WO2011098746A1 - Crystalline acid addition salts of ( 5r) -enanti0mer of pioglitazone - Google Patents
Crystalline acid addition salts of ( 5r) -enanti0mer of pioglitazone Download PDFInfo
- Publication number
- WO2011098746A1 WO2011098746A1 PCT/GB2010/050200 GB2010050200W WO2011098746A1 WO 2011098746 A1 WO2011098746 A1 WO 2011098746A1 GB 2010050200 W GB2010050200 W GB 2010050200W WO 2011098746 A1 WO2011098746 A1 WO 2011098746A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pioglitazone
- salt
- ray powder
- powder diffraction
- melting point
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCC(C=[N+])=CC=*CCOCC Chemical compound CCC(C=[N+])=CC=*CCOCC 0.000 description 1
- HYAFETHFCAUJAY-UHFFFAOYSA-N CCc1cnc(CCOc2ccc(CC(C(N3)=O)SC3=O)cc2)cc1 Chemical compound CCc1cnc(CCOc2ccc(CC(C(N3)=O)SC3=O)cc2)cc1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 1
- XLJQTZALGFITPY-UHFFFAOYSA-N CCc1cnc(CCOc2ccc(CC(C(N3O)=O)SC3=O)cc2)cc1 Chemical compound CCc1cnc(CCOc2ccc(CC(C(N3O)=O)SC3=O)cc2)cc1 XLJQTZALGFITPY-UHFFFAOYSA-N 0.000 description 1
- HYAFETHFCAUJAY-QGZVFWFLSA-N CCc1cnc(CCOc2ccc(C[C@H](C(N3)=O)SC3=O)cc2)cc1 Chemical compound CCc1cnc(CCOc2ccc(C[C@H](C(N3)=O)SC3=O)cc2)cc1 HYAFETHFCAUJAY-QGZVFWFLSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
Definitions
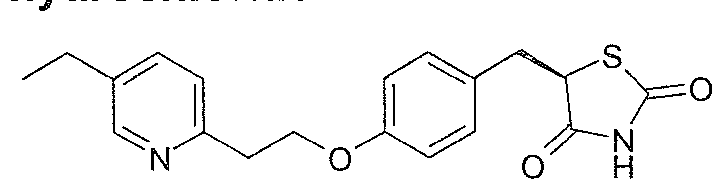
- This invention relates to salts of the substantially pure 5R enantiomer of 5- ⁇ 4-[2-(5- ethylpyridin-2-yl)ethoxy]benzyl ⁇ -1 ,3-thiazolidine-2,4-ciione (pioglitazone), and their use for treatment of inflammatory respiratory diseases by pulmonary administration by inhalation.
- Pio litazone has structural formula (I)
- Pioglitazone and rosiglitazone are members of the glitazone class of Peroxisome Proliferation Receptor gamma receptor (PPARy) agonists, which is a class of drugs which increase sensitivity to glucose in diabetic patients and have utility for the treatment of diabetes.
- PPARy agonists may also be useful for the treatment of inflammatory respiratory disorders including asthma, COPD, cystic fibrosis and pulmonary fibrosis. See WO0053601 , WO0213812 and WO0062766. These suggestions include administration by both the systemic oral and pulmonary inhalation routes.
- the 5S enantiomer of rosiglitazone has a higher binding affinity for the PPARy receptor than the 5R enantiomer (30nM vs 2 ⁇ , Parks et al., 1998, Bioorg. Med. Chem. Lett. 8(24):3657-8).
- Rivoglitazone the 5S enantiomer aiso has higher receptor binding affinity than the 5R enantiomer (see page 13 of WO2007100027).
- pioglitazone and rosiglitazone are administered for treatment of diabetes as a mixture of 5R and 5S enantiomers (a 1 :1 racemic mixture) by the oral route for systemic delivery.
- the individual enantiomers of these compounds, and members of the glitazone family generally, are known to equilibrate rapidly in vivo after oral administration (see for example J. Clin. Pharmacol. 2007, 47, 323-33; Rapid
- (Pioglitazone) contains one asymmetric carbon, and the compound is synthesized and used as the racemic mixture. The two enantiomers of pioglitazone interconvert in vivo. No differences were found in the
- the present invention provides a crystalline acid addition salt of 5R-pioglitazone, said salt being selected from the hydrochloride, hydrobromide, (-)-0,0'-dibenzoyl-L- tartrate, phosphate, hydroxy-ethanesulfonic acid and the naphthalene-1 ,5-disulfonic acid salts thereof, wherein the naphthalene-1 ,5-disulfonic acid salt contains two molecules of pioglitazone for each molecule of naphthalene-1 ,5-disulfonic acid.
- 5R-pioglitazone means the compound 5- ⁇ 4-[2-(5- ethylpyridin-2-yl)ethoxy]benzyl ⁇ -1 ,3-thiazolidine-2,4-dione, of which at least 95% by weight is the 5R enantiomer and less than 5% by weight is the 5S enantiomer.
- the 5R-pioglitazone salt of the invention preferably contains as little of the 5S enantiomer as possible.
- the 5R enantiomer may constitute at least 97%, or at least 98%, or at least 99% by weight.
- the (-)-0,0'-dibenzoyl-L-tartrate pioglitazone salt as described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 1 and/or a melting point of 153°C.
- the (-)-0,0'-dibenzoyl-L-tartrate pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 7.30, 9.45 and 17.49 degree 2-theta and/or a melting point of 153°C.
- -dibenzoyI-L-tartrate pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 7.30, 8.75, 9.45, 17.49 and 19.39 degree 2-theta and/or a melting point of 153°C.
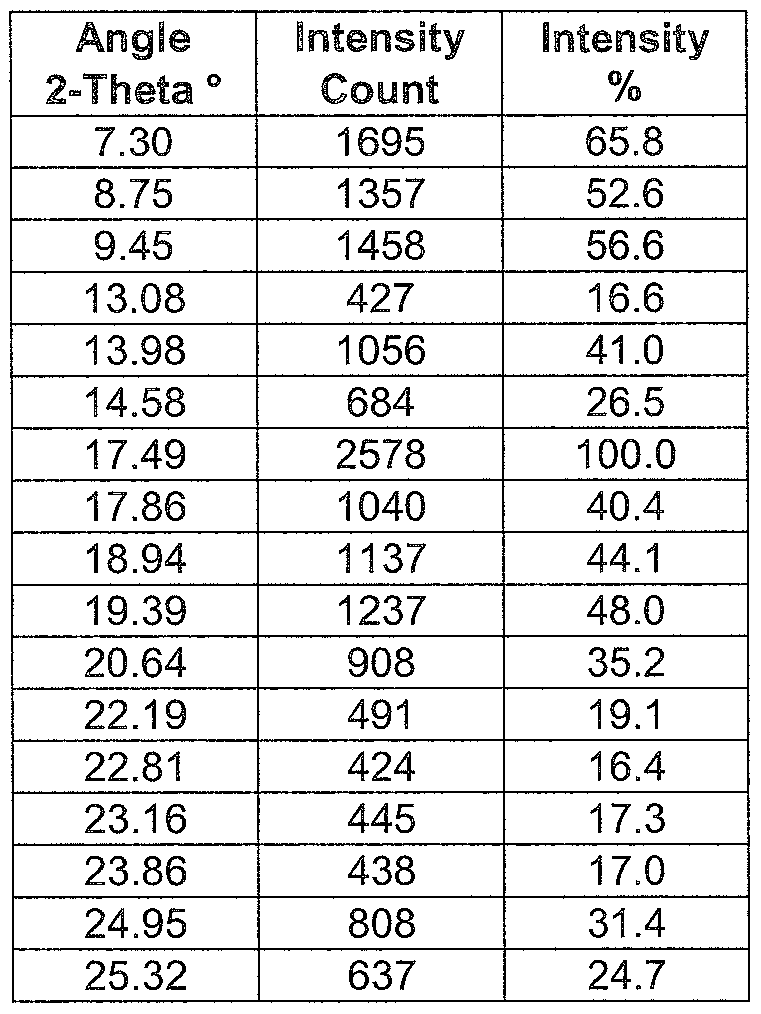
- the (-)-0,0'-dibenzoyl-L-tartrate pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 7.30, 8.75, 9.45, 13.98, 17.49, 17.86, 18.94, 19.39, 20.64 and 24.95 degree 2-theta and/or a melting point of 153°C.
- the (-)-0,0'-dibenzoyl-L-tartrate pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks expressed as an angle 2- theta as shown in the following Table 1 and/or a melting point of 153°C
- the hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 2, and/or a melting point of 201°C.
- the hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 8.84, 20.04 and 26.04 degree 2-theta and/or a melting point of 201 °C.
- the hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 8.68, 8.84, 20.04, 22.89 and 26.04 degree 2-theta and/or a melting point of 201 °C.
- the hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 8.68, 8.84, 12.82, 17.63, 20.04, 20.89, 22.89, 26.04, 26.53 and 28.17 degree 2-theta and/or a melting point of 201 °C.
- the hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks expressed as an angle 2-theta as shown in the following Table 2, and/or a melting point of 201 °C.
- the hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 3, and/or a melting point of 178°C.
- the hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks expressed as an angle 2-theta as shown in Table 3 in Example 8, and/or a melting point of 178°C.
- the hydrobromide pioglitazone salt as described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 4, and/or a melting point of 207°C.
- the hydrobromide pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 20.01 , 22.50 and 25.99 degree 2-theta, and/or a melting point of 207°C.
- the hydrobromide pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 20.01 , 22.50, 25.99, 27.26 and 28.1 1 degree 2-theta, and/or a melting point of 207°C.
- the hydrobromide pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 8.75, 18.81 , 20.01 , 21.09, 22.50, 25.99, 27.26, 28.1 1 , 29.91 and 32.00 degree 2-theta, and/or a melting point of 207°C.
- the hydrobromide pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks expressed as an angle 2-theta as shown in the following Table 4, and/or a melting point of 207°C.
- the naphthalene-1 ,5-disulfonic acid (2:1 ) pioglitazone salt as described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 5, and/or a melting point of 240°C.
- the naphthalene-1 ,5-disulfonic acid (2:1 ) pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 5.59, 16.83 and 24.05 degree 2-theta, and/or a melting point of 240°C.
- the naphthalene-1 ,5-disulfonic acid (2:1 ) pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 5.59, 12.48, 16.83, 21.50 and 24.05 degree 2-theta, and/or a melting point of 240°C.
- the naphthalene-1 , 5-disulfonic acid (2:1 ) pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks expressed as an angle 2-theta as shown in the following Table 5, and/or a melting point of 240°C.
- the phosphate pioglitazone salt described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 6, and/or a melting point of
- the phosphate pioglitazone salt described herein may have an X-ray powder diffraction pattern comprising main peaks at 16.39, 17.60 and 22.01 degree 2-theta, and/or a melting point of 177°C.
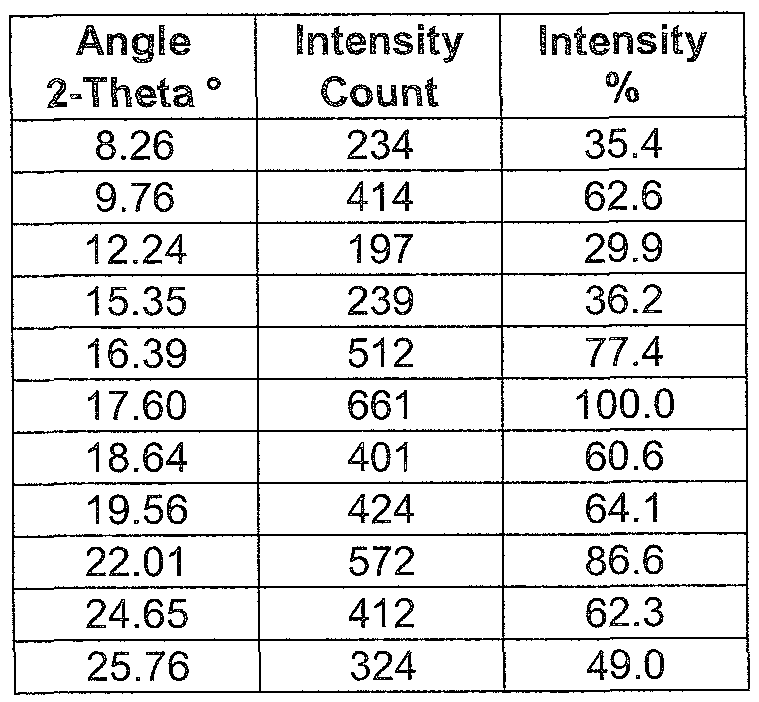
- the phosphate pioglitazone salt described herein may have an X-ray powder diffraction pattern comprising main peaks at 9.76, 16.39, 17.60, 19.56 and 22.01 degree 2-theta, and/or a melting point of 177°C.
- the phosphate pioglitazone salt described herein may have an X-ray powder diffraction pattern comprising main peaks at 8.26, 9.76, 15.39, 16.39, 17.60, 18.64, 19.56, 22.01 , 24.65 and 25.76 degree 2-theta, and/or a melting point of 177°C.
- the phosphate pioglitazone salt described herein may have an X-ray powder diffraction pattern prising main peaks at comprising main peaks expressed as an angle 2-theta as shown in the following Table 6, and/or a melting point of 177°C.
- the 2-hydroxy-ethanesulfonic acid pioglitazone salt described herein may have an X- ray powder diffraction pattern corresponding to that shown in Fig 7, and/or a melting point of 106°C.
- the 2-hydroxy-ethanesulfonic acid pioglitazone salt described herein may have an X- ray powder diffraction pattern comprising main peaks at 17.86, 20.44 and 23.05 degree 2-theta, and/or a melting point of 106°C.
- the 2-hydroxy-ethanesulfonic acid pioglitazone salt described herein may have an X- ray powder diffraction pattern comprising main peaks at 17.86, 18.41 , 20.44, 23.05 and 24.39 degree 2-theta, and/or a melting point of 106°C.
- the 2-hydroxy-ethanesulfonic acid pioglitazone salt described herein may have an X- ray powder diffraction pattern comprising main peaks at 8.85, 17.04, 17.86, 18.41 , 20.44, 23.05, 24.39, 25.21 , 26.45 and 28.05 degree 2-theta, and/or a melting point of 106°C.
- the 2-hydroxy-ethanesulfonic acid pioglitazone salt described herein may have an X- ray powder diffraction pattern comprising main peaks expressed as an angle 2-theta as shown in the following Table 7, and/or a melting point of 106°C. Table 7
- compositions comprising a pioglitazone salt as specified above, together with one or more pharmaceutically acceptable carriers and/or excipients.
- a composition which is adapted for pulmonary administration by inhalation.
- the X-ray powder diffraction patterns are obtained using Cu Ku radiation.
- the X-rays produced using Cu Ka radiation has a wavelength of 1 .5406 Angstroms.
- the X-ray powder diffraction pattern comprises main peaks expressed in terms of an angle 2-theta that have a margin of error of ⁇ 0.5 degree; more preferably a margin of error of ⁇ 0.4 degree; more preferably a margin of error of ⁇ 0.3 degree; more preferably a margin of error of ⁇ 0.2 degree; more preferably a margin of error of ⁇ 0.1 degree.
- the piogiitazone salts of the invention are useful for treatment of inflammatory respiratory disease by pulmonary administration by inhalation.
- the piogiitazone salt may be accompanied by, or administered sequentially or concurrently with, one or more respiratory disorder treatment agents useful for the purpose of preventing and treating respiratory disorders, other than a PPARy agonist.
- the inflammatory respiratory disease may be selected from, for example, mild asthma, moderate asthma, severe asthma, steroid resistant asthma, bronchitis, chronic obstructive pulmonary disease (COPD), cystic fibrosis, pulmonary edema, pulmonary embolism, pneumonia, pulmonary sarcoisosis, silicosis, pulmonary fibrosis, respiratory failure, acute respiratory distress syndrome, emphysema, chronic bronchitis, tuberculosis, and lung cancer.
- COPD chronic obstructive pulmonary disease
- compositions of the invention are useful for treatment of inflammatory respiratory disorders, for example asthma (mild, moderate or severe), e.g., bronchial, allergic, intrinsic, extrinsic, exercise-induced, drug-induced (including aspirin and NSAID- induced) and dust-induced asthma, steroid resistant asthma, bronchitis including infectious and eosinophilic bronchitis, chronic obstructive pulmonary disease
- asthma mild, moderate or severe
- bronchial allergic, intrinsic, extrinsic
- exercise-induced including aspirin and NSAID- induced
- dust-induced asthma steroid resistant asthma
- bronchitis including infectious and eosinophilic bronchitis
- COPD cystic fibrosis
- pulmonary fibrosis including cryptogenic fibrosing alveolitis, idiopathic pulmonary fibrosis, idiopathic interstitial pneumonias, fibrosis complicating anti-neoplastic therapy and chronic infection, including tuberculosis and aspergillosis and other fungal infections; complications of lung transplantation; vasculitic and thrombotic disorders of the lung vasculature, and pulmonary hypertension (including pulmonary arterial hypertension); antitussive activity including treatment of chronic cough associated with inflammatory and secretory conditions of the airways, and iatrogenic cough; acute and chronic rhinitis including rhinitis medicamentosa, and vasomotor rhinitis; perennial and seasonal allergic rhinitis including rhinitis nervosa (hay fever); nasal polyposis; acute viral infection including the common cold, and infection due to respiratory syncytial virus, influenza, coronavirus (including SARS) and adeno
- COPD chronic obstructive pulmonary disease
- COPD refers to a set of physiological symptoms including chronic bronchitis, chronic cough, expectoration, exertional dyspnea and a significant, progressive reduction in airflow that may or may not be partly reversible.
- Emphysema may also be present in the lungs.
- COPD is a disease characterized by a progressive airflow limitation caused by an abnormal inflammatory reaction to the chronic inhalation of particles.
- COPD chronic obstructive bronchitis and emphysema.
- compositions suitable for administration by inhalation via the mouth or the nose are known, and may include carriers and/or diluents that are known for use in such compositions.
- the composition may contain 0.01-99% by weight of the pioglitazone salt.
- a unit dose comprises the pioglitazone salt in an amount of 1 ⁇ g to 15+ mg.
- the most suitable dosage level may be determined by any suitable method known to one skilled in the art. It will be understood, however, that the specific amount for any particular patient will depend upon a variety of factors, including the activity of the specific compound that is used, the age, body weight, diet, general health and sex of the patient, time of administration, the route of administration, the rate of excretion, the use of any other drugs, and the severity of the disease undergoing treatment. Optimum dosages will be determined by clinical trial, as is required in the art. Compositions of the invention may be used in combination with other therapeutic agents that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which present compounds are useful.
- Such other therapeutic agents may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with the glitazone component, particularly the pioglitazone or rosiglitazone component.
- a pharmaceutical composition containing such other therapeutic agents in addition to the pioglitazone salt is preferred.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to the pioglitazone salt.
- Suitable therapeutic agents for a combination therapy with the pioglitazone compositions of the invention include: (1 ) a steroid drug such as a corticosteroid, for example beclomethasone, (e.g., as the mono or the dipropionate ester), flunisolide, fluticasone (e.g., as the propionate or furoate ester), ciclesonide, mometasone (e.g., as the furoate ester), mometasone desonide, rofleponide, hydrocortisone, prednisone, prednisolone, methyl prednisolone, naflocort, deflazacort, halopredone acetate, fluocinolone acetonide, fluocinonide, clocortolone, tipredane, prednicarbate, alclometasone dipropionate, halometasone, rimexolone, depro
- Steroid drugs can additionally include steroids in clinical or pre-clinical development for respiratory diseases such as GW-685698, GW-799943, GSK 870086, QAE397, NCX-1010, NCX-1020, NO-dexamethasone, PL-2146, NS- 126 (formerly ST- 126) and compounds referred to in international patent applications WO0212265, WO0212266, WO02100879, WO03062259, WO03048181 and WO03042229.
- respiratory diseases such as GW-685698, GW-799943, GSK 870086, QAE397, NCX-1010, NCX-1020, NO-dexamethasone, PL-2146, NS- 126 (formerly ST- 126) and compounds referred to in international patent applications WO0212265, WO0212266, WO02100879, WO03062259, WO03048181 and WO03042229.
- Steroid drugs can also additionally include next generation molecules in development with reduced side effect profiles such as selective glucocorticoid receptor agonists (SEGRAs), including ZK-216348 and compounds referred to in international patent applications WO-00032585, WO-000210143, WO-2005034939, WO-2005003098, WO-2005035518 and WO-2005035502 and functional equivalents and functional derivatives thereof; (2) a p2-adrenoreceptor agonist, such as albuterol, bambuterol, terbutaline, fenoterol, formoterol, formoterol fumarate, salmeterol, salmeterol xinafoate, arformoterol, arfomoterol tartrate, indacaterol (QAB-149), carmoterol, picumeterol, Bl 1744 CL, GSK159797, GSK59790, GSK159802, GSK642444, GSK678007, GSK96
- the invention provides for the use of inhaled administration of the pioglitazone salt of the invention in combination with other anti-inflammatory drugs and bronchodilator drug combinations (i.e. triple combination product), including but not limited to salmeterol xinafoate/fluticasone propionate (Advair/Seretide®), formoterol fumarate/budesonide (Symbicort®), formoterol fumarate/mometasone furoate, formoterol fumarate/beclometasone dipropionate (Foster®), formoterol fumarate/fluticasone propionate (FlutiForm®), Indacaterol/mometasone furoate, lndacaterol/QAE-397, GSK159797/GSK 685698, GSK159802/GSK 685698,
- the invention provides for the use of inhaled administration of the pioglitazone salt of the invention in combination with other bronchodilator drug combinations, particularly B2 agonist/M3 antagonist combinations (i.e. triple combination product), including but not limited to salmeterol xinafoate/tiotropium bromide, formoterol fumarate/tiotropium bromide, Bl 1744 CL/tiotropium bromide, indacaterol/NVA237, indacterol/QAT-370, formoterol/ LAS34273, GSK159797/GSK 573719, GSK159802/GSK 573719, GSK642444/GSK 573719, GSK159797/GSK 233705, GSK159802/GSK 233705, GSK642444/GSK 233705, and compounds which possess both ⁇ 2 agonist and M3 antagonist activity in the same molecule (dual functionality) such as GSK 961081.
- the invention provides a kit for treatment of respiratory disorders in a subject, the kit comprising one dosage form comprising a pioglitazone salt composition of the invention, and a second dosage form comprising another therapeutic agent, for example as discussed above, selected from anti-inflammatory agents, bronchodilators, mucolytic agents, antitussive agents, leukotriene
- the pioglitazone salt is preferably in the form of
- microparticles These may be prepared by a variety of techniques, including spray- drying, freeze-drying and micronisation. Following size reduction to produce microparticles, particle size distribution (PSD) of the compound is examined and generally described in the art by specifying d10, d50 and d90 values.
- PSD particle size distribution
- the average particle size i.e. the average equivalent diameter, is defined as the diameter where 50 mass-% (of the particles) of the powder have a larger equivalent diameter, and the other 50 mass-% have a smaller equivalent diameter. Hence the average particle size is denoted as equivalent d50.
- a d50 of less than 10 microns, preferably less than 5 microns is desired.
- a composition of the invention may be prepared as a suspension for delivery from a nebuliser or as an aerosol in a liquid propellant, for example for use in a pressurised metered dose inhaler (PMDI).
- PMDI pressurised metered dose inhaler
- Propellants suitable for use in a PMDI are known to the skilled person, and include CFC-12, H FA- 134a, HFA-227, HCFC-22 (CCI 2 F 2 ) and HFA-152 (CH 4 F 2 and isobutane).
- a composition of the invention is in dry powder form, for delivery using a dry powder inhaler (DPI).
- DPI dry powder inhaler
- Microparticles for delivery by inhalation may be formulated with excipients that aid delivery and release.
- microparticles may be formulated with large carrier particles that aid flow from the DPI into the lung.
- Suitable carrier particles are known, and include lactose particles; they may have a mass median aerodynamic diameter of greater than 90 ⁇ .
- Aerosol generation can be carried out using, for example, pressure-driven jet atomizers or ultrasonic atomizers, preferably using propellant-d riven metered aerosols or propellant-free administration of micronized active compounds from, for example, inhalation capsules or other "dry powder" delivery systems.
- the compositions may be dosed as described depending on the inhaler system used.
- the administration forms may additionally contain excipients, such as, for example, propellants (e.g., Frigen in the case of metered aerosols), surface-active substances, emulsifiers, stabilizers, preservatives, flavourings, fillers (e.g. lactose in the case of powder inhalers) or, if appropriate, further active compounds.
- propellants e.g., Frigen in the case of metered aerosols
- surface-active substances e.g., emulsifiers, stabilizers, preservatives, flavourings, fillers (e.g. lactose in the case of powder inhalers) or, if appropriate, further active compounds.
- propellants e.g., Frigen in the case of metered aerosols
- surface-active substances e.g., emulsifiers, stabilizers, preservatives, flavourings
- fillers e.g. lactose in the case of powder inhalers
- Glitazones can be separated using chiral HPLC.
- Chiral columns include CHIRALPAK AD, AD-H, AS-V, 50801 , IA, IC, OD, OF, OG, OJ, OK, and OZ.
- Preferred chiral columns for HPLC are CHIRALPAK AD-H and CHIRALPAK IA using elution with ethanol and varying portions of TFA, preferably 0.05-0.2% TFA.
- the isolated R-pioglitazone L-DBTA obtained in this manner has high e.e. (see Example 4 below) and is used to seed resolution reactions described below.
- chiral acid resolving agents include, without limitation, dibenzoyl tartaric acid, dianisoyl tartaric acid, ditoluyl tartaric acid, tartaric acid, phencyphos, chlocyphos, anicyphos, 1 ,1 '-binaphthyl-2,2'-diyl hydrogen phosphate,
- camphorsulfonic acid bromo camphorsulfonic acid, camphoric acid, phenylethylsulfonic acid, malic acid, mandelic acid, 4-bromomandelic acid, 4- methylmande!ic acid, lactic acid and chalcone sulfonic acids.
- a more preferred set of chiral acid resolving agents include (-)-0,0'-dibenzoyl-L- tartaric acid (anhydrous), (-)-0,0'-dibenzoyl-L-tartaric acid monohydrate, (-)-di-O-p- toluoyl-L-tartaric acid, L-(+)-tartaric acid and (-)-di-p-anisolyl-L-tartaric acid.
- the most preferred chiral acid resolving agent is (-)-0,0'-dibenzoyl-L-tartaric acid (anhydrous) with addition of seed crystals (5R)-5- ⁇ 4-[2-(5-ethylpyridin-2- yl)ethoxy]benzyl ⁇ -1 ,3-thiazolidine-2,4-dione (-)-0,0'-dibenzoyl-L-tartrate that have high e.e and are prepared as described in example 4.
- Chiral acid resolving agents can be used in different stoichiometries to effect resolutions.
- the above resolving agents can be used in a ratio of 1 part (I) to 10 parts of chiral acids (II), more preferably in a ratio of 1 part (I) to 5 parts of chiral acids (II), and most preferably in a ratio of 1 part (I) to 2 part of chiral acids (II). Even more preferred is a ratio of 1 part (I) to 0.6 part of chiral acids (II).
- solvents may be useful to form diastereomeric salts (III) with chiral acids (II).
- Preferred solvents will include ethyl acetate, dichloromethane, tetrahydrofuran, 1 ,4-dioxane, acetone, acetonitrile, MeOH, EtOH, IPA, 1 -propanol, 1 ,2- dimethoxyethane, diethyl ether, dichloroethane, ferf-butylmethyl ether, 1-butanol, 2- butanol, t-butanol, 2-butanone, toluene, cyclohexane, heptane, hexane, H2O, DMF, petroleum ethers and CHCI 3.
- the most preferred solvent is acetonitrile.
- Solvents may be used in combination with each other and preferred combinations include 10% HCI in H2O, 10% H2O in DMF, 10% H2O in EtOH, 10% H2O in IPA, methanol and water in varying portions, methanol and water in varying portions with HCI, CHCI 3 and ethyl acetate in varying portions, EtOH and water in varying portions, IPA and water in varying portions, 1 -propanol and water in varying portions and CHCI 3 and dioxane in varying portions.
- More preferred solvent combinations include CHCi 3 and dioxane in varying portions, CHCI 3 and ethyl acetate in varying portions, methanol and water in varying portions with HCI and methanol and water in varying portions.
- the product from resolution described in scheme 2 gives the R-Pio L-DBTA salt that is substantially enriched as the R enantiomer. Further enrichment to >90% e.e can be obtained by recrystallisation. Conditions for recrystallisation have surprisingly been found to require HCI in solvents, preferably MeOH and water. If only MeOH and water are used, extensive conversion to racemic pioglitazone free base occurs.
- the isolated R-pioglitazone L-DBTA obtained in this manner has high e.e. (see Example 6 below).
- Preferred solvents for conversion of R-pioglitazone L-DBTA to R-pioglitazone HCI are MeOH containing cone HCI followed by addition of EtOAc.
- the isolated R- pioglitazone HCI obtained in this manner is crystalline and has high e.e. (see Example 7 below). It has been found that only some crystallisation methods are suitable for recrystallisation of the R-pioglitazone HCI salts to give a high e.e. whilst retaining the salt.
- Preferred solvents are acetic acid-water with addition of HCI-water (see example 8), more preferably MeOH-Et 2 0 and most preferably MeOH-EtOAc.
- R-pioglitazone L-DBTA preferred solvents for conversion of R-pioglitazone L-DBTA to R- pioglitazone HBr are MeOH containing cone HBr followed by addition of EtOAc.
- the isolated R-pioglitazone HBr obtained in this manner is crystalline and has high e.e. (see Example 9 below).
- Preferred solvent for conversion of R-pioglitazone L-DBTA to R-pioglitazone naphthalene- 1 ,5-disulfonic acid is MeOH and the salt forms a complex with 2 Piogiitazone units as determined by NMR.
- the isolated R-pioglitazone naphthalene- 1 ,5-disulfonic acid obtained in this manner is crystalline and has high e.e. (see Example 10 below).
- Preferred solvent for conversion of R-pioglitazone L-DBTA to R-pioglitazone phosphate is acetone.
- the isolated R-pioglitazone phosphate obtained in this manner is crystalline and has high e.e. (see Example 1 1 below).
- Preferred solvents for conversion of R-pioglitazone L-DBTA to R-pioglitazone isethionate are I PA and isopropyi acetate.
- the isolated R-pioglitazone phosphate obtained in this manner is crystalline and has high e.e. (see Example 12 below).
- Figure 1 Illustrative X-ray powder diffraction (XRPD) pattern of (5R)-5- ⁇ 4-[2-(5- ethylpyridin-2-yl)ethoxy]benzyl ⁇ -1 ,3 thiazolidine-2,4-dione (-)-0,0'-dibenzoyI-L- tartrate (example 6);
- Figure 2 Illustrative XRPD pattern of (5R)-5- ⁇ 4-[2-(5-Ethylpyridin-2-yl)ethoxy]benzyl ⁇ - 1 ,3-thiazolidine-2,4-dione hydrochloride (example 7);
- Figure 3 Illustrative XRPD pattern of (5R)-5- ⁇ 4-[2-(5-Ethylpyridin-2-yl)ethoxy]benzyl ⁇ - 1 ,3-thiazolidine-2,4-dione hydrochloride (example 8);
- Figure 7 Illustrative XRPD pattern of (R)-5- ⁇ 4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]- benzyl ⁇ -thiazolidine-2,4-dione 2-hydroxy-ethanesulfonic acid (example 12).
- NMR spectra were obtained on a Varian Unity I nova 400 spectrometer with a 5mm inverse detection triple resonance probe operating at 400MHz or on a Bruker Avance DRX 400 spectrometer with a 5mm inverse detection triple resonance TXI probe operating at 400MHz or on a Bruker Avance DPX 300 spectrometer with a standard 5mm dual frequency probe operating at 300MHz. Shifts are given in ppm relative to
- XRPD X-ray powder diffraction
- DSC Differential Scanning Calorimetry
- CHIRALPAK AD-H 250 x 30 mm, 5pm
- EtOH +0.05%TFA - flow rate 30 ml/min.
- Detection -In-line UV detection set at 250 nM wavelength
- CHIRALPAK 1A 250 x 4.6 mm, 5 ⁇
- Detection - In-line DAD set at 280 nM wavelength
- MS ionisation method Electrospray (positive ion)
- Figure 1 provides an illustrative XRPD pattern of (5R)-5- ⁇ 4-[2-(5-Ethylpyridin-2- yl)ethoxy]benzyl ⁇ -1 ,3-thiazolidine-2,4-dione (-)-0,0'-dibenzoyl-L-tartrate.
- the main diffraction peaks and relative intensities for the peaks shown in Figure 1 are listed in Table 1 .
- Pioglitazone.L-DBTA salt from example 6 (976.75 g, 1.366mol) was split into 2 batches and dissolved in premixed MeOH (2.5 L) and concentrated HCI (148 mL) at 40-45 °C (internal temperature). The reaction was allowed to stir over ⁇ 5 min to give a clear solution and was then filtered quickly through a glass fibre filter paper into a 20 L flange top reactor flask containing seed crystals from example 3 (300 mg, e.e. 94.66%). The procedure was repeated with the other Pioglitazone.L-DBTA batch. EtOAc (7.5 L) was added to the flange top reactor containing both filtered solutions and the mixture was stirred mechanically.
- Figure 2 provides an illustrative XRPD pattern of (5R)-5- ⁇ 4-[2-(5-Ethylpyridin-2- yl)ethoxy]benzyl ⁇ -1 ,3-thiazolidine-2,4-dione hydrochloride .
- Figure 3 provides an illustrative XRPD pattern of (5R)-5- ⁇ 4-[2-(5-Ethylpyridin-2- yl)ethoxy]benzyl ⁇ -1 ,3-thiazolidine-2,4-dione hydrochloride.
- Figure 4 provides an illustrative XRPD pattern of ( R)-5- ⁇ 4-[2-( 5-Ethyl-pyrid in-2-yl )- ethoxy]-benzyl ⁇ -thiazolidine-2,4-dione hydrobromide.
- Figure 5 provides an illustrative XRPD pattern of (R)-5- ⁇ 4-[2-(5-Ethyl-pyridin-2-yl)- ethoxy]-benzyl ⁇ -thiazolidine-2,4-dione naphthalene-1 ,5-disulfonic acid (2:1 ).
- Figure 6 provides an illustrative XRPD pattern of (R)-5- ⁇ 4-[2-(5-Ethyl-pyridin-2-yl)- ethoxy]-benzyl ⁇ -thiazolidine-2,4-dione phosphate.
- Figure 7 provides an illustrative XRPD pattern of (R)-5- ⁇ 4-[2-(5-Ethyl-pyridin-2-yl)- ethoxy]-benzyl ⁇ -thiazolidine-2,4-dione 2-hydroxy-ethanesulfonic acid .
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
A crystalline acid addition salt of 5R-pioglitazone, said salt being selected from the hydrochloride, hydrobromide, (-)-O,O'-dibenzoyl-L-tartrate, phosphate, hydroxy-ethanesulfonic acid and the naphthalene-1,5-disulfonic acid (2:1 ) salts thereof, wherein the naphthalene-1,5-disulfonic acid salt contains 2 molecules of (5R)-5-{4-[2- (5-ethylpyrϊdin-2-yl)ethoxy]benzyI}-1,3-thiazoIidine-2,4-dione for each molecule of naphthalene-1,5-disulfonic acid.
Description
CRYSTALLINE ACID ADDITION SALTS OF (5R) -ENANTIOMER OF PIOGLITAZONE
This invention relates to salts of the substantially pure 5R enantiomer of 5-{4-[2-(5- ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-ciione (pioglitazone), and their use for treatment of inflammatory respiratory diseases by pulmonary administration by inhalation.
Background to the invention
Pio litazone has structural formula (I)

Another member of the glitazone class of drugs is rosiglitazone 5-(4-{2-[methyl (pyridin-2-yl)amino]ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione). It too is asymmetric at the 5-position of the thiazolidine-dione ring and has two enantiomers, the 5R and 5S enantiomers.
Pioglitazone and rosiglitazone are members of the glitazone class of Peroxisome Proliferation Receptor gamma receptor (PPARy) agonists, which is a class of drugs which increase sensitivity to glucose in diabetic patients and have utility for the treatment of diabetes. Separately from their utility in treatment of diabetes, based on observations of anti-inflammatory activity in cells relevant to the lung, it has been suggested that PPARy agonists may also be useful for the treatment of inflammatory respiratory disorders including asthma, COPD, cystic fibrosis and pulmonary fibrosis. See WO0053601 , WO0213812 and WO0062766. These suggestions include administration by both the systemic oral and pulmonary inhalation routes.
The 5S enantiomer of rosiglitazone has a higher binding affinity for the PPARy receptor than the 5R enantiomer (30nM vs 2μΜ, Parks et al., 1998, Bioorg. Med. Chem. Lett. 8(24):3657-8). For another member of the glitazone class, Rivoglitazone,
the 5S enantiomer aiso has higher receptor binding affinity than the 5R enantiomer (see page 13 of WO2007100027).
In practice, pioglitazone and rosiglitazone are administered for treatment of diabetes as a mixture of 5R and 5S enantiomers (a 1 :1 racemic mixture) by the oral route for systemic delivery. The individual enantiomers of these compounds, and members of the glitazone family generally, are known to equilibrate rapidly in vivo after oral administration (see for example J. Clin. Pharmacol. 2007, 47, 323-33; Rapid
Commun. Mass Spectrom. 2005, 19, 1 125-9; J. Chromatography, 835 (2006), 40-46; Biopharmaceutics and Drug Disposition 1997, 18 (4), 305-24; Chem. Pharm. Bull
1984, 32, (1 1 ) 4460-65; T. J. Med. Chem. 1991 , 34, 319-25) so there is no difference in practice between oral administration of either substantially pure isomer and oral administration of the racemic mixture. Specifically in relation to pioglitazone, it has been stated in a submission to the Federal Drug Administration (FDA) that there was no difference in activity following oral administration either of the racemate or the individual enantiomers in a rodent diabetes model
(www.fda.aov/medwatch/SAFETY/2007/Sep Pl/Actoplus Met Pl.pdf):
"(Pioglitazone) contains one asymmetric carbon, and the compound is synthesized and used as the racemic mixture. The two enantiomers of pioglitazone interconvert in vivo. No differences were found in the
pharmacologic activity between the two enantiomers".
Our International patent application no. PCT/GB2009/001920 presents evidence that, for treatment of inflammatory respiratory disease by inhalation, the 5R-enantiomer of a glitazone is more effective than the 5S enantiomer. As demonstrated in that application, proof of principle drives from an animal model of treatment of
inflammatory respiratory disease by inhalation, in which the SR-enantiomers of pioglitazone and rosiglitazone were shown to be active, whereas the 5S enantiomers were essentially inactive. That finding led to the conclusion that inhaled pulmonary administration of the 5R enantiomer of a glitazone, in particular the 5R-enantiomer of pioglitazone or rosiglitazone, allows the anti-inflammatory effect of the compound to be achieved more efficiently than by similar administration of the racemate, with all the concomitant reduced side effect benefits of lower systemic exposure than oral administration.
For pulmonary administration of 5R-pioglitazone, to obtain the advantages disclosed in PCT/GB2009/001920 for that 5R enantiomer over the 5S or racemic forms, it is desirable that it be available in a stable crystalline form, which is stable under the high energy input conditions of micronisation.
Description of the invention
The present invention provides a crystalline acid addition salt of 5R-pioglitazone, said salt being selected from the hydrochloride, hydrobromide, (-)-0,0'-dibenzoyl-L- tartrate, phosphate, hydroxy-ethanesulfonic acid and the naphthalene-1 ,5-disulfonic acid salts thereof, wherein the naphthalene-1 ,5-disulfonic acid salt contains two molecules of pioglitazone for each molecule of naphthalene-1 ,5-disulfonic acid.
As used herein the term "5R-pioglitazone" means the compound 5-{4-[2-(5- ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione, of which at least 95% by weight is the 5R enantiomer and less than 5% by weight is the 5S enantiomer.
The 5R-pioglitazone salt of the invention preferably contains as little of the 5S enantiomer as possible. For example, the 5R enantiomer may constitute at least 97%, or at least 98%, or at least 99% by weight.
As used herein, the term "enantiomeric excess" or its abbreviation "e.e." is defined as the percentage:
((R-S)/(R+S)) x 100 percent where R and S are the respective weight fractions of the R and S enantiomers in a sample. Thus for a glitazone sample containing 95% by weight of the 5R enantiomer and 5% of the 5S enantiomer, the enantiomeric excess of R over S enantiomer is ((95-5)/95+5)) x 100 = 90%.
The (-)-0,0'-dibenzoyl-L-tartrate pioglitazone salt as described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 1 and/or a melting point of 153°C. The (-)-0,0'-dibenzoyl-L-tartrate pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 7.30, 9.45 and 17.49 degree 2-theta and/or a melting point of 153°C.
The (-)-0,0!-dibenzoyI-L-tartrate pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 7.30, 8.75, 9.45, 17.49 and 19.39 degree 2-theta and/or a melting point of 153°C.
The (-)-0,0'-dibenzoyl-L-tartrate pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 7.30, 8.75, 9.45, 13.98, 17.49, 17.86, 18.94, 19.39, 20.64 and 24.95 degree 2-theta and/or a melting point of 153°C.
The (-)-0,0'-dibenzoyl-L-tartrate pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks expressed as an angle 2- theta as shown in the following Table 1 and/or a melting point of 153°C
Table 1
The hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 2, and/or a melting point of 201°C.
The hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 8.84, 20.04 and 26.04 degree 2-theta and/or a melting point of 201 °C.
The hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 8.68, 8.84, 20.04, 22.89 and 26.04 degree 2-theta and/or a melting point of 201 °C.
The hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 8.68, 8.84, 12.82, 17.63, 20.04, 20.89, 22.89, 26.04, 26.53 and 28.17 degree 2-theta and/or a melting point of 201 °C.
The hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks expressed as an angle 2-theta as shown in the following Table 2, and/or a melting point of 201 °C.
Table 2

The hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 3, and/or a melting point of 178°C.
The hydrochloride pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks expressed as an angle 2-theta as shown in Table 3 in Example 8, and/or a melting point of 178°C. The hydrobromide pioglitazone salt as described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 4, and/or a melting point of 207°C.
The hydrobromide pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 20.01 , 22.50 and 25.99 degree 2-theta, and/or a melting point of 207°C.
The hydrobromide pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 20.01 , 22.50, 25.99, 27.26 and 28.1 1 degree 2-theta, and/or a melting point of 207°C.
The hydrobromide pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 8.75, 18.81 , 20.01 , 21.09, 22.50, 25.99, 27.26, 28.1 1 , 29.91 and 32.00 degree 2-theta, and/or a melting point of 207°C.
The hydrobromide pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks expressed as an angle 2-theta as shown in the following Table 4, and/or a melting point of 207°C.
Table 4
Angle Intensity Intensity
2-Theta ° Count %
8.75 646 32.3
11 .85 317 15.9
12.64 329 16.5
15.80 356 17.8
17.53 480 24.0
18.81 676 33.8
20.01 1998 100.0
20.50 531 26.6
21 .09 574 28.7
22.50 1060 53.0
25.47 429 21 .5
25.99 1 180 59.1
26.45 516 25.8
27.26 830 41.5

The naphthalene-1 ,5-disulfonic acid (2:1 ) pioglitazone salt as described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 5, and/or a melting point of 240°C.
The naphthalene-1 ,5-disulfonic acid (2:1 ) pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 5.59, 16.83 and 24.05 degree 2-theta, and/or a melting point of 240°C. The naphthalene-1 ,5-disulfonic acid (2:1 ) pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks at 5.59, 12.48, 16.83, 21.50 and 24.05 degree 2-theta, and/or a melting point of 240°C.
The naphthalene-1 , 5-disulfonic acid (2:1 ) pioglitazone salt as described herein may have an X-ray powder diffraction pattern comprising main peaks expressed as an angle 2-theta as shown in the following Table 5, and/or a melting point of 240°C.
Table 5

The phosphate pioglitazone salt described herein may have an X-ray powder diffraction pattern corresponding to that shown in Fig 6, and/or a melting point of
177°C. The phosphate pioglitazone salt described herein may have an X-ray powder diffraction pattern comprising main peaks at 16.39, 17.60 and 22.01 degree 2-theta, and/or a melting point of 177°C.
The phosphate pioglitazone salt described herein may have an X-ray powder diffraction pattern comprising main peaks at 9.76, 16.39, 17.60, 19.56 and 22.01 degree 2-theta, and/or a melting point of 177°C.
The phosphate pioglitazone salt described herein may have an X-ray powder diffraction pattern comprising main peaks at 8.26, 9.76, 15.39, 16.39, 17.60, 18.64, 19.56, 22.01 , 24.65 and 25.76 degree 2-theta, and/or a melting point of 177°C.
The phosphate pioglitazone salt described herein may have an X-ray powder diffraction pattern prising main peaks at comprising main peaks expressed as an angle 2-theta as shown in the following Table 6, and/or a melting point of 177°C.
Table 6
The 2-hydroxy-ethanesulfonic acid pioglitazone salt described herein may have an X- ray powder diffraction pattern corresponding to that shown in Fig 7, and/or a melting point of 106°C. The 2-hydroxy-ethanesulfonic acid pioglitazone salt described herein may have an X- ray powder diffraction pattern comprising main peaks at 17.86, 20.44 and 23.05 degree 2-theta, and/or a melting point of 106°C.
The 2-hydroxy-ethanesulfonic acid pioglitazone salt described herein may have an X- ray powder diffraction pattern comprising main peaks at 17.86, 18.41 , 20.44, 23.05 and 24.39 degree 2-theta, and/or a melting point of 106°C.
The 2-hydroxy-ethanesulfonic acid pioglitazone salt described herein may have an X- ray powder diffraction pattern comprising main peaks at 8.85, 17.04, 17.86, 18.41 , 20.44, 23.05, 24.39, 25.21 , 26.45 and 28.05 degree 2-theta, and/or a melting point of 106°C.
The 2-hydroxy-ethanesulfonic acid pioglitazone salt described herein may have an X- ray powder diffraction pattern comprising main peaks expressed as an angle 2-theta as shown in the following Table 7, and/or a melting point of 106°C. Table 7

Another aspect of the invention is a pharmaceutical composition comprising a pioglitazone salt as specified above, together with one or more pharmaceutically acceptable carriers and/or excipients. Especially preferred is a composition which is adapted for pulmonary administration by inhalation.
Preferably the X-ray powder diffraction patterns are obtained using Cu Ku radiation. The X-rays produced using Cu Ka radiation has a wavelength of 1 .5406 Angstroms.
Preferably the X-ray powder diffraction pattern comprises main peaks expressed in terms of an angle 2-theta that have a margin of error of ± 0.5 degree; more preferably a margin of error of ± 0.4 degree; more preferably a margin of error of ± 0.3 degree; more preferably a margin of error of ± 0.2 degree; more preferably a margin of error of ± 0.1 degree.
The piogiitazone salts of the invention are useful for treatment of inflammatory respiratory disease by pulmonary administration by inhalation.
In all aspects of the invention, the piogiitazone salt may be accompanied by, or administered sequentially or concurrently with, one or more respiratory disorder treatment agents useful for the purpose of preventing and treating respiratory disorders, other than a PPARy agonist.
In all aspects of the invention, the inflammatory respiratory disease may be selected from, for example, mild asthma, moderate asthma, severe asthma, steroid resistant asthma, bronchitis, chronic obstructive pulmonary disease (COPD), cystic fibrosis, pulmonary edema, pulmonary embolism, pneumonia, pulmonary sarcoisosis, silicosis, pulmonary fibrosis, respiratory failure, acute respiratory distress syndrome, emphysema, chronic bronchitis, tuberculosis, and lung cancer.
Compositions of the invention are useful for treatment of inflammatory respiratory disorders, for example asthma (mild, moderate or severe), e.g., bronchial, allergic, intrinsic, extrinsic, exercise-induced, drug-induced (including aspirin and NSAID- induced) and dust-induced asthma, steroid resistant asthma, bronchitis including infectious and eosinophilic bronchitis, chronic obstructive pulmonary disease
(COPD), cystic fibrosis, pulmonary fibrosis including cryptogenic fibrosing alveolitis, idiopathic pulmonary fibrosis, idiopathic interstitial pneumonias, fibrosis complicating anti-neoplastic therapy and chronic infection, including tuberculosis and aspergillosis and other fungal infections; complications of lung transplantation; vasculitic and thrombotic disorders of the lung vasculature, and pulmonary hypertension (including pulmonary arterial hypertension); antitussive activity including treatment of chronic cough associated with inflammatory and secretory conditions of the airways, and iatrogenic cough; acute and chronic rhinitis including rhinitis medicamentosa, and vasomotor rhinitis; perennial and seasonal allergic rhinitis including rhinitis nervosa (hay fever); nasal polyposis; acute viral infection including the common cold, and infection due to respiratory syncytial virus, influenza, coronavirus (including SARS) and adenovirus, pulmonary edema, pulmonary embolism, pneumonia, pulmonary sarcoidosis, silicosis, farmer's lung and related diseases; hypersensitivity
pneumonitis, respiratory failure, acute respiratory distress syndrome, emphysema, chronic bronchitis, tuberculosis, and lung cancer. In particular, the methods and compositions of the present invention encompass the prevention and treatment of the respiratory disorder, COPD.
As used herein, the term "chronic obstructive pulmonary disease" or "COPD" refers to a set of physiological symptoms including chronic bronchitis, chronic cough, expectoration, exertional dyspnea and a significant, progressive reduction in airflow that may or may not be partly reversible. Emphysema may also be present in the lungs. COPD is a disease characterized by a progressive airflow limitation caused by an abnormal inflammatory reaction to the chronic inhalation of particles.
In subjects with the disorder, poor gas exchange in the lungs leads to decreased oxygen levels in the blood, increased levels of carbon dioxide and shortness of breath. Chronic airflow obstruction in COPD is complicated by the loss of lung elasticity resulting from enzymatic destruction of the lung parenchyma. Rather than a single pathologic condition, COPD is an umbrella term encompassing chronic obstructive bronchitis and emphysema.
Compositions suitable for administration by inhalation via the mouth or the nose are known, and may include carriers and/or diluents that are known for use in such compositions. The composition may contain 0.01-99% by weight of the pioglitazone salt. Preferably, a unit dose comprises the pioglitazone salt in an amount of 1 μg to 15+ mg.
The most suitable dosage level may be determined by any suitable method known to one skilled in the art. It will be understood, however, that the specific amount for any particular patient will depend upon a variety of factors, including the activity of the specific compound that is used, the age, body weight, diet, general health and sex of the patient, time of administration, the route of administration, the rate of excretion, the use of any other drugs, and the severity of the disease undergoing treatment. Optimum dosages will be determined by clinical trial, as is required in the art. Compositions of the invention may be used in combination with other therapeutic agents that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which present compounds are useful. Such other therapeutic agents may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with the glitazone component, particularly the pioglitazone or rosiglitazone component. When a compound of the invention is used contemporaneously with one or more other therapeutic agents, a pharmaceutical composition containing such other therapeutic agents in addition to
the pioglitazone salt is preferred. Accordingly, the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to the pioglitazone salt. Suitable therapeutic agents for a combination therapy with the pioglitazone compositions of the invention include: (1 ) a steroid drug such as a corticosteroid, for example beclomethasone, (e.g., as the mono or the dipropionate ester), flunisolide, fluticasone (e.g., as the propionate or furoate ester), ciclesonide, mometasone (e.g., as the furoate ester), mometasone desonide, rofleponide, hydrocortisone, prednisone, prednisolone, methyl prednisolone, naflocort, deflazacort, halopredone acetate, fluocinolone acetonide, fluocinonide, clocortolone, tipredane, prednicarbate, alclometasone dipropionate, halometasone, rimexolone, deprodone propionate, triamcinolone, betamethasone, fludrocortisone, desoxycorticosterone, etiprendnol dicloacetate and the like. Steroid drugs can additionally include steroids in clinical or pre-clinical development for respiratory diseases such as GW-685698, GW-799943, GSK 870086, QAE397, NCX-1010, NCX-1020, NO-dexamethasone, PL-2146, NS- 126 (formerly ST- 126) and compounds referred to in international patent applications WO0212265, WO0212266, WO02100879, WO03062259, WO03048181 and WO03042229. Steroid drugs can also additionally include next generation molecules in development with reduced side effect profiles such as selective glucocorticoid receptor agonists (SEGRAs), including ZK-216348 and compounds referred to in international patent applications WO-00032585, WO-000210143, WO-2005034939, WO-2005003098, WO-2005035518 and WO-2005035502 and functional equivalents and functional derivatives thereof; (2) a p2-adrenoreceptor agonist, such as albuterol, bambuterol, terbutaline, fenoterol, formoterol, formoterol fumarate, salmeterol, salmeterol xinafoate, arformoterol, arfomoterol tartrate, indacaterol (QAB-149), carmoterol, picumeterol, Bl 1744 CL, GSK159797, GSK59790, GSK159802, GSK642444, GSK678007, GSK96108, clenbuterol, procaterol, bitolterol, and broxaterol,TA-2005 and also compounds of EP1440966, JP05025045, WO93/ 8007, WO99/64035, US2002/0055651 , US2005/0133417, US2005/5159448, WO00/0751 14, WO01/42193, WO01/83462, WO02/66422, WO02/70490, WO02/76933, WO03/24439, WO03/42160, WO03/42164, WO03/72539, WO03/91204, WO03/99764, WO04/16578, WO04/016601 , WO04/22547, WO04/32921 , WO04/33412, WO04/37768, WO04/37773, WO04/37807, WO0439762, WO04/39766, WO04/45618, WO04/46083, WO04/71388, WO04/80964, EP1460064, WO04/087142, WO04/89892, EP01477167, US2004/0242622, US2004/0229904, WO04/108675, WO04/108676, WO05/033121 ,
WO05/040103, WOQ5/044787, WO04/071388, WO05/058299, WO05/058867, WO05/065650, WO05/066140, WO05/070908, WO05/092840, WO05/092841 , WO05/092860, WO05/092887, WO05/092861 , WO05/090288, WO05/092087, WOQ5/Q80324, WO05/080313, US20050182091 , US20050171 147, WO05/092870, WO05/077361 , DE10258695, WO05/1 1 1002, WO05/1 1 1005, WO05/1 10990, US2005/0272769 WO05/1 10359, WO05/121065, US2006/0019991 , WO06/016245, WO06/014704, WO06/031556, WO06/032627, US2006/0106075, US2006/0106213, WO06/051373, WO06/056471 , WO08/0961 12, WO08/104790, WO08/0961 9, WO08/0961 2; (3) a leukotriene modulator, for example, montelukast or pranlukast; (4) anticholinergic agents, for example, selective muscarinic- 3 (M3) receptor antagonists such as ipratropium bromide, tiotropium, tiotropium bromide (Spiriva®), glycopyrollate, NVA237, LAS34273, GSK656398, GSK233705, GSK 573719, LAS35201 , QAT370 and oxytropium bromide; (5) phosphodiesterase-IV (PDE-IV) inhibitors, for example, roflumilast or cilomilast; (6) an antitussive agent, such as codeine or dextramorphan; (7) a non-steroidal anti-inflammatory agent (NSAID), for example, ibuprofen or ketoprofen; (8) a mucolytic, for example, N acetyl cysteine or fudostein; (9) a expectorant/mucokinetic modulator, for example, ambroxol, hypertonic solutions (e.g., saline or mannitol) or surfactant; (10) a peptide mucolytic, for example, recombinant human deoxyribonoclease I (dornase-alfa and rhDNase) or helicidin; (1 1 ) antibiotics, for example, azithromycin, tobramycin and aztreonam; and (12) p38 MAP kinase inhibitors such as GSK 856553 and GSK 681323.
In one aspect, the invention provides for the use of inhaled administration of the pioglitazone salt of the invention in combination with other anti-inflammatory drugs and bronchodilator drug combinations (i.e. triple combination product), including but not limited to salmeterol xinafoate/fluticasone propionate (Advair/Seretide®), formoterol fumarate/budesonide (Symbicort®), formoterol fumarate/mometasone furoate, formoterol fumarate/beclometasone dipropionate (Foster®), formoterol fumarate/fluticasone propionate (FlutiForm®), Indacaterol/mometasone furoate, lndacaterol/QAE-397, GSK159797/GSK 685698, GSK159802/GSK 685698,
GSK642444/GSK 685698, formoterol fumarate/ciclesonide, arformoterol
tartrate/ciclesonide.
In another aspect, the invention provides for the use of inhaled administration of the pioglitazone salt of the invention in combination with other bronchodilator drug combinations, particularly B2 agonist/M3 antagonist combinations (i.e. triple combination product), including but not limited to salmeterol xinafoate/tiotropium
bromide, formoterol fumarate/tiotropium bromide, Bl 1744 CL/tiotropium bromide, indacaterol/NVA237, indacterol/QAT-370, formoterol/ LAS34273, GSK159797/GSK 573719, GSK159802/GSK 573719, GSK642444/GSK 573719, GSK159797/GSK 233705, GSK159802/GSK 233705, GSK642444/GSK 233705, and compounds which possess both β2 agonist and M3 antagonist activity in the same molecule (dual functionality) such as GSK 961081.
Thus in another aspect, the invention provides a kit for treatment of respiratory disorders in a subject, the kit comprising one dosage form comprising a pioglitazone salt composition of the invention, and a second dosage form comprising another therapeutic agent, for example as discussed above, selected from anti-inflammatory agents, bronchodilators, mucolytic agents, antitussive agents, leukotriene
inhibitors, and antibiotics. For delivery by inhalation, the pioglitazone salt is preferably in the form of
microparticles. These may be prepared by a variety of techniques, including spray- drying, freeze-drying and micronisation. Following size reduction to produce microparticles, particle size distribution (PSD) of the compound is examined and generally described in the art by specifying d10, d50 and d90 values. The average particle size, i.e. the average equivalent diameter, is defined as the diameter where 50 mass-% (of the particles) of the powder have a larger equivalent diameter, and the other 50 mass-% have a smaller equivalent diameter. Hence the average particle size is denoted as equivalent d50. For inhaled use a d50 of less than 10 microns, preferably less than 5 microns is desired.
By way of example, a composition of the invention may be prepared as a suspension for delivery from a nebuliser or as an aerosol in a liquid propellant, for example for use in a pressurised metered dose inhaler (PMDI). Propellants suitable for use in a PMDI are known to the skilled person, and include CFC-12, H FA- 134a, HFA-227, HCFC-22 (CCI2F2) and HFA-152 (CH4F2 and isobutane).
In a preferred embodiment of the invention, a composition of the invention is in dry powder form, for delivery using a dry powder inhaler (DPI). Many types of DPI are known.
Microparticles for delivery by inhalation may be formulated with excipients that aid delivery and release. For example, in a dry powder formulation, microparticles may
be formulated with large carrier particles that aid flow from the DPI into the lung. Suitable carrier particles are known, and include lactose particles; they may have a mass median aerodynamic diameter of greater than 90 μηι. Aerosol generation can be carried out using, for example, pressure-driven jet atomizers or ultrasonic atomizers, preferably using propellant-d riven metered aerosols or propellant-free administration of micronized active compounds from, for example, inhalation capsules or other "dry powder" delivery systems. The compositions may be dosed as described depending on the inhaler system used. In addition to the active compounds, the administration forms may additionally contain excipients, such as, for example, propellants (e.g., Frigen in the case of metered aerosols), surface-active substances, emulsifiers, stabilizers, preservatives, flavourings, fillers (e.g. lactose in the case of powder inhalers) or, if appropriate, further active compounds.
For the purposes of inhalation, a large number of systems are available with which aerosols of optimum particle size can be generated and administered, using an inhalation technique which is appropriate for the patient. In addition to the use of adaptors (spacers, expanders) and pear-shaped containers (e.g. Nebulator®,
Volumatic®), and automatic devices emitting a puffer spray (Autohaler®), for metered aerosols, in particular in the case of powder inhalers, a number of technical solutions are available (e.g. Diskhaler®, Rotadisk®, Turbohaler® or the inhalers for example as described in EP-A-0505321 ).
Methods of preparation of qlitazone salts
Glitazones can be separated using chiral HPLC. A method of resolution, using pioglitazone as an example, is shown in Example 1 below. Chiral columns include CHIRALPAK AD, AD-H, AS-V, 50801 , IA, IC, OD, OF, OG, OJ, OK, and OZ.
Preferred chiral columns for HPLC are CHIRALPAK AD-H and CHIRALPAK IA using elution with ethanol and varying portions of TFA, preferably 0.05-0.2% TFA.
It will be recognised that chiral HPLC separation using this method generates the TFA acid salt of the R-pioglitazone and that racemic pioglitazone can easily form during attempts to form the corresponding R-pioglitazone as the free base if aqueous base is used. It has been identified that use of a resin bound base, preferably
macroporous triethylammonium methyipolystyrene carbonate, is important to convert R-pioglitazone TFA salt to R-pioglitazone free base using MeOH as the preferred solvent (see Example 2 below). It has been found that addition of hydrochloric acid to this suspension of the R-pioglitazone free base in MeOH gives conversion to the HCI salt (see Example 3 below). The isolated R-pioglitazone HCI obtained in this manner is crystalline and has high e.e.
A surprising observation is that a solution of R-pioglitazone HCI in MeOH
interconverts to the R-pioglitazone L-DBTA salt when L-DBTA is added. Addition of water is important to give conversion and crystallisation of the R-pioglitazone DBTA salt. The isolated R-pioglitazone L-DBTA obtained in this manner has high e.e. (see Example 4 below) and is used to seed resolution reactions described below.
An alternative method of resolution, using pioglitazone as an example, is shown in Scheme 2 (see also Example 5).
Scheme 2

Recent review articles on optical resolution methods: E. Fogassy et. al., Tetrahedron: asymmetry, 2008, 19, 519-536; S.H.Wilen, Topics in Stereochemistry, Wiley- Interscience: NY, 1972, 6, 107 Eds. E.L. Eliel, N.L. Allinger; P.Newman, Optical resolution Procedures of Chiral Compounds 1-3, Resolution Information Center, NY, 1978-1984; J.Jaques, S.H.Wilen, A. Collett, Enantiomers Racemates and
Resolutions, Wiley-lnterscience: NY, 1991 ; R.A. Scheldon, Chirotechnology, Marcel Dekker, NY, 1993; Optical Resolutions via diastereomeric salt formation, CRC Press, 2002, Ed. David Kozma. Commonly used chiral acid resolving agents include, without limitation, dibenzoyl tartaric acid, dianisoyl tartaric acid, ditoluyl tartaric acid, tartaric acid, phencyphos, chlocyphos, anicyphos, 1 ,1 '-binaphthyl-2,2'-diyl hydrogen phosphate,
camphorsulfonic acid, bromo camphorsulfonic acid, camphoric acid,
phenylethylsulfonic acid, malic acid, mandelic acid, 4-bromomandelic acid, 4- methylmande!ic acid, lactic acid and chalcone sulfonic acids.
A more preferred set of chiral acid resolving agents include (-)-0,0'-dibenzoyl-L- tartaric acid (anhydrous), (-)-0,0'-dibenzoyl-L-tartaric acid monohydrate, (-)-di-O-p- toluoyl-L-tartaric acid, L-(+)-tartaric acid and (-)-di-p-anisolyl-L-tartaric acid.
The most preferred chiral acid resolving agent is (-)-0,0'-dibenzoyl-L-tartaric acid (anhydrous) with addition of seed crystals (5R)-5-{4-[2-(5-ethylpyridin-2- yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione (-)-0,0'-dibenzoyl-L-tartrate that have high e.e and are prepared as described in example 4.
Chiral acid resolving agents can be used in different stoichiometries to effect resolutions. The above resolving agents can be used in a ratio of 1 part (I) to 10 parts of chiral acids (II), more preferably in a ratio of 1 part (I) to 5 parts of chiral acids (II), and most preferably in a ratio of 1 part (I) to 2 part of chiral acids (II). Even more preferred is a ratio of 1 part (I) to 0.6 part of chiral acids (II).
A variety of solvents may be useful to form diastereomeric salts (III) with chiral acids (II). Preferred solvents will include ethyl acetate, dichloromethane, tetrahydrofuran, 1 ,4-dioxane, acetone, acetonitrile, MeOH, EtOH, IPA, 1 -propanol, 1 ,2- dimethoxyethane, diethyl ether, dichloroethane, ferf-butylmethyl ether, 1-butanol, 2- butanol, t-butanol, 2-butanone, toluene, cyclohexane, heptane, hexane, H2O, DMF, petroleum ethers and CHCI3.
The most preferred solvent is acetonitrile.
Solvents may be used in combination with each other and preferred combinations include 10% HCI in H2O, 10% H2O in DMF, 10% H2O in EtOH, 10% H2O in IPA, methanol and water in varying portions, methanol and water in varying portions with HCI, CHCI3 and ethyl acetate in varying portions, EtOH and water in varying portions, IPA and water in varying portions, 1 -propanol and water in varying portions and CHCI3 and dioxane in varying portions.
More preferred solvent combinations include CHCi3 and dioxane in varying portions, CHCI3 and ethyl acetate in varying portions, methanol and water in varying portions with HCI and methanol and water in varying portions. The product from resolution described in scheme 2 gives the R-Pio L-DBTA salt that is substantially enriched as the R enantiomer. Further enrichment to >90% e.e can be obtained by recrystallisation. Conditions for recrystallisation have surprisingly been found to require HCI in solvents, preferably MeOH and water. If only MeOH and water are used, extensive conversion to racemic pioglitazone free base occurs. The isolated R-pioglitazone L-DBTA obtained in this manner has high e.e. (see Example 6 below).
Methods for preparation of other salts of R-pioglitazone from pioglitazone L-DBTA are shown in scheme 3.
Scheme 3
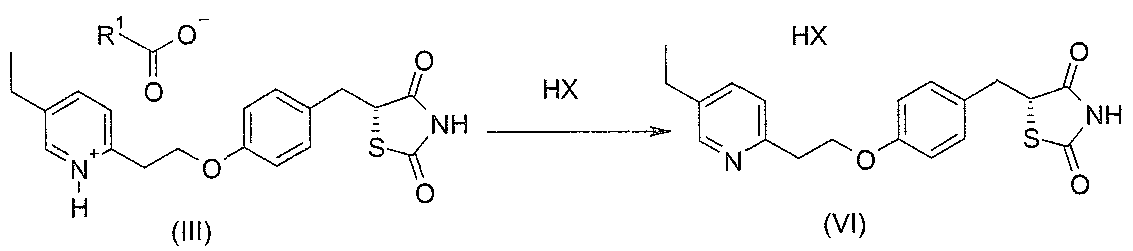
Similarly, preferred solvents for conversion of R-pioglitazone L-DBTA to R- pioglitazone HBr are MeOH containing cone HBr followed by addition of EtOAc. The isolated R-pioglitazone HBr obtained in this manner is crystalline and has high e.e. (see Example 9 below).
Preferred solvent for conversion of R-pioglitazone L-DBTA to R-pioglitazone naphthalene- 1 ,5-disulfonic acid is MeOH and the salt forms a complex with 2
Piogiitazone units as determined by NMR. The isolated R-pioglitazone naphthalene- 1 ,5-disulfonic acid obtained in this manner is crystalline and has high e.e. (see Example 10 below). Preferred solvent for conversion of R-pioglitazone L-DBTA to R-pioglitazone phosphate is acetone. The isolated R-pioglitazone phosphate obtained in this manner is crystalline and has high e.e. (see Example 1 1 below).
Preferred solvents for conversion of R-pioglitazone L-DBTA to R-pioglitazone isethionate are I PA and isopropyi acetate. The isolated R-pioglitazone phosphate obtained in this manner is crystalline and has high e.e. (see Example 12 below).
The invention will now be described in more detail with reference to the following figures:
Figure 1 : Illustrative X-ray powder diffraction (XRPD) pattern of (5R)-5-{4-[2-(5- ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3 thiazolidine-2,4-dione (-)-0,0'-dibenzoyI-L- tartrate (example 6);
Figure 2: Illustrative XRPD pattern of (5R)-5-{4-[2-(5-Ethylpyridin-2-yl)ethoxy]benzyl}- 1 ,3-thiazolidine-2,4-dione hydrochloride (example 7);
Figure 3: Illustrative XRPD pattern of (5R)-5-{4-[2-(5-Ethylpyridin-2-yl)ethoxy]benzyl}- 1 ,3-thiazolidine-2,4-dione hydrochloride (example 8);
Figure 4: Illustrative XRPD pattern of (R)-5-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]- benzyl}-thiazolidine-2,4-dione hydrobromide (example 9);
Figure 5: Illustrative XRPD pattern of (R)-5-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]- benzyl}-thiazolidine-2,4-dione naphthalene-1 ,5-disulfonic acid (2:1 ) (example 10);
Figure 6: Illustrative XRPD pattern of (R)-5-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]- benzyl}-thiazolidine-2,4-dione phosphate (example 1 1 ); and
Figure 7: Illustrative XRPD pattern of (R)-5-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]- benzyl}-thiazolidine-2,4-dione 2-hydroxy-ethanesulfonic acid (example 12).
CHEMICAL EXAMPLES
General Experimental Details
Abbreviations used in the experimental section:
c = concentration; h = hour; min = minutes; H20 = distilled water; HPLC = high performance liquid chromatography; LCMS = liquid chromatography mass
spectrometry; RT = room temperature; Rt = retention time; e.e. = enantiomeric excess; d.e. = diastereomeric excess; MP-Carbonate = macroporous
triethylammonium methylpolystyrene carbonate (0.5% inorganic antistatic agent); MeOH = methanol; TFA = trifluoroacetic acid; DMSO = dimethyl sulphoxide; HCI = hydrogen chloride; EtOH = ethanol; EtOAc = ethyl acetate; IPA = isopropanol; L- DBTA = (-)-dibenzoyl-L-tartaric acid, anhydrous; (±) 5-{4-[2-(5-ethylpyridin-2- yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione hydrochloride = (±) Pioglitazone hydrochloride; (5R) 5-{4-[2-(5-ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4- dione hydrochloride = (5R) Pioglitazone hydrochloride; 5-{4-[2-(5-ethylpyridin-2- yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione (-)-0,0'-dibenzoyl-L-tartrate = (5R)- Pioglitazone.L-DBTA.
The nomenclature of structures was assigned using ACD Labs version 10. NMR spectra were obtained on a Varian Unity I nova 400 spectrometer with a 5mm inverse detection triple resonance probe operating at 400MHz or on a Bruker Avance DRX 400 spectrometer with a 5mm inverse detection triple resonance TXI probe operating at 400MHz or on a Bruker Avance DPX 300 spectrometer with a standard 5mm dual frequency probe operating at 300MHz. Shifts are given in ppm relative to
tetramethylsilane. Optical rotations were measured using an AA-10R automatic polarimeter with 5x25 mm jacketed sample cell. X-ray powder diffraction (XRPD) patterns were collected on a Siemens D5000 diffractometer using Cu Ka radiation (40kV, 40mA), Θ-Θ goniometer, divergence of V20 and receiving slits, a graphite secondary monochromator and a scintillation counter. The instrument was performance checked using a certified Corundum standard (NIST 1976). The software used for data collection was Diffrac Plus XRD Commander v2.3.1 and the data were analysed and presented using Diffrac Plus EVA v 1 1.0.0.3. Samples were run under ambient conditions as flat plate specimens using powder as received. The sample was gently packed into a cavity cut into polished, zero-background (510) silicon wafer. The sample was rotated in its own plane during analysis. The details of the data collection are: Angular range: 2 to 42 '29; Step size: 0.05 "2Θ; Collection time: 4 s.step"1.
Differential Scanning Calorimetry (DSC) data were collected on a Mettler Toledo DSC823e equipped with a 34 position auto-sampler. The calibration for energy and temperature was carried out using certified indium. Typically 0.5-3 mg of each sample, in a pin-holed aluminium pan, was heated at 10 °C.min"1 from 30 °C
to 300°C. A purge of dry nitrogen at 50 mi.rnin"1 was maintained over the sample. The instrument control software was STARe software version 9.2. Data quoted are the peaks with a heating rate of 10°C/min. All solvents and commercial reagents were used as received.
The Liquid Chromatography Mass Spectroscopy (LC/MS) systems used:
Method 1
CHIRALPAK AD-H (250 x 30 mm, 5pm), elution with EtOH +0.05%TFA - flow rate 30 ml/min. Detection -In-line UV detection set at 250 nM wavelength
Method 2
CHIRALPAK 1A (250 x 4.6 mm, 5μΜ), elution with EtOH +0.05%TFA - flow rate 0.7 ml/min. Detection - In-line DAD set at 280 nM wavelength
Method 3
CHIRALCEL OD-RH (150 X 4.6 mm), elution with 90% MeOH + 10% H20 - flow rate 0.5 ml/min. Detection - In-line UV detection set at 254 nM wavelength Method 4
Waters Micromass ZQ2000 with a C18-reverse-phase column (100 χ 3.0 mm Higgins Clipeus with 5 pm particle size), elution with A: H20 + 0.1 % formic acid; B:
acetonitrile + 0.1 % formic acid. Gradient:
Gradient - Time flow mL/min %A %B
0.00 1.0 95 5
1.00 1.0 95 5
15.00 1.0 5 95
20.00 1.0 5 95
22.00 1.0 95 5
25.00 1.0 95 5
Detection - MS, ELS, UV (100 pi split to MS with in-line UV detector). MS ionisation method - Electrospray (positive ion)
Method 5
CHIRALPAK IA (250 x 4.6 mm, 5μΜ), eiution with A, 0.05% TFA in EtOH; B, heptane; D, IPA (A:B:D = 40:30:30), - flow rate 0.7 ml/min. Detection - In-line DAD set at 225 nM wavelength. Detection - MS, ELS, UV PDA. MS ionisation method - Electrospray (positive/negative ion).
Method 6
Waters Micromass ZQ2000 quadrupole mass spectrometer linked to a Waters Acquity UPLC system with a PDA UV detector. The spectrometer has an
electrospray source operating in positive and negative ion mode. LC Column - Acquity BEH C18 1.7um 100 x 2.1 mm, maintained at 40°C or Acquity BEH Shield RP18 1.7um 100 x 2.1 mm, maintained at 40°C.
Gradient - Time flow ml/min
%A %B
0.00 0.4 95 5
0.40 0.4 95 5
6.00 0.4 5 95
6.80 0.4 5 95
7.00 0.4 95 5
8.00 0.4 95 5
Detection - MS, UV PDA
MS ionisation method - Electrospray (positive/negative ion)
All reactions were carried out under an atmosphere of nitrogen unless specified otherwise.
Example 1
(5R)-5-{4-[2-(5-ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione trifluoroacetate
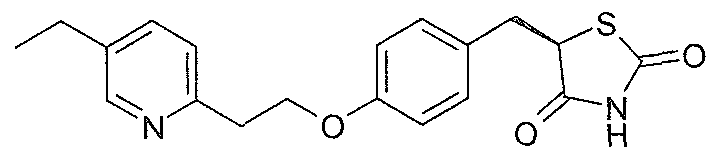
CF3C02H
The title compound (480 mg) was isolated using method 1. LCMS (Method 4): Rt 6.00 min, m/z 357 [M-CF3C02H+]. [a]D 25 +104° (c 1.0, MeOH). e.e. (Method 2) > 98%, Rt 4.69 min. 1H NMR (400 MHz, DMSO-d6): δ 12.02-1 1.88 (1 H, bs), 8.68-8.60 (1 H, d, J 1.7), 8.32-8.23 (1 H, d, J 7.7), 7.90-7.82(1 H, d, J 8.4), 7.14-7.06 (2 H, d, J 8.7),
6.85-6.78 (2 H, d, J 8.7), 4.85-4.78(1 H, dd, J 4.4, 8.9), 4.35-4.27 (2 H, t, J 6.2), 3.40- 3.34 (2 H, t, J 6.1 ), 3.28-3.21 (1 H, dd, J 4.3, 14.3), 3.05-2.97 (1 H, dd, J 9.0, 14.3), 2.77-2.67 (2 H, q, J 7.6), 1.22-1.14 (3 H, q, J 7.5). Example 2
(5R)-5-{4-[2-{5-ethyipyridin-2-y!)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione

MP-Carbonate (389 mg, 1.06 mmol) was added to a solution of the product from example 1 (100 mg, 0.21 mmol) in MeOH (100 mL) and stirred at RT for 2 h, filtered and the resin washed with MeOH (3 X 0 mL). The filtrate was concentrated in vacuo to afford the title compound (35 mg, 47%). e.e. (Method 2) 92.90%, Rt 6.27 min. 1H NMR (400 MHz, DMSO-d6): δ 12.44-1 1.1 1 (1 H, bs), 8.34-8.29 (1 H, d, J 1.9 Hz), 7.55-7.49 (1 H, dd, J 2.2, 7.9 Hz), 7.24-7.20 (1 H, d, J 7.8 Hz), 7.12-7.05 (2 H, d, J 8.6 Hz), 6.84-6.77 (2 H, d, J 8.6 Hz), 4.78-4.71 (1 H, dd, J 4.3, 9.1 Hz), 4.30-4.19 (1 H, d, J 4.3 Hz), 3.24-3.18 (2 H, m), 3.1 1 -3.03 (2 H, t, J 6.6 Hz), 3.00-2.92 (1 H, dd, J 9.2, 14.2 Hz), 2.59-2.50 (2 H, q, J 7.6 Hz), 1.17-1 .09 (3 H, t, J 7.7 Hz). Example 3
(5R)-5-{4-[2-(5-ethylpyridin-2-yl)ethoxy]benzyl}»1 ,3-thiazolidine-2,4-dione hydrochloride
HCI
-1 .25 M HCI in MeOH (0.33 mL, 0.33 mmol) was added to a suspension of the product from example 2 (30 mg, 0.084 mmol) in MeOH (5 mL) and stirred at RT for 1 h. The solvent was removed in vacuo to afford the title compound (32.4 mg, 100%). LCMS (Method 4): Rt 5.95 min, m/z 357 [M-HCf]. e.e (Method 3) 93.2%, Rt 12.10 min. Stereochemisty at C-5 was assigned (R) configuration by single crystal X-ray diffraction analysis. [u]D 24 +108° (c 1.0, MeOH). 1H NMR (400 MHz, DMSO-d6): δ 12.03-1 1.88 (1 H, bs), 8.68-8.62 (1 H, d, J 1.7 Hz), 8.34-8.25 (1 H, d, J 7.9 Hz), 7.91 - 7.83 (1 H, d, J 8.3 Hz), 7.14-7.05 (2 H, d, J 8.7 Hz), 6.86-6.77 (2 H, d, J 8.7 Hz), 4.85-4.77 (1 H, dd, J 4.3, 8.9 Hz), 4.38-4.28 (2 H, t, J 6.0 Hz), 3.42-3.36 (2 H, t, J 6.2 Hz), 3.28-3.20 (1 H, dd, J 9.0, 14.2 Hz), 3.06-2.96 (1 H, dd, J 9.0, 14.2 Hz), 2.77-2.67
(2 H, q, J 7.7 Hz), 1.23-1.15 (3 H, t, J 7.7 Hz). Recrystallisation using MeOH-EtOAc or MeOH-Et20 gave the title compound with an e.e. >97%.
Example 4

(5R)-5-{4-t2-(5-ethylpyridin-2-yl)ethoxy]benzyl}-1,3-thiazolidine-2,4-dione (-)- 0,0'-dibenzoyl-L-tartrate
To a mixture of the product from example 3 (91 .8% d.e.) (50 mg) and L-DBTA (50 mg) was added MeOH (1 .5 mL). The clear solution was rapidly stirred whilst adding H20 dropwise until a cloudiness persisted. The reaction was allowed to stir at RT for 48h and the solid collected by filtration to give the title compound as seed crystals (43mg). d.e. (Method 5) 99.01 % Rt 10.83 min; 0.98% Rt 15.83 min; 98.03% d.e.
Example 5

(5R)-5-{4-[2-(5-Ethylpyridin-2-yl)ethoxy]benzyl}-1,3-thiazolidine-2,4-dione (-)- 0,0'-dibenzoyl-L-tartrate
To a stirred mixture of MeOH (6.636 L) and H20 (2.5 L) was added DBTA (577.5 g, 1.61 mol) and pioglitazone. hydrochloride (1.05 Kg, 2.68 mol). The mixture was stirred until a solution was obtained. H20 (1.896 L) was added in a steady flow into the vortex of the stirred reaction followed by seed crystals from example 4 (300 mg). Further H20 (3.0 L) was added dropwise over 1 1 h (~5ml/min delivered by HPLC pump). The suspension was stirred for 44 h and the granular solid was collected by filtration, washed with 330 mL premixed H20/MeOH (2: 1 ) and dried at 40 °C under
vacuum to give the title compound (919.29g, 48%). e.e. (Method 5) 84.56%, Rt 10.75 min.; 15.44%, Rt 16.75 min.; 69.13% d.e. 1H N R (400 MHz, DMSO-d6): δ 1 1 .98 (1 H, bs), 8.37 (1 H, d, J 2.0 Hz), 8.02 (4 H, bd, J 8.4 Hz), 7.74 (2H, tt, 7.4,1.2 Hz) 7.63- 7.57 (5H, m) 7.29 (1 H, d, J 7.9 Hz), 7.13, 6.86 (4 H, A2B2q, J 8.6 Hz), 5.87 (2H, s), 4.86 (1 H, dd, J 4.4, 9.1 Hz), 4.30 (2 H, t, J 6.7 Hz), 3.29 (1 H, dd, J 4.3, 14.1 Hz), 3.12 (2 H, t, J 6.7 Hz), 3.04 (1 H, dd, J 9.0, 14.1 Hz), 2.59 (2 H, q, J 7.6 Hz), 1.18 (3 H, t, J 7.6 Hz).
Example 6

(5R)-5-{4-[2-(5-Ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3»thiazolidine-2,4-dione (-)- Ο,Ο'-dibenzoyl-L-tartrate
To the product from example 5 (1380.4 g) was added premixed MeOH (6.62 L) and 1 M HCI (1 .93 L). The solution was warmed to 30°C and filtered to remove undissolved starting material. The clear solution was stirred and water (1 L) was added over 1 min into the vortex of the reaction. Seed crystals from example 4 (300 mg) were added followed by dropwise addition of further H20 (2.5 L) over 14 h at 3ml/min using an HPLC pump. The slurry was stirred for 44 h then the solid was collected by filtration, washed with 330 mL premixed H20 /MeOH (2:1 ) and dried at 40 °C under vacuum to give the title compound (976.55g, 71 %). d.e. (Method 5) 96.69%, Rt 10.48 min.; 3.30%, Rt 16.73 min.; 93.39% d.e. DSC 153°C. 1H NMR (400 MHz, DMSO-d6): δ 1 1.98 (1 H, bs), 8.37 (1 H, d, J 2.0 Hz), 8.02 (4 H, bd, J 8.4 Hz), 7.74 (2H, tt, 7.4, 1.2 Hz) 7.63-7.57 (5H, m) 7.29 (1 H, d, J 7.9 Hz), 7.13, 6.86 (4 H, A2B2q, J 8.6 Hz), 5.87 (2H, s), 4.86 (1 H, dd, J 4.4, 9.1 Hz), 4.30 (2 H, t, J 6.7 Hz), 3.29 (1 H, dd, J 4.3, 14.1 Hz), 3.12 (2 H, t, J 6.7 Hz), 3.04 (1 H, dd, J 9.0, 14.1 Hz), 2.59 (2 H, q, J 7.6 Hz), 1.18 (3 H, t, J 7.6 Hz)
Figure 1 provides an illustrative XRPD pattern of (5R)-5-{4-[2-(5-Ethylpyridin-2- yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione (-)-0,0'-dibenzoyl-L-tartrate.
The main diffraction peaks and relative intensities for the peaks shown in Figure 1 are listed in Table 1 .
Example 7

HCI
(5R)-5-{4-[2-(5-Ethyipyridin-2-yl)ethoxy]ben2yl}~1 ,3-thiazoiidine-2,4-dione hydrochloride
Pioglitazone.L-DBTA salt from example 6 (976.75 g, 1.366mol) was split into 2 batches and dissolved in premixed MeOH (2.5 L) and concentrated HCI (148 mL) at 40-45 °C (internal temperature). The reaction was allowed to stir over <5 min to give a clear solution and was then filtered quickly through a glass fibre filter paper into a 20 L flange top reactor flask containing seed crystals from example 3 (300 mg, e.e. 94.66%). The procedure was repeated with the other Pioglitazone.L-DBTA batch. EtOAc (7.5 L) was added to the flange top reactor containing both filtered solutions and the mixture was stirred mechanically. The remaining 2.5L EtOAc was added in a steady stream into the vortex and stirring continued over 3 h. The solid was collected by filtration through a sintered funnel, washed with EtOAc (1 L) and dried for 18 h at 40 °C under vacuum to give the title compound (418.75g, 78%). (Method 5) 97.45%, Rt 10.06min.; 2.54%, Rt 14.67min.; e.e. 94.91 %. (Method 3) 96.69%, Rt 15.72min.; 3.31 %, Rt 17.78min.; e.e. 93.38%. DSC 201 °C. LCMS (Method 6) Rt 2.88min, m/z 357 [MH+-HCI], 99.55%. 1H NMR (400 MHz, DMSO-cfe): δ 12.0 (1 H, s), 8.70 (1 H, d, J 1.7 Hz), 8.36 (1 H, bd, J 8.3 Hz), 7.93 (1 H, d, J 8.2 Hz), 7.15, 6.87 (4 H, A2B2q, J 8.7 Hz), 4.86 (1 H, dd, J 4.4, 8.9 Hz), 4.38 (2 H, t, J 6.3 Hz), 3.44 (2 H, t, J 6.2 Hz), 3.29 (1 H, dd, J 4.3, 14.2 Hz), 3.06 (1 H, dd, J 9.0, 14.3 Hz), 2.78 (2 H, q, J 7.6 Hz), 1 .23 (3 H, t, J 7.6 Hz)
Figure 2 provides an illustrative XRPD pattern of (5R)-5-{4-[2-(5-Ethylpyridin-2- yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione hydrochloride .
The main diffraction peaks and relative intensities for the peaks shown in Figure 2 are listed in Table 2.

HCI
(5R)-5-{4-[2-(5"Ethylpyr3din-2-yl)ethoxy]benzyi}-1 ,3«thiazolid!ne-2,4 -dione hydrochloride
To the product from example 7 (0.50g) (e.e. 92.6 %) was added acetic acid (1 mL) and water (1 mL) and the mixture was warmed to give a solution. 1 M HCI (1 mL) and water (0.5mL) were added and the warm solution filtered. On standing solid crystallised. After standing at room temperature for 18h the solid was filtered off and washed with water, then dried at 40°C under vacuum. After 6 h the solid was ground in a pestle and mortar then re-dried at 40°C under vacuum for 18 h to give the title compound (0.32g, 64%). (Method 5) 98.45%, Rt 10.06min.; 1.56%, Rt 14.67min.; e.e. 96.9 %. DSC 178°C. 1H NMR (400 MHz, DMSO-d6): δ 12.0 (1 H, s), 8.65 (1 H, d, J 1.7 Hz), 8.36 (1 H, bd, J 8.5 Hz), 7.82 (1 H, d, J 8.2 Hz), 7.15, 6.87 (4 H, A2B2q, J 8.5 Hz), 4.86 (1 H, dd, J 4.6, 8.9 Hz), 4.36 (2 H, t, J 6.3 Hz), 3.39 (2 H, t, J 6.2 Hz), 3.29 (1 H, dd, J 4.4, 14.2 Hz), 3.05 (1 H, dd, J 8.9, 14.2 Hz), 2.75 (2 H, q, J 7.6 Hz), 1.22 (3 H, t, J 7.6 Hz)
Figure 3 provides an illustrative XRPD pattern of (5R)-5-{4-[2-(5-Ethylpyridin-2- yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione hydrochloride.
The main diffraction peaks and relative intensities for the peaks shown in Figure 3 are listed in Table 3 below.
Intensity
Angle d value Intensity
%
2-Theta ° Angstrom Count
9.04 9.77 711 34.9
13.06 6.77 678 33.2
16.10 5.50 172 8.5
17.86 4.96 166 8.2
19.03 4.66 616 30.2
20.24 4.38 1512 74.2
21.06 4.22 066 52.3
23.05 3.86 2039 100
26.25 3.39 939 46.1
27.62 3.23 407 19.9
28.37 3.14 650 31.9
30.01 2.98 360 17.7
31 .51 2.84 603 29.6
32.32 2.77 523 25.7
Example 9
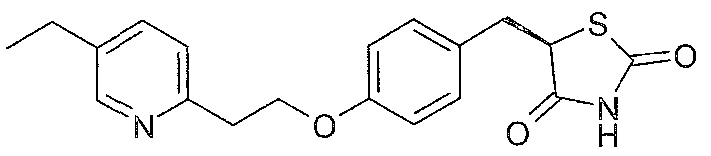
HBr
(R)-5-{4-[2-{5-Ethyl-pyridin-2-yl)-ethoxy]»benzyl}-thiazo!idine~2,4-dione hydrobromide
To a solution of 48% HBr (1.58mL) in MeOH (20mL) at 40°C was added (5R)-5-{4-[2- (5-ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione (-)-O.O'-dibenzoyl-L- tartrate (4.0g, 5.6mmol) (86.27% d.e.) to give a clear solution. EtOAc (40mL) was added rapidly to give a solid. The reaction was allowed to cool and stirred for 30 min before the solid was collected by filtration, washed with EtOAc (20mL) and dried at 40°C for 64h (1.72g). The title compound was obtained by recrystallisation from MeOH-EtOAc. (Method 5) 98.8%, Rt 10.39min.; 1.17%, Rt 16.93min.; e.e. 97.6 %. LCMS (Method 6) Rt 2.89min, m/z 357 [MhT HBr], >99%%. DSC 207°C. 1H NMR (400 MHz, DMSO-d6): δ 12.0 (1 H, s), 8.65 (1 H, d, J 1.7 Hz), 8.36 (1 H, bd, J 8.5 Hz), 7.82 (1 H, d, J 8.2 Hz), 7.15, 6.87 (4 H, A2B2q, J 8.5 Hz), 4.86 (1 H, dd, J 4.6, 8.9 Hz), 4.36 (2 H, t, J 6.3 Hz), 3.39 (2 H, t, J 6.2 Hz), 3.29 (1 H, dd, J 4.4, 14.2 Hz), 3.05 (1 H, dd, J 8.9, 14.2 Hz), 2.75 (2 H, q, J 7.6 Hz), 1 .22 (3 H, t, J 7.6 Hz)
Figure 4 provides an illustrative XRPD pattern of ( R)-5-{4-[2-( 5-Ethyl-pyrid in-2-yl )- ethoxy]-benzyl}-thiazolidine-2,4-dione hydrobromide.
The main diffraction peaks and relative intensities for the peaks shown in Figure 4 are listed in Table 4.
Example 10

(R)-5-{4-[2-(5-Ethyi-pyridin-2-yl)-ethoxyi-benzyl}-thiazo!idine-2,4-dione naphthalene-1 ,5-disulfonic acid (2:1)
To a stirred solution of naphthalene-1 ,5-disulfonic acid (0.44g) in methanol (32mL) at 40 °C was added (5R)-5-{4-[2-(5-ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4- dione (-)-0,0'-dibenzoyl-L-tartrate (1.6g) (87.27% d.e.). The solid dissolved then a solid crystallised. After stirring at room temperature for 20 min the solid was collected by filtration, washed with MeOH (10 mL), EtOAc (10 mL) and dried under vacuum at
40 °C to give the title compound (1.0 g, 89%), (Method 5) 95.84%, Rt 10.06min.; 4.16%, Rt 14.67min.; d.e. 91.68 %. LCMS (Method 6) Rt 2.89min, m/z 357 [0.5(M- naphthalene-1 ,5-disulfonic acid)H+], 99.52%. DSC 240°C. 1H NMR (400 MHz, DMSO-d6): δ 1 1.94, (1 H, s), 8.82 (1 H, bd, J 8.2 Hz), 8.67 (1 H, d, J 1.95 Hz), 8.26 (1 H, dd, J 8.6, 1 .95 Hz), 7.91-7.86 (2 H, m), 7.35, (1 H, dd, J 8.4, 7.0 Hz), 7.10, 6.81 (4 H, A2B2q, J 8.8 Hz), 4.82 (1 H, dd, J 4.5, 9.0 Hz), 4.30 (2 H, t, J 6.2 Hz), 3.35 (2 H, t, J 6.2 Hz), 3.25 (1 H, dd, J 4.3, 14.2 Hz), 3.01 (1 H, dd, J 8.8, 14.2 Hz), 2.73 (2 H, q, J 7.6 Hz), 1.19 (3 H, t, J 7.6 Hz).
Figure 5 provides an illustrative XRPD pattern of (R)-5-{4-[2-(5-Ethyl-pyridin-2-yl)- ethoxy]-benzyl}-thiazolidine-2,4-dione naphthalene-1 ,5-disulfonic acid (2:1 ).
The main diffraction peaks and relative intensities for the peaks shown in Figure 5 are listed in Table 5.
Example 11

H3P04
(R)-5^4-[2-(5-Ethyi-pyridin-2-yl)-ethoxy]-b3nzy!}-thia2oiidine-2,4-dione
phosphate
(5R)-5^4-[2-(5-ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione (-)-Ο,Ο'- dibenzoyl-L-tartrate (2.1 g, 2.94mmol) (98.07% d.e.) was added to a solution of phosphoric acid (720mg) in acetone (210ml) at 40°C. The solution was partially concentrated and allowed to cool. The solution was stirred for 1 h before the solid was collected by filtration, washed with acetone and dried at 35°C for 64h to give the title compound. (Method 3) 98.67%, Rt 15.67min.; 1.33%, Rt 17.87min.; d.e. 97.34 %. LCMS (Method 6) Rt 2.89min, m/z 357 [(M-phosphate)H+], 98.9%. DSC 177°C. 1H NMR (400 MHz, DMSO-d6): δ 12.0 (1 H, s), 8.65 (1 H, d, J 1 .7 Hz), 8.36 (1 H, bd, J 8.5 Hz), 7.82 (1 H, d, J 8.2 Hz), 7.15, 6.87 (4 H, A2B2q, J 8.5 Hz), 4.86 (1 H, dd, J 4.6, 8.9 Hz), 4.36 (2 H, t, J 6.3 Hz), 3.39 (2 H, t, J 6.2 Hz), 3.29 (1 H, dd, J 4.4, 14.2 Hz), 3.05 (1 H, dd, J 8.9, 14.2 Hz), 2.75 (2 H, q, J 7.6 Hz), 1.22 (3 H, t, J 7.6 Hz). Figure 6 provides an illustrative XRPD pattern of (R)-5-{4-[2-(5-Ethyl-pyridin-2-yl)- ethoxy]-benzyl}-thiazolidine-2,4-dione phosphate.
The main diffraction peaks and relative intensities for the peaks shown in Figure 6 are listed in Table 6.
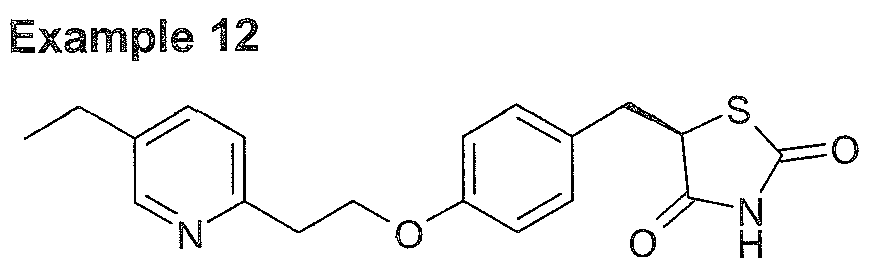
HO' "
o
(R)-5-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-thiazolidine-2,4-dione 2- hydroxy-ethanesulfonic acid
(5R)-5-{4-[2-(5-Ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione (-)-Ο,Ο'- dibenzoyl-L-tartrate (98.07% d.e.) (2.7g, 3.78mmol) was added to a solution of hydroxy-ethanesulfonic acid (1.19g) in I PA (27ml_) at 45°C. Heating was stopped and isopropyl acetate (60mL) was added to the stirred solution to give some solid which was removed by filtration. The solids were collected by filtration after 1 h, washed with isopropyl acetate and dried in vacuo at 35°C for 64h to give the title compound (1.28g). (Method 3) 98.56%, Rt 15.57min.; 1.44%, Rt 17.78min.; d.e. 97.12 %. LCMS (Method 6) Rt 2.86min, m/z 357 [(M- hydroxy-ethanesulfonic acid )H+], 99.4%. DSC 106°C. 1H NMR (400 MHz, DMSO- /6): δ 1.23 ( 3H, t, J 8 Hz), 2.61 (2H, t, J 8 Hz),
2.77 (2H, q, J 8 Hz), 3.05 (1 H, dd, J 12, 16 Hz), 3.29 (1 H, dd, J 4, 16 Hz), 3.40 (2H, t, J 8 Hz), 3.62 (2H, t, J = 4 Hz), 4.35 (2H, t, J = 4 Hz), 4.86 (1 H, dd, J = 4, 8 Hz), 6.86 (2H, d, J 8 Hz), 7.15 (2H, d, J = 8 Hz), 7.94 (1 H, J 8 Hz), 8.33-8.40 (1 H, m), 8.70- 8.73 (1 H, m), 1 1.98 (1 H, br s).
Figure 7 provides an illustrative XRPD pattern of (R)-5-{4-[2-(5-Ethyl-pyridin-2-yl)- ethoxy]-benzyl}-thiazolidine-2,4-dione 2-hydroxy-ethanesulfonic acid .
The main diffraction peaks and relative intensities for the peaks shown in Figure 7 are listed in Table 7.
Claims
1. A crystalline acid addition salt of SR-pioglitazone, said salt being selected from the hydrochloride, hydrobromide, (-)-0,0'-dibenzoyl-L-tartrate, phosphate, hydroxy-ethanesulfonic acid and the naphthalene-1 ,5-disulfonic acid (2:1 ) salts thereof, wherein the naphthalene- ,5-disulfonic acid salt contains 2 molecules of (5R)-5-{4-[2-(5-ethylpyridin-2-yl)ethoxy]benzyl}-1 ,3-thiazolidine-2,4-dione for each molecule of naphthalene-1 ,5-disulfonic acid.
2. A crystalline form of the (-)-0,0'-dibenzoyl-L-tartrate salt as claimed in claim 1 that is characterized by having an X-ray powder diffraction pattern comprising main peaks at 7.30, 9.45 and 17.49 degree 2-theta, and/or a melting point of 153°C.
3. A crystalline form of the hydrochloride salt as claimed in claim 1 that is characterized by having an X-ray powder diffraction pattern comprising main peaks at 8.84, 20.04 and 26.04 degree 2-theta, and/or a melting point of 201 °C.
4. A crystalline form of the hydrobromide salt as claimed in claim 1 that is characterized by having an X-ray powder diffraction pattern comprising main peaks at 20.01 , 22.50 and 25.99 degree 2-theta, and/or a melting point of 207°C.
5. A crystalline form of the naphthalene-1 ,5-disulfonic acid (2:1 ) salt as claimed in claim 1 that is characterized by having an X-ray powder diffraction pattern comprising main peaks at 5.59, 16.83 and 24.05 degree 2-theta, and/or a melting point of 240°C.
6. A crystalline form of the phosphate salt as claimed in claim 1 that is characterized by having an X-ray powder diffraction pattern comprising main peaks at 16.39, 17.60 and 22.01 degree 2-theta, and/or a melting point of 177°C.
7. A crystalline form of the 2-hydroxy-ethanesulfonic acid salt as claimed in claim 1 that is characterized by having an X-ray powder diffraction pattern comprising main peaks at 17.86, 20.44 and 23.05 degree 2-theta, and/or a melting point of 106°C.
8. A pharmaceutical composition comprising a salt as claimed in any of the preceding claims, together with one or more pharmaceutically acceptable carriers and/or excipients.
9. A pharmaceutical composition as claimed in claim 8, which is adapted for pulmonary administration by inhalation.
10. A salt as claimed in any of claims 1 to 7 for use in the treatment of inflammatory respiratory disease by pulmonary administration by inhalation.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/GB2010/050200 WO2011098746A1 (en) | 2010-02-09 | 2010-02-09 | Crystalline acid addition salts of ( 5r) -enanti0mer of pioglitazone |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/GB2010/050200 WO2011098746A1 (en) | 2010-02-09 | 2010-02-09 | Crystalline acid addition salts of ( 5r) -enanti0mer of pioglitazone |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011098746A1 true WO2011098746A1 (en) | 2011-08-18 |
Family
ID=42238220
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2010/050200 Ceased WO2011098746A1 (en) | 2010-02-09 | 2010-02-09 | Crystalline acid addition salts of ( 5r) -enanti0mer of pioglitazone |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011098746A1 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013092269A1 (en) | 2011-12-19 | 2013-06-27 | Ares Trading S.A. | Pharmaceutical compositions comprising glitazones and nrf2 activators |
| US9504679B2 (en) | 2011-12-19 | 2016-11-29 | Bjoern Colin Kahrs | Pharmaceutical compositions comprising glitazones and Nrf2 activators |
| US9585855B2 (en) | 2008-06-19 | 2017-03-07 | The University Of Utah Research Foundation | Use of nitrated lipids for treatment of side effects of toxic medical therapies |
| US9663444B2 (en) | 2009-10-02 | 2017-05-30 | Complexa, Inc. | Heteroatom containing substituted fatty acids |
| US9700534B2 (en) | 2007-08-01 | 2017-07-11 | University of Pittsburgh—of the Commonwealth System of Higher Education | Nitrated-fatty acids modulation of type II diabetes |
| US9750725B2 (en) | 2009-07-31 | 2017-09-05 | University of Pittsburgh—of the Commonwealth System of Higher Education | Fatty acids as anti-inflammatory agents |
| US9790167B2 (en) | 2008-05-01 | 2017-10-17 | Complexa, Inc. | Vinyl substituted fatty acids |
| US10010532B2 (en) | 2011-08-19 | 2018-07-03 | The University Of Utah Research Foundation | Combination therapy with nitrated lipids and inhibitors of the renin-angiotensin-aldosterone system |
| US10369125B2 (en) | 2008-06-19 | 2019-08-06 | The University Of Utah Research Foundation | Method of treating renal system damage |
| US10537541B2 (en) | 2015-10-02 | 2020-01-21 | Complexa Inc. | Treatment of focal segmental glomerular sclerosis (FSGS) using therapeutically effective oral doses of 10-nitro-9(E)-octadec-9-enoic acid |
| US11608342B2 (en) | 2015-07-07 | 2023-03-21 | H. Lundbeck A/S | PDE9 inhibitors with imidazo triazinone backbone and imidazo pyrazinone backbone for treatment of peripheral diseases |
| JP2023532950A (en) * | 2020-06-30 | 2023-08-01 | ポクセル・ソシエテ・アノニム | Crystalline form of deuterium-enriched pioglitazone |
| US12006319B2 (en) | 2018-05-25 | 2024-06-11 | Cardurion Pharmaceuticals, Inc. | Monohydrate and crystalline forms of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl-7H-imidazo[1,5-a]pyrazin-8-one |
| US12213975B2 (en) | 2018-08-31 | 2025-02-04 | Cardurion Pharmaceuticals, Inc. | PDE9 inhibitors for treating sickle cell disease |
Citations (92)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0147716A2 (en) | 1983-12-24 | 1985-07-10 | ANT Nachrichtentechnik GmbH | Method and apparatus for the encipherable transmission of a series of binary information signals with authenticity check |
| EP0505321A2 (en) | 1991-03-21 | 1992-09-23 | Ciba-Geigy Ag | Inhaler |
| US5159448A (en) | 1990-11-20 | 1992-10-27 | Sony Corporation | Highly efficient coding apparatus |
| JPH0525045B2 (en) | 1985-08-09 | 1993-04-09 | Noritoshi Nakabachi | |
| WO1993018007A1 (en) | 1992-03-13 | 1993-09-16 | Tokyo Tanabe Company Limited | Novel carbostyril derivative |
| WO1999064035A1 (en) | 1998-06-08 | 1999-12-16 | Advanced Medicine, Inc. | β2-ADRENERGIC RECEPTOR AGONISTS |
| WO2000021014A2 (en) | 1998-10-06 | 2000-04-13 | Spalding Sports Worldwide, Inc. | Computerized game ball customization system |
| WO2000032585A1 (en) | 1998-12-02 | 2000-06-08 | Lek Pharmaceutical And Chemical Company D.D. | Process for the preparation of simvastatin and analogs thereof |
| WO2000053601A1 (en) | 1999-03-08 | 2000-09-14 | The University Of Mississippi | 1,2-dithiolane derivatives |
| WO2000062766A2 (en) | 1999-04-15 | 2000-10-26 | Smithkline Beecham Plc | Uses of ppar-gamma agonists in neutrophil-induced diseases |
| WO2000075114A1 (en) | 1999-06-04 | 2000-12-14 | Novartis Ag | Beta2-adrenoceptor agonists |
| WO2001042193A1 (en) | 1999-12-08 | 2001-06-14 | Theravance, Inc. | β2-ADRENERGIC RECEPTOR AGONISTS |
| WO2001083462A1 (en) | 2000-04-27 | 2001-11-08 | Boehringer Ingelheim Pharma Kg | Novel, slow-acting betamimetics, a method for their production and their use as medicaments |
| WO2002012265A1 (en) | 2000-08-05 | 2002-02-14 | Glaxo Group Limited | 6.ALPHA., 9.ALPHA.-DIFLUORO-17.ALPHA.-`(2-FURANYLCARBOXYL) OXY!-11.BETA.-HYDROXY-16.ALPHA.-METHYL-3-OXO-ANDROST-1,4,-DIENE-17-CARBOTHIOIC ACID S-FLUOROMETHYL ESTER AS AN ANTI-INFLAMMATORY AGENT |
| WO2002013812A1 (en) | 2000-08-17 | 2002-02-21 | Pershadsingh Harrihar A | Methods for treating inflammatory diseases |
| US20020055651A1 (en) | 1999-06-02 | 2002-05-09 | Moran Edmund J. | Beta2-adrenergic receptor agonists |
| WO2002066422A1 (en) | 2001-02-14 | 2002-08-29 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2002070490A1 (en) | 2001-03-08 | 2002-09-12 | Glaxo Group Limited | Agonists of beta-adrenoceptors |
| WO2002076933A1 (en) | 2001-03-22 | 2002-10-03 | Glaxo Group Limited | Formailide derivatives as beta2-adrenoreceptor agonists |
| WO2002100879A1 (en) | 2001-06-12 | 2002-12-19 | Glaxo Group Limited | Novel anti-inflammatory 17.alpha.-heterocyclic-esters of 17.beta.carbothioate androstane derivatives |
| WO2003024439A1 (en) | 2001-09-14 | 2003-03-27 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2003042160A1 (en) | 2001-11-13 | 2003-05-22 | Theravance, Inc. | Aryl aniline beta-2 adrenergic receptor agonists |
| WO2003042229A1 (en) | 2001-11-12 | 2003-05-22 | Glaxo Group Limited | Non-aromatic heterocyclic esters of furan-2-one-esters of 17.beta.-carboxyl or 17.beta.-carbothio glucocorticoids |
| WO2003042164A1 (en) | 2001-11-13 | 2003-05-22 | Theravance, Inc | Aryl aniline beta-2 adrenergic receptor agonists |
| WO2003048181A1 (en) | 2001-12-01 | 2003-06-12 | Glaxo Group Limited | 17.alpha. -cyclic esters of 16-methylpregnan-3,20-dione as anti-inflammatory agents |
| WO2003062259A2 (en) | 2002-01-21 | 2003-07-31 | Glaxo Group Limited | Non-aromatic 17.alpha.-esters of androstane-17.beta.-carboxylate esters as anti-inflammatory agents |
| WO2003072539A1 (en) | 2002-02-28 | 2003-09-04 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2003091204A1 (en) | 2002-04-25 | 2003-11-06 | Glaxo Group Limited | Phenethanolamine derivatives |
| WO2003099764A1 (en) | 2002-05-28 | 2003-12-04 | Theravance, Inc. | ALKOXY ARYL β2 ADRENERGIC RECEPTOR AGONISTS |
| WO2004016578A2 (en) | 2002-07-25 | 2004-02-26 | Glaxo Group Limited | Arylethanolamine beta2-adrenoreceptor agonist compounds |
| WO2004016601A1 (en) | 2002-08-09 | 2004-02-26 | Novartis Ag | Benzothiazole derivatives having beta-2-adrenoreceptor agonist activity |
| WO2004022547A1 (en) | 2002-09-06 | 2004-03-18 | Glaxo Group Limited | Phenethanolamine derivatives and their use in the treatment of respiratory diseases |
| WO2004032921A1 (en) | 2002-10-11 | 2004-04-22 | Pfizer Limited | Indole derivatives as beta-2 agonists |
| WO2004033412A1 (en) | 2002-10-04 | 2004-04-22 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel beta mimetics with extended duration of action, method for production and use thereof as medicaments |
| WO2004037807A2 (en) | 2002-10-22 | 2004-05-06 | Glaxo Group Limited | Medicinal arylethanolamine compounds |
| WO2004037773A1 (en) | 2002-10-28 | 2004-05-06 | Glaxo Group Limited | Phenethanolamine derivative for the treatment of respiratory diseases |
| WO2004037768A2 (en) | 2002-10-28 | 2004-05-06 | Glaxo Group Limited | Phenethanolamine derivatives |
| WO2004039762A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenethanolamine derivatives for the treatment of respiratory diseases |
| WO2004039766A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenylethanolamine derivatives for the treatment of respiratory diseases |
| WO2004045618A2 (en) | 2002-11-15 | 2004-06-03 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel medicaments for the treatment of chronic obstructive pulmonary diseases |
| WO2004046083A1 (en) | 2002-11-15 | 2004-06-03 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel dihydroxy-methylphenyl derivatives, method for the production and use thereof as medicaments |
| DE10258695A1 (en) | 2002-12-16 | 2004-06-24 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | New 2-(2-hydroxy-2-(3-methylsulfonylamino-4-hydroxyphenyl)ethylamino-4-(pyrid-2-ylamino)-2-methylbutane used e.g. for treating asthma and chronic obstructive pulmonary disease |
| EP1440966A1 (en) | 2003-01-10 | 2004-07-28 | Pfizer Limited | Indole derivatives useful for the treatment of diseases |
| WO2004071388A2 (en) | 2003-02-14 | 2004-08-26 | Glaxo Group Limited | Medicinal compounds |
| EP1460064A1 (en) | 2003-03-14 | 2004-09-22 | Pfizer Limited | Indole-2-carboxamide derivatives useful as beta-2 agonists |
| WO2004087142A1 (en) | 2003-04-04 | 2004-10-14 | Novartis Ag | Quinoline-2-one-derivatives for the treatment of airways diseases |
| WO2004089892A2 (en) | 2003-04-01 | 2004-10-21 | Theravance, Inc. | Diarylmethyl and related compounds having beta2 andrenergic receptor agonist and muscarinic receptor antagonist activity |
| US20040229904A1 (en) | 2003-05-15 | 2004-11-18 | Pfizer Inc | Compounds useful for the treatment of diseases |
| US20040242622A1 (en) | 2003-05-28 | 2004-12-02 | Mathai Mammen | Azabicycloalkane compounds |
| WO2004108675A1 (en) | 2003-06-04 | 2004-12-16 | Pfizer Limited | 2-amino-pyridine derivatives as beta-2 adrenoreceptor agonists |
| WO2004108676A1 (en) | 2003-06-04 | 2004-12-16 | Pfizer Limited | 2-(6-amino-pyridin-3-yl)-2-hydroxyethylamine derivatives as beta 2-adrenoceptors agonists |
| WO2005003098A1 (en) | 2003-07-01 | 2005-01-13 | Schering Aktiengesellschaft | Heterocyclically substituted pentanol derivatives, method for the production thereof, and use thereof as anti-inflammatory agents |
| WO2005033121A2 (en) | 2003-10-03 | 2005-04-14 | King Pharmaceuticals Research & Development, Inc. | Synthesis of 2-aralkyloxyadenosines, 2-alkoxyadenosines, and their analogs |
| WO2005035518A1 (en) | 2003-10-08 | 2005-04-21 | Schering Aktiengesellschaft | Rearranged pentanols, a method for the production thereof, and their use as antiphlogistics |
| WO2005034939A1 (en) | 2003-10-08 | 2005-04-21 | Schering Aktiengesellschaft | 1-amino-2-oxy-substituted tetrahydronaphtalene derivatives, methods for the production thereof, and their use as antiphlogistics |
| WO2005035502A1 (en) | 2003-10-06 | 2005-04-21 | Schering Aktiengesellschaft | 1-(quinoline amino) and 1-(isoquinoline amino)-substituted pentan-2-ols, method for their production and their use as anti-inflammatories |
| WO2005040103A1 (en) | 2003-10-22 | 2005-05-06 | Glaxo Group Limited | Medicinal compounds |
| WO2005044787A1 (en) | 2003-10-24 | 2005-05-19 | Glaxo Group Limited | Phenetanolamine derivatives |
| US20050133417A1 (en) | 2003-12-19 | 2005-06-23 | Bhan Opinder K. | Systems, methods, and catalysts for producing a crude product |
| WO2005058867A1 (en) | 2003-12-17 | 2005-06-30 | Glaxo Group Limited | Benzothiophene-and thiochromene containing phenethanolamine derivatives for the treatment of respiratory disorders |
| WO2005058299A1 (en) | 2003-12-09 | 2005-06-30 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2005066140A1 (en) | 2004-01-09 | 2005-07-21 | Boehringer Ingelheim International Gmbh | 3-hydroxymethyl-4-hydroxy-phenyl derivatives for the treatment of respiratory illnesses |
| WO2005065650A2 (en) | 2003-10-09 | 2005-07-21 | Glaxo Group Limited | Aerosol formulations comprising a carboxylic acid surfactant |
| WO2005070908A1 (en) | 2004-01-23 | 2005-08-04 | Boehringer Ingelheim International Gmbh | Novel long-working beta-2-agonists and use thereof as medicaments |
| US20050171147A1 (en) | 2004-01-22 | 2005-08-04 | Brown Alan D. | Sulfonamide derivatives for the treatment of diseases |
| US20050182091A1 (en) | 2004-01-22 | 2005-08-18 | Brown Alan D. | Sulfonamide derivatives for the treatment of diseases |
| WO2005077361A1 (en) | 2004-02-14 | 2005-08-25 | Boehringer Ingelheim International Gmbh | Novel, sustained-action beta-2-agonists and their use as medicaments |
| WO2005090288A1 (en) | 2004-03-17 | 2005-09-29 | Pfizer Limited | Phenylaminoethanol derivates as beta2 receptor agonists |
| WO2005092841A1 (en) | 2004-03-23 | 2005-10-06 | Pfizer Limited | Compounds having beta-agonist activity |
| WO2005092861A1 (en) | 2004-03-11 | 2005-10-06 | Pfizer Limited | Quinolinone derivatives pharmaceutical compositions containing them and their use |
| WO2005092860A1 (en) | 2004-03-23 | 2005-10-06 | Pfizer Limited | Compounds for the treatment of diseases |
| WO2005092087A2 (en) | 2004-03-22 | 2005-10-06 | Mars Incorporated | Animal chew |
| WO2005092870A1 (en) | 2004-03-17 | 2005-10-06 | Boehringer Ingelheim International Gmbh | Novel benzoxazinone derivatives used as long-acting betamimetics for treating respiratory illnesses |
| WO2005092840A1 (en) | 2004-03-23 | 2005-10-06 | Pfizer Limited | Formamide derivatives useful as adrenoceptor |
| WO2005092887A1 (en) | 2004-03-23 | 2005-10-06 | Pfizer Limited | Compounds for the treatment of diseases |
| WO2005110359A1 (en) | 2004-05-14 | 2005-11-24 | Boehringer Ingelheim International Gmbh | Inhalation powder formulations containing enantiomerically pure beta-agonists |
| WO2005110990A1 (en) | 2004-05-13 | 2005-11-24 | Boehringer Ingelheim International Gmbh | Hydroxy-substituted benzo-condensed heterocycles for use as beta agonists in the treatment of respiratory diseases |
| WO2005111005A1 (en) | 2004-05-14 | 2005-11-24 | Boehringer Ingelheim International Gmbh | Novel enantiomerically pure beta-agonists, method for the production and the use thereof in the form of a drug |
| WO2005111002A2 (en) | 2004-05-13 | 2005-11-24 | Boehringer Ingelheim International Gmbh | Substituted cycloalkyl derivatives for use in the treatment of respiratory diseases |
| US20050272769A1 (en) | 2004-06-03 | 2005-12-08 | Theravance, Inc. | Diamine beta2 adrenergic receptor agonists |
| US20060019991A1 (en) | 2004-07-21 | 2006-01-26 | Theravance, Inc. | Diaryl ether beta2 adrenergic receptor agonists |
| WO2006016245A1 (en) | 2004-08-05 | 2006-02-16 | Ranbaxy Laboratories Limited | Muscarinic receptor antagonists |
| WO2006031556A2 (en) | 2004-09-10 | 2006-03-23 | Theravance. Inc. | Amidine substituted aryl aniline compounds |
| WO2006032627A1 (en) | 2004-09-21 | 2006-03-30 | Boehringer Ingelheim International Gmbh | Heteroaryl compounds for use as betamimetics in the treatment of respiratory diseases |
| US20060106213A1 (en) | 2002-11-15 | 2006-05-18 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | New Medicaments for the Treatment of Chronic Obstructive Pulmonary Disease |
| WO2006051373A1 (en) | 2004-11-12 | 2006-05-18 | Pfizer Limited | Compounds for the treatment of diseases |
| WO2006056471A1 (en) | 2004-11-29 | 2006-06-01 | Novartis Ag | 5-hydroxy-benzothiazole derivatives having beta-2-adrenorecptor agonist activity |
| WO2007100027A1 (en) | 2006-03-02 | 2007-09-07 | Daiichi Sankyo Company, Limited | Optically active thiazolidinedione derivative |
| WO2008096112A1 (en) | 2007-02-08 | 2008-08-14 | Astrazeneca Ab | Salts 668 |
| WO2008096119A1 (en) | 2007-02-08 | 2008-08-14 | Astrazeneca Ab | Salts 669 |
| WO2008104790A1 (en) | 2007-03-01 | 2008-09-04 | Astrazeneca Ab | Salts of a selective beta-2 andrenoceptor agonist |
| WO2010015818A1 (en) * | 2008-08-07 | 2010-02-11 | Argenta Discovery Limited | Respiratory disease treatment |
-
2010
- 2010-02-09 WO PCT/GB2010/050200 patent/WO2011098746A1/en not_active Ceased
Patent Citations (99)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0147716A2 (en) | 1983-12-24 | 1985-07-10 | ANT Nachrichtentechnik GmbH | Method and apparatus for the encipherable transmission of a series of binary information signals with authenticity check |
| JPH0525045B2 (en) | 1985-08-09 | 1993-04-09 | Noritoshi Nakabachi | |
| US5159448A (en) | 1990-11-20 | 1992-10-27 | Sony Corporation | Highly efficient coding apparatus |
| EP0505321A2 (en) | 1991-03-21 | 1992-09-23 | Ciba-Geigy Ag | Inhaler |
| WO1993018007A1 (en) | 1992-03-13 | 1993-09-16 | Tokyo Tanabe Company Limited | Novel carbostyril derivative |
| WO1999064035A1 (en) | 1998-06-08 | 1999-12-16 | Advanced Medicine, Inc. | β2-ADRENERGIC RECEPTOR AGONISTS |
| WO2000021014A2 (en) | 1998-10-06 | 2000-04-13 | Spalding Sports Worldwide, Inc. | Computerized game ball customization system |
| WO2000032585A1 (en) | 1998-12-02 | 2000-06-08 | Lek Pharmaceutical And Chemical Company D.D. | Process for the preparation of simvastatin and analogs thereof |
| WO2000053601A1 (en) | 1999-03-08 | 2000-09-14 | The University Of Mississippi | 1,2-dithiolane derivatives |
| WO2000062766A2 (en) | 1999-04-15 | 2000-10-26 | Smithkline Beecham Plc | Uses of ppar-gamma agonists in neutrophil-induced diseases |
| US20020055651A1 (en) | 1999-06-02 | 2002-05-09 | Moran Edmund J. | Beta2-adrenergic receptor agonists |
| WO2000075114A1 (en) | 1999-06-04 | 2000-12-14 | Novartis Ag | Beta2-adrenoceptor agonists |
| WO2001042193A1 (en) | 1999-12-08 | 2001-06-14 | Theravance, Inc. | β2-ADRENERGIC RECEPTOR AGONISTS |
| WO2001083462A1 (en) | 2000-04-27 | 2001-11-08 | Boehringer Ingelheim Pharma Kg | Novel, slow-acting betamimetics, a method for their production and their use as medicaments |
| WO2002012265A1 (en) | 2000-08-05 | 2002-02-14 | Glaxo Group Limited | 6.ALPHA., 9.ALPHA.-DIFLUORO-17.ALPHA.-`(2-FURANYLCARBOXYL) OXY!-11.BETA.-HYDROXY-16.ALPHA.-METHYL-3-OXO-ANDROST-1,4,-DIENE-17-CARBOTHIOIC ACID S-FLUOROMETHYL ESTER AS AN ANTI-INFLAMMATORY AGENT |
| WO2002012266A1 (en) | 2000-08-05 | 2002-02-14 | Glaxo Group Limited | 17.beta.-carbothioate 17.alpha.-arylcarbonyloxyloxy androstane derivative as anti-inflammatory agents |
| WO2002013812A1 (en) | 2000-08-17 | 2002-02-21 | Pershadsingh Harrihar A | Methods for treating inflammatory diseases |
| WO2002066422A1 (en) | 2001-02-14 | 2002-08-29 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2002070490A1 (en) | 2001-03-08 | 2002-09-12 | Glaxo Group Limited | Agonists of beta-adrenoceptors |
| WO2002076933A1 (en) | 2001-03-22 | 2002-10-03 | Glaxo Group Limited | Formailide derivatives as beta2-adrenoreceptor agonists |
| WO2002100879A1 (en) | 2001-06-12 | 2002-12-19 | Glaxo Group Limited | Novel anti-inflammatory 17.alpha.-heterocyclic-esters of 17.beta.carbothioate androstane derivatives |
| WO2003024439A1 (en) | 2001-09-14 | 2003-03-27 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2003042229A1 (en) | 2001-11-12 | 2003-05-22 | Glaxo Group Limited | Non-aromatic heterocyclic esters of furan-2-one-esters of 17.beta.-carboxyl or 17.beta.-carbothio glucocorticoids |
| WO2003042164A1 (en) | 2001-11-13 | 2003-05-22 | Theravance, Inc | Aryl aniline beta-2 adrenergic receptor agonists |
| WO2003042160A1 (en) | 2001-11-13 | 2003-05-22 | Theravance, Inc. | Aryl aniline beta-2 adrenergic receptor agonists |
| WO2003048181A1 (en) | 2001-12-01 | 2003-06-12 | Glaxo Group Limited | 17.alpha. -cyclic esters of 16-methylpregnan-3,20-dione as anti-inflammatory agents |
| WO2003062259A2 (en) | 2002-01-21 | 2003-07-31 | Glaxo Group Limited | Non-aromatic 17.alpha.-esters of androstane-17.beta.-carboxylate esters as anti-inflammatory agents |
| WO2003072539A1 (en) | 2002-02-28 | 2003-09-04 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2003091204A1 (en) | 2002-04-25 | 2003-11-06 | Glaxo Group Limited | Phenethanolamine derivatives |
| WO2003099764A1 (en) | 2002-05-28 | 2003-12-04 | Theravance, Inc. | ALKOXY ARYL β2 ADRENERGIC RECEPTOR AGONISTS |
| WO2004016578A2 (en) | 2002-07-25 | 2004-02-26 | Glaxo Group Limited | Arylethanolamine beta2-adrenoreceptor agonist compounds |
| WO2004016601A1 (en) | 2002-08-09 | 2004-02-26 | Novartis Ag | Benzothiazole derivatives having beta-2-adrenoreceptor agonist activity |
| US20060106075A1 (en) | 2002-08-09 | 2006-05-18 | Bernard Cuenoud | Benzothiazole derivatives having beta-2-adrenoreceptor agonist activity |
| WO2004022547A1 (en) | 2002-09-06 | 2004-03-18 | Glaxo Group Limited | Phenethanolamine derivatives and their use in the treatment of respiratory diseases |
| WO2004033412A1 (en) | 2002-10-04 | 2004-04-22 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel beta mimetics with extended duration of action, method for production and use thereof as medicaments |
| WO2004032921A1 (en) | 2002-10-11 | 2004-04-22 | Pfizer Limited | Indole derivatives as beta-2 agonists |
| WO2004037807A2 (en) | 2002-10-22 | 2004-05-06 | Glaxo Group Limited | Medicinal arylethanolamine compounds |
| WO2004037773A1 (en) | 2002-10-28 | 2004-05-06 | Glaxo Group Limited | Phenethanolamine derivative for the treatment of respiratory diseases |
| WO2004037768A2 (en) | 2002-10-28 | 2004-05-06 | Glaxo Group Limited | Phenethanolamine derivatives |
| WO2004039762A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenethanolamine derivatives for the treatment of respiratory diseases |
| WO2004039766A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenylethanolamine derivatives for the treatment of respiratory diseases |
| WO2004045618A2 (en) | 2002-11-15 | 2004-06-03 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel medicaments for the treatment of chronic obstructive pulmonary diseases |
| WO2004046083A1 (en) | 2002-11-15 | 2004-06-03 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel dihydroxy-methylphenyl derivatives, method for the production and use thereof as medicaments |
| US20060106213A1 (en) | 2002-11-15 | 2006-05-18 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | New Medicaments for the Treatment of Chronic Obstructive Pulmonary Disease |
| DE10258695A1 (en) | 2002-12-16 | 2004-06-24 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | New 2-(2-hydroxy-2-(3-methylsulfonylamino-4-hydroxyphenyl)ethylamino-4-(pyrid-2-ylamino)-2-methylbutane used e.g. for treating asthma and chronic obstructive pulmonary disease |
| EP1440966A1 (en) | 2003-01-10 | 2004-07-28 | Pfizer Limited | Indole derivatives useful for the treatment of diseases |
| WO2004071388A2 (en) | 2003-02-14 | 2004-08-26 | Glaxo Group Limited | Medicinal compounds |
| EP1460064A1 (en) | 2003-03-14 | 2004-09-22 | Pfizer Limited | Indole-2-carboxamide derivatives useful as beta-2 agonists |
| WO2004080964A1 (en) | 2003-03-14 | 2004-09-23 | Pfizer Limited | Indole derivatives useful for the treatment of diseases |
| WO2004089892A2 (en) | 2003-04-01 | 2004-10-21 | Theravance, Inc. | Diarylmethyl and related compounds having beta2 andrenergic receptor agonist and muscarinic receptor antagonist activity |
| WO2004087142A1 (en) | 2003-04-04 | 2004-10-14 | Novartis Ag | Quinoline-2-one-derivatives for the treatment of airways diseases |
| US20040229904A1 (en) | 2003-05-15 | 2004-11-18 | Pfizer Inc | Compounds useful for the treatment of diseases |
| US20040242622A1 (en) | 2003-05-28 | 2004-12-02 | Mathai Mammen | Azabicycloalkane compounds |
| WO2004108676A1 (en) | 2003-06-04 | 2004-12-16 | Pfizer Limited | 2-(6-amino-pyridin-3-yl)-2-hydroxyethylamine derivatives as beta 2-adrenoceptors agonists |
| WO2004108675A1 (en) | 2003-06-04 | 2004-12-16 | Pfizer Limited | 2-amino-pyridine derivatives as beta-2 adrenoreceptor agonists |
| WO2005003098A1 (en) | 2003-07-01 | 2005-01-13 | Schering Aktiengesellschaft | Heterocyclically substituted pentanol derivatives, method for the production thereof, and use thereof as anti-inflammatory agents |
| WO2005033121A2 (en) | 2003-10-03 | 2005-04-14 | King Pharmaceuticals Research & Development, Inc. | Synthesis of 2-aralkyloxyadenosines, 2-alkoxyadenosines, and their analogs |
| WO2005035502A1 (en) | 2003-10-06 | 2005-04-21 | Schering Aktiengesellschaft | 1-(quinoline amino) and 1-(isoquinoline amino)-substituted pentan-2-ols, method for their production and their use as anti-inflammatories |
| WO2005034939A1 (en) | 2003-10-08 | 2005-04-21 | Schering Aktiengesellschaft | 1-amino-2-oxy-substituted tetrahydronaphtalene derivatives, methods for the production thereof, and their use as antiphlogistics |
| WO2005035518A1 (en) | 2003-10-08 | 2005-04-21 | Schering Aktiengesellschaft | Rearranged pentanols, a method for the production thereof, and their use as antiphlogistics |
| WO2005065650A2 (en) | 2003-10-09 | 2005-07-21 | Glaxo Group Limited | Aerosol formulations comprising a carboxylic acid surfactant |
| WO2005040103A1 (en) | 2003-10-22 | 2005-05-06 | Glaxo Group Limited | Medicinal compounds |
| WO2005044787A1 (en) | 2003-10-24 | 2005-05-19 | Glaxo Group Limited | Phenetanolamine derivatives |
| WO2005058299A1 (en) | 2003-12-09 | 2005-06-30 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2005058867A1 (en) | 2003-12-17 | 2005-06-30 | Glaxo Group Limited | Benzothiophene-and thiochromene containing phenethanolamine derivatives for the treatment of respiratory disorders |
| US20050133417A1 (en) | 2003-12-19 | 2005-06-23 | Bhan Opinder K. | Systems, methods, and catalysts for producing a crude product |
| WO2005066140A1 (en) | 2004-01-09 | 2005-07-21 | Boehringer Ingelheim International Gmbh | 3-hydroxymethyl-4-hydroxy-phenyl derivatives for the treatment of respiratory illnesses |
| US20050182091A1 (en) | 2004-01-22 | 2005-08-18 | Brown Alan D. | Sulfonamide derivatives for the treatment of diseases |
| WO2005080324A1 (en) | 2004-01-22 | 2005-09-01 | Pfizer Limited | Sulfonamide derivatives for the treatment of diseases |
| WO2005080313A2 (en) | 2004-01-22 | 2005-09-01 | Pfizer Limited | Sulfonamide derivatives for the treatment of diseases |
| US20050171147A1 (en) | 2004-01-22 | 2005-08-04 | Brown Alan D. | Sulfonamide derivatives for the treatment of diseases |
| WO2005070908A1 (en) | 2004-01-23 | 2005-08-04 | Boehringer Ingelheim International Gmbh | Novel long-working beta-2-agonists and use thereof as medicaments |
| WO2005077361A1 (en) | 2004-02-14 | 2005-08-25 | Boehringer Ingelheim International Gmbh | Novel, sustained-action beta-2-agonists and their use as medicaments |
| WO2005092861A1 (en) | 2004-03-11 | 2005-10-06 | Pfizer Limited | Quinolinone derivatives pharmaceutical compositions containing them and their use |
| WO2005090288A1 (en) | 2004-03-17 | 2005-09-29 | Pfizer Limited | Phenylaminoethanol derivates as beta2 receptor agonists |
| WO2005092870A1 (en) | 2004-03-17 | 2005-10-06 | Boehringer Ingelheim International Gmbh | Novel benzoxazinone derivatives used as long-acting betamimetics for treating respiratory illnesses |
| WO2005092087A2 (en) | 2004-03-22 | 2005-10-06 | Mars Incorporated | Animal chew |
| WO2005092860A1 (en) | 2004-03-23 | 2005-10-06 | Pfizer Limited | Compounds for the treatment of diseases |
| WO2005092887A1 (en) | 2004-03-23 | 2005-10-06 | Pfizer Limited | Compounds for the treatment of diseases |
| WO2005092840A1 (en) | 2004-03-23 | 2005-10-06 | Pfizer Limited | Formamide derivatives useful as adrenoceptor |
| WO2005092841A1 (en) | 2004-03-23 | 2005-10-06 | Pfizer Limited | Compounds having beta-agonist activity |
| WO2005110990A1 (en) | 2004-05-13 | 2005-11-24 | Boehringer Ingelheim International Gmbh | Hydroxy-substituted benzo-condensed heterocycles for use as beta agonists in the treatment of respiratory diseases |
| WO2005111002A2 (en) | 2004-05-13 | 2005-11-24 | Boehringer Ingelheim International Gmbh | Substituted cycloalkyl derivatives for use in the treatment of respiratory diseases |
| WO2005110359A1 (en) | 2004-05-14 | 2005-11-24 | Boehringer Ingelheim International Gmbh | Inhalation powder formulations containing enantiomerically pure beta-agonists |
| WO2005111005A1 (en) | 2004-05-14 | 2005-11-24 | Boehringer Ingelheim International Gmbh | Novel enantiomerically pure beta-agonists, method for the production and the use thereof in the form of a drug |
| US20050272769A1 (en) | 2004-06-03 | 2005-12-08 | Theravance, Inc. | Diamine beta2 adrenergic receptor agonists |
| WO2005121065A2 (en) | 2004-06-03 | 2005-12-22 | Theravance, Inc. | DIAMINE β2 ADRENERGIC RECEPTOR AGONISTS |
| US20060019991A1 (en) | 2004-07-21 | 2006-01-26 | Theravance, Inc. | Diaryl ether beta2 adrenergic receptor agonists |
| WO2006014704A1 (en) | 2004-07-21 | 2006-02-09 | Theravance, Inc. | DIARYL ETHER β2 ADRENERGIC RECEPTOR AGONISTS |
| WO2006016245A1 (en) | 2004-08-05 | 2006-02-16 | Ranbaxy Laboratories Limited | Muscarinic receptor antagonists |
| WO2006031556A2 (en) | 2004-09-10 | 2006-03-23 | Theravance. Inc. | Amidine substituted aryl aniline compounds |
| WO2006032627A1 (en) | 2004-09-21 | 2006-03-30 | Boehringer Ingelheim International Gmbh | Heteroaryl compounds for use as betamimetics in the treatment of respiratory diseases |
| WO2006051373A1 (en) | 2004-11-12 | 2006-05-18 | Pfizer Limited | Compounds for the treatment of diseases |
| WO2006056471A1 (en) | 2004-11-29 | 2006-06-01 | Novartis Ag | 5-hydroxy-benzothiazole derivatives having beta-2-adrenorecptor agonist activity |
| WO2007100027A1 (en) | 2006-03-02 | 2007-09-07 | Daiichi Sankyo Company, Limited | Optically active thiazolidinedione derivative |
| WO2008096112A1 (en) | 2007-02-08 | 2008-08-14 | Astrazeneca Ab | Salts 668 |
| WO2008096119A1 (en) | 2007-02-08 | 2008-08-14 | Astrazeneca Ab | Salts 669 |
| WO2008104790A1 (en) | 2007-03-01 | 2008-09-04 | Astrazeneca Ab | Salts of a selective beta-2 andrenoceptor agonist |
| WO2010015818A1 (en) * | 2008-08-07 | 2010-02-11 | Argenta Discovery Limited | Respiratory disease treatment |
Non-Patent Citations (15)
| Title |
|---|
| B. JAMALI ET AL: "Investigation of racemisation of the enantiomers of glitazone drug compounds at different pH using chiral HPLC and chiral CE", JOURNAL OF PHARMACEUTICAL AND BIOCHEMICAL ANALYSIS, vol. 46, 2008, pages 82 - 87, XP002587887 * |
| BIOPHARMACEUTICS AND DRUG DISPOSITION, vol. 18, no. 4, 1997, pages 305 - 24 |
| CHEM. PHARM. BULL, vol. 32, no. 11, 1984, pages 4460 - 65 |
| DAVID KOZMA: "Optical Resolutions via diastereomeric salt formation", 2002, CRC PRESS |
| E. FOGASSYET, TETRAHEDRON: ASYMMETRY, vol. 19, 2008, pages 519 - 536 |
| J. CHROMATOGRAPHY, vol. 835, 2006, pages 40 - 46 |
| J.CLIN. PHARMACOL., vol. 47, 2007, pages 323 - 33 |
| J.JAQUES; S.H.WILEN; A. COLLETT: "Enantiomers Racemates and Resolutions", 1991, WILEY-INTERSCIENCE |
| NARALA VENKATA R ET AL: "Pioglitazone is as effective as dexamethasone in a cockroach allergen-induced murine model of asthma", RESPIRATORY RESEARCH, BIOMED CENTRAL LTD., LONDON, GB, vol. 8, no. 1, 4 December 2007 (2007-12-04), pages 90, XP021031353, ISSN: 1465-9921 * |
| P.NEWMAN: "Optical resolution Procedures ofChiral Compounds", 1978, RESOLUTION INFORMATION CENTER, pages: 1 - 3 |
| PARKS ET AL., BIOORG. MED. CHEM. LETT., vol. 8, no. 24, 1998, pages 3657 - 8 |
| R.A. SCHELDON: "Chirotechnology", 1993, MARCEL DEKKER |
| RAPID COMMUN. MASS SPECTROM., vol. 19, 2005, pages 1125 - 9 |
| S.H.WILEN: "Topics in Stereochemistry", vol. 6, 1972, WILEY-INTERSCIENCE, pages: 107 |
| T. J. MED. CHEM., vol. 34, 1991, pages 319 - 25 |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9700534B2 (en) | 2007-08-01 | 2017-07-11 | University of Pittsburgh—of the Commonwealth System of Higher Education | Nitrated-fatty acids modulation of type II diabetes |
| US10576051B2 (en) | 2007-08-01 | 2020-03-03 | University of Pittsburgh—of the Commonwealth System of Higher Education | Nitrated-fatty acids modulation of type II diabetes |
| US10869850B2 (en) | 2007-08-01 | 2020-12-22 | University of Pittsburgh—of the Commonwealth System of Higher Education | Nitrated-fatty acids modulation of type II diabetes |
| US10258589B2 (en) | 2007-08-01 | 2019-04-16 | University of Pittsburgh—of the Commonwealth System of Higher Education | Nitrated-fatty acids modulation of type II diabetes |
| US9790167B2 (en) | 2008-05-01 | 2017-10-17 | Complexa, Inc. | Vinyl substituted fatty acids |
| US10369125B2 (en) | 2008-06-19 | 2019-08-06 | The University Of Utah Research Foundation | Method of treating renal system damage |
| US9585855B2 (en) | 2008-06-19 | 2017-03-07 | The University Of Utah Research Foundation | Use of nitrated lipids for treatment of side effects of toxic medical therapies |
| US10568857B2 (en) | 2008-06-19 | 2020-02-25 | The University Of Utah Research Foundation | Method of treating renal system damage |
| US9750725B2 (en) | 2009-07-31 | 2017-09-05 | University of Pittsburgh—of the Commonwealth System of Higher Education | Fatty acids as anti-inflammatory agents |
| US10213417B2 (en) | 2009-07-31 | 2019-02-26 | University of Pittsburgh—of the Commonwealth System of Higher Education | Fatty acids as anti-inflammatory agents |
| US10835518B2 (en) | 2009-07-31 | 2020-11-17 | University of Pittsburgh—of the Commonwealth System of Higher Education | Fatty acids as anti-inflammatory agents |
| US11723897B2 (en) | 2009-07-31 | 2023-08-15 | University of Pittsburgh—of the Commonwealth System of Higher Education | Fatty acids as anti-inflammatory agents |
| US9663444B2 (en) | 2009-10-02 | 2017-05-30 | Complexa, Inc. | Heteroatom containing substituted fatty acids |
| US10709690B2 (en) | 2011-08-19 | 2020-07-14 | The University Of Utah Research Foundation | Combination therapy with nitrated lipids and inhibitors of the renin-angiotensin-aldosterone system |
| US10010532B2 (en) | 2011-08-19 | 2018-07-03 | The University Of Utah Research Foundation | Combination therapy with nitrated lipids and inhibitors of the renin-angiotensin-aldosterone system |
| US11484530B2 (en) | 2011-12-19 | 2022-11-01 | Bjoern Colin Kahrs | Pharmaceutical compositions comprising the PPAR agonist INT-131 and Nrf2 activators |
| US10426763B2 (en) | 2011-12-19 | 2019-10-01 | Bjoern Colin Kahrs | Pharmaceutical compositions comprising glitazones and NRF2 activators |
| WO2013092269A1 (en) | 2011-12-19 | 2013-06-27 | Ares Trading S.A. | Pharmaceutical compositions comprising glitazones and nrf2 activators |
| US9504679B2 (en) | 2011-12-19 | 2016-11-29 | Bjoern Colin Kahrs | Pharmaceutical compositions comprising glitazones and Nrf2 activators |
| US12083107B2 (en) | 2011-12-19 | 2024-09-10 | Bjoern Colin Kahrs | Pharmaceutical compositions comprising glitazones and Nrf2 activators |
| US11608342B2 (en) | 2015-07-07 | 2023-03-21 | H. Lundbeck A/S | PDE9 inhibitors with imidazo triazinone backbone and imidazo pyrazinone backbone for treatment of peripheral diseases |
| US10537541B2 (en) | 2015-10-02 | 2020-01-21 | Complexa Inc. | Treatment of focal segmental glomerular sclerosis (FSGS) using therapeutically effective oral doses of 10-nitro-9(E)-octadec-9-enoic acid |
| US12006319B2 (en) | 2018-05-25 | 2024-06-11 | Cardurion Pharmaceuticals, Inc. | Monohydrate and crystalline forms of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl-7H-imidazo[1,5-a]pyrazin-8-one |
| US12466832B2 (en) | 2018-05-25 | 2025-11-11 | Cardurion Pharmaceuticals, Inc. | Monohydrate and crystalline forms of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl-7H-imidazo[1,5-a]pyrazin-8-one |
| US12213975B2 (en) | 2018-08-31 | 2025-02-04 | Cardurion Pharmaceuticals, Inc. | PDE9 inhibitors for treating sickle cell disease |
| JP2023532950A (en) * | 2020-06-30 | 2023-08-01 | ポクセル・ソシエテ・アノニム | Crystalline form of deuterium-enriched pioglitazone |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011098746A1 (en) | Crystalline acid addition salts of ( 5r) -enanti0mer of pioglitazone | |
| CA2733440C (en) | Respiratory disease treatment | |
| US9078885B2 (en) | Respiratory disease treatment | |
| JP4484522B2 (en) | Novel fluorene carboxylic acid ester, process for its production and use as a drug | |
| JP4554936B2 (en) | Tropenol and scopine xanthenecarboxylic acid esters as M3 antagonists, processes for their preparation and their use as drugs | |
| JP4484521B2 (en) | Anticholinergics, methods for their production and their use as drugs | |
| WO2010150014A1 (en) | 5r- 5 -deuterated glitazones for respiratory disease treatment | |
| JP2005516067A6 (en) | Novel fluorene carboxylic acid ester, process for its production and use as a drug | |
| PT1981872T (en) | Succinic acid salt of biphenyl-2-ylcarbamic acid 1-[2-(2-chloro-4-{[ (r)-2-hydroxy-2- (8-hydroxy-2-oxo-1, 2-dihydroquinolin-5-yl) eth ylaminoimethyl]} -5-methoxyphenylcarbamoyl) ethyl]piperidin-4-yl ester and its use for the treatment of pulmonary disorders | |
| JP2015155470A (en) | Crystalline oxalate salts of diamide compound | |
| WO2011081937A1 (en) | Corticosteroid-beta-agonist-muscarinic antagonist compounds for use in therapy | |
| WO2011098801A1 (en) | Inflammatory disease treatment | |
| WO2010132743A1 (en) | Corticosteroid beta-agonist compounds for use in therapy | |
| WO2011098799A2 (en) | Respiratory disease treatment | |
| JP2009514779A (en) | Pharmaceutical formulation | |
| CN103249418A (en) | Novel combinations | |
| JP4589006B2 (en) | Novel esters of hydroxy-substituted nitrogen heterocycles as antagonists of muscarinic M3 receptors, their preparation and their use as pharmaceutical compositions | |
| JP2009513531A (en) | Inhalable pharmaceutical formulations containing sugar esters | |
| JP2005520832A (en) | Difurylglycolic acid tropenol ester for use as an anticholinesterase drug | |
| HK1156501B (en) | Respiratory disease treatment | |
| HK1082376B (en) | Fluorene carboxylic acid esters, methods for the production thereof, and use of the same for preparing pharmaceuticals |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10703947 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10703947 Country of ref document: EP Kind code of ref document: A1 |