CN109369467B - 一种n2-多氟烷基酰基胍类化合物及其制备方法 - Google Patents
一种n2-多氟烷基酰基胍类化合物及其制备方法 Download PDFInfo
- Publication number
- CN109369467B CN109369467B CN201811390684.5A CN201811390684A CN109369467B CN 109369467 B CN109369467 B CN 109369467B CN 201811390684 A CN201811390684 A CN 201811390684A CN 109369467 B CN109369467 B CN 109369467B
- Authority
- CN
- China
- Prior art keywords
- polyfluoroalkyl
- compound
- guanidine
- hydrochloride
- compounds
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 17
- 238000002360 preparation method Methods 0.000 title claims abstract description 17
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims abstract description 48
- -1 acyl guanidine compound Chemical class 0.000 claims abstract description 36
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 30
- 239000012043 crude product Substances 0.000 claims abstract description 28
- 238000006243 chemical reaction Methods 0.000 claims abstract description 25
- 239000003960 organic solvent Substances 0.000 claims abstract description 22
- 238000000034 method Methods 0.000 claims abstract description 21
- 239000000047 product Substances 0.000 claims abstract description 19
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 claims abstract description 18
- ZRALSGWEFCBTJO-UHFFFAOYSA-N anhydrous guanidine Natural products NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 claims abstract description 16
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 claims abstract description 16
- 239000012074 organic phase Substances 0.000 claims abstract description 15
- 238000012544 monitoring process Methods 0.000 claims abstract description 14
- 238000010898 silica gel chromatography Methods 0.000 claims abstract description 14
- 150000004820 halides Chemical class 0.000 claims abstract description 10
- 150000002357 guanidines Chemical class 0.000 claims abstract description 6
- 150000002462 imidazolines Chemical class 0.000 claims abstract description 5
- 238000003756 stirring Methods 0.000 claims abstract description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical group CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 45
- 239000007795 chemical reaction product Substances 0.000 claims description 14
- 229960004198 guanidine Drugs 0.000 claims description 13
- PGRFXXCKHGIFSV-UHFFFAOYSA-N 1,1,1,2,2,3,3,4,4-nonafluoro-4-iodobutane Chemical group FC(F)(F)C(F)(F)C(F)(F)C(F)(F)I PGRFXXCKHGIFSV-UHFFFAOYSA-N 0.000 claims description 11
- 238000001035 drying Methods 0.000 claims description 8
- 238000005286 illumination Methods 0.000 claims description 7
- KYVBNYUBXIEUFW-UHFFFAOYSA-N 1,1,3,3-tetramethylguanidine Chemical compound CN(C)C(=N)N(C)C KYVBNYUBXIEUFW-UHFFFAOYSA-N 0.000 claims description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 4
- BULLJMKUVKYZDJ-UHFFFAOYSA-N 1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluoro-6-iodohexane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)I BULLJMKUVKYZDJ-UHFFFAOYSA-N 0.000 claims description 3
- XTGYEAXBNRVNQU-UHFFFAOYSA-N 1,1,1,2,2,3,3-heptafluoro-3-iodopropane Chemical compound FC(F)(F)C(F)(F)C(F)(F)I XTGYEAXBNRVNQU-UHFFFAOYSA-N 0.000 claims description 3
- XNIBSTIKPHNKSO-UHFFFAOYSA-N 1-chloro-1,1,2,2-tetrafluoro-2-iodoethane Chemical compound FC(F)(Cl)C(F)(F)I XNIBSTIKPHNKSO-UHFFFAOYSA-N 0.000 claims description 3
- VKAZCPSILJQPFT-UHFFFAOYSA-N 2-(1H-pyrazol-5-yl)guanidine hydrochloride Chemical compound N1N=C(C=C1)NC(=N)N.Cl VKAZCPSILJQPFT-UHFFFAOYSA-N 0.000 claims description 3
- XNHWVEFREAKDHV-UHFFFAOYSA-N 2-piperidin-1-ylguanidine hydrochloride Chemical compound C1CCN(CC1)N=C(N)N.Cl XNHWVEFREAKDHV-UHFFFAOYSA-N 0.000 claims description 3
- UHWHYRJXUNAUJR-UHFFFAOYSA-N 2-propan-2-ylguanidine;hydrochloride Chemical compound Cl.CC(C)NC(N)=N UHWHYRJXUNAUJR-UHFFFAOYSA-N 0.000 claims description 3
- DISXFZWKRTZTRI-UHFFFAOYSA-N 4,5-dihydro-1h-imidazol-2-amine Chemical compound NC1=NCCN1 DISXFZWKRTZTRI-UHFFFAOYSA-N 0.000 claims description 3
- YOKMPPFIHYUXJD-UHFFFAOYSA-N Cl.O1CCN(CC1)NC(=N)N Chemical compound Cl.O1CCN(CC1)NC(=N)N YOKMPPFIHYUXJD-UHFFFAOYSA-N 0.000 claims description 3
- FHOQBEFKSSAXDW-UHFFFAOYSA-N carbamimidoyl(phenyl)azanium;chloride Chemical compound [Cl-].NC([NH3+])=NC1=CC=CC=C1 FHOQBEFKSSAXDW-UHFFFAOYSA-N 0.000 claims description 3
- 229960000789 guanidine hydrochloride Drugs 0.000 claims description 3
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 claims description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 2
- 238000004809 thin layer chromatography Methods 0.000 claims 1
- 239000002904 solvent Substances 0.000 abstract description 14
- 150000003672 ureas Chemical class 0.000 abstract description 4
- 239000003386 histamine H2 receptor agonist Substances 0.000 abstract 1
- 238000001308 synthesis method Methods 0.000 abstract 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 48
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 36
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 27
- 239000003480 eluent Substances 0.000 description 24
- 239000000203 mixture Substances 0.000 description 23
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 20
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 13
- 239000000741 silica gel Substances 0.000 description 13
- 229910002027 silica gel Inorganic materials 0.000 description 13
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 12
- 238000004293 19F NMR spectroscopy Methods 0.000 description 12
- 238000005160 1H NMR spectroscopy Methods 0.000 description 12
- 238000002844 melting Methods 0.000 description 12
- 230000008018 melting Effects 0.000 description 12
- 239000003208 petroleum Substances 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- 238000002791 soaking Methods 0.000 description 10
- 239000002253 acid Substances 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 239000013078 crystal Substances 0.000 description 4
- 239000002994 raw material Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- FOBJABJCODOMEO-UHFFFAOYSA-N 2,2,3,3,4,4,4-heptafluorobutanamide Chemical compound NC(=O)C(F)(F)C(F)(F)C(F)(F)F FOBJABJCODOMEO-UHFFFAOYSA-N 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 238000006880 cross-coupling reaction Methods 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 2
- 125000002757 morpholinyl group Chemical group 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- UDWBMXSQHOHKOI-UHFFFAOYSA-N 1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10-henicosafluoro-10-iododecane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)I UDWBMXSQHOHKOI-UHFFFAOYSA-N 0.000 description 1
- KWXGJTSJUKTDQU-UHFFFAOYSA-N 1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8-heptadecafluoro-8-iodooctane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)I KWXGJTSJUKTDQU-UHFFFAOYSA-N 0.000 description 1
- NQQVLNFSVPLPII-UHFFFAOYSA-N 1,1-dimethylguanidine;hydrochloride Chemical compound [Cl-].CN(C)C([NH3+])=N NQQVLNFSVPLPII-UHFFFAOYSA-N 0.000 description 1
- KSESOTWVQYBRLJ-UHFFFAOYSA-N 1,3-dimethylimidazolidin-2-imine Chemical compound CN1CCN(C)C1=N KSESOTWVQYBRLJ-UHFFFAOYSA-N 0.000 description 1
- GVIYXAGLNHCQQX-UHFFFAOYSA-N 2,2,3,3,4,4,4-heptafluoro-n'-(3,4,5,6-tetrahydro-2h-azepin-7-yl)butanehydrazide Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(=O)NNC1=NCCCCC1 GVIYXAGLNHCQQX-UHFFFAOYSA-N 0.000 description 1
- QKLHCAXBFMBYPX-UHFFFAOYSA-N 2-chloro-1,3-dimethylimidazolidine Chemical class CN1CCN(C)C1Cl QKLHCAXBFMBYPX-UHFFFAOYSA-N 0.000 description 1
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 239000002879 Lewis base Substances 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 150000001266 acyl halides Chemical class 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001409 amidines Chemical class 0.000 description 1
- 150000001412 amines Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- ZIPLUEXSCPLCEI-UHFFFAOYSA-N cyanamide group Chemical group C(#N)[NH-] ZIPLUEXSCPLCEI-UHFFFAOYSA-N 0.000 description 1
- 239000012024 dehydrating agents Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- UXPOJVLZTPGWFX-UHFFFAOYSA-N pentafluoroethyl iodide Chemical compound FC(F)(F)C(F)(F)I UXPOJVLZTPGWFX-UHFFFAOYSA-N 0.000 description 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C279/00—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C279/20—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups containing any of the groups, X being a hetero atom, Y being any atom, e.g. acylguanidines
- C07C279/22—Y being a hydrogen or a carbon atom, e.g. benzoylguanidines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C277/00—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C277/08—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups of substituted guanidines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/04—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D233/28—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/44—Nitrogen atoms not forming part of a nitro radical
- C07D233/48—Nitrogen atoms not forming part of a nitro radical with acyclic hydrocarbon or substituted acyclic hydrocarbon radicals, attached to said nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/20—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carbonic acid, or sulfur or nitrogen analogues thereof
- C07D295/215—Radicals derived from nitrogen analogues of carbonic acid
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明涉及一种N2‑多氟烷基酰基胍类化合物及其制备方法。所述的N2‑多氟烷基酰基胍类化合物具有通式(Ⅰ)或(Ⅱ)的结构。该类化合物的合成方法包括以下步骤:将胍类化合物或脲类化合物或咪唑啉类化合物置于反应瓶中,加入有机溶剂和多氟烷基卤,在室温和光照下搅拌6‑15小时,TLC监测反应终点;将所得产物倒入水中,用二氯甲烷萃取,收集有机相,干燥,减压蒸馏除去溶剂;所得粗品通过硅胶柱层析的方法进行分离纯化,即得到N2‑多氟烷基酰基胍产物。本发明合成路线具有方法简单、成本低、范围宽,条件温和,反应路线安全、操作简便等优点。该类化合物可以作为潜在的组胺H2受体激动剂使用。 或
或
Description
技术领域
本发明属于有机合成技术领域,特别涉及一种N2-多氟烷基酰基胍类化合物的制备方法。
背景技术
胍基是一个常见的结构单元,广泛存在许多药物以及天然产物中。并且,胍基路易斯碱也可以作为催化剂,用在许多有机化学反应中。本发明涉及的N2-多氟烷基酰基胍由于其独特的结构特点,相信会更加吸引人们对其理论以及实际应用的探究。到目前对N2-多氟烷基酰基胍这类化合物的合成研究报道较少,其中一些文献报道过的酰基胍衍生物的主要合成方法包括:1)使用脱水剂,2-氯-1,3-二甲基咪唑啉盐,在三乙胺做碱,二氯甲烷为溶剂的条件下,与酰胺发生亲核取代反应,得到酰基胍产物(Isobe,T.;Ishikawa,T.J.Org.Chem., 1999,64,6984)。2)α位全氟烷基取代的酰氯(RFC=OCl)与氰胺、胺经由两步亲核取代,最终得到酰化产物,全氟烷基酰基胍(Lebedev,V.N.et.al.,Zh.Vses.Khim.1981,26, 465)。3)取代酰卤与脒在三乙胺、乙醚、零下20℃条件下,发生酰化反应得到含氟酰基胍 (Leonov,O.N.;Kryukova,L.Y.et.al.,Zh.Obshch.Khim.,1992,62,1592)。
总结现有的合成酰基胍衍生物的方法,不难看出,大多数方法需要使用有毒的酰卤试剂。有些方法涉及原料制备,或者多步,或者低温。因此,发展绿色的N2-多氟烷基酰基胍类化合物的制备方法很有必要。
发明内容
本发明的目的是提供一种,使用胍类化合物或脲类化合物或咪唑啉类化合物和多氟烷基卤为原料,在光照的条件下,经由C-N自由基交叉偶联,再水解得到N2-多氟烷基酰基胍类化合物的合成方法。
本发明采用的技术方案是:一种N2-多氟烷基酰基胍类化合物,所述的N2-多氟烷基酰基胍类化合物具有如(Ⅰ)或(Ⅱ)所示的结构通式,
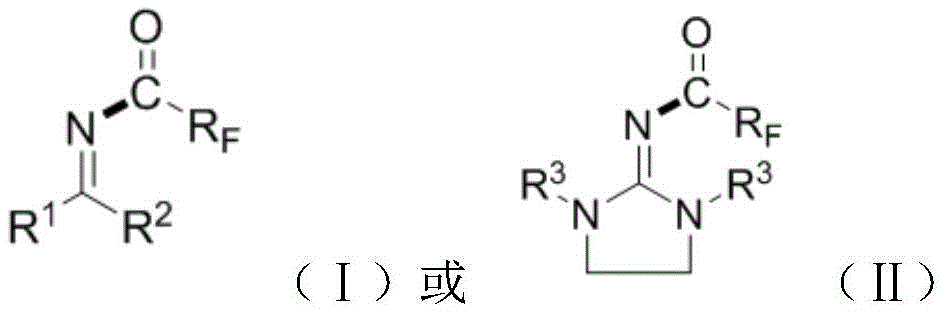
其中,R1为氨基、甲胺基、二甲胺基、异丙胺基、苯胺基、环己胺基、环戊胺基、哌啶基、吗啉基、吡唑基;R2为氨基或取代胺基;R3为氢或甲基;RF为多氟烷基CnF2n+1或CnF2nX,n=2~10的整数,X=Cl,Br,I。
一种N2-多氟烷基酰基胍类化合物的制备方法,包括如下步骤:
1)将胍类化合物或脲类化合物或咪唑啉类化合物投入反应瓶中,加入有机溶剂和多氟烷基卤,在室温光照下搅拌6-15小时,TLC监测反应终点;将得到的反应产物倒入水中,用二氯甲烷萃取,收集有机相,干燥,减压蒸馏除去有机溶剂,得粗品;
2)将粗品用硅胶柱层析的方法,分离纯化,得到目标产物N2-多氟烷基酰基胍类化合物。
优选的,上述的一种N2-多氟烷基酰基胍类化合物的制备方法,所述的胍类化合物的结构式如(Ⅲ)或(Ⅳ)所示:

其中,R1为氨基、甲胺基、二甲胺基、异丙胺基、苯胺基、环己胺基、环戊胺基、哌啶基、吗啉基、吡唑基;R2为氨基或取代胺基;R3为氢或甲基。
更优选的,上述的一种N2-多氟烷基酰基胍类化合物的制备方法,所述的胍类化合物是盐酸胍、四甲基胍、盐酸1-甲基胍、盐酸1,1-二甲基胍、盐酸N-吡唑基胍、盐酸哌啶基胍、盐酸吗啉基胍、盐酸N-苯基胍或盐酸异丙基胍。
更优选的,上述的一种N2-多氟烷基酰基胍类化合物的制备方法,所述的脲类化合物是异硫脲;所述的咪唑啉类化合物是2-亚胺基-1,3-二甲基咪唑啉或2-亚氨基咪唑啉。
更优选的,所述的胍类化合物用碱预处理后,中和除酸,得到游离的胍类化合物。
优选的,上述的一种N2-多氟烷基酰基胍类化合物的制备方法,所述的有机溶剂为乙腈、N,N-二甲基甲酰胺或二甲基亚砜;所述的有机溶剂中含水量为0.05-0.1%。更优选的,所述的有机溶剂为乙腈和N,N-二甲基甲酰胺。
优选的,上述的一种N2-多氟烷基酰基胍类化合物的制备方法,所述的多氟烷基卤为 CnF2n+1I或CnF2nX I,n=2~10的整数,X=Cl,Br,I。
更优选的,上述的一种N2-多氟烷基酰基胍类化合物的制备方法,所述的多氟烷基卤为全氟碘代丙烷、全氟碘代丁烷、全氟碘代己烷、全氟碘代辛烷、五氟碘代乙烷、全氟碘代癸烷或1-氯-2-碘四氟乙烷。
优选的,上述的一种N2-多氟烷基酰基胍类化合物的制备方法,所述的光照的光源来自于太阳光、室内灯光、家用节能灯、蓝色LED或黑灯。更优选的,光照的光源为36W家用节能灯和26W黑灯。
优选的,上述的一种N2-多氟烷基酰基胍类化合物的制备方法,按摩尔比,胍类化合物或脲类化合物或咪唑啉类化合物:多氟烷基卤=1.0:1.1~3.1。
本发明所述的N2-多氟烷基酰基胍类化合物的制备方法,反应式如下:

其中,RF-X为多氟烷基卤,通式为CnF2n+1I或CnF2nX I,n=2~10的整数,X=Cl,Br,I。
本发明的有益效果是:
1.本发明的N2-多氟烷基酰基胍类化合物,结构新颖,产物立体选择性专一。并且由于本身的结构特点,可以作为潜在的组胺H2受体激动剂使用。
2.本发明的制备方法,与现有的方法相比,有以下优点:(1)通过光和氧气催化,光源绿色,条件温和;(2)原料廉价易得,底物范围宽、反应效率较高;(3)反应路线安全,条件简单,操作简便。
3.本发明的制备方法,胍类化合物或脲类化合物或咪唑啉类化合物和多氟烷基卤,在光照的条件下,经由C-N自由基交叉偶联,再在反应体系中存在或产生的微量水的作用下,水解即可得到目标产物。
附图说明
图1为实施例1制备的产物的单晶结构图。
图2为实施例2制备的产物的单晶结构图。
具体实施方式
下面以具体实施例来说明本发明技术方案,但本发明的保护范围不限于此:
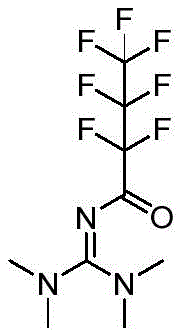
实施例1氮-(双(二甲氨基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺
1.在室温下,将四甲基胍(1.0mmol)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂。然后,加入全氟碘代丁烷(1.1eq)。36W CFL灯照射下反应,TLC监测直至反应完全(约10h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品。
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为产物氮-(双(二甲氨基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺,收率为76%,熔点46-47℃。结构式如下:
1H NMR(500MHz,CDCl3):δ2.99(s,12H);
13C NMR(125MHz,CDCl3):δ40.3,106.4-107.5(m),108.2,108.8,109.3,111.6-110.6(m), 114.6-116.9(m),114.5(t,J=34.0Hz),116.8(t,J=34.0Hz),119.1(t,J=34.0Hz),121.3(t,J= 33.9Hz),161.5(t,J=24.6Hz),168.4;
19F NMR(470MHz,CDCl3):δ-128.3(s,2F),-120.0(q,J=4.5Hz,2F),-82.6(t,J=8.9Hz, 3F)。
HRMS(ESI)(m/z):C9H12F7N3O(M+H)+:计算值312.0947,实测值312.0943。
氮-(双(二甲氨基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺的单晶结构图如图1所示。
实施例2(E)-氮-(氨基(二甲氨基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺
1.在室温下,将盐酸1,1-二甲基胍(1.0mmol)和氢氧化钠(1.0eq)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,中和4h除酸,然后,加入全氟碘代丁烷 (1.1eq)。36W CFL灯光照射下反应,TLC监测直至反应完全(约12h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品。
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为产物(E)-氮-(氨基(二甲氨基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺,收率为73%,熔点83-84℃。结构式如下:

1H NMR(300MHz,CDCl3):δ1.87(s,6H),3.10(d,J=28.1Hz,2H);
13C NMR(150MHz,CDCl3):δ35.6,37.7,106.8-107.2(m),108.4-109.0(m),109.9,110.2- 110.7(m),115.0(t,J=33.6Hz),117.0(t,J=33.9Hz),118.8(t,J=34.2Hz),120.7(t,J=33.8 Hz),160.6,166.3(t,J=24.8Hz);
19F NMR(376MHz,DMSO-d6):δ-125.9(s,2F),-117.3(q,J=4.3Hz,2F),-80.1(t,J=8.7 Hz,3F)。
HRMS(ESI)(m/z):C7H8F7N3O(M+H)+:计算值284.0634,实测值284.0637。
(E)-氮-(氨基(二甲氨基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺的单晶结构图如图2所示。
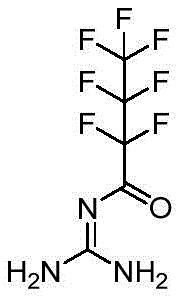
实施例3氮-(二氨基亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺
1.在室温下,将盐酸胍(1.0mmol)和氢氧化钠(1.0eq)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,中和4h除酸,然后,加入全氟碘代丁烷(1.1 eq)。36W CFL灯光照射下反应,TLC监测直至反应完全(约14h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品。
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为产物氮-(二氨基亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺,收率为64%,熔点85-86℃。结构式如下:
1H NMR(300MHz,DMSO-d6):δ7.17(s,2H),7.99(s,2H);
13C NMR(125MHz,DMSO-d6):δ108.9,116.6-121.1(m),163.3,165.7(t,J=23.0Hz);
19F NMR(470MHz,DMSO-d6):δ-123.3(s,2F),-114.6(q,J=4.2Hz,2F),-77.4(t,J=4.2 Hz,3F)。
HRMS(ESI)(m/z):C5H4F7N3O(M+H)+:计算值256.0321,实测值256.0325。
实施例4(E)-氮-(氨基(异丙氨基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺
1.在室温下,将盐酸异丙基胍(1.0mmol)和氢氧化钠(1.1eq)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,中和4h除酸,然后,加入全氟碘代丁烷 (1.1eq)。36WCFL灯光照射下反应,TLC监测直至反应完全(约13h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品。
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为产物(E)-氮-(氨基(异丙氨基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺,收率为74%,熔点126-127℃。结构式如下:

1HNMR(300MHz,CDCl3):δ1.30(d,J=3.0Hz,8H),3.57(s,1H),7.88(s,1H);
13C NMR(150MHz,DMSO-d6):δ22.3,42.8,104.0,104.2,105.7,106.8,108.6,109.1,110.0, 110.6,117.0,118.9,120.8,160.4,165.1(t,J=23.4Hz);
19F NMR(376MHz,DMSO-d6):δ-125.9(s,2F),-117.5(q,J=4.1Hz,1F),-117.2(q,J=4.1 Hz,1F),-80.2(t,J=8.6Hz,3F)。
HRMS(ESI)(m/z):C8H10F7N3O(M+H)+:计算值298.0790,实测值298.0786。
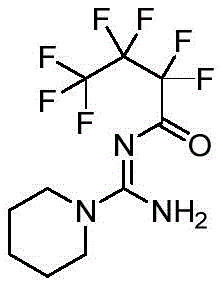
实施例5(E)-氮-(氨基(哌啶基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺
1.在室温下,将盐酸哌啶基胍(1.0mmol)和氢氧化钠(1.0eq)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,中和4h除酸,然后,加入全氟碘代丁烷 (1.1eq)。36WCFL灯光照射下反应,TLC监测直至反应完全(约11h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品。
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为(E)-氮-(氨基(哌啶基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺,收率为75%,熔点109-110℃。结构式如下:
1H NMR(300MHz,CDCl3):δ1.65(m,6H),3.57(s,4H),7.45(s,2H);
13C NMR(150MHz,CDCl3):δ23.9,25.2,45.2,106.7-107.4(m),108.4,108.7,108.9,110.2- 110.9(m),115.0(t,J=34.1Hz),116.9(t,J=34.1Hz),118.8(t,J=33.9Hz),120.7(t,J=34.2Hz), 159.1,166.4(t,J=24.6Hz);
19F NMR(376MHz,CDCl3):δ-126.4(s,2F),-118.7(q,J=4.1Hz,2F),-80.7(t,J=9.0Hz, 3F)。
HRMS(ESI)(m/z):C10H12F7N3O(M+H)+:计算值324.0947,实测值324.0950。
实施例6(E)-氮-(氨基(吗啉)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺
1.在室温下,将盐酸吗啉基胍(1.0mmol)和氢氧化钠(1.0eq)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,中和4h除酸,然后,加入全氟碘代丁烷 (1.1eq)。36WCFL灯光照射下反应,TLC监测直至反应完全(约15h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品。
2.将粗品通过硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为(E)-氮-(氨基(吗啉)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺,收率为77%,熔点81-82℃。结构式如下:
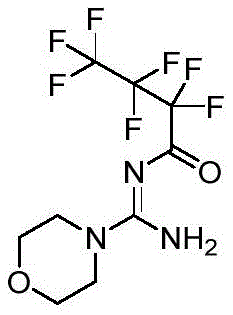
1H NMR(300MHz,CDCl3):δ1.59(s,2H),3.62(s,3H),3.76(t,J=4.5Hz,4H),7.53(s,2H);
13C NMR(100MHz,):δ44.0,66.0,106.2,108.3-109.0(m),111.7(t,J=30.6Hz),113.6, 116.4(t,J=34.0Hz),119.3(t,J=33.8Hz),122.2(t,J=33.4Hz),128.8,131.0,159.8,167.0(t,J= 24.9Hz);
19F NMR(376MHz,CDCl3):δ-126.4(s,2F),-118.7(q,J=4.5Hz,2F),-80.7(t,J=8.6Hz, 3F)。
HRMS(ESI)(m/z):C9H10F7N3O2(M+H)+:计算值326.0739,实测值326.0743。
实施例7(E)-氮-(氨基(苯胺基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺
1.在室温下,将盐酸N-苯基胍(1.0mmol)和氢氧化钠(1.0eq)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,中和4h除酸,然后,加入全氟碘代丁烷 (1.1eq)。36WCFL灯光照射下反应,TLC监测直至反应完全(约14h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品;
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为(E)-氮-(氨基(苯胺基)亚甲基)-2,2,3,3,4,4,4-七氟丁酰胺,收率为68%,熔点71-72℃。结构式如下:

1H NMR(300MHz,CDCl3):δ5.45(s,1H),7.29(d,J=3.9Hz,2H),7.40(t,J=7.5Hz,1H), 7.50(t,J=7.2Hz,2H),8.31(s,1H),9.88(s,1H);
13C NMR(150MHz,CDCl3):δ106.9(t,J=30.9Hz),108.4-109.0(m),110.4(t,J=30.8Hz), 114.8,116.7(t,J=33.8Hz),118.7(t,J=33.9Hz),120.6,120.8,121.0,125.5,128.5,130.4,134.0, 168.3(t,J=24.5Hz);
19F NMR(376MHz,CDCl3):δ-126.5(s,2F),-119.2(q,J=8.3Hz,2F),-80.7(t,J=8.6Hz, 3F)。
HRMS(ESI)(m/z):C11H8F7N3O(M+H)+:计算值332.0634,实测值332.0630。
实施例8氮-(双(二甲氨基)亚甲基)-2,2,3,3,4,4,5,5,6,6,6-十一氟己酰胺
1.在室温下,将四甲基胍(1.0mmol)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,然后,加入全氟碘代己烷(1.1eq)。36W CFL灯光照射下反应,TLC 监测直至反应完全(约10h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3 次,并有机相,干燥,减压蒸馏除去有机溶剂,得粗品;
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为产物氮-(双(二甲氨基)亚甲基)-2,2,3,3,4,4,5,5,6,6,6-十一氟己酰胺,收率为79%,熔点51-52 ℃。结构式如下:
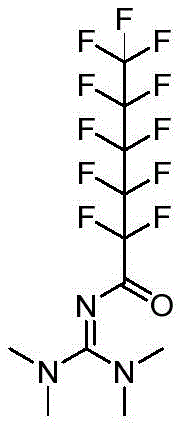
1H NMR(500MHz,CDCl3):δ2.99(s,12H);
13C NMR(75.0MHz,CDCl3):δ40.3,105.1-107.9(m),106.4-107.5(m),108.3,108.7,109.3, 109.8,110.2,110.5,111.0-114.1(m),114.9,115.8,116.3,119.2(t,J=32.2Hz),121.3(t,J=33.9 Hz),161.5(t,J=24.7Hz),168.3;
19F NMR(376MHz,CDCl3):δ-126.2(q,J=5.5Hz,2F),-122.4(s,2F),-122.0(s,2F),- 117.1(t,J=12.8Hz,2F),-80.8(m,3F)。
HRMS(ESI)(m/z):C11H12F11N3O(M+H)+:计算值412.0883,实测值412.0880。
实施例9氮-(双(二甲氨基)亚甲基)-2-氯-2,2-二氟乙酰胺
1.在室温下,将四甲基胍(1.0mmol)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,然后,加入1-氯-2-碘四氟乙烷(1.1eq)。36W CFL灯光照射下反应,TLC 监测直至反应完全(约12h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3 次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品;
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为产物氮-(双(二甲氨基)亚甲基)-2-氯-2,2-二氟乙酰胺,收率为70%,熔点178-179℃。结构式如下:

1H NMR(300MHz,CDCl3):δ2.99(s,12H);
13C NMR(100MHz,CDCl3):δ39.8,100.0,120.4,123.5,162.2,162.9;
19F NMR(376MHz,CDCl3):δ-62.3(s,1F),-61.5(s,1F)。
HRMS(ESI)(m/z):C7H12ClF2N3O(M+H)+:计算值228.0715,实测值228.0717。
实施例10(E)-氮(氨基(1-氢吡唑基)亚甲基)2,2,3,3,4,4,4-七氟丁酰胺
1.在室温下,将盐酸N-吡唑基胍(1.0mmol)和氢氧化钠(1.0eq)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,中和4h除酸,然后,加入全氟碘代丁烷 (1.1eq)。36W CFL灯光照射下反应,TLC监测直至反应完全(约15h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品;
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为产物(E)-氮(氨基-1-氢吡唑-1-基)亚甲基)2,2,3,3,4,4,4-七氟丁酰胺,收率为70%,熔点127-128℃。结构式如下:

1H NMR(300MHz,CDCl3):δ6.37(s,1H),6.54(s,1H),6.85(s,1H),7.87(s,1H),8.60(d,J =1.5Hz,1H);
13C NMR(100MHz,DMSO-d6):δ104.1,105.2,105.5,105.9,106.3,107.1-108.9(m),110.8, 111.2-113.4(m),114.7,116.0(t,J=33.6Hz),118.8(t,J=33.6Hz),121.6(t,J=33.5Hz),130.0, 144.9,164.9(t,J=24.9Hz),167.4;
19F NMR(376MHz,CDCl3):δ-126.1(s,2F),-118.4(q,J=4.5Hz,2F),-80.3(t,J=9.0Hz, 3F)。
HRMS(ESI)(m/z):C8H5F7N4O(M+H)+:计算值307.0430,实测值307.0433。
实施例11氮-(双(二甲氨基)亚甲基)-2,2,3,3,3-五氟丙酰胺
1.在室温下,将四甲基胍(1.0mmol)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,然后,加入全氟碘代丙烷(1.1eq)。36W CFL灯光照射下反应,TLC监测直至反应完全(约13h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取3次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品;
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,收集洗脱液,减压蒸馏,得到白色固体,即为产物氮-(双(二甲氨基)亚甲基)-2,2,3,3,3-五氟丙酰胺,收率为77%,熔点50-51℃。结构式如下:

1H NMR(400MHz,CDCl3):δ2.99(s,12H);
13C NMR(100MHz,CDCl3):δ40.2,104.5,104.9,105.2,105.6(q,J=36.9Hz),107.1,107.4, 107.8,108.2(q,J=37.0Hz),109.7,110.0,110.4(t,J=37.3Hz),114.2,114.5,114.9(t,J=36.1 Hz),117.2,117.4,117.7(t,J=35.7Hz),119.9,120.2,120.6(t,J=35.7Hz),122.7,123.1,123.4(t, J=35.8Hz),161.2,161.4,161.7(t,J=24.5Hz),168.4;
19F NMR(CDCl3,376MHz):δ-120.6(s,2F),-82.3(s,3F)。
HRMS(ESI)(m/z):C8H12F5N3O(M+H)+:计算值262.0979,实测值262.0981。
实施例12 2,2,3,3,4,4,4-氮-(咪唑烷酮-2-叉)-七氟丁酰胺
1.在室温下,将2-亚氨基咪唑啉(1.0mmol)投入到25mL圆底烧瓶中,注入2mL乙腈(含水量0.1%)溶剂,然后,加入全氟碘代丁烷(1.1eq)。36W CFL灯光照射下反应, TLC监测直至反应完全(约11h)。将所得反应产物倒入100mL水中,用二氯甲烷萃取 3次,合并有机相,干燥,减压蒸馏除去有机溶剂,得粗品;
2.将粗品进行硅胶柱层析(硅胶需要用Et3N/PE=1:9浸泡过夜预处理),用洗脱剂(石油醚:乙酸乙酯=1:1(V:V))进行洗脱,得到白色固体,即为产物2,2,3,3,4,4,4-氮-(咪唑烷酮-2-叉)-七氟丁酰胺,收率为60%,熔点157-158℃。结构式如下:

1H NMR(400MHz,DMSO-d6):δ3.58(s,4H),8.44(s,2H);
13C NMR(100MHz,DMSO-d6):δ41.6,105.8,106.1,106.4(t,J=30.0Hz),108.1-109.2(m), 110.6-111.8(m),113.0,113.3,133.7(t,J=35.0Hz),115.8,116.2,116.5(t,J=35.0Hz),118.7, 119.0,119.4(t,J=35.0Hz),121.6,121.9,121.2(t,J=30.0Hz),164.5,164.7,164.9,165.1(t,J= 20.0Hz);
19F NMR(376MHz,DMSO-d6):δ-126.1(s,2F),-117.5(q,J=8.2Hz,2F),-80.3(t,J=8.5Hz, 2F)。
HRMS(ESI)(m/z):C7H6F7N3O(M+H)+:计算值282.0477,实测值282.0479。
Claims (4)
1.一种N2-多氟烷基酰基胍类化合物的制备方法,其特征在于,包括如下步骤:
1)将胍类化合物或咪唑啉类化合物投入反应瓶中,加入有机溶剂和多氟烷基卤,在室温光照下搅拌6-15小时,TLC监测反应终点,将所得反应产物倒入水中,用二氯甲烷萃取,收集有机相,干燥,减压蒸馏除去有机溶剂,得粗品;
所述胍类化合物是四甲基胍、盐酸1,1-二甲基胍、盐酸胍、盐酸异丙基胍、盐酸哌啶基胍、盐酸吗啉基胍、盐酸N-苯基胍、盐酸N-吡唑基胍;
所述咪唑啉类化合物是2-亚氨基咪唑啉;
所述多氟烷基卤是全氟碘代丁烷、全氟碘代己烷、1-氯-2-碘四氟乙烷、全氟碘代丙烷;
2)将粗品用硅胶柱层析的方法,分离纯化,得到目标产物N2-多氟烷基酰基胍类化合物。
2.根据权利要求1所述的一种N2-多氟烷基酰基胍类化合物的制备方法,其特征在于,所述的有机溶剂为乙腈、N,N-二甲基甲酰胺或二甲基亚砜;所述的有机溶剂中含水量为0.05-0.1%。
3.根据权利要求1所述的一种N2-多氟烷基酰基胍类化合物的制备方法,其特征在于,所述的光照的光源来自于太阳光、室内灯光、家用节能灯、蓝色LED或黑灯。
4.根据权利要求1所述的一种N2-多氟烷基酰基胍类化合物的制备方法,其特征在于,按摩尔比, 胍类化合物或咪唑啉类化合物 : 多氟烷基卤 = 1.0 : 1.1~3.1。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201811390684.5A CN109369467B (zh) | 2018-11-21 | 2018-11-21 | 一种n2-多氟烷基酰基胍类化合物及其制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201811390684.5A CN109369467B (zh) | 2018-11-21 | 2018-11-21 | 一种n2-多氟烷基酰基胍类化合物及其制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN109369467A CN109369467A (zh) | 2019-02-22 |
| CN109369467B true CN109369467B (zh) | 2021-05-28 |
Family
ID=65376637
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201811390684.5A Active CN109369467B (zh) | 2018-11-21 | 2018-11-21 | 一种n2-多氟烷基酰基胍类化合物及其制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN109369467B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025163139A1 (en) * | 2024-02-02 | 2025-08-07 | Syngenta Crop Protection Ag | Process for the preparation of microbiocidal oxadiazole derivatives |
-
2018
- 2018-11-21 CN CN201811390684.5A patent/CN109369467B/zh active Active
Non-Patent Citations (3)
| Title |
|---|
| New Hexanuclear Niobium Cluster Compounds with Perfluorinated Ligands Made Using Ionic Liquids;Daniel Holger Weiβ et al.;《Journal of Inorgain and General Chemistry》;20171231;第643卷;第345-351页 * |
| Preparation of Imidazolones from N-cyano-N"-methylcarboxyguanidines. An Unusual C-N Formation in the Hydrogenolysis of a Benzyl Ester in an Attempted Synthesis of an Inhibitor of Carboxypeptidase A;Peter J. Garratt et al.;《Tetrahedron》;19931231;第49卷(第31期);第6885-6898页 * |
| Synthesis and Hypotensive Activity of N-Alkyl-N’’-cyano-N’-pyridylguanidines;Hans Jφrgen Petersen et al.;《Journal of Medicinal Chemistry》;19781231;第21卷(第8期);第773-781页 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025163139A1 (en) * | 2024-02-02 | 2025-08-07 | Syngenta Crop Protection Ag | Process for the preparation of microbiocidal oxadiazole derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| CN109369467A (zh) | 2019-02-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN104447396B (zh) | 一种安息香肟衍生物及其制备方法 | |
| CN103408445B (zh) | 一种芳胺类衍生物及其制备方法 | |
| CN107445857A (zh) | 一种具有co2响应性能的长链烷基酸酰胺丙基二甲胺的制备方法 | |
| CN109369467B (zh) | 一种n2-多氟烷基酰基胍类化合物及其制备方法 | |
| CN103240034B (zh) | 磺酸盐型及磺酸内盐型氟碳表面活性剂及其制备和应用 | |
| CN104591959B (zh) | 一种二苯乙烯类化合物的制备方法 | |
| CN110003081B (zh) | 一种多氟烷基取代的吲哚啉和四氢异喹啉的合成方法 | |
| CN109293589B (zh) | 一种6-多氟烷基-1,3,5-三嗪类化合物及其合成方法和应用 | |
| CN104447336B (zh) | 一种三碟烯衍生物及其制备方法 | |
| CN102942463A (zh) | 一种二苯甲酮类化合物的制备方法 | |
| CN106117222A (zh) | 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉二元化合物盘状液晶材料合成方法 | |
| CN107573263B (zh) | 一种ω-取代缩二脲类化合物的合成方法 | |
| CN102285935B (zh) | 2,2’-(1,3-亚苯基)二{5-[4-(1,1-二甲基乙基)苯基]}-1,3,4-噁二唑的合成方法 | |
| CN106117225A (zh) | 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法 | |
| CN108424416B (zh) | 一种合成吲哚[1,2-c]喹唑啉类化合物的方法 | |
| CN112851456B (zh) | 一种碱性条件下制备烯烃化合物的方法 | |
| CN104529802A (zh) | 一种合成n,n′-双取代氟代丙二酰胺化合物的方法 | |
| CN116621762A (zh) | 一种3-硝基吲哚类似物及其制备方法 | |
| TWI670255B (zh) | 用於製備鹵代苯之方法 | |
| CN102643221B (zh) | 西洛多辛中间体及其制备方法 | |
| CN106118688A (zh) | 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法 | |
| CN108250087B (zh) | 一种4-异丁氧基苄胺的合成方法 | |
| CN115626861B (zh) | 合成三氟甲基芳香化合物的方法 | |
| CN103897427B (zh) | 一类有机小分子荧光染料的制备方法及其应用 | |
| CN116924940B (zh) | 一种邻碘代苯胺衍生物的合成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |