RU2683279C1 - Новый способ синтеза агомелатина - Google Patents
Новый способ синтеза агомелатина Download PDFInfo
- Publication number
- RU2683279C1 RU2683279C1 RU2016126661A RU2016126661A RU2683279C1 RU 2683279 C1 RU2683279 C1 RU 2683279C1 RU 2016126661 A RU2016126661 A RU 2016126661A RU 2016126661 A RU2016126661 A RU 2016126661A RU 2683279 C1 RU2683279 C1 RU 2683279C1
- Authority
- RU
- Russia
- Prior art keywords
- formula
- compound
- methoxynaphthalen
- synthesis
- iii
- Prior art date
Links
- YJYPHIXNFHFHND-UHFFFAOYSA-N agomelatine Chemical compound C1=CC=C(CCNC(C)=O)C2=CC(OC)=CC=C21 YJYPHIXNFHFHND-UHFFFAOYSA-N 0.000 title claims abstract description 51
- 238000000034 method Methods 0.000 title claims abstract description 45
- 230000015572 biosynthetic process Effects 0.000 title claims abstract description 40
- 238000003786 synthesis reaction Methods 0.000 title claims abstract description 38
- 229960002629 agomelatine Drugs 0.000 title claims abstract description 28
- 150000001875 compounds Chemical class 0.000 claims abstract description 139
- 238000006243 chemical reaction Methods 0.000 claims abstract description 43
- -1 (2,5-dioxopyrrolidin-1-yl)methyl Chemical group 0.000 claims abstract description 20
- 239000007787 solid Substances 0.000 claims abstract description 20
- UNFNRIIETORURP-UHFFFAOYSA-N 7-methoxynaphthalen-2-ol Chemical compound C1=CC(O)=CC2=CC(OC)=CC=C21 UNFNRIIETORURP-UHFFFAOYSA-N 0.000 claims abstract description 11
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 claims abstract description 11
- 239000003638 chemical reducing agent Substances 0.000 claims abstract description 11
- 238000006392 deoxygenation reaction Methods 0.000 claims abstract description 10
- 229910052723 transition metal Inorganic materials 0.000 claims abstract description 9
- 150000003624 transition metals Chemical class 0.000 claims abstract description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 25
- LEQAOMBKQFMDFZ-UHFFFAOYSA-N glyoxal Chemical compound O=CC=O LEQAOMBKQFMDFZ-UHFFFAOYSA-N 0.000 claims description 15
- 230000009471 action Effects 0.000 claims description 14
- 239000000543 intermediate Substances 0.000 claims description 13
- 229910052763 palladium Inorganic materials 0.000 claims description 13
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 12
- 238000005694 sulfonylation reaction Methods 0.000 claims description 11
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 9
- 239000001257 hydrogen Substances 0.000 claims description 9
- 230000006103 sulfonylation Effects 0.000 claims description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 8
- 229940015043 glyoxal Drugs 0.000 claims description 7
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 claims description 6
- 150000004678 hydrides Chemical class 0.000 claims description 6
- 238000001308 synthesis method Methods 0.000 claims description 6
- 229910052784 alkaline earth metal Inorganic materials 0.000 claims description 5
- 150000001342 alkaline earth metals Chemical class 0.000 claims description 5
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 claims description 5
- 229910052759 nickel Inorganic materials 0.000 claims description 5
- DSBYYLKTYXWZNW-UHFFFAOYSA-N 2-(2-hydroxy-7-methoxynaphthalen-1-yl)acetonitrile Chemical compound OC1=C(C2=CC(=CC=C2C=C1)OC)CC#N DSBYYLKTYXWZNW-UHFFFAOYSA-N 0.000 claims description 4
- PMQSYXOMEORYPV-UHFFFAOYSA-N [1-(2-acetamidoethyl)-7-methoxynaphthalen-2-yl] acetate Chemical compound C(C)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CCNC(C)=O PMQSYXOMEORYPV-UHFFFAOYSA-N 0.000 claims description 4
- 125000005425 toluyl group Chemical group 0.000 claims description 3
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 claims description 3
- 150000008064 anhydrides Chemical class 0.000 claims description 2
- JHRWWRDRBPCWTF-OLQVQODUSA-N captafol Chemical compound C1C=CC[C@H]2C(=O)N(SC(Cl)(Cl)C(Cl)Cl)C(=O)[C@H]21 JHRWWRDRBPCWTF-OLQVQODUSA-N 0.000 claims description 2
- 230000004048 modification Effects 0.000 claims description 2
- 238000012986 modification Methods 0.000 claims description 2
- 230000008707 rearrangement Effects 0.000 claims description 2
- 125000001424 substituent group Chemical group 0.000 claims description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 claims description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 claims 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N dimethylmethane Natural products CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 claims 1
- 239000001294 propane Substances 0.000 claims 1
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 abstract description 6
- 239000000126 substance Substances 0.000 abstract description 6
- 230000000694 effects Effects 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 204
- 239000000047 product Substances 0.000 description 97
- 238000005481 NMR spectroscopy Methods 0.000 description 87
- 239000002904 solvent Substances 0.000 description 53
- 239000000243 solution Substances 0.000 description 52
- 238000003756 stirring Methods 0.000 description 49
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 45
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 44
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 39
- 239000012043 crude product Substances 0.000 description 39
- 238000001704 evaporation Methods 0.000 description 34
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 33
- 229910052938 sodium sulfate Inorganic materials 0.000 description 33
- 235000011152 sodium sulphate Nutrition 0.000 description 33
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 32
- 230000008020 evaporation Effects 0.000 description 32
- 239000012267 brine Substances 0.000 description 28
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 28
- 239000000203 mixture Substances 0.000 description 27
- 238000010898 silica gel chromatography Methods 0.000 description 26
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 24
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical group [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 20
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 18
- 239000012074 organic phase Substances 0.000 description 18
- 239000003208 petroleum Substances 0.000 description 17
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 16
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 14
- 239000007864 aqueous solution Substances 0.000 description 14
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 12
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 12
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 11
- 229910052786 argon Inorganic materials 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- 239000012279 sodium borohydride Substances 0.000 description 10
- 229910000033 sodium borohydride Inorganic materials 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- 238000002844 melting Methods 0.000 description 9
- 230000008018 melting Effects 0.000 description 9
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- 238000000926 separation method Methods 0.000 description 7
- 239000001632 sodium acetate Substances 0.000 description 7
- 235000017281 sodium acetate Nutrition 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- 238000001816 cooling Methods 0.000 description 5
- 239000012280 lithium aluminium hydride Substances 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- VDIFLYQCJAFOLQ-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)ethanol Chemical compound C1=CC=C(CCO)C2=CC(OC)=CC=C21 VDIFLYQCJAFOLQ-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 4
- 235000011114 ammonium hydroxide Nutrition 0.000 description 4
- 239000008241 heterogeneous mixture Substances 0.000 description 4
- 238000004949 mass spectrometry Methods 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- DIOHEXPTUTVCNX-UHFFFAOYSA-N 1,1,1-trifluoro-n-phenyl-n-(trifluoromethylsulfonyl)methanesulfonamide Chemical compound FC(F)(F)S(=O)(=O)N(S(=O)(=O)C(F)(F)F)C1=CC=CC=C1 DIOHEXPTUTVCNX-UHFFFAOYSA-N 0.000 description 3
- PYJMGUQHJINLLD-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)acetonitrile Chemical compound C1=CC=C(CC#N)C2=CC(OC)=CC=C21 PYJMGUQHJINLLD-UHFFFAOYSA-N 0.000 description 3
- FQOHMDPQTPNTEK-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)ethyl methanesulfonate Chemical compound C1=CC=C(CCOS(C)(=O)=O)C2=CC(OC)=CC=C21 FQOHMDPQTPNTEK-UHFFFAOYSA-N 0.000 description 3
- GABLTKRIYDNDIN-UHFFFAOYSA-N 7-methoxy-3,4-dihydro-2h-naphthalen-1-one Chemical compound C1CCC(=O)C2=CC(OC)=CC=C21 GABLTKRIYDNDIN-UHFFFAOYSA-N 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 3
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 150000002815 nickel Chemical class 0.000 description 3
- LAIZPRYFQUWUBN-UHFFFAOYSA-L nickel chloride hexahydrate Chemical compound O.O.O.O.O.O.[Cl-].[Cl-].[Ni+2] LAIZPRYFQUWUBN-UHFFFAOYSA-L 0.000 description 3
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- LGYBVRIYYBHYCX-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)acetamide Chemical compound C1=CC=C(CC(N)=O)C2=CC(OC)=CC=C21 LGYBVRIYYBHYCX-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- ARZLNQHDDHORBP-UHFFFAOYSA-N [1-(2,3-dihydroxypropyl)-7-methoxynaphthalen-2-yl] 4-methylbenzenesulfonate Chemical compound CC1=CC=C(C=C1)S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CC(CO)O ARZLNQHDDHORBP-UHFFFAOYSA-N 0.000 description 2
- KBBKQHNFLXIYFT-UHFFFAOYSA-N [1-(2-acetamidoethyl)-7-methoxynaphthalen-2-yl] 4-methylbenzenesulfonate Chemical compound CC1=CC=C(C=C1)S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CCNC(C)=O KBBKQHNFLXIYFT-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical group [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 2
- 238000005899 aromatization reaction Methods 0.000 description 2
- 238000010533 azeotropic distillation Methods 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 239000003610 charcoal Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 150000002596 lactones Chemical class 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- NNFCIKHAZHQZJG-UHFFFAOYSA-N potassium cyanide Chemical compound [K+].N#[C-] NNFCIKHAZHQZJG-UHFFFAOYSA-N 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- KPBSJEBFALFJTO-UHFFFAOYSA-N propane-1-sulfonyl chloride Chemical compound CCCS(Cl)(=O)=O KPBSJEBFALFJTO-UHFFFAOYSA-N 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- QAGNSTKCENIUAN-UHFFFAOYSA-N 1-(2-hydroxyethyl)-7-methoxynaphthalen-2-ol Chemical compound OCCC1=C(C=CC2=CC=C(C=C12)OC)O QAGNSTKCENIUAN-UHFFFAOYSA-N 0.000 description 1
- AFBKLVYGFYSANF-UHFFFAOYSA-N 1-[(dimethylamino)methyl]-7-methoxynaphthalen-2-ol Chemical compound CN(C)CC1=C(C=CC2=CC=C(C=C12)OC)O AFBKLVYGFYSANF-UHFFFAOYSA-N 0.000 description 1
- RXLLTHYDNNNDMU-UHFFFAOYSA-N 1-[2-(dibenzylamino)ethyl]-7-methoxynaphthalen-2-ol Chemical compound C(C1=CC=CC=C1)N(CCC1=C(C=CC2=CC=C(C=C12)OC)O)CC1=CC=CC=C1 RXLLTHYDNNNDMU-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- WQKQWNPVKYRGLQ-UHFFFAOYSA-N 2-(2-hydroxy-7-methoxynaphthalen-1-yl)acetamide Chemical compound OC1=C(C2=CC(=CC=C2C=C1)OC)CC(=O)N WQKQWNPVKYRGLQ-UHFFFAOYSA-N 0.000 description 1
- CEEVJGQXRADXHD-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)acetaldehyde Chemical compound C1=CC=C(CC=O)C2=CC(OC)=CC=C21 CEEVJGQXRADXHD-UHFFFAOYSA-N 0.000 description 1
- BDBCLSMMNQUYLQ-UHFFFAOYSA-N 2-methoxy-7-prop-2-enoxynaphthalene Chemical compound COC1=CC2=CC(=CC=C2C=C1)OCC=C BDBCLSMMNQUYLQ-UHFFFAOYSA-N 0.000 description 1
- FXKXNQOUYQHCOD-UHFFFAOYSA-N 3-(7-methoxynaphthalen-1-yl)propane-1,2-diol Chemical compound COC1=CC=C2C=CC=C(C2=C1)CC(CO)O FXKXNQOUYQHCOD-UHFFFAOYSA-N 0.000 description 1
- WAZPLXZGZWWXDQ-UHFFFAOYSA-N 4-methyl-4-oxidomorpholin-4-ium;hydrate Chemical compound O.C[N+]1([O-])CCOCC1 WAZPLXZGZWWXDQ-UHFFFAOYSA-N 0.000 description 1
- 102000006902 5-HT2C Serotonin Receptor Human genes 0.000 description 1
- 108010072553 5-HT2C Serotonin Receptor Proteins 0.000 description 1
- OJWQHXQSHREWPU-UHFFFAOYSA-N 7-methoxy-1-prop-2-enylnaphthalen-2-ol Chemical compound COC1=CC=C2C=CC(=C(C2=C1)CC=C)O OJWQHXQSHREWPU-UHFFFAOYSA-N 0.000 description 1
- GULUUGIJMTZFSB-UHFFFAOYSA-N 8-methoxy-1,2-dihydrobenzo[e][1]benzofuran-1,2-diol Chemical compound COC1=CC=C2C=CC=3OC(C(C=3C2=C1)O)O GULUUGIJMTZFSB-UHFFFAOYSA-N 0.000 description 1
- KAQUFGAVNNXEAH-UHFFFAOYSA-N 8-methoxy-1H-benzo[e][1]benzofuran-2-one Chemical compound COC1=CC=C2C=CC=3OC(CC=3C2=C1)=O KAQUFGAVNNXEAH-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 238000006683 Mannich reaction Methods 0.000 description 1
- XPBJEZONNGSONE-UHFFFAOYSA-N N,N-dibenzyl-2-(2-hydroxy-7-methoxynaphthalen-1-yl)acetamide Chemical compound C(C1=CC=CC=C1)N(C(CC1=C(C=CC2=CC=C(C=C12)OC)O)=O)CC1=CC=CC=C1 XPBJEZONNGSONE-UHFFFAOYSA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- HDTBLGVRVPXOKZ-UHFFFAOYSA-N [1-(2-acetamidoethyl)-7-methoxynaphthalen-2-yl] propane-1-sulfonate Chemical compound C(CC)S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CCNC(C)=O HDTBLGVRVPXOKZ-UHFFFAOYSA-N 0.000 description 1
- SQCXQFYDRBROMJ-UHFFFAOYSA-N [1-(2-amino-2-oxoethyl)-7-methoxynaphthalen-2-yl] 4-methylbenzenesulfonate Chemical compound CC1=CC=C(C=C1)S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CC(=O)N SQCXQFYDRBROMJ-UHFFFAOYSA-N 0.000 description 1
- KPQJQWGVJDNARZ-UHFFFAOYSA-N [1-(2-hydroxyethyl)-7-methoxynaphthalen-2-yl] 4-methylbenzenesulfonate Chemical compound CC1=CC=C(C=C1)S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CCO KPQJQWGVJDNARZ-UHFFFAOYSA-N 0.000 description 1
- YMJVOESMHSUIMA-UHFFFAOYSA-N [1-(cyanomethyl)-7-methoxynaphthalen-2-yl] 4-methylbenzenesulfonate Chemical compound CC1=CC=C(C=C1)S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CC#N YMJVOESMHSUIMA-UHFFFAOYSA-N 0.000 description 1
- VBYZHFWEHWLHBL-UHFFFAOYSA-N [1-(cyanomethyl)-7-methoxynaphthalen-2-yl] trifluoromethanesulfonate Chemical compound FC(S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CC#N)(F)F VBYZHFWEHWLHBL-UHFFFAOYSA-N 0.000 description 1
- FTRPKSCLAPSBAY-UHFFFAOYSA-N [1-[(dimethylamino)methyl]-7-methoxynaphthalen-2-yl] 4-methylbenzenesulfonate Chemical compound CC1=CC=C(C=C1)S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CN(C)C FTRPKSCLAPSBAY-UHFFFAOYSA-N 0.000 description 1
- YKGAOVOWQYTTNB-UHFFFAOYSA-N [1-[2-(2,5-dioxopyrrolidin-1-yl)ethyl]-7-methoxynaphthalen-2-yl] propane-1-sulfonate Chemical compound C(CC)S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CCN1C(CCC1=O)=O YKGAOVOWQYTTNB-UHFFFAOYSA-N 0.000 description 1
- VHUAFBNEZYDITG-UHFFFAOYSA-N [1-[2-(dibenzylamino)ethyl]-7-methoxynaphthalen-2-yl] trifluoromethanesulfonate Chemical compound FC(S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)CCN(CC1=CC=CC=C1)CC1=CC=CC=C1)(F)F VHUAFBNEZYDITG-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 150000001343 alkyl silanes Chemical class 0.000 description 1
- TVJORGWKNPGCDW-UHFFFAOYSA-N aminoboron Chemical compound N[B] TVJORGWKNPGCDW-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- RJTANRZEWTUVMA-UHFFFAOYSA-N boron;n-methylmethanamine Chemical compound [B].CNC RJTANRZEWTUVMA-UHFFFAOYSA-N 0.000 description 1
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- GFPFBSQSFSUKBH-UHFFFAOYSA-N chlorosulfonylsulfonylmethane Chemical compound CS(=O)(=O)S(Cl)(=O)=O GFPFBSQSFSUKBH-UHFFFAOYSA-N 0.000 description 1
- 238000010908 decantation Methods 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 210000002249 digestive system Anatomy 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- XYYQWMDBQFSCPB-UHFFFAOYSA-N dimethoxymethylsilane Chemical compound COC([SiH3])OC XYYQWMDBQFSCPB-UHFFFAOYSA-N 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 206010022437 insomnia Diseases 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- QEUMNQFVAMSSNS-UHFFFAOYSA-N n-benzyl-1-phenylmethanamine;hydrochloride Chemical compound [Cl-].C=1C=CC=CC=1C[NH2+]CC1=CC=CC=C1 QEUMNQFVAMSSNS-UHFFFAOYSA-N 0.000 description 1
- CWCMQKWNHUXBFQ-UHFFFAOYSA-N naphthalen-2-yl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OC1=CC=C(C=CC=C2)C2=C1 CWCMQKWNHUXBFQ-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000012285 osmium tetroxide Substances 0.000 description 1
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 1
- 150000002940 palladium Chemical class 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 208000012672 seasonal affective disease Diseases 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 208000019116 sleep disease Diseases 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- PODWXQQNRWNDGD-UHFFFAOYSA-L sodium thiosulfate pentahydrate Chemical compound O.O.O.O.O.[Na+].[Na+].[O-]S([S-])(=O)=O PODWXQQNRWNDGD-UHFFFAOYSA-L 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/02—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions involving the formation of amino groups from compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B43/00—Formation or introduction of functional groups containing nitrogen
- C07B43/06—Formation or introduction of functional groups containing nitrogen of amide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/12—Preparation of carboxylic acid amides by reactions not involving the formation of carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/56—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms
- C07C217/58—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms with amino groups and the six-membered aromatic ring, or the condensed ring system containing that ring, bound to the same carbon atom of the carbon chain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/56—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms
- C07C217/60—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms linked by carbon chains having two carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/02—Preparation of carboxylic acid amides from carboxylic acids or from esters, anhydrides, or halides thereof by reaction with ammonia or amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/06—Preparation of carboxylic acid amides from nitriles by transformation of cyano groups into carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/06—Preparation of carboxylic acid amides from nitriles by transformation of cyano groups into carboxamide groups
- C07C231/065—By hydration using metals or metallic ions as catalyst
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/10—Preparation of carboxylic acid amides from compounds not provided for in groups C07C231/02 - C07C231/08
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/16—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms
- C07C233/17—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
- C07C233/18—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom having the carbon atom of the carboxamide group bound to a hydrogen atom or to a carbon atom of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/32—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings
- C07C235/34—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/30—Preparation of carboxylic acid nitriles by reactions not involving the formation of cyano groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/01—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms
- C07C255/32—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms having cyano groups bound to acyclic carbon atoms of a carbon skeleton containing at least one six-membered aromatic ring
- C07C255/37—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms having cyano groups bound to acyclic carbon atoms of a carbon skeleton containing at least one six-membered aromatic ring the carbon skeleton being further substituted by etherified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/26—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids
- C07C303/28—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids by reaction of hydroxy compounds with sulfonic acids or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/26—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids
- C07C303/30—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids by reactions not involving the formation of esterified sulfo groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/63—Esters of sulfonic acids
- C07C309/64—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms
- C07C309/65—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/63—Esters of sulfonic acids
- C07C309/64—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms
- C07C309/65—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms of a saturated carbon skeleton
- C07C309/66—Methanesulfonates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/63—Esters of sulfonic acids
- C07C309/72—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/73—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton to carbon atoms of non-condensed six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/16—Preparation of ethers by reaction of esters of mineral or organic acids with hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/18—Preparation of ethers by reactions not forming ether-oxygen bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/18—Preparation of ethers by reactions not forming ether-oxygen bonds
- C07C41/26—Preparation of ethers by reactions not forming ether-oxygen bonds by introduction of hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/32—Preparation of ethers by isomerisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/20—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring
- C07C43/215—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring having unsaturation outside the six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/20—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring
- C07C43/23—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring containing hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/27—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation
- C07C45/30—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation with halogen containing compounds, e.g. hypohalogenation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C47/00—Compounds having —CHO groups
- C07C47/20—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms
- C07C47/277—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/92—Naphthofurans; Hydrogenated naphthofurans
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
- Furan Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Изобретение относится к способу промышленного синтеза соединения формулы (I). Способ характеризуется тем, что в реакцию вводят 7-метоксинафталин-2-ол формулы (II), в который в положение 1 соединения формулы (II) вводят группу -СН-Х, в которой X представляет собой -N(CH), -CO-N(CH-Ph), -СН-ОН, -СН=СНили -CO-NH, с получением соединения формулы (III), в которой X представляет собой группу -N(CH), -CO-N(CH-Ph), -СН-ОН, -СН=СНили -CO-NH. При этом соединение формулы (III) подвергают реакции сульфонилирования по ароматическому спирту и заместитель X соединения формулы (III) является модифицированным до или после стадии сульфонилирования ароматического спирта при помощи обычных химических реакций с получением соединения формулы (IV), в которой X' представляет собой группу -CN, -CO-NH, -СН-ОН, -СНО, -CH-N(CH-Ph), -CH-NH-CO-CH, -СН(ОН)-СН-ОН или (2,5-диоксопирролидин-1-ил)метил и R представляет собой группу -СН, -(СН)-СН, -CFили толуил. Соединение формулы (IV) подвергают реакции дезоксигенирования в присутствии переходного металла и восстановителя с получением непосредственно соединения формулы (I), которое выделяют в виде твердого вещества, если X' представляет собой группу -CH-NH-CO-CH, или соединения формулы (V), в которой X'' представляет собой группу -CN, -CH-N(CH-Ph), -СН-ОН, -СН(ОН)-СН-ОН, -CO-NHили (2,5-диоксопирролидин-1-ил)метил. Соединение формулы (V) подвергают обычным химическим реакциям с получением соединения формулы (I), которое выделяют в виде твердого вещества. Предлагаемый способ позволяет получать агомелатин формулы (I) с высоким выходом и превосходной чистотой. Изобретение относится также к промежуточным соединениям и их применению в синтезе агомелатина формулы (I) указанным способом. 14 н. и 15 з.п. ф-лы, 11 пр.
Description
Настоящее изобретение относится к новому способу промышленного синтеза агомелатина или N-[2-(7-метокси-1-нафтил)этил]ацетамида формулы (I):

Агомелатин, или N-[2-(7-метокси-1-нафтил)этил]ацетамид, обладает ценными фармакологическими свойствами.
В действительности, он обладает двойственными свойствами, являясь, с одной стороны, агонистом рецепторов мелатонинэргической системы и, с другой стороны, антагонистом 5-НТ2С рецептора. Эти свойства придают ему активность в центральной нервной системе и, более конкретно в лечении глубокой депрессии, сезонных аффективных расстройств, нарушений сна, патологий сердечно-сосудистой системы, патологий пищеварительной системы, бессонницы и утомляемости вследствие нарушения биоритмов в связи с перелетом через несколько часовых поясов, нарушений аппетита и ожирения.
Агомелатин, его получение и терапевтическое применение описаны в Европейских патентах ЕР 0447285 и ЕР 1564202
Принимая во внимание фармацевтическую ценность данного соединения, важно, чтобы его получали при помощи высокопроизводительного способа промышленного синтеза, который легко переводится в промышленный масштаб и обеспечивает получение агомелатина с высоким выходом и с превосходной чистотой.
В описании патента ЕР 0447285 раскрыто получение агомелатина за восемь стадий, исходя из 7-метокси-1-тетралона. В описании патента ЕР 1564202, заявитель разработал намного более эффективный и применимый в промышленности способ синтеза всего лишь за четыре стадии, исходя из 7-метокси-1-тетралона, позволяя получить агомелатин чрезвычайно репродуктивным способом в хорошо определенной кристаллической форме. Тем не менее, все еще продолжаются поиски новых способов синтеза, в особенности исходя из исходных веществ менее дорогостоящих, нежели 7-метокси-1-тетралон.
Заявителем были продолжены его исследования, и был разработан новый способ синтеза агомелатина, исходя из 7-метокси-нафталин-2-ола: это новое исходное вещество обладает преимуществом в том, что его можно получать просто и без особого труда в больших количествах с меньшими затратами. Преимущество 7-метокси-нафталин-2-ола также заключается в том, что в своей структуре он имеет нафталиновую кольцевую систему, которая позволяет избежать в синтезе введения стадии ароматизации - стадии, которая всегда является проблематичной с промышленной точки зрения.
Кроме того, этот новый способ обеспечивает получение агомелатина воспроизводимым способом и без потребности в лабораторной очистке, с чистотой, которая сопоставима с его применением в качестве фармацевтического действующего вещества. http://www-iab.ujf-grenoble.fr/clientzone/plforme10/img/tomo_xfluo.jpg
Более конкретно, настоящее изобретение относится к способу промышленного синтеза соединения формулы (I):

отличающемуся тем, что в реакцию вводят 7-метокси-нафталин-2-ол формулы (II):

в который в положении 1 соединения формулы (II) вводят группу -СН2-Х, в которой X представляет собой группу -N(CH3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или -CO-NH2,
чтобы получить соединение формулы (III):

в которой X представляет собой -N(СН3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или группу -CO-NH2;
соединение формулы (III) подвергают реакции сульфонилирования по ароматическому спирту и заместитель X соединения формулы (III) является модифицированным, до или после стадии сульфонилирования ароматического спирта, при помощи обычных химических реакций с получением соединения формулы (IV):
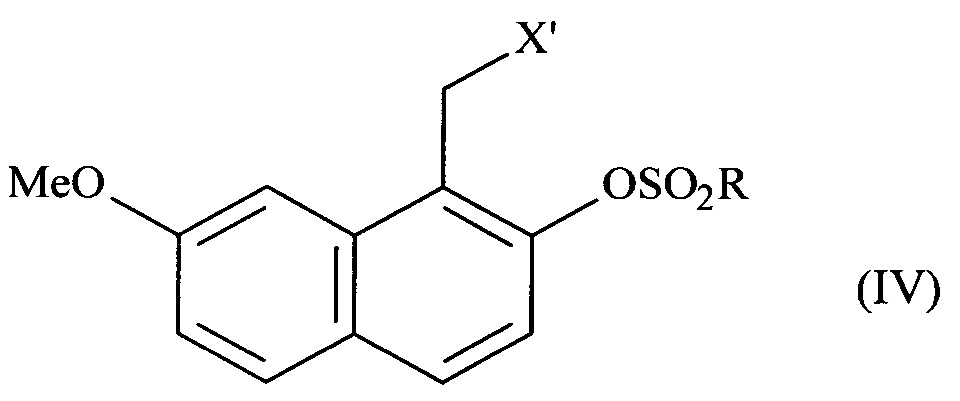
в которой X' представляет собой -CN, -CO-NH2, -СН2-ОН, -CH2-N(CH2-Ph)2, -CH2-NH-CO-CH3, -СН(ОН)-СН2-ОН, -СНО или группу (2,5-диоксопирролидин-1-ил)метил и R представляет собой группу -СН3, -(СН2)2-СН3, -CF3 или толуил;
соединение формулы (IV) подвергают реакции дезоксигенирования в присутствии переходного металла и восстановителя с получением:
- или, если X' представляет собой группу -CH2-NH-CO-CH3, соединения формулы (I) непосредственно, которое выделяют в виде твердого вещества;
- или соединения формулы (V):

в которой X'' представляет собой группу -CN, -CH2-N(CH2-Ph)2, -СН2-ОН, -CO-NH2, -СН(ОН)-СН2-ОН или (2,5-диоксопирролидин-1-ил)метил;
затем соединение формулы (V) подвергают обычным химическим реакциям с получением соединения формулы (I), которое выделяют в виде твердого вещества.
Вариант способа промышленного синтеза состоит в том, что во время превращения соединения формулы (III) в соединение формулы (IV) группа X не модифицирована. Полученное соединение, сульфонилированное по его ароматическому спирту, затем подвергают реакции дезоксигенирования посредством действия переходного металла и восстановителя. Группу X впоследствии модифицируют при помощи обычных химических реакций с получением соединения формулы (I), которое выделяют в виде твердого вещества.
Соединение формулы (II) имеется в продаже или может быть легко получено специалистом в данной области техники, используя химические реакции, которые являются стандартными или описаны в литературных источниках.
В способе, предлагаемом в изобретении, превращение соединения формулы (II) в соединение формулы (III), в которой X представляет собой группу -N(CH3)2 осуществляют в соответствии с реакцией Манниха посредством действия формальдегида в присутствии диметиламина.
В способе, предлагаемом в изобретении, превращение соединения формулы (II) в соединение формулы (III), в которой X представляет собой группу -СН2-ОН, состоит в том, что соединение формулы (II) подвергают действию глиоксаля (или этан-1,2-диона), за которым следует действие восстановителя. Преимущественно, восстановитель представляет собой алюмогидрид лития, гидрид диизобутилалюминия, триэтилгидрид лития или диметилсульфид борана. Предпочтительно, восстановитель представляет собой алюмогидрид лития.
В способе, предлагаемом в изобретении, превращение соединения формулы (II) в соединение формулы (III), в которой X представляет собой группу -СО-NH2 или -CO-N(CH2-Ph)2 осуществляют посредством действия глиоксаля, за которым следует действие, в нагретой среде, соединения формулы NHR'R', в которой R' представляет собой Н или группу -СН2-Ph.
Указанную реакцию с глиоксалем, приводящую к образованию промежуточного лактона формулы (VI):

предпочтительно осуществляют в две стадии.
На первой стадии соединение формулы (II) растворяют в основной среде в присутствии глиоксаля. Предпочтительно основание представляет собой гидроксид натрия или гидроксид калия, и более конкретно гидроксид калия.
На второй стадии, промежуточный продукт, а именно 8-метокси-1,2-дигидронафто[2,1-б]фуран-1,2-диол, растворяют непосредственно в кислотной среде, предпочтительно хлористоводородной кислоте, с получением промежуточного лактона формулы (VI).
В способе, предлагаемом в изобретении, превращение соединения формулы (II) в соединение формулы (III), в которой X представляет собой группу -СН=СН2, осуществляют в соответствии с сигматропной перегруппировкой Клайзена посредством действия аллилбромида в основной среде, после чего следует термическая перегруппировка. Действие аллилбромида осуществляют в присутствии основания, такого как гидрид натрия, трет-бутилат калия, метилат натрия, гидроксид калия, гидроксид натрия, карбонат калия или карбонат натрия. Преимущественный вариант осуществления состоит в использовании карбоната калия в качестве основания в стадии, которая включает реакцию с аллилбромидом.
В способе, предлагаемом в изобретении, превращение соединения формулы (III) в соединение формулы (IV) состоит в том, что стадия сульфонилирования ароматического спирта сопровождается модификацией группы X при помощи обычных химических реакций, X определен как указано выше. В соответствии с другим преимущественным вариантом осуществления, превращение соединения формулы (III) в соединение формулы (IV) состоит в модификации группы X при помощи обычных химических реакций, после чего следует стадия сульфонилирования ароматического спирта, X определен как указано выше.
Указанную стадию сульфонилирования осуществляют посредством действия сульфонилхлорида, сульфонового ангидрида или сульфонимида. В соответствии с предпочтительным вариантом осуществления, стадию сульфонилирования осуществляют посредством действия тозилхлорида, н-пропилсульфонилхлорида, трифторметансульфонового ангидрида или фенилтрифлимида (или N,N-бис(трифторметилсульфонил)анилин).
В способе, предлагаемом в изобретении, превращение соединения формулы (IV) в соединение формулы (V) состоит из стадии дезоксигенирования в присутствии переходного металла и восстановителя.
Предпочтительно, переходный металл представляет собой никель, палладий или платину. Переходный металл может находиться или в виде соли, или в виде простого вещества. Предпочтительно, соль переходного металла представляет собой соль никеля или соль палладия, более предпочтительно соль никеля.
Преимущественно, восстановитель представляет собой или гидрид, такой как борогидрид натрия или алюмогидрид лития; или аминоборан, такой как диметиламин-боран; или алкоксисилан, такой как диметоксиметилсилан; или алкилсилан, такой как триэтилсилан; или щелочноземельный металл, такой как магний; или водород.
В соответствии с другим предпочтительным вариантом осуществления, превращение соединения формулы (IV) в соединение формулы (V) состоит из стадии дезоксигенирования в присутствии никеля, в частности соли никеля, и гидрида, предпочтительно борогидрида натрия.
В соответствии с другим предпочтительным вариантом осуществления, превращение соединения формулы (IV) в соединение формулы (V) состоит из стадии дезоксигенирования в присутствии палладия и водорода. Водород применяют непосредственно в его газообразном виде или его непрямо получают путем разложения формиата аммония.
В соответствии с другим предпочтительным вариантом осуществления, превращение соединения формулы (IV) в соединение формулы (V) состоит из стадии дезоксигенирования в присутствии палладия и щелочноземельного металла, предпочтительно магния.
В соответствии с другим предпочтительным вариантом осуществления, превращение соединения формулы (IV) в соединение формулы (V) состоит из стадии дезоксигенирования в присутствии переходного металла, восстановителя и лиганда. Лиганд предпочтительно представляет собой фосфиновый лиганд, и более конкретно трифенилфосфин.
В соответствии с конкретным вариантом осуществления, стадия дезоксигенирования соединения формулы (IV), в которой X' представляет собой группу -CH2-NH-CO-CH3, которую осуществляют:
- или в присутствии никеля, в частности соли никеля, и гидрида, предпочтительно борогидрида натрия;
- или в присутствии палладия и водорода;
- или в присутствии палладия и щелочноземельного металла; приводит непосредственно к образованию соединения формулы (I).
Этот способ является в особенности выгодным по нижеследующим причинам:
- обеспечивает возможность получения соединения формулы (I) в промышленных масштабах с хорошим выходом, исходя из простого и недорогого исходного вещества;
- дает возможность избежать реакции ароматизации - стадии, которая всегда является проблематичной с промышленной точки зрения - потому что в исходном субстрате присутствует нафталиновая кольцевая система;
- обеспечивает возможность получения агомелатина, исходя из 7-метокси-нафталин-2-ола с уменьшенным количеством стадий.
Соединения формул (III), (IV) и (VI), полученные в соответствии со способом, предлагаемым в изобретении, являются новыми и применимыми в качестве промежуточных соединений в синтезе агомелатина.
Соединения формулы (V), полученные в соответствии со способом, предлагаемым в изобретении, пригодны в качестве промежуточных соединений в синтезе агомелатина. Соединения формулы (V), полученные в соответствии со способом, предлагаемым в изобретении, являются новыми, за исключением (7-метоксинафталин-1-ил)ацетонитрила, N,N-дибензил-2-(7-метоксинафталин-1-ил)этанамида, 2-(7-метоксинафталин-1-ил)этанола и 2-(7-метоксинафталин-1-ил)ацетамида.
Предпочтительными соединениями формулы (III) являются следующие:
- 1-[(диметиламино)метил]-7-метоксинафталин-2-ол;
- N,N-дибензил-2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид;
- 1-(2-гидроксиэтил)-7-метоксинафталин-2-ол;
- 2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид;
- 7-метокси-1-(проп-2-ен-1-ил)нафталин-2-ол.
Предпочтительными соединениями формулы (IV) являются следующие:
- трифторметансульфонат 1-(цианометил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила;
- трифторметансульфонат 1-[2-(дибензиламино)этил]-7-метоксинафталин-2-ила;
- пропан-1-сульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила;
- пропан-1-сульфонат 1-[2-(2,5-диоксопирролидин-1-ил)этил]-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-(2-гидроксиэтил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-(2-амино-2-оксоэтил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 7-метокси-1-(2-оксоэтил)нафталин-2-ила;
- 4-метилбензолсульфонат 1-(2,3-дигидроксипропил)-7-метоксинафталин-2-ила.
(2-Гидрокси-7-метоксинафталин-1-ил)ацетонитрил, 4-метилбензолсульфонат 7-метокси-1-(2-{[(4-метилфенил)сульфонил]окси}этил)нафталин-2-ила, пропан-1-сульфонат 7-метокси-1-{2-[(пропилсульфонил)окси]этил}нафталин-2-ила и ацетат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила являются новыми и пригодны в качестве промежуточных соединений в синтезе агомелатина.
Приведенные ниже примеры демонстрируют изобретение, но никоим образом не ограничивают его.
Для того чтобы подтвердить правильность реакционных способов, промежуточные продукты синтеза были систематически выделены и охарактеризованы. Тем не менее, можно существенно оптимизировать процедуры путем ограничения количества выделенных промежуточных соединений.
Структуры описанных соединений были подтверждены при помощи стандартных спектроскопических методик: протонный ЯМР (s = синглет; bs = широкий синглет; d = дублет; t = триплет; dd = дублет дублетов; m = мультиплет); углеродный ЯМР (s = синглет; d = дублет; t = триплет; q = квадруплет); масс-спектрометрия с ионизацией электрораспылением (ESI).
Пример 1: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 1-[(диметиламино)метил]-7-метоксинафталин-2-ол
К раствору 7-метокси-нафталин-2-ола (1.74 г; 10 ммоль) в этаноле (10 мл) при температуре окружающей среды добавляют диметиламин (40% в воде; 1.52 мл; 12 ммоль) и затем формальдегид (37% в воде; 0.78 мл; 10.5 ммоль). После перемешивания в течение одного часа, растворитель выпаривают. Полученный сырой продукт (количественный выход) применяют непосредственно в следующей стадии без дополнительной очистки.
1Н ЯМР спектроскопический анализ (ДМСО-d6, 300.13 МГц, δ в част. на млн.): 11.18 (bs, 1Н); 7.68 (d, J=8.8 Гц, 1Н); 7.6 (d, J=8.8 Гц, 1Н); 7.23 (d, J=2.3 Гц, 1Н); 6.93 (dd, J=8.8 и 2.3 Гц, 1H); 6.89 (d, J=8.8 Гц, 1Н); 3.96 (s, 2Н); 3.86 (s, 3Н); 2.3 (s, 6Н).
13С ЯМР спектроскопический анализ (ДМСО-d6, 75.5 МГц, δ в част. на млн.): 157.7 (s); 156.1 (s); 134.3 (s); 129.9 (d); 128.5 (d); 123.3 (s); 116.0 (d); 114.2 (d); 112.0 (s); 101.6 (d); 55.8 (t); 55.0 (q); 44.3 (2 x q).
Стадия В: (2-гидрокси-7-метоксинафталин-1-ил)ацетонитрил
Раствор продукта из стадии А выше (1.155 г; 5 ммоль) в диметилформамиде (5 мл) в присутствии цианида калия (390 мг; 6 ммоль) нагревают при 80°С в течение 30 часов. После разбавления с этилацетатом, добавляют 2М водный раствор HCl (5 мл). Смесь перемешивают и затем нейтрализуют посредством добавления раствора разбавленного NaHCO3. Две фазы разделяют и органическую фракцию промывают три раза рассолом, сушат над сульфатом натрия и фильтруют.После выпаривания растворителя получают сырой продукт, который затем очищают хроматографией на силикагелевой колонке (элюант : простой эфир/петролейный эфир 40/60) с получением целевого продукта.
1Н ЯМР спектроскопический анализ (ацетон-d6, 300.13 МГц, δ в част. на млн.): 9.25 (bs, 1Н, ОН); 7. 74 (d, J=9.1 Гц, 1Н); 7. 71 (d, J=8.9 Гц, 1Н); 7.31 (d, J=2.5 Гц, 1Н); 7.13 (d, J=8.9 Гц, 1Н); 7.03 (dd, J=9.1 и 2.5 Гц, 1Н); 4.21 (s, 2Н); 3.96 (s, 3Н).
13С ЯМР спектроскопический анализ (ацетон-d6, 75.5 МГц, δ в част. на млн.): 159.9 (s); 154.1 (s); 135.1 (s); 131.1 (d); 130.4 (d); 124.9 (s); 119.1 (s); 116.2 (d); 115. 7 (d); 108. 7 (s); 102.1 (d); 55.6 (q); 13.6 (t).
Стадия С: трифторметансульфонат 1-(цианометил)-7-метоксинафталин-2-ила
К раствору продукта из стадии В выше (120 мг; 0.52 ммоль) в дихлорметане (5 мл) добавляют N,N-бис(трифторметилсульфонил)анилин (204 мг; 0.571 ммоль) и триэтиламин (72 мкл; 0.52 ммоль). После перемешивания в течение 16 часов, растворители выпаривают, и остаток очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир, градиент от 10/90 до 20/80) с получением указанного в заголовке продукта (125 мг; 70%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.85-7.78 (m, 2Н); 7.3-7.22 (m, 3Н); 4.13 (s, 2Н); 3.98 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 159.9 (s); 145.6 (s); 133.2 (s); 131.2 (d); 130.8 (d); 128.0 (s); 125.0 (s); 120.3 (d); 118.6 (s, J=318 Гц); 116.9 (s); 116.8 (d); 116.0 (s); 102.3 (d); 55.6 (q); 15.2 (t). Масс-спектрометрия (ESI; m/z(%)): 345(45) [M]+•; 212(100); 184(15); 169(34); 140(18).
Стадия D: (7-метоксинафталин-1-ил)ацетонитрил
Раствор продукта из стадии С выше (73 мг; 0.212 ммоль) в абсолютном этаноле (4 мл) в присутствии палладия 10% на угле (4.5 мг; 0.004 ммоль) и триэтиламина (150 мкл) гидрируют (4 бар) при температуре окружающей среды в течение 20 часов. После фильтрации через целит, промывания этилацетатом и выпаривания растворителей, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 15/85) с получением указанного в заголовке продукта (28 мг; 67%).
Точка плавления: 86-87°С.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.78 (d, J=8.9 Гц, 1Н); 7.77 (d, J=7.8 Гц, 1Н); 7.52 (d, J=7.1 Гц, 1Н); 7.32 (dd, J=7.8 и 7.1 Гц, 1Н); 7.21 (dd, J=8.9 и 2.4 Гц, 1Н); 7.03 (d, J=2.4 Гц, 1Н); 4.0 (s, 2Н); 3.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.5 (s); 132.0 (s); 130.6 (d); 129.1 (s); 128.8 (d); 127.1 (d); 124.4 (s); 123.2 (d); 118.8 (d); 117.7 (s); 101.3 (d); 55.4 (q); 21.9 (t).
Стадия Е: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
136 г никеля Ренея, 2.06 л этанола и 0.23 л воды вводят в 8-литровый реактор. В ходе перемешивания при 70°С и под 30 бар водорода медленно добавляют соединение, полученное в стадии D выше (0.8 кг), растворенное в уксусном ангидриде (2.4 л). После завершения добавления реакционную смесь перемешивают в течение 1 часа под атмосферой водорода при 30 бар, и затем в реакторе сбрасывают давление, а жидкости фильтруют. После концентрирования смеси остаток кристаллизуют из смеси этанол/вода 35/65 с получением указанного в заголовке продукта с выходом в 89% и с химической чистотой более чем 99%.
Точка плавления: 108°С.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1H); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1H); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 2: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 4-метилбензолсульфонат 1-[(диметиламино)метил]-7-метоксинафталин-2-ила
К раствору продукта, полученного в стадии А примера 1 (2.042 г; 8.84 ммоль) в диметилформамиде (8 мл) добавляют трет-бутппат калия (1.091 г; 9.724 ммоль) и затем, через 5 минут, тозилхлорид (1.684 г; 8.84 ммоль). После перемешивания в течение 6 часов, раствор разбавляют с этилацетататом и два раза промывают водой, а затем рассолом. Органическую фазу высушивают над сульфатом натрия, и фильтруют, а растворитель выпаривают. Полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир, градиент от 30/70 до 70/30) с получением целевого продукта в виде твердого вещества (1.94 г; 57%).
Точка плавления: 115-118°С
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.78 (d, J=8.1 Гц, 2Н); 7.7 (d, J=9.0 Гц, 1Н); 7.62 (d, J=8.8 Гц, 1Н); 7.53 (d, J=2.3 Гц, 1Н); 7.34 (d, J=8.1 Гц, 2Н); 7.14 (dd, J=9.0 и 2.3 Гц, 1Н); 6.96 (d, J=8.8 Гц, 1Н); 3.93 (s, 3Н); 3.67 (s, 2Н); 2.47 (s, 3Н); 2.24 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.4 (s); 146.9 (s); 145.5 (s); 135.1 (s); 133.2 (s); 130.0 (2 x d); 129.9 (d); 129.1 (d); 128.6 (2 x d); 127.8 (s); 125.7 (s); 118.5 (d); 117.9 (d); 104.5 (d); 55.3 (q); 54.2 (t); 45.6 (2 x q); 21.9 (q).
Стадия В: 4-метилбензолсульфонат 1-(цианометил)-7-метоксинафталин-2-ила
Йодметан (267 мкл; 4.29 ммоль) добавляют к раствору продукта из стадии А выше (1.5 г; 9.9 ммоль) в диметилформамиде (8 мл). После перемешивания в течение 4 часов при температуре окружающей среды добавляют цианид калия (304 мг; 4.68 ммоль). Раствор перемешивают в течение еще 16 часов и затем разбавляют с этилацетататом, промывают водой и три раза рассолом, сушат над сульфатом натрия и фильтруют.Выпаривание растворителя обеспечивает сырой продукт, который затем очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 40/60) с получением целевого продукта (1.276 г; 89%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.77 (d, J=8.3 Гц, 2Н); 7.75 (d, J=8.9 Гц, 1Н); 7.69 (d, J=8.9 Гц, 1Н); 7.35 (d, J=8.3 Гц, 2Н); 7.22-7.15 (m, 2Н); 7.01 (d, J=8.9 Гц, 1Н); 3.98 (s, 2Н); 3.95 (s, 3Н); 2.45 (s, 3Н).
Стадия С: 4-метилбензолсульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила
Продукт из стадии В выше (54 мг; 0.147 ммоль), хлорид никеля гексагидрат (35 мг; 0.147 ммоль), дихлорметан (1.5 мл) и метанол (1.5 мл) вводят в герметичную колбу. Затем аргон кипятят в растворе в течение 5 минут и после этого осторожно, маленькими порциями добавляют борогидрид натрия (100 мг; 2.94 ммоль). После перемешивания в течение 30 минут под аргоном и при температуре окружающей среды добавляют воду. После перемешивания в течение 15 минут, смесь фильтруют через целит, и затем промывают дихлорметаном. Органическую фракцию высушивают над сульфатом натрия и затем фильтруют. После выпаривания растворителей, полученный сырой продукт помещают в присутствии уксусного ангидрида (1 мл) и ацетата натрия (50 мг) при перемешивании при температуре окружающей среды в течение 30 минут. Смесь выливают в разбавленный раствор Na2CO3, и продукт экстрагируют три раза при помощи этилацетата. Органические фракции промывают рассолом, сушат над сульфатом натрия и фильтруют. Выпаривание растворителя обеспечивает сырой продукт, который затем очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением целевого продукта в виде твердого вещества (24 мг; 40%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.81 (d, J=8.2 Гц, 2Н); 7.68 (d, J=8.9 Гц, 1Н); 7.62 (d, J=2.4 Гц, 1Н); 7.55 (d, J=8.9 Гц, 1Н); 7.36 (d, J=8.2 Гц, 2Н); 7.14 (dd, J=8.9 и 2.4 Гц, 1Н); 6.89 (d, J=8.9 Гц, 1Н); 5.97 (m, 1H); 4.01 (s, 3Н); 3.53-3.46 (m, 2Н); 3.22-3.17 (m, 2Н); 2.46 (s, 3Н); 1.95 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 170.9 (s); 158.9 (s); 146.4 (s); 145.7 (s); 134.6 (s); 133.3 (s); 130.1 (2 x d); 130.0 (d); 128.5 (2 x d); 128.2 (d); 127. 7 (s); 126.2 (s); 119.2 (d); 117.8 (d); 103.3 (d); 55.8 (q); 39.4 (t); 26.4 (t); 23.4 (q); 21.9 (q).
Стадия D: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид Продукт из стадии С выше (83 мг; 0.2 ммоль), хлорид никеля (26 мг; 0.2 ммоль) и метанол (4 мл) вводят в герметичную колбу. Затем барботируют аргоном в растворе в течение 5 минут, и затем маленькими порциями осторожно добавляют натрия борогидрид (136 мг; 4 ммоль). После перемешивания в течение 30 минут под аргоном и при температуре окружающей среды, смесь фильтруют через целит, затем ее промывают при помощи этилацетата и растворители выпаривают. Полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением целевого продукта (39 мг; 80%).
1H ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1H); 7.74 (d, J=8.9 Гц, 1H); 7.65 (d, J=8.0 Гц, 1H); 7.52 (d, J=2.5 Гц, 1H); 7.31-7.2 (m, 2H); 7.11 (dd, J=8.9 и 2.5 Гц, 1H); 3.96 (s, 3H); 3.52-3.44 (m, 2H); 3.23-3.18 (m, 2H); 1.94 (s, 3H).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 3: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 8-метоксинафто[2,1-b]фуран-2(1H)-он
Водный раствор 85% гидроксида калия (2.76 г; 40 ммоль) и 7-метокси-нафталин-2-ола (6.96 г; 40 ммоль) в воде (80 мл) добавляют по каплям при температуре окружающей среды к раствору глиоксаля (40% в воде; 28 мл; 240 ммоль). После перемешивания в течение 3 часов, осадок белого цвета собирают фильтрацией и промывают водой. Полученный твердый (8-метокси-1,2-дигидронафто[2,1-6]фуран-1,2-диол) растворяют в 1,2-дихлорэтан (160 мл) и затем добавляют водный раствор 3М HCl (300 мл). Гетерогенную смесь нагревают при 50°С с интенсивным перемешиванием. Через 1,5 часа все твердые вещества растворяют и две фазы разделяют. Органическую фазу собирают и растворители выпаривают. Сырой продукт сушат азеотропной перегонкой с толуолом с получением указанного в заголовке продукта (8.69 г), который будут использовать непосредственно в следующей стадии без дополнительной очистки.
1Н ЯМР спектроскопический анализ (ДМСО-d6, 300.13 МГц, δ в част. на млн.): 7.88 (d, J=8.8 Гц, 1Н); 7.85 (d, J=8.6 Гц, 1Н); 7.28 (d, J=8.8 Гц, 1Н); 7.12-7.06 (m, 2Н); 4.15 (s, 2Н); 3.89 (s, 3Н).
Стадия В: N,N-дибензил-2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид
Продукт из стадии А выше (1.976 г; 9.23 ммоль) и дибензиламин (4 мл; 20.3 ммоль) вводят в колбу и затем нагревают при 120°С в течение 2 часов. После охлаждения остаток разбавляют с этилацетататом (200 мл). Добавление водного раствора 2М HCl дает в осадке гидрохлоридную соль дибензиламина, которую затем фильтруют через целит. Органическую фазу промывают водным раствором 2М HCl, затем промывают рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 30/70) с получением целевого продукта в виде твердого вещества (2.88 г; 76%).
Точка плавления: 155-157°С
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 9.85 (bs, 1Н); 7.66 (d, J=8.7 Гц, 1Н); 7.62 (d, J=8.7 Гц, 1Н); 7.4-7.24 (m, 6Н); 7.18-7.14 (m, 4Н); 7.08 (d, J=8.7 Гц, 1Н); 6.95-6.91 (m, 2Н); 4.72 (s, 2Н); 4.64 (s, 2Н); 4.21 (s, 2Н); 3.55 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 174.6 (s); 158.6 (s); 155.8 (s); 136.4 (s); 135.5 (s); 134.1 (s); 130.6 (d); 129.3 (d); 129.2 (2 x d); 128.9 (2 x d); 128.4 (2 x d); 128.1 (d); 127.8 (d); 126.4 (2 x d); 124.6 (s); 117.5 (d); 114.9 (d); 111.5 (s); 101.2 (d); 55.9 (q); 50.8 (t); 49.0 (t); 31.5 (t).
Стадия С: 1-[2-(дибензиламино)этил]-7-метоксинафталин-2-ол
1M раствор комплекса боран-тетрагидрофуран в тетрагидрофуране (1.7 мл; 1.7 ммоль) добавляют к раствору продукта из стадии В выше (230 мг; 0.56 ммоль) в тетрагидрофуране (10 мл). Смесь нагревают с обратным холодильником в течение 2 часов, и после чего добавляют водный раствор 2М HCl (10 мл). После перемешивания в течение ночи добавляют насыщенный раствор NaHCO3 для достижения нейтрального рН, и продукт экстрагируют три раза при помощи этилацетата. Органические фракции сушат над сульфатом натрия и фильтруют, а растворитель выпаривают. Полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 30/70) с получением целевого продукта (150 мг; 72%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 12.09 (bs, 1Н); 7.69 (d, J=8.9 Гц, 1Н); 7.61 (d, J=8.8 Гц, 1Н); 7.44-7.26 (m, 10Н); 7.14 (d, J=8.8 Гц, 1Н); 7.12 (d, J=2.4 Гц, 1Н); 7.0 (dd, J=8.8 и 2.4 Гц, 1Н); 3.93 (s, 3Н); 3.79 (s, 4Н); 3.21-3.18 (m, 2Н); 3.01-2.98 (m, 2Н).
Стадия D: трифторметансульфонат 1-[2-(дибензиламино)этил]-7-метоксинафталин-2-ила
Трифторметансульфоновый ангидрид (135 мкл; 0.801 ммоль) добавляют при 0°С к раствору продукта из стадии С выше (303 мг; 0.763 ммоль) в дихлорметане (10 мл). После перемешивания в течение 2 часов при этой температуре, растворитель выпаривают. Остаток ресуспендируют в смеси из диэтиловый простой эфир/водный полунасыщенный раствор NaHCO3. После разделения, органическую фазу высушивают над сульфатом натрия и фильтруют. После выпаривания растворителей, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 10/90) с получением целевого продукта (306 мг; 76%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.73 (d, J=8.9 Гц, 1Н); 7.69 (d, J=8.9 Гц, 1Н); 7.44-7.4 (m, 4Н); 7.35-7.22 (m, 7Н); 7.15 (dd, J=8.9 и 2.3 Гц, 1Н); 6.99 (d, J=2.3 Гц, 1Н); 3.78 (s, 4Н); 3.6 (s, 3Н); 3.43-3.37 (m, 2Н); 2.91-2.86 (m, 2Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.9 (s); 146.1 (s); 139.8 (2 х s); 134.3 (s); 130.3 (d); 128.7 (4 x d); 128.6 (d); 128.3 (4 x d); 128.1 (s); 127.4 (s); 127.0 (2 x d); 119.5 (d); 118.7 (s, JC-F = 318 Гц); 116.8 (d); 102.8 (d); 58.3 (2 x t); 55.3 (q); 52.5 (t); 24.6 (t).
Стадия E: N,N-дибензил-2-(7-метоксинафталин-1-ил)этанамид
Продукт из стадии D выше (130 мг; 0.25 ммоль), ацетат палладия (5.6 мг; 0.025 ммоль), трифенилфосфин (20 мг; 0.075 ммоль), формиат аммония (142 мг; 2.25 ммоль) и диметилформамид (1 мл) вводят в колбу. После перемешивания в течение 16 часов при 60°С, раствор разбавляют с этилацетататом, промывают водой, два раза промывают рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 10/90) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (93 мг; 98%).
Точка плавления: 92-93°С.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.74 (d, J=8.9 Гц, 1Н); 7.65 (m, 1Н); 7.43-7.39 (m, 4Н); 7.35-7.22 (m, 8Н); 7.11 (dd, J=8.9 и 2.4 Гц, 1Н); 7.05 (d, J=2.4 Гц, 1H); 3. 76 (s, 4Н); 3.67 (s, 3Н); 3.29-3.23 (m, 2Н); 2.92-2.86 (m, 2Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 157.6 (s); 139.9 (2 х s); 135.3 (s); 133.2 (s); 130.3 (d); 129.3 (s); 128.7 (4 x d); 128.3 (4 x d); 127.3 (d); 127.0 (2 x d); 126.5 (d); 123.3(d); 118.2 (d); 102.2 (d); 58.6 (2 t); 55.2 (q); 54.2 (t); 31.5 (t).
Стадия F: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Продукт из стадии Е выше (66 мг; 0.173 ммоль), гидроксид палладия на угле (20% Pd, 60% влажность; 10 мг), этанол (2 мл) и этилацетат (2 мл) вводят в колбу, помещенную в автоклав. Автоклав заполняют водородом под давлением (5 бар), и смесь перемешивают в течение 30 часов. Раствор затем фильтруют через целит и промывают этанолом, а растворители выпаривают. К сырому реакционному продукту добавляют уксусный ангидрид (500 мкл) и ацетат натрия (100 мг). После перемешивания в течение 4 часов, смесь разбавляют с этилацетататом. Органическую фазу промывают два раза водным раствором 2М гидроксида натрия и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, целевой продукт получают в чистом виде (41 мг; 98%).
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 4: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия A: 1-(2-гидроксиэтил)-7-метоксинафталин-2-ол
Продукт, полученный в стадии А примера 3 (8.96 г; 40 ммоль) растворяют в тетрагидрофуране (160 мл), и затем добавляют алюмогидрид лития (1.52; 40 ммоль) порциями при 0°С под потоком азота. Смесь перемешивают в течение 16 часов при температуре окружающей среды и затем реакцию останавливают при 0°С посредством добавления этилацетата, а потом и воды. Добавляют водный 1М раствор серной кислоты (80 мл). После перемешивания в течение одного часа, гетерогенную смесь фильтруют через целит, и промывают при помощи этилацетата. После сцеживания и разделения, органическую фазу промывают водой, а затем рассолом. Раствор снова фильтруют через тонкий слой кремнезема, и растворители выпаривают с получением указанного в заголовке продукта без дополнительной очистки (8.21 г; 94% исходя из 7-метокси-нафталин-2-ола).
1Н ЯМР спектроскопический анализ (ДМСО-d6, 300.13 МГц, δ в част. на млн.): 7.6 (d, J=8.8 Гц, 1Н); 7.49 (d, J=8.8 Гц, 1Н); 7.25 (d, J=2.4 Гц, 1Н); 6.94 (d, J=8.8 Гц, 1Н); 6.9 (dd, J=8.8 и 2.4 Гц, 1Н); 3.9 (s, 3Н); 3.79-3.73 (m, 2Н); 3.31-3.25 (m, 2Н).
Стадия В: 4-метилбензолсульфонат 7-метокси-1-(2-{[(4-метилфенил)сульфонил]окси}этил)нафталин-2-ила
Продукт из стадии А выше (436 мг; 2 ммоль), дихлорметан (10 мл), триэтиламин (670 мкл; 4.8 ммоль) и 4-диметиламинопиридин (12 мг; 0.1 ммоль) вводят в колбу. После охлаждения до 0°С, тозилхлорид (800 мг; 4 ммоль) добавляют за один раз, и раствор перемешивают в течение 2 часов при 0°С и затем в течение 14 часов при температуре окружающей среды. После выпаривание растворителя, остаток ресуспендируют в этилацетате и воде. Органическую фракцию промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир, градиент от 20/80 до 30/70) с получением указанного в заголовке продукта (728 мг; 69%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.78 (d, J=8.3 Гц, 2Н); 7.69 (d, J=9.1 Гц, 1Н); 7.66 (d, J=8.3 Гц, 2Н); 7.59 (d, J=8.9 Гц, 1Н); 7.35 (d, J=8.3 Гц, 2Н); 7.26 (d, J=2.4 Гц, 1Н); 7.23 (d, J=8.3 Гц, 2Н); 7.15 (dd, J=8.9 и 2.4 Гц, 1Н); 7.03 (d, J=9.1 Гц, 1Н); 4.2 (t, J=7.7 Гц, 2Н); 3.94 (s, 3Н); 3.34 (d, J=7.7 Гц, 2Н); 2.47 (s, 3Н); 2.39 (s, 3Н).
Стадия С: 4-метилбепзолсулъфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила
Продукт из стадии В выше (124 мг; 0.236 ммоль), ацетонитрил (1 мл) и 35% водный раствор аммиака (1 мл) вводят в герметичную колбу. Колбу помещают в баню, нагретую при 110°С, и раствор перемешивают в течение 3.5 часов. После охлаждения, раствор разбавляют с этилацетататом, промывают разбавленным раствором NaHCO3, промывают рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, сырой продукт растворяют в уксусном ангидриде (1 мл), и после чего добавляют ацетат натрия (300 мг). После перемешивания в течение 14 часов, смесь выливают в разбавленный раствор NaHCO3. После перемешивания в течение 30 минут, продукт экстрагируют три раза при помощи этилацетата. Органическую фазу промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (84 мг; 86%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.81 (d, J=8.2 Гц, 2Н); 7.68 (d, J=8.9 Гц, 1Н); 7.62 (d, J=2.4 Гц, 1Н); 7.55 (d, J=8.9 Гц, 1Н); 7.36 (d, J=8.2 Гц, 2Н); 7.14 (dd, J=8.9 и 2.4 Гц, 1Н); 6.89 (d, 8.9 Гц, 1H); 5.97 (m, 1Н); 4.01 (s, 3Н); 3.53-3.46 (m, 2Н); 3.22-3.17 (m, 2Н); 2.46 (s, 3Н); 1.95 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 170.9 (s); 158.9 (s); 146.4 (s); 145. 7 (s); 134.6 (s); 133.3 (s); 130.1 (2 x d); 130.0 (d); 128.5 (2 x d); 128.2 (d); 127.7 (s); 126.2 (s); 119.2 (d); 117.8 (d); 103.3 (d); 55.8 (q); 39.4 (t); 26.4 (t); 23.4 (q); 21.9 (q).
Стадия D: N-[2-(7-метоксинафталин-1-ил)этил)ацетамид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии D примера 2, используя продукт из стадии С выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1H); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1H); 7.31-7.20 (m, 2H); 7.11 (dd, J=8.9 и 2.5 Гц, 1H); 3.96 (s, 3H); 3.52-3.44 (m, 2H); 3.23-3.18 (m, 2H); 1.94 (s, 3H).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 5: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: пропан-1-сульфонат 7-метокси-1-{2-[(пропилсулъфонил)окси]этил}нафталин-2-ила
Продукт, полученный в стадии А примера 4 (2.45 г; 11.238 ммоль), растворяют в дихлорметане (60 мл), и после чего добавляют триэтиламин (3.7 мл; 26.4 ммоль). После охлаждения до 0°С, по каплям добавляют n-пропилсульфонилхлорид (2.8 мл; 24.6 ммоль). После перемешивания в течение 2 часов при температуре окружающей среды, растворитель выпаривают. Полученный остаток ресуспендируют в простом диэтиловом эфире и воде. После разделения, органическую фазу промывают разбавленным водным раствором HCl, водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 20/80) с получением указанного в заголовке продукта в виде твердого вещества (3.335 г; 66% в 3 стадии, исходя из 7-метокси-нафталин-2-ола).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (m, 2Н); 7.37 (d, J=2.4 Гц, 1Н); 7.31 (d, J=9.0 Гц, 1Н); 7.17 (dd, J=9.0 и 2.4 Гц, 1Н); 4.48 (t, J=8.0 Гц, 2Н); 3.96 (s, 3Н); 3.6 (t, J=8.0 Гц, 2Н); 3.45-3.4 (m, 2Н); 3.04-2.98 (m, 2Н); 2.1 (m, 2Н); 1.8 (m, 2Н); 1.17 (t, J=7.3 Гц, 3Н); 0.99 (t, J=7.3 Гц, 3Н).
Стадия В: пропан-1-сульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила
Продукт из стадии А выше (532 мг; 1.237 ммоль), ацетонитрил (18 мл) и водный 35% раствор аммиака (18 мл) вводят в герметичную колбу. Затем смесь помещают в баню, нагретую при 110°С в течение 3 часов. После этого растворители выпаривают под сниженным давлением, осуществляют азеотропную перегонку с этанолом. Сырой продукт растворяют в уксусном ангидриде (5 мл) и впоследствии добавляют ацетат натрия (500 мг). После перемешивания в течение одного часа, раствор разбавляют с этилацетататом и затем осторожно выливают в насыщенный водный раствор NaHCO3. После перемешивания в течение 15 минут две фазы разделяют и водную фазу экстрагируют два раза этилацетататом. Органические фракции объединяют, промывают водой и затем рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (289 мг; 64%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.73 (d, J=8.9 Гц, 1Н); 7.68 (d, J=8.9 Гц, 1Н); 7.62 (d, J=2.5 Гц, 1Н); 7.27 (d, J=8.9 Гц, 1Н); 7.16 (dd, J=8.9 и 2.5 Гц, 1Н); 5.95 (m, 1Н); 4.02 (s, 3Н); 3.62-3.54 (m, 2Н); 3.43-3.32 (m, 2Н); 2.09 (m, 2Н); 1.17 (t, J=7.4 Гц, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 170.9 (s); 159.0 (s); 145.6 (s); 134.7 (s); 130.2 (d); 128.6 (d); 127.7 (s); 126.0 (s); 119.2 (d); 118.1 (d); 103.3 (d); 55.9 (q); 53.6 (t); 39.4 (t); 26.5 (t); 23.4 (q); 17.6 (t); 13.1 (q).
Стадия С: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Продукт из стадии В выше (327 мг; 0.896 ммоль), ацетат аммония (1.19 г; 15.54 ммоль), палладий 10% на угле (95 мг; 0.09 ммоль), этанол (3 мл) и порошковый магний (83 мг; 3.584 ммоль) вводят в колбу. После перемешивания в течение 16 часов, гетерогенную смесь фильтруют через целит, и промывают этилацетататом. После выпаривания растворителей, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (190 мг; 87%).
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1H); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 6: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: пропан-1-сульфонат 1-[2-(2,5-диоксопирролидин-1-ил)этил]-7-метоксинафталин-2-ила
Продукт, полученный в стадии А примера 5 (240 мг; 0.558 ммоль), калия карбонат (231 мг; 1.674 ммоль), сукцинимид (66 мг; 0.67 ммоль) и диметилформамид (2 мл) вводят в колбу. После перемешивания в течение 16 часов при 100°С, раствор разбавляют с этилацетататом, промывают разбавленным водным раствором HCl, водой и рассолом и затем сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 70/30) с получением указанного в заголовке продукта (123 мг; 57%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.74 (d, J=9.0 Гц, 1Н); 7.7 (d, J=8.9 Гц, 1Н); 7.6 (d, J=2.4 Гц, 1Н); 7.38 (d, J=9.0 Гц, 1Н); 7.16 (dd, J=8.9 и 2.4 Гц, 1H); 4.05 (s, 3Н); 3.86-3.8 (m, 2Н); 3.58-3.52 (m, 2Н); 3.33-3.27 (m, 2Н); 2.72 (s, 4Н); 2.14 (m, 2Н); 1.2 (t, J=7.5 Гц, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 177.3 (2 х s); 159.1 (s); 145.3 (s); 134.4 (s); 130.4 (d); 128.8 (d); 127.7 (s); 124.2 (s); 119.2 (d); 118.4 (d); 102.4 (d); 55.8 (q); 53.5 (t); 37.7 (t); 28.4 (2 x t); 24. 7 (t); 17.5 (t); 13.1 (q).
Стадия В: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Продукт из стадии А выше (65 мг; 0.16 ммоль), ацетат аммония (240 мг; 3.12 ммоль), палладий 10% на угле (16.5 мг; 0.016 ммоль), метанол (0.7 мл) и порошковый магний (8 мг; 0.36 ммоль) одной порцией вводят в колбу. После перемешивания в течение 12 часов при 30°С, гетерогенную смесь фильтруют через целит, и затем промывают этилацетататом. После выпаривания растворителей, сырой продукт растворяют в этаноле (2 мл) в герметично закрытой пробирке, и затем добавляют водный раствор гидроксида натрия (180 мг; 4.5 ммоль в 1 мл воды). После этого смесь разбавляют с уксусным ангидрид (5 мл), и вводят ацетат натрия (500 мг). После перемешивания в течение одного часа, раствор разбавляют с этилацетататом и затем осторожно выливают в насыщенный водный раствор NaHCO3. После перемешивания в течение 15 минут, Две фазы разделяют и водную фазу экстрагируют два раза с этилацетататом. Органические фракции объединяют, промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (14.4 мг; 37%).
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1H); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 7: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 4-метилбензолсульфонат 1-(2-гидроксиэтил)-7-метоксинафталин-2-ила
К раствору продукта, полученного в стадии А примера 4 (109 мг; 0.5 ммоль) в тетрагидрофуране (2.5 мл) добавляют при 0°С трет-бутилат калия (56.4 мг; 0.5 ммоль) и затем, через 5 минут, тозилхлорид (95 мг; 0.5 ммоль). После перемешивания в течение 3 часов, раствору дают достичь температуры окружающей среды и перемешивают в течение еще 15 часов. Растворитель выпаривают, и полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 50/50) с получением смеси из двух диастереоизомеров (116 мг; 62%) в соотношении 88:12. Смесь применяют как таковую без дополнительного очищения.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.83 (d, J=8.2 Гц, 2Н); 7.71 (d, J=9.1 Гц, 1Н); 7.59 (d, J=8.9 Гц, 1Н); 7.35 (d, J=8.2 Гц, 2Н); 7.29 (d, J=2.4 Гц, 1Н); 7.15 (dd, J=8.9 и 2.4 Гц, 1H); 7.04 (d, J=9.1 Гц, 1H); 3.91 (s, 3Н); 3.87 (t, J=7.1 Гц, 2Н); 3.23 (t, J=7.1 Гц, 2Н); 2.46 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.6 (s); 146.7 (s); 145.7 (s); 134.5 (s); 133.3 (s); 130.3 (d); 130.1 (2 x d); 128.5 (2 x d); 128.2 (d); 127.8 (s); 125.7 (s); 118.5 (d); 118.3 (d); 103.4 (d); 62.1 (t); 55.5 (q); 29.7 (t); 21.9 (q).
Стадия В: 2-(7-метоксинафталин-1-ил)этанол
Смесь из диастереоизомеров, полученную в стадии А выше (80 мг; 0.176 ммоль соответствующего изомера), хлорид никеля гексагидрат (51 мг; 0.215 ммоль), дихлорметан (2 мл) и метанол (2 мл) вводят в герметичную колбу. Затем барботируют аргоном в растворе в течение 5 минут, и после этого маленькими порциями осторожно добавляют борогидрид натрия (146 мг; 4.3 ммоль). После перемешивания в течение одного часа при температуре окружающей среды под аргоном, добавляют разбавленный водный раствор HCl. После перемешивания в течение 4 часов, смесь фильтруют через целит и промывают этанолом. Растворитель выпаривают, и полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 40/60) с получением указанного в заголовке продукта (22 мг; 62%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1Н); 7.63 (d, J=8.1 Гц, 1Н); 7.3-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.6 Гц, 1H); 3.93 (t, J=6.7 Гц, 2Н); 3.88 (s, 3Н); 3.24 (d, J=6.7 Гц, 2Н); 1.99 (bs, 1Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 157.8 (s); 133.2 (s); 133.0 (s); 130.4 (d); 129.4 (s); 127.7 (d); 127.1 (d); 123.3 (d); 118.1 (d); 102.5 (d); 62.7 (t); 55.4 (q); 36.4 (t).
Масс-спектрометрия (ESI; m/z(%)): 202(29) [M]+•; 171(100).
Стадия С: метансульфонат 2-(7-метоксинафталин-1-ил)этила
К раствору продукта из стадии В выше (275 мг; 1.361 ммоль) в дихлорметане (7 мл) добавляют при 0°С триэтиламин (227 мкл; 1.633 ммоль) и мезилсульфонилхлорид (116 мкл; 1.498 ммоль). После перемешивания в течение одного часа, растворители выпаривают и остаток ресуспендируют в простом диэтиловом эфире и воде. После разделения, органическую фазу промывают три раза водой и рассолом, сушат над сульфатом натрия и фильтруют. Выпаривание растворителя обеспечивает чистый целевой продукт без любой дополнительной очистки (356 мг; 93%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1Н); 7.66 (d, J=8.9 Гц, 1Н); 7.31-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.5 Гц, 1Н); 4.47 (t, J=7.4 Гц, 2Н); 3.92 (s, 3Н); 3.45 (d, J=7.4 Гц, 2Н); 2. 77 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.2 (s); 133.0 (s); 133.0 (s); 130.6 (s); 130.4 (d); 129.3 (s); 128.0 (d); 127.7 (d); 123.2 (d); 118.4 (d); 101.9 (d); 67.9 (t); 55.5 (q); 37.4 (q); 33.3 (t).
Стадия D: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Продукт из стадии С выше (356 мг; 1.271 ммоль), ацетонитрил (7 мл) и водный 35% раствор аммиака (5 мл) вводят в колбу. Колбу помещают в баню, нагретую до 110°С, и раствор перемешивают в течение 3 часов. Раствор разбавляют с этилацетататом и промывают водным 2М раствором гидроксида натрия, водой и рассолом, и затем сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт растворяют в уксусном ангидриде (2 мл) в присутствии ацетата натрия (500 мг). После перемешивания в течение одного часа добавляют воду и этилацетат и после перемешивания в течение 15 минут, органическую фазу промывают два раза водным 2М раствором гидроксида натрия, промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. Растворитель выпаривают, и полученный сырой продукт затем очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (230 мг; 74%).
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 8: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид
7N метанольный раствор аммиака (10 мл; 70 ммоль) и продукт, полученный в стадии А примера 3 (1.5 г; 7 ммоль) вводят в герметичную колбу. После перемешивания в течение 18 часов при 100°С, растворитель выпаривают с получением сырого продукта, указанного в заголовке (1.58 г; 98%), который применяют непосредственно без всякой дополнительной очистки.
1Н ЯМР спектроскопический анализ (ДМСО-d6, 300.13 МГц, δ в част. на млн.): 9.87 (bs, 1H); 7.68 (d, J=9.0 Гц, 1Н); 7.59 (d, J=8.7 Гц, 1Н); 7.36 (bs, 1Н); 7.18 (d, J=2.4 Гц, 1Н); 7.01 (d, J=8.7 Гц, 1Н); 7.01 (bs, 1Н); 6.94 (dd, J=9.0 и 2.4 Гц, 1Н); 3.84 (s, 3Н); 3.79 (s, 2Н).
Стадия В: ацетат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила
Алюмогидрид лития (396 мг; 10.432 ммоль) добавляют к суспензии продукта из стадии А выше (1.205 г; 5.216 ммоль) в тетрагидрофуране (25 мл). После перемешивания при кипячении с обратным холодильником в течение δ часов, смесь охлаждают до 0°С и добавляют воду (40 мл), а после этого лимонную кислоту (10 г). После перемешивания в течение 14 часов, смесь нейтрализуют с использованием насыщенного водного раствора NaHCO3 и продукт экстрагируют три раза при помощи этилацетата. Органические фазы объединяют, промывают водой и рассолом, и затем сушат над сульфатом натрия и фильтруют. После выпаривания сырой продукт (802 мг) растворяют в уксусном ангидриде (3 мл) в присутствии ацетата натрия (500 мг). Смесь перемешивают в течение 16 часов и затем выливают в разбавленный водный раствор NaHCO3. Продукт экстрагируют три раза при помощи этилацетата и органические фазы объединяют, промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/метанол, градиент от 100/0 до 95/5) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (337 мг; 21%). Точка плавления: 149-151°С
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.73 (d, J=8.9 Гц, 1Н); 7.68 (d, J=8.8 Гц, 1Н); 7.42 (d, J=2.2 Гц, 1Н); 7.14 (dd, J=8.8 и 2.2 Гц, 1Н); 7.01 (d, J=8.9 Гц, 1H); 5. 75 (bs, 1H); 3.97 (s, 3H); 3.52 (m, 2H); 3.19 (t, J=6.7 Гц, 2H); 2.39 (s, 3H); 1.9 (s, 3H).
l3C ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 170.7 (s); 170.6 (s); 158.6 (s); 147.5 (s); 134.3 (s); 130.3 (d); 128.2 (d); 127.5 (s); 124.1 (s); 119.0 (d); 118.2 (d); 102.9 (d); 55.6 (q); 39.1 (t); 25.9 (t); 23.4 (q); 21.1 (q).
Стадия С: 4-метилбензолсульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила
Продукт из стадии В выше (230 мг; 0.764 ммоль) добавляют к раствору гидроксида натрия (61 мг; 1.528 ммоль) в абсолютном этаноле (10 мл). После перемешивания в течение одного часа, растворитель выпаривают. Остаток ресуспендируют в смеси из этилацетат/водный раствор разбавленной хлористоводородной кислоты. После разделения, органическую фазу промывают водой и рассолом, затем сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт растворяют в дихлорметане (4 мл) в присутствии триэтиламина (150 мкл; 2.47 ммоль) и тозилхлорид (174 мг; 0.917 ммоль). После перемешивания в течение 16 часов при температуре окружающей среды, растворитель выпаривают. Остаток ресуспендируют в этилацетате и воде. После разделения, органическую фазу промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (261 мг; 83%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.81 (d, J=8.2 Гц, 2Н); 7.68 (d, J=8.9 Гц, 1Н); 7.62 (d, J=2.4 Гц, 1Н); 7.55 (d, J=8.9 Гц, 1Н); 7.36 (d, J=8.2 Гц, 2Н); 7.14 (dd, J=8.9 и 2.4 Гц, 1Н); 6.89 (d, 8.9 Гц, 1Н); 5.97 (m, 1Н); 4.01 (s, 3Н); 3.53-3.46 (m, 2Н); 3.22-3.17 (m, 2Н); 2.46 (s, 3Н); 1.95 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 170.9 (s); 158.9 (s); 146.4 (s); 145.7 (s); 134.6 (s); 133.3 (s); 130.1 (2 x d); 130.0 (d); 128.5 (2xd); 128.2 (d); 127.7 (s); 126.2 (s); 119.2 (d); 117.8 (d); 103.3 (d); 55.8 (q); 39.4 (t); 26.4 (t); 23.4 (q); 21.9 (q).
Стадия D: N-[2-(7-метоксинафталии-1-ил)этил]ацетамид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии D примера 2, используя продукт из стадии С выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1H); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 9: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 4-метилбензолсульфонат 1-(2-амино-2-оксоэтил)-7-метоксинафталин-2-ила
Гидрид натрия (60% в минеральном масле; 78 мг; 1.953 ммоль) добавляют при 0°С к раствору продукта, полученного в стадии А примера δ (376 мг; 1.628 ммоль) в диметилформамиде (3 мл). После перемешивания в течение 30 минут, тозилхлорид (341 мг; 1.79 ммоль) вводят одной порцией. После перемешивания в течение 2 часов реакционную смесь разбавляют с этилацетататом, а после этого два раза промывают водой и два раза рассолом. Органическую фазу высушивают над сульфатом натрия и фильтруют. После выпаривания растворителей, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/дихлорметан 30/70) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (260 мг; 40%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.89 (d, J=7.9 Гц, 2Н); 7.72 (d, J=9.0 Гц, 1Н); 7.66 (d, J=9.0 Гц, 1Н); 7.4 (d, J=7.9 Гц, 2Н); 7.37 (d, J=2.4 Гц, 1H); 7.17 (dd, J=9.0 и 2.4 Гц, 1Н); 7.01 (d, J=9.0 Гц, 1Н); 6.02 (bs, 1Н); 5.48 (bs, 1Н); 3.93 (s, 2Н); 3.91 (s, 3Н); 2.50 (s, 3Н).
Стадия В: 2-(7-метоксинафталин-1-ил)ацетамид
Продукт, полученный в стадии А выше (50 мг; 0.13 ммоль), хлорид никеля гексагидрат (31 мг; 0.13 ммоль), дихлорметан (1.3 мл) и метанол (1.3 мл) вводят в герметичную колбу. Затем барботируют аргоном в растворе в течение 5 минут и после чего маленькими порциями осторожно добавляют борогидрид натрия (88 мг; 2.6 ммоль). После перемешивания в течение 1 часа под аргоном при температуре окружающей среды, добавляют воду. После перемешивания в течение 15 минут, смесь фильтруют через целит, и затем промывают дихлорметаном и этилацетатом. Органическую фракцию высушивают над сульфатом натрия и затем фильтруют. После выпаривания растворителей, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (8 мг; 29%).
1Н ЯМР спектроскопический анализ (ДМСО-d6, 300.13 МГц, δ в част. на млн.): 7.84 (d, J=9.0 Гц, 1Н); 7.73 (d, J=8.0 Гц, 1Н); 7.62 (bs, 1Н); 7.41-7.36 (m, 2Н); 7.28 (dd, J=8.0 и 7.1 Гц, 1Н); 7.18 (dd, J=9.0 и 2.4 Гц, 1Н); 7.02 (bs, 1Н); 3.88 (s, 3Н); 3.82 (s, 2Н).
13С ЯМР спектроскопический анализ (ДМСО-d6, 75.5 МГц, δ в част. на млн.): 172.3 (s); 157.3 (s); 133.2 (s); 131.8 (s); 130.0 (d); 128.7 (s); 128.4 (d); 126.7 (d); 123.1 (d); 117.7 (d); 103.4 (d); 55.2 (q); 40.2 (t).
Стадия С: (7-метоксинафталин-1-ил)ацетонитрил
Указанный в заголовке продукт получают в соответствии с протоколом, описанным в стадии F получения 1 описания к патенту ЕР 0447285, используя продукт из стадии В выше в качестве исходного реагента.
Точка плавления: 86-87°С.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.78 (d, J=8.9 Гц, 1Н); 7.77 (d, J=7.8 Гц, 1Н); 7.52 (d, J=7.1 Гц, 1Н); 7.32 (dd, J=7.8 и 7.1 Гц, 1Н); 7.21 (dd, J=8.9 и 2.4 Гц, 1H); 7.03 (d, J=2.4 Гц, 1Н); 4.0 (s, 2Н); 3.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.5 (s); 132.0 (s); 130.6(d); 129.1 (s); 128.8 (d); 127.1 (d); 124.4 (s); 123.2 (d); 118.8 (d); 117.7 (s); 101,3 (d); 55.4 (q); 21.9 (t).
Стадия D: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии Е примера 1, используя продукт из стадии С выше. Точка плавления: 108°С.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1H); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173,4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 10: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 2-метокси-7-(проп-2-ен-1-илокси)нафталин
Аллилбромид (4.4 мл; 50.56 ммоль) добавляют к раствору 7-метокси-нафталин-2-ола (5.865 г; 33.71 ммоль) и карбоната калия (13.96 г; 101.12 ммоль) в ацетоне (33 мл). После перемешивания при 65°С в течение 16 часов, смесь охлаждают до температуры окружающей среды и добавляют воду (60 мл). После перемешивания в течение 3 часов, продукт экстрагируют три раза при помощи этилацетата. Органические фракции объединяют, два раза промывают водой и затем рассолом, сушат над сульфатом натрия и фильтруют. Выпаривание растворителей обеспечивает сырой продукт (7.963 г), который применяют непосредственно в следующей стадии без дополнительной очистки.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.68 (d, J=8.9 Гц, 1Н); 7.67 (d, J=8.9 Гц, 1Н); 7.08-6.99 (m, 3Н); 6.13 (m, 1Н); 5.48 (m, 1H); 5.34 (m, 1Н); 4.65 (m, 1Н); 3.92 (s, 3Н).
Стадия В: 7-метокси-1-(проп-2-ен-1-ил)нафталин-2-ол
Используя колбу, продукт из стадии А выше (7.963 г; 33.71 ммоль) помещают в баню, нагретую до 200°С в течение 2,5 часов. После охлаждения, сырой продукт применяют непосредственно в следующей стадии без дополнительной очистки.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.69 (d, J=8.9 Гц, 1Н); 7.6 (d, J=8.7 Гц, 1Н); 7.2 (d, J=2.4 Гц, 1Н); 7.03 (dd, J=8.9 и 2.4 Гц, 1H); 6.95 (d, J=8.7 Гц, 1Н); 6.08 (m, 1Н); 5.28 (s, 1Н); 5.14 (m, 1H); 5.1 (m, 1Н); 3.93 (s, 3Н); 3.8 (m, 1Н).
Стадия С: 4-метилбензолсульфонат 7-метокси-1-(проп-2-ен-1-ил)нафталин-2-ила
Триэтиламин (1.95 мл; 14.02 ммоль) и тозилхлорид (2.23 г; 11.68 ммоль) добавляют к раствору продукта из стадии В выше (2.5 г; 11.68 ммоль) в дихлорметане (22 мл). После перемешивания в течение 16 часов при температуре окружающей среды, растворитель выпаривают и остаток ресуспендируют в этилацетате и воде. После разделения, органическую фазу промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир, градиент от 10/90 до 20/80) с получением указанного в заголовке продукта (3.79 г; 88%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.8 (d, J=8.3 Гц, 2Н); 7.71 (d, J=8.9 Гц, 1Н); 7.6 (d, J=8.9 Гц, 1Н); 7.33 (d, J=8.3 Гц, 2Н); 7.24 (d, J=2.5 Гц, 1Н); 7.14 (dd, J=8.9 и 2.5 Гц); 7.1 (d, J=8.9 Гц, 1Н); 5. 79 (m, 1H); 5.02-4.95 (m, 2Н); 3.88 (s, 3Н); 3.68 (d, J=5.9 Гц, 2Н); 2.46 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.3 (s); 146.1 (s); 145.5 (s); 135.5 (d); 134.4 (s); 133.3 (s); 130.1 (d); 130.0 (2 x d); 128.5 (2 x d); 128.0 (d); 127.7 (s); 126.6 (s); 118.5 (d); 118.4 (d); 116.1 (t); 103.9 (d); 55.4 (q); 30.5 (t); 21.8 (q).
Стадия D: 4-метилбензолсульфонат 1-(2,3-дигидроксипропил)-7-метоксинафталин-2-ила
4-Метилморфолина N-оксид гидрат (1.666 г; 12.35 ммоль) и тетроксид осмия (4% в воде; 653 мкл; 0.01 ммоль) добавляют к раствору продукта из стадии С выше (3.786 г; 10.29 ммоль) в ацетоне (40 мл) и деионизированную воду (10 мл). После перемешивания при температуре окружающей среды в течение 20 часов, добавляют тиосульфат натрия пентагидрат (1 г). После еще дополнительного часа перемешивания растворители выпаривают. Остаток ресуспендируют в этилацетате и органическую фазу промывают рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (3.617 г; 87%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.81 (d, J=8.3 Гц, 2Н); 7.69 (d, J=9.0 Гц, 1Н); 7.58 (d, J=9.0 Гц, 1Н); 7.33 (d, J=8.3 Гц, 2Н); 7.13 (dd, J=9.0 и 2.4, 1Н); 7.02 (d, J=9.0 Гц, 1Н); 4.01 (m, 1Н); 3.88 (s, 3Н); 3.63-3.47 (m, 2Н); 3.11 (d, J=7.0 Гц, 2Н); 2.9 (bs, 1Н); 2.62 (bs, 1Н); 2.44 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.6 (s); 146.8 (s); 145.8 (s); 134.6 (s); 133.1 (s); 130.2 (d); 130.1 (2 x d); 128.4 (2 x d); 128.3 (d); 127.7 (s); 125.4 (s); 118.6 (d); 118.0 (d); 103.6 (d); 72.0 (d); 65.8 (t); 55.5 (q); 30.1 (t); 21.8 (q).
Стадия E: 4-метилбензолсульфонат 7-метокси-1-(2-оксоэтил)нафталин-2-ила
Периодат натрия (785 мг; 3.671 ммоль) и водный 2М раствор HCl (1.8 мл) добавляют к раствору продукта из стадии D выше (1.23 г; 3.06 ммоль) в тетрагидрофуране (12 мл) и воде (3 мл). Через 30 минут, смесь нейтрализуют разбавленным водным раствором NaHCO3, и продукт затем экстрагируют при помощи этилацетата. Органические фракции объединяют, сушат над сульфатом натрия и фильтруют. Выпаривание растворителей обеспечивает указанный в заголовке продукт в виде чистого твердого вещества белого цвета (1.13 г; 100%), которое применяют непосредственно в следующей стадии.
Точка плавления: 101-103°С.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 9.43 (t, J=2.6 Гц, 1Н); 7.78 (d, J=8.3 Гц, 2Н); 7. 74 (d, J=8.9 Гц, 1Н); 7.68 (d, J=8.9 Гц, 1Н); 7.35 (d, J=8.3 Гц, 2Н); 7.17 (dd, J=8.9 и 2.4 Гц, 1Н); 7.04 (d, J=8.9 Гц, 1Н); 6.97 (d, J=2.4 Гц, 1Н); 3.98 (d, J=2.6 Гц, 2Н); 3.88 (s, 3Н); 2.46 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 199.2 (d); 159.1 (s); 146.8 (s); 146.0 (s); 134.7 (s); 132.7 (s); 130.4 (d); 130.2 (2 x d); 129.4 (d); 128.6 (2 x d); 127.6 (s); 120.1 (s); 119.1 (d); 118.6 (d); 102.9 (d); 55.5 (q); 41.7 (t); 21.9 (q).
Стадия F: 2-(7-метоксинафталин-1-ил)этанол
К раствору продукта из стадии Е выше (950 мг; 2.567 ммоль) в метаноле (13 мл) добавляют под аргоном, маленькими порциями хлорид никеля (366 мг; 2.824 ммоль) и борогидрид натрия (1.3 г; 38.2 ммоль). После перемешивания в течение одного часа, смесь подвергают гидролизу водным раствором 2М HCl (80 мл) и затем перемешивают в течение 30 минут в присутствии этилацетата. Гетерогенный раствор фильтруют через целит, и промывают этилацетататом. Две фазы разделяют и органическую фракцию промывают рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 40/60) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (442 мг; 85%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1Н); 7.63 (d, J=8.1 Гц, 1Н); 7.3-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.6 Гц, 1Н); 3.93 (t, J=6.7 Гц, 2Н); 3.88 (s, 3Н); 3.24 (d, J=6.7 Гц, 2Н); 1.99 (bs, 1H).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 157.8 (s); 133.2 (s); 133.0 (s); 130.4 (d); 129.4 (s); 127.7 (d); 127.1 (d); 123.3 (d); 118.1 (d); 102.5 (d); 62.7 (t); 55.4 (q); 36.4 (t).
Масс-спектрометрия (ESI; m/z(%)): 202(29) [M]+•; 171(100).
Стадия G: метансульфонат 2-(7-метоксинафталин-1-ил)этила
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии С примера 7, используя продукт из стадии F выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1Н); 7.66 (d, J=8.9 Гц, 1Н); 7.31-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.5 Гц, 1H); 4.47 (t, J=7.4 Гц, 2Н); 3.92 (s, 3Н); 3.45 (d, J=7.4 Гц, 2Н); 2.77 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.2 (s); 133.0 (s); 133.0 (s); 130.6 (s); 130.4 (d); 129.3 (s); 128.0 (d); 127.7 (d); 123.2 (d); 118.4 (d); 101.9 (d); 67.9 (t); 55.5 (q); 37.4 (q); 33.3 (t).
Стадия H: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии D примера 7, используя продукт из стадии G выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 11: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 3-(7-метоксинафталин-1-ил)пропан-1,2-диол
Продукт, полученный в стадии D примера 10 (44 мг; 0.11 ммоль), хлорид никеля (16 мг; 0.12 ммоль) и метанол (1 мл) вводят, под аргоном, в герметичную колбу. После чего маленькими порциями осторожно добавляют борогидрид натрия (74 мг; 2.19 ммоль). После перемешивания в течение 2 часов, добавляют воду (1 мл) и затем пероксид водорода (35% в воде; 1 мл), и смесь перемешивают в течение 2 часов. После чего добавляют рассол, и водную фазу экстрагируют три раза при помощи этилацетата. Органические фазы объединяют, сушат над сульфатом натрия и фильтруют. Выпаривание растворителей обеспечивает чистый продукт (22 мг; 86%) без потребности в дополнительной очистке.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.75 (d, J=9.1 Гц, 1H); 7.68 (d, J=8.1 Гц, 1Н); 7.32-7.23 (m, 3Н); 7.16 (dd, J=9.1 и 2.4 Гц, 1H); 4.11 (m, 1H); 3.92 (s, 3Н); 3. 72-3.55 (m, 2Н); 3.25-3.1 (m, 2Н); 2.54 (bs, 2Н, ОН).
Стадия В: (7-метоксинафталин-1-ил)ацетальдегид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии Е примера 10, используя продукт из стадии А выше вместо 4-метилбензолсульфоната 1-(2,3-дигидроксипропил)-7-метоксинафталин-2-ила.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 9.74 (t, J=2.6 Гц, 1Н); 7.74-7.84 (m, 2Н); 7.38 (d, J=6.0 Гц, 1Н); 7.32 (t, J=7.5 Гц, 1Н); 7.19 (dd, J=9.0 Гц и 2.4 Гц, 1Н); 7.1 (d, J=2.4 Гц, 1H); 4.05 (d, J=2.6 Гц, 2Н); 3.92 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 199.85; 158.29; 133.54; 130.44; 129.32; 129.07; 128.23; 126.83; 123.36; 118.63; 102.12; 55.36; 48.83.
Стадия С: 2-(7-метоксинафталин-1-ил)этанол
Борогидрид натрия (38 мг; 1 ммоль) добавляют к раствору продукта из стадии В выше (20 мг; 0.1 ммоль) в этаноле (1 мл). После перемешивания в течение 2 часов при температуре окружающей среды, смесь подвергают гидролизу посредством водного раствора 2М HCl (2 мл) и затем перемешивают в течение 30 минут в присутствии этилацетата (1 мл). Раствор экстрагируют 4 раза при помощи этилацетата. Органические фракции объединяют, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 20/80) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (19.8 мг; 98%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1Н); 7.63 (d, J=8.1 Гц, 1H); 7.3-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.6 Гц, 1Н); 3.93 (t, J=6.7 Гц, 2Н); 3.88 (s, 3Н); 3.24 (d, J=6.7 Гц, 2Н); 1.99 (bs, 1Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 157.8 (s); 133.2 (s); 133.0 (s); 130.4 (d); 129.4 (s); 127.7 (d); 127.1 (d); 123.3 (d); 118.1 (d); 102.5 (d); 62.7 (t); 55.4 (q); 36.4 (t).
Масс-спектрометрия (ESI; m/z(%)): 202(29) [M]+•; 171(100).
Стадия D: метансульфонат 2-(7-метоксинафталин-1-ил)этила
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии С примера 7, используя продукт из стадии С выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1H); 7.66 (d, J=8.9 Гц, 1Н); 7.31-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.5 Гц, 1Н); 4.47 (t, J=7.4 Гц, 2Н); 3.92 (s, 3Н); 3.45 (d, J=7.4 Гц, 2Н); 2.77 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.2 (s); 133.0 (s); 133.0 (s); 130.6 (s); 130.4 (d); 129.3 (s); 128.0 (d); 127.7 (d); 123.2 (d); 118.4 (d); 101.9 (d); 67.9 (t); 55.5 (q); 37.4 (q); 33.3 (t).
Стадия E: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии D примера 7, используя продукт из стадии D выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
Claims (74)
1. Способ промышленного синтеза соединения формулы (I)

отличающийся тем, что в реакцию вводят 7-метоксинафталин-2-ол формулы (II)

в который в положение 1 соединения формулы (II) вводят группу -СН2-Х, в которой X представляет собой группу -N(CH3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или -CO-NH2,
с получением соединения формулы (III)

в которой X представляет собой группу -N(CH3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или -CO-NH2;
причем соединение формулы (III) подвергают реакции сульфонилирования по ароматическому спирту и заместитель X соединения формулы (III) является модифицированным до или после стадии сульфонилирования ароматического спирта при помощи обычных химических реакций с получением соединения формулы (IV)

в которой X' представляет собой группу -CN, -CO-NH2, -СН2-ОН, -СНО, -CH2-N(CH2-Ph)2, -CH2-NH-CO-CH3, -СН(ОН)-СН2-ОН или (2,5-диоксопирролидин-1-ил)метил и R представляет собой группу -СН3, -(СН2)2-СН3, -CF3 или толуил;
причем соединение формулы (IV) подвергают реакции дезоксигенирования в присутствии переходного металла и восстановителя с получением:
- или, если X' представляет собой группу -CH2-NH-CO-CH3, непосредственно соединения формулы (I), которое выделяют в виде твердого вещества;
- или соединения формулы (V)

в которой X'' представляет собой группу -CN, -CH2-N(CH2-Ph)2, -СН2-ОН, -СН(ОН)-СН2-ОН, -CO-NH2 или (2,5-диоксопирролидин-1-ил)метил;
причем соединение формулы (V) подвергают обычным химическим реакциям с получением соединения формулы (I), которое выделяют в виде твердого вещества.
2. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (II) в соединение формулы (III) осуществляют посредством действия формальдегида и диметиламина с получением соединения формулы (III), в которой X представляет собой -N(СН3)2.
3. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (II) в соединение формулы (III) осуществляют посредством действия глиоксаля с последующим действием восстановителя с получением соединения формулы (III), в которой X представляет собой -CH2-ОН.
4. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (II) в соединение формулы (III) осуществляют посредством действия глиоксаля с последующим действием соединения формулы NHR'R', в которой R' представляет собой Н или группу -CH2-Ph, с получением соединения формулы (III), в которой X представляет собой -CO-NH2 или -CO-N(CH2-Ph)2.
5. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (II) в соединение формулы (III) осуществляют посредством действия аллилбромида с последующей термической перегруппировкой с получением соединения формулы (III), в которой X представляет собой -СН=СН2.
6. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что при превращении соединения формулы (III) в соединение формулы (IV) стадию сульфонилирования осуществляют посредством действия сульфонилхлорида, сульфонового ангидрида или сульфонимида.
7. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (III) в соединение формулы (IV) состоит из стадии сульфонилирования ароматического спирта с последующей модификацией группы X при помощи обычных химических реакций, X является таким, как определен в формуле (III).
8. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (III) в соединение формулы (IV) состоит в модификации группы X при помощи обычных химических реакций, после чего следует стадия сульфонилирования ароматического спирта, X является таким, как определен в формуле (III).
9. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV) в соединение формулы (V) осуществляют в присутствии никеля и гидрида.
10. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV) в соединение формулы (V) осуществляют в присутствии палладия и водорода.
11. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV) в соединение формулы (V) осуществляют в присутствии палладия и щелочноземельного металла.
12. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV), в которой X' представляет собой группу -CH2-NH-CO-CH3, в соединение формулы (I) осуществляют в присутствии никеля и гидрида.
13. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV), в которой X' представляет собой группу -CH2-NH-CO-CH3, в соединение формулы (I) осуществляют в присутствии палладия и водорода.
14. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV), в которой X' представляет собой группу -CH2-NH-CO-CH3, в соединение формулы (I) осуществляют в присутствии палладия и щелочноземельного металла.
15. Соединение формулы (III)

в которой X представляет собой группу -N(СН3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или -CO-NH2;
для применения в качестве промежуточного соединения в синтезе агомелатина формулы (I) по п.1.
16. Соединение формулы (III) по п.15, выбранное из следующих соединений:
- 1-[(диметиламино)метил]-7-метоксинафталин-2-ол;
- N,N-дибензил-2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид;
- 1-(2-гидроксиэтил)-7-метоксинафталин-2-ол;
- 2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид;
- 7-метокси-1-(проп-2-ен-1-ил)нафталин-2-ол.
17. Применение соединения формулы (III) по п.15 или 16 в синтезе агомелатина формулы (I) по п.1.
18. Соединение формулы (IV)

в которой X' представляет собой группу -CN, -CO-NH2, -СН2-ОН, -СНО, -CH2-N(CH2-Ph)2, -CH2-NH-CO-CH3, -СН(ОН)-СН2-ОН или (2,5-диоксопирролидин-1-ил)метил и R представляет собой группу -СН3, -(СН2)2-СН3, -CF3 или толуил;
для применения в качестве промежуточного соединения в синтезе агомелатина формулы (I) по п.1.
19. Соединение формулы (IV) по п.18, выбранное из следующих соединений:
- трифторметансульфонат 1-(цианометил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила;
- трифторметансульфонат 1-[2-(дибензиламино)этил]-7-метоксинафталин-2-ила;
- пропан-1-сульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила;
- пропан-1-сульфонат 1-[2-(2,5-диоксопирролидин-1-ил)этил]-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-(2-гидроксиэтил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-(2-амино-2-оксоэтил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 7-метокси-1-(2-оксоэтил)нафталин-2-ила;
- 4-метилбензолсульфонат 1-(2,3-дигидроксипропил)-7-метоксинафталин-2-ила.
20. Применение соединения формулы (IV) по п.18 или 19 в синтезе агомелатина формулы (I) по п.1.
21. Соединение формулы (V)

в которой X'' представляет собой группу -СН(ОН)-СН2-ОН или (2,5-диоксопирролидин-1-ил)метил, выбранное из следующих соединений:
- 1-[2-(7-метоксинафталин-1-ил)этил]пирролидин-2,5-дион;
- 3-(7-метоксинафталин-1-ил)пропан-1,2-диол;
для применения в качестве промежуточных соединений в синтезе агомелатина формулы (I) по п.1.
22. Применение соединения формулы (V) по п.21 в синтезе агомелатина формулы (I) по п.1.
23. (2-Гидрокси-7-метоксинафталин-1-ил)ацетонитрил, 4-метилбензолсульфонат 7-метокси-1-(2-{[(4-метилфенил)сульфонил]окси}этил)нафталин-2-ила, пропан-1-сульфонат 7-метокси-1-{2-[(пропилсульфонил)окси]этил}нафталин-2-ила и ацетат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила для применения в качестве промежуточных соединений в синтезе агомелатина формулы (I) по п.1.
24. Применение (2-гидрокси-7-метоксинафталин-1-ил)ацетонитрила, 4-метилбензолсульфоната 7-метокси-1-(2-{[(4-метилфенил)сульфонил]окси}этил)нафталин-2-ила, пропан-1-сульфоната 7-метокси-1-{2-[(пропилсульфонил)окси]этил}нафталин-2-ила и ацетата 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила по п.23 в синтезе агомелатина формулы (I) по п.1.
25. Соединение формулы (VI)

для применения в качестве промежуточного соединения в синтезе агомелатина формулы (I) по п.1.
26. Применение соединения формулы (VI) по п.25 в синтезе агомелатина формулы (I) по п.1.
27. Применение соединения формулы (II)

в синтезе агомелатина формулы (I) по п.1.
28. Способ синтеза агомелатина по п.1 исходя из соединения формулы (III), отличающийся тем, что соединение формулы (III) получают способом синтеза по любому из пп.2-5.
29. Способ синтеза агомелатина по п.1 исходя из соединения формулы (IV), отличающийся тем, что соединение формулы (IV) получают способом синтеза по любому из пп.2-8.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR13/62198 | 2013-12-05 | ||
| FR1362198A FR3014437B1 (fr) | 2013-12-05 | 2013-12-05 | Nouveau procede de synthese de l'agomelatine |
| PCT/FR2014/053157 WO2015082847A1 (fr) | 2013-12-05 | 2014-12-04 | Nouveau procede de synthese de l'agomelatine |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| RU2683279C1 true RU2683279C1 (ru) | 2019-03-27 |
Family
ID=50482958
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2016126661A RU2683279C1 (ru) | 2013-12-05 | 2014-12-04 | Новый способ синтеза агомелатина |
Country Status (27)
| Country | Link |
|---|---|
| US (1) | US9604911B2 (ru) |
| EP (1) | EP3077354B1 (ru) |
| JP (1) | JP2017504580A (ru) |
| KR (1) | KR20160093698A (ru) |
| CN (1) | CN105793224B (ru) |
| AU (1) | AU2014358965B2 (ru) |
| CA (1) | CA2932193C (ru) |
| CY (1) | CY1121116T1 (ru) |
| DK (1) | DK3077354T3 (ru) |
| EA (1) | EA030800B1 (ru) |
| ES (1) | ES2661262T3 (ru) |
| FR (1) | FR3014437B1 (ru) |
| HR (1) | HRP20180251T1 (ru) |
| HU (1) | HUE036562T2 (ru) |
| LT (1) | LT3077354T (ru) |
| MA (1) | MA39063B1 (ru) |
| MD (1) | MD20160071A2 (ru) |
| ME (1) | ME02968B (ru) |
| MX (1) | MX2016007186A (ru) |
| NO (1) | NO3077354T3 (ru) |
| PL (1) | PL3077354T3 (ru) |
| PT (1) | PT3077354T (ru) |
| RS (1) | RS56768B1 (ru) |
| RU (1) | RU2683279C1 (ru) |
| SI (1) | SI3077354T1 (ru) |
| WO (1) | WO2015082847A1 (ru) |
| ZA (1) | ZA201603457B (ru) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106831467B (zh) * | 2016-12-08 | 2019-03-01 | 江苏豪森药业集团有限公司 | 阿戈美拉汀的制备方法 |
| CN108821989A (zh) * | 2017-05-18 | 2018-11-16 | 郑州蛋壳信息科技有限公司 | 阿戈美拉汀中间体的制备方法 |
| CN107353229B (zh) * | 2017-08-08 | 2019-04-30 | 许昌恒生制药有限公司 | 一种阿戈美拉汀中间体的制备方法 |
| CN114560785B (zh) * | 2022-03-04 | 2023-06-13 | 郑州大学 | 一种伯酰胺类化合物的合成方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0447285A1 (fr) * | 1990-02-27 | 1991-09-18 | Adir Et Compagnie | Nouveaux dérivés à structure naphtalénique, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent |
| EP1564202A1 (fr) * | 2004-02-13 | 2005-08-17 | Les Laboratoires Servier | Nouveau procédé de synthèse et nouvelle forme cristalline de l'agomelatine ainsi que les compositions pharmaceutiques qui la contiennent |
| EP2151427A1 (fr) * | 2008-08-05 | 2010-02-10 | Les Laboratoires Servier | Nouveau procédé de synthése de l'agomélatine |
| EP2322508A1 (en) * | 2008-07-29 | 2011-05-18 | NHWA Pharma. Corporation | Process for the manufacture of agomelatine and its intermediate |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2713636B1 (fr) * | 1993-12-07 | 1996-01-05 | Adir | Nouveaux dérivés naphtaléniques, leur procédé de préparation, et les compositions pharmaceutiques qui les contiennent. |
| FR2771739B1 (fr) * | 1997-11-28 | 2001-04-20 | Adir | Nouveaux composes naphtaleniques, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| JP4798924B2 (ja) * | 1999-12-13 | 2011-10-19 | 中外製薬株式会社 | ヒドロキシカルボニル−ハロゲノアルキル側鎖を有する化合物 |
| KR100849559B1 (ko) * | 2004-01-22 | 2008-07-31 | 일라이 릴리 앤드 캄파니 | 혈관운동 증상의 치료를 위한 선택적 에스트로겐 수용체조절제 |
| FR2934859B1 (fr) * | 2008-08-05 | 2010-08-13 | Servier Lab | Nouveau procede de synthese de l'agomelatine |
| FR2934855B1 (fr) * | 2008-08-05 | 2010-08-13 | Servier Lab | Nouveau procede de synthese de l'agomelatine |
-
2013
- 2013-12-05 FR FR1362198A patent/FR3014437B1/fr not_active Expired - Fee Related
-
2014
- 2014-12-04 RU RU2016126661A patent/RU2683279C1/ru active
- 2014-12-04 US US15/100,782 patent/US9604911B2/en active Active
- 2014-12-04 CA CA2932193A patent/CA2932193C/fr not_active Expired - Fee Related
- 2014-12-04 MX MX2016007186A patent/MX2016007186A/es unknown
- 2014-12-04 DK DK14821794.6T patent/DK3077354T3/en active
- 2014-12-04 PT PT148217946T patent/PT3077354T/pt unknown
- 2014-12-04 EP EP14821794.6A patent/EP3077354B1/fr active Active
- 2014-12-04 RS RS20180042A patent/RS56768B1/sr unknown
- 2014-12-04 MA MA39063A patent/MA39063B1/fr unknown
- 2014-12-04 WO PCT/FR2014/053157 patent/WO2015082847A1/fr active Application Filing
- 2014-12-04 JP JP2016536741A patent/JP2017504580A/ja active Pending
- 2014-12-04 EA EA201600440A patent/EA030800B1/ru not_active IP Right Cessation
- 2014-12-04 LT LTEP14821794.6T patent/LT3077354T/lt unknown
- 2014-12-04 SI SI201430587T patent/SI3077354T1/en unknown
- 2014-12-04 PL PL14821794T patent/PL3077354T3/pl unknown
- 2014-12-04 CN CN201480066211.3A patent/CN105793224B/zh not_active Expired - Fee Related
- 2014-12-04 NO NO14821794A patent/NO3077354T3/no unknown
- 2014-12-04 AU AU2014358965A patent/AU2014358965B2/en not_active Ceased
- 2014-12-04 ES ES14821794.6T patent/ES2661262T3/es active Active
- 2014-12-04 MD MDA20160071A patent/MD20160071A2/ru not_active Application Discontinuation
- 2014-12-04 ME MEP-2017-287A patent/ME02968B/me unknown
- 2014-12-04 HU HUE14821794A patent/HUE036562T2/hu unknown
- 2014-12-04 KR KR1020167017813A patent/KR20160093698A/ko not_active Application Discontinuation
-
2016
- 2016-05-20 ZA ZA201603457A patent/ZA201603457B/en unknown
-
2017
- 2017-12-01 CY CY171101262T patent/CY1121116T1/el unknown
-
2018
- 2018-02-08 HR HRP20180251TT patent/HRP20180251T1/hr unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0447285A1 (fr) * | 1990-02-27 | 1991-09-18 | Adir Et Compagnie | Nouveaux dérivés à structure naphtalénique, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent |
| EP1564202A1 (fr) * | 2004-02-13 | 2005-08-17 | Les Laboratoires Servier | Nouveau procédé de synthèse et nouvelle forme cristalline de l'agomelatine ainsi que les compositions pharmaceutiques qui la contiennent |
| EP2322508A1 (en) * | 2008-07-29 | 2011-05-18 | NHWA Pharma. Corporation | Process for the manufacture of agomelatine and its intermediate |
| EP2151427A1 (fr) * | 2008-08-05 | 2010-02-10 | Les Laboratoires Servier | Nouveau procédé de synthése de l'agomélatine |
| EA015545B1 (ru) * | 2008-08-05 | 2011-08-30 | Ле Лаборатуар Сервье | Новый способ синтеза агомелатина |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2683279C1 (ru) | Новый способ синтеза агомелатина | |
| CN102659658A (zh) | 一种高度官能化的吡咯类化合物的合成方法 | |
| RU2550812C2 (ru) | Новый способ синтеза агомелатина | |
| CN102993040B (zh) | 一种合成阿戈美拉汀的新方法 | |
| CN109942442A (zh) | 一种盐酸达泊西汀有关物质i的制备方法 | |
| RU2680243C1 (ru) | Новый способ синтеза 7-метокси-нафталин-1-карбальдегида и его применение в синтезе агомелатина | |
| RU2522460C1 (ru) | Способ получения алкенилантрахинонов | |
| RU2804710C2 (ru) | Способ получения антигельминтных 4-амино-хинолин-3-карбоксамидных производных | |
| TW201217324A (en) | Preparation of bicyclo[2.2.2]octan-2-one compounds | |
| CN116478052A (zh) | 一种铜催化合成1-萘胺衍生物的方法 | |
| JP4750286B2 (ja) | 反応活性な基を有する新規なビフェニル化合物の製造方法 | |
| JP2006508156A (ja) | 触媒として白金を用いたテルビナフィンの製造方法 | |
| WO2015082848A2 (fr) | Nouveau procede de synthese du 7-methoxy-naphtalene-1-carbaldehyde et application a la synthese de l'agomelatine | |
| JP2012006921A (ja) | リン酸オセルタミビルの製造法 | |
| CN106883140A (zh) | 一种含叠氮基小分子化合物的制备方法 | |
| ITMI962363A1 (it) | Procedimento migliorato per la preparazione di 1,1-dimetil-2-(n- metil-n-(3,3-difenil propil)amminoetanolo |