazol-2-yl or pyrazol-1-yl
- X 1 and X 2 are N
- each R X is independently C1-C6 alkoxy or C3-C8 cycloalkyl
- each R Z is independently C1-C6 alkyl or C1-C6 haloalkyl
- G is -CH2-;
- R 1 is C 1 -C 6 alkyl;
- R 2 ⁇ and R 3 ⁇ are hydrogen and R 2 and R 3 are taken together to form a C1-C3 alkylene bridge; or
- R 2 and R 3 are hydrogen and R 2 ⁇ and R 3 ⁇ are taken together to form a C1-C3 alkylene bridge;
- subscript p is 2
- subscript m is
- the present application provides compounds of formula (IB), or pharmaceutically a . [0047] In some embodiments, the present application provides compounds of formula (IC), or pharmaceutic . [0048] In some embodiments, the present application provides compounds of formula (ID), or pharmaceutically acceptable salts thereof or stereoisomers thereof.
- the present application provides compounds of formula (IE), or pharmaceutic [0050] In some embodiments, the present application provides compounds of formula (IF), or pharmaceutic [0051] In some embodiments, the present application provides compounds of formula (IG), or pharma ceut ca y accepta e sa ts t ereo or stereo somers t ereof, wherein the bridge L G is -(CH2)1-3- or -CH2-O-CH2-.
- ring Z is imidazolyl or pyrazolyl; X 1 and X 2 are N; Y 1 to Y 4 are independently N or C, wherein 1-2 of Y 1 to Y 4 are N; each R X is independently C 1 -C 6 alkoxy or C 3 -C 8 cycloalkyl; each R Z is independently C1-C6 alkyl or halo(C1-C6)alkyl; L G is -(CH 2 ) 1-3 -; G is -C(R 4 R 4 ⁇ )-; R 1 is hydrogen, C 1 -C 6 alkyl, or halo(C 1 -C 6 )alkyl; R 4 and R 4 ⁇ are hydrogen or R 4 and R 4 ⁇ , taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; subscript p
- ring Z is imidazol-2-yl or pyrazol-1-yl; X 1 and X 2 are N; each R X is independently C 1 -C 6 alkoxy or C 3 -C 8 cycloalkyl; each R Z is independently C1-C6 alkyl or halo(C1-C6)alkyl; L G is -(CH 2 ) 1-3 -; G is -CH2-; R 1 is C 1 -C 6 alkyl; subscript p is 2; and subscript n is 2.
- the present application provides compounds of formula (IH), or pharmac eutically acceptable salts thereof or stereoisomers thereof, wherein the bridge L G is -(CH 2 ) 1-3 - or -CH 2 -O-CH 2 -.
- the present application provides fused pyrazole derivatives of formula (I), and pharmaceutically acceptable salts and stereoisomers thereof, wherein: ring ; ring Y is , wherein the asterisk marks the point of attachment to ring Z; ring Z is 5- to 14-membered heteroaryl; X 1 and X 2 are independently N or C; Y 1 to Y 4 are independently N or C, wherein 0-2 of Y 1 to Y 4 are N; each R X is independently C 1 -C 6 alkoxy or C 3 -C 8 cycloalkyl; each R Z is independently hydrogen, halo, C1-C6 alkyl or C1-C6 haloalkyl; G is -[C(R G2 R G3 )] q - R 1 is C1-C6 alkyl, halo(C1-C6)alkyl or C3-C8 cycloalkyl; R 2 , R 2 ⁇ , R 3
- ring X i In some embodiments, ring X i . [0057] In some embodiments, ring X i . [0058] In some embodiments, ring Y i , wherein the asterisk marks the point of attachment to ring Z. [0059] In some embodiments, ring Y i , int e [0061] In some embodiments, ring Z is a 5-membered heteroaryl. [0062] In some embodiments, rin ein the asterisk marks the point of attac [0063] In some embodiments, ring Z is imidazolyl or pyrazolyl.
- Substituents R X , R Y and R Z may be present on any atom of ring X, ring Y and ring Z. In some embodiments, any one or more of R X , R Y and R Z is present on a carbon atom of ring X, ring Y and ring Z. In some embodiments, any one or more of R X , R Y and R Z is present on a nitrogen atom of ring X, ring Y and ring Z. [0065] In some embodiments, R 1 is C 1 -C 6 alkyl or C 3 -C 8 cycloalkyl. In some embodiments, R 1 is C1-C6 alkyl.
- R 1 is C3-C8 cycloalkyl.
- R 2 is C1-C6 alkyl.
- R 3 is C1-C6 alkyl.
- R X is C1-C6 alkyl. In some embodiments, R X is methyl or isopropyl.
- R X is C 1 -C 6 alkoxy. In some embodiments, R X is methoxy or ethoxy.
- R X is C3-C8 cycloalkyl. In some embodiments, R X is cyclopropyl.
- each R X is independently C1-C6 alkoxy or C3-C8 cycloalkyl. In some embodiments, each R X is independently methoxy or cyclopropyl. [0072] In some embodiments, G is -C(R 4 R 4 ⁇ )-. In some embodiments, G is -[C(R G2 R G3 )] q - or -C(R 4 R 4 ⁇ )-, and q is 2.
- R 2 and R 3 are taken together to form a C 1 -C 3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S.
- R 4 and R 3 are taken together to form a C 1 -C 3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S.
- R 4 and R 4 ⁇ are taken together along with the carbon atom to which they are attached to form C3-C8 cycloalkyl.
- R Y is halo. In some embodiments, R Y is C1-C6 alkoxy. [0077] In some embodiments, R Z is C1-C6 alkyl. In some embodiments, R Z is C1-C6 haloalkyl. In some embodiments, R Z is halo. In some embodiments, R Z is C 1 -C 6 alkoxy. In some embodiments, R Z is C3-C8 cycloalkyl. [0078] In some embodiments, each R Z is independently C1-C6 alkyl or halo(C1-C6)alkyl.
- each R Z is independently methyl, ethyl, isopropyl, or trifluoromethyl. [0079] In some embodiments, each R Z is independently C 3 -C 8 cycloalkyl or halo(C1-C6)alkyl. [0080] In some embodiments, compounds of formula (I) are provided, wherein R X is C1-C6 alkoxy or C 3 -C 8 cycloalkyl, subscript m is 0, R Z is C 1 -C 6 alkyl or halo(C 1 -C 6 )alkyl, R 1 is C 1 - C6 alkyl and, G is -C(R 4 R 4 ⁇ )-.
- compounds of formula (I) are provided, wherein ring X is pyrimidinyl, ring Y is pyridinyl and ring Z is imidazolyl.
- subscript m is 1. In some embodiments, subscript m is 0.
- subscript n is 2. In some embodiments, subscript p is 2. In some embodiments, subscript q is 2.
- the present application provides a compound selected from the group consisting of: Comp 7- 7- 7- Comp ound Compound Structure Compound Name 8- Comp ound Compound Structure Compound Name - Comp ound Compound Structure Compound Name 7- 7- - 7- Comp ound Compound Structure Compound Name 7- 2- 7- )- 7- - 7- Comp ound Compound Structure Compound Name 7- Comp ound Compound Structure Compound Name - 6- 7- 7- Comp ound Compound Structure Compound Name Comp ound Compound Structure Compound Name - - Comp ound Compound Structure Compound Name - 7- 7- Comp ound Compound Structure Compound Name 7- - 7- or [0085]
- the present disclosure provides a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof or a stereoisomer thereof as described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent
- the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein.
- compounds described in the present disclosure may be admixed with a pharmaceutically acceptable excipient (such as a carrier or a diluent) or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
- a pharmaceutically acceptable excipient such as a carrier or a diluent
- Compounds of formula (I) are useful as USP1 inhibitors.
- the present disclosure provides pharmaceutical compositions for use in treating and/or preventing a disease and/or disorder responsive to the inhibition of USP1 protein expression and/or USP1 activity.
- the present disclosure provides a method of inhibiting USP1 in a subject, comprising administering to the subject in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof.
- the present disclosure provides a method of inhibiting USP1 in a subject, comprising administering to the subject an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof.
- the present disclosure provides a method of treating diseases and/or disorders mediated by USP1 comprising administering to a subject in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof.
- the subject is a human or other mammal.
- the compounds of the disclosure are typically administered in the form of a pharmaceutical composition. Such compositions can be prepared using procedures well known in the pharmaceutical art.
- the pharmaceutical composition typically includes one or more compounds described herein and one or more pharmaceutically acceptable excipients. Typically, the pharmaceutically acceptable excipients are approved by regulatory authorities or are generally regarded as safe for human or animal use.
- Examples of pharmaceutically acceptable excipients include, but are not limited to, carriers, diluents, glidants and lubricants, preservatives, buffering agents, chelating agents, polymers, gelling agents, viscosifying agents, solvents and the like.
- the pharmaceutical composition may include, for example, one or more of water, salt solutions, alcohols, polyethylene glycols, peanut oil, olive oil, sugars (e.g., lactose, sucrose, and the like), dextrin, sugar, amylose, lower alkyl ethers of cellulose, magnesium carbonate, magnesium stearate, talc, terra alba, silicic acid, gelatin, agar, pectin, acacia, fatty acids (e.g., stearic acid), fatty acid amides, and fatty acid esters including fatty acid monoglycerides and diglycerides, or the like.
- sugars e.g., lactose, sucrose, and the like
- dextrin sugar
- amylose lower alkyl ethers of cellulose
- magnesium carbonate magnesium carbonate
- magnesium stearate magnesium carbonate
- talc magnesium stearate
- talc magnesium stearate
- terra alba silicic acid
- the pharmaceutical composition may further include one or more pharmaceutically acceptable auxiliary agents, wetting agents, suspending agents, preserving agents, buffers, sweetening agents, flavouring agents, colorants or any combination of the foregoing.
- the pharmaceutical compositions may be in conventional forms, for example, tablets, capsules, solutions, suspensions, injectables or products for topical application. Further, the pharmaceutical composition of the present disclosure may be formulated so as to provide a desired release profile.
- Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges.
- Liquid formulations include, but are not limited to, syrups, emulsions and sterile injectable liquids, such as suspensions or solutions.
- Topical dosage forms of the compounds include ointments, pastes, creams, lotions, powders, solutions, eye or ear drops, impregnated dressings and may contain appropriate conventional additives such as preservatives, solvents to assist drug penetration.
- Administration of the compounds of the disclosure, in pure form or in an appropriate pharmaceutical composition can be carried out using any of the accepted routes of administration of pharmaceutical compositions.
- the route of administration may be any route which effectively transports the active compound of the present disclosure to the appropriate or desired site of action.
- Suitable routes of administration include, but are not limited to, oral, nasal, buccal, dermal, intradermal, transdermal, parenteral, rectal, subcutaneous, intravenous, intraurethral, intramuscular or topical.
- the composition is administered by oral, parenteral or inhalation routes.
- parenteral administration include administration by injection, as well as percutaneous, transmucosal, transnasal and transpulmonary administration.
- Suitable doses of the compounds for use in treating the diseases or disorders described herein can be determined by those skilled in the relevant art. Therapeutic doses are generally identified through dose ranging studies in humans based on preliminary evidence derived from animal studies. Preferably, doses are sufficient to result in a desired therapeutic benefit without causing unwanted side effects.
- the present disclosure provides a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof, for use as a medicament and for use in the manufacture of medicaments.
- the disclosure provides the use of compounds of Formula (I) in the treatment and prevention of diseases and/or disorders responsive to the inhibition of USP1 protein expression and USP1 activity, as well as in the manufacture of medicaments for treating such diseases and/or disorders. Typically, the symptoms of the disease are treated, improved, diminished and/or prevented by inhibition of USP1.
- the USP1 mediated disease and/or disorder is cancer. Accordingly, compounds of formula (I) for use in the treatment of cancer and in the manufacture of medicaments for treatment of cancer are also provided. [0101] In related embodiments, the disclosure provides a method of treating a disease or disorder mediated by USP1 in a subject comprising administering to the subject a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof. In some embodiments, the disease and/or disorder is cancer.
- the cancer is selected from a hematological cancer, a lymphatic cancer, a DNA damage repair pathway deficient cancer, a homologous- recombination deficient cancer, a cancer comprising cancer cells with a mutation in a gene encoding p53, and a cancer comprising cancer cells with a loss of function mutation in a gene encoding p53.
- the cancer is a hematological cancer or a lymphatic cancer.
- the cancer is a DNA damage repair pathway deficient cancer and/or a homologous-recombination deficient cancer.
- the cancer is a DNA damage repair pathway deficient cancer.
- the cancer is a homologous-recombination deficient cancer.
- the cancer is selected from a hematological cancer, a lymphatic cancer, a cancer comprising cancer cells with a mutation in a gene encoding p53, and a cancer comprising cancer cells with a loss of function mutation in a gene encoding p53.
- the cancer comprises cancer cells with a mutation in a gene encoding p53.
- the mutation in the gene encoding p53 is a loss of function mutation.
- methods for treating cancer and other diseases include administering to a subject in need thereof a therapeutically effective amount of a compound of the present disclosure along with one or more additional chemotherapeutic agents independently selected from anti-proliferative agents, anti-cancer agents, immunosuppressant agents and pain-relieving agents.
- the method(s) of treatment of the present disclosure generally comprises administering a safe and effective amount of a compound according to formula (I) or a pharmaceutically acceptable salt thereof to a patient (particularly a human) in need thereof.
- Compounds of the disclosure are indicated both in the therapeutic and/or prophylactic treatment of the above-mentioned conditions.
- the dosage administered will typically vary with the compound employed, the mode of administration, the treatment desired and the disorder or disease indicated.
- the compounds of the present disclosure may be used as single drug or as a pharmaceutical composition in which the compound is mixed with various pharmacologically acceptable materials, as described above.
- the compounds of the present disclosure can also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds.
- the present disclosure also embraces isotopically-labeled variants of the present disclosure which are identical to those recited herein, but for the fact that one or more atoms of the compound are replaced by an atom having the atomic mass or mass number different from the predominant atomic mass or mass number usually found in nature for the atom. All isotopes of any particular atom or element as specified are contemplated within the scope of the compounds of the disclosure and their uses.
- Exemplary isotopes that can be incorporated in to compounds of the disclosure include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine, chlorine and iodine, such as 2 H (“D”), 3 H, 11 C, 13 C, 14 C, 13 N, 15 N, 15 O, 17 O, 18 O, 32 P, 33 P, 35 S, 18 F, 36 Cl, 123 I and 125 I.
- Isotopically labeled compounds of the present disclosure can generally be prepared by following procedures analogous to those disclosed in the schemes and/or in the examples herein below, by substituting an isotopically labeled reagent for a non-isotopically labeled reagent.
- work-up includes distribution of the reaction mixture between the organic and aqueous phases, separation of layers and drying the organic layer over anhydrous sodium sulphate, filtration and evaporation of the solvent.
- Purification includes purification by silica gel chromatographic techniques, generally using ethyl acetate/petroleum ether mixtures of a suitable polarity as the mobile phase. Some of the intermediates were taken to the next step based on TLC results, without further characterization, unless otherwise specified. Analysis for the compounds of the present disclosure, unless otherwise specified, was conducted using general methods well known to a person skilled in the art.
- GENERAL SYNTHETIC SCHEMES General Scheme I: [0118] Some intermediates of the present invention are generally synthesized utilizing the process outlined in General Scheme I.
- the commercially available or synthesized GS-IA is reacted with dimethylformamide dimethyl acetal in the presence of suitable reagents and solvents (DMF, 100 °C, 16 h) to obtain GS-IB which upon reacting with hydrazine hydrate in presence of suitable reagents and solvents (EtOH, 80 °C, 16 h) affords GS-IC.
- GS-IC Treatment of GS-IC with iodine in the presence of suitable reagents and solvent(s) (K2CO3, DMF, 80 °C, 16 h) provides GS-ID.
- This GS-ID upon reacting with R 1 -halide (R 1 X) in the presence of a base such as cesium carbonate and a solvent such as 1,4-dioxane, affords GS-IE.
- Coupling GS-IE with an appropriate boronic acid or boronate ester in the presence of a suitable catalyst, base and solvent e.g., Pd(dppf)Cl2 ⁇ DCM, K2CO3, 1,4-dioxane:H2O, 100°C, 5h affords GS-IF.
- GS-IIA The commercially available or synthesized GS-IIA is reacted with a functionalized (3,3-dihalo)propan-2-one (e.g., 3,3-dibromo-1,1,1-trifluoropropan-2-one) in the presence of suitable reagents and solvents (e.g., NaOAc, H 2 O, 100 °C, 1 h; MeOH, NH 4 OH, RT, 40 min then 100 °C, 4 h) to obtain GS-IIB which upon reacting with R Z -halide in the presence of suitable reagents and solvents (K 2 CO 3 , DMF) affords GS-IIC.
- suitable reagents and solvents e.g., NaOAc, H 2 O, 100 °C, 1 h; MeOH, NH 4 OH, RT, 40 min then 100 °C, 4 h
- GS-IIB treatment of GS-IIB with an appropriate RZ-boronic acid in presence of suitable reagents and solvent(s) [e.g., Cu(OAc)2, O 2 , 2,2’-bipyridin, Na 2 CO 3 , 1,2-DCE, 70 °C, 16 h] also provides GS-IIC.
- suitable reagents and solvent(s) e.g., Cu(OAc)2, O 2 , 2,2’-bipyridin, Na 2 CO 3 , 1,2-DCE, 70 °C, 16 h
- suitable reagents and solvent(s) e.g., Cu(OAc)2, O 2 , 2,2’-bipyridin, Na 2 CO 3 , 1,2-DCE, 70 °C, 16 h
- suitable reagents and solvent(s) e.g., Cu(OAc)2, O 2 , 2,2’-bipyridin, Na 2 CO 3 , 1,2-DCE, 70
- GS-IIIA reacts with GS-IIIB in presence of suitable reagents and solvents (e.g., AcOH, reflux) to obtain GS-IIIC which upon reacting with R Z ”’-halide in presence of suitable reagents and solvents (e.g., NaH, DMF) gives intermediate GS-IIID.
- suitable reagents and solvents e.g., AcOH, reflux
- suitable reagents and solvents e.g., NaH, DMF
- GS-IG is reacted with appropriate intermediates GS-IIC or GS-IIIC or GS-IIID, in the presence of suitable reagents and solvents under Buchwald coupling conditions (e.g., Pd2(dba)3, RuPhos, Cs2CO3, PhMe, 100°C, 2h) to afford respective compounds of formula (I).
- suitable reagents and solvents e.g., Pd2(dba)3, RuPhos, Cs2CO3, PhMe, 100°C, 2h
- S ess outlined in General Scheme V.
- the commercially available or synthesized GS-IG is reacted with an appropriate intermediate GS-IIC in the presence of suitable reagents and solvents (e.g., DIPEA, NMP, 130 o C, 12h) to afford respective compounds of formula (I).
- Table VI The compounds in Table VI were prepared according to procedures similar to the procedure described for Compound 1, using appropriate starting materials and intermediates as set forth above.
- the final concentrations of USP1-UAF1 complex protein and substrate Ubiquitin- Rhodamine-110 (R&D systems, Catalog U-555-050) used in the assay were 0.45 and 150 nM respectively.50 mM HEPES pH.7.5, 100 mM NaCl, 0.5 mM EDTA, 1 mM TCEP, 10% BSA, 0.01% Tween 20 buffer was used in the assay. The total assay volume was 20 ⁇ L. The compounds were initially prepared in 100% DMSO and appropriate dilutions were made by 1/3 rd serial dilutions from the stock, for use in determination of IC50 values. The final DMSO concentration in the assay was 1%.
- IC50 values of the compounds are summarized in Table VIII below, wherein “A” refers to an IC 50 value less than 0.05 ⁇ M, “B” refers to an IC50 value in range of 0.05 ⁇ M to 0.5 ⁇ M and “C” refers to an IC50 value greater than 0.5 ⁇ M.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present disclosure provides fused pyrazole derivatives of formula (I), which are therapeutically useful as USP1 inhibitors. These compounds are useful in the treatment and/or prevention of diseases and/or disorders responsive to the inhibition of USP1 protein expression and USP1 activity. Compounds of the present disclosure are especially useful for treating cancer. The present disclosure also provides processes for preparation of the compounds and pharmaceutical formulations comprising at least one of the compounds of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof.
Description
FUSED PYRAZOLE DERIVATIVES AS USP1 INHIBITORS CROSS-REFERENCES TO RELATED APPLICATIONS [0001] The present application claims priority to Indian provisional application no. 202341045361, filed July 6, 2023, the entire contents of which are incorporated herein by reference. TECHNICAL FIELD [0002] The present application is directed to fused pyrazole derivatives of formula (I) as USP1 inhibitors, useful for the treatment of cancer and inflammatory diseases or disorders. The disclosure also provides pharmaceutically acceptable compositions comprising compounds of the present application and methods of using said compositions in the treatment of diseases associated with USP1. BACKGROUND [0003] Deubiquitinases (DUBs) are a class of enzymes that act on ubiquitinated substrates to catalyze the removal of ubiquitin moieties. The human genome contains approximately 100 genes that encode DUBs. Human DUBs are classified into five different families (Nijman, S.M. et al. (2005) Cell 123, 773-86, Nalepa, G. et al. (2006) Nat Rev Drug Discov 5, 596-613). USP1 (ubiquitin specific protease 1) belongs to the USP subfamily of DUBs. The USP1 gene encodes a 785 amino acid protein having a conserved USP domain amino-terminal Cys box motif and a carboxy-terminal His box motif (Nijman, S.M. et al. (2005) Mol Cell 17, 331-9). The interaction of USP1 with UAF1, a WD40 repeat-containing protein, leads to formation of an activated USP1/UAF1 complex, which is required for the deubiquitinase activity of USP1 (Cohn, M.A. et al. (2007) Mol Cell 28, 786-97, Cohn, M.A. et al. (2009) J Biol Chem 284, 5343-51). USP1 gene transcription is regulated in a cell cycle- dependent manner. mRNA levels of USP1 remain low during G1 phase and reach a peak during S phase (Nijman, S.M. et al. (2005) Mol Cell 17, 331-9). The expression of USP1 is also regulated at the protein level by proteasomal degradation (Cataldo F, Mol Cell Biol (2013), 33(12):2485–2496). [0004] USP1 is a nuclear protein and localizes to chromatin where it is specifically associated with Fanconi anemia protein FANCD2. USP1 regulates several important steps in the DNA damage response pathway, including the Fanconi anemia (FA) pathway and the
process of translesion synthesis (TLS). USP1 deubiquitinates monoubiquitinated FANCD2, which plays an important role in DNA damage repair (Nijman, S.M. et al. (2005) Mol Cell 17, 331-9, Guervilly, J.H. et al. (2011) Hum Mol Genet). While DNA-dependent mono- ubiquitination of FANCD2 facilitates DNA repair, it is deubiquitinated by USP1 to block the DNA-repairing response. USP1 is also critical for the deubiquitination of monoubiquitinated PCNA and thus negatively regulates PCNA-mediated TLS during DNA repair (Huang TT. et al. Nat Cell Biol (2006), 8(4):339–347). The expression of USP1 is significantly increased in several cancers (Das DS. et al. Clin Cancer Res. (2017) 23:4280–9, Xin Xu, et al. Front Oncol. (2019) 9:1406). Inhibition of USP1 inhibited DNA repair and induced cell death in multiple myeloma cells (Das DS. Et al. Clin Cancer Res. (2017) 23:4280–9). Lung cancer cells are sensitized to cisplatin upon inhibition of USP1 activity (Chen J, et al. Chem Biol. (2011) 18:1390–400). The results from these studies indicate that USP1 is a promising anti-cancer target. [0005] Certain USP1 inhibitors are reported in the literature, including compounds described in WO2007/149484, WO2011/137320, WO2014/105952, WO2017/087837, WO2019/089216, WO2020/132269, WO2020/139988, WO2021/163530, WO2021/247606, WO2022/174184, WO2022/197892, WO2022/199652, WO2022/214053, WO2022/216820, WO2022/228399, WO2022/233263, WO2022/253188, WO2023/066299, WO2023/030295, WO2023/083285, WO2023/083286, and WO2023/083297. SUMMARY [0006] Inhibition of USP1 with small molecule inhibitors has the potential to be a treatment for cancers and other disorders. It is, therefore, an object of this disclosure to provide compounds useful in the treatment of such diseases and/or disorders responsive to the inhibition of USP1 protein expression and USP1 activity. [0007] Provided herein are fused pyrazole derivatives of formula (I) and pharmaceutical compositions thereof, which are capable of inhibiting USP1.
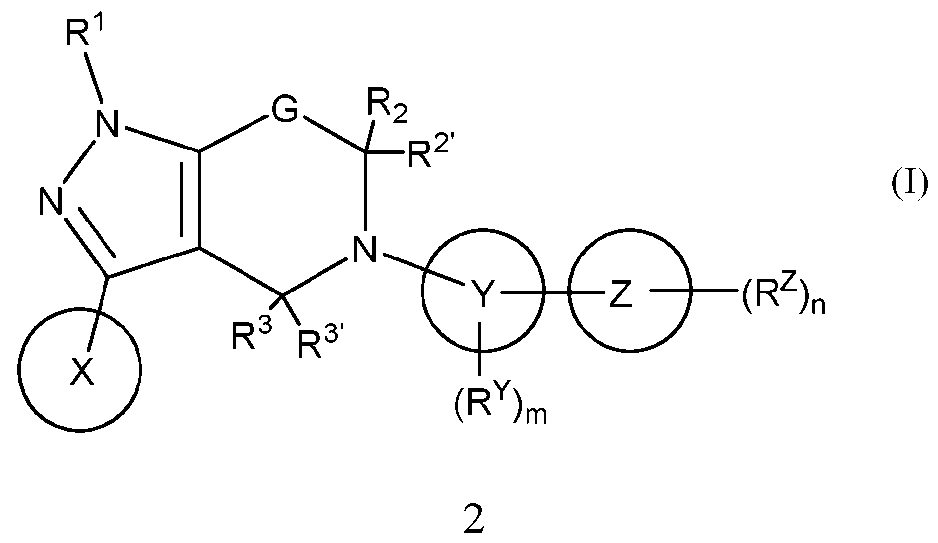 [0008] In compounds of formula (I) and pharmaceutically acceptable salts or stereoisomers thereof ;
[0008] In compounds of formula (I) and pharmaceutically acceptable salts or stereoisomers thereof ;
 ring Y is C3-C8 cycloalkyl, C5-C10 cycloalkeny , wherein the asterisk marks the point of attachment to rin
ring Y is C3-C8 cycloalkyl, C5-C10 cycloalkeny , wherein the asterisk marks the point of attachment to rin
 ring Z is 3- to 15-membered heterocycloalkyl or 5- to 14-membered heteroaryl; X1, X2 and X3 are independently N or C; Y1 to Y4 are independently N or C, wherein 0-2 of Y1 to Y4 are N; Y5 and Y6 are independently N, C, O or S; each RX is independently C1-C6 alkyl, C1-C6 alkoxy or C3-C8 cycloalkyl; RY is halo, C1-C6 alkyl, C1-C6 alkoxy or C1-C6 alkylamino; each RZ is independently hydrogen, halo, C1-C6 alkyl, halo(C1-C6)alkyl, C1-C6 alkoxy, C3-C8 cycloalkyl, (C1-C6)alkylamino-(C1-C6)alkyl, (C1-C6)alkoxy-(C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl or (C1-C6 alkyl)-substituted 3- to 15-membered heterocycloalkyl, wherein halo and C1-C6 alkoxy are bonded to carbon atoms in the Z ring; G is -O-, -N(RG1)-, *-C(RG2RG3)-N(RG1)-, *-N(RG1)-C(RG2RG3)-, *-C(RG2RG3)-O-, -[C(RG2RG3)]q- or -C(R4R4 ^)-, wherein the asterisk marks the point of attachment to the pyrazole ring; RG1, each RG2 and each RG3 are independently hydrogen or C1-C6 alkyl; R1 is hydrogen, C1-C6 alkyl, halo(C1-C6)alkyl, C3-C8 cycloalkyl, (C1-C6)alkylamino- (C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl, (C1-C6)alkyl-substituted 3- to 15-membered heterocycloalkyl, optionally substituted (C6-C14)aryl(C1-C6)alkyl, or optionally substituted (5- to 14-membered heteroaryl)-(C1-C6)alkyl, wherein the substituents on (C6- C14)aryl(C1-C6)alkyl and (5- to 14-membered heteroaryl)-(C1-C6 )alkyl are independently halo, C1-C6 alkyl or C1-C6 alkoxy; R2, R2 ^, R3 and R3 ^ are independently hydrogen, C1-C6 alkyl, halo, cyano or amino; or R2 and R2 ^, taken together, form an oxo group; or
R2 and R2 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; or R2 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; or R3 and RG2 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; R4 and R4 ^ are: i. taken together along with the carbon atom to which they are attached to form a C3-C8 cycloalkyl, ii. independently hydroxy or C1-C6 alkoxy, or iii. independently hydrogen or C1-C6 alkyl, with the proviso that R4 and R4 ^ are not hydrogen or alkyl when: (a) each of R2, R2 ^, R3, R3 ^ is hydrogen, (b) ring Y is phenyl, thiophenyl, thiazolyl, 6-membered heteroaryl or cubanyl, and (c) ring Z is imidazolyl; or R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; subscript p is 1, 2 or 3; subscript q is 2, 3 or 4; subscript m is 0 or 1; and subscript n is 1, 2 or 3. [0009] Also provided are pharmaceutical compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and at least one pharmaceutically acceptable excipient, such as a pharmaceutically acceptable carrier or diluent. [0010] Also provided are processes for the preparation of compounds of formula (I). [0011] Compounds of formula (I) are useful for the inhibition of USP1, and the therapeutic use of such compounds is also provided.
DETAILED DESCRIPTION [0012] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as is commonly understood by one of skill in art to which the subject matter herein belongs. As used in the specification and the appended claims, unless specified to the contrary, the following terms have the meaning indicated to facilitate the understanding of the present disclosure. [0013] As used herein, the term “alkyl,” alone or in combination with other terms, refers to saturated aliphatic hydrocarbon chains, including C1-C10 straight or C1-C10 branched alkyl chains, (e.g., C1-C6 straight or branched alkyl chains). Examples of “alkyl” include but are not limited to methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, isopentyl or neopentyl and the like. The term “alkylene” refers to an alkyl group, as defined above, linking at least two other groups (i.e., a divalent alkyl radical). The two moieties linked to the alkylene group can be linked to the same carbon atom or different carbon atoms of the alkylene group. The alkylene group preferably has 1-6 carbon atoms and, more preferably, 1- 3 carbon atoms. [0014] As used herein, the terms “halo” and “halogen,” alone or in combination with other terms, are used interchangeably to refer to fluorine, chlorine, bromine or iodine. [0015] As used herein, the term “haloalkyl,” alone or in combination with other terms, refers to alkyl substituted with one or more halogen atoms, wherein the alkyl groups are as defined above. Examples of “haloalkyl” include but are not limited to fluoromethyl, difluoromethyl, chloromethyl, trifluoromethyl, 2,2,2-trifluoroethyl and the like. [0016] As used herein, the term “alkoxy,” alone or in combination with other terms, refers to the group alkyl-O- or -O-alkyl, where the alkyl groups are as defined above. Examples of “alkoxy” include but are not limited to methoxy, ethoxy, n-propoxy, n-butoxy, t-butoxy and the like. [0017] As used herein, the term “alkoxyalkyl,” alone or in combination with other terms, refers to the “alkoxy” as defined above linked to the rest of the molecule via an alkyl moiety. Examples of “alkoxyalkyl” include but not limited to -CH2-OCH3, -C2H4-OCH3, -CH2-OCH2CH3, -CH2-OC3H7, -C2H4-OCH2CH3 and the like. [0018] The terms “amino” and “amine,” alone or in combination with other terms, refer to a primary amine (–NH2), a secondary amine (wherein ‘N’ is substituted with two substituents
other than hydrogen) or a tertiary amine (wherein ‘N’ is substituted with three substituents other than hydrogen). [0019] As used herein, the term “alkylamino,” alone or in combination with other terms, refers to an amino group substituted with one or more alkyl groups, wherein the alkyl group and amino group are as defined above. Examples of “alkylamino” groups include but are not limited to -NHCH3, -NHCH2CH3, -N(CH3)2, -N(CH3)(CH2CH3) and the like. [0020] As used herein, the term “alkylaminoalkyl,” alone or in combination with other terms, refers to an alkylamino group as defined above linked to the rest of the molecule via an alkyl moiety. Examples of “alkylaminoalkyl” include but are not limited to -CH2-NHCH3, -C2H4-NHCH3, -CH2-NHCH2CH3, -CH2-N(CH3)2, -CH2-N(CH3)(CH2CH3) and the like. [0021] As used herein, the term “cycloalkyl,” alone or in combination with other terms, refers to a C3-C10 saturated cyclic hydrocarbon ring. A cycloalkyl may be monocyclic, typically containing from 3 to 7 carbon ring atoms. Examples of monocyclic cycloalkyls include but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and the like. A cycloalkyl may alternatively be polycyclic, containing more than one ring. Examples of polycyclic cycloalkyls include bridged, fused and spirocyclic cycloalkyls and the like. [0022] As used herein, the term “cycloalkenyl,” alone or in combination with other terms, refers to a partially unsaturated C5-C12 cyclic hydrocarbon ring with at least one C-C double bond. A cycloalkenyl may be monocyclic, typically containing from 5 to 10 carbon ring atoms. Examples of monocyclic cycloalkenyls include but are not limited to, cyclopentenyl, cyclohexenyl, cyclohepentyl, 1,3-cyclohexadienyl and the like. A cycloalkenyl may alternatively be a polycyclic moiety containing more than one ring. Examples of polycyclic cycloalkenyls include, but are not limited to, bridged, fused and spirocyclic ring systems such as bicyclo[2.2.1]hept-2-enyl, bicyclo[4.4.0]dec-1-enyl and the like. [0023] As used herein, the term “aryl,” alone or in combination with other terms, refers to an unsubstituted or substituted monocyclic, bicyclic or polycyclic aromatic hydrocarbon ring system of about 6 to 14 carbon atoms. Examples of “aryl” include, but are not limited to phenyl, naphthyl, anthryl, and biphenyl. An aryl group may be unsubstituted or substituted with one or more suitable groups.
[0024] As used herein, the term “arylalkyl,” alone or in combination with other terms, refers to an aryl group as defined above linked to the rest of the molecule via an alkyl moiety. Examples of “arylalkyl” include but are not limited to (phenyl)alkyl-, (naphthyl)alkyl-, (anthryl)alkyl- and the like. [0025] The term “heteroatom” as used herein refers to a sulfur, nitrogen or oxygen atom. [0026] The term “heterocycloalkyl,” alone or in combination with other terms, refers to a non-aromatic, saturated or partially saturated monocyclic or polycyclic ring system of 3 to 15 members having at least one heteroatom or heterogroup selected from O, N, S, S(O), S(O)2and NH, with the remaining ring atoms being independently selected from carbon, oxygen, nitrogen and sulfur. A monocyclic heterocycloalkyl typically contains 4 to 7 ring atoms. Examples of “heterocycloalkyl” include, but are not limited to azetidinyl, oxetanyl, imidazolidinyl, pyrrolidinyl, oxazolidinyl, thiazolidinyl, pyrazolidinyl, tetrahydrofuranyl, piperidinyl, piperazinyl, tetrahydropyranyl, morpholinyl, oxopiperazinyl, oxopiperidinyl, tetrahydrothiophenyl, dihydropyranyl, indolinyl, azepanyl and N-oxides thereof. Attachment of a heterocycloalkyl substituent can occur via either a carbon atom or a heteroatom. A heterocycloalkyl group can be unsubstituted or substituted with one or more suitable groups. [0027] As used herein, the term “heteroaryl” alone or in combination with other terms, refers to an aromatic unsaturated ring system containing a total of 5 to 14 ring atoms. At least one of the ring atoms is a heteroatom (oxygen, nitrogen, or sulfur), with the remaining ring atoms/groups being independently selected from carbon, oxygen, nitrogen and sulfur. A heteroaryl may be a single-ring (monocyclic) or polycyclic ring system. Examples of “heteroaryl” include but are not limited to pyridyl, indolyl, benzimidazolyl, benzothiazolyl, pyrrolyl, pyrazolyl, imidazolyl, pyrimidinyl, pyrazinyl, pyridazinyl and the like. [0028] As used herein, the term “heteroarylalkyl” alone or in combination with other terms, refers to the heteroaryl as defined above is linked to the rest of the molecule via an alkyl moiety. Examples of “heteroarylalkyl” include but are not limited to (pyridine)alkyl-, (indole)alkyl-, (benzimidazole)alkyl-, (pyrrole)alkyl-, (pyrazole)alkyl-, (imidazole)alkyl-, (pyrimidine)- alkyl-, (pyrazine)alkyl-, (pyridazine)alkyl- and the like. [0029] As used in the above definitions, the term “optionally substituted” or “substituted” or “optionally substituted with suitable groups” refers to replacement of one or more hydrogen radicals in a given structure with a radical of a substituent such as halo, alkyl,
alkenyl, alkynyl, aryl, heteroaryl, heterocycloalkyl, thiol, alkylthio, arylthio, alkylthioalkyl, arylthioalkyl, alkylsulfonyl, alkylsulfonylalkyl, arylsulfonylalkyl, alkoxy, aryloxy, arylalkoxy, aminocarbonyl, alkylaminocarbonyl, arylaminocarbonyl, alkoxycarbonyl, aryloxycarbonyl, haloalkyl (e.g., trifluoromethyl), amino, cyano, nitro, alkylamino, arylamino, alkylaminoalkyl, arylaminoalkyl, aminoalkylamino, hydroxy, alkoxyalkyl, carboxyalkyl, alkoxycarbonylalkyl, aminocarbonylalkyl, acyl, arylalkoxycarbonyl, carboxylate, sulfonate, sulfonyl, phosphate, phosphonate, phosphinate, or the like. It is understood that any substituent may be further substituted. [0030] As used herein, the term “including” as well as other forms, such as “include”, “includes” and “included,” is not limiting. [0031] As used herein, the terms “comprise” and “comprising” are generally used in the sense of “include” and “including,” i.e., permitting the presence of one or more features or components. [0032] As used herein, the term “or” means “and/or” unless stated otherwise. [0033] As used herein, the term “composition” is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts. [0034] As used herein, the terms “treat”, “treating” and “treatment” refer to the alleviation or abrogation of a disease and/or its attendant symptoms. [0035] As used herein, the terms “prevent”, “preventing” and “prevention” refer to preventing the onset of a disease and/or its attendant symptoms or barring a subject from acquiring a disease. As used herein, “prevent”, “preventing” and “prevention” also include delaying the onset of a disease and/or its attendant symptoms and reducing a subject's risk of acquiring a disease. [0036] As used herein, the term “therapeutically effective amount” refers to the amount of the compound being administered sufficient to prevent development of or alleviate to some extent one or more of the symptoms of the disease or disorder being treated. [0037] “Pharmaceutically acceptable” refers to substances that are generally safe, non-toxic and neither biologically undesirable nor otherwise undesirable, including substances which
are acceptable for human pharmaceutical use and veterinary use. A pharmaceutically acceptable excipient, for example, is compatible with the other ingredients of the formulation and not deleterious to the recipient thereof. [0038] As used herein, the phrase “pharmaceutically acceptable excipient” refers to a pharmaceutically acceptable material that aids the administration of an active agent to a subject. Examples of excipients include, but are not limited to, liquid or solid fillers, diluents, solvents, encapsulating materials, disintegrants, lubricants, glidants, sweeteners, flavors, and colors. See, e.g., Remington: The Science and Practice of Pharmacy, 23rd ed.; Academic Press: Cambridge, Mass., 2020; Handbook of Pharmaceutical Excipients, 9th ed.; Sheskey et al, Eds.; Pharmaceutical Press 2020; Handbook of Pharmaceutical Additives, 3rd ed.; Ash and Ash Eds.; Gower Publishing Company: 2007; Pharmaceutical Preformulation and Formulation, 2nd ed.; Gibson Ed.; CRC Press LLC: Boca Raton, Fla., 2009. [0039] As used herein, “pharmaceutically acceptable salt(s)” refers to a parent compound which is modified by converting an existing acid or base moiety to its salt form. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like. The pharmaceutically acceptable salts of the present disclosure include conventional non-toxic salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. The pharmaceutically acceptable salts of the present disclosure can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, non-aqueous media like ether, ethyl acetate, alcohols or acetonitrile (ACN) are preferred. [0040] The term “stereoisomers” refers to any enantiomers, diastereoisomers, or geometric isomers of the compounds of Formula (I), (IA), (IB), (IC), (ID), (IE), (IF), (IG) and (IH), wherever they are chiral or when they bear one or more double bonds. When the compounds of formula (I), (IA), (IB), (IC), (ID), (IE), (IF), (IG) and (IH) are chiral, they can exist in racemic form or in optically active form. It should be understood that the disclosure encompasses all stereochemical isomeric forms, including diastereomeric, enantiomeric and epimeric forms, including (R) isomers, (S) isomers, d-isomers and l-isomers and mixtures thereof. Individual stereoisomers of compounds can be prepared synthetically from
commercially available starting materials which contain chiral centers or by preparation of mixtures of enantiomeric products followed by separation, e.g., via conversion to a mixture of diastereomers followed by separation or recrystallization, chromatographic techniques (including direct separation of enantiomers on chiral chromatographic columns), or any other appropriate method known in the art. Starting compounds of particular stereochemistry are either commercially available or can be made and resolved by techniques known in the art. Additionally, the compounds of the present disclosure may exist as geometric isomers. The present disclosure includes all cis and trans, syn and anti, entgegen (E) and zusammen (Z) isomers as well as the appropriate mixtures thereof. [0041] The present disclosure provides fused pyrazole derivatives of formula (I), which are useful for the inhibition of USP1. The present disclosure further provides pharmaceutical compositions comprising the compounds of formula (I), and their derivatives as therapeutic agents. It will be apparent to those skilled in the art that various modifications and variations can be made to the compounds, compositions, and methods described herein without departing from the scope or spirit of various embodiments disclosed herein. For instance, features illustrated or described as part of one embodiment can be applied to another embodiment to yield a still further embodiment. Thus, it is intended that the present application includes such modifications and variations and their equivalents. Other objects, features, and aspects of the present application are disclosed in the following detailed description. It is to be understood by one of ordinary skill in the art that the present discussion is a description of exemplary embodiments and is not to be construed as limiting the broader aspects of the present disclosure. The embodiments below are illustrative of the present disclosure and are not intended to limit the claims to the specific embodiments exemplified. [0042] Provided herein are fused pyrazole derivatives of formula (I), and pharmaceuticall
ring Z is 3- to 15-membered heterocycloalkyl or 5- to 14-membered heteroaryl; X1, X2 and X3 are independently N or C; Y1 to Y4 are independently N or C, wherein 0-2 of Y1 to Y4 are N; Y5 and Y6 are independently N, C, O or S; each RX is independently C1-C6 alkyl, C1-C6 alkoxy or C3-C8 cycloalkyl; RY is halo, C1-C6 alkyl, C1-C6 alkoxy or C1-C6 alkylamino; each RZ is independently hydrogen, halo, C1-C6 alkyl, halo(C1-C6)alkyl, C1-C6 alkoxy, C3-C8 cycloalkyl, (C1-C6)alkylamino-(C1-C6)alkyl, (C1-C6)alkoxy-(C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl or (C1-C6 alkyl)-substituted 3- to 15-membered heterocycloalkyl, wherein halo and C1-C6 alkoxy are bonded to carbon atoms in the Z ring; G is -O-, -N(RG1)-, *-C(RG2RG3)-N(RG1)-, *-N(RG1)-C(RG2RG3)-, *-C(RG2RG3)-O-, -[C(RG2RG3)]q- or -C(R4R4 ^)-, wherein the asterisk marks the point of attachment to the pyrazole ring; RG1, each RG2 and each RG3 are independently hydrogen or C1-C6 alkyl; R1 is hydrogen, C1-C6 alkyl, halo(C1-C6)alkyl, C3-C8 cycloalkyl, (C1-C6)alkylamino- (C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl, (C1-C6)alkyl-substituted 3- to 15-membered heterocycloalkyl, optionally substituted (C6-C14)aryl(C1-C6)alkyl, or optionally substituted (5- to 14-membered heteroaryl)-(C1-C6)alkyl, wherein the substituents on (C6- C14)aryl(C1-C6)alkyl and (5- to 14-membered heteroaryl)-(C1-C6 )alkyl are independently halo, C1-C6 alkyl or C1-C6 alkoxy; R2, R2 ^, R3 and R3 ^ are independently hydrogen, C1-C6 alkyl, halo, cyano or amino; or R2 and R2 ^, taken together, form an oxo group; or
R2 and R2 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; or R2 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; or R3 and RG2 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; R4 and R4 ^ are: i. taken together along with the carbon atom to which they are attached to form a C3-C8 cycloalkyl, ii. independently hydroxy or C1-C6 alkoxy, or iii. independently hydrogen or C1-C6 alkyl, with the proviso that R4 and R4 ^ are not hydrogen or alkyl when: (a) each of R2, R2 ^, R3, R3 ^ is hydrogen, (b) ring Y is phenyl, thiophenyl, thiazolyl, 6-membered heteroaryl or cubanyl, and (c) ring Z is imidazolyl; or R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; subscript p is 1, 2 or 3; subscript q is 2, 3 or 4; subscript m is 0 or 1; and subscript n is 1, 2 or 3. [0009] Also provided are pharmaceutical compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and at least one pharmaceutically acceptable excipient, such as a pharmaceutically acceptable carrier or diluent. [0010] Also provided are processes for the preparation of compounds of formula (I). [0011] Compounds of formula (I) are useful for the inhibition of USP1, and the therapeutic use of such compounds is also provided.
DETAILED DESCRIPTION [0012] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as is commonly understood by one of skill in art to which the subject matter herein belongs. As used in the specification and the appended claims, unless specified to the contrary, the following terms have the meaning indicated to facilitate the understanding of the present disclosure. [0013] As used herein, the term “alkyl,” alone or in combination with other terms, refers to saturated aliphatic hydrocarbon chains, including C1-C10 straight or C1-C10 branched alkyl chains, (e.g., C1-C6 straight or branched alkyl chains). Examples of “alkyl” include but are not limited to methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, isopentyl or neopentyl and the like. The term “alkylene” refers to an alkyl group, as defined above, linking at least two other groups (i.e., a divalent alkyl radical). The two moieties linked to the alkylene group can be linked to the same carbon atom or different carbon atoms of the alkylene group. The alkylene group preferably has 1-6 carbon atoms and, more preferably, 1- 3 carbon atoms. [0014] As used herein, the terms “halo” and “halogen,” alone or in combination with other terms, are used interchangeably to refer to fluorine, chlorine, bromine or iodine. [0015] As used herein, the term “haloalkyl,” alone or in combination with other terms, refers to alkyl substituted with one or more halogen atoms, wherein the alkyl groups are as defined above. Examples of “haloalkyl” include but are not limited to fluoromethyl, difluoromethyl, chloromethyl, trifluoromethyl, 2,2,2-trifluoroethyl and the like. [0016] As used herein, the term “alkoxy,” alone or in combination with other terms, refers to the group alkyl-O- or -O-alkyl, where the alkyl groups are as defined above. Examples of “alkoxy” include but are not limited to methoxy, ethoxy, n-propoxy, n-butoxy, t-butoxy and the like. [0017] As used herein, the term “alkoxyalkyl,” alone or in combination with other terms, refers to the “alkoxy” as defined above linked to the rest of the molecule via an alkyl moiety. Examples of “alkoxyalkyl” include but not limited to -CH2-OCH3, -C2H4-OCH3, -CH2-OCH2CH3, -CH2-OC3H7, -C2H4-OCH2CH3 and the like. [0018] The terms “amino” and “amine,” alone or in combination with other terms, refer to a primary amine (–NH2), a secondary amine (wherein ‘N’ is substituted with two substituents
other than hydrogen) or a tertiary amine (wherein ‘N’ is substituted with three substituents other than hydrogen). [0019] As used herein, the term “alkylamino,” alone or in combination with other terms, refers to an amino group substituted with one or more alkyl groups, wherein the alkyl group and amino group are as defined above. Examples of “alkylamino” groups include but are not limited to -NHCH3, -NHCH2CH3, -N(CH3)2, -N(CH3)(CH2CH3) and the like. [0020] As used herein, the term “alkylaminoalkyl,” alone or in combination with other terms, refers to an alkylamino group as defined above linked to the rest of the molecule via an alkyl moiety. Examples of “alkylaminoalkyl” include but are not limited to -CH2-NHCH3, -C2H4-NHCH3, -CH2-NHCH2CH3, -CH2-N(CH3)2, -CH2-N(CH3)(CH2CH3) and the like. [0021] As used herein, the term “cycloalkyl,” alone or in combination with other terms, refers to a C3-C10 saturated cyclic hydrocarbon ring. A cycloalkyl may be monocyclic, typically containing from 3 to 7 carbon ring atoms. Examples of monocyclic cycloalkyls include but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and the like. A cycloalkyl may alternatively be polycyclic, containing more than one ring. Examples of polycyclic cycloalkyls include bridged, fused and spirocyclic cycloalkyls and the like. [0022] As used herein, the term “cycloalkenyl,” alone or in combination with other terms, refers to a partially unsaturated C5-C12 cyclic hydrocarbon ring with at least one C-C double bond. A cycloalkenyl may be monocyclic, typically containing from 5 to 10 carbon ring atoms. Examples of monocyclic cycloalkenyls include but are not limited to, cyclopentenyl, cyclohexenyl, cyclohepentyl, 1,3-cyclohexadienyl and the like. A cycloalkenyl may alternatively be a polycyclic moiety containing more than one ring. Examples of polycyclic cycloalkenyls include, but are not limited to, bridged, fused and spirocyclic ring systems such as bicyclo[2.2.1]hept-2-enyl, bicyclo[4.4.0]dec-1-enyl and the like. [0023] As used herein, the term “aryl,” alone or in combination with other terms, refers to an unsubstituted or substituted monocyclic, bicyclic or polycyclic aromatic hydrocarbon ring system of about 6 to 14 carbon atoms. Examples of “aryl” include, but are not limited to phenyl, naphthyl, anthryl, and biphenyl. An aryl group may be unsubstituted or substituted with one or more suitable groups.
[0024] As used herein, the term “arylalkyl,” alone or in combination with other terms, refers to an aryl group as defined above linked to the rest of the molecule via an alkyl moiety. Examples of “arylalkyl” include but are not limited to (phenyl)alkyl-, (naphthyl)alkyl-, (anthryl)alkyl- and the like. [0025] The term “heteroatom” as used herein refers to a sulfur, nitrogen or oxygen atom. [0026] The term “heterocycloalkyl,” alone or in combination with other terms, refers to a non-aromatic, saturated or partially saturated monocyclic or polycyclic ring system of 3 to 15 members having at least one heteroatom or heterogroup selected from O, N, S, S(O), S(O)2and NH, with the remaining ring atoms being independently selected from carbon, oxygen, nitrogen and sulfur. A monocyclic heterocycloalkyl typically contains 4 to 7 ring atoms. Examples of “heterocycloalkyl” include, but are not limited to azetidinyl, oxetanyl, imidazolidinyl, pyrrolidinyl, oxazolidinyl, thiazolidinyl, pyrazolidinyl, tetrahydrofuranyl, piperidinyl, piperazinyl, tetrahydropyranyl, morpholinyl, oxopiperazinyl, oxopiperidinyl, tetrahydrothiophenyl, dihydropyranyl, indolinyl, azepanyl and N-oxides thereof. Attachment of a heterocycloalkyl substituent can occur via either a carbon atom or a heteroatom. A heterocycloalkyl group can be unsubstituted or substituted with one or more suitable groups. [0027] As used herein, the term “heteroaryl” alone or in combination with other terms, refers to an aromatic unsaturated ring system containing a total of 5 to 14 ring atoms. At least one of the ring atoms is a heteroatom (oxygen, nitrogen, or sulfur), with the remaining ring atoms/groups being independently selected from carbon, oxygen, nitrogen and sulfur. A heteroaryl may be a single-ring (monocyclic) or polycyclic ring system. Examples of “heteroaryl” include but are not limited to pyridyl, indolyl, benzimidazolyl, benzothiazolyl, pyrrolyl, pyrazolyl, imidazolyl, pyrimidinyl, pyrazinyl, pyridazinyl and the like. [0028] As used herein, the term “heteroarylalkyl” alone or in combination with other terms, refers to the heteroaryl as defined above is linked to the rest of the molecule via an alkyl moiety. Examples of “heteroarylalkyl” include but are not limited to (pyridine)alkyl-, (indole)alkyl-, (benzimidazole)alkyl-, (pyrrole)alkyl-, (pyrazole)alkyl-, (imidazole)alkyl-, (pyrimidine)- alkyl-, (pyrazine)alkyl-, (pyridazine)alkyl- and the like. [0029] As used in the above definitions, the term “optionally substituted” or “substituted” or “optionally substituted with suitable groups” refers to replacement of one or more hydrogen radicals in a given structure with a radical of a substituent such as halo, alkyl,
alkenyl, alkynyl, aryl, heteroaryl, heterocycloalkyl, thiol, alkylthio, arylthio, alkylthioalkyl, arylthioalkyl, alkylsulfonyl, alkylsulfonylalkyl, arylsulfonylalkyl, alkoxy, aryloxy, arylalkoxy, aminocarbonyl, alkylaminocarbonyl, arylaminocarbonyl, alkoxycarbonyl, aryloxycarbonyl, haloalkyl (e.g., trifluoromethyl), amino, cyano, nitro, alkylamino, arylamino, alkylaminoalkyl, arylaminoalkyl, aminoalkylamino, hydroxy, alkoxyalkyl, carboxyalkyl, alkoxycarbonylalkyl, aminocarbonylalkyl, acyl, arylalkoxycarbonyl, carboxylate, sulfonate, sulfonyl, phosphate, phosphonate, phosphinate, or the like. It is understood that any substituent may be further substituted. [0030] As used herein, the term “including” as well as other forms, such as “include”, “includes” and “included,” is not limiting. [0031] As used herein, the terms “comprise” and “comprising” are generally used in the sense of “include” and “including,” i.e., permitting the presence of one or more features or components. [0032] As used herein, the term “or” means “and/or” unless stated otherwise. [0033] As used herein, the term “composition” is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts. [0034] As used herein, the terms “treat”, “treating” and “treatment” refer to the alleviation or abrogation of a disease and/or its attendant symptoms. [0035] As used herein, the terms “prevent”, “preventing” and “prevention” refer to preventing the onset of a disease and/or its attendant symptoms or barring a subject from acquiring a disease. As used herein, “prevent”, “preventing” and “prevention” also include delaying the onset of a disease and/or its attendant symptoms and reducing a subject's risk of acquiring a disease. [0036] As used herein, the term “therapeutically effective amount” refers to the amount of the compound being administered sufficient to prevent development of or alleviate to some extent one or more of the symptoms of the disease or disorder being treated. [0037] “Pharmaceutically acceptable” refers to substances that are generally safe, non-toxic and neither biologically undesirable nor otherwise undesirable, including substances which
are acceptable for human pharmaceutical use and veterinary use. A pharmaceutically acceptable excipient, for example, is compatible with the other ingredients of the formulation and not deleterious to the recipient thereof. [0038] As used herein, the phrase “pharmaceutically acceptable excipient” refers to a pharmaceutically acceptable material that aids the administration of an active agent to a subject. Examples of excipients include, but are not limited to, liquid or solid fillers, diluents, solvents, encapsulating materials, disintegrants, lubricants, glidants, sweeteners, flavors, and colors. See, e.g., Remington: The Science and Practice of Pharmacy, 23rd ed.; Academic Press: Cambridge, Mass., 2020; Handbook of Pharmaceutical Excipients, 9th ed.; Sheskey et al, Eds.; Pharmaceutical Press 2020; Handbook of Pharmaceutical Additives, 3rd ed.; Ash and Ash Eds.; Gower Publishing Company: 2007; Pharmaceutical Preformulation and Formulation, 2nd ed.; Gibson Ed.; CRC Press LLC: Boca Raton, Fla., 2009. [0039] As used herein, “pharmaceutically acceptable salt(s)” refers to a parent compound which is modified by converting an existing acid or base moiety to its salt form. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like. The pharmaceutically acceptable salts of the present disclosure include conventional non-toxic salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. The pharmaceutically acceptable salts of the present disclosure can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, non-aqueous media like ether, ethyl acetate, alcohols or acetonitrile (ACN) are preferred. [0040] The term “stereoisomers” refers to any enantiomers, diastereoisomers, or geometric isomers of the compounds of Formula (I), (IA), (IB), (IC), (ID), (IE), (IF), (IG) and (IH), wherever they are chiral or when they bear one or more double bonds. When the compounds of formula (I), (IA), (IB), (IC), (ID), (IE), (IF), (IG) and (IH) are chiral, they can exist in racemic form or in optically active form. It should be understood that the disclosure encompasses all stereochemical isomeric forms, including diastereomeric, enantiomeric and epimeric forms, including (R) isomers, (S) isomers, d-isomers and l-isomers and mixtures thereof. Individual stereoisomers of compounds can be prepared synthetically from
commercially available starting materials which contain chiral centers or by preparation of mixtures of enantiomeric products followed by separation, e.g., via conversion to a mixture of diastereomers followed by separation or recrystallization, chromatographic techniques (including direct separation of enantiomers on chiral chromatographic columns), or any other appropriate method known in the art. Starting compounds of particular stereochemistry are either commercially available or can be made and resolved by techniques known in the art. Additionally, the compounds of the present disclosure may exist as geometric isomers. The present disclosure includes all cis and trans, syn and anti, entgegen (E) and zusammen (Z) isomers as well as the appropriate mixtures thereof. [0041] The present disclosure provides fused pyrazole derivatives of formula (I), which are useful for the inhibition of USP1. The present disclosure further provides pharmaceutical compositions comprising the compounds of formula (I), and their derivatives as therapeutic agents. It will be apparent to those skilled in the art that various modifications and variations can be made to the compounds, compositions, and methods described herein without departing from the scope or spirit of various embodiments disclosed herein. For instance, features illustrated or described as part of one embodiment can be applied to another embodiment to yield a still further embodiment. Thus, it is intended that the present application includes such modifications and variations and their equivalents. Other objects, features, and aspects of the present application are disclosed in the following detailed description. It is to be understood by one of ordinary skill in the art that the present discussion is a description of exemplary embodiments and is not to be construed as limiting the broader aspects of the present disclosure. The embodiments below are illustrative of the present disclosure and are not intended to limit the claims to the specific embodiments exemplified. [0042] Provided herein are fused pyrazole derivatives of formula (I), and pharmaceuticall
 wherein:
;
wherein:
;
 ring Y is C3-C8 cycloalkyl, C5-C10 cycloalkeny , wherein the asterisk marks the point of attachment to rin
ring Y is C3-C8 cycloalkyl, C5-C10 cycloalkeny , wherein the asterisk marks the point of attachment to rin
 ring Z is 3- to 15-membered heterocycloalkyl or 5- to 14-membered heteroaryl; X1, X2 and X3 are independently N or C; Y1 to Y4 are independently N or C, wherein 0-2 of Y1 to Y4 are N; Y5 and Y6 are independently N, C, O or S; each RX is independently C1-C6 alkyl, C1-C6 alkoxy or C3-C8 cycloalkyl; RY is halo, C1-C6 alkyl, C1-C6 alkoxy or C1-C6 alkylamino; each RZ is independently hydrogen, halo, C1-C6 alkyl, halo(C1-C6)alkyl, C1-C6 alkoxy, C3-C8 cycloalkyl, (C1-C6)alkylamino-(C1-C6)alkyl, (C1-C6)alkoxy-(C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl or (C1-C6 alkyl)-substituted 3- to 15-membered heterocycloalkyl, wherein halo and C1-C6 alkoxy are bonded to carbon atoms in the Z ring; G is -O-, -N(RG1)-, *-C(RG2RG3)-N(RG1)-, *-N(RG1)-C(RG2RG3)-, *-C(RG2RG3)-O-, -[C(RG2RG3)]q- or -C(R4R4 ^)-, wherein the asterisk marks the point of attachment to the pyrazole ring; RG1, each RG2 and each RG3 are independently hydrogen or C1-C6 alkyl; R1 is hydrogen, C1-C6 alkyl, halo(C1-C6)alkyl, C3-C8 cycloalkyl, (C1-C6)alkylamino- (C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl, (C1-C6)alkyl-substituted 3- to 15-membered heterocycloalkyl, optionally substituted (C6-C14)aryl(C1-C6)alkyl, or optionally substituted (5- to 14-membered heteroaryl)-(C1-C6)alkyl, wherein the substituents on (C6- C14)aryl(C1-C6)alkyl and (5- to 14-membered heteroaryl)-(C1-C6 )alkyl are independently halo, C1-C6 alkyl or C1-C6 alkoxy; R2, R2 ^, R3 and R3 ^ are independently hydrogen, C1-C6 alkyl, halo, cyano or amino; or R2 and R2 ^, taken together, form an oxo group; or R2 and R2 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; or
R2 and R3, taken together, form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; or R3 and RG2, taken together, form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; R4 and R4 ^ are: i. taken together along with the carbon atom to which they are attached to form a C3-C8 cycloalkyl, or ii. independently hydroxy or C1-C6 alkoxy, or iii. independently hydrogen or C1-C6 alkyl, with the proviso that R4 and R4 ^ are not hydrogen or alkyl when: (a) each of R2, R2 ^, R3, R3 ^ is hydrogen, (b) ring Y is phenyl, thiophenyl, thiazolyl, 6-membered heteroaryl or cubanyl, and (c) ring Z is imidazolyl; or R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; subscript p is 1, 2 or 3; subscript q is 2, 3 or 4; subscript m is 0 or 1; and subscript n is 1, 2 or 3. [0043] In some embodiments, the present application provides compounds of formula (IA), or pharmaceutically
ring Z is 3- to 15-membered heterocycloalkyl or 5- to 14-membered heteroaryl; X1, X2 and X3 are independently N or C; Y1 to Y4 are independently N or C, wherein 0-2 of Y1 to Y4 are N; Y5 and Y6 are independently N, C, O or S; each RX is independently C1-C6 alkyl, C1-C6 alkoxy or C3-C8 cycloalkyl; RY is halo, C1-C6 alkyl, C1-C6 alkoxy or C1-C6 alkylamino; each RZ is independently hydrogen, halo, C1-C6 alkyl, halo(C1-C6)alkyl, C1-C6 alkoxy, C3-C8 cycloalkyl, (C1-C6)alkylamino-(C1-C6)alkyl, (C1-C6)alkoxy-(C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl or (C1-C6 alkyl)-substituted 3- to 15-membered heterocycloalkyl, wherein halo and C1-C6 alkoxy are bonded to carbon atoms in the Z ring; G is -O-, -N(RG1)-, *-C(RG2RG3)-N(RG1)-, *-N(RG1)-C(RG2RG3)-, *-C(RG2RG3)-O-, -[C(RG2RG3)]q- or -C(R4R4 ^)-, wherein the asterisk marks the point of attachment to the pyrazole ring; RG1, each RG2 and each RG3 are independently hydrogen or C1-C6 alkyl; R1 is hydrogen, C1-C6 alkyl, halo(C1-C6)alkyl, C3-C8 cycloalkyl, (C1-C6)alkylamino- (C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl, (C1-C6)alkyl-substituted 3- to 15-membered heterocycloalkyl, optionally substituted (C6-C14)aryl(C1-C6)alkyl, or optionally substituted (5- to 14-membered heteroaryl)-(C1-C6)alkyl, wherein the substituents on (C6- C14)aryl(C1-C6)alkyl and (5- to 14-membered heteroaryl)-(C1-C6 )alkyl are independently halo, C1-C6 alkyl or C1-C6 alkoxy; R2, R2 ^, R3 and R3 ^ are independently hydrogen, C1-C6 alkyl, halo, cyano or amino; or R2 and R2 ^, taken together, form an oxo group; or R2 and R2 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; or
R2 and R3, taken together, form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; or R3 and RG2, taken together, form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; R4 and R4 ^ are: i. taken together along with the carbon atom to which they are attached to form a C3-C8 cycloalkyl, or ii. independently hydroxy or C1-C6 alkoxy, or iii. independently hydrogen or C1-C6 alkyl, with the proviso that R4 and R4 ^ are not hydrogen or alkyl when: (a) each of R2, R2 ^, R3, R3 ^ is hydrogen, (b) ring Y is phenyl, thiophenyl, thiazolyl, 6-membered heteroaryl or cubanyl, and (c) ring Z is imidazolyl; or R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; subscript p is 1, 2 or 3; subscript q is 2, 3 or 4; subscript m is 0 or 1; and subscript n is 1, 2 or 3. [0043] In some embodiments, the present application provides compounds of formula (IA), or pharmaceutically
 acceptable salts thereof or stereoisomers thereof. [0044] In some embodiments, compounds of formula (IA) and pharmaceutically acceptable salts thereof and stereoisomers thereof are provided, wherein:
ring Y is , wherein the asterisk marks the point of attachment to ring Z;
acceptable salts thereof or stereoisomers thereof. [0044] In some embodiments, compounds of formula (IA) and pharmaceutically acceptable salts thereof and stereoisomers thereof are provided, wherein:
ring Y is , wherein the asterisk marks the point of attachment to ring Z;
 ring Z is imidazolyl or pyrazolyl; X1 and X2 are N; Y1 to Y4 are independently N or C, wherein 1-2 of Y1 to Y4 are N; each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl; each RZ is independently C1-C6 alkyl or halo(C1-C6)alkyl; G is -C(R4R4 ^)-; R1 is hydrogen, C1-C6 alkyl, or halo(C1-C6)alkyl; R2 ^ and R3 ^ are hydrogen and R2 and R3 are taken together to form a C1-C3 alkylene bridge; or R2 and R3 are hydrogen and R2 ^ and R3 ^ are taken together to form a C1-C3 alkylene bridge; R4 and R4 ^ are hydrogen or R4 and R4 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; subscript p is 1 or 2; subscript m is 0; and subscript n is 1 or 2. [0045] In some embodiments, compounds of formula (IA) and pharmaceutically acceptable salts thereof and stereoisomers thereof are provided, wherein: ring Y is pyridin-2-yl, pyridin-3-yl, or pyrazin-2-yl; ring Z is imidazol-2-yl or pyrazol-1-yl; X1 and X2 are N; each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl; each RZ is independently C1-C6 alkyl or C1-C6 haloalkyl; G is -CH2-; R1 is C1-C6 alkyl; R2 ^ and R3 ^ are hydrogen and R2 and R3 are taken together to form a C1-C3 alkylene bridge; or R2 and R3 are hydrogen and R2 ^ and R3 ^ are taken together to form a C1-C3 alkylene bridge;
subscript p is 2; subscript m is 0; and subscript n is 2. [0046] In some embodiments, the present application provides compounds of formula (IB), or pharmaceutically a
ring Z is imidazolyl or pyrazolyl; X1 and X2 are N; Y1 to Y4 are independently N or C, wherein 1-2 of Y1 to Y4 are N; each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl; each RZ is independently C1-C6 alkyl or halo(C1-C6)alkyl; G is -C(R4R4 ^)-; R1 is hydrogen, C1-C6 alkyl, or halo(C1-C6)alkyl; R2 ^ and R3 ^ are hydrogen and R2 and R3 are taken together to form a C1-C3 alkylene bridge; or R2 and R3 are hydrogen and R2 ^ and R3 ^ are taken together to form a C1-C3 alkylene bridge; R4 and R4 ^ are hydrogen or R4 and R4 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; subscript p is 1 or 2; subscript m is 0; and subscript n is 1 or 2. [0045] In some embodiments, compounds of formula (IA) and pharmaceutically acceptable salts thereof and stereoisomers thereof are provided, wherein: ring Y is pyridin-2-yl, pyridin-3-yl, or pyrazin-2-yl; ring Z is imidazol-2-yl or pyrazol-1-yl; X1 and X2 are N; each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl; each RZ is independently C1-C6 alkyl or C1-C6 haloalkyl; G is -CH2-; R1 is C1-C6 alkyl; R2 ^ and R3 ^ are hydrogen and R2 and R3 are taken together to form a C1-C3 alkylene bridge; or R2 and R3 are hydrogen and R2 ^ and R3 ^ are taken together to form a C1-C3 alkylene bridge;
subscript p is 2; subscript m is 0; and subscript n is 2. [0046] In some embodiments, the present application provides compounds of formula (IB), or pharmaceutically a
 . [0047] In some embodiments, the present application provides compounds of formula (IC), or pharmaceutic
. [0047] In some embodiments, the present application provides compounds of formula (IC), or pharmaceutic
 . [0048] In some embodiments, the present application provides compounds of formula (ID),
. [0048] In some embodiments, the present application provides compounds of formula (ID),
 or pharmaceutically acceptable salts thereof or stereoisomers thereof. [0049] In some embodiments, the present application provides compounds of formula (IE),
or pharmaceutic
or pharmaceutically acceptable salts thereof or stereoisomers thereof. [0049] In some embodiments, the present application provides compounds of formula (IE),
or pharmaceutic
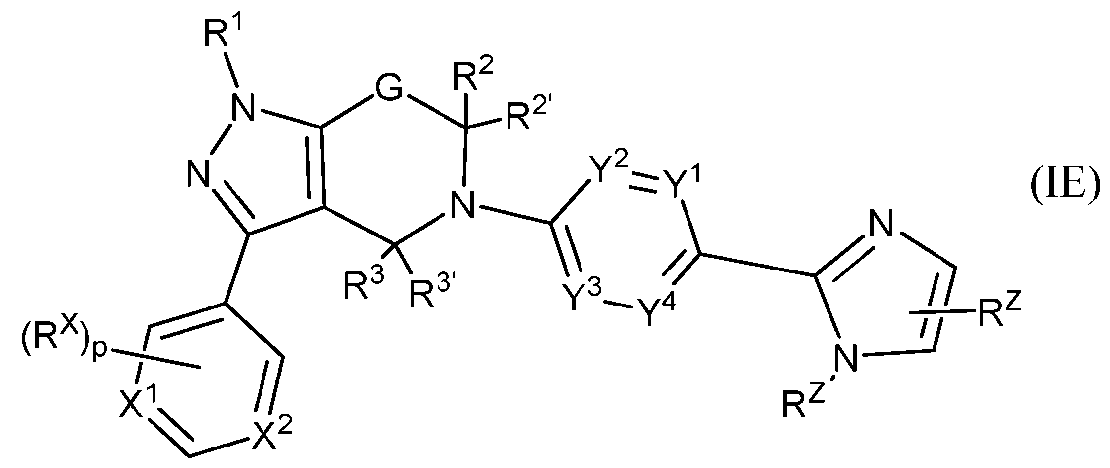 [0050] In some embodiments, the present application provides compounds of formula (IF), or pharmaceutic
[0050] In some embodiments, the present application provides compounds of formula (IF), or pharmaceutic
 [0051] In some embodiments, the present application provides compounds of formula (IG), or pharma
[0051] In some embodiments, the present application provides compounds of formula (IG), or pharma
 ceut ca y accepta e sa ts t ereo or stereo somers t ereof, wherein the bridge LG is -(CH2)1-3- or -CH2-O-CH2-. [0052] In some embodiments, compounds of formula (IG) and pharmaceutically acceptable salts thereof and stereoisomers thereof are provided, wherein: ring Z is imidazolyl or pyrazolyl; X1 and X2 are N; Y1 to Y4 are independently N or C, wherein 1-2 of Y1 to Y4 are N; each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl;
each RZ is independently C1-C6 alkyl or halo(C1-C6)alkyl; LG is -(CH2)1-3-; G is -C(R4R4 ^)-; R1 is hydrogen, C1-C6 alkyl, or halo(C1-C6)alkyl; R4 and R4 ^ are hydrogen or R4 and R4 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; subscript p is 1 or 2; and subscript n is 1 or 2. [0053] In some embodiments, compounds of formula (IG) and pharmaceutically acceptable salts thereof and stereoisomers thereof are provided, wherein: ring Z is imidazol-2-yl or pyrazol-1-yl; X1 and X2 are N; each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl; each RZ is independently C1-C6 alkyl or halo(C1-C6)alkyl; LG is -(CH2)1-3-; G is -CH2-; R1 is C1-C6 alkyl; subscript p is 2; and subscript n is 2. [0054] In some embodiments, the present application provides compounds of formula (IH), or pharmac
ceut ca y accepta e sa ts t ereo or stereo somers t ereof, wherein the bridge LG is -(CH2)1-3- or -CH2-O-CH2-. [0052] In some embodiments, compounds of formula (IG) and pharmaceutically acceptable salts thereof and stereoisomers thereof are provided, wherein: ring Z is imidazolyl or pyrazolyl; X1 and X2 are N; Y1 to Y4 are independently N or C, wherein 1-2 of Y1 to Y4 are N; each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl;
each RZ is independently C1-C6 alkyl or halo(C1-C6)alkyl; LG is -(CH2)1-3-; G is -C(R4R4 ^)-; R1 is hydrogen, C1-C6 alkyl, or halo(C1-C6)alkyl; R4 and R4 ^ are hydrogen or R4 and R4 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; subscript p is 1 or 2; and subscript n is 1 or 2. [0053] In some embodiments, compounds of formula (IG) and pharmaceutically acceptable salts thereof and stereoisomers thereof are provided, wherein: ring Z is imidazol-2-yl or pyrazol-1-yl; X1 and X2 are N; each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl; each RZ is independently C1-C6 alkyl or halo(C1-C6)alkyl; LG is -(CH2)1-3-; G is -CH2-; R1 is C1-C6 alkyl; subscript p is 2; and subscript n is 2. [0054] In some embodiments, the present application provides compounds of formula (IH), or pharmac
 eutically acceptable salts thereof or stereoisomers thereof, wherein the bridge LG is -(CH2)1-3- or -CH2-O-CH2-.
[0055] In some embodiments, the present application provides fused pyrazole derivatives of formula (I), and pharmaceutically acceptable salts and stereoisomers thereof, wherein: ring ;
eutically acceptable salts thereof or stereoisomers thereof, wherein the bridge LG is -(CH2)1-3- or -CH2-O-CH2-.
[0055] In some embodiments, the present application provides fused pyrazole derivatives of formula (I), and pharmaceutically acceptable salts and stereoisomers thereof, wherein: ring ;
 ring Y is , wherein the asterisk marks the point of attachment to ring Z;
ring Y is , wherein the asterisk marks the point of attachment to ring Z;
 ring Z is 5- to 14-membered heteroaryl; X1 and X2 are independently N or C; Y1 to Y4 are independently N or C, wherein 0-2 of Y1 to Y4 are N; each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl; each RZ is independently hydrogen, halo, C1-C6 alkyl or C1-C6 haloalkyl; G is -[C(RG2RG3)]q- R1 is C1-C6 alkyl, halo(C1-C6)alkyl or C3-C8 cycloalkyl; R2, R2 ^, R3 and R3 ^ is independently hydrogen or C1-C6 alkyl; R2 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; R4 and R4 ^ are hydrogen or R4 and R4 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl, with the proviso that R4 and R4 ^ are not hydrogen or alkyl when: (a) each of R2, R2 ^, R3, R3 ^ is hydrogen, (b) ring Y is phenyl, thiophenyl, thiazolyl, 6-membered heteroaryl or cubanyl, and (c) ring Z is imidazolyl; or R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; subscript p is 1, 2 or 3; subscript q is 2; subscript m is 0; and subscript n is 1, 2 or 3.
[0056] In some embodiments, ring X i .
ring Z is 5- to 14-membered heteroaryl; X1 and X2 are independently N or C; Y1 to Y4 are independently N or C, wherein 0-2 of Y1 to Y4 are N; each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl; each RZ is independently hydrogen, halo, C1-C6 alkyl or C1-C6 haloalkyl; G is -[C(RG2RG3)]q- R1 is C1-C6 alkyl, halo(C1-C6)alkyl or C3-C8 cycloalkyl; R2, R2 ^, R3 and R3 ^ is independently hydrogen or C1-C6 alkyl; R2 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; R4 and R4 ^ are hydrogen or R4 and R4 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl, with the proviso that R4 and R4 ^ are not hydrogen or alkyl when: (a) each of R2, R2 ^, R3, R3 ^ is hydrogen, (b) ring Y is phenyl, thiophenyl, thiazolyl, 6-membered heteroaryl or cubanyl, and (c) ring Z is imidazolyl; or R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; subscript p is 1, 2 or 3; subscript q is 2; subscript m is 0; and subscript n is 1, 2 or 3.
[0056] In some embodiments, ring X i .
 [0057] In some embodiments, ring X i .
[0057] In some embodiments, ring X i .
 [0058] In some embodiments, ring Y i , wherein the asterisk marks the point of attachment to ring Z.
[0058] In some embodiments, ring Y i , wherein the asterisk marks the point of attachment to ring Z.
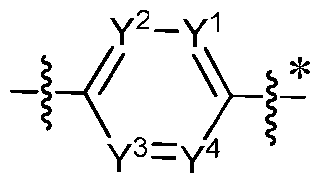 [0059] In some embodiments, ring Y i ,
[0059] In some embodiments, ring Y i ,
 int e
int e
 [0061] In some embodiments, ring Z is a 5-membered heteroaryl. [0062] In some embodiments, rin ein the asterisk marks the point of attac
[0061] In some embodiments, ring Z is a 5-membered heteroaryl. [0062] In some embodiments, rin ein the asterisk marks the point of attac
 [0063] In some embodiments, ring Z is imidazolyl or pyrazolyl. [0064] Substituents RX, RY and RZ may be present on any atom of ring X, ring Y and ring Z. In some embodiments, any one or more of RX, RY and RZ is present on a carbon atom of ring X, ring Y and ring Z. In some embodiments, any one or more of RX, RY and RZ is present on a nitrogen atom of ring X, ring Y and ring Z. [0065] In some embodiments, R1 is C1-C6 alkyl or C3-C8 cycloalkyl. In some embodiments, R1 is C1-C6 alkyl. In some embodiments, R1 is C3-C8 cycloalkyl.
[0066] In some embodiments, R2 is C1-C6 alkyl. [0067] In some embodiments, R3 is C1-C6 alkyl. [0068] In some embodiments, RX is C1-C6 alkyl. In some embodiments, RX is methyl or isopropyl. [0069] In some embodiments, RX is C1-C6 alkoxy. In some embodiments, RX is methoxy or ethoxy. [0070] In some embodiments, RX is C3-C8 cycloalkyl. In some embodiments, RX is cyclopropyl. [0071] In some embodiments, each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl. In some embodiments, each RX is independently methoxy or cyclopropyl. [0072] In some embodiments, G is -C(R4R4 ^)-. In some embodiments, G is -[C(RG2RG3)]q- or -C(R4R4 ^)-, and q is 2. [0073] In some embodiments, R2 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S. [0074] In some embodiments, R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S. [0075] In some embodiments, R4 and R4 ^ are taken together along with the carbon atom to which they are attached to form C3-C8 cycloalkyl. [0076] In some embodiments, RY is halo. In some embodiments, RY is C1-C6 alkoxy. [0077] In some embodiments, RZ is C1-C6 alkyl. In some embodiments, RZ is C1-C6 haloalkyl. In some embodiments, RZ is halo. In some embodiments, RZ is C1-C6 alkoxy. In some embodiments, RZ is C3-C8 cycloalkyl. [0078] In some embodiments, each RZ is independently C1-C6 alkyl or halo(C1-C6)alkyl. In some embodiments, each RZ is independently methyl, ethyl, isopropyl, or trifluoromethyl. [0079] In some embodiments, each RZ is independently C3-C8 cycloalkyl or halo(C1-C6)alkyl.
[0080] In some embodiments, compounds of formula (I) are provided, wherein RX is C1-C6 alkoxy or C3-C8 cycloalkyl, subscript m is 0, RZ is C1-C6 alkyl or halo(C1-C6)alkyl, R1 is C1- C6 alkyl and, G is -C(R4R4 ^)-. [0081] In some embodiments, compounds of formula (I) are provided, wherein ring X is pyrimidinyl, ring Y is pyridinyl and ring Z is imidazolyl. [0082] In some embodiments, subscript m is 1. In some embodiments, subscript m is 0. [0083] In some embodiments, subscript n is 2. In some embodiments, subscript p is 2. In some embodiments, subscript q is 2. [0084] In certain embodiments, the present application provides a compound selected from the group consisting of: Comp 7- 7- 7-
[0063] In some embodiments, ring Z is imidazolyl or pyrazolyl. [0064] Substituents RX, RY and RZ may be present on any atom of ring X, ring Y and ring Z. In some embodiments, any one or more of RX, RY and RZ is present on a carbon atom of ring X, ring Y and ring Z. In some embodiments, any one or more of RX, RY and RZ is present on a nitrogen atom of ring X, ring Y and ring Z. [0065] In some embodiments, R1 is C1-C6 alkyl or C3-C8 cycloalkyl. In some embodiments, R1 is C1-C6 alkyl. In some embodiments, R1 is C3-C8 cycloalkyl.
[0066] In some embodiments, R2 is C1-C6 alkyl. [0067] In some embodiments, R3 is C1-C6 alkyl. [0068] In some embodiments, RX is C1-C6 alkyl. In some embodiments, RX is methyl or isopropyl. [0069] In some embodiments, RX is C1-C6 alkoxy. In some embodiments, RX is methoxy or ethoxy. [0070] In some embodiments, RX is C3-C8 cycloalkyl. In some embodiments, RX is cyclopropyl. [0071] In some embodiments, each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl. In some embodiments, each RX is independently methoxy or cyclopropyl. [0072] In some embodiments, G is -C(R4R4 ^)-. In some embodiments, G is -[C(RG2RG3)]q- or -C(R4R4 ^)-, and q is 2. [0073] In some embodiments, R2 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S. [0074] In some embodiments, R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S. [0075] In some embodiments, R4 and R4 ^ are taken together along with the carbon atom to which they are attached to form C3-C8 cycloalkyl. [0076] In some embodiments, RY is halo. In some embodiments, RY is C1-C6 alkoxy. [0077] In some embodiments, RZ is C1-C6 alkyl. In some embodiments, RZ is C1-C6 haloalkyl. In some embodiments, RZ is halo. In some embodiments, RZ is C1-C6 alkoxy. In some embodiments, RZ is C3-C8 cycloalkyl. [0078] In some embodiments, each RZ is independently C1-C6 alkyl or halo(C1-C6)alkyl. In some embodiments, each RZ is independently methyl, ethyl, isopropyl, or trifluoromethyl. [0079] In some embodiments, each RZ is independently C3-C8 cycloalkyl or halo(C1-C6)alkyl.
[0080] In some embodiments, compounds of formula (I) are provided, wherein RX is C1-C6 alkoxy or C3-C8 cycloalkyl, subscript m is 0, RZ is C1-C6 alkyl or halo(C1-C6)alkyl, R1 is C1- C6 alkyl and, G is -C(R4R4 ^)-. [0081] In some embodiments, compounds of formula (I) are provided, wherein ring X is pyrimidinyl, ring Y is pyridinyl and ring Z is imidazolyl. [0082] In some embodiments, subscript m is 1. In some embodiments, subscript m is 0. [0083] In some embodiments, subscript n is 2. In some embodiments, subscript p is 2. In some embodiments, subscript q is 2. [0084] In certain embodiments, the present application provides a compound selected from the group consisting of: Comp 7- 7- 7-
 Comp ound Compound Structure Compound Name 8-
Comp ound Compound Structure Compound Name 8-
 Comp ound Compound Structure Compound Name -
Comp ound Compound Structure Compound Name -
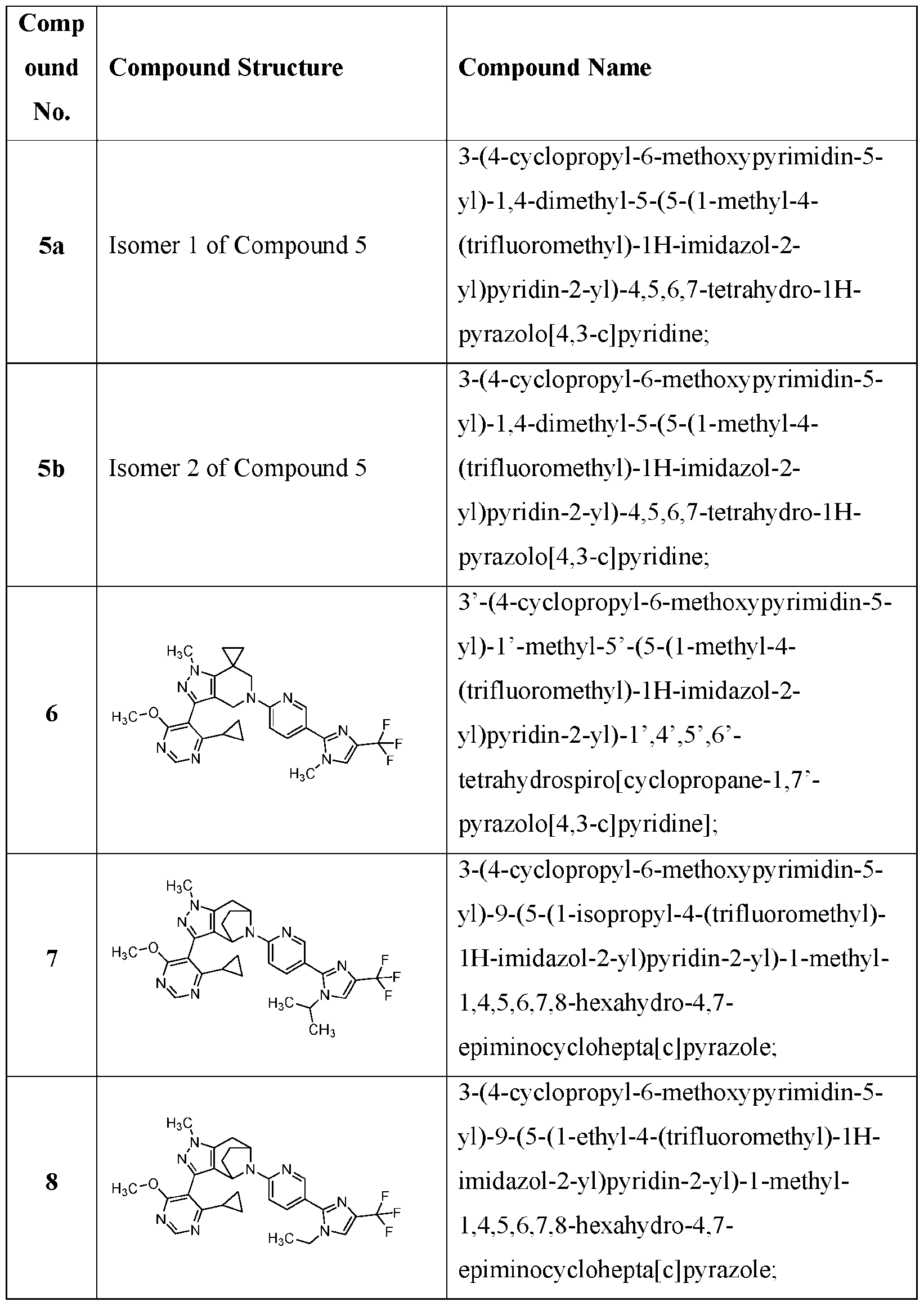 Comp ound Compound Structure Compound Name 7- 7- - 7-
Comp ound Compound Structure Compound Name 7- 7- - 7-
 Comp ound Compound Structure Compound Name 7- 2- 7- )- 7- - 7-
Comp ound Compound Structure Compound Name 7- 2- 7- )- 7- - 7-
 Comp ound Compound Structure Compound Name 7-
Comp ound Compound Structure Compound Name 7-
 Comp ound Compound Structure Compound Name - 6- 7- 7-
Comp ound Compound Structure Compound Name - 6- 7- 7-
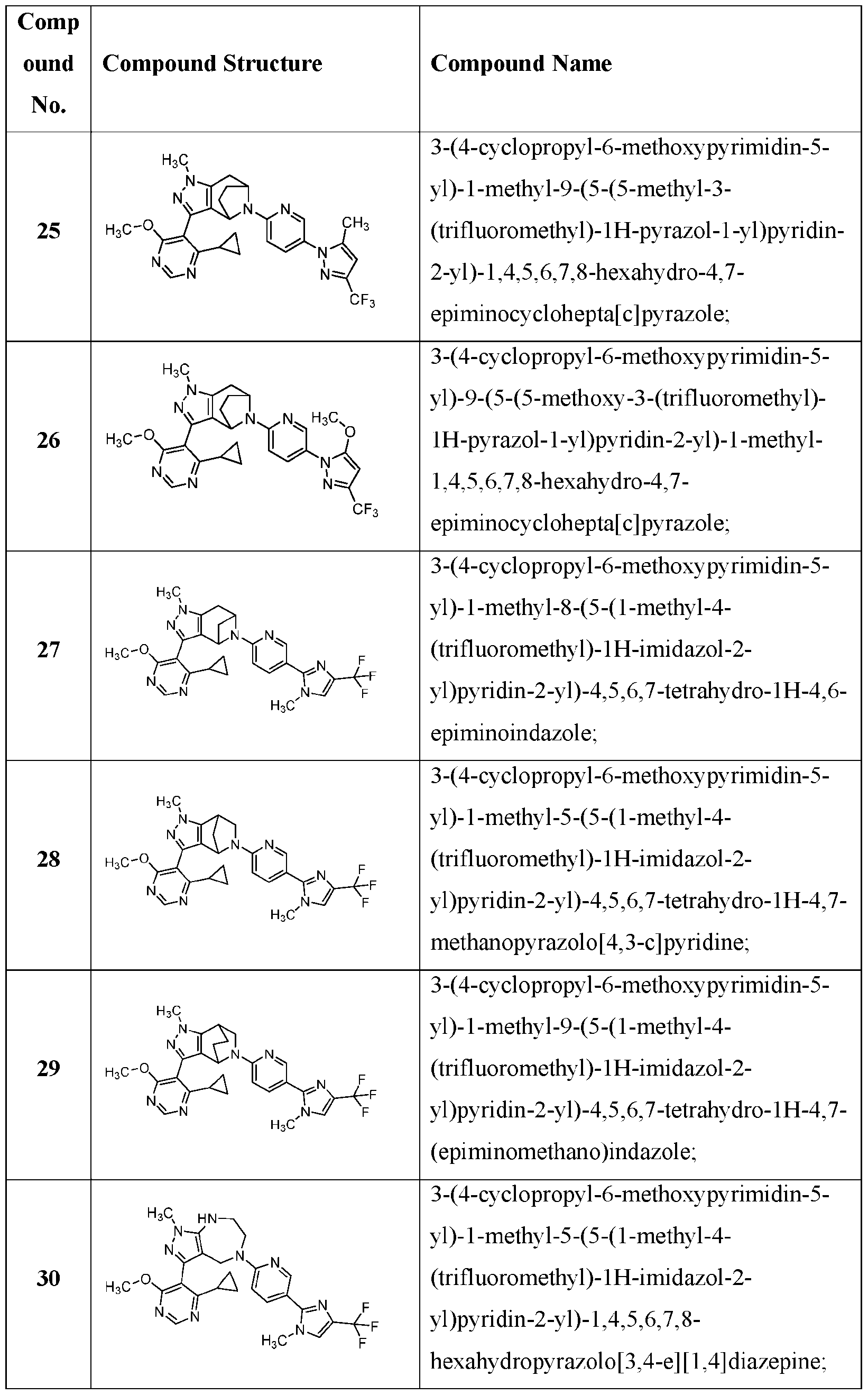 Comp ound Compound Structure Compound Name
Comp ound Compound Structure Compound Name
 Comp ound Compound Structure Compound Name - -
Comp ound Compound Structure Compound Name - -
 Comp ound Compound Structure Compound Name - 7- 7-
Comp ound Compound Structure Compound Name - 7- 7-
 Comp ound Compound Structure Compound Name 7- - 7- or
Comp ound Compound Structure Compound Name 7- - 7- or
 [0085] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof or a stereoisomer thereof as described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent). Preferably, the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein. As described in more detail below, compounds described in the present disclosure may be admixed with a pharmaceutically acceptable excipient (such as a carrier or a diluent) or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container. [0086] Compounds of formula (I) are useful as USP1 inhibitors. In some embodiments, the present disclosure provides pharmaceutical compositions for use in treating and/or preventing a disease and/or disorder responsive to the inhibition of USP1 protein expression and/or USP1 activity. [0087] In some embodiments, the present disclosure provides a method of inhibiting USP1 in a subject, comprising administering to the subject in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof.
[0088] In some embodiments, the present disclosure provides a method of inhibiting USP1 in a subject, comprising administering to the subject an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof. [0089] In some embodiments, the present disclosure provides a method of treating diseases and/or disorders mediated by USP1 comprising administering to a subject in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof. In some embodiments, the subject is a human or other mammal. [0090] The compounds of the disclosure are typically administered in the form of a pharmaceutical composition. Such compositions can be prepared using procedures well known in the pharmaceutical art. The pharmaceutical composition typically includes one or more compounds described herein and one or more pharmaceutically acceptable excipients. Typically, the pharmaceutically acceptable excipients are approved by regulatory authorities or are generally regarded as safe for human or animal use. Examples of pharmaceutically acceptable excipients include, but are not limited to, carriers, diluents, glidants and lubricants, preservatives, buffering agents, chelating agents, polymers, gelling agents, viscosifying agents, solvents and the like. [0091] The pharmaceutical composition may include, for example, one or more of water, salt solutions, alcohols, polyethylene glycols, peanut oil, olive oil, sugars (e.g., lactose, sucrose, and the like), dextrin, sugar, amylose, lower alkyl ethers of cellulose, magnesium carbonate, magnesium stearate, talc, terra alba, silicic acid, gelatin, agar, pectin, acacia, fatty acids (e.g., stearic acid), fatty acid amides, and fatty acid esters including fatty acid monoglycerides and diglycerides, or the like. The pharmaceutical composition may further include one or more pharmaceutically acceptable auxiliary agents, wetting agents, suspending agents, preserving agents, buffers, sweetening agents, flavouring agents, colorants or any combination of the foregoing. [0092] The pharmaceutical compositions may be in conventional forms, for example, tablets, capsules, solutions, suspensions, injectables or products for topical application. Further, the pharmaceutical composition of the present disclosure may be formulated so as to provide a desired release profile.
[0093] Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges. [0094] Liquid formulations include, but are not limited to, syrups, emulsions and sterile injectable liquids, such as suspensions or solutions. [0095] Topical dosage forms of the compounds include ointments, pastes, creams, lotions, powders, solutions, eye or ear drops, impregnated dressings and may contain appropriate conventional additives such as preservatives, solvents to assist drug penetration. [0096] Administration of the compounds of the disclosure, in pure form or in an appropriate pharmaceutical composition, can be carried out using any of the accepted routes of administration of pharmaceutical compositions. The route of administration may be any route which effectively transports the active compound of the present disclosure to the appropriate or desired site of action. Suitable routes of administration include, but are not limited to, oral, nasal, buccal, dermal, intradermal, transdermal, parenteral, rectal, subcutaneous, intravenous, intraurethral, intramuscular or topical. In some embodiments, the composition is administered by oral, parenteral or inhalation routes. Examples of parenteral administration include administration by injection, as well as percutaneous, transmucosal, transnasal and transpulmonary administration. [0097] Suitable doses of the compounds for use in treating the diseases or disorders described herein can be determined by those skilled in the relevant art. Therapeutic doses are generally identified through dose ranging studies in humans based on preliminary evidence derived from animal studies. Preferably, doses are sufficient to result in a desired therapeutic benefit without causing unwanted side effects. Varying modes of administration, dosage forms and suitable pharmaceutical excipients can be well used and adjusted by those skilled in the art. All changes and modifications are envisioned within the scope of the present disclosure. [0098] In some embodiments, the present disclosure provides a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof, for use as a medicament and for use in the manufacture of medicaments. [0099] In some embodiments, the disclosure provides the use of compounds of Formula (I) in the treatment and prevention of diseases and/or disorders responsive to the inhibition of
USP1 protein expression and USP1 activity, as well as in the manufacture of medicaments for treating such diseases and/or disorders. Typically, the symptoms of the disease are treated, improved, diminished and/or prevented by inhibition of USP1. [0100] In some embodiments, the USP1 mediated disease and/or disorder is cancer. Accordingly, compounds of formula (I) for use in the treatment of cancer and in the manufacture of medicaments for treatment of cancer are also provided. [0101] In related embodiments, the disclosure provides a method of treating a disease or disorder mediated by USP1 in a subject comprising administering to the subject a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof. In some embodiments, the disease and/or disorder is cancer. [0102] In some embodiments, the cancer is selected from a hematological cancer, a lymphatic cancer, a DNA damage repair pathway deficient cancer, a homologous- recombination deficient cancer, a cancer comprising cancer cells with a mutation in a gene encoding p53, and a cancer comprising cancer cells with a loss of function mutation in a gene encoding p53. [0103] In some embodiments, the cancer is a hematological cancer or a lymphatic cancer. [0104] In some embodiments, the cancer is a DNA damage repair pathway deficient cancer and/or a homologous-recombination deficient cancer. [0105] In some embodiments, the cancer is a DNA damage repair pathway deficient cancer. [0106] In some embodiments, the cancer is a homologous-recombination deficient cancer. [0107] In some embodiments, the cancer is selected from a hematological cancer, a lymphatic cancer, a cancer comprising cancer cells with a mutation in a gene encoding p53, and a cancer comprising cancer cells with a loss of function mutation in a gene encoding p53. [0108] In some embodiments, the cancer comprises cancer cells with a mutation in a gene encoding p53. In some embodiments, the mutation in the gene encoding p53 is a loss of function mutation. [0109] In some embodiments, methods for treating cancer and other diseases include administering to a subject in need thereof a therapeutically effective amount of a compound
of the present disclosure along with one or more additional chemotherapeutic agents independently selected from anti-proliferative agents, anti-cancer agents, immunosuppressant agents and pain-relieving agents. [0110] The method(s) of treatment of the present disclosure generally comprises administering a safe and effective amount of a compound according to formula (I) or a pharmaceutically acceptable salt thereof to a patient (particularly a human) in need thereof. [0111] Compounds of the disclosure are indicated both in the therapeutic and/or prophylactic treatment of the above-mentioned conditions. For the above-mentioned therapeutic uses the dosage administered will typically vary with the compound employed, the mode of administration, the treatment desired and the disorder or disease indicated. [0112] The compounds of the present disclosure may be used as single drug or as a pharmaceutical composition in which the compound is mixed with various pharmacologically acceptable materials, as described above. [0113] In some embodiments, the compounds of the present disclosure can also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds. For example, the present disclosure also embraces isotopically-labeled variants of the present disclosure which are identical to those recited herein, but for the fact that one or more atoms of the compound are replaced by an atom having the atomic mass or mass number different from the predominant atomic mass or mass number usually found in nature for the atom. All isotopes of any particular atom or element as specified are contemplated within the scope of the compounds of the disclosure and their uses. Exemplary isotopes that can be incorporated in to compounds of the disclosure include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine, chlorine and iodine, such as 2H (“D”), 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 32P, 33P, 35S, 18F, 36Cl, 123I and 125I. Isotopically labeled compounds of the present disclosure can generally be prepared by following procedures analogous to those disclosed in the schemes and/or in the examples herein below, by substituting an isotopically labeled reagent for a non-isotopically labeled reagent. [0114] The present application provides methods for the preparation of compounds of formula (I) according to the description provided herein using appropriate methods and/or materials. It is to be understood by those skilled in the art that known variations of the conditions and processes of the following procedures can be used to prepare these
intermediates and compounds. Moreover, by utilizing the procedures described in detail, one of ordinary skill in the art can prepare additional compounds of the present disclosure. [0115] Compounds of this disclosure may be made by synthetic chemical processes, examples of which are shown herein. It is meant to be understood that the order of the steps in the processes may be varied; that reagents, solvents and reaction conditions may be substituted for those specifically mentioned; and that reactive moieties may be protected and deprotected, as necessary. [0116] Unless otherwise stated, work-up includes distribution of the reaction mixture between the organic and aqueous phases, separation of layers and drying the organic layer over anhydrous sodium sulphate, filtration and evaporation of the solvent. Purification, unless otherwise mentioned, includes purification by silica gel chromatographic techniques, generally using ethyl acetate/petroleum ether mixtures of a suitable polarity as the mobile phase. Some of the intermediates were taken to the next step based on TLC results, without further characterization, unless otherwise specified. Analysis for the compounds of the present disclosure, unless otherwise specified, was conducted using general methods well known to a person skilled in the art. [0117] Having described the disclosure with reference to certain preferred embodiments, other embodiments will become apparent to one skilled in the art from consideration of the specification. The disclosure is further defined by reference to the following examples, describing in detail the analysis of the compounds of the disclosure, which are not construed to be viewed as limiting the scope of the disclosure. It will be apparent to those skilled in the art that many modifications, both to materials and methods, may be practiced without departing from the scope of the disclosure.
[0085] In some embodiments, the present disclosure provides a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof or a stereoisomer thereof as described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent). Preferably, the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein. As described in more detail below, compounds described in the present disclosure may be admixed with a pharmaceutically acceptable excipient (such as a carrier or a diluent) or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container. [0086] Compounds of formula (I) are useful as USP1 inhibitors. In some embodiments, the present disclosure provides pharmaceutical compositions for use in treating and/or preventing a disease and/or disorder responsive to the inhibition of USP1 protein expression and/or USP1 activity. [0087] In some embodiments, the present disclosure provides a method of inhibiting USP1 in a subject, comprising administering to the subject in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof.
[0088] In some embodiments, the present disclosure provides a method of inhibiting USP1 in a subject, comprising administering to the subject an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof. [0089] In some embodiments, the present disclosure provides a method of treating diseases and/or disorders mediated by USP1 comprising administering to a subject in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof. In some embodiments, the subject is a human or other mammal. [0090] The compounds of the disclosure are typically administered in the form of a pharmaceutical composition. Such compositions can be prepared using procedures well known in the pharmaceutical art. The pharmaceutical composition typically includes one or more compounds described herein and one or more pharmaceutically acceptable excipients. Typically, the pharmaceutically acceptable excipients are approved by regulatory authorities or are generally regarded as safe for human or animal use. Examples of pharmaceutically acceptable excipients include, but are not limited to, carriers, diluents, glidants and lubricants, preservatives, buffering agents, chelating agents, polymers, gelling agents, viscosifying agents, solvents and the like. [0091] The pharmaceutical composition may include, for example, one or more of water, salt solutions, alcohols, polyethylene glycols, peanut oil, olive oil, sugars (e.g., lactose, sucrose, and the like), dextrin, sugar, amylose, lower alkyl ethers of cellulose, magnesium carbonate, magnesium stearate, talc, terra alba, silicic acid, gelatin, agar, pectin, acacia, fatty acids (e.g., stearic acid), fatty acid amides, and fatty acid esters including fatty acid monoglycerides and diglycerides, or the like. The pharmaceutical composition may further include one or more pharmaceutically acceptable auxiliary agents, wetting agents, suspending agents, preserving agents, buffers, sweetening agents, flavouring agents, colorants or any combination of the foregoing. [0092] The pharmaceutical compositions may be in conventional forms, for example, tablets, capsules, solutions, suspensions, injectables or products for topical application. Further, the pharmaceutical composition of the present disclosure may be formulated so as to provide a desired release profile.
[0093] Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges. [0094] Liquid formulations include, but are not limited to, syrups, emulsions and sterile injectable liquids, such as suspensions or solutions. [0095] Topical dosage forms of the compounds include ointments, pastes, creams, lotions, powders, solutions, eye or ear drops, impregnated dressings and may contain appropriate conventional additives such as preservatives, solvents to assist drug penetration. [0096] Administration of the compounds of the disclosure, in pure form or in an appropriate pharmaceutical composition, can be carried out using any of the accepted routes of administration of pharmaceutical compositions. The route of administration may be any route which effectively transports the active compound of the present disclosure to the appropriate or desired site of action. Suitable routes of administration include, but are not limited to, oral, nasal, buccal, dermal, intradermal, transdermal, parenteral, rectal, subcutaneous, intravenous, intraurethral, intramuscular or topical. In some embodiments, the composition is administered by oral, parenteral or inhalation routes. Examples of parenteral administration include administration by injection, as well as percutaneous, transmucosal, transnasal and transpulmonary administration. [0097] Suitable doses of the compounds for use in treating the diseases or disorders described herein can be determined by those skilled in the relevant art. Therapeutic doses are generally identified through dose ranging studies in humans based on preliminary evidence derived from animal studies. Preferably, doses are sufficient to result in a desired therapeutic benefit without causing unwanted side effects. Varying modes of administration, dosage forms and suitable pharmaceutical excipients can be well used and adjusted by those skilled in the art. All changes and modifications are envisioned within the scope of the present disclosure. [0098] In some embodiments, the present disclosure provides a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof, for use as a medicament and for use in the manufacture of medicaments. [0099] In some embodiments, the disclosure provides the use of compounds of Formula (I) in the treatment and prevention of diseases and/or disorders responsive to the inhibition of
USP1 protein expression and USP1 activity, as well as in the manufacture of medicaments for treating such diseases and/or disorders. Typically, the symptoms of the disease are treated, improved, diminished and/or prevented by inhibition of USP1. [0100] In some embodiments, the USP1 mediated disease and/or disorder is cancer. Accordingly, compounds of formula (I) for use in the treatment of cancer and in the manufacture of medicaments for treatment of cancer are also provided. [0101] In related embodiments, the disclosure provides a method of treating a disease or disorder mediated by USP1 in a subject comprising administering to the subject a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or a stereoisomer thereof. In some embodiments, the disease and/or disorder is cancer. [0102] In some embodiments, the cancer is selected from a hematological cancer, a lymphatic cancer, a DNA damage repair pathway deficient cancer, a homologous- recombination deficient cancer, a cancer comprising cancer cells with a mutation in a gene encoding p53, and a cancer comprising cancer cells with a loss of function mutation in a gene encoding p53. [0103] In some embodiments, the cancer is a hematological cancer or a lymphatic cancer. [0104] In some embodiments, the cancer is a DNA damage repair pathway deficient cancer and/or a homologous-recombination deficient cancer. [0105] In some embodiments, the cancer is a DNA damage repair pathway deficient cancer. [0106] In some embodiments, the cancer is a homologous-recombination deficient cancer. [0107] In some embodiments, the cancer is selected from a hematological cancer, a lymphatic cancer, a cancer comprising cancer cells with a mutation in a gene encoding p53, and a cancer comprising cancer cells with a loss of function mutation in a gene encoding p53. [0108] In some embodiments, the cancer comprises cancer cells with a mutation in a gene encoding p53. In some embodiments, the mutation in the gene encoding p53 is a loss of function mutation. [0109] In some embodiments, methods for treating cancer and other diseases include administering to a subject in need thereof a therapeutically effective amount of a compound
of the present disclosure along with one or more additional chemotherapeutic agents independently selected from anti-proliferative agents, anti-cancer agents, immunosuppressant agents and pain-relieving agents. [0110] The method(s) of treatment of the present disclosure generally comprises administering a safe and effective amount of a compound according to formula (I) or a pharmaceutically acceptable salt thereof to a patient (particularly a human) in need thereof. [0111] Compounds of the disclosure are indicated both in the therapeutic and/or prophylactic treatment of the above-mentioned conditions. For the above-mentioned therapeutic uses the dosage administered will typically vary with the compound employed, the mode of administration, the treatment desired and the disorder or disease indicated. [0112] The compounds of the present disclosure may be used as single drug or as a pharmaceutical composition in which the compound is mixed with various pharmacologically acceptable materials, as described above. [0113] In some embodiments, the compounds of the present disclosure can also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds. For example, the present disclosure also embraces isotopically-labeled variants of the present disclosure which are identical to those recited herein, but for the fact that one or more atoms of the compound are replaced by an atom having the atomic mass or mass number different from the predominant atomic mass or mass number usually found in nature for the atom. All isotopes of any particular atom or element as specified are contemplated within the scope of the compounds of the disclosure and their uses. Exemplary isotopes that can be incorporated in to compounds of the disclosure include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine, chlorine and iodine, such as 2H (“D”), 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 32P, 33P, 35S, 18F, 36Cl, 123I and 125I. Isotopically labeled compounds of the present disclosure can generally be prepared by following procedures analogous to those disclosed in the schemes and/or in the examples herein below, by substituting an isotopically labeled reagent for a non-isotopically labeled reagent. [0114] The present application provides methods for the preparation of compounds of formula (I) according to the description provided herein using appropriate methods and/or materials. It is to be understood by those skilled in the art that known variations of the conditions and processes of the following procedures can be used to prepare these
intermediates and compounds. Moreover, by utilizing the procedures described in detail, one of ordinary skill in the art can prepare additional compounds of the present disclosure. [0115] Compounds of this disclosure may be made by synthetic chemical processes, examples of which are shown herein. It is meant to be understood that the order of the steps in the processes may be varied; that reagents, solvents and reaction conditions may be substituted for those specifically mentioned; and that reactive moieties may be protected and deprotected, as necessary. [0116] Unless otherwise stated, work-up includes distribution of the reaction mixture between the organic and aqueous phases, separation of layers and drying the organic layer over anhydrous sodium sulphate, filtration and evaporation of the solvent. Purification, unless otherwise mentioned, includes purification by silica gel chromatographic techniques, generally using ethyl acetate/petroleum ether mixtures of a suitable polarity as the mobile phase. Some of the intermediates were taken to the next step based on TLC results, without further characterization, unless otherwise specified. Analysis for the compounds of the present disclosure, unless otherwise specified, was conducted using general methods well known to a person skilled in the art. [0117] Having described the disclosure with reference to certain preferred embodiments, other embodiments will become apparent to one skilled in the art from consideration of the specification. The disclosure is further defined by reference to the following examples, describing in detail the analysis of the compounds of the disclosure, which are not construed to be viewed as limiting the scope of the disclosure. It will be apparent to those skilled in the art that many modifications, both to materials and methods, may be practiced without departing from the scope of the disclosure.
GENERAL SYNTHETIC SCHEMES: General Scheme I:
 [0118] Some intermediates of the present invention are generally synthesized utilizing the process outlined in General Scheme I. The commercially available or synthesized GS-IA is reacted with dimethylformamide dimethyl acetal in the presence of suitable reagents and solvents (DMF, 100 °C, 16 h) to obtain GS-IB which upon reacting with hydrazine hydrate in presence of suitable reagents and solvents (EtOH, 80 °C, 16 h) affords GS-IC. Treatment of GS-IC with iodine in the presence of suitable reagents and solvent(s) (K2CO3, DMF, 80 °C, 16 h) provides GS-ID. This GS-ID, upon reacting with R1-halide (R1X) in the presence of a base such as cesium carbonate and a solvent such as 1,4-dioxane, affords GS-IE. Coupling GS-IE with an appropriate boronic acid or boronate ester in the presence of a suitable catalyst, base and solvent (e.g., Pd(dppf)Cl2·DCM, K2CO3, 1,4-dioxane:H2O, 100°C, 5h) affords GS-IF. This GS-IF, upon deprotection using trifluoroacetic acid in an appropriate solvent (e.g., DCM), affords intermediate GS-IG.
General Scheme II:
[0118] Some intermediates of the present invention are generally synthesized utilizing the process outlined in General Scheme I. The commercially available or synthesized GS-IA is reacted with dimethylformamide dimethyl acetal in the presence of suitable reagents and solvents (DMF, 100 °C, 16 h) to obtain GS-IB which upon reacting with hydrazine hydrate in presence of suitable reagents and solvents (EtOH, 80 °C, 16 h) affords GS-IC. Treatment of GS-IC with iodine in the presence of suitable reagents and solvent(s) (K2CO3, DMF, 80 °C, 16 h) provides GS-ID. This GS-ID, upon reacting with R1-halide (R1X) in the presence of a base such as cesium carbonate and a solvent such as 1,4-dioxane, affords GS-IE. Coupling GS-IE with an appropriate boronic acid or boronate ester in the presence of a suitable catalyst, base and solvent (e.g., Pd(dppf)Cl2·DCM, K2CO3, 1,4-dioxane:H2O, 100°C, 5h) affords GS-IF. This GS-IF, upon deprotection using trifluoroacetic acid in an appropriate solvent (e.g., DCM), affords intermediate GS-IG.
General Scheme II:
 p g y y izing the process outlined in General Scheme II, wherein ring Z is imidazolyl. The commercially available or synthesized GS-IIA is reacted with a functionalized (3,3-dihalo)propan-2-one (e.g., 3,3-dibromo-1,1,1-trifluoropropan-2-one) in the presence of suitable reagents and solvents (e.g., NaOAc, H2O, 100 °C, 1 h; MeOH, NH4OH, RT, 40 min then 100 °C, 4 h) to obtain GS-IIB which upon reacting with RZ-halide in the presence of suitable reagents and solvents (K2CO3, DMF) affords GS-IIC. Alternatively, treatment of GS-IIB with an appropriate RZ-boronic acid in presence of suitable reagents and solvent(s) [e.g., Cu(OAc)2, O2, 2,2’-bipyridin, Na2CO3, 1,2-DCE, 70 °C, 16 h] also provides GS-IIC. General Scheme III:
p g y y izing the process outlined in General Scheme II, wherein ring Z is imidazolyl. The commercially available or synthesized GS-IIA is reacted with a functionalized (3,3-dihalo)propan-2-one (e.g., 3,3-dibromo-1,1,1-trifluoropropan-2-one) in the presence of suitable reagents and solvents (e.g., NaOAc, H2O, 100 °C, 1 h; MeOH, NH4OH, RT, 40 min then 100 °C, 4 h) to obtain GS-IIB which upon reacting with RZ-halide in the presence of suitable reagents and solvents (K2CO3, DMF) affords GS-IIC. Alternatively, treatment of GS-IIB with an appropriate RZ-boronic acid in presence of suitable reagents and solvent(s) [e.g., Cu(OAc)2, O2, 2,2’-bipyridin, Na2CO3, 1,2-DCE, 70 °C, 16 h] also provides GS-IIC. General Scheme III:
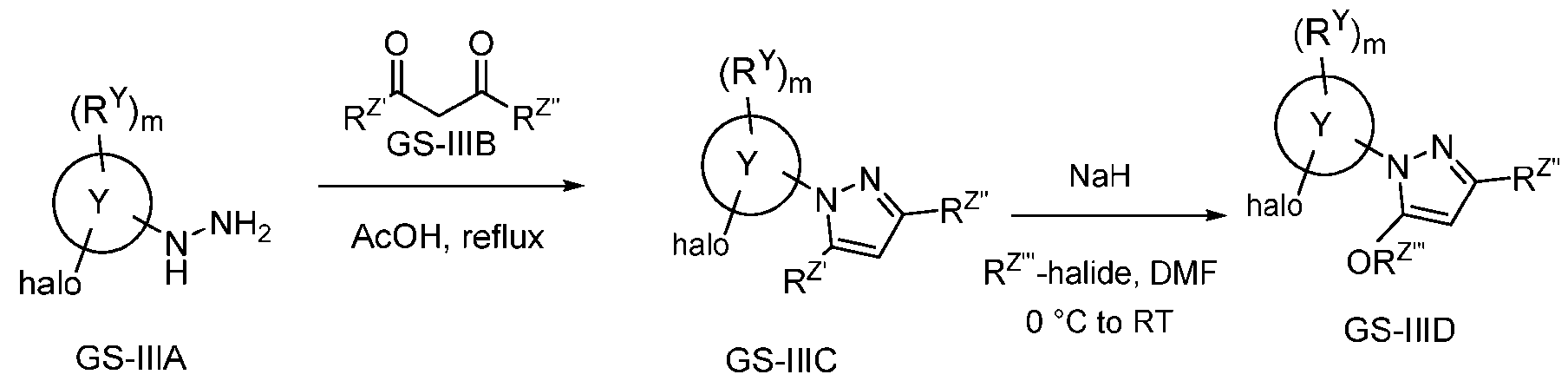 [0120] Some intermediates of the present invention may be synthesized utilizing the process outlined in General Scheme III wherein ring Z is pyrazole. The commercially
available or synthesized GS-IIIA reacts with GS-IIIB in presence of suitable reagents and solvents (e.g., AcOH, reflux) to obtain GS-IIIC which upon reacting with RZ”’-halide in presence of suitable reagents and solvents (e.g., NaH, DMF) gives intermediate GS-IIID. General Scheme IV:
[0120] Some intermediates of the present invention may be synthesized utilizing the process outlined in General Scheme III wherein ring Z is pyrazole. The commercially
available or synthesized GS-IIIA reacts with GS-IIIB in presence of suitable reagents and solvents (e.g., AcOH, reflux) to obtain GS-IIIC which upon reacting with RZ”’-halide in presence of suitable reagents and solvents (e.g., NaH, DMF) gives intermediate GS-IIID. General Scheme IV:
 [0121] Some compounds of the present invention are generally synthesized utilizing the process outlined in General Scheme IV. The commercially available or synthesized GS-IG is reacted with appropriate intermediates GS-IIC or GS-IIIC or GS-IIID, in the presence of suitable reagents and solvents under Buchwald coupling conditions (e.g., Pd2(dba)3, RuPhos, Cs2CO3, PhMe, 100°C, 2h) to afford respective compounds of formula (I).
General Scheme V: [0122] S
[0121] Some compounds of the present invention are generally synthesized utilizing the process outlined in General Scheme IV. The commercially available or synthesized GS-IG is reacted with appropriate intermediates GS-IIC or GS-IIIC or GS-IIID, in the presence of suitable reagents and solvents under Buchwald coupling conditions (e.g., Pd2(dba)3, RuPhos, Cs2CO3, PhMe, 100°C, 2h) to afford respective compounds of formula (I).
General Scheme V: [0122] S
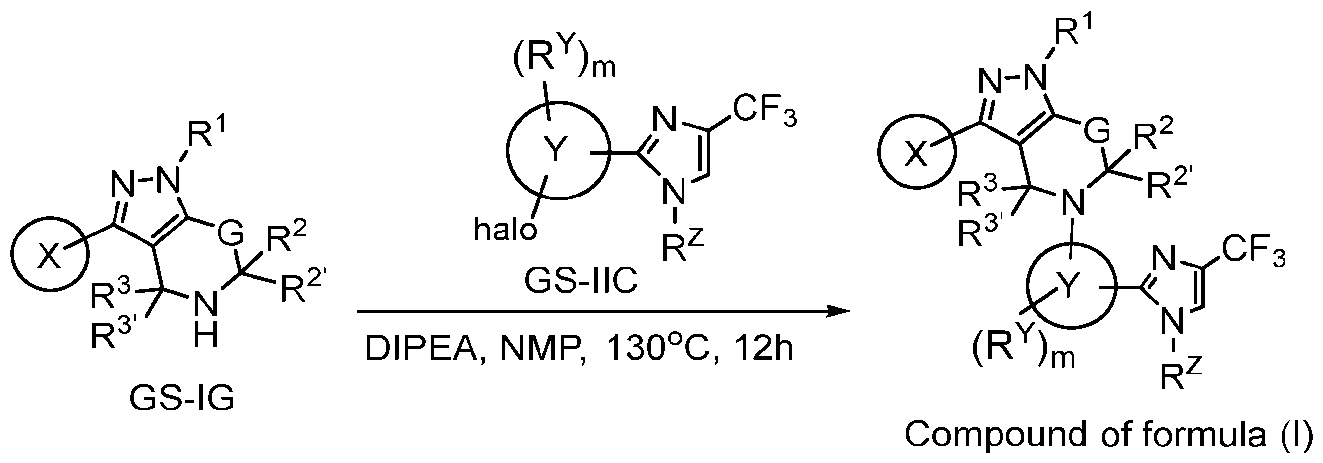 ess outlined in General Scheme V. The commercially available or synthesized GS-IG is reacted with an appropriate intermediate GS-IIC in the presence of suitable reagents and solvents (e.g., DIPEA, NMP, 130oC, 12h) to afford respective compounds of formula (I). EXPERIMENTAL [0123] The following general guidelines apply to all experimental procedures described here. Unless otherwise stated, reactions were performed under positive pressure of nitrogen, and temperatures described are external temperatures (oil bath temperature). Reagents and solvents received from vendors were used as such without any further drying or purification. Molarities mentioned here for reagents in solutions are approximate, as they were not verified by a prior titration with a standard. All reactions were stirred with a magnetic stir bar. Cooling to negative temperatures was achieved with acetone / dry ice or wet ice / salt baths. Magnesium sulfate and sodium sulfate were used as solvent drying agent after reaction work up and are interchangeable. Removal of solvents under reduced pressure/vacuum was conducted using a rotary evaporator. The specifics of the processes for preparing compounds of the present disclosure are detailed in the experimental section. Abbreviations: [0124] DMF-N,N-Dimethylformamide; ACN-Acetonitrile; EtOAc-Ethyl acetate; THF- Tetrahydrofuran; DCM-Dichloromethane; MeOH-Methanol; EtOH-Ethanol; DMSO- dimethyl sulfoxide; MeI-Methyl iodide; EtI-Ethyl iodide; TEA (or) Et3N-Triethylamine; DIPEA- N,N-Diisopropylethylamine; K2CO3-Potassium carbonate; NaOAc-Sodium acetate; KOAc-Potassium acetate; NH4OH-Ammonium hydroxide; NH4Cl-Ammonium chloride; HATU-1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate; RuPhos-2-Dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl; TFA-
Trifluoroacetic acid; AcOH-Acetic acid; RT/rt-Room temperature; Pd(dppf)Cl2·DCM-[1,1′- Bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane; TLC-Thin layer chromatography; LCMS-Liquid chromatography–mass spectrometry; HPLC- High-performance liquid chromatography; DMSO-d6-Deuterated dimethyl sulfoxide; Chloroform-d-Deuterated chloroform; NMR-Nuclear magnetic resonance; s-Singlet; d- Doublet; t-Triplet; q-Quartet; app. quint-Apparent quintet; dd-doublet of doublets; td-Triplet of doublets; SM-starting material; Int-Intermediate. SYNTHESIS OF INTERMEDIATES Synthesis of tert-butyl 1,4,5,6,7,8-hexahydro-4,7-epiminocyclohepta[c]pyrazole-9- carboxylate (I-1) [0125] A mixture o
ess outlined in General Scheme V. The commercially available or synthesized GS-IG is reacted with an appropriate intermediate GS-IIC in the presence of suitable reagents and solvents (e.g., DIPEA, NMP, 130oC, 12h) to afford respective compounds of formula (I). EXPERIMENTAL [0123] The following general guidelines apply to all experimental procedures described here. Unless otherwise stated, reactions were performed under positive pressure of nitrogen, and temperatures described are external temperatures (oil bath temperature). Reagents and solvents received from vendors were used as such without any further drying or purification. Molarities mentioned here for reagents in solutions are approximate, as they were not verified by a prior titration with a standard. All reactions were stirred with a magnetic stir bar. Cooling to negative temperatures was achieved with acetone / dry ice or wet ice / salt baths. Magnesium sulfate and sodium sulfate were used as solvent drying agent after reaction work up and are interchangeable. Removal of solvents under reduced pressure/vacuum was conducted using a rotary evaporator. The specifics of the processes for preparing compounds of the present disclosure are detailed in the experimental section. Abbreviations: [0124] DMF-N,N-Dimethylformamide; ACN-Acetonitrile; EtOAc-Ethyl acetate; THF- Tetrahydrofuran; DCM-Dichloromethane; MeOH-Methanol; EtOH-Ethanol; DMSO- dimethyl sulfoxide; MeI-Methyl iodide; EtI-Ethyl iodide; TEA (or) Et3N-Triethylamine; DIPEA- N,N-Diisopropylethylamine; K2CO3-Potassium carbonate; NaOAc-Sodium acetate; KOAc-Potassium acetate; NH4OH-Ammonium hydroxide; NH4Cl-Ammonium chloride; HATU-1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate; RuPhos-2-Dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl; TFA-
Trifluoroacetic acid; AcOH-Acetic acid; RT/rt-Room temperature; Pd(dppf)Cl2·DCM-[1,1′- Bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane; TLC-Thin layer chromatography; LCMS-Liquid chromatography–mass spectrometry; HPLC- High-performance liquid chromatography; DMSO-d6-Deuterated dimethyl sulfoxide; Chloroform-d-Deuterated chloroform; NMR-Nuclear magnetic resonance; s-Singlet; d- Doublet; t-Triplet; q-Quartet; app. quint-Apparent quintet; dd-doublet of doublets; td-Triplet of doublets; SM-starting material; Int-Intermediate. SYNTHESIS OF INTERMEDIATES Synthesis of tert-butyl 1,4,5,6,7,8-hexahydro-4,7-epiminocyclohepta[c]pyrazole-9- carboxylate (I-1) [0125] A mixture o
 e - u y -o o- -a a cyc o . . oc a e- -carboxylate (50 g, 221.29 mmol, 1.0 equiv.) and dimethylformamide dimethyl acetal (66.67 g, 554.84 mmol, 2.5 equiv.) in DMF (200 mL) was stirred at 100 °C for 16 hours. After that, the reaction mixture was cooled to room temperature and ethanol (50 mL) was added followed by hydrazine hydrate (99%, 44.44 g, 887.74 mmol, 4 equiv.). The reaction was then heated to 100 °C for 16 hours. The reaction mixture was cooled to room temperature and poured into ice cold water then extracted with ethyl acetate twice. Combined organic layers were dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to obtain a crude product. The crude material was suspended in 10 % ethyl acetate in hexane (1 L) and stirred for 30 minutes at room temperature then filtered to provide crude tert-butyl 1,4,5,6,7,8- hexahydro-4,7-epiminocyclohepta[c]pyrazole-9-carboxylate (I-1) (55 g). LC-MS: m/z 250.2 (M+H)+ [0126] The intermediates in Table I were prepared according to the procedure described in I-1 and using appropriate starting material as given here.
Table I: Int. No. SM Int. Structure Analytical Data
e - u y -o o- -a a cyc o . . oc a e- -carboxylate (50 g, 221.29 mmol, 1.0 equiv.) and dimethylformamide dimethyl acetal (66.67 g, 554.84 mmol, 2.5 equiv.) in DMF (200 mL) was stirred at 100 °C for 16 hours. After that, the reaction mixture was cooled to room temperature and ethanol (50 mL) was added followed by hydrazine hydrate (99%, 44.44 g, 887.74 mmol, 4 equiv.). The reaction was then heated to 100 °C for 16 hours. The reaction mixture was cooled to room temperature and poured into ice cold water then extracted with ethyl acetate twice. Combined organic layers were dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to obtain a crude product. The crude material was suspended in 10 % ethyl acetate in hexane (1 L) and stirred for 30 minutes at room temperature then filtered to provide crude tert-butyl 1,4,5,6,7,8- hexahydro-4,7-epiminocyclohepta[c]pyrazole-9-carboxylate (I-1) (55 g). LC-MS: m/z 250.2 (M+H)+ [0126] The intermediates in Table I were prepared according to the procedure described in I-1 and using appropriate starting material as given here.
Table I: Int. No. SM Int. Structure Analytical Data
 Synthesis of tert-butyl (4R,7S)-3-iodo-1,4,5,6,7,8-hexahydro-4,7- epiminocyclohepta[c]pyrazole-9-carboxylate (II-1):
Synthesis of tert-butyl (4R,7S)-3-iodo-1,4,5,6,7,8-hexahydro-4,7- epiminocyclohepta[c]pyrazole-9-carboxylate (II-1):
 [0127] To a mixture of pyrazole I-1 (55 g, 220.60 mmol, 1.0 equiv.) and potassium carbonate (91.46 g, 661.82 mmol, 3.0 equiv.) in acetonitrile (400 mL) was added iodine (111.98 g, 441.21 mmol, 2.0 equiv.). The mixture was heated to 50 °C and stirred for 16 hours. The reaction mixture was quenched with addition of aq. sodium thiosulphate and extracted with ethyl acetate (3x). Combined organic layers were washed with brine and dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to obtain a crude product, which was purified by combi-flash column chromatography using ethyl
acetate in hexane to afford the pure product iodopyrazole II-1 (37.0 g). LC-MS: m/z 376.0 (M+H)+ [0128] The intermediates in Table II were prepared according to the procedure described in II-1 and using appropriate starting material as given here. Table II: Int. No. SM Int. Structure Analytical Data
[0127] To a mixture of pyrazole I-1 (55 g, 220.60 mmol, 1.0 equiv.) and potassium carbonate (91.46 g, 661.82 mmol, 3.0 equiv.) in acetonitrile (400 mL) was added iodine (111.98 g, 441.21 mmol, 2.0 equiv.). The mixture was heated to 50 °C and stirred for 16 hours. The reaction mixture was quenched with addition of aq. sodium thiosulphate and extracted with ethyl acetate (3x). Combined organic layers were washed with brine and dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to obtain a crude product, which was purified by combi-flash column chromatography using ethyl
acetate in hexane to afford the pure product iodopyrazole II-1 (37.0 g). LC-MS: m/z 376.0 (M+H)+ [0128] The intermediates in Table II were prepared according to the procedure described in II-1 and using appropriate starting material as given here. Table II: Int. No. SM Int. Structure Analytical Data
 Synthesis of tert-butyl 3-iodo-1-methyl-1,4,5,6,7,8-hexahydro-4,7- epiminocyclohepta[c]pyrazole-9-carboxylate (III-1):
Synthesis of tert-butyl 3-iodo-1-methyl-1,4,5,6,7,8-hexahydro-4,7- epiminocyclohepta[c]pyrazole-9-carboxylate (III-1):
 [0129] To a stirred solution of iodopyrazole II-1 (35 g, 93.28 mmol, 1.0 equiv.) in 1,4- dioxane (350 mL) were added cesium carbonate (91.17 g, 279.84 mmol, 3.0 equiv.) followed
by methyl iodide (19.86 g, 139.91 mmol, 1.5 equiv.) at room temperature and the mixture was stirred for 16 hours. The reaction mixture was diluted with addition of ice-cold water and then extracted with ethyl acetate (3x). Combined organic layers were washed with brine and dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to obtain a crude product. which was purified by combi-flash column chromatography using ethyl acetate in hexane to afford the pure product III-1 (17.0 g). LC-MS: m/z 390.1 (M+H)+ [0130] The intermediates in Table III were prepared according to the procedure described in III-1 and using appropriate starting material as given here. Table III: Int. No. SM Int. Structure Analytical Data
[0129] To a stirred solution of iodopyrazole II-1 (35 g, 93.28 mmol, 1.0 equiv.) in 1,4- dioxane (350 mL) were added cesium carbonate (91.17 g, 279.84 mmol, 3.0 equiv.) followed
by methyl iodide (19.86 g, 139.91 mmol, 1.5 equiv.) at room temperature and the mixture was stirred for 16 hours. The reaction mixture was diluted with addition of ice-cold water and then extracted with ethyl acetate (3x). Combined organic layers were washed with brine and dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to obtain a crude product. which was purified by combi-flash column chromatography using ethyl acetate in hexane to afford the pure product III-1 (17.0 g). LC-MS: m/z 390.1 (M+H)+ [0130] The intermediates in Table III were prepared according to the procedure described in III-1 and using appropriate starting material as given here. Table III: Int. No. SM Int. Structure Analytical Data
 Synthesis of tert-butyl 3-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-methyl-1,4,5,6,7,8- hexahydro-4,7-epiminocyclohepta[c]pyrazole-9-carboxylate (IV-1): [0131] A mix
Synthesis of tert-butyl 3-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-methyl-1,4,5,6,7,8- hexahydro-4,7-epiminocyclohepta[c]pyrazole-9-carboxylate (IV-1): [0131] A mix
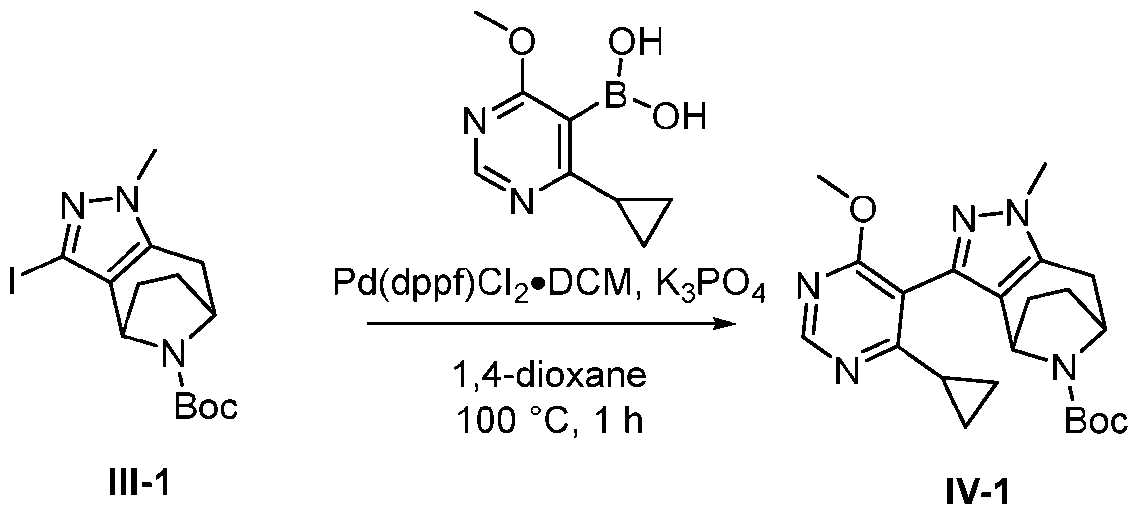 , , , 4-cyclopropyl-6- methoxypyrimidin-5-yl)boronic acid (21.18 g, 109.18 mmol, 2.5 equiv.) and tripotassium phosphate (27.81 g, 131.02 mmol, 3.0 equiv.) in 1,4-dioxane (150 mL) was degassed with argon for 10 min. After that, Pd(dppf)Cl2·DCM (3.56 g, 4.36 mmol, 0.1 equiv.) was added and then the reaction mixture was heated to 100 °C for 1 hour. The reaction was cooled to room temperature and quenched with the addition of ice-cold water, then extracted with ethyl acetate (3x). The combined organic layers were washed with brine and dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to a give crude product which was purified by combi-flash column chromatography using ethyl acetate in hexane as an eluent to afford the coupled product IV-1 (15 g). LC-MS: m/z 412.3 (M+H)+ [0132] The intermediates in Table IV were prepared according to the procedure described in IV-1 and using appropriate starting material as given here. Table IV: Int. No. SM Int. Structure Analytical Data
, , , 4-cyclopropyl-6- methoxypyrimidin-5-yl)boronic acid (21.18 g, 109.18 mmol, 2.5 equiv.) and tripotassium phosphate (27.81 g, 131.02 mmol, 3.0 equiv.) in 1,4-dioxane (150 mL) was degassed with argon for 10 min. After that, Pd(dppf)Cl2·DCM (3.56 g, 4.36 mmol, 0.1 equiv.) was added and then the reaction mixture was heated to 100 °C for 1 hour. The reaction was cooled to room temperature and quenched with the addition of ice-cold water, then extracted with ethyl acetate (3x). The combined organic layers were washed with brine and dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to a give crude product which was purified by combi-flash column chromatography using ethyl acetate in hexane as an eluent to afford the coupled product IV-1 (15 g). LC-MS: m/z 412.3 (M+H)+ [0132] The intermediates in Table IV were prepared according to the procedure described in IV-1 and using appropriate starting material as given here. Table IV: Int. No. SM Int. Structure Analytical Data
 Int. No. SM Int. Structure Analytical Data
Int. No. SM Int. Structure Analytical Data
 Synthesis of 3-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-methyl-1,4,5,6,7,8- hexahydro-4,7-epiminocyclohepta[c]pyrazole (V-1):
Synthesis of 3-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-methyl-1,4,5,6,7,8- hexahydro-4,7-epiminocyclohepta[c]pyrazole (V-1):


 [0133] To a stirred solution of N-Boc-piperidine IV-1 (15 g, 36.45 mmol, 1.0 equiv.) in dichloromethane (150 mL) was added trifluoroacetic acid (30 mL, 2 V) at 0 °C. The reaction mixture was allowed to warm to room temperature and was stirred for 16 hours. After that, the reaction mixture was concentrated under reduced pressure and then quenched with addition of sat. sodium bicarbonate solution and extracted with EtOAc (3x). Combined organic layers were washed with brine and dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to give a crude product V-1 (11 g), which was taken to the next step. LC-MS: m/z 312.1 (M+H)+ [0134] The intermediates in Table V were prepared according to the procedure described in V-1 and using appropriate starting material as given here.
Table V: Int. No. SM Int. Structure Analytical Data
[0133] To a stirred solution of N-Boc-piperidine IV-1 (15 g, 36.45 mmol, 1.0 equiv.) in dichloromethane (150 mL) was added trifluoroacetic acid (30 mL, 2 V) at 0 °C. The reaction mixture was allowed to warm to room temperature and was stirred for 16 hours. After that, the reaction mixture was concentrated under reduced pressure and then quenched with addition of sat. sodium bicarbonate solution and extracted with EtOAc (3x). Combined organic layers were washed with brine and dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to give a crude product V-1 (11 g), which was taken to the next step. LC-MS: m/z 312.1 (M+H)+ [0134] The intermediates in Table V were prepared according to the procedure described in V-1 and using appropriate starting material as given here.
Table V: Int. No. SM Int. Structure Analytical Data
 Synthesis of 2-bromo-5-(4-(trifluoromethyl)-1H-imidazol-2-yl)pyridine (VI-1):
Synthesis of 2-bromo-5-(4-(trifluoromethyl)-1H-imidazol-2-yl)pyridine (VI-1):
 [0135] To a stirred solution of 3,3-dibromo-1,1,1-trifluoropropan-2-one (1.34 kg, 4.946 mol, 2.3 equiv.) in water (1.5 L) was added NaOAc (0.71 kg, 8.602 mol, 4.0 equiv.), and the mixture was stirred at 100 °C for 1 hour and then cooled to 0 °C. A pre-mixed solution of 6- bromonicotinaldehyde (0.4 kg, 2.150 mol, 1.0 equiv.) and aq. ammonia (25% in water, 1.5 L) in MeOH (4.0 L) was added at 0 °C, and the reaction mixture was then stirred at room
temperature for 40 min. After that, the reaction mixture was heated to 100 °C with stirring for 4 hours. The reaction mixture was concentrated to remove methanol. The obtained crude solid was filtered from the aqueous phase and dried under high vacuum to give a crude product. The crude solid was washed with 5% ethyl acetate in hexane to remove some unreacted starting material that provided sufficiently pure imidazole VI-1 (0.33 kg).1H NMR (400 MHz, DMSO-d6): δ 13.49 (bs, 1H), 8.91 (d, J = 2.4 Hz, 1H), 8.22 (dd, J = 8.4, 2,4 Hz, 1H), 8.02 (s, 1H), 7.77 (d, J = 8.4 Hz, 1H); LC-MS: m/z 291.9 (M+H)+ Synthesis of 2-bromo-5-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)pyridine (VII- 1): [0136] To a s
[0135] To a stirred solution of 3,3-dibromo-1,1,1-trifluoropropan-2-one (1.34 kg, 4.946 mol, 2.3 equiv.) in water (1.5 L) was added NaOAc (0.71 kg, 8.602 mol, 4.0 equiv.), and the mixture was stirred at 100 °C for 1 hour and then cooled to 0 °C. A pre-mixed solution of 6- bromonicotinaldehyde (0.4 kg, 2.150 mol, 1.0 equiv.) and aq. ammonia (25% in water, 1.5 L) in MeOH (4.0 L) was added at 0 °C, and the reaction mixture was then stirred at room
temperature for 40 min. After that, the reaction mixture was heated to 100 °C with stirring for 4 hours. The reaction mixture was concentrated to remove methanol. The obtained crude solid was filtered from the aqueous phase and dried under high vacuum to give a crude product. The crude solid was washed with 5% ethyl acetate in hexane to remove some unreacted starting material that provided sufficiently pure imidazole VI-1 (0.33 kg).1H NMR (400 MHz, DMSO-d6): δ 13.49 (bs, 1H), 8.91 (d, J = 2.4 Hz, 1H), 8.22 (dd, J = 8.4, 2,4 Hz, 1H), 8.02 (s, 1H), 7.77 (d, J = 8.4 Hz, 1H); LC-MS: m/z 291.9 (M+H)+ Synthesis of 2-bromo-5-(1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl)pyridine (VII- 1): [0136] To a s
 e so u o o e e a e - . g, . o , .0 equiv.) in ACN (2.0 L) at RT were added Cs2CO3 (0.74 kg, 2.259 mol, 2.0 equiv.) followed by methyl iodide (0.24 kg, 1.694 mol, 1.5 equiv.). The reaction mixture was stirred for 16 hours at room temperature. The reaction was quenched with addition of ice-cold water (1 L), then the mixture was extracted with ethyl acetate (3 x 2 L). Combined organic layers were washed with brine and dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to a give crude product VII-1 (0.32 kg, rr approximately 93:7) which was taken to the next step without further purification. 1H NMR (400 MHz, DMSO-d6): δ 8.74 (d, J = 2.8 Hz, 1H), 8.11 (dd, J = 8.2, 2.6 Hz, 1H), 8.03 (s, 1H), 7.81 (d, J = 8.4 Hz, 1H), 3.82 (s, 3H); LC-MS: m/z 306.0 (M+H)+ Example 1: Synthesis of 3-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-methyl-9-(5-(1- methyl-4-(trifluoromethyl)-1H-imidazol2-yl)pyridin-2-yl)-1,4,5,6,7,8-hexahydro-4,7- epiminocyclohepta[c]pyrazole (Compound 1)
[0137] A
e so u o o e e a e - . g, . o , .0 equiv.) in ACN (2.0 L) at RT were added Cs2CO3 (0.74 kg, 2.259 mol, 2.0 equiv.) followed by methyl iodide (0.24 kg, 1.694 mol, 1.5 equiv.). The reaction mixture was stirred for 16 hours at room temperature. The reaction was quenched with addition of ice-cold water (1 L), then the mixture was extracted with ethyl acetate (3 x 2 L). Combined organic layers were washed with brine and dried over anhydrous sodium sulphate. The filtered organic layer was concentrated to a give crude product VII-1 (0.32 kg, rr approximately 93:7) which was taken to the next step without further purification. 1H NMR (400 MHz, DMSO-d6): δ 8.74 (d, J = 2.8 Hz, 1H), 8.11 (dd, J = 8.2, 2.6 Hz, 1H), 8.03 (s, 1H), 7.81 (d, J = 8.4 Hz, 1H), 3.82 (s, 3H); LC-MS: m/z 306.0 (M+H)+ Example 1: Synthesis of 3-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-methyl-9-(5-(1- methyl-4-(trifluoromethyl)-1H-imidazol2-yl)pyridin-2-yl)-1,4,5,6,7,8-hexahydro-4,7- epiminocyclohepta[c]pyrazole (Compound 1)
[0137] A
 ridine derivative VII-1 (16.21 g, 52.98 mmol, 1.5 equiv.), RuPhos (3.23 g, 7.06 mmol, 0.2 equiv.) and cesium carbonate (34.53 g, 105.97 mmol, 3.0 eq) in toluene (120 mL) was degassed with argon for 10 minutes. After that, Pd2(dba)3 (3.23 g, 3.53 mmol, 0.1 equiv.) was added and the reaction mixture was heated to 100 °C for 3 hours. The reaction mixture was cooled to room temperature and filtered through a Celite® bed. The filtered organic layer was concentrated to a give crude product which was purified by combi-flash column chromatography using ethyl acetate in hexane as an eluent to afford the racemic Compound 1 (11 g). LC-MS: m/z 537.3 (M+H)+. [0138] The racemic mixture was subjected to chiral HPLC purification to afford Compound 1a (2.3 g) and Compound 1b (2.35 g). (-) Compound 1a (Isomer 1 of Compound 1): 1H NMR (400 MHz, DMSO-d6): ^ 8.63 (s, 1H), 8.37 (d, J = 2.4 Hz, 1H), 7.87 (t, J = 1.2 Hz, 1H), 7.80 (dd, J = 8.8, 2.4 Hz, 1H), 6.79 (d, J = 8.8 Hz, 1H), 4.96 (d, J = 3.2 Hz, 2H), 3.91 (s, 3H), 3.73 (s, 3H), 3.63 (s, 3H), 3.24-3.19 (m, 1H), 2.62 (d, J = 16 Hz, 1H), 2.34-2.30 (m, 1H), 2.15-2.10 (m, 1H), 2.07-1.94 (m, 2H), 1.85-1.76 (m, 1H), 1.10-0.90 (m, 2H), 0.92-0.88 (m, 2H); LC-MS: m/z 537.3 (M+H)+; [α]20 D –210.06 (c = 0.1, EtOH). (+) Compound 1b (Isomer 2 of Compound 1): 1H NMR (400 MHz, DMSO-d6): ^ 8.63 (s, 1H), 8.37 (d, J = 2.4 Hz, 1H), 7.87 (d, J = 1.2 Hz, 1H), 7.80 (dd, J = 8.8, 2.4 Hz, 1H), 6.79 (d, J = 8.8 Hz, 1H), 4.96 (d, J = 3.6 Hz, 2H), 3.91 (s, 3H), 3.73 (s, 3H), 3.63 (s, 3H), 3.24-3.19 (m, 1H), 2.62 (d, J = 16 Hz, 1H), 2.34-2.30 (m, 1H), 2.15-2.10 (m, 1H), 2.07-1.94 (m, 2H), 1.85-1.76 (m, 1H), 1.10-0.90 (m, 2H), 0.92-0.88 (m, 2H); LC-MS: m/z 537.3 (M+H)+; [α]20 D +207.79 (c = 0.1, EtOH).
[0139] The compounds in Table VI were prepared according to procedures similar to the procedure described for Compound 1, using appropriate starting materials and intermediates as set forth above. Table VI: Int. Int. No. Compound Structure Analytical Data No. No. ^ ), z, s, = J 70 10 ), ), ); ^ ), = z, 74 .8 90 85 +
ridine derivative VII-1 (16.21 g, 52.98 mmol, 1.5 equiv.), RuPhos (3.23 g, 7.06 mmol, 0.2 equiv.) and cesium carbonate (34.53 g, 105.97 mmol, 3.0 eq) in toluene (120 mL) was degassed with argon for 10 minutes. After that, Pd2(dba)3 (3.23 g, 3.53 mmol, 0.1 equiv.) was added and the reaction mixture was heated to 100 °C for 3 hours. The reaction mixture was cooled to room temperature and filtered through a Celite® bed. The filtered organic layer was concentrated to a give crude product which was purified by combi-flash column chromatography using ethyl acetate in hexane as an eluent to afford the racemic Compound 1 (11 g). LC-MS: m/z 537.3 (M+H)+. [0138] The racemic mixture was subjected to chiral HPLC purification to afford Compound 1a (2.3 g) and Compound 1b (2.35 g). (-) Compound 1a (Isomer 1 of Compound 1): 1H NMR (400 MHz, DMSO-d6): ^ 8.63 (s, 1H), 8.37 (d, J = 2.4 Hz, 1H), 7.87 (t, J = 1.2 Hz, 1H), 7.80 (dd, J = 8.8, 2.4 Hz, 1H), 6.79 (d, J = 8.8 Hz, 1H), 4.96 (d, J = 3.2 Hz, 2H), 3.91 (s, 3H), 3.73 (s, 3H), 3.63 (s, 3H), 3.24-3.19 (m, 1H), 2.62 (d, J = 16 Hz, 1H), 2.34-2.30 (m, 1H), 2.15-2.10 (m, 1H), 2.07-1.94 (m, 2H), 1.85-1.76 (m, 1H), 1.10-0.90 (m, 2H), 0.92-0.88 (m, 2H); LC-MS: m/z 537.3 (M+H)+; [α]20 D –210.06 (c = 0.1, EtOH). (+) Compound 1b (Isomer 2 of Compound 1): 1H NMR (400 MHz, DMSO-d6): ^ 8.63 (s, 1H), 8.37 (d, J = 2.4 Hz, 1H), 7.87 (d, J = 1.2 Hz, 1H), 7.80 (dd, J = 8.8, 2.4 Hz, 1H), 6.79 (d, J = 8.8 Hz, 1H), 4.96 (d, J = 3.6 Hz, 2H), 3.91 (s, 3H), 3.73 (s, 3H), 3.63 (s, 3H), 3.24-3.19 (m, 1H), 2.62 (d, J = 16 Hz, 1H), 2.34-2.30 (m, 1H), 2.15-2.10 (m, 1H), 2.07-1.94 (m, 2H), 1.85-1.76 (m, 1H), 1.10-0.90 (m, 2H), 0.92-0.88 (m, 2H); LC-MS: m/z 537.3 (M+H)+; [α]20 D +207.79 (c = 0.1, EtOH).
[0139] The compounds in Table VI were prepared according to procedures similar to the procedure described for Compound 1, using appropriate starting materials and intermediates as set forth above. Table VI: Int. Int. No. Compound Structure Analytical Data No. No. ^ ), z, s, = J 70 10 ), ), ); ^ ), = z, 74 .8 90 85 +
 Int. Int. No. Compound Structure Analytical Data No. No. 5 al ^ ), = z, d, s, ), m, 0- C- ^ ), = z, d, 75 J 16 m, /z
Int. Int. No. Compound Structure Analytical Data No. No. 5 al ^ ), = z, d, s, ), m, 0- C- ^ ), = z, d, 75 J 16 m, /z
 Int. Int. No. Compound Structure Analytical Data No. No. ^ ), z, d, z, 75 78 J ), .3 ^ ), z, d, z, 7- ), z, m, ^ ), = z, 82 ), ), ).
Int. Int. No. Compound Structure Analytical Data No. No. ^ ), z, d, z, 75 78 J ), .3 ^ ), z, d, z, 7- ), z, m, ^ ), = z, 82 ), ), ).
 [0140] Although the present application has been illustrated by certain of the preceding examples, it is not to be construed as being limited thereby; but rather, the present application encompasses the generic area as hereinbefore disclosed. For example, the compounds in the below Table VII which can be prepared by following similar procedure as described in above Schemes/Examples with suitable modifications known to the one ordinary skilled in the art are also included in the scope of the present application: Table VII:
[0140] Although the present application has been illustrated by certain of the preceding examples, it is not to be construed as being limited thereby; but rather, the present application encompasses the generic area as hereinbefore disclosed. For example, the compounds in the below Table VII which can be prepared by following similar procedure as described in above Schemes/Examples with suitable modifications known to the one ordinary skilled in the art are also included in the scope of the present application: Table VII:
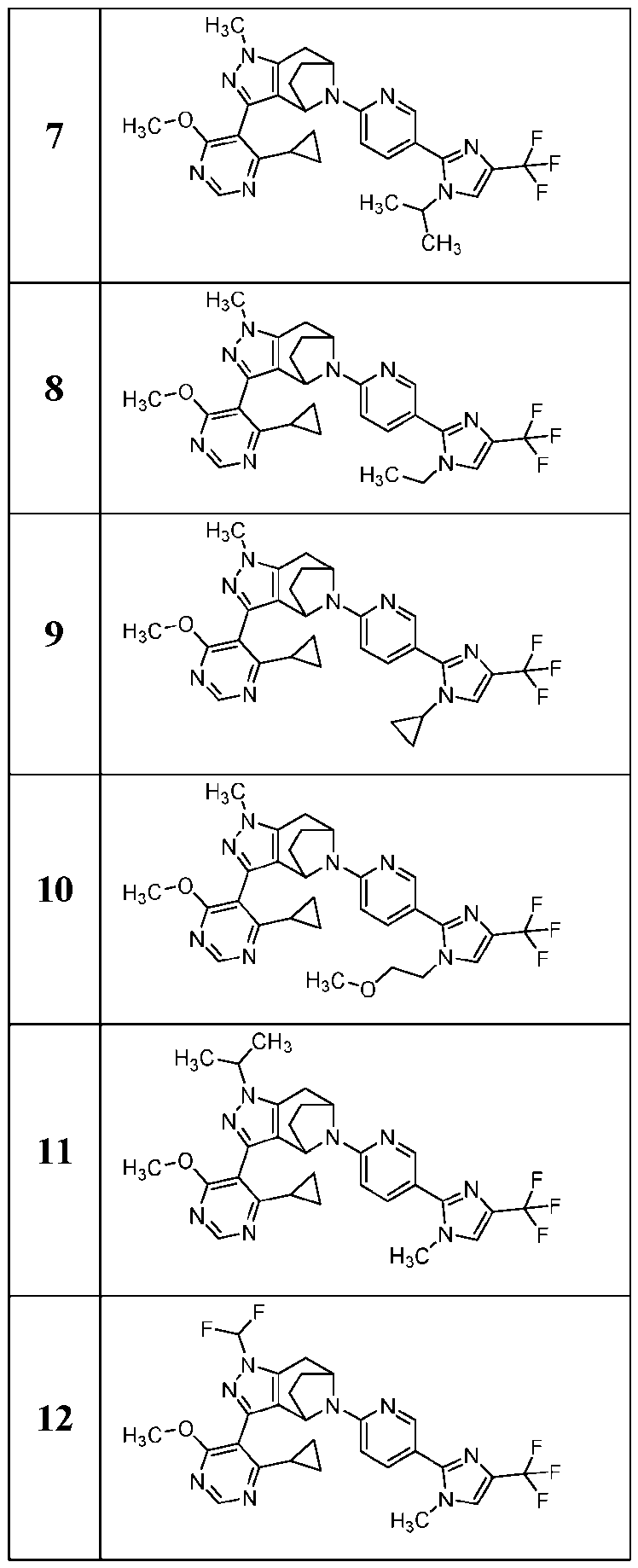
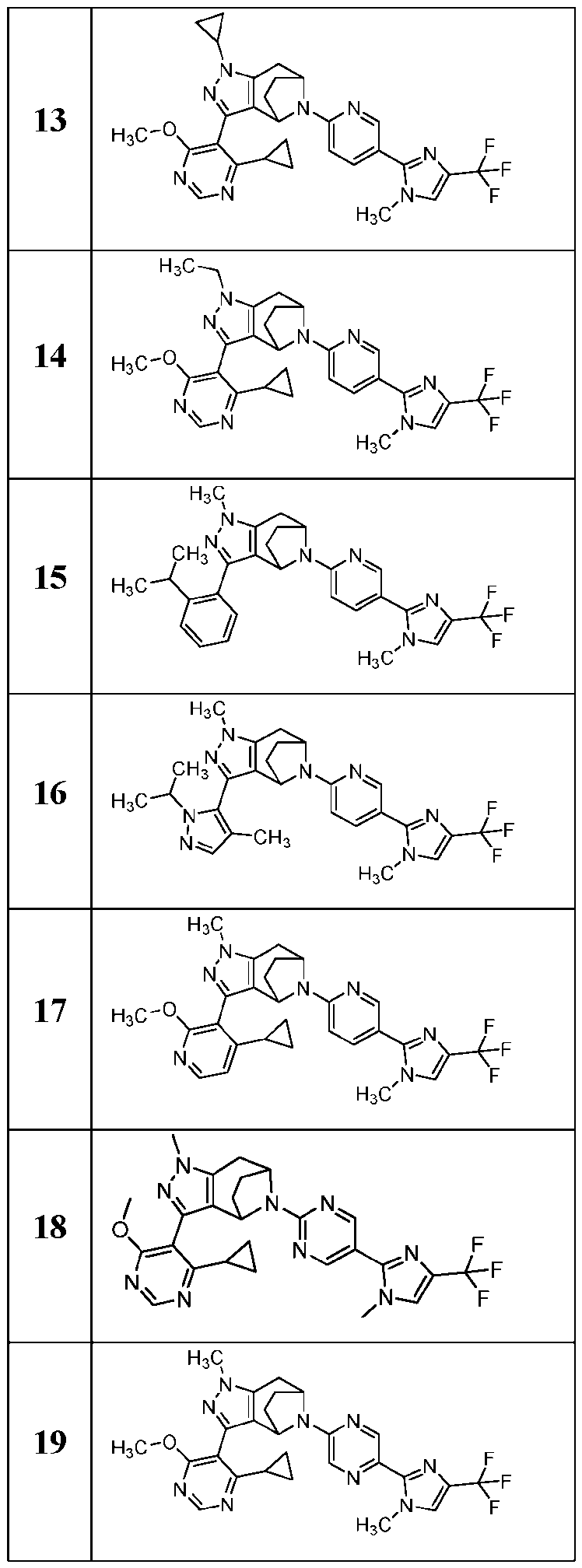 20 28
20 28

 42
42

 BIOLOGY [0141] Biochemical Assay: The compounds were evaluated for their potential to inhibit USP1-UAF1 complex (Boston Biochem, Catalog: E568-050) using a fluorescence-based assay. The final concentrations of USP1-UAF1 complex protein and substrate Ubiquitin- Rhodamine-110 (R&D systems, Catalog U-555-050) used in the assay were 0.45 and 150 nM
respectively.50 mM HEPES pH.7.5, 100 mM NaCl, 0.5 mM EDTA, 1 mM TCEP, 10% BSA, 0.01% Tween 20 buffer was used in the assay. The total assay volume was 20 µL. The compounds were initially prepared in 100% DMSO and appropriate dilutions were made by 1/3rd serial dilutions from the stock, for use in determination of IC50 values. The final DMSO concentration in the assay was 1%. The compounds were pre-incubated with USP1-UAF1 complex at 25°C for 15 min. After preincubation, the required concentration of substrate was added and incubated at 25°C for 60 min. Fluorescence at Excitation: 485 nm, Emission: 535 nm was measured in Victor-5 from Perkin Elmer. [0142] To determine IC50 values, dose response curves were generated by plotting percentage inhibition as a function of inhibitor concentration, and the data was fitted to a sigmoidal non-linear regression equation (variable slope) using Graph Pad prism software V8. [0143] The compounds were screened by the above-mentioned assay procedure. Greater than 97% inhibition of USP1-UAF1 was observed when combined with Compounds 1a, 2, 3, 4a, 4b, 5b, and 6 at 10 μM. Less than 20% inhibition of USP1-UAF1 was observed when combined with Compounds 1b and 5a at 10 μM. IC50 values of the compounds are summarized in Table VIII below, wherein “A” refers to an IC50 value less than 0.05 μM, “B” refers to an IC50 value in range of 0.05 μM to 0.5 μM and “C” refers to an IC50 value greater than 0.5 μM. Table VIII: USP1-UAF1 IC50 values for compounds of the disclosure Compound USP1-UAF1 IC50 (µM)
BIOLOGY [0141] Biochemical Assay: The compounds were evaluated for their potential to inhibit USP1-UAF1 complex (Boston Biochem, Catalog: E568-050) using a fluorescence-based assay. The final concentrations of USP1-UAF1 complex protein and substrate Ubiquitin- Rhodamine-110 (R&D systems, Catalog U-555-050) used in the assay were 0.45 and 150 nM
respectively.50 mM HEPES pH.7.5, 100 mM NaCl, 0.5 mM EDTA, 1 mM TCEP, 10% BSA, 0.01% Tween 20 buffer was used in the assay. The total assay volume was 20 µL. The compounds were initially prepared in 100% DMSO and appropriate dilutions were made by 1/3rd serial dilutions from the stock, for use in determination of IC50 values. The final DMSO concentration in the assay was 1%. The compounds were pre-incubated with USP1-UAF1 complex at 25°C for 15 min. After preincubation, the required concentration of substrate was added and incubated at 25°C for 60 min. Fluorescence at Excitation: 485 nm, Emission: 535 nm was measured in Victor-5 from Perkin Elmer. [0142] To determine IC50 values, dose response curves were generated by plotting percentage inhibition as a function of inhibitor concentration, and the data was fitted to a sigmoidal non-linear regression equation (variable slope) using Graph Pad prism software V8. [0143] The compounds were screened by the above-mentioned assay procedure. Greater than 97% inhibition of USP1-UAF1 was observed when combined with Compounds 1a, 2, 3, 4a, 4b, 5b, and 6 at 10 μM. Less than 20% inhibition of USP1-UAF1 was observed when combined with Compounds 1b and 5a at 10 μM. IC50 values of the compounds are summarized in Table VIII below, wherein “A” refers to an IC50 value less than 0.05 μM, “B” refers to an IC50 value in range of 0.05 μM to 0.5 μM and “C” refers to an IC50 value greater than 0.5 μM. Table VIII: USP1-UAF1 IC50 values for compounds of the disclosure Compound USP1-UAF1 IC50 (µM)
 Incorporation by Reference [0144] All publications and patents mentioned herein are hereby incorporated by reference in their entirety as if each individual publication or patent were specifically and individually indicated to be incorporated by reference. In case of conflict, the present application, including any definitions herein, will control. Equivalents [0145] While specific embodiments of the subject disclosure have been discussed, the above specification is illustrative and not restrictive. Many variations of the disclosure will become apparent to those skilled in the art upon review of this specification and the claims below. The full scope of the disclosure should be determined by reference to the claims, along with their full scope of equivalents, and the specification, along with such variations.
Incorporation by Reference [0144] All publications and patents mentioned herein are hereby incorporated by reference in their entirety as if each individual publication or patent were specifically and individually indicated to be incorporated by reference. In case of conflict, the present application, including any definitions herein, will control. Equivalents [0145] While specific embodiments of the subject disclosure have been discussed, the above specification is illustrative and not restrictive. Many variations of the disclosure will become apparent to those skilled in the art upon review of this specification and the claims below. The full scope of the disclosure should be determined by reference to the claims, along with their full scope of equivalents, and the specification, along with such variations.
Claims
WHAT IS CLAIMED IS: 1. A compound of formula (I): or a pharmaceutical
 wherein: ;
wherein: ;
 ring Y is C3-C8 cycloalkyl, C5-C10 cycloalkeny , wherein the asterisk marks the point of attachment to rin
ring Y is C3-C8 cycloalkyl, C5-C10 cycloalkeny , wherein the asterisk marks the point of attachment to rin
 ring Z is 3- to 15-membered heterocycloalkyl or 5- to 14-membered heteroaryl; X1, X2 and X3 are independently N or C; Y1 to Y4 are independently N or C, wherein 0-2 of Y1 to Y4 are N; Y5 and Y6 are independently N, C, O or S; each RX is independently C1-C6 alkyl, C1-C6 alkoxy or C3-C8 cycloalkyl; RY is halo, C1-C6 alkyl, C1-C6 alkoxy or C1-C6 alkylamino; each RZ is independently hydrogen, halo, C1-C6 alkyl, halo(C1-C6)alkyl, C1-C6 alkoxy, C3-C8 cycloalkyl, (C1-C6)alkylamino-(C1-C6)alkyl, (C1-C6)alkoxy-(C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl or (C1-C6 alkyl)-substituted 3- to 15-membered heterocycloalkyl, wherein halo and C1-C6 alkoxy are bonded to carbon atoms in the Z ring; G is -O-, -N(RG1)-, *-C(RG2RG3)-N(RG1)-, *-N(RG1)-C(RG2RG3)-, *-C(RG2RG3)-O-, -[C(RG2RG3)]q- or -C(R4R4 ^)-, wherein the asterisk marks the point of attachment to the pyrazole ring; RG1, each RG2 and each RG3 are independently hydrogen or C1-C6 alkyl;
R1 is hydrogen, C1-C6 alkyl, halo(C1-C6)alkyl, C3-C8 cycloalkyl, (C1-C6)alkylamino- (C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl, (C1-C6)alkyl-substituted 3- to 15-membered heterocycloalkyl, optionally substituted (C6-C14)aryl(C1-C6)alkyl, or optionally substituted (5- to 14-membered heteroaryl)-(C1-C6)alkyl, wherein the substituents on (C6- C14)aryl(C1-C6)alkyl and (5- to 14-membered heteroaryl)-(C1-C6 )alkyl are independently halo, C1-C6 alkyl or C1-C6 alkoxy; R2, R2 ^, R3 and R3 ^ are independently hydrogen, C1-C6 alkyl, halo, cyano or amino; or R2 and R2 ^, taken together, form an oxo group; or R2 and R2 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; or R2 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; or R3 and RG2 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; R4 and R4 ^ are: i. taken together along with the carbon atom to which they are attached to form a C3-C8 cycloalkyl, or ii. independently hydroxy or C1-C6 alkoxy, or iii. independently hydrogen or C1-C6 alkyl, with the proviso that R4 and R4 ^ are not hydrogen or alkyl when: (a) each of R2, R2 ^, R3, R3 ^ is hydrogen, (b) ring Y is phenyl, thiophenyl, thiazolyl, 6-membered heteroaryl or cubanyl, and (c) ring Z is imidazolyl; or R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; subscript p is 1,
ring Z is 3- to 15-membered heterocycloalkyl or 5- to 14-membered heteroaryl; X1, X2 and X3 are independently N or C; Y1 to Y4 are independently N or C, wherein 0-2 of Y1 to Y4 are N; Y5 and Y6 are independently N, C, O or S; each RX is independently C1-C6 alkyl, C1-C6 alkoxy or C3-C8 cycloalkyl; RY is halo, C1-C6 alkyl, C1-C6 alkoxy or C1-C6 alkylamino; each RZ is independently hydrogen, halo, C1-C6 alkyl, halo(C1-C6)alkyl, C1-C6 alkoxy, C3-C8 cycloalkyl, (C1-C6)alkylamino-(C1-C6)alkyl, (C1-C6)alkoxy-(C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl or (C1-C6 alkyl)-substituted 3- to 15-membered heterocycloalkyl, wherein halo and C1-C6 alkoxy are bonded to carbon atoms in the Z ring; G is -O-, -N(RG1)-, *-C(RG2RG3)-N(RG1)-, *-N(RG1)-C(RG2RG3)-, *-C(RG2RG3)-O-, -[C(RG2RG3)]q- or -C(R4R4 ^)-, wherein the asterisk marks the point of attachment to the pyrazole ring; RG1, each RG2 and each RG3 are independently hydrogen or C1-C6 alkyl;
R1 is hydrogen, C1-C6 alkyl, halo(C1-C6)alkyl, C3-C8 cycloalkyl, (C1-C6)alkylamino- (C1-C6)alkyl, unsubstituted 3- to 15-membered heterocycloalkyl, (C1-C6)alkyl-substituted 3- to 15-membered heterocycloalkyl, optionally substituted (C6-C14)aryl(C1-C6)alkyl, or optionally substituted (5- to 14-membered heteroaryl)-(C1-C6)alkyl, wherein the substituents on (C6- C14)aryl(C1-C6)alkyl and (5- to 14-membered heteroaryl)-(C1-C6 )alkyl are independently halo, C1-C6 alkyl or C1-C6 alkoxy; R2, R2 ^, R3 and R3 ^ are independently hydrogen, C1-C6 alkyl, halo, cyano or amino; or R2 and R2 ^, taken together, form an oxo group; or R2 and R2 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl; or R2 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; or R3 and RG2 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; R4 and R4 ^ are: i. taken together along with the carbon atom to which they are attached to form a C3-C8 cycloalkyl, or ii. independently hydroxy or C1-C6 alkoxy, or iii. independently hydrogen or C1-C6 alkyl, with the proviso that R4 and R4 ^ are not hydrogen or alkyl when: (a) each of R2, R2 ^, R3, R3 ^ is hydrogen, (b) ring Y is phenyl, thiophenyl, thiazolyl, 6-membered heteroaryl or cubanyl, and (c) ring Z is imidazolyl; or R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S; subscript p is 1,
2 or 3; subscript q is 2,
3 or 4; subscript m is 0 or 1; and subscript n is 1, 2 or 3. 2. The compound of claim 1, having a formula (IA):
or a pharmaceutica
 3. The compound of claim 1, having a formula (IB): or a pharmaceutically
3. The compound of claim 1, having a formula (IB): or a pharmaceutically
 f.
f.
4. The compound of claim 1, having a formula (IC): or a pharmaceut

5. The compound of claim 1, having a formula (ID): or a pharmaceut
 .
.
6. The compound of claim 1, having a formula (IE): or a pharmaceut

7. The compound of claim 1, having a formula (IF): or a pharmaceuti

8. The compound of claim 1, having a formula (IG): or pharma
 f, wherein the bridge LG is -(CH2)1-3- or -CH2-O-CH2-.
f, wherein the bridge LG is -(CH2)1-3- or -CH2-O-CH2-.
9. The compound of claim 1, having a formula (IH): or pharm f, wherein the G
 bridge L is -(CH2)1-3- or -CH2-O-CH2-.
bridge L is -(CH2)1-3- or -CH2-O-CH2-.
10. The compound of any one of claims 1 to 9, wherein ring Z is a 5-membered heteroaryl.
11. The compound of any one of claims 1 to 10, wherein ring Z is imidazolyl or pyrazolyl.
12. The compound of any one of claims 1 to 9, wherein at least one RX is C1-C6 alkyl.
13. The compound of any one of claims 1 to 9, wherein each RX is independently C1-C6 alkoxy or C3-C8 cycloalkyl.
14. The compound of any one of claims 1 to 9, wherein G is -C(R4R4 ^)-.
15. The compound of any one of claims 1 to 9, wherein G is -[C(RG2RG3)]q- or -C(R4R4 ^)-, and q is 2.
16. The compound of any one of claims 1 to 9, wherein R1 is C1-C6 alkyl.
17. The compound of any one of claims 1 to 9, wherein both R4 and R4 ^ are hydrogen.
18. The compound of any one of claims 1 to 9, wherein R2 is C1-C6 alkyl.
19. The compound of any one of claims 1 to 9, wherein R3 is C1-C6 alkyl.
20. The compound of any one of claims 1 to 9, wherein R2 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S.
21. The compound of any one of claims 1 to 9, wherein R4 and R3 are taken together to form a C1-C3 alkylene bridge, wherein 1 to 2 carbon atom(s) of the alkylene bridge is/are optionally replaced by heteroatom(s) independently selected from N, O and S.
22. The compound of any one of claims 1 to 9, wherein R4 and R4 ^, taken together along with the carbon atom to which they are attached, form C3-C8 cycloalkyl.
23. The compound of any one of claims 1 to 9, wherein each RZ is independently C1-C6 alkyl or halo(C1-C6)alkyl.
24. The compound of any one of claims 1 to 9, wherein subscript m is 0.
25. The compound of any one of claims 1 to 9, wherein subscript n is 2.
26. The compound of any one of claims 1 to 9, wherein subscript p is 2.
27. The compound of any one of claims 1 to 9, wherein subscript q is 2.
28. A compound selected from: Compound Compound Structure Compound Name 7- 7- 7-
 Compound Compound Structure Compound Name No. 8-
Compound Compound Structure Compound Name No. 8-
 Compound Compound Structure Compound Name No. - -
Compound Compound Structure Compound Name No. - -
 Compound Compound Structure Compound Name No. 7- 7- - 7- 7- 2- 7-
Compound Compound Structure Compound Name No. 7- 7- - 7- 7- 2- 7-
 Compound Compound Structure Compound Name No. )- 7- - 7- 7-
Compound Compound Structure Compound Name No. )- 7- - 7- 7-
 Compound Compound Structure Compound Name No. -
Compound Compound Structure Compound Name No. -
 Compound Compound Structure Compound Name No. 6- 7- 7-
Compound Compound Structure Compound Name No. 6- 7- 7-
 Compound Compound Structure Compound Name No. -
Compound Compound Structure Compound Name No. -
 Compound Compound Structure Compound Name No. - - 7- 7-
Compound Compound Structure Compound Name No. - - 7- 7-
 Compound Compound Structure Compound Name No. 7- - 7-
Compound Compound Structure Compound Name No. 7- - 7-

29. A pharmaceutical composition comprising a compound of any one of claims 1 to 28, or a pharmaceutically acceptable salt thereof or a stereoisomer thereof, and one or more pharmaceutically acceptable excipients.
30. The compound of any one of claims 1 to 28, or a pharmaceutically acceptable salt or a stereoisomer thereof, for use as a medicament.
31. The compound of any one of claims 1 to 28, or a pharmaceutically acceptable salt or a stereoisomer thereof, for use in the treatment of cancer.
32. The compound of claim 31, wherein the cancer is a hematological cancer or a lymphatic cancer.
33. The compound of any one of claims 31 or 32, wherein the cancer is a DNA damage repair pathway deficient cancer and/or a homologous-recombination deficient cancer.
34. The compound of any one of claims 31 or 32, wherein the cancer comprises cancer cells with a mutation in a gene encoding p53.
35. The compound of claim 34, wherein the mutation in the gene encoding p53 is a loss of function mutation.
36. A method of inhibiting USP1 in a subject comprising administering to the subject an effective amount of a compound of any one of claims 1 to 28 or a pharmaceutically acceptable salt or a stereoisomer thereof.
37. A method of treating a disease or disorder mediated by USP1 in a subject comprising administering to the subject a therapeutically effective amount of a compound according to any one of claims 1 to 28 or a pharmaceutically acceptable salt or a stereoisomer thereof.
38. The method of claim 37, wherein the disease or disorder is cancer.
39. The method of claim 38, wherein the cancer is a hematological cancer or a lymphatic cancer.
40. The method of any one of the claims 37 or 38, wherein the cancer is a DNA damage repair pathway deficient cancer and/or a homologous-recombination deficient cancer.
41. The method of any one of the claims 37 to 40, wherein the cancer comprises cancer cells with a mutation in a gene encoding p53.
42. The method of claim 41, wherein the mutation in the gene encoding p53 is a loss of function mutation.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN202341045361 | 2023-07-06 | ||
| IN202341045361 | 2023-07-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025010245A1 true WO2025010245A1 (en) | 2025-01-09 |
Family
ID=92108666
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/036492 Pending WO2025010245A1 (en) | 2023-07-06 | 2024-07-02 | Fused pyrazole derivatives as usp1 inhibitors |
Country Status (3)
| Country | Link |
|---|---|
| AR (1) | AR133186A1 (en) |
| TW (1) | TW202506110A (en) |
| WO (1) | WO2025010245A1 (en) |
Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007149484A2 (en) | 2006-06-20 | 2007-12-27 | Dana-Farber Cancer Institute | Inhibitors of usp1 deubiquitinating enzyme complex |
| WO2011137320A2 (en) | 2010-04-30 | 2011-11-03 | Dana-Farber Cancer Institute, Inc. | Small molecule inhibitors of usp1 deubiquitinating enzyme activity |
| WO2014105952A2 (en) | 2012-12-28 | 2014-07-03 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Inhibitors of the usp1/uaf1 deubiquitinase complex and uses thereof |
| WO2017087837A1 (en) | 2015-11-20 | 2017-05-26 | Forma Therapeutics, Inc. | Purinones as ubiquitin-specific protease 1 inhibitors |
| WO2019089216A1 (en) | 2017-11-01 | 2019-05-09 | Dana-Farber Cancer Institute, Inc. | Methods of treating cancers |
| WO2020132269A1 (en) | 2018-12-20 | 2020-06-25 | KSQ Therapeutics, Inc. | Substituted pyrazolopyrimidines and substituted purines and their use as ubiquitin-specific-processing protease 1 (usp1) inhibitors |
| WO2020139988A1 (en) | 2018-12-28 | 2020-07-02 | Forma Therapeutics, Inc. | Compositions for inhibiting ubiquitin specific protease 1 |
| WO2021163530A1 (en) | 2020-02-14 | 2021-08-19 | KSQ Therapeutics, Inc. | Therapeutic combinations comprising ubiquitin-specific-processing protease 1 (usp1) inhibitors and poly (adp-ribose) polymerase (parp) inhibitors |
| WO2021247606A1 (en) | 2020-06-02 | 2021-12-09 | KSQ Therapeutics, Inc. | Nitrogen-containing fused bicyclic compounds and their use as ubiquitin-specific-processing protease 1 (usp1) inhibitors |
| WO2022174184A1 (en) | 2021-02-15 | 2022-08-18 | Tango Therapeutics, Inc. | Pyrrolo[3,2-d]pyrimidine compounds and methods of use in the treatment of cancer |
| WO2022197892A1 (en) | 2021-03-17 | 2022-09-22 | Tango Therapeutics, Inc. | Purine derivatives as anticancer agents |
| WO2022199652A1 (en) | 2021-03-24 | 2022-09-29 | Impact Therapeutics (Shanghai) , Inc | Five-membered heteroaryl-pyrimidine compounds as usp1 inhibitors and the use thereof |
| WO2022214053A1 (en) | 2021-04-09 | 2022-10-13 | 海南耀臻生物医药科技有限公司 | Ubiquitin-specific protease 1 (usp1) inhibitor |
| WO2022216820A1 (en) | 2021-04-07 | 2022-10-13 | Forma Therapeutics, Inc. | Inhibiting ubiquitin-specific protease 1 (usp1) |
| WO2022228399A1 (en) | 2021-04-27 | 2022-11-03 | 山东轩竹医药科技有限公司 | Tricyclic usp1 inhibitor and use thereof |
| WO2022233263A1 (en) | 2021-05-03 | 2022-11-10 | 山东轩竹医药科技有限公司 | Tricyclic ubiquitin specific protease 1 inhibitor and use thereof |
| WO2022253188A1 (en) | 2021-05-31 | 2022-12-08 | Impact Therapeutics (Shanghai) , Inc | Nitrogen-containing fused heteroaromatic bicyclic compounds as usp1 inhibitors and the use thereof |
| WO2023030295A1 (en) | 2021-09-01 | 2023-03-09 | 先声再明医药有限公司 | Ubiquitin specific protease 1 (usp1) inhibitor |
| WO2023066299A1 (en) | 2021-10-19 | 2023-04-27 | Impact Therapeutics (Shanghai) , Inc | Substituted triazoloheteroaryl compounds as usp1 inhibitors and the use thereof |
| WO2023083285A1 (en) | 2021-11-12 | 2023-05-19 | Insilico Medicine Ip Limited | Small molecule inhibitors of ubiquitin specific protease 1 (usp1) and uses thereof |
| WO2023083297A1 (en) | 2021-11-12 | 2023-05-19 | Insilico Medicine Ip Limited | Small molecule inhibitors of ubiquitin specific protease 1 (usp1) and uses thereof |
| WO2023083286A1 (en) | 2021-11-12 | 2023-05-19 | Insilico Medicine Ip Limited | Small molecule inhibitors of ubiquitin specific protease 1 (usp1) and uses thereof |
| WO2024086790A1 (en) * | 2022-10-21 | 2024-04-25 | Exelixis, Inc. | 4,5,6,7-TETRAHYDRO-1H-PYRAZOLO[4,3-c]PYRIDINE COMPOUNDS AND DERIVATIVES AS USP1 INHIBITORS |
-
2024
- 2024-07-02 WO PCT/US2024/036492 patent/WO2025010245A1/en active Pending
- 2024-07-05 TW TW113125228A patent/TW202506110A/en unknown
- 2024-07-05 AR ARP240101764A patent/AR133186A1/en unknown
Patent Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007149484A2 (en) | 2006-06-20 | 2007-12-27 | Dana-Farber Cancer Institute | Inhibitors of usp1 deubiquitinating enzyme complex |
| WO2011137320A2 (en) | 2010-04-30 | 2011-11-03 | Dana-Farber Cancer Institute, Inc. | Small molecule inhibitors of usp1 deubiquitinating enzyme activity |
| WO2014105952A2 (en) | 2012-12-28 | 2014-07-03 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Inhibitors of the usp1/uaf1 deubiquitinase complex and uses thereof |
| WO2017087837A1 (en) | 2015-11-20 | 2017-05-26 | Forma Therapeutics, Inc. | Purinones as ubiquitin-specific protease 1 inhibitors |
| WO2019089216A1 (en) | 2017-11-01 | 2019-05-09 | Dana-Farber Cancer Institute, Inc. | Methods of treating cancers |
| WO2020132269A1 (en) | 2018-12-20 | 2020-06-25 | KSQ Therapeutics, Inc. | Substituted pyrazolopyrimidines and substituted purines and their use as ubiquitin-specific-processing protease 1 (usp1) inhibitors |
| WO2020139988A1 (en) | 2018-12-28 | 2020-07-02 | Forma Therapeutics, Inc. | Compositions for inhibiting ubiquitin specific protease 1 |
| WO2021163530A1 (en) | 2020-02-14 | 2021-08-19 | KSQ Therapeutics, Inc. | Therapeutic combinations comprising ubiquitin-specific-processing protease 1 (usp1) inhibitors and poly (adp-ribose) polymerase (parp) inhibitors |
| WO2021247606A1 (en) | 2020-06-02 | 2021-12-09 | KSQ Therapeutics, Inc. | Nitrogen-containing fused bicyclic compounds and their use as ubiquitin-specific-processing protease 1 (usp1) inhibitors |
| WO2022174184A1 (en) | 2021-02-15 | 2022-08-18 | Tango Therapeutics, Inc. | Pyrrolo[3,2-d]pyrimidine compounds and methods of use in the treatment of cancer |
| WO2022197892A1 (en) | 2021-03-17 | 2022-09-22 | Tango Therapeutics, Inc. | Purine derivatives as anticancer agents |
| WO2022199652A1 (en) | 2021-03-24 | 2022-09-29 | Impact Therapeutics (Shanghai) , Inc | Five-membered heteroaryl-pyrimidine compounds as usp1 inhibitors and the use thereof |
| WO2022216820A1 (en) | 2021-04-07 | 2022-10-13 | Forma Therapeutics, Inc. | Inhibiting ubiquitin-specific protease 1 (usp1) |
| WO2022214053A1 (en) | 2021-04-09 | 2022-10-13 | 海南耀臻生物医药科技有限公司 | Ubiquitin-specific protease 1 (usp1) inhibitor |
| WO2022228399A1 (en) | 2021-04-27 | 2022-11-03 | 山东轩竹医药科技有限公司 | Tricyclic usp1 inhibitor and use thereof |
| WO2022233263A1 (en) | 2021-05-03 | 2022-11-10 | 山东轩竹医药科技有限公司 | Tricyclic ubiquitin specific protease 1 inhibitor and use thereof |
| WO2022253188A1 (en) | 2021-05-31 | 2022-12-08 | Impact Therapeutics (Shanghai) , Inc | Nitrogen-containing fused heteroaromatic bicyclic compounds as usp1 inhibitors and the use thereof |
| WO2023030295A1 (en) | 2021-09-01 | 2023-03-09 | 先声再明医药有限公司 | Ubiquitin specific protease 1 (usp1) inhibitor |
| WO2023066299A1 (en) | 2021-10-19 | 2023-04-27 | Impact Therapeutics (Shanghai) , Inc | Substituted triazoloheteroaryl compounds as usp1 inhibitors and the use thereof |
| WO2023083285A1 (en) | 2021-11-12 | 2023-05-19 | Insilico Medicine Ip Limited | Small molecule inhibitors of ubiquitin specific protease 1 (usp1) and uses thereof |
| WO2023083297A1 (en) | 2021-11-12 | 2023-05-19 | Insilico Medicine Ip Limited | Small molecule inhibitors of ubiquitin specific protease 1 (usp1) and uses thereof |
| WO2023083286A1 (en) | 2021-11-12 | 2023-05-19 | Insilico Medicine Ip Limited | Small molecule inhibitors of ubiquitin specific protease 1 (usp1) and uses thereof |
| WO2024086790A1 (en) * | 2022-10-21 | 2024-04-25 | Exelixis, Inc. | 4,5,6,7-TETRAHYDRO-1H-PYRAZOLO[4,3-c]PYRIDINE COMPOUNDS AND DERIVATIVES AS USP1 INHIBITORS |
Non-Patent Citations (14)
| Title |
|---|
| "Handbook of Pharmaceutical Additives", 2007, GOWER PUBLISHING COMPANY |
| "Pharmaceutical Preformulation and Formulation", 2009, CRC PRESS LLC |
| "Remington: The Science and Practice of Pharmacy", 2020, PHARMACEUTICAL PRESS |
| CATALDO F, MOL CELL BIOL, vol. 33, no. 12, 2013, pages 2485 - 2496 |
| CHEN J ET AL., CHEM BIOL., vol. 18, 2011, pages 1390 - 400 |
| COHN, M.A. ET AL., J BIOL CHEM, vol. 284, 2009, pages 5343 - 51 |
| COHN, M.A. ET AL., MOL CELL, vol. 28, 2007, pages 786 - 97 |
| DAS DS. ET AL., CLIN CANCER RES., vol. 23, 2017, pages 4280 - 9 |
| GUERVILLY, J.H. ET AL., HUM MOL GENET, 2011 |
| HUANG TT. ET AL., NAT CELL BIOL, vol. 8, no. 4, 2006, pages 339 - 347 |
| NALEPA, G. ET AL., NAT REV DRUG DISCOV, vol. 5, 2006, pages 596 - 613 |
| NIJMAN, S.M. ET AL., CELL, vol. 123, 2005, pages 773 - 86 |
| NIJMAN, S.M. ET AL., MOL CELL, vol. 17, 2005, pages 331 - 9 |
| XIN XU ET AL., FRONT ONCOL., vol. 9, 2019, pages 1406 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202506110A (en) | 2025-02-16 |
| AR133186A1 (en) | 2025-09-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7044769B2 (en) | Cyano-substituted heterocycle with activity as an inhibitor of USP30 | |
| EP3302448B1 (en) | Substituted heterocyclyl derivatives as cdk inhibitors | |
| JP7604457B2 (en) | 2-Hydroxycycloalkane-1-carbamoyl derivatives | |
| EP2358698B1 (en) | Tetrasubstituted pyridazine as hedgehog pathway antagonists | |
| CN110831936B (en) | Substituted cyanopyrrolidines with DUB inhibitor activity | |
| EP3019482B1 (en) | Trisubstituted benzotriazole derivatives as dihydroorotate oxygenase inhibitors | |
| EP3331884B1 (en) | Urea derivative or pharmacologically acceptable salt thereof | |
| WO2015077193A1 (en) | Inhibitors of lysine methyl transferase | |
| WO2023148643A1 (en) | Fused bicyclic heterocyclyl compounds as usp1 inhibitors | |
| EP4291556B1 (en) | Hydroxyheterocycloalkane-carbamoyl derivatives | |
| CN111484491B (en) | Substituted pyrido-cyclic compounds, process for their preparation and their use | |
| EP4206197A1 (en) | Preparation method for novel rho-related protein kinase inhibitor and intermediate in preparation method | |
| WO2024086790A1 (en) | 4,5,6,7-TETRAHYDRO-1H-PYRAZOLO[4,3-c]PYRIDINE COMPOUNDS AND DERIVATIVES AS USP1 INHIBITORS | |
| EP3971176A1 (en) | Aromatic amine ar and bet targeting protein degradation chimera compound and use | |
| EP3490984A1 (en) | 2-pyrrolidine phenylhydrazides antibacterial agents | |
| US11970493B2 (en) | Autotaxin inhibitor compounds | |
| IL323884A (en) | Disubstituted pyrimidine compounds for ketohexokinase inhibition | |
| WO2025010245A1 (en) | Fused pyrazole derivatives as usp1 inhibitors | |
| EP3904348A1 (en) | Aminopyridine compound, preparation method therefor and use thereof | |
| CN112876419A (en) | Allylamine derivatives, process for producing the same and use thereof | |
| CN115894376B (en) | Aromatic amide compound, pharmaceutical composition and use thereof | |
| WO2022255399A1 (en) | G9a inhibitor | |
| WO2024258591A2 (en) | Novel inhibitors of histone demethylases and compositions and methods thereof | |
| HK40101885A (en) | Hydroxyheterocycloalkane-carbamoyl derivatives | |
| HK40101885B (en) | Hydroxyheterocycloalkane-carbamoyl derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24748732 Country of ref document: EP Kind code of ref document: A1 |