ES2925720T3 - Un proceso de polimerización para producir polímeros a base de etileno - Google Patents
Un proceso de polimerización para producir polímeros a base de etileno Download PDFInfo
- Publication number
- ES2925720T3 ES2925720T3 ES16778178T ES16778178T ES2925720T3 ES 2925720 T3 ES2925720 T3 ES 2925720T3 ES 16778178 T ES16778178 T ES 16778178T ES 16778178 T ES16778178 T ES 16778178T ES 2925720 T3 ES2925720 T3 ES 2925720T3
- Authority
- ES
- Spain
- Prior art keywords
- atoms
- independently
- hydrocarbyl
- mmol
- carbazolyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 title claims abstract description 40
- 238000006116 polymerization reaction Methods 0.000 title claims abstract description 37
- 239000005977 Ethylene Substances 0.000 title claims abstract description 35
- 229920000642 polymer Polymers 0.000 title claims description 29
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 22
- 125000003118 aryl group Chemical group 0.000 claims abstract description 19
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 16
- 239000004711 α-olefin Substances 0.000 claims abstract description 14
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 13
- 125000001153 fluoro group Chemical group F* 0.000 claims abstract description 11
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 5
- -1 perfluoro Chemical group 0.000 claims description 101
- 239000003446 ligand Substances 0.000 claims description 93
- 125000001183 hydrocarbyl group Chemical group 0.000 claims description 63
- 125000004432 carbon atom Chemical group C* 0.000 claims description 47
- 125000004429 atom Chemical group 0.000 claims description 46
- 239000003054 catalyst Substances 0.000 claims description 42
- 125000000743 hydrocarbylene group Chemical group 0.000 claims description 30
- 229910020008 S(O) Inorganic materials 0.000 claims description 29
- 125000005842 heteroatom Chemical group 0.000 claims description 27
- 230000007935 neutral effect Effects 0.000 claims description 27
- 230000000052 comparative effect Effects 0.000 claims description 17
- 229910052760 oxygen Inorganic materials 0.000 claims description 15
- 229910052717 sulfur Inorganic materials 0.000 claims description 15
- 229910052799 carbon Inorganic materials 0.000 claims description 14
- 229910052739 hydrogen Inorganic materials 0.000 claims description 12
- 239000001257 hydrogen Substances 0.000 claims description 12
- 125000005843 halogen group Chemical group 0.000 claims description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 8
- 229910052735 hafnium Chemical group 0.000 claims description 8
- VBJZVLUMGGDVMO-UHFFFAOYSA-N hafnium atom Chemical group [Hf] VBJZVLUMGGDVMO-UHFFFAOYSA-N 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 8
- 230000003647 oxidation Effects 0.000 claims description 8
- 238000007254 oxidation reaction Methods 0.000 claims description 8
- 230000009257 reactivity Effects 0.000 claims description 8
- 125000006413 ring segment Chemical group 0.000 claims description 8
- 125000001424 substituent group Chemical group 0.000 claims description 8
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical group [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 claims description 7
- 229910052726 zirconium Inorganic materials 0.000 claims description 7
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical group [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 claims description 6
- 238000006467 substitution reaction Methods 0.000 claims description 6
- 239000010936 titanium Substances 0.000 claims description 6
- 229910052719 titanium Inorganic materials 0.000 claims description 6
- 229910052698 phosphorus Inorganic materials 0.000 claims description 5
- 125000006702 (C1-C18) alkyl group Chemical group 0.000 claims description 4
- 230000000379 polymerizing effect Effects 0.000 claims description 2
- 229910052731 fluorine Inorganic materials 0.000 abstract description 4
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 abstract 1
- 239000011737 fluorine Substances 0.000 abstract 1
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 119
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 108
- 238000006243 chemical reaction Methods 0.000 description 61
- 239000000243 solution Substances 0.000 description 60
- 239000000203 mixture Substances 0.000 description 51
- 239000007787 solid Substances 0.000 description 50
- 238000002360 preparation method Methods 0.000 description 44
- UHOVQNZJYSORNB-MZWXYZOWSA-N benzene-d6 Chemical compound [2H]C1=C([2H])C([2H])=C([2H])C([2H])=C1[2H] UHOVQNZJYSORNB-MZWXYZOWSA-N 0.000 description 40
- 239000000047 product Substances 0.000 description 40
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 39
- 238000003756 stirring Methods 0.000 description 39
- 238000005481 NMR spectroscopy Methods 0.000 description 38
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 38
- 238000005160 1H NMR spectroscopy Methods 0.000 description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 32
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 28
- 239000002904 solvent Substances 0.000 description 28
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 27
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 27
- 239000012267 brine Substances 0.000 description 25
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 25
- 238000000034 method Methods 0.000 description 24
- 238000004009 13C{1H}-NMR spectroscopy Methods 0.000 description 23
- 239000000725 suspension Substances 0.000 description 20
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 17
- 239000004215 Carbon black (E152) Substances 0.000 description 16
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 16
- 230000003213 activating effect Effects 0.000 description 16
- 229930195733 hydrocarbon Natural products 0.000 description 16
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- 239000000463 material Substances 0.000 description 15
- 239000000706 filtrate Substances 0.000 description 14
- 239000011541 reaction mixture Substances 0.000 description 14
- 125000002947 alkylene group Chemical group 0.000 description 13
- 239000010410 layer Substances 0.000 description 13
- 239000003039 volatile agent Substances 0.000 description 13
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 12
- 229910052681 coesite Inorganic materials 0.000 description 12
- 150000001875 compounds Chemical class 0.000 description 12
- 229910052906 cristobalite Inorganic materials 0.000 description 12
- 235000019439 ethyl acetate Nutrition 0.000 description 12
- 239000000178 monomer Substances 0.000 description 12
- 239000012044 organic layer Substances 0.000 description 12
- 239000000377 silicon dioxide Substances 0.000 description 12
- 235000012239 silicon dioxide Nutrition 0.000 description 12
- 229910052682 stishovite Inorganic materials 0.000 description 12
- 229910052905 tridymite Inorganic materials 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- 238000010528 free radical solution polymerization reaction Methods 0.000 description 11
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 11
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 11
- 239000000843 powder Substances 0.000 description 11
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 10
- 125000002993 cycloalkylene group Chemical group 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 10
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 10
- 239000012071 phase Substances 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 150000003254 radicals Chemical class 0.000 description 10
- 238000002390 rotary evaporation Methods 0.000 description 10
- 125000004122 cyclic group Chemical group 0.000 description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 9
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 9
- 229920006395 saturated elastomer Polymers 0.000 description 9
- KWKAKUADMBZCLK-UHFFFAOYSA-N 1-octene Chemical compound CCCCCCC=C KWKAKUADMBZCLK-UHFFFAOYSA-N 0.000 description 8
- 229910004298 SiO 2 Inorganic materials 0.000 description 8
- 230000009977 dual effect Effects 0.000 description 8
- DLYUQMMRRRQYAE-UHFFFAOYSA-N tetraphosphorus decaoxide Chemical compound O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 8
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 8
- 239000004698 Polyethylene Substances 0.000 description 7
- 150000001721 carbon Chemical group 0.000 description 7
- 239000000460 chlorine Substances 0.000 description 7
- 125000000753 cycloalkyl group Chemical group 0.000 description 7
- 238000003818 flash chromatography Methods 0.000 description 7
- 239000012634 fragment Substances 0.000 description 7
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- 229920000573 polyethylene Polymers 0.000 description 7
- 125000006657 (C1-C10) hydrocarbyl group Chemical group 0.000 description 6
- 125000006659 (C1-C20) hydrocarbyl group Chemical group 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 239000002841 Lewis acid Substances 0.000 description 6
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 229910052782 aluminium Inorganic materials 0.000 description 6
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 6
- 229910052801 chlorine Inorganic materials 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 6
- 150000007517 lewis acids Chemical class 0.000 description 6
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 6
- 235000019341 magnesium sulphate Nutrition 0.000 description 6
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- OBAJXDYVZBHCGT-UHFFFAOYSA-N tris(pentafluorophenyl)borane Chemical compound FC1=C(F)C(F)=C(F)C(F)=C1B(C=1C(=C(F)C(F)=C(F)C=1F)F)C1=C(F)C(F)=C(F)C(F)=C1F OBAJXDYVZBHCGT-UHFFFAOYSA-N 0.000 description 6
- HUHXLHLWASNVDB-UHFFFAOYSA-N 2-(oxan-2-yloxy)oxane Chemical compound O1CCCCC1OC1OCCCC1 HUHXLHLWASNVDB-UHFFFAOYSA-N 0.000 description 5
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 5
- 229910007926 ZrCl Inorganic materials 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 150000001336 alkenes Chemical class 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 125000000524 functional group Chemical group 0.000 description 5
- 125000004474 heteroalkylene group Chemical group 0.000 description 5
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 5
- 229910052751 metal Inorganic materials 0.000 description 5
- 239000002184 metal Substances 0.000 description 5
- CPOFMOWDMVWCLF-UHFFFAOYSA-N methyl(oxo)alumane Chemical compound C[Al]=O CPOFMOWDMVWCLF-UHFFFAOYSA-N 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 5
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 4
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 4
- 238000012644 addition polymerization Methods 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 238000007334 copolymerization reaction Methods 0.000 description 4
- 239000013058 crude material Substances 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- 239000012065 filter cake Substances 0.000 description 4
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 4
- 229910021482 group 13 metal Inorganic materials 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- IVSZLXZYQVIEFR-UHFFFAOYSA-N m-xylene Chemical group CC1=CC=CC(C)=C1 IVSZLXZYQVIEFR-UHFFFAOYSA-N 0.000 description 4
- 150000002736 metal compounds Chemical class 0.000 description 4
- TVMXDCGIABBOFY-UHFFFAOYSA-N n-Octanol Natural products CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 4
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 4
- 125000003367 polycyclic group Chemical group 0.000 description 4
- 239000002685 polymerization catalyst Substances 0.000 description 4
- 235000015320 potassium carbonate Nutrition 0.000 description 4
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 238000004809 thin layer chromatography Methods 0.000 description 4
- VOITXYVAKOUIBA-UHFFFAOYSA-N triethylaluminium Chemical compound CC[Al](CC)CC VOITXYVAKOUIBA-UHFFFAOYSA-N 0.000 description 4
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 3
- PBKONEOXTCPAFI-UHFFFAOYSA-N 1,2,4-trichlorobenzene Chemical compound ClC1=CC=C(Cl)C(Cl)=C1 PBKONEOXTCPAFI-UHFFFAOYSA-N 0.000 description 3
- KKYXVYJJSMTMHP-UHFFFAOYSA-N 2-bromo-3,4,5-trifluorophenol Chemical compound OC1=CC(F)=C(F)C(F)=C1Br KKYXVYJJSMTMHP-UHFFFAOYSA-N 0.000 description 3
- MRWWWZLJWNIEEJ-UHFFFAOYSA-N 4,4,5,5-tetramethyl-2-propan-2-yloxy-1,3,2-dioxaborolane Chemical compound CC(C)OB1OC(C)(C)C(C)(C)O1 MRWWWZLJWNIEEJ-UHFFFAOYSA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 3
- 239000004743 Polypropylene Substances 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- 239000007983 Tris buffer Substances 0.000 description 3
- 239000012190 activator Substances 0.000 description 3
- 125000002015 acyclic group Chemical group 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 229910000085 borane Inorganic materials 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 238000005227 gel permeation chromatography Methods 0.000 description 3
- 150000004820 halides Chemical class 0.000 description 3
- 125000004404 heteroalkyl group Chemical group 0.000 description 3
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 239000012535 impurity Substances 0.000 description 3
- 125000000593 indol-1-yl group Chemical group [H]C1=C([H])C([H])=C2N([*])C([H])=C([H])C2=C1[H] 0.000 description 3
- 150000007527 lewis bases Chemical group 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 239000012452 mother liquor Substances 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000001624 naphthyl group Chemical group 0.000 description 3
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 3
- 229920000098 polyolefin Polymers 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- 235000011152 sodium sulphate Nutrition 0.000 description 3
- 239000011343 solid material Substances 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 239000003760 tallow Substances 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 3
- QXALIERKYGCHHA-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl)borane Chemical compound BC1=C(F)C(F)=C(F)C(F)=C1F QXALIERKYGCHHA-UHFFFAOYSA-N 0.000 description 2
- 125000003837 (C1-C20) alkyl group Chemical group 0.000 description 2
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 description 2
- 125000006651 (C3-C20) cycloalkyl group Chemical group 0.000 description 2
- 125000006736 (C6-C20) aryl group Chemical group 0.000 description 2
- ULTHEAFYOOPTTB-UHFFFAOYSA-N 1,4-dibromobutane Chemical compound BrCCCCBr ULTHEAFYOOPTTB-UHFFFAOYSA-N 0.000 description 2
- QTLLJEKQHSHZBV-UHFFFAOYSA-N 2-[2-iodo-4-(2,4,4-trimethylpentan-2-yl)phenoxy]oxane Chemical compound IC1=CC(C(C)(C)CC(C)(C)C)=CC=C1OC1OCCCC1 QTLLJEKQHSHZBV-UHFFFAOYSA-N 0.000 description 2
- BINAYPRCRGPNHS-UHFFFAOYSA-N 2-iodo-4-(2,4,4-trimethylpentan-2-yl)phenol Chemical compound CC(C)(C)CC(C)(C)C1=CC=C(O)C(I)=C1 BINAYPRCRGPNHS-UHFFFAOYSA-N 0.000 description 2
- OYFFSPILVQLRQA-UHFFFAOYSA-N 3,6-ditert-butyl-9h-carbazole Chemical compound C1=C(C(C)(C)C)C=C2C3=CC(C(C)(C)C)=CC=C3NC2=C1 OYFFSPILVQLRQA-UHFFFAOYSA-N 0.000 description 2
- LZPWAYBEOJRFAX-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2$l^{2}-dioxaborolane Chemical compound CC1(C)O[B]OC1(C)C LZPWAYBEOJRFAX-UHFFFAOYSA-N 0.000 description 2
- YMQPKONILWWJQG-UHFFFAOYSA-N 4-bromo-1,2-difluorobenzene Chemical compound FC1=CC=C(Br)C=C1F YMQPKONILWWJQG-UHFFFAOYSA-N 0.000 description 2
- ISAVYTVYFVQUDY-UHFFFAOYSA-N 4-tert-Octylphenol Chemical compound CC(C)(C)CC(C)(C)C1=CC=C(O)C=C1 ISAVYTVYFVQUDY-UHFFFAOYSA-N 0.000 description 2
- ZIRVQSRSPDUEOJ-UHFFFAOYSA-N 9-bromoanthracene Chemical compound C1=CC=C2C(Br)=C(C=CC=C3)C3=CC2=C1 ZIRVQSRSPDUEOJ-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 2
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 2
- OLTRLWKSSDBMGW-UHFFFAOYSA-N CC1(OB(OC1(C)C)C=1C(=C(C=C(C=1)C(C)(CC(C)(C)C)C)C1=CC(=CC(=C1)C1=CC=CC=C1)C1=CC=CC=C1)OC1OCCCC1)C Chemical compound CC1(OB(OC1(C)C)C=1C(=C(C=C(C=1)C(C)(CC(C)(C)C)C)C1=CC(=CC(=C1)C1=CC=CC=C1)C1=CC=CC=C1)OC1OCCCC1)C OLTRLWKSSDBMGW-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- 239000007818 Grignard reagent Substances 0.000 description 2
- 239000002879 Lewis base Substances 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- 229910019093 NaOCl Inorganic materials 0.000 description 2
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- CKUAXEQHGKSLHN-UHFFFAOYSA-N [C].[N] Chemical compound [C].[N] CKUAXEQHGKSLHN-UHFFFAOYSA-N 0.000 description 2
- JXBAVRIYDKLCOE-UHFFFAOYSA-N [C].[P] Chemical compound [C].[P] JXBAVRIYDKLCOE-UHFFFAOYSA-N 0.000 description 2
- HMDDXIMCDZRSNE-UHFFFAOYSA-N [C].[Si] Chemical compound [C].[Si] HMDDXIMCDZRSNE-UHFFFAOYSA-N 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- 150000001499 aryl bromides Chemical class 0.000 description 2
- 125000000732 arylene group Chemical group 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 150000001639 boron compounds Chemical class 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 2
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 238000000113 differential scanning calorimetry Methods 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 230000004907 flux Effects 0.000 description 2
- 150000004795 grignard reagents Chemical class 0.000 description 2
- 150000004678 hydrides Chemical class 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 2
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- NSPJNIDYTSSIIY-UHFFFAOYSA-N methoxy(methoxymethoxy)methane Chemical compound COCOCOC NSPJNIDYTSSIIY-UHFFFAOYSA-N 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 125000000538 pentafluorophenyl group Chemical group FC1=C(F)C(F)=C(*)C(F)=C1F 0.000 description 2
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Natural products C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 2
- YNPNZTXNASCQKK-UHFFFAOYSA-N phenanthrene Chemical compound C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 2
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 2
- 235000009518 sodium iodide Nutrition 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- NBRKLOOSMBRFMH-UHFFFAOYSA-N tert-butyl chloride Chemical compound CC(C)(C)Cl NBRKLOOSMBRFMH-UHFFFAOYSA-N 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 2
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 2
- 235000019798 tripotassium phosphate Nutrition 0.000 description 2
- XCPQUQHBVVXMRQ-UHFFFAOYSA-N (-)-7,7-Dimethyl-2-methylenebicyclo[2.2.1]heptane Chemical compound C1CC2C(=C)CC1C2(C)C XCPQUQHBVVXMRQ-UHFFFAOYSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000006747 (C2-C10) heterocycloalkyl group Chemical group 0.000 description 1
- 125000006652 (C3-C12) cycloalkyl group Chemical group 0.000 description 1
- 125000003626 1,2,4-triazol-1-yl group Chemical group [*]N1N=C([H])N=C1[H] 0.000 description 1
- DYEQHQNRKZJUCT-UHFFFAOYSA-N 1,2-dimethylidenecyclohexane Chemical compound C=C1CCCCC1=C DYEQHQNRKZJUCT-UHFFFAOYSA-N 0.000 description 1
- 125000004509 1,3,4-oxadiazol-2-yl group Chemical group O1C(=NN=C1)* 0.000 description 1
- 125000004521 1,3,4-thiadiazol-2-yl group Chemical group S1C(=NN=C1)* 0.000 description 1
- VEFLKXRACNJHOV-UHFFFAOYSA-N 1,3-dibromopropane Chemical compound BrCCCBr VEFLKXRACNJHOV-UHFFFAOYSA-N 0.000 description 1
- CTDOPUVVTOMREJ-UHFFFAOYSA-N 1,3-dimethylidenecyclohexane Chemical compound C=C1CCCC(=C)C1 CTDOPUVVTOMREJ-UHFFFAOYSA-N 0.000 description 1
- AFKMSJURYYBPGF-UHFFFAOYSA-N 1-(methoxymethoxy)-4-(2,4,4-trimethylpentan-2-yl)benzene Chemical compound COCOC1=CC=C(C(C)(C)CC(C)(C)C)C=C1 AFKMSJURYYBPGF-UHFFFAOYSA-N 0.000 description 1
- 125000004173 1-benzimidazolyl group Chemical group [H]C1=NC2=C([H])C([H])=C([H])C([H])=C2N1* 0.000 description 1
- MUUAQFJJUGVBGB-UHFFFAOYSA-N 1-bromo-2,3,4-trifluorobenzene Chemical compound FC1=CC=C(Br)C(F)=C1F MUUAQFJJUGVBGB-UHFFFAOYSA-N 0.000 description 1
- IOPQERQQZZREDR-UHFFFAOYSA-N 1-bromo-3,5-diphenylbenzene Chemical compound C=1C(Br)=CC(C=2C=CC=CC=2)=CC=1C1=CC=CC=C1 IOPQERQQZZREDR-UHFFFAOYSA-N 0.000 description 1
- QDFKKJYEIFBEFC-UHFFFAOYSA-N 1-bromo-3-fluorobenzene Chemical compound FC1=CC=CC(Br)=C1 QDFKKJYEIFBEFC-UHFFFAOYSA-N 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- IJGSULQFKYOYEU-UHFFFAOYSA-N 2,3,4-trifluorophenol Chemical compound OC1=CC=C(F)C(F)=C1F IJGSULQFKYOYEU-UHFFFAOYSA-N 0.000 description 1
- WEOIMVGRKQBCKD-UHFFFAOYSA-N 2,3-dimethylidenebicyclo[2.2.2]octane Chemical compound C1CC2CCC1C(=C)C2=C WEOIMVGRKQBCKD-UHFFFAOYSA-N 0.000 description 1
- OSBFCGMTUOCRNH-UHFFFAOYSA-N 2-(2,3,4-trifluorophenyl)-4-(2,4,4-trimethylpentan-2-yl)phenol Chemical compound FC1=C(C=CC(=C1F)F)C=1C(=CC=C(C=1)C(C)(CC(C)(C)C)C)O OSBFCGMTUOCRNH-UHFFFAOYSA-N 0.000 description 1
- NMXLXQGHBSPIDR-UHFFFAOYSA-N 2-(2-methylpropyl)oxaluminane Chemical compound CC(C)C[Al]1CCCCO1 NMXLXQGHBSPIDR-UHFFFAOYSA-N 0.000 description 1
- SAPJEJJHYDWQBR-UHFFFAOYSA-N 2-[2-(3,5-diphenylphenyl)-4-(2,4,4-trimethylpentan-2-yl)phenoxy]oxane Chemical compound C1(=CC=CC=C1)C=1C=C(C=C(C=1)C1=C(C=CC(=C1)C(C)(CC(C)(C)C)C)OC1OCCCC1)C1=CC=CC=C1 SAPJEJJHYDWQBR-UHFFFAOYSA-N 0.000 description 1
- FOSMIDNVYAHVJE-UHFFFAOYSA-N 2-[4-(2,4,4-trimethylpentan-2-yl)phenoxy]oxane Chemical compound CC(C)(C)CC(C)(C)C1=CC=C(OC2CCCCO2)C=C1 FOSMIDNVYAHVJE-UHFFFAOYSA-N 0.000 description 1
- AQBHOUJTUWPBJS-UHFFFAOYSA-N 2-bromo-1-[3-(2-bromo-3,4,5-trifluorophenoxy)propoxy]-3,4,5-trifluorobenzene Chemical compound BrC1=C(OCCCOC2=C(C(=C(C(=C2)F)F)F)Br)C=C(C(=C1F)F)F AQBHOUJTUWPBJS-UHFFFAOYSA-N 0.000 description 1
- SKDCOCNNWBLAMY-UHFFFAOYSA-N 2-bromo-1-[4-(2-bromo-3,4,5-trifluorophenoxy)butoxy]-3,4,5-trifluorobenzene Chemical compound BrC1=C(OCCCCOC2=C(C(=C(C(=C2)F)F)F)Br)C=C(C(=C1F)F)F SKDCOCNNWBLAMY-UHFFFAOYSA-N 0.000 description 1
- FCYZOOHWUOEAOX-UHFFFAOYSA-N 2-bromo-4,5-difluorophenol Chemical compound OC1=CC(F)=C(F)C=C1Br FCYZOOHWUOEAOX-UHFFFAOYSA-N 0.000 description 1
- MEYRABVEYCFHHB-UHFFFAOYSA-N 2-bromo-4-fluorophenol Chemical compound OC1=CC=C(F)C=C1Br MEYRABVEYCFHHB-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- AQZWEFBJYQSQEH-UHFFFAOYSA-N 2-methyloxaluminane Chemical compound C[Al]1CCCCO1 AQZWEFBJYQSQEH-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- VDEDIYDCJTUMNX-UHFFFAOYSA-N 3,6-ditert-butyl-9-[2-(oxan-2-yloxy)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5-(2,4,4-trimethylpentan-2-yl)phenyl]carbazole Chemical compound C1CCCOC1OC=1C(N2C3=CC=C(C=C3C3=CC(=CC=C32)C(C)(C)C)C(C)(C)C)=CC(C(C)(C)CC(C)(C)C)=CC=1B1OC(C)(C)C(C)(C)O1 VDEDIYDCJTUMNX-UHFFFAOYSA-N 0.000 description 1
- ABMRGRMWIYEOLT-UHFFFAOYSA-N 3,6-ditert-butyl-9-[2-(oxan-2-yloxy)-5-(2,4,4-trimethylpentan-2-yl)phenyl]carbazole Chemical compound C12=CC=C(C(C)(C)C)C=C2C2=CC(C(C)(C)C)=CC=C2N1C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OC1CCCCO1 ABMRGRMWIYEOLT-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- AFECMSFINAAEGN-UHFFFAOYSA-N 4-fluoro-1-[4-(4-fluoro-2-iodophenoxy)butoxy]-2-iodobenzene Chemical compound FC1=CC(=C(OCCCCOC2=C(C=C(C=C2)F)I)C=C1)I AFECMSFINAAEGN-UHFFFAOYSA-N 0.000 description 1
- FTTVHYAABUMDNX-UHFFFAOYSA-N 4-fluoro-2-iodophenol Chemical compound OC1=CC=C(F)C=C1I FTTVHYAABUMDNX-UHFFFAOYSA-N 0.000 description 1
- 125000004920 4-methyl-2-pentyl group Chemical group CC(CC(C)*)C 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- VXEHRIAYBTWNGI-UHFFFAOYSA-N 9-[2-(methoxymethoxy)-5-(2,4,4-trimethylpentan-2-yl)phenyl]anthracene Chemical compound COCOC1=C(C=C(C=C1)C(C)(CC(C)(C)C)C)C=1C2=CC=CC=C2C=C2C=CC=CC=12 VXEHRIAYBTWNGI-UHFFFAOYSA-N 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 239000007848 Bronsted acid Substances 0.000 description 1
- MBJRUELITDCEGQ-UHFFFAOYSA-N C1=CC=CC2=CC3=CC=CC=C3C(=C12)C=1C(=C(C=C(C=1)C(C)(CC(C)(C)C)C)B1OC(C(O1)(C)C)(C)C)OCOC Chemical compound C1=CC=CC2=CC3=CC=CC=C3C(=C12)C=1C(=C(C=C(C=1)C(C)(CC(C)(C)C)C)B1OC(C(O1)(C)C)(C)C)OCOC MBJRUELITDCEGQ-UHFFFAOYSA-N 0.000 description 1
- YZTKZBVSPLVTKK-UHFFFAOYSA-N CC(C)(C)CC(C)(C)C1=CC(B2OC(C)(C)C(C)(C)O2)=C(OC2CCCCO2)C=C1 Chemical compound CC(C)(C)CC(C)(C)C1=CC(B2OC(C)(C)C(C)(C)O2)=C(OC2CCCCO2)C=C1 YZTKZBVSPLVTKK-UHFFFAOYSA-N 0.000 description 1
- HOSDDHCEVXRYKZ-UHFFFAOYSA-N COCOC1=C(C=C(C=C1)C(C)(CC(C)(C)C)C)B1OC(C(O1)(C)C)(C)C Chemical compound COCOC1=C(C=C(C=C1)C(C)(CC(C)(C)C)C)B1OC(C(O1)(C)C)(C)C HOSDDHCEVXRYKZ-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical class S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 239000006057 Non-nutritive feed additive Substances 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 239000005708 Sodium hypochlorite Substances 0.000 description 1
- 229910052775 Thulium Inorganic materials 0.000 description 1
- 239000012963 UV stabilizer Substances 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 125000004559 acridin-9-yl group Chemical group C1=CC=CC2=NC3=CC=CC=C3C(=C12)* 0.000 description 1
- 238000007605 air drying Methods 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000005234 alkyl aluminium group Chemical group 0.000 description 1
- 125000005037 alkyl phenyl group Chemical group 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 239000002216 antistatic agent Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 238000006254 arylation reaction Methods 0.000 description 1
- 125000004266 aziridin-1-yl group Chemical group [H]C1([H])N(*)C1([H])[H] 0.000 description 1
- 150000005347 biaryls Chemical group 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- GPRLTFBKWDERLU-UHFFFAOYSA-N bicyclo[2.2.2]octane Chemical compound C1CC2CCC1CC2 GPRLTFBKWDERLU-UHFFFAOYSA-N 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical group C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 1
- 239000007844 bleaching agent Substances 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 238000006758 bulk electrolysis reaction Methods 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000001716 carbazoles Chemical class 0.000 description 1
- 125000005517 carbenium group Chemical group 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000007806 chemical reaction intermediate Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- JZPDBTOWHLZQFC-UHFFFAOYSA-N chloro-di(propan-2-yl)phosphane Chemical compound CC(C)P(Cl)C(C)C JZPDBTOWHLZQFC-UHFFFAOYSA-N 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 150000001993 dienes Chemical class 0.000 description 1
- 125000004639 dihydroindenyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- NKDDWNXOKDWJAK-UHFFFAOYSA-N dimethoxymethane Chemical compound COCOC NKDDWNXOKDWJAK-UHFFFAOYSA-N 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- HQQADJVZYDDRJT-UHFFFAOYSA-N ethene;prop-1-ene Chemical group C=C.CC=C HQQADJVZYDDRJT-UHFFFAOYSA-N 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 238000012685 gas phase polymerization Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- PDPJQWYGJJBYLF-UHFFFAOYSA-J hafnium tetrachloride Chemical compound Cl[Hf](Cl)(Cl)Cl PDPJQWYGJJBYLF-UHFFFAOYSA-J 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 125000003037 imidazol-2-yl group Chemical group [H]N1C([*])=NC([H])=C1[H] 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000001282 iso-butane Substances 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004254 isoquinolin-1-yl group Chemical group [H]C1=C([H])C2=C([H])C([H])=C([H])C([H])=C2C(*)=N1 0.000 description 1
- 125000004501 isothiazol-5-yl group Chemical group S1N=CC=C1* 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- KVKFRMCSXWQSNT-UHFFFAOYSA-N n,n'-dimethylethane-1,2-diamine Chemical compound CNCCNC KVKFRMCSXWQSNT-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- VFLWKHBYVIUAMP-UHFFFAOYSA-N n-methyl-n-octadecyloctadecan-1-amine Chemical compound CCCCCCCCCCCCCCCCCCN(C)CCCCCCCCCCCCCCCCCC VFLWKHBYVIUAMP-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003145 oxazol-4-yl group Chemical group O1C=NC(=C1)* 0.000 description 1
- 125000004274 oxetan-2-yl group Chemical group [H]C1([H])OC([H])(*)C1([H])[H] 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- GTCCGKPBSJZVRZ-UHFFFAOYSA-N pentane-2,4-diol Chemical compound CC(O)CC(C)O GTCCGKPBSJZVRZ-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 229920013716 polyethylene resin Polymers 0.000 description 1
- 208000003383 pontocerebellar hypoplasia type 3 Diseases 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 239000012041 precatalyst Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 1
- 125000004353 pyrazol-1-yl group Chemical group [H]C1=NN(*)C([H])=C1[H] 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 125000004159 quinolin-2-yl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C([H])C(*)=NC2=C1[H] 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000004819 silanols Chemical class 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- SCABQASLNUQUKD-UHFFFAOYSA-N silylium Chemical class [SiH3+] SCABQASLNUQUKD-UHFFFAOYSA-N 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 125000005717 substituted cycloalkylene group Chemical group 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 125000004523 tetrazol-1-yl group Chemical group N1(N=NN=C1)* 0.000 description 1
- 125000004299 tetrazol-5-yl group Chemical group [H]N1N=NC(*)=N1 0.000 description 1
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- WYXIGTJNYDDFFH-UHFFFAOYSA-Q triazanium;borate Chemical compound [NH4+].[NH4+].[NH4+].[O-]B([O-])[O-] WYXIGTJNYDDFFH-UHFFFAOYSA-Q 0.000 description 1
- 229940062627 tribasic potassium phosphate Drugs 0.000 description 1
- NVLRFXKSQQPKAD-UHFFFAOYSA-N tricarbon Chemical group [C]=C=[C] NVLRFXKSQQPKAD-UHFFFAOYSA-N 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 238000007738 vacuum evaporation Methods 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 1
- DUNKXUFBGCUVQW-UHFFFAOYSA-J zirconium tetrachloride Chemical compound Cl[Zr](Cl)(Cl)Cl DUNKXUFBGCUVQW-UHFFFAOYSA-J 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/16—Copolymers of ethene with alpha-alkenes, e.g. EP rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/14—Monomers containing five or more carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/64003—Titanium, zirconium, hafnium or compounds thereof the metallic compound containing a multidentate ligand, i.e. a ligand capable of donating two or more pairs of electrons to form a coordinate or ionic bond
- C08F4/64168—Tetra- or multi-dentate ligand
- C08F4/64186—Dianionic ligand
- C08F4/64193—OOOO
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2500/00—Characteristics or properties of obtained polyolefins; Use thereof
- C08F2500/01—High molecular weight, e.g. >800,000 Da.
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2500/00—Characteristics or properties of obtained polyolefins; Use thereof
- C08F2500/03—Narrow molecular weight distribution, i.e. Mw/Mn < 3
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2500/00—Characteristics or properties of obtained polyolefins; Use thereof
- C08F2500/08—Low density, i.e. < 0.91 g/cm3
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2500/00—Characteristics or properties of obtained polyolefins; Use thereof
- C08F2500/12—Melt flow index or melt flow ratio
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65908—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with an ionising compound other than alumoxane, e.g. (C6F5)4B-X+
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65912—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with an organoaluminium compound
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Crystallography & Structural Chemistry (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
La descripción proporciona un procatalizador para la polimerización de etileno y, opcionalmente, una o más alfa-olefinas que tienen la estructura que se muestra en la fórmula (I) a continuación: en la que al menos dos de Y1-Y3 y al menos dos de Y4-Y6 son átomos de flúor y cuando solo dos de Y1-Y3 y solo dos de Y4-Y6 son átomos de flúor, los Y1-Y6 que no son de flúor se seleccionan del grupo que consiste en un átomo de H, grupos alquilo, grupos arilo, grupos heteroarilo y grupos alcoxi. También se proporciona un proceso de polimerización usando el procatalizador. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Un proceso de polimerización para producir polímeros a base de etileno
Campo de invención
La descripción se refiere a un procatalizador para la polimerización de etileno y, opcionalmente, una o más alfa-olefinas (a-olefinas) y un proceso de polimerización que lo utiliza.
Antecedentes de la invención
Los polímeros a base de olefinas, tales como el polietileno y/o el polipropileno, se producen a través de varios sistemas catalíticos. La selección de dichos sistemas catalíticos usados en el proceso de polimerización de los polímeros a base de olefinas es un factor importante que contribuye a las características y propiedades de dichos polímeros a base de olefinas.
El polietileno es conocido por su uso en la fabricación de una amplia variedad de artículos. El proceso de polimerización de polietileno se puede variar en varios aspectos para producir una amplia variedad de resinas de polietileno resultantes con diferentes propiedades físicas que hacen que las diversas resinas sean adecuadas para su uso en diferentes aplicaciones. En general, se sabe que el polietileno se puede producir en uno o más reactores convencionales, por ejemplo reactores de bucle, reactores esféricos, reactores isotérmicos, reactores de lecho fluidizado de fase gaseosa, reactores de tanque agitado, reactores discontinuos, en paralelo, en serie y/o cualquier combinación de los mismos. En los reactores de fase de disolución, los monómeros de etileno y, opcionalmente, uno o más comonómeros y el sistema catalítico están presentes en un diluyente líquido, tal como un alcano o isoalcano, por ejemplo, isobutano.
El Documento de Patente de Número WO2012/027448 se refiere a un proceso que copolimeriza dos o más olefinas polimerizables y a un catalizador que comprende un complejo metal-ligando (precatalizador).
El Documento de Patente de Número WO2014/209927 se refiere a oligómeros a base de etileno hiperramificados. A pesar de los esfuerzos de investigación en el desarrollo de sistemas catalíticos adecuados para las poliolefinas, tales como la polimerización de polietileno y/o polipropileno, todavía existe la necesidad de un procatalizador y un sistema catalítico que muestre una respuesta mejorada de a-olefina (es decir, una relación de reactividad más baja).
Sumario de la invención
En una realización, la presente descripción proporciona un procatalizador para la polimerización de etileno y, opcionalmente, una o más alfa-olefinas con la estructura que se muestra en la fórmula (I) dada a continuación:

en donde:
M es titanio, zirconio o hafnio, estando cada uno independientemente en un estado de oxidación formal de 2, 3 o 4; y n es un número entero de desde 0 a 3, y en donde cuando n es 0, X está ausente; y
cada X es independientemente un ligando monodentado que es neutro, monoaniónico o dianiónico; o se juntan dos X para formar un ligando bidentado que es neutro, monoaniónico o dianiónico; y X y n se eligen de tal manera que el complejo metal-ligando de la fórmula (I) sea globalmente neutro; y
cada Z es independientemente O, S, N hidrocarbilo de (C1-C40), o P hidrocarbilo de (C1-C40);
L es hidrocarbileno de (C2-C40) o heterohidrocarbileno de (C2-C40), en donde el hidrocarbileno de (C2-C40) tiene una parte que comprende una cadena principal conectora de 2 átomos de carbono a 10 átomos de carbono que une los átomos Z en la fórmula (I) (a la que está unido L) y el heterohidrocarbileno de (C2-C40) tiene una parte que comprende una cadena principal conectora de 3 átomos a 10 átomos que une los átomos Z en la fórmula
(I), en donde cada uno de los 3 a 10 átomos de la cadena principal conectora de 3 átomos a 10 átomos del heterohidrocarbileno de (C2-C40) es independientemente un átomo de carbono o heteroátomo, en donde cada heteroátomo es independientemente O, S, S(O), S(O)2, Si(RC)2, Ge(RC)2, P(RP), o N(RN), en donde cada RC se selecciona independientemente del grupo que consiste en hidrocarbilo de (C1-C40), cada RP es hidrocarbilo de (C1-C40); y cada RN es hidrocarbilo de (C1-C40) o está ausente; y
R1-10 se seleccionan cada uno independientemente del grupo que consiste en un hidrocarbilo de (C1-C40), heterohidrocarbilo de (C1-C40), Si(RC)3 , Ge(RC)3, P(Rp)2, N(Rn)2, ORc , SRc , NO2, CN, C3F, RCS(O)-, RCS(O)2-, (Rc)2C=N-, Rc C(O)O-, Rc o C(O)-, Rc C(O)n (R)-, (Rc)2NC(O)-, átomo de halógeno, átomo de hidrógeno y cualquier combinación de los mismos, y
al menos dos de Y1-Y3 y al menos dos de Y4-Y6 son átomos de flúor y cuando sólo dos de Y1-Y3 y solo dos de Y4-Y6 son átomos de flúor, los no fluorados Y1-Y6 se seleccionan del grupo que consiste en átomo de H, grupos alquilo, grupos arilo, grupos heteroarilo y grupos alcoxi; y
cada uno de los grupos hidrocarbilo, heterohidrocarbilo, Si(RC)3, Ge(RC)3, P(RP)2, N(RN)2, ORC, SRC , RCS(O)-, RcS(O)2-, (Rc)2C=N-, RcC(O)O-, RcOC(O)-, Rc C(O)N(R)-, (Rc)2NC(O)-, hidrocarbileno y heterohidrocarbileno no están sustituidos o están sustituidos independientemente con uno o más sustituyentes RS, cada RS es independientemente un átomo de halógeno, sustitución de polifluoro, sustitución perfluoro, alquilo de (C1- C18) no sustituido, F3C-, FCH2O-, F2HCO-, F3CO-, R3SK R3Ge-, RO-, RS-, RS(O)-, RS(O)2-, R2P-, R2N-, R2C=N-, Nc -, RC(O)O-, ROC(O)-, RC(O)N(R)-, o R2NC(O)-, o dos de los RS se juntan para formar un hidrocarbileno de (C1-C18) no sustituido en donde cada R es independientemente un hidrocarbilo de (C1- C18);
opcionalmente dos o más grupos R de los grupos R1-10 (por ejemplo, de R1-4, R5-8) se pueden combinar juntos en estructuras de anillo teniendo tales estructuras de anillo de 2 a 50 átomos en el anillo excluyendo cualquier átomo de hidrógeno; y
en donde cuando el heterohidrocarbilo de (C1-C40) es un heteroarilo de (C1-C40), este se selecciona de 2,7-di(terciario-butil)-carbazolilo, 3,6-di(terciario-butil)-carbazolilo, 2,7-di(terciario-octil)-carbazolilo, 3,6-di(terciario-octil)-carbazolilo, 2,7-difenilcarbazolilo, 2,7-bis(2,4,6-trimetilfenil)-carbazolilo o 3,6-bis(2,4,6-trimetilfenil)-carbazolilo. En otra realización, la descripción proporciona un proceso de polimerización para producir polímeros a base de etileno que comprende polimerizar etileno y, opcionalmente, una o más a-olefinas en presencia de un sistema catalítico en un sistema reactor que tiene uno o más reactores, en donde el sistema catalítico comprende uno o más procatalizadores de la invención.
Descripción detallada de la invención
En una primera realización, la descripción proporciona un procatalizador para la polimerización de etileno y, opcionalmente, una o más alfa-olefinas con la estructura que se muestra en la fórmula (I) dada a continuación:

M es titanio, zirconio o hafnio, estando cada uno independientemente en un estado de oxidación formal de 2, 3 o 4; y n es un número entero de desde 0 a 3, y cuando n es 0, X está ausente; y
cada X es independientemente un ligando monodentado que es neutro, monoaniónico o dianiónico; o se juntan dos X para formar un ligando bidentado que es neutro, monoaniónico o dianiónico; y X y n se eligen de tal manera que el complejo metal-ligando de la fórmula (I) sea globalmente neutro; y
cada Z es independientemente O, S, N hidrocarbilo de (C1-C40), o P hidrocarbilo de (C1-C40);
L es hidrocarbileno de (C2-C40) o heterohidrocarbileno de (C2-C40), en donde el hidrocarbileno de (C2-C40) tiene una parte que comprende una cadena principal conectora de 2 átomos de carbono a 10 átomos de carbono que une los átomos Z en la fórmula (I) (a la que está unido L) y el heterohidrocarbileno de (C2-C40) tiene una parte que comprende una cadena principal conectora de 3 átomos a 10 átomos que une los átomos Z en la fórmula (I), en donde cada
uno de los 3 a 10 átomos de la cadena principal conectora de 3 átomos a 10 átomos del heterohidrocarbileno de (C2-C40) es independientemente un átomo de carbono o heteroátomo, en donde cada heteroátomo es independientemente O, S, S(O), S(O)2, Si(RC)2, Ge(RC)2, P(RP), o N(RN), en donde cada RC se selecciona independientemente del grupo formado por hidrocarbilo de (C1-C40). Como se usa en la presente invención, RC incluye la situación en donde dos grupos RC se unen para formar un anillo dirradical y en donde el Si está dentro del anillo. Cada RP es hidrocarbilo de (C1-C40); y cada RN es hidrocarbilo de (C1-C40) o está ausente; y
R1-10 se seleccionan cada uno independientemente del grupo que consiste en un hidrocarbilo de (C1-C40), heterohidrocarbilo de (C1-C40), Si(RC)3, Ge(RC)3, P(Rp)2, N(Rn)2, ORc , SRc , NO2, CN, CF3, RCS(O)-, RCS(O)2-, (RC)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O)-, átomo de halógeno, átomo de hidrógeno y cualquier combinación de los mismos, y
al menos dos de Y1-Y3 y al menos dos de Y4-Y6 son átomos de flúor y cuando sólo dos de Y 1-Y3 y solo dos de Y4-Y6 son átomos de flúor, los no fluorados Y 1-Y6 se seleccionan del grupo que consiste en un átomo de H, grupos alquilo, grupos arilo, grupos heteroarilo y grupos alcoxi,
opcionalmente dos o más grupos R de los grupos R1-10 (por ejemplo, de R1-4, R5-8) se pueden combinar juntos en estructuras de anillo teniendo tales estructuras de anillo de 2 a 50 átomos en el anillo excluyendo cualquier átomo de hidrógeno; y
en donde cuando el heterohidrocarbilo de (C1-C40) es un heteroarilo de (C1-C40) se selecciona de 2,7-di(terciariobutil)-carbazolilo, 3,6-di(terciario-butil)-carbazolilo, 2,7-di(terciario-octil)-carbazolilo, 3,6-di (terciario-octil)-carbazolilo, 2,7-difenilcarbazolilo, 2,7-bis(2,4,6-trimetilfenil)-carbazolilo o 3,6-bis(2,4,6-trimetilfenil)-carbazolilo.
Como se usa en la presente invención, el término "hidrocarbilo de (Cx-Cy)" significa un radical hidrocarbonado de desde x a y átomos de carbono y el término "hidrocarbileno de (Cx-Cy)" significa un dirradical hidrocarbonado de desde x a y átomos de carbono y el término "alquilo de (Cx-Cy)" significa un grupo alquilo de desde x a y átomos de carbono y el término "cicloalquilo de (Cx-Cy)" significa un grupo cicloalquilo de desde x a y átomos de carbono.
Como se usa en la presente invención, el término "hidrocarbilo de (C1-C40)" significa un radical hidrocarbonado de desde 1 a 40 átomos de carbono y el término "hidrocarbileno de (C2-C40)" significa un dirradical hidrocarbonado de desde 2 a 40 átomos de carbono, en donde cada radical hidrocarbonado y dirradical es independientemente aromático (6 átomos de carbono o más) o no aromático, saturado o insaturado, de cadena lineal o cadena ramificada, cíclico (incluyendo mono - y policíclicos, policíclicos condensados y no condensados, incluidos los bicíclicos; 3 átomos de carbono o más) o acíclicos, o una combinación de dos o más de los mismos; y cada radical hidrocarbonado y dirradical es independientemente igual o diferente de otro hidrocarbonado radical y dirradical, respectivamente, y no está sustituido o está sustituido independientemente por uno o más RS.
Preferiblemente, un hidrocarbilo de (C1-C40) es independientemente un alquilo de (C1-C40) sustituido o no sustituido, cicloalquilo de (C3-C40), cicloalquilo de (C3-C20)-alquileno de (C1-C20), arilo de (C6-C40), o arilo de (C6-C20)-alquileno de (C1-C20). Más preferiblemente, cada uno de los mencionados anteriormente grupos hidrocarbilo de (C1-C40) tienen independientemente un máximo de 20 átomos de carbono (es decir, hidrocarbilo de (C1-C20)), y aún más preferiblemente un máximo de 12 átomos de carbono.
Los términos "alquilo de (C1-C40)" y "alquilo de (C1-C18)" significa un radical hidrocarbonado lineal o ramificado saturado de desde 1 a 40 átomos de carbono o de 1 a 18 átomos de carbono, respectivamente, que no está sustituido o está sustituido por uno o más RS. Ejemplos de alquilo de (C1-C40) no sustituido son alquilo de (C1-C20) no sustituido; alquilo de (C1-C10) no sustituido; alquilo de (C1-C5) no sustituido; metilo; etilo; 1 -propilo; 2-propilo; 1 -butilo; 2-butilo; 2-metilpropilo; 1, 1 -dimetiletilo; 1 -pentilo; 1 -hexilo; 1 -heptilo; 1 -nonilo; y 1 -decilo. Ejemplos de alquilo de (C1-C40) sustituido son alquilo de (C1-C20) sustituido, alquilo de (C1-C10) sustituido, trifluorometilo y alquilo de (C45). El alquilo de (C45) es, por ejemplo, un alquilo de (C27-C40) sustituido por un RS, que es un alquilo de (C18-C5), respectivamente. Preferiblemente, cada alquilo de (C1-C5) es independientemente metilo, trifluorometilo, etilo, 1 -propilo, 1 -metiletilo o 1, 1 -dimetiletilo.
El término "arilo de (C6-C40)" significa un radical hidrocarbonado aromático mono-, bi- o tricíclico de desde 6 a 40 átomos de carbono, de los cuales al menos de 6 a 14 de los átomos de carbono son átomos de carbono del anillo aromático, no sustituido o sustituido (por uno o más RS) y el radical mono-, bi- o tricíclico comprende 1, 2 o 3 anillos, respectivamente; en donde el 1 anillo es aromático y los 2 o 3 anillos están fusionados o no fusionados independientemente y al menos uno de los 2 o 3 anillos es aromático. Ejemplos de arilo de (C6-C40) no sustituido son arilo de (C6-C20) no sustituido; arilo de (C6-C18) no sustituido; 2-alquilo(C1-C5)-fenilo; 2,4-bisalquilo de (C1-C5)-fenilo; fenilo; fluorenilo; tetrahidrofluorenilo; indacenilo; hexahidroindacenilo; indenilo; dihidroindenilo; naftilo; tetrahidronaftilo; y fenantreno. Ejemplos de arilo de (C6-C40) sustituido son arilo de (C6-C20) sustituido; arilo de (C6-C18) sustituido; 2,4-bis[alquilo de (C20)]-fenilo; polifluorofenilo; pentafluorofenilo; y fluoren-9-ona-1-ilo.
El término "cicloalquilo de (C3-C40)" significa un radical hidrocarbonado cíclico saturado de desde 3 a 40 átomos de carbono que no está sustituido o está sustituido por uno o más RS. Otros grupos cicloalquilo (por ejemplo, cicloalquilo de (C3-C12)) se definen de manera análoga. Ejemplos de cicloalquilo de (C3-C40) no sustituido son cicloalquilo de (C3-C20) no sustituido, cicloalquilo de (C3-C10) no sustituido, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo,
ciclooctilo, ciclononilo y ciclodecilo. Ejemplos de cicloalquilo de (C3-C40) sustituido son cicloalquilo de (C3-C20) sustituido, cicloalquilo de (C3-C10) sustituido, ciclopentanon-2-ilo y 1-fluorociclohexilo.
Ejemplos de hidrocarbileno de (C1-C40) son arileno de (C6-C40) sustituido o no sustituido, cicloalquileno de (C3-C40), y alquileno de (C1-C40) (por ejemplo, alquileno de (C1-C20)). En algunas realizaciones, los dirradicales están en el mismo átomo de carbono (por ejemplo, -CH2-) o en átomos de carbono adyacentes (es decir, 1,2-dirradicales), o están separados por uno, dos o más átomos de carbono intermedios (por ejemplo, los respectivos 1,3-dirradicales, 1,4-dirradicales, etc.). Se prefiere un 1,2-, 1,3-, 1,4-, o un alfa, omega-dirradical, y más preferiblemente un 1,2-dirradical. El alfa, omega-dirradical es un dirradical que tiene un espaciamiento máximo de la cadena principal de átomos de carbono entre los carbonos radicales. Más preferida es una versión 1,2-dirradical, 1,3-dirradical o 1,4-dirradical de arileno de (C6-C18), cicloalquileno de (C3-C20), o alquileno de (C2-C20).
El término "cicloalquileno de (C3-C40)" significa un dirradical cíclico (es decir, los radicales están en los átomos del anillo) de desde 3 a 40 átomos de carbono que no está sustituido o está sustituido por uno o más RS. Ejemplos de cicloalquileno de (C3-C40) no sustituido son 1,3-ciclopropileno, 1,1-ciclopropileno y 1,2-ciclohexileno. Ejemplos de cicloalquileno de (C3-C40) sustituido son 2-oxo-1,3-ciclopropileno y 1,2-dimetil-1,2-ciclohexileno.
El término "heterohidrocarbilo de (C1-C40)" significa un radical heterohidrocarbonado de desde 1 a 40 átomos de carbono y el término "heterohidrocarbileno de (C1-C40)" significa un dirradical heterohidrocarbonado de desde 1 a 40 átomos de carbono, y cada heterohidrocarbonado tiene independientemente uno o más heteroátomos O; S; S(O); S(O)2; Si(RC)2; Ge(RC)2; P(RP); y N(Rn), en donde cada RC es independientemente hidrocarbilo de (C1-C40) no sustituido, cada RP es hidrocarbilo de (C1-C40) no sustituido; y cada RN es hidrocarbilo de (C1-C40) no sustituido o está ausente (por ejemplo, ausente cuando N comprende -N= o N sustituido con tricarbono). El radical heterohidrocarbonado y cada uno de los dirradicales heterohidrocarbonados está independientemente en un átomo de carbono o en un heteroátomo del mismo, aunque preferentemente está en un átomo de carbono cuando está unidos a un heteroátomo en la fórmula (I) o a un heteroátomo de otro heterohidrocarbilo o heterohidrocarbileno. Cada heterohidrocarbilo de (C1-C40) y heterohidrocarbileno de (C1-C40) no está sustituido o está sustituido independientemente (por uno o más RS), aromático o no aromático, saturado o insaturado, de cadena lineal o de cadena ramificada, cíclico (incluyendo mono y policíclico, policíclico condensado y no condensado) o acíclico, o una combinación de dos o más de los mismos; y cada uno es respectivamente igual o diferente de otro.
Preferiblemente, el heterohidrocarbilo de (C1-C40) es independientemente heteroalquilo de (C1-C40) sustituido o no sustituido, hidrocarbilo de (C1-C40)-O-, hidrocarbilo de (C1-C«)-S-, hidrocarbilo de (C1-C40)-S(O)-, hidrocarbilo de (C1-C40)-S(O)2-, hidrocarbilo de (C1-C40)-Si(RC)2-, hidrocarbilo de (C1-C40)-Ge(RC)2-, hidrocarbilo de (C1-C40)-N(RN)-, hidrocarbilo de (C1-C40)-P(Rp)-, heterocicloalquilo de (C2-C40), heterocicloalquil de (C2-C19)-alquileno de (C1-C20), cicloalquil de (C3-C20)-heteroalquileno de (C1-C19), heterocicloalquil de (C2-C19)-heteroalquileno de (C1-C20), heteroarilo de (C1-C40), heteroaril de (C1-C19)-alquileno de (C1-C20), aril de (C6-C20)-heteroalquileno de (C1-C19), o heteroaril de (C1-C19)- heteroalquileno de (C1-C20).
El término "heteroarilo de (C1-C40)" significa un radical hidrocarbonado heteroaromático mono-, bi- o tricíclico de desde 1 a 40 átomos de carbono totales y de desde 1 a 4 heteroátomos, no sustituido o sustituido (por uno o más RS) y el radical mono-, bi- o tricíclico comprende 1, 2 o 3 anillos, respectivamente, en donde los 2 o 3 anillos están independientemente fusionados o no fusionados y al menos uno de los 2 o 3 anillos es heteroaromático. Otros grupos heteroarilo (por ejemplo, heteroarilo de (C4-C12))) se definen en una manera análoga. El radical hidrocarbonado heteroaromático monocíclico es un anillo de 5 o 6 miembros. El anillo de 5 miembros tiene de 1 a 4 átomos de carbono y de 4 a 1 heteroátomos, respectivamente, siendo cada heteroátomo O, S, N o P, y preferiblemente O, S o N. Ejemplos de radical hidrocarbonado heteroaromático de anillo de 5 miembros son pirrol-1-ilo; pirrol-2-ilo; furan-3-ilo; tiofen-2-ilo; pirazol-1-ilo; isoxazol-2-ilo; isotiazol-5-ilo; imidazol-2-ilo; oxazol-4-ilo; tiazol-2-ilo; 1,2,4-triazol-1 -ilo; 1,3,4-oxadiazol-2-ilo; 1,3,4-tiadiazol-2-ilo; tetrazol-1-ilo; tetrazol-2-ilo; y tetrazol-5-ilo. El anillo de 6 miembros tiene 4 ó 5 átomos de carbono y 2 ó 1 heteroátomos, siendo los heteroátomos N o P, y preferiblemente N. Ejemplos de radical hidrocarbonado heteroaromático de anillo de 6 miembros son piridin-2-ilo; pirimidin-2-ilo; y pirazin-2-ilo. El radical hidrocarbonado heteroaromático bicíclico es preferiblemente un sistema de anillos fusionados -5,6- o -6,6. Ejemplos del radical hidrocarbonado heteroaromático bicíclico del sistema de anillos fusionados -5,6 son indol-1-ilo; y bencimidazol-1-ilo. Ejemplos del radical hidrocarbonado heteroaromático bicíclico del sistema de anillos fusionados -6.6 son quinolin-2-ilo; e isoquinolin-1-ilo. El radical hidrocarbonado heteroaromático tricíclico es preferiblemente un sistema de anillos fusionados -5,6,5; -5,6,6; -6,5,6; o -6,6,6. Un ejemplo del sistema de anillos fusionados -5,6,5 es 1,7-dihidropirrolo[3,2-í]indol-1-ilo. Un ejemplo del sistema de anillos fusionados -5,6,6 es 1H-benzo[í]indol-1-ilo. Un ejemplo del sistema de anillos fusionados -6,5,6 es 9H-carbazol-9-ilo. Un ejemplo del sistema de anillos fusionados -6.6.6 es acridin-9-ilo.
En algunas realizaciones, el heteroarilo de (C1-C40) es carbazolilo 2,7-disustituido o carbazolilo 3,6-disustituido o carbazoles no sustituidos, más preferiblemente en donde cada RS es independientemente fenilo, metilo, etilo, isopropilo o butilo terciario, aún más preferiblemente 2,7-di(terc-butil)-carbazolilo, 3,6-di(terc-butil)-carbazolilo, 2,7-di(terc-octil)-carbazolilo, 3,6-di(terc-octilo)-carbazolilo, 2,7-difenilcarbazolilo, 2,7-bis(2,4,6-trimetilfenil)-carbazolilo o 3,6-bis( 2,4,6-trimetilfenil)-carbazolilo.
Los grupos heteroalquilo y heteroalquileno antes mencionados son radicales saturados o dirradicales de cadena lineal
o ramificada, respectivamente, que contienen átomos de carbono (C1-C40), o menos átomos de carbono según sea el caso, y uno o más de los heteroátomos Si(RC)2, Ge(RC)2, P(RP), N(RN), N, O, S, S(O) y S(O)2 como se definió anteriormente, en donde cada uno de los grupos heteroalquilo y heteroalquileno está independientemente sustituido o no sustituido por uno o más RS.
Ejemplos de heterocicloalquilo de (C2-C40) no sustituido son heterocicloalquilo de (C2-C40) no sustituido , heterocicloalquilo de (C2-C10) no sustituido, aziridin-1-ilo, oxetan-2-ilo, tetrahidrofuran-3-ilo, pirrolidin-1-ilo, tetrahidrotiofeno-S,S-dióxido-2-ilo, morfolin-4-ilo, 1,4-dioxano-2-ilo, hexahidroazepin-4-ilo, 3-oxa-ciclooctilo, 5-tiociclononilo y 2-aza-ciclodecilo.
El término “átomo de halógeno” significa radical de átomo de flúor (F), átomo de cloro (Cl), átomo de bromo (Br) o átomo de yodo (I). Preferiblemente cada átomo de halógeno es independientemente el radical del Br, F o Cl, y más preferiblemente el radical del F o Cl. El término ''haluro'' significa anión fluoruro (F-), cloruro (Cl-), bromuro (Br-), o yoduro (I-).
A menos que se indique lo contrario en la presente invención, el término "heteroátomo" significa O, S, S(O), S(O)2, Si(RC)2, Ge(RC)2, P(RP), o N(RN), en donde cada RC es independientemente hidrocarbilo de (C1-C40) no sustituido, cada RP es hidrocarbilo de (C1-C40) no sustituido; y cada RN es hidrocarbilo de (C1-C40) no sustituido o está ausente (ausente cuando N comprende -N=). Preferiblemente, no hay enlaces O-O, S-S u O-S, distintos de los enlaces O-S en un grupo funcional dirradical S(O) o S(O)2, en el complejo metal-ligando de la fórmula (I). Más preferiblemente, no hay enlaces O-O, N-N, P-P, N-P, S-S u O-S, distintos de los enlaces O-S en un grupo funcional dirradical S(O) o S(O)2, en el complejo metal-ligando de la fórmula (I).
Preferiblemente, no hay enlaces O-O, S-S u O-S, distintos de los enlaces O-S en un grupo funcional dirradical S(O) o S(O)2, en el complejo metal-ligando de la fórmula (I). Más preferiblemente, no hay enlaces O-O, N-N, P-P, N-P, S-S u O-S, distintos de los enlaces O-S en un grupo funcional dirradical S(O) o S(O)2, en el complejo metal-ligando de la fórmula (I).
El término "saturado" significa que carece de enlaces dobles carbono-carbono, enlaces triples carbono-carbono y enlaces dobles o triples carbono-nitrógeno, carbono-fósforo y carbono-silicio (en grupos que contienen heteroátomos). Cuando un grupo químico saturado está sustituido por uno o más sustituyentes RS, uno o más enlaces dobles y/o triples pueden estar presentes o no opcionalmente en los sustituyentes RS. El término "insaturado" significa que contiene uno o más dobles enlaces carbono-carbono, triples enlaces carbono-carbono y dobles o triples enlaces carbono-nitrógeno, carbono-fósforo y carbono-silicio (en grupos que contienen heteroátomos), sin incluir cualquier doble enlace que pueda estar presente en los sustituyentes RS, si los hay, o en anillos (hetero)aromáticos, si los hay.
M es titanio, circonio o hafnio. En una realización, M es circonio o hafnio, y en otra realización M es hafnio. En algunas realizaciones, M está en un estado de oxidación formal de 2, 3 o 4. En algunas realizaciones, n es 0, 1, 2 ó 3. Cada X es independientemente un ligando monodentado que es neutro, monoaniónico o dianiónico; o se juntan dos X para formar un ligando bidentado que es neutro, monoaniónico o dianiónico. X y n se eligen de tal manera que el complejo metal-ligando de la fórmula (I) sea, globalmente, neutro. En algunas realizaciones, cada X es independientemente el ligando monodentado. En una realización, cuando hay dos o más ligandos monodentados X, cada X es el mismo. En algunas realizaciones, el ligando monodentado es el ligando monoaniónico. El ligando monoaniónico tiene un estado de oxidación formal neto de -1. Cada ligando monoaniónico puede ser independientemente hidruro, carbanión de hidrocarbilo de (C1-C40), carbanión de heterohidrocarbilo de (C1-C40), haluro, nitrato, carbonato, fosfato, sulfato, HC(O)O-, hidrocarbilo de (Cr C40)C(O)O-, HC(O)N(H)-, hidrocarbilo de (C1-C40)C(O)N(H)-, hidrocarbilo de (C1-C40)C(O)N(hidrocarbilo de (C1-C20))-, RKRLB-, RKRLN-, RKO-, RKS-, RKRLP-, o RMRKRLSi-, donde cada RK, RL y RM es independientemente hidrógeno, hidrocarbilo de (C1-C40), o heterohidrocarbilo de (C1-C40), o RK y RL se unen para formar un hidrocarbileno de (C2-C40) o heterohidrocarbileno de (C1-C40) y RM es como se define anteriormente.
En algunas realizaciones, al menos un ligando monodentado de X es independientemente el ligando neutro. En una realización, el ligando neutro es un grupo base de Lewis neutro que es RXNRKRL, RKOL, RKRSL, o RXPRKRL, en donde cada RX es independientemente hidrógeno, hidrocarbilo de (C1-C40), [hidrocarbilo de (C1-C10)]3Si, [hidrocarbilo de (C1-C10)]aSi hidrocarbilo de (C1-C10), o heterohidrocarbilo de (C1-C40) y cada RK y RL es independientemente como se define anteriormente.
En algunas realizaciones, cada X es un ligando monodentado que es independientemente un átomo de halógeno, hidrocarbilo de (C1-C20) no sustituido, hidrocarbilo (C1-C20) no sustituido C(O)O-, o RKRLN- en donde cada uno de RK y RL es independientemente un hidrocarbilo de (C1-C20) no sustituido. En algunas realizaciones, cada ligando monodentado X es un átomo de cloro, hidrocarbilo de (C1-C10) (por ejemplo, alquilo de (C1-C6) o bencilo), hidrocarbilo de (C1-C10) no sustituido C(O)O-, o RKRLN- en donde cada uno de RK y RL es independientemente un hidrocarbilo de (C1-C10) no sustituido.
En algunas realizaciones hay al menos dos X y los dos X se juntan para formar el ligando bidentado. En algunas realizaciones, el ligando bidentado es un ligando bidentado neutro. En una realización, el ligando bidentado neutro es un dieno de la fórmula (RD)2C=C(RD)-C(RD)=C(RD)2, en donde cada RD es independientemente H, alquilo de (C1-C6)
no sustituido, fenilo o naftilo. En algunas realizaciones, el ligando bidentado es un ligando monoaniónico-mono (base de Lewis). El ligando monoaniónico-mono (base de Lewis) puede ser un 1,3-dionato de la fórmula (D): RE-CO-)=CH-C(=O)-RE (D), donde cada RD es independientemente H, alquilo de (C1-C6) no sustituido, fenilo o naftilo. En algunas realizaciones, el ligando bidentado es un ligando dianiónico. El ligando dianiónico tiene un estado de oxidación formal neto de -2. En una realización, cada ligando dianiónico es independientemente carbonato, oxalato (es decir, -O2CC(O)O-), dicarbanión de hidrocarbileno de (C2-C40) , dicarbanión de heterohidrocarbileno de (C1-C40), fosfato o sulfato.
Como se mencionó anteriormente, el número y la carga (neutro, monoaniónico, dianiónico) de X se seleccionan dependiendo del estado de oxidación formal de M, de manera que el complejo metal-ligando de la fórmula (I) es, en general, neutro.
En algunas realizaciones, cada X es el mismo, en donde cada X es metilo; etilo; 1 -propilo; 2-propilo; 1 -butilo; 2,2,-dimetilpropilo; trimetilsililmetilo; fenilo; bencilo; o cloro. En algunas realizaciones, n es 2 y cada X es el mismo.
En algunas realizaciones, al menos dos X son diferentes. En algunas realizaciones, n es 2 y cada X es uno diferente de metilo; etilo; 1 -propilo; 2-propilo; 1 -butilo; 2,2,-dimetilpropilo; trimetilsililmetilo; fenilo; bencilo; y cloro.
El número entero n indica el número de X. En una realización, n es 2 ó 3 y al menos dos X son independientemente ligandos monodentados monoaniónicos y un tercer X, si está presente, es un ligando monodentado neutro. En algunas realizaciones, n es 2 y dos X se toman juntos para formar un ligando bidentado. En algunas realizaciones, el ligando bidentado es 2,2-dimetil-2-silapropano-1,3-diilo o 1,3-butadieno.
Cada Z es independientemente O, S, N hidrocarbilo de (C1-C40), o P hidrocarbilo de (C1-C40). En algunas realizaciones, cada Z es diferente. En algunas realizaciones, un Z es O y un Z es NCH3. En algunas realizaciones, un Z es O y un Z es S. En algunas realizaciones, un Z es S y un Z es N hidrocarbilo de (C1-C40) (por ejemplo, NCH3). En algunas realizaciones, cada Z es el mismo. En algunas realizaciones cada Z es O. En algunas realizaciones cada Z es S. En algunas realizaciones cada Z es N hidrocarbilo de (C1-C40) (por ejemplo, NCH3). En algunas realizaciones al menos uno, y en algunas realizaciones cada Z es P hidrocarbilo de (C1-C40) (por ejemplo, PCH3).
L es hidrocarbileno de (C2-C40) o heterohidrocarbileno de (C2-C40), en donde el hidrocarbileno de (C2-C40) tiene una parte que comprende una cadena principal conectora de 2 átomos de carbono a 10 átomos de carbono que une los átomos Z en la fórmula (I) (a la que está unido L) y el heterohidrocarbileno de (C2-C40) tiene una parte que comprende un cadena principal conectora de 3 átomos a 10 átomos que une los átomos Z en la fórmula (I), en donde cada uno de los 3 a 10 átomos de la cadena principal conectora de 3 átomos a 10 átomos del heterohidrocarbileno de (C2-C40) es independientemente un átomo de carbono o heteroátomo, en donde cada heteroátomo es independientemente , O, S, S(O), S(O)2, Si(RC)2, Ge(RC)2, P(RP), o N(RN), en donde cada RC es independientemente hidrocarbilo de (C1-C40), cada RP es hidrocarbilo de (C1-C40); y cada Rn es hidrocarbilo de (C1-C40) o está ausente. Por ejemplo, cuando L tiene tres átomos, al menos dos de los átomos son átomos de carbono y un átomo es O o S.
En algunas realizaciones, L es el hidrocarbileno de (C3-C40). Preferiblemente, la parte antes mencionada que comprende una cadena principal conectora de 3 átomos de carbono a 10 átomos de carbono del hidrocarbileno de (C3-C40) de L comprende una cadena principal conectora de 3 carbonos de carbono a 10 átomos de carbonos, y más preferiblemente una de 3 átomos de carbono o de 4 átomos de carbono que une los átomos Z en la fórmula (I) a la que está unida L. En algunas realizaciones, L comprende la cadena principal conectora de 3 átomos de carbono (por ejemplo, L es -CH2CH2CH2-; -CH(CH3)CH2CH(CH3)-; -CH(CH3)CH(CH3)CH(CH3)-; -CH2C(CH3)2CH2-); 1,3-ciclopentano-diilo; o 1,3-ciclohexano-diilo. En algunas realizaciones, L comprende la cadena principal conectora de 4 átomos de carbono (por ejemplo, L es -CH2CH2CH2CH2-; -CH2C(CH3^C(CH3)2CH2-; 1,2-bis(metilen)ciclohexano; o 2,3-bis(metileno)-biciclo[2.2.2]octano). En algunas realizaciones, L comprende la cadena principal conectora de 5 átomos de carbono (por ejemplo, L es -CH2CH2CH2CH2CH2- o 1,3-bis(metileno)ciclohexano). En algunas realizaciones, L comprende la cadena principal conectora de 6 átomos de carbono (por ejemplo, L es -CH2CH2CH2CH2CH2CH2- o 1,2-bis(etileno)ciclohexano).
En algunas realizaciones, L es el hidrocarbileno de (C2-C40) y el hidrocarbileno de (C2-C40) de L es un hidrocarbileno de (C2-C12), y más preferiblemente hidrocarbileno de (C2-C8). En algunas realizaciones, el hidrocarbileno de (C2-C40) es un alquileno de (C2-C40) no sustituido. En algunas realizaciones, el hidrocarbileno de (C2-C40) es un alquileno de (C2-C40) sustituido. En algunas realizaciones, el hidrocarbileno de (C2-C40) es un cicloalquileno de (C2-C40) no sustituido o cicloalquileno de (C2-C40) sustituido, en donde cada sustituyente es independientemente RS, en donde preferiblemente el RS es independientemente alquilo de (C1-C4).
En algunas realizaciones, L es un alquileno de (C2-C40) no sustituido, y en algunas otras realizaciones, L es un alquileno de (C2-C40) acíclico no sustituido, y aún más preferiblemente el alquileno de (C2-C40) acíclico no sustituido es, -CH2CH2CH2-, cis-CH(CH3)CH2CH(CH3)-, trans-CH(CH3)CH2CH(CH3)-, -CH(CH3)CH2CH(CH3)2-, -CH(CH3)CH(CH3)CH(CH3)-, -CH2C(CH3)2CH2-, -CH2CH2CH2CH2-, o -CH2C(CH3)2C(CH3)2CH2-. En algunas realizaciones, L es trans-1,2-bis(metileno)ciclopentano, cis-1,2-bis(metileno)ciclopentano, trans-1,2-bis(metileno)ciclohexano, o cis-1,2-bis(metileno)ciclohexano. En algunas realizaciones, el alquileno de (C2-C40) sustituido con alquileno de (C1-C40)alquileno es exo-2,3-bis(metileno)biciclo[2.2.2]octano o exo-2,3-bis(metileno)-7,7-dimetilbiciclo[2.2.1]heptano. En algunas realizaciones, L es el cicloalquileno de (C3-C40) no sustituido, y en algunas otras
realizaciones, L es cis-1,3-ciclopentano-diilo o cis-1,3-ciclohexano-diilo. En algunas realizaciones, L es el cicloalquileno de (C2-C40) sustituido, y más preferiblemente L es un cicloalquileno de (C2-C40) sustituido con alquileno de (C1-C40), y en algunas otras realizaciones, L es el cicloalquileno de (C2-C40) sustituido con alquileno de (C1-C40) que es exobiciclo[2.2.2]octan-2,3-diilo.
En algunas realizaciones, L es el heterohidrocarbileno de (C2-C40). En algunas realizaciones, la parte antes mencionada que comprende una cadena principal conectora de 3 átomos a 6 átomos del heterohidrocarbileno de (C2-C40) de L comprende un cadena principal conectora de 3 átomos a 5 átomos y, en algunas otras realizaciones, un cadena principal conectora de 3 átomos o 4 átomos que une los átomos Z en la fórmula (I) a la que está unido L. En algunas realizaciones, L comprende la cadena principal conectora de 3 átomos (por ejemplo, L es -CH2CH2CH(OCH3)-, -CH2Si(CH3)2CH2-, o -CH2Ge(CH3)2CH2-). La "CH2Si(CH3)2CH2-" se puede denominar en la presente invención como un 1,3-dirradical del 2,2-dimetil-2-silapropano. En algunas realizaciones, L comprende la cadena principal conectora de 4 átomos (por ejemplo, L es -CH2CH2OCH2- o -CH2P(CH3)CH2CH2-). En algunas realizaciones, L comprende la cadena principal conectora de 5 átomos (por ejemplo, L es -CH2CH2OCH2CH2- o -CH2CH2N(CH3)CH2CH2-). En algunas realizaciones, L comprende la cadena principal conectora de 6 átomos (por ejemplo, L es -CH2CH2C(OCH3)2CH2CH2CH2-, -CH2CH2CH2SO)2CH2CH2-, o -CH2CH2S(O)CH2CH2CH2-)-. En algunas realizaciones, cada uno de los 3 a 6 átomos de la cadena principal conectora de 3 a 6 átomos es un átomo de carbono. En algunas realizaciones, al menos un heteroátomo es el Si(RC)2. En algunas realizaciones, al menos un heteroátomo es el O. En algunas realizaciones, al menos un heteroátomo es el N(RN). En algunas realizaciones, no hay enlaces O-O, S-S u O-S, distintos de los enlaces O-S en el grupo funcional dirradical S(O) o S(O)2, en -Z-L-Z-. En algunas otras realizaciones, no hay enlaces O-O, N-N, P-P, N-P, S-S u O-S, distintos de los enlaces O-S en un grupo funcional dirradical S(O) o S(O)2, en -Z-L-Z-. En algunas realizaciones, el heterohidrocarbileno de (C2-C40) es heterohidrocarbileno de (C2-C10), y en algunas otras realizaciones heterohidrocarbileno de(C2-C8). En algunas realizaciones, el heterohidrocarbileno de (C2-C8) de L es -CH2Si(CH3)2CH2-; -CH2CH2Si(CH3)2CH2-; o CH2Si(CH3)CH2CH2-. En algunas realizaciones, el heterohidrocarbileno de (C2-C8) de L es -CH2Si(CH3)2CH2-, -CH2Si(CH2CH3)2CH2-, -CH2Si(isopropilo)2CH2-, -CH2Si(tetrametileno)CH2-, o -CH2Si(pentametileno)CH2-. El CH2Si(tetrametileno)CH2- se denomina 1-silaciclopentan-1,1-dimetileno. El CH2Si(pentametileno)CH2- se denomina 1 -silaciclohexano-1,1-dimetileno.
En algunas realizaciones, el complejo metal-ligando de la fórmula (I) es un complejo metal-ligando de cualquiera de las siguientes fórmulas:


En algunas realizaciones, los ligandos de la invención se pueden preparar usando procedimientos conocidos. Específicamente, los ligandos de la invención se pueden preparar usando una variedad de rutas sintéticas, dependiendo de la variación deseada en el ligando. En general, se preparan bloques constituyentes que luego se unen con un grupo puente. Se pueden introducir variaciones en los sustituyentes del grupo R en la síntesis de los bloques constituyentes. Se pueden introducir variaciones en el puente con la síntesis del grupo puente. Los ligandos específicos dentro del alcance de esta invención se pueden preparar según los esquemas generales que se muestran a continuación, donde primero se preparan los bloques constituyentes y luego se acoplan entre sí. Hay varias maneras diferentes de usar estos bloques constituyentes. En una realización, generalmente, cada uno de los anillos de fenilo opcionalmente sustituidos se prepara como un bloque de constitución separado. Los fenilos opcionalmente sustituidos deseados se combinan luego en bloques constituyentes de bifenilo, que luego se unen entre sí. En otra realización, los bloques constituyentes de fenilo opcionalmente sustituidos se unen entre sí y luego se añaden bloques constituyentes de fenilo opcionalmente sustituidos adicionales para formar las estructuras de biarilo con puente. Los materiales de partida o los reactivos usados generalmente están disponibles comercialmente o se preparan mediante métodos sintéticos de rutina, como se describe en la presente invención.
En los esquemas siguientes, el término ligando se refiere al precursor orgánico del procatalizador. El procatalizador se deriva de una reacción del ligando con un precursor metálico adecuado (titanio, zirconio o hafnio). Las abreviaturas comunes se enumeran en el sistema de claves dado a continuación.
PG: grupo protector genérico, los ejemplos comunes incluyen:

éter metoximetílico (MOM) éter tetrahidropiranílico (THP)
R, L, M, Z, X: como se define anteriormente
Me: metilo; Et: etilo; Ph: fenilo; i-Pr: iso-propilo;
t-Bu: tero-butilo; t-Oct: terc-octilo;
Ts: sulfonato de tolueno; THF: tetrahidrofurano; Et2O: éter dietílico; DMF: dimetilformamida; CH2Cl2: diclorometano; MeOH: metanol; CH3CN: acetonitrilo;
EtOAc: acetato de etilo; C6D6 : benceno deuterado; CDCh : cloroformo deuterado; DIAD: azodicarboxilato de diisopropilo; PPhe: trifenilfosfina; NaI: yoduro de sodio; NaOCl: hipoclorito de sodio; Na2S2O3 : tiosulfato de sodio; NaHSO3 : bisulfito de sodio; CuI: yoduro de cobre(I); SiO2: gel de sílice; NaOH: hidróxido de sodio; K2CO3: carbonato de potasio; Na2CO3 : carbonato de sodio; K3PO4: fosfato de potasio tribásico; salmuera: disolución acuosa saturada de cloruro de sodio; Na2SO4 : sulfato de sodio; MgSO4 : sulfato de magnesio; HCl: cloruro de
hidrogeno; NH4CI sat. ac.: disolución acuosa saturada de cloruro de amonio; PTSA: ácido para-toluenosulfónico; NIS: W-yodosuccinimida; P2O5: pentóxido de fósforo; Pd(PPh3)4 : tetrakis(trifenilfosfina)paladio(0); Pd(Pf-Bu3)2 : bis(tri-íerc-butilfosfina)paladio(0); n-BuLi: n-Butil litio; HfCl4 : cloruro de hafnio (IV); ZrCl4 : cloruro de circonio (IV); N2: nitrógeno gas;
CG: cromatografía de gases; LC: cromatografía líquida; TLC: cromatografía de capa fina; RMN: resonancia magnética nuclear; mmol: milimoles; ml: mililitros; M: molar; N: normal; min: minutos; h: horas
Componente cocatalizador
El procatalizador que comprende el complejo metal-ligando de la fórmula (I) se puede volver catalíticamente activo, en algunas realizaciones, poniéndolo en contacto o combinándolo con el cocatalizador activador o usando una técnica activadora tales como las que se conocen en la técnica para su uso con reacciones de polimerización de olefinas basadas en metales. Los cocatalizadores activadores adecuados para usar en la presente invención incluyen alquilaluminios; alumoxanos poliméricos u oligoméricos (también conocidos como aluminoxanos); ácidos de Lewis neutros; y compuestos formadores de iones no poliméricos, no coordinantes (incluyendo el uso de dichos compuestos en condiciones oxidantes). Una técnica activadora adecuada es la electrólisis en masa. También se contemplan combinaciones de uno o más de los cocatalizadores y técnicas activadoras anteriores. El término “alquilaluminio” significa un dihidruro de monoalquilaluminio o dihaluro de monoalquilaluminio, un hidruro de dialquilaluminio o haluro de dialquilaluminio, o un trialquilaluminio. Los aluminoxanos y sus preparaciones se conocen, por ejemplo, en el Documento de Patente de los Estados Unidos de Número (USPN, por sus siglas en inglés) US 6.103.657. Ejemplos de alumoxanos poliméricos u oligoméricos preferidos son metilalumoxano, metilalumoxano modificado con triisobutilaluminio, e isobutilalumoxano.
Ejemplos de cocatalizadores activadores de ácido de Lewis son compuestos de metales del Grupo 13 que contienen de 1 a 3 sustituyentes hidrocarbilo como se describe en la presente invención. En algunas realizaciones, los ejemplos de compuestos de metales del Grupo 13 son compuestos de aluminio sustituido con tri(hidrocarbilo) o compuesto de boro sustituidos con tri(hidrocarbilo). En algunas otras realizaciones, los ejemplos de compuestos de metales del Grupo 13 son compuesto de aluminio sustituido con tri(hidrocarbilo) o compuestos de boro sustituidos con tri(hidrocarbilo) son tri(alquilo de (C1-C10)) aluminio o tri(arilo de (C6-C18)) boro y sus derivados halogenados (incluidos los perhalogenados). En algunas otras realizaciones, los ejemplos de compuestos de metal del Grupo 13 son tris(fenil fluoro sustituido) boranos, en otras realizaciones, tris (pentafluorofenil) borano. En algunas realizaciones, el cocatalizador activador es un tris(hidrocarbilo de (C1-C20)) borato (por ejemplo, tetrafluoroborato de tritilo) o un tri(hidrocarbilo de (C1-C20)) amonio tetra(hidrocarbilo de (C1-C20)) borano (por ejemplo, bis(octadecil) metilamonio tetrakis(pentafluorofenil) borano). Como se usa en la presente invención, el término “amonio” significa un catión de nitrógeno que es un (hidrocarbilo de (C1-C2ü))4N+, un (hidrocarbilo de (C1-C2ü))3NH)+, un (hidrocarbilo de (C1-C2ü))2NH)2+, hidrocarbilo de (C1-C2ü)N(H)3+, o N(H)4+, donde cada hidrocarbilo de (C1-C20) puede ser igual o diferente.
Ejemplos de combinaciones de cocatalizadores activadores de ácido de Lewis neutro incluyen mezclas que comprenden una combinación de un compuesto de tri(alquilo de (C1-C4)) aluminio y un compuesto de tri(arilo de (C6-C18)) boro, especialmente un tris(pentafluorofenil) borano. Otras realizaciones ejemplares son combinaciones de tales mezclas de ácidos de Lewis neutros con un alumoxano polimérico u oligomérico, y combinaciones de un solo ácido de Lewis neutro, especialmente tris(pentafluorofenil) borano con un alumoxano polimérico u oligomérico. Ejemplos de realizaciones de relaciones de los números de moles de (complejo metal-ligando):tris(pentafluorofenilborano):(alumoxano) [por ejemplo, (complejo metal-ligando del Grupo 4):(tris(pentafluorofenilborano):(alumoxano)] son de 1:1:1 a 1:10:30, otras realizaciones ejemplares son de 1:1:1,5 a 1:5:10.
Muchos cocatalizadores activadores y técnicas activadoras se han enseñado previamente con respecto a diferentes complejos metal-ligando en los siguientes Documentos de Patente de los EE.UU. de Números: US 5.064.802; US 5.153.157; US 5.296.433; US 5.321.106; US 5.350.723; US 5.425.872; US 5.625.087; US 5.721.185; US 5.783.512;. US 5.883.204; US 5.919.983; US 6.696.379; y US 7.163.907. Ejemplos de hidrocarbilóxidos adecuados se describen en el Documento de Patente de los EE.UU. de Número US 5.296.433. Ejemplos de sales de ácido de Bronsted adecuadas para catalizadores de polimerización por adición se describen en los Documentos de Patente de los EE.UU. de Números US 5.064.802; US 5.919.983; US 5.783.512. Ejemplos de sales adecuadas de un agente oxidante catiónico y un anión compatible no coordinante como cocatalizadores activadores para catalizadores de polimerización por adición se describen en el Documento de Patente de los EE.UU. de Número US 5.321.106. Ejemplos de sales de carbenio adecuadas como cocatalizadores activadores para catalizadores de polimerización por adición se describen en el Documento de Patente de los EE.UU. de Número US 5.350.723. Ejemplos de sales de sililio adecuadas como cocatalizadores activadores para catalizadores de polimerización por adición se describen en el Documento de Patente de los EE.UU. de Número US 5.625.087. Ejemplos de complejos adecuados de alcoholes, mercaptanos, silanoles y oximas con tris(pentafluorofenil) borano se describen en el Documento de Patente de los EE.UU. de Número US 5.296.433. Algunos de estos catalizadores también se describen en una parte del Documento de Patente de los EE.UU. de Número US 6.515.155 B1 comenzando en la columna 50, en la línea 39, y llega hasta la columna 56, en la línea 55, solo la parte de la cual se incorpora en la presente invención como referencia.
En algunas realizaciones, el procatalizador que comprende el complejo metal-ligando de la fórmula (I) se puede activar para formar una composición de catalizador activo mediante combinación con uno o más cocatalizadores tales como un
cocatalizador formador de cationes, un ácido de Lewis fuerte o una combinación de los mismos. Cocatalizadores adecuados para su uso incluyen aluminoxanos poliméricos u oligoméricos, especialmente metil aluminoxano, así como compuestos formadores de iones, inertes, compatibles, y no coordinantes. Cocatalizadores adecuados ejemplares incluyen, pero no se limitan a, metil aluminoxano modificado (MMAO, por sus siglas en inglés), bis(alquilo de sebo hidrogenado) metilo, tetrakis(pentafluorofenil) borato (1-) amina (RIBS-2), trietilaluminio (TEA) y cualquier combinación de los mismos.
En algunas realizaciones, se usan uno o más de los cocatalizadores activadores anteriores en combinación entre sí. Una combinación especialmente preferida es una mezcla de un tri(hidrocarbilo de (C1-C4)) aluminio, tri(hidrocarbilo de (C1 -C4)) borano, o un borato de amonio con un compuesto de alumoxano oligomérico o polimérico.
La relación del número total de moles de los complejos metal-ligando de la fórmula (I) al número total de moles de uno o más de los cocatalizadores activadores es de 1:10.000 a 100:1. En algunas realizaciones, la relación es de al menos 1:5.000, en algunas otras realizaciones, de al menos 1:1.000; y 10:1 o menos, y en algunas otras realizaciones, 1:1 o menos. Cuando se usa un alumoxano solo como cocatalizador activador, preferiblemente el número de moles del alumoxano que se emplea es al menos 100 veces el número de moles del complejo metal-ligando de la fórmula (I). Cuando se usa tris(pentafluorofenil) borano solo como cocatalizador activador, en algunas otras realizaciones, el número de moles del tris(pentafluorofenil) borano que se emplean con respecto al número total de moles de los complejos metalligando de la fórmula (I) es de 0,5:1 a 10:1, en algunas otras realizaciones, de 1:1 a 6:1, en algunas otras realizaciones, de 1:1 a 5:1. Los cocatalizadores activadores restantes se emplean generalmente en cantidades molares aproximadamente iguales a las cantidades molares totales de uno o más complejos metal-ligando de la fórmula (I).
Condiciones del proceso de polimerización
Se puede emplear cualquier proceso de polimerización convencional para llevar a cabo el proceso de la invención. Dichos procesos de polimerización convencionales incluyen, pero no se limitan a, proceso de polimerización en disolución, proceso de polimerización en fase gaseosa, proceso de polimerización en fase de suspensión y combinaciones de los mismos usando uno o más reactores convencionales, por ejemplo reactores de bucle, reactores isotérmicos, reactores de lecho fluidizado de fase gaseosa, reactores de tanque agitado, reactores discontinuos en paralelo, en serie y/o cualquier combinación de los mismos.
El proceso de la invención puede ocurrir en un proceso de polimerización en fase de disolución usando uno o más reactores de bucle, reactores isotérmicos y combinaciones de los mismos.
En general, el proceso de polimerización en fase de disolución ocurre en uno o más reactores bien agitados tales como uno o más reactores de bucle o en uno o más reactores isotérmicos esféricos a una temperatura en el intervalo de 120 a 300°C; por ejemplo, de 160 a 215°C, y a presiones en el intervalo de 2.068 kPa a 10.342 kPa (300 a 1.500 psi); por ejemplo, de 2.754 kPa a 5.171 kPa (400 a 750 psi). El tiempo de residencia en el proceso de polimerización en fase de disolución está típicamente en el intervalo de desde 2 a 30 min; por ejemplo, de 10 a 20 min. El etileno, uno o más disolventes, uno o más sistemas catalíticos, por ejemplo un sistema catalítico de la invención, opcionalmente uno o más cocatalizadores y opcionalmente uno o más comonómeros se alimentan continuamente al uno o más reactores. Disolventes ejemplares incluyen, pero no se limitan a, isoparafinas. Por ejemplo, dichos disolventes están disponibles comercialmente con el nombre ISOPAR E de ExxonMobil Chemical Co., Houston, Texas. A continuación, se retira del reactor la mezcla resultante del polímero a base de etileno y el disolvente y se aísla el polímero a base de etileno. El disolvente generalmente se recupera a través de una unidad de recuperación de disolvente, es decir, intercambiadores de calor y tambor separador de vapor líquido, y luego se recicla nuevamente al sistema de polimerización.
En una realización, el proceso de la invención ocurre en un reactor de polimerización en disolución en un sistema reactor dual, por ejemplo, un sistema de reactor de bucle dual, en donde el etileno y, opcionalmente, una o más aolefinas se polimerizan en presencia del sistema catalítico de la invención, como se describe en la presente invención, y opcionalmente uno o más cocatalizadores. En una realización, el polímero a base de etileno se puede producir mediante polimerización en disolución en un sistema reactor dual, por ejemplo, un sistema de reactor de bucle dual, en donde el etileno y, opcionalmente, una o más a-olefinas se polimerizan en presencia del sistema catalítico de la invención, como se describe en la presente invención, y opcionalmente uno o más catalizadores. El sistema catalítico de la invención, como se describe en la presente invención, se puede usar en el primer reactor o en el segundo reactor, opcionalmente en combinación con uno o más catalizadores. En una realización, el polímero a base de etileno se puede producir mediante polimerización en disolución en un sistema de reactor dual, por ejemplo, un sistema de reactor de buble dual, en donde el etileno y, opcionalmente, una o más a-olefinas se polimerizan en presencia del sistema catalítico de la invención, como se describe en la presente invención, en ambos reactores.
En otra realización, el proceso de la invención ocurre en un reactor de polimerización en disolución en un sistema de reactor único, por ejemplo, un sistema de reactor de bucle único, en donde el etileno y, opcionalmente, una o más aolefinas se polimerizan en presencia del sistema catalítico de la invención, como se describe en la presente invención, y opcionalmente uno o más cocatalizadores.
En otra realización, el proceso de la invención ocurre en una polimerización en disolución en un sistema reactor único, por ejemplo, un sistema reactor de buble único, en donde el etileno y, opcionalmente, una o más a-olefinas se
polimerizan en presencia del sistema catalítico de la invención, como se describe en la presente invención, opcionalmente uno o más catalizadores y opcionalmente uno o más cocatalizadores.
El procatalizador que comprende el complejo metal-ligando de la fórmula (I) se puede activar para formar una composición de catalizador activo mediante la combinación con uno o más cocatalizadores, como se describe anteriormente, por ejemplo, un cocatalizador formador de cationes, un ácido de Lewis fuerte o una combinación del mismo. Cocatalizadores adecuados para su uso incluyen aluminoxanos poliméricos u oligoméricos, especialmente metil aluminoxano, así como compuestos formadores de iones, inertes, compatibles, no coordinantes. Cocatalizadores adecuados ejemplares incluyen, pero no se limitan a, metil aluminoxano modificado (MMAO), bis(alquilo de sebo hidrogenado) metilo, tetrakis(pentafluorofenil) borato (1-) amina (RIBS-2), trietilaluminio (TEA) y combinaciones de los mismos.
En otra realización, el proceso de la invención puede ocurrir en un reactor de polimerización en disolución en un sistema reactor dual, por ejemplo, un sistema reactor esférico o de bucle dual, en donde el etileno y, opcionalmente, una o más a-olefinas se polimerizan en presencia de uno o más sistemas catalíticos.
En otra realización, el proceso de la invención puede ocurrir en un reactor de polimerización en disolución en un sistema de reactores múltiples en donde los reactores están conectados en serie.
En otra realización, el proceso de la invención puede ocurrir en un reactor de polimerización en disolución en un sistema de reactores múltiples en donde los reactores están conectados en paralelo.
En otra realización, el proceso de la invención puede ocurrir en un reactor de polimerización en disolución en un sistema reactor único, por ejemplo, un sistema reactor de bucle único, donde el etileno y opcionalmente una o más aolefinas se polimerizan en presencia de uno o más sistemas catalíticos.
El proceso de la invención puede ocurrir además en presencia de uno o más aditivos. Dichos aditivos incluyen, pero no se limitan a, agentes antiestáticos, potenciadores del color, tintes, lubricantes, pigmentos, antioxidantes primarios, antioxidantes secundarios, auxiliares de procesamiento, estabilizadores UV y combinaciones de los mismos.
Ejemplos
Los siguientes Ejemplos ilustran la presente invención pero no pretenden limitar el alcance de la invención.
Ejemplo Comparativo C1
C1-1. Preparación del 2-(4-(2,4,4-trimetilpentan-2-il)fenoxi)tetrahidro-2H-pirano

A una disolución agitada de 10,0 g (48,5 mmol) de 4-(2,4,4-trimetilpentan-2-il)fenol y en 24 ml de CH2G 2, a 0°C, se añadieron 0,092 g (0,48 mmol) de monohidrato de PTSA y luego se añadieron gota a gota 5,31 ml (58,2 mmol) de 3,4-dihidropirano. La disolución resultante se agitó a 0°C durante aproximadamente 30 min. Luego se añadió trietilamina (0,1 ml), y la mezcla resultante se concentró y luego se diluyó con 100 ml de Et2O, y se lavó sucesivamente con 100 ml de cada uno de NaOH 2 M, agua y salmuera. A continuación, la parte orgánica resultante se secó sobre MgSO4, se filtró y se concentró, proporcionando el producto como un sólido amarillo claro (14,05 g, rendimiento cuantitativo). RMN 1H (400 MHz, CDCta) 57,26 (d, J = 8,8 Hz, 2H), 6,96 (d, J = 8,8 Hz, 2H), 5,37 (t, J = 3,4 Hz, 1H), 3,93 (ddd, J = 11,4, 9,2, 3,1 Hz, 1H), 3,59 (dtd, J = 11,4, 4,3, 1,5 Hz, 1H), 2,00 (m, 1 H), 1,84 (m, 2H), 1,68 - 1,50 (m, 3H), 1,34 (s, 6H), 0,71 (s, 9H). RMN 13C{1H} (101 MHz, CDCta) 5154,86, 143,25, 127,07, 115,78, 96,69, 62,19, 57,15, 38,12, 32,44, 31,91,31,79, 31,75, 30,63, 25,41, 19,09.
C1 -2. Preparación del 4,4,5,5-tetrametil-2-(2-((tetrahidro-2H-piran-2-il)oxi)-5-(2,4,4-trimetilpentan-2-il)fenil)-1,3,2-dioxaborolano

Se cargó un matraz de 1 l secado al horno bajo N2 con el sustrato fenólico protegido (10,0 g, 34,4 mmol) y 344 ml de THF anhidro. La disolución resultante se enfrió a 0°C y se añadió lentamente n-BuLi (17,9 ml, disolución 2,5 M en hexano, 44,8 mmol) mediante una jeringa. La disolución resultante se dejó calentar lentamente a temperatura ambiente con agitación durante 2 h, momento en donde se añadió 2-isopropoxi-4,4,5,5-tetrametil-1,3,2-dioxaborolano (9,13 ml, 44,8 mmol) mediante jeringa. Luego, esta mezcla se agitó durante la noche a temperatura ambiente, luego se detuvo la reacción con 4 ml de H2O. Los volátiles se eliminaron bajo vacío, teniendo cuidado de no calentar la mezcla en exceso. El residuo resultante se disolvió en 200 ml de CH2Cl2, luego se lavó con salmuera (3 x 200 ml). La capa orgánica se secó sobre MgSO4, se filtró y se concentró. El producto bruto se purificó por cromatografía en columna sobre SiO2, eluyendo con un gradiente de EtOAc/hexano al 1 —>10 %. Se recogieron 8,04 g (56 % de rendimiento) de producto como un aceite claro que eventualmente solidificó. RMN 1H (400 MHz, CDCh) 57,60 (d, J = 2,6 Hz, 1H), 7,35 (dd, J = 8,7, 2,7 Hz, 1 H), 7,00 (d, J = 8,7 Hz, 1 H), 5,44 (t, J = 2,9 Hz, 1H), 4,00 (td, J = 11,1,2,8 Hz, 1H), 3,56 (m, 1 H), 2,17 (m, 1H), 1,94 (m, 1H), 1,84 (m, 1H), 1,70 (s, 2H), 1,69 - 1,57 (m, 3H), 1,35 (br s, 18H), 0,71 (s, 9H). RMN 13C{1H} (101 MHz, CDCh) 5159,48, 142,90, 133,68, 130,18, 127,07, 115,25, 96,81,83,30, 61,60, 57,08, 38,15, 32,46, 31,98, 31,79, 31,65, 30,44, 25,74, 25,11,25,00, 18,38.
C1-3. Preparación del 2-((5'-fenil-5-(2,4,4-trimetilpentan-2-il)-[1,1':3',1"-terfenil]-2-il)oxi)tetrahidro- 2H-pirano

A un matraz de fondo redondo de 100 ml que contiene el sustrato borilado (5,00 g, 12,0 mmol) bajo N2 se añadió 3,5-difenil-1-bromobenceno (3,90 g, 12,6 mmol), NaOH (2,88 g, 72,0 mmol), Pd(PPh3)4 (278 mg, 0,24 mmol), 50 ml de tolueno desgasificado y 10 ml de agua desgasificada. El sistema se burbujeó con N2 y luego se calentó a 110°C durante 24 h. La reacción se enfrió y los volátiles se eliminaron por evaporación rotatoria. El residuo se recogió en Et2O, se lavó con salmuera, se secó sobre MgSO4 anhidro, se filtró a través de un lecho de SiO2 y luego se concentró para proporcionar el material crudo. Este se purificó aún más por cromatografía en SiO2, eluyendo con hexano al 100 % seguido de EtOAc/hexano al 5—10 %. El producto se aisló como un sólido blanco (5,1 g, 81,9 % de rendimiento). RMN 1H (400 MHz, CDCh) 57,79 (m, 3H), 7,71 (m, 4H), 7,48 (m, 4H), 7,43 (d, J = 2,5 Hz, 1H), 7,39 (m, 2H), 7,32 (dd, J = 8,7, 2,5 Hz, 1H), 7,18 (d, J = 8,7 Hz, 1H), 5,43 (t, J = 2,7 Hz, 1H), 3,88 (td, J = 10,7, 10,2, 2,7 Hz, 1 H), 3,59 (m, 1H), 1,86 - 1,75 (m, 3H), 1,74 (s, 2H), 1,63 (m, 1H), 1,54 - 1,44 (m, 2H), 1,40 (s, 6H), 0,76 (s, 9H). RMN 13C{1H} (101 MHz, CDCh) 5 151,79, 143,81, 141,53, 141,35, 140,40, 130,63, 128,93, 128,89, 127,76, 127,46, 127,41, 126,58, 124,59, 115,37, 97,20, 62,17, 57,20, 38,30, 32,57, 32,02, 31,77, 30,59, 25,40, 18,76.
C1-4. Preparación del 4,4,5,5-tetrametil-2-(5'-fenil-2-((tetrahidro-2H-piran-2-il)oxi)-5-(2,4,4-trimetilpentan-2- il)-[1,1':3',1"-terfenil]-3-il)-1,3,2-dioxaborolano

El sustrato (8,05 g, 15,5 mmol) se disolvió en 155 ml de THF anhidro en un matraz de 500 ml secado en horno bajo N2 y se enfrió a 0°C. A esto se añadió n-BuLi (8,07 ml, disolución 2,5 M en hexano, 20,2 mmol) gota a gota con una jeringa. La disolución se dejó calentar lentamente a temperatura ambiente con agitación durante 2 h, momento en el que se añadió 2-isopropoxi-4,4,5,5-tetrametil-1,3,2-dioxaborolano (4,12 ml, 20,2 mmol) a través de una jeringa y la mezcla resultante se agitó durante la noche a temperatura ambiente. La reacción se detuvo con 4 ml de H2O, y se eliminaron los volátiles bajo vacío, teniendo cuidado de no calentar la mezcla en exceso. El residuo crudo se disolvió en 200 ml de CH2G 2 y se lavó con salmuera (3 x 200 ml). La capa orgánica se secó sobre MgSO4, se filtró y se concentró, proporcionando el producto (9,0 g, 90 % de rendimiento) como un polvo blanco. RMN 1H (400 MHz, CDCh) 5 7,81 (d, J = 1,8 Hz, 2H), 7,76 - 7,69 (m, 6H), 7,50 - 7,44 (m, 5H), 7,40 - 7,34 (m, 2H), 5,02 (t, J = 3,1 Hz, 1H), 3,08 - 2,94 (m, 2H), 1,99 (m, 1H), 1,75 (ap d, J = 6,0 Hz, 2H), 1,72 - 1,58 (m, 2H), 1,45 - 1,31 (m, 3H), 1,41 (s, 6H), 1,38 (s, 6H), 1,37 (s, 6H), 0,74 (s, 9 H). RMN 13C{1H} (101 MHz, CDCh) 5 157,24, 145,08, 141,50, 141,46, 141,43, 134,73, 134,07, 132,05, 128,90, 128,16, 127,48, 127,40, 124,42, 102,88, 83,75, 62,22, 57,20, 38,42, 32,59, 32,04, 31,82, 31,47 30,58, 25,49, 25,21,24,79, 19,05.
C1-5. Preparación del meso-4,4'-pentano-2,4-diilbis(oxi))bis(3-bromo-1-fluorobenceno)

Un matraz de fondo redondo de tres bocas de 2 l, equipado con un termómetro, un agitador magnético, un embudo de adición y atmósfera de N2, se cargó con 2,4-pentanodiol (30,46 g, 292,5 mmol, 1 equivalente), 2-bromo-4-fluorofenol (114,39 g, 598,9 mmol, 2,04 equivalentes), trifenilfosfina (157,12 g, 599,0 mmol, 2,04 equivalentes) y THF (600 ml), y el contenido se enfrió a 2°C en un baño de agua con hielo. Se añadió una disolución de DIAD (121,11 g, 598,9 mmol, 2,04 equivalentes) en THF (130 ml), en el embudo de adición, a una velocidad tal que se mantuviera la reacción por debajo de 5°C (la adición tardó aproximadamente 4 h). La mezcla resultante se agitó a 2-5°C durante 1 h más y se tomó una muestra para análisis Gc -MS, lo que indicó que la reacción estaba a punto de completarse. Después de agitar durante la noche, a temperatura ambiente, se eliminaron los volátiles a presión reducida. Se añadió ciclohexano (700 ml) al residuo y la suspensión se agitó a temperatura ambiente durante 30 min. El sólido insoluble se filtró, se enjuagó con ciclohexano (100 ml x 3). La disolución de ciclohexano se lavó con NaOH 1N (200 ml), agua (200 ml), HCl 1N (200 ml), agua (500 ml x 2) y luego se concentró, a presión reducida, para dar un residuo oleoso. El residuo oleoso se disolvió en hexano (100 ml) y luego se pasó a través de un lecho de SiO2 (315 g), eluyendo con hexano (300 ml) y hexano-EtOAc (20:1 en volumen, 2 l de hexano 100 ml de EtOAc), se concentró y se secó para dar el grupo inferior deseado (123,8 gramos, 94 % de rendimiento). RMN 1H (400 MHz, CDCh) ó 7,14 (dd, J = 8,4, 3.9 Hz, 2H), 6,64 (dt, J = 9,1,3,9 Hz, 2H), 6,48 (dd, J = 9,0, 3,7 Hz, 2H), 4,22 (m, 2H), 2,17 (dt, J = 13,6, 6,5 Hz, 1H), 1,45 (dt, J = 13,6, 5,6 Hz, 1 H) y 0,98 (d, J = 6,1 Hz, 6H). RMN 13C{1H} (101 MHz, CDCh) 5156,9 (d, Jcf = 235,8 Hz), 150,9 (d, Jcf = 2,8 Hz), 120,9 (d, Jcf = 25,8 Hz), 115,62 (d, Jcf = 7,7 Hz), 114,9 (d, Jcf = 21,5 Hz), 113,7 (d, Jcf = 10,1 Hz), 72,8, 42,7 y 19,6. RMN 19F{1H} (376 MHz, CDCh) 5: -121,33.
C1-6. Preparación del Ligando Comparativo C1

A un matraz de 250 ml que contiene el éster de boronato (3,15 g, 4,89 mmol) bajo N2 se añadió el bromuro de arilo (1,00 g, 2,22 mmol), NaOH (0,533 g, 13,3 mmol), Pd(PPh3)4 (0,103 g, 0,089 mmol), 9,3 ml de tolueno desgasificado y 1,8 ml de agua desgasificada. El sistema se burbujeó con N2. La reacción se calentó a 110°C durante 24 h. La reacción se enfrió y los volátiles se eliminaron por evaporación rotatoria. El residuo se recogió en Et2O (100 ml), se lavó con salmuera (100 ml), se secó sobre MgSO4, se filtró a través de un lecho de SiO2, luego se concentró para dar el ligando protegido. Para la etapa de desprotección del THP, este material se disolvió en 20 ml de THF y se añadieron 20 ml de MeOH, seguido de aproximadamente 5 gotas de HCl concentrado. Esta mezcla se calentó a reflujo durante 1 hora, luego se enfrió y se concentró al vacío. Se añadió Et2O (80 ml) y la disolución resultante se lavó con agua (50 ml). La parte orgánica se separó y se secó sobre MgSO4, se filtró y se concentró bajo vacío. Este material crudo se purificó adicionalmente por cromatografía de columna sobre SiO2, eluyendo con un gradiente de EtOAc/hexano al 1 ^ 8 %, proporcionando el producto (2,46 g, 95,5 % de rendimiento) como un sólido blanco. RMN 1H (400 MHz, CDCh) 57,80 (t, J = 1,7 Hz, 2H), 7,76 (d, J = 1,7 Hz, 4H), 7,68 (m, 8H), 7,45 (m, 10H), 7,37 (m, 4H), 7,17 (d, J = 2,4 Hz, 2H), 7,03 (dd, J = 9,0, 3,0 Hz, 2H), 6,78 (m, 4H), 6,33 (s, 2H), 4,32 (q, J = 6,2 Hz, 2H), 1,74 (s, 4H), 1,39 (s, 6H), 1,38 (s, 6H), 1,04 (d, J = 6,1 Hz, 6H), 0,75 (s, 18H). RMN 13C{1H} (101 MHz, CDCh) 5 159,25, 156,85, 149,73, 149,70, 148,27, 142,62, 141,82, 141,37, 140,18, 131,69, 131,61, 129,54, 129,06, 128,95, 128,75, 127,65, 127,56, 127,49, 125,78, 125,76, 125,01, 119,11, 118,88, 117,63, 117,54, 115,69, 115,47, 74,45, 57,19, 42,45, 38,29, 32,59, 32,09, 31,88, 31,85, 19,82. RMN 19F{1 H} (376 MHz, CDCh) 5 -120,52.
C1-6. Preparación del Ejemplo Comparativo C1

A un vial equipado con una barra de agitación en una caja de guantes se añadió ZrCU (0,201 g, 0,864 mmol) y tolueno anhidro (22 ml). La mezcla se enfrió a -30°C y a esta se le añadió MeMgBr (1,21 ml, disolución 3,0 M en Et2O, 3,63 mmol). La suspensión resultante se agitó durante 2 minutos, después de lo cual se añadió el ligando C1 (1,00 g, 0,864 mmol) a la suspensión fría con agitación. La reacción se calentó a temperatura ambiente y se agitó durante la noche. Luego, la reacción se concentró para proporcionar un sólido oscuro, que se lavó con hexano (100 ml). Al material sólido resultante se le añadió tolueno (100 ml) y se filtró la suspensión resultante. El filtrado se concentró para proporcionar el producto como un sólido marrón (0,634 g, 58 %). RMN 1H (400 MHz, C6D6) 58,36 (s, 2H), 8,26 (s, 2H), 7,97 (t, J = 1,8 Hz, 1H), 7,92 (t, J = 1,7 Hz, 1 H), 7,73 (d, J = 2,5 Hz, 1H), 7,69 (d, J = 2,5 Hz, 1H), 7,66 (m, 8H), 7,22 (m, 9H), 7,12 (m, 6H), 7,03 (dd, J = 8,9, 3,1 Hz, 1H), 6,62 (m, 2H), 5,81 (dd, J = 8,9, 5,1 Hz, 1H), 5,56 (dd, J = 8,9, 5,1 Hz, 1 H), 4,33 (p, J = 6,8 Hz, 1H), 3,61 (m, 1 H), 1,78 - 1,54 (m, 4H), 1,34 (s, 6H), 1,33 (s, 2H), 1,31 (s, 3H), 1,23 (m, 1H), 0,80 (s, 9H), 0,72 (s, 9H), 0,65 (d, J = 6,0 Hz, 3H), 0,61 (m, 1 H), 0,40 (d, J = 6,8 Hz, 3H), -0,08 (s, 3H), -0,24 (s, 3H). RMN 19F{1H} (376 MHz, CeDe) 5 -114,27, -114,44.
Ejemplo Comparativo C2
C2-1. Preparación del 3,6-bis(1,1-dimetiletil)-9H-carbazol

Un matraz de fondo redondo de tres bocas de 500 ml, equipado con un agitador superior, burbujeador de N2 y un embudo de adición, se cargó con 20,02 g (120,8 mmol) de carbazol, 49,82 g (365,5 mmol) de ZnCl2, y 300 ml de nitrometano a temperatura ambiente. A la suspensión de color marrón oscuro resultante, se añadieron gota a gota desde el embudo de adición, 49,82 g (365,5 mmol) de 2-cloro-2-metilpropano (también conocido como cloruro de butilo terciario), durante un período de 2,5 h. Después de completar la adición, la suspensión resultante se agitó durante 18 h adicionales y la mezcla de reacción se vertió en 800 ml de agua helada y se extrajo con CH2Cl2 (3 x 500 ml). Los extractos combinados se secaron con MgSO4 anhidro, se filtraron y se concentraron, primero por evaporación rotatoria y luego por evaporación a alto vacío para eliminar el nitrometano. El residuo resultante se disolvió en CH2Cl2 caliente (70 ml), seguido de hexanos calientes (50 ml), y la disolución resultante se enfrió a temperatura ambiente y luego se colocó en un refrigerador durante la noche. Se aislaron los sólidos resultantes que se formaron, se lavaron con hexanos fríos y luego se secaron a alto vacío para producir 10,80 g (32,0 %) del producto deseado en forma de cristales blanquecinos. RMN 1H (400 MHz, CDCh) 58,11 (d, J = 1,6 Hz, 2H), 7,75 (s, 1H), 7,48 (dd, J = 8,5, 1,9 Hz, 2H), 7,31 (d, J = 8,5 Hz, 2H), 1,48 (s, 18H). RMN 13C{1H} (101 MHz, CDCh) 5 142,17 (s), 137,96 (s), 123,45 (s), 123,28 (s), 116,11 (s), 109,97 (s), 34,73 (s), 32,09 (s).
C2-2. Preparación del 2-yodo-4-(2,4,4-trimetilpentan-2-il)fenol

A una disolución agitada de 10,30 g (50,00 mmol) de 4-(2,4,4-trimetilpentan-2-il)fenol en 125 ml de metanol a 0°C, se añadieron 7,48 g (50,00 mmol) de Nal y 2,00 g (50,0 mmol) de NaOH. A la mezcla resultante se añadieron 86 ml de disolución acuosa de NaOCl al 5 % (blanqueador comercial) durante un período de una hora. La suspensión resultante se agitó durante una hora más a 0°C, luego se añadieron 30 ml de disolución acuosa de Na2S2O3 al 10 % , y la mezcla de reacción resultante se acidificó con la adición de ácido clorhídrico diluido. La mezcla resultante se extrajo con CH2G 2, y la capa orgánica resultante se lavó con salmuera y luego se secó sobre MgSO4 anhidro. Los volátiles se eliminaron al vacío y el residuo resultante se purificó mediante cromatografía ultrarrápida en SiO2, eluyendo con acetato de etilo en hexanos al 5 por ciento en volumen (% en volumen) para producir 11,00 g (66 %) del producto deseado como un aceite viscoso. RMN 1H (400 MHz, CDCh) 57,60 (d, J = 2,5 Hz, 1H), 7,25 (dd, J = 8,5 y 2,2 Hz, 1H),
6,90 (d, J = 8,5 Hz, 1 H), 5,13 (s, 1 H), 1,69 (s, 2H), 1,32 (s, 6H) y 0,74 (s, 9H). RMN 13C{1H} (101 MHz, CDCI3) 5152,21, 144,52, 135,56, 128,03, 114,17, 85,36, 56,92, 38,01,32,43, 31,90 y 31,64, GC/MS (m/e): 332 (M+).
C2-3. Preparación del 2-(2-yodo-4-(2,4,4-trimetilpentan-2-il)fenoxi)tetrahidro-2H-pirano

A una disolución agitada de 4,91 g (14,8 mmol) de 2-yodo-4-(2,4,4-trimetilpentan-2-il)fenol y 1,50 g (17,8 mmol) de 3,4-dihidropirano, en 5 ml de CH2G 2, a 0°C, se añadieron 0,039 g (0,205 mmol) de monohidrato de PTSA. La disolución resultante se dejó calentar a temperatura ambiente y se agitó durante aproximadamente 10 min. Luego se añadió trietilamina (0,018 g, 0,178 mmol) y la mezcla resultante se diluyó con 50 ml de CH2G 2 y se lavó sucesivamente con 50 ml de cada uno de NaOH 1 M, agua y salmuera. La fase orgánica se secó con MgSO4 anhidro, se filtró y se concentró para dar un material crudo, que se purificó por cromatografía ultrarrápida en SiO2, usando acetato de etilo al 5 % en volumen en hexanos, para producir 5,18 g (93,12 %) del producto deseado como un aceite dorado. RMN 1H (400 MHz, CDCl3) 57.74 (d, J = 2,3 Hz, 1 H), 7,27 (dd, J = 2,3 y 8,6 Hz, 1H), 6,99 (d, J = 8,6 Hz, 1H), 5,49 (m, 1H), 3,91 (m, 1H), 3,61 (m,1 H), 2,20-1,60 (m, 6H), 1,69 (s, 2H), 1,34 (s, 6H) y 0,75 (s, 9H), RMN 13C{1 H} (101 MHz, CDCh) 5153,27, 145,49, 136,98, 127,08, 114,44, 96,72, 87,09, 61,69, 56,91,37,95, 32,33, 31,81,31,52, 31,44, 30,26, 25,27, 18,36.
C2-4. Preparación del 3,6-di-terc-butil-9-(2-(tetrahidro-2H-piran-2-iloxi)-5-(2,4,4-trimetil-pentan-2-il)fenil)-9H -carbazol

A un matraz de fondo redondo de tres bocas de 50 ml, equipado con una barra de agitación y un condensador, bajo atmósfera de N2, se añadió lo siguiente: 20 ml de tolueno seco, 5,00 g (12,01 mmol) de 2-(2-yodo-4-(2,4,4-trimetilpentan-2-il)fenoxi)tetrahidro-2H-pirano; 3,56 g (12,01 mmol) de 3,6-di-terc-butil carbazol, 0,488 g (2,56 mmol) de Cul, 7,71 g (36,2 mmol) de K3PO4 y 0,338 g (3,84 mmol) de N,N'-dimetiletilendiamina. La mezcla de reacción resultante se calentó a reflujo durante 48 h, se enfrió y se filtró a través de un lecho de SiO2. El SiO2 se enjuagó con THF y la disolución resultante se concentró para dar un residuo crudo. La purificación se realizó por recristalización en CH3CN, para producir 4,57 g (67,0 %) del producto deseado como un sólido blanco. RMN 1H (400 MHz, CDCh) 5 8,13 (t, J = 1,71 Hz, 1 H), 7,48 (d, J = 2,4 Hz, 1H), 7,40 (m, 3H), 7,31 (d, J = 8,68 Hz, 1H), 7,14 (d, J = 8,68 Hz, 1H), 7,08 (d, J = 8,56 Hz, 1 H), 5,22 (t, J = 2,81 Hz, 1H), 3,72 (td, J = 11,12 y 2,8 Hz, 1H), 3,47 (dt, J = 11,12 y 3,47 Hz, 1 H), 1,75 (s, 2H), 1,474 (s, 9H), 1,472 (s, 9H), 1,394 (s, 3H), 1,391 (s, 3H), 1,37-1,12 (m, 6H), 0,82 (s, 9H), RMN 13C{1H} (101 MHz, CDCl3) 5150,96, 144,22, 142,07, 140,02, 127,49, 126,60, 126,56, 123,14, 123,12, 122,96, 116,37, 115,88, 115,72, 110,18, 109,52, 97,02, 61,56, 57,03, 38,23, 34,69, 32,41,32,07, 31,86, 31,72, 31,50, 29,98, 25,06, 17,61.
C2-5. Preparación del 3,6-di-terc-butil-9-(2-((tetrahidro-2H-piran-2-il)oxi)-3-(4,4,5,5-tetrametil-1,3,2 -dioxaborolan-2-il)-5-(2,4,4-trimetilpentan-2-il)fenil)-9H-carbazol

A una disolución agitada de 2,5 g (4,4 mmol) de derivado de carbazol, en 40 ml de THF, a 0°C, bajo atmósfera de N2, se añadieron 2,8 ml (7,0 mmol) de n-Butil litio (disolución 2,5 M en hexanos), durante un periodo de 5 min. La disolución se agitó a 0°C durante 3 horas más. A esto se le añadió 2-isopropoxi-4,4,5,5-tetrametil-1,3,2-dioxaborolano (1,44 ml, 7,0 mmol) y se continuó agitando a 0°C durante 1 hora más. La mezcla de reacción se calentó lentamente a temperatura ambiente y se agitó durante 18 horas. La mezcla de reacción se concentró a sequedad mediante evaporación rotatoria y se añadieron 100 ml de agua helada. La mezcla se extrajo con CH2G 2. La capa orgánica se lavó con salmuera y se secó sobre MgSO4 anhidro. La eliminación del disolvente, seguida de recristalización en CH3CN, dio 2,4 g (78,6 %) del producto titulado como un sólido blanco. RMN 1H (400 MHz, CDCh) 58,30-7,96 (m,
2H), 7,81 (d, J = 2,5 Hz, 1 H), 7,58-7,32 (m, 3H), 7,14 (d, J = 8,6 Hz, 2H), 4,85 (d, J = 2,8 Hz, 1H), 2,76 (td, J = 11,0, 2,7 Hz, 1 H), 2,59 (dd, J = 7,9, 3,5 Hz, 1 H), 1,73 (s, 2H), 1,67-0,87 (m, 6H), 1,46 (s, 9H), 1,45 (s, 9H), 1,38 (s, 9H), 1,37 (s, 9H), 0,78 (s, 9H); RMN 13C{1H} (101 MHz, CDCI3) 5156,25, 145,86, 142,05, 142,01, 139,79, 139,78, 133,82, 130,61, 129,72, 123,39, 123,37, 123,05, 115,59, 115,55, 110,20, 110,11, 101,41, 83,64, 61,20, 56,95, 38,37, 34,68, 32,42, 32,08, 31,90, 31,45, 29,97, 25,06, 25,04, 24,79, 18,16.
C2-6. Preparación del 1,4-bis(4-fluoro-2-yodofenoxi)butano

Se añadieron 4-fluoro-2-yodofenol (25,0 g, 105 mmol), 1,4-dibromobutano (11,3 g, 52,5 mmol) y K2CO3 (18,1 g, 131 mmol) a un matraz de 500 ml que contenía 250 ml de acetona. La mezcla de reacción se calentó a reflujo durante la noche. La GC/MS de la mezcla mostró que la reacción era de ~4:1 de bis a producto monosustituido. La mezcla se filtró mientras estaba caliente y los sólidos filtrados se recogieron en acetona, se calentaron a 50°C durante 15 min y se filtraron de nuevo. Los filtrados combinados se redujeron a un sólido por evaporación rotatoria. El sólido así obtenido se disolvió en CH3CN caliente, y se enfrió para dar 14,0 g de cristales blancos. Las aguas madres se colocaron en el congelador (-13°C) y se recogieron 3,0 g adicionales de producto. Las aguas madres de este material se recristalizaron dos veces a baja temperatura y se obtuvieron 3,0 g adicionales de producto para un total de 20,0 g (72 % de rendimiento). RMN 1H (400 MHz, CDCh) 57,47 (dd, J = 7,6, 3,0 Hz, 2 H), 6,99 (ddd, J = 8.8, 8.0, 3.0 Hz, 2 H), 6,73 (dd, J = 9,0, 4,6 Hz, 2 H), 4,06 (m, 4 H), 2,08 (m, 4 H). RMN 13C{1H} (101 MHz, CDCh) 5156,7 (d, J = 243,4 Hz), 154,1 (d, J = 2,5 Hz), 126,0 (d, J = 25,1 Hz), 115,6 (d, J = 22,6 Hz), 112,2 (d, J = 8,1 Hz), 86,0 (d, J = 8,6 Hz), 69,4, 26,1.
C2-7. Preparación del Ligando Comparativo C2

El fragmento borilado (20,8 g, 30,0 mmol) se disolvió en 65 ml de 1,2-dimetoxietano. A continuación, la disolución se trató con una disolución de NaOH (3,5 g, 86,6 mmol) en 45 ml de agua, 65 ml de THF y el compuesto de diyodo 7 (7,65 g, 14,4 mmol). El sistema se burbujeó durante 30 min con N2 y se añadió el Pd(PPh3)4 (0,58 g, 0,51 mmol). La mezcla se calentó a 85°C durante 36 h bajo atmósfera de N2. La mezcla de reacción se enfrió y los volátiles se eliminaron por evaporación rotatoria. El residuo se recogió en 225 ml de CH2G 2 y se lavó con 200 ml de salmuera. La capa orgánica se secó sobre MgSO4 anhidro, se filtró y se pasó a través de un pequeño lecho de SiO2 y se concentró por evaporación rotatoria. El residuo se disolvió en 170 ml de THF y se trató con 170 ml de metanol y 2,15 ml de HCl concentrado. A continuación, la disolución amarilla se calentó a 50°C durante 30 min. La disolución se redujo a un sólido amarillo (20,38 g). Este material se recristalizó en Et2O/MeOH a -15°C, proporcionando el producto como un sólido blanco (14,44 g, 81 %). RMN 1H (400 MHz, CDCh) 58,12 (d, J = 1,7 Hz, 4H), 7,40 - 7,34 (m, 6H), 7,31 (d, J = 2,4 Hz, 2H), 7,10 (dd, J = 8,9, 3,3 Hz, 2H), 7,02 (d, J = 8,6 Hz, 4H), 6,96 - 6,90 (m, 2H), 6,72 (dd, J = 9,0, 4,5 Hz, 2H), 5,89 (s, 2 H), 3,73 (br m, 4H), 1,68 (s, 4H), 1,59 (br m, 4H), 1,41 (s, 36H), 1,32 (s, 12H), 0,76 (s, 18H).
C2-8. Elaboración del Ejemplo Comparativo C2

A un frasco equipado con una barra de agitación en una caja de guantes se añadió ZrCU (0,059 g, 0,254 mmol, 1,05 equivalentes) disuelto en tolueno (10 ml). La frasco se enfrió a -30°C y a este se añadió MeMgBr (0,32 ml, 0,966 mmol,
4 equivalentes). La disolución resultante se agitó durante 15 minutos, después de lo cual se añadió el ligando C2 (0,300 g, 0,241 mmol, 1 equivalente) a la suspensión fría con agitación. La reacción se calentó a 20-25°C y se agitó durante 3 h. La reacción se filtró y se concentró y luego se extrajo con hexanos. Las capas de hexanos se combinaron y se concentraron para proporcionar el catalizador como un sólido blanquecino (0,181 g, 55 %). RMN 1H (400 MHz, C6D6) ó 8,47 - 8,41 (m, 2H), 8,26 (dd, J = 1,9, 0,6 Hz, 2H), 7,57 - 7,45 (m, 8H), 7,30 (dd, J = 8,8, 1,9 Hz, 2H), 7,12 (d, J = 2,4 Hz, 2H), 7,00 (p, J = 1,0 Hz, 6H), 6,79 (dd, J = 8,9, 3,1 Hz, 2H), 6,51 - 6,42 (m, 2H), 4,83 (dd, J = 9,0, 4,8 Hz, 2H), 3,96 (t, J = 9,7 Hz, 2H), 3,34 (d, J = 11,7 Hz, 2H), 1,46 (s, 4H), 1,33 (s, 14H), 1,23 - 0,99 (m, 30H), 0,83 - 0,70 (m, 8H), 0,68 (s, 12H), -0,92 (s, 6H). RMN 19F{1H} (376 MHz, C6D6) 5: -115,93.
Ejemplo Comparativo C3
C3-1. Preparación del 1-(metoximetoxi)-4-(2,4,4-trimetilpentan-2-il)benceno

En un matraz de 500 ml se disolvieron 4-terc-octilfenol (10,0 g, 48,5 mmol) y dimetoximetano (42,9 ml, 485 mmol) en 240 ml de CH2Cl2 bajo N2. Se añadió P2O5 en porciones de aproximadamente de 3 g en 0,5 h. La mezcla resultante se agitó durante 2,5 h adicionales. Reacción se filtró a través de un tapón de SiO2 para eliminar el P2O5. Se lavó el filtrado con 150 ml de H2O, y luego con 150 ml de salmuera secuencialmente. La capa orgánica se secó sobre MgSO4 , se filtró y se concentró. Se purificó por cromatografía en columna sobre SiO2, eluyendo con un gradiente de CH2Cl2/hexano al 30^50 %. Se recogieron 9,07 g de producto (74,7 % de rendimiento) como un aceite transparente. RMN 1H (400 MHz, CDCh) 57,27 (d, J = 8,8 Hz, 2H), 6,94 (d, J = 8,8 Hz, 2H), 5,16 (s, 2H), 3,48 (s, 3H), 1,70 (s, 2H), 1,34 (s, 6H), 0,72 (s, 9H). RMN 13C{1H} (101 MHz, CDCta) 5 155,04, 143,74, 127,22, 115,61, 94,79, 57,15, 56,10, 38,18, 32,48, 31,93, 31,76.]
C3-2. Preparación del 2-(2-(metoximetoxi)-5-(2,4,4-trimetilpentan-2-il)fenil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano

En un matraz de 1 l secado al horno bajo N2 se disolvió el sustrato fenólico protegido (9,07 g, 36,2 mmol) en THF (362 ml, desgasificado y almacenado sobre alúmina bajo N2). Esta disolución se enfrió a 0°C y se añadió n-BuLi (18,8 ml, disolución 2,5 M en hexano, 47,1 mmol) lentamente mediante una jeringa. La disolución resultante se dejó calentar lentamente a temperatura ambiente con agitación durante 2 horas. Luego, se añadió isopropoxi(pinacolborano) (10,3 ml, 50,7 mmol) mediante una jeringa y la mezcla se agitó durante la noche a temperatura ambiente. La reacción se detuvo con H2O (6 ml), y los volátiles se eliminaron al vacío, teniendo cuidado de no calentar en exceso la mezcla. El residuo resultante se disolvió en CH2G 2 (300 ml), y esta disolución se lavó con salmuera (3 x 300 ml). La capa orgánica se secó sobre MgSO4 , se filtró y se concentró para dar un aceite de color amarillo claro. Mediante RMN 1H el material es una mezcla de producto y material de partida, aproximadamente una relación 10:1. Este material se usó sin purificación adicional. RMN 1H (400 MHz, CDCb) 57,63 (d, J = 2,6 Hz, 1H), 7,36 (dd, J = 8,7, 2,7 Hz, 1H), 6,93 (d, J = 8,7 Hz, 1 H), 5,15 (s, 2H), 3,51 (s, 3H), 1,71 (s, 2H), 1,36 (s, 6H), 1,35 (s, 12H), 0,73 (s, 9H). RMN 13C{1H} (101 MHz, CDCl3) 5159,68, 143,46, 133,92, 130,22, 115,33, 95,73, 83,46, 57,05, 56,21,38,20, 32,48, 31,99, 31,70, 24,98.
C3-3. Preparación del 9-(2-(metoximetoxi)-5-(2,4,4-trimetilpentan-2-il)fenil)antraceno

A un matraz de 200 ml que contiene el éster de boronato (5,00 g, 13,3 mmol) bajo N2 se añadió 9-bromoantraceno (3,11 g, 12,1 mmol), NaOH (2,90 g, 72,5 mmol), Pd (PPh3)4 (279 mg, 0,242 mmol), 50 ml de tolueno desgasificado y 10 ml de agua desgasificada. El sistema se burbujeó con N2. La reacción se calentó a 110°C durante 72 h. Luego, la reacción se enfrió y los volátiles se eliminaron mediante evaporación rotatoria. El residuo se recogió en Et2O, se lavó
con salmuera, se secó sobre MgSÜ4 anhidro, se filtró a través de un lecho de SiÜ2 y luego se concentró para proporcionar el producto bruto. Este material se purificó por recristalización en CH3CN, proporcionando el producto como agujas de color amarillo anaranjado (3,93 g, 76,3 % de rendimiento). RMN 1H (400 MHz, CDCh) ó 8,49 (s, 1H), 8.05 (d, J = 8,3 Hz, 2H), 7,64 (d, J = 8,8 Hz, 2H), 7,49 (dd, J = 8,7, 2,6 Hz, 1H), 7,45 (ddd, J = 8,5, 6,5, 1,2 Hz, 2H), 7,33 (ddd, J = 8,8, 6,5, 1,3 Hz, 2H), 7,26 (m, 2H), 4,87 (s, 2H), 3,01 (s, 3H), 1,72 (s, 2H), 1,37 (s, 6H), 0,80 (s, 9H).
C3-3. Preparación del 2-(3-(antracen-9-il)-2-(metoximetoxi)-5-(2,4,4-trimetilpentan-2-il)fenil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano

En un matraz de 250 ml secado al horno bajo N2 se disolvió el sustrato (3,93 g, 9,21 mmol) en 92 ml de THF anhidro y la disolución resultante se enfrió a 0°C. A esto se añadió n-BuLi (4,79 ml, disolución 2,5 M en hexano, 12,0 mmol) gota a gota con una jeringa. La disolución se dejó calentar lentamente a temperatura ambiente con agitación durante 2 h, momento en el que se añadió isopropoxi(pinacolborano) (2,63 ml, 12,9 mmol) mediante una jeringa y la mezcla resultante se agitó durante la noche a temperatura ambiente. La reacción se detuvo con 2 ml de H2O, y luego se eliminaron los disolventes al vacío, cuidando de no calentar en exceso la mezcla. El residuo resultante se disolvió en 100 ml de CH2G 2, se lavó con 100 ml de salmuera, luego se secó sobre MgSÜ4, se filtró y se concentró para dar un sólido amarillo. Este material se trituró con MeÜH. El sólido se ablandó y se convirtió en un aceite tras la adición inicial de MeÜH, luego el producto precipitó como un polvo amarillo claro (3,55 g de producto, 69,7 % de rendimiento). RMN 1H (400 MHz, CDCl3) 58,47 (s, 1H), 8,01 (d, J = 9,1 Hz, 2H), 7,92 (d, J = 2,7 Hz, 1H), 7,65 (d, J = 8,8 Hz, 2H), 7,43 (m, 3H), 7,34 (ddd, J = 8,8, 6,5, 1,3 Hz, 2H), 4,63 (s, 2H), 1,86 (s, 3H), 1,75 (s, 2H), 1,40 (s, 6H), 1,37 (s, 12H), 0,81 (s, 9 H). RMN 13C{1H} (101 MHz, CDCh) 5 159,09, 145,39, 135,02, 134,56, 134,26, 131,51, 131,41, 130,88, 128,17, 127,51,126,39, 125,34, 125,19, 100,22, 83,79, 57,03, 55,37, 38,53, 32,61,32,17, 31,86, 25,03.
C3-4. Preparación del Ligando Comparativo C3

A un matraz de 100 ml que contiene el éster de boronato del grupo superior (1,08 g, 1,96 mmol) bajo N2 se añadió el fragmento di-bromo (0,400 g, 0,889 mmol), Na2CÜ3 (0,565 g, 5,33 mmol), Pd(ptBu3)2 (0,018 g, 0,036 mmol), 30 ml de THF desgasificado y 10 ml de agua desgasificada. El sistema se burbujeó con N2 y luego se calentó a reflujo durante 24 h. La reacción se enfrió y los volátiles se eliminaron por evaporación rotatoria. El residuo se recogió en Et2Ü, se lavó con salmuera, se secó sobre MgSÜ4 anhidro, se filtró a través de un lecho de SiÜ2 y luego se concentró para dar el ligando protegido. Para la desprotección del THP, se añadió THF (20 ml) y la mezcla se agitó hasta que se disolvió el ligando. A esto se le añadieron 20 ml de MeÜH y 2 gotas de HCl concentrado. Esta mezcla se calentó a reflujo durante 1 hora, luego se retiró del calor y se concentró al vacío. Se añadieron 50 ml de agua y se extrajo con 80 ml de Et2Ü. La capa orgánica se recogió y se secó sobre MgSÜ4, se filtró y se concentró al vacío. Este material se purificó por cromatografía en columna, eluyendo con EtÜAc/hexano al 1 —>15 %; sin embargo, el material aislado contenía una impureza, posiblemente un producto monoacoplado. Este material se sometió a una segunda cromatografía en columna, eluyendo con acetona/hexano al 1—15 %, proporcionando el producto deseado como un polvo amarillo claro (0,228 g, 24,4 % de rendimiento). RMN 1H (400 MHz, CDCh) 58,52 (s, 2H), 8,04 (dd, J = 15,5, 8,5 Hz, 4H), 7,68 (dd, J = 13,9, 8,8 Hz, 4H), 7,42 (m, 2H), 7,38 (d, J = 2,5 Hz, 2H), 7,34 (m, 2H), 7,30 (d, J = 2,4 Hz, 2H), 7,26 (m, 4H), 7,16 (dd, J = 9,1,3,1 Hz, 2H), 6,68 (td, J = 8,4, 8,0, 3,2 Hz, 2H), 6,54 (dd, J = 9,0, 4,7 Hz, 2H), 5,96 (s, 2H), 4,39 (q, J = 6,1 Hz, 2H), 2,08 (dt, J = 14,2, 7,0 Hz, 1H), 1,74 (s, 4H), 1,62 (dt, J = 14,3, 5,4 Hz, 1H), 1,40 (s, 6H), 1,39 (s, 6H), 1,03 (d, J = 6,1 Hz, 6H), 0,81 (s, 18H). RMN 13C{1H} (101 MHz, CDCh) 5 159,07, 156,68, 149,59, 149,57, 142,29, 133,69, 131,74, 131,70, 131,68, 130,71, 130,67, 129,22, 128,67, 126,94, 126,77, 126,69, 126,16, 125,55, 125,52, 125,28, 125,23, 118,98, 118,75, 116,78, 116,69, 115,42, 115,20, 73,92, 57,32, 42,65, 38,32, 32,62, 32,13, 31,98, 31,94, 19,57. RMN 19F{1H} (376 MHz, CDCh) 5 -121,15.
C3-4. Preparación del Procatalizador Comparativo C3

En una caja de guantes, se suspendió ZrCU (0,077 g, 0,332 mmol) en 8,3 ml de tolueno (anhidro) en un vial de centelleo de 40 ml secado al horno con una barra de agitación. La suspensión se enfrió a -30°C. Se añadió MeMgBr (0,465 ml, disolución 3,0 M en Et2O, 1,40 mmol) a la suspensión fría con agitación. Después de 2 min, la disolución se oscureció y se añadió el ligando C3 (0,350 g, 0,332 mmol). Esta mezcla se dejó calentar a temperatura ambiente y se agitó durante la noche. El disolvente se eliminó al vacío para producir un sólido marrón, que se lavó con aproximadamente 80 ml de hexano, luego se extrajo con aproximadamente 80 ml de tolueno y se filtró. El filtrado se concentró para dar el producto como un polvo marrón claro (0,229 g, 58,8 % de rendimiento). RMN 1H (400 MHz, C6D6) 58,37 - 8,25 (m, 4H), 8,21 (d, J = 8,8 Hz, 2H), 7,95 (m, 2H), 7,80 (dd, J = 8,5, 4,1 Hz, 2H), 7,55 (dd, J = 4,9, 2,5 Hz, 2H), 7,40 (d, J = 2,5 Hz, 1H), 7,34 (d, J = 2,5 Hz, 1H), 7,32 - 7,19 (m, 4H), 7,14 - 6,99 (m, 6H), 6,48 (dddd, J = 9,0, 7,4, 3,2, 1,6 Hz, 2H), 4,27 (dd, J = 8,9, 5,3 Hz, 1H), 4,13 (dd, J = 8,9, 5,0 Hz, 1H), 4,00 (m, 2H), 1,65 - 1,56 (m, 4H), 1,47 (dt, J = 16,8, 8,9 Hz, 1H), 1,31 (s, 3H), 1,29 (s, 3H), 1,27 (s, 3H), 1,25 (s, 3H), 1,12 (d, J = 15,7 Hz, 1H), 0,88 (s, 9H), 0,83 (s, 9H), 0,43 (d, J = 6,3 Hz, 3H), 0,29 (d, J = 6,5 Hz, 3H), -1,21 (s, 3H), -1,29 (s, 3H). RMN 19F{1H} (376 MHz, CeDe) 5 -115,36, -115,68.
Ejemplo Comparativo C4
C4-1. Preparación del Ejemplo Comparativo C4

A un vial equipado con una barra de agitación en una caja de guantes se añadió HfCU (0,277 g, 0,864 mmol) y tolueno anhidro (22 ml). La mezcla se enfrió a -30°C y a esta se le añadió MeMgBr (1,21 ml, disolución 3,0 M en Et2O, 3,63 mmol). La suspensión resultante se agitó durante 10 min, después de lo cual se añadió el ligando C1 (1,00g, 0,864 mmol) a la suspensión fría con agitación. La reacción se calentó a temperatura ambiente y se agitó durante la noche. Luego, la reacción se concentró para proporcionar un sólido oscuro, que se lavó con hexano (100 ml). Al material sólido resultante se le añadió tolueno (100 ml) y se filtró la suspensión resultante. El filtrado se concentró para proporcionar el producto como un sólido marrón (0,801 g, 68 %). RMN 1H (400 MHz, C6D6) 58,34 (s, 2H), 8,23 (s, 2H), 7,98 (t, J = 1,8 Hz, 1H), 7,92 (t, J = 1,7 Hz, 1 H), 7,75 (d, J = 2,5 Hz, 1H), 7,70 (d, J = 2,6 Hz, 1H), 7,66 (m, 8H), 7,23 (m, 9H), 7,12 (m, 5H), 7,02 (dd, J = 8,9, 3,1 Hz, 1H), 6,63 (m, 2H), 5,82 (dd, J = 8,9, 5,1 Hz, 1H), 5,57 (dd, J = 8,9, 5,1 Hz, 1 H), 4,40 (p, J = 7,1 Hz, 1H), 3,65 (m, 1 H), 1,78 - 1,53 (m, 4H), 1,42 (m, 1H), 1,34 (s, 6H), 1,32 (s, 3H), 1,31 (s, 3H), 0,80 (s, 9H), 0,72 (s, 9H), 0,67 (d, J = 6,0 Hz, 3H), 0,63 (m, 1 H), 0,40 (d, J = 6,8 Hz, 3H), -0,28 (s, 3H), -0,44 (s, 3H). RMN 19F{1H} (376 MHz, C6D6) 5 -114,21, -114,35.
Ejemplo Comparativo C5
C5-1. Preparación del Ejemplo Comparativo C5.

Se suspendieron el ligando C2 (7,41 g, 5,96 mmol) y HfCU (1,91 g, 5,96 mmol) en 280 ml de tolueno en un frasco en una caja de guantes. El reactivo de Grignard (3,0 M en Et2O, 11,9 ml, 35,8 mmol) se añadió gota a gota lentamente a la mezcla amarilla mediante una jeringa. Después de la adición del reactivo de Grignard, la disolución se volvió marrón ligeramente oscuro. La disolución se agitó a temperatura ambiente durante la noche (de color negro a la mañana siguiente). La disolución se redujo a sequedad bajo vacío y los sólidos se extrajeron con 250 ml de hexano. La mezcla se filtró y los sólidos oscuros restantes se lavaron con 250 ml de hexano adicionales. Los filtrados combinados se filtraron una vez más y se redujeron a sequedad para dar el producto puro (6,957 g, 80,5 %). RMN 1H (400 MHz, CDCh) 58,31 (d, J = 1,6 Hz, 2 H), 8,07 (d, J = 1,5 Hz, 2 H), 7,59 (d, J = 2,3 Hz, 2 H), 7,45 (dd, J = 8,6, 1,7 Hz, 2 H), 7,37 (dd, J = 8,8, 1,8 Hz, 2 H), 7,34 (d, J = 8,6 Hz, 2 H), 7,25 (d, J = 2,3 Hz, 2 H), 7,22 (d, J = 8,6 Hz, 2 H), 6,92 (dd, J = 9,1,3,1 Hz, 2 H), 6,36 (m, 2 H), 4,59 (dd, J = 9,1,4,9 Hz, 2 H), 4,02 (br m, 2 H) 3,46 (d, J = 12,2 Hz, 2 H), 1,75 (s, 4H), 1,55 (s, 18 H), 1,40 (s, 6 H), 1,38 (s, 18 H), 1,33 (s, 6 H), 1,12 (br s, 4 H), 0,79 (s, 18 H), -1,79 (s, 6 H).
Ejemplo Comparativo C6
La preparación de este compuesto se informó previamente en Solicitud Internacional PCT de Número de Publicación WO2011025784.
Ejemplo de la Invención I1 a
I1a-1. Preparación del 5,5'-(((meso)-pentano-2,4-diil)bis(oxi))bis(4-bromo-1,2-difluorobenceno)

Se añadió THF (25 ml) a un matraz de fondo redondo de 250 ml cargado con 2-bromo-4,5-difluorofenol (2,53 g, 12,1 mmol) y (meso)-pentano-2,4-diol (0,6 g, 5,76 mmol). La mezcla se enfrió a 0°C y luego se añadieron secuencialmente el PPh3 (3,17 g, 12,1 mmol) y DIAD (2,38 ml, 12,1 mmol). La reacción se agitó durante 18 h mientras se agotaba lentamente el baño frío. Luego, los volátiles se eliminaron a presión reducida. Se añadió pentano (10 ml) al residuo bruto, que posteriormente se eliminó a presión reducida y luego se repitió este proceso. Se añadió de nuevo pentano (25 ml) y precipitó óxido de trifenilfosfina sólido. El sólido se eliminó por filtración y luego se lavó con pentano (25 ml). Las capas de pentano se combinaron y el disolvente se eliminó a presión reducida. El producto deseado se cristalizó en pentano (8 ml) para dar 1,75 gramos del producto. Las aguas madres se concentraron y el material resultante se trató con pentano/Et2O (3:1,2 ml) y se enfrió a -15°C durante 17 h. Se eliminó el disolvente amarillo, dejando cristales blancos (300 mg). Los cristales se lavaron con pentano frío (2 ml) para proporcionar un total combinado de 2 g (71 %) del compuesto del título como un sólido blanco. RMN 1H (400 MHz, CDCh) 57,40 (dd, J = 9,5, 8,4 Hz, 2H), 6,89 (dd, J = 11,9, 7,0 Hz, 2H), 4,54 (h, J = 6,2 Hz, 2H), 2,37 (dt, J = 13,7, 6,7 Hz, 1H), 1,87 (dt, J = 14,3, 5,9 Hz, 1H), 1,38 (d, J = 6,1 Hz, 6H). RMN 13C{1H} (101 MHz, CDCh) 5150,49 (dd, Jcf = 7,6, 2,7 Hz), 149,61 (dd, Jcf = 248,7, 13,5 Hz), 144,49 (dd, Jcf = 245,3, 13,5 Hz), 121,51 (dd, Jcf = 20,8, 1,4 Hz), 106,48 (dd, Jcf = 7,0, 3,9 Hz), 104,25 (d, Jcf = 21,3 Hz), 73,27, 42,41,19,69. RMN 19F{1 H} (376 MHz, CDCh) 5 -135,23 (d, J = 21,8 Hz), -145,32 (d, J = 21,7 Hz).
I1a-2. Preparación del ligando de la Invención I1a

Se añadieron tolueno desgasificado (10 ml) y agua desgasificada (2 ml) a un vial de 40 ml cargado con 4,4,5,5-tetrametil-2-(5'-fenil-2-((tetrahidro-2H-piran-2-il)oxi)-5-(2,4,4-trimetilpentan-2-il)-[1,1':3',1"-terfenil]-3-il)-1,3,2-dioxaborolano (2,51 g, 3,89 mmol), 5,5'-(((meso)-pentano-2,4-diil)bis(oxi))bis(4-bromo-1,2-difluorobenceno) (0,84 g, 1,73 mmol) y NaOH sólido (0,410 g, 10,4 mmol). La mezcla se burbujeó con N2 durante 5 min y luego se añadió Pd(PPh3)4 sólido (0,100 g, 0,090 mmol) al vial. La reacción se calentó a 85°C y se mantuvo a esta temperatura durante 17 h mientras se agitaba vigorosamente. Pasado este tiempo, la reacción se enfrió a temperatura ambiente. La mezcla se transfirió a un embudo de decantación, se añadieron tolueno (10 ml) y agua (8 ml) y se separaron las capas. Los extractos orgánicos se lavaron con agua (5 ml), salmuera (5 ml), se secaron (Na2SO4), y se filtraron. Se añadió MeOH (10 ml) a la disolución de tolueno anterior y luego se añadió HCl concentrado (8 gotas desde una pipeta de vidrio). El
matraz se equipó con un condensador, luego se calentó la mezcla a 75°C y se mantuvo a esta temperatura durante 3 h. Después de este tiempo, la reacción se enfrió a temperatura ambiente y aproximadamente la mitad del volumen de disolvente se eliminó a presión reducida. Al residuo crudo se añadieron agua (10 ml) y CH2Cl2 (15 ml), luego se transfirieron las fases a un embudo de decantación y se separaron. La fase acuosa se extrajo adicionalmente con CH2G 2 (15 ml) y los extractos orgánicos combinados se lavaron con salmuera (10 ml), se secaron (Na2SO4) y luego se pasaron a través de un tapón de SiO2. El tapón se lavó con CH2G 2 (65 ml) para asegurarse de que se recolectó todo el material (controlado por TLC). Fueron necesarias tres purificaciones para obtener el producto. Se añadió CH2G 2 (15 ml) al concentrado y luego se añadió CELITE a la fase orgánica. El disolvente se eliminó a presión reducida y el material sólido resultante se cargó directamente en una precolumna y se purificó mediante cromatografía en columna ultrarrápida (80 g SiO2, 0 % de EtOAc a 20 % de EtOAc en hexanos). Luego, el material aislado se purificó usando cromatografía de SiO2 (40 g SiO2, 0 % de acetona a 10 % de acetona en hexanos). Una purificación final por cromatografía C18 de fase inversa, comenzando con agua/CH3CN, luego cambiando a CH3CN/THF proporcionó 1,00 g (49 %) del ligando como un polvo blanco. RMN 1H (400 MHz, CDCh) ó 7,79 (t, J = 1,7 Hz, 2H), 7,73 (d, J = 1,7 Hz, 4H), 7,70 - 7,63 (m, 8H), 7,48 - 7,42 (m, 10H), 7,39 - 7,33 (m, 4H), 7,17 - 7,10 (m, 4H), 6,63 (dd, J = 11,5, 6,8 Hz, 2H), 5,83 (s, 2H), 4,32 - 4,20 (m, 2H), 2,08 - 1,92 (m, 1H), 1,73 (s, 4H), 1,65 - 1,46 (m, 1H), 1,38 (app d, J = 6,1 Hz, 12H), 1,06 (app d, J = 6,1 Hz, 6H), 0,76 (s, 18H). RMN 13C{1H} (101 MHz, CDCh) 5149,83 (dd, Jcf = 250,1, 13,7 Hz), 149,76 (dd, Jcf = 7,4, 2,0 Hz), 147,87, 145,41 (dd, Jcf = 243,5, 12,5 Hz), 142,43, 141,91, 141,10, 139,61, 128,99, 128,80, 128,75, 128,61, 127,47, 127,32, 127,28, 125,77 (dd, Jcf = 5,3, 3,6 Hz), 125,08, 124,33, 120,24 (d, Jcf = 18,4 Hz), 105,27 (d, Jcf = 19,7 Hz), 74,06, 57,03, 42,03, 38,12, 32,45, 31,93, 31,72, 31,57, 19,51. RMN 19F{1H} (376 MHz, CDCh) 5 -134,76 (d, J = 22,4 Hz), -145,15 (d, J = 22,4 Hz).
I1a-3. Preparación del Catalizador de la Invención I1a

F F F F F f 11a F F
En una caja de guantes, en un vial de centelleo de 40 ml secado en un horno con una barra de agitación, se suspendió ZrCl4 (0,06 g, 0,25 mmol) en tolueno (8 ml, anhidro). Se enfrió la mezcla a -30°C en un congelador, se retiró del congelador y luego se añadió MeMgBr 3M en Et2O (0,38 ml) con agitación. La disolución se agitó durante 5 min, luego se añadió el ligando sólido I1a (0,3 g, 0,25 mmol). La reacción se agitó mientras se calentaba lentamente a temperatura ambiente y se continuó agitando durante 18 h. La reacción se filtró a través de un embudo fritado que contenía un lecho de CELITE y el sólido se lavó con tolueno (5 ml). El disolvente se eliminó a presión reducida para producir un sólido marrón. Se añadió hexano (15 ml) al sólido, luego la disolución se filtró a través de un embudo fritado que contenía un lecho de CELITE. La torta del filtro se lavó con hexanos (10 ml), luego la capa de hexanos combinados se concentró a presión reducida para proporcionar 80 mg del procatalizador. Luego, la torta de filtración se extrajo con CH2G 2 (10 ml). El extracto de CH2G 2 se concentró a sequedad para dar un polvo. Este polvo se trató nuevamente con CH2G 2 (10 ml), y la disolución se pasó a través de un filtro de jeringa para eliminar los insolubles. El disolvente se eliminó para proporcionar 100 mg de procatalizador para un total de 0,18 g (53 %) de 11 a como un polvo blanquecino. RMN 1H (400 MHz, C6D6) 58,39 (br s, 2H), 8,29 (br s, 2H), 8,23 - 8,17 (m, 1H), 8,17 - 8,12 (m, 1H), 7,85 - 7,70 (m, 10H), 7,28 - 7,18 (m , 8H), 7,14 - 7,09 (m, 5H), 7,05 - 6,92 (m, 2H), 6,87 (dd, J = 10,6, 8,9 Hz, 1H), 5,52 (dd, J = 10,6, 7,2 Hz, 1H), 5,33 (dd, J = 10,6, 7,1 Hz, 1H), 4,29 - 4,21 (m, 1H), 3,60 - 3,47 (m, 1H), 1,77 (dd, J = 14,6, 7,6 Hz, 2H), 1,63 (dd, J = 28,7, 14,6 Hz, 2H), 1,41 - 1,24 (m, 14H), 0,81 (s, 9H), 0,72 (s, 9H), 0,53 (d, J = 6,1 Hz, 3H), 0,28 (d, J = 6,8 Hz, 3H), -0,10 (s, 3H), -0,25 (s, 3H). RMN 19F{1H} (376 MHz, CeDe) 5 -136,63 (d, J = 22,8 Hz), -137,27 (d, J = 23,0 Hz), -137,67 (d, J = 22,9 Hz), -137,91 (d, J = 22,7 Hz).
Ejemplo de la Invención I1b
I1b-1. Preparación del 2-bromo-3,4,5-trifluorofenol

A un frasco equipado con una barra de agitación se añadió el trifluorofenol (25 g, 168 mmol, 1 equivalente) disuelto en CH2G 2 (500 ml). Se añadió bromo (12,9 ml, 253 mmol, 1,5 equivalentes) gota a gota a temperatura ambiente y la disolución se dejó en agitación durante la noche. La reacción se monitoreó por NMR 1H, y se detuvo con NaHSO3 acuoso saturado cuando está se completó. La disolución bifásica se agitó hasta que el color rojo se disipó por completo
y luego se separaron las capas. La capa orgánica se lavó con salmuera, se filtró a través de un tapón de SiÜ2 y se eluyó con CH2Cl2. El filtrado se concentró para dar el producto como un compuesto transparente e incoloro (36,5 g, 95 %). RMN 1H (400 MHz, C6D6) 55,98 (ddd, J = 11,3, 6,5, 2,3 Hz, 1H) y 4,65 (s, 1H). RMN 19F{1H} (376 MHz, C6D6) 5 -126,15 (dd, J = 21,8, 6,0 Hz), -134,45 (dd, J = 22,0, 5,4 Hz), y -167,51 (t, J = 20,8 Hz). RMN 13C{1H} (101 MHz, CDCh) 5151,02 (ddd, Jcf = 248, 11,5 Hz), 148,82 (ddd, Jcf = 249, 12, 6 Hz), 148,70 (dt, Jcf = 13, 4 Hz), 135,20 (dt, Jcf = 249, 16 Hz), 99,85 (dd, Jcf = 22, 4, 2 Hz), 94,00 (dd, Jcf = 21,4 Hz).
I1b-2. Preparación del 5,5'-(((meso)-pentano-2,4-diil)bis(oxi))bis(4-bromo-1,2,3-trifluorobenceno)

En un matraz de fondo redondo de 100 ml se colocó el diol (0,5 g, 4,8 mmol, 1 equivalente) disuelto en THF (30 ml). El matraz se enfrió a 0°C, se añadió el fenol (2,3 g, 10,1 mmol, 2,1 equivalentes) seguido de PPh3 (2,64 g, 10,1 mmol, 2,1 equivalentes) y DIAD (1,96 g, 10,1 mmol, 2,1 equivalentes). La reacción se dejó calentar a 23°C y se agitó durante la noche. Cuando se completó por TLC, la reacción se detuvo con una disolución saturada acuosa de NH4G, se extrajo con Et2Ü, se secó con salmuera y MgSÜ4, se filtró y se concentró. La trituración con pentanos para eliminar el óxido de trifenilfosfina seguida de concentración para eliminar los pentanos proporcionó el grupo inferior como un sólido blanco. (61 % de rendimiento). RMN 1H (400 MHz, C6D6) 56,20 (ddd, J = 11,9, 6,3, 2,3 Hz, 2H), 3,89 (sexteto, J = 6,2 Hz, 2H), 1,92 (dt, J = 14,2, 6,6 Hz, 1H), 1,25 (dt, J = 14,2, 5,8 Hz, 1H), 0,83 (d, J = 6,0 Hz, 6H). RMN 19F{1H} (376 MHz, C6D6) 5 -123,98 (dd, J = 22,3, 4,3 Hz), -134,54 (dd, J = 22,8, 5,5 Hz) y -166,82 (t, J = 22,3 Hz). RMN 13C{1H} (101 MHz, CDCl3) 5 151,57, 151,52, 151,47, 151,42, 150,96, 150,90, 150,84, 150,78, 150,10, 150,06, 150,02, 150,00, 149,96, 149,93, 149,11, 149,06, 149,00, 148,95, 148,50, 148,45, 148,39,148,33, 136,35, 136,19, 133,89, 133,73, 98,21,98,18, 97,99, 97,96, 96,81,96,76, 96,62, 96,57, 72,62, 41,55, 18,91.
I1b-3. Preparación del Ligando de la Invención I1b

A un matraz de 250 ml que contiene el éster de boronato (1,78 g, 2,95 mmol) bajo N2 se añadió el bromuro de arilo (0,70 g, 1,34 mmol), NaÜH (0,322 g, 8,04 mmol), Pd(PPh3)4 (0,062 g, 0,054 mmol), 5,6 ml de tolueno desgasificado y 1,1 ml de agua desgasificada. El sistema se burbujeó con N2. La reacción se calentó a 110°C durante 72 h. La reacción se enfrió y los volátiles se eliminaron por evaporación rotatoria. El residuo se recogió en Et2Ü (100 ml), se lavó con salmuera (100 ml), se secó sobre MgSÜ4, se filtró a través de un lecho de SiÜ2, y luego se concentró para dar el ligando protegido. Para la etapa de desprotección del THP, este material se disolvió en 20 ml de THF y se añadieron 20 ml de MeÜH, seguido de aproximadamente 5 gotas de HCl concentrado. Esta mezcla se calentó a reflujo durante 1 hora, luego se enfrió y se concentró al vacío. Se añadió Et2Ü (250 ml) y la disolución resultante se lavó con agua (200 ml). La parte orgánica se separó y se secó sobre MgSÜ4, se filtró y se concentró al vacío. Este material crudo se purificó adicionalmente por cromatografía en columna sobre SiÜ2, eluyendo con un gradiente de EtÜAc/hexano al 0—>10 %, proporcionando el producto (1,18 g, 72 % de rendimiento) como un sólido blanco. RMN 1H (400 MHz, CDCh) 5 7,81 (m, 2H), 7,67 (m, 12H), 7,42 (m, 14H), 7,11 (m, 2H), 6,41 (m, 2H), 5,25 (m, 2H), 4,25 (m, 2H), 1,96 (m, 1H), 1,72 (m, 4H), 1,50 (m, 1H), 1,36 (m, 12H), 1,08 (m, 6H), 0,75 (m, 18H). RMN 19F{1H} (376 MHz, CDCh) 5 -132,22 (m, 2F), -133,50 (m, 1F), -133,77 (m, 1F), -168,33 (m, 1F), -169,14 (m, 1F).
I1b-4. Preparación del Ejemplo de la Invención I1b

A un vial equipado con una barra de agitación en una caja de guantes se añadió ZrCl4 (0,147 g, 0,631 mmol) y tolueno anhidro (16 ml). La mezcla se enfrió a -30°C y a esta se le añadió MeMgBr (0,884 ml, disolución 3,0 M en Et2O, 2,65 mmol). La suspensión resultante se agitó durante 2 minutos, después de lo cual se añadió el ligando I1b (0,776 g, 0,631 mmol) a la suspensión fría con agitación. La reacción se calentó a temperatura ambiente y se agitó durante la noche. Luego, la reacción se concentró para proporcionar un sólido oscuro, que se lavó con hexano (100 ml) y se filtró. El filtrado se concentró para proporcionar el producto como un sólido amarillo, que se lavó con hexano (20 ml), proporcionando el producto como un polvo blanco (0,171 g). La extracción de la torta del filtro restante con tolueno (100 ml), seguida de filtración y secado del filtrado al vacío, proporcionó 0,485 g adicionales del producto como un sólido marrón. El rendimiento combinado fue de 0,656 g (77 %). RMN 1H (400 MHz, C6D6) ó 8,34 (br s, 2H), 8,23 (br s, 2H), 8,16 (s, 1H), 8,11 (s, 1H), 7,80 (d, J = 2,5 Hz, 1H), 7,74 (m, 9H), 7,39 - 7,17 (m, 12H), 7,15 (m, 2H), 5,28 (m, 1H), 5,06 (m, 1H), 4,15 (m, 1 H), 3,53 (m, 1H), 1,86 - 1,60 (m, 4H), 1,41 (s, 3H), 1,40 (s, 3H), 1,35 (s, 3H), 1,32 (s, 3H), 1,05 (ddd, J = 16,3, 11,6, 8,7 Hz, 1 H), 0,86 (s, 9H), 0,77 (s, 9H), 0,52 (d, J = 6,1 Hz, 3H), 0,50 (m, 1 H), 0,26 (d, J = 6,7 Hz, 3H), -0,15 (s, 3H), -0,28 (s, 3H). RMN 19F{1H} (376 MHz, CeDe) 5-133,93 (dd, J = 22,5, 5,1 Hz, 1F), -134,57 (m, 2F), -135,35 (dd, J = 22,7, 4,9 Hz, 1F), -158,99 (t, J = 22,7 Hz, 1F), -159,49 (t, J = 22,6 Hz, 1F).
Ejemplo de la Invención I2
I2-1. Preparación del 1,4-bis(2-bromo-3,4,5-trifluorofenoxi)butano

A un matraz de fondo redondo de 500 ml se añadió 2-bromo-3,4,5-trifluorofenol (15,00 g, 66,09 mmol), 1,4-dibromobutano (2,631 ml, 22,03 mmol), K2CO3 (12,178 g, 88,11 mmol) y DMSO (240 ml). El matraz se fijó con el condensador Stevens y se calentó a 50°C durante la noche. Al día siguiente se calentó a 100°C durante 1 hora. La mezcla de reacción se añadió al agua que formó una emulsión. La suspensión acuosa se extrajo con CH2G 2. La capa orgánica se secó sobre MgSO4 , se filtró y se concentró. El sólido se lavó con hexano para eliminar las impurezas, produciendo un polvo blanquecino. Se recogieron 7,68 g, 68,7 % de rendimiento. RMN 1H (400 MHz, C6D6) 55,92 ((ddd, J = 11,1,6,1, 1,8 Hz, 2H), 3,12 (m, 4H) y 1,50 (m, 4H). RMN 19F{1H} (376 MHz, C6D6) 5 -124,7 (dd, J = 22,8, 5,8 Hz), -134,79 (dd, J = 22,0, 5,9 Hz) y -167,39 (t, J = 21,5 Hz).
I2-2. Preparación del Ligando de la invención I2

A un vial de 40 ml equipado con una barra de agitación se le añadió el fragmento borilado (6,009 g, 8,66 mmol) y el fragmento dibromado (2,00 g, 3,94 mmol). Se añadieron al vial tolueno burbujeado con N2 (15 ml) y una disolución de NaOH (0,945 g, 23,62 mmol) disuelta en agua (1 ml), seguido rápidamente por el Pd(PPh3)4 (0,182 g, 0,16 mmol) disuelto en tolueno burbujeado con N2 (1 ml). El vial se calentó bajo N2 a 80°C y se monitoreó por LCMS hasta completarse la reacción. Cuando se completó la reacción, se eliminó la capa inferior acuosa y se añadió una disolución acuosa de HCl al 20 % (1 ml). La mezcla de reacción se calentó a 80°C durante la noche. Cuando se completó la reacción, se enfrió a temperatura ambiente, se eliminó la capa acuosa, luego se lavó con salmuera, se secó sobre MgSO4 , se filtró y se concentró al vacío. El aceite espeso se recogió en MeOH y se filtró para recoger el sólido. Se
purificó mediante cromatografía ultrarrápida en SiÜ2, eluyendo con hexano:acetona 9:1. Se recogieron 2,73 g (52,8 % de rendimiento) como un sólido blanco. RMN 1H (400 MHz, C6Ü6) ó 8,38 (ddd, J = 11,1,6,1, 1,8 Hz, 2H), 7,55 - 7,22 (m, 20H), 3,33 (tq, J = 9,0, 4,0 Hz, 4H), 1,61 - 1,32 (m, 36H), 1,31 - 1,11 (m, 14H), 0,93 - 0,78 (m, 21H), RMN 13C{1H} (101 MHz, CDCh) 5148,77, 148,71,143,54, 143,49, 143,41,143,35, 142,53, 142,46, 139,98, 139,87, 130,22, 124,65, 124,52, 124,45, 124,37, 124,28, 124,24, 124,01,123,67, 118,23, 116,80, 116,73, 116,51,109,80, 109,39, 68,42, 56,74, 37,85, 34,50, 34,47, 32,14, 32,11, 31,74, 31,71, 31,67, 31,64, 31,34, 31,12, 31,02, 25,44. RMN 19F{1H} (376 MHz, CeDe) 5 -133,62 (m), -134,52 (m), y -169,9 (m)
I2-3. Preparación del Ejemplo de la Invención I2.

A un frasco equipado con una barra de agitación en una caja de guantes se añadió ZrCU (0,056 g, 0,228 mmol) disuelto en (10 ml). La frasco se enfrió a -30°C y se le añadió MeMgBr (0,304 ml, 0,913 mmol) y la disolución resultante se agitó durante 15 min, después de lo cual se añadió el ligando I2 (0,300 g, 0,228 mmol) a la suspensión fría con agitación. La reacción se calentó a 20-25°C y se agitó durante 3 h. La reacción se filtró y se concentró y luego se extrajo con hexanos. Las capas de hexanos se combinaron y concentraron para proporcionar el procatalizador como un sólido blanquecino (0,28 g, 84 %). RMN 1H (400 MHz, C6D6) 58,71 - 8,66 (m, 2H), 8,44 - 8,38 (m, 2H), 7,73 - 7,49 (m, 8H), 7,38 (dd, J = 8,8, 1,9 Hz, 4H), 7,31 (dd, J = 3,7, 2,3 Hz, 2H), 7,16 (p, J = 1,1 Hz, 4H), 4,84 - 4,74 (m, 2H), 3,88 - 3,80 (m, 2H), 3,32 (d, J = 11,7 Hz, 2H), 1,57 (s, 24H), 1,40 - 1,16 (m, 28H), 0,94 - 0,79 (m, 16H), -0,76 (s, 6H). RMN 19F{1H} (376 MHz, C6D6) 5 -131,4 (dd, J = 22,4, 5,0 Hz), -133,47 (dd, J = 22,6, 5,4 Hz) y -160,99 (t, J = 22,9 Hz).
Ejemplo de la Invención I3
I3-1. Preparación del 6',6"'-(((meso)-pentano-2,4-diil)bis(oxi))bis(2',3',4'-trifluoro-5-(2,4,4-trimetilpentan-2-il)-[1,1'-bifenil]-2-ol)

A un matraz de fondo redondo de 500 ml de dos bocas equipado con una barra de agitación se añadió el fragmento protegido con THP (9,16 g, 22,0 mmol) seguido del fragmento multifluorado (5,22 g, 10,0 mmol). Se añadieron el tolueno (30 ml) y el NaÜH acuoso (2,40 g, 60,0 mmol, en 8 ml H2O) al matraz seguido de burbujeo de N2 durante 15 min. Pd(PPh3)4 (0,462 g, 0,40 mmol) se disolvió en 17 ml de tolueno desgasificado seco y se inyectó en la mezcla de reacción. La reacción se calentó en una atmósfera de N2 a 80°C durante 18 h. Los análisis de HPLC y GC-MS indicaron la formación de producto, pero una conversión incompleta. Se añadió una cantidad adicional del fragmento protegido con THP (4,50 g, 1,08 equivalentes) en 10 ml de tolueno seco y desgasificado, junto con 0,02 equivalentes adicionales de Pd(PPh3)4 , también en 109 ml de tolueno seco y desgasificado. La mezcla resultante se calentó a 80°C hasta la conversión completa según lo indicado por GC-MS y HPLC. La reacción se trató con la adición de EtOAc y agua. Se separó la capa orgánica y se lavó a fondo con salmuera, se secó con MgSÜ4 anhidro, se filtró y se concentró. El residuo resultante se disolvió en 60 ml de THF y se añadió a 60 ml de agua que contenía varias gotas de HCl concentrado, seguido de calentamiento a 80°C durante 3 h. La mezcla de reacción enfriada se trató con la adición de EtÜAc y salmuera. Se separó la capa orgánica y se lavó a fondo con salmuera, se secó con MgSÜ4 anhidro, se filtró y se concentró. La cromatografía en columna ultrarrápida en SiÜ2 sobre la mezcla concentrada usando una mezcla en gradiente de EtOAc y hexanos proporcionó 7,67 g del producto deseado con un rendimiento del 99 %. RMN 1H (400 MHz, C6D6) 57,37 - 7,28 (m, 2H), 7,15 - 7,03 (m, 2H), 6,98 - 6,88 (m, 2H), 6,61 - 6,50 (m, 1 H), 6,45 - 6,36 (m, 1H), 5,33 - 5,04 (m, 2H), 4,37 - 4,23 (m, 1 H), 4,20 - 4,06 (m, 1 H), 1,75 - 1,64 (m, 4H), 1,41 - 1,20 (m, 16H), 1,10 - 1,01 (m, 3H), 0,93 - 0,81 (m, 3H), 0,80 - 0,59 (m, 18H). RMN 19F{1H} (376 MHz, C6D6) 5 -131,97, -132,74 (m, 2F), -133,23, -133,77 (m, 2F), -167,57 (dt, J = 83,7, 22,4 Hz, 1F), -168,24 (dt, J = 109,9, 22,4 Hz, 1F).
I3-2. Preparación del Ligando de la Invención I3

Se añadió una disolución de clorodiisopropilfosfina (0,197 g, 0,5 equivalentes) y precursor de Rh (0,25 equivalentes de monómero Rh o 0,125 equivalentes de dímero de Rh) en m-xileno (2 ml) a una mezcla de 9-bromoantraceno (2,00 g, 7,78 mmol), el fragmento bisfenólico (2,00 g, 2,59 mmol) y K3PO4 (2,20 g, 10,37 mmol) en 8 ml de m-xileno. La mezcla se calentó a 140°C durante 15 h. El análisis HPLC de la mezcla de reacción indicó la formación del producto deseado junto con una cantidad menor del intermedio de reacción (orto-arilación en solo un lado del sustrato bisfenólico). La reacción se enfrió a temperatura ambiente y se secó al vacío. El residuo bruto se sometió a cromatografía en columna ultrarrápida de fase normal usando una mezcla de gradiente de acetona y hexanos (hasta aproximadamente un 15 % de acetona), produciendo 1,08 g (37 % de rendimiento) del ligando I3. RMN 1H (400 MHz, CeDe) 58,62 - 8,49 (m, 2H), 8,15 - 7,97 (m, 4H), 7,80 - 7,19 (m, 15H), 7,17 - 7,08 (m, 1 H), 6,52 - 6,40 (m, 1 H), 6,30 -6,13 (m, 1 H), 4,73 - 4,51 (m, 2H), 4,44 - 4,26 (m, 2H), 2,16 - 1,98 (m, 2H), 1,84 - 1,61 (m, 4H), 1,49 - 1,28 (m, 12H) , 1,25 - 0,95 (m, 6 H), 0,92 - 0,72 (m, 18H). RMN 19F{1H} (376 MHz, C6D6) 5 -132,86 (ddd, J = 44,6, 23,0, 6,0 Hz, 1F), -133,39 (ddd, J = 23,1, 17,1,5,9 Hz, 1F), -134,36 (ddd, J = 22,0, 10,5, 5,9 Hz, 1 F), -135,03 (ddd, J = 23,2, 18,1,5,9 Hz, 1F), -169,22--169,73 (m, 2F).
I3-3. Preparación del Procatalizador de la Invención I3

En una caja de guantes, se suspendió ZrCl4 (0,025 g, 0,107 mmol) en 2,8 ml de tolueno anhidro en un vial de centelleo equipado con una barra de agitación. Luego, el vial de reacción se enfrió a -36°C en el congelador. A la mezcla enfriada se añadió MeMgBr (0,15 ml de disolución 3,0 M en éter, 0,44 mmol) con agitación. La mezcla se volvió oscura después de 3 a 4 min. Se añadió el ligando I3 (0,120 g, 0,107 mmol) a la mezcla seguido de agitación durante 3 h. Los volátiles se eliminaron al vacío y el residuo marrón se lavó a fondo con hexanos y se separó por filtración al vacío. El filtrado se concentró para producir 0,022 g del complejo deseado como un sólido de color amarillo claro. El residuo marrón del filtro se lavó con tolueno. Tras la eliminación del tolueno del filtrado al vacío, se aislaron 0,101 g de un sólido amarillo y posteriormente se identificó como el producto deseado. El rendimiento combinado fue de 0,123 g (93 %). RMN 1H (400 MHz, C6D6) 58,54 - 8,46 (m, 2H), 8,32 - 8,25 (m, 1H), 8,24 - 8,13 (m, 5H), 7,84 - 7,78 (m, 2H), 7,60 -7,54 (m, 2H), 7,48 - 7,19 (m, 7H), 7,13 - 6,93 (m, 5H), 4,07 - 3,88 (m, 3H), 3,84 - 3,74 (m, 1 H), 1,67 - 1,54 (m, 4H), 1,50 - 1,35 (m, 2H) , 1,35 - 1,21 (m, 12H), 0,86 (d, J = 17,8 Hz, 18H), 0,35 (d, J = 6,3 Hz, 3H), 0,22 (d, J = 6,3 Hz, 3H), -1,14 (s , 3H), -1,22 (s, 3H). RMN 19F{1H} (376 MHz, C6D6) 5 -133,57 (ddd, J = 46,6, 22,4, 5,4 Hz, 2F), -134,68 (dd, J = 22,4, 5,1 Hz, 1F), -135,13 (dd, J = 22,5, 5,4 Hz, 1 F), -160,58 (t, J = 22,4 Hz, 1 F), -161,02 (t, J = 22,5 Hz, 1F).
Ejemplo de la Invención I4
I1b-4. Preparación del Ejemplo de la Invención I4
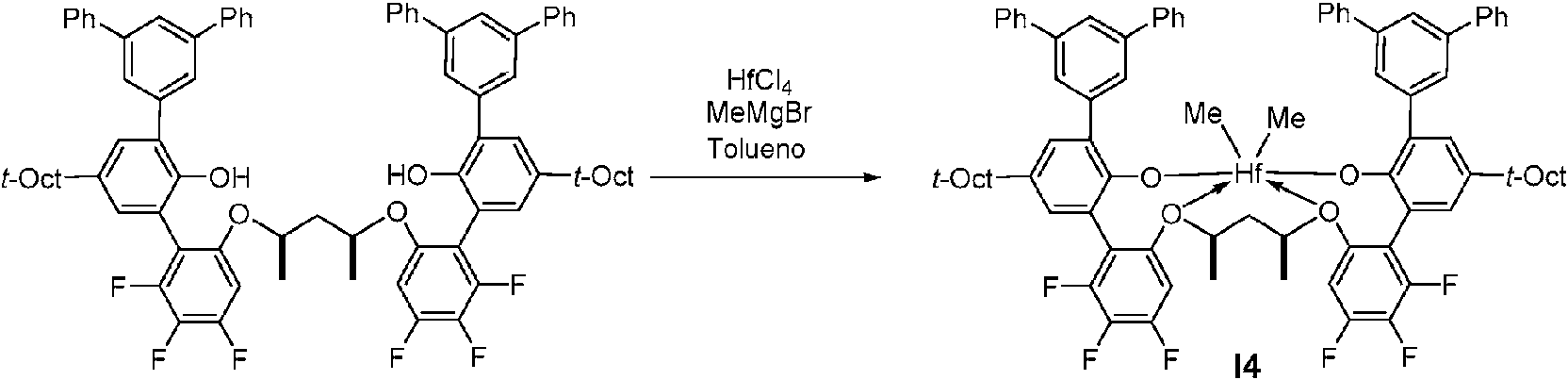
A un vial equipado con una barra de agitación en una caja de guantes se añadió HfCU (0,078 g, 0,244 mmol) y tolueno anhidro (6 ml). La mezcla se enfrió a -30°C y a esto se le añadió MeMgBr (0,342 ml, disolución 3,0 M en Et2O, 1,02
mmol). La suspensión resultante se agitó durante 10 min, después de lo cual se añadió el ligando I1b (0,300 g, 0,244 mmol) a la suspensión fría con agitación. La reacción se calentó a temperatura ambiente y se agitó durante 4 h. Luego, la reacción se concentró para proporcionar un sólido oscuro, que se lavó con hexano (100 ml) y se filtró. El filtrado se secó, proporcionando el producto como un polvo marrón (0,126 g). La extracción de la torta del filtro restante con tolueno (80 ml), seguida de filtración y secado del filtrado al vacío, proporcionó 0,217 g adicionales del producto en forma de un sólido marrón. El rendimiento combinado fue de 0,343 g (98 %). RMN 1H (400 MHz, C6D6) ó 8,32 (s, 2H), 8,19 (s, 2H), 8,17 (s, 1 H), 8,11 (s, 1H), 7,82 (d, J = 2,5 Hz, 1 H), 7,80 - 7,68 (m, 9H), 7,38 - 7,17 (m, 11H), 7,13 (m, 3H), 5,29 (m, 1 H), 5,08 (m, 1 H), 4,22 (p, J = 7,3 Hz, 1H), 3,58 (m, 1H), 1,85 - 1,60 (m, 4H), 1,41 (s, 3H), 1,40 (s, 3H), 1,35 (s, 3H), 1,32 (s, 3H) , 1,12 (m, 1H), 0,86 (s, 9H), 0,76 (s, 9H), 0,53 (d, J = 5,8 Hz, 3H), 0,50 (m, 1H), 0,27 (d, J = 6,7 Hz, 3H), -0,35 (s, 3H), -0,49 (s, 3H). RMN 19F{1H} (376 MHz, C6D6) 5 -133,92 (dd, J = 22,5, 5,0 Hz, 1F), -134,64 (d, J = 22,7 Hz, 2F), -135,45 (dd, J = 22,9, 4,9 Hz, 1 F), -158,81 (t, J = 22,4 Hz, 1F), -159,36 (t, J = 22,3 Hz, 1F).
Ejemplo de la Invención I5
I5-1. Preparación del Ejemplo de la Invención I5.

A un frasco equipado con una barra de agitación en una caja de guantes se añadió HfCU (0,066 g, 0,205 mmol) disueltos en tolueno (10 ml). La frasco se enfrió a -30°C y a esto se le añadió MeMgBr (0,26 ml, 0,779 mmol). La disolución resultante se agitó durante 15 minutos, después de lo cual se añadió el ligando I2 (0,256 g, 0,195 mmol) a la suspensión fría con agitación. La reacción se calentó a 20-25°C y se agitó durante 3 h. La reacción se filtró y se concentró y luego se extrajo con hexanos. Las capas de hexanos se combinaron y se concentraron para proporcionar el catalizador como un sólido blanquecino (0,27 g, 91 %). RMN 1H (400 MHz, C6D6) 58,69 (d, J = 1,8 Hz, 2H), 8,44 -8,38 (m, 2H), 7,73 - 7,65 (m, 4H), 7,61 (d, J = 8,5 Hz, 2H), 7,49 (dd, J = 8,7, 0,7 Hz, 2H), 7,40 - 7,27 (m, 4H), 7,15 (p, J = 1,0 Hz, 6H), 4,80 (ddd, J = 10,7, 6,5, 1,9 Hz, 2H), 3,94 (dd, J = 11,1,5,6 Hz, 2H), 3,40 (d, J = 11,5 Hz, 2H), 1,57 (s, 24H), 1,43 - 1,17 (m, 28H), 0,96 - 0,76 (m, 16H), -0,99 (s, 6H). RMN 19F{1H} (376 MHz, CeDe) 5 -131,12 (dd, J = 23,3, 5,2 Hz), -133,38 (dd, J = 22,4, 5,4 Hz) y -160,81 (t, J = 22,4 Hz).
Ejemplo de la Invención I6
I6-1. Preparación del 1,3-bis(2-bromo-3,4,5-trifluorofenoxi)propano

A un vial de 40 ml equipado con una barra de agitación se añadió 2-bromo-3,4,5-trifluorofenol (5,86 g, 25,8 mmol), 1,3-dibromopropano (0,874 ml, 8,6 mmol), K2CO3 (4,76 g, 34,4 mmol) y DMSO (15 ml). El vial se calentó a 50°C durante la noche, luego se calentó a 100°C durante 1 hora. Al día siguiente, la mezcla de reacción se enfrió y se añadió agua (8 ml) que dio como resultado la formación de un precipitado blanco. El sólido se filtró, se lavó con agua y se disolvió en CH2Cl2 donde se secó sobre MgSO4. Luego, el filtrado se recogió y se concentró al vacío. El sólido rosa pálido resultante se pasó a través de un tapón de SiO2 con tolueno como eluyente para proporcionar el compuesto deseado como un sólido blanco (3,57 g, 84 %). RMN 1H (400 MHz, C6D6) 55,92 (m, 2H), 3,32 (t, J = 5,9 Hz, 4H) y 1,59 (p, J = 5,9 Hz, 2H). RMN 19F{1H} (376 MHz, C6D6) 5 -124.68, (dd, J = 22,7, 6,1 Hz), - 134,5 (dd, J = 22,1,5,8 Hz) y -167,05 (t, J = 22,1 Hz). RMN 13C{1H} (101 MHz, CDCb) 5 151,76-151,87 (m), 151,34-151,71 (m), 150,99-151,11 (m), 149,35-149,4 (m), 148,54-148,71 (m), 136,59-136,92 (m), 134,13-134,46 (m) , 97,30-97,07 (m), 95,87-96,12 (m), 65,71 y 28,39.
I6-1. Preparación del Ligando de la invención I6

A un vial de 40 ml equipado con una barra de agitación se añadió el fragmento borilado (1,88 g, 4,45 mmol) y el compuesto dibromado (1,00 g, 2,02 mmol). Se añadieron al vial tolueno burbujeado con N2 (15 ml) y una disolución de NaOH (0,486 g, 12,1 mmol) disuelta en agua (1 ml), seguido rápidamente por el Pd(PPh3)4 (0,094 g, 0,08 mmol) disuelto en tolueno burbujeado con N2 (1 ml). El vial se calentó bajo N2 a 80°C y se monitoreó por LCMS hasta completar la reacción. Cuando se completó la reacción, se eliminó la capa inferior acuosa y se añadió una disolución acuosa al 20 % de HCl (1,00 ml). La mezcla de reacción se calentó a 80°C durante la noche. Cuando se completó la reacción, se enfrió a temperatura ambiente, se eliminó la capa acuosa, luego se lavó con salmuera, se secó sobre MgSO4 , se filtró y se concentró en el rotoevaporador. La purificación se realizó mediante cromatografía ultrarrápida en SiO2 usando un gradiente de hexano:acetona 9:1 seguido de una columna C18 de fase inversa con agua:CH3CN, gradiente de CH3CN del 70 al 100 %. Se recogieron 1,37 g (89,4 % de rendimiento) de producto como un sólido blanco. RMN 1H (400 MHz, C6D6) 57,55-7,54 (m, 2H), 7,47 - 7,37 (m, 4H), 7,23 - 7,17 (m, 4H), 6,87 (td, J = 5,7, 2,1 Hz, 2H), 6,07 (dtd, J = 7,9, 5,9, 2,9 Hz, 2H), 5,26 (d, J = 2,4 Hz, 2H), 3,35 - 3,21 (m, 4H), 2,17 - 2,11 (m, 6H), 1,40 (d, J = 3,6 Hz, 2H), 1,37 - 1,29 (m, 2H), 1,29 - 1,17 (m, 32H). RMN 19F{1 H} (376 MHz, C6D6) 5 -132,48 (m), -134,52 (m) y -170,32 (m). RMN 13C{1H} (101 MHz, CDCb) 5 151,79, 151,76, 148,76, 148,68, 136,88, 136,84, 136,82, 131,80, 131,72, 131,50, 131,43, 129,95, 129,93, 129,86, 129,25, 129,22, 129,19, 128,31, 127,91, 127,79, 127,67, 127,55, 127,43, 123,49, 121,73, 117,33, 117,28, 113,32, 96,83, 65,54, 64,96, 64,79, 34,67, 34,60, 34,49, 31,58, 31,28, 31,14, 27,91, 27,77, 27,61,26,87, 25,27, 22,67, 20,51,20,11,20,06, 13,97.
I6-1. Preparación de Procatalizador de la Invención I6

A un frasco equipado con una barra de agitación en una caja de guantes se añadió ZrCL (0,079 g, 0,34 mmol) y tolueno (10 ml) y se enfrió a -30°C. A esto se le añadió MeMgBr (0,432 ml de una disolución 3M en Et2O, 1,29 mmol) y la disolución resultante se agitó durante 15 min, después de lo cual se añadió el ligando (0,300 g, 0,324 mmol) a la suspensión fría con agitación. La reacción se calentó a 20-25°C y se agitó durante 3 h. La reacción se filtró y se concentró y luego se extrajo con hexanos. Las capas de hexanos se combinaron y se concentraron para proporcionar el procatalizador como un sólido blanquecino (0,22 g, 59 %). RMN 1H (400 MHz, CeDe) 57,78 (t, J = 1,9 Hz, 2H), 7,56 (s, 1 H), 7,31 (d, J = 2,3 Hz, 2H), 7,16 (t, J = 1,3 Hz, 3H), 6,91 (t, J = 2,8 Hz, 2H), 4,96 (ddd, J = 9,9, 6,7, 2,0 Hz, 2H), 3,40 (dt, J = 9,8, 4,7 Hz, 2H), 2,96 (dt, J = 10,6, 5,6 Hz, 2H) , 2,16 (s, 6H), 1,49 - 1,21 (m, 33H), 0,93 - 0,85 (m, 1H), 0,60 - 0,50 (m, 2H), 0,14 (s, 6H). RMN 19F{1H} (376 MHz, C6D6) 5 -132,64 (dd, J = 22,1,5,3 Hz), -133,65 (dd, J = 22,5, 5.1 Hz) y -160,19 (t, J = 22,7 Hz).
Ejemplos de la invención


Polimerización
Se usaron los Catalizadores Comparativos 1-6 y los Catalizadores de la Invención 1-6 para polimerizar etileno y 1 octeno en un reactor de polimerización por lotes de 2 litros de Parr. Todas las alimentaciones se pasaron a través de columnas de alúmina y catalizador Q-5 (disponible de Engelhard Chemicals Inc.) antes de la introducción en el reactor de polimerización. Las disoluciones de procatalizador y de activador se manipularon en una caja de guantes bajo atmósfera de N2 antes de la adición al reactor de polimerización.
La Tabla 1 ilustra las polimerizaciones por lotes y la información del polímero resultante en las siguientes condiciones: temperatura del reactor: 147°C; ISOPAR-E: 1.325 ml; 1-octeno: 250 g; presión de etileno: 3,1 MPa (450 psi); Tiempo de reacción: 10 min. bis(alquil de sebo hidrogenado)metil, tetrakis(pentafluorofenil)borato(1-)amina: 1,1 equivalentes; MMAO: 50 equivalentes ISOPAR-E es un disolvente isoparafínico que está comercialmente disponible en ExxonMobil Chemical Company (Bayport TX). La Tabla 1 proporciona los resultados de la polimerización para dicha polimerización por lotes.
Tabla 1

Se llevaron a cabo reacciones de polimerización adicionales en un proceso continuo bajo las siguientes condiciones: Las materias primas (etileno, 1-octeno) y el disolvente del proceso (un disolvente isoparafínico de alta pureza de intervalo de ebullición estrecho de marca comercial ISOPAR E disponible comercialmente de ExxonMobil Corporation) se purifican con tamices moleculares antes de introducirlos en el medio de reacción. El hidrógeno se suministra en cilindros presurizados como un grado de alta pureza y no se purifica más. La corriente de alimentación de monómero (etileno) del reactor se presuriza mediante de un compresor mecánico a una presión de reacción superior a 3,61 MPa (525 psig). La alimentación de disolvente y comonómero (1-octeno) se presuriza mediante una bomba mecánica de desplazamiento positivo a una presión de reacción superior a 3,61 MPa (525 psig). El metilaluminoxano modificado (MMAO), disponible comercialmente de AkzoNobel, se usa como eliminador de impurezas. Los componentes individuales del catalizador (cocatalizador y procatalizador) se diluyen manualmente por lotes a las concentraciones
de componentes especificadas con disolvente purificado (Isopar E) y se presurizan a una presión superior a la de reacción a 3,61 MPa (525 psig). El cocatalizador es [HNMe(C18H37)2][B(C6F5)4], comercialmente disponible de Boulder Scientific, y se usa en una relación molar de 1,2 con respecto al procatalizador. Todos los flujos de alimentación de reacción se miden con medidores de flujo másico y se controlan de forma independiente con sistemas de control de válvulas automatizadas por computadora.
Las polimerizaciones en disolución en continuo se llevan a cabo en un reactor de tipo tanque agitado continuo (CSTR, por sus siglas en ingles) de 3,785 l (1 galón). La alimentación combinada de disolvente, monómero, comonómero e hidrógeno al reactor tiene una temperatura controlada entre 5°C y 30°C y es típicamente 15°C. Todos estos materiales se alimentan al reactor de polimerización con la alimentación de disolvente. El catalizador se alimenta al reactor para alcanzar una conversión específica de etileno. El cocatalizador se alimenta por separado en base a una relación molar específica calculada (1,2 equivalentes molares) al componente catalizador. El MMAO comparte la misma línea que el cocatalizador y el flujo se basa en una concentración de Al en el reactor o en una relación molar específica a componente del catalizador. El efluente del reactor de polimerización (que contiene disolvente, monómero, comonómero, hidrógeno, componentes del catalizador y polímero fundido) sale del reactor y se pone en contacto con agua para terminar la polimerización. Además, en este punto se pueden añadir varios aditivos, tales como antioxidantes. Luego, la corriente pasa a través de un mezclador estático para dispersar uniformemente la eliminación del catalizador y los aditivos.
Luego de la adición de los aditivos, el efluente (que contiene disolvente, monómero, comonómero, hidrógeno, componentes del catalizador y polímero fundido) pasa a través de un intercambiador de calor para elevar la temperatura de la corriente en preparación para la separación del polímero de los otros componentes de reacción de menor punto de ebullición. Luego, la corriente pasa a través de la válvula de control de presión del reactor, a través de la cual se recure considerablemente la presión. Desde allí, entra a un sistema de separación de dos etapas que consta de un desvolatizador y una extrusora de vacío, donde se eliminan del polímero el disolvente y el hidrógeno sin reaccionar, el monómero, el comonómero y el agua. A la salida de la extrusora, la hebra de polímero fundido formada pasa por un baño de agua fría, donde solidifica. Luego, la hebra se alimenta a través de una cortadora de hebras, donde el polímero se corta en gránulos después de secarse al aire.

Métodos de prueba
Los métodos de prueba incluyen lo siguiente:
Densidad
Las muestras cuya densidad se mide se preparan según la norma ASTM D-1928. Las mediciones se realizan dentro de una hora posterior al prensado de la muestra usando la norma ASTM D-792, Método B.
Índices de fluidez se miden según la norma ASTM D-1238.
Relación de reactividad
Las relaciones de reactividad de los catalizadores se pueden obtener por métodos conocidos, por ejemplo, la técnica descrita en "Linear Method for Determining Monomer Reactivity in Copolymerization", M. Fineman y S. D. Ross, J. Polymer Science, 5, 259 (1.950) o "Copolymerization", F. R. Mayo y C. Walling, Chem. Rev., 46, 191 (1.950). Un modelo de copolimerización ampliamente usado se basa en las siguientes ecuaciones:

en donde M1 se refiere a una molécula de monómero que se designa arbitrariamente como "i" en donde i = 1,2; y M2* se refiere a una cadena de polímero en crecimiento a la que el monómero i se ha unido más recientemente.
Los valores de K son las constantes de velocidad para las reacciones indicadas. Por ejemplo, en la copolimerización de etileno/propileno, K11 representa la velocidad a la que una unidad de etileno se inserta en una cadena de polímero en crecimiento en la que la unidad de monómero previamente insertada también era etileno. Las relaciones de reactividad siguen como: n=Kn/K 12 y r2=K22/K21 donde K11, K12 y K21 son las constantes de velocidad para la adición de etileno (1) o de propileno (2) a un sitio de catalizador donde el último monómero polimerizado es un etileno (K1x) o propileno (K2x)
Determinación de Tc y Tm
La calorimetría diferencial de barrido (DSC, por sus siglas en inglés) se puede usar para medir el comportamiento de fusión y de cristalización de un polímero en un amplio intervalo de temperatura. Por ejemplo, para realizar este análisis se usa el DSC Q1000 de TA Instruments, equipado con un RCS (sistema de enfriamiento refrigerado) y un muestreador automático. Durante la prueba, se usa un flujo de gas de purga de N2 de 50 ml/min. Cada muestra se prensa en estado fundido en una película delgada a aproximadamente 175°C; la muestra fundida luego se enfría al aire a temperatura ambiente (25°C). Se extrae una muestra de 3-10 mg y 6 mm de diámetro del polímero enfriado, se pesa, se coloca en una bandeja de aluminio ligera (aproximadamente 50 mg) y se cierra por presión. Luego se realiza un análisis para determinar sus propiedades térmicas.
El comportamiento térmico de la muestra se determina aumentando y disminuyendo la temperatura de la muestra para crear un perfil de flujo de calor versus temperatura. Primero, la muestra se calienta rápidamente a 180°C y se mantiene isoterma durante 3 min para eliminar su historial térmico. A continuación, la muestra se enfría a -40°C a una velocidad de enfriamiento de 10°C/minuto y se mantiene isoterma a -40°C durante 3 min. A continuación, la muestra se calienta a 150°C (esta es la rampa del "segundo calentamiento") a una velocidad de calentamiento de 10°C/minuto. Se registran las curvas de enfriamiento y del segundo calentamiento. La curva del frío se analiza estableciendo puntos finales de referencia desde el comienzo de la cristalización hasta -20°C. La curva del calor se analiza estableciendo puntos finales de referencia desde -20°C hasta el final de la fusión. Los valores determinados son la temperatura máxima de fusión (Tm) y la temperatura máxima de cristalización (Tc).
Eficiencia del catalizador (Eficiencia)
La eficiencia del catalizador se calcula dividiendo el número de gramos del copolímero de poliolefina preparado por el número total de gramos de metal M del ingrediente (a) empleado (es decir, el metal M del al menos un complejo metalligando de la fórmula (I) (es decir, eficiencia del catalizador = g de copolímero de poliolefina preparado/g del metal M del (de los) complejo(s) metal-ligando(s) de fórmula (I) empleado(s)).
Determinación de Mw y MWD
Los pesos moleculares promedio en número y en peso (Mi y Mw, respectivamente) de los polímeros se determinaron mediante cromatografía de permeación en gel (GPC, por sus siglas en inglés). El sistema cromatográfico consistía en un Modelo PL-210 de Polymer Laboratories o un Modelo PL-220 de Polymer Laboratories. Los compartimentos de las columnas y del carrusel se operaron a 140°C. Se usaron tres columnas Mixed-B de 10 micras de Polymer Laboratories con un disolvente de 1,2,4-triclorobenceno. Las muestras se prepararon a una concentración de 0,1 g de polímero en
50 ml de solvente. El disolvente usado para preparar las muestras contenía 200 ppm de hidroxitolueno butilado (BHT). Las muestras se prepararon agitando ligeramente durante 2 horas a 160°C. El volumen de inyección usado fue de 100 pl y el caudal fue de 1,0 ml/min. La calibración del conjunto de columnas de GPC se realizó con patrones de poliestireno de MWD estrecho adquiridos de Polymer Laboratories. Los pesos moleculares máximos estándar de poliestireno se convirtieron a pesos moleculares de polietileno usando:
Mpolietileno = A(Mpoliestireno)B
Dónde M es el peso molecular, A tiene un valor de 0,4316, y B es igual a 1,0. Los cálculos del peso molecular del equivalente de polietileno se realizaron usando el programa informático Viscotek TriSEC, versión 3.0. La distribución del peso molecular (MWD) se define como Mw/Mn.
La presente invención se puede realizar de otras formas sin apartarse del espíritu y los atributos esenciales de la misma y, en consecuencia, se debe hacer referencia a las reivindicaciones adjuntas, en lugar de a la especificación anterior, como indicación del alcance de la invención.
Claims (12)
1. Un procatalizador para la polimerización de etileno y, opcionalmente, una o más alfa olefinas con la estructura que se muestra en la fórmula (I) a continuación:

en donde:
M es titanio, zirconio o hafnio, estando cada uno independientemente en un estado de oxidación formal de 2,
3 o 4; y n es un número entero de desde 0 a 3, y en donde cuando n es 0, X está ausente; y
cada X es independientemente un ligando monodentado que es neutro, monoaniónico o dianiónico; o se juntan dos X para formar un ligando bidentado que es neutro, monoaniónico o dianiónico; y X y n se eligen de tal manera que el complejo metal-ligando de la fórmula (I) sea globalmente neutro; y
cada Z es independientemente O, S, N hidrocarbilo de (C1-C40) , o P hidrocarbilo de (C1-C40);
L es hidrocarbileno de (C2-C40) o heterohidrocarbileno de (C2-C40), en donde el hidrocarbileno de (C2-C40) tiene una parte que comprende un cadena principal conectora de 2 átomos de carbono a 10 átomos de carbono que une los átomos Z en la fórmula (I) (a la que está unido L) y el heterohidrocarbileno de (C2-C40) tiene una parte que comprende un cadena principal conectora de 3 átomos a 10 átomos que une los átomos Z en la fórmula
(I), en donde cada uno de los 3 a 10 átomos de la cadena principal conectora de 3 átomos a 10 átomos del heterohidrocarbileno de (C2-C40) es independientemente un átomo de carbono o heteroátomo, en donde cada heteroátomo es independientemente O, S, S(O), S(O)2, Si(RC)2, Ge(RC)2, P(RP), o N(Rn), en donde independientemente cada RC se selecciona del grupo que consiste en hidrocarbilo de (C1-C40), cada RP es hidrocarbilo de (C1-C40); y cada RN es hidrocarbilo de (C1-C40) o está ausente; y
R1-10 se seleccionan cada uno independientemente del grupo que consiste en un hidrocarbilo de (C1-C40), heterohidrocarbilo de (C1-C40), Si(RC)s, Ge(RC)s, P(RP)2, N(RN)2, ORC, SRC, NO2, CN, CF3, RCS(O)-, RCS( (Rc)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O)-, átomo de halógeno, átomo de hidrógeno y cualquier combinación de los mismos, y
al menos dos de Y 1-Y3 y al menos dos de Y4-Y6 son átomos de flúor y cuando sólo dos de Y 1-Y3 y solo dos de
Y4-Y6 son átomos de flúor, los Y 1-Y6 no fluorados se seleccionan del grupo que consiste en átomo de H, grupos alquilo, grupos arilo, grupos heteroarilo y grupos alcoxi; y
cada uno de los grupos hidrocarbilo, heterohidrocarbilo, Si(RC)3, Ge(RC)3, P(RP)2, N(RN)2, ORC, SRC, RCS(O)-, RCS(O)2-, (Rc)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O)-, hidrocarbileno y heterohidrocarbileno no están o están sustituidos independientemente con uno o más sustituyentes RS, cada RS es independientemente un átomo de halógeno, sustitución de polifluoro, sustitución de perfluoro, alquilo de (C1-C18) no sustituido, F3C-, FCH2O-, F2HCO-, F3CO-, R3Si-, R3Ge-, RO-, RS-, RS(O)-, RS(O)2-, R2P-, R2N-, R2C=N-, NC-, RC(O)O-, ROC(O)-, RC(O)N(R)-, o R2NC(o )-, o dos de los RS se toman juntos para formar un hidrocarbileno de (C1-C18) no sustituido en donde cada R es independientemente un hidrocarbilo de (C1-C18); opcionalmente dos o más grupos R de los grupos R1-10 (por ejemplo, de R1-4, R5-8) se pueden combinar juntos en estructuras de anillo con tales estructuras de anillo teniendo de 2 a 50 átomos en el anillo excluyendo cualquier átomo de hidrógeno; y
en donde cuando el heterohidrocarbilo de (C1-C40) es un heteroarilo de (C1-C40) se selecciona de 2,7-di(terciario-butil)-carbazolilo, 3,6-di(terciario-butil)-carbazolilo, 2,7-di(terciario-octil)-carbazolilo, 3,6-di (terciariooctil)-carbazolilo, 2,7-difenilcarbazolilo, 2,7-bis(2,4,6-trimetilfenil)-carbazolilo o 3,6-bis(2,4,6-trimetilfenil)-carbazolilo.2
2. El procatalizador según la reivindicación 1, con la estructura que se muestra a continuación:

3. El procatalizador según la reivindicación 1, con tiene la estructura que se muestra a continuación:

4. El procatalizador según la reivindicación 1, con la estructura que se muestra a continuación:

5. El procatalizador según la reivindicación 1, con la estructura que se muestra a continuación:

6. El procatalizador según la reivindicación 1, con la estructura que se muestra a continuación:

7. El procatalizador según la reivindicación 1, con la estructura que se muestra a continuación:

8. El procatalizador según la reivindicación 1, con la estructura que se muestra a continuación:

9. Un proceso de polimerización para producir polímeros a base de etileno que comprende polimerizar etileno y, opcionalmente, una o más a-olefinas en presencia de un sistema catalítico en un sistema reactor con uno o más reactores, en donde el sistema catalítico comprende;
(a) uno o más procatalizadores que comprenden un complejo metal-ligando de la fórmula (I) dada a continuación:

en donde:
M es titanio, zirconio o hafnio, estando cada uno independientemente en un estado de oxidación formal de 2, 3 o 4; y n es un número entero de 0 a 3, y en donde cuando n es 0, X está ausente; y
cada X es independientemente un ligando monodentado que es neutro, monoaniónico o dianiónico; o se juntan dos X para formar un ligando bidentado que es neutro, monoaniónico o dianiónico; y X y n se eligen de tal manera que el complejo metal-ligando de la fórmula (I) sea globalmente neutro; y
cada Z es independientemente O, S, N hidrocarbilo de (C1-C40), o P hidrocarbilo de (C1-C40); L es hidrocarbileno de (C2-C40) o heterohidrocarbileno de (C2-C40), en donde el hidrocarbileno de (C2-C40) tiene una parte que comprende un cadena principal conectora de 2 átomos de carbono a 10 átomos de carbono que une los átomos Z en la fórmula (I) (a la que está unido L) y el heterohidrocarbileno de (C2-C40) tiene una parte que comprende un cadena principal conectora de 3 átomos a 10 átomos que une los átomos Z en la fórmula (I), en donde cada uno de los 3 a 10 átomos de la cadena principal conectora de 3 átomos a 10 átomos del heterohidrocarbileno de (C2-C40) es independientemente un átomo de carbono o heteroátomo, en donde cada heteroátomo es independientemente O, S, S(O), S(O)2, Si(RC)2, Ge(RC)2, P(RP), o N(RN), en donde cada RC se selecciona independientemente del grupo que consiste en hidrocarbilo de(C1-C40), cada RP es hidrocarbilo de (C1-C40); y cada RN es hidrocarbilo de (C1-C40) o está ausente; y
R1-10 se seleccionan cada uno independientemente del grupo que consiste en un hidrocarbilo de (C1-C40), heterohidrocarbilo de (C1-C40), Si(RC)3, Ge(RC)3, P(Rp)2, N(Rn)2, ORc , SRc , NO2, CN, C3F, RCS(O)-, RCS(O)2-, (Rc )2C=N-, Rc C(O)O-, Rc OC(O)-, Rc C(O)N(R)-, (Rc)2NC(O)-, átomo de halógeno, átomo de hidrógeno y cualquier combinación de los mismos, y
al menos dos de Y1-Y3 y al menos dos de Y4-Y6 son átomos de flúor y cuando sólo dos de Y1-Y3 y solo dos de Y4-Y6 son átomos de flúor, los Y1-Y6 no fluorados se seleccionan del grupo que consiste en átomo de H, grupos alquilo, grupos arilo, grupos heteroarilo y grupos alcoxi; y
cada uno de los grupos hidrocarbilo, heterohidrocarbilo, Si(RC)3, Ge(RC)3, P(RP)2, N(RN)2, ORC, SRC, RCS(O)-, Rc SO)2-, (Rc )2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O), hidrocarbileno y heterohidrocarbileno no están sustituidos o están sustituidos independientemente con uno o más sustituyentes RS, cada RS es independientemente un átomo de halógeno, sustitución de polifluoro, sustitución de perfluoro, alquilo de (C1-C18) no sustituido, F3C-, FCH2O-, F2HCO-, F3CO-, R3Si-, R3Ge-, RO-, RS-, RS(O)-, RS(O)2-, R2P-, R2N-, R2C=N-, NC-, RC(O)O-, ROC(O)-, RC(O)N(R)-, o R2NC(O)-, o dos de los RS se juntan para formar un hidrocarbileno de (C1-C18) no sustituido en donde cada R es independientemente un hidrocarbilo de (C1-C18);
opcionalmente dos o más grupos R de los grupos R1-10 (por ejemplo, de R1-4, R5-8) se pueden combinar juntos en estructuras de anillo teniendo tales estructuras de anillo de 2 a 50 átomos en el anillo excluyendo cualquier átomo de hidrógeno; y
en donde cuando el heterohidrocarbilo de (C1-C40) es un heteroarilo de (C1-C40) se selecciona de 2,7-di(terciario-butil)-carbazolilo, 3,6-di(terciario-butil)-carbazolilo, 2,7-di(terciario-octil)-carbazolilo, 3,6-di (terciariooctil)-carbazolilo, 2,7-difenilcarbazolilo, 2,7-bis(2,4,6-trimetilfenil)-carbazolilo o 3,6-bis(2,4,6-trimetilfenil)-carbazolilo.
10. El proceso de polimerización según la reivindicación 9, en donde una o más alfa-olefinas están presentes en uno o más reactores.
11. El proceso de polimerización según la reivindicación 10, en donde el procatalizador presenta una relación de reactividad de menos de o igual a 8.
12. El proceso de polimerización según la reivindicación 10, en donde el procatalizador presenta una relación de reactividad que es menor de una relación de reactividad de un catalizador comparativo con una estructura idéntica al procatalizador, excepto que el catalizador comparativo no incluye dos o más átomos de F en cada uno de Y1-Y3 y Y4-Y6.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562234910P | 2015-09-30 | 2015-09-30 | |
| PCT/US2016/054088 WO2017058858A1 (en) | 2015-09-30 | 2016-09-28 | A polymerization process for producing ethylene based polymers |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2925720T3 true ES2925720T3 (es) | 2022-10-19 |
Family
ID=57104215
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16778178T Active ES2925720T3 (es) | 2015-09-30 | 2016-09-28 | Un proceso de polimerización para producir polímeros a base de etileno |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US10647797B2 (es) |
| EP (1) | EP3356430B1 (es) |
| KR (1) | KR102589562B1 (es) |
| CN (1) | CN108377649B (es) |
| BR (1) | BR112018005909B1 (es) |
| ES (1) | ES2925720T3 (es) |
| WO (1) | WO2017058858A1 (es) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2983941T3 (es) | 2016-09-29 | 2024-10-28 | Dow Global Technologies Llc | (Pro)catalizador y sistema ziegler-natta modificados |
| US11008412B2 (en) | 2016-09-29 | 2021-05-18 | Dow Global Technologies Llc | Method of polymerizing an olefin |
| KR102449474B1 (ko) | 2016-09-29 | 2022-10-11 | 다우 글로벌 테크놀로지스 엘엘씨 | 마그네슘 할라이드-담지 티탄 (전)촉매 |
| WO2018183700A1 (en) | 2017-03-31 | 2018-10-04 | Dow Global Technologies Llc | Bis-biphenyl-phenoxy catalysts for olefin polymerization |
| BR112019020416B1 (pt) | 2017-03-31 | 2023-05-16 | Dow Global Technologies Llc | Sistema de catalisador e processo de polimerização para produzir um polímero à base de etileno |
| EP3642248B1 (en) * | 2017-06-20 | 2024-12-18 | Dow Global Technologies LLC | Biaryl phenoxy group iv transition metal catalysts for olefin polymerization |
| KR102100142B1 (ko) * | 2017-12-21 | 2020-04-14 | 사빅 에스케이 넥슬렌 컴퍼니 피티이 엘티디 | 금속-리간드 착체, 이를 포함하는 에틸렌계 중합용 촉매 조성물 및 이를 이용한 에틸렌계 중합체의 제조방법 |
| ES2971111T3 (es) | 2018-08-30 | 2024-06-03 | Dow Global Technologies Llc | Procatalizadores de poliolefina bis-fenil-fenoxi que tienen dos ligandos de antracenilo |
| JP7706366B2 (ja) * | 2018-12-20 | 2025-07-11 | ダウ グローバル テクノロジーズ エルエルシー | オレフィン重合のためのビアリールフェノキシiv族遷移金属触媒 |
| US12297303B2 (en) * | 2019-10-01 | 2025-05-13 | Nova Chemicals (International) S.A. | High productivity polymerization with aryloxy ether ligand catalyst |
| KR20220097924A (ko) | 2019-11-04 | 2022-07-08 | 다우 글로벌 테크놀로지스 엘엘씨 | 바이모달 촉매 시스템 |
| CA3156388A1 (en) | 2019-11-04 | 2021-05-14 | Angela I. Padilla-Acevedo | Biphenylphenol polymerization catalysts |
| KR20220097897A (ko) * | 2019-11-04 | 2022-07-08 | 다우 글로벌 테크놀로지스 엘엘씨 | 티타늄 바이페닐페놀 중합 촉매 |
| WO2021155158A1 (en) | 2020-01-31 | 2021-08-05 | Dow Global Technologies Llc | Group iii and lanthanide bis-phenyl-phenoxy metal-ligand complexes |
| WO2021242801A1 (en) | 2020-05-29 | 2021-12-02 | Dow Global Technologies Llc | Attenuated hybrid catalysts |
| JP7774578B2 (ja) | 2020-05-29 | 2025-11-21 | ダウ グローバル テクノロジーズ エルエルシー | 弱化ポストメタロセン触媒 |
| US20240150502A1 (en) | 2021-02-15 | 2024-05-09 | Dow Global Technologies Llc | Biphenylphenol polymerization catalysts |
| CA3207943A1 (en) | 2021-02-15 | 2022-08-18 | Ruth Figueroa | Method for making a poly(ethylene-co-1-alkene) copolymer with reverse comonomer distribution |
| WO2024074937A1 (en) * | 2022-10-07 | 2024-04-11 | Sabic Sk Nexlene Company Pte. Ltd. | Transition metal compound, catalyst composition containing the same, and method for preparing olefin polymer using the same |
Family Cites Families (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1069848A (en) | 1965-05-17 | 1967-05-24 | Imp Chemical Imdustries Ltd | Process for the polymerisation of vinyl chloride |
| US7163907B1 (en) | 1987-01-30 | 2007-01-16 | Exxonmobil Chemical Patents Inc. | Aluminum-free monocyclopentadienyl metallocene catalysts for olefin polymerization |
| US5153157A (en) | 1987-01-30 | 1992-10-06 | Exxon Chemical Patents Inc. | Catalyst system of enhanced productivity |
| US5064802A (en) | 1989-09-14 | 1991-11-12 | The Dow Chemical Company | Metal complex compounds |
| JP2545006B2 (ja) | 1990-07-03 | 1996-10-16 | ザ ダウ ケミカル カンパニー | 付加重合触媒 |
| US5721185A (en) | 1991-06-24 | 1998-02-24 | The Dow Chemical Company | Homogeneous olefin polymerization catalyst by abstraction with lewis acids |
| US5296433A (en) | 1992-04-14 | 1994-03-22 | Minnesota Mining And Manufacturing Company | Tris(pentafluorophenyl)borane complexes and catalysts derived therefrom |
| US5350723A (en) | 1992-05-15 | 1994-09-27 | The Dow Chemical Company | Process for preparation of monocyclopentadienyl metal complex compounds and method of use |
| US5372682A (en) | 1993-06-24 | 1994-12-13 | The Dow Chemical Company | Electrochemical preparation of addition polymerization catalysts |
| US5625087A (en) | 1994-09-12 | 1997-04-29 | The Dow Chemical Company | Silylium cationic polymerization activators for metallocene complexes |
| JP3990458B2 (ja) | 1996-03-27 | 2007-10-10 | ダウ グローバル テクノロジーズ インコーポレーテッド | 溶解度の高いオレフィン重合触媒活性剤 |
| BR9708335A (pt) | 1996-03-27 | 1999-08-03 | Dow Chemical Co | Processo de polimerização em solução para polimerizar olefinas |
| BR9709302A (pt) | 1996-05-02 | 1999-08-10 | Equistar Chem Lp | Sistema catalisador contendo um produto de silicone líquido e poliamina |
| US5783512A (en) | 1996-12-18 | 1998-07-21 | The Dow Chemical Company | Catalyst component dispersion comprising an ionic compound and solid addition polymerization catalysts containing the same |
| US6103657A (en) | 1997-07-02 | 2000-08-15 | Union Carbide Chemicals & Plastics Technology Corporation | Catalyst for the production of olefin polymers |
| US6696379B1 (en) | 1997-09-19 | 2004-02-24 | The Dow Chemical Company | Supported modified alumoxane catalyst activator |
| JP2000159829A (ja) | 1998-11-27 | 2000-06-13 | Tosoh Corp | オレフィン重合用触媒およびそれを用いたポリオレフィンの製造方法 |
| CA2393793C (en) | 1999-12-10 | 2009-09-29 | Jerzy Klosin | Substituted group 4 metal complexes, catalysts and olefin polymerization process |
| US7060848B2 (en) | 2002-04-24 | 2006-06-13 | Symyx Technologies, Inc. | Bridged bi-aromatic catalysts, complexes, and methods of using the same |
| CA2483192A1 (en) | 2002-04-24 | 2003-11-06 | Symyx Technologies, Inc. | Bridged bi-aromatic ligands, complexes, catalysts and processes for polymerizing and poymers therefrom |
| MXPA06010481A (es) | 2004-03-17 | 2006-12-19 | Dow Global Technologies Inc | Composicion catalizadora que comprende agente de enlace para la formacion de copolimeros de multiples bloques de olefina superior. |
| WO2007136497A2 (en) | 2006-05-17 | 2007-11-29 | Dow Global Technologies Inc. | High temperature solution polymerization process |
| CN102015874B (zh) | 2008-02-29 | 2014-08-27 | 陶氏环球技术有限责任公司 | 包括乙烯/α-烯烃嵌段互聚物的取向膜 |
| US8372930B2 (en) | 2008-06-20 | 2013-02-12 | Exxonmobil Chemical Patents Inc. | High vinyl terminated propylene based oligomers |
| WO2010061630A1 (ja) | 2008-11-28 | 2010-06-03 | 三菱レイヨン株式会社 | 発泡成形用加工助剤、発泡成形用塩化ビニル系樹脂組成物及び発泡成形体 |
| ES2638913T3 (es) | 2009-08-31 | 2017-10-24 | Dow Global Technologies Llc | Catalizador y procedimiento para polimerizar una olefina y poliolefina preparada mediante el mismo |
| JP5806218B2 (ja) | 2009-10-02 | 2015-11-10 | ダウ グローバル テクノロジーズ エルエルシー | ブロック複合材及び耐衝撃性改質組成物 |
| US8476366B2 (en) | 2009-10-02 | 2013-07-02 | Dow Global Technologies, Llc | Block compositions in thermoplastic vulcanizate applications |
| BR112012020720B1 (pt) | 2010-02-19 | 2020-01-21 | Dow Global Technologies Llc | processo para polimerizar um monômero olefínico |
| KR20130004585A (ko) | 2010-03-02 | 2013-01-11 | 다우 글로벌 테크놀로지스 엘엘씨 | 에틸렌-기재 중합체 조성물 |
| WO2011146044A1 (en) | 2010-05-17 | 2011-11-24 | Dow Global Technologies Llc | Process for selectively polymerizing ethylene and catalyst therefor |
| MX375966B (es) | 2010-05-17 | 2025-03-06 | Dow Global Technologies Llc | Proceso para polimerizacion selectiva de etileno y un catalizador del mismo. |
| EP2591021A2 (en) | 2010-07-06 | 2013-05-15 | Ticona GmbH | Ultra-high molecular weight polyethylene, its production and use |
| WO2012027448A1 (en) * | 2010-08-25 | 2012-03-01 | Dow Global Technologies Llc | Process for polymerizing a polymerizable olefin and catalyst therefor |
| US20120116034A1 (en) | 2010-11-08 | 2012-05-10 | Dow Global Technologies, Inc. | Solution polymerization process and procatalyst carrier systems useful therein |
| KR101869997B1 (ko) | 2010-12-03 | 2018-06-22 | 다우 글로벌 테크놀로지스 엘엘씨 | 에틸렌-기재 중합체 조성물의 제조 방법 |
| US9643900B2 (en) | 2011-03-25 | 2017-05-09 | Dow Global Technologies Llc | Hyperbranched ethylene-based oils and greases |
| CA2861504C (en) | 2011-12-29 | 2019-11-05 | Dow Global Technologies Llc | Process for producing low molecular weight ethylene- and alpha-olefin-based materials |
| EP2797963B1 (en) | 2011-12-29 | 2019-07-03 | Dow Global Technologies LLC | Hyperbranched olefin oil-based dielectric fluid |
| CN104735995B (zh) | 2012-10-18 | 2018-10-12 | 陶氏环球技术有限公司 | 基于油酸和中等链长甘油三酯、低粘度、高闪点的介电流体 |
| EP2938649B1 (en) | 2012-12-27 | 2019-04-10 | Dow Global Technologies LLC | A polymerization process for producing ethylene based polymers |
| ES2750524T3 (es) | 2012-12-27 | 2020-03-26 | Dow Global Technologies Llc | Procedimiento para la polimerización de etileno y octeno |
| EP2938648B1 (en) | 2012-12-27 | 2019-01-30 | Dow Global Technologies LLC | A polymerization process for producing ethylene based polymers |
| KR20150100844A (ko) | 2012-12-27 | 2015-09-02 | 다우 글로벌 테크놀로지스 엘엘씨 | 에틸렌계 중합체 |
| EP2938643B1 (en) | 2012-12-27 | 2018-01-31 | Dow Global Technologies LLC | Catalyst systems for olefin polymerization |
| WO2014209917A1 (en) * | 2013-06-23 | 2014-12-31 | Reterro, Inc. | Controlling processes for evaporative desorption processes |
| MX374936B (es) * | 2013-06-28 | 2025-03-06 | Dow Global Technologies Llc | Oligomeros hiper-ramificados a base de etileno. |
| CN105283474A (zh) | 2013-06-28 | 2016-01-27 | 陶氏环球技术有限责任公司 | 使用卤化双苯基苯氧基催化剂控制聚烯烃的分子量 |
| JP6327471B2 (ja) | 2013-09-25 | 2018-05-23 | 三菱ケミカル株式会社 | 電線被覆材及び被覆された電線 |
| EP3050922A4 (en) | 2013-09-25 | 2016-09-28 | Mitsubishi Rayon Co | SOFT VINYL CHLORIDE RESIN COMPOSITION, FORM BODY, ELECTRIC CABLE COATING MATERIAL AND COATED ELECTRICAL CABLE |
| CN107787336B (zh) | 2015-06-30 | 2021-05-28 | 陶氏环球技术有限责任公司 | 用于制备基于乙烯的聚合物的聚合方法 |
| WO2017004456A1 (en) | 2015-06-30 | 2017-01-05 | Dow Global Technologies Llc | A polymerization process for producing ethylene based polymers |
-
2016
- 2016-09-28 ES ES16778178T patent/ES2925720T3/es active Active
- 2016-09-28 BR BR112018005909-9A patent/BR112018005909B1/pt active IP Right Grant
- 2016-09-28 CN CN201680066247.0A patent/CN108377649B/zh active Active
- 2016-09-28 WO PCT/US2016/054088 patent/WO2017058858A1/en not_active Ceased
- 2016-09-28 US US15/763,937 patent/US10647797B2/en active Active
- 2016-09-28 EP EP16778178.0A patent/EP3356430B1/en active Active
- 2016-09-28 KR KR1020187010477A patent/KR102589562B1/ko active Active
Also Published As
| Publication number | Publication date |
|---|---|
| BR112018005909A2 (pt) | 2018-10-16 |
| US10647797B2 (en) | 2020-05-12 |
| CN108377649A (zh) | 2018-08-07 |
| KR102589562B1 (ko) | 2023-10-16 |
| US20180282452A1 (en) | 2018-10-04 |
| EP3356430B1 (en) | 2022-06-22 |
| EP3356430A1 (en) | 2018-08-08 |
| BR112018005909B1 (pt) | 2022-05-03 |
| WO2017058858A1 (en) | 2017-04-06 |
| CN108377649B (zh) | 2021-04-23 |
| KR20180063135A (ko) | 2018-06-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2925720T3 (es) | Un proceso de polimerización para producir polímeros a base de etileno | |
| US11242422B2 (en) | Polymerizations for olefin-based polymers | |
| ES2796870T3 (es) | Sistemas catalíticos para la polimerización de olefina | |
| ES2956113T3 (es) | Catalizadores de bis-bifenil-fenoxi con puente de germanio para la polimerización de olefinas | |
| ES2785628T3 (es) | Un proceso de polimerización para producir polímeros basados en etileno | |
| ES2665586T3 (es) | Sistemas catalizadores para polimerización de olefinas | |
| BR112018006183B1 (pt) | Pró-catalisador e processo de polimerização | |
| ES2971111T3 (es) | Procatalizadores de poliolefina bis-fenil-fenoxi que tienen dos ligandos de antracenilo | |
| ES2811135T3 (es) | Un procedimiento de polimerización para producir polímeros a base de etileno | |
| ES3044042T3 (en) | Group iii and lanthanide bis-phenyl-phenoxy metal-ligand complexes | |
| BR112019019130B1 (pt) | Sistema catalisador de polimerização de olefina e processo para preparar um copolímero de múltiplos blocos | |
| ES2988765T3 (es) | Catalizadores de polimerización de metales de transición del grupo IV de biarilhidroxitiofeno con capacidad de transferencia de cadena | |
| ES2979233T3 (es) | Sistema de catalizador para la formación de copolímero multibloque | |
| ES2919998T3 (es) | Un catalizador de polimerización de olefinas | |
| CN106029675A (zh) | 吡啶基二氨基过渡金属络合物、其制备和用途 | |
| JP7550164B2 (ja) | オレフィン重合触媒活性化剤のための弱配位性アニオンとしてのアニオン性iii族錯体 | |
| ES2911503T3 (es) | Catalizadores de bis-fenil-fenoxi-poliolefina que tienen un ligando de metilentrialquilsilicio en el metal para mejorar la solubilidad | |
| ES2880630T3 (es) | Complejos de metales de transición de fenolato, producción y uso de los mismos | |
| BR112021002240B1 (pt) | Sistema de catalisador, e, processo de polimerização |