JP2025505208A - ナファモスタット、カモスタット及びそれらの誘導体の生成のためのプロセス - Google Patents
ナファモスタット、カモスタット及びそれらの誘導体の生成のためのプロセス Download PDFInfo
- Publication number
- JP2025505208A JP2025505208A JP2024547007A JP2024547007A JP2025505208A JP 2025505208 A JP2025505208 A JP 2025505208A JP 2024547007 A JP2024547007 A JP 2024547007A JP 2024547007 A JP2024547007 A JP 2024547007A JP 2025505208 A JP2025505208 A JP 2025505208A
- Authority
- JP
- Japan
- Prior art keywords
- group
- compound
- fragment
- nafamostat
- mesylate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images







Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C277/00—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C277/08—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups of substituted guanidines
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
この発明は、トリハロトリアジンをカップリング剤として使用し、酸及びアルコール官能性からエステルを合成するための、新規で経済的かつ実用的なルートを提供する。具体的には、この発明は、ナファモスタット及びカモスタットの生成のための新規で経済的かつ実用的なルートを提供する。また、カップリング試薬としてトリハロトリアジンを使用することにより、チオ尿素及びp-アミノ安息香酸からp-グアニジノ安息香酸(A1)の合成も達成された。
Description
本発明は、一般式(F)の化合物、又はその塩もしくは異性体に関するプロセスを提供する。より詳細には、本発明は、高度に官能化されたエステルベースの薬、即ちナファモスタット、カモスタット及びそれらの誘導体の製造のための効果的かつ経済的な方法を提供する。
FUT-175又は6’-アミジノ-2-ナフチル-4-グアニジノベンゾエート二塩酸塩としても知られるメシル酸ナファモスタットは、Fujiiら(Biochim Biophys Acta,1981,661,342)によって合成され、膵炎(Surgery,2001,130,175)、播種性血管内凝固症候群(DIC)、及び全身性炎症反応症候群などの炎症関連疾患の治療のために、1986年に日本たばこ産業株式会社によって市販された広域セリンプロテアーゼ阻害剤である。
別のグアニジン類似体であるカモスタットは、もともとはFOY-305として知られており、1977年に小野薬品工業株式会社によって初めて報告され、そのメシル酸塩は逆流性食道炎及び慢性膵炎の治療薬として1985年に日本で承認された(Org Process Res Dev,2020,24,940;Digestion,2004,30,171;
ナファモスタット及びカモスタットの合成については、いくつかの方法が公開されている。ナファモスタット及びカモスタットの主な合成戦略は、p-グアニジノ安息香酸としてのフラグメントA1や、6-ヒドロキシ-2-ナフトイミダミド(6-hydroxy-2-naphthimidamide)としてのフラグメントB1や、2-(ジメチルアミノ)-2-オキソエチル2-(4-ヒドロキシフェニル)アセテートとしてのフラグメントC1など、複数のフラグメントの生成、そしてナファモスタットの生成のための2つのフラグメント(A1及びB1)のカップリング、及びカモスタットの合成のための2つの主要フラグメント(A1及びC1)のカップリングに基づいている。
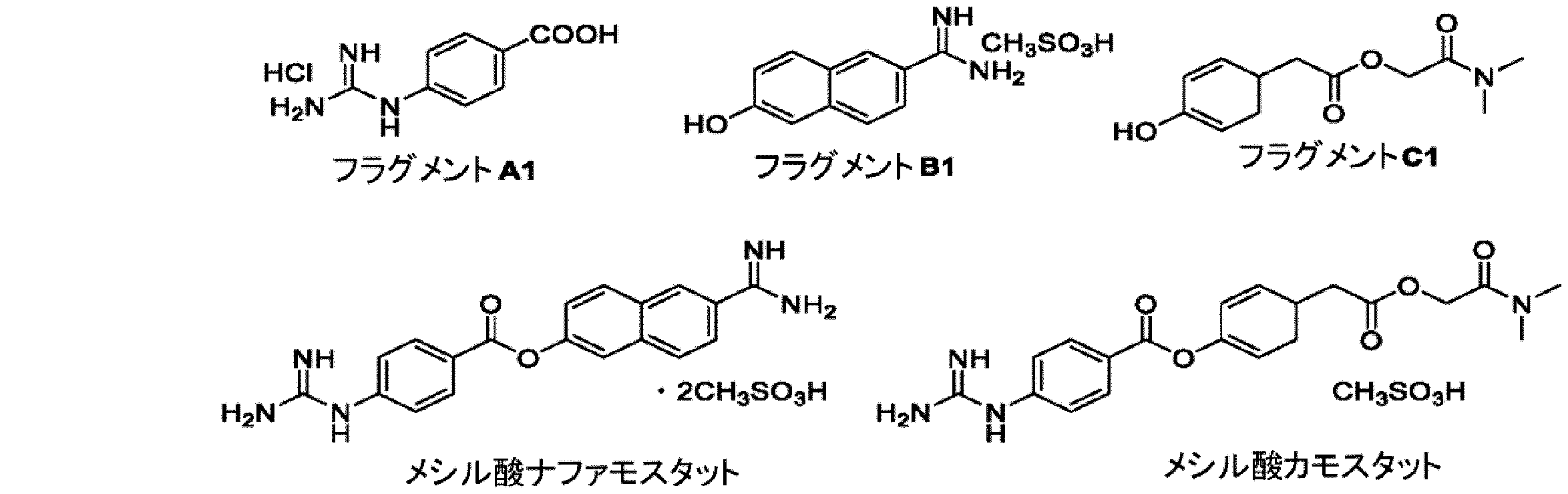
前述のプロセスのほとんどは、フラグメントA1及びB1の生成のための類似した基本的な経路に従うものであった。フラグメントA1及びB1からのナファモスタットの生成には、特定のカップリング剤が必要である。
中国特許出願公開第103012214A号には、アミジノ-β-ナフトールメタンスルホン酸塩とグアニジンラジカル塩化ベンゾイル塩酸塩(guanidine radicals benzoyl chloride hydrochloride salt)から、ピリジン中0~5℃でメシル酸ナファモスタット塩酸塩(Nafamostat mesylate hydrochloride)を生成し、最終工程でメシル酸ナファモスタットに変換する方法が記載されている。
2013年の中国特許第103641749B号及び中国特許第103641749B号には、メチレンジクロリド混合中で、4,5-ジシアノイミダゾール(DCI)をカップリング剤として使用して、メシル酸ナファモスタットを、p-グアニジノ安息香酸塩酸塩、6-アミジノ-β-ナフトールから生成する方法が記載されており、0~5℃は1時間撹拌され、その後温度を18~22℃に高め、断熱反応8~12時間後、最終的にメシル酸塩として得られる。
大韓民国登録特許第101595747B1号は、p-グアニジノ安息香酸塩酸塩と6-アミジノ-2-ナフトールメタンスルホン酸塩から、N,N’-ジイソプロピルカルボジイミド(DIC)と4-ジメチルアミノピリジン(DMAP)の存在下、カップリング剤としてのピリジン中で、メシル酸ナファモスタットを生成する方法を特許請求の範囲としている。
欧州特許第0465913B1号には、ジシクロヘキシルカルボジイミド(DCC)、N,N’-カルボニルジイミダゾール(CDI)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド(EDC)などの縮合剤を用いて、ジアミノトリフルオロメチルピリミジン誘導体を生成するための方法が記載されている。
中国特許出願公開第113999145A号には、p-クロロ安息香酸と6-アミジノ-2-ナフトール塩酸塩から、カルボジイミドベースのDCCをカップリング試薬として利用して、ナファモスタットを生成する方法が記載されている。
欧州特許第0048433B1号にも、DCCを4-グアニジノ安息香酸塩酸塩と反応させ、次いで6-アミジノ-2-ナフトールメタンスルホン酸塩とカップリングさせる、メシル酸ナファモスタットを生成する方法が記載されている。
同様に、別のグアニジン類似体であるカモスタットも、DIC、EDC、DCCなどの有害な化学物質又はカップリング試薬を用いて合成した。
Fujiらは1977年に米国特許第4,021,472号において、N,N-ジメチルカルバモイルメチル-p-ヒドロキシベンゾエートとp-グアニジノ安息香酸の酸塩化物誘導体を、ピリジン中で室温にて2時間反応させることによる、メシル酸カモスタットの合成を報告している。重炭酸ナトリウム溶液を添加し、濾過し、次いでメタノールに溶解し、メタンスルホン酸で酸性にすることによって結晶が生成された。ジエチルエーテルを加えると、メシル酸カモスタットが沈殿する。
上述した分子の合成に関する先行技術の方法のほとんどは、カルボジイミドベースのカップリング試薬を用いたフラグメントA1とフラグメントB1(又はフラグメントC1)のカップリングを要し、これはプロセスを非常に高額にし、またワークアップが面倒になる。
これらの薬の重要性を考慮すると、安価で作業が容易な、効果的なカップリング条件が強く求められる。この方向で、本発明は、安価で効果的なカップリング試薬としてトリハロトリアジンを利用してナファモスタット及びカモスタットを合成するための代替的かつ効果的な方法を提供し、さらに、ワークアップに関する作業の容易さに対してさらなる利点も提供するものである。
発明の目的
本発明の目的は、一般式(F)の化合物に関するプロセスを提供することである。
本発明の目的は、一般式(F)の化合物に関するプロセスを提供することである。
本発明の別の目的は、ナファモスタット及びカモスタット薬並びにそれらの誘導体を生成するための、新規で実用的かつ経済的なルートを提供することである。
本発明のさらに別の目的は、エステルを生成するための新規かつ極めて経済的なカップリング剤としてトリハロトリアジン、より好ましくはトリクロロトリアジン(TCT)を研究し、そしてナファモスタット、カモスタット薬及びその誘導体の合成における利用を調査することである。
従って本発明は、一般式Fの化合物を生成するためのプロセスを提供するものであって、

式中、ZはO、S、NHであり、
環X及びYは、アリール、ヘテロアリール、拡張された、ナフタレン、フェナントレン、キノリン、イソキノリンからなる群から選択される環であり、
R1は、グアニジニル(guanidinyl)、アミジニル(amidinyl)、2-(ジメチルアミノ)-2-オキソエチルアセチル、ハロゲン、アルキル基、アミド基、シアノ基、ニトロ基、アミノ基、メトキシ基、O-ベンジルエステル、N-ベンジルエステル、ヒドロキシル基、アリール基、ヘテロアリール基であり、
R2は、グアニジニル、アミジニル、2-(ジメチルアミノ)-2-オキソエチルアセチル、ハロゲン、アルキル基、アミド基、シアノ基、ニトロ基、アミノ基、メトキシ基、O-ベンジルエステル、N-ベンジルエステル、ヒドロキシル基、アリール基、ヘテロアリール基、置換ベンゼンであり、
前記プロセスは以下の各工程、即ち、
i.2,4,6-トリハロ-1,3,5-トリアジンと塩基を、25~400Cで5分間反応させ、活性複合体を得る工程と、
ii.工程iで得られた活性複合体とフラグメントAを、40~60℃で4時間反応させ、中間複合体を得る工程と、
iii.工程iiで得られた中間複合体を、0~1の範囲の比率でフラグメントBと、0~1の範囲の比率でフラグメントCと、反応させ、化合物Dを得る工程と、
iv.工程iiiで得られた化合物Dを重炭酸ナトリウム水溶液で処理する工程と、
v.工程ivで得られた化合物Dの得られた重炭酸塩を、任意に精製する工程と、
vi.工程iv及び工程vで得られた化合物Dを、メタノール存在下にて、メタンスルホン酸で処理して、式Fを得る工程と、を含む。
環X及びYは、アリール、ヘテロアリール、拡張された、ナフタレン、フェナントレン、キノリン、イソキノリンからなる群から選択される環であり、
R1は、グアニジニル(guanidinyl)、アミジニル(amidinyl)、2-(ジメチルアミノ)-2-オキソエチルアセチル、ハロゲン、アルキル基、アミド基、シアノ基、ニトロ基、アミノ基、メトキシ基、O-ベンジルエステル、N-ベンジルエステル、ヒドロキシル基、アリール基、ヘテロアリール基であり、
R2は、グアニジニル、アミジニル、2-(ジメチルアミノ)-2-オキソエチルアセチル、ハロゲン、アルキル基、アミド基、シアノ基、ニトロ基、アミノ基、メトキシ基、O-ベンジルエステル、N-ベンジルエステル、ヒドロキシル基、アリール基、ヘテロアリール基、置換ベンゼンであり、
前記プロセスは以下の各工程、即ち、
i.2,4,6-トリハロ-1,3,5-トリアジンと塩基を、25~400Cで5分間反応させ、活性複合体を得る工程と、
ii.工程iで得られた活性複合体とフラグメントAを、40~60℃で4時間反応させ、中間複合体を得る工程と、
iii.工程iiで得られた中間複合体を、0~1の範囲の比率でフラグメントBと、0~1の範囲の比率でフラグメントCと、反応させ、化合物Dを得る工程と、
iv.工程iiiで得られた化合物Dを重炭酸ナトリウム水溶液で処理する工程と、
v.工程ivで得られた化合物Dの得られた重炭酸塩を、任意に精製する工程と、
vi.工程iv及び工程vで得られた化合物Dを、メタノール存在下にて、メタンスルホン酸で処理して、式Fを得る工程と、を含む。
本発明の一実施形態において、代表的な化合物は、メシル酸ナファモスタット及びメシル酸カモスタットから選択される。
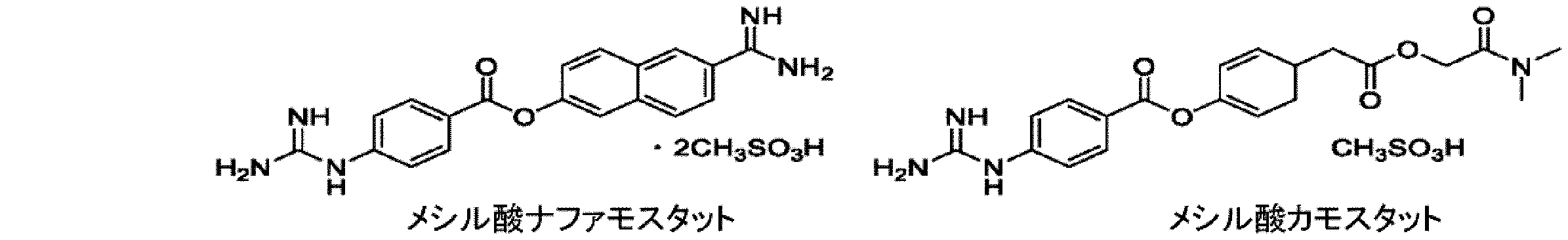
本発明の別の実施形態では、フラグメントAは、エタノール中、塩基の存在下でp-アミノ安息香酸(1)をチオ尿素(4)と反応させることによって生成される。
本発明の別の実施形態では、塩基は、ピリジン(py)、N-メチルモルホリン(NMM)トリエチルアミン(Et3N)ジアザビシクロウンデセン(DBU)からなる群から選択され、より好ましくはN-メチルモルホリン(NMM)である。
本発明の別の実施形態では、トリハロトリアジンが、トリクロロトリアジン、トリブロモトリアジン、トリフルオロトリアジンからなる群から選択される。
本発明の別の実施形態では、塩基は、炭酸ナトリウム、炭酸セシウム、炭酸アンモニウム、より好ましくは炭酸カリウムから選択される。
本発明の別の実施形態において、メシル酸ナファモスタットの収率は54~70%であり、メシル酸カモスタットの収率は17~28%である。
本発明の一実施形態において、代表的な化合物は以下から選択される。

本発明の別の実施形態では、塩基は、ピリジン(py)、N-メチルモルホリン(NMM)トリエチルアミン(Et3N)ジアザビシクロウンデセン(DBU)からなる群から選択され、より好ましくはN-メチルモルホリン(NMM)である。
本発明の別の実施形態では、トリハロトリアジンは、トリクロロトリアジン、トリブロモトリアジン、トリフルオロトリアジンからなる群から選択される。
使用した略語のリスト

本発明の主な態様は、一般式(F)の化合物のためのプロセスを提供することであって、

式中、ZはO、S、NHであり、
環X及びYは、アリール、ヘテロアリール、拡張された、ナフタレン、フェナントレン、キノリン、イソキノリンからなる群から選択される環であり、
R1は、グアニジニル、アミジニル、2-(ジメチルアミノ)-2-オキソエチルアセチル、ハロゲン、アルキル基、アミド基、シアノ基、ニトロ基、アミノ基、メトキシ基、O-ベンジルエステル、N-ベンジルエステル、ヒドロキシル基、アリール基、ヘテロアリール基であり、
R2は、グアニジニル、アミジニル、2-(ジメチルアミノ)-2-オキソエチルアセチル、ハロゲン、アルキル基、アミド基、シアノ基、ニトロ基、アミノ基、メトキシ基、O-ベンジルエステル、N-ベンジルエステル、ヒドロキシル基、アリール基、ヘテロアリール基、置換ベンゼンであって、
i.2,4,6-トリハロ-1,3,5-トリアジンと塩基を、25~40℃で5分間反応させて活性複合体を得る工程と、
ii.工程iで得られた活性複合体を、フラグメントAと40~60℃で4時間反応させて中間複合体を得る工程と、
iii.工程iiで得られた中間化合物を、0~1の範囲の比率でフラグメントBと、0~1の範囲の比率でフラグメントCと反応させて、化合物Dを得る工程と、
iv.工程iiiで得られた化合物Dを重炭酸ナトリウム水溶液で処理する工程と、
v.工程ivで得られた化合物Dの、得られた重炭酸塩を任意に精製する工程と、
vi.工程iv及び工程vで得られた化合物Dを、メタノール存在下にて、メタンスルホン酸で処理して、式Fを得る工程と、を含む。
環X及びYは、アリール、ヘテロアリール、拡張された、ナフタレン、フェナントレン、キノリン、イソキノリンからなる群から選択される環であり、
R1は、グアニジニル、アミジニル、2-(ジメチルアミノ)-2-オキソエチルアセチル、ハロゲン、アルキル基、アミド基、シアノ基、ニトロ基、アミノ基、メトキシ基、O-ベンジルエステル、N-ベンジルエステル、ヒドロキシル基、アリール基、ヘテロアリール基であり、
R2は、グアニジニル、アミジニル、2-(ジメチルアミノ)-2-オキソエチルアセチル、ハロゲン、アルキル基、アミド基、シアノ基、ニトロ基、アミノ基、メトキシ基、O-ベンジルエステル、N-ベンジルエステル、ヒドロキシル基、アリール基、ヘテロアリール基、置換ベンゼンであって、
i.2,4,6-トリハロ-1,3,5-トリアジンと塩基を、25~40℃で5分間反応させて活性複合体を得る工程と、
ii.工程iで得られた活性複合体を、フラグメントAと40~60℃で4時間反応させて中間複合体を得る工程と、
iii.工程iiで得られた中間化合物を、0~1の範囲の比率でフラグメントBと、0~1の範囲の比率でフラグメントCと反応させて、化合物Dを得る工程と、
iv.工程iiiで得られた化合物Dを重炭酸ナトリウム水溶液で処理する工程と、
v.工程ivで得られた化合物Dの、得られた重炭酸塩を任意に精製する工程と、
vi.工程iv及び工程vで得られた化合物Dを、メタノール存在下にて、メタンスルホン酸で処理して、式Fを得る工程と、を含む。
この実施形態の文脈において、カップリング剤トリハロトリアジンは、トリクロロトリアジン、トリブロモトリアジン、トリフルオロトリアジンから選択され、より好ましくは安価なトリクロロトリアジンであり、そして塩基は、ピリジン(py)、N-メチルモルホリン(NMM)トリエチルアミン(Et3N)ジアザビシクロウンデセン(DBU)から選択され、より好ましくはN-メチルモルホリン(NMM)である。
本発明の一実施形態では、フラグメントAは、エタノール中、塩基の存在下でp-アミノ安息香酸(1)をチオ尿素(4)と反応させることによって生成される。

この実施形態の文脈において、塩基は、ピリジン(py)、N-メチルモルホリン(NMM)トリエチルアミン(Et3N)ジアザビシクロウンデセン(DBU)のリストより選択され、より好ましくはN-メチルモルホリン(NMM)である。
別の実施形態によると、本発明は、ピリジン(py)、N-メチルモルホリン(NMM)トリエチルアミン(Et3N)ジアザビシクロウンデセン(DBU)、より好ましくはN-メチルモルホリン(NMM)などの塩基の存在下で、カップリング試薬としてトリクロロトリアジン、トリブロモトリアジン、トリフルオロトリアジン、より好ましくは安価なトリクロロトリアジン(TCT)を使用する、フラグメントA1、即ちp-グアニジノ安息香酸と、フラグメントB1、即ち6-ヒドロキシ-2-ナフトイミダミドとのカップリングによるナファモスタットの生成プロセスを提供する。ナファモスタット(D1)を重炭酸塩で処理した後に、炭酸塩として沈殿させ、続いてメタンスルホン酸(MSA)で酸性化してメシル酸ナファモスタット(F)を得る。
別の実施形態によれば、本発明は、炭酸ナトリウム、炭酸セシウム、炭酸アンモニウム、より好ましくは炭酸カリウムから選択される塩基存在下での、p-アミノ安息香酸1と三酸化チオ尿素(thioureatrioxide)2との反応によるフラグメントA1の生成プロセスを提供する。

別の実施形態によれば、本発明は、過酸化水素/過酢酸系を用いた酸化による、チオ尿素4からの三酸化チオ尿素2の合成プロセスを提供する。

別の実施形態によれば、本発明は、シアノのアミジンへの酸触媒による変換を介した、化合物5からのフラグメントB1の合成プロセスを提供する。
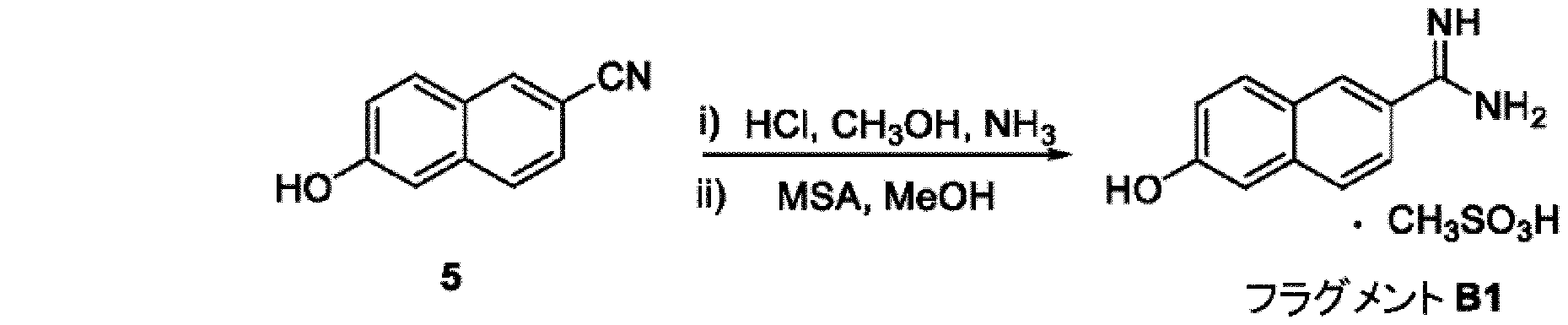
別の実施形態によると、本発明は、ブロモ基からシアノ基への変換を介した、化合物6からの化合物5の合成プロセスを提供する。

別の実施形態によれば、本発明は、2段階反応を介した、β-ナフトール7からの化合物6の合成プロセスを提供する。

別の実施形態によると、本発明は、ピリジン(py)、N-メチルモルホリン(NMM)トリエチルアミン(Et3N)ジアザビシクロウンデセン(DBU)、より好ましくはN-メチルモルホリン(NMM)などの塩基存在下で、カップリング試薬としてトリクロロトリアジン、トリブロモトリアジン、トリフルオロトリアジン、より好ましくは安価なトリクロロトリアジンを用いる、フラグメントA1、即ちp-グアニジノ安息香酸と、フラグメントC1、即ち2-(ジメチルアミノ)-2-オキソエチル2-(4-ヒドロキシフェニル)アセテートとのカップリングによるカモスタットの生成プロセスを提供する。カモスタット(D2)を重炭酸塩で処理した後に炭酸塩として沈殿させ、続いてメタンスルホン酸(MSA)で酸性化してメシル酸カモスタット(F)を得る

別の実施形態によれば、本発明は、4-ヒドロキシフェニル酢酸8とN,N-ジメチルブロモアセトアミド9とのカップリングによる、フラグメントC1の合成プロセスを提供する。

別の実施形態によると、本発明は、ブロモアセチルブロミド10とN,N-ジメチルアミン塩酸塩とのカップリングからの、化合物9の合成プロセスを提供する。

別の実施形態によれば、本発明は、カップリング試薬としてトリクロロトリアジン、トリブロモトリアジン、トリフルオロトリアジン、より好ましくは安価なトリクロロトリアジンを用いた、p-アミノ安息香酸1とチオ尿素4とのカップリングからの、フラグメントAの合成プロセスを提供する。

すべての生成物混合物を薄層クロマトグラフィーによって分析した。UV不活性化合物を染色溶液中で可視化し、UV活性化合物をUVランプ(λ=254nm)で検出した。すべての反応は、必要に応じて不活性雰囲気下で行われた。
NMRスペクトル(1HNMR、13C、DEPT)を、CDCl3及びCD3OD溶媒を用いて、400MHz分光計で記録した。
ES1-MS及びHRMSスペクトルは、LC-MS/MS及びHRMS-6540-UHD装置で記録した。旋光度をパーキン・エルマーの偏光計で測定した。
カラムクロマトグラフィーはシリカゲル(60~120、230~400メッシュ)を用いて実施した。
トリクロロトリアジン(TCT)は、安価で安定したカップリング試薬であり、これを用いることで、ナファモスタット、カモスタット及びそれらの誘導体の、費用対効果の高い合成ルートが得られる。
カップリング試薬としてTCTを消費することで、ナファモスタットやカモスタットの生成が容易になり、収率が高く、不純物が少なく、工業生産に適したものになる。
フラグメントA1、B1、C1、並びにメシル酸ナファモスタット及びメシル酸カモスタットの合成スキームが、実施例に示されている。表1及び表2は、使用した反応物質及び反応条件を、得られた生成物とそれらの収率と共に示す。


例
以下の実施例は例示の目的で示すものであり、本発明の範囲を限定するものと解釈されるべきではない。
以下の実施例は例示の目的で示すものであり、本発明の範囲を限定するものと解釈されるべきではない。
例1:フラグメントA1(p-グアニジノ安息香酸塩酸塩)の合成

工程1:二酸化チオ尿素(3)と三酸化チオ尿素(2)の合成:

チオ尿素(4)50g(0.657mol、1当量)を40℃の温水200mLに加え、十分に撹拌してチオ尿素を完全に溶解させた。このようにして作成したチオ尿素水溶液を冷却し、次いで、371.42mLの過酸化水素(濃度:30%)(4.6mol、7当量)を、溶液温度が10℃未満に保たれるほどの速度で、ゆっくり添加した。その後、溶液を0℃まで冷却し、約30分間撹拌して結晶を成長させた。結晶成長後、0℃の固液混合物を速やかに濾過し、分画した結晶を50℃で乾燥させた。さらに、反応混合物をエタノールで処理して、未反応のチオ尿素を除去した。このようにして得られた結晶の収率は64%であり、45gの二酸化チオ尿素(3)がそれぞれ得られた。過酢酸(48.54mL、0.664mol)を0℃で0.5時間撹拌した後、水(100mL)中の二酸化チオ尿素(3)(18g、0.664mol)を添加し、これを室温にゆっくりと温め、終夜撹拌した。沈殿物(2)(三酸化チオ尿素)を濾過によって取り除き、さらに得られた溶液をロタバポア(rotavapor)で蒸発させ、さらにエタノールで洗浄することで、三酸化チオ尿素(2)(17g、収率:67%)をそれぞれ得た。m.p.=132~1340C、MS(ESI+)m/z calcd. for CH5N2O3S,125.01(M+H);found 125.12。
工程2:4-グアニジノ安息香酸(A1)の合成

炭酸カリウム(6.01g、0.0435mol)を、水(40ml)中の4-アミノ安息香酸(1)(6g、0.0435mol)に加えた。混合物を室温で0.5時間撹拌し、次いで、三酸化チオ尿素(2)(8.14g、0.0653mol)をゆっくりと添加した。白色固体が沈殿し、混合物を室温でさらに1時間撹拌した。濾過後、白色固体を1N塩化水素のメタノール溶液(50mL)に溶解し、0.5時間撹拌した。溶媒をロタバポアで除去し、生成物がその塩酸塩(6.5g、収率:83%)として得られた。m.p.=280~2850C。1H NMR(400 MHz,DMSO-d6)δ 11.16(s,1H),10.19(s,1H),7.97-7.99(d,J=8Hz,2H),7.70(s,3H),7.33-7.35(d,J=8Hz,2H)、MS(ESI+)m/z calcd for C8H10N3O2,180.07(M+H);found 180.18。
例2:フラグメントB1(6-ヒドロキシ-2-ナフトイミダミドメタンスルホン酸塩)の合成

工程1:6-ブロモナフタレン-2-オール(6)の合成:

β-ナフトール(7)40g(1モル)及び氷酢酸100mlを、滴下漏斗及び還流冷却器を取り付けた丸底フラスコに入れた。酢酸30ml中の臭素88g(2モル)の溶液を、滴下漏斗を通して15~30分間かけて添加した。添加中にフラスコを穏やかに振盪し、β-ナフトールがこの時間中に溶解し、熱が発生した。混合物を、臭化水素の過剰なロスを避けるために、添加の終わりに向けていくらか冷却し、次いで、30mlの水を添加し、混合物を沸騰するまで加熱した。続いて、反応混合物を100℃に冷却し、5gの花状スズ(mossy tin)を添加し、金属が溶解するまで沸騰させた。次いで、2回目の分の5gのスズを添加し、煮沸させることで溶解させ、最後に3回目の分の32g(合計42g)のスズを投入した。混合物を3時間沸騰させ、50℃に冷却し、吸引濾過した。このようにして取り除かれた結晶質のスズ塩を、漏斗上で、冷酢酸100mlで洗浄し、洗液は濾液の主要部分に添加された。この濾液を冷水中で撹拌した。沈殿した6-ブロモ-2-ナフトール(6)を吸引濾過し、漏斗から取り出し、冷水100mlと攪拌することで洗浄した。再度濾過し、100℃で乾燥させた後、6-ブロモ-2-ナフトール(6)60g(収率91%)を得た。この粗生成物は123~127℃で融解し、若干のスズを含んでいるが、ほとんどの目的には十分な純度である。1H NMR(400 MHz,CDCl3)δ 10.08(s,1H),7.96(s,1H),7.72-7.70(d,1H,J=8 Hz),7.62-7.60(d,1H,J=8Hz),7.45-7.42(m,1H),7.13-7.11(m,2H)、MS(ESI+)m/z calcd for C10H8BrO,222.97(M+H);found 224.08。
工程2:6-ヒドロキシ-2-ナフトニトリル(5)の合成:

DMF中の、30gの6-ブロモ-2-ナフトール(6)及び15gのCuCNの混合物を、150℃で5~6時間、勢いよく撹拌した。室温まで冷却した後、混合物に10%NaOHを添加し、5分間撹拌した。反応物を濾過し、濾液を水で洗浄した。次いで、3N HClを用いてpHを2~3に調整した。沈殿物を濾過によって回収し、6-ヒドロキシ-2-ナフトニトリル(5)を褐色固体(20g、87%)として得た。m.p.=165~1700C。1H NMR(400 MHz,CDCl3)δ 10.05(s,1H),8.50(s,1H),8.07-8.05(d,1H,J=8 Hz),7.95(m,1H)7.50-7.45(m,2H),7.04-7.02(m、1H)、MS(ESI+)m/z calcd for C11H8NO,170.06(M+H);found 170.19。
工程3:6-ヒドロキシ-2-ナフトイミダミドメタンスルホン酸塩(B1)の合成:

6-ヒドロキシ-2-ナフトニトリル(5)10g(50mmol)を冷却した飽和MeOH-HCl溶液(50ml)に加え、混合物を室温で終夜撹拌し、濃縮した。残渣をMeOH(50ml)に溶解し、気体のNH3を50℃で3時間、溶液に導入した。混合物を真空中で濃縮し、残渣に飽和NaHCO3溶液を、撹拌しながら添加した。沈殿物を回収し、水、次いでアセトンで洗浄し、MeOH(10ml)中の沈殿物の懸濁液を、MSA(5.8g、60mmol)で処理した。Et2Oを溶液に添加し、沈殿物を回収して、6-ヒドロキシ-2-ナフトイミダミドメタンスルホン酸塩(B1)(9.2g、65%)を得た。EtOHからの再結晶により、淡黄色粉末として分析試料が得られた。mp 227~228℃。1H NMR(400 MHz,CDCl3)δ 9.20(s,2H),8.95(s,2H),8.37(s,1H),7.92-7.90(d,J=8Hz 1H),7.85-7.83(d,J=8Hz,1H),7.72-7.70(d,J=8Hz,1H),7.23(s,2H),2.49(s,1H),2.45(s,3H)、MS(ESI+)m/z calcd for C11H11N2O,187.08(M+H);found 187.22。
例3:フラグメントC1(2-(ジメチルアミノ)-2-オキソエチル2-(4-ヒドロキシフェニル)アセテート)の合成

工程1:2-ブロモ-N,N-ジメチルアセトアミド(9)の合成。

ブロモアセチルブロミド(10)(1.52mL、17.28mmol)をCH2Cl2(20mL)に溶解し、ジメチルアンモニウム塩化物(1g、12.34mmol)を0℃で添加した。反応混合物を室温で終夜撹拌した。次いで、反応混合物を、DCM及び水で後処理した。有機層を乾燥させ(Na2SO4)、真空中で濃縮した。粗生成物(9)は黄色オイル(0.450g、54%)として得られ、精製することなくさらに使用することができる。1H NMR(400 MHz,DMSO-d6)δ 4.34(s,2H),3.04(s,3H),2.89(s,3H)、m/z=164.97,MS(ESI+)m/z calcd for C4H9BrNO,165.98(M+H);found 167.02。
工程2:2-(ジメチルアミノ)-2-オキソエチル2-(4-ヒドロキシフェニル)アセテート(C1)の合成:

2-ブロモ-N,N-ジメチルアセトアミド(9)(0.50g、3.01mmol)及びp-ヒドロキシフェニル酢酸(8)(0.450g、3.01mmol)をアセトニトリル10mlに溶解し、トリエチルアミン(0.425g、4.21mmol)を溶液に加えた。得られた混合物を7時間還流した。反応が完了し次第(TLCで確認)、溶媒を蒸発させ、フラグメントC1をカラムクロマトグラフィーによって精製した。TLC(ヘキサン/EtOAc,2:8)Rf=0.4;収量(0.60g、81%);白色固体;m.p.:103~107℃。1H NMR(400 MHz,DMSO-d6)δ 9.39(s,1H),7.10-7.08(d,J=8Hz,2H),6.74-6.72(d,J=8Hz,2H),4.75(s,2H),3.61(s,2H),2.85(s,3H),2.79(s,3H)、MS(ESI+)m/z calcd for C12H16NO4 238.10(M+H);found 238.26。
例4:6-カルバミミドイルナフタレン-2-イル4-グアニジノベンゾエートジメシラート(メシル酸ナファモスタット)の合成:

2,4,6-トリクロロ-1,3,5-トリアジン(5g、26.88mmol)及び4-メチルモルホリン(70ml)を丸底フラスコに取り、室温で5分間撹拌した。反応混合物に、4-グアニジノ安息香酸、即ちフラグメントA1(9.62g、53.76mmol)を添加し、40℃で4時間撹拌した。次いで、6-ヒドロキシ-2-ナフトイミダミド、即ちフラグメントB1(10g、53.76mmol)を添加し、反応混合物を室温で終夜撹拌した。反応、即ち生成物D1の生成が完了し次第(TLCで観察)、重炭酸塩溶液を反応混合物に添加し、生成物の沈殿のために10分間撹拌した。沈殿物を濾過によって回収し、続いて水、アセトン及びメタノールで洗浄した。次いで、沈殿物をメタノールに溶解し、メタンスルホン酸で処理した。ジエチルエーテルの添加で、メシル酸ナファモスタット(F)が沈殿する(13g、収率70%)。m.p.=257~2620C。1H NMR(400 MHz,DMSO-d6)δ 10.33(s,1H),9.50(s,2H),9.29(s,2H),8.60(s,1H),8.25-8.17(m,4H),8.02-7.88(m,6H),7.66-7.64(d,J=8 Hz,1H),7.47-7.45(d,J=8Hz,2H),2.47(s,6H)。13C NMR(101 MHz,DMSO-d6)δ 166.0,164.4,155.8,150.9,142.1,136.1,132.0,131.4,130.2,129.9,128.9,125.9,125.3,124.9,123.8,123.0,119.4、MS(ESI+)m/z calcd for C19H18N5O2 348.14(M+H);found 348.38。
例5:6-カルバミミドイルナフタレン-2-イル4-グアニジノベンゾエートジメシラート(メシル酸ナファモスタット)の合成:

2,4,6-トリクロロ-1,3,5-トリアジン(5g、26.88mmol)及び4-メチルモルホリン(70ml)を丸底フラスコに取り、室温で5分間撹拌した。反応混合物に、4-グアニジノ安息香酸、即ちフラグメントA1(9.62g、53.76mmol)を添加し、40℃で4時間撹拌した。次いで、6-ヒドロキシ-2-ナフトイミダミド、即ちフラグメントB1(10g、53.76mmol)を添加し、反応混合物を室温で終夜撹拌した。反応、即ち生成物D1の生成が完了し次第(TLCで観察)、重炭酸塩溶液を反応混合物に添加し、生成物の沈殿のために10分間撹拌した。沈殿物を濾過によって回収し、続いて水、アセトン及びメタノールで洗浄した。次いで、沈殿物をメタノールに溶解し、メタンスルホン酸で処理した。ジエチルエーテルの添加で、メシル酸ナファモスタット(F)が沈殿する(11.7g、収率63%)。
例6:6-カルバミミドイルナフタレン-2-イル4-グアニジノベンゾエートジメシラート(メシル酸ナファモスタット)の合成:

2,4,6-トリクロロ-1,3,5-トリアジン(5g、26.88mmol)及び4-メチルモルホリン(70ml)を丸底フラスコに取り、室温で5分間撹拌した。反応混合物に、4-グアニジノ安息香酸、即ちフラグメントA1(9.62g、53.76mmol)を添加し、40℃で4時間撹拌した。次いで、6-ヒドロキシ-2-ナフトイミダミド、即ちフラグメントB1(10g、53.76mmol)を添加し、反応混合物を室温で終夜撹拌した。反応、即ち生成物D1の生成が完了し次第(TLCで観察)、重炭酸塩溶液を反応混合物に添加し、生成物の沈殿のために10分間撹拌した。沈殿物を濾過によって回収し、続いて水、アセトン及びメタノールで洗浄した。次いで、沈殿物をメタノールに溶解し、メタンスルホン酸で処理した。ジエチルエーテルの添加で、メシル酸ナファモスタット(F)が沈殿する(11g、収率60%)。
例7:6-カルバミミドイルナフタレン-2-イル4-グアニジノベンゾエートジメシラート(メシル酸ナファモスタット)の合成:

2,4,6-トリクロロ-1,3,5-トリアジン(5g、26.88mmol)及び4-メチルモルホリン(70ml)を丸底フラスコに取り、室温で5分間撹拌した。反応混合物に、4-グアニジノ安息香酸、即ちフラグメントA1(9.62g、53.76mmol)を添加し、40℃で4時間撹拌した。次いで、6-ヒドロキシ-2-ナフトイミダミド、即ちフラグメントB1(10g、53.76mmol)を添加し、反応混合物を室温で終夜撹拌した。反応、即ち生成物D1の生成が完了し次第(TLCで観察)、重炭酸塩溶液を反応混合物に添加し、生成物の沈殿のために10分間撹拌した。沈殿物を濾過によって回収し、続いて水、アセトン及びメタノールで洗浄した。次いで、沈殿物をメタノールに溶解し、メタンスルホン酸で処理した。ジエチルエーテルの添加で、メシル酸ナファモスタット(F)が沈殿する(10g、収率54%)。
例8:4-(2-(2-(ジメチルアミノ)-2-オキソエトキシ)-2-オキソエチル)フェニル4-グアニジノベンゾエートメタンスルホナート(メシル酸カモスタット)の合成:

2,4,6-トリクロロ-1,3,5-トリアジン(TCT)(0.250g、1.39mmol)及び4-メチルモルホリン(20ml)を丸底フラスコに加え、室温で5分間撹拌した。反応混合物に、4-グアニジノ安息香酸A1(0.500g、2.79mmol)を添加し、40℃で4時間撹拌した。次に、2-(ジメチルアミノ)-2-オキソエチル2-(4-ヒドロキシフェニル)アセテートC1(0.662g、2.79mmol)を添加し、反応混合物を室温で終夜撹拌した。反応、即ち生成物D2の生成が完了し次第(TLCによって確認)、溶媒を蒸発させ、カラムクロマトグラフィーによって精製した。次いで、生成物D2をメタノールに溶解し、メタンスルホン酸で処理し、ジエチルエーテルの添加でメシル酸カモスタット(F)を沈殿させた。TLC(DCM/MeOH,7:3)Rf=0.6;(0.75g、収率28%);白色固体;m.p.:153~158℃。1H NMR(400 MHz,DMSO-d6)δ 8.28-8.26 9(d,J=8Hz,2H),7.48-7.43(m,4H),7.21-7.19(d,J=8Hz,2H),4.87(s,2H),3.85(s,2H),3.02(s,3H),2.97(s,3H),2.72(s,3H)。13C{1H} NMR(101 MHz,CD3OD)δ 171.4,167.6,164.2,156.2,149.9,140.4,131.9,131.4,130.4,127.2,123.4,121.3,61.3,39.2,38.0,34.7,34.4、MS(ESI+)m/z calcd for C20H23N4O5 399.16(M+H);found 399.42。
例9:4-(2-(2-(ジメチルアミノ)-2-オキソエトキシ)-2-オキソエチル)フェニル4-グアニジノベンゾエートメタンスルホナート(メシル酸カモスタット)の合成:

2,4,6-トリクロロ-1,3,5-トリアジン(TCT)(0.250g、1.39mmol)及び4-メチルモルホリン(20ml)を丸底フラスコに加え、室温で5分間撹拌した。反応混合物に、4-グアニジノ安息香酸A1(0.500g、2.79mmol)を添加し、40℃で4時間撹拌した。次に、2-(ジメチルアミノ)-2-オキソエチル2-(4-ヒドロキシフェニル)アセテートC1(0.662g、2.79mmol)を添加し、反応混合物を室温で終夜撹拌した。反応、即ち生成物D2の生成が完了し次第(TLCによって確認)、溶媒を蒸発させ、カラムクロマトグラフィーによって精製した。次いで、生成物D2をメタノールに溶解し、メタンスルホン酸で処理し、ジエチルエーテルの添加でメシル酸カモスタット(F)を沈殿させた(0.20g、収率23%)。
例10:4-(2-(2-(ジメチルアミノ)-2-オキソエトキシ)-2-オキソエチル)フェニル4-グアニジノベンゾエートメタンスルホナート(メシル酸カモスタット)の合成:

2,4,6-トリクロロ-1,3,5-トリアジン(TCT)(0.250g、1.39mmol)及び4-メチルモルホリン(20ml)を丸底フラスコに加え、室温で5分間撹拌した。反応混合物に、4-グアニジノ安息香酸A1(0.500g、2.79mmol)を添加し、40℃で4時間撹拌した。次に、2-(ジメチルアミノ)-2-オキソエチル2-(4-ヒドロキシフェニル)アセテートC1(0.662g、2.79mmol)を添加し、反応混合物を室温で終夜撹拌した。反応、即ち生成物D2の生成が完了し次第(TLCによって確認)、溶媒を蒸発させ、カラムクロマトグラフィーによって精製した。次いで、生成物D2をメタノールに溶解し、メタンスルホン酸で処理し、ジエチルエーテルの添加でメシル酸カモスタット(F)を沈殿させた(0.17g、収率20%)。
例11:4-(2-(2-(ジメチルアミノ)-2-オキソエトキシ)-2-オキソエチル)フェニル4-グアニジノベンゾエートメタンスルホナート(メシル酸カモスタット)の合成:

2,4,6-トリクロロ-1,3,5-トリアジン(TCT)(0.250g、1.39mmol)及び4-メチルモルホリン(20ml)を丸底フラスコに加え、室温で5分間撹拌した。反応混合物に、4-グアニジノ安息香酸A1(0.500g、2.79mmol)を添加し、40℃で4時間撹拌した。次に、2-(ジメチルアミノ)-2-オキソエチル2-(4-ヒドロキシフェニル)アセテートC1(0.662g、2.79mmol)を添加し、反応混合物を室温で終夜撹拌した。反応、即ち生成物D2の生成が完了し次第(TLCによって確認)、溶媒を蒸発させ、カラムクロマトグラフィーによって精製した。次いで、生成物D2をメタノールに溶解し、メタンスルホン酸で処理し、ジエチルエーテルの添加でメシル酸カモスタット(F)を沈殿させた(0.15g、収率17%)。
例12:p-グアニジノ安息香酸(A)の合成:

エタノール中のTCT(1当量)とチオ尿素(4)(12g、1当量)を丸底フラスコに取り、2時間撹拌して、両方の反応物質を消費させた。ロータリーエバポレーターを用いて溶媒を除去し、白色固体を得た。得られた固体1.5gをp-アミノ安息香酸(1)0.699gと共に10mL丸底フラスコに取り、15分間撹拌した。反応混合物の中にN-メチルモルホリン3.56mLを添加した。1時間後、両反応物質は消費され(TLC分析)、4-グアニジノ安息香酸Aが他のスポットと共に形成された。N-メチルモルホリンを真空下で除去し、生成物を、カラムクロマトグラフィーを用いて単離した(0.15g、収率15%)。
本発明の利点
1.安価で安定なカップリング試薬としてのトリクロロトリアジン(TCT)の使用が、先行技術で用いられているもの以上の利点を提供する。
2.本発明はまた、先行技術とは対照的にワークアップ中の操作の容易さを提供し、繰り返しの溶媒処理と洗浄を回避する。
3.トリクロロトリアジン(TCT)の使用も、ナファモスタット、カモスタット及びそれらの誘導体の合成のための費用対効果の高いルートを提供する。
4.カップリング試薬としてTCTを消費することで、ナファモスタットやカモスタットの生成が容易になり、収率が高く、不純物が少なく、工業生産に適したものになる。
1.安価で安定なカップリング試薬としてのトリクロロトリアジン(TCT)の使用が、先行技術で用いられているもの以上の利点を提供する。
2.本発明はまた、先行技術とは対照的にワークアップ中の操作の容易さを提供し、繰り返しの溶媒処理と洗浄を回避する。
3.トリクロロトリアジン(TCT)の使用も、ナファモスタット、カモスタット及びそれらの誘導体の合成のための費用対効果の高いルートを提供する。
4.カップリング試薬としてTCTを消費することで、ナファモスタットやカモスタットの生成が容易になり、収率が高く、不純物が少なく、工業生産に適したものになる。
Claims (7)
- 式Fの化合物の生成プロセスであって、

式中、ZはO、S、又はNHであり、
環X及びYは、アリール、ヘテロアリール、又は拡張された、ナフタレン、フェナントレン、キノリン及びイソキノリンからなる群から選択される環であり、
R1は、グアニジニル(guanidinyl)、アミジニル(amidinyl)、2-(ジメチルアミノ)-2-オキソエチルアセチル、ハロゲン、アルキル基、アミド基、シアノ基、ニトロ基、アミノ基、メトキシ基、O-ベンジルエステル、N-ベンジルエステル、ヒドロキシル基、アリール基又はヘテロアリール基であり、
R2は、グアニジニル、アミジニル、2-(ジメチルアミノ)-2-オキソエチルアセチル、ハロゲン、アルキル基、アミド基、シアノ基、ニトロ基、アミノ基、メトキシ基、O-ベンジルエステル、N-ベンジルエステル、ヒドロキシル基、アリール基、ヘテロアリール基又は置換ベンゼンであり、
前記プロセスは以下の工程、
i.2,4,6-トリハロ-1,3,5-トリアジンと塩基を、25~40℃で5分間反応させ、活性複合体を得る工程と、
ii.工程iで得られた前記活性複合体とフラグメントAを、40~60℃で4時間反応させ、中間化合物を得る工程と、

iii.工程iiで得られた前記中間化合物を、0~1の範囲の比率でフラグメントBと、0~1の範囲の比率でフラグメントCと反応させ、化合物Dを得る工程と、

iv.工程iiiで得られた前記化合物Dを重炭酸ナトリウム水溶液で処理し、前記化合物Dの重炭酸塩を生成する工程と、
v.工程ivで得られた化合物Dの前記重炭酸塩を、任意に精製する工程と、
vi.工程iv及び工程vで得られた前記化合物Dを、メタノール存在下にて、メタンスルホン酸で処理し、式Fの前記化合物を得る工程と、を含むプロセス。 - 前記化合物Dがメシル酸ナファモスタット及びメシル酸カモスタットから選択される、請求項1に記載のプロセス。

- フラグメントAは、エタノール中、塩基の存在下でp-アミノ安息香酸(1)をチオ尿素(4)と反応させることによって生成される、請求項1に記載のプロセス。

- 前記塩基が、ピリジン(py)、N-メチルモルホリン(NMM)トリエチルアミン(Et3N)ジアザビシクロウンデセン(DBU)からなる群から選択され、より好ましくはN-メチルモルホリン(NMM)である、請求項1に記載のプロセス。
- 前記トリハロトリアジンが、トリクロロトリアジン、トリブロモトリアジン、トリフルオロトリアジンからなる群から選択される、請求項1に記載のプロセス。
- 前記塩基が、炭酸ナトリウム、炭酸セシウム、炭酸アンモニウム、及び、より好ましくは炭酸カリウムから選択される、請求項1に記載のプロセス。
- メシル酸ナファモスタットの収率が54~70%であり、メシル酸カモスタットの収率が17~28%である、請求項1に記載のプロセス。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN202211006599 | 2022-02-07 | ||
| IN202211006599 | 2022-02-07 | ||
| PCT/IN2023/050123 WO2023148783A1 (en) | 2022-02-07 | 2023-02-07 | Process for the preparation of nafamostat, camostat and their derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2025505208A true JP2025505208A (ja) | 2025-02-21 |
Family
ID=87553285
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2024547007A Pending JP2025505208A (ja) | 2022-02-07 | 2023-02-07 | ナファモスタット、カモスタット及びそれらの誘導体の生成のためのプロセス |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20250188022A1 (ja) |
| EP (1) | EP4475835A4 (ja) |
| JP (1) | JP2025505208A (ja) |
| WO (1) | WO2023148783A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117986161A (zh) * | 2024-02-01 | 2024-05-07 | 华仁医学研究(安徽)有限公司 | 一种高纯度抗血凝药物的合成方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU527371B2 (en) * | 1980-09-16 | 1983-03-03 | Torii & Co., Ltd. | Amidine |
| JPH09309873A (ja) * | 1996-05-22 | 1997-12-02 | Jiyunsei Kagaku Kk | メシル酸カモスタットの新規な製造法 |
| CN111574409A (zh) * | 2020-05-14 | 2020-08-25 | 河北省医疗器械与药品包装材料检验研究院(河北省医疗器械技术审评中心) | 一种甲磺酸萘莫司他的重结晶工艺方法 |
| KR102314436B1 (ko) * | 2021-01-27 | 2021-10-19 | (주)국전약품 | 나파모스타트 메실산염 및 그의 중간체의 제조방법 |
-
2023
- 2023-02-07 JP JP2024547007A patent/JP2025505208A/ja active Pending
- 2023-02-07 US US18/836,241 patent/US20250188022A1/en active Pending
- 2023-02-07 EP EP23749463.8A patent/EP4475835A4/en active Pending
- 2023-02-07 WO PCT/IN2023/050123 patent/WO2023148783A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| WO2023148783A1 (en) | 2023-08-10 |
| EP4475835A1 (en) | 2024-12-18 |
| US20250188022A1 (en) | 2025-06-12 |
| EP4475835A4 (en) | 2026-01-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4303726B2 (ja) | ウレア誘導体およびその製造方法 | |
| CZ305085B6 (cs) | Způsob přípravy dabigatranu | |
| JP2025505208A (ja) | ナファモスタット、カモスタット及びそれらの誘導体の生成のためのプロセス | |
| CA3044654A1 (en) | Method for producing triazolopyridine compound | |
| EP4452921A1 (en) | Process of the preparation of hydroxylamine derivatives | |
| KR101728443B1 (ko) | 2-아미노니코틴산벤질에스테르 유도체의 제조 방법 | |
| KR20170129191A (ko) | (4s)-4-[4-사이아노-2-(메틸설폰일)페닐]-3,6-다이메틸-2-옥소-1-[3-(트라이플루오로메틸)페닐]-1,2,3,4-테트라하이드로 피리미딘-5-카보나이트릴의 생성 방법 | |
| KR101139431B1 (ko) | 이매티닙 염기의 신규한 제조방법 | |
| CN115745874A (zh) | 一种邻吡咯烷基苯甲酰胺类化合物的制备方法 | |
| CN1251574A (zh) | O-(3-氨基-2-羟基-丙基)-肟酰卤类的制备方法 | |
| RU2556004C2 (ru) | Способ получения производного ароматического амида карбоновой кислоты | |
| RU2571417C2 (ru) | Способ получения n-замещенной 2-амино-4-(гидроксиметилфосфинил)-2-бутеновой кислоты | |
| EP2094661A2 (en) | Process for the preparation of atazanavir | |
| RU2193034C2 (ru) | Способ получения производных 2-аминотиазолкарбоксамида | |
| CN107629039B (zh) | 氘代丙烯酰胺的制备方法和中间体 | |
| RU2852723C2 (ru) | Способ получения джактиниба дигидрохлорида моногидрата | |
| JP5234856B2 (ja) | Npyy5受容体拮抗作用を有する化合物の結晶 | |
| JP4551763B2 (ja) | 3,4−ジクロロ−n−(2−シアノフェニル)−5−イソチアゾールカルボキサミドの製造方法 | |
| CN115850258B (zh) | 一种马赛替尼的合成方法 | |
| RU2825395C1 (ru) | Способ получения гидрохлорида 2-(1,2,3-тиадиазол-4-ил)бензиламина | |
| HU201516B (en) | Process for producing 2-cyano-2-oximinoacetamide derivatives | |
| JPS61158962A (ja) | 1,4−ジヒドロピリジン誘導体の製造法 | |
| KR0142140B1 (ko) | 2-벤조일-3-아미노아크릴레이트 유도체의 제조방법 | |
| KR20220145941A (ko) | 구아니디노-벤조에이트 설폰산 화합물의 제조 방법 | |
| EP0259140B1 (en) | Cyanoguanidine derivative and process for preparation thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20260109 |