JP2010004760A - 糸状菌プロテアーゼの生産方法 - Google Patents
糸状菌プロテアーゼの生産方法 Download PDFInfo
- Publication number
- JP2010004760A JP2010004760A JP2008165094A JP2008165094A JP2010004760A JP 2010004760 A JP2010004760 A JP 2010004760A JP 2008165094 A JP2008165094 A JP 2008165094A JP 2008165094 A JP2008165094 A JP 2008165094A JP 2010004760 A JP2010004760 A JP 2010004760A
- Authority
- JP
- Japan
- Prior art keywords
- protease
- self
- cloning
- filamentous fungi
- vector
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 108091005804 Peptidases Proteins 0.000 title claims abstract description 123
- 239000004365 Protease Substances 0.000 title claims abstract description 93
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 35
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 title claims abstract 5
- 230000002538 fungal effect Effects 0.000 title abstract description 5
- 241000233866 Fungi Species 0.000 claims abstract description 108
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 65
- 239000013604 expression vector Substances 0.000 claims abstract description 43
- 239000013599 cloning vector Substances 0.000 claims abstract description 30
- 239000003550 marker Substances 0.000 claims abstract description 27
- 108700007698 Genetic Terminator Regions Proteins 0.000 claims abstract description 4
- 239000002773 nucleotide Substances 0.000 claims description 8
- 125000003729 nucleotide group Chemical group 0.000 claims description 8
- 125000003275 alpha amino acid group Chemical group 0.000 claims 2
- 238000000034 method Methods 0.000 abstract description 36
- 239000013598 vector Substances 0.000 abstract description 19
- 238000012258 culturing Methods 0.000 abstract description 14
- 102000035195 Peptidases Human genes 0.000 description 94
- 235000019419 proteases Nutrition 0.000 description 76
- 238000010367 cloning Methods 0.000 description 58
- 239000012228 culture supernatant Substances 0.000 description 56
- 240000006439 Aspergillus oryzae Species 0.000 description 39
- 235000002247 Aspergillus oryzae Nutrition 0.000 description 39
- 238000012181 QIAquick gel extraction kit Methods 0.000 description 30
- 239000002609 medium Substances 0.000 description 30
- 238000002835 absorbance Methods 0.000 description 27
- 230000000694 effects Effects 0.000 description 26
- 241000228212 Aspergillus Species 0.000 description 23
- 238000005259 measurement Methods 0.000 description 22
- 239000013600 plasmid vector Substances 0.000 description 20
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 20
- 108020004414 DNA Proteins 0.000 description 19
- 239000000047 product Substances 0.000 description 18
- 238000002360 preparation method Methods 0.000 description 17
- 238000012408 PCR amplification Methods 0.000 description 16
- 108091005658 Basic proteases Proteins 0.000 description 15
- 238000000246 agarose gel electrophoresis Methods 0.000 description 15
- 239000012634 fragment Substances 0.000 description 14
- 239000013612 plasmid Substances 0.000 description 13
- 239000007787 solid Substances 0.000 description 13
- 238000004458 analytical method Methods 0.000 description 12
- 101100096168 Aspergillus oryzae (strain ATCC 42149 / RIB 40) sodB gene Proteins 0.000 description 10
- 101150108358 GLAA gene Proteins 0.000 description 10
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 10
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 10
- 235000013305 food Nutrition 0.000 description 10
- 101150087539 sodA gene Proteins 0.000 description 10
- 101150107789 sodM gene Proteins 0.000 description 10
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 9
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 9
- 150000001413 amino acids Chemical class 0.000 description 9
- 238000001035 drying Methods 0.000 description 9
- 239000006228 supernatant Substances 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 210000004027 cell Anatomy 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 239000000523 sample Substances 0.000 description 8
- 241000894007 species Species 0.000 description 8
- 241000132177 Aspergillus glaucus Species 0.000 description 7
- 241000228257 Aspergillus sp. Species 0.000 description 7
- 239000007983 Tris buffer Substances 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 7
- 241000588724 Escherichia coli Species 0.000 description 6
- 108010041102 azocasein Proteins 0.000 description 6
- 239000008103 glucose Substances 0.000 description 6
- 235000015097 nutrients Nutrition 0.000 description 6
- 108090000765 processed proteins & peptides Proteins 0.000 description 6
- VWDWKYIASSYTQR-UHFFFAOYSA-N sodium nitrate Chemical compound [Na+].[O-][N+]([O-])=O VWDWKYIASSYTQR-UHFFFAOYSA-N 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 230000009466 transformation Effects 0.000 description 6
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 6
- 102000005572 Cathepsin A Human genes 0.000 description 5
- 108010059081 Cathepsin A Proteins 0.000 description 5
- 239000008351 acetate buffer Substances 0.000 description 5
- 230000003321 amplification Effects 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 235000019341 magnesium sulphate Nutrition 0.000 description 5
- 238000003199 nucleic acid amplification method Methods 0.000 description 5
- 239000001103 potassium chloride Substances 0.000 description 5
- 235000011164 potassium chloride Nutrition 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 230000001131 transforming effect Effects 0.000 description 5
- 241000351920 Aspergillus nidulans Species 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 108010073178 Glucan 1,4-alpha-Glucosidase Proteins 0.000 description 4
- 244000061456 Solanum tuberosum Species 0.000 description 4
- 235000002595 Solanum tuberosum Nutrition 0.000 description 4
- 239000011575 calcium Substances 0.000 description 4
- 229910052791 calcium Inorganic materials 0.000 description 4
- 229940041514 candida albicans extract Drugs 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000008121 dextrose Substances 0.000 description 4
- 229940088598 enzyme Drugs 0.000 description 4
- 238000009630 liquid culture Methods 0.000 description 4
- 244000005700 microbiome Species 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 239000012138 yeast extract Substances 0.000 description 4
- 229920001817 Agar Polymers 0.000 description 3
- 241001513093 Aspergillus awamori Species 0.000 description 3
- 241000228245 Aspergillus niger Species 0.000 description 3
- 241000131386 Aspergillus sojae Species 0.000 description 3
- 241000134719 Aspergillus tamarii Species 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 229910002651 NO3 Inorganic materials 0.000 description 3
- 101100205189 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) leu-5 gene Proteins 0.000 description 3
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 3
- 108091028043 Nucleic acid sequence Proteins 0.000 description 3
- 239000001888 Peptone Substances 0.000 description 3
- 108010080698 Peptones Proteins 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 239000008272 agar Substances 0.000 description 3
- 108090000637 alpha-Amylases Proteins 0.000 description 3
- 239000012225 czapek media Substances 0.000 description 3
- 235000021245 dietary protein Nutrition 0.000 description 3
- 101150051757 glaB gene Proteins 0.000 description 3
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 3
- 229910000358 iron sulfate Inorganic materials 0.000 description 3
- BAUYGSIQEAFULO-UHFFFAOYSA-L iron(2+) sulfate (anhydrous) Chemical compound [Fe+2].[O-]S([O-])(=O)=O BAUYGSIQEAFULO-UHFFFAOYSA-L 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 229910017604 nitric acid Inorganic materials 0.000 description 3
- 235000019319 peptone Nutrition 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- -1 promoters Proteins 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 235000010344 sodium nitrate Nutrition 0.000 description 3
- 239000004317 sodium nitrate Substances 0.000 description 3
- 238000010561 standard procedure Methods 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 229920000936 Agarose Polymers 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- 241000228197 Aspergillus flavus Species 0.000 description 2
- 241000122821 Aspergillus kawachii Species 0.000 description 2
- 101100101264 Aspergillus oryzae (strain ATCC 42149 / RIB 40) melO gene Proteins 0.000 description 2
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 102000005367 Carboxypeptidases Human genes 0.000 description 2
- 108010006303 Carboxypeptidases Proteins 0.000 description 2
- QNAYBMKLOCPYGJ-UHFFFAOYSA-N D-alpha-Ala Natural products CC([NH3+])C([O-])=O QNAYBMKLOCPYGJ-UHFFFAOYSA-N 0.000 description 2
- 102100022624 Glucoamylase Human genes 0.000 description 2
- QNAYBMKLOCPYGJ-UWTATZPHSA-N L-Alanine Natural products C[C@@H](N)C(O)=O QNAYBMKLOCPYGJ-UWTATZPHSA-N 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241001608711 Melo Species 0.000 description 2
- 240000007594 Oryza sativa Species 0.000 description 2
- 235000007164 Oryza sativa Nutrition 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 108091058545 Secretory proteins Proteins 0.000 description 2
- 102000040739 Secretory proteins Human genes 0.000 description 2
- 241000209140 Triticum Species 0.000 description 2
- 235000021307 Triticum Nutrition 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 229960003767 alanine Drugs 0.000 description 2
- 108010028144 alpha-Glucosidases Proteins 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 230000036772 blood pressure Effects 0.000 description 2
- 239000001110 calcium chloride Substances 0.000 description 2
- 229910001628 calcium chloride Inorganic materials 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000013611 chromosomal DNA Substances 0.000 description 2
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 2
- 238000001962 electrophoresis Methods 0.000 description 2
- 238000004520 electroporation Methods 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 101150087199 leuA gene Proteins 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- BDRTVPCFKSUHCJ-UHFFFAOYSA-N molecular hydrogen;potassium Chemical compound [K].[H][H] BDRTVPCFKSUHCJ-UHFFFAOYSA-N 0.000 description 2
- 101150095344 niaD gene Proteins 0.000 description 2
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 2
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 108091008146 restriction endonucleases Proteins 0.000 description 2
- 235000009566 rice Nutrition 0.000 description 2
- 235000013555 soy sauce Nutrition 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 238000011426 transformation method Methods 0.000 description 2
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 108010039636 3-isopropylmalate dehydrogenase Proteins 0.000 description 1
- 108010011619 6-Phytase Proteins 0.000 description 1
- 108010031025 Alanine Dehydrogenase Proteins 0.000 description 1
- 241001225321 Aspergillus fumigatus Species 0.000 description 1
- 101100001186 Aspergillus oryzae (strain ATCC 42149 / RIB 40) agdA gene Proteins 0.000 description 1
- 101100425074 Aspergillus oryzae (strain ATCC 42149 / RIB 40) thiA gene Proteins 0.000 description 1
- 241000228251 Aspergillus phoenicis Species 0.000 description 1
- 108090000145 Bacillolysin Proteins 0.000 description 1
- 241000193830 Bacillus <bacterium> Species 0.000 description 1
- 101100277447 Bacillus subtilis (strain 168) degQ gene Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- 108010076119 Caseins Proteins 0.000 description 1
- 108010059892 Cellulase Proteins 0.000 description 1
- 108010008885 Cellulose 1,4-beta-Cellobiosidase Proteins 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 101710121765 Endo-1,4-beta-xylanase Proteins 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 108010093031 Galactosidases Proteins 0.000 description 1
- 102000002464 Galactosidases Human genes 0.000 description 1
- 108010017544 Glucosylceramidase Proteins 0.000 description 1
- 102000004547 Glucosylceramidase Human genes 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 101100295959 Halobacterium salinarum (strain ATCC 700922 / JCM 11081 / NRC-1) arcB gene Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 108090001060 Lipase Proteins 0.000 description 1
- 102000004882 Lipase Human genes 0.000 description 1
- 239000004367 Lipase Substances 0.000 description 1
- 102100024295 Maltase-glucoamylase Human genes 0.000 description 1
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 1
- 108010011756 Milk Proteins Proteins 0.000 description 1
- 102000014171 Milk Proteins Human genes 0.000 description 1
- 244000294411 Mirabilis expansa Species 0.000 description 1
- 235000015429 Mirabilis expansa Nutrition 0.000 description 1
- YBHQCJILTOVLHD-YVMONPNESA-N Mirin Chemical compound S1C(N)=NC(=O)\C1=C\C1=CC=C(O)C=C1 YBHQCJILTOVLHD-YVMONPNESA-N 0.000 description 1
- 241000235395 Mucor Species 0.000 description 1
- 241000588652 Neisseria gonorrhoeae Species 0.000 description 1
- 241000221960 Neurospora Species 0.000 description 1
- 102000035092 Neutral proteases Human genes 0.000 description 1
- 108091005507 Neutral proteases Proteins 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 108700026244 Open Reading Frames Proteins 0.000 description 1
- 108010029182 Pectin lyase Proteins 0.000 description 1
- 102000015439 Phospholipases Human genes 0.000 description 1
- 108010064785 Phospholipases Proteins 0.000 description 1
- 101710180319 Protease 1 Proteins 0.000 description 1
- 241000235527 Rhizopus Species 0.000 description 1
- 101710137710 Thioesterase 1/protease 1/lysophospholipase L1 Proteins 0.000 description 1
- 241000223259 Trichoderma Species 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- 229930003451 Vitamin B1 Natural products 0.000 description 1
- 108010027199 Xylosidases Proteins 0.000 description 1
- 102000004139 alpha-Amylases Human genes 0.000 description 1
- 229940024171 alpha-amylase Drugs 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- 229960000723 ampicillin Drugs 0.000 description 1
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 1
- 101150009288 amyB gene Proteins 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 101150008194 argB gene Proteins 0.000 description 1
- 229940091771 aspergillus fumigatus Drugs 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 239000003788 bath preparation Substances 0.000 description 1
- 235000015278 beef Nutrition 0.000 description 1
- RIOXQFHNBCKOKP-UHFFFAOYSA-N benomyl Chemical compound C1=CC=C2N(C(=O)NCCCC)C(NC(=O)OC)=NC2=C1 RIOXQFHNBCKOKP-UHFFFAOYSA-N 0.000 description 1
- MITFXPHMIHQXPI-UHFFFAOYSA-N benzoxaprofen Natural products N=1C2=CC(C(C(O)=O)C)=CC=C2OC=1C1=CC=C(Cl)C=C1 MITFXPHMIHQXPI-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 235000019658 bitter taste Nutrition 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 210000005056 cell body Anatomy 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 230000001079 digestive effect Effects 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 235000011194 food seasoning agent Nutrition 0.000 description 1
- 235000013376 functional food Nutrition 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 238000003898 horticulture Methods 0.000 description 1
- 239000003262 industrial enzyme Substances 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 230000031891 intestinal absorption Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 229960000318 kanamycin Drugs 0.000 description 1
- 229930027917 kanamycin Natural products 0.000 description 1
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 1
- 229930182823 kanamycin A Natural products 0.000 description 1
- 235000019421 lipase Nutrition 0.000 description 1
- 238000001638 lipofection Methods 0.000 description 1
- 101150039489 lysZ gene Proteins 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- 239000011572 manganese Substances 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 235000021239 milk protein Nutrition 0.000 description 1
- 235000013536 miso Nutrition 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 125000001477 organic nitrogen group Chemical group 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- 101150060364 ptrA gene Proteins 0.000 description 1
- 101150054232 pyrG gene Proteins 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 235000019992 sake Nutrition 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 101150017120 sod gene Proteins 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012089 stop solution Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 235000019640 taste Nutrition 0.000 description 1
- RYCLIXPGLDDLTM-UHFFFAOYSA-J tetrapotassium;phosphonato phosphate Chemical compound [K+].[K+].[K+].[K+].[O-]P([O-])(=O)OP([O-])([O-])=O RYCLIXPGLDDLTM-UHFFFAOYSA-J 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229960003495 thiamine Drugs 0.000 description 1
- DPJRMOMPQZCRJU-UHFFFAOYSA-M thiamine hydrochloride Chemical compound Cl.[Cl-].CC1=C(CCO)SC=[N+]1CC1=CN=C(C)N=C1N DPJRMOMPQZCRJU-UHFFFAOYSA-M 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- JREYOWJEWZVAOR-UHFFFAOYSA-N triazanium;[3-methylbut-3-enoxy(oxido)phosphoryl] phosphate Chemical compound [NH4+].[NH4+].[NH4+].CC(=C)CCOP([O-])(=O)OP([O-])([O-])=O JREYOWJEWZVAOR-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 239000010455 vermiculite Substances 0.000 description 1
- 235000019354 vermiculite Nutrition 0.000 description 1
- 229910052902 vermiculite Inorganic materials 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 239000011691 vitamin B1 Substances 0.000 description 1
- 235000010374 vitamin B1 Nutrition 0.000 description 1
Images







Landscapes
- Enzymes And Modification Thereof (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
【解決手段】糸状菌体内で機能する選択マーカー遺伝子配列、糸状菌体内で機能するプロモーター配列、特定の塩基配列からなる群より選択されるいずれか一つの塩基配列からなるプロテアーゼ遺伝子、および糸状菌体内で機能するターミネーター配列を有する糸状菌用発現ベクター(好ましくはセルフクローニングベクター)、前記ベクターで形質転換された糸状菌および前記糸状菌を培養し回収するプロテアーゼの生産方法。
【選択図】なし
Description
British Journal of Nutrition, 94, 84-91 (2005)
(I)糸状菌用発現ベクター
(I-1)糸状菌体内で機能する選択マーカー遺伝子配列、糸状菌体内で機能するプロモーター配列、配列番号1〜10に記載される塩基配列からなる群より選択されるいずれか一つの塩基配列からなるプロテアーゼ遺伝子、および糸状菌体内で機能するターミネーター配列を有する糸状菌用発現ベクター。
(I-2)実質的に糸状菌に由来するヌクレオチドだけからなるセルフクローニングベクターである、(I-1)に記載する糸状菌用発現ベクター。
(I-3)直鎖状であることを特徴とする、(I-1)または(I-2)に記載する糸状菌用発現ベクター。
(I-4)糸状菌がアスペルギルス属に属する糸状菌である、(I-1)乃至(I-3)のいずれかに記載する糸状菌用発現ベクター。
(II-1)(I-1)乃至(I-4)のいずれかに記載する糸状菌用発現ベクターで形質転換されてなる糸状菌。
(II-2)アスペルギルス属に属する糸状菌である、(II-1)に記載する糸状菌。
(III-1)(II-1)または(II-2)に記載する形質転換された糸状菌を培地で培養し、当該培地から配列番号11〜20に記載されるアミノ酸配列からなる群より選択されるいずれかの一つのアミノ酸配列からなるプロテアーゼを回収する工程を含む、プロテアーゼの生産方法。
本発明の糸状菌用発現ベクターは、糸状菌またはその変異株を宿主として、当該宿主内で配列番号11〜20のいずれかに記載するアミノ酸配列を有する糸状菌プロテアーゼを発現し産生することのできるベクターである。かかるベクターは、上記プロテアーゼをコードする配列番号1〜10のいずれかに記載される塩基配列を有するプロテアーゼ遺伝子に加えて、糸状菌体内で機能するプロモーター配列、糸状菌体内で機能するターミネーター配列、および糸状菌体内で機能し、形質転換体の選択に好適に使用される選択マーカー遺伝子配列を有する。
本発明が対象とするプロテアーゼは、配列番号11〜20に記載するいずれかのアミノ酸配列を有する糸状菌由来、特に麹菌由来のプロテアーゼである。これらのプロテアーゼのうち、配列番号11〜13に示すプロテアーゼはアルカリ性プロテアーゼ、配列番号14〜17に示す酸性カルボキシペプチダーゼ、配列番号18に示すプロテアーゼは酸性プロテアーゼ、および配列番号19〜20に示すプロテアーゼは中性プロテアーゼに分類することができる。
選択マーカー遺伝子は、糸状菌体内、特にアスペルギルス属糸状菌体内で選択マーカーとして機能するもの、すなわち発現できるものであればよいが、好ましくは糸状菌、特にアスペルギルス属糸状菌に由来する選択マーカー遺伝子である。かかる選択マーカー遺伝子として、例えば、niaD(Biosci.Biotechnol.Biochem.,59,1795-1797(1995))、argB(Enzyme Microbiol Technol, 6, 386-389, (1984))、sC(Gene, 84, 329-334, (1989))、ptrA(Biosci Biotechnol Biochem, 64, 1416-1421, (2000))、pyrG(Biochem Biophys Res Commun, 112, 284-289, (1983)),amdS(Gene, 26, 205-221, (1983))、オーレオバシジン耐性遺伝子(Mol Gen Genet, 261, 290-296, (1999))、ベノミル耐性遺伝子(Proc Natl Acad Sci USA, 83, 4869-4873, (1986))、及びハイグロマイシン耐性遺伝子(Gene, 57, 21-26, (1987))から選ばれるマーカー遺伝子を挙げることができる。
プロモーターは、糸状菌体内、特にアスペルギルス属糸状菌体内でプロモーターとして機能するものであればよいが、好ましくは糸状菌、特にアスペルギルス属糸状菌に由来するプロモーターである。かかるプロモーターとして具体的には、例えばα−アミラーゼ遺伝子プロモーターamyB(Biosci Biotechnol Biochem,56,1849-1853,(1992)), グルコアミラーゼ遺伝子プロモーターglaA(Gene,108,145-150,(1992))、α−グルコシダーゼ遺伝子プロモーターagdA(Curr Genet, 30,432-438,(1996))、マンガンSOD遺伝子プロモーターsodM(特開2000-224381号公報)、チロシナーゼ遺伝子プロモーターmelO(特開2001−046078号公報)、グルコアミラーゼ遺伝子プロモーターglaB(特開2000−245465号公報、特開平11−243965号公報)等が挙げられる。液体培養に特異的な高発現プロモーターとして、好ましくはsodM、およびmelOを挙げることができ、また固体培養に特異的な高発現プロモーターとして、好ましくはglaBを挙げることができる。
ターミネーターは、糸状菌体内、特にアスペルギルス属糸状菌体内でターミネーターとして機能するものであればよいが、好ましくは糸状菌、特にアスペルギルス属糸状菌に由来するターミネーターである。好ましくはアスペルギルス属、より好ましくはアスペルギルス・オリゼに由来する、例えばα−アミラーゼ遺伝子のターミネーター、またはグルコアミラーゼ(glaB)ターミネーター(Gene. 207, 127-134,(1998))等を挙げることができる。
本発明の糸状菌用発現ベクターは、これらの選択マーカー遺伝子、プロモーター、プロテアーゼ遺伝子、およびターミネーターが、直接連結されてなるものであってもよいが、それらの間に1〜2000塩基程度のヌクレオチドが挟まれていてもよい。但し、これらのヌクレオチドの配列は糸状菌由来、好ましくはアスペルギルス属糸状菌に由来する配列であることが望ましい。例えば、目的とするプロテアーゼが菌体内タンパク質として生産されるものである場合は、目的とするプロテアーゼの遺伝子のオープンリーディングフレームに隣接して、上流に糸状菌、好ましくはアスペルギルス属糸状菌の既知分泌タンパク質の遺伝子を配置してもよく、こうすることにより、目的とするプロテアーゼは、上記分泌タンパク質との融合タンパク質として発現産生され、斯くして産生された融合タンパク質は菌体外に分泌される。また、その他のヌクレオチドとして、通常ベクターが備える制限酵素切断部位などを備えていてもよい。
本発明の形質転換体は、前述する本発明の糸状菌用発現ベクターで糸状菌を形質転換したものである。
本発明のプロテアーゼの生産方法は、前述した本発明の糸状菌の形質転換体を培養する工程と、培養物から目的タンパク質を回収する工程を含む。
本発明において、好ましい糸状菌発現ベクターは、糸状菌に由来する選択マーカー遺伝子、糸状菌に由来するプロモーター、糸状菌に由来するプロテアーゼ遺伝子、および糸状菌に由来するターミネーターを含む、実質的に糸状菌に由来するヌクレオチドだけからなるセルフクローニングベクターである。また本発明において、好ましい形質転換体は、上記セルフクローニングベクターで形質転換してなる糸状菌である。糸状菌のなかでも好ましくは麹菌であり、麹菌のなかでも好ましくはアスペルギルス属に属する麹菌である。
(1)選択マーカー遺伝子(配列番号21)の調製
麹菌(Aspergillus oryzae)のゲノムDNAを鋳型として、下記のプライマーを用いて下記条件でPCRにより増幅した。得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いて選択マーカー遺伝子断片を切り出した。
プライマー1:5’- gcgtggttta ctagctttag tgctaccaaa-3’(配列番号22)
プライマー2:5’- ccgtacgcggggagtgtgcttaaggcgatg -3’ (配列番号23)
<PCR条件>
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃ 5分)を30サイクル
・72℃(7分)を1サイクル。
麹菌(Aspergillus oryzae)のゲノムDNAを鋳型として、下記のプライマーを用いて下記条件でPCRにより増幅し。得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いてターミネーター断片を切り出した。
プライマー3:5’- ATGTACTTTCCAGTGCGTGTAGTCTACTC-3’(配列番号25)
プライマー4:5’- CTGCAGATCGGCTGAAGTTAGGAGCGGCCATTGTC -3’(配列番号26)
<PCR条件>
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(1分)を30サイクル
・72℃(7分)を1サイクル。
麹菌(Aspergillus oryzae)のゲノムDNAを鋳型として、下記のプライマーを用いて下記条件でPCRにより増幅し。得られたPCR増幅産物についてアガロースゲル電気泳動を行い、得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いてsodMプロモーター断片を切り出した。
プライマー5:5’-CTGCAGTTATGTACTCCGTACTCGGTTGAATTAT-3’(配列番号28)
プライマー6:5’-TTTGGGTGGTTTGGTTGGTATTCTGGTTGAGGGTTTTGAG-3’(配列番号29)
<PCR条件>
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(1分)を30サイクル
・72℃(7分)を1サイクル。
麹菌(Aspergillus oryzae)のゲノムDNAを鋳型として、下記のプライマーを用いて下記条件でPCRにより増幅し。得られたPCR増幅産物についてアガロースゲル電気泳動を行い、得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いてglaAプロモーター断片を切り出した。
プライマー7:5’- GAATTCTGTA GCTGCTCTAT TTCTATTACT -3’(配列番号31)
プライマー8:5’- CTTGCTTCGACTTCGTTTGCTGATGTG -3’(配列番号32)
<PCR条件>
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(1分)を30サイクル
・72℃(7分)を1サイクル。
麹菌用発現ベクターを、pUC118(タカラバイオ、日本)を基本プラスミドとして用いて、次のようにして作成した。
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(7分)を30サイクル
・72℃(7分)を1サイクル。
pUCLT1に、上記で調製したsodMプロモーター(配列番号27)をサブクローニングした。具体的には、上記pUCLT1を鋳型として、プライマー2(配列番号23)とプライマー3(配列番号25)を用いてPCRを行い、得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いて目的断片を切り出し、ベクターを調製した。
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(8分)を30サイクル
・72℃(7分)を1サイクル。
pUCLT1に、上記で調製したglaAプロモーター(配列番号30)をサブクローニングした。具体的には、上記pUCLT1を鋳型として、プライマー2(配列番号23)とプライマー3(配列番号25)を用いてPCRを行い、得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いて目的断片を切り出し、ベクターを調製した。
・96℃(5分)を1サイクル
・96℃(20秒)、60℃ (30秒)、72℃(8分)を30サイクル
・72℃(7分)を1サイクル。
麹菌(Aspergillus oryzae)のアルカリ性プロテアーゼ(AO090003001036)(便宜上、これを「プロテアーゼ1」とする)を、麹菌を用いたセルフクローニング法によって発現させ、産生させた。
アルカリ性プロテアーゼのアミノ酸配列を配列番号11に、それをコードするアルカリプロテアーゼ遺伝子の塩基配列を配列番号1に示す。このアルカリ性プロテアーゼ遺伝子は、下記の方法によって調製した。
プライマー10:5’- ATGCAGTCCATCAAGCGTACCTTGCTCCTCCTCGG-3’(配列番号34)
プライマー11:5’- TTAAGCGTTACCGTTGTAGGCAAGCAGGTT -3’ (配列番号35)
<PCR条件>
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(2分)を30サイクル
・72℃(7分)を1サイクル。
(2-1)プラスミドベクター
まず上記pUCLT1を鋳型として、プライマー3(配列番号25)とプライマー6(配列番号29)を用いてPCRを行い、得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いて目的断片を切り出し、これをベクターとした。
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(9分)を30サイクル
・72℃(7分)を1サイクル。
上記で得られたプラスミドベクター(IE-232)を鋳型として、プライマー1(配列番号22)とプライマー4(配列番号26)を用いてPCRを行い、得られたPCR増幅産物をQIAquick Gel Extraction kit(QIAGEN)を用いて精製し、これをセルフクローニングベクターとした。
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(9分)を30サイクル
・72℃(7分)を1サイクル。
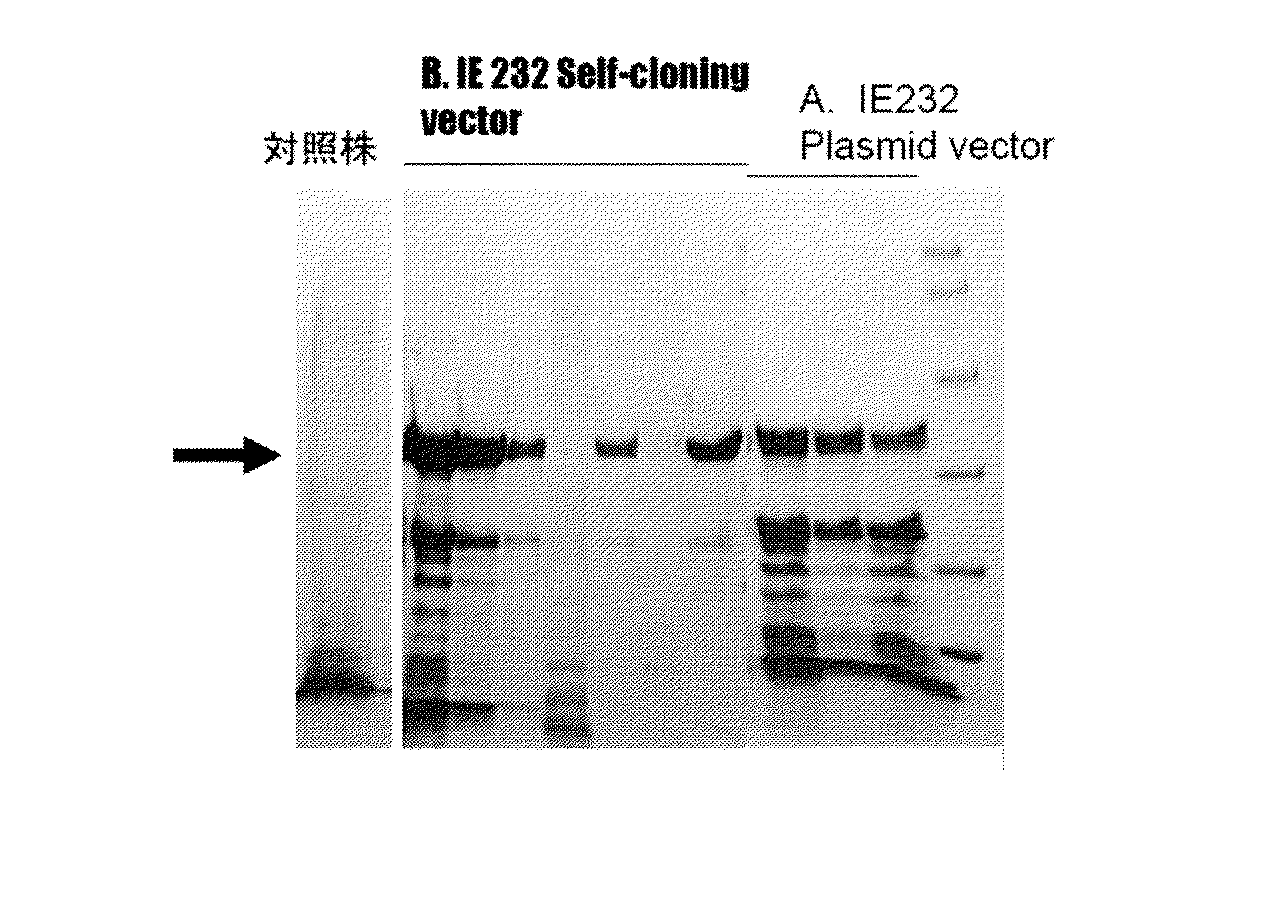
定法であるPEG−カルシウム法(Mol Gen Genet,218,99-104,(1989))に従って、上記で調製したプラスミドベクター(IE-232)またはセルフクローニングベクター(IE-232)をそれぞれ用いて、ロイシン要求性変異麹菌株を形質転換した。なお、当該ロイシン要求性変異麹菌株は、麹菌(Aspergillus oryzae)のロイシンをコードする遺伝子を変異させて正常なロイシンが合成できないようにした菌株であり、ロイシン要求性を相補する選択マーカー遺伝子が組み込まれなければ、通常、ロイシンを含まない培地では増殖することができない。当該ロイシン要求性変異麹菌株は、Aspergillus oryzae leu-5として、茨城県つくば市東1−1−1つくばセンター中央第6に住所を有する独立行政法人産業技術総合研究所 特許生物寄託センターに寄託されている(寄託番号FERM P-20079)。
形質転換効率は、プラスミドベクター(IE-232)が1±0.5/μg-DNAであったのに対して、セルフクローニングベクターは、8.6±3.4/μg-DNAであった。このことから、セルフクローニングベクター(IE-232)を用いると、形質転換効率が、そうでないベクターに比して約8倍も上昇することが確認された。
サザン解析にて、遺伝子導入の有無を調べた。
次に、サザン解析を用いて、得られた形質転換体に大腸菌に由来する配列が存在するか否かを確認した。
上記で調製した各形質転換体を、ポテトデキストロース培地で胞子形成させ、30℃で3日間、GPY液体培地(2%グルコース、1%ペプトン、0.5%酵母エキス、0.1%リン酸1水素2カリウム、0.05%塩化カリウム、0.05%硫酸マグネシウム、0.001%硫酸鉄、0.3%硝酸ナトリウム)で培養した。次いで、培養上清100μlを濃縮乾燥して、SDS−PAGEに供した。また、対照試験として、実施例1で作成したプラスミドpUCLTS1を用いて、麹菌ロイシン要求性変異株を形質転換した株を用いて同様に培養し、SDS−PAGEに供した。
麹菌に由来するプロテアーゼ6種類(プロテアーゼ2〜7)を、麹菌を用いたセルフクローニング法によって発現させ産生させた(sodMプロモーター使用)。
表2に示す6種類のプロテアーゼの遺伝子を、麹菌(Aspergillus oryzae)ゲノムDNAを鋳型に、同表に示す各2本のプライマーを用いて、次のPCR条件で増幅して調製した。得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いて目的断片を切り出し、プロテアーゼ遺伝子とした。
・96℃ (5分)を1サイクル
・96℃ (20秒)、 60℃ (30秒)、 72℃ (2分)を30サイクル
・72℃ (7分)を1サイクル。
(2-1)プラスミドベクター
まずpUCLTS1を鋳型として、プライマー3(配列番号25)とプライマー6(配列番号29)を用いて下記条件でPCRを行い、得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いて目的断片を切り出し、これをベクターとした。
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(9分)を30サイクル
・72℃(7分)を1サイクル。
上記6種類のプラスミド(IE233、IE292、IE338、IE339、IE340、IE341)をそれぞれ鋳型として、プライマー1(配列番号22)とプライマー4(配列番号26)を用いて、下記条件でPCRを行って、麹菌の遺伝子部分のみ増幅した。得られたPCR増幅産物を、QIAquick Gel Extraction kit(QIAGEN)を用いて精製し、これをセルフクローニングベクター(IE233、IE292、IE338、IE339、IE340、IE341)とした。
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(9分)を30サイクル
・72℃(7分)を1サイクル。
定法であるPEG−カルシウム法に従って、上記で調製した各セルフクローニングベクターを用いてロイシン要求性変異麹菌株(Aspergillus oryzae leu-5(寄託番号:FERM P-20079))を形質転換した。これを、硝酸を単一窒素源とするツアペクドックス(Czapek-Dox)培地で培養し、この培地で生育できる菌株を選択することにより、各セルフクローニングベクターにつきそれぞれ10個の形質転換体を取得した。これらすべての形質転換体についてサザン解析を行い、いずれもが大腸菌由来の遺伝子が含まれないセルフクローニング株であることを確認した。
上記で調製した各形質転換体を、ポテトデキストロース培地で胞子形成させ、30℃で3日間、GPY液体培地(2%グルコース、1%ペプトン、0.5%酵母エキス、0.1%リン酸1水素2カリウム、0.05%塩化カリウム、0.05%硫酸マグネシウム、0.001%硫酸鉄、0.3%硝酸ナトリウム)で培養した。次いで、その培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した。また、対照試験として、実施例1で作成したpUCLTS1を用いて、麹菌ロイシン要求性変異株を形質転換した株を用いて同様に培養し、培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した(対照株)。その結果、対照株は、菌体外(培養上清)にプロテアーゼは確認できなかったのに対して、形質転換体(セルフクローニング株)はいずれも菌体外(培養上清)にプロテアーゼが生成していることが明確に確認できた。
(a) プロテアーゼの産生
上記方法で培養して得られた培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した結果を、対照株の結果と併せて図4に示す。図4に示すように、セルフクローニング株(IE-233)では、対照株にないプロテアーゼのバンドが大量に検出された。
培養上清についてプロテアーゼ活性を測定した。測定は、50mMトリス緩衝液(pH7.0)に溶解した1.25%アゾカゼイン溶液400μlと、培養上清100μlを混合し、37℃で1時間反応させた後、1mlの10%トリクロロ酢酸を加え、激しく混合した。次いで19000xgで5分間遠心分離を行い、上清の吸光度(335nm)を測定した。ブランク値として、培養上清100μlに、1mlの10%トリクロロ酢酸を加え、さらに50mMトリス緩衝液(pH7.0)に溶解した1.25%アゾカゼイン溶液400μlを加え、激しく混合した後、19000xgで5分間遠心分離を行い、上清の吸光度(335nm)を測定した値を用いた。ブランク値を差し引いた対照株の培養上清の吸光度(OD335)は、非検出であったのに対して、ブランク値を差し引いたセルフクローニング株の培養上清の吸光度(OD335)は、最大で3.42であった。
(a) プロテアーゼの産生
上記方法で培養して得られた培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した結果を、対照株の結果と併せて図5に示す。図5に示すように、セルフクローニング株(IE-292)では、対照株にないプロテアーゼのバンドが大量に検出された。
培養上清についてプロテアーゼ活性を測定した。測定は、50mMトリス緩衝液(pH7.0)に代えて50mM酢酸緩衝液(pH5.0)を使用する以外は、上記セルフクローニング株(IE-233)と同様に行った。その結果、ブランク値を差し引いた対照株の培養上清の吸光度(OD335)は、非検出であったのに対して、ブランク値を差し引いたセルフクローニング株の培養上清の吸光度(OD335)は、最大で1.22であった。
(a) プロテアーゼの産生
上記方法で培養して得られた培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した結果を図6に示す。図6に示すように、セルフクローニング株(IE-338)ではバンドが大量に検出された。一方、対照株では対応するバンドは検出されなかった(結果示さず)。
培養上清についてプロテアーゼ活性を測定した。測定は、キッコーマン醸造分析キットである「酸性カルボキシペプチダーゼ測定キット」を用いて行った。この原理は、基質のカルボベンゾキシ-L-チロシルL-アラニンが酸性カルボキシペプチダーゼによって分解されると、L-アラニンを生じる。生成したL-アラニンは、NAD+の存在下でアラニンデヒドロゲナーゼを添加することによって特異的に分解され、NADHが生成する。そこで生成したNADHをテトラゾリウム塩、PMSで発色させ、吸光度(460nm)で定量するというものである。
(a) プロテアーゼの産生
上記方法で培養して得られた培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した結果を図6に示す。図6に示すように、セルフクローニング株(IE-339)ではバンドが大量に検出された。一方、対照株では対応するバンドは検出されなかった(結果示さず)。
培養上清100μlについてプロテアーゼ活性を測定した。測定は、キッコーマン醸造分析キットである「酸性カルボキシペプチダーゼ測定キット」を用いて、セルフクローニング株(IE-338)と同様にして行った。ブランク値を差し引いた対照株の培養上清の吸光度(OD460)は、非検出であったのに対して、ブランク値を差し引いたセルフクローニング株(IE-339)の吸光度(OD460)は、最大で0.89であった。
(a) プロテアーゼの産生
上記方法で培養して得られた培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した結果を図6に示す。図6に示すように、セルフクローニング株(IE-340)ではバンドが大量に検出された。一方、対照株では対応するバンドは検出されなかった(結果示さず)。
培養上清100μlについてプロテアーゼ活性を測定した。測定は、キッコーマン醸造分析キットである「酸性カルボキシペプチダーゼ測定キット」を用いて、セルフクローニング株(IE-338)と同様にして行った。ブランク値を差し引いた対照株の培養上清の吸光度(OD460)は、非検出であったのに対して、ブランク値を差し引いたセルフクローニング株(IE340)の吸光度(OD460)は、最大で0.89であった。
(a) プロテアーゼの産生
上記方法で培養して得られた培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した結果を図6に示す。図6に示すように、セルフクローニング株(IE-341)ではバンドが大量に検出された。一方、対照株では対応するバンドは検出されなかった(結果示さず)。
培養上清100μlについてプロテアーゼ活性を測定した。測定は、キッコーマン醸造分析キットである「酸性カルボキシペプチダーゼ測定キット」を用いて、セルフクローニング株(IE-338)と同様にして行った。ブランク値を差し引いた対照株の培養上清の吸光度(OD460)は、非検出であったのに対して、ブランク値を差し引いたセルフクローニング株(IE341)の吸光度(OD460)は、最大で0.76であった。
麹菌に由来するプロテアーゼ3種類(プロテアーゼ8〜10)を、麹菌を用いたセルフクローニング法によって発現させ産生させた(glaAプロモーター(配列番号30)使用)。
表4に示す3種類のプロテアーゼの遺伝子を、麹菌(Aspergillus oryzae)ゲノムDNAを鋳型に、同表に示す各2本のプライマーを用いて、次のPCR条件で増幅して調製した。得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いて目的断片を切り出し、プロテアーゼ遺伝子とした。
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(2分)を30サイクル
・72℃(7分)を1サイクル。
(2-1)プラスミドベクター
まずpUCLTA1を鋳型として、プライマー3(配列番号25)とプライマー8(配列番号32)を用いて下記条件でPCRを行い、得られたPCR増幅産物についてアガロースゲル電気泳動を行い、QIAquick Gel Extraction kit(QIAGEN)を用いて目的断片を切り出し、これをベクターとした。
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(9分)を30サイクル
・72℃ (7分)を1サイクル。
上記3種類のプラスミド(IE326、IE327、IE328)をそれぞれ鋳型として、プライマー1(配列番号22)とプライマー4(プライマー26)を用いて、下記条件でPCRを行って、麹菌の遺伝子部分のみ増幅した。得られたPCR増幅産物を、QIAquick Gel Extraction kit(QIAGEN)を用いて精製し、これをセルフクローニングベクター(IE-326、IE-327、IE-328)とした。
・96℃(5分)を1サイクル
・96℃(20秒)、60℃(30秒)、72℃(9分)を30サイクル
・72℃(7分)を1サイクル。
定法であるPEG−カルシウム法に従って、上記で調製した各セルフクローニングベクターを用いてロイシン要求性変異麹菌株(Aspergillus oryzae leu-5(寄託番号:FERM P-20079))を形質転換した。これを、硝酸を単一窒素源とするツアペクドックス(Czapek-Dox)培地で培養し、この培地で生育できる菌株を選択することにより、各セルフクローニングベクターにつきそれぞれ10個の形質転換体を取得した。これらすべての形質転換体についてサザン解析を行い、いずれもが大腸菌由来の遺伝子が含まれないセルフクローニング株であることを確認した。
上記で調製した各形質転換体を、ポテトデキストロース培地で胞子形成させ、30℃で3日間、GPY液体培地(2%グルコース、1%ペプトン、0.5%酵母エキス、0.1%リン酸1水素2カリウム、0.05%塩化カリウム、0.05%硫酸マグネシウム、0.001%硫酸鉄、0.3%硝酸ナトリウム)で培養した。次いで、その培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した。また、対照試験として、実施例1で作成したpUCLTS1を用いて、麹菌ロイシン要求性変異株を形質転換した株を用いて同様に培養し、培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した(対照株)。その結果、対照株は、菌体外(培養上清)にプロテアーゼは確認できなかったのに対して、形質転換体(セルフクローニング株)はいずれも菌体外(培養上清)にプロテアーゼが生成していることが明確に確認できた。
(a) プロテアーゼの産生
上記方法で培養して得られた培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した結果を、対照株の結果と併せて図7に示す。図7に示すように、セルフクローニング株(IE-326)では、対照株にないプロテアーゼのバンドが大量に検出された。
培養上清についてプロテアーゼ活性を測定した。測定は、50mM酢酸緩衝液(pH3.0)に溶解した1.25%アゾカゼイン溶液400μlと、培養上清100μlを混合し、37℃で1時間反応させた後、1mlの10%トリクロロ酢酸を加え、激しく混合した。次いで19000xgで5分間遠心分離を行い、上清の吸光度(335nm)を測定した。ブランク値として、培養上清100μlに、1mlの10%トリクロロ酢酸を加え、さらに50mM酢酸緩衝液(pH3.0)に溶解した1.25%アゾカゼイン溶液400μlを加え、激しく混合した後、19000xgで5分間遠心分離を行い、上清の吸光度(335nm)を測定した値を用いた。ブランク値を差し引いた対照株の培養上清の吸光度(OD335)は、非検出であったのに対して、ブランク値を差し引いたセルフクローニング株(IE-326)の培養上清の吸光度(OD335)は、最大で0.84であった。
(a) プロテアーゼの産生
上記方法で培養して得られた培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した結果を、対照株の結果と併せて図7に示す。図7に示すように、セルフクローニング株(IE-327)では、対照株にないプロテアーゼのバンドが大量に検出された。
培養上清についてプロテアーゼ活性を測定した。測定は、50mM酢酸緩衝液(pH3.0)に代えて50mMトリス緩衝液(pH7.0)を使用する以外は、上記セルフクローニング株(IE-326)と同様に行った。その結果、ブランク値を差し引いた対照株の培養上清の吸光度(OD335)は、非検出であったのに対して、ブランク値を差し引いたセルフクローニング株(IE-327)の培養上清の吸光度(OD335)は、最大で0.45であった。
(a) プロテアーゼの産生
上記方法で培養して得られた培養上清100μlを濃縮乾燥させて、SDS−PAGEに供した結果を図7に示す。図7に示すように、セルフクローニング株(IE-328)では、対照株にないプロテアーゼのバンドが大量に検出された。
培養上清についてプロテアーゼ活性を測定した。測定は、上記セルフクローニング株(IE-327)と同様に、50mM酢酸緩衝液(pH3.0)に代えて50mMトリス緩衝液(pH7.0)を使用する以外は、上記セルフクローニング株(IE-326)と同様の方法で行った。その結果、ブランク値を差し引いた対照株の培養上清の吸光度(OD335)は、非検出であったのに対して、ブランク値を差し引いたセルフクローニング株(IE-328)の培養上清の吸光度(OD335)は、最大で0.63であった。
配列番号24は、ターミネーターの塩基配列、配列番号27はsodMプロモーターの塩基配列、および配列番号30はglaAプロモーターの塩基配列をそれぞれ示す。
Claims (5)
- 糸状菌体内で機能する選択マーカー遺伝子配列、糸状菌体内で機能するプロモーター配列、配列番号1〜10に記載される塩基配列からなる群より選択されるいずれか一つの塩基配列からなるプロテアーゼ遺伝子、および糸状菌体内で機能するターミネーター配列を有する糸状菌用発現ベクター。
- 実質的に糸状菌に由来するヌクレオチドだけからなるセルフクローニングベクターである、請求項1に記載する糸状菌用発現ベクター。
- 直鎖状であることを特徴とする、請求項1または2に記載する糸状菌用発現ベクター。
- 請求項1〜3のいずれかに記載する糸状菌用発現ベクターで形質転換されてなる糸状菌。
- 請求項4に記載する形質転換された糸状菌を培地で培養し、当該培地から配列番号11〜20に記載されるアミノ酸配列からなる群より選択されるいずれかの一つのアミノ酸配列からなるプロテアーゼを回収する工程を含む、プロテアーゼの生産方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008165094A JP5344857B2 (ja) | 2008-06-24 | 2008-06-24 | 糸状菌プロテアーゼの生産方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008165094A JP5344857B2 (ja) | 2008-06-24 | 2008-06-24 | 糸状菌プロテアーゼの生産方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2010004760A true JP2010004760A (ja) | 2010-01-14 |
| JP2010004760A5 JP2010004760A5 (ja) | 2011-02-24 |
| JP5344857B2 JP5344857B2 (ja) | 2013-11-20 |
Family
ID=41585965
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008165094A Expired - Fee Related JP5344857B2 (ja) | 2008-06-24 | 2008-06-24 | 糸状菌プロテアーゼの生産方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5344857B2 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012065552A (ja) * | 2010-09-21 | 2012-04-05 | Gekkeikan Sake Co Ltd | 糸状菌ペプチダーゼの生産方法 |
| WO2012110564A1 (en) * | 2011-02-16 | 2012-08-23 | Novozymes A/S | Detergent compositions comprising m7 or m35 metalloproteases |
| CN103476915A (zh) * | 2011-02-16 | 2013-12-25 | 诺维信公司 | 包含金属蛋白酶的去污剂组合物 |
| WO2017142080A1 (ja) * | 2016-02-18 | 2017-08-24 | 天野エンザイム株式会社 | 腸内菌叢改善剤 |
| WO2019181961A1 (ja) | 2018-03-20 | 2019-09-26 | 三菱商事ライフサイエンス株式会社 | β―NMNの製造方法およびその含有組成物 |
| US11123410B2 (en) | 2016-02-18 | 2021-09-21 | Amanzo Enzyme Inc. | Intestinal flora improvement agent |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0394690A (ja) * | 1989-09-08 | 1991-04-19 | Shokuhin Sangyo Kouso Kinou Henkan Gijutsu Kenkyu Kumiai | アルカリプロテアーゼプロモーター、同ターミネーター及びこれらからなる遺伝子発現用ユニット、並びにアルカリプロテアーゼのゲノム遺伝子 |
| JPH08275780A (ja) * | 1995-04-07 | 1996-10-22 | Meiji Milk Prod Co Ltd | β−ガラクトシダーゼ遺伝子 |
| WO1997022705A1 (en) * | 1995-12-15 | 1997-06-26 | Novo Nordisk A/S | A FUNGUNS WHEREIN THE areA, pepC AND/OR pepE GENES HAVE BEEN INACTIVATED |
| JP2000245465A (ja) * | 1999-02-25 | 2000-09-12 | Gekkeikan Sake Co Ltd | 高発現ベクタープラスミド用dna断片 |
| JP2004329143A (ja) * | 2003-05-09 | 2004-11-25 | Gekkeikan Sake Co Ltd | アスペルギルス属糸状菌由来の環状ヌクレオチド、アスペルギルス属糸状菌セルフクローニング株の製造方法及びセルフクローニング株 |
| JP2006345817A (ja) * | 2005-06-20 | 2006-12-28 | Gekkeikan Sake Co Ltd | アスペルギルス・オリザの新規変異株及び選択マーカー |
| JP2007174962A (ja) * | 2005-12-27 | 2007-07-12 | Bio Energy Kk | 糸状菌においてタンパク質を分泌生産する方法 |
| JP2008109861A (ja) * | 2006-10-27 | 2008-05-15 | Kirin Holdings Co Ltd | 発酵麦芽飲料製造用麦汁の製造方法 |
| JP2008516592A (ja) * | 2004-10-15 | 2008-05-22 | ディーエスエム アイピー アセッツ ビー.ブイ. | 真核細胞における化合物の生成方法 |
-
2008
- 2008-06-24 JP JP2008165094A patent/JP5344857B2/ja not_active Expired - Fee Related
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0394690A (ja) * | 1989-09-08 | 1991-04-19 | Shokuhin Sangyo Kouso Kinou Henkan Gijutsu Kenkyu Kumiai | アルカリプロテアーゼプロモーター、同ターミネーター及びこれらからなる遺伝子発現用ユニット、並びにアルカリプロテアーゼのゲノム遺伝子 |
| JPH08275780A (ja) * | 1995-04-07 | 1996-10-22 | Meiji Milk Prod Co Ltd | β−ガラクトシダーゼ遺伝子 |
| WO1997022705A1 (en) * | 1995-12-15 | 1997-06-26 | Novo Nordisk A/S | A FUNGUNS WHEREIN THE areA, pepC AND/OR pepE GENES HAVE BEEN INACTIVATED |
| JP2000245465A (ja) * | 1999-02-25 | 2000-09-12 | Gekkeikan Sake Co Ltd | 高発現ベクタープラスミド用dna断片 |
| JP2004329143A (ja) * | 2003-05-09 | 2004-11-25 | Gekkeikan Sake Co Ltd | アスペルギルス属糸状菌由来の環状ヌクレオチド、アスペルギルス属糸状菌セルフクローニング株の製造方法及びセルフクローニング株 |
| JP2008516592A (ja) * | 2004-10-15 | 2008-05-22 | ディーエスエム アイピー アセッツ ビー.ブイ. | 真核細胞における化合物の生成方法 |
| JP2006345817A (ja) * | 2005-06-20 | 2006-12-28 | Gekkeikan Sake Co Ltd | アスペルギルス・オリザの新規変異株及び選択マーカー |
| JP2007174962A (ja) * | 2005-12-27 | 2007-07-12 | Bio Energy Kk | 糸状菌においてタンパク質を分泌生産する方法 |
| JP2008109861A (ja) * | 2006-10-27 | 2008-05-15 | Kirin Holdings Co Ltd | 発酵麦芽飲料製造用麦汁の製造方法 |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012065552A (ja) * | 2010-09-21 | 2012-04-05 | Gekkeikan Sake Co Ltd | 糸状菌ペプチダーゼの生産方法 |
| WO2012110564A1 (en) * | 2011-02-16 | 2012-08-23 | Novozymes A/S | Detergent compositions comprising m7 or m35 metalloproteases |
| CN103476915A (zh) * | 2011-02-16 | 2013-12-25 | 诺维信公司 | 包含金属蛋白酶的去污剂组合物 |
| WO2017142080A1 (ja) * | 2016-02-18 | 2017-08-24 | 天野エンザイム株式会社 | 腸内菌叢改善剤 |
| JPWO2017142080A1 (ja) * | 2016-02-18 | 2018-12-20 | 天野エンザイム株式会社 | 腸内菌叢改善剤 |
| US11123410B2 (en) | 2016-02-18 | 2021-09-21 | Amanzo Enzyme Inc. | Intestinal flora improvement agent |
| US11167016B2 (en) | 2016-02-18 | 2021-11-09 | Amanoenzyme Inc. | Intestinal flora improvement agent |
| JP7072151B2 (ja) | 2016-02-18 | 2022-05-20 | 天野エンザイム株式会社 | 腸内菌叢改善剤 |
| US11833192B2 (en) | 2016-02-18 | 2023-12-05 | Amano Enzyme Inc. | Method for improving intestinal flora |
| WO2019181961A1 (ja) | 2018-03-20 | 2019-09-26 | 三菱商事ライフサイエンス株式会社 | β―NMNの製造方法およびその含有組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5344857B2 (ja) | 2013-11-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9512409B2 (en) | Thermostable catalase | |
| JP5344857B2 (ja) | 糸状菌プロテアーゼの生産方法 | |
| Arasaratnam et al. | Supplementation of whey with glucose and different nitrogen sources for lactic acid production by Lactobacillus delbrueckii | |
| Lin et al. | High-level expression and characterization of the thermostable leucine aminopeptidase Thelap from the thermophilic fungus Thermomyces lanuginosus in Aspergillus niger and its application in soy protein hydrolysis | |
| Matsushita‐Morita et al. | Characterization of recombinant prolyl aminopeptidase from Aspergillus oryzae | |
| CN116855387A (zh) | 一种高产菌体蛋白文氏镰刀真菌突变株及其应用 | |
| JP5297656B2 (ja) | 新規枯草菌変異株及びタンパク質の製造方法 | |
| JPWO1997000944A1 (ja) | Pf1022物質を産生する形質転換体、及び糸状菌綱に属する菌の形質転換方法 | |
| CN109666657B (zh) | 提升耐热性的葡萄糖氧化酶 | |
| JP6599583B1 (ja) | 多重遺伝子破壊アスペルギルス属微生物及びその製造方法 | |
| WO1996041866A1 (en) | Microbial process for producing transglutaminase | |
| JP5759132B2 (ja) | 糸状菌ペプチダーゼの生産方法 | |
| CN112639117A (zh) | 谷胱甘肽的制造方法 | |
| JP2003250588A (ja) | タンナーゼ、その遺伝子及びタンナーゼの製造法 | |
| JP2009118783A (ja) | 糸状菌タンパク質分泌生産の改善 | |
| JPH10210967A (ja) | 高活性変異株及びそれを用いる蛋白加水分解物の製造法 | |
| JP2006320294A (ja) | β−ヒドロキシアミノ酸の製造方法およびこれに用いる酵素 | |
| Sánchez et al. | Microbial synthesis of secondary metabolites and strain improvement: Current trends and future prospects | |
| JP6656919B2 (ja) | 5−オキソプロリナーゼ、5−オキソプロリナーゼ遺伝子、および5−オキソプロリナーゼの製造方法 | |
| JP2000050883A (ja) | 酵母におけるリボフラビンの過剰生産 | |
| JP3942718B2 (ja) | 酒類、食品の製造方法 | |
| JP4029927B2 (ja) | 転写因子、転写因子遺伝子、転写因子遺伝子を含む組み換えベクター、該ベクターによって形質転換された麹菌及び麹菌の使用法 | |
| JP5116270B2 (ja) | 鉄により生産が抑制される生物生産物の製造方法 | |
| JP2009296958A (ja) | 高発現プロモーター | |
| WO2015064517A1 (ja) | ギ酸脱水素酵素とその利用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100720 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110112 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110617 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130326 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130502 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20130502 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130528 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130628 |
|
| TRDD | Decision of grant or rejection written | ||
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20130628 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130723 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130813 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5344857 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |