CN1066847A - 血管紧张素ii受体阻断剂2,3,6取代的喹唑啉酮 - Google Patents
血管紧张素ii受体阻断剂2,3,6取代的喹唑啉酮 Download PDFInfo
- Publication number
- CN1066847A CN1066847A CN92100545A CN92100545A CN1066847A CN 1066847 A CN1066847 A CN 1066847A CN 92100545 A CN92100545 A CN 92100545A CN 92100545 A CN92100545 A CN 92100545A CN 1066847 A CN1066847 A CN 1066847A
- Authority
- CN
- China
- Prior art keywords
- carbon atoms
- lower alkyl
- quinazolinone
- phenyl
- butyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/08—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/88—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/88—Oxygen atoms
- C07D239/91—Oxygen atoms with aryl or aralkyl radicals attached in position 2 or 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/081—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te
- C07F7/0812—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Manufacture Of Tobacco Products (AREA)
- Fats And Perfumes (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
如下式的新颖的喹唑啉酮化合物:
(其中,R5、R6、R7、R8、R及X如在说明书中所定义)
具有血管紧张素II(AII)拮抗活性,用于制备该化合
物的中间体,生产和使用该化合物于减轻哺乳动物中
由血管紧张素诱发的高血压及治疗充血性心力衰
竭。
Description
本发明涉及某些新颖的2,3,6取代的喹唑啉酮化合物,该化合物显示了作为血管紧张素Ⅱ(AⅡ)阻断剂的增强了的活体内活性,因而可用于减轻血管紧张素诱发的高血压及治疗充血性心力衰竭。
血管紧张素原酶作用于血浆α2-球蛋白,血管紧张素原,以产生血管紧张素Ⅰ,该血管紧张素Ⅰ然后又被血管紧张素转化酶转化为AⅡ。该AⅡ物质为一有力的血管升压剂,通常即指用于在哺乳动物中产生高血压的致病剂。因此,抑制荷尔蒙血管紧张素Ⅱ(AⅡ)作用的化合物可用于减轻血管紧张素诱发的高血压。
古川等人在第4,340,598号美国专利中揭示了下式的降压的血管紧张素Ⅱ受体阻断剂的咪唑衍生物及其盐:
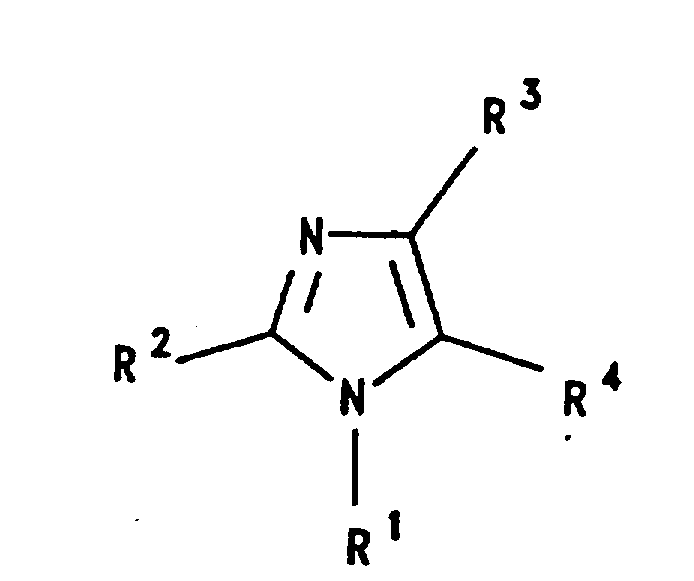
式中,R1为低级烷基或可选择地由卤素或硝基取代的苯基C1-2烷基,R2为低级烷基、环烷基或可有选择地取代的苯基;R3和R4之一为其中的R5是氨基、低级烷氧基或羟基而n为0、1、2的-(CH2)nCOR5基团,R3和R4之另一个为氢或卤素;当R1为低级烷基或苯乙基,R3为氢、n=1,而R5为低级烷氧基或羟基。
古川等人在欧洲第103,647号申请专利中揭示了如下式的用于治疗水肿和高血压并具有血管紧张素Ⅱ受体阻断活性的4-氯-2-苯基咪唑-5-乙酸衍生物及其盐:

其中,R表示低级烷基。
D.J.Carini等人在已公开的、申请号为87109919.8,申请日为1987年9月7日的欧洲申请专利,及在申请号为89100144.8,申请日为1989年5月1日的欧洲申请专利中,揭示了如下式的血管紧张素Ⅱ受体阻断剂的咪唑:
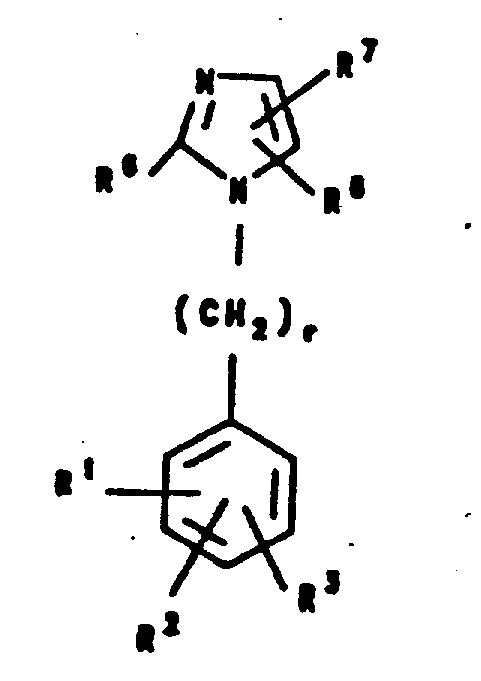
其中,该取代物的定义可在该申请中找到。
P.Aldrich等人在1989年10月17日发布的4,874,867号美国专利中,描述了下式的四唑中间体:
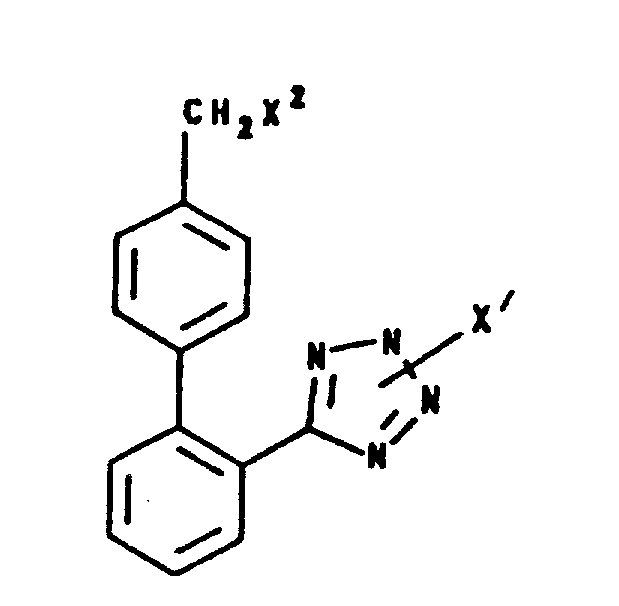
其中,X2及X1如该专利中所定义。这些中间体被描述为用于制造可作为荷尔蒙血管紧张素Ⅱ(AⅡ)的抑制剂的化合物。
D.J.Carini等人在1989年11月24日发布的4,880,804号美国专利中,描述了下式的、可用作荷尔蒙血管紧张素Ⅱ(AⅡ)抑制剂的、取代的苯并咪唑:
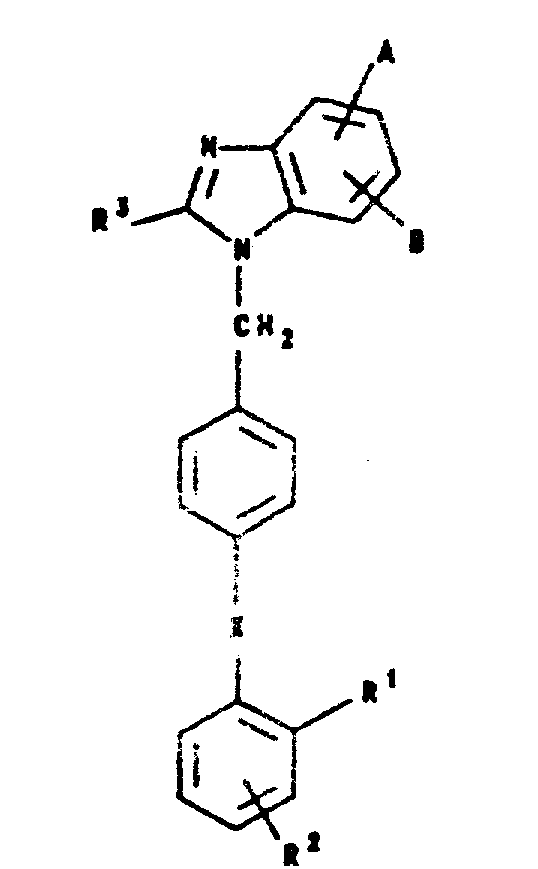
其中,R1为-CO2H、-NHSO2CF3、或;

R2为H、卤素、NO2,甲氧基,或1至4个碳原子的烷基;R3为1至6个碳原子的烷基、或3至6个碳原子的、其每个基团皆可有选择地被卤原子取代的,链烯基或炔基,-OR4或两个-CO2R4基团;但其条件是:当R3为甲基,它须被-OR4或-CO2R4取代;R4为H,或1-4个碳原子的烷基;A为H,1至10个碳原子的烷基,CrF2r+1(其中r=1-6),C6F5,卤素,1-6个碳原子的烷氧基;
(CH2)nOR4, 
COR5,或
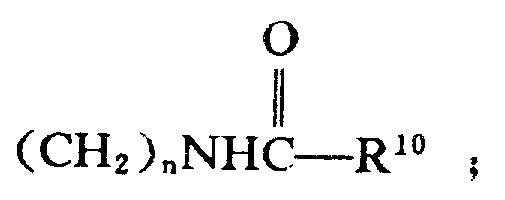
B为H、1-10个碳原子的烷基、CrF2r+1(r=1-6)、C5F5、卤素或1-6个碳原子的烷氧基;X为碳-碳单键、-CO-、-O-、-NHCO-、或-OCH2-。
D.J.Carini等人在已公开的、申请号为89100142.2、申请日为1989年5月1日的欧洲申请专利中,揭示了如下的血管紧张素Ⅱ受体阻断剂的吡咯、吡唑及三唑:

至此,现有的文献即无明示,也无启示;上述已知的AⅡ的阻断剂具有喹唑啉酮结构。
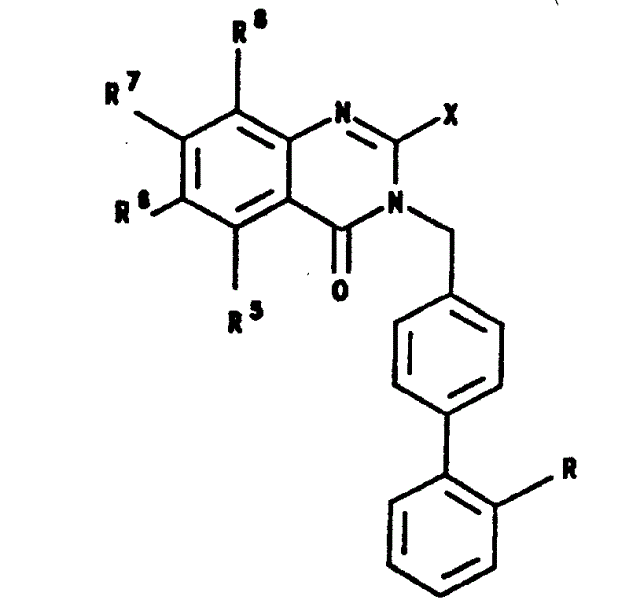
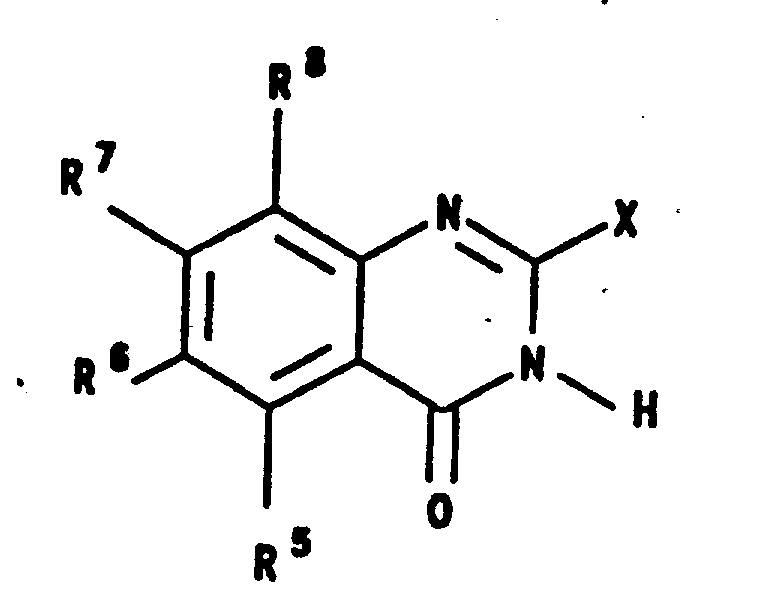
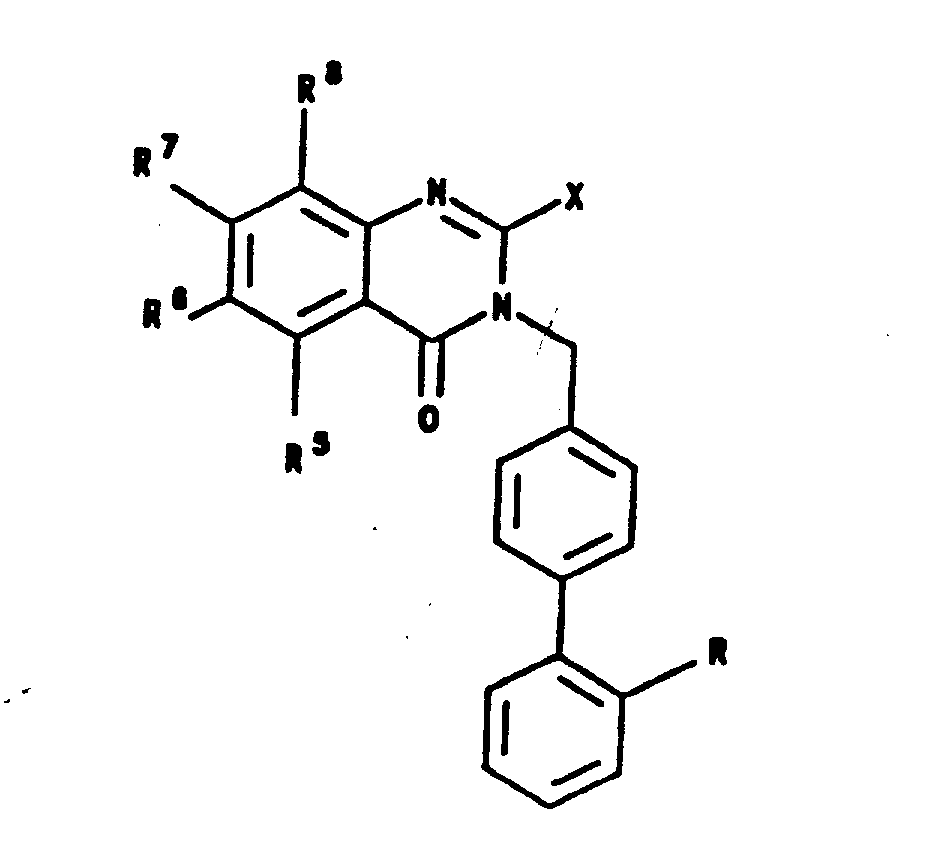
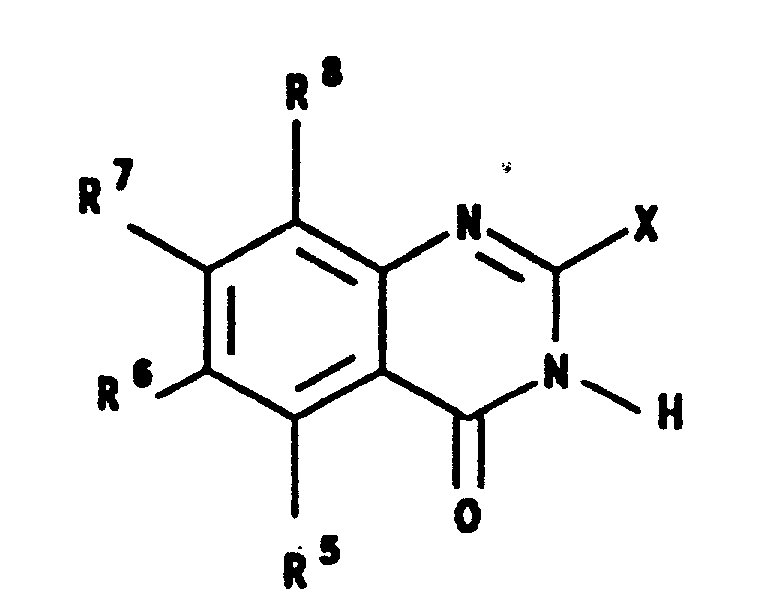
根据本发明,揭供了式Ⅰ的新颖化合物及这些化合物的药学上可接受的盐,它具有增强了的活体内的血管紧张素Ⅱ的拮抗性能,并可用作抗高血压药物:

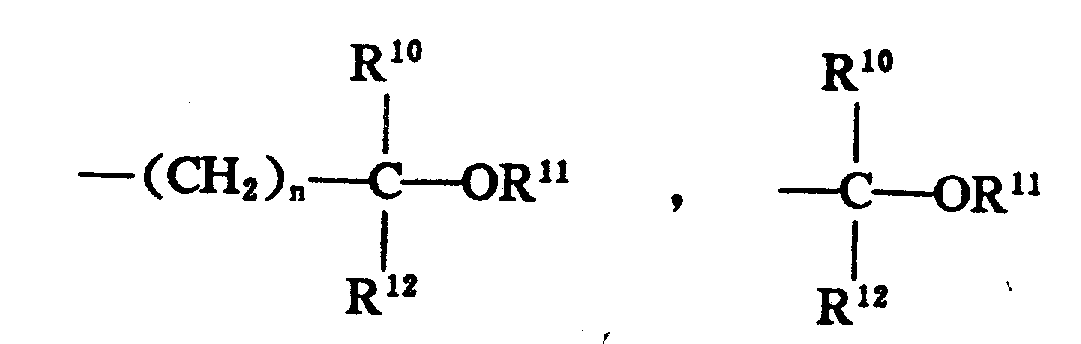
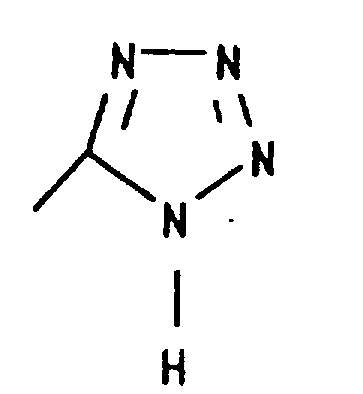
其中,R为
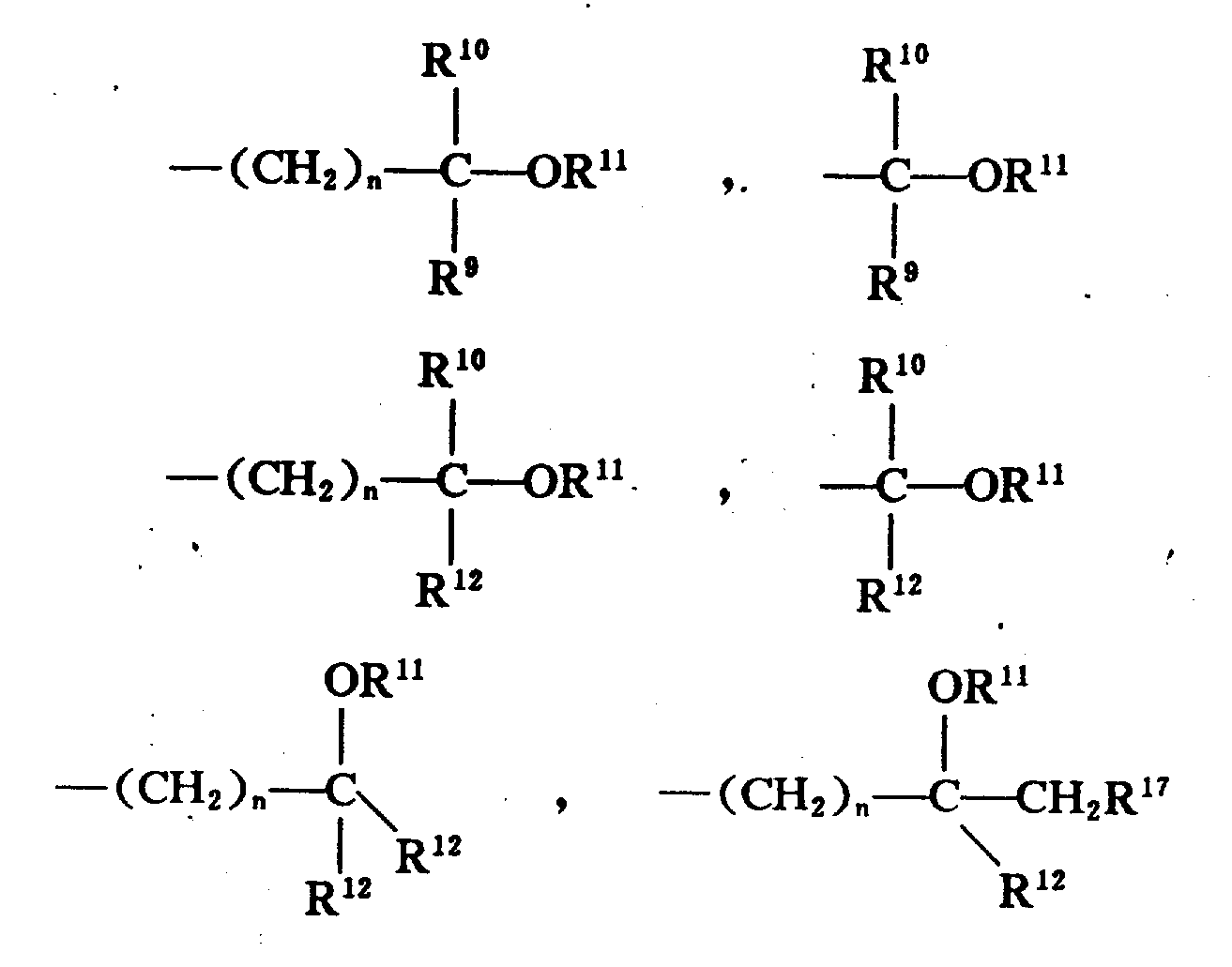
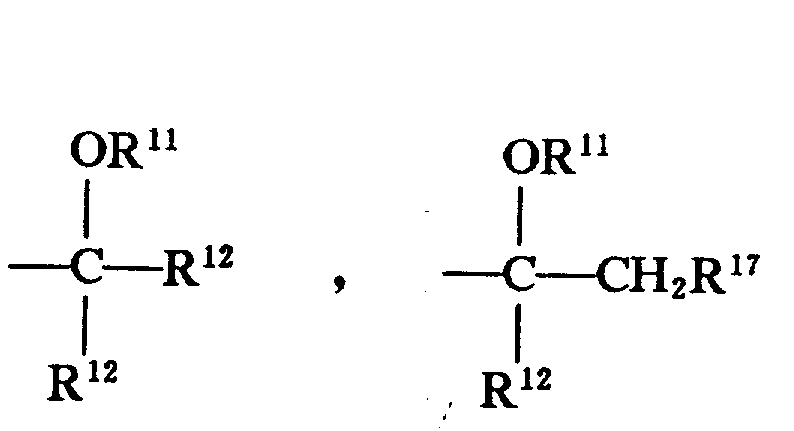
X为3-5个碳原子的直链或支链烷基;n为1-3;R5为H;R6为;

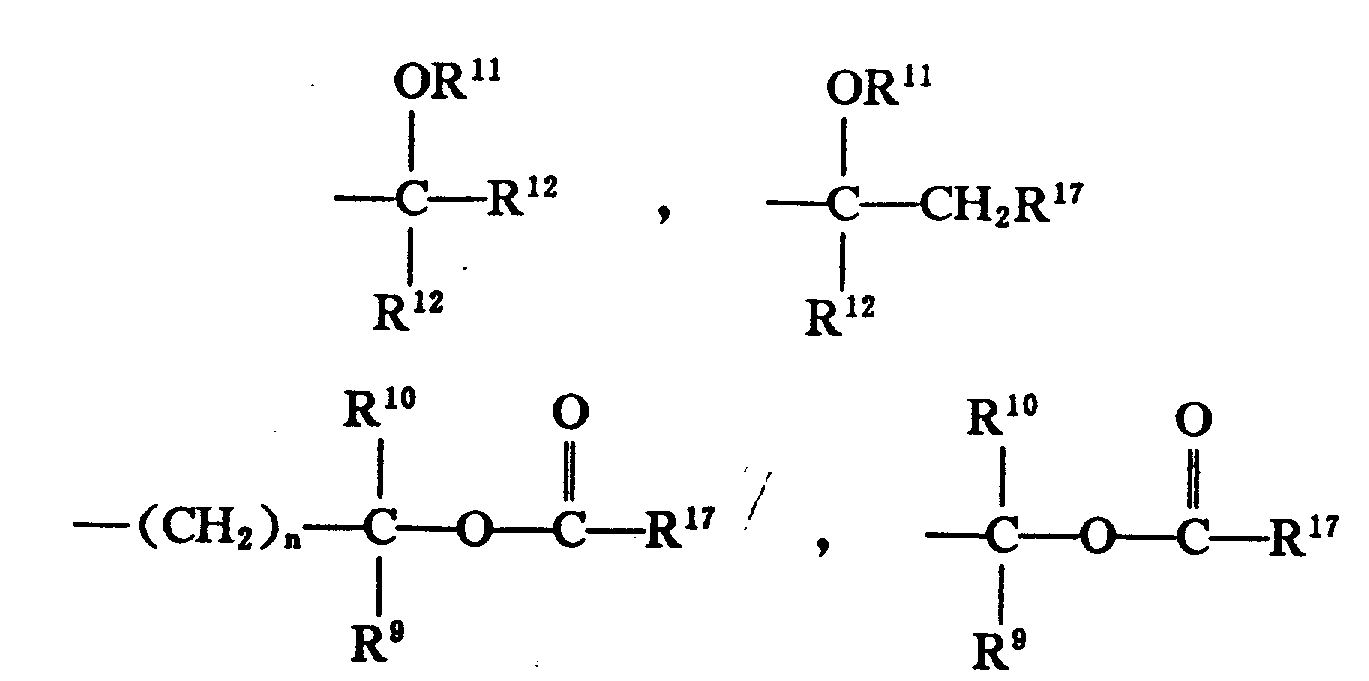
其中,R9为H,1-4个碳原子的直链低级烷基,苯基,取代的苯基(选自1-3个碳原子的单个低级烷基、-CF3、硝基、1-3个碳原子的邻位烷基、NH2的取代基),吡啶,噻吩,或呋喃;
R10为H,1-4个碳原子的直链低级烷基,苯基,取代的苯基(选自1-3个碳原子的单个低级烷基、-CF3、硝基、1-3个碳原子的邻位烷基、NH2的取代基),吡啶,噻吩或呋喃;但是R9和R10不可同时为H;
R11为H,1-4个碳原子的直链或支链低级烷基;
R12为1-4个碳原子的直链低级烷基,苯基,取代的苯基(选自1-3个碳原子的单个低级烷基、-CF3、硝基、1-3个碳原子的邻位烷基、NH2的取代基),吡啶,噻吩,或呋喃;
R17为1-4个碳原子的直链或支链低级烷基;
R7和R8为H。
本发明也提供了新颖的中间体化合物,制造新颖的2,3,6取代的喹唑啉酮的血管紧张素Ⅱ拮抗化合物的方法,制造该新颖的中间体的方法,使用该新颖的喹唑啉酮的血管紧张素Ⅱ拮抗化合物治疗高血压、充血性心力衰竭及拮抗血管紧张素Ⅱ的效应的方法。
图1所示数据显示了血管紧张素Ⅱ对自发性高血压的大鼠的血管升压反应的拮抗效果。
图2-7所示为对主动脉缩窄的高血压的大鼠的平均血压反应数据。
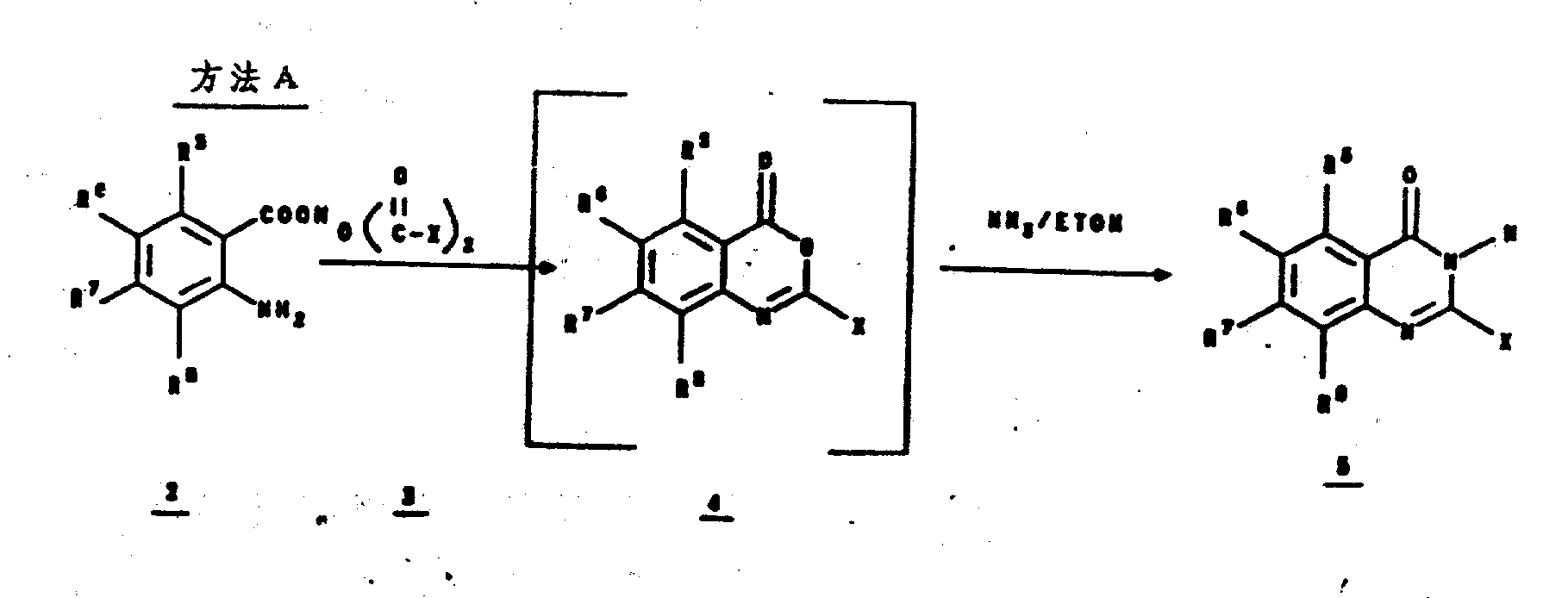
本发明的新颖化合物根据以下反应图式制得。
参见方法A,式5的喹唑啉酮中间体由相应取代的邻氨基苯甲酸2制得。其中,取代基R5、R6、R7和R8如上述,但不同的是,R6不可是:
该相应的邻氨基苯甲酸2在其中X为3-5个碳原子的烷基的烷基酸酐3中被加热回流,以给出由浓缩该反应混合物而分离出的、不须进一步提供即可使用的4H-3,1-苯并噁嗪-4-酮4。
当在含氨、或氢氧化铵溶液的乙醇中回流该4H-3,1-苯并噁嗪-4-酮时,可获得该喹唑啉酮中间体5。为由此方法制备其中不包括R6的化合物,请参见此处的图式Ⅰ至Ⅹ。
参照方法B,B.Baker等人的方法J.Org.Chem 17,157(1952),被用于将适当取代的苯胺6转化为喹唑啉酮5中,在该喹唑啉酮5中,取代基R5、R6、R7及R8由以上所述,不同之处在于,它们不可如下式:
将该取代的苯胺6与氯醛和盐酸羟胺反应,以提供一有硫酸存在时被环化为靛红8的肟7。然后使用30%的过氧化氢水溶液和氢氧化钠水溶液将该靛红8水解至邻氨基苯甲酸9。方法A中的进一步反应生成该喹唑啉酮中间体5。为用此方法制备不包括R6的化合物,请参见此处的图Ⅰ至Ⅹ。
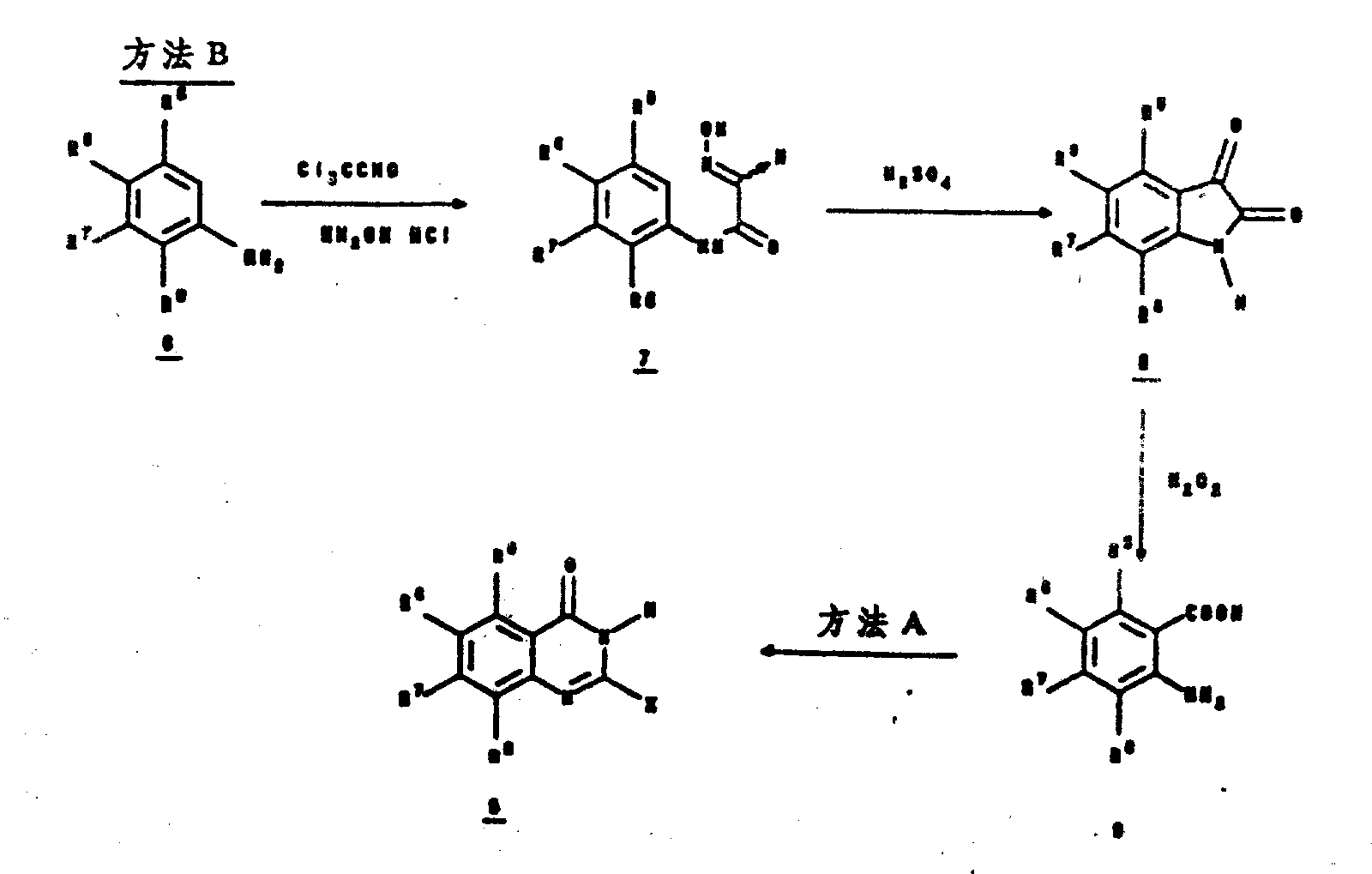
在“杂环化合物化学,稠合嘧啶,第一部分:喹唑啉酮”,W.L.F.Armarego;Interscience Publishers(1967),pp.74-94中给出了对于3,4-二氢-4-氧代-喹唑啉酮5的合成的参考。在“杂环化合物”,Vol,6,P.334,R.C.Elderfield(编者),Wiley and Sons,1957中,描述了另外一些参考资料。
接着根据下图将喹唑啉酮中间体5改性,以获得本发明的新颖的2,3,6取代的喹唑啉酮血管紧张素Ⅱ拮抗化合物。
在图1中,以N-溴丁二酰亚胺溴化由方法A制得的6-甲基喹唑啉酮10,以生成该溴甲基化合物11。在二甲亚砜中用碳酸钾水溶液水解该溴化物生成伯醇12。
在N,N-二甲基甲酰胺中以吡啶鎓重铬酸盐氧化醇12得到醛13。用各种格利雅试剂R12MgBr或锂试剂R12Li在四氢呋喃中,与该醛13反应,给出所希望的仲醇14。在该锂试剂中R12选自1-4个碳原子的直链或支链烷基,苯基,取代的苯基,吡啶基,噻吩及呋喃。
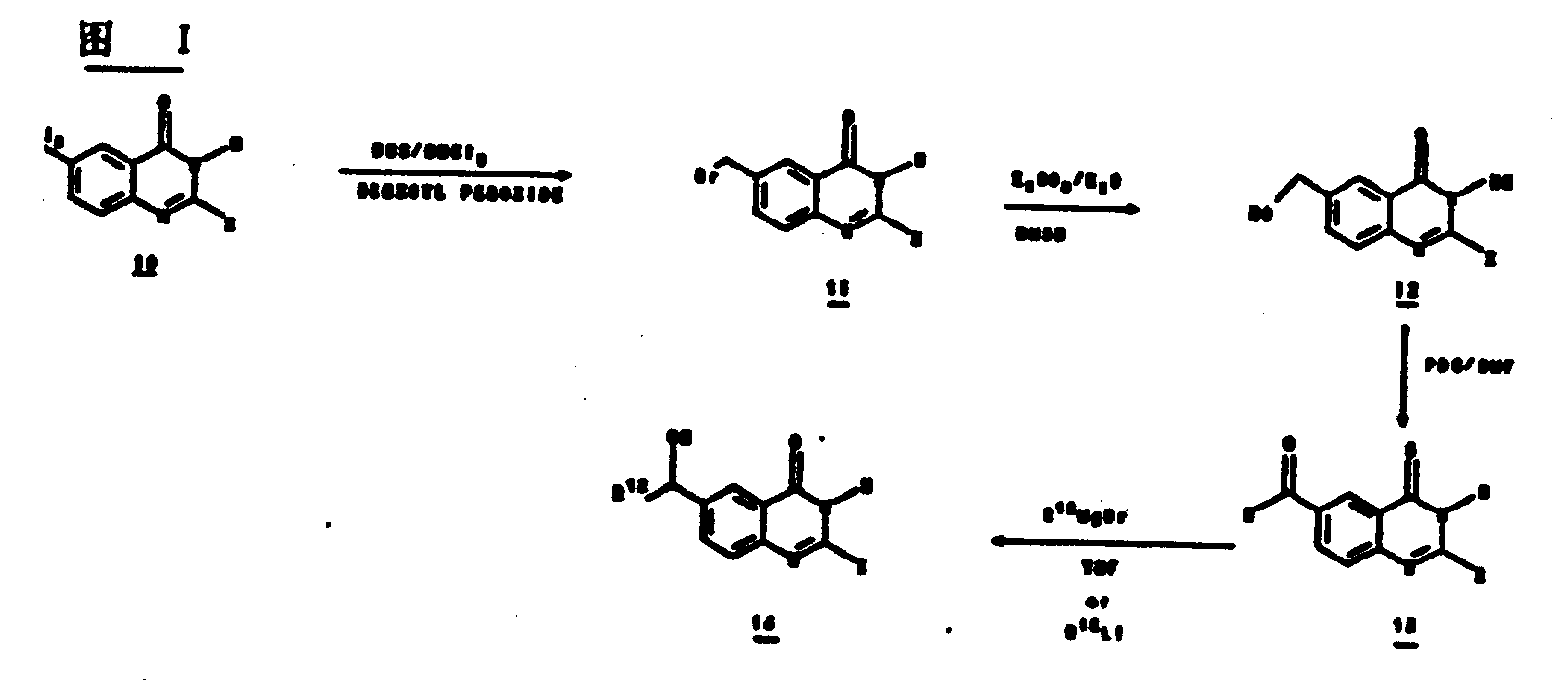
在另一种到达13的方法中,如图Ⅱ所示,由方法A制备的2-烷基取代-6-碘-4(1H)-喹唑啉酮15,通过一钯催化的羰基化反应给出醛13。
用一氧化碳和甲醇由2-烷基取代-6-碘-4(1H)-喹唑啉酮15的钯(Ⅱ)催化偶合形成酯16。该酯16用过量的格利雅试剂R12MgX或锂试剂R12Li的进一步衍生得到其中R12如下限定的叔醇17。

合成至叔醇取代的喹唑啉酮的合成途径示于图Ⅲ。
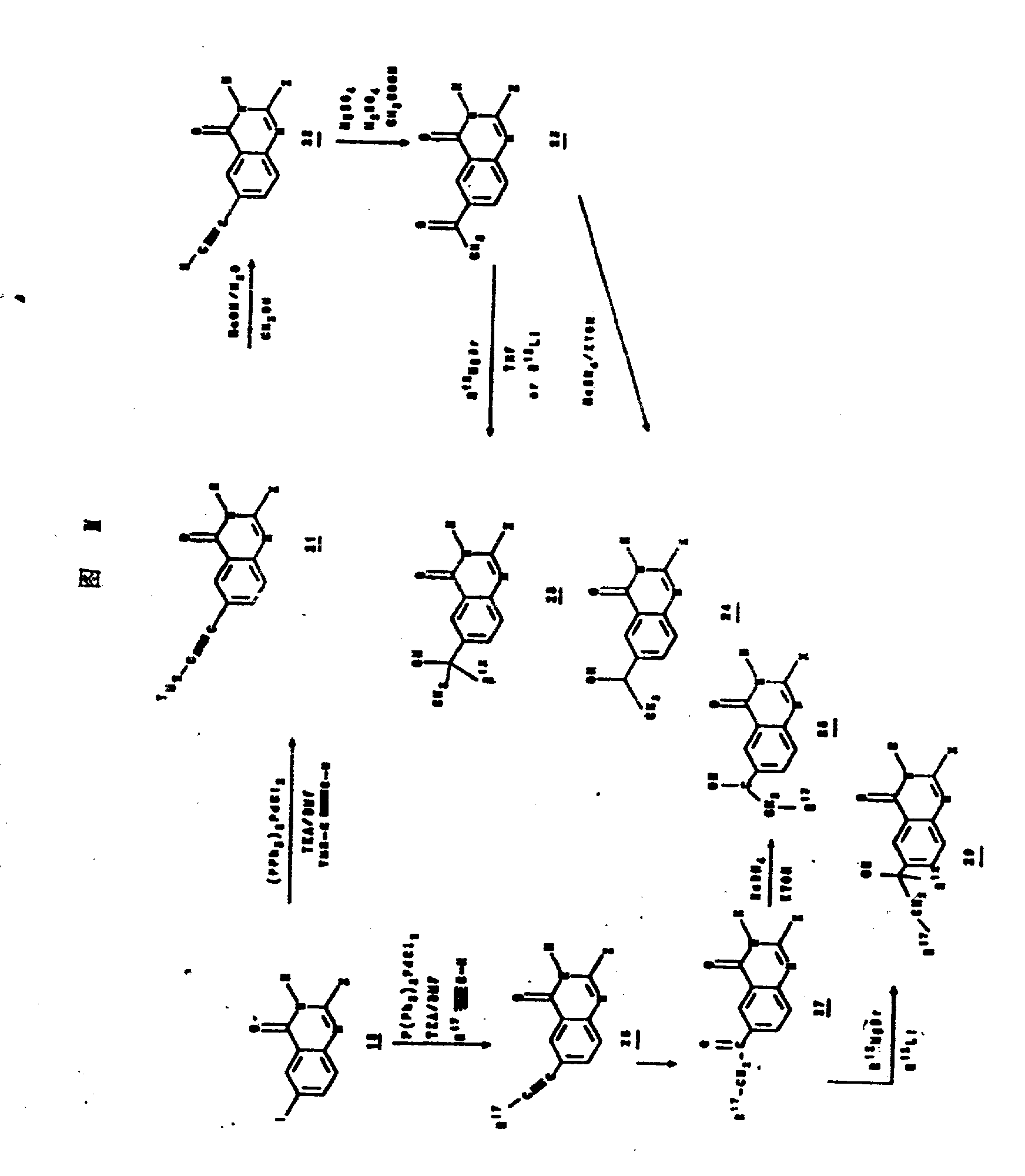
如图Ⅲ所示,(三甲基甲硅基)乙炔和2-烷基取代-6-碘代-4(1H)-喹唑啉酮15的钯(Ⅱ)催化偶合生成炔属喹唑啉酮21。以氢氧化钠在水-甲醇中对该乙炔的脱甲硅基处理给出最终的炔烃22。在乙醇中以催化量的硫酸汞-硫酸进行水合提供了甲基酮23。以氢硼化钠在乙醇中还原酮23给出仲醇24。或者,使甲基酮23与其中R12如上定义的格利雅试剂R12MgBr或锂试剂R12Li反应,生成叔醇25。R17定义为1-4个碳原子的直链或支链低级烷基的取代的乙炔与2-烷基取代-6-碘-4(1H)-喹唑啉酮15的钯(Ⅱ)催化偶合生成炔属喹唑啉酮26。26在乙醇中与催化量的硫酸汞-硫酸进行水合给出甲酮27。用氢硼化钠在乙醇中还原甲酮27给出仲醇28。甲酮27与格利雅试剂R12MgBr或锂试剂R12Li(其中R12如上定义)的反应生成醇29。
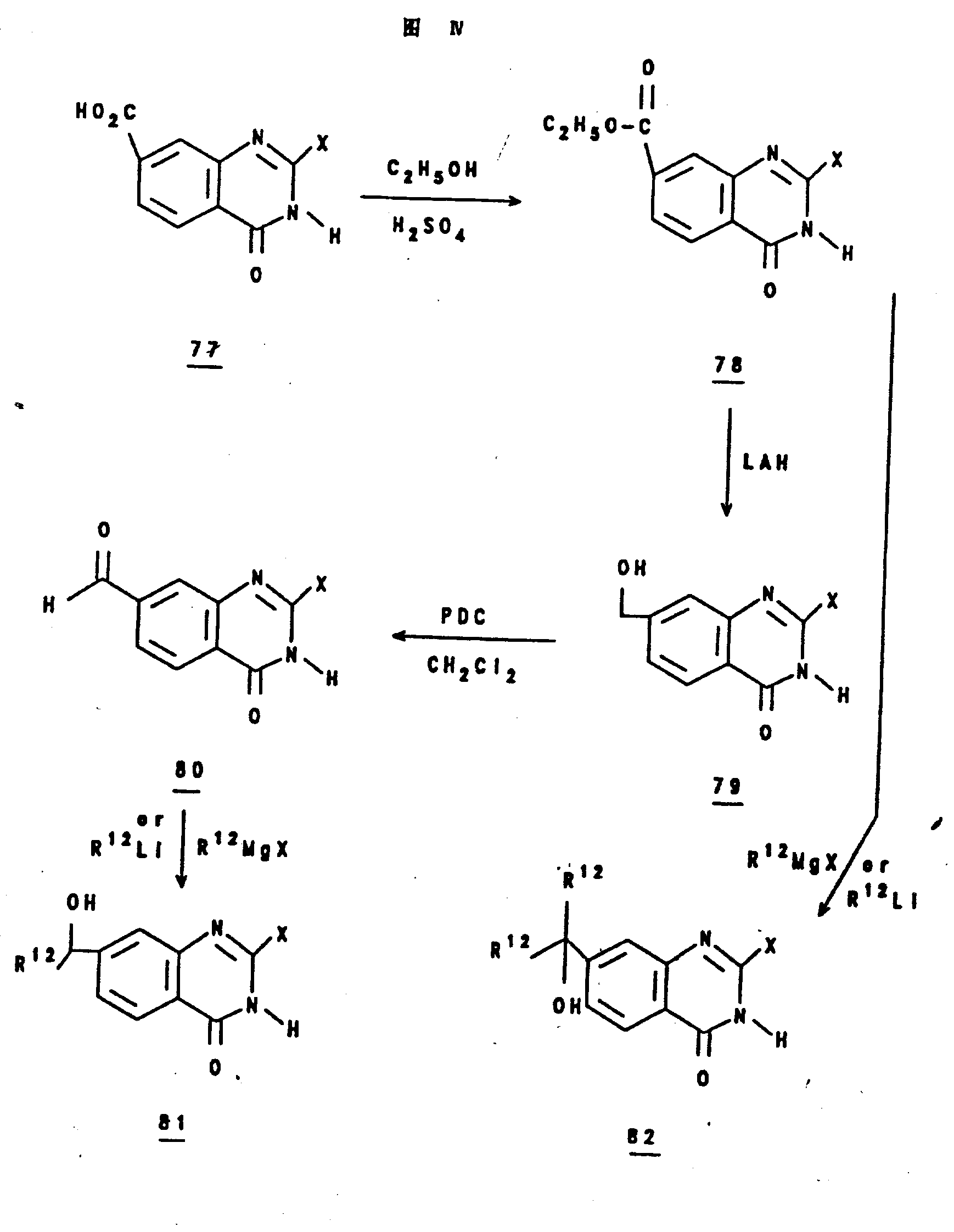
以方法A制备的,如图Ⅳ所示,羧酸77,由与含一催化量的硫酸的乙醇反应被转化为乙酯78。该乙酯78与氢化铝锂在四氢呋喃中反应得到醇79。以吡啶鎓的重铬酸盐氧化醇79生成醛80。
醛80与格利雅试剂R12MgBr或锂试剂R12Li(R12皆如上定义)反应提供了醇81。同样,乙酯78与格利雅试剂R12MgBr或锂试剂R12Li反应给出醇82。
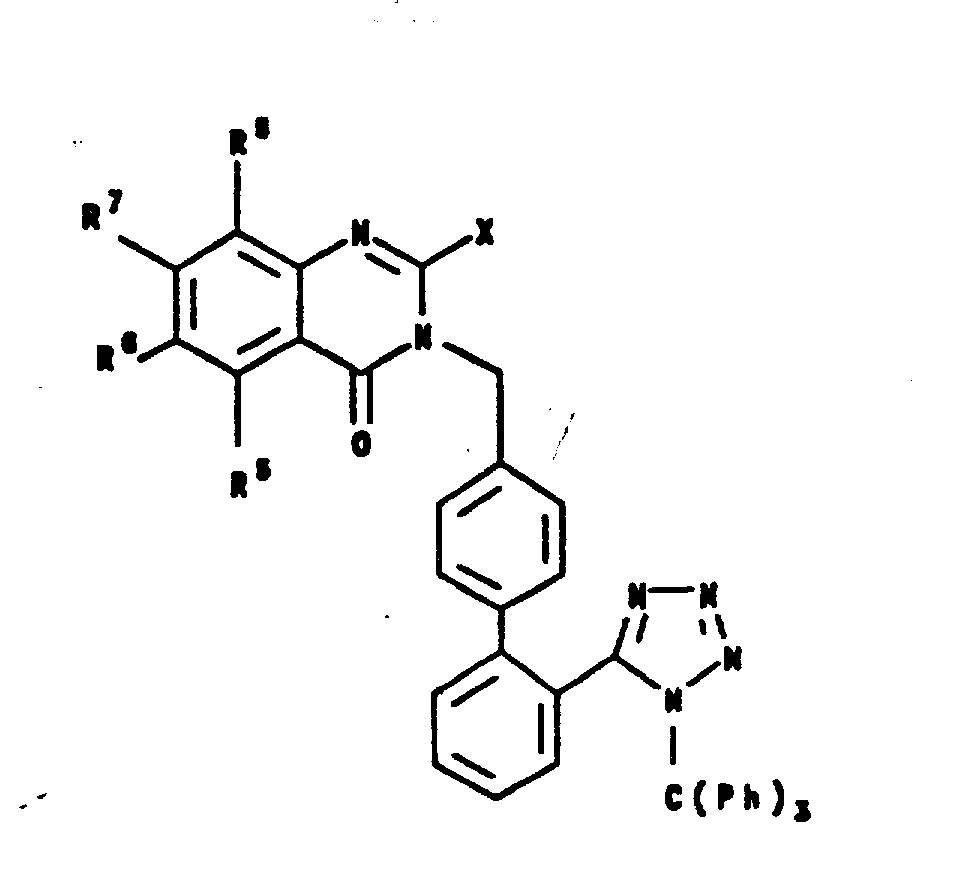
图Ⅴ说明了一喹唑啉酮中间体5和二苯四唑84(其中R如上定义)的偶合,它们由P.E.Aldrich等人的、1989年10月17日发布的4,874,867号美国专利的方法制备。
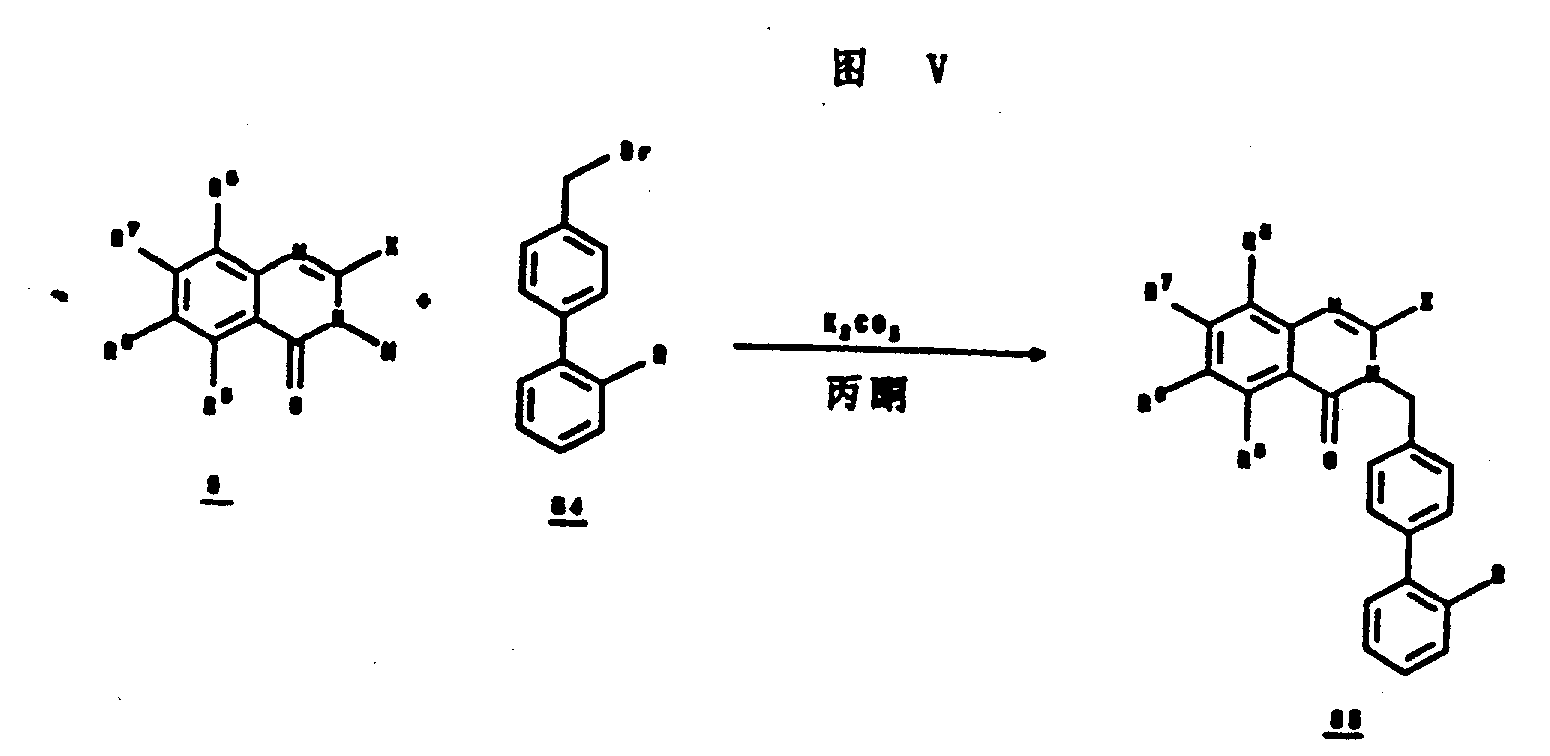
在20-60℃,存有过量的碳酸钾或另一种合适的碱,如,碳酸钠、碳酸铯、氢化钠、氢化钾、甲氧基钠、乙氧基钠、叔-丁氧基钠或叔丁氧基钾时,将该喹唑啉酮5和二苯四唑84溶于丙酮或其它合适的溶剂,如,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮,甲醇,乙醇,叔丁醇,四氢呋喃,二噁烷或二甲基亚砜中2-24个小时。所得的烷基化喹唑啉酮85可用色谱法予以提纯或被用于如进一步的转化和/或脱保护基中。
在这些场合,此时,该烷基化喹唑啉酮85中的R为三苯甲基四唑,该三苯甲基的脱保护基反应,如图Ⅵ所示,附带有用一催化量的盐酸或其它合适的酸,如硫酸、三氟乙醇或氯化氢回流该烷基化喹唑啉酮86的丙酮水溶液2-24个小时。生成的四唑87由闪蒸色谱法或用醚研碎分离后,过滤收集。

如图Ⅶ所示,在有碱,如氢化钠存在时,醇93与一烷基化剂R17I(其中,R17为1-4个碳原子的烷基,n为0-3而I为碘化物)反应,得到醚98。将该中间产物醚98通过稀酸在丙酮中解封给出四唑99。

如图Ⅷ所示,由用稀酸水解除去三苯甲基保护基从93制得醇100,在有如吡啶的碱的存在下,与所希望的酸酐(R17CO)2O(其中,R17定义为1-4个碳原子的直链或支链低级烷基)或-酰基氯R17COCl(其中n为0-3)反应,得到酯101。
由图Ⅰ、Ⅱ和ⅩⅤ的方法制得醇93。
图Ⅸ说明了另一种从5中制备87的合成方法。

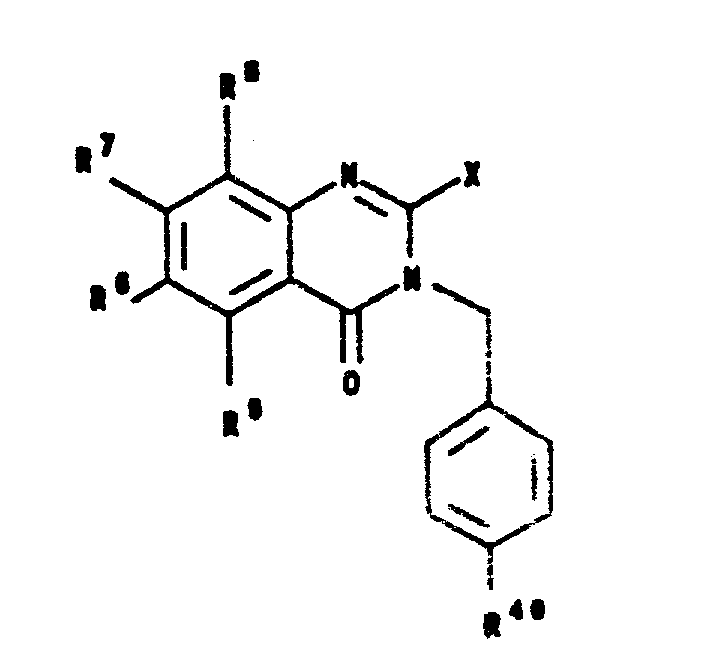
喹唑啉酮5在有碳酸钾存在时,用下式化合物在如丙酮的溶剂中被烷基化,给出108:

式中,R40选自I、Br或-OSO2CF3,B选自合适的离去基团,如I、Br、Cl、-OMS、-OTS或-OSO2CF3。108和其中M可以是-MgBr,-Sn(1-4个碳原子的低级烷基或苯基)的109、Li或-Zn配位化合物的钯或镍的催化偶合提供了110,110经脱保护基给出87。
图Ⅹ说明了制备109的方法。邻位溴苄腈和三-正丁基-亭叠氮化合物反应提供了111。111与氯化氢及三苯甲基氯的进一步反应给出112。112在与ZnCl2或(Me)3SnCl反应之后再与金属M,如,镁,或正丁基锂、或仲丁基锂反应,提供了109。

可取的是,在如图Ⅴ所示的烷基化反应之后,伴以一如图Ⅰ-Ⅹ所示的R5、R6、R7及R8的化学处理。另外,在如图Ⅴ所示的烷基化反应之前,伴以一如图Ⅶ-Ⅷ所示的R5、R6、R7及R8的化学处理也是可达到的。这些反应在适合于所用的试剂和材料的溶剂中进行,并有效的适用于转化反应中。本有机合成领域的技术人员可以理解到,存在于该分子上的各种官能度须与作出的化学转变相一致。这往往需要判断出合成步骤、保护基团,如需要的话及脱保护基的条件的次序。起始材料上的取代基可能与某些反应条件不相一致。这样的一些对可与反应条件相容的取代基的限制,对本领域的技术人员来说是显见的。
药物学上合适的盐即包括金属(无机)盐,也包括有机盐;在Remington′s Pharmaceutical Sciences,17th Edition,Pg.1418(1985)中给出了它们的一览表。本领域技术人员公知,合适的盐的形式根据其物理和化学稳定性,流动性,含湿度及溶解性而选定。由于上述理由,本发明的优选的盐包括钾、钠、钙、镁及铵盐。
上述图中的一些化合物具有不对称中心,从而,该些化合物可以具有至少两个,常为四个立体异构体。本发明包括了所有这些化合物的立体异构体,而不论该化合物是否不具有其它立体异构体,或与其它立体异构以任何比率混合。这样,本发明包括了,例如,对映体的外消旋混合物以及异构体的非对映异构体的混合物。任何化合物的绝对构型可由通常的X-光结晶学测定。
当使用三苯甲基保护基于该四唑化合物以说明本发明时,对本领域技术人员来说,显然也可以利用其它的氮保护基。期望的相当的保护基有:苄基、对硝基苄基、丙腈基或其它任何适用于保护该四唑氮的保护基。此外,对本领域技术人员来说,显见的是:脱去除三苯甲基之外的各种氮保护基,可能需要除了稀酸以外的方法。
由以下的实施例可进一步理解本发明的化合物及其制备方法,但这并不成为对本发明的限制。
实施例1
2-丁基-6-(甲基)-4(1H)-喹唑啉酮
方法A:
将60ml的戊酸酐加入20.0g的2-氨基-5-甲基苯甲酸中。回流加热该混合物18小时后在减压下提浓该混合物。生成的棕色固体残余物溶解于-200ml的30%氢氧化氨溶液和300ml的乙醇的混合物液中。回流加热该混合液5小时后使其冷却至室温。冷却后过滤收集沉淀物。用乙醇及水洗涤该滤饼,在真空下干燥给出8.92g白色固体的喹唑啉酮。
方法B:
使用了B·Baker等人在J·Org·Chem 17 157(1952)上及Sandmeyer在Helv.Chim.Acta 2,234(1919)上所述的过程。
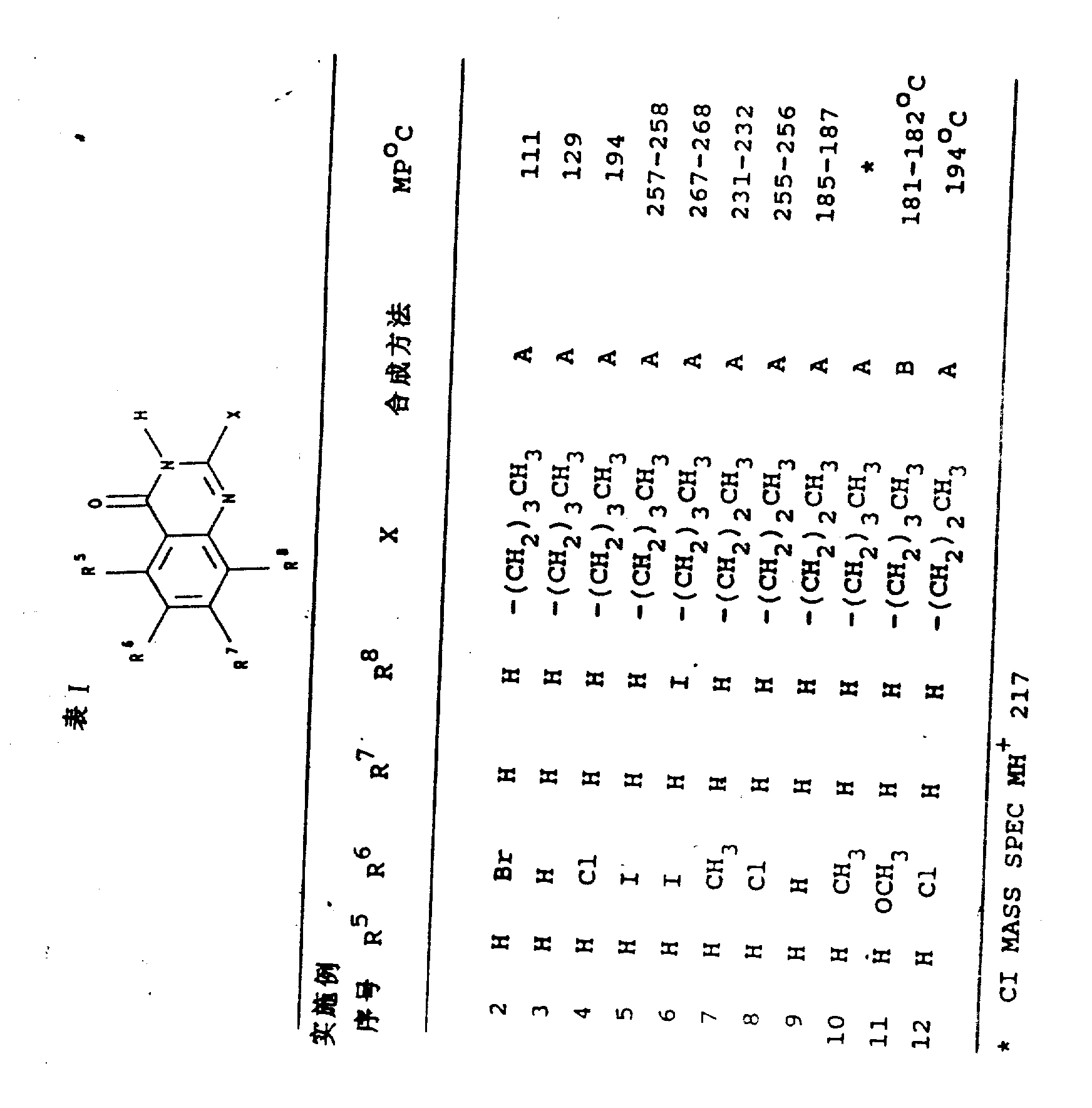
表1中的实施例2-34由使用适当取代了的邻氨基苯甲酸制得。该邻氨基苯甲酸的取代使用了上述合成方法A或B。
实施例13
2-丁基-7-乙羧基-4(1H)-喹唑啉酮
回流含2ml硫酸的100ml的无水乙醇中的2-丁基-7-羧基-4-(1H)-喹唑啉酮的5.0g混合物48小时。真空蒸发该溶剂,该残余物在水和氯仿之间分配。以饱和碳酸氢钠水溶液洗涤该有机层,用无水硫酸钠干燥,过滤蒸发出残余物。将该残余物从乙酸乙酯-己烷中结晶,得4.5g的所希望的产物,其mp为145℃。
实施例14
2-丁基-6-(溴甲基)-4(1H)-喹唑啉酮
将3.39g的N-溴丁二酰亚胺和0.25g的过氧化苯酰加于100ml氯仿中的3.50g的6-甲基喹唑酮的悬浮液。回流加热该反应混合物18小时,趁热过滤。得到所希望的溴化物和起始6-甲基喹唑啉酮的不可分的混合物2.21g的沉淀物,并在不经进一步的提纯下将其用于下面步骤。
实施例15
2-丁基-6-(羟甲基)-4(1H)-喹唑啉酮
将1.0g的碳酸钾加于35ml的二甲基亚砜和20ml的水中的2.0g不纯的2-丁基-6-(溴甲基)-4-(1H)-喹唑啉酮的悬浮液中。回流加热该反应混合液6小时,生成一完全溶液。缓慢冷却至室温后,过滤收集形成的白色沉淀物。以硅胶闪蒸色谱法提纯该滤饼,用9∶1的氯仿-甲醇洗脱,给出0.67g的目的产物的白色固体。
CI MASS SPEC 233(M+H)。
实施例16
2-丁基-1,4-二氯-4-氧代-6-喹唑啉酮-甲醛
将1.7g的吡啶鎓重铬酸盐加入3.5ml的无水N,N-二甲基甲酰胺中的0.3g2-丁基-6-(羟甲基)-4(1H)-喹唑啉酮的溶液中。在室温下搅拌该反应混合液16个小时,倒入125ml的水中。滤去生成的沉淀物,以9∶1的氯仿-甲醇萃取滤液。在硫酸镁上干燥该合并的有机萃取物,将其过滤,真空下浓缩,并与上述沉淀物混合。以硅胶闪蒸色谱法提纯,用1∶1的乙酸乙酯-己烷洗脱,给出0.27g的目的产物。CI MASS SPEC 231(M+H)。
实施例17
2-丁基-6-(1-羟乙基)-4(1H)-喹唑啉酮
将3.0M溴化甲基镁的二乙醚溶液2.61ml滴加入30ml无水四氢呋喃中的0.60g的2-丁基-1,4-二氢-4-氧代-6-喹唑啉酮-甲醛(carboxaldehyde)的、冷却至0℃的溶液中。在0℃下搅拌该反应混合液30分钟后,用氯化铵的水溶液10ml骤冷。用10ml水稀释后,以9∶1的氯仿-甲醇萃取该反应混合液。以硫酸镁干燥该合并的萃取物,过滤、浓缩生成0.64g的目的产物。CI MASS SPEC 247(MH+)。
实施例18
2-丁基-6-(1-羟丙基)-4(1H)-喹唑啉酮。
将溶液四氢呋喃中2.0M溴化乙基镁的1.63ml加入10ml无水四氢呋喃中的0.25g的2-丁基-1,4-二氢-4-氧代-6-喹唑啉酮-甲醛(carboxaldehyde)的冷却至0℃的溶液中。在0℃下搅拌该反应混合液30分钟,以20ml的饱和氯化铵溶液和20ml水骤冷。用9∶1的氯仿-甲醇萃取该反应混合物,硫酸镁干燥,过滤并在真空下蒸发,给出0.26g的目的产物。CI MASS SPEC 261(MH+)。
实施例19
2-丁基-1,4-二氢-4-氧代-6-喹唑啉-甲醛
将2.5ml甲苯中的1.40g的三正丁基锡氢化物的溶液通过喷雾泵,以6小时加入在15ml的四氢呋喃和5ml的N,N-二甲基甲酰胺中的、1.0g的2-丁基-1,4-二氢-4-氧代-6-碘-喹唑啉和0.355g的四个-(三苯基膦)合钯的、在一氧化碳气氛中加热至55℃的溶液中。上述添加完成后,使反应冷却至室温,以盐水稀释,氯仿萃取。真空浓缩该合并的有机物,用乙醚研磨生成的残余物,过滤收集沉淀物并以硅胶闪蒸色谱法提纯,用1∶1的乙酸乙酯-己烷洗脱,给出0.35g的目的产物,其m.p.242-244℃。
实施例20
2-丁基-6-〔(三甲基甲硅烷基)乙炔基〕-4-(1H)-喹唑啉酮
将0.36g的(三甲基甲硅烷基)乙炔加入5.0ml的N,N-二甲基甲酰胺和5.0ml的三乙胺中的、1.0g的2-丁基-1,4-二氢-4-氧代-6-碘-喹唑啉酮和0.043g的氯化双(三苯基膦)合钯(Ⅱ)、5.8mg的碘化亚铜(Ⅰ)的溶液。在45℃下加热生成的反应混合液1小时,再在65℃下加热5小时。冷却后,真空浓缩该反应混合液,以硅胶闪蒸色谱法提纯该残余物,1∶3的乙酸乙酯-己烷洗脱后,生成0.75g的目的产物的白色固体。CI MASS SPEC 299(M+)。
实施例21
2-丁基-6-〔(三甲基甲硅烷基)乙炔基〕-7-氟-4-(1H)-喹唑啉酮。
使用实施例20的实验条件,由7-氟-6-溴-2-丁基-4(1H)-喹唑啉酮制得该化合物,其m.p.为192℃。
实施例22
2-丁基-6-乙炔基-4(1H)-喹唑啉酮
将10.0ml的1.0N氢氧化钠溶液加入20ml的甲醇和20ml的四氢呋喃中的0.70g的2-丁基-6-〔(三甲基甲硅烷基)乙炔基〕-4(1H)-喹唑啉酮溶液中。在室温下搅拌该反应混合液2小时,然后,以5%的盐酸溶液稀释,直至pH为2。过滤收集产生的黄褐色沉淀物,真空干燥,生成0.50g的目的产物。CI MASS SPEC 227(MH+)。
实施例23
6-乙酰基-2-丁基-4(1H)-喹唑啉酮
将0.45g的硫酸汞、0.9ml的水及0.3ml的硫酸加入90ml乙酸中的1.20g的2-丁基-6-乙炔基-4(1H)-喹唑啉酮溶液中。回流加热该反应混合物5小时,冷却至室温,并以150ml水骤冷。真空浓缩生成的混合物,以150ml水稀释、并以6∶1的氯仿-甲醇萃取。在硫酸镁上干燥、过滤并真空浓缩该合并的有机物。以硅胶闪蒸色谱法提纯残余物,以1∶1的乙酸乙酯-己烷洗脱,给出0.67g的目的产物的白色固体。CI MASS SPEC 245(MH+)。
实施例24
2-丁基-6-(1-羟基-1-甲基乙基)-4(1H)-喹唑啉酮。
对冷却至0℃的、250ml无水四氢呋喃中的4.00g的6-乙酰基-2-丁基-4(1H)-喹唑啉酮溶液,滴加入16.4ml的3.0M的溴甲基镁二乙醚溶液。在0℃下搅拌该反应混合液0.5小时,然后,使其升温至室温,再以100ml的饱和氯化铵溶液骤冷。用50ml的水稀释该混合物,以乙酸乙酯萃取。盐水洗涤,无水硫酸镁干燥,过滤并真空浓缩该合并的有机层。用硅胶闪蒸色谱法提纯残余物,以100∶0.25的氯仿-甲醇洗脱,给出2.75g的目的产物的白色固体。CI MASS SPEC261(MH+)。
实施例25
2-丁基-6-(1-羟乙基)-4(1H)-喹唑啉酮
对10.0ml乙醇中的0.102g 6-乙酰基-2-丁基-4(1H)-喹唑啉酮悬浮液,加入0.015g的氢硼化钠。室温下搅拌该反应混合物1.5小时,然后用50ml水稀释。以5∶1氯仿-甲醇萃取该水层,硫酸镁干燥该合并的有机物,过滤并真空浓缩后生成0.103g的目的产物。CI MASS SPEC 247(MH+)。
实施例26
2-丁基-6-乙基-4(1H)-喹唑啉酮
对8ml吡啶中的0.278g2-丁基-6-乙炔基-4(1H)-喹唑啉酮的悬浮液,加入0.080g的5%载持在硫酸钡上的钯。在氢气氛中搅拌该反应混合液48小时,过滤,真空浓缩。以硅胶闪蒸色谱法提纯残余物,以乙酸乙酯-己烷洗脱,给出0.179g的目的产物。CI MASS SPEC 231(MH+)。
实施例27
2-丁基-1,4-二氢-4-氧代-6-喹唑啉羧酸甲酯
对25ml甲醇和5mlN,N-二甲基甲酸胺中的1.00g2-丁基1,4-二氢-4-氧代-6-碘代喹唑啉和6.0ml三乙胺的溶液,加入0.275g的氯化双(三苯基膦)合钯(Ⅱ)。在一氧化碳气氛下回流加热该反应混合液16小时,然后冷却,真空浓缩。以硅胶闪蒸色谱法提纯残余物,以1∶1的乙酸乙酯-己烷洗脱,给出0.389g目的产物的白色固体。CI MASS SPEC 261(MH+)。
实施例28
2-丁基-6-(1-羟基-1-甲基乙基)-4(1H)-喹唑啉酮
对冷却至0℃的、5ml无水四氢呋喃中的0.075g2-丁基-1,4-二氢-4-氧代-6-喹唑啉羧酸甲酯溶液,滴加入3.0M溴甲基镁的二乙醚溶液0.51ml。在0℃下搅拌该反应混合物0.5小时,然后在室温下搅拌1小时。再以饱和氯化铵溶液10ml骤冷。以10ml水稀释生成的反应混合液,以乙酸乙酯萃取。以硫酸镁干燥该合并的有机物,过滤并真空浓缩。以硅胶闪蒸色谱法提纯残余物,用100∶0.25的氯仿-甲醇洗脱,生成0.055g的目的产物的白色固体,其m.p.为190-192℃。
实施例29
2-丁基-6-(1-甲基乙烯基)-4(1H)-喹唑啉酮
对冷却至-78℃的、30ml无水四氢呋喃中的3.66g溴化甲基三苯基鏻的悬浮液,滴加入1.73m正丁基锂鎓己烷溶液5.9ml。滴加完成后,使该反应混合液升温至室温并搅拌15分钟,直至所有的溴化鏻溶解。然后冷却该反应混合液至-78℃,加入15ml无水四氢呋喃中的6-乙酰基-2-丁基-4(1H)-喹唑啉酮悬浮液。使该反应混合物升温至室温,搅拌24小时后,以饱和的氯化铵溶液骤冷。以10ml水稀释后,用氯仿萃取该含水层,硫酸镁干燥该合并的有机物,过滤并真空浓缩。以硅胶闪蒸色谱法提纯残余物,以1∶2的乙酸乙酯-己烷洗脱后,给出0.23g目的产物的白色固体。CI MASS SPEC 234(MH+)。
实施例30
2-丁基-6-(羟基苯基甲基)-4(1H)-喹唑啉酮
对冷却至0℃的100ml四氢呋喃中的2.00g2-丁基-1,4-二氢-4-氧代-6-喹唑啉-甲醛(carboxaldehyde)的搅拌溶液,加入13.0ml2.0M的苯基锂,并继续搅拌1小时。停止冷却,使反应物升温至室温,在室温下搅拌30分钟,用饱和氯化铵溶液稀释,再以乙酸乙酯萃取该反应混合液,干燥并蒸发该有机层至一残余物,以硅胶色谱法提纯,以0.25∶100的甲醇-氯防洗脱。给出0.932g的目的产物。CI MASS SPEC 309(MH+)。
实施例31
2-丁基-6-(1-羟乙基)-3-〔(2′-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1′-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮
将2.50g2-丁基-6-(1-羟乙基)-4(1H)-喹唑啉酮、6.79g5-〔(4′-(溴甲基)〔1,1′-二苯基〕-2-基〕-1-(三苯基-甲基)-1H-四唑及4.20g无水碳酸钾在225ml的无水丙酮中的悬浮液回流加热16小时。使该反应混合物冷却至室温,过滤,真空蒸发滤液。由硅胶上高压液相色谱法提纯残余物,1∶2的乙酸乙酯-己烷洗脱,得到4.25g目的产物的白色固体,FABM+H723。
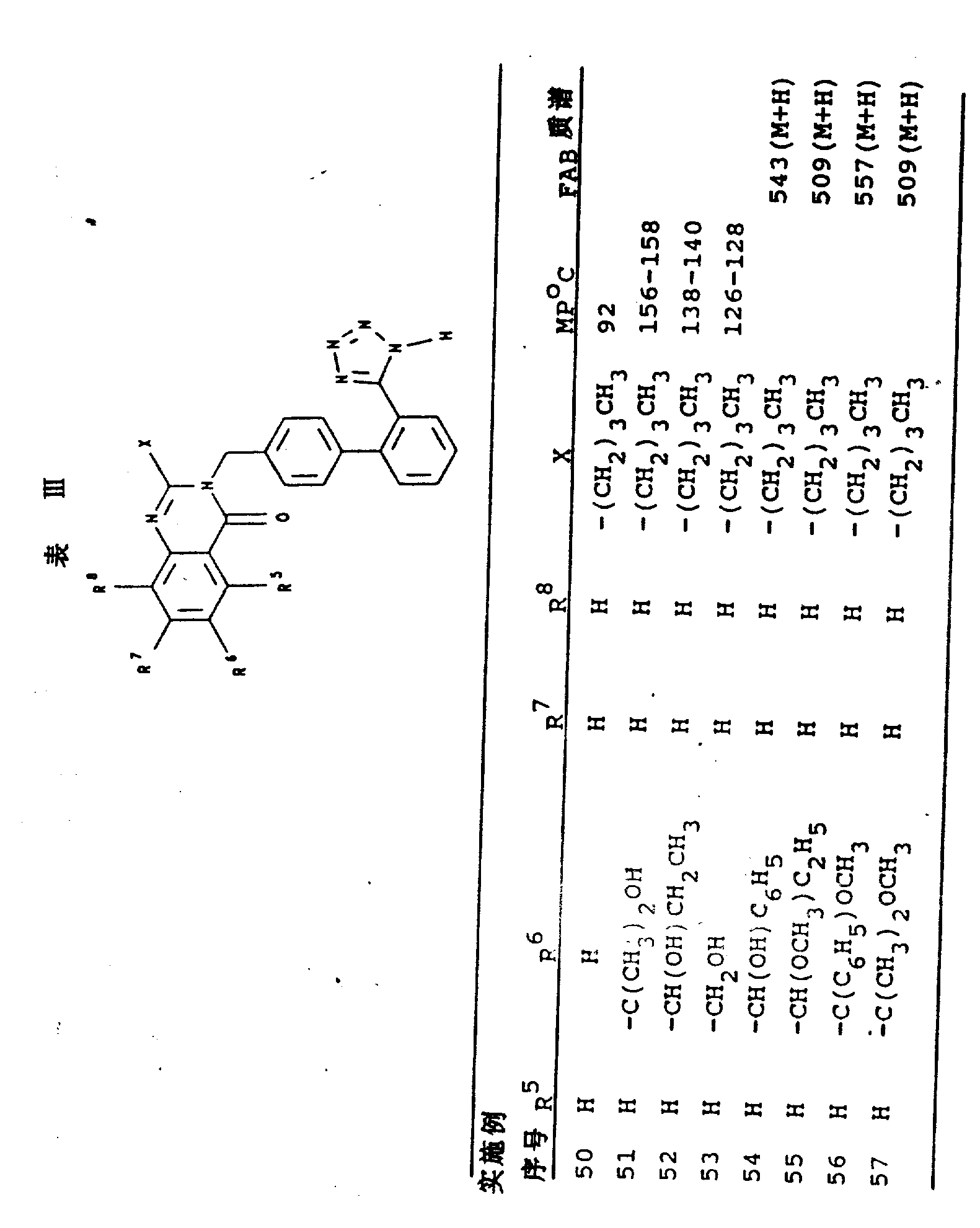
在基本如同实施例31的烷基化条件下,从合适的取代喹唑啉酮起始材料,制得表Ⅱ中的实施例32-48。
实施例49
2-丁基-6-(1-羟乙基)-3-〔〔2′-(1H-四唑-5-基)-〔1,1′-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮
对2.00g2-丁基-6-(1-羟乙基)-3-〔〔2′-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1′-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮在70ml的3∶1丙酮-水悬浮液,加入一滴5%的盐酸水溶液。然后,回流加热该混合物16小时。冷却后,真空浓缩该反应混合物,以硅胶闪蒸色谱法提纯残余物,以9∶1的氯仿-甲醇洗脱,得到0.915g目的产物的白色固体,其m.p.146-147℃。
在基本如同实施例49的条件下,从合适的取代喹唑啉酮起始材料制备表Ⅲ中的实施例50-57。
实施例58
6-〔1-(乙酰氧基)乙基〕-2-丁基-3-〔〔2′-(1H-四唑-5-基)〔1,1′-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮
在室温下将2滴无水吡啶加到1.00g2-丁基-6-(1-羟乙基)-3-〔〔2′-(1H-四唑-5-基)-〔1,1′-二苯基〕4-基〕甲基-4(3H)-喹唑啉酮在2.0ml的乙酸酐中的悬浮液,在室温下搅拌该反应混合物16小时后,真空蒸发。以硅胶闪蒸色谱法提纯残余物,以95∶5的氯仿-甲醇洗脱,生成0.059g目的产物的白色固体。
FAB MASS SPEC 523(M+H)。
实施例59
2-丁基-6-(1-甲氧基乙基)-4-〔〔2′-(1H-四唑)-5-基)〔1,1′-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮
将0.300g2-丁基-6-(1-甲氧基乙基)-3-〔〔2′-〔1-(三苯基甲基)-1H-四唑-5-基〕-〔1,1′-二苯基〕-4-基〕-甲基-4(3H)-喹唑啉酮在20ml、含1滴5%盐酸的3∶1丙酮-水中的混合液回流加热16小时,蒸发出残余物。以硅胶闪蒸色谱法提纯残余物,以95∶5的氯仿-甲醇洗脱,提供0.171g白色固体的产物,其m.p.154-156℃。
实施例60
2-丁基-6-(1-甲氧基乙基)-3-〔〔2′-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1′-二苯基〕-4-基〕-甲基-4(3H)-喹唑啉酮
将矿物油中分散有0.044g的60%氢化钠及5.0ml无水四氢呋喃中的0.345ml的甲基碘的悬浮液,在其一部分中加入0.400g的2-丁基-6-(1-羟乙基)-3-〔〔2′-〔1-三苯基甲基)-1H-四唑-5-基〕-〔1,1′-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮。在室温下搅拌该反应混合液18小时后,倒于一硅胶板上。以1∶3的乙酸乙酯-己烷洗脱,蒸发含所需产物的馏份,得到0.356g白色固体。FAB MASS SPEC 737(M+H)。
实施例61
2-丁基-6-(1-羟基-1-甲基乙基)-3-〔〔2′-(1H-四唑-5-基)〔1,1′-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮单钠盐
将0.400g2-丁基-6-(1-羟基-1-甲基乙基)-3-〔〔2′-(1H-四唑-5-基)〔1,1′-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮单钠盐、10ml的甲醇及0.810ml 1N的氢氧化钠的混合物在室温下搅拌1小时,真空蒸发生成残余物。用醚研磨该残余物,过滤后,空气干燥滤饼,给出固体的目的产物。FAB MASS SPEC 517(M+H)。
实施例62
2-丁基-6-(羟甲基)-3-〔〔2’-(1H-四唑-5-基)〔1,1’-二苯基〕-4-基-4(3H)-喹唑啉酮单钠盐
按实施例182的方法,使用2-丁基-6-(1-羟基-1-甲基乙基)-3-〔〔2’-(1H-四唑-5-基)〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮,得到本实施例的产物。
实施例63
2-丁基-6-(1-甲氧基-1-甲基乙基)-3-〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基-4(3H)-喹唑啉酮
将0.76ml的甲基碘加入0.049g的60%油分散的氢化钠在4.5ml四氢呋喃中的悬浮液,再加入0.45g的2-丁基-6-(1-羟基-1-甲基乙基)-3-〔〔2’-(1H-四唑-5-基)-〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮。在室温下搅拌该反应混合物24小时,然后再加入0.05g的60%氢化钠及0.80ml的甲基碘。再在室温下搅拌该反应混合物24小时后,以饱和氯化铵溶液骤冷,以醚萃取。以MgSO4干燥该有机物,过滤并真空浓缩。由硅胶闪蒸色谱法提纯该残余物,以1∶5乙酸乙酯/己烷洗脱,得到0.397g目的产物的白色固体。FAB MASS SPEC 751(M+H)。
实施例64
2-丁基-6-(1-甲氧基-1-甲基乙基)-3-〔〔2’-(1H-四唑-5-基)〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮。
使用实施例111的条件和0.397g的2-丁基-6-(1-甲氧基-1-甲基乙基)-3-〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基-4(3H)-喹唑啉酮,给出0.188g目的产物的白色固体,接着由硅胶色谱法提纯以50∶50∶5∶0.1的乙酸乙酯/己烷/甲醇/乙酸洗脱,制得本实施例的产物。FAB MASS SPEC 509(M+H)。
实施例65
2-丁基-6-(甲氧基苯基甲基)-3-〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基-(3H)-喹唑啉酮。
将0.851g60%的氢化钠加入到0.398ml的甲基碘在5.0ml的THF中的溶液,再加入0.500g的2-丁基-6-(羟基苯基甲基)3-〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮。在室温下搅拌该反应混合物16小时,以饱和NH4Cl溶液骤冷,用醚萃取。在MgSO4上干燥该有机物,过滤并真空浓缩。由硅闪蒸色谱法提纯残余物,以乙酸乙酯/己烷(1∶5)洗脱,得到0.434g目的产物的白色固体。FAB MASS SPEC 799(M+H)
实施例66
2-丁基-6-(甲氧基苯基甲基)-3-〔〔2’-(1H-四唑-5-基)〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮。
由使用实施例49的条件和0.413g2-丁基-6-(甲氧基苯基甲基)-3-〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基〕-(3H)-喹唑啉酮,给出0.192g目的产物的白色固体,再经以50∶50∶5∶0.1的乙酸乙酯/己烷/甲基醇/乙酸洗脱并用硅胶色谱法提纯制得本实施例的产物。FAB MASS SPEC 557(M+H)。
实施例67
2-丁基-6-(1-甲氧基丙基)-3-〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮。
对0.556ml甲基碘在5.0ml THF中的溶液,加入0.071g的60%氢化钠,再加入0.657g2-丁基-6-(1-羟丙基)-3-〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基〕4(3H)-喹唑啉酮。在室温下搅拌该反应混合物16小时后,以饱和氯化铵溶液骤冷,用醚萃取。在MgSO4上干燥该有机物质,过滤并在真空下浓缩。以硅胶闪蒸色谱法提纯该残余物,以1∶5乙酸乙酯/己烷洗脱,得到0.59g的目的产物的白色固体。FAB MASS SPEC 751(M+H)。
实施例68
2-丁基-6-(1-甲氧基丙基)-3-〔〔2’-(1H-四唑-5-基)〔〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮。
由使用实施例111的条件和0.58g2-丁基-6-(1-甲氧基丙基)-3-〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮,给出0.326g目的产物的白色固体,再经由50∶50∶5∶0.1的乙酸乙酯/己烷/甲基醇/乙酸洗脱并用硅胶色谱法,制得本实施例的产物。FAB MASS SPEC 509(M+H)。
实施例69
1-氨基-3-氟-4-溴-苯甲酸
对在250ml的冰醋酸中的3.0g的4-氟邻氨基苯甲酸的溶液,缓慢加入10ml乙酸中的3.2g溴的溶液。在室温下搅拌该反应混合物8小时后,倒入500ml水中。滤去生成的沉淀物,从甲醇中结晶滤饼,给出3.4g目的产物,其m.p.180℃。
实施例70
6-溴-2-丁基-7-氟-4(1H)-喹唑啉酮
按实施例1的方法,使用3.0g1-氨基-3-氟-4-溴-苯甲酸,100ml的戊酸酐和200ml的氢氧化铵,得到本实施例的目的产物,其m.p.225℃。
实施例71
2-丁基-7-氟-6-〔(三甲基硅烷基)乙炔基〕喹唑啉酮4(1H)-喹唑啉酮。
使用实施例20的方法和6-溴-2-丁基-7-氟-4(1H)-喹唑啉酮,得到本实施例的产物,其m.p.为192℃。
实施例72
2-丁基-6-乙炔基-7-氟-4(1H)-喹唑啉酮
在60℃下加热1.0g的2-丁基-7-氟-6-〔(三甲基硅烷基)乙炔基〕-4(1H)-喹唑啉酮,20ml的1N氢氧化钠和25ml的甲醇的混合物5小时,然后真空蒸发。将残余物溶于100ml水中并予以酸化。收集干燥生成的固体物,给出700mg目的产物的黄色固体,其m.p.为218℃。
实施例73
3-〔(4-溴苯基)甲基〕-2-丁基-6-(1-羟基-1-甲基乙基)-4(3H)-喹唑啉酮
对1.37g2-丁基-6-(1-羟基-1-甲基乙基)-4(1H)-喹唑啉酮在115ml丙酮中的溶液,加入1.58g的4-溴苄基溴化物和2.18g的无水碳酸钾。回流加热产生的悬浮液16小时。然后,使反应混合物冷却至室温,过滤并真空浓缩该滤液。由以乙酸乙酯/己烷(1∶3)洗脱并用HPLC提纯残余物,提供目的产物。
在大致如同实施例73的烷基化条件下,从合适的取代喹唑啉酮作起始材料制备表Ⅵ中的实施例74-84。
实施例85
3-〔(4-溴苯基)甲基〕-2-丁基-6-(1-甲氧基-1-甲基乙基)-4(3H)-喹唑啉酮
对0.186g60%氢化钠和2.90ml碘代甲烷在THF的溶液,在室温下加入1.00g的3-〔(4-溴苯基)甲基〕-2-丁基-6-(1-羟基-1-甲基乙基)-4(3H)-喹唑啉酮。在室温下,搅拌该反应混合物过夜,然后,用氯化铵溶液骤冷,用水稀释,用醚萃取该含水层,在MgSO4干燥该合并的有机物,过滤并在真空下浓缩。由以乙酸乙酯/己烷(1∶3)洗脱,用闪蒸色谱法提纯残余物,提供目的产物。
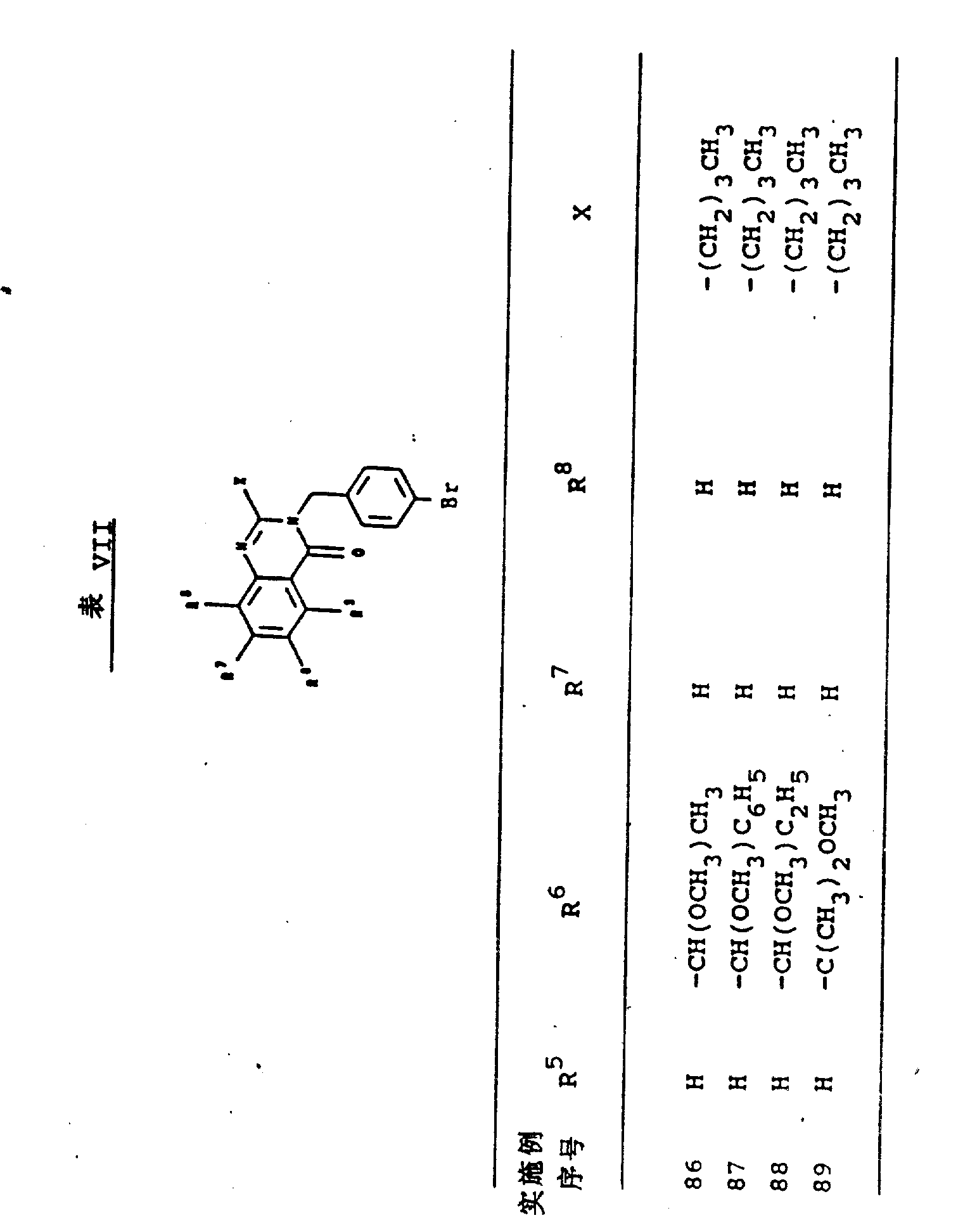
在基本上如同实施例85的烷基化条件下,从合适的取代喹唑啉酮起始材料制备了表Ⅶ中的实施例86-89。
实施例90
2-丁基-6-(1-甲氧基-1-甲基乙基)-3-〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮
对0.41g的镁镟片(屑)在50ml THF中的悬浮液,加入一催化量的碘,再加入在10ml THF中的1.00g的5-(2-溴苯基)-1-(三苯基甲基)-1H-四唑。回流加热该反应混合液,直至如碘的颜色消失所显示出的,引发格利雅形成反应。接着,以一足以保持缓慢回流的速度,在反应液中加入剩余的70ml THF中的6.94g溴化物。添加完该溴化物后,在室温下搅拌反应混合物4小时。
按Kumada(Tet.Letters,52,5319(1981)的方法,对4.08g3-〔(4-溴苯基)甲基〕-2-丁基-6-(1-甲氧基-1-甲基乙基)-4(3H)-喹唑啉酮和71mg氯化1,4-双(二苯基膦基)丁烷合钯(Ⅱ)在100ml THF中的溶液,加入上述的格利雅溶液,回流加热该生成的溶液2小时,冷却至室温后,以水和稀氢氧化钠溶液骤冷该反应混合物,然后,以醚萃取。在MgSO4上干燥该合并的有机物,过滤并在真空下浓缩。由以乙酸乙酯/己烷(1∶3)洗脱,用硅胶闪蒸色谱法提纯残余物,提供目的产物。
在基本上如同实施例90的偶合条件,从合适的取代喹唑啉酮起始作材料制备表Ⅷ中的实施例91-94。
实施例95
3-〔(4-溴苯基)甲基〕-2-丁基-6-〔-〔〔(1,1-二甲基乙基)二甲基硅烷基〕氧〕-1-甲基乙基〕-4(3H)-喹唑啉酮
对1.00g3-〔(4-溴苯基)甲基〕-2-丁基-6-(1-羟基-1-甲基乙基)-4(3H)-喹唑啉酮在2.0mlDMF中的溶液,加入0.438g叔丁基二甲基硅烷基氯化物,再加入0.4g的咪唑。在室温下搅拌该反应混合液18小时后,以水稀释,用醚萃取。在MgSO4上干燥该合并的醚萃取物,过滤,在真空下浓缩。由以乙酸乙酯/己烷(1∶5)洗脱,用闪蒸色谱法提纯残余物,生成目的产物的白色固体。
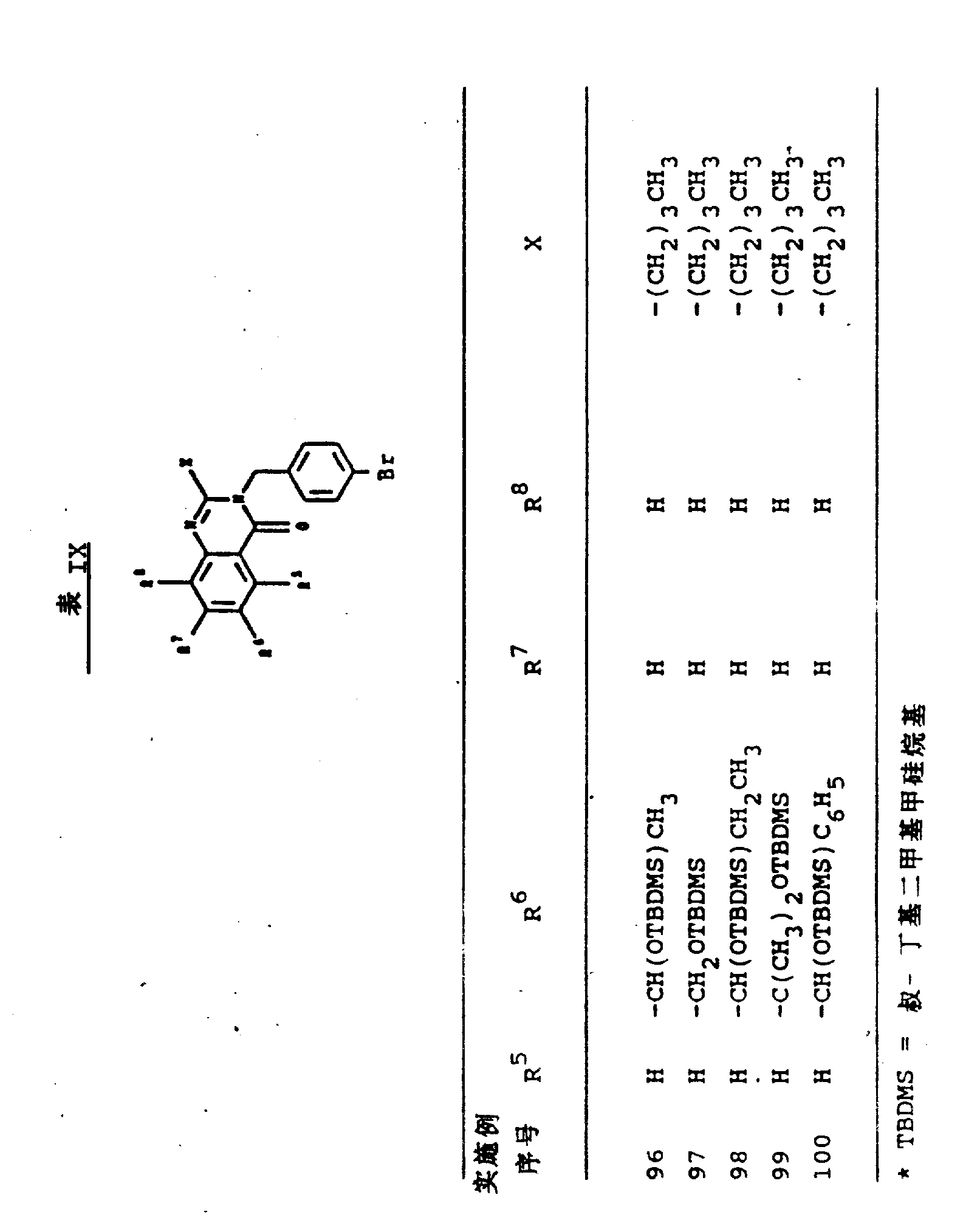
在基本上如同实施例95的条件下,从合适的取代喹唑啉酮起始材料制备了表Ⅸ中的实施例69-100。
实施例101
2-丁基-6-〔1-〔〔(1,1-二甲基乙基)二甲基甲硅烷基)氧基〕-1-甲基乙基〕-3-〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮
对0.41g镁镟屑(片)在50mlTHF中的悬浮液,加入一催化量的碘,再加入10mlTHF中的1.00g5-(2-溴苯基)-1-(三苯基甲基)-1H-四唑。回流加热该反应混合液,直至如碘的颜色消失所显示的,引发格利雅试剂形成反应。再以一足以维持缓慢回流的速率,将剩余70mlTHF中的6.94g的溴化物加入反应。然后,在加完该溴化物后,在室温下搅拌该反应混合液4小时。
根据Kumade(Tet.Letters,52,5319(1981))的方法对5.00g的3-〔(4-溴苯基)甲基〕-2-丁基-6-〔1-〔〔(1,1-二甲基乙基)二甲基甲硅烷基〕氧〕-1-甲基乙基〕-4(3H)-喹唑啉酮和71mg1,4-双(二苯膦)丁烷合钯(Ⅱ)氯化物在100mlTHF中的溶液,加入上述的格利雅溶液,回流加热生成的溶液2小时。冷却至室温后,用水和稀氢氧化钠溶液骤冷该反应混合物,然后,用醚萃取。在MgSO4上干燥该合并的有机物,过滤并在真空下浓缩,由以乙酸乙酯/己烷(1/∶3)洗脱,用硅胶色谱法提纯残余物,提供目的产物。
在基本上如同实施例101的条件下,从合适的取代喹唑啉酮起始材料制备了表Ⅹ中的实施例102-106。
实施例107
2-丁基-6-(1-羟基-1-甲基乙基)-3-〔〔2’-〔1-三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮
对0.50g2-丁基-6-〔1-〔〔(1,1-二甲基乙基)二甲基甲硅烷基〕氧〕-1-甲基乙基〕-3〔〔2’-〔1-(三苯基甲基)-1H-四唑-5-基〕〔1,1’-二苯基〕-4-基〕甲基〕-4(3H)-喹唑啉酮在15.0mlTHF中的溶液,加入0.88ml1.0M的氟化四丁基铵在THF中的溶液。在室温下搅拌该反应混合物1小时后,倒入100ml水中。用醚萃取该水层,盐水洗涤该合并的有机物,在MgSO4上干燥,过滤并在真空下浓缩。由以乙酸乙酯-己烷(1∶3)洗脱,用硅胶闪蒸色谱法提纯残余物,提供目的产物。
在基本上如同实施例107的条件下,从合适的取代喹唑啉酮起始材料制备了表Ⅺ中的实施例108-112。
实施例113
5-(2-溴苯基)-1-(三甲基甲锡烷基)-1H-四唑
对1.50g邻溴苄腈在8.0ml甲苯中的溶液,加入1.70g三甲基锡叠氮化合物。回流加热该反应混合物18小时,使其冷却至室温。过滤分离出生成的白色沉淀物,并不经提纯用于下一步(反应)。
实施例114
5-(2-溴苯基)-1H-四唑
对1.0g5-(2-溴苯基)-1-(三甲基甲锡烷基)-1H-四唑在甲苯/THF(10∶1)的溶液,在室温下,通过一鼓泡装置加入HCl气体。出现沉淀物之后再继续鼓入气体5分钟,过滤分离出该固体并以己烷洗涤。
实施例115
5-(2-溴苯基)-1-(三苯基甲基)-1H-四唑
对0.50g5-(2-溴苯基)-1H-四唑在20mlCH2Cl2中的溶液加入0.65g的三苯基氯甲烷;再加入0.37ml的三乙胺。回流该溶液2.5小时,冷却至室温后,水洗,在MgSO4上干燥并在真空下浓缩。以乙酸乙酯/己烷(1∶5)洗脱,用闪蒸色谱法提纯残余物,提供目的产物。
实施例116
2-丁基-6-(1-甲氧基-1-甲基乙基)-3-〔〔2’-(1H-四唑-5-基)〔1,1’-二苯基)-4-基〕甲基〕-4(3H)-喹唑啉酮,钠盐
对2.488g从实施例57中分离出的四唑在60ml甲醇中的溶液加入5.036ml 1N的氢氧化钠溶液。在室温下搅拌该混合物1小时后真空浓缩。用己烷滴定该残余物,过滤,真空干燥,提供2.29g白色固体产物。
实施例117
2-丁基-6-(1-甲氧基乙基)-4-〔〔2’-(1H-四唑)-5-基)〔1,1’-二苯基〕-4-基〕甲基-4(3H)-喹唑啉酮,钠盐
对2.156g从实施例59中分离出的四唑在60ml甲醇中的溶液,加入4.359ml 1N的氢氧化钠溶液。在室温下搅拌该混合液1小时后真空浓缩。己烷滴定在该残余物,过滤,真空干燥,提供2.09g白色固体产物。
实施例118
2-丁基-6-(甲氧基甲基)-4-〔〔2’-(1H-四唑)-5-基)〔1,1’-二苯基)-4-基〕甲基-4(3H)-喹唑啉酮
对0℃的、从实施例53所得的NaH(500mg)和伯醇的搅拌过的在干THF(35ml)中的溶液,加入CH3I(1ml)。在室温下搅拌该反应混合液4小时。然后,将该反应混合液小心倒于碎冰上,氯仿萃取。用水充分洗涤有机层,干燥并浓缩。将所得的海绵状固体溶于丙酮(25ml),加入5N HCl(3ml)。在室温下搅拌该反应混合液3小时。然后浓缩该反应混合液,柱色谱法提纯产物。产率:8.5mg;m.p.85℃。
本发明的新颖化合物的性能示于以下的体外试验中。用于本发明的有代表性化合物的试验结果示于表Ⅳ。
血管紧张素Ⅱ拮抗剂体外试验
在筛选中所用的血管紧张素Ⅱ受体的来源是大鼠肾上腺皮质微粒体。将皮质放在冰冷却的蔗糖缓冲液(0.2m蔗糖、1mmEDTA、10mm Trizma碱,pH7.4)中,在冷的磨砂玻璃组织研磨器中研成匀浆。匀浆于300xg离心10分钟。将所得的上清液倾倒通过粗平布,并于12000xg离心3分钟,然后将清液1000,000xg离心60分钟。将微粒再悬浮于试验缓冲液(0.25%牛血清白蛋白、5mm MgCl2、50mmTrizma碱,pH7.2)。在没有或有化合物存在(40μM终浓度)下,培养等分的新鲜微粒体以进行结合试验。10分钟后,将3H-血管紧张素Ⅱ加到每个管中(2nM终浓度),于27℃培养60分钟。加入3ml无白蛋白的冷的试验缓冲液以终止反应,并迅速通过用试验缓冲液预先湿润的玻璃纤维滤纸来分离结合的和游离的放射活性。再用3ml缓冲液淋洗二次后,将滤纸放在闪烁液中,在闪烁计数器中计数以测定捕获的放射活性。置换50%标记血管紧张素的化合物被认为是活性化合物,然后在浓度-相应实验中进行评估,以测得IC50值。结果见表Ⅳ。
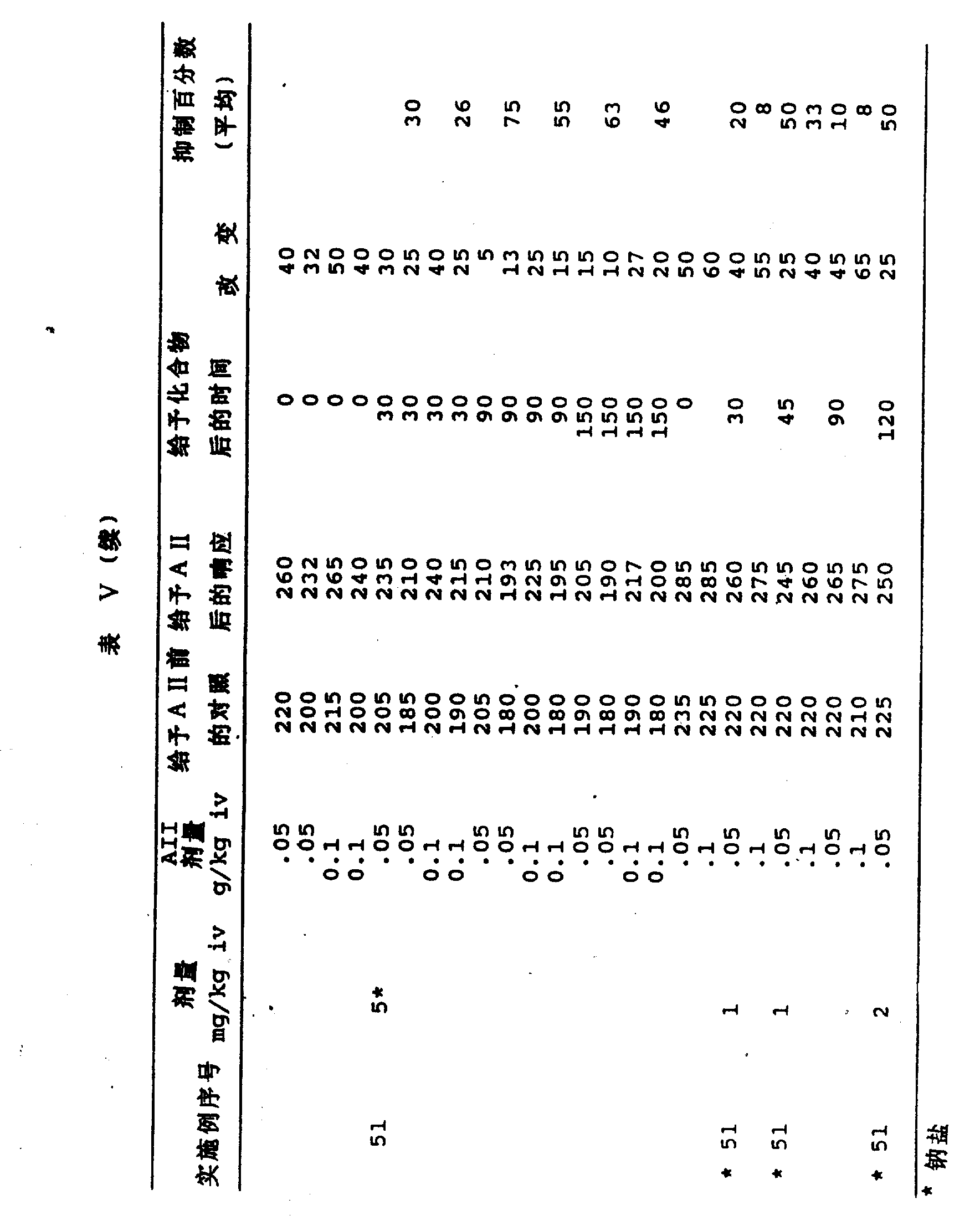
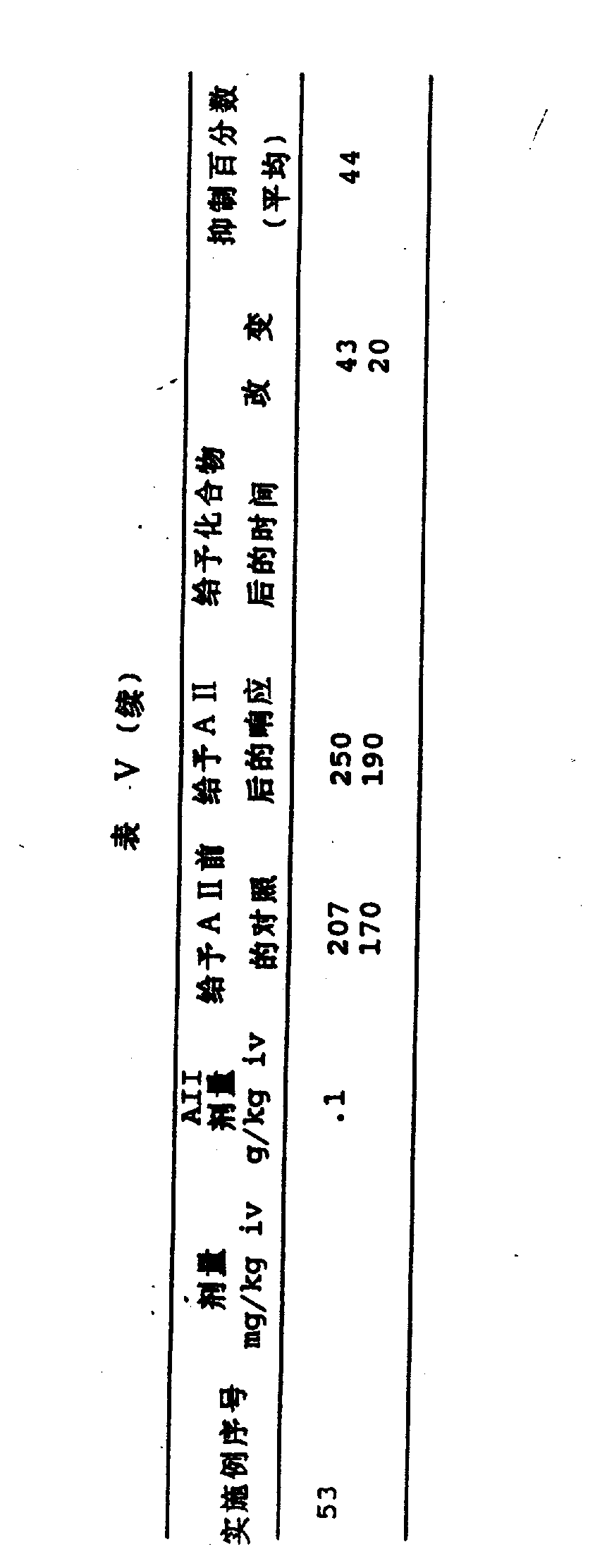
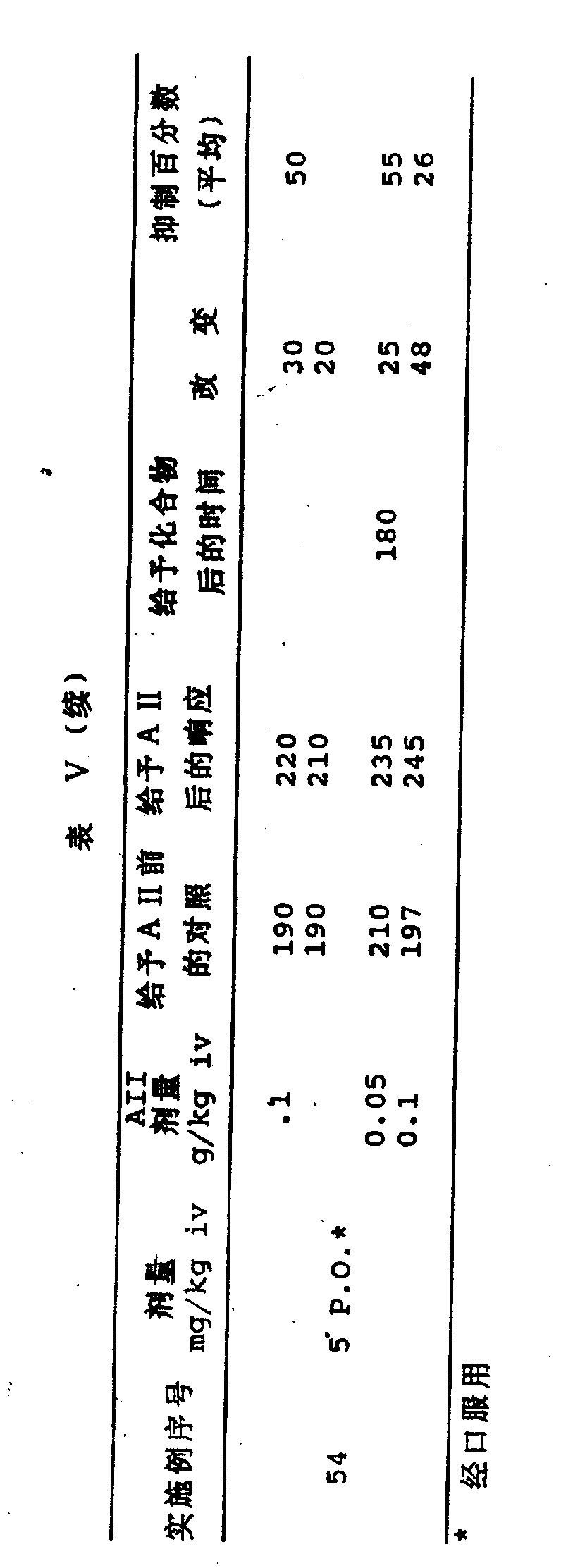
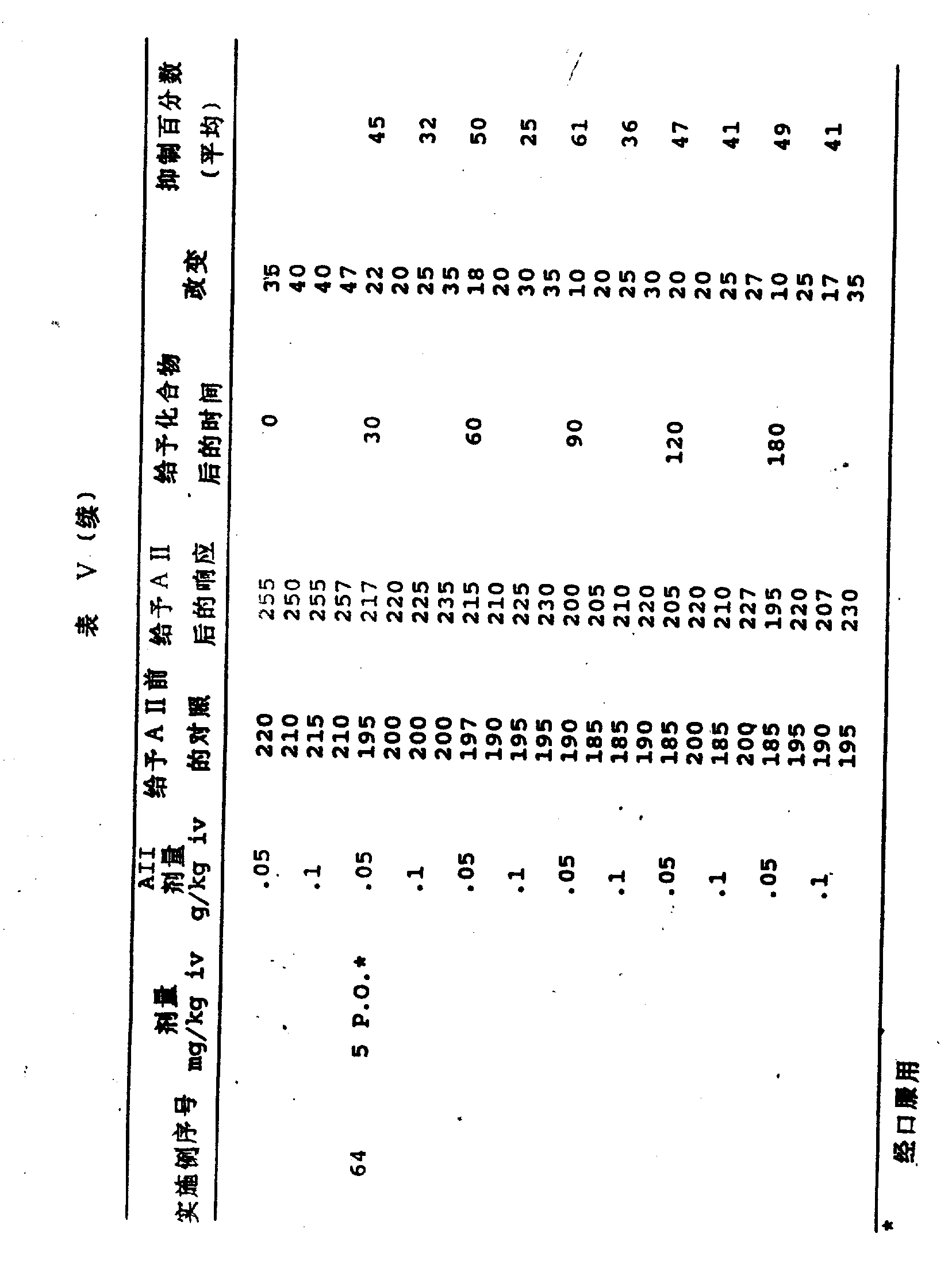
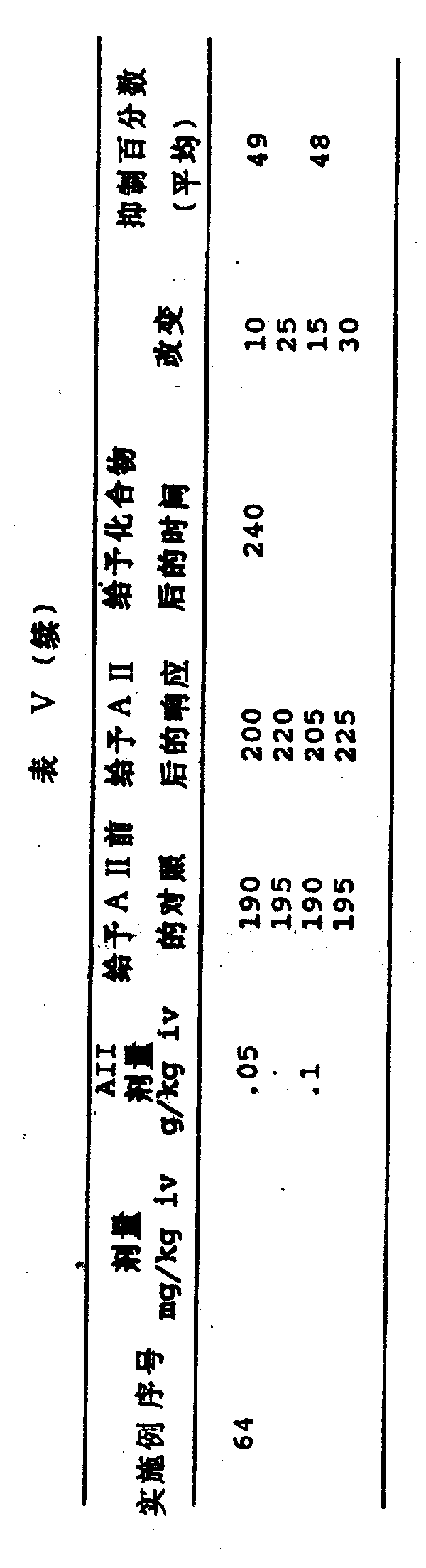
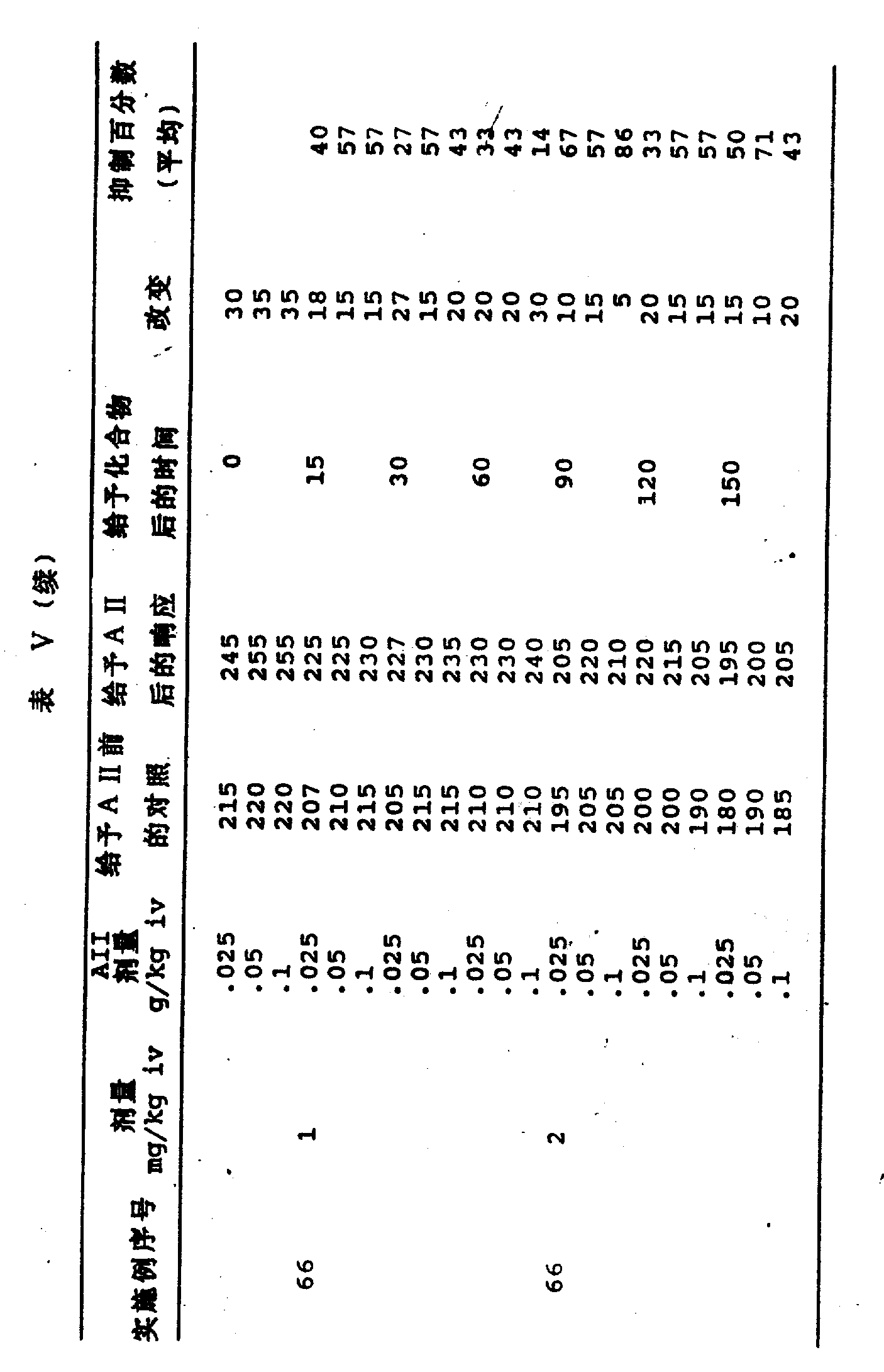
本发明的化合物抑制AⅡ的作用。对大鼠投予本发明的化合物,然后改用血管紧张素Ⅱ,可看到血管升压反应的阻断。对本发明的代表性化合物的此项试验结果见表Ⅴ。
AII功击
清醒的雄性Okamoto-Aoki自发性高血压大鼠,16-20周龄,体重约330g,购得Charles River Labs(Wilmington,MA)。用松紧带将清醒大鼠仰位固定,尾根部用2%普鲁卡因皮下浸润作局部麻醉。分离腹侧尾动脉和静脉。将聚乙烯(PE)制成的10-20管系(用热融合在一起)导管分别插入下腹主动脉和腔静脉。此管是安全的,肝素化的(1,000I.U./ml),密闭的。缝合伤口。将动物放在处于垂直位的塑料约束笼内。导管连接在Statham P23D6压力传感器上,用Gould Brush记录仪记录10-15分钟搏动的血压(Chan et al.,Drug Development Res.,18:75-94,1989)。
将血管紧张素Ⅱ(人类,Sigma Chem,Co.,St,Louis,Mo)0.05和0.1μg/kg给所有大鼠静脉注射(ⅳ)(给药前反应)。然后给各组大鼠静脉注射、腹腔注射或口服受试化合物、赋形剂或已知的血管紧张素Ⅱ拮抗剂。在给予化合物或赋形剂后30、90和150分钟,每只大鼠再给予二个剂量血管紧张素Ⅱ。以收缩压(mmHg)的升高测得;知管紧张素Ⅱ的血管升压响应。将给化合物前血管紧张素Ⅱ对每只大鼠的血管升压响应(收缩压的升高)作为100%,计算化合物对血管紧张素Ⅱ血管升压响应的拮抗或阻断百分数。化合物若在30mg/kg静脉注射时拮抗至少有50%的响应,就被认为有活性。
结果见表Ⅴ和图1。
在清醒的主动脉缩窄的血管紧张素
原酶/血管紧张素Ⅱ依赖性肾性高
血压大鼠上的抗高血压效应
按照Chan等(Drug Development Res,18:75-94,1989)报告的根据Rojo-ortega和Genest(Cam,J.Physio Pharmacol 46:883-885,1968)和Fernandes等(J.Lab,Clin.Med.87:561-567,1976)的方法加以改良的外科常规,通过在两侧肾动脉始端之间完全结扎主动脉而引起高血压。雄性Sprague-Dawley大鼠(Charles River Labs,Inc.,Wilmington,MA),体重350-400g,用甲己炔马比妥钠(brevital sodium,Eli Lilly and Co)60mg/kg腹腔注射进行麻醉。在平行于肋骨的左侧切开,用3-0号丝线在两侧肾动脉始端之间完全结扎主动脉。缝合伤口,将动物放回各自的笼子。大鼠在主动脉缩窄后第7天使用。用松紧带将大鼠仰位固定,轻轻地固定头部使其不动。用2%利多卡因皮下浸润,进行颈腹测局部麻醉。分离左颈动脉,插入一根PE50导管,此导管随后被连接到statham p23D6压力传感器-Beckman Dynagraph记录系统。在某些研究中,为了长期监测血压而将导管从颈背部引出。记录进行15-20分钟,给大鼠服用受试化合物或赋形剂(生理盐水)。给药后,连续监测血压。结果见图2-7。
为了上述效用而使用化合物时,它们可与一种或一种以上药学上可接受的载体(如溶剂、稀释剂等)合用,以片剂、胶囊剂、可分散粉剂、颗粒剂的形式,或混悬剂(含有例如0.05-5%悬浮剂)、糖浆剂(含有例如约10-50%蔗糖)和酏剂(含有例如约20-50%乙醇)等形式口服给药,或以灭菌可注射溶液或混悬液(在等渗介质中含约0.05-5%悬浮剂)的形式非经口给药。这样的药制剂可含有从约0.05直至90%(重量)活性成分,合用载体,更通常地在约5%和60%之间(重量)活性成分。
所用活性成分的有效剂量可随所用的具体化合物、给药方式和要治疗的病情的严重性而改变。然而,一般来说,当本发明化合物以每天约0.5-约500mg/kg动物体重的剂量(最好分2-4次/天给药),或以持续释放的方式给予时,可获得满意的结果。对于大多数大哺乳动物,每天总剂量为约1-100mg,以约2-80mg为较佳。适合于内服的剂量形式包括约0.5-500mg活性化合物与药学上可接受的固体或液体载体密切混合。这一剂量形式可被调整成提供最适的治疗反应。例如,按治疗情况的紧急程度所示,每天可给几个分开的剂量,或可按比例减少剂量。
这些活性化合物可经口服,也可经静脉内、肌肉或皮下途径给药。按适合于活性成分的本质和所需的具体给药方式,固体载体可包括淀粉、乳糖、磷酸二钙、微晶纤维素、蔗糖和高岭土,而液体载体包括灭菌水、聚乙二醇、非离子型表面活性剂和食用油,例如玉米、花生和芝麻油。在药物组合物的制备上常规采用的佐剂可有益地包括诸如矫味剂、着色剂、防腐剂和抗氧化剂,如维生素E、抗坏血酸、BHT和BHA。
从便于制备和给药的观点出发,较佳的药物组合物为固体组合物,特别是片剂和硬充填或液体充填的胶囊剂。化合物的口服给药是可取的。
这些活性化合物也可非胃肠道或腹腔内给药。可在适当混有表面活性剂(如羟丙基纤维素)的水中这些以游离碱或药理学上可接受的盐的形式存在的化合物配制成溶液或混悬剂。还可在甘油、液状聚乙二醇及其在油中的混合物中配制分散剂。在通常的贮存和使用条件下,这些制剂含有防腐剂以预防微生物的生长。
适于注射使用的药物形式包括灭菌水溶液或分散剂和用于临时配制灭菌可注射溶液或分散剂的灭菌粉剂。在各种情况下,制剂必须是灭菌的,必须流动到存在易注射性的程度。它在生产和贮存条件下必须是稳定的,并且必须抗微生物如细菌和真菌的污染作用以便保藏。载体可以是溶剂或分散介质,包括诸如水、乙醇、多羟基化合物(如甘油、丙二醇和液状聚乙二醇)及其合适的混合物,以及植物油。





















Claims (13)
1、一种具下式的喹唑啉酮化合物及其药学上可接受的盐:
其中,R为;
X为3-5个碳原子的直链或支链烷基;
n为1~3;
R5为H;
R6为


其中,R9为H,1-4个碳原子的直链低级烷基,苯基,取代苯基(选自1~3个碳原子的单个低级烷基、-CF3、硝基、1-3个碳原子的邻位烷基、或-NH2的取代基),吡啶,噻吩,或呋喃;
R10为H,1-4个碳原子的直链低级烷基,苯基,取代的苯基(选自1-3个碳原子的单个低级烷基、-CF3、硝基、1-3个碳原子的邻位烷基、或-NH2的取代),吡啶,噻吩或呋南;但R9和R10不同时为H;
R11为H,1-4个碳原子的直链或支链低级烷基;
R12为1-4个碳原子的直链低级烷基,苯基,取代的苯基(选自1-3个碳原子的单个低级烷基,-CF3、硝基、1-3个碳原子的邻位烷基、或-NH2的取代基),吡啶,噻吩,或呋南;
R17为1-4个碳原子的直链或支链低级烷基;
R7和R8为H。
2、如权利要求1所述的化合物,其特征在于,其中R6为:R6是,
X为4个碳原子的烷基。
3、如权利要求1所述的化合物,其特征在于,其中,X为4个碳原子的直链或支链烷基:
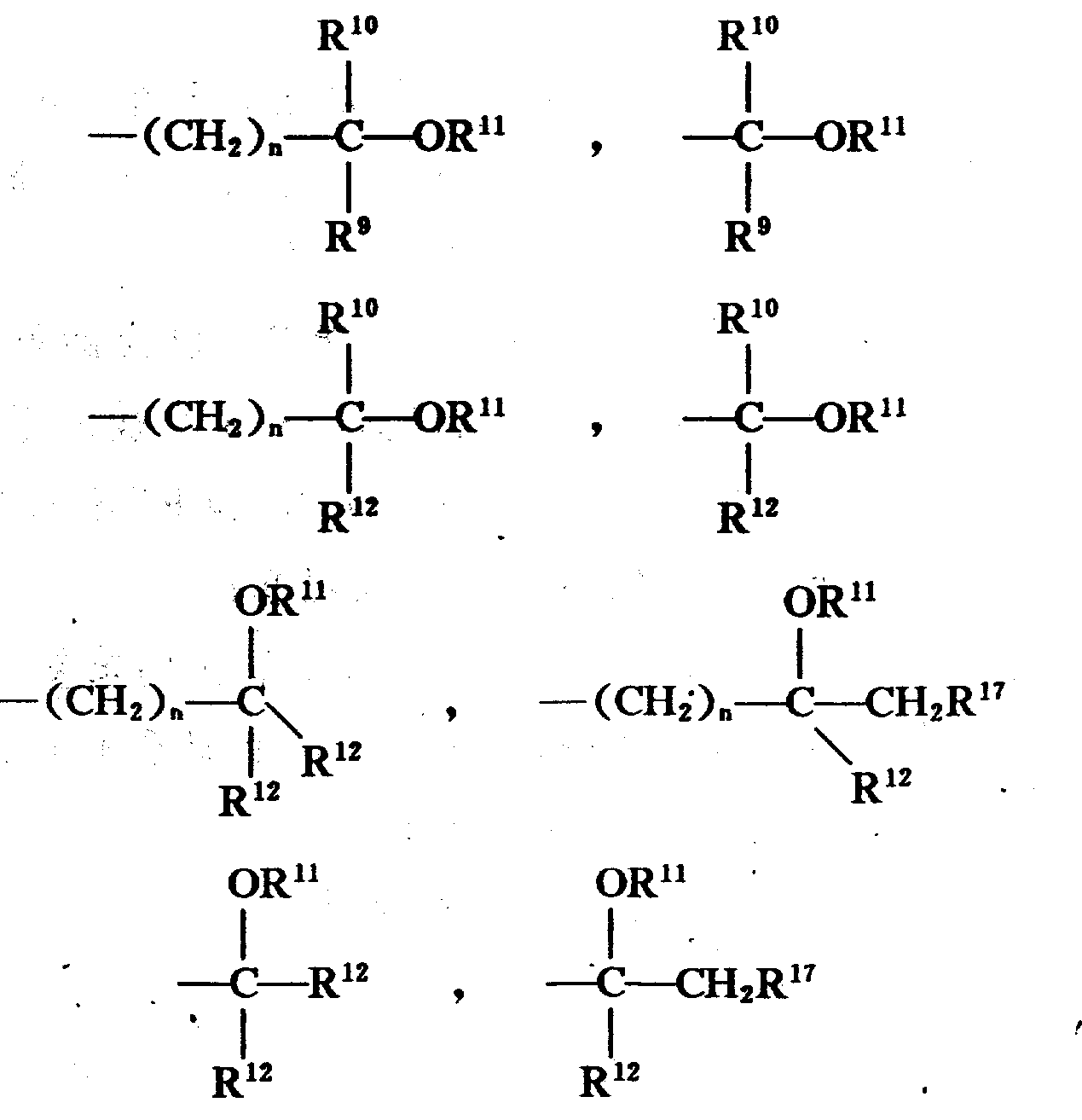
R9为H,1-4个碳原子的直链低级烷基,苯基或1-3个碳原子的单个低级烷基取代的苯基;
R10为H,1-4个碳原子的直链低级烷基,苯基或1-3个碳原子的单个低级烷基取代的苯基;
R11为H,1-4个碳原子的直链或支链低级烷基;
R12为1-4个碳原子的直链低级烷基,苯基或1-3个碳原子的单个低级烷基取代的苯基;
R17为1-4个碳原子的直链或支链低级烷基;
4、一种具下式的喹唑啉酮化合物:
其中:
X为3-5个碳原子的直链或支链低级烷基;
n为1-3;
R5为H;
R6为:


其中,R9为H,1-4个碳原子的直链低级烷基,苯基,取代苯基(选自1~3个碳原子的单个低级烷基、-CF3、硝基、1-3个碳原子的邻位烷基、或-NH2的取代基),吡啶,噻吩,或呋喃;
R10为H,1-4个碳原子的直链低级烷基,苯基,取代的苯基(选自1-3个碳原子的单个低级烷基、-CF3、硝基、1-3个碳原子的邻位烷基、或-NH2的取代基),吡啶,噻吩或呋喃;但R9和R10不同时为H;
R11为H,1-4个碳原子的直链或支链低级烷基;
R12为1-4个碳原子的直链低级烷基,苯基,取代的苯基(选自1-3个碳原子的单个低级烷基,-CF3、硝基、1-3个碳原子的邻位烷基、或-NH2的取代),吡啶,噻吩,或呋喃;
R17为1-4个碳原子的直链或支链低级烷基;
R7和R8为H。
5、一种具下式的喹唑啉酮化合物:
其特征在于,其中:
R40为-I,-Br或-OSO2CF3;
X为3-5个碳原子的直链或支链烷基;
n为1~3;
R5为H;
R6为

其中,R9为H,1-4个碳原子的直链低级烷基,苯基,取代苯基(选自1~3个碳原子的单个低级烷基、-CF3、硝基、1-3个碳原子的邻位烷基、或-NH2的取代),吡啶,噻吩,或呋喃;
R10为H,1-4个碳原子的直链低级烷基,苯基,取代的苯基(选自1-3个碳原子的单个低级烷基、-CF3、硝基、1-3个碳原子的邻位烷基、或-NH2的取代),吡啶,噻吩或呋喃;但R9和R10不同时为H;R11为H,1-4个碳原子的直链或支链低级烷基;
R12为1-4个碳原子的直链低级烷基,苯基,取代的苯基(选自1-3个碳原子的单个低级烷基,-CF3、硝基、1-3个碳原子的邻位烷基、或-NH2的取代),吡啶,噻吩,或呋喃;
R17为1-4个碳原子的直链或支链低级烷基;
R7和R8为H。
6、如权利要求4所述的下式的喹唑啉酮化合物:

其特征在于,其中:
X为3-5个碳原子的直链或支链烷基;
R6为:
R9为H,1-4个碳原子的直链低级烷基,苯基或1-3个碳原子的单个低级烷基取代的苯基;
R10为H,1-4个碳原子的直链低级烷基,苯基或1-3个碳原子的单个低级烷基取代的苯基;
R11为H,1-4个碳原子的直链或支链低级烷基;
R12为1-4个碳原子的直链低级烷基,苯基或1-3个碳原子的单个低级烷基取代的苯基;
7、如权利要求5所述的下式的喹唑啉酮化合物:

其特征在于,其中:
R40为-I,-Br或-OSO2CF3;
X为3-5个碳原子的直链或支链烷基:
R9为H,1-4个碳原子的直链低级烷基,苯基或1-3个碳原子的单个低级烷基取代的苯基;
R10为H,1-4个碳原子的直链低级烷基,苯基或1-3个碳原子的单个低级烷基取代的苯基;
R11为H,1-4个碳原子的直链或支链低级烷基;
R12为1-4个碳原子的直链低级烷基,苯基或1-3个碳原子的单个低级烷基取代的苯基;
8、如权利要求1所述的化合物,包括:2-丁基-6-(1-羟乙基)-3-[[2′-(1H-四唑-5-基)-[1,1’-二苯基]-4-基]甲基]-4(3H)-喹唑啉酮;
2-丁基-6-(1-羟基-1-甲基乙基)-3-[[2’-(1H-四唑-5-基)-[1,1’-二苯基]-4-基]甲基]-4(3H)-喹唑啉酮;
2-丁基-6-(1-羟基-1-甲基乙基)-3-[[2’-(1H-四唑-5-基)-[1,1’-二苯基]-4-基]甲基]-4(3H)-喹唑啉酮,钠盐;
2-丁基-6-(1-羟丙基)-3-[[2’-(1H-四唑-5-基)[1,1’-二苯基]-4-基]甲基]-4(3H)-喹唑啉酮;
2-丁基-7-(1-羟基-1-甲基乙基)-3-[[2’-(1H-四唑-5-基)[1,1’-二苯基]-4-基]甲基]-4(3H)-喹唑啉酮;
2-丁基-6-(1-甲氧基丙基)-3-[[2’-(1H-四唑-5-基)[[1,1’-二苯基]-4-基]甲基-4(3H)-喹唑啉酮;
2-丁基-6-(甲氧基苯基甲基)-3-[[2’-(1H-四唑-5-基)[1,1’-二苯基]-4-基]甲基]-4(3H)-喹唑啉酮;
2-丁基-6-(1-甲氧基-1-甲基乙基)-3-[[2-(1H-四唑-5-基)[1,1’-二苯基]-4-基]甲基]-4(3H)-喹唑啉酮;
6-[1-(乙酰氧基)乙基]-2-丁基-3-[[2’-(1H-四唑-5-基)[1,1’-二苯基]-4-基]甲基]-4(3H)-喹唑啉酮;
2-丁基-6-(羟基苯基甲基)-3-[[2’-(1H-四唑-5-基)-[1,1’-二苯基]-4-基]甲基]-4(3H)-喹唑啉酮;或,
2-丁基-6-(1-羟基-1-甲基乙基)-3-[[2’-(1H-四唑-5-基)[1,1’-二苯基]-4-基]甲基]-4(3H)-喹唑啉酮单钠盐。
9、如权利要求4所述的化合物,其特征在于,所述的化合物包括:
2-丁基-6-(1-羟乙基)-4(1H)-喹唑啉酮;
2-丁基-6-[(1-羟基(2,2,2-三甲基)乙基]-4(1H)-喹唑啉酮;
2-丁基-6-(1-羟丙基)-4(1H)-喹唑啉酮;
2-丁基-6-(1-羟基-1-甲基乙基)-4(1H)-喹唑啉酮;
2-丁基-6-(1-羟乙基)-4(1H)-喹唑啉酮;
2-丁基-6-(1-羟基-1-甲基乙基)-4(1H)-喹唑啉酮;或
2-丁基-6-(羟基苯基甲基)-4(1H)-喹唑啉酮;
10、一种用于治疗哺乳动物的血管紧张素诱发的高血压或充血性心力衰竭的药学组合物,其特征在于,所述的药学组合物包括一合适的药学载体和一有效量的权利要求1的化合物。
11、一种治疗哺乳动物的血管紧张素诱发的高血压的方法,其特征在于,将权利要求1的化合物以一有效量给所述的哺乳动物服用,以降低血管紧张素诱发的高血压。
12、一种制备权利要求1的化合物的方法,其特征在于,所述的方法包括:
用下式的化合物:

其中,R为:

在合适的溶剂中,有碱存在时,由在约20℃加热至60℃,将下式的化合物烷基化:
(其中,R5、R6、R7、R8和X如权利要求1中所定义)给出下式的中间体:
其中,R为

用稀酸搅拌该中间体,得到权利要求1的化合物。
13、一种制备权利要求1的化合物的方法,其特征在于,所述的方法包括:
用下式的化合物:
(其中,R4O选自I、Br、或-OSO2CF3,B选自合适的离去基团,而该离去基团选自I、Br、Cl、-OMS、-OTS或-OSO2CF3)有碱存在时,在一合适极性溶剂中,与下式化合物反应:
(其中,R5、R6、R7、R8和X如权利要求中所定义)给出下式的中间产物:
再将该中间产物与下式化合物反应,
其中,M选自-MgBr,-Sn(1-4个碳原子的低级烷基或苯基),Li或Zn配位化合物,
给出下式中间产物:
用稀酸与如此形成的中间产物反应,得到权利要求1的化合物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US64849291A | 1991-01-30 | 1991-01-30 | |
| US07/648,492 | 1991-01-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1066847A true CN1066847A (zh) | 1992-12-09 |
| CN1037440C CN1037440C (zh) | 1998-02-18 |
Family
ID=24601009
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN92100545A Expired - Fee Related CN1037440C (zh) | 1991-01-30 | 1992-01-30 | 血管紧张素ⅱ受体阻断剂2,3,6取代的喹唑啉酮的制造方法 |
Country Status (23)
| Country | Link |
|---|---|
| US (2) | US5290780A (zh) |
| EP (1) | EP0497150B1 (zh) |
| JP (1) | JP2758766B2 (zh) |
| KR (1) | KR920014805A (zh) |
| CN (1) | CN1037440C (zh) |
| AT (1) | ATE164158T1 (zh) |
| AU (2) | AU642573B2 (zh) |
| CA (1) | CA2060154A1 (zh) |
| CZ (1) | CZ281429B6 (zh) |
| DE (1) | DE69224763T2 (zh) |
| DK (1) | DK0497150T3 (zh) |
| ES (1) | ES2117014T3 (zh) |
| FI (1) | FI920217A7 (zh) |
| GR (1) | GR3026592T3 (zh) |
| HU (1) | HUT61748A (zh) |
| IE (1) | IE920142A1 (zh) |
| IL (1) | IL100711A0 (zh) |
| NO (1) | NO920298L (zh) |
| NZ (1) | NZ241343A (zh) |
| PL (1) | PL293316A1 (zh) |
| SK (1) | SK279004B6 (zh) |
| TW (1) | TW282467B (zh) |
| ZA (1) | ZA92392B (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1073110C (zh) * | 1995-06-07 | 2001-10-17 | 美国家用产品公司 | 联苯衍生物的制备方法 |
| CN107056715A (zh) * | 2017-04-07 | 2017-08-18 | 江南大学 | 一种增强TRPV4‑KCa2.3复合体耦联度的化合物及其在抗高血压中的应用 |
Families Citing this family (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5290780A (en) * | 1991-01-30 | 1994-03-01 | American Cyanamid Co. | Angiotensin II receptor blocking 2,3,6 substituted quinazolinones |
| US5385894A (en) * | 1991-03-06 | 1995-01-31 | Merck & Co., Inc. | Disubstituted 6-aminoquinazolinones |
| ES2126061T3 (es) * | 1993-03-24 | 1999-03-16 | American Home Prod | Piridopirimidinas substituidas como antihipertensores. |
| US5358951A (en) * | 1993-04-23 | 1994-10-25 | American Cyanamid Company | Angiotensin II receptor blocking 2, 3, 6 substituted quinazolinones |
| US5610301A (en) * | 1993-07-01 | 1997-03-11 | American Cyanamid Company | Process for the regioselective n-alkylation of quinazolinones |
| US5342944A (en) * | 1993-07-19 | 1994-08-30 | American Cyanamid Company | Process for the preparation of 2-alkyl-3,5,6,7- or 8-substituted-4(3H)-quinazolinones |
| US5338736A (en) * | 1993-10-07 | 1994-08-16 | American Cyanamid Company | Angiotensin II receptor blocking 2,3,6-substituted quinazolinones |
| DE69507887T2 (de) * | 1994-09-20 | 1999-06-17 | Wakunaga Seiyaku K.K., Osaka | Verfahren zur herstellung von n-biphenylmethylthiadiazolin derivaten oder deren salzen sowie dafür benötigte zwischen verbindungen |
| US5760220A (en) * | 1995-06-07 | 1998-06-02 | American Home Products Corporation | Process for preparation of biphenyl derivatives |
| US5739330A (en) * | 1996-02-05 | 1998-04-14 | Hoechst Celanese Corporation | Process for preparing quinazolones |
| US5763608A (en) * | 1996-02-05 | 1998-06-09 | Hoechst Celanese Corporation | Process for preparing pyrimidine derivatives |
| US6638937B2 (en) | 1998-07-06 | 2003-10-28 | Bristol-Myers Squibb Co. | Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists |
| SE9903028D0 (sv) | 1999-08-27 | 1999-08-27 | Astra Ab | New use |
| GB0223730D0 (en) * | 2002-10-11 | 2002-11-20 | Novartis Ag | Organic compounds |
| EP1557023A2 (en) * | 2002-10-18 | 2005-07-27 | Koninklijke Philips Electronics N.V. | Method and system for metadata protection in tv-anytime |
| US20060052345A1 (en) * | 2002-11-04 | 2006-03-09 | Nps Pharmaceuticals, Inc. | Quinazolinone compounds as calcilytics |
| GB0412769D0 (en) * | 2004-06-08 | 2004-07-07 | Novartis Ag | Organic compounds |
| KR101129868B1 (ko) * | 2006-10-04 | 2012-04-12 | 화이자 프로덕츠 인코포레이티드 | 칼슘 수용체 길항제로서의 피리도[4,3-d]피리미딘-4(3H)-온 유도체 |
| US8969514B2 (en) | 2007-06-04 | 2015-03-03 | Synergy Pharmaceuticals, Inc. | Agonists of guanylate cyclase useful for the treatment of hypercholesterolemia, atherosclerosis, coronary heart disease, gallstone, obesity and other cardiovascular diseases |
| ES2559319T3 (es) | 2007-06-04 | 2016-02-11 | Synergy Pharmaceuticals Inc. | Agonistas de guanilato cliclasa útiles para el tratamiento de trastornos gastrointestinales, inflamación, cáncer y otros trastornos |
| CA2726917C (en) | 2008-06-04 | 2018-06-26 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of gastrointestinal disorders, inflammation, cancer and other disorders |
| WO2010009319A2 (en) | 2008-07-16 | 2010-01-21 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of gastrointestinal, inflammation, cancer and other disorders |
| US9616097B2 (en) | 2010-09-15 | 2017-04-11 | Synergy Pharmaceuticals, Inc. | Formulations of guanylate cyclase C agonists and methods of use |
| JP6258928B2 (ja) | 2012-06-13 | 2018-01-10 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | 新規ジアザスピロシクロアルカンおよびアザスピロシクロアルカン |
| DK2900669T3 (da) | 2012-09-25 | 2019-11-04 | Hoffmann La Roche | Hexahydropyrrolo[3,4-C]pyrrolderivater og relaterede forbindelser som autotaxin (ATX)-inhibitorer og som inhibitorer af lysophosphatidsyre (LPA)-produktion til behandling af f.eks. nyresygdomme |
| AR095079A1 (es) | 2013-03-12 | 2015-09-16 | Hoffmann La Roche | Derivados de octahidro-pirrolo[3,4-c]-pirrol y piridina-fenilo |
| CA2905438A1 (en) | 2013-03-15 | 2014-09-25 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase and their uses |
| AU2014235209B2 (en) | 2013-03-15 | 2018-06-14 | Bausch Health Ireland Limited | Guanylate cyclase receptor agonists combined with other drugs |
| WO2014197720A2 (en) | 2013-06-05 | 2014-12-11 | Synergy Pharmaceuticals, Inc. | Ultra-pure agonists of guanylate cyclase c, method of making and using same |
| CA2923523A1 (en) | 2013-11-26 | 2015-06-04 | F. Hoffmann-La Roche Ag | New octahydro-cyclobuta [1,2-c;3,4-c']dipyrrol-2-yl |
| UA118582C2 (uk) | 2014-03-26 | 2019-02-11 | Ф. Хоффманн-Ля Рош Аг | Біциклічні сполуки як інгібітори продукції аутотаксину (atx) і лізофосфатидилової кислоти (lpa) |
| AU2015238541B2 (en) | 2014-03-26 | 2019-09-19 | F. Hoffmann-La Roche Ag | Condensed [1,4]diazepine compounds as autotaxin (ATX) and lysophosphatidic acid (LPA) production inhibitors |
| MA41898A (fr) | 2015-04-10 | 2018-02-13 | Hoffmann La Roche | Dérivés de quinazolinone bicyclique |
| CA2992889A1 (en) | 2015-09-04 | 2017-03-09 | F. Hoffmann-La Roche Ag | Phenoxymethyl derivatives |
| KR20180054634A (ko) | 2015-09-24 | 2018-05-24 | 에프. 호프만-라 로슈 아게 | 이중 오토탁신(atx)/탄산 무수화효소(ca) 억제제로서 신규한 이환형 화합물 |
| MX2018001890A (es) | 2015-09-24 | 2018-06-20 | Hoffmann La Roche | Compuestos biciclicos como inhibidores de autotaxina (atx). |
| BR112017026682A2 (pt) | 2015-09-24 | 2018-08-14 | Hoffmann La Roche | novos compostos bicíclicos como inibidores de dupla ação de atx/ca |
| AU2016328535A1 (en) | 2015-09-24 | 2017-11-09 | F. Hoffmann-La Roche Ag | Bicyclic compounds as ATX inhibitors |
| CN110382484B (zh) | 2017-03-16 | 2022-12-06 | 豪夫迈·罗氏有限公司 | 新的作为atx抑制剂的二环化合物 |
| SG11201908560SA (en) | 2017-03-16 | 2019-10-30 | Hoffmann La Roche | Heterocyclic compounds useful as dual atx/ca inhibitors |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8607683D0 (en) * | 1986-03-27 | 1986-04-30 | Ici Plc | Anti-tumor agents |
| MX7829A (es) * | 1986-08-21 | 1993-08-01 | Pfizer | Quinazolindionas y piridopirimindionas y procedimiento para su preparacion |
| CS272018B1 (en) * | 1988-11-01 | 1990-12-13 | Fisnerova Ludmila | Ethereal derivatives of 4/3h/-quinazolinone |
| US5240928A (en) * | 1989-07-03 | 1993-08-31 | Merck & Co., Inc. | Substituted quinazolinones as angiotensin II antagonists |
| CA2020073A1 (en) * | 1989-07-03 | 1991-01-04 | Eric E. Allen | Substituted quinazolinones as angiotensin ii antagonists |
| EP0407342A3 (en) * | 1989-07-06 | 1991-07-10 | Ciba-Geigy Ag | Pyrimidine derivatives |
| CA2037630C (en) * | 1990-03-07 | 2001-07-03 | Akira Morimoto | Nitrogen-containing heterocylic compounds, their production and use |
| CA2053148A1 (en) * | 1990-10-16 | 1992-04-17 | Karnail Atwal | Dihydropyrimidine derivatives |
| US5290780A (en) * | 1991-01-30 | 1994-03-01 | American Cyanamid Co. | Angiotensin II receptor blocking 2,3,6 substituted quinazolinones |
-
1992
- 1992-01-14 US US07/818,721 patent/US5290780A/en not_active Expired - Lifetime
- 1992-01-16 DE DE69224763T patent/DE69224763T2/de not_active Expired - Fee Related
- 1992-01-16 EP EP92100640A patent/EP0497150B1/en not_active Expired - Lifetime
- 1992-01-16 AT AT92100640T patent/ATE164158T1/de not_active IP Right Cessation
- 1992-01-16 DK DK92100640.9T patent/DK0497150T3/da active
- 1992-01-16 ES ES92100640T patent/ES2117014T3/es not_active Expired - Lifetime
- 1992-01-17 IE IE014292A patent/IE920142A1/en not_active Application Discontinuation
- 1992-01-17 FI FI920217A patent/FI920217A7/fi unknown
- 1992-01-20 IL IL100711A patent/IL100711A0/xx unknown
- 1992-01-20 ZA ZA92392A patent/ZA92392B/xx unknown
- 1992-01-21 NZ NZ241343A patent/NZ241343A/en unknown
- 1992-01-22 NO NO92920298A patent/NO920298L/no unknown
- 1992-01-22 AU AU10397/92A patent/AU642573B2/en not_active Ceased
- 1992-01-28 CA CA002060154A patent/CA2060154A1/en not_active Abandoned
- 1992-01-29 CZ CS92255A patent/CZ281429B6/cs unknown
- 1992-01-29 PL PL29331692A patent/PL293316A1/xx unknown
- 1992-01-29 JP JP4038746A patent/JP2758766B2/ja not_active Expired - Lifetime
- 1992-01-29 SK SK255-92A patent/SK279004B6/sk unknown
- 1992-01-30 HU HU9200278A patent/HUT61748A/hu unknown
- 1992-01-30 KR KR1019920001413A patent/KR920014805A/ko not_active Ceased
- 1992-01-30 CN CN92100545A patent/CN1037440C/zh not_active Expired - Fee Related
- 1992-01-31 TW TW081100762A patent/TW282467B/zh active
-
1993
- 1993-08-25 AU AU44906/93A patent/AU647992B2/en not_active Ceased
- 1993-10-29 US US08/145,458 patent/US5405849A/en not_active Expired - Lifetime
-
1998
- 1998-04-10 GR GR980400781T patent/GR3026592T3/el unknown
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1073110C (zh) * | 1995-06-07 | 2001-10-17 | 美国家用产品公司 | 联苯衍生物的制备方法 |
| CN107056715A (zh) * | 2017-04-07 | 2017-08-18 | 江南大学 | 一种增强TRPV4‑KCa2.3复合体耦联度的化合物及其在抗高血压中的应用 |
| CN107056715B (zh) * | 2017-04-07 | 2019-07-30 | 江南大学 | 一种增强TRPV4-KCa2.3复合体耦联度的化合物及其在抗高血压中的应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| GR3026592T3 (en) | 1998-07-31 |
| CZ281429B6 (cs) | 1996-09-11 |
| AU642573B2 (en) | 1993-10-21 |
| CS25592A3 (en) | 1992-08-12 |
| ATE164158T1 (de) | 1998-04-15 |
| EP0497150A1 (en) | 1992-08-05 |
| ES2117014T3 (es) | 1998-08-01 |
| KR920014805A (ko) | 1992-08-25 |
| HU9200278D0 (en) | 1992-04-28 |
| FI920217L (fi) | 1992-07-31 |
| IL100711A0 (en) | 1992-09-06 |
| AU1039792A (en) | 1992-08-06 |
| JP2758766B2 (ja) | 1998-05-28 |
| FI920217A7 (fi) | 1992-07-31 |
| AU647992B2 (en) | 1994-03-31 |
| FI920217A0 (fi) | 1992-01-17 |
| EP0497150B1 (en) | 1998-03-18 |
| AU4490693A (en) | 1993-11-18 |
| TW282467B (zh) | 1996-08-01 |
| DK0497150T3 (da) | 1998-06-02 |
| PL293316A1 (en) | 1993-08-23 |
| IE920142A1 (en) | 1992-07-29 |
| US5405849A (en) | 1995-04-11 |
| HUT61748A (en) | 1993-03-01 |
| US5290780A (en) | 1994-03-01 |
| ZA92392B (en) | 1992-10-28 |
| SK279004B6 (sk) | 1998-05-06 |
| NO920298D0 (no) | 1992-01-22 |
| NO920298L (no) | 1992-07-31 |
| DE69224763D1 (de) | 1998-04-23 |
| CN1037440C (zh) | 1998-02-18 |
| JPH0578332A (ja) | 1993-03-30 |
| DE69224763T2 (de) | 1998-07-09 |
| CA2060154A1 (en) | 1992-07-31 |
| NZ241343A (en) | 1994-06-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1066847A (zh) | 血管紧张素ii受体阻断剂2,3,6取代的喹唑啉酮 | |
| CN1253449C (zh) | 作为蛋白激酶c抑制剂的吲哚基马来酰亚胺衍生物 | |
| CN1036456C (zh) | 氨基取代的吡唑 | |
| CN1036341C (zh) | 咪唑衍生物的制备方法 | |
| CN1048486C (zh) | 苯并咪唑衍生物的制备方法 | |
| CN1190432C (zh) | 作为5-htib拮抗药的哌嗪衍生物 | |
| CN1072682A (zh) | 取代嘧啶 | |
| CN1031940C (zh) | 取代的唑类化合物的制备方法 | |
| CN1102648A (zh) | 取代的单-和双-吡啶基甲基吡啶酮类化合物 | |
| CN1149214C (zh) | 三唑并衍生物和含有它们作为有效组分的趋化因子抑制剂 | |
| CN1079648A (zh) | 制备含咪唑基链烯酸的药物组合物的方法 | |
| CN1444570A (zh) | 非甾体抗炎药 | |
| CN1193963A (zh) | 芳族化合物和含有它们的药物组合物 | |
| CN1072219C (zh) | 二氮杂䓬酮、其生产和用途 | |
| CN1694708A (zh) | TGFβ的抑制剂 | |
| CN1478080A (zh) | 作为谷氨酸受体拮抗剂的苯基乙烯基或苯基乙炔基衍生物 | |
| CN1073174A (zh) | 杂环衍生物 | |
| CN1234025A (zh) | 1-苯基-苯并咪唑类化合物及其作为baga-a受体调节剂的用途 | |
| CN1127498C (zh) | 吡啶衍生物、制备吡啶衍生物的方法及其中间体 | |
| CN1960984A (zh) | 作为mglu5受体拮抗剂的吡啶-4-基-乙炔基-咪唑和吡唑 | |
| CN1040435C (zh) | 联苯基吡啶酮及其制备方法以及它们在药物中的用途 | |
| CN1802367A (zh) | 作为谷氨酸受体拮抗剂的咪唑衍生物 | |
| CN1044243C (zh) | 联苯基取代的喹啉衍生物及其药物组合物和用途 | |
| CN1051555A (zh) | 取代的n-(咪唑基)烷基丙氨酸衍生物 | |
| CN1503797A (zh) | 吡啶并嘧啶或二氮杂萘衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |