JP2018531919A - B型肝炎ウイルスコアタンパク質モジュレーター - Google Patents
B型肝炎ウイルスコアタンパク質モジュレーター Download PDFInfo
- Publication number
- JP2018531919A JP2018531919A JP2018513789A JP2018513789A JP2018531919A JP 2018531919 A JP2018531919 A JP 2018531919A JP 2018513789 A JP2018513789 A JP 2018513789A JP 2018513789 A JP2018513789 A JP 2018513789A JP 2018531919 A JP2018531919 A JP 2018531919A
- Authority
- JP
- Japan
- Prior art keywords
- oxo
- dihydrodibenzo
- carboxamide
- benzyl
- thiazepine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- JZXDFQGCZQDITH-UHFFFAOYSA-N C/C=S(\c1c2cccc1)/c(ccc(C(NCc1cccc(Sc3cccc(OC)c3)c1)=O)c1)c1NC2=O Chemical compound C/C=S(\c1c2cccc1)/c(ccc(C(NCc1cccc(Sc3cccc(OC)c3)c1)=O)c1)c1NC2=O JZXDFQGCZQDITH-UHFFFAOYSA-N 0.000 description 1
- CJQRGUWCFOGZMI-UHFFFAOYSA-N C/C=S(\c1ccccc11)/c(ccc(C(NCc2cccc(Oc(cc3)ccc3OC)c2)=O)c2)c2NC1=O Chemical compound C/C=S(\c1ccccc11)/c(ccc(C(NCc2cccc(Oc(cc3)ccc3OC)c2)=O)c2)c2NC1=O CJQRGUWCFOGZMI-UHFFFAOYSA-N 0.000 description 1
- VODDZYBGNFJCNV-UHFFFAOYSA-N C/C=S(\c1ccccc11)/c(ccc(C(NCc2cccc(Sc3cccc(O)c3)c2)=O)c2)c2NC1=O Chemical compound C/C=S(\c1ccccc11)/c(ccc(C(NCc2cccc(Sc3cccc(O)c3)c2)=O)c2)c2NC1=O VODDZYBGNFJCNV-UHFFFAOYSA-N 0.000 description 1
- VHMAUNMPTKYAPJ-UHFFFAOYSA-N CC1(C2=CC1)Nc(cc(cc1)C(NCc3cccc(Oc(cc4)ccc4O)c3)=O)c1/S2=C/C Chemical compound CC1(C2=CC1)Nc(cc(cc1)C(NCc3cccc(Oc(cc4)ccc4O)c3)=O)c1/S2=C/C VHMAUNMPTKYAPJ-UHFFFAOYSA-N 0.000 description 1
- NVWXYGOBYIWHCH-UHFFFAOYSA-N CCN(c(cccc1)c1C(OC)=O)c(ccc(C(OC)=O)c1)c1N=O Chemical compound CCN(c(cccc1)c1C(OC)=O)c(ccc(C(OC)=O)c1)c1N=O NVWXYGOBYIWHCH-UHFFFAOYSA-N 0.000 description 1
- UPYKRPHLOALCAO-UHFFFAOYSA-N CCN(c1c2cccc1)c(ccc(C(OC)=O)c1)c1NC2=O Chemical compound CCN(c1c2cccc1)c(ccc(C(OC)=O)c1)c1NC2=O UPYKRPHLOALCAO-UHFFFAOYSA-N 0.000 description 1
- 0 CCN(c1c2cccc1)c(ccc([*+])c1)c1NC2=O Chemical compound CCN(c1c2cccc1)c(ccc([*+])c1)c1NC2=O 0.000 description 1
- GUZZSMRXTALNOA-UHFFFAOYSA-N CN(c1c2cccc1)c(ccc(C(OC)=O)c1)c1NC2=O Chemical compound CN(c1c2cccc1)c(ccc(C(OC)=O)c1)c1NC2=O GUZZSMRXTALNOA-UHFFFAOYSA-N 0.000 description 1
- WNAZYCXCUUJPGQ-UHFFFAOYSA-N COC(c(cc1)cc([N+]([O-])=O)c1Nc(cccc1)c1C(OC)=O)=O Chemical compound COC(c(cc1)cc([N+]([O-])=O)c1Nc(cccc1)c1C(OC)=O)=O WNAZYCXCUUJPGQ-UHFFFAOYSA-N 0.000 description 1
- HWTNNFFYEDWPFE-UHFFFAOYSA-N COC(c(cc1)cc([N+]([O-])=O)c1Oc1ccccc1C(OC)=O)=O Chemical compound COC(c(cc1)cc([N+]([O-])=O)c1Oc1ccccc1C(OC)=O)=O HWTNNFFYEDWPFE-UHFFFAOYSA-N 0.000 description 1
- PTNUAECXOSDHBF-UHFFFAOYSA-N COC(c(cc1N)ccc1Oc(cccc1)c1C(OC)=O)=O Chemical compound COC(c(cc1N)ccc1Oc(cccc1)c1C(OC)=O)=O PTNUAECXOSDHBF-UHFFFAOYSA-N 0.000 description 1
- ULVPVGZDKBSNNT-UHFFFAOYSA-N COc(cc1)ccc1Oc1cccc(CNC(c(cc2)cc(N3)c2Oc2ccccc2C3=O)=O)c1 Chemical compound COc(cc1)ccc1Oc1cccc(CNC(c(cc2)cc(N3)c2Oc2ccccc2C3=O)=O)c1 ULVPVGZDKBSNNT-UHFFFAOYSA-N 0.000 description 1
- QGJIZMFVWMOZTI-UHFFFAOYSA-N COc(cc1)ccc1Sc1cc(CNC(c(cc2)cc(N3)c2Oc2ccccc2C3=O)=O)ccc1 Chemical compound COc(cc1)ccc1Sc1cc(CNC(c(cc2)cc(N3)c2Oc2ccccc2C3=O)=O)ccc1 QGJIZMFVWMOZTI-UHFFFAOYSA-N 0.000 description 1
- UQNWCFFGOGLDKD-UHFFFAOYSA-N N#Cc1cc(Cc2cccc(O)c2)ccc1 Chemical compound N#Cc1cc(Cc2cccc(O)c2)ccc1 UQNWCFFGOGLDKD-UHFFFAOYSA-N 0.000 description 1
- CVKOOKPNCVYHNY-UHFFFAOYSA-N N#Cc1cccc(CBr)c1 Chemical compound N#Cc1cccc(CBr)c1 CVKOOKPNCVYHNY-UHFFFAOYSA-N 0.000 description 1
- WFWQWTPAPNEOFE-UHFFFAOYSA-N OB(c1cc(O)ccc1)O Chemical compound OB(c1cc(O)ccc1)O WFWQWTPAPNEOFE-UHFFFAOYSA-N 0.000 description 1
- KJXVVTDFXONOKC-UHFFFAOYSA-N Oc(cc1)ccc1Oc1cc(CNC(c(cc2N3)ccc2Oc(cccc2)c2C3=O)=O)ccc1 Chemical compound Oc(cc1)ccc1Oc1cc(CNC(c(cc2N3)ccc2Oc(cccc2)c2C3=O)=O)ccc1 KJXVVTDFXONOKC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/554—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one sulfur as ring hetero atoms, e.g. clothiapine, diltiazem
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D243/00—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms
- C07D243/06—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4
- C07D243/10—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems
- C07D243/38—[b, e]- or [b, f]-condensed with six-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D267/00—Heterocyclic compounds containing rings of more than six members having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D267/02—Seven-membered rings
- C07D267/08—Seven-membered rings having the hetero atoms in positions 1 and 4
- C07D267/12—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems
- C07D267/16—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems condensed with two six-membered rings
- C07D267/20—[b, f]-condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D281/00—Heterocyclic compounds containing rings of more than six members having one nitrogen atom and one sulfur atom as the only ring hetero atoms
- C07D281/02—Seven-membered rings
- C07D281/04—Seven-membered rings having the hetero atoms in positions 1 and 4
- C07D281/08—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems
- C07D281/12—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems condensed with two six-membered rings
- C07D281/16—[b, f]-condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Virology (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description
本願は、2015年9月15日に出願された米国仮出願第62/218,815号に対する優先権を主張する。この米国仮出願は、その全体が本明細書中に参考として援用される。
B型肝炎ウイルス(HBV)は、ウイルス性肝炎を引き起こし、この肝炎はさらに、慢性肝疾患を招き得、肝硬変および肝臓がん(肝細胞癌)のリスクを高め得る。世界的に、約20億人がHBVに感染しており、約3億6千万人が慢性的に感染しており、毎年、HBV感染症による死亡者は50万人を超える(2009年;WHO、2009年)。HBVは、体液によって母親から子どもへ、性交によって、および血液製剤を介して伝播し得る。また、HBV陽性の母親から生まれた子どもは、誕生時にワクチン接種しない限り、感染し得る。
本明細書では、下記のものなどの特性を有することができる化合物が提供され、この化合物は、一部の実施形態では、
ここで、本開示の特徴および他の詳細を、より具体的に記載する。本発明をさらに記載する前に、本明細書、実施例および添付の特許請求の範囲に用いられているある特定の用語を、ここにまとめる。これらの定義は、本開示の残りを考慮し、当業者によって理解される通りに読み取られるべきである。別段定義されない限り、本明細書で使用されるあらゆる技術的および科学的用語は、当業者によって一般に理解されるものと同じ意味を有する。
定義
Yは、S(O)y、C=O、C(R11)2、NRYおよびOからなる群から選択され、yは、0、1、または2であり、R11は、HまたはC1〜6アルキルであり、RYは、H、メチル、エチル、プロピル、プロペニル(proprenyl)、ブチル、フェニルおよびベンジルからなる群から選択され、RYは、Hでない場合、ヒドロキシルによって任意選択で置換されていてよく、
RZは、H、メチル、エチル、プロピル、フェニルおよびベンジルからなる群から選択され、
Rm’およびRmは、それぞれ独立に、HおよびC1〜6アルキル(ハロゲンおよびヒドロキシルからそれぞれ独立に選択される1つ、2つまたは3つの置換基によって任意選択で置換されている)からなる群から選択され、
R21は、出現するごとに、H、ハロゲンおよびC1〜6アルキルからなる群から選択され、
qは、0、1、または2であり、
R22は、出現するごとに、H、ハロゲン、ヒドロキシル、ニトロ、シアノ、カルボキシ、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C1〜6アルコキシ、NR’R’’、−C(O)−C1〜6アルキル、−C(O)−C1〜6アルコキシ、−C(O)−NR’R’’、−C(=NH)−NR’R’’、X2−フェニル(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、フェニル(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、5〜6員の単環式ヘテロアリール(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、9〜10員の二環式ヘテロアリール(R73によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、C3〜6シクロアルキル、−S(O)w−C1〜6アルキル(wは、0、1または2である)、−S(O)w−NR’R’’(wは、0、1または2である)、および−NR’−S(O)w(wは、0、1または2である)からなる群から選択され、
R’は、出現するごとに独立に、H、メチル、エチル、およびプロプルから選択され、
R’’は、出現するごとに独立に、H、メチル、エチル、プロピル、ブチル、−C(O)−メチルおよび−C(O)−エチルから選択されるか、またはR’およびR’’は、それらが結合する窒素と一緒になって、4〜7員の複素環を形成することができ、
R4、R5、R6、R7、R8、R9、およびR10部分のそれぞれは、出現するごとに独立に、水素、C1〜6アルキル、C2〜6アルキニル、C2〜6アルケニル、ハロゲン、ヒドロキシル、ニトロ、シアノ、およびNR’R’’からなる群から選択され、
R63は、出現するごとに独立に、H、ハロゲン、ヒドロキシル、ニトロ、シアノ、カルボキシ、C1〜6アルキル、−C(O)−O−C1〜6アルキル、複素環(ハロゲンまたはNR’R’によって任意選択で置換されている)、−C(O)−NR’R’’、−C(=NH)−NR’R’’、ヘテロアリール、フェニル、ベンジル、C2〜6アルケニル、C2〜6アルキニル、C1〜6アルコキシ、カルボキシ、NR’R’’、−C(O)−C1〜6アルキル、−C(O)−C1〜6アルコキシ、C3〜6シクロアルキル、−S(O)w−C1〜6アルキル(wは、0、1または2である)、−S(O)w−NR’R’’(wは、0、1または2である)、−NR’−S(O)w−C1〜6アルキル(wは、0、1または2である)、X2−R69からなる群から選択され、
R69は、H、ハロゲン、ヒドロキシル、ニトロ、シアノ、C1〜6アルキル、−C(O)−O−C1〜6アルキル、複素環(ハロゲンまたはNR’R’によって任意選択で置換されている)、)、−C(O)−NR’R’’、−C(=NH)−NR’R’’、ヘテロアリール、フェニル、C2〜6アルケニル、C2〜6アルキニル、C1〜6アルコキシ、カルボキシ、NR’R’’、−C(O)−C1〜6アルキル、−C(O)−C1〜6アルコキシ、C3〜6シクロアルキル、−S(O)w−C1〜6アルキル(wは、0、1または2である)、−S(O)w−NR’R’’(wは、0、1または2である)、および−NR’−S(O)w−C1〜6アルキル(wは、0、1または2である)からなる群から選択され、
X2は、S(O)w(wは、0、1、または2である)、O、CH2、またはNR’から選択され、
ここで、出現するごとに、C1〜6アルキルは、ハロゲン、ヒドロキシル、ニトロ、シアノ、カルボキシ、C2〜6アルケニル、C2〜6アルキニル、C1〜6アルコキシ、NR’R’’、−NR’−S(O)w−C1〜6アルキル(wは、0、1または2である)、およびS(O)w−NR’R’’(wは、0、1または2である)からなる群から選択される1つ、2つ、3つまたはそれよりも多い置換基で任意選択で置換されていてよく、C1〜6アルコキシは、ハロゲン、ヒドロキシル、ニトロ、シアノ、カルボキシ、C1〜6アルキル、NR’R’’、−NR’−S(O)w−C1〜6アルキル(wは、0、1または2である)、およびS(O)w−NR’R’’(wは、0、1または2である)からなる群から選択される1つ、2つ、3つまたはそれよりも多い置換基で任意選択で置換されていてよい]
が提供される。
によって表されてよい。
例えば、一部の実施形態では、化合物は、
によって表される。
によって表される。
(実施例1)
11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボン酸(9)の合成:共通中間体
メチル3−アミノ−4−((2−(メトキシカルボニル)フェニル)チオ)ベンゾエート(4)の合成:
3−アミノ−4−((2−カルボキシフェニル)チオ)安息香酸(5)の合成:
11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボン酸(6)の合成:
メチル11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキシレート(7)の合成:
メチル11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキシレート5,5−ジオキシド(8)の合成:
11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボン酸5,5−ジオキシド(9)の合成:
(実施例2)
11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボン酸(14)の合成:共通中間体
メチル3−アミノ−4−(2−(メトキシカルボニル)フェノキシ)ベンゾエート(12)の合成:
3−アミノ−4−(2−カルボキシフェノキシ)安息香酸(13)の合成:
11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボン酸(14)の合成:
(実施例3)
11−オキソ−10,11−ジヒドロ−5H−ジベンゾ[b,e][1,4]ジアゼピン−8−カルボン酸(19)の合成:共通中間体
メチル3−アミノ−4−((2−メトキシカルボニル)フェニル)アミノ)ベンゾエート(17)の合成:
3−アミノ−4((2−カルボキシフェニル)アミノ)安息香酸(18)の合成:
11−オキソ−10,11−ジヒドロ−5H−ジベンゾ[b,e][1,4]ジアゼピン−8−カルボン酸(19)の合成:
(実施例4)
5−メチル−11−オキソ−10,11−ジヒドロ−5H−ジベンゾ[b,e][1,4]ジアゼピン−8−カルボン酸(22)の合成:共通中間体
メチル5−メチル−11−オキソ−10,11−ジヒドロ−5H−ジベンゾ[b,e][1,4]ジアゼピン−8−カルボキシレート(21)の合成:
5−メチル−11−オキソ−10,11−ジヒドロ−5H−ジベンゾ[b,e][1,4]ジアゼピン−8−カルボン酸(22)の合成:
(実施例5)
5−エチル−11−オキソ−10,11−ジヒドロ−5H−ジベンゾ[b,e][1,4]ジアゼピン−8−カルボン酸(25)の合成:共通中間体
メチル5−エチル−11−オキソ−10,11−ジヒドロ−5H−ジベンゾ[b,e][1,4]ジアゼピン−8−カルボキシレート(24)の合成:
5−エチル−11−オキソ−10,11−ジヒドロ−5H−ジベンゾ[b,e][1,4]ジアゼピン−8−カルボン酸(25)の合成:
(実施例6)
市販アミン:
(実施例7)
4−(2−アミノエチル)−N,N−ジメチルベンゼンスルホンアミド塩酸塩(33)の合成:
4−(2−ブロモエチル)−N,N−ジメチルベンゼンスルホンアミド(31)の合成:
4−(2−アジドエチル)−N,N−ジメチルベンゼンスルホンアミド(32)の合成:
4−(2−アミノエチル)−N,N−ジメチルベンゼンスルホンアミド塩酸塩(33)の合成:
(実施例8)
2−(4−フルオロフェノキシ)エタン−1−アミン塩酸塩(37)の合成:
(実施例9)
ベンジル(4−(4−(2−アミノエチル)フェノキシ)ブチル)カルバメート塩酸塩(44)の合成:
ベンジル(4−ヒドロキシブチル)カルバメート(42)の合成:
tert−ブチル(4−(4−(((ベンジルオキシ)カルボニル)アミノ)ブトキシ)フェネチル)カルバメート(43)の合成:
ベンジル(4−(4−(2−アミノエチル)フェノキシ)ブチル)カルバメート塩酸塩(44)の合成:
(実施例10)
2−フェニルブタン−1−アミン塩酸塩(47)の合成:
2−フェニルブタン−1−アミン塩酸塩(47)の合成:
(実施例11)
2−フェニル−2−(ピロリジン−1−イル)エタン−1−アミン塩酸塩(51)の合成:
2−フェニル−2−(ピロリジン−1−イル)エタン−1−アミン塩酸塩(51)の合成:
(実施例12)
3−フェニル−3−(ピロリジン−1−イル)プロパン−1−アミン(54)の合成:
3−フェニル−3−(ピロリジン−1−イル)プロパン−1−アミン(54)の合成:
(実施例13)
(3−(2H−1,2,3−トリアゾール−2−イル)フェニル)メタンアミン塩酸塩(58)の合成:
(3−2H−1,2,3−トリアゾール−2−イル)フェニル)メタンアミン塩酸塩(58)の合成:
(実施例14)
(4−(2H−1,2,3−トリアゾール−2−イル)フェニル)メタンアミン(62)の合成:
TLC:20%EtOAc/ヘキサン(Rf:0.8);1H-NMR (DMSO-d6, 400 MHz): δ 10.05 (s, 1H), 8.25 (d, J = 10.0 Hz, 4H), 8.11 (d, J = 8.8 Hz, 2H).
化合物60A分析データ:
TLC:20%EtOAc/ヘキサン(Rf:0.3);1H-NMR (DMSO-d6, 400 MHz): δ 10.07 (s, 1H), 8.98 (s, 1H), 8.19 (d, J = 8.8 Hz, 2H), 8.13 (d, J = 8.8 Hz, 2H), 8.04 (s, 1H).
4−(2H−1,2,3−トリアゾール−2−イル)ベンズアルデヒドオキシム(61)の合成:
(4−(2H−1,2,3−トリアゾール−2−イル)フェニル)メタンアミン(62)の合成:
(実施例15)
(4−(1−ベンジル−1H−1,2,3−トリアゾール−5−イル)フェニル)メタンアミン(70)の合成:
tert−ブチル(4−((トリメチルシリル)エチニル)ベンジル)カルバメート(65)の合成:
tert−ブチル(4−エチニルベンジル)カルバメート(66)の合成:
(アジドメチル)ベンゼン(68)の合成:
tert−ブチル(4−(1−ベンジル−1H−1,2,3−トリアゾール−5−イル)ベンジル)カルバメート(69)の合成:
(4−(1−ベンジル−1H−1,2,3−トリアゾール−5−イル)フェニル)メタンアミン(70)の合成:
(実施例16)
(4−(1H−1,2,3−トリアゾール−5−イル)フェニル)メタンアミンTFA塩(72)の合成:
(4−(1H−1,2,3−トリアゾール−5−イル)フェニル)メタンアミンTFA塩(72)の合成:
(実施例17)
(3−(1H−1,2,3−トリアゾール−5−イル)フェニル)メタンアミン(78)の合成:
tert−ブチル(3−((トリメチルシリル)エチニル)ベンジル)カルバメート(75)の合成:
tert−ブチル(3−エチニルベンジル)カルバメート(76)の合成:
tert−ブチル(3−(1H−1,2,3−トリアゾール−5−イル)ベンジル)カルバメート(77)の合成:
(3−(1H−1,2,3−トリアゾール−5−イル)フェニル)メタンアミン(78)の合成:
(実施例18)
(4−(ピリミジン−5−イル)フェニル)メタンアミン(83)の合成:
4−(ピリミジン−5−イル)ベンジルメタンスルホネート(81)の合成:
5−(4−アジドメチル)フェニル)ピリミジン(82)の合成:
(4−(ピリミジン−5−イル)フェニル)メタンアミン(83)の合成:
(実施例19)
[1,1’−ビフェニル]−3−イルメタンアミン塩酸塩(86)の合成:
[1,1’−ビフェニル]−3−イルメタンアミン塩酸塩(86)の合成:
(実施例20)
4’−(アミノメチル)−N,N−ジメチル−[1,1’−ビフェニル]−4−アミン塩酸塩(91)の合成:
tert−ブチル((4’−アミノ−[1,1’−ビフェニル]−4−イル)メチル)カルバメート(89)の合成:
tert−ブチル((4’−(ジメチルアミノ)−[1,1’−ビフェニル]−4−イル)メチル)カルバメート(90)の合成:
4’−(アミノメチル)−N,N−ジメチル−[1,1’−ビフェニル]−4−アミン塩酸塩(91)の合成:
(実施例21)
(4’−フルオロ−[1,1’−ビフェニル]−4−イル)メタンアミン塩酸塩(94)の合成:
(4’−フルオロ−[1,1’−ビフェニル]−4−イル)メタンアミン塩酸塩(94)の合成:
(実施例22)
(4’−フルオロ−[1,1’−ビフェニル]−3−イル)メタンアミン塩酸塩(96)の合成:
(4’−フルオロ−[1,1’−ビフェニル]−3−イル)メタンアミン塩酸塩(96)の合成:
(実施例23)
4’−(アミノメチル)−[1,1’−ビフェニル]−3−オール塩酸塩(99)の合成:
4’−(アミノメチル)−[1,1’−ビフェニル]−3−オール塩酸塩(99)の合成:
(実施例24)
4’−(アミノメチル)−[1,1’−ビフェニル]−4−オール塩酸塩(102)の合成:
4’−(アミノメチル)−[1,1’−ビフェニル]−4−オール塩酸塩(102)の合成:
(実施例25)
4’−((メチルアミノ)メチル)−[1,1’−ビフェニル]−4−オール塩酸塩(105)の合成:
tert−ブチル((4’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)メチル)(メチル)カルバメート(104)の合成:
4’−((メチルアミノ)メチル)−[1,1’−ビフェニル]−4−オール塩酸塩(105)の合成:
(実施例26)
4’−(アミノメチル)−2−クロロ−[1,1’−ビフェニル]−4−オール(109)の合成:
4’−(アミノメチル)−2−クロロ−[1,1’−ビフェニル]−4−オール(109)の合成:
(実施例27)
4’−(アミノメチル)−2’−クロロ−[1,1’−ビフェニル]−4−オール(112)の合成:
4’−(アミノメチル)−2’−クロロ−[1,1’−ビフェニル]−4−オール(112)の合成:
(実施例28)
(R)−4’−(1−アミノエチル)−[1,1’−ビフェニル]−4−オール(114)の合成:
(実施例29)
(S)−4’−(1−アミノエチル)−[1,1’−ビフェニル]−4−オール(116)の合成:
(実施例30)
2−((4’−(アミノメチル)−[1,1’−ビフェニル]−4−イル)オキシ)−N,N−ジメチルエタン−1−アミン塩酸塩(120)の合成:
tert−ブチル((4’−(2−(ジメチルアミノ)エトキシ)−[1,1’−ビフェニル]−4−イル)メチル)カルバメート(119)の合成:
2−((4’−(アミノメチル)−[1,1’−ビフェニル]−4−イル)オキシ)−N,N−ジメチルエタン−1−アミン塩酸塩(120)の合成:
(実施例31)
tert−ブチル((3’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)メチル)カルバメート(122)の合成:
2−((4’−(アミノメチル)−[1,1’−ビフェニル]−3−イル)オキシ)−N,N−ジメチルエタン−1−アミン塩酸塩(122)の合成:
(実施例32)
3−(3−(アミノメチル)ベンジル)フェノール塩酸塩(125)の合成:
3−(3−(アミノメチル)ベンジル)フェノール塩酸塩(125)の合成:
(実施例33)
(3−(3−メトキシベンジル)フェニル)メタンアミン塩酸塩(129)の合成:
(3−(3−メトキシベンジル)フェニル)メタンアミン塩酸塩(129)の合成:
(実施例34)
3−(4−メトキシベンジル)フェニル)メタンアミン塩酸塩(132)の合成:
(3−(4−メトキシベンジル)フェニル)メタンアミン塩酸塩(132)の合成:
(実施例35)
(3−(3−メトキシフェノキシ)フェニル)メタンアミン塩酸塩(136)の合成:
(実施例36)
(3−(4−メトキシフェノキシ)フェニル)メタンアミン(139)の合成:
(3−(4−メトキシフェノキシ)フェニル)メタンアミン塩酸塩(139)の合成:
(実施例37)
(3−((3−メトキシフェニル)チオ)フェニル)メタンアミン塩酸塩(143)の合成:
(実施例38)
(3−((4−メトキシフェニル)チオ)フェニル)メタンアミン塩酸塩(146)の合成:
(3−((4−メトキシフェニル)チオ)フェニル)メタンアミン塩酸塩(146)の合成:
(実施例39)
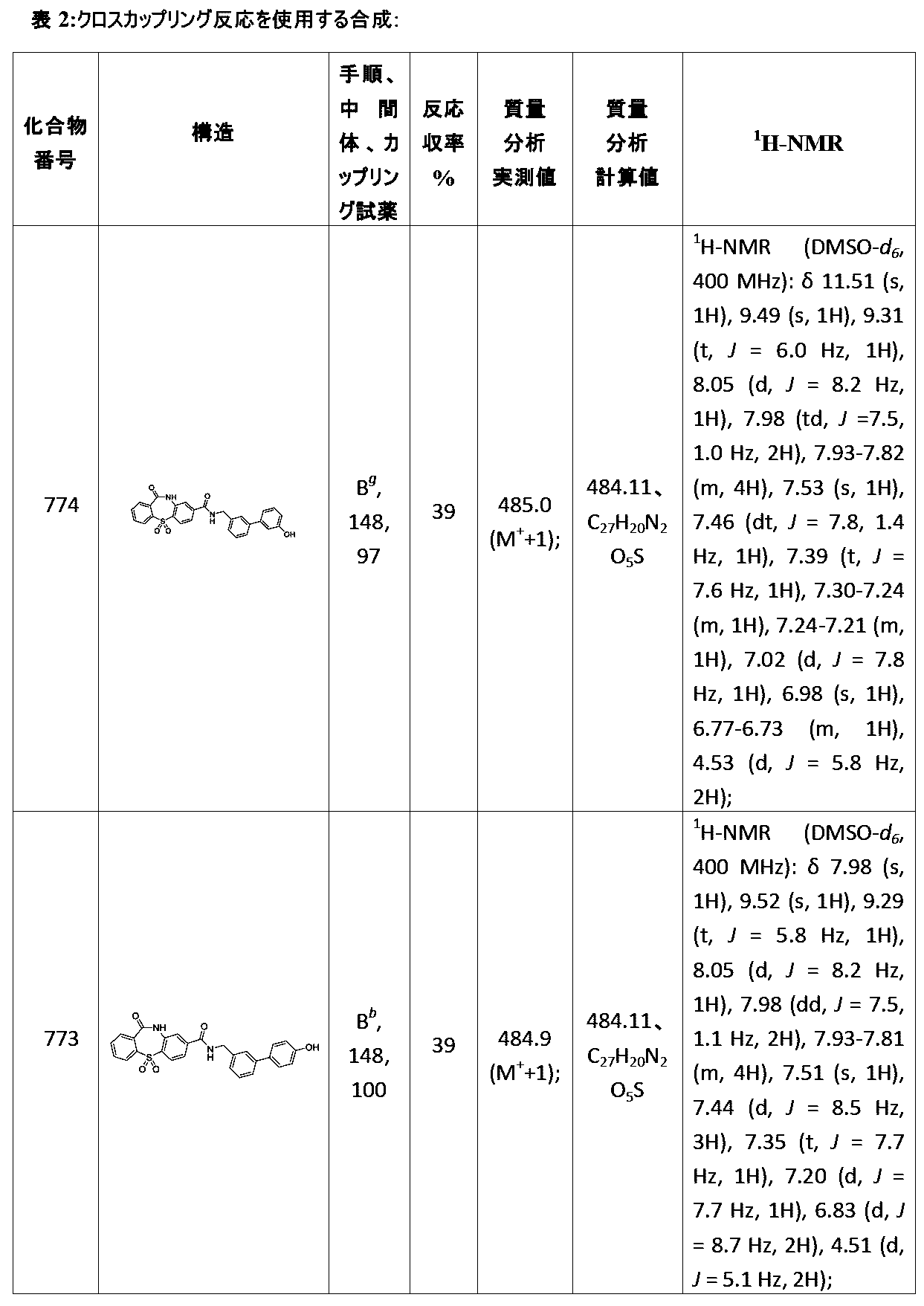
化合物調製
(実施例40)
N−(4−ブロモベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド(147)の合成:−共通中間体
(実施例41)
N−(3−ブロモベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド(148)の合成:共通中間体
(実施例42)
N−(4−ブロモベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド(149)の合成:共通中間体
(実施例43)
市販のボロン酸およびボロン酸誘導体:
化合物調製
典型的な手順B:
典型的な手順C:
Bc:Pd(PPh3)4(0.1当量)、密封管内で行った反応;Bd:Na2CO3(4当量);Be:Pd(PPh3)4(0.1当量)、ボロン酸(1当量)、Na2CO3(2当量)、12時間密封管;Bf:密封管内で行った反応;Bg:DME:H2O(3:1)、Na2CO3(3当量)、ボロン酸/エステル(1.2当量)、密封管内で行った反応、反応温度120℃、16時間;
Ca:反応時間12時間
(実施例45)
482の合成
(実施例46)
776の合成
(実施例47)
652−Aおよび652の合成
4’−(アミノメチル)−[1,1’−ビフェニル]−4−カルボニトリル塩酸塩(156)の合成:
N−((4’−シアノ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド(652−A)の合成:
N−((4’−(アミノメチル)−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド塩酸塩(652)の合成:
(実施例48)
765の合成
(実施例49)
676の合成
(実施例50)
808の合成:
(実施例51)
809の合成
(実施例52)
810の合成
(実施例53)
811の合成
(実施例54)
812の合成
(実施例55)
726の合成
(実施例56)
741の合成
(実施例57)
744の合成
(実施例58)
745の合成
(実施例59)
742の合成
(実施例60)
743の合成
(実施例61)
813の合成:
N−(3−((4−メトキシフェニル)チオ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド(813)の合成:
(実施例62)
475の合成:
tert−ブチル(4−((トリメチルシリル)エチニル)フェネチル)カルバメート(159)の合成:
tert−ブチル(4−エチニルフェネチル)カルバメート(160)の合成:
tert−ブチル(4−(1−ベンジル−1H−1,2,3−トリアゾール−5−イル)フェネチル)カルバメート(161)の合成:
2−(4−(1−ベンジル−1H−1,2,3−トリアゾール−5−イル)フェニル)エタン−1−アミン(162)の合成:
N−(4−(1−ベンジル−1H−1,2,3−トリアゾール−5−イル)フェネチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド(163)の合成:
N−(4−(1H−1,2,3−トリアゾール−5−イル)フェネチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド(475)の合成:
(実施例63)
AD38細胞におけるウイルス産生およびAD38細胞の生存率に対する化合物の活性を測定するアッセイ
均等論
Claims (23)
- 以下によって表される化合物
Yは、S(O)y、C=O、C(R11)2、NRYおよびOからなる群から選択され、yは、0、1、または2であり、R11は、HまたはC1〜6アルキルであり、RYは、H、メチル、エチル、プロピル、プロペニル、ブチル、フェニルおよびベンジルからなる群から選択され、RYは、Hでない場合、ヒドロキシルによって任意選択で置換されていてよく、
RZは、H、メチル、エチル、プロピル、フェニルおよびベンジルからなる群から選択され、
Rm’およびRmは、それぞれ独立に、HおよびC1〜6アルキル(ハロゲンおよびヒドロキシルからそれぞれ独立に選択される1つ、2つまたは3つの置換基によって任意選択で置換されている)からなる群から選択され、
R21は、出現するごとに、H、ハロゲンおよびC1〜6アルキルからなる群から選択され、
qは、0、1、または2であり、
R22は、出現するごとに、H、ハロゲン、ヒドロキシル、ニトロ、シアノ、カルボキシ、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C1〜6アルコキシ、NR’R’’、−C(O)−C1〜6アルキル、−C(O)−C1〜6アルコキシ、−C(O)−NR’R’’、−C(=NH)−NR’R’’、X2−フェニル(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、フェニル(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、5〜6員の単環式ヘテロアリール(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、9〜10員の二環式ヘテロアリール(R73によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、C3〜6シクロアルキル、−S(O)w−C1〜6アルキル(wは、0、1または2である)、−S(O)w−NR’R’’(wは、0、1または2である)、および−NR’−S(O)w(wは、0、1または2である)からなる群から選択され、
R’は、出現するごとに独立に、H、メチル、エチル、およびプロプルから選択され、
R’’は、出現するごとに独立に、H、メチル、エチル、プロピル、ブチル、−C(O)−メチルおよび−C(O)−エチルから選択されるか、またはR’およびR’’は、それらが結合する窒素と一緒になって、4〜7員の複素環を形成することができ、
R4、R5、R6、R7、R8、R9、およびR10部分のそれぞれは、出現するごとに独立に、水素、C1〜6アルキル、C2〜6アルキニル、C2〜6アルケニル、ハロゲン、ヒドロキシル、ニトロ、シアノ、およびNR’R’’からなる群から選択され、
R63は、出現するごとに独立に、H、ハロゲン、ヒドロキシル、ニトロ、シアノ、カルボキシ、C1〜6アルキル、−C(O)−O−C1〜6アルキル、複素環(ハロゲンまたはNR’R’によって任意選択で置換されている)、−C(O)−NR’R’’、−C(=NH)−NR’R’’、ヘテロアリール、フェニル、ベンジル、C2〜6アルケニル、C2〜6アルキニル、C1〜6アルコキシ、カルボキシ、NR’R’’、−C(O)−C1〜6アルキル、−C(O)−C1〜6アルコキシ、C3〜6シクロアルキル、−S(O)w−C1〜6アルキル(wは、0、1または2である)、−S(O)w−NR’R’’(wは、0、1または2である)、−NR’−S(O)w−C1〜6アルキル(wは、0、1または2である)、X2−R69からなる群から選択され、
R69は、H、ハロゲン、ヒドロキシル、ニトロ、シアノ、C1〜6アルキル、−C(O)−O−C1〜6アルキル、複素環(ハロゲンまたはNR’R’によって任意選択で置換されている)、)、−C(O)−NR’R’’、−C(=NH)−NR’R’’、ヘテロアリール、フェニル、C2〜6アルケニル、C2〜6アルキニル、C1〜6アルコキシ、カルボキシ、NR’R’’、−C(O)−C1〜6アルキル、−C(O)−C1〜6アルコキシ、C3〜6シクロアルキル、−S(O)w−C1〜6アルキル(wは、0、1または2である)、−S(O)w−NR’R’’(wは、0、1または2である)、および−NR’−S(O)w−C1〜6アルキル(wは、0、1または2である)からなる群から選択され、
X2は、S(O)w(wは、0、1、または2である)、O、CH2、またはNR’から選択され、
ここで、出現するごとに、C1〜6アルキルは、ハロゲン、ヒドロキシル、ニトロ、シアノ、カルボキシ、C2〜6アルケニル、C2〜6アルキニル、C1〜6アルコキシ、NR’R’’、−NR’−S(O)w−C1〜6アルキル(wは、0、1または2である)、およびS(O)w−NR’R’’(wは、0、1または2である)からなる群から選択される1つ、2つ、3つまたはそれよりも多い置換基で任意選択で置換されていてよく、C1〜6アルコキシは、ハロゲン、ヒドロキシル、ニトロ、シアノ、カルボキシ、C1〜6アルキル、NR’R’’、−NR’−S(O)w−C1〜6アルキル(wは、0、1または2である)、およびS(O)w−NR’R’’(wは、0、1または2である)からなる群から選択される1つ、2つ、3つまたはそれよりも多い置換基で任意選択で置換されていてよい]、
およびその薬学的に許容される塩。 - Yが、S(O)yである、請求項1に記載の化合物。
- yが、1または2である、請求項1または2に記載の化合物。
- yが、0である、請求項1または2に記載の化合物。
- Yが、NHである、請求項1に記載の化合物。
- R22が、出現するごとに、NR’R’’、−C(O)−C1〜6アルキル、−C(O)−C1〜6アルコキシ、−C(O)−NR’R’’、−C(=NH)−NR’R’’、X2−フェニル(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、フェニル(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、C3〜6シクロアルキル、−S(O)w−C1〜6アルキル(wは、0、1または2である)、−S(O)w−NR’R’’(wは、0、1または2である)、および−NR’−S(O)w(wは、0、1または2である)からなる群から選択される、請求項1〜5のいずれか一項に記載の化合物。
- 1つのR22が、X2−フェニル(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、フェニル(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、5〜6員の単環式ヘテロアリール(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)、および9〜10員の二環式ヘテロアリール(R73によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)からなる群から選択される、請求項1〜6のいずれか一項に記載の化合物。
- R22が、−X2−フェニル(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)である、請求項1〜7のいずれか一項に記載の化合物。
- R22が、フェニル(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)である、請求項1〜8のいずれか一項に記載の化合物。
- R22が、5〜6員の単環式ヘテロアリール(R63によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)または9〜10員の二環式ヘテロアリール(R73によって表される1つ、2つまたは3つの置換基によって任意選択で置換されている)である、請求項1〜8のいずれか一項に記載の化合物。
- qが、0である、請求項1〜10のいずれか一項に記載の化合物。
-
によって表される、請求項1〜10のいずれか一項に記載の化合物。 - 前記化合物が、
によって表される、請求項11に記載の化合物。 - 前記化合物が、
によって表される、請求項11に記載の化合物。 - R22が、
からなる群から選択される置換基によって表される、請求項1〜7のいずれか一項に記載の化合物。 - R63が、出現するごとに独立に、H、ハロゲン、ヒドロキシル、ニトロ、シアノ、C1〜6アルキル、C1〜6ハロアルキル、−C1〜6アルキル−OH、−C(O)−O−C1〜6アルキル、−C(O)−NR’R’’、−C(=NH)−NR’R’’、C2〜6アルケニル、C2〜6アルキニル、C1〜6アルコキシ、カルボキシ、NR’R’’、−C(O)−C1〜6アルキル、−C(O)−C1〜6アルコキシ、C3〜6シクロアルキル、−S(O)w−C1〜6アルキル(wは、0、1または2である)、−S(O)w−NR’R’’(wは、0、1または2である)、−NR’−S(O)w−C1〜6アルキル(wは、0、1または2である)、および−NR’−S(O)w(wは、0、1または2である)およびX2−C1〜6アルキレン−R69からなる群から選択される、請求項1〜15のいずれか一項に記載の化合物。
- R63が、出現するごとに独立に、H、ハロゲン、ヒドロキシル、シアノ、C1〜6アルキル、C1〜6ハロアルキル、C1〜6アルキル−OH、C1〜6アルキル−NR’R’’、−O−C1〜6アルキル−NR’R’’、−C1〜6アルキル−OH、−C(O)−NR’R’’、C1〜6アルコキシ、カルボキシ、NR’R’’、およびベンジルからなる群から選択される、請求項1〜15のいずれか一項に記載の化合物。
- 11−オキソ−N−(4−(ピリミジン−5−イル)ベンジル)−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド、
N−([1,1’−ビフェニル]−3−イルメチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−([1,1’−ビフェニル]−4−イルメチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((4’−フルオロ−[1,1’−ビフェニル]−3−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((4’−フルオロ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((3’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((4’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((4’−(ジメチルアミノ)−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−(2H−1,2,3−トリアゾール−2−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(4−(2H−1,2,3−トリアゾール−2−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−(1H−1,2,3−トリアゾール−5−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(4−(1H−1,2,3−トリアゾール−4−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(4−(1−ベンジル−1H−1,2,3−トリアゾール−4−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
(R)−N−(1−(4’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)エチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
(S)−N−(1−(4’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)エチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((4’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)メチル)−N−メチル−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((4’−(2−(ジメチルアミノ)エトキシ)−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−(3−メトキシベンジル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−(4−メトキシベンジル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−(3−メトキシフェノキシ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−(4−メトキシフェノキシ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−((3−メトキシフェニル)チオ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((2’−クロロ−4’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−((2−クロロ−4’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−((3’−(2−(ジメチルアミノ)エトキシ)−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−(3−ヒドロキシベンジル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−(4−メトキシベンジル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−(3−メトキシフェノキシ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−(4−メトキシフェノキシ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−((3−メトキシフェニル)チオ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−((4−メトキシフェニル)チオ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−((3’−ヒドロキシ−[1,1’−ビフェニル]−3−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((4’−ヒドロキシ−[1,1’−ビフェニル]−3−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(4−(6−メトキシピリジン−3−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(4−(2−アミノピリミジン−5−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(4−(1H−インドール−5−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(4−(1H−インダゾール−5−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(4−(1H−ベンゾ[d]イミダゾール−5−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((3’−シアノ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((3’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−((4’−ヒドロキシ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−((3’−シアノ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−((4’−シアノ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(4−(1H−インドール−5−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(4−(1H−ベンゾ[d]イミダゾール−5−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(4−(6−ヒドロキシピリジン−3−イル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((4’−シアノ−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((4’−(アミノメチル)−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド塩酸塩、
N−((4’−カルバモイル−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((3’−カルバムイミドイル−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド塩酸塩、
N−(3−(3−ヒドロキシベンジル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−(4−ヒドロキシベンジル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−(3−ヒドロキシフェノキシ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−(4−ヒドロキシフェノキシ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(3−((3−ヒドロキシフェニル)チオ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−((4’−(アミノメチル)−[1,1’−ビフェニル]−4−イル)メチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−(4−ヒドロキシベンジル)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−(3−ヒドロキシフェノキシ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−(4−ヒドロキシフェノキシ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−((3−ヒドロキシフェニル)チオ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−((4−ヒドロキシフェニル)チオ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]オキサゼピン−8−カルボキサミド、
N−(3−((4−ヒドロキシフェニル)チオ)ベンジル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド、
N−(4−(1H−1,2,3−トリアゾール−5−イル)フェネチル)−11−オキソ−10,11−ジヒドロジベンゾ[b,f][1,4]チアゼピン−8−カルボキサミド5,5−ジオキシド;
からなる群から選択される化合物、およびその薬学的に許容される塩。 - 請求項1〜18のいずれか一項に記載の化合物、および薬学的に許容される添加剤を含む、薬学的に許容される組成物。
- B型肝炎感染症を処置することを必要とする患者のB型肝炎感染症を処置する方法であって、有効量の請求項1〜18のいずれか一項に記載の化合物を投与するステップを含む、方法。
- B型肝炎感染症を処置することを必要とする患者のB型肝炎感染症を処置する方法であって、請求項1〜18に記載の化合物から選択される第1の化合物を投与するステップ、および任意選択で1つまたは複数のさらなる化合物を投与するステップを含む、方法。
- B型肝炎感染症を処置することを必要とする患者のB型肝炎感染症を処置する方法であって、ある量の請求項1〜18のいずれか一項に記載の化合物を投与するステップ、および別のHBVカプシド集合促進剤を投与するステップを含む、方法。
- B型肝炎感染症を処置することを必要とする患者のB型肝炎感染症を処置する方法であって、請求項1〜18のいずれか一項から選択される第1の化合物、ならびにHBVカプシド集合促進剤、HBFウイルスポリメラーゼ干渉ヌクレオシド、ウイルス侵入阻害剤、HBsAg分泌阻害剤、ヌクレオカプシド形成の撹乱物質、cccDNA形成阻害剤、抗ウイルスコアタンパク質変異体、HBcを対象とするトランスボディ、RNAi標的HBV RNA、免疫賦活薬、TLR−7/9アゴニスト、シクロフィリン阻害剤、HBVワクチン、SMAC模倣薬、エピジェネティックモジュレーター、キナーゼ阻害剤およびSTINGアゴニストからなる群からそれぞれ選択される他の1つまたは複数のHBV薬剤を投与するステップを含む、方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562218815P | 2015-09-15 | 2015-09-15 | |
| US62/218,815 | 2015-09-15 | ||
| PCT/US2016/051949 WO2017048962A1 (en) | 2015-09-15 | 2016-09-15 | Hepatitis b core protein modulators |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2018531919A true JP2018531919A (ja) | 2018-11-01 |
Family
ID=58289869
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018513837A Pending JP2018531921A (ja) | 2015-09-15 | 2016-09-15 | B型肝炎ウイルスコアタンパク質モジュレーター |
| JP2018513789A Pending JP2018531919A (ja) | 2015-09-15 | 2016-09-15 | B型肝炎ウイルスコアタンパク質モジュレーター |
| JP2018513782A Expired - Fee Related JP7023838B2 (ja) | 2015-09-15 | 2016-09-15 | B型肝炎ウイルスコアタンパク質モジュレーター |
| JP2021005706A Active JP7150903B2 (ja) | 2015-09-15 | 2021-01-18 | B型肝炎ウイルスコアタンパク質モジュレーター |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018513837A Pending JP2018531921A (ja) | 2015-09-15 | 2016-09-15 | B型肝炎ウイルスコアタンパク質モジュレーター |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018513782A Expired - Fee Related JP7023838B2 (ja) | 2015-09-15 | 2016-09-15 | B型肝炎ウイルスコアタンパク質モジュレーター |
| JP2021005706A Active JP7150903B2 (ja) | 2015-09-15 | 2021-01-18 | B型肝炎ウイルスコアタンパク質モジュレーター |
Country Status (31)
| Country | Link |
|---|---|
| US (6) | US10377748B2 (ja) |
| EP (4) | EP3782626A1 (ja) |
| JP (4) | JP2018531921A (ja) |
| KR (3) | KR20180050405A (ja) |
| CN (4) | CN108347943B (ja) |
| AR (3) | AR106040A1 (ja) |
| AU (4) | AU2016323297A1 (ja) |
| CA (3) | CA2998803A1 (ja) |
| CL (2) | CL2018000684A1 (ja) |
| CY (1) | CY1123570T1 (ja) |
| DK (1) | DK3349580T3 (ja) |
| DO (1) | DOP2018000074A (ja) |
| EA (3) | EA201890735A1 (ja) |
| ES (2) | ES2905950T3 (ja) |
| HK (2) | HK1258697A1 (ja) |
| HR (1) | HRP20201812T1 (ja) |
| HU (1) | HUE051872T2 (ja) |
| IL (3) | IL258099A (ja) |
| LT (1) | LT3349580T (ja) |
| MX (4) | MX392309B (ja) |
| MY (1) | MY198974A (ja) |
| PH (3) | PH12018500571B1 (ja) |
| PL (1) | PL3349580T3 (ja) |
| PT (1) | PT3349580T (ja) |
| RS (1) | RS61064B1 (ja) |
| SG (1) | SG10202011103UA (ja) |
| SI (1) | SI3349580T1 (ja) |
| SM (1) | SMT202000630T1 (ja) |
| TW (4) | TWI786639B (ja) |
| WO (3) | WO2017048954A1 (ja) |
| ZA (1) | ZA201802456B (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018531918A (ja) * | 2015-09-15 | 2018-11-01 | アッセンブリー バイオサイエンシズ,インコーポレイテッド | B型肝炎ウイルスコアタンパク質モジュレーター |
Families Citing this family (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102363174B1 (ko) | 2014-03-13 | 2022-02-15 | 인디애나 유니버시티 리서치 앤드 테크놀로지 코포레이션 | B형 간염 코어 단백질 알로스테릭 조정제 |
| CN109937201A (zh) * | 2016-09-15 | 2019-06-25 | 组装生物科学股份有限公司 | 乙型肝炎核心蛋白调节剂 |
| CN117402852A (zh) | 2016-10-14 | 2024-01-16 | 精密生物科学公司 | 对乙肝病毒基因组中的识别序列特异性的工程化大范围核酸酶 |
| US10821103B2 (en) | 2016-11-07 | 2020-11-03 | Arbutus Biopharma Corporation | Substituted pyridinone-containing trycyclic compounds, and methods using same |
| LT3587420T (lt) | 2017-02-23 | 2021-07-12 | Fujian Cosunter Pharmaceutical Co., Ltd. | Triciklis junginys ir jo naudojimas |
| WO2018160878A1 (en) | 2017-03-02 | 2018-09-07 | Assembly Biosciences, Inc. | Cyclic sulfamide compounds and methods of using same |
| AU2018236188B2 (en) | 2017-03-13 | 2022-01-27 | Assembly Biosciences, Inc. | Process for making hepatitis B core protein modulators |
| CA3056886A1 (en) | 2017-03-21 | 2018-09-27 | Arbutus Biopharma Corporation | Substituted dihydroindene-4-carboxamides and analogs thereof, and methods using same |
| AR115131A1 (es) | 2017-11-02 | 2020-12-02 | Aicuris Gmbh & Co Kg | Altamente activas indolo-2-carboxamidas sustituidas con pirazolo-pirimidina activas contra el virus de la hepatitis b (vhb) |
| TW201930315A (zh) | 2017-11-02 | 2019-08-01 | 德商艾庫瑞斯公司 | 具抗b型肝炎病毒(hbv)活性之新穎高活性胺基-噻唑取代之吲哚-2-甲醯胺 |
| AU2018392212B9 (en) | 2017-12-20 | 2021-03-18 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3' cyclic dinucleotides with phosphonate bond activating the STING adaptor protein |
| AU2018392213B2 (en) | 2017-12-20 | 2021-03-04 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3' cyclic dinucleotides with phosphonate bond activating the STING adaptor protein |
| JP7050165B2 (ja) | 2018-02-26 | 2022-04-07 | ギリアード サイエンシーズ, インコーポレイテッド | Hbv複製阻害剤としての置換ピロリジン化合物 |
| WO2019195181A1 (en) | 2018-04-05 | 2019-10-10 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis b virus protein x |
| TWI833744B (zh) | 2018-04-06 | 2024-03-01 | 捷克科學院有機化學與生物化學研究所 | 3'3'-環二核苷酸 |
| TW202005654A (zh) | 2018-04-06 | 2020-02-01 | 捷克科學院有機化學與生物化學研究所 | 2,2,─環二核苷酸 |
| TWI818007B (zh) | 2018-04-06 | 2023-10-11 | 捷克科學院有機化學與生物化學研究所 | 2'3'-環二核苷酸 |
| TW201945388A (zh) | 2018-04-12 | 2019-12-01 | 美商精密生物科學公司 | 對b型肝炎病毒基因體中之識別序列具有特異性之最佳化之經工程化巨核酸酶 |
| IL278460B2 (en) * | 2018-05-03 | 2025-04-01 | Univ Emory | Regulators of orphan nuclear receptors for NASH and other metabolic diseases |
| TW202014193A (zh) | 2018-05-03 | 2020-04-16 | 捷克科學院有機化學與生物化學研究所 | 包含碳環核苷酸之2’3’-環二核苷酸 |
| TWI815887B (zh) * | 2018-05-15 | 2023-09-21 | 美商愛彼特生物製藥股份有限公司 | 經取代的2,2'-雙嘧啶基化合物、其類似物及其使用方法 |
| TWI826492B (zh) | 2018-07-27 | 2023-12-21 | 加拿大商愛彼特生物製藥公司 | 經取代四氫環戊[c]吡咯、經取代二氫吡咯,其類似物及使用其之方法 |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| US12030875B2 (en) | 2018-09-07 | 2024-07-09 | PIC Therapeutics, Inc. | EIF4E inhibitors and uses thereof |
| SG11202103839UA (en) | 2018-10-31 | 2021-05-28 | Gilead Sciences Inc | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| KR102658602B1 (ko) | 2018-10-31 | 2024-04-19 | 길리애드 사이언시즈, 인코포레이티드 | Hpk1 억제 활성을 갖는 치환된 6-아자벤즈이미다졸 화합물 |
| UY38434A (es) | 2018-11-02 | 2020-05-29 | Aicuris Gmbh & Co Kg | Nuevas 6,7-dihidro-4h-pirazolo[1,5-a]pirazin indol-2-carboxamidas activas contra el virus de la hepatitis b (hbv) |
| UY38439A (es) | 2018-11-02 | 2020-05-29 | Aicuris Gmbh & Co Kg | Novedosas urea 6,7-dihidro-4h-pirazolo[4,3-c]piridinas activas contra el virus de la hepatitis b (vhb) |
| AR117188A1 (es) | 2018-11-02 | 2021-07-21 | Aicuris Gmbh & Co Kg | Derivados de urea 6,7-dihidro-4h-pirazolo[1,5-a]pirazinas activas contra el virus de la hepatitis b (vhb) |
| AR116948A1 (es) | 2018-11-02 | 2021-06-30 | Aicuris Gmbh & Co Kg | Derivados de urea 6,7-dihidro-4h-pirazolo[1,5-a]pirazinas activas contra el virus de la hepatitis b (vhb) |
| EA202191221A1 (ru) | 2018-11-02 | 2021-08-04 | Айкурис Гмбх Унд Ко. Кг | НОВЫЕ МОЧЕВИНО-6,7-ДИГИДРО-4H-ТИАЗОЛО[5,4-c]ПИРИДИНЫ, АКТИВНЫЕ ПРОТИВ ВИРУСА ГЕПАТИТА В (HBV) |
| AR116947A1 (es) | 2018-11-02 | 2021-06-30 | Aicuris Gmbh & Co Kg | Derivados de urea 6,7-dihidro-4h-pirazolo[1,5-a]pirazinas-indol-2-carboxamidas activas contra el virus de la hepatitis b (vhb) |
| TW202415643A (zh) | 2018-12-12 | 2024-04-16 | 加拿大商愛彼特生物製藥公司 | 經取代之芳基甲基脲類及雜芳基甲基脲類、其類似物及其使用方法 |
| JP2022517799A (ja) * | 2019-01-17 | 2022-03-10 | エフ.ホフマン-ラ ロシュ アーゲー | B型肝炎ウイルス感染症の処置および予防のための置換されたクロメン-4-オン |
| CA3129011C (en) | 2019-03-07 | 2023-12-19 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| CA3129022C (en) | 2019-03-07 | 2023-08-01 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| EP3935065A1 (en) | 2019-03-07 | 2022-01-12 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| TWI751517B (zh) | 2019-04-17 | 2022-01-01 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| TW202210480A (zh) | 2019-04-17 | 2022-03-16 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| SG11202111236UA (en) | 2019-04-30 | 2021-11-29 | Aicuris Gmbh & Co Kg | Novel indole-2-carboxamides active against the hepatitis b virus (hbv) |
| CA3138385A1 (en) | 2019-04-30 | 2020-11-05 | Aicuris Gmbh & Co. Kg | Novel indolizine-2-carboxamides active against the hepatitis b virus (hbv) |
| MX2021013086A (es) | 2019-04-30 | 2021-11-17 | Aicuris Gmbh & Co Kg | Nuevas fenil y piridil ureas activas contra el virus de la hepatitis b (vhb). |
| MX2021013105A (es) | 2019-04-30 | 2021-11-17 | Aicuris Gmbh & Co Kg | Nuevas oxalil piperazinas activas contra el virus de la hepatitis b (vhb). |
| US11453681B2 (en) | 2019-05-23 | 2022-09-27 | Gilead Sciences, Inc. | Substituted eneoxindoles and uses thereof |
| MX2021014379A (es) * | 2019-05-24 | 2022-01-06 | Assembly Biosciences Inc | Composiciones farmaceuticas para el tratamiento de hbv. |
| CN114245807B (zh) | 2019-06-25 | 2025-05-02 | 吉利德科学公司 | Flt3l-fc融合蛋白和使用方法 |
| US20220296619A1 (en) | 2019-08-19 | 2022-09-22 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| KR20220074917A (ko) | 2019-09-30 | 2022-06-03 | 길리애드 사이언시즈, 인코포레이티드 | Hbv 백신 및 hbv를 치료하는 방법 |
| CN114728973B (zh) * | 2019-11-22 | 2024-06-28 | 正大天晴药业集团股份有限公司 | 一种核蛋白抑制剂的晶型及其应用 |
| WO2021113765A1 (en) | 2019-12-06 | 2021-06-10 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| EP4114529A1 (en) | 2020-03-03 | 2023-01-11 | PIC Therapeutics, Inc. | Eif4e inhibitors and uses thereof |
| WO2021183639A1 (en) * | 2020-03-11 | 2021-09-16 | Nocion Therapeutics, Inc. | Charged ion channel blockers and methods for use |
| US12162851B2 (en) | 2020-03-11 | 2024-12-10 | Nocion Therapeutics, Inc. | Charged ion channel blockers and methods for use |
| KR20220156884A (ko) | 2020-03-20 | 2022-11-28 | 길리애드 사이언시즈, 인코포레이티드 | 4'-c-치환된-2-할로-2'-데옥시아데노신 뉴클레오시드의 프로드러그 및 이의 제조 및 사용 방법 |
| WO2021216656A1 (en) * | 2020-04-22 | 2021-10-28 | Assembly Biosciences, Inc. | 5-membered heteroaryl carboxamide compounds for treatment of hbv |
| CN113929648A (zh) * | 2020-07-14 | 2022-01-14 | 浙江晖石药业有限公司 | 一种环丁烷-1,2-二甲酸酐及其中间体的制备方法 |
| WO2022031894A1 (en) | 2020-08-07 | 2022-02-10 | Gilead Sciences, Inc. | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| TW202406932A (zh) | 2020-10-22 | 2024-02-16 | 美商基利科學股份有限公司 | 介白素2-Fc融合蛋白及使用方法 |
| WO2022241134A1 (en) | 2021-05-13 | 2022-11-17 | Gilead Sciences, Inc. | COMBINATION OF A TLR8 MODULATING COMPOUND AND ANTI-HBV siRNA THERAPEUTICS |
| AU2022298639C1 (en) | 2021-06-23 | 2025-07-17 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| KR20240005901A (ko) | 2021-06-23 | 2024-01-12 | 길리애드 사이언시즈, 인코포레이티드 | 디아실글리세롤 키나제 조절 화합물 |
| KR20240025616A (ko) | 2021-06-23 | 2024-02-27 | 길리애드 사이언시즈, 인코포레이티드 | 다이아실글리세롤 키나제 조절 화합물 |
| JP7654118B2 (ja) | 2021-06-23 | 2025-03-31 | ギリアード サイエンシーズ, インコーポレイテッド | ジアシルグリセロールキナーゼ調節化合物 |
| US12157732B2 (en) | 2021-08-25 | 2024-12-03 | PIC Therapeutics, Inc. | eIF4E inhibitors and uses thereof |
| CN118103368A (zh) | 2021-08-25 | 2024-05-28 | 皮克医疗公司 | Eif4e抑制剂及其用途 |
| CN115521238B (zh) * | 2022-07-02 | 2024-05-31 | 甘肃瀚聚药业有限公司 | 一种n-甲基-2-(2-氯乙基)吡咯烷的制备方法 |
| WO2025240242A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| US20250345390A1 (en) | 2024-05-13 | 2025-11-13 | Gilead Sciences, Inc. | Combination therapies |
| US20250345389A1 (en) | 2024-05-13 | 2025-11-13 | Gilead Sciences, Inc. | Combination therapies |
| WO2025240246A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070105835A1 (en) * | 2005-11-07 | 2007-05-10 | Kazantsev Aleksey G | Compositions and methods for modulating poly(ADP-ribose) polymerase activity |
| JP2011529053A (ja) * | 2008-07-23 | 2011-12-01 | アメリカ合衆国 | 抗マラリア薬としてのプラスモジウム表面アニオンチャンネル阻害剤 |
| WO2013006394A1 (en) * | 2011-07-01 | 2013-01-10 | Institute For Hepatitis And Virus Research | Sulfamoylbenzamide derivatives as antiviral agents against hbv infection |
| WO2015017412A1 (en) * | 2013-07-29 | 2015-02-05 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | 11-oxo-10,11-dihydrodibenzo[b,f][1,4]thiazepine s-oxide derivatives and their use as dopamine d2 receptor antagonists |
| WO2015138895A1 (en) * | 2014-03-13 | 2015-09-17 | Indiana University Research And Technology Corporation | Hepatitis b core protein allosteric modulators |
| JP2018531921A (ja) * | 2015-09-15 | 2018-11-01 | アッセンブリー バイオサイエンシズ,インコーポレイテッド | B型肝炎ウイルスコアタンパク質モジュレーター |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US688762A (en) * | 1901-06-19 | 1901-12-10 | Charles Weber | Toy. |
| GB1480553A (en) | 1976-06-03 | 1977-07-20 | Pfizer Ltd | Tricyclic sulphonamides |
| JPS58225074A (ja) | 1982-06-25 | 1983-12-27 | Chugai Pharmaceut Co Ltd | ジベンゾオキサゼピン誘導体 |
| GB9109557D0 (en) | 1991-05-02 | 1991-06-26 | Wellcome Found | Chemical compounds |
| US5512563A (en) | 1993-07-29 | 1996-04-30 | American Cyanamid Company | Tricyclic benzazepine vasopressin antagonists |
| DE19624659A1 (de) * | 1996-06-20 | 1998-01-08 | Klinge Co Chem Pharm Fab | Neue Pyridylalken- und Pyridylalkinsäureamide |
| DE102004004928A1 (de) | 2004-01-31 | 2005-08-18 | Bayer Healthcare Ag | Dibenzoxazepine II |
| CA2625423A1 (en) | 2005-10-17 | 2007-04-26 | Acadia Pharmaceuticals Inc. | Cb-1 modulating compounds and their use |
| JP4042065B1 (ja) | 2006-03-10 | 2008-02-06 | 健治 吉田 | 情報処理装置への入力処理システム |
| WO2008036139A2 (en) * | 2006-06-07 | 2008-03-27 | The Regents Of The University Of California | Inhibitors of mshc and homologs thereof, and methods of identifying same |
| WO2008118141A2 (en) | 2006-10-17 | 2008-10-02 | Acadia Pharmaceuticals Inc. | Use of cannabinoid modulating compounds in combination with other therapeutic compounds for adjunctive therapy |
| CN106336400A (zh) * | 2008-12-08 | 2017-01-18 | 萌蒂制药国际有限公司 | 酪氨酸激酶蛋白受体拮抗剂 |
| CN104341401B (zh) * | 2009-12-18 | 2017-02-15 | 北京凯因科技股份有限公司 | C型肝炎病毒复制的新型抑制剂 |
| WO2012045194A1 (en) | 2010-10-09 | 2012-04-12 | Abbott Laboratories | Benzodiazepinones as fak inhibitors for treatment of cancer |
| WO2013010069A1 (en) | 2011-07-13 | 2013-01-17 | Indiana University Research And Technology Corporation | Modified viral structural protein with antiviral activity |
| JP6162144B2 (ja) | 2012-01-06 | 2017-07-19 | ヤンセン・サイエンシズ・アイルランド・ユーシー | 4,4−二置換−1,4−ジヒドロピリミジン、及びb型肝炎の処置のための医薬としてのその使用 |
| CN104902885A (zh) | 2012-08-28 | 2015-09-09 | 爱尔兰詹森科学公司 | 氨磺酰基-芳基酰胺和其作为药物用于治疗乙型肝炎的用途 |
| MX358015B (es) | 2012-08-28 | 2018-08-02 | Janssen Sciences Ireland Uc | Derivados biciclicos fusionados de sulfamoilo y su uso como medicamentos para el tratamiento de la hepatitis b. |
| HK1210152A1 (en) | 2012-09-10 | 2016-04-15 | F. Hoffmann-La Roche Ag | 6-amino acid heteroaryldihydropyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| CA2889892A1 (en) | 2012-11-09 | 2014-05-15 | Indiana University Research And Technology Corporation | Alternative uses for hbv assembly effectors |
| WO2014089296A2 (en) | 2012-12-06 | 2014-06-12 | Institute For Hepatitis And Virus Research | Functionalized benzamide derivatives as antiviral agents against hbv infection |
| JP6409001B2 (ja) | 2012-12-27 | 2018-10-17 | ドレクセル ユニバーシティ | Hbv感染に対する新規抗ウイルス剤 |
| RU2519546C1 (ru) | 2013-01-16 | 2014-06-10 | Общество С Ограниченной Ответственностью "Биоинтегратор" (Ооо "Биоинтегратор") | КОНЪЮГАТЫ И МАЛЫЕ МОЛЕКУЛЫ, ВЗАИМОДЕЙСТВУЮЩИЕ С РЕЦЕПТОРОМ CD16а |
| PL2961732T3 (pl) | 2013-02-28 | 2017-09-29 | Janssen Sciences Ireland Uc | Sulfamoilo-aryloamidy i ich stosowanie jako leków do leczenia wirusowego zapalenia wątroby typu B |
| AR095426A1 (es) | 2013-03-14 | 2015-10-14 | Onyx Therapeutics Inc | Inhibidores tripeptídicos de la epoxicetona proteasa |
| CA2907243C (en) | 2013-03-15 | 2021-12-28 | Celgene Avilomics Research, Inc. | Substituted dihydropyrimidopyrimidinone compounds and pharmaceutical compositions thereof use fgfr4 inhibitor |
| EA027068B1 (ru) | 2013-04-03 | 2017-06-30 | Янссен Сайенсиз Айрлэнд Юси | Производные n-фенилкарбоксамида и их применение в качестве лекарственных препаратов для лечения гепатита b |
| US9266904B2 (en) | 2013-05-17 | 2016-02-23 | Hoffmann-La Roche Inc. | 6-bridged heteroaryldihydropyrimidines for the treatment and prophylaxis of hepatitis B virus infection |
| JO3603B1 (ar) | 2013-05-17 | 2020-07-05 | Janssen Sciences Ireland Uc | مشتقات سلفامويل بيرولاميد واستخدامها كادوية لمعالجة التهاب الكبد نوع بي |
| CN105960400B (zh) | 2013-05-17 | 2019-05-31 | 爱尔兰詹森科学公司 | 氨磺酰基噻吩酰胺衍生物及其作为药物用于治疗乙型肝炎的用途 |
| WO2014205138A1 (en) | 2013-06-19 | 2014-12-24 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulator and uses thereof |
| CR20170008A (es) | 2014-06-10 | 2017-06-13 | Basf Se | Compuestos de [1,2,4] triazol e imidazol sustituidos |
-
2016
- 2016-09-14 TW TW110118046A patent/TWI786639B/zh not_active IP Right Cessation
- 2016-09-14 TW TW105130076A patent/TW201720802A/zh unknown
- 2016-09-14 TW TW105130078A patent/TWI730985B/zh not_active IP Right Cessation
- 2016-09-14 TW TW105130074A patent/TWI721016B/zh not_active IP Right Cessation
- 2016-09-15 CA CA2998803A patent/CA2998803A1/en not_active Abandoned
- 2016-09-15 LT LTEP16847295.9T patent/LT3349580T/lt unknown
- 2016-09-15 HK HK19101078.2A patent/HK1258697A1/zh unknown
- 2016-09-15 ES ES16847305T patent/ES2905950T3/es active Active
- 2016-09-15 KR KR1020187010461A patent/KR20180050405A/ko not_active Withdrawn
- 2016-09-15 WO PCT/US2016/051940 patent/WO2017048954A1/en not_active Ceased
- 2016-09-15 MX MX2018003198A patent/MX392309B/es unknown
- 2016-09-15 CN CN201680065785.8A patent/CN108347943B/zh not_active Expired - Fee Related
- 2016-09-15 EP EP20185808.1A patent/EP3782626A1/en not_active Withdrawn
- 2016-09-15 AU AU2016323297A patent/AU2016323297A1/en not_active Abandoned
- 2016-09-15 CN CN201680065137.2A patent/CN108348529A/zh active Pending
- 2016-09-15 US US15/760,284 patent/US10377748B2/en active Active
- 2016-09-15 CA CA2998793A patent/CA2998793A1/en not_active Abandoned
- 2016-09-15 JP JP2018513837A patent/JP2018531921A/ja active Pending
- 2016-09-15 SG SG10202011103UA patent/SG10202011103UA/en unknown
- 2016-09-15 AR ARP160102819A patent/AR106040A1/es unknown
- 2016-09-15 SM SM20200630T patent/SMT202000630T1/it unknown
- 2016-09-15 CA CA2998741A patent/CA2998741C/en active Active
- 2016-09-15 KR KR1020187010462A patent/KR20180050406A/ko not_active Withdrawn
- 2016-09-15 AR ARP160102820A patent/AR106041A1/es unknown
- 2016-09-15 PT PT168472959T patent/PT3349580T/pt unknown
- 2016-09-15 KR KR1020187010455A patent/KR20180053363A/ko not_active Ceased
- 2016-09-15 US US15/760,343 patent/US10392379B2/en not_active Expired - Fee Related
- 2016-09-15 CN CN201680065139.1A patent/CN108347942B/zh not_active Expired - Fee Related
- 2016-09-15 PL PL16847295.9T patent/PL3349580T3/pl unknown
- 2016-09-15 EA EA201890735A patent/EA201890735A1/ru unknown
- 2016-09-15 PH PH1/2018/500571A patent/PH12018500571B1/en unknown
- 2016-09-15 AR ARP160102816A patent/AR106037A1/es unknown
- 2016-09-15 EA EA201890731A patent/EA201890731A1/ru unknown
- 2016-09-15 RS RS20201384A patent/RS61064B1/sr unknown
- 2016-09-15 WO PCT/US2016/051949 patent/WO2017048962A1/en not_active Ceased
- 2016-09-15 MX MX2018003207A patent/MX2018003207A/es unknown
- 2016-09-15 HU HUE16847295A patent/HUE051872T2/hu unknown
- 2016-09-15 SI SI201630982T patent/SI3349580T1/sl unknown
- 2016-09-15 EA EA201890736A patent/EA201890736A1/ru unknown
- 2016-09-15 WO PCT/US2016/051934 patent/WO2017048950A1/en not_active Ceased
- 2016-09-15 AU AU2016323305A patent/AU2016323305A1/en not_active Abandoned
- 2016-09-15 US US15/760,387 patent/US10766890B2/en active Active
- 2016-09-15 ES ES16847295T patent/ES2831333T3/es active Active
- 2016-09-15 JP JP2018513789A patent/JP2018531919A/ja active Pending
- 2016-09-15 AU AU2016323293A patent/AU2016323293B2/en not_active Ceased
- 2016-09-15 DK DK16847295.9T patent/DK3349580T3/da active
- 2016-09-15 EP EP16847305.6A patent/EP3349581B8/en not_active Not-in-force
- 2016-09-15 EP EP16847298.3A patent/EP3349761B1/en not_active Not-in-force
- 2016-09-15 MY MYPI2018000361A patent/MY198974A/en unknown
- 2016-09-15 HK HK19101106.8A patent/HK1259141A1/zh unknown
- 2016-09-15 CN CN202010540566.9A patent/CN112094264A/zh active Pending
- 2016-09-15 MX MX2018003206A patent/MX2018003206A/es unknown
- 2016-09-15 EP EP16847295.9A patent/EP3349580B1/en active Active
- 2016-09-15 HR HRP20201812TT patent/HRP20201812T1/hr unknown
- 2016-09-15 JP JP2018513782A patent/JP7023838B2/ja not_active Expired - Fee Related
-
2018
- 2018-03-13 IL IL258099A patent/IL258099A/en unknown
- 2018-03-13 IL IL258097A patent/IL258097A/en unknown
- 2018-03-14 IL IL258124A patent/IL258124B/en active IP Right Grant
- 2018-03-14 MX MX2022005899A patent/MX2022005899A/es unknown
- 2018-03-15 DO DO2018000074A patent/DOP2018000074A/es unknown
- 2018-03-15 PH PH12018500573A patent/PH12018500573A1/en unknown
- 2018-03-15 PH PH12018500572A patent/PH12018500572A1/en unknown
- 2018-03-15 CL CL2018000684A patent/CL2018000684A1/es unknown
- 2018-04-13 ZA ZA2018/02456A patent/ZA201802456B/en unknown
-
2019
- 2019-05-28 CL CL2019001433A patent/CL2019001433A1/es unknown
- 2019-05-30 US US16/426,727 patent/US10968211B2/en not_active Expired - Fee Related
-
2020
- 2020-11-16 CY CY20201101088T patent/CY1123570T1/el unknown
-
2021
- 2021-01-04 US US17/140,800 patent/US11814376B2/en active Active
- 2021-01-18 JP JP2021005706A patent/JP7150903B2/ja active Active
- 2021-04-01 AU AU2021202065A patent/AU2021202065B2/en not_active Ceased
-
2023
- 2023-09-01 US US18/460,337 patent/US20240025890A1/en not_active Abandoned
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070105835A1 (en) * | 2005-11-07 | 2007-05-10 | Kazantsev Aleksey G | Compositions and methods for modulating poly(ADP-ribose) polymerase activity |
| JP2011529053A (ja) * | 2008-07-23 | 2011-12-01 | アメリカ合衆国 | 抗マラリア薬としてのプラスモジウム表面アニオンチャンネル阻害剤 |
| WO2013006394A1 (en) * | 2011-07-01 | 2013-01-10 | Institute For Hepatitis And Virus Research | Sulfamoylbenzamide derivatives as antiviral agents against hbv infection |
| WO2015017412A1 (en) * | 2013-07-29 | 2015-02-05 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | 11-oxo-10,11-dihydrodibenzo[b,f][1,4]thiazepine s-oxide derivatives and their use as dopamine d2 receptor antagonists |
| WO2015138895A1 (en) * | 2014-03-13 | 2015-09-17 | Indiana University Research And Technology Corporation | Hepatitis b core protein allosteric modulators |
| JP2018531921A (ja) * | 2015-09-15 | 2018-11-01 | アッセンブリー バイオサイエンシズ,インコーポレイテッド | B型肝炎ウイルスコアタンパク質モジュレーター |
| JP2018531918A (ja) * | 2015-09-15 | 2018-11-01 | アッセンブリー バイオサイエンシズ,インコーポレイテッド | B型肝炎ウイルスコアタンパク質モジュレーター |
Non-Patent Citations (6)
| Title |
|---|
| "CAS REGISTRY NO. 443670-41-5", DATABASE REGISTRY, [ONLINE], JPN7020002104, 12 August 2002 (2002-08-12), ISSN: 0004670005 * |
| "CAS REGISTRY NO. 688762-67-6", DATABASE REGISTRY, [ONLINE], JPN7020002103, 3 June 2004 (2004-06-03), ISSN: 0004670004 * |
| "CAS REGISTRY NO. 688762-71-2", DATABASE REGISTRY, [ONLINE], JPN7020002102, 3 June 2004 (2004-06-03), ISSN: 0004670003 * |
| "CAS REGISTRY NO. 903147-56-8", DATABASE REGISTRY, [ONLINE], JPN7020002101, 22 August 2006 (2006-08-22), ISSN: 0004670002 * |
| "CAS REGISTRY NO. 931950-46-8", DATABASE REGISTRY, [ONLINE], JPN7020002100, 23 April 2007 (2007-04-23), ISSN: 0004670001 * |
| HALL, PR ET AL.: "Small molecule inhibitors of hantavirus infection", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 20, JPN6020026075, 2010, pages 7085 - 7091, ISSN: 0004520891 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018531918A (ja) * | 2015-09-15 | 2018-11-01 | アッセンブリー バイオサイエンシズ,インコーポレイテッド | B型肝炎ウイルスコアタンパク質モジュレーター |
| JP2018531921A (ja) * | 2015-09-15 | 2018-11-01 | アッセンブリー バイオサイエンシズ,インコーポレイテッド | B型肝炎ウイルスコアタンパク質モジュレーター |
| JP2021054872A (ja) * | 2015-09-15 | 2021-04-08 | アッセンブリー バイオサイエンシズ,インコーポレイテッド | B型肝炎ウイルスコアタンパク質モジュレーター |
| JP7023838B2 (ja) | 2015-09-15 | 2022-03-31 | アッセンブリー バイオサイエンシズ,インコーポレイテッド | B型肝炎ウイルスコアタンパク質モジュレーター |
| JP7150903B2 (ja) | 2015-09-15 | 2022-10-11 | アッセンブリー バイオサイエンシズ,インコーポレイテッド | B型肝炎ウイルスコアタンパク質モジュレーター |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2018531919A (ja) | B型肝炎ウイルスコアタンパク質モジュレーター | |
| JP6602311B2 (ja) | B型肝炎コアタンパク質アロステリックモジュレーター | |
| US10987360B2 (en) | Hepatitis B core protein modulators | |
| HK1258761B (en) | Hepatitis b core protein modulators | |
| HK1231697A1 (en) | Hepatitis b core protein allosteric modulators | |
| HK1231697B (en) | Hepatitis b core protein allosteric modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190912 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20200716 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200720 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20201012 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210120 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210604 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20211224 |