CN115448849B - Preparation method of 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester - Google Patents
Preparation method of 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester Download PDFInfo
- Publication number
- CN115448849B CN115448849B CN202211408228.5A CN202211408228A CN115448849B CN 115448849 B CN115448849 B CN 115448849B CN 202211408228 A CN202211408228 A CN 202211408228A CN 115448849 B CN115448849 B CN 115448849B
- Authority
- CN
- China
- Prior art keywords
- reaction
- fluoro
- benzyloxy
- solvent
- synthesis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 16
- VRXGVVAMXZXKNA-UHFFFAOYSA-N phenyl 5-(dibenzylamino)-3-fluoro-2-methyl-6-phenylmethoxybenzoate Chemical compound C=1C=CC=CC=1COC1=C(C(=O)OC=2C=CC=CC=2)C(C)=C(F)C=C1N(CC=1C=CC=CC=1)CC1=CC=CC=C1 VRXGVVAMXZXKNA-UHFFFAOYSA-N 0.000 title claims description 9
- 238000006243 chemical reaction Methods 0.000 claims abstract description 46
- 238000004519 manufacturing process Methods 0.000 claims abstract description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 36
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 36
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical group C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 36
- 239000002904 solvent Substances 0.000 claims description 26
- 230000015572 biosynthetic process Effects 0.000 claims description 22
- 238000003786 synthesis reaction Methods 0.000 claims description 22
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 21
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 claims description 20
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 20
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-diisopropylethylamine Substances CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 19
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 17
- 239000003513 alkali Substances 0.000 claims description 16
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 12
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 12
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical group [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 claims description 12
- 238000000034 method Methods 0.000 claims description 12
- 239000001569 carbon dioxide Substances 0.000 claims description 10
- 229910002092 carbon dioxide Inorganic materials 0.000 claims description 10
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 10
- 238000005660 chlorination reaction Methods 0.000 claims description 9
- 238000006482 condensation reaction Methods 0.000 claims description 9
- 238000005886 esterification reaction Methods 0.000 claims description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 8
- 239000007818 Grignard reagent Substances 0.000 claims description 8
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 8
- AHVYPIQETPWLSZ-UHFFFAOYSA-N N-methyl-pyrrolidine Natural products CN1CC=CC1 AHVYPIQETPWLSZ-UHFFFAOYSA-N 0.000 claims description 8
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical group BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 claims description 8
- -1 benzyl halide Chemical class 0.000 claims description 8
- 150000004795 grignard reagents Chemical class 0.000 claims description 8
- AVFZOVWCLRSYKC-UHFFFAOYSA-N 1-methylpyrrolidine Chemical compound CN1CCCC1 AVFZOVWCLRSYKC-UHFFFAOYSA-N 0.000 claims description 7
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical group ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 claims description 6
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 claims description 6
- WLXMPPVZSJTUDG-UHFFFAOYSA-N 5-(dibenzylamino)-3-fluoro-2-methyl-6-phenylmethoxybenzoic acid Chemical compound CC1=C(C=C(C(=C1C(=O)O)OCC2=CC=CC=C2)N(CC3=CC=CC=C3)CC4=CC=CC=C4)F WLXMPPVZSJTUDG-UHFFFAOYSA-N 0.000 claims description 5
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical group [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 5
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 5
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 5
- DBTNVRCCIDISMV-UHFFFAOYSA-L lithium;magnesium;propane;dichloride Chemical compound [Li+].[Mg+2].[Cl-].[Cl-].C[CH-]C DBTNVRCCIDISMV-UHFFFAOYSA-L 0.000 claims description 5
- 239000011777 magnesium Substances 0.000 claims description 5
- 229910052749 magnesium Inorganic materials 0.000 claims description 5
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 4
- 239000012320 chlorinating reagent Substances 0.000 claims description 4
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 claims description 3
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims description 3
- 239000003153 chemical reaction reagent Substances 0.000 claims description 3
- 150000001263 acyl chlorides Chemical class 0.000 claims description 2
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 claims description 2
- 229940073608 benzyl chloride Drugs 0.000 claims description 2
- 230000002194 synthesizing effect Effects 0.000 claims description 2
- 239000012535 impurity Substances 0.000 abstract description 4
- 150000001875 compounds Chemical class 0.000 abstract description 3
- 229940095102 methyl benzoate Drugs 0.000 abstract 1
- 238000000746 purification Methods 0.000 abstract 1
- 238000000926 separation method Methods 0.000 abstract 1
- 239000000543 intermediate Substances 0.000 description 82
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 42
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 36
- 230000035484 reaction time Effects 0.000 description 27
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 25
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 23
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 17
- 238000003756 stirring Methods 0.000 description 16
- 238000004128 high performance liquid chromatography Methods 0.000 description 15
- 239000007787 solid Substances 0.000 description 14
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 12
- 238000001914 filtration Methods 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- 239000000243 solution Substances 0.000 description 9
- RVYGYYVGWSCWGY-UHFFFAOYSA-N 4-fluoro-3-methylphenol Chemical compound CC1=CC(O)=CC=C1F RVYGYYVGWSCWGY-UHFFFAOYSA-N 0.000 description 7
- 238000005893 bromination reaction Methods 0.000 description 6
- 239000003638 chemical reducing agent Substances 0.000 description 6
- 229910052739 hydrogen Inorganic materials 0.000 description 6
- 238000006396 nitration reaction Methods 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 239000002994 raw material Substances 0.000 description 6
- JVBXVOWTABLYPX-UHFFFAOYSA-L sodium dithionite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])=O JVBXVOWTABLYPX-UHFFFAOYSA-L 0.000 description 6
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 238000001228 spectrum Methods 0.000 description 5
- 229940072172 tetracycline antibiotic Drugs 0.000 description 5
- 238000001291 vacuum drying Methods 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- WPXFJBPJUGMYOD-UHFFFAOYSA-N 5-fluoro-2-methoxybenzoic acid Chemical compound COC1=CC=C(F)C=C1C(O)=O WPXFJBPJUGMYOD-UHFFFAOYSA-N 0.000 description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- SXAMGRAIZSSWIH-UHFFFAOYSA-N 2-[3-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,2,4-oxadiazol-5-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NOC(=N1)CC(=O)N1CC2=C(CC1)NN=N2 SXAMGRAIZSSWIH-UHFFFAOYSA-N 0.000 description 3
- CONKBQPVFMXDOV-QHCPKHFHSA-N 6-[(5S)-5-[[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]methyl]-2-oxo-1,3-oxazolidin-3-yl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C[C@H]1CN(C(O1)=O)C1=CC2=C(NC(O2)=O)C=C1 CONKBQPVFMXDOV-QHCPKHFHSA-N 0.000 description 3
- 238000003747 Grignard reaction Methods 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 239000004098 Tetracycline Substances 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 238000006555 catalytic reaction Methods 0.000 description 3
- 239000012295 chemical reaction liquid Substances 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 230000032050 esterification Effects 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 239000011259 mixed solution Substances 0.000 description 3
- 238000006722 reduction reaction Methods 0.000 description 3
- 235000010288 sodium nitrite Nutrition 0.000 description 3
- HYHCSLBZRBJJCH-UHFFFAOYSA-N sodium polysulfide Chemical compound [Na+].S HYHCSLBZRBJJCH-UHFFFAOYSA-N 0.000 description 3
- 235000019364 tetracycline Nutrition 0.000 description 3
- 150000003522 tetracyclines Chemical class 0.000 description 3
- KZEVSDGEBAJOTK-UHFFFAOYSA-N 1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-2-[5-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]ethanone Chemical compound N1N=NC=2CN(CCC=21)C(CC=1OC(=NN=1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)=O KZEVSDGEBAJOTK-UHFFFAOYSA-N 0.000 description 2
- JQMFQLVAJGZSQS-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-N-(2-oxo-3H-1,3-benzoxazol-6-yl)acetamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)NC1=CC2=C(NC(O2)=O)C=C1 JQMFQLVAJGZSQS-UHFFFAOYSA-N 0.000 description 2
- YJLUBHOZZTYQIP-UHFFFAOYSA-N 2-[5-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NN=C(O1)CC(=O)N1CC2=C(CC1)NN=N2 YJLUBHOZZTYQIP-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- 125000006278 bromobenzyl group Chemical group 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 230000032798 delamination Effects 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 229910052979 sodium sulfide Inorganic materials 0.000 description 2
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 229960002180 tetracycline Drugs 0.000 description 2
- 229930101283 tetracycline Natural products 0.000 description 2
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- SWHOVFGSZQZYIM-UHFFFAOYSA-N 3-fluoro-6-methoxy-2-methylbenzoic acid Chemical compound COC1=CC=C(F)C(C)=C1C(O)=O SWHOVFGSZQZYIM-UHFFFAOYSA-N 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 241000606161 Chlamydia Species 0.000 description 1
- 241000204031 Mycoplasma Species 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 241000606701 Rickettsia Species 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 208000012839 conversion disease Diseases 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- FVWDBVACVTXVJN-UHFFFAOYSA-L dipotassium;propan-2-one;carbonate Chemical compound [K+].[K+].CC(C)=O.[O-]C([O-])=O FVWDBVACVTXVJN-UHFFFAOYSA-L 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- LVKCSZQWLOVUGB-UHFFFAOYSA-M magnesium;propane;bromide Chemical compound [Mg+2].[Br-].C[CH-]C LVKCSZQWLOVUGB-UHFFFAOYSA-M 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 230000001035 methylating effect Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 230000001546 nitrifying effect Effects 0.000 description 1
- 229910000510 noble metal Inorganic materials 0.000 description 1
- LJGRYUWTKQWGBX-UHFFFAOYSA-N phenyl 3-fluoro-2-methyl-5-nitro-6-phenylmethoxybenzoate Chemical compound C=1C=CC=CC=1OC(=O)C=1C(C)=C(F)C=C([N+]([O-])=O)C=1OCC1=CC=CC=C1 LJGRYUWTKQWGBX-UHFFFAOYSA-N 0.000 description 1
- WCJBQCKZTRUDDM-UHFFFAOYSA-N phenyl 3-fluoro-6-hydroxy-2-methylbenzoate Chemical compound CC1=C(F)C=CC(O)=C1C(=O)OC1=CC=CC=C1 WCJBQCKZTRUDDM-UHFFFAOYSA-N 0.000 description 1
- RSDPUBRHTSGERI-UHFFFAOYSA-N phenyl 3-fluoro-6-methoxy-2-methylbenzoate Chemical compound COC1=CC=C(F)C(C)=C1C(=O)OC1=CC=CC=C1 RSDPUBRHTSGERI-UHFFFAOYSA-N 0.000 description 1
- DKVGPXIDZBAHMN-UHFFFAOYSA-N phenyl 5-amino-3-fluoro-2-methyl-6-phenylmethoxybenzoate Chemical compound C=1C=CC=CC=1OC(=O)C=1C(C)=C(F)C=C(N)C=1OCC1=CC=CC=C1 DKVGPXIDZBAHMN-UHFFFAOYSA-N 0.000 description 1
- ZUCMYHKPYDFZHO-UHFFFAOYSA-N phenyl 5-fluoro-2-hydroxy-6-methyl-3-nitrobenzoate Chemical compound CC1=C(F)C=C([N+]([O-])=O)C(O)=C1C(=O)OC1=CC=CC=C1 ZUCMYHKPYDFZHO-UHFFFAOYSA-N 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 238000005496 tempering Methods 0.000 description 1
- 229940040944 tetracyclines Drugs 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/02—Formation of carboxyl groups in compounds containing amino groups, e.g. by oxidation of amino alcohols
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C201/00—Preparation of esters of nitric or nitrous acid or of compounds containing nitro or nitroso groups bound to a carbon skeleton
- C07C201/06—Preparation of nitro compounds
- C07C201/10—Preparation of nitro compounds by substitution of functional groups by nitro groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C201/00—Preparation of esters of nitric or nitrous acid or of compounds containing nitro or nitroso groups bound to a carbon skeleton
- C07C201/06—Preparation of nitro compounds
- C07C201/12—Preparation of nitro compounds by reactions not involving the formation of nitro groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/02—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions involving the formation of amino groups from compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/08—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions not involving the formation of amino groups, hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/14—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof
- C07C227/18—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof by reactions involving amino or carboxyl groups, e.g. hydrolysis of esters or amides, by formation of halides, salts or esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C37/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring
- C07C37/62—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by introduction of halogen; by substitution of halogen atoms by other halogen atoms
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The application provides a preparation method of 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methyl benzoate, and relates to the technical field of compound preparation. The preparation method provided by the application has the advantages of low production cost, high yield, mild reaction conditions, low requirements on equipment, convenience in separation and purification of products, very high purity, single impurity controllability and the like.
Description
Technical Field
The invention relates to the technical field of compound preparation, and particularly relates to a preparation method of 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester.
Background
Tetracycline antibiotics are widely used for various bacteria and infections caused by rickettsia, chlamydia, mycoplasma and the like. However, the increased use of tetracyclines in human and veterinary medicine has resulted in the development of resistance by many organisms that were previously susceptible to tetracycline antibiotics. With the development of technology in recent years, it has become possible to synthesize novel tetracycline antibiotics with high efficiency. The 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoate can be used for synthesizing various novel tetracycline antibiotics (such as compounds 1-6) and is an important drug intermediate.






The prior patent WO2010126607A2 discloses the synthesis of tetracycline and its intermediate, the synthesis method is:
methylating 5-fluoro-2-methoxybenzoic acid in tetrahydrofuran by LDA and MeI to generate 3-fluoro-6-methoxy-2-methylbenzoic acid, esterifying the 3-fluoro-6-methoxy-2-methylbenzoic acid phenyl ester with phenol in dichloromethane, demethoxylating in dichloromethane by boron tribromide to generate 3-fluoro-6-hydroxy-2-methylbenzoic acid phenyl ester, nitrifying in water with nitric acid to generate 3-fluoro-6-hydroxy-2-methyl-5-nitrobenzoic acid phenyl ester, protecting the hydroxy by benzyl in benzyl bromide and potassium carbonate acetone to generate 2-benzyloxy-5-fluoro-6-methyl-3-nitrobenzoic acid phenyl ester, reducing the nitro in tetrahydrofuran and water by sodium hydrosulfite to generate 3-amino-2-benzyloxy-5-fluoro-6-methylbenzoic acid phenyl ester, and protecting the amino by benzyl bromide and DIPEA in NMP to generate 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester.
The specific synthetic route is as follows:


the starting material 5-fluoro-2-methoxybenzoic acid in the route has high price, low reaction conversion rate of only 50-60%, and auxiliary material MeI has high price and six equivalent weight. The process according to WO2010126607A2 patent reproduces an overall yield of only 28.9%. Ultra-low temperature conditions such as-30 ℃ and-78 ℃ need to be used, and the requirement on large-scale production equipment is high; and the intermediates in multiple steps are oily substances, which are not beneficial to purifying and controlling impurities.
In addition, the prior art (Chinese Journal of Pharmaceuticals 2017, 48 (4), p.506-509) discloses an optimized synthetic route, which comprises the following specific synthetic routes:

the route mainly aims at the low conversion rate of the first step of the patent WO2010126607A2 and recovers the raw material 5-fluoro-2-methoxybenzoic acid, but the step has more impurities, and does not describe how to purify and recycle the recovered raw material, the yield calculated by directly deducting the corresponding recovery amount is not accurate, and the actual total yield is 35.4%. In addition, the nitro-group reducing condition is changed into Pd/C and H 2 The final overall cost is higher due to the high price of 10% Pd/C.
In addition, the prior art (Journal of Organic Chemistry, 2017, vol. 82, # 2, p 936-943) discloses a synthetic method using 3-methyl-4-fluorophenol as a raw material, and the specific synthetic route is as follows:


the method takes 4-fluoro-3-methylphenol as a raw material, and although the unit price of the raw material is reduced by about 50%, the total yield of the route is still not high and is only 23.8%. The product of the route is only suitable for elysin (the compound 1) in new tetracycline medicines, and the application range is narrow. And the reaction condition of the last step of re-esterification of carboxyl by Grignard exchange/carbon dioxide is denied because of low yield, etc., and the condition of catalytic carbonyl insertion by a pressure vessel is changed to Pd (OAc) 2 The unit price is very high, and the total cost is not superior to that of the patent.
Therefore, the development of a method for preparing 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester with high yield, low requirements on production equipment and greatly reduced overall cost is required
Disclosure of Invention
Based on the defects in the prior art, the application aims to provide the preparation method of the 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester, which has high yield, low requirement on production equipment and low overall cost.
In order to achieve the technical effect, the following technical scheme is adopted in the application:
a preparation method of 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester comprises the following steps:
step A: synthesis of intermediate 1:
carrying out bromination reaction on the 4-fluoro-3-methylphenol to obtain an intermediate 1 reaction solution;
the reaction equation is as follows:

and B: synthesis of intermediate 2:
carrying out nitration reaction on the reaction liquid of the intermediate 1 prepared in the step A to obtain an intermediate 2;
the reaction equation is as follows:

and C: synthesis of intermediate 3:
carrying out substitution reaction on the intermediate 2 obtained in the step B to obtain an intermediate 3;
the reaction equation is:

step D: synthesis of intermediate 4:
c, carrying out reduction reaction on the intermediate 3 obtained in the step C to obtain an intermediate 4;
the reaction equation is as follows:

step E: synthesis of intermediate 5:
carrying out condensation reaction on the intermediate 4 obtained in the step D to obtain an intermediate 5, namely N, N-dibenzyl-2-benzyloxy-3-bromo-5-fluoro-4-methylaniline;
the reaction equation is:

step F: synthesis of intermediate 6:
performing Grignard reaction or Grignard exchange on the intermediate 5 obtained in the step E; then reacting with carbon dioxide to obtain an intermediate 6, namely 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid;
the reaction equation is:

step G: synthesis of intermediate 7:
carrying out chlorination reaction on the intermediate 6 obtained in the step F; then carrying out esterification reaction to obtain an intermediate 7, namely 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoate;
the reaction equation is:

the bromination reaction in the step A is specifically carried out as follows: dissolving 4-fluoro-3-methylphenol in acetic acid, and then dropwise adding liquid bromine into the acetic acid solution to carry out bromination reaction to obtain an intermediate 1 reaction solution; wherein the mol ratio of the 4-fluoro-3-methylphenol to the liquid bromine is 1.0-2.2; preferably 1.0.
The bromination reaction temperature in the step A is 20-30 ℃, and the reaction time is 2-5h; preferably, the bromination reaction temperature is 23-28 ℃, and the reaction time is 2-4h; still preferably, the bromination reaction temperature is 25 ℃ and the reaction time is 3h.
The reaction solution of the intermediate 1 prepared in the step A is directly used for the next reaction without treatment.
The operation of the nitration reaction in the step B is as follows: b, dripping a sodium nitrite aqueous solution into the reaction liquid of the intermediate 1 prepared in the step A, and carrying out nitration reaction to obtain an intermediate 2; wherein, the molar ratio of the intermediate 1 to the sodium nitrite is 1.0; preferably 1.0; still more preferably 1.0.
The nitration reaction temperature in the step B is 10-30 ℃, and the reaction time is 2-5h; preferably, the nitration reaction temperature is 20-30 ℃, and the reaction time is 2-4h; more preferably, the nitration reaction temperature is 25 ℃ and the reaction time is 3 hours.
The substitution reaction in the step C is specifically performed by: adding the intermediate 2 obtained in the step B, alkali, benzyl bromide and potassium iodide into a solvent, and reacting to obtain an intermediate 3; wherein the molar ratio of the intermediate 2, benzyl bromide, base and potassium iodide is 1.0-1.2; preferably 1.0:1.0:1.5:0.1
The reaction temperature in the step C is 50-85 ℃, and the reaction time is 3-8h; preferably, the reaction temperature is 60-80 ℃, and the reaction time is 5-7h; more preferably, the reaction temperature is 70 ℃ and the reaction time is 6h.
The alkali in the step C is one or more of potassium carbonate, sodium carbonate, cesium carbonate, potassium hydroxide, sodium hydroxide, triethylamine and N, N-diisopropylethylamine; preferably, the alkali is one or more of potassium carbonate, sodium carbonate and cesium carbonate; preferably, the alkali is one or more of potassium carbonate and sodium carbonate; further preferably, the base is potassium carbonate.
The solvent in the step C is one or more of acetone, acetonitrile, N-methylpyrrolidine and N, N-dimethylformamide; preferably, the solvent is one or more of acetone, acetonitrile and N-methylpyrrolidine; preferably, the solvent is one or more of acetone and acetonitrile; further preferably, the solvent is acetonitrile.
The reduction reaction in the step D is specifically performed by: adding the intermediate 3 obtained in the step C and a reducing agent into a mixed system of a solvent and water, and carrying out reduction reaction to obtain an intermediate 4; wherein, the molar ratio of the intermediate 3 to the reducing agent is 1.0-6.0; preferably 1.0; still more preferably 1.0.
The reducing agent is one or more of sodium hydrosulfite, sodium sulfide, sodium polysulfide and sodium thiosulfate; preferably, the reducing agent is one or more of sodium hydrosulfite, sodium sulfide and sodium polysulfide; preferably, the reducing agent is one or more of sodium hydrosulfite and sodium polysulfide; further preferably, the reducing agent is sodium hydrosulfite.
The solvent is one or more of tetrahydrofuran, methanol, ethanol, acetonitrile and ethyl acetate; preferably, the solvent is one or more of tetrahydrofuran, methanol, ethanol and acetonitrile; preferably, the solvent is one or more of tetrahydrofuran and acetonitrile; further preferably, the solvent is tetrahydrofuran
The reaction temperature in the step D is 10-40 ℃, and the reaction time is 2-8 h; preferably, the reaction temperature is 20-30 ℃, and the reaction time is 4-6h; more preferably, the reaction temperature is 25 ℃ and the reaction time is 5h.
The condensation reaction in the step E is specifically performed by: d, adding the intermediate 4 obtained in the step D, benzyl halide and alkali into a solvent, and carrying out condensation reaction to obtain an intermediate 5, namely N, N-dibenzyl-2-benzyloxy-3-bromo-5-fluoro-4-methylaniline; wherein the molar ratio of the intermediate 4, the benzyl halide and the base is 1.0:2.5-10.0:3.5-11.0; preferably 1.0:3.0:5.0;
the halogenated benzyl is selected from benzyl bromide or/and benzyl chloride.
The alkali is one or more of triethylamine, N-diisopropylethylamine, sodium carbonate, potassium carbonate and cesium carbonate; preferably, the base is one or more of triethylamine, N-diisopropylethylamine, sodium carbonate and potassium carbonate; preferably, the alkali is one or more of N, N-diisopropylethylamine and potassium carbonate; further preferably, the base is N, N-diisopropylethylamine.
The solvent is one or more of N-methylpyrrolidine, N-dimethylformamide, N-dimethylacetamide, dimethyl sulfoxide and acetonitrile; preferably, the solvent is one or more of N-methylpyrrolidine, N-dimethylformamide, N-dimethylacetamide and dimethyl sulfoxide; preferably, the solvent is one or more of N-methylpyrrolidine and dimethyl sulfoxide; further preferably, the solvent is N-methylpyrrolidine
The condensation reaction temperature in the step E is 70-130 ℃, and the reaction time is 6-48h; preferably, the condensation reaction temperature is 90-120 ℃, and the reaction time is 12-22h; still preferably, the condensation reaction temperature is 105 ℃ and the reaction time is 16h.
The specific operation in the step F is as follows: dissolving the intermediate 5 obtained in the step E in a solvent, and carrying out Grignard reaction or Grignard exchange with a Grignard reagent; then reacting with carbon dioxide to obtain an intermediate 6; wherein, the Grignard reagent is one or more of magnesium, isopropyl magnesium chloride, isopropyl magnesium bromide and isopropyl magnesium chloride lithium chloride; preferably one or more of magnesium, isopropyl magnesium chloride and isopropyl magnesium chloride lithium chloride; preferably one or more of isopropyl magnesium chloride and isopropyl magnesium chloride lithium chloride; further preferably isopropyl magnesium chloride.
The solvent is tetrahydrofuran or/and 2-methyltetrahydrofuran;
the Grignard reaction or Grignard exchange temperature is 30-70 ℃, and the reaction time is 2-8 h; preferably, the temperature is 40-60 ℃, and the reaction time is 4-6h; more preferably, the temperature is 50 ℃ and the reaction time is 5h.
The molar ratio of the intermediate 5, the grignard reagent and the carbon dioxide in the step F is 1.0:2.0-5.0:2.5-10.0; preferably 1.0:3.0:5.0.
the reaction temperature with the carbon dioxide in the step F is 0-50 ℃, and the reaction time is 1-3 h; preferably, the reaction temperature is 10-25 ℃, and the reaction time is 1-2h; more preferably, the reaction temperature is 15 ℃ and the reaction time is 1.5h.
The specific operation of the step G is as follows: f, firstly, carrying out chlorination reaction on the intermediate 6 obtained in the step F and a chlorination reagent to generate acyl chloride; then carrying out esterification reaction with phenol and alkali in a solvent to obtain an intermediate 7; wherein the molar ratio of the intermediate 6, the chlorinating reagent, the phenol and the alkali is 1.0:1.0-30.0:1.05-1.3:1.5-2.5; preferably 1.0:1.5:1.05:2.0;
the chlorinating agent is oxalyl chloride or/and thionyl chloride;
the solvent is one or more of dichloromethane, dichloroethane and chloroform; preferably, the solvent is dichloromethane or/and dichloroethane;
the alkali is triethylamine or/and N, N-diisopropylethylamine;
the chlorination reaction temperature in the step G is 20-80 ℃, and the reaction time is 1-4 h; preferably, the chlorination temperature is 20-30 ℃, and the reaction time is 1.5-2.5h; still preferably, the chlorination temperature is 25 ℃ and the reaction time is 2h.
The esterification reaction temperature in the step G is 5-35 ℃, and the reaction time is 1-4 h; preferably, the esterification temperature is 20-30 ℃, and the reaction time is 1.5-2.5h; still preferably, the esterification temperature is 25 ℃ and the reaction time is 2h.
Compared with the prior art, the beneficial effect of this application lies in:
(1) The production cost is low: the method takes 4-fluoro-3-methylphenol as the starting material, the unit price is reduced by about 50 percent compared with that of 5-fluoro-2-methoxybenzoic acid, LDA, meI and noble metal catalysts with higher prices are not used, and the cost of the whole material is reduced by 50 percent;
(2) The yield is high: the total yield of the preparation method route provided by the application is 51.1%, and is improved by 15.7-22.2% compared with the yield of the existing (mentioned in the background art) three schemes;
(3) The reaction condition is mild, and the requirement on equipment is low: the preparation method provided by the application does not need low temperature of-78 ℃ and pressure kettle conditions, and has low requirements on equipment;
(4) The product of each step of the preparation method provided by the application is solid, is convenient to separate and purify, and has very high purity and controllable single impurity;
(5) The preparation method provided by the application relates to the preparation of the intermediate 4, and the intermediate is converted into the intermediate 7 with wider application range by using methods of condensation, grignard and esterification with low cost and mild conditions, and can be used for the synthesis of various new tetracycline antibiotics.
Drawings
FIG. 1 is a reaction scheme of phenyl 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoate according to the present invention;
FIG. 2 is a LC-MS diagram of intermediate 7 prepared in example 7-1;
FIG. 3 is a nuclear magnetic hydrogen spectrum of intermediate 7 produced in example 7-1.
Detailed Description
Example 1 synthesis of intermediate 1:
the reaction equation is as follows:

adding 4-fluoro-3-methylphenol (100.0 g,1.0 eq.) into acetic acid (1000 mL, 10V), dropwise adding a mixed solution of liquid bromine (266.1 g,2.1 eq.) and acetic acid (300 mL, 3V) at 25 ℃, and stirring for 3 hours at the constant temperature; finishing the reaction of the HPLC middle control raw materials; the reaction solution was used in the next step without further treatment, and the material was calculated in 100% yield.
Example 2 synthesis of intermediate 2:
the reaction equation is:

adjusting the temperature of the intermediate 1 reaction liquid prepared in example 1 to 5 ℃, dropwise adding a mixed liquid of sodium nitrite (158.6 g,2.9 eq.) and water (450 mL, 4.5V), slowly heating to 25 ℃, keeping the temperature and stirring for 3h, and controlling the intermediate 1 to react completely by HPLC; the temperature is reduced to 10 ℃, and ice water (1600 g, 16V) is added dropwise; then, a mixed solution of sodium bisulfite (55.0 g,0.67 eq.) and water (500 mL, 5V) is added dropwise, and the starch potassium iodide test paper detects no color change; cooling to 0 deg.C, filtering, washing with water (300 mL, 3V) for three times; returning the wet product to the kettle, adding n-heptane (170 mL, 1.7V), stirring at 5 ℃ for 2h, cooling to 5 ℃, filtering, washing with n-heptane (100 mL, 1V), and drying at 50 ℃ to obtain 150.4 g of orange solid with purity of 95.6%, water content of 0.07% and yield of 76.0% in two steps.
Example 3 synthesis of intermediate 3:
the reaction equation is as follows:

intermediate 2 prepared in example 2 (150.0 g,1.0 eq.), anhydrous potassium carbonate (124.4 g,1.5 eq.), potassium iodide (10.0 g,0.1 eq.), benzyl bromide (102.6 g,1.0 eq.) were added to acetonitrile (1200 mL, 8V), warmed to 70 ℃ and stirred for 6h, and intermediate 2 was controlled to react out by HPLC; the temperature is adjusted to 25 ℃, the mixture is filtered, the filter cake is washed once by acetonitrile (600 mL, 4V), the combined filtrates are concentrated and dried to obtain 213.4 g of solid with the crude yield of over hundred percent and the purity of 95.6 percent according to 100 percent.
Example 4 synthesis of intermediate 4:
the reaction equation is as follows:

intermediate 3 (213.4 g, 100% yield from previous step, 204 g,1.0 eq.) prepared in example 3 was added to tetrahydrofuran (2000 ml, 10V), and a mixture of sodium dithionite (593.3 g,5.0 eq.) and water (2000 ml, 10V) was added dropwise at 25 ℃ and stirred for 5h with constant temperature; the intermediate 3 is controlled by HPLC to react completely; THF in the reaction solution was concentrated under reduced pressure at 40 ℃ or lower; adding methyl tert-butyl ether (2000 mL, 10V), stirring, standing, demixing, adding water (800 mL, 4V) and washing an organic phase; 4M hydrogen chloride/ethanol solution (158.3 g,1.1 eq.) was added dropwise at 20 ℃ to precipitate a solid; filtration at 20 ℃ and rinsing with methyl tert-butyl ether (600 mL, 3V) followed by vacuum drying at 45 ℃ gave 179.6 g of an off-white solid with 98.4% purity in 86.4% yield
Hydrogen spectrum: 1H NMR (400 MHz, DMSO-d 6) 7.32-7.56 (m, 5H), 6.62 (d, J =11.6 Hz, 1H), 5.43 (s, 3H), 4.80 (s, 2H), 2.11 (d, J =2.4 Hz, 3H)
Example 5 synthesis of intermediate 5:
the reaction equation is as follows:

example 5-1
Intermediate 4 (165.0 g,1.0 eq.), bromobenzyl (48.5 g,3.0 eq.), and N, N-diisopropylethylamine (313.0 g,5.0 eq.) prepared in example 4 were added to N-methylpyrrolidone (1320 mL,8 w/w) and stirred for 16h at 105 ℃; after the intermediate 4 is completely reacted in HPLC, adding water (2000 mL, 12V), separating out a solid, and filtering; the wet product is returned to the kettle, methanol (495 mL,3 w/w) is added, stirring is carried out for 16h at the temperature of 25 ℃, the temperature is adjusted to 0 ℃, filtration is carried out, methanol (165 mL, 1V) is adopted for rinsing, vacuum drying is carried out at the temperature of 45 ℃, 221.4 g of off-white solid is obtained, the purity is 99.9 percent, and the yield is 93.3 percent.
Hydrogen spectrum: 1H NMR (400 MHz, DMSO-d 6): 7.23-7.58 (m, 15H), 6.80 (d, J =12.0 Hz, 1H), 5.02 (s, 2H), 4.34 (s, 4H), 2.19 (d, J =2.4 Hz, 3H)
Example 5-2
Intermediate 4 (4.5 g,1.0 eq.) prepared in example 4, benzyl bromide (22.2 g,10.0 eq.) and cesium carbonate (46.5 g,11.0 eq.) were added to N-methylpyrrolidone (45 g,10 w/w) and stirred for 16h at 100 ℃ with tempering; after the reaction of the intermediate 4 in HPLC is finished, the intermediate is cooled to 25 ℃, filtered, rinsed by ethyl acetate (45 mL, 10V), added with water (180 mL, 40V), stirred, kept stand for layering, concentrated at 45 ℃ to dry an organic phase, added with methanol (60 g,13.3 w/w), adjusted to 0 ℃, stirred for 0.5 h, filtered, rinsed by methanol (15 mL, 3.3V) and dried in vacuum at 45 ℃ to obtain 5.0 g of off-white solid with the purity of 98.0 percent and the yield of 78.5 percent.
Examples 5 to 3
Intermediate 4 (2.0 g,1.0 eq.) prepared in example 4, bromobenzyl (9.9 g,10.0 eq.) and potassium carbonate (8.8 g,11.0 eq.) were added with acetonitrile (20 g,10 w/w) and stirred at 80 ℃ for 40 h; after the reaction of the intermediate 4 in HPLC is finished, the intermediate is cooled to 25 ℃, filtered, rinsed by ethyl acetate (20 mL, 10V), added with water (20 mL, 10V), stirred, kept stand for layering, concentrated at 45 ℃ to dry the organic phase, added with methanol (20 g,10 w/w), adjusted to 0 ℃, stirred for 0.5 h, filtered, rinsed by methanol (5 mL, 2.5V) and dried in vacuum at 45 ℃ to obtain 2.44 g of off-white solid with the purity of 99.0 percent and the yield of 86.2 percent.
Example 6 synthesis of intermediate 6:
the reaction equation is:

example 6-1
Intermediate 5 (215.0 g,1.0 eq.) prepared in example 5-1 was added to tetrahydrofuran (2.2L, 10V), 2.0M solution of isopropyl magnesium chloride in tetrahydrofuran (660 mL,3.0 eq.) was added, the temperature was adjusted to 50 ℃ and stirred for 5.0 h, and the reaction of intermediate 5 was controlled to completion by HPLC; adjusting the temperature to 15 ℃, introducing carbon dioxide gas (49.1L, 5.0 eq.) for 1.5h, and finishing the intermediate state reaction in HPLC; adjusting the temperature to 5 ℃, dropwise adding 2N hydrochloric acid (250 mL, 1.2V) to adjust the pH value to 7, concentrating the dried tetrahydrofuran under reduced pressure below 40 ℃, adding water (430 mL, 2V), adding ethyl acetate (1290 mL, 6V), adjusting the temperature to 20 ℃, dropwise adding 2N hydrochloric acid (450 mL, 2.1V) to adjust the pH value to 2-3, extracting and demixing, extracting the water phase once with ethyl acetate (537.5 mL, 2.5V), combining the ethyl acetate phases, and washing once with water (430 mL, 2V); concentrating the organic phase under reduced pressure below 45 deg.C to paste, adding n-heptane (400 mL, 1.9V), concentrating under reduced pressure below 45 deg.C to paste, dripping n-heptane (860 mL, 4V), stirring at 25 deg.C for 1h, adjusting temperature to 5 deg.C, stirring for 1h, filtering, rinsing with n-heptane (200 mL, 1V), and vacuum drying at 45 deg.C to obtain 174.7 g white solid with purity of 99.8% and yield of 87.5%
Hydrogen spectrum: 1H NMR (400 MHz, DMSO-d 6): 13.42 (s, 1H), 7.19-7.45 (m, 15H), 6.69 (d, J =12.0 Hz, 1H), 5.05 (s, 2H), 4.31 (s, 2H), 2.01 (s, 3H).
Example 6-2
Intermediate 5 (5.0 g,1.0 eq.) prepared in example 5-2 was added to 2-methyltetrahydrofuran (50 mL, 10V), 1.0M solution of isopropylmagnesium chloride lithium chloride in tetrahydrofuran (20.4 mL,2.0 eq.) was added, the temperature was adjusted to 65 ℃ and the mixture was stirred for 3.0 h, and the reaction of intermediate 5 was controlled to completion by HPLC; adjusting the temperature to 20 ℃, introducing carbon dioxide gas (1.1L, 5.0 eq.) for 1.5h, and finishing the intermediate state reaction in HPLC; the temperature was adjusted to 15 ℃, 2N hydrochloric acid (10 mL, 2V) was added dropwise to adjust pH 7, and dry tetrahydrofuran was concentrated under reduced pressure at a temperature below 40 ℃. Adding water (10 mL, 2V), adding ethyl acetate (30 mL, 6V), adjusting temperature to 20 deg.C, dropwise adding 2N hydrochloric acid (10 mL, 2V) to adjust pH 2-3, extracting for layering, extracting water phase with ethyl acetate (15 mL, 3V) once, mixing ethyl acetate phases, washing with water (10 mL, 2V) once, concentrating organic phase under reduced pressure below 45 deg.C to paste, adding N-heptane (10 mL, 2V), concentrating under reduced pressure below 45 deg.C to paste, dropwise adding N-heptane (20 mL, 4V), stirring at 25 deg.C for 1h, adjusting temperature to 5 deg.C, stirring for 1h, filtering, rinsing with N-heptane (5 mL, 1V), vacuum drying at 45 deg.C to obtain 3.74 g yellow solid with purity of 91.4%, yield of 80.5%
Examples 6 to 3
Adding magnesium (0.5 g, 2eq.) into tetrahydrofuran (30 mL, 6V), adjusting the temperature to 60 ℃, adding iodine, and adding 1, 2-dibromoethane (0.5 mL, 0.1V) for initiation; a mixed solution of the intermediate 5 (5.0 g,1.0 eq.) and tetrahydrofuran (20 mL, 4V) in the example 5-1 is added dropwise, the mixture is stirred for 2h at 60 ℃, and the intermediate 5 is controlled to react completely by HPLC; adjusting the temperature to 20 ℃, introducing carbon dioxide gas (2.3L, 10.0 eq.) for 1.5h, and finishing the intermediate state reaction in HPLC; adjusting temperature to 15 deg.C, adding 2N hydrochloric acid (10 mL, 2V) to adjust pH 7, concentrating dry tetrahydrofuran under reduced pressure below 40 deg.C, adding water (10 mL, 2V) and ethyl acetate (30 mL, 6V), adjusting temperature to 20 deg.C, adding 2N hydrochloric acid (10 mL, 2V) to adjust pH 2-3, extracting for layering, extracting water phase with ethyl acetate (15 mL, 3V), mixing ethyl acetate phases, washing with water (10 mL, 2V) once, concentrating organic phase under reduced pressure below 45 deg.C to paste, adding N-heptane (10 mL, 2V), and concentrating under reduced pressure below 45 deg.C to paste. N-heptane (20 mL, 4V) was added dropwise, stirring was carried out at 25 ℃ for 1h, the temperature was adjusted to 5 ℃ and stirring was carried out for 1h, filtration was carried out, n-heptane (5 mL, 1V) rinsing was carried out, vacuum drying was carried out at 45 ℃ to obtain 3.61 g of yellow solid, purity was 90.6%, and yield was 77.7%.
Example 7 synthesis of intermediate 7:
the reaction equation is:

example 7-1
Adding the intermediate 6 (170.0 g,1.0 eq.) prepared in example 6-1 into dichloromethane (1700 mL, 10V), adding two drops of DMF for catalysis, adjusting the temperature to 25 ℃, adding oxalyl chloride (71.1 g,1.5 eq.), keeping the temperature and stirring for 2h, concentrating under reduced pressure below 40 ℃, adding dichloromethane (1020 mL, 6V), concentrating under reduced pressure below 40 ℃, adding dichloromethane (1700 mL, 10V), adding DMAP (460 mg,0.01 eq.) for catalysis, adding phenol (36.9 g,1.05 eq.), adjusting the temperature to 25 ℃, adding triethylamine (75.5 g,2.0 eq.) and stirring for 2h at 25 ℃, and controlling the intermediate 6 to react completely by HPLC; adjusting the temperature to 15 ℃, adding water (680 mL, 4V) for extraction and delamination, adding water (510 mL, 3V), then dripping 2N hydrochloric acid (120 g,0.7 w/w) for adjusting pH to 4, extracting and delaminating, concentrating 2/3 dichloromethane under reduced pressure below 35 ℃, adding methanol (680 mL, 4V), concentrating under reduced pressure below 45 ℃ until the total volume is remained to be 3V, adding methanol (510 mL, 3V), adjusting the temperature to 0 ℃, stirring for 2h, filtering, rinsing with methanol (250 mL, 1.5V), and drying under vacuum at 45 ℃ to obtain 189.0g of off-white solid with the purity of 99.9 percent and the yield of 95.3 percent.
Mass spectrum: ESI-MS (m/z): 532[ M ] +H ] +
Hydrogen spectrum: 1H NMR (400 MHz, DMSO-d 6) 7.33-7.43 (m, 7H), 7.18-7.31 (m, 11H), 7.05 (d, J =8.8 Hz, 2H), 6.87 (d, J =12.0 Hz, 1H), 5.19 (s, 2H), 4.40 (s, 4H), 2.18 (d, J =1.6 Hz, 3H).
Example 7-2
Adding the intermediate 6 (3.0 g,1.0 eq.) prepared in example 6-2 into thionyl chloride (23.5 g,30.0 eq.) in portions at 25 ℃, heating to 80 ℃, refluxing for 2h, concentrating under reduced pressure below 50 ℃, adding dichloromethane (20 mL, 7V), concentrating under reduced pressure below 40 ℃, adding dichloromethane (20 mL, 7V), adding DMAP (8 mg,0.01 eq.) for catalysis, adding phenol (0.8 g,1.3 eq.), adjusting the temperature to 25 ℃, dropping N, N-diisopropylethylamine (1.7 g,2.0 eq.) and stirring for 4h at 25 ℃, and controlling the intermediate 6 to react in HPLC; adjusting the temperature to 15 ℃, adding water (12 mL, 4V) for extraction and delamination, adding water (9 mL, 3V), then dripping 2N hydrochloric acid (2.1 g,0.7 w/w) to adjust the pH value to 4, extracting and delaminating, concentrating 2/3 dichloromethane under reduced pressure below 35 ℃, adding methanol (12 mL, 4V), concentrating under reduced pressure below 45 ℃ to the residual 3V of the total volume, adding methanol (9 mL, 3V), adjusting the temperature to 0 ℃, stirring for 2h, filtering, rinsing with methanol (6 mL, 2V), and drying under vacuum at 45 ℃ to obtain 3.32 g of white-like solid with the purity of 100.0 percent and the yield of 94.8 percent.
Claims (9)
1. A preparation method of 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoate is characterized by comprising the following steps: the method comprises the following steps:
step E: synthesis of intermediate 5:
intermediate 4 Condensation reaction is carried out to obtain an intermediate 5
Condensation reaction is carried out to obtain an intermediate 5 I.e., N-dibenzyl-2-benzyloxy-3-bromo-5-fluoro-4-methylaniline;
I.e., N-dibenzyl-2-benzyloxy-3-bromo-5-fluoro-4-methylaniline;
step F: synthesis of intermediate 6:
the intermediate 5 is To prepare a Grignard reagent or an intermediate 5
To prepare a Grignard reagent or an intermediate 5 Performing Grignard exchange; then reacting with carbon dioxide to obtain intermediate 6
Performing Grignard exchange; then reacting with carbon dioxide to obtain intermediate 6 I.e., 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid;
I.e., 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid;
wherein, the reagent used for preparing the Grignard reagent is magnesium, and the Grignard reagent used for Grignard exchange is isopropyl magnesium chloride or isopropyl magnesium chloride lithium chloride;
g: and (3) synthesizing an intermediate 7:
intermediate 6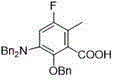 Firstly, carrying out chlorination reaction; then carrying out esterification reaction to obtain an intermediate 7
Firstly, carrying out chlorination reaction; then carrying out esterification reaction to obtain an intermediate 7 I.e., 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester.
I.e., 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester.
2. The method of claim 1, wherein: the condensation reaction in step E is specifically performed by: and adding the intermediate 4, benzyl halide and alkali into a solvent, and carrying out condensation reaction to obtain an intermediate 5, namely the N, N-dibenzyl-2-benzyloxy-3-bromo-5-fluoro-4-methylaniline.
3. The method of claim 2, wherein: the benzyl halide is selected from benzyl bromide or/and benzyl chloride;
the alkali is one or more of triethylamine, N-diisopropylethylamine, sodium carbonate, potassium carbonate and cesium carbonate;
the solvent is one or more of N-methylpyrrolidine, N-dimethylformamide, N-dimethylacetamide, dimethyl sulfoxide and acetonitrile.
4. The production method according to claim 2, characterized in that: the molar ratio of the intermediate 4, the benzyl halide and the alkali is 1.0:2.5-10.0:3.5-11.0.
5. The method of claim 1, wherein: the specific operation in the step F is as follows: dissolving the intermediate 5 obtained in the step E in a solvent, and preparing a Grignard reagent with magnesium or carrying out Grignard exchange with the Grignard reagent; and then reacted with carbon dioxide to afford intermediate 6.
6. The method of claim 5, wherein: the solvent is tetrahydrofuran or/and 2-methyltetrahydrofuran.
7. The production method according to claim 1, characterized in that: the concrete operation of the step G is as follows: f, carrying out chlorination reaction on the intermediate 6 obtained in the step F and a chlorination reagent to generate acyl chloride; then carrying out esterification reaction on the intermediate 7, phenol and alkali in a solvent;
the chlorinating agent is oxalyl chloride or/and thionyl chloride;
the solvent is one or more of dichloromethane, dichloroethane and chloroform;
the alkali is triethylamine or/and N, N-diisopropylethylamine.
8. The method of claim 7, wherein: the molar ratio of the intermediate 6, the chlorinating reagent, the phenol and the alkali is 1.0:1.0-30.0:1.05-1.3:1.5-2.5.
9. The method of claim 1, wherein: the preparation method further comprises the following steps:
step A, synthesis of intermediate 1 ;
;
The reaction equation is:

step B, synthesis of intermediate 2 ;
;
The reaction equation is:

step C, synthesis of intermediate 3 ;
;
The reaction equation is:

step D, synthesis of intermediate 4 ;
;
The reaction equation is:

Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202211408228.5A CN115448849B (en) | 2022-11-10 | 2022-11-10 | Preparation method of 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202211408228.5A CN115448849B (en) | 2022-11-10 | 2022-11-10 | Preparation method of 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN115448849A CN115448849A (en) | 2022-12-09 |
| CN115448849B true CN115448849B (en) | 2023-02-03 |
Family
ID=84295384
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202211408228.5A Active CN115448849B (en) | 2022-11-10 | 2022-11-10 | Preparation method of 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN115448849B (en) |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9073829B2 (en) * | 2009-04-30 | 2015-07-07 | President And Fellows Of Harvard College | Synthesis of tetracyclines and intermediates thereto |
| CA2883238C (en) * | 2012-08-31 | 2021-11-23 | Tetraphase Pharmaceuticals, Inc. | Tetracycline compounds |
-
2022
- 2022-11-10 CN CN202211408228.5A patent/CN115448849B/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN115448849A (en) | 2022-12-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101279997B (en) | Novel preparation of budesonide | |
| CN106316967B (en) | The preparation method of West pa lattice intermediate and West pa lattice | |
| CN111320548A (en) | Synthesis method of anticancer drug intermediate 2-fluoro-3-methyl aminobenzoate | |
| CN114409566A (en) | Preparation method of ioversol hydrolysate | |
| CN115448849B (en) | Preparation method of 2-benzyloxy-3-dibenzylamino-5-fluoro-6-methylbenzoic acid phenyl ester | |
| CN105367399B (en) | A kind of preparation method of 9,10-phenanthrene quinone compound | |
| EP3643714B1 (en) | 4,5-disubstituted-1-hydro-pyrrole(2,3-f)quinolone-2,7,9-tricarboxylate compound and applications | |
| CN102321016A (en) | Synthesis method of 5-bromo-2-methyl 4-hydroxypyridinecarboxylate | |
| EP3527556B1 (en) | Method for preparing deuterated imidazole diketone compound | |
| CN110698352B (en) | A kind of synthetic method of 3-bromo-5-aminocatechol dimethyl ether | |
| WO2023174062A1 (en) | Method for preparing nilutamide and intermediate thereof | |
| CN108689957A (en) | A kind of preparation method and application of 2R/2S- trifluoromethyls morpholine and its hydrochloride | |
| CN116621754A (en) | The preparation method of pyrrolidone-3-β'-amino derivatives | |
| CN114621109A (en) | Synthetic method of apatamide and intermediate thereof | |
| CN114163380A (en) | Alavazepam intermediate, preparation method and application thereof | |
| CN119118913B (en) | A preparation method of anti-fibrosis drug pirfenidone | |
| CN108014112B (en) | Application of o-toluidine amino acetamido benzo [ d ] aza-based quinazoline compound in preparation of drugs for treating lung cancer | |
| CN114181152B (en) | A kind of preparation method of arylpyrazole drug intermediate | |
| CN104418805B (en) | Dabigatran etexilate intermediate as well as preparation method and application thereof | |
| CN110922411B (en) | Synthetic method of drug intermediate | |
| CN117186010A (en) | A kind of synthesis method of atipamezole hydrochloride | |
| CN116987022A (en) | Preparation method of compound containing azabicyclo- [3.1.0] -hexane-2-one | |
| CN108250185B (en) | 6-(2-(o-tolylamino)acetamido)quinazoline compounds and their preparation and application | |
| CN105111217A (en) | Method for synthesizing isoindole dihyroquinazoline derivative | |
| CN116345509A (en) | The preparation method of 3-hydroxyl-n-halobenzoic acid methyl ester compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |