CN111018778A - 一种喹诺酮类衍生物及其制备方法和应用 - Google Patents
一种喹诺酮类衍生物及其制备方法和应用 Download PDFInfo
- Publication number
- CN111018778A CN111018778A CN201911400744.1A CN201911400744A CN111018778A CN 111018778 A CN111018778 A CN 111018778A CN 201911400744 A CN201911400744 A CN 201911400744A CN 111018778 A CN111018778 A CN 111018778A
- Authority
- CN
- China
- Prior art keywords
- quinolone
- formula
- preparation
- reaction
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 42
- 150000007660 quinolones Chemical class 0.000 title claims abstract description 32
- 238000006243 chemical reaction Methods 0.000 claims abstract description 29
- 239000002253 acid Substances 0.000 claims abstract description 21
- 239000011230 binding agent Substances 0.000 claims abstract description 19
- 239000003054 catalyst Substances 0.000 claims abstract description 12
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 claims abstract description 12
- 239000013067 intermediate product Substances 0.000 claims abstract description 6
- 150000001875 compounds Chemical class 0.000 claims description 40
- -1 trifluoromethoxy, difluoromethyl Chemical group 0.000 claims description 25
- 229910052739 hydrogen Inorganic materials 0.000 claims description 18
- 239000001257 hydrogen Substances 0.000 claims description 18
- 150000002431 hydrogen Chemical class 0.000 claims description 17
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 13
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 12
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 claims description 10
- 229920001661 Chitosan Polymers 0.000 claims description 10
- 229910052736 halogen Inorganic materials 0.000 claims description 10
- 150000002367 halogens Chemical class 0.000 claims description 10
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 10
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 9
- 239000002841 Lewis acid Substances 0.000 claims description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 8
- 150000007517 lewis acids Chemical class 0.000 claims description 8
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims description 7
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 7
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 claims description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 6
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 claims description 6
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 6
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 6
- 150000007530 organic bases Chemical class 0.000 claims description 6
- 239000003960 organic solvent Substances 0.000 claims description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 6
- 235000011181 potassium carbonates Nutrition 0.000 claims description 6
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 5
- 150000007529 inorganic bases Chemical class 0.000 claims description 5
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 5
- 239000002994 raw material Substances 0.000 claims description 5
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 4
- 125000003545 alkoxy group Chemical group 0.000 claims description 4
- 239000002246 antineoplastic agent Substances 0.000 claims description 4
- 229940041181 antineoplastic drug Drugs 0.000 claims description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 3
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 claims description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 3
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 3
- 229910000363 nickel(II) sulfate Inorganic materials 0.000 claims description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 3
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 claims description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 3
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 2
- OCJBOOLMMGQPQU-UHFFFAOYSA-N 1,4-dichlorobenzene Chemical compound ClC1=CC=C(Cl)C=C1 OCJBOOLMMGQPQU-UHFFFAOYSA-N 0.000 claims description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 2
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 claims description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 2
- MQRWBMAEBQOWAF-UHFFFAOYSA-N acetic acid;nickel Chemical group [Ni].CC(O)=O.CC(O)=O MQRWBMAEBQOWAF-UHFFFAOYSA-N 0.000 claims description 2
- 229940124350 antibacterial drug Drugs 0.000 claims description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 2
- 238000006555 catalytic reaction Methods 0.000 claims description 2
- 239000000460 chlorine Substances 0.000 claims description 2
- 229910052801 chlorine Inorganic materials 0.000 claims description 2
- 229940117389 dichlorobenzene Drugs 0.000 claims description 2
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 claims description 2
- 229910052731 fluorine Inorganic materials 0.000 claims description 2
- 239000011737 fluorine Substances 0.000 claims description 2
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 claims description 2
- 229910052751 metal Inorganic materials 0.000 claims description 2
- 239000002184 metal Substances 0.000 claims description 2
- 229940078494 nickel acetate Drugs 0.000 claims description 2
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 claims description 2
- LGQLOGILCSXPEA-UHFFFAOYSA-L nickel sulfate Chemical compound [Ni+2].[O-]S([O-])(=O)=O LGQLOGILCSXPEA-UHFFFAOYSA-L 0.000 claims description 2
- 229910000028 potassium bicarbonate Inorganic materials 0.000 claims description 2
- 235000015497 potassium bicarbonate Nutrition 0.000 claims description 2
- 239000011736 potassium bicarbonate Substances 0.000 claims description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 claims description 2
- 238000007363 ring formation reaction Methods 0.000 claims description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 2
- 239000008096 xylene Substances 0.000 claims description 2
- PQIOSYKVBBWRRI-UHFFFAOYSA-N methylphosphonyl difluoride Chemical group CP(F)(F)=O PQIOSYKVBBWRRI-UHFFFAOYSA-N 0.000 claims 2
- 150000002825 nitriles Chemical class 0.000 claims 2
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical class [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 claims 2
- VSTXCZGEEVFJES-UHFFFAOYSA-N 1-cycloundecyl-1,5-diazacycloundec-5-ene Chemical compound C1CCCCCC(CCCC1)N1CCCCCC=NCCC1 VSTXCZGEEVFJES-UHFFFAOYSA-N 0.000 claims 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 claims 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims 1
- 229910052759 nickel Inorganic materials 0.000 claims 1
- UQPSGBZICXWIAG-UHFFFAOYSA-L nickel(2+);dibromide;trihydrate Chemical compound O.O.O.Br[Ni]Br UQPSGBZICXWIAG-UHFFFAOYSA-L 0.000 claims 1
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 claims 1
- 238000000034 method Methods 0.000 abstract description 18
- 230000000259 anti-tumor effect Effects 0.000 abstract description 16
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 abstract description 14
- 230000000844 anti-bacterial effect Effects 0.000 abstract description 9
- 238000005516 engineering process Methods 0.000 abstract description 3
- 230000000975 bioactive effect Effects 0.000 abstract description 2
- 238000004519 manufacturing process Methods 0.000 abstract description 2
- 229910052757 nitrogen Inorganic materials 0.000 abstract description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 abstract description 2
- 238000005580 one pot reaction Methods 0.000 abstract description 2
- 238000000746 purification Methods 0.000 abstract description 2
- 230000035484 reaction time Effects 0.000 abstract description 2
- 238000000926 separation method Methods 0.000 abstract description 2
- 125000001424 substituent group Chemical group 0.000 abstract description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 24
- 239000007787 solid Substances 0.000 description 21
- 210000004027 cell Anatomy 0.000 description 16
- 230000000694 effects Effects 0.000 description 14
- 239000000047 product Substances 0.000 description 12
- 238000012360 testing method Methods 0.000 description 12
- FINHMKGKINIASC-UHFFFAOYSA-N Tetramethylpyrazine Chemical compound CC1=NC(C)=C(C)N=C1C FINHMKGKINIASC-UHFFFAOYSA-N 0.000 description 11
- 238000001953 recrystallisation Methods 0.000 description 10
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000007864 aqueous solution Substances 0.000 description 9
- 210000004881 tumor cell Anatomy 0.000 description 9
- 230000001580 bacterial effect Effects 0.000 description 8
- 239000003814 drug Substances 0.000 description 8
- 230000002401 inhibitory effect Effects 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- MQLACMBJVPINKE-UHFFFAOYSA-N 10-[(3-hydroxy-4-methoxyphenyl)methylidene]anthracen-9-one Chemical compound C1=C(O)C(OC)=CC=C1C=C1C2=CC=CC=C2C(=O)C2=CC=CC=C21 MQLACMBJVPINKE-UHFFFAOYSA-N 0.000 description 6
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- GSDSWSVVBLHKDQ-JTQLQIEISA-N Levofloxacin Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-JTQLQIEISA-N 0.000 description 5
- 238000012258 culturing Methods 0.000 description 5
- IHXAZYUYQYFLSG-UHFFFAOYSA-N ethyl 7-chloro-6-fluoro-1-[4-(4-methoxyphenoxy)phenyl]-4-oxoquinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=C(C=C1)OC)C(=O)OCC)=O)F IHXAZYUYQYFLSG-UHFFFAOYSA-N 0.000 description 5
- VSGGZXKWADCCCW-UHFFFAOYSA-N ethyl 7-chloro-6-fluoro-4-oxo-1-[4-[4-(trifluoromethoxy)phenoxy]phenyl]quinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=C(C=C1)OC(F)(F)F)C(=O)OCC)=O)F VSGGZXKWADCCCW-UHFFFAOYSA-N 0.000 description 5
- 229960003376 levofloxacin Drugs 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 238000011160 research Methods 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- 102000007537 Type II DNA Topoisomerases Human genes 0.000 description 4
- 108010046308 Type II DNA Topoisomerases Proteins 0.000 description 4
- 230000004071 biological effect Effects 0.000 description 4
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 4
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 4
- XUBOMFCQGDBHNK-JTQLQIEISA-N (S)-gatifloxacin Chemical compound FC1=CC(C(C(C(O)=O)=CN2C3CC3)=O)=C2C(OC)=C1N1CCN[C@@H](C)C1 XUBOMFCQGDBHNK-JTQLQIEISA-N 0.000 description 3
- DQXKOHDUMJLXKH-PHEQNACWSA-N (e)-n-[2-[2-[[(e)-oct-2-enoyl]amino]ethyldisulfanyl]ethyl]oct-2-enamide Chemical compound CCCCC\C=C\C(=O)NCCSSCCNC(=O)\C=C\CCCCC DQXKOHDUMJLXKH-PHEQNACWSA-N 0.000 description 3
- 102000003964 Histone deacetylase Human genes 0.000 description 3
- 108090000353 Histone deacetylase Proteins 0.000 description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 3
- 241000191967 Staphylococcus aureus Species 0.000 description 3
- 239000007810 chemical reaction solvent Substances 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- ZENGMFUWXTVEEH-UHFFFAOYSA-N ethyl 1-[4-(4-tert-butylphenoxy)phenyl]-7-chloro-6-fluoro-4-oxoquinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=C(C=C1)C(C)(C)C)C(=O)OCC)=O)F ZENGMFUWXTVEEH-UHFFFAOYSA-N 0.000 description 3
- SDCJHHZDLOGKTA-UHFFFAOYSA-N ethyl 7-chloro-1-[4-(3,4-dichlorophenoxy)phenyl]-6-fluoro-4-oxoquinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC(=C(C=C1)Cl)Cl)C(=O)OCC)=O)F SDCJHHZDLOGKTA-UHFFFAOYSA-N 0.000 description 3
- IARYXJPOEFPPEH-UHFFFAOYSA-N ethyl 7-chloro-1-[4-(3,4-dimethylphenoxy)phenyl]-6-fluoro-4-oxoquinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC(=C(C=C1)C)C)C(=O)OCC)=O)F IARYXJPOEFPPEH-UHFFFAOYSA-N 0.000 description 3
- GGPZGWXHTPOYGO-UHFFFAOYSA-N ethyl 7-chloro-1-[[4-(4-chlorophenoxy)phenyl]methyl]-6-fluoro-4-oxoquinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)CC1=CC=C(C=C1)OC1=CC=C(C=C1)Cl)C(=O)OCC)=O)F GGPZGWXHTPOYGO-UHFFFAOYSA-N 0.000 description 3
- TUVJNFMDSNDJPL-UHFFFAOYSA-N ethyl 7-chloro-6-fluoro-1-[4-(4-fluorophenoxy)phenyl]-4-oxoquinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=C(C=C1)F)C(=O)OCC)=O)F TUVJNFMDSNDJPL-UHFFFAOYSA-N 0.000 description 3
- BGLGVJJLZYCDMA-UHFFFAOYSA-N ethyl 7-chloro-6-fluoro-1-[4-(4-methylphenoxy)phenyl]-4-oxoquinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=C(C=C1)C)C(=O)OCC)=O)F BGLGVJJLZYCDMA-UHFFFAOYSA-N 0.000 description 3
- HJFOXDBZCAACOT-UHFFFAOYSA-N ethyl 7-chloro-6-fluoro-1-[[4-(2-methylphenoxy)phenyl]methyl]-4-oxoquinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)CC1=CC=C(C=C1)OC1=C(C=CC=C1)C)C(=O)OCC)=O)F HJFOXDBZCAACOT-UHFFFAOYSA-N 0.000 description 3
- JBOCXNXFYOQEBO-UHFFFAOYSA-N ethyl 7-chloro-6-fluoro-4-oxo-1-(4-phenoxyphenyl)quinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=CC=C1)C(=O)OCC)=O)F JBOCXNXFYOQEBO-UHFFFAOYSA-N 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 229960003923 gatifloxacin Drugs 0.000 description 3
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 3
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 3
- 201000007270 liver cancer Diseases 0.000 description 3
- 208000014018 liver neoplasm Diseases 0.000 description 3
- 201000005202 lung cancer Diseases 0.000 description 3
- 208000020816 lung neoplasm Diseases 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 3
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 2
- YJILUBVUPBRFOH-UHFFFAOYSA-N 7-chloro-1-[4-(4-chlorophenoxy)phenyl]-6-fluoro-4-oxoquinoline-3-carboxylic acid Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=C(C=C1)Cl)C(=O)O)=O)F YJILUBVUPBRFOH-UHFFFAOYSA-N 0.000 description 2
- WIHHBOAWOMPISM-UHFFFAOYSA-N 7-chloro-6-fluoro-1-[4-(4-methoxyphenoxy)phenyl]-4-oxoquinoline-3-carboxylic acid Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=C(C=C1)OC)C(=O)O)=O)F WIHHBOAWOMPISM-UHFFFAOYSA-N 0.000 description 2
- LQAJVGZMDWICRG-UHFFFAOYSA-N 7-chloro-6-fluoro-4-oxo-1-[4-[4-(trifluoromethoxy)phenoxy]phenyl]quinoline-3-carboxylic acid Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=C(C=C1)OC(F)(F)F)C(=O)O)=O)F LQAJVGZMDWICRG-UHFFFAOYSA-N 0.000 description 2
- 244000063299 Bacillus subtilis Species 0.000 description 2
- 235000014469 Bacillus subtilis Nutrition 0.000 description 2
- LQJMZBWXJVRNHA-UHFFFAOYSA-N C(=O)=O.CNC Chemical compound C(=O)=O.CNC LQJMZBWXJVRNHA-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- 241000192125 Firmicutes Species 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 229910021578 Iron(III) chloride Inorganic materials 0.000 description 2
- RJQXTJLFIWVMTO-TYNCELHUSA-N Methicillin Chemical compound COC1=CC=CC(OC)=C1C(=O)N[C@@H]1C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 RJQXTJLFIWVMTO-TYNCELHUSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- 229910021585 Nickel(II) bromide Inorganic materials 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 125000004414 alkyl thio group Chemical group 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- INTYQACVZWQWNV-UHFFFAOYSA-N ethyl 7-chloro-6-fluoro-4-oxo-1-[4-[4-(trifluoromethyl)phenoxy]phenyl]quinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=C(C=C1)C(F)(F)F)C(=O)OCC)=O)F INTYQACVZWQWNV-UHFFFAOYSA-N 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 125000004785 fluoromethoxy group Chemical group [H]C([H])(F)O* 0.000 description 2
- 229940124307 fluoroquinolone Drugs 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical compound Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 description 2
- 238000003760 magnetic stirring Methods 0.000 description 2
- 229960003085 meticillin Drugs 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- BFSQJYRFLQUZKX-UHFFFAOYSA-L nickel(ii) iodide Chemical compound I[Ni]I BFSQJYRFLQUZKX-UHFFFAOYSA-L 0.000 description 2
- 125000002560 nitrile group Chemical group 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 239000008213 purified water Substances 0.000 description 2
- 239000002516 radical scavenger Substances 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 241001225321 Aspergillus fumigatus Species 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 229940124087 DNA topoisomerase II inhibitor Drugs 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 229910021576 Iron(III) bromide Inorganic materials 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 229910021588 Nickel(II) iodide Inorganic materials 0.000 description 1
- 241000080590 Niso Species 0.000 description 1
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium chloride Substances Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 229940091771 aspergillus fumigatus Drugs 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 230000003385 bacteriostatic effect Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229960001338 colchicine Drugs 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- HMYJBCDVWUWYEI-UHFFFAOYSA-N ethyl 7-chloro-1-[4-(4-chlorophenoxy)phenyl]-6-fluoro-4-oxoquinoline-3-carboxylate Chemical compound ClC1=C(C=C2C(C(=CN(C2=C1)C1=CC=C(C=C1)OC1=CC=C(C=C1)Cl)C(=O)OCC)=O)F HMYJBCDVWUWYEI-UHFFFAOYSA-N 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- DZZPXUQERKWECE-UHFFFAOYSA-N formyl acetate;sodium Chemical compound [Na].CC(=O)OC=O DZZPXUQERKWECE-UHFFFAOYSA-N 0.000 description 1
- 230000000855 fungicidal effect Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- CAOOISJXWZMLBN-PPHPATTJSA-N htn0d03vrz Chemical compound Cl.C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 CAOOISJXWZMLBN-PPHPATTJSA-N 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000009630 liquid culture Methods 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- IPLJNQFXJUCRNH-UHFFFAOYSA-L nickel(2+);dibromide Chemical compound [Ni+2].[Br-].[Br-] IPLJNQFXJUCRNH-UHFFFAOYSA-L 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 238000007790 scraping Methods 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 238000007711 solidification Methods 0.000 description 1
- 230000008023 solidification Effects 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229960000237 vorinostat Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D215/54—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3
- C07D215/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3 with oxygen atoms in position 4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本发明涉及一种喹诺酮类衍生物及其制备方法和应用,对喹喏酮的结构进行改造,将1号位氮原子上的取代基进行衍生,引入具有生物活性的二苯醚类结构单元,同时适当改变主环3位、6位和7位,形成新型喹喏酮类衍生物。本发明创新了喹诺酮类化合物结构,采用生物活性分子拼接方法,通过C‑N键,将二苯醚结构单元引入喹诺酮分子中,制得了一系列新型的喹诺酮类衍生物,扩展了该结构的范围;另外本发明制备的喹诺酮类衍生物兼具抑菌活性和抗肿瘤活性,有望扩展其应用范围;本发明引入高效的缚酸剂和催化剂,采用“一锅法”技术,成功实现了该系列衍生物的制备,避免了中间产物的分离纯化,减少了反应工序,极大地缩短反应时间了,提高了生产效率。
Description
技术领域
本发明涉及一种喹诺酮类衍生物及其制备方法和应用。
背景技术
氟喹诺酮类药物在1962问世,经过数十年的发展,得到了不断的更新和改进,形成了第二代、第三代、第四代氟喹诺酮类药物,具有抗菌谱广、杀菌活性强、毒性低及疗效高等诸多优点,在临床上广泛用于各种细菌感染的治疗。近十余年来,通过结构修饰人们发现这类化合物也具有很多其它生物活性,如在抗肿瘤、抗病毒、抗焦虑等方面的研究已取得一定进展,某些候选化合物己进入临床研究阶段。喹诺酮类物是细菌拓扑异构酶II抑制剂,研究发现,细菌拓扑构酶II和哺乳动物的拓扑异构酶II在活性部位酪氨酸周围的序列有同源性,某些喹诺酮类化合物对哺乳动物拓扑异构酶II有较强的抑制作用。目前,通过结构改造,已有数百个具有抗肿瘤活性的喹诺酮衍生物被报道,其有可能成为一类以拓扑异构酶II为靶标的新型抗肿瘤药物。因此,以喹诺酮类药物为结构基础来设计新型的抗肿瘤药物已经成为肿瘤化学的一个热门领域。
卢颖洁等人[华中科技大学学报(医学版),2018,47,4,410-416]设计、合成左氧氟沙星与川芎嗪缀合物并研究其抑制微管蛋白聚合作用和抗肿瘤活性。以左氧氟沙星为原料,在C3位羧基上引入川芎嗪单元,并采用微管蛋白聚合试剂盒和MTT法测试所合成目标缀合物对微管蛋白聚合的抑制活性和体外抗A549、HepG2、MCF7、MDAMB231、PC3这5种肿瘤细胞的活性。初步的生物活性结果测试表明,所合成的左氧氟沙星川芎嗪缀合物对微管蛋白聚合的抑制作用与阳性药物秋水仙碱相当,并对A549、HepG2、MCF7、MDAMB231、PC3这5种肿瘤细胞均有较强的抑制活性;同时,这些左氧氟沙星川芎嗪缀合物对正常乳腺上皮细胞MCF10A均没有毒性。因此,川芎嗪是一个抗肿瘤药效团,能够增强喹诺酮类药物的抗肿瘤活性。
杨科等人[中国抗生素杂志,2019,44,7,811-819]以喹诺酮母体设计合成加替沙星-组蛋白去乙酰化酶抑制剂(histone deacetylase inhibitor,HDACi)缀合物,并探讨其抗肿瘤活性。以加替沙星为原料,对其进行结构修饰,分别在其C-7哌嗪基和C-3位羧基引入辛二酰苯胺异羟肟酸(SAHA)单元,并采用HDACs试剂盒、微管蛋白试剂盒和MTT法对所合成的缀合物进行酶和抗肿瘤抑制活性测试。初步的生物活性研究表明目标化合物能有效地抑制微管蛋白聚合和HDACs,其中SAHA类似物单元的侧链为6个亚甲基的缀合物10b和13对微管蛋白聚合和HDACs的抑制活性以及抗肿瘤活性强于其他衍生物。因此,将组蛋白去乙酰化酶抑制剂(HDACi)单元引入到加替沙星上,能提高其抗肿瘤活性。
本发明就是在此背景下,对喹喏酮的结构进行改造,将1号位氮原子上的取代基进行衍生,引入具有生物活性的二苯醚类结构单元,形成新型喹喏酮类衍生物,并进行了抗菌活性和抗肿瘤活性初步研究。
发明内容
本发明要解决的技术问题是在喹诺酮主环的1位引入二苯醚结构单元,同时适当改变主环3位、6位和7位,得到一类结构新型的兼具抗菌活性和抗肿瘤活性的喹诺酮衍生物,并公开其制备方法。
本发明采用的技术方案如下:
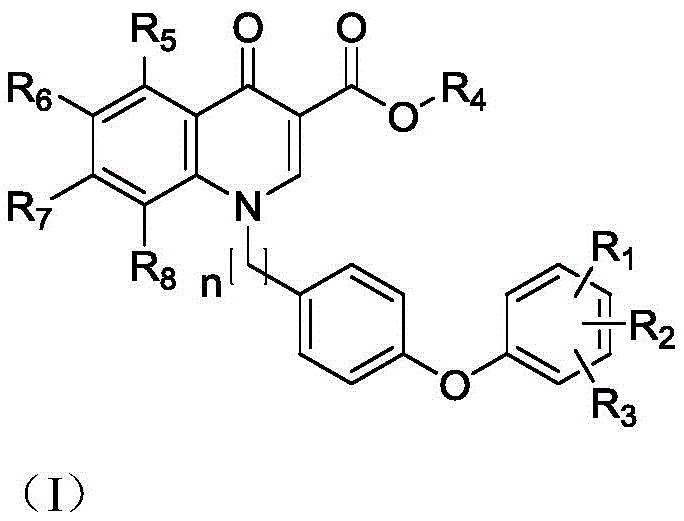
一种喹诺酮类衍生物,其特征在于结构式如(I)所示:
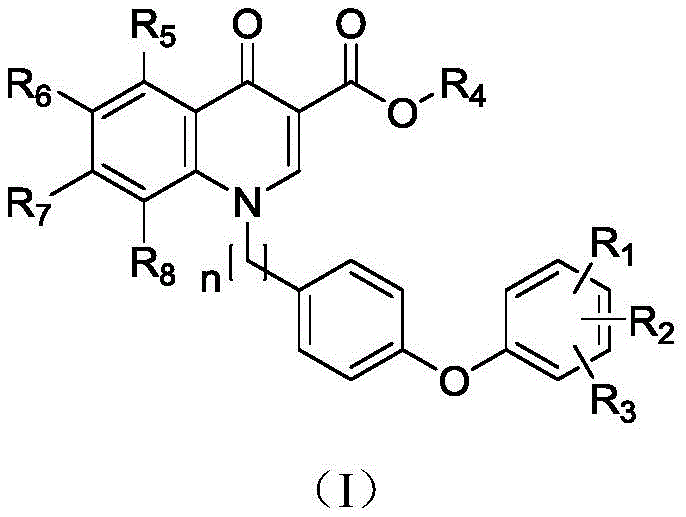
其中,n为数字0或1;
R1、R2、R3分别为氢、C1~C6烷基、C1~C6烷氧基、C5~C6环烷基、C1~C6烷基氧基、C1~C6羟基亚烷基、C1~C6烷基巯基、C1~C6巯基亚烷基、C1~C6烷基氨基、卤素、羟基、巯基、氰基、硝基、三氟甲基、三氟甲氧基、二氟甲基、二氟甲氧基、一氟甲基、一氟甲氧基中的一种;
R4为氢、C1~C6烷基、C5~C6环烷基、苯基、取代苯基、苄基、取代苄基中的一种;
R5、R6、R7、R8分别为氢、C1~C3烷基、卤素、腈基、硝基中的一种。
作为优选,其结构式相应于式(I-a)所示:
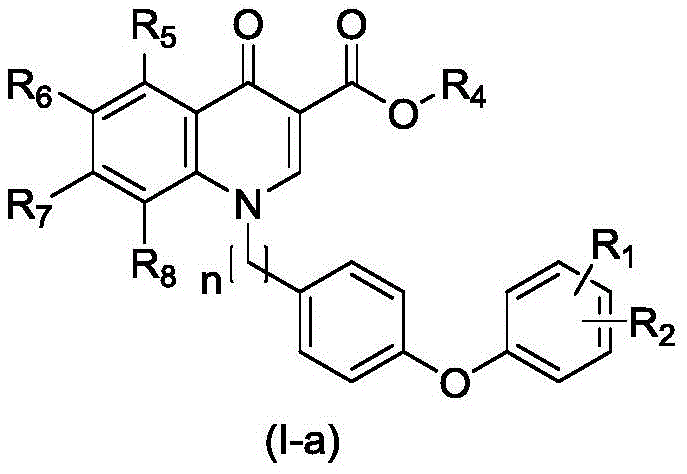
其中,R1、R2分别为氢、C1~C6烷基、C1~C6烷氧基、C5~C6环烷基、C1~C6烷基氧基、C1~C6羟基亚烷基、C1~C6烷基巯基、C1~C6巯基亚烷基、C1~C6烷基氨基、卤素、羟基、巯基、氰基、硝基、三氟甲基、三氟甲氧基、二氟甲基、二氟甲氧基、一氟甲基、一氟甲氧基中的一种;
R4为氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、苯基中的一种;
R5、R6、R7和R8分别为氢、卤素、腈基、硝基中的一种。
作为进一步优选,其结构式相应于式(I-b)所示:

其中,R1、R2分别为氢、C1~C6烷基、C1~C6烷氧基、卤素、氰基、硝基、三氟甲基、三氟甲氧基、二氟甲基、二氟甲氧基中的一种;R4为氢、甲基、乙基中的一种;R6为氟,R7为氯。
本发明还提供了上述喹诺酮类衍生物的制备方法,包括下述步骤:
(a)在有机溶剂中,路易斯酸催化下化合物式(II)与水反应,得到中间产物A;
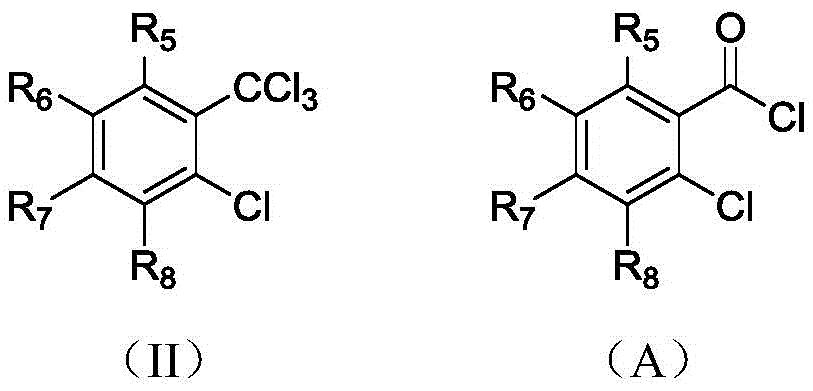
(b)反应体系中先加入第一缚酸剂和式(III)所示的原料混合反应,然后加入式(IV)所示的二苯醚胺类化合物,反应得到中间产物B;
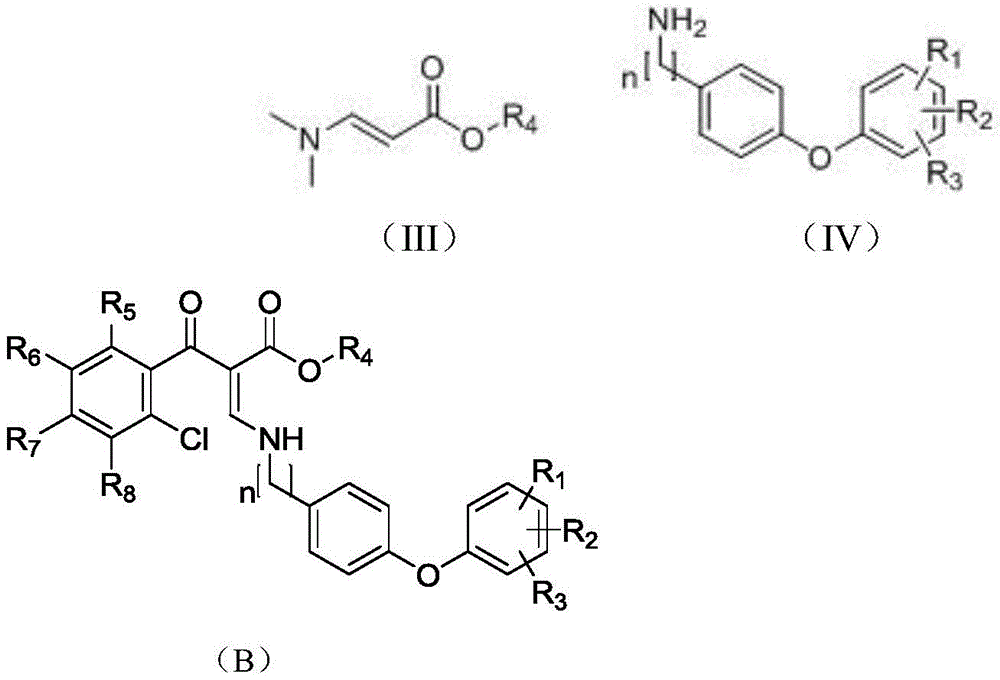
(c)上述反应步骤b)制得的反应体系中再加入第二缚酸剂和负载催化剂,进行环合反应得到目标化合物式(I)。
反应产生的副产物二甲胺经二氧化碳吸收得到二甲胺二氧化碳络合物(VI),然后在液碱存在下,该络合物与甲酰基乙酸酯钠盐(VII)反应,重新得到式(III),实现二甲胺的循环利用。过程如下Scheme 1所示:

作为优选,步骤(a)所述路易斯酸为FeCl3、FeBr3或AlCl3,化合物式(II)与水的摩尔配比为1:1~10;所述有机溶剂为苯、甲苯、二甲苯、氯苯、二氯苯、四氢呋喃、1,4-二氧六环、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮、二甲基亚砜、环丁砜、六甲基磷酰胺、乙腈、二乙二醇二甲醚中的一种或二种以上任意组合。
更为优选的,所述路易斯酸为FeCl3,化合物式(II)与水的摩尔配比为1:3~6。
更为优选的,有机溶剂选自氯苯、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮、二甲基亚砜、环丁砜中的一种;进一步优选,有机溶剂选自N-甲基吡咯烷酮或二甲基亚砜。
作为优选,步骤(b)所述第一缚酸剂选自有机碱类,所述有机碱选自如式(V)所示的化合物、吡啶、4-二甲氨基吡啶、N,N-二异丙基乙胺、1,8-二氮杂二环十一碳-7-烯、四甲基乙二胺、中的一种或二种以上任意组合;
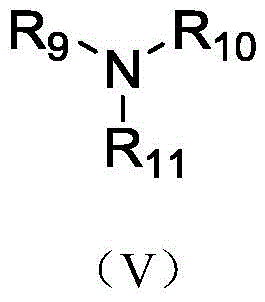
其中,R9、R10和R11分别为氢(H)、甲基(Methly)、乙基(Ethyl)、正丙基(n-Propyl)、异丙基(i-Propyl)、正丁基(n-Butyl)、异丁基(i-Butyl)、叔丁基(t-Butyl)中的一种、两种或三种组合。
更为优选的,所述有机碱为结构式(V)所示的化合物,其中,R9、R10和R11分别为乙基(Ethyl)、正丙基(n-Propyl)、异丙基(i-Propyl)、正丁基(n-Butyl)、异丁基(i-Butyl)中的一种。所述有机碱还可以是吡啶、4-二甲氨基吡啶(DMAP)中的一种。
作为优选,步骤(c)所述第二缚酸剂选自无机碱类,所述无机碱为氢氧化锂、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸铯、碳酸氢钠、碳酸氢钾中的一种或二种以上任意组合。
更为优选的,所述无机碱为碳酸钠或碳酸钾。
作为优选,步骤(c)所述负载催化剂的载体为壳聚糖(chitosan,CS),金属催化剂为醋酸镍[Ni(OAc)2]、硫酸镍(NiSO4)、氯化镍(NiCl2)、溴化镍(NiBr2)或碘化镍(NiI2);制备的负载催化剂为CS@Ni(OAc)2、CS@NiSO4、CS@NiCl2、CS@NiBr2和CS@NiI2,所述负载催化剂与式(II)的质量比为1:1.0~100。
更为优选的,所述负载催化剂为CS@Ni(OAc)2,与式(II)的质量比为1:1.0~20。
作为优选,式(II)、(III)、(IV)、路易斯酸、第一缚酸剂和第二缚酸剂的摩尔配比为1:1~3.0:1~3.0:0.05~1.0:1.0~5.0:1.0~5.0;步骤(a)的反应条件为:0~100℃下,反应0~3小时;步骤(b)的反应条件为:0~100℃下,反应1~10小时;步骤(c)的反应条件为:0~150℃下,反应1~20小时。
更为优选的,式(II)、(III)、(IV)、路易斯酸、第一缚酸剂和第二缚酸剂的摩尔配比为1:1~1.1.5:1~1.5:0.1~0.5:1.0~3.0:1.0~3.0;步骤(a)的反应条件为:50~80℃下,反应0~2小时;步骤(b)的反应条件为:10~30℃下,反应3~6小时;步骤(c)的反应条件为:60~100℃下,反应3~10小时。
本发明另外还提供了上述喹诺酮类衍生物在抗菌药物、抗肿瘤药物中的应用。
本发明制得的新型喹诺酮类衍生物(I),具有抗菌活性应用,能够抑制革兰氏阳性菌的活性,所述的格兰氏阳性菌包括枯草芽孢杆菌、金黄色葡萄球菌、烟曲霉等,特别对耐甲氧西林的金黄色葡萄球菌(MRSA)表现出较好的抑制效果。适合用于抑制枯草芽孢杆菌的活性。
本发明制得的新型喹诺酮类衍生物(I),还具有抗肿瘤细胞活性应用。所述的抗肿瘤细胞包括非小细胞肺癌细胞(HCC827)、肺癌细胞(A549)和肝癌细胞(HepG2),特别适合用于抑制非小细胞肺癌细胞(HCC827)的活性。
与现有技术相比,本发明的有益效果主要体现在:
首先,本发明创新了喹诺酮类化合物结构,采用生物活性分子拼接方法,通过C-N键,将二苯醚结构单元引入喹诺酮分子中,制得了一系列新型的喹诺酮类衍生物,扩展了该结构的范围,并进行了生物活性测试,取得了良好的活性数据,本发明制备的喹诺酮类衍生物兼具抑菌活性和抗肿瘤活性,有望扩展其应用范围;其次,引入高效的缚酸剂和催化剂,采用“一锅法”技术,成功实现了该系列衍生物的制备,避免了中间产物的分离纯化,减少了反应工序,极大地缩短反应时间了,提高了生产效率。
具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此。
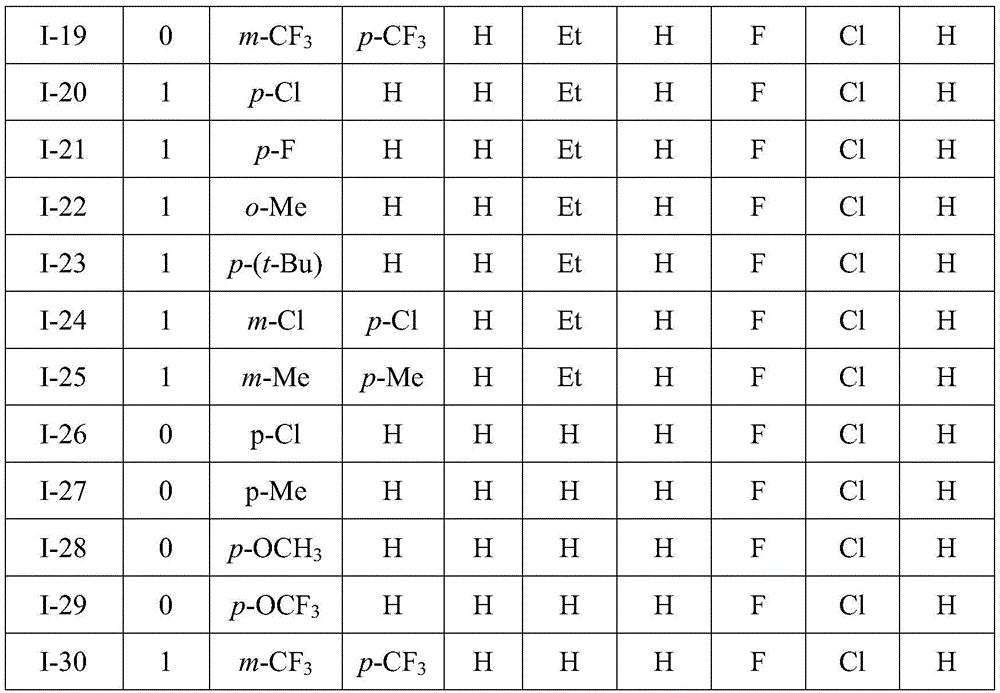
由于本发明涉及的物质较多,但制备方法相似,只是相关基团的替换,故仅选取其中几个代表性化合物对其制备进行描述。所制得喹诺酮衍生物(I)如表1所示:

表1化合物库表

实施例1
7-氯-1-(4-(4-氯苯氧基)苯基)-6-氟-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-3)
a)100mL三口瓶中加入II-1(2.82g,10mmol)、三氯化铁(0.16g,1mmol)、纯净水(0.54g,30mmol)和溶剂N,N-二甲基乙酰胺(30g),磁力搅拌下升温至80℃,保温1h,得到中间体A-1;
b)上述反应液中加入式(III)(1.71g,12mmol)、三乙胺(2.02g,20mmol),30℃保温3h,TLC跟踪反应至A-1转化完全;再加入(IV-3,2.20g,10mmol),升温至60℃,保温3h;
c)上述反应b)制得的反应液中再加入碳酸钾(2.76g,20mmol)、CS@Ni(OAc)2(0.45g),升温至100℃,反应6h,反应完成。降至室温后,加入纯净水150g,过滤,烘干,得到淡黄色固体4.05g,经5%乙醇水溶液重结晶得到目标产物Ⅰ-3,白色粉末3.26g,理论产量4.72g,总收率69.1%。
1H NMR(CDCl3,600MHz):δ8.46(s,1H),8.21(d,J=12Hz,1H),7.42-7.40(m,4H),7.20(d,J=12Hz,2H),7.11-7.07(m,3H),4.38(q,J=6Hz,2H),1.39(t,J=6Hz,3H)。
实施例2
7-氯-6-氟-4-氧-1-(4-苯氧基苯基)-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-4)
本实施例的操作步骤同实施例1,区别在于:在制备过程中,改变反应溶剂,由实施例1的N,N-二甲基乙酰胺变为甲苯。得到粗品淡黄色固体3.85g,经5%乙醇水溶液重结晶得到目标产物I-4,白色粉末2.80g,理论产量4.38g,总收率63.9%。
实施例3
7-氯-6-氟-1-(4-(4-氟苯氧基)苯基)-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-7)
本实施例的操作步骤同实施例1,区别在于:在制备过程中,改变反应溶剂,由实施例1的N,N-二甲基乙酰胺变为1,4-二氧六环。得到粗品棕黄色固体3.35g,经5%乙醇水溶液重结晶得到目标产物I-7,棕黄色固体2.10g,理论产量4.56g,总收率46.1%。
实施例4
7-氯-6-氟-1-(4-(4-甲苯氧基)苯基)-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-8)
本实施例的操作步骤同实施例1,区别在于:在制备过程中,改变反应溶剂,由实施例1的N,N-二甲基乙酰胺变为二甲基亚砜。得到粗品浅黄色固体4.35g,经5%乙醇水溶液重结晶得到目标产物I-8,米黄色固体3.15g,理论产量4.52g,总收率69.7%。
实施例5
7-氯-6-氟-1-(4-(4-叔丁基苯氧基)苯基)-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-10)
本实施例的操作步骤同实施例1,区别在于:在制备过程中,改变反应第一缚酸剂,由实施例1的三乙胺变为三正丁胺。得到粗品淡黄色固体3.75g,经5%乙醇水溶液重结晶得到目标产物I-10,白色固体3.05g,理论产量4.94g,总收率61.7%。
实施例6
7-氯-6-氟-1-(4-(4-甲氧基苯氧基)苯基)-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-11)
本实施例的操作步骤同实施例1,区别在于:在制备过程中,改变第一缚酸剂,由实施例1的三乙胺变为4-二甲氨基吡啶。得到粗品淡黄色固体3.80g,经5%乙醇水溶液重结晶得到目标产物I-11,白色固体2.85g,理论产量4.68g,总收率60.9%。
1H NMR(CDCl3,600MHz):δ8.46(s,1H),8.21(d,J=9.0Hz,1H),7.35-7.33(m,2H),7.14-7.08(m,5H),6.98-6.97(m,2H),4.38(q,J=7.2Hz,2H),3.85(s,3H),1.39(t,J=7.2Hz,3H)。
实施例7
7-氯-6-氟-1-(4-(4-三氟甲氧基苯氧基)苯基)-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-12)
本实施例的操作步骤同实施例1,区别在于:在制备过程中,改变第二缚酸剂,由实施例1的碳酸钾变为碳酸钠。得到粗品淡黄色固体4.10g,经5%乙醇水溶液重结晶得到目标产物I-12,白色固体3.35g,理论产量5.22g,总收率64.2%。
实施例8
7-氯-6-氟-1-(4-(4-三氟甲基苯氧基)苯基)-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-14)
本实施例的操作步骤同实施例1,区别在于:在制备过程中,改变第二缚酸剂,由实施例1的碳酸钾变为氢氧化钠。得到粗品黄色固体3.35g,经5%乙醇水溶液重结晶得到目标产物I-14,浅黄色固体2.32g,理论产量5.06g,总收率45.8%。
实施例9
7-氯-1-(4-(3,4-二氯苯氧基)苯基)-6-氟-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-15)
本实施例的操作步骤同实施例1,区别在于:在制备过程中,改变第一缚酸剂用量,由实施例1的(2.02g,20mmol)变为(1.21g,12mmol)。得到粗品淡黄色固体4.05g,经5%乙醇水溶液重结晶得到目标产物I-15,淡黄色固体3.32g,理论产量5.07g,总收率65.5%。
1H NMR(CDCl3,600MHz):δ8.46(s,1H),8.21(d,J=8.4Hz,1H),7.51(d,J=9.0Hz,1H),7.45(d,J=9.0Hz,2H),7.27-7.23(m,3H),7.07(d,J=6.0Hz,1H),7.04-7.02(m,1H),4.38(q,J=7.2Hz,2H),1.39(t,J=7.2Hz,3H)。
实施例10
7-氯-1-(4-(3,4-二甲基苯氧基)苯基)-6-氟-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-17)
本实施例的操作步骤同实施例1,区别在于:在制备过程中,改变步骤c)的反应温度用量,由实施例1的100℃变为60℃。得到粗品米黄色固体3.80g,经5%乙醇水溶液重结晶得到目标产物I-17,白色固体2.74g,理论产量4.66g,总收率58.8%。
实施例11
7-氯-1-(4-(4-氯苯氧基)苄基)-6-氟-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-20)
本实施例的操作步骤和投料量参照实施例1,得到粗品3.90g,经5%乙醇水溶液重结晶得到目标产物I-20,白色固体3.50g,理论产量4.86g,总收率72.0%。
实施例12
7-氯-1-(4-(2-甲基苯氧基)苄基)-6-氟-4-氧-1,4-二氢喹啉-3-羧酸乙酯的制备(化合物I-22)
本实施例的操作步骤和投料量参照实施例1,得到米黄色粗品3.50g,经5%乙醇水溶液重结晶得到目标产物I-22,白色固体3.05g,理论产量4.66g,总收率65.5%。
实施例13
7-氯-1-(4-(4-氯苯氧基)苯基)-6-氟-4-氧-1,4-二氢喹啉-3-羧酸的制备(化合物I-26)
以上述制备的化合物I-3为原料,具体实施过程如下:
50ml单口玻璃瓶中加入化合物I-3(1.89g,4mmol),5%氢氧化钠溶液(16.0g),磁力搅拌下升温回流3h,反应液变澄清。降温至30℃以下,滴加15%的盐酸调节至中性,过滤,滤饼经真空干燥后得白色粉末1.47g,即为化合物I-20,理论产量1.78g,收率82.6%。
1H NMR(CDCl3,600MHz):δ14.37(brs,1H),8.76(s,1H),8.28(d,J=8.4Hz,1H),7.44-7.42(m,2H),7.38-7.36(m,2H),7.25-7.21(m,3H),7.12-7.10(m,2H)。
实施例14
7-氯-1-(4-(4-甲氧基苯氧基)苯基)-6-氟-4-氧-1,4-二氢喹啉-3-羧酸的制备(化合物I-28)
以上述制备的化合物I-11为原料,方法同实施例13,区别在于:在制备过程中,改变氢氧化钠的浓度,由实施例13的5%溶液16.0g变为10%溶液16.0g。
化合物I-11投料量为(1.87g,4mmol)。得到白色粉末1.34g,即为化合物I-28,理论产量1.76g,收率76.1%。
1H NMR(DMSO-d6,600MHz):δ14.79(brs,0.51H),8.61(s,1H),8.20(d,J=8.4Hz,1H),7.58-7.56(m,2H),7.13-7.01(m,7H)。
实施例15
7-氯-1-(4-(4-三氟甲氧基苯氧基)苯基)-6-氟-4-氧-1,4-二氢喹啉-3-羧酸的制备(化合物I-29)
以上述制备的化合物I-12为原料,方法同实施例13,区别在于:在制备过程中,改变反应回流时间,由实施例13的3h变为6h。
化合物I-12的投料量为2.09g,4mmol。得白色粉末1.48g,即为化合物I-29,理论产量2.08g,收率71.2%。
1H NMR(DMSO-d6,600MHz):δ14.65(brs,0.32H),8.67(s,1H),8.21(d,J=8.4Hz,1H),7.70-7.68(m,2H),7.46-7.44(m,2H),7.33-7.29(m,5H)。
实施例16
杀菌活性测试
为了明确所合成化合物的实用性,选取部分化合物为代表,采用常规方法进行杀菌活性测定。具体操作程序如下:
在超净台里,将保存菌种转接至斜面试管,置37℃温箱培养24h,然后取出低温保藏,备抗菌实验用。用接种环轻刮转接后生长良好的菌落,至液体培养基中,摇晃,混匀,置37℃温箱培养24h。将培养基倒入平板,每板约15mL左右,水平放置,凝固后倒置放入37℃温箱中,2d后观察是否有菌落生长,若没有则可供下一步实验使用,否则需重新倒平板。用移液管移取0.5ml菌悬液注入平板,放置5min左右后用涂布棒涂布均匀,每种菌设置两个重复平板。在超净台里,将平板平均分区,设置平行组。每张滤纸片(直径6mm)为一贴,用镊子夹着滤纸片在样品中轻轻蘸一下后,取出贴在平板的指定位置,轻摁一下使滤纸片牢固。放入(倒置)培养箱中培养,设定温度37℃,培养24h后观察并记录结果。抗菌活性测试具体结果见表2:
表2化合物抑菌活性测试
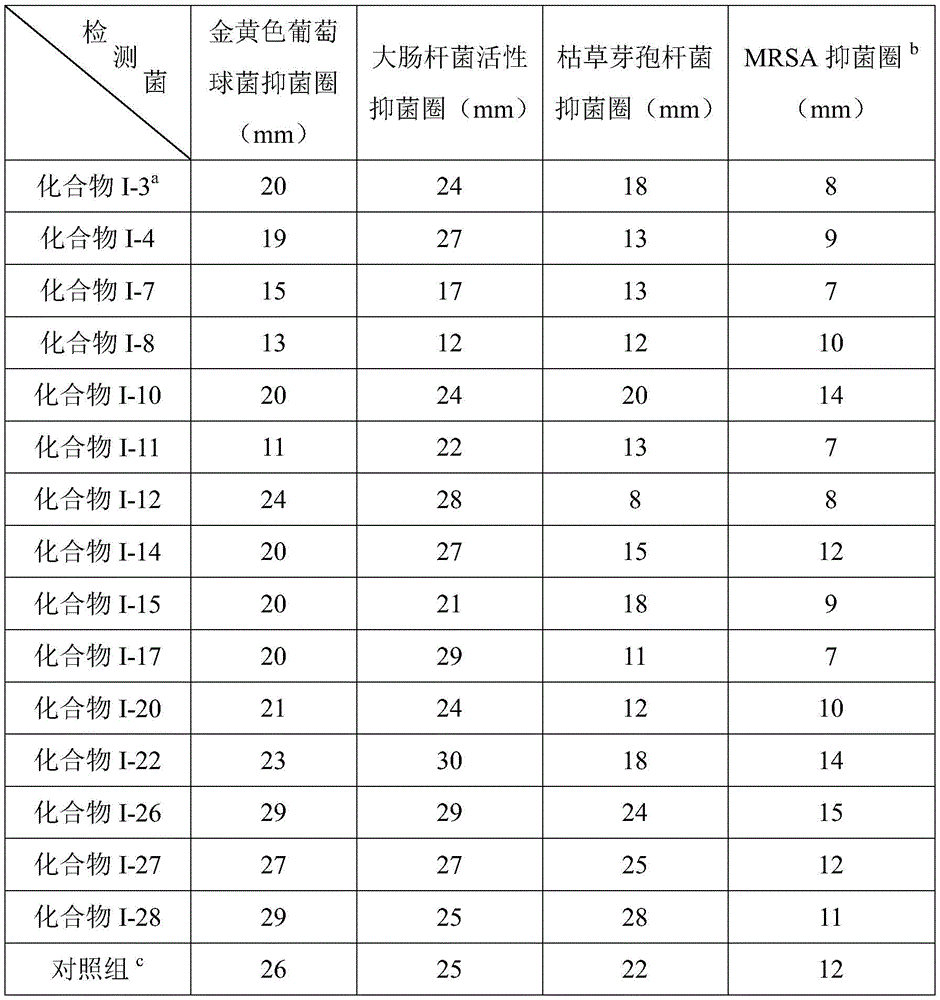
a:滤纸片的直径为6mm,样品浓度为10mg/mL;b:耐甲氧西林的金黄色葡萄球菌;c:对照组为盐酸左氧氟沙星*,对照浓度为5mg/mL。
实施例17
抗肿瘤活性测试
对于该类化合物的抗肿瘤活性测试,以常见的三种肿瘤细胞为研究对象,分别为非小细胞肺癌细胞(HCC827)、肺癌细胞(A549)和肝癌细胞(HepG2),在制备的新型喹诺酮类衍生物的作用下观察细胞的生长情况,并用MTT法测定肿瘤细胞的增殖情况。
具体操作如下:将肿瘤细胞按一定的细胞量接种于96孔培养板内,细胞密度为2×104个/ml;37℃,CO2浓度5%的培养箱中过夜后,加入所筛样品(样品浓度参照表1,加药组加10μL/孔相应浓度药物,对照组加10μL/孔PBS),培养44h后,加入10μL/孔MTT继续培养4h,用DMSO溶解,震摇,570nm酶标仪下进行检测。
以实施例1、7和13制得的新型喹诺酮衍生物以及对照组阳性药紫杉醇,测试对三种肿瘤细胞的半数抑制浓度IC50,测试结果如表3所示。
表3抗肿瘤活性测试
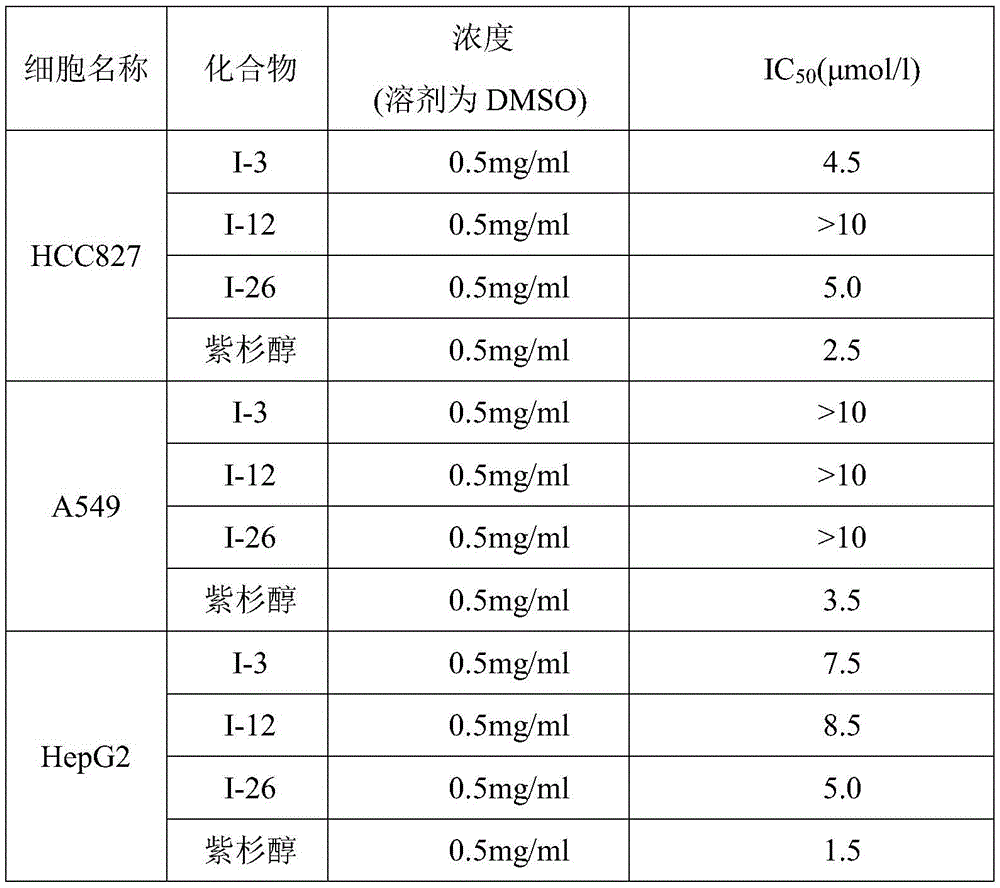
通过实验我们发现合成的新型喹诺酮类衍生物具有抑制HCC827(非小细胞肺癌细胞)和HepG2(人肝癌细胞)肿瘤细胞的功能,对A549(肺癌细胞)抑制效果不大。与抑制该肿瘤细胞的目前通用的阳性药紫杉醇有一定的差距,但也可以反应出我们设计的新型喹诺酮类衍生物存在抑制功能,对其它肿瘤细胞也有可能存在抑制活性,具有一定的应用前景。
Claims (10)
1.一种喹诺酮类衍生物,其特征在于结构式如(I)所示:
其中,n为数字0或1;
R1、R2、R3分别为氢、C1~C6烷基、C1~C6烷氧基、C5~C6环烷基、C1~C6烷基氧基、C1~C6羟基亚烷基、C1~C6烷基巯基、C1~C6巯基亚烷基、C1~C6烷基氨基、卤素、羟基、巯基、氰基、硝基、三氟甲基、三氟甲氧基、二氟甲基、二氟甲氧基、一氟甲基、一氟甲氧基中的一种;
R4为氢、C1~C6烷基、C5~C6环烷基、苯基、取代苯基、苄基、取代苄基中的一种;
R5、R6、R7、R8分别为氢、C1~C3烷基、卤素、腈基、硝基中的一种。
2.根据权利要求1所述的喹诺酮类衍生物,其特征在于其结构式相应于式(I-a)所示:

其中,R1、R2分别为氢、C1~C6烷基、C1~C6烷氧基、C5~C6环烷基、C1~C6烷基氧基、C1~C6羟基亚烷基、C1~C6烷基巯基、C1~C6巯基亚烷基、C1~C6烷基氨基、卤素、羟基、巯基、氰基、硝基、三氟甲基、三氟甲氧基、二氟甲基、二氟甲氧基、一氟甲基、一氟甲氧基中的一种;
R4为氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、苯基中的一种;
R5、R6、R7和R8分别为氢、卤素、腈基、硝基中的一种。
3.根据权利要求1所述的喹诺酮类衍生物,其特征在于其结构式相应于式(I-b)所示:
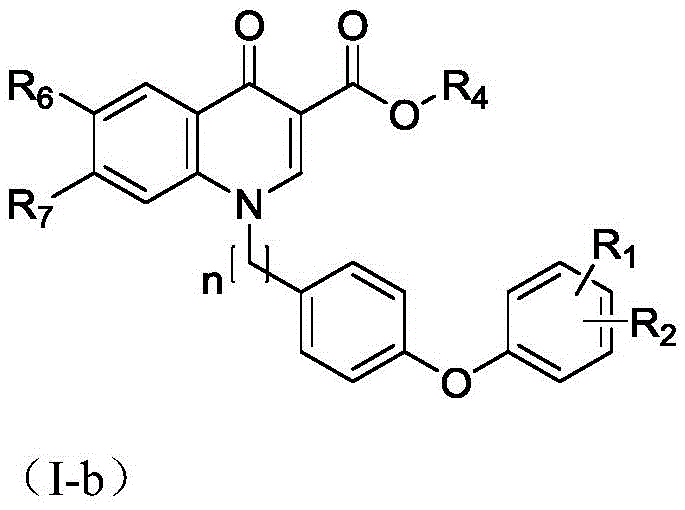
其中,R1、R2分别为氢、C1~C6烷基、C1~C6烷氧基、卤素、氰基、硝基、三氟甲基、三氟甲氧基、二氟甲基、二氟甲氧基中的一种;R4为氢、甲基、乙基中的一种;R6为氟,R7为氯。
4.一种权利要求1所述喹诺酮类衍生物的制备方法,其特征在于包括下述步骤:
(a)在有机溶剂中,路易斯酸催化下化合物式(II)与水反应,得到中间产物A;

(b)上述反应体系中先加入第一缚酸剂和式(III)所示的原料混合反应,然后加入式(IV)所示的二苯醚胺类化合物,反应得到中间产物B;

(c)上述反应步骤(b)制得的反应体系中再加入第二缚酸剂和负载催化剂,进行环合反应得到目标化合物式(I)。
5.根据权利要求4所述喹诺酮类衍生物的制备方法,其特征在于:步骤(a)所述路易斯酸为FeCl3、FeBr3或AlCl3,化合物式(II)与水的摩尔配比为1:1~10;所述有机溶剂为苯、甲苯、二甲苯、氯苯、二氯苯、四氢呋喃、1,4-二氧六环、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮、二甲基亚砜、环丁砜、六甲基磷酰胺、乙腈、二乙二醇二甲醚中的一种或二种以上任意组合。
6.根据权利要求4所述喹诺酮类衍生物的制备方法,其特征在于:步骤(b)所述第一缚酸剂选自有机碱类,所述有机碱选自如式(V)所示的化合物、吡啶、4-二甲氨基吡啶、N,N-二异丙基乙胺、1,8-二氮杂二环十一碳-7-烯、四甲基乙二胺、中的一种或二种以上任意组合;
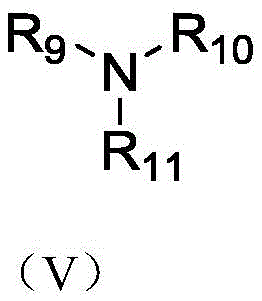
其中,R9、R10和R11分别为氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基中的一种或二种以上任意组合。
7.根据权利要求4所述喹诺酮类衍生物的制备方法,其特征在于:步骤(c)所述第二缚酸剂选自无机碱类,所述无机碱为氢氧化锂、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸铯、碳酸氢钠、碳酸氢钾中的一种或二种以上任意组合。
8.根据权利要求4所述喹诺酮类衍生物的制备方法,其特征在于:所述负载催化剂的载体为壳聚糖,金属催化剂为醋酸镍、硫酸镍、氯化镍、溴化镍、碘化镍中的一种;制备的负载催化剂为CS@Ni(OAc)2、CS@NiSO4、CS@NiCl2、CS@NiBr2或CS@NiI2;所述负载催化剂与式(II)的质量比为1:1.0~100。
9.根据权利要求4所述喹诺酮类衍生物的制备方法,其特征在于:式(II)、(III)、(IV)、路易斯酸、第一缚酸剂和第二缚酸剂的摩尔配比为1:1~3.0:1~3.0:0.05~1.0:1.0~5.0:1.0~5.0;步骤(a)的反应条件为:0~100℃下,反应0~3小时;步骤(b)的反应条件为:0~100℃下,反应1~10小时;步骤(c)的反应条件为:0~150℃下,反应1~20小时。
10.权利要求1所述喹诺酮类衍生物在抗菌药物、抗肿瘤药物中的应用。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201911400744.1A CN111018778B (zh) | 2019-12-30 | 2019-12-30 | 一种喹诺酮类衍生物及其制备方法和应用 |
| US16/828,446 US11161818B2 (en) | 2019-12-30 | 2020-03-24 | Quinolone derivatives, preparation methods and application thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201911400744.1A CN111018778B (zh) | 2019-12-30 | 2019-12-30 | 一种喹诺酮类衍生物及其制备方法和应用 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN111018778A true CN111018778A (zh) | 2020-04-17 |
| CN111018778B CN111018778B (zh) | 2020-11-06 |
Family
ID=70196525
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201911400744.1A Active CN111018778B (zh) | 2019-12-30 | 2019-12-30 | 一种喹诺酮类衍生物及其制备方法和应用 |
Country Status (2)
| Country | Link |
|---|---|
| US (1) | US11161818B2 (zh) |
| CN (1) | CN111018778B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116444491A (zh) * | 2023-03-30 | 2023-07-18 | 浙江工业大学 | 一种喹诺酮类衍生物及其制备方法和应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101638385A (zh) * | 2009-08-25 | 2010-02-03 | 杭州师范大学 | 一种新型喹诺酮类衍生物及其制备和应用 |
| WO2013157018A1 (en) * | 2012-04-18 | 2013-10-24 | Indian Institute Of Technology Madras | A process for the preparation of the core structure in quinolone and napthyridone class of antibiotics |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE475650T1 (de) * | 2005-12-05 | 2010-08-15 | Merck Sharp & Dohme | Positive allosterische chinolon-m1- rezeptormodulatoren |
-
2019
- 2019-12-30 CN CN201911400744.1A patent/CN111018778B/zh active Active
-
2020
- 2020-03-24 US US16/828,446 patent/US11161818B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101638385A (zh) * | 2009-08-25 | 2010-02-03 | 杭州师范大学 | 一种新型喹诺酮类衍生物及其制备和应用 |
| WO2013157018A1 (en) * | 2012-04-18 | 2013-10-24 | Indian Institute Of Technology Madras | A process for the preparation of the core structure in quinolone and napthyridone class of antibiotics |
Non-Patent Citations (1)
| Title |
|---|
| 刘娟 等: "新型喹诺酮类化合物的合成及生物活性研究", 《中国化学会第二十七届学术年会论文集》 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116444491A (zh) * | 2023-03-30 | 2023-07-18 | 浙江工业大学 | 一种喹诺酮类衍生物及其制备方法和应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| US11161818B2 (en) | 2021-11-02 |
| US20210198207A1 (en) | 2021-07-01 |
| CN111018778B (zh) | 2020-11-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103275018B (zh) | 4-[3-氯-4-取代苯胺基]-6-取代甲酰氨基喹唑啉类化合物及制备和应用 | |
| CN104955811B (zh) | 氘代的苯基氨基嘧啶化合物以及包含该化合物的药物组合物 | |
| CN109761960B (zh) | 抗耐药抗肿瘤egfr抑制剂的制备方法 | |
| WO2015007249A1 (zh) | N-烷基色胺酮衍生物及其制备方法和应用 | |
| JPWO2014017515A1 (ja) | 4−[5−(ピリジン−4−イル)−1h−1,2,4−トリアゾール−3−イル]ピリジン−2−カルボニトリルの結晶多形およびその製造方法 | |
| CN114524799B (zh) | 一种hdac抑制剂及其制备方法和用途 | |
| WO2018214866A1 (zh) | 一种氮杂芳基衍生物、其制备方法和在药学上的应用 | |
| CN107573327B (zh) | 吲唑-甲酰胺-吡啶酮衍生物及其制备方法和用途 | |
| CN106674128A (zh) | 一类具抗肿瘤活性的甘草查尔酮a硫脲嘧啶类衍生物及其合成方法 | |
| CN102249997A (zh) | 一组具有抗肿瘤活性的4-取代苯氨基喹啉化合物 | |
| CN111018778A (zh) | 一种喹诺酮类衍生物及其制备方法和应用 | |
| CN103922992B (zh) | 一种抗癌活性吲哚酮衍生物、合成方法及其用途 | |
| CN106674129A (zh) | 一类具抗肿瘤活性的甘草查尔酮a二氢氨基嘧啶类化合物及其合成方法 | |
| CN103896918A (zh) | 化合物及其制备方法和用途 | |
| CN104586842B (zh) | 一种抗癌活性吲哚衍生物、合成方法及其用途 | |
| CN103910643B (zh) | 一种抗癌活性甲酮衍生物、合成方法及其用途 | |
| CN104059062B (zh) | 含苯并噻唑和三唑双杂环的稠环化合物及其应用 | |
| CN103254141A (zh) | 4-[4-(2-二丙氨基乙酰氨基)苯胺基]-6-取代喹唑啉类化合物及制备和应用 | |
| CN104817535A (zh) | 一种喹啉酮衍生物及其合成方法及应用 | |
| CN115490689B (zh) | 不可逆krasg12c抑制剂的制备及其应用 | |
| CN103254143B (zh) | 4-[4-(2-二乙氨基乙酰氨基)苯胺基]-6-取代喹唑啉类化合物及制备和应用 | |
| CN106008559A (zh) | 取代吡啶并嘧啶类化合物的合成工艺 | |
| CN104230786B (zh) | 一种具有抗肿瘤活性的含吲哚结构的化合物及其合成方法 | |
| CN115385896B (zh) | 一种csf-1r抑制剂中间体或其酸式盐的制备方法 | |
| CN109516957A (zh) | Nl-101多晶型及其制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |