WO2022130206A1 - TGFβr1 INHIBITOR COMBINATION THERAPIES - Google Patents
TGFβr1 INHIBITOR COMBINATION THERAPIES Download PDFInfo
- Publication number
- WO2022130206A1 WO2022130206A1 PCT/IB2021/061713 IB2021061713W WO2022130206A1 WO 2022130206 A1 WO2022130206 A1 WO 2022130206A1 IB 2021061713 W IB2021061713 W IB 2021061713W WO 2022130206 A1 WO2022130206 A1 WO 2022130206A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- antibody
- pharmaceutically acceptable
- inhibitor
- acceptable salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/455—Nicotinic acids, e.g. niacin; Derivatives thereof, e.g. esters, amides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
Definitions
- the .txt file contains a sequence listing entitled “PC072670SEQLISTING_ST25.txt created on November 15, 2021 and having a size of 85 KB.
- the sequence listing contained in this .txt file is part of the specification and is herein incorporated by reference in its entirety.
- the present invention relates to methods and combination therapies useful for the treatment of cancer.
- the invention relates to method of treating cancer comprising administering to the subject an amount of a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, or a pharmaceutically acceptable salt thereof and an immune checkpoint inhibitor (e.g., PD-1 axis binding antagonists, 0X40 agonists, 4-1 BB agonists and CTLA4 antagonists, or combinations thereof).
- TGFprl transforming growth factor beta receptor type 1
- an immune checkpoint inhibitor e.g., PD-1 axis binding antagonists, 0X40 agonists, 4-1 BB agonists and CTLA4 antagonists, or combinations thereof.
- Embodiments of the present invention relate to associated combinations for use in treatment of cancer, pharmaceutical compositions and uses thereof.
- TGFp signalling is an emerging pathway in cancer progression and has a role in modulating immune response, and in many other cancer pathways including metastasis and angiogenesis. Elevated TGFp expression by tumor and stromal cells in the tumor microenvironment and activation of TGFp receptor intracellular signalling is observed in many cancers (Massague J., TGFbeta in Cancer. Cell 2008, 134(2):215-30; Neuzillet C, et al., Targeting the TGFp pathway for cancer therapy, Pharmacol Ther 2015, 147:22- 31 ).
- the TGFp signalling pathway can be activated upon interaction of dimeric TGFp ligand with its specific cell-surface transmembrane serine/threonine kinase receptors.
- TGFpr2 TGFp type II receptors
- TGFprl TGFp type I receptors
- ALK5 activin receptor-like kinase
- EMT epithelial-to- mesenchymal transition
- epithelial cells lose their apico-basal polarity and cell-cell adhesion, to become highly migratory mesenchymal cells, leading to metastasis.
- EMT has also been linked to tumor cell evasion of immune surveillance (Akalay I, et al., Epithelial-to- mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis, Cancer Res 2013, 73(8):2418-27).
- TGF is a potent immunosuppressive agent on both innate and adaptive immune cells, including dendritic cells, macrophages, natural killer cells, and CD4+ and CD8+ T cells.
- TGFp has a key role stimulating the differentiation of immune-suppressive regulatory T (Treg) cells and myeloid derived suppressor cells (MDSCs) (Akalay I, et al., 2013).
- TGF pathways have key roles in disease progression and resistance to therapy in a broad spectrum of tumors (Neuzillet C., et. al., 2015; Colak S, et. al., Targeting TGF- B signaling in cancer, Trends in Cancer 2017, 3(1 ):56-71 ).
- High TGFp signatures and EMT gene expression are found in a variety of tumors (Mak MP, etal., A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition, Clin Cancer Res 2016, 22(3):609-20.).
- TGFp is an important regulator of the tumor microenvironment by inducing expression of extracellular matrix (ECM) proteins and suppressing expression of chemokines and cytokines required for T cell tumor infiltration, creating a reactive stroma with dense ECM and a T cell excluded infiltrate phenotype, with peritumoral or stromal T cell localization (Hegde PS, et. al., The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition, Clin Cancer F?es 2016, 22(8):1865-74).
- ECM extracellular matrix
- TGFprl inhibitor PF- 06952229 transforming growth factor beta receptor type 1
- PF-06952229 transforming growth factor beta receptor type 1
- the programmed death 1 (PD-1) receptor and PD-1 ligands 1 and 2 play integral roles in immune regulation.
- PD-1 is a key immune checkpoint receptor expressed by activated T and B cells.
- PD-1 is a member of the CD28 family of receptors, which includes CD28, CTLA4, ICOS, PD-1 , and BTLA.
- Two cell surface glycoprotein ligands for PD-1 have been identified, Programmed Death Ligand- 1 (PD-L1 ) and Programmed Death Ligand-2 (PD-L2), that are expressed on antigen- presenting cells as well as many human cancers and have been shown to down-regulate T cell activation and cytokine secretion upon binding to PD-1 .
- PD-1 axis binding antagonists including the anti-PD-1 antibodies nivolumab (Opdivo), and pembrolizumab (Keytruda) were approved by the U.S. Food and Drug Administration (FDA) for the treatment of oncology indications in recent years.
- FDA Food and Drug Administration
- the 0X40 receptor (0X40, also known as CD134, TNFRSF4, ACT-4, ACT35, and TXGP1 L) is a member of the TNF receptor super family. 0X40 is found to be expressed on activated CD4+ and CD8+ T-cells. High numbers of 0X40+ T cells have been demonstrated within tumors (tumor infiltrating lymphocytes) and in the draining lymph nodes of cancer patients (Weinberg, A. et a!., Engagement of the OX-40 receptor in vivo enhances antitumor immunity, J. Immunol. 2000,164: 2160-69; Petty, J.
- 4-1 BB (also known as CD137 and TNFRSF9), which was first identified as an inducible costimulatory receptor expressed on activated T cells, is a membrane spanning glycoprotein of the Tumor Necrosis Factor (TNF) receptor superfamily.
- TNF Tumor Necrosis Factor
- Current understanding of 4-1 BB indicates that expression is generally activation dependent and encompasses a broad subset of immune cells including activated NK and NKT cells; regulatory T cells; dendritic cells (DC) including follicular DC; stimulated mast cells, differentiating myeloid cells, monocytes, neutrophils, eosinophils, and activated B cells. 4-1 BB expression has also been demonstrated on tumor vasculature (19-20) and atherosclerotic endothelium.
- the ligand that stimulates 4-1 BB (4-1 BBL) is expressed on activated antigen presenting cells (APCs), myeloid progenitor cells and hematopoietic stem cells.
- APCs activated antigen presenting cells
- 4-1 BB agonist mAbs increase costimulatory molecule expression and markedly enhance cytolytic T lymphocyte responses, resulting in anti-tumor efficacy in various models.
- 4-1 BB agonist mAbs have demonstrated efficacy in prophylactic and therapeutic settings and both monotherapy and combination therapy tumor models and have established durable anti-tumor protective T cell memory responses.
- CTLA4 Cytotoxic T lymphocyte associated antigen 4
- CTLA4 is expressed on activated T cells and serves as a co-inhibitor to keep T cell responses in check following CD28- mediated T cell activation.
- CTLA4 is believed to regulate the amplitude of the early activation of naive and memory T cells following TCR engagement and to be part of a central inhibitory pathway that affects both antitumor immunity and autoimmunity.
- CTLA4 is expressed exclusively on T cells, and the expression of its ligands CD80 (B7.1 ) and CD86 (B7.2), is largely restricted to antigen-presenting cells, T cells, and other immune mediating cells.
- Antagonistic anti-CTLA4 antibodies that block the CTLA4 signaling pathway have been reported to enhance T cell activation.
- ipilimumab was approved by the FDA in 2011 for the treatment of metastatic melanoma.
- Another anti-CTLA4 antibody, tremelimumab was tested in phase III trials for the treatment of advanced melanoma but did not significantly increase the overall survival of patients compared to the standard of care (temozolomide or dacarbazine) at that time.
- the methods, combinations, uses and pharmaceutical compositions of the present invention have been demonstrated greater efficacy than treatment with either therapeutic agent alone.
- the methods, combinations, uses and pharmaceutical compositions of the present invention are believed to have one or more advantages, such as potential to reduce drug-drug interactions; potential to enable an improved dosing schedule; potential to reduce side effects; potential to overcome resistance mechanisms and the like.
- kits comprising one or more of the compositions of the invention.
- the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject:
- 3r1 transforming growth factor beta receptor type 1
- the invention provides a method further comprising administering to the subject (c) an amount of an additional anti-cancer agent; wherein the amounts in (a), (b) and (c) together are effective in treating cancer.
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof.
- the PD-1 axis binding antagonist is an anti-PD-1 antibody.
- the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-011 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001 ), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, Bl- 754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009,
- the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-PD-1 antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
- TGFprl transforming growth factor beta receptor type 1
- the anti-PD-1 antibody is sasanlimab (PF-6801591), and wherein the amounts together are effective in treating cancer.
- the TGFprl inhibitor and the PD-1 axis binding antagonist are administered sequentially, simultaneously, or concurrently.
- the invention provides a combination comprising:
- TGFprl transforming growth factor beta receptor type 1
- a PD-1 axis binding antagonist for use in treating cancer.
- the combination for use further comprises
- the TGF rl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2- (5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof.
- the PD-1 axis binding antagonist is an anti-PD-1 antibody.
- the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-01 1), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD
- the anti-PD- 1 antibody is sasanlimab (PF-6801591 ).
- the invention provides a combination comprising a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-PD-1 antibody, wherein the TGF[3r1 inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and wherein the anti-PD-1 antibody is sasanlimab (PF-6801591), for use in treating cancer.
- TGFprl transforming growth factor beta receptor type 1
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a PD-1 axis binding antagonist, for treating cancer.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a PD-1 axis binding antagonist, for use in combination with 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer.
- the PD-1 axis binding antagonist is an anti-PD-1 antibody.
- the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-01 1 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ),
- the anti- PD-1 antibody is sasanlimab (PF-6801591 ).
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
- an anti-PD-1 antibody wherein the anti-PD-1 antibody is sasanlimab (PF-6801591 ), for treating cancer.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising an anti-PD-1 antibody, for use in combination with 4-(2-(5-chloro- 2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the anti-PD-1 antibody is sasanlimab (PF-6801591 ), for treating cancer.
- the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject: (a) an amount of a transforming growth factor beta receptor type 1 (TGF[3r1) inhibitor, or a pharmaceutically acceptable salt thereof, and;
- TGF[3r1) inhibitor transforming growth factor beta receptor type 1
- the invention provides a method further comprising administering to the subject (c) an amount of an additional anti-cancer agent; wherein the amounts in (a), (b) and (c) together are effective in treating cancer.
- the TGF rl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof.
- the 0X40 agonist is selected from the group consisting of an anti-OX40 antibody, an OX40L agonist fragment, an 0X40 oligomeric receptor, a trimeric OX40L-Fc protein and an 0X40 immunoadhesin, or a combination thereof.
- the 0X40 agonist is an anti-OX40 antibody.
- the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR-8383, and ABBV-368, or combinations thereof.
- the anti-OX40 antibody PF-04518600.
- the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-OX40 antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof, wherein the anti-OX40 antibody is PF- 04518600, and wherein the amounts together are effective in treating cancer.
- TGFprl transforming growth factor beta receptor type 1
- the TGFprl inhibitor and the 0X40 agonist are administered sequentially, simultaneously, or concurrently.
- the invention provides a combination comprising:
- TGFprl transforming growth factor beta receptor type 1
- the combination for use further comprises
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2- (5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof.
- 3r1 inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof.
- the 0X40 agonist is an anti-OX40 antibody, an OX40L agonist fragment, an 0X40 oligomeric receptor, a trimeric OX40L-Fc protein or an 0X40 immunoadhesin, or combinations thereof.
- the 0X40 agonist is an anti-OX40 antibody.
- the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG- 7888, GSK-3174998, BMS-986178, GBR-8383, and ABBV-368, or combinations thereof.
- the anti-OX40 antibody is PF-04518600.
- the invention provides a combination comprising a transforming growth factor beta receptor type 1 (TGF rl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-OX40 antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and wherein the anti-OX40 antibody is PF- 04518600, for use in treating cancer.
- TGF rl transforming growth factor beta receptor type 1
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with an 0X40 agonist, for treating cancer.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising an 0X40 agonist, for use in combination with 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer.
- the 0X40 agonist is an anti-OX40 antibody.
- the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR- 8383, and ABBV-368, or combinations thereof.
- the anti- 0X40 antibody is PF-04518600.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with an anti-OX40 antibody, wherein the anti-OX40 antibody is PF-04518600, for treating cancer.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising an anti-OX40 antibody, for use in combination with 4-(2-(5- chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the anti-OX40 antibody is PF-04518600, for treating cancer.
- the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject:
- TGFprl transforming growth factor beta receptor type 1
- the invention provides a method further comprising administering to the subject (c) an amount of an additional anti-cancer agent; wherein the amounts in (a), (b) and (c) together are effective in treating cancer.
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGF[3r1 inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof.
- the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4- 1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No.
- the 4-1 BB agonist is utomilumab.
- the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-4-1 BB antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof, wherein the anti-4-1 BB antibody is utomilumab, and wherein the amounts together are effective in treating cancer.
- TGFprl transforming growth factor beta receptor type 1
- the TGFprl inhibitor and the 4-1 BB agonist are administered sequentially, simultaneously, or concurrently.
- the invention provides a combination comprising:
- TGFprl transforming growth factor beta receptor type 1
- the combination for use further comprises
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof.
- the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC
- the 4-1 BB agonist is utomilumab.
- the invention provides a combination comprising a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of a 4-1 BB agonist, wherein the TGF[3r1 inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan- 2-yl)nicotinamide having the structure:
- TGFprl transforming growth factor beta receptor type 1
- the 4-1 BB agonist is utomilumab, for use in treating cancer.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a 4-1 BB agonist, for treating cancer.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a 4-1 BB agonist, for use in combination with 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer.
- the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No.
- the 4-1 BB agonist is utomilumab.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a 4-1 BB agonist, wherein the 4-1 BB agonist is utomilumab, for treating cancer.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a 4-1 BB agonist, for use in combination with 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the 4-1 BB agonist is utomilumab, for treating cancer.
- the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject:
- TGFprl transforming growth factor beta receptor type 1
- the invention provides a method further comprising administering to the subject (c) an amount of an additional anti-cancer agent; wherein the amounts in (a), (b) and (c) together are effective in treating cancer.
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof.
- the CTLA4 antagonist is an anti-CTLA4 antibody.
- the anti-CTLA4 antibody is selected from the group consisting of ipilimumab (1 ODI), tremelimumab, and AGEN-1884, or combinations thereof.
- the anti-CTLA4 antibody is ipilimumab (10DI).
- the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-CTLA4 antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof, wherein the anti-CTLA4 antibody is ipilimumab (1 ODI), and wherein the amounts together are effective in treating cancer.
- TGFprl transforming growth factor beta receptor type 1
- the TGFprl inhibitor and the CTLA4 antagonist are administered sequentially, simultaneously, or concurrently.
- the invention provides a combination comprising:
- TGFprl transforming growth factor beta receptor type 1
- the combination for use further comprises
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure: or a pharmaceutically acceptable salt thereof.
- the anti-CTLA4 antibody is selected from the group consisting of ipilimumab (1 ODI), tremelimumab, and AGEN-1884, or combinations thereof.
- the anti-CTLA4 antibody is ipilimumab (1 ODI).
- the invention provides a combination comprising a transforming growth factor beta receptor type 1 (TGF rl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-CTLA4 antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and wherein the anti-CTLA4 antibody is ipilimumab (1 ODI), for use in treating cancer.
- TGF rl transforming growth factor beta receptor type 1
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a CTLA4 antagonist.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a CTLA4 antagonist, for use in combination with 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer.
- the CTLA4 antagonist is an anti-CTLA4 antibody.
- the anti- CTLA4 antibody is selected from the group consisting of ipilimumab (10DI), tremelimumab, and AGEN-1884, or combinations thereof.
- the anti-CTLA4 antibody is ipilimumab (1 GDI).
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with an anti-CTLA4 antibody, wherein the anti-CTLA4 antibody is ipilimumab (1 GDI), for treating cancer.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising an anti-CTLA4 antibody, for use in combination with 4-(2-(5- chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide having the structure: or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the anti-CTLA4 antibody is ipilimumab (10DI), for treating cancer.
- the additional anti-cancer agent is selected from the group consisting of a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and anti-angiogenic agent.
- the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma.
- the cancer is prostate cancer.
- the prostate cancer is hormone sensitive prostate cancer.
- the prostate cancer is castration resistant prostate cancer.
- the prostate cancer is metastatic.
- the prostate cancer is non-metastatic.
- the combination is a synergistic combination.
- Embodiments of each of the aspects described herein, including the methods, combinations, uses and pharmaceutical compositions of the invention, may be combined with one or more other embodiments of the present invention described herein which is not inconsistent with the embodiment(s) with which it is combined.
- FIG. 1 Kaplan Meier survival curve on Day 63 (study termination) demonstrating the effects of combining a TGFprl inhibitor (PF-06952229) and an anti-PD-1 antibody in the MC38 syngeneic tumor model (Panel A: Survival Curve; Panel B)
- FIG. 2 Shows effects of combining a TGFprl inhibitor (PF-06952229) and an anti-4-1 BB antibody; and a TGF rl inhibitor (PF-06952229) and an anti-OX40 antibody, on tumor growth inhibition in the 4T1 orthotopic syngeneic mouse tumor model.
- FIG. 3 Shows the effects of combining a TGFprl inhibitor (PF-06952229) and an anti- CTLA4 antibody (D) on tumor growth delay relative to PF-06952229 monotherapy treatment (C), anti-CTLA4 antibody, (B) monotherapy treatment, in the CT26 syngeneic tumor model. Control treatment has no effect (A).
- a dose of about 5 mg/kg should be understood to mean that the dose may vary between 4.5 mg/kg and 5.5 mg/kg.
- a “disorder” is any condition that would benefit from treatment with the compounds of the present invention. This includes chronic and acute disorders or diseases including those pathological conditions which predispose the subject to the disorder in question.
- administration refers to contact of an exogenous pharmaceutical, therapeutic or diagnostic agent, or composition, to the animal, human, experimental subject, cell, tissue, organ or biological fluid.
- Treatment of a cell encompasses contact of a reagent to the cell, as well as contact of a reagent to a fluid, where the fluid is in contact with the cell.
- administering also means in vitro and ex vivo treatment, e.g., of a cell, by a reagent, diagnostic, binding compound, or by another cell.
- drug including, but not limited to, “drug,” “agent,” “component,” “composition,” “compound,” “substance,” “targeted agent,” “targeted therapeutic agent,” “therapeutic agent,” and “medicament” may be used interchangeably to refer to the small molecule compounds of the present invention, e.g., a TGF[3r inhibitor or a TGF[3r1 inhibitor.
- drugs including, but not limited to, “drug,” “agent,” “component,” “composition,” “compound,” “substance,” “targeted agent,” “targeted therapeutic agent,” “therapeutic agent,” therapeutic antibody,” and “medicament” may be used interchangeably to refer to the antibodies of the present invention, e.g., an anti-PD-1 antibody, an anti-OX40 antibody, an anti-4-1 BB antibody, and an anti-CTLA4 antibody or combinations thereof.
- antibody refers to an immunoglobulin molecule capable of specific binding to a target, such as a carbohydrate, polynucleotide, lipid, polypeptide, etc., through at least one antigen recognition site, located in the variable region of the immunoglobulin molecule.
- the term encompasses a polyclonal antibody, a monoclonal antibody, a chimeric antibody, a bispecific antibody, a dual-specific antibody, bifunctional antibody, a trispecific antibody, a multispecific antibody, a bispecific heterodimeric diabody, a bispecific heterodimeric IgG, a labeled antibody, a humanized antibody, a human antibody, and fragments thereof (such as Fab, Fab’, F(ab’)2, Fv), single chain (ScFv) and domain antibodies (including, for example, shark and camelid antibodies), fusion proteins comprising an antibody, any other modified configuration of the immunoglobulin molecule that comprises an antigen recognition site, and antibody like binding peptidomimetics (ABiPs).
- ABSiPs binding peptidomimetics
- An antibody includes an antibody of any class, such as IgG, IgA, or IgM (or sub-class thereof), and the antibody need not be of any particular class.
- immunoglobulins can be assigned to different classes. There are five major classes of immunoglobulins: IgA, IgD, IgE, IgG, and IgM, and several of these may be further divided into subclasses (isotypes), e.g., lgG-1 , IgG- 2, lgG-3, lgG-4, Ig A1 and lgA2.
- the heavy-chain constant regions that correspond to the different classes of immunoglobulins are called alpha, delta, epsilon, gamma, and mu, respectively.
- the subunit structures and three-dimensional configurations of different classes of immunoglobulins are well known.
- a “bispecific antibody,” “dual-specific antibody,” “bifunctional antibody,” “heteromultimer,” “heteromultimeric complex,” “bispecific heterodimeric diabody” or a “heteromultimeric polypeptide” is a molecule comprising at least a first polypeptide and a second polypeptide, wherein the second polypeptide differs in amino acid sequence from the first polypeptide by at least one amino acid residue.
- the bispecific is an artificial hybrid antibody having two different heavy chain region and light chain region.
- the bispecific antibody has binding specificity for at least two different ligands, antigens or binding sites. Accordingly, the bispecific antibodies can bind simultaneously to two different antigens.
- the two antigen binding sites of a bispecific antibody bind to two different epitopes, which may reside on the same or different protein targets, e.g., tumor target.
- the bispecific antibody, dual-specific antibody, bifunctional antibody, heteromultimer, heteromultimeric complex, bispecific heterodimeric diabody or the heteromultimeric polypeptide can be prepared by constructing sFv fragments with short linkers (e.g., about 3-10 residues) between the VH and VL regions such that inter-chain but not intra-chain pairing of the V regions is achieved, resulting in a bivalent fragment, i.e. , fragment having two antigen-binding sites.
- Bispecific antibodies can be derived from full length antibodies or antibody fragments (e.g., F(ab')2 bispecific antibodies).
- Diabodies are described more fully in, for example, EP 404,097; WO 1993/011161 ; and Hollinger et al., A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity, Proc. Natl. Acad. Sci. 1993, 90:6444-6448.
- Bispecific antibodies are heterodimers of two "crossover" sFv fragments in which the VH and VL regions of the two antibodies are present on different polypeptide chains.
- a bispecific antibody may comprise one antigenbinding site that recognizes an epitope on one protein (e.g., PD-1 , 0X40, 4-1 BB, and/or CTLA4) and further comprise a second, different antigen-binding site that recognizes a different epitope on a second protein (e.g., PD-1 , 0X40, 4-1 BB, and/or CTLA4).
- one protein e.g., PD-1 , 0X40, 4-1 BB, and/or CTLA4
- second protein e.g., PD-1 , 0X40, 4-1 BB, and/or CTLA4
- reference to binding means specific binding.
- therapeutic antibody refers to an antibody that is used in the treatment of a disease or a disorder.
- a therapeutic antibody may have various mechanisms of action.
- a therapeutic antibody may bind and neutralize the normal function of a target associated with an antigen.
- a monoclonal antibody that blocks the activity of the of protein needed for the survival of a cancer cell causes the cell's death.
- Another therapeutic antibody may bind and activate the normal function of a target associated with an antigen.
- a monoclonal antibody can bind to a protein on a cell and trigger an apoptosis signal.
- Yet another monoclonal antibody may bind to a target antigen expressed only on diseased tissue, conjugation of a toxic payload (effective agent), such as a chemotherapeutic or radioactive agent, to the monoclonal antibody can create an agent for specific delivery of the toxic payload to the diseased tissue, reducing harm to healthy tissue.
- a toxic payload such as a chemotherapeutic or radioactive agent
- a “biologically functional fragment” of a therapeutic antibody will exhibit at least one if not some or all of the biological functions attributed to the intact antibody, the function comprising at least specific binding to the target antigen.
- the therapeutic antibody may bind to any protein, including, without limitation, a PD-1 , an 0X40, a 4-1 BB and/or a CTLA4 antigen. Accordingly, therapeutic antibodies include, without limitation, anti-PD-1 antibodies, anti-OX40 antibodies, anti-4-1 BB antibodies, and anti-CTLA4 antibodies or combinations thereof.
- biotherapeutic agent means a biological molecule, such as an antibody or fusion protein, that blocks ligand/receptor signaling in any biological pathway that supports tumor maintenance and/or growth or suppresses the anti-tumor immune response.
- immune response refers to any detectable response to a particular substance (such as an antigen or immunogen) by the immune system of a host vertebrate animal, including, but not limited to, innate immune responses (e.g., activation of Toll-like receptor signalling cascade), cell-mediated immune responses (e.g., responses mediated by T cells, such as antigen-specific T cells, and non-specific cells of the immune system), and humoral immune responses (e.g., responses mediated by B cells, such as generation and secretion of antibodies into the plasma, lymph, and/or tissue fluids).
- innate immune responses e.g., activation of Toll-like receptor signalling cascade
- cell-mediated immune responses e.g., responses mediated by T cells, such as antigen-specific T cells, and non-specific cells of the immune system
- humoral immune responses e.g., responses mediated by B cells, such as generation and secretion of antibodies into the plasma, lymph, and/or tissue fluids.
- immune responses include an alteration (e.g., increase) in Toll-like receptor activation, lymphokine (e.g., cytokine (e.g., Th1 , Th2 or Th17 type cytokines) or chemokine) expression or secretion, macrophage activation, dendritic cell activation, T cell (e.g., CD4+ or CD8+ T cell) activation, NK cell activation, B cell activation (e.g., antibody generation and/or secretion), binding of an immunogen (e.g., antigen, immunogenic polypeptide) to an MHC molecule, induction of a cytotoxic T lymphocyte ("CTL") response, induction of a B cell response (e.g., antibody production), and expansion (e.g., growth of a population of cells) of cells of the immune system (e.g., T cells and B cells), and increased processing and presentation of antigen by antigen presenting cells.
- lymphokine e.g., cytokine
- immunogen refers to a substance that is immunogenic.
- immunogenic refers to the ability of a substance upon administration to a mammal (such as a human) to cause, elicit, stimulate, or induce an immune response, or to improve, enhance, increase or prolong a pre-existing immune response, against a particular antigen in the mammal, whether alone or when linked to a carrier, in the presence or absence of an adjuvant.
- the term "immunoglobulin” or “Ig” is used interchangeably with “antibody” herein.
- the basic 4-chain antibody unit is a heterotetrameric glycoprotein composed of two identical light (L) chains and two identical heavy (H) chains.
- An IgM antibody consists of 5 of the basic heterotetramer units along with an additional polypeptide called a J chain, and contains 10 antigen binding sites, while IgA antibodies comprise from 2-5 of the basic 4-chain units which can polymerize to form polyvalent assemblages in combination with the J chain.
- the 4-chain unit is generally about 150,000 Daltons.
- Each L chain is linked to an H chain by one covalent disulfide bond, while the two H chains are linked to each other by one or more disulfide bonds depending on the H chain isotype.
- Each H and L chain also has regularly spaced intrachain disulfide bridges.
- Each H chain has at the N-terminus, a variable domain (VH) followed by three constant domains (CH) for each of the a and y chains and four CH domains for p and £ isotypes.
- Each L chain has at the N-terminus, a variable domain (VL) followed by a constant domain at its other end. The VL is aligned with the VH and the CL is aligned with the first constant domain of the heavy chain (CHI).
- Particular amino acid residues are believed to form an interface between the light chain and heavy chain variable domains.
- the pairing of a VH and VL together forms a single antigen-binding site.
- the L chain from any vertebrate species can be assigned to one of two clearly distinct types, called kappa and lambda, based on the amino acid sequences of their constant domains.
- immunoglobulins can be assigned to different classes or isotypes.
- full-length antibody “intact antibody” or “whole antibody” are used interchangeably to refer to an antibody in its substantially intact form, as opposed to an antibody fragment.
- whole antibodies include those with heavy and light chains including an Fc region.
- the constant domains may be native sequence constant domains (e.g., human native sequence constant domains) or amino acid sequence variants thereof.
- the intact antibody may have one or more effector functions.
- antibody fragment comprises a portion of an intact antibody, preferably the antigen binding and/or the variable region of the intact antibody.
- antibody fragments suitable for use in this invention include, without limitation: (i) the Fab fragment, consisting of VL, VH, CL, and CH1 domains; (ii) the “Fd” fragment consisting of the VH and CH1 domains; (iii) the “Fv” fragment consisting of the VL and VH domains of a single antibody; (iv) the “dAb” fragment, which consists of a VH domain; (v) isolated complementarity determining (CDR) regions; (vi) F(ab')2 fragments, a bivalent fragment comprising two linked Fab fragments; (vii) single chain Fv molecules (scFv), wherein a VH domain and a VL domain are linked by a peptide linker that allows the two domains to associate to form a binding domain; (viii) bi-specific single chain Fv dimers (e.g.,
- Fv, scFv, or diabody molecules may be stabilized by the incorporation of disulphide bridges linking the VH and VL domains.
- Minibodies comprising a scFv joined to a CH3 domain may also be made (Hu et al., Minibodies are minimized antibody-like proteins comprising a scFv joined to a CH3 domain, Cancer Res. 1996, 56:3055-3061 )).
- isolated antibody or “isolated antibody fragment” refers to the purification status and in such context means the named molecule is substantially free of other biological molecules such as nucleic acids, proteins, lipids, carbohydrates, or other material such as cellular debris and growth media. Generally, the term “isolated” is not intended to refer to a complete absence of such material or to an absence of water, buffers, or salts, unless they are present in amounts that substantially interfere with experimental or therapeutic use of the binding compound described herein.
- conventional (polyclonal) antibody preparations typically include a multitude of different antibodies having different amino acid sequences in their variable domains, particularly their CDRs, which are often specific for different epitopes.
- the term “monoclonal” indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies and is not to be construed as requiring production of the antibody by any particular method.
- the monoclonal antibodies to be used in accordance with the present invention may be made by the hybridoma method first described by Kohler etal., Continuous cultures of fused cells secreting antibody of predefined specificity, Nature 1975, 256: 495; or may be made by recombinant DNA methods (e.g., U.S. Patent No. 4,816,567).
- the "monoclonal antibodies” may also be isolated from phage antibody libraries using the techniques described in Clackson et al., Making antibody fragments using phage display libraries, Nature 1991 , 352: 624-628 and Marks et al., By-passing immunization: human antibodies from V-gene libraries displayed on phage, J. Mol. Biol. 1991 , 222: 581 -597, for example. See also Presta, Selection, design, and engineering of therapeutic antibodies, J. Allergy Clin. Immunol. 2005,116:731 .
- Chimeric antibody refers to an antibody in which a portion of the heavy and/or light chain is identical with or homologous to corresponding sequences in an antibody derived from a particular species (e.g., human) or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is identical with or homologous to corresponding sequences in an antibody derived from another species (e.g., mouse) or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit the desired biological activity.
- a particular species e.g., human
- another species e.g., mouse
- Human antibody refers to an antibody that comprises human immunoglobulin protein sequences only.
- a human antibody may contain murine carbohydrate chains if produced in a mouse, in a mouse cell, or in a hybridoma derived from a mouse cell.
- mouse antibody or rat antibody refer to an antibody that comprises only mouse or rat immunoglobulin sequences, respectively.
- Humanized antibody refers to forms of antibodies that contain sequences from non-human (e.g., murine) antibodies as well as human antibodies. Such antibodies contain minimal sequence derived from non-human immunoglobulin.
- the humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin and all or substantially all of the FR regions are those of a human immunoglobulin sequence.
- the humanized antibody optionally also will comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin.
- Fc immunoglobulin constant region
- the prefix “hum,” “hu” or “h” is added to antibody clone designations when necessary to distinguish humanized antibodies from parental rodent antibodies.
- the humanized forms of rodent antibodies will generally comprise the same CDR sequences of the parental rodent antibodies, although certain amino acid substitutions may be included to increase affinity, increase stability of the humanized antibody, or for other reasons.
- variable region of an antibody refers to the variable region of the antibody light chain or the variable region of the antibody heavy chain, either alone or in combination.
- variable regions of the heavy and light chain each consist of four framework regions (FR) connected by three complementarity determining regions (CDRs) also known as hypervariable regions.
- hypervariable region refers to the regions of an antibody variable domain which are hypervariable in sequence and/or form structurally defined loops.
- antibodies comprise six HVRs; three in the VH (H1 , H2, H3), and three in the VL (L1 , L2, L3).
- H3 and L3 display the most diversity of the six HVRs, and H3 in particular is believed to play a unique role in conferring fine specificity to antibodies.
- HVR delineations are in use and are encompassed herein.
- the Kabat Complementarity Determining Regions are based on sequence variability and are the most commonly used (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, 1991 ). Chothia refers instead to the location of the structural loops (Chothia and Lesk, Canonical structures for the hypervariable regions of immunoglobulins, J. Mol. Biol. 1987, 196:901 - 917).
- the AbM HVRs represent a compromise between the Kabat HVRs and Chothia structural loops, are used by Oxford Molecular's AbM antibody modeling software.
- the "contact" HVRs are based on an analysis of the available complex crystal structures.
- a “complementarity determining region” or “CDR” of a variable domain are amino acid residues within the variable region that are identified in accordance with the definitions of the Kabat, Chothia, the accumulation of both Kabat and Chothia, AbM, contact, and/or conformational definitions or any method of CDR determination well known in the art.
- Antibody CDRs may be identified as the hypervariable regions originally defined by Kabat et al. See, e.g., Kabat et al., Sequences of Proteins of Immunological Interest, 5th ed., Public Health Service, NIH, 1992. The positions of the CDRs may also be identified as the structural loop structures originally described by Chothia and others.
- the positions of the CDRs may be identified as the residues that make enthalpic contributions to antigen binding. See, e.g., Makabe et al., Thermodynamic consequences of mutations in vernier zone residues of a humanized anti-human epidermal growth factor receptor murine antibody, Journal of Biological Chemistry 2008, 283:1 156-1166. Still other CDR boundary definitions may not strictly follow one of the above approaches but will nonetheless overlap with at least a portion of the Kabat CDRs, although they may be shortened or lengthened in light of prediction or experimental findings that particular residues or groups of residues or even entire CDRs do not significantly impact antigen binding.
- a CDR may refer to CDRs defined by any approach known in the art, including combinations of approaches.
- the methods used herein may utilize CDRs defined according to any of these approaches.
- the CDRs may be defined in accordance with any of Kabat, Chothia, extended, AbM, contact, and/or conformational definitions.
- Kabat numbering unless otherwise stated, is defined as the numbering of the residues in, e.g., an IgG heavy chain antibody using the EU index as in Kabat et al. (Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991 )), expressly incorporated herein by reference. "Framework” or "FR" residues are those variable-domain residues other than the HVR residues as herein defined.
- a "human consensus framework” or "acceptor human framework” is a framework that represents the most commonly occurring amino acid residues in a selection of human immunoglobulin VL or VH framework sequences. Generally, the selection of human immunoglobulin VL or VH sequences is from a subgroup of variable domain sequences.
- the subgroup of sequences is a subgroup as in Kabat eta!., Sequences of Proteins of Immunological Interest, 5 lh Ed. Public Health Service, National Institutes of Health, 1991.
- the subgroup may be subgroup kappa I, kappa II, kappa III or kappa IV as in Kabat eta!., supra.
- the subgroup may be subgroup I, subgroup II, or subgroup III as in Kabat et al., supra.
- a human consensus framework can be derived from the above in which particular residues, such as when a human framework residue is selected based on its homology to the donor framework by aligning the donor framework sequence with a collection of various human framework sequences.
- An acceptor human framework "derived from" a human immunoglobulin framework or a human consensus framework may comprise the same amino acid sequence thereof, or it may contain pre-existing amino acid sequence changes. In some embodiments, the number of pre-existing amino acid changes are 10 or less, 9 or less, 8 or less, 7 or less, 6 or less, 5 or less, 4 or less, 3 or less, or 2 or less.
- amino-acid modification at a specified position, e.g., of the Fc region, refers to the substitution or deletion of the specified residue, or the insertion of at least one amino acid residue adjacent the specified residue. Insertion "adjacent" to a specified residue means insertion within one to two residues thereof. The insertion may be N- terminal or C-terminal to the specified residue.
- the preferred amino acid modification herein is a substitution.
- Constantly modified variants or “conservative substitution” refers to substitutions of amino acids in a protein with other amino acids having similar characteristics (e.g., charge, side-chain size, hydrophobicity/hydrophilicity, backbone conformation and rigidity, etc.), such that the changes can frequently be made without altering the biological activity or other desired property of the protein, such as antigen affinity and/or specificity.
- Those of skill in this art recognize that, in general, single amino acid substitutions in non-essential regions of a polypeptide do not substantially alter biological activity (e.g., Watson et al., Molecular Biology of the Gene (4th Ed.), 1987, p.
- an “affinity-matured” antibody is one with one or more alterations in one or more HVRs thereof that result in an improvement in the affinity of the antibody for antigen, compared to a parent antibody that does not possess those alteration(s).
- an affinity-matured antibody has nanomolar or even picomolar affinities for the target antigen.
- Affinity-matured antibodies are produced by procedures known in the art. For example, Marks et al., By-passing immunization: Building high affinity human antibodies by chain shuffling, Bio/Technology 1992, 10:779-783, describes affinity maturation by VH- and VL-domain shuffling.
- Random mutagenesis of HVR and/or framework residues is described by, for example: Barbas et al., In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity, Proc Nat. Acad. Sci. 1994, 91 :3809-3813; Schier et al., Identification of functional and structural amino-acid residues by parsimonious mutagenesis, Gene 1995, 169: 147- 155; Yelton et al., Affinity maturation of the BR96 anti-carcinoma antibody by codon-based mutagenesis, J. Immunol. 1995, 155: 1994- 2004; Jackson et al., In vitro antibody maturation.
- Fc region herein is used to define a C-terminal region of an immunoglobulin heavy chain, including native-sequence Fc regions and variant Fc regions.
- the human IgG heavy-chain Fc region is usually defined to stretch from an amino acid residue at position Cys226, or from Pro230, to the carboxyl-terminus thereof.
- the C-terminal lysine (residue 447 according to the EU numbering system) of the Fc region may be removed, for example, during production or purification of the antibody, or by recombinantly engineering the nucleic acid encoding a heavy chain of the antibody.
- a composition of intact antibodies may comprise antibody populations with all K447 residues removed, antibody populations with no K447 residues removed, and antibody populations having a mixture of antibodies with and without the K447 residue.
- Suitable native-sequence Fc regions for use in the antibodies of the invention include human lgG-1 , lgG-2 (lgG2A, lgG2B), lgG-3 and lgG-4.
- Fc receptor or “FcR” describes a receptor that binds to the Fc region of an antibody.
- the preferred FcR is a native sequence human FcR.
- a preferred FcR is one which binds an IgG antibody (a gamma receptor) and includes receptors of the FcyRI, FcyRII, and FeyRIII subclasses, including allelic variants and alternatively spliced forms of these receptors, FcyRII receptors include FcyRIIA (an “activating receptor”) and FcyRIIB (an “inhibiting receptor”), which have similar amino acid sequences that differ primarily in the cytoplasmic domains thereof.
- Activating receptor FcyRIIA contains an immunoreceptor tyrosine-based activation motif (ITAM) in its cytoplasmic domain.
- Inhibiting receptor FcyRIIB contains an immunoreceptor tyrosinebased inhibition motif (ITFM) in its cytoplasmic domain, (e.g., M. Daeron, Fc RECEPTOR BIOLOGY, Anna. Rev. Immunol. J 1997, 5 :203-234.
- ITFM immunoreceptor tyrosinebased inhibition motif
- FcR FcR
- Fc receptor or FcR also includes the neonatal receptor, FcRn, which is responsible for the transfer of maternal IgGs to the fetus.
- FcRn the neonatal receptor
- Guyer et al. Immunoglobulin binding by mouse intestinal epithelial cell receptors, J. Immunol. 1976, 1 17: 587, and Tokoyama etal., How do natural killer cells find self to achieve tolerance? Immunity 1994, 24, 249-257.
- Methods of measuring binding to FcRn are known (e.g., Ghetie and Ward, FcRn: the MHC class l-related receptor that is more than an IgG transporter, Immunol.
- Binding to FcRn in vivo and serum half-life of human FcRn high-affinity binding polypeptides can be assayed, e.g., in transgenic mice or transfected human cell lines expressing human FcRn, or in primates to which the polypeptides having a variant Fc region are administered.
- WO 2004/042072 (Presta) describes antibody variants which improved or diminished binding to FcRs. See also, e.g., Shields etal., High Resolution Mapping of the Binding Site on Human lgG1 for FcyRI, FcyRII, FcyRIII, and FcRn and Design of lgG1 Variants with Improved Binding to the FcyR, J. Biol. Chem. 2001 , 9(2): 6591 -6604.
- substantially reduced denotes a sufficiently high degree of difference between two numeric values (generally one associated with a molecule and the other associated with a reference/comparator molecule) such that one of skill in the art would consider the difference between the two values to be of statistical significance within the context of the biological characteristic measured by said values (e.g., Kd values).
- the difference between said two values is, for example, greater than about 10%, greater than about 20%, greater than about 30%, greater than about 40%, and/or greater than about 50% as a function of the value for the reference/comparator molecule.
- the term "substantially similar” or “substantially the same” denotes a sufficiently high degree of similarity between two numeric values (for example, one associated with an antibody of the invention and the other associated with a reference/comparator antibody), such that one of skill in the art would consider the difference between the two values to be of little or no biological and/or statistical significance within the context of the biological characteristic measured by said values (e.g., Kd values).
- the difference between said two values is, for example, less than about 50%, less than about 40%, less than about 30%, less than about 20%, and/or less than about 10% as a function of the reference/comparator value.
- the term “specifically binds to” or is “specific for” refers to measurable and reproducible interactions such as binding between a target and an antibody, which is determinative of the presence of the target in the presence of a heterogeneous population of molecules including biological molecules.
- an antibody that specifically binds to a target (which can be an epitope) is an antibody that binds this target with greater affinity, avidity, more readily, and/or with greater duration than it binds to other targets.
- the extent of binding of an antibody to an unrelated target is less than about 10 percent of the binding of the antibody to the target as measured, e.g., by a radioimmunoassay (RIA).
- an antibody that specifically binds to a target has a dissociation constant (Kd) of ⁇ 1 pM, ⁇ 100 nM, ⁇ 10 nM, ⁇ 1 nM, or ⁇ 0.1 nM.
- Kd dissociation constant
- an antibody specifically binds to an epitope on a protein that is conserved among the protein from different species.
- specific binding can include, but does not require exclusive binding.
- immunoadhesin designates antibody-like molecules which combine the binding specificity of a heterologous protein (an “adhesin”) with the effector functions of immunoglobulin constant domains.
- the immunoadhesins comprise a fusion of an amino acid sequence with the desired binding specificity which is other than the antigen recognition and binding site of an antibody (/.e., is “heterologous”), and an immunoglobulin constant domain sequence.
- the adhesin part of an immunoadhesin molecule typically is a contiguous amino acid sequence comprising at least the binding site of a receptor or a ligand.
- the immunoglobulin constant domain sequence in the immunoadhesin may be obtained from any immunoglobulin, such as lgG-1 , lgG-2 (including lgG2A and lgG2B), lgG-3, or lgG-4 subtypes, IgA (including IgA- 1 and IgA-2), IgE, IgD or IgM.
- the Ig fusions preferably include the substitution of a domain of a polypeptide or antibody described herein in the place of at least one variable region within an Ig molecule.
- the immunoglobulin fusion includes the hinge, CH2 and CH3, or the hinge, CHI, CH2 and CH3 regions of an lgG-1 molecule.
- Immunoadhesin combinations of Ig Fc and ECD of cell surface receptors are sometimes termed soluble receptors.
- peptide As used herein, the terms “peptide,” “polypeptide,” and “protein” are used interchangeably herein, and refer to a polymeric form of amino acids of any length, which can include coded and non-coded amino acids, chemically, or biochemically modified or derivatized amino acids, and polypeptides having modified polypeptide backbones.
- fusion protein and a “fusion polypeptide” refer to a polypeptide having two portions covalently linked together, where each of the portions is a polypeptide having a different property.
- the property may be a biological property, such as activity in vitro or in vivo.
- the property may also be simple chemical or physical property, such as binding to a target molecule, catalysis of a reaction, etc.
- the two portions may be linked directly by a single peptide bond or through a peptide linker but are in reading frame with each other.
- the term "antagonist” antibody or a “blocking” antibody is one that inhibits or reduces a biological activity of the antigen it binds. In some embodiments, blocking antibodies or antagonist antibodies substantially or completely inhibit the biological activity of the antigen.
- the anti-PD-L1 antibodies of the invention block the signaling through PD-1 so as to restore a functional response by T-cells (e.g., proliferation, cytokine production, target cell killing) from a dysfunctional state to antigen stimulation.
- agonist or “activating antibody” is one that enhances or initiates signaling by the antigen to which it binds.
- agonist antibodies cause or activate signaling without the presence of the natural ligand.
- the term “dysfunction” in the context of immune dysfunction refers to a state of reduced immune responsiveness to antigenic stimulation.
- the term includes the common elements of both exhaustion and/or anergy in which antigen recognition may occur, but the ensuing immune response is ineffective to control infection or tumor growth.
- the term “dysfunctional” also includes refractory or unresponsive to antigen recognition, specifically, impaired capacity to translate antigen recognition into down-stream T-cell effector functions, such as proliferation, cytokine production and/or target cell killing.
- the term "anergy” refers to the state of unresponsiveness to antigen stimulation resulting from incomplete or insufficient signals delivered through the T-cell receptor (e.g., increase in intracellular Ca+2 in the absence of ras-activation). T cell anergy can also result upon stimulation with antigen in the absence of co- stimulation, resulting in the cell becoming refractory to subsequent activation by the antigen even in the context of co stimulation.
- the unresponsive state can often be overridden by the presence of lnterleukin-2. Anergic T-cells do not undergo clonal expansion and/or acquire effector functions.
- exhaustion refers to T cell exhaustion as a state of T cell dysfunction that arises from sustained TCR signaling that occurs during many chronic infections and cancer. It is distinguished from anergy in that it arises not through incomplete or deficient signaling, but from sustained signaling. It is defined by poor effector function, sustained expression of inhibitory receptors and a transcriptional state distinct from that of functional effector or memory T cells. Exhaustion prevents optimal control of infection and tumors. Exhaustion can result from both extrinsic negative regulatory pathways (e.g., immunoregulatory cytokines) as well as cell intrinsic negative regulatory (co-stimulatory) pathways.
- extrinsic negative regulatory pathways e.g., immunoregulatory cytokines
- Enhancing T-cell function means to induce, cause or stimulate a T-cell to have a sustained or amplified biological function, or renew or reactivate exhausted or dysfunctional T-cells.
- Examples of enhancing T-cell function include increased secretion of y-interferon from CD4+ or CD8+ T-cells, increased proliferation, increased survival, increased differentiation, increased antigen responsiveness (e.g., viral, pathogen, or tumor clearance) relative to such levels before the intervention.
- the level of enhancement is as least 50%, alternatively 60%, 70%, 80%, 90%, 100%, 120%, 150%, 200%. The manner of measuring this enhancement is known to one of ordinary skill in the art.
- abnormal cell growth refers to cell growth that is independent of normal regulatory mechanisms (e.g., loss of contact inhibition). Abnormal cell growth may be benign (not cancerous), or malignant (cancerous).
- cancer cancer
- cancer cancer
- cancer refers to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth.
- cancer refers to any malignant and/or invasive growth or tumor caused by abnormal cell growth.
- cancer refers to solid tumors named for the type of cells that form them, as well as cancer of blood, bone marrow, or the lymphatic system. Examples of solid tumors include but not limited to sarcomas and carcinomas.
- cancers of the blood include but not limited to leukemias, lymphomas and myeloma.
- cancer includes, but is not limited to, a primary cancer that originates at a specific site in the body, a metastatic cancer that has spread from the place in which it started to other parts of the body, a recurrence from the original primary cancer after remission, and a second primary cancer that is a new primary cancer in a person with a history of previous cancer of a different type from latter one.
- prostate cancer and "locally advanced prostate cancer” mean prostate cancers that have extended through the prostate capsule, and are meant to include stage C disease under the American Urological Association (AUA) system, stage Cl - C2 disease under the Whitmore-Jewett system, and stage T3 - T4 and N+ disease under the TNM (tumor, node, metastasis) system.
- AUA American Urological Association
- stage Cl - C2 disease under the Whitmore-Jewett system
- T3 - T4 and N+ disease under the TNM (tumor, node, metastasis) system stage C disease under the American Urological Association (AUA) system
- TNM tumor, node, metastasis
- Locally advanced prostate cancer is clinically identified by palpable evidence of induration beyond the lateral border of the prostate, or asymmetry or induration above the prostate base.
- Metastasis refers to a type of cancer that originates in one tissue type, but then spreads to one or more tissues outside of the (primary) cancer’s origin. Cancer cells can break away from a primary tumor, penetrate into lymphatic and blood vessels, circulate through the bloodstream, and grow in a distant focus (metastasize) in normal tissues elsewhere in the body. Metastasis can be local or distant. Metastasis is a sequential process, contingent on tumor cells breaking off from the primary tumor, traveling through the bloodstream, and stopping at a distant site. At the new site, the cells establish a blood supply and can grow to form a life-threatening mass. Both stimulatory and inhibitory molecular pathways within the tumor cell regulate this behavior, and interactions between the tumor cell and host cells in the distant site are also significant.
- metastatic prostate cancer means prostate cancer that has spread to regional lymph nodes or to distant sites and is meant to include stage D disease under the AUA system and stage TxNxM+ under the TNM system.
- surgery is generally not indicated for patients with metastatic disease, and hormonal (androgen ablation) therapy is a preferred treatment modality.
- Patients with metastatic prostate cancer eventually develop an androgen-refractory state within 12 to 18 months of treatment initiation. Approximately half of these androgen-refractory patients die within 6 months.
- the most common site for prostate cancer metastasis is bone. Prostate cancer bone metastases are often osteoblastic rather than osteolytic (/.e., resulting in net bone formation).
- Bone metastases are found most frequently in the spine, followed by the femur, pelvis, rib cage, skull and humerus. Other common sites for metastasis include lymph nodes, lung, liver and brain. Metastatic prostate cancer is typically diagnosed by open or laparoscopic pelvic lymphadenectomy, whole body radionuclide scans, skeletal radiography, and/or bone lesion biopsy.
- TGF expressing cancer is one that produces sufficient levels of TGFp at the surface of cells thereof, such that an anti-TGFp antibody can bind thereto and have a therapeutic effect with respect to the cancer.
- a cancer “characterized by excessive activation” of a TGFp receptor is one in which the extent of TGFp receptor activation in cancer cells significantly exceeds the level of activation of that receptor in non-cancerous cells of the same tissue type. Such excessive activation may result from overexpression of the TGFp receptor and/or greater than normal levels of a TGFp ligand available for activating the TGFp receptor in the cancer cells. Such excessive activation may cause and/or be caused by the malignant state of a cancer cell.
- the cancer will be subjected to a diagnostic or prognostic assay to determine whether amplification and/or overexpression of a TGFp receptor is occurring that results in such excessive activation of the TGFp receptor.
- the cancer may be subjected to a diagnostic or prognostic assay to determine whether amplification and/or overexpression of a TGFp ligand is occurring in the cancer that attributes to excessive activation of the receptor.
- a diagnostic or prognostic assay to determine whether amplification and/or overexpression of a TGFp ligand is occurring in the cancer that attributes to excessive activation of the receptor.
- excessive activation of the receptor may result from an autocrine-stimulatory pathway.
- self-stimulation occurs by virtue of the cancer cell producing both a TGFp ligand and its cognate TGFp receptor.
- the cancer may express or overexpress TGFp receptor and also express or overexpress a TGFp ligand (e.g., TGFpi ligand).
- a cancer that “overexpresses” a TGFp receptor is one that has significantly higher levels of a TGFp receptor, at the cell surface thereof, compared to a non-cancerous cell of the same tissue type. Such overexpression may be caused by gene amplification or by increased transcription or translation. TGFp receptor overexpression may be determined in a diagnostic or prognostic assay by evaluating increased levels of the TGFp protein present on the surface of a cell (e.g., via an immunohistochemistry assay; IHC).
- TGFp encoding nucleic acid in the cell, e.g., via fluorescent in situ hybridization (FISH; see WO 1998/045479 published October, 1998), southern blotting, or polymerase chain reaction (PCR) techniques, such as real-time quantitative PCR (RT-PCR).
- FISH fluorescent in situ hybridization
- PCR polymerase chain reaction
- RT-PCR real-time quantitative PCR
- ELISA for Quantitation of the Extracellular Domain of p185HER2 in Biological Fluids, J. Immunol. Methods 1990, 132: 73-80.
- various in vivo assays are available to the skilled practitioner. For example, one may expose cells within the body of the patient to an antibody that is optionally labeled with a detectable label, e.g., a radioactive isotope, and binding of the antibody to cells in the patient can be evaluated, e.g., by external scanning for radioactivity or by analyzing a biopsy taken from a patient previously exposed to the antibody.
- a detectable label e.g., a radioactive isotope
- a cancer that is “not characterized by overexpression of the TGFp receptor” is one that, in a diagnostic assay, does not express higher than normal levels of TGFp receptor compared to a non-cancerous cell of the same tissue type.
- a cancer that “overexpresses” a TGFp ligand is one that produces significantly higher levels of that ligand compared to a non-cancerous cell of the same tissue type. Such overexpression may be caused by gene amplification or by increased transcription or translation. Overexpression of the TGF ligand may be determined diagnostically by evaluating levels of the ligand (or nucleic acid encoding it) in the patient, e.g., in a tumor biopsy or by various diagnostic assays such as the IHC, FISH, southern blotting, PCR, enzyme-linked immunosorbent assay (ELISA) or in vivo assays described above.
- diagnostic assays such as the IHC, FISH, southern blotting, PCR, enzyme-linked immunosorbent assay (ELISA) or in vivo assays described above.
- a “hormone-independent” cancer is one in which proliferation thereof is not dependent on the presence of a hormone that binds to a receptor expressed by cells in the cancer. Such cancers do not undergo clinical regression upon administration of pharmacological or surgical strategies that reduce the hormone concentration in or near the tumor.
- hormone-independent cancers include androgen-independent prostate cancer, estrogen-independent breast cancer, endometrial cancer, and ovarian cancer. Such cancers may begin as hormone-dependent tumors and progress from a hormone-sensitive stage to a hormone-refractory tumor following anti-hormonal therapy.
- in combination with or “in conjunction with” refers to administration of one agent in addition to at least one other agent.
- in combination with or “in conjunction with” refers to administration of one agent before, during, or after administration of at least one other agent to the individual.
- Any of the combinations, methods and uses of the present invention provided may be used to treat a subject (e.g., human) who has been diagnosed with or is suspected of having cancer.
- subject means a mammal being assessed for treatment and/or being treated.
- the mammal is a human.
- the terms “subject,” “individual,” and “patient” thus encompass individuals having cancer (e.g., prostate cancer), including those who have undergone or are candidates for resection (surgery) to remove cancerous tissue.
- mammal refers to any animal species of the Mammalia class. Examples of mammals include: humans; non-human primates such as monkeys; laboratory animals such as rats, mice, guinea pigs; domestic animals such as cats, dogs, rabbits, cattle, sheep, goats, horses, and pigs; and captive wild animals such as lions, tigers, elephants, and the like.
- the subject is a human and may be referred to as a patient. In some embodiments, the subject is a human child between the ages of birth and 18. In a preferred embodiment, the subject is a male having a prostate cancer. In a preferred embodiment, the subject may be a human who exhibits one or more symptoms associated with cancer.
- the subject may be a human who is at risk, or genetically or otherwise predisposed (e.g., risk factor) to developing cancer who has or has not been diagnosed.
- an “at risk” subject is a subject who is at risk of developing cancer.
- the subject may or may not have detectable disease, and may or may not have displayed detectable disease prior to the treatment methods described herein.
- An at-risk subject may have one or more so-called risk factors, which are measurable parameters that correlate with development of cancer, which are described herein.
- a subject having one or more of these risk factors has a higher probability of developing cancer than an individual without these risk factor(s).
- risk factors may include, for example, age, sex, race, diet, history of previous disease, presence of precursor disease, genetic (e.g., hereditary) considerations, and environmental exposure.
- the subjects at risk for cancer include, for example, those having relatives who have experienced the disease, and those whose risk is determined by analysis of genetic or biochemical markers.
- the subject is at an early stage of a cancer. In other embodiments, the subject is at an advanced stage of cancer. In a more preferred embodiment, the cancer is metastatic, non-metastatic or benign. In a preferred embodiment, the cancer is non-metastatic. In particular embodiments, the cancer is a metastatic cancer. In certain embodiments, the cancer is identified as being at risk for or having a propensity for metastasis or there is no indication that the cancer has yet metastasized. In certain embodiments, identification of a cancer at risk of metastasis is based on assessment of a tumor biopsy. In addition, the subject may be a human who is undergoing one or more standard therapies, such as chemotherapy, radiotherapy, immunotherapy, surgery, or combination thereof. Accordingly, one or more of the combinations of the present invention may be administered before, during, or after administration of chemotherapy, radiotherapy, immunotherapy, surgery or combination thereof.
- standard therapies such as chemotherapy, radiotherapy, immunotherapy, surgery, or combination thereof. Accordingly, one or more of the combinations of the present invention may be administered
- the subject may be a human who is (i) substantially refractory to at least one chemotherapy treatment, or (ii) is in relapse after treatment with chemotherapy, or both (i) and (ii). In some of embodiments, the subject is refractory to at least two, at least three, or at least four chemotherapy treatments (including standard or experimental chemotherapies).
- treat or “treating” or “treatment” of a cancer as used herein means to administer a combination therapy according to the present invention to a subject having cancer, or diagnosed with cancer, to achieve at least one positive therapeutic effect, such as, for example, reduced number of cancer cells, reduced tumor size, reduced rate of cancer cell infiltration into peripheral organs, or reduced rate of tumor metastases or tumor growth, reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition.
- treatment refers to the act of treating as "treating” is defined immediately above.
- the term “treating” also includes adjuvant and neo-adjuvant treatment of a subject.
- beneficial or desired clinical results include, but are not limited to, one or more of the following: reducing the proliferation of (or destroying) neoplastic or cancerous cell; inhibiting metastasis or neoplastic cells; shrinking or decreasing the size of a tumor; remission of the cancer; decreasing symptoms resulting from the cancer; increasing the quality of life of those suffering from the cancer; decreasing the dose of other medications required to treat the cancer; delaying the progression of the cancer; curing the cancer; overcoming one or more resistance mechanisms of the cancer; and/or prolonging survival of patients the cancer.
- Positive therapeutic effects in cancer can be measured in a number of ways (see, for example, W. A. Weber, Assessing tumor response to therapy, J. Nucl. Med.
- T/C tumor growth inhibition
- NCI National Cancer Institute
- CR complete response
- complete response means the disappearance of all signs of cancer (e.g., disappearance of all target lesions) in response to treatment. This does not always mean the cancer has been cured.
- the term “disease-free survival” means the length of time after primary treatment for a cancer ends that the patient survives without any signs or symptoms of that cancer.
- the term “duration of response” means the length of time that a tumor continues to respond to treatment without the cancer growing or spreading. Treatments that demonstrate improved DoR can produce a durable, meaningful delay in disease progression.
- the terms “objective response” and “overall response” refer to a measurable response, including complete response (CR) or partial response (PR).
- the term “overall response rate” (ORR) refers to the sum of the complete response (CR) rate and the partial response (PR) rate.
- OS all survival
- partial response refers to a decrease in the size of one or more tumors or lesions, or in the extent of cancer in the body, in response to treatment.
- PR refers to at least a 30% decrease in the sum of the longest diameters (SLD) of target lesions, taking as reference the baseline SLD.
- progression free survival refers to the length of time during and after treatment during which the disease being treated (e.g., cancer) does not get worse.
- PFS also referred to as “Time to Tumor Progression,” may include the amount of time patients have experienced a CR or PR, as well as the amount of time patients have experienced SD.
- PR refers to at least a 20% increase in the SLD of target lesions, taking as reference the smallest SLD recorded since the treatment started, or to the presence of one or more new lesions.
- stable disease or “SD” refers to a cancer that is neither decreasing nor increasing in extent or severity.
- the term "sustained response" refers to the sustained effect on reducing tumor growth after cessation of a treatment.
- the tumor size may be the same size or smaller as compared to the size at the beginning of the medicament administration phase.
- the sustained response has a duration of at least the same as the treatment duration, at least 1.5x, 2x, 2.5x, or 3x length of the treatment duration, or longer.
- the anti-cancer effect of the method of treating cancer may be defined and assessed by the investigators using RECIST v1 .1 (Eisenhauer et al., New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1 ), Eur J of Cancer 2009, 45(2):228-47).
- immune checkpoint inhibitor refers to a molecule, compound, or composition that binds to an immune checkpoint protein and blocks its activity and/or inhibits the function of the immune regulatory cell expressing the immune checkpoint protein that it binds (e.g., Treg cells, tumor-associated macrophages, etc.), immune checkpoint modulator includes any compound, including but not limited to antibodies and small molecules.
- Immune checkpoint proteins may include, but are not limited to, PD1 (also known as PD-1 ; Programmed Death 1 receptor), 0X40, 4-1 BB, CTLA4 (Cytotoxic T-Lymphocyte- Associated protein 4, CD152), PD-L1 , PD-L2, LAG-3 (Lymphocyte Activation Gene-3), A2AR (Adenosine A2A receptor), B7-H3 (CD276), B7-H4 (VTCN1), BTLA (B and T Lymphocyte Attenuator, CD272), IDO (Indoleamine 2,3-dioxygenase), KIR (Killer-cell Immunoglobulin-like Receptor), TIM 3 (T-cell Immunoglobulin domain and Mucin domain 3), VISTA (V-domain Ig suppressor of T cell activation), and IL-2R (interleukin-2 receptor).
- PD1 also known as PD-1 ; Programmed Death 1 receptor
- CTLA4 Cy
- Immune checkpoint inhibitors are well known in the art and are commercially or clinically available. These include, but are not limited to, agonists, antagonists, or antibodies that modulate immune checkpoint proteins. Illustrative examples of checkpoint inhibitors, referenced by their target immune checkpoint protein, are provided as follows.
- Immune checkpoint inhibitors comprising anti-PD-1 antibody include, but are not limited to, sasanlimab (PF-6801591), nivolumab (MDX 1106), pembrolizumab (MK- 3475), pidilizumab (CT-011 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001 ), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP- 3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD-100, camrelizumab (SHR- 1210), Sym-021 , ABBV-181 , AK
- Immune checkpoint inhibitors comprising an anti-OX40 antibody include, but are not limited to, PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR-8383, and ABBV-368.
- Immune checkpoint inhibitors comprising a 4-1 BB agonist 4-1 BB agonist include, but are not limited to, utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No.
- Immune checkpoint inhibitors comprising anti-CTLA4 antibody include, but are not limited to, ipilimumab (10DI), tremelimumab, and AGEN-1884.
- Immune checkpoint inhibitors comprising a B7-H3 inhibitor include, but are not limited to, MGA271 .
- Immune checkpoint inhibitors comprising an LAG3 inhibitor include, but are not limited to, IMP321 , BMS-986016.
- Immune checkpoint inhibitors comprising a KIR inhibitor include, but are not limited to, IPH2101 (lirilumab).
- An immune checkpoint inhibitor targeting IL-2R, for preferentially depleting Treg cells comprises IL-2-toxin fusion proteins, which include, but are not limited to, denileukin diftitox (Ontak).
- the therapeutic effect achieved by the TGFprl inhibitor or a pharmaceutically acceptable salt, solvate or polymorph thereof in combination with and/or an additional anti-cancer agent as further described herein, e.g., a further immune checkpoint inhibitor is defined by reference to any of the following: complete response (OR), disease free survival (DFS), duration of response (DoR), overall response rate (ORR), overall survival (OS), partial response (PR), or progression free survival (PFS).
- response to a combination of the invention is any of PR, CR, PFS, DFS, OR or OS that is assessed using Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 response criteria.
- the invention relates to neoadjuvant therapy, adjuvant therapy, first-line therapy, second-line therapy, or third-line or later lines of therapy.
- the cancer may be localized, advanced or metastatic, and the intervention may occur at point along the disease continuum (/.e., at any stage of the cancer).
- the treatment regimen for a method, combination, uses and pharmaceutical compositions of the invention that is effective to treat cancer in a subject may vary according to factors such as the disease state, age, and weight of the subject, and the ability of the therapy to elicit an anti-cancer response in the subject. While an embodiment of any of the aspects of the invention may not be effective in achieving a positive therapeutic effect in every subject, it should do so in a statistically significant number of subjects as determined by any statistical test known in the art such as the Student’s t-test, the chi2-test the U-test according to Mann and Whitney, the Kruskal- Wallis test (H-test), Jonckheere-Terpstrat-test and the Wilcon on-test.
- any statistical test known in the art such as the Student’s t-test, the chi2-test the U-test according to Mann and Whitney, the Kruskal- Wallis test (H-test), Jonckheere-Terpstrat-test and the Wilcon on-test.
- treatment regimen may be used interchangeably to refer to the dose and timing of administration of each therapeutic agent in a combination of the invention.
- “Ameliorating” means a lessening or improvement of one or more symptoms upon treatment with a combination described herein, as compared to not administering the combination. “Ameliorating” also includes shortening or reduction in duration of a symptom.
- an “effective dosage” or “effective amount” of a compound or a pharmaceutical composition is an amount sufficient to affect any one or more beneficial or desired outcomes, including biochemical, histological and/or behavioural symptoms, of the disease, its complications and intermediate pathological phenotypes presenting during development of the disease.
- a “therapeutically effective amount” refers to that amount of a compound being administered which will relieve to some extent one or more of the symptoms of the disorder being treated.
- a therapeutically effective amount refers to that amount which has the effect of (1 ) reducing the size of the tumor, (2) inhibiting (that is, slowing to some extent, preferably stopping) tumor metastasis, (3) inhibiting to some extent (that is, slowing to some extent, preferably stopping) tumor growth or tumor invasiveness, (4) relieving to some extent (or, preferably, eliminating) one or more signs or symptoms associated with the cancer, (5) decreasing the dose of other medications required to treat the disease, and/or (6) enhancing the effect of another medication, and/or (7) delaying the progression of the disease in a patient.
- an effective dosage can be administered in one or more administrations.
- an effective dosage of drug, compound, or pharmaceutical composition is an amount sufficient to accomplish prophylactic or therapeutic treatment either directly or indirectly.
- an effective dosage of drug, compound or pharmaceutical composition may or may not be achieved in conjunction with another drug, compound or pharmaceutical composition.
- Tumor as it applies to a subject diagnosed with, or suspected of having, a cancer refers to a malignant or potentially malignant neoplasm or tissue mass of any size and includes primary tumors and secondary neoplasms.
- a solid tumor is an abnormal growth or mass of tissue that usually does not contain cysts or liquid areas. Examples of solid tumors are sarcomas, carcinomas, and lymphomas. Leukaemia’s (cancers of the blood) generally do not form solid tumors (National Cancer Institute, Dictionary of Cancer Terms).
- Tumor burden refers to the total amount of tumorous material distributed throughout the body. Tumor burden refers to the total number of cancer cells or the total size of tumor(s), throughout the body, including lymph nodes and bone marrow. T umor burden can be determined by a variety of methods known in the art, such as, e.g., using callipers, or while in the body using imaging techniques, e.g., ultrasound, bone scan, computed tomography (CT), or magnetic resonance imaging (MRI) scans.
- imaging techniques e.g., ultrasound, bone scan, computed tomography (CT), or magnetic resonance imaging (MRI) scans.
- tumor size refers to the total size of the tumor which can be measured as the length and width of a tumor. Tumor size may be determined by a variety of methods known in the art, such as, e.g., by measuring the dimensions of tumor(s) upon removal from the subject, e.g., using callipers, or while in the body using imaging techniques, e.g., bone scan, ultrasound, CR or MRI scans.
- imaging techniques e.g., bone scan, ultrasound, CR or MRI scans.
- additive is used to mean that the result of the combination of two compounds, components or targeted agents is no greater than the sum of each compound, component or targeted agent individually.
- concentration or “synergistic” are used to mean that the result of the combination of two or more compounds, components or targeted agents is greater than the sum of each compound, component or targeted agent individually.
- This improvement in the disease, condition or disorder being treated is a “synergistic” effect and combinations providing a synergistic effect may be referred to as synergistic combinations.
- a “synergistic amount” is an amount of the combination of the two compounds, components or targeted agents that results in a synergistic effect, as “synergistic” is defined herein.
- a synergistic effect can be calculated, for example, using suitable methods such as the Sigmoid-Emax equation (Holford et al., Understanding the does-effect relationship: Clinical application of pharmacokinetic-pharmacodynamic models. Clin. Pharmacokinet. 1981 , 6: 429-453), the equation of Loewe additivity (Loewe, S. et al., Effect of combinations: mathematical basis of problem, Exp. Pathol Pharmacol. 1926, 114: 313-326) and the median-effect equation (Chou, T. C., et al., Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors, Adv. Enzyme Regul. 1984, 22: 27-55).
- suitable methods such as the Sigmoid-Emax equation (Holford et al., Understanding the does-effect relationship: Clinical application of pharmacokinetic-pharmacodynamic models. Clin. Pharmacokinet. 1981 , 6: 429-453), the equation of Loewe addit
- Each equation referred to above can be applied to experimental data to generate a corresponding graph to aid in assessing the effects of the drug combination.
- the corresponding graphs associated with the equations referred to above are the concentration-effect curve, isobologram curve and combination index curve, respectively.
- the term "pharmaceutically acceptable” refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication,
- the term “pharmaceutically acceptable salt” refers to those salts which retain the biological effectiveness and properties of the parent compound.
- pharmaceutically acceptable salt(s),” as used herein, unless otherwise indicated, includes salts of acidic or basic groups which may be present in the compounds of the formulae disclosed herein.
- the compounds of the invention that are basic in nature may be capable of forming a wide variety of salts with various inorganic and organic acids.
- anions suitable for mono- and di- acid addition salts include, but are not limited to, acetate, asparatate, benzenesulfonate, benzoate, besylate, bicarbonate, bisulfate, bitartrate, bromide, calcium edetate, camsylate, carbonate, chloride, citrate, decanoate, edetate, edislyate, estolate, esylate, fumarate, gluceptate, gluconate, glutamate, glycollate, hexanoate, hexylresorcinate, hydrabamine, hydroxynaphthoate, iodide, isethionate, lactate, lactobionate, malate, maleate, mandelate, mesylate, methylsulfate, mucate, napsylate, nitrate, octanoate, oleate, pamoate (embonate), pantothenate,
- compounds that are acidic in nature may be capable of forming base salts with various pharmacologically acceptable cations which form non-toxic base salts.
- non-toxic base salts include, but are not limited to, those derived from such pharmacologically acceptable cations such as alkali metal cations (e.g., potassium and sodium) and alkaline earth metal cations (e.g., calcium and magnesium), ammonium or water-soluble amine addition salts such as N-methylglucamine-(meglumine), and the lower alkanolammonium and other base salts of pharmaceutically acceptable organic amines.
- salts examples include alkali metal or alkaline-earth metal salts and other cations, including aluminium, arginine, benzathine, calcium, chloroprocaine, choline, diethanolamine, ethanolamine, ethylenediamine, lysine, magnesium, histidine, lithium, meglumine, potassium, procaine, sodium, triethyamine and zinc.
- Salts may be prepared by conventional techniques. Hemisalts of acids and bases may also be formed, for example, hemisulphate and hemicalcium salts.
- suitable salts see Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002). Methods for making pharmaceutically acceptable salts are known to those of skill in the art.
- Tumor burden also referred to as “tumor load” refers to the total amount of tumor material distributed throughout the body. Tumor burden refers to the total number of cancer cells or the total size of tumor(s), throughout the body, including lymph nodes and bone narrow. Tumor burden can be determined by a variety of methods known in the art, such as, e.g., by measuring the dimensions of tumor(s) upon removal from the subject, e.g., using calipers, or while in the body using imaging techniques, e.g., magnetic resonance imaging (MRI) scans, computed tomography (CT), multi-detector CT (MDCT), positron emission tomography (PET), X-ray, ultrasound, or bone scan.
- imaging techniques e.g., magnetic resonance imaging (MRI) scans, computed tomography (CT), multi-detector CT (MDCT), positron emission tomography (PET), X-ray, ultrasound, or bone scan.
- tumor size refers to the total size of the tumor which can be measured as the length and width of a tumor. Tumor size may be determined by a variety of methods known in the art, such as, e.g., by measuring the dimensions of tumor(s) upon removal from the subject, e.g., using calipers, or while in the body using imaging techniques, e.g., MRI scans, bone scan, ultrasound, or CT.
- imaging techniques e.g., MRI scans, bone scan, ultrasound, or CT.
- regulatory agency refers to a country's agency for the approval of the medical use of pharmaceutical agents with the country.
- FDA U.S. Food and Drug Administration
- Transforming growth factor p is a multifunctional cytokine having three forms designated TGFpl , TGFp2 and TGFp3. TGFpl to 3 form a subfamily of highly similar proteins within the TGFp superfamily of cytokines.
- TGFpl Human TGFpl (NCBI Reference Sequence: NP_000651.3) is a 390 amino acid protein, whilst TGFp2 (NCBI Reference Sequence: NP_001129071.1 ) is a 442 amino acid protein, and TGFP3 (NCBI Reference Sequence: NP_001316868.1 ) contains 412 amino acids.
- TGFpi-3 have a 20-30 amino acid signal peptide at the N-terminus which is necessary for secretion, a pro-region called latency associated peptide (LAP), and a 112-114 amino acid C-terminal region that becomes the mature TGFp molecule following proteolytic cleavage from the pro-region (Khalil et al., TGF-beta: from latent to active, Microbes Infect. 1999, 1 (15): 1255-63). Mature monomeric TGp dimerize to produce the biologically active 25 KDa protein.
- LAP latency associated peptide
- TGFpl to 3 can form homodimers with the same type of TGFp, or can form heterodimers with another type of TGFp (e.g., a TGFpi :TGFp2 heterodimer).
- TGFp comprises nine conserved cysteine residues, eight of which form disulfide bonds to form the cysteine knot structure which is characteristic of the TGFp superfamily. The remaining cysteine is involved interacts with that of another TGFp monomer to form the dimer.
- the surface-exposed region between the fifth and sixth conserved cysteine residues is the region, which is least conserved between TGFpl to 3 proteins, and is thought to be important for receptor binding and specificity of TGFp.
- TGFp refers to TGFp from any species and includes isoforms, fragments, variants or homologues of a TGFp from any species.
- the TGFp is a human TGF[3, primate TGFp, non-human primate TGFp, rodent TGFp, murine TGFp, or mammalian TGF
- TGFp exerts its functional consequences through binding to and activating signaling through TGFp receptors.
- TGFp receptors comprising an extracellular domain having a TGFp binding region, a single pass transmembrane domain and an intracellular domain comprising a serine/threonine kinase domain.
- TGFpRI NCBI Reference Sequence: NP_004603.1
- TGFPR2 NCBI Reference Sequence: NP_001020018.1
- TGFPR3 NCBI Reference Sequence: NP-003234.2
- a TGFp ligand binds to and activates TGFp receptor.
- TGFp receptor refers to a TGFp receptor from any species and includes isoforms, fragments, variants or homologues of a TGFp receptor from any species.
- the TGFp receptor is a human TGFp receptor, primate TGFp receptor, non-human primate TGFp receptor, rodent TGFp receptor, murine TGFp receptor, or mammalian TGFp receptor.
- the TGFp receptor is TGFp receptor 1 (TGFprl ), TGFp receptor 2 (TGFpr2), or TGFp receptor 3 (TGFpr3).
- TGFp receptor unless otherwise indicated, refers to any receptor that binds at least one TGFp receptor.
- TGFp family is a class within the TGFp superfamily and in human contains three members: TGFpl , TGFp2, and TGFp3, which are structurally similar. The three growth factors are known to signal via the same receptors.
- TGFpi was originally defined by its ability to cause the phenotypic transformation of rat fibroblasts.
- TGFpi is a multipotent cytokine with cell- and dose-dependent activities. Although TGFpi is a growth inhibitor for most cell types, it can act as a stimulator for some cell types. TGFpi has ubiquitous distribution. For reviews on TGFpi , see Massague, The transforming growth factor-beta family, J. Ann. Rev. Cell Biol. 1990, 6, 597; Letterio etal., Regulation of immune responses by TGF-beta, Ann. Rev. Immunol. 1998, 16, 137.
- TGFpi demonstrates regulatory effects on a wide range of cell types, and modulates embryonic development, bone formation, mammary development, wound healing, haematopoiesis, angiogenesis, cell cycle progression and the production of the extracellular matrix.
- TGFpi inhibits T and B cell proliferation and acts as an anti-inflammatory molecule both in vitro and in vivo.
- TGFpi inhibits macrophage maturation and activation, and also inhibits the activity of natural killer cells and lymphokine-activated killer (LAK) cells and blocks cytokine production.
- LAK lymphokine-activated killer
- TGFpl -positive cancer refers to a cancer or tumor with aberrant TGF[31 expression (overexpression).
- Many human cancer/tumor types show predominant expression of the TGF
- TGFB is sometimes used to refer to the gene as opposed to protein
- cancer/tumor may show co-dominant expression of another isoform, such as TGF 3.
- a number of epithelial cancers e.g., carcinoma may co-express TGFpl and TGFP3.
- TGFpl may arise from multiple sources, including, for example, cancer cells, tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), regulatory ? cells (Tregs), myeloid-derived suppressor cells (MDSCs), and the surrounding extracellular matrix (ECM).
- TAMs tumor-associated macrophages
- CAFs cancer-associated fibroblasts
- Regs regulatory ? cells
- MDSCs myeloid-derived suppressor cells
- ECM extracellular matrix
- preclinical cancer or tumor models that recapitulate human conditions are TGFpl -positive cancer or tumor.
- TGFp inhibitor or “TGF[3r inhibitor” refers to any agent capable of antagonizing biological activities, signaling or function of TGFp growth factor (e.g., TGFP1 (TGFprl), TGFP2 (TGFpr2), and/or TGFP3 (TGFpr3)).
- TGFprl TGFP1
- TGFpr2 TGFpr2
- TGFpr3 TGFpr3
- the term is not intended to limit its mechanism of action and includes, for example, neutralizing inhibitors, small molecule inhibitors, receptor antagonists, soluble ligand traps, and activation inhibitors of TGFp.
- TGFpr inhibitors also include antibodies that are capable of reducing the availability of latent proTGFp which can be activated in the niche, for example, by inducing antibody-dependent cell mediated cytotoxicity (ADCC), and/or antibodydependent cellular phagocytosis (ADPC), as well as antibodies that result in internalization of cell-surface complex comprising latent proTGFp, thereby removing the precursor from the plasma membrane without depleting the cells themselves.
- Internalization may be a suitable mechanism of action for LRRC33-containing protein complexes (such as human LRRC33-proTGFpi ) which results in reduced levels of cells expressing LRRC33-containing protein complexes on cell surface.
- Table 2 below provides exemplary TGFpr inhibitors useful in the each of the methods, combinations, uses and pharmaceutical compositions described herein.
- 3r1 inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGF rl inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
- TGFprl inhibitor PF-06952229 is a potent and selective TGF rl (transforming growth factor beta receptor type 1) inhibitor, having the structure:
- TGFprl inhibitor references to pharmaceutically acceptable salts, solvates, hydrates and complexes thereof, and to solvates, hydrates and complexes of pharmaceutically acceptable salts thereof, and include amorphous and polymorphic forms, stereoisomers, and isotopically labelled versions thereof.
- Table 3 below provides a list of the amino acid sequences of exemplary PD-1 axis binding antagonists useful in the each of the methods, combinations, uses and pharmaceutical compositions described herein.
- the mAB7 is also known as sasanlimab, PF-6801591 , or RN888.
- mAb7 and mAb15 are disclosed in PCT Publication No. WO 2011/001100600A1
- the PD-1 axis binding antagonist is sasanlimab (PF-6801591 ) and will be administered subcutaneously at a dose of about 1 , 2, 3, 4, 5, 6, 7 or 8 mg/kg at intervals of about 14 days ( ⁇ 2 days) or about 21 days ( ⁇ 2 days) or about 30 days ( ⁇ 2 days) throughout the course of treatment.
- sasanlimab (PF-6801591 ) is administer as a flat dose of about 80, 150, 160, 200, 240, 250, 300, 320, 350, 400, preferably 300mg at intervals of about 14 days ( ⁇ 2 days) or about 21 days ( ⁇ 2 days) or about 30 days ( ⁇ 2 days).
- sasanlimab (PF-6801591 ) is administered subcutaneously in an amount of 300 mg Q4W.
- PD-1 axis binding antagonist means any chemical compound or biological molecule that blocks binding of PD-L1 expressed on a cancer cell to PD-1 expressed on an immune cell (T cell, B cell or natural killer (NK) cell) and preferably also blocks binding of PD-L2 expressed on a cancer cell to the immune-cell expressed PD-1.
- PD-1 and its ligands include: PDCD1 , PD1 , CD279 and SLEB2 for PD-1 ; PDCD1 L1 , PD-L1 , B7H1 , B7-4, CD274 and B7-H for PD-L1 ; and PDCD1 L2, PDL2, B7-DC, Btdc and CD273 for PD-L2.
- the PD-1 antagonist may block binding of human PD-L1 to human PD-1 , and block binding of both human PD-L1 and PD-L2 to human PD-1.
- Exemplary human PD-1 amino acid sequences can be found in NCBI Locus No.: NP_005009.
- Exemplary human PD-L1 and PD-L2 amino acid sequences can be found in NCBI Locus No.: NP_054862 and NP_079515, respectively.
- PD-1 antagonists useful in any of the other embodiments of each of the methods, combinations, uses and pharmaceutical compositions described herein, include a monoclonal antibody (mAb), or antigen binding fragment thereof, which specifically binds to PD-1 or PD-L1 , and preferably specifically binds to human PD-1 or human PD-L1 .
- the mAb may be a human antibody, a humanized antibody or a chimeric antibody, and may include a human constant region.
- the human constant region is selected from the group consisting of lgG1 , lgG2, lgG3 and lgG4 constant regions, and in some embodiments, the human constant region is an lgG1 or lgG4 constant region.
- the antigen binding fragment is selected from the group consisting of Fab, Fab'-SH, F(ab')2, scFv and Fv fragments.
- Specific anti-human PD-1 mAbs useful as the PD-1 antagonist in the methods, combinations, uses and pharmaceutical compositions of the present invention include: nivolumab (MDX 1 106), pembrolizumab (MK-3475), pidilizumab (CT-01 1 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), sasanlimab (PF-6801591 ), mAb15, MEDI-0680 (AMP-514), BGB-108, or AGEN-2034, or combinations thereof.
- sasanlimab (also referred to as PF-6801591 , mAb7, or RN888) is a monoclonal antibody that targets and inhibits programmed cell death 1 ligand (PDCD1 ).
- PDCD1 programmed cell death 1 ligand
- nivolumab is a human lgG4 PD-1 antibody transiently expressed by applicants in 293 HEK cells that utilizes the heavy chain and light chain sequences from Proposed INN: List 107 (CAS#946414-94-4).
- pembrolizumab is a human lgG4 PD-1 antibody transiently expressed by applicants in 293 HEK cells that utilizes the heavy chain and light chain sequences from Proposed INN: List 72.
- Table 4 below provides exemplary anti-PD-1 antibody sequences useful in the each of the methods, combinations, uses and pharmaceutical compositions described herein. Table 4
- 0X40 agonist or “0X40 binding agonist,” as used herein, means, any chemical compound or biological molecule, as defined herein, which upon binding to 0X40, (1) stimulates or activates 0X40, (2) enhances, increases, promotes, induces, or prolongs an activity, function, or presence of 0X40, or (3) enhances, increases, promotes, or induces the expression of 0X40.
- 0X40 agonists useful in the any of the methods, combinations, uses and pharmaceutical compositions of the present invention include a monoclonal antibody (mAb), or antigen binding fragment thereof, which specifically binds to 0X40.
- mAb monoclonal antibody
- the 0X40 agonists increase an OX40-mediated response.
- 0X40 agonists markedly enhance cytotoxic T-cell responses, resulting in antitumor activity in several models.
- An 0X40 agonist includes, for example, an 0X40 agonist antibody (e.g., an antihuman 0X40 agonist antibody), an OX40L agonist fragment, an 0X40 oligomeric receptor, and an 0X40 immunoadhesin.
- an 0X40 agonist antibody e.g., an antihuman 0X40 agonist antibody
- an OX40L agonist fragment e.g., an antihuman 0X40 agonist antibody
- OX40L agonist fragment e.g., an antihuman 0X40 agonist antibody
- OX40L agonist fragment e.g., an antihuman 0X40 agonist antibody
- OX40L agonist fragment e.g., an antihuman 0X40 agonist antibody
- OX40L agonist fragment e.g., an OX40L agonist fragment
- an 0X40 oligomeric receptor e.g., an antihuman 0X40 agonist antibody
- 0X40 antibody means an antibody, as defined herein, capable of binding to 0X40 receptor (e.g., human 0X40 receptor).
- 0X40 and 0X40 receptor are used interchangeably in the present application, and refer to any form of 0X40 receptor, as well as variants, isoforms, and species homologs thereof that retain at least a part of the activity of 0X40 receptor. Accordingly, a binding molecule, as defined and disclosed herein, may also bind 0X40 from species other than human. In other cases, a binding molecule may be completely specific for the human 0X40 and may not exhibit species or other types of crossreactivity. Unless indicated differently, such as by specific reference to human 0X40, 0X40 includes all mammalian species of native sequence 0X40, e.g., human, canine, feline, equine and bovine. One exemplary human 0X40 is a 277 amino acid protein (UniProt Accession No. P43489).
- An 0X40 agonist antibody means, any antibody, as defined herein, which upon binding to 0X40, (1) stimulates or activates 0X40, (2) enhances, increases, promotes, induces, or prolongs an activity, function, or presence of 0X40, or (3) enhances, increases, promotes, or induces the expression of 0X40.
- 0X40 agonists useful in the any of the methods, combinations, uses and pharmaceutical compositions of the present invention include a monoclonal antibody (mAb) which specifically binds to 0X40 (e.g., anti-OX40 agonist antibody).
- mAb monoclonal antibody
- the 0X40 agonist antibody increases CD4+ effector T cell proliferation and/or increases cytokine production by the CD4+ effector T cell as compared to proliferation and/or cytokine production prior to treatment with the 0X40 agonist antibody.
- the cytokine is IFN-y.
- the 0X40 agonist antibody increases memory T cell proliferation and/or increasing cytokine production by the memory cell.
- the cytokine is IFN-y.
- the 0X40 agonist antibody inhibits Treg suppression of effector T cell function.
- effector T cell function is effector T cell proliferation and/or cytokine production.
- the effector T cell is a CD4+ effector T cell.
- the 0X40 agonist antibody increases 0X40 signal transduction in a target cell that expresses 0X40. In some embodiments, 0X40 signal transduction is detected by monitoring NFkB downstream signaling.
- the anti-human 0X40 agonist antibody is a depleting antihuman 0X40 antibody (e.g., depletes cells that express human 0X40).
- the human 0X40 expressing cells are CD4+ effector T cells.
- the human 0X40 expressing cells are Treg cells.
- depleting is by ADCC and/or phagocytosis.
- the antibody mediates ADCC by binding FcyR expressed by a human effector cell and activating the human effector cell function.
- the antibody mediates phagocytosis by binding FcyR expressed by a human effector cell and activating the human effector cell function.
- Exemplary human effector cells include, e.g., macrophage, natural killer (NK) cells, monocytes, neutrophils.
- the human effector cell is macrophage.
- the anti-human 0X40 agonist antibody has a functional Fc region.
- effector function of a functional Fc region is ADCC.
- effector function of a functional Fc region is phagocytosis.
- effector function of a functional Fc region is ADCC and phagocytosis.
- the Fc region is a human lgG-1. In some embodiments, the Fc region is a human lgG-4.
- the anti-human 0X40 agonist antibody is a human or humanized antibody.
- an anti-OX40 antibody useful in the methods, combinations, uses and pharmaceutical compositions disclosed herein is a fully human agonist monoclonal antibody comprising a heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 54 and SEQ ID NO: 55, respectively.
- the anti-OX40 antibody is a fully human lgG-2 or lgG-1 antibody.
- the anti-OX40 antibody is PF- 04518600.
- Table 5 below provides exemplary anti-OX40 monoclonal antibody sequences useful in each of the methods, combinations, uses and pharmaceutical compositions described herein.
- 4-1 BB binding agonist or “4-1 BB agonist,” as used herein, means, any chemical compound or biological molecule, as defined herein, which upon binding to 4- 1 BB, (1 ) stimulates or activates 4-1 BB, (2) enhances, increases, promotes, induces, or prolongs an activity, function, or presence of 4-1 BB, or (3) enhances, increases, promotes, or induces the expression of 4-1 BB.
- 4-1 BB agonists useful in any of the methods, combinations, uses and pharmaceutical compositions of the present invention include a monoclonal antibody (mAb), or antigen binding fragment thereof, which specifically binds to 4-1 BB.
- mAb monoclonal antibody
- Alternative names or synonyms for 4-1 BB include CD137 and TNFRSF9.
- the 4- 1 BB agonists increase a 4-1 BB-mediated response.
- 41 BB agonists markedly enhance cytotoxic T-cell responses, resulting in antitumor activity in several models.
- the 4-1 BB agonist is utomilumab.
- 4-1 BB antibody means an antibody, as defined herein, capable of binding to 4-1 BB receptor (e.g., human 4-1 BB receptor).
- 4-1 BB and 4-1 BB receptor are used interchangeably in the present application and refer to any form of 4-1 BB receptor, as well as variants, isoforms, and species homologs thereof that retain at least a part of the activity of 4-1 BB receptor. Accordingly, a binding molecule, as defined and disclosed herein, may also bind 4-1 BB from species other than human. In other cases, a binding molecule may be completely specific for the human 4-1 BB and may not exhibit species or other types of crossreactivity. Unless indicated differently, such as by specific reference to human 4-1 BB.
- 4- 1 BB includes all mammalian species of native sequence of 4-1 BB, e.g., human, canine, feline, equine and bovine.
- One exemplary human 4-1 BB is a 255 amino acid protein (Accession No. NM_001561 ; NP_001552).
- 4-1 BB comprises a signal sequence (amino acid residues 1 -17), followed by an extracellular domain (169 amino acids), a transmembrane region (27 amino acids), and an intracellular domain (42 amino acids) (Cheuk ATC etal., Cancer Gene Therapy 2004, 11 : 215-226).
- the receptor is expressed on the cell surface in monomer and dimer forms and likely trimerizes with 4-1 BB ligand to signal.
- Human 4-1 BB comprises a signal sequence (amino acid residues 1 -17), followed by an extracellular domain (169 amino acids), a transmembrane region (27 amino acids), and an intracellular domain (42 amino acids) (Cheuk ATC et al., Cancer Gene Therapy 2004, 11 : 215-226).
- the receptor is expressed on the cell surface in monomer and dimer forms and likely trimerizes with 4-1 BB ligand to signal.
- an anti-4- 1 BB antibody useful in the treatment, method, medicaments and uses disclosed herein is a fully humanized lgG-2 agonist monoclonal antibody comprising a heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 64 and SEQ ID NO: 65, respectively.
- Table 6 below provides exemplary anti-4-1 BB monoclonal antibody sequences useful in each of the methods, combinations, uses and pharmaceutical compositions described herein.
- Cytotoxic T lymphocyte associated antigen 4 (CTLA4) closely relates to CD28 in gene structure, chromosomal localization, homology of sequences and gene expression, and both of them are receptors of costimulatory molecule B7, and mainly expresses on the cell surface of activated T cells. Interaction of CTLA4 and B7 inhibits the activation of T cells in mice and human, and negatively regulates the activation of T cells. Certain embodiments of the present invention comprise an anti-cytotoxic T- lymphocyte associated antigen 4 (anti-CTLA4) antibody.
- anti-CTLA4 antibody anti-cytotoxic T- lymphocyte associated antigen 4
- Suitable anti-CTLA4 antibodies for use in the methods of the invention include, without limitation, anti-CTLA4 antibodies, human anti-CTLA4 antibodies, mouse anti-CTLA4 antibodies, mammalian anti-CTLA4 antibodies, humanized anti-CTLA4 antibodies, monoclonal anti-CTLA4 antibodies, polyclonal anti-CTLA4 antibodies, chimeric anti-CTLA4 antibodies, ipilimumab, tremelimumab, anti-CD28 antibodies, anti-CTLA4 adnectins, anti-CTLA4 domain antibodies, single chain anti-CTLA4 fragments, heavy chain anti-CTLA4 fragments, light chain anti-CTLA4 fragments, inhibitors of CTLA4 that agonize the co-stimulatory pathway, the antibodies disclosed in PCT Publication No.
- Ipilimumab (marketed as YERVOY®; also known as MEX-010, MDX-101 , or by its CAS Registry No. 477202-00-9) is disclosed as antibody 10DI in PCT Publication No. WO 01/014424.
- Examples of pharmaceutical composition comprising Ipilimumab are provided in PCT Publication No. WO 07/067959.
- Ipilimumab is approved in the U.S. for the treatment of unresectable or metastatic melanoma.
- Ipilimumab may be administered intradermally or subcutaneously.
- the effective amount of Ipilimumab administered locally is typically in the range of 5 - 200 mg/dose per person.
- the effective amount of Ipilimumab is in the range of 10 - 150 mg/dose per person per dose. In some particular embodiments, the effective amount of Ipilimumab is about 10, 25, 50, 75, 100, 125, 150, 175, or 200 mg/dose per person.
- Tremelimumab (also known as CP-675,206) is a fully human lgG2 monoclonal antibody and has the CAS number 745013-59-6. Tremelimumab is disclosed as antibody 11.2.1 in U.S. Patent No: 6,682,736.
- tremelimumab may be administered intravenously, intradermally, or subcutaneously.
- the effective amount of tremelimumab administered intradermally or subcutaneously is typically in the range of 5-200 mg/dose per person.
- the effective amount of tremelimumab is in the range of 10 - 150 mg/dose per person per dose.
- the effective amount of tremelimumab is about 10, 25, 50, 75, 100, 125, 150, 175, or 200 mg/dose per person.
- Table 7 below provides exemplary anti-CTLA4 monoclonal antibody sequences useful in each of the methods, combinations, uses and pharmaceutical compositions described herein Table 7
- the present invention provides methods, combinations and uses that may be useful for treating cancer. Some embodiments provided herein result in one or more of the following effects: (1 ) inhibiting cancer cell proliferation; (2) inhibiting cancer cell invasiveness; (3) inducing apoptosis of cancer cells; (4) inhibiting cancer cell metastasis;
- the invention provides a combination comprising a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof; and a PD-1 axis binding antagonist, for use in the manufacture of a medicament for treating cancer in a subject.
- the invention provides use of a combination comprising a TGF r1 inhibitor or a pharmaceutically acceptable salt thereof; and a PD-1 axis binding antagonist, in the manufacture of a medicament for treating cancer in a subject.
- the combination further comprises an additional anticancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an antiandrogen and/or anti-angiogenic agent, for use in the manufacture of a medicament.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, antimetabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the anti-androgen inhibitor selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides a combination comprising a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof; and an 0X40 agonist, for use in the manufacture of a medicament for treating cancer in a subject.
- the invention provides use of a combination comprising a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; and an 0X40 agonist, in the manufacture of a medicament for treating cancer in a subject.
- the combination further comprises an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, for use in the manufacture of a medicament.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides a combination comprising a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof; and a 4-1 BB agonist, for use in the manufacture of a medicament for treating cancer in a subject.
- the invention provides use of a combination comprising a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; and a 4-1 BB agonist, in the manufacture of a medicament for treating cancer in a subject.
- the combination further comprises an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, for use in the manufacture of a medicament.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 - oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4- naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2- propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides a combination comprising a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof; and a CTLA4 antagonist, for use in the manufacture of a medicament for treating cancer in a subject.
- the invention provides use of a combination comprising a TGF r1 inhibitor or a pharmaceutically acceptable salt thereof; and a CTLA4 antagonist, in the manufacture of a medicament for treating cancer in a subject.
- the combination further comprises an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, for use in the manufacture of a medicament.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a PD-1 axis binding antagonist.
- the invention provides a TGFprl inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a PD-1 axis binding antagonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a PD-1 axis binding antagonist.
- the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a PD-1 axis binding antagonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides a TGFprl inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with an 0X40 agonist.
- the invention provides a TGFprl inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with an 0X40 agonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or antihormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with an 0X40 agonist.
- the invention provides use of a TGF rl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with an 0X40 agonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or antihormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a 4-1 BB agonist.
- the invention provides a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a 4-1 BB agonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or antihormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a 4-1 BB agonist.
- the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a 4-1 BB agonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a CTLA4 antagonist.
- the invention provides a TGFprl inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a CTLA4 antagonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or antihormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a CTLA4 antagonist.
- the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a CTLA4 antagonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, antimetabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the present invention provides methods, combinations and uses comprising a TGFprl inhibitor or a pharmaceutically acceptable salt, and an immune checkpoint inhibitor, wherein the immune checkpoint inhibitor is a PD-1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, and/or a CTLA4 antagonist.
- the present invention provides methods comprising administering to a subject in need thereof an amount of a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of a programmed cell death protein 1 (PD-1 ) axis binding antagonist, wherein the amounts together are effective in treating cancer.
- TGFprl transforming growth factor beta receptor type 1
- PD-1 programmed cell death protein 1
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide
- the present invention provides methods, combinations and uses comprising the compound, 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4- ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (also referred to as “PF-06952229” or “PF-‘2229”).
- PF-06952229 is a potent and selective TGFprl (transforming growth factor beta receptor type 1) inhibitor, having the structure:
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
- the PD-1 axis binding antagonist is an anti- PD-1 antibody.
- the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-06801591 ) (see WO2016/092419), nivolumab ((Opdivo®), MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-011 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001 ), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, Bl- 754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4- ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof; and the anti-PD-1 antibody is sasanlimab (PF- 6801591 ), wherein the amounts together are effective in treating cancer.
- the TGFprl inhibitor and the PD-1 axis binding antagonist are administered sequentially, simultaneously, or concurrently.
- the TGF rl inhibitor and the PD-1 axis binding antagonist are administered sequentially, simultaneously, or concurrently.
- the present invention provides methods comprising administering to a subject in need thereof an amount of a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an 0X40 agonist, wherein the amounts together are effective in treating cancer.
- TGFprl transforming growth factor beta receptor type 1
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
- the 0X40 agonist is an anti-OX40 antibody, an OX40L agonist fragment, an 0X40 oligomeric receptor, a trimeric OX40L-Fc protein or an 0X40 immunoadhesin, or combinations thereof.
- the 0X40 agonist is an anti-OX40 antibody.
- the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS- 986178, GBR-8383, and ABBV-368, or combinations thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof; and the anti-OX40 antibody is PF-04518600, and wherein the amounts together are effective in treating cancer.
- the TGFprl inhibitor and the 0X40 agonist are administered sequentially, simultaneously, or concurrently.
- the present invention provides methods comprising administering to a subject in need thereof an amount of a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an amount of a 4-1 BB agonist, wherein the amounts together are effective in treating cancer.
- TGFprl transforming growth factor beta receptor type 1
- the TGF rl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
- the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No.
- the 4-1 BB agonist is utomilumab.
- the TGFprI inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof; and the 4-1 BB agonist is utomilumab, wherein the amounts together are effective in treating cancer.
- the TGFprI inhibitor and the 4-1 BB agonist are administered sequentially, simultaneously, or concurrently.
- the present invention provides methods comprising administering to a subject in need thereof an amount of a transforming growth factor beta receptor (TGF r) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of a CTLA4 antagonist, wherein the amounts together are effective in treating cancer.
- TGF r transforming growth factor beta receptor
- the TGF rI inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGFprI inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
- the CTLA4 antagonist is an anti-CTLA4 antibody.
- the anti-CTLA4 antibody is selected from the group consisting of ipilimumab (Yervoy®), tremelimumab (CP-675,206), and AGEN-1884, or combinations thereof.
- the anti-CTLA4 antibody is ipilimumab (Yervoy®).
- the TGFprI inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof; and the anti-CTLA4 antibody is ipilimumab (Yervoy®), wherein the amounts together are effective in treating cancer.
- the TGFprI inhibitor and the CTLA4 antagonist are administered sequentially, simultaneously, or concurrently.
- the invention provides methods, combinations and uses, wherein the subject is a human.
- the combination further comprises an additional anti-cancer agent; wherein the combination is effective in treating cancer.
- the additional anti-cancer agent is selected from the group consisting of a further immune checkpoint inhibitor, an anti-tumor agent, an antiandrogen and anti-angiogenic agent.
- the anti-tumor agent is selected from the group consisting of mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics, antihormones, androgen deprivation therapy and anti-androgens.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 - oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4- naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2- propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
- the cancer is prostate cancer.
- the prostate cancer is hormone sensitive prostate cancer.
- the prostate cancer is castration resistant prostate cancer.
- the prostate cancer is metastatic.
- the prostate cancer is non-metastatic.
- the present invention further provides pharmaceutical compositions, medicaments and kits comprising a TGF0r inhibitor, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor and/or an additional anticancer agent, as further described below.
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
- 3r1 inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable carrier or excipient, for use in combination with an immune checkpoint inhibitor for use in treating cancer.
- the immune checkpoint inhibitor is a PD-1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, and/or a CTLA4 antagonist.
- the pharmaceutical composition further comprises an additional anti-cancer agent.
- the invention provides a first pharmaceutical composition comprising a TGF rl inhibitor or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable carrier or excipient, and a second pharmaceutical composition comprising a PD-1 axis binding antagonist, wherein the first and second pharmaceutical compositions are administered sequentially, simultaneously, or concurrently.
- Some embodiments of this aspect further comprise a third pharmaceutical composition comprising an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, and a pharmaceutically acceptable carrier or excipient, wherein the first, second and third pharmaceutical compositions are administered sequentially, simultaneously, or concurrently.
- a third pharmaceutical composition comprising an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, and a pharmaceutically acceptable carrier or excipient, wherein the first, second and third pharmaceutical compositions are administered sequentially, simultaneously, or concurrently.
- the anti-tumor agent is selected from the group consisting of mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics, antihormones, androgen deprivation therapy and anti-androgens.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 - oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4- naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2- propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides a first pharmaceutical composition comprising a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable carrier or excipient, and a second pharmaceutical composition comprising an 0X40 agonist, wherein the first and second pharmaceutical compositions are administered sequentially, simultaneously, or concurrently.
- a third pharmaceutical composition comprising an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, and a pharmaceutically acceptable carrier or excipient, wherein the first, second and third pharmaceutical compositions are administered sequentially, simultaneously, or concurrently.
- the anti-tumor agent is selected from the group consisting of mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics, antihormones, androgen deprivation therapy and anti-androgens.
- the anti-androgen inhibitor selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 - oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4- naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2- propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention provides a first pharmaceutical composition comprising a TGF r1 inhibitor or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable carrier or excipient, and a second pharmaceutical composition comprising an anti-4-1 BB antibody, wherein the first and second pharmaceutical compositions are administered sequentially, simultaneously, or concurrently.
- a third pharmaceutical composition comprising an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, and a pharmaceutically acceptable carrier or excipient, wherein the first, second and third pharmaceutical compositions are administered sequentially, simultaneously, or concurrently.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the antiandrogen inhibitor selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5- methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6- yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidom
- the invention provides a first pharmaceutical composition comprising a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable carrier or excipient, and a second pharmaceutical composition comprising a CTLA4 antagonist, wherein the first and second pharmaceutical compositions are administered sequentially, simultaneously, or concurrently.
- a third pharmaceutical composition comprising an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, and a pharmaceutically acceptable carrier or excipient, wherein the first, second and third pharmaceutical compositions are administered sequentially, simultaneously, or concurrently.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
- the PD-1 axis binding antagonist is an anti-PD- 1 antibody.
- the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1 106), pembrolizumab (MK-3475), pidilizumab (CT-011), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP- 3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM
- the 0X40 agonist is an anti-OX40 antibody, an OX40L agonist fragment, an 0X40 oligomeric receptor, a trimeric OX40L-Fc protein or an 0X40 immunoadhesin, or combinations thereof.
- the 0X40 agonist is an anti-OX40 antibody.
- the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR- 8383, and ABBV-368, or combinations thereof.
- the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No.
- the 4-1 BB agonist is utomilumab.
- the CTLA4 antagonist is an anti-CTLA4 antibody.
- the anti-CTLA4 antibody is selected from the group consisting of ipilimumab (10DI), tremelimumab, and AGEN-1884, or combinations thereof.
- the invention provides a kit comprising a first container, a second container and a package insert, wherein the first container comprises at least one dose of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, as further described herein; the second container comprises at least one dose of a PD-1 axis binding antagonist; at least one dose of and 0X40 agonist, at least one dose of 4-1 BB agonist, or at least one dose of CTLA4 antagonist, or combination thereof , and the package insert comprises instructions for treating cancer in a subject using the medicaments.
- the invention provides a kit comprising a first container, a second container, a third container, and a package insert, wherein the first container comprises at least one dose of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; the second container comprises at least one dose of a PD-1 axis binding antagonist; at least one dose of an 0X40 agonist, at least one dose of a 4-1 BB agonist, or at least one dose of CTLA4 antagonist, or combination thereof; the third container comprises at least one dose of an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent; and the package insert comprises instructions for treating cancer in a subject using the medicaments.
- the first container comprises at least one dose of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof
- the second container comprises at least one dose of a PD-1 axis binding antagonist
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the antiandrogen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5- methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6- yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidom
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
- the PD-1 axis binding antagonist is an anti-PD-1 antibody.
- the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1 106), pembrolizumab (MK-3475), pidilizumab (CT-01 1 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS- 010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105
- the 0X40 agonist is an anti-OX40 antibody, an OX40L agonist fragment, an 0X40 oligomeric receptor, a trimeric OX40L- Fc protein or an 0X40 immunoadhesin, or combinations thereof. In some such embodiments, the 0X40 agonist is an anti-OX40 antibody.
- the anti-OX40 antibody is selected from the group consisting of PF- 04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR-8383, and ABBV-368, or combinations thereof.
- the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No.
- the 4-1 BB agonist is utomilumab.
- the CTLA4 antagonist is an anti-CTLA4 antibody.
- the anti-CTLA4 antibody is selected from the group consisting of ipilimumab (10DI), tremelimumab, and AGEN-1884, or combinations thereof.
- the additional anti-cancer agent is a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti- angiogenic agent.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the antiandrogen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5- methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6- yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidom
- compositions, medicaments and kits described herein may be useful for treating the cancers described above with respect to the methods, combinations and uses of the invention.
- the pharmaceutical compositions, medicaments and kits may be useful for treating cancer is selected from the group consisting of prostate cancer, colorectal cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, kidney cancer, stomach cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
- the invention provides methods and uses comprising a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor and/or an additional anti-cancer agent, as further described below.
- TGFprl transforming growth factor beta receptor type 1
- the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2- (5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
- the TGFprl inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof.
- the immune checkpoint inhibitor is a PD-1 axis binding antagonist.
- the PD-1 axis binding antagonist is an anti-PD-1 antibody.
- the anti-PD-1 antibody is sasanlimab, nivolumab (MDX 1 106), pembrolizumab (MK-3475), pidilizumab (CT-01 1 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS- 010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolim
- the immune checkpoint inhibitor is an 0X40 agonist.
- the 0X40 agonist is an is an anti-OX40 antibody.
- the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR- 8383, and ABBV-368, or combinations thereof.
- the anti- 0X40 antibody is PF-04518600.
- the immune checkpoint inhibitor is a 4-1 BB agonist.
- the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No.
- the 4-1 BB agonist is utomilumab.
- the immune checkpoint inhibitor is a CTLA4 antagonist.
- the CTLA4 antagonist is an anti-CTLA4 antibody.
- the anti- CTLA4 antibody is selected from the group consisting of ipilimumab (10DI), tremelimumab, and AGEN-1884, or combinations thereof.
- the anti-CTLA4 antibody is ipilimumab (10DI).
- the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
- the invention provides a method of treating prostate cancer in a subject in need thereof comprising administering to the subject an amount of 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yljnicotinamide, or a pharmaceutically acceptable salt thereof; and an amount of an immune checkpoint inhibitor, wherein the amounts together are effective in treating cancer.
- the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor, for use in treating prostate cancer.
- the prostate cancer is hormone sensitive prostate cancer. In a preferred embodiment of this aspect, the prostate cancer is castration resistant prostate cancer. In a preferred embodiment of this aspect, the prostate cancer is metastatic. In a preferred embodiment of this aspect, the prostate cancer is non-metastatic.
- the invention provides a method of treating lung cancer in a subject in need thereof comprising administering to the subject an amount of 4-(2- (5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yljnicotinamide, or a pharmaceutically acceptable salt thereof; and an amount of an immune checkpoint inhibitor, wherein the amounts together are effective in treating cancer.
- the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor, for use in treating lung cancer.
- the lung cancer is small cell lung cancer (SCLC).
- SCLC SCLC is RB-negative.
- the lung cancer is non-small cell lung cancer (NSCLC).
- NSCLC non-small cell lung cancer
- the lung cancer is advanced or metastatic lung cancer.
- the lung cancer is advanced or metastatic SCLC.
- the lung cancer is advanced or metastatic NSCLC.
- the invention provides a method of treating ovarian cancer, peritoneal cancer, or fallopian tube cancer in a subject in need thereof comprising administering to the subject an amount of 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and an amount of an immune checkpoint inhibitor, wherein the amounts together are effective in treating cancer.
- the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4- ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor, for use in treating ovarian cancer, peritoneal cancer, or fallopian tube cancer.
- the cancer is ovarian cancer.
- the ovarian cancer is epithelial ovarian cancer (EOC).
- the ovarian cancer is advanced or metastatic ovarian cancer (including EOC).
- the ovarian cancer is platinum resistant ovarian cancer (including EOC).
- the ovarian cancer is platinum resistant advanced or metastatic ovarian cancer (including EOC).
- the cancer is peritoneal cancer.
- the peritoneal cancer is primary peritoneal carcinomatosis (PPC).
- the cancer is fallopian tube cancer (FTC).
- the invention provides a method of treating breast cancer in a subject in need thereof comprising administering to the subject an amount of 4-(2- (5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and an amount of an immune checkpoint inhibitor, wherein the amounts together are effective in treating cancer.
- the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor, for use in treating breast cancer.
- the breast cancer is HR+/HER2- breast cancer.
- the breast cancer is HR+/HER2- advanced or metastatic breast cancer.
- the breast cancer is triple negative breast cancer (TNBC).
- TNBC triple negative breast cancer
- the TNBC is locally recurrent, advanced or metastatic TNBC.
- the breast cancer is HR+/HER2- breast cancer or TNBC, which may be advanced or metastatic, and the subject is a woman of any menopausal status or a man.
- the method comprises administering to the subject an amount of 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, an amount of an immune checkpoint inhibitor, and an amount of additional anti-cancer agent, wherein the amount of 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, the amount of an immune checkpoint inhibitor, and the amount of the additional anti-cancer agent together are effective in treating cancer.
- the additional anticancer agent is an endocrine therapeutic agent.
- the endocrine therapeutic agent is an aromatase inhibitor, a selective estrogen receptor degrader (SERD), or a selective estrogen receptor modulator (SERM).
- the endocrine therapeutic agent is letrozole.
- the endocrine therapeutic agent is fulvestrant.
- the breast cancer is HR+/HER2- breast cancer.
- the breast cancer is HR+/HER2- advanced or metastatic breast cancer.
- the breast cancer is triple negative breast cancer (TNBC).
- TNBC is locally recurrent, advanced or metastatic TNBC.
- the breast cancer is HR+/HER2- breast cancer or TNBC, which may be advanced or metastatic, and the subject is a woman of any menopausal status or a man.
- the immune checkpoint inhibitor is a PD- 1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, and/or a CTLA4 antagonist.
- the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof for use in treating cancer.
- the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4- ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof for use in treating cancer, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
- the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestin
- the invention provides use of a combination comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide (PF-06952229), having the structure: or a pharmaceutically acceptable salt thereof; and a PD-1 axis binding antagonist, wherein the use of the combination is effective in treating cancer.
- the PD-1 axis binding antagonist is an anti-PD-1 antibody.
- the anti-PD-1 antibody is sasanlimab, nivolumab (MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-01 1), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105, BAT- 1306, or AGEN-2034
- the invention provides use of a combination comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide (PF-06952229), having the structure: or a pharmaceutically acceptable salt thereof; and an 0X40 agonist, wherein the combination is effective in treating cancer.
- the 0X40 agonist is an is an anti-OX40 antibody.
- the anti-OX40 antibody is PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR-8383, or ABBV-368, or combinations thereof.
- the anti-OX40 antibody is PF- 04518600.
- the invention provides use of a combination comprising
- the 4-1 BB agonist is selected from the group consisting of utomilumab (PF- 05082566), 1 D8, SEIor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No.
- the 4-1 BB agonist is utomilumab.
- the invention provides use of a combination comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide (PF-06952229), having the structure: or a pharmaceutically acceptable salt thereof; and CTLA4 antagonist, wherein the combination is effective in treating cancer.
- the CTLA4 antagonist is an anti-CTLA4 antibody.
- the anti-CTLA4 antibody is ipilimumab (10DI), tremelimumab or AGEN-1884, or combinations thereof.
- the anti-CTLA4 antibody is ipilimumab (1 GDI).
- the invention provides use of 4-(2-(5- chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof in combination with immune checkpoint inhibitor, for treating cancer, wherein 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof is administered in combination with an additional anti-cancer agent.
- the immune checkpoint inhibitor is a PD-1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, or and/or CTLA4 antagonist.
- the additional anticancer agent is a further immune checkpoint inhibitor, an anti-tumor agent, an antiandrogen and/or anti-angiogenic agent.
- the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones.
- the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®).
- the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 - oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4- naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2- propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenali
- the invention further provides methods and uses comprising a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient in combination with an additional anticancer agent, as further described below.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating cancer.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in treating cancer, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
- the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating lung cancer.
- the lung cancer is small cell lung cancer (SCLC).
- SCLC is RB-negative.
- the lung cancer is non-small cell lung cancer (NSCLC).
- NSCLC non-small cell lung cancer
- the lung cancer is advanced or metastatic lung cancer.
- the lung cancer is advanced or metastatic SCLC.
- the lung cancer is advanced or metastatic NSCLC.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating ovarian cancer, peritoneal cancer, or fallopian tube cancer.
- the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating ovarian cancer.
- the ovarian cancer is epithelial ovarian cancer (EOC).
- the ovarian cancer is advanced or metastatic ovarian cancer (including EOC).
- the ovarian cancer is platinum resistant ovarian cancer (including EOC).
- the ovarian cancer is platinum resistant advanced or metastatic ovarian cancer (including EOC).
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating peritoneal cancer.
- the peritoneal cancer is primary peritoneal carcinomatosis (PPC).
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient, , and an immune checkpoint inhibitor, for use in treating the fallopian tube cancer (FTC).
- FTC fallopian tube cancer
- the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in treating breast cancer.
- the breast cancer is HR+/HER2- breast cancer.
- the breast cancer is HR+/HER2- advanced or metastatic breast cancer.
- the breast cancer is triple negative breast cancer (TNBC).
- TNBC is locally recurrent, advanced or metastatic TNBC.
- the breast cancer is HR+/HER2- breast cancer or TNBC, which may be advanced or metastatic, and the subject is a woman of any menopausal status or a man.
- the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating cancer, wherein the pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient is administered in combination with
- the pharmaceutical composition comprising 4-(2-(5- chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, the immune checkpoint inhibitor, and the additional anti-cancer agent together are effective in treating cancer.
- the additional anti-cancer agent is an endocrine therapeutic agent.
- the endocrine therapeutic agent is an aromatase inhibitor, a selective estrogen receptor degrader (SERD), or a selective estrogen receptor modulator (SERM).
- the endocrine therapeutic agent is letrozole. In other such embodiments, the endocrine therapeutic agent is fulvestrant.
- the invention provides use of a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating cancer.
- the invention provides use of a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating cancer
- the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
- the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating lung cancer.
- the lung cancer is small cell lung cancer (SCLC).
- SCLC is RB-negative.
- the lung cancer is non-small cell lung cancer (NSCLC).
- NSCLC non-small cell lung cancer
- the lung cancer is advanced or metastatic lung cancer.
- the lung cancer is advanced or metastatic SCLC.
- the lung cancer is advanced or metastatic NSCLC.
- the invention provides use of a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating ovarian cancer, peritoneal cancer, or fallopian tube cancer.
- the invention provides use of a pharmaceutical composition
- a pharmaceutical composition comprising 4- (2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient for treating ovarian cancer.
- the ovarian cancer is epithelial ovarian cancer (EOC).
- the ovarian cancer is advanced or metastatic ovarian cancer (including EOC).
- the ovarian cancer is platinum resistant ovarian cancer (including EOC).
- the ovarian cancer is platinum resistant advanced or metastatic ovarian cancer (including EOC).
- the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating peritoneal cancer.
- the peritoneal cancer is primary peritoneal carcinomatosis (PPC).
- the invention provides use of a pharmaceutical composition
- a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating the fallopian tube cancer (FTC).
- FTC fallopian tube cancer
- the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating breast cancer.
- the breast cancer is HR+/HER2- breast cancer.
- the breast cancer is HR+/HER2- advanced or metastatic breast cancer.
- the breast cancer is triple negative breast cancer (TNBC).
- TNBC is locally recurrent, advanced or metastatic TNBC.
- the breast cancer is HR+/HER2- breast cancer or TNBC, which may be advanced or metastatic, and the subject is a woman of any menopausal status or a man.
- the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating cancer, wherein the pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient is administered in combination with an additional anti-cancer agent.
- the pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, an immune checkpoint inhibitor, and a pharmaceutically acceptable excipient and the additional anti-cancer agent together are effective in treating cancer.
- the additional anti-cancer agent is an endocrine therapeutic agent.
- the endocrine therapeutic agent is an aromatase inhibitor, a selective estrogen receptor degrader (SERD), or a selective estrogen receptor modulator (SERM).
- the endocrine therapeutic agent is letrozole. In other such embodiments, the endocrine therapeutic agent is fulvestrant. In some embodiments of the foregoing, the invention provides further comprises treating the subject with chemotherapy, surgery and/or radiation therapy.
- the cancer stage includes but is not limited to early, advanced, locally advanced, remission, refractory, reoccurred after remission and progressive.
- Each therapeutic agent of the methods and combination therapies of the present invention may be administered in a medicament (also referred to herein as a pharmaceutical composition) which comprises the therapeutic agent and one or more pharmaceutically acceptable carriers, excipients, or diluents, according to pharmaceutical practice.
- excipient means the substances used to formulate active pharmaceutical ingredients (API) into pharmaceutical formulations.
- Excipients e.g., mannitol, Captisol®, lactose, starch, magnesium stearate, sodium saccharine, talcum, cellulose, sodium crosscarmellose, glucose, gelatin, sucrose, magnesium carbonate, and the like
- Excipients are an intergral part of pharmaceutical development and help to achieve the desired product profile including but not limited to an aid in manufacturing, modify a drug's stability, and efficacy.
- Acceptable excipients are non-toxic and do not adversely affect the therapeutic benefit of at least one chemical entity described herein.
- excipient may be any solid, liquid, semi-solid or, in the case of an aerosol composition, gaseous excipient that is generally available to one of skill in the art.
- excipient encompasses solubilizing agents, stabilizers, carriers, diluents, bulking agents, pH buffering agents, tonicifying agents, antimicrobial agents, wetting agents, and emulsifying agents e.g., sodium acetate, sodium citrate, cyclodextrine derivatives, sorbitan monolaurate, triethanolamine acetate, triethanolamine oleate, and the like).
- excipients are approved for or considered to be safe for human and animal administration.
- the pharmaceutical composition will contain about 0.005% to 95%; in certain embodiments, about 0.5% to 50% by weight of a chemical entity.
- lyophilization refers to a process by which the material to be dried is first frozen and then the ice or frozen solvent is removed by sublimation in a vacuum environment.
- lyophilized powder or "lyophilized preparation” refers to any solid material obtained by lyophilization, i.e., freeze-drying of an aqueous solution.
- the aqueous solution may contain non-aqueous solvents, i.e., a solution composed of aqueous and one or more non-aqueous solvent(s).
- a lyophilized preparation is one in which the solid material is obtained by freeze-drying a solution composed of water as a pharmaceutically acceptable excipient.
- co-administration refers to administration of two or more agents to the same subject during a treatment period.
- combination or “combination therapy” refer to the administration of each therapeutic agent of the combination therapy of the invention, in the form of a pharmaceutical composition or medicament, either sequentially, simultaneously, or concurrently.
- sequential refers to the administration of each therapeutic agent of the combination therapy of the invention, either alone or in a medicament, one after the other, wherein each therapeutic agent can be administered in any order. Sequential administration may be particularly useful when the therapeutic agents in the combination therapy are in different dosage forms, for example, one agent is a tablet and another agent is a sterile liquid, and/or the agents are administered according to different dosing schedules, for example, one agent is administered daily, and the second agent is administered less frequently such as weekly.
- the term “administered simultaneously,” simultaneously,” “simultaneous” or “simultaneous administration” means that the administration of the first therapeutic agent and that of a second therapeutic agent overlap in time with each other.
- the term “simultaneous” further refers to the administration of each therapeutic agent of the combination therapy of the invention in the same medicament.
- the term “concurrently” refers to the administration of each therapeutic agent in the combination therapy of the invention, either alone or in separate medicaments, wherein the second therapeutic agent is administered immediately after the first therapeutic agent, but that the therapeutic agents can be administered in any order.
- the therapeutic agents are administered concurrently.
- the two or more therapeutic agents may be encompassed in a single formulation and thus be administered simultaneously.
- the two or more therapeutic agents may be in separate physical formulations and administered separately, either sequentially, simultaneously, or concurrentlyto the subject.
- the combination therapy may be usefully administered to a subject during different stages of their treatment.
- the combination therapy is administered to a subject who is previously untreated, i.e. is treatment naive.
- the combination therapy is administered to a subject who has failed to achieve a sustained response after a prior therapy with a biotherapeutic or chemotherapeutic agent, i.e. is treatment experienced.
- the combination therapy may be administered prior to of following surgery to remove a tumor and I or may be used prior to, during or after radiation therapy, and / or may be used prior to, during or after chemotherapy.
- the invention relates to neoadjuvant therapy, adjuvant therapy, first-line therapy, second-line therapy, or third-line or later therapy, in each case for treating cancer as further described herein.
- the cancer may be localized, advanced or metastatic, and the intervention may occur at point along the disease continuum (i.e., at any stage of the cancer).
- the efficacy of combinations described herein in certain tumors may be enhanced by combination with other approved or experimental cancer therapies, e.g., radiation, surgery, chemotherapeutic agents, targeted therapies, agents that inhibit other signaling pathways that are dysregulated in tumors, and other immune enhancing agents, such as PD-1 axis binding antagonists, 0X40 agonists, 4-1 BB agonists, and/or CTLA4 antagonists.
- cancer therapies e.g., radiation, surgery, chemotherapeutic agents, targeted therapies, agents that inhibit other signaling pathways that are dysregulated in tumors, and other immune enhancing agents, such as PD-1 axis binding antagonists, 0X40 agonists, 4-1 BB agonists, and/or CTLA4 antagonists.
- the methods, combinations and uses of the current invention may further comprise one or more additional anti-cancer agents.
- Dosage regimens may be adjusted to provide the optimum desired response.
- a therapeutic agent of the combination therapy of the present invention may be administered as a single bolus, as several divided doses administered over time, or the dose may be proportionally reduced or increased as indicated by the exigencies of the therapeutic situation. It may be particularly advantageous to formulate a therapeutic agent in a dosage unit form for ease of administration and uniformity of dosage.
- Dosage unit form refers to physically discrete units suited as unitary dosages for the mammalian subjects to be treated; each unit containing a predetermined quantity of active compound calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier.
- the specification for the dosage unit forms of the invention may be dictated by and directly dependent on (a) the unique characteristics of the chemotherapeutic agent and the particular therapeutic or prophylactic effect to be achieved, and (b) the limitations inherent in the art of compounding such an active compound for the treatment of sensitivity in individuals.
- the dose and dosing regimen is adjusted in accordance with methods well- known in the therapeutic arts. That is, the maximum tolerable dose may be readily established, and the effective amount providing a detectable therapeutic benefit to a subject may also be determined, as can the temporal requirements for administering each agent to provide a detectable therapeutic benefit to the subject. Accordingly, while certain dose and administration regimens are exemplified herein, these examples in no way limit the dose and administration regimen that may be provided to a subject in practicing the present invention.
- dosage values may vary with the type and severity of the condition to be alleviated and may include single or multiple doses. It is to be further understood that for any particular subject, specific dosage regimens should be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the compositions, taking into consideration factors such as the severity of the disorder or condition, the rate of administration, the disposition of the compound and the discretion of the prescribing physician.
- the dosage ranges set forth herein are exemplary only and are not intended to limit the scope or practice of the claimed composition. For example, doses may be adjusted based on pharmacokinetic or pharmacodynamic parameters, which may include clinical effects such as toxic effects and/or laboratory values.
- the present invention encompasses intra-patient dose-escalation as determined by the skilled artisan. Determining appropriate dosages and regimens for administration of the chemotherapeutic agent are well-known in the relevant art and would be understood to be encompassed by the skilled artisan once provided the teachings disclosed herein.
- At least one of the therapeutic agents in the combination therapy is administered using the same dosage regimen (dose, frequency and duration of treatment) that is typically employed when the agent is used as a monotherapy for treating the same cancer.
- the subject received a lower total amount of at least one of the therapeutic agents in the combination therapy than when the same agent is used as a monotherapy, for example a lower dose of therapeutic agent, a reduced frequency of dosing and / or a shorter duration of dosing.
- An effective dosage of a small molecule inhibitor is typically in the range of from about 0.001 to about 100 mg per kg body weight per day, preferably about 1 to about 35 mg/kg/day, in single or divided doses. For a 70 kg human, this would amount to about 0.01 to about 7 g/day, preferably about 0.02 to about 2.5 g/day, and more preferably from about 0.02 to about 1 .0 g/day. In some instances, dosage levels at the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several small doses for administration throughout the day.
- the dosage may be administered as a single dose (QD), or optionally may be subdivided into smaller doses, suitable for BID (twice daily), TID (three times daily) or QID (four times daily) administration.
- An effective amount of the TGF[3r inhibitor, a PD-1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, or a CTLA4 antagonist may be administered for prevention or treatment of disease.
- the appropriate dosage of the TGFpr inhibitor, the PD-1 axis binding antagonist, the 0X40 agonist, the 4-1 BB agonist, and/or the CTLA4 antagonist may be determined based on the type of disease to be treated, the TGFpr inhibitor, the PD-1 axis binding antagonist, the 0X40 agonist, the 4-1 BB agonist, and/or the CTLA4 antagonist, the severity and course of the disease, the clinical condition of the subject, the subject's clinical history and response to the treatment, and the discretion of the attending physician.
- combination treatment with the TGF r inhibitor, the PD-1 axis binding antagonist (e.g., anti- PD-1 antibody), the 0X40 agonist (e.g., anti-human 0X40 agonist antibody), the 4-1 BB agonist (e.g., anti-human 4-1 BB agonist antibody), and/or the CTLA4 antagonist (anti-CTLA4 antibody) are synergistic, whereby an efficacious dose of the TGF[3r inhibitor, the PD-1 axis binding antagonist, the 0X40 agonist, the 4-1 BB agonist, and/or the CTLA4 antagonist in the combination is reduced relative to efficacious dose of the each of the TGFpr inhibitor, the PD-1 axis binding antagonist, the 0X40 agonist, the 4-1 BB agonist, and/or the CTLA4 antagonist as a single agent.
- the PD-1 axis binding antagonist e.g., anti- PD-1 antibody
- the 0X40 agonist e.g., anti-
- Dosage units for a PD-1 axis binding antagonist may be expressed as a flat dose, i.e., 100 mg, 200 mg, 300 mg, or as a patient-specific dose, i.e., mg/kg (mg therapeutic agent/kg of body weight) or mg/m 2 (quantity in milligrams per square meter of body surface area).
- a flat dose i.e., 100 mg, 200 mg, 300 mg
- a patient-specific dose i.e., mg/kg (mg therapeutic agent/kg of body weight) or mg/m 2 (quantity in milligrams per square meter of body surface area).
- Some embodiments that employ an antibody, antibody fragment or fusion soluble receptor as the PD-1 axis binding antagonist in the combination therapy may comprise administering the antibody at a dose of about 0.5, 1 , 2, 3, 5 or 10 mg/kg at intervals of about 7 days ( ⁇ 2 days) or 14 days ( ⁇ 2 days) or about 21 days ( ⁇ 2 days) or about 30 days ( ⁇ 2 days) throughout the course of treatment.
- the dosing regimen will comprise administering the antibody a dose of from about 0.005 mg/kg to about 10 mg/kg, with intra-patient dose escalation.
- the interval between doses will be progressively shortened, e.g., about 30 days ( ⁇ 2 days) between the first and second dose, about 14 days ( ⁇ 2 days) between the second and third doses.
- the dosing interval will be about 14 days ( ⁇ 2 days), for doses subsequent to the second dose.
- the dosing interval will be about 7 days ( ⁇ 2 days), for doses subsequent to the second dose.
- a subject will be administered an intravenous (IV) infusion of a medicament comprising any of the PD-1 axis binding antagonists described herein.
- IV intravenous
- the PD-1 axis binding antagonist in the combination therapy is sasanlimab (PF-6801591 ), nivolumab or pembrolizumab, which is administered intravenously or in a liquid dosage form at a dose selected from the group consisting of any one of : 1 mg/kg Q2W, 2 mg/kg Q2W, 3 mg/kg Q2W, 5 mg/kg Q2W, 10 mg Q2W, 1 mg/kg Q3W, 2 mg/kg Q3W, 3 mg/kg Q3W, 5 mg/kg Q3W, and 10 mg Q3W.
- the optimal dose for a PD-1 axis binding antagonist in combination with a TGFprl inhibitor may be identified by dose escalation of one or both of these agents.
- the TGF[3r1 inhibitor may be administered orally (PO), either once daily (QD) or twice daily (BID), with or without food on a continuous schedule starting on Cycle 1 Day 1 .
- a PD-1 axis binding antagonist may be administered as a 30-minute to 1 -hr intravenous (IV) infusion every 2 weeks (Q2W), every 3 weeks (Q3W) or in case of dose reduction, every 4 weeks (Q4W), starting on Cycle 1 Day 1 , except in the case of TGF[3r1 inhibitor lead-in.
- the TGFprl inhibitor may be given prior to or after administration of the PD-1 axis binding antagonist.
- an TGFprl inhibitor can be administered at25 mg, 50 mg, 75 mg, 100 mg, 125 mg, 150 mg, 200 mg, 250 mg, 375 mg, 500 mg, 625 mg, 1000 mg, 1200 mg, 1500 mg or 2000 mg on a BID or QD schedule, and the PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg, or 5 mg/kg or 10 mg/kg, at a dosing interval of Q2W, Q3W or alternately Q4W
- the TGF rl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg BID or QD for a 3-week lead-in period and then the PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg Q3W or 200 mg Q3W after the lead-in period.
- the TGFprl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg BID or QD and the PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg Q4W. In another embodiment, the TGFprl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg BID or QD and PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg Q3W. In another embodiment, the TGFprl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg BID or QD and the PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg Q4W.
- the TGFprl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg QD and sasanlimab (PF-6801591 ) is administered at a starting dose of 2 mg/kg Q3W. In another embodiment, the TGFprl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg QD and the PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg Q4W. In some embodiments, the patient is treated with a 3-week lead-in period of single-agent TGFprl inhibitor directly preceding the combination administration of the TGFprl inhibitor and the PD-1 axis binding antagonist.
- a treatment cycle begins with the first day of combination treatment and last for 3 weeks.
- the combination therapy is preferably administered for at least 18 weeks (6 cycles of treatment), more preferably at least 24 weeks (8 cycles of treatment), and even more preferably at least 2 weeks after the patient achieves a CR.
- the 4-1 BB agonist in the combination therapy comprises an anti-4-1 BB monoclonal antibody comprising heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 64 and SEQ ID NO: 65, respectively, and is administered in a liquid medicament at a dose selected from the group consisting of 1 mg/kg Q2W, 2 mg/kg Q2W, 3 mg/kg Q2W, 5 mg/kg Q2W, 10 mg Q2W, 1 mg/kg Q3W, 2 mg/kg Q3W, 3 mg/kg Q3W, 5 mg/kg Q3W, and 10 mg Q3W.
- the anti-4-1 BB monoclonal antibody is administered as a liquid medicament, and the selected dose of the medicament is administered by IV infusion over a time period of about 60 minutes.
- the anti-4-1 BB monoclonal antibody is administered at a starting dose of about 0.6 mg/kg Q4W and a PD-1 axis binding antagonist is administered at a starting dose of 10 mg/kg Q2W, and if the starting dose combination is not tolerated by the patient, then the dose of the PD-1 axis binding antagonist is reduced to 5 mg/kg Q2W and/or the dose of the anti-4-1 BB monoclonal antibody is reduced to 0.3 mg/kg Q4W.
- 3r1 inhibitor, or a pharmaceutically acceptable salt thereof is in the range of from about 0.001 to about 100 mg per kg body weight per day, preferably about 1 to about 35 mg/kg/day, in single or divided doses. For example, for a 70 kg human, this would amount to about 0.01 to about 7 g/day, preferably about 0.02 to about 2.5 g/day. In some instances, dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several small doses for administration throughout the day.
- the dose of TGF r1 inhibitor is increased up to a maximum dose of 250 mg BID if the patient tolerates the combination treatment at a lower total dose of TGFprl inhibitor.
- the TGFpd inhibitor is administered at a daily dosage of from about 50 mg to about 2000 mg per day, about 50 mg per day, about 100 mg per day, about 150 mg per day, about 200 mg per day, about 250 mg per day, about 300 mg per day, about 350 mg per day, about 400 mg per day, about 450 mg per day, about 500 mg per day, about 550 mg per day, about 600 mg per day, about 650 mg per day, about 700 mg per day, about 750 mg per day, about 800 mg per day, about 850 mg per day, about 900 mg per day, about 950 mg per day, about 1000 mg per day, about 1100 mg per day, about 1200 mg per day, about 1300 mg per day, about 1400 mg per day, or about 1500 mg per day.
- This dose may optionally be sub-divided into small doses, for example a dosage of 150 mg per day could be dosed as 75 mg dose twice per day.
- Dosage units for a TGF rl inhibitor may be expressed as a flat dose, i.e., 25 mg, 50 mg, 75 mg, 100 mg, 125 mg, etc. or as a patient-specific dose, i.e., mg/kg (mg therapeutic agent/kg of body weight) or mg/m 2 (quantity in milligrams per square meter of body surface area).
- Some embodiments may comprise administering the TGFprl inhibitor in a dose of about: 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, 70 mg, 75 mg, 80 mg, 85 mg, 90 mg, 95 mg, 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, 250 mg, or more than 250 mg, wherein the amounts can be administered once a day (q.d.), twice a day (b.i.d), three times a day (t.i.d.), four times a day (q.i.d.), or on some other dosing schedule.
- a “continuous dosing schedule” as used herein is an administration or dosing regimen without dose interruptions, e.g., without days off treatment. Repetition of 21 or 28 day treatment cycles without dose interruptions between the treatment cycles is an example of a continuous dosing schedule.
- the compounds of the combination of the present invention can be administered in a continuous dosing schedule.
- the TGFprl inhibitor is PF-06952229.
- PF-06952229, or a pharmaceutically acceptable salt thereof is administered at a daily dosage of about 125 mg once daily, about 100 mg once daily, about 75 mg once daily, or about 50 mg daily. In an embodiment, which is the recommended starting dose or standard clinical dose, PF-06952229, or a pharmaceutically acceptable salt thereof, is administered at a daily dosage of about 125 mg once a day. In an embodiment, PF-06952229, or a pharmaceutically acceptable salt thereof, is administered at a non-standard clinical dose. In an embodiment, a non- standard clinical dose is a low-dose amount of PF-06952229, or a pharmaceutically acceptable salt thereof.
- PF-06952229 is administered at a dose of about 100 mg once daily, about 75 mg once daily, or about 50 mg once daily.
- PF-06952229, or a pharmaceutically acceptable salt thereof is administered at a dose of about 100 mg once daily.
- PF-06952229, or a pharmaceutically acceptable salt thereof is administered at a dose of about 75 mg once daily.
- PF-06952229, or a pharmaceutically acceptable salt thereof is administered at a dose of about 50 mg once daily.
- Dosage amounts provided herein refer to the dose of the free base form of PF-06952229, or are calculated as the free base equivalent of an administered PF- 06952229 salt form.
- a dosage or amount of PF-06952229 such as 100 mg, 75 mg or 50 mg, refers to the free base equivalent.
- This dosage regimen may be adjusted to provide the optimal therapeutic response. For example, the dose may be proportionally reduced or increased as indicated by the exigencies of the therapeutic situation.
- the practice of the method of this invention may be accomplished through various administration or dosing regimens.
- the compounds of the combination of the present invention can be administered sequentially, simultaneously, or concurrently.
- the compounds of the combination of the present invention can be administered in a concurrent dosing regimen.
- a “continuous dosing schedule,” as used herein, is an administration or dosing regimen without dose interruptions, e.g., without days off treatment. Repetition of 21 or 28 day treatment cycles without dose interruptions between the treatment cycles is an example of a continuous dosing schedule.
- the compounds of the combination of the present invention can be administered in a continuous dosing schedule. In an embodiment, the compounds of the combination of the present invention can be administered concurrently in a continuous dosing schedule.
- Administration of combinations of the invention may be affected by any method that enables delivery of the compounds to the site of action. These methods include oral routes, intraduodenal routes, parenteral injection, topical, and rectal administration. In some embodiments, therapeutic agents of the combination therapies of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ. Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular, subcutaneous infusion. Suitable devices for parenteral administration include needle (including micro needle) injectors, needle-free injectors and infusion techniques.
- Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile nonaqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
- excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9)
- a suitable vehicle such as sterile, pyrogen-free water.
- parenteral formulations under sterile conditions may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- solubility of therapeutic agents used in the preparation of parenteral solutions may potentially be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents.
- Formulations for parenteral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- therapeutic agents of the combination therapies of the invention may potentially be formulated as a solid, semisolid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug-coated stents and PGLA microspheres.
- the therapeutic agents of the combination therapies of the invention may also potentially be administered topically to the skin or mucosa, that is, dermally or transdermally.
- Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibers, bandages and microemulsions. Liposomes may also be used.
- Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol.
- Penetration enhancers may be incorporated; see, for example, Finnin and Morgan, Transdermal penetration enhancers: applications, limitations, and potential, J Pharm Sci 1999, 88 (10), 955-958.
- Other means of topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and micro needle or needle-free (e.g., PowderjectTM, BiojectTM, etc.) injection.
- the disclosures of these references are incorporated herein by reference in their entireties.
- Formulations for topical administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- Therapeutic agents of the combination therapies of the invention may also potentially be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurized container, pump, spray, atomizer (preferably an atomizer using electrohydrodynamics to produce a fine mist), or nebulizer, with or without the use of a suitable propellant, such as 1 , 1 ,1 ,2- tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3-heptafluoropropane.
- the powder may include a bioadhesive agent, for example, chitosan or cyclodextrin.
- the pressurized container, pump, spray, atomizer, or nebulizer may contain a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilizing, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilizing, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- the compound Prior to use in a dry powder or suspension formulation, the compound may be micronized to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray drying.
- comminuting method such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray drying.
- Capsules made, for example, from gelatin or HPMC
- blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the therapeutic agent, a suitable powder base such as lactose or starch and a performance modifier such as l-leucine, mannitol, or magnesium stearate.
- the lactose may be anhydrous or in the form of the monohydrate, preferably the latter.
- Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
- a suitable solution formulation for use in an atomizer using electrohydrodynamics to produce a fine mist may contain from 1 g to 20mg of the therapeutic agent per actuation and the actuation volume may vary from 1 pL to 100pL.
- a typical formulation includes a therapeutic agent, propylene glycol, sterile water, ethanol and sodium chloride.
- Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
- Suitable flavors such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations intended for inhaled/intranasal administration.
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, poly(DL-lactic-coglycolic acid (PGLA).
- Modified release formulations include delayed-, sustained-, pulsed-, controlled- , targeted and programmed release.
- the dosage unit is determined by means of a valve which delivers a metered amount.
- Units in accordance with the invention are typically arranged to administer a metered dose or “puff” containing a desired mount of the therapeutic agent.
- the overall daily dose may be administered in a single dose or, more usually, as divided doses throughout the day.
- Therapeutic agents of the combination therapies of the invention may potentially be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
- Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- Therapeutic agents of the combination therapies of the invention may also potentially be administered directly to the eye or ear, typically in the form of drops of a micronized suspension or solution in isotonic, pH-adjusted, sterile saline.
- Other formulations suitable for ocular and aural administration may include ointments, biodegradable (e.g., absorbable gel sponges, collagen) and non-biodegradable (e.g., silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes.
- a polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride.
- a preservative such as benzalkonium chloride.
- Such formulations may also be delivered by iontophoresis.
- Example 1 TGFprl Inhibitor, PF-06952229, Anti-tumor Efficacy in Combination with an anti-PD-1 Antibody
- the TGFprl inhibitor, PF-06952229 anti-tumor efficacy in combination with an anti-PD-1 antibody was studied in the syngeneic MC38 tumor model to assess efficacy on primary tumor growth and effects on survival.
- PF-06952229 combination with an anti- PD-1 antibody led to a significant increase in tumor growth inhibition and to significant increased survival relative to PF-06952229 or PD-1 monotherapy.
- vehicle 50% PEG4000 in 10 mM citric acid
- PF-06952229 rat polyclonal clone 2A3 control antibody
- PF-06952229 anti-PD-1 antibody
- anti-PD-1 antibody mouse RMP1 -14 BioxCell
- the MC38 syngeneic colon cancer cell line was obtained from the National Cancer Institute (NCI).
- PF-06952229 was administered orally (PO) continuously twice daily (BID) at 30 mg/kg, or intermittently (7 days on/ 7 days off) at 10 or 30 mg/kg for 5 cycles.
- Antibodies were administered at 5 mg/kg intraperitoneally (IP) twice weekly for two weeks (Biwk x2).
- T umors were measured using calipers twice a week.
- T reatment efficacy was determined based on mean tumor volumes of animals remaining on the last day of the study (Day 63) and from the incidence and magnitude of tumor regression responses observed during the study.
- a Kaplan Meier survival curve was constructed to show the percentage of animals in each group remaining in the study as a function of time. Animals were eliminated from study when tumor volume reached the 1500 mm3 as a survival endpoint.
- TGFprl inhibitor PF-06952229 combination with an anti-PD1 antibody in the MC38 syngeneic tumor model led to greater tumor growth inhibition and improvement in survival relative to PF-06952229 monotherapy or anti-PD1 antibody monotherapies in the MC38 syngeneic mouse tumor model (Table 9 and FIG. 1A and FIG. 1 B).
- Table 9 shows the results of PF-06952229 single agent and in combination with anti-PD-1 antibody in syngeneic MC38 murine tumor Model.
- Example 2 TGFprl Inhibitor, PF-06952229, Anti-tumor Efficacy in Combination with an Anti-4-1 BB Antibody or an Anti-OX40 Antibody
- the TGFprl inhibitor, PF-06952229 was evaluated in the 4T1 orthotopic syngeneic mouse tumor model in combination with an anti-4-1 BB antibody or in combination with an anti-OX40 antibody to assess effect on primary tumor growth.
- PF06952229 combination with 4-1 BB antibody led to a significant increase in tumor growth inhibition relative to PF-06952229 monotherapy and to 4-1 BB monotherapy treatments.
- PF-06952229 combination treatment with the anti-OX40 antibody led to greater tumor growth inhibition relative to PF-06952229 monotherapy and to 0X40 antibody monotherapy treatment.
- mice Female Balb/cJ mice were obtained from Jackson Laboratories and were implanted with 0.1 x 10 6 4T1 cells (American Tissue Culture Collection) in the mammary fat pad implanted. Tumor bearing mice were randomized into six treatment groups based on average tumor sizes of approximately 70-80 mm 3 per group. Study groups included vehicle, 30 mg/kg PF-06952229, 1 mg/kg 4-1 BB antibody (MAB9371 , mouse IgG 1 , R&D Systems), 0X40 antibody (0X86, mouse lgG1 ), combination of PF-06952229 + 4-1 BB antibody and of PF-06952229 + 0X40 antibody.
- PF-06952229 was administered orally (po) twice daily (BID) continuously, until the end of the study (Day 25).
- 4-1 BB and 0X40 antibodies were administered intraperitoneally (ip) every four days, for 3 cycles (Q4Dx3).
- Tumor volumes were measured two times a week. Tumor volume was calculated based on two dimensional caliper measurement with cubic millimeter volume calculated using the formula (length x width 2 ) x 0.5. Tumor growth results were plotted using GraphPad Prism 7 software.
- the treatment groups and dose regimen information are summarized in Table 10.
- Tumor growth results on Day 25 post-treatment initiation showed that treatment with the TGF[3r1 inhibitor (PF-06952229) monotherapy, anti-4-1 BB mAb, and anti-OX40 mAb did not significantly inhibit primary tumor growth in the 4T1 syngeneic tumor model compared to vehicle-treated control group with mean tumor growth inhibition of 18%, 8%, 0.5% respectively, relative to the vehicle control group; however, PF-06952229 treatment in combination with the anti-4-1 BB antibody led to a significant decrease in tumor growth relative to PF-06952229 monotherapy or anti-4-1 BB mAB monotherapy, with mean tumor growth inhibition of 41% relative to vehicle-treated control group.
- TGFprl inhibitor (PF-06952229) combination with anti-4-1 BB antibody or anti- 0X40 antibody led to greater tumor growth inhibition relative to PF-06952229 or antibody monotherapy treatments in the 4T 1 orthotopic syngeneic tumor model. Accordingly, the TGF rl inhibitor (PF-06952229) combination with anti-4-1 BB antibody or with anti-OX40 antibody led to a greater efficacy than PF-06952229 monotherapy, anti-4-1 BB antibody monotherapy, or anti-OX40 antibody monotherapy.
- Example 3 TGFprl Inhibitor, PF-06952229, Anti-tumor Efficacy in Combination with an Anti-CTLA4 antibody
- mice Female Balb/c mice (Charles River Laboratories) were implanted with 3 x 10 5 CT26 cells (American Tissue Culture Collection) in the right flank on Day 0 and treatments started on Day 3. Study groups included vehicle, control antibody (hamster polyclonal IgG), 30 mg/kg PF-06952229, 5 and 2.5 mg/kg anti-CTLA4 antibody (UC10-4F10-11 , BioXcell), and combination of PF-06952229 + anti-CTLA4 antibody. PF-06952229 was administered orally (po) twice daily (BID) continuously, for 28 days. Anti-CTLA4 antibody was administered intraperitoneally (ip) on Days 8, 11 and 14. The treatment groups and dose regimen information are summarized in Table 12.
- TTE Time to endpoint
- the data set consisted of the first observation that exceeded the endpoint volume used in analysis and the three consecutive observations that immediately preceded the attainment of this endpoint volume.
- the calculated TTE is usually less than the tumor progression (TP) date, the day on which the animal was eliminated from study for tumor size. Animals with tumors that did not reach the endpoint volume were assigned a TTE value equal to the last day of the study. In instances in which the log-transformed calculated TTE preceded the day prior to reaching endpoint or exceeded the day of reaching tumor volume endpoint, a linear interpolation was performed to approximate TTE.
- TTD tumor growth delay
- Tumor growth results were plotted using GraphPad Prism software.
- Tumor growth results on Day 60 show that treatment with the TGF[3r1 inhibitor (PF-06952229) monotherapy did not significantly inhibit primary tumor growth in the CT26 syngeneic tumor model, and treatment with anti-CTLA4 antibody as a monotherapy did show significant tumor growth delay compared to PF-06952229 monotherapy treatment.
- combination treatment of PF-0692225 with the anti-CTLA4 antibody led to a significant combinatorial effect regarding delay of tumor growth relative to PF-06952229 monotherapy (p ⁇ 0.001 ) and anti-CTLA4 antibody monotherapy (p ⁇ 0.05).
- Table 13 shows asummary of results from the combination study in the CT26.
- TGF[3r1 inhibitor (PF-06952229) combination with the anti-CTLA4 antibody led to greater tumor growth delay relative to PF-06952229 or anti-CTLA4 antibody monotherapy treatment in the CT26 syngeneic tumor model. Accordingly, the TGF[3r1 inhibitor (PF-06952229) combination with anti-CTLA4 antibody led to a greater efficacy than PF-06952229 monotherapy, or anti-CTLA4 antibody monotherapy.
Landscapes
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
This invention relates to TGFβr1 inhibitors for use in treating cancer in combination with immune checkpoint inhibitors, PD-1 axis binding antagonists, OX40 agonists, 4-1BB agonists and CTLA4 antagonists, or combinations thereof and associated methods of treatment, combinations, pharmaceutical compositions and uses thereof.
Description
TGFprl INHIBITOR COMBINATION THERAPIES
REFERENCE TO SEQUENCE LISTING
This application is being filed electronically via EFSWeb and includes an electronically submitted sequence listing in .txt format. The .txt file contains a sequence listing entitled “PC072670SEQLISTING_ST25.txt created on November 15, 2021 and having a size of 85 KB. The sequence listing contained in this .txt file is part of the specification and is herein incorporated by reference in its entirety.
FIELD OF THE INVENTION
The present invention relates to methods and combination therapies useful for the treatment of cancer. In particular, the invention relates to method of treating cancer comprising administering to the subject an amount of a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, or a pharmaceutically acceptable salt thereof and an immune checkpoint inhibitor (e.g., PD-1 axis binding antagonists, 0X40 agonists, 4-1 BB agonists and CTLA4 antagonists, or combinations thereof). Embodiments of the present invention relate to associated combinations for use in treatment of cancer, pharmaceutical compositions and uses thereof.
BACKGROUND
TGFp signalling is an emerging pathway in cancer progression and has a role in modulating immune response, and in many other cancer pathways including metastasis and angiogenesis. Elevated TGFp expression by tumor and stromal cells in the tumor microenvironment and activation of TGFp receptor intracellular signalling is observed in many cancers (Massague J., TGFbeta in Cancer. Cell 2008, 134(2):215-30; Neuzillet C, et al., Targeting the TGFp pathway for cancer therapy, Pharmacol Ther 2015, 147:22- 31 ). The TGFp signalling pathway can be activated upon interaction of dimeric TGFp ligand with its specific cell-surface transmembrane serine/threonine kinase receptors. The activated TGFp ligand interacts with TGFp type II receptors (TGFpr2), which recruit and phosphorylate TGFp type I receptors (TGFprl , also known as activin receptor-like kinase (ALK5)) at specific serine and threonine residues (Principe et al., TGF-B: duality of function between tumor prevention and carcinogenesis, J Natl Cancer Inst 2014, 106(2): djt369).
Activation of the TGF pathway in cancer cells can induce epithelial-to- mesenchymal transition (EMT) in which epithelial cells lose their apico-basal polarity and cell-cell adhesion, to become highly migratory mesenchymal cells, leading to metastasis. In addition to importance in tumor cell migration and metastasis, EMT has also been linked to tumor cell evasion of immune surveillance (Akalay I, et al., Epithelial-to- mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis, Cancer Res 2013, 73(8):2418-27). TGF is a potent immunosuppressive agent on both innate and adaptive immune cells, including dendritic cells, macrophages, natural killer cells, and CD4+ and CD8+ T cells. Conversely, TGFp has a key role stimulating the differentiation of immune-suppressive regulatory T (Treg) cells and myeloid derived suppressor cells (MDSCs) (Akalay I, et al., 2013).
TGF pathways have key roles in disease progression and resistance to therapy in a broad spectrum of tumors (Neuzillet C., et. al., 2015; Colak S, et. al., Targeting TGF- B signaling in cancer, Trends in Cancer 2017, 3(1 ):56-71 ). High TGFp signatures and EMT gene expression are found in a variety of tumors (Mak MP, etal., A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition, Clin Cancer Res 2016, 22(3):609-20.).
TGFp is an important regulator of the tumor microenvironment by inducing expression of extracellular matrix (ECM) proteins and suppressing expression of chemokines and cytokines required for T cell tumor infiltration, creating a reactive stroma with dense ECM and a T cell excluded infiltrate phenotype, with peritumoral or stromal T cell localization (Hegde PS, et. al., The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition, Clin Cancer F?es 2016, 22(8):1865-74).
The compound, 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide (also referred to as “TGFprl inhibitor PF- 06952229,” “PF-06952229” or “PF-‘2229”), is a potent and selective TGFprl (transforming growth factor beta receptor type 1 ) inhibitor, having the structure:

PF-06952229 and pharmaceutically acceptable salts thereof are disclosed in International Application No. PCT/US2014/072922, which published as International Publication No. WO 2015/103355 on 9 July 2015, and U.S. Patent No. 10,030,004 issued on 10 December 2019. The contents of each of the foregoing references are incorporated herein by reference in their entirety.
The programmed death 1 (PD-1) receptor and PD-1 ligands 1 and 2 (PD-L1 and PD-L2, respectively), play integral roles in immune regulation. PD-1 is a key immune checkpoint receptor expressed by activated T and B cells. PD-1 is a member of the CD28 family of receptors, which includes CD28, CTLA4, ICOS, PD-1 , and BTLA. Two cell surface glycoprotein ligands for PD-1 have been identified, Programmed Death Ligand- 1 (PD-L1 ) and Programmed Death Ligand-2 (PD-L2), that are expressed on antigen- presenting cells as well as many human cancers and have been shown to down-regulate T cell activation and cytokine secretion upon binding to PD-1 . Several PD-1 axis binding antagonists, including the anti-PD-1 antibodies nivolumab (Opdivo), and pembrolizumab (Keytruda) were approved by the U.S. Food and Drug Administration (FDA) for the treatment of oncology indications in recent years.
The 0X40 receptor (0X40, also known as CD134, TNFRSF4, ACT-4, ACT35, and TXGP1 L) is a member of the TNF receptor super family. 0X40 is found to be expressed on activated CD4+ and CD8+ T-cells. High numbers of 0X40+ T cells have been demonstrated within tumors (tumor infiltrating lymphocytes) and in the draining lymph nodes of cancer patients (Weinberg, A. et a!., Engagement of the OX-40 receptor in vivo enhances antitumor immunity, J. Immunol. 2000,164: 2160-69; Petty, J. etal., Survival in human colorectal cancer correlates with expression of the T-cell costimulatory molecule
OX-40 (CD134), Am. J. Surg. 2002, 183: 512-518). Signaling through 0X40 on activated CD4+ and CD8+ T cells leads to enhanced cytokine production, granzyme and perforin release, and expansion of effector and memory T-cell pools. In addition, 0X40 signaling on Treg cells inhibits expansion of Tregs, shuts down the induction of Tregs, and blocks Treg-suppressive function. It was shown in tumor models in mice that engagement of 0X40 in vivo during tumor priming significantly delayed and prevented the appearance of tumors as compared to control treated mice (Weinberg et al., 2000). Therefore, it has been contemplated to enhance the immune response of a mammal to an antigen by engaging 0X40 through the use of an 0X40 binding agent (WO 1999/042585; Weinberg et al., 2000).
4-1 BB (also known as CD137 and TNFRSF9), which was first identified as an inducible costimulatory receptor expressed on activated T cells, is a membrane spanning glycoprotein of the Tumor Necrosis Factor (TNF) receptor superfamily. Current understanding of 4-1 BB indicates that expression is generally activation dependent and encompasses a broad subset of immune cells including activated NK and NKT cells; regulatory T cells; dendritic cells (DC) including follicular DC; stimulated mast cells, differentiating myeloid cells, monocytes, neutrophils, eosinophils, and activated B cells. 4-1 BB expression has also been demonstrated on tumor vasculature (19-20) and atherosclerotic endothelium. The ligand that stimulates 4-1 BB (4-1 BBL) is expressed on activated antigen presenting cells (APCs), myeloid progenitor cells and hematopoietic stem cells. 4-1 BB agonist mAbs increase costimulatory molecule expression and markedly enhance cytolytic T lymphocyte responses, resulting in anti-tumor efficacy in various models. 4-1 BB agonist mAbs have demonstrated efficacy in prophylactic and therapeutic settings and both monotherapy and combination therapy tumor models and have established durable anti-tumor protective T cell memory responses.
Cytotoxic T lymphocyte associated antigen 4 (CTLA4) is expressed on activated T cells and serves as a co-inhibitor to keep T cell responses in check following CD28- mediated T cell activation. CTLA4 is believed to regulate the amplitude of the early activation of naive and memory T cells following TCR engagement and to be part of a central inhibitory pathway that affects both antitumor immunity and autoimmunity. CTLA4 is expressed exclusively on T cells, and the expression of its ligands CD80 (B7.1 ) and CD86 (B7.2), is largely restricted to antigen-presenting cells, T cells, and other immune mediating cells. Antagonistic anti-CTLA4 antibodies that block the CTLA4 signaling
pathway have been reported to enhance T cell activation. One such antibody, ipilimumab, was approved by the FDA in 2011 for the treatment of metastatic melanoma. Another anti-CTLA4 antibody, tremelimumab, was tested in phase III trials for the treatment of advanced melanoma but did not significantly increase the overall survival of patients compared to the standard of care (temozolomide or dacarbazine) at that time.
Nevertheless, there remains a need for improved therapies for the treatment of cancers. The methods, combinations, uses and pharmaceutical compositions of the present invention have been demonstrated greater efficacy than treatment with either therapeutic agent alone. In addition to greater efficacy than treatment with either therapeutic agent alone, the methods, combinations, uses and pharmaceutical compositions of the present invention are believed to have one or more advantages, such as potential to reduce drug-drug interactions; potential to enable an improved dosing schedule; potential to reduce side effects; potential to overcome resistance mechanisms and the like.
SUMMARY OF THE INVENTION
This invention relates to methods, combinations, uses and pharmaceutical compositions for treating cancer in a subject in need thereof. Also provided are kits comprising one or more of the compositions of the invention.
In one aspect, the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject:
(a) an amount of a transforming growth factor beta receptor type 1 (TGF|3r1 ) inhibitor, or a pharmaceutically acceptable salt thereof, and;
(b) an amount of a programmed cell death protein 1 (PD-1 ) axis binding antagonist; wherein the amounts in (a) and (b) together are effective in treating cancer.
In some embodiments of this aspect, the invention provides a method further comprising administering to the subject (c) an amount of an additional anti-cancer agent; wherein the amounts in (a), (b) and (c) together are effective in treating cancer.
In a preferred embodiment of the methods described herein, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2-
fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof. In a more preferred embodiment, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
In some embodiments of the methods described herein, the PD-1 axis binding antagonist is an anti-PD-1 antibody. In some such emdodiments, the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-011 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001 ), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, Bl- 754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105, BAT-1306, and AGEN-2034, or combinations thereof. In a specific embodiment, the anti-PD-1 antibody is sasanlimab (PF-6801591).
In one preferred aspect, the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-PD-1 antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
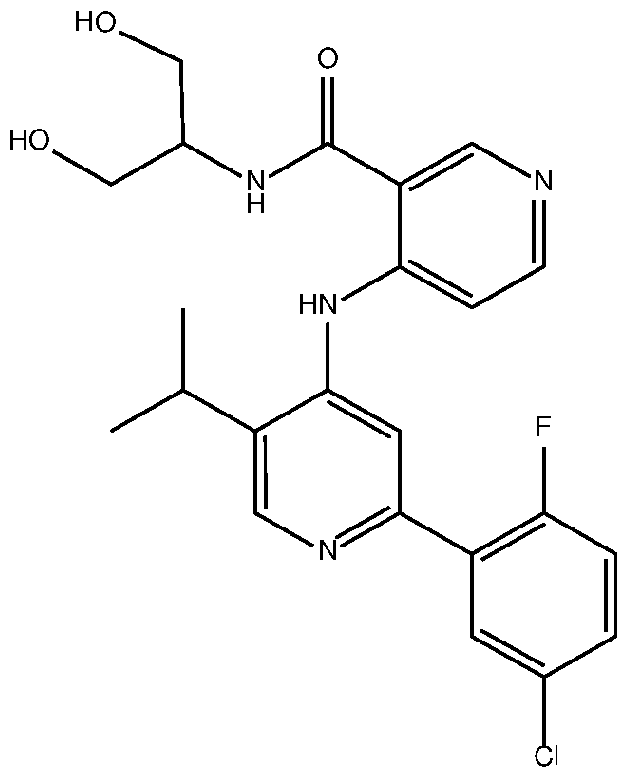
In some embodiments of the methods described herein, the TGFprl inhibitor and the PD-1 axis binding antagonist are administered sequentially, simultaneously, or concurrently.
In one aspect, the invention provides a combination comprising:
(a) an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof, and;
(b) a PD-1 axis binding antagonist; for use in treating cancer.
In some embodiments of this aspect, the combination for use further comprises
(c) an additional anti-cancer agent.
In a preferred embodiment of the combinations described herein, the TGF rl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2- (5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof. In a specific embodiment, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
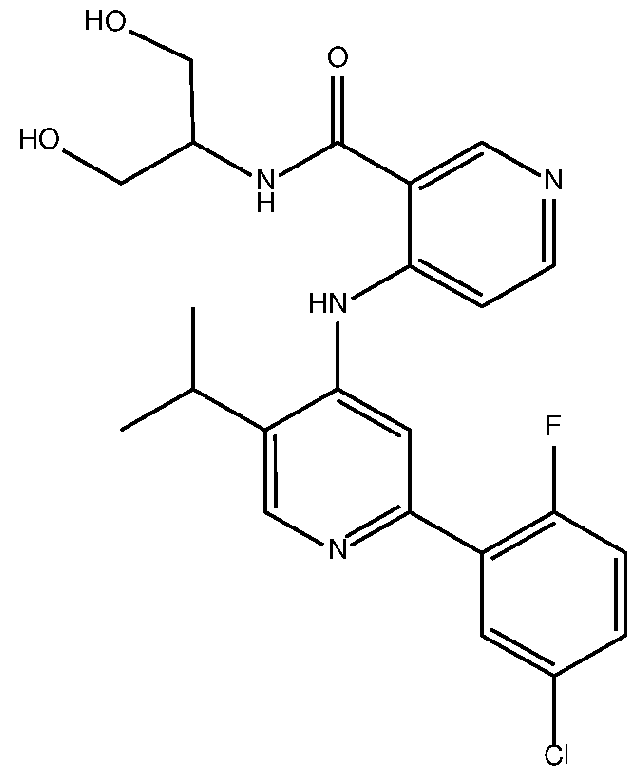 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of the combinations described herein, the PD-1 axis binding antagonist is an anti-PD-1 antibody. In some such emdodiments, the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-01 1), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105, BAT-
1306, and AGEN-2034, or combinations thereof. In a specific embodiment, the anti-PD- 1 antibody is sasanlimab (PF-6801591 ).
In one preferred aspect, the invention provides a combination comprising a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-PD-1 antibody, wherein the TGF[3r1 inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure:
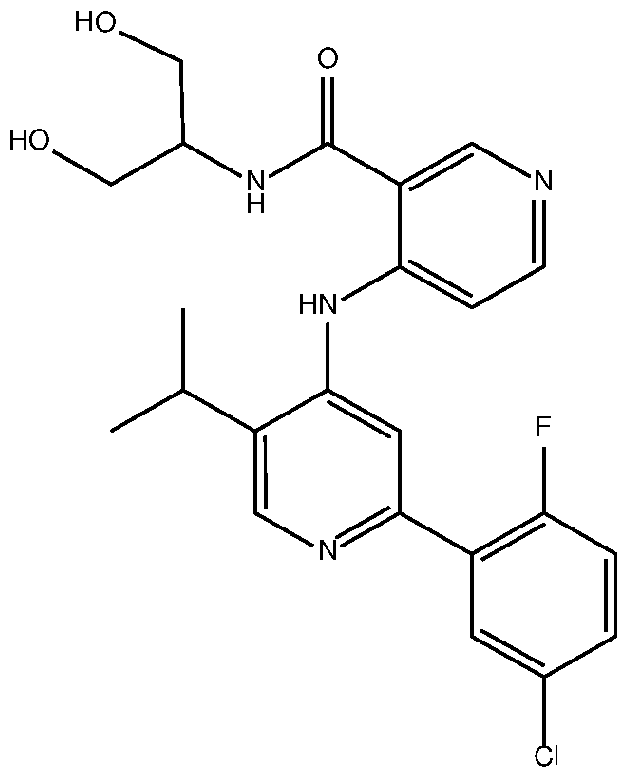 or a pharmaceutically acceptable salt thereof; and wherein the anti-PD-1 antibody is sasanlimab (PF-6801591), for use in treating cancer.
or a pharmaceutically acceptable salt thereof; and wherein the anti-PD-1 antibody is sasanlimab (PF-6801591), for use in treating cancer.
In one preferred aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
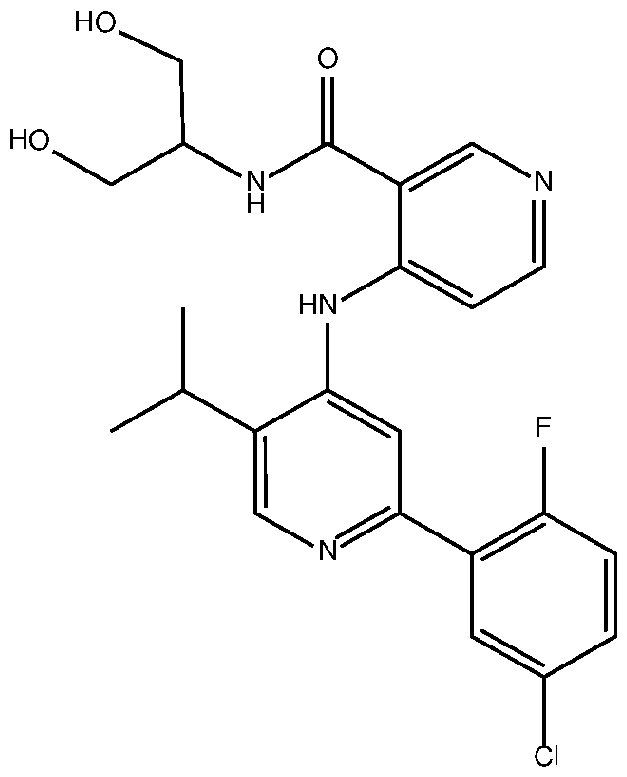 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a PD-1 axis binding antagonist, for treating cancer. In one preferred aspect, the invention provides a pharmaceutical composition comprising a PD-1 axis binding antagonist, for use in combination with 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a PD-1 axis binding antagonist, for treating cancer. In one preferred aspect, the invention provides a pharmaceutical composition comprising a PD-1 axis binding antagonist, for use in combination with 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
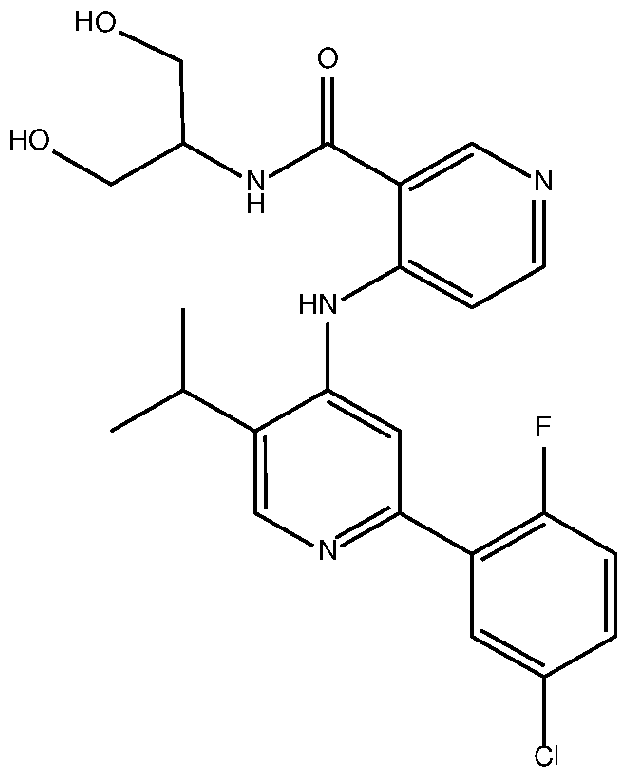 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer.
In a preferred embodiment of the pharmaceutical composition described herein, the PD-1 axis binding antagonist is an anti-PD-1 antibody. In some such emdodiments, the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-01 1 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ),
LZM-009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105, BAT- 1306, and AGEN-2034, or combinations thereof. In some such emdodiments, the anti- PD-1 antibody is sasanlimab (PF-6801591 ).
In a preferred embodiment of this aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
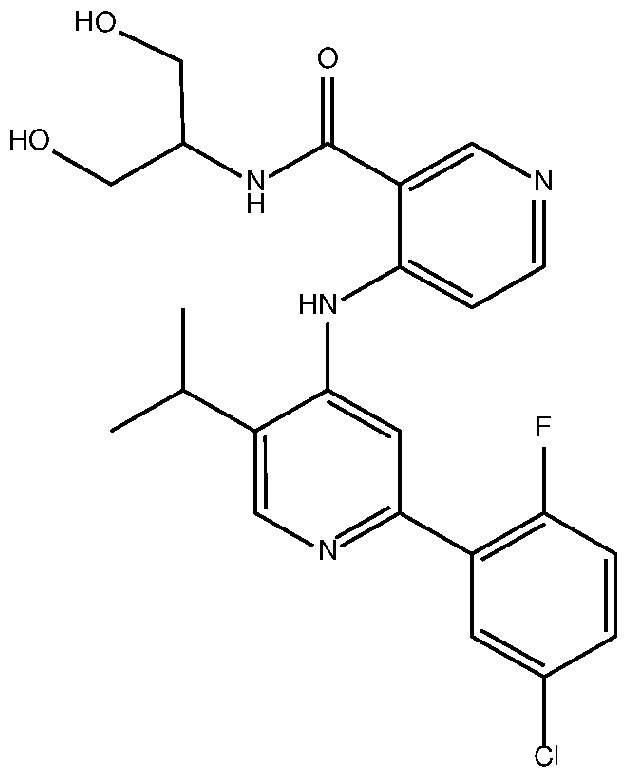
In a preferred embodiment of this aspect, the invention provides a pharmaceutical composition comprising an anti-PD-1 antibody, for use in combination with 4-(2-(5-chloro- 2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the anti-PD-1 antibody is sasanlimab (PF-6801591 ), for treating cancer.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the anti-PD-1 antibody is sasanlimab (PF-6801591 ), for treating cancer.
In one aspect, the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject:
(a) an amount of a transforming growth factor beta receptor type 1 (TGF[3r1) inhibitor, or a pharmaceutically acceptable salt thereof, and;
(b) an amount of an 0X40 agonist, wherein the amounts in (a) and (b) together are effective in treating cancer.
In some embodiments of this aspect, the invention provides a method further comprising administering to the subject (c) an amount of an additional anti-cancer agent; wherein the amounts in (a), (b) and (c) together are effective in treating cancer.
In a preferred embodiment of the methods described herein, the TGF rl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof. In a specific embodiment, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
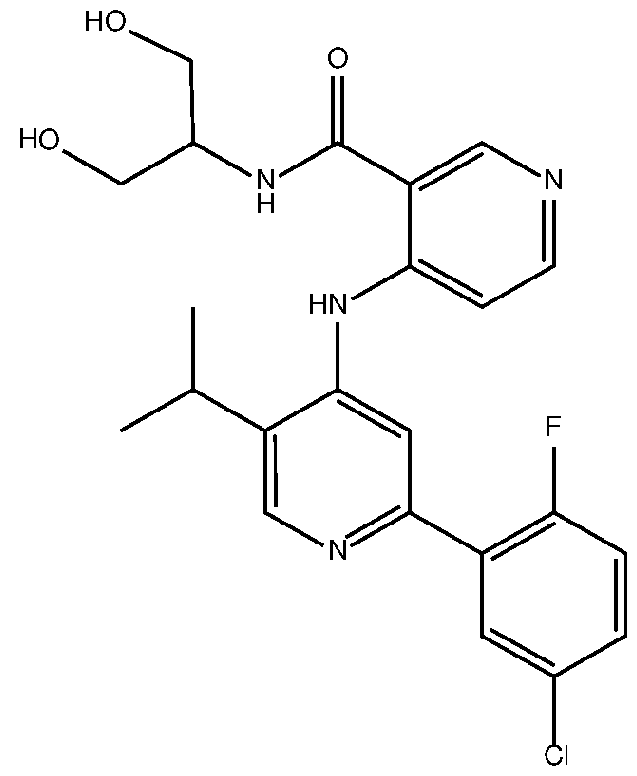 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
In some embodiments of the methods described herein, the 0X40 agonist is selected from the group consisting of an anti-OX40 antibody, an OX40L agonist fragment, an 0X40 oligomeric receptor, a trimeric OX40L-Fc protein and an 0X40 immunoadhesin, or a combination thereof. In some such emdodiments, the 0X40 agonist is an anti-OX40 antibody. In some such emdodiments, the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383,
MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR-8383, and ABBV-368, or combinations thereof. In a specific embodiment, the anti-OX40 antibody PF-04518600.
In one preferred aspect, the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-OX40 antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
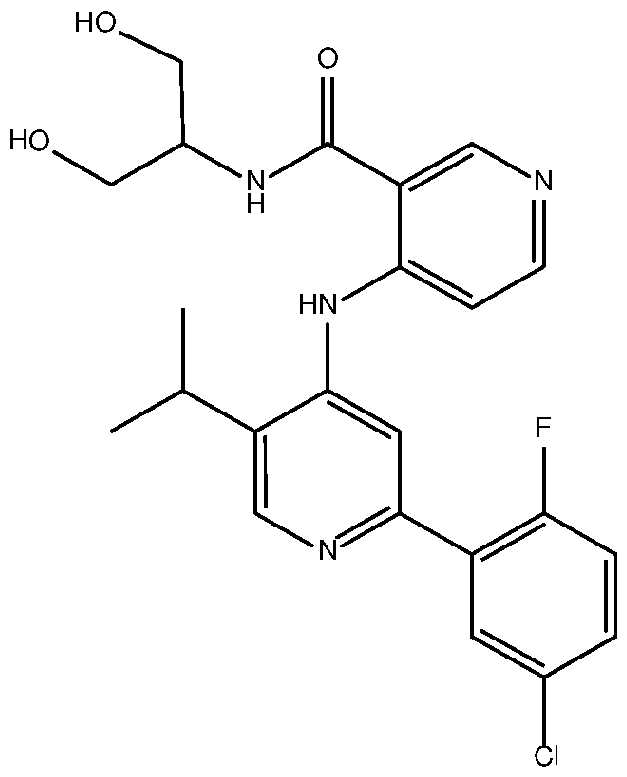 or a pharmaceutically acceptable salt thereof, wherein the anti-OX40 antibody is PF- 04518600, and wherein the amounts together are effective in treating cancer.
or a pharmaceutically acceptable salt thereof, wherein the anti-OX40 antibody is PF- 04518600, and wherein the amounts together are effective in treating cancer.
In some embodiments of the methods described herein, the TGFprl inhibitor and the 0X40 agonist are administered sequentially, simultaneously, or concurrently.
In one aspect, the invention provides a combination comprising:
(a) an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof, and;
(b) an 0X40 agonist; for use in treating cancer.
In some embodiments of this aspect, the combination for use further comprises
(c) an additional anti-cancer agent.
In a preferred embodiment of the combinations described herein, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334,
SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2- (5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof. In a specific embodiment, the TGF|3r1 inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of the combinations described herein, the 0X40 agonist is an anti-OX40 antibody, an OX40L agonist fragment, an 0X40 oligomeric receptor, a trimeric OX40L-Fc protein or an 0X40 immunoadhesin, or combinations thereof. In some such emdodiments, the 0X40 agonist is an anti-OX40 antibody. In some such emdodiments, the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG- 7888, GSK-3174998, BMS-986178, GBR-8383, and ABBV-368, or combinations thereof. In a specific embodiment, the anti-OX40 antibody is PF-04518600.
In one preferred aspect, the invention provides a combination comprising a transforming growth factor beta receptor type 1 (TGF rl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-OX40 antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure:
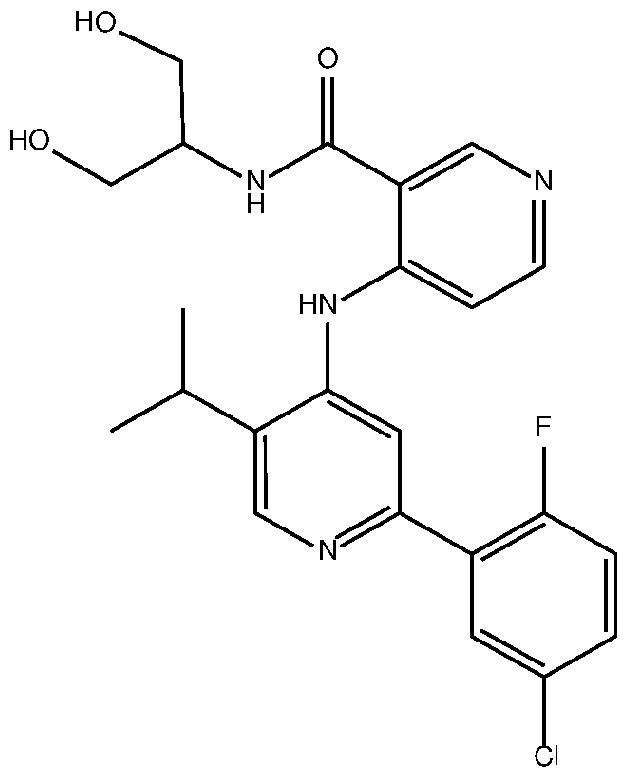 or a pharmaceutically acceptable salt thereof; and wherein the anti-OX40 antibody is PF- 04518600, for use in treating cancer.
or a pharmaceutically acceptable salt thereof; and wherein the anti-OX40 antibody is PF- 04518600, for use in treating cancer.
In one preferred aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
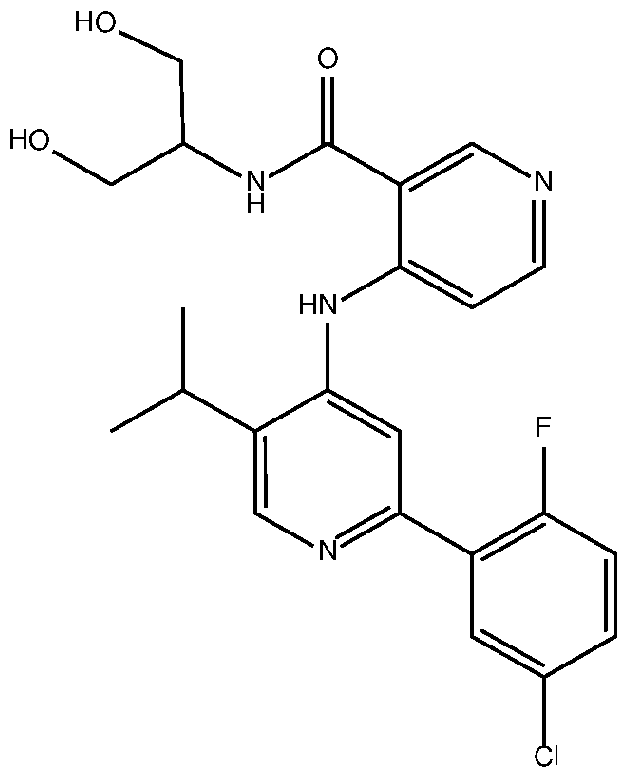 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with an 0X40 agonist, for treating cancer. In one preferred aspect, the invention provides a pharmaceutical composition comprising an 0X40 agonist, for use in combination with 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with an 0X40 agonist, for treating cancer. In one preferred aspect, the invention provides a pharmaceutical composition comprising an 0X40 agonist, for use in combination with 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
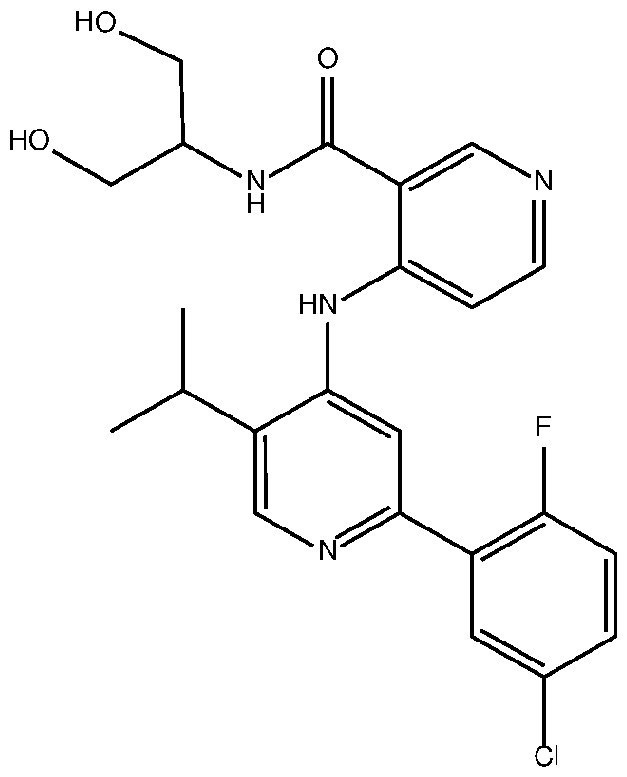 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer.
In a preferred embodiment of the pharmaceutical composition described herein, the 0X40 agonist is an anti-OX40 antibody. In some such emdodiments, the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR- 8383, and ABBV-368, or combinations thereof. In some such emdodiments, the anti- 0X40 antibody is PF-04518600. In a preferred embodiment of this aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with an anti-OX40 antibody, wherein the anti-OX40 antibody is PF-04518600, for treating cancer.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with an anti-OX40 antibody, wherein the anti-OX40 antibody is PF-04518600, for treating cancer.
In a preferred embodiment of this aspect, the invention provides a pharmaceutical composition comprising an anti-OX40 antibody, for use in combination with 4-(2-(5- chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide having the structure:
 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the anti-OX40 antibody is PF-04518600, for treating cancer.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the anti-OX40 antibody is PF-04518600, for treating cancer.
In one aspect, the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject:
(a) an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof, and;
(b) an amount of a 4-1 BB agonist, wherein the amounts in (a) and (b) together are effective in treating cancer.
In some embodiments of this aspect, the invention provides a method further comprising administering to the subject (c) an amount of an additional anti-cancer agent; wherein the amounts in (a), (b) and (c) together are effective in treating cancer.
In a preferred embodiment of the methods described herein, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2-
fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof. In a specific embodiment, the TGF[3r1 inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
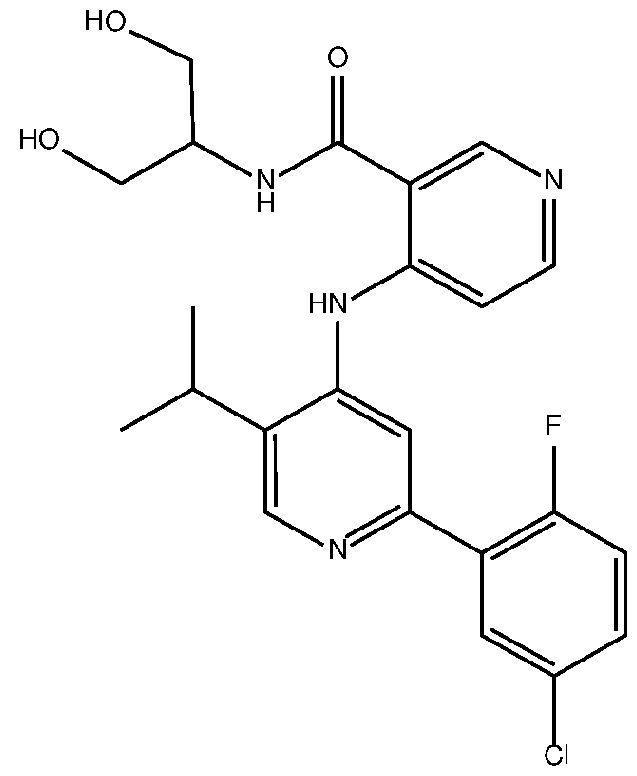 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of the methods described herein, the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4- 1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No. HB- 11248, 5F4, C65-485, urelumab (BMS-663513), 20H4.9-lgG-1 (BMS-663031 ), 4E9, BMS-554271 , BMS-469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR-7480, MOR-7480.1 , MOR-7480.2, MOR-7483, MOR-7483.1 , MOR-7483.2, 3EI, 53A2, 1 D8, and 3B8, or combinations thereof. In a specific embodiment, the 4-1 BB agonist is utomilumab.
In one preferred aspect, the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-4-1 BB antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure:
 or a pharmaceutically acceptable salt thereof, wherein the anti-4-1 BB antibody is utomilumab, and wherein the amounts together are effective in treating cancer.
or a pharmaceutically acceptable salt thereof, wherein the anti-4-1 BB antibody is utomilumab, and wherein the amounts together are effective in treating cancer.
In some embodiments of the methods described herein, the TGFprl inhibitor and the 4-1 BB agonist are administered sequentially, simultaneously, or concurrently.
In one aspect, the invention provides a combination comprising:
(a) an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof, and;
(b) a 4-1 BB agonist; for use in treating cancer.
In some embodiments of this aspect, the combination for use further comprises
(c) an additional anti-cancer agent.
In a preferred embodiment of the combinations described herein, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-
(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof. In a specific embodiment, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
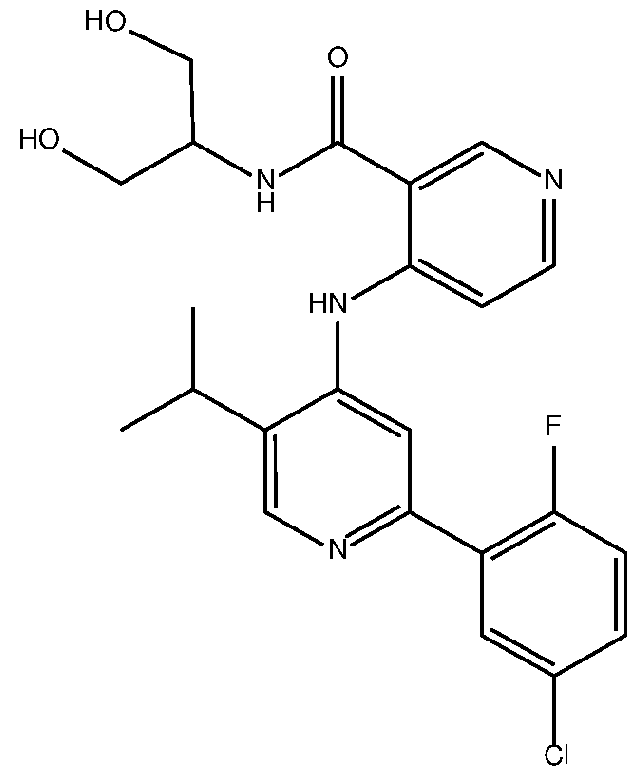 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of the combinations described herein, the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC
No. HB-1 1248, 5F4, C65-485, urelumab (BMS-663513), 20H4.9-lgG-1 (BMS-663031 ), 4E9, BMS-554271 , BMS-469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR- 7480, MOR-7480.1 , MOR-7480.2, MOR-7483, MOR-7483.1 , MOR-7483.2, 3EI, 53A2, 1 D8, and 3B8, or combinations thereof. In a specific embodiment, the 4-1 BB agonist is utomilumab.
In one preferred aspect, the invention provides a combination comprising a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of a 4-1 BB agonist, wherein the TGF[3r1 inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan- 2-yl)nicotinamide having the structure:

In one preferred aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure:
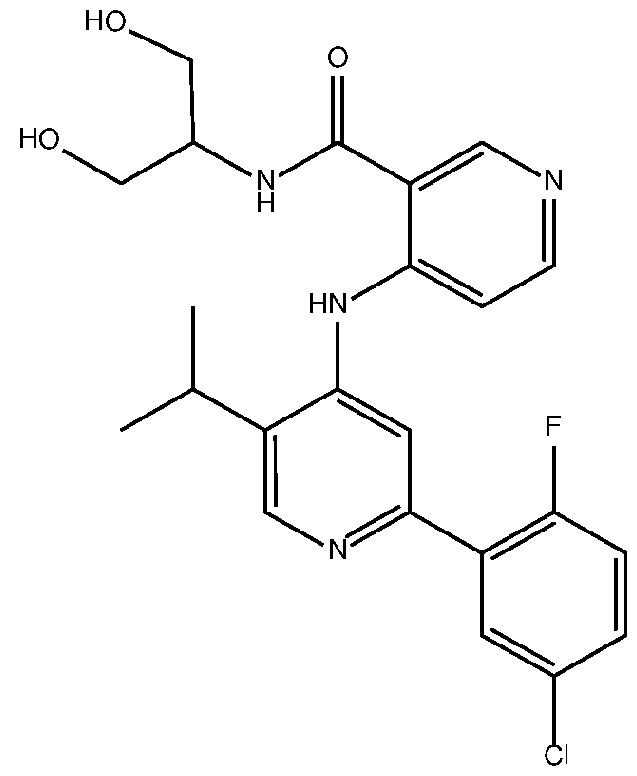 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a 4-1 BB agonist, for treating cancer. In one preferred aspect, the invention provides a pharmaceutical composition comprising a 4-1 BB agonist, for use in combination with 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a 4-1 BB agonist, for treating cancer. In one preferred aspect, the invention provides a pharmaceutical composition comprising a 4-1 BB agonist, for use in combination with 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
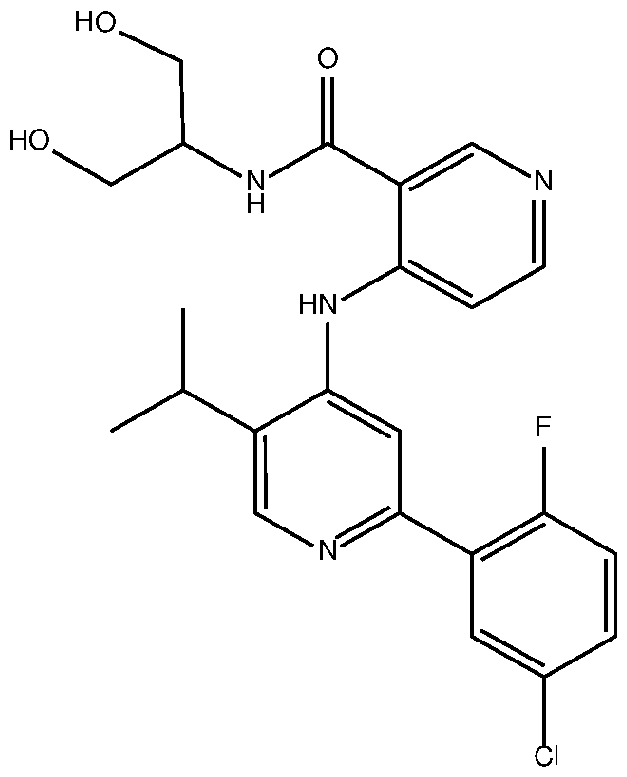 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer. In a preferred embodiment of the pharmaceutical composition described herein, the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No. HB-1 1248, 5F4, C65-485, urelumab (BMS- 663513), 20H4.9-lgG-1 (BMS-663031 ), 4E9, BMS-554271 , BMS-469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR-7480, MOR-7480.1 , MOR-7480.2, MOR-7483, MOR-7483.1 , MOR-7483.2, 3EI, 53A2, 1 D8, and 3B8, or combinations thereof. In some such emdodiments, the 4-1 BB agonist is utomilumab.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer. In a preferred embodiment of the pharmaceutical composition described herein, the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No. HB-1 1248, 5F4, C65-485, urelumab (BMS- 663513), 20H4.9-lgG-1 (BMS-663031 ), 4E9, BMS-554271 , BMS-469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR-7480, MOR-7480.1 , MOR-7480.2, MOR-7483, MOR-7483.1 , MOR-7483.2, 3EI, 53A2, 1 D8, and 3B8, or combinations thereof. In some such emdodiments, the 4-1 BB agonist is utomilumab.
In a preferred embodiment of this aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a 4-1 BB agonist, wherein the 4-1 BB agonist is utomilumab, for treating cancer.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a 4-1 BB agonist, wherein the 4-1 BB agonist is utomilumab, for treating cancer.
In a preferred embodiment of this aspect, the invention provides a pharmaceutical composition comprising a 4-1 BB agonist, for use in combination with 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
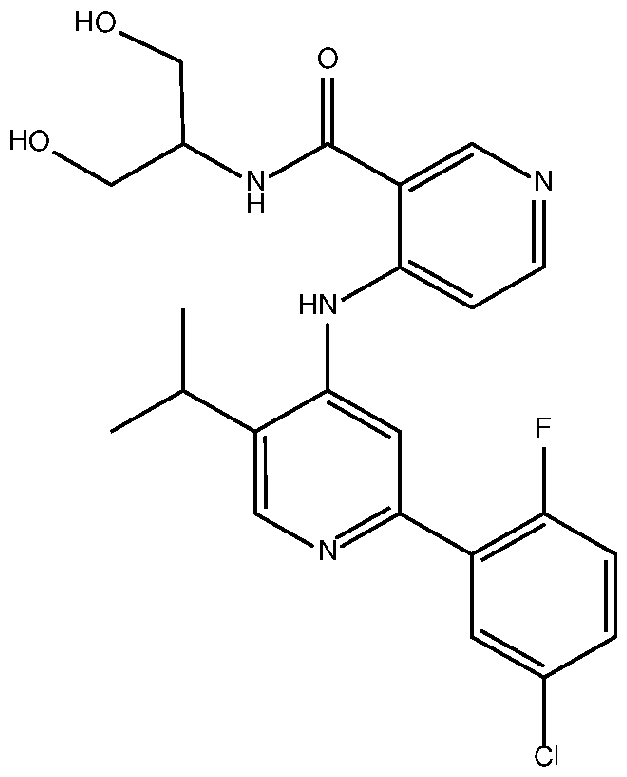 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the 4-1 BB agonist is utomilumab, for treating cancer.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the 4-1 BB agonist is utomilumab, for treating cancer.
In one aspect, the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject:
(a) an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof, and;
(b) an amount of a CTLA4 antagonist, wherein the amounts in (a) and (b) together are effective in treating cancer.
In some embodiments of this aspect, the invention provides a method further comprising administering to the subject (c) an amount of an additional anti-cancer agent; wherein the amounts in (a), (b) and (c) together are effective in treating cancer.
In a preferred embodiment of the methods described herein, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2-
fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof. In a specific embodiment, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of the methods described herein, the CTLA4 antagonist is an anti-CTLA4 antibody. In some such emdodiments, the anti-CTLA4 antibody is selected from the group consisting of ipilimumab (1 ODI), tremelimumab, and AGEN-1884, or combinations thereof. In a specific embodiment, the anti-CTLA4 antibody is ipilimumab (10DI).
In one preferred aspect, the invention provides a method of treating cancer in a subject in need thereof, comprising administering to the subject an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-CTLA4 antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure:
 or a pharmaceutically acceptable salt thereof, wherein the anti-CTLA4 antibody is ipilimumab (1 ODI), and wherein the amounts together are effective in treating cancer.
or a pharmaceutically acceptable salt thereof, wherein the anti-CTLA4 antibody is ipilimumab (1 ODI), and wherein the amounts together are effective in treating cancer.
In some embodiments of the methods described herein, the TGFprl inhibitor and the CTLA4 antagonist are administered sequentially, simultaneously, or concurrently.
In one aspect, the invention provides a combination comprising:
(a) an amount of a transforming growth factor beta receptor type 1 (TGFprl) inhibitor, or a pharmaceutically acceptable salt thereof, and;
(b) an CTLA4 antagonist; for use in treating cancer.
In some embodiments of this aspect, the combination for use further comprises
(c) an additional anti-cancer agent.
In a preferred embodiment of the combinations described herein, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-
(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof. In a specific embodiment, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229) having the structure:
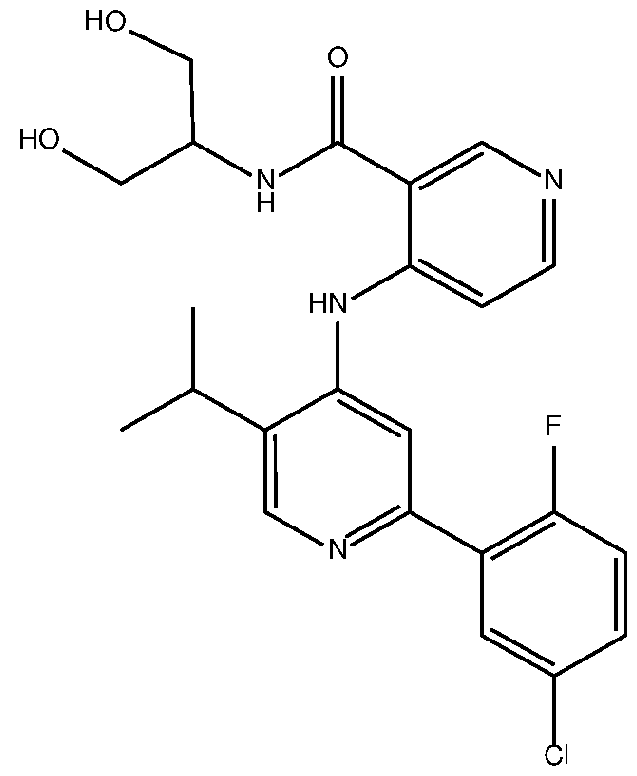 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of the combinations described herein, the anti-CTLA4 antibody is selected from the group consisting of ipilimumab (1 ODI), tremelimumab, and AGEN-1884, or combinations thereof. In a specific embodiment, the anti-CTLA4 antibody is ipilimumab (1 ODI).
In one preferred aspect, the invention provides a combination comprising a transforming growth factor beta receptor type 1 (TGF rl) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an anti-CTLA4 antibody, wherein the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure:
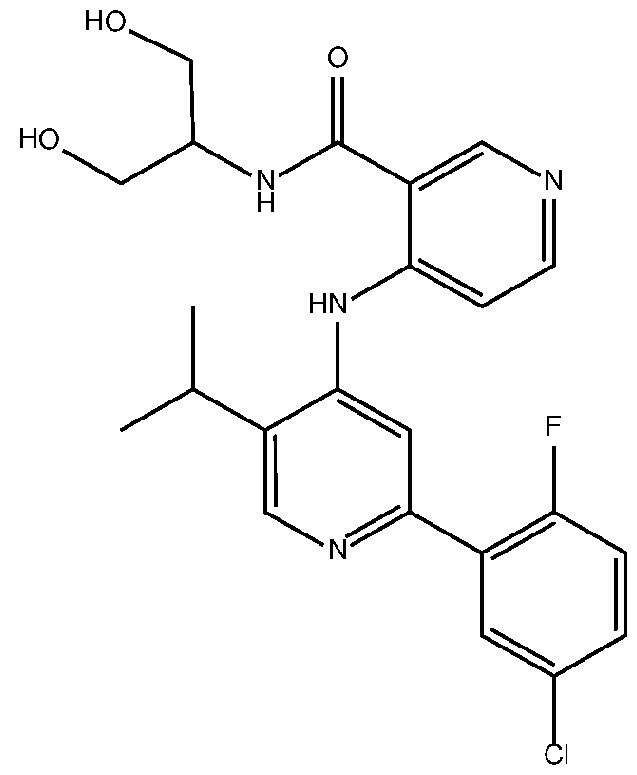 or a pharmaceutically acceptable salt thereof; and wherein the anti-CTLA4 antibody is ipilimumab (1 ODI), for use in treating cancer.
In one preferred aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure:
or a pharmaceutically acceptable salt thereof; and wherein the anti-CTLA4 antibody is ipilimumab (1 ODI), for use in treating cancer.
In one preferred aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide having the structure:
 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a CTLA4 antagonist.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with a CTLA4 antagonist.
In one preferred aspect, the invention provides a pharmaceutical composition comprising a CTLA4 antagonist, for use in combination with 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
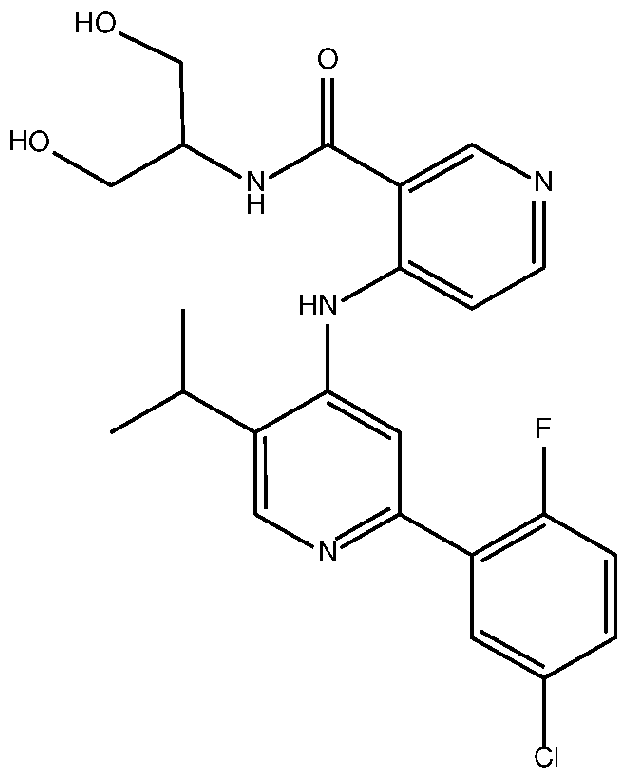 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer.
In a preferred embodiment of the pharmaceutical composition described herein, the CTLA4 antagonist is an anti-CTLA4 antibody. In some such embodiments, the anti- CTLA4 antibody is selected from the group consisting of ipilimumab (10DI), tremelimumab, and AGEN-1884, or combinations thereof. In a specific embodiment, the anti-CTLA4 antibody is ipilimumab (1 GDI).
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for treating cancer.
In a preferred embodiment of the pharmaceutical composition described herein, the CTLA4 antagonist is an anti-CTLA4 antibody. In some such embodiments, the anti- CTLA4 antibody is selected from the group consisting of ipilimumab (10DI), tremelimumab, and AGEN-1884, or combinations thereof. In a specific embodiment, the anti-CTLA4 antibody is ipilimumab (1 GDI).
In a preferred embodiment of this aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide having the structure:
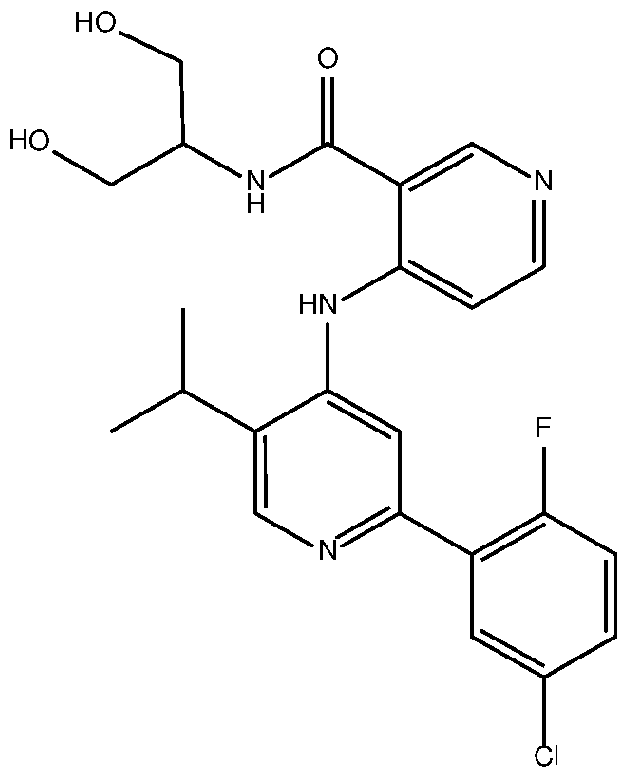 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with an anti-CTLA4 antibody, wherein the anti-CTLA4 antibody is ipilimumab (1 GDI), for treating cancer.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in combination with an anti-CTLA4 antibody, wherein the anti-CTLA4 antibody is ipilimumab (1 GDI), for treating cancer.
In a preferred embodiment of this aspect, the invention provides a pharmaceutical composition comprising an anti-CTLA4 antibody, for use in combination with 4-(2-(5- chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide having the structure:
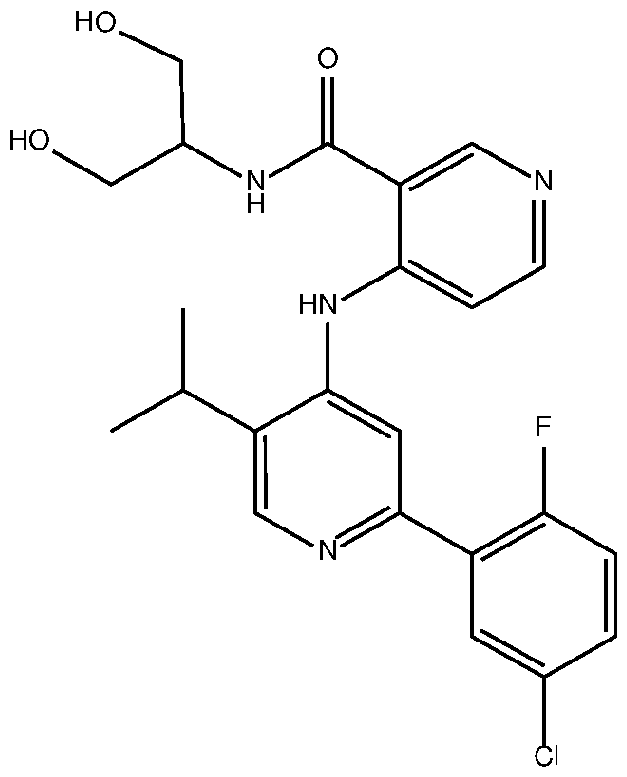 or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the anti-CTLA4 antibody is ipilimumab (10DI), for treating cancer.
or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, wherein the anti-CTLA4 antibody is ipilimumab (10DI), for treating cancer.
In some embodiments of each of the methods, combinations, uses and pharmaceutical compositions described herein, the additional anti-cancer agent is selected from the group consisting of a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and anti-angiogenic agent.
In some embodiments of each of the methods, combinations, uses and pharmaceutical compositions described herein, the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma. In a preferred embodiment, the cancer is prostate cancer. In some such embodiments, the prostate cancer is hormone sensitive prostate cancer. In some such embodiments, the prostate cancer is castration resistant prostate cancer. In a preferred embodiment, the prostate cancer is metastatic. In some embodiments, the prostate cancer is non-metastatic.
In a preferred embodiment of each of the combinations described herein, the combination is a synergistic combination.
Embodiments of each of the aspects described herein, including the methods, combinations, uses and pharmaceutical compositions of the invention, may be combined
with one or more other embodiments of the present invention described herein which is not inconsistent with the embodiment(s) with which it is combined.
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1. Kaplan Meier survival curve on Day 63 (study termination) demonstrating the effects of combining a TGFprl inhibitor (PF-06952229) and an anti-PD-1 antibody in the MC38 syngeneic tumor model (Panel A: Survival Curve; Panel B)
FIG. 2. Shows effects of combining a TGFprl inhibitor (PF-06952229) and an anti-4-1 BB antibody; and a TGF rl inhibitor (PF-06952229) and an anti-OX40 antibody, on tumor growth inhibition in the 4T1 orthotopic syngeneic mouse tumor model.
FIG. 3. Shows the effects of combining a TGFprl inhibitor (PF-06952229) and an anti- CTLA4 antibody (D) on tumor growth delay relative to PF-06952229 monotherapy treatment (C), anti-CTLA4 antibody, (B) monotherapy treatment, in the CT26 syngeneic tumor model. Control treatment has no effect (A).
DETAILED DESCRIPTION
The present invention may be understood more readily by reference to the following detailed description of the preferred embodiments of the invention and the Examples included herein. It is to be understood that the terminology used herein is for the purpose of describing specific embodiments only and is not intended to be limiting. It is further to be understood that unless specifically defined herein, the terminology used herein is to be given its traditional meaning as known in the relevant art.
The following abbreviations are used herein:
ANCOVA analysis of covariance
BID twice daily
Biwk biweekly
BW body weight
BWL body weight loss
CDR complementarity determining region
CHO Chinese hamster ovary
CR complete response
DFS disease free survival
DMSO dimethylsulfoxide
DTR dose limiting toxicity
FR framework region
IgG immunoglobulin G
IHC immunohistochemistry or immunohistochemical
IP intraperitoneal
MPK milligram per kilogram (mg/kg or mg drug per kg body weight of animal)
MTD maximum tolerated dose
N or n number of subjects
NCBI National Center for Biotechnology Information
NCI National Cancer Institute
NOD/SCID nonobese diabetic / severe combination immunodeficiency
NS not significant
NSG NOD scid gamma
PCR polymerase chain reaction
PD progressive disease
PFS progression free survival
PO oral dosing
PR partial response
Q2W one dose every two weeks
Q3W one dose every three weeks
Q4W one dose every four weeks
QD one dose per day
QD3 1 dose every 3 days
RECIST response Evaluation Criteria in Solid Tumors
RPMI Roswell Park Memorial Institute
QD once Daily qRT quantitative real time
RT reverse transcription
SD stable disease
SEM standard error of the mean
TGI tumor growth inhibition
TTE time-to-event w/w weight per weight
WT wild type
As used herein, the singular form "a", "an", and "the" include plural references unless indicated otherwise. For example, "a" substituent includes one or more substituents.
The invention described herein suitably may be practiced in the absence of any element(s) not specifically disclosed herein. Thus, for example, in each instance herein any of the terms "comprising", "consisting essentially of", and "consisting of" may be replaced with either of the other two terms.
The term “about” which used to modify a numerically defined parameter means that the parameter may vary by as much as 10% above or below the stated numerical value for that parameter (± 10%). For example, a dose of about 5 mg/kg should be understood to mean that the dose may vary between 4.5 mg/kg and 5.5 mg/kg.
A “disorder” is any condition that would benefit from treatment with the compounds of the present invention. This includes chronic and acute disorders or diseases including those pathological conditions which predispose the subject to the disorder in question.
The term “administration” and “treatment” as it applies to an animal, human, experimental subject, cell, tissue, organ or biological fluid, refers to contact of an exogenous pharmaceutical, therapeutic or diagnostic agent, or composition, to the animal, human, experimental subject, cell, tissue, organ or biological fluid. Treatment of a cell encompasses contact of a reagent to the cell, as well as contact of a reagent to a fluid, where the fluid is in contact with the cell. “Administration” and “treatment” also
means in vitro and ex vivo treatment, e.g., of a cell, by a reagent, diagnostic, binding compound, or by another cell.
As used herein, terms, including, but not limited to, “drug,” “agent,” “component,” “composition,” “compound,” “substance,” “targeted agent,” “targeted therapeutic agent,” “therapeutic agent,” and "medicament” may be used interchangeably to refer to the small molecule compounds of the present invention, e.g., a TGF[3r inhibitor or a TGF[3r1 inhibitor.
As used herein, terms, including, but not limited to, “drug,” “agent," “component," “composition," “compound," “substance," “targeted agent," “targeted therapeutic agent," “therapeutic agent," therapeutic antibody,” and “medicament” may be used interchangeably to refer to the antibodies of the present invention, e.g., an anti-PD-1 antibody, an anti-OX40 antibody, an anti-4-1 BB antibody, and an anti-CTLA4 antibody or combinations thereof.
The term “antibody,” as used herein, refers to an immunoglobulin molecule capable of specific binding to a target, such as a carbohydrate, polynucleotide, lipid, polypeptide, etc., through at least one antigen recognition site, located in the variable region of the immunoglobulin molecule. As used herein, the term encompasses a polyclonal antibody, a monoclonal antibody, a chimeric antibody, a bispecific antibody, a dual-specific antibody, bifunctional antibody, a trispecific antibody, a multispecific antibody, a bispecific heterodimeric diabody, a bispecific heterodimeric IgG, a labeled antibody, a humanized antibody, a human antibody, and fragments thereof (such as Fab, Fab’, F(ab’)2, Fv), single chain (ScFv) and domain antibodies (including, for example, shark and camelid antibodies), fusion proteins comprising an antibody, any other modified configuration of the immunoglobulin molecule that comprises an antigen recognition site, and antibody like binding peptidomimetics (ABiPs). An antibody includes an antibody of any class, such as IgG, IgA, or IgM (or sub-class thereof), and the antibody need not be of any particular class. Depending on the antibody amino acid sequence of the constant region of its heavy chains, immunoglobulins can be assigned to different classes. There are five major classes of immunoglobulins: IgA, IgD, IgE, IgG, and IgM, and several of these may be further divided into subclasses (isotypes), e.g., lgG-1 , IgG- 2, lgG-3, lgG-4, Ig A1 and lgA2. The heavy-chain constant regions that correspond to the different classes of immunoglobulins are called alpha, delta, epsilon, gamma, and mu,
respectively. The subunit structures and three-dimensional configurations of different classes of immunoglobulins are well known.
As used herein, a “bispecific antibody," “dual-specific antibody," “bifunctional antibody," "heteromultimer," "heteromultimeric complex," “bispecific heterodimeric diabody” or a "heteromultimeric polypeptide” is a molecule comprising at least a first polypeptide and a second polypeptide, wherein the second polypeptide differs in amino acid sequence from the first polypeptide by at least one amino acid residue. In some instances, the bispecific is an artificial hybrid antibody having two different heavy chain region and light chain region. Preferably, the bispecific antibody has binding specificity for at least two different ligands, antigens or binding sites. Accordingly, the bispecific antibodies can bind simultaneously to two different antigens. The two antigen binding sites of a bispecific antibody bind to two different epitopes, which may reside on the same or different protein targets, e.g., tumor target.
The bispecific antibody, dual-specific antibody, bifunctional antibody, heteromultimer, heteromultimeric complex, bispecific heterodimeric diabody or the heteromultimeric polypeptide can be prepared by constructing sFv fragments with short linkers (e.g., about 3-10 residues) between the VH and VL regions such that inter-chain but not intra-chain pairing of the V regions is achieved, resulting in a bivalent fragment, i.e. , fragment having two antigen-binding sites. Bispecific antibodies can be derived from full length antibodies or antibody fragments (e.g., F(ab')2 bispecific antibodies). Diabodies are described more fully in, for example, EP 404,097; WO 1993/011161 ; and Hollinger et al., A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity, Proc. Natl. Acad. Sci. 1993, 90:6444-6448. Bispecific antibodies are heterodimers of two "crossover" sFv fragments in which the VH and VL regions of the two antibodies are present on different polypeptide chains.
By way of non-limiting example, a bispecific antibody may comprise one antigenbinding site that recognizes an epitope on one protein (e.g., PD-1 , 0X40, 4-1 BB, and/or CTLA4) and further comprise a second, different antigen-binding site that recognizes a different epitope on a second protein (e.g., PD-1 , 0X40, 4-1 BB, and/or CTLA4). Generally, but not necessarily, reference to binding means specific binding.
The term “therapeutic antibody” refers to an antibody that is used in the treatment of a disease or a disorder. A therapeutic antibody may have various mechanisms of action. A therapeutic antibody may bind and neutralize the normal function of a target associated with an antigen. For example, a monoclonal antibody that blocks the activity
of the of protein needed for the survival of a cancer cell causes the cell's death. Another therapeutic antibody may bind and activate the normal function of a target associated with an antigen. For example, a monoclonal antibody can bind to a protein on a cell and trigger an apoptosis signal. Yet another monoclonal antibody may bind to a target antigen expressed only on diseased tissue, conjugation of a toxic payload (effective agent), such as a chemotherapeutic or radioactive agent, to the monoclonal antibody can create an agent for specific delivery of the toxic payload to the diseased tissue, reducing harm to healthy tissue. A “biologically functional fragment” of a therapeutic antibody will exhibit at least one if not some or all of the biological functions attributed to the intact antibody, the function comprising at least specific binding to the target antigen.
The therapeutic antibody may bind to any protein, including, without limitation, a PD-1 , an 0X40, a 4-1 BB and/or a CTLA4 antigen. Accordingly, therapeutic antibodies include, without limitation, anti-PD-1 antibodies, anti-OX40 antibodies, anti-4-1 BB antibodies, and anti-CTLA4 antibodies or combinations thereof.
The term “biotherapeutic agent" means a biological molecule, such as an antibody or fusion protein, that blocks ligand/receptor signaling in any biological pathway that supports tumor maintenance and/or growth or suppresses the anti-tumor immune response.
The term "immune response" refers to any detectable response to a particular substance (such as an antigen or immunogen) by the immune system of a host vertebrate animal, including, but not limited to, innate immune responses (e.g., activation of Toll-like receptor signalling cascade), cell-mediated immune responses (e.g., responses mediated by T cells, such as antigen-specific T cells, and non-specific cells of the immune system), and humoral immune responses (e.g., responses mediated by B cells, such as generation and secretion of antibodies into the plasma, lymph, and/or tissue fluids). Examples of immune responses include an alteration (e.g., increase) in Toll-like receptor activation, lymphokine (e.g., cytokine (e.g., Th1 , Th2 or Th17 type cytokines) or chemokine) expression or secretion, macrophage activation, dendritic cell activation, T cell (e.g., CD4+ or CD8+ T cell) activation, NK cell activation, B cell activation (e.g., antibody generation and/or secretion), binding of an immunogen (e.g., antigen, immunogenic polypeptide) to an MHC molecule, induction of a cytotoxic T lymphocyte ("CTL") response, induction of a B cell response (e.g., antibody production), and expansion (e.g., growth of a population of cells) of cells of the immune system (e.g., T cells and B cells), and increased processing and presentation of antigen by antigen
presenting cells. The term “immune response” also encompasses any detectable response to a particular substance (such as an antigen or immunogen) by one or more components of the immune system of a vertebrate animal in vitro.
The term “immunogen” refers to a substance that is immunogenic. The term “immunogenic” refers to the ability of a substance upon administration to a mammal (such as a human) to cause, elicit, stimulate, or induce an immune response, or to improve, enhance, increase or prolong a pre-existing immune response, against a particular antigen in the mammal, whether alone or when linked to a carrier, in the presence or absence of an adjuvant.
The term "immunoglobulin" or “Ig” is used interchangeably with "antibody" herein. The basic 4-chain antibody unit is a heterotetrameric glycoprotein composed of two identical light (L) chains and two identical heavy (H) chains. An IgM antibody consists of 5 of the basic heterotetramer units along with an additional polypeptide called a J chain, and contains 10 antigen binding sites, while IgA antibodies comprise from 2-5 of the basic 4-chain units which can polymerize to form polyvalent assemblages in combination with the J chain. In the case of IgGs, the 4-chain unit is generally about 150,000 Daltons. Each L chain is linked to an H chain by one covalent disulfide bond, while the two H chains are linked to each other by one or more disulfide bonds depending on the H chain isotype. Each H and L chain also has regularly spaced intrachain disulfide bridges. Each H chain has at the N-terminus, a variable domain (VH) followed by three constant domains (CH) for each of the a and y chains and four CH domains for p and £ isotypes. Each L chain has at the N-terminus, a variable domain (VL) followed by a constant domain at its other end. The VL is aligned with the VH and the CL is aligned with the first constant domain of the heavy chain (CHI). Particular amino acid residues are believed to form an interface between the light chain and heavy chain variable domains. The pairing of a VH and VL together forms a single antigen-binding site. For the structure and properties of the different classes of antibodies, e.g., Daniel P. Sties, Abba I. Terr and Tristram G. Parsolw (eds), Basic and Clinical Immunology, 8th Edition, 1994, page 71 and Chapter 6. The L chain from any vertebrate species can be assigned to one of two clearly distinct types, called kappa and lambda, based on the amino acid sequences of their constant domains. Depending on the amino acid sequence of the constant domain of their heavy chains (CH), immunoglobulins can be assigned to different classes or isotypes.
The terms "full-length antibody,” "intact antibody" or "whole antibody" are used interchangeably to refer to an antibody in its substantially intact form, as opposed to an antibody fragment. Specifically, whole antibodies include those with heavy and light chains including an Fc region. The constant domains may be native sequence constant domains (e.g., human native sequence constant domains) or amino acid sequence variants thereof. In some cases, the intact antibody may have one or more effector functions.
An "antibody fragment" comprises a portion of an intact antibody, preferably the antigen binding and/or the variable region of the intact antibody. Examples of antibody fragments suitable for use in this invention include, without limitation: (i) the Fab fragment, consisting of VL, VH, CL, and CH1 domains; (ii) the “Fd” fragment consisting of the VH and CH1 domains; (iii) the “Fv” fragment consisting of the VL and VH domains of a single antibody; (iv) the “dAb” fragment, which consists of a VH domain; (v) isolated complementarity determining (CDR) regions; (vi) F(ab')2 fragments, a bivalent fragment comprising two linked Fab fragments; (vii) single chain Fv molecules (scFv), wherein a VH domain and a VL domain are linked by a peptide linker that allows the two domains to associate to form a binding domain; (viii) bi-specific single chain Fv dimers (e.g., U.S. Pat. No. 5,091 ,513); and (ix) diabodies, multivalent or multispecific fragments constructed by gene fusion (US Patent App. Pub. 2005/0214860). Fv, scFv, or diabody molecules may be stabilized by the incorporation of disulphide bridges linking the VH and VL domains. Minibodies comprising a scFv joined to a CH3 domain may also be made (Hu et al., Minibodies are minimized antibody-like proteins comprising a scFv joined to a CH3 domain, Cancer Res. 1996, 56:3055-3061 )).
Murali et al., Antibody like peptidomimetics as large scale immunodetection probes, Cell Mol Biol 2003, 49:209-216, describe a methodology for reducing antibodies into smaller peptidomimetics, they term “antibody like binding peptidomimetics” (ABiP) which may also be useful as an alternative to antibodies.
"Isolated antibody" or “isolated antibody fragment” refers to the purification status and in such context means the named molecule is substantially free of other biological molecules such as nucleic acids, proteins, lipids, carbohydrates, or other material such as cellular debris and growth media. Generally, the term "isolated" is not intended to refer to a complete absence of such material or to an absence of water, buffers, or salts, unless they are present in amounts that substantially interfere with experimental or therapeutic use of the binding compound described herein.
"Monoclonal antibody" or “mAb” or “Mab,” as used herein, refers to a population of substantially homogeneous antibodies, i.e., the antibody molecules comprising the population are identical in amino acid sequence except for possible naturally occurring mutations that may be present in minor amounts. In contrast, conventional (polyclonal) antibody preparations typically include a multitude of different antibodies having different amino acid sequences in their variable domains, particularly their CDRs, which are often specific for different epitopes. The term "monoclonal" indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies and is not to be construed as requiring production of the antibody by any particular method. For example, the monoclonal antibodies to be used in accordance with the present invention may be made by the hybridoma method first described by Kohler etal., Continuous cultures of fused cells secreting antibody of predefined specificity, Nature 1975, 256: 495; or may be made by recombinant DNA methods (e.g., U.S. Patent No. 4,816,567). The "monoclonal antibodies" may also be isolated from phage antibody libraries using the techniques described in Clackson et al., Making antibody fragments using phage display libraries, Nature 1991 , 352: 624-628 and Marks et al., By-passing immunization: human antibodies from V-gene libraries displayed on phage, J. Mol. Biol. 1991 , 222: 581 -597, for example. See also Presta, Selection, design, and engineering of therapeutic antibodies, J. Allergy Clin. Immunol. 2005,116:731 .
"Chimeric antibody" refers to an antibody in which a portion of the heavy and/or light chain is identical with or homologous to corresponding sequences in an antibody derived from a particular species (e.g., human) or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is identical with or homologous to corresponding sequences in an antibody derived from another species (e.g., mouse) or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit the desired biological activity.
“Human antibody” refers to an antibody that comprises human immunoglobulin protein sequences only. A human antibody may contain murine carbohydrate chains if produced in a mouse, in a mouse cell, or in a hybridoma derived from a mouse cell. Similarly, “mouse antibody” or “rat antibody” refer to an antibody that comprises only mouse or rat immunoglobulin sequences, respectively.
"Humanized antibody" refers to forms of antibodies that contain sequences from non-human (e.g., murine) antibodies as well as human antibodies. Such antibodies contain minimal sequence derived from non-human immunoglobulin. In general, the
humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin and all or substantially all of the FR regions are those of a human immunoglobulin sequence. The humanized antibody optionally also will comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. The prefix “hum,” “hu” or “h” is added to antibody clone designations when necessary to distinguish humanized antibodies from parental rodent antibodies. The humanized forms of rodent antibodies will generally comprise the same CDR sequences of the parental rodent antibodies, although certain amino acid substitutions may be included to increase affinity, increase stability of the humanized antibody, or for other reasons.
A “variable region” of an antibody refers to the variable region of the antibody light chain or the variable region of the antibody heavy chain, either alone or in combination. As known in the art, the variable regions of the heavy and light chain each consist of four framework regions (FR) connected by three complementarity determining regions (CDRs) also known as hypervariable regions.
The term "hypervariable region," "HVR," or "HV" when used herein refers to the regions of an antibody variable domain which are hypervariable in sequence and/or form structurally defined loops. Generally, antibodies comprise six HVRs; three in the VH (H1 , H2, H3), and three in the VL (L1 , L2, L3). In native antibodies, H3 and L3 display the most diversity of the six HVRs, and H3 in particular is believed to play a unique role in conferring fine specificity to antibodies. See, e.g., Xu et al, Disruption of Early Tumor Necrosis Factor Alpha Signaling Prevents Classical Activation of Dendritic Cells in Lung- Associated Lymph Nodes and Development of Protective Immunity against Cryptococcal Infection, Immunity 2000, J-3:37-45; Johnson and Wu, Antibody Engineering Methods and Protocols Methods in Molecular Biology 2003, 248: 1 -25. Indeed, naturally occurring camelid antibodies consisting of a heavy chain only are functional and stable in the absence of light chain. See, e.g., Hamers-Casterman et al., Naturally occurring antibodies devoid of light chains, Nature 1993, 363:446-448; Sheriff et al., Similarity between C2 domain jaws and immunoglobulin CDRs, Nature Struct. Biol 1996, 3:733- 736.
A number of HVR delineations are in use and are encompassed herein. The Kabat Complementarity Determining Regions (CDRs) are based on sequence variability and are the most commonly used (Kabat et al., Sequences of Proteins of Immunological
Interest, 5th Ed. Public Health Service, National Institutes of Health, 1991 ). Chothia refers instead to the location of the structural loops (Chothia and Lesk, Canonical structures for the hypervariable regions of immunoglobulins, J. Mol. Biol. 1987, 196:901 - 917). The AbM HVRs represent a compromise between the Kabat HVRs and Chothia structural loops, are used by Oxford Molecular's AbM antibody modeling software. The "contact" HVRs are based on an analysis of the available complex crystal structures.
A “complementarity determining region” or “CDR” of a variable domain are amino acid residues within the variable region that are identified in accordance with the definitions of the Kabat, Chothia, the accumulation of both Kabat and Chothia, AbM, contact, and/or conformational definitions or any method of CDR determination well known in the art. Antibody CDRs may be identified as the hypervariable regions originally defined by Kabat et al. See, e.g., Kabat et al., Sequences of Proteins of Immunological Interest, 5th ed., Public Health Service, NIH, 1992. The positions of the CDRs may also be identified as the structural loop structures originally described by Chothia and others. See, e.g., Chothia et al., Conformations of immunoglobulin hypervariable regions, Nature, 1989, 342:877-883. Other approaches to CDR identification include the “AbM definition,” which is a compromise between Kabat and Chothia and is derived using Oxford Molecular's AbM antibody modeling software (now Accelrys®), or the “contact definition” of CDRs based on observed antigen contacts, set forth in MacCallum et al., Antibody-antigen interactions: contact analysis and binding site topography, J. Mol. Biol. 1996, 262:732-745. In another approach, referred to herein as the “conformational definition” of CDRs, the positions of the CDRs may be identified as the residues that make enthalpic contributions to antigen binding. See, e.g., Makabe et al., Thermodynamic consequences of mutations in vernier zone residues of a humanized anti-human epidermal growth factor receptor murine antibody, Journal of Biological Chemistry 2008, 283:1 156-1166. Still other CDR boundary definitions may not strictly follow one of the above approaches but will nonetheless overlap with at least a portion of the Kabat CDRs, although they may be shortened or lengthened in light of prediction or experimental findings that particular residues or groups of residues or even entire CDRs do not significantly impact antigen binding. As used herein, a CDR may refer to CDRs defined by any approach known in the art, including combinations of approaches. The methods used herein may utilize CDRs defined according to any of these approaches. For any given embodiment containing more than one CDR, the CDRs may be defined in
accordance with any of Kabat, Chothia, extended, AbM, contact, and/or conformational definitions.
The term “Kabat numbering” unless otherwise stated, is defined as the numbering of the residues in, e.g., an IgG heavy chain antibody using the EU index as in Kabat et al. (Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991 )), expressly incorporated herein by reference. "Framework" or "FR" residues are those variable-domain residues other than the HVR residues as herein defined.
A "human consensus framework” or "acceptor human framework" is a framework that represents the most commonly occurring amino acid residues in a selection of human immunoglobulin VL or VH framework sequences. Generally, the selection of human immunoglobulin VL or VH sequences is from a subgroup of variable domain sequences.
Generally, the subgroup of sequences is a subgroup as in Kabat eta!., Sequences of Proteins of Immunological Interest, 5lh Ed. Public Health Service, National Institutes of Health, 1991. Examples for the VL, the subgroup may be subgroup kappa I, kappa II, kappa III or kappa IV as in Kabat eta!., supra. Additionally, for the VH, the subgroup may be subgroup I, subgroup II, or subgroup III as in Kabat et al., supra. Alternatively, a human consensus framework can be derived from the above in which particular residues, such as when a human framework residue is selected based on its homology to the donor framework by aligning the donor framework sequence with a collection of various human framework sequences. An acceptor human framework "derived from" a human immunoglobulin framework or a human consensus framework may comprise the same amino acid sequence thereof, or it may contain pre-existing amino acid sequence changes. In some embodiments, the number of pre-existing amino acid changes are 10 or less, 9 or less, 8 or less, 7 or less, 6 or less, 5 or less, 4 or less, 3 or less, or 2 or less.
An "amino-acid modification" at a specified position, e.g., of the Fc region, refers to the substitution or deletion of the specified residue, or the insertion of at least one amino acid residue adjacent the specified residue. Insertion "adjacent" to a specified residue means insertion within one to two residues thereof. The insertion may be N- terminal or C-terminal to the specified residue. The preferred amino acid modification herein is a substitution.
"Conservatively modified variants" or "conservative substitution" refers to substitutions of amino acids in a protein with other amino acids having similar characteristics (e.g., charge, side-chain size, hydrophobicity/hydrophilicity, backbone
conformation and rigidity, etc.), such that the changes can frequently be made without altering the biological activity or other desired property of the protein, such as antigen affinity and/or specificity. Those of skill in this art recognize that, in general, single amino acid substitutions in non-essential regions of a polypeptide do not substantially alter biological activity (e.g., Watson et al., Molecular Biology of the Gene (4th Ed.), 1987, p.
224). In addition, substitutions of structurally or functionally similar amino acids are less likely to disrupt biological activity. Exemplary conservative substitutions are set forth in Table 1 below.
Table 1

An "affinity-matured" antibody is one with one or more alterations in one or more HVRs thereof that result in an improvement in the affinity of the antibody for antigen,
compared to a parent antibody that does not possess those alteration(s). In one embodiment, an affinity-matured antibody has nanomolar or even picomolar affinities for the target antigen. Affinity-matured antibodies are produced by procedures known in the art. For example, Marks et al., By-passing immunization: Building high affinity human antibodies by chain shuffling, Bio/Technology 1992, 10:779-783, describes affinity maturation by VH- and VL-domain shuffling. Random mutagenesis of HVR and/or framework residues is described by, for example: Barbas et al., In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity, Proc Nat. Acad. Sci. 1994, 91 :3809-3813; Schier et al., Identification of functional and structural amino-acid residues by parsimonious mutagenesis, Gene 1995, 169: 147- 155; Yelton et al., Affinity maturation of the BR96 anti-carcinoma antibody by codon-based mutagenesis, J. Immunol. 1995, 155: 1994- 2004; Jackson et al., In vitro antibody maturation. Improvement of a high affinity, neutralizing antibody against IL-1 beta, J. Immunol. 1995, 154(7):33 10-9; and Hawkins et al., Selection of phage antibodies by binding affinity: mimicking affinity maturation, J. Mol. Biol. 1992, 226:889-896.
The term "Fc region" herein is used to define a C-terminal region of an immunoglobulin heavy chain, including native-sequence Fc regions and variant Fc regions. Although the boundaries of the Fc region of an immunoglobulin heavy chain might vary, the human IgG heavy-chain Fc region is usually defined to stretch from an amino acid residue at position Cys226, or from Pro230, to the carboxyl-terminus thereof. The C-terminal lysine (residue 447 according to the EU numbering system) of the Fc region may be removed, for example, during production or purification of the antibody, or by recombinantly engineering the nucleic acid encoding a heavy chain of the antibody. Accordingly, a composition of intact antibodies may comprise antibody populations with all K447 residues removed, antibody populations with no K447 residues removed, and antibody populations having a mixture of antibodies with and without the K447 residue. Suitable native-sequence Fc regions for use in the antibodies of the invention include human lgG-1 , lgG-2 (lgG2A, lgG2B), lgG-3 and lgG-4.
"Fc receptor" or "FcR" describes a receptor that binds to the Fc region of an antibody. The preferred FcR is a native sequence human FcR. Moreover, a preferred FcR is one which binds an IgG antibody (a gamma receptor) and includes receptors of the FcyRI, FcyRII, and FeyRIII subclasses, including allelic variants and alternatively spliced forms of these receptors, FcyRII receptors include FcyRIIA (an "activating
receptor") and FcyRIIB (an "inhibiting receptor"), which have similar amino acid sequences that differ primarily in the cytoplasmic domains thereof. Activating receptor FcyRIIA contains an immunoreceptor tyrosine-based activation motif (ITAM) in its cytoplasmic domain. Inhibiting receptor FcyRIIB contains an immunoreceptor tyrosinebased inhibition motif (ITFM) in its cytoplasmic domain, (e.g., M. Daeron, Fc RECEPTOR BIOLOGY, Anna. Rev. Immunol. J 1997, 5 :203-234. FcRs are reviewed in Ravetch and Kinet, Fc receptors, Annu. Rev. Immunol. 1991 , 9: 457-92; Capel etal., Heterogeneity of human IgG Fc receptors, Immunomethods 1994, 4: 25-34; and de Haas et al., Fey receptors of phagocytes, J. Lab. Clin. Med. 1995, 126: 330-41. Other FcRs, including those to be identified in the future, are encompassed by the term "FcR" herein.
The term Fc receptor or FcR also includes the neonatal receptor, FcRn, which is responsible for the transfer of maternal IgGs to the fetus. Guyer et al., Immunoglobulin binding by mouse intestinal epithelial cell receptors, J. Immunol. 1976, 1 17: 587, and Tokoyama etal., How do natural killer cells find self to achieve tolerance? Immunity 1994, 24, 249-257. Methods of measuring binding to FcRn are known (e.g., Ghetie and Ward, FcRn: the MHC class l-related receptor that is more than an IgG transporter, Immunol. Today 1997, 1 8: (12): 592-8; Ghetie et al., Increasing the serum persistence of an IgG fragment by random mutagenesis, Nat Biotechnol. Jul. 1997,15(7):637-40; Hinton et al., Engineered human IgG antibodies with longer serum half-lives in primates, J. Biol. Chem. 2004, 279 (8): 6213-6; WO 2004/092219 (Hinton et al.). Binding to FcRn in vivo and serum half-life of human FcRn high-affinity binding polypeptides can be assayed, e.g., in transgenic mice or transfected human cell lines expressing human FcRn, or in primates to which the polypeptides having a variant Fc region are administered. WO 2004/042072 (Presta) describes antibody variants which improved or diminished binding to FcRs. See also, e.g., Shields etal., High Resolution Mapping of the Binding Site on Human lgG1 for FcyRI, FcyRII, FcyRIII, and FcRn and Design of lgG1 Variants with Improved Binding to the FcyR, J. Biol. Chem. 2001 , 9(2): 6591 -6604.
The phrase "substantially reduced," "substantially different," or “substantially inhibit,” as used herein, denotes a sufficiently high degree of difference between two numeric values (generally one associated with a molecule and the other associated with a reference/comparator molecule) such that one of skill in the art would consider the difference between the two values to be of statistical significance within the context of the biological characteristic measured by said values (e.g., Kd values). The difference between said two values is, for example, greater than about 10%, greater than about
20%, greater than about 30%, greater than about 40%, and/or greater than about 50% as a function of the value for the reference/comparator molecule.
As used herein, the term "substantially similar" or "substantially the same" denotes a sufficiently high degree of similarity between two numeric values (for example, one associated with an antibody of the invention and the other associated with a reference/comparator antibody), such that one of skill in the art would consider the difference between the two values to be of little or no biological and/or statistical significance within the context of the biological characteristic measured by said values (e.g., Kd values). The difference between said two values is, for example, less than about 50%, less than about 40%, less than about 30%, less than about 20%, and/or less than about 10% as a function of the reference/comparator value.
As used herein, the term "specifically binds to" or is "specific for" refers to measurable and reproducible interactions such as binding between a target and an antibody, which is determinative of the presence of the target in the presence of a heterogeneous population of molecules including biological molecules. For example, an antibody that specifically binds to a target (which can be an epitope) is an antibody that binds this target with greater affinity, avidity, more readily, and/or with greater duration than it binds to other targets. In one embodiment, the extent of binding of an antibody to an unrelated target is less than about 10 percent of the binding of the antibody to the target as measured, e.g., by a radioimmunoassay (RIA). In certain embodiments, an antibody that specifically binds to a target has a dissociation constant (Kd) of < 1 pM, < 100 nM, < 10 nM, < 1 nM, or < 0.1 nM. In certain embodiments, an antibody specifically binds to an epitope on a protein that is conserved among the protein from different species. In another embodiment, specific binding can include, but does not require exclusive binding.
As used herein, the term "immunoadhesin" designates antibody-like molecules which combine the binding specificity of a heterologous protein (an "adhesin") with the effector functions of immunoglobulin constant domains. Structurally, the immunoadhesins comprise a fusion of an amino acid sequence with the desired binding specificity which is other than the antigen recognition and binding site of an antibody (/.e., is "heterologous"), and an immunoglobulin constant domain sequence. The adhesin part of an immunoadhesin molecule typically is a contiguous amino acid sequence comprising at least the binding site of a receptor or a ligand. The immunoglobulin constant domain sequence in the immunoadhesin may be obtained from any immunoglobulin, such as
lgG-1 , lgG-2 (including lgG2A and lgG2B), lgG-3, or lgG-4 subtypes, IgA (including IgA- 1 and IgA-2), IgE, IgD or IgM. The Ig fusions preferably include the substitution of a domain of a polypeptide or antibody described herein in the place of at least one variable region within an Ig molecule. In a particularly preferred embodiment, the immunoglobulin fusion includes the hinge, CH2 and CH3, or the hinge, CHI, CH2 and CH3 regions of an lgG-1 molecule. For the production of immunoglobulin fusions, see also US Patent No. 5,428,130 issued June 27, 1995. Immunoadhesin combinations of Ig Fc and ECD of cell surface receptors are sometimes termed soluble receptors.
As used herein, the terms "peptide," "polypeptide," and "protein" are used interchangeably herein, and refer to a polymeric form of amino acids of any length, which can include coded and non-coded amino acids, chemically, or biochemically modified or derivatized amino acids, and polypeptides having modified polypeptide backbones.
As used herein, the terms "fusion protein" and a "fusion polypeptide" refer to a polypeptide having two portions covalently linked together, where each of the portions is a polypeptide having a different property. The property may be a biological property, such as activity in vitro or in vivo. The property may also be simple chemical or physical property, such as binding to a target molecule, catalysis of a reaction, etc. The two portions may be linked directly by a single peptide bond or through a peptide linker but are in reading frame with each other.
As used herein, the term "antagonist” antibody or a "blocking" antibody is one that inhibits or reduces a biological activity of the antigen it binds. In some embodiments, blocking antibodies or antagonist antibodies substantially or completely inhibit the biological activity of the antigen. The anti-PD-L1 antibodies of the invention block the signaling through PD-1 so as to restore a functional response by T-cells (e.g., proliferation, cytokine production, target cell killing) from a dysfunctional state to antigen stimulation.
As used herein, the term "agonist" or “activating antibody” is one that enhances or initiates signaling by the antigen to which it binds. In some embodiments, agonist antibodies cause or activate signaling without the presence of the natural ligand.
As used herein, the term "dysfunction" in the context of immune dysfunction, refers to a state of reduced immune responsiveness to antigenic stimulation. The term includes the common elements of both exhaustion and/or anergy in which antigen recognition may occur, but the ensuing immune response is ineffective to control infection or tumor growth.
As used herein, the term "dysfunctional" also includes refractory or unresponsive to antigen recognition, specifically, impaired capacity to translate antigen recognition into down-stream T-cell effector functions, such as proliferation, cytokine production and/or target cell killing.
As used herein, the term "anergy" refers to the state of unresponsiveness to antigen stimulation resulting from incomplete or insufficient signals delivered through the T-cell receptor (e.g., increase in intracellular Ca+2 in the absence of ras-activation). T cell anergy can also result upon stimulation with antigen in the absence of co- stimulation, resulting in the cell becoming refractory to subsequent activation by the antigen even in the context of co stimulation. The unresponsive state can often be overridden by the presence of lnterleukin-2. Anergic T-cells do not undergo clonal expansion and/or acquire effector functions.
As used herein, the term "exhaustion" refers to T cell exhaustion as a state of T cell dysfunction that arises from sustained TCR signaling that occurs during many chronic infections and cancer. It is distinguished from anergy in that it arises not through incomplete or deficient signaling, but from sustained signaling. It is defined by poor effector function, sustained expression of inhibitory receptors and a transcriptional state distinct from that of functional effector or memory T cells. Exhaustion prevents optimal control of infection and tumors. Exhaustion can result from both extrinsic negative regulatory pathways (e.g., immunoregulatory cytokines) as well as cell intrinsic negative regulatory (co-stimulatory) pathways.
"Enhancing T-cell function" means to induce, cause or stimulate a T-cell to have a sustained or amplified biological function, or renew or reactivate exhausted or dysfunctional T-cells. Examples of enhancing T-cell function include increased secretion of y-interferon from CD4+ or CD8+ T-cells, increased proliferation, increased survival, increased differentiation, increased antigen responsiveness (e.g., viral, pathogen, or tumor clearance) relative to such levels before the intervention. In some embodiments, the level of enhancement is as least 50%, alternatively 60%, 70%, 80%, 90%, 100%, 120%, 150%, 200%. The manner of measuring this enhancement is known to one of ordinary skill in the art.
As used herein, the term “abnormal cell growth,” unless otherwise indicated, refers to cell growth that is independent of normal regulatory mechanisms (e.g., loss of contact inhibition). Abnormal cell growth may be benign (not cancerous), or malignant (cancerous).
The terms “cancer,” “cancerous” or “malignant” refer to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth. As used herein “cancer” refers to any malignant and/or invasive growth or tumor caused by abnormal cell growth. As used herein “cancer” refers to solid tumors named for the type of cells that form them, as well as cancer of blood, bone marrow, or the lymphatic system. Examples of solid tumors include but not limited to sarcomas and carcinomas. Examples of cancers of the blood include but not limited to leukemias, lymphomas and myeloma. The term “cancer” includes, but is not limited to, a primary cancer that originates at a specific site in the body, a metastatic cancer that has spread from the place in which it started to other parts of the body, a recurrence from the original primary cancer after remission, and a second primary cancer that is a new primary cancer in a person with a history of previous cancer of a different type from latter one.
The terms "advanced prostate cancer" and "locally advanced prostate cancer" mean prostate cancers that have extended through the prostate capsule, and are meant to include stage C disease under the American Urological Association (AUA) system, stage Cl - C2 disease under the Whitmore-Jewett system, and stage T3 - T4 and N+ disease under the TNM (tumor, node, metastasis) system. In general, surgery is not recommended for patients with locally advanced prostate cancer and these patients have substantially less favorable outcomes compared to patients having clinically localized (organ- confined) prostate cancer. Locally advanced prostate cancer is clinically identified by palpable evidence of induration beyond the lateral border of the prostate, or asymmetry or induration above the prostate base. Locally advanced prostate cancer is presently diagnosed pathologically following radical prostatectomy if the tumor invades or penetrates the prostatic capsule, extends into the surgical margin, or invades the seminal vesicles.
The term, "metastasis," “metastatic cancer” or “metastatic” (also known as “secondary cancer”) as used herein, refers to a type of cancer that originates in one tissue type, but then spreads to one or more tissues outside of the (primary) cancer’s origin. Cancer cells can break away from a primary tumor, penetrate into lymphatic and blood vessels, circulate through the bloodstream, and grow in a distant focus (metastasize) in normal tissues elsewhere in the body. Metastasis can be local or distant. Metastasis is a sequential process, contingent on tumor cells breaking off from the primary tumor, traveling through the bloodstream, and stopping at a distant site. At the new site, the
cells establish a blood supply and can grow to form a life-threatening mass. Both stimulatory and inhibitory molecular pathways within the tumor cell regulate this behavior, and interactions between the tumor cell and host cells in the distant site are also significant.
For example, the term "metastatic prostate cancer" means prostate cancer that has spread to regional lymph nodes or to distant sites and is meant to include stage D disease under the AUA system and stage TxNxM+ under the TNM system. As is the case with locally advanced prostate cancer, surgery is generally not indicated for patients with metastatic disease, and hormonal (androgen ablation) therapy is a preferred treatment modality. Patients with metastatic prostate cancer eventually develop an androgen-refractory state within 12 to 18 months of treatment initiation. Approximately half of these androgen-refractory patients die within 6 months. The most common site for prostate cancer metastasis is bone. Prostate cancer bone metastases are often osteoblastic rather than osteolytic (/.e., resulting in net bone formation). Bone metastases are found most frequently in the spine, followed by the femur, pelvis, rib cage, skull and humerus. Other common sites for metastasis include lymph nodes, lung, liver and brain. Metastatic prostate cancer is typically diagnosed by open or laparoscopic pelvic lymphadenectomy, whole body radionuclide scans, skeletal radiography, and/or bone lesion biopsy.
A “TGF expressing cancer” is one that produces sufficient levels of TGFp at the surface of cells thereof, such that an anti-TGFp antibody can bind thereto and have a therapeutic effect with respect to the cancer.
A cancer “characterized by excessive activation” of a TGFp receptor (TGFpr) is one in which the extent of TGFp receptor activation in cancer cells significantly exceeds the level of activation of that receptor in non-cancerous cells of the same tissue type. Such excessive activation may result from overexpression of the TGFp receptor and/or greater than normal levels of a TGFp ligand available for activating the TGFp receptor in the cancer cells. Such excessive activation may cause and/or be caused by the malignant state of a cancer cell. In some embodiments, the cancer will be subjected to a diagnostic or prognostic assay to determine whether amplification and/or overexpression of a TGFp receptor is occurring that results in such excessive activation of the TGFp receptor. Alternatively, or additionally, the cancer may be subjected to a diagnostic or prognostic assay to determine whether amplification and/or overexpression
of a TGFp ligand is occurring in the cancer that attributes to excessive activation of the receptor. In a subset of such cancers, excessive activation of the receptor may result from an autocrine-stimulatory pathway.
In an autocrine-stimulatory pathway, self-stimulation occurs by virtue of the cancer cell producing both a TGFp ligand and its cognate TGFp receptor. For example, the cancer may express or overexpress TGFp receptor and also express or overexpress a TGFp ligand (e.g., TGFpi ligand).
A cancer that “overexpresses” a TGFp receptor is one that has significantly higher levels of a TGFp receptor, at the cell surface thereof, compared to a non-cancerous cell of the same tissue type. Such overexpression may be caused by gene amplification or by increased transcription or translation. TGFp receptor overexpression may be determined in a diagnostic or prognostic assay by evaluating increased levels of the TGFp protein present on the surface of a cell (e.g., via an immunohistochemistry assay; IHC). Alternatively, or additionally, one may measure levels of TGFp encoding nucleic acid in the cell, e.g., via fluorescent in situ hybridization (FISH; see WO 1998/045479 published October, 1998), southern blotting, or polymerase chain reaction (PCR) techniques, such as real-time quantitative PCR (RT-PCR). One may also study TGFp receptor overexpression by measuring shed antigen e.g., TGFp extracellular domain) in a biological fluid such as serum (e.g., U.S. Pat. No. 4,933,294 issued Jun. 12, 1990; WO 1991/005264 published Apr. 18, 1991 ; U.S. Pat. No. 5,401 ,638 issued Mar. 28, 1995; and Sias et al., ELISA for Quantitation of the Extracellular Domain of p185HER2 in Biological Fluids, J. Immunol. Methods 1990, 132: 73-80). Aside from the above assays, various in vivo assays are available to the skilled practitioner. For example, one may expose cells within the body of the patient to an antibody that is optionally labeled with a detectable label, e.g., a radioactive isotope, and binding of the antibody to cells in the patient can be evaluated, e.g., by external scanning for radioactivity or by analyzing a biopsy taken from a patient previously exposed to the antibody.
Conversely, a cancer that is “not characterized by overexpression of the TGFp receptor” is one that, in a diagnostic assay, does not express higher than normal levels of TGFp receptor compared to a non-cancerous cell of the same tissue type.
A cancer that “overexpresses” a TGFp ligand is one that produces significantly higher levels of that ligand compared to a non-cancerous cell of the same tissue type. Such overexpression may be caused by gene amplification or by increased transcription
or translation. Overexpression of the TGF ligand may be determined diagnostically by evaluating levels of the ligand (or nucleic acid encoding it) in the patient, e.g., in a tumor biopsy or by various diagnostic assays such as the IHC, FISH, southern blotting, PCR, enzyme-linked immunosorbent assay (ELISA) or in vivo assays described above.
A “hormone-independent” cancer is one in which proliferation thereof is not dependent on the presence of a hormone that binds to a receptor expressed by cells in the cancer. Such cancers do not undergo clinical regression upon administration of pharmacological or surgical strategies that reduce the hormone concentration in or near the tumor. Examples of hormone-independent cancers include androgen-independent prostate cancer, estrogen-independent breast cancer, endometrial cancer, and ovarian cancer. Such cancers may begin as hormone-dependent tumors and progress from a hormone-sensitive stage to a hormone-refractory tumor following anti-hormonal therapy.
As used herein, “in combination with” or "in conjunction with" refers to administration of one agent in addition to at least one other agent. As such, “in combination with” or "in conjunction with" refers to administration of one agent before, during, or after administration of at least one other agent to the individual.
Any of the combinations, methods and uses of the present invention provided may be used to treat a subject (e.g., human) who has been diagnosed with or is suspected of having cancer.
The terms "subject," "individual," and "patient" are used interchangeably herein to refer to a mammal being assessed for treatment and/or being treated. In an embodiment, the mammal is a human. The terms "subject," "individual," and "patient" thus encompass individuals having cancer (e.g., prostate cancer), including those who have undergone or are candidates for resection (surgery) to remove cancerous tissue.
The term “mammal” refers to any animal species of the Mammalia class. Examples of mammals include: humans; non-human primates such as monkeys; laboratory animals such as rats, mice, guinea pigs; domestic animals such as cats, dogs, rabbits, cattle, sheep, goats, horses, and pigs; and captive wild animals such as lions, tigers, elephants, and the like.
In a preferred embodiment, the subject is a human and may be referred to as a patient. In some embodiments, the subject is a human child between the ages of birth and 18. In a preferred embodiment, the subject is a male having a prostate cancer.
In a preferred embodiment, the subject may be a human who exhibits one or more symptoms associated with cancer.
In a preferred embodiment, the subject may be a human who is at risk, or genetically or otherwise predisposed (e.g., risk factor) to developing cancer who has or has not been diagnosed. As used herein, an “at risk” subject is a subject who is at risk of developing cancer. The subject may or may not have detectable disease, and may or may not have displayed detectable disease prior to the treatment methods described herein. An at-risk subject may have one or more so-called risk factors, which are measurable parameters that correlate with development of cancer, which are described herein. A subject having one or more of these risk factors has a higher probability of developing cancer than an individual without these risk factor(s). These risk factors may include, for example, age, sex, race, diet, history of previous disease, presence of precursor disease, genetic (e.g., hereditary) considerations, and environmental exposure. In a preferred embodiment, the subjects at risk for cancer include, for example, those having relatives who have experienced the disease, and those whose risk is determined by analysis of genetic or biochemical markers.
In a preferred embodiment, the subject is at an early stage of a cancer. In other embodiments, the subject is at an advanced stage of cancer. In a more preferred embodiment, the cancer is metastatic, non-metastatic or benign. In a preferred embodiment, the cancer is non-metastatic. In particular embodiments, the cancer is a metastatic cancer. In certain embodiments, the cancer is identified as being at risk for or having a propensity for metastasis or there is no indication that the cancer has yet metastasized. In certain embodiments, identification of a cancer at risk of metastasis is based on assessment of a tumor biopsy. In addition, the subject may be a human who is undergoing one or more standard therapies, such as chemotherapy, radiotherapy, immunotherapy, surgery, or combination thereof. Accordingly, one or more of the combinations of the present invention may be administered before, during, or after administration of chemotherapy, radiotherapy, immunotherapy, surgery or combination thereof.
In certain embodiments, the subject may be a human who is (i) substantially refractory to at least one chemotherapy treatment, or (ii) is in relapse after treatment with chemotherapy, or both (i) and (ii). In some of embodiments, the subject is refractory to at
least two, at least three, or at least four chemotherapy treatments (including standard or experimental chemotherapies).
The terms “treat” or “treating” or “treatment” of a cancer as used herein means to administer a combination therapy according to the present invention to a subject having cancer, or diagnosed with cancer, to achieve at least one positive therapeutic effect, such as, for example, reduced number of cancer cells, reduced tumor size, reduced rate of cancer cell infiltration into peripheral organs, or reduced rate of tumor metastases or tumor growth, reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition. The term "treatment", as used herein, unless otherwise indicated, refers to the act of treating as "treating" is defined immediately above. The term “treating” also includes adjuvant and neo-adjuvant treatment of a subject.
For the purposes of this invention, beneficial or desired clinical results include, but are not limited to, one or more of the following: reducing the proliferation of (or destroying) neoplastic or cancerous cell; inhibiting metastasis or neoplastic cells; shrinking or decreasing the size of a tumor; remission of the cancer; decreasing symptoms resulting from the cancer; increasing the quality of life of those suffering from the cancer; decreasing the dose of other medications required to treat the cancer; delaying the progression of the cancer; curing the cancer; overcoming one or more resistance mechanisms of the cancer; and/or prolonging survival of patients the cancer. Positive therapeutic effects in cancer can be measured in a number of ways (see, for example, W. A. Weber, Assessing tumor response to therapy, J. Nucl. Med. 2009, 50 Suppl. 1 :1 S- 10S). For example, with respect to tumor growth inhibition (T/C), according to the National Cancer Institute (NCI) standards, a T/C less than or equal to 42% is the minimum level of anti-tumor activity. A T/C <10% is considered a high anti-tumor activity level, with T/C (%) = median tumor volume of the treated / median tumor volume of the control x 100.
As used herein, the term "complete response" or "CR" means the disappearance of all signs of cancer (e.g., disappearance of all target lesions) in response to treatment. This does not always mean the cancer has been cured.
As used herein, the term “disease-free survival” (DFS) means the length of time after primary treatment for a cancer ends that the patient survives without any signs or symptoms of that cancer.
As used herein, the term “duration of response” (DoR) means the length of time that a tumor continues to respond to treatment without the cancer growing or spreading. Treatments that demonstrate improved DoR can produce a durable, meaningful delay in disease progression.
As used herein, the terms "objective response" and “overall response” refer to a measurable response, including complete response (CR) or partial response (PR). The term "overall response rate" (ORR) refers to the sum of the complete response (CR) rate and the partial response (PR) rate.
As used herein, the term “overall survival” (OS) means the length of time from either the date of diagnosis or the start of treatment for a disease, such as cancer, that patients diagnosed with the disease are still alive. OS is typically measured as the prolongation in life expectancy in patients who receive a certain treatment as compared to patients in a control group (/.e., taking either another drug or a placebo).
As used herein, the term "partial response" or "PR" refers to a decrease in the size of one or more tumors or lesions, or in the extent of cancer in the body, in response to treatment. For example, in a preferred embodiment, PR refers to at least a 30% decrease in the sum of the longest diameters (SLD) of target lesions, taking as reference the baseline SLD.
As used herein, the term "progression free survival" or “PFS” refers to the length of time during and after treatment during which the disease being treated (e.g., cancer) does not get worse. PFS, also referred to as “Time to Tumor Progression,” may include the amount of time patients have experienced a CR or PR, as well as the amount of time patients have experienced SD.
As used herein, the term "progressive disease" or "PD" refers to a cancer that is growing, spreading or getting worse. In a preferred embodiment, PR refers to at least a 20% increase in the SLD of target lesions, taking as reference the smallest SLD recorded since the treatment started, or to the presence of one or more new lesions.
As used herein, the term “stable disease” or “SD” refers to a cancer that is neither decreasing nor increasing in extent or severity.
As used herein, the term "sustained response" refers to the sustained effect on reducing tumor growth after cessation of a treatment. For example, the tumor size may be the same size or smaller as compared to the size at the beginning of the medicament
administration phase. In some embodiments, the sustained response has a duration of at least the same as the treatment duration, at least 1.5x, 2x, 2.5x, or 3x length of the treatment duration, or longer.
The anti-cancer effect of the method of treating cancer, including “objective response,” “complete response,” “partial response,” “progressive disease,” “stable disease,” “progression free survival,” and “duration of response,” as used herein, may be defined and assessed by the investigators using RECIST v1 .1 (Eisenhauer et al., New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1 ), Eur J of Cancer 2009, 45(2):228-47).
As used herein, the term “immune checkpoint inhibitor” or “immune checkpoint modulator” refers to a molecule, compound, or composition that binds to an immune checkpoint protein and blocks its activity and/or inhibits the function of the immune regulatory cell expressing the immune checkpoint protein that it binds (e.g., Treg cells, tumor-associated macrophages, etc.), immune checkpoint modulator includes any compound, including but not limited to antibodies and small molecules. Immune checkpoint proteins may include, but are not limited to, PD1 (also known as PD-1 ; Programmed Death 1 receptor), 0X40, 4-1 BB, CTLA4 (Cytotoxic T-Lymphocyte- Associated protein 4, CD152), PD-L1 , PD-L2, LAG-3 (Lymphocyte Activation Gene-3), A2AR (Adenosine A2A receptor), B7-H3 (CD276), B7-H4 (VTCN1), BTLA (B and T Lymphocyte Attenuator, CD272), IDO (Indoleamine 2,3-dioxygenase), KIR (Killer-cell Immunoglobulin-like Receptor), TIM 3 (T-cell Immunoglobulin domain and Mucin domain 3), VISTA (V-domain Ig suppressor of T cell activation), and IL-2R (interleukin-2 receptor).
Immune checkpoint inhibitors are well known in the art and are commercially or clinically available. These include, but are not limited to, agonists, antagonists, or antibodies that modulate immune checkpoint proteins. Illustrative examples of checkpoint inhibitors, referenced by their target immune checkpoint protein, are provided as follows.
Immune checkpoint inhibitors comprising anti-PD-1 antibody include, but are not limited to, sasanlimab (PF-6801591), nivolumab (MDX 1106), pembrolizumab (MK- 3475), pidilizumab (CT-011 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001 ), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP- 3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD-100, camrelizumab (SHR-
1210), Sym-021 , ABBV-181 , AK-105, BAT-1306, and AGEN-2034, or combinations thereof.
Immune checkpoint inhibitors comprising an anti-OX40 antibody include, but are not limited to, PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR-8383, and ABBV-368.
Immune checkpoint inhibitors comprising a 4-1 BB agonist 4-1 BB agonist include, but are not limited to, utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No. HB-11248, 5F4, C65-485, urelumab (BMS-663513), 20H4.9-lgG-1 (BMS-663031 ), 4E9, BMS-554271 , BMS-469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR-7480, MOR-7480.1 , MOR-7480.2, MOR-7483, MOR-7483.1 , MOR-7483.2, 3EI, 53A2, 1 D8, and 3B8
Immune checkpoint inhibitors comprising anti-CTLA4 antibody include, but are not limited to, ipilimumab (10DI), tremelimumab, and AGEN-1884.
Immune checkpoint inhibitors comprising a B7-H3 inhibitor include, but are not limited to, MGA271 .
Immune checkpoint inhibitors comprising an LAG3 inhibitor include, but are not limited to, IMP321 , BMS-986016.
Immune checkpoint inhibitors comprising a KIR inhibitor include, but are not limited to, IPH2101 (lirilumab). An immune checkpoint inhibitor targeting IL-2R, for preferentially depleting Treg cells (e.g., FoxP-3+ CD4+ cells), comprises IL-2-toxin fusion proteins, which include, but are not limited to, denileukin diftitox (Ontak).
In some embodiments of each of the methods, combinations, uses and pharmaceutical compositions disclosed herein, the therapeutic effect achieved by the TGFprl inhibitor or a pharmaceutically acceptable salt, solvate or polymorph thereof in combination with and/or an additional anti-cancer agent as further described herein, e.g., a further immune checkpoint inhibitor (for example, a PD-1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, and/or a CTLA4 antagonist), is defined by reference to any of the following: complete response (OR), disease free survival (DFS), duration of response (DoR), overall response rate (ORR), overall survival (OS), partial response (PR), or progression free survival (PFS). In some embodiments, response to a combination of the invention is any of PR, CR, PFS, DFS, OR or OS that is assessed using Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 response criteria.
In some embodiments of each of the methods, combinations, uses and pharmaceutical compositions herein, the invention relates to neoadjuvant therapy, adjuvant therapy, first-line therapy, second-line therapy, or third-line or later lines of therapy. In each case as further described herein, the cancer may be localized, advanced or metastatic, and the intervention may occur at point along the disease continuum (/.e., at any stage of the cancer).
The treatment regimen for a method, combination, uses and pharmaceutical compositions of the invention that is effective to treat cancer in a subject may vary according to factors such as the disease state, age, and weight of the subject, and the ability of the therapy to elicit an anti-cancer response in the subject. While an embodiment of any of the aspects of the invention may not be effective in achieving a positive therapeutic effect in every subject, it should do so in a statistically significant number of subjects as determined by any statistical test known in the art such as the Student’s t-test, the chi2-test the U-test according to Mann and Whitney, the Kruskal- Wallis test (H-test), Jonckheere-Terpstrat-test and the Wilcon on-test.
The terms “treatment regimen,” “dosing protocol” and “dosing regimen” may be used interchangeably to refer to the dose and timing of administration of each therapeutic agent in a combination of the invention.
“Ameliorating” means a lessening or improvement of one or more symptoms upon treatment with a combination described herein, as compared to not administering the combination. “Ameliorating” also includes shortening or reduction in duration of a symptom.
As used herein, an “effective dosage” or “effective amount” of a compound or a pharmaceutical composition is an amount sufficient to affect any one or more beneficial or desired outcomes, including biochemical, histological and/or behavioural symptoms, of the disease, its complications and intermediate pathological phenotypes presenting during development of the disease. For therapeutic use, a “therapeutically effective amount” refers to that amount of a compound being administered which will relieve to some extent one or more of the symptoms of the disorder being treated. In reference to the treatment of cancer, a therapeutically effective amount refers to that amount which has the effect of (1 ) reducing the size of the tumor, (2) inhibiting (that is, slowing to some extent, preferably stopping) tumor metastasis, (3) inhibiting to some extent (that is, slowing to some extent, preferably stopping) tumor growth or tumor invasiveness, (4)
relieving to some extent (or, preferably, eliminating) one or more signs or symptoms associated with the cancer, (5) decreasing the dose of other medications required to treat the disease, and/or (6) enhancing the effect of another medication, and/or (7) delaying the progression of the disease in a patient.
An effective dosage can be administered in one or more administrations. For the purposes of this invention, an effective dosage of drug, compound, or pharmaceutical composition is an amount sufficient to accomplish prophylactic or therapeutic treatment either directly or indirectly. As is understood in the clinical context, an effective dosage of drug, compound or pharmaceutical composition may or may not be achieved in conjunction with another drug, compound or pharmaceutical composition.
“Tumor” as it applies to a subject diagnosed with, or suspected of having, a cancer refers to a malignant or potentially malignant neoplasm or tissue mass of any size and includes primary tumors and secondary neoplasms. A solid tumor is an abnormal growth or mass of tissue that usually does not contain cysts or liquid areas. Examples of solid tumors are sarcomas, carcinomas, and lymphomas. Leukaemia’s (cancers of the blood) generally do not form solid tumors (National Cancer Institute, Dictionary of Cancer Terms).
“Tumor burden” or “tumor load’, refers to the total amount of tumorous material distributed throughout the body. Tumor burden refers to the total number of cancer cells or the total size of tumor(s), throughout the body, including lymph nodes and bone marrow. T umor burden can be determined by a variety of methods known in the art, such as, e.g., using callipers, or while in the body using imaging techniques, e.g., ultrasound, bone scan, computed tomography (CT), or magnetic resonance imaging (MRI) scans.
The term “tumor size” refers to the total size of the tumor which can be measured as the length and width of a tumor. Tumor size may be determined by a variety of methods known in the art, such as, e.g., by measuring the dimensions of tumor(s) upon removal from the subject, e.g., using callipers, or while in the body using imaging techniques, e.g., bone scan, ultrasound, CR or MRI scans.
The term “additive” is used to mean that the result of the combination of two compounds, components or targeted agents is no greater than the sum of each compound, component or targeted agent individually.
The term “synergy” or “synergistic” are used to mean that the result of the combination of two or more compounds, components or targeted agents is greater than the sum of each compound, component or targeted agent individually. This improvement in the disease, condition or disorder being treated is a “synergistic” effect and combinations providing a synergistic effect may be referred to as synergistic combinations. A “synergistic amount” is an amount of the combination of the two compounds, components or targeted agents that results in a synergistic effect, as “synergistic” is defined herein. A synergistic effect can be calculated, for example, using suitable methods such as the Sigmoid-Emax equation (Holford et al., Understanding the does-effect relationship: Clinical application of pharmacokinetic-pharmacodynamic models. Clin. Pharmacokinet. 1981 , 6: 429-453), the equation of Loewe additivity (Loewe, S. et al., Effect of combinations: mathematical basis of problem, Exp. Pathol Pharmacol. 1926, 114: 313-326) and the median-effect equation (Chou, T. C., et al., Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors, Adv. Enzyme Regul. 1984, 22: 27-55). Each equation referred to above can be applied to experimental data to generate a corresponding graph to aid in assessing the effects of the drug combination. The corresponding graphs associated with the equations referred to above are the concentration-effect curve, isobologram curve and combination index curve, respectively. (Ma & Motsinger-Reif, Current Method for Quantifying Drug Synergism, Proteom, Bioinform 2019, 1 (2):43-48; Tang et a/., What is Synergy? The Saariselka Agreement Revisited, Front Pharmacol. 2015, Article 181 , 6: 1 -5).
As used herein, the term "pharmaceutically acceptable" refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication,
As used herein, the term “pharmaceutically acceptable salt” refers to those salts which retain the biological effectiveness and properties of the parent compound. The phrase “pharmaceutically acceptable salt(s),” as used herein, unless otherwise indicated, includes salts of acidic or basic groups which may be present in the compounds of the formulae disclosed herein. For example, the compounds of the invention that are basic in nature may be capable of forming a wide variety of salts with various inorganic and organic acids. The acids that may be used to prepare pharmaceutically acceptable acid
addition salts of such basic compounds of those that form non-toxic acid addition salts, i.e. , salts containing pharmacologically acceptable anions. Examples of anions suitable for mono- and di- acid addition salts include, but are not limited to, acetate, asparatate, benzenesulfonate, benzoate, besylate, bicarbonate, bisulfate, bitartrate, bromide, calcium edetate, camsylate, carbonate, chloride, citrate, decanoate, edetate, edislyate, estolate, esylate, fumarate, gluceptate, gluconate, glutamate, glycollate, hexanoate, hexylresorcinate, hydrabamine, hydroxynaphthoate, iodide, isethionate, lactate, lactobionate, malate, maleate, mandelate, mesylate, methylsulfate, mucate, napsylate, nitrate, octanoate, oleate, pamoate (embonate), pantothenate, phosphate, polygalacturonate, propionate, salicylate, stearate, subacetate, succinate, sulfate, tannate, tartrate, teoclate, tosylate, triethiodode, and valerate salts. Alternatively, compounds that are acidic in nature may be capable of forming base salts with various pharmacologically acceptable cations which form non-toxic base salts. Such non-toxic base salts include, but are not limited to, those derived from such pharmacologically acceptable cations such as alkali metal cations (e.g., potassium and sodium) and alkaline earth metal cations (e.g., calcium and magnesium), ammonium or water-soluble amine addition salts such as N-methylglucamine-(meglumine), and the lower alkanolammonium and other base salts of pharmaceutically acceptable organic amines. Examples of cations suitable for such salts include alkali metal or alkaline-earth metal salts and other cations, including aluminium, arginine, benzathine, calcium, chloroprocaine, choline, diethanolamine, ethanolamine, ethylenediamine, lysine, magnesium, histidine, lithium, meglumine, potassium, procaine, sodium, triethyamine and zinc. Salts may be prepared by conventional techniques. Hemisalts of acids and bases may also be formed, for example, hemisulphate and hemicalcium salts. For a review on suitable salts, see Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002). Methods for making pharmaceutically acceptable salts are known to those of skill in the art.
“Tumor burden" also referred to as "tumor load", refers to the total amount of tumor material distributed throughout the body. Tumor burden refers to the total number of cancer cells or the total size of tumor(s), throughout the body, including lymph nodes and bone narrow. Tumor burden can be determined by a variety of methods known in the art, such as, e.g., by measuring the dimensions of tumor(s) upon removal from the subject, e.g., using calipers, or while in the body using imaging techniques, e.g., magnetic
resonance imaging (MRI) scans, computed tomography (CT), multi-detector CT (MDCT), positron emission tomography (PET), X-ray, ultrasound, or bone scan.
The term "tumor size" refers to the total size of the tumor which can be measured as the length and width of a tumor. Tumor size may be determined by a variety of methods known in the art, such as, e.g., by measuring the dimensions of tumor(s) upon removal from the subject, e.g., using calipers, or while in the body using imaging techniques, e.g., MRI scans, bone scan, ultrasound, or CT.
The term "regulatory agency" refers to a country's agency for the approval of the medical use of pharmaceutical agents with the country. For example, a non-limiting example of a regulatory agency is the U.S. Food and Drug Administration (FDA).
TGFprl inhibitor
Transforming growth factor p (TGFp) is a multifunctional cytokine having three forms designated TGFpl , TGFp2 and TGFp3. TGFpl to 3 form a subfamily of highly similar proteins within the TGFp superfamily of cytokines.
Human TGFpl (NCBI Reference Sequence: NP_000651.3) is a 390 amino acid protein, whilst TGFp2 (NCBI Reference Sequence: NP_001129071.1 ) is a 442 amino acid protein, and TGFP3 (NCBI Reference Sequence: NP_001316868.1 ) contains 412 amino acids. Each of TGFpi-3 have a 20-30 amino acid signal peptide at the N-terminus which is necessary for secretion, a pro-region called latency associated peptide (LAP), and a 112-114 amino acid C-terminal region that becomes the mature TGFp molecule following proteolytic cleavage from the pro-region (Khalil et al., TGF-beta: from latent to active, Microbes Infect. 1999, 1 (15): 1255-63). Mature monomeric TGp dimerize to produce the biologically active 25 KDa protein. TGFpl to 3 can form homodimers with the same type of TGFp, or can form heterodimers with another type of TGFp (e.g., a TGFpi :TGFp2 heterodimer). TGFp comprises nine conserved cysteine residues, eight of which form disulfide bonds to form the cysteine knot structure which is characteristic of the TGFp superfamily. The remaining cysteine is involved interacts with that of another TGFp monomer to form the dimer. The surface-exposed region between the fifth and sixth conserved cysteine residues is the region, which is least conserved between TGFpl to 3 proteins, and is thought to be important for receptor binding and specificity of TGFp.
The term “TGFp” refers to TGFp from any species and includes isoforms, fragments, variants or homologues of a TGFp from any species. In some embodiments,
the TGFp is a human TGF[3, primate TGFp, non-human primate TGFp, rodent TGFp, murine TGFp, or mammalian TGF|3.
TGFp exerts its functional consequences through binding to and activating signaling through TGFp receptors. TGFp receptors comprising an extracellular domain having a TGFp binding region, a single pass transmembrane domain and an intracellular domain comprising a serine/threonine kinase domain. There are three main types of TGFp receptor; TGFpRI (NCBI Reference Sequence: NP_004603.1 ) TGFPR2 (NCBI Reference Sequence: NP_001020018.1 ) and TGFPR3 (NCBI Reference Sequence: NP-003234.2).
In some embodiments, a TGFp ligand binds to and activates TGFp receptor. The term “TGFp receptor” refers to a TGFp receptor from any species and includes isoforms, fragments, variants or homologues of a TGFp receptor from any species. In some embodiments, the TGFp receptor is a human TGFp receptor, primate TGFp receptor, non-human primate TGFp receptor, rodent TGFp receptor, murine TGFp receptor, or mammalian TGFp receptor. In some embodiments, the TGFp receptor is TGFp receptor 1 (TGFprl ), TGFp receptor 2 (TGFpr2), or TGFp receptor 3 (TGFpr3). The term “TGFp receptor,” unless otherwise indicated, refers to any receptor that binds at least one TGFp receptor.
The “TGFp family” is a class within the TGFp superfamily and in human contains three members: TGFpl , TGFp2, and TGFp3, which are structurally similar. The three growth factors are known to signal via the same receptors.
TGFpi was originally defined by its ability to cause the phenotypic transformation of rat fibroblasts. TGFpi is a multipotent cytokine with cell- and dose-dependent activities. Although TGFpi is a growth inhibitor for most cell types, it can act as a stimulator for some cell types. TGFpi has ubiquitous distribution. For reviews on TGFpi , see Massague, The transforming growth factor-beta family, J. Ann. Rev. Cell Biol. 1990, 6, 597; Letterio etal., Regulation of immune responses by TGF-beta, Ann. Rev. Immunol. 1998, 16, 137. TGFpi demonstrates regulatory effects on a wide range of cell types, and modulates embryonic development, bone formation, mammary development, wound healing, haematopoiesis, angiogenesis, cell cycle progression and the production of the extracellular matrix. With respect to the immune system, TGFpi inhibits T and B cell proliferation and acts as an anti-inflammatory molecule both in vitro and in vivo. TGFpi
inhibits macrophage maturation and activation, and also inhibits the activity of natural killer cells and lymphokine-activated killer (LAK) cells and blocks cytokine production.
The term “TGFpl -positive cancer” or “TGFpl -positive tumor,” as used herein, refers to a cancer or tumor with aberrant TGF[31 expression (overexpression). Many human cancer/tumor types show predominant expression of the TGF|31 (note that “TGFB” is sometimes used to refer to the gene as opposed to protein) isoform. In some embodiments, such cancer/tumor may show co-dominant expression of another isoform, such as TGF 3. A number of epithelial cancers (e.g., carcinoma) may co-express TGFpl and TGFP3. Within the tumor environment of TGF 1 -positive tumors, TGFpl may arise from multiple sources, including, for example, cancer cells, tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), regulatory ? cells (Tregs), myeloid-derived suppressor cells (MDSCs), and the surrounding extracellular matrix (ECM). In the context of the present disclosure, preclinical cancer or tumor models that recapitulate human conditions are TGFpl -positive cancer or tumor.
The term “TGFp inhibitor” or “TGF[3r inhibitor” refers to any agent capable of antagonizing biological activities, signaling or function of TGFp growth factor (e.g., TGFP1 (TGFprl), TGFP2 (TGFpr2), and/or TGFP3 (TGFpr3)). The term is not intended to limit its mechanism of action and includes, for example, neutralizing inhibitors, small molecule inhibitors, receptor antagonists, soluble ligand traps, and activation inhibitors of TGFp. TGFpr inhibitors also include antibodies that are capable of reducing the availability of latent proTGFp which can be activated in the niche, for example, by inducing antibody-dependent cell mediated cytotoxicity (ADCC), and/or antibodydependent cellular phagocytosis (ADPC), as well as antibodies that result in internalization of cell-surface complex comprising latent proTGFp, thereby removing the precursor from the plasma membrane without depleting the cells themselves. Internalization may be a suitable mechanism of action for LRRC33-containing protein complexes (such as human LRRC33-proTGFpi ) which results in reduced levels of cells expressing LRRC33-containing protein complexes on cell surface.
Table 2 below provides exemplary TGFpr inhibitors useful in the each of the methods, combinations, uses and pharmaceutical compositions described herein.
Table 2


In some embodiments of each of the methods, combinations, uses and pharmaceutical compositions described herein, the TGF|3r1 inhibitor is selected from the
group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
In some preferred embodiments of each of the methods, combinations, uses and pharmaceutical compositions described herein, the TGF rl inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
The compound, 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide (“TGFprl inhibitor PF-06952229,” “PF- 06952229” or “PF-‘2229”), is a potent and selective TGF rl (transforming growth factor beta receptor type 1) inhibitor, having the structure:
Formula
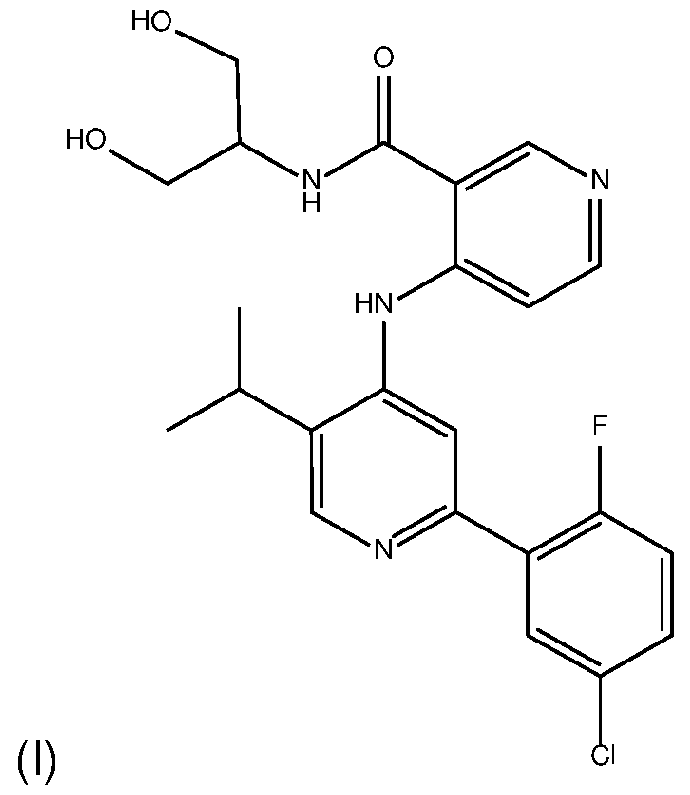
PF-06952229 and pharmaceutically acceptable salts thereof are disclosed in International Application No. PCT/US2014/072922, which published as International Publication No. WO 2015/103355 on 9 July 2015, and U.S. Patent No. 10,030,004 which issued on 10 December 2019. The contents of each of the foregoing references are incorporated herein by reference in their entirety.
Unless indicated otherwise, all references herein to TGFprl inhibitor include references to pharmaceutically acceptable salts, solvates, hydrates and complexes thereof, and to solvates, hydrates and complexes of pharmaceutically acceptable salts thereof, and include amorphous and polymorphic forms, stereoisomers, and isotopically labelled versions thereof.
PD-1 Axis Binding Antagonist
Table 3 below provides a list of the amino acid sequences of exemplary PD-1 axis binding antagonists useful in the each of the methods, combinations, uses and pharmaceutical compositions described herein. The mAB7 is also known as sasanlimab, PF-6801591 , or RN888. mAb7 and mAb15 are disclosed in PCT Publication No. WO
2016/092419, the disclosure of which is hereby incorporated by reference in its entirety. Table 3










In other embodiments of each of the methods, combinations, uses and pharmaceutical compositions described herein, the PD-1 axis binding antagonist is sasanlimab (PF-6801591 ) and will be administered subcutaneously at a dose of about 1 , 2, 3, 4, 5, 6, 7 or 8 mg/kg at intervals of about 14 days (± 2 days) or about 21 days (± 2 days) or about 30 days (± 2 days) throughout the course of treatment. In a preferred embodiment, sasanlimab (PF-6801591 ) is administer as a flat dose of about 80, 150, 160, 200, 240, 250, 300, 320, 350, 400, preferably 300mg at intervals of about 14 days (± 2 days) or about 21 days (± 2 days) or about 30 days (± 2 days). In a preferred embodiment, sasanlimab (PF-6801591 ) is administered subcutaneously in an amount of 300 mg Q4W.
In one embodiment, "PD-1 axis binding antagonist" means any chemical compound or biological molecule that blocks binding of PD-L1 expressed on a cancer
cell to PD-1 expressed on an immune cell (T cell, B cell or natural killer (NK) cell) and preferably also blocks binding of PD-L2 expressed on a cancer cell to the immune-cell expressed PD-1. Alternative names or synonyms for PD-1 and its ligands include: PDCD1 , PD1 , CD279 and SLEB2 for PD-1 ; PDCD1 L1 , PD-L1 , B7H1 , B7-4, CD274 and B7-H for PD-L1 ; and PDCD1 L2, PDL2, B7-DC, Btdc and CD273 for PD-L2. In any of the methods, combinations, uses and pharmaceutical compositions of the present invention in which a human individual is being treated, the PD-1 antagonist may block binding of human PD-L1 to human PD-1 , and block binding of both human PD-L1 and PD-L2 to human PD-1. Exemplary human PD-1 amino acid sequences can be found in NCBI Locus No.: NP_005009. Exemplary human PD-L1 and PD-L2 amino acid sequences can be found in NCBI Locus No.: NP_054862 and NP_079515, respectively.
PD-1 antagonists useful in any of the other embodiments of each of the methods, combinations, uses and pharmaceutical compositions described herein, include a monoclonal antibody (mAb), or antigen binding fragment thereof, which specifically binds to PD-1 or PD-L1 , and preferably specifically binds to human PD-1 or human PD-L1 . The mAb may be a human antibody, a humanized antibody or a chimeric antibody, and may include a human constant region. In some embodiments the human constant region is selected from the group consisting of lgG1 , lgG2, lgG3 and lgG4 constant regions, and in some embodiments, the human constant region is an lgG1 or lgG4 constant region. In some embodiments, the antigen binding fragment is selected from the group consisting of Fab, Fab'-SH, F(ab')2, scFv and Fv fragments.
Examples of mAbs that bind to human PD-1 , and useful in the methods, combinations, uses and pharmaceutical compositions of the present invention, are described in U.S. Patent Nos. 7,488,802, 7,521 ,051 , 8,008,449, 8,354,509, 8,168,757, PCT Patent Publication Nos. WO 2004/004771 , WO 2004/072286, WO 2004/056875, WO201 6/092419, and US Patent Publication No. 201 1 0271358. Specific anti-human PD-1 mAbs useful as the PD-1 antagonist in the methods, combinations, uses and pharmaceutical compositions of the present invention include: nivolumab (MDX 1 106), pembrolizumab (MK-3475), pidilizumab (CT-01 1 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), sasanlimab (PF-6801591 ), mAb15, MEDI-0680 (AMP-514), BGB-108, or AGEN-2034, or combinations thereof.
As used herein, sasanlimab (also referred to as PF-6801591 , mAb7, or RN888) is a monoclonal antibody that targets and inhibits programmed cell death 1 ligand (PDCD1 ).
As used herein, nivolumab is a human lgG4 PD-1 antibody transiently expressed by applicants in 293 HEK cells that utilizes the heavy chain and light chain sequences from Proposed INN: List 107 (CAS#946414-94-4).
As used herein, pembrolizumab is a human lgG4 PD-1 antibody transiently expressed by applicants in 293 HEK cells that utilizes the heavy chain and light chain sequences from Proposed INN: List 72.
Table 4 below provides exemplary anti-PD-1 antibody sequences useful in the each of the methods, combinations, uses and pharmaceutical compositions described herein. Table 4

0X40 Agonists
Certain embodiments of the present invention comprise an 0X40 agonist. The term “0X40 agonist” or “0X40 binding agonist,” as used herein, means, any chemical compound or biological molecule, as defined herein, which upon binding to 0X40, (1) stimulates or activates 0X40, (2) enhances, increases, promotes, induces, or prolongs an activity, function, or presence of 0X40, or (3) enhances, increases, promotes, or induces the expression of 0X40. 0X40 agonists useful in the any of the methods, combinations, uses and pharmaceutical compositions of the present invention include a monoclonal antibody (mAb), or antigen binding fragment thereof, which specifically binds to 0X40. In any of the methods, combinations, uses and pharmaceutical compositions of the present invention in which a human individual is being treated, the 0X40 agonists increase an OX40-mediated response. In some embodiments of the methods,
combinations, uses and pharmaceutical compositions of the present invention, 0X40 agonists markedly enhance cytotoxic T-cell responses, resulting in antitumor activity in several models.
An 0X40 agonist includes, for example, an 0X40 agonist antibody (e.g., an antihuman 0X40 agonist antibody), an OX40L agonist fragment, an 0X40 oligomeric receptor, and an 0X40 immunoadhesin.
The term “0X40 antibody,” “0X40 agonist antibody,” “anti-OX40 monoclonal antibody,” “aOX40” or “anti-OX40 antibody,” as used herein, means an antibody, as defined herein, capable of binding to 0X40 receptor (e.g., human 0X40 receptor).
The terms “0X40” and “0X40 receptor” are used interchangeably in the present application, and refer to any form of 0X40 receptor, as well as variants, isoforms, and species homologs thereof that retain at least a part of the activity of 0X40 receptor. Accordingly, a binding molecule, as defined and disclosed herein, may also bind 0X40 from species other than human. In other cases, a binding molecule may be completely specific for the human 0X40 and may not exhibit species or other types of crossreactivity. Unless indicated differently, such as by specific reference to human 0X40, 0X40 includes all mammalian species of native sequence 0X40, e.g., human, canine, feline, equine and bovine. One exemplary human 0X40 is a 277 amino acid protein (UniProt Accession No. P43489).
An 0X40 agonist antibody, as used herein, means, any antibody, as defined herein, which upon binding to 0X40, (1) stimulates or activates 0X40, (2) enhances, increases, promotes, induces, or prolongs an activity, function, or presence of 0X40, or (3) enhances, increases, promotes, or induces the expression of 0X40.
0X40 agonists useful in the any of the methods, combinations, uses and pharmaceutical compositions of the present invention include a monoclonal antibody (mAb) which specifically binds to 0X40 (e.g., anti-OX40 agonist antibody).
In some embodiments, the 0X40 agonist antibody increases CD4+ effector T cell proliferation and/or increases cytokine production by the CD4+ effector T cell as compared to proliferation and/or cytokine production prior to treatment with the 0X40 agonist antibody. In some embodiments, the cytokine is IFN-y.
In some embodiments, the 0X40 agonist antibody increases memory T cell proliferation and/or increasing cytokine production by the memory cell. In some embodiments, the cytokine is IFN-y. [0211] In some embodiments, the 0X40 agonist antibody inhibits Treg suppression of effector T cell function. In some embodiments,
effector T cell function is effector T cell proliferation and/or cytokine production. In some embodiments, the effector T cell is a CD4+ effector T cell.
In some embodiments, the 0X40 agonist antibody increases 0X40 signal transduction in a target cell that expresses 0X40. In some embodiments, 0X40 signal transduction is detected by monitoring NFkB downstream signaling.
In some embodiments, the anti-human 0X40 agonist antibody is a depleting antihuman 0X40 antibody (e.g., depletes cells that express human 0X40). In some embodiments, the human 0X40 expressing cells are CD4+ effector T cells. In some embodiments, the human 0X40 expressing cells are Treg cells. In some embodiments, depleting is by ADCC and/or phagocytosis. In some embodiments, the antibody mediates ADCC by binding FcyR expressed by a human effector cell and activating the human effector cell function. In some embodiments, the antibody mediates phagocytosis by binding FcyR expressed by a human effector cell and activating the human effector cell function. Exemplary human effector cells include, e.g., macrophage, natural killer (NK) cells, monocytes, neutrophils. In some embodiments, the human effector cell is macrophage.
In some embodiments, the anti-human 0X40 agonist antibody has a functional Fc region. In some embodiments, effector function of a functional Fc region is ADCC. In some embodiments, effector function of a functional Fc region is phagocytosis. In some embodiments, effector function of a functional Fc region is ADCC and phagocytosis. In some embodiments, the Fc region is a human lgG-1. In some embodiments, the Fc region is a human lgG-4.
In some embodiments, the anti-human 0X40 agonist antibody is a human or humanized antibody.
Examples of 0X40 agonist antibody, and useful in the methods, combinations, uses and pharmaceutical compositions described herein, for example, U.S. Patent No. 7,960,515, PCT Patent Application Publication Nos. WO 2013/028231 and WO 2013/1 19202, and U.S. Patent Application Publication No. 2015/0190506.
In a preferred embodimentan anti-OX40 antibody useful in the methods, combinations, uses and pharmaceutical compositions disclosed herein is a fully human agonist monoclonal antibody comprising a heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 54 and SEQ ID NO: 55, respectively. In a preferred embodiment, the anti-OX40 antibody is a fully
human lgG-2 or lgG-1 antibody. In certain embodiments, the anti-OX40 antibody is PF- 04518600.
Table 5 below provides exemplary anti-OX40 monoclonal antibody sequences useful in each of the methods, combinations, uses and pharmaceutical compositions described herein.
Table 5

4-1 BB Agonist
Certain embodiments of the present invention comprise a 4-1 BB binding agonist. The term “4-1 BB binding agonist” or “4-1 BB agonist,” as used herein, means, any chemical compound or biological molecule, as defined herein, which upon binding to 4- 1 BB, (1 ) stimulates or activates 4-1 BB, (2) enhances, increases, promotes, induces, or prolongs an activity, function, or presence of 4-1 BB, or (3) enhances, increases, promotes, or induces the expression of 4-1 BB. 4-1 BB agonists useful in any of the methods, combinations, uses and pharmaceutical compositions of the present invention include a monoclonal antibody (mAb), or antigen binding fragment thereof, which specifically binds to 4-1 BB. Alternative names or synonyms for 4-1 BB include CD137 and TNFRSF9. In any of the methods, combinations, uses and pharmaceutical compositions of the present invention in which a human individual is being treated, the 4- 1 BB agonists increase a 4-1 BB-mediated response. In some embodiments of the methods, combinations, uses and pharmaceutical compositions of the present invention, 41 BB agonists markedly enhance cytotoxic T-cell responses, resulting in antitumor activity in several models. In certain embodiments, the the 4-1 BB agonist is utomilumab.
The term “4-1 BB antibody,” “4-1 BB agonist antibody,” “anti-4-1 BB monoclonal antibody,” “a4-1 BB” or “anti-4-1 BB antibody,” as used herein, means an antibody, as defined herein, capable of binding to 4-1 BB receptor (e.g., human 4-1 BB receptor).
The terms “4-1 BB” and “4-1 BB receptor” are used interchangeably in the present application and refer to any form of 4-1 BB receptor, as well as variants, isoforms, and species homologs thereof that retain at least a part of the activity of 4-1 BB receptor. Accordingly, a binding molecule, as defined and disclosed herein, may also bind 4-1 BB from species other than human. In other cases, a binding molecule may be completely
specific for the human 4-1 BB and may not exhibit species or other types of crossreactivity. Unless indicated differently, such as by specific reference to human 4-1 BB. 4- 1 BB includes all mammalian species of native sequence of 4-1 BB, e.g., human, canine, feline, equine and bovine. One exemplary human 4-1 BB is a 255 amino acid protein (Accession No. NM_001561 ; NP_001552).
4-1 BB comprises a signal sequence (amino acid residues 1 -17), followed by an extracellular domain (169 amino acids), a transmembrane region (27 amino acids), and an intracellular domain (42 amino acids) (Cheuk ATC etal., Cancer Gene Therapy 2004, 11 : 215-226). The receptor is expressed on the cell surface in monomer and dimer forms and likely trimerizes with 4-1 BB ligand to signal.
Human 4-1 BB comprises a signal sequence (amino acid residues 1 -17), followed by an extracellular domain (169 amino acids), a transmembrane region (27 amino acids), and an intracellular domain (42 amino acids) (Cheuk ATC et al., Cancer Gene Therapy 2004, 11 : 215-226). The receptor is expressed on the cell surface in monomer and dimer forms and likely trimerizes with 4-1 BB ligand to signal.
Examples of mAbs that bind to human 4-1 BB, and useful in the methods, combinations, uses and pharmaceutical compositions of the present invention, are described in US 8,337,850 and US20130078240. In a preferred embodiment, an anti-4- 1 BB antibody useful in the treatment, method, medicaments and uses disclosed herein is a fully humanized lgG-2 agonist monoclonal antibody comprising a heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 64 and SEQ ID NO: 65, respectively.
Table 6 below provides exemplary anti-4-1 BB monoclonal antibody sequences useful in each of the methods, combinations, uses and pharmaceutical compositions described herein.
Table 6

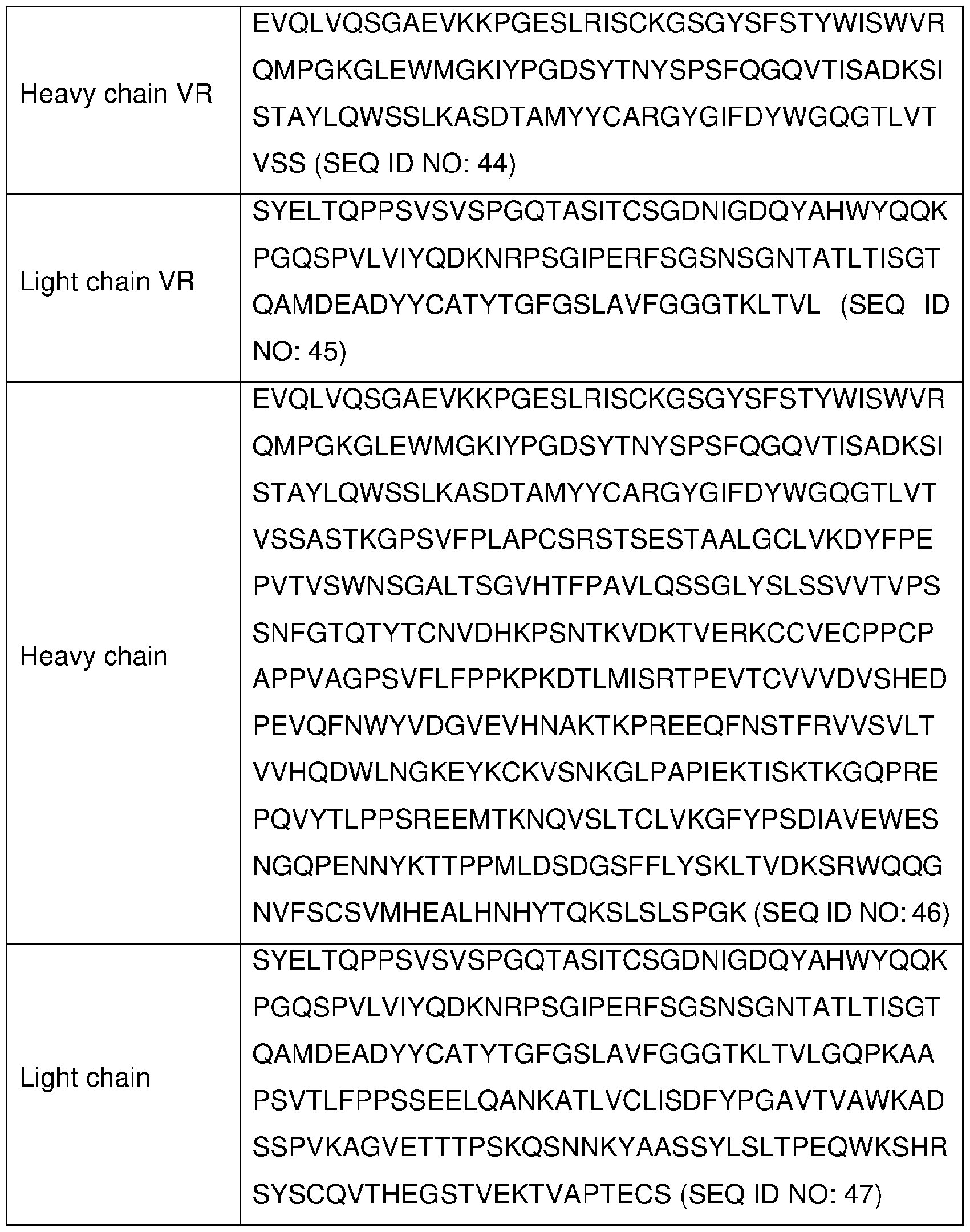
CTLA4
Cytotoxic T lymphocyte associated antigen 4 (CTLA4) closely relates to CD28 in gene structure, chromosomal localization, homology of sequences and gene expression, and both of them are receptors of costimulatory molecule B7, and mainly expresses on the cell surface of activated T cells. Interaction of CTLA4 and B7 inhibits the activation of T cells in mice and human, and negatively regulates the activation of T cells.
Certain embodiments of the present invention comprise an anti-cytotoxic T- lymphocyte associated antigen 4 (anti-CTLA4) antibody. Suitable anti-CTLA4 antibodies for use in the methods of the invention, include, without limitation, anti-CTLA4 antibodies, human anti-CTLA4 antibodies, mouse anti-CTLA4 antibodies, mammalian anti-CTLA4 antibodies, humanized anti-CTLA4 antibodies, monoclonal anti-CTLA4 antibodies, polyclonal anti-CTLA4 antibodies, chimeric anti-CTLA4 antibodies, ipilimumab, tremelimumab, anti-CD28 antibodies, anti-CTLA4 adnectins, anti-CTLA4 domain antibodies, single chain anti-CTLA4 fragments, heavy chain anti-CTLA4 fragments, light chain anti-CTLA4 fragments, inhibitors of CTLA4 that agonize the co-stimulatory pathway, the antibodies disclosed in PCT Publication No. WO 2001/014424, the antibodies disclosed in PCT Publication No. WO 2004/035607, the antibodies disclosed in U.S. Published Application No. 2005/0201994, and the antibodies disclosed in granted European Patent No. EP 1212422B1 . Additional CTLA4 antibodies are described in U.S. Pat. Nos. 5,811 ,097, 5,855,887, 6,051 ,227, and 6,984,720; in PCT Publication Nos. WO 01/014424 and WO 00/037504; and in U.S. Publication Nos. 02/0039581 and 02/086014. Other anti-CTLA4 antibodies that can be used in a method of the present invention include, for example, those disclosed in: WO 98/042752; U.S. Pat. Nos. 6,682,736 and 6,207,156; Hurwitz et al., CTLA4 blockade synergizes with tumor-derived granulocytemacrophage colony-stimulating factor for treatment of an experimental mammary carcinoma, Proc. Natl. Acad. Sci. 1998, 95(17): 10067-10071 ; Camacho et al., Abstract No. 2505, antibody CP-675206, J. Clin. Oncology, 2004, 22(145); Mokyr et al., Realization of the therapeutic potential of CTLA-4 blockade in low-dose chemo-therapy- treated tumor-bearing mice, Cancer Res. 1998, 58:5301 -5304, and U.S. Pat. Nos. 5,977,318, 6,682,736, 7,109,003, and 7,132,281 .
Ipilimumab (marketed as YERVOY®; also known as MEX-010, MDX-101 , or by its CAS Registry No. 477202-00-9) is disclosed as antibody 10DI in PCT Publication No. WO 01/014424. Examples of pharmaceutical composition comprising Ipilimumab are provided in PCT Publication No. WO 07/067959. Ipilimumab is approved in the U.S. for the treatment of unresectable or metastatic melanoma. In the methods provided by the present invention, Ipilimumab may be administered intradermally or subcutaneously. The effective amount of Ipilimumab administered locally is typically in the range of 5 - 200 mg/dose per person. In some embodiments, the effective amount of Ipilimumab is in the range of 10 - 150 mg/dose per person per dose. In some particular embodiments, the
effective amount of Ipilimumab is about 10, 25, 50, 75, 100, 125, 150, 175, or 200 mg/dose per person.
Tremelimumab (also known as CP-675,206) is a fully human lgG2 monoclonal antibody and has the CAS number 745013-59-6. Tremelimumab is disclosed as antibody 11.2.1 in U.S. Patent No: 6,682,736. In the VBIR for cancer provided by the present invention, tremelimumab may be administered intravenously, intradermally, or subcutaneously. The effective amount of tremelimumab administered intradermally or subcutaneously is typically in the range of 5-200 mg/dose per person. In some embodiments, the effective amount of tremelimumab is in the range of 10 - 150 mg/dose per person per dose. In some particular embodiments, the effective amount of tremelimumab is about 10, 25, 50, 75, 100, 125, 150, 175, or 200 mg/dose per person.
Table 7 below provides exemplary anti-CTLA4 monoclonal antibody sequences useful in each of the methods, combinations, uses and pharmaceutical compositions described herein Table 7


Therapeutic Methods, Combinations and Uses
The present invention provides methods, combinations and uses that may be useful for treating cancer. Some embodiments provided herein result in one or more of the following effects: (1 ) inhibiting cancer cell proliferation; (2) inhibiting cancer cell invasiveness; (3) inducing apoptosis of cancer cells; (4) inhibiting cancer cell metastasis;
(5) inhibiting angiogenesis; or (6) overcoming one or more resistance mechanisms relating to a cancer treatment.
In another aspect, the invention provides a combination comprising a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof; and a PD-1 axis binding antagonist, for use in the manufacture of a medicament for treating cancer in a subject.
In another aspect, the invention provides use of a combination comprising a TGF r1 inhibitor or a pharmaceutically acceptable salt thereof; and a PD-1 axis binding antagonist, in the manufacture of a medicament for treating cancer in a subject. In some embodiments of these aspects, the combination further comprises an additional anticancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an antiandrogen and/or anti-angiogenic agent, for use in the manufacture of a medicament. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, antimetabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the anti-androgen inhibitor selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides a combination comprising a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof; and an 0X40 agonist, for use in the manufacture of a medicament for treating cancer in a subject. In another aspect, the invention provides use of a combination comprising a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; and an 0X40 agonist, in the manufacture of a medicament for treating cancer in a subject. In some embodiments of these aspects, the combination further comprises an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, for use in the manufacture of a medicament. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase
inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides a combination comprising a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof; and a 4-1 BB agonist, for use in the manufacture of a medicament for treating cancer in a subject. In another aspect, the invention provides use of a combination comprising a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; and a 4-1 BB agonist, in the manufacture of a medicament for treating cancer in a subject. In some embodiments of these aspects, the combination further comprises an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, for use in the manufacture of a medicament. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 - oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4- naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2- propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides a combination comprising a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof; and a CTLA4 antagonist, for use in the manufacture of a medicament for treating cancer in a subject. In another aspect, the invention provides use of a combination comprising a TGF r1 inhibitor or a pharmaceutically acceptable salt thereof; and a CTLA4 antagonist, in the manufacture of a medicament for treating cancer in a subject. In some embodiments of these aspects, the combination further comprises an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, for use in the manufacture of a medicament. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a PD-1 axis binding antagonist. In another aspect, the invention provides a TGFprl inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a PD-1 axis binding antagonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies,
cytotoxics or anti-hormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a PD-1 axis binding antagonist. In another aspect, the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a PD-1 axis binding antagonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides a TGFprl inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer,
wherein the medicament is adapted for use in combination with an 0X40 agonist. In another aspect, the invention provides a TGFprl inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with an 0X40 agonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or antihormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with an 0X40 agonist. In another aspect, the invention provides use of a TGF rl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with an 0X40 agonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or antihormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such
embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a 4-1 BB agonist. In another aspect, the invention provides a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a 4-1 BB agonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or antihormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a 4-1 BB agonist. In another aspect, the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein
the medicament is adapted for use in combination with a 4-1 BB agonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides a TGF[3r1 inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a CTLA4 antagonist. In another aspect, the invention provides a TGFprl inhibitor or a pharmaceutically acceptable salt thereof for use in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a CTLA4 antagonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or antihormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin,
which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In another aspect, the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a CTLA4 antagonist. In another aspect, the invention provides use of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer, wherein the medicament is adapted for use in combination with a CTLA4 antagonist, and an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, antimetabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
The present invention provides methods, combinations and uses comprising a TGFprl inhibitor or a pharmaceutically acceptable salt, and an immune checkpoint inhibitor, wherein the immune checkpoint inhibitor is a PD-1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, and/or a CTLA4 antagonist.
In one aspect the present invention provides methods comprising administering to a subject in need thereof an amount of a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of a
programmed cell death protein 1 (PD-1 ) axis binding antagonist, wherein the amounts together are effective in treating cancer.
In a preferred embodiment of this aspect, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof. In a specific embodiment, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide
In one preferred aspect, the present invention provides methods, combinations and uses comprising the compound, 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4- ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (also referred to as “PF-06952229” or “PF-‘2229”). PF-06952229 is a potent and selective TGFprl (transforming growth factor beta receptor type 1) inhibitor, having the structure:
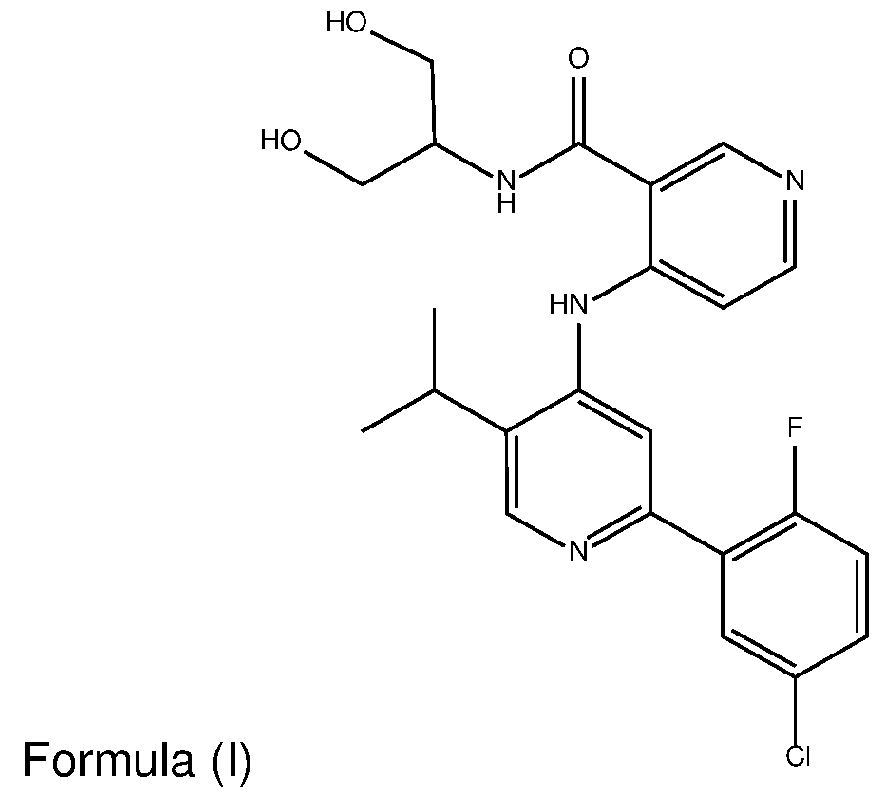
In a specific embodiment, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
In some embodiments of this aspect, the PD-1 axis binding antagonist is an anti- PD-1 antibody. In some embodiments, the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-06801591 ) (see WO2016/092419), nivolumab ((Opdivo®), MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-011 ), cemiplimab (REGN2810),
tislelizumab (BGB-A317), spartalizumab (PDR001 ), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, Bl- 754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105, BAT-1306, and AGEN-2034, or combinations thereof. In a specific embodiment, the anti-PD-1 antibody is sasanlimab (PF-6801591).
In some embodiments of each of the methods, combinations and uses described herein, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4- ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof; and the anti-PD-1 antibody is sasanlimab (PF- 6801591 ), wherein the amounts together are effective in treating cancer.
In some embodiments of each of the methods, combinations and uses described herein, the TGFprl inhibitor and the PD-1 axis binding antagonist are administered sequentially, simultaneously, or concurrently.
In some embodiments of each of the methods, combinations and uses and described herein, the TGF rl inhibitor and the PD-1 axis binding antagonist, are administered sequentially, simultaneously, or concurrently.
In one aspect the present invention provides methods comprising administering to a subject in need thereof an amount of a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an 0X40 agonist, wherein the amounts together are effective in treating cancer.
In a preferred embodiment of this aspect, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
In a specific embodiment of this aspect, the TGFprl inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
In some embodiments of this aspect, the 0X40 agonist is an anti-OX40 antibody, an OX40L agonist fragment, an 0X40 oligomeric receptor, a trimeric OX40L-Fc protein or an 0X40 immunoadhesin, or combinations thereof. In a particular embodiment, the
0X40 agonist is an anti-OX40 antibody. In a preferred embodiment of this aspect, the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS- 986178, GBR-8383, and ABBV-368, or combinations thereof.
In a preferred embodiment of each of the methods, combinations, uses and pharmaceutical compositions described herein, the TGFprl inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof; and the anti-OX40 antibody is PF-04518600, and wherein the amounts together are effective in treating cancer.
In some embodiments of each of the methods, combinations and uses described herein, the TGFprl inhibitor and the 0X40 agonist are administered sequentially, simultaneously, or concurrently.
In one aspect the present invention provides methods comprising administering to a subject in need thereof an amount of a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of an amount of a 4-1 BB agonist, wherein the amounts together are effective in treating cancer.
In a preferred embodiment of this aspect, the TGF rl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
In a specific embodiment of this aspect, the TGFprl inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of this aspect, the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No. HB-1 1248, 5F4, C65- 485, urelumab (BMS-663513), 20H4.9-lgG-1 (BMS-663031 ), 4E9, BMS-554271 , BMS- 469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR-7480, MOR-7480.1 , MOR- 7480.2, MOR-7483, MOR-7483.1 , MOR-7483.2, 3EI, 53A2, 1 D8, and 3B8, or combinations thereof. In a specific embodiment, the 4-1 BB agonist is utomilumab.
In a preferred embodiment of each of the methods, combinations and uses described herein, the TGFprI inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof; and the 4-1 BB agonist is utomilumab, wherein the amounts together are effective in treating cancer.
In some embodiments of each of the methods, combinations and described herein, the TGFprI inhibitor and the 4-1 BB agonist are administered sequentially, simultaneously, or concurrently.
In one aspect the present invention provides methods comprising administering to a subject in need thereof an amount of a transforming growth factor beta receptor (TGF r) inhibitor, or a pharmaceutically acceptable salt thereof; and an amount of a CTLA4 antagonist, wherein the amounts together are effective in treating cancer.
In a preferred embodiment of this aspect, the TGF rI inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof. In a specific embodiment of this aspect, the TGFprI inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of this aspect, the CTLA4 antagonist is an anti-CTLA4 antibody. In a preferred embodiment, the anti-CTLA4 antibody is selected from the group consisting of ipilimumab (Yervoy®), tremelimumab (CP-675,206), and AGEN-1884, or combinations thereof. In a specific embodiment, the anti-CTLA4 antibody is ipilimumab (Yervoy®).
In a preferred embodiment of each of the methods, combinations and uses described herein, the TGFprI inhibitor is 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof; and the anti-CTLA4 antibody is ipilimumab (Yervoy®), wherein the amounts together are effective in treating cancer.
In a preferred embodiment of each of the methods, combinations and uses described herein, the TGFprI inhibitor and the CTLA4 antagonist are administered
sequentially, simultaneously, or concurrently.
In a preferred embodiment of this aspect, the invention provides methods, combinations and uses, wherein the subject is a human.
In a preferred embodiment of this aspect, the combination further comprises an additional anti-cancer agent; wherein the combination is effective in treating cancer.
In a specific embodiment, the additional anti-cancer agent is selected from the group consisting of a further immune checkpoint inhibitor, an anti-tumor agent, an antiandrogen and anti-angiogenic agent.
In one such embodiments, the anti-tumor agent is selected from the group consisting of mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics, antihormones, androgen deprivation therapy and anti-androgens. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 - oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4- naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2- propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In some embodiments of each of the methods, combinations, uses and pharmaceutical compositions described herein, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof. In a preferred embodiment, the cancer is prostate cancer. In a preferred embodiment, the prostate cancer is hormone sensitive prostate cancer. In a preferred embodiment, the prostate cancer is castration
resistant prostate cancer. In a preferred embodiment, the prostate cancer is metastatic. In some embodiments, the prostate cancer is non-metastatic.
Pharmaceutical Compositions, Medicaments and Kits
The present invention further provides pharmaceutical compositions, medicaments and kits comprising a TGF0r inhibitor, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor and/or an additional anticancer agent, as further described below.
In a preferred embodiment of each of the pharmaceutical compositions, medicaments and kits described herein, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of each of the pharmaceutical compositions, medicaments and kits described herein, the TGF|3r1 inhibitor is 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
In another aspect, the invention provides a pharmaceutical composition comprising a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable carrier or excipient, for use in combination with an immune checkpoint inhibitor for use in treating cancer. In some embodiments of the foregoing, the immune checkpoint inhibitor is a PD-1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, and/or a CTLA4 antagonist. In some embodiments of this aspect, the pharmaceutical composition further comprises an additional anti-cancer agent.
In some embodiments of each of the pharmaceutical compositions, medicaments and kits described herein, the invention provides a first pharmaceutical composition comprising a TGF rl inhibitor or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable carrier or excipient, and a second pharmaceutical composition comprising a PD-1 axis binding antagonist, wherein the first and second pharmaceutical compositions are administered sequentially, simultaneously, or concurrently. Some embodiments of this aspect further comprise a third pharmaceutical composition comprising an additional anti-cancer agent, e.g., a further immune
checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, and a pharmaceutically acceptable carrier or excipient, wherein the first, second and third pharmaceutical compositions are administered sequentially, simultaneously, or concurrently. In some such embodiments, the anti-tumor agent is selected from the group consisting of mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics, antihormones, androgen deprivation therapy and anti-androgens. In some preferred embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 - oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4- naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2- propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In some embodiments of each of the pharmaceutical compositions, medicaments and kits described herein, the invention provides a first pharmaceutical composition comprising a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable carrier or excipient, and a second pharmaceutical composition comprising an 0X40 agonist, wherein the first and second pharmaceutical compositions are administered sequentially, simultaneously, or concurrently. Some embodiments of this aspect further comprise a third pharmaceutical composition comprising an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, and a pharmaceutically acceptable carrier or excipient, wherein the first, second and third pharmaceutical compositions are administered sequentially, simultaneously, or concurrently. In some such embodiments, the anti-tumor agent is selected from the group consisting of mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics, antihormones, androgen deprivation therapy and anti-androgens. In some such
embodiments, the anti-androgen inhibitor selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 - oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4- naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2- propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In some embodiments of each of the pharmaceutical compositions, medicaments and kits described herein, the invention provides a first pharmaceutical composition comprising a TGF r1 inhibitor or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable carrier or excipient, and a second pharmaceutical composition comprising an anti-4-1 BB antibody, wherein the first and second pharmaceutical compositions are administered sequentially, simultaneously, or concurrently. Some embodiments of this aspect further comprise a third pharmaceutical composition comprising an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, and a pharmaceutically acceptable carrier or excipient, wherein the first, second and third pharmaceutical compositions are administered sequentially, simultaneously, or concurrently. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the antiandrogen inhibitor selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5- methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6- yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In some embodiments of each of the pharmaceutical compositions, medicaments and kits described herein, the invention provides a first pharmaceutical composition comprising a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable carrier or excipient, and a second pharmaceutical composition comprising a CTLA4 antagonist, wherein the first and second pharmaceutical compositions are administered sequentially, simultaneously, or concurrently. Some embodiments of this aspect further comprise a third pharmaceutical composition comprising an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent, and a pharmaceutically acceptable carrier or excipient, wherein the first, second and third pharmaceutical compositions are administered sequentially, simultaneously, or concurrently. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2, 4,6,8- decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3- methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4- methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4- dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In a preferred embodiment of each of the pharmaceutical compositions, medicaments and kits described herein, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
In some such embodiments, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of each of the pharmaceutical compositions, medicaments and kits described herein, the PD-1 axis binding antagonist is an anti-PD- 1 antibody. In some such embodiments, the anti-PD-1 antibody is selected from the
group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1 106), pembrolizumab (MK-3475), pidilizumab (CT-011), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP- 3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD-100, camrelizumab (SHR- 1210), Sym-021 , ABBV-181 , AK-105, BAT-1306, and AGEN-2034, or combinations thereof. In a specific embodiment, the anti-PD-1 antibody is sasanlimab (PF-6801591).
In a preferred embodiment of each of the pharmaceutical compositions, medicaments and kits described herein, the 0X40 agonist is an anti-OX40 antibody, an OX40L agonist fragment, an 0X40 oligomeric receptor, a trimeric OX40L-Fc protein or an 0X40 immunoadhesin, or combinations thereof. In some such embodiments, the 0X40 agonist is an anti-OX40 antibody. In some specific embodiments, the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR- 8383, and ABBV-368, or combinations thereof.
In a preferred embodiment of each of the pharmaceutical compositions, medicaments and kits described herein, the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No. HB-1 1248, 5F4, C65- 485, urelumab (BMS-663513), 20H4.9-lgG-1 (BMS-663031 ), 4E9, BMS-554271 , BMS- 469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR-7480, MOR-7480.1 , MOR- 7480.2, MOR-7483, MOR-7483.1 , MOR-7483.2, 3EI, 53A2, 1 D8, and 3B8, or combinations thereof. In some such embodiments, the 4-1 BB agonist is utomilumab.
In a preferred embodiment of each of the pharmaceutical compositions, medicaments and kits described herein, the CTLA4 antagonist is an anti-CTLA4 antibody. In some such embodiments, the anti-CTLA4 antibody is selected from the group consisting of ipilimumab (10DI), tremelimumab, and AGEN-1884, or combinations thereof.
In another aspect, the invention provides a kit comprising a first container, a second container and a package insert, wherein the first container comprises at least one dose of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof, as further described herein; the second container comprises at least one dose of a PD-1 axis binding antagonist; at least one dose of and 0X40 agonist, at least one dose of 4-1 BB
agonist, or at least one dose of CTLA4 antagonist, or combination thereof , and the package insert comprises instructions for treating cancer in a subject using the medicaments. In another aspect, the invention provides a kit comprising a first container, a second container, a third container, and a package insert, wherein the first container comprises at least one dose of a TGFprl inhibitor or a pharmaceutically acceptable salt thereof; the second container comprises at least one dose of a PD-1 axis binding antagonist; at least one dose of an 0X40 agonist, at least one dose of a 4-1 BB agonist, or at least one dose of CTLA4 antagonist, or combination thereof; the third container comprises at least one dose of an additional anti-cancer agent, e.g., a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti-angiogenic agent; and the package insert comprises instructions for treating cancer in a subject using the medicaments. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the antiandrogen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5- methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6- yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
In a preferred embodiment of the kits herein, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof, or combinations thereof.
In a preferred embodiment, the TGFprl inhibitor is 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
In a preferred embodiment of the kits herein, the PD-1 axis binding antagonist is an anti-PD-1 antibody. In some such embodiments, the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1 106), pembrolizumab (MK-3475), pidilizumab (CT-01 1 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS- 010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105, BAT-1306, and AGEN-2034, or combinations thereof. In a specific embodiment, the anti-PD-1 antibody is sasanlimab (PF-6801591 ).
In a preferred embodiment of the kits herein, the 0X40 agonist is an anti-OX40 antibody, an OX40L agonist fragment, an 0X40 oligomeric receptor, a trimeric OX40L- Fc protein or an 0X40 immunoadhesin, or combinations thereof. In some such embodiments, the 0X40 agonist is an anti-OX40 antibody. In some specific embodiments, the anti-OX40 antibody is selected from the group consisting of PF- 04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR-8383, and ABBV-368, or combinations thereof.
In a preferred embodiment of the kits herein, the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No. HB-11248, 5F4, C65-485, urelumab (BMS-663513), 20H4.9-lgG-1 (BMS-663031 ), 4E9, BMS-554271 , BMS-469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR-7480, MOR-7480.1 , MOR-7480.2, MOR-7483, MOR-7483.1 , MOR-7483.2, 3EI, 53A2, 1 D8, and 3B8, or combinations thereof. In some such embodiments, the 4-1 BB agonist is utomilumab.
In some embodiments of the kits herein, the CTLA4 antagonist is an anti-CTLA4 antibody. In some such embodiments, the anti-CTLA4 antibody is selected from the group consisting of ipilimumab (10DI), tremelimumab, and AGEN-1884, or combinations thereof.
In embodiments of the pharmaceutical compositions, medicaments, and kits comprising an additional anti-cancer agent, the additional anti-cancer agent is a further immune checkpoint inhibitor, an anti-tumor agent, an anti-androgen and/or anti- angiogenic agent. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors,
radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the antiandrogen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5- methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 -oxaspiro[2.5]oct-6- yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4-naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2-propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
The pharmaceutical compositions, medicaments and kits described herein may be useful for treating the cancers described above with respect to the methods, combinations and uses of the invention. In some embodiments, the pharmaceutical compositions, medicaments and kits may be useful for treating cancer is selected from the group consisting of prostate cancer, colorectal cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, kidney cancer, stomach cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
Additional Methods, Combinations and Uses
The invention provides methods and uses comprising a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor and/or an additional anti-cancer agent, as further described below.
In a preferred embodimentof the foregoing methods and uses, the TGFprl inhibitor is selected from the group consisting of galunisertib, LY2109761 , SB525334, SP505124, GW788388, LY364947, RepSox, SD-208, vactosertib, LY3200882 and 4-(2- (5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide (PF-06952229), or a pharmaceutically acceptable salt thereof.
In a preferred embodimentof the foregoing methods and uses, In a preferred embodimentof the foregoing methods and uses, the TGFprl inhibitor is 4-(2-(5-chloro-2-
fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof.
In a preferred embodimentof the foregoing methods and uses, the immune checkpoint inhibitor is a PD-1 axis binding antagonist. In a preferred embodiment of the foregoing, the PD-1 axis binding antagonist is an anti-PD-1 antibody. In some such emdodiments, the anti-PD-1 antibody is sasanlimab, nivolumab (MDX 1 106), pembrolizumab (MK-3475), pidilizumab (CT-01 1 ), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS- 010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM-009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105, BAT-1306, or AGEN-2034, or a combination thereof. In some such emdodiments, the anti-PD-1 antibody is sasanlimab.
In a preferred embodimentof the foregoing methods and uses, the immune checkpoint inhibitor is an 0X40 agonist. In a preferred embodiment of the foregoing, the 0X40 agonist is an is an anti-OX40 antibody. In some such emdodiments, the anti-OX40 antibody is selected from the group consisting of PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR- 8383, and ABBV-368, or combinations thereof. In some such emdodiments, the anti- 0X40 antibody is PF-04518600.
In a preferred embodimentof the foregoing methods and uses, the immune checkpoint inhibitor is a 4-1 BB agonist. In a preferred embodiment of the foregoing, the 4-1 BB agonist is selected from the group consisting of utomilumab (PF-05082566), 1 D8, 3Elor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No. HB-11248, 5F4, C65-485, urelumab (BMS-663513), 20H4.9-lgG-1 (BMS- 663031), 4E9, BMS-554271 , BMS-469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR-7480, MOR-7480.1 , MOR-7480.2, MOR-7483, MOR-7483.1 , MOR-7483.2, 3EI, 53A2, 1 D8, and 3B8, or combinations thereof. In a preferred embodiment of this aspect, the 4-1 BB agonist is utomilumab.
In a preferred embodimentof the foregoing methods and uses, the immune checkpoint inhibitor is a CTLA4 antagonist. In a preferred embodiment of the foregoing, the CTLA4 antagonist is an anti-CTLA4 antibody. In some such embodiments, the anti- CTLA4 antibody is selected from the group consisting of ipilimumab (10DI),
tremelimumab, and AGEN-1884, or combinations thereof. In some such embodiments, the anti-CTLA4 antibody is ipilimumab (10DI).
In some embodiments of the foregoing methods and uses, the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
In one preferred aspect, the invention provides a method of treating prostate cancer in a subject in need thereof comprising administering to the subject an amount of 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yljnicotinamide, or a pharmaceutically acceptable salt thereof; and an amount of an immune checkpoint inhibitor, wherein the amounts together are effective in treating cancer. In one preferred aspect, the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor, for use in treating prostate cancer.
In a preferred embodiment of these aspects, the prostate cancer is hormone sensitive prostate cancer. In a preferred embodiment of this aspect, the prostate cancer is castration resistant prostate cancer. In a preferred embodiment of this aspect, the prostate cancer is metastatic. In a preferred embodiment of this aspect, the prostate cancer is non-metastatic.
In one preferred aspect, the invention provides a method of treating lung cancer in a subject in need thereof comprising administering to the subject an amount of 4-(2- (5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yljnicotinamide, or a pharmaceutically acceptable salt thereof; and an amount of an immune checkpoint inhibitor, wherein the amounts together are effective in treating cancer. In one preferred aspect, the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor, for use in treating lung cancer.
In some embodiments of these aspects, the lung cancer is small cell lung cancer (SCLC). In some such embodiments the SCLC is RB-negative. In some embodiments of this aspect, the lung cancer is non-small cell lung cancer (NSCLC). In some embodiments of this aspect, the lung cancer is advanced or metastatic lung cancer. In some such embodiments, the lung cancer is advanced or metastatic SCLC. In some such embodiments, the lung cancer is advanced or metastatic NSCLC.
In one preferred aspect, the invention provides a method of treating ovarian cancer, peritoneal cancer, or fallopian tube cancer in a subject in need thereof comprising administering to the subject an amount of 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and an amount of an immune checkpoint inhibitor, wherein the amounts together are effective in treating cancer. In one preferred aspect, the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4- ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor, for use in treating ovarian cancer, peritoneal cancer, or fallopian tube cancer.
In some embodiments of these aspects, the cancer is ovarian cancer. In some such embodiments, the ovarian cancer is epithelial ovarian cancer (EOC). In some such embodiments, the ovarian cancer is advanced or metastatic ovarian cancer (including EOC). In some such embodiments, the ovarian cancer is platinum resistant ovarian cancer (including EOC). In some such embodiments, the ovarian cancer is platinum resistant advanced or metastatic ovarian cancer (including EOC). In some embodiments, the cancer is peritoneal cancer. In some such embodiments, the peritoneal cancer is primary peritoneal carcinomatosis (PPC). In some embodiments, the cancer is fallopian tube cancer (FTC).
In one preferred aspect, the invention provides a method of treating breast cancer in a subject in need thereof comprising administering to the subject an amount of 4-(2- (5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and an amount of an immune checkpoint inhibitor, wherein the amounts together are effective in treating cancer. In another aspect, the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a
pharmaceutically acceptable salt thereof, in combination with an immune checkpoint inhibitor, for use in treating breast cancer.
In some embodiments of these aspects, the breast cancer is HR+/HER2- breast cancer. In some such embodiments, the breast cancer is HR+/HER2- advanced or metastatic breast cancer. In some embodiments, the breast cancer is triple negative breast cancer (TNBC). In some such embodiments, the TNBC is locally recurrent, advanced or metastatic TNBC. In some embodiments of the foregoing, the breast cancer is HR+/HER2- breast cancer or TNBC, which may be advanced or metastatic, and the subject is a woman of any menopausal status or a man.
In a preferred embodiment of the foregoing methods, the method comprises administering to the subject an amount of 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, an amount of an immune checkpoint inhibitor, and an amount of additional anti-cancer agent, wherein the amount of 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, the amount of an immune checkpoint inhibitor, and the amount of the additional anti-cancer agent together are effective in treating cancer.
In a preferred embodiment wherein the cancer is breast cancer, the additional anticancer agent is an endocrine therapeutic agent. In some such embodiments, the endocrine therapeutic agent is an aromatase inhibitor, a selective estrogen receptor degrader (SERD), or a selective estrogen receptor modulator (SERM). In some such embodiments, the endocrine therapeutic agent is letrozole. In other such embodiments, the endocrine therapeutic agent is fulvestrant. In a preferred embodiment, the breast cancer is HR+/HER2- breast cancer. In a preferred embodiment, the breast cancer is HR+/HER2- advanced or metastatic breast cancer. In some embodiments, the breast cancer is triple negative breast cancer (TNBC). In a preferred embodiment, the TNBC is locally recurrent, advanced or metastatic TNBC. In a preferred embodiment of the foregoing, the breast cancer is HR+/HER2- breast cancer or TNBC, which may be advanced or metastatic, and the subject is a woman of any menopausal status or a man.
In some embodiments of the foregoing, the immune checkpoint inhibitor is a PD- 1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, and/or a CTLA4 antagonist.
In one preferred aspect, the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof for use in treating cancer. In a preferred embodiment, the invention provides 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4- ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof for use in treating cancer, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
In one preferred aspect, the invention provides use of a combination comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide (PF-06952229), having the structure:
 or a pharmaceutically acceptable salt thereof; and a PD-1 axis binding antagonist, wherein the use of the combination is effective in treating cancer. In a preferred embodiment of the foregoing uses, the PD-1 axis binding antagonist is an anti-PD-1 antibody. In some such emdodiments, the anti-PD-1 antibody is sasanlimab, nivolumab (MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-01 1), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ),
LZM-009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105, BAT- 1306, or AGEN-2034, or a combination thereof. In some such emdodiments, the anti-PD- 1 antibody is sasanlimab.
or a pharmaceutically acceptable salt thereof; and a PD-1 axis binding antagonist, wherein the use of the combination is effective in treating cancer. In a preferred embodiment of the foregoing uses, the PD-1 axis binding antagonist is an anti-PD-1 antibody. In some such emdodiments, the anti-PD-1 antibody is sasanlimab, nivolumab (MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-01 1), cemiplimab (REGN2810), tislelizumab (BGB-A317), spartalizumab (PDR001), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, BI-754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ),
LZM-009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105, BAT- 1306, or AGEN-2034, or a combination thereof. In some such emdodiments, the anti-PD- 1 antibody is sasanlimab.
In one preferred aspect, the invention provides use of a combination comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide (PF-06952229), having the structure:
 or a pharmaceutically acceptable salt thereof; and an 0X40 agonist, wherein the combination is effective in treating cancer. In a preferred embodiment of the foregoing uses, the 0X40 agonist is an is an anti-OX40 antibody. In some such emdodiments, the anti-OX40 antibody is PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR-8383, or ABBV-368, or combinations thereof. In some such emdodiments, the anti-OX40 antibody is PF- 04518600. In one preferred aspect, the invention provides use of a combination comprising
or a pharmaceutically acceptable salt thereof; and an 0X40 agonist, wherein the combination is effective in treating cancer. In a preferred embodiment of the foregoing uses, the 0X40 agonist is an is an anti-OX40 antibody. In some such emdodiments, the anti-OX40 antibody is PF-04518600, MEDI6469, MEDI0562 (tavolixizumab), MEDI6383, MOXR0916, RG-7888, GSK-3174998, BMS-986178, GBR-8383, or ABBV-368, or combinations thereof. In some such emdodiments, the anti-OX40 antibody is PF- 04518600. In one preferred aspect, the invention provides use of a combination comprising
4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide (PF-06952229), having the structure:
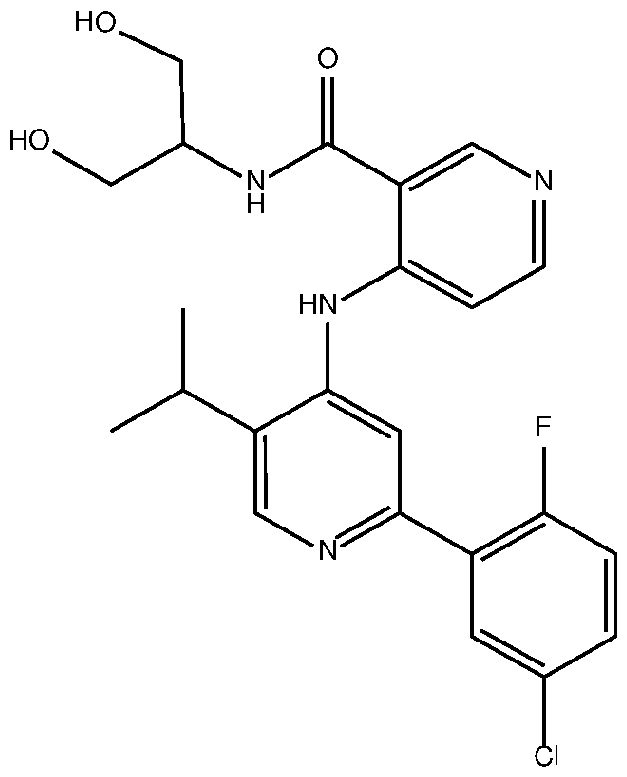 or a pharmaceutically acceptable salt thereof; and a 4-1 BB agonist, wherein the combination is effective in treating cancer. In a preferred embodiment of the foregoing uses, the 4-1 BB agonist is selected from the group consisting of utomilumab (PF- 05082566), 1 D8, SEIor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No. HB-1 1248, 5F4, C65-485, urelumab (BMS-663513), 20H4.9- lgG-1 (BMS-663031 ), 4E9, BMS-554271 , BMS-469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR-7480, MOR-7480.1 , MOR-7480.2, MOR-7483, MOR-7483.1 , MOR- 7483.2, 3EI, 53A2, 1 D8, and 3B8, or combinations thereof. In a preferred embodiment of this aspect, the 4-1 BB agonist is utomilumab.
or a pharmaceutically acceptable salt thereof; and a 4-1 BB agonist, wherein the combination is effective in treating cancer. In a preferred embodiment of the foregoing uses, the 4-1 BB agonist is selected from the group consisting of utomilumab (PF- 05082566), 1 D8, SEIor, 4B4, H4-1 BB-M127, BBK2, 145501 , antibody produced by cell line deposited as ATCC No. HB-1 1248, 5F4, C65-485, urelumab (BMS-663513), 20H4.9- lgG-1 (BMS-663031 ), 4E9, BMS-554271 , BMS-469492, 3H3, BMS- 469497, MOR-6032, MOR-7361 , MOR-7480, MOR-7480.1 , MOR-7480.2, MOR-7483, MOR-7483.1 , MOR- 7483.2, 3EI, 53A2, 1 D8, and 3B8, or combinations thereof. In a preferred embodiment of this aspect, the 4-1 BB agonist is utomilumab.
In one preferred aspect, the invention provides use of a combination comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide (PF-06952229), having the structure:
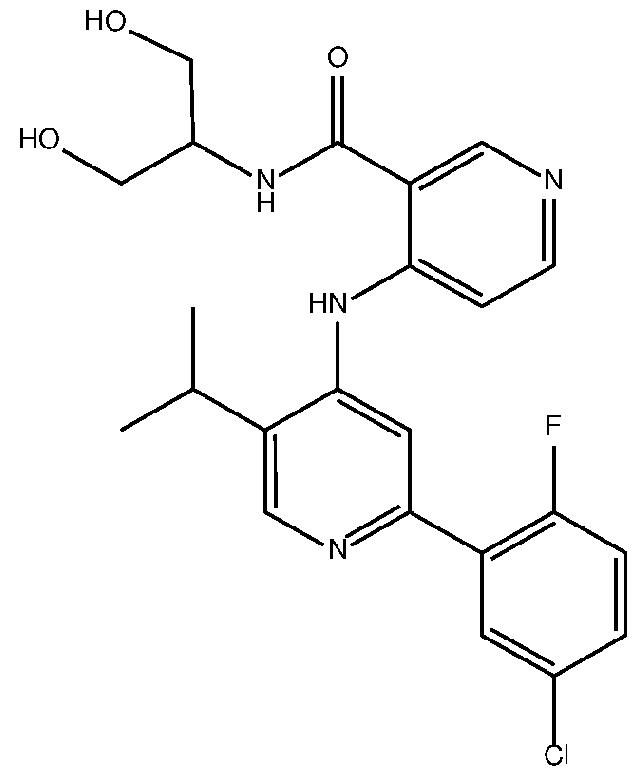 or a pharmaceutically acceptable salt thereof; and CTLA4 antagonist, wherein the combination is effective in treating cancer. In a preferred embodiment of the foregoing uses, the CTLA4 antagonist is an anti-CTLA4 antibody. In some such embodiments of this aspect, the anti-CTLA4 antibody is ipilimumab (10DI), tremelimumab or AGEN-1884, or combinations thereof. In some such embodiment, the anti-CTLA4 antibody is ipilimumab (1 GDI).
or a pharmaceutically acceptable salt thereof; and CTLA4 antagonist, wherein the combination is effective in treating cancer. In a preferred embodiment of the foregoing uses, the CTLA4 antagonist is an anti-CTLA4 antibody. In some such embodiments of this aspect, the anti-CTLA4 antibody is ipilimumab (10DI), tremelimumab or AGEN-1884, or combinations thereof. In some such embodiment, the anti-CTLA4 antibody is ipilimumab (1 GDI).
In a preferred embodiment of the foregoing, the invention provides use of 4-(2-(5- chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof in combination with immune checkpoint inhibitor, for treating cancer, wherein 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof is administered in combination with an additional anti-cancer agent. In some such embodiments, use of 4-(2-(5-chloro-2- fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and the additional anti-cancer agent together are effective in treating cancer. In some embodiments of the foregoing, the immune checkpoint inhibitor is a PD-1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, or and/or CTLA4 antagonist.
In some embodiments wherein the cancer is prostate cancer, the additional anticancer agent is a further immune checkpoint inhibitor, an anti-tumor agent, an antiandrogen and/or anti-angiogenic agent. In some such embodiments, the anti-tumor agent is mitotic inhibitors, alkylating agents, anti-metabolites, intercalating antibiotics, growth factor inhibitors, radiation, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, antibodies, cytotoxics or anti-hormones. In some such embodiments, the anti-androgen inhibitor is selected from the group consisting of enzalutamide (Xtandi®), apalutamide (ERLEADA®), darolutamide (NUBEQA®), bicalutamide (CASODEX®) and flutamide (Eulexin®). In some such embodiments, the anti-angiogenic agent is Fumagillin, which is known as 2,4,6,8-decatetraenedioic acid; mono[3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-butenyl)oxi-ranyl]-1 - oxaspiro[2.5]oct-6-yl]ester, (2E,4E,6E,8E)-(9CI); Shikonin, which is also known as 1 ,4- naphthalenedione, 5,8-dihydroxy-2-[(1 R)-1 -hydroxy-4-methyl-3-pentenyl]-(9CI); Tranilast, which is also known as benzoic acid, 2-[[3-(3,4-dimethoxyphenyl)-1 -oxo-2- propenyl]amino]-(9CI); ursolic acid; suramin; thalidomide or lenalidomide (REVLIMID).
The invention further provides methods and uses comprising a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient in combination with an additional anticancer agent, as further described below.
In one preferred aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating cancer. In a preferred embodiment the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in treating cancer, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
In one preferred aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating lung cancer. In some embodiments of this aspect, the lung cancer is small cell lung cancer (SCLC). In some such embodiments the SCLC is RB-negative. In some embodiments of this aspect, the lung cancer is non-small cell lung cancer (NSCLC). In some embodiments of this aspect, the lung cancer is advanced or metastatic lung cancer. In some such embodiments, the lung cancer is advanced or metastatic SCLC. In some such embodiments, the lung cancer is advanced or metastatic NSCLC.
In one preferred aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a
pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating ovarian cancer, peritoneal cancer, or fallopian tube cancer.
In a preferred embodiment, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating ovarian cancer. In some such embodiments, the ovarian cancer is epithelial ovarian cancer (EOC). In some such embodiments, the ovarian cancer is advanced or metastatic ovarian cancer (including EOC). In some such embodiments, the ovarian cancer is platinum resistant ovarian cancer (including EOC). In some such embodiments, the ovarian cancer is platinum resistant advanced or metastatic ovarian cancer (including EOC).
In a preferred embodiment the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating peritoneal cancer. In some such embodiments, the peritoneal cancer is primary peritoneal carcinomatosis (PPC). In a preferred embodiment, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient, , and an immune checkpoint inhibitor, for use in treating the fallopian tube cancer (FTC).
In one preferred aspect, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3- dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, for use in treating breast cancer. In some embodiments, the breast cancer is HR+/HER2- breast cancer. In some such embodiments, the breast cancer is HR+/HER2- advanced or metastatic breast cancer. In some embodiments, the breast cancer is triple negative breast cancer (TNBC). In some such embodiments, the TNBC is locally recurrent, advanced or metastatic TNBC. In some embodiments of the foregoing, the breast cancer is HR+/HER2- breast cancer or TNBC, which may be advanced or metastatic, and the subject is a woman of any menopausal status or a man.
In a preferred embodiment of the foregoing, the invention provides a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for use in treating cancer, wherein the pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient is administered in combination with an additional anti-cancer agent.
In some such embodiments, the pharmaceutical composition comprising 4-(2-(5- chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, the immune checkpoint inhibitor, and the additional anti-cancer agent together are effective in treating cancer. In some preferred embodiments wherein the cancer is breast cancer, the additional anti-cancer agent is an endocrine therapeutic agent. In some such embodiments, the endocrine therapeutic agent is an aromatase inhibitor, a selective estrogen receptor degrader (SERD), or a selective estrogen receptor modulator (SERM). In some such embodiments, the endocrine therapeutic agent is letrozole. In other such embodiments, the endocrine therapeutic agent is fulvestrant.
In one preferred aspect, the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating cancer. In a preferred embodiment, the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating cancer, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer,
endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
In one preferred aspect, the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating lung cancer. In some embodiments of this aspect, the lung cancer is small cell lung cancer (SCLC). In some such embodiments the SCLC is RB-negative. In some embodiments of this aspect, the lung cancer is non-small cell lung cancer (NSCLC). In some embodiments of this aspect, the lung cancer is advanced or metastatic lung cancer. In some such embodiments, the lung cancer is advanced or metastatic SCLC. In some such embodiments, the lung cancer is advanced or metastatic NSCLC.
In one preferred aspect, the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating ovarian cancer, peritoneal cancer, or fallopian tube cancer. In a preferred embodiment, the invention provides use of a pharmaceutical composition comprising 4- (2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2- yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient for treating ovarian cancer. In some such embodiments, the ovarian cancer is epithelial ovarian cancer (EOC). In some such embodiments, the ovarian cancer is advanced or metastatic ovarian cancer (including EOC). In some such embodiments, the ovarian cancer is platinum resistant ovarian cancer (including EOC). In some such embodiments, the ovarian cancer is platinum resistant advanced or metastatic ovarian cancer (including EOC). In a preferred embodiment, the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)- 5-isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating peritoneal cancer. In some such embodiments, the peritoneal cancer is primary peritoneal carcinomatosis (PPC). In a preferred embodiment, the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(1 ,3-
dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating the fallopian tube cancer (FTC).
In one preferred aspect, the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating breast cancer. In some embodiments, the breast cancer is HR+/HER2- breast cancer. In some such embodiments, the breast cancer is HR+/HER2- advanced or metastatic breast cancer. In some embodiments, the breast cancer is triple negative breast cancer (TNBC). In some such embodiments, the TNBC is locally recurrent, advanced or metastatic TNBC. In some embodiments of the foregoing, the breast cancer is HR+/HER2- breast cancer or TNBC, which may be advanced or metastatic, and the subject is a woman of any menopausal status or a man.
In a preferred embodiment of the foregoing, the invention provides use of a pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5- isopropylpyridin-4-ylamino)-N-(1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient, and an immune checkpoint inhibitor, for treating cancer, wherein the pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof; and a pharmaceutically acceptable excipient is administered in combination with an additional anti-cancer agent. In some such embodiments, use of the pharmaceutical composition comprising 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N- (1 ,3-dihydroxypropan-2-yl)nicotinamide, or a pharmaceutically acceptable salt thereof, an immune checkpoint inhibitor, and a pharmaceutically acceptable excipient and the additional anti-cancer agent together are effective in treating cancer. In some embodiments wherein the cancer is breast cancer, the additional anti-cancer agent is an endocrine therapeutic agent. In some such embodiments, the endocrine therapeutic agent is an aromatase inhibitor, a selective estrogen receptor degrader (SERD), or a selective estrogen receptor modulator (SERM). In some such embodiments, the endocrine therapeutic agent is letrozole. In other such embodiments, the endocrine therapeutic agent is fulvestrant.
In some embodiments of the foregoing, the invention provides further comprises treating the subject with chemotherapy, surgery and/or radiation therapy.
Any of the methods provided may be used to treat cancer at various stages. By way of example, the cancer stage includes but is not limited to early, advanced, locally advanced, remission, refractory, reoccurred after remission and progressive.
Dosage Forms and Regimens
Each therapeutic agent of the methods and combination therapies of the present invention may be administered in a medicament (also referred to herein as a pharmaceutical composition) which comprises the therapeutic agent and one or more pharmaceutically acceptable carriers, excipients, or diluents, according to pharmaceutical practice.
As used herein, the term "excipient" means the substances used to formulate active pharmaceutical ingredients (API) into pharmaceutical formulations. Excipients (e.g., mannitol, Captisol®, lactose, starch, magnesium stearate, sodium saccharine, talcum, cellulose, sodium crosscarmellose, glucose, gelatin, sucrose, magnesium carbonate, and the like) are an intergral part of pharmaceutical development and help to achieve the desired product profile including but not limited to an aid in manufacturing, modify a drug's stability, and efficacy. Acceptable excipients are non-toxic and do not adversely affect the therapeutic benefit of at least one chemical entity described herein. Such excipient may be any solid, liquid, semi-solid or, in the case of an aerosol composition, gaseous excipient that is generally available to one of skill in the art.
Further the term "excipient' encompasses solubilizing agents, stabilizers, carriers, diluents, bulking agents, pH buffering agents, tonicifying agents, antimicrobial agents, wetting agents, and emulsifying agents e.g., sodium acetate, sodium citrate, cyclodextrine derivatives, sorbitan monolaurate, triethanolamine acetate, triethanolamine oleate, and the like). Preferably, excipients are approved for or considered to be safe for human and animal administration. Generally, depending on the intended mode of administration, the pharmaceutical composition will contain about 0.005% to 95%; in certain embodiments, about 0.5% to 50% by weight of a chemical entity. Actual methods of preparing such dosage forms are known, or will be apparent, to those skilled in this art; for example, see Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pennsylvania.
As used here in, "lyophilization", "lyophilized," and "freeze-dried" refers to a process by which the material to be dried is first frozen and then the ice or frozen solvent is removed by sublimation in a vacuum environment. The term "lyophilized powder" or "lyophilized preparation" refers to any solid material obtained by lyophilization, i.e., freeze-drying of an aqueous solution. The aqueous solution may contain non-aqueous solvents, i.e., a solution composed of aqueous and one or more non-aqueous solvent(s). Preferably, a lyophilized preparation is one in which the solid material is obtained by freeze-drying a solution composed of water as a pharmaceutically acceptable excipient.
As used herein, the term "co-administration" refers to administration of two or more agents to the same subject during a treatment period.
As used herein, the terms “combination” or “combination therapy” refer to the administration of each therapeutic agent of the combination therapy of the invention, in the form of a pharmaceutical composition or medicament, either sequentially, simultaneously, or concurrently.
As used herein, the term “sequential” or “sequentially” refers to the administration of each therapeutic agent of the combination therapy of the invention, either alone or in a medicament, one after the other, wherein each therapeutic agent can be administered in any order. Sequential administration may be particularly useful when the therapeutic agents in the combination therapy are in different dosage forms, for example, one agent is a tablet and another agent is a sterile liquid, and/or the agents are administered according to different dosing schedules, for example, one agent is administered daily, and the second agent is administered less frequently such as weekly.
As used herein, the term “administered simultaneously,” simultaneously,” “simultaneous” or “simultaneous administration” means that the administration of the first therapeutic agent and that of a second therapeutic agent overlap in time with each other. The term “simultaneous” further refers to the administration of each therapeutic agent of the combination therapy of the invention in the same medicament.
As used herein, the term “concurrently” refers to the administration of each therapeutic agent in the combination therapy of the invention, either alone or in separate medicaments, wherein the second therapeutic agent is administered immediately after the first therapeutic agent, but that the therapeutic agents can be administered in any order. In a preferred embodiment the therapeutic agents are administered concurrently.
The two or more therapeutic agents may be encompassed in a single formulation and thus be administered simultaneously. Alternatively, the two or more therapeutic agents may be in separate physical formulations and administered separately, either sequentially, simultaneously, or concurrentlyto the subject.
As will be understood by those skilled in the art, the combination therapy may be usefully administered to a subject during different stages of their treatment.
In some embodiments of each of the methods, combinations and uses herein, the combination therapy is administered to a subject who is previously untreated, i.e. is treatment naive.
In some embodiments of each of the methods, combinations and uses herein, the combination therapy is administered to a subject who has failed to achieve a sustained response after a prior therapy with a biotherapeutic or chemotherapeutic agent, i.e. is treatment experienced.
In some embodiments of each of the methods, combinations and uses herein, the combination therapy may be administered prior to of following surgery to remove a tumor and I or may be used prior to, during or after radiation therapy, and / or may be used prior to, during or after chemotherapy.
In some embodiments of each of the methods, combinations and uses herein, the invention relates to neoadjuvant therapy, adjuvant therapy, first-line therapy, second-line therapy, or third-line or later therapy, in each case for treating cancer as further described herein. In each of the foregoing embodiments, the cancer may be localized, advanced or metastatic, and the intervention may occur at point along the disease continuum (i.e., at any stage of the cancer).
The efficacy of combinations described herein in certain tumors may be enhanced by combination with other approved or experimental cancer therapies, e.g., radiation, surgery, chemotherapeutic agents, targeted therapies, agents that inhibit other signaling pathways that are dysregulated in tumors, and other immune enhancing agents, such as PD-1 axis binding antagonists, 0X40 agonists, 4-1 BB agonists, and/or CTLA4 antagonists. The methods, combinations and uses of the current invention may further comprise one or more additional anti-cancer agents.
Dosage regimens may be adjusted to provide the optimum desired response. For example, a therapeutic agent of the combination therapy of the present invention may be
administered as a single bolus, as several divided doses administered over time, or the dose may be proportionally reduced or increased as indicated by the exigencies of the therapeutic situation. It may be particularly advantageous to formulate a therapeutic agent in a dosage unit form for ease of administration and uniformity of dosage. Dosage unit form, as used herein, refers to physically discrete units suited as unitary dosages for the mammalian subjects to be treated; each unit containing a predetermined quantity of active compound calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier. The specification for the dosage unit forms of the invention may be dictated by and directly dependent on (a) the unique characteristics of the chemotherapeutic agent and the particular therapeutic or prophylactic effect to be achieved, and (b) the limitations inherent in the art of compounding such an active compound for the treatment of sensitivity in individuals.
Thus, the skilled artisan would appreciate, based upon the disclosure provided herein, that the dose and dosing regimen is adjusted in accordance with methods well- known in the therapeutic arts. That is, the maximum tolerable dose may be readily established, and the effective amount providing a detectable therapeutic benefit to a subject may also be determined, as can the temporal requirements for administering each agent to provide a detectable therapeutic benefit to the subject. Accordingly, while certain dose and administration regimens are exemplified herein, these examples in no way limit the dose and administration regimen that may be provided to a subject in practicing the present invention.
It is to be noted that dosage values may vary with the type and severity of the condition to be alleviated and may include single or multiple doses. It is to be further understood that for any particular subject, specific dosage regimens should be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the compositions, taking into consideration factors such as the severity of the disorder or condition, the rate of administration, the disposition of the compound and the discretion of the prescribing physician. The dosage ranges set forth herein are exemplary only and are not intended to limit the scope or practice of the claimed composition. For example, doses may be adjusted based on pharmacokinetic or pharmacodynamic parameters, which may include clinical effects such as toxic effects and/or laboratory values. Thus, the present invention encompasses intra-patient dose-escalation as determined by the skilled artisan.
Determining appropriate dosages and regimens for administration of the chemotherapeutic agent are well-known in the relevant art and would be understood to be encompassed by the skilled artisan once provided the teachings disclosed herein.
In some embodiments, at least one of the therapeutic agents in the combination therapy is administered using the same dosage regimen (dose, frequency and duration of treatment) that is typically employed when the agent is used as a monotherapy for treating the same cancer. In other embodiments, the subject received a lower total amount of at least one of the therapeutic agents in the combination therapy than when the same agent is used as a monotherapy, for example a lower dose of therapeutic agent, a reduced frequency of dosing and / or a shorter duration of dosing.
An effective dosage of a small molecule inhibitor is typically in the range of from about 0.001 to about 100 mg per kg body weight per day, preferably about 1 to about 35 mg/kg/day, in single or divided doses. For a 70 kg human, this would amount to about 0.01 to about 7 g/day, preferably about 0.02 to about 2.5 g/day, and more preferably from about 0.02 to about 1 .0 g/day. In some instances, dosage levels at the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several small doses for administration throughout the day. The dosage may be administered as a single dose (QD), or optionally may be subdivided into smaller doses, suitable for BID (twice daily), TID (three times daily) or QID (four times daily) administration.
An effective amount of the TGF[3r inhibitor, a PD-1 axis binding antagonist, an 0X40 agonist, a 4-1 BB agonist, or a CTLA4 antagonist may be administered for prevention or treatment of disease. The appropriate dosage of the TGFpr inhibitor, the PD-1 axis binding antagonist, the 0X40 agonist, the 4-1 BB agonist, and/or the CTLA4 antagonist may be determined based on the type of disease to be treated, the TGFpr inhibitor, the PD-1 axis binding antagonist, the 0X40 agonist, the 4-1 BB agonist, and/or the CTLA4 antagonist, the severity and course of the disease, the clinical condition of the subject, the subject's clinical history and response to the treatment, and the discretion of the attending physician. In some embodiments, combination treatment with the TGF r inhibitor, the PD-1 axis binding antagonist (e.g., anti- PD-1 antibody), the 0X40 agonist (e.g., anti-human 0X40 agonist antibody), the 4-1 BB agonist (e.g., anti-human 4-1 BB agonist antibody), and/or the CTLA4 antagonist (anti-CTLA4 antibody) are synergistic,
whereby an efficacious dose of the TGF[3r inhibitor, the PD-1 axis binding antagonist, the 0X40 agonist, the 4-1 BB agonist, and/or the CTLA4 antagonist in the combination is reduced relative to efficacious dose of the each of the TGFpr inhibitor, the PD-1 axis binding antagonist, the 0X40 agonist, the 4-1 BB agonist, and/or the CTLA4 antagonist as a single agent. Dosage units for a PD-1 axis binding antagonist (e.g., pembrolizumab, nivolumab) may be expressed as a flat dose, i.e., 100 mg, 200 mg, 300 mg, or as a patient-specific dose, i.e., mg/kg (mg therapeutic agent/kg of body weight) or mg/m2 (quantity in milligrams per square meter of body surface area).
Some embodiments that employ an antibody, antibody fragment or fusion soluble receptor as the PD-1 axis binding antagonist in the combination therapy, may comprise administering the antibody at a dose of about 0.5, 1 , 2, 3, 5 or 10 mg/kg at intervals of about 7 days (± 2 days) or 14 days (± 2 days) or about 21 days (± 2 days) or about 30 days (± 2 days) throughout the course of treatment. Alternately, in some embodiments that employ an antibody, antibody fragment or fusion soluble receptor as the PD-1 axis binding antagonist in the combination therapy, the dosing regimen will comprise administering the antibody a dose of from about 0.005 mg/kg to about 10 mg/kg, with intra-patient dose escalation. In other escalating dose embodiments, the interval between doses will be progressively shortened, e.g., about 30 days (± 2 days) between the first and second dose, about 14 days (± 2 days) between the second and third doses. In certain embodiments, the dosing interval will be about 14 days (± 2 days), for doses subsequent to the second dose. In certain embodiments, the dosing interval will be about 7 days (± 2 days), for doses subsequent to the second dose.
In certain embodiments, a subject will be administered an intravenous (IV) infusion of a medicament comprising any of the PD-1 axis binding antagonists described herein.
In one embodiment of the invention, the PD-1 axis binding antagonist in the combination therapy is sasanlimab (PF-6801591 ), nivolumab or pembrolizumab, which is administered intravenously or in a liquid dosage form at a dose selected from the group consisting of any one of : 1 mg/kg Q2W, 2 mg/kg Q2W, 3 mg/kg Q2W, 5 mg/kg Q2W, 10 mg Q2W, 1 mg/kg Q3W, 2 mg/kg Q3W, 3 mg/kg Q3W, 5 mg/kg Q3W, and 10 mg Q3W.
The optimal dose for a PD-1 axis binding antagonist in combination with a TGFprl inhibitor may be identified by dose escalation of one or both of these agents. The TGF[3r1 inhibitor may be administered orally (PO), either once daily (QD) or twice daily (BID), with
or without food on a continuous schedule starting on Cycle 1 Day 1 . A PD-1 axis binding antagonist may be administered as a 30-minute to 1 -hr intravenous (IV) infusion every 2 weeks (Q2W), every 3 weeks (Q3W) or in case of dose reduction, every 4 weeks (Q4W), starting on Cycle 1 Day 1 , except in the case of TGF[3r1 inhibitor lead-in. On the day of TGFprl inhibitor administration, the TGFprl inhibitor may be given prior to or after administration of the PD-1 axis binding antagonist. In another embodiment, an TGFprl inhibitor can be administered at25 mg, 50 mg, 75 mg, 100 mg, 125 mg, 150 mg, 200 mg, 250 mg, 375 mg, 500 mg, 625 mg, 1000 mg, 1200 mg, 1500 mg or 2000 mg on a BID or QD schedule, and the PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg, or 5 mg/kg or 10 mg/kg, at a dosing interval of Q2W, Q3W or alternately Q4W In one embodiment, the TGF rl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg BID or QD for a 3-week lead-in period and then the PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg Q3W or 200 mg Q3W after the lead-in period. In another embodiment, the TGFprl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg BID or QD and the PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg Q4W. In another embodiment, the TGFprl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg BID or QD and PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg Q3W. In another embodiment, the TGFprl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg BID or QD and the PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg Q4W. In another embodiment, the TGFprl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg QD and sasanlimab (PF-6801591 ) is administered at a starting dose of 2 mg/kg Q3W. In another embodiment, the TGFprl inhibitor is administered at 25 mg, 50 mg, 75 mg or 100 mg QD and the PD-1 axis binding antagonist is administered at a starting dose of 2 mg/kg Q4W. In some embodiments, the patient is treated with a 3-week lead-in period of single-agent TGFprl inhibitor directly preceding the combination administration of the TGFprl inhibitor and the PD-1 axis binding antagonist.
In some embodiments, a treatment cycle begins with the first day of combination treatment and last for 3 weeks. In such embodiments, the combination therapy is preferably administered for at least 18 weeks (6 cycles of treatment), more preferably at least 24 weeks (8 cycles of treatment), and even more preferably at least 2 weeks after the patient achieves a CR.
In a preferred embodiment, the 4-1 BB agonist in the combination therapy comprises an anti-4-1 BB monoclonal antibody comprising heavy chain variable region
and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 64 and SEQ ID NO: 65, respectively, and is administered in a liquid medicament at a dose selected from the group consisting of 1 mg/kg Q2W, 2 mg/kg Q2W, 3 mg/kg Q2W, 5 mg/kg Q2W, 10 mg Q2W, 1 mg/kg Q3W, 2 mg/kg Q3W, 3 mg/kg Q3W, 5 mg/kg Q3W, and 10 mg Q3W. In some embodiments, the anti-4-1 BB monoclonal antibody is administered as a liquid medicament, and the selected dose of the medicament is administered by IV infusion over a time period of about 60 minutes.
In some embodiments, the anti-4-1 BB monoclonal antibody is administered at a starting dose of about 0.6 mg/kg Q4W and a PD-1 axis binding antagonist is administered at a starting dose of 10 mg/kg Q2W, and if the starting dose combination is not tolerated by the patient, then the dose of the PD-1 axis binding antagonist is reduced to 5 mg/kg Q2W and/or the dose of the anti-4-1 BB monoclonal antibody is reduced to 0.3 mg/kg Q4W.
An effective dosage of a TGF|3r1 inhibitor, or a pharmaceutically acceptable salt thereof, is in the range of from about 0.001 to about 100 mg per kg body weight per day, preferably about 1 to about 35 mg/kg/day, in single or divided doses. For example, for a 70 kg human, this would amount to about 0.01 to about 7 g/day, preferably about 0.02 to about 2.5 g/day. In some instances, dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several small doses for administration throughout the day.
In some embodiments, the dose of TGF r1 inhibitor is increased up to a maximum dose of 250 mg BID if the patient tolerates the combination treatment at a lower total dose of TGFprl inhibitor.
In some embodiments, the TGFpd inhibitor, or a pharmaceutically acceptable salt thereof, is administered at a daily dosage of from about 50 mg to about 2000 mg per day, about 50 mg per day, about 100 mg per day, about 150 mg per day, about 200 mg per day, about 250 mg per day, about 300 mg per day, about 350 mg per day, about 400 mg per day, about 450 mg per day, about 500 mg per day, about 550 mg per day, about 600 mg per day, about 650 mg per day, about 700 mg per day, about 750 mg per day, about 800 mg per day, about 850 mg per day, about 900 mg per day, about 950 mg per day, about 1000 mg per day, about 1100 mg per day, about 1200 mg per day, about 1300 mg per day, about 1400 mg per day, or about 1500 mg per day. This dose may optionally
be sub-divided into small doses, for example a dosage of 150 mg per day could be dosed as 75 mg dose twice per day.
Dosage units for a TGF rl inhibitor (e.g., PF-06952229) may be expressed as a flat dose, i.e., 25 mg, 50 mg, 75 mg, 100 mg, 125 mg, etc. or as a patient-specific dose, i.e., mg/kg (mg therapeutic agent/kg of body weight) or mg/m2 (quantity in milligrams per square meter of body surface area).
Some embodiments may comprise administering the TGFprl inhibitor in a dose of about: 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, 70 mg, 75 mg, 80 mg, 85 mg, 90 mg, 95 mg, 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, 250 mg, or more than 250 mg, wherein the amounts can be administered once a day (q.d.), twice a day (b.i.d), three times a day (t.i.d.), four times a day (q.i.d.), or on some other dosing schedule.
Repetition of the administration or dosing regimens, or adjustment of the administration or dosing regimen may be conducted as necessary to achieve the desired treatment. A “continuous dosing schedule” as used herein is an administration or dosing regimen without dose interruptions, e.g., without days off treatment. Repetition of 21 or 28 day treatment cycles without dose interruptions between the treatment cycles is an example of a continuous dosing schedule. In an embodiment, the compounds of the combination of the present invention can be administered in a continuous dosing schedule.
In a specific embodiment, the TGFprl inhibitor is PF-06952229.
Those skilled in the art will be able to determine, according to known methods, the appropriate amount, dose or dosage of each compound, as used in the combination of the present invention, to administer to a patient, taking into account factors such as age, weight, general health, the compound administered, the route of administration, the nature and advancement of breast cancer, requiring treatment, and the presence of other medications.
In an embodiment, PF-06952229, or a pharmaceutically acceptable salt thereof, is administered at a daily dosage of about 125 mg once daily, about 100 mg once daily, about 75 mg once daily, or about 50 mg daily. In an embodiment, which is the recommended starting dose or standard clinical dose, PF-06952229, or a pharmaceutically acceptable salt thereof, is administered at a daily dosage of about 125 mg once a day. In an embodiment, PF-06952229, or a pharmaceutically acceptable salt thereof, is administered at a non-standard clinical dose. In an embodiment, a non-
standard clinical dose is a low-dose amount of PF-06952229, or a pharmaceutically acceptable salt thereof. For example, PF-06952229, or a pharmaceutically acceptable salt thereof, is administered at a dose of about 100 mg once daily, about 75 mg once daily, or about 50 mg once daily. In an embodiment, PF-06952229, or a pharmaceutically acceptable salt thereof, is administered at a dose of about 100 mg once daily. In an embodiment, PF-06952229, or a pharmaceutically acceptable salt thereof, is administered at a dose of about 75 mg once daily. In an embodiment, PF-06952229, or a pharmaceutically acceptable salt thereof, is administered at a dose of about 50 mg once daily. Dosage amounts provided herein refer to the dose of the free base form of PF-06952229, or are calculated as the free base equivalent of an administered PF- 06952229 salt form. For example, a dosage or amount of PF-06952229, such as 100 mg, 75 mg or 50 mg, refers to the free base equivalent. This dosage regimen may be adjusted to provide the optimal therapeutic response. For example, the dose may be proportionally reduced or increased as indicated by the exigencies of the therapeutic situation.
The practice of the method of this invention may be accomplished through various administration or dosing regimens. The compounds of the combination of the present invention can be administered sequentially, simultaneously, or concurrently. In an embodiment, the compounds of the combination of the present invention can be administered in a concurrent dosing regimen.
Repetition of the administration or dosing regimens may be conducted as necessary to achieve the desired reduction or diminution of cancer cells. A “continuous dosing schedule,” as used herein, is an administration or dosing regimen without dose interruptions, e.g., without days off treatment. Repetition of 21 or 28 day treatment cycles without dose interruptions between the treatment cycles is an example of a continuous dosing schedule. In an embodiment, the compounds of the combination of the present invention can be administered in a continuous dosing schedule. In an embodiment, the compounds of the combination of the present invention can be administered concurrently in a continuous dosing schedule.
Administration of combinations of the invention may be affected by any method that enables delivery of the compounds to the site of action. These methods include oral routes, intraduodenal routes, parenteral injection, topical, and rectal administration.
In some embodiments, therapeutic agents of the combination therapies of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ. Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular, subcutaneous infusion. Suitable devices for parenteral administration include needle (including micro needle) injectors, needle-free injectors and infusion techniques.
Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile nonaqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
The preparation of parenteral formulations under sterile conditions, for example, by lyophilization, may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
The solubility of therapeutic agents used in the preparation of parenteral solutions may potentially be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents.
Formulations for parenteral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release. Thus, therapeutic agents of the combination therapies of the invention may potentially be formulated as a solid, semisolid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug-coated stents and PGLA microspheres.
The therapeutic agents of the combination therapies of the invention may also potentially be administered topically to the skin or mucosa, that is, dermally or transdermally. Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibers, bandages and microemulsions. Liposomes may also be used. Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers
may be incorporated; see, for example, Finnin and Morgan, Transdermal penetration enhancers: applications, limitations, and potential, J Pharm Sci 1999, 88 (10), 955-958. Other means of topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and micro needle or needle-free (e.g., Powderject™, Bioject™, etc.) injection. The disclosures of these references are incorporated herein by reference in their entireties.
Formulations for topical administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
Therapeutic agents of the combination therapies of the invention may also potentially be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurized container, pump, spray, atomizer (preferably an atomizer using electrohydrodynamics to produce a fine mist), or nebulizer, with or without the use of a suitable propellant, such as 1 , 1 ,1 ,2- tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3-heptafluoropropane. For intranasal use, the powder may include a bioadhesive agent, for example, chitosan or cyclodextrin.
The pressurized container, pump, spray, atomizer, or nebulizer may contain a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilizing, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
Prior to use in a dry powder or suspension formulation, the compound may be micronized to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray drying.
Capsules (made, for example, from gelatin or HPMC), blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the therapeutic agent, a suitable powder base such as lactose or starch and a performance modifier such as l-leucine, mannitol, or magnesium stearate. The lactose may be
anhydrous or in the form of the monohydrate, preferably the latter. Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
A suitable solution formulation for use in an atomizer using electrohydrodynamics to produce a fine mist may contain from 1 g to 20mg of the therapeutic agent per actuation and the actuation volume may vary from 1 pL to 100pL. A typical formulation includes a therapeutic agent, propylene glycol, sterile water, ethanol and sodium chloride. Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
Suitable flavors, such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations intended for inhaled/intranasal administration.
Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, poly(DL-lactic-coglycolic acid (PGLA). Modified release formulations include delayed-, sustained-, pulsed-, controlled- , targeted and programmed release.
In the case of dry powder inhalers and aerosols, the dosage unit is determined by means of a valve which delivers a metered amount. Units in accordance with the invention are typically arranged to administer a metered dose or “puff” containing a desired mount of the therapeutic agent. The overall daily dose may be administered in a single dose or, more usually, as divided doses throughout the day.
Therapeutic agents of the combination therapies of the invention may potentially be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
Therapeutic agents of the combination therapies of the invention may also potentially be administered directly to the eye or ear, typically in the form of drops of a micronized suspension or solution in isotonic, pH-adjusted, sterile saline. Other formulations suitable for ocular and aural administration may include ointments,
biodegradable (e.g., absorbable gel sponges, collagen) and non-biodegradable (e.g., silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes. A polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride. Such formulations may also be delivered by iontophoresis.
These and other aspects of the invention, including the exemplary specific embodiments listed below, will be apparent from the teachings contained herein.
EXAMPLES
Example 1 : TGFprl Inhibitor, PF-06952229, Anti-tumor Efficacy in Combination with an anti-PD-1 Antibody
Overview:
The TGFprl inhibitor, PF-06952229 anti-tumor efficacy in combination with an anti-PD-1 antibody was studied in the syngeneic MC38 tumor model to assess efficacy on primary tumor growth and effects on survival. PF-06952229 combination with an anti- PD-1 antibody led to a significant increase in tumor growth inhibition and to significant increased survival relative to PF-06952229 or PD-1 monotherapy.
Materials and Methods:
Female C57BL/6 mice (Charles River Laboratories) bearing established MC38 tumors (mean volume ~ 100 mm3) were sorted into 9 groups (n = 10 mice/group) and dosed with vehicle (50% PEG4000 in 10 mM citric acid), rat polyclonal clone 2A3 control antibody, PF-06952229, anti-PD-1 antibody (mouse RMP1 -14 BioxCell), or the combination of PF-06952229 and anti-PD-1 antibody as described in Table 8. The MC38 syngeneic colon cancer cell line was obtained from the National Cancer Institute (NCI).
PF-06952229 was administered orally (PO) continuously twice daily (BID) at 30 mg/kg, or intermittently (7 days on/ 7 days off) at 10 or 30 mg/kg for 5 cycles. Antibodies were administered at 5 mg/kg intraperitoneally (IP) twice weekly for two weeks (Biwk x2). T umors were measured using calipers twice a week. T reatment efficacy was determined based on mean tumor volumes of animals remaining on the last day of the study (Day 63) and from the incidence and magnitude of tumor regression responses observed
during the study. A Kaplan Meier survival curve was constructed to show the percentage of animals in each group remaining in the study as a function of time. Animals were eliminated from study when tumor volume reached the 1500 mm3 as a survival endpoint. Group mean tumor volume was plotted over time (to Day 63). Prism 7.04 (GraphPad) for Windows was used for graphical presentations and statistical analyses. Statistical analyses of the differences between two groups were accomplished using the ANOVA- Dunnett test and Logrank (Mantel-Cox) Test. Tests results are reported as not significant (ns) at P > 0.05, significant (symbolized by “*”) at 0.01 < P < 0.05, very significant (“**”) at 0.001 < P < 0.01 , and extremely significant (““*”) at P < 0.001 (Table 8). Table 8
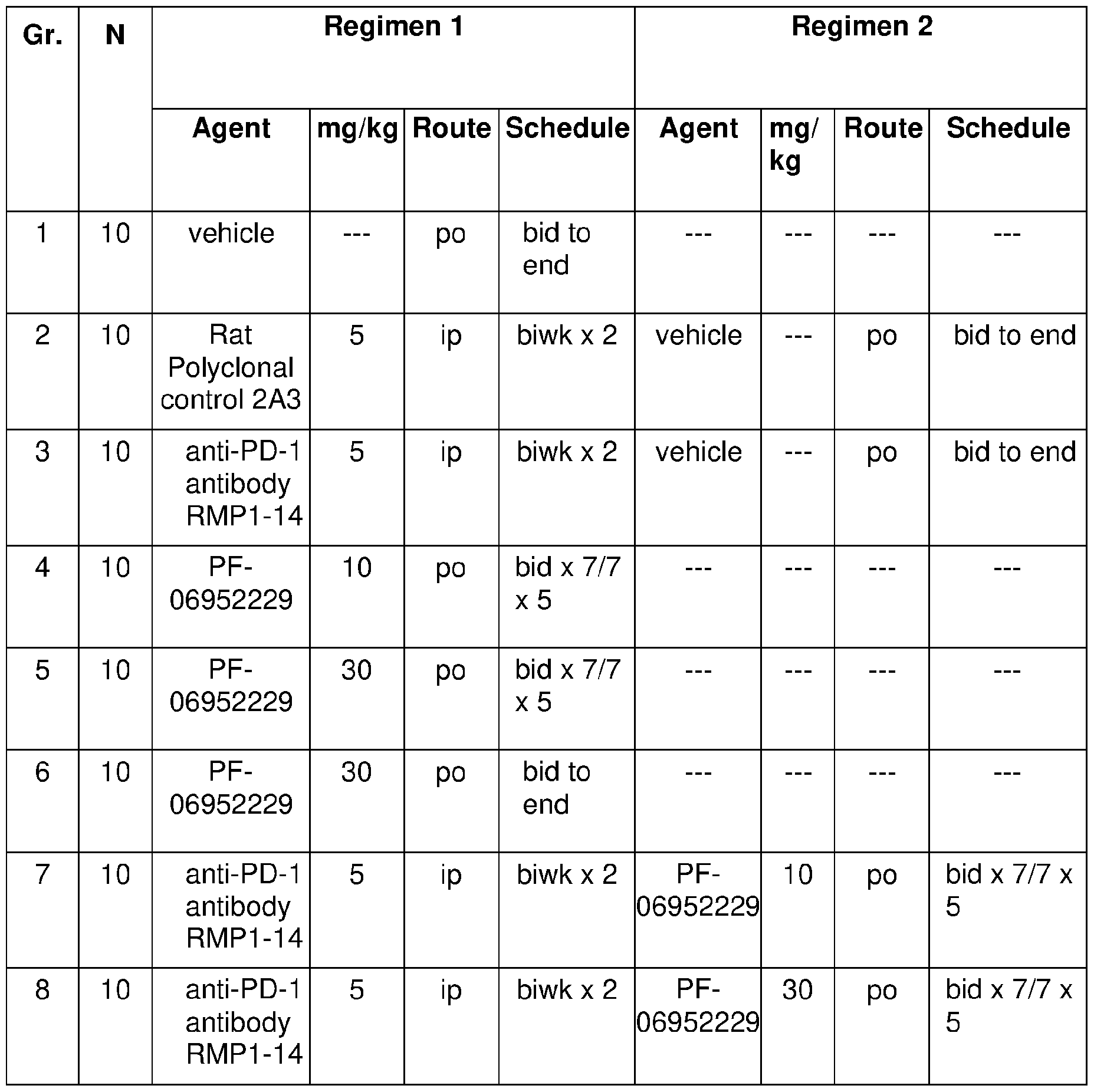

Results:
Survival and tumor growth were evaluated on Day 63 (FIG. 1). Statistical analysis indicated that all treatment groups, except PF-06952229 10 mg/kg (bid x 7/7 x 5) monotherapy, led to significant improvement of survival (Table 8) compared to vehicle and to control antibody. PF-06952229 at 30 mg/kg delivered intermittently (bid x in x 5) as a monotherapy achieved 7 complete responses (CRs), and 5 complete responses when delivered continuously (bid to end). An anti-PD-1 antibody treatment achieved 2 complete responses as a monotherapy. When anti-PD-1 antibody was combined with PF06952229 at 10 mg/kg or delivered continuously (bid to end) a significant combinatorial effect was reached compared to PF-06952229 as a single agent. The number of complete responses for these combination groups reached 6 and 9 total. Combination of anti-PD-1 antibody with PF-06952229 at 30 mg/kg delivered intermittently (bid x 7/7 x 5) resulted in 10 complete responses (CRs), with all animals surviving up to Day 63 of the study, demonstrating a combinatorial effect between these treatments.
Conclusions
TGFprl inhibitor PF-06952229 combination with an anti-PD1 antibody in the MC38 syngeneic tumor model led to greater tumor growth inhibition and improvement in survival relative to PF-06952229 monotherapy or anti-PD1 antibody monotherapies in the MC38 syngeneic mouse tumor model (Table 9 and FIG. 1A and FIG. 1 B). Table 9 shows the results of PF-06952229 single agent and in combination with anti-PD-1 antibody in syngeneic MC38 murine tumor Model.
Table 9
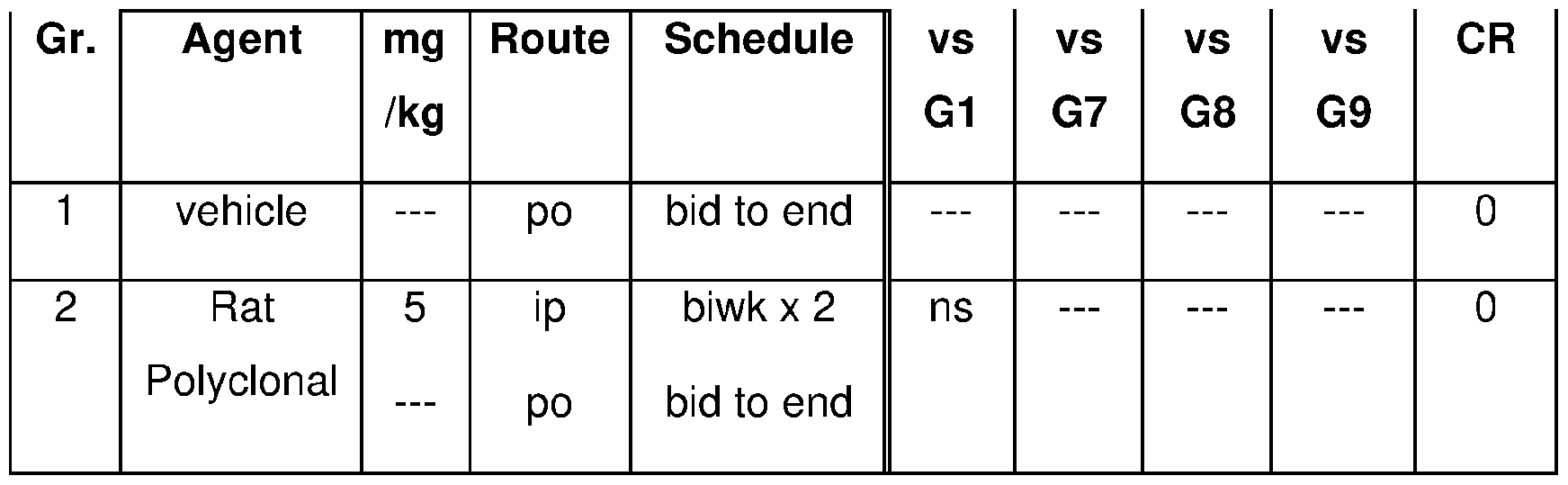

CR = Complete response; ns = Not significant; 7/7 seven days on treatment/7 days off treatment; bid: twice a day; biwk : twice a week; P > 0.05, significant (symbolized by “*”) at 0.01 < P < 0.05, very significant (“**”) at 0.001 < P < 0.01 , and extremely significant (“***”) at P < 0.001
Accordingly, the TGF r1 inhibitor (PF-06952229) combination with anti-4-1 BB antibody or with anti-OX40 antibody led to a greater efficacy than PF-06952229 monotherapy, or an anti-PD1 antibody monotherapy.
Example 2: TGFprl Inhibitor, PF-06952229, Anti-tumor Efficacy in Combination with an Anti-4-1 BB Antibody or an Anti-OX40 Antibody
Overview
The TGFprl inhibitor, PF-06952229 was evaluated in the 4T1 orthotopic syngeneic mouse tumor model in combination with an anti-4-1 BB antibody or in combination with an anti-OX40 antibody to assess effect on primary tumor growth. PF06952229 combination with 4-1 BB antibody led to a significant increase in tumor growth inhibition relative to PF-06952229 monotherapy and to 4-1 BB monotherapy treatments. PF-06952229 combination treatment with the anti-OX40 antibody led to greater tumor growth inhibition relative to PF-06952229 monotherapy and to 0X40 antibody monotherapy treatment.
Materials and Methods
Female Balb/cJ mice were obtained from Jackson Laboratories and were implanted with 0.1 x 1064T1 cells (American Tissue Culture Collection) in the mammary fat pad implanted. Tumor bearing mice were randomized into six treatment groups based on average tumor sizes of approximately 70-80 mm3 per group. Study groups included vehicle, 30 mg/kg PF-06952229, 1 mg/kg 4-1 BB antibody (MAB9371 , mouse IgG 1 , R&D Systems), 0X40 antibody (0X86, mouse lgG1 ), combination of PF-06952229 + 4-1 BB antibody and of PF-06952229 + 0X40 antibody. PF-06952229 was administered orally (po) twice daily (BID) continuously, until the end of the study (Day 25). 4-1 BB and 0X40 antibodies were administered intraperitoneally (ip) every four days, for 3 cycles (Q4Dx3). Tumor volumes were measured two times a week. Tumor volume was calculated based on two dimensional caliper measurement with cubic millimeter volume calculated using the formula (length x width2) x 0.5. Tumor growth results were plotted using GraphPad Prism 7 software. The treatment groups and dose regimen information are summarized in Table 10.
Table 10
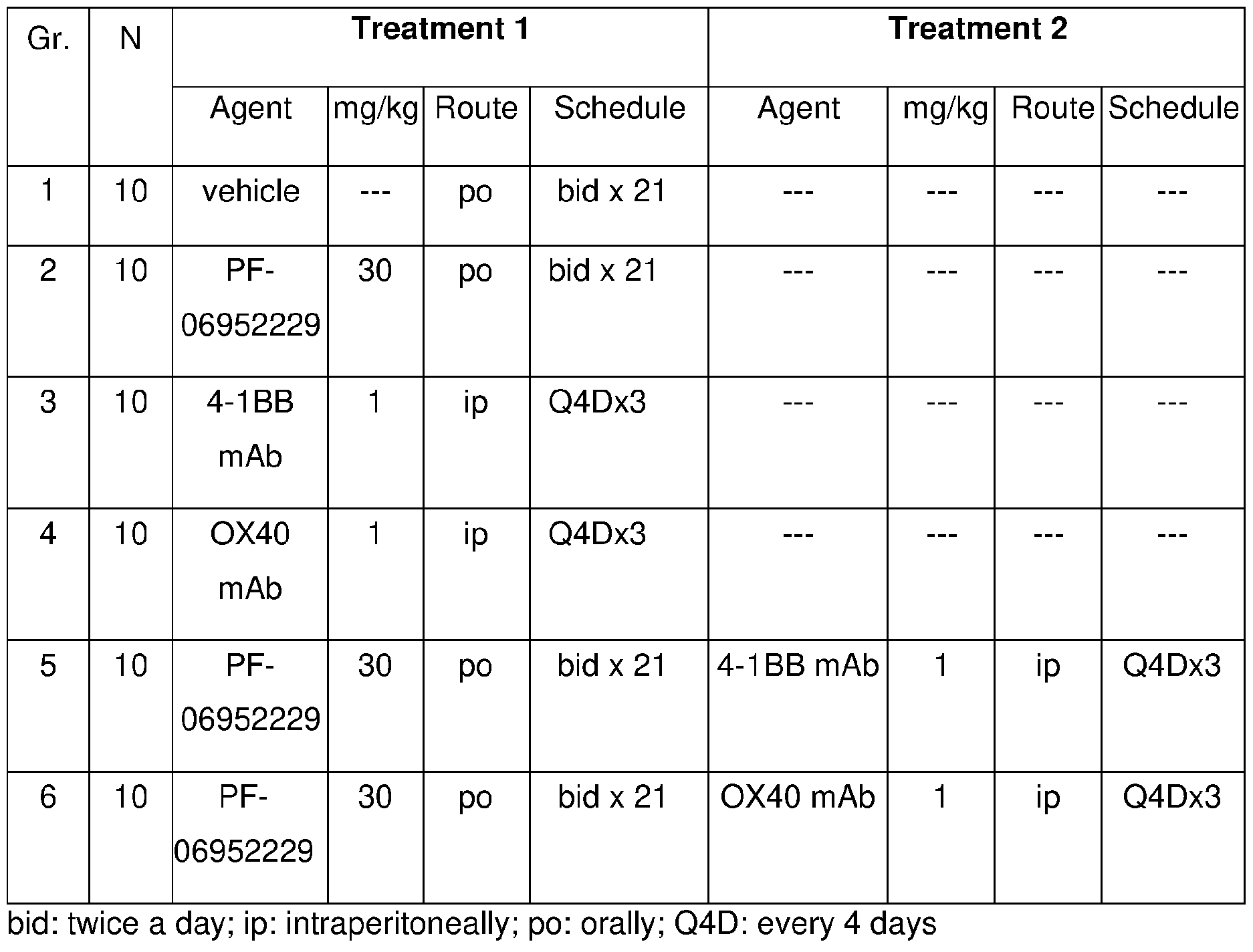
Results:
Tumor growth results on Day 25 post-treatment initiation showed that treatment with the TGF[3r1 inhibitor (PF-06952229) monotherapy, anti-4-1 BB mAb, and anti-OX40 mAb did not significantly inhibit primary tumor growth in the 4T1 syngeneic tumor model compared to vehicle-treated control group with mean tumor growth inhibition of 18%, 8%, 0.5% respectively, relative to the vehicle control group; however, PF-06952229 treatment in combination with the anti-4-1 BB antibody led to a significant decrease in tumor growth relative to PF-06952229 monotherapy or anti-4-1 BB mAB monotherapy, with mean tumor growth inhibition of 41% relative to vehicle-treated control group. A combinatorial effect was also observed when the TGF[3r1 inhibitor (PF-06952229) was combined with the anti-OX40 antibody, leading to tumor growth inhibition of 29.5% relative to the vehicle control group (FIG. 2 and Table 11 ): Table 11 shows tumor growth inhibition relative to control (Vehicle Group). Table 11

Conclusions
TGFprl inhibitor (PF-06952229) combination with anti-4-1 BB antibody or anti- 0X40 antibody led to greater tumor growth inhibition relative to PF-06952229 or antibody monotherapy treatments in the 4T 1 orthotopic syngeneic tumor model. Accordingly, the TGF rl inhibitor (PF-06952229) combination with anti-4-1 BB antibody or with anti-OX40 antibody led to a greater efficacy than PF-06952229 monotherapy, anti-4-1 BB antibody monotherapy, or anti-OX40 antibody monotherapy.
Example 3: TGFprl Inhibitor, PF-06952229, Anti-tumor Efficacy in Combination with an Anti-CTLA4 antibody
Overview
Efficacy of PF-06952229 in combination with an anti-CTLA4 antibody was evaluated in the syngeneic CT26 mouse tumor model.
Materials and Methods
Female Balb/c mice (Charles River Laboratories) were implanted with 3 x 105 CT26 cells (American Tissue Culture Collection) in the right flank on Day 0 and treatments started on Day 3. Study groups included vehicle, control antibody (hamster polyclonal IgG), 30 mg/kg PF-06952229, 5 and 2.5 mg/kg anti-CTLA4 antibody (UC10-4F10-11 , BioXcell), and combination of PF-06952229 + anti-CTLA4 antibody. PF-06952229 was administered orally (po) twice daily (BID) continuously, for 28 days. Anti-CTLA4 antibody
was administered intraperitoneally (ip) on Days 8, 11 and 14. The treatment groups and dose regimen information are summarized in Table 12.
Table 12
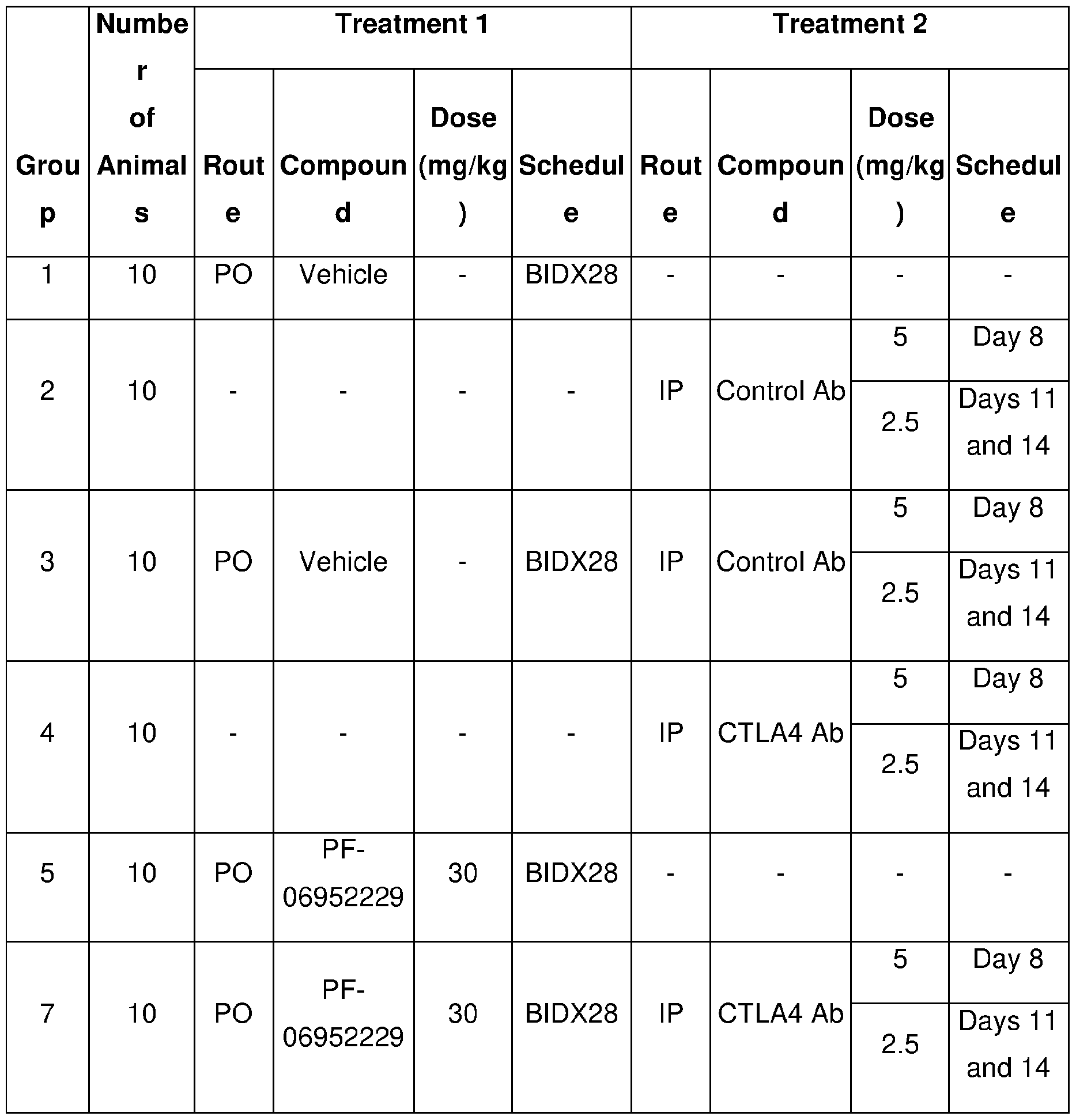 Tumor volumes were measured two times a week. T umor volume was calculated based on two-dimensional calliper measurement with cubic millimeter volume calculated using the formula (length x width2) x 0.5. Animals that reached the pre-determined study endpoint tumor volume of 1500 mm3 were removed from study. The study ended on Day 59. The time to endpoint (TTE) for analysis was calculated for each mouse by the
following equation: TTE=Log10[(endpoint volume)-b]/m, where TTE is expressed in days, endpoint volume is expressed in mm3, b is the intercept, and m is the slope of the line obtained by linear regression of a log-transformed tumor growth data set. The data set consisted of the first observation that exceeded the endpoint volume used in analysis and the three consecutive observations that immediately preceded the attainment of this endpoint volume. The calculated TTE is usually less than the tumor progression (TP) date, the day on which the animal was eliminated from study for tumor size. Animals with tumors that did not reach the endpoint volume were assigned a TTE value equal to the last day of the study. In instances in which the log-transformed calculated TTE preceded the day prior to reaching endpoint or exceeded the day of reaching tumor volume endpoint, a linear interpolation was performed to approximate TTE. Treatment outcome was evaluated from tumor growth delay (TGD), which is defined as the increase in the median time to endpoint (TTE) in a treatment group compared to the control group: %TGD = [(T - C)/C] x 100 where: T = median TTE for a treatment group, and C = median TTE for the designated control group. Tumor growth results were plotted using GraphPad Prism software. Statistical analyses were performed using the logrank test analyzed the individual TTEs for all animals in a group. For statistical analyses, two-tailed tests were conducted at significance level P = 0.05.
Tumor volumes were measured two times a week. T umor volume was calculated based on two-dimensional calliper measurement with cubic millimeter volume calculated using the formula (length x width2) x 0.5. Animals that reached the pre-determined study endpoint tumor volume of 1500 mm3 were removed from study. The study ended on Day 59. The time to endpoint (TTE) for analysis was calculated for each mouse by the
following equation: TTE=Log10[(endpoint volume)-b]/m, where TTE is expressed in days, endpoint volume is expressed in mm3, b is the intercept, and m is the slope of the line obtained by linear regression of a log-transformed tumor growth data set. The data set consisted of the first observation that exceeded the endpoint volume used in analysis and the three consecutive observations that immediately preceded the attainment of this endpoint volume. The calculated TTE is usually less than the tumor progression (TP) date, the day on which the animal was eliminated from study for tumor size. Animals with tumors that did not reach the endpoint volume were assigned a TTE value equal to the last day of the study. In instances in which the log-transformed calculated TTE preceded the day prior to reaching endpoint or exceeded the day of reaching tumor volume endpoint, a linear interpolation was performed to approximate TTE. Treatment outcome was evaluated from tumor growth delay (TGD), which is defined as the increase in the median time to endpoint (TTE) in a treatment group compared to the control group: %TGD = [(T - C)/C] x 100 where: T = median TTE for a treatment group, and C = median TTE for the designated control group. Tumor growth results were plotted using GraphPad Prism software. Statistical analyses were performed using the logrank test analyzed the individual TTEs for all animals in a group. For statistical analyses, two-tailed tests were conducted at significance level P = 0.05.
Results:
Tumor growth results on Day 60 show that treatment with the TGF[3r1 inhibitor (PF-06952229) monotherapy did not significantly inhibit primary tumor growth in the CT26 syngeneic tumor model, and treatment with anti-CTLA4 antibody as a monotherapy did show significant tumor growth delay compared to PF-06952229 monotherapy treatment. However, combination treatment of PF-0692225 with the anti-CTLA4 antibody led to a significant combinatorial effect regarding delay of tumor growth relative to PF-06952229 monotherapy (p <0.001 ) and anti-CTLA4 antibody monotherapy (p<0.05). (FIG. 3A to FIG. 3D and Table 13). Table 13 shows asummary of results from the combination study in the CT26.
Table 13

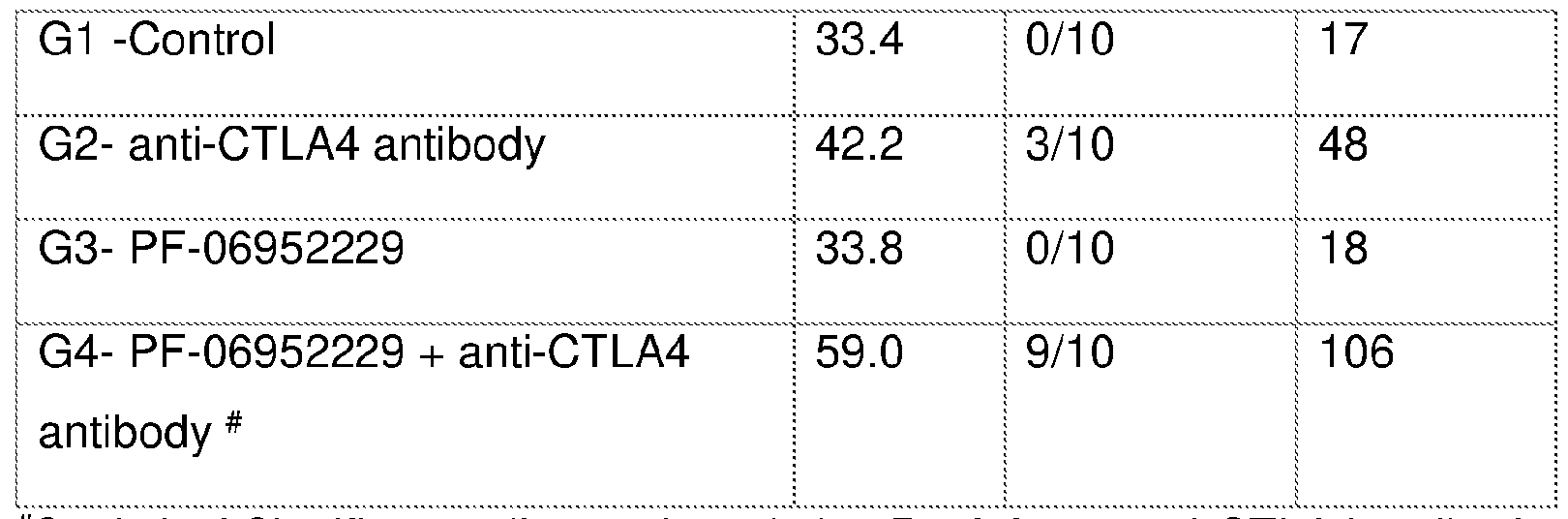
***
^Statistical Significance (Logrank test): * = P < 0.05 vs anli-CTLA4 antibody; P <
0.001 vs PF-06952229
Conclusions
TGF[3r1 inhibitor (PF-06952229) combination with the anti-CTLA4 antibody led to greater tumor growth delay relative to PF-06952229 or anti-CTLA4 antibody monotherapy treatment in the CT26 syngeneic tumor model. Accordingly, the TGF[3r1 inhibitor (PF-06952229) combination with anti-CTLA4 antibody led to a greater efficacy than PF-06952229 monotherapy, or anti-CTLA4 antibody monotherapy.
All publications and patents/patent applications cited in the specification are herein incorporated by reference in their entirety. Although the foregoing invention has been described in some detail by way of illustration and example, it will be readily apparent to those of ordinary skill in the art in light of the teachings of this invention that certain changes and modifications may be made thereto without departing from the spirit or scope of the appended claims.
Claims
We Claim:
1 . A method of treating cancer in a subject in need thereof, comprising administering to the subject:
(a) an amount of a transforming growth factor beta receptor type 1 (TGF|3r1 ) inhibitor, having the structure:
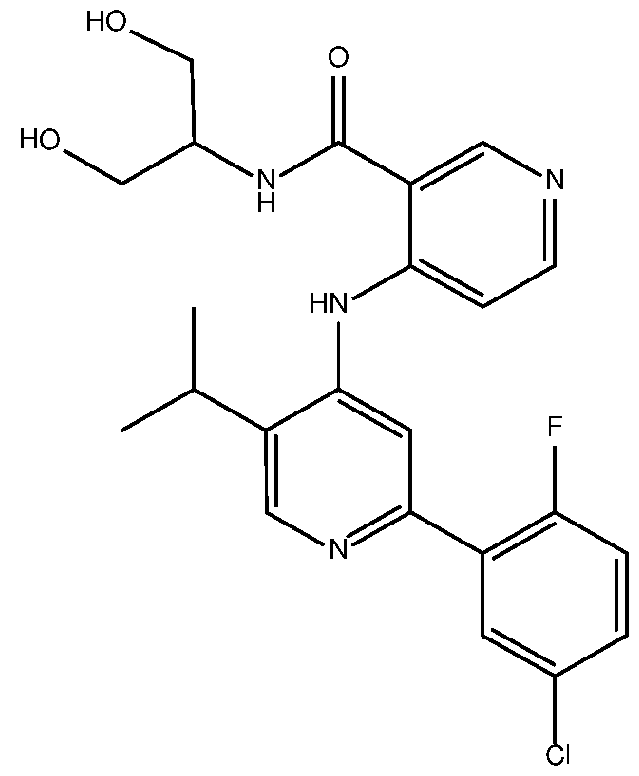 or a pharmaceutically acceptable salt thereof; and
or a pharmaceutically acceptable salt thereof; and
(b) an amount an anti-PD-1 antibody; wherein the amounts in (a) and (b) together are effective in treating cancer.
2. The method of claim 1 , wherein the anti-PD-1 antibody is selected from the group consisting of sasanlimab (PF-6801591 ), nivolumab (MDX 1106), pembrolizumab (MK-3475), pidilizumab (CT-011), cemiplimab (REGN2810), tislelizumab (BGB- A317), spartalizumab (PDR001 ), mAb15, MEDI-0680 (AMP-514), BGB-108, GLS-010 (WBP-3055), AK-103 (HX-008), CS-1003, HLX-10, MGA-012, Bl- 754091 , JS-001 (toripalimab), JNJ-63723283, genolimzumab (CBT-501 ), LZM- 009, BCD-100, camrelizumab (SHR-1210), Sym-021 , ABBV-181 , AK-105, BAT- 1306, and AGEN-2034, or combinations thereof.
3. The method of claim 2, wherein the anti-PD-1 antibody is sasanlimab (PF- 6801591).
The method of any one of claims 1 to 3, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma. A combination comprising:
(a) a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, having the structure:
 or a pharmaceutically acceptable salt thereof; and
or a pharmaceutically acceptable salt thereof; and
(b) sasanlimab (PF-6801591 ); for use in treating cancer. The combination of claim 5, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma, and metastases thereof.
147
7. A method of treating cancer in a subject in need thereof, comprising administering to the subject:
(a) an amount of a transforming growth factor beta receptor type 1 (TGF(3r1 ) inhibitor, having the structure:
 or a pharmaceutically acceptable salt thereof; and
or a pharmaceutically acceptable salt thereof; and
(b) an amount of an anti-OX40 antibody, wherein the amounts in (a) and (b) together are effective in treating cancer.
8. The method of claim 7, wherein the anti-OX40 antibody is PF-04518600. 9. The method of claim 7 or 8, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma.
10. A combination comprising:
(a) a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, having the structure:
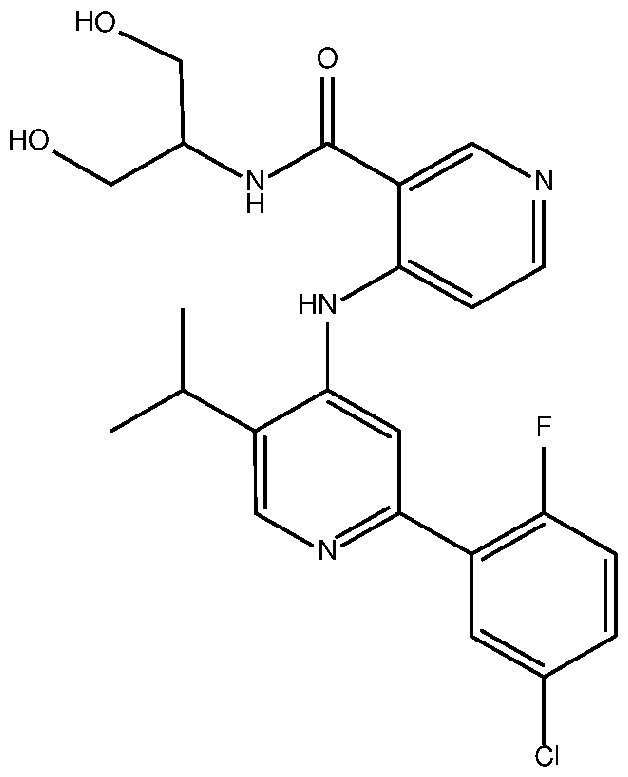 or a pharmaceutically acceptable salt thereof; and
or a pharmaceutically acceptable salt thereof; and
(b) PF-04518600, for use in treating cancer. 11. A method of treating cancer in a subject in need thereof, comprising administering to the subject:
(a) an amount of a transforming growth factor beta receptor type 1 (TGF|3r1 ) inhibitor, having the structure:
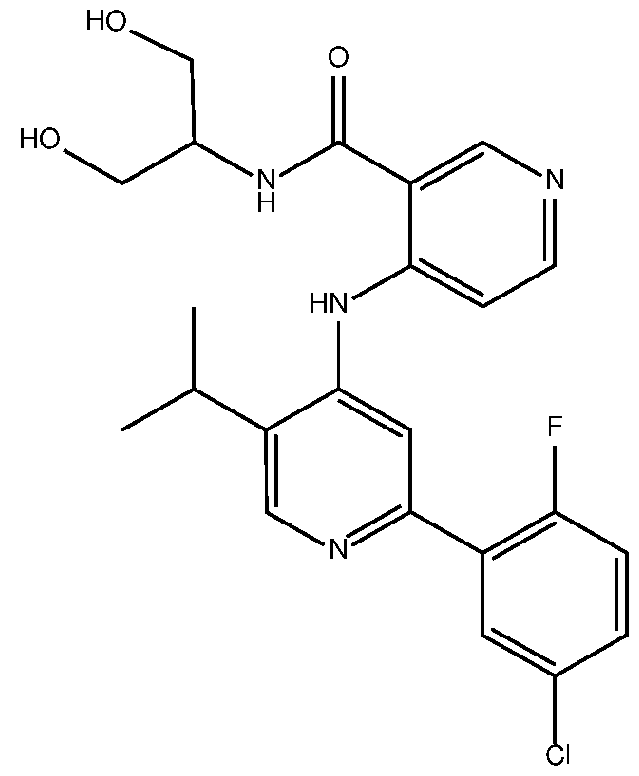 or a pharmaceutically acceptable salt thereof; and
or a pharmaceutically acceptable salt thereof; and
(b) an amount of a 4-1 BB agonist, wherein the amounts in (a) and (b) together are effective in treating cancer.
12. The method of claim 11 , wherein the 4-1 BB agonist is utomilumab.
3. The method of claim 1 1 or 12, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma. 4. A combination comprising:
(a) a transforming growth factor beta receptor type 1 (TGF rl ) inhibitor, having the structure:
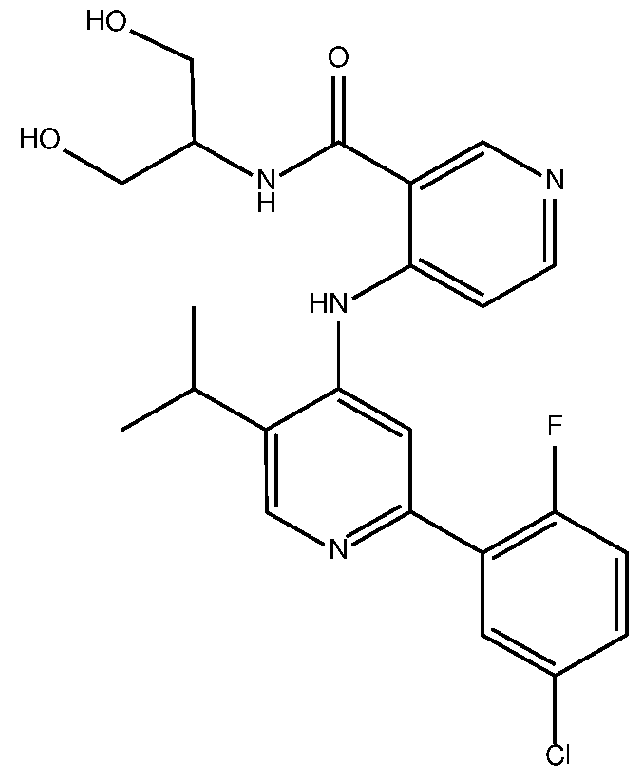 or a pharmaceutically acceptable salt thereof; and
or a pharmaceutically acceptable salt thereof; and
(b) utomilumab; for use in treating cancer. 15. A method of treating cancer in a subject in need thereof, comprising administering to the subject:
(a) an amount of a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, having the structure:
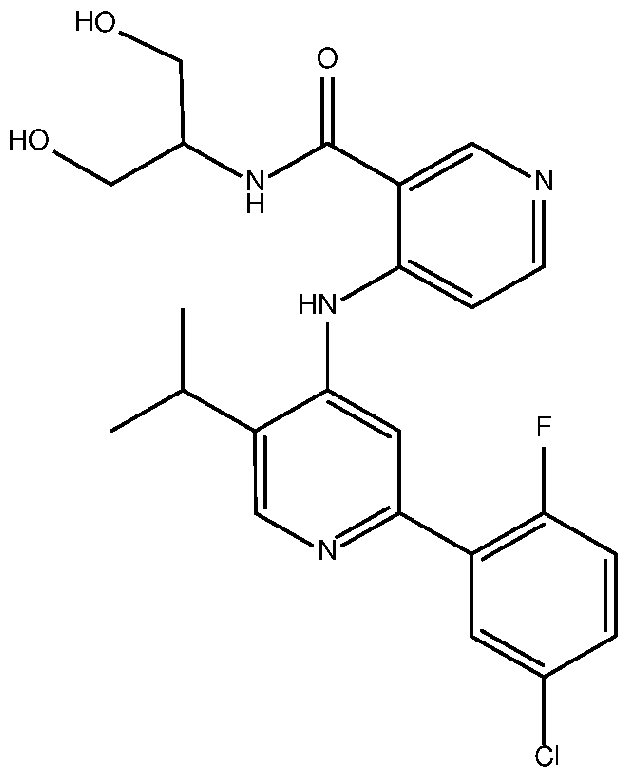
(b) an amount of a CTLA4 antagonist, wherein the amounts in (a) and (b) together are effective in treating cancer.
16. The method of claim 15, further comprising administering to the subject: (c) an amount of an additional anti-cancer agent; wherein the amounts in (a), (b) and (c) together are effective in treating cancer.
17. The method of claim 15 or 16, wherein the CTLA4 antagonist is an anti-CTLA4 antibody.
18. The method of claim 17, wherein the anti-CTLA4 antibody is ipilimumab (10DI).
19. The method of any one of claims 15 to 18, wherein the cancer is selected from the group consisting of prostate cancer, testicular cancer, colon cancer, colorectal cancer, small intestine cancer, esophageal cancer, breast cancer, lung cancer, ovarian cancer, cervical cancer, peritoneal cancer, fallopian tube cancer, bladder cancer, uterine cancer, liver cancer, pancreatic cancer, bile duct cancer, kidney cancer, stomach cancer, endometrial cancer, gallbladder cancer, glioblastoma, neuroblastoma, melanoma, and hepatocellular carcinoma.
20. A combination comprising:
(a) a transforming growth factor beta receptor type 1 (TGFprl ) inhibitor, having the structure:
151
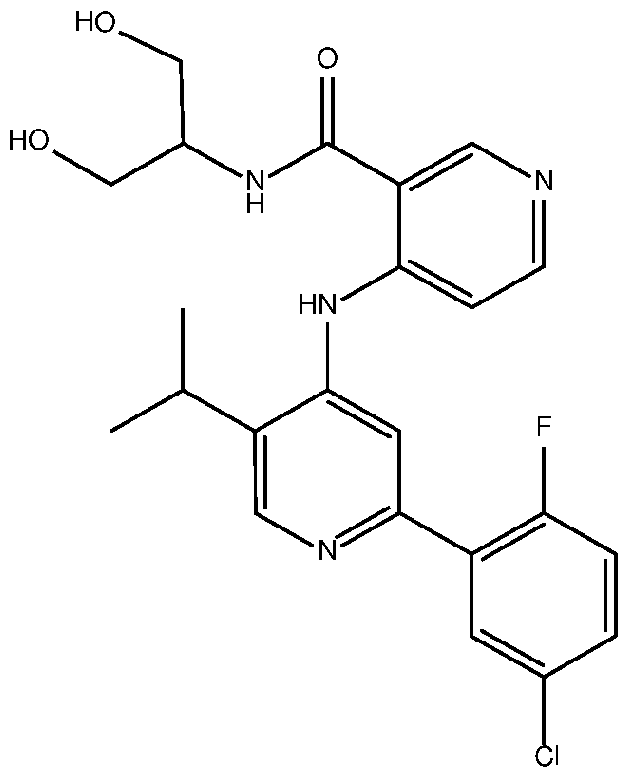 or a pharmaceutically acceptable salt thereof; and
or a pharmaceutically acceptable salt thereof; and
(b) an CTLA4 antagonist; for use in treating cancer.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202063126429P | 2020-12-16 | 2020-12-16 | |
| US63/126,429 | 2020-12-16 | ||
| US202163280862P | 2021-11-18 | 2021-11-18 | |
| US63/280,862 | 2021-11-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022130206A1 true WO2022130206A1 (en) | 2022-06-23 |
Family
ID=79018783
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2021/061713 Ceased WO2022130206A1 (en) | 2020-12-16 | 2021-12-14 | TGFβr1 INHIBITOR COMBINATION THERAPIES |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2022130206A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025193774A1 (en) * | 2024-03-12 | 2025-09-18 | The Regents Of The University Of California | Methods of identifying and treating an immune poor cancer |
Citations (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4933294A (en) | 1984-01-30 | 1990-06-12 | Icrf Patents Limited | Method of detecting truncated epidermal growth factor receptors |
| EP0404097A2 (en) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispecific and oligospecific, mono- and oligovalent receptors, production and applications thereof |
| WO1991005264A1 (en) | 1989-09-29 | 1991-04-18 | Oncogenetics Partners | Detection and quantification of neu related proteins in the biological fluids of humans |
| US5091513A (en) | 1987-05-21 | 1992-02-25 | Creative Biomolecules, Inc. | Biosynthetic antibody binding sites |
| WO1993011161A1 (en) | 1991-11-25 | 1993-06-10 | Enzon, Inc. | Multivalent antigen-binding proteins |
| US5401638A (en) | 1986-06-04 | 1995-03-28 | Oncogene Science, Inc. | Detection and quantification of neu related proteins in the biological fluids of humans |
| US5428130A (en) | 1989-02-23 | 1995-06-27 | Genentech, Inc. | Hybrid immunoglobulins |
| US5811097A (en) | 1995-07-25 | 1998-09-22 | The Regents Of The University Of California | Blockade of T lymphocyte down-regulation associated with CTLA-4 signaling |
| WO1998042752A1 (en) | 1997-03-21 | 1998-10-01 | Brigham And Women's Hospital Inc. | Immunotherapeutic ctla-4 binding peptides |
| WO1998045479A1 (en) | 1997-04-04 | 1998-10-15 | Albany Medical College | Method for assessing prostate cancer |
| US5855887A (en) | 1995-07-25 | 1999-01-05 | The Regents Of The University Of California | Blockade of lymphocyte down-regulation associated with CTLA-4 signaling |
| WO1999042585A1 (en) | 1998-02-24 | 1999-08-26 | Sisters Of Providence In Oregon | Compositions containing an ox-40 receptor binding agent or a nucleic acid encoding the same and methods for enhancing antigen-specific immune response |
| US5977318A (en) | 1991-06-27 | 1999-11-02 | Bristol Myers Squibb Company | CTLA4 receptor and uses thereof |
| US6051227A (en) | 1995-07-25 | 2000-04-18 | The Regents Of The University Of California, Office Of Technology Transfer | Blockade of T lymphocyte down-regulation associated with CTLA-4 signaling |
| WO2000037504A2 (en) | 1998-12-23 | 2000-06-29 | Pfizer Inc. | Human monoclonal antibodies to ctla-4 |
| WO2001014424A2 (en) | 1999-08-24 | 2001-03-01 | Medarex, Inc. | Human ctla-4 antibodies and their uses |
| US20020039581A1 (en) | 2000-01-27 | 2002-04-04 | Carreno Beatriz M. | Antibodies against CTLA4 and uses therefor |
| US20020086014A1 (en) | 1999-08-24 | 2002-07-04 | Korman Alan J. | Human CTLA-4 antibodies and their uses |
| WO2004004771A1 (en) | 2002-07-03 | 2004-01-15 | Ono Pharmaceutical Co., Ltd. | Immunopotentiating compositions |
| US6682736B1 (en) | 1998-12-23 | 2004-01-27 | Abgenix, Inc. | Human monoclonal antibodies to CTLA-4 |
| WO2004035607A2 (en) | 2002-10-17 | 2004-04-29 | Genmab A/S | Human monoclonal antibodies against cd20 |
| WO2004042072A2 (en) | 2002-11-01 | 2004-05-21 | The Regents Of The University Of Colorado, A Body Corporate | Quantitative analysis of protein isoforms using matrix-assisted laser desorption/ionization time of flight mass spectrometry |
| WO2004056875A1 (en) | 2002-12-23 | 2004-07-08 | Wyeth | Antibodies against pd-1 and uses therefor |
| WO2004072286A1 (en) | 2003-01-23 | 2004-08-26 | Ono Pharmaceutical Co., Ltd. | Substance specific to human pd-1 |
| WO2004092219A2 (en) | 2003-04-10 | 2004-10-28 | Protein Design Labs, Inc | Alteration of fcrn binding affinities or serum half-lives of antibodies by mutagenesis |
| US20050214860A1 (en) | 1999-01-29 | 2005-09-29 | Zhenping Zhu | Antibodies specific to KDR and uses thereof |
| US7109003B2 (en) | 1998-12-23 | 2006-09-19 | Abgenix, Inc. | Methods for expressing and recovering human monoclonal antibodies to CTLA-4 |
| WO2007067959A2 (en) | 2005-12-07 | 2007-06-14 | Medarex, Inc. | Ctla-4 antibody dosage escalation regimens |
| US7960515B2 (en) | 2007-12-14 | 2011-06-14 | Bristol-Myers Squibb Company | Binding molecules to the human OX40 receptor |
| US8008449B2 (en) | 2005-05-09 | 2011-08-30 | Medarex, Inc. | Human monoclonal antibodies to programmed death 1 (PD-1) and methods for treating cancer using anti-PD-1 antibodies alone or in combination with other immunotherapeutics |
| US20110271358A1 (en) | 2008-09-26 | 2011-11-03 | Dana-Farber Cancer Institute, Inc. | Human anti-pd-1, pd-l1, and pd-l2 antibodies and uses therefor |
| US8168757B2 (en) | 2008-03-12 | 2012-05-01 | Merck Sharp & Dohme Corp. | PD-1 binding proteins |
| US8337850B2 (en) | 2010-09-09 | 2012-12-25 | Pfizer Inc. | 4-1BB binding molecules |
| US8354509B2 (en) | 2007-06-18 | 2013-01-15 | Msd Oss B.V. | Antibodies to human programmed death receptor PD-1 |
| WO2013028231A1 (en) | 2011-08-23 | 2013-02-28 | Board Of Regents, The University Of Texas System | Anti-ox40 antibodies and methods of using the same |
| WO2013119202A1 (en) | 2012-02-06 | 2013-08-15 | Providence Health & Services - Oregon | Cancer treatment and monitoring methods using ox40 agonists |
| WO2015103355A1 (en) | 2014-01-01 | 2015-07-09 | Medivation Technologies, Inc. | Compounds and methods of use |
| US20150190506A1 (en) | 2013-12-17 | 2015-07-09 | Genentech, Inc. | Combination therapy comprising ox40 binding agonists and pd-1 axis binding antagonists |
| WO2016092419A1 (en) | 2014-12-09 | 2016-06-16 | Rinat Neuroscience Corp. | Anti-pd-1 antibodies and methods of use thereof |
| WO2020128893A1 (en) * | 2018-12-21 | 2020-06-25 | Pfizer Inc. | Combination treatments of cancer comprising a tlr agonist |
| US20200199104A1 (en) * | 2018-12-20 | 2020-06-25 | Pfizer Inc. | Novel Polymorphic Forms of a TGFBeta Inhibitor |
| WO2021123902A1 (en) * | 2019-12-20 | 2021-06-24 | Novartis Ag | Combination of anti tim-3 antibody mbg453 and anti tgf-beta antibody nis793, with or without decitabine or the anti pd-1 antibody spartalizumab, for treating myelofibrosis and myelodysplastic syndrome |
-
2021
- 2021-12-14 WO PCT/IB2021/061713 patent/WO2022130206A1/en not_active Ceased
Patent Citations (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4933294A (en) | 1984-01-30 | 1990-06-12 | Icrf Patents Limited | Method of detecting truncated epidermal growth factor receptors |
| US5401638A (en) | 1986-06-04 | 1995-03-28 | Oncogene Science, Inc. | Detection and quantification of neu related proteins in the biological fluids of humans |
| US5091513A (en) | 1987-05-21 | 1992-02-25 | Creative Biomolecules, Inc. | Biosynthetic antibody binding sites |
| US5428130A (en) | 1989-02-23 | 1995-06-27 | Genentech, Inc. | Hybrid immunoglobulins |
| EP0404097A2 (en) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispecific and oligospecific, mono- and oligovalent receptors, production and applications thereof |
| WO1991005264A1 (en) | 1989-09-29 | 1991-04-18 | Oncogenetics Partners | Detection and quantification of neu related proteins in the biological fluids of humans |
| US5977318A (en) | 1991-06-27 | 1999-11-02 | Bristol Myers Squibb Company | CTLA4 receptor and uses thereof |
| WO1993011161A1 (en) | 1991-11-25 | 1993-06-10 | Enzon, Inc. | Multivalent antigen-binding proteins |
| US5811097A (en) | 1995-07-25 | 1998-09-22 | The Regents Of The University Of California | Blockade of T lymphocyte down-regulation associated with CTLA-4 signaling |
| US6051227A (en) | 1995-07-25 | 2000-04-18 | The Regents Of The University Of California, Office Of Technology Transfer | Blockade of T lymphocyte down-regulation associated with CTLA-4 signaling |
| US5855887A (en) | 1995-07-25 | 1999-01-05 | The Regents Of The University Of California | Blockade of lymphocyte down-regulation associated with CTLA-4 signaling |
| WO1998042752A1 (en) | 1997-03-21 | 1998-10-01 | Brigham And Women's Hospital Inc. | Immunotherapeutic ctla-4 binding peptides |
| US6207156B1 (en) | 1997-03-21 | 2001-03-27 | Brigham And Women's Hospital, Inc. | Specific antibodies and antibody fragments |
| WO1998045479A1 (en) | 1997-04-04 | 1998-10-15 | Albany Medical College | Method for assessing prostate cancer |
| WO1999042585A1 (en) | 1998-02-24 | 1999-08-26 | Sisters Of Providence In Oregon | Compositions containing an ox-40 receptor binding agent or a nucleic acid encoding the same and methods for enhancing antigen-specific immune response |
| WO2000037504A2 (en) | 1998-12-23 | 2000-06-29 | Pfizer Inc. | Human monoclonal antibodies to ctla-4 |
| US7132281B2 (en) | 1998-12-23 | 2006-11-07 | Amgen Fremont Inc. | Methods and host cells for producing human monoclonal antibodies to CTLA-4 |
| US7109003B2 (en) | 1998-12-23 | 2006-09-19 | Abgenix, Inc. | Methods for expressing and recovering human monoclonal antibodies to CTLA-4 |
| US6682736B1 (en) | 1998-12-23 | 2004-01-27 | Abgenix, Inc. | Human monoclonal antibodies to CTLA-4 |
| US20050214860A1 (en) | 1999-01-29 | 2005-09-29 | Zhenping Zhu | Antibodies specific to KDR and uses thereof |
| WO2001014424A2 (en) | 1999-08-24 | 2001-03-01 | Medarex, Inc. | Human ctla-4 antibodies and their uses |
| US20050201994A1 (en) | 1999-08-24 | 2005-09-15 | Medarex, Inc. | Human CTLA-4 antibodies and their uses |
| US20020086014A1 (en) | 1999-08-24 | 2002-07-04 | Korman Alan J. | Human CTLA-4 antibodies and their uses |
| US6984720B1 (en) | 1999-08-24 | 2006-01-10 | Medarex, Inc. | Human CTLA-4 antibodies |
| EP1212422B1 (en) | 1999-08-24 | 2007-02-21 | Medarex, Inc. | Human ctla-4 antibodies and their uses |
| US20020039581A1 (en) | 2000-01-27 | 2002-04-04 | Carreno Beatriz M. | Antibodies against CTLA4 and uses therefor |
| WO2004004771A1 (en) | 2002-07-03 | 2004-01-15 | Ono Pharmaceutical Co., Ltd. | Immunopotentiating compositions |
| WO2004035607A2 (en) | 2002-10-17 | 2004-04-29 | Genmab A/S | Human monoclonal antibodies against cd20 |
| WO2004042072A2 (en) | 2002-11-01 | 2004-05-21 | The Regents Of The University Of Colorado, A Body Corporate | Quantitative analysis of protein isoforms using matrix-assisted laser desorption/ionization time of flight mass spectrometry |
| WO2004056875A1 (en) | 2002-12-23 | 2004-07-08 | Wyeth | Antibodies against pd-1 and uses therefor |
| US7521051B2 (en) | 2002-12-23 | 2009-04-21 | Medimmune Limited | Methods of upmodulating adaptive immune response using anti-PD-1 antibodies |
| US7488802B2 (en) | 2002-12-23 | 2009-02-10 | Wyeth | Antibodies against PD-1 |
| WO2004072286A1 (en) | 2003-01-23 | 2004-08-26 | Ono Pharmaceutical Co., Ltd. | Substance specific to human pd-1 |
| WO2004092219A2 (en) | 2003-04-10 | 2004-10-28 | Protein Design Labs, Inc | Alteration of fcrn binding affinities or serum half-lives of antibodies by mutagenesis |
| US8008449B2 (en) | 2005-05-09 | 2011-08-30 | Medarex, Inc. | Human monoclonal antibodies to programmed death 1 (PD-1) and methods for treating cancer using anti-PD-1 antibodies alone or in combination with other immunotherapeutics |
| WO2007067959A2 (en) | 2005-12-07 | 2007-06-14 | Medarex, Inc. | Ctla-4 antibody dosage escalation regimens |
| US8354509B2 (en) | 2007-06-18 | 2013-01-15 | Msd Oss B.V. | Antibodies to human programmed death receptor PD-1 |
| US7960515B2 (en) | 2007-12-14 | 2011-06-14 | Bristol-Myers Squibb Company | Binding molecules to the human OX40 receptor |
| US8168757B2 (en) | 2008-03-12 | 2012-05-01 | Merck Sharp & Dohme Corp. | PD-1 binding proteins |
| US20110271358A1 (en) | 2008-09-26 | 2011-11-03 | Dana-Farber Cancer Institute, Inc. | Human anti-pd-1, pd-l1, and pd-l2 antibodies and uses therefor |
| US20130078240A1 (en) | 2010-09-09 | 2013-03-28 | Pfizer Inc | 4-1bb binding molecules |
| US8337850B2 (en) | 2010-09-09 | 2012-12-25 | Pfizer Inc. | 4-1BB binding molecules |
| WO2013028231A1 (en) | 2011-08-23 | 2013-02-28 | Board Of Regents, The University Of Texas System | Anti-ox40 antibodies and methods of using the same |
| WO2013119202A1 (en) | 2012-02-06 | 2013-08-15 | Providence Health & Services - Oregon | Cancer treatment and monitoring methods using ox40 agonists |
| US20150190506A1 (en) | 2013-12-17 | 2015-07-09 | Genentech, Inc. | Combination therapy comprising ox40 binding agonists and pd-1 axis binding antagonists |
| WO2015103355A1 (en) | 2014-01-01 | 2015-07-09 | Medivation Technologies, Inc. | Compounds and methods of use |
| US10030004B2 (en) | 2014-01-01 | 2018-07-24 | Medivation Technologies Llc | Compounds and methods of use |
| US20180208577A1 (en) * | 2014-01-01 | 2018-07-26 | Medivation Technologies Llc | Compounds and Methods of Use |
| WO2016092419A1 (en) | 2014-12-09 | 2016-06-16 | Rinat Neuroscience Corp. | Anti-pd-1 antibodies and methods of use thereof |
| US20200199104A1 (en) * | 2018-12-20 | 2020-06-25 | Pfizer Inc. | Novel Polymorphic Forms of a TGFBeta Inhibitor |
| WO2020128893A1 (en) * | 2018-12-21 | 2020-06-25 | Pfizer Inc. | Combination treatments of cancer comprising a tlr agonist |
| WO2021123902A1 (en) * | 2019-12-20 | 2021-06-24 | Novartis Ag | Combination of anti tim-3 antibody mbg453 and anti tgf-beta antibody nis793, with or without decitabine or the anti pd-1 antibody spartalizumab, for treating myelofibrosis and myelodysplastic syndrome |
Non-Patent Citations (66)
| Title |
|---|
| "NCBI", Database accession no. NP_054862 |
| AKALAY I ET AL.: "Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis", CANCER RES, vol. 73, no. 8, 2013, pages 2418 - 27 |
| AKALAY I, TGFΒ PATHWAYS HAVE KEY ROLES IN DISEASE PROGRESSION AND RESISTANCE TO THERAPY IN A BROAD SPECTRUM OF TUMORS, 2013 |
| ANONYMOUS: "IPILIMUMAB, Wikipedia, the free encyclopedia", 12 December 2017 (2017-12-12), pages 1 - 11, XP055826813, Retrieved from the Internet <URL:https://en.wikipedia.org/w/index.php?title=Ipilimumab&oldid=815038002> [retrieved on 20210722] * |
| BARBAS ET AL.: "In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity", PROC NAT. ACAD. SCI., vol. 91, 1994, pages 3809 - 3813 |
| CAMACHO ET AL.: "Abstract No. 2505, antibody CP-675206", J. CLIN. ONCOLOGY, vol. 22, 2004, pages 145 |
| CAPEL ET AL.: "Heterogeneity of human IgG Fc receptors", IMMUNOMETHODS, vol. 4, 1994, pages 25 - 34, XP000604919, DOI: 10.1006/immu.1994.1004 |
| CAS, no. 477202-00-9 |
| CAS, no. 745013-59-6 |
| CHEUK ATC ET AL., CANCER GENE THERAPY, vol. 11, 2004, pages 215 - 226 |
| CHOTHIA ET AL.: "Conformations of immunoglobulin hypervariable regions", NATURE, vol. 342, 1989, pages 877 - 883, XP002030586, DOI: 10.1038/342877a0 |
| CHOTHIALESK: "Canonical structures for the hypervariable regions of immunoglobulins", J. MOL. BIOL., vol. 196, 1987, pages 901 - 917 |
| CHOU, T. C. ET AL.: "Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors", ADV. ENZYME REGUL., vol. 22, 1984, pages 27 - 55, XP023796270, DOI: 10.1016/0065-2571(84)90007-4 |
| CLACKSON ET AL.: "Making antibody fragments using phage display libraries", NATURE, vol. 352, 1991, pages 624 - 628, XP002101159, DOI: 10.1038/352624a0 |
| COLAK S: "Targeting TGF-B signaling in cancer", TRENDS IN CANCER, vol. 3, no. 1, 2017, pages 56 - 71 |
| DE HAAS ET AL.: "Fcy receptors of phagocytes", J. LAB. CLIN. MED., vol. 126, 1995, pages 330 - 41 |
| DREW M. PARDOLL: "The blockade of immune checkpoints in cancer immunotherapy", NATURE REVIEWS CANCER, vol. 12, no. 4, 1 April 2012 (2012-04-01), London, pages 252 - 264, XP055415943, ISSN: 1474-175X, DOI: 10.1038/nrc3239 * |
| EISENHAUER ET AL.: "New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1", EUR J OF CANCER, vol. 45, no. 2, 2009, pages 228 - 47, XP025841550, DOI: 10.1016/j.ejca.2008.10.026 |
| FINNINMORGAN: "Transdermal penetration enhancers: applications, limitations, and potential", J PHARM SCI, vol. 88, no. 10, 1999, pages 955 - 958, XP000851879, DOI: 10.1021/js990154g |
| GHETIE ET AL.: "Increasing the serum persistence of an IgG fragment by random mutagenesis", NAT BIOTECHNOL., vol. 15, no. 7, July 1997 (1997-07-01), pages 637 - 40, XP000876642, DOI: 10.1038/nbt0797-637 |
| GHETIEWARD: "FcRn: the MHC class I-related receptor that is more than an IgG transporter", IMMUNOL. TODAY, vol. 18, no. 12, 1997, pages 592 - 8 |
| GUYER ET AL.: "Immunoglobulin binding by mouse intestinal epithelial cell receptors", J. IMMUNOL., vol. 1, no. 17, 1976, pages 587 |
| HAMERS-CASTERMAN ET AL.: "Naturally occurring antibodies devoid of light chains", NATURE, vol. 363, 1993, pages 446 - 448, XP002535892, DOI: 10.1038/363446a0 |
| HAWKINS ET AL.: "Selection of phage antibodies by binding affinity: mimicking affinity maturation", J. MOL. BIOL., vol. 226, 1992, pages 889 - 896, XP024015348, DOI: 10.1016/0022-2836(92)90639-2 |
| HEGDE PS: "The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition", CLIN CANCER RES, vol. 22, no. 8, 2016, pages 1865 - 74, XP055749543, DOI: 10.1158/1078-0432.CCR-15-1507 |
| HINTON ET AL.: "Engineered human IgG antibodies with longer serum half-lives in primates", J. BIOL. CHEM., vol. 279, no. 8, 2004, pages 6213 - 6, XP002482523, DOI: 10.1074/jbc.C300470200 |
| HOLFORD ET AL.: "Understanding the does-effect relationship: Clinical application of pharmacokinetic-pharmacodynamic models", CLIN. PHARMACOKINET, vol. 6, 1981, pages 429 - 453 |
| HOLLINGER ET AL.: "A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity", PROC. NATL. ACAD. SCI., vol. 90, 1993, pages 6444 - 6448 |
| HU ET AL.: "Minibodies are minimized antibody-like proteins comprising a scFv joined to a CH3 domain", CANCER RES., vol. 56, 1996, pages 3055 - 3061 |
| HURWITZ ET AL.: "CTLA4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma", PROC. NATL. ACAD. SCI., vol. 95, no. 17, 1998, pages 10067 - 10071, XP002133809, DOI: 10.1073/pnas.95.17.10067 |
| JACKSON ET AL.: "In vitro antibody maturation. Improvement of a high affinity, neutralizing antibody against IL-1 beta", J. IMMUNOL., vol. 154, no. 7, 1995, pages 10 - 9, XP002355265 |
| JOHNSONWU: "Antibody Engineering Methods and Protocols", METHODS IN MOLECULAR BIOLOGY, vol. 248, 2003, pages 1 - 25 |
| KABAT ET AL.: "Public Health Service", 1991, NATIONAL INSTITUTES OF HEALTH, article "Sequences of Proteins of Immunological Interest" |
| KHALIL ET AL.: "TGF-beta: from latent to active", MICROBES INFECT., vol. 1, no. 15, 1999, pages 1255 - 63 |
| KOHLER ET AL.: "Continuous cultures of fused cells secreting antibody of predefined specificity", NATURE, vol. 256, 1975, pages 495, XP037052082, DOI: 10.1038/256495a0 |
| LETTERIO ET AL.: "Regulation of immune responses by TGF-beta", ANN. REV. IMMUNOL., vol. 16, 1998, pages 137 |
| LIN XIN ET AL: "Progress in PD-1/PD-L1 pathway inhibitors: From biomacromolecules to small molecules", EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, ELSEVIER, AMSTERDAM, NL, vol. 186, 15 November 2019 (2019-11-15), XP086035817, ISSN: 0223-5234, [retrieved on 20191115], DOI: 10.1016/J.EJMECH.2019.111876 * |
| LOEWE, S. ET AL.: "Effect of combinations: mathematical basis of problem", EXP. PATHOL PHARMACOL, vol. 114, 1926, pages 313 - 326 |
| M. DAERON: "Fc RECEPTOR BIOLOGY", ANNU. REV. IMMUNOL. J, vol. 5, 1997, pages 203 - 234, XP001010547, DOI: 10.1146/annurev.immunol.15.1.203 |
| MACCALLUM ET AL.: "Antibody-antigen interactions: contact analysis and binding site topography", J. MOL. BIOL., vol. 262, 1996, pages 732 - 745, XP002242391, DOI: 10.1006/jmbi.1996.0548 |
| MAK MP: "A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition", CLIN CANCER RES, vol. 22, no. 3, 2016, pages 609 - 20, XP055731459, DOI: 10.1158/1078-0432.CCR-15-0876 |
| MAKABE ET AL.: "Thermodynamic consequences of mutations in vernier zone residues of a humanized anti-human epidermal growth factor receptor murine antibody", JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 283, 2008, pages 1156 - 1166, XP055110460, DOI: 10.1074/jbc.M706190200 |
| MAMOTSINGER-REIF: "Current Method for Quantifying Drug Synergism, Proteom", BIOINFORM, vol. 1, no. 2, 2019, pages 43 - 48 |
| MARKS ET AL.: "By-passing immunization: Building high affinity human antibodies by chain shuffling", BIOLTECHNOLOGY, vol. 10, 1992, pages 779 - 783, XP002917376, DOI: 10.1038/nbt0792-779 |
| MARKS ET AL.: "By-passing immunization: human antibodies from V-gene libraries displayed on phage", J. MOL. BIOL., vol. 222, 1991, pages 581 - 597, XP024010124, DOI: 10.1016/0022-2836(91)90498-U |
| MASSAGUE J.: "TGFbeta in Cancer", GE1, vol. 134, no. 2, 2008, pages 215 - 30 |
| MASSAGUE: "The transforming growth factor-beta family", J. ANN. REV. CELL BIOL., vol. 6, 1990, pages 597, XP002110188 |
| MOKYR ET AL.: "Realization of the therapeutic potential of CTLA-4 blockade in low-dose chemo-therapy-treated tumor-bearing mice", CANCER RES., vol. 58, 1998, pages 5301 - 5304 |
| MURALI ET AL.: "Antibody like peptidomimetics as large scale immunodetection probes", CELL MOL BIOL, vol. 49, 2003, pages 209 - 216, XP008098779 |
| NEUZILLET C ET AL.: "Targeting the TGF¡3 pathway for cancer therapy", PHARMACOL THER, vol. 147, 2015, pages 22 - 31, XP002765095, DOI: 10.1016/j.pharmthera.2014.11.001 |
| PETTY, J. ET AL.: "Survival in human colorectal cancer correlates with expression of the T-cell costimulatory molecule OX-40 (CD134", AM. J. SURG., vol. 183, 2002, pages 512 - 518, XP055361225, DOI: 10.1016/S0002-9610(02)00831-0 |
| PRESTA: "Selection, design, and engineering of therapeutic antibodies", J. ALLERGY CLIN. IMMUNOL., vol. 116, 2005, pages 731, XP005094459, DOI: 10.1016/j.jaci.2005.08.003 |
| PRINCIPE ET AL.: "TGF-B: duality of function between tumor prevention and carcinogenesis", J NATL CANCER INST, vol. 106, no. 2, 2014, pages djt369 |
| RAVETCHKINET: "Fc receptors", ANNU. REV. IMMUNOL., vol. 9, 1991, pages 457 - 92 |
| SCHIER ET AL.: "Identification of functional and structural amino-acid residues by parsimonious mutagenesis", GENE, vol. 169, 1995, pages 147 - 155, XP004042894, DOI: 10.1016/0378-1119(95)00821-7 |
| SHERIFF ET AL.: "Similarity between C2 domain jaws and immunoglobulin CDRs", NATURE STRUCT. BIOL, vol. 3, 1996, pages 733 - 736 |
| SHIELDS ET AL.: "High Resolution Mapping of the Binding Site on Human IgG1 for FcyRI, FcyRII, FcyRIII, and FcRn and Design of IgG1 Variants with Improved Binding to the FcyR", J. BIOL. CHEM., vol. 9, no. 2, 2001, pages 6591 - 6604 |
| SIAS ET AL.: "ELISA for Quantitation of the Extracellular Domain of p185HER2 in Biological Fluids", J. IMMUNOL. METHODS, vol. 132, 1990, pages 73 - 80, XP023986949, DOI: 10.1016/0022-1759(90)90400-P |
| STAHLWERMUTH: "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VCH |
| TANG ET AL.: "What is Synergy? The Saariselka Agreement Revisited", FRONT PHARMACOL., vol. 181, no. 6, pages 1 - 5 |
| TOKOYAMA ET AL.: "How do natural killer cells find self to achieve tolerance?", IMMUNITY, vol. 24, 1994, pages 249 - 257 |
| W. A. WEBER: "Assessing tumor response to therapy", J. NUCL. MED., vol. 50, no. 1, 2009 |
| WATSON ET AL.: "Molecular Biology of the Gene", 1987, pages: 224 |
| WEINBERG, A. ET AL.: "Engagement of the OX-40 receptor in vivo enhances antitumor immunity", J. IMMUNOL., vol. 164, 2000, pages 2160 - 69, XP002224132 |
| XU ET AL.: "Disruption of Early Tumor Necrosis Factor Alpha Signaling Prevents Classical Activation of Dendritic Cells in Lung-Associated Lymph Nodes and Development of Protective Immunity against Cryptococcal Infection", IMMUNITY, vol. J-3, 2000, pages 37 - 45 |
| YELTON ET AL.: "Affinity maturation of the BR96 anti-carcinoma antibody by codon-based mutagenesis", J. IMMUNOL., vol. 155, 1995, pages 1994 - 2004, XP002096491 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025193774A1 (en) * | 2024-03-12 | 2025-09-18 | The Regents Of The University Of California | Methods of identifying and treating an immune poor cancer |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20220281979A1 (en) | Anti-pd-1 antibodies for treatment of lung cancer | |
| KR102464774B1 (en) | Methods of treating skin cancer by administering a pd-1 inhibitor | |
| US20190382491A1 (en) | Treatment of cancer using a combination of an anti-pd-1 antibody and anti-cd137 | |
| KR20210006405A (en) | Inhibition of the combination of PD-1/PD-L1, TGFβ and DNA-PK for the treatment of cancer | |
| CN110494161A (en) | Combination of anti-PD-L1 antibody and DNA-PK inhibitor for the treatment of cancer | |
| CN110366562A (en) | Methods of using anti-PD-L1 antibodies and anti-androgens to treat cancer | |
| KR20210044183A (en) | Methods of treating cancer using a combination of anti-PD-1 antibody and anti-tissue factor antibody-drug conjugate | |
| JP7352582B2 (en) | Administration of SUMO activating enzyme inhibitor and anti-CD20 antibody | |
| CN113874036A (en) | Combination therapy with CDK inhibitors | |
| KR20200112904A (en) | Compositions and methods for treating cancer | |
| JP2024529451A (en) | Methods and Compositions for Treating Cancer | |
| WO2022130206A1 (en) | TGFβr1 INHIBITOR COMBINATION THERAPIES | |
| JP2022535609A (en) | Combination therapy including anti-CD19 antibody drug conjugate and PI3K inhibitor or secondary agent | |
| US20220227862A1 (en) | Treatment of b cell malignancies using afucosylated pro-apoptotic anti-cd19 antibodies in combination with anti cd20 antibodies or chemotherapeutics | |
| RU2771759C2 (en) | Antibodies against pd-1 for treatment of lung cancer | |
| TWI910495B (en) | Methods of treating skin cancer by administering a pd-1 inhibitor | |
| EA049388B1 (en) | METHODS OF TREATMENT OF BASAL CELL CARCINOMA BY ADMINISTRATION OF AN ANTIBODY OR ITS ANTIGEN-BINDING FRAGMENT THAT SPECIFICALLY BIND TO PD-1 | |
| EA045900B1 (en) | COMBINATION OF ANTI-PD-1 ANTIBODIES AND RADIATION FOR THE TREATMENT OF MALIGNANT TUMOR | |
| EA040891B1 (en) | METHODS FOR TREATMENT OR INHIBITION OF GROWTH OF SQUAT CELL SKIN CARCINOMA USING ANTIBODY AGAINST PD-1 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21830337 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 21830337 Country of ref document: EP Kind code of ref document: A1 |