CN102491317B - Preparation method of graphene - Google Patents
Preparation method of graphene Download PDFInfo
- Publication number
- CN102491317B CN102491317B CN201110412972.8A CN201110412972A CN102491317B CN 102491317 B CN102491317 B CN 102491317B CN 201110412972 A CN201110412972 A CN 201110412972A CN 102491317 B CN102491317 B CN 102491317B
- Authority
- CN
- China
- Prior art keywords
- graphene
- graphite oxide
- zinc
- preparation
- colloid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 title claims abstract description 138
- 229910021389 graphene Inorganic materials 0.000 title claims abstract description 66
- 238000002360 preparation method Methods 0.000 title claims abstract description 21
- 229910002804 graphite Inorganic materials 0.000 claims abstract description 70
- 239000010439 graphite Substances 0.000 claims abstract description 70
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims abstract description 50
- 229910052725 zinc Inorganic materials 0.000 claims abstract description 42
- 239000011701 zinc Substances 0.000 claims abstract description 42
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 34
- 239000000084 colloidal system Substances 0.000 claims abstract description 31
- 239000003638 chemical reducing agent Substances 0.000 claims abstract description 23
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 34
- 238000006243 chemical reaction Methods 0.000 claims description 28
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 18
- 239000003513 alkali Substances 0.000 claims description 10
- 230000035484 reaction time Effects 0.000 claims description 6
- 238000000034 method Methods 0.000 abstract description 24
- 230000002378 acidificating effect Effects 0.000 abstract description 20
- 125000000524 functional group Chemical group 0.000 abstract description 8
- 230000007613 environmental effect Effects 0.000 abstract description 6
- 229910052751 metal Inorganic materials 0.000 abstract description 5
- 239000002184 metal Substances 0.000 abstract description 5
- 231100000252 nontoxic Toxicity 0.000 abstract description 5
- 230000003000 nontoxic effect Effects 0.000 abstract description 5
- 230000036541 health Effects 0.000 abstract description 4
- 238000006722 reduction reaction Methods 0.000 description 33
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 31
- 230000009467 reduction Effects 0.000 description 24
- 239000000243 solution Substances 0.000 description 14
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 10
- 239000000203 mixture Substances 0.000 description 10
- 239000002253 acid Substances 0.000 description 8
- 230000008901 benefit Effects 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- 230000000694 effects Effects 0.000 description 4
- 238000000921 elemental analysis Methods 0.000 description 4
- 239000011229 interlayer Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 238000002441 X-ray diffraction Methods 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 229910044991 metal oxide Inorganic materials 0.000 description 3
- 150000004706 metal oxides Chemical class 0.000 description 3
- 239000002994 raw material Substances 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 239000003575 carbonaceous material Substances 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 239000004020 conductor Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 229910000000 metal hydroxide Inorganic materials 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 238000002371 ultraviolet--visible spectrum Methods 0.000 description 2
- DIIIISSCIXVANO-UHFFFAOYSA-N 1,2-Dimethylhydrazine Chemical compound CNNC DIIIISSCIXVANO-UHFFFAOYSA-N 0.000 description 1
- XMWRBQBLMFGWIX-UHFFFAOYSA-N C60 fullerene Chemical class C12=C3C(C4=C56)=C7C8=C5C5=C9C%10=C6C6=C4C1=C1C4=C6C6=C%10C%10=C9C9=C%11C5=C8C5=C8C7=C3C3=C7C2=C1C1=C2C4=C6C4=C%10C6=C9C9=C%11C5=C5C8=C3C3=C7C1=C1C2=C4C6=C2C9=C5C3=C12 XMWRBQBLMFGWIX-UHFFFAOYSA-N 0.000 description 1
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 1
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 238000005452 bending Methods 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 238000001354 calcination Methods 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 239000002041 carbon nanotube Substances 0.000 description 1
- 229910021393 carbon nanotube Inorganic materials 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000003922 charged colloid Substances 0.000 description 1
- 238000005229 chemical vapour deposition Methods 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 229910003460 diamond Inorganic materials 0.000 description 1
- 239000010432 diamond Substances 0.000 description 1
- 238000002050 diffraction method Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 125000003700 epoxy group Chemical group 0.000 description 1
- 238000004299 exfoliation Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229910003472 fullerene Inorganic materials 0.000 description 1
- 229910052733 gallium Inorganic materials 0.000 description 1
- 229910052732 germanium Inorganic materials 0.000 description 1
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 238000012844 infrared spectroscopy analysis Methods 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000002122 magnetic nanoparticle Substances 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 150000004692 metal hydroxides Chemical class 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012805 post-processing Methods 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- 238000000634 powder X-ray diffraction Methods 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000004065 semiconductor Substances 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000003313 weakening effect Effects 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Images





Landscapes
- Carbon And Carbon Compounds (AREA)
Abstract
本发明提供一种石墨烯的制备方法,包括:在超声波振荡的条件下将氧化石墨分散于水中,得到胶体;在酸性或碱性的条件下,以锌为还原剂还原所述氧化石墨,得到石墨烯;所述氧化石墨与锌的重量比为1∶(5.5~6.5)。本发明使用锌作为还原剂,锌对氧化石墨具有较好的还原性能,且其廉价无毒,不会损害操作人员的身体健康,环保性能较高,以锌作为还原剂还不会引入其他官能团,有利于保持石墨烯良好的电学性能。更为重要的是,锌是一种两性金属,使得上述方法既可在酸性条件下又可在碱性条件下制备石墨烯,因此,采用上述方法可根据需求灵活选择制备条件。The invention provides a preparation method of graphene, comprising: dispersing graphite oxide in water under the condition of ultrasonic vibration to obtain a colloid; under acidic or alkaline conditions, using zinc as a reducing agent to reduce the graphite oxide to obtain Graphene; the weight ratio of graphite oxide to zinc is 1: (5.5-6.5). The present invention uses zinc as a reducing agent. Zinc has good reducing performance on graphite oxide, is cheap and non-toxic, does not damage the health of operators, and has high environmental protection performance. Using zinc as a reducing agent will not introduce other functional groups , which is conducive to maintaining the good electrical properties of graphene. More importantly, zinc is an amphoteric metal, so that the above method can prepare graphene under both acidic and alkaline conditions. Therefore, the above method can be used to flexibly select the preparation conditions according to requirements.
Description
技术领域 technical field
本发明涉及石墨烯领域,特别涉及一种石墨烯的制备方法。 The invention relates to the field of graphene, in particular to a preparation method of graphene. the
背景技术 Background technique
石墨烯是单层碳原子紧密堆积成二维蜂窝状结构的一种新型碳材料,其是构件其他维度碳质材料,如零维富勒烯、一维纳米碳管、三维石墨等的基本单元。 Graphene is a new type of carbon material in which a single layer of carbon atoms is tightly packed into a two-dimensional honeycomb structure. It is the basic unit of other dimensional carbon materials, such as zero-dimensional fullerenes, one-dimensional carbon nanotubes, and three-dimensional graphite. . the
石墨烯除了具有特殊的结构外,还具有诸多独特性质,最为显著是导热性和机械强度。石墨烯是一种良好的导体,可以很快的散发热量,而电子穿过石墨烯几乎没有任何阻力,所产生的热量也非常少。石墨烯非常牢固坚硬,其硬度高于钻石,强度优于钢铁,理想强度可达110~130GPa。同时,石墨烯又是一种非常优异的半导体材料,具有比硅高很多的载流子迁移率。石墨烯还是目前已知在常温下导电性能最优秀的材料,电子在其中的运动速度高于一般导体,这一特性使得其在纳米电子元件、传感器、晶体管和电池领域中有着巨大的应用前景。石墨烯还具有良好的透光性,是传统ITO膜潜在的替代产品。 In addition to its special structure, graphene also has many unique properties, most notably thermal conductivity and mechanical strength. Graphene is a good conductor that can dissipate heat quickly, and electrons pass through graphene with almost no resistance, and the heat generated is very small. Graphene is very strong and hard, its hardness is higher than that of diamond, and its strength is better than that of steel. The ideal strength can reach 110-130GPa. At the same time, graphene is a very excellent semiconductor material with a much higher carrier mobility than silicon. Graphene is also currently known as the material with the best electrical conductivity at room temperature, and the movement speed of electrons in it is higher than that of ordinary conductors. This feature makes it have great application prospects in the fields of nanoelectronic components, sensors, transistors and batteries. Graphene also has good light transmission and is a potential substitute for traditional ITO films. the
石墨烯的制备方法主要有化学还原法,微机械剥离法和化学气相沉淀法,后两种方法产量较低,对工艺要求较高。因此目前主要采用化学还原法。化学还原法以氧化石墨为原料,通常用二甲肼、对苯二酚和硼氢化钠等为还原剂还原氧化石墨得到石墨烯。但是这些还原剂毒性较大或者易燃,不利于操作人员的健康,操作也较为不便,而且会引入其他官能团影响石墨烯的电学性能。 The preparation methods of graphene mainly include chemical reduction method, micro-mechanical exfoliation method and chemical vapor deposition method. The latter two methods have low output and high process requirements. Therefore, the chemical reduction method is mainly used at present. The chemical reduction method uses graphite oxide as a raw material, and usually uses dimethylhydrazine, hydroquinone, and sodium borohydride as reducing agents to reduce graphite oxide to obtain graphene. However, these reducing agents are highly toxic or flammable, which is not conducive to the health of operators and is inconvenient to operate, and will introduce other functional groups to affect the electrical properties of graphene. the
发明内容 Contents of the invention
本发明解决的技术问题在于提供一种石墨烯的制备方法,该方法不使用有毒原料,环保性较高,且不会引入其他官能团影响石墨烯的电学性能。 The technical problem solved by the present invention is to provide a preparation method of graphene, which does not use toxic raw materials, has high environmental protection, and does not introduce other functional groups to affect the electrical properties of graphene. the
有鉴于此,本发明提供一种石墨烯的制备方法,包括: In view of this, the invention provides a kind of preparation method of graphene, comprising:
a)、在超声波振荡的条件下将氧化石墨分散于水中,得到胶体; a), under the condition of ultrasonic vibration, graphite oxide is dispersed in water to obtain a colloid;
b)、在酸性或碱性的条件下,以锌为还原剂还原所述氧化石墨,得到石墨烯;所述氧化石墨与锌的重量比为1∶(5.5~6.5)。 b) Under acidic or alkaline conditions, using zinc as a reducing agent to reduce the graphite oxide to obtain graphene; the weight ratio of the graphite oxide to zinc is 1: (5.5-6.5). the
优选的,所述步骤b)在碱性条件下进行,具体为: Preferably, the step b) is carried out under alkaline conditions, specifically:
b1)、向所述胶体中依次加入摩尔比为1∶(10~15)的锌和碱,得到石墨烯;所述碱为氢氧化钠或氢氧化钾,所述氧化石墨与锌的重量比为1∶(5.5~6.5)。 b1), in the colloid, adding zinc and alkali successively in a molar ratio of 1: (10~15) to obtain graphene; the alkali is sodium hydroxide or potassium hydroxide, and the weight ratio of graphite oxide to zinc is It is 1: (5.5~6.5). the
优选的,所述氢氧化钠或氢氧化钾与氧化石墨的重量比为(15~30)∶1。 Preferably, the weight ratio of sodium hydroxide or potassium hydroxide to graphite oxide is (15-30):1. the
优选的,所述步骤b1的反应时间为5h~8h。 Preferably, the reaction time of the step b1 is 5h-8h. the
优选的,所述步骤b1的反应温度为80℃~100℃。 Preferably, the reaction temperature in step b1 is 80°C to 100°C. the
优选的,所述步骤b)在酸性条件下进行,具体为: Preferably, the step b) is carried out under acidic conditions, specifically:
b2)、向所述胶体中依次加入锌和酸液,得到石墨烯;所述酸液为浓度为0.5mol/L~2mol/L的盐酸或硫酸溶液,所述氧化石墨与锌的重量比为1∶(5.5~6.5)。 b2), in the colloid, add zinc and acid solution successively, obtain graphene; Described acid solution is the hydrochloric acid or the sulfuric acid solution that concentration is 0.5mol/L~2mol/L, and the weight ratio of described graphite oxide and zinc is 1: (5.5~6.5). the
所述盐酸中的HCl或硫酸与氧化石墨的重量比为(15~50)∶1。 The weight ratio of HCl or sulfuric acid in the hydrochloric acid to graphite oxide is (15-50):1. the
所述步骤b2的反应时间为5h~8h。 The reaction time of the step b2 is 5h-8h. the
所述胶体中氧化石墨的浓度为0.8g/L 1.2g/L。 The concentration of graphite oxide in the colloid is 0.8g/L-1.2g/L. the
本发明提供一种石墨烯的制备方法,其是先将氧化石墨分散于水中得到氧化石墨胶体,然后以锌为还原剂,在酸性或碱性条件下还原氧化石墨制得。本发明使用锌作为还原剂,锌对氧化石墨具有较好的还原性能,且其廉价无毒,不会损害操作人员的身体健康,环保性能较高,以锌作为还原剂还不会引入其他官能团,有利于保持石墨烯良好的电学性能。更为重要的是,锌是一种两性金属,使得上述方法既可在酸性条件下又可在碱性条件下制备石墨烯,在酸性条件下,产物 纯度较高,纯化步骤简单,后处理容易;在碱性条件下制备,还原程度较高,且可实现还原氧化石墨的同时实现对石墨烯的修饰。因此,采用上述方法可根据需求灵活选择制备条件。 The invention provides a preparation method of graphene, which is obtained by firstly dispersing graphite oxide in water to obtain graphite oxide colloid, and then using zinc as a reducing agent to reduce graphite oxide under acidic or alkaline conditions. The present invention uses zinc as a reducing agent. Zinc has good reducing performance on graphite oxide, is cheap and non-toxic, does not damage the health of operators, and has high environmental protection performance. Using zinc as a reducing agent will not introduce other functional groups , which is conducive to maintaining the good electrical properties of graphene. More importantly, zinc is an amphoteric metal, so that the above method can prepare graphene under acidic conditions and alkaline conditions. Under acidic conditions, the product has higher purity, simple purification steps, and easy post-treatment. ; Prepared under alkaline conditions, the degree of reduction is high, and the modification of graphene can be realized while reducing graphite oxide. Therefore, the above method can be used to flexibly select the preparation conditions according to the needs. the
附图说明 Description of drawings
图1为氧化石墨溶液的紫外-可见光谱; Fig. 1 is the ultraviolet-visible spectrum of graphite oxide solution;
图2为a、b、c、d的XRD图; Fig. 2 is the XRD figure of a, b, c, d;
图3为e、f、g、h的XRD图; Fig. 3 is the XRD figure of e, f, g, h;
图4为a、b、c、d的红外光谱图; Fig. 4 is the infrared spectrogram of a, b, c, d;
图5为e、f、g、h的红外光谱图。 Fig. 5 is the infrared spectrogram of e, f, g, h. the
具体实施方式 Detailed ways
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。 In order to further understand the present invention, the preferred embodiments of the present invention are described below in conjunction with examples, but it should be understood that these descriptions are only to further illustrate the features and advantages of the present invention, rather than limiting the claims of the present invention. the
本发明实施例公开了一种石墨烯的制备方法,包括如下步骤: The embodiment of the present invention discloses a kind of preparation method of graphene, comprises the following steps:
a)、在超声波振荡的条件下将氧化石墨分散于水中,得到胶体; a), under the condition of ultrasonic vibration, graphite oxide is dispersed in water to obtain a colloid;
b)、在酸性或碱性条件下,以锌为还原剂还原所述氧化石墨,得到石墨烯;所述氧化石墨与锌的重量比为1∶(5.5~6.5)。 b) Under acidic or alkaline conditions, using zinc as a reducing agent to reduce the graphite oxide to obtain graphene; the weight ratio of the graphite oxide to zinc is 1: (5.5-6.5). the
上述方法是先采用超声波振荡将氧化石墨分散于水中,得到胶体,然后以锌作为还原剂来还原氧化石墨,由于锌是一种两性金属,在酸性条件或碱性条件均能发挥还原作用,使得该还原反应在酸性条件和碱性条件下均能进行。在酸性条件下制备石墨烯的优势在于产物纯度较高,纯化步骤简单,后处理容易;在碱性条件下制备石墨烯的优势在于还原程度较高,且可实现还原氧化石墨烯的同时实现对石墨烯进行修饰。因此,采用上述方法可根据需求灵活选择制备条件。并且采用的锌还原剂无毒,使用较为环保,且不会引入其他官能团影响石墨烯的电学性能。 The above method is to disperse graphite oxide in water by ultrasonic vibration to obtain colloid, and then use zinc as a reducing agent to reduce graphite oxide. Since zinc is an amphoteric metal, it can play a reducing role in acidic or alkaline conditions. The reduction reaction can be carried out under both acidic and basic conditions. The advantage of preparing graphene under acidic conditions is that the product has high purity, simple purification steps, and easy post-treatment; the advantage of preparing graphene under alkaline conditions is that the degree of reduction is high, and the reduction of graphene oxide can be achieved at the same time. Graphene is modified. Therefore, the above method can be used to flexibly select the preparation conditions according to the needs. And the zinc reducing agent used is non-toxic, more environmentally friendly, and will not introduce other functional groups to affect the electrical properties of graphene. the
上述制备方法中步骤a是将石墨烯分散在水介质的过程,氧化石 墨烯可以按照Hummer法制备,为了保证石墨充分氧化,氧化石墨优选按照如下方法制备: In the above-mentioned preparation method, step a is the process of dispersing graphene in an aqueous medium. Graphene oxide can be prepared according to the Hummer method. In order to ensure that graphite is fully oxidized, graphite oxide is preferably prepared according to the following method:
S1)、将石墨与浓硫酸混合后置于冰浴中搅拌15min~20min; S1), mix the graphite and concentrated sulfuric acid and place in an ice bath to stir for 15min to 20min;
S2)、向步骤S1得到的混合物中加入高锰酸钾,在30℃~40℃保温25min~40min; S2), adding potassium permanganate to the mixture obtained in step S1, and incubating at 30°C to 40°C for 25min to 40min;
S3)、向步骤S2得到的混合物中加入蒸馏水稀释,置于95℃~100℃的油浴中保温10~20min,再加入过氧化氢溶液; S3), adding distilled water to the mixture obtained in step S2 for dilution, placing it in an oil bath at 95° C. to 100° C. for 10 to 20 minutes, and then adding hydrogen peroxide solution;
S4)、将产品离心分离,用盐酸洗涤后,再用去离子水清洗至pH值到5,干燥。 S4), the product is centrifuged, washed with hydrochloric acid, and then washed with deionized water until the pH value reaches 5, and dried. the
为了使步骤b的还原反应更为充分,本发明优选控制胶体中氧化石墨的浓度为0.8g/L 1.2g/L。 In order to make the reduction reaction of step b more fully, the present invention preferably controls the concentration of graphite oxide in the colloid to be 0.8g/L-1.2g/L. the
步骤b是以锌为还原剂将氧化石墨进行还原的工序,本发明选择锌作为还原剂的原因在于:首先,锌是一种廉价无毒的还原剂,使用其作为还原剂不会操作人员的健康构成伤害,环保性能好;其次,锌是一种两性金属,无论在酸性条件还是在碱性条件下其均能够对氧化石墨进行还原,制备条件可灵活选择;相对于其他两性金属,镓、锗和铟单质价格较为昂贵,生产成本过高;锡的电极电势较低,还原性不及锌,若以锡为还原剂会造成反应条件过于苛刻,或产物还原不充分,产物纯度低;铝的电极电势虽然较高,但本发明人经研究发现:将其应用于氧化石墨的还原,其还原性能不及锌。为此,本发明选择以锌作为还原剂,以提高反应的环保性能、制备条件的灵活选择性和产物纯度。 Step b is a process of reducing graphite oxide with zinc as a reducing agent. The reason why the present invention selects zinc as a reducing agent is that: first, zinc is a cheap and non-toxic reducing agent, and using it as a reducing agent will not cause operator inconvenience. It poses harm to health and has good environmental protection performance; secondly, zinc is an amphoteric metal, which can reduce graphite oxide no matter in acidic or alkaline conditions, and the preparation conditions can be flexibly selected; compared with other amphoteric metals, gallium, The price of germanium and indium is relatively expensive, and the production cost is too high; the electrode potential of tin is low, and the reducibility is not as good as that of zinc. If tin is used as the reducing agent, the reaction conditions will be too harsh, or the product reduction will be insufficient, and the product purity will be low; Although the electrode potential is relatively high, the inventors have found through research that: when it is applied to the reduction of graphite oxide, its reducing performance is inferior to that of zinc. For this reason, the present invention selects zinc as the reducing agent, to improve the environmental performance of the reaction, the flexible selectivity of the preparation conditions and the product purity. the
在还原氧化石墨的过程中,为了保证使氧化石墨被充分还原,本发明控制氧化石墨与锌的重量比为1∶(5.5~6.5),优选为1∶(5.6~6.0),更优选为1∶(5.7~5.8)。 In the process of reducing graphite oxide, in order to ensure that graphite oxide is fully reduced, the present invention controls the weight ratio of graphite oxide to zinc to be 1: (5.5-6.5), preferably 1: (5.6-6.0), more preferably 1 : (5.7~5.8). the
锌即可在酸性条件下又可在碱性条件下实现对氧化石墨的还原,若反应在碱性条件下进行,则步骤b优选按照如下方式进行: Zinc can realize the reduction of graphite oxide under acidic conditions and under alkaline conditions, if the reaction is carried out under alkaline conditions, then step b is preferably carried out as follows:
b1)、在60℃~100℃,向上述胶体中依次加入锌和碱,所述碱为 NaOH或KOH,氧化石墨与锌的重量比为1∶(5.5~6.5),得到石墨烯。 b1), at 60°C to 100°C, add zinc and alkali successively to the colloid, the alkali is NaOH or KOH, and the weight ratio of graphite oxide to zinc is 1: (5.5 to 6.5) to obtain graphene. the
上述步骤中,为了提高产物的纯度,优选设置反应温度为80℃~100℃,还原反应的时间优选设置为5h~8h。碱一方面用于与锌反应,另一方面用于分散胶体,碱加入量过低易造成反应速率过慢,还原不够充分,由此需要延长反应时间;碱加入量过高则会造成过多的碱残留,增加了后续对产物进行洗涤的次数。为此,本发明优选控制碱和锌的摩尔比为(11~13)∶1。氧化石墨与氢氧化钠或氢氧化钾的重量比优选为1∶(15~30)。 In the above steps, in order to improve the purity of the product, the reaction temperature is preferably set at 80° C. to 100° C., and the reduction reaction time is preferably set at 5 h to 8 h. On the one hand, the alkali is used to react with zinc, and on the other hand, it is used to disperse the colloid. If the amount of alkali added is too low, the reaction rate will be too slow and the reduction will not be sufficient, thus prolonging the reaction time; Alkali residues increase the number of times the product is subsequently washed. For this reason, the present invention preferably controls the molar ratio of alkali and zinc to be (11~13):1. The weight ratio of graphite oxide to sodium hydroxide or potassium hydroxide is preferably 1: (15-30). the
碱性条件下还原氧化石墨的优点在于:还原程度较高,且便于对石墨烯进行修饰。在碱性条件下,氧化石墨更容易剥离得到稳定的带负电的胶体,可与带正电的片状分子进行静电组装,而这种分子层次的组装比现有的物理混合效果好。因此,采用上述方法通过在还原反应过程中引入客体分子而实现一步制备出修饰效果优异的石墨烯。 The advantage of reducing graphite oxide under alkaline conditions is that the degree of reduction is high and it is convenient to modify graphene. Under alkaline conditions, graphite oxide is easier to exfoliate to obtain a stable negatively charged colloid, which can be electrostatically assembled with positively charged sheet molecules, and this molecular level assembly is better than the existing physical mixing effect. Therefore, the graphene with excellent modification effect can be prepared in one step by introducing guest molecules during the reduction reaction by using the above method. the
引入的客体分子是在碱性条件下稳定存在的,且能与氧化石墨表面的-OH、-O-、-COOH等反应的化合物,可以为:金属氧化物,如Fe3O4磁性纳米粒子、ZnO或TiO2等;生物小分子,如寡核苷酸等;有机小分子,如吡啶等;层状化合物,如层状双金属氢氧化物等。若客体分子为金属氧化物或氢氧化物,在高温真空煅烧还原后得到金属氧化物与石墨烯的复合材料。 The introduced guest molecule is a compound that exists stably under alkaline conditions and can react with -OH, -O-, -COOH on the surface of graphite oxide, which can be: metal oxides, such as Fe 3 O 4 magnetic nanoparticles , ZnO or TiO2 , etc.; small biological molecules, such as oligonucleotides, etc.; small organic molecules, such as pyridine, etc.; layered compounds, such as layered double metal hydroxides, etc. If the guest molecules are metal oxides or hydroxides, a composite material of metal oxides and graphene can be obtained after high-temperature vacuum calcination and reduction.
此外,在碱性条件下且在80℃~100℃的温度范围制备石墨烯还可以提高氧化石墨烯的还原程度,产物纯度较高。 In addition, preparing graphene under alkaline conditions and at a temperature range of 80° C. to 100° C. can also increase the reduction degree of graphene oxide, and the product has a higher purity. the
若还原反应在酸性条件下进行,则步骤b优选按照如下方式进行: If the reduction reaction is carried out under acidic conditions, then step b is preferably carried out as follows:
b2)、向上述胶体中依次加入锌和酸液,得到石墨烯,所述酸液为浓度为0.5mol/L~2mol/L的盐酸或硫酸溶液,氧化石墨与锌的重量比为1∶(5.5~6.5)。 b2), add zinc and acid solution successively to above-mentioned colloid, obtain graphene, and described acid solution is the hydrochloric acid or sulfuric acid solution that concentration is 0.5mol/L~2mol/L, and the weight ratio of graphite oxide and zinc is 1:( 5.5~6.5). the
酸性条件下制备石墨烯的优点在于:产物纯度较高,纯化步骤简单,后处理容易。上述步骤中,为了使氧化石墨还原充分,还原反应的温度优选设置为60℃~100℃,时间优选设置为5h~8h。酸液浓度过 低易造成反应速率过慢,还原不够充分,由此需要延长反应时间;酸液浓度过高则会造成过多的酸残留,增加了后续对产物进行洗涤的次数。为此,本发明优选控制酸液的浓度为0.5mol/L~2mol/L。盐酸中的HCl或硫酸与氧化石墨的重量比优选为(15~50)∶1。 The advantages of preparing graphene under acidic conditions are: high product purity, simple purification steps, and easy post-processing. In the above steps, in order to fully reduce the graphite oxide, the temperature of the reduction reaction is preferably set at 60° C. to 100° C., and the time is preferably set at 5 h to 8 h. If the concentration of the acid solution is too low, the reaction rate is too slow and the reduction is not sufficient, so the reaction time needs to be prolonged; if the concentration of the acid solution is too high, there will be too much acid residue, which increases the number of subsequent washings of the product. For this reason, the present invention preferably controls the concentration of the acid solution to be 0.5 mol/L-2 mol/L. The weight ratio of HCl in hydrochloric acid or sulfuric acid to graphite oxide is preferably (15-50):1. the
按照上述方法将反应液过滤后,用盐酸洗涤去除未反应的锌,再用水洗涤附着于产物表面的锌盐,干燥,即得石墨烯。 After filtering the reaction solution according to the above method, washing with hydrochloric acid to remove unreacted zinc, washing with water the zinc salt attached to the surface of the product, and drying to obtain graphene. the
由上述结果可知,采用本发明提供的方法制备石墨烯,其采用廉价无毒的锌作为还原剂,环保性能好;不会引入其他官能团而影响石墨烯电学性能;同时,其既可在酸性条件下又可在碱性条件下制备石墨烯,在酸性条件下产物纯度较高,纯化步骤简单,后处理容易;在碱性条件下可实现还原氧化石墨的同时实现对石墨烯进行修饰。因此,采用上述方法可根据需求灵活选择制备条件。 As can be seen from the above results, the method provided by the present invention is used to prepare graphene, which uses cheap and non-toxic zinc as a reducing agent, and has good environmental protection performance; it will not introduce other functional groups to affect the electrical properties of graphene; at the same time, it can be used in acidic conditions. Graphene can be prepared under alkaline conditions, and the product purity is higher under acidic conditions, the purification steps are simple, and the post-treatment is easy; under alkaline conditions, graphite oxide can be reduced and graphene can be modified at the same time. Therefore, the above method can be used to flexibly select the preparation conditions according to the needs. the
为了进一步理解本发明,下面结合实施例对本发明提供的石墨烯的制备方法进行描述,本发明的保护范围不受以下实施例的限制。 In order to further understand the present invention, the preparation method of graphene provided by the present invention is described below in conjunction with examples, and the protection scope of the present invention is not limited by the following examples. the
实施例1制备氧化石墨
1、蒸馏水冲洗石墨粉末,倾去上层悬浮物,过滤,干燥; 1. Rinse the graphite powder with distilled water, pour off the suspended matter in the upper layer, filter and dry;
2、取干燥后的石墨5g、98%浓硫酸230mL混合置于冰浴中,搅拌15min,使其充分混合.称取30g KMnO4加入上述混合液继续搅拌2h后,移入35℃中温水浴中继续搅拌30min,将460ml蒸馏水缓缓加入反应体系中,然后置于98℃油浴中保温15min,之后加入1400ml温水,再加入100ml 30%H2O2,将产品离心,用2L 5%HCl溶液洗涤,再用水洗至PH≈5,干燥,得到氧化石墨。 2. Take 5g of dried graphite and 230mL of 98% concentrated sulfuric acid, mix them in an ice bath, stir for 15 minutes, and make them fully mixed. Weigh 30g of KMnO 4 and add the above mixture to continue stirring for 2 hours, then transfer it to a medium-temperature water bath at 35°C Continue to stir for 30 minutes, slowly add 460ml of distilled water into the reaction system, then place it in a 98°C oil bath for 15 minutes, then add 1400ml of warm water, then add 100ml of 30% H 2 O 2 , centrifuge the product, and wash it with 2L of 5% HCl solution Washing, and then washing with water to pH ≈ 5, and drying to obtain graphite oxide.
采用超声波振荡将120mg氧化石墨分散于150ml水中,如图1所示为该氧化石墨溶液的紫外-可见光谱,氧化石墨溶液在波长230nm处有一个特征峰,在300nm左右有一个特征峰,由此可知氧化石墨成功制备。 Adopt ultrasonic vibration to disperse 120mg graphite oxide in 150ml water, as shown in Figure 1, be the ultraviolet-visible spectrum of this graphite oxide solution, graphite oxide solution has a characteristic peak at wavelength 230nm place, has a characteristic peak at about 300nm, thus It can be seen that graphite oxide was successfully prepared. the
以下实施例所使用的氧化石墨均由本实施例制备。 The graphite oxide used in the following examples is all prepared by this example. the
实施例2 Example 2
1、采用超声波振荡将120mg氧化石墨分散于120ml水中,得到胶体; 1. Use ultrasonic vibration to disperse 120mg graphite oxide in 120ml water to obtain a colloid;
2、向步骤1得到的胶体中依次加入655mg锌粉和30ml 2.0mol/L盐酸,室温反应6小时后结束反应,将反应得到的混合物过滤,水洗,干燥,得到石墨烯。
2. Add 655mg zinc powder and 30ml 2.0mol/L hydrochloric acid successively to the colloid obtained in
实施例3 Example 3
1、采用超声波振荡将120mg氧化石墨分散于120ml水中,得到胶体; 1. Use ultrasonic vibration to disperse 120mg graphite oxide in 120ml water to obtain a colloid;
2、向步骤1得到的胶体中依次加入658mg锌粉和30ml 2.0mol/L盐酸,在60℃反应6小时后结束反应,将反应得到的混合物过滤,水洗,干燥,得到石墨烯。
2. Add 658mg zinc powder and 30ml 2.0mol/L hydrochloric acid successively to the colloid obtained in
实施例4 Example 4
1、采用超声波振荡将120mg氧化石墨分散于120ml水中,得到胶体; 1. Use ultrasonic vibration to disperse 120mg graphite oxide in 120ml water to obtain a colloid;
2、向步骤1得到的胶体中依次加入662mg锌粉和30ml 2.0mol/L盐酸,在80℃反应6小时后结束反应,将反应得到的混合物过滤,水洗,干燥,得到石墨烯。
2. Add 662mg zinc powder and 30ml 2.0mol/L hydrochloric acid successively to the colloid obtained in
实施例5 Example 5
1、采用超声波振荡将120mg氧化石墨分散于120ml水中,得到胶体; 1. Use ultrasonic vibration to disperse 120mg graphite oxide in 120ml water to obtain a colloid;
2、向步骤1得到的胶体中依次加入658mg锌粉和30ml 2.0mol/L盐酸,100℃反应6小时后结束反应,将反应得到的混合物过滤,水洗,干燥,得到石墨烯。
2. Add 658mg of zinc powder and 30ml of 2.0mol/L hydrochloric acid successively to the colloid obtained in
实施例6 Example 6
1、采用超声波振荡将120mg氧化石墨分散于150ml水中,得到胶体; 1. Use ultrasonic vibration to disperse 120mg graphite oxide in 150ml water to obtain a colloid;
2、向步骤1得到的胶体中依次加入654mg锌粉和4.8g NaOH,在室温恒温反应6小时后结束反应,将反应得到的混合物过滤,依次 用盐酸和水进行洗涤,干燥,得到石墨烯。
2. Add 654mg zinc powder and 4.8g NaOH successively to the colloid obtained in
实施例7 Example 7
1、采用超声波振荡将120mg氧化石墨分散于150ml水中,得到胶体; 1. Use ultrasonic vibration to disperse 120mg graphite oxide in 150ml water to obtain a colloid;
2、向步骤1得到的胶体中依次加入655mg锌粉和4.8g NaOH,在60℃恒温反应6小时后结束反应,将反应得到的混合物过滤,依次用盐酸和水进行洗涤,干燥,得到石墨烯。
2. Add 655 mg of zinc powder and 4.8 g of NaOH to the colloid obtained in
实施例8 Example 8
1、采用超声波振荡将120mg氧化石墨分散于150ml水中,得到胶体; 1. Use ultrasonic vibration to disperse 120mg graphite oxide in 150ml water to obtain a colloid;
2、向步骤1得到的胶体中依次加入655mg锌粉和4.8g NaOH,在80℃恒温反应6小时后结束反应,将反应得到的混合物过滤,依次用盐酸和水进行洗涤,干燥,得到石墨烯。
2. Add 655mg of zinc powder and 4.8g of NaOH to the colloid obtained in
实施例9 Example 9
1、采用超声波振荡将120mg氧化石墨分散于150ml水中,得到胶体; 1. Use ultrasonic vibration to disperse 120mg graphite oxide in 150ml water to obtain a colloid;
2、向步骤1得到的胶体中依次加入655mg锌粉和4.8g NaOH,在100℃恒温反应6小时后结束反应,将反应得到的混合物过滤,依次用盐酸和水进行洗涤,干燥,得到石墨烯。
2. Add 655mg of zinc powder and 4.8g of NaOH to the colloid obtained in
分别取实施例2~5制备的石墨烯粉末,编号依次为a、b、c、d,取实施例6~9制备的石墨烯粉末,编号依次为e、f、g、h;将上述材料依次进行X射线粉末衍射分析、红外光谱分析和定量元素分析,分析结果如下: Get respectively the graphene powder prepared by embodiment 2~5, numbering is successively a, b, c, d, get the graphene powder prepared by embodiment 6~9, numbering is successively e, f, g, h; above-mentioned material Carry out X-ray powder diffraction analysis, infrared spectroscopic analysis and quantitative elemental analysis in turn, and the analysis results are as follows:
[XRD粉末衍射分析] [XRD Powder Diffraction Analysis]
如图2为a、b、c、d的XRD图,从图中可以看到,随着温度的升高,2θ=11.8°(对应的d=0.75nm)处的衍射峰逐渐变弱,当温度为100℃时完全消失;而在2θ=23.5°(对应的d=0.38nm)处的衍射峰逐渐增强,当温度为100℃时到达极值。图3为e,f,g,h的XRD图, 温度由室温增加到100℃的过程中,2θ=11.8°处均无衍射峰,只有2θ=23.5°处的单峰。由于氧化石墨的层间含有羰基、环氧基等官能团,层间距能达到0.60~0.90nm,还原成石墨烯后,层间官能团消失,层间距缩小为0.30nm左右。结合XRD图可以说明,在酸性条件下,随反应温度的升高,氧化石墨的还原程度增大,在100℃时最充分,可判定氧化石墨被已还原为石墨烯,而在碱性条件下,室温即可达到良好的还原效果。 Figure 2 is the XRD pattern of a, b, c, and d, as can be seen from the figure, as the temperature increases, the diffraction peak at 2θ=11.8° (corresponding to d=0.75nm) gradually becomes weaker, when It disappears completely when the temperature is 100°C; while the diffraction peak at 2θ=23.5° (corresponding to d=0.38nm) gradually increases, and reaches the extreme value when the temperature is 100°C. Figure 3 shows the XRD patterns of e, f, g, and h. During the temperature increase from room temperature to 100°C, there is no diffraction peak at 2θ=11.8°, only a single peak at 2θ=23.5°. Since the interlayers of graphite oxide contain functional groups such as carbonyl groups and epoxy groups, the interlayer spacing can reach 0.60-0.90nm. After being reduced to graphene, the interlayer functional groups disappear, and the interlayer spacing is reduced to about 0.30nm. Combined with the XRD pattern, it can be shown that under acidic conditions, with the increase of reaction temperature, the reduction degree of graphite oxide increases, and it is the most sufficient at 100 ° C. It can be determined that graphite oxide has been reduced to graphene, while under alkaline conditions , a good reduction effect can be achieved at room temperature. the
[红外光谱分析] [Infrared spectrum analysis]
图4为a、b、c、d的红外光谱图,图5为e、f、g、h的红外光谱图。由a、b、c、d对比可以看出,在酸性条件下还原GO,温度对还原程度的影响较大,室温下,波数在3430cm-1,1614cm-1分别代表羟基的伸缩和弯曲振动几乎没有减小,波数在1727cm-1、1384cm-1、1226cm-1、1064cm-1分别代表C=O、C-O、C-O-C、醇的C-O伸缩振动依然明显存在,说明室温时GO还原不完全;而随着温度的升高,80℃时,3430cm-1,1614cm-1处强度变弱,分别代表羟基的伸缩和弯曲振动减少,吸水性减弱,波数在1727cm-1、1384cm-1、1226cm-1、1064cm-1的吸收峰明显消失或减少;100℃时,吸收强度继续减弱,表明还原程度进一步增大;通过Zn-NaOH和Zn-HCl还原GO对比,可以看出Zn-HCl还原的极值出现在100℃时,而Zn-NaOH在常温下即可很好的还原,在100℃还原最为彻底。 Figure 4 is the infrared spectrograms of a, b, c, and d, and Figure 5 is the infrared spectrograms of e, f, g, and h. From the comparison of a, b, c, and d, it can be seen that the reduction of GO under acidic conditions has a great influence on the degree of reduction by temperature. There is no decrease, and the wave numbers at 1727cm -1 , 1384cm -1 , 1226cm -1 , and 1064cm -1 respectively represent the CO stretching vibrations of C=O, CO, COC, and alcohols still obviously exist, indicating that the reduction of GO is not complete at room temperature; As the temperature rises, at 80°C, the strength at 3430cm -1 and 1614cm -1 becomes weaker, respectively representing the reduction of the stretching and bending vibration of the hydroxyl group, and the weakening of water absorption. The wave numbers are at 1727cm -1 , 1384cm -1 , 1226cm -1 , The absorption peak at 1064cm -1 disappeared or decreased obviously; at 100°C, the absorption intensity continued to weaken, indicating that the degree of reduction further increased; through the comparison of Zn-NaOH and Zn-HCl reduction of GO, it can be seen that the extreme value of Zn-HCl reduction appeared At 100°C, Zn-NaOH can be reduced very well at room temperature, and the reduction is most thorough at 100°C.
[定量元素分析] [Quantitative elemental analysis]
测试结果列于表1: The test results are listed in Table 1:
表1元素分析测试结果 Table 1 elemental analysis test results
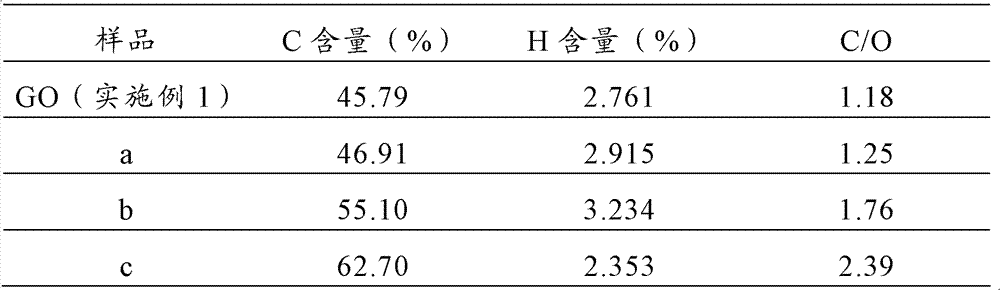

元素分析的结果表明,相比于原料GO、a、b、c、d数据表明,在酸性条件下,随温度的升高,GO还原越来越充分,e、f、g、h说明,在碱性条件下还原常温便具有较好的还原效果,在100℃还原最彻底。 The results of elemental analysis show that compared with the raw material GO, a, b, c, d data show that under acidic conditions, with the increase of temperature, GO is more and more fully reduced, e, f, g, h show that in Reduction at room temperature under alkaline conditions has a better reduction effect, and the reduction is most thorough at 100°C. the
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。 The descriptions of the above embodiments are only used to help understand the method and core idea of the present invention. It should be pointed out that for those skilled in the art, without departing from the principle of the present invention, some improvements and modifications can be made to the present invention, and these improvements and modifications also fall within the protection scope of the claims of the present invention. the
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。 The above description of the disclosed embodiments is provided to enable any person skilled in the art to make or use the invention. Various modifications to these embodiments will be readily apparent to those skilled in the art, and the general principles defined herein may be implemented in other embodiments without departing from the spirit or scope of the invention. Therefore, the present invention will not be limited to the embodiments shown herein, but is to be accorded the widest scope consistent with the principles and novel features disclosed herein. the
Claims (5)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201110412972.8A CN102491317B (en) | 2011-12-12 | 2011-12-12 | Preparation method of graphene |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201110412972.8A CN102491317B (en) | 2011-12-12 | 2011-12-12 | Preparation method of graphene |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102491317A CN102491317A (en) | 2012-06-13 |
| CN102491317B true CN102491317B (en) | 2014-01-22 |
Family
ID=46183188
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201110412972.8A Expired - Fee Related CN102491317B (en) | 2011-12-12 | 2011-12-12 | Preparation method of graphene |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102491317B (en) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102826540B (en) * | 2012-08-06 | 2016-01-13 | 常州第六元素材料科技股份有限公司 | A kind of method preparing reduced graphene or graphene film |
| CN103896257A (en) * | 2012-12-26 | 2014-07-02 | 海洋王照明科技股份有限公司 | Preparation method for graphene |
| CN103578796A (en) * | 2013-11-15 | 2014-02-12 | 复旦大学 | Preparation method of super-capacitor electrode without adhesives |
| MY194301A (en) | 2016-11-03 | 2022-11-27 | Karex Holdings Sdn Bhd | Polyisoprene latex graphene composites and method of making them |
| CN107611392A (en) * | 2017-09-07 | 2018-01-19 | 南京汉尔斯生物科技有限公司 | A kind of graphene composite material for improving lithium polymer battery mechanical strength |
| CN115650214A (en) * | 2022-10-15 | 2023-01-31 | 四川大学 | Low-energy-consumption extremely-fast efficient graphene oxide reduction method |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006320853A (en) * | 2005-05-20 | 2006-11-30 | Hitachi Powdered Metals Co Ltd | Graphite-based hydrogen storage material and method for producing the same |
| CN101857222A (en) * | 2010-05-28 | 2010-10-13 | 常州大学 | A method for preparing a large-area, continuous graphene/zinc oxide composite structure |
| CN101864098A (en) * | 2010-06-03 | 2010-10-20 | 四川大学 | In situ reduction preparation method of polymer/graphene composites |
| CN102040217A (en) * | 2009-10-26 | 2011-05-04 | 国家纳米科学中心 | Method for preparing graphene |
| JP2011105569A (en) * | 2009-11-20 | 2011-06-02 | Fuji Electric Holdings Co Ltd | Method for manufacturing graphene thin film |
-
2011
- 2011-12-12 CN CN201110412972.8A patent/CN102491317B/en not_active Expired - Fee Related
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006320853A (en) * | 2005-05-20 | 2006-11-30 | Hitachi Powdered Metals Co Ltd | Graphite-based hydrogen storage material and method for producing the same |
| CN102040217A (en) * | 2009-10-26 | 2011-05-04 | 国家纳米科学中心 | Method for preparing graphene |
| JP2011105569A (en) * | 2009-11-20 | 2011-06-02 | Fuji Electric Holdings Co Ltd | Method for manufacturing graphene thin film |
| CN101857222A (en) * | 2010-05-28 | 2010-10-13 | 常州大学 | A method for preparing a large-area, continuous graphene/zinc oxide composite structure |
| CN101864098A (en) * | 2010-06-03 | 2010-10-20 | 四川大学 | In situ reduction preparation method of polymer/graphene composites |
Non-Patent Citations (2)
| Title |
|---|
| Ultrasonication-assisted ultrafast reduction of graphene oxide by zinc powder at room temperature;Xiaoguang Mei et. al.;《carbon》;20110822;5389-5397 * |
| Xiaoguang Mei et. al..Ultrasonication-assisted ultrafast reduction of graphene oxide by zinc powder at room temperature.《carbon》.2011,5389-5397. |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102491317A (en) | 2012-06-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102491317B (en) | Preparation method of graphene | |
| CN102619080B (en) | Preparation method of graphene coating polyacrylonitrile fiber composite material | |
| CN103014683B (en) | Preparation method of graphene-based nano-silver composite | |
| CN101549864A (en) | Method for simply and innoxiously preparing single-layer graphene | |
| CN101585555A (en) | Preparation method of monolayer manganese dioxide nano-plates | |
| CN101709147A (en) | Method for preparing composite material of graphene and graphene poly-p-phenylenediamine | |
| CN103833008A (en) | Method for preparing graphene at normal temperature | |
| CN103990444A (en) | Preparation method of graphene-homemade TiO2 nanowire photocatalyst | |
| CN105344380A (en) | Metal organic frame/graphene carried palladium nano-composite catalyst and preparing method and application thereof | |
| CN102633256A (en) | Preparation method of graphene colloid dispersion solution | |
| CN103801298A (en) | Hydrothermal rapid synthesis method of graphene load nickel nanoparticle composite material | |
| CN104058399B (en) | A kind of direct preparation method of high-purity and high-quality graphene | |
| CN104098087B (en) | A kind of metal/tea polyphenols is as the method for reducing agent redox graphene | |
| CN107140623B (en) | A kind of method for preparing graphene | |
| CN101966989B (en) | Method for realizing photocatalytic reduction of graphene oxide by quadrangular zinc oxide | |
| CN108504049A (en) | A kind of preparation method of the compound thermal electric film of macromolecule | |
| CN105502370B (en) | A kind of solid-phase reduction process of graphene oxide | |
| CN104986802B (en) | Platy nanometer material and preparation method thereof | |
| CN103483589A (en) | Two-dimensional polyphosphazene nanosheet layer and preparation and application method | |
| CN103342355B (en) | Method for preparing graphene and composite material thereof | |
| CN103056380B (en) | Method for self-assembling and preparing bismuth metal nanowire through octa-amino silsesquioxane | |
| CN104445340B (en) | By the preparation method of the octahedra cerium oxide of nanometer blocks self-assembly | |
| CN103387228B (en) | Preparation method for graphene scrolls | |
| CN110508299A (en) | A rapid heating method for preparing two-dimensional localized oxidation of transition metal fluoride catalysts | |
| CN106238080B (en) | Phosphorus-doped porous graphene and preparation method thereof and method for catalyzing benzylamine oxidation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20140122 Termination date: 20191212 |
|
| CF01 | Termination of patent right due to non-payment of annual fee |