CN1060095A - 氟代乙基喜树碱衍生物 - Google Patents
氟代乙基喜树碱衍生物 Download PDFInfo
- Publication number
- CN1060095A CN1060095A CN91108649A CN91108649A CN1060095A CN 1060095 A CN1060095 A CN 1060095A CN 91108649 A CN91108649 A CN 91108649A CN 91108649 A CN91108649 A CN 91108649A CN 1060095 A CN1060095 A CN 1060095A
- Authority
- CN
- China
- Prior art keywords
- compound
- residue
- general formula
- group
- changing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/22—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains four or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
本发明的目的是提供一种具有抗溃疡活性的低
毒性的喜树碱衍生物。
构成本发明的特征是由下面通式(I)所示的新
型的氟代乙基喜树碱衍生物:
Description
本发明涉及具有抗溃疡活性的新型化合物及其制备方法。
喜树碱是从旱莲木的树皮、根、果实和叶中分离出的五环的生物碱(Wall等人在J.Am.Chem.Soc.,88,3888-3890(1966)上有报导),现已知道它具有抗溃疡活性,因为它能阻止核酸的合成(Lown等人在Biochem.Pharmacol.,29,905~915(1980)上有报导)。可是在美国的临床试验结果表明,存在着毒性方面的问题,因而中止了作为医药品的开发。
其后,世界上曾进行着对于以减轻毒性或增加活性为目的衍生物的研究,但至今仍没有关于可赋予能满足临床要求的结果的衍生物的报导。
迄今为止,所报导的大多数的喜树碱衍生物均是内酯环4位的烷基是乙基的产物,仅仅由管沢等人(在J.Med.Chem.,19,675~679(1975)上报导过在烯丙基、炔丙基和苄基等取代基上显示了活性,但是,在毒性方面,吸收性等的体内动态方面的改进没有成功。
在这样的现实状况下本发明者们为了得到作用优良、安全性较高,而且在吸收性等的体内动态方面均有所改进的喜树碱衍生物而进行深入研究的结果发现,上述内酯环4位的烷基变换为2-氟代乙基的化合物显示出超过喜树碱的特性,于是完成了本发明。
按照本发明的目的是提供一种用下列通式(Ⅰ)所示的化合物及其盐:
【化13】

{式中R1表示氢原子、C1-C6的烷基、羟甲基、酰羟甲基、甲酰基、R2、R3、R4分别是独立的并且代表氢原子、羟基、C1-C6的烷基、C2-C6的链烯基、C2-C6的炔基、C1-C6烷氧基、卤原子、氨基、C2-C7的酰胺基、C1-C6的烷氨基、NR5(R6)[R5和R6是相同的或不同的、并代表C1-C6的烷基、或者与其中结合的氮原子一起再构成环的一部分。该环可以含有0、S、N-R7(R7代表氢原子、C1-C6的烷基或氨基的保护基)原子,并且所构成的环是任何一个成环的碳原子均可被C1-C3的烷基、氨基或氨甲基取代的5环或6环]基、苯基、萘基、吡啶基,另外R1和R2它们也可以在一起形成由-(CH2)m-Z-(CH2)n-所示的交联结构[Z表示0、S、CH-R8(R8代表氢原子或C1-C6的烷基或N-R7′(R7′代表氢原子、C1-C6的烷基或氨基的保护基)、并且m和n分别代表0、1、或2,但是m和n不能同时为零]}。
另外,本发明的其它目的则是提供一种制备上述式(Ⅰ)所示化合物的方法。
上述式(Ⅰ)所示化合物的定义中,R1、R2、R3、R4、R7和R8的烷基、链烯基、炔基和它们的取代基的其它官能基或取代基可以被组合的取代基中所对应部分的碳数为1~6,而作为较理想的烷基特别是甲基、乙基等。作为氨基的保护基可以列举的有甲酰基、乙酰基、三苯甲基、叔丁氧基羰基、对-甲氧基苄氧基羰基等。
R1、R2、R3和R4的具体实例有:甲基、乙基、羟基、甲氧基、氟、氯、溴、硝基、氨基、二甲氨基、3-氨基-1-吡咯烷基、3-氨甲基-1-吡咯烷基、1-哌嗪基、3-甲基-1-哌嗪基、4-甲基-1-哌嗪基、4-吗啉基、苯基、1-萘基、3-吡啶基等。
另外在式(Ⅰ)中关于内酯环的不对称碳的配位为具有S型的化合物的作用较好。
本发明的化合物,例如可以采用示于下列反应式的方法进行制造,
【化14】


(式中R1,R2,R3,和R4的定义同前)
即采用弗里德伦德反应(Organic Reactions,28,37-202,John Wiley & Sons INc.,New york(1982))使氨基酮化合物(Ⅴ)与吡喃并吲哚嗪化合物(Ⅱ1)进行缩合而制得化合物(Ⅰ)。
氨基酮(Ⅴ)是公知的或是用公知的标准方法很容易制得的化合物。
化合物(Ⅴ)与(Ⅱ1)的缩合闭环反应的条件是酸或碱的存在下,从常温到加热的条件中进行适宜的选择为好。
作为所用的溶剂,如果对于反应是不活性的那么就没有其它特别的限制了,但是可列举的实例有:苯、甲苯、二甲苯等芳香族烃、氯仿、二氯甲烷、1,1-二氯乙烷、1,2-二氯乙烷等卤代烃;二乙基醚、二异丙基醚、四氢呋喃、乙二醇二甲醚、乙二醇二乙基醚、二乙二醇二甲醚等醚类;甲醇、乙醇、丙醇、叔丁醇等低级醇类;乙酰胺、二甲基乙酰胺、N,N-二甲基甲酰胺等酰胺类;醋酸等,最好是苯、甲苯、醋酸。
无机酸、有机酸均可使用的酸,但是作为无机酸的实例可列举的有:盐酸、硫酸等。作为有机酸的实例可列举的有:甲磺酸、三氟代甲磺酸、苯磺酸、对-甲苯磺酸等磺酸类,醋酸等羧酸类,但是最好的是对-甲苯磺酸或醋酸,当使用醋酸时还可以兼做溶剂。
所用的碱可以是无机碱也可以是有机碱,但是作为无机碱的例子可列举的有:氢氧化锂、氢氧化钠、氢氧化钾、碳酸锂、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾等碱金属氢氧化物、碳酸盐、碳酸氢盐等或氢化钠。作为有机碱可列举的实例有:甲醇钠、乙醇钠、叔丁醇钾等碱金属醇盐类;三乙胺、N,N-二异丙基乙胺等叔烷基胺类;N,N-二甲基苯胺、N,N-二乙基苯胺、N,N-二甲基氨基吡啶等芳香族叔胺类或吡啶,1,8-二氮杂二环十一碳烯等,但是作为无机碱中最好的是碳酸钾,而作为有机碱中最好的是三乙胺。
反应温度通常为20~150℃,最好为80~120℃。
反应时间以1-48小时为好,通常以1~24小时完成。作为代表性的条件,可列举的实例有使之在醋酸中进行加热回流或在对-甲苯磺酸的存在下,在苯或甲苯中进行加热回流的方法。另外当R1、R2、R3和R4等为具有保护基的氨基的情况下,可用通常使用的方法除去保护基,例如使用酸或碱的加水分解或采用还原反应可以除去保护基。
另外含有烷氧基的化合物在甲苯或苯等惰性的溶剂中用氯化铝、溴化铝等进行处理,或在溴化氢酸溶液中进行加热可以导致产生对应的羟基化合物。
含有迭氮基或硝基的化合物,通过使用白金或钯进行接触还原,可以使之成为对应的氨基化合物。含有氨基的化合物在酸性溶剂中在低温下,用亚硝酸钠等进行重氮化,再使得到的重氮盐进行加水分解可以导致产生对应的羟基化合物。
另外,用上述相同方法处理氨基的化合物可以导致产生重氮盐,然后通过进行桑德米耶反应可以导致产生对应的卤化物。
桑德米耶反应可以在通用的条件下使用氯化亚铜或溴化亚铜来进行。
另一方面,吡喃吲哚嗪化合物(Ⅱ1)是一种新型化合物,可以采用下述方法由化合物(Ⅳ)来制备。
【化15】


(式中R10代表甲酰基、乙酰基、苯酰基或(R)-1-(对-甲苯磺(酰)脯胺酰基)。
所说的氟代乙基化是使化合物(Ⅳ)与例如氢化钠、叔丁醇钾等碱在N,N-二甲酰胺、1,2-二甲氧基乙烷等与反应无关的溶剂中进行反应,然后通过加入溴化氟代乙基、碘化氟代乙基等氟代乙基卤化物或氟代乙基甲苯磺酸酯等氟代乙醇的磺酸酯,就可以制得化合物(Ⅲ3)。特别是,R10是(R)-1-(对甲苯磺酰)脯胺酰基时可以优先得到S体。通常,反应是在-78~150℃,最好是在0~80℃的温度下和10分~48小时,最好是1~24小时的条件下进行的。
氨基的还原,是使化合物(Ⅲ3)在有醋酸酐存在下的氢气流中,使用拉奈镍,如果需要再一面使用钨灯照射一面进行反应,就可以制得化合物(Ⅲ2)。反应通常是在10~100℃,最好20~60℃的温度下进行10分~8小时,最好是30分~5小时。
经亚硝基的转位反应,使化合物(Ⅲ2)在醋酸酐和醋酸的混合溶剂中、与亚硝酸钠等亚硝基化试剂在0~50℃最好0~30℃的温度下反应30分~15小时,最好1-~5小时,然后将得到的亚硝基体在50~120℃,最好在60~90℃的温度下加热30分~12小时,最好1~5小时,通过搅拌即可得到化合物(Ⅲ1)。
闭环反应,使化合物(Ⅲ1)在乙醇等醇类,二噁烷等与反应无关的溶剂中,使用氢氧化锂、氢氧化钠或氢氧化钾等碱性水溶液进行加水分解以后,再在用醋酸、柠檬酸、盐酸等调制的酸性溶剂中进行处理,就可得到化合物(Ⅱ2)。加水分解是在0~50℃,最好是20~40℃进行5分~5小时、最好10分~3小时;在酸性溶剂中的反应是在0~70℃,最好10~40℃进行1-72小时,最好是12~24小时进行反应。
脱酮缩醇化反应,是使化合物(Ⅱ2)在用盐酸、硫酸、三氟醋酸等调配的酸性溶剂中进行处理,就可以得到化合物(Ⅱ1)。反应通常是在0~100℃,最好10~40℃进行10分~24小时,最好10分~5小时。
本发明的化合物(Ⅰ)中的某些化合物可以制成生理上允许的各种盐,如根据需要可以由碱金属或碱土金属的氢氧化物转变成它们的盐或由具有氨基的碱性化合物转变成盐酸、硫酸、磷酸等无机酸的盐或甲酸、醋酸等有机酸的盐。
通过本发明提供的新型氟代乙基喜树碱衍生物、由于其能作为抗溃疡物质,所以它在医药和医药中间体的使用方面是一种很有用的化合物。
作为抗溃疡药剂的给药方法可以采用静脉注射、肌肉注射、皮下注射等各种注射剂的注射法,或经口服用等各种方法进行给药。
[实施例]
举出下面的实施例用来更详细的说明本发明。
实施例1:6-氰基-1,1-(亚乙二氧基)-α-(2-氟代乙基)-α-[(R)-1-(对-甲苯磺酰基)-吡咯烷-2-基-羰氧基]-5-羰基-1,2,3,5-四氢化吲哚嗪-7-乙酸乙酯的合成:
将920mg的6-氰基-1,1-(亚乙二氧基)-α-[(R)-1-(对-甲苯磺酰基)-吡咯烷-2-基-羰氧基]-5-羰基-1,2,3,5-四氢化吲哚嗪-7-乙酸乙酯加入到80mg60%氢化钠悬浮在7ml的二甲基甲酰胺的溶液中,在室温下搅拌1小时。向该溶液加入2.26克的2-氟代溴乙烷,在氩气保护下,在室温下搅拌4小时,60℃下搅拌2小时,进而在80℃下再搅拌2小时,将反应液进行浓缩,并使残渣溶解在60ml氯仿中,接着先用水后用饱和食盐水进行洗净,再用无水芒硝干燥后进行浓缩。残渣用硅胶柱子(溶出溶剂,氯仿)进行精制,最后可以得到750mg黄色油状的粗制品。该粗制品再次用硅胶柱(溶出溶剂为苯∶乙酸乙酯=7∶3)和制备薄层色谱法(プレパラティプTLC)(展开溶剂为苯∶醋酸乙酯=7∶3)进行精制,最终将得到335mg淡黄色树脂状的目的产物[化17]
H-NMR(在CDCl3.δ.ppm)
【化16】
1.31(3H.t.COOCH2 CH 3)
1.60~1.75,1.92~2.06(2H.m.  )
)
1.92~2.18(2H.m.  )
)
2.39~2.45(5H.m.  ,C2-H)
,C2-H)
2.90~3.10(2H.m.  )
)
3.12~3.25,3.50~3.60(2H.m.  )
)
4.08~4.12,4.23~4.26(4H.m. )
4.12~4.21(2H.m.C3-H)
4.26~4.32(2H.q.COOCH2CH3)
4.43~4.52(1H.m.  )
)
4.52~4.72(2H.m.  )
)
6.66(1H.s.C8-H)
7.34(2H.d.  )
)
7.87(2H.d  )
)
【化17】

实施例2:6-(乙酰胺甲基)-1,1-(亚乙二氧基)-α-(2-氟代乙基)-α-[(R)-1-(对-甲苯磺酰基)吡咯烷-2-基-羰氧基]-5-羰基-1,2,3,5-四氢化吲哚嗪-7-乙酸乙酯的合成
将实施例1制得的化合物280mg加入到含有由1120mg拉奈镍合金调配而成的拉奈镍W-2催化剂的11ml醋酸酐和3ml醋酸的混合物中,然后在氢气流存在下于室温搅拌2小时,并进而再于钨灯照射下搅拌1小时。过滤除去反应液中催化剂,并将滤液进行浓缩。再将残渣溶解在氯仿中并进行水洗,然后用无水芒硝干燥之后再进行浓缩,将残渣用硅胶柱(溶出溶剂为氯仿∶甲醇=50∶1)进行精制,最好得318mg黄色油状的目的产物[化19]。
H-NMR(在CDCl3.δ.ppm)
【化18】
1.27(3H.t.COOCH2CH3)
1.71~1.73,1.95~1.98(2H.m.  )
)
1.95(3H.s.  )
)
1.95~1.98,2.07~2.15(2H.m.  )
)
2.41~2.48(5H.m.  ,C2-H)
,C2-H)
2.95~3.06(2H.m. )
3.17~3.28,3.50~3.55(2H.m.  )
)
4.04~4.33(9H.m.  ,COOCH2CH3,C3-H,
,COOCH2CH3,C3-H, 
4.40~4.44(1H.m. )
4.50~4.70(2H.m.  )
)
4.77~4.87(1H.m.  )
)
6.79(1H.s.C8-H)
7.11~7.14(1H.br.-CONH-)
7.33(2H.d.  )
)
7.55(2H.d.  )
)
【化19】

实施例3:6-(乙酰氧甲基)-1,1-(亚乙二氧基)-α-(2-氟代乙基)-α-[(R)-1-(对-甲苯磺酰基)吡咯烷-2-基-羰氧基]-5-羰基-1,2,3,5-四氢化吲哚嗪-7-乙酸乙酯的合成
将实施例2中制得的化合物318mg溶解于3.2ml醋酸酐和1ml醋酸的混合液中,然后一面在食盐-冰浴中冷却一面慢慢加入140mg亚硝酸钠,在同此温度下搅拌5.5小时(并在第2小时和第4小时分别追加140mg亚硝酸钠),进而再于室温下搅拌30分钟。过滤除去反应液中的不溶物,再对滤液进行浓缩,再将8ml四氯化碳加入到残渣中,加热回流4小时,在反应液中加入40ml氯仿进行烯释,再先用水后用饱和食盐水进行洗净,再用无水芒硝干燥后进行浓缩。用硅胶柱(溶出溶剂为氯仿∶甲醇=50∶1)对残渣进行精制,最后制得189mg无色油状的目的产物[化21]。
H-NMR(在CDCl3.δ.ppm)
【化20】
1.35(3H.t.COOCH2CH3)
1.66~1.72,1.92~2.01(2H.m.  )
)
1.92~2.03(2H.m. )
2.41(2H.t.C2-H)
2.45(3H.s. )
2.70~2.80,3.04~3.17(2H.m.  )
)
3.17~3.24,3.51~3.59(2H.m.  )
)
4.06~4.28(8H.m. ,COOCH2CH3,C3-H)
4.29~4.36(1H.m. )
4.56(2H.dt.  ,J=46.9Hz)
,J=46.9Hz)
5.28~5.31(2H.m.  )
)
6.70(1H.s.C8-H)
7.35(2H.d.  )
)
7.76(2H.d. )
【化21】

实施例4:6,6-(亚乙二氧基)-4-(2-氟代乙基)-1,4,7,8-四氢-4-羟基吡喃并[3,4-f]吲哚嗪-3,10(6H)-二酮的合成
将实施例3制得的化合物3.5mg溶于0.1ml乙醇和0.05ml水的混合液中,加入1.0mg氢氧化锂一水合物,然后在室温下搅拌1小时,将反应液浓缩,然后残渣中加入0.3ml水、1.0ml二氯甲烷和0.2ml醋酸,并在室温下搅拌18小时,分离出二氯甲烷层,并用1ml二氯甲烷对水层抽提2次。合并二氯甲烷层,再用饱和食盐水洗净,后用无水芒硝干燥,再进行浓缩。残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得0.89mg白色油状的目的产物[化23]。
H-NMR(在CDCl3.δ.ppm)
【化22】
2.13~2.15,2.19~2.22(2H.m.  ,J=26.8Hz)
,J=26.8Hz)
2.42(2H.t.C7-H)
4.08~4.24(6H.m. ,C8-H)
4.49~4.78(2H.m. ,J=46.4Hz)
5.20,5.66(2H.ABq.C1-H.J=16.1Hz)
6.59(1H.s.C5-H)
【化23】

实施例5:4-(2-氟代乙基)-1,4,7,8-四氢-4-羟基吡喃并[3,4,f]吲哚嗪-3,6,10-三酮的合成。
将0.89mg实施例4制得的化合物溶于0.1ml80%的三氟醋酸水溶液中,在氩气流存在的室温下搅拌1小时,将反应液浓缩,用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)对残渣进行精制,最后制得0.55mg红色油状的目的产物[化25]
H-NMR(在CDCl3.δ.ppm)
【化24】
2.07~2.30(2H.m.  )
)
2.98(2H.t.C7-H)
4.31~4.38(2H.m.C8-H)
4.55~4.65,4.67~4.77(2H.m.  ,J=46.9Hz)
,J=46.9Hz)
5.28,5.73(2H.ABq.C1-H.J=17.1Hz)
7.26(1H.s.C5-H)
【化25】

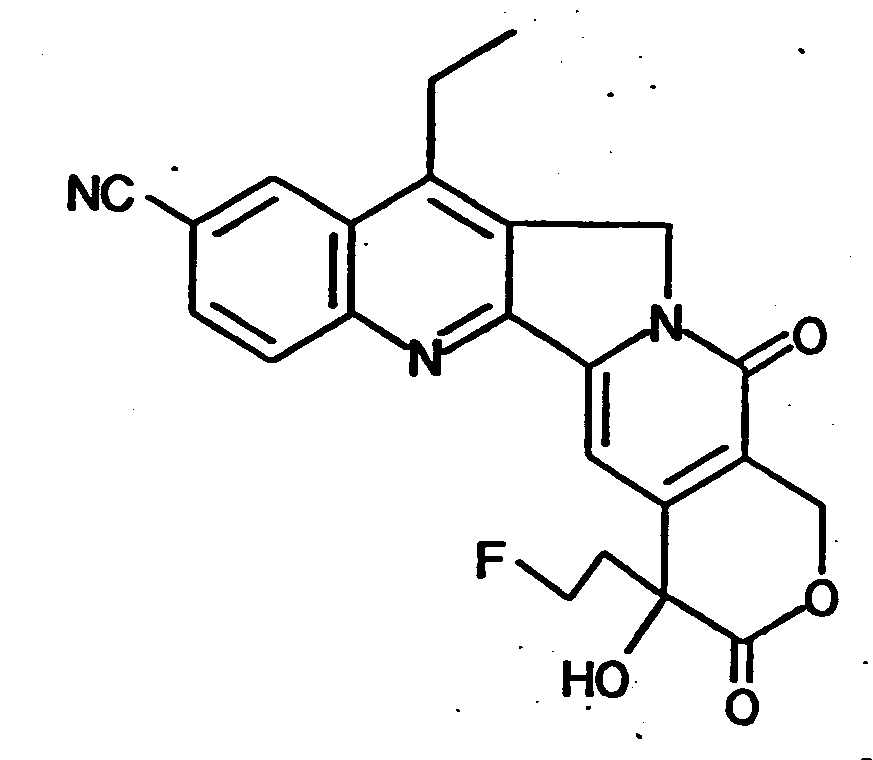
实施例6:11-乙基-4-(2-氟代乙基)-4-羟基-9-甲氧基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将例5制得的化合物2.03mg和1.12mg2-氨基-5-甲氧丙酰苯溶于0.6ml甲苯中,再加入催化剂量的对-甲苯磺酸,加热回流2小时,将反应液进行浓缩,其残渣用制备薄层色谱法(展开溶剂为乙酸乙酯∶正-己烷=4∶1)精制,最后得到1.32mg白色结晶的目的产物[化27]。
Mass m/z=424(M+)
H-NMR(在CDCl3.δ.ppm)
【化26】
1.41(3H.t.C11-CH2CH3.J=7.8Hz)
2.22~2.36(2H.m.  )
)
3.16(2H.q.C11-CH2CH3,J=7.8Hz)
4.01(3H.s.OCH3)
4.56~4.62,4.67~4.73,4.79~4.83(2H.m.  ,J=46.9Hz)
,J=46.9Hz)
5.25(2H.s.C12-H)
5.34,5.81(2H.ABq.C1-H)
7.33(1H.d.C10-H,J=2.4Hz)
7.48(1H.dd.C8-H,J=2.9Hz,9.3Hz)
7.61(1H.s.C5-H)
8.14(1H.d.C7-H,J=9.3Hz)
[化27]

实施例7:11-乙基-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3-14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,在有氩气流存在的室温下搅拌1小时。浓缩反应液,再将残渣溶于二氯甲烷中,并进行水洗,然后进行浓缩干固,将2.2mg的2-氨基丙酰苯、0.5ml甲苯和催化剂量的对-甲苯磺酸加到残渣中,并加热回流1.5小时。浓缩反应液,再用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)精制残渣,最后制得2.7mg淡黄色结晶的目的产物
mass m/z=394(M+)
H-NMR(在CDCl3.δ.ppm)
[化28]
1.47(3H.t.C11-CH2CH3,J=7.8Hz)
2.22~2.37(2H.m.  )
)
3.22(2H.q.C11-CH2CH3)
4.57~4.62,4.67~4.74,4.79~4.84(2H.m.

5.27(2H.s.C12-H)
5.33,5.81(2H.ABq.C1-H,J=16,1Hz)
7.68(1H.s.C5-H)
7.64~7.71,7.97~8.34(1H×2.m×2.C8,C9-H)
8.14.8.24(1H×2.dd×2.C7,C10-H)
【化29】

实施例8:11-乙基-9-氟-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5mg80%的三氟代乙酸水溶液中,在有氩气流存在的室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,再于水洗之后进行浓缩。再向残渣中加入2.5mg的2-氨基-5-氟代丙酰苯、0.5ml甲苯和催化剂量的对-甲苯磺酰,并加热回流1.5小时。浓缩反应液,再用制备性薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1),精制残渣,最后制得3.3mg淡黄色结晶的目的产物(化31)。
mass m/z=412(M+)
H-NMR(在CDCl3.δ.ppm)
【化30】
1.41(3H.t.C11-CH2CH3)
2.21~2.35(2H.m.  )
)
3.16(2H.q.C11-CH2CH3)
4.59~4.63,4.67~4.74,4.78~4.82(2H.m.  )
)
5.27(2H.s.C12-H)
5.34,5.80(2H.ABq.C1-H)
7.52~7.57(1H.m.C8-H)
7.65(1H.s.C5-H)
7.72(1H.dd.C10-H)
8.24(1H.dd.C7-H)
[化31]

实施例9:9-氯-11-乙基-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′、4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,于氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,再于水洗之后进行浓缩干固,向残渣加入2.8mg的2-氨基-5-氯代丙酰基苯、催化剂量的对-甲苯磺酸和0.5ml甲苯,并加热回流1小时,浓缩反应液,并用制备薄层色谱法(展开溶剂为乙酸乙酯)精制残渣,最后制得3.2mg淡褐色结晶的目的产物[化32]。
H-NMR(在CDCl3,δ,ppm)
1.41(3H,t,C11-CH2CH3)
2.21~2.35(2H,m,CH2CH2F)
3.17(3H,q,C11-CH2CH3)
4.58~4.82(2H,m,CH2CH2F)
5.27(2H,s,C12-H)
5.34,5.80(2H,ABq,C1-H)
7.66(1H,s,C5-H)
7.75(1H,dd,C8-H)
8.08(1H,d,C10-H)
8.16(1H,d,C7-H)
【化32】

实施例10:9-溴-11-乙基-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4中制得的化合物5.0mg溶于0.5ml80%的三氟乙酸水溶液中,并在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,再在水洗后进行浓缩干固,向残渣加入3.4mg的2-氨基-5-溴代丙酰基苯、催化剂量的对-甲苯磺酸和0.5ml甲苯,并加热回流1.5小时,浓缩反应液,残渣用硅胶柱(溶出溶剂为氯仿→氯仿∶甲醇=10∶1)进行精制,最后得到3.7mg红色油状的目的产物[化33]
H-NMR(在CDCl3.δ.ppm)
1.41(3H,t,C11-CH2CH3)
2.17~2.33(2H,m,-CH2CH2F)
3.16(2H,q,C11-CH2CH3)
4.59~4.82(2H,m,CH2CH2,F)
5.27(2H,s,C12-H)
5.34,5.80(2H,ABq,C1-H)
7.66(1H,s,C5-H)
7.87(1H,dd,C8-H)
8.09(1H,d,C7-H)
8.26(1H,d,C10-H)
【化33】

实施例11:11-乙基-4-(2-氟代乙基)-4-羟基-9-甲基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,并在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,再在水洗后进行浓缩干固,向残渣加入2.5mg2-氨基-5-甲基丙酰基苯、催化剂量的对-甲苯磺酸和0.5ml甲苯,并在加热下回流1小时。浓缩反应液,并用硅胶柱(溶出溶剂)为氯仿→氯仿∶甲醇=25∶1)精制残渣,最后制得3.0mg黄色结晶的目的产物[化34]
H-NMR(在CDCl3,δ,ppm)
1.40(3H,t,C11-CH2CH3)
2.17~2.34(2H,m,CH2CH2F)
2.62(3H,s,C9-CH3)
3.18(2H,q,C11-CH2CH3)
4.57~4.82(2H,m,CH2CH2F)
5.24(1H,s,C12-H)
5.34,5.79(2H,ABq,C1-H)
7.64(1H,dd,C8-H)
7.64(1H,s,C5-H)
7.86(1H,d,C10-H)
8.11(1H,d,C7-H)
【化34】

实施例12:11-乙基-4-(2-氟代乙基)-4-羟基-9-苯基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,并在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,再在水洗后进行浓缩干固,向残渣加入3.4mg的2-氨基-5-苯基丙酰基苯、催化剂量的对-甲苯磺酸和0.5ml甲苯,并于加热下回流1.5小时,浓缩反应液,再用硅胶柱(溶出溶剂为氯仿→氯仿∶甲醇=25∶1)精制残渣,最后得到5.2mg的目的产物[化36]。
H-NMR(在CDCl3.δ.ppm)
【化35】
1.46(3H.t.C11-CH2CH3)
2.23~2.36(2H,m,CH2CH2F)
3.27(2H,q,C11CH2CH3)
4.60~4.84(2H,m,CH2CH2F)
5.30(2H,s,C12-H)
5.36,5.82(2H,ABq,C1-H)
7.48(1H.t.  )
)
7.52~7.57(2H,t,  )
)
7.69(1H,s,C5-H)
7.75(2H.d.  )
)
8.08(1H,dd,C8-H)
8.27(1H,d,C10-H)
8.30(1H,d,C7-H)
【化36】

实施例13:11-乙基-4-(2-氟代乙基)-4-羟基-9-(1-萘基)-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,并在氩气流和室温下搅拌1小时,浓缩反应液,再将残渣溶于二氯甲烷中并在水洗后浓缩干固。向残渣加入4.1mg2-氨基-5-(1-萘基)-丙酰基苯、催化剂量的对-甲苯磺酸和0.5ml甲苯,然后加热回流1.5小时,浓缩反应液,并用硅胶柱(溶出溶剂为氯仿→氯仿∶甲醇=25∶1)精制残渣,最后制得4.2mg黄色油状的目的产物[化38]
H-NMR(在CDCl3.δ.ppm)
【化37】
1.41(3H,t,C11-CH2CH3)
2.24~2.36(2H,m,CH2CH2F)
3.21(2H,q,C11-CH2CH3)
4.61~4.85(2H,m,CH2CH2F)
5.31(2H,s,C12-H)
5.37,5.83(2H,ABq,C1-H)
7.46~7.63(4H,m, )
7.73(1H,s,C5-H)
7.91(1H,dd,C8-H)
7.95~7.99(3H,m, )
8.23(1H,d,C10-H)
8.35(1H,d,C7-H)
[化38]

实施例14:11-乙基-4-(2-氟代乙基)-4-羟基-9-(2-苯基乙烯基)-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,并于氩气流和室温下搅拌1.5小时,浓缩反应液,并将残渣溶于二氯甲烷中,再在水洗后进行浓缩干固,向残渣加入3.6mg的2-氨基-5-(2-苯基乙烯基)丙酰基苯,催化剂量的对-甲苯磺酸和0.5ml甲苯,并加热回流2小时,浓缩反应液,并用硅胶柱(溶出溶剂为氯仿→氯仿∶甲醇=25∶1)精制残渣,最后制得4.6mg的目的产物[化40]
H-NMR(在CDCl3.δ.ppm)
【化39】
1.45(3H,t,C11-CH2CH3)
2.21~2.42(2H,m,CH2CH2F)
3.23(2H,q,C11-CH2CH3)
4.60~4.80(2H,m,CH2CH2F)
5.27(2H,s,C12-H)
5.35,5.81(2H,ABq,C1-H)
7.31~7.35(3H,m, )
7.42(2H,t,  )
)
7.61(2H,d, )
7.66(1H,s,C5-H)
8.06(1H,d,C10-H)
8.10(1H,dd,C8-H)
8.20(1H,d,C7-H)
【化40】

实施例15:9-氰基-11-乙基-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入2.6mg2-氨基-5-氰基丙酰基苯、催化剂量的对-甲苯磺酸和0.5ml甲苯,并进行加热回流2小时,浓缩反应液,再用硅胶柱(溶出溶剂为氯仿→氯仿∶甲醇=10∶1)精制残渣,最后制得2.5mg淡褐色结晶的目的产物[化41]。
H-NMR(在CDCl3.δ.ppm)
1.44(3H,t,CH2CH3)
2.25~2.35(2H,m,CH2CH2F)
3.22(2H,q,CH2CH3
4.54~4.84(2H,m,CH2CH2F)
5.27,5.81(2H,ABq,C1-H)
5.30(2H,s,C12-H)
7.65(1H,s,C5-H)
7.95(1H,dd,C8-H)
8.32(1H,d,C7-H)
8.52(1H,d,C10-H)
[化41]
实施例16:11-乙基-4-(2-氟代乙基)-4-羟基-9-硝基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制备的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,并在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣中加入2.9mg2-氨基-5-硝基丙酰基苯,催化剂量的对-甲苯磺酸和0.5ml甲苯,并加热回流2小时,浓缩反应液,并用硅胶柱(溶出溶剂为氯仿→氯仿∶甲醇=50∶1)精制残渣,最后制得3.1mg的目的产物[化42]。
H-NMR(在CDCl3,δ,ppm)
1.47(3H,t,CH2CH3)
2.21~2.35(2H,m,CH2CH2F)
3.30(2H,q,CH2CH3)
4.63~4.81(2H,m,CH2CH2F)
5.33(2H,s,C12-H)
5.35,5.81(2H,ABq,C1-H)
7.73(1H,s,C5-H)
8.37(1H,d,C7-H)
8.57(1H,dd,C8-H)
9.08(1H,d,C10-H)
【化42】

实施例17:9-氨基-11-乙基-4-(2-氰代乙基-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制备的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,并于氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,再在水洗后进行浓缩干固,向残渣加入2.5mg的2,5-二氨基丙酰基苯,催化剂量的对-甲苯磺酸和0.5ml甲苯,并加热回流1小时,浓缩反应液,并用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)精制残渣,最后制得1.1mg褐色结晶的目的产物[化43]。
MS m/z=409(M+)
H-NMR(在CDCl3,δ,ppm)
1.37(3H,t,CH2CH3)
2.21~2.38(2H,m,CH2CH2F)
3.09(2H,q,CH2CH3)
4.55~4.84(2H,m,CH2CH2F)
5.20(2H,s,C12-H)
5.33,5.79(2H,ABq,C1-H)
7.14(1H,d,C10-H)
7.24(1H,dd,C8-H)
7.57(1H,s,C5-H)
8.03(1H,d,C7-H)
【化43】

实施例18:11-乙基-4-(2-氟代乙基)-4-羟基-9-(N,N-二甲胺基)-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制备的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,并在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,再在水洗后进行浓缩干固,向残渣中加入2.6mg2-氨基-5-(N,N-二甲胺基)丙酰基苯,催化剂量的对-甲苯磺酰和0.5ml甲苯,再加热回流1小时,浓缩反应液,并用制备薄层色谱法(展开溶液为氯仿∶甲醇=10∶1)精制残渣,最后制得2.5mg黄色结晶的目的产物[化44]。
MS m/z=437(M+)
H-NMR(在CDCl3,δ,ppm)
1.38(3H,t,CH2CH3)
2.20~2.32(2H,m,CH2CH2F)
3.11(2H,q,CH2CH3)
3.17(6H,s,N(CH3)2)
4.54~4.84(2H,m,CH2CH2F)
5.19(2H,s,C12-H)
5.33,5.78(2H,ABq,C1-H)
7.14(1H,d,C10-H)
7.24(1H,dd,C8-H)
7.57(1H,s,C5-H)
8.03(1H,d,C7-H)
[化44]

实施例19:11-乙基-4-(2-氟代乙基)-4-羟基-9-(4-甲基-1-哌嗪基)-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟乙酸水溶液中,并在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,再在水洗后进行浓缩干固,向残渣加入3.8mg2-氨基-5-(4-甲基-1-哌嗪基)丙酰基苯,催化剂量的对-甲苯磺酸和0.5ml甲苯,然后加热回流1小时,浓缩反应液,再用制备薄层色谱法(展开溶液为氯仿∶甲醇=10∶1)精制残渣,最后制得1.1mg黄褐色结晶的目的产物[化45]。
MS m/z=492(M+)
H-NMR(在CDCl3,δ,ppm)
1.18(3H,t,CH2CH3)
2.20~2.36(2H,m,CH2CH2F)
2.43(3H,s,>NCH3)
2.73(4H,m,ピペラジン-H)
3.06(2H,q,CH2CH3)
3.42(4H,m,ピペラジン-H)
4.55~4.85(2H,m,CH2CH2F)
5.16(2H,s,C12-H)
5.27,5.73(2H,ABq,C1-H)
7.22(1H,d,C10-H)
7.56(1H,dd,C8-H)
7.58(1H,s,C5-H)
8.08(1H,d,C12-H)
【化45】
实施例20:11-乙基-4-(2-氟代乙基)-4-羟基-9-(3-(N,N-二甲胺基甲基)-1-吡咯烷基)-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,并在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,再在水洗后进行浓缩干固,向残渣加入4.1mg2-氨基-5-(3-(N,N-二甲胺基甲基)-1-吡咯烷基)丙酰基苯和0.2ml醋酸,并在90℃下搅拌3小时,浓缩反应液,再向残渣加入二氯甲烷和水并用浓氨水中和之后,分离收取二氯甲烷层,二氯甲烷层在水洗之后进行浓缩,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)进行精制,最后制得3.2mg黄红色结晶的目的产物[化47]。
H-NMR(在CDCl3.δ.ppm)
【化46】
1.36(3H.t.CH2CH3)
1.81~1.89,2.18~2.66(5H,m,CH2CH2F  )
)
2.32(6H,s,N(CH3)2)
3.08(2H,q,CH2CH3)
3.21~3.25,3.49~3.70(6H,m, )
4.57~4.85(2H,m,CH2CH2F)
5.18(2H,s.C12-H)
5.32,5.78(2H,ABq.C1-H)
6.76(1H.d.C10-H)
7.27(1H,dd,C8-H)
7.53(1H,s,C5-H)
8.04(1H,d,C7-H)
[化47]

实施例21:合成3-苄基-9-(2-氟代乙基)-1,2-二氢化-9-羟基-4-甲氧基-3H,12H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-C]苯并[i,J][2,7]萘啶-10,13[9H,15H)-二酮
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,再在水洗后进行浓缩干固,向残渣加入4.2mg的5-氨基-1-苄基-1,2,3,4-四氢化-8-甲氧基-4-喹啉酮,催化剂量的对-甲苯磺酸和0.5ml甲苯,再加热回流1.5小时,浓缩反应液,残渣用硅胶柱(溶出溶剂为氯仿→氯仿∶甲醇=25∶1~10∶1)进行精制,最后制得1.8mg黄色结晶的目的产物[化49]
H-NMR(在CDCl3.δ.ppm)
【化48】
2.21~2.32(2H,m,CH2CH2F)
3.00,3.41(2H×2,t×2,C1,C2-H)
4.08(3H,s,OCH3)
4.43(2H,s,CH2Ph)
4.59~4.72(2H,m,CH2CH2F)
5.15(2H,s,C15-H)
5.34,5.79(2H,ABq,C12-H)
7.29~7.38(3H,m, 
7.48(2H,d,  )
)
7.62(1H,d,C5-H)
7.63(1H,s,C8-H)
7.91(1H,d,C6-H)
[化49]

实施例22:9-(2-氟代乙基)-1,2-二氢化-9-羟基-4-甲氧基-12H-噻吩并[4,3,2-de]吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-10,13(9H,15H)-二酮的合成
将实施例4制得的化合物4.3mg溶于0.5ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入3.6mg5-氨基-8-甲氧基-4-硫色满酮、催化量的对-甲苯磺酸和0.5ml甲苯并加热回流1小时,浓缩反应液,并用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1精制残渣,最后制得0.8mg黄色结晶的目的产物[化50]
H-NMR(在CDCl3.δ.ppm)
2.20~2.34(2H,m,CH2CH2F)
3.21,3.45(2H×2,t×2,C1C2-H)
4.05(3H,s,OCH3)
4.50~4.80(2H,m,CH2CH2F)
5.17(2H,s,C15-H)
5.32,5.74(2H,ABq,C12-H)
7.51(1H,d,C5-H)
7.63(1H,s,C8-H)
7.98(1H,d,C6-H)
[化50]

实施例23:合成9-(2-氟代乙基)-1,2-二氢化-9-羟基-4-甲氧基-12H-吡喃并[4,3,2-de]吡喃并[3′,4∶6,7]吲哚嗪并[1,2-b]喹啉-10,13(9H,15H)-二酮
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,并在氩气流和室温下搅拌1小时,浓缩反应液并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入5-氨基-8-甲氧基-4-色满酮4.3mg,催化量的对-甲苯磺酸和0.7ml甲苯并加热回流1小时,浓缩反应液,并用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)精制残渣,最后制得1.6mg黄色结晶的目的产物[化51]。
H-NMR(在CDCl3,δ,ppm)
2.18~2.35(2H,m,CH2CH2F)
3.32(2H,t,C1-H)
4.07(3H,s,OCH3)
4.59(2H,t,C2-H)
4.59~4.81(2H,m,CH2CH2F)
5.19(2H,s,C15-H)
5.34,5.79(2H,ABq,C12-H)
7.60(1H,d,C5-H)
7.64(1H,S,C8-H)
7.85(1H,d,C6-H)
[化51]

实施例24:合成4-氯-9-(2-氟代乙基)-2,3-二氢-9-羟基-1H,12H-苯并[d,e]吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-10,13(9H,15H)-二酮
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液并将残渣于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入3.0mg的8-氨基-5-氯-1,2,3,4-四氢化-1-萘酮,催化量的对-甲苯磺酸和0.5ml甲苯,加热回流1小时,浓缩反应液,并用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)精制残渣,最后制得3.2mg红色结晶的目的产物[化52]。
H-NMR(在CDCl3,δ,ppm)
2.19~2.41(4H,m,CH2CH2F,C2-H)
3.13,3.22(2H×2,t×2,C1,C3-H)
4.56~4.79(2H,m,CH2CH2F)
5.20(2H,s,C15-H)
5.32,5.75(2H,ABq,C12-H)
7.66(1H,s,C8-H)
7.73(1H,d,C5-H)
7.97(1H,d,C6-H)
[化52]

实施例25:合成9,11-二乙基-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入3.9mg的2-氨基-5-乙基丙酰基苯,催化量的对-甲苯磺酸和0.7ml甲苯,加热回流2小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得3.8mg淡褐色结晶的目的产物[化53]。
H-NMR(在CDCl3,δ,ppm)
1.37~1.43(6H,m,CH2CH3×2)
2.18~2.33(2H,m,CH2CH2F)
2.92(2H,q,C9-CH2CH3)
3.20(2H,q,C11-CH2CH3)
4.58~4.82(2H,m,CH2CH2F)
5.25(2H,s,C12-H)
5.34,5.80(2H,ABq,C1-H)
7.66(1H,s,C5-H)
7.68(1H,dd,C8-H)
7.87(1H,d,C10-H)
8.15(1H,d,C7-H)
【化53】
实施例26:9-乙氧基-11-乙基-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入4.3mg2-氨基-5-乙氧基丙酰基苯,催化量的对-甲苯磺酸和0.7ml甲苯,在加热下回流2小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得3.6mg淡褐色结晶的目的产物[化54]
H-NMR(在CDCl3,δ,ppm)
1.39(3H,t,CH2CH3)
1.54(3H,t,OCH2CH3)
2.21~2.32(2H,m,CH2CH2F)
3.13(2H,q,CH2CH3)
4.22(2H,q,OCH2CH3)
4.57~4.81(2H,m,CH2CH2F)
5.23(2H,s,C12-H)
5.33,5.79(2H,ABq,C1-H)
7.30(1H,d,C10-H)
7.46(1H,dd,C8-H)
7.60(1H,s,C5-H)
8.12(1H,d,C7-H)
[化54]

实施例27:合成11-乙基-4-(2-氟代乙基)-4-羟基-8-甲基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮
将实施例4制得的化合物6.1mg溶于0.5ml80%三氟乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣中加入3.1mg2-氨基-4-甲基丙酰基苯、催化量的对-甲苯磺酸和0.5ml甲苯,加热回流1小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得2.9mg淡褐色结晶的目的产物[化55]
H-NMR(在CDCl3,δ,ppm)
1.40(3H,t,CH2CH3)
2.23~2.30(2H,m,CH2CH2F)
2.61(3H,s,CH3)
3.18(2H,q,CH2CH3)
4.59~4.81(2H,m,CH2CH2F)
5.24(2H,s,C12-H)
5.34,5.80(2H,ABq,C1-H)
7.51(1H,dd,C9-H)
7.65(1H,S,C5-H)
8.00~8.03(2H,m,C7,C10-H)
【化55】
实施例28:合成11-乙基-4-(2-氟化乙基-4-羟基-8-甲氧基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,4(4H,12H)-2酮
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入2.5mg的2-氨基-4-甲氧基丙酰基苯,催化量的对-甲苯磺酸和0.5ml甲苯,并加热回流3小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得4.4mg淡褐色结晶的目的产物[化56]。
H-NMR(在CDCl3,δ,ppm)
1.39(3H,t,CH2CH3)
2.21~2.32(2H,m,CH2CH2F)
3.17(2H,q,CH2CH3)
4.01(3H,s,OCH3)
4.58~4.83(2H,m,CH2CH2F)
5.23(2H,s,C12-H)
5.34,5.80(2H,ABq,C1-H)
7.33(1H,dd,C9-H)
7.54(1H,d,C7-H)
7.65(1H,s,C5-H)
8.01(1H,d,C10-H)
【化56】

实施例29:合成11-乙基-8-氟代-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入3.7mg2-氨基-4-氟代丙酰基苯,催化量的对-甲苯磺酸和0.7ml甲苯,在加热下回流6小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得3.7mg淡褐色结晶的目的产物[化57]。
H-NMR(在CDCl3,δ,ppm)
1.41(3H,t,CH2CH3)
2.21~2.33(2H,m,CH2CH2F)
3.20(2H,q,CH2CH3)
4.59~4.81(2H,m,CH2CH2F)
5.26(2H,s,C12-H)
5.34,5.80(2H,ABq,C1-H)
7.47(1H,d,d,d,C9-H)
7.66(1H,s,C5-H)
7.84(1H,dd,C7-H)
8.13(1H,dd,C10-H)
【化57】

实施例30:合成8-氯代-11-乙基-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,水洗后进行浓缩干固,向残渣加入4.1mg2-氨基-4-氯代丙酰基苯,催化量的对-甲苯酰磺酸和0.7ml甲苯,加热回流6小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)精制,最后制得3.7mg淡褐色结晶的目的产物[化58]。
H-NMR(在CDCl3,δ,ppm)
1.40(3H,t,CH2CH3)
2.21~2.33(2H,m,CH2CH2F)
3.19(2H,q,CH2CH3)
4.59~4.81(2H,m,CH2CH2F)
5.25(2H,s,C12-H)
5.34,5.80(2H,ABq,C1-H)
7.61(1H,dd,C9-H)
7.64(1H,s,C5-H)
8.05(1H,d,C10-H)
8.20(1H,d,C7-H)
【化58】

实施例31:合成8-溴-11-乙基-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,水洗后进行浓缩干固,向残渣加入5.0mg2-氨基-4-溴代丙酰基苯,催化量的对-甲苯磺酸和0.7ml甲苯,再加热回流6小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得4.3mg淡褐色结晶的目的产物[化59]
H-NMR(在CDCl3.δ.ppm)
1.40(3H,t,CH2CH3)
2.21~2.33(2H,m,CH2CH2F)
3.19(2H,q,CH2CH3)
4.59~4.81(2H,m,CH2CH2F)
5.24(2H,s,C12-H)
5.34,5.79(2H,ABq,C1-H)
7.64(1H,s,C5-H)
7.74(1H,d,d,C9-H)
7.98(1H,d,C10-H)
8.39(1H,d,C7-H)
[化59]

实施例32:11-乙基-4-(2-氟代乙基)-4-羟基-8-(N,N-二甲胺基)-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,水洗后进行浓缩干固,向残渣加入2.7mg2-氨基-4-(N,N-二甲胺基)丙酰基苯、催化量的对-甲苯磺酸和0.5ml甲苯,加热回流3小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得3.4mg橙色结晶的目的产物[化60]
H-NMR(在CDCl3,δ,ppm)
1.37(3H,t,CH2CH3)
2.22~2.31(2H,m,CH2CH2F)
3.12(2H,q,CH2CH3)
3.16(6H,s,N(CH3)2)
4.56~4.79(2H,m,CH2CH2F)
5.18(2H,s,C12-H)
5.32,5.79(2H,ABq,C1-H)
7.23(1H,d,C7-H)
7.27(1H,dd,C9-H)
7.62(1H,s,C5-H)
7.95(1H,d,C10-H)
【化60】

实施例33:11-乙基-4-(2-氟代乙基)-4-羟基-10-甲氧基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,水洗后进行浓缩干固,向残渣加入3.9mg2-氨基-6-甲氧基丙酰基苯,催化量的对-甲苯磺酸和0.5ml甲苯,加热回流5小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得4.2mg淡褐色结晶的目的产物[化61]。
H-NMR(在CDCl3,δ,ppm)
1.36(3H,t,CH2CH3)
2.21~2.32(2H,m,CH2CH2F)
3.35~3.43(2H,m,CH2CH3)
4.03(3H,s,OCH3)
4.57~4.83(2H,m,CH2CH2F)
5.25(2H,s,C12-H)
5.34,5.80(2H,ABq,C1-H)
6.90(1H,dd,C9-H)
7.63(1H,S,C5-H)
7.68(1H,t,C8-H)
7.80(1H,dd,C7-H)
【化61】

实施例34:合成11-乙基-4-(2-氟代乙基)-4-羟基-10-(N,N-二甲胺基)-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液并将残渣溶于二氯甲烷中,水洗后浓缩干固,向残渣加入4.2mg2-氨基-6-(N,N-二甲胺基)丙酰基苯,催化量的对-甲苯磺酸和0.7ml甲苯,加热回流5小时,浓缩反应液,残渣用制备薄层法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得4.4mg淡褐色结晶的目的产物[化62]。
H-NMR(在CDCl3,δ,ppm)
1.23(3H,t,CH2CH3)
2.21~2.33(2H,m,CH2CH2F)
2.78,2.79(3H×2,s×2,N(CH3)2)
3.52~3.60(2H,m,CH2CH3)
4.58~4.82(2H,m,CH2CH2F)
5.30(2H,s,C12-H)
5.34,5.81(2H,ABq,C1-H)
7.41(1H,dd,C9-H)
7.64(1H,s,C5-H)
7.69(1H,dd,C8-H)
7.95(1H,dd,C7-H)
【化62】

实施例35:合成4-(2-氟代乙基)-4-羟基-9-甲氧基-11-甲基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,水洗后进行浓缩干固,向残渣加入2.8mg2-氨基-5-甲氧基乙酰基苯,催化量的对-甲苯磺酸和0.5ml甲苯,加热回流1小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)进行精制,最后制得3.8mg淡褐色结晶的目的产物[化63]。
H-NMR(在CDCl3,δ,ppm)
2.17~2.33(2H,m,CH2CH2F)
2.76(3H,s,CH3)
4.01(3H,s,OCH3)
4.58~4.81(2H,m,CH2CH2F)
5.21(2H,s,C12-H)
5.35,5.81(2H,ABq,C1-H)
7.28(1H,d,C10-H)
7.48(1H,dd,C8-H)
7.61(1H,s,C5-H)
8.13(1H,d,C7-H)
【化63】

实施例36:合成4-(2-氟代乙基)-4-羟基-9-甲氧基-11-丙基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液并将残渣溶于二氯甲烷中,水洗后进行浓缩干固,向残渣加入4.3mg2-氨基-5-甲氧基丁酰苯、催化量的对-甲苯磺酸和0.7ml甲苯,加热回流3小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得4.2mg黄褐色结晶的目的产物[化64]。
H-NMR(在CDCl3,δ,ppm)
1.09(3H,t,CH2CH2CH3)
1.81~1.87(2H,m,CH2CH2CH3)
2.21~2.32(2H,m,CH2CH2F)
3.11(2H,t,CH2CH2CH3)
4.00(3H,s,OCH3)
4.57~4.81(2H,m,CH2CH2F)
5.24(2H,s,C12-H)
5.33,5.80(2H,ABq,C1-H)
7.30(1H,d,C10-H)
7.46(1H,dd,C8-H)
7.61(1H,s,C5-H)
8.13(1H,d,C7-H)
【化64】
实施例37:合成4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮
将实施例4制得的化合物31.5mg溶于1.5ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入11.8mg2-氨基苯甲醛和1.5ml醋酸并于100℃下搅拌3小时,浓缩反应液,并将残渣悬浮于乙腈中并过滤收集,制得13.9mg淡褐色结晶的目的产物[化65]。滤液再用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)进行精制并制得6.5mg目的产物。
H-NMR(在CDCl3,δ,ppm)
2.22~2.35(2H,m,CH2CH2F)
4.59~4.82(2H,m,CH2CH2F)
5.31(2H,s,C12-H)
5.35,5.81(2H,ABq,C1-H)
7.68(1H,ddd,C8orC9-H)
7.70(1H,s,C5-H)
7.84(1H,ddd,C8orC9-H)
7.94(1H,dd,C7orC10-H)
8.24(1H,dd,C7orC10-H)
8.41(1H,s,C11-H)
【化65】
[化65]

实施例38:合成4-(2-氟代乙基)-4-羟基-11-羟甲基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮
将实施例37制得的化合物20.0mg悬浮于0.6ml甲醇中,再加入0.5ml75%的硫酸和0.5ml水使其溶解,再加入15.3mg硫酸亚铁7水合物,在冰冷却条件下慢慢加入0.1ml35%的过氧化氢,然后在室温下搅拌5小时后再放置一夜,向反应液加入冰水并过滤收集析出的结晶,水洗后进行减压干燥制得13.3mg淡褐色结晶的目的产物[化66]。
H-NMR(在DMSO-d6,δ,ppm)
2.29(2H,d,t,CH2CH2F)
4.49~4.73(2H,m,CH2CH2F)
5.28,5.43,5.47(2H×3,s×3,CH2OH,C1-H,C12-H)
7.36(1H,s,C5-H)
7.71,7.86(1H×2,ddd×2,C8,C9-H)
8.16~8.20(2H,m,C7,C10-H)
【化66】

实施例39:4-(2-氟代乙基)-11-甲酰基-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例38制得的化合物8.3mg悬浮于4ml醋酸中并加热回流3小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)进行精制,将制得的黄色结晶悬浮于约1ml冰水中,加入0.6ml浓盐酸并在室温下搅拌10小时再于50℃下搅拌1小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)精制得到3.1mg黄色结晶的目的产物[化67]。
H-NMR(在CDCl3)
2.22~2.36(2H,m,CH2CH2F)
4.60~4.81(2H,m,CH2CH2F)
5.35,5.82(2H,ABq,C1-H)
5.62(2H,s,C12-H)
7.70(1H,s,C5-H)
7.87,7.93(1H×2,ddd×2,C8,C9-H)
8.37(1H,dd,C7-H)
8.77(1H,dd,C10-H)
11.2(1H,s,CHO)
【化67】

实施例40:11-乙酸基甲基-4-(2-氟代乙基)-4-羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例38制得的化合物9.4mg悬浮于1ml吡啶与1ml二甲基甲酰胺的混合液中,再加入20μl醋酸酐,将此混合物加热溶解后于室温下搅拌1小时,加入10μl醋酸酐并且再搅拌1小时后,对反应液进行浓缩,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)精制得到1.9mg淡褐色结晶的目的产物[化68]
H-NMR(在CDCl3,δ,ppm)
2.19(3H,s,COCH3)
2.22~2.35(2H,m,CH2CH2F)
4.59~4.83(2H,m,CH2CH2F)
5.34,5.80(2H,ABq,C1-H)
5.47(2H,s,C12-H)
5.73(2H,s,C11CH2OAc)
7.54(1H,s,C5-H)
7.72,7.85(1H×2,ddd×2,C8,C9-H)
8.14,8.26(1H×2,d,C7,C10-H)
【化68】
实施例41:4-(2-氟代乙基)-4-羟基-11-甲基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液,在氩气流和室温下搅拌1小时,浓缩反应液并将残渣溶于二氯甲烷中,水洗后进行浓缩干固,向残渣加入1.9mg2-氨基乙酰苯,催化量的对-甲苯磺酸和0.5ml甲苯,在加热回流2小时,浓缩反应液,并用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制残渣,制得4.6mg淡褐色结晶的目的产物[化69]。
H-NMR(在CDCl3,δ,ppm)
2.22~2.35(2H,m,CH2CH2F)
2.83(3H,s,C11-CH3)
4.59~4.82(2H,m,CH2CH2F)
5.28(2H,s,C12-H)
5.35,5.81(2H,ABq,C1-H)
7.68(1H,s,C5-H)
7.70,7.83(1H×2,ddd×2,C8,C9-H)
8.13,8.23(1H×2,dd×2,C7,C10-H)
【化69】

实施例42:4-(2-氟代乙基)-4-羟基-11-丙基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物5.0mg溶于0.5ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入2-氨基丁酰苯2.6mg,催化量的对-甲苯磺酸和0.5ml甲苯,加热回流2小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=25∶1)进行精制,最后制得3.5mg黄色结晶的目的产物[化70]。
H-NMR(在CDCl3.δ.ppm)
1.10(3H,t,C11-CH2CH2CH3)
1.81~1.87(2H,m,C11-CH2CH2CH3)
2.22~2.35(2H,m,CH2CH2F)
3.17(3H,t,C11CH2CH2CH3)
4.58~4.82(2H,m,CH2CH2F)
5.28(2H,s,C12-H)
5.34,5.80(2H,ABq,C1-H)
7.68,7.82(1H×2,ddd×2,C8,C9-H)
7.70(1H,s,C5-H)
8.13,8.25(1H×2,dd×2,C7,C10-H)
[化70]

实施例43:11-乙基-4-(2-氟代乙基)-4,9-二羟基-1H-吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-3,14(4H,12H)-二酮的合成
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,并在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,在水洗后浓缩干固,向残渣加入3.7mg的2-氨基-5-羟基丙酰苯和0.7ml乙酸,并于100℃下搅拌2小时,浓缩反应液并用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)进行精制残渣,制得4.1mg淡褐色结晶的目的产物[化71]。
H-NMR(在DMSO-d6,δ,ppm)
1.30(3H,t,CH2CH3)
2.23~2.32(2H,m,CH2CH2F)
3.09(2H,q,CH2CH3)
4.48~4.72(2H,m,CH2CH2F)
5.29(2H,s,C12-H)
5.46(2H,s,C1-H)
6.80(1H,s,OH)
7.27(1H,s,C5-H)
7.41(1H,d,C10-H)
7.42(1H,d,C8-H)
8.03(1H,d,C12-H)
10.3(1H,br,OH)
【化71】
实施例44:9-(2-氟代乙基)-2,3-二氢-9-羟基-4-甲基-1H,12H-苯并[d,e]吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-10,13(9H,15H)-二酮的合成
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入4.1mg的8-氨基-5-甲氧基-1,2,3,4-四氢-1-萘酮,催化量的对-甲苯磺酸以及0.5ml甲苯,加热回流1小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)进行精制,最后制得7.4mg淡褐色结晶的目的产物[化76]。
H-NMR(在DMSO-d6.δ.ppm)
2.05~2.07(2H,m,C2-H)
2.31~2.39(2H,m,CH2CH2F)
3.03,3.14(2H×2,m×2,C1,C3-H)
4.01(3H,s,OCH3)
4.52~4.79(2H,m,CH2CH2F)
5.23(2H,s,C15-H)
5.51(2H,s,C12-H)
6.86(1H,s,OH)
7.33(1H,s,C8-H)
7.76(1H,d,C5-H)
8.06(1H,d,C6-H)
[化76]

实施例45:4-乙氧基-9-(2-氟代乙基)-2,3-二氢-9-羟基-1H,12H-苯并[d,e]吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-10,13-(9H,15H)-二酮的合成
将实施例4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,在水洗后浓缩干固,向残渣加入4.4mg的8-氨基-5-乙氧基-1,2,3,4-四氢-1-萘酮,催化量的对-甲苯磺酸以及0.5ml甲苯,加热回流1小时,浓缩反应液并将残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)进行精制,最后得到5.0mg淡褐色结晶的目的产物[化77]。
H-NMR(在DMSO-d6.δ.ppm)
1.40(3H,t,OCH2CH3)
2.01~2.02(2H,m,C2-H)
2.26~2.32(2H,m,CH2CH2F)
2.99,3.10(2H×2,m×2,C1,C3-H)
4.14(2H,q,OCH2CH3)
4.50~4.73(2H,m,CH2CH2F)
5.19(2H,s,C15-H)
5.46(2H,s,C12-H)
6.81(1H,s,OH)
7.29(1H,s,C8-H)
7.70(1H,d,C5-H)
7.98(1H,d,C6-H)
【化77】
实施例46:9-(2-氟代乙基)-2,3-二氢-9-羟基-4-甲基-1H,12H-苯并[d,e]吡喃并[3′,4′∶6,7]吲哚嗪并[1,2-b]喹啉-10,13(9H,15H)-二酮的合成
将实施4制得的化合物7.0mg溶于0.7ml80%的三氟代乙酸水溶液中,在氩气流和室温下搅拌1小时,浓缩反应液,并将残渣溶于二氯甲烷中,在水洗后进行浓缩干固,向残渣加入3.8mg的8-氨基-5-甲基-1,2,3,4-四氢-1-萘酮,催化量的对-甲苯磺酸以及0.5ml甲苯,加热回流1小时,浓缩反应液,残渣用制备薄层色谱法(展开溶剂为氯仿∶甲醇=10∶1)进行精制,最后得到5.4mg淡褐色结晶的目的产物[化78]。
H-NMR(在DMSO-d6.δ.ppm)
2.05~2.08(2H,m,C2-H)
2.26~2.32(2H,m,CH2CH2F)
2.42(3H,s,CH3)
3.00,3.12(2H×2,m×2,C1,C3-H)
4.51~4.71(2H,m,CH2CH2F)
5.18(2H,s,C15-H)
5.46(2H,s,C12-H)
6.83(1H,s,OH)
7.30(1H,s,C8-H)
7.64(1H,d,C5-H)
7.87(1H,d,C6-H)
【化78】
试验例1
p388白血病大白鼠体内的抗溃疡效果:
将106个p388细胞移植到9周令左右的CDF1大白鼠(雌的,未给药(被检测化合物)的一群是5~8只,给药(被检测化合物)的一群是1~5只)的腹腔内,移植后的第一天、第五天和第九天每天一次(共计3次)将溶于或悬浮于1%吐温80(关东化学制)/生理食盐水中的被检测的化合物送入腹腔内,然后用由下列公式示出的T/C值(%)来表示被检测化合物的抗溃疡效果。
[数1]
T/C(%)= (给予被检测化合物的一群的平均生存天数)/(未给予被检测化合物的一群的平均生存天数) ×100
[表1]
表1在p388白血病大白鼠体内的抗溃疡效果
| 被检测的化合物 总给予量(mg/Kg) T/C(%)实施例11 20 483实施例16 20 345实施例24 20 385实施例25 30 345实施例26 30 583实施例41 30 386喜树碱 20 218本发明的化合物显示出极好的抗溃疡效果 |
Claims (8)
1、一种由下列通式(Ⅰ)表示的化合物及其盐:
[化1]
{式中R1表示氢原子、C1-C6的烷基、羟甲基、酰羟甲基、甲酰基、R2、R3、R4各自为独立的并代表氢原子、羟基、C1-C6的烷基、C2-C6的链烯基、C2-C6的炔基、C1-C6烷氧基、卤原子、氨基、C2-C7的酰胺基、C1-C6的烷氨基、NR5(R6)[R5和R6是相同或不同的并代表C1-C6的烷基、或者与其中结合的氮原子一起再构成环的一部分。该环可以含有O、S、N-R7(R7代表氢原子、C1-C6的烷基或氨基的保护基)原子,并且所构成的环是任何一个成环的碳原子均可被C1-C3的烷基、氨基或氨甲基取代的5环或6环]基、苯基、萘基、吡啶基,另外R1和R2他们还可以在一起构成由-(CH2)m-Z-(CH2)n-所示的交联结构[Z表示O、S、CHR8(R8代表氢原子或C1-C6的烷基)或N-R7′(R7′代表氢原子、C1-C6的烷基或氨基的保护基),并且m和n分别代表0、1或2,但是m和n不能同时为零]}。
2、按照权利要求1的化合物及其盐,其特征在于通式(Ⅰ)所示的化合物的4位立体配置为S配位。
3、一种由下列通式(Ⅱ)表示的吡喃并吲哚嗪衍生物:
[化2]
式中Q表示 
4、按照权利要求3的化合物,其特征在于通式(Ⅱ)所示的4位的立体配置为S配位。
5、一种由下列通式(Ⅲ)所示的吡喃并吲哚嗪衍生物:
[化3]

式中R9是氰基、乙酰胺甲基、乙酰氧基甲基,R10是甲酰基、乙酰基、苯酰基或(R)-1-(对-甲苯磺酰基)脯氨酰基。
6、按照权利要求5的化合物,其特征在于通式(Ⅲ)的7位乙酸基的α位的立体配置为S配位。
7、一种由下列通式(Ⅱ1)表示的吡喃并吲哚嗪衍生物的制备方法,
[化9]

其特征在于首先将下面通式(Ⅳ)所示的化合物:
[化4]

(式中R10的代表甲酰基、乙酰基、苯酰基或(R)-1-(对-甲苯磺(酰)脯胺酰基)。
进行2-氟代乙基化制成下面通式(Ⅲ3)所示的化合物:
[化5]

接着将(Ⅲ3)还原成下面通式(Ⅲ2)所示的化合物。
[化6]

然后再将相应的化合物在亚硝基化试剂的存在下进行转位反应,使其成为下面通式(Ⅲ1)所示的化合物:
[化7]
接着再使(Ⅲ1)进行闭环反应而成为下面通式(Ⅱ2)所示的化合物:
[化8]

然后再将相应的化合物进行酮缩醇化反应,使成为(Ⅱ1)所示的吡喃并吲哚嗪衍生物。
8、一种权利要求1或2所述的由下列通式(Ⅰ)所示的化合物:
[化12]

(式中R1,R2,R3和R4的定义如前所述)的制备方法,其特征在于使下面通式(Ⅱ1)所示的化合物
[化10]

与下面通式(Ⅴ)所示的化合物:
[化11]

(式中R1,R2,R3和R4的定义如前所述)进行缩合来制备(Ⅰ)所示的化合物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP21521490 | 1990-08-14 | ||
| JP215214/90 | 1990-08-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1060095A true CN1060095A (zh) | 1992-04-08 |
| CN1031941C CN1031941C (zh) | 1996-06-05 |
Family
ID=16668588
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN91108649A Expired - Fee Related CN1031941C (zh) | 1990-08-14 | 1991-08-14 | 氟代乙基喜树碱衍生物的制备方法 |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP0471358B1 (zh) |
| KR (1) | KR960002854B1 (zh) |
| CN (1) | CN1031941C (zh) |
| CA (1) | CA2048896C (zh) |
| DE (1) | DE69119323T2 (zh) |
| ES (1) | ES2086442T3 (zh) |
| HU (1) | HU213136B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100473654C (zh) * | 1996-10-30 | 2009-04-01 | 田边三菱制药株式会社 | S型4-取代羟基吡喃并吲哚哩啶化合物和其衍生物以及其制备方法 |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU664830B2 (en) * | 1990-09-28 | 1995-12-07 | Smithkline Beecham Corporation | Water soluble camptothecin analogues, processes and methods |
| AP9300587A0 (en) * | 1992-11-12 | 1995-05-05 | Glaxo Inc | Water soluble camptothecin derivatives. |
| US5663177A (en) * | 1995-05-31 | 1997-09-02 | Smithkline Beecham Corporation | Water soluble camptothecin analogs |
| JPH08333370A (ja) * | 1995-06-08 | 1996-12-17 | Kyorin Pharmaceut Co Ltd | 水に可溶な新規フルオロエチルカンプトテシン誘導体、及びその製造方法 |
| IN189180B (zh) * | 1997-07-09 | 2003-01-04 | Chong Kun Dang Corp | |
| AU2002356720B2 (en) | 2001-11-30 | 2009-01-29 | Chugai Seiyaku Kabushiki Kaisha | Hexacyclic compounds |
| BRPI0509732A (pt) | 2004-04-09 | 2007-09-25 | Chugai Pharmaceutical Co Ltd | pró-fármaco solúvel em água, composição farmacêutica, agente terapêutico, agente antifungo e uso do referido pró-fármaco |
| TW200744603A (en) | 2005-08-22 | 2007-12-16 | Chugai Pharmaceutical Co Ltd | Novel anticancer concomitant drug |
| US8093265B2 (en) | 2007-03-09 | 2012-01-10 | Renovis, Inc. | Bicycloheteroaryl compounds as P2X7 modulators and uses thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0220601B1 (en) * | 1985-10-21 | 1991-12-11 | Daiichi Seiyaku Co., Ltd. | Pyranoindolizine derivatives and preparation process thereof |
| JPH0615547B2 (ja) * | 1988-01-20 | 1994-03-02 | 株式会社ヤクルト本社 | 新規なカンプトテシン誘導体 |
-
1991
- 1991-08-07 HU HU912639A patent/HU213136B/hu not_active IP Right Cessation
- 1991-08-09 CA CA002048896A patent/CA2048896C/en not_active Expired - Fee Related
- 1991-08-14 ES ES91113652T patent/ES2086442T3/es not_active Expired - Lifetime
- 1991-08-14 DE DE69119323T patent/DE69119323T2/de not_active Expired - Fee Related
- 1991-08-14 CN CN91108649A patent/CN1031941C/zh not_active Expired - Fee Related
- 1991-08-14 KR KR1019910014063A patent/KR960002854B1/ko not_active Expired - Fee Related
- 1991-08-14 EP EP91113652A patent/EP0471358B1/en not_active Expired - Lifetime
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100473654C (zh) * | 1996-10-30 | 2009-04-01 | 田边三菱制药株式会社 | S型4-取代羟基吡喃并吲哚哩啶化合物和其衍生物以及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| HU213136B (en) | 1997-02-28 |
| DE69119323T2 (de) | 1996-11-14 |
| AU638347B2 (en) | 1993-06-24 |
| HU912639D0 (en) | 1992-01-28 |
| KR960002854B1 (ko) | 1996-02-27 |
| ES2086442T3 (es) | 1996-07-01 |
| CA2048896C (en) | 1999-11-16 |
| EP0471358B1 (en) | 1996-05-08 |
| KR920004392A (ko) | 1992-03-27 |
| EP0471358A1 (en) | 1992-02-19 |
| DE69119323D1 (de) | 1996-06-13 |
| CA2048896A1 (en) | 1992-02-15 |
| AU8161891A (en) | 1992-02-20 |
| HUT63425A (en) | 1993-08-30 |
| CN1031941C (zh) | 1996-06-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1268133A (zh) | 含有稠合环取代基的作为nos抑制剂的2-氨基吡啶 | |
| CN1310705A (zh) | 吲哚衍生物及其用于治疗恶性肿瘤和其它基于病理细胞增生的疾病的用途 | |
| CN1711265A (zh) | 新型氨基-取代的二氢嘧啶并[4,5-d]嘧啶酮衍生物、其制备方法及其作为药剂的应用 | |
| CN1556705A (zh) | 7,8,9,10-四氢-6H-氮杂卓并、6,7,8,9-四氢-吡啶并和2,3-二氢-2H-吡咯并[2,1-b]-喹唑啉酮衍生物 | |
| CN86100090A (zh) | 萘衍生物的制备方法 | |
| CN87101285A (zh) | 8-取代的-2-氨基-1,2,3,4-四氢化荼 | |
| CN1282743A (zh) | 新颖的β-咔啉化合物、它们的制备方法和含有它们的药物组合物 | |
| CN1212692A (zh) | 取代的氮-及二氮杂环庚烷及-环辛烷化合物及其用途 | |
| CN1768058A (zh) | 作为黑皮质素受体激动剂的咪唑并吡啶衍生物 | |
| CN1077954A (zh) | 新的苯并吡喃衍生物 | |
| CN1017707B (zh) | 新的噻唑化合物的制备方法 | |
| CN1092425A (zh) | 吲哚衍生物、其制造方法及其医药用途 | |
| CN1646528A (zh) | 作为有效的α2-肾上腺素受体拮抗剂的多环化合物 | |
| CN1031941C (zh) | 氟代乙基喜树碱衍生物的制备方法 | |
| CN1413205A (zh) | 具有抗肿瘤活性的2-(1h-吲哚-3-基)-2-氧代-乙酰胺 | |
| CN1157394C (zh) | 二氢呋喃并[3,4-b]喹啉-1-酮类化合物、其制备方法和含有这些化合物的药物组合物 | |
| CN1054600A (zh) | 新的胆碱酯酶抑制剂三环—环胺 | |
| CN1079745A (zh) | 新的9-氟-7-氧代-7H-吡啶并[1,2,3-d,e][1,4]苯并嗪-6-羧酸及其酯 | |
| CN1036758A (zh) | 双环胺化合物及其制备方法 | |
| CN1246120A (zh) | 咪唑并哒嗪类化合物 | |
| CN1124026A (zh) | 新型哌啶基硫代吲哚衍生物,它们的制备方法,和含有它们的药物组合物,作为止痛药的用途 | |
| CN1066449C (zh) | 新的三环肟醚,其制备方法和含它们的药物组合物 | |
| CN1276920C (zh) | 噻唑烷二酮衍生物以及包含该衍生物的药物组合物 | |
| CN87103087A (zh) | 作为血小板活化因子—拮抗剂的哌嗪化合物及其制备方法 | |
| CN1181075C (zh) | 新型嘧啶-4-酮化合物、其制备方法和含有它们的药物组合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C15 | Extension of patent right duration from 15 to 20 years for appl. with date before 31.12.1992 and still valid on 11.12.2001 (patent law change 1993) | ||
| OR01 | Other related matters | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |