RU2181722C2 - Способ получения и промежуточные соединения для получения ингибиторов 5-липоксигеназы - Google Patents
Способ получения и промежуточные соединения для получения ингибиторов 5-липоксигеназы Download PDFInfo
- Publication number
- RU2181722C2 RU2181722C2 RU99124413/04A RU99124413A RU2181722C2 RU 2181722 C2 RU2181722 C2 RU 2181722C2 RU 99124413/04 A RU99124413/04 A RU 99124413/04A RU 99124413 A RU99124413 A RU 99124413A RU 2181722 C2 RU2181722 C2 RU 2181722C2
- Authority
- RU
- Russia
- Prior art keywords
- formula
- compound
- alkyl
- acid
- och
- Prior art date
Links
- 0 *C(Nc(cc1)ccc1Sc1cccc(C2(CCOCC2)C#N)c1)=NCC(O*)O* Chemical compound *C(Nc(cc1)ccc1Sc1cccc(C2(CCOCC2)C#N)c1)=NCC(O*)O* 0.000 description 3
- VHRPXUJEVHGFFC-UHFFFAOYSA-N Nc(cc1)ccc1Sc1cccc(C2(CCOCC2)C#N)c1 Chemical compound Nc(cc1)ccc1Sc1cccc(C2(CCOCC2)C#N)c1 VHRPXUJEVHGFFC-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pyrane Compounds (AREA)
Abstract
Изобретение относится к новому способу получения соединений формулы I

где А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, - ингибиторов 5-липоксигеназы, полезных для лечения или облегчения воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний. Изобретение также относится к новым соединениям формулы I, где А - С1-C6, и промежуточным соединениям формулы VIII

где Х - Br или Cl, необходимым для получения соединений формулы I. Технический результат состоит в разработке нового более простого и дешевого способа получения ингибиторов 5-липоксигеназы. 3 с. и 23 з.п. ф-лы.
где А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, - ингибиторов 5-липоксигеназы, полезных для лечения или облегчения воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний. Изобретение также относится к новым соединениям формулы I, где А - С1-C6, и промежуточным соединениям формулы VIII
где Х - Br или Cl, необходимым для получения соединений формулы I. Технический результат состоит в разработке нового более простого и дешевого способа получения ингибиторов 5-липоксигеназы. 3 с. и 23 з.п. ф-лы.
Description
Предпосылки к созданию изобретения
Данное изобретение относится к способу и промежуточным соединениям для получения ингибиторов 5-липоксигеназы. Ингибиторы 5-липоксигеназы, полученные в соответствии с настоящим изобретением, раскрыты в патентной заявке США серийный номер 09/0200140, которая представляет собой продолжение 08/809901, поданной 13 июня 1997, в настоящее время отозвана. Данная находящаяся на рассмотрении заявка озаглавлена "ингибиторы 5-липоксигеназы" и включена ссылкой в описание в полном объеме.
Данное изобретение относится к способу и промежуточным соединениям для получения ингибиторов 5-липоксигеназы. Ингибиторы 5-липоксигеназы, полученные в соответствии с настоящим изобретением, раскрыты в патентной заявке США серийный номер 09/0200140, которая представляет собой продолжение 08/809901, поданной 13 июня 1997, в настоящее время отозвана. Данная находящаяся на рассмотрении заявка озаглавлена "ингибиторы 5-липоксигеназы" и включена ссылкой в описание в полном объеме.
Ингибиторы 5-липоксигеназы, полученные в соответствии с настоящим изобретением, представляют собой селективные ингибиторы действия фермента липоксигеназы и полезны при лечении или облегчении воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний у млекопитающих.
Краткое описание изобретения
Настоящее изобретение относится к способу получения соединения формулы

в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, который включает взаимодействие соединения формулы II

в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с сульфокислотой в C1-C5 алкиловом спирте; и осаждение соединения формулы I путем добавления органического растворителя, менее полярного, чем спирт.
Настоящее изобретение относится к способу получения соединения формулы
в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, который включает взаимодействие соединения формулы II
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с сульфокислотой в C1-C5 алкиловом спирте; и осаждение соединения формулы I путем добавления органического растворителя, менее полярного, чем спирт.
Кислота представляет собой метансульфокислоту, и органический растворитель является диизопропиловым эфиром или этилацетатом.
В следующем аспекте настоящего изобретения соединение формулы II получают взаимодействием соединения формулы III

в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с гидроксидом в спиртовом растворителе.
в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с гидроксидом в спиртовом растворителе.
Гидроксид представляет собой гидроксид калия, и спирт является трет-бутиловым спиртом.
В следующем аспекте настоящего изобретения соединение формулы III получают взаимодействием соединения формулы IV

в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с органической или минеральной кислотой.
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с органической или минеральной кислотой.
Кислота представляет собой уксусную кислоту, серную кислоту, муравьиную кислоту или п-толуолсульфокислоту. Предпочтительной кислотой является муравьиная кислота.
В следующем аспекте настоящего изобретения соединение формулы IV получают при взаимодействии соединения формулы V

в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, и в которой Х является Cl, Br, I или ОСН3, с избытком ацеталя аминоацетальдегида.
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, и в которой Х является Cl, Br, I или ОСН3, с избытком ацеталя аминоацетальдегида.
В следующем аспекте настоящего изобретения соединение формулы V, в которой Х является Cl, Br или I, получают взаимодействием соединения формулы VI
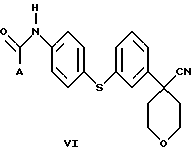
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с пентагалогенидом фосфора в инертном растворителе. Соединение формулы V можно также получить взаимодействием соединения формулы VI с (CH3)3O+BF4 - с образованием промежуточного соединения, в котором Х представляет собой ОСН3.
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с пентагалогенидом фосфора в инертном растворителе. Соединение формулы V можно также получить взаимодействием соединения формулы VI с (CH3)3O+BF4 - с образованием промежуточного соединения, в котором Х представляет собой ОСН3.
Пентагалогенид представляет собой пятихлористый фосфор(III), пятииодистый фосфор (III) или пятибромистый фосфор (III), а растворитель представляет собой толуол. Предпочтительным А является СН3.
В дополнительном аспекте настоящего изобретения соединение формулы VI получают взаимодействием соединения формулы VIII

в которой Х является Сl, Br или I, с избытком 4-аминотиофенола с основанием в инертном растворителе с образованием соединения формулы VII

и последующей обработкой соединения формулы VII путем ацилирования галогенангидридом или ангидридом кислоты.
в которой Х является Сl, Br или I, с избытком 4-аминотиофенола с основанием в инертном растворителе с образованием соединения формулы VII
и последующей обработкой соединения формулы VII путем ацилирования галогенангидридом или ангидридом кислоты.
Другой, наиболее предпочтительный путь получения соединения формулы VI состоит во взаимодействии соединения формулы VIII

в которой Х является Сl, Br или I, с избытком 4-амидотиофенола с основанием в инертном растворителе.
в которой Х является Сl, Br или I, с избытком 4-амидотиофенола с основанием в инертном растворителе.
4-амидотиофенол представляет собой 4-ацетамидотиофенол. Растворитель представляет собой N-метилпирролидон (NMP) или диметисульфоксид (DMSO).
Основание представляет собой карбонат натрия/карбонат цезия.
В следующем аспекте настоящего изобретения соединение формулы VIII можно получить взаимодействием соединения формулы

в которой Х является Сl, Br или I, с бис 2-хлорэтиловым эфиром, щелочным основанием и межфазным катализатором в инертном растворителе.
в которой Х является Сl, Br или I, с бис 2-хлорэтиловым эфиром, щелочным основанием и межфазным катализатором в инертном растворителе.
Межфазным катализатором является кислый сульфат тетрабутиламмония. Основание представляет собой гидроксид натрия.
Инертный растворитель представляет собой смесь тетрагидрофурана с водой.
Изобретение относится также к новому соединению формулы

Изобретение также относится к новому соединению формулы

в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом.
Изобретение также относится к новому соединению формулы
в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом.
Изобретение относится также к новому соединению формулы

в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, и в которой Х является Cl, Br, I или ОСН3.
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, и в которой Х является Cl, Br, I или ОСН3.
Изобретение относится также к новому соединению формулы

в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, и в которой R является C1-С6 алкилом.
в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, и в которой R является C1-С6 алкилом.
Изобретение относится также к новому соединению формулы

в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Сl, Br, ОСН3, C1-С3-алкилом или бензилом.
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Сl, Br, ОСН3, C1-С3-алкилом или бензилом.
Предпочтительным соединением является

Эти новые соединения используются при получении ингибиторов 5-липоксигеназы и их фармацевтической композиции, полезных при лечении или облегчении воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний у млекопитающих.
Эти новые соединения используются при получении ингибиторов 5-липоксигеназы и их фармацевтической композиции, полезных при лечении или облегчении воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний у млекопитающих.
Подробное описание изобретения
Новый способ синтеза представлен на реакционной схеме 1.
Новый способ синтеза представлен на реакционной схеме 1.
3-Бромфенилацетонитрил в тетрагидрофуране обрабатывают водным NaOH, кислым сульфатом тетрабутиламмония и бис 2-хлорэтиловым эфиром, получая арилбромидное соединение формулы VIII.
Арилбромидное соединение формулы VIII обрабатывают либо 4-аминотиофенолом, получая анилиновое соединение VII, с последующим ацилированием, либо 4-амидотиофенолом, получая амидное соединение VI. Имидазольную функцию вводят при превращении амидогруппы формулы VI путем нагревания соединения VI с пентагалогенидом фосфора (III), получая соединение V, которое обрабатывают алкилацеталем аминоацетальдегида, что приводит к амидиновому соединению IV. Амидиновое соединение IV существует в виде смеси таутомеров, которые не выделяют и немедленно подвергают кислотно-индуцированной циклизации, получая имидазольное соединение III. Последующий гидролиз нитрильной функции имидазольного соединения III приводит к соединению II - ингибитору 5-липоксигеназы. Найдена предпочтительная солевая форма путем обработки соединения II метансульфокислотой, что приводит к соединению I.
В новом способе настоящего изобретения исключены две предыдущие дорогостоящие реакции сочетания на палладии (0) для введения в молекулу сульфидной связи, как описано в патентной заявке США 09/0200140, которая включена ссылкой. Кроме того, упомянутый предпочтительный атом серы ранее вводили с использованием ТИПС-тиольного реагента (ТИПС представляет собой триизопропилсилил), получаемого из токсичного сероводорода и дорогостоящего ТИПС-хлорида.
Соединение I, в котором А представляет собой СН3, является предпочтительной солевой формой ингибитора 5-липоксигеназы, полезного при лечении или облегчении воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний у млекопитающих. В особенности соединение I полезно при лечении или облегчении воспалительных заболеваний.
Эти полезные ингибиторы 5-липоксигеназы можно вводить в виде самых разнообразных лекарственных форм.
Для лечения различных описанных выше состояний эти соединения и их фармацевтически приемлемые соли можно вводить человеку либо сами по себе, либо предпочтительно в сочетании с фармацевтически приемлемыми носителями или разбавителями в фармацевтической композиции в соответствии со стандартной фармацевтической практикой. Соединения можно вводить перорально или парентерально обычным образом.
Когда соединения вводят человеку для предотвращения или лечения воспалительного заболевания, пероральная дозировка составит от примерно 0,1 до 10 мг/кг на вес тела субъекта, подлежащего лечению, в день, предпочтительно от примерно 0,1 до 4 мг/кг в день в виде одной или разделенной доз. Если желательно парентеральное введение, тогда эффективная доза составит от примерно 0,05 до 5 мг/кг на вес тела субъекта, подлежащего лечению, в день. В некоторых случаях может быть необходимым применять дозировки, выходящие за эти пределы, поскольку дозировки будут неизбежно меняться в соответствии с возрастом, весом и реакцией индивидуального пациента, а также с серьезностью симптомов заболевания и эффективностью определенного вводимого соединения.
Для перорального введения соединения данного изобретения и их фармацевтически приемлемые соли можно вводить, например, в виде таблеток, порошков, лепешек, сиропов, или капсул, или в виде водного раствора или суспензии. В случае таблеток для перорального применения обычно используемые носители включают лактозу и кукурузный крахмал. Кроме этого, обычно добавляют смазывающие агенты, такие как стеарат магния. В случае капсул полезными разбавителями являются лактоза и высушенный кукурузный крахмал. Когда для перорального применения требуются водные суспензии, активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. При желании можно добавить определенные подслащивающие и/или ароматизирующие агенты. Для внутримышечного, внутрибрюшинного, подкожного и внутривенного применения обычно готовят стерильные растворы активного ингредиента и рН этих растворов должен быть соответствующим образом установлен и забуферен. При внутривенном применении следует контролировать общую концентрацию растворенного вещества для того, чтобы получить препарат изотоническим.
Кроме того, особенно для лечения астмы, соединения формулы I данного изобретения можно вводить человеку путем ингаляции. Для этой цели их вводят в виде спрея или аэрозоля в соответствии со стандартной практикой.
Настоящее изобретение иллюстрируется следующими примерами, но не ограничивается их деталями.
Пример 1
Нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты

3-Бромфенилацетонитрил (51 г) в ТГФ (300 мл) обработали 40%-ным водным NaOH (470 мл), кислым сульфатом тетрабутиламмония (9 г) и добавили по каплям бис 2-хлорэтиловый эфир (32 мл). Реакционную смесь нагревали при температуре образования флегмы 4 часа и затем охладили. Смесь разбавили EtOAc (400 мл), промыли 5%-ной НСl (200 мл), водой (200 мл) и насыщенным раствором NаНСО3. После высушивания над Mg2SO4 растворитель удалили, получив сырой СР-399,554 в виде воскообразного твердого вещества (75,4 г). Это твердое вещество суспендировали в смеси 1:1 изопропилового эфира и гексана (100 мл), получив нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты (55,3 г, выход 80%).
Нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты
3-Бромфенилацетонитрил (51 г) в ТГФ (300 мл) обработали 40%-ным водным NaOH (470 мл), кислым сульфатом тетрабутиламмония (9 г) и добавили по каплям бис 2-хлорэтиловый эфир (32 мл). Реакционную смесь нагревали при температуре образования флегмы 4 часа и затем охладили. Смесь разбавили EtOAc (400 мл), промыли 5%-ной НСl (200 мл), водой (200 мл) и насыщенным раствором NаНСО3. После высушивания над Mg2SO4 растворитель удалили, получив сырой СР-399,554 в виде воскообразного твердого вещества (75,4 г). Это твердое вещество суспендировали в смеси 1:1 изопропилового эфира и гексана (100 мл), получив нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты (55,3 г, выход 80%).
Пример 2
Нитрил 4-[3-(4-аминофенилсульфанил)-фенил]-тетрагидропиран-4-карбоновой кислоты

Нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты (133,4 г), Na2СО3 (363,6 г), Сs2СО3 (223,1 г) и аминотиофенол (62,8 г) нагревали в N-метилпирролидиноне (2,3 л) при 130oС в течение 24 часов. Добавили еще аминотиофенол (35,6 г) и продолжали нагревание в течение еще 8 часов. Смесь охладили до комнатной температуры, вылили в ледяную воду (6,8 л) и отфильтровали. Продукт суспендировали в воде (2,5 л), снова отфильтровали и промыли водой (1,5 л). После этого продукт суспендировали в EtOH (0,5 л), отфильтровали и высушили при 40oС/20 мбар, получив в результате нитрил 4-[3-(4-аминофенилсульфанил)-фенил] -тетрагидропиран-4-карбоновой кислоты (134,3 г, 86%).
Нитрил 4-[3-(4-аминофенилсульфанил)-фенил]-тетрагидропиран-4-карбоновой кислоты
Нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты (133,4 г), Na2СО3 (363,6 г), Сs2СО3 (223,1 г) и аминотиофенол (62,8 г) нагревали в N-метилпирролидиноне (2,3 л) при 130oС в течение 24 часов. Добавили еще аминотиофенол (35,6 г) и продолжали нагревание в течение еще 8 часов. Смесь охладили до комнатной температуры, вылили в ледяную воду (6,8 л) и отфильтровали. Продукт суспендировали в воде (2,5 л), снова отфильтровали и промыли водой (1,5 л). После этого продукт суспендировали в EtOH (0,5 л), отфильтровали и высушили при 40oС/20 мбар, получив в результате нитрил 4-[3-(4-аминофенилсульфанил)-фенил] -тетрагидропиран-4-карбоновой кислоты (134,3 г, 86%).
Пример 3
N-{4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]-фенил}ацетамид
Нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты (1,33 г) смешали с Na2СО3 (1,59 г), Сs2СО3 (0,651 г) и 4-ацетамидотиофенолом (1 г) в N-метилпирролидиноне (15 мл). Реакционную смесь нагревали при 130oС в течение ночи. После охлаждения смесь вылили в ледяную воду. Продукт, выпавший в виде твердого вещества, собрали при помощи фильтрования с отсасыванием. Твердое вещество перекристаллизовали из смеси EtOAc и гексана, получив при этом N-{ 4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]-фенил}ацетамид (1,4 г, выход 80%).
N-{4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]-фенил}ацетамид
Нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты (1,33 г) смешали с Na2СО3 (1,59 г), Сs2СО3 (0,651 г) и 4-ацетамидотиофенолом (1 г) в N-метилпирролидиноне (15 мл). Реакционную смесь нагревали при 130oС в течение ночи. После охлаждения смесь вылили в ледяную воду. Продукт, выпавший в виде твердого вещества, собрали при помощи фильтрования с отсасыванием. Твердое вещество перекристаллизовали из смеси EtOAc и гексана, получив при этом N-{ 4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]-фенил}ацетамид (1,4 г, выход 80%).
Объединенные примеры 2 и 3
N-{4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]фенил}ацетамид

Нитрил 4-[3-(4-аминофенилсульфанил)фенил] -тетрагидропиранкарбоновой кислоты (93,57 г) и Et3N (53,1 мл) растворили в EtOAc (1,23 л) и нагрели до 50-60oС. К этому раствору в течение 30 минут добавили хлористый ацетил (27,7 мл) в EtOAc (73 мл). Полученную суспензию отфильтровали и промыли фильтровальную лепешку EtOAc (3•150 мл). Объединенные растворы EtOAc промыли водой (0,5 л), наполовину насыщенным водным раствором Na2СО3 (2•0,5 л), водой (0,5 л) и насыщенным водным раствором NaCl (0,25 л). Органические слои высушили над Na2SO4 и выпарили при 40oС. Сырой продукт перекристаллизовали из кипящего EtOH (0,52 л), получив после охлаждения, фильтрования и высушивания при 40oС/20 мбар N-{4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]-фенил} ацетамид (55,27 г, выход 52%).
N-{4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]фенил}ацетамид
Нитрил 4-[3-(4-аминофенилсульфанил)фенил] -тетрагидропиранкарбоновой кислоты (93,57 г) и Et3N (53,1 мл) растворили в EtOAc (1,23 л) и нагрели до 50-60oС. К этому раствору в течение 30 минут добавили хлористый ацетил (27,7 мл) в EtOAc (73 мл). Полученную суспензию отфильтровали и промыли фильтровальную лепешку EtOAc (3•150 мл). Объединенные растворы EtOAc промыли водой (0,5 л), наполовину насыщенным водным раствором Na2СО3 (2•0,5 л), водой (0,5 л) и насыщенным водным раствором NaCl (0,25 л). Органические слои высушили над Na2SO4 и выпарили при 40oС. Сырой продукт перекристаллизовали из кипящего EtOH (0,52 л), получив после охлаждения, фильтрования и высушивания при 40oС/20 мбар N-{4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]-фенил} ацетамид (55,27 г, выход 52%).
Пример 4
Нитрил 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил}-тетрагидропиран-4-карбоновой кислоты

Нитрил N-4-[3-(4-аминофенилсульфанил)фенил] -тетрагидропиранкарбоновой кислоты (49,77 г) растворили в толуоле (545 мл) и нагрели до 60oС в условиях азеотропа. Из этого раствора изеотронно отогнали 20 мл растворителя для удаления оставшейся воды. К этому раствору в виде нескольких порций добавили PCl5 (35,0 г). После перемешивания в течение 1 часа при 60oС растворитель отогнали. Остаток охладили до 10oС и добавили смесь Еt3N (19,8 мл) и диметилацеталя аминоацетальдегида (15,2 мл) в EtOAc (500 мл). Полученную суспензию перемешивали в течение 30 минут при 10oС и после этого добавили еще EtOAc (150 мл). Смесь промыли водой (360 мл), а затем насыщенным водным раствором NaCl (150 мл). Органические слои высушили над Na2SO4 (52 г) и выпарили при 50oС. Остаток растворили в муравьиной кислоте (250 мл) и нагревали до температуры образования флегмы в течение 1 часа. Реакционную смесь сконцентрировали при 50oС/100 мбар до состояния масла. Это масло растворили в 10%-ной лимонной кислоте (400 мл) и EtOAc (200 мл). Водный слой экстрагировали EtOAc (350 мл). рН водного слоя довели до 9-10 при помощи наполовину насыщенного раствора К2СО3 (175 мл) и экстрагировали этот раствор EtOAc (200 мл). Экстракт высушили над Na2SO4 (48 г) и выпарили при 50oС/100 мбар, получив после фильтрования через слой двуокиси кремния с использованием в качестве элюента СН2Сl2/ 10% МеОН, нитрил 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил} -тетрагидропиран-4-карбоновой кислоты (27,6 г, общий выход 55%).
Нитрил 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил}-тетрагидропиран-4-карбоновой кислоты
Нитрил N-4-[3-(4-аминофенилсульфанил)фенил] -тетрагидропиранкарбоновой кислоты (49,77 г) растворили в толуоле (545 мл) и нагрели до 60oС в условиях азеотропа. Из этого раствора изеотронно отогнали 20 мл растворителя для удаления оставшейся воды. К этому раствору в виде нескольких порций добавили PCl5 (35,0 г). После перемешивания в течение 1 часа при 60oС растворитель отогнали. Остаток охладили до 10oС и добавили смесь Еt3N (19,8 мл) и диметилацеталя аминоацетальдегида (15,2 мл) в EtOAc (500 мл). Полученную суспензию перемешивали в течение 30 минут при 10oС и после этого добавили еще EtOAc (150 мл). Смесь промыли водой (360 мл), а затем насыщенным водным раствором NaCl (150 мл). Органические слои высушили над Na2SO4 (52 г) и выпарили при 50oС. Остаток растворили в муравьиной кислоте (250 мл) и нагревали до температуры образования флегмы в течение 1 часа. Реакционную смесь сконцентрировали при 50oС/100 мбар до состояния масла. Это масло растворили в 10%-ной лимонной кислоте (400 мл) и EtOAc (200 мл). Водный слой экстрагировали EtOAc (350 мл). рН водного слоя довели до 9-10 при помощи наполовину насыщенного раствора К2СО3 (175 мл) и экстрагировали этот раствор EtOAc (200 мл). Экстракт высушили над Na2SO4 (48 г) и выпарили при 50oС/100 мбар, получив после фильтрования через слой двуокиси кремния с использованием в качестве элюента СН2Сl2/ 10% МеОН, нитрил 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил} -тетрагидропиран-4-карбоновой кислоты (27,6 г, общий выход 55%).
Пример 5
Амид 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил]фенил}-тетрагидропиран-4-карбоновой кислоты

Нитрил 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил}-тетрагидропиран-4-карбоновой кислоты (27,35 г) растворили в трет-BuOH (280 мл) при 50oС. К этому раствору добавили КОН (12,28 г) и перемешивали смесь в течение ночи. Суспензию охладили до комнатной температуры и добавили воду (180 мл). Полученную суспензию отфильтровали, а фильтровальную лепешку высушили при 50oС, получив амид 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил]фенил}-тетрагидропиран-4-карбоновой кислоты (17,52 г, выход 55%).
Амид 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил]фенил}-тетрагидропиран-4-карбоновой кислоты
Нитрил 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил}-тетрагидропиран-4-карбоновой кислоты (27,35 г) растворили в трет-BuOH (280 мл) при 50oС. К этому раствору добавили КОН (12,28 г) и перемешивали смесь в течение ночи. Суспензию охладили до комнатной температуры и добавили воду (180 мл). Полученную суспензию отфильтровали, а фильтровальную лепешку высушили при 50oС, получив амид 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил]фенил}-тетрагидропиран-4-карбоновой кислоты (17,52 г, выход 55%).
Пример 6
Метилсульфонат амида 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил]фенил}-тетрагидропиран-4-карбоновой кислоты

Амид 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил}-тетрагидропиран-4-карбоновой кислоты (5,05 г) суспендировали в МеОН (39 мл) при комнатной температуре. К этой суспензии по каплям добавляли метансульфокислоту до тех пор, пока все вещество не растворилось. Полученный раствор отфильтровали и промыли фильтрат МеОН (20 мл). Объединенные растворы МеОН обработали диизопропиловым эфиром (280 мл) при комнатной температуре. После перемешивания в течение ночи образовались кристаллы, которые собрали при фильтровании и высушили при 40oС/19 мбар, получив при этом метилсульфонат амида 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил} -тетрагидропиран-4-карбоновой кислоты (4,85 г, выход 77%).
Метилсульфонат амида 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил]фенил}-тетрагидропиран-4-карбоновой кислоты
Амид 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил}-тетрагидропиран-4-карбоновой кислоты (5,05 г) суспендировали в МеОН (39 мл) при комнатной температуре. К этой суспензии по каплям добавляли метансульфокислоту до тех пор, пока все вещество не растворилось. Полученный раствор отфильтровали и промыли фильтрат МеОН (20 мл). Объединенные растворы МеОН обработали диизопропиловым эфиром (280 мл) при комнатной температуре. После перемешивания в течение ночи образовались кристаллы, которые собрали при фильтровании и высушили при 40oС/19 мбар, получив при этом метилсульфонат амида 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил} -тетрагидропиран-4-карбоновой кислоты (4,85 г, выход 77%).
Claims (25)
1. Способ получения соединения формулы I

в которой А представляет C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом,
а) включающий взаимодействие соединения формулы

в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с сульфокислотой в C1-C5-алкиловом спирте;
в) осаждение соединения формулы I путем добавления органического растворителя, полярность которого меньше, чем у упомянутого спирта.
в которой А представляет C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом,
а) включающий взаимодействие соединения формулы
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с сульфокислотой в C1-C5-алкиловом спирте;
в) осаждение соединения формулы I путем добавления органического растворителя, полярность которого меньше, чем у упомянутого спирта.
2. Способ по п. 1, где кислота представляет собой метансульфокислоту.
3. Способ по п. 1, где органический растворитель представляет собой диизопропиловый эфир или этилацетат.
4. Способ по п. 1, где упомянутое соединение формулы II получают взаимодействием соединения формулы

в которой А представляет C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом,
с гидроксидом в спиртовом растворителе.
в которой А представляет C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом,
с гидроксидом в спиртовом растворителе.
5. Способ по п. 4, где гидроксид представляет собой гидроксид калия.
6. Способ по п. 4, где спирт представляет собой третбутиловый спирт.
7. Способ по п. 4, где упомянутое соединение формулы III получают взаимодействием соединения формулы

в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом,
с органической или минеральной кислотой.
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом,
с органической или минеральной кислотой.
8. Способ по п. 7, где кислота представляет собой уксусную кислоту, серную кислоту, муравьиную кислоту, или п-толуолсульфокислоту.
9. Способ по п. 8, где предпочтительной кислотой является муравьиная кислота.
10. Способ по п. 7, где упомянутое соединение формулы IV получают взаимодействием соединения формулы

в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Сl, Br, ОСН3, C1-С3 алкилом или бензилом;
Х представляет Сl, Br, I или ОСН3, с избытком ацеталя аминоацетальдегида.
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Сl, Br, ОСН3, C1-С3 алкилом или бензилом;
Х представляет Сl, Br, I или ОСН3, с избытком ацеталя аминоацетальдегида.
11. Способ по п. 10, где ацеталь аминоацетальдегида представляет собой диметилацеталь аминоацетальдегида или диэтилацеталь аминоацетальдегида.
12. Способ по п. 10, где упомянутое соединение формулы V, в которой Х представляет Сl, Br, или I, получают взаимодействием соединения формулы
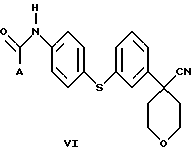
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом, с пентагалогенидом фосфора в инертном растворителе; или в которой Х представляет ОСН3 в формуле V, с (CH3)3O+BF4 - с образованием промежуточного соединения.
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом, с пентагалогенидом фосфора в инертном растворителе; или в которой Х представляет ОСН3 в формуле V, с (CH3)3O+BF4 - с образованием промежуточного соединения.
13. Способ по п. 12, где пентагалогенид представляет собой пятихлористый фосфор, пятииодистый фосфор или пятибромистый фосфор, а растворитель представляет собой толуол.
14. Способ по п. 12, где упомянутое соединение формулы VI получают взаимодействием соединения формулы

в которой Х представляет Cl, Br, или I, с избытком 4-аминотиофенола с основанием в инертном растворителе, с образованием соединения формулы

и последующей обработкой соединения формулы VII ацилированием с использованием галогенангидрида или ангидрида кислоты.
в которой Х представляет Cl, Br, или I, с избытком 4-аминотиофенола с основанием в инертном растворителе, с образованием соединения формулы
и последующей обработкой соединения формулы VII ацилированием с использованием галогенангидрида или ангидрида кислоты.
15. Способ по п. 14, где ацилирующий агент представляет собой хлористый ацетил.
16. Способ по п. 12, где упомянутое соединение формулы VI получают взаимодействием соединения формулы VIII

в которой Х представляет Сl, Br, или I, с избытком 4-амидотиофенола с основанием в инертном растворителе.
в которой Х представляет Сl, Br, или I, с избытком 4-амидотиофенола с основанием в инертном растворителе.
17. Способ по п. 16, где 4-амидотиофенол представляет собой 4-ацетамидотиофенол.
18. Способ по п. 16, где растворитель представляет собой N-метилпирролидон или диметилсульфоксид.
19. Способ по п. 16, где основание представляет собой карбонат натрия/карбонат цезия.
20. Способ по п. 14, где основание представляет собой карбонат натрия/карбонат цезия.
21. Способ по п. 14, где упомянутый растворитель представляет собой диметилсульфоксид или N-метилпирролидон.
22. Способ по п. 16, где упомянутое соединение формулы VIII можно получить взаимодействием соединения формулы

в которой Х представляет Сl, Br, или I, с бис 2-хлорэтиловым эфиром, щелочным основанием и межфазным катализатором в инертном растворителе.
в которой Х представляет Сl, Br, или I, с бис 2-хлорэтиловым эфиром, щелочным основанием и межфазным катализатором в инертном растворителе.
23. Способ по п. 22, где инертный растворитель представляет собой смесь тетрагидрофурана и воды, основание является гидроксидом натрия, а межфазный катализатор представляет собой кислый сульфат тетрабутиламмония.
24. Соединение формулы

в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом.
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом.
25. Соединение по п. 24, где А представляет СН3

26. Соединение формулы

в которой Х представляет Br или Cl.
26. Соединение формулы
в которой Х представляет Br или Cl.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11322198P | 1998-12-22 | 1998-12-22 | |
| US60/113221 | 1998-12-22 | ||
| US60/113,221 | 1998-12-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU99124413A RU99124413A (ru) | 2001-08-10 |
| RU2181722C2 true RU2181722C2 (ru) | 2002-04-27 |
Family
ID=22348241
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU99124413/04A RU2181722C2 (ru) | 1998-12-22 | 1999-11-16 | Способ получения и промежуточные соединения для получения ингибиторов 5-липоксигеназы |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US6194585B1 (ru) |
| EP (1) | EP1029865B1 (ru) |
| JP (2) | JP3468729B2 (ru) |
| KR (1) | KR100362789B1 (ru) |
| CN (1) | CN1258675A (ru) |
| AR (1) | AR019497A3 (ru) |
| AT (1) | ATE265460T1 (ru) |
| AU (1) | AU6539199A (ru) |
| BR (1) | BR9905918A (ru) |
| CA (1) | CA2289422A1 (ru) |
| CZ (1) | CZ453699A3 (ru) |
| DE (1) | DE69916778D1 (ru) |
| HU (1) | HUP9904282A3 (ru) |
| ID (1) | ID24014A (ru) |
| IL (1) | IL132897A0 (ru) |
| IN (1) | IN187165B (ru) |
| PL (1) | PL337397A1 (ru) |
| RU (1) | RU2181722C2 (ru) |
| TR (1) | TR199903204A2 (ru) |
| TW (1) | TW455586B (ru) |
| YU (1) | YU59399A (ru) |
| ZA (1) | ZA997113B (ru) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4569187B2 (ja) * | 2004-06-17 | 2010-10-27 | 宇部興産株式会社 | 4−シアノテトラヒドロピランの製法 |
| US7674811B2 (en) | 2006-03-14 | 2010-03-09 | Ranbaxy Laboratories Limited | 5-lipoxygenase inhibitors |
| JP4469924B2 (ja) * | 2006-11-27 | 2010-06-02 | ファイザー・プロダクツ・インク | ピラゾール類似体 |
| SG175390A1 (en) | 2009-04-29 | 2011-12-29 | Amarin Corp Plc | Pharmaceutical compositions comprising epa and a cardiovascular agent and methods of using the same |
| WO2021133689A2 (en) * | 2019-12-23 | 2021-07-01 | Sri International | Lipoxygenase inhibitors |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0462830B1 (en) * | 1990-06-21 | 1995-10-18 | Zeneca Limited | Cyclic ether derivatives |
| IE913866A1 (en) * | 1990-11-28 | 1992-06-03 | Ici Plc | Aryl derivatives |
| EP0505122A1 (en) * | 1991-03-21 | 1992-09-23 | Zeneca Limited | Alpha, alpha-dialkylbenzyl derivatives |
| EP0540165A1 (en) * | 1991-10-03 | 1993-05-05 | Zeneca Limited | Alkanoic acid derivatives |
| EP0703913B1 (en) * | 1993-06-14 | 1997-06-11 | Pfizer Inc. | Imidazoles as lipoxygenase inhibitors |
| SK281577B6 (sk) * | 1994-10-18 | 2001-05-10 | Pfizer Inc. | Heterocyklické zlúčeniny a farmaceutický prostriedok na ich báze |
| US5883106A (en) * | 1994-10-18 | 1999-03-16 | Pfizer Inc. | 5-lipoxygenase inhibitors |
-
1999
- 1999-08-24 US US09/387,707 patent/US6194585B1/en not_active Expired - Fee Related
- 1999-11-11 IL IL13289799A patent/IL132897A0/xx unknown
- 1999-11-12 AT AT99309046T patent/ATE265460T1/de not_active IP Right Cessation
- 1999-11-12 CA CA002289422A patent/CA2289422A1/en not_active Abandoned
- 1999-11-12 DE DE69916778T patent/DE69916778D1/de not_active Expired - Lifetime
- 1999-11-12 EP EP99309046A patent/EP1029865B1/en not_active Expired - Lifetime
- 1999-11-15 ZA ZA9907113A patent/ZA997113B/xx unknown
- 1999-11-16 JP JP32599999A patent/JP3468729B2/ja not_active Expired - Fee Related
- 1999-11-16 HU HU9904282A patent/HUP9904282A3/hu unknown
- 1999-11-16 RU RU99124413/04A patent/RU2181722C2/ru not_active IP Right Cessation
- 1999-11-17 YU YU59399A patent/YU59399A/sh unknown
- 1999-11-17 TW TW088120072A patent/TW455586B/zh not_active IP Right Cessation
- 1999-11-18 IN IN814BO1999 patent/IN187165B/en unknown
- 1999-11-18 AR ARP990105879A patent/AR019497A3/es unknown
- 1999-11-22 ID IDP991082D patent/ID24014A/id unknown
- 1999-12-14 CZ CZ19994536A patent/CZ453699A3/cs unknown
- 1999-12-21 KR KR1019990059661A patent/KR100362789B1/ko not_active IP Right Cessation
- 1999-12-21 AU AU65391/99A patent/AU6539199A/en not_active Abandoned
- 1999-12-21 CN CN99126450A patent/CN1258675A/zh active Pending
- 1999-12-21 BR BR9905918-5A patent/BR9905918A/pt not_active IP Right Cessation
- 1999-12-22 PL PL99337397A patent/PL337397A1/xx not_active Application Discontinuation
- 1999-12-22 TR TR1999/03204A patent/TR199903204A2/xx unknown
-
2003
- 2003-05-13 JP JP2003134568A patent/JP2004002415A/ja not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| JP2004002415A (ja) | 2004-01-08 |
| ID24014A (id) | 2000-06-22 |
| EP1029865A2 (en) | 2000-08-23 |
| CN1258675A (zh) | 2000-07-05 |
| DE69916778D1 (de) | 2004-06-03 |
| HUP9904282A3 (en) | 2001-04-28 |
| AU6539199A (en) | 2000-06-29 |
| JP3468729B2 (ja) | 2003-11-17 |
| IN187165B (ru) | 2002-02-23 |
| US6194585B1 (en) | 2001-02-27 |
| YU59399A (sh) | 2002-11-15 |
| AR019497A3 (es) | 2002-02-20 |
| KR100362789B1 (ko) | 2002-11-30 |
| HU9904282D0 (en) | 2000-01-28 |
| JP2000191654A (ja) | 2000-07-11 |
| TW455586B (en) | 2001-09-21 |
| TR199903204A2 (xx) | 2000-07-21 |
| PL337397A1 (en) | 2000-07-03 |
| ZA997113B (en) | 2001-05-15 |
| CZ453699A3 (cs) | 2000-07-12 |
| ATE265460T1 (de) | 2004-05-15 |
| BR9905918A (pt) | 2000-09-26 |
| EP1029865A3 (en) | 2000-11-08 |
| IL132897A0 (en) | 2001-03-19 |
| HUP9904282A2 (hu) | 2000-12-28 |
| MX9912076A (ru) | 2000-04-30 |
| KR20000052537A (ko) | 2000-08-25 |
| EP1029865B1 (en) | 2004-04-28 |
| CA2289422A1 (en) | 2000-06-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100339935B1 (ko) | 치환된 인다졸 유도체, 및 포스포디에스테라제 4형 및 종양괴사 인자 형성의 억제제로서의 그의 용도 | |
| EP0672031B1 (en) | Catechol diethers as selective pde iv inhibitors | |
| US5869511A (en) | Isoxazoline compounds as inhibitors of TNF release | |
| JPH0272136A (ja) | ナフタレン誘導体、その製法及びその合成中間体 | |
| JP2599665B2 (ja) | ピラジン誘導体 | |
| KR100276933B1 (ko) | 아릴사이클로알킬 유도체, 이의 제조방법 및 이를 함유하는 약제학적 조성물 | |
| EP1641758B1 (fr) | Derives de diphenylpyridine, leur preparation et leur application therapeutique | |
| JPS5989625A (ja) | 芳香族置換環状アミジン止瀉剤 | |
| US5169854A (en) | N-substituted-furylalkenyl hydroxamic acid and N-hydroxyurea compounds having lipoxygenase inhibitory activity | |
| EP0525111B1 (en) | Antiinflammatory hydroxamic acids and n-hydroxyureas | |
| WO2000002860A1 (fr) | Derives de 2-aminopyridines, leur utilisation en tant que medicaments et compositions pharmaceutiques les contenant | |
| JP3218243B2 (ja) | ベンズイミダゾール、その製造および使用法 | |
| JPS6222986B2 (ru) | ||
| WO1994010148A1 (en) | Aryl and heteroarylmethoxyphenyl inhibitors of leukotriene biosynthesis | |
| RU2181722C2 (ru) | Способ получения и промежуточные соединения для получения ингибиторов 5-липоксигеназы | |
| JPH06502390A (ja) | シクロペンタン誘導体、その製法及びその薬剤学的使用 | |
| US5420282A (en) | Thiopyrano(2,3,4-c,d) indolyloxime ether alkylcarboxylates | |
| JPH04503074A (ja) | リポキシゲナーゼ阻止化合物 | |
| JPH0536436B2 (ru) | ||
| FI70572C (fi) | Foerfarande foer framstaellning av farmaceutiskt anvaendbara pyrrolidinderivat samt vid deras framstaellning anvaendbara foereningar | |
| JP2661841B2 (ja) | インドリン誘導体 | |
| JPS604183A (ja) | イミダゾリジンジオン誘導体 | |
| CA1210392A (fr) | Procede pour la preparation d'amides n-substitues, et les amides ainsi obtenus | |
| JPH01313460A (ja) | N‐置換されたn‐アミノ‐ピロール | |
| US5432194A (en) | (4-alkoxypyran-4-yl) substituted arylalkylaryl-, aryalkenylaryl-, and aryalkynylarylurea inhibitors of 5-lipoxygenase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20041117 |