WO2023056468A1 - Extracellular vesicle comprising cholesterol tagged sting-agonist - Google Patents
Extracellular vesicle comprising cholesterol tagged sting-agonist Download PDFInfo
- Publication number
- WO2023056468A1 WO2023056468A1 PCT/US2022/077424 US2022077424W WO2023056468A1 WO 2023056468 A1 WO2023056468 A1 WO 2023056468A1 US 2022077424 W US2022077424 W US 2022077424W WO 2023056468 A1 WO2023056468 A1 WO 2023056468A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fold
- days
- aspects
- sting agonist
- exosome
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/554—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being a steroid plant sterol, glycyrrhetic acid, enoxolone or bile acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6901—Conjugates being cells, cell fragments, viruses, ghosts, red blood cells or viral vectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4702—Regulators; Modulating activity
- C07K14/4705—Regulators; Modulating activity stimulating, promoting or activating activity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/515—Animal cells
- A61K2039/5156—Animal cells expressing foreign proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
- A61K2039/575—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2 humoral response
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/70—Multivalent vaccine
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/20011—Coronaviridae
- C12N2770/20023—Virus like particles [VLP]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/20011—Coronaviridae
- C12N2770/20034—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
Definitions
- Stimulator of Interferon Genes is a cytosolic sensor of cyclic dinucleotides that is typically produced by bacteria. Upon activation, it leads to the production of type I interferons and initiates an immune response. Agonism of STING has been shown as a promising approach for generating an immune response against tumors pre-clinically.
- systemic delivery of STING agonists leads to systemic inflammation. This limits the dose that can be given which in turn limits the therapeutic efficacy.
- An alternative approach to systemic delivery is to inject the STING agonist directly into the tumor. Intra-tumoral injections are quite effective; however, they are limited to solid tumors that can be reached with a needle and lead to tissue damage. Improved methods of delivering STING agonists are therefore needed.
- the present disclosure provides an extracellular vesicle (EV) comprising a cholesterol or derivative thereof on the surface, wherein the cholesterol is linked to a stimulator of interferon genes protein (STING) agonist through a cleavable peptide linker.
- EV extracellular vesicle
- STING interferon genes protein
- the cleavable peptide linker comprises a linker cleavable by cathepsin.
- the cleavable peptide linker comprises a Valine- Alanine linker and/or a Valine-Citrulline linker.
- the EV is an exosome, a nanovesicle, an apoptotic body, a microvesicle, a lysosome, an endosome, a liposome, a lipid nanoparticle, a micelle, a multilamellar structure, a revesiculated vesicle, or an extruded cell.
- the EV is an exosome.
- the EV overexpresses a PTGFRN protein.
- the EV is produced by a cell that overexpresses a PTGFRN protein.
- the extracellular vesicle further comprises a ligand, a cytokine, an antigen, or an antibody.
- the antibody comprises an antagonistic antibody and/or an agonistic antibody.
- the STING agonist is a cyclic dinucleotide. In some aspects, the STING agonist is a non-cyclic dinucleotide. In some aspects, the STING agonist is modified such that a polarity and/or a charge different from the corresponding unmodified STING agonist. In some aspects, the STING agonist comprises:
- Xi is H, OH, or F
- X 2 is H, OH, or F
- Z is OH, ORi, SH or SRi, wherein: i) Ri is Na or NH4, or ii) Ri is an enzyme-labile group which provides OH or SH in vivo such as pivaloyloxymethyl;
- Bi and B2 are bases chosen from:
- the STING agonist is selected from the group consisting of: and a pharmaceutically acceptable salt thereof.
- the cholesterol or derivative thereof comprises:
- the EV described herein comprises:
- the EVs described herein further comprises a scaffold moiety.
- the scaffold moiety comprises Scaffold X, which comprises an amino acid sequence as set forth in SEQ ID NO: 302.
- the Scaffold X comprises an amino acid sequence at least 50%, at least 60%, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or about 100% identical to SEQ ID NO:302.
- the scaffold moiety comprises Scaffold Y, which comprises a BASP1 protein or a functional fragment thereof.
- Scaffold Y comprises an amino acid sequence as set forth in GGKLSKK (SEQ ID NO: 411). In some aspects, Scaffold Y comprises an amino acid sequence as set forth in i) GGKLSKKK (SEQ ID NO: 438), (ii) GGKLSKKS (SEQ ID NO: 439), (iii) GAKLSKKK (SEQ ID NO: 440), (iv) GAKLSKKS (SEQ ID NO: 441), (v) GGKQSKKK (SEQ ID NO: 442), (vi) GGKQSKKS (SEQ ID NO: 443), (vii) GGKLAKKK (SEQ ID NO: 444), (viii) GGKLAKKS (SEQ ID NO: 445), or (ix) any combination thereof.
- the present disclosure provides a pharmaceutical composition comprising the EV described herein and a pharmaceutically acceptable carrier.
- the pharmaceutical composition when administered to a mammal, the composition does not deplete T cells and/or macrophages in the mammal.
- the pharmaceutical composition when administered to a mammal, the composition depletes T cells and/or macrophages in the mammal at a lesser degree than the free STING agonist.
- the present disclosure also provides a kit comprising the compositions described herein and instructions for use.
- the present disclosure also provides a method of producing an EV, e.g., exosome, comprising a cholesterol or derivative thereof, which is linked to a STING agonist via a cleavable peptide linker, the method comprising:
- the cleavable peptide linker comprises a linker cleavable by cathepsin. In some aspects, the cleavable peptide linker comprises a Valine-Alanine linker and/or a Valine- Citrulline linker.
- the present disclosure provides a method of inducing or modulating an immune response and/or an inflammatory response in a subject in need thereof, the method comprising administering to the subject the EVs described herein or the compositions described herein.
- the present disclosure provides a method of treating a tumor in a subject in need thereof, the method comprising administering to the subject the EVs described herein or the compositions described herein.
- the administering induces or modulates the immune response and/or the inflammatory response in the subject.
- the administering activates Dendritic Cells.
- the administering activates myeloid Dendritic Cells.
- the administering results in reduced monocyte cell activation compared to the free STING agonist. In some aspects, the administering does not induce monocyte cell activation. In some aspects, the administering induces interferon-P (IFN-P) production. In some aspects, the administering results in reduced systemic inflammation compared to the free STING agonist. In some aspects, the administering results in insubstantial amounts of systemic inflammation. In some aspects, the administration is parenterally, orally, intravenously, intramuscularly, intra-tumorally, intraperitoneally, or via any other appropriate administration route. In some aspects, the administration is intravenous. In some aspects, the immune response is an anti-tumor response. In some aspects, the methods further comprises administering an additional therapeutic agent.
- IFN-P interferon-P
- the additional therapeutic agent is an immunomodulating agent. In some aspects, the additional therapeutic agent is an antibody or antigen-binding fragment thereof. In some aspects, the antibody or antigen-binding fragment thereof is an inhibitor of CTLA- 4, PD-1, PD-L1, PD-L2, TIM-3, or LAG3. In some aspects, the administering prevents metastasis of the tumor in the subject.
- the tumor comprises or is derived from a chondroid sarcoma, a cutaneous squamous cell carcinoma (cSCC), a head and neck squamous cell carcinoma of the oral cavity, a hepatocellular cancer (HCC), a cervical cancer, an eye melanoma, a choroidal eye melanoma, a gastric spindle cell sarcoma, a hemangioendothelioma, a mesothelioma, a parotoid gland cancer, a renal cancer, a triple-negative breast cancer (TNBC), or any combination thereof.
- a chondroid sarcoma a cutaneous squamous cell carcinoma (cSCC), a head and neck squamous cell carcinoma of the oral cavity
- HCC hepatocellular cancer
- cervical cancer an eye melanoma
- a gastric spindle cell sarcoma
- the EV is thermostable at about 20°C for at least about 7 days, at least about 8 days, at least about 9 days, at least about 10 days, at least about 11 days, at least about 12 days, at least about 13 days, at least about 14 days, at least about 15 days, at least about 16 days, at least about 17 days, at least about 18 days, at least about 19 days, at least about 20 days, at least about 21 days, at least about 22 days, at least about 23 days, at least about 24 days, at least about 25 days, at least about 26 days, at least about 27 days, at least about 28 days, at least about 29 days, or at least about 30 days.
- the EV is thermostable at about 37°C for at least about 7 days, at least about 8 days, at least about 9 days, at least about 10 days, at least about 11 days, at least about 12 days, at least about 13 days, at least about 14 days, at least about 15 days, at least about 16 days, at least about 17 days, at least about 18 days, at least about 19 days, at least about 20 days, at least about 21 days, at least about 22 days, at least about 23 days, at least about 24 days, at least about 25 days, at least about 26 days, at least about 27 days, at least about 28 days, at least about 29 days, or at least about 30 days.
- the EV is capable of inducing an immune response in a subject.
- FIG. 1A shows Interferon-P expression of cholesterol tagged STING (ECC-STING) exosomes (exoSTING ECC) on hPBMCs in vitro compared to exoSTING and soluble (free) STING.
- ECC-STING cholesterol tagged STING
- ExoSTING is a mixture of the STING agonist and exosomes (i.e., exosomes loaded with STING agonist, such as by diffusion).
- FIG. IB shows cholesterol tagged STING (ECC-STING) exosomes (exoSTING ECC) retain in vivo activity, as measured by IFN-P expression in spleen, for at least 1 month at 37°C compared to exoSTING. For each day, left bar is exoSTING and right bar is exoSTING-ECC.
- FIGs. 2A-2B are bar graphs illustrating anti-SARS CoV2 RBD IgG titers (FIG. 2A) and anti-SARS CoV2 RBD neutralizing antibodies (FIG.
- exosomes following subcutaneous administration of exosomes expressing the SARS CoV2 RBD and stably loaded with STING-ECC ("exoRBD/STING-ECC"; FIGs. 2A-2B) or exosomes expressing SARS CoV2 RBD alone ("exoRBD”; FIG. 2A) in mice.
- exosomes Prior to administration, exosomes were stored for 30 days at 37°C or -20°C, as indicated (FIGs. 2A-2B).
- FIGs. 3A-3D show neutralizing antibody production (FIG. 3A), anti-RBD antibody induction (FIG. 3B), and splenocytes expression of IFNy (FIG. 3C) and TNFa (FIG. 3D) in mice subcutaneously administered varying doses of exosomes expressing the RBDs from both SARS CoVl and CoV2 (both fused to PTGFRN) and loaded with cholesterol tagged STING (ECC STING), as indicated.
- ECC STING cholesterol tagged STING
- a range of 1 to 10 is understood to include any number, combination of numbers, or sub-range from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10.
- a value is explicitly recited, it is to be understood that values which are about the same quantity or amount as the recited value are also within the scope of the disclosure.
- a combination is disclosed, each subcombination of the elements of that combination is also specifically disclosed and is within the scope of the disclosure. Conversely, where different elements or groups of elements are individually disclosed, combinations thereof are also disclosed.
- Nucleotides are referred to by their commonly accepted single-letter codes. Unless otherwise indicated, nucleotide sequences are written left to right in 5' to 3' orientation. Nucleotides are referred to herein by their commonly known one-letter symbols recommended by the IUPAC- IUB Biochemical Nomenclature Commission. Accordingly, A represents adenine, C represents cytosine, G represents guanine, T represents thymine, and U represents uracil.
- Amino acid sequences are written left to right in amino to carboxy orientation. Amino acids are referred to herein by either their commonly known three letter symbols or by the one- letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission.
- the term "about” or “approximately” is used herein to mean approximately roughly, around, or in the region of. When the term “about” is used in conjunction with a numerical range, it modifies that range by extending the boundaries above and below the numerical values set forth.
- the term used herein means up to 10% of the referenced amount, e.g., about 50% is understood to encompass a range of values from 45% to 55%. In some aspects, the term means more than or less than 10% of the reference amount, e.g., ⁇ 9%, ⁇ 8%, ⁇ 7%, ⁇ 6%, ⁇ 5%, ⁇ 4%, ⁇ 3%, ⁇ 2%, ⁇ 1%, or ⁇ 0.1%.
- extracellular vesicle refers to a cell-derived vesicle comprising a membrane that encloses an internal space.
- Extracellular vesicles comprise all membrane-bound vesicles e.g., exosomes, nanovesicles) that have a smaller diameter than the cell from which they are derived.
- extracellular vesicles range in diameter from 20 nm to 1000 nm, and can comprise various macromolecular payload either within the internal space (i.e., lumen), displayed on the external surface of the extracellular vesicle, and/or spanning the membrane.
- Said payload can comprise nucleic acids, proteins, carbohydrates, lipids, small molecules, and/or combinations thereof.
- an extracellular vesicle comprises a scaffold moiety.
- extracellular vesicles include apoptotic bodies, fragments of cells, vesicles derived from cells by direct or indirect manipulation (e.g., by serial extrusion or treatment with alkaline solutions), vesiculated organelles, and vesicles produced by living cells (e.g., by direct plasma membrane budding or fusion of the late endosome with the plasma membrane).
- Extracellular vesicles can be derived from a living or dead organism, explanted tissues or organs, prokaryotic or eukaryotic cells, and/or cultured cells. In some aspects, extracellular vesicles are produced by cells that express one or more transgene products.
- exosome refers to a cell-derived small (between 20-300 nm in diameter, more preferably 40-200 nm in diameter) vesicle comprising a membrane that encloses an internal space (i.e., lumen), and which is generated from said cell by direct plasma membrane budding or by fusion of the late endosome with the plasma membrane.
- the exosome is a species of extracellular vesicle.
- the exosome comprises lipid or fatty acid and polypeptide and optionally comprises a payload (e.g., a therapeutic agent), a receiver (e.g., a targeting moiety), a polynucleotide (e.g., a nucleic acid, RNA, or DNA), a sugar (e.g., a simple sugar, polysaccharide, or glycan) or other molecules.
- a payload e.g., a therapeutic agent
- a receiver e.g., a targeting moiety
- a polynucleotide e.g., a nucleic acid, RNA, or DNA
- a sugar e.g., a simple sugar, polysaccharide, or glycan
- an exosome comprises a scaffold moiety.
- the exosome can be derived from a producer cell, and isolated from the producer cell based on its size, density, biochemical parameters, or a combination thereof.
- the term "nanovesicle” refers to a cell-derived small (between 20-250 nm in diameter, more preferably 30-150 nm in diameter) vesicle comprising a membrane that encloses an internal space, and which is generated from said cell by direct or indirect manipulation such that said nanovesicle would not be produced by said producer cell without said manipulation.
- Appropriate manipulations of said producer cell include but are not limited to serial extrusion, treatment with alkaline solutions, sonication, or combinations thereof. The production of nanovesicles may, in some instances, result in the destruction of said producer cell.
- populations of nanovesicles are substantially free of vesicles that are derived from producer cells by way of direct budding from the plasma membrane or fusion of the late endosome with the plasma membrane.
- the nanovesicle comprises lipid or fatty acid and polypeptide, and optionally comprises a payload (e.g., a therapeutic agent), a receiver (e.g., a targeting moiety), a polynucleotide (e.g., a nucleic acid, RNA, or DNA), a sugar (e.g., a simple sugar, polysaccharide, or glycan) or other molecules.
- a nanovesicle comprises a scaffold moiety.
- the nanovesicle once it is derived from a producer cell according to said manipulation, may be isolated from the producer cell based on its size, density, biochemical parameters, or a combination thereof.
- modified when used in the context of exosomes described herein, refers to an alteration or engineering of an EV, such that the modified EV is different from a naturally- occurring EV.
- a modified EV described herein comprises a membrane that differs in composition of a protein, a lipid, a small molecular, a carbohydrate, etc.
- membrane compared to the membrane of a naturally-occurring EV (e.g., membrane comprises higher density or number of natural EV proteins and/or membrane comprises proteins that are not naturally found in EVs.
- modifications to the membrane changes the exterior surface of the EV.
- such modifications to the membrane changes the lumen of the EV.
- a scaffold moiety refers to a molecule that can be used to anchor STING agonists disclosed herein or any other compound of interest (e.g., payload) to the EV either on the luminal surface or on the exterior surface of the EV.
- a scaffold moiety comprises a synthetic molecule.
- a scaffold moiety comprises a nonpolypeptide moiety.
- a scaffold moiety comprises a lipid, carbohydrate, or protein that naturally exists in the EV.
- a scaffold moiety comprises a lipid, carbohydrate, or protein that does not naturally exist in the exosome.
- a scaffold moiety is Scaffold X.
- a scaffold moiety is Scaffold Y.
- a scaffold moiety comprises both Scaffold X and Scaffold Y.
- Scaffold X refers to exosome proteins that have recently been identified on the surface of exosomes. See, e.g., U.S. Pat. No. 10,195,290, which is incorporated herein by reference in its entirety.
- Non-limiting examples of Scaffold X proteins include: prostaglandin F2 receptor negative regulator (“the PTGFRN protein”); basigin (“the BSG protein”); immunoglobulin superfamily member 2 (“the IGSF2 protein”); immunoglobulin superfamily member 3 (“the IGSF3 protein”); immunoglobulin superfamily member 8 (“the IGSF8 protein”); integrin beta-1 ("the ITGB1 protein); integrin alpha-4 (“the ITGA4 protein”); 4F2 cellsurface antigen heavy chain (“the SLC3 A2 protein”); and a class of ATP transporter proteins (“the ATP1A1 protein,” “the ATP1A2 protein,” “the ATP1A3 protein,” “the ATP1A4 protein,” “the ATP1B3 protein,” “the ATP2B1 protein,” “the ATP2B2 protein,” “the ATP2B3 protein,” “the ATP2B protein”).
- a Scaffold X protein can be a whole protein or a fragment thereof (e.g, functional fragment, e.g., the smallest fragment that is capable of anchoring another moiety on the exterior surface or on the luminal surface of the EV, e.g., exosome,).
- a Scaffold X can anchor a moiety (e.g, STING agonist) to the external surface or the luminal surface of the EVs, e.g., exosomes,.
- STING agonist e.g, STING agonist
- Non-limiting examples of Scaffold Y proteins include: myristoylated alanine rich Protein Kinase C substrate ("the MARCKS protein”); myristoylated alanine rich Protein Kinase C substrate like 1 (“the MARCKSL1 protein”); and brain acid soluble protein 1 (“the BASP1 protein”).
- a Scaffold Y protein can be a whole protein or a fragment thereof (e.g., functional fragment, e.g., the smallest fragment that is capable of anchoring a moiety on the luminal surface of the EVs, e.g., exosomes,).
- a Scaffold Y can anchor a moiety (e.g., STING agonist) to the lumen of the EVs, e.g., exosomes,.
- fragment of a protein refers to an amino acid sequence of a protein that is shorter than the naturally- occurring sequence, N- and/or C-terminally deleted or any part of the protein deleted in comparison to the naturally occurring protein.
- functional fragment refers to a protein fragment that retains protein function. Accordingly, in some aspects, a functional fragment of a Scaffold X protein retains the ability to anchor a moiety on the luminal surface and/or on the exterior surface of the EV.
- a functional fragment of a Scaffold Y protein retains the ability to anchor a moiety on the luminal surface of the EV. Whether a fragment is a functional fragment can be assessed by any art known methods to determine the protein content of EVs including Western Blots, FACS analysis and fusions of the fragments with autofluorescent proteins like, e.g., GFP.
- a functional fragment of a Scaffold X protein retains at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90% or at least about 100% of the ability, e.g., an ability to anchor a moiety, of the naturally occurring Scaffold X protein.
- a functional fragment of a Scaffold Y protein retains at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90% or at least about 100% of the ability, e.g., an ability to anchor another molecule, of the naturally occurring Scaffold Y protein.
- variant of a molecule refers to a molecule that shares certain structural and functional identities with another molecule upon comparison by a method known in the art.
- a variant of a protein can include a substitution, insertion, deletion, frameshift or rearrangement in another protein.
- a "conservative amino acid substitution” is one in which the amino acid residue is replaced with an amino acid residue having a similar side chain.
- Families of amino acid residues having similar side chains have been defined in the art, including basic side chains (e.g., lysine, arginine, histidine), acidic side chains (e.g., aspartic acid, glutamic acid), uncharged polar side chains (e.g., glycine, asparagine, glutamine, serine, threonine, tyrosine, cysteine), nonpolar side chains (e.g., alanine, valine, leucine, isoleucine, proline, phenylalanine, methionine, tryptophan), beta-branched side chains (e.g., threonine, valine, isoleucine) and aromatic side chains (e.g., tyrosine, phenylalanine, tryptophan, histidine).
- basic side chains e.g., lysine, arginine, histidine
- acidic side chains e.g., aspartic acid
- a string of amino acids can be conservatively replaced with a structurally similar string that differs in order and/or composition of side chain family members.
- percent sequence identity or “percent identity” between two polynucleotide or polypeptide sequences refers to the number of identical matched positions shared by the sequences over a comparison window, taking into account additions or deletions (i.e., gaps) that must be introduced for optimal alignment of the two sequences.
- a matched position is any position where an identical nucleotide or amino acid is presented in both the target and reference sequence. Gaps presented in the target sequence are not counted since gaps are not nucleotides or amino acids. Likewise, gaps presented in the reference sequence are not counted since target sequence nucleotides or amino acids are counted, not nucleotides or amino acids from the reference sequence.
- the percentage of sequence identity is calculated by determining the number of positions at which the identical amino-acid residue or nucleic acid base occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison and multiplying the result by 100 to yield the percentage of sequence identity.
- the comparison of sequences and determination of percent sequence identity between two sequences may be accomplished using readily available software both for online use and for download. Suitable software programs are available from various sources, and for alignment of both protein and nucleotide sequences. One suitable program to determine percent sequence identity is bl2seq, part of the BLAST suite of programs available from the U.S.
- B12seq performs a comparison between two sequences using either the BLASTN or BLASTP algorithm.
- BLASTN is used to compare nucleic acid sequences
- BLASTP is used to compare amino acid sequences.
- Other suitable programs are, e.g., Needle, Stretcher, Water, or Matcher, part of the EMBOSS suite of bioinformatics programs and also available from the European Bioinformatics Institute (EBI) at www.ebi.ac.uk/Tools/psa.
- Different regions within a single polynucleotide or polypeptide target sequence that aligns with a polynucleotide or polypeptide reference sequence can each have their own percent sequence identity. It is noted that the percent sequence identity value is rounded to the nearest tenth. For example, 80.11, 80.12, 80.13, and 80.14 are rounded down to 80.1, while 80.15, 80.16, 80.17, 80.18, and 80.19 are rounded up to 80.2. It also is noted that the length value will always be an integer.
- sequence alignments are not limited to binary sequence-sequence comparisons exclusively driven by primary sequence data. Sequence alignments can be derived from multiple sequence alignments.
- One suitable program to generate multiple sequence alignments is ClustalW2, available from www.clustal.org.
- Another suitable program is MUSCLE, available from www.drive5.com/muscle/.
- ClustalW2 and MUSCLE are alternatively available, e.g., from the EBI.
- sequence alignments can be generated by integrating sequence data with data from heterogeneous sources such as structural data (e.g., crystallographic protein structures), functional data (e.g., location of mutations), or phylogenetic data.
- a suitable program that integrates heterogeneous data to generate a multiple sequence alignment is T-Coffee, available at www.tcoffee.org, and alternatively available, e.g., from the EBI. It will also be appreciated that the final alignment used to calculate percent sequence identity may be curated either automatically or manually.
- the polynucleotide variants can contain alterations in the coding regions, non-coding regions, or both. In some aspects, the polynucleotide variants contain alterations which produce silent substitutions, additions, or deletions, but do not alter the properties or activities of the encoded polypeptide. In some aspects, nucleotide variants are produced by silent substitutions due to the degeneracy of the genetic code. In other aspects, variants in which 5-10, 1-5, or 1-2 amino acids are substituted, deleted, or added in any combination.
- Polynucleotide variants can be produced for a variety of reasons, e.g., to optimize codon expression for a particular host (change codons in the human mRNA to others, e.g., a bacterial host such as E. colt).
- Naturally occurring variants are called "allelic variants," and refer to one of several alternate forms of a gene occupying a given locus on a chromosome of an organism (Genes II, Lewin, B., ed., John Wiley & Sons, New York (1985)). These allelic variants can vary at either the polynucleotide and/or polypeptide level and are included in the present disclosure. Alternatively, non-naturally occurring variants can be produced by mutagenesis techniques or by direct synthesis. [0049] Using known methods of protein engineering and recombinant DNA technology, variants can be generated to improve or alter the characteristics of the polypeptides.
- one or more amino acids can be deleted from the N-terminus or C-terminus of the secreted protein without substantial loss of biological function.
- interferon gamma exhibited up to ten times higher activity after deleting 8-10 amino acid residues from the carboxy terminus of this protein. (Dobeli et al, J. Biotechnology 7: 199-216 (1988), incorporated herein by reference in its entirety.)
- polypeptide variants include, e.g., modified polypeptides.
- Modifications include, e.g., acetylation, acylation, ADP-ribosylation, amidation, covalent attachment of flavin, covalent attachment of a heme moiety, covalent attachment of a nucleotide or nucleotide derivative, covalent attachment of a lipid or lipid derivative, covalent attachment of phosphotidylinositol, cross-linking, cyclization, disulfide bond formation, demethylation, formation of covalent cross-links, formation of cysteine, formation of pyroglutamate, formylation, gamma-carboxylation, glycosylation, GPI anchor formation, hydroxylation, iodination, methylation, myristoylation, oxidation, pegylation (Mei et al., Blood 116:21Q-'19 (2010), which is incorporated herein by reference in its entirety
- Scaffold X and/or Scaffold Y is modified at any convenient location.
- the term "producer cell” refers to a cell used for generating an EV.
- a producer cell can be a cell cultured in vitro, or a cell in vivo.
- a producer cell includes, but not limited to, a cell known to be effective in generating EVs, e.g., exosomes, e.g., HEK293 cells, Chinese hamster ovary (CHO) cells, mesenchymal stem cells (MSCs), BJ human foreskin fibroblast cells, s9f cells, fHDF fibroblast cells, AGE.HN® neuronal precursor cells, CAP® amniocyte cells, adipose mesenchymal stem cells, and RPTEC/TERT1 cells.
- a producer cell is an antigen-presenting cell.
- the producer cell is a bacterial cell.
- a producer cell is a dendritic cell, a B cell, a mast cell, a macrophage, a neutrophil, a Kupffer-Browicz cell, or a cell derived from any of these cells, or any combination thereof.
- the producer cell is not a bacterial cell. In other aspects, the producer cell is not an antigen-presenting cell.
- the term "linked to” or “conjugated to” are used interchangeably and refer to a covalent or non-covalent bond formed between a first moiety and a second moiety, e.g., a STING agonist and an extracellular vesicle, respectively, e.g., a scaffold moiety expressed in or on the extracellular vesicle and a STING agonist, e.g., Scaffold X (e.g., a PTGFRN protein), respectively, on the luminal surface of or on the external surface of the extracellular vesicle.
- a first moiety and a second moiety e.g., a STING agonist and an extracellular vesicle, respectively, e.g., a scaffold moiety expressed in or on the extracellular vesicle and a STING agonist, e.g., Scaffold X (e.g., a PTGFRN protein), respectively, on the luminal surface of or on the external surface
- isolating or purifying is the process of removing, partially removing (e.g., a fraction) of the EVs from a sample containing producer cells.
- an isolated EV composition has no detectable undesired activity or, alternatively, the level or amount of the undesired activity is at or below an acceptable level or amount. In some aspects, an isolated EV composition has an amount and/or concentration of desired EVs at or above an acceptable amount and/or concentration. In some aspects, the isolated EV composition is enriched as compared to the starting material (e.g., producer cell preparations) from which the composition is obtained. This enrichment can be by 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 96%, 97%, 98%, 99%, 99.9%, 99.99%, 99.999%, 99.9999%, or greater than 99.9999% as compared to the starting material.
- the starting material e.g., producer cell preparations
- isolated EV preparations are substantially free of residual biological products.
- the isolated EV preparations are 100% free, 99% free, 98% free, 97% free, 96% free, 95% free, 94% free, 93% free, 92% free, 91% free, or 90% free of any contaminating biological matter.
- Residual biological products can include abiotic materials (including chemicals) or unwanted nucleic acids, proteins, lipids, or metabolites.
- Substantially free of residual biological products can also mean that the EV composition contains no detectable producer cells and that only EVs are detectable.
- agonist refers to a molecule that binds to a receptor and activates the receptor to produce a biological response.
- Receptors can be activated by either an endogenous or an exogenous agonist.
- endogenous agonist include hormones, neurotransmitters, and cyclic dinucleotides.
- exogenous agonist include drugs, small molecules, and cyclic dinucleotides.
- the agonist can be a full, partial, or inverse agonist.
- antagonist refers to a molecule that blocks or dampens an agonist mediated response rather than provoking a biological response itself upon bind to a receptor.
- Many antagonists achieve their potency by competing with endogenous ligands or substrates at structurally defined binding sites on the receptors.
- Non-limiting examples of antagonists include alpha blockers, beta-blocker, and calcium channel blockers.
- the antagonist can be a competitive, non-competitive, or uncompetitive antagonist.
- free STING agonist means a STING agonist not associated with an extracellular vesicle, but otherwise identical to the STING agonist associated with the extracellular vesicle. Especially when compared to an extracellular vesicle associated with a STING agonist, the free STING agonist is the same STING agonist associated with the extracellular vesicle.
- the amount of the free STING agonist compared to the STING agonist associated with the extracellular vesicle is the same as the amount of the STING agonist associated with the EV.
- the term “ligand” refers to a molecule that binds to a receptor and modulates the receptor to produce a biological response. Modulation can be activation, deactivation, blocking, or damping of the biological response mediated by the receptor.
- Receptors can be modulated by either an endogenous or an exogenous ligand.
- Non-limiting examples of endogenous ligands include antibodies and peptides.
- Non-limiting examples of exogenous agonist include drugs, small molecules, and cyclic dinucleotides.
- the ligand can be a full, partial, or inverse ligand.
- antibody encompasses an immunoglobulin whether natural or partly or wholly synthetically produced, and fragments thereof. The term also covers any protein having a binding domain that is homologous to an immunoglobulin binding domain. “Antibody” further includes a polypeptide comprising a framework region from an immunoglobulin gene or fragments thereof that specifically binds and recognizes an antigen.
- antibody is meant to include whole antibodies, polyclonal, monoclonal and recombinant antibodies, fragments thereof, and further includes single-chain antibodies, humanized antibodies, murine antibodies, chimeric, mouse-human, mouse-primate, primate-human monoclonal antibodies, anti-idiotype antibodies, antibody fragments, such as, e.g., scFv, (scFv)2, Fab, Fab', and F(ab')2, F(abl)2, Fv, dAb, and Fd fragments, diabodies, and antibody-related polypeptides.
- Antibody includes bispecific antibodies and multispecific antibodies so long as they exhibit the desired biological activity or function.
- terapéuticaally effective amount is the amount of reagent or pharmaceutical compound that is sufficient to a produce a desired therapeutic effect, pharmacologic and/or physiologic effect on a subject in need thereof.
- a therapeutically effective amount can be a “prophylactically effective amount” as prophylaxis can be considered therapy.
- the term “pharmaceutical composition” refers to one or more of the compounds described herein, such as, e.g., an EV mixed or intermingled with, or suspended in one or more other chemical components, such as pharmaceutically-acceptable carriers and excipients.
- a pharmaceutical composition is to facilitate administration of preparations of EVs to a subject.
- excipient or “carrier” refers to an inert substance added to a pharmaceutical composition to further facilitate administration of a compound.
- pharmaceutically-acceptable carrier or “pharmaceutically-acceptable excipient” and grammatical variations thereof, encompasses any of the agents approved by a regulatory agency of the US Federal government or listed in the US Pharmacopeia for use in animals, including humans, as well as any carrier or diluent that does not cause the production of undesirable physiological effects to a degree that prohibits administration of the composition to a subject and does not abrogate the biological activity and properties of the administered compound. Included are excipients and carriers that are useful in preparing a pharmaceutical composition and are generally safe, non-toxic, and desirable.
- administration refers to introducing a composition, such as an EV, or agent into a subject and includes concurrent and sequential introduction of a composition or agent.
- the introduction of a composition or agent into a subject is by any suitable route, including intratumorally, orally (e.g., sublingually), pulmonarily, intranasally, parenterally (intravenously, intra-arterially, intramuscularly, intraperitoneally, or subcutaneously), rectally, intralymphatically, intrathecally, periocularly, topically, intradermally, or transdermally.
- Administration includes self-administration and the administration by another.
- a suitable route of administration allows the composition or the agent to perform its intended function. For example, if a suitable route is intravenous, the composition is administered by introducing the composition or agent into a vein of the subject.
- the administering comprises the use of a device, e.g., a pump or a patch.
- treat refers to, e.g., the reduction in severity of a disease or condition; the reduction in the duration of a disease course; the amelioration or elimination of one or more symptoms associated with a disease or condition; the provision of beneficial effects to a subject with a disease or condition, without necessarily curing the disease or condition.
- the term also include prophylaxis or prevention of a disease or condition or its symptoms thereof.
- treating or “treatment” means inducing an immune response in a subject against an antigen.
- prevent refers to decreasing or reducing the occurrence or severity of a particular outcome. In some aspects, preventing an outcome is achieved through prophylactic treatment.
- the term “modulate,” “modulating”, “modify,” and/or “modulator” generally refers to the ability to alter, by increase or decrease, e.g., directly or indirectly promoting/stimulating/up-regulating or interfering with/inhibiting/down-regulating a specific concentration, level, expression, function or behavior, such as, e.g., to act as an antagonist or agonist.
- a modulator can increase and/or decrease a certain concentration, level, activity or function relative to a control, or relative to the average level of activity that would generally be expected or relative to a control level of activity.
- a mammalian subject includes all mammals, including without limitation, humans, domestic animals (e.g., dogs, cats and the like), farm animals (e.g., cows, sheep, pigs, horses and the like) and laboratory animals (e.g., monkey, rats, mice, rabbits, guinea pigs and the like).
- domestic animals e.g., dogs, cats and the like
- farm animals e.g., cows, sheep, pigs, horses and the like
- laboratory animals e.g., monkey, rats, mice, rabbits, guinea pigs and the like.
- the terms “individual,” “subject,” “host,” and “patient,” are used interchangeably herein and refer to any mammalian subject for whom diagnosis, treatment, or therapy is desired, particularly humans.
- the methods described herein are applicable to both human therapy and veterinary applications.
- the subject is a mammal, and in other aspects the subject is a human.
- the term “substantially free” means that the sample comprising EVs comprise less than 10% of macromolecules by mass/volume (m/v) percentage concentration. Some fractions may contain less than 0.001%, less than 0.01%, less than 0.05%, less than 0.1%, less than 0.2%, less than 0.3 %, less than 0.4%, less than 0.5%, less than 0.6%, less than 0.7%, less than 0.8%, less than 0.9%, less than 1%, less than 2%, less than 3%, less than 4%, less than 5%, less than 6%, less than 7%, less than 8%, less than 9%, or less than 10% (m/v) of macromolecules.
- macromolecule means nucleic acids, exogenous proteins, lipids, carbohydrates, metabolites, or a combination thereof.
- the term “insubstantial,” “reduced,” or “negligible” refers to the presence, level, or amount of an inflammation response in a subject after administration of the composition comprising EVs comprising a cholesterol moiety linked to a STING agonist via a cleavable peptide linker, e.g., Valine- Alanine linker or Valine-Citrulline linker, relative to the baseline inflammation response in the subject or compared to the subject inflammation response to the administration of a free STING agonist.
- a cleavable peptide linker e.g., Valine- Alanine linker or Valine-Citrulline linker
- a negligible or insubstantial presence, level or amount of systemic inflammation may be less than 0.001%, less than 0.01%, less than 0.1%, less than 0.2%, less than 0.3 %, less than 0.4%, less than 0.5%, less than 0.6%, less than 0.7%, less than 0.8%, less than 0.9%, less than 1%, less than 2%, less than 3%, less than 4%, less than 5%, less than 6%, less than 7%, less than 8%, less than 9%, less than 10%, less than 12%, less than 15%, less than 17%, less than 20%, or less than 25% of systemic inflammation as relative to the baseline inflammation in the subject or compared to the subject immune response to the administration of a free STING agonist.
- a level or amount of a systemic inflammation may be less than 0.1-fold, less than 0.5-fold, less than 0.5-fold, less than 1-fold, less than 1.5-fold, less than 2- fold relative to the baseline or compared to the inflammation response to the administration of a free STING agonist.
- Ranges recited herein are understood to be shorthand for all of the values within the range, inclusive of the recited endpoints.
- a range of 1 to 50 is understood to include any number, combination of numbers, or sub-range from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, and 50.
- Present disclosure comprises EVs comprising a cholesterol moiety linked to a STING agonist via a cleavable peptide linker e.g., Valine- Alanine or Valine-Citrulline (“cholesterol tagged STING agonist”).
- a cleavable peptide linker e.g., Valine- Alanine or Valine-Citrulline (“cholesterol tagged STING agonist”).
- the cholesterol moiety is embedded into the surface of the EVs.
- the cholesterol moiety comprises a cholesterol or a derivative thereof.
- the cholesterol moiety comprises:
- the EV is an exosome, a nanovesicle, an apoptotic body, a microvesicle, a lysosome, an endosome, a liposome, a lipid nanoparticle, a micelle, a multilamellar structure, a revesiculated vesicle, or an extruded cell.
- the EV comprises an exosome.
- the EV overexpresses a PTGFRN protein. In other aspects, the EV is produced by a cell that overexpresses a PTGFRN protein.
- the extracellular vesicle further comprises a therapeutic agent, e.g., a ligand, a cytokine, an antigen, or an antibody.
- a therapeutic agent e.g., a ligand, a cytokine, an antigen, or an antibody.
- the antibody comprises an antagonistic antibody and/or an agonistic antibody.
- the STING agonist is a cyclic dinucleotide. In some aspects, the STING agonist is a non-cyclic dinucleotide.
- Cyclic dinucleotides were first identified as bacterial signaling molecules characterized by two 3’, 5’ phosphodiester bonds, such as in the molecule c-di-GMP. While STING can be activated by bacterial CDNs, the innate immune response in mammalian cells is also mediated by the CDN signaling molecule cGAMP which is produced by cGAS. cGAMP is characterized by a mixed 2’, 5’ and 3’, 5’ phosphodiester linkage. Both bacterial and mammalian CDNs directly interact with STING to induce the pro-inflammatory signaling cascade that results in the production of type I IFNs, such as IFNa and IFN-p.
- the linker is conjugated into the phosphate backbone of a STING agonist, e.g., a cyclic dinucleotide (CDN). In some aspects, the linker is conjugated to the base of a CDN (STING agonist).
- STING agonist e.g., a cyclic dinucleotide
- the EV comprises:
- the EV is thermostable at about 20°C for at least about 7 days, at least about 8 days, at least about 9 days, at least about 10 days, at least about 11 days, at least about 12 days, at least about 13 days, at least about 14 days, at least about 15 days, at least about 16 days, at least about 17 days, at least about 18 days, at least about 19 days, at least about 20 days, at least about 21 days, at least about 22 days, at least about 23 days, at least about 24 days, at least about 25 days, at least about 26 days, at least about 27 days, at least about 28 days, at least about 29 days, at least about 30 days, at least about 35 days, at least about 40 days, at least about 45 days, at least about 50 days, at least about 55 days, at least about 60 days, at least about 65 days, at least about 70 days, at least about 75 days, at least about 80 days, at least about 85 days, at least about 90 days, at least about 95 days, or at least about 100 days.
- the EV is thermostable at about 20°C for at least about 30 days. In some aspects, the EV is thermostable at about 37°C for at least about 7 days, at least about 8 days, at least about 9 days, at least about 10 days, at least about 11 days, at least about 12 days, at least about 13 days, at least about 14 days, at least about 15 days, at least about 16 days, at least about 17 days, at least about 18 days, at least about 19 days, at least about 20 days, at least about 21 days, at least about 22 days, at least about 23 days, at least about 24 days, at least about 25 days, at least about 26 days, at least about 27 days, at least about 28 days, at least about 29 days, at least about 30 days, at least about 35 days, at least about 40 days, at least about 45 days, at least about 50 days, at least about 55 days, at least about 60 days, at least about 65 days, at least about 70 days, at least about 75 days, at least about 80 days, at least about 85 days, at least about 90 days
- STING agonists used in this disclosure can be cyclic dinucleotides (CDNs) or non- cyclic dinucleotide agonists.
- Cyclic purine dinucleotides such as, but not limited to, cGMP, cyclic di-GMP (c-di-GMP), cAMP, cyclic di-AMP (c-di-AMP), cyclic-GMP-AMP (cGAMP), cyclic di-
- IMP c-di-IMP
- cAIMP cyclic AMP-IMP
- any analogue thereof are known to stimulate or enhance an immune or inflammation response in a patient.
- the CDNs may have 2’2’, 2’3’, 2’5’, 3’3’, or 3’5’ bonds linking the cyclic dinucleotides, or any combination thereof.
- Cyclic purine dinucleotides may be modified via standard organic chemistry techniques to produce analogues of purine dinucleotides.
- Suitable purine dinucleotides include, but are not limited to, adenine, guanine, inosine, hypoxanthine, xanthine, isoguanine, or any other appropriate purine dinucleotide known in the art.
- the cyclic dinucleotides may be modified analogues. Any suitable modification known in the art may be used, including, but not limited to, phosphorothioate, biphosphorothioate, fluorinate, and difluorinate modifications.
- Non cyclic dinucleotide agonists may also be used, such as 5,6-Dimethylxanthenone- 4-acetic acid (DMXAA), or any other non-cyclic dinucleotide agonist known in the art.
- DMXAA 5,6-Dimethylxanthenone- 4-acetic acid
- any STING agonist may be used.
- the STING agonists are DMXAA, STING agonist-1, ML RR-S2 CDA, ML RR-S2c-di-GMP, ML-RR-S2 cGAMP, 2’3’-c-di-AM(PS)2, 2’3’-cGAMP, 2’3’-cGAMPdFHS, 3'3'-cGAMP, 3'3'-cGAMPdFSH, cAIMP, cAIM(PS)2, 3’3’-cAIMP, 3’3’-cAIMPdFSH, 2’2’-cGAMP, 2’3’-cGAM(PS)2, 3'3'-cGAMP, c-di- AMP, 2'3 '-c-di-AMP, 2’3’-c-di-AM(PS)2, c-di-GMP, 2’3’-c-di-GMP
- the STING agonist useful for the present disclosure comprises a compound having the following formula:
- Xi is H, OH, or F
- X 2 is H, OH, or F
- Z is OH, ORi, SH or SRi, wherein: i) Ri is Na or NH4, or ii) Ri is an enzyme-labile group which provides OH or SH in vivo such as pivaloyloxymethyl;
- Bi and B2 are bases chosen from:
- the STING agonist useful for the present disclosure comprises:
- CL-659 c-[2FdAMP(S)-2FdIMP(S)](POM) 2 and a pharmaceutically acceptable salt thereof. See WO 2016/096174A1.
- the STING agonist useful for the present disclosure comprises a compound having the following formula: or any pharmaceutically acceptable salts thereof.
- the STING agonist useful for the present disclosure comprises a compound disclosed in WO 2014/093936, a compound in WO 2014/189805, a compound in WO 2015/077354, c-di-AMP, c-di-GMP, c-di-IMP, c-AMP-GMP, c-AMP-IMP, and c-GMP-IMP, described in WO 2013/185052 and/or Sci. Transl. Med.
- the STING agonist useful for the present disclosure is CL606, CL611, CL602, CL655, CL604, CL609, CL614, CL656, CL647, CL626, CL629, CL603, CL632, CL633, CL659, or a pharmaceutically acceptable salt thereof.
- the STING agonist useful for the present disclosure is CL606 or a pharmaceutically acceptable salt thereof.
- the STING agonist useful for the present disclosure is CL611 or a pharmaceutically acceptable salt thereof.
- the STING agonist useful for the present disclosure is CL602 or a pharmaceutically acceptable salt thereof.
- the STING agonist useful for the present disclosure is CL655 or a pharmaceutically acceptable salt thereof.
- the STING agonist useful for the present disclosure is CL604 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL609 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL614 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL656 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL647 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL626 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL629 or a pharmaceutically acceptable salt thereof.
- the STING agonist useful for the present disclosure is CL603 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL632 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL633 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL659 or a pharmaceutically acceptable salt thereof.
- the EV e.g., exosome
- the EV comprises a cyclic dinucleotide STING agonist and/or a non-cyclic dinucleotide STING agonist.
- STING agonists can be the same or they can be different.
- non-cyclic dinucleotide STING agonists can be the same or they can be different.
- an EV, e.g., exosome, composition of the present disclosure can comprise two or more populations of EVs, e.g., exosomes, wherein each population of EVs, e.g., exosomes, comprises a different STING agonist or combination thereof.
- the STING agonists can also be modified to increase their association (e.g., linkage) to cholesterol on the surface of an extracellular vesicle or EV (e.g., either unbound in the lumen).
- the modification allows better expression of the STING agonist on the exterior surface of the EV, e.g., exosome, (e.g., linked to a scaffold moiety disclosed herein, e.g., Scaffold X).
- This modification can include a chemical or enzyme, or by physically or chemically altering the polarity or charge of the STING agonist.
- the STING agonist may be modified by a single treatment, or by a combination of treatments, e.g., adding a lipid binding tag only, or adding a lipid binding tag and altering the polarity.
- the previous example is meant to be a non-limiting illustrative instance. It is contemplated that any combination of modifications may be practiced.
- the modification may increase linkage of the agonist to the cholesterol moiety on the surface of the EV by between 2-fold and 10,000 fold, between 10-fold and 1,000 fold, or between 100-fold and 500- fold compared to an unmodified agonist.
- the modification may increase linkage of the agonist to the cholesterol moiety in the EV by at least 2-fold, 5-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50- fold, 60-fold, 70-fold, 80-fold, 90-fold, 100-fold, 200-fold, 300-fold, 400-fold, 500-fold, 600-fold, 700-fold, 800-fold, 900-fold, 1000-fold, 2000-fold, 3000-fold, 4000-fold, 5000-fold, 6000-fold, 7000-fold, 8000-fold, 9000-fold, or 10,000-fold compared to an unmodified agonist.
- STING agonists can be modified to allow for better expression of the agonists on the exterior surface of the EV, e.g., exosome, (e.g., linked to a cholesterol moiety disclosed herein). Any of the modifications described above can be used.
- the modification may increase linkage of the agonist to the cholesterol moiety in the EV, e.g., exosome, by about between 2-fold and 10,000 fold, about between 10-fold and 1,000 fold, or about between 100-fold and 500- fold compared to an unmodified agonist.
- the modification can increase expression of the agonist on the exterior surface of the EV, e.g., exosome, by at least about 2-fold, 5-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50-fold, 60-fold, 70-fold, 80-fold, 90-fold, 100-fold, 200-fold, 300-fold, 400-fold, 500-fold, 600-fold, 700-fold, 800-fold, 900-fold, 1000-fold, 2000-fold, 3000-fold, 4000-fold, 5000-fold, 6000-fold, 7000-fold, 8000-fold, 9000-fold, or 10,000-fold compared to expression of an unmodified agonist.
- EVs of the present disclosure comprise a membrane modified in its composition.
- their membrane compositions can be modified by changing the protein, lipid, or glycan content of the membrane.
- the surface-engineered EVs are generated by chemical and/or physical methods, such as PEG-induced fusion and/or ultrasonic fusion.
- the surface- engineered EVs, e.g., exosomes are generated by genetic engineering. EVs produced from a genetically-modified producer cell or a progeny of the genetically-modified cell can contain modified membrane compositions.
- surface-engineered EVs, e.g., exosomes have one or more exosome proteins at a higher or lower density (e.g., higher number) or include a variant or a fragment of the exosome proteins.
- surface-engineered EVs can be produced from a cell e.g., HEK293 cells) transformed with an exogenous sequence encoding one or more exosome proteins, e.g., PTGFRN protein) or a variant or a fragment thereof.
- EVs including one or more exosome proteins expressed from the exogenous sequence can include modified membrane compositions.
- exosome proteins modified to have enhanced affinity to a binding agent can be used for generating surface-engineered EVs that can be purified using the binding agent.
- Exosome proteins modified to be more effectively targeted to EVs, e.g., exosomes, and/or membranes can be used.
- Exosome proteins modified to comprise a minimal fragment required for specific and effective targeting to EVs, e.g., exosomes, membranes can be also used.
- the surface-engineered EVs described herein demonstrate superior characteristics compared to EVs, e.g., exosomes, known in the art.
- surface-engineered EVs contain modified proteins more highly enriched on their surface than naturally occurring EVs, e.g., exosomes, or the EVs, e.g., exosomes, produced using conventional exosome proteins.
- the surface-engineered EVs of the present invention can have greater, more specific, or more controlled biological activity compared to naturally occurring EVs, e.g., exosomes, or the EVs, e.g., exosomes, produced using conventional exosome proteins.
- exosome proteins useful for the present disclosure comprise Prostaglandin F2 receptor negative regulator (the PTGFRN polypeptide).
- the PTGFRN protein can be also referred to as CD9 partner 1 (CD9P-1), Glu-Trp-Ile EWI motif-containing protein F (EWI-F), Prostaglandin F2-alpha receptor regulatory protein, Prostaglandin F2-alpha receptor- associated protein, or CD315.
- CD9P-1 CD9 partner 1
- EWI-F Glu-Trp-Ile EWI motif-containing protein F
- Prostaglandin F2-alpha receptor regulatory protein Prostaglandin F2-alpha receptor- associated protein
- CD315 The full length amino acid sequence of the human PTGFRN protein (Uniprot Accession No. Q9P2B2) is shown at Table 1 as SEQ ID NO: 1.
- the PTGFRN polypeptide contains a signal peptide (amino acids 1 to 25 of SEQ ID NO: 1), the extracellular domain (amino acids 26 to 832 of SEQ ID NO: 1), a transmembrane domain (amino acids 833 to 853 of SEQ ID NO: 1), and a cytoplasmic domain (amino acids 854 to 879 of SEQ ID NO: 1).
- the mature PTGFRN polypeptide consists of SEQ ID NO: 1 without the signal peptide, i.e., amino acids 26 to 879 of SEQ ID NO: 1.
- a PTGFRN polypeptide fragment useful for the present disclosure comprises a transmembrane domain of the PTGFRN polypeptide.
- a PTGFRN polypeptide fragment useful for the present disclosure comprises the transmembrane domain of the PTGFRN polypeptide and (i) at least five, at least 10, at least 15, at least 20, at least 25, at least 30, at least 40, at least 50, at least 70, at least 80, at least 90, at least 100, at least 110, at least 120, at least 130, at least 140, at least 150 amino acids at the N terminus of the transmembrane domain, (ii) at least five, at least 10, at least 15, at least 20, or at least 25 amino acids at the C terminus of the transmembrane domain, or both (i) and (ii).
- the fragments of PTGFRN polypeptide lack one or more functional or structural domains, such as IgV.
- the PTGFRN polypeptide comprises an amino acid sequence at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or about 100% identical to amino acids 26 to 879 of SEQ ID NO: 1.
- the PTGFRN polypeptide comprises an amino acid sequence at least about at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or about 100% identical to SEQ ID NO: 33.
- the PTGFRN polypeptide comprises the amino acid sequence of SEQ ID NO: 33, except one amino acid mutation, two amino acid mutations, three amino acid mutations, four amino acid mutations, five amino acid mutations, six amino acid mutations, or seven amino acid mutations.
- the mutations can be a substitution, an insertion, a deletion, or any combination thereof.
- the PTGFRN polypeptide comprises the amino acid sequence of SEQ ID NO: 33 and 1 amino acid, two amino acids, three amino acids, four amino acids, five amino acids, six amino acids, seven amino acids, eight amino acids, nine amino acids, ten amino acids, 11 amino acids, 12 amino acids, 13 amino acids, 14 amino acids, 15 amino acids, 16 amino acids, 17 amino acids, 18 amino acids, 19 amino acids, or 20 amino acids or longer at the N terminus and/or C terminus of SEQ ID NO: 33.
- the PTGFRN polypeptide comprises an amino acid sequence at least about at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or about 100% identical to SEQ ID NO: 2, 3, 4, 5, 6, or 7.
- the PTGFRN polypeptide comprises the amino acid sequence of SEQ ID NO: 2, 3, 4, 5, 6, or 7, except one amino acid mutation, two amino acid mutations, three amino acid mutations, four amino acid mutations, five amino acid mutations, six amino acid mutations, or seven amino acid mutations.
- the PTGFRN polypeptide comprises the amino acid sequence of SEQ ID NO: 2, 3, 4, 5, 6, or 7 and 1 amino acid, two amino acids, three amino acids, four amino acids, five amino acids, six amino acids, seven amino acids, eight amino acids, nine amino acids, ten amino acids, 11 amino acids, 12 amino acids, 13 amino acids, 14 amino acids, 15 amino acids, 16 amino acids, 17 amino acids, 18 amino acids, 19 amino acids, or 20 amino acids or longer at the N terminus and/or C terminus of SEQ ID NO: 2, 3, 4, 5, 6, or 7.
- Non-limiting examples of other Scaffold X proteins that can be used to link a STING agonist to the surface ofEVs, e.g., exosomes, can be found at US Patent No. 10,195,290 Bl, issued Feb. 5, 2019, which is incorporated by reference in its entirety.
- a Scaffold X protein useful for the present disclosure lacks at least about 5, 10, 50, 100, 200, 300, 400, 500, 600, 700, or 800 amino acids from the N-terminus of the native protein. In some aspects, a Scaffold X lacks at least about 5, 10, 50, 100, 200, 300, 400, 500, 600, 700, or 800 amino acids from the C-terminus of the native protein. In some aspects, a Scaffold X lacks at least about 5, 10, 50, 100, 200, 300, 400, 500, 600, 700, or 800 amino acids from both the N-terminus and C-terminus of the native protein. In some aspects, a Scaffold X lacks one or more functional or structural domains of the native protein.
- Scaffold X described herein can also be used to link an additional payload on the luminal surface and/or on the exterior surface of the EVs, e.g., exosomes, at the same time.
- the PTGFRN polypeptide can be used to link a payload inside the lumen in addition to the surface of the EV, e.g., exosome.
- EVs, e.g., exosomes, of the present disclosure comprise an internal space (i.e., lumen) that is different from that of the naturally occurring EVs, e.g., exosomes.
- the EV, e.g., exosome can be changed such that the composition in the luminal side of the EV, e.g., exosome, has the protein, lipid, or glycan content different from that of the naturally- occurring EVs, e.g., exosomes.
- engineered EVs e.g., exosomes
- a scaffold moiety e.g., exosome proteins, e.g., Scaffold Y
- modification or a fragment of the scaffold moiety that changes the composition or content of the luminal side of the EV, e.g., exosome.
- modifications or fragments of the exosome protein that can be expressed in the luminal side of the EV, e.g., exosome can be used for the aspects of the present disclosure.
- EVs disclosed herein comprise an additional payload (e.g., an antigen) in the lumen of the EV, e.g., encapsulated).
- a payload e.g., antigen
- a payload is linked to the luminal surface of the EV, e.g., exosome.
- a molecule e.g., antigen or adjuvant
- exosome e.g., associated
- a payload is expressed on the luminal surface of the EV, e.g., exosome as a fusion molecule, e.g., fusion molecule of a payload to a scaffold moiety (e.g., Scaffold Y).
- Scaffold Y comprises the MARCKS protein, MARCKSL1 protein, BASP1 protein, or any combination thereof.
- the EVs, e.g., exosomes, of the present disclosure comprise a cholesterol tagged STING agonist, e.g., cholesterol linked to a STING agonist via a cleavable peptide linker, e.g., Valine-Alanine linker, Valine-Citrulline linker, and a payload linked to a Scaffold Y.
- a cholesterol tagged STING agonist e.g., cholesterol linked to a STING agonist via a cleavable peptide linker, e.g., Valine-Alanine linker, Valine-Citrulline linker, and a payload linked to a Scaffold Y.
- the EVs, e.g., exosomes, of the present disclosure comprise cholesterol linked to a STING agonist via a cleavable peptide linker, e.g., Valine-Alanine linker, Valine-Citrulline linker, and a payload linked to a Scaffold X.
- a cleavable peptide linker e.g., Valine-Alanine linker, Valine-Citrulline linker, and a payload linked to a Scaffold X.
- scaffold moi eties that can change the luminal side of the EVs, e.g., exosomes
- Scaffold Y comprises Brain Acid Soluble Protein 1 (the BASP1 protein).
- the BASP1 protein is also known as 22 kDa neuronal tissue-enriched acidic protein or neuronal axonal membrane protein NAP-22.
- the full-length human BASP1 protein sequence is shown in Table 2. An isomer produced by an alternative splicing is missing amino acids 88 to 141 from SEQ ID NO: XX (isomer 1).
- the mature BASP1 protein sequence is missing the first Met from SEQ ID NO: 49 and thus contains amino acids 2 to 227 of SEQ ID NO: 49.
- a Scaffold Y that can be used to express a payload in the luminal surface of an EV, e.g., exosome, comprises an amino acid sequence at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or about 100% identical to GGKLSKK (SEQ ID NO: XX).
- a Scaffold Y that can be used to express a payload in the luminal surface of an EV, e.g., exosome, comprises an amino acid sequence at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or about 100% identical to the amino acid sequence as set forth in i) GGKLSKKK (SEQ ID NO: XX), (ii) GGKLSKKS (SEQ ID NO: XX), (iii) GAKLSKKK (SEQ ID NO: XX), (iv) GAKLSKKS (SEQ ID NO: XX), (v) GGKQSKKK (SEQ ID NO: XX), (vi) GGKQSKKS (SEQ ID NO: XX), (vii) GGKLAKKK (SEQ ID NO: XX)
- a Scaffold Y comprises a full-length BASP1 protein.
- the EVs of the present disclosure can comprises one or more linkers that link the payload to EVs or to a scaffold moiety, e.g., Scaffold X on the exterior surface of the EVs.
- the payload is linked to the EVs directly or in a scaffold moiety on the EVs by a linker.
- the linker can be any chemical moiety known in the art.
- linker refers to a peptide or polypeptide sequence (e.g., a synthetic peptide or polypeptide sequence) or to a non-polypeptide.
- two or more linkers can be linked in tandem.
- linkers provide flexibility or prevent/ameliorate steric hindrances. Linkers are not typically cleaved; however in certain aspects, such cleavage can be desirable.
- a linker can comprise one or more protease-cleavable sites, which can be located within the sequence of the linker or flanking the linker at either end of the linker sequence.
- the linker is a peptide linker.
- the peptide linker can comprise at least about two, at least about three, at least about four, at least about five, at least about 10, at least about 15, at least about 20, at least about 25, at least about 30, at least about 35, at least about 40, at least about 45, at least about 50, at least about 55, at least about 60, at least about 65, at least about 70, at least about 75, at least about 80, at least about 85, at least about 90, at least about 95, or at least about 100 amino acids.
- the peptide linker is synthetic, i.e., non-naturally occurring.
- a peptide linker includes peptides (or polypeptides) (e.g., natural or non-naturally occurring peptides) which comprise an amino acid sequence that links or genetically fuses a first linear sequence of amino acids to a second linear sequence of amino acids to which it is not naturally linked or genetically fused in nature.
- the peptide linker can comprise non-naturally occurring polypeptides which are modified forms of naturally occurring polypeptides (e.g., comprising a mutation such as an addition, substitution or deletion).
- Linkers may be susceptible to cleavage ("cleavable linker") thereby facilitating release of the payloads.
- the linker is a "reduction-sensitive linker.”
- the reduction-sensitive linker contains a disulfide bond.
- the linker is an "acid labile linker.”
- the acid labile linker contains hydrazone.
- Suitable acid labile linkers also include, for example, a cis-aconitic linker, a hydrazide linker, a thiocarbamoyl linker, or any combination thereof.
- the linker comprises a non-cleavable liker.
- EVs can be produced from a cell grown in vitro or a body fluid of a subject.
- various producer cells e.g., HEK293 cells
- Additional cell types that can be used for the production of the lumen-engineered EVs, e.g., exosomes, described herein include, without limitation, mesenchymal stem cells, T-cells, B-cells, dendritic cells, macrophages, and cancer cell lines.
- a producer cell is not a dendritic cell, macrophage, B cell, mast cell, neutrophil, Kupffer-Browicz cell, cell derived from any of these cells, or any combination thereof.
- Some aspects may also include genetically modifying the EV, e.g., exosome, to comprise one or more exogenous sequences to produce modified EVs that express exogenous proteins on the vesicle surface.
- the exogenous sequences can comprise a sequence encoding the EV, e.g., exosome, protein or a modification or a fragment of the EV protein.
- An extra copy of the sequence encoding the EV, e.g., exosome, protein can be introduced to produce a surface- engineered EV having a higher density of the EV protein.
- An exogenous sequence encoding a modification or a fragment of the EV, e.g., exosome, protein can be introduced to produce a modified EV containing the modification or the fragment of the EV protein.
- An exogenous sequence encoding an affinity tag can be introduced to produce a modified EV, e.g., exosome, containing a fusion protein comprising the affinity tag attached to the EV protein.
- the exogenous sequence encodes for Scaffold X (e.g., a PTGFRN protein, a BSG protein, an IGSF2 protein, an IGSF3 protein, an IGSF8 protein, an ITGB1 protein, an ITGA4 protein, a SLC3A2 protein, an ATP transporter protein, or a fragment or a variant thereof).
- Scaffold X e.g., a PTGFRN protein, a BSG protein, an IGSF2 protein, an IGSF3 protein, an IGSF8 protein, an ITGB1 protein, an ITGA4 protein, a SLC3A2 protein, an ATP transporter protein, or a fragment or a variant thereof.
- the modified EV overexpresses Scaffold X (e.g., a PTGFRN protein, a BSG protein, an IGSF2 protein, an IGSF3 protein, an IGSF8 protein, an ITGB 1 protein, an ITGA4 protein, a SLC3A2 protein, an ATP transporter protein, or a fragment or a variant thereof).
- Scaffold X e.g., a PTGFRN protein, a BSG protein, an IGSF2 protein, an IGSF3 protein, an IGSF8 protein, an ITGB 1 protein, an ITGA4 protein, a SLC3A2 protein, an ATP transporter protein, or a fragment or a variant thereof.
- the EV e.g., exosome
- the EV is produced by a cell that overexpresses Scaffold X (e.g.
- a PTGFRN protein a BSG protein, an IGSF2 protein, an IGSF3 protein, an IGSF8 protein, an ITGB1 protein, an ITGA4 protein, a SLC3A2 protein, an ATP transporter protein, or a fragment or a variant thereof).
- the exogenous sequence encodes for Scaffold Y (e.g., the MARCKS protein, MARCKSL1 protein, BASP1 protein, or a fragment or variant thereof).
- the modified EV, e.g., exosome overexpresses Scaffold Y (e.g., the MARCKS protein, MARCKSL1 protein, BASP1 protein, or a fragment or variant thereof).
- the EV, e.g., exosome is produced by a cell that overexpresses Scaffold Y (e.g., the MARCKS protein, MARCKSL1 protein, BASP1 protein, or a fragment or variant thereof).
- the exogenous sequence may be transiently or stably expressed in the producer cell or cell line via transfection, transformation, transduction, electroporation, or any other appropriate method of gene delivery or combination thereof known in the art.
- the exogenous sequence may be integrated into the producer cell genome, or remain extra chromosomal.
- the exogenous sequence can be transformed as a plasmid.
- the exogenous sequences can be stably integrated into a genomic sequence of the producer cell, at a targeted site or in a random site.
- the exogenous sequences can be inserted into a genomic sequence of the producer cell, located within, upstream (5’-end) or downstream (3’-end) of an endogenous sequence encoding the EV, e.g., exosome, protein.
- cells modified using various gene editing methods e.g., methods using a homologous recombination, transposon-mediated system, loxP-Cre system, CRISPR/Cas9 CRISPR/Cfpl, CRISPR/C2cl, C2c2, or C2c3, CRISPR/CasY or CasX, TAL- effector nuclease or TALEN, or zinc finger nuclease (ZFN) systems
- gene editing methods e.g., methods using a homologous recombination, transposon-mediated system, loxP-Cre system, CRISPR/Cas9 CRISPR/Cfpl, CRISPR/C2cl, C2c2, or C2c3, CRISPR/CasY or CasX, TAL- effector nuclease or TALEN, or zinc finger nuclease (ZFN) systems
- ZFN zinc finger nuclease
- the producer cell is further modified to comprise an additional exogenous sequence.
- an additional exogenous sequence can be included to modulate endogenous gene expression, modulate the immune response or immune signaling, or produce an EV, e.g., exosome, including a certain polypeptide as a payload or additional surface expressed ligand.
- the producer cell can be further modified to comprise an additional exogenous sequence conferring additional functionalities to EVs, e.g., exosomes, for example, specific targeting capabilities, delivery functions, enzymatic functions, increased or decreased halflife in vivo, etc.
- the producer cell is modified to comprise two exogenous sequences, one encoding the exosome protein or a modification or a fragment of the exosome protein, and the other encoding a protein conferring the additional functionalities to exosomes.
- the EV, e.g., exosome, of the present can be produced from a cell transformed with a sequence encoding one or more additional exogenous proteins including, but not limited to ligands, cytokines, or antibodies, or any combination thereof. These additional exogenous proteins may enable activation or modulation of additional immune stimulatory signals in combination with the STING agonist.
- the EV, e.g., exosome is further modified with a ligand comprising CD40L, OX40L, or CD27L.
- the EV, e.g., exosome is further modified with a cytokine comprising IL-7, IL-12, or IL-15.
- Any of the one or more exosome proteins described herein can be expressed from a plasmid, an exogenous sequence inserted into the genome or other exogenous nucleic acid such as a synthetic messenger RNA (mRNA).
- mRNA synthetic messenger RNA
- the EV e.g., exosome
- the EV is further modified to display an antagonistic antibody or an agonistic antibody or a fragment thereof on the EV, e.g., exosome, surface to direct EV uptake, activate, or block cellular pathways to enhance the combinatorial effect of the STING agonist.
- the antibody or fragment thereof is an antibody against DEC205, CLEC9A, CLEC6, DCIR, DC-SIGN, LOX-1, or Langerin.
- the producer cell may be modified to comprise an additional exogenous sequence encoding for an antagonistic antibody or an agonistic antibody.
- the antagonistic antibody or agonistic antibody may be covalently linked or conjugated to the EV, e.g., exosome, via any appropriate linking chemistry known in the art.
- suitable linking chemistry include amine-reactive groups, carboxylreactive groups, sulfhydryl-reactive groups, aldehyde-reactive groups, photoreactive groups, ClickIT chemistry, biotin-streptavidin or other avidin conjugation, or any combination thereof.
- the present disclosure comprises a method of producing an EV, e.g., exosome, comprising a cholesterol or derivative thereof, which is linked to a STING agonist via a cleavable peptide linker, e.g., a Valine- Alanine linker or Valine-Citrulline linker, the method comprising:
- the buffer conditions of the solution of EVs can also be altered to optimize cholesterol-cleavable peptide linker-a STING agonist into the EVs, e.g., exosomes.
- the buffer can be a phosphate buffered saline (PBS) with sucrose.
- PBS is a well- known buffer to those skilled in the art. Additional buffer modifications can also be used, such as shear protectants, viscosity modifiers, and/or solutes that affect vesicle structural properties.
- Excipients can also be added to improve the efficiency of the cholesterol-cleavable peptide linker- a STING agonist into the surface (membrane) of EVs, such as membrane softening materials and molecular crowding agents.
- Other modifications to the buffer can include specific pH ranges and/or concentrations of salts, organic solvents, small molecules, detergents, zwitterions, amino acids, polymers, and/or any combination of the above including multiple concentrations.
- the one or more moieties can be introduced into suitable producer cells using synthetic macromolecules, such as cationic lipids and polymers (Papapetrou et al., Gene Therapy 12: SI 18-S130 (2005)).
- the cationic lipids form complexes with the one or more moieties through charge interactions.
- the positively charged complexes bind to the negatively charged cell surface and are taken up by the cell by endocytosis.
- a cationic polymer can be used to transfect producer cells.
- the cationic polymer is polyethylenimine (PEI).
- chemicals such as calcium phosphate, cyclodextrin, or polybrene, can be used to introduce the one or more moieties to the producer cells.
- the one or more moieties can also be introduced into a producer cell using a physical method such as particle- mediated transfection, “gene gun”, biolistics, or particle bombardment technology (Papapetrou et al., Gene Therapy 12: S118-S130 (2005)).
- a reporter gene such as, for example, beta- galactosidase, chloramphenicol acetyltransferase, luciferase, or green fluorescent protein can be used to assess the transfection efficiency of the producer cell.
- the EVs, e.g., exosomes, prepared for the present disclosure can be isolated from the producer cells. It is contemplated that all known manners of isolation of EVs, e.g., exosomes, are deemed suitable for use herein.
- physical properties of EVs, e.g., exosomes may be employed to separate them from a medium or other source material, including separation on the basis of electrical charge (e.g., electrophoretic separation), size (e.g., filtration, molecular sieving, etc), density (e.g., regular or gradient centrifugation), Svedberg constant (e.g., sedimentation with or without external force, etc).
- isolation may be based on one or more biological properties, and include methods that may employ surface markers (e.g., for precipitation, reversible binding to solid phase, FACS separation, specific ligand binding, nonspecific ligand binding, etc.).
- the EVs e.g., exosomes
- the EVs may also be fused using chemical and/or physical methods, including PEG-induced fusion and/or ultrasonic fusion.
- the EVs may also be purified after incubation with the cholesterol tagged STING agonist to remove free cholesterol tagged STING agonists from the composition. All manners of previously disclosed methods are also deemed suitable for use herein, including separation on the basis of physical or biological properties of EVs, e.g., exosomes.
- Isolation, purification, and enrichment can be done in a general and non-selective manner (typically including serial centrifugation). Alternatively, isolation, purification, and enrichment can be done in a more specific and selective manner (e.g., using producer cell-specific surface markers). For example, specific surface markers may be used in immunoprecipitation, FACS sorting, affinity purification, bead-bound ligands for magnetic separation etc.
- size exclusion chromatography can be utilized to isolate or purify the EVs, e.g., exosomes. Size exclusion chromatography techniques are known in the art. Exemplary, non-limiting techniques are provided herein.
- a void volume fraction is isolated and comprises the EVs, e.g., exosomes, of interest.
- density gradient centrifugation can be utilized to further isolate the EVs, e.g., exosomes.
- the producer cell-derived EVs e.g., exosomes
- the non-producer cell-derived EVs e.g., exosomes
- immunosorbent capture using an antigen antibody specific for the producer cell.
- the isolation of EVs may involve size exclusion chromatography or ion chromatography, such as anion exchange, cation exchange, or mixed mode chromatography.
- the isolation of EVs, e.g., exosomes may involve desalting, dialysis, tangential flow filtration, ultrafiltration, or diafiltration, or any combination thereofO.
- the isolation of EVs, e.g., exosomes may involve combinations of methods that include, but are not limited to, differential centrifugation, size-based membrane filtration, concentration and/or rate zonal centrifugation.
- the isolation of EVs may involve one or more centrifugation steps.
- the centrifugation may be performed at about 50,000 to 150,000 x g.
- the centrifugation may be performed at about 50,000 x g, 75,000 x g, 100,000 x g, 125,000 x g, or 150,000 x g.
- EV e.g., exosome
- DCs Dendritic cells
- pDCs plasmacytoid DCs
- mDCs myeloid DCs
- eDCs conventional DCs
- a third population of DCs are monocyte-derived DCs (moDCs) which arise from a monocyte precursor, not a DC progenitor like pDCs and eDCs.
- moDCs develop after receiving inflammatory cues.
- Immature DCs reside in peripheral tissue before maturation.
- PRRs pattern recognition receptors
- Each subset of immature DCs varies in the protein expression patterns of PRRs which allows the immature DC populations to respond differently upon activation of the same PRR. This results in modulation of the immune response mediated by DCs.
- PRRs present in DCs include Toll-like receptors (TLRs), C-type lectin receptors, retinoic-acid inducible gene (RIG)-I-like receptors (RLRs), NOD-like receptors (NLRs), and STING.
- TLRs Toll-like receptors
- C-type lectin receptors C-type lectin receptors
- RLRs retinoic-acid inducible gene
- NLRs NOD-like receptors
- the STING pathway is the dominant DNA sensing pathway in both mDCs and pDCs. Activation of the STING pathway in DCs results in Type I IFN and pro inflammatory cytokine production via TBK1, IRF3, and NF-KB signaling. Binding of IFN to their receptors on cells results in activation of IFN-stimulated response elements and the transcription of IFN-sensitive genes that result in the immune and inflammatory response. IFN signaling also cross-primes DCs to promote antigen persistence, alters the antigen repertoire available for MHCI presentation, enhances MHCI presentation of antigens, and increases the overall surface expression of MHCI, MHCII, and costimulatory molecules CD40, CD80, and CD86. These actions result in increased priming of tumor specific CD8+ T cells and initiation of the adaptive immune response.
- the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist to a subject in need thereof activates or induces dendritic cells, thereby inducing or modulating an immune or inflammatory response in the subject.
- the dendritic cells activated are myeloid dendritic cells.
- the dendritic cells are plasmacytoid dendritic cells.
- the EV is administered as an adjuvant.
- the method induces interferon (IFN)-P production.
- IFN interferon
- Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist (e.g., expressed on the exterior surface) may result in between 2-fold and 10,000-fold greater IFN-P induction compared to administration of a STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation).
- Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the surface may result in between about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500- 600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000- 9000 fold, or 9000-10,000 fold greater IFN-P induction compared to administration of a STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation).
- exoSTING e.g., a combination of exosome
- Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in greater than about 2-fold, >5 fold, >10-fold, >20-fold, >30-fold, >40-fold, >50-fold, >60-fold, >70-fold, >80-fold, >90-fold, >100- fold, >200-fold, >300-fold, >400-fold, >500-fold, >600-fold, >700-fold, >800-fold, >900-fold, >1000-fold, >2000-fold, >3000-fold, >4000-fold, >5000-fold, >6000-fold, >7000-fold, >8000- fold, >9000-fold, or > 10,000-fold IFN-P induction compared to administration of a cholesterol tagged STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation).
- exoSTING e.g.,
- EVs e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface
- EVs e.g., exosomes
- a cholesterol tagged STING agonist expressed on the exterior surface may result in between 2-fold and 10,000-fold greater IFN-P induction compared to the subject’s baseline IFN-P production.
- Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in between about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30- 40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200- 300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000-9000 fold, or 9000-10,000 fold greater IFN-p induction compared to the subject’s baseline IFN-P production.
- Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist may result in greater than about 2-fold, >5 fold, >10-fold, >20-fold, >30-fold, >40-fold, >50-fold, >60-fold, >70-fold, >80-fold, >90-fold, >100- fold, >200-fold, >300-fold, >400-fold, >500-fold, >600-fold, >700-fold, >800-fold, >900-fold, >1000-fold, >2000-fold, >3000-fold, >4000-fold, >5000-fold, >6000-fold, >7000-fold, >8000- fold, >9000-fold, or >10, 000-fold IFN-P induction compared to the subject’s baseline IFN-P production.
- administering an EV, e.g., exosome comprising a cholesterol tagged STING agonist expressed on the exterior surface, disclosed herein to a subject can also regulate the levels of other immune modulators e.g., cytokines or chemokines).
- the method disclosed herein can increase the level of IFN-y, CXCL9, and/or CXCL10.
- administration of EVs e.g., exosomes comprising a cholesterol tagged STING agonist expressed on the exterior surface, described herein can result in between about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000-9000 fold, or 9000-10,000 fold greater amount of IFN-y, CXCL9, and/or CXCL10 compared to a free STING agonist or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation).
- the method induces myeloid dendritic cell (mDC) activation.
- EVs e.g., exosomes
- a cholesterol tagged STING agonist expressed on the exterior surface may result in between 2-fold and 50,000-fold greater mDC activation compared to administration of a STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation).
- EVs e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface
- Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in between about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000- 8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20, GOO- 25, 000 fold, 25,000-30,000 fold, 30,000-35,000 fold, 35,000-40,000 fold, 40,000
- EVs e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface
- Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in greater than about 2-fold, >5 fold, >10-fold, >20-fold, >30-fold, >40-fold, >50-fold, >60-fold, >70-fold, >80-fold, >90-fold, >100-fold, >200- fold, >300-fold, >400-fold, >500-fold, >600-fold, >700-fold, >800-fold, >900-fold, >1000-fold, >2000-fold, >3000-fold, >4000-fold, >5000-fold, >6000-fold, >7000-fold, >8000-fold, >9000- fold, >10, 000-fold, >15, 000-fold, >20, 000-fold, >25, 000-fold, >30, 000-fold, >35, 000-fold
- EVs e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface
- EVs e.g., exosomes
- a cholesterol tagged STING agonist expressed on the exterior surface may result in between 2-fold and 10,000-fold greater mDC activation compared to the subject’s baseline mDC activation.
- Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in between about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000- 8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20, GOO- 25, 000 fold, 25,000-30,000 fold, 30,000-35,000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold greater mDC activation compared to the subject’s baseline mDC activation.
- EVs e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface
- Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in greater than about 2-fold, >5 fold, > 10-fold, >20-fold, >30- fold, >40-fold, >50-fold, >60-fold, >70-fold, >80-fold, >90-fold, >100-fold, >200-fold, >300-fold, >400-fold, >500-fold, >600-fold, >700-fold, >800-fold, >900-fold, > 1000-fold, >2000-fold, >3000-fold, >4000-fold, >5000-fold, >6000-fold, >7000-fold, >8000-fold, >9000-fold, >10, 000- fold, >15, 000-fold, >20, 000-fold, >25, 000-fold, >30, 000-fold, >35, 000-fold,
- the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface does not induce monocyte activation as compared to the subject’s baseline monocyte activation.
- an EV e.g., exosome
- a cholesterol tagged STING agonist expressed on the exterior surface results in less than less than about 2-fold, ⁇ 5 fold, ⁇ 10-fold, ⁇ 20-fold, ⁇ 30- fold, ⁇ 40-fold, ⁇ 50-fold, ⁇ 60-fold, ⁇ 70-fold, ⁇ 80-fold, ⁇ 90-fold, ⁇ 100-fold, ⁇ 200-fold, ⁇ 300-fold, ⁇ 400-fold, ⁇ 500-fold, ⁇ 600-fold, ⁇ 700-fold, ⁇ 800-fold, ⁇ 900-fold, ⁇ 1000-fold, ⁇ 2000-fold, ⁇ 3000-fold, ⁇ 4000-fold, ⁇ 5000-fold, ⁇ 6000-fold, ⁇ 7000-fold, ⁇ 8000-fold, ⁇ 9000-fold, ⁇ 10, 000- fold, ⁇ 15, 000-fold, ⁇ 20, 000-fold, ⁇ 25, 000-fold, ⁇ 30, 000-fold, ⁇
- an EV e.g., exosome
- a cholesterol tagged STING agonist expressed on the exterior surface results in less than about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000- 8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20, GOO- 25, 000 fold, 25,000-30,000 fold, 30,000-35,000 fold, 35,000-40,000 fold, 40,000-45,000 fold, 45,000-50,000 fold, 55,000-60,000 fold, 60,000-65,000 fold, 65,000-
- the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface to a subject does not induce monocyte activation as compared to administration of the STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation).
- an exoSTING e.g., a combination of exosome and the STING agonist that can be prepared by incubation.
- an EV e.g., exosome
- a cholesterol tagged STING agonist expressed on the exterior surface results in less than less than about 2-fold, ⁇ 5 fold, ⁇ 10- fold, ⁇ 20-fold, ⁇ 30-fold, ⁇ 40-fold, ⁇ 50-fold, ⁇ 60-fold, ⁇ 70-fold, ⁇ 80-fold, ⁇ 90-fold, ⁇ 100-fold, ⁇ 200-fold, ⁇ 300-fold, ⁇ 400-fold, ⁇ 500-fold, ⁇ 600-fold, ⁇ 700-fold, ⁇ 800-fold, ⁇ 900-fold, ⁇ 1000- fold, ⁇ 2000-fold, ⁇ 3000-fold, ⁇ 4000-fold, ⁇ 5000-fold, ⁇ 6000-fold, ⁇ 7000-fold, ⁇ 8000-fold, ⁇ 9000-fold, ⁇ 10, 000-fold, ⁇ 15, 000-fold, ⁇ 20, 000-fold, ⁇ 25, 000-fold, ⁇ 30, 000-fold, ⁇ 10, 000-fold,
- an EV e.g., exosome
- a cholesterol tagged STING agonist expressed on the exterior surface results in less than about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90- 100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20,000-25,000 fold, 25,000-30,000 fold, 30, GOO- 35, 000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold induction of monocyte activation relative to the amount of monocyte activ
- lower dosages of the EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface can be delivered compared to the free STING agonist or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation).
- an exoSTING e.g., a combination of exosome and the STING agonist that can be prepared by incubation.
- non-selective delivery of high doses of STING agonists can attenuate desirable immune stimulatory responses.
- the EVs e.g., exosomes, described herein can be administered at lower doses, in some aspects, they can operate in a wider therapeutic window and reduce the liabilities e.g., systemic toxicity, immune cell killing, lack of cell selectivity) observed with free STING agonists.
- compositions described herein may be administered in a dosage sufficient to ameliorate the disease, disorder, condition, or symptom of the subject in need thereof.
- the dosage of the EV, e.g., exosome, comprising a cholesterol tagged STING agonist administered to a subject in need is between about 0.01 to 0.1 pM, 0.1 to 1 pM, 1 to 10 pM, 10 to 100 pM, or 100 to 1000 pM.
- the dosage of the EV, e.g., exosome, comprising a cholesterol tagged STING agonist administered to a subject in need is about 0.01 pM, 0.05 pM, 0.1 pM, 0.2 pM, 0.3 pM, 0.4 pM, 0.5 pM, 0.6 pM, 0.7 pM, 0.8 pM, 0.9 pM, 1 pM, 2 pM, 3 pM, 4 pM, 5 pM, 6 pM, 7 pM, 8 pM, 9 pM, 10 pM, 11 pM, 12 pM, 13 pM, 14 pM, 15 pM, 16 pM, 17 pM, 18 pM, 19 pM, 20 pM, 25 pM, 30 pM, 35 pM, 40 pM, 45 pM, 40 pM, 55 pM, 60 pM, 65 pM, 70 p
- the dosage of the EV, e.g., exosome, comprising a cholesterol tagged STING agonist administered to a subject in need is about 0.3 pg, about 1 pg, about 3 pg, about 6 pg, or about 12 pg.
- STING agonist expressed on the exterior surface administered to a subject in need is less than 2- fold, ⁇ 5 fold, ⁇ 10-fold, ⁇ 20-fold, ⁇ 30-fold, ⁇ 40-fold, ⁇ 50-fold, ⁇ 60-fold, ⁇ 70-fold, ⁇ 80-fold, ⁇ 90- fold, ⁇ 100-fold, ⁇ 200-fold, ⁇ 300-fold, ⁇ 400-fold, ⁇ 500-fold, ⁇ 600-fold, ⁇ 700-fold, ⁇ 800-fold, ⁇ 900-fold, ⁇ 1000-fold, ⁇ 2000-fold, ⁇ 3000-fold, ⁇ 4000-fold, ⁇ 5000-fold, ⁇ 6000-fold, ⁇ 7000-fold, ⁇ 8000-fold, ⁇ 9000-fold, ⁇ 10, 000-fold, ⁇ 15, 000-fold, ⁇ 20, 000-fold, ⁇ 25, 000-fold, ⁇ 30, 000-fold, ⁇ 35,000-fold, ⁇ 40, 000-fold, ⁇ 45, 000-fold, or
- the amount of the EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface administered to a subject in need is between less than about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000- 4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20,000-25,000 fold, 25,000-30,000 fold, 30,000-35,000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold less relative to the amount of a free STING agonist or an
- the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist does not induce systemic inflammation as compared to the subject’s baseline systemic inflammation.
- the administration of an EV, e.g., exosome, comprising a cholesterol tagged STING agonist results in less than less than about 2- fold, ⁇ 5 fold, ⁇ 10-fold, ⁇ 20-fold, ⁇ 30-fold, ⁇ 40-fold, ⁇ 50-fold, ⁇ 60-fold, ⁇ 70-fold, ⁇ 80-fold, ⁇ 90- fold, ⁇ 100-fold, ⁇ 200-fold, ⁇ 300-fold, ⁇ 400-fold, ⁇ 500-fold, ⁇ 600-fold, ⁇ 700-fold, ⁇ 800-fold, ⁇ 900-fold, ⁇ 1000-fold, ⁇ 2000-fold, ⁇ 3000-fold, ⁇ 4000-fold, ⁇ 5000-fold, ⁇ 6000-fold, ⁇ 7000-fold,
- an EV e.g., exosome
- a cholesterol tagged STING agonist results in less than about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500- 600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000- 9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20,000-25,000 fold, 25,000- 30,000 fold, 30,000-35,000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold induction of systemic inflammation relative to the subject’s baseline systemic inflammation.
- the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface to a subject does not induce systemic inflammation as compared to administration of the STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation).
- an exoSTING e.g., a combination of exosome and the STING agonist that can be prepared by incubation.
- an EV e.g., exosome
- a cholesterol tagged STING agonist expressed on the exterior surface results in less than about 2-fold, ⁇ 5 fold, ⁇ 10-fold, ⁇ 20- fold, ⁇ 30-fold, ⁇ 40-fold, ⁇ 50-fold, ⁇ 60-fold, ⁇ 70-fold, ⁇ 80-fold, ⁇ 90-fold, ⁇ 100-fold, ⁇ 200-fold, ⁇ 300-fold, ⁇ 400-fold, ⁇ 500-fold, ⁇ 600-fold, ⁇ 700-fold, ⁇ 800-fold, ⁇ 900-fold, ⁇ 1000-fold, ⁇ 2000- fold, ⁇ 3000-fold, ⁇ 4000-fold, ⁇ 5000-fold, ⁇ 6000-fold, ⁇ 7000-fold, ⁇ 8000-fold, ⁇ 9000-fold, ⁇ 10, 000-fold, ⁇ 15, 000-fold, ⁇ 20, 000-fold, ⁇ 25, 000-fold, ⁇ 30, 000-fold, ⁇ 35
- an EV e.g., exosome
- a cholesterol tagged STING agonist expressed on the exterior surface results in less than about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90- 100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20,000-25,000 fold, 25,000-30,000 fold, 30, GOO- 35, 000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold induction of systemic inflammation relative to the amount of systemic inflammation after administration of
- the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface to a subject additionally comprises administering an additional therapeutic agent.
- the additional therapeutic agent is an immunomodulating agent.
- the immunomodulating component is an inhibitor for a negative checkpoint regulator or an inhibitor for a binding partner of a negative checkpoint regulator.
- the negative checkpoint regulator is selected from the group consisting of: cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1), lymphocyte-activated gene 3 (LAG-3), T-cell immunoglobulin mucin-containing protein 3 (TIM-3), B and T lymphocyte attenuator (BTLA), T cell immunoreceptor with Ig and ITIM domains (TIGIT), V-domain Ig suppressor of T cell activation (VISTA), adenosine A2a receptor (A2aR), killer cell immunoglobulin like receptor (KIR), indoleamine 2,3-dioxygenase (IDO), CD20, CD39, and CD73.
- CTLA-4 cytotoxic T-lymphocyte-associated protein 4
- PD-1 programmed cell death protein 1
- LAG-3 lymphocyte-activated gene 3
- TIM-3 T-cell immunoglobulin mucin-containing protein 3
- the additional therapeutic agent is an antibody or antigen-binding fragment thereof.
- the antibody or antigen-binding fragment thereof is one or more whole antibodies, polyclonal, monoclonal and recombinant antibodies, fragments thereof, and further includes single-chain antibodies, humanized antibodies, murine antibodies, chimeric, mouse-human, mouse-primate, primate-human monoclonal antibodies, anti-idiotype antibodies, antibody fragments, such as, e.g., scFv, (scFv)2, Fab, Fab', and F(ab')2, F(abl)2, Fv, dAb, and Fd fragments, diabodies, and antibody- related polypeptides.
- the term antibody includes bispecific antibodies and multispecific antibodies so long as they exhibit the desired biological activity or function.
- the additional therapeutic agent is a therapeutic antibody or antigen-binding fragment thereof that is an inhibitor of CTLA-4, PD-1, PD-L1, PD-L2, TIM-3, or LAG3.
- the additional therapeutic agent is an agent that prevents or treats T cell exhaustion.
- Such agents may increase, decrease, or modulate the expression of genes associated with T cell exhaustion, including Prdml, Bhlhe40, Irf4. Ikzf2, Zeb2, Lass6, Egr2, Tox, Eomes, Nfatcl, Nfatc2, Zbtb32, Rbpj, Hifla, Lag3, Tnfrsf9, Ptger2, Haver 2, A team.
- Therapeutic agents may also increase, decrease, or modulate a protein associated with T cell exhaustion, including NFAT-1 or NF AT-2.
- compositions disclosed herein are capable of up-regulating a STING-mediated immune response in the subject, thereby enhancing the tumor targeting of the subject’s immune system.
- the composition is administered intra-tumorally to the subject.
- the composition is administered parenterally, orally, intravenously, intramuscularly, intraperitoneally, or via any other appropriate administration route.
- compositions disclosed herein comprising administering to the subject a therapeutically effective amount of the compositions disclosed herein, wherein the composition is capable of preventing one or more tumors at one location in the subject from promoting the growth of one or more tumors at another location in the subject.
- the composition is administered intratumorally in a first tumor in one location, and the composition administered in a first tumor prevents metastasis of one or more tumors at a second location.
- administering an EV, e.g., exosome, disclosed herein inhibits and/or reduces tumor growth in a subject.
- the tumor growth e.g., tumor volume or weight
- the tumor growth is reduced by at least about 5%, at least about 10%, at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, or about 100% compared to a reference (e.g., tumor volume in a corresponding subject after administration of free STING agonist or an EV, e.g., exosome, without the STING agonist).
- the cancer being treated is characterized by infiltration of leukocytes (T-cells, B-cells, macrophages, dendritic cells, monocytes) into the tumor microenvironment, or so-called “hot tumors” or “inflammatory tumors”.
- the cancer being treated is characterized by low levels or undetectable levels of leukocyte infiltration into the tumor microenvironment, or so-called “cold tumors” or “non-inflammatory tumors”.
- an EV e.g., exosome
- an EV is administered in an amount and for a time sufficient to convert a “cold tumor” into a “hot tumor”, z.e., said administering results in the infiltration of leukocytes (such as T-cells) into the tumor microenvironment.
- cancer comprises bladder cancer, cervical cancer, renal cell cancer, testicular cancer, colorectal cancer, lung cancer, head and neck cancer, and ovarian, lymphoma, liver cancer, glioblastoma, melanoma, myeloma, leukemia, pancreatic cancers, or combinations thereof.
- distal tumor refers to a tumor that has spread from the original (or primary) tumor to distant organs or distant tissues, e.g., lymph nodes.
- the EVs, e.g., exosomes, of the disclosure treats a tumor after the metastatic spread.
- Non-limiting examples of cancers (or tumors) that can be treated with methods disclosed herein include squamous cell carcinoma, small-cell lung cancer (SCLC), non-small cell lung cancer, squamous non-small cell lung cancer (NSCLC), nonsquamous NSCLC, gastrointestinal cancer, renal cancer (e.g., clear cell carcinoma), ovarian cancer, liver cancer (e.g., hepatocellular carcinoma), colorectal cancer, endometrial cancer, kidney cancer (e.g., renal cell carcinoma (RCC)), prostate cancer (e.g., hormone refractory prostate adenocarcinoma), thyroid cancer, pancreatic cancer, cervical cancer, stomach cancer, bladder cancer, hepatoma, breast cancer, colon carcinoma, and head and neck cancer (or carcinoma), gastric cancer, germ cell tumor, pediatric sarcoma, sinonasal natural killer, melanoma (e.g., metastatic malignant melanoma, such as cutaneous or intraocular malignant melanom
- a cancer (or tumor) that can be treated comprises a breast cancer, head and neck cancer, uterine cancer, brain cancer, skin cancer, renal cancer, lung cancer, colorectal cancer, prostate cancer, liver cancer, bladder cancer, kidney cancer, peritoneal cancer, pancreatic cancer, thyroid cancer, esophageal cancer, eye cancer, stomach (gastric) cancer, gastrointestinal cancer, carcinoma, sarcoma, leukemia, lymphoma, myeloma, or a combination thereof.
- a cancer that can be treated with the present disclosure is a pancreatic cancer and/or a peritoneal cancer.
- a cancer (or tumor) that can be treated comprises a chondroid sarcoma (also referred to as a chondrosarcoma).
- a cancer (or tumor) that can be treated comprises a cutaneous squamous cell carcinoma (cSCC).
- a cancer (or tumor) that can be treated comprises a head and neck squamous cell carcinoma of the oral cavity.
- a cancer (or tumor) that can be treated comprises a hepatocellular cancer (HCC).
- a cancer (or tumor) that can be treated comprises a cervical cancer.
- a cancer (or tumor) that can be treated comprises an eye melanoma.
- a cancer (or tumor) that can be treated comprises a choroidal eye melanoma.
- a cancer (or tumor) that can be treated comprises a gastric spindle cell sarcoma.
- a cancer (or tumor) that can be treated comprises a hemangioendothelioma.
- a cancer (or tumor) that can be treated comprises a mesothelioma.
- a cancer (or tumor) that can be treated comprises a parotoid gland cancer.
- a cancer (or tumor) that can be treated comprises a renal cancer.
- a cancer (or tumor) that can be treated comprises a triple-negative breast cancer (TNBC).
- TNBC triple-negative breast cancer
- the methods described herein can also be used for treatment of metastatic cancers, unresectable, refractory cancers (e.g., cancers refractory to previous cancer therapy), and/or recurrent cancers.
- EVs e.g., exosomes
- additional anti-cancer and/or immunomodulating agents can include, for example, chemotherapy drugs, small molecule drugs, or antibodies that stimulate the immune response to a given cancer.
- the methods described herein are used in combination with a standard of care treatment (e.g., surgery, radiation, and chemotherapy).
- a method for treating a cancer disclosed herein can comprise administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface with one or more immuno-oncology agents, such that multiple elements of the immune pathway can be targeted.
- Non-limiting of such combinations include: a therapy that enhances tumor antigen presentation (e.g., dendritic cell vaccine, GM-CSF secreting cellular vaccines, CpG oligonucleotides, imiquimod); a therapy that inhibits negative immune regulation e.g., by inhibiting CTLA-4 and/or PD1/PD-L1/PD-L2 pathway and/or depleting orblocking Tregs or other immune suppressing cells (e.g., myeloid-derived suppressor cells); a therapy that stimulates positive immune regulation, e.g., with agonists that stimulate the CD-137, OX-40, and/or CD40 or GITR pathway and/or stimulate T cell effector function; a therapy that increases systemically the frequency of anti-tumor T cells; a therapy that depletes or inhibits Tregs, such as Tregs in the tumor, e.g., using an antagonist of CD25 (e.g., daclizumab) or by ex vivo anti-CD25
- an immuno-oncology agent that can be used in combination with EVs, e.g., exosomes, disclosed herein comprises an immune checkpoint inhibitor (i.e., blocks signaling through the particular immune checkpoint pathway).
- immune checkpoint inhibitors that can be used in the present methods comprise a CTLA-4 antagonist (e.g., anti-CTLA-4 antibody), PD-1 antagonist (e.g., anti-PD-1 antibody, anti-PD-Ll antibody), TIM-3 antagonist (e.g., anti-TIM-3 antibody), or combinations thereof.
- an immuno-oncology agent comprises an immune checkpoint activator i.e., promotes signaling through the particular immune checkpoint pathway).
- immune checkpoint activator comprises 0X40 agonist (e.g., anti-OX40 antibody), LAG-3 agonist (e.g. anti-LAG-3 antibody), 4-1BB (CD137) agonist (e.g., anti-CD137 antibody), GITR agonist (e.g., anti-GITR antibody), or any combination thereof.
- a combination of an EV, e.g., exosome, disclosed herein and a second agent discussed herein can be administered concurrently as a single composition in a pharmaceutically acceptable carrier.
- a combination of an EV, e.g., exosome, and a second agent discussed herein can be administered concurrently as separate compositions.
- a combination of an EV, e.g., exosome, and a second agent discussed herein (e.g., immune checkpoint inhibitor) can be administered sequentially.
- an EV, e.g., exosome is administered prior to the administration of a second agent (e.g., immune checkpoint inhibitor).
- compositions comprising EVs, e.g., exosomes, that are suitable for administration to a subject.
- the pharmaceutical compositions generally comprise a plurality of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface and a pharmaceutically-acceptable excipient or carrier in a form suitable for administration to a subject.
- Pharmaceutically-acceptable excipients or carriers are determined in part by the particular composition being administered, as well as by the particular method used to administer the composition. Accordingly, there is a wide variety of suitable formulations of pharmaceutical compositions comprising a plurality of EVs, e.g., exosomes,.
- compositions are generally formulated sterile and in full compliance with all Good Manufacturing Practice (GMP) regulations of the U.S. Food and Drug Administration.
- GMP Good Manufacturing Practice
- the pharmaceutical composition comprises one or more STING agonist and the EVs, e.g., exosomes, described herein.
- compositions include excipients that are generally safe (GRAS), non-toxic, and desirable, including excipients that are acceptable for veterinary use as well as for human pharmaceutical use.
- GRAS generally safe
- excipients that are acceptable for veterinary use as well as for human pharmaceutical use.
- Examples of carriers or diluents include, but are not limited to, water, saline, Ringer's solutions, dextrose solution, and 5% human serum albumin.
- the use of such media and compounds for pharmaceutically active substances is well known in the art. Except insofar as any conventional media or compound is incompatible with the EVs, e.g., exosomes, described herein, use thereof in the compositions is contemplated. Supplementary therapeutic agents may also be incorporated into the compositions.
- a pharmaceutical composition is formulated to be compatible with its intended route of administration.
- the EVs can be administered by intratumoral, parenteral, topical, intravenous, oral, subcutaneous, intraarterial, intradermal, transdermal, rectal, intracranial, intraperitoneal, intranasal; intramuscular route or as inhalants.
- the pharmaceutical composition comprising EVs, e.g., exosomes is administered intravenously, e.g. by injection.
- the EVs, e.g., exosomes can optionally be administered in combination with other therapeutic agents that are at least partly effective in treating the disease, disorder or condition for which the EVs, e.g., exosomes, are intended.
- Solutions or suspensions can include the following components: a sterile diluent such as water, saline solution, fixed oils, polyethylene glycols, glycerin, propylene glycol or other synthetic solvents; antibacterial compounds such as benzyl alcohol or methyl parabens; antioxidants such as ascorbic acid or sodium bisulfite; chelating compounds such as ethylenediaminetetraacetic acid (EDTA); buffers such as acetates, citrates or phosphates, and compounds for the adjustment of tonicity such as sodium chloride or dextrose.
- the pH can be adjusted with acids or bases, such as hydrochloric acid or sodium hydroxide.
- the preparation can be enclosed in ampoules, disposable syringes or multiple dose vials made of glass or plastic.
- compositions suitable for injectable use include sterile aqueous solutions (if water soluble) or dispersions and sterile powders.
- suitable carriers include physiological saline, bacteriostatic water, Cremophor ELTM (BASF, Parsippany, N.J.) or phosphate buffered saline (PBS).
- the composition is generally sterile and fluid to the extent that easy syringeability exists.
- the carrier can be a solvent or dispersion medium containing, e.g., water, ethanol, polyol (e.g., glycerol, propylene glycol, and liquid polyethylene glycol, and the like), and suitable mixtures thereof.
- the proper fluidity can be maintained, e.g., by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants.
- Prevention of the action of microorganisms can be achieved by various antibacterial and antifungal compounds, e.g., parabens, chlorobutanol, phenol, ascorbic acid, thimerosal, and the like.
- isotonic compounds e.g., sugars, polyalcohols such as mannitol, sorbitol, sodium chloride can be added to the composition.
- Prolonged absorption of the injectable compositions can be brought about by including in the composition a compound which delays absorption, e.g., aluminum monostearate and gelatin.
- Sterile injectable solutions can be prepared by incorporating the EVs, e.g., exosomes, in an effective amount and in an appropriate solvent with one or a combination of ingredients enumerated herein, as desired.
- dispersions are prepared by incorporating the EVs, e.g., exosomes, into a sterile vehicle that contains a basic dispersion medium and any desired other ingredients.
- methods of preparation are vacuum drying and freeze-drying that yields a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
- the EVs can be administered in the form of a depot injection or implant preparation which can be formulated in such a manner to permit a sustained or pulsatile release of the EVs, e.g., exosomes.
- the EVs, e.g., exosomes are administered by way of a pump.
- the EVs, e.g., exosomes are administered by way of a patch.
- compositions comprising EVs can also be by transmucosal or transdermal means.
- penetrants appropriate to the barrier to be permeated are used in the formulation.
- penetrants are generally known in the art, and include, e.g., for transmucosal administration, detergents, bile salts, and fusidic acid derivatives.
- Transmucosal administration can be accomplished through the use of nasal sprays, suppositories, sublingual spray, drops, or fast dissolve strips.
- the modified EVs, e.g., exosomes are formulated into ointments, salves, gels, creams, or patches as generally known in the art.
- HEK293SF cells were grown to high density in chemically defined medium for 7 days.
- Conditioned cell culture media was collected and centrifuged at 300 - 800 x g for 5 minutes at room temperature to remove cells and large debris.
- Media supernatant was then supplemented with 1000 U/L BENZONASE® and incubated at 37 °C for 1 hour in a water bath.
- Supernatant was collected and centrifuged at 16,000 x g for 30 minutes at 4 °C to remove residual cell debris and other large contaminants.
- Supernatant was then ultracentrifuged at 133,900 x g for 3 hours at 4 °C to pellet the exosomes.
- Supernatant was discarded and any residual media was aspirated from the bottom of the tube.
- the pellet was resuspended in 200 - 1000 pL PBS (-Ca -Mg).
- the pellet was processed via density gradient purification (sucrose or OPTIPREPTM).
- sucrose gradient purification the exosome pellet was layered on top of a sucrose gradient as defined in Table 3 below.
- the exosome layer was gently removed from the top layer and diluted in -32.5 mL PBS in a 38.5 mL Ultra-Clear (344058) tube and ultracentrifuged again at 133,900 x g for 3 hours at 4 °C to pellet the purified exosomes. The resulting pellet was resuspended in a minimal volume of PBS (-200 pL) and stored at 4 °C.
- OPTIPREPTM gradient a 3-tier sterile gradient is prepared with equal volumes of 10%, 30%, and 45% OPTIPREPTMin a 12 mL Ultra-Clear (344059) tube for a SW 41 Ti rotor. The pellet was added to the OPTIPREPTM gradient and ultracentrifuged at 200,000 x g for 16 hours at 4 °C to separate the exosome fraction. The exosome layer was then gently collected from the top -3 mL of the tube.
- the exosome fraction was diluted in -32 mL PBS in a 38.5 mL Ultra-Clear (344058) tube and ultracentrifuged at 133,900 x g for 3 hours at 4 °C to pellet the purified exosomes. The pelleted exosomes were then resuspended in a minimal volume of PBS (-200 pL) and store at 4°C.
- exoSTING ECC, exoSTING and free STING agonist were tested for activity in human peripheral blood mononuclear cells (PBMCs).
- PBMCs peripheral blood mononuclear cells
- PBMCs were isolated from fresh human blood by centrifugation over a layer of Lymphoprep at 1000 x g for 15 minutes. The resulting buffy coat was washed in PBS and counted.
- PBMCs were plated in 96-well U-bottom plates. Titrations of the exoSTING ECC, exoSTING, or free STING agonist were prepared in a separate U-bottom plate to perform dose-response studies. The exoSTING ECC, exoSTING or free STING agonist was added to the PBMCs and incubated at 37°C overnight.
- exoSTING ECC and exoSTING were incubated at 37°C for different times (3 days, 7 days, 14 days, or 30 days) and -80°C for a control.
- RNAs were isolated by using RNeasy Lipid Tissue Mini Kit (Thermo Fisher Scientific), according to manufacturer’s instructions.
- cDNA was synthesized by using Superscript IV VILO Master Mix (Thermo Fisher Scientific).
- IFNP mRNA expression was measured by using TaqManTM Universal PCR Master Mix (Thermo Fisher Scientific) and QuantStudioTM 3 & 5 Real-Time PCR System (Thermo Fisher Scientific). Ct values were normalized to Ct values of the housekeeping gene RPS13. As shown in Fig. IB, activity of exoSTING-ECC was stable up to 1 month at 37°C without any reduction of activity, whereas activity of exoSTING was almost gone from 3 days of incubation at 37°C. This data shows that superior stability of exoSTING ECC in 37°C.
- Exosomes expressing SARS CoV-2 RBD (receptor binding domain) fused to PTGFRN (exoRBD) and exosomes expressing SARS CoV-2 RBD fused to PTGFRN (exoRBD) and loaded with cholesterol tagged STING (exoRBD/ECC-STING) were stored at either 37°C or -20°C degrees for 30 days.
- Wild type C57BL/6 mice were immunized subcutaneously with exoRBD and exoRBD/ECC STING from the two storage conditions on days 0 and 14.
- Sera anti-RBD IgG antibody titers (FIG. 2 A) and neutralizing antibody titers (FIG. 2B) were measured 14 days after the second immunization.
- Exosomes expressing the RBDs from both SARS CoVl and CoV2 were loaded with cholesterol tagged STING (ECC STING).
- ECC STING cholesterol tagged STING
- Wild type C57BL/6 mice were immunized subcutaneously with three different doses of exosomes (4E11, 2E11, and 1E11) or PBS on days 0 and 14.
- sera neutralizing antibody titers and anti-RBD IgG antibody titers were measured.
- Sera from human subjects obtained 14 days after the second dose of an mRNA vaccine Human mRNA Vax
- Neutralizing antibody responses against RBDs from the wild type (Washington) SARS CoV-2 strain, SARS CoV-1, WIV-1 bat coronavirus, and the Omicron variant of SARS CoV-2 were measured; and anti-RBD IgG antibody responses against RBDs from the wild type (Washington) SARS CoV-2 strain, SARS CoV-1, the Omicron variant of SARS CoV-2, and the bat coronavirus RaTG13 were measured.
- the present disclosure provides, inter alia, compositions of exosomes comprising cholesterol tagged STING agonists for use as therapeutics.
- the present disclosure also provides methods of producing exosomes comprising cholesterol tagged STING agonists and methods of administering such exosomes as therapeutics. While various specific aspects have been illustrated and described, the above specification is not restrictive. It will be appreciated that various changes can be made without departing from the spirit and scope of the invention(s). Many variations will become apparent to those skilled in the art upon review of this specification.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Molecular Biology (AREA)
- Virology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Oncology (AREA)
- Toxicology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Cell Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Communicable Diseases (AREA)
- Hematology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Botany (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Provided herein are EVs, e.g., exosomes, carrying STING agonists linked to cholesterol through a cleavable peptide linker, e.g., Valine-Alanine linker or Valine-Citrulline linker, and methods of producing the compositions described. Also provided herein are methods of modulating an immune response via administration of a therapeutic amount of EVs, e.g., exosomes carrying STING agonists linked to cholesterol through a cleavable peptide linker, e.g., Valine-Alanine linker or Valine-Citrulline linker,. Also provided herein are methods of modulating an immune response that does not induce systemic inflammation via administration of exosomes carrying STING agonists linked to cholesterol through a cleavable peptide linker, e.g., Valine-Alanine linker or Valine-Citrulline linker.
Description
EXTRACELLULAR VESICLE COMPRISING CHOLESTEROL TAGGED
STING-AGONIST
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority benefit of U.S. Provisional Application Nos. 63/261,918, filed September 30, 2021; and 63/364,875 filed May 17, 2022, each of which is herein incorporated by reference in its entirety.
REFERENCE TO SEQUENCE LISTING SUBMITTED ELECTRONICALLY VIA EFS-WEB
[0002] The content of the electronically submitted sequence listing in ASCII text file (Name: 4000_130PC02_Seqlisting_ST26; Size: 1,487,294 bytes; and Date of Creation: September 28, 2022) filed with the application is herein incorporated by reference in its entirety.
BACKGROUND OF THE DISCLOSURE
[0003] Stimulator of Interferon Genes (STING) is a cytosolic sensor of cyclic dinucleotides that is typically produced by bacteria. Upon activation, it leads to the production of type I interferons and initiates an immune response. Agonism of STING has been shown as a promising approach for generating an immune response against tumors pre-clinically. Unfortunately, given the broad expression profile of STING, systemic delivery of STING agonists leads to systemic inflammation. This limits the dose that can be given which in turn limits the therapeutic efficacy. An alternative approach to systemic delivery is to inject the STING agonist directly into the tumor. Intra-tumoral injections are quite effective; however, they are limited to solid tumors that can be reached with a needle and lead to tissue damage. Improved methods of delivering STING agonists are therefore needed.
SUMMARY OF THE DISCLOSURE
[0004] In some aspects, the present disclosure provides an extracellular vesicle (EV) comprising a cholesterol or derivative thereof on the surface, wherein the cholesterol is linked to a stimulator of interferon genes protein (STING) agonist through a cleavable peptide linker. In some aspects, the cleavable peptide linker comprises a linker cleavable by cathepsin. In some aspects, the cleavable peptide linker comprises a Valine- Alanine linker and/or a Valine-Citrulline linker. In some aspects, the EV is an exosome, a nanovesicle, an apoptotic body, a microvesicle, a lysosome, an endosome,
a liposome, a lipid nanoparticle, a micelle, a multilamellar structure, a revesiculated vesicle, or an extruded cell. In some aspects, the EV is an exosome. In some aspects, the EV overexpresses a PTGFRN protein. In some aspects, the EV is produced by a cell that overexpresses a PTGFRN protein. In some aspects, the extracellular vesicle further comprises a ligand, a cytokine, an antigen, or an antibody. In some aspects, the antibody comprises an antagonistic antibody and/or an agonistic antibody. In some aspects, the STING agonist is a cyclic dinucleotide. In some aspects, the STING agonist is a non-cyclic dinucleotide. In some aspects, the STING agonist is modified such that a polarity and/or a charge different from the corresponding unmodified STING agonist. In some aspects, the STING agonist comprises:
Formula 1 Formula 2 Formula 3

Xi is H, OH, or F;
X2 is H, OH, or F;
Z is OH, ORi, SH or SRi, wherein: i) Ri is Na or NH4, or ii) Ri is an enzyme-labile group which provides OH or SH in vivo such as pivaloyloxymethyl;
Bi and B2 are bases chosen from:
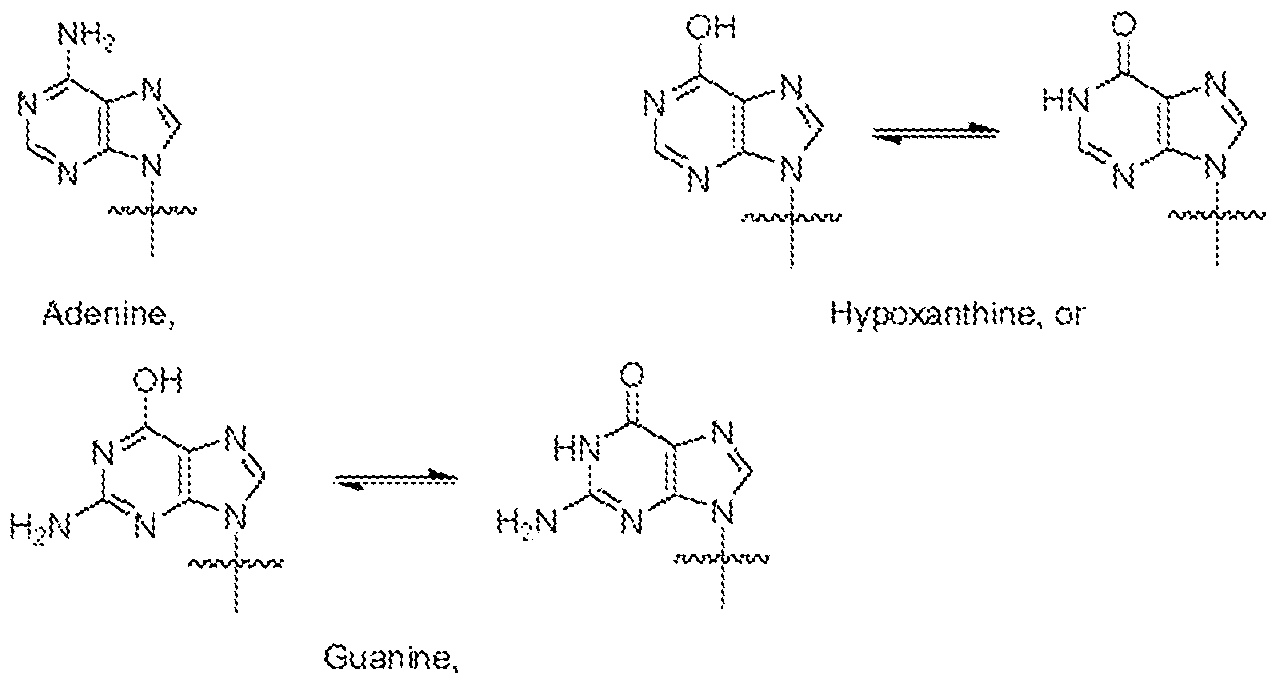
With the proviso that:
- in Formula (I): Xi and X2 are not OH,
- in Formula (II): when Xi and X2 are OH, Bi is not Adenine and B2 is not Guanine, and
- in Formula (III): when Xi and X2 are OH, Bi is not Adenine, B2 is not Guanine and Z is not OH, or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist is selected from the group consisting of:

 and a pharmaceutically acceptable salt thereof.
and a pharmaceutically acceptable salt thereof.
[0005] In some aspects, the cholesterol or derivative thereof comprises:


[0006] In some aspects, the EV described herein comprises:

Valine-Alanine Cholesterol linker
(Formula II),
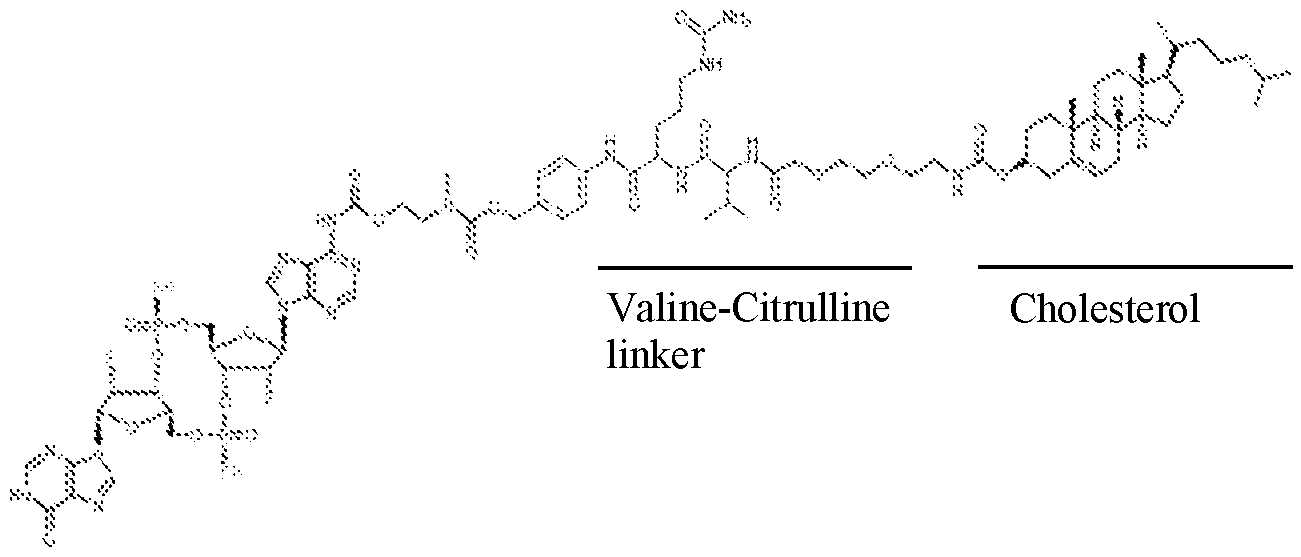 (Forumula III), or
(Forumula III), or
 a ne- ru ne Cholesterol linker
a ne- ru ne Cholesterol linker
(Formula IV).
[0007] In some aspects, the EVs described herein further comprises a scaffold moiety. In some aspects, the scaffold moiety comprises Scaffold X, which comprises an amino acid sequence as set forth in SEQ ID NO: 302. In some aspects, the Scaffold X comprises an amino acid sequence at least 50%, at least 60%, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or about 100% identical to SEQ ID NO:302. In some aspects, the scaffold moiety comprises Scaffold Y, which comprises a BASP1 protein or a functional fragment thereof. In some aspects, Scaffold Y comprises an amino acid sequence as set forth in GGKLSKK (SEQ ID NO: 411). In some aspects, Scaffold Y comprises an amino acid sequence as set forth in i) GGKLSKKK (SEQ ID NO: 438), (ii) GGKLSKKS (SEQ ID NO: 439), (iii) GAKLSKKK (SEQ ID NO: 440), (iv) GAKLSKKS (SEQ ID NO: 441), (v) GGKQSKKK (SEQ ID NO: 442), (vi) GGKQSKKS (SEQ ID NO: 443), (vii) GGKLAKKK (SEQ ID NO: 444), (viii) GGKLAKKS (SEQ ID NO: 445), or (ix) any combination thereof.
[0008] In some aspects, the present disclosure provides a pharmaceutical composition comprising the EV described herein and a pharmaceutically acceptable carrier. In some aspects, the pharmaceutical composition, when administered to a mammal, the composition does not deplete T cells and/or macrophages in the mammal. In some aspects, the pharmaceutical composition, when administered to a mammal, the composition depletes T cells and/or macrophages in the mammal at a lesser degree than the free STING agonist.
[0009] In some aspects, the present disclosure also provides a kit comprising the compositions described herein and instructions for use.
[0010] In some aspects, the present disclosure also provides a method of producing an EV, e.g., exosome, comprising a cholesterol or derivative thereof, which is linked to a STING agonist via a cleavable peptide linker, the method comprising:
(i) Obtaining an EV, e.g., exosome;
(ii) Mixing the EV, e.g., exosome with cholesterol linked to a STING agonist via a cleavable peptide linker in a solution;
(iii) Incubating the mixture of the EV, e.g., exosome and the cholesterol linked to the STING agonist in a solution comprising a buffer under suitable conditions; and
(iv) Purifying the EV, e.g., exosome.
[0011] In some aspects, the cleavable peptide linker comprises a linker cleavable by cathepsin. In some aspects, the cleavable peptide linker comprises a Valine-Alanine linker and/or a Valine- Citrulline linker.
[0012] In some aspects, the present disclosure provides a method of inducing or modulating an immune response and/or an inflammatory response in a subject in need thereof, the method comprising administering to the subject the EVs described herein or the compositions described herein. In some aspects, the present disclosure provides a method of treating a tumor in a subject in need thereof, the method comprising administering to the subject the EVs described herein or the compositions described herein. In some aspects, the administering induces or modulates the immune response and/or the inflammatory response in the subject. In some aspects, the administering activates Dendritic Cells. In some aspects, the administering activates myeloid Dendritic Cells. In some aspects, the administering results in reduced monocyte cell activation compared to the free STING agonist. In some aspects, the administering does not induce monocyte cell activation. In some aspects, the administering induces interferon-P (IFN-P) production. In some aspects, the administering results in reduced systemic inflammation compared to the free STING agonist. In some aspects, the administering results in insubstantial amounts of systemic inflammation. In some aspects, the administration is parenterally, orally, intravenously, intramuscularly, intra-tumorally, intraperitoneally, or via any other appropriate administration route. In some aspects, the administration is intravenous. In some aspects, the immune response is an anti-tumor response. In some aspects, the methods further comprises administering an additional
therapeutic agent. In some aspects, the additional therapeutic agent is an immunomodulating agent. In some aspects, the additional therapeutic agent is an antibody or antigen-binding fragment thereof. In some aspects, the antibody or antigen-binding fragment thereof is an inhibitor of CTLA- 4, PD-1, PD-L1, PD-L2, TIM-3, or LAG3. In some aspects, the administering prevents metastasis of the tumor in the subject.
[0013] In some aspects, the tumor comprises or is derived from a chondroid sarcoma, a cutaneous squamous cell carcinoma (cSCC), a head and neck squamous cell carcinoma of the oral cavity, a hepatocellular cancer (HCC), a cervical cancer, an eye melanoma, a choroidal eye melanoma, a gastric spindle cell sarcoma, a hemangioendothelioma, a mesothelioma, a parotoid gland cancer, a renal cancer, a triple-negative breast cancer (TNBC), or any combination thereof.
[0014] In some aspects, the EV is thermostable at about 20°C for at least about 7 days, at least about 8 days, at least about 9 days, at least about 10 days, at least about 11 days, at least about 12 days, at least about 13 days, at least about 14 days, at least about 15 days, at least about 16 days, at least about 17 days, at least about 18 days, at least about 19 days, at least about 20 days, at least about 21 days, at least about 22 days, at least about 23 days, at least about 24 days, at least about 25 days, at least about 26 days, at least about 27 days, at least about 28 days, at least about 29 days, or at least about 30 days. In some aspects, the EV is thermostable at about 37°C for at least about 7 days, at least about 8 days, at least about 9 days, at least about 10 days, at least about 11 days, at least about 12 days, at least about 13 days, at least about 14 days, at least about 15 days, at least about 16 days, at least about 17 days, at least about 18 days, at least about 19 days, at least about 20 days, at least about 21 days, at least about 22 days, at least about 23 days, at least about 24 days, at least about 25 days, at least about 26 days, at least about 27 days, at least about 28 days, at least about 29 days, or at least about 30 days.
[0015] In some aspects, the EV is capable of inducing an immune response in a subject.
BRIEF DESCRIPTION OF THE DRAWINGS
[0016] FIG. 1A shows Interferon-P expression of cholesterol tagged STING (ECC-STING) exosomes (exoSTING ECC) on hPBMCs in vitro compared to exoSTING and soluble (free) STING. ExoSTING is a mixture of the STING agonist and exosomes (i.e., exosomes loaded with STING agonist, such as by diffusion).
[0017] FIG. IB shows cholesterol tagged STING (ECC-STING) exosomes (exoSTING ECC) retain in vivo activity, as measured by IFN-P expression in spleen, for at least 1 month at 37°C compared to exoSTING. For each day, left bar is exoSTING and right bar is exoSTING-ECC.
[0018] FIGs. 2A-2B are bar graphs illustrating anti-SARS CoV2 RBD IgG titers (FIG. 2A) and anti-SARS CoV2 RBD neutralizing antibodies (FIG. 2B) following subcutaneous administration of exosomes expressing the SARS CoV2 RBD and stably loaded with STING-ECC ("exoRBD/STING-ECC"; FIGs. 2A-2B) or exosomes expressing SARS CoV2 RBD alone ("exoRBD"; FIG. 2A) in mice. Prior to administration, exosomes were stored for 30 days at 37°C or -20°C, as indicated (FIGs. 2A-2B).
[0019] FIGs. 3A-3D show neutralizing antibody production (FIG. 3A), anti-RBD antibody induction (FIG. 3B), and splenocytes expression of IFNy (FIG. 3C) and TNFa (FIG. 3D) in mice subcutaneously administered varying doses of exosomes expressing the RBDs from both SARS CoVl and CoV2 (both fused to PTGFRN) and loaded with cholesterol tagged STING (ECC STING), as indicated.
DETAILED DESCRIPTION OF THE DISCLOSURE
[0020] Before the present invention is described in greater detail, it is to be understood that this invention is not limited to particular aspects described, as such can, of course, vary. It is also to be understood that the terminology used herein is for the purpose of describing particular aspects only, and is not intended to be limiting, since the scope of the present invention will be limited only by the appended claims.
[0021] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. Although any methods and materials similar or equivalent to those described herein can also be used in the practice or testing of the present invention, representative illustrative methods and materials are now described.
[0022] All publications and patents cited in this specification are herein incorporated by reference as if each individual publication or patent were specifically and individually indicated to be incorporated by reference and are incorporated herein by reference to disclose and describe the methods and/or materials in connection with which the publications are cited.
[0023] As will be apparent to those of skill in the art upon reading this disclosure, each of the individual aspects described and illustrated herein has discrete components and features which can be readily separated from or combined with the features of any of the other several aspects without departing from the scope or spirit of the present invention. Any recited method can be carried out in the order of events recited or in any other order which is logically possible.
I. Definitions
[0024] It is noted that, as used herein and in the appended claims, the singular forms "a," "an," and "the" include plural referents unless the context clearly dictates otherwise. As such, the terms "a" (or "an"), "one or more," and "at least one" can be used interchangeably herein. It is further noted that the claims can be drafted to exclude any optional element. As such, this statement is intended to serve as antecedent basis for use of such exclusive terminology as "solely," "only" and the like in connection with the recitation of claim elements, or use of a negative limitation.
[0025] Furthermore, "and/or" where used herein is to be taken as specific disclosure of each of the two specified features or components with or without the other. Thus, the term "and/or" as used in a phrase such as "A and/or B" herein is intended to include "A and B," "A or B," "A" (alone), and "B" (alone). Likewise, the term "and/or" as used in a phrase such as "A, B, and/or C" is intended to encompass each of the following aspects: A, B, and C; A, B, or C; A or C; A or B; B or C; A and C; A and B; B and C; A (alone); B (alone); and C (alone).
[0026] It is understood that wherever aspects are described herein with the language "comprising," otherwise analogous aspects described in terms of "consisting of and/or "consisting essentially of are also provided.
[0027] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this disclosure is related. For example, the Concise Dictionary of Biomedicine and Molecular Biology, Juo, Pei- Show, 2nd ed., 2002, CRC Press; The Dictionary of Cell and Molecular Biology, 3rd ed., 1999, Academic Press; and the Oxford Dictionary Of Biochemistry And Molecular Biology, Revised, 2000, Oxford University Press, provide one of skill with a general dictionary of many of the terms used in this disclosure.
[0028] Units, prefixes, and symbols are denoted in their Systeme International de Unites (SI) accepted form. Numeric ranges are inclusive of the numbers defining the range. Where a range of values is recited, it is to be understood that each intervening integer value, and each fraction thereof, between the recited upper and lower limits of that range is also specifically disclosed, along with each subrange between such values. The upper and lower limits of any range can independently be included in or excluded from the range, and each range where either, neither or both limits are included is also encompassed within the disclosure. Thus, ranges recited herein are understood to be shorthand for all of the values within the range, inclusive of the recited endpoints. For example, a range of 1 to 10 is understood to include any number, combination of numbers, or sub-range from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10.
[0029] Where a value is explicitly recited, it is to be understood that values which are about the same quantity or amount as the recited value are also within the scope of the disclosure. Where a combination is disclosed, each subcombination of the elements of that combination is also specifically disclosed and is within the scope of the disclosure. Conversely, where different elements or groups of elements are individually disclosed, combinations thereof are also disclosed. Where any element of a disclosure is disclosed as having a plurality of alternatives, examples of that disclosure in which each alternative is excluded singly or in any combination with the other alternatives are also hereby disclosed; more than one element of a disclosure can have such exclusions, and all combinations of elements having such exclusions are hereby disclosed.
[0030] Nucleotides are referred to by their commonly accepted single-letter codes. Unless otherwise indicated, nucleotide sequences are written left to right in 5' to 3' orientation. Nucleotides are referred to herein by their commonly known one-letter symbols recommended by the IUPAC- IUB Biochemical Nomenclature Commission. Accordingly, A represents adenine, C represents cytosine, G represents guanine, T represents thymine, and U represents uracil.
[0031] Amino acid sequences are written left to right in amino to carboxy orientation. Amino acids are referred to herein by either their commonly known three letter symbols or by the one- letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission.
[0032] The term "about" or "approximately" is used herein to mean approximately roughly, around, or in the region of. When the term "about" is used in conjunction with a numerical range, it modifies that range by extending the boundaries above and below the numerical values set forth. The term used herein means up to 10% of the referenced amount, e.g., about 50% is understood to encompass a range of values from 45% to 55%. In some aspects, the term means more than or less than 10% of the reference amount, e.g., ± 9%, ± 8%, ± 7%, ± 6%, ± 5%, ± 4%, ± 3%, ± 2%, ± 1%, or ± 0.1%.
[0033] As used herein, the term "extracellular vesicle" or "EV" refers to a cell-derived vesicle comprising a membrane that encloses an internal space. Extracellular vesicles comprise all membrane-bound vesicles e.g., exosomes, nanovesicles) that have a smaller diameter than the cell from which they are derived. Generally extracellular vesicles range in diameter from 20 nm to 1000 nm, and can comprise various macromolecular payload either within the internal space (i.e., lumen), displayed on the external surface of the extracellular vesicle, and/or spanning the membrane. Said payload can comprise nucleic acids, proteins, carbohydrates, lipids, small molecules, and/or combinations thereof. In some aspects, an extracellular vesicle comprises a scaffold moiety. By way of example and without limitation, extracellular vesicles include apoptotic
bodies, fragments of cells, vesicles derived from cells by direct or indirect manipulation (e.g., by serial extrusion or treatment with alkaline solutions), vesiculated organelles, and vesicles produced by living cells (e.g., by direct plasma membrane budding or fusion of the late endosome with the plasma membrane). Extracellular vesicles can be derived from a living or dead organism, explanted tissues or organs, prokaryotic or eukaryotic cells, and/or cultured cells. In some aspects, extracellular vesicles are produced by cells that express one or more transgene products.
[0034] As used herein the term "exosome" refers to a cell-derived small (between 20-300 nm in diameter, more preferably 40-200 nm in diameter) vesicle comprising a membrane that encloses an internal space (i.e., lumen), and which is generated from said cell by direct plasma membrane budding or by fusion of the late endosome with the plasma membrane. The exosome is a species of extracellular vesicle. The exosome comprises lipid or fatty acid and polypeptide and optionally comprises a payload (e.g., a therapeutic agent), a receiver (e.g., a targeting moiety), a polynucleotide (e.g., a nucleic acid, RNA, or DNA), a sugar (e.g., a simple sugar, polysaccharide, or glycan) or other molecules. In some aspects, an exosome comprises a scaffold moiety. The exosome can be derived from a producer cell, and isolated from the producer cell based on its size, density, biochemical parameters, or a combination thereof. In some aspects, the exosomes of the present disclosure are produced by cells that express one or more transgene products.
[0035] As used herein, the term "nanovesicle" refers to a cell-derived small (between 20-250 nm in diameter, more preferably 30-150 nm in diameter) vesicle comprising a membrane that encloses an internal space, and which is generated from said cell by direct or indirect manipulation such that said nanovesicle would not be produced by said producer cell without said manipulation. Appropriate manipulations of said producer cell include but are not limited to serial extrusion, treatment with alkaline solutions, sonication, or combinations thereof. The production of nanovesicles may, in some instances, result in the destruction of said producer cell. Preferably, populations of nanovesicles are substantially free of vesicles that are derived from producer cells by way of direct budding from the plasma membrane or fusion of the late endosome with the plasma membrane. The nanovesicle comprises lipid or fatty acid and polypeptide, and optionally comprises a payload (e.g., a therapeutic agent), a receiver (e.g., a targeting moiety), a polynucleotide (e.g., a nucleic acid, RNA, or DNA), a sugar (e.g., a simple sugar, polysaccharide, or glycan) or other molecules. In some aspects, a nanovesicle comprises a scaffold moiety. The nanovesicle, once it is derived from a producer cell according to said manipulation, may be isolated from the producer cell based on its size, density, biochemical parameters, or a combination thereof.
[0036] The term "modified," when used in the context of exosomes described herein, refers to an alteration or engineering of an EV, such that the modified EV is different from a naturally- occurring EV. In some aspects, a modified EV described herein comprises a membrane that differs in composition of a protein, a lipid, a small molecular, a carbohydrate, etc. compared to the membrane of a naturally-occurring EV (e.g., membrane comprises higher density or number of natural EV proteins and/or membrane comprises proteins that are not naturally found in EVs. In certain aspects, such modifications to the membrane changes the exterior surface of the EV. In certain aspects, such modifications to the membrane changes the lumen of the EV.
[0037] As used herein, the term "scaffold moiety" refers to a molecule that can be used to anchor STING agonists disclosed herein or any other compound of interest (e.g., payload) to the EV either on the luminal surface or on the exterior surface of the EV. In certain aspects, a scaffold moiety comprises a synthetic molecule. In some aspects, a scaffold moiety comprises a nonpolypeptide moiety. In other aspects, a scaffold moiety comprises a lipid, carbohydrate, or protein that naturally exists in the EV. In some aspects, a scaffold moiety comprises a lipid, carbohydrate, or protein that does not naturally exist in the exosome. In certain aspects, a scaffold moiety is Scaffold X. In some aspects, a scaffold moiety is Scaffold Y. In further aspects, a scaffold moiety comprises both Scaffold X and Scaffold Y.
[0038] As used herein, the term "Scaffold X" refers to exosome proteins that have recently been identified on the surface of exosomes. See, e.g., U.S. Pat. No. 10,195,290, which is incorporated herein by reference in its entirety. Non-limiting examples of Scaffold X proteins include: prostaglandin F2 receptor negative regulator ("the PTGFRN protein"); basigin ("the BSG protein"); immunoglobulin superfamily member 2 ("the IGSF2 protein"); immunoglobulin superfamily member 3 ("the IGSF3 protein"); immunoglobulin superfamily member 8 ("the IGSF8 protein"); integrin beta-1 ("the ITGB1 protein); integrin alpha-4 ("the ITGA4 protein"); 4F2 cellsurface antigen heavy chain ("the SLC3 A2 protein"); and a class of ATP transporter proteins ("the ATP1A1 protein," "the ATP1A2 protein," "the ATP1A3 protein," "the ATP1A4 protein," "the ATP1B3 protein," "the ATP2B1 protein," "the ATP2B2 protein," "the ATP2B3 protein," "the ATP2B protein"). In some aspects, a Scaffold X protein can be a whole protein or a fragment thereof (e.g, functional fragment, e.g., the smallest fragment that is capable of anchoring another moiety on the exterior surface or on the luminal surface of the EV, e.g., exosome,). In some aspects, a Scaffold X can anchor a moiety (e.g, STING agonist) to the external surface or the luminal surface of the EVs, e.g., exosomes,.
[0039] As used herein, the term "Scaffold Y" refers to exosome proteins that were newly identified within the luminal surface of exosomes. See, e.g., International Appl. No. PCT/US2018/061679, which is incorporated herein by reference in its entirety. Non-limiting examples of Scaffold Y proteins include: myristoylated alanine rich Protein Kinase C substrate ("the MARCKS protein"); myristoylated alanine rich Protein Kinase C substrate like 1 ("the MARCKSL1 protein"); and brain acid soluble protein 1 ("the BASP1 protein"). In some aspects, a Scaffold Y protein can be a whole protein or a fragment thereof (e.g., functional fragment, e.g., the smallest fragment that is capable of anchoring a moiety on the luminal surface of the EVs, e.g., exosomes,). In some aspects, a Scaffold Y can anchor a moiety (e.g., STING agonist) to the lumen of the EVs, e.g., exosomes,.
[0040] As used herein, the term "fragment" of a protein (e.g., therapeutic protein, Scaffold X, or Scaffold Y) refers to an amino acid sequence of a protein that is shorter than the naturally- occurring sequence, N- and/or C-terminally deleted or any part of the protein deleted in comparison to the naturally occurring protein. As used herein, the term "functional fragment" refers to a protein fragment that retains protein function. Accordingly, in some aspects, a functional fragment of a Scaffold X protein retains the ability to anchor a moiety on the luminal surface and/or on the exterior surface of the EV. Similarly, in certain aspects, a functional fragment of a Scaffold Y protein retains the ability to anchor a moiety on the luminal surface of the EV. Whether a fragment is a functional fragment can be assessed by any art known methods to determine the protein content of EVs including Western Blots, FACS analysis and fusions of the fragments with autofluorescent proteins like, e.g., GFP. In certain aspects, a functional fragment of a Scaffold X protein retains at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90% or at least about 100% of the ability, e.g., an ability to anchor a moiety, of the naturally occurring Scaffold X protein. In some aspects, a functional fragment of a Scaffold Y protein retains at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90% or at least about 100% of the ability, e.g., an ability to anchor another molecule, of the naturally occurring Scaffold Y protein.
[0041] As used herein, the term "variant" of a molecule (e.g., functional molecule, antigen, Scaffold X and/or Scaffold Y) refers to a molecule that shares certain structural and functional identities with another molecule upon comparison by a method known in the art. For example, a variant of a protein can include a substitution, insertion, deletion, frameshift or rearrangement in another protein.
[0042] A "conservative amino acid substitution" is one in which the amino acid residue is replaced with an amino acid residue having a similar side chain. Families of amino acid residues having similar side chains have been defined in the art, including basic side chains (e.g., lysine, arginine, histidine), acidic side chains (e.g., aspartic acid, glutamic acid), uncharged polar side chains (e.g., glycine, asparagine, glutamine, serine, threonine, tyrosine, cysteine), nonpolar side chains (e.g., alanine, valine, leucine, isoleucine, proline, phenylalanine, methionine, tryptophan), beta-branched side chains (e.g., threonine, valine, isoleucine) and aromatic side chains (e.g., tyrosine, phenylalanine, tryptophan, histidine). Thus, if an amino acid in a polypeptide is replaced with another amino acid from the same side chain family, the substitution is considered to be conservative. In another aspect, a string of amino acids can be conservatively replaced with a structurally similar string that differs in order and/or composition of side chain family members.
[0043] The term "percent sequence identity" or "percent identity" between two polynucleotide or polypeptide sequences refers to the number of identical matched positions shared by the sequences over a comparison window, taking into account additions or deletions (i.e., gaps) that must be introduced for optimal alignment of the two sequences. A matched position is any position where an identical nucleotide or amino acid is presented in both the target and reference sequence. Gaps presented in the target sequence are not counted since gaps are not nucleotides or amino acids. Likewise, gaps presented in the reference sequence are not counted since target sequence nucleotides or amino acids are counted, not nucleotides or amino acids from the reference sequence.
[0044] The percentage of sequence identity is calculated by determining the number of positions at which the identical amino-acid residue or nucleic acid base occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison and multiplying the result by 100 to yield the percentage of sequence identity. The comparison of sequences and determination of percent sequence identity between two sequences may be accomplished using readily available software both for online use and for download. Suitable software programs are available from various sources, and for alignment of both protein and nucleotide sequences. One suitable program to determine percent sequence identity is bl2seq, part of the BLAST suite of programs available from the U.S. government's National Center for Biotechnology Information BLAST web site (blast.ncbi.nlm.nih.gov). B12seq performs a comparison between two sequences using either the BLASTN or BLASTP algorithm. BLASTN is used to compare nucleic acid sequences, while BLASTP is used to compare amino acid sequences. Other suitable programs are, e.g., Needle,
Stretcher, Water, or Matcher, part of the EMBOSS suite of bioinformatics programs and also available from the European Bioinformatics Institute (EBI) at www.ebi.ac.uk/Tools/psa. Different regions within a single polynucleotide or polypeptide target sequence that aligns with a polynucleotide or polypeptide reference sequence can each have their own percent sequence identity. It is noted that the percent sequence identity value is rounded to the nearest tenth. For example, 80.11, 80.12, 80.13, and 80.14 are rounded down to 80.1, while 80.15, 80.16, 80.17, 80.18, and 80.19 are rounded up to 80.2. It also is noted that the length value will always be an integer.
[0045] One skilled in the art will appreciate that the generation of a sequence alignment for the calculation of a percent sequence identity is not limited to binary sequence-sequence comparisons exclusively driven by primary sequence data. Sequence alignments can be derived from multiple sequence alignments. One suitable program to generate multiple sequence alignments is ClustalW2, available from www.clustal.org. Another suitable program is MUSCLE, available from www.drive5.com/muscle/. ClustalW2 and MUSCLE are alternatively available, e.g., from the EBI. [0046] It will also be appreciated that sequence alignments can be generated by integrating sequence data with data from heterogeneous sources such as structural data (e.g., crystallographic protein structures), functional data (e.g., location of mutations), or phylogenetic data. A suitable program that integrates heterogeneous data to generate a multiple sequence alignment is T-Coffee, available at www.tcoffee.org, and alternatively available, e.g., from the EBI. It will also be appreciated that the final alignment used to calculate percent sequence identity may be curated either automatically or manually.
[0047] The polynucleotide variants can contain alterations in the coding regions, non-coding regions, or both. In some aspects, the polynucleotide variants contain alterations which produce silent substitutions, additions, or deletions, but do not alter the properties or activities of the encoded polypeptide. In some aspects, nucleotide variants are produced by silent substitutions due to the degeneracy of the genetic code. In other aspects, variants in which 5-10, 1-5, or 1-2 amino acids are substituted, deleted, or added in any combination. Polynucleotide variants can be produced for a variety of reasons, e.g., to optimize codon expression for a particular host (change codons in the human mRNA to others, e.g., a bacterial host such as E. colt).
[0048] Naturally occurring variants are called "allelic variants," and refer to one of several alternate forms of a gene occupying a given locus on a chromosome of an organism (Genes II, Lewin, B., ed., John Wiley & Sons, New York (1985)). These allelic variants can vary at either the
polynucleotide and/or polypeptide level and are included in the present disclosure. Alternatively, non-naturally occurring variants can be produced by mutagenesis techniques or by direct synthesis. [0049] Using known methods of protein engineering and recombinant DNA technology, variants can be generated to improve or alter the characteristics of the polypeptides. For instance, one or more amino acids can be deleted from the N-terminus or C-terminus of the secreted protein without substantial loss of biological function. Ron et al.. J. Biol. Chem. 268: 2984-2988 (1993), incorporated herein by reference in its entirety, reported variant KGF proteins having heparin binding activity even after deleting 3, 8, or 27 amino-terminal amino acid residues. Similarly, interferon gamma exhibited up to ten times higher activity after deleting 8-10 amino acid residues from the carboxy terminus of this protein. (Dobeli et al, J. Biotechnology 7: 199-216 (1988), incorporated herein by reference in its entirety.)
[0050] Moreover, ample evidence demonstrates that variants often retain a biological activity similar to that of the naturally occurring protein. For example, Gayle and coworkers (J. Biol. Chem 265:22105-22111 (1993), incorporated herein by reference in its entirety) conducted extensive mutational analysis of human cytokine IL- la. They used random mutagenesis to generate over 3,500 individual IL-la mutants that averaged 2.5 amino acid changes per variant over the entire length of the molecule. Multiple mutations were examined at every possible amino acid position. The investigators found that "[m]ost of the molecule could be altered with little effect on either [binding or biological activity]." (See Abstract.) In fact, only 23 unique amino acid sequences, out of more than 3,500 nucleotide sequences examined, produced a protein that significantly differed in activity from wild-type.
[0051] As stated above, polypeptide variants include, e.g., modified polypeptides. Modifications include, e.g., acetylation, acylation, ADP-ribosylation, amidation, covalent attachment of flavin, covalent attachment of a heme moiety, covalent attachment of a nucleotide or nucleotide derivative, covalent attachment of a lipid or lipid derivative, covalent attachment of phosphotidylinositol, cross-linking, cyclization, disulfide bond formation, demethylation, formation of covalent cross-links, formation of cysteine, formation of pyroglutamate, formylation, gamma-carboxylation, glycosylation, GPI anchor formation, hydroxylation, iodination, methylation, myristoylation, oxidation, pegylation (Mei et al., Blood 116:21Q-'19 (2010), which is incorporated herein by reference in its entirety), proteolytic processing, phosphorylation, prenylation, racemization, sei enoyl ati on, sulfation, transfer-RNA mediated addition of amino acids to proteins such as arginylation, and ubiquitination. In some aspects, Scaffold X and/or Scaffold Y is modified at any convenient location.
[0052] As used herein the term "producer cell" refers to a cell used for generating an EV. A producer cell can be a cell cultured in vitro, or a cell in vivo. A producer cell includes, but not limited to, a cell known to be effective in generating EVs, e.g., exosomes, e.g., HEK293 cells, Chinese hamster ovary (CHO) cells, mesenchymal stem cells (MSCs), BJ human foreskin fibroblast cells, s9f cells, fHDF fibroblast cells, AGE.HN® neuronal precursor cells, CAP® amniocyte cells, adipose mesenchymal stem cells, and RPTEC/TERT1 cells. In certain aspects, a producer cell is an antigen-presenting cell. In some aspects, the producer cell is a bacterial cell. In some aspects, a producer cell is a dendritic cell, a B cell, a mast cell, a macrophage, a neutrophil, a Kupffer-Browicz cell, or a cell derived from any of these cells, or any combination thereof. In some aspects, the producer cell is not a bacterial cell. In other aspects, the producer cell is not an antigen-presenting cell.
[0053] As used herein the term "linked to" or "conjugated to" are used interchangeably and refer to a covalent or non-covalent bond formed between a first moiety and a second moiety, e.g., a STING agonist and an extracellular vesicle, respectively, e.g., a scaffold moiety expressed in or on the extracellular vesicle and a STING agonist, e.g., Scaffold X (e.g., a PTGFRN protein), respectively, on the luminal surface of or on the external surface of the extracellular vesicle.
[0054] As used herein, the terms "isolate," "isolated," and "isolating" or "purify," "purified," and "purifying" as well as "extracted" and "extracting" are used interchangeably and refer to the state of a preparation (e.g., a plurality of known or unknown amount and/or concentration) of desired EVs, that have undergone one or more processes of purification, e.g., a selection or an enrichment of the desired EV preparation. In some aspects, isolating or purifying as used herein is the process of removing, partially removing (e.g., a fraction) of the EVs from a sample containing producer cells. In some aspects, an isolated EV composition has no detectable undesired activity or, alternatively, the level or amount of the undesired activity is at or below an acceptable level or amount. In some aspects, an isolated EV composition has an amount and/or concentration of desired EVs at or above an acceptable amount and/or concentration. In some aspects, the isolated EV composition is enriched as compared to the starting material (e.g., producer cell preparations) from which the composition is obtained. This enrichment can be by 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 96%, 97%, 98%, 99%, 99.9%, 99.99%, 99.999%, 99.9999%, or greater than 99.9999% as compared to the starting material. In some aspects, isolated EV preparations are substantially free of residual biological products. In some aspects, the isolated EV preparations are 100% free, 99% free, 98% free, 97% free, 96% free, 95% free, 94% free, 93% free, 92% free, 91% free, or 90% free of any contaminating biological matter. Residual biological
products can include abiotic materials (including chemicals) or unwanted nucleic acids, proteins, lipids, or metabolites. Substantially free of residual biological products can also mean that the EV composition contains no detectable producer cells and that only EVs are detectable.
[0055] As used herein, the term "agonist" refers to a molecule that binds to a receptor and activates the receptor to produce a biological response. Receptors can be activated by either an endogenous or an exogenous agonist. Non-limiting examples of endogenous agonist include hormones, neurotransmitters, and cyclic dinucleotides. Non-limiting examples of exogenous agonist include drugs, small molecules, and cyclic dinucleotides. The agonist can be a full, partial, or inverse agonist.
[0056] As used herein, the term “antagonist” refers to a molecule that blocks or dampens an agonist mediated response rather than provoking a biological response itself upon bind to a receptor. Many antagonists achieve their potency by competing with endogenous ligands or substrates at structurally defined binding sites on the receptors. Non-limiting examples of antagonists include alpha blockers, beta-blocker, and calcium channel blockers. The antagonist can be a competitive, non-competitive, or uncompetitive antagonist.
[0057] The term "free STING agonist" as used herein means a STING agonist not associated with an extracellular vesicle, but otherwise identical to the STING agonist associated with the extracellular vesicle. Especially when compared to an extracellular vesicle associated with a STING agonist, the free STING agonist is the same STING agonist associated with the extracellular vesicle. In some aspects, when a free STING agonist is compared to an extracellular vesicle comprising the STING agonist in its efficacy, toxicity, and/or any other characteristics, the amount of the free STING agonist compared to the STING agonist associated with the extracellular vesicle is the same as the amount of the STING agonist associated with the EV.
[0058] As used herein, the term “ligand” refers to a molecule that binds to a receptor and modulates the receptor to produce a biological response. Modulation can be activation, deactivation, blocking, or damping of the biological response mediated by the receptor. Receptors can be modulated by either an endogenous or an exogenous ligand. Non-limiting examples of endogenous ligands include antibodies and peptides. Non-limiting examples of exogenous agonist include drugs, small molecules, and cyclic dinucleotides. The ligand can be a full, partial, or inverse ligand.
[0059] As used herein, the term “antibody” encompasses an immunoglobulin whether natural or partly or wholly synthetically produced, and fragments thereof. The term also covers any protein having a binding domain that is homologous to an immunoglobulin binding domain. “Antibody”
further includes a polypeptide comprising a framework region from an immunoglobulin gene or fragments thereof that specifically binds and recognizes an antigen. Use of the term antibody is meant to include whole antibodies, polyclonal, monoclonal and recombinant antibodies, fragments thereof, and further includes single-chain antibodies, humanized antibodies, murine antibodies, chimeric, mouse-human, mouse-primate, primate-human monoclonal antibodies, anti-idiotype antibodies, antibody fragments, such as, e.g., scFv, (scFv)2, Fab, Fab', and F(ab')2, F(abl)2, Fv, dAb, and Fd fragments, diabodies, and antibody-related polypeptides. Antibody includes bispecific antibodies and multispecific antibodies so long as they exhibit the desired biological activity or function.
[0060] As used herein the term “therapeutically effective amount” is the amount of reagent or pharmaceutical compound that is sufficient to a produce a desired therapeutic effect, pharmacologic and/or physiologic effect on a subject in need thereof. A therapeutically effective amount can be a “prophylactically effective amount” as prophylaxis can be considered therapy.
[0061] As used herein, the term “pharmaceutical composition” refers to one or more of the compounds described herein, such as, e.g., an EV mixed or intermingled with, or suspended in one or more other chemical components, such as pharmaceutically-acceptable carriers and excipients. One purpose of a pharmaceutical composition is to facilitate administration of preparations of EVs to a subject. The term “excipient” or “carrier” refers to an inert substance added to a pharmaceutical composition to further facilitate administration of a compound. The term “pharmaceutically-acceptable carrier” or “pharmaceutically-acceptable excipient” and grammatical variations thereof, encompasses any of the agents approved by a regulatory agency of the US Federal government or listed in the US Pharmacopeia for use in animals, including humans, as well as any carrier or diluent that does not cause the production of undesirable physiological effects to a degree that prohibits administration of the composition to a subject and does not abrogate the biological activity and properties of the administered compound. Included are excipients and carriers that are useful in preparing a pharmaceutical composition and are generally safe, non-toxic, and desirable.
[0062] The terms “administration,” “administering” and variants thereof refer to introducing a composition, such as an EV, or agent into a subject and includes concurrent and sequential introduction of a composition or agent. The introduction of a composition or agent into a subject is by any suitable route, including intratumorally, orally (e.g., sublingually), pulmonarily, intranasally, parenterally (intravenously, intra-arterially, intramuscularly, intraperitoneally, or subcutaneously), rectally, intralymphatically, intrathecally, periocularly, topically, intradermally,
or transdermally. Administration includes self-administration and the administration by another. A suitable route of administration allows the composition or the agent to perform its intended function. For example, if a suitable route is intravenous, the composition is administered by introducing the composition or agent into a vein of the subject. In some aspects, the administering comprises the use of a device, e.g., a pump or a patch.
[0063] The term "treat," "treatment," or "treating," as used herein refers to, e.g., the reduction in severity of a disease or condition; the reduction in the duration of a disease course; the amelioration or elimination of one or more symptoms associated with a disease or condition; the provision of beneficial effects to a subject with a disease or condition, without necessarily curing the disease or condition. The term also include prophylaxis or prevention of a disease or condition or its symptoms thereof. In one aspect, the term "treating" or "treatment" means inducing an immune response in a subject against an antigen.
[0064] The term "prevent" or "preventing," as used herein, refers to decreasing or reducing the occurrence or severity of a particular outcome. In some aspects, preventing an outcome is achieved through prophylactic treatment.
[0065] As used herein, the term “modulate,” “modulating”, “modify,” and/or “modulator” generally refers to the ability to alter, by increase or decrease, e.g., directly or indirectly promoting/stimulating/up-regulating or interfering with/inhibiting/down-regulating a specific concentration, level, expression, function or behavior, such as, e.g., to act as an antagonist or agonist. In some instances a modulator can increase and/or decrease a certain concentration, level, activity or function relative to a control, or relative to the average level of activity that would generally be expected or relative to a control level of activity.
[0066] As used herein, “a mammalian subject” includes all mammals, including without limitation, humans, domestic animals (e.g., dogs, cats and the like), farm animals (e.g., cows, sheep, pigs, horses and the like) and laboratory animals (e.g., monkey, rats, mice, rabbits, guinea pigs and the like).
[0067] The terms “individual,” “subject,” “host,” and “patient,” are used interchangeably herein and refer to any mammalian subject for whom diagnosis, treatment, or therapy is desired, particularly humans. The methods described herein are applicable to both human therapy and veterinary applications. In some aspects, the subject is a mammal, and in other aspects the subject is a human.
[0068] As used herein, the term “substantially free” means that the sample comprising EVs comprise less than 10% of macromolecules by mass/volume (m/v) percentage concentration.
Some fractions may contain less than 0.001%, less than 0.01%, less than 0.05%, less than 0.1%, less than 0.2%, less than 0.3 %, less than 0.4%, less than 0.5%, less than 0.6%, less than 0.7%, less than 0.8%, less than 0.9%, less than 1%, less than 2%, less than 3%, less than 4%, less than 5%, less than 6%, less than 7%, less than 8%, less than 9%, or less than 10% (m/v) of macromolecules. [0069] As used herein, the term “macromolecule” means nucleic acids, exogenous proteins, lipids, carbohydrates, metabolites, or a combination thereof.
[0070] As used herein, the term “insubstantial,” “reduced,” or “negligible” refers to the presence, level, or amount of an inflammation response in a subject after administration of the composition comprising EVs comprising a cholesterol moiety linked to a STING agonist via a cleavable peptide linker, e.g., Valine- Alanine linker or Valine-Citrulline linker, relative to the baseline inflammation response in the subject or compared to the subject inflammation response to the administration of a free STING agonist. For example, a negligible or insubstantial presence, level or amount of systemic inflammation may be less than 0.001%, less than 0.01%, less than 0.1%, less than 0.2%, less than 0.3 %, less than 0.4%, less than 0.5%, less than 0.6%, less than 0.7%, less than 0.8%, less than 0.9%, less than 1%, less than 2%, less than 3%, less than 4%, less than 5%, less than 6%, less than 7%, less than 8%, less than 9%, less than 10%, less than 12%, less than 15%, less than 17%, less than 20%, or less than 25% of systemic inflammation as relative to the baseline inflammation in the subject or compared to the subject immune response to the administration of a free STING agonist. A level or amount of a systemic inflammation may be less than 0.1-fold, less than 0.5-fold, less than 0.5-fold, less than 1-fold, less than 1.5-fold, less than 2- fold relative to the baseline or compared to the inflammation response to the administration of a free STING agonist.
[0071] Ranges recited herein are understood to be shorthand for all of the values within the range, inclusive of the recited endpoints. For example, a range of 1 to 50 is understood to include any number, combination of numbers, or sub-range from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, and 50.
[0072] Unless otherwise indicated, reference to a compound that has one or more stereocenters intends each stereoisomer, and all combinations of stereoisomers, thereof.
II. Extracellular Vesicles with STING Agonist
[0073] Present disclosure comprises EVs comprising a cholesterol moiety linked to a STING agonist via a cleavable peptide linker e.g., Valine- Alanine or Valine-Citrulline (“cholesterol tagged
STING agonist”). In some aspects, the cholesterol moiety is embedded into the surface of the EVs.
In some aspects, the cholesterol moiety comprises a cholesterol or a derivative thereof.
[0074] In some aspects, the cholesterol moiety comprises:
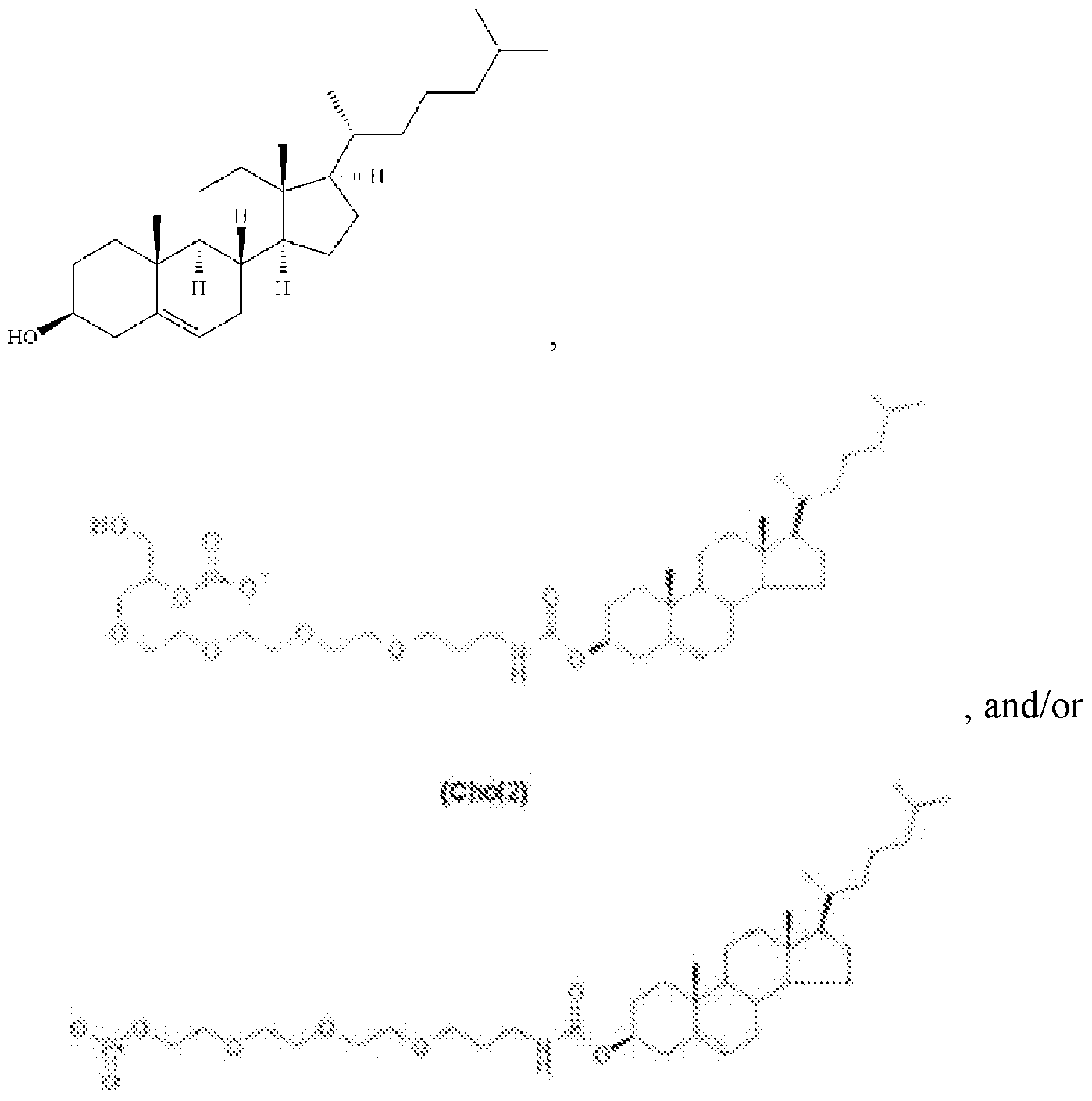
[0075] In some aspects, the EV is an exosome, a nanovesicle, an apoptotic body, a microvesicle, a lysosome, an endosome, a liposome, a lipid nanoparticle, a micelle, a multilamellar structure, a revesiculated vesicle, or an extruded cell. In some aspects, the EV comprises an exosome.
[0076] In some aspects, the EV overexpresses a PTGFRN protein. In other aspects, the EV is produced by a cell that overexpresses a PTGFRN protein.
[0077] In some aspects, the extracellular vesicle further comprises a therapeutic agent, e.g., a ligand, a cytokine, an antigen, or an antibody. In some aspects, the antibody comprises an antagonistic antibody and/or an agonistic antibody.
[0078] In some aspects, the STING agonist is a cyclic dinucleotide. In some aspects, the STING agonist is a non-cyclic dinucleotide. Cyclic dinucleotides (CDNs) were first identified as bacterial signaling molecules characterized by two 3’, 5’ phosphodiester bonds, such as in the molecule c-di-GMP. While STING can be activated by bacterial CDNs, the innate immune
response in mammalian cells is also mediated by the CDN signaling molecule cGAMP which is produced by cGAS. cGAMP is characterized by a mixed 2’, 5’ and 3’, 5’ phosphodiester linkage. Both bacterial and mammalian CDNs directly interact with STING to induce the pro-inflammatory signaling cascade that results in the production of type I IFNs, such as IFNa and IFN-p.
[0079] In some aspects, the linker is conjugated into the phosphate backbone of a STING agonist, e.g., a cyclic dinucleotide (CDN). In some aspects, the linker is conjugated to the base of a CDN (STING agonist).
[0080] In some aspects, the EV comprises:
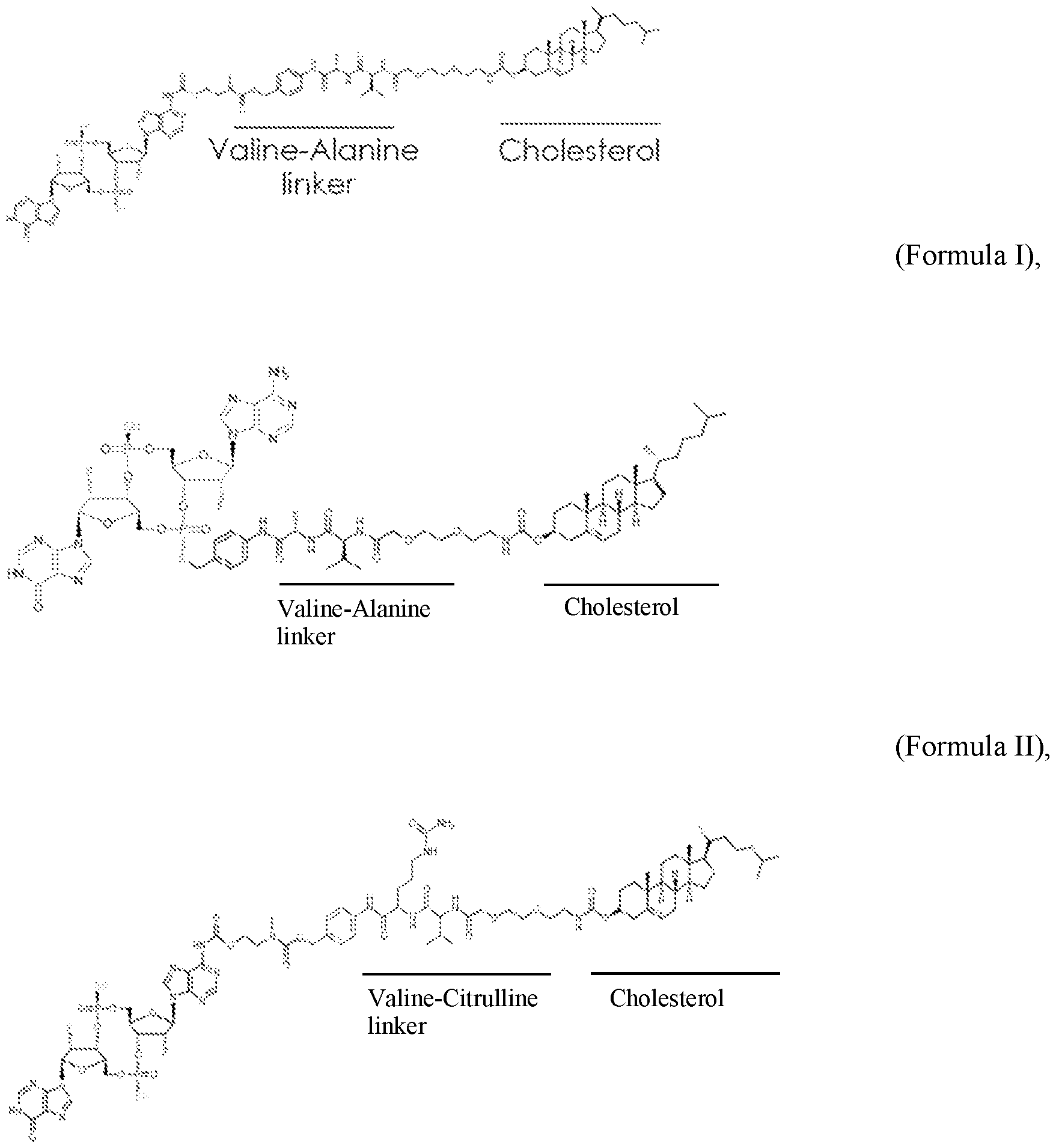
(Formula III), or
Hi
 u Cholesterol (Formula IV). linker
u Cholesterol (Formula IV). linker
[0081] In some aspects, the EV is thermostable at about 20°C for at least about 7 days, at least about 8 days, at least about 9 days, at least about 10 days, at least about 11 days, at least about 12 days, at least about 13 days, at least about 14 days, at least about 15 days, at least about 16 days, at least about 17 days, at least about 18 days, at least about 19 days, at least about 20 days, at least about 21 days, at least about 22 days, at least about 23 days, at least about 24 days, at least about 25 days, at least about 26 days, at least about 27 days, at least about 28 days, at least about 29 days, at least about 30 days, at least about 35 days, at least about 40 days, at least about 45 days, at least about 50 days, at least about 55 days, at least about 60 days, at least about 65 days, at least about 70 days, at least about 75 days, at least about 80 days, at least about 85 days, at least about 90 days, at least about 95 days, or at least about 100 days. In some aspects, the EV is thermostable at about 20°C for at least about 30 days. In some aspects, the EV is thermostable at about 37°C for at least about 7 days, at least about 8 days, at least about 9 days, at least about 10 days, at least about 11 days, at least about 12 days, at least about 13 days, at least about 14 days, at least about 15 days, at least about 16 days, at least about 17 days, at least about 18 days, at least about 19 days, at least about 20 days, at least about 21 days, at least about 22 days, at least about 23 days, at least about 24 days, at least about 25 days, at least about 26 days, at least about 27 days, at least about 28 days, at least about 29 days, at least about 30 days, at least about 35 days, at least about 40 days, at least about 45 days, at least about 50 days, at least about 55 days, at least about 60 days, at least about 65 days, at least about 70 days, at least about 75 days, at least about 80 days, at least about 85 days, at least about 90 days, at least about 95 days, or at least about 100 days. In some aspects, the EV is thermostable at about 37°C for at least about 30 days.
ILA. STING Agonists
[0082] STING agonists used in this disclosure can be cyclic dinucleotides (CDNs) or non- cyclic dinucleotide agonists. Cyclic purine dinucleotides such as, but not limited to, cGMP, cyclic di-GMP (c-di-GMP), cAMP, cyclic di-AMP (c-di-AMP), cyclic-GMP-AMP (cGAMP), cyclic di-
IMP (c-di-IMP), cyclic AMP-IMP (cAIMP), and any analogue thereof, are known to stimulate or enhance an immune or inflammation response in a patient. The CDNs may have 2’2’, 2’3’, 2’5’, 3’3’, or 3’5’ bonds linking the cyclic dinucleotides, or any combination thereof.
[0083] Cyclic purine dinucleotides may be modified via standard organic chemistry techniques to produce analogues of purine dinucleotides. Suitable purine dinucleotides include, but are not limited to, adenine, guanine, inosine, hypoxanthine, xanthine, isoguanine, or any other appropriate purine dinucleotide known in the art. The cyclic dinucleotides may be modified analogues. Any suitable modification known in the art may be used, including, but not limited to, phosphorothioate, biphosphorothioate, fluorinate, and difluorinate modifications.
[0084] Non cyclic dinucleotide agonists may also be used, such as 5,6-Dimethylxanthenone- 4-acetic acid (DMXAA), or any other non-cyclic dinucleotide agonist known in the art.
[0085] It is contemplated that any STING agonist may be used. Among the STING agonists are DMXAA, STING agonist-1, ML RR-S2 CDA, ML RR-S2c-di-GMP, ML-RR-S2 cGAMP, 2’3’-c-di-AM(PS)2, 2’3’-cGAMP, 2’3’-cGAMPdFHS, 3'3'-cGAMP, 3'3'-cGAMPdFSH, cAIMP, cAIM(PS)2, 3’3’-cAIMP, 3’3’-cAIMPdFSH, 2’2’-cGAMP, 2’3’-cGAM(PS)2, 3'3'-cGAMP, c-di- AMP, 2'3 '-c-di-AMP, 2’3’-c-di-AM(PS)2, c-di-GMP, 2’3’-c-di-GMP, c-di-IMP, c-di-UMP or any combination thereof. In a preferred aspect, the STING agonist is 3’3’-cAIMPdFSH, alternatively named 3-3 cAIMPdFSH. Additional STING agonists known in the art may also be used.
[0086] In some aspects, the STING agonist useful for the present disclosure comprises a compound having the following formula:
Formula 1 Formula 2 Formula 3

Xi is H, OH, or F;
X2 is H, OH, or F;
Z is OH, ORi, SH or SRi, wherein:
i) Ri is Na or NH4, or ii) Ri is an enzyme-labile group which provides OH or SH in vivo such as pivaloyloxymethyl;
Bi and B2 are bases chosen from:

Guanine,
With proviso that:
- in Formula (I): Xi and X2 are not OH,
- in Formula (II): when Xi and X2 are OH, Bi is not Adenine and B2 is not Guanine, and
- in Formula (III): when Xi and X2 are OH, Bi is not Adenine, B2 is not Guanine and Z is not OH. See WO 2016/096174, the content of which is incorporated herein by reference in its entirety.
[0087] In some aspects, the STING agonist useful for the present disclosure comprises:

CL-656 c-[2FdAMP-2FdIMP], c-[2FdAMP(S)-2FdIMP(S)],

CL-797 CL-655 c-[2FdAMP(S)-IMP], c-[AMP(S)-IMP(S)],

CL-659 c-[2FdAMP(S)-2FdIMP(S)](POM)2 and a pharmaceutically acceptable salt thereof. See WO 2016/096174A1.
[0088] In other aspects, the STING agonist useful for the present disclosure comprises a compound having the following formula:

 or any pharmaceutically acceptable salts thereof.
or any pharmaceutically acceptable salts thereof.
[0089] In some aspects, the STING agonist useful for the present disclosure comprises a compound disclosed in WO 2014/093936, a compound in WO 2014/189805, a compound in WO 2015/077354, c-di-AMP, c-di-GMP, c-di-IMP, c-AMP-GMP, c-AMP-IMP, and c-GMP-IMP, described in WO 2013/185052 and/or Sci. Transl. Med. 283,283ra52 (2015), a compound in WO 2014/189806, a compound in WO 2015/185565, a compound in WO 2014/179760, a compound in WO 2014/179335, a compound in WO 2015/017652, a compound in WO 2016/096577, a compound in WO 2016/120305, a compound in WO 2016/145102, a compound in WO 2017/027646, a compound in WO 2017/075477, a compound in WO 2017/027645, a compound in WO 2018/100558, a compound in WO 2017/175147, or a compound in WO 2017/175156, each of which is incorporated herein by reference in its entirety.
[0090] In some aspects, the STING agonist useful for the present disclosure is CL606, CL611, CL602, CL655, CL604, CL609, CL614, CL656, CL647, CL626, CL629, CL603, CL632, CL633, CL659, or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL606 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL611 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL602 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL655 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL604 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL609 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL614 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL656 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL647 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL626 or a
pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL629 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL603 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL632 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL633 or a pharmaceutically acceptable salt thereof. In some aspects, the STING agonist useful for the present disclosure is CL659 or a pharmaceutically acceptable salt thereof.
[0091] In some aspects, the EV, e.g., exosome, comprises a cyclic dinucleotide STING agonist and/or a non-cyclic dinucleotide STING agonist. In some aspects, when several cyclic dinucleotide STING agonist are present on an EV, e.g., exosome, disclosed herein, such STING agonists can be the same or they can be different. In some aspects, when several non-cyclic dinucleotide STING agonist are present, such STING agonists can be the same or they can be different. In some aspects, an EV, e.g., exosome, composition of the present disclosure can comprise two or more populations of EVs, e.g., exosomes, wherein each population of EVs, e.g., exosomes, comprises a different STING agonist or combination thereof.
[0092] The STING agonists can also be modified to increase their association (e.g., linkage) to cholesterol on the surface of an extracellular vesicle or EV (e.g., either unbound in the lumen). In some aspects, the modification allows better expression of the STING agonist on the exterior surface of the EV, e.g., exosome, (e.g., linked to a scaffold moiety disclosed herein, e.g., Scaffold X). This modification can include a chemical or enzyme, or by physically or chemically altering the polarity or charge of the STING agonist. The STING agonist may be modified by a single treatment, or by a combination of treatments, e.g., adding a lipid binding tag only, or adding a lipid binding tag and altering the polarity. The previous example is meant to be a non-limiting illustrative instance. It is contemplated that any combination of modifications may be practiced. The modification may increase linkage of the agonist to the cholesterol moiety on the surface of the EV by between 2-fold and 10,000 fold, between 10-fold and 1,000 fold, or between 100-fold and 500- fold compared to an unmodified agonist. The modification may increase linkage of the agonist to the cholesterol moiety in the EV by at least 2-fold, 5-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50- fold, 60-fold, 70-fold, 80-fold, 90-fold, 100-fold, 200-fold, 300-fold, 400-fold, 500-fold, 600-fold, 700-fold, 800-fold, 900-fold, 1000-fold, 2000-fold, 3000-fold, 4000-fold, 5000-fold, 6000-fold, 7000-fold, 8000-fold, 9000-fold, or 10,000-fold compared to an unmodified agonist.
[0093] In some aspects, STING agonists can be modified to allow for better expression of the agonists on the exterior surface of the EV, e.g., exosome, (e.g., linked to a cholesterol moiety
disclosed herein). Any of the modifications described above can be used. The modification may increase linkage of the agonist to the cholesterol moiety in the EV, e.g., exosome, by about between 2-fold and 10,000 fold, about between 10-fold and 1,000 fold, or about between 100-fold and 500- fold compared to an unmodified agonist. The modification can increase expression of the agonist on the exterior surface of the EV, e.g., exosome, by at least about 2-fold, 5-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50-fold, 60-fold, 70-fold, 80-fold, 90-fold, 100-fold, 200-fold, 300-fold, 400-fold, 500-fold, 600-fold, 700-fold, 800-fold, 900-fold, 1000-fold, 2000-fold, 3000-fold, 4000-fold, 5000-fold, 6000-fold, 7000-fold, 8000-fold, 9000-fold, or 10,000-fold compared to expression of an unmodified agonist.
II.B. Scaffold-X-Engineered EVs, e.g., Exosomes
[0094] In some aspects, EVs of the present disclosure comprise a membrane modified in its composition. For example, their membrane compositions can be modified by changing the protein, lipid, or glycan content of the membrane.
[0095] In some aspects, the surface-engineered EVs are generated by chemical and/or physical methods, such as PEG-induced fusion and/or ultrasonic fusion. In other aspects, the surface- engineered EVs, e.g., exosomes, are generated by genetic engineering. EVs produced from a genetically-modified producer cell or a progeny of the genetically-modified cell can contain modified membrane compositions. In some aspects, surface-engineered EVs, e.g., exosomes, have one or more exosome proteins at a higher or lower density (e.g., higher number) or include a variant or a fragment of the exosome proteins.
[0096] For example, surface-engineered EVs can be produced from a cell e.g., HEK293 cells) transformed with an exogenous sequence encoding one or more exosome proteins, e.g., PTGFRN protein) or a variant or a fragment thereof. EVs including one or more exosome proteins expressed from the exogenous sequence can include modified membrane compositions.
[0097] Various modifications or fragments of the exosome proteins can be used for the aspects of the present disclosure. For example, exosome proteins modified to have enhanced affinity to a binding agent can be used for generating surface-engineered EVs that can be purified using the binding agent. Exosome proteins modified to be more effectively targeted to EVs, e.g., exosomes, and/or membranes can be used. Exosome proteins modified to comprise a minimal fragment required for specific and effective targeting to EVs, e.g., exosomes, membranes can be also used. [0098] In some aspects, the surface-engineered EVs described herein demonstrate superior characteristics compared to EVs, e.g., exosomes, known in the art. For example, surface- engineered EVs contain modified proteins more highly enriched on their surface than naturally
occurring EVs, e.g., exosomes, or the EVs, e.g., exosomes, produced using conventional exosome proteins. Moreover, the surface-engineered EVs of the present invention can have greater, more specific, or more controlled biological activity compared to naturally occurring EVs, e.g., exosomes, or the EVs, e.g., exosomes, produced using conventional exosome proteins.
[0099] In some aspects, exosome proteins useful for the present disclosure comprise Prostaglandin F2 receptor negative regulator (the PTGFRN polypeptide). The PTGFRN protein can be also referred to as CD9 partner 1 (CD9P-1), Glu-Trp-Ile EWI motif-containing protein F (EWI-F), Prostaglandin F2-alpha receptor regulatory protein, Prostaglandin F2-alpha receptor- associated protein, or CD315. The full length amino acid sequence of the human PTGFRN protein (Uniprot Accession No. Q9P2B2) is shown at Table 1 as SEQ ID NO: 1. The PTGFRN polypeptide contains a signal peptide (amino acids 1 to 25 of SEQ ID NO: 1), the extracellular domain (amino acids 26 to 832 of SEQ ID NO: 1), a transmembrane domain (amino acids 833 to 853 of SEQ ID NO: 1), and a cytoplasmic domain (amino acids 854 to 879 of SEQ ID NO: 1). The mature PTGFRN polypeptide consists of SEQ ID NO: 1 without the signal peptide, i.e., amino acids 26 to 879 of SEQ ID NO: 1. In some aspects, a PTGFRN polypeptide fragment useful for the present disclosure comprises a transmembrane domain of the PTGFRN polypeptide. In other aspects, a PTGFRN polypeptide fragment useful for the present disclosure comprises the transmembrane domain of the PTGFRN polypeptide and (i) at least five, at least 10, at least 15, at least 20, at least 25, at least 30, at least 40, at least 50, at least 70, at least 80, at least 90, at least 100, at least 110, at least 120, at least 130, at least 140, at least 150 amino acids at the N terminus of the transmembrane domain, (ii) at least five, at least 10, at least 15, at least 20, or at least 25 amino acids at the C terminus of the transmembrane domain, or both (i) and (ii).
[0100] In some aspects, the fragments of PTGFRN polypeptide lack one or more functional or structural domains, such as IgV.
[0101] In other aspects, the PTGFRN polypeptide comprises an amino acid sequence at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or about 100% identical to amino acids 26 to 879 of SEQ ID NO: 1. In other aspects, the PTGFRN polypeptide comprises an amino acid sequence at least about at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or about 100% identical to SEQ ID NO: 33. In other aspects, the PTGFRN polypeptide comprises the amino acid sequence of SEQ ID NO: 33, except one amino acid mutation, two amino acid mutations, three amino acid mutations,
four amino acid mutations, five amino acid mutations, six amino acid mutations, or seven amino acid mutations. The mutations can be a substitution, an insertion, a deletion, or any combination thereof. In some aspects, the PTGFRN polypeptide comprises the amino acid sequence of SEQ ID NO: 33 and 1 amino acid, two amino acids, three amino acids, four amino acids, five amino acids, six amino acids, seven amino acids, eight amino acids, nine amino acids, ten amino acids, 11 amino acids, 12 amino acids, 13 amino acids, 14 amino acids, 15 amino acids, 16 amino acids, 17 amino acids, 18 amino acids, 19 amino acids, or 20 amino acids or longer at the N terminus and/or C terminus of SEQ ID NO: 33.
[0102] In other aspects, the PTGFRN polypeptide comprises an amino acid sequence at least about at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or about 100% identical to SEQ ID NO: 2, 3, 4, 5, 6, or 7. In other aspects, the PTGFRN polypeptide comprises the amino acid sequence of SEQ ID NO: 2, 3, 4, 5, 6, or 7, except one amino acid mutation, two amino acid mutations, three amino acid mutations, four amino acid mutations, five amino acid mutations, six amino acid mutations, or seven amino acid mutations. The mutations can be a substitution, an insertion, a deletion, or any combination thereof. In some aspects, the PTGFRN polypeptide comprises the amino acid sequence of SEQ ID NO: 2, 3, 4, 5, 6, or 7 and 1 amino acid, two amino acids, three amino acids, four amino acids, five amino acids, six amino acids, seven amino acids, eight amino acids, nine amino acids, ten amino acids, 11 amino acids, 12 amino acids, 13 amino acids, 14 amino acids, 15 amino acids, 16 amino acids, 17 amino acids, 18 amino acids, 19 amino acids, or 20 amino acids or longer at the N terminus and/or C terminus of SEQ ID NO: 2, 3, 4, 5, 6, or 7.
Table 1.


[0103] Non-limiting examples of other Scaffold X proteins that can be used to link a STING agonist to the surface ofEVs, e.g., exosomes, can be found at US Patent No. 10,195,290 Bl, issued Feb. 5, 2019, which is incorporated by reference in its entirety.
[0104] In some aspects, a Scaffold X protein useful for the present disclosure lacks at least about 5, 10, 50, 100, 200, 300, 400, 500, 600, 700, or 800 amino acids from the N-terminus of the native protein. In some aspects, a Scaffold X lacks at least about 5, 10, 50, 100, 200, 300, 400, 500, 600, 700, or 800 amino acids from the C-terminus of the native protein. In some aspects, a Scaffold X lacks at least about 5, 10, 50, 100, 200, 300, 400, 500, 600, 700, or 800 amino acids from both the N-terminus and C-terminus of the native protein. In some aspects, a Scaffold X lacks one or more functional or structural domains of the native protein.
[0105] In some aspects, Scaffold X described herein can also be used to link an additional payload on the luminal surface and/or on the exterior surface of the EVs, e.g., exosomes, at the same time. For example, the PTGFRN polypeptide can be used to link a payload inside the lumen in addition to the surface of the EV, e.g., exosome.
ILC. Scaffold-Y-Engineered EVs, e.g., Exosomes
[0106] In some aspects, EVs, e.g., exosomes, of the present disclosure comprise an internal space (i.e., lumen) that is different from that of the naturally occurring EVs, e.g., exosomes. For example, the EV, e.g., exosome, can be changed such that the composition in the luminal side of the EV, e.g., exosome, has the protein, lipid, or glycan content different from that of the naturally- occurring EVs, e.g., exosomes.
[0107] In some aspects, engineered EVs, e.g., exosomes, can be produced from a cell transformed with an exogenous sequence encoding a scaffold moiety e.g., exosome proteins, e.g., Scaffold Y) or a modification or a fragment of the scaffold moiety that changes the composition or content of the luminal side of the EV, e.g., exosome. Various modifications or fragments of the
exosome protein that can be expressed in the luminal side of the EV, e.g., exosome, can be used for the aspects of the present disclosure.
[0108] In some aspects, EVs disclosed herein comprise an additional payload (e.g., an antigen) in the lumen of the EV, e.g., encapsulated). In some aspects, a payload, e.g., antigen, is linked to the luminal surface of the EV, e.g., exosome. As used herein, when a molecule (e.g., antigen or adjuvant) is described as "in the lumen" of the EV, e.g., exosome, it means that the molecule is located within the EV, e.g., exosome (e.g., associated), but is not linked to any molecule on the luminal surface of EVs. In other aspects, a payload is expressed on the luminal surface of the EV, e.g., exosome as a fusion molecule, e.g., fusion molecule of a payload to a scaffold moiety (e.g., Scaffold Y). In certain aspects, Scaffold Y comprises the MARCKS protein, MARCKSL1 protein, BASP1 protein, or any combination thereof.
[0109] In other aspects, the EVs, e.g., exosomes, of the present disclosure comprise a cholesterol tagged STING agonist, e.g., cholesterol linked to a STING agonist via a cleavable peptide linker, e.g., Valine-Alanine linker, Valine-Citrulline linker, and a payload linked to a Scaffold Y. In other aspects, the EVs, e.g., exosomes, of the present disclosure comprise cholesterol linked to a STING agonist via a cleavable peptide linker, e.g., Valine-Alanine linker, Valine-Citrulline linker, and a payload linked to a Scaffold X.
[0110] In some aspects, scaffold moi eties (e.g., Scaffold Y) that can change the luminal side of the EVs, e.g., exosomes, include, but are not limited to the MARCKS protein, MARCKSL1 protein, BASP1 protein, or any combination thereof. In some aspects, Scaffold Y comprises Brain Acid Soluble Protein 1 (the BASP1 protein). The BASP1 protein is also known as 22 kDa neuronal tissue-enriched acidic protein or neuronal axonal membrane protein NAP-22. The full-length human BASP1 protein sequence (isomer 1) is shown in Table 2. An isomer produced by an alternative splicing is missing amino acids 88 to 141 from SEQ ID NO: XX (isomer 1).
Table 2.
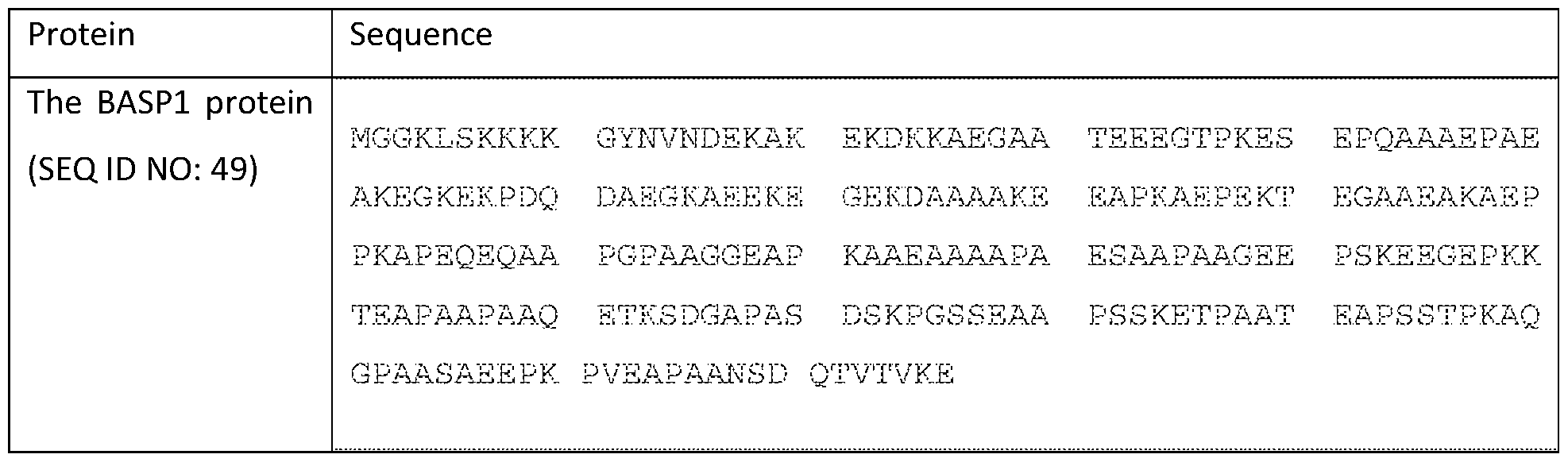

[0111] The mature BASP1 protein sequence is missing the first Met from SEQ ID NO: 49 and thus contains amino acids 2 to 227 of SEQ ID NO: 49.
[0112] In some aspects, a Scaffold Y that can be used to express a payload in the luminal surface of an EV, e.g., exosome, comprises an amino acid sequence at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or about 100% identical to GGKLSKK (SEQ ID NO: XX).
[0113] In some aspects, a Scaffold Y that can be used to express a payload in the luminal surface of an EV, e.g., exosome, comprises an amino acid sequence at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or about 100% identical to the amino acid sequence as set forth in i) GGKLSKKK (SEQ ID NO: XX), (ii) GGKLSKKS (SEQ ID NO: XX), (iii) GAKLSKKK (SEQ ID NO: XX), (iv) GAKLSKKS (SEQ ID NO: XX), (v) GGKQSKKK (SEQ ID NO: XX), (vi) GGKQSKKS (SEQ ID NO: XX), (vii) GGKLAKKK (SEQ ID NO: XX), (viii) GGKLAKKS (SEQ ID NO: XX), or (ix) any combination thereof.
[0114] In some aspects, a Scaffold Y comprises a full-length BASP1 protein.
II.C. Linker
[0115] The EVs of the present disclosure can comprises one or more linkers that link the payload to EVs or to a scaffold moiety, e.g., Scaffold X on the exterior surface of the EVs. In some aspects, the payload is linked to the EVs directly or in a scaffold moiety on the EVs by a linker. The linker can be any chemical moiety known in the art.
[0116] In some aspects, the term “linker” refers to a peptide or polypeptide sequence (e.g., a synthetic peptide or polypeptide sequence) or to a non-polypeptide. In some aspects, two or more linkers can be linked in tandem. Generally, linkers provide flexibility or prevent/ameliorate steric hindrances. Linkers are not typically cleaved; however in certain aspects, such cleavage can be desirable. Accordingly, in some aspects a linker can comprise one or more protease-cleavable sites, which can be located within the sequence of the linker or flanking the linker at either end of the linker sequence.
[0117] In some aspects, the linker is a peptide linker. In some aspects, the peptide linker can comprise at least about two, at least about three, at least about four, at least about five, at least
about 10, at least about 15, at least about 20, at least about 25, at least about 30, at least about 35, at least about 40, at least about 45, at least about 50, at least about 55, at least about 60, at least about 65, at least about 70, at least about 75, at least about 80, at least about 85, at least about 90, at least about 95, or at least about 100 amino acids.
[0118] In some aspects, the peptide linker is synthetic, i.e., non-naturally occurring. In one aspect, a peptide linker includes peptides (or polypeptides) (e.g., natural or non-naturally occurring peptides) which comprise an amino acid sequence that links or genetically fuses a first linear sequence of amino acids to a second linear sequence of amino acids to which it is not naturally linked or genetically fused in nature. For example, in one aspect the peptide linker can comprise non-naturally occurring polypeptides which are modified forms of naturally occurring polypeptides (e.g., comprising a mutation such as an addition, substitution or deletion).
[0119] Linkers may be susceptible to cleavage ("cleavable linker") thereby facilitating release of the payloads. In some aspects, the linker is a "reduction-sensitive linker." In some aspects, the reduction-sensitive linker contains a disulfide bond. In some aspects, the linker is an "acid labile linker." In some aspects, the acid labile linker contains hydrazone. Suitable acid labile linkers also include, for example, a cis-aconitic linker, a hydrazide linker, a thiocarbamoyl linker, or any combination thereof. In some aspects, the linker comprises a non-cleavable liker.
II.D. Producer Cells and Modifications
[0120] EVs, e.g., exosomes, can be produced from a cell grown in vitro or a body fluid of a subject. When EVs, e.g., exosomes, are produced from in vitro cell culture, various producer cells, e.g., HEK293 cells, can be used. Additional cell types that can be used for the production of the lumen-engineered EVs, e.g., exosomes, described herein include, without limitation, mesenchymal stem cells, T-cells, B-cells, dendritic cells, macrophages, and cancer cell lines. Further examples include: Chinese hamster ovary (CHO) cells, mesenchymal stem cells (MSCs), BJ human foreskin fibroblast cells, fHDF fibroblast cells, AGE.HN® neuronal precursor cells, CAP® amniocyte cells, adipose mesenchymal stem cells, and RPTEC/TERT1 cells. In certain aspects, a producer cell is not a dendritic cell, macrophage, B cell, mast cell, neutrophil, Kupffer-Browicz cell, cell derived from any of these cells, or any combination thereof.
[0121] Some aspects may also include genetically modifying the EV, e.g., exosome, to comprise one or more exogenous sequences to produce modified EVs that express exogenous proteins on the vesicle surface. The exogenous sequences can comprise a sequence encoding the EV, e.g., exosome, protein or a modification or a fragment of the EV protein. An extra copy of the sequence encoding the EV, e.g., exosome, protein can be introduced to produce a surface-
engineered EV having a higher density of the EV protein. An exogenous sequence encoding a modification or a fragment of the EV, e.g., exosome, protein can be introduced to produce a modified EV containing the modification or the fragment of the EV protein. An exogenous sequence encoding an affinity tag can be introduced to produce a modified EV, e.g., exosome, containing a fusion protein comprising the affinity tag attached to the EV protein.
[0122] In some aspects, the exogenous sequence encodes for Scaffold X (e.g., a PTGFRN protein, a BSG protein, an IGSF2 protein, an IGSF3 protein, an IGSF8 protein, an ITGB1 protein, an ITGA4 protein, a SLC3A2 protein, an ATP transporter protein, or a fragment or a variant thereof). In some aspects the modified EV, e.g., exosome, overexpresses Scaffold X (e.g., a PTGFRN protein, a BSG protein, an IGSF2 protein, an IGSF3 protein, an IGSF8 protein, an ITGB 1 protein, an ITGA4 protein, a SLC3A2 protein, an ATP transporter protein, or a fragment or a variant thereof). In other aspects, the EV, e.g., exosome, is produced by a cell that overexpresses Scaffold X (e.g. , a PTGFRN protein, a BSG protein, an IGSF2 protein, an IGSF3 protein, an IGSF8 protein, an ITGB1 protein, an ITGA4 protein, a SLC3A2 protein, an ATP transporter protein, or a fragment or a variant thereof).
[0123] In some aspects, the exogenous sequence encodes for Scaffold Y (e.g., the MARCKS protein, MARCKSL1 protein, BASP1 protein, or a fragment or variant thereof). In some aspects, the modified EV, e.g., exosome, overexpresses Scaffold Y (e.g., the MARCKS protein, MARCKSL1 protein, BASP1 protein, or a fragment or variant thereof). In other aspects, the EV, e.g., exosome, is produced by a cell that overexpresses Scaffold Y (e.g., the MARCKS protein, MARCKSL1 protein, BASP1 protein, or a fragment or variant thereof).
[0124] The exogenous sequence may be transiently or stably expressed in the producer cell or cell line via transfection, transformation, transduction, electroporation, or any other appropriate method of gene delivery or combination thereof known in the art. The exogenous sequence may be integrated into the producer cell genome, or remain extra chromosomal. The exogenous sequence can be transformed as a plasmid. The exogenous sequences can be stably integrated into a genomic sequence of the producer cell, at a targeted site or in a random site. The exogenous sequences can be inserted into a genomic sequence of the producer cell, located within, upstream (5’-end) or downstream (3’-end) of an endogenous sequence encoding the EV, e.g., exosome, protein. Various methods known in the art can be used for the introduction of the exogenous sequences into the producer cell. For example, cells modified using various gene editing methods (e.g., methods using a homologous recombination, transposon-mediated system, loxP-Cre system, CRISPR/Cas9 CRISPR/Cfpl, CRISPR/C2cl, C2c2, or C2c3, CRISPR/CasY or CasX, TAL-
effector nuclease or TALEN, or zinc finger nuclease (ZFN) systems) are within the scope of various aspects.
[0125] In some aspects, the producer cell is further modified to comprise an additional exogenous sequence. For example, an additional exogenous sequence can be included to modulate endogenous gene expression, modulate the immune response or immune signaling, or produce an EV, e.g., exosome, including a certain polypeptide as a payload or additional surface expressed ligand. In some aspects, the producer cell can be further modified to comprise an additional exogenous sequence conferring additional functionalities to EVs, e.g., exosomes, for example, specific targeting capabilities, delivery functions, enzymatic functions, increased or decreased halflife in vivo, etc. In some aspects, the producer cell is modified to comprise two exogenous sequences, one encoding the exosome protein or a modification or a fragment of the exosome protein, and the other encoding a protein conferring the additional functionalities to exosomes.
[0126] More specifically, the EV, e.g., exosome, of the present can be produced from a cell transformed with a sequence encoding one or more additional exogenous proteins including, but not limited to ligands, cytokines, or antibodies, or any combination thereof. These additional exogenous proteins may enable activation or modulation of additional immune stimulatory signals in combination with the STING agonist. In some aspects, the EV, e.g., exosome, is further modified with a ligand comprising CD40L, OX40L, or CD27L. In some aspects, the EV, e.g., exosome, is further modified with a cytokine comprising IL-7, IL-12, or IL-15. Any of the one or more exosome proteins described herein can be expressed from a plasmid, an exogenous sequence inserted into the genome or other exogenous nucleic acid such as a synthetic messenger RNA (mRNA).
[0127] In some aspects, the EV, e.g., exosome, is further modified to display an antagonistic antibody or an agonistic antibody or a fragment thereof on the EV, e.g., exosome, surface to direct EV uptake, activate, or block cellular pathways to enhance the combinatorial effect of the STING agonist. In some aspects, the antibody or fragment thereof is an antibody against DEC205, CLEC9A, CLEC6, DCIR, DC-SIGN, LOX-1, or Langerin. The producer cell may be modified to comprise an additional exogenous sequence encoding for an antagonistic antibody or an agonistic antibody. Alternatively, the antagonistic antibody or agonistic antibody may be covalently linked or conjugated to the EV, e.g., exosome, via any appropriate linking chemistry known in the art. Non-limiting examples of appropriate linking chemistry include amine-reactive groups, carboxylreactive groups, sulfhydryl-reactive groups, aldehyde-reactive groups, photoreactive groups, ClickIT chemistry, biotin-streptavidin or other avidin conjugation, or any combination thereof.
III. Method of Producing EVs with STING Agonists
[0128] The present disclosure comprises a method of producing an EV, e.g., exosome, comprising a cholesterol or derivative thereof, which is linked to a STING agonist via a cleavable peptide linker, e.g., a Valine- Alanine linker or Valine-Citrulline linker, the method comprising:
(i) obtaining an EV, e.g., exosome;
(ii) mixing the EV, e.g., exosome with cholesterol linked to a STING agonist via a cleavable peptide linker, e.g., a Valine- Alanine linker or Valine-Citrulline linker, in a solution;
(iii) incubating the mixture of the EV, e.g., exosome and the cholesterol linked to the STING agonist in a solution comprising a buffer under suitable conditions; and
(iv) purifying the EV, e.g., exosome.
[0129] The buffer conditions of the solution of EVs, e.g., exosomes, can also be altered to optimize cholesterol-cleavable peptide linker-a STING agonist into the EVs, e.g., exosomes. In some aspects, the buffer can be a phosphate buffered saline (PBS) with sucrose. PBS is a well- known buffer to those skilled in the art. Additional buffer modifications can also be used, such as shear protectants, viscosity modifiers, and/or solutes that affect vesicle structural properties. Excipients can also be added to improve the efficiency of the cholesterol-cleavable peptide linker- a STING agonist into the surface (membrane) of EVs, such as membrane softening materials and molecular crowding agents. Other modifications to the buffer can include specific pH ranges and/or concentrations of salts, organic solvents, small molecules, detergents, zwitterions, amino acids, polymers, and/or any combination of the above including multiple concentrations.
[0130] In some aspects, the one or more moieties can be introduced into suitable producer cells using synthetic macromolecules, such as cationic lipids and polymers (Papapetrou et al., Gene Therapy 12: SI 18-S130 (2005)). In some aspects, the cationic lipids form complexes with the one or more moieties through charge interactions. In some aspects, the positively charged complexes bind to the negatively charged cell surface and are taken up by the cell by endocytosis. In some aspects, a cationic polymer can be used to transfect producer cells. In some aspects, the cationic polymer is polyethylenimine (PEI). In aspects, chemicals such as calcium phosphate, cyclodextrin, or polybrene, can be used to introduce the one or more moieties to the producer cells. The one or more moieties can also be introduced into a producer cell using a physical method such as particle- mediated transfection, “gene gun”, biolistics, or particle bombardment technology (Papapetrou et al., Gene Therapy 12: S118-S130 (2005)). A reporter gene such as, for example, beta-
galactosidase, chloramphenicol acetyltransferase, luciferase, or green fluorescent protein can be used to assess the transfection efficiency of the producer cell.
IV. EV Purification
[0131] The EVs, e.g., exosomes, prepared for the present disclosure can be isolated from the producer cells. It is contemplated that all known manners of isolation of EVs, e.g., exosomes, are deemed suitable for use herein. For example, physical properties of EVs, e.g., exosomes, may be employed to separate them from a medium or other source material, including separation on the basis of electrical charge (e.g., electrophoretic separation), size (e.g., filtration, molecular sieving, etc), density (e.g., regular or gradient centrifugation), Svedberg constant (e.g., sedimentation with or without external force, etc). Alternatively, or additionally, isolation may be based on one or more biological properties, and include methods that may employ surface markers (e.g., for precipitation, reversible binding to solid phase, FACS separation, specific ligand binding, nonspecific ligand binding, etc.). In yet further contemplated methods, the EVs, e.g., exosomes, may also be fused using chemical and/or physical methods, including PEG-induced fusion and/or ultrasonic fusion.
[0132] The EVs, e.g., exosomes, may also be purified after incubation with the cholesterol tagged STING agonist to remove free cholesterol tagged STING agonists from the composition. All manners of previously disclosed methods are also deemed suitable for use herein, including separation on the basis of physical or biological properties of EVs, e.g., exosomes.
[0133] Isolation, purification, and enrichment can be done in a general and non-selective manner (typically including serial centrifugation). Alternatively, isolation, purification, and enrichment can be done in a more specific and selective manner (e.g., using producer cell-specific surface markers). For example, specific surface markers may be used in immunoprecipitation, FACS sorting, affinity purification, bead-bound ligands for magnetic separation etc.
[0134] In some aspects, size exclusion chromatography can be utilized to isolate or purify the EVs, e.g., exosomes. Size exclusion chromatography techniques are known in the art. Exemplary, non-limiting techniques are provided herein. In some aspects, a void volume fraction is isolated and comprises the EVs, e.g., exosomes, of interest. In some aspects, for example, density gradient centrifugation can be utilized to further isolate the EVs, e.g., exosomes. Still further, in some aspects, it can be desirable to further separate the producer cell-derived EVs, e.g., exosomes, from EVs of other origin. For example, the producer cell-derived EVs, e.g., exosomes, can be separated
from non-producer cell-derived EVs, e.g., exosomes, by immunosorbent capture using an antigen antibody specific for the producer cell.
[0135] In some aspects, the isolation of EVs, e.g., exosomes, may involve size exclusion chromatography or ion chromatography, such as anion exchange, cation exchange, or mixed mode chromatography. In some aspects, the isolation of EVs, e.g., exosomes, may involve desalting, dialysis, tangential flow filtration, ultrafiltration, or diafiltration, or any combination thereofO. In some aspects, the isolation of EVs, e.g., exosomes, may involve combinations of methods that include, but are not limited to, differential centrifugation, size-based membrane filtration, concentration and/or rate zonal centrifugation. In some aspects, the isolation of EVs, e.g., exosomes, may involve one or more centrifugation steps. The centrifugation may be performed at about 50,000 to 150,000 x g. The centrifugation may be performed at about 50,000 x g, 75,000 x g, 100,000 x g, 125,000 x g, or 150,000 x g.
V. Therapeutic Administration
V.A. Immune Modulation and Dosage
[0136] Provided herein are methods for inducing and/or modulating an immune or inflammatory response in a subject by administering a pharmaceutically effective amount of an EV, e.g., exosome, comprising a STING agonist.
[0137] Dendritic cells (DCs) are a population of antigen present cells derived from a hematopoietic cell lineage that link the innate and adaptive immune systems. DCs share a common myeloid precursor with monocytes and macrophages and are generally separated into two major groups: plasmacytoid DCs (pDCs) and myeloid DCs (mDCs), which are also known as conventional DCs (eDCs). mDCs are further classified based on their development from myeloid or lymphoid precursors and expression levels of CD8a, CD4, and Cl lb. A third population of DCs are monocyte-derived DCs (moDCs) which arise from a monocyte precursor, not a DC progenitor like pDCs and eDCs. moDCs develop after receiving inflammatory cues. Immature DCs reside in peripheral tissue before maturation. Several signaling pathways lead to DC maturation, including the signaling cascades induced by pattern recognition receptors (PRRs). Each subset of immature DCs varies in the protein expression patterns of PRRs which allows the immature DC populations to respond differently upon activation of the same PRR. This results in modulation of the immune response mediated by DCs. PRRs present in DCs include Toll-like receptors (TLRs), C-type lectin
receptors, retinoic-acid inducible gene (RIG)-I-like receptors (RLRs), NOD-like receptors (NLRs), and STING.
[0138] The STING pathway is the dominant DNA sensing pathway in both mDCs and pDCs. Activation of the STING pathway in DCs results in Type I IFN and pro inflammatory cytokine production via TBK1, IRF3, and NF-KB signaling. Binding of IFN to their receptors on cells results in activation of IFN-stimulated response elements and the transcription of IFN-sensitive genes that result in the immune and inflammatory response. IFN signaling also cross-primes DCs to promote antigen persistence, alters the antigen repertoire available for MHCI presentation, enhances MHCI presentation of antigens, and increases the overall surface expression of MHCI, MHCII, and costimulatory molecules CD40, CD80, and CD86. These actions result in increased priming of tumor specific CD8+ T cells and initiation of the adaptive immune response.
[0139] In some aspects, the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist to a subject in need thereof activates or induces dendritic cells, thereby inducing or modulating an immune or inflammatory response in the subject. In some aspects, the dendritic cells activated are myeloid dendritic cells. In some aspects, the dendritic cells are plasmacytoid dendritic cells. In some aspects, the EV is administered as an adjuvant.
[0140] In some aspects, the method induces interferon (IFN)-P production. Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist (e.g., expressed on the exterior surface) may result in between 2-fold and 10,000-fold greater IFN-P induction compared to administration of a STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the surface may result in between about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500- 600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000- 9000 fold, or 9000-10,000 fold greater IFN-P induction compared to administration of a STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in greater than about 2-fold, >5 fold, >10-fold, >20-fold, >30-fold, >40-fold, >50-fold, >60-fold, >70-fold, >80-fold, >90-fold, >100- fold, >200-fold, >300-fold, >400-fold, >500-fold, >600-fold, >700-fold, >800-fold, >900-fold, >1000-fold, >2000-fold, >3000-fold, >4000-fold, >5000-fold, >6000-fold, >7000-fold, >8000-
fold, >9000-fold, or > 10,000-fold IFN-P induction compared to administration of a cholesterol tagged STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in between 2-fold and 10,000-fold greater IFN-P induction compared to the subject’s baseline IFN-P production. Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in between about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30- 40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200- 300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000-9000 fold, or 9000-10,000 fold greater IFN-p induction compared to the subject’s baseline IFN-P production. Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist may result in greater than about 2-fold, >5 fold, >10-fold, >20-fold, >30-fold, >40-fold, >50-fold, >60-fold, >70-fold, >80-fold, >90-fold, >100- fold, >200-fold, >300-fold, >400-fold, >500-fold, >600-fold, >700-fold, >800-fold, >900-fold, >1000-fold, >2000-fold, >3000-fold, >4000-fold, >5000-fold, >6000-fold, >7000-fold, >8000- fold, >9000-fold, or >10, 000-fold IFN-P induction compared to the subject’s baseline IFN-P production.
[0141] In some aspects, administering an EV, e.g., exosome comprising a cholesterol tagged STING agonist expressed on the exterior surface, disclosed herein to a subject can also regulate the levels of other immune modulators e.g., cytokines or chemokines). In certain aspects, the method disclosed herein can increase the level of IFN-y, CXCL9, and/or CXCL10. In some aspects, administration of EVs, e.g., exosomes comprising a cholesterol tagged STING agonist expressed on the exterior surface, described herein can result in between about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000-9000 fold, or 9000-10,000 fold greater amount of IFN-y, CXCL9, and/or CXCL10 compared to a free STING agonist or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). [0142] In some aspects, the method induces myeloid dendritic cell (mDC) activation. Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in between 2-fold and 50,000-fold greater mDC activation
compared to administration of a STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in between about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000- 8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20, GOO- 25, 000 fold, 25,000-30,000 fold, 30,000-35,000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold greater mDC activation compared to administration of a STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in greater than about 2-fold, >5 fold, >10-fold, >20-fold, >30-fold, >40-fold, >50-fold, >60-fold, >70-fold, >80-fold, >90-fold, >100-fold, >200- fold, >300-fold, >400-fold, >500-fold, >600-fold, >700-fold, >800-fold, >900-fold, >1000-fold, >2000-fold, >3000-fold, >4000-fold, >5000-fold, >6000-fold, >7000-fold, >8000-fold, >9000- fold, >10, 000-fold, >15, 000-fold, >20, 000-fold, >25, 000-fold, >30, 000-fold, >35, 000-fold, >40, 000-fold, >45, 000-fold, or >50, 000-fold mDC activation compared to administration of a STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation).
[0143] Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in between 2-fold and 10,000-fold greater mDC activation compared to the subject’s baseline mDC activation. Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in between about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000- 8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20, GOO- 25, 000 fold, 25,000-30,000 fold, 30,000-35,000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold greater mDC activation compared to the subject’s baseline mDC activation. Administration of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface may result in greater than about 2-fold, >5 fold, > 10-fold, >20-fold, >30-
fold, >40-fold, >50-fold, >60-fold, >70-fold, >80-fold, >90-fold, >100-fold, >200-fold, >300-fold, >400-fold, >500-fold, >600-fold, >700-fold, >800-fold, >900-fold, > 1000-fold, >2000-fold, >3000-fold, >4000-fold, >5000-fold, >6000-fold, >7000-fold, >8000-fold, >9000-fold, >10, 000- fold, >15, 000-fold, >20, 000-fold, >25, 000-fold, >30, 000-fold, >35, 000-fold, >40, 000-fold, >45, 000-fold, or >50, 000-fold mDC activation compared to the subject’s baseline mDC activation. [0144] In some aspects, the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface does not induce monocyte activation as compared to the subject’s baseline monocyte activation. In some aspects, the administration of an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface results in less than less than about 2-fold, <5 fold, < 10-fold, <20-fold, <30- fold, <40-fold, <50-fold, <60-fold, <70-fold, <80-fold, <90-fold, <100-fold, <200-fold, <300-fold, <400-fold, <500-fold, <600-fold, <700-fold, <800-fold, <900-fold, < 1000-fold, <2000-fold, <3000-fold, <4000-fold, <5000-fold, <6000-fold, <7000-fold, <8000-fold, <9000-fold, <10, 000- fold, <15, 000-fold, <20, 000-fold, <25, 000-fold, <30, 000-fold, <35, 000-fold, <40, 000-fold, <45, 000-fold, <50, 000-fold, <55, 000-fold, <60, 000-fold, <65, 000-fold, <70, 000-fold, <75, 000- fold, <80, 000-fold, <85, 000-fold, <90, 000-fold, <95, 000-fold, < 100,000-fold, <200, 000-fold, <300, 000-fold, <400, 000-fold, <500, 000-fold, <600, 000-fold, <700, 000-fold, <800, 000-fold, <900, 000-fold, or <1,000, 000-fold induction of monocyte activation relative to the subject’s baseline monocyte activation. In some aspects, the administration of an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface to a subject results in less than about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000- 8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20, GOO- 25, 000 fold, 25,000-30,000 fold, 30,000-35,000 fold, 35,000-40,000 fold, 40,000-45,000 fold, 45,000-50,000 fold, 55,000-60,000 fold, 60,000-65,000 fold, 65,000-70,000 fold, 70,000-75,000 fold, 75,000-80,000 fold, 80,000-85,000 fold, 85,000-90,000 fold, 90,000-95,000 fold, 95,000- 100,000 fold, 100,000-200,000 fold, 200,000-300,000 fold, 300,000-400,000 fold, 400,000- 500,000 fold, 500,000-600,000 fold, 600,000-700,000 fold, 700,000-800,000 fold, 800,000- 900,000 fold, or 900,000-1,000,000 fold induction of monocyte activation relative to the subject’s baseline monocyte activation.
[0145] In some aspects, the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface to a subject does not induce monocyte activation as compared to administration of the STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). In some aspects, the administration of an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface results in less than less than about 2-fold, <5 fold, <10- fold, <20-fold, <30-fold, <40-fold, <50-fold, <60-fold, <70-fold, <80-fold, <90-fold, <100-fold, <200-fold, <300-fold, <400-fold, <500-fold, <600-fold, <700-fold, <800-fold, <900-fold, <1000- fold, <2000-fold, <3000-fold, <4000-fold, <5000-fold, <6000-fold, <7000-fold, <8000-fold, <9000-fold, <10, 000-fold, <15, 000-fold, <20, 000-fold, <25, 000-fold, <30, 000-fold, <35, 000-fold, <40, 000-fold, <45, 000-fold, or <50, 000-fold induction of monocyte activation relative to the amount of monocyte activation after administration of the free STING agonist or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). In some aspects, the administration of an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface to a subject results in less than about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90- 100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20,000-25,000 fold, 25,000-30,000 fold, 30, GOO- 35, 000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold induction of monocyte activation relative to the amount of monocyte activation after administration of the free STING agonist or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). Monocyte activation may be measured by the surface expression of CD86 on the monocyte, or by any other appropriate monocyte activation marker known in the art.
[0146] Because of the improved therapeutic effects associated with EVs, e.g., exosomes, described herein, in some aspects, lower dosages of the EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface can be delivered compared to the free STING agonist or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). Moreover, non-selective delivery of high doses of STING agonists can attenuate desirable immune stimulatory responses. Accordingly, because the EVs, e.g., exosomes, described herein can be administered at lower doses, in some aspects, they can
operate in a wider therapeutic window and reduce the liabilities e.g., systemic toxicity, immune cell killing, lack of cell selectivity) observed with free STING agonists.
[0147] The compositions described herein may be administered in a dosage sufficient to ameliorate the disease, disorder, condition, or symptom of the subject in need thereof. In some aspects, the dosage of the EV, e.g., exosome, comprising a cholesterol tagged STING agonist administered to a subject in need is between about 0.01 to 0.1 pM, 0.1 to 1 pM, 1 to 10 pM, 10 to 100 pM, or 100 to 1000 pM. In certain aspects, the dosage of the EV, e.g., exosome, comprising a cholesterol tagged STING agonist administered to a subject in need is about 0.01 pM, 0.05 pM, 0.1 pM, 0.2 pM, 0.3 pM, 0.4 pM, 0.5 pM, 0.6 pM, 0.7 pM, 0.8 pM, 0.9 pM, 1 pM, 2 pM, 3 pM, 4 pM, 5 pM, 6 pM, 7 pM, 8 pM, 9 pM, 10 pM, 11 pM, 12 pM, 13 pM, 14 pM, 15 pM, 16 pM, 17 pM, 18 pM, 19 pM, 20 pM, 25 pM, 30 pM, 35 pM, 40 pM, 45 pM, 40 pM, 55 pM, 60 pM, 65 pM, 70 pM, 75 pM, 80 pM, 85 pM, 90 pM, 95 pM, 100 pM, 150 pM, 200 pM, 250 pM, 300 pM, 350 pM, 400 pM, 450 pM, 500 pM, 550 pM, 600 pM, 650 pM, 700 pM, 750 pM, 800 pM, 850 pM, 900 pM, 950 pM, or 1000 pM. In some aspects, the dosage of the EV, e.g., exosome, comprising a cholesterol tagged STING agonist administered to a subject in need is about 0.3 pg, about 1 pg, about 3 pg, about 6 pg, or about 12 pg.
[0148] In some aspects, the amount of the EV, e.g., exosome, comprising a cholesterol tagged
STING agonist expressed on the exterior surface administered to a subject in need is less than 2- fold, <5 fold, <10-fold, <20-fold, <30-fold, <40-fold, <50-fold, <60-fold, <70-fold, <80-fold, <90- fold, <100-fold, <200-fold, <300-fold, <400-fold, <500-fold, <600-fold, <700-fold, <800-fold, <900-fold, <1000-fold, <2000-fold, <3000-fold, <4000-fold, <5000-fold, <6000-fold, <7000-fold, <8000-fold, <9000-fold, <10, 000-fold, <15, 000-fold, <20, 000-fold, <25, 000-fold, <30, 000-fold, <35,000-fold, <40, 000-fold, <45, 000-fold, or <50, 000-fold relative to the amount of a free STING agonist or an exoSTING required to effect the same ameliorative results in a subject in need. In some aspects, the amount of the EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface administered to a subject in need is between less than about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000- 4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20,000-25,000 fold, 25,000-30,000 fold, 30,000-35,000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold less
relative to the amount of a free STING agonist or an exoSTING required to effect the same ameliorative results in a subject in need.
[0149] In some aspects, the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist does not induce systemic inflammation as compared to the subject’s baseline systemic inflammation. In some aspects, the administration of an EV, e.g., exosome, comprising a cholesterol tagged STING agonist results in less than less than about 2- fold, <5 fold, <10-fold, <20-fold, <30-fold, <40-fold, <50-fold, <60-fold, <70-fold, <80-fold, <90- fold, <100-fold, <200-fold, <300-fold, <400-fold, <500-fold, <600-fold, <700-fold, <800-fold, <900-fold, <1000-fold, <2000-fold, <3000-fold, <4000-fold, <5000-fold, <6000-fold, <7000-fold, <8000-fold, <9000-fold, <10, 000-fold, <15, 000-fold, <20, 000-fold, <25, 000-fold, <30, 000-fold, <35,000-fold, <40, 000-fold, <45, 000-fold, or <50, 000-fold induction of systemic inflammation relative to the subject’s baseline systemic inflammation. In some aspects, the administration of an EV, e.g., exosome, comprising a cholesterol tagged STING agonist to a subject results in less than about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90-100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500- 600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000- 9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20,000-25,000 fold, 25,000- 30,000 fold, 30,000-35,000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold induction of systemic inflammation relative to the subject’s baseline systemic inflammation.
[0150] In some aspects, the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface to a subject does not induce systemic inflammation as compared to administration of the STING agonist alone or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). In some aspects, the administration of an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface results in less than about 2-fold, <5 fold, <10-fold, <20- fold, <30-fold, <40-fold, <50-fold, <60-fold, <70-fold, <80-fold, <90-fold, <100-fold, <200-fold, <300-fold, <400-fold, <500-fold, <600-fold, <700-fold, <800-fold, <900-fold, <1000-fold, <2000- fold, <3000-fold, <4000-fold, <5000-fold, <6000-fold, <7000-fold, <8000-fold, <9000-fold, <10, 000-fold, <15, 000-fold, <20, 000-fold, <25, 000-fold, <30, 000-fold, <35, 000-fold, <40, 000- fold, <45, 000-fold, or <50, 000-fold induction of systemic inflammation relative to the amount of systemic inflammation after administration of the free STING agonist or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). In some
aspects, the administration of an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface to subject results in less than about 2-5 fold, 5-10 fold, 10-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-60 fold, 60-70 fold, 70-80 fold, 80-90 fold, 90- 100 fold, 100-200 fold, 200-300 fold, 300-400 fold, 400-500 fold, 500-600 fold, 600-700 fold, 700-800 fold, 800-900 fold, 900-1000 fold, 1000-2000 fold, 2000-3000 fold, 3000-4000 fold, 4000-5000 fold, 5000-6000 fold, 6000-7000 fold, 7000-8000 fold, 8000-9000 fold, 9000-10,000 fold, 10,000-15,000 fold, 15,000-20,000 fold, 20,000-25,000 fold, 25,000-30,000 fold, 30, GOO- 35, 000 fold, 35,000-40,000 fold, 40,000-45,000 fold, or 45,000-50,000 fold induction of systemic inflammation relative to the amount of systemic inflammation after administration of the free STING agonist or an exoSTING (e.g., a combination of exosome and the STING agonist that can be prepared by incubation). Systemic inflammation may be quantified or measured by any appropriate method known in the art.
[0151] In some aspects, the method of administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface to a subject additionally comprises administering an additional therapeutic agent. In some aspects, the additional therapeutic agent is an immunomodulating agent. In some aspects, the immunomodulating component is an inhibitor for a negative checkpoint regulator or an inhibitor for a binding partner of a negative checkpoint regulator. In some of these aspects, the negative checkpoint regulator is selected from the group consisting of: cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1), lymphocyte-activated gene 3 (LAG-3), T-cell immunoglobulin mucin-containing protein 3 (TIM-3), B and T lymphocyte attenuator (BTLA), T cell immunoreceptor with Ig and ITIM domains (TIGIT), V-domain Ig suppressor of T cell activation (VISTA), adenosine A2a receptor (A2aR), killer cell immunoglobulin like receptor (KIR), indoleamine 2,3-dioxygenase (IDO), CD20, CD39, and CD73. In various aspects, the additional therapeutic agent is an antibody or antigen-binding fragment thereof. In some aspects, the antibody or antigen-binding fragment thereof is one or more whole antibodies, polyclonal, monoclonal and recombinant antibodies, fragments thereof, and further includes single-chain antibodies, humanized antibodies, murine antibodies, chimeric, mouse-human, mouse-primate, primate-human monoclonal antibodies, anti-idiotype antibodies, antibody fragments, such as, e.g., scFv, (scFv)2, Fab, Fab', and F(ab')2, F(abl)2, Fv, dAb, and Fd fragments, diabodies, and antibody- related polypeptides. The term antibody includes bispecific antibodies and multispecific antibodies so long as they exhibit the desired biological activity or function. In some aspects, the additional
therapeutic agent is a therapeutic antibody or antigen-binding fragment thereof that is an inhibitor of CTLA-4, PD-1, PD-L1, PD-L2, TIM-3, or LAG3.
[0152] In some aspects, the additional therapeutic agent is an agent that prevents or treats T cell exhaustion. Such agents may increase, decrease, or modulate the expression of genes associated with T cell exhaustion, including Prdml, Bhlhe40, Irf4. Ikzf2, Zeb2, Lass6, Egr2, Tox, Eomes, Nfatcl, Nfatc2, Zbtb32, Rbpj, Hifla, Lag3, Tnfrsf9, Ptger2, Haver 2, A team. j Tigit, Ctla4, Ptger4, Tnfrsflb, Ccl4, CD 109, CD200, Tnfsf9, Nrpl, Sema4c, Ptprj, 1121, Tspan2, Rgsl6, Sh2d2a, Nucbl, Plscrl, Ptpnll, Prkca, Plscr4, Casp3, Gpd2, Gas2, Sh3rfl, Nhedc2, Plek, Tnfaip2, and Ctsb, or any combination thereof. Therapeutic agents may also increase, decrease, or modulate a protein associated with T cell exhaustion, including NFAT-1 or NF AT-2.
V.B. Method of Treating Cancer
[0153] Provided herein are methods of treating cancer in a subject. The method comprises administering to the subject a therapeutically effective amount of the compositions disclosed herein, wherein the composition is capable of up-regulating a STING-mediated immune response in the subject, thereby enhancing the tumor targeting of the subject’s immune system. In some aspects, the composition is administered intra-tumorally to the subject. In some aspects, the composition is administered parenterally, orally, intravenously, intramuscularly, intraperitoneally, or via any other appropriate administration route.
[0154] Also provided herein are methods of preventing metastasis of cancer in a subject. The method comprises administering to the subject a therapeutically effective amount of the compositions disclosed herein, wherein the composition is capable of preventing one or more tumors at one location in the subject from promoting the growth of one or more tumors at another location in the subject. In some aspects, the composition is administered intratumorally in a first tumor in one location, and the composition administered in a first tumor prevents metastasis of one or more tumors at a second location.
[0155] In some aspects, administering an EV, e.g., exosome, disclosed herein inhibits and/or reduces tumor growth in a subject. In some aspects, the tumor growth (e.g., tumor volume or weight) is reduced by at least about 5%, at least about 10%, at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, or about 100% compared to a reference (e.g., tumor volume in a corresponding subject after administration of free STING agonist or an EV, e.g., exosome, without the STING agonist).
[0156] In some aspects, the cancer being treated is characterized by infiltration of leukocytes (T-cells, B-cells, macrophages, dendritic cells, monocytes) into the tumor microenvironment, or so-called “hot tumors” or “inflammatory tumors”. In some aspects, the cancer being treated is characterized by low levels or undetectable levels of leukocyte infiltration into the tumor microenvironment, or so-called “cold tumors” or “non-inflammatory tumors”. In some aspects, an EV, e.g., exosome, is administered in an amount and for a time sufficient to convert a “cold tumor” into a “hot tumor”, z.e., said administering results in the infiltration of leukocytes (such as T-cells) into the tumor microenvironment. In certain aspects, cancer comprises bladder cancer, cervical cancer, renal cell cancer, testicular cancer, colorectal cancer, lung cancer, head and neck cancer, and ovarian, lymphoma, liver cancer, glioblastoma, melanoma, myeloma, leukemia, pancreatic cancers, or combinations thereof. The term "distal tumor" or "distant tumor" as used herein refers to a tumor that has spread from the original (or primary) tumor to distant organs or distant tissues, e.g., lymph nodes. In some aspects, the EVs, e.g., exosomes, of the disclosure treats a tumor after the metastatic spread.
[0157] Non-limiting examples of cancers (or tumors) that can be treated with methods disclosed herein include squamous cell carcinoma, small-cell lung cancer (SCLC), non-small cell lung cancer, squamous non-small cell lung cancer (NSCLC), nonsquamous NSCLC, gastrointestinal cancer, renal cancer (e.g., clear cell carcinoma), ovarian cancer, liver cancer (e.g., hepatocellular carcinoma), colorectal cancer, endometrial cancer, kidney cancer (e.g., renal cell carcinoma (RCC)), prostate cancer (e.g., hormone refractory prostate adenocarcinoma), thyroid cancer, pancreatic cancer, cervical cancer, stomach cancer, bladder cancer, hepatoma, breast cancer, colon carcinoma, and head and neck cancer (or carcinoma), gastric cancer, germ cell tumor, pediatric sarcoma, sinonasal natural killer, melanoma (e.g., metastatic malignant melanoma, such as cutaneous or intraocular malignant melanoma), bone cancer, skin cancer, uterine cancer, cancer of the anal region, testicular cancer, carcinoma of the fallopian tubes, carcinoma of the endometrium, carcinoma of the cervix, carcinoma of the vagina, carcinoma of the vulva, cancer of the esophagus (e.g., gastroesophageal junction cancer), cancer of the small intestine, cancer of the endocrine system, cancer of the parathyroid gland, cancer of the adrenal gland, sarcoma of soft tissue, cancer of the urethra, cancer of the penis, solid tumors of childhood, cancer of the ureter, carcinoma of the renal pelvis, tumor angiogenesis, pituitary adenoma, Kaposi's sarcoma, epidermoid cancer, squamous cell cancer, T-cell lymphoma, environmentally-induced cancers including those induced by asbestos, virus-related cancers or cancers of viral origin (e.g., human papilloma virus (HPV-related or -originating tumors)), and hematologic malignancies derived from
either of the two major blood cell lineages, i.e., the myeloid cell line (which produces granulocytes, erythrocytes, thrombocytes, macrophages and mast cells) or lymphoid cell line (which produces B, T, NK and plasma cells), such as all types of leukemias, lymphomas, and myelomas, e.g., acute, chronic, lymphocytic and/or myelogenous leukemias, such as acute leukemia (ALL), acute myelogenous leukemia (AML), chronic lymphocytic leukemia (CLL), and chronic myelogenous leukemia (CML), undifferentiated AML (MO), myeloblastic leukemia (Ml), myeloblastic leukemia (M2; with cell maturation), promyelocytic leukemia (M3 or M3 variant [M3V]), myelomonocytic leukemia (M4 or M4 variant with eosinophilia [M4E]), monocytic leukemia (M5), erythroleukemia (M6), megakaryoblastic leukemia (M7), isolated granulocytic sarcoma, and chloroma; lymphomas, such as Hodgkin's lymphoma (HL), non-Hodgkin's lymphoma (NHL), B cell hematologic malignancy, e.g., B-cell lymphomas, T-cell lymphomas, lymphoplasmacytoid lymphoma, monocytoid B-cell lymphoma, mucosa-associated lymphoid tissue (MALT) lymphoma, anaplastic (e.g., Ki 1+) large-cell lymphoma, adult T-cell lymphoma/leukemia, mantle cell lymphoma, angio immunoblastic T-cell lymphoma, angiocentric lymphoma, intestinal T-cell lymphoma, primary mediastinal B-cell lymphoma, precursor T-lymphoblastic lymphoma, T- lymphoblastic; and lymphoma/leukaemia (T-Lbly/T-ALL), peripheral T- cell lymphoma, lymphoblastic lymphoma, post-transplantation lymphoproliferative disorder, true histiocytic lymphoma, primary effusion lymphoma, B cell lymphoma, lymphoblastic lymphoma (LBL), hematopoietic tumors of lymphoid lineage, acute lymphoblastic leukemia, diffuse large B-cell lymphoma, Burkitt's lymphoma, follicular lymphoma, diffuse histiocytic lymphoma (DHL), immunoblastic large cell lymphoma, precursor B-lymphoblastic lymphoma, cutaneous T-cell lymphoma (CTLC) (also called mycosis fungoides or Sezary syndrome), and lymphoplasmacytoid lymphoma (LPL) with Waldenstrom's macroglobulinemia; myelomas, such as IgG myeloma, light chain myeloma, nonsecretory myeloma, smoldering myeloma (also called indolent myeloma), solitary plasmocytoma, and multiple myelomas, chronic lymphocytic leukemia (CLL), hairy cell lymphoma; hematopoietic tumors of myeloid lineage, tumors of mesenchymal origin, including fibrosarcoma and rhabdomyoscarcoma; seminoma, teratocarcinoma, tumors of mesenchymal origin, including fibrosarcoma, rhabdomyoscaroma, and osteosarcoma; and other tumors, including melanoma, xeroderma pigmentosum, keratoacanthoma, seminoma, thyroid follicular cancer and teratocarcinoma, hematopoietic tumors of lymphoid lineage, for example T-cell and B- cell tumors, including but not limited to T-cell disorders such as T-prolymphocytic leukemia (T- PLL), including of the small cell and cerebriform cell type; large granular lymphocyte leukemia (LGL) of the T-cell type; a/d T-NHL hepatosplenic lymphoma; peripheral/post-thymic T cell
lymphoma (pleomorphic and immunoblastic subtypes); angiocentric (nasal) T-cell lymphoma; cancer of the head or neck, renal cancer, rectal cancer, cancer of the thyroid gland; acute myeloid lymphoma, and any combinations thereof.
[0158] In some aspects, a cancer (or tumor) that can be treated comprises a breast cancer, head and neck cancer, uterine cancer, brain cancer, skin cancer, renal cancer, lung cancer, colorectal cancer, prostate cancer, liver cancer, bladder cancer, kidney cancer, peritoneal cancer, pancreatic cancer, thyroid cancer, esophageal cancer, eye cancer, stomach (gastric) cancer, gastrointestinal cancer, carcinoma, sarcoma, leukemia, lymphoma, myeloma, or a combination thereof. In certain aspects, a cancer that can be treated with the present disclosure is a pancreatic cancer and/or a peritoneal cancer.
[0159] In some aspects, a cancer (or tumor) that can be treated comprises a chondroid sarcoma (also referred to as a chondrosarcoma). In some aspects, a cancer (or tumor) that can be treated comprises a cutaneous squamous cell carcinoma (cSCC). In some aspects, a cancer (or tumor) that can be treated comprises a head and neck squamous cell carcinoma of the oral cavity. In some aspects, a cancer (or tumor) that can be treated comprises a hepatocellular cancer (HCC). In some aspects, a cancer (or tumor) that can be treated comprises a cervical cancer. In some aspects, a cancer (or tumor) that can be treated comprises an eye melanoma. In some aspects, a cancer (or tumor) that can be treated comprises a choroidal eye melanoma. In some aspects, a cancer (or tumor) that can be treated comprises a gastric spindle cell sarcoma. In some aspects, a cancer (or tumor) that can be treated comprises a hemangioendothelioma. In some aspects, a cancer (or tumor) that can be treated comprises a mesothelioma. In some aspects, a cancer (or tumor) that can be treated comprises a parotoid gland cancer. In some aspects, a cancer (or tumor) that can be treated comprises a renal cancer. In some aspects, a cancer (or tumor) that can be treated comprises a triple-negative breast cancer (TNBC).
[0160] In some aspects, the methods described herein can also be used for treatment of metastatic cancers, unresectable, refractory cancers (e.g., cancers refractory to previous cancer therapy), and/or recurrent cancers.
[0161] In some aspects, EVs, e.g., exosomes, disclosed herein can be used in combination with one or more additional anti-cancer and/or immunomodulating agents. Such agents can include, for example, chemotherapy drugs, small molecule drugs, or antibodies that stimulate the immune response to a given cancer. In some aspects, the methods described herein are used in combination with a standard of care treatment (e.g., surgery, radiation, and chemotherapy).
[0162] In some aspects, a method for treating a cancer disclosed herein can comprise administering an EV, e.g., exosome, comprising a cholesterol tagged STING agonist expressed on the exterior surface with one or more immuno-oncology agents, such that multiple elements of the immune pathway can be targeted. Non-limiting of such combinations include: a therapy that enhances tumor antigen presentation (e.g., dendritic cell vaccine, GM-CSF secreting cellular vaccines, CpG oligonucleotides, imiquimod); a therapy that inhibits negative immune regulation e.g., by inhibiting CTLA-4 and/or PD1/PD-L1/PD-L2 pathway and/or depleting orblocking Tregs or other immune suppressing cells (e.g., myeloid-derived suppressor cells); a therapy that stimulates positive immune regulation, e.g., with agonists that stimulate the CD-137, OX-40, and/or CD40 or GITR pathway and/or stimulate T cell effector function; a therapy that increases systemically the frequency of anti-tumor T cells; a therapy that depletes or inhibits Tregs, such as Tregs in the tumor, e.g., using an antagonist of CD25 (e.g., daclizumab) or by ex vivo anti-CD25 bead depletion; a therapy that impacts the function of suppressor myeloid cells in the tumor; a therapy that enhances immunogenicity of tumor cells (e.g., anthracyclines); adoptive T cell or NK cell transfer including genetically modified cells, e.g., cells modified by chimeric antigen receptors (CAR-T therapy); a therapy that inhibits a metabolic enzyme such as indoleamine dioxigenase (IDO), dioxigenase, arginase, or nitric oxide synthetase; a therapy that reverses/prevents T cell anergy or exhaustion; a therapy that triggers an innate immune activation and/or inflammation at a tumor site; administration of immune stimulatory cytokines; or blocking of immuno repressive cytokines.
[0163] In some aspects, an immuno-oncology agent that can be used in combination with EVs, e.g., exosomes, disclosed herein comprises an immune checkpoint inhibitor (i.e., blocks signaling through the particular immune checkpoint pathway). Non-limiting examples of immune checkpoint inhibitors that can be used in the present methods comprise a CTLA-4 antagonist (e.g., anti-CTLA-4 antibody), PD-1 antagonist (e.g., anti-PD-1 antibody, anti-PD-Ll antibody), TIM-3 antagonist (e.g., anti-TIM-3 antibody), or combinations thereof.
[0164] In some aspects, an immuno-oncology agent comprises an immune checkpoint activator i.e., promotes signaling through the particular immune checkpoint pathway). In certain aspects, immune checkpoint activator comprises 0X40 agonist (e.g., anti-OX40 antibody), LAG-3 agonist (e.g. anti-LAG-3 antibody), 4-1BB (CD137) agonist (e.g., anti-CD137 antibody), GITR agonist (e.g., anti-GITR antibody), or any combination thereof.
[0165] In some aspects, a combination of an EV, e.g., exosome, disclosed herein and a second agent discussed herein (e.g., immune checkpoint inhibitor) can be administered concurrently as a
single composition in a pharmaceutically acceptable carrier. In other aspects, a combination of an EV, e.g., exosome, and a second agent discussed herein (e.g., immune checkpoint inhibitor) can be administered concurrently as separate compositions. In further aspects, a combination of an EV, e.g., exosome, and a second agent discussed herein (e.g., immune checkpoint inhibitor) can be administered sequentially. In some aspects, an EV, e.g., exosome, is administered prior to the administration of a second agent (e.g., immune checkpoint inhibitor).
V.C. Pharmaceutical Compositions
[0166] Provided herein are pharmaceutical compositions comprising EVs, e.g., exosomes, that are suitable for administration to a subject. The pharmaceutical compositions generally comprise a plurality of EVs, e.g., exosomes, comprising a cholesterol tagged STING agonist expressed on the exterior surface and a pharmaceutically-acceptable excipient or carrier in a form suitable for administration to a subject. Pharmaceutically-acceptable excipients or carriers are determined in part by the particular composition being administered, as well as by the particular method used to administer the composition. Accordingly, there is a wide variety of suitable formulations of pharmaceutical compositions comprising a plurality of EVs, e.g., exosomes,. (See, e.g., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa. 18th ed. (1990)). The pharmaceutical compositions are generally formulated sterile and in full compliance with all Good Manufacturing Practice (GMP) regulations of the U.S. Food and Drug Administration.
[0167] In some aspects, the pharmaceutical composition comprises one or more STING agonist and the EVs, e.g., exosomes, described herein.
[0168] Pharmaceutically-acceptable excipients include excipients that are generally safe (GRAS), non-toxic, and desirable, including excipients that are acceptable for veterinary use as well as for human pharmaceutical use.
[0169] Examples of carriers or diluents include, but are not limited to, water, saline, Ringer's solutions, dextrose solution, and 5% human serum albumin. The use of such media and compounds for pharmaceutically active substances is well known in the art. Except insofar as any conventional media or compound is incompatible with the EVs, e.g., exosomes, described herein, use thereof in the compositions is contemplated. Supplementary therapeutic agents may also be incorporated into the compositions. Typically, a pharmaceutical composition is formulated to be compatible with its intended route of administration. The EVs, e.g., exosomes, can be administered by intratumoral, parenteral, topical, intravenous, oral, subcutaneous, intraarterial, intradermal, transdermal, rectal, intracranial, intraperitoneal, intranasal; intramuscular route or as inhalants. In one aspect, the
pharmaceutical composition comprising EVs, e.g., exosomes, is administered intravenously, e.g. by injection. The EVs, e.g., exosomes, can optionally be administered in combination with other therapeutic agents that are at least partly effective in treating the disease, disorder or condition for which the EVs, e.g., exosomes, are intended.
[0170] Solutions or suspensions can include the following components: a sterile diluent such as water, saline solution, fixed oils, polyethylene glycols, glycerin, propylene glycol or other synthetic solvents; antibacterial compounds such as benzyl alcohol or methyl parabens; antioxidants such as ascorbic acid or sodium bisulfite; chelating compounds such as ethylenediaminetetraacetic acid (EDTA); buffers such as acetates, citrates or phosphates, and compounds for the adjustment of tonicity such as sodium chloride or dextrose. The pH can be adjusted with acids or bases, such as hydrochloric acid or sodium hydroxide. The preparation can be enclosed in ampoules, disposable syringes or multiple dose vials made of glass or plastic.
[0171] Pharmaceutical compositions suitable for injectable use include sterile aqueous solutions (if water soluble) or dispersions and sterile powders. For intravenous administration, suitable carriers include physiological saline, bacteriostatic water, Cremophor EL™ (BASF, Parsippany, N.J.) or phosphate buffered saline (PBS). The composition is generally sterile and fluid to the extent that easy syringeability exists. The carrier can be a solvent or dispersion medium containing, e.g., water, ethanol, polyol (e.g., glycerol, propylene glycol, and liquid polyethylene glycol, and the like), and suitable mixtures thereof. The proper fluidity can be maintained, e.g., by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants. Prevention of the action of microorganisms can be achieved by various antibacterial and antifungal compounds, e.g., parabens, chlorobutanol, phenol, ascorbic acid, thimerosal, and the like. If desired, isotonic compounds, e.g., sugars, polyalcohols such as mannitol, sorbitol, sodium chloride can be added to the composition. Prolonged absorption of the injectable compositions can be brought about by including in the composition a compound which delays absorption, e.g., aluminum monostearate and gelatin.
[0172] Sterile injectable solutions can be prepared by incorporating the EVs, e.g., exosomes, in an effective amount and in an appropriate solvent with one or a combination of ingredients enumerated herein, as desired. Generally, dispersions are prepared by incorporating the EVs, e.g., exosomes, into a sterile vehicle that contains a basic dispersion medium and any desired other ingredients. In the case of sterile powders for the preparation of sterile injectable solutions, methods of preparation are vacuum drying and freeze-drying that yields a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof. The EVs,
e.g., exosomes, can be administered in the form of a depot injection or implant preparation which can be formulated in such a manner to permit a sustained or pulsatile release of the EVs, e.g., exosomes. In some aspects, the EVs, e.g., exosomes, are administered by way of a pump. In some aspects, the EVs, e.g., exosomes, are administered by way of a patch.
[0173] Systemic administration of compositions comprising EVs, e.g., exosomes, can also be by transmucosal or transdermal means. For transmucosal or transdermal administration, penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art, and include, e.g., for transmucosal administration, detergents, bile salts, and fusidic acid derivatives. Transmucosal administration can be accomplished through the use of nasal sprays, suppositories, sublingual spray, drops, or fast dissolve strips. For transdermal administration, the modified EVs, e.g., exosomes, are formulated into ointments, salves, gels, creams, or patches as generally known in the art.
EXAMPLES
[0174] The following examples are provided for illustrative purposes only, and are not to be construed as limiting the scope or content of the invention in any way. The practice of the current invention will employ, unless otherwise indicated, conventional methods of protein chemistry, biochemistry, recombinant DNA techniques and pharmacology, within the skill of the art. Such techniques are explained fully in the literature. See, e.g., T.E. Creighton, Proteins: Structures and Molecular Properties (W.H. Freeman and Company, 1993); Green & Sambrook et al., Molecular Cloning: A Laboratory Manual, 4th Edition (Cold Spring Harbor Laboratory Press, 2012); Colowick & Kaplan, Methods In Enzymology (Academic Press); Remington: The Science and Practice of Pharmacy, 22nd Edition (Pharmaceutical Press, 2012); Sundberg & Carey, Advanced Organic Chemistry: Parts A and B, 5th Edition (Springer, 2007).
Methods
Exosome Purification
[0175] HEK293SF cells were grown to high density in chemically defined medium for 7 days. Conditioned cell culture media was collected and centrifuged at 300 - 800 x g for 5 minutes at room temperature to remove cells and large debris. Media supernatant was then supplemented with 1000 U/L BENZONASE® and incubated at 37 °C for 1 hour in a water bath. Supernatant was collected and centrifuged at 16,000 x g for 30 minutes at 4 °C to remove residual cell debris and other large contaminants. Supernatant was then ultracentrifuged at 133,900 x g for 3 hours at 4 °C
to pellet the exosomes. Supernatant was discarded and any residual media was aspirated from the bottom of the tube. The pellet was resuspended in 200 - 1000 pL PBS (-Ca -Mg).
[0176] To further enrich exosome populations, the pellet was processed via density gradient purification (sucrose or OPTIPREP™). For sucrose gradient purification, the exosome pellet was layered on top of a sucrose gradient as defined in Table 3 below.
Table 3:
WORKING 65% MILLI-Q
PERCENTAGE STOCK VOL. (ML)
(%) VOL. (ML)
50. 3.85. 1.15.
40 § 3.08 1.92
25 1.92 3.08
10 § 0.46 2.54
[0177] The gradient was spun at 200,000 x g for 16 hours at 4 °C in a 12 mL Ultra-Clear (344059) tube placed in a SW 41 Ti rotor to separate the exosome fraction.
[0178] The exosome layer was gently removed from the top layer and diluted in -32.5 mL PBS in a 38.5 mL Ultra-Clear (344058) tube and ultracentrifuged again at 133,900 x g for 3 hours at 4 °C to pellet the purified exosomes. The resulting pellet was resuspended in a minimal volume of PBS (-200 pL) and stored at 4 °C.
[0179] For OPTIPREP™ gradient, a 3-tier sterile gradient is prepared with equal volumes of 10%, 30%, and 45% OPTIPREP™in a 12 mL Ultra-Clear (344059) tube for a SW 41 Ti rotor. The pellet was added to the OPTIPREP™ gradient and ultracentrifuged at 200,000 x g for 16 hours at 4 °C to separate the exosome fraction. The exosome layer was then gently collected from the top -3 mL of the tube.
[0180] The exosome fraction was diluted in -32 mL PBS in a 38.5 mL Ultra-Clear (344058) tube and ultracentrifuged at 133,900 x g for 3 hours at 4 °C to pellet the purified exosomes. The pelleted exosomes were then resuspended in a minimal volume of PBS (-200 pL) and store at 4°C.
Example 1: Potency of exoSTING ECC (Exosome-Cholesterol Conjugates)
Loading of cholesterol tagged STING agonist into exosomes
[0181] Cholesterol tagged STING agonist (0.1 mM) was incubated with exosomes for 1 h at 37°C. The mixture was then washed once in PBS and purified by ultra-centrifugation at 100,000 x g-
In vitro PBMC assay
[0182] exoSTING ECC, exoSTING and free STING agonist were tested for activity in human peripheral blood mononuclear cells (PBMCs). PBMCs were isolated from fresh human blood by centrifugation over a layer of Lymphoprep at 1000 x g for 15 minutes. The resulting buffy coat was washed in PBS and counted. PBMCs were plated in 96-well U-bottom plates. Titrations of the exoSTING ECC, exoSTING, or free STING agonist were prepared in a separate U-bottom plate to perform dose-response studies. The exoSTING ECC, exoSTING or free STING agonist was added to the PBMCs and incubated at 37°C overnight. General activation of PBMCs was detected by measuring the amount of IFNP in the supernatant. As shown in FIG. 1 A, both exoSTING ECC and exoSTING were superior to free STING agonist in terms of ECso for IFNP production. In addition, exoSTING ECC showed enhanced activity over exoSTING.
In vivo potency assay to assess stability of exoSTING ECC and exoSTING
[0183] exoSTING ECC and exoSTING were incubated at 37°C for different times (3 days, 7 days, 14 days, or 30 days) and -80°C for a control. exoSTING ECC and exoSTING from each time point were injected into naive C57BL/6 mice intravenously at 20 ng dose (n = 3). After 4 h, mice were euthanized and 14 of spleens were collected. Spleens were lysed Trizol solution with mechanical disruption. RNAs were isolated by using RNeasy Lipid Tissue Mini Kit (Thermo Fisher Scientific), according to manufacturer’s instructions. cDNA was synthesized by using Superscript IV VILO Master Mix (Thermo Fisher Scientific). IFNP mRNA expression was measured by using TaqMan™ Universal PCR Master Mix (Thermo Fisher Scientific) and QuantStudio™ 3 & 5 Real-Time PCR System (Thermo Fisher Scientific). Ct values were normalized to Ct values of the housekeeping gene RPS13. As shown in Fig. IB, activity of exoSTING-ECC was stable up to 1 month at 37°C without any reduction of activity, whereas activity of exoSTING was almost gone from 3 days of incubation at 37°C. This data shows that superior stability of exoSTING ECC in 37°C.
[0184] ExoRBD ECC STING thermal stability
[0185] Exosomes expressing SARS CoV-2 RBD (receptor binding domain) fused to PTGFRN (exoRBD) and exosomes expressing SARS CoV-2 RBD fused to PTGFRN (exoRBD) and loaded
with cholesterol tagged STING (exoRBD/ECC-STING) were stored at either 37°C or -20°C degrees for 30 days. Wild type C57BL/6 mice were immunized subcutaneously with exoRBD and exoRBD/ECC STING from the two storage conditions on days 0 and 14. Sera anti-RBD IgG antibody titers (FIG. 2 A) and neutralizing antibody titers (FIG. 2B) were measured 14 days after the second immunization. Storage of exoRBD and exoRBD/ ECC-STING at 37°C for 30 days did not affect the RBD antigen immunogenicity (FIG. 2A). Further, storage of exoRBD/ ECC-STING did not alter the adjuvant activity as measured by both anti-RBD IgG antibody (FIG. 2A) and neutralizing antibody (FIG. 2B) responses.
[0186] ExoRBD (SARS CoV-1 & SARS CoV-2) dose response
[0187] Exosomes expressing the RBDs from both SARS CoVl and CoV2 (both fused to PTGFRN) were loaded with cholesterol tagged STING (ECC STING). Wild type C57BL/6 mice were immunized subcutaneously with three different doses of exosomes (4E11, 2E11, and 1E11) or PBS on days 0 and 14. Fourteen days after the second dose, sera neutralizing antibody titers and anti-RBD IgG antibody titers were measured. Sera from human subjects obtained 14 days after the second dose of an mRNA vaccine (Human mRNA Vax) was used as a comparator. Sera from each mouse group (N=5) and human subject sera (N=5) were pooled for the analyses. Neutralizing antibody responses against RBDs from the wild type (Washington) SARS CoV-2 strain, SARS CoV-1, WIV-1 bat coronavirus, and the Omicron variant of SARS CoV-2 were measured; and anti-RBD IgG antibody responses against RBDs from the wild type (Washington) SARS CoV-2 strain, SARS CoV-1, the Omicron variant of SARS CoV-2, and the bat coronavirus RaTG13 were measured.
[0188] Immunization of mice with exosomes expressing both SARS CoV-1 & CoV-2 RBDs induced neutralizing antibodies against the RBDs from SARS CoV-1 and SARS CoV-2 including the omicron variant in a dose dependent manner (FIG. 3A). Furthermore cross-neutralizing antibody responses were generated sufficient to neutralize a different beta coronavirus (WIV-1) suggesting a pan-beta CoV immune response was generated.
[0189] Immunization of mice with exosomes expressing both SARS CoV-1 & CoV-2 RBDs and loaded with ECC STING induced anti-RBD antibodies against the RBDs from SARS CoV-1 and SARS CoV-2 including the omicron variant in a dose dependent manner (FIG. 3B). Furthermore cross-neutralizing antibody responses were generated sufficient to bind an RBD from a different beta coronavirus (RaTG13) suggesting a pan-beta CoV immune response was generated.
[0190] Fourteen days after the 2nd dose, sera splenocytes were assayed for T cell responses against SARS CoV-2 WT RBD using overlapping RBD peptides and culturing for 48 hours. Supernatants from the splenocyte cultures were measured for fFNy expression (FIG. 3C) and TNFa expression (FIG. 3D) using Alphalisa. Immunization of mice with exosomes expressing both SARS CoV-1 & CoV-2 RBDs induced T cell responses against the RBD from SARS CoV-2 in a dose dependent manner.
INCORPORATION BY REFERENCE
[0191] All publications, patents, patent applications and other documents cited in this application are hereby incorporated by reference in their entireties for all purposes to the same extent as if each individual publication, patent, patent application or other document were individually indicated to be incorporated by reference for all purposes.
EQUIVALENTS
[0192] The present disclosure provides, inter alia, compositions of exosomes comprising cholesterol tagged STING agonists for use as therapeutics. The present disclosure also provides methods of producing exosomes comprising cholesterol tagged STING agonists and methods of administering such exosomes as therapeutics. While various specific aspects have been illustrated and described, the above specification is not restrictive. It will be appreciated that various changes can be made without departing from the spirit and scope of the invention(s). Many variations will become apparent to those skilled in the art upon review of this specification.
Claims
64
CLAIMS An extracellular vesicle (EV) comprising a cholesterol or derivative thereof on the surface, wherein the cholesterol is linked to a stimulator of interferon genes protein (STING) agonist through a cleavable peptide linker. The EV of claim 1, wherein the cleavable peptide linker comprises a linker cleavable by cathepsin. The EV of claim 1 or 2, wherein the cleavable peptide linker comprises a Valine- Alanine linker and/or a Valine-Citrulline linker. The EV of any one of claims 1 to 3, wherein the EV is an exosome, a nanovesicle, an apoptotic body, a microvesicle, a lysosome, an endosome, a liposome, a lipid nanoparticle, a micelle, a multilamellar structure, a revesiculated vesicle, or an extruded cell. The EV of claim 4, wherein the EV is an exosome. The EV of any one of claims 1 to 5, wherein the EV overexpresses a PTGFRN protein. The EV of any one of claims 1 to 6, wherein the EV is produced by a cell that overexpresses a PTGFRN protein. The EV of any one of claims 1 to 7, wherein the extracellular vesicle further comprises a ligand, a cytokine, an antigen, or an antibody. The EV of claim 8, wherein the antibody comprises an antagonistic antibody and/or an agonistic antibody. The EV of any one of claims 1 to 9, wherein the STING agonist is a cyclic dinucleotide. The EV of any one of claims 1 to 9, wherein the STING agonist is a non-cyclic dinucleotide. The EV of claim 10 or 11, wherein the STING agonist is modified such that a polarity and/or a charge different from the corresponding unmodified STING agonist. The EV of any one of claims 1 to 12, wherein the STING agonist comprises:
65
Formula 1 Formula 2 Formula 3

Xi is H, OH, or F;
X2 is H, OH, or F;
Z is OH, ORi, SH or SRi, wherein: i) Ri is Na or NH4, or ii) Ri is an enzyme-labile group which provides OH or SH in vivo such as pivaloyloxymethyl;
Bi and B2 are bases chosen from:

Guanine, with the proviso that:
- in Formula (I): Xi and X2 are not OH,
- in Formula (II): when Xi and X2 are OH, Bi is not Adenine and B2 is not Guanine, and
- in Formula (III): when Xi and X2 are OH, Bi is not Adenine, B2 is not Guanine and Z is not OH, or a pharmaceutically acceptable salt thereof.
14. The EV of claim 13, wherein the STING agonist is selected from the group consisting of:
66

 and a pharmaceutically acceptable salt thereof.
and a pharmaceutically acceptable salt thereof.
15. The EV of any one of claims 1 to 14, wherein the cholesterol or derivative thereof comprises:

16. The EV of claim 1, which comprises:
68

(Formula I),
 u Cholesterol linker
u Cholesterol linker
(Formula IV).
17. The EV of any one of claims 1 to 16, which further comprises a scaffold moiety.
The EV of claim 17, wherein the scaffold moiety comprises Scaffold X, which comprises an amino acid sequence as set forth in SEQ ID NO: 302. The EV of claim 18, wherein the Scaffold X comprises an amino acid sequence at least 50%, at least 60%, at least 70%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or about 100% identical to SEQ ID NO:302. The EV of claim 19, wherein the scaffold moiety comprises Scaffold Y, which comprises a BASP1 protein or a functional fragment thereof. The EV of claim 20, wherein Scaffold Y comprises an amino acid sequence as set forth in GGKLSKK (SEQ ID NO: 411). The EV of claim 21, wherein Scaffold Y comprises an amino acid sequence as set forth in i) GGKLSKKK (SEQ ID NO: 438), (ii) GGKLSKKS (SEQ ID NO: 439), (iii) GAKLSKKK (SEQ ID NO: 440), (iv) GAKLSKKS (SEQ ID NO: 441), (v) GGKQSKKK (SEQ ID NO: 442), (vi) GGKQSKKS (SEQ ID NO: 443), (vii) GGKLAKKK (SEQ ID NO: 444), (viii) GGKLAKKS (SEQ ID NO: 445), or (ix) any combination thereof. A pharmaceutical composition comprising the EV of any one of claims 1 to 22 and a pharmaceutically acceptable carrier. The composition of claim 23, wherein, when administered to a mammal, the composition does not deplete T cells and/or macrophages in the mammal. The composition of claim 23, wherein, when administered to a mammal, the composition depletes T cells and/or macrophages in the mammal at a lesser degree than the free STING agonist. A kit comprising the composition of any one of claim 23 to 25 and instructions for use. A method of producing an EV, e.g., exosome, comprising a cholesterol or derivative thereof, which is linked to a STING agonist via a cleavable peptide linker, the method comprising: a. Obtaining an EV, e.g., exosome; b. Mixing the EV, e.g., exosome with cholesterol linked to a STING agonist via a cleavable peptide linker in a solution;
c. Incubating the mixture of the EV, e.g., exosome and the cholesterol linked to the STING agonist in a solution comprising a buffer under suitable conditions; and d. Purifying the EV, e.g., exosome. The method of claim 27, wherein the cleavable peptide linker comprises a linker cleavable by cathepsin. The method of claim 28, wherein the cleavable peptide linker comprises a Valine-Alanine linker and/or a Valine-Citrulline linker. A method of inducing or modulating an immune response and/or an inflammatory response in a subject in need thereof, the method comprising administering to the subject the EV of any one of claims 1 to 22 or the composition of any one of claims 23 to 25. A method of treating a tumor in a subject in need thereof, the method comprising administering to the subject the EV of any one of claims 1 to 22 or the composition of any one of claims 23 to 25. The method of claim 30 or 31, wherein the administering induces or modulates the immune response and/or the inflammatory response in the subject. The method of any one of claims 30 to 32, wherein the administering activates dendritic cells. The method of any one of claims 30 to 33, wherein the administering activates myeloid dendritic cells. The method of any one of claims 30 to 34, wherein the administering results in reduced monocyte cell activation compared to the free STING agonist. The method of any one of claims 30 to 35, wherein the administering does not induce monocyte cell activation. The method of any one of claims 30 to 36, wherein the administering induces interferon-P (IFN-P) production. The method of any one of claims 30 to 37, wherein the administering results in reduced systemic inflammation compared to the free STING agonist. The method of any one of claims 30 to 38, wherein the administering results in insubstantial amounts of systemic inflammation.
The method of any one of claims 30 to 39, wherein the administration is parenterally, orally, intravenously, intramuscularly, intra-tumorally, intraperitoneally, or via any other appropriate administration route. The method of any one of claims 30 to 40, wherein the administration is intravenous. The method of any one of claims 30 to 41, wherein the immune response is an anti-tumor response. The method of any one of claims 30 to 42, further comprising administering an additional therapeutic agent. The method of claim 43, wherein the additional therapeutic agent is an immunomodulating agent. The method of claim 43 or 44, wherein the additional therapeutic agent is an antibody or antigen-binding fragment thereof. The method of claim 45, wherein the antibody or antigen-binding fragment thereof is an inhibitor of CTLA-4, PD-1, PD-L1, PD-L2, TIM-3, or LAG3. The method of any one of claims 30 to 46, wherein the administering prevents metastasis of the tumor in the subject. The method of any one of claims 31 to 47, wherein the tumor comprises or is derived from a chondroid sarcoma, a cutaneous squamous cell carcinoma (cSCC), a head and neck squamous cell carcinoma of the oral cavity, a hepatocellular cancer (HCC), a cervical cancer, an eye melanoma, a choroidal eye melanoma, a gastric spindle cell sarcoma, a hemangioendothelioma, a mesothelioma, a parotoid gland cancer, a renal cancer, a triple-negative breast cancer (TNBC), or any combination thereof. The EV of any one of claims 1 to 22, which is thermostable at about 20°C for at least about 7 days, at least about 8 days, at least about 9 days, at least about 10 days, at least about 11 days, at least about 12 days, at least about 13 days, at least about 14 days, at least about 15 days, at least about 16 days, at least about 17 days, at least about 18 days, at least about 19 days, at least about 20 days, at least about 21 days, at least about 22 days, at least about 23 days, at least about 24 days, at least about 25 days, at least about 26 days, at least about 27 days, at least about 28 days, at least about 29 days, or at least about 30 days.
72 The EV of any one of claims 1 to 22 or 49, which is thermostable at about 37°C for at least about 7 days, at least about 8 days, at least about 9 days, at least about 10 days, at least about 11 days, at least about 12 days, at least about 13 days, at least about 14 days, at least about 15 days, at least about 16 days, at least about 17 days, at least about 18 days, at least about 19 days, at least about 20 days, at least about 21 days, at least about 22 days, at least about 23 days, at least about 24 days, at least about 25 days, at least about 26 days, at least about 27 days, at least about 28 days, at least about 29 days, or at least about 30 days. The EV of any one of claims 1 to 22, 49, and 50, which is capable of inducing an immune response in a subject.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202163261918P | 2021-09-30 | 2021-09-30 | |
| US63/261,918 | 2021-09-30 | ||
| US202263364875P | 2022-05-17 | 2022-05-17 | |
| US63/364,875 | 2022-05-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023056468A1 true WO2023056468A1 (en) | 2023-04-06 |
Family
ID=83903314
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2022/077424 Ceased WO2023056468A1 (en) | 2021-09-30 | 2022-09-30 | Extracellular vesicle comprising cholesterol tagged sting-agonist |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023056468A1 (en) |
Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013185052A1 (en) | 2012-06-08 | 2013-12-12 | Aduro Biotech | Compostions and methods for cancer immunotherapy |
| WO2014093936A1 (en) | 2012-12-13 | 2014-06-19 | Aduro Biotech, Inc. | Compositions comprising cyclic purine dinucleotides having defined stereochemistries and methods for their preparation and use |
| WO2014179760A1 (en) | 2013-05-03 | 2014-11-06 | The Regents Of The University Of California | Cyclic di-nucleotide induction of type i interferon |
| WO2014179335A1 (en) | 2013-04-29 | 2014-11-06 | Memorial Sloan Kettering Cancer Center | Compositions and methods for altering second messenger signaling |
| WO2014189806A1 (en) | 2013-05-18 | 2014-11-27 | Aduro Biotech, Inc. | Compositions and methods for inhibiting "stimulator of interferon gene" dependent signalling |
| WO2014189805A1 (en) | 2013-05-18 | 2014-11-27 | Auro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene"-dependent signalling |
| WO2015017652A1 (en) | 2013-07-31 | 2015-02-05 | Memorial Sloan-Kettering Cancer Center | Sting crystals and modulators |
| WO2015077354A1 (en) | 2013-11-19 | 2015-05-28 | The University Of Chicago | Use of sting agonist as cancer treatment |
| WO2015185565A1 (en) | 2014-06-04 | 2015-12-10 | Glaxosmithkline Intellectual Property Development Limited | Cyclic di-nucleotides as modulators of sting |
| WO2016096577A1 (en) | 2014-12-16 | 2016-06-23 | Invivogen | Combined use of a chemotherapeutic agent and a cyclic dinucleotide for cancer treatment |
| WO2016096174A1 (en) | 2014-12-16 | 2016-06-23 | Invivogen | Fluorinated cyclic dinucleotides for cytokine induction |
| WO2016120305A1 (en) | 2015-01-29 | 2016-08-04 | Glaxosmithkline Intellectual Property Development Limited | Cyclic dinucleotides useful for the treatment of inter alia cancer |
| WO2016145102A1 (en) | 2015-03-10 | 2016-09-15 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene" -dependent signalling |
| WO2017027645A1 (en) | 2015-08-13 | 2017-02-16 | Merck Sharp & Dohme Corp. | Cyclic di-nucleotide compounds as sting agonists |
| WO2017075477A1 (en) | 2015-10-28 | 2017-05-04 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene"-dependent signalling |
| WO2017175147A1 (en) | 2016-04-07 | 2017-10-12 | Glaxosmithkline Intellectual Property Development Limited | Heterocyclic amides useful as protein modulators |
| WO2017175156A1 (en) | 2016-04-07 | 2017-10-12 | Glaxosmithkline Intellectual Property Development Limited | Heterocyclic amides useful as protein modulators |
| WO2018100558A2 (en) | 2016-12-01 | 2018-06-07 | Takeda Pharmaceutical Company Limited | Cyclic dinucleotide |
| US10195290B1 (en) | 2017-08-25 | 2019-02-05 | Codiak Biosciences, Inc. | Preparation of therapeutic exosomes using membrane proteins |
| WO2019183578A1 (en) * | 2018-03-23 | 2019-09-26 | Codiak Biosciences, Inc. | Extracellular vesicles comprising sting-agonist |
| WO2020191377A1 (en) * | 2019-03-21 | 2020-09-24 | Codiak Biosciences, Inc. | Extracellular vesicle conjugates and uses thereof |
| WO2021062060A1 (en) * | 2019-09-25 | 2021-04-01 | Codiak Biosciences, Inc. | Sting agonist comprising exosomes combined with il-12 displaying exosomes for treating a tumour |
| WO2021189047A2 (en) * | 2020-03-20 | 2021-09-23 | Codiak Biosciences, Inc. | Extracellular vesicles for therapy |
-
2022
- 2022-09-30 WO PCT/US2022/077424 patent/WO2023056468A1/en not_active Ceased
Patent Citations (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013185052A1 (en) | 2012-06-08 | 2013-12-12 | Aduro Biotech | Compostions and methods for cancer immunotherapy |
| WO2014093936A1 (en) | 2012-12-13 | 2014-06-19 | Aduro Biotech, Inc. | Compositions comprising cyclic purine dinucleotides having defined stereochemistries and methods for their preparation and use |
| WO2014179335A1 (en) | 2013-04-29 | 2014-11-06 | Memorial Sloan Kettering Cancer Center | Compositions and methods for altering second messenger signaling |
| WO2014179760A1 (en) | 2013-05-03 | 2014-11-06 | The Regents Of The University Of California | Cyclic di-nucleotide induction of type i interferon |
| WO2014189806A1 (en) | 2013-05-18 | 2014-11-27 | Aduro Biotech, Inc. | Compositions and methods for inhibiting "stimulator of interferon gene" dependent signalling |
| WO2014189805A1 (en) | 2013-05-18 | 2014-11-27 | Auro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene"-dependent signalling |
| WO2015017652A1 (en) | 2013-07-31 | 2015-02-05 | Memorial Sloan-Kettering Cancer Center | Sting crystals and modulators |
| WO2015077354A1 (en) | 2013-11-19 | 2015-05-28 | The University Of Chicago | Use of sting agonist as cancer treatment |
| WO2015185565A1 (en) | 2014-06-04 | 2015-12-10 | Glaxosmithkline Intellectual Property Development Limited | Cyclic di-nucleotides as modulators of sting |
| WO2016096577A1 (en) | 2014-12-16 | 2016-06-23 | Invivogen | Combined use of a chemotherapeutic agent and a cyclic dinucleotide for cancer treatment |
| WO2016096174A1 (en) | 2014-12-16 | 2016-06-23 | Invivogen | Fluorinated cyclic dinucleotides for cytokine induction |
| WO2016120305A1 (en) | 2015-01-29 | 2016-08-04 | Glaxosmithkline Intellectual Property Development Limited | Cyclic dinucleotides useful for the treatment of inter alia cancer |
| WO2016145102A1 (en) | 2015-03-10 | 2016-09-15 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene" -dependent signalling |
| WO2017027645A1 (en) | 2015-08-13 | 2017-02-16 | Merck Sharp & Dohme Corp. | Cyclic di-nucleotide compounds as sting agonists |
| WO2017027646A1 (en) | 2015-08-13 | 2017-02-16 | Merck Sharp & Dohme Corp. | Cyclic di-nucleotide compounds as sting agonists |
| WO2017075477A1 (en) | 2015-10-28 | 2017-05-04 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene"-dependent signalling |
| WO2017175147A1 (en) | 2016-04-07 | 2017-10-12 | Glaxosmithkline Intellectual Property Development Limited | Heterocyclic amides useful as protein modulators |
| WO2017175156A1 (en) | 2016-04-07 | 2017-10-12 | Glaxosmithkline Intellectual Property Development Limited | Heterocyclic amides useful as protein modulators |
| WO2018100558A2 (en) | 2016-12-01 | 2018-06-07 | Takeda Pharmaceutical Company Limited | Cyclic dinucleotide |
| US10195290B1 (en) | 2017-08-25 | 2019-02-05 | Codiak Biosciences, Inc. | Preparation of therapeutic exosomes using membrane proteins |
| WO2019183578A1 (en) * | 2018-03-23 | 2019-09-26 | Codiak Biosciences, Inc. | Extracellular vesicles comprising sting-agonist |
| WO2020191377A1 (en) * | 2019-03-21 | 2020-09-24 | Codiak Biosciences, Inc. | Extracellular vesicle conjugates and uses thereof |
| WO2021062060A1 (en) * | 2019-09-25 | 2021-04-01 | Codiak Biosciences, Inc. | Sting agonist comprising exosomes combined with il-12 displaying exosomes for treating a tumour |
| WO2021189047A2 (en) * | 2020-03-20 | 2021-09-23 | Codiak Biosciences, Inc. | Extracellular vesicles for therapy |
Non-Patent Citations (13)
| Title |
|---|
| "Genes II", 1985, JOHN WILEY & SONS |
| "Oxford Dictionary Of Biochemistry And Molecular Biology", 2000, OXFORD UNIVERSITY PRESS |
| "Remington's Pharmaceutical Sciences", 1990, MACK PUBLISHING CO. |
| "The Dictionary of Cell and Molecular Biology", 1999, ACADEMIC PRESS |
| COLOWICKKAPLAN: "Advanced Organic Chemistry: Parts A and B", 2007, ACADEMIC PRESS |
| DOBELI ET AL., J. BIOTECHNOLOGY, vol. 7, 1988, pages 199 - 216 |
| GAYLE, J. BIOL. CHEM, vol. 268, 1993, pages 22105 - 22111 |
| GREENSAMBROOK ET AL.: "Remington: The Science and Practice of Pharmacy", 2012, COLD SPRING HARBOR LABORATORY PRESS |
| JUO, PEI-SHOW: "Concise Dictionary of Biomedicine and Molecular Biology", 2002, CRC PRESS |
| MEI ET AL., BLOOD, vol. 116, 2010, pages 270 - 79 |
| PAPAPETROU ET AL., GENE THERAPY, vol. 12, 2005, pages S118 - S130 |
| RON ET AL., J. BIOL. CHEM., vol. 268, 1993, pages 2984 - 2988 |
| T.E. CREIGHTON: "Proteins: Structures and Molecular Properties", 1993, W.H. FREEMAN AND COMPANY |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20210322327A1 (en) | Extracellular vesicles comprising sting-agonist | |
| EP4678184A2 (en) | Engineered chimeric fusion protein compositions and methods of use thereof | |
| US20230181758A1 (en) | Extracellular vesicles targeting dendritic cells and uses thereof | |
| US20220395465A1 (en) | Sting agonist comprising exosomes combined with il-12 displaying exosomes for treating a tumour | |
| US20250249072A1 (en) | Methods of treating cancer | |
| US20250129170A1 (en) | Epo receptor agonists and antagonists | |
| US20250381248A1 (en) | Methods of treating a tumor | |
| WO2023056468A1 (en) | Extracellular vesicle comprising cholesterol tagged sting-agonist | |
| EP3860622A1 (en) | Methods for treating cancer with double stranded rna sensor activators and adoptive cell therapy | |
| US20250381292A1 (en) | Methods of administering a sting agonist | |
| HK40119152A (en) | Methods of administering a sting agonist | |
| CN117836327A (en) | Engineered chimeric fusion protein compositions and methods of use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22793358 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 22793358 Country of ref document: EP Kind code of ref document: A1 |