WO2024186626A1 - Aza-bicyclic sting agonist immunoconjugates, and uses thereof - Google Patents
Aza-bicyclic sting agonist immunoconjugates, and uses thereof Download PDFInfo
- Publication number
- WO2024186626A1 WO2024186626A1 PCT/US2024/018051 US2024018051W WO2024186626A1 WO 2024186626 A1 WO2024186626 A1 WO 2024186626A1 US 2024018051 W US2024018051 W US 2024018051W WO 2024186626 A1 WO2024186626 A1 WO 2024186626A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyldiyl
- immunoconjugate
- och
- peg
- antibody
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the invention relates generally to an immunoconjugate comprising an antibody conjugated to one or more aza-bicyclic STING agonist moieties.
- STING Stimulator of Interferon Genes
- TMEM173 transmembrane protein 173
- MPYS/MITA/ERIS transmembrane protein 173
- STING is broadly expressed, particularly in immune cells, lung, and ovary.
- STING plays a role in innate immunity by inducing type I interferon production when cells are infected with intracellular pathogens, such as viruses, mycobacteria, and intracellular parasites.
- Type I interferon mediated by STING, protects infected cells and nearby cells from local infection by binding to the same cell that secretes it by autocrine signaling and nearby cells by paracrine signaling.
- STING works as both a direct cytosolic DNA sensor (CDS) and an adaptor protein in Type I interferon signaling through different molecular mechanisms.
- STING has been shown to activate downstream transcription factors STAT6 and IRF3 through TBK1, which are responsible for antiviral response and innate immune response against intracellular pathogens.
- Compounds that bind to STING and act as an agonist have been shown to induce secretion of proinflammatory cytokines including type 1 interferons on incubation with human PBMCs (WO 2017/175147).
- STING modulators may be useful in the treatment of various disorders, for example, allergic diseases, neurodegenerative diseases, pre-cancerous syndromes, and cancer, and may also be useful in immunogenic compositions or vaccine adjuvants.
- the invention is generally directed to immunoconjugates comprising an antibody covalently attached to one or more STING agonist moieties by a linker, and having Formula I: Ab ⁇ [L ⁇ D] p I or a pharmaceutically acceptable salt thereof, wherein: Ab is the antibody; L is the linker; p is an integer from 1 to 8; D is the STING agonist moiety having the formula: where the dashed lines are optional double bonds and the substituents are defined herein.
- the invention is further directed to use of such an immunoconjugates in the treatment of an illness, in particular cancer.
- Another aspect of the invention is an aza-bicyclic-linker compound.
- Another aspect of the invention is a method for treating cancer comprising administering a therapeutically effective amount of an immunoconjugate comprising an antibody linked by conjugation to one or more aza-bicyclic STING agonist moieties.
- Another aspect of the invention is a use of an immunoconjugate comprising an antibody linked by conjugation to one or more aza-bicyclic STING agonist moieties for treating cancer.
- Another aspect of the invention is a method of preparing an immunoconjugate by conjugation of one or more aza-bicyclic STING agonist moieties with an antibody.
- immunoconjugate refers to an antibody construct that is covalently bonded to an adjuvant moiety via a linker.
- immunoconjugate refers to an antibody construct that is covalently bonded to an adjuvant moiety via a linker.
- immunoconjugate refers to a moiety, substance or adjuvant capable of eliciting an immune response in a subject exposed to the immunostimulatory moiety or the immunostimulatory compound after in vivo cleavage of the linker.
- adjuvant moiety refers to an adjuvant, alternatively referred to as a “payload”, that is covalently bonded to a cell-binding agent, such as an antibody construct, through an elastase-substrate, peptide linker, as described herein.
- the adjuvant moiety can elicit the immune response while bonded to the antibody construct or after cleavage (e.g., enzymatic cleavage) from the antibody construct following administration of an immunoconjugate to the subject.
- Immunoconjugates allow targeted delivery of an active adjuvant moiety while the target antigen is bound.
- Adjuvant refers to a substance capable of eliciting an immune response in a subject exposed to the adjuvant.
- adjuvant moiety refers to an adjuvant that is covalently bonded to an antibody construct, e.g., through a linker, as described herein. The adjuvant moiety can elicit the immune response while bonded to the antibody construct or after cleavage (e.g., enzymatic cleavage) from the antibody construct following administration of an immunoconjugate to the subject.
- antibody is used in the broadest sense and specifically encompasses monoclonal antibodies (including full length monoclonal antibodies), polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired biological activity.
- Antibody fragment and all grammatical variants thereof as used herein are defined as a portion of an intact antibody comprising the antigen binding site or variable region of the intact antibody, wherein the portion is free of the constant heavy chain domains (i.e., CH2, CH3, and CH4, depending on antibody isotype) of the Fc region of the intact antibody.
- antibody fragments include Fab,Fab',Fab'-SH, F(ab' ) 2 , and Fv fragments; diabodies; any antibody fragment that is a polypeptide having a primary structure consisting of one uninterrupted sequence of contiguous amino acid residues (referred to herein as a “single-chain antibody fragment” or “single chain polypeptide”), including without limitation (1) single-chain Fv (scFv) molecules; (2) single chain polypeptides containing only one light chain variable domain, or a fragment thereof that contains the three CDRs of the light chain variable domain, without an associated heavy chain moiety; (3) single chain polypeptides containing only one heavy chain variable region, or a fragment thereof containing the three CDRs of the heavy chain variable region, without an associated light chain moiety; (4) nanobodies comprising single Ig domains from non-human species or other specific single-domain binding modules; and (5) multispecific or multivalent structures formed from antibody fragments.
- the heavy chain(s) can contain any constant domain sequence (e.g., CH1 in the IgG isotype) found in a non-Fc region of an intact antibody, and/or can contain any hinge region sequence found in an intact antibody, and/or can contain a leucine zipper sequence fused to or situated in the hinge region sequence or the constant domain sequence of the heavy chain(s).
- any constant domain sequence e.g., CH1 in the IgG isotype
- Antibody refers to a polypeptide comprising an antigen binding region (including the complementarity determining region (CDRs)) from an immunoglobulin gene or fragments thereof.
- the term “antibody” specifically encompasses monoclonal antibodies (including full length monoclonal antibodies), polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments that exhibit the desired biological activity.
- An exemplary immunoglobulin (antibody) structural unit comprises a tetramer. Each tetramer is composed of two identical pairs of polypeptide chains, each pair having one “light” (about 25 kDa) and one “heavy” chain (about 50-70 kDa) connected by disulfide bonds.
- Each chain is composed of structural domains, which are referred to as immunoglobulin domains. These domains are classified into different categories by size and function, e.g., variable domains or regions on the light and heavy chains (VL and VH, respectively) and constant domains or regions on the light and heavy chains (CL and CH, respectively).
- the N-terminus of each chain defines a variable region of about 100 to 110 or more amino acids, referred to as the paratope, primarily responsible for antigen recognition, i.e., the antigen binding domain.
- Light chains are classified as either kappa or lambda.
- Heavy chains are classified as gamma, mu, alpha, delta, or epsilon, which in turn define the immunoglobulin classes, IgG, IgM, IgA, IgD and IgE, respectively.
- IgG antibodies are large molecules of about 150 kDa composed of four peptide chains.
- IgG antibodies contain two identical class ⁇ heavy chains of about 50 kDa and two identical light chains of about 25 kDa, thus a tetrameric quaternary structure. The two heavy chains are linked to each other and to a light chain each by disulfide bonds. The resulting tetramer has two identical halves, which together form the Y-like shape. Each end of the fork contains an identical antigen binding domain.
- IgG1 is the most abundant.
- antigen binding domain of an antibody will be most critical in specificity and affinity of binding to cancer cells.
- An antibody that targets a particular antigen includes a bispecific or multispecific antibody with at least one antigen binding region that targets the particular antigen.
- the targeted monoclonal antibody is a bispecific antibody with at least one antigen binding region that targets tumor cells.
- antigens include but are not limited to: mesothelin, prostate specific membrane antigen (PSMA), HER2, TROP2, CEA, EGFR, 5T4,Nectin4, CD 19, CD20, CD22, CD30, CD70, B7H3, B7H4 (also known as 08E), protein tyrosine kinase 7 (PTK7), glypi can-3, RG1, fucosyl-GMl, CTLA-4, and CD44 (WO 2017/196598).
- Antibody construct refers to an antibody or a fusion protein comprising (i) an antigen binding domain and (ii) an Fc domain.
- the binding agent is an antigen-binding antibody “fragment,” which is a construct that comprises at least an antigen-binding region of an antibody, alone or with other components that together constitute the antigen-binding construct.
- fragment is a construct that comprises at least an antigen-binding region of an antibody, alone or with other components that together constitute the antigen-binding construct.
- antibody “fragments” are known in the art, including, for instance, (i) a Fab fragment, which is a monovalent fragment consisting of the V L , V H , C L , and CHi domains, (ii) a F(ab’) 2 fragment, which is a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region, (iii) a Fv fragment consisting of the V L and V H domains of a single arm of an antibody, (iv) a Fab’ fragment, which results from breaking the disulfide bridge of an F(ab’) 2 fragment using mild reducing conditions
- the antibody or antibody fragments can be part of a larger construct, for example, a conjugate or fusion construct of the antibody fragment to additional regions.
- the antibody fragment can be fused to an Fc region as described herein.
- the antibody fragment e.g., a Fab or scFv
- the antibody fragment can be part of a chimeric antigen receptor or chimeric T-cell receptor, for instance, by fusing to a transmembrane domain (optionally with an intervening linker or “stalk” (e.g., hinge region)) and optional intercellular signaling domain.
- the antibody fragment can be fused to the gamma and/or delta chains of a T-cell receptor, so as to provide a T-cell receptor like construct that binds TROP2.
- the antibody fragment is part of a bispecific T-cell engager (BiTEs) comprising a CD1 or CD3 binding domain and linker.
- BiTEs bispecific T-cell engager
- Epitope means any antigenic determinant or epitopic determinant of an antigen to which an antigen binding domain binds (i.e., at the paratope of the antigen binding domain).
- Antigenic determinants usually consist of chemically active surface groupings of molecules, such as amino acids or sugar side chains, and usually have specific three dimensional structural characteristics, as well as specific charge characteristics.
- Fc receptor refers to a receptor that binds to the Fc region of an antibody.
- Fc ⁇ R which binds to IgG
- FcaR which binds to IgA
- FcaR which binds to IgE.
- the Fc ⁇ R family includes several members, such as Fc ⁇ l (CD64), Fc ⁇ RIIA (CD32A), Fc ⁇ RIIB (CD32B), Fc ⁇ RIIIA (CD16A), and Fc ⁇ RIIIB (CD16B).
- the Fey receptors differ in their affinity for IgG and also have different affinities for the IgG subclasses (e.g., IgG1, IgG2, IgG3, and IgG4).
- Immune checkpoint inhibitor refers to any modulator that inhibits the activity of the immune checkpoint molecule.
- Immune checkpoint inhibitors can include, but are not limited to, immune checkpoint molecule binding proteins, small molecule inhibitors, antibodies (including bispecific and multispecific antibodies with at least one antigen binding region that targets an immune checkpoint protein, e.g., bispecific or multispecific antibodies that do not exclusively target immune checkpoint proteins, as well as antibodies that are dual immunomodulators (simultaneous targeting two immunomodulating targets), which result in blockade of inhibitory targets, depletion of suppressive cells, and/or activation of effector cells; tumor-targeted immunomodulators (directs potent costimulation to the tumor- infiltrating immune cells by targeting a tumor antigen and costimulatory molecules such as CD40 or 4- IBB); NK-cell redirectors (redirects NK cells to malignant cells by targeting a tumor antigen and CD16A); or T-cell redirectors (redirects T cells
- Nucleic acid or amino acid sequence “identity,” as referenced herein, can be determined by comparing a nucleic acid or amino acid sequence of interest to a reference nucleic acid or amino acid sequence.
- the percent identity is the number of nucleotides or amino acid residues that are the same (i.e., that are identical) as between the optimally aligned sequence of interest and the reference sequence divided by the length of the longest sequence (i.e., the length of either the sequence of interest or the reference sequence, whichever is longer). Alignment of sequences and calculation of percent identity can be performed using available software programs.
- Such programs include CLUSTAL-W, T-Coffee, and ALIGN (for alignment of nucleic acid and amino acid sequences), BLAST programs (e.g., BLAST 2.1, BL2SEQ, BLASTp, BLASTn, and the like) and FASTA programs (e.g., FASTA3x, FASTM, and SSEARCH) (for sequence alignment and sequence similarity searches). Sequence alignment algorithms also are disclosed in, for example, Altschul et al., J. Molecular Biol., 215(3): 403-410 (1990), Beigert et al., Proc. Natl. Acad. Sci.
- Percent (%) identity of sequences can be also calculated, for example, as 100 x [(identical positions)/min(TGA, TGB)], where TGA and TGB are the sum of the number of residues and internal gap positions in peptide sequences A and B in the alignment that minimizes TG A and TG B . See, e.g., Russell et al., J. Mol Biol., 244: 332-350 (1994).
- the binding agent comprises Ig heavy and light chain variable region polypeptides that together form the antigen binding site.
- Each of the heavy and light chain variable regions are polypeptides comprising three complementarity determining regions (CDR1, CDR2, and CDR3) connected by framework regions.
- the binding agent can be any of a variety of types of binding agents known in the art that comprise Ig heavy and light chains.
- the binding agent can be an antibody, an antigen-binding antibody “fragment,” or a T-cell receptor.
- Amino acid refers to any monomeric unit that can be incorporated into a peptide, polypeptide, or protein. Amino acids include naturally-occurring ⁇ -amino acids and their stereoisomers, as well as unnatural (non-naturally occurring) amino acids and their stereoisomers.
- “Stereoisomers” of a given amino acid refer to isomers having the same molecular formula and intramolecular bonds but different three-dimensional arrangements of bonds and atoms (e.g., an L-amino acid and the corresponding D-amino acid).
- the amino acids can be glycosylated (e.g., N-linked glycans, O-linked glycans, phosphoglycans, C-linked glycans, or glypication) or deglycosylated.
- Amino acids may be referred to herein by either the commonly known three letter symbols or by the one-letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission.
- Naturally-occurring amino acids are those encoded by the genetic code, as well as those amino acids that are later modified, e.g., hydroxyproline, ⁇ -carboxyglutamate, and O-phosphoserine.
- Naturally-occurring ⁇ -amino acids include, without limitation, alanine (Ala), cysteine (Cys), aspartic acid (Asp), glutamic acid (Glu), phenylalanine (Phe), glycine (Gly), histidine (His), isoleucine (Ile), arginine (Arg), lysine (Lys), leucine (Leu), methionine (Met), asparagine (Asn), proline (Pro), glutamine (Gln), serine (Ser), threonine (Thr), valine (Val), tryptophan (Trp), tyrosine (Tyr), and combinations thereof.
- Stereoisomers of naturally- occurring ⁇ -amino acids include, without limitation, D-alanine (D-Ala), D-cysteine (D-Cys), D-aspartic acid (D-Asp), D-glutamic acid (D-Glu), D-phenylalanine (D-Phe), D-histidine (D-His), D-isoleucine (D-Ile), D-arginine (D-Arg), D-lysine (D-Lys), D-leucine (D-Leu), D-methionine (D-Met), D-asparagine (D-Asn), D-proline (D-Pro), D-glutamine (D-Gln), D-serine (D-Ser), D-threonine (D-Thr), D-valine (D-Val), D-tryptophan (D-Trp), D-tyrosine (D-Tyr), and combinations thereof.
- D-Ala D-c
- Naturally-occurring amino acids include those formed in proteins by post-translational modification, such as citrulline (Cit).
- Unnatural (non-naturally occurring) amino acids include, without limitation, amino acid analogs, amino acid mimetics, synthetic amino acids, N-substituted glycines, and N-methyl amino acids in either the L- or D-configuration that function in a manner similar to the naturally- occurring amino acids.
- amino acid analogs can be unnatural amino acids that have the same basic chemical structure as naturally-occurring amino acids (i.e., a carbon that is bonded to a hydrogen, a carboxyl group, an amino group) but have modified side-chain groups or modified peptide backbones, e.g., homoserine, norleucine, methionine sulfoxide, and methionine methyl sulfonium.
- Amino acid mimetics refer to chemical compounds that have a structure that is different from the general chemical structure of an amino acid, but that functions in a manner similar to a naturally-occurring amino acid.
- Linker refers to a functional group that covalently bonds two or more moieties in a compound or material.
- the linker serves to covalently bond an adjuvant moiety to an antibody construct in an immunoconjugate.
- Linking moiety refers to a functional group that covalently bonds two or more moieties in a compound or material.
- the linking moiety can serve to covalently bond an adjuvant moiety to an antibody in an immunoconjugate.
- Useful bonds for connecting linking moieties to proteins and other materials include, but are not limited to, amides, amines, esters, carbamates, ureas, thioethers, thiocarbamates, thiocarbonates, and thioureas.
- Divalent refers to a chemical moiety that contains two points of attachment for linking two functional groups; polyvalent linking moieties can have additional points of attachment for linking further functional groups.
- Divalent radicals may be denoted with the suffix “diyl”.
- divalent linking moieties include divalent polymer moieties such as divalent poly(ethylene glycol), divalent cycloalkyl, divalent heterocycloalkyl, divalent aryl, and divalent heteroaryl group.
- a “divalent cycloalkyl, heterocycloalkyl, aryl, or heteroaryl group” refers to a cycloalkyl, heterocycloalkyl, aryl, or heteroaryl group having two points of attachment for covalently linking two moieties in a molecule or material. Cycloalkyl, heterocycloalkyl, aryl, or heteroaryl groups can be substituted or unsubstituted. Cycloalkyl, heterocycloalkyl, aryl, or heteroaryl groups can be substituted with one or more groups selected from halo, hydroxy, amino, alkylamino, amido, acyl, nitro, cyano, and alkoxy.
- a wavy line (“ ”) represents a point of attachment of the specified chemical moiety. If the specified chemical moiety has two wavy lines (“ ”) present, it will be understood that the chemical moiety can be used bilaterally, i.e., as read from left to right or from right to left. In some embodiments, a specified moiety having two wavy lines (“ ”) present is considered to be used as read from left to right.
- Alkyl refers to a straight (linear) or branched, saturated, aliphatic radical having the number of carbon atoms indicated. Alkyl can include any number of carbons, for example from one to six, one to eight, one to twelve or one to twenty.
- alkyl groups include, but are not limited to, methyl (Me, -CH 3 ), ethyl (Et, -CH 2 CH 3 ), 1-propyl (n-Pr, n-propyl, - CH 2 CH 2 CH 3 ), 2-propyl (i-Pr, i-propyl, -CH(CH 3 ) 2 ), 1-butyl (n-Bu, n-butyl, -CH 2 CH 2 CH 2 CH 3 ), 2-methyl-1-propyl (i-Bu, i-butyl, -CH 2 CH(CH 3 ) 2 ), 2-butyl (s-Bu, s-butyl, -CH(CH 3 )CH 2 CH 3 ), 2- methyl-2-propyl (t-Bu, t-butyl, -C(CH 3 ) 3 ), 1-pentyl (n-pentyl, -CH 2 CH 2 CH 2 CH 3 ), 2-pentyl (-CH(CH 3
- alkyldiyl refers to a divalent alkyl radical.
- alkyldiyl groups include, but are not limited to, methylene (-CH 2 -), ethylene (-CH 2 CH 2 -), propylene (- CH 2 CH 2 CH 2 -), and the like.
- An alkyldiyl group may also be referred to as an “alkylene” group.
- alkyldiyl groups can be geminally substituted where a carbon atom of the alkyl forms a spiro, cycloalkyl group such as cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl.
- Alkenyl refers to a straight (linear) or branched, unsaturated, aliphatic radical having the number of carbon atoms indicated and at least one carbon-carbon double bond, sp2. Alkenyl can include from two to about 12 or more carbons atoms.
- Alkenyl groups are radicals having “cis” and “trans” orientations, or alternatively, “E” and “Z” orientations.
- alkenylene or “alkenyldiyl” refer to a linear or branched-chain divalent hydrocarbon radical.
- Alkynyl refers to a straight (linear) or branched, unsaturated, aliphatic radical having the number of carbon atoms indicated and at least one carbon-carbon triple bond, sp. Alkynyl can include from two to about 12 or more carbons atoms.
- C 2 -C 6 alkynyl includes, but is not limited to ethynyl (-C ⁇ CH), propynyl (propargyl, -CH 2 C ⁇ CH), butynyl, pentynyl, hexynyl, and isomers thereof
- alkynylene or “alkynyldiyl” refer to a divalent alkynyl radical.
- "Heteroalkyl” or “heteroalkylene” refer to a monovalent, straight or branched chain alkyl group, as defined above, comprising at least one heteroatom including but not limited to Si, N, O, P or S within the alkyl chain or at a terminus of the alkyl chain. In some embodiments, a heteroatom is within the alkyl chain. In other embodiments, a heteroatom is at a terminus of the alkylene and thus serves to join the alkyl to the remainder of the molecule. Unless stated otherwise specifically in the specification, a heteroalkyl group is optionally substituted.
- heteroalkyl groups can be substituted with 1-6 fluoro (F) substituents, for example, on the carbon backbone (as ⁇ CHF ⁇ or ⁇ CF 2 ⁇ ) or on terminal carbons of straight chain or branched heteroalkyls (such as ⁇ CHF 2 or ⁇ CF 3 ).
- F fluoro
- a terminal polyethylene glycol (PEG) moiety is a type of heteroalkyl group.
- exemplary heteroalkyl groups also include ethylene oxide (e.g., polyethylene oxide), propylene oxide, amino acid chains (i.e., short to medium length peptides such as containing 1-15 amino acids), and alkyl chains connected via a variety of functional groups such as amides, disulfides, ketones, phosphonates, phosphates, sulfates, sulfones, sulfonamides, esters, ethers, -S-, carbamates, ureas, thioureas, anhydrides, or the like (including combinations thereof).
- a heteroalkyl group includes a polyamino acid having 1-10 amino acids. In some embodiments, a heteroalkyl group includes a polyamino acid having 1-5 amino acids.
- exemplary heteroalkyl groups include a solubilizing group (SolG) comprising one or more units of polyglycine, polysarcosine, polyethyleneoxy (PEG), and a glycoside, or combinations thereof.
- SolG solubilizing group
- Heteroalkenyl refers to a heteroalkyl group, as defined above, that contains at least one carbon-carbon double bond.
- Heteroalkynyl refers to a heteroalkyl group, as defined above, that contains at least one carbon-carbon triple bond.
- Heteroalkyldiyl refers to a divalent form of a heteroalkyl group as defined above.
- a divalent polyethylene glycol (PEG) moiety with one to about 50 units of ⁇ OCH 2 CH 2 ⁇ is a type of heteroalkyldiyl group.
- Heteroalkenyldiyl refers to a divalent form of a heteroalkenyl group.
- Heteroalkynyldiyl refers to a divalent form of a heteroalkynyl group.
- carrier refers to a saturated or partially unsaturated, monocyclic, fused bicyclic, spiro, or bridged polycyclic ring assembly containing from 3 to 12 ring atoms, or the number of atoms indicated.
- Saturated monocyclic carbocyclic rings include, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cyclooctyl.
- Saturated bicyclic and polycyclic carbocyclic rings include, for example, norbornane, [2.2.2] bicyclooctane, decahydronaphthalene and adamantane.
- Carbocyclic groups can also be partially unsaturated, having one or more double or triple bonds in the ring.
- carbocyclic groups that are partially unsaturated include, but are not limited to, cyclobutene, cyclopentene, cyclohexene, cyclohexadiene (1,3- and 1,4-isomers), cycloheptene, cycloheptadiene, cyclooctene, cyclooctadiene (1,3-, 1,4- and 1,5-isomers), norbornene, and norbornadiene.
- cycloalkyldiyl refers to a divalent cycloalkyl radical.
- Aryl refers to a monovalent aromatic hydrocarbon radical of 6-20 carbon atoms (C 6 ⁇ C 20 ) derived by the removal of one hydrogen atom from a single carbon atom of a parent aromatic ring system.
- Aryl groups can be monocyclic, fused to form bicyclic or tricyclic groups, or linked by a bond to form a biaryl group.
- Representative aryl groups include phenyl, naphthyl and biphenyl.
- Other aryl groups include benzyl, having a methylene linking group.
- Some aryl groups have from 6 to 12 ring members, such as phenyl, naphthyl or biphenyl.
- aryl groups have from 6 to 10 ring members, such as phenyl or naphthyl.
- arylene or “aryldiyl” mean a divalent aromatic hydrocarbon radical of 6-20 carbon atoms (C 6 ⁇ C 20 ) derived by the removal of two hydrogen atom from a two carbon atoms of a parent aromatic ring system.
- Some aryldiyl groups are represented in the exemplary structures as “Ar”.
- Aryldiyl includes bicyclic radicals comprising an aromatic ring fused to a saturated, partially unsaturated ring, or aromatic carbocyclic ring.
- Typical aryldiyl groups include, but are not limited to, radicals derived from benzene (phenyldiyl), substituted benzenes, naphthalene, anthracene, biphenylene, indenylene, indanylene, 1,2-dihydronaphthalene, 1,2,3,4- tetrahydronaphthyl, and the like.
- Aryldiyl groups are also referred to as “arylene”, and are optionally substituted with one or more substituents described herein.
- heterocycle refers to a saturated or a partially unsaturated (i.e., having one or more double and/or triple bonds within the ring) carbocyclic radical of 3 to about 20 ring atoms in which at least one ring atom is a heteroatom selected from nitrogen, oxygen, phosphorus and sulfur, the remaining ring atoms being C, where one or more ring atoms is optionally substituted independently with one or more substituents described below.
- a heterocycle may be a monocycle having 3 to 7 ring members (2 to 6 carbon atoms and 1 to 4 heteroatoms selected from N, O, P, and S) or a bicycle having 7 to 10 ring members (4 to 9 carbon atoms and 1 to 6 heteroatoms selected from N, O, P, and S), for example: a bicyclo [4,5], [5,5], [5,6], or [6,6] system.
- Heterocycles are described in Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W.A.
- Heterocyclyl also includes radicals where heterocycle radicals are fused with a saturated, partially unsaturated ring, or aromatic carbocyclic or heterocyclic ring.
- heterocyclic rings include, but are not limited to, morpholin-4-yl, piperidin-1-yl, piperazinyl, piperazin-4-yl-2-one, piperazin-4-yl-3-one, pyrrolidin-1-yl, thiomorpholin-4-yl, S- dioxothiomorpholin-4-yl, azocan-1-yl, azetidin-1-yl, octahydropyrido[1,2-a]pyrazin-2-yl, [1,4]diazepan-1-yl, pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl, homopiperazinyl, aze
- Spiro heterocyclyl moieties are also included within the scope of this definition.
- spiro heterocyclyl moieties include azaspiro[2.5]octanyl and azaspiro[2.4]heptanyl.
- the heterocycle groups herein are optionally substituted independently with one or more substituents described herein.
- heterocyclyldiyls examples include morpholinyldiyl, piperidinyldiyl, piperazinyldiyl, pyrrolidinyldiyl, dioxanyldiyl, thiomorpholinyldiyl, and S- dioxothiomorpholinyldiyl.
- heteroaryl refers to a monovalent aromatic radical of 5-, 6-, or 7-membered rings, and includes fused ring systems (at least one of which is aromatic) of 5-20 atoms, containing one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur.
- heteroaryl groups are pyridinyl (including, for example, 2-hydroxypyridinyl), imidazolyl, imidazopyridinyl, pyrimidinyl (including, for example, 4-hydroxypyrimidinyl), pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxadiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazol
- Heteroaryl groups are optionally substituted independently with one or more substituents described herein.
- heteroaryldiyl refers to a divalent aromatic radical of 5-, 6-, or 7-membered rings, and includes fused ring systems (at least one of which is aromatic) of 5-20 atoms, containing one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur.
- Examples of 5-membered and 6-membered heteroaryldiyls include pyridyldiyl, imidazolyldiyl, pyrimidinyldiyl, pyrazolyldiyl, triazolyldiyl, pyrazinyldiyl, tetrazolyldiyl, furyldiyl, thienyldiyl, isoxazolyldiyldiyl, thiazolyldiyl, oxadiazolyldiyl, oxazolyldiyl, isothiazolyldiyl, and pyrrolyldiyl.
- the heterocycle or heteroaryl groups may be carbon (carbon-linked), or nitrogen (nitrogen-linked) bonded where such is possible.
- carbon bonded heterocycles or heteroaryls are bonded at position 2, 3, 4, 5, or 6 of a pyridine, position 3, 4, 5, or 6 of a pyridazine, position 2, 4, 5, or 6 of a pyrimidine, position 2, 3, 5, or 6 of a pyrazine, position 2, 3, 4, or 5 of a furan, tetrahydrofuran, thiofuran, thiophene, pyrrole or tetrahydropyrrole, position 2, 4, or 5 of an oxazole, imidazole or thiazole, position 3, 4, or 5 of an isoxazole, pyrazole, or isothiazole, position 2 or 3 of an aziridine, position 2, 3, or 4 of an azetidine, position 2, 3, 4, 5, 6, 7, or 8 of a quinoline or position 1, 3, 4, 5, 6,
- nitrogen bonded heterocycles or heteroaryls are bonded at position 1 of an aziridine, azetidine, pyrrole, pyrrolidine, 2-pyrroline, 3-pyrroline, imidazole, imidazolidine, 2-imidazoline, 3-imidazoline, pyrazole, pyrazoline, 2-pyrazoline, 3- pyrazoline, piperidine, piperazine, indole, indoline, 1H-indazole, position 2 of a isoindole, or isoindoline, position 4 of a morpholine, and position 9 of a carbazole, or ⁇ -carboline.
- halo and halogen refer to a fluorine, chlorine, bromine, or iodine atom.
- quaternary ammonium salt refers to a tertiary amine that has been quaternized with an alkyl substituent (e.g., a C 1 -C4 alkyl such as methyl, ethyl, propyl, or butyl).
- treat refers to any indicia of success in the treatment or amelioration of an injury, pathology, condition (e.g., cancer), or symptom (e.g., cognitive impairment), including any objective or subjective parameter such as abatement; remission; diminishing of symptoms or making the symptom, injury, pathology, or condition more tolerable to the patient; reduction in the rate of symptom progression; decreasing the frequency or duration of the symptom or condition; or, in some situations, preventing the onset of the symptom.
- the treatment or amelioration of symptoms can be based on any objective or subjective parameter, including, for example, the result of a physical examination.
- cancer refers to cells which exhibit autonomous, unregulated growth, such that the cells exhibit an aberrant growth phenotype characterized by a significant loss of control over cell proliferation.
- Cells of interest for detection, analysis, and/or treatment in the context of the invention include cancer cells (e.g., cancer cells from an individual with cancer), malignant cancer cells, pre-metastatic cancer cells, metastatic cancer cells, and non-metastatic cancer cells. Cancers of virtually every tissue are known.
- cancer burden refers to the quantum of cancer cells or cancer volume in a subject. Reducing cancer burden accordingly refers to reducing the number of cancer cells or the cancer cell volume in a subject.
- cancer cell refers to any cell that is a cancer cell (e.g., from any of the cancers for which an individual can be treated, e.g., isolated from an individual having cancer) or is derived from a cancer cell, e.g., clone of a cancer cell.
- a cancer cell can be from an established cancer cell line, can be a primary cell isolated from an individual with cancer, can be a progeny cell from a primary cell isolated from an individual with cancer, and the like.
- the term can also refer to a portion of a cancer cell, such as a sub-cellular portion, a cell membrane portion, or a cell lysate of a cancer cell.
- cancers are known to those of skill in the art, including solid tumors such as carcinomas, sarcomas, glioblastomas, melanomas, lymphomas, and myelomas, and circulating cancers such as leukemias.
- solid tumors such as carcinomas, sarcomas, glioblastomas, melanomas, lymphomas, and myelomas
- circulating cancers such as leukemias.
- cancer includes any form of cancer, including but not limited to, solid tumor cancers (e.g., skin, lung, prostate, breast, gastric, bladder, colon, ovarian, pancreas, kidney, liver, glioblastoma, medulloblastoma, leiomyosarcoma, head & neck squamous cell carcinomas, melanomas, and neuroendocrine) and liquid cancers (e.g., hematological cancers); carcinomas; soft tissue tumors; sarcomas; teratomas; melanomas; leukemias; lymphomas; and brain cancers, including minimal residual disease, and including both primary and metastatic tumors.
- solid tumor cancers e.g., skin, lung, prostate, breast, gastric, bladder, colon, ovarian
- pancreas kidney, liver, glioblastoma, medulloblastoma, leiomyosarcoma, head & neck squamous cell carcinomas, melan
- the “pathology” of cancer includes all phenomena that compromise the well-being of the patient. This includes, without limitation, abnormal or uncontrollable cell growth, metastasis, interference with the normal functioning of neighboring cells, release of cytokines or other secretory products at abnormal levels, suppression or aggravation of inflammatory or immunological response, neoplasia, premalignancy, malignancy, and invasion of surrounding or distant tissues or organs, such as lymph nodes.
- cancer recurrence and “tumor recurrence,” and grammatical variants thereof, refer to further growth of neoplastic or cancerous cells after diagnosis of cancer. Particularly, recurrence may occur when further cancerous cell growth occurs in the cancerous tissue.
- Tumor spread similarly, occurs when the cells of a tumor disseminate into local or distant tissues and organs, therefore, tumor spread encompasses tumor metastasis.
- Tuor invasion occurs when the tumor growth spread out locally to compromise the function of involved tissues by compression, destruction, or prevention of normal organ function.
- metastasis refers to the growth of a cancerous tumor in an organ or body part, which is not directly connected to the organ of the original cancerous tumor. Metastasis will be understood to include micrometastasis, which is the presence of an undetectable amount of cancerous cells in an organ or body part that is not directly connected to the organ of the original cancerous tumor.
- Metastasis can also be defined as several steps of a process, such as the departure of cancer cells from an original tumor site, and migration and/or invasion of cancer cells to other parts of the body.
- effective amount and “therapeutically effective amount” refer to a dose or amount of a substance such as an immunoconjugate that produces therapeutic effects for which it is administered.
- the therapeutically effective amount of the immunoconjugate may reduce the number of cancer cells; reduce the tumor size; inhibit (i.e., slow to some extent and preferably stop) cancer cell infiltration into peripheral organs; inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; inhibit, to some extent, tumor growth; and/or relieve to some extent one or more of the symptoms associated with the cancer.
- the immunoconjugate may prevent growth and/or kill existing cancer cells, it may be cytostatic and/or cytotoxic.
- efficacy can, for example, be measured by assessing the time to disease progression (TTP) and/or determining the response rate (RR)
- TTP time to disease progression
- RR response rate
- Recipient “individual,” “subject,” “host,” and “patient” are used interchangeably and refer to any mammalian subject for whom diagnosis, treatment, or therapy is desired (e.g., humans).
- “Mammal” for purposes of treatment refers to any animal classified as a mammal, including humans, domestic and farm animals, and zoo, sports, or pet animals, such as dogs, horses, cats, cows, sheep, goats, pigs, camels, etc. In certain embodiments, the mammal is human.
- the phrase “synergistic adjuvant” or “synergistic combination” in the context of this invention includes the combination of two immune modulators such as a receptor agonist, cytokine, and adjuvant polypeptide, that in combination elicit a synergistic effect on immunity relative to either administered alone.
- the immunoconjugates disclosed herein comprise synergistic combinations of the claimed adjuvant and antibody construct. These synergistic combinations upon administration elicit a greater effect on immunity, e.g., relative to when the antibody construct or adjuvant is administered in the absence of the other moiety.
- a decreased amount of the immunoconjugate may be administered (as measured by the total number of antibody constructs or the total number of adjuvants administered as part of the immunoconjugate) compared to when either the antibody construct or adjuvant is administered alone.
- administering refers to parenteral, intravenous, intraperitoneal, intramuscular, intratumoral, intralesional, intranasal, or subcutaneous administration, oral administration, administration as a suppository, topical contact, intrathecal administration, or the implantation of a slow-release device, e.g., a mini-osmotic pump, to the subject.
- the immunoconjugate of the invention comprises an antibody. Included in the scope of the embodiments of the invention are functional variants of the antibody constructs or antigen binding domain described herein.
- the term “functional variant” as used herein refers to an antibody construct having an antigen binding domain with substantial or significant sequence identity or similarity to a parent antibody construct or antigen binding domain, which functional variant retains the biological activity of the antibody construct or antigen binding domain of which it is a variant.
- Functional variants encompass, for example, those variants of the antibody constructs or antigen binding domain described herein (the parent antibody construct or antigen binding domain) that retain the ability to recognize target cells expressing a tumor-associated antigen or cell surface receptor to a similar extent, the same extent, or to a higher extent, as the parent antibody construct or antigen binding domain.
- the functional variant can, for instance, be at least about 30%, about 50%, about 75%, about 80%, about 85%, about 90%, about 91%, about 92%, about 93%, about 94%, about 95%, about 96%, about 97%, about 98%, about 99% or more identical in amino acid sequence to the antibody construct or antigen binding domain.
- a functional variant can, for example, comprise the amino acid sequence of the parent antibody construct or antigen binding domain with at least one conservative amino acid substitution.
- the functional variants can comprise the amino acid sequence of the parent antibody construct or antigen binding domain with at least one non- conservative amino acid substitution.
- the non-conservative amino acid substitution may enhance the biological activity of the functional variant, such that the biological activity of the functional variant is increased as compared to the parent antibody construct or antigen binding domain.
- the antibodies comprising the immunoconjugates of the invention include Fc engineered variants.
- the mutations in the Fc region that result in modulated binding to one or more Fc receptors can include one or more of the following mutations: SD (S239D), SDIE (S239D/I332E), SE (S267E), SELF (S267E/L328F), SDIE (S239D/I332E), SDIEAL (S239D/I332E/A330L), GA (G236A), ALIE (A330L/I332E), GASDALIE (G236A/S239D/A330L/I332E), V9 (G237D/P238D/P271G/A330R), and V11 (G237D/P238D/H268D/P271G/A330R), and/or one or more mutations at the following amino acids: E345R, E233, G237, P238, H268, P271, L328 and A330.
- the antibodies comprising the immunoconjugates of the invention include glycan variants, such as afucosylation.
- the Fc region of the binding agents are modified to have an altered glycosylation pattern of the Fc region compared to the native non-modified Fc region.
- the antibodies in the immunoconjugates contain a modified Fc region, wherein the modification modulates the binding of the Fc region to one or more Fc receptors.
- the antibodies in the immunoconjugates contain one or more modifications (e.g., amino acid insertion, deletion, and/or substitution) in the Fc region that results in modulated binding (e.g., increased binding or decreased binding) to one or more Fc receptors (e.g., Fc ⁇ RI (CD64), Fc ⁇ RIIA (CD32A), Fc ⁇ RIIB (CD32B), Fc ⁇ RIIIA (CD16a), and/or Fc ⁇ RIIIB (CD16b)) as compared to the native antibody lacking the mutation in the Fc region.
- modifications e.g., amino acid insertion, deletion, and/or substitution
- Fc receptors e.g., Fc ⁇ RI (CD64), Fc ⁇ RIIA (CD32A), Fc ⁇ RIIB (CD32B), Fc ⁇ RIIIA (CD16a), and/or Fc ⁇ RIIIB (CD16b)
- the antibodies in the immunoconjugates contain one or more modifications (e.g., amino acid insertion, deletion, and/or substitution) in the Fc region that reduce the binding of the Fc region of the antibody to Fc ⁇ RIIB. In some embodiments, the antibodies in the immunoconjugates contain one or more modifications (e.g., amino acid insertion, deletion, and/or substitution) in the Fc region of the antibody that reduce the binding of the antibody to Fc ⁇ RIIB while maintaining the same binding or having increased binding to Fc ⁇ RI (CD64), Fc ⁇ RIIA (CD32A), and/or FcR ⁇ IIIA (CD16a) as compared to the native antibody lacking the mutation in the Fc region.
- modifications e.g., amino acid insertion, deletion, and/or substitution
- the antibodies in the immunoconjugates contain one of more modifications in the Fc region that increase the binding of the Fc region of the antibody to Fc ⁇ RIIB.
- the modulated binding is provided by mutations in the Fc region of the antibody relative to the native Fc region of the antibody.
- the mutations can be in a CH2 domain, a CH 3 domain, or a combination thereof.
- a “native Fc region” is synonymous with a “wild-type Fc region” and comprises an amino acid sequence that is identical to the amino acid sequence of an Fc region found in nature or identical to the amino acid sequence of the Fc region found in the native antibody (e.g., cetuximab).
- Native sequence human Fc regions include a native sequence human IgG1 Fc region, native sequence human IgG2 Fc region, native sequence human IgG3 Fc region, and native sequence human IgG4 Fc region, as well as naturally occurring variants thereof.
- Native sequence Fc includes the various allotypes of Fcs (Jefferis et al., (2009) mAbs, 1(4):332-338).
- the Fc region of the antibodies of the immunoconjugates are modified to have an altered glycosylation pattern of the Fc region compared to the native non-modified Fc region.
- Human immunoglobulin is glycosylated at the Asn297 residue in the C ⁇ 2 domain of each heavy chain.
- This N-linked oligosaccharide is composed of a core heptasaccharide, N-acetylglucosamine4Mannose3 (GlcNAc4Man3). Removal of the heptasaccharide with endoglycosidase or PNGase F is known to lead to conformational changes in the antibody Fc region, which can significantly reduce antibody-binding affinity to activating Fc ⁇ R and lead to decreased effector function.
- the core heptasaccharide is often decorated with galactose, bisecting GlcNAc, fucose, or sialic acid, which differentially impacts Fc binding to activating and inhibitory Fc ⁇ R.
- the modification to alter the glycosylation pattern is a mutation. For example, a substitution at Asn297. In some embodiments, Asn297 is mutated to glutamine (N297Q).
- the antibodies of the immunoconjugates are modified to contain an engineered Fab region with a non-naturally occurring glycosylation pattern.
- hybridomas can be genetically engineered to secrete afucosylated mAb, desialylated mAb or deglycosylated Fc with specific mutations that enable increased FcR ⁇ IIIa binding and effector function.
- the antibodies of the immunoconjugates are engineered to be afucosylated.
- the entire Fc region of an antibody in the immunoconjugates is exchanged with a different Fc region, so that the Fab region of the antibody is conjugated to a non-native Fc region.
- the Fab region of cetuximab which normally comprises an IgG1 Fc region
- the Fab region of nivolumab which normally comprises an IgG4 Fc region
- the Fc modified antibody with a non-native Fc domain also comprises one or more amino acid modification, such as the S228P mutation within the IgG4 Fc, that modulate the stability of the Fc domain described.
- the Fc modified antibody with a non-native Fc domain also comprises one or more amino acid modifications described herein that modulate Fc binding to FcR.
- the modifications that modulate the binding of the Fc region to FcR do not alter the binding of the Fab region of the antibody to its antigen when compared to the native non-modified antibody.
- the modifications that modulate the binding of the Fc region to FcR also increase the binding of the Fab region of the antibody to its antigen when compared to the native non-modified antibody.
- the antibodies in the immunoconjugates contain a modified Fc region, wherein the modification modulates the binding of the Fc region to one or more Fc receptors.
- the Fc region is modified by inclusion of a transforming growth factor beta 1 (TGF ⁇ 1) receptor, or a fragment thereof, that is capable of binding TGF ⁇ 1.
- the receptor can be TGF ⁇ receptor II (TGF ⁇ RII).
- TGF ⁇ receptor is a human TGF ⁇ receptor.
- the IgG has a C-terminal fusion to a TGF ⁇ RII extracellular domain (ECD) as described in US 9676863, incorporated herein.
- An “Fc linker” may be used to attach the IgG to the TGF ⁇ RII extracellular domain.
- the Fc linker may be a short, flexible peptide that allows for the proper three-dimensional folding of the molecule while maintaining the binding-specificity to the targets.
- the N-terminus of the TGF ⁇ receptor is fused to the Fc of the antibody construct (with or without an Fc linker).
- the C-terminus of the antibody construct heavy chain is fused to the TGF ⁇ receptor (with or without an Fc linker).
- the C-terminal lysine residue of the antibody construct heavy chain is mutated to alanine.
- the antibodies in the immunoconjugates are glycosylated.
- the antibodies in the immunoconjugates are a cysteine-engineered antibody which provides for site-specific conjugation of an adjuvant, label, or drug moiety to the antibody through cysteine substitutions at sites where the engineered cysteines are available for conjugation but do not perturb immunoglobulin folding and assembly or alter antigen binding and effector functions (Junutula, et al., (2008) Nature Biotech., 26(8):925-932; Dornan et al.
- a “cysteine engineered antibody” or “cysteine engineered antibody variant” is an antibody in which one or more residues of an antibody are substituted with cysteine residues.
- Cysteine-engineered antibodies can be conjugated to the aza-bicyclic (AzaBi) moiety as a AzaBi-linker compound with uniform stoichiometry (e.g., up to two AzaBi moieties per antibody in an antibody that has a single engineered cysteine site).
- cysteine-engineered antibodies are used to prepare immunoconjugates.
- Immunoconjugates may have a reactive cysteine thiol residue introduced at a site on the light chain, such as the 149-lysine site (LC K149C), or on the heavy chain such as the 122-serine site (HC S122C), as numbered by Kabat numbering.
- the cysteine-engineered antibodies have a cysteine residue introduced at the 118-alanine site (EU numbering) of the heavy chain (HC A118C). This site is alternatively numbered 121 by Sequential numbering or 114 by Kabat numbering.
- the cysteine- engineered antibodies have a cysteine residue introduced in: (i) the light chain at G64C, R142C, K188C, L201C, T129C, S114C, or E105C according to Kabat numbering; (ii) the heavy chain at D101C, V184C, T205C, or S122C according to Kabat numbering; or (iii) other cysteine-mutant antibodies, and as described in Bhakta, S.
- the antibody of an immunoconjugate is an immune checkpoint inhibitor.
- the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins.
- the immune checkpoint inhibitor reduces the interaction between one or more immune checkpoint proteins and their ligands.
- Immune checkpoint inhibitors nivolumab and atezolizumab can be modified to include an IgG1 Fc, and subsequently converted into an immunoconjugate of the invention.
- Most checkpoint antibodies are designed not to have effector function to kill cells, but rather to block the signalling.
- Immunoconjugates of the present invention can add back the "effector functionality" needed to activate myeloid immunity.
- the immune checkpoint inhibitor is cytotoxic T-lymphocyte antigen 4 (CTLA4, also known as CD152), T cell immunoreceptor with Ig and ITIM domains (TIGIT), glucocorticoid-induced TNFR-related protein (GITR, also known as TNFRSF18), inducible T cell costimulatory (ICOS, also known as CD278), CD96, poliovirus receptor-related 2 (PVRL2, also known as CD112R, programmed cell death protein 1 (PD-1, also known as CD279), programmed cell death 1 ligand 1 (PD-L1, also known as B7-H3 and CD274), programmed cell death ligand 2 (PD-L2, also known as B7-DC and CD273), lymphocyte activation gene-3 (LAG-3, also known as CD223), B7-H4, killer immunoglobulin receptor (KIR), Tumor Necrosis Factor Receptor superfamily member 4 (TNFRST4, also known as OX40 and CD134
- the immune checkpoint inhibitor is an inhibitor of CTLA4, PD-1, or PD-L1.
- the antibody is selected from: ipilimumab (also known as YERVOY®) pembrolizumab (also known as KEYTRUDA®), nivolumab (also known as OPDIVO®), atezolizumab (also known as TECENTRIQ®), avelumab (also known as BAVENCIO®), and durvalumab (also known as IMFINZI®).
- the immune checkpoint inhibitor is an inhibitor of CTLA4.
- the immune checkpoint inhibitor is an antibody against CTLA4.
- the immune checkpoint inhibitor is a monoclonal antibody against CTLA4. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against CTLA4. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as CTLA4. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-1. In some embodiments, the immune checkpoint inhibitor is an antibody against PD-1. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against PD-1. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against PD-1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as PD-1.
- the immune checkpoint inhibitor is an inhibitor of PD-L1. In some embodiments, the immune checkpoint inhibitor is an antibody against PD-L1. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against PD-L1. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against PD-L1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as PD-L1. In some embodiments, the immune checkpoint inhibitor reduces the interaction between PD-1 and PD-L1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-L2. In some embodiments, the immune checkpoint inhibitor is an antibody against PD-L2.
- the immune checkpoint inhibitor is a monoclonal antibody against PD-L2. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against PD-L2. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as PD-L2. In some embodiments, the immune checkpoint inhibitor reduces the interaction between PD-1 and PD-L2. In some embodiments, the immune checkpoint inhibitor is an inhibitor of LAG-3. In some embodiments, the immune checkpoint inhibitor is an antibody against LAG-3. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against LAG-3. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against LAG-3.
- the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as LAG-3.
- the immune checkpoint inhibitor is an inhibitor of B7-H4.
- the immune checkpoint inhibitor is an antibody against B7-H4.
- the immune checkpoint inhibitor is a monoclonal antibody against B7-H4.
- the immune checkpoint inhibitor is a human or humanized antibody against B7-H4.
- the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as B7-H4.
- the immune checkpoint inhibitor is an inhibitor of KIR.
- the immune checkpoint inhibitor is an antibody against KIR.
- the immune checkpoint inhibitor is a monoclonal antibody against KIR. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against KIR. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as KIR. In some embodiments, the immune checkpoint inhibitor is an inhibitor of TNFRSF4. In some embodiments, the immune checkpoint inhibitor is an antibody against TNFRSF4. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against TNFRSF4. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against TNFRSF4. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as TNFRSF4.
- the immune checkpoint inhibitor is an inhibitor of OX40L. In some embodiments, the immune checkpoint inhibitor is an antibody against OX40L. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against OX40L. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against OX40L. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as OX40L. In some embodiments, the immune checkpoint inhibitor reduces the interaction between TNFRSF4 and OX40L. In some embodiments, the immune checkpoint inhibitor is an inhibitor of IDO-1. In some embodiments, the immune checkpoint inhibitor is an antibody against IDO-1.
- the immune checkpoint inhibitor is a monoclonal antibody against IDO-1, in some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against IDO-1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as IDO-1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of IDO-2. In some embodiments, the immune checkpoint inhibitor is an antibody against IDO-2. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against IDO-2. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against IDO-2. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as IDO-2.
- the immune checkpoint inhibitor is an inhibitor of CEACAM1. In some embodiments, the immune checkpoint inhibitor is an antibody against CEACAM1. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against CEACAM1. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against CEACAM1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as CEACAM1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of BTLA. In some embodiments, the immune checkpoint inhibitor is an antibody against BTLA. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against BTLA.
- the immune checkpoint inhibitor is a human or humanized antibody against BMA. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as BTLA. In some embodiments, the immune checkpoint inhibitor is an inhibitor of TIM3. In some embodiments, the immune checkpoint inhibitor is an antibody against TIM3. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against TIM3. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against TIM3. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as TIM3. In some embodiments, the immune checkpoint inhibitor is an inhibitor of A2Ar.
- the immune checkpoint inhibitor is an antibody against A2Ar. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against A2Ar. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against A2Ar. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as A2Ar. In some embodiments, the immune checkpoint inhibitor is an inhibitor of VISTA protein. In some embodiments, the immune checkpoint inhibitor is an antibody against VISTA protein. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against VISTA protein. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against VISTA protein.
- the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as VISTA protein.
- the antibody of an immunoconjugate is capable of binding one or more targets selected from (e.g., specifically binds to a target selected from) 5T4, ABL, ABCF1, ACVR1, ACVR1B, ACVR2, ACVR2B, ACVRL1, ADORA2A, Aggrecan, AGR2, AICDA, AIF1, AIGI, AKAP1, AKAP2, AMH, AMHR2, ANGPT1, ANGPT2, ANGPTL3, ANGPTL4, ANPEP, APC, APOC 1 , AR, aromatase, ATX, AX1, AZGP1 (zinc-a-glycoprotein), B7.1, B7.2, B7-H1, BAD, BAFF, BAG1, BAI1, BCR, BCL2, BCL6, BDNF, BLNK, BLR1 (MDR15),
- FGF20 FGF21, FGF22, FGF23, FGF3 (int-2), FGF4 (HST), FGF5, FGF6 (HST- 2), FGF7 (KGF), FGF8, FGF9, FGFR3, FIGF (VEGFD), FILI (EPSILON), FBL1 (ZETA), FLJ12584, FLJ25530, FLRT1 (fibronectin), FLT1, FLT-3, FOS, FOSL1 (FRA-1), FY (DARC), GABRP (GABAa), GAGEB1, GAGEC1, GALNAC4S-6ST, GATA3, GD2, GDF5, GFI1, GGT1, GM-CSF, GNAS1, GNRH1, GPR2 (CCR10), GPR31, GPR44, GPR81 (FKSG80), GRCC1O (C1O), GRP, GSN (Gelsolin), GSTP1, HAVCR2, HDAC, HDAC4, HDAC5, HDAC7A, HDAC9, Hedgehog, HGF, H
- TNFSF6 FasL
- TNFSF7 CD27 ligand
- TNFSF8 CD30 ligand
- TNFSF9 4-1BB ligand
- TOLLIP Toll-like receptors
- TOP2A topoisomerase 1ia
- TP53 TPM1, TPM2, TRADD, TRAF1, TRAF2, TRAF3, TRAF4, TRAF5, TRAF6, TRKA, TREM1, TREM2, TROP2, TRPC6, TSLP, TWEAK, Tyrosinase, uPAR, VEGF, VEGFB, VEGFC, versican, VHL C5, VLA-4, Wnt-1, XCL1 (tymphotactin), XCL2 (SCM-Ib), XCRI (GPR5/CCXCR1), YYI, ZFPM2, CLEC4C (BDCA-2, DLEC, CD303, CLECSF7), CLEC4D (MCL, CLECSF8), CLEC4E (Mincle), CLEC6A
- CLEC5A MDL-1, CLECSF5), CLEC1B (CLEC-2), CLEC9A (DNGR-1), CLEC7A (Dectin-1), PDGFRa, SLAMF7, GP6 (GPVI), LILRA1 (CD85I), LILRA2 (CD85H, ILT1), LILRA4 (CD85G, ILT7), LILRA5 (CD85F, ILT11), LILRA6 (CD85b, ILT8), NCR1 (CD335, LY94, NKp46), NCR3 (CD335, LY94, NKp46), NCR3 (CD337, NKp30), OSCAR, TARM1, CD300C, CD300E, CD300LB (CD300B), CD300LD (CD300D), KIR2DL4 (CD158D), KIR2DS, KLRC2 (CD159C, NKG2C), KLRK1 (CD314, NKG2D), NCR2 (CD336, NKp44), PILRB,
- the antibody binds to an FcR.gamma-coupled receptor.
- the FcR.gamma-coupled receptor is selected from the group consisting of GP6 (GPVI), LILRA1 (CD85I), LILRA2 (CD85H, ILT1), LILRA4 (CD85G, ILT7), LILRA5 (CD85F, ILT11), LILRA6 (CD85b, ILT8), NCR1 (CD335, LY94, NKp46), NCR3 (CD335, LY94, NKp46), NCR3 (CD337, NKp30), OSCAR, and TARM1.
- the antibody binds to a DAP12-coupled receptor.
- the DAP12-coupled receptor is selected from the group consisting of CD300C, CD300E, CD300LB (CD300B), CD300LD (CD300D), KIR2DL4 (CD158D), KIR2DS, KLRC2 (CD159C, NKG2C), KLRK1 (CD314, NKG2D), NCR2 (CD336, NKp44).
- PILRB SIGLEC1 (CD169, SN), SIGLEC14, SIGLEC15 (CD33L3), SIGLEC16, SIRPB1 (CD172B), TREM1 (CD354), and TREM2.
- the antibody binds to a hemITAM-bearing receptor.
- the hemITAM-bearing receptor is KLRF1 (NKp80).
- the antibody is capable of binding one or more targets selected from CLEC4C (BDCA-2, DLEC, CD303, CLECSF7), CLEC4D (MCL, CLECSF8), CLEC4E (Mincle), CLEC6A (Dectin-2), CLEC5A (MDL-1, CLECSF5), CLEC1B (CLEC-2), CLEC9A (DNGR-1), and CLEC7A (Dectin-1).
- the antibody is capable of binding CLEC6A (Dectin-2) or CLEC5A.
- the antibody is capable of binding CLEC6A (Dectin-2).
- the antibody is capable of binding one or more targets selected from (e.g., specifically binds to a target selected from): ATP5I (Q06185), OAT (P29758), AIFM1 (Q9Z0X1), AOFA (Q64133), MTDC (P18155), CMC1 (Q8BH59), PREP (Q8K411), YMEL1 (O88967), LPPRC (Q6PB66), LONM (Q8CGK3), ACON (Q99KI0), ODO1 (Q60597), IDHP (P54071), ALDH2 (P47738), ATPB (P56480), AATM (P05202), TMM93 (Q9CQW0), ERGI3 (Q9CQE7), RTN4 (Q99P72), CL041 (Q8BQR4), ERLN2 (Q8BFZ9), TERA (Q01853), DAD1 (P61804), CALX (P35564)
- the antibody binds to an antigen selected from CDH1, CD19, CD20, CD29, CD30, CD38, CD40, CD47, EpCAM, MUC1, MUC16, EGFR, Her2, SLAMF7, and gp75.
- the antigen is selected from CD19, CD20, CD47, EpCAM, MUC1, MUC16, EGFR, and Her2.
- the antibody binds to an antigen selected from the Tn antigen and the Thomsen-Friedenreich antigen.
- the antibody or Fc fusion protein is selected from: abagovomab, abatacept (also known as ORENCIA®), abciximab (also known as REOPRO®), c7E3 Fab), adalimumab (also known as HUMIRA®), adecatumumab, alemtuzumab (also known as CAMPATH®), MabCampath or Campath-1H), altumomab, afelimomab, anatumomab mafenatox, anetumumab, anrukizumab, apolizumab, arcitumomab, aselizumab, atlizumab, atorolimumab, bapineuzumab, basiliximab (also known as SIMULECT®), bavituximab, bectumomab (also known as LYMPHOSCAN®), belimumab (also known as
- the antibody is rituximab.
- the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds PD-L1.
- Programmed Death-Ligand 1 (PD-L1, cluster of differentiation 274, CD274, B7- homolog 1, or B7-H1) belongs to the B7 protein superfamily, and is a ligand of programmed cell death protein 1 (PD-1, PDCD1, cluster of differentiation 279, or CD279).
- PD-L1 can also interact with B7.1 (CD80) and such interaction is believed to inhibit T cell priming.
- the PD- L1/PD-1 axis plays a large role in suppressing the adaptive immune response.
- PD-L1/PD-1 pathway also contributes to preventing autoimmunity and therefore agonistic agents against PD-L1 or agents that deliver immune inhibitory payloads may help treatment of autoimmune disorders.
- PD-L1-binding agents including agents that bind PD-L1 with high affinity and effectively prevent PD-L1/PD-1 signaling and agents that can deliver therapeutic payloads to PD-L1 expressing cells.
- new PD-L1-binding agents to treat autoimmune disorders and infections.
- the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds HER2.
- immunoconjugates of the invention comprise anti-HER2 antibodies.
- an anti-HER2 antibody of an immunoconjugate of the invention comprises a humanized anti-HER2 antibody, e.g., huMAb4D5-1, huMAb4D5- 2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7 and huMAb4D5- 8, as described in Table 3 of US 5821337, which is specifically incorporated by reference herein.
- Those antibodies contain human framework regions with the complementarity- determining regions of a murine antibody (4D5) that binds to HER2.
- the humanized antibody huMAb4D5-8 is also referred to as trastuzumab, commercially available under the tradename HERCEPTINTM (Genentech, Inc.).
- the antibody construct or antigen binding domain comprises the CDR regions of trastuzumab.
- the anti-HER2 antibody further comprises the framework regions of the trastuzumab.
- the anti-HER2 antibody further comprises one or both variable regions of trastuzumab.
- an anti-HER2 antibody of an immunoconjugate of the invention comprises a humanized anti-HER2 antibody, e.g., humanized 2C4, as described in US 7862817.
- An exemplary humanized 2C4 antibody is pertuzumab (CAS Reg. No.380610- 27-5), PERJETATM (Genentech, Inc.).
- Pertuzumab is a HER dimerization inhibitor (HDI) and functions to inhibit the ability of HER2 to form active heterodimers or homodimers with other HER receptors (such as EGFR/HER1, HER2, HER3 and HER4). See, for example, Harari and Yarden, Oncogene 19:6102-14 (2000); Yarden and Sliwkowski. Nat Rev Mol Cell Biol 2:127-37 (2001); Sliwkowski Nat Struct Biol 10:158-9 (2003); Cho et al. Nature 421:756-60 (2003); and Malik et al. Pro Am Soc Cancer Res 44:176-7 (2003).
- PERJETATM is approved for the treatment of breast cancer.
- the antibody construct or antigen binding domain comprises the CDR regions of pertuzumab.
- the anti-HER2 antibody further comprises the framework regions of the pertuzumab.
- the anti-HER2 antibody further comprises one or both variable regions of pertuzumab.
- the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds CEA.
- Carcinoembryonic antigen-related cell adhesion molecule 5 also known as CD66e (Cluster of Differentiation 66e)
- CD66e Cluster of Differentiation 66e
- the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds CEA.
- Elevated expression of carcinoembryonic antigen (CEA, CD66e, CEACAM5) has been implicated in various biological aspects of neoplasia, especially tumor cell adhesion, metastasis, the blocking of cellular immune mechanisms, and having antiapoptosis functions.
- CEA is also used as a blood marker for many carcinomas. Labetuzumab (CEA-CIDE TM , Immunomedics, CAS Reg.
- No.219649-07-7 also known as MN-14 and hMN14, is a humanized IgG1 monoclonal antibody and has been studied for the treatment of colorectal cancer (Blumenthal, R. et al (2005) Cancer Immunology Immunotherapy 54(4):315-327).
- Labetuzumab conjugated to a camptothecin analog targets carcinoembryonic antigen- related cell adhesion mol.5 (CEACAM5) and is being studied in patients with relapsed or refractory metastatic colorectal cancer (Sharkey, R.
- the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Trop2.
- Tumor-associated calcium signal transducer 2 (TROP-2) is a transmembrane glycoprotein encoded by the TACSTD2 gene (Linnenbach AJ, et al (1993) Mol Cell Biol.13(3): 1507–15; Calabrese G, et al (2001) Cytogenet Cell Genet.92(1–2): 164–5).
- Trop2 is an intracellular calcium signal transducer that is differentially expressed in many cancers and signals cells for self-renewal, proliferation, invasion, and survival. Trop2 is considered a stem cell marker and is expressed in many normal tissues, though in contrast, it is overexpressed in many cancers (Ohmachi T, et al., (2006) Clin. Cancer Res., 12(10), 3057-3063; Muhlmann G, et al., (2009) J. Clin. Pathol., 62(2), 152-158; Fong D, et al., (2008) Br. J. Cancer, 99(8), 1290- 1295; Fong D, et al., (2008) Mod. Pathol., 21(2), 186-191; Ning S, et al., (2013) Neurol.
- Trop2 Overexpression of Trop2 is of prognostic significance.
- TACSTD2 tumor-associated calcium signal transducer 2, GA733-1, EGP-1, M1S1; hereinafter, referred to as hTrop2
- hTrop2 Human Trop2
- hTrop2 tumor-associated calcium signal transducer 2, GA733-1, EGP-1, M1S1; hereinafter, referred to as hTrop2
- an antigen molecule recognized by a monoclonal antibody against a cell membrane protein in a human choriocarcinoma cell line was identified and designated as Trop2 as one of the molecules expressed in human trophoblasts (Lipinski M, et al., Proc. Natl. Acad. Sci.78(8), 5147-5150 (1981)).
- This molecule was also designated as tumor antigen GA733-1 recognized by a mouse monoclonal antibody GA733 (Linnenbach A J, et al., Proc. Natl. Acad.
- hTrop2 The DNA sequence and amino acid sequence of hTrop2 are available on a public database and can be referred to, for example, under Accession Nos. NM_002353 and NP_002344 (NCBI). In response to such information suggesting the association with cancer, a plurality of anti-hTrop2 antibodies have been established so far and studied for their antitumor effects.
- an unconjugated antibody that exhibits in itself antitumor activity in nude mouse xenograft models WO 2008/144891; WO 2011/145744; WO 2011/155579; WO 2013/077458
- an antibody that exhibits antitumor activity as ADC with a cytotoxic drug WO 2003/074566; WO 2011/068845; WO 2013/068946; US 7999083.
- TROP2 expression in cancer cells has been correlated with drug resistance.
- the Trop2 antibody in sacituzumab govitecan is conjugated to SN-38, the active metabolite of irinotecan (US 2016/0297890; WO 2015/098099).
- the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Caprin-1 (Ellis JA, Luzio JP (1995) J Biol Chem.270(35):20717–23; Wang B, et al (2005) J Immunol.175 (7):4274–82; Solomon S, et al (2007) Mol Cell Biol.27(6):2324–42).
- Caprin-1 is also known as GPIAP1, GPIP137, GRIP137, M11S1, RNG105, p137GPI, and cell cycle associated protein 1.
- Cytoplasmic activation/proliferation-associated protein-1 (caprin-1) is an RNA-binding protein that participates in the regulation of cell cycle control-associated genes. Caprin-1 selectively binds to c-Myc and cyclin D2 mRNAs, which accelerates cell progression through the G1 phase into the S phase, enhances cell viability and promotes cell growth, indicating that it may serve an important role in tumorigenesis (Wang B, et al (2005) J Immunol.175:4274– 4282).
- Caprin-1 acts alone or in combination with other RNA-binding proteins, such as RasGAP SH3-domain-binding protein 1 and fragile X mental retardation protein.
- caprin-1 primarily functions by activating cell proliferation and upregulating the expression of immune checkpoint proteins.
- caprin-1 is also involved in the process by which tumor cells adapt to adverse conditions, which contributes to radiation and chemotherapy resistance. Given its role in various clinical malignancies, caprin-1 holds the potential to be used as a biomarker and a target for the development of novel therapeutics (Yang, Z-S, et al (2019) Oncology Letters 18:15-21).
- the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Claudin-1.
- Claudin-1 is a member of the transmembrane protein family claudins located in cell-cell tight junctions and it acts as a co-receptor for HCV entry into hepatic cells (Kniesel U, et al (2000). Cell. Mol. Neurobiol.20(1):57–76; Furuse M, et al (1998). J. Cell Biol.141(7):1539–50; Swisshelm K, et al (2005) Adv. Drug Deliv. Rev.57(6):919–28).
- Claudin 1 is also known as Senescence-associated epithelial membrane protein, senescence-associated epithelial membrane protein 1, CLDN1, CLD1, ILVASC, SEMP1.
- the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Nectin-4.
- the nectins are a protein family of cell adhesion molecules involved in calcium-dependent cell adhesion (Takai Y. et al (2003) Cancer Science 94(8):655-67; Fuchs, A. et al (2006) Seminars in Cancer Biology 16(5):359-366; Miyoshi J.
- the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds LRRC15 (Leucine rich repeat containing 15).
- LRRC15 is a cell membrane-expressed protein that in humans is encoded by the LRRC15 gene.
- LRRC15 is expressed on stromal fibroblasts in many solid tumors (e.g., breast, head and neck, lung, pancreatic) as well as directly on a subset of cancer cells of mesenchymal origin (e.g., sarcoma, melanoma, glioblastoma). LRRC15 may have utility as a therapeutic target for the treatment of cancers with LRRC15-positive stromal desmoplasia or cancers of mesenchymal origin (Purcell, JW et al (2016) Cancer Res., 78(14):4059-4072).
- AZA-BICYCLIC ADJUVANT COMPOUNDS The immunoconjugate of the invention comprises an aza-bicyclic adjuvant moiety.
- the adjuvant moiety described herein is a compound that elicits an immune response (i.e., an immunostimulatory agent).
- an immunostimulatory agent i.e., an immunostimulatory agent
- the adjuvant moiety described herein is a STING agonist.
- Certain amido benzimidazole compounds are demonstrated STING receptor agonists with systemic activity (Ramanjulu, J.M. et al (2018 ) Nature 564:439–443; Barber, G.N. (2015) Nature Rev Immunol 15:760–770; US 2019/0300513).
- STING is a dimeric structure with a large and symmetrical binding pocket.
- the aza- bicyclic compounds (AzaBi) of Tables 1a and 1b when conjugated to a targeting antibody were designed to target and bind to the open conformation of the binding pocket of STING. Binding to a small molecule agonist usually induces a closed conformation of the STING protein. This introduces the risk that a linker, particularly if “noncleavable”, will interfere with binding and activation.
- the bis-benzimidazole STING agonists are reported to bind and activate through an open conformation, which are predicted to be more amenable to attachment of a linker (Ramanjulu, J.M. et al (2016) Nature 564:439–443; Barber, G.N. (2015) Nature Rev Immunol 15:760–770).
- Table 1a Aza-bicyclic compounds (AzaBi) Table 1b. Aza-bicyclic compounds (AzaBi)
- the immunoconjugates of the invention are prepared by conjugation of an antibody with an aza-bicyclic linker compound, such as those exemplified in Table 2.
- the aza-bicyclic linker compounds (AzaBi-L) comprise a nitrogen-substituted, bis-benzimidazole motif, or analog thereof, covalently attached to a linker unit (L), and described as the Formula II compounds.
- the linker units comprise functional groups and subunits which affect stability, permeability, solubility, and other pharmacokinetic, safety, and efficacy properties of the immunoconjugates.
- the linker unit includes a reactive functional group which reacts, i.e.
- a nucleophilic group such as a lysine side chain amino of the antibody reacts with an electrophilic reactive functional group such as an activated ester, including but not limited to tetrafluorophenyl or sulfotetrafluorophenyl, of the AzaBi-L compound to form the immunoconjugate.
- an electrophilic reactive functional group such as an activated ester, including but not limited to tetrafluorophenyl or sulfotetrafluorophenyl, of the AzaBi-L compound to form the immunoconjugate.
- a cysteine thiol of the antibody reacts with a maleimide or bromoacetamide group of the AzaBi-L compound to form the immunoconjugate.
- Other chemistries can effect covalent attachment of an aza-bicyclic linker compound to an antibody such as glycan remodeling.
- Linker refers to a functional group or unit that covalently bonds two or more moieties in a compound.
- the linker can serve to covalently bond a STING agonist moiety to an antibody construct in an immunoconjugate.
- Useful bonds for connecting linking moieties to proteins and other materials include, but are not limited to, amides, amines, esters, carbamates, ureas, thioethers, thiocarbamates, thiocarbonates, and thioureas.
- a linker can be divalent, connecting the STING agonist moiety to the antibody.
- a linker can also be trivalent, connecting the STING agonist moiety to the antibody and including a branch site with a group that effects additional properties on the immunoconjugate.
- the design of the immunoconjugates of the invention include: (1) preventing the premature release of the aza-bicyclic (AzaBi) moiety during in vivo circulation and (2) ensuring that a biologically active form of the AzaBi moiety is released at the desired site of action at an adequate rate.
- the complex structure of the immunoconjugate together with its functional properties requires careful design and selection of every component of the molecule including antibody, conjugation site, linker structure, and the AzaBi compound.
- the linker determines the mechanism and rate of adjuvant release.
- the linker unit (L) may be cleavable or non-cleavable.
- Cleavable linker units may include a peptide sequence which is a substrate for certain proteases such as Cathepsins which recognize and cleave the peptide linker unit, separating the STING agonist from the antibody (Caculitan NG, et al (2017) Cancer Res.77(24):7027-7037).
- Cleavable linker units may include labile functionality such as an acid-sensitive disulfide group (Kellogg, BA et al (2011) Bioconjugate Chem.22, 717 ⁇ 727; Jamaicart, A. D. et al (2011) Clin. Cancer Res.17, 6417 ⁇ 6427; Pillow, T., et al (2017) Chem.
- the linker is non-cleavable under physiological conditions .
- physiological conditions refers to a temperature range of 20-40 degrees Celsius , atmospheric pressure (i.e. , 1 atm ) , a pH of about 6 to about 8 , and the one or more physiological enzymes, proteases, acids , and bases.
- the linker comprises a trivalent, branch point as part of an amino acid unit (e.g., lysine) wherein additional linker units are attached via the side chain amine of lysine or linked to other sites of an amino acid unit (US 11,173,214).
- an amino acid unit e.g., lysine
- additional linker units are attached via the side chain amine of lysine or linked to other sites of an amino acid unit (US 11,173,214).
- a similar motif could be utilized with a glutamic acid of an amino acid unit.
- the linker comprises a trivalent, branch point as part of an aryl group that attaches the STING agonist moiety, the antibody, and an additional linker unit (WO 2023/173121).
- An exemplary additional linker unit is a monovalent solubilizing group (SolG), such as one or more units of polyglycine, polysarcosine, polyethyleneoxy (PEG), and a glycoside, or combinations thereof.
- SolG monovalent solubilizing group
- the solubilizing unit may bear a group at the terminus such as an amino acid, carboxylic acid, glycerol, or a sugar such as pentaerythritol, maltitol, sorbitol, xylitol, erythritol, isomalt, or combinations thereof.
- the trivalent branch point is part of an amino acid unit (e.g., lysine) wherein additional units are attached via the side chain amine of the lysine and other additional units are linked to the amino acid element via one or more peptide bonds to the peptide backbone carbon of a lysine.
- an amino acid unit comprises one of the following trivalent, branching structures: . wherein * indicates the attachment site of an additional linker unit such as a monovalent solubilizing unit, and the wavy lines indicate the attachment sites to the antibody and to the STING agonist D moiety.
- the monovalent solubilizing unit of a trivalent branched linker is a a polysarcosine with one of the following structures: where n is independently an integer from 1 to 20. In some embodiments, the monovalent solubilizing unit of a trivalent branched linker has one of the following structures, or a combination thereof: where n is independently an integer from 1 to 20, and R is independently selected from H, and C 1 -C 6 alkyl. In some embodiments, the monovalent solubilizing unit of a trivalent branched linker has one of the following structures:
- the invention includes a peptide linking unit, i.e. L or linker, between the antibody and the STING agonist moiety D, comprising a peptide radical based on a linear sequence of specific amino acid residues which can be selectively cleaved by a protease such as a cathepsin, a tumor-associated elastase enzyme or an enzyme with protease-like or elastase-like activity.
- the peptide radical may be about two to about twelve amino acids. Enzymatic cleavage of a bond within the peptide linker releases an active form of the immunostimulatory moiety.
- an amino acid unit or peptide unit comprises one or more amino acids selected from the group consisting of glycine, alanine, serine, threonine, cysteine, valine, leucine, isoleucine, methionine, proline, phenylalanine, tyrosine, tryptophan, aspartic acid, glutamic acid, asparagine, glutamine, histidine, lysine, arginine, sarcosine, and beta-alanine.
- the invention includes an amino acid unit or a peptide linking unit, i.e. L or linker, between the antibody and the immunostimulatory moiety, comprising a peptide comprising a linear sequence of specific amino acid residues which can be selectively cleaved by a protease such as a cathepsin, caspase, a tumor-associated elastase enzyme or an enzyme with protease-like or elastase-like activity.
- the peptide radical may be two to about twelve amino acids. Enzymatic cleavage of a bond within the peptide linker releases an active form of the immunostimulatory moiety.
- lysosomal proteases such as cathepsin and plasmin which may be present at elevated levels in certain tumor tissues.
- the lysosomal enzyme can be, for example, cathepsin B, ⁇ -glucuronidase, or ⁇ -galactosidase.
- a cleavable peptide of a peptide linker unit can be selected from tetrapeptides such as Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, tripeptides such as Glu-Val-Cit, or dipeptides such as Val- Cit, Val-Ala, Ala-Ala, and Phe-Lys.
- the linker provides sufficient stability of the immunoconjugate in biological media, e.g. culture medium or serum and, at the same time, the desired intracellular action within tumor tissue as a result of its specific enzymatic or hydrolytic cleavability with release of the immunostimulatory moiety, i.e. “payload”.
- the enzymatic activity of a protease, cathepsin, or elastase can catalyze cleavage of a covalent bond of the immunoconjugate under physiological conditions.
- the enzymatic activity being the expression product of cells associated with tumor tissue.
- the enzymatic activity on the cleavage site of the targeting peptide converts the immunoconjugate to an active immunostimulatory drug free of targeting peptide and linking group.
- the cleavage site may be specifically recognized by the enzyme.
- Cathepsin or elastase may catalyze the cleavage of a specific peptidic bond between the C-terminal amino acid residue of the specific peptide and the immunostimulatory moiety of the immunoconjugate.
- the invention includes a linking unit, i.e. L or linker, between the cell-binding agent and the immunostimulatory moiety, comprising a substrate for glucuronidase (Jeffrey SC, et al (2006) Bioconjug Chem.17(3):831-40), or sulfatase (Bargh JD, et al (2020) Chem Sci.11(9):2375-2380) cleavage.
- glucuronidase Jeffrey SC, et al (2006) Bioconjug Chem.17(3):831-40
- sulfatase Bargh JD, et al (2020) Chem Sci.11(9):2375-2380
- L include a Gluc unit and comprise a formula selected from:
- Reactive electrophilic reactive functional groups (Q in Formula II) suitable for the aza- bicyclic linker compounds include, but are not limited to, N-hydroxysuccinimidyl (NHS) esters and N-hydroxysulfosuccinimidyl (sulfo-NHS) esters (amine reactive); carbodiimides (amine and carboxyl reactive); hydroxymethyl phosphines (amine reactive); maleimides (thiol reactive); halogenated acetamides such as N-iodoacetamides (thiol reactive); aryl azides (primary amine reactive); fluorinated aryl azides (reactive via carbon-hydrogen (C- H) insertion); pentafluorophenyl (PFP) esters (amine reactive); tetrafluorophenyl (TFP) and sulfotetrafluorophenyl (STP) esters (amine reactive); imidoesters (amine reactive); is
- linkers such as those comprising peptide units and substrates for protease may be labile in the blood stream, thereby releasing unacceptable amounts of the drug prior to internalization in a target cell (Khot, A. et al (2015) Bioanalysis 7(13):1633–1648).
- Other linkers may provide stability in the bloodstream, but intracellular release effectiveness may be negatively impacted.
- Linkers that provide for desired intracellular release may have poor stability in the bloodstream.
- the amount of adjuvant/drug moiety loaded on the antibody i.e.
- cleavable linkers for example with protease-substrate peptide units or immolative units such as para-aminobenzyloxycarbonyl, can provide certain advantages, linkers need not be cleavable.
- azaBz adjuvant moiety release may not depend on the differential properties between the plasma and some cytoplasmic compartments.
- the release of a adjuvant moiety or its metabolite can occur after internalization of the immunoconjugate of via antigen-mediated endocytosis and delivery to lysosomal compartment, where the targeting moiety (or binding fragment thereof) can be degraded to the level of amino acids through intracellular proteolytic degradation. This process can release an adjuvant moiety or its metabolite.
- the released adjuvant moiety or metabolite thereof may be more hydrophilic and less membrane permeable, which can lead to less bystander effects and less non-specific toxicities compared to conjugates with a cleavable linker.
- Immunoconjugates with non-cleavable linkers can have greater stability in circulation than immunoconjugates with cleavable linkers.
- Non-cleavable linkers can include alkylene chains, or can be polymeric, such as, for example, based upon polyalkylene glycol polymers (PEG), amide polymers, or can include segments of alkylene chains, polyalkylene glycols and/or amide polymers.
- the linker can contain a PEG having from 2 to 50 ethylene glycol (PEG) units, or from 2 to 10 ethylene glycol (PEG) units.
- Conjugation of the adjuvant azaBi moiety to a glycan group of an antibody may improve linkage stability, homogeneity, aggregation, and various pharmacokinetic properties of the immunoconjugate relative to conjugation to a native or engineered cysteine residue (Zhou, Q., et al (2014) Bioconjugate Chem.25(3), 510-520; Okeley, N.M., et al (2013) Bioconjugate Chem. 24(10):1650-1655; US 10,072,096; WO2015057063; WO2021248048).
- Some glycan remodeling methods use recombinant microbial transglutaminase to enable efficient, site- specific conjugation of drug-linker intermediates to position HC-Q295 of native, fully glycosylated IgG-type antibodies (Dickgeisser, S., et al (2020) Bioconjugate Chemistry 31(4), 1070-1076).
- the native glycan and modified glycan groups and the methods of conjugation may be those taught in Qasba, P.K. (2015) Bioconjugate Chem.26:2170 ⁇ 2175; Jaramillo, M.L. et al, (2023) MABS, VOL.15, NO.1:1-15; Zhang, X., et al (2021) ACS Chem.
- linkers may provide stability in the bloodstream, but intracellular release effectiveness may be negatively impacted. Linkers that provide for desired intracellular release typically have poor stability in the bloodstream.
- the amount of adjuvant/drug moiety loaded on the antibody i.e. drug loading
- Exemplary embodiments include a STING agonist-linker intermediate compound having Formula II: where the dashed lines are optional double bonds; X a and X b are independently selected from a five-membered heteroaryl, optionally substituted with R 1 or R 4 ; R 1 is independently H or C 1 -C 6 alkyl; Y 1a and Y 1b are independently selected from N, NH, O, and S; Y 2a and Y 2b are independently selected from N, NH, and S; each of X 1 , X 2 , X 3 , X 4 , X 5 , X 6 , X 7 , and X 8 are independently selected from N, NR 2a , CR 2b and C(R 2b ) 2 ; q and r are independently selected from 0, 1, 2, and 3; one or two of X 1 , X 2 , X 3 , X 4 , X 5 , X 6 , X 7 , and X 8 are
- An exemplary embodiment of the STING agonist-linker intermediate compound having Formula II includes wherein L is selected from the structures: where the wavy line indicates the attachment to D.
- the invention includes all reasonable combinations, and permutations of the features, of the Formula II embodiments.
- Exemplary embodiments of the Aza-bicyclic linker (AzaBi-L) intermediate compound are shown in Tables 2a and 2b. Each AzaBi-L compound was prepared and characterized by mass spectrometry and shown to have the mass indicated. The AzaBi-L compounds of Tables 2a and 2b demonstrate the surprising and unexpected property of STING agonist selectivity which may predict useful therapeutic activity to treat cancer and other disorders when conjugated to an antibody. Table 2a. Aza-bicyclic linker (AzaBi-L) Formula II compounds
- the immunoconjugates of the invention induce target-specific activation of immune effector cells such as myeloid cells as well as tumor cells expressing STING themselves.
- immune effector cells such as myeloid cells
- tumor targeting brings specificity to minimize off-target STING activation, and the immunoconjugate enables phagocytosis to not only increase activation of the effector cells but also immune complex uptake and subsequent tumor antigen processing and presentation.
- immunoconjugates comprise an antibody covalently attached to one or more aza-bicyclic STING agonist moieties by a linker, and having Formula I: Ab ⁇ [L ⁇ D] p I or a pharmaceutically acceptable salt thereof, wherein: Ab is the antibody; L is the linker; p is an integer from 1 to 8; D is the STING agonist moiety having the formula: where the dashed lines are optional double bonds; X a and X b are independently selected from a five-membered heteroaryl, optionally substituted with R 1 or R 4 ; R 1 is independently H or C 1 -C 6 alkyl; Y 1a and Y 1b are independently selected from N, NH, O, and S; Y 2a and Y 2b are independently selected from N, NH, and S; each of X 1 , X 2 , X 3 , X 4 , X 5 , X 6 , X 7 , and X 8 are independently selected from N
- Antibodies target tumor-specific and/or immune-specific (e.g. PD-L1) antigens to bring specificity into the targeting of the immunoconjugate to enable safe and systemic delivery
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein the antibody is an antibody construct that has an antigen binding domain that binds PD-L1, such as atezolizumab, durvalumab, and avelumab.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein the antibody is an antibody construct that has an antigen binding domain that binds HER2, such as trastuzumab and pertuzumab.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein the antibody is an antibody construct that has an antigen binding domain that binds CEA, such as labetuzumab.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein the antibody is an antibody construct that has an antigen binding domain that binds TROP2, such as sacituzumab.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein L is a divalent or a branched, trivalent linker.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein X a and X b are independently selected from the group consisting of imidazolyl, pyrazolyl, triazolyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxadiazolyl, oxazolyl, isothiazolyl, pyrrolyl, oxadiazolyl, and thiadiazolyl.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein X a or X b is substituted with R 4 .
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein Y 1a and Y 1b are each N.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein Y 1a is S and Y 1b is N.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein Y 2a and Y 2b are each N.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein X 1 is N; X 2 is CR 5b ; X 3 is CR 5b ; and X 4 is CR 5b , and q is 1.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein X 1 is CR 5b ; X 2 is N; X 3 is CR 5b ; and X 4 is CR 5b , and q is 1.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein X 1 is CR 5b ; X 2 is CR 5b ; X 3 is N; and X 4 is CR 5b , and q is 1.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein X 1 is CR 5b ; X 2 is CR 5b ; X 3 is CR 5b ; and X 4 is N, and q is 1.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein X 5 , X 6 , X 7 , and X 8 are each CR 2b ; r is 1; and one of CR 2b is R 4 .
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein one of X 1 , X 2 , X 3 , X 4 , X 5 , X 6 , X 7 , and X 8 is N or NR 2a .
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein two of X 1 , X 2 , X 3 , X 4 , X 5 , X 6 , X 7 , and X 8 are N or NR 2a .
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein R 2b is selected from the group consisting of ⁇ OCH 3 , ⁇ OCH 2 CH 3 , ⁇ OCH 2 CH 2 OCH 3 , ⁇ OCH 2 CH 2 OH, and ⁇ OCH 2 CH 2 N(CH 3 ) 2 .
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein R 3 is C 2 -C 4 alkenyldiyl, substituted with one or more groups selected from F, ⁇ OH, and ⁇ OCH 3 .
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein R 4 is ⁇ O ⁇ (C 1 -C 12 alkyldiyl) ⁇ (C 2 -C 20 heterocyclyldiyl) ⁇ *.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein C 1 - C 12 alkyldiyl is propyldiyl and C 2 -C 20 heterocyclyldiyl is piperidiyl.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein L is attached to a cysteine thiol of the antibody.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein for the PEG, m is 1 or 2, and n is an integer from 1 to 10.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein n is 1, 2, 3, 4, or 5.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein n is 3, 4, or 5.
- An exemplary embodiment of the immunoconjugate of Formula I includes wherein wherein n is 10.
- the invention includes all reasonable combinations, and permutations of the features, of the Formula I embodiments.
- the immunoconjugate compounds of the invention include those with immunostimulatory activity.
- the antibody-drug conjugates of the invention selectively deliver an effective dose of an aza-bicyclic drug to tumor tissue, whereby greater selectivity (i.e., a lower efficacious dose) may be achieved while increasing the therapeutic index (“therapeutic window”) relative to unconjugated aza-bicyclic drug.
- Drug loading is represented by p, the number of AzaBi moieties per antibody in an immunoconjugate of Formula I.
- Drug (AzaBi) loading may range from 1 to about 8 drug moieties (D) per antibody.
- Immunoconjugates of Formula I include mixtures or collections of antibodies conjugated with a range of drug moieties, from 1 to about 8.
- the number of drug moieties that can be conjugated to an antibody is limited by the number of reactive or available amino acid side chain residues such as lysine and cysteine.
- free cysteine residues are introduced into the antibody amino acid sequence by the methods described herein.
- p may be 1, 2, 3, 4, 5, 6, 7, or 8, and ranges thereof, such as from 1 to 8 or from 2 to 5.
- Exemplary immunoconjugates of Formula I include, but are not limited to, antibodies that have 1, 2, 3, or 4 engineered cysteine amino acids (Lyon, R. et al.
- one or more free cysteine residues are already present in an antibody forming intra-chain and inter-chain disulfide bonds (native disulfide groups), without the use of engineering, in which case the existing free, reduced cysteine residues may be used to conjugate the antibody to a drug.
- an antibody is exposed to reducing conditions prior to conjugation of the antibody in order to generate one or more free cysteine residues.
- p may be limited by the number of attachment sites on the antibody.
- an antibody may have only one or a limited number of cysteine thiol groups, or may have only one or a limited number of sufficiently reactive thiol groups, to which the drug may be attached.
- one or more lysine amino groups in the antibody may be available and reactive for conjugation with a AzaBi-linker compound of Formula II.
- higher drug loading e.g. p >5, may cause aggregation, insolubility, toxicity, or loss of cellular permeability of certain antibody-drug conjugates.
- the average drug loading for an immunoconjugate ranges from 1 to about 8; from about 2 to about 6; or from about 3 to about 5.
- an antibody is subjected to denaturing conditions to reveal reactive nucleophilic groups such as lysine or cysteine.
- the loading (drug/antibody ratio, DAR) of an immunoconjugate may be controlled in different ways, and for example, by: (i) limiting the molar excess of the AzaBi-linker intermediate compound relative to antibody, (ii) limiting the conjugation reaction time or temperature, and (iii) partial or limiting reductive denaturing conditions for optimized antibody reactivity.
- the resulting product is a mixture of immunoconjugate compounds with a distribution of one or more drug moieties attached to an antibody.
- the average number of drugs per antibody may be calculated from the mixture by a dual ELISA antibody assay, which is specific for antibody and specific for the drug.
- Individual immunoconjugate molecules may be identified in the mixture by mass spectroscopy and separated by HPLC, for example hydrophobic interaction chromatography, HIC (McDonagh et al. (2006) Prot. Engr. Design & Selection 19(7):299-307; Hamblett et al. (2004) Clin.
- a homogeneous immunoconjugate with a single loading value may be isolated from the conjugation mixture by electrophoresis or chromatography.
- An exemplary embodiment of the immunoconjugate of Formula I is selected from the Table 3 Immunoconjugates.
- Assessment of Immunoconjugate Activity In Vitro may be conducted according to the methods of Example 202. STING activation is canonically associated with induction of type I/III IFNs (interferons) through IRF3 (interferon regulatory factor 3) signaling, but can also induce proinflammatory cytokines such as TNF ⁇ (tumor necrosis factor alpha) through the NF- ⁇ B (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway.
- TNF ⁇ tumor necrosis factor alpha
- NF- ⁇ B nuclear factor kappa-light-chain-enhancer of activated B cells
- immunoconjugates demonstrate the ability to elicit IFN ⁇ 1 (interferon lambda 1) as well as TNF ⁇ , consistent with STING activation.
- trastuzumab elicited essentially no TNF ⁇ or IFN ⁇ 1 in the PBMC tumor co-culture assay (Example 202).
- compositions e.g., a pharmaceutically or pharmacologically acceptable composition or formulation, comprising a plurality of immunoconjugates as described herein and optionally a carrier therefor, e.g., a pharmaceutically or pharmacologically acceptable carrier.
- the immunoconjugates can be the same or different in the composition, i.e., the composition can comprise immunoconjugates that have the same number of AzaBi adjuvants linked to the same positions on the antibody construct and/or immunoconjugates that have the same number of AzaBi adjuvants linked to different positions on the antibody construct, that have different numbers of adjuvants linked to the same positions on the antibody construct, or that have different numbers of adjuvants linked to different positions on the antibody construct.
- a composition comprising the immunoconjugate compounds comprises a mixture of the immunoconjugate compounds, wherein the average drug (AzaBi) loading per antibody in the mixture of immunoconjugate compounds is about 2 to about 5.
- a composition of immunoconjugates of the invention can have an average adjuvant to antibody construct ratio (DAR) of about 0.4 to about 10.
- DAR adjuvant to antibody construct ratio
- the adjuvant to antibody construct (e.g., antibody) ratio can be assessed by any suitable means, many of which are known in the art.
- the immunoconjugates of the invention can be formulated for parenteral administration, such as IV administration or administration into a body cavity or lumen of an organ.
- the immunoconjugates can be injected into the tumor (intra-tumorally).
- Compositions for injection will commonly comprise a solution of the immunoconjugate dissolved in a pharmaceutically acceptable carrier.
- acceptable vehicles and solvents that can be employed are water and an isotonic solution of one or more salts such as sodium chloride, e.g., Ringer's solution.
- sterile fixed oils can conventionally be employed as a solvent or suspending medium.
- any bland fixed oil can be employed, including synthetic monoglycerides or diglycerides.
- compositions desirably are sterile and generally free of undesirable matter.
- These compositions can be sterilized by conventional, well known sterilization techniques.
- the compositions can contain pharmaceutically acceptable auxiliary substances as required to approximate physiological conditions such as pH adjusting and buffering agents, toxicity adjusting agents, e.g., sodium acetate, sodium chloride, potassium chloride, calcium chloride, sodium lactate and the like.
- the composition can contain any suitable concentration of the immunoconjugate.
- the concentration of the immunoconjugate in the composition can vary widely, and will be selected primarily based on fluid volumes, viscosities, body weight, and the like, in accordance with the particular mode of administration selected and the patient's needs. In certain embodiments, the concentration of an immunoconjugate in a solution formulation for injection will range from about 0.1% (w/w) to about 10% (w/w).
- METHOD OF TREATING CANCER WITH IMMUNOCONJUGATES The invention provides a method for treating cancer. The method includes administering a therapeutically effective amount of an immunoconjugate as described herein (e.g., as a composition as described herein) to a subject in need thereof, e.g., a subject that has cancer and is in need of treatment for the cancer.
- the method includes administering a therapeutically effective amount of an immunoconjugate (IC) selected from Table 3.
- IC immunoconjugate
- the immunoconjugate of the present invention may be used to treat various hyperproliferative diseases or disorders, e.g. characterized by the overexpression of a tumor antigen.
- hyperproliferative disorders include benign or malignant solid tumors and hematological disorders such as leukemia and lymphoid malignancies.
- an immunoconjugate for use as a medicament is provided.
- the invention provides an immunoconjugate for use in a method of treating an individual comprising administering to the individual an effective amount of the immunoconjugate.
- the method further comprises administering to the individual an effective amount of at least one additional therapeutic agent, e.g., as described herein.
- the invention provides for the use of an immunoconjugate in the manufacture or preparation of a medicament.
- the medicament is for treatment of cancer, the method comprising administering to an individual having cancer an effective amount of the medicament.
- the method further comprises administering to the individual an effective amount of at least one additional therapeutic agent, e.g., as described herein.
- Carcinomas are malignancies that originate in the epithelial tissues. Epithelial cells cover the external surface of the body, line the internal cavities, and form the lining of glandular tissues.
- carcinomas include, but are not limited to, adenocarcinoma (cancer that begins in glandular (secretory) cells such as cancers of the breast, pancreas, lung, prostate, stomach, gastroesophageal junction, and colon) adrenocortical carcinoma; hepatocellular carcinoma; renal cell carcinoma; ovarian carcinoma; carcinoma in situ; ductal carcinoma; carcinoma of the breast; basal cell carcinoma; squamous cell carcinoma; transitional cell carcinoma; colon carcinoma; nasopharyngeal carcinoma; multilocular cystic renal cell carcinoma; oat cell carcinoma; large cell lung carcinoma; small cell lung carcinoma; non-small cell lung carcinoma; and the like.
- adenocarcinoma cancer that begins in glandular (secretory) cells such as cancers of the breast, pancreas, lung, prostate, stomach, gastroesophageal junction, and colon
- adrenocortical carcinoma hepatocellular carcinoma
- renal cell carcinoma ovarian carcinoma
- carcinoma in situ duct
- Carcinomas may be found in prostrate, pancreas, colon, brain (usually as secondary metastases), lung, breast, and skin.
- Soft tissue tumors are a highly diverse group of rare tumors that are derived from connective tissue. Examples of soft tissue tumors include, but are not limited to, alveolar soft part sarcoma; angiomatoid fibrous histiocytoma; chondromyoxid fibroma; skeletal chondrosarcoma; extraskeletal myxoid chondrosarcoma; clear cell sarcoma; desmoplastic small round-cell tumor; dermatofibrosarcoma protuberans; endometrial stromal tumor; Ewing’s sarcoma; fibromatosis (Desmoid); fibrosarcoma, infantile; gastrointestinal stromal tumor; bone giant cell tumor; tenosynovial giant cell tumor; inflammatory myofibroblastic tumor; uterine leiomyoma; leiomyosarcom
- a sarcoma is a rare type of cancer that arises in cells of mesenchymal origin, e.g., in bone or in the soft tissues of the body, including cartilage, fat, muscle, blood vessels, fibrous tissue, or other connective or supportive tissue.
- Different types of sarcoma are based on where the cancer forms. For example, osteosarcoma forms in bone, liposarcoma forms in fat, and rhabdomyosarcoma forms in muscle.
- sarcomas include, but are not limited to, Askin's tumor; sarcoma botryoides; chondrosarcoma; Ewing's sarcoma; malignant hemangioendothelioma; malignant schwannoma; osteosarcoma; and soft tissue sarcomas (e.g., alveolar soft part sarcoma; angiosarcoma; cystosarcoma phyllodesdermatofibrosarcoma protuberans (DFSP); desmoid tumor; desmoplastic small round cell tumor; epithelioid sarcoma; extraskeletal chondrosarcoma; extraskeletal osteosarcoma; fibrosarcoma; gastrointestinal stromal tumor (GIST); hemangiopericytoma; hemangiosarcoma (more commonly referred to as “angiosarcoma”); Kaposi’s sarcoma; leiomyosarcoma; liposarcom
- a teratoma is a type of germ cell tumor that may contain several different types of tissue (e.g., can include tissues derived from any and/or all of the three germ layers: endoderm, mesoderm, and ectoderm), including, for example, hair, muscle, and bone. Teratomas occur most often in the ovaries in women, the testicles in men, and the tailbone in children.
- Melanoma is a form of cancer that begins in melanocytes (cells that make the pigment melanin). Melanoma may begin in a mole (skin melanoma), but can also begin in other pigmented tissues, such as in the eye or in the intestines.
- Merkel cell carcinoma is a rare type of skin cancer that usually appears as a flesh-colored or bluish-red nodule on the face, head or neck. Merkel cell carcinoma is also called neuroendocrine carcinoma of the skin.
- methods for treating Merkel cell carcinoma include administering an immunoconjugate containing an antibody construct that is capable of binding TROP2 (e.g., sacituzumab, sacituzumab govitecan, or a biosimilar thereof).
- the Merkel cell carcinoma has metastasized when administration occurs.
- Leukemias are cancers that start in blood-forming tissue, such as the bone marrow, and cause large numbers of abnormal blood cells to be produced and enter the bloodstream.
- leukemias can originate in bone marrow-derived cells that normally mature in the bloodstream.
- Leukemias are named for how quickly the disease develops and progresses (e.g., acute versus chronic) and for the type of white blood cell that is affected (e.g., myeloid versus lymphoid).
- Myeloid leukemias are also called myelogenous or myeloblastic leukemias.
- Lymphoid leukemias are also called lymphoblastic or lymphocytic leukemia. Lymphoid leukemia cells may collect in the lymph nodes, which can become swollen.
- lymphomas are cancers that begin in cells of the immune system.
- lymphomas can originate in bone marrow-derived cells that normally mature in the lymphatic system.
- lymphomas There are two basic categories of lymphomas.
- One category of lymphoma is Hodgkin lymphoma (HL), which is marked by the presence of a type of cell called the Reed-Sternberg cell.
- HL Hodgkin lymphoma
- Hodgkin lymphomas examples include nodular sclerosis classical Hodgkin lymphoma (CHL), mixed cellularity CHL, lymphocyte- depletion CHL, lymphocyte-rich CHL, and nodular lymphocyte predominant HL.
- CHL classical Hodgkin lymphoma
- NHL non-Hodgkin lymphomas
- Non-Hodgkin lymphomas can be further divided into cancers that have an indolent (slow-growing) course and those that have an aggressive (fast-growing) course.
- non-Hodgkin lymphomas include, but are not limited to, AIDS-related Lymphomas, anaplastic large-cell lymphoma, angioimmunoblastic lymphoma, blastic NK-cell lymphoma, Burkitt’s lymphoma, Burkitt-like lymphoma (small non- cleaved cell lymphoma), chronic lymphocytic leukemia/small lymphocytic lymphoma, cutaneous T-Cell lymphoma, diffuse large B-Cell lymphoma, enteropathy-type T-Cell lymphoma, follicular lymphoma, hepatosplenic gamma-delta T-Cell lymphomas, T-Cell leukemias, lymphoblastic lymphoma, mantle cell lymphoma, marginal zone lymphoma, nasal T- Cell lymphoma, pediatric lymphoma, peripheral T-Cell lymphomas, primary central nervous system lymphoma, transformed lymphomas
- Brain cancers include any cancer of the brain tissues.
- Examples of brain cancers include, but are not limited to, gliomas (e.g., glioblastomas, astrocytomas, oligodendrogliomas, ependymomas, and the like), meningiomas, pituitary adenomas, and vestibular schwannomas, primitive neuroectodermal tumors (medulloblastomas).
- Immunoconjugates of the invention can be used either alone or in combination with other agents in a therapy. For instance, an immunoconjugate may be co-administered with at least one additional therapeutic agent, such as a chemotherapeutic agent.
- Such combination therapies encompass combined administration (where two or more therapeutic agents are included in the same or separate formulations), and separate administration, in which case, administration of the immunoconjugate can occur prior to, simultaneously, and/or following, administration of the additional therapeutic agent and/or adjuvant.
- Immunoconjugates can also be used in combination with radiation therapy.
- the immunoconjugates of the invention (and any additional therapeutic agent) can be administered by any suitable means, including parenteral, intrapulmonary, and intranasal, and, if desired for local treatment, intralesional administration.
- Parenteral infusions include intramuscular, intravenous, intraarterial, intraperitoneal, or subcutaneous administration. Dosing can be by any suitable route, e.g.
- the immunoconjugate described herein can be used to treat the same types of cancers as sacituzumab, sacituzumab govitecan, or a biosimilar thereof, particularly breast cancer, especially triple negative (test negative for estrogen receptors, progesterone receptors, and excess HER2 protein) breast cancer, bladder cancer, and Merkel cell carcinoma.
- the immunoconjugates described herein may be effective in the treatment of bladder cancer, salivary gland cancer, endometrial cancer, urinary tract cancer, urothelial carcinoma, lung cancer, non-small cell lung cancer, Merkel cell carcinoma, colon cancer, colorectal cancer, gastric cancer, and breast cancer.
- the immunoconjugate is administered to a subject in need thereof in any therapeutically effective amount using any suitable dosing regimen, such as the dosing regimens utilized for sacituzumab, sacituzumab govitecan, or a biosimilar thereof.
- the methods can include administering the immunoconjugate to provide a dose of from about 100 ng/kg to about 50 mg/kg to the subject.
- the immunoconjugate dose can range from about 5 mg/kg to about 50 mg/kg, from about 10 ⁇ g/kg to about 5 mg/kg, or from about 100 ⁇ g/kg to about 1 mg/kg.
- the immunoconjugate dose can be about 100, 200, 300, 400, or 500 ⁇ g/kg.
- the immunoconjugate dose can be about 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 mg/kg.
- the immunoconjugate dose can also be outside of these ranges, depending on the particular conjugate as well as the type and severity of the cancer being treated. Frequency of administration can range from a single dose to multiple doses per week, or more frequently. In some embodiments, the immunoconjugate is administered from about once per month to about five times per week.
- the immunoconjugate is administered once per week.
- the invention provides a method for preventing cancer.
- the method comprises administering a therapeutically effective amount of an immunoconjugate (e.g., as a composition as described above) to a subject.
- the subject is susceptible to a certain cancer to be prevented.
- the methods can include administering the immunoconjugate to provide a dose of from about 100 ng/kg to about 50 mg/kg to the subject.
- the immunoconjugate dose can range from about 5 mg/kg to about 50 mg/kg, from about 10 ⁇ g/kg to about 5 mg/kg, or from about 100 ⁇ g/kg to about 1 mg/kg.
- the immunoconjugate dose can be about 100, 200, 300, 400, or 500 ⁇ g/kg.
- the immunoconjugate dose can be about 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 mg/kg.
- the immunoconjugate dose can also be outside of these ranges, depending on the particular conjugate as well as the type and severity of the cancer being treated. Frequency of administration can range from a single dose to multiple doses per week, or more frequently.
- the immunoconjugate is administered from about once per month to about five times per week.
- the immunoconjugate is administered once per week.
- the immunoconjugates of the invention can be used for treating ductal carcinoma in situ; invasive ductal carcinoma (e.g., tubular carcinoma; medullary carcinoma; mucinous carcinoma; papillary carcinoma; or cribriform carcinoma of the breast); lobular carcinoma in situ; invasive lobular carcinoma; inflammatory breast cancer; and other forms of breast cancer such as triple negative (test negative for estrogen receptors, progesterone receptors, and excess HER2 protein) breast cancer.
- the cancer is susceptible to a pro-inflammatory response induced by STING.
- reaction mixture was quenched by addition of H 2 O (50 mL) at 0 °C, and then extracted with EtOAc (30 mL x 3). The combined organic layers were washed with brine (10 mL x 3), dried over Na 2 SO 4 , filtered and concentrated under reduced pressure to give a residue.
- tert-butyl 4-[3-(5- carbamoyl-2-chloro-3-nitro-phenoxy)propyl]piperazine-1-carboxylate (253 mg, 572 umol, 0.6 eq) was added, and then stirred at 120°C for 15 hours under N 2 .
- the reaction mixture was cooled to 20°C and concentrated in reduced pressure.
- the residue was diluted with ice-water (15 mL).
- the aqueous phase was extracted with ethyl acetate (10 mL x 3).
- the reaction mixture was poured into water (30 mL) and extracted with DCM (20 mL x 3). The combined organic phase was concentrated. The residue was diluted with MeOH (20 mL), and K 2 CO 3 (1.20 g, 8.68 mmol, 8.0 eq) was added, then stirred at 25°C for another 0.5h. After that, it was filtered and the filtrate was concentrated, the residue was purified by flash silica gel chromatography (ISCO®; 20 g SepaFlash® Silica Flash Column, Eluent of 0 ⁇ 60% Ethylacetate/Petroleum ether gradient @ 60 mL/min) to give 8h (350 mg, 693 umol, 63.9% yield) as light yellow oil.
- ISCO® 20 g SepaFlash® Silica Flash Column, Eluent of 0 ⁇ 60% Ethylacetate/Petroleum ether gradient @ 60 mL/min
- reaction mixture was filtered and purified by prep-HPLC (column: Phenomenex Luna C1875*30mm*3um;mobile phase: [TFA-ACN];B%: 15.000%-45.000%, 8.000min) to give AzaBi-L-4 (32.5 mg, 22.5 umol, 42.1% yield) as light yellow solid.
- Example 201 Preparation of Immunoconjugates (IC)
- an antibody is buffer exchanged into a conjugation buffer containing 100 mM Borate, 50 mM sodium chloride, 1 mM ethylenediaminetetraacetic acid at pH 8.3 using ZebaTM Spin Desalting Columns (Thermo Fisher Scientific).
- the concentration of the buffer-exchanged antibody was adjusted to approximately 5 – 25 mg/ml using the conjugation buffer and sterile-filtered.
- the Aza-bicyclic linker (AzaBi-L) intermediate compound of Formula II is either dissolved in dimethylsulfoxide (DMSO) or dimethylacetamide (DMA) to a concentration of 5 – 20 mM.
- DMSO dimethylsulfoxide
- DMA dimethylacetamide
- the antibody is mixed with 4 – 20 molar equivalents of AzaBi-L.
- additional DMA or DMSO up to 20% (v/v) was added to improve the solubility of AzaBi-L in the conjugation buffer.
- the reaction is allowed to proceed for approximately 30 min to 4 hours at 20 °C or 30 °C or 37 °C.
- the resulting conjugate is purified away from the unreacted AzaBi-L using two successive ZebaTM Spin Desalting Columns.
- the columns are pre-equilibrated with phosphate-buffered saline (PBS), pH 7.2.
- Adjuvant to antibody ratio (DAR) is estimated by liquid chromatography mass spectrometry analysis using a C4 reverse phase column on an ACQUITY TM UPLC H-class (Waters Corporation, Milford, MA) connected to a XEVO TM G2- XS TOF mass spectrometer (Waters Corporation).
- an antibody is buffer exchanged into a conjugation buffer containing PBS, pH 7.2 with 2 mM EDTA using ZebaTM Spin Desalting Columns (Thermo Fisher Scientific).
- the interchain disulfides are reduced using 2 – 4 molar excess of Tris (2-carboxyethyl) phosphine (TCEP) or dithiothreitol (DTT) at 37 °C for 30 min – 2 hours. Excess TCEP or DTT was removed using a ZebaTM Spin Desalting column pre-equilibrated with the conjugation buffer.
- the concentration of the buffer- exchanged antibody was adjusted to approximately 5 – 20 mg/ml using the conjugation buffer and sterile-filtered.
- the AzaBi-L is either dissolved in dimethylsulfoxide (DMSO) or dimethylacetamide (DMA) to a concentration of 5 – 20 mM.
- DMSO dimethylsulfoxide
- DMA dimethylacetamide
- the antibody is mixed with about 10 to 20 molar equivalents of AzaBi-L.
- additional DMA or DMSO up to 20% (v/v), was added to improve the solubility of the AzaBi-L in the conjugation buffer.
- the reaction is allowed to proceed for approximately 30 min to 4 hours at 20 °C.
- the resulting conjugate is purified away from the unreacted BBI-L using two successive ZebaTM Spin Desalting Columns.
- the columns are pre-equilibrated with phosphate-buffered saline (PBS), pH 7.2.
- Adjuvant to antibody ratio (DAR) is estimated by liquid chromatography mass spectrometry analysis using a C4 reverse phase column on an ACQUITY TM UPLC H-class (Waters Corporation, Milford, MA) connected to a XEVO TM G2-XS TOF mass spectrometer (Waters Corporation).
- the conjugates may be purified further using size exclusion chromatography, hydrophobic interaction chromatography, ion exchange chromatography, chromatofocusing, ultrafiltration, centrifugal ultrafiltration, tangential flow filtration, and combinations thereof.
- an antibody is buffer exchanged into a conjugation buffer containing 100 mM boric acid, 50 mM sodium chloride, 1 mM ethylenediaminetetraacetic acid at pH 8.3, using G-25 SEPHADEX TM desalting columns (Sigma-Aldrich, St. Louis, MO).
- the eluates are then each adjusted to a concentration of about 1-10 mg/ml using the buffer and then sterile filtered.
- the antibody is pre-warmed to 20-30 °C and rapidly mixed with 2-20 (e.g., 7-10) molar equivalents of AzaBi-L intermediate compound of Formula II.
- the reaction is allowed to proceed for about 16 hours at 30 °C and the immunoconjugate (IC) is separated from reactants by running over two successive G-25 desalting columns equilibrated in phosphate buffered saline (PBS) at pH 7.2 to provide the Immunoconjugate (IC) of Table 3.
- PBS phosphate buffered saline
- Adjuvant-antibody ratio is determined by liquid chromatography mass spectrometry analysis using a C4 reverse phase column on an ACQUITY TM UPLC H-class (Waters Corporation, Milford, MA) connected to a XEVO TM G2- XS TOF mass spectrometer (Waters Corporation).
- the antibody may be dissolved in a aqueous buffer system known in the art that will not adversely impact the stability or antigen-binding specificity of the antibody. Phosphate buffered saline may be used.
- the AzaBi-L is dissolved in a solvent system comprising at least one polar aprotic solvent as described elsewhere herein.
- the AzaBi-L is dissolved to a concentration of about 5 mM, about 10 mM, about 20 mM, about 30 mM, about 40 mM or about 50 mM, and ranges thereof such as from about 5 mM to about 50mM or from about 10 mM to about 30 mM in pH 8 Tris buffer (e.g., 50 mM Tris).
- the AzaBi-L is dissolved in DMSO (dimethylsulfoxide), DMA (dimethylacetamide) or acetonitrile, or another suitable dipolar aprotic solvent.
- DMSO dimethylsulfoxide
- DMA dimethylacetamide
- acetonitrile acetonitrile
- an equivalent excess of AzaBi-L solution may be diluted and combined with antibody solution.
- the AzaBi-L solution may suitably be diluted with at least one polar aprotic solvent and at least one polar protic solvent, examples of which include water, methanol, ethanol, n-propanol, and acetic acid.
- the molar equivalents of bis- benzimidazole-linker intermediate to antibody may be about 1.5:1, about 3:1, about 5:1, about 10:1, about 15:1, or about 20:1, and ranges thereof, such as from about 1.5:1 to about 20:1 from about 1.5:1 to about 15:1, from about 1.5:1 to about 10:1,from about 3:1 to about 15:1, from about 3:1 to about 10:1, from about 5:1 to about 15:1 or from about 5:1 to about 10:1.
- the reaction may suitably be monitored for completion by methods known in the art, such as LC- MS.
- the conjugation reaction is typically complete in a range from about 1 hour to about 16 hours.
- a reagent may be added to the reaction mixture to quench the reaction. If antibody thiol groups are reacting with a thiol-reactive group such as maleimide of the AzaBi-L, unreacted antibody thiol groups may be reacted with a capping reagent.
- a capping reagent is ethylmaleimide.
- the immunoconjugates may be purified and separated from unconjugated reactants and/or conjugate aggregates by purification methods known in the art such as, for example and not limited to, size exclusion chromatography, hydrophobic interaction chromatography, ion exchange chromatography, chromatofocusing, ultrafiltration, centrifugal ultrafiltration, tangential flow filtration, and combinations thereof.
- purification may be preceded by diluting the immunoconjugate, such in 20 mM sodium succinate, pH 5.
- the diluted solution is applied to a cation exchange column followed by washing with, e.g., at least 10 column volumes of 20 mM sodium succinate, pH 5.
- the conjugate may be suitably eluted with a buffer such as PBS.
- the immunoconjugates of the invention may be prepared, analyzed, purified, characterized, and tested according to the protocols, where applicable, in “Antibody-Drug Conjugates Methods and Protocols”, Humana Press, Editor: L. Nathan Tumey, 2020.
- Example 202 Functional Assessment of Immunoconjugates The immunoconjugates of the invention can be assessed in a co-culture assay using primary human peripheral blood mononuclear cells (PBMC) co-cultured with target antigen- expressing tumor cells. Briefly, PBMCs are freshly isolated from healthy human donor blood (Stanford Blood Center) by density centrifugation.
- PBMC peripheral blood mononuclear cells
- PBMCs are then co-cultured with antigen- expressing tumor cells at a 10:1 effector to target ratio in complete medium (RPMI supplemented with 10% FBS) and incubated overnight with a range of concentrations of the indicated test articles.
- Activation is measured by secretion of pro-inflammatory cytokines, such as IFN ⁇ 1 and TNF ⁇ , by BioLegend LEGENDPLEX TM cytokine bead array.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Immunology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Cell Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention provides immunoconjugates of Formula I comprising an antibody linked by conjugation to one or more STING agonist moieties. The invention also provides STING agonist-linker intermediate compounds comprising a reactive functional group. Such intermediate compositions are suitable substrates for formation of the immunoconjugates through a linker or linking moiety. The invention further provides methods of treating cancer with the immunoconjugates.
Description
AZA-BICYCLIC STING AGONIST IMMUNOCONJUGATES, AND USES THEREOF CROSS REFERENCE TO RELATED APPLICATIONS This non-provisional application claims the benefit of priority to U.S. Provisional Application No.63/449,659 filed 3 March 2023, which is incorporated by reference in its entirety. FIELD OF THE INVENTION The invention relates generally to an immunoconjugate comprising an antibody conjugated to one or more aza-bicyclic STING agonist moieties. BACKGROUND OF THE INVENTION STING (Stimulator of Interferon Genes), also known as transmembrane protein 173 (TMEM173) and MPYS/MITA/ERIS, is a protein encoded in humans by the STING1 gene. STING is broadly expressed, particularly in immune cells, lung, and ovary. STING plays a role in innate immunity by inducing type I interferon production when cells are infected with intracellular pathogens, such as viruses, mycobacteria, and intracellular parasites. Type I interferon, mediated by STING, protects infected cells and nearby cells from local infection by binding to the same cell that secretes it by autocrine signaling and nearby cells by paracrine signaling. STING works as both a direct cytosolic DNA sensor (CDS) and an adaptor protein in Type I interferon signaling through different molecular mechanisms. STING has been shown to activate downstream transcription factors STAT6 and IRF3 through TBK1, which are responsible for antiviral response and innate immune response against intracellular pathogens. Compounds that bind to STING and act as an agonist have been shown to induce secretion of proinflammatory cytokines including type 1 interferons on incubation with human PBMCs (WO 2017/175147). STING modulators may be useful in the treatment of various disorders, for example, allergic diseases, neurodegenerative diseases, pre-cancerous syndromes, and cancer, and may also be useful in immunogenic compositions or vaccine adjuvants. New compositions and methods for the delivery of antibodies and immune adjuvants are needed in order to reach inaccessible tumors and/or to expand treatment options for cancer patients and other subjects. SUMMARY OF THE INVENTION The invention is generally directed to immunoconjugates comprising an antibody covalently attached to one or more STING agonist moieties by a linker, and having Formula I:
Ab−[L−D]p I or a pharmaceutically acceptable salt thereof, wherein: Ab is the antibody; L is the linker; p is an integer from 1 to 8; D is the STING agonist moiety having the formula:
 where the dashed lines are optional double bonds and the substituents are defined herein. The invention is further directed to use of such an immunoconjugates in the treatment of an illness, in particular cancer. Another aspect of the invention is an aza-bicyclic-linker compound. Another aspect of the invention is a method for treating cancer comprising administering a therapeutically effective amount of an immunoconjugate comprising an antibody linked by conjugation to one or more aza-bicyclic STING agonist moieties. Another aspect of the invention is a use of an immunoconjugate comprising an antibody linked by conjugation to one or more aza-bicyclic STING agonist moieties for treating cancer. Another aspect of the invention is a method of preparing an immunoconjugate by conjugation of one or more aza-bicyclic STING agonist moieties with an antibody. DETAILED DESCRIPTION OF THE INVENTION Reference will now be made in detail to certain embodiments of the invention, examples of which are illustrated in the accompanying structures and formulas. While the invention will be described in conjunction with the enumerated embodiments, it will be understood that they are not intended to limit the invention to those embodiments. On the contrary, the invention is
intended to cover all alternatives, modifications, and equivalents, which may be included within the scope of the invention as defined by the claims. One skilled in the art will recognize many methods and materials similar or equivalent to those described herein, which could be used in the practice of the present invention. The invention is in no way limited to the methods and materials described. DEFINITIONS The term “immunoconjugate” refers to an antibody construct that is covalently bonded to an adjuvant moiety via a linker. The terms “immunostimulant” and “immunostimulatory” are used equivalently and refer to a moiety, substance or adjuvant capable of eliciting an immune response in a subject exposed to the immunostimulatory moiety or the immunostimulatory compound after in vivo cleavage of the linker. The terms “adjuvant moiety” or ”immunostimulatory moiety” refer to an adjuvant, alternatively referred to as a “payload”, that is covalently bonded to a cell-binding agent, such as an antibody construct, through an elastase-substrate, peptide linker, as described herein. The adjuvant moiety can elicit the immune response while bonded to the antibody construct or after cleavage (e.g., enzymatic cleavage) from the antibody construct following administration of an immunoconjugate to the subject. Immunoconjugates allow targeted delivery of an active adjuvant moiety while the target antigen is bound. “Adjuvant” refers to a substance capable of eliciting an immune response in a subject exposed to the adjuvant. The phrase “adjuvant moiety” refers to an adjuvant that is covalently bonded to an antibody construct, e.g., through a linker, as described herein. The adjuvant moiety can elicit the immune response while bonded to the antibody construct or after cleavage (e.g., enzymatic cleavage) from the antibody construct following administration of an immunoconjugate to the subject. The term “antibody” is used in the broadest sense and specifically encompasses monoclonal antibodies (including full length monoclonal antibodies), polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired biological activity. “Antibody fragment” and all grammatical variants thereof as used herein are defined as a portion of an intact antibody comprising the antigen binding site or variable region of the intact antibody, wherein the portion is free of the constant heavy chain domains (i.e., CH2, CH3, and CH4, depending on antibody isotype) of the Fc region of the intact antibody. Examples of antibody fragments include Fab,Fab',Fab'-SH, F(ab' )2, and Fv fragments; diabodies; any antibody fragment that is a polypeptide having a primary structure consisting of one uninterrupted sequence of contiguous amino acid residues
(referred to herein as a “single-chain antibody fragment” or “single chain polypeptide”), including without limitation (1) single-chain Fv (scFv) molecules; (2) single chain polypeptides containing only one light chain variable domain, or a fragment thereof that contains the three CDRs of the light chain variable domain, without an associated heavy chain moiety; (3) single chain polypeptides containing only one heavy chain variable region, or a fragment thereof containing the three CDRs of the heavy chain variable region, without an associated light chain moiety; (4) nanobodies comprising single Ig domains from non-human species or other specific single-domain binding modules; and (5) multispecific or multivalent structures formed from antibody fragments. In an antibody fragment comprising one or more heavy chains, the heavy chain(s) can contain any constant domain sequence (e.g., CH1 in the IgG isotype) found in a non-Fc region of an intact antibody, and/or can contain any hinge region sequence found in an intact antibody, and/or can contain a leucine zipper sequence fused to or situated in the hinge region sequence or the constant domain sequence of the heavy chain(s).
where the dashed lines are optional double bonds and the substituents are defined herein. The invention is further directed to use of such an immunoconjugates in the treatment of an illness, in particular cancer. Another aspect of the invention is an aza-bicyclic-linker compound. Another aspect of the invention is a method for treating cancer comprising administering a therapeutically effective amount of an immunoconjugate comprising an antibody linked by conjugation to one or more aza-bicyclic STING agonist moieties. Another aspect of the invention is a use of an immunoconjugate comprising an antibody linked by conjugation to one or more aza-bicyclic STING agonist moieties for treating cancer. Another aspect of the invention is a method of preparing an immunoconjugate by conjugation of one or more aza-bicyclic STING agonist moieties with an antibody. DETAILED DESCRIPTION OF THE INVENTION Reference will now be made in detail to certain embodiments of the invention, examples of which are illustrated in the accompanying structures and formulas. While the invention will be described in conjunction with the enumerated embodiments, it will be understood that they are not intended to limit the invention to those embodiments. On the contrary, the invention is
intended to cover all alternatives, modifications, and equivalents, which may be included within the scope of the invention as defined by the claims. One skilled in the art will recognize many methods and materials similar or equivalent to those described herein, which could be used in the practice of the present invention. The invention is in no way limited to the methods and materials described. DEFINITIONS The term “immunoconjugate” refers to an antibody construct that is covalently bonded to an adjuvant moiety via a linker. The terms “immunostimulant” and “immunostimulatory” are used equivalently and refer to a moiety, substance or adjuvant capable of eliciting an immune response in a subject exposed to the immunostimulatory moiety or the immunostimulatory compound after in vivo cleavage of the linker. The terms “adjuvant moiety” or ”immunostimulatory moiety” refer to an adjuvant, alternatively referred to as a “payload”, that is covalently bonded to a cell-binding agent, such as an antibody construct, through an elastase-substrate, peptide linker, as described herein. The adjuvant moiety can elicit the immune response while bonded to the antibody construct or after cleavage (e.g., enzymatic cleavage) from the antibody construct following administration of an immunoconjugate to the subject. Immunoconjugates allow targeted delivery of an active adjuvant moiety while the target antigen is bound. “Adjuvant” refers to a substance capable of eliciting an immune response in a subject exposed to the adjuvant. The phrase “adjuvant moiety” refers to an adjuvant that is covalently bonded to an antibody construct, e.g., through a linker, as described herein. The adjuvant moiety can elicit the immune response while bonded to the antibody construct or after cleavage (e.g., enzymatic cleavage) from the antibody construct following administration of an immunoconjugate to the subject. The term “antibody” is used in the broadest sense and specifically encompasses monoclonal antibodies (including full length monoclonal antibodies), polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired biological activity. “Antibody fragment” and all grammatical variants thereof as used herein are defined as a portion of an intact antibody comprising the antigen binding site or variable region of the intact antibody, wherein the portion is free of the constant heavy chain domains (i.e., CH2, CH3, and CH4, depending on antibody isotype) of the Fc region of the intact antibody. Examples of antibody fragments include Fab,Fab',Fab'-SH, F(ab' )2, and Fv fragments; diabodies; any antibody fragment that is a polypeptide having a primary structure consisting of one uninterrupted sequence of contiguous amino acid residues
(referred to herein as a “single-chain antibody fragment” or “single chain polypeptide”), including without limitation (1) single-chain Fv (scFv) molecules; (2) single chain polypeptides containing only one light chain variable domain, or a fragment thereof that contains the three CDRs of the light chain variable domain, without an associated heavy chain moiety; (3) single chain polypeptides containing only one heavy chain variable region, or a fragment thereof containing the three CDRs of the heavy chain variable region, without an associated light chain moiety; (4) nanobodies comprising single Ig domains from non-human species or other specific single-domain binding modules; and (5) multispecific or multivalent structures formed from antibody fragments. In an antibody fragment comprising one or more heavy chains, the heavy chain(s) can contain any constant domain sequence (e.g., CH1 in the IgG isotype) found in a non-Fc region of an intact antibody, and/or can contain any hinge region sequence found in an intact antibody, and/or can contain a leucine zipper sequence fused to or situated in the hinge region sequence or the constant domain sequence of the heavy chain(s).
“Antibody” refers to a polypeptide comprising an antigen binding region (including the complementarity determining region (CDRs)) from an immunoglobulin gene or fragments thereof. The term “antibody” specifically encompasses monoclonal antibodies (including full length monoclonal antibodies), polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments that exhibit the desired biological activity. An exemplary immunoglobulin (antibody) structural unit comprises a tetramer. Each tetramer is composed of two identical pairs of polypeptide chains, each pair having one “light” (about 25 kDa) and one “heavy” chain (about 50-70 kDa) connected by disulfide bonds. Each chain is composed of structural domains, which are referred to as immunoglobulin domains. These domains are classified into different categories by size and function, e.g., variable domains or regions on the light and heavy chains (VL and VH, respectively) and constant domains or regions on the light and heavy chains (CL and CH, respectively). The N-terminus of each chain defines a variable region of about 100 to 110 or more amino acids, referred to as the paratope, primarily responsible for antigen recognition, i.e., the antigen binding domain. Light chains are classified as either kappa or lambda. Heavy chains are classified as gamma, mu, alpha, delta, or epsilon, which in turn define the immunoglobulin classes, IgG, IgM, IgA, IgD and IgE, respectively. IgG antibodies are large molecules of about 150 kDa composed of four peptide chains. IgG antibodies contain two identical class γ heavy chains of about 50 kDa and two identical light chains of about 25 kDa, thus a tetrameric quaternary structure. The two heavy chains are linked to each other and to a light chain each by disulfide bonds. The resulting tetramer has two identical halves, which together form the Y-like shape. Each end of the fork contains an identical antigen binding domain. There are four IgG subclasses (IgG1, IgG2, IgG3, and IgG4)
in humans, named in order of their abundance in serum (i.e., IgG1 is the most abundant). Typically, the antigen binding domain of an antibody will be most critical in specificity and affinity of binding to cancer cells.
An antibody that targets a particular antigen includes a bispecific or multispecific antibody with at least one antigen binding region that targets the particular antigen. In some embodiments, the targeted monoclonal antibody is a bispecific antibody with at least one antigen binding region that targets tumor cells. Such antigens include but are not limited to: mesothelin, prostate specific membrane antigen (PSMA), HER2, TROP2, CEA, EGFR, 5T4,Nectin4, CD 19, CD20, CD22, CD30, CD70, B7H3, B7H4 (also known as 08E), protein tyrosine kinase 7 (PTK7), glypi can-3, RG1, fucosyl-GMl, CTLA-4, and CD44 (WO 2017/196598).
“Antibody construct” refers to an antibody or a fusion protein comprising (i) an antigen binding domain and (ii) an Fc domain.
In some embodiments, the binding agent is an antigen-binding antibody “fragment,” which is a construct that comprises at least an antigen-binding region of an antibody, alone or with other components that together constitute the antigen-binding construct. Many different types of antibody “fragments” are known in the art, including, for instance, (i) a Fab fragment, which is a monovalent fragment consisting of the VL, VH, CL, and CHi domains, (ii) a F(ab’)2 fragment, which is a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region, (iii) a Fv fragment consisting of the VL and VH domains of a single arm of an antibody, (iv) a Fab’ fragment, which results from breaking the disulfide bridge of an F(ab’)2 fragment using mild reducing conditions, (v) a disulfide-stabilized Fv fragment (dsFv), and (vi) a single chain Fv (scFv), which is a monovalent molecule consisting of the two domains of the Fv fragment (i.e., VL and VH) joined by a synthetic linker which enables the two domains to be synthesized as a single polypeptide chain.
The antibody or antibody fragments can be part of a larger construct, for example, a conjugate or fusion construct of the antibody fragment to additional regions. For instance, in some embodiments, the antibody fragment can be fused to an Fc region as described herein. In other embodiments, the antibody fragment (e.g., a Fab or scFv) can be part of a chimeric antigen receptor or chimeric T-cell receptor, for instance, by fusing to a transmembrane domain (optionally with an intervening linker or “stalk” (e.g., hinge region)) and optional intercellular signaling domain. For instance, the antibody fragment can be fused to the gamma and/or delta chains of a T-cell receptor, so as to provide a T-cell receptor like construct that binds TROP2. In yet another embodiment, the antibody fragment is part of a bispecific T-cell engager (BiTEs) comprising a CD1 or CD3 binding domain and linker.
“Epitope” means any antigenic determinant or epitopic determinant of an antigen to which an antigen binding domain binds (i.e., at the paratope of the antigen binding domain). Antigenic determinants usually consist of chemically active surface groupings of molecules, such as amino acids or sugar side chains, and usually have specific three dimensional structural characteristics, as well as specific charge characteristics.
The terms “Fc receptor” or “FcR” refer to a receptor that binds to the Fc region of an antibody. There are three main classes of Fc receptors: (1) FcγR which bind to IgG, (2) FcaR which binds to IgA, and (3) FcaR which binds to IgE. The FcγR family includes several members, such as Fcγl (CD64), FcγRIIA (CD32A), FcγRIIB (CD32B), FcγRIIIA (CD16A), and FcγRIIIB (CD16B). The Fey receptors differ in their affinity for IgG and also have different affinities for the IgG subclasses (e.g., IgG1, IgG2, IgG3, and IgG4).
As used herein, the phrase “immune checkpoint inhibitor” refers to any modulator that inhibits the activity of the immune checkpoint molecule. Immune checkpoint inhibitors can include, but are not limited to, immune checkpoint molecule binding proteins, small molecule inhibitors, antibodies (including bispecific and multispecific antibodies with at least one antigen binding region that targets an immune checkpoint protein, e.g., bispecific or multispecific antibodies that do not exclusively target immune checkpoint proteins, as well as antibodies that are dual immunomodulators (simultaneous targeting two immunomodulating targets), which result in blockade of inhibitory targets, depletion of suppressive cells, and/or activation of effector cells; tumor-targeted immunomodulators (directs potent costimulation to the tumor- infiltrating immune cells by targeting a tumor antigen and costimulatory molecules such as CD40 or 4- IBB); NK-cell redirectors (redirects NK cells to malignant cells by targeting a tumor antigen and CD16A); or T-cell redirectors (redirects T cells to malignant cells by targeting a tumor antigen and CD3)), antibody-derivatives (including Fc fusions, Fab fragments, and scFvs), antibody-drug conjugates, antisense oligonucleotides, siRNA, aptamers, peptides and peptide mimetics.
Nucleic acid or amino acid sequence “identity,” as referenced herein, can be determined by comparing a nucleic acid or amino acid sequence of interest to a reference nucleic acid or amino acid sequence. The percent identity is the number of nucleotides or amino acid residues that are the same (i.e., that are identical) as between the optimally aligned sequence of interest and the reference sequence divided by the length of the longest sequence (i.e., the length of either the sequence of interest or the reference sequence, whichever is longer). Alignment of sequences and calculation of percent identity can be performed using available software programs. Examples of such programs include CLUSTAL-W, T-Coffee, and ALIGN (for alignment of nucleic acid and amino acid sequences), BLAST programs (e.g., BLAST 2.1,
BL2SEQ, BLASTp, BLASTn, and the like) and FASTA programs (e.g., FASTA3x, FASTM, and SSEARCH) (for sequence alignment and sequence similarity searches). Sequence alignment algorithms also are disclosed in, for example, Altschul et al., J. Molecular Biol., 215(3): 403-410 (1990), Beigert et al., Proc. Natl. Acad. Sci. USA, 106(10): 3770-3775 (2009), Durbin et al., eds., Biological Sequence Analysis: Probalistic Models of Proteins and Nucleic Acids, Cambridge University Press, Cambridge, UK (2009), Soding, Bioinformatics, 21(7): 951- 960 (2005), Altschul et al., Nucleic Acids Res., 25(17): 3389-3402 (1997), and Gusfield, Algorithms on Strings, Trees and Sequences, Cambridge University Press, Cambridge UK (1997)). Percent (%) identity of sequences can be also calculated, for example, as 100 x [(identical positions)/min(TGA, TGB)], where TGA and TGB are the sum of the number of residues and internal gap positions in peptide sequences A and B in the alignment that minimizes TGA and TGB. See, e.g., Russell et al., J. Mol Biol., 244: 332-350 (1994). The binding agent comprises Ig heavy and light chain variable region polypeptides that together form the antigen binding site. Each of the heavy and light chain variable regions are polypeptides comprising three complementarity determining regions (CDR1, CDR2, and CDR3) connected by framework regions. The binding agent can be any of a variety of types of binding agents known in the art that comprise Ig heavy and light chains. For instance, the binding agent can be an antibody, an antigen-binding antibody “fragment,” or a T-cell receptor. “Amino acid” refers to any monomeric unit that can be incorporated into a peptide, polypeptide, or protein. Amino acids include naturally-occurring α-amino acids and their stereoisomers, as well as unnatural (non-naturally occurring) amino acids and their stereoisomers. “Stereoisomers” of a given amino acid refer to isomers having the same molecular formula and intramolecular bonds but different three-dimensional arrangements of bonds and atoms (e.g., an L-amino acid and the corresponding D-amino acid). The amino acids can be glycosylated (e.g., N-linked glycans, O-linked glycans, phosphoglycans, C-linked glycans, or glypication) or deglycosylated. Amino acids may be referred to herein by either the commonly known three letter symbols or by the one-letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. Naturally-occurring amino acids are those encoded by the genetic code, as well as those amino acids that are later modified, e.g., hydroxyproline, γ-carboxyglutamate, and O-phosphoserine. Naturally-occurring α-amino acids include, without limitation, alanine (Ala), cysteine (Cys), aspartic acid (Asp), glutamic acid (Glu), phenylalanine (Phe), glycine (Gly), histidine (His), isoleucine (Ile), arginine (Arg), lysine (Lys), leucine (Leu), methionine (Met), asparagine (Asn), proline (Pro), glutamine (Gln), serine (Ser), threonine (Thr), valine (Val), tryptophan (Trp), tyrosine (Tyr), and combinations thereof. Stereoisomers of naturally-
occurring α-amino acids include, without limitation, D-alanine (D-Ala), D-cysteine (D-Cys), D-aspartic acid (D-Asp), D-glutamic acid (D-Glu), D-phenylalanine (D-Phe), D-histidine (D-His), D-isoleucine (D-Ile), D-arginine (D-Arg), D-lysine (D-Lys), D-leucine (D-Leu), D-methionine (D-Met), D-asparagine (D-Asn), D-proline (D-Pro), D-glutamine (D-Gln), D-serine (D-Ser), D-threonine (D-Thr), D-valine (D-Val), D-tryptophan (D-Trp), D-tyrosine (D-Tyr), and combinations thereof. Naturally-occurring amino acids include those formed in proteins by post-translational modification, such as citrulline (Cit). Unnatural (non-naturally occurring) amino acids include, without limitation, amino acid analogs, amino acid mimetics, synthetic amino acids, N-substituted glycines, and N-methyl amino acids in either the L- or D-configuration that function in a manner similar to the naturally- occurring amino acids. For example, “amino acid analogs” can be unnatural amino acids that have the same basic chemical structure as naturally-occurring amino acids (i.e., a carbon that is bonded to a hydrogen, a carboxyl group, an amino group) but have modified side-chain groups or modified peptide backbones, e.g., homoserine, norleucine, methionine sulfoxide, and methionine methyl sulfonium. “Amino acid mimetics” refer to chemical compounds that have a structure that is different from the general chemical structure of an amino acid, but that functions in a manner similar to a naturally-occurring amino acid. “Linker” refers to a functional group that covalently bonds two or more moieties in a compound or material. The linker serves to covalently bond an adjuvant moiety to an antibody construct in an immunoconjugate. “Linking moiety” refers to a functional group that covalently bonds two or more moieties in a compound or material. For example, the linking moiety can serve to covalently bond an adjuvant moiety to an antibody in an immunoconjugate. Useful bonds for connecting linking moieties to proteins and other materials include, but are not limited to, amides, amines, esters, carbamates, ureas, thioethers, thiocarbamates, thiocarbonates, and thioureas. “Divalent” refers to a chemical moiety that contains two points of attachment for linking two functional groups; polyvalent linking moieties can have additional points of attachment for linking further functional groups. Divalent radicals may be denoted with the suffix “diyl”. For example, divalent linking moieties include divalent polymer moieties such as divalent poly(ethylene glycol), divalent cycloalkyl, divalent heterocycloalkyl, divalent aryl, and divalent heteroaryl group. A “divalent cycloalkyl, heterocycloalkyl, aryl, or heteroaryl group” refers to a cycloalkyl, heterocycloalkyl, aryl, or heteroaryl group having two points of attachment for covalently linking two moieties in a molecule or material. Cycloalkyl, heterocycloalkyl, aryl, or heteroaryl groups can be substituted or unsubstituted. Cycloalkyl, heterocycloalkyl, aryl, or
heteroaryl groups can be substituted with one or more groups selected from halo, hydroxy, amino, alkylamino, amido, acyl, nitro, cyano, and alkoxy. A wavy line (“
 ”) represents a point of attachment of the specified chemical moiety. If the specified chemical moiety has two wavy lines (“
”) represents a point of attachment of the specified chemical moiety. If the specified chemical moiety has two wavy lines (“
 ”) present, it will be understood that the chemical moiety can be used bilaterally, i.e., as read from left to right or from right to left. In some embodiments, a specified moiety having two wavy lines (“
”) present, it will be understood that the chemical moiety can be used bilaterally, i.e., as read from left to right or from right to left. In some embodiments, a specified moiety having two wavy lines (“
 ”) present is considered to be used as read from left to right. “Alkyl” refers to a straight (linear) or branched, saturated, aliphatic radical having the number of carbon atoms indicated. Alkyl can include any number of carbons, for example from one to six, one to eight, one to twelve or one to twenty. Examples of alkyl groups include, but are not limited to, methyl (Me, -CH3), ethyl (Et, -CH2CH3), 1-propyl (n-Pr, n-propyl, - CH2CH2CH3), 2-propyl (i-Pr, i-propyl, -CH(CH3)2), 1-butyl (n-Bu, n-butyl, -CH2CH2CH2CH3), 2-methyl-1-propyl (i-Bu, i-butyl, -CH2CH(CH3)2), 2-butyl (s-Bu, s-butyl, -CH(CH3)CH2CH3), 2- methyl-2-propyl (t-Bu, t-butyl, -C(CH3)3), 1-pentyl (n-pentyl, -CH2CH2CH2CH2CH3), 2-pentyl (-CH(CH3)CH2CH2CH3), 3-pentyl (-CH(CH2CH3)2), 2-methyl-2-butyl (-C(CH3)2CH2CH3), 3- methyl-2-butyl (-CH(CH3)CH(CH3)2), 3-methyl-1-butyl (-CH2CH2CH(CH3)2), 2-methyl-1-butyl (-CH2CH(CH3)CH2CH3), 1-hexyl (-CH2CH2CH2CH2CH2CH3), 2-hexyl (- CH(CH3)CH2CH2CH2CH3), 3-hexyl (-CH(CH2CH3)(CH2CH2CH3)), 2-methyl-2-pentyl (- C(CH3)2CH2CH2CH3), 3-methyl-2-pentyl (-CH(CH3)CH(CH3)CH2CH3), 4-methyl-2-pentyl (- CH(CH3)CH2CH(CH3)2), 3-methyl-3-pentyl (-C(CH3)(CH2CH3)2), 2-methyl-3-pentyl (- CH(CH2CH3)CH(CH3)2), 2,3-dimethyl-2-butyl (-C(CH3)2CH(CH3)2), 3,3-dimethyl-2-butyl (- CH(CH3)C(CH3)3, 1-heptyl, 1-octyl, and the like. Alkyl groups can be substituted or unsubstituted. Substituted alkyl groups can be substituted with one or more groups selected from halo, hydroxy, amino, oxo (=O), alkylamino, amido, acyl, nitro, cyano, and alkoxy. Substituted alkyl groups can be geminally substituted where a carbon atom of the alkyl forms a spiro, cycloalkyl group such as cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. The term “alkyldiyl” refers to a divalent alkyl radical. Examples of alkyldiyl groups include, but are not limited to, methylene (-CH2-), ethylene (-CH2CH2-), propylene (- CH2CH2CH2-), and the like. An alkyldiyl group may also be referred to as an “alkylene” group. Alkyldiyl groups can be substituted or unsubstituted. Substituted alkyldiyl groups can be substituted with one or more groups selected from halo, hydroxy, amino, oxo (=O), alkylamino, amido, acyl, nitro, cyano, and alkoxy. Substituted alkyldiyl groups can be geminally substituted where a carbon atom of the alkyl forms a spiro, cycloalkyl group such as cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl.
“Alkenyl” refers to a straight (linear) or branched, unsaturated, aliphatic radical having the number of carbon atoms indicated and at least one carbon-carbon double bond, sp2. Alkenyl can include from two to about 12 or more carbons atoms. Alkenyl groups are radicals having “cis” and “trans” orientations, or alternatively, “E” and “Z” orientations. Examples include, but are not limited to, ethylenyl or vinyl (-CH=CH2), allyl (-CH2CH=CH2). butenyl, pentenyl, and isomers thereof. Alkenyl groups can be substituted or unsubstituted. “Substituted alkenyl” groups can be substituted with one or more groups selected from halo, hydroxy, amino, oxo (=O), alkylamino, amido, acyl, nitro, cyano, and alkoxy. The terms “alkenylene” or “alkenyldiyl” refer to a linear or branched-chain divalent hydrocarbon radical. Examples include, but are not limited to, ethylenylene or vinylene (- CH=CH-), allyl (-CH2CH=CH-), and the like. “Alkynyl” refers to a straight (linear) or branched, unsaturated, aliphatic radical having the number of carbon atoms indicated and at least one carbon-carbon triple bond, sp. Alkynyl can include from two to about 12 or more carbons atoms. For example, C2-C6 alkynyl includes, but is not limited to ethynyl (-C ≡CH), propynyl (propargyl, -CH2C ≡CH), butynyl, pentynyl, hexynyl, and isomers thereof Alkynyl groups can be substituted or unsubstituted. “Substituted alkynyl” groups can be substituted with one or more groups selected from halo, hydroxy, amino, oxo (=O), alkylamino, amido, acyl, nitro, cyano, and alkoxy. The term “alkynylene” or “alkynyldiyl” refer to a divalent alkynyl radical. "Heteroalkyl" or “heteroalkylene” refer to a monovalent, straight or branched chain alkyl group, as defined above, comprising at least one heteroatom including but not limited to Si, N, O, P or S within the alkyl chain or at a terminus of the alkyl chain. In some embodiments, a heteroatom is within the alkyl chain. In other embodiments, a heteroatom is at a terminus of the alkylene and thus serves to join the alkyl to the remainder of the molecule. Unless stated otherwise specifically in the specification, a heteroalkyl group is optionally substituted. For example, heteroalkyl groups can be substituted with 1-6 fluoro (F) substituents, for example, on the carbon backbone (as − CHF− or −CF2−) or on terminal carbons of straight chain or branched heteroalkyls (such as −CHF2 or −CF3). Examples of heteroalkyl groups include, but are not limited to, − CH2CH2OCH3, −CH2CH2NHCH3, −CH2CH2N(CH3)2, −C(=O)NHCH2CH2NHCH3, − C(=O)N(CH3)CH2CH2N(CH3)2, −C(=O)NHCH2CH2NHC(=O)CH2CH3, − C(=O)N(CH3)CH2CH2N(CH3)C(=O)CH2CH3, −OCH2CH2CH2NH(CH3), − OCH2CH2CH2N(CH3)2, −OCH2CH2CH2NHC(=O)CH2CH3, −
OCH2CH2CH2N(CH3)C(=O)CH2CH3, −CH2CH2CH2NH(CH3), −OCH2CH2CH2N(CH3)2, −CH2CH2CH2NHC(=O)CH2CH3, −CH2CH2CH2N(CH3)C(=O)CH2CH3, −CH2SCH2CH3, −CH2CH2S(O)CH3, −NHCH2CH2NHC(=O)CH2CH3, −CH2CH2S(O)2CH3, − CH2CH2OCF3, and −Si(CH3)3. Up to two heteroatoms may be consecutive, such as, for example, −CH2NHOCH3 and −CH2OSi(CH3)3. A terminal polyethylene glycol (PEG) moiety is a type of heteroalkyl group. Exemplary heteroalkyl groups also include ethylene oxide (e.g., polyethylene oxide), propylene oxide, amino acid chains (i.e., short to medium length peptides such as containing 1-15 amino acids), and alkyl chains connected via a variety of functional groups such as amides, disulfides, ketones, phosphonates, phosphates, sulfates, sulfones, sulfonamides, esters, ethers, -S-, carbamates, ureas, thioureas, anhydrides, or the like (including combinations thereof). In some embodiments, a heteroalkyl group includes a polyamino acid having 1-10 amino acids. In some embodiments, a heteroalkyl group includes a polyamino acid having 1-5 amino acids. Exemplary heteroalkyl groups include a solubilizing group (SolG) comprising one or more units of polyglycine, polysarcosine, polyethyleneoxy (PEG), and a glycoside, or combinations thereof. "Heteroalkenyl" refers to a heteroalkyl group, as defined above, that contains at least one carbon-carbon double bond. "Heteroalkynyl" refers to a heteroalkyl group, as defined above, that contains at least one carbon-carbon triple bond. “Heteroalkyldiyl” refers to a divalent form of a heteroalkyl group as defined above. Examples of heteroalkylene groups include, but are not limited to, − CH2CH2OCH2−, −CH2CH2OCF2−, −CH2CH2NHCH2−, −C(=O)NHCH2CH2NHCH2−, − C(=O)N(CH3)CH2CH2N(CH3)CH2−, −C(=O)NHCH2CH2NHC(=O)CH2CH2−, − C(=O)N(CH3)CH2CH2N(CH3)C(=O)CH2CH2−, −OCH2CH2OCH2CH2−, − OCH2CH2OCH2C(=O)−, −OCH2CH2OCH2CH2C(=O)−, −OCH2CH2NHCH2−, − OCH2CH2N(CH3)CH2−, −OCH2CH2CH2NHCH2−, −OCH2CH2CH2N(CH3)CH2−, − OCH2CH2CH2NHC(=O)CH2CH2−, −OCH2CH2CH2N(CH3)C(=O)CH2CH2−, − CH2CH2CH2NHCH2−-, −CH2CH2CH2N(CH3)CH2−, −CH2CH2CH2NHC(=O)CH2CH2−, −CH2CH2CH2N(CH3)C(=O)CH2CH2−, −CH2CH2NHC(=O)−, −CH2CH2N(CH3)CH2−, − CH2CH2N+(CH3)2−, −NHCH2CH2(NH2)CH2−, and −NHCH2CH2(NHCH3)CH2−. A divalent polyethylene glycol (PEG) moiety with one to about 50 units of −OCH2CH2− is a
type of heteroalkyldiyl group. “Heteroalkenyldiyl” refers to a divalent form of a heteroalkenyl group. “Heteroalkynyldiyl” refers to a divalent form of a heteroalkynyl group. The terms “carbocycle”, “carbocyclyl”, “carbocyclic ring” and “cycloalkyl” refer to a saturated or partially unsaturated, monocyclic, fused bicyclic, spiro, or bridged polycyclic ring assembly containing from 3 to 12 ring atoms, or the number of atoms indicated. Saturated monocyclic carbocyclic rings include, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cyclooctyl. Saturated bicyclic and polycyclic carbocyclic rings include, for example, norbornane, [2.2.2] bicyclooctane, decahydronaphthalene and adamantane. Carbocyclic groups can also be partially unsaturated, having one or more double or triple bonds in the ring. Representative carbocyclic groups that are partially unsaturated include, but are not limited to, cyclobutene, cyclopentene, cyclohexene, cyclohexadiene (1,3- and 1,4-isomers), cycloheptene, cycloheptadiene, cyclooctene, cyclooctadiene (1,3-, 1,4- and 1,5-isomers), norbornene, and norbornadiene. The term “cycloalkyldiyl” refers to a divalent cycloalkyl radical. “Aryl” refers to a monovalent aromatic hydrocarbon radical of 6-20 carbon atoms (C6− C20) derived by the removal of one hydrogen atom from a single carbon atom of a parent aromatic ring system.. Aryl groups can be monocyclic, fused to form bicyclic or tricyclic groups, or linked by a bond to form a biaryl group. Representative aryl groups include phenyl, naphthyl and biphenyl. Other aryl groups include benzyl, having a methylene linking group. Some aryl groups have from 6 to 12 ring members, such as phenyl, naphthyl or biphenyl. Other aryl groups have from 6 to 10 ring members, such as phenyl or naphthyl. The terms “arylene” or “aryldiyl” mean a divalent aromatic hydrocarbon radical of 6-20 carbon atoms (C6−C20) derived by the removal of two hydrogen atom from a two carbon atoms of a parent aromatic ring system. Some aryldiyl groups are represented in the exemplary structures as “Ar”. Aryldiyl includes bicyclic radicals comprising an aromatic ring fused to a saturated, partially unsaturated ring, or aromatic carbocyclic ring. Typical aryldiyl groups include, but are not limited to, radicals derived from benzene (phenyldiyl), substituted benzenes, naphthalene, anthracene, biphenylene, indenylene, indanylene, 1,2-dihydronaphthalene, 1,2,3,4- tetrahydronaphthyl, and the like. Aryldiyl groups are also referred to as “arylene”, and are optionally substituted with one or more substituents described herein. The terms “heterocycle”, “heterocyclyl”, and “heterocyclic ring” are used interchangeably herein and refer to a saturated or a partially unsaturated (i.e., having one or more double and/or triple bonds within the ring) carbocyclic radical of 3 to about 20 ring atoms in which at least one ring atom is a heteroatom selected from nitrogen, oxygen, phosphorus and
sulfur, the remaining ring atoms being C, where one or more ring atoms is optionally substituted independently with one or more substituents described below. A heterocycle may be a monocycle having 3 to 7 ring members (2 to 6 carbon atoms and 1 to 4 heteroatoms selected from N, O, P, and S) or a bicycle having 7 to 10 ring members (4 to 9 carbon atoms and 1 to 6 heteroatoms selected from N, O, P, and S), for example: a bicyclo [4,5], [5,5], [5,6], or [6,6] system. Heterocycles are described in Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W.A. Benjamin, New York, 1968), particularly Chapters 1, 3, 4, 6, 7, and 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28; and J. Am. Chem. Soc. (1960) 82:5566. “Heterocyclyl” also includes radicals where heterocycle radicals are fused with a saturated, partially unsaturated ring, or aromatic carbocyclic or heterocyclic ring. Examples of heterocyclic rings include, but are not limited to, morpholin-4-yl, piperidin-1-yl, piperazinyl, piperazin-4-yl-2-one, piperazin-4-yl-3-one, pyrrolidin-1-yl, thiomorpholin-4-yl, S- dioxothiomorpholin-4-yl, azocan-1-yl, azetidin-1-yl, octahydropyrido[1,2-a]pyrazin-2-yl, [1,4]diazepan-1-yl, pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl, homopiperazinyl, azetidinyl, oxetanyl, thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl, diazepinyl, thiazepinyl, 2-pyrrolinyl, 3- pyrrolinyl, indolinyl, 2H-pyranyl, 4H-pyranyl, dioxanyl, 1,3-dioxolanyl, pyrazolinyl, dithianyl, dithiolanyl, dihydropyranyl, dihydrothienyl, dihydrofuranyl, pyrazolidinylimidazolinyl, imidazolidinyl, 3-azabicyco[3.1.0]hexanyl, 3-azabicyclo[4.1.0]heptanyl, azabicyclo[2.2.2]hexanyl, 3H-indolyl quinolizinyl and N-pyridyl ureas. Spiro heterocyclyl moieties are also included within the scope of this definition. Examples of spiro heterocyclyl moieties include azaspiro[2.5]octanyl and azaspiro[2.4]heptanyl. Examples of a heterocyclic group wherein 2 ring atoms are substituted with oxo (=O) moieties are pyrimidinonyl and 1,1- dioxo-thiomorpholinyl. The heterocycle groups herein are optionally substituted independently with one or more substituents described herein. The term “heterocyclyldiyl” refers to a divalent, saturated or a partially unsaturated (i.e., having one or more double and/or triple bonds within the ring) carbocyclic radical of 3 to about 20 ring atoms in which at least one ring atom is a heteroatom selected from nitrogen, oxygen, phosphorus and sulfur, the remaining ring atoms being C, where one or more ring atoms is optionally substituted independently with one or more substituents as described. Examples of 5- membered and 6-membered heterocyclyldiyls include morpholinyldiyl, piperidinyldiyl, piperazinyldiyl, pyrrolidinyldiyl, dioxanyldiyl, thiomorpholinyldiyl, and S- dioxothiomorpholinyldiyl.
The term “heteroaryl” refers to a monovalent aromatic radical of 5-, 6-, or 7-membered rings, and includes fused ring systems (at least one of which is aromatic) of 5-20 atoms, containing one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur. Examples of heteroaryl groups are pyridinyl (including, for example, 2-hydroxypyridinyl), imidazolyl, imidazopyridinyl, pyrimidinyl (including, for example, 4-hydroxypyrimidinyl), pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxadiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl, thiadiazolyl, thiadiazolyl, furazanyl, benzofurazanyl, benzothiophenyl, benzothiazolyl, benzoxazolyl, quinazolinyl, quinoxalinyl, naphthyridinyl, and furopyridinyl. Heteroaryl groups are optionally substituted independently with one or more substituents described herein. The term “heteroaryldiyl” refers to a divalent aromatic radical of 5-, 6-, or 7-membered rings, and includes fused ring systems (at least one of which is aromatic) of 5-20 atoms, containing one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur. Examples of 5-membered and 6-membered heteroaryldiyls include pyridyldiyl, imidazolyldiyl, pyrimidinyldiyl, pyrazolyldiyl, triazolyldiyl, pyrazinyldiyl, tetrazolyldiyl, furyldiyl, thienyldiyl, isoxazolyldiyldiyl, thiazolyldiyl, oxadiazolyldiyl, oxazolyldiyl, isothiazolyldiyl, and pyrrolyldiyl. The heterocycle or heteroaryl groups may be carbon (carbon-linked), or nitrogen (nitrogen-linked) bonded where such is possible. By way of example and not limitation, carbon bonded heterocycles or heteroaryls are bonded at position 2, 3, 4, 5, or 6 of a pyridine, position 3, 4, 5, or 6 of a pyridazine, position 2, 4, 5, or 6 of a pyrimidine, position 2, 3, 5, or 6 of a pyrazine, position 2, 3, 4, or 5 of a furan, tetrahydrofuran, thiofuran, thiophene, pyrrole or tetrahydropyrrole, position 2, 4, or 5 of an oxazole, imidazole or thiazole, position 3, 4, or 5 of an isoxazole, pyrazole, or isothiazole, position 2 or 3 of an aziridine, position 2, 3, or 4 of an azetidine, position 2, 3, 4, 5, 6, 7, or 8 of a quinoline or position 1, 3, 4, 5, 6, 7, or 8 of an isoquinoline. By way of example and not limitation, nitrogen bonded heterocycles or heteroaryls are bonded at position 1 of an aziridine, azetidine, pyrrole, pyrrolidine, 2-pyrroline, 3-pyrroline, imidazole, imidazolidine, 2-imidazoline, 3-imidazoline, pyrazole, pyrazoline, 2-pyrazoline, 3- pyrazoline, piperidine, piperazine, indole, indoline, 1H-indazole, position 2 of a isoindole, or isoindoline, position 4 of a morpholine, and position 9 of a carbazole, or β-carboline. The terms “halo” and “halogen,” by themselves or as part of another substituent, refer to a fluorine, chlorine, bromine, or iodine atom.
The term “carbonyl,” by itself or as part of another substituent, refers to C(=O) or – C(=O)–, i.e., a carbon atom double-bonded to oxygen and bound to two other groups in the moiety having the carbonyl. As used herein, the phrase “quaternary ammonium salt” refers to a tertiary amine that has been quaternized with an alkyl substituent (e.g., a C1-C4 alkyl such as methyl, ethyl, propyl, or butyl). The terms “treat,” “treatment,” and “treating” refer to any indicia of success in the treatment or amelioration of an injury, pathology, condition (e.g., cancer), or symptom (e.g., cognitive impairment), including any objective or subjective parameter such as abatement; remission; diminishing of symptoms or making the symptom, injury, pathology, or condition more tolerable to the patient; reduction in the rate of symptom progression; decreasing the frequency or duration of the symptom or condition; or, in some situations, preventing the onset of the symptom. The treatment or amelioration of symptoms can be based on any objective or subjective parameter, including, for example, the result of a physical examination. The terms “cancer,” “neoplasm,” and “tumor” are used herein to refer to cells which exhibit autonomous, unregulated growth, such that the cells exhibit an aberrant growth phenotype characterized by a significant loss of control over cell proliferation. Cells of interest for detection, analysis, and/or treatment in the context of the invention include cancer cells (e.g., cancer cells from an individual with cancer), malignant cancer cells, pre-metastatic cancer cells, metastatic cancer cells, and non-metastatic cancer cells. Cancers of virtually every tissue are known. The phrase “cancer burden” refers to the quantum of cancer cells or cancer volume in a subject. Reducing cancer burden accordingly refers to reducing the number of cancer cells or the cancer cell volume in a subject. The term “cancer cell” as used herein refers to any cell that is a cancer cell (e.g., from any of the cancers for which an individual can be treated, e.g., isolated from an individual having cancer) or is derived from a cancer cell, e.g., clone of a cancer cell. For example, a cancer cell can be from an established cancer cell line, can be a primary cell isolated from an individual with cancer, can be a progeny cell from a primary cell isolated from an individual with cancer, and the like. In some embodiments, the term can also refer to a portion of a cancer cell, such as a sub-cellular portion, a cell membrane portion, or a cell lysate of a cancer cell. Many types of cancers are known to those of skill in the art, including solid tumors such as carcinomas, sarcomas, glioblastomas, melanomas, lymphomas, and myelomas, and circulating cancers such as leukemias. As used herein, the term “cancer” includes any form of cancer, including but not limited to, solid tumor cancers (e.g., skin, lung, prostate, breast, gastric, bladder, colon, ovarian, pancreas, kidney, liver, glioblastoma, medulloblastoma, leiomyosarcoma, head & neck
squamous cell carcinomas, melanomas, and neuroendocrine) and liquid cancers (e.g., hematological cancers); carcinomas; soft tissue tumors; sarcomas; teratomas; melanomas; leukemias; lymphomas; and brain cancers, including minimal residual disease, and including both primary and metastatic tumors. The “pathology” of cancer includes all phenomena that compromise the well-being of the patient. This includes, without limitation, abnormal or uncontrollable cell growth, metastasis, interference with the normal functioning of neighboring cells, release of cytokines or other secretory products at abnormal levels, suppression or aggravation of inflammatory or immunological response, neoplasia, premalignancy, malignancy, and invasion of surrounding or distant tissues or organs, such as lymph nodes. As used herein, the phrases “cancer recurrence” and “tumor recurrence,” and grammatical variants thereof, refer to further growth of neoplastic or cancerous cells after diagnosis of cancer. Particularly, recurrence may occur when further cancerous cell growth occurs in the cancerous tissue. “Tumor spread,” similarly, occurs when the cells of a tumor disseminate into local or distant tissues and organs, therefore, tumor spread encompasses tumor metastasis. “Tumor invasion” occurs when the tumor growth spread out locally to compromise the function of involved tissues by compression, destruction, or prevention of normal organ function. As used herein, the term “metastasis” refers to the growth of a cancerous tumor in an organ or body part, which is not directly connected to the organ of the original cancerous tumor. Metastasis will be understood to include micrometastasis, which is the presence of an undetectable amount of cancerous cells in an organ or body part that is not directly connected to the organ of the original cancerous tumor. Metastasis can also be defined as several steps of a process, such as the departure of cancer cells from an original tumor site, and migration and/or invasion of cancer cells to other parts of the body. The phrases “effective amount” and “therapeutically effective amount” refer to a dose or amount of a substance such as an immunoconjugate that produces therapeutic effects for which it is administered. The exact dose will depend on the purpose of the treatment, and will be ascertainable by one skilled in the art using known techniques (see, e.g., Lieberman, Pharmaceutical Dosage Forms (vols.1-3, 1992); Lloyd, The Art, Science and Technology of Pharmaceutical Compounding (1999); Pickar, Dosage Calculations (1999); Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th Edition (McGraw-Hill, 2006); and Remington: The Science and Practice of Pharmacy, 22nd Edition, (Pharmaceutical Press, London, 2012)). In the case of cancer, the therapeutically effective amount of the immunoconjugate may reduce the number of cancer cells; reduce the tumor size; inhibit (i.e.,
slow to some extent and preferably stop) cancer cell infiltration into peripheral organs; inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; inhibit, to some extent, tumor growth; and/or relieve to some extent one or more of the symptoms associated with the cancer. To the extent the immunoconjugate may prevent growth and/or kill existing cancer cells, it may be cytostatic and/or cytotoxic. For cancer therapy, efficacy can, for example, be measured by assessing the time to disease progression (TTP) and/or determining the response rate (RR) “Recipient,” “individual,” “subject,” “host,” and “patient” are used interchangeably and refer to any mammalian subject for whom diagnosis, treatment, or therapy is desired (e.g., humans). “Mammal” for purposes of treatment refers to any animal classified as a mammal, including humans, domestic and farm animals, and zoo, sports, or pet animals, such as dogs, horses, cats, cows, sheep, goats, pigs, camels, etc. In certain embodiments, the mammal is human. The phrase “synergistic adjuvant” or “synergistic combination” in the context of this invention includes the combination of two immune modulators such as a receptor agonist, cytokine, and adjuvant polypeptide, that in combination elicit a synergistic effect on immunity relative to either administered alone. Particularly, the immunoconjugates disclosed herein comprise synergistic combinations of the claimed adjuvant and antibody construct. These synergistic combinations upon administration elicit a greater effect on immunity, e.g., relative to when the antibody construct or adjuvant is administered in the absence of the other moiety. Further, a decreased amount of the immunoconjugate may be administered (as measured by the total number of antibody constructs or the total number of adjuvants administered as part of the immunoconjugate) compared to when either the antibody construct or adjuvant is administered alone. As used herein, the term “administering” refers to parenteral, intravenous, intraperitoneal, intramuscular, intratumoral, intralesional, intranasal, or subcutaneous administration, oral administration, administration as a suppository, topical contact, intrathecal administration, or the implantation of a slow-release device, e.g., a mini-osmotic pump, to the subject. The terms “about” and “around,” as used herein to modify a numerical value, indicate a close range surrounding the numerical value. Thus, if “X” is the value, “about X” or “around X” indicates a value of from 0.9X to 1.1X, e.g., from 0.95X to 1.05X or from 0.99X to 1.01X. A reference to “about X” or “around X” specifically indicates at least the values X, 0.95X, 0.96X, 0.97X, 0.98X, 0.99X, 1.01X, 1.02X, 1.03X, 1.04X, and 1.05X. Accordingly, “about X” and “around X” are intended to teach and provide written description support for a claim limitation of, e.g., “0.98X.”
ANTIBODIES The immunoconjugate of the invention comprises an antibody. Included in the scope of the embodiments of the invention are functional variants of the antibody constructs or antigen binding domain described herein. The term “functional variant” as used herein refers to an antibody construct having an antigen binding domain with substantial or significant sequence identity or similarity to a parent antibody construct or antigen binding domain, which functional variant retains the biological activity of the antibody construct or antigen binding domain of which it is a variant. Functional variants encompass, for example, those variants of the antibody constructs or antigen binding domain described herein (the parent antibody construct or antigen binding domain) that retain the ability to recognize target cells expressing a tumor-associated antigen or cell surface receptor to a similar extent, the same extent, or to a higher extent, as the parent antibody construct or antigen binding domain. In reference to the antibody construct or antigen binding domain, the functional variant can, for instance, be at least about 30%, about 50%, about 75%, about 80%, about 85%, about 90%, about 91%, about 92%, about 93%, about 94%, about 95%, about 96%, about 97%, about 98%, about 99% or more identical in amino acid sequence to the antibody construct or antigen binding domain. A functional variant can, for example, comprise the amino acid sequence of the parent antibody construct or antigen binding domain with at least one conservative amino acid substitution. Alternatively, or additionally, the functional variants can comprise the amino acid sequence of the parent antibody construct or antigen binding domain with at least one non- conservative amino acid substitution. In this case, it is preferable for the non-conservative amino acid substitution to not interfere with or inhibit the biological activity of the functional variant. The non-conservative amino acid substitution may enhance the biological activity of the functional variant, such that the biological activity of the functional variant is increased as compared to the parent antibody construct or antigen binding domain. The antibodies comprising the immunoconjugates of the invention include Fc engineered variants. In some embodiments, the mutations in the Fc region that result in modulated binding to one or more Fc receptors can include one or more of the following mutations: SD (S239D), SDIE (S239D/I332E), SE (S267E), SELF (S267E/L328F), SDIE (S239D/I332E), SDIEAL (S239D/I332E/A330L), GA (G236A), ALIE (A330L/I332E), GASDALIE (G236A/S239D/A330L/I332E), V9 (G237D/P238D/P271G/A330R), and V11 (G237D/P238D/H268D/P271G/A330R), and/or one or more mutations at the following amino acids: E345R, E233, G237, P238, H268, P271, L328 and A330. Additional Fc region modifications for modulating Fc receptor binding are described in, for example, US
2016/0145350, US 7416726 and US 5624821, which are hereby incorporated by reference in their entireties herein. The antibodies comprising the immunoconjugates of the invention include glycan variants, such as afucosylation. In some embodiments, the Fc region of the binding agents are modified to have an altered glycosylation pattern of the Fc region compared to the native non-modified Fc region. In some embodiments, the antibodies in the immunoconjugates contain a modified Fc region, wherein the modification modulates the binding of the Fc region to one or more Fc receptors. In some embodiments, the antibodies in the immunoconjugates (e.g., antibodies conjugated to at least two adjuvant moieties) contain one or more modifications (e.g., amino acid insertion, deletion, and/or substitution) in the Fc region that results in modulated binding (e.g., increased binding or decreased binding) to one or more Fc receptors (e.g., FcγRI (CD64), FcγRIIA (CD32A), FcγRIIB (CD32B), FcγRIIIA (CD16a), and/or FcγRIIIB (CD16b)) as compared to the native antibody lacking the mutation in the Fc region. In some embodiments, the antibodies in the immunoconjugates contain one or more modifications (e.g., amino acid insertion, deletion, and/or substitution) in the Fc region that reduce the binding of the Fc region of the antibody to FcγRIIB. In some embodiments, the antibodies in the immunoconjugates contain one or more modifications (e.g., amino acid insertion, deletion, and/or substitution) in the Fc region of the antibody that reduce the binding of the antibody to FcγRIIB while maintaining the same binding or having increased binding to FcγRI (CD64), FcγRIIA (CD32A), and/or FcRγIIIA (CD16a) as compared to the native antibody lacking the mutation in the Fc region. In some embodiments, the antibodies in the immunoconjugates contain one of more modifications in the Fc region that increase the binding of the Fc region of the antibody to FcγRIIB. In some embodiments, the modulated binding is provided by mutations in the Fc region of the antibody relative to the native Fc region of the antibody. The mutations can be in a CH2 domain, a CH3 domain, or a combination thereof. A “native Fc region” is synonymous with a “wild-type Fc region” and comprises an amino acid sequence that is identical to the amino acid sequence of an Fc region found in nature or identical to the amino acid sequence of the Fc region found in the native antibody (e.g., cetuximab). Native sequence human Fc regions include a native sequence human IgG1 Fc region, native sequence human IgG2 Fc region, native sequence human IgG3 Fc region, and native sequence human IgG4 Fc region, as well as naturally occurring variants thereof. Native sequence Fc includes the various allotypes of Fcs (Jefferis et al., (2009) mAbs, 1(4):332-338).
In some embodiments, the Fc region of the antibodies of the immunoconjugates are modified to have an altered glycosylation pattern of the Fc region compared to the native non-modified Fc region. Human immunoglobulin is glycosylated at the Asn297 residue in the Cγ2 domain of each heavy chain. This N-linked oligosaccharide is composed of a core heptasaccharide, N-acetylglucosamine4Mannose3 (GlcNAc4Man3). Removal of the heptasaccharide with endoglycosidase or PNGase F is known to lead to conformational changes in the antibody Fc region, which can significantly reduce antibody-binding affinity to activating FcγR and lead to decreased effector function. The core heptasaccharide is often decorated with galactose, bisecting GlcNAc, fucose, or sialic acid, which differentially impacts Fc binding to activating and inhibitory FcγR. Additionally, it has been demonstrated that α2,6-sialyation enhances anti-inflammatory activity in vivo, while afucosylation leads to improved FcγRIIIa binding and a 10-fold increase in antibody-dependent cellular cytotoxicity and antibody-dependent phagocytosis. Specific glycosylation patterns, therefore, can be used to control inflammatory effector functions. In some embodiments, the modification to alter the glycosylation pattern is a mutation. For example, a substitution at Asn297. In some embodiments, Asn297 is mutated to glutamine (N297Q). Methods for controlling immune response with antibodies that modulate FcγR- regulated signaling are described, for example, in US 7416726, US 2007/0014795 and US 2008/0286819, which are hereby incorporated by reference in their entireties. In some embodiments, the antibodies of the immunoconjugates are modified to contain an engineered Fab region with a non-naturally occurring glycosylation pattern. For example, hybridomas can be genetically engineered to secrete afucosylated mAb, desialylated mAb or deglycosylated Fc with specific mutations that enable increased FcRγIIIa binding and effector function. In some embodiments, the antibodies of the immunoconjugates are engineered to be afucosylated. In some embodiments, the entire Fc region of an antibody in the immunoconjugates is exchanged with a different Fc region, so that the Fab region of the antibody is conjugated to a non-native Fc region. For example, the Fab region of cetuximab, which normally comprises an IgG1 Fc region, can be conjugated to IgG2, IgG3, IgG4, or IgA, or the Fab region of nivolumab, which normally comprises an IgG4 Fc region, can be conjugated to IgG1, IgG2, IgG3, IgA1, or IgG2. In some embodiments, the Fc modified antibody with a non-native Fc domain also comprises one or more amino acid modification, such as the S228P mutation within the IgG4 Fc, that modulate the stability of the Fc domain described. In some embodiments, the Fc modified
antibody with a non-native Fc domain also comprises one or more amino acid modifications described herein that modulate Fc binding to FcR. In some embodiments, the modifications that modulate the binding of the Fc region to FcR do not alter the binding of the Fab region of the antibody to its antigen when compared to the native non-modified antibody. In other embodiments, the modifications that modulate the binding of the Fc region to FcR also increase the binding of the Fab region of the antibody to its antigen when compared to the native non-modified antibody. In some embodiments, the antibodies in the immunoconjugates contain a modified Fc region, wherein the modification modulates the binding of the Fc region to one or more Fc receptors. In some embodiments, the Fc region is modified by inclusion of a transforming growth factor beta 1 (TGFβ1) receptor, or a fragment thereof, that is capable of binding TGFβ1. For example, the receptor can be TGFβ receptor II (TGFβRII). In some embodiments, theTGFβ receptor is a human TGFβ receptor. In some embodiments, the IgG has a C-terminal fusion to a TGFβRII extracellular domain (ECD) as described in US 9676863, incorporated herein. An “Fc linker” may be used to attach the IgG to the TGFβRII extracellular domain. The Fc linker may be a short, flexible peptide that allows for the proper three-dimensional folding of the molecule while maintaining the binding-specificity to the targets. In some embodiments, the N-terminus of the TGFβ receptor is fused to the Fc of the antibody construct (with or without an Fc linker). In some embodiments, the C-terminus of the antibody construct heavy chain is fused to the TGFβ receptor (with or without an Fc linker). In some embodiments, the C-terminal lysine residue of the antibody construct heavy chain is mutated to alanine. In some embodiments, the antibodies in the immunoconjugates are glycosylated. In some embodiments, the antibodies in the immunoconjugates are a cysteine-engineered antibody which provides for site-specific conjugation of an adjuvant, label, or drug moiety to the antibody through cysteine substitutions at sites where the engineered cysteines are available for conjugation but do not perturb immunoglobulin folding and assembly or alter antigen binding and effector functions (Junutula, et al., (2008) Nature Biotech., 26(8):925-932; Dornan et al. (2009) Blood 114(13):2721-2729; US 7521541; US 7723485; US 2012/0121615; WO 2009/052249). A “cysteine engineered antibody” or “cysteine engineered antibody variant” is an antibody in which one or more residues of an antibody are substituted with cysteine residues. Cysteine-engineered antibodies can be conjugated to the aza-bicyclic (AzaBi) moiety as a AzaBi-linker compound with uniform stoichiometry (e.g., up to two AzaBi moieties per antibody in an antibody that has a single engineered cysteine site).
In some embodiments, cysteine-engineered antibodies are used to prepare immunoconjugates. Immunoconjugates may have a reactive cysteine thiol residue introduced at a site on the light chain, such as the 149-lysine site (LC K149C), or on the heavy chain such as the 122-serine site (HC S122C), as numbered by Kabat numbering. In other embodiments, the cysteine-engineered antibodies have a cysteine residue introduced at the 118-alanine site (EU numbering) of the heavy chain (HC A118C). This site is alternatively numbered 121 by Sequential numbering or 114 by Kabat numbering. In other embodiments, the cysteine- engineered antibodies have a cysteine residue introduced in: (i) the light chain at G64C, R142C, K188C, L201C, T129C, S114C, or E105C according to Kabat numbering; (ii) the heavy chain at D101C, V184C, T205C, or S122C according to Kabat numbering; or (iii) other cysteine-mutant antibodies, and as described in Bhakta, S. et al, (2013) “Engineering THIOMABs for Site- Specific Conjugation of Thiol-Reactive Linkers”, Laurent Ducry (ed.), Antibody-Drug Conjugates, Methods in Molecular Biology, vol.1045, pages 189-203; WO 2011/156328; US 9000130. IMMUNE CHECKPOINT INHIBITORS In some embodiments, the antibody of an immunoconjugate is an immune checkpoint inhibitor. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins. In another embodiment, the immune checkpoint inhibitor reduces the interaction between one or more immune checkpoint proteins and their ligands. Inhibitory nucleic acids that decrease the expression and/or activity of immune checkpoint molecules can also be used in the methods disclosed herein. Immune checkpoint inhibitors nivolumab and atezolizumab can be modified to include an IgG1 Fc, and subsequently converted into an immunoconjugate of the invention. Most checkpoint antibodies are designed not to have effector function to kill cells, but rather to block the signalling. Immunoconjugates of the present invention can add back the "effector functionality" needed to activate myeloid immunity. In some embodiments, the immune checkpoint inhibitor is cytotoxic T-lymphocyte antigen 4 (CTLA4, also known as CD152), T cell immunoreceptor with Ig and ITIM domains (TIGIT), glucocorticoid-induced TNFR-related protein (GITR, also known as TNFRSF18), inducible T cell costimulatory (ICOS, also known as CD278), CD96, poliovirus receptor-related 2 (PVRL2, also known as CD112R, programmed cell death protein 1 (PD-1, also known as CD279), programmed cell death 1 ligand 1 (PD-L1, also known as B7-H3 and CD274), programmed cell death ligand 2 (PD-L2, also known as B7-DC and CD273), lymphocyte activation gene-3 (LAG-3, also known as CD223), B7-H4, killer immunoglobulin receptor
(KIR), Tumor Necrosis Factor Receptor superfamily member 4 (TNFRST4, also known as OX40 and CD134) and its ligand OX40L (CD252), indoleamine 2,3-dioxygenase 1 (IDO-1), indoleamine 2,3-dioxygenase 2 (IDO-2), carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), B and T lymphocyte attenuator (BTLA, also known as CD272), T-cell membrane protein 3 (TIM3), the adenosine A2A receptor (A2Ar), and V-domain Ig suppressor of T cell activation (VISTA protein). In some embodiments, the immune checkpoint inhibitor is an inhibitor of CTLA4, PD-1, or PD-L1. In some embodiments, the antibody is selected from: ipilimumab (also known as YERVOY®) pembrolizumab (also known as KEYTRUDA®), nivolumab (also known as OPDIVO®), atezolizumab (also known as TECENTRIQ®), avelumab (also known as BAVENCIO®), and durvalumab (also known as IMFINZI®). In some embodiments, the immune checkpoint inhibitor is an inhibitor of CTLA4. In some embodiments, the immune checkpoint inhibitor is an antibody against CTLA4. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against CTLA4. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against CTLA4. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as CTLA4. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-1. In some embodiments, the immune checkpoint inhibitor is an antibody against PD-1. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against PD-1. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against PD-1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as PD-1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-L1. In some embodiments, the immune checkpoint inhibitor is an antibody against PD-L1. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against PD-L1. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against PD-L1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as PD-L1. In some embodiments, the immune checkpoint inhibitor reduces the interaction between PD-1 and PD-L1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-L2. In some embodiments, the immune checkpoint inhibitor is an antibody against PD-L2. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against PD-L2. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against PD-L2. In some embodiments, the immune checkpoint inhibitor reduces the expression or
activity of one or more immune checkpoint proteins, such as PD-L2. In some embodiments, the immune checkpoint inhibitor reduces the interaction between PD-1 and PD-L2. In some embodiments, the immune checkpoint inhibitor is an inhibitor of LAG-3. In some embodiments, the immune checkpoint inhibitor is an antibody against LAG-3. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against LAG-3. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against LAG-3. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as LAG-3. In some embodiments, the immune checkpoint inhibitor is an inhibitor of B7-H4. In some embodiments, the immune checkpoint inhibitor is an antibody against B7-H4. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against B7-H4. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against B7-H4. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as B7-H4. In some embodiments, the immune checkpoint inhibitor is an inhibitor of KIR. In some embodiments, the immune checkpoint inhibitor is an antibody against KIR. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against KIR. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against KIR. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as KIR. In some embodiments, the immune checkpoint inhibitor is an inhibitor of TNFRSF4. In some embodiments, the immune checkpoint inhibitor is an antibody against TNFRSF4. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against TNFRSF4. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against TNFRSF4. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as TNFRSF4. In some embodiments, the immune checkpoint inhibitor is an inhibitor of OX40L. In some embodiments, the immune checkpoint inhibitor is an antibody against OX40L. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against OX40L. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against OX40L. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as OX40L. In some embodiments, the immune checkpoint inhibitor reduces the interaction between TNFRSF4 and OX40L.In some embodiments, the immune checkpoint inhibitor is an inhibitor of IDO-1. In some embodiments, the immune checkpoint inhibitor is an antibody against IDO-1. In some embodiments, the
immune checkpoint inhibitor is a monoclonal antibody against IDO-1, in some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against IDO-1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as IDO-1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of IDO-2. In some embodiments, the immune checkpoint inhibitor is an antibody against IDO-2. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against IDO-2. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against IDO-2. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as IDO-2. In some embodiments, the immune checkpoint inhibitor is an inhibitor of CEACAM1. In some embodiments, the immune checkpoint inhibitor is an antibody against CEACAM1. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against CEACAM1. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against CEACAM1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as CEACAM1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of BTLA. In some embodiments, the immune checkpoint inhibitor is an antibody against BTLA. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against BTLA. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against BMA. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as BTLA. In some embodiments, the immune checkpoint inhibitor is an inhibitor of TIM3. In some embodiments, the immune checkpoint inhibitor is an antibody against TIM3. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against TIM3. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against TIM3. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as TIM3. In some embodiments, the immune checkpoint inhibitor is an inhibitor of A2Ar. In some embodiments, the immune checkpoint inhibitor is an antibody against A2Ar. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against A2Ar. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against A2Ar. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as A2Ar.
In some embodiments, the immune checkpoint inhibitor is an inhibitor of VISTA protein. In some embodiments, the immune checkpoint inhibitor is an antibody against VISTA protein. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against VISTA protein. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against VISTA protein. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as VISTA protein. ANTIBODY TARGETS In some embodiments, the antibody of an immunoconjugate is capable of binding one or more targets selected from (e.g., specifically binds to a target selected from) 5T4, ABL, ABCF1, ACVR1, ACVR1B, ACVR2, ACVR2B, ACVRL1, ADORA2A, Aggrecan, AGR2, AICDA, AIF1, AIGI, AKAP1, AKAP2, AMH, AMHR2, ANGPT1, ANGPT2, ANGPTL3, ANGPTL4, ANPEP, APC, APOC1, AR, aromatase, ATX, AX1, AZGP1 (zinc-a-glycoprotein), B7.1, B7.2, B7-H1, BAD, BAFF, BAG1, BAI1, BCR, BCL2, BCL6, BDNF, BLNK, BLR1 (MDR15), BIyS, BMP1, BMP2, BMP3B (GDFIO), BMP4, BMP6, BMP8, BMPRTA, BMPR1B, BMPR2, BPAG1 (plectin), BRCA1, C19orflO (IL27w), C3, C4A, C5, C5R1, CANT1, CAPRIN-1, CASP1, CASP4, CAV1, CCBP2 (D6/JAB61), CCLI (1-309), CCLI1 (eotaxin), CCL13 (MCP- 4), CCL15 (MIP-Id), CCL16 (HCC-4), CCL17 (TARC), CCL18 (PARC), CCL19 (MIP-3b), CCL2 (MCP-1), MCAF, CCL20 (MIP-3a), CCL21 (MEP-2), SLC, exodus-2, CCL22(MDC/STC-1), CCL23 (MPIF-I), CCL24 (MPIF-2/eotaxin-2), CCL25 (TECK), CCL26 (eotaxin-3), CCL27 (CTACK/ILC), CCL28, CCL3 (MIP-Ia), CCL4 (MIPIb), CCL5 (RANTES), CCL7 (MCP-3), CCL8 (mcp-2), CCNA1, CCNA2, CCND1, CCNE1, CCNE2, CCR1 (CKR1/HM145), CCR2 (mcp-IRB/RA), CCR3 (CKR3/CMKBR3), CCR4, CCR5 (CMKBR5/ChemR13), CCR6 (CMKBR6/CKR-L3/STRL22/DRY6), CCR7 (CKR7/EBI1), CCR8 (CMKBR8/TERI/CKR-L1), CCR9 (GPR-9-6), CCRL1 (VSHK1), CCRL2 (L-CCR), CD164, CD19, CDIC, CD2, CD20, CD21, CD200, CD-22, CD24, CD27, CD28, CD3, CD33, CD35, CD37, CD38, CD3E, CD3G, CD3Z, CD4, CD38, CD40, CD40L, CD44, CD45RB, CD47, CD52, CD69, CD72, CD74, CD79A, CD79B, CD8, CD80, CD81, CD83, CD86, CD137, CD152, CD274, CDH1 (Ecadherin), CDH1O, CDH12, CDH13, CDH18, CDH19, CDH2O, CDH5, CDH7, CDH8, CDH9, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK9, CDKN1A (p21Wap1/Cip1), CDKN1B (p27Kip1), CDKN1C, CDKN2A (p16INK4a), CDKN2B, CDKN2C, CDKN3, CEBPB, CERI, CHGA, CHGB, Chitinase, CHST1O, CKLFSF2, CKLFSF3, CKLFSF4, CKLFSF5, CKLFSF6, CKLFSF7, CKLFSF8, CLDN3, CLDN7 (claudin-7), CLDN18.2 (claudin 18.2), CLN3, CLU (clusterin), CMKLR1, CMKOR1 (RDC1),
CNR1, COL18A1, COLIA1, COL4A3, COL6A1, CR2, Cripto, CRP, CSF1 (M-CSF), CSF2 (GM-CSF), CSF3 (GCSF), CTL8, CTNNB1 (b-catenin), CTSB (cathepsin B), CX3CL1 (SCYD1), CX3CR1 (V28), CXCL1 (GRO1), CXCL1O (IP-IO), CXCLI1 (1-TAC/IP-9), CXCL12 (SDF1), CXCL13, CXCL14, CXCL16, CXCL2 (GRO2), CXCL3 (GRO3), CXCL5 (ENA-78/LIX), CXCL6 (GCP-2), CXCL9 (MIG), CXCR3 (GPR9/CKR-L2), CXCR4, CXCR6 (TYMSTR/STRL33/Bonzo), CYB5, CYC1, CYSLTR1, DAB2IP, DES, DKFZp451J0118, DNCL1, DPP4, E2F1, Engel, Edge, Fennel, EFNA3, EFNB2, EGF, EGFR, ELAC2, ENG, Enola, ENO2, ENO3, EPHA1, EPHA2, EPHA3, EPHA4, EPHA5, EPHA6, EPHA7, EPHA8, EPHA9, EPRA10, EPHB1, EPHB2, EPHB3, EPHB4, EPHB5, EPHB6, EPHRIN-A1, EPHRIN- A2, EPHRINA3, EPHRIN-A4, EPHRIN-A5, EPHRIN-A6, EPHRIN-B1, EPHRIN-B2, EPHRIN-B3, EPHB4, EPG, ERBB2 (Her-2), EREG, ERK8, Estrogen receptor, Earl, ESR2, F3 (TF), FADD, famesyltransferase, FasL, FASNf, FCER1A, FCER2, FCGR3A, FGF, FGF1 (aFGF), FGF10, FGF11, FGF12, FGF12B, FGF13, FGF14, FGF16, FGF17, FGF18, FGF19, FGF2 (bFGF). FGF20, FGF21, FGF22, FGF23, FGF3 (int-2), FGF4 (HST), FGF5, FGF6 (HST- 2), FGF7 (KGF), FGF8, FGF9, FGFR3, FIGF (VEGFD), FILI (EPSILON), FBL1 (ZETA), FLJ12584, FLJ25530, FLRT1 (fibronectin), FLT1, FLT-3, FOS, FOSL1 (FRA-1), FY (DARC), GABRP (GABAa), GAGEB1, GAGEC1, GALNAC4S-6ST, GATA3, GD2, GDF5, GFI1, GGT1, GM-CSF, GNAS1, GNRH1, GPR2 (CCR10), GPR31, GPR44, GPR81 (FKSG80), GRCC1O (C1O), GRP, GSN (Gelsolin), GSTP1, HAVCR2, HDAC, HDAC4, HDAC5, HDAC7A, HDAC9, Hedgehog, HGF, HIF1A, HIP1, histamine and histamine receptors, HLA- A, HLA-DRA, HLA-E, HM74, HMOXI, HSP90, HUMCYT2A, ICEBERG, ICOSL, ID2, IFN- a, IFNA1, IFNA2, IFNA4, IFNA5, EFNA6, BFNA7, IFNB1, IFNgamma, IFNW1, IGBP1, IGF1, IGFIR, IGF2, IGFBP2, IGFBP3, IGFBP6, DL-1, ILIO, ILIORA, ILIORB, IL-1, IL1R1 (CD121a), IL1R2 (CD121b), IL-IRA, IL-2, IL2RA (CD25), IL2RB (CD122), IL2RG (CD132), IL-4, IL-4R (CD123), IL-5, IL5RA (CD125), IL3RB (CD131), IL-6, IL6RA, (CD126), IR6RB (CD130), IL-7, IL7RA (CD127), IL-8, CXCR1 (IL8RA), CXCR2, (IL8RB/CD128), IL-9, IL9R (CD129), IL-10, IL10RA (CD210), IL10RB (CDW210B), IL-11, IL11RA, IL-12, IL-12A, IL- 12B, IL-12RB1, IL-12RB2, IL-13, IL13RA1, IL13RA2, IL14, IL15, IL15RA, IL16, IL17, IL17A, IL17B, IL17C, IL17R, IL18, IL18BP, IL18R1, IL18RAP, IL19, ILIA, ILIB, ILIF10, ILIF5, IL1F6, ILIF7, IL1F8, DL1F9, ILIHYI, ILIR1, ILIR2, ILIRAP, ILIRAPLI, ILIRAPL2, ILIRL1, IL1RL2, ILIRN, IL2, IL20, IL20RA, IL21R, IL22, IL22R, IL22RA2, IL23, DL24, IL25, IL26, IL27, IL28A, IL28B, IL29, IL2RA, IL2RB, IL2RG, IL3, IL30, IL3RA, IL4, IL4, IL6ST (glycoprotein 130), ILK, INHA, INHBA, INSL3, INSL4, IRAK1, IRAK2, ITGA1, ITGA2, ITGA3, ITGA6 (.alpha.6 integrin), ITGAV, ITGB3, ITGB4 (.beta.4 integrin), JAG1, JAK1, JAK3, JTB, JUN, K6HF, KAI1, KDR, KITLG, KLF5 (GC Box BP), KLF6, KLK10,
KLK12, KLK13, KLK14, KLK15, KLK3, KLK4, KLK5, KLK6, KLK9, KRT1, KRT19 (Keratin 19), KRT2A, KRTHB6 (hair-specific type II keratin), LAMA5, LEP (leptin), Lingo- p75, Lingo-Troy, LPS, LRRC15, LTA (TNF-b)), LTB, LTB4R (GPR16), LTB4R2, LTBR, MACMARCKS, MAG or OMgp, MAP2K7 (c-Jun), MCP-1, MDK, MIB1, midkine, MIF, MISRII, MJP-2, MK, MKI67 (Ki-67), MMP2, MMP9, MS4A1, MSMB, MT3 (metallothionectin-UI), mTOR, MTSS1, MUC1 (mucin), MYC, MYD88, NCK2, neurocan, Nectin-4, NFKBI, NFKB2, NGFB (NGF), NGFR, NgR-Lingo, NgRNogo66, (Nogo), NgR-p75, NgR-Troy, NMEI (NM23A), NOTCH, NOTCH1, NOX5, NPPB, NROB1, NROB2, NRID1, NR1D2, NR1H2, NR1H3, NR1H4, NR112, NR113, NR2C1, NR2C2, NR2E1, NR2E3, NR2F1, NR2F2, NR2F6, NR3C1, NR3C2, NR4A1, NR4A2, NR4A3, NR5A1, NR5A2, NR6A1, NRP1, NRP2, NT5E, NTN4, ODZI, OPRDI, P2RX7, PAP, PART1, PATE, PAWR, PCA3, PCDGF, PCNA, PDGFA, PDGFB, PDGFRA, PDGFRB, PECAMI, peg-asparaginase, PF4 (CXCL4), PGF, PGR, phosphacan, PIAS2, PI3 Kinase, PIK3CG, PLAU (uPA), PLG, PLXDCI, PKC, PKC-beta, PPBP (CXCL7), PPID, PR1, PRKCQ, PRKD1, PRL, PROC, PROK2, PSAP, PSCA, PTAFR, PTEN, PTGS2 (COX-2), PIN, RAC2 (P21Rac2), RANK, RANK ligand, RARB, RGS1, RGS13, RGS3, RNFI1O (ZNF144), Ron, ROBO2, RXR, S100A2, SCGB 1D2 (lipophilin B), SCGB2A1 (mammaglobin 2), SCGB2A2 (mammaglobin 1), SCYE1 (endothelial Monocyte-activating cytokine), SDF2, SERPENA1, SERPINA3, SERPINB5 (maspin), SERPINEI (PAI-I), SERPINFI, SHIP-1, SHIP-2, SHB1, SHB2, SHBG, SfcAZ, SLC2A2, SLC33A1, SLC43A1, SLIT2, SPP1, SPRR1B (Spr1), ST6GAL1, STAB1, STATE, STEAP, STEAP2, TB4R2, TBX21, TCP1O, TDGF1, TEK, TGFA, TGFB1, TGFB1I1, TGFB2, TGFB3, TGFBI, TGEBR1, TGFBR2, TGFBR3, THIL, THBS1 (thrombospondin-1), THBS2, THBS4, THPO, TIE (Tie-1), TIMP3, tissue factor, TLR1, TLR2, TLR3, TLR4, TLR5, TLR6, TLR7, TLR8, TLR9, TLR10, TLR11, TNF, TNF-a, TNFAIP2 (B94), TNFAIP3, TNFRSF11A, TNFRSF1A, TNFRSF1B, TNFRSF21, TNFRSF5, TNFRSF6 (Fas), TNFRSF7, TNFRSF8, TNFRSF9, TNFSF1O (TRAIL), TNFSF11 (TRANCE), TNFSF12 (APO3L), TNFSF13 (April), TNFSF13B, TNSF14 (HVEM-L), TNFRSF14 (HVEM), TNFSF15 (VEGI), TNFSF18, TNFSF4 (OX40 ligand), TNFSF5 (CD40 ligand). TNFSF6 (FasL), TNFSF7 (CD27 ligand), TNFSF8 (CD30 ligand), TNFSF9 (4-1BB ligand), TOLLIP, Toll-like receptors, TOP2A (topoisomerase 1ia), TP53, TPM1, TPM2, TRADD, TRAF1, TRAF2, TRAF3, TRAF4, TRAF5, TRAF6, TRKA, TREM1, TREM2, TROP2, TRPC6, TSLP, TWEAK, Tyrosinase, uPAR, VEGF, VEGFB, VEGFC, versican, VHL C5, VLA-4, Wnt-1, XCL1 (tymphotactin), XCL2 (SCM-Ib), XCRI (GPR5/CCXCR1), YYI, ZFPM2, CLEC4C (BDCA-2, DLEC, CD303, CLECSF7), CLEC4D (MCL, CLECSF8), CLEC4E (Mincle), CLEC6A (Dectin-2). CLEC5A (MDL-1, CLECSF5), CLEC1B (CLEC-2), CLEC9A (DNGR-1), CLEC7A (Dectin-1), PDGFRa,
SLAMF7, GP6 (GPVI), LILRA1 (CD85I), LILRA2 (CD85H, ILT1), LILRA4 (CD85G, ILT7), LILRA5 (CD85F, ILT11), LILRA6 (CD85b, ILT8), NCR1 (CD335, LY94, NKp46), NCR3 (CD335, LY94, NKp46), NCR3 (CD337, NKp30), OSCAR, TARM1, CD300C, CD300E, CD300LB (CD300B), CD300LD (CD300D), KIR2DL4 (CD158D), KIR2DS, KLRC2 (CD159C, NKG2C), KLRK1 (CD314, NKG2D), NCR2 (CD336, NKp44), PILRB, SIGLEC1 (CD169, SN), SIGLEC14, SIGLEC15 (CD33L3), SIGLEC16, SIRPB1 (CD172B), TREM1 (CD354), TREM2, and KLRF1 (NKp80). In some embodiments, the antibody binds to an FcR.gamma-coupled receptor. In some embodiments, the FcR.gamma-coupled receptor is selected from the group consisting of GP6 (GPVI), LILRA1 (CD85I), LILRA2 (CD85H, ILT1), LILRA4 (CD85G, ILT7), LILRA5 (CD85F, ILT11), LILRA6 (CD85b, ILT8), NCR1 (CD335, LY94, NKp46), NCR3 (CD335, LY94, NKp46), NCR3 (CD337, NKp30), OSCAR, and TARM1. In some embodiments, the antibody binds to a DAP12-coupled receptor. In some embodiments, the DAP12-coupled receptor is selected from the group consisting of CD300C, CD300E, CD300LB (CD300B), CD300LD (CD300D), KIR2DL4 (CD158D), KIR2DS, KLRC2 (CD159C, NKG2C), KLRK1 (CD314, NKG2D), NCR2 (CD336, NKp44). PILRB, SIGLEC1 (CD169, SN), SIGLEC14, SIGLEC15 (CD33L3), SIGLEC16, SIRPB1 (CD172B), TREM1 (CD354), and TREM2. In some embodiments, the antibody binds to a hemITAM-bearing receptor. In some embodiments, the hemITAM-bearing receptor is KLRF1 (NKp80). In some embodiments, the antibody is capable of binding one or more targets selected from CLEC4C (BDCA-2, DLEC, CD303, CLECSF7), CLEC4D (MCL, CLECSF8), CLEC4E (Mincle), CLEC6A (Dectin-2), CLEC5A (MDL-1, CLECSF5), CLEC1B (CLEC-2), CLEC9A (DNGR-1), and CLEC7A (Dectin-1). In some embodiments, the antibody is capable of binding CLEC6A (Dectin-2) or CLEC5A. In some embodiments, the antibody is capable of binding CLEC6A (Dectin-2). In some embodiments, the antibody is capable of binding one or more targets selected from (e.g., specifically binds to a target selected from): ATP5I (Q06185), OAT (P29758), AIFM1 (Q9Z0X1), AOFA (Q64133), MTDC (P18155), CMC1 (Q8BH59), PREP (Q8K411), YMEL1 (O88967), LPPRC (Q6PB66), LONM (Q8CGK3), ACON (Q99KI0), ODO1 (Q60597), IDHP (P54071), ALDH2 (P47738), ATPB (P56480), AATM (P05202), TMM93 (Q9CQW0), ERGI3 (Q9CQE7), RTN4 (Q99P72), CL041 (Q8BQR4), ERLN2 (Q8BFZ9), TERA (Q01853), DAD1 (P61804), CALX (P35564), CALU (O35887), VAPA (Q9WV55), MOGS (Q80UM7), GANAB (Q8BHN3), ERO1A (Q8R180), UGGG1 (Q6P5E4), P4HA1 (Q60715), HYEP (Q9D379), CALR (P14211), AT2A2 (O55143), PDIA4 (P08003), PDIA1 (P09103), PDIA3
(P27773), PDIA6 (Q922R8), CLH (Q68FD5), PPIB (P24369), TCPG (P80318), MOT4 (P57787), NICA (P57716), BASI (P18572), VAPA (Q9WV55), ENV2 (P11370), VAT1 (Q62465), 4F2 (P10852), ENOA (P17182), ILK (O55222), GPNMB (Q99P91), ENV1 (P10404), ERO1A (Q8R180), CLH, (Q68FD5), DSG1A (Q61495), AT1A1 (Q8VDN2), HYOU1 (Q9JKR6), TRAP1 (Q9CQN1), GRP75 (P38647), ENPL (P08113), CH60 (P63038), and CH10 (Q64433). In the preceding list, accession numbers are shown in parentheses. In some embodiments, the antibody binds to an antigen selected from CDH1, CD19, CD20, CD29, CD30, CD38, CD40, CD47, EpCAM, MUC1, MUC16, EGFR, Her2, SLAMF7, and gp75. In some embodiments, the antigen is selected from CD19, CD20, CD47, EpCAM, MUC1, MUC16, EGFR, and Her2. In some embodiments, the antibody binds to an antigen selected from the Tn antigen and the Thomsen-Friedenreich antigen. In some embodiments, the antibody or Fc fusion protein is selected from: abagovomab, abatacept (also known as ORENCIA®), abciximab (also known as REOPRO®), c7E3 Fab), adalimumab (also known as HUMIRA®), adecatumumab, alemtuzumab (also known as CAMPATH®), MabCampath or Campath-1H), altumomab, afelimomab, anatumomab mafenatox, anetumumab, anrukizumab, apolizumab, arcitumomab, aselizumab, atlizumab, atorolimumab, bapineuzumab, basiliximab (also known as SIMULECT®), bavituximab, bectumomab (also known as LYMPHOSCAN®), belimumab (also known as LYMPHO-STAT- B®), bertilimumab, besilesomab, bevacizumab (also known as AVASTIN®), biciromab brallobarbital, bivatuzumab mertansine, campath, canakinumab (also known as ACZ885), cantuzumab mertansine, capromab (also known as PROSTASCINT®), catumaxomab (also known as REMOVAB®), cedelizumab (also known as CIMZIA®), certolizumab pegol, cetuximab (also known as ERBITUX®), clenoliximab, dacetuzumab, dacliximab, daclizumab (also known as ZENAPAX®), denosumab (also known as AMG 162), detumomab, dorlimomab aritox, dorlixizumab, duntumumab, durimulumab, durmulumab, ecromeximab, eculizumab (also known as SOLIRIS®), edobacomab, edrecolomab (also known as Mab17-1A, PANOREX®), efalizumab (also known as RAPTIVA®), efungumab (also known as MYCOGRAB®), elsilimomab, enlimomab pegol, epitumomab cituxetan, efalizumab, epitumomab, epratuzumab, erlizumab, ertumaxomab (also known as REXOMUN®), etanercept (also known as ENBREL®), etaracizumab (also known as etaratuzumab, VITAXIN®, ABEGRIN®), exbivirumab, fanolesomab (also known as NEUTROSPEC®), faralimomab, felvizumab, fontolizumab (also known as HUZAF®), galiximab, gantenerumab, gavilimomab (also known as ABXCBL®), gemtuzumab ozogamicin (also known as MYLOTARG®), golimumab (also known as CNTO 148), gomiliximab, ibalizumab (also known as TNX-355), ibritumomab tiuxetan (also known as ZEVALIN®), igovomab, imciromab, infliximab (also known as
REMICADE®), inolimomab, inotuzumab ozogamicin, ipilimumab (also known as MDX-010, MDX-101), iratumumab, keliximab, labetuzumab, lemalesomab, lebrilizumab, lerdelimumab, lexatumumab (also known as, HGS-ETR2, ETR2-ST01), lexitumumab, libivirumab, lintuzumab, lucatumumab, lumiliximab, mapatumumab (also known as HGSETR1, TRM-1), maslimomab, matuzumab (also known as EMD72000), mepolizumab (also known as BOSATRIA®), metelimumab, milatuzumab, minretumomab, mitumomab, morolimumab, motavizwnab (also known as NUMAX®), muromonab (also known as OKT3), nacolomab tafenatox, naptumomab estafenatox, natalizumab (also known as TYSABRI®, ANTEGREN®), nebacumab, nerelimomab, nimotuzumab (also known as THERACIM hR3®, THERA-CIM- hR3®, THERALOC®), nofetumomab merpentan (also known as VERLUMA®), ocrelizumab, odulimomab, ofatumumab, omalizumab (also known as XOLAIR®), oregovomab (also known as OVAREX®), otelixizumab, pagibaximab, palivizumab (also known as SYNAGIS®), panitumumab (also known as ABX-EGF, VECTIBIX®), pascolizumab, pemtumomab (also known as THERAGYN®), pertuzumab (also known as 2C4, OMNITARG®), pexelizumab, pintumomab, priliximab, pritumumab, ranibizumab (also known as LUCENTIS®), raxibacumab, regavirumab, reslizumab, rituximab (also known as RITUXAN®, MabTHERA®), rovelizumab, ruplizumab, satumomab, sevirumab, sibrotuzumab, siplizumab (also known as MEDI-507), sontuzumab, stamulumab (also known as MYO-029), sulesomab (also known as LEUKOSCAN®), tacatuzumab tetraxetan, tadocizumab, talizumab, taplitumomab paptox, tefibazumab (also known as AUREXIS®), telimomab aritox, teneliximab, teplizumab, ticilimumab, tocilizumab (also known as ACTEMRA®), toralizumab, tositumomab, trastuzumab (also known as HERCEPTIN®), tremelimumab (also known as CP-675,206), tucotuzumab celmoleukin, tuvirumab, urtoxazumab, ustekinumab (also known as CNTO 1275), vapaliximab, veltuzumab, vepalimomab, visilizumab (also known as NUVION®), volociximab (also known as M200), votumumab (also known as HUMASPECT®), zalutumumab, zanolimumab (also known as HuMAX-CD4), ziralimumab, zolimomab aritox, daratumumab, elotuxumab, obintunzumab, olaratumab, brentuximab vedotin, afibercept, abatacept, belatacept, afibercept, etanercept, romiplostim, SBT-040 (sequences listed in US 2017/0158772. In some embodiments, the antibody is rituximab. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds PD-L1. Programmed Death-Ligand 1 (PD-L1, cluster of differentiation 274, CD274, B7- homolog 1, or B7-H1) belongs to the B7 protein superfamily, and is a ligand of programmed cell death protein 1 (PD-1, PDCD1, cluster of differentiation 279, or CD279). PD-L1 can also
interact with B7.1 (CD80) and such interaction is believed to inhibit T cell priming. The PD- L1/PD-1 axis plays a large role in suppressing the adaptive immune response. More specifically, it is believed that engagement of PD-L1 with its receptor, PD-1, delivers a signal that inhibits activation and proliferation of T-cells. Agents that bind to PD-L1 and prevent the ligand from binding to the PD-1 receptor prevent this immunosuppression, and can, therefore, enhance an immune response when desired, such as for the treatment of cancers, or infections. PD-L1/PD-1 pathway also contributes to preventing autoimmunity and therefore agonistic agents against PD-L1 or agents that deliver immune inhibitory payloads may help treatment of autoimmune disorders. Several antibodies targeting PD-L1 have been developed for the treatment of cancer, including atezolizumab (TECENTRIQTM), durvalumab (IMFINZITM), and avelumab (BAVENCIOTM). Nevertheless, there continues to be a need for new PD-L1-binding agents, including agents that bind PD-L1 with high affinity and effectively prevent PD-L1/PD-1 signaling and agents that can deliver therapeutic payloads to PD-L1 expressing cells. In addition, there is a need for new PD-L1-binding agents to treat autoimmune disorders and infections. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds HER2. In certain embodiments, immunoconjugates of the invention comprise anti-HER2 antibodies. In one embodiment of the invention, an anti-HER2 antibody of an immunoconjugate of the invention comprises a humanized anti-HER2 antibody, e.g., huMAb4D5-1, huMAb4D5- 2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7 and huMAb4D5- 8, as described in Table 3 of US 5821337, which is specifically incorporated by reference herein. Those antibodies contain human framework regions with the complementarity- determining regions of a murine antibody (4D5) that binds to HER2. The humanized antibody huMAb4D5-8 is also referred to as trastuzumab, commercially available under the tradename HERCEPTIN™ (Genentech, Inc.). Trastuzumab (CAS 180288-69-1, HERCEPTIN ^, huMAb4D5-8, rhuMAb HER2, Genentech) is a recombinant DNA-derived, IgG1 kappa, monoclonal antibody that is a humanized version of a murine anti-HER2 antibody (4D5) that selectively binds with high affinity in a cell-based assay (Kd = 5 nM) to the extracellular domain of HER2 (US 5677171; US 5821337; US 6054297; US 6165464; US 6339142; US 6407213; US 6639055; US 6719971; US 6800738; US 7074404; Coussens et al (1985) Science 230:1132-9; Slamon et al (1989) Science 244:707-12; Slamon et al (2001) New Engl. J. Med.344:783-792).
In an embodiment of the invention, the antibody construct or antigen binding domain comprises the CDR regions of trastuzumab. In an embodiment of the invention, the anti-HER2 antibody further comprises the framework regions of the trastuzumab. In an embodiment of the invention, the anti-HER2 antibody further comprises one or both variable regions of trastuzumab. In another embodiment of the invention, an anti-HER2 antibody of an immunoconjugate of the invention comprises a humanized anti-HER2 antibody, e.g., humanized 2C4, as described in US 7862817. An exemplary humanized 2C4 antibody is pertuzumab (CAS Reg. No.380610- 27-5), PERJETA™ (Genentech, Inc.). Pertuzumab is a HER dimerization inhibitor (HDI) and functions to inhibit the ability of HER2 to form active heterodimers or homodimers with other HER receptors (such as EGFR/HER1, HER2, HER3 and HER4). See, for example, Harari and Yarden, Oncogene 19:6102-14 (2000); Yarden and Sliwkowski. Nat Rev Mol Cell Biol 2:127-37 (2001); Sliwkowski Nat Struct Biol 10:158-9 (2003); Cho et al. Nature 421:756-60 (2003); and Malik et al. Pro Am Soc Cancer Res 44:176-7 (2003). PERJETA™ is approved for the treatment of breast cancer. In an embodiment of the invention, the antibody construct or antigen binding domain comprises the CDR regions of pertuzumab. In an embodiment of the invention, the anti-HER2 antibody further comprises the framework regions of the pertuzumab. In an embodiment of the invention, the anti-HER2 antibody further comprises one or both variable regions of pertuzumab. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds CEA. Carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5) also known as CD66e (Cluster of Differentiation 66e), is a member of the carcinoembryonic antigen (CEA) gene family. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds CEA. Elevated expression of carcinoembryonic antigen (CEA, CD66e, CEACAM5) has been implicated in various biological aspects of neoplasia, especially tumor cell adhesion, metastasis, the blocking of cellular immune mechanisms, and having antiapoptosis functions. CEA is also used as a blood marker for many carcinomas. Labetuzumab (CEA-CIDETM, Immunomedics, CAS Reg. No.219649-07-7), also known as MN-14 and hMN14, is a humanized IgG1 monoclonal antibody and has been studied for the treatment of colorectal cancer (Blumenthal, R. et al (2005) Cancer Immunology Immunotherapy 54(4):315-327). Labetuzumab conjugated to a
camptothecin analog (labetuzumab govitecan, IMMU-130) targets carcinoembryonic antigen- related cell adhesion mol.5 (CEACAM5) and is being studied in patients with relapsed or refractory metastatic colorectal cancer (Sharkey, R. et al, (2018), Molecular Cancer Therapeutics 17(1):196-203; Cardillo, T. et al (2018) Molecular Cancer Therapeutics 17(1):150-160). In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Trop2. Tumor-associated calcium signal transducer 2 (TROP-2) is a transmembrane glycoprotein encoded by the TACSTD2 gene (Linnenbach AJ, et al (1993) Mol Cell Biol.13(3): 1507–15; Calabrese G, et al (2001) Cytogenet Cell Genet.92(1–2): 164–5). Trop2 is an intracellular calcium signal transducer that is differentially expressed in many cancers and signals cells for self-renewal, proliferation, invasion, and survival. Trop2 is considered a stem cell marker and is expressed in many normal tissues, though in contrast, it is overexpressed in many cancers (Ohmachi T, et al., (2006) Clin. Cancer Res., 12(10), 3057-3063; Muhlmann G, et al., (2009) J. Clin. Pathol., 62(2), 152-158; Fong D, et al., (2008) Br. J. Cancer, 99(8), 1290- 1295; Fong D, et al., (2008) Mod. Pathol., 21(2), 186-191; Ning S, et al., (2013) Neurol. Sci., 34(10), 1745-1750). Overexpression of Trop2 is of prognostic significance. Several ligands have been proposed that interact with Trop2. Trop2 signals the cells via different pathways and it is transcriptionally regulated by a complex network of several transcription factors. Human Trop2 (TACSTD2: tumor-associated calcium signal transducer 2, GA733-1, EGP-1, M1S1; hereinafter, referred to as hTrop2) is a single-pass transmembrane type 1 cell membrane protein consisting of 323 amino acid residues. While the presence of a cell membrane protein involved in immune resistance, which is common to human trophoblasts and cancer cells (Faulk W P, et al., Proc. Natl. Acad. Sci.75(4):1947-1951 (1978)), has previously been suggested, an antigen molecule recognized by a monoclonal antibody against a cell membrane protein in a human choriocarcinoma cell line was identified and designated as Trop2 as one of the molecules expressed in human trophoblasts (Lipinski M, et al., Proc. Natl. Acad. Sci.78(8), 5147-5150 (1981)). This molecule was also designated as tumor antigen GA733-1 recognized by a mouse monoclonal antibody GA733 (Linnenbach A J, et al., Proc. Natl. Acad. Sci.86(1), 27- 31 (1989)) obtained by immunization with a gastric cancer cell line or an epithelial glycoprotein (EGP-1; Basu A, et al., Int. J. Cancer, 62 (4), 472-479 (1995)) recognized by a mouse monoclonal antibody RS7-3G11 obtained by immunization with non-small cell lung cancer cells. In 1995, however, the Trop2 gene was cloned, and all of these molecules were confirmed to be identical molecules (Fornaro M, et al., Int. J. Cancer, 62(5), 610-618 (1995)). The DNA
sequence and amino acid sequence of hTrop2 are available on a public database and can be referred to, for example, under Accession Nos. NM_002353 and NP_002344 (NCBI). In response to such information suggesting the association with cancer, a plurality of anti-hTrop2 antibodies have been established so far and studied for their antitumor effects. Among these antibodies, there is disclosed, for example, an unconjugated antibody that exhibits in itself antitumor activity in nude mouse xenograft models (WO 2008/144891; WO 2011/145744; WO 2011/155579; WO 2013/077458) as well as an antibody that exhibits antitumor activity as ADC with a cytotoxic drug (WO 2003/074566; WO 2011/068845; WO 2013/068946; US 7999083). However, the strength or coverage of their activity is still insufficient, and there are unsatisfied medical needs for hTrop2 as a therapeutic target. TROP2 expression in cancer cells has been correlated with drug resistance. Several strategies target TROP2 on cancer cells that include antibodies, antibody fusion proteins, chemical inhibitors, nanoparticles, etc. The in vitro studies and pre-clinical studies, using these various therapeutic treatments, have resulted in significant inhibition of tumor cell growth both in vitro and in vivo in mice. Clinical studies have explored the potential application of Trop2 as both a prognostic biomarker and as a therapeutic target to reverse resistance. Sacituzumab govitecan (TRODELVY®, Immunomedics, IMMU-132), an antibody-drug conjugate comprising a Trop2-directed antibody linked to a topoisomerase inhibitor drug, is indicated for the treatment of metastatic triple-negative breast cancer (mTNBC) in adult patients that have received at least two prior therapies. The Trop2 antibody in sacituzumab govitecan is conjugated to SN-38, the active metabolite of irinotecan (US 2016/0297890; WO 2015/098099). In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Caprin-1 (Ellis JA, Luzio JP (1995) J Biol Chem.270(35):20717–23; Wang B, et al (2005) J Immunol.175 (7):4274–82; Solomon S, et al (2007) Mol Cell Biol.27(6):2324–42). Caprin-1 is also known as GPIAP1, GPIP137, GRIP137, M11S1, RNG105, p137GPI, and cell cycle associated protein 1. Cytoplasmic activation/proliferation-associated protein-1 (caprin-1) is an RNA-binding protein that participates in the regulation of cell cycle control-associated genes. Caprin-1 selectively binds to c-Myc and cyclin D2 mRNAs, which accelerates cell progression through the G1 phase into the S phase, enhances cell viability and promotes cell growth, indicating that it may serve an important role in tumorigenesis (Wang B, et al (2005) J Immunol.175:4274– 4282). Caprin-1 acts alone or in combination with other RNA-binding proteins, such as RasGAP SH3-domain-binding protein 1 and fragile X mental retardation protein. In the tumorigenesis process, caprin-1 primarily functions by activating cell proliferation and upregulating the
expression of immune checkpoint proteins. Through the formation of stress granules, caprin-1 is also involved in the process by which tumor cells adapt to adverse conditions, which contributes to radiation and chemotherapy resistance. Given its role in various clinical malignancies, caprin-1 holds the potential to be used as a biomarker and a target for the development of novel therapeutics (Yang, Z-S, et al (2019) Oncology Letters 18:15-21). Antibodies that target caprin-1 for treatment and detection have been described (WO 2011/096519; WO 2013/125654; WO 2013/125636; WO 2013/125640; WO 2013/125630; WO 2013/018889; WO 2013/018891; WO 2013/018883; WO 2013/018892; WO 2014/014082; WO 2014/014086; WO 2015/020212; WO 2018/079740). In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Claudin-1. Claudin-1 is a member of the transmembrane protein family claudins located in cell-cell tight junctions and it acts as a co-receptor for HCV entry into hepatic cells (Kniesel U, et al (2000). Cell. Mol. Neurobiol.20(1):57–76; Furuse M, et al (1998). J. Cell Biol.141(7):1539–50; Swisshelm K, et al (2005) Adv. Drug Deliv. Rev.57(6):919–28). Claudin 1 is also known as Senescence-associated epithelial membrane protein, senescence-associated epithelial membrane protein 1, CLDN1, CLD1, ILVASC, SEMP1. Claudins are abundant in luminal epithelial sheets where they maintain epithelial cell polarity. Claudin-1 is expressed in most tissues such as bladder, fallopian tube, liver, pancreas, prostate, and skin. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Nectin-4. The nectins are a protein family of cell adhesion molecules involved in calcium- dependent cell adhesion (Takai Y. et al (2003) Cancer Science 94(8):655-67; Fuchs, A. et al (2006) Seminars in Cancer Biology 16(5):359-366; Miyoshi J. et al (2007) American journal of nephrology 27(6):590-604). Nectins play an important role in the bonding between cells in many different tissues, including the intermediate junction of epithelial cells or the chemical synapse of nerve cells. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds LRRC15 (Leucine rich repeat containing 15). LRRC15 is a cell membrane-expressed protein that in humans is encoded by the LRRC15 gene. LRRC15 is expressed on stromal fibroblasts in many solid tumors (e.g., breast,
head and neck, lung, pancreatic) as well as directly on a subset of cancer cells of mesenchymal origin (e.g., sarcoma, melanoma, glioblastoma). LRRC15 may have utility as a therapeutic target for the treatment of cancers with LRRC15-positive stromal desmoplasia or cancers of mesenchymal origin (Purcell, JW et al (2018) Cancer Res., 78(14):4059-4072). AZA-BICYCLIC ADJUVANT COMPOUNDS The immunoconjugate of the invention comprises an aza-bicyclic adjuvant moiety. The adjuvant moiety described herein is a compound that elicits an immune response (i.e., an immunostimulatory agent). Generally, the adjuvant moiety described herein is a STING agonist. Certain amido benzimidazole compounds are demonstrated STING receptor agonists with systemic activity (Ramanjulu, J.M. et al (2018 ) Nature 564:439–443; Barber, G.N. (2015) Nature Rev Immunol 15:760–770; US 2019/0300513). STING is a dimeric structure with a large and symmetrical binding pocket. The aza- bicyclic compounds (AzaBi) of Tables 1a and 1b when conjugated to a targeting antibody were designed to target and bind to the open conformation of the binding pocket of STING. Binding to a small molecule agonist usually induces a closed conformation of the STING protein. This introduces the risk that a linker, particularly if “noncleavable”, will interfere with binding and activation. The bis-benzimidazole STING agonists are reported to bind and activate through an open conformation, which are predicted to be more amenable to attachment of a linker (Ramanjulu, J.M. et al (2018) Nature 564:439–443; Barber, G.N. (2015) Nature Rev Immunol 15:760–770).
”) present is considered to be used as read from left to right. “Alkyl” refers to a straight (linear) or branched, saturated, aliphatic radical having the number of carbon atoms indicated. Alkyl can include any number of carbons, for example from one to six, one to eight, one to twelve or one to twenty. Examples of alkyl groups include, but are not limited to, methyl (Me, -CH3), ethyl (Et, -CH2CH3), 1-propyl (n-Pr, n-propyl, - CH2CH2CH3), 2-propyl (i-Pr, i-propyl, -CH(CH3)2), 1-butyl (n-Bu, n-butyl, -CH2CH2CH2CH3), 2-methyl-1-propyl (i-Bu, i-butyl, -CH2CH(CH3)2), 2-butyl (s-Bu, s-butyl, -CH(CH3)CH2CH3), 2- methyl-2-propyl (t-Bu, t-butyl, -C(CH3)3), 1-pentyl (n-pentyl, -CH2CH2CH2CH2CH3), 2-pentyl (-CH(CH3)CH2CH2CH3), 3-pentyl (-CH(CH2CH3)2), 2-methyl-2-butyl (-C(CH3)2CH2CH3), 3- methyl-2-butyl (-CH(CH3)CH(CH3)2), 3-methyl-1-butyl (-CH2CH2CH(CH3)2), 2-methyl-1-butyl (-CH2CH(CH3)CH2CH3), 1-hexyl (-CH2CH2CH2CH2CH2CH3), 2-hexyl (- CH(CH3)CH2CH2CH2CH3), 3-hexyl (-CH(CH2CH3)(CH2CH2CH3)), 2-methyl-2-pentyl (- C(CH3)2CH2CH2CH3), 3-methyl-2-pentyl (-CH(CH3)CH(CH3)CH2CH3), 4-methyl-2-pentyl (- CH(CH3)CH2CH(CH3)2), 3-methyl-3-pentyl (-C(CH3)(CH2CH3)2), 2-methyl-3-pentyl (- CH(CH2CH3)CH(CH3)2), 2,3-dimethyl-2-butyl (-C(CH3)2CH(CH3)2), 3,3-dimethyl-2-butyl (- CH(CH3)C(CH3)3, 1-heptyl, 1-octyl, and the like. Alkyl groups can be substituted or unsubstituted. Substituted alkyl groups can be substituted with one or more groups selected from halo, hydroxy, amino, oxo (=O), alkylamino, amido, acyl, nitro, cyano, and alkoxy. Substituted alkyl groups can be geminally substituted where a carbon atom of the alkyl forms a spiro, cycloalkyl group such as cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. The term “alkyldiyl” refers to a divalent alkyl radical. Examples of alkyldiyl groups include, but are not limited to, methylene (-CH2-), ethylene (-CH2CH2-), propylene (- CH2CH2CH2-), and the like. An alkyldiyl group may also be referred to as an “alkylene” group. Alkyldiyl groups can be substituted or unsubstituted. Substituted alkyldiyl groups can be substituted with one or more groups selected from halo, hydroxy, amino, oxo (=O), alkylamino, amido, acyl, nitro, cyano, and alkoxy. Substituted alkyldiyl groups can be geminally substituted where a carbon atom of the alkyl forms a spiro, cycloalkyl group such as cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl.
“Alkenyl” refers to a straight (linear) or branched, unsaturated, aliphatic radical having the number of carbon atoms indicated and at least one carbon-carbon double bond, sp2. Alkenyl can include from two to about 12 or more carbons atoms. Alkenyl groups are radicals having “cis” and “trans” orientations, or alternatively, “E” and “Z” orientations. Examples include, but are not limited to, ethylenyl or vinyl (-CH=CH2), allyl (-CH2CH=CH2). butenyl, pentenyl, and isomers thereof. Alkenyl groups can be substituted or unsubstituted. “Substituted alkenyl” groups can be substituted with one or more groups selected from halo, hydroxy, amino, oxo (=O), alkylamino, amido, acyl, nitro, cyano, and alkoxy. The terms “alkenylene” or “alkenyldiyl” refer to a linear or branched-chain divalent hydrocarbon radical. Examples include, but are not limited to, ethylenylene or vinylene (- CH=CH-), allyl (-CH2CH=CH-), and the like. “Alkynyl” refers to a straight (linear) or branched, unsaturated, aliphatic radical having the number of carbon atoms indicated and at least one carbon-carbon triple bond, sp. Alkynyl can include from two to about 12 or more carbons atoms. For example, C2-C6 alkynyl includes, but is not limited to ethynyl (-C ≡CH), propynyl (propargyl, -CH2C ≡CH), butynyl, pentynyl, hexynyl, and isomers thereof Alkynyl groups can be substituted or unsubstituted. “Substituted alkynyl” groups can be substituted with one or more groups selected from halo, hydroxy, amino, oxo (=O), alkylamino, amido, acyl, nitro, cyano, and alkoxy. The term “alkynylene” or “alkynyldiyl” refer to a divalent alkynyl radical. "Heteroalkyl" or “heteroalkylene” refer to a monovalent, straight or branched chain alkyl group, as defined above, comprising at least one heteroatom including but not limited to Si, N, O, P or S within the alkyl chain or at a terminus of the alkyl chain. In some embodiments, a heteroatom is within the alkyl chain. In other embodiments, a heteroatom is at a terminus of the alkylene and thus serves to join the alkyl to the remainder of the molecule. Unless stated otherwise specifically in the specification, a heteroalkyl group is optionally substituted. For example, heteroalkyl groups can be substituted with 1-6 fluoro (F) substituents, for example, on the carbon backbone (as − CHF− or −CF2−) or on terminal carbons of straight chain or branched heteroalkyls (such as −CHF2 or −CF3). Examples of heteroalkyl groups include, but are not limited to, − CH2CH2OCH3, −CH2CH2NHCH3, −CH2CH2N(CH3)2, −C(=O)NHCH2CH2NHCH3, − C(=O)N(CH3)CH2CH2N(CH3)2, −C(=O)NHCH2CH2NHC(=O)CH2CH3, − C(=O)N(CH3)CH2CH2N(CH3)C(=O)CH2CH3, −OCH2CH2CH2NH(CH3), − OCH2CH2CH2N(CH3)2, −OCH2CH2CH2NHC(=O)CH2CH3, −
OCH2CH2CH2N(CH3)C(=O)CH2CH3, −CH2CH2CH2NH(CH3), −OCH2CH2CH2N(CH3)2, −CH2CH2CH2NHC(=O)CH2CH3, −CH2CH2CH2N(CH3)C(=O)CH2CH3, −CH2SCH2CH3, −CH2CH2S(O)CH3, −NHCH2CH2NHC(=O)CH2CH3, −CH2CH2S(O)2CH3, − CH2CH2OCF3, and −Si(CH3)3. Up to two heteroatoms may be consecutive, such as, for example, −CH2NHOCH3 and −CH2OSi(CH3)3. A terminal polyethylene glycol (PEG) moiety is a type of heteroalkyl group. Exemplary heteroalkyl groups also include ethylene oxide (e.g., polyethylene oxide), propylene oxide, amino acid chains (i.e., short to medium length peptides such as containing 1-15 amino acids), and alkyl chains connected via a variety of functional groups such as amides, disulfides, ketones, phosphonates, phosphates, sulfates, sulfones, sulfonamides, esters, ethers, -S-, carbamates, ureas, thioureas, anhydrides, or the like (including combinations thereof). In some embodiments, a heteroalkyl group includes a polyamino acid having 1-10 amino acids. In some embodiments, a heteroalkyl group includes a polyamino acid having 1-5 amino acids. Exemplary heteroalkyl groups include a solubilizing group (SolG) comprising one or more units of polyglycine, polysarcosine, polyethyleneoxy (PEG), and a glycoside, or combinations thereof. "Heteroalkenyl" refers to a heteroalkyl group, as defined above, that contains at least one carbon-carbon double bond. "Heteroalkynyl" refers to a heteroalkyl group, as defined above, that contains at least one carbon-carbon triple bond. “Heteroalkyldiyl” refers to a divalent form of a heteroalkyl group as defined above. Examples of heteroalkylene groups include, but are not limited to, − CH2CH2OCH2−, −CH2CH2OCF2−, −CH2CH2NHCH2−, −C(=O)NHCH2CH2NHCH2−, − C(=O)N(CH3)CH2CH2N(CH3)CH2−, −C(=O)NHCH2CH2NHC(=O)CH2CH2−, − C(=O)N(CH3)CH2CH2N(CH3)C(=O)CH2CH2−, −OCH2CH2OCH2CH2−, − OCH2CH2OCH2C(=O)−, −OCH2CH2OCH2CH2C(=O)−, −OCH2CH2NHCH2−, − OCH2CH2N(CH3)CH2−, −OCH2CH2CH2NHCH2−, −OCH2CH2CH2N(CH3)CH2−, − OCH2CH2CH2NHC(=O)CH2CH2−, −OCH2CH2CH2N(CH3)C(=O)CH2CH2−, − CH2CH2CH2NHCH2−-, −CH2CH2CH2N(CH3)CH2−, −CH2CH2CH2NHC(=O)CH2CH2−, −CH2CH2CH2N(CH3)C(=O)CH2CH2−, −CH2CH2NHC(=O)−, −CH2CH2N(CH3)CH2−, − CH2CH2N+(CH3)2−, −NHCH2CH2(NH2)CH2−, and −NHCH2CH2(NHCH3)CH2−. A divalent polyethylene glycol (PEG) moiety with one to about 50 units of −OCH2CH2− is a
type of heteroalkyldiyl group. “Heteroalkenyldiyl” refers to a divalent form of a heteroalkenyl group. “Heteroalkynyldiyl” refers to a divalent form of a heteroalkynyl group. The terms “carbocycle”, “carbocyclyl”, “carbocyclic ring” and “cycloalkyl” refer to a saturated or partially unsaturated, monocyclic, fused bicyclic, spiro, or bridged polycyclic ring assembly containing from 3 to 12 ring atoms, or the number of atoms indicated. Saturated monocyclic carbocyclic rings include, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cyclooctyl. Saturated bicyclic and polycyclic carbocyclic rings include, for example, norbornane, [2.2.2] bicyclooctane, decahydronaphthalene and adamantane. Carbocyclic groups can also be partially unsaturated, having one or more double or triple bonds in the ring. Representative carbocyclic groups that are partially unsaturated include, but are not limited to, cyclobutene, cyclopentene, cyclohexene, cyclohexadiene (1,3- and 1,4-isomers), cycloheptene, cycloheptadiene, cyclooctene, cyclooctadiene (1,3-, 1,4- and 1,5-isomers), norbornene, and norbornadiene. The term “cycloalkyldiyl” refers to a divalent cycloalkyl radical. “Aryl” refers to a monovalent aromatic hydrocarbon radical of 6-20 carbon atoms (C6− C20) derived by the removal of one hydrogen atom from a single carbon atom of a parent aromatic ring system.. Aryl groups can be monocyclic, fused to form bicyclic or tricyclic groups, or linked by a bond to form a biaryl group. Representative aryl groups include phenyl, naphthyl and biphenyl. Other aryl groups include benzyl, having a methylene linking group. Some aryl groups have from 6 to 12 ring members, such as phenyl, naphthyl or biphenyl. Other aryl groups have from 6 to 10 ring members, such as phenyl or naphthyl. The terms “arylene” or “aryldiyl” mean a divalent aromatic hydrocarbon radical of 6-20 carbon atoms (C6−C20) derived by the removal of two hydrogen atom from a two carbon atoms of a parent aromatic ring system. Some aryldiyl groups are represented in the exemplary structures as “Ar”. Aryldiyl includes bicyclic radicals comprising an aromatic ring fused to a saturated, partially unsaturated ring, or aromatic carbocyclic ring. Typical aryldiyl groups include, but are not limited to, radicals derived from benzene (phenyldiyl), substituted benzenes, naphthalene, anthracene, biphenylene, indenylene, indanylene, 1,2-dihydronaphthalene, 1,2,3,4- tetrahydronaphthyl, and the like. Aryldiyl groups are also referred to as “arylene”, and are optionally substituted with one or more substituents described herein. The terms “heterocycle”, “heterocyclyl”, and “heterocyclic ring” are used interchangeably herein and refer to a saturated or a partially unsaturated (i.e., having one or more double and/or triple bonds within the ring) carbocyclic radical of 3 to about 20 ring atoms in which at least one ring atom is a heteroatom selected from nitrogen, oxygen, phosphorus and
sulfur, the remaining ring atoms being C, where one or more ring atoms is optionally substituted independently with one or more substituents described below. A heterocycle may be a monocycle having 3 to 7 ring members (2 to 6 carbon atoms and 1 to 4 heteroatoms selected from N, O, P, and S) or a bicycle having 7 to 10 ring members (4 to 9 carbon atoms and 1 to 6 heteroatoms selected from N, O, P, and S), for example: a bicyclo [4,5], [5,5], [5,6], or [6,6] system. Heterocycles are described in Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W.A. Benjamin, New York, 1968), particularly Chapters 1, 3, 4, 6, 7, and 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28; and J. Am. Chem. Soc. (1960) 82:5566. “Heterocyclyl” also includes radicals where heterocycle radicals are fused with a saturated, partially unsaturated ring, or aromatic carbocyclic or heterocyclic ring. Examples of heterocyclic rings include, but are not limited to, morpholin-4-yl, piperidin-1-yl, piperazinyl, piperazin-4-yl-2-one, piperazin-4-yl-3-one, pyrrolidin-1-yl, thiomorpholin-4-yl, S- dioxothiomorpholin-4-yl, azocan-1-yl, azetidin-1-yl, octahydropyrido[1,2-a]pyrazin-2-yl, [1,4]diazepan-1-yl, pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl, homopiperazinyl, azetidinyl, oxetanyl, thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl, diazepinyl, thiazepinyl, 2-pyrrolinyl, 3- pyrrolinyl, indolinyl, 2H-pyranyl, 4H-pyranyl, dioxanyl, 1,3-dioxolanyl, pyrazolinyl, dithianyl, dithiolanyl, dihydropyranyl, dihydrothienyl, dihydrofuranyl, pyrazolidinylimidazolinyl, imidazolidinyl, 3-azabicyco[3.1.0]hexanyl, 3-azabicyclo[4.1.0]heptanyl, azabicyclo[2.2.2]hexanyl, 3H-indolyl quinolizinyl and N-pyridyl ureas. Spiro heterocyclyl moieties are also included within the scope of this definition. Examples of spiro heterocyclyl moieties include azaspiro[2.5]octanyl and azaspiro[2.4]heptanyl. Examples of a heterocyclic group wherein 2 ring atoms are substituted with oxo (=O) moieties are pyrimidinonyl and 1,1- dioxo-thiomorpholinyl. The heterocycle groups herein are optionally substituted independently with one or more substituents described herein. The term “heterocyclyldiyl” refers to a divalent, saturated or a partially unsaturated (i.e., having one or more double and/or triple bonds within the ring) carbocyclic radical of 3 to about 20 ring atoms in which at least one ring atom is a heteroatom selected from nitrogen, oxygen, phosphorus and sulfur, the remaining ring atoms being C, where one or more ring atoms is optionally substituted independently with one or more substituents as described. Examples of 5- membered and 6-membered heterocyclyldiyls include morpholinyldiyl, piperidinyldiyl, piperazinyldiyl, pyrrolidinyldiyl, dioxanyldiyl, thiomorpholinyldiyl, and S- dioxothiomorpholinyldiyl.
The term “heteroaryl” refers to a monovalent aromatic radical of 5-, 6-, or 7-membered rings, and includes fused ring systems (at least one of which is aromatic) of 5-20 atoms, containing one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur. Examples of heteroaryl groups are pyridinyl (including, for example, 2-hydroxypyridinyl), imidazolyl, imidazopyridinyl, pyrimidinyl (including, for example, 4-hydroxypyrimidinyl), pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxadiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl, thiadiazolyl, thiadiazolyl, furazanyl, benzofurazanyl, benzothiophenyl, benzothiazolyl, benzoxazolyl, quinazolinyl, quinoxalinyl, naphthyridinyl, and furopyridinyl. Heteroaryl groups are optionally substituted independently with one or more substituents described herein. The term “heteroaryldiyl” refers to a divalent aromatic radical of 5-, 6-, or 7-membered rings, and includes fused ring systems (at least one of which is aromatic) of 5-20 atoms, containing one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur. Examples of 5-membered and 6-membered heteroaryldiyls include pyridyldiyl, imidazolyldiyl, pyrimidinyldiyl, pyrazolyldiyl, triazolyldiyl, pyrazinyldiyl, tetrazolyldiyl, furyldiyl, thienyldiyl, isoxazolyldiyldiyl, thiazolyldiyl, oxadiazolyldiyl, oxazolyldiyl, isothiazolyldiyl, and pyrrolyldiyl. The heterocycle or heteroaryl groups may be carbon (carbon-linked), or nitrogen (nitrogen-linked) bonded where such is possible. By way of example and not limitation, carbon bonded heterocycles or heteroaryls are bonded at position 2, 3, 4, 5, or 6 of a pyridine, position 3, 4, 5, or 6 of a pyridazine, position 2, 4, 5, or 6 of a pyrimidine, position 2, 3, 5, or 6 of a pyrazine, position 2, 3, 4, or 5 of a furan, tetrahydrofuran, thiofuran, thiophene, pyrrole or tetrahydropyrrole, position 2, 4, or 5 of an oxazole, imidazole or thiazole, position 3, 4, or 5 of an isoxazole, pyrazole, or isothiazole, position 2 or 3 of an aziridine, position 2, 3, or 4 of an azetidine, position 2, 3, 4, 5, 6, 7, or 8 of a quinoline or position 1, 3, 4, 5, 6, 7, or 8 of an isoquinoline. By way of example and not limitation, nitrogen bonded heterocycles or heteroaryls are bonded at position 1 of an aziridine, azetidine, pyrrole, pyrrolidine, 2-pyrroline, 3-pyrroline, imidazole, imidazolidine, 2-imidazoline, 3-imidazoline, pyrazole, pyrazoline, 2-pyrazoline, 3- pyrazoline, piperidine, piperazine, indole, indoline, 1H-indazole, position 2 of a isoindole, or isoindoline, position 4 of a morpholine, and position 9 of a carbazole, or β-carboline. The terms “halo” and “halogen,” by themselves or as part of another substituent, refer to a fluorine, chlorine, bromine, or iodine atom.
The term “carbonyl,” by itself or as part of another substituent, refers to C(=O) or – C(=O)–, i.e., a carbon atom double-bonded to oxygen and bound to two other groups in the moiety having the carbonyl. As used herein, the phrase “quaternary ammonium salt” refers to a tertiary amine that has been quaternized with an alkyl substituent (e.g., a C1-C4 alkyl such as methyl, ethyl, propyl, or butyl). The terms “treat,” “treatment,” and “treating” refer to any indicia of success in the treatment or amelioration of an injury, pathology, condition (e.g., cancer), or symptom (e.g., cognitive impairment), including any objective or subjective parameter such as abatement; remission; diminishing of symptoms or making the symptom, injury, pathology, or condition more tolerable to the patient; reduction in the rate of symptom progression; decreasing the frequency or duration of the symptom or condition; or, in some situations, preventing the onset of the symptom. The treatment or amelioration of symptoms can be based on any objective or subjective parameter, including, for example, the result of a physical examination. The terms “cancer,” “neoplasm,” and “tumor” are used herein to refer to cells which exhibit autonomous, unregulated growth, such that the cells exhibit an aberrant growth phenotype characterized by a significant loss of control over cell proliferation. Cells of interest for detection, analysis, and/or treatment in the context of the invention include cancer cells (e.g., cancer cells from an individual with cancer), malignant cancer cells, pre-metastatic cancer cells, metastatic cancer cells, and non-metastatic cancer cells. Cancers of virtually every tissue are known. The phrase “cancer burden” refers to the quantum of cancer cells or cancer volume in a subject. Reducing cancer burden accordingly refers to reducing the number of cancer cells or the cancer cell volume in a subject. The term “cancer cell” as used herein refers to any cell that is a cancer cell (e.g., from any of the cancers for which an individual can be treated, e.g., isolated from an individual having cancer) or is derived from a cancer cell, e.g., clone of a cancer cell. For example, a cancer cell can be from an established cancer cell line, can be a primary cell isolated from an individual with cancer, can be a progeny cell from a primary cell isolated from an individual with cancer, and the like. In some embodiments, the term can also refer to a portion of a cancer cell, such as a sub-cellular portion, a cell membrane portion, or a cell lysate of a cancer cell. Many types of cancers are known to those of skill in the art, including solid tumors such as carcinomas, sarcomas, glioblastomas, melanomas, lymphomas, and myelomas, and circulating cancers such as leukemias. As used herein, the term “cancer” includes any form of cancer, including but not limited to, solid tumor cancers (e.g., skin, lung, prostate, breast, gastric, bladder, colon, ovarian, pancreas, kidney, liver, glioblastoma, medulloblastoma, leiomyosarcoma, head & neck
squamous cell carcinomas, melanomas, and neuroendocrine) and liquid cancers (e.g., hematological cancers); carcinomas; soft tissue tumors; sarcomas; teratomas; melanomas; leukemias; lymphomas; and brain cancers, including minimal residual disease, and including both primary and metastatic tumors. The “pathology” of cancer includes all phenomena that compromise the well-being of the patient. This includes, without limitation, abnormal or uncontrollable cell growth, metastasis, interference with the normal functioning of neighboring cells, release of cytokines or other secretory products at abnormal levels, suppression or aggravation of inflammatory or immunological response, neoplasia, premalignancy, malignancy, and invasion of surrounding or distant tissues or organs, such as lymph nodes. As used herein, the phrases “cancer recurrence” and “tumor recurrence,” and grammatical variants thereof, refer to further growth of neoplastic or cancerous cells after diagnosis of cancer. Particularly, recurrence may occur when further cancerous cell growth occurs in the cancerous tissue. “Tumor spread,” similarly, occurs when the cells of a tumor disseminate into local or distant tissues and organs, therefore, tumor spread encompasses tumor metastasis. “Tumor invasion” occurs when the tumor growth spread out locally to compromise the function of involved tissues by compression, destruction, or prevention of normal organ function. As used herein, the term “metastasis” refers to the growth of a cancerous tumor in an organ or body part, which is not directly connected to the organ of the original cancerous tumor. Metastasis will be understood to include micrometastasis, which is the presence of an undetectable amount of cancerous cells in an organ or body part that is not directly connected to the organ of the original cancerous tumor. Metastasis can also be defined as several steps of a process, such as the departure of cancer cells from an original tumor site, and migration and/or invasion of cancer cells to other parts of the body. The phrases “effective amount” and “therapeutically effective amount” refer to a dose or amount of a substance such as an immunoconjugate that produces therapeutic effects for which it is administered. The exact dose will depend on the purpose of the treatment, and will be ascertainable by one skilled in the art using known techniques (see, e.g., Lieberman, Pharmaceutical Dosage Forms (vols.1-3, 1992); Lloyd, The Art, Science and Technology of Pharmaceutical Compounding (1999); Pickar, Dosage Calculations (1999); Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th Edition (McGraw-Hill, 2006); and Remington: The Science and Practice of Pharmacy, 22nd Edition, (Pharmaceutical Press, London, 2012)). In the case of cancer, the therapeutically effective amount of the immunoconjugate may reduce the number of cancer cells; reduce the tumor size; inhibit (i.e.,
slow to some extent and preferably stop) cancer cell infiltration into peripheral organs; inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; inhibit, to some extent, tumor growth; and/or relieve to some extent one or more of the symptoms associated with the cancer. To the extent the immunoconjugate may prevent growth and/or kill existing cancer cells, it may be cytostatic and/or cytotoxic. For cancer therapy, efficacy can, for example, be measured by assessing the time to disease progression (TTP) and/or determining the response rate (RR) “Recipient,” “individual,” “subject,” “host,” and “patient” are used interchangeably and refer to any mammalian subject for whom diagnosis, treatment, or therapy is desired (e.g., humans). “Mammal” for purposes of treatment refers to any animal classified as a mammal, including humans, domestic and farm animals, and zoo, sports, or pet animals, such as dogs, horses, cats, cows, sheep, goats, pigs, camels, etc. In certain embodiments, the mammal is human. The phrase “synergistic adjuvant” or “synergistic combination” in the context of this invention includes the combination of two immune modulators such as a receptor agonist, cytokine, and adjuvant polypeptide, that in combination elicit a synergistic effect on immunity relative to either administered alone. Particularly, the immunoconjugates disclosed herein comprise synergistic combinations of the claimed adjuvant and antibody construct. These synergistic combinations upon administration elicit a greater effect on immunity, e.g., relative to when the antibody construct or adjuvant is administered in the absence of the other moiety. Further, a decreased amount of the immunoconjugate may be administered (as measured by the total number of antibody constructs or the total number of adjuvants administered as part of the immunoconjugate) compared to when either the antibody construct or adjuvant is administered alone. As used herein, the term “administering” refers to parenteral, intravenous, intraperitoneal, intramuscular, intratumoral, intralesional, intranasal, or subcutaneous administration, oral administration, administration as a suppository, topical contact, intrathecal administration, or the implantation of a slow-release device, e.g., a mini-osmotic pump, to the subject. The terms “about” and “around,” as used herein to modify a numerical value, indicate a close range surrounding the numerical value. Thus, if “X” is the value, “about X” or “around X” indicates a value of from 0.9X to 1.1X, e.g., from 0.95X to 1.05X or from 0.99X to 1.01X. A reference to “about X” or “around X” specifically indicates at least the values X, 0.95X, 0.96X, 0.97X, 0.98X, 0.99X, 1.01X, 1.02X, 1.03X, 1.04X, and 1.05X. Accordingly, “about X” and “around X” are intended to teach and provide written description support for a claim limitation of, e.g., “0.98X.”
ANTIBODIES The immunoconjugate of the invention comprises an antibody. Included in the scope of the embodiments of the invention are functional variants of the antibody constructs or antigen binding domain described herein. The term “functional variant” as used herein refers to an antibody construct having an antigen binding domain with substantial or significant sequence identity or similarity to a parent antibody construct or antigen binding domain, which functional variant retains the biological activity of the antibody construct or antigen binding domain of which it is a variant. Functional variants encompass, for example, those variants of the antibody constructs or antigen binding domain described herein (the parent antibody construct or antigen binding domain) that retain the ability to recognize target cells expressing a tumor-associated antigen or cell surface receptor to a similar extent, the same extent, or to a higher extent, as the parent antibody construct or antigen binding domain. In reference to the antibody construct or antigen binding domain, the functional variant can, for instance, be at least about 30%, about 50%, about 75%, about 80%, about 85%, about 90%, about 91%, about 92%, about 93%, about 94%, about 95%, about 96%, about 97%, about 98%, about 99% or more identical in amino acid sequence to the antibody construct or antigen binding domain. A functional variant can, for example, comprise the amino acid sequence of the parent antibody construct or antigen binding domain with at least one conservative amino acid substitution. Alternatively, or additionally, the functional variants can comprise the amino acid sequence of the parent antibody construct or antigen binding domain with at least one non- conservative amino acid substitution. In this case, it is preferable for the non-conservative amino acid substitution to not interfere with or inhibit the biological activity of the functional variant. The non-conservative amino acid substitution may enhance the biological activity of the functional variant, such that the biological activity of the functional variant is increased as compared to the parent antibody construct or antigen binding domain. The antibodies comprising the immunoconjugates of the invention include Fc engineered variants. In some embodiments, the mutations in the Fc region that result in modulated binding to one or more Fc receptors can include one or more of the following mutations: SD (S239D), SDIE (S239D/I332E), SE (S267E), SELF (S267E/L328F), SDIE (S239D/I332E), SDIEAL (S239D/I332E/A330L), GA (G236A), ALIE (A330L/I332E), GASDALIE (G236A/S239D/A330L/I332E), V9 (G237D/P238D/P271G/A330R), and V11 (G237D/P238D/H268D/P271G/A330R), and/or one or more mutations at the following amino acids: E345R, E233, G237, P238, H268, P271, L328 and A330. Additional Fc region modifications for modulating Fc receptor binding are described in, for example, US
2016/0145350, US 7416726 and US 5624821, which are hereby incorporated by reference in their entireties herein. The antibodies comprising the immunoconjugates of the invention include glycan variants, such as afucosylation. In some embodiments, the Fc region of the binding agents are modified to have an altered glycosylation pattern of the Fc region compared to the native non-modified Fc region. In some embodiments, the antibodies in the immunoconjugates contain a modified Fc region, wherein the modification modulates the binding of the Fc region to one or more Fc receptors. In some embodiments, the antibodies in the immunoconjugates (e.g., antibodies conjugated to at least two adjuvant moieties) contain one or more modifications (e.g., amino acid insertion, deletion, and/or substitution) in the Fc region that results in modulated binding (e.g., increased binding or decreased binding) to one or more Fc receptors (e.g., FcγRI (CD64), FcγRIIA (CD32A), FcγRIIB (CD32B), FcγRIIIA (CD16a), and/or FcγRIIIB (CD16b)) as compared to the native antibody lacking the mutation in the Fc region. In some embodiments, the antibodies in the immunoconjugates contain one or more modifications (e.g., amino acid insertion, deletion, and/or substitution) in the Fc region that reduce the binding of the Fc region of the antibody to FcγRIIB. In some embodiments, the antibodies in the immunoconjugates contain one or more modifications (e.g., amino acid insertion, deletion, and/or substitution) in the Fc region of the antibody that reduce the binding of the antibody to FcγRIIB while maintaining the same binding or having increased binding to FcγRI (CD64), FcγRIIA (CD32A), and/or FcRγIIIA (CD16a) as compared to the native antibody lacking the mutation in the Fc region. In some embodiments, the antibodies in the immunoconjugates contain one of more modifications in the Fc region that increase the binding of the Fc region of the antibody to FcγRIIB. In some embodiments, the modulated binding is provided by mutations in the Fc region of the antibody relative to the native Fc region of the antibody. The mutations can be in a CH2 domain, a CH3 domain, or a combination thereof. A “native Fc region” is synonymous with a “wild-type Fc region” and comprises an amino acid sequence that is identical to the amino acid sequence of an Fc region found in nature or identical to the amino acid sequence of the Fc region found in the native antibody (e.g., cetuximab). Native sequence human Fc regions include a native sequence human IgG1 Fc region, native sequence human IgG2 Fc region, native sequence human IgG3 Fc region, and native sequence human IgG4 Fc region, as well as naturally occurring variants thereof. Native sequence Fc includes the various allotypes of Fcs (Jefferis et al., (2009) mAbs, 1(4):332-338).
In some embodiments, the Fc region of the antibodies of the immunoconjugates are modified to have an altered glycosylation pattern of the Fc region compared to the native non-modified Fc region. Human immunoglobulin is glycosylated at the Asn297 residue in the Cγ2 domain of each heavy chain. This N-linked oligosaccharide is composed of a core heptasaccharide, N-acetylglucosamine4Mannose3 (GlcNAc4Man3). Removal of the heptasaccharide with endoglycosidase or PNGase F is known to lead to conformational changes in the antibody Fc region, which can significantly reduce antibody-binding affinity to activating FcγR and lead to decreased effector function. The core heptasaccharide is often decorated with galactose, bisecting GlcNAc, fucose, or sialic acid, which differentially impacts Fc binding to activating and inhibitory FcγR. Additionally, it has been demonstrated that α2,6-sialyation enhances anti-inflammatory activity in vivo, while afucosylation leads to improved FcγRIIIa binding and a 10-fold increase in antibody-dependent cellular cytotoxicity and antibody-dependent phagocytosis. Specific glycosylation patterns, therefore, can be used to control inflammatory effector functions. In some embodiments, the modification to alter the glycosylation pattern is a mutation. For example, a substitution at Asn297. In some embodiments, Asn297 is mutated to glutamine (N297Q). Methods for controlling immune response with antibodies that modulate FcγR- regulated signaling are described, for example, in US 7416726, US 2007/0014795 and US 2008/0286819, which are hereby incorporated by reference in their entireties. In some embodiments, the antibodies of the immunoconjugates are modified to contain an engineered Fab region with a non-naturally occurring glycosylation pattern. For example, hybridomas can be genetically engineered to secrete afucosylated mAb, desialylated mAb or deglycosylated Fc with specific mutations that enable increased FcRγIIIa binding and effector function. In some embodiments, the antibodies of the immunoconjugates are engineered to be afucosylated. In some embodiments, the entire Fc region of an antibody in the immunoconjugates is exchanged with a different Fc region, so that the Fab region of the antibody is conjugated to a non-native Fc region. For example, the Fab region of cetuximab, which normally comprises an IgG1 Fc region, can be conjugated to IgG2, IgG3, IgG4, or IgA, or the Fab region of nivolumab, which normally comprises an IgG4 Fc region, can be conjugated to IgG1, IgG2, IgG3, IgA1, or IgG2. In some embodiments, the Fc modified antibody with a non-native Fc domain also comprises one or more amino acid modification, such as the S228P mutation within the IgG4 Fc, that modulate the stability of the Fc domain described. In some embodiments, the Fc modified
antibody with a non-native Fc domain also comprises one or more amino acid modifications described herein that modulate Fc binding to FcR. In some embodiments, the modifications that modulate the binding of the Fc region to FcR do not alter the binding of the Fab region of the antibody to its antigen when compared to the native non-modified antibody. In other embodiments, the modifications that modulate the binding of the Fc region to FcR also increase the binding of the Fab region of the antibody to its antigen when compared to the native non-modified antibody. In some embodiments, the antibodies in the immunoconjugates contain a modified Fc region, wherein the modification modulates the binding of the Fc region to one or more Fc receptors. In some embodiments, the Fc region is modified by inclusion of a transforming growth factor beta 1 (TGFβ1) receptor, or a fragment thereof, that is capable of binding TGFβ1. For example, the receptor can be TGFβ receptor II (TGFβRII). In some embodiments, theTGFβ receptor is a human TGFβ receptor. In some embodiments, the IgG has a C-terminal fusion to a TGFβRII extracellular domain (ECD) as described in US 9676863, incorporated herein. An “Fc linker” may be used to attach the IgG to the TGFβRII extracellular domain. The Fc linker may be a short, flexible peptide that allows for the proper three-dimensional folding of the molecule while maintaining the binding-specificity to the targets. In some embodiments, the N-terminus of the TGFβ receptor is fused to the Fc of the antibody construct (with or without an Fc linker). In some embodiments, the C-terminus of the antibody construct heavy chain is fused to the TGFβ receptor (with or without an Fc linker). In some embodiments, the C-terminal lysine residue of the antibody construct heavy chain is mutated to alanine. In some embodiments, the antibodies in the immunoconjugates are glycosylated. In some embodiments, the antibodies in the immunoconjugates are a cysteine-engineered antibody which provides for site-specific conjugation of an adjuvant, label, or drug moiety to the antibody through cysteine substitutions at sites where the engineered cysteines are available for conjugation but do not perturb immunoglobulin folding and assembly or alter antigen binding and effector functions (Junutula, et al., (2008) Nature Biotech., 26(8):925-932; Dornan et al. (2009) Blood 114(13):2721-2729; US 7521541; US 7723485; US 2012/0121615; WO 2009/052249). A “cysteine engineered antibody” or “cysteine engineered antibody variant” is an antibody in which one or more residues of an antibody are substituted with cysteine residues. Cysteine-engineered antibodies can be conjugated to the aza-bicyclic (AzaBi) moiety as a AzaBi-linker compound with uniform stoichiometry (e.g., up to two AzaBi moieties per antibody in an antibody that has a single engineered cysteine site).
In some embodiments, cysteine-engineered antibodies are used to prepare immunoconjugates. Immunoconjugates may have a reactive cysteine thiol residue introduced at a site on the light chain, such as the 149-lysine site (LC K149C), or on the heavy chain such as the 122-serine site (HC S122C), as numbered by Kabat numbering. In other embodiments, the cysteine-engineered antibodies have a cysteine residue introduced at the 118-alanine site (EU numbering) of the heavy chain (HC A118C). This site is alternatively numbered 121 by Sequential numbering or 114 by Kabat numbering. In other embodiments, the cysteine- engineered antibodies have a cysteine residue introduced in: (i) the light chain at G64C, R142C, K188C, L201C, T129C, S114C, or E105C according to Kabat numbering; (ii) the heavy chain at D101C, V184C, T205C, or S122C according to Kabat numbering; or (iii) other cysteine-mutant antibodies, and as described in Bhakta, S. et al, (2013) “Engineering THIOMABs for Site- Specific Conjugation of Thiol-Reactive Linkers”, Laurent Ducry (ed.), Antibody-Drug Conjugates, Methods in Molecular Biology, vol.1045, pages 189-203; WO 2011/156328; US 9000130. IMMUNE CHECKPOINT INHIBITORS In some embodiments, the antibody of an immunoconjugate is an immune checkpoint inhibitor. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins. In another embodiment, the immune checkpoint inhibitor reduces the interaction between one or more immune checkpoint proteins and their ligands. Inhibitory nucleic acids that decrease the expression and/or activity of immune checkpoint molecules can also be used in the methods disclosed herein. Immune checkpoint inhibitors nivolumab and atezolizumab can be modified to include an IgG1 Fc, and subsequently converted into an immunoconjugate of the invention. Most checkpoint antibodies are designed not to have effector function to kill cells, but rather to block the signalling. Immunoconjugates of the present invention can add back the "effector functionality" needed to activate myeloid immunity. In some embodiments, the immune checkpoint inhibitor is cytotoxic T-lymphocyte antigen 4 (CTLA4, also known as CD152), T cell immunoreceptor with Ig and ITIM domains (TIGIT), glucocorticoid-induced TNFR-related protein (GITR, also known as TNFRSF18), inducible T cell costimulatory (ICOS, also known as CD278), CD96, poliovirus receptor-related 2 (PVRL2, also known as CD112R, programmed cell death protein 1 (PD-1, also known as CD279), programmed cell death 1 ligand 1 (PD-L1, also known as B7-H3 and CD274), programmed cell death ligand 2 (PD-L2, also known as B7-DC and CD273), lymphocyte activation gene-3 (LAG-3, also known as CD223), B7-H4, killer immunoglobulin receptor
(KIR), Tumor Necrosis Factor Receptor superfamily member 4 (TNFRST4, also known as OX40 and CD134) and its ligand OX40L (CD252), indoleamine 2,3-dioxygenase 1 (IDO-1), indoleamine 2,3-dioxygenase 2 (IDO-2), carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), B and T lymphocyte attenuator (BTLA, also known as CD272), T-cell membrane protein 3 (TIM3), the adenosine A2A receptor (A2Ar), and V-domain Ig suppressor of T cell activation (VISTA protein). In some embodiments, the immune checkpoint inhibitor is an inhibitor of CTLA4, PD-1, or PD-L1. In some embodiments, the antibody is selected from: ipilimumab (also known as YERVOY®) pembrolizumab (also known as KEYTRUDA®), nivolumab (also known as OPDIVO®), atezolizumab (also known as TECENTRIQ®), avelumab (also known as BAVENCIO®), and durvalumab (also known as IMFINZI®). In some embodiments, the immune checkpoint inhibitor is an inhibitor of CTLA4. In some embodiments, the immune checkpoint inhibitor is an antibody against CTLA4. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against CTLA4. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against CTLA4. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as CTLA4. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-1. In some embodiments, the immune checkpoint inhibitor is an antibody against PD-1. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against PD-1. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against PD-1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as PD-1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-L1. In some embodiments, the immune checkpoint inhibitor is an antibody against PD-L1. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against PD-L1. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against PD-L1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as PD-L1. In some embodiments, the immune checkpoint inhibitor reduces the interaction between PD-1 and PD-L1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of PD-L2. In some embodiments, the immune checkpoint inhibitor is an antibody against PD-L2. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against PD-L2. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against PD-L2. In some embodiments, the immune checkpoint inhibitor reduces the expression or
activity of one or more immune checkpoint proteins, such as PD-L2. In some embodiments, the immune checkpoint inhibitor reduces the interaction between PD-1 and PD-L2. In some embodiments, the immune checkpoint inhibitor is an inhibitor of LAG-3. In some embodiments, the immune checkpoint inhibitor is an antibody against LAG-3. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against LAG-3. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against LAG-3. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as LAG-3. In some embodiments, the immune checkpoint inhibitor is an inhibitor of B7-H4. In some embodiments, the immune checkpoint inhibitor is an antibody against B7-H4. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against B7-H4. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against B7-H4. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as B7-H4. In some embodiments, the immune checkpoint inhibitor is an inhibitor of KIR. In some embodiments, the immune checkpoint inhibitor is an antibody against KIR. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against KIR. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against KIR. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as KIR. In some embodiments, the immune checkpoint inhibitor is an inhibitor of TNFRSF4. In some embodiments, the immune checkpoint inhibitor is an antibody against TNFRSF4. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against TNFRSF4. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against TNFRSF4. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as TNFRSF4. In some embodiments, the immune checkpoint inhibitor is an inhibitor of OX40L. In some embodiments, the immune checkpoint inhibitor is an antibody against OX40L. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against OX40L. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against OX40L. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as OX40L. In some embodiments, the immune checkpoint inhibitor reduces the interaction between TNFRSF4 and OX40L.In some embodiments, the immune checkpoint inhibitor is an inhibitor of IDO-1. In some embodiments, the immune checkpoint inhibitor is an antibody against IDO-1. In some embodiments, the
immune checkpoint inhibitor is a monoclonal antibody against IDO-1, in some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against IDO-1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as IDO-1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of IDO-2. In some embodiments, the immune checkpoint inhibitor is an antibody against IDO-2. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against IDO-2. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against IDO-2. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as IDO-2. In some embodiments, the immune checkpoint inhibitor is an inhibitor of CEACAM1. In some embodiments, the immune checkpoint inhibitor is an antibody against CEACAM1. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against CEACAM1. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against CEACAM1. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as CEACAM1. In some embodiments, the immune checkpoint inhibitor is an inhibitor of BTLA. In some embodiments, the immune checkpoint inhibitor is an antibody against BTLA. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against BTLA. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against BMA. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as BTLA. In some embodiments, the immune checkpoint inhibitor is an inhibitor of TIM3. In some embodiments, the immune checkpoint inhibitor is an antibody against TIM3. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against TIM3. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against TIM3. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as TIM3. In some embodiments, the immune checkpoint inhibitor is an inhibitor of A2Ar. In some embodiments, the immune checkpoint inhibitor is an antibody against A2Ar. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against A2Ar. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against A2Ar. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as A2Ar.
In some embodiments, the immune checkpoint inhibitor is an inhibitor of VISTA protein. In some embodiments, the immune checkpoint inhibitor is an antibody against VISTA protein. In some embodiments, the immune checkpoint inhibitor is a monoclonal antibody against VISTA protein. In some embodiments, the immune checkpoint inhibitor is a human or humanized antibody against VISTA protein. In some embodiments, the immune checkpoint inhibitor reduces the expression or activity of one or more immune checkpoint proteins, such as VISTA protein. ANTIBODY TARGETS In some embodiments, the antibody of an immunoconjugate is capable of binding one or more targets selected from (e.g., specifically binds to a target selected from) 5T4, ABL, ABCF1, ACVR1, ACVR1B, ACVR2, ACVR2B, ACVRL1, ADORA2A, Aggrecan, AGR2, AICDA, AIF1, AIGI, AKAP1, AKAP2, AMH, AMHR2, ANGPT1, ANGPT2, ANGPTL3, ANGPTL4, ANPEP, APC, APOC1, AR, aromatase, ATX, AX1, AZGP1 (zinc-a-glycoprotein), B7.1, B7.2, B7-H1, BAD, BAFF, BAG1, BAI1, BCR, BCL2, BCL6, BDNF, BLNK, BLR1 (MDR15), BIyS, BMP1, BMP2, BMP3B (GDFIO), BMP4, BMP6, BMP8, BMPRTA, BMPR1B, BMPR2, BPAG1 (plectin), BRCA1, C19orflO (IL27w), C3, C4A, C5, C5R1, CANT1, CAPRIN-1, CASP1, CASP4, CAV1, CCBP2 (D6/JAB61), CCLI (1-309), CCLI1 (eotaxin), CCL13 (MCP- 4), CCL15 (MIP-Id), CCL16 (HCC-4), CCL17 (TARC), CCL18 (PARC), CCL19 (MIP-3b), CCL2 (MCP-1), MCAF, CCL20 (MIP-3a), CCL21 (MEP-2), SLC, exodus-2, CCL22(MDC/STC-1), CCL23 (MPIF-I), CCL24 (MPIF-2/eotaxin-2), CCL25 (TECK), CCL26 (eotaxin-3), CCL27 (CTACK/ILC), CCL28, CCL3 (MIP-Ia), CCL4 (MIPIb), CCL5 (RANTES), CCL7 (MCP-3), CCL8 (mcp-2), CCNA1, CCNA2, CCND1, CCNE1, CCNE2, CCR1 (CKR1/HM145), CCR2 (mcp-IRB/RA), CCR3 (CKR3/CMKBR3), CCR4, CCR5 (CMKBR5/ChemR13), CCR6 (CMKBR6/CKR-L3/STRL22/DRY6), CCR7 (CKR7/EBI1), CCR8 (CMKBR8/TERI/CKR-L1), CCR9 (GPR-9-6), CCRL1 (VSHK1), CCRL2 (L-CCR), CD164, CD19, CDIC, CD2, CD20, CD21, CD200, CD-22, CD24, CD27, CD28, CD3, CD33, CD35, CD37, CD38, CD3E, CD3G, CD3Z, CD4, CD38, CD40, CD40L, CD44, CD45RB, CD47, CD52, CD69, CD72, CD74, CD79A, CD79B, CD8, CD80, CD81, CD83, CD86, CD137, CD152, CD274, CDH1 (Ecadherin), CDH1O, CDH12, CDH13, CDH18, CDH19, CDH2O, CDH5, CDH7, CDH8, CDH9, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK9, CDKN1A (p21Wap1/Cip1), CDKN1B (p27Kip1), CDKN1C, CDKN2A (p16INK4a), CDKN2B, CDKN2C, CDKN3, CEBPB, CERI, CHGA, CHGB, Chitinase, CHST1O, CKLFSF2, CKLFSF3, CKLFSF4, CKLFSF5, CKLFSF6, CKLFSF7, CKLFSF8, CLDN3, CLDN7 (claudin-7), CLDN18.2 (claudin 18.2), CLN3, CLU (clusterin), CMKLR1, CMKOR1 (RDC1),
CNR1, COL18A1, COLIA1, COL4A3, COL6A1, CR2, Cripto, CRP, CSF1 (M-CSF), CSF2 (GM-CSF), CSF3 (GCSF), CTL8, CTNNB1 (b-catenin), CTSB (cathepsin B), CX3CL1 (SCYD1), CX3CR1 (V28), CXCL1 (GRO1), CXCL1O (IP-IO), CXCLI1 (1-TAC/IP-9), CXCL12 (SDF1), CXCL13, CXCL14, CXCL16, CXCL2 (GRO2), CXCL3 (GRO3), CXCL5 (ENA-78/LIX), CXCL6 (GCP-2), CXCL9 (MIG), CXCR3 (GPR9/CKR-L2), CXCR4, CXCR6 (TYMSTR/STRL33/Bonzo), CYB5, CYC1, CYSLTR1, DAB2IP, DES, DKFZp451J0118, DNCL1, DPP4, E2F1, Engel, Edge, Fennel, EFNA3, EFNB2, EGF, EGFR, ELAC2, ENG, Enola, ENO2, ENO3, EPHA1, EPHA2, EPHA3, EPHA4, EPHA5, EPHA6, EPHA7, EPHA8, EPHA9, EPRA10, EPHB1, EPHB2, EPHB3, EPHB4, EPHB5, EPHB6, EPHRIN-A1, EPHRIN- A2, EPHRINA3, EPHRIN-A4, EPHRIN-A5, EPHRIN-A6, EPHRIN-B1, EPHRIN-B2, EPHRIN-B3, EPHB4, EPG, ERBB2 (Her-2), EREG, ERK8, Estrogen receptor, Earl, ESR2, F3 (TF), FADD, famesyltransferase, FasL, FASNf, FCER1A, FCER2, FCGR3A, FGF, FGF1 (aFGF), FGF10, FGF11, FGF12, FGF12B, FGF13, FGF14, FGF16, FGF17, FGF18, FGF19, FGF2 (bFGF). FGF20, FGF21, FGF22, FGF23, FGF3 (int-2), FGF4 (HST), FGF5, FGF6 (HST- 2), FGF7 (KGF), FGF8, FGF9, FGFR3, FIGF (VEGFD), FILI (EPSILON), FBL1 (ZETA), FLJ12584, FLJ25530, FLRT1 (fibronectin), FLT1, FLT-3, FOS, FOSL1 (FRA-1), FY (DARC), GABRP (GABAa), GAGEB1, GAGEC1, GALNAC4S-6ST, GATA3, GD2, GDF5, GFI1, GGT1, GM-CSF, GNAS1, GNRH1, GPR2 (CCR10), GPR31, GPR44, GPR81 (FKSG80), GRCC1O (C1O), GRP, GSN (Gelsolin), GSTP1, HAVCR2, HDAC, HDAC4, HDAC5, HDAC7A, HDAC9, Hedgehog, HGF, HIF1A, HIP1, histamine and histamine receptors, HLA- A, HLA-DRA, HLA-E, HM74, HMOXI, HSP90, HUMCYT2A, ICEBERG, ICOSL, ID2, IFN- a, IFNA1, IFNA2, IFNA4, IFNA5, EFNA6, BFNA7, IFNB1, IFNgamma, IFNW1, IGBP1, IGF1, IGFIR, IGF2, IGFBP2, IGFBP3, IGFBP6, DL-1, ILIO, ILIORA, ILIORB, IL-1, IL1R1 (CD121a), IL1R2 (CD121b), IL-IRA, IL-2, IL2RA (CD25), IL2RB (CD122), IL2RG (CD132), IL-4, IL-4R (CD123), IL-5, IL5RA (CD125), IL3RB (CD131), IL-6, IL6RA, (CD126), IR6RB (CD130), IL-7, IL7RA (CD127), IL-8, CXCR1 (IL8RA), CXCR2, (IL8RB/CD128), IL-9, IL9R (CD129), IL-10, IL10RA (CD210), IL10RB (CDW210B), IL-11, IL11RA, IL-12, IL-12A, IL- 12B, IL-12RB1, IL-12RB2, IL-13, IL13RA1, IL13RA2, IL14, IL15, IL15RA, IL16, IL17, IL17A, IL17B, IL17C, IL17R, IL18, IL18BP, IL18R1, IL18RAP, IL19, ILIA, ILIB, ILIF10, ILIF5, IL1F6, ILIF7, IL1F8, DL1F9, ILIHYI, ILIR1, ILIR2, ILIRAP, ILIRAPLI, ILIRAPL2, ILIRL1, IL1RL2, ILIRN, IL2, IL20, IL20RA, IL21R, IL22, IL22R, IL22RA2, IL23, DL24, IL25, IL26, IL27, IL28A, IL28B, IL29, IL2RA, IL2RB, IL2RG, IL3, IL30, IL3RA, IL4, IL4, IL6ST (glycoprotein 130), ILK, INHA, INHBA, INSL3, INSL4, IRAK1, IRAK2, ITGA1, ITGA2, ITGA3, ITGA6 (.alpha.6 integrin), ITGAV, ITGB3, ITGB4 (.beta.4 integrin), JAG1, JAK1, JAK3, JTB, JUN, K6HF, KAI1, KDR, KITLG, KLF5 (GC Box BP), KLF6, KLK10,
KLK12, KLK13, KLK14, KLK15, KLK3, KLK4, KLK5, KLK6, KLK9, KRT1, KRT19 (Keratin 19), KRT2A, KRTHB6 (hair-specific type II keratin), LAMA5, LEP (leptin), Lingo- p75, Lingo-Troy, LPS, LRRC15, LTA (TNF-b)), LTB, LTB4R (GPR16), LTB4R2, LTBR, MACMARCKS, MAG or OMgp, MAP2K7 (c-Jun), MCP-1, MDK, MIB1, midkine, MIF, MISRII, MJP-2, MK, MKI67 (Ki-67), MMP2, MMP9, MS4A1, MSMB, MT3 (metallothionectin-UI), mTOR, MTSS1, MUC1 (mucin), MYC, MYD88, NCK2, neurocan, Nectin-4, NFKBI, NFKB2, NGFB (NGF), NGFR, NgR-Lingo, NgRNogo66, (Nogo), NgR-p75, NgR-Troy, NMEI (NM23A), NOTCH, NOTCH1, NOX5, NPPB, NROB1, NROB2, NRID1, NR1D2, NR1H2, NR1H3, NR1H4, NR112, NR113, NR2C1, NR2C2, NR2E1, NR2E3, NR2F1, NR2F2, NR2F6, NR3C1, NR3C2, NR4A1, NR4A2, NR4A3, NR5A1, NR5A2, NR6A1, NRP1, NRP2, NT5E, NTN4, ODZI, OPRDI, P2RX7, PAP, PART1, PATE, PAWR, PCA3, PCDGF, PCNA, PDGFA, PDGFB, PDGFRA, PDGFRB, PECAMI, peg-asparaginase, PF4 (CXCL4), PGF, PGR, phosphacan, PIAS2, PI3 Kinase, PIK3CG, PLAU (uPA), PLG, PLXDCI, PKC, PKC-beta, PPBP (CXCL7), PPID, PR1, PRKCQ, PRKD1, PRL, PROC, PROK2, PSAP, PSCA, PTAFR, PTEN, PTGS2 (COX-2), PIN, RAC2 (P21Rac2), RANK, RANK ligand, RARB, RGS1, RGS13, RGS3, RNFI1O (ZNF144), Ron, ROBO2, RXR, S100A2, SCGB 1D2 (lipophilin B), SCGB2A1 (mammaglobin 2), SCGB2A2 (mammaglobin 1), SCYE1 (endothelial Monocyte-activating cytokine), SDF2, SERPENA1, SERPINA3, SERPINB5 (maspin), SERPINEI (PAI-I), SERPINFI, SHIP-1, SHIP-2, SHB1, SHB2, SHBG, SfcAZ, SLC2A2, SLC33A1, SLC43A1, SLIT2, SPP1, SPRR1B (Spr1), ST6GAL1, STAB1, STATE, STEAP, STEAP2, TB4R2, TBX21, TCP1O, TDGF1, TEK, TGFA, TGFB1, TGFB1I1, TGFB2, TGFB3, TGFBI, TGEBR1, TGFBR2, TGFBR3, THIL, THBS1 (thrombospondin-1), THBS2, THBS4, THPO, TIE (Tie-1), TIMP3, tissue factor, TLR1, TLR2, TLR3, TLR4, TLR5, TLR6, TLR7, TLR8, TLR9, TLR10, TLR11, TNF, TNF-a, TNFAIP2 (B94), TNFAIP3, TNFRSF11A, TNFRSF1A, TNFRSF1B, TNFRSF21, TNFRSF5, TNFRSF6 (Fas), TNFRSF7, TNFRSF8, TNFRSF9, TNFSF1O (TRAIL), TNFSF11 (TRANCE), TNFSF12 (APO3L), TNFSF13 (April), TNFSF13B, TNSF14 (HVEM-L), TNFRSF14 (HVEM), TNFSF15 (VEGI), TNFSF18, TNFSF4 (OX40 ligand), TNFSF5 (CD40 ligand). TNFSF6 (FasL), TNFSF7 (CD27 ligand), TNFSF8 (CD30 ligand), TNFSF9 (4-1BB ligand), TOLLIP, Toll-like receptors, TOP2A (topoisomerase 1ia), TP53, TPM1, TPM2, TRADD, TRAF1, TRAF2, TRAF3, TRAF4, TRAF5, TRAF6, TRKA, TREM1, TREM2, TROP2, TRPC6, TSLP, TWEAK, Tyrosinase, uPAR, VEGF, VEGFB, VEGFC, versican, VHL C5, VLA-4, Wnt-1, XCL1 (tymphotactin), XCL2 (SCM-Ib), XCRI (GPR5/CCXCR1), YYI, ZFPM2, CLEC4C (BDCA-2, DLEC, CD303, CLECSF7), CLEC4D (MCL, CLECSF8), CLEC4E (Mincle), CLEC6A (Dectin-2). CLEC5A (MDL-1, CLECSF5), CLEC1B (CLEC-2), CLEC9A (DNGR-1), CLEC7A (Dectin-1), PDGFRa,
SLAMF7, GP6 (GPVI), LILRA1 (CD85I), LILRA2 (CD85H, ILT1), LILRA4 (CD85G, ILT7), LILRA5 (CD85F, ILT11), LILRA6 (CD85b, ILT8), NCR1 (CD335, LY94, NKp46), NCR3 (CD335, LY94, NKp46), NCR3 (CD337, NKp30), OSCAR, TARM1, CD300C, CD300E, CD300LB (CD300B), CD300LD (CD300D), KIR2DL4 (CD158D), KIR2DS, KLRC2 (CD159C, NKG2C), KLRK1 (CD314, NKG2D), NCR2 (CD336, NKp44), PILRB, SIGLEC1 (CD169, SN), SIGLEC14, SIGLEC15 (CD33L3), SIGLEC16, SIRPB1 (CD172B), TREM1 (CD354), TREM2, and KLRF1 (NKp80). In some embodiments, the antibody binds to an FcR.gamma-coupled receptor. In some embodiments, the FcR.gamma-coupled receptor is selected from the group consisting of GP6 (GPVI), LILRA1 (CD85I), LILRA2 (CD85H, ILT1), LILRA4 (CD85G, ILT7), LILRA5 (CD85F, ILT11), LILRA6 (CD85b, ILT8), NCR1 (CD335, LY94, NKp46), NCR3 (CD335, LY94, NKp46), NCR3 (CD337, NKp30), OSCAR, and TARM1. In some embodiments, the antibody binds to a DAP12-coupled receptor. In some embodiments, the DAP12-coupled receptor is selected from the group consisting of CD300C, CD300E, CD300LB (CD300B), CD300LD (CD300D), KIR2DL4 (CD158D), KIR2DS, KLRC2 (CD159C, NKG2C), KLRK1 (CD314, NKG2D), NCR2 (CD336, NKp44). PILRB, SIGLEC1 (CD169, SN), SIGLEC14, SIGLEC15 (CD33L3), SIGLEC16, SIRPB1 (CD172B), TREM1 (CD354), and TREM2. In some embodiments, the antibody binds to a hemITAM-bearing receptor. In some embodiments, the hemITAM-bearing receptor is KLRF1 (NKp80). In some embodiments, the antibody is capable of binding one or more targets selected from CLEC4C (BDCA-2, DLEC, CD303, CLECSF7), CLEC4D (MCL, CLECSF8), CLEC4E (Mincle), CLEC6A (Dectin-2), CLEC5A (MDL-1, CLECSF5), CLEC1B (CLEC-2), CLEC9A (DNGR-1), and CLEC7A (Dectin-1). In some embodiments, the antibody is capable of binding CLEC6A (Dectin-2) or CLEC5A. In some embodiments, the antibody is capable of binding CLEC6A (Dectin-2). In some embodiments, the antibody is capable of binding one or more targets selected from (e.g., specifically binds to a target selected from): ATP5I (Q06185), OAT (P29758), AIFM1 (Q9Z0X1), AOFA (Q64133), MTDC (P18155), CMC1 (Q8BH59), PREP (Q8K411), YMEL1 (O88967), LPPRC (Q6PB66), LONM (Q8CGK3), ACON (Q99KI0), ODO1 (Q60597), IDHP (P54071), ALDH2 (P47738), ATPB (P56480), AATM (P05202), TMM93 (Q9CQW0), ERGI3 (Q9CQE7), RTN4 (Q99P72), CL041 (Q8BQR4), ERLN2 (Q8BFZ9), TERA (Q01853), DAD1 (P61804), CALX (P35564), CALU (O35887), VAPA (Q9WV55), MOGS (Q80UM7), GANAB (Q8BHN3), ERO1A (Q8R180), UGGG1 (Q6P5E4), P4HA1 (Q60715), HYEP (Q9D379), CALR (P14211), AT2A2 (O55143), PDIA4 (P08003), PDIA1 (P09103), PDIA3
(P27773), PDIA6 (Q922R8), CLH (Q68FD5), PPIB (P24369), TCPG (P80318), MOT4 (P57787), NICA (P57716), BASI (P18572), VAPA (Q9WV55), ENV2 (P11370), VAT1 (Q62465), 4F2 (P10852), ENOA (P17182), ILK (O55222), GPNMB (Q99P91), ENV1 (P10404), ERO1A (Q8R180), CLH, (Q68FD5), DSG1A (Q61495), AT1A1 (Q8VDN2), HYOU1 (Q9JKR6), TRAP1 (Q9CQN1), GRP75 (P38647), ENPL (P08113), CH60 (P63038), and CH10 (Q64433). In the preceding list, accession numbers are shown in parentheses. In some embodiments, the antibody binds to an antigen selected from CDH1, CD19, CD20, CD29, CD30, CD38, CD40, CD47, EpCAM, MUC1, MUC16, EGFR, Her2, SLAMF7, and gp75. In some embodiments, the antigen is selected from CD19, CD20, CD47, EpCAM, MUC1, MUC16, EGFR, and Her2. In some embodiments, the antibody binds to an antigen selected from the Tn antigen and the Thomsen-Friedenreich antigen. In some embodiments, the antibody or Fc fusion protein is selected from: abagovomab, abatacept (also known as ORENCIA®), abciximab (also known as REOPRO®), c7E3 Fab), adalimumab (also known as HUMIRA®), adecatumumab, alemtuzumab (also known as CAMPATH®), MabCampath or Campath-1H), altumomab, afelimomab, anatumomab mafenatox, anetumumab, anrukizumab, apolizumab, arcitumomab, aselizumab, atlizumab, atorolimumab, bapineuzumab, basiliximab (also known as SIMULECT®), bavituximab, bectumomab (also known as LYMPHOSCAN®), belimumab (also known as LYMPHO-STAT- B®), bertilimumab, besilesomab, bevacizumab (also known as AVASTIN®), biciromab brallobarbital, bivatuzumab mertansine, campath, canakinumab (also known as ACZ885), cantuzumab mertansine, capromab (also known as PROSTASCINT®), catumaxomab (also known as REMOVAB®), cedelizumab (also known as CIMZIA®), certolizumab pegol, cetuximab (also known as ERBITUX®), clenoliximab, dacetuzumab, dacliximab, daclizumab (also known as ZENAPAX®), denosumab (also known as AMG 162), detumomab, dorlimomab aritox, dorlixizumab, duntumumab, durimulumab, durmulumab, ecromeximab, eculizumab (also known as SOLIRIS®), edobacomab, edrecolomab (also known as Mab17-1A, PANOREX®), efalizumab (also known as RAPTIVA®), efungumab (also known as MYCOGRAB®), elsilimomab, enlimomab pegol, epitumomab cituxetan, efalizumab, epitumomab, epratuzumab, erlizumab, ertumaxomab (also known as REXOMUN®), etanercept (also known as ENBREL®), etaracizumab (also known as etaratuzumab, VITAXIN®, ABEGRIN®), exbivirumab, fanolesomab (also known as NEUTROSPEC®), faralimomab, felvizumab, fontolizumab (also known as HUZAF®), galiximab, gantenerumab, gavilimomab (also known as ABXCBL®), gemtuzumab ozogamicin (also known as MYLOTARG®), golimumab (also known as CNTO 148), gomiliximab, ibalizumab (also known as TNX-355), ibritumomab tiuxetan (also known as ZEVALIN®), igovomab, imciromab, infliximab (also known as
REMICADE®), inolimomab, inotuzumab ozogamicin, ipilimumab (also known as MDX-010, MDX-101), iratumumab, keliximab, labetuzumab, lemalesomab, lebrilizumab, lerdelimumab, lexatumumab (also known as, HGS-ETR2, ETR2-ST01), lexitumumab, libivirumab, lintuzumab, lucatumumab, lumiliximab, mapatumumab (also known as HGSETR1, TRM-1), maslimomab, matuzumab (also known as EMD72000), mepolizumab (also known as BOSATRIA®), metelimumab, milatuzumab, minretumomab, mitumomab, morolimumab, motavizwnab (also known as NUMAX®), muromonab (also known as OKT3), nacolomab tafenatox, naptumomab estafenatox, natalizumab (also known as TYSABRI®, ANTEGREN®), nebacumab, nerelimomab, nimotuzumab (also known as THERACIM hR3®, THERA-CIM- hR3®, THERALOC®), nofetumomab merpentan (also known as VERLUMA®), ocrelizumab, odulimomab, ofatumumab, omalizumab (also known as XOLAIR®), oregovomab (also known as OVAREX®), otelixizumab, pagibaximab, palivizumab (also known as SYNAGIS®), panitumumab (also known as ABX-EGF, VECTIBIX®), pascolizumab, pemtumomab (also known as THERAGYN®), pertuzumab (also known as 2C4, OMNITARG®), pexelizumab, pintumomab, priliximab, pritumumab, ranibizumab (also known as LUCENTIS®), raxibacumab, regavirumab, reslizumab, rituximab (also known as RITUXAN®, MabTHERA®), rovelizumab, ruplizumab, satumomab, sevirumab, sibrotuzumab, siplizumab (also known as MEDI-507), sontuzumab, stamulumab (also known as MYO-029), sulesomab (also known as LEUKOSCAN®), tacatuzumab tetraxetan, tadocizumab, talizumab, taplitumomab paptox, tefibazumab (also known as AUREXIS®), telimomab aritox, teneliximab, teplizumab, ticilimumab, tocilizumab (also known as ACTEMRA®), toralizumab, tositumomab, trastuzumab (also known as HERCEPTIN®), tremelimumab (also known as CP-675,206), tucotuzumab celmoleukin, tuvirumab, urtoxazumab, ustekinumab (also known as CNTO 1275), vapaliximab, veltuzumab, vepalimomab, visilizumab (also known as NUVION®), volociximab (also known as M200), votumumab (also known as HUMASPECT®), zalutumumab, zanolimumab (also known as HuMAX-CD4), ziralimumab, zolimomab aritox, daratumumab, elotuxumab, obintunzumab, olaratumab, brentuximab vedotin, afibercept, abatacept, belatacept, afibercept, etanercept, romiplostim, SBT-040 (sequences listed in US 2017/0158772. In some embodiments, the antibody is rituximab. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds PD-L1. Programmed Death-Ligand 1 (PD-L1, cluster of differentiation 274, CD274, B7- homolog 1, or B7-H1) belongs to the B7 protein superfamily, and is a ligand of programmed cell death protein 1 (PD-1, PDCD1, cluster of differentiation 279, or CD279). PD-L1 can also
interact with B7.1 (CD80) and such interaction is believed to inhibit T cell priming. The PD- L1/PD-1 axis plays a large role in suppressing the adaptive immune response. More specifically, it is believed that engagement of PD-L1 with its receptor, PD-1, delivers a signal that inhibits activation and proliferation of T-cells. Agents that bind to PD-L1 and prevent the ligand from binding to the PD-1 receptor prevent this immunosuppression, and can, therefore, enhance an immune response when desired, such as for the treatment of cancers, or infections. PD-L1/PD-1 pathway also contributes to preventing autoimmunity and therefore agonistic agents against PD-L1 or agents that deliver immune inhibitory payloads may help treatment of autoimmune disorders. Several antibodies targeting PD-L1 have been developed for the treatment of cancer, including atezolizumab (TECENTRIQTM), durvalumab (IMFINZITM), and avelumab (BAVENCIOTM). Nevertheless, there continues to be a need for new PD-L1-binding agents, including agents that bind PD-L1 with high affinity and effectively prevent PD-L1/PD-1 signaling and agents that can deliver therapeutic payloads to PD-L1 expressing cells. In addition, there is a need for new PD-L1-binding agents to treat autoimmune disorders and infections. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds HER2. In certain embodiments, immunoconjugates of the invention comprise anti-HER2 antibodies. In one embodiment of the invention, an anti-HER2 antibody of an immunoconjugate of the invention comprises a humanized anti-HER2 antibody, e.g., huMAb4D5-1, huMAb4D5- 2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7 and huMAb4D5- 8, as described in Table 3 of US 5821337, which is specifically incorporated by reference herein. Those antibodies contain human framework regions with the complementarity- determining regions of a murine antibody (4D5) that binds to HER2. The humanized antibody huMAb4D5-8 is also referred to as trastuzumab, commercially available under the tradename HERCEPTIN™ (Genentech, Inc.). Trastuzumab (CAS 180288-69-1, HERCEPTIN ^, huMAb4D5-8, rhuMAb HER2, Genentech) is a recombinant DNA-derived, IgG1 kappa, monoclonal antibody that is a humanized version of a murine anti-HER2 antibody (4D5) that selectively binds with high affinity in a cell-based assay (Kd = 5 nM) to the extracellular domain of HER2 (US 5677171; US 5821337; US 6054297; US 6165464; US 6339142; US 6407213; US 6639055; US 6719971; US 6800738; US 7074404; Coussens et al (1985) Science 230:1132-9; Slamon et al (1989) Science 244:707-12; Slamon et al (2001) New Engl. J. Med.344:783-792).
In an embodiment of the invention, the antibody construct or antigen binding domain comprises the CDR regions of trastuzumab. In an embodiment of the invention, the anti-HER2 antibody further comprises the framework regions of the trastuzumab. In an embodiment of the invention, the anti-HER2 antibody further comprises one or both variable regions of trastuzumab. In another embodiment of the invention, an anti-HER2 antibody of an immunoconjugate of the invention comprises a humanized anti-HER2 antibody, e.g., humanized 2C4, as described in US 7862817. An exemplary humanized 2C4 antibody is pertuzumab (CAS Reg. No.380610- 27-5), PERJETA™ (Genentech, Inc.). Pertuzumab is a HER dimerization inhibitor (HDI) and functions to inhibit the ability of HER2 to form active heterodimers or homodimers with other HER receptors (such as EGFR/HER1, HER2, HER3 and HER4). See, for example, Harari and Yarden, Oncogene 19:6102-14 (2000); Yarden and Sliwkowski. Nat Rev Mol Cell Biol 2:127-37 (2001); Sliwkowski Nat Struct Biol 10:158-9 (2003); Cho et al. Nature 421:756-60 (2003); and Malik et al. Pro Am Soc Cancer Res 44:176-7 (2003). PERJETA™ is approved for the treatment of breast cancer. In an embodiment of the invention, the antibody construct or antigen binding domain comprises the CDR regions of pertuzumab. In an embodiment of the invention, the anti-HER2 antibody further comprises the framework regions of the pertuzumab. In an embodiment of the invention, the anti-HER2 antibody further comprises one or both variable regions of pertuzumab. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds CEA. Carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5) also known as CD66e (Cluster of Differentiation 66e), is a member of the carcinoembryonic antigen (CEA) gene family. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds CEA. Elevated expression of carcinoembryonic antigen (CEA, CD66e, CEACAM5) has been implicated in various biological aspects of neoplasia, especially tumor cell adhesion, metastasis, the blocking of cellular immune mechanisms, and having antiapoptosis functions. CEA is also used as a blood marker for many carcinomas. Labetuzumab (CEA-CIDETM, Immunomedics, CAS Reg. No.219649-07-7), also known as MN-14 and hMN14, is a humanized IgG1 monoclonal antibody and has been studied for the treatment of colorectal cancer (Blumenthal, R. et al (2005) Cancer Immunology Immunotherapy 54(4):315-327). Labetuzumab conjugated to a
camptothecin analog (labetuzumab govitecan, IMMU-130) targets carcinoembryonic antigen- related cell adhesion mol.5 (CEACAM5) and is being studied in patients with relapsed or refractory metastatic colorectal cancer (Sharkey, R. et al, (2018), Molecular Cancer Therapeutics 17(1):196-203; Cardillo, T. et al (2018) Molecular Cancer Therapeutics 17(1):150-160). In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Trop2. Tumor-associated calcium signal transducer 2 (TROP-2) is a transmembrane glycoprotein encoded by the TACSTD2 gene (Linnenbach AJ, et al (1993) Mol Cell Biol.13(3): 1507–15; Calabrese G, et al (2001) Cytogenet Cell Genet.92(1–2): 164–5). Trop2 is an intracellular calcium signal transducer that is differentially expressed in many cancers and signals cells for self-renewal, proliferation, invasion, and survival. Trop2 is considered a stem cell marker and is expressed in many normal tissues, though in contrast, it is overexpressed in many cancers (Ohmachi T, et al., (2006) Clin. Cancer Res., 12(10), 3057-3063; Muhlmann G, et al., (2009) J. Clin. Pathol., 62(2), 152-158; Fong D, et al., (2008) Br. J. Cancer, 99(8), 1290- 1295; Fong D, et al., (2008) Mod. Pathol., 21(2), 186-191; Ning S, et al., (2013) Neurol. Sci., 34(10), 1745-1750). Overexpression of Trop2 is of prognostic significance. Several ligands have been proposed that interact with Trop2. Trop2 signals the cells via different pathways and it is transcriptionally regulated by a complex network of several transcription factors. Human Trop2 (TACSTD2: tumor-associated calcium signal transducer 2, GA733-1, EGP-1, M1S1; hereinafter, referred to as hTrop2) is a single-pass transmembrane type 1 cell membrane protein consisting of 323 amino acid residues. While the presence of a cell membrane protein involved in immune resistance, which is common to human trophoblasts and cancer cells (Faulk W P, et al., Proc. Natl. Acad. Sci.75(4):1947-1951 (1978)), has previously been suggested, an antigen molecule recognized by a monoclonal antibody against a cell membrane protein in a human choriocarcinoma cell line was identified and designated as Trop2 as one of the molecules expressed in human trophoblasts (Lipinski M, et al., Proc. Natl. Acad. Sci.78(8), 5147-5150 (1981)). This molecule was also designated as tumor antigen GA733-1 recognized by a mouse monoclonal antibody GA733 (Linnenbach A J, et al., Proc. Natl. Acad. Sci.86(1), 27- 31 (1989)) obtained by immunization with a gastric cancer cell line or an epithelial glycoprotein (EGP-1; Basu A, et al., Int. J. Cancer, 62 (4), 472-479 (1995)) recognized by a mouse monoclonal antibody RS7-3G11 obtained by immunization with non-small cell lung cancer cells. In 1995, however, the Trop2 gene was cloned, and all of these molecules were confirmed to be identical molecules (Fornaro M, et al., Int. J. Cancer, 62(5), 610-618 (1995)). The DNA
sequence and amino acid sequence of hTrop2 are available on a public database and can be referred to, for example, under Accession Nos. NM_002353 and NP_002344 (NCBI). In response to such information suggesting the association with cancer, a plurality of anti-hTrop2 antibodies have been established so far and studied for their antitumor effects. Among these antibodies, there is disclosed, for example, an unconjugated antibody that exhibits in itself antitumor activity in nude mouse xenograft models (WO 2008/144891; WO 2011/145744; WO 2011/155579; WO 2013/077458) as well as an antibody that exhibits antitumor activity as ADC with a cytotoxic drug (WO 2003/074566; WO 2011/068845; WO 2013/068946; US 7999083). However, the strength or coverage of their activity is still insufficient, and there are unsatisfied medical needs for hTrop2 as a therapeutic target. TROP2 expression in cancer cells has been correlated with drug resistance. Several strategies target TROP2 on cancer cells that include antibodies, antibody fusion proteins, chemical inhibitors, nanoparticles, etc. The in vitro studies and pre-clinical studies, using these various therapeutic treatments, have resulted in significant inhibition of tumor cell growth both in vitro and in vivo in mice. Clinical studies have explored the potential application of Trop2 as both a prognostic biomarker and as a therapeutic target to reverse resistance. Sacituzumab govitecan (TRODELVY®, Immunomedics, IMMU-132), an antibody-drug conjugate comprising a Trop2-directed antibody linked to a topoisomerase inhibitor drug, is indicated for the treatment of metastatic triple-negative breast cancer (mTNBC) in adult patients that have received at least two prior therapies. The Trop2 antibody in sacituzumab govitecan is conjugated to SN-38, the active metabolite of irinotecan (US 2016/0297890; WO 2015/098099). In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Caprin-1 (Ellis JA, Luzio JP (1995) J Biol Chem.270(35):20717–23; Wang B, et al (2005) J Immunol.175 (7):4274–82; Solomon S, et al (2007) Mol Cell Biol.27(6):2324–42). Caprin-1 is also known as GPIAP1, GPIP137, GRIP137, M11S1, RNG105, p137GPI, and cell cycle associated protein 1. Cytoplasmic activation/proliferation-associated protein-1 (caprin-1) is an RNA-binding protein that participates in the regulation of cell cycle control-associated genes. Caprin-1 selectively binds to c-Myc and cyclin D2 mRNAs, which accelerates cell progression through the G1 phase into the S phase, enhances cell viability and promotes cell growth, indicating that it may serve an important role in tumorigenesis (Wang B, et al (2005) J Immunol.175:4274– 4282). Caprin-1 acts alone or in combination with other RNA-binding proteins, such as RasGAP SH3-domain-binding protein 1 and fragile X mental retardation protein. In the tumorigenesis process, caprin-1 primarily functions by activating cell proliferation and upregulating the
expression of immune checkpoint proteins. Through the formation of stress granules, caprin-1 is also involved in the process by which tumor cells adapt to adverse conditions, which contributes to radiation and chemotherapy resistance. Given its role in various clinical malignancies, caprin-1 holds the potential to be used as a biomarker and a target for the development of novel therapeutics (Yang, Z-S, et al (2019) Oncology Letters 18:15-21). Antibodies that target caprin-1 for treatment and detection have been described (WO 2011/096519; WO 2013/125654; WO 2013/125636; WO 2013/125640; WO 2013/125630; WO 2013/018889; WO 2013/018891; WO 2013/018883; WO 2013/018892; WO 2014/014082; WO 2014/014086; WO 2015/020212; WO 2018/079740). In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Claudin-1. Claudin-1 is a member of the transmembrane protein family claudins located in cell-cell tight junctions and it acts as a co-receptor for HCV entry into hepatic cells (Kniesel U, et al (2000). Cell. Mol. Neurobiol.20(1):57–76; Furuse M, et al (1998). J. Cell Biol.141(7):1539–50; Swisshelm K, et al (2005) Adv. Drug Deliv. Rev.57(6):919–28). Claudin 1 is also known as Senescence-associated epithelial membrane protein, senescence-associated epithelial membrane protein 1, CLDN1, CLD1, ILVASC, SEMP1. Claudins are abundant in luminal epithelial sheets where they maintain epithelial cell polarity. Claudin-1 is expressed in most tissues such as bladder, fallopian tube, liver, pancreas, prostate, and skin. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds Nectin-4. The nectins are a protein family of cell adhesion molecules involved in calcium- dependent cell adhesion (Takai Y. et al (2003) Cancer Science 94(8):655-67; Fuchs, A. et al (2006) Seminars in Cancer Biology 16(5):359-366; Miyoshi J. et al (2007) American journal of nephrology 27(6):590-604). Nectins play an important role in the bonding between cells in many different tissues, including the intermediate junction of epithelial cells or the chemical synapse of nerve cells. In an exemplary embodiment, the immunoconjugates of the invention comprise an antibody construct that comprises an antigen binding domain that specifically recognizes and binds LRRC15 (Leucine rich repeat containing 15). LRRC15 is a cell membrane-expressed protein that in humans is encoded by the LRRC15 gene. LRRC15 is expressed on stromal fibroblasts in many solid tumors (e.g., breast,
head and neck, lung, pancreatic) as well as directly on a subset of cancer cells of mesenchymal origin (e.g., sarcoma, melanoma, glioblastoma). LRRC15 may have utility as a therapeutic target for the treatment of cancers with LRRC15-positive stromal desmoplasia or cancers of mesenchymal origin (Purcell, JW et al (2018) Cancer Res., 78(14):4059-4072). AZA-BICYCLIC ADJUVANT COMPOUNDS The immunoconjugate of the invention comprises an aza-bicyclic adjuvant moiety. The adjuvant moiety described herein is a compound that elicits an immune response (i.e., an immunostimulatory agent). Generally, the adjuvant moiety described herein is a STING agonist. Certain amido benzimidazole compounds are demonstrated STING receptor agonists with systemic activity (Ramanjulu, J.M. et al (2018 ) Nature 564:439–443; Barber, G.N. (2015) Nature Rev Immunol 15:760–770; US 2019/0300513). STING is a dimeric structure with a large and symmetrical binding pocket. The aza- bicyclic compounds (AzaBi) of Tables 1a and 1b when conjugated to a targeting antibody were designed to target and bind to the open conformation of the binding pocket of STING. Binding to a small molecule agonist usually induces a closed conformation of the STING protein. This introduces the risk that a linker, particularly if “noncleavable”, will interfere with binding and activation. The bis-benzimidazole STING agonists are reported to bind and activate through an open conformation, which are predicted to be more amenable to attachment of a linker (Ramanjulu, J.M. et al (2018) Nature 564:439–443; Barber, G.N. (2015) Nature Rev Immunol 15:760–770).
Table 1a. Aza-bicyclic compounds (AzaBi)




 Table 1b. Aza-bicyclic compounds (AzaBi)
Table 1b. Aza-bicyclic compounds (AzaBi)















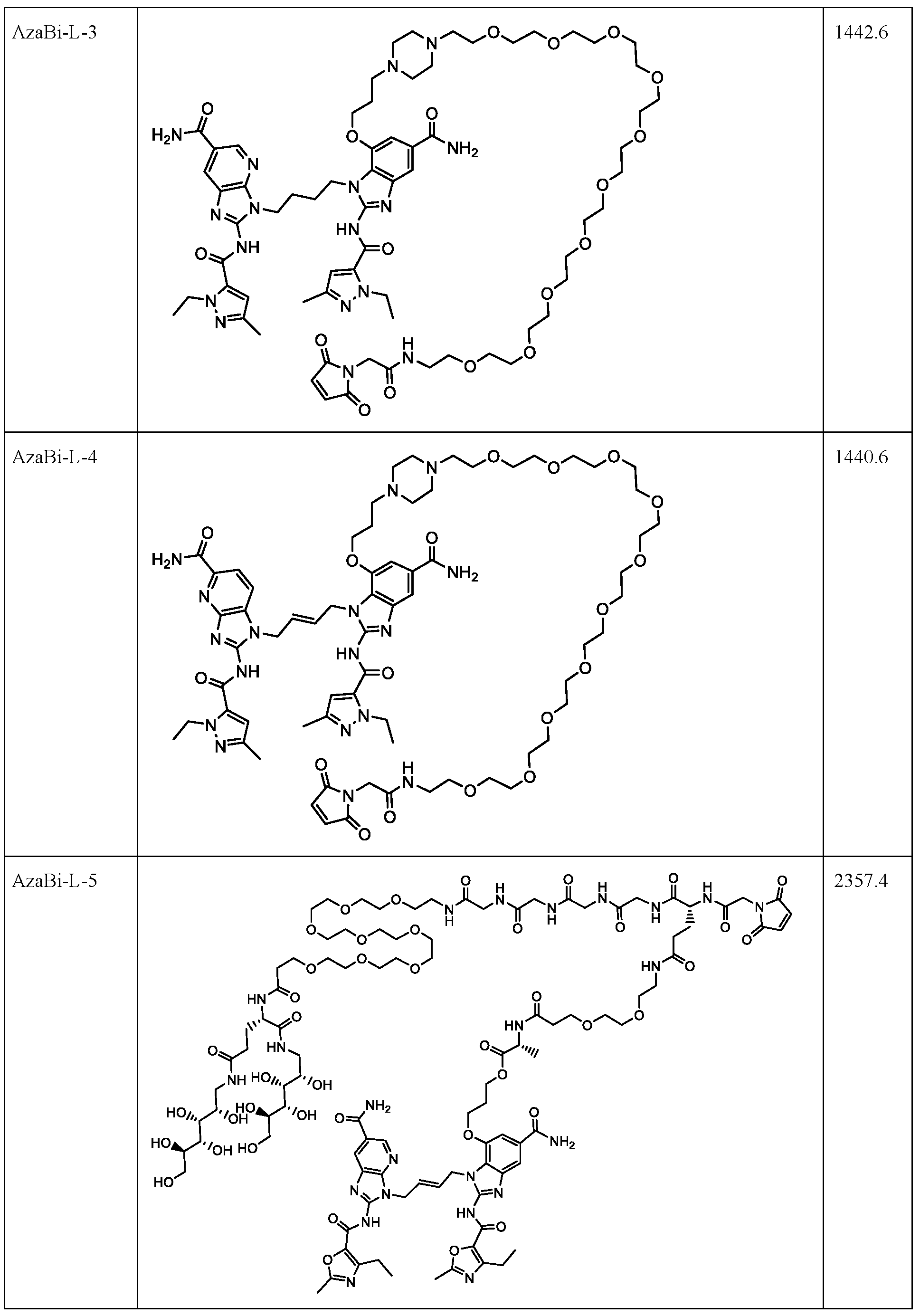


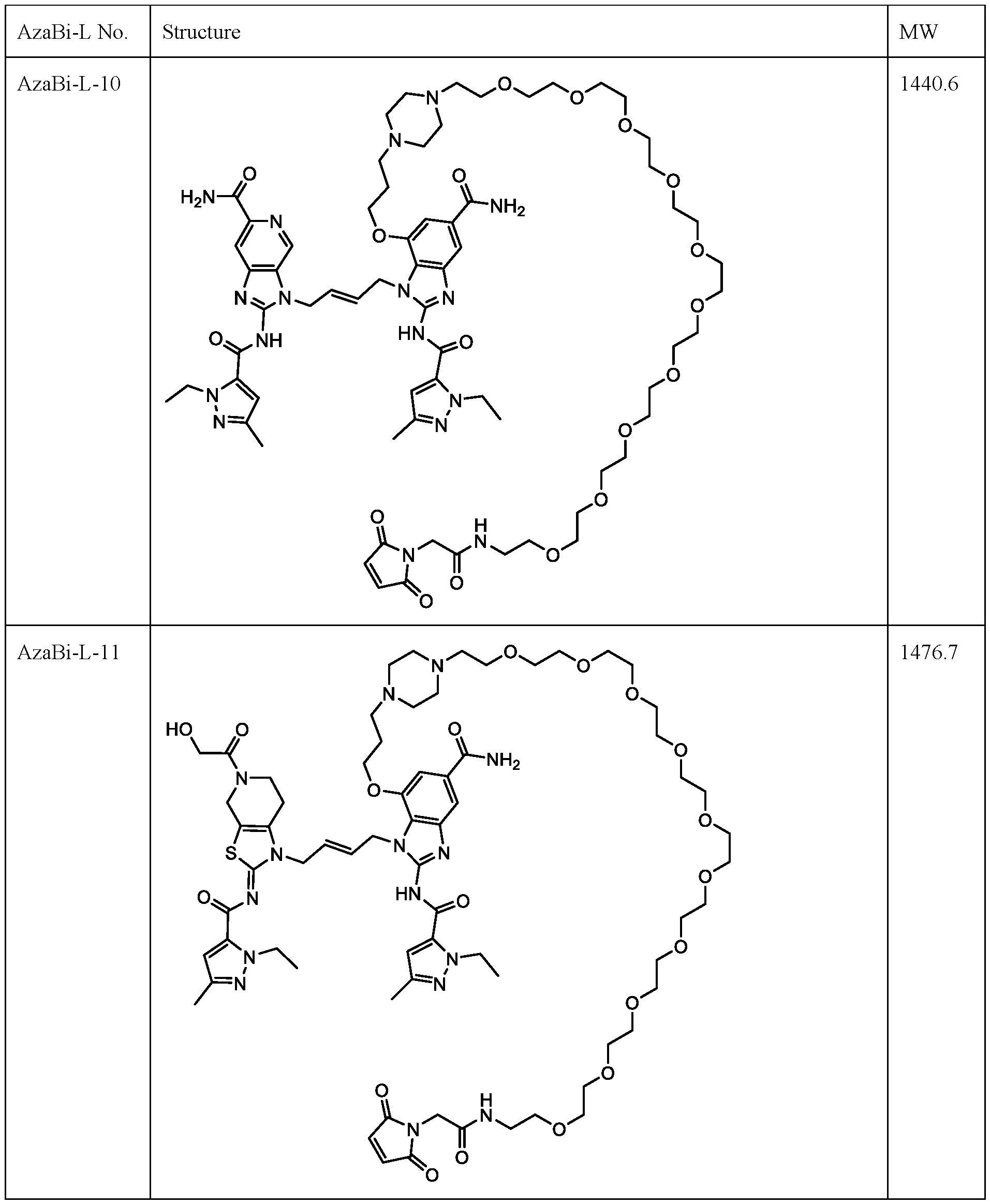







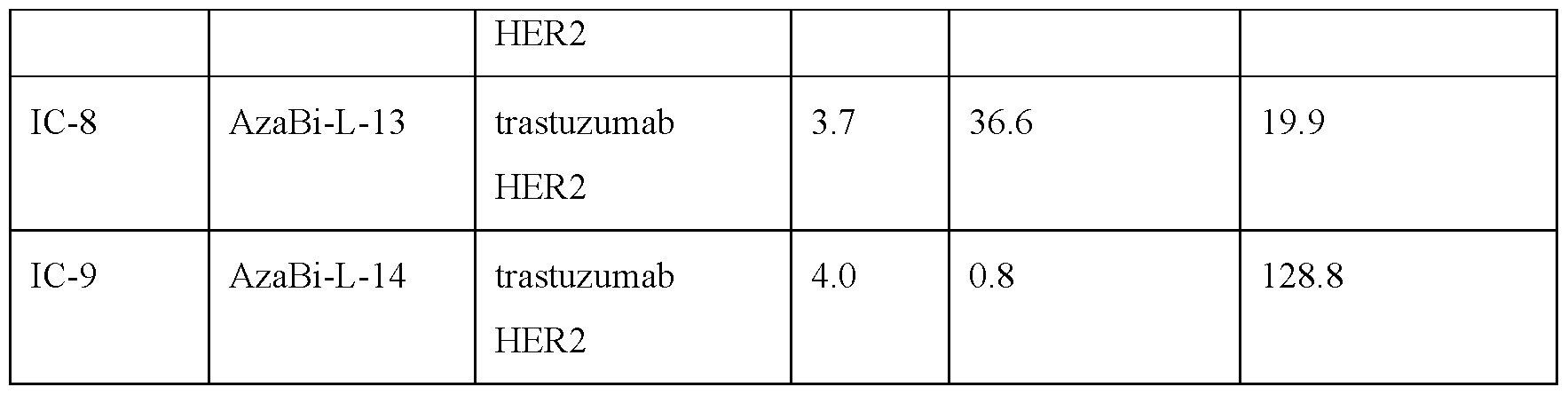















Claims
CLAIMS: 1. An immunoconjugate comprising an antibody covalently attached to one or more STING agonist moieties by a linker, and having Formula I: Ab−[L−D]p I or a pharmaceutically acceptable salt thereof, wherein: Ab is the antibody; L is the linker; p is an integer from 1 to 8; D is the STING agonist moiety having the formula:
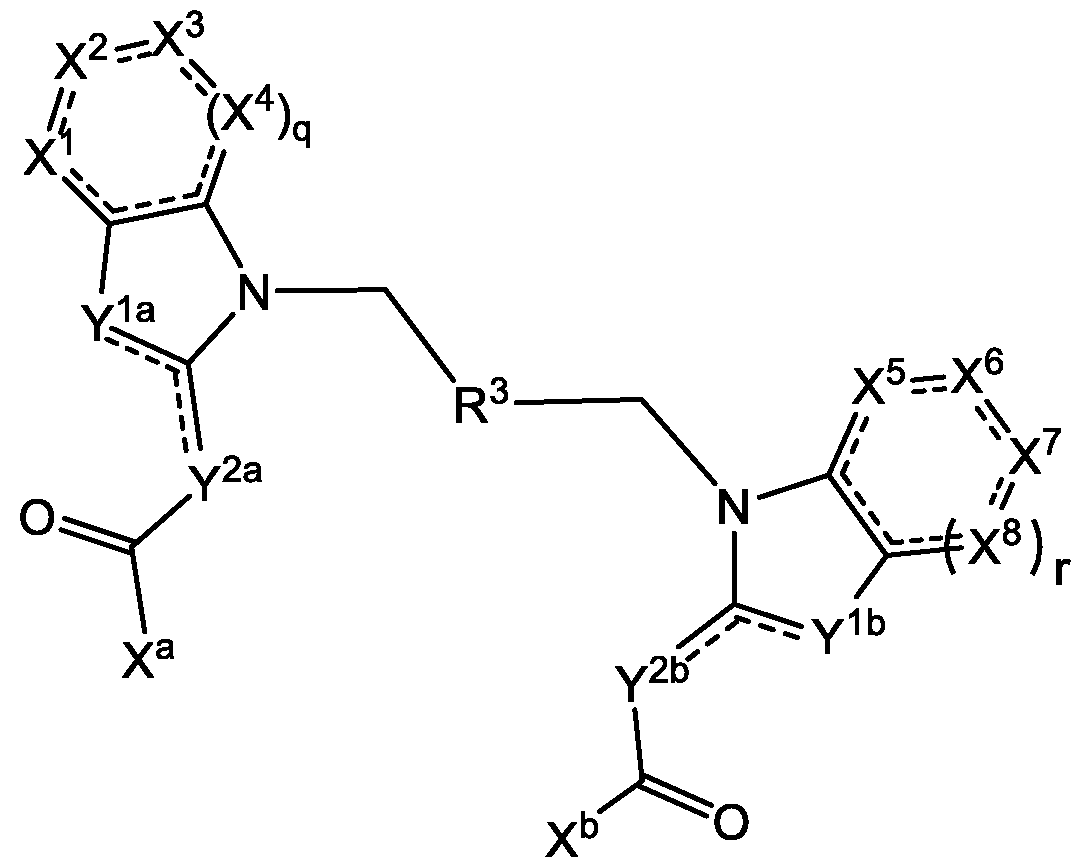 where the dashed lines are optional double bonds; Xa and Xb are independently selected from a five-membered heteroaryl, optionally substituted with R1 or R4; R1 is independently H or C1-C6 alkyl; Y1a and Y1b are independently selected from N, NH, O, and S; Y2a and Y2b are independently selected from N, NH, and S; each of X1, X2, X3, X4, X5, X6, X7, and X8 are independently selected from N, NR2a, CR2b and C(R2b)2; q and r are independently selected from 0, 1, 2, and 3; one or two of X1, X2, X3, X4, X5, X6, X7, and X8 are N or NR2a; one of X1, X2, X3, X4, X5, X6, X7, and X8 is selected from NR2a, CR2b and C(R2b)2 where R2a or R2b is R4;
R2a is independently selected from H, C1-C6 alkyl, −S(O)2N(R1)2, −C(=O)N(R1)2, − C(=O)R1, and R4; R2b is independently selected from H, F, Cl, CN, C1-C6 alkyl, −O(C1-C6 alkyl), C1-C6 heteroalkyl, −C(=O)N(R1)2, and R4; R3 is selected from C1−C6 alkyldiyl, −(C1−C3 alkyldiyl)−O−(C1−C3 alkyldiyl)−, C2−C6 alkenyldiyl and C2−C6 alkynyldiyl, optionally substituted with one or more groups selected from F, Cl, −OH, −OCH3, −OCH2CH3, −OCH2CH2OCH3, −OCH2CH2OH, −OCH2CH2N(CH3)2; R4 is selected from the group consisting of: −(C1-C12 alkyldiyl)−*; −(C1-C12 alkyldiyl)−N(R1)−*; −(C1-C12 alkyldiyl)−O−*; −(C1-C12 alkyldiyl)−(C2-C20 heterocyclyldiyl)−*; −(C1-C20 heteroalkyldiyl)−N(R1)−*; −(C1-C12 heteroalkyldiyl)−O−*; −O−(C1-C12 alkyldiyl)−*; −O−(C1-C12 alkyldiyl)−N(R1)−*; −O−(C1-C12 alkyldiyl)−O−*; −O−(C1-C12 alkyldiyl)−(C2-C20 heterocyclyldiyl)−*; −O−(C1-C12 alkyldiyl)−(C2-C20 heterocyclyldiyl)−N(R1)−*; −OC(=O)N(R1)−*; −OC(=O)N(R1)−(C1-C12 alkyldiyl)−N(R1)−*; −N(R1)−*; −N(R1)−(C1-C12 alkyldiyl)−*; −N(R1)−(C1-C12 alkyldiyl)−N(R4)−*; −N(R1)−(C1-C12 alkyldiyl)−O−*; −N(R1)−(C1-C12 alkyldiyl)−(C2-C20 heterocyclyldiyl)−*; −C(=O)N(R1)−*; −C(=O)N(R1)−(C1-C12 alkyldiyl)−*; −C(=O)N(R1)−(C1-C12 alkyldiyl)−N(R1)−*; −C(=O)N(R1)−(C1-C12 alkyldiyl)−O−*; −(C2-C20 heterocyclyldiyl)−*; −S(=O)2−(C2-C20 heterocyclyldiyl)−*; and −S(=O)2−(C2-C20 heterocyclyldiyl)−(C1-C12 alkyldiyl)−N(R1)−*;
where the asterisk * indicates the attachment site of L; and alkyl, alkyldiyl, alkenyl, alkenyldiyl, alkynyl, alkynyldiyl, heteroalkyl, heteroalkyldiyl, aryl, aryldiyl, carbocyclyl, carbocyclyldiyl, heterocyclyl, heterocyclyldiyl, heteroaryl, and heteroaryldiyl are independently and optionally substituted with one or more groups independently selected from F, Cl, Br, I, −CN, −CH3, −CH2CH3, −CH=CH2, −C ≡CH, − C ≡CCH3, −CH2CH2CH3, −CH(CH3)2, −CH2CH(CH3)2, −CH2OH, −CH2OCH3, −CH2CH2OH, − C(CH3)2OH, −CH(OH)CH(CH3)2, −C(CH3)2CH2OH, −CH2CH2SO2CH3, −CH2OP(O)(OH)2, − CH2F, −CHF2, −CF3, −CH2CF3, −CH2CHF2, −CH(CH3)CN, −C(CH3)2CN, −CH2CN, − CH2NH2, −CH2NHSO2CH3, −CH2NHCH3, −CH2N(CH3)2, −CO2H, −COCH3, −CO2CH3, − CO2C(CH3)3, −COCH(OH)CH3, −CONH2, −CONHCH3, −CON(CH3)2, −C(CH3)2CONH2, − NH2, −NHCH3, −N(CH3)2, −NHCOCH3, −N(CH3)COCH3, −NHS(O)2CH3, − N(CH3)C(CH3)2CONH2, −N(CH3)CH2CH2S(O)2CH3, − NHC(=NH)H, −NHC(=NH)CH3, − NHC(=NH)NH2, −NHC(=O)NH2, −NO2, =O, −OH, −OCH3, −OCH2CH3, −OCH2CH2OCH3, − OCH2CH2OH, −OCH2CH2N(CH3)2, −O(CH2CH2O)n−(CH2)mCO2H, −O(CH2CH2O)nH, − OCH2F, −OCHF2, −OCF3, −OP(O)(OH)2, −S(O)2N(CH3)2, −SCH3, −S(O)2CH3, and −S(O)3H.
where the dashed lines are optional double bonds; Xa and Xb are independently selected from a five-membered heteroaryl, optionally substituted with R1 or R4; R1 is independently H or C1-C6 alkyl; Y1a and Y1b are independently selected from N, NH, O, and S; Y2a and Y2b are independently selected from N, NH, and S; each of X1, X2, X3, X4, X5, X6, X7, and X8 are independently selected from N, NR2a, CR2b and C(R2b)2; q and r are independently selected from 0, 1, 2, and 3; one or two of X1, X2, X3, X4, X5, X6, X7, and X8 are N or NR2a; one of X1, X2, X3, X4, X5, X6, X7, and X8 is selected from NR2a, CR2b and C(R2b)2 where R2a or R2b is R4;
R2a is independently selected from H, C1-C6 alkyl, −S(O)2N(R1)2, −C(=O)N(R1)2, − C(=O)R1, and R4; R2b is independently selected from H, F, Cl, CN, C1-C6 alkyl, −O(C1-C6 alkyl), C1-C6 heteroalkyl, −C(=O)N(R1)2, and R4; R3 is selected from C1−C6 alkyldiyl, −(C1−C3 alkyldiyl)−O−(C1−C3 alkyldiyl)−, C2−C6 alkenyldiyl and C2−C6 alkynyldiyl, optionally substituted with one or more groups selected from F, Cl, −OH, −OCH3, −OCH2CH3, −OCH2CH2OCH3, −OCH2CH2OH, −OCH2CH2N(CH3)2; R4 is selected from the group consisting of: −(C1-C12 alkyldiyl)−*; −(C1-C12 alkyldiyl)−N(R1)−*; −(C1-C12 alkyldiyl)−O−*; −(C1-C12 alkyldiyl)−(C2-C20 heterocyclyldiyl)−*; −(C1-C20 heteroalkyldiyl)−N(R1)−*; −(C1-C12 heteroalkyldiyl)−O−*; −O−(C1-C12 alkyldiyl)−*; −O−(C1-C12 alkyldiyl)−N(R1)−*; −O−(C1-C12 alkyldiyl)−O−*; −O−(C1-C12 alkyldiyl)−(C2-C20 heterocyclyldiyl)−*; −O−(C1-C12 alkyldiyl)−(C2-C20 heterocyclyldiyl)−N(R1)−*; −OC(=O)N(R1)−*; −OC(=O)N(R1)−(C1-C12 alkyldiyl)−N(R1)−*; −N(R1)−*; −N(R1)−(C1-C12 alkyldiyl)−*; −N(R1)−(C1-C12 alkyldiyl)−N(R4)−*; −N(R1)−(C1-C12 alkyldiyl)−O−*; −N(R1)−(C1-C12 alkyldiyl)−(C2-C20 heterocyclyldiyl)−*; −C(=O)N(R1)−*; −C(=O)N(R1)−(C1-C12 alkyldiyl)−*; −C(=O)N(R1)−(C1-C12 alkyldiyl)−N(R1)−*; −C(=O)N(R1)−(C1-C12 alkyldiyl)−O−*; −(C2-C20 heterocyclyldiyl)−*; −S(=O)2−(C2-C20 heterocyclyldiyl)−*; and −S(=O)2−(C2-C20 heterocyclyldiyl)−(C1-C12 alkyldiyl)−N(R1)−*;
where the asterisk * indicates the attachment site of L; and alkyl, alkyldiyl, alkenyl, alkenyldiyl, alkynyl, alkynyldiyl, heteroalkyl, heteroalkyldiyl, aryl, aryldiyl, carbocyclyl, carbocyclyldiyl, heterocyclyl, heterocyclyldiyl, heteroaryl, and heteroaryldiyl are independently and optionally substituted with one or more groups independently selected from F, Cl, Br, I, −CN, −CH3, −CH2CH3, −CH=CH2, −C ≡CH, − C ≡CCH3, −CH2CH2CH3, −CH(CH3)2, −CH2CH(CH3)2, −CH2OH, −CH2OCH3, −CH2CH2OH, − C(CH3)2OH, −CH(OH)CH(CH3)2, −C(CH3)2CH2OH, −CH2CH2SO2CH3, −CH2OP(O)(OH)2, − CH2F, −CHF2, −CF3, −CH2CF3, −CH2CHF2, −CH(CH3)CN, −C(CH3)2CN, −CH2CN, − CH2NH2, −CH2NHSO2CH3, −CH2NHCH3, −CH2N(CH3)2, −CO2H, −COCH3, −CO2CH3, − CO2C(CH3)3, −COCH(OH)CH3, −CONH2, −CONHCH3, −CON(CH3)2, −C(CH3)2CONH2, − NH2, −NHCH3, −N(CH3)2, −NHCOCH3, −N(CH3)COCH3, −NHS(O)2CH3, − N(CH3)C(CH3)2CONH2, −N(CH3)CH2CH2S(O)2CH3, − NHC(=NH)H, −NHC(=NH)CH3, − NHC(=NH)NH2, −NHC(=O)NH2, −NO2, =O, −OH, −OCH3, −OCH2CH3, −OCH2CH2OCH3, − OCH2CH2OH, −OCH2CH2N(CH3)2, −O(CH2CH2O)n−(CH2)mCO2H, −O(CH2CH2O)nH, − OCH2F, −OCHF2, −OCF3, −OP(O)(OH)2, −S(O)2N(CH3)2, −SCH3, −S(O)2CH3, and −S(O)3H.
2. The immunoconjugate of claim 1 wherein the linker L is a divalent or a branched, trivalent linker.
3. The immunoconjugate of claim 1 wherein the linker L is the linker selected from the group consisting of: −C(=O)−PEG−; −C(=O)−PEG−O−; −C(=O)−PEG−O−C(=O)−; −C(=O)−PEG−C(=O)−; −C(=O)−PEG−N(R1)−; −C(=O)−PEG−SS−(C1-C12 alkyldiyl)−OC(=O)−; −C(=O)−PEG−SS−(C1-C12 alkyldiyl)−C(=O)−; −C(=O)−(C1-C20 heteroalkyldiyl)−; −succinimidyl−(CH2)m−C(=O)N(R1)−(C1-C20 heteroalkyldiyl)−; −succinimidyl−(CH2)m−C(=O)N(R1)−(SolG)−(C1-C20 heteroalkyldiyl)−; −succinimidyl−(CH2)m−C(=O)N(R1)−PEG−; −succinimidyl−(CH2)m−C(=O)N(R1)−PEG−O−; −succinimidyl−(CH2)m−C(=O)N(R1)−PEG−O−C(=O)−; −succinimidyl−(CH2)m−C(=O)N(R1)−PEG−C(=O)−;
−succinimidyl−(CH2)m−C(=O)N(R1)−PEG−N(R4)−; −succinimidyl−(CH2)m−C(=O)N(R1)−PEG−N(R4)−C(=O)−; and −succinimidyl−(CH2)m−C(=O)N(R1)−PEG−SS−(C1-C12 alkyldiyl)−OC(=O)−; PEG has the formula: −(CH2CH2O)n−(CH2)m−; m is an integer from 1 to 5, n is an integer from 1 to 50; and SolG is a solubilizing group selected from the group consisting of polyglycine, polysarcosine, PEG, and a glycoside, or combinations thereof.
4. The immunoconjugate of claim 1 wherein the antibody is an antibody construct that has an antigen binding domain that binds to a target selected from PD-L1, HER2, CEA, and TROP2.
5. The immunoconjugate of claim 4 wherein the antibody is selected from the group consisting of atezolizumab, durvalumab, avelumab, trastuzumab, pertuzumab, labetuzumab, and sacituzumab, or a cysteine-engineered mutant thereof.
6. The immunoconjugate of claim 1 wherein L is attached to a cysteine thiol of the antibody.
7. The immunoconjugate of any one of claims 1 to 6 wherein D has the formula:
 wherein one of R2b is R4.
wherein one of R2b is R4.
8. The immunoconjugate of claim 7 wherein D is selected from the formulas a-f:


9. The immunoconjugate of claim 7 wherein D is selected from the formulas g-n:


10. The immunoconjugate of any one of claims 1 to 6 wherein Xa and Xb are independently selected from the group consisting of imidazolyl, pyrazolyl, triazolyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxadiazolyl, oxazolyl, isothiazolyl, pyrrolyl, oxadiazolyl, and thiadiazolyl.
11. The immunoconjugate of claim 10 wherein Xa and Xb are each pyrazolyl, substituted with one or more groups selected from −CH3, −CH2CH3, −CH=CH2, −C ≡CH, − C ≡CCH3, −CH2CH2CH3, −CH(CH3)2, and −CH2CH(CH3)2.
12. The immunoconjugate of claim 10 wherein Xa or Xb is substituted with R4.
13. The immunoconjugate of any one of claims 1 to 6 wherein Y1a and Y1b are each N.
14. The immunoconjugate of any one of claims 1 to 6 wherein Y1a is S and Y1b is N.
15. The immunoconjugate of any one of claims 1 to 6 wherein Y2a and Y2b are each N.
16. The immunoconjugate of any one of claims 1 to 6 wherein X1 is N; X2 is CR5b; X3 is CR5b; and X4 is CR5b, and q is 1.
17. The immunoconjugate of any one of claims 1 to 6 wherein X1 is CR5b; X2 is N; X3 is CR5b; and X4 is CR5b, and q is 1.
18. The immunoconjugate of any one of claims 1 to 6 wherein X1 is CR5b; X2 is CR5b; X3 is N; and X4 is CR5b, and q is 1.
19. The immunoconjugate of any one of claims 1 to 6 wherein X1 is CR5b; X2 is CR5b; X3 is CR5b; and X4 is N, and q is 1.
20. The immunoconjugate of any one of claims 1 to 6 wherein X5, X6, X7, and X8 are each CR2b; r is 1; and one of CR2b is R4.
21. The immunoconjugate of any one of claims 1 to 6 wherein one of X1, X2, X3, X4, X5, X6, X7, and X8 is N or NR2a.
22. The immunoconjugate of any one of claims 1 to 6 wherein two of X1, X2, X3, X4, X5, X6, X7, and X8 are N or NR2a.
23. The immunoconjugate of any one of claims 1 to 6 wherein one or two R2a are − C(=O)N(R1)2 or −C(=O)R1.
24. The immunoconjugate of any one of claims 1 to 6 wherein one or two R2b are − C(=O)N(R1)2.
25. The immunoconjugate of any one of claims 1 to 6 wherein R2b is selected from the group consisting of −OCH3, −OCH2CH3, −OCH2CH2OCH3, −OCH2CH2OH, and − OCH2CH2N(CH3)2.
26. The immunoconjugate of any one of claims 1 to 6 wherein R3 is selected from − CH2CH2−, −CH=CH−, and −C ≡C−.
27. The immunoconjugate of any one of claims 1 to 6 wherein R3 is C2-C4 alkenyldiyl, substituted with one or more groups selected from F, −OH, and −OCH3.
28. The immunoconjugate of any one of claims 1 to 6 wherein R4 is −O−(C1-C12 alkyldiyl)−(C2-C20 heterocyclyldiyl)−*.
29. The immunoconjugate of claim 28 wherein C1-C12 alkyldiyl is propyldiyl and C2- C20 heterocyclyldiyl is piperidiyl.
30. The immunoconjugate of any one of claims 1 to 6 wherein L is −C(=O)−PEG− or −C(=O)−PEG−C(=O)−.
31. The immunoconjugate of any one of claims 1 to 6 wherein L is attached to a cysteine thiol of the antibody.
32. The immunoconjugate any one of claims 1 to 6 wherein L is −succinimidyl− (CH2)m−C(=O)N(R1)−PEG−.
33. The immunoconjugate of any one of claims 1 to 6 wherein for the PEG, m is 1 or 2, and n is an integer from 1 to 10.
34. The immunoconjugate of claim 33 wherein n is 1, 2, 3, 4, or 5.
35. The immunoconjugate of claim 34 wherein n is 3, 4, or 5.
36. The immunoconjugate of claim 33 wherein n is 10.
37. The immunoconjugate of any one of claims 1 to 6 wherein L attached to the antibody is selected from the structures:
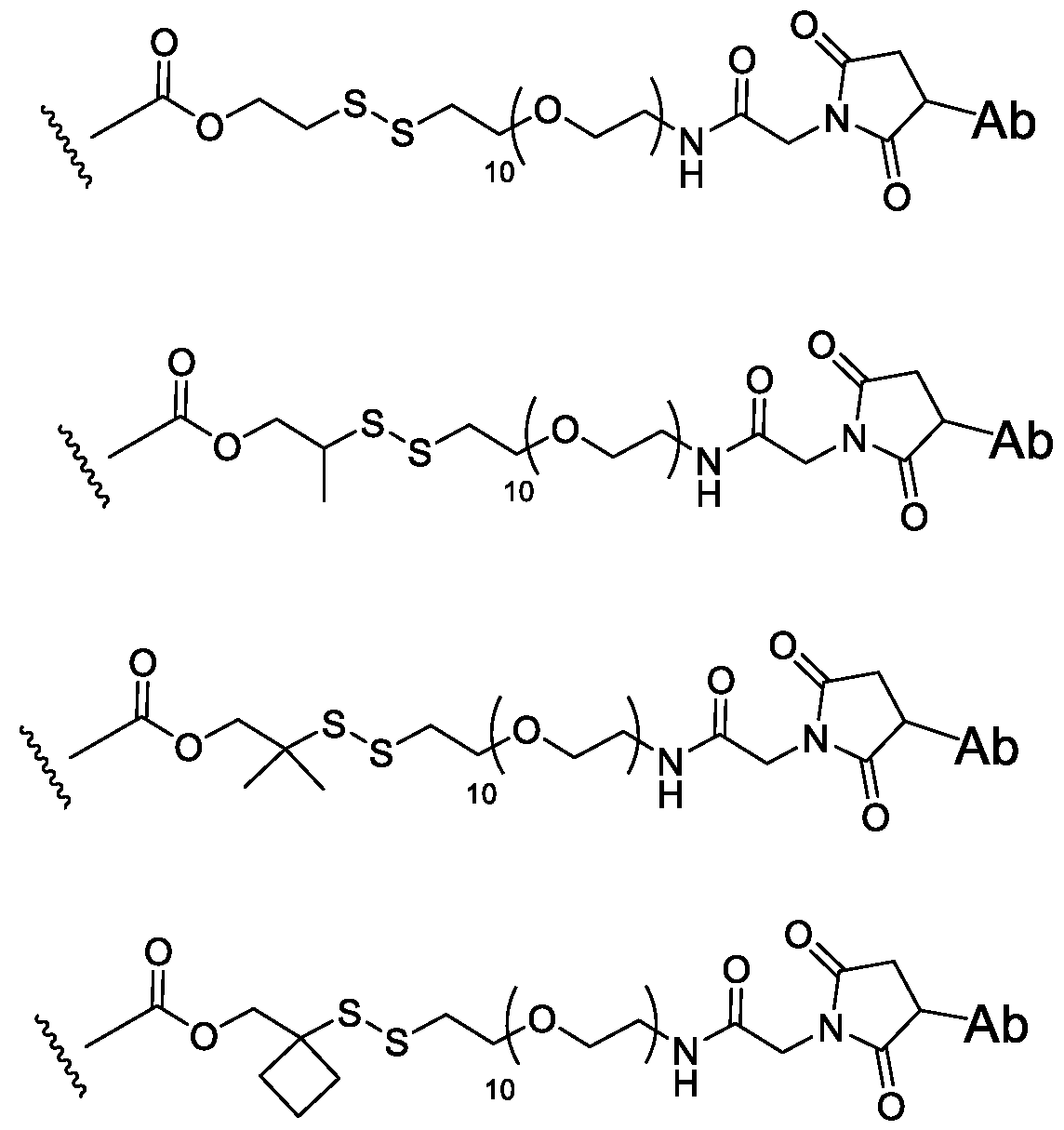 where the wavy line indicates the attachment to R4.
where the wavy line indicates the attachment to R4.
38. A STING agonist-linker intermediate compound having Formula II:

39. The STING agonist-linker intermediate compound of claim 38 wherein Q is selected from:

40. The STING agonist-linker intermediate compound of claim 38 wherein L is selected from the structures:
 where the wavy line indicates the attachment to D.
where the wavy line indicates the attachment to D.
41. A STING agonist-linker intermediate compound selected from Tables 2a and 2b.
42. An immunoconjugate prepared by conjugation of an antibody with a STING agonist-linker intermediate compound of any one of claims 38 to 41.
43. A pharmaceutical composition comprising a therapeutically effective amount of an immunoconjugate of any one of claims 1 to 37, and one or more pharmaceutically acceptable diluent, vehicle, carrier or excipient.
44. A method for treating cancer comprising administering a therapeutically effective amount of a pharmaceutical composition according to claim 43, to a patient in need thereof, wherein the cancer is selected from bladder cancer, salivary gland cancer, endometrial cancer, urinary tract cancer, urothelial carcinoma, lung cancer, non-small cell lung cancer, Merkel cell carcinoma, colon cancer, colorectal cancer, gastric cancer, and breast cancer.
45. The method of claim 44, wherein the cancer is susceptible to a pro-inflammatory response induced by STING agonism.
46. Use of an immunoconjugate according to any one of claims 1 to 37 for treating cancer.
47. A method of preparing an immunoconjugate of any one of claims 1 to 37 wherein a STING agonist-linker intermediate compound of claim 35 is conjugated with the antibody.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363449659P | 2023-03-03 | 2023-03-03 | |
| US63/449,659 | 2023-03-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024186626A1 true WO2024186626A1 (en) | 2024-09-12 |
Family
ID=90716980
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/018051 Ceased WO2024186626A1 (en) | 2023-03-03 | 2024-03-01 | Aza-bicyclic sting agonist immunoconjugates, and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024186626A1 (en) |
Citations (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5624821A (en) | 1987-03-18 | 1997-04-29 | Scotgen Biopharmaceuticals Incorporated | Antibodies with altered effector functions |
| US5677171A (en) | 1988-01-12 | 1997-10-14 | Genentech, Inc. | Monoclonal antibodies directed to the HER2 receptor |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| US6054297A (en) | 1991-06-14 | 2000-04-25 | Genentech, Inc. | Humanized antibodies and methods for making them |
| US6339142B1 (en) | 1998-05-06 | 2002-01-15 | Genentech, Inc. | Protein purification |
| WO2003074566A2 (en) | 2002-03-01 | 2003-09-12 | Immunomedics, Inc. | Rs7 antibodies |
| US6800738B1 (en) | 1991-06-14 | 2004-10-05 | Genentech, Inc. | Method for making humanized antibodies |
| US20070014795A1 (en) | 2004-12-30 | 2007-01-18 | Dhodapkar Madhav V | Compositions and methods for enhanced dendritic cell maturation and function |
| US7416726B2 (en) | 2000-04-13 | 2008-08-26 | The Rockefeller University | Enhancement of antibody-mediated immune responses |
| US20080286819A1 (en) | 2005-11-07 | 2008-11-20 | Ravetch Jeffrey V | Reagents, Methods and Systems for Selecting a Cytotoxic Antibody or Variant Thereof |
| WO2008144891A1 (en) | 2007-05-30 | 2008-12-04 | F. Hoffmann-La Roche Ag | Humanized and chimeric anti-trop-2 antibodies that mediate cancer cell cytotoxicity |
| US7521541B2 (en) | 2004-09-23 | 2009-04-21 | Genetech Inc. | Cysteine engineered antibodies and conjugates |
| WO2009052249A1 (en) | 2007-10-19 | 2009-04-23 | Genentech, Inc. | Cysteine engineered anti-tenb2 antibodies and antibody drug conjugates |
| US7723485B2 (en) | 2007-05-08 | 2010-05-25 | Genentech, Inc. | Cysteine engineered anti-MUC16 antibodies and antibody drug conjugates |
| US7862817B2 (en) | 1999-06-25 | 2011-01-04 | Genentech, Inc. | Humanized anti-ErbB2 antibodies and treatment with anti-ErbB2 antibodies |
| WO2011068845A1 (en) | 2009-12-02 | 2011-06-09 | Immunomedics, Inc. | Combining radioimmunotherapy and antibody-drug conjugates for improved cancer therapy |
| WO2011096519A1 (en) | 2010-02-04 | 2011-08-11 | 東レ株式会社 | Medicinal composition for treating and/or preventing cancer |
| US7999083B2 (en) | 2002-12-13 | 2011-08-16 | Immunomedics, Inc. | Immunoconjugates with an intracellularly-cleavable linkage |
| WO2011145744A1 (en) | 2010-05-17 | 2011-11-24 | 株式会社リブテック | Anti-human trop-2 antibody having antitumor activity in vivo |
| WO2011155579A1 (en) | 2010-06-10 | 2011-12-15 | 北海道公立大学法人札幌医科大学 | ANTI-Trop-2 ANTIBODY |
| WO2011156328A1 (en) | 2010-06-08 | 2011-12-15 | Genentech, Inc. | Cysteine engineered antibodies and conjugates |
| US20120121615A1 (en) | 2010-11-17 | 2012-05-17 | Flygare John A | Alaninyl maytansinol antibody conjugates |
| WO2013018892A1 (en) | 2011-08-04 | 2013-02-07 | 東レ株式会社 | Drug composition for cancer treatment and/or prevention |
| WO2013018891A1 (en) | 2011-08-04 | 2013-02-07 | 東レ株式会社 | Pharmaceutical composition for treatment and/or prophylaxis of cancer |
| WO2013018883A1 (en) | 2011-08-04 | 2013-02-07 | 東レ株式会社 | Cancer treatment and/or prevention drug composition |
| WO2013018889A1 (en) | 2011-08-04 | 2013-02-07 | 東レ株式会社 | Cancer treatment and/or prevention drug composition |
| WO2013068946A2 (en) | 2011-11-11 | 2013-05-16 | Rinat Neuroscience Corp. | Antibodies specific for trop-2 and their uses |
| WO2013077458A1 (en) | 2011-11-22 | 2013-05-30 | 株式会社リブテック | Anti-human trop-2 antibody exhibiting antitumor activity in vivo |
| WO2013125640A1 (en) | 2012-02-21 | 2013-08-29 | 東レ株式会社 | Pharmaceutical composition for treatment and/or prevention of cancer |
| WO2013125654A1 (en) | 2012-02-21 | 2013-08-29 | 東レ株式会社 | Medicinal composition for treating and/or preventing cancer |
| WO2013125636A1 (en) | 2012-02-21 | 2013-08-29 | 東レ株式会社 | Pharmaceutical composition for treatment and/or prevention of cancer |
| WO2013125630A1 (en) | 2012-02-21 | 2013-08-29 | 東レ株式会社 | Pharmaceutical composition for treatment and/or prevention of cancer |
| WO2014014082A1 (en) | 2012-07-19 | 2014-01-23 | 東レ株式会社 | Cancer detection method |
| WO2014014086A1 (en) | 2012-07-19 | 2014-01-23 | 東レ株式会社 | Cancer detection method |
| WO2015020212A1 (en) | 2013-08-09 | 2015-02-12 | 東レ株式会社 | Pharmaceutical composition for treatment and/or prevention of cancer |
| WO2015057063A1 (en) | 2013-10-14 | 2015-04-23 | Synaffix B.V. | Modified glycoprotein, protein-conjugate and process for the preparation thereof |
| WO2015098099A1 (en) | 2013-12-25 | 2015-07-02 | 第一三共株式会社 | Anti-trop2 antibody-drug conjugate |
| US20160145350A1 (en) | 2014-11-21 | 2016-05-26 | Bristol-Myers Squibb Company | Antibodies against cd73 and uses thereof |
| US20170158772A1 (en) | 2015-12-07 | 2017-06-08 | Opi Vi - Ip Holdco Llc | Compositions of antibody construct - agonist conjugates and methods of use thereof |
| US9676863B2 (en) | 2014-02-10 | 2017-06-13 | Merck Patent Gmbh | Targeted TGFβ inhibitors |
| WO2017175147A1 (en) | 2016-04-07 | 2017-10-12 | Glaxosmithkline Intellectual Property Development Limited | Heterocyclic amides useful as protein modulators |
| WO2017196598A1 (en) | 2016-05-10 | 2017-11-16 | Bristol-Myers Squibb Company | Antibody-drug conjugates of tubulysin analogs with enhanced stability |
| WO2018079740A1 (en) | 2016-10-28 | 2018-05-03 | 東レ株式会社 | Pharmaceutical composition for cancer treatment and/or prevention |
| WO2019134705A1 (en) * | 2018-01-08 | 2019-07-11 | 成都先导药物开发股份有限公司 | Immunomodulator |
| US20190300513A1 (en) | 2018-04-03 | 2019-10-03 | Merck Sharp & Dohme Corp. | Sting agonist compounds |
| WO2020010451A1 (en) * | 2018-07-10 | 2020-01-16 | Trillium Therapeutics Inc. | Heteroaromatic-fused imidazolyl amides, compositions and uses thereof as sting agonists |
| WO2020202091A1 (en) * | 2019-04-05 | 2020-10-08 | Glaxosmithkline Intellectual Property Development Limited | Chemical compounds |
| US20210032269A1 (en) * | 2019-08-02 | 2021-02-04 | Mersana Therapeutics, Inc. | Sting agonist compounds and methods of use |
| WO2021202984A1 (en) * | 2020-04-02 | 2021-10-07 | Mersana Therapeutics, Inc. | Antibody drug conjugates comprising sting agonists |
| US11173214B2 (en) | 2015-11-25 | 2021-11-16 | Legochem Biosciences, Inc. | Antibody-drug conjugates comprising branched linkers and methods related thereto |
| WO2021248048A2 (en) | 2020-06-05 | 2021-12-09 | Development Center For Biotechnology | Antibody-drug conjugates containing an anti-mesothelin antibody and uses thereof |
| WO2022147532A1 (en) * | 2021-01-04 | 2022-07-07 | Mersana Therapeutics, Inc. | B7h4-targeted antibody-drug conjugates and methods of use thereof |
| WO2022246597A1 (en) * | 2021-05-24 | 2022-12-01 | Forever Millets Limited | Imidazopyridine derivatives as sting agonists |
| WO2023025256A1 (en) * | 2021-08-26 | 2023-03-02 | 成都先导药物开发股份有限公司 | Sting agonist suitable for use as effector molecule of antibody drug conjugate |
| WO2023078464A1 (en) * | 2021-11-08 | 2023-05-11 | 上海弼领生物技术有限公司 | Epsilon-poly-l-lysine-based drug conjugate, intermediate thereof, and application thereof |
| WO2023137403A2 (en) * | 2022-01-12 | 2023-07-20 | Purdue Research Foundation | Compound comprising raltitrexed or 5-mthf linked to a therapeutic agent, composition, and method of use |
| WO2023173121A1 (en) | 2022-03-11 | 2023-09-14 | Firefly Bio, Inc. | Phenyl maleimide linker agents |
| WO2023178038A1 (en) * | 2022-03-14 | 2023-09-21 | Fulgent Genetics, Inc. | Nanoencapsulated pharmaceutical composition and use thereof |
| WO2024100452A2 (en) * | 2022-11-08 | 2024-05-16 | Legochem Biosciences, Inc. | Heterocyclic compounds as sting agonists |
-
2024
- 2024-03-01 WO PCT/US2024/018051 patent/WO2024186626A1/en not_active Ceased
Patent Citations (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5624821A (en) | 1987-03-18 | 1997-04-29 | Scotgen Biopharmaceuticals Incorporated | Antibodies with altered effector functions |
| US6165464A (en) | 1988-01-12 | 2000-12-26 | Genetech, Inc. | Monoclonal antibodies directed to the HER2 receptor |
| US5677171A (en) | 1988-01-12 | 1997-10-14 | Genentech, Inc. | Monoclonal antibodies directed to the HER2 receptor |
| US6407213B1 (en) | 1991-06-14 | 2002-06-18 | Genentech, Inc. | Method for making humanized antibodies |
| US6054297A (en) | 1991-06-14 | 2000-04-25 | Genentech, Inc. | Humanized antibodies and methods for making them |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| US6639055B1 (en) | 1991-06-14 | 2003-10-28 | Genentech, Inc. | Method for making humanized antibodies |
| US6719971B1 (en) | 1991-06-14 | 2004-04-13 | Genentech, Inc. | Method for making humanized antibodies |
| US6800738B1 (en) | 1991-06-14 | 2004-10-05 | Genentech, Inc. | Method for making humanized antibodies |
| US6339142B1 (en) | 1998-05-06 | 2002-01-15 | Genentech, Inc. | Protein purification |
| US7074404B2 (en) | 1998-05-06 | 2006-07-11 | Genentech, Inc. | Protein purification |
| US7862817B2 (en) | 1999-06-25 | 2011-01-04 | Genentech, Inc. | Humanized anti-ErbB2 antibodies and treatment with anti-ErbB2 antibodies |
| US7416726B2 (en) | 2000-04-13 | 2008-08-26 | The Rockefeller University | Enhancement of antibody-mediated immune responses |
| WO2003074566A2 (en) | 2002-03-01 | 2003-09-12 | Immunomedics, Inc. | Rs7 antibodies |
| US7999083B2 (en) | 2002-12-13 | 2011-08-16 | Immunomedics, Inc. | Immunoconjugates with an intracellularly-cleavable linkage |
| US7521541B2 (en) | 2004-09-23 | 2009-04-21 | Genetech Inc. | Cysteine engineered antibodies and conjugates |
| US20070014795A1 (en) | 2004-12-30 | 2007-01-18 | Dhodapkar Madhav V | Compositions and methods for enhanced dendritic cell maturation and function |
| US20080286819A1 (en) | 2005-11-07 | 2008-11-20 | Ravetch Jeffrey V | Reagents, Methods and Systems for Selecting a Cytotoxic Antibody or Variant Thereof |
| US7723485B2 (en) | 2007-05-08 | 2010-05-25 | Genentech, Inc. | Cysteine engineered anti-MUC16 antibodies and antibody drug conjugates |
| WO2008144891A1 (en) | 2007-05-30 | 2008-12-04 | F. Hoffmann-La Roche Ag | Humanized and chimeric anti-trop-2 antibodies that mediate cancer cell cytotoxicity |
| WO2009052249A1 (en) | 2007-10-19 | 2009-04-23 | Genentech, Inc. | Cysteine engineered anti-tenb2 antibodies and antibody drug conjugates |
| WO2011068845A1 (en) | 2009-12-02 | 2011-06-09 | Immunomedics, Inc. | Combining radioimmunotherapy and antibody-drug conjugates for improved cancer therapy |
| WO2011096519A1 (en) | 2010-02-04 | 2011-08-11 | 東レ株式会社 | Medicinal composition for treating and/or preventing cancer |
| WO2011145744A1 (en) | 2010-05-17 | 2011-11-24 | 株式会社リブテック | Anti-human trop-2 antibody having antitumor activity in vivo |
| US9000130B2 (en) | 2010-06-08 | 2015-04-07 | Genentech, Inc. | Cysteine engineered antibodies and conjugates |
| WO2011156328A1 (en) | 2010-06-08 | 2011-12-15 | Genentech, Inc. | Cysteine engineered antibodies and conjugates |
| WO2011155579A1 (en) | 2010-06-10 | 2011-12-15 | 北海道公立大学法人札幌医科大学 | ANTI-Trop-2 ANTIBODY |
| US20120121615A1 (en) | 2010-11-17 | 2012-05-17 | Flygare John A | Alaninyl maytansinol antibody conjugates |
| WO2013018892A1 (en) | 2011-08-04 | 2013-02-07 | 東レ株式会社 | Drug composition for cancer treatment and/or prevention |
| WO2013018891A1 (en) | 2011-08-04 | 2013-02-07 | 東レ株式会社 | Pharmaceutical composition for treatment and/or prophylaxis of cancer |
| WO2013018883A1 (en) | 2011-08-04 | 2013-02-07 | 東レ株式会社 | Cancer treatment and/or prevention drug composition |
| WO2013018889A1 (en) | 2011-08-04 | 2013-02-07 | 東レ株式会社 | Cancer treatment and/or prevention drug composition |
| WO2013068946A2 (en) | 2011-11-11 | 2013-05-16 | Rinat Neuroscience Corp. | Antibodies specific for trop-2 and their uses |
| WO2013077458A1 (en) | 2011-11-22 | 2013-05-30 | 株式会社リブテック | Anti-human trop-2 antibody exhibiting antitumor activity in vivo |
| WO2013125654A1 (en) | 2012-02-21 | 2013-08-29 | 東レ株式会社 | Medicinal composition for treating and/or preventing cancer |
| WO2013125630A1 (en) | 2012-02-21 | 2013-08-29 | 東レ株式会社 | Pharmaceutical composition for treatment and/or prevention of cancer |
| WO2013125640A1 (en) | 2012-02-21 | 2013-08-29 | 東レ株式会社 | Pharmaceutical composition for treatment and/or prevention of cancer |
| WO2013125636A1 (en) | 2012-02-21 | 2013-08-29 | 東レ株式会社 | Pharmaceutical composition for treatment and/or prevention of cancer |
| WO2014014082A1 (en) | 2012-07-19 | 2014-01-23 | 東レ株式会社 | Cancer detection method |
| WO2014014086A1 (en) | 2012-07-19 | 2014-01-23 | 東レ株式会社 | Cancer detection method |
| WO2015020212A1 (en) | 2013-08-09 | 2015-02-12 | 東レ株式会社 | Pharmaceutical composition for treatment and/or prevention of cancer |
| WO2015057063A1 (en) | 2013-10-14 | 2015-04-23 | Synaffix B.V. | Modified glycoprotein, protein-conjugate and process for the preparation thereof |
| US10072096B2 (en) | 2013-10-14 | 2018-09-11 | Synaffix B.V. | Modified glycoprotein, protein-conjugate and process for the preparation thereof |
| WO2015098099A1 (en) | 2013-12-25 | 2015-07-02 | 第一三共株式会社 | Anti-trop2 antibody-drug conjugate |
| US20160297890A1 (en) | 2013-12-25 | 2016-10-13 | Daiichi Sankyo Company, Limited | Anti-trop2 antibody-drug conjugate |
| US9676863B2 (en) | 2014-02-10 | 2017-06-13 | Merck Patent Gmbh | Targeted TGFβ inhibitors |
| US20160145350A1 (en) | 2014-11-21 | 2016-05-26 | Bristol-Myers Squibb Company | Antibodies against cd73 and uses thereof |
| US11173214B2 (en) | 2015-11-25 | 2021-11-16 | Legochem Biosciences, Inc. | Antibody-drug conjugates comprising branched linkers and methods related thereto |
| US20170158772A1 (en) | 2015-12-07 | 2017-06-08 | Opi Vi - Ip Holdco Llc | Compositions of antibody construct - agonist conjugates and methods of use thereof |
| WO2017175147A1 (en) | 2016-04-07 | 2017-10-12 | Glaxosmithkline Intellectual Property Development Limited | Heterocyclic amides useful as protein modulators |
| WO2017196598A1 (en) | 2016-05-10 | 2017-11-16 | Bristol-Myers Squibb Company | Antibody-drug conjugates of tubulysin analogs with enhanced stability |
| WO2018079740A1 (en) | 2016-10-28 | 2018-05-03 | 東レ株式会社 | Pharmaceutical composition for cancer treatment and/or prevention |
| WO2019134705A1 (en) * | 2018-01-08 | 2019-07-11 | 成都先导药物开发股份有限公司 | Immunomodulator |
| US20190300513A1 (en) | 2018-04-03 | 2019-10-03 | Merck Sharp & Dohme Corp. | Sting agonist compounds |
| WO2020010451A1 (en) * | 2018-07-10 | 2020-01-16 | Trillium Therapeutics Inc. | Heteroaromatic-fused imidazolyl amides, compositions and uses thereof as sting agonists |
| US20200031825A1 (en) * | 2018-07-10 | 2020-01-30 | Trillium Therapeutics Inc. | Heteroaromatic-fused imidazolyl amides, compositions and uses thereof as sting agonists |
| WO2020202091A1 (en) * | 2019-04-05 | 2020-10-08 | Glaxosmithkline Intellectual Property Development Limited | Chemical compounds |
| US20210032269A1 (en) * | 2019-08-02 | 2021-02-04 | Mersana Therapeutics, Inc. | Sting agonist compounds and methods of use |
| WO2021202984A1 (en) * | 2020-04-02 | 2021-10-07 | Mersana Therapeutics, Inc. | Antibody drug conjugates comprising sting agonists |
| WO2021248048A2 (en) | 2020-06-05 | 2021-12-09 | Development Center For Biotechnology | Antibody-drug conjugates containing an anti-mesothelin antibody and uses thereof |
| WO2022147532A1 (en) * | 2021-01-04 | 2022-07-07 | Mersana Therapeutics, Inc. | B7h4-targeted antibody-drug conjugates and methods of use thereof |
| WO2022246597A1 (en) * | 2021-05-24 | 2022-12-01 | Forever Millets Limited | Imidazopyridine derivatives as sting agonists |
| WO2023025256A1 (en) * | 2021-08-26 | 2023-03-02 | 成都先导药物开发股份有限公司 | Sting agonist suitable for use as effector molecule of antibody drug conjugate |
| WO2023078464A1 (en) * | 2021-11-08 | 2023-05-11 | 上海弼领生物技术有限公司 | Epsilon-poly-l-lysine-based drug conjugate, intermediate thereof, and application thereof |
| WO2023137403A2 (en) * | 2022-01-12 | 2023-07-20 | Purdue Research Foundation | Compound comprising raltitrexed or 5-mthf linked to a therapeutic agent, composition, and method of use |
| WO2023173121A1 (en) | 2022-03-11 | 2023-09-14 | Firefly Bio, Inc. | Phenyl maleimide linker agents |
| WO2023178038A1 (en) * | 2022-03-14 | 2023-09-21 | Fulgent Genetics, Inc. | Nanoencapsulated pharmaceutical composition and use thereof |
| WO2023177629A1 (en) * | 2022-03-14 | 2023-09-21 | Anp Technologies, Inc. | Polymer nanoaggregate pharmaceutical composition and use thereof |
| WO2024100452A2 (en) * | 2022-11-08 | 2024-05-16 | Legochem Biosciences, Inc. | Heterocyclic compounds as sting agonists |
Non-Patent Citations (71)
| Title |
|---|
| "NCBI", Database accession no. NP_002344 |
| "The Chemistry of Heterocyclic Compounds, A series of Monographs", vol. 13, 14, 16, 19, 28, 1950, JOHN WILEY & SONS |
| ALTSCHUL ET AL., J. MOLECULAR BIOL., vol. 215, no. 3, 1990, pages 403 - 410 |
| ALTSCHUL ET AL., NUCLEIC ACIDS RES., vol. 25, no. 17, 1997, pages 3389 - 3402 |
| BARBER, G.N., NATURE REV IMMUNOL, vol. 15, 2015, pages 760 - 770 |
| BARGH JD ET AL., CHEM SCI., vol. 11, no. 9, 2020, pages 2375 - 2380 |
| BEIGERT ET AL., PROC. NATL. ACAD. SCI. USA, vol. 106, no. 10, 2009, pages 3770 - 3775 |
| BHAKTA, S. ET AL.: "Antibody-Drug Conjugates, Methods in Molecular Biology", vol. 1045, 2013, article "Engineering THIOMABs for Site-Specific Conjugation of Thiol-Reactive Linkers", pages: 189 - 203 |
| BLUMENTHAL, R. ET AL., CANCER IMMUNOLOGY IMMUNOTHERAPY, vol. 54, no. 219649-07-7, 2005, pages 315 - 327 |
| CACULITAN NG ET AL., CANCER RE., ., vol. 77, no. 24, 2017, pages 7027 - 7037 |
| CALABRESE G ET AL., CYTOGENET CELL GENET., vol. 92, no. 1-2, 2001, pages 164 - 5 |
| CARDILLO, T. ET AL., MOLECULAR CANCER THERAPEUTICS, vol. 17, no. 1, 2018, pages 150 - 160 |
| CHO ET AL., NATURE, vol. 421, 2003, pages 756 - 60 |
| COUSSENS ET AL., SCIENCE, vol. 230, 1985, pages 1132 - 9 |
| DICKGEISSER, S. ET AL., BIOCONJUGATE CHEMISTRY, vol. 31, no. 4, 2020, pages 1070 - 1076 |
| DOMAN, BLOOD, vol. 114, no. 13, 2009, pages 2721 - 2729 |
| ELLIS JALUZIO JP, J BIOL CHEM., vol. 270, no. 35, 1995, pages 20717 - 23 |
| FAULK W P ET AL., PROC. NATL. ACAD. SCI., vol. 75, no. 4, 1978, pages 1947 - 1951 |
| FONG D ET AL., BR. J. CANCER, vol. 99, no. 8, 2008, pages 1290 - 1295 |
| FONG D ET AL., MOD. PATHOL., vol. 21, no. 2, 2008, pages 186 - 191 |
| FORNARO M ET AL., INT. J. CANCER, vol. 62, no. 5, 1995, pages 610 - 618 |
| FUCHS, A. ET AL., SEMINARS IN CANCER BIOLOGY, vol. 16, no. 5, 2006, pages 359 - 366 |
| FURUSE M ET AL., J. CELL BIOL., vol. 141, no. 7, 1998, pages 1539 - 50 |
| GUSFIELD: "Algorithms on Strings, Trees and Sequences", 1997, CAMBRIDGE UNIVERSITY PRESS |
| HAMBLETT ET AL., CLIN. CANCER RES., vol. 10, 2004, pages 7063 - 7070 |
| HAMBLETT, K.J. ET AL.: "2004 Annual Meeting", vol. 45, 27 March 2004, AMERICAN ASSOCIATION FOR CANCER RESEARCH, article "Effect of drug loading on the pharmacology, pharmacokinetics, and toxicity of an anti-CD30 antibody-drug conjugate" |
| HARARIYARDEN, ONCOGENE, vol. 19, no. 380610-27-5, 2000, pages 6102 - 14 |
| HERMANSON: "Bioconjugate Techniques", 2008, ACADEMIC PRESS |
| J. AM. CHEM. SOC., vol. 82, 1960, pages 5566 |
| JARAMILLO, M.L. ET AL., MABS, vol. 15, no. 1, 2023, pages 1 - 15 |
| JEFFERIS ET AL., MABS, vol. 1, no. 4, 2009, pages 332 - 338 |
| JEFFREY SC ET AL., BIOCONJUG CHEM., vol. 17, no. 3, 2006, pages 831 - 40 |
| JUNUTULA ET AL., NATURE BIOTECH., vol. 26, no. 8, 2008, pages 925 - 932 |
| KELLOGG, BA ET AL., BIOCONJUGATE CHEM., vol. 22, 2011, pages 717 - 727 |
| KHOT, A. ET AL., BIOANALYSIS, vol. 7, no. 13, 2015, pages 1633 - 1648 |
| KNIESEL U ET AL., CELL. MOL. NEUROBIOL., vol. 20, no. 1, 2000, pages 57 - 76 |
| LIEBERMAN: "Pharmaceutical Dosage Forms", vol. 1-3, 1992 |
| LINNENBACH A J ET AL., PROC. NATL. ACAD. SCI., vol. 86, no. 1, 1989, pages 27 - 31 |
| LINNENBACH AJ ET AL., MOL CELL BIOL., vol. 13, no. 3, 1993, pages 1507 - 15 |
| LIPINSKI M ET AL., PROC. NATL. ACAD. SCI., vol. 78, no. 8, 1981, pages 5147 - 5150 |
| LLOYD, THE ART, SCIENCE AND TECHNOLOGY OF PHARMACEUTICAL COMPOUNDING, 1999 |
| LYON, R. ET AL., METHODS IN ENZYM., vol. 502, 2012, pages 123 - 138 |
| MALIK ET AL., PRO AM SOC CANCER RES, vol. 44, 2003, pages 176 - 7 |
| MCDONAGH ET AL., PROT. ENGR. DESIGN & SELECTION, vol. 19, no. 7, 2006, pages 299 - 307 |
| MIYOSHI ET AL., AMERICAN JOURNAL OF NEPHROLOGY, vol. 27, no. 6, 2007, pages 590 - 604 |
| MUHLMANN G ET AL., J. CLIN. PATHOL., vol. 62, no. 2, 2009, pages 152 - 158 |
| NING S ET AL., NEUROL. SCI., vol. 34, no. 10, 2013, pages 1745 - 1750 |
| no. 76513-69-4 |
| OHMACHI T ET AL., CLIN. CANCER RES., vol. 12, no. 10, 2006, pages 3057 - 3063 |
| OKELEY, N.M. ET AL., BIOCONJUGATE CHEM., vol. 24, no. 10, 2013, pages 1650 - 1655 |
| PAQUETTE, LEO A.: "Principles of Modern Heterocyclic Chemistry", 1968, W.A. BENJAMIN |
| PICKAR, DOSAGE CALCULATIONS, 1999 |
| PILLOW, T. ET AL., CHEM. SCI., vol. 8, 2017, pages 366 - 370 |
| PURCELL, JW ET AL., CANCER RES., vol. 78, no. 14, 2018, pages 4059 - 4072 |
| QASBA, P.K., BIOCONJUGATE CHEM., vol. 26, 2015, pages 2170 - 2175 |
| RAMANJULU, J.M. ET AL., NATURE, vol. 564, 2018, pages 439 - 443 |
| RICART, A. D. ET AL., CLIN. CANCER RES., vol. 17, 2011, pages 6417 - 6427 |
| RUSSELL ET AL., J. MOL BIOL., vol. 244, 1994, pages 332 - 350 |
| SLAMON ET AL., NEW ENGL. J. MED., vol. 344, 2001, pages 783 - 792 |
| SLAMON ET AL., SCIENCE, vol. 244, 1989, pages 707 - 12 |
| SLIWKOWSKI, NAT STRUCT BIOL, vol. 10, 2003, pages 158 - 9 |
| SODING, BIOINFORMATICS, vol. 21, no. 7, 2005, pages 951 - 960 |
| SOLOMON S ET AL., MOL CELL BIOL., vol. 27, no. 6, 2007, pages 2324 - 42 |
| SWISSHELM K ET AL., ADV. DRUG DELIV. REV., vol. 57, no. 6, 2005, pages 919 - 28 |
| TAKAI Y. ET AL., CANCER SCIENCE, vol. 94, no. 8, 2003, pages 655 - 67 |
| WANG B ET AL., J IMMUNOL., vol. 175, no. 7, 2005, pages 4274 - 4282 |
| YANG, Z-S ET AL., ONCOLOGY LETTERS, vol. 18, 2019, pages 15 - 21 |
| YARDENSLIWKOWSKI, NAT REV MOL CELL BIO, vol. 12, 2001, pages 127 - 37 |
| ZHANG D ET AL., ACS MED CHEM LEFT., vol. 7, no. 11, 2016, pages 988 - 993 |
| ZHANG, X. ET AL., ACS CHEM. BIOL., vol. 16, 2021, pages 2502 - 2514 |
| ZHOU, Q. ET AL., BIOCONJUGATE CHEM., vol. 25, no. 3, 2014, pages 510 - 520 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11547761B1 (en) | Antibody adjuvant conjugates | |
| EP3609540B1 (en) | Immunoconjugate synthesis method | |
| US20210154316A1 (en) | Immunoconjugates | |
| US20240335557A1 (en) | Bis-benzimidazole sting agonist immunoconjugates, and uses thereof | |
| CA3046790A1 (en) | Antibody adjuvant conjugates | |
| US20220347312A1 (en) | Immunoconjugate Synthesis Method | |
| US20250000995A1 (en) | Tlr agonist immunoconjugates with cysteine-mutant antibodies, and uses thereof | |
| US20230263903A1 (en) | Pyrazoloazepine immunoconjugates, and uses thereof | |
| WO2024186626A1 (en) | Aza-bicyclic sting agonist immunoconjugates, and uses thereof | |
| EP4412660A1 (en) | Asymmetric bis-benzimidazole sting agonist immunoconjugates and uses thereof | |
| WO2024129956A1 (en) | Thienoazepine immunoconjugates, and uses thereof | |
| WO2024173387A1 (en) | Aza-benzazepine immunoconjugates, and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24716021 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |