ES2743153T3 - Métodos para tratar el síndrome de fibromialgia - Google Patents
Métodos para tratar el síndrome de fibromialgia Download PDFInfo
- Publication number
- ES2743153T3 ES2743153T3 ES10828479T ES10828479T ES2743153T3 ES 2743153 T3 ES2743153 T3 ES 2743153T3 ES 10828479 T ES10828479 T ES 10828479T ES 10828479 T ES10828479 T ES 10828479T ES 2743153 T3 ES2743153 T3 ES 2743153T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- enantiomer
- compounds
- administration
- fibromyalgia
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/22—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acyclic acids, e.g. pravastatin
- A61K31/223—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acyclic acids, e.g. pravastatin of alpha-aminoacids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/27—Esters, e.g. nitroglycerine, selenocyanates of carbamic or thiocarbamic acids, meprobamate, carbachol, neostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Emergency Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compuesto que tiene la fórmula estructural (1) o sal farmacéuticamente aceptable del mismo para su uso en el tratamiento de síntomas asociados con síndrome de fibromialgia que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto que tiene la fórmula estructural (1) o una sal farmacéuticamente aceptable del mismo, en el que el compuesto que tiene la fórmula estructural (1) o una sal farmacéuticamente aceptable del mismo es un enantiómero (R) libre de otro enantiómero o una mezcla enantiomérica en la que el enantiómero (R) del compuesto que tiene la fórmula estructural (1) predomina hasta el grado del 90% o más y el otro enantiómero es el 10% o menos, a un mamífero que padece síndrome de fibromialgia que necesita tratamiento, en el que los síntomas asociados con síndrome de fibromialgia se seleccionan del grupo que consiste en dolor y alteración del sueño:**Fórmula** en la que, R, R1 y R2 son hidrógeno y x=1.
Description
DESCRIPCIÓN
Métodos para tratar el síndrome de fibromialgia
[Campo técnico]
La presente invención se refiere al compuesto O-carbamoilfenilalaninol para su uso en el tratamiento de dolor y alteraciones del sueño como síntomas del síndrome de fibromialgia en un mamífero que padece síndrome de fibromialgia.
[Antecedentes de la técnica]
El síndrome de fibromialgia es una enfermedad crónica que implica dolor generalizado, rigidez y dolor a la palpación en tejidos relacionados con el músculo esquelético incluyendo músculos, tendones y ligamentos (Bennett, 2009). Los pacientes con fibromialgia muestran alteraciones del sueño, fatiga, ansiedad y/o fibroniebla. La fibroniebla abarca la incapacidad para concentrarse, pérdida de memoria y depresión. Se produce fibromialgia en aproximadamente el 2% de la población general de los Estados Unidos con una mayor incidencia en mujeres (3,4%) en comparación con los hombres (0,5%) (Mease, 2005; Arnold et al., 2006).
El síntoma central de la fibromialgia es el dolor generalizado descrito como que surge del músculo y las articulaciones (Bennett, 2009). Muchos pacientes con fibromialgia tienen una piel dolorosa a la palpación. El dolor normalmente aumenta y disminuye en intensidad con exacerbaciones que acompañan a un ejercicio inusual, inactividad prolongada, lesiones de tejidos blandos, cirugía, sueño insuficiente, exposición al frío, largos viajes en coche y estrés. El dolor tiene predominantemente una ubicación axial pero también puede producirse en manos y pies.
La fatiga es un síntoma común para los pacientes con fibromialgia. La fatiga, aunque usada de manera intercambiable con somnolencia, se ha descrito como un cansancio de la mente y el cuerpo que perjudica la productividad y el disfrute de la vida (Bennett, 2009). El tratamiento con antidepresivos da como resultado sólo una modesta mejora de la fatiga, lo que sugiere que hay más en el síntoma de fatiga que la depresión. El aumento del sueño reparador no es suficiente solo para reducir la fatiga.
Los patrones del sueño comúnmente están alterados en pacientes con fibromialgia (Bennett, 2009). Hay problemas con el inicio del sueño y el mantenimiento del sueño pero los pacientes se sienten cansados cuando se despiertan, por tanto la excesiva somnolencia diurna da como resultado una mayor incompatibilidad con el funcionamiento diario que el inicio y mantenimiento del sueño.
Los pacientes con fibromialgia normalmente notifican dolor a la palpación en donde hay sensibilidad al tacto y se experimenta dolor incluso tras un contacto mínimo (Bennett, 2009). La duración del dolor generalizado debe ser de al menos tres meses y el dolor a la palpación debe producirse en 11 o más de 18 puntos de dolor a la palpación específicos con el fin de clasificarse como fibromialgia (Mease, 2005; Arnold et al., 2006). Además, dificultades con la memoria, concentración y realización de dos tareas son problemas notificados por pacientes con fibromialgia. Se ha mostrado que la fibromialgia es comórbida con trastorno bipolar, trastorno depresivo mayor, cualquier trastorno de ansiedad, cualquier trastorno de la alimentación y cualquier trastorno por consumo de sustancias (Arnold et al, 2006). Estas enfermedades pueden compartir vínculos fisiopatológicos subyacentes y el tratamiento debe elegirse teniendo en cuenta las características de presentación comórbidas.
Aunque la etiología de la fibromialgia se desconoce, hay pruebas que sugieren algunas anomalías en la neurotransmisión monoaminérgica central incluyendo los sistemas de serotonina y norepinefrina (Verdu et al., 2008). Se han explorado antidepresivos tricíclicos (TCA) y SNRI como tratamiento para la fibromialgia. Los TCA incluyendo amitriptilina y ciclobezaprina mostraron una modesta eficacia con mejoras en las autoclasificaciones de dolor, rigidez, fatiga, sueño y dolor a la palpación. Se obtuvieron resultados limitados y contradictorios para SRI incluyendo fluoxetina y citalopram. Se obtuvieron resultados positivos para SNRI incluyendo duloxetina y milnacipran con una mayor mejora en el dolor a la palpación en comparación con TCA y estos compuestos se aprobaron específicamente para el tratamiento de la fibromialgia por la FDA en 2007 y 2009, respectivamente.
Los antidepresivos usados para tratar la fibromialgia muestran reacciones adversas en un porcentaje significativo de los pacientes (Verdu et al., 2008). Se ha mostrado que la introducción progresiva de antidepresivos aumentaba la tolerabilidad. Los TCA muestran sequedad de la boca, estreñimiento y dificultades en el vaciado urinario y del intestino; efectos asociados con propiedades anticolinérgicas. También son comunes sedación, adormecimiento e hipotensión ortostática. Los efectos secundarios comunes de SRI están asociados con las acciones de la serotonina e incluyen náuseas, molestias gástricas, vómitos, anorexia, diarrea e hiperhidrosis cutánea. Además, existe el riesgo de dependencia física y abstinencia con el cese abrupto del tratamiento con antidepresivos. Además, los antidepresivos normalmente interfieren con la función sexual y el deseo. Se evaluó la tolerabilidad de la duloxetina a través de cinco ensayos clínicos de más de 6 meses de duración y los efectos secundarios más comunes incluían nauseas, cefaleas, boca seca, insomnio, fatiga, estreñimiento, diarrea y mareo (Choy et al., 2009), abandonando aproximadamente el 20% de los pacientes debido a los efectos adversos. Aunque el milnacipran generalmente se
toleraba bien, aproximadamente el doble de pacientes en los grupos tratados se retiraron del estudio debido a efectos adversos incluyendo nauseas, estreñimiento, palpitaciones y sofoco en comparación con aquellos en el grupo tratado con placebo (Harris & Clauw, 2008).
Se han recetado fármacos antiepilépticos incluyendo gabapentina y pregabalina a pacientes con subtipos de dolor neuropático y fibromialgia (Mease, 2005). La pregabalina fue el primer fármaco aprobado para la fibromialgia en 2007 por la FDA. La pregabalina ha demostrado eficacia en varios ensayos recientes de fibromialgia con puntuaciones de dolor, calidad del sueño y fatiga mejoradas (Kim et al., 2009). Los efectos secundarios comunes de la pregabalina incluían mareo, somnolencia y aumento de peso. Efectos secundarios menos comunes son dificultades para concentrarse y prestar atención, boca seca y visión borrosa. La pregabalina es también un compuesto incluido en la lista V lo que sugiere cierto potencial de abuso y síntomas de abstinencia. Por tanto, aunque la pregabalina mostró dolor reducido en algunos pacientes, hay todavía espacio para la mejora de tanto la eficacia como el perfil de efectos secundarios.
Otros compuestos que pueden ser útiles en el tratamiento de la fibromialgia incluyen inhibidores de monoamina oxidasa (es decir, pirlindol), antagonistas de 5-HT3 (es decir, tropisetrón), opioides, tramadol, relajantes musculares, antagonistas del receptor de NMDA y agonistas de dopamina (es decir, pramipexol) (Mease, 2005). Muchos de estos compuestos muestran o bien una eficacia débil, una eficacia de espectro menos amplio para los síntomas o bien perfiles de efectos secundarios intolerables incluyendo drogodependencia.
El documento WO-A-98/15526 da a conocer compuestos según la fórmula (I) y su utilidad en el tratamiento del dolor y la depresión.
El documento US-A-2008/0039529 da a conocer compuestos según la fórmula (I) como útiles para tratar la depresión.
El documento WO-A-2006/133393 da a conocer compuestos según la fórmula (I) y su utilidad para tratar la “somnolencia diurna excesiva” (EDS) y la narcolepsia.
El documento US-A-2008/090902 da a conocer la utilidad de los compuestos según la fórmula (I) en el tratamiento de dolor neuropático.
El documento US-A-5955499 da a conocer la actividad de compuestos según la fórmula (I) en un modelo de natación forzada.
[Divulgación]
[Problema técnico]
Por consiguiente, existe la necesidad en el tratamiento del síndrome de fibromialgia que mejorara la eficacia en el tratamiento de dolor, sueño, fatiga y otras comorbilidades incluyendo depresión y redujera los perfiles de efectos secundarios.
[Solución técnica]
La presente invención se define mediante las reivindicaciones en el presente documento.
Se da a conocer un método de tratamiento del dolor y la alteración del sueño como síntomas del síndrome de fibromialgia que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto que tiene la fórmula estructural (1) o una sal farmacéuticamente aceptable del mismo, a un mamífero que necesita tratamiento:

en la que,
R, R1 y R2 son hidrógeno, y x es 1.
El compuesto que tiene la fórmula estructural (1) en la que R, R1 y R2 son hidrógeno y x=1 es un enantiómero (R) sustancialmente libre de otros enantiómeros o una mezcla enantiomérica en la que predomina el enantiómero (R) del compuesto que tiene la fórmula estructural (1). El enantiómero (R) predomina hasta el grado del 90% o más, y preferiblemente el 98% o más.
El enantiómero dado a conocer es el enantiómero (S) o (L) tal como se representa mediante la fórmula estructural (1a) o el enantiómero (R) o (D) según la invención, tal como se representa mediante la fórmula estructural (1b):

R, R1 y R2 se seleccionan todos de hidrógeno y x es 1, que se muestran en la siguiente fórmula:
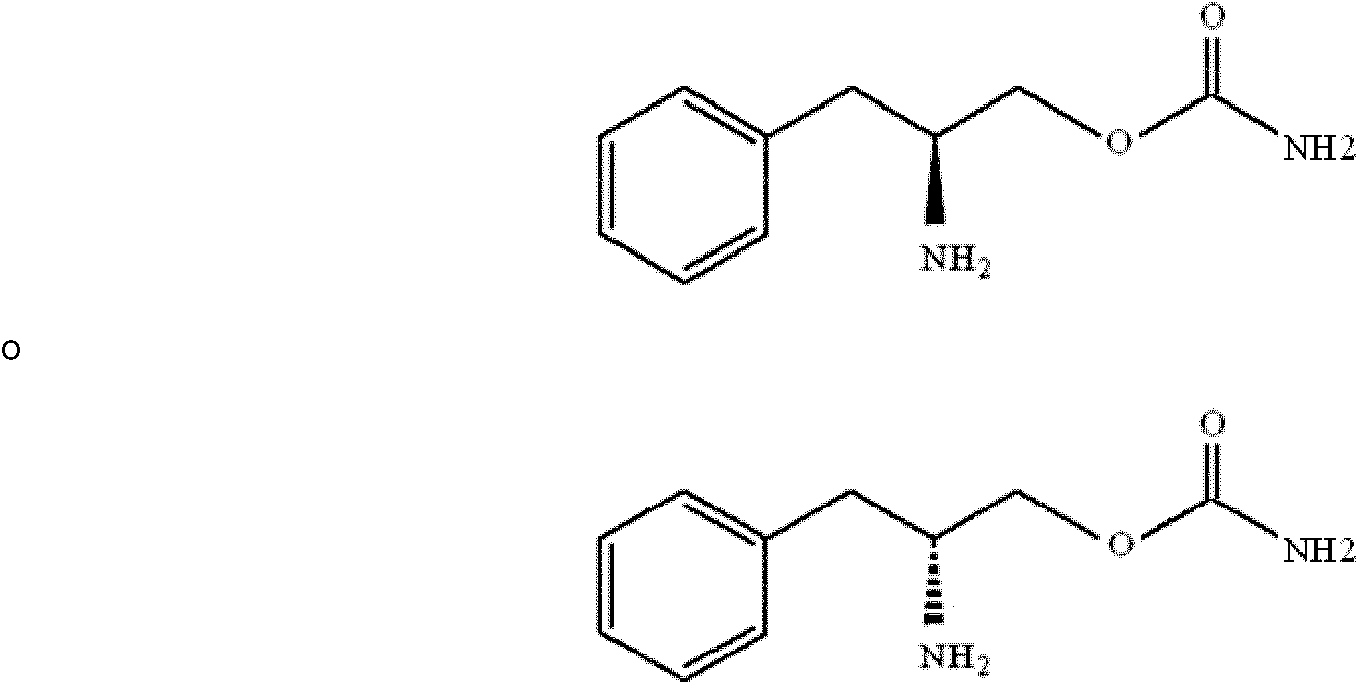
(nota: en la fórmula estructural de la fórmula 1b a continuación el grupo amino unido al carbono beta sobresale en el plano del papel. Este es el enantiómero dextrorrotatorio (D) que es de configuración absoluta (R).
[Efectos ventajosos]
La presente invención se basa en parte en el descubrimiento de que los carbamatos de fenilalquilamino de fórmula 1 comentados anteriormente tienen propiedades farmacológicas novedosas y únicas. Se ha mostrado en varios modelos animales que estos compuestos tienen la capacidad de tratar el dolor y la alteración del sueño como síntomas asociados con el síndrome de fibromialgia.
Aunque el mecanismo de acción preciso no se entiende completamente, se sabe que estos compuestos no funcionan mediante los mismos mecanismos que la mayoría de los otros tratamientos conocidos para el síndrome de fibromialgia. Por estos motivos, los compuestos de fórmula 1 son especialmente adecuados para su uso como único tratamiento o tratamiento adjunto para la fibromialgia y la modificación de los síntomas asociados con el síndrome de fibromialgia.
[Mejor modo]
Estos y otros objetos de la invención se entenderán más completamente a partir de la siguiente descripción de la invención y las reivindicaciones adjuntas a la misma.
La presente invención se refiere a un compuesto que tiene la fórmula estructural (I) para su uso en un método de tratamiento del dolor y la alteración del sueño como síntomas del síndrome de fibromialgia que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto que tiene la fórmula estructural (1) incluyendo ambos enantiómeros, y sales farmacéuticamente aceptables del mismo, a un mamífero que padece síndrome de fibromialgia y que necesita tratamiento:

en la que,
R, Ri y R2 son hidrógeno y x=1.
La presente invención también incluye el uso de un compuesto seleccionado del grupo que consiste en la fórmula 1a de la divulgación o 1b de la invención, o sales farmacéuticamente aceptable sales del mismo:

en las que Rx, R1 y R2 son los mismos que se definieron anteriormente.
En la fórmula estructural de la fórmula 1b, el grupo amino unido al carbono beta sobresale en el plano del papel. Este es el enantiómero dextrorrotatorio (D) que es de configuración absoluta (R).
O-Carbamoil-(D)-fenilalaninol también se denomina carbamato de (R)-(beta-amino-bencenopropilo).
Los compuestos de fórmula 1 pueden sintetizarse mediante métodos conocidos por un experto en la técnica. Se han descrito algunos esquemas de reacción para sintetizar compuestos de fórmula (1) en la patente estadounidense n.° 5705640, patente estadounidense n.° 5756817, patente estadounidense n.° 5955499 y patente estadounidense n.° 6140532 publicadas. Se han descrito detalles de los esquemas de reacción anteriores así como ejemplos representativos de la preparación de compuestos específicos en la patente estadounidense n.° 5705640, patente estadounidense n.° 5756817, patente estadounidense n.° 5955499 y patente estadounidense n.° 6140532 publicadas.
Las sales de los compuestos de fórmula (1) pueden producirse tratando el compuesto con un ácido (HX) en un disolvente adecuado o por medios bien conocidos por los expertos en la técnica.
A partir de la fórmula estructural 1, es evidente que hay un átomo de carbono asimétrico. Se pretende que la presente invención incluya dentro de su alcance las formas isoméricas estereoquímicamente puras de los compuestos así como sus racematos. Pueden obtenerse formas isoméricas estereoquímicamente puras mediante la aplicación de principios conocidos en la técnica. Pueden separarse diaestereoisómeros mediante métodos de separación física tales como cristalización fraccionada y técnicas cromatográficas, y pueden separase enantiómeros entre sí mediante la cristalización selectiva de las sales diastereoméricas con ácidos o bases ópticamente activos o mediante cromatografía quiral. También pueden prepararse estereoisómeros puros de manera sintética a partir de materiales de partida estereoquímicamente puros apropiados, o usando reacciones estereoselectivas.
Durante cualquiera de los procedimientos para la preparación de los compuestos de la presente invención, puede ser necesario y/o deseable proteger grupos sensibles o reactivos en cualquiera de las moléculas afectadas. Esto puede lograrse por medio de grupos protectores convencionales, tales como los descritos en Protective Grupos in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973; y T.W. Greene & P.G.M. Wuts, Protective Grupos in Organic Synthesis, tercera edición, John Wiley & Sons, 1999. Los grupos protectores pueden retirarse en una fase posterior conveniente usando métodos conocidos de la técnica.
La presente invención se basa en parte en el descubrimiento de que los carbamatos de fenilalquilamino de fórmula 1 comentados anteriormente tienen propiedades farmacológicas novedosas y únicas. Se ha mostrado en varios modelos animales que estos compuestos tienen la capacidad de tratar el síndrome de fibromialgia y la modificación de síntomas asociados con el síndrome de fibromialgia.
Aunque el mecanismo de acción preciso no se entiende completamente, se sabe que estos compuestos no funcionan mediante los mismos mecanismos que la mayoría de los demás tratamientos conocidos para el síndrome de fibromialgia. Por estos motivos, los compuestos de fórmula 1 son especialmente adecuados para su uso como único tratamiento o tratamiento adjunto para la fibromialgia y la modificación de los síntomas asociados con el síndrome de fibromialgia.
Por tanto, estos compuestos pueden usarse de manera segura solos o en combinación con otros medicamentos útiles para proporcionar eficacia potenciada y efectos secundarios reducidos porque podrían usarse dosis más pequeñas de cada fármaco.
Esta invención se refiere al compuesto O-carbamoilfenilalaninol para su uso en el tratamiento de dolor y alteraciones del sueño como síntomas del síndrome de fibromialgia en un mamífero que padece síndrome de fibromialgia; comprendiendo el método administrar al sujeto una cantidad terapéuticamente eficaz de uno o más de los compuestos de carbamato de la invención o una sal farmacéuticamente aceptable de los mismos y un portador, diluyente o excipiente farmacéuticamente aceptable.
Definiciones
Por conveniencia, en el presente documento se recopilan ciertos términos empleados en la memoria descriptiva, los ejemplos y las reivindicaciones adjuntas.
Debe entenderse que la terminología utilizada en el presente documento tiene el propósito de describir realizaciones particulares.
Tal como se usa en el presente documento, el término “sujeto” se refiere a un animal, preferiblemente un mamífero, y lo más preferiblemente un ser humano, tanto masculino como femenino, que ha sido objeto de tratamiento, observación o experimento.
El término “cantidad terapéuticamente efectiva” tal como se usa en el presente documento, significa la cantidad de compuesto activo o agente farmacéutico que provoca la respuesta biológica o médica en un sistema de tejido, animal o ser humano que está buscando un investigador, veterinario, doctor u otro médico, que incluye el alivio de uno o más de los signos o síntomas de la enfermedad o trastorno que está tratándose.
El término “cantidad profilácticamente eficaz” se refiere a la cantidad de un fármaco farmacéutico que evitará o reducirá el riesgo de aparición del evento biológico o médico que se busca prevenir de un tejido, sistema, animal o ser humano siendo buscado por un investigador, veterinario, doctor u otro médico.
El término “sales farmacéuticamente aceptables” significará sales no tóxicas de los compuestos empleados en esta invención que generalmente se preparan haciendo reaccionar el ácido libre con una base orgánica o inorgánica adecuada. Los ejemplos de tales sales incluyen acetato, bencenosulfonato, benzoato, bicarbonato, bisulfato, bitartrato, borato, bromuro, calcio, edetato de calcio, camsilato, carbonato, cloruro, clavulanato, citrato, diclorhidrato, edetato, edisilato, estolato, esilato, fumarato, gluceptato, gluconato, glutamato, glicolilarsanilato, hexilresorcinato, hidrabamina, bromhidrato, clorhidarto, hidroxinaftoato, yoduro, isotionato, lactato, lactobionato, laurato, malato, maleato, mandelato, mesilato, metilbromuro, metilnitrato, metilsulfato, mucato, napsilato, nitrato, oleato, oxalato, pamoato, palmitato, pantotenato, fosfato/difosfato, poligalacturonato, potasio, salicilato, sodio, estearato, subacetato, succinato, tanato, tartrato, teoclato, tosilato, trietyoduro, valerato.
Por tanto, el término “un paciente que necesita tratamiento”, tal como se usa en el presente documento, se referirá a cualquier sujeto o paciente que actualmente tiene o puede desarrollar cualquiera de los síndromes o trastornos anteriores.
El término “tratar” o “tratamiento” tal como se usa en el presente documento se refiere a cualquier indicio de éxito en la prevención o mejora de una lesión, patología o estado del síndrome de fibromialgia y la modificación de los síntomas del síndrome de fibromialgia dentro del alcance de las reivindicaciones, incluyendo cualquier parámetro objetivo o subjetivo tal como reducción; remisión; disminución de los síntomas o hacer que la lesión, patología o estado sea más tolerable para el paciente; ralentización en la tasa de degeneración o disminución o empeoramiento de la enfermedad; hacer que el punto final de empeoramiento sea menos debilitante; o mejorar el bienestar físico o mental de un sujeto. El tratamiento o la mejoría de los síntomas puede basarse en parámetros objetivos o subjetivos; incluyendo los resultados de un examen físico, examen neurológico y/o evaluaciones psiquiátricas. Por consiguiente, el término “tratar” o “tratamiento” incluye la administración de los compuestos o agentes de la presente invención para el tratamiento de cualquier forma de síndrome de fibromialgia tanto en hombres como en mujeres dentro del alcance de las reivindicaciones. En algunos casos, el tratamiento con los compuestos de la presente invención se
realizará en combinación con otros compuestos para prevenir, inhibir o detener la progresión del síndrome de fibromialgia.
El término “efecto terapéutico” tal como se usa en el presente documento se refiere a la mejora eficaz en o la reducción de los síntomas del síndrome de fibromialgia. El término “una cantidad terapéuticamente eficaz” tal como se usa en el presente documento significa una cantidad suficiente de uno o más de los compuestos de la invención para producir un efecto terapéutico, tal como se definió anteriormente, en un sujeto o paciente que necesita tal tratamiento de fibromialgia.
Los términos “sujeto” o “paciente” se usan en el presente documento de manera intercambiable y, tal como se usan en el presente documento, significan cualquier mamífero incluyendo seres humanos incluyendo un paciente o sujeto humano al que pueden administrarse las composiciones de la invención. El término mamíferos incluye pacientes humanos, tanto masculinos como femeninos, y primates no humanos, así como animales experimentales tales como conejos, ratas y ratones y otros animales.
Se conocen métodos en la técnica para determinar dosis terapéuticas y profilácticamente eficaces para la presente composición farmacéutica. Por ejemplo, el compuesto puede emplearse a una dosis diaria en el intervalo de 0,1 mg a 400 mg, habitualmente en un régimen de 1 a 2 veces al día, para un humano adulto promedio. La cantidad eficaz, sin embargo, puede variar dependiendo del compuesto particular usado, el modo de administración, la concentración de la preparación, el modo de administración y el avance del estado patológico. Además, factores asociados con el paciente particular que está tratándose, incluyendo la edad, el peso, la dieta y el tiempo de administración del paciente, darán como resultado la necesidad de ajustar las dosificaciones.
El compuesto puede administrarse a un sujeto por cualquier vía de administración convencional, incluyendo intravenosa, oral, subcutánea, intramuscular, intradérmica y parenteral. Dependiendo de la vía de administración, los compuestos de fórmula (1) pueden constituirse en cualquier forma. Por ejemplo, las formas adecuadas para la administración oral incluyen formas sólidas, como píldoras, cápsulas de gel, comprimidos, comprimidos oblongos, cápsulas (incluyendo cada uno formulaciones de liberación inmediata, liberación programada y liberación sostenida), gránulos y polvos. Las formas adecuadas para administración oral también incluyen formas líquidas, tales como disoluciones, jarabes, elixires, emulsiones y suspensiones. Además, las formas útiles para la administración parenteral incluyen disoluciones estériles, emulsiones y suspensiones.
Para preparar las composiciones farmacéuticas de esta invención, uno o más compuestos de fórmula (1) o sal de los mismos como principio activo se mezclan íntimamente con un portador farmacéutico según técnicas convencionales de composición farmacéutica. Son necesarios portadores y pueden emplearse excipientes farmacéuticos inertes, incluyendo aglutinantes, agentes de suspensión, lubricantes, aromatizantes, edulcorantes, conservantes, colorantes y recubrimientos. En la preparación de composiciones en forma de dosificación oral, puede emplearse cualquiera de los portadores farmacéuticos habituales. Por ejemplo, para preparaciones orales líquidas, los portadores y aditivos adecuados incluyen agua, glicoles, aceites, alcoholes, agentes aromatizantes, conservantes y agentes colorantes; para preparaciones orales sólidas, los portadores y aditivos adecuados incluyen almidones, azúcares, diluyentes, agentes de granulación, lubricantes, aglutinantes y agentes disgregantes. Para uso parenteral, el portador comprenderá generalmente agua estéril, aunque pueden incluirse otros componentes, por ejemplo, para fines tales como ayudar en la solubilidad o la conservación. También pueden prepararse suspensiones inyectables, en cuyo caso pueden emplearse agentes de suspensión y portadores líquidos apropiados.
Debido a su facilidad de administración, los comprimidos y las cápsulas representan la forma unitaria de dosificación oral más ventajosa, en cuyo caso obviamente se emplean portadores farmacéuticos sólidos. Si se desea, los comprimidos pueden estar recubiertos con azúcar o con recubrimiento entérico mediante técnicas convencionales. Pueden prepararse supositorios, en cuyo caso podría usarse manteca de cacao como portador. Los comprimidos o píldoras pueden recubrirse o combinarse de otra forma para proporcionar una forma de dosificación que proporcione la ventaja de una acción prolongada. Por ejemplo, el comprimido o las píldoras pueden comprender una dosificación interna y un componente de dosificación externa, estando esta última en forma de una envoltura sobre la primera. Los dos componentes pueden estar separados por una capa entérica, que sirve para resistir la disgregación en el estómago y permite que el componente interno pase intacto al duodeno o se retrase su liberación. Puede usarse una variedad de materiales para tales capas o recubrimientos entéricos, incluyendo tales materiales varios ácidos poliméricos con materiales tales como laca, alcohol cetílico y acetato de celulosa.
El fármaco activo también puede administrarse en forma de sistemas de administración de liposomas, tales como vesículas unilamelares pequeñas, vesículas unilamelares grandes y vesículas multilamelares. Los liposomas pueden formarse a partir de una variedad de fosfolípidos, tales como colesterol, estearilamina o fosfatidilcolinas.
El fármaco activo también puede administrarse mediante el uso de anticuerpos monoclonales como portadores individuales a los que se acoplan las moléculas de compuesto. El fármaco activo también puede acoplarse con polímeros solubles como portadores de fármacos dirigibles. Tales polímeros pueden incluir polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamida-fenol, polihidroxietil-aspartamida-fenol o poli(óxido de etileno)-polilisina sustituido con residuos de palmitoílo. Además, el fármaco activo puede acoplarse a una clase de polímeros biodegradables útiles para lograr la liberación controlada de un fármaco, por ejemplo, poli(ácido láctico), poli(ácido
glicólico), copolímeros de poli(ácido láctico) y poli(ácido glicólico), poliépsilon caprolactona, ácido polihidroxibutírico, poliortoésteres, poliacetales, polidihidropiranos, policianoacrilatos y copolímeros de bloque de hidrogeles reticulados o anfipáticos.
Preferiblemente, estas composiciones están en formas de dosificación unitarias tales como comprimidos, píldoras, cápsulas, polvos, gránulos, disoluciones o suspensiones parenterales estériles, aerosoles medidos o pulverizaciones líquidas, gotas, ampollas, dispositivos de autoinyector o supositorios, para administración parenteral oral, intranasal, sublingual o rectal, o para administración por inhalación o insuflación.
Alternativamente, la composición puede presentarse en una forma adecuada para administración una vez a la semana o una vez al mes; por ejemplo, una sal insoluble del compuesto activo, tal como la sal de decanoato, puede adaptarse para proporcionar una preparación de depósito para inyección intramuscular.
Las composiciones farmacéuticas en el presente documento contendrán, por unidad de dosificación, por ejemplo, comprimido, cápsula, polvo, inyección, cucharadita y supositorio, una cantidad del principio activo necesaria para administrar una dosis eficaz tal como se describió anteriormente. Por ejemplo, las composiciones farmacéuticas en el presente documento pueden contener, por unidad de dosificación unitaria, desde 25 hasta 400 mg del principio activo. Preferiblemente, el intervalo es de desde 50 hasta 200 mg del principio activo.
En algunas realizaciones de la presente invención, se administrarán compuestos de carbamato adecuados para su uso en la práctica de esta invención o bien individualmente o bien de manera concomitante con al menos uno o más de otros compuestos o agentes terapéuticos. Esto incluye la etapa de administrar a un paciente que necesita tratamiento una cantidad eficaz de uno de los compuestos de carbamato descritos en el presente documento en combinación con una cantidad eficaz de uno o más de otros compuestos o agentes terapéuticos.
Se entiende que los sustituyentes y patrones de sustitución en los compuestos de la presente invención puede seleccionarlos un experto habitual en la técnica para proporcionar compuestos que son químicamente estables y que pueden sintetizarse fácilmente mediante técnicas conocidas en la técnica, así como los métodos proporcionados en el presente documento.
La presente descripción incluye el uso del enantiómero (R) aislado de fórmula 1. En una realización, una composición farmacéutica que comprende el enantiómero R aislado de fórmula 1 se usa para proporcionar un tratamiento del dolor y la alteración del sueño como síntomas del síndrome de fibromialgia en un sujeto.
En la presente invención, predomina el enantiómero (R) de fórmula 1. Un enantiómero que predomina en la mezcla es uno que está presente en la mezcla en una cantidad mayor que el otro enantiómero presente en la mezcla, por ejemplo, en una cantidad mayor del 90%. En un aspecto, un enantiómero predominará hasta el grado del 90% o hasta el grado del 91%, el 92%, el 93%, el 94%, el 95%, el 96%, el 97% o el 98% o más. El enantiómero que predomina en una composición que comprende un compuesto de fórmula 1 es el enantiómero R de fórmula 1. La presente invención incluye métodos de uso de enantiómeros (R) y mezclas enantioméricas, en los que el enantiómero (R) predomina hasta el grado del 90% o más, de los compuestos representados por la fórmula 1. Un enantiómero de carbamato de fórmula 1 contiene un centro quiral en el segundo carbono alifático adyacente al anillo de fenilo. Un enantiómero que está aislado es uno que está sustancialmente libre del enantiómero correspondiente. Por tanto, un enantiómero aislado se refiere a un compuesto que se separa por medio de técnicas de separación o se prepara sin el enantiómero correspondiente. El término “sustancialmente libre”, tal como se usa en el presente documento, significa que el compuesto está constituido por una proporción significativamente mayor de un enantiómero. Según la invención, el compuesto incluye al menos el 90% en peso de un enantiómero preferido. En otras realizaciones de la invención, el compuesto incluye al menos el 99% en peso de un enantiómero preferido. Los enantiómeros preferidos pueden aislarse de mezclas racémicas por cualquier método conocido por los expertos en la técnica, incluyendo cromatografía de líquidos de alta resolución (HPLC) y la formación y cristalización de sales quirales, o los enantiómeros preferidos pueden prepararse mediante métodos descritos en el presente documento. Compuestos de carbamato como productos farmacéuticos:
La presente invención proporciona enantiómeros (R), mezclas enantioméricas en las que predomina el enantiómero (R) hasta el grado del 90% o más como productos farmacéuticos. Los compuestos de carbamato se formulan como productos farmacéuticos para proporcionar acción contra la fibromialgia en un sujeto.
En general, los compuestos de carbamato de la presente invención pueden administrarse como composiciones farmacéuticas mediante cualquier método conocido en la técnica para administrar fármacos terapéuticos incluyendo administración oral, bucal, tópica, sistémica (por ejemplo, transdérmica, intranasal o mediante supositorio) o parenteral (por ejemplo, inyección intramuscular, subcutánea o intravenosa). La administración de los compuestos directamente al sistema nervioso puede incluir, por ejemplo, administración a las vías de administración intracerebral, intraventricular, intacerebroventricular, intratecal, intracisternal, intraespinal o periespinal mediante administración por medio de catéteres o agujas intracraneales o intravertebrales con o sin dispositivos de bomba.
Las composiciones pueden adoptar la forma de comprimidos, píldoras, cápsulas, semisólidos, polvos, formulaciones de liberación sostenida, disoluciones, suspensiones, emulsiones, jarabes, elixires, aerosoles o cualquier otra composición apropiada; y comprender al menos un compuesto de esta invención en combinación con al menos un excipiente farmacéuticamente aceptable. Tales excipientes los conocen bien los expertos habituales en la técnica, y pueden encontrarse, y los métodos de formulación de las composiciones, en referencias convencionales tales como Alfonso AR: Remington’s Pharmaceutical Sciences, 17a ed., Mack Publishing Company, Easton PA, 1985. Los portadores líquidos adecuados, especialmente para disoluciones inyectables, incluyen agua, solución salina acuosa, disolución de dextrosa acuosa y glicoles.
Los compuestos de carbamato pueden proporcionarse como suspensiones acuosas. Las suspensiones acuosas de la invención pueden contener un compuesto de carbamato en mezcla con excipientes adecuados para la fabricación de suspensiones acuosas. Tales excipientes pueden incluir, por ejemplo, un agente de suspensión, tal como carboximetilcelulosa de sodio, metilcelulosa, hidroxipropilmetilcelulosa, alginato de sodio, polivinilpirrolidona, goma tragacanto y goma arábiga, y agentes humectantes o de dispensación tales como un fosfátido que se produce de manera natural (por ejemplo, lecitina), un producto de condensación de un óxido de alquileno con un ácido graso (por ejemplo, poli(estearato de oxietileno), un producto de condensación de óxido de etileno con un alcohol alifático de cadena larga (por ejemplo, heptadecaetilenoxicetanol), un producto de condensación de óxido de etileno con un éster parcial derivado de un ácido graso y un hexitol (por ejemplo, mono-oleato de polioxietilensorbitol), o un producto de condensación de óxido de etileno con un éster parcial derivado de ácido graso y un anhídrido de hexitol (por ejemplo, mono-oleato de polioxietilensorbitano).
La suspensión acuosa puede contener también uno o más conservantes tales como p-hidroxibenzoato de etilo o npropilo, uno o más agentes colorantes, uno más agentes aromatizantes y uno o más agentes edulcorantes, tales como sacarosa, aspartamo o sacarina. Las formulaciones pueden ajustarse para la osmolaridad.
Pueden formularse suspensiones oleosas para su uso en los presentes métodos suspendiendo un compuesto de carbamato en un aceite vegetal, tal como aceite de maní, aceite de oliva, aceite de sésamo o aceite de coco, o en un aceite mineral tal como parafina líquida; o una mezcla de estos. Las suspensiones oleosas pueden contener un agente espesante, tal como cera de abejas, parafina dura o alcohol cetílico. Pueden añadirse agentes edulcorantes para proporcionar una preparación oral agradable, tales como glicerol, sorbitol o sacarosa. Estas formulaciones pueden conservarse mediante la adición de un antioxidante tal como ácido ascórbico. Como ejemplo de un vehículo oleoso inyectable, véase Minto, J. Pharmacol. Exp. Ther. 281:93-102, 1997. Las formulaciones farmacéuticas de la invención pueden estar también en forma de emulsiones de aceite en agua. La fase oleosa puede ser un aceite vegetal o un aceite mineral, descrito anteriormente, o una mezcla de estos.
Los agentes emulsionantes adecuados incluyen gomas que se producen de manera natural, tales como goma arábiga y goma tragacanto, fosfátidos que se producen de manera natural tales como lecitina de soja, ésteres o ésteres parciales derivados de ácidos grasos y anhídridos de hexitol, tales como mono-oleato de sorbitano, y productos de condensación de estos ésteres parciales con óxido de etileno, tales como mono-oleato de polioxietilensorbitano. La emulsión puede contener también agentes edulcorantes y agentes aromatizantes, como en la formulación de jarabes y elixires. Tales formulaciones pueden contener también un demulcente, un conservante o un agente colorante.
El compuesto de elección, solo o en combinación con otros componentes adecuados, puede prepararse en formulaciones de aerosol (es decir, pueden “nebulizarse”) para administrarse por medio de inhalación. Las formulaciones de aerosol pueden colocarse en propelentes aceptables presurizados, tales como diclorodifluorometano, propano y nitrógeno.
Las formulaciones de la presente invención adecuadas para administración parenteral, tal como, por ejemplo, por las vías intraarticular (en las articulaciones), intravenosa, intramuscular, intradérmica, intraperitoneal y subcutánea, pueden incluir disoluciones de inyección estériles isotónicas, acuosas y no acuosas, que pueden contener antioxidantes, tampones, bacteriostáticos y solutos que hacen que la formulación sea isotónica con la sangre del receptor previsto, y suspensiones estériles acuosas y no acuosas que pueden incluir agentes de suspensión, solubilizantes, agentes espesantes, estabilizadores y conservantes. Entre los vehículos y disolventes aceptables que pueden emplearse están agua y disolución de Ringer, un cloruro de sodio isotónico. Además, pueden emplearse convencionalmente aceites fijos estériles como disolvente o medio de suspensión. Para este fin, puede emplearse cualquier aceite fijo insípido incluyendo mono- o diglicéridos sintéticos. Además, pueden usarse asimismo ácidos grasos tales como ácido oleico en la preparación de inyectables. Estas disoluciones son estériles y generalmente libres de materia no deseada.
Cuando los compuestos son suficientemente solubles, pueden disolverse directamente en solución salina normal con o sin el uso de disolventes orgánicos adecuados, tales como propilenglicol o polietilenglicol. Pueden prepararse dispersiones de los compuestos finamente divididos en disolución acuosa de almidón o carboximetilcelulosa de sodio, o en un aceite adecuado, como el aceite de maní. Estas formulaciones pueden esterilizarse mediante técnicas de esterilización convencionales y bien conocidas. Las formulaciones pueden contener sustancias auxiliares farmacéuticamente aceptables según se requiera para aproximarse a las condiciones fisiológicas tales como
agentes de ajuste y tamponamiento del pH, agentes de ajuste de la toxicidad, por ejemplo, acetato de sodio, cloruro de sodio, cloruro de potasio, cloruro de calcio y lactato de sodio.
La concentración de un compuesto de carbamato en estas formulaciones puede variar ampliamente, y se seleccionará principalmente basándose en los volúmenes de fluido, viscosidades, peso corporal y similares, según el modo particular de administración seleccionado y las necesidades del paciente. Para la administración IV, la formulación puede ser una preparación inyectable estéril, tal como una suspensión acuosa u oleaginosa inyectable estéril. Esta suspensión puede formularse según la técnica conocida usando aquellos agentes dispersantes o humectantes y agentes de suspensión adecuados. La preparación inyectable estéril también puede ser una disolución o suspensión inyectable estéril en un diluyente o disolvente parenteralmente aceptable no tóxico, tal como una disolución de 1,3-butanodiol. Las formulaciones de recomendaciones pueden presentarse en recipientes sellados de dosis unitaria o de múltiples dosis, tales como ampollas y viales. Las disoluciones y suspensiones de inyección pueden prepararse a partir de polvos, gránulos y comprimidos estériles del tipo descrito anteriormente. Un compuesto de carbamato adecuado para su uso en la práctica de esta invención puede administrarse y se administra preferiblemente por vía oral. La cantidad de un compuesto de la presente invención en la composición puede variar ampliamente dependiendo del tipo de composición, el tamaño de una dosis unitaria, el tipo de excipientes y otros factores bien conocidos por los expertos habituales en la técnica. En general, la composición final puede comprender, por ejemplo, del 0,000001% en peso (% en peso) hasta el 50% en peso del compuesto de carbamato, preferiblemente del 0,00001% en peso al 25% en peso, siendo el resto el excipiente o excipientes. Pueden administrarse formulaciones farmacéuticas para administración oral usando portadores farmacéuticamente aceptables bien conocidos en la técnica en dosificaciones adecuadas para administración oral. Tales portadores permiten que las formulaciones farmacéuticas se formulen en formas de dosificación unitarias como comprimidos, píldoras, polvos, comprimidos recubiertos de azúcar, cápsulas, líquidos, pastillas para chupar, geles, jarabes, suspensiones espesas y suspensiones adecuadas para su ingestión por el paciente.
Las formulaciones adecuadas para administración oral pueden consistir en (a) disolución líquida, tal como una cantidad eficaz de la formulación farmacéutica suspendida en diluyentes, tales como agua, solución salina o polietilenglicol (PEG) 400; (b) cápsulas, bolsitas o comprimidos, conteniendo cada uno una cantidad predeterminada del principio activo, como líquidos, sólidos, gránulos o gelatina; (c) suspensiones en un líquido apropiado; y (d) emulsiones adecuadas.
Pueden obtenerse preparaciones farmacéuticas para uso oral mediante la combinación de los compuestos de la presente invención con un excipiente sólido, triturando opcionalmente una mezcla resultante y procesando la mezcla de gránulos, tras añadir compuestos adicionales adecuados, si se desea, para obtener núcleos de comprimidos o comprimidos recubiertos de azúcar. Excipientes sólidos adecuados son cargas de hidratos de carbono o proteínas e incluyen azúcares, incluyendo lactosa, sacarosa, manitol o sorbitol; almidón de maíz, trigo, arroz, patata u otras plantas; celulosa tal como metilcelulosa, hidroximetilcelulosa, hidroxipropilmetilcelulosa o carboximetilcelulosa de sodio; y gomas incluyendo arábiga y tragacanto; así como proteínas tales como gelatina y colágeno.
Si se desea, pueden añadirse agentes disgregantes o solubilizantes, tales como polivinilpirrolidona reticulada, agar, ácido algínico o una sal del mismo, tal como alginato de sodio. Las formas de comprimidos pueden incluir uno o más de lactosa, sacarosa, manitol, sorbitol, fosfatos de calcio, almidón de maíz, almidón de patata, celulosa microcristalina, gelatina, dióxido de silicio coloidal, talco, estearato de magnesio, ácido esteárico y otros excipientes, colorantes, cargas, aglutinantes, diluyentes, agentes tamponantes, agentes humectantes, conservantes, agentes aromatizantes, colorantes, agentes disgregantes y portadores farmacéuticamente compatibles. Las formas de pastilla para chupar pueden comprender el principio activo en un sabor, por ejemplo, sacarosa, así como pastillas que comprenden el principio activo en una base inerte, como gelatina y glicerina o emulsiones y geles de sacarosa y goma arábiga que contienen, además del principio activo, portadores conocido en la técnica.
Los compuestos de la presente invención también pueden administrarse en forma de supositorios para la administración rectal del fármaco. Estas formulaciones pueden prepararse mezclando el fármaco con un excipiente adecuado no irritante que es sólido a temperaturas normales pero líquido a las temperaturas rectales y, por tanto, se fundirá en el recto liberando el fármaco. Tales materiales son manteca de cacao y polietilenglicoles.
Los compuestos de la presente invención también pueden administrarse por vía intranasal, intraocular, intravaginal e intrarrectal, incluyendo supositorios, insuflación, polvos y formulaciones en aerosol (para ejemplos de inhalantes de esteroides, véase Rohatagi, J. Clin. Pharmacol 35: 1187-1193, 1995; Tjwa, Ann. Alergia Asma Immunol. 75: 107 111, 1995).
Los compuestos de la presente invención pueden administrarse por vía transdérmica, por una vía tópica, formulados como barras aplicadoras, disoluciones, suspensiones, emulsiones, geles, cremas, pomadas, pastas, jaleas, pinturas, polvos y aerosoles.
También pueden emplearse materiales encapsulantes con los compuestos de la presente invención y el término “composición” puede incluir el principio activo en combinación con un material encapsulante como una formulación, con o sin otros portadores. Por ejemplo, los compuestos de la presente invención también pueden administrarse
como microesferas para liberación lenta en el cuerpo. En una realización, pueden administrarse microesferas por medio de inyección intradérmica de microesferas que contienen fármaco (por ejemplo, mifepristona), que se liberan lentamente por vía subcutánea (véase Rao, J. Biomater Sci. Polym. Ed. 7: 623-645, 1995); como formulaciones de gel inyectables y biodegradables (véase, por ejemplo, Gao, Pharm. Res. 12: 857-863, 1995); o como microesferas para administración oral (véase, por ejemplo, Eyles, J. Pharm. Pharmacol 49: 669-674, 1997). Tanto las vías transdérmicas como las intradérmicas permiten una administración constante durante semanas o meses. También pueden usarse comprimidos oblongos en la administración de los compuestos de la presente invención.
En otra realización, los compuestos de la presente invención pueden administrarse mediante el uso de liposomas que se fusionan con la membrana celular o experimentan endocitosis, es decir, mediante el empleo de ligandos unidos al liposoma que se unen a receptores de proteínas de membrana de la superficie de la célula dando como resultado endocitosis. Mediante el uso de liposomas, particularmente cuando la superficie del liposoma porta ligandos específicos para células diana, o de lo contrario preferentemente dirigidos a un órgano específico, puede enfocarse la administración del compuesto de carbamato a células diana in vivo. (Véase, por ejemplo, Al-Muhammed, J. Microencapsul. 13: 293-306, 1996; Chonn, Curr. Opin. Biotechnol. 6: 698-708, 1995; Ostro, am. J. Hosp. Pharm 46: 1576-1587, 1989).
Las formulaciones farmacéuticas de la invención pueden proporcionarse como una sal y pueden formarse con muchos ácidos, incluyendo clorhídrico, sulfúrico, acético, láctico, tartárico, málico y succínico. Las sales tienden a ser más solubles en disolventes acuosos u otros disolventes protónicos que las formas de base libre correspondientes. En otros casos, la preparación preferida puede ser un polvo liofilizado que puede contener, por ejemplo, cualquiera o todos de los siguientes: histidina 1 mM-50 mM, sacarosa al 0,1%-2%, manitol al 2%-7%, a un intervalo de pH de 4,5 a 5,5, que se combina con el tampón antes de su uso.
Las sales farmacéuticamente aceptables se refieren a sales que son farmacéuticamente aceptables y tienen las propiedades farmacológicas deseadas. Tales sales incluyen sales que pueden formarse cuando los protones ácidos presentes en los compuestos son capaces de reaccionar con bases inorgánicas u orgánicas. Las sales inorgánicas adecuadas incluyen las formadas con los metales alcalinos, por ejemplo sodio y potasio, magnesio, calcio y aluminio. Las sales orgánicas adecuadas incluyen las formadas con bases orgánicas tales como las bases de amina, por ejemplo etanolamina, dietanolamina, trietanolamina, trometamina y N-metilglucamina. Las sales farmacéuticamente aceptables también pueden incluir sales de adición de ácido formadas a partir de la reacción de restos de amina en el compuesto original con ácidos inorgánicos (por ejemplo, ácidos clorhídrico y bromhídrico) y ácidos orgánicos (por ejemplo, ácido acético, ácido cítrico, ácido maleico y los ácido alcano- y areno-sulfónicos tales como ácido metanosulfónico y ácido bencenosulfónico). Cuando hay dos grupos ácidos presentes, una sal farmacéuticamente aceptable puede ser una mono-sal de mono-acido o una di-sal; y de manera similar, cuando hay más de dos grupos ácidos presentes, algunos o todos de tales grupos pueden estar salificados.
Los compuestos nombrados en esta invención pueden estar presentes en forma no salificada, o en forma salificada. La presente invención incluye formas de sal farmacéuticamente aceptables de fórmula (1). Puede existir más de una forma cristalina de un enantiómero (R) de fórmula 1 y, como tal, también se incluyen en la presente invención.
Una composición farmacéutica de la divulgación puede contener opcionalmente, además de un compuesto de carbamato, al menos otro agente terapéutico útil en el tratamiento del síndrome de fibromialgia. Por ejemplo, los compuestos de carbamato de fórmula 1 pueden combinarse físicamente con otros tratamientos de fibromialgia en combinaciones de dosis fijas para simplificar su administración.
Se han descrito métodos para formular composiciones farmacéuticas en numerosas publicaciones, tales como Pharmaceutical Dosage Forms: Tablets. Segunda edicion. Revisado y ampliado. Volúmenes 1-3, editado por Lieberman et al; Pharmaceutical Dosage Forms: Parenteral Medications. Volúmenes 1-2, editado por Avis et al; y Pharmaceutical Dosage Forms: Disperse Systems. Volúmenes 1-2, editado por Lieberman et al; publicado por Marcel Dekker, Inc.
Las composiciones farmacéuticas generalmente se formulan como estériles, sustancialmente isotónicas y en total conformidad con todas las regulaciones de Buenas prácticas de fabricación (GMP) de la Food and Drug Administration de los EE. UU.
Regímenes de dosificación
La presente invención implica métodos para proporcionar acción anti-fibromialgia en un mamífero usando compuestos de carbamato. La cantidad del compuesto de carbamato necesaria para reducir o tratar el/los síntoma (s) del síndrome de fibromialgia se define como una dosis terapéutica o farmacéuticamente eficaz. El programa de dosificación y las cantidades eficaces para su uso, es decir, el régimen de dosis o dosificación, dependerá de una variedad de factores incluyendo el estadio de la enfermedad, el estado físico del paciente y la edad. En el cálculo del régimen de dosificación para un paciente, el modo de administración también se tiene en cuenta.
Un experto habitual en la técnica será capaz, sin experimentación excesiva, teniendo en cuenta su experiencia y esta divulgación, de determinar una cantidad terapéuticamente eficaz de un compuesto de carbamato sustituido particular para la práctica de esta invención (véase, por ejemplo, Lieberman, Pharmaceutical Dosage Forms (vol. 1
3, 1992); Lloyd, 1999, The art, Science and Technology of Pharmaceutical Compounding; y Pickar, 1999, Dosage Calculations). Una dosis terapéuticamente eficaz es también una en la que cualquier efecto secundario tóxico o perjudicial del agente activo se compensa en términos clínicos por los efectos terapéuticamente beneficiosos. Ha de indicarse además que para cada sujeto particular, deben evaluarse regímenes de dosificación específicos y ajustarse a lo largo del tiempo según la necesidad individual y el criterio profesional de la persona que administra o supervisa la administración de los compuestos.
Para fines de tratamiento, las composiciones o los compuestos dados a conocer en el presente documento pueden administrarse al sujeto en una única administración en bolo, por medio de administración continua a lo largo de un periodo de tiempo prolongado, o en un protocolo de administración repetida (por ejemplo, mediante un protocolo de administración repetida cada hora, diariamente o semanalmente). Las formulaciones farmacéuticas de la presente invención pueden administrarse, por ejemplo, una o más veces al día, 3 veces a la semana, o semanalmente. En una realización de la presente invención, las formulaciones farmacéuticas de la presente invención se administran por vía oral una o dos veces al día.
En este contexto, una dosificación terapéuticamente eficaz de los compuestos de carbamato puede incluir dosis repetidas dentro de un régimen de tratamiento prolongado que producirá resultados clínicamente significativos para tratar el síndrome de fibromialgia. La determinación de dosificaciones eficaces en este contexto se basa normalmente en estudios en modelos animales seguido por ensayos clínicos en seres humanos y se guía por la determinación de dosis eficaces y protocolos de administración que reducen significativamente la aparición o gravedad de los síntomas o estados de exposición dirigida en el sujeto. Los modelos adecuados a este respecto incluyen, por ejemplo, murinos, ratas, porcinos, felinos, primates no humanos y otros sujetos de modelos animales aceptados conocidos en la técnica. Alternativamente, pueden determinarse dosificaciones eficaces usando modelos in vitro (por ejemplo, ensayos inmunológicos e histopatológicos). Al usar tales modelos, solo se requieren cálculos y ajustes habituales para determinar una concentración y dosis apropiadas para administrar una cantidad terapéuticamente eficaz del/de los agente(s) biológicamente activo(s) (por ejemplo, cantidades que son eficaces por vía intranasal, eficaces por vía transdérmica, eficaces por vía intravenosa o por vía intramuscular para provocar una respuesta deseada).
En una realización a modo de ejemplo de la presente invención, se preparan formas de dosificación unitarias de los compuestos para regímenes de administración convencionales. De esta manera, la composición puede subdividirse fácilmente en dosis más pequeñas según el criterio del médico. Por ejemplo, pueden constituirse dosificaciones unitarias en polvos, viales o ampollas envasados y preferiblemente en forma de cápsula o comprimido.
El compuesto activo presente en estas formas de dosificación unitarias de la composición puede estar presente en una cantidad de, por ejemplo, desde 10 mg hasta un gramo o más, para administración diaria única o múltiple, según la necesidad particular del paciente. Al iniciar el régimen de tratamiento con una dosis mínima diaria de un gramo, los niveles en sangre de los compuestos de carbamato pueden usarse para determinar si está indicada una dosis mayor o menor.
La administración eficaz de los compuestos de carbamato de esta invención puede administrarse, por ejemplo, a una dosis oral o parenteral de desde 0,01 mg/kg/dosis hasta 150 mg/kg/dosis. Preferiblemente, la administración será de desde 0,1 mg/kg/dosis hasta 25 mg/kg/dosis, más preferiblemente desde 0,2 hasta 18 mg/kg/dosis. Por tanto, la cantidad terapéuticamente eficaz del principio activo contenido por unidad de dosificación tal como se describe en el presente documento puede ser, por ejemplo, de desde 1 mg/día hasta 7000 mg/día para un sujeto que tiene, por ejemplo, un peso promedio de 70 kg.
Aunque la invención anterior se ha descrito en detalle a modo de ejemplo con fines de claridad de comprensión, resultará evidente para el experto que la divulgación comprende ciertos cambios y modificaciones y que puede ponerse en práctica sin excesiva experimentación dentro del alcance de las reivindicaciones adjuntas. Los siguientes ejemplos se proporcionan para ilustrar aspectos específicos de la invención.
Puede obtenerse una mejor comprensión de la presente invención a la luz de los siguientes ejemplos que se exponen para ilustrar la presente invención.
[Modo para la invención]
EJEMPLO 1
El compuesto de prueba (O-carbamoil-(D)-fenilalaninol) administrado a 30 mg/kg por vía intraperitoneal (i.p.) aumentó significativamente la latencia de retirada de la pata a un estímulo térmico en ratas con ligamiento del nervio ciático. Estos datos sugieren que el compuesto de prueba muestra propiedades hiperalgésicas antitérmicas.
(Métodos)
Se anestesiaron inicialmente ratas Sprague-Dawley macho adultas jóvenes (CD(SD)IGS, 150-200 g) con isoflurano en O2 mediante una mascarilla y se realizó el procedimiento quirúrgico según el método descrito por Bennett y Xie (1988). En resumen, los nervios ciáticos derechos se ataron sin apretar usando ligaduras. Todos los experimentos
se realizaron según las directrices de la International Association for the Study of Pain. Se realizaron las pruebas de comportamiento al menos 14 días de manera posoperatoria.
Se disolvió el compuesto de prueba en solución salina y se administró por vía intraperitoneal a 30 mg/kg a los animales en un volumen de 3 ml/kg de peso corporal.
Para someter a prueba la hiperalgesia térmica, se determinó la respuesta térmica mediante la latencia de retirada de la pata trasera, usando un medidor plantar (UGO BASILE, Italia), y un método modificado de Hargreaves et al. (1988). Se permitió que las ratas se aclimataran dentro de recintos de plástico sobre una placa de vidrio transparente mantenida a temperatura ambiente. Se controló una fuente de calor radiante (intensidad 90) con un cronómetro y se enfocó sobre la superficie plantar de la plata trasera derecha de la rata que abarca la piel lampiña. La retirada de la pata detuvo tanto la fuente de calor como el cronómetro. Se usó un punto de corte máximo de 30 s para evitar el daño tisular. Se evaluaron las ratas para determinar la hiperalgesia térmica antes de la dosis (0 h) y a las 1,4 y 8 horas después de la administración de compuesto de prueba 30 mg/kg.
Los datos se expresan como la media ± error estándar de la media (EEM). Se compararon los valores de signos de comportamiento de dolor neuropático a diversos puntos de tiempo posoperatorios con los del periodo de control preoperatorio mediante un análisis de la varianza de una vía de medidas repetidas (seguido por prueba a posteriori de Dunnett). Valores de P menores de 0,05 se consideró que eran significativos.
(Resultados)
Los efectos hiperalgésicos anti-térmicos del compuesto de prueba en las ratas con lesión nerviosa se presentan en la tabla 1 a continuación. El compuesto de prueba, administrado por vía intraperitoneal a ratas, aumentó significativamente la latencia de retirada de la pata a estímulos térmicos nocivos en comparación con la latencia de retirada antes de la inyección (0 h) a 30 mg/kg, i.p.
[Tabla 1] Efectos hiperalgésicos anti-térmicos (latencia de retirada de la pata media en segundos) del compuesto de prueba

EJEMPLO 2
Se evaluaron los efectos del compuesto de prueba (50-150 mg/kg, p.o.) sobre diversos parámetros del sueño en 8 ratones narcolépticos con supresión de células de hipocretina (transgénicos para prepororexina/ataxina-3) y sus ratones de tipo natural compañeros de camada, y se compararon los efectos con los de modafinilo, un compuesto que promueve el despertar de referencia. El compuesto de prueba mostró episodios de vigilia significativamente aumentados en tanto ratones narcolépticos como de tipo natural y podía normalizar los patrones de sueño de ratones narcolépticos.
(Métodos)
Se capturó la señal del polígrafo (EEG y EMG) con SleepSign (Kissei Comtech), y se puntuó visualmente el estadio del sueño con periodos de 10 s para la vigilia, sueño no REM y sueño REM. Los criterios de puntuación eran: la vigilia se caracteriza por actividad de EMG alta y EEG de frecuencia mixta, de baja amplitud desincronizada (>4 Hz). También pueden aparecer ondas alfa rítmicas (8-9Hz) (con actividades de EMG alta). El sueño no REM se caracteriza EEG sincronizada, de alta amplitud, de baja frecuencia (0,25-4 Hz) con actividad de EMG reducida (en comparación con la vigilia). La actividad de EEG en el sueño REM es similar a la de la vigilia, ondas desincronizadas, de frecuencia mixta y baja amplitud. También puede aparecer una onda alfa rítmica (8-9 Hz) con actividades de EMG reducidas. La actividad de EEG durante el sueño REM se reduce incluso adicionalmente y, en muchos casos, está completamente ausente. Puede ser evidente cierta contracción muscular en el perfil de EMG durante el sueño REM.
Tres dosis de fármaco del compuesto de prueba (50, 100 y 150 mg/kg p.o.) más vehículo se administraron por vía oral ZT 2 (2 horas tras encender la luz) o ZT14 (2 horas tras apagar la luz), y se monitorizaron los efectos sobre el sueño durante 6 horas tras la administración del fármaco (los datos de sueño se recogieron durante 30 horas tras la inyección del fármaco, y estaban disponibles para su análisis adicional). Las dosis de modafinilo fueron de 50 y 200 mg/kg p.o. (más vehículo), y también se administró el modafinilo a ZT 2 y ZT14.
Si las señales de polígrafo de algunos ratones no eran suficientes para puntuar el estadio del sueño con precisión (especialmente EMG malo), se excluyeron los datos de estos animales, y se incluyó el mínimo de 5 animales (excepto para la dosis más alta del compuesto de prueba en ratones de tipo natural en el periodo de oscuridad, n=4) para el análisis de datos y el número de animales se indicó en las figuras.
Se analizaron los efectos del compuesto de prueba y modafinilo sobre la cantidad vigilia, sueño no REM, sueño REM (segundos acumulados), número de episodios para cada estadio del sueño durante 6 horas, longitudes (segundos) de episodios sueño/vigilia medias en cada animal y se calculó la media de cada parámetro en cada genotipo. Los efectos de los compuestos sobre las cantidades de vigilia y sueño son útiles para evaluar la potencia de promoción de la vigilia, y el número de episodios para cada estadio del sueño y la longitud de episodios de sueño/vigilia media son parámetros para evaluar la fragmentación del sueño. La anfetamina y el modafinilo, dos compuestos principales que promueven la vigilia actualmente usados para el tratamiento de EDS asociada con diversas etiologías (narcolepsia, hipersomnia idiopática y EDS secundaria), se sabe que aumentan en tiempo de vigilia y prolongan la longitud del episodio de vigilia en estados de EDS y normales.
Con estos análisis de datos, se evaluaron los efectos terapéuticos y de promoción de la vigilia del compuesto de prueba en narcolepsia, y se compararon los efectos con los de modafinilo. Una comparación de los efectos entre ratones de tipo natural y deficientes en hipocretina es muy útil en la determinación de si la promoción de la vigilia del compuesto de prueba depende de la disponibilidad de hipocretina, y si hay un posible cambio en la sensibilidad de los mecanismos receptivos del compuesto de prueba en ratones narcolépticos debido a la deficiencia del ligando hipocretina.
(Resultados)
Efectos sobre el sueño durante el periodo de reposo:
Se observaron efectos de promoción de la vigilia muy potentes del compuesto de prueba en tanto ratones narcolépticos de tipo natural como deficientes en hipocretina. Los efectos eran dependientes de la dosis, y administraciones de 50, 100, 150 mg/kg p.o. de compuesto de prueba indujeron una vigilia continua en la mayoría de los ratones de tipo natural y narcolépticos durante hasta 3, 4 y 5 horas, respectivamente. Durante este periodo, el sueño no REM y sueño REM se suprimieron completamente. No hubo patrones de EEG anómalos tras la administración del compuesto de prueba, y el sueño que se produjo tras la vigilia prolongada era normal mediante evaluaciones poligráficas.
En cambio, los efectos de promoción de la vigilia del modafinilo eran modestos, y el efecto de promoción de la vigilia de 200 mg/kg de modafinilo se corresponde aproximadamente con 50 mg/kg de compuesto de prueba. Sin embargo, el modafinilo no redujo fuertemente el sueño REM tras la administración de 50 mg/kg de compuesto de prueba. Además, el compuesto de prueba redujo potentemente el sueño REM, y esto contrasta con los efectos del modafinilo.
Efectos sobre el sueño durante el periodo activo:
Se repitió el mismo experimento administrando compuestos en el periodo activo. Durante el periodo activo, los ratones narcolépticos pasan más tiempo durmiendo que los ratones de tipo natural. Los animales de tipo natural normalmente permanecen despiertos durante casi tres horas tras la administración del vehículo. Similar a los efectos observados durante el periodo de luz, el compuesto de prueba aumentó la vigilia de manera dependiente de la dosis en ratones tanto de tipo natural como narcolépticos. Sin embargo, los efectos de promoción de la vigilia en ratones de tipo natural fueron sutiles durante el periodo de oscuridad debido a la alta cantidad de vigilia en el nivel inicial, y sólo se observaron pequeños efectos. En cambio, se observaron efectos de promoción de la vigilia muy pronunciados en ratones narcolépticos, y las cantidades de vigilia en estos ratones tras la administración de compuesto de prueba 100 y 150 mg/kg administración aumentaron hasta los niveles de ratones de tipo natural, sugiriendo que este compuesto normaliza la cantidad de sueño/vigilia de ratones narcolépticos. De manera similar, el sueño no REM y REM se redujeron en ratones narcolépticos mediante compuesto de prueba y las cantidades de sueño no REM y REM también disminuyeron hasta los niveles de ratones de tipo natural. Se observaron también efectos similares pero mucho más débiles tras la administración de modafinilo en estos ratones. Aunque el modafinilo aumenta la vigilia de manera dependiente de la dosis en ratones narcolépticos, la dosis alta de modafinilo (200 mg/kg) no llevó la cantidad de vigilia hasta la de los niveles iniciales de tipo natural.
EJEMPLO 3 (referencia)
Se sometió a prueba el compuesto de prueba para determinar los efectos sobre la prueba de natación forzada, un modelo animal de depresión, tanto en ratones como en ratas. Tras dosis individuales del compuesto de prueba, la duración media de la inmovilidad se redujo con una DE50 de 16,6 mg/kg p.o. en ratones y 18,5 mg/kg p.o. en ratas. El compuesto de prueba era incluso más potente tras la dosificación múltiple en ratones con una DE50 de 5,5 mg/kg p.o. Estos datos sugieren que el compuesto de prueba muestra propiedades antidepresivas.
(Métodos)
Se utilizaron ratones CD-1 macho (16-24 g) y ratas Wistar macho (90-125 g) en estos experimentos. El compuesto de prueba (10, 15 y 30 mg/kg) se disolvió en solución salina fisiológica (0,9%) y se administró por vía oral p.o. en un volumen de 1 ml/100 g de peso corporal.
Se colocaron los ratones y las ratas en cilindros de vidrio (vasos de precipitados de 1000 ml; altura 14 cm, diámetro 11,5 cm) y (vasos de precipitados de 4000 ml; altura 24,5 cm, diámetro 18,0 cm) respectivamente, que contenían agua (25°C) hasta una altura de 9,0 cm para ratones y 19,0 para ratas. Se colocó cada ratón o rata en el cilindro de vidrio y se permitió que nadara durante 2 minutos, tras lo cual se observaron durante un periodo de 4 minutos para detectar signos de inmovilidad. Se definió la inmovilidad como falta de movimiento, tal como flotar en el agua con poco o ningún movimiento de las patas traseras. Se cronometró la duración de la inmovilidad con un cronómetro y se registró. En algunos experimentos, se permitió que los ratones o las ratas nadaran durante 6 o 10 minutos, respectivamente, un día antes del experimento de natación forzada.
En la prueba de dosis individual, se les administró a los ratones o las ratas compuesto de prueba o NaCl al 0,9% y se colocaron en cilindros de vidrio 1 hora o 4 horas tras el tratamiento, respectivamente. En los experimentos de múltiples dosis, se dosificó a los ratones dos veces al día durante 3 días y se les administró una dosis adicional el día 4. Además, se colocaron los ratones en los cilindros de vidrio que contenían agua a 25°C y se les permitió nadar durante seis minutos el día 3. Se realizó la evaluación estadística usando un programa informático basado en análisis probit. Se determinó la significación estadística usando la prueba de la t de Student a un valor de P de < 0,05.
(Resultados)
El compuesto de prueba, administrado en una dosis individual a ratones, redujo la duración media de la inmovilidad de una manera dependiente de la dosis para dosis de 10, 15 y 30 mg/kg p.o. 10 mg/kg del compuesto de prueba redujeron la duración media de la inmovilidad hasta 101 s en comparación con 131 s para el control. Dosis de 15 y 30 mg/kg produjeron reducciones significativas del tiempo de inmovilidad medio desde 154 s hasta 80 s y desde 132 s hasta 30 s, respectivamente. El valor de DE50 (reducción del 50% en el tiempo de inmovilidad medio) para el compuesto de prueba fue de 16,6 mg/kg.
El compuesto de prueba, tras la dosificación múltiple ratones, redujo la duración media de la inmovilidad de una manera dependiente de la dosis para dosis de 3, 5 y 8 mg/kg p.o. A 3 mg/kg de compuesto de prueba, la duración media de la inmovilidad se redujo hasta 63 s desde 85 s para el control. Dosis de 5 y 8 mg/kg produjeron reducciones significativas del tiempo de inmovilidad medio desde 136 s hasta 73 s y desde 114 s hasta 39 s, respectivamente. El valor de DE50 para el compuesto de prueba fue de 5,5 mg/kg p.o.
En ratas, el compuesto de prueba administrado a 30 mg/kg redujo significativamente la duración media de la inmovilidad desde 38 s hasta 9 s a las 4 horas tras el tratamiento. Dosis de compuesto de prueba a 10 y 15 mg/kg también redujeron la duración de la inmovilidad desde 74 s hasta 62 s y de 65 s a 39 s, respectivamente, pero estas diferencias no fueron estadísticamente significativas. La DE50 fue de 18,5 mg/kg p.o. que es similar al valor de DE50 en ratones anteriormente.
Referencias citadas
La discusión de referencias en el presente documento pretende meramente resumir las afirmaciones realizadas por los autores y no se hace ninguna admisión de que cualquier referencia constituya técnica anterior. Los solicitantes se reservan el derecho a cuestionar la exactitud y pertinencia de las referencias citadas.
La presente invención se define mediante los términos de las reivindicaciones adjuntas.
Claims (3)
1. Compuesto que tiene la fórmula estructural (1) o sal farmacéuticamente aceptable del mismo para su uso en el tratamiento de síntomas asociados con síndrome de fibromialgia que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto que tiene la fórmula estructural (1) o una sal farmacéuticamente aceptable del mismo, en el que el compuesto que tiene la fórmula estructural (1) o una sal farmacéuticamente aceptable del mismo es un enantiómero (R) libre de otro enantiómero o una mezcla enantiomérica en la que el enantiómero (R) del compuesto que tiene la fórmula estructural (1) predomina hasta el grado del 90% o más y el otro enantiómero es el 10% o menos, a un mamífero que padece síndrome de fibromialgia que necesita tratamiento, en el que los síntomas asociados con síndrome de fibromialgia se seleccionan del grupo que consiste en dolor y alteración del sueño:

en la que,
R, R1 y R2 son hidrógeno y x=1.
2. Compuesto o sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de síntomas asociados con síndrome de fibromialgia según la reivindicación 1, en el que el enantiómero (R) predomina hasta el grado del 98% o más y el otro enantiómero es el 2% o menos.
3. Compuesto o sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de síntomas asociados con síndrome de fibromialgia según la reivindicación 1, en el que el enantiómero (R) es un compuesto representado por la fórmula estructural (1 b) libre del otro enantiómero

en la que R, x, R1 y R2 son tal como se definieron en la reivindicación 1.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US25867209P | 2009-11-06 | 2009-11-06 | |
| PCT/KR2010/007603 WO2011055944A2 (en) | 2009-11-06 | 2010-11-01 | Methods for treating fibromyalgia syndrome |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2743153T3 true ES2743153T3 (es) | 2020-02-18 |
Family
ID=43970515
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES10828479T Active ES2743153T3 (es) | 2009-11-06 | 2010-11-01 | Métodos para tratar el síndrome de fibromialgia |
Country Status (12)
| Country | Link |
|---|---|
| US (3) | US8927602B2 (es) |
| EP (1) | EP2496227B1 (es) |
| JP (1) | JP5901528B2 (es) |
| KR (1) | KR101783633B1 (es) |
| CN (1) | CN102781436B (es) |
| AU (2) | AU2010316172B2 (es) |
| BR (1) | BR112012010648A2 (es) |
| CA (1) | CA2779442A1 (es) |
| ES (1) | ES2743153T3 (es) |
| MX (1) | MX350745B (es) |
| RU (1) | RU2557533C2 (es) |
| WO (1) | WO2011055944A2 (es) |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5024635B2 (ja) | 2005-06-08 | 2012-09-12 | エスケー バイオファーマスティカルズ カンパニー リミテッド | 睡眠覚醒障害の治療 |
| KR20120098578A (ko) | 2009-06-22 | 2012-09-05 | 에스케이바이오팜 주식회사 | 피로의 치료 또는 예방 방법 |
| KR101783632B1 (ko) | 2009-11-06 | 2017-10-10 | 에스케이바이오팜 주식회사 | 주의력 결핍/과잉행동 장애(adhd)의 치료 방법 |
| WO2011055944A2 (en) | 2009-11-06 | 2011-05-12 | Sk Holdings Co., Ltd. | Methods for treating fibromyalgia syndrome |
| US9610274B2 (en) | 2010-06-30 | 2017-04-04 | Sk Biopharmaceuticals Co., Ltd. | Methods for treating bipolar disorder |
| JP6530741B2 (ja) | 2013-03-13 | 2019-06-12 | エアリアル,バイオファーマ,エルエルシー | カタプレキシーの治療 |
| PL3021838T3 (pl) | 2013-07-18 | 2021-03-08 | Jazz Pharmaceuticals Ireland Limited | Leczenie otyłości |
| US10888542B2 (en) | 2014-02-28 | 2021-01-12 | Sk Biopharmaceuticals Co., Ltd. | Aminocarbonylcarbamate compounds |
| TWI698415B (zh) | 2014-02-28 | 2020-07-11 | 南韓商愛思開生物製藥股份有限公司 | 胺基羰基胺基甲酸酯化合物 |
| KR102489052B1 (ko) * | 2016-05-19 | 2023-01-16 | 에스케이바이오팜 주식회사 | 섬유근육통 또는 섬유근육통의 연관된 기능적 증후군을 예방 또는 치료하기 위한 카바메이트 화합물의 용도 |
| US10195151B2 (en) | 2016-09-06 | 2019-02-05 | Jazz Pharmaceuticals International Iii Limited | Formulations of (R)-2-amino-3-phenylpropyl carbamate |
| FI3509582T3 (fi) | 2016-09-06 | 2024-02-08 | Axsome Malta Ltd | (r)-2-amino-3-fenyylipropyylikarbamaatin solvaattimuoto |
| US10710958B2 (en) | 2016-10-06 | 2020-07-14 | Jazz Pharmaceuticals Ireland Limited | Carbamoyl phenylalaninol compounds and uses thereof |
| PL3592392T3 (pl) * | 2017-03-10 | 2024-03-18 | University Of Louisville Research Foundation, Inc. | Biomateriały modyfikowane fasl o funkcji immunomodulacyjnej |
| BR112019025286A2 (pt) | 2017-06-02 | 2020-06-23 | Jazz Pharmaceuticals Ireland Limited | Métodos e composições para tratamento de sonolência excessiva |
| AU2018312328B2 (en) | 2017-07-31 | 2022-12-15 | Jazz Pharmaceuticals Ireland Limited | Carbamoyl phenylalaninol analogs and uses thereof |
| KR101972333B1 (ko) | 2017-08-07 | 2019-04-25 | 김재일 | 섬유근육통 진단용 조성물, 그를 포함하는 마이크로어레이 및 키트 |
| US20220370468A1 (en) * | 2019-07-23 | 2022-11-24 | Astellas Pharma Global Development, Inc. | Methods of treating sleep disorders associated with pain |
| US10940133B1 (en) | 2020-03-19 | 2021-03-09 | Jazz Pharmaceuticals Ireland Limited | Methods of providing solriamfetol therapy to subjects with impaired renal function |
| WO2025006965A1 (en) | 2023-06-29 | 2025-01-02 | Axsome Therapeutics, Inc. | Methods of administering solriamfetol to lactating women |
Family Cites Families (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL109872A (en) | 1993-06-10 | 2000-12-06 | Lilly Co Eli | Tetrahydrobenz ¬CD¾ indole-6-carboxamides for use in preventing emesis and pharmaceutical compositions comprising same |
| GB9406857D0 (en) * | 1994-04-07 | 1994-06-01 | Sandoz Ltd | Improvements in or relating to organic compounds |
| KR100197892B1 (ko) | 1994-09-09 | 1999-06-15 | 남창우 | 신규한 페닐알킬아미노 카바메이트 화합물과 그의 제조방법 |
| KR0173862B1 (ko) * | 1995-02-11 | 1999-04-01 | 조규향 | O-카바모일-(d)-페닐알라닌올 화합물과 그의 약제학적으로 유용한 염 및 이들의 제조방법 |
| US5756817C1 (en) | 1995-02-11 | 2001-04-17 | Sk Corp | O-carbamoyl-phenylananinol compounds their pharmaceutically useful salts and process for preparing the same |
| IT1275903B1 (it) | 1995-03-14 | 1997-10-24 | Boehringer Ingelheim Italia | Esteri e ammidi della 1,4-piperidina disostituita |
| KR0173863B1 (ko) | 1995-04-10 | 1999-04-01 | 조규향 | 페닐에 치환체가 있는 o-카바모일-페닐알라닌올 화합물과 그의 약제학적으로 유용한 염 및 이들의 제조방법 |
| EP0873308B1 (en) * | 1996-10-10 | 2002-01-02 | SK Corporation | O-carbamoyl-phenylalaninol compounds and their pharmaceutically useful salts |
| AU4674797A (en) | 1996-10-22 | 1998-05-15 | Ortho Pharmaceutical Corporation | Phenylalaninol derivatives for the treatment of central nervous system disorders |
| TW402591B (en) | 1997-07-11 | 2000-08-21 | Janssen Pharmaceutica Nv | Monocyclic benzamides of 3- or 4-substituted 4-(aminomethyl)-piperidine derivatives |
| SK15852000A3 (sk) | 1998-04-28 | 2001-06-11 | Dainippon Pharmaceutical Co., Ltd. | Derivát 1-[(1-substituovaného-4-piperidinyl)metyl]-4-piperidínu, spôsob jeho prípravy, farmaceutický prípravok obsahujúci tento derivát a medziprodukt tohto derivátu |
| US20010029262A1 (en) | 1998-06-29 | 2001-10-11 | Sethi Kapil Dev | Method of treatment or prophylaxis |
| TW518218B (en) * | 1999-05-27 | 2003-01-21 | Merck Patent Gmbh | Pharmaceutical compositions comprising 1-[4-(5-cyanoindol-3-yl)butyl]-4-(2-carbamoylbenzofuran-5-yl)piperazine or its physiologically acceptable salts for use in the treatment of sub-type anxiety disorders |
| US20010034320A1 (en) | 2000-01-18 | 2001-10-25 | Hans-Michael Brecht | NK1-receptor antagonists for treating restless legs syndrome |
| DE60117605T2 (de) | 2000-12-21 | 2006-12-07 | Bristol-Myers Squibb Co. | Thiazolyl-inhibitoren von tyrosinkinasen der tec-familie |
| DK1404312T3 (da) * | 2001-02-27 | 2011-03-14 | Ortho Mcneil Pharm Inc | Carbamatforbindelser til anvendelse ved smertebehandling |
| DE60221670T2 (de) | 2001-02-27 | 2008-04-30 | Ortho-Mcneil Pharmaceutical, Inc. | Carbamatverbindungen zur vorbeugung und behandlung von bewegungsstörungen |
| JP2005511478A (ja) | 2001-04-03 | 2005-04-28 | メルク エンド カムパニー インコーポレーテッド | N−置換非アリール複素環アミジル系nmda/nr2b拮抗薬 |
| JP2004537526A (ja) | 2001-06-12 | 2004-12-16 | メルク エンド カムパニー インコーポレーテッド | 片頭痛の治療又は予防用nr2b受容体拮抗薬 |
| US20040115263A1 (en) | 2002-08-26 | 2004-06-17 | Robertson David W. | Use of bupropion for treating restless legs syndrome |
| WO2004026868A1 (en) | 2002-09-20 | 2004-04-01 | Pfizer Japan Inc. | N-substituted piperidinyl-imidazopyridine compounds as 5-ht4 receptor modulators |
| WO2004094418A1 (en) | 2003-04-21 | 2004-11-04 | Pfizer Inc. | Imidazopyridine compounds having 5-ht4 receptor agonistic activity and 5-ht3 receptor antagonistic activity |
| PT1664036E (pt) | 2003-09-03 | 2012-02-16 | Pfizer | Compostos de benzimidazolona que possuem actividade agonística do receptor 5-ht4 |
| US20050080268A1 (en) | 2003-10-08 | 2005-04-14 | Yong-Moon Choi | Process of preparing O-carbamoyl compounds in the presence of active amine group |
| EP1548011A1 (en) * | 2003-12-23 | 2005-06-29 | Neuro3D | Benzo[1,4]diazepin-2-one derivatives as phosphodiesterase PDE2 inhibitors, preparation and therapeutic use thereof |
| WO2005092882A1 (en) | 2004-03-01 | 2005-10-06 | Pfizer Japan, Inc. | 4-amino-5-halogeno-benzamide derivatives as 5-ht4 receptor agonists for the treatment of gastrointestinal, cns, neurological and cardiovascular disorders |
| WO2006050037A1 (en) | 2004-10-28 | 2006-05-11 | Sk Corporation | Adjunctive therapy for depression |
| EP1869019B1 (en) | 2005-04-08 | 2014-08-27 | Pfizer Products Inc. | Bicyclic [3.1.0] heteroaryl amides as type i glycine transport inhibitors |
| US7598279B2 (en) | 2005-04-22 | 2009-10-06 | Sk Holdings Co., Ltd. | Neurotherapeutic azole compounds |
| JP5024635B2 (ja) | 2005-06-08 | 2012-09-12 | エスケー バイオファーマスティカルズ カンパニー リミテッド | 睡眠覚醒障害の治療 |
| DE602006012670D1 (de) | 2005-06-22 | 2010-04-15 | Sk Holdings Co Ltd | Behandlung der sexuellen dysfunktion |
| WO2007018496A1 (en) | 2005-07-26 | 2007-02-15 | Janssen Pharmaceutica, N.V. | Methods for treating substance-related disorders |
| EP1754476A1 (en) * | 2005-08-18 | 2007-02-21 | Schwarz Pharma Ag | Lacosamide (SPM 927) for treating myalgia, e.g. fibromyalgia |
| KR20090082213A (ko) | 2006-10-13 | 2009-07-29 | 얀센 파마슈티카 엔.브이. | 페닐알킬아미노 카바메이트 조성물 |
| WO2008048770A1 (en) | 2006-10-17 | 2008-04-24 | Lipothera, Inc. | Methods, compositions, and formulations for the treatment of thyroid eye disease |
| US8642772B2 (en) | 2008-10-14 | 2014-02-04 | Sk Biopharmaceuticals Co., Ltd. | Piperidine compounds, pharmaceutical composition comprising the same and its use |
| KR20120098578A (ko) * | 2009-06-22 | 2012-09-05 | 에스케이바이오팜 주식회사 | 피로의 치료 또는 예방 방법 |
| US8232315B2 (en) | 2009-06-26 | 2012-07-31 | Sk Biopharmaceuticals Co., Ltd. | Methods for treating drug addiction and improving addiction-related behavior |
| KR101783632B1 (ko) | 2009-11-06 | 2017-10-10 | 에스케이바이오팜 주식회사 | 주의력 결핍/과잉행동 장애(adhd)의 치료 방법 |
| WO2011055944A2 (en) | 2009-11-06 | 2011-05-12 | Sk Holdings Co., Ltd. | Methods for treating fibromyalgia syndrome |
| US8623913B2 (en) | 2010-06-30 | 2014-01-07 | Sk Biopharmaceuticals Co., Ltd. | Methods for treating restless legs syndrome |
-
2010
- 2010-11-01 WO PCT/KR2010/007603 patent/WO2011055944A2/en active Application Filing
- 2010-11-01 RU RU2012123154/15A patent/RU2557533C2/ru active
- 2010-11-01 MX MX2012005257A patent/MX350745B/es active IP Right Grant
- 2010-11-01 BR BR112012010648-1A patent/BR112012010648A2/pt not_active Application Discontinuation
- 2010-11-01 JP JP2012537802A patent/JP5901528B2/ja active Active
- 2010-11-01 CA CA2779442A patent/CA2779442A1/en not_active Abandoned
- 2010-11-01 AU AU2010316172A patent/AU2010316172B2/en active Active
- 2010-11-01 US US13/508,145 patent/US8927602B2/en active Active
- 2010-11-01 KR KR1020127014769A patent/KR101783633B1/ko active IP Right Grant
- 2010-11-01 EP EP10828479.5A patent/EP2496227B1/en active Active
- 2010-11-01 ES ES10828479T patent/ES2743153T3/es active Active
- 2010-11-01 CN CN201080061045.XA patent/CN102781436B/zh active Active
-
2014
- 2014-09-24 US US14/495,416 patent/US9688620B2/en active Active
-
2016
- 2016-02-05 AU AU2016200765A patent/AU2016200765B2/en active Active
-
2017
- 2017-06-26 US US15/632,847 patent/US20170349541A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| WO2011055944A3 (en) | 2011-11-03 |
| EP2496227B1 (en) | 2019-05-22 |
| CN102781436B (zh) | 2014-01-08 |
| JP2013510142A (ja) | 2013-03-21 |
| US9688620B2 (en) | 2017-06-27 |
| AU2010316172A1 (en) | 2012-05-24 |
| US20170349541A1 (en) | 2017-12-07 |
| KR101783633B1 (ko) | 2017-10-10 |
| CA2779442A1 (en) | 2011-05-12 |
| BR112012010648A2 (pt) | 2020-12-01 |
| MX350745B (es) | 2017-09-14 |
| AU2010316172B2 (en) | 2015-11-12 |
| AU2016200765B2 (en) | 2017-06-15 |
| KR20120083518A (ko) | 2012-07-25 |
| US8927602B2 (en) | 2015-01-06 |
| AU2016200765A1 (en) | 2016-02-25 |
| RU2557533C2 (ru) | 2015-07-20 |
| WO2011055944A2 (en) | 2011-05-12 |
| MX2012005257A (es) | 2012-06-19 |
| EP2496227A2 (en) | 2012-09-12 |
| EP2496227A4 (en) | 2013-04-24 |
| US20150011625A1 (en) | 2015-01-08 |
| RU2012123154A (ru) | 2013-12-27 |
| CN102781436A (zh) | 2012-11-14 |
| US20120252892A1 (en) | 2012-10-04 |
| JP5901528B2 (ja) | 2016-04-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2743153T3 (es) | Métodos para tratar el síndrome de fibromialgia | |
| ES2638074T3 (es) | Carbamatos para utilizar en el síndrome de piernas inquietas | |
| ES2927675T3 (es) | Tratamiento de la cataplexia | |
| ES2435403T3 (es) | Tratamiento de trastornos del sueño y vigilia | |
| US8822492B2 (en) | Use of huperzine for disorders | |
| ES2447295T3 (es) | Métodos para el tratamiento del trastorno de déficit de atención/hiperactividad | |
| ES2853730T3 (es) | Composiciones para tratar la adicción a las drogas y mejorar el comportamiento relacionado con la adicción | |
| ES2654615T3 (es) | Compuestos de carbamato para el tratamiento del trastorno bipolar | |
| ES2344469T3 (es) | Tratamiento de disfuncion sexual. | |
| ES2291454T3 (es) | Utilizacion de carbamatos para prevenir o tratar trastornos del movimiento. |