CN112313748A - 病毒基因突变模式的测量和预测 - Google Patents
病毒基因突变模式的测量和预测 Download PDFInfo
- Publication number
- CN112313748A CN112313748A CN201980041733.0A CN201980041733A CN112313748A CN 112313748 A CN112313748 A CN 112313748A CN 201980041733 A CN201980041733 A CN 201980041733A CN 112313748 A CN112313748 A CN 112313748A
- Authority
- CN
- China
- Prior art keywords
- prevalence
- virus
- mutation
- time period
- period
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images








Classifications
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B5/00—ICT specially adapted for modelling or simulations in systems biology, e.g. gene-regulatory networks, protein interaction networks or metabolic networks
- G16B5/30—Dynamic-time models
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- G—PHYSICS
- G06—COMPUTING OR CALCULATING; COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F30/00—Computer-aided design [CAD]
- G06F30/20—Design optimisation, verification or simulation
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B10/00—ICT specially adapted for evolutionary bioinformatics, e.g. phylogenetic tree construction or analysis
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/20—Allele or variant detection, e.g. single nucleotide polymorphism [SNP] detection
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/50—Mutagenesis
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16H—HEALTHCARE INFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR THE HANDLING OR PROCESSING OF MEDICAL OR HEALTHCARE DATA
- G16H10/00—ICT specially adapted for the handling or processing of patient-related medical or healthcare data
- G16H10/40—ICT specially adapted for the handling or processing of patient-related medical or healthcare data for data related to laboratory analysis, e.g. patient specimen analysis
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16H—HEALTHCARE INFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR THE HANDLING OR PROCESSING OF MEDICAL OR HEALTHCARE DATA
- G16H50/00—ICT specially adapted for medical diagnosis, medical simulation or medical data mining; ICT specially adapted for detecting, monitoring or modelling epidemics or pandemics
- G16H50/50—ICT specially adapted for medical diagnosis, medical simulation or medical data mining; ICT specially adapted for detecting, monitoring or modelling epidemics or pandemics for simulation or modelling of medical disorders
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16H—HEALTHCARE INFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR THE HANDLING OR PROCESSING OF MEDICAL OR HEALTHCARE DATA
- G16H50/00—ICT specially adapted for medical diagnosis, medical simulation or medical data mining; ICT specially adapted for detecting, monitoring or modelling epidemics or pandemics
- G16H50/80—ICT specially adapted for medical diagnosis, medical simulation or medical data mining; ICT specially adapted for detecting, monitoring or modelling epidemics or pandemics for detecting, monitoring or modelling epidemics or pandemics, e.g. flu
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/16011—Orthomyxoviridae
- C12N2760/16111—Influenzavirus A, i.e. influenza A virus
- C12N2760/16121—Viruses as such, e.g. new isolates, mutants or their genomic sequences
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/16011—Orthomyxoviridae
- C12N2760/16111—Influenzavirus A, i.e. influenza A virus
- C12N2760/16122—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Physics & Mathematics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medical Informatics (AREA)
- General Health & Medical Sciences (AREA)
- Theoretical Computer Science (AREA)
- Biotechnology (AREA)
- Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Bioinformatics & Computational Biology (AREA)
- Evolutionary Biology (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Analytical Chemistry (AREA)
- Epidemiology (AREA)
- Primary Health Care (AREA)
- Biomedical Technology (AREA)
- Physiology (AREA)
- Animal Behavior & Ethology (AREA)
- Organic Chemistry (AREA)
- Data Mining & Analysis (AREA)
- Databases & Information Systems (AREA)
- Pathology (AREA)
- General Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- Geometry (AREA)
- Computer Hardware Design (AREA)
- Evolutionary Computation (AREA)
- General Physics & Mathematics (AREA)
- Virology (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
Abstract
本发明公开了一种通过鉴定病毒(例如流感病毒)氨基酸序列中的有效突变和有效突变期,从而测量和预测所述病毒突变模式的方法。在有效突变期期间,所述突变使病毒能够逃避人体免疫。基于对现有病毒组成和人群感染率的分析,该方法可度量病毒基因突变活性(“g‑度量”)并优化表现病毒基因活性的一个或多个参数。本发明可用于预测所述病毒的未来基因活性,突变、筛选病毒疫苗毒株和/或预测感染性疾病爆发。
Description
相关申请的交叉引用
本申请要求获得2018年6月20日提交的第62/687,645号美国临时申请的权益,所述美国临时申请的公开内容通过引用以其整体并入。
背景
本发明总体上涉及病毒感染性疾病(例如,流行性感冒)的遗传流行病学,尤其涉及引起感染性疾病的病毒的病毒基因(或氨基酸)突变模式的测量和预测。
流行性感冒,也称为“流感”,是一种已经困扰人类几个世纪的传染性呼吸系统疾病。当人们发现流感是由病毒(流行性感冒病毒,即流感病毒)引起时,便希望产生有效的疫苗。经过多年的研究,流感疫苗现在得到广泛的应用。然而,流感病毒会迅速突变为新毒株,并且针对一种毒株有效的疫苗可能针对其它(突变的)毒株无效。因此,在制备流感疫苗时使用的流感病毒株的“配方”会根据对未来有效毒株的预测而定期修改,政府鼓励个人每年接种新的流感疫苗,以帮助他们的免疫系统跟上突变的流感病毒。
目前,每年流感疫苗生产和分配的方案包括需要决定在下一轮的疫苗接种中防御哪些流感病毒毒株。目前,这项决定是基于对来自世界各地的流感病毒样本、已知的抗原位点(例如,病毒序列中的特定氨基酸)的研究,以及从经验中学到的关于病毒突变模式的教训得出的。其目的是预测在未来约18个月至2年内哪些流感病毒毒株将有效对抗人类免疫系统(即,产生疾病)。流感疫苗就是根据这一预测研制的。
预测并不总是准确的,因此,每年流感疫苗的有效性差异很大。这使得个体不太愿意接种流感疫苗,从而损害了当大多数人针对某传染原进行免疫时获得的“群体免疫”效应。
因此,改进用于预测病毒突变,且特别是用于预测在未来至少两年的时间范围内哪些突变将会有效对抗人类免疫系统的技术将是尤其重要的。
概述
本发明的某些实施方案涉及基于病毒序列(例如氨基酸序列)和群体流行水平测量和预测病毒突变模式的技术。预测是基于鉴定“有效突变”,即有助于病毒逃避人类免疫的具有进化优势的突变(氨基酸序列或核酸序列的变异),这与对病毒存活和繁殖的能力没有(或有可忽略的)影响的“不重要突变”相反。预测还基于人类免疫将最终学会识别和阻止有效突变(在疫苗的帮助下或不在疫苗的帮助下)的假设。这意味着有效突变具有“有效突变期”,这是突变使病毒能够逃避人类免疫的时间段。使用本文所述的技术,鉴定有效突变和确定有效突变期,可更精准的预测给定病毒的哪些毒株(即,哪些突变)将在未来的时间段中流行。这种预测可实现多种实际目的,包括:(1)帮助选择用于疫苗生产的病毒毒株;(2)提供关于给定版本的疫苗的可能功效的实时信息;和/或(3)预测病毒活性(例如,由病毒引起的感染性疾病的发病率)。
本文所用的一些说明性技术依赖于流感病毒组成(氨基酸序列)和感染率的纵向群组分析,以计算流感病毒的基因突变活性的度量,在本文中称为“g-度量”。下面将从至少两个方面来更具体地描述g-度量如何模拟基因活性。第一是单一突变是否应被认为是重要的。假设更适应的突变将在新出现后广泛地扩散,而不重要的突变将不会扩散,则单一残基的流行率将导致更高的g-度量。基因活性的第二方面体现在同时突变的基因数目,g-度量可捕获同时具有多个残基取代的潜在抗原性转变;在给定的流行率下,较高数量的有效突变将增加g-度量。因此,g-度量反映了突变的适应性和同时有效突变的数目。此外,如果某一位点在研究期内出现多于一个的有效突变期,则g-度量将涵盖随后的有效突变期。计算g-度量还包括优化进一步表征流感病毒基因活性的参数,如优势阈值(残基被认为是有效突变所需的最小流行率)和延长的有效期(代表有效突变在获得优势后保持有效对抗人类免疫的时间)。g-度量和/或相关参数可以用于预测流感病毒的未来基因活性,这可以有助于为下一轮流感疫苗选择病毒毒株和/或预测流感爆发。类似的技术可以应用于其它病毒和相关的感染性疾病。
以下详述与附图一起提供了对所要求保护的发明的性质和优势的更好的理解。
附图简述
图1A-1C示出了根据本发明实施方案的编码序列构造的简化实例。图1A显示了在一段时间内观察到的四种示例性氨基酸序列。图1B显示了根据本发明的实施方案可以定义研究期内的标签序列。图1C显示了对应于图1A的氨基酸序列和图1B的标签序列的编码序列。
图1D显示了根据本发明的实施方案从图1C的编码序列计算的流行率向量。
图2显示了根据本发明的实施方案从流行率向量鉴定有效突变和有效突变期的简化实例。
图3和图4是显示了g-度量与在群体中观察到的流感感染变化的相关性的图。图3显示了获自1996年至2015年香港的流感病毒活性的观察结果的数据。图4显示了获自2003年至2016年纽约的流感病毒活性的观察结果的数据。
图5显示了根据本发明实施方案用于测量和预测流感病毒活性的方法的流程图。
详述
本发明所述的用于模拟病毒活性的技术依赖于病毒组成(氨基酸序列)和感染率的纵向群组分析,以计算病毒的基因突变活性的度量,在本文中称为“g-度量”。在被分成一组相等持续时间的时间段的“研究期”中进行分析。在一些实施方案中,每个时间段可以是一年;其它实施方案可以定义更短的时间段(例如,三个月、一个月、一周)或更长的时间段(例如,两年、五年等)。为了说明的目的,提及了流行性感冒或“流感”病毒;然而,所描述的技术可以应用于其它病毒。
对于给定的时间段t,收集了nt数目的流感病毒(或其它目标病毒)的样本。对于时间段t内的每个样本i,确定病毒的氨基酸序列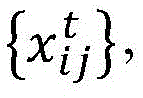 其中指数j表示氨基酸序列内的特定位点,且x是特定氨基酸的标识符。可以使用常规技术或其它技术确定流感病毒给定样本的氨基酸序列,并且特定的测序技术对于理解本发明不是关键的。通常,nt数目的氨基酸序列
其中指数j表示氨基酸序列内的特定位点,且x是特定氨基酸的标识符。可以使用常规技术或其它技术确定流感病毒给定样本的氨基酸序列,并且特定的测序技术对于理解本发明不是关键的。通常,nt数目的氨基酸序列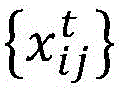 的实例是已被确定的。
的实例是已被确定的。
假定病毒可以在研究期期间突变,并且在相同时间段内收集的流感病毒的不同样本可以具有不同的突变。为了便于突变的分析,定义研究期内的“标签序列”是有帮助的,所述“标签序列”可以用于以统一的格式表示每个样本。对于k=1,…,K,标签序列可以是氨基酸序列{ak},其中K被定义为:

其中J是病毒的总氨基酸序列长度,并且qj是在整个研究期期间在位点j观察到的独特氨基酸的数目。标签序列{ak}是由连接在氨基酸序列的每个位点j处观察到的所有独特的氨基酸组成的。标签序列使得能够在不建立参考序列(建立参考序列是常规操作)的情况下对突变进行评估;因此,标签序列不是序列的比较,而是提供了一种工具来捕获每种可能残基的动态。
给定标签序列{ak},每个观察到的氨基酸序列 可以表示为编码序列
可以表示为编码序列 编码序列可以是K个指示符(例如,位数)的序列,标签序列中每个位置k对应一个指示符;如果在位置j处的相应氨基酸存在于样本i中,则可以将在第k位指示符设置为第一值(例如1),如果不存在,则设置为第二值(例如0)。
编码序列可以是K个指示符(例如,位数)的序列,标签序列中每个位置k对应一个指示符;如果在位置j处的相应氨基酸存在于样本i中,则可以将在第k位指示符设置为第一值(例如1),如果不存在,则设置为第二值(例如0)。
图1A-1C示出了根据本发明实施方案的编码序列 的构造的简化实例。图1A显示了在时间段t(例如一年)期间观察到的四个示例性氨基酸序列101、102、103、104;使用标准的IUPAC单字母编码方案,通过单字母代码表示氨基酸。可以看出,在观察的序列101-104中,第一个位点(j=1)具有氨基酸N或K;第二个位点(j=2)具有氨基酸S;第三个位点(j=3)具有氨基酸E或K;第四个位点(j=4)具有氨基酸N;并且第五个位点(j=5)具有氨基酸A或T。
的构造的简化实例。图1A显示了在时间段t(例如一年)期间观察到的四个示例性氨基酸序列101、102、103、104;使用标准的IUPAC单字母编码方案,通过单字母代码表示氨基酸。可以看出,在观察的序列101-104中,第一个位点(j=1)具有氨基酸N或K;第二个位点(j=2)具有氨基酸S;第三个位点(j=3)具有氨基酸E或K;第四个位点(j=4)具有氨基酸N;并且第五个位点(j=5)具有氨基酸A或T。
在该实例中,假定在研究期期间的其它时间段(例如,多年)中也观察到氨基酸序列,并且在那些时间段中的至少一个时间段内观察到一些位点上出现其它氨基酸。具体来说,假定进行以下观察:对于位点j=1,观察到氨基酸V、I、N或K;对于位点j=2,观察到氨基酸S;对于位点j=3,观察到氨基酸E或K;对于位点j=4,观察到氨基酸N或D;并且对于位点j=5,观察到氨基酸A或T。图1B显示了根据本发明实施方案可以定义研究期内的标签序列120。在该实例中,对标签序列120的位数进行排序,使得前四个标签序列位置对应于在j=1处观察到的氨基酸,下一个标签序列位置对应于在j=2处观察到的氨基酸,等等。当标签序列的多个位数对应于氨基酸序列中的相同位点时,可以基于第一次观察的时间段对位数进行排序。如果需要,可以使用其它排序。
图1C显示了分别对应于氨基酸序列101、102、103、104的编码序列131、132、133、134。编码序列131-134提供了与原始氨基酸序列101-104相同的信息,但采用了如下所述的计算分析的格式。应理解,流感病毒的氨基酸序列比该简化实例中的氨基酸序列长得多,并且在一段时间内获得的序列样本的数目可以比所示的四个实例大得多。还应理解,图1A-1C中的特定序列仅用于说明的目的,并且可以对应或不对应于现有的病毒。
给定一组与时间段t期间观察到的样本i相对应的nt个编码序列 在t时间段内的流行率向量
在t时间段内的流行率向量 可以被定义为:
可以被定义为:

流行率向量pt的每个分量都可以理解为在氨基酸序列中特定位点处的特定氨基酸的流行率。图1D示出了根据等式(2)的从图1C的编码序列计算的流行率向量pt。
为了鉴定有效突变,即提供对抗人类免疫的进化优势的突变,可以在研究期内的整个时间段中分析流行率向量pt。可以通过检测标签位置k处的流行率从时间段t0的零到随后时间段t0+1的非零的变化等,来鉴定突变。假定有效突变将增加流行率并最终至少达到阈值流行率,阈值流行率在本文中被称为“优势阈值”并表示为θ。为了便于分析,如果在研究期内存在时间t0和时间tθ,使得
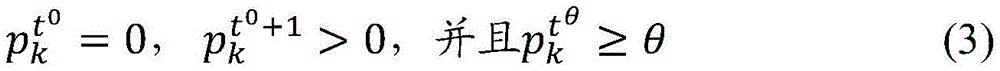
则将标签序列的位置ak处的突变定义为有效。如下所述,可以凭经验确定优势阈值θ的值。
定义有效突变期(EMP,在本文中用ω表示)也是有用的,所述有效突变期表示有效突变保持其进化优势的时间长度。该时间段包括转变时间tθ-t0(即,从第一次出现突变的时间到突变达到优势阈值的时间)。EMP还包括表示为h的“延长的有效突变期”,其对应于突变在达到优势之后保持其进化优势的时间长度。因此,对于位点k处的给定突变,总EMP被定义为:
ωk(θ,h)={t0<t≤tθ+h|θ,h,k}。 (4)
时间段t期间的有效突变的集合(在本文中用Wt表示)可以表示为:
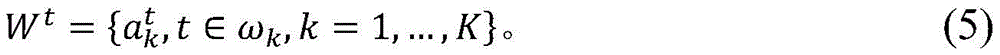
θ和h的最佳值可以使用以下所述的拟合程序凭经验确定。原则上,标签序列{ak}中不同的位点k拥有其特定的θ和h的值;然而,在实践中,收集足够的数据以确定每个位置的拟合有时是不可行的,故可以假定所有突变共用相同的θ和h值。在一个具体的实例中,θ=0.8且h=2。
图2显示了根据本发明实施方案用流行率向量鉴定有效突变和EMP的简化实例。假设来自图1B的标签序列{ak},并且假设图1D的流行率向量p是时间段t=1的流行率向量。图中还展示了时间段t=2至t=7的流行率向量pt;这些向量可以以上述的方式确定。为了便于说明,假设θ=0.8且h=2。对于每个有效突变(即,满足等式(2)的条件的突变),转变时间内的流行率值以浅灰色显示,延长的有效突变期内的流行率值以黑色显示,总EMP以粗黑线概述。应注意,尽管θ和h的值被假定为与位点无关,但总EMP可能由于转变时间的差异而变化。在该分析中,在位点k=6和k=8处的突变没有被鉴定为有效突变,即使它们在至少一些时间段内确实满足优势阈值亦如此,这是因为从零流行率到非零流行率的转变发生在t=1之前。
在鉴定有效突变和EMP后,可以计算反应基因突变活性的度量(本文称为“g-度量”)。具体来说,对于每个时间段t,K分量的指示符向量mt被定义为:

其中根据等式(4)定义ω(θ,h)。g-度量可以被定义为:

图2展示了每个时间段的根据等式(7)计算的gt。g-度量向量g=[gt]表示不同时间段内突变活性的趋势。
g-度量可以被理解为在给定时间段内所有有效突变的流行率的函数(例如,总和)。它模拟了基因活性的两个相关方面。第一是突变是否应被认为是重要的。假设更适应的突变将在新出现后广泛地扩散,而不重要的突变将不会扩散,则单一残基的流行将导致更高的g-度量。第二个方面是同时突变的数目,其捕获同时具有多个残基取代的潜在抗原性转变;在给定的流行率下,较高数量的有效突变将增加g-度量。因此,g-度量反映了突变的适应性和同时有效突变的数目。此外,如果某一位点在研究期内出现多于一个的有效突变期,则g-度量将涵盖所有的有效突变期。g-度量可以用于各种目的,包括:(1)预测流行病学;(2)基于有效突变和EMP选择用于下一轮流感疫苗的病毒毒株;(3)基于对当前的有效突变与疫苗毒株进行比较来评估当前可用的流感疫苗毒株。
如上所述,g-度量取决于两个参数:优势阈值θ和延长的有效突变期h。在一些实施方案中,可以基于群体水平流行性变量,如亚型的血清阳性率、一段时间内诊断的病毒感染病例的数目或该段时间内病毒感染的住院率,凭经验确定这些参数的值。预期g-度量的时间变化应与群体水平流行性变量的时间变化相关,因为新的有效突变的扩散将导致群体中更多的感染。
因此,在本发明的一些实施方案中,以下拟合程序可以用于确定θ和h的值。群体水平流行性变量(例如,诊断病例的数目或住院治疗的数目)被定义为向量f=[ft],其中指数t表示研究期内的任一时间段。选择测量向量g和f之间的匹配质量的函数S(f,g)。例如,S可以是广义线性模型的拟合优度统计量的p值,其中f是反应变量,且g是预测变量。在这种情况下,较小的S值表示反应和预测之间的更好匹配。θ和h的最佳值可以被定义为使S最小化的值 即:
即:

其中H={0、1、2、..},并且θ=[0.5,1]。
通过说明的方式,图3和图4显示了g-度量与在群体中观察到的流感感染变化的相关性的图。图3显示了获自1996年至2015年香港的流感病毒活性的观察结果的数据。通过虚线连接的菱形数据点对应于每年诊断出的甲型流行性感冒病例的数目。通过实线连接的圆形数据点表示使用如上所述计算的g-度量预测的病例的数目。类似地,图4显示了获自2003年至2016年纽约的流感病毒活性的观察结果的数据。通过虚线连接的菱形数据点显示了在给定年份中归因于病毒的H3毒株的流行性感冒病例的百分比。通过实线连接的圆形数据点表示使用如上所述计算的g-度量预测的此类病例的数目。如从图3和图4中可以看出的,具有θ和h的最佳值的g-度量可以模拟群体中流感发生率的变化。
如本文所述的g-度量可以用于进行对未来流感病毒活性的预测。在一些实施方案中,可以对流感的未来发生率进行预测。例如,如果拟合函数S(f,g)是泊松回归模型的拟合优度统计量的p值,则可以从现有数据获得以下拟合模型:
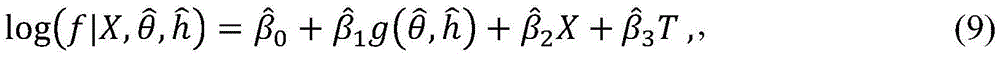
其中X是与流行病相关的环境协变量(例如,温度和湿度),并且T是时间变量;通过拟合确定系数 至
至 当样本量足够时,也可以使用更复杂的拟合函数,如系统动态模型。
当样本量足够时,也可以使用更复杂的拟合函数,如系统动态模型。
当时间段t+1的病毒序列样本可用时,可以根据等式(7),使用pt+1和 计算g-度量。当序列样本不可用时(例如,当t+1对应于未来的时间段时),可以基于现有数据中的条件流行率分布
计算g-度量。当序列样本不可用时(例如,当t+1对应于未来的时间段时),可以基于现有数据中的条件流行率分布
 来前瞻性地估计pt+1;在时间段t+1的流行率的估计是:
来前瞻性地估计pt+1;在时间段t+1的流行率的估计是:

其中E表示从条件流行率分布 确定的期望值。可以以上述方式从pt+1进行mt+1和gt+1的预测,并且预测的流行性水平由下式给出:
确定的期望值。可以以上述方式从pt+1进行mt+1和gt+1的预测,并且预测的流行性水平由下式给出:

在一些实施方案中,可以对下一个优势流行性感冒亚型进行预测。例如,可以获得每种亚型的g-度量,并且具有最高 的亚型是预测的下一时间段的优势亚型。通常,g-度量的变化,即基于突变流行率的函数,可以用于预测下一个优势亚型和未来流感趋势。
的亚型是预测的下一时间段的优势亚型。通常,g-度量的变化,即基于突变流行率的函数,可以用于预测下一个优势亚型和未来流感趋势。
在一些实施方案中,还可以进行有效突变的预测。等式(5)定义了时间段t的有效突变Wt的集合。可以从Wt等式(10)开始进行Wt+1的预测,并且优势阈值 可以用于鉴定时间段t+1内可能变成占优势的突变。延长的有效突变期
可以用于鉴定时间段t+1内可能变成占优势的突变。延长的有效突变期 可以用于鉴定Wt中的有效突变,其可能在时间段t+1中失去有效性。预测的有效突变Wt+1的集合可以用于疫苗抗原设计。例如,对于使用基因工程改造的疫苗,Wt+1可以鉴定疫苗中需要包括的氨基酸。
可以用于鉴定Wt中的有效突变,其可能在时间段t+1中失去有效性。预测的有效突变Wt+1的集合可以用于疫苗抗原设计。例如,对于使用基因工程改造的疫苗,Wt+1可以鉴定疫苗中需要包括的氨基酸。
在一些实施方案中,可以定义时间段t的代表性病毒序列 例如,对于每个氨基酸位点j,在该位点具有最高流行率的氨基酸可以被定义为代表性的氨基酸。为了便于说明,请参考图1B的标签序列和图1D的流行率向量,对于位点j=1,氨基酸K具有最高的流行率(p=0.75);对于位点j=2,氨基酸S具有最高的流行率(p=1);对于位点j=3,氨基酸E和K具有相同的流行率(p=0.5),因此可以选择任一种;对于位点j=4,氨基酸N具有最高的流行率(p=1);并且对于位点j=5,氨基酸T具有最高的流行率(p=0.75)。更普遍的来说,如上所述,标签序列{ak}包括对应于氨基酸序列中的每个位点的氨基酸的数目qj。在该情况下,代表性病毒序列
例如,对于每个氨基酸位点j,在该位点具有最高流行率的氨基酸可以被定义为代表性的氨基酸。为了便于说明,请参考图1B的标签序列和图1D的流行率向量,对于位点j=1,氨基酸K具有最高的流行率(p=0.75);对于位点j=2,氨基酸S具有最高的流行率(p=1);对于位点j=3,氨基酸E和K具有相同的流行率(p=0.5),因此可以选择任一种;对于位点j=4,氨基酸N具有最高的流行率(p=1);并且对于位点j=5,氨基酸T具有最高的流行率(p=0.75)。更普遍的来说,如上所述,标签序列{ak}包括对应于氨基酸序列中的每个位点的氨基酸的数目qj。在该情况下,代表性病毒序列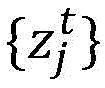 的每个氨基酸将是:
的每个氨基酸将是:

其中r0是产生以下的指数r的值:

其中,对于序列位点j,范围(rL,rU)通过以下定义:

rU=rL+qj。 (14b)
代表性病毒序列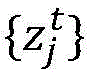 是在时间t时包括的所有有效突变的病毒的概率性总结。将代表性病毒序列与当前可用的流感疫苗中包括的毒株进行比较允许评估疫苗的可能有效性。例如,可以计算代表性病毒序列
是在时间t时包括的所有有效突变的病毒的概率性总结。将代表性病毒序列与当前可用的流感疫苗中包括的毒株进行比较允许评估疫苗的可能有效性。例如,可以计算代表性病毒序列 和当前可用的流感疫苗中包括的毒株之间的距离。为了实现这个目的,可以根据常规的序列相似性度量来定义序列间的距离,如氨基酸的p-距离或汉明距离(Hamming distance)。距离越小,匹配越好(并且疫苗可能更有效地用于保护患者免受流感感染)。
和当前可用的流感疫苗中包括的毒株之间的距离。为了实现这个目的,可以根据常规的序列相似性度量来定义序列间的距离,如氨基酸的p-距离或汉明距离(Hamming distance)。距离越小,匹配越好(并且疫苗可能更有效地用于保护患者免受流感感染)。
在一些实施方案中,未来时间段的代表性病毒序列 可以以相同的方式,使用在等式(10)中定义的前瞻性流行率向量来预测。在以现有野生型病毒制备流感疫苗的情况下,可以通过鉴定与预测的代表性病毒序列
可以以相同的方式,使用在等式(10)中定义的前瞻性流行率向量来预测。在以现有野生型病毒制备流感疫苗的情况下,可以通过鉴定与预测的代表性病毒序列 具有最近距离的现有野生型病毒来选择下一轮疫苗的最佳候选病毒毒株。如上所述,可以根据常规的序列相似性度量来定义距离,如氨基酸的p-距离。当在野生型毒株中未发现代表性病毒序列的预测的有效突变时,可以将基因工程技术应用于野生型序列,以使其与预测的序列完全相同或尽可能相似。
具有最近距离的现有野生型病毒来选择下一轮疫苗的最佳候选病毒毒株。如上所述,可以根据常规的序列相似性度量来定义距离,如氨基酸的p-距离。当在野生型毒株中未发现代表性病毒序列的预测的有效突变时,可以将基因工程技术应用于野生型序列,以使其与预测的序列完全相同或尽可能相似。
本文所述的分析方法可以应用于特定地区的序列和流行病数据、全球数据,或地区和全球数据的组合。候选疫苗病毒的预测可以是特定地区(例如,国家、大陆或半球)特异性的或制作用于全球使用。
本文所述的分析方法可以应用于流感病毒的任何或所有基因区段。由于每个基因可能具有不同的θ和h参数,因此当样本量足够大时,可以同时进行不同基因的多个g-度量的拟合(全局估计),或者可以首先估计重要基因的θ和h参数(例如,血凝素和神经氨酸酶-最常见的突变区段),然后有条件地估计剩余基因区段的θ和h参数(局部优化)。
本文所述的分析方法可以应用于任何流行性感冒病毒亚型,如H3N2、大流行的H1N1、B/Yamagata、B/Victoria。同样的方法也可以应用于其它已知的引起感染性疾病的病毒,如A-EV71病毒(手足口病的原因)、鼻病毒(普通感冒的原因)或者新出现的可引起流行病或大流行病的病原体。
可以使用任何可用的测序技术(包括但不限于第一代测序(Sanger)、下一代测序(Illumina平台)或第三代测序(PacBio平台或Nanopore平台))获得在本文所述种类的分析中使用的测序数据。
本文所述的分析方法可以用于计算机实施的预测流感病毒活性的方法中。图5显示了根据本发明实施方案用于测量和预测流感病毒活性的过程500的流程图。图5可以使用常规设计的计算机系统来实施。该过程的输入可以包括在研究期内收集的真实世界数据,包括关于流感报告病例的发病率或比率的数据和在研究期内观察到的流感病毒的序列数据。
在框502处,定义了研究期。研究期可以与期望的一样长,例如10年、15年、20年等。研究期可以分成多个相等长度的时间段(例如,一年、三个月等)。研究期的选择和每个时间段的长度可以基于可用于确定流感病毒中特定突变的流行率的数据的可及性。
在框504处,获得每个时间段内的群体水平的流行性变量。如上所述,这可以是代表群体中流感病毒感染发生的次数或频率的变量。根据哪些数据源是可用的,群体水平流行性变量可以基于报告的流感诊断病例数和/或报告的流感住院治疗病例数。这样的数据可以从多年以前的公共健康记录中获得。另外,也可以使用来自前瞻性纵向群组的采样替代,并且可以对回顾性获取的和/或从正在进行的采样中获取的数据的任何组合实施过程500。
在框506处,获得每个时间段内的流感病毒样本的氨基酸序列。例如,可以定期收集流感病毒样本并对其进行测序。可以从感染的患者、从环境表面或以任何其它方式收集样本。可以使用常规技术测定流感病毒样本的氨基酸序列。注意,流感病毒的获得和测序已经在世界上的至少一些地方成为常规实践,允许使用先前获取的和目前获取的并记录的数据来实施过程500。
在框508处,确定了所有时间段内流感病毒的每个样本的编码序列。如上所述,编码序列可以通过首先产生代表在整个研究期的每个序列位置处观察到的每个氨基酸的标签序列来确定,并且特定样本的编码序列可以基于在该特定样本的每个序列位置中存在哪个观察到的氨基酸来确定。
在框510处,对于每个时间段,从与该时间段有关的编码序列中确定流行率向量。可以按上述方式计算流行率向量。
在框512处,可以基于研究期内所有时间段的流行率向量,鉴定一个或多个有效突变,并且对于每个有效突变,可以鉴定其有效突变期。如上所述,有效突变的鉴定可以基于该突变是否在第一个时间段之后首次出现以及该突变是否达到优势阈值θ。有效突变期可以被鉴定为该突变从第一次出现到达到优势阈值的时间加上延长的有效突变期h。
在框514处,基于在框512处鉴定的一个或多个有效突变和在框504处获得的群体水平流行性变量来优化g-度量。例如,如上所述,可以定义相似性函数S(f,g),使得较小的S表示f(代表观察到的群体水平流行性变量的向量)和g之间的更接近的匹配。可以使用θ和h的值的不同组合来计算向量g-度量,并且对于每个g(θ,h),可以确定S的值。通过对θ和h的值的不同组合进行迭代,可以确定使S最小化的值。
在框516处,对未来流感病毒活性(即,在研究期的最后一个时间段之后的至少一个“未来”时间段t+1期间的活性)进行预测。可以基于在流行率向量中观察到的g-度量和/或模式来进行预测。可以使用上述预测方法。例如,可以使用等式(10)和(11)预测未来的流行性水平。未来的有效突变可以使用等式(10)和等式(5)处有效突变的定义来预测。未来的代表性病毒序列可以使用等式(10)和(12)-(14b)进行预测。疫苗匹配评分可以基于当前的代表性病毒序列(如上所述)和疫苗中包括的病毒毒株之间的距离来计算。
可以将在框516处做出的预测报告给医疗专业人员以用于各种用途。实例包括:为流感病毒的预计增加做准备(包括发布公共健康公告、生产用于治疗流感患者的另外的药物等);选择要包括在流感疫苗中的流感毒株(野生型或基因工程改造的序列);和/或评估当前可用的流感疫苗的可能有效性。
虽然已经参考具体实施方案描述了本发明,但本领域技术人员将可以进行改变和修改。以上所述的所有过程都是说明性的并且可以进行修改。可以将描述为单独的框的处理操作进行组合,可以将操作的顺序修改到逻辑允许的程度,可以改变或省略上述的处理操作,并且可以添加没有具体描述的另外的处理操作。可以根据需要修改特定的定义和数据格式。
根据数据的可用性,研究期可以和期望的一样长或者和期望的一样短。在一些实施方案中,可以将病毒样本和群体水平数据定位到特定区域(例如,国家、州或地区、城市),从而允许对病毒活性的地理变化进行建模。
此外,尽管上述实施方案具体涉及流感病毒,但本领域技术人员将理解,相同的分析方法可以应用于与其它感染性疾病相关的其它病毒,并且本发明不限于任何特定的病毒。
本发明描述的数据分析和计算操作可以在常规设计的计算机系统,如台式计算机、膝上型计算机、平板计算机、移动设备(例如智能电话)等中实现。计算集群和/或基于云的计算系统可以用于增加计算能力。这样的系统包括执行程序代码的一个或多个处理器(例如,可用作中央处理单元(CPU)的通用微处理器和/或诸如图形处理器(GPU)的专用处理器,其可以提供增强的并行处理能力);存储程序代码和数据的存储器和其它存储设备;用户输入设备(例如,键盘、诸如鼠标或触摸板的定点设备、麦克风);用户输出设备(例如,显示设备、扬声器、打印机);组合的输入/输出设备(例如,触摸屏显示器);信号输入/输出端口;网络通信接口(例如,有线网络接口,如以太网接口和/或无线网络通信接口,如Wi-Fi);等等。并入本发明的各种特征的计算机程序可以被编码并存储在各种计算机可读存储介质上;合适的介质包括磁盘或磁带、诸如光盘(CD)或DVD(数字通用光盘)的光学存储介质、闪存和其它非暂时性介质上。(应理解,数据的“存储”与使用诸如载波的暂时性介质的数据传播不同。)可以将用程序代码编码的计算机可读介质与兼容的计算机系统或其它电子设备打包在一起,或者可以将程序代码与电子设备分开提供(例如,经由因特网下载或作为单独打包的计算机可读存储介质)。输入数据和/或输出数据可以以安全形式提供,例如使用区块链或其它加密技术。
因此,尽管本发明已经针对特定实施方案进行了描述,但应理解本发明旨在覆盖所附权利要求范围内的所有修改和等同物。
Claims (19)
1.一种用于模拟病毒活性的方法,所述方法包括:
对于研究期间内的多个时间段中的每一时间段,确定病毒的基因活性的定量度量(“g-度量”),其中所述g-度量模拟有效突变的流行率和同时发生的有效突变的数目的组合;以及
使用一个或多个所述g-度量以及一个或多个单独突变的流行率来预测所述病毒在所述研究期之后的未来时间段内的活性。
2.如权利要求1所述的方法,其中所述病毒是流感病毒。
3.如权利要求1所述的方法,其中所述突变包括所述病毒的氨基酸序列中的突变。
4.如权利要求1所述的方法,其中所述g-度量是基于来自特定地区的数据,并且所述病毒的活性的预测是针对所述特定地区的。
5.如权利要求1所述的方法,其中所述g-度量是基于全球数据,并且所述病毒的活性的预测是全球预测。
6.如权利要求1所述的方法,其中确定所述g-度量包括:
对于研究期内的每个时间段,获得所述病毒的若干样本的氨基酸序列数据;
基于所述氨基酸序列数据,确定所述病毒的每个样本的编码序列;
对于每个时间段,基于所述病毒的每个样本的编码序列,确定流行率向量,所述流行率向量是指每个序列位置上的每种氨基酸的流行率;
根据所有时间段的流行率向量鉴定一个或多个有效突变;
对于每个有效突变,鉴定有效突变期;以及
基于在该时间段中鉴定的有效突变计算每个时间段的g-度量。
7.如权利要求6所述的方法,其中鉴定有效突变包括选择优势阈值,使得有效突变在至少第一时间段的流行率为零,并且在所述第一时间段后的至少一个时间段内的流行率至少等于所述优势阈值。
8.如权利要求7所述的方法,其中鉴定有效突变期包括鉴定延长的有效突变期,其中有效突变期包括:
从有效突变的第一个非零的流行率至有效突变的流行率至少等于优势阈值的最早时间段的所有时间段;以及
延长的有效突变期。
9.如权利要求8所述的方法,其中基于优化g-度量和指示在所述研究期内的时间段期间由所述病毒引起的感染的人群水平的流行变量之间的拟合来确定所述优势阈值和所述延长的有效突变期。
10.如权利要求6所述的方法,其中计算每个时间段的g-度量包括计算在该时间段内鉴定的每个有效突变的相应流行率的总和。
11.如权利要求6所述的方法,其中使用一个或多个所述g-度量以及一个或多个单独突变的流行率来预测所述病毒在所述研究期之后的未来时间段中的活性包括:
基于一个或多个单独突变的流行率和一个时间段内的突变流行率与随后时间段内的流行率相关联的条件流行率分布来预测所述一个或多个单独突变的未来流行率;
基于预测的所述一个或多个单独突变的未来流行率来预测所述未来时间段的g-度量的值;以及
至少部分基于所述g-度量的预测值来预测由所述病毒引起的感染的人群水平流行性变量的未来值。
12.如权利要求6所述的方法,其中使用一个或多个所述g-度量以及一个或多个单独突变的流行率来预测所述病毒在所述研究期之后的未来时间段中的活性包括:
基于一个或多个单独突变的流行率和一个时间段中的突变流行率与随后时间段中的流行率相关联的条件流行率分布来预测所述一个或多个单独突变的未来流行率;以及
基于预测的所述一个或多个单独突变的未来流行率来预测所述一个或多个突变中的至少一个突变将在未来时间段内成为优势突变。
13.如权利要求12所述的方法,其还包括:
选择要包括在疫苗中的氨基酸,其中所述选择包括预测在所述未来的时间段中变得占优势的一个或多个突变中的至少一个突变。
14.如权利要求6所述的方法,其中使用一个或多个所述g-度量以及一个或多个单独突变的流行率来预测所述病毒在所述研究期之后的未来时间段内的活性包括:
基于一个或多个单独突变的流行率和一个时间段内的突变流行率与随后时间段内的流行率相关联的条件流行率分布来预测所述一个或多个单独突变的未来流行率;以及
在所述随后时间段,基于预测的所述一个或多个单独突变的未来流行率来定义代表性病毒序列。
15.如权利要求14所述的方法,其中使用一个或多个所述g-度量和一个或多个单独突变的流行率来预测所述病毒在所述研究期之后的未来时间段中的活性还包括:
基于所述一个或多个单独突变的流行率来预测未来代表性毒株的病毒基因区段。
16.如权利要求14所述的方法,其还包括:
筛选一种现有的病毒毒株作为要包括在疫苗中的病毒毒株,所述现有病毒毒株比任何其它现有病毒毒株更接近在随后时间段内的代表性病毒序列。
17.如权利要求6所述的方法,其还包括:
基于当前时间段的流行率向量,定义当前时间段的代表性病毒序列;
确定所述代表性病毒序列和包括在疫苗中的一种或多种病毒毒株之间的距离测度;以及
至少部分地基于所述距离测度确定所述疫苗的可能功效。
18.系统,其包括:
存储数据的存储器;和
处理器,其偶联到所述存储器且经配置以实施权利要求1-17中任一项所述的方法。
19.计算机可读的存储介质,其上存储有程序代码指令,所述程序代码指令在由计算机系统的处理器执行时,使所述处理器实施权利要求1-17中任一项所述的方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862687645P | 2018-06-20 | 2018-06-20 | |
| US62/687,645 | 2018-06-20 | ||
| PCT/CN2019/091652 WO2019242597A1 (en) | 2018-06-20 | 2019-06-18 | Measurement and prediction of virus genetic mutation patterns |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN112313748A true CN112313748A (zh) | 2021-02-02 |
| CN112313748B CN112313748B (zh) | 2025-04-04 |
Family
ID=68982769
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980041733.0A Active CN112313748B (zh) | 2018-06-20 | 2019-06-18 | 病毒基因突变模式的测量和预测 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20210233606A1 (zh) |
| EP (1) | EP3810796A4 (zh) |
| CN (1) | CN112313748B (zh) |
| WO (1) | WO2019242597A1 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113284555A (zh) * | 2021-06-11 | 2021-08-20 | 中山大学 | 一种基因突变网络的构建方法、装置、设备及存储介质 |
| CN115798578A (zh) * | 2022-12-06 | 2023-03-14 | 中国人民解放军军事科学院军事医学研究院 | 一种分析与检测病毒新流行变异株的装置及方法 |
| CN117352053A (zh) * | 2022-06-27 | 2024-01-05 | 复旦大学 | 一种流感病毒未来突变的预测方法 |
| CN117352053B (zh) * | 2022-06-27 | 2026-02-13 | 复旦大学 | 一种流感病毒未来突变的预测方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111243662B (zh) * | 2020-01-15 | 2023-04-21 | 云南大学 | 基于改进XGBoost的泛癌症基因通路预测方法、系统和存储介质 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006033691A2 (en) * | 2004-07-02 | 2006-03-30 | Niman Henry L | Copy choice recombination and uses thereof |
| CN101847179A (zh) * | 2010-04-13 | 2010-09-29 | 中国疾病预防控制中心病毒病预防控制所 | 通过模型预测流感抗原的方法及应用 |
| WO2011028897A1 (en) * | 2009-09-03 | 2011-03-10 | Ordway Research Institute, Inc. | Methods for identifying a virulent strain of virus |
| US20110280907A1 (en) * | 2008-11-25 | 2011-11-17 | Max-Planck-Gesellschaft Zur Forderung Der Wissenschaften E.V. | Method and system for building a phylogeny from genetic sequences and using the same for recommendation of vaccine strain candidates for the influenza virus |
| CN106939355A (zh) * | 2017-03-01 | 2017-07-11 | 苏州系统医学研究所 | 一种流感病毒弱毒活疫苗毒株的筛选和鉴定方法 |
| CN107949636A (zh) * | 2015-06-04 | 2018-04-20 | 香港大学 | 活减毒病毒以及生产和使用方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2491499A4 (en) * | 2009-10-19 | 2016-05-18 | Theranos Inc | INTEGRATED SYSTEM FOR COLLECTION AND ANALYSIS OF HEALTH DATA |
| CA2898633C (en) * | 2013-02-07 | 2021-07-13 | Massachusetts Institute Of Technology | Human adaptation of h5 influenza |
-
2019
- 2019-06-18 WO PCT/CN2019/091652 patent/WO2019242597A1/en not_active Ceased
- 2019-06-18 EP EP19822710.0A patent/EP3810796A4/en active Pending
- 2019-06-18 CN CN201980041733.0A patent/CN112313748B/zh active Active
- 2019-06-18 US US17/252,698 patent/US20210233606A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006033691A2 (en) * | 2004-07-02 | 2006-03-30 | Niman Henry L | Copy choice recombination and uses thereof |
| US20110280907A1 (en) * | 2008-11-25 | 2011-11-17 | Max-Planck-Gesellschaft Zur Forderung Der Wissenschaften E.V. | Method and system for building a phylogeny from genetic sequences and using the same for recommendation of vaccine strain candidates for the influenza virus |
| WO2011028897A1 (en) * | 2009-09-03 | 2011-03-10 | Ordway Research Institute, Inc. | Methods for identifying a virulent strain of virus |
| CN101847179A (zh) * | 2010-04-13 | 2010-09-29 | 中国疾病预防控制中心病毒病预防控制所 | 通过模型预测流感抗原的方法及应用 |
| CN107949636A (zh) * | 2015-06-04 | 2018-04-20 | 香港大学 | 活减毒病毒以及生产和使用方法 |
| CN106939355A (zh) * | 2017-03-01 | 2017-07-11 | 苏州系统医学研究所 | 一种流感病毒弱毒活疫苗毒株的筛选和鉴定方法 |
Non-Patent Citations (2)
| Title |
|---|
| JOHN P.BARTON 等: "Relative rate and location of intra-host HIV evolution to evade cellular immunity are predictable", 《NATURE COMMUNICATIONS》, vol. 7, no. 1, 1 September 2016 (2016-09-01), pages 1 - 10, XP055902673, DOI: 10.1038/ncomms11660 * |
| 孙海波;刘双;宋亦春;孙柏红;宋歌;王璐璐;孙英伟;: "辽宁省2016―2017年H3N2亚型流感病毒HA1基因特征分析", 中国微生态学杂志, no. 01, 15 January 2018 (2018-01-15) * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113284555A (zh) * | 2021-06-11 | 2021-08-20 | 中山大学 | 一种基因突变网络的构建方法、装置、设备及存储介质 |
| CN113284555B (zh) * | 2021-06-11 | 2023-08-22 | 中山大学 | 一种基因突变网络的构建方法、装置、设备及存储介质 |
| CN117352053A (zh) * | 2022-06-27 | 2024-01-05 | 复旦大学 | 一种流感病毒未来突变的预测方法 |
| CN117352053B (zh) * | 2022-06-27 | 2026-02-13 | 复旦大学 | 一种流感病毒未来突变的预测方法 |
| CN115798578A (zh) * | 2022-12-06 | 2023-03-14 | 中国人民解放军军事科学院军事医学研究院 | 一种分析与检测病毒新流行变异株的装置及方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3810796A1 (en) | 2021-04-28 |
| CN112313748B (zh) | 2025-04-04 |
| EP3810796A4 (en) | 2024-01-31 |
| US20210233606A1 (en) | 2021-07-29 |
| WO2019242597A1 (en) | 2019-12-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Dilthey et al. | Strain-level metagenomic assignment and compositional estimation for long reads with MetaMaps | |
| Lindgreen et al. | An evaluation of the accuracy and speed of metagenome analysis tools | |
| Lewitus et al. | Characterizing and comparing phylogenies from their Laplacian spectrum | |
| Du et al. | Evolution-informed forecasting of seasonal influenza A (H3N2) | |
| Mostafavi et al. | Normalizing RNA-sequencing data by modeling hidden covariates with prior knowledge | |
| Zhang et al. | Time series modelling of syphilis incidence in China from 2005 to 2012 | |
| Saw et al. | Alignment-free method for DNA sequence clustering using Fuzzy integral similarity | |
| Smith et al. | Rapid incidence estimation from SARS-CoV-2 genomes reveals decreased case detection in Europe during summer 2020 | |
| Lau et al. | Inferring influenza dynamics and control in households | |
| Acera Mateos et al. | PACIFIC: a lightweight deep-learning classifier of SARS-CoV-2 and co-infecting RNA viruses | |
| McCloskey et al. | A model-based clustering method to detect infectious disease transmission outbreaks from sequence variation | |
| Pappas et al. | Virus bioinformatics | |
| Chen et al. | Approaches and challenges to inferring the geographical source of infectious disease outbreaks using genomic data | |
| CN112313748A (zh) | 病毒基因突变模式的测量和预测 | |
| Laenen et al. | Identifying the patterns and drivers of Puumala hantavirus enzootic dynamics using reservoir sampling | |
| Loka et al. | Reliable variant calling during runtime of Illumina sequencing | |
| Berman et al. | MutaGAN: A sequence-to-sequence GAN framework to predict mutations of evolving protein populations | |
| Pitot et al. | Conservative taxonomy and quality assessment of giant virus genomes with GVClass | |
| Zhang et al. | Monitoring real-time transmission heterogeneity from incidence data | |
| Shi et al. | Influenza vaccine strain selection with an AI-based evolutionary and antigenicity model | |
| Gonzalez-Isunza et al. | Using machine learning to detect coronaviruses potentially infectious to humans | |
| Sun et al. | Phylogenetic-informed graph deep learning to classify dynamic transmission clusters in infectious disease epidemics | |
| Li et al. | Machine learning early detection of SARS‐CoV‐2 high‐risk variants | |
| Tang et al. | Comparative subgenomic mRNA profiles of SARS-CoV-2 Alpha, Delta and Omicron BA. 1, BA. 2 and BA. 5 sub-lineages using Danish COVID-19 genomic surveillance data | |
| Yu et al. | Utilizing profile hidden Markov model databases for discovering viruses from metagenomic data: A comprehensive review |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |