CN101468985A - 5-(3-aromatic heterocyclic substituted phenyl) tetrazole compounds and anti-HIV/AIDS use thereof - Google Patents
5-(3-aromatic heterocyclic substituted phenyl) tetrazole compounds and anti-HIV/AIDS use thereof Download PDFInfo
- Publication number
- CN101468985A CN101468985A CNA2007103069390A CN200710306939A CN101468985A CN 101468985 A CN101468985 A CN 101468985A CN A2007103069390 A CNA2007103069390 A CN A2007103069390A CN 200710306939 A CN200710306939 A CN 200710306939A CN 101468985 A CN101468985 A CN 101468985A
- Authority
- CN
- China
- Prior art keywords
- cooh
- coor
- phenyl
- tetrazole
- hydrocarbyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 title claims abstract description 16
- -1 tetrazole compounds Chemical class 0.000 title claims description 52
- 230000036436 anti-hiv Effects 0.000 title description 8
- 208000030507 AIDS Diseases 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 64
- 150000003839 salts Chemical class 0.000 claims abstract description 21
- 239000003814 drug Substances 0.000 claims abstract description 16
- 238000000034 method Methods 0.000 claims abstract description 13
- 208000031886 HIV Infections Diseases 0.000 claims abstract description 6
- 201000010099 disease Diseases 0.000 claims abstract description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 6
- 208000037357 HIV infectious disease Diseases 0.000 claims abstract description 5
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 5
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 claims abstract description 5
- 125000001424 substituent group Chemical group 0.000 claims abstract description 5
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 51
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 36
- 229910052736 halogen Inorganic materials 0.000 claims description 36
- 150000002367 halogens Chemical class 0.000 claims description 34
- 238000006243 chemical reaction Methods 0.000 claims description 21
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims description 20
- 125000001183 hydrocarbyl group Chemical group 0.000 claims description 19
- 229910006069 SO3H Inorganic materials 0.000 claims description 16
- 229910052799 carbon Inorganic materials 0.000 claims description 12
- 229910052760 oxygen Inorganic materials 0.000 claims description 12
- 229910052717 sulfur Inorganic materials 0.000 claims description 12
- 125000000623 heterocyclic group Chemical class 0.000 claims description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 10
- 125000003545 alkoxy group Chemical group 0.000 claims description 8
- 125000004185 ester group Chemical group 0.000 claims description 8
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 claims description 6
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims description 6
- 125000004122 cyclic group Chemical group 0.000 claims description 6
- 239000008194 pharmaceutical composition Substances 0.000 claims description 6
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Substances C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 claims description 5
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 5
- 229910052757 nitrogen Inorganic materials 0.000 claims description 5
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 claims description 4
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzenecarbonitrile Natural products N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 claims description 4
- 239000003153 chemical reaction reagent Substances 0.000 claims description 4
- 238000005658 halogenation reaction Methods 0.000 claims description 4
- 125000001072 heteroaryl group Chemical group 0.000 claims description 4
- IYPXPGSELZFFMI-UHFFFAOYSA-N 1-phenyltetrazole Chemical compound C1=NN=NN1C1=CC=CC=C1 IYPXPGSELZFFMI-UHFFFAOYSA-N 0.000 claims description 3
- GFISDBXSWQMOND-UHFFFAOYSA-N 2,5-dimethoxyoxolane Chemical compound COC1CCC(OC)O1 GFISDBXSWQMOND-UHFFFAOYSA-N 0.000 claims description 3
- QAKHTYQLHKUQHY-UHFFFAOYSA-N 3-[3-(2h-tetrazol-5-yl)phenyl]-5-(trifluoromethyl)-1,2,4-oxadiazole Chemical compound O1C(C(F)(F)F)=NC(C=2C=C(C=CC=2)C2=NNN=N2)=N1 QAKHTYQLHKUQHY-UHFFFAOYSA-N 0.000 claims description 3
- LEXDTPFPHRIPNN-UHFFFAOYSA-N 5-(3-pyrrol-1-ylphenyl)-2h-tetrazole Chemical compound C1=CC=CN1C1=CC=CC(C=2NN=NN=2)=C1 LEXDTPFPHRIPNN-UHFFFAOYSA-N 0.000 claims description 3
- HGXBKEFXSPFLOP-UHFFFAOYSA-N 5-[3-(2,5-dimethyl-1-pyrrolyl)phenyl]-2H-tetrazole Chemical compound CC1=CC=C(C)N1C1=CC=CC(C2=NNN=N2)=C1 HGXBKEFXSPFLOP-UHFFFAOYSA-N 0.000 claims description 3
- 238000006069 Suzuki reaction reaction Methods 0.000 claims description 3
- 239000004327 boric acid Substances 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 3
- 239000002994 raw material Substances 0.000 claims description 3
- FOJZCIXBTZKNBJ-UHFFFAOYSA-N 2-[5-[3-[5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl]phenyl]tetrazol-1-yl]acetic acid Chemical compound OC(=O)CN1N=NN=C1C1=CC=CC(C=2N=C(ON=2)C(F)(F)F)=C1 FOJZCIXBTZKNBJ-UHFFFAOYSA-N 0.000 claims description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 2
- 238000006086 Paal-Knorr synthesis reaction Methods 0.000 claims description 2
- 230000010933 acylation Effects 0.000 claims description 2
- 238000005917 acylation reaction Methods 0.000 claims description 2
- 125000003172 aldehyde group Chemical group 0.000 claims description 2
- 150000001336 alkenes Chemical class 0.000 claims description 2
- 150000001345 alkine derivatives Chemical class 0.000 claims description 2
- 125000006615 aromatic heterocyclic group Chemical group 0.000 claims description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 2
- 229910052794 bromium Inorganic materials 0.000 claims description 2
- 125000004432 carbon atom Chemical group C* 0.000 claims description 2
- 238000009833 condensation Methods 0.000 claims description 2
- 230000005494 condensation Effects 0.000 claims description 2
- 150000002148 esters Chemical class 0.000 claims description 2
- 150000008282 halocarbons Chemical class 0.000 claims description 2
- 230000026030 halogenation Effects 0.000 claims description 2
- 150000005826 halohydrocarbons Chemical class 0.000 claims description 2
- 229910052739 hydrogen Inorganic materials 0.000 claims description 2
- 239000001257 hydrogen Substances 0.000 claims description 2
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical group C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 claims description 2
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 2
- 150000003536 tetrazoles Chemical group 0.000 claims description 2
- 229930194542 Keto Natural products 0.000 claims 1
- 125000000468 ketone group Chemical group 0.000 claims 1
- 230000002265 prevention Effects 0.000 claims 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims 1
- 229940079593 drug Drugs 0.000 abstract description 9
- 239000000203 mixture Substances 0.000 abstract description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 48
- 239000000243 solution Substances 0.000 description 23
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- 210000004027 cell Anatomy 0.000 description 20
- 239000007787 solid Substances 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 18
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 18
- 241000725303 Human immunodeficiency virus Species 0.000 description 17
- 101800001690 Transmembrane protein gp41 Proteins 0.000 description 16
- 241000700605 Viruses Species 0.000 description 16
- 238000005160 1H NMR spectroscopy Methods 0.000 description 13
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 13
- 150000003254 radicals Chemical class 0.000 description 13
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 12
- 230000004927 fusion Effects 0.000 description 12
- 239000002904 solvent Substances 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- 230000000694 effects Effects 0.000 description 11
- 230000002401 inhibitory effect Effects 0.000 description 11
- 230000003612 virological effect Effects 0.000 description 11
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 10
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- 239000003208 petroleum Substances 0.000 description 10
- 108090000765 processed proteins & peptides Proteins 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 229960000583 acetic acid Drugs 0.000 description 9
- 210000000170 cell membrane Anatomy 0.000 description 9
- 239000012528 membrane Substances 0.000 description 9
- 150000003384 small molecules Chemical class 0.000 description 9
- 230000008569 process Effects 0.000 description 8
- 230000010076 replication Effects 0.000 description 8
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 7
- 239000007864 aqueous solution Substances 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 239000005457 ice water Substances 0.000 description 7
- 238000003556 assay Methods 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 238000004237 preparative chromatography Methods 0.000 description 6
- FLGISLVJQPPAMV-UHFFFAOYSA-N 3-(2h-tetrazol-5-yl)aniline Chemical compound NC1=CC=CC(C2=NNN=N2)=C1 FLGISLVJQPPAMV-UHFFFAOYSA-N 0.000 description 5
- 108010041397 CD4 Antigens Proteins 0.000 description 5
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 5
- PEASPLKKXBYDKL-FXEVSJAOSA-N enfuvirtide Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(C)=O)[C@@H](C)O)[C@@H](C)CC)C1=CN=CN1 PEASPLKKXBYDKL-FXEVSJAOSA-N 0.000 description 5
- 229940125777 fusion inhibitor Drugs 0.000 description 5
- 238000001819 mass spectrum Methods 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 239000012954 diazonium Substances 0.000 description 4
- 150000001989 diazonium salts Chemical class 0.000 description 4
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 4
- 239000002835 hiv fusion inhibitor Substances 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- 230000029812 viral genome replication Effects 0.000 description 4
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 3
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 3
- 108010032976 Enfuvirtide Proteins 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000002259 anti human immunodeficiency virus agent Substances 0.000 description 3
- 230000000840 anti-viral effect Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000012362 glacial acetic acid Substances 0.000 description 3
- WJRBRSLFGCUECM-UHFFFAOYSA-N hydantoin Chemical compound O=C1CNC(=O)N1 WJRBRSLFGCUECM-UHFFFAOYSA-N 0.000 description 3
- 150000002576 ketones Chemical group 0.000 description 3
- YDCHPLOFQATIDS-UHFFFAOYSA-N methyl 2-bromoacetate Chemical compound COC(=O)CBr YDCHPLOFQATIDS-UHFFFAOYSA-N 0.000 description 3
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 3
- 150000003233 pyrroles Chemical class 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- VGGUVGVAVAAODK-ZROIWOOFSA-N (5z)-2-amino-5-[(3-cyclopentyloxy-4-methoxyphenyl)methylidene]-1,3-thiazol-4-one Chemical compound C1=C(OC2CCCC2)C(OC)=CC=C1\C=C1/SC(=N)NC1=O VGGUVGVAVAAODK-ZROIWOOFSA-N 0.000 description 2
- OJVAMHKKJGICOG-UHFFFAOYSA-N 2,5-hexanedione Chemical compound CC(=O)CCC(C)=O OJVAMHKKJGICOG-UHFFFAOYSA-N 0.000 description 2
- 239000005725 8-Hydroxyquinoline Substances 0.000 description 2
- 102100035875 C-C chemokine receptor type 5 Human genes 0.000 description 2
- 101710149870 C-C chemokine receptor type 5 Proteins 0.000 description 2
- 101710205625 Capsid protein p24 Proteins 0.000 description 2
- 208000035473 Communicable disease Diseases 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 102100034349 Integrase Human genes 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- 229940122313 Nucleoside reverse transcriptase inhibitor Drugs 0.000 description 2
- 101710177166 Phosphoprotein Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 101710149279 Small delta antigen Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 2
- 102100022563 Tubulin polymerization-promoting protein Human genes 0.000 description 2
- 108010003533 Viral Envelope Proteins Proteins 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 150000001299 aldehydes Chemical group 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- VQFAIAKCILWQPZ-UHFFFAOYSA-N bromoacetone Chemical compound CC(=O)CBr VQFAIAKCILWQPZ-UHFFFAOYSA-N 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 239000002975 chemoattractant Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000011162 core material Substances 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 210000000805 cytoplasm Anatomy 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 238000002451 electron ionisation mass spectrometry Methods 0.000 description 2
- 229960002062 enfuvirtide Drugs 0.000 description 2
- WANUOLLLVOSMFL-UHFFFAOYSA-N ethyl 2-acetyl-4-oxopentanoate Chemical compound CCOC(=O)C(C(C)=O)CC(C)=O WANUOLLLVOSMFL-UHFFFAOYSA-N 0.000 description 2
- 239000012737 fresh medium Substances 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 210000004698 lymphocyte Anatomy 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 230000034217 membrane fusion Effects 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 229960003540 oxyquinoline Drugs 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 238000012746 preparative thin layer chromatography Methods 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- MCJGNVYPOGVAJF-UHFFFAOYSA-N quinolin-8-ol Chemical compound C1=CN=C2C(O)=CC=CC2=C1 MCJGNVYPOGVAJF-UHFFFAOYSA-N 0.000 description 2
- 238000010839 reverse transcription Methods 0.000 description 2
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- JUWYQISLQJRRNT-UHFFFAOYSA-N (5-formylfuran-2-yl)boronic acid Chemical compound OB(O)C1=CC=C(C=O)O1 JUWYQISLQJRRNT-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- BBVIDBNAYOIXOE-UHFFFAOYSA-N 1,2,4-oxadiazole Chemical group C=1N=CON=1 BBVIDBNAYOIXOE-UHFFFAOYSA-N 0.000 description 1
- ZOBPZXTWZATXDG-UHFFFAOYSA-N 1,3-thiazolidine-2,4-dione Chemical compound O=C1CSC(=O)N1 ZOBPZXTWZATXDG-UHFFFAOYSA-N 0.000 description 1
- TUSDEZXZIZRFGC-UHFFFAOYSA-N 1-O-galloyl-3,6-(R)-HHDP-beta-D-glucose Natural products OC1C(O2)COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC1C(O)C2OC(=O)C1=CC(O)=C(O)C(O)=C1 TUSDEZXZIZRFGC-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- UPHOPMSGKZNELG-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=C(O)C=CC2=C1 UPHOPMSGKZNELG-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- HZSYDUGIMVHNFF-UHFFFAOYSA-N 2-sulfanylideneimidazolidin-4-one Chemical compound N1C(=S)NC(=O)C1.S=C1NCC(N1)=O HZSYDUGIMVHNFF-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 239000001263 FEMA 3042 Substances 0.000 description 1
- 101710114816 Gene 41 protein Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 101000922348 Homo sapiens C-X-C chemokine receptor type 4 Proteins 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 108010061833 Integrases Proteins 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- LRBQNJMCXXYXIU-PPKXGCFTSA-N Penta-digallate-beta-D-glucose Natural products OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-PPKXGCFTSA-N 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 241000589970 Spirochaetales Species 0.000 description 1
- 101710172711 Structural protein Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 229940040526 anhydrous sodium acetate Drugs 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 230000034303 cell budding Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 229940121657 clinical drug Drugs 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- NKNDPYCGAZPOFS-UHFFFAOYSA-M copper(i) bromide Chemical compound Br[Cu] NKNDPYCGAZPOFS-UHFFFAOYSA-M 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 230000000120 cytopathologic effect Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 238000002784 cytotoxicity assay Methods 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- XYIBRDXRRQCHLP-UHFFFAOYSA-N ethyl acetoacetate Chemical compound CCOC(=O)CC(C)=O XYIBRDXRRQCHLP-UHFFFAOYSA-N 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 235000013861 fat-free Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 229960002598 fumaric acid Drugs 0.000 description 1
- 238000007499 fusion processing Methods 0.000 description 1
- 230000000799 fusogenic effect Effects 0.000 description 1
- 229940099052 fuzeon Drugs 0.000 description 1
- LRBQNJMCXXYXIU-QWKBTXIPSA-N gallotannic acid Chemical compound OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@H]2[C@@H]([C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-QWKBTXIPSA-N 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 229940091173 hydantoin Drugs 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229940099690 malic acid Drugs 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 210000004779 membrane envelope Anatomy 0.000 description 1
- 230000037323 metabolic rate Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- ACTNHJDHMQSOGL-UHFFFAOYSA-N n',n'-dibenzylethane-1,2-diamine Chemical compound C=1C=CC=CC=1CN(CCN)CC1=CC=CC=C1 ACTNHJDHMQSOGL-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229940042402 non-nucleoside reverse transcriptase inhibitor Drugs 0.000 description 1
- 239000002726 nonnucleoside reverse transcriptase inhibitor Substances 0.000 description 1
- 235000003715 nutritional status Nutrition 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 229940064914 retrovir Drugs 0.000 description 1
- KIWUVOGUEXMXSV-UHFFFAOYSA-N rhodanine Chemical compound O=C1CSC(=S)N1 KIWUVOGUEXMXSV-UHFFFAOYSA-N 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- 230000001502 supplementing effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 235000015523 tannic acid Nutrition 0.000 description 1
- 229920002258 tannic acid Polymers 0.000 description 1
- 229940033123 tannic acid Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000007502 viral entry Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D257/04—Five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/10—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Virology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Tropical Medicine & Parasitology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- AIDS & HIV (AREA)
- General Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention relates to medical salts for 5-(3-aromic hetero ring substituted benzene)-tetrazole compounds in a formula I, wherein various substituents are defined in the patent claim. The invention also relates to a method for preparing the compounds, a medicine composition containing the compounds, and application of the compounds in preparing medicines for treating or preventing diseases related to HIV infection.
Description
Technical Field
The invention relates to 5- (3-aromatic heterocycle substituted benzene) -tetrazole compounds, a preparation method thereof, a pharmaceutical composition containing the same and application thereof in preparing medicines for treating or preventing diseases or symptoms related to HIV infection.
Background
AIDS is a serious infectious disease caused by Human Immunodeficiency Virus (HIV). HIV is an RNA retrovirus that selectively infects cells of the human immune system that have CD4 receptors on their surface, such as lymphocytes, monocytes, macrophages, dendritic cells, and the like. The surface of the virus is a double lipid membrane, and two important glycoproteins are arranged on the membrane: gp120 and gp41, gp120 being outside the membrane, for recognizing the CD4 receptor on the cell surface; gp41 crosses the viral membrane and serves primarily to fuse the viral membrane to the cell membrane, thereby releasing the viral internal core material into the host cytoplasm. The viral membrane is enveloped with 2 single-stranded RNAs and some important enzymes (e.g., reverse transcriptase, proteolytic enzyme, integrase) and structural proteins (p24, p17, p7, etc.). HIV cannot reproduce in vitro, and must be replicated and regenerated by human cells, and the replication process is roughly divided into 7 steps: viral attack (binding), fusion (fusing), reverse transcription (reverse transcription), integration (integration), transcription (transcription), translation (translation) and recombination and flooding (assembly & budding) of cells. AIDS virus is continuously replicated in such a cyclic process, so that immune cells of a human body are infected, the immune system of the human body is damaged, and finally, the complete loss of the immune function of the human body is caused, so that patients are in danger of various infections without resistance, and various infectious diseases and tumors are caused, and finally, the patients die. Theoretically, the drug can achieve the purposes of inhibiting virus and treating diseases only by blocking any link in the virus replication process.
To date, there are 20 clinical drugs approved by the FDA for the treatment of aids, which are classified into four categories (Erik dc. antiviral drugs in current clinical use. j Clin Virol, 2004, 30 (2): 115-: (1) nucleoside Reverse Transcriptase Inhibitors (NRTIs), 8; (2) 3 non-nucleoside reverse transcriptase inhibitors (NNRTIs); (3) protease Inhibitors (PIs), 8; (4) fusion Inhibitors (FIs), 1. Different mechanisms of action are most clinically used in combination with drugs, such as two reverse transcriptase inhibitors and one protease inhibitor (Robb's GK, De GV, Shafer RW, et al. Complex of sequence enzyme-primer as primer for HIV-1 primer. N Eng 1J Med, 2003, 349: 2293303. and Shafer RW, Smeaton LM, Robbins GK, et al. Complex of four-primer and pairs of sequence enzyme-primer as primer for HIV-1 primer. N Eng 1J Med, 2003, 349: 230415). The treatment can effectively inhibit the viral load in the body of an infected person and reduce the morbidity and mortality of the infected person, but still has the problems of easy generation of drug resistance, great toxic and side effects and the like. Therefore, it is urgently needed to search for new drug targets in the virus replication process and develop anti-HIV drugs with new action mechanisms.
The existing anti-HIV drugs all play an inhibiting role in the replication process of viruses after entering cells. However, with the progress of the research on the mechanism of fusion between HIV virus and cell, people are paying more attention to the research on drugs which can prevent the virus from invading cells and play an antiviral role in the early stage of virus replication. The medicine can inhibit virus infected cell and virus replication, and is expected to provide effective novel anti-H IV therapeutic medicine with different action mechanisms for patients.
The process of HIV invading cells is mainly composed of 3 steps: adhesion, binding to co-receptors, membrane fusion. The viral envelope glycoprotein GP120 first binds to the cell surface CD4 molecule (GallaherWR, Ball JM, Garry RF, et al. A general model for the surf aceroloproteins of HIV and other retroviruses. AIDS Res Hum Retrovir, 1995, 11: 191-202), changes in conformation and then binds to the co-receptor (chemoattractant, such as CXCR4 or CCR5) (Dragon T, Litwin V, Allaway GP, et al. HIV-1entry into O CD4+ cells is mediated by the chemoattractant CC-CKR-5.Nature, 1996, 381: 66773); then gp41 is inserted into cell membrane to form 6 spirochetes, and the virus membrane is drawn close to the cell membrane to fuse. In this process, gp120 and gp41, the CD4 receptor, and the co-receptors are considered possible drug targets. Of these, gp41 plays a crucial role throughout the fusion process.
The amino acid sequence of Gp41 has four functional regions. The transmembrane domain (TM) at the C-terminus immobilizes gp41 on the viral membrane; CHR segment (C-tertiary header repeat, CHR) and NHR segment (N-tertiary header repeat, NHR) are functional parts of gp41 structure; fusion Peptide (FP) is a highly hydrophobic sequence located at the N-terminus, the main function of which is to insert into the host Cell membrane (Melikyan GB, Markosyn RM, Hemmati H, et al. evaluation which the transition of HIV-1gp41 into a six-helix bundle, not the bundling configuration, indeces membrane fusion. J Cell Biol, 2000, 151: 41323 and Munoz-Barhos I, Salzdel K, Hunter E, et al. roll soft membrane-promoter domain in the interaction stage of a human tissue virus, type 1 expression protein-mediator J, 608992).
The three NHR helices of gp41 on the surface of the virus were arranged in parallel in the center and the three CHR helices were wrapped around in antiparallel, surrounded by 3 gp 120. When The virus infects cells, The conformation of gp120 on The surface of The virus changes after binding with CD4 receptor and co-receptor on The surface of The cell, and The NHR helix of gp41 extends out from The center, and The N-terminal fusogenic peptide is inserted into The cell membrane (Coleman CI, music BL and Ross J. Enfuvirtide: The first fusion inhibitor for The treatment of diseases with HIV-1infection. formulation, 2003, 38: 204222). Subsequently, NHR and CHR come together and reform into parallel six-membered helical bundles. This conformational change provides the energy required to bring the hydrated surfaces of the viral envelope and host cell membrane into close proximity, thereby drawing the viral membrane and cell membrane together and encouraging fusion to occur. (Cooley LA and Lewis SR, HIV-1 cell entry and development in viral entry therapy. J Clin Virol, 2003, 26: 121132 and Moore JP and Doms RW. the entry of entry inhibition torrs: A fusion of science and medicine. Proc Natl Acad Sci U S A, 2003, 100: 1059810602). The surfaces of the viruses are provided with a plurality of gp41 films to form fusion holes between the two films, the fluidity of the films enables the films to be rapidly expanded, and finally the complete fusion of the HIV envelope and the host cell film is realized, and the virus core substances are released into the cytoplasm of the host.
Both functional regions NHR and CHR of gp41 can be the target of HIV fusion inhibitor. The first fusion inhibitor drug, T-20(Fuzeon), approved by the FDA in the United states is a 36 amino acid polypeptide that mimics the helix structure sequence of the CHR. It blocks the formation of six-membered helix bundle by combining with NHR, thus achieving the purpose of inhibiting the Fusion of virus and cell membrane (Fung HB, BCPS and Guo Y. Enfuvirtide: A Fusion Inhibitor for the Treatment of HIV infection. Clin Ther, 2004, 26 (3): 352-378). Because T20 is a peptide drug and has the defects of poor oral bioavailability, high production cost and the like, the search for a high-efficiency and low-toxicity non-peptide small-molecule HIV fusion inhibitor primer is one of the main directions for the research of new anti-HIV drugs.
By performing targeted activity screening on a small molecule compound library with diversified structures, two N-aryl carboxylic acid Substituted Pyrrole small molecule compounds NB-2 and NB-64(Jiang Sh-B, Lu H, Liu Sh-W, et al.N-understituted Pyrrole Derivatives as Novel Humanimmunity details Virus Type 1Entry inhibition That with gp41 Six-helium Bundle formation and Block Virus fusion. anti-cancer Agents Chemother, 2004, 48: 4349-4359) not only show good anti-HIV replication activity in a cell model (EC-2 and NB-64)50Values of 1.04. mu.M and 2.21. mu.M, respectively), and in the test of fusion of virus with cell membrane (EC)50The values were 6.74. mu. respectivelyM and 29.92. mu.M) and gp41 in six-membered helix bundle formation assay [ IC50(6-HB) values were 13.48. mu.M and 15.69. mu.M, respectively]All have obvious inhibitory activity. The results of these experiments indicate that NB-2 and NB-64 are indeed small molecule active compounds acting on gp 41. Based on the basic principle that the structure is related to the biological activity, the small molecule fusion inhibitor with better activity can be found by modifying the structure of the compound.

Disclosure of Invention
The invention relates to a compound with a 5-substituted phenyl-tetrazole skeleton structure shown in a formula I, which can effectively inhibit the formation of a hexamer of HIV-1 surface glycoprotein gp41, thereby inhibiting HIV replication. The intensive research on the compounds can possibly discover a novel non-peptide small molecule HIV fusion inhibitor, and the inhibitor becomes a novel AI DS resistant medicament.
The first aspect of the invention relates to a tetrazole aryl heterocyclic compound shown in the formula I or a medicinal salt thereof:

wherein,
R1=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6a hydrocarbyl group;
R2-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Alkyl radical, C1-6Alkoxy, -CF3、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
ar is a five membered heteroaromatic ring containing 1 to 3 heteroatoms selected from N, O, S, selected from:


wherein R is3=CHO、COR’、COOR’、COOH、CF3、CH2R', halogen, C1-6Hydrocarbyl radical, C1-6Alkoxy, -NH2、-OH、-NO2、-CN、-HC=CH-CN、-CH=CH2-C ≡ CH, -C ≡ CR ', -CH ═ CHR ', -CH ═ CHCOR ', or optionally bearing an ester group, a carboxyl group, C ≡ C optionally at available positions in its ring structure1-6Hydrocarbyl, phenyl-substituted heterocyclic groups:

wherein X and Y are each independently selected from C, O, S and NH;
R4=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6hydrocarbyl, phenyl;
R5-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Hydrocarbyl radical, C1-6Alkoxy, -CF3、CHO、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
the five-membered heteroaromatic ring optionally carries a substituent selected from the group consisting of aldehyde, ketone, ester, carboxyl, cyano, alpha, beta unsaturated ketone, alkene, alkyne, C, at an available position on the ring1-6Hydrocarbyl radical, C1-6Alkoxy, halogen, -NH2、-OH、-NO2and-CF3A substituent of (1);
R’=C1-6a hydrocarbyl group; and is
R' is halogen, OH or C1-6An alkoxy group.
The term "hydrocarbyl" as used herein includes alkyl, alkenyl and alkynyl groups.
Para substituent R of the invention3The "following heterocyclic groups" referred to in the description include, but are not limited to, 2, 4-thiazolidinedione, 2-thio-2, 4-thiazolidinedione (Rhodanine), succinimide, 2, 4-imidazolidinedione (hydantoin ), 2-Thiohydantoin (2-Thiohydantoin), Pseudothiohydantoin (Pseudothiohydantoin), and the like.
In a second aspect, the present invention relates to a process for the preparation of a compound of formula I or a pharmaceutically acceptable salt thereof.
A third aspect of the present invention relates to a pharmaceutical composition comprising at least one compound of formula I or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable carriers or excipients.
A fourth aspect of the present invention relates to the use of a compound of formula I as described above, or a pharmaceutically acceptable salt thereof, for the manufacture of a medicament for the treatment of a disease or condition associated with HIV infection.
According to a preferred embodiment of the invention, Ar is a substituted pyrrole, represented by formula II:

wherein,
R1=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6a hydrocarbyl group;
R2-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Alkyl radical, C1-6Alkoxy, -CF3、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R3=CHO、COR’、COOR’、COOH、CF3、CH2r', halogen, C1-6Hydrocarbyl radical, C1-6Alkoxy, -NH2、-OH、-NO2、-CN、-HC=CH-CN、-CH=CH2-C ≡ CH, -C ≡ CR ', -CH ═ CHR', -CH ═ CHCOR ', or optionally bearing an ester group, a carboxyl group, C ≡ CH', or a cyclic structure thereof1-6Hydrocarbyl, phenyl-substituted heterocyclic groups:

wherein X and Y are each independently selected from C, O, S and NH;
R4=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6hydrocarbyl, phenyl;
R5-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Hydrocarbyl radical, C1-6Alkoxy, -CF3、CHO、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R6=H、CH3、CF3halogen or C2-4A hydrocarbyl group;
R’=C1-6a hydrocarbyl group; and is
R' is halogen, OH or C1-6An alkoxy group.
According to another preferred embodiment of the invention, Ar is 1, 2, 4-oxadiazole, as shown in formula III below:

wherein,
R1=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6a hydrocarbyl group;
R2-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Alkyl radical, C1-6Alkoxy, -CF3、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R3=CHO、COR’、COOR’、COOH、CF3、CH2r', halogen, C1-6Hydrocarbyl radical, C1-6Alkoxy, -NH2、-OH、-NO2、-CN、-HC=CH-CN、-CH=CH2-C ≡ CH, -C ≡ CR ', -CH ═ CHR', -CH ═ CHCOR ', or optionally bearing an ester group, a carboxyl group, C ≡ CH', or a cyclic structure thereof1-6Hydrocarbyl, phenyl-substituted heterocyclic groups:
wherein X and Y are each independently selected from C, O, S and NH;
R4=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6hydrocarbyl, phenyl;
R5-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Hydrocarbyl radical, C1-6Alkoxy, -CF3、CHO、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R’=C1-6a hydrocarbyl group; and is
R' is halogen, OH or C1-6An alkoxy group.
According to another preferred embodiment of the invention, Ar is a 5-substituted furan, as shown in formula IV below:
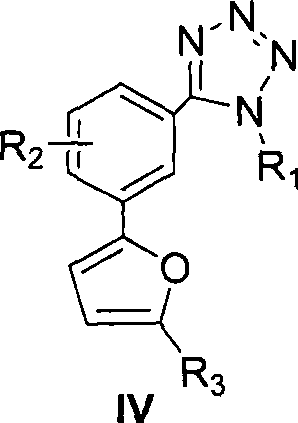
wherein,
R1=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6a hydrocarbyl group;
R2-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Alkyl radical, C1-6Alkoxy, -CF3、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R3=CHO、COR’、COOR’、COOH、CF3、CH2r', halogen, C1-6Hydrocarbyl radical, C1-6Alkoxy, -NH2、-OH、-NO2、-CN、-HC=CH-CN、-CH=CH2-C ≡ CH, -C ≡ CR ', -CH ═ CHR', -CH ═ CHCOR ', or optionally bearing an ester group, a carboxyl group, C ≡ CH', or a cyclic structure thereof1-6Hydrocarbyl, phenyl-substituted heterocyclic groups:
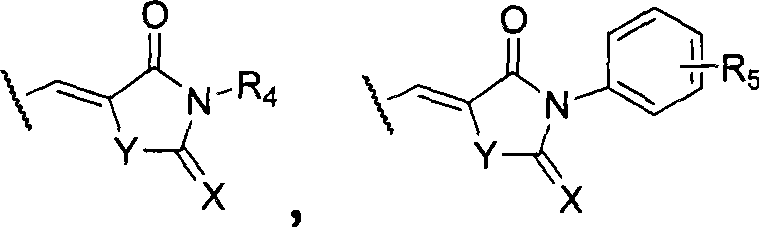
wherein X and Y are each independently selected from C, O, S and NH;
R4=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6hydrocarbyl, phenyl;
R5-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Hydrocarbyl radical, C1-6Alkoxy, -CF3、CHO、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R’=C1-6a hydrocarbyl group; and is
R' is halogen, OH or C1-6An alkoxy group.
More preferred in the present invention are the following compounds:
5- (3- (2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole (II-1 a);
5- (2-hydroxy-5- (2, 5-dimethyl-1H-pyrrole-1-yl) phenyl) -1H-tetrazole (II-1 b);
5- (2-chloro-5- (2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole (II-1 c);
5- (3- (3-ethoxycarbonyl-2, 5-dimethyl-1H-pyrrole-1-yl) phenyl) -1H-tetrazole (II-1 d);
5- (3- (3-carboxy-2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole (II-1 e);
5- (3- (1H-pyrrol-1-yl) phenyl) -1H-tetrazole (II-1 f);
1-carboxymethyl-5- (3- (2, 5-dimethyl-1H-pyrrole-1-yl) phenyl) -1H-tetrazole (II-2 b);
1-carboxymethyl-5- (3- (3-carboxyl-2, 5-dimethyl-1H-pyrrole-1-yl) phenyl) -1H-tetrazole (II-2d)
5- (3- (5- (trifluoromethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazole (III-1 a);
5- (3- (5- (chloromethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazole (III-1 b);
5- (3- (5- (hydroxymethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazolium (III-1 c);
1-carboxymethyl-5- (3- (5- (trifluoromethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazole (III-2 b);
1-ethyl-5- (3- (5-methine- (rhodanin-5-yl) -furan-2-yl) phenyl) -1H-tetrazolium (IV-2 a); and
1-carboxymethyl-5- (3- (5-methine- (rhodanin-5-yl) -furan-2-yl) phenyl) -1H-tetrazole (IV-2 b).
The compounds of the present invention can be prepared by a variety of reaction schemes and methods, as shown in the figure:
route a: the general method comprises the following steps:
reaction conditions are as follows: (i) hydrochloride (such as amine chloride, lithium chloride and the like) and DMF (dimethyl formamide) are taken as a solvent, and the mixture is refluxed at room temperature for 4 to 24 hours; (ii) sodium alkoxide in methanol or ethanol at room temperature to 100 deg.C for 4-32 hr
Route B: for compounds of formula I wherein Ar is a pyrrole ring:

reaction conditions are as follows: (iii) microwave reaction, acetic acid is used as solvent or no solvent, at the temperature of 120 ℃ and 160 ℃, for 5-20 minutes; (ii) the same reaction conditions as in the second step of scheme A.
Route C: for compounds of formula I wherein Ar is an oxadiazole ring:

reaction conditions are as follows: (iv) in the presence of 8-hydroxyquinoline, sodium carbonate or potassium carbonate is added at 80-100 ℃ for 2-8 hours, and ethanol is used as a solvent; (v) pyridine or tetrahydrofuran as solvent at room temperature to 120 deg.c for 2-6 hr; (ii) the same reaction conditions as in the second step of scheme A.
Route D: for compounds of formula I wherein Ar is a five-membered heterocyclic (e.g., pyrrole, furan, etc.) aldehyde:

reaction conditions are as follows: (vi) suzuki coupling reaction, organic boric acid reagent and palladium catalyst; (vii) condensation reaction (with ketone reagents shown or related in the figure), alkaline conditions: organic amine, inorganic strong base or weak acid strong base salt, and mixed solvent of methanol, ethanol, acetic acid, DMF or DMF and water at room temperature of-160 deg.C for 1-44 hr.
Ar and R in the above reaction scheme1-R4、R6And X, Y are as previously described for formula I, each U, V, W independently represents a heteroatom selected from N, O, S or is a carbon atom. In particular, the present invention relates to a method for producing,
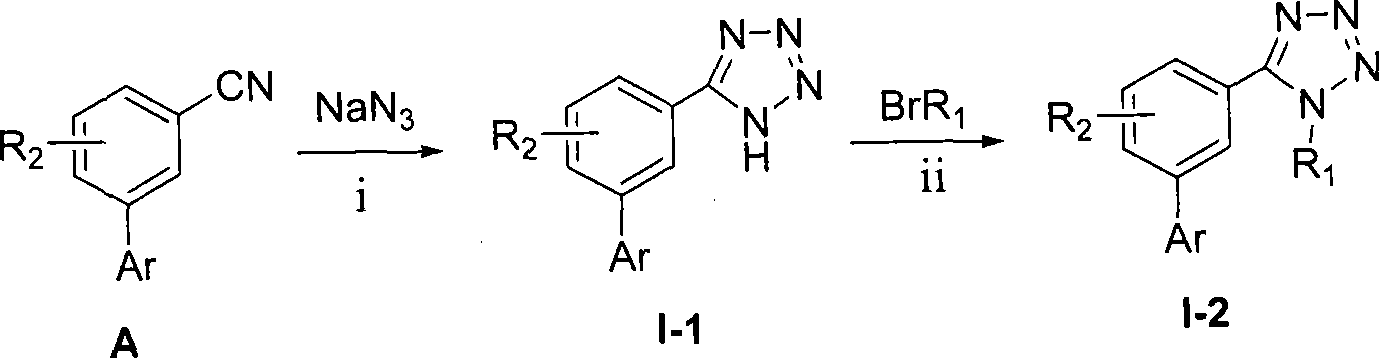
scheme a: the cyano group of the substituted benzonitrile compound (A) can react with sodium azide to synthesize a 1H-tetrazole ring to obtain a target compound I-1, and the 1-site nitrogen hydrogen on the tetrazole ring reacts with halohydrocarbon to obtain a compound I-2.
Scheme B: the substituted 5- (3-amino) phenyl-1H-tetrazole (B) and 2, 5-dimethoxytetrahydrofuran or beta-substituted 1, 4-diketone are subjected to Paal-Knorr reaction to obtain an N-arylpyrrole compound (II-1), and then the N-arylpyrrole compound (II-1) is reacted with halogenated hydrocarbon to generate a 1-substituted 1H-tetrazole-N-arylpyrrole compound (II-2).
Scheme C: substituted 5- (3-cyano) phenyl-1H-tetrazole (C) is used as a raw material, reacts with hydroxylamine hydrochloride to generate an amino oxime intermediate (D), then reacts with an acylation reagent to obtain a 3-aryl-1, 2, 4-oxadiazole compound (III-1), and then undergoes a nitrogen halogenation reaction to obtain a target compound III-2.
Scheme D: the substituted 5- (3-bromine) phenyl-1H-tetrazole (E) and the aromatic heterocyclic boric acid compound are coupled to generate the target compound IV-1 containing the aromatic heterocyclic through Suzuki reaction. When the aromatic heterocycle contains aldehyde group, the target compound IV-2 can be obtained by condensation and halogenation.
The invention relates to a substituted 5- (3-aromatic heterocycle substituted phenyl) tetrazole compound (formula I), which is an anti-HIV active compound with a novel skeleton structure. The compounds act on HIV-1gp41, and are expected to be developed into a novel anti-HIV drug with a specific target: non-peptide small molecule fusion inhibitors. The test results of the invention show that: IC for inhibiting 6-HB binding by Compound II-1a50The value was 25.61. mu.M, which is higher than that of the positive control compound NB-64 (IC) used in the same assay5058.74 μ M). The inhibitory Activity of this Compound against wild type HIV in a cellular assay (MT-2 lymphocytes), EC50The value was 7.70. mu.M (SI)>32) And has inhibitory activity on replication of various clinically isolated HIV strains, which is obviously superior to the known active compound NB-64. The activity data for some of the related compounds are shown in tables 1-2. The result of the invention shows that the compound represented by II-1a is used as HIV-1gp41 target, has a wide antiviral spectrum and is a novel anti-HIV active compound, and can be developed into a non-peptide small molecule anti-HIV fusion inhibitor.
The compounds of the present invention may be used either as such or in the form of their pharmaceutically acceptable salts or solvates. Pharmaceutically acceptable salts of the compounds of formula I include conventional salts with pharmaceutically acceptable inorganic or organic acids or bases. Examples of suitable acid addition salts include salts with hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid, perchloric acid, fumaric acid, acetic acid, propionic acid, succinic acid, glycolic acid, formic acid, lactic acid, maleic acid, tartaric acid, citric acid, pamoic acid, malonic acid, hydroxymaleic acid, phenylacetic acid, glutamic acid, benzoic acid, salicylic acid, fumaric acid, toluenesulfonic acid, methanesulfonic acid, naphthalene-2-sulfonic acid, benzenesulfonic acid, hydroxynaphthoic acid, hydroiodic acid, malic acid, tannic acid, and the like. Examples of suitable base addition salts include salts with sodium, lithium, potassium, magnesium, aluminum, calcium, zinc, N' -dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, N-methylglucamine, procaine, and the like. Reference herein to the compounds of the present invention includes compounds of formula I and pharmaceutically acceptable salts or solvates thereof.
According to the invention, the compounds of formula I according to the invention can be combined with customary pharmaceutical carriers or excipients to form pharmaceutical compositions. The pharmaceutical composition can be administered by oral or parenteral route. The pharmaceutical composition of the present invention can be prepared into various dosage forms including, but not limited to, tablets, capsules, solutions, suspensions, granules or injections, etc. according to conventional methods in the art, and can be administered orally or parenterally.
It is further noted that the dosage and method of administration of the compounds of the present invention will depend upon a variety of factors including the age, weight, sex, physical condition, nutritional status, the activity level of the compound, time of administration, metabolic rate, severity of the condition, and the subjective judgment of the treating physician. The preferable dosage is 0.01-100 mg/kg body weight/day.
Detailed Description
The following examples are intended to further illustrate the invention, but are not intended to limit the invention thereto.
Example 1: 5- (3- (2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazol (II-1a) (scheme B)
2, 5-hexanedione (0.13mL) was added to a solution of 3- (1H-tetrazol-5-yl) aniline (1mmol) in glacial acetic acid (3mL) and reacted at 150 ℃ for 10 minutes under microwave conditions. Cooling to room temperature, pouring the reaction product into ice water, and collecting solidWashing with water to neutrality, and separating with preparative chromatography (petroleum ether/ethyl acetate/acetic acid) to obtain white solid 124mg with yield of 52%, mp147-148 deg.C;1H NMR(DMSO-D6)δ ppm 8.14(1H,d,J=8.4Hz,ArH-6),7.87(1H,s,ArH-2),7.76(1H,t,J=8.4Hz,ArH-5),7.53(1H,d,J=8.4Hz,ArH-4),5.85(2H,s,PyH),2.02(6H,s,Py-CH3x 2) mass spectrum (EI-MS): m/z (%) 239 (M)+,76),211(M-2×N,100).
Example 2: 5- (3- (1H-pyrrole-1-yl) phenyl) -1H-tetrazole (II-1f)
The preparation method is the same as II-1a (route B). 3- (1H-tetrazol-5-yl) aniline (1mmo l) and 2, 5-dimethoxy tetrahydrofuran (0.14mL) react to obtain a compound II-1f, white solid 186mg, yield 88%, mp 210-;1H NMR(DMSO-D6)δ ppm 8.19(1H,d,J=2.0Hz,ArH-2),7.92(1H,d,J=8.4Hz,ArH-6),7.83(1H,dd,J=8.4 &2.0Hz, ArH-4), 7.71(1H, t, J ═ 8.4Hz, ArH-5), 7.47(2H, m, PyH-2, 5), 6.35(2H, t, J ═ 2.2Hz, PyH-3, 4); mass spectrum (EI-MS): m/z (%) 211 (M)+,100),183(M-2×N,98),168(M-2×N-NH,37).
Example 3: 5- (3- (3-ethoxycarbonyl-2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazol (II-1d) (scheme B)
Sodium methoxide (1.65g, 30.6mmol) was added in portions to a 0 ℃ solution of ethyl acetoacetate (4g, 30.6mmol) in anhydrous methanol (20mL), the mixture was allowed to react for 30 minutes while maintaining the temperature, and bromoacetone (2.2mL, 25.5mmol) was slowly added dropwise to the mixture, followed by reaction at room temperature for 10 hours. Adjusting pH to neutral with hydrochloric acid aqueous solution, extracting with ethyl acetate, drying, removing solvent, separating with chromatographic column (petroleum ether/ethyl acetate ratio is 8: 1) to obtain 3-ethoxycarbonyl-2, 5-hexanedione as light yellow liquid 2.9g, and obtaining product yield of 61%.
The newly prepared 3-ethoxycarbonyl-2, 5-hexanedione (205mg) and 3- (1H-tetrazol-5-yl) aniline (1mmol) react under the same conditions as in the preparation of II-1a to obtain a product II-1d, which is 256mg of light yellow oily liquid with yield of 78%, mp121-123℃。1H-NMR(CDCl3)δppm 11.66(1H,br,Tetrazole-H),8.33(1H,d,J=8.0Hz,ArH-6),7.99(1H,s,ArH-2),7.68(1H,t,J=8.0Hz,ArH-5),7.34(1H,d,J=8.0Hz,ArH-4),6.36(1H,s,Py-H),4.29(2H,q,-OCH 2 CH3),2.24(3H,s,Py-CH3),1.97(3H,s,Py-CH3),1.35(3H,t,-CH2 CH 3 ).
Example 4: 5- (3- (3-carboxy-2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazol-e (II-1e) (scheme B)
II-1d (100mg, 0.32mmol) was reacted in 1N aqueous NaOH (6mL) for 48 h at room temperature, adjusted to pH 3 with aqueous hydrochloric acid, the solid was collected, washed to neutrality with water and separated by preparative chromatography (petroleum ether/ethyl acetate/acetic acid) to give 63mg of white solid in 69% yield, decomposed at mp 250 ℃.1H-NMR(CDCl3)δ ppm 11.77(1H,br,Tetrazole-H),8.16(1H,d,J=8.0Hz,ArH-6),7.86(1H,s,ArH-2),7.78(1H,t,J=8.0Hz,ArH-5),7.55(1H,d,J=8.0Hz,ArH-4),6.24(1H,s,Py-H),2.21(3H,s,Py-CH3),1.87(3H,s,Py-CH3).
Example 5: 1-methoxycarbonylmethyl-5- (3- (2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazolium (II-2a) (synthetic route B or A)
Sodium methoxide (270mg, 5mmol) and methyl bromoacetate (0.3mL, 3.3mmol) were added to a solution of II-1a (231mg, 1mmol) in methanol (10mL), and the reaction was refluxed for 27 hours. The solvent was evaporated, the residue was dissolved in ethyl acetate, washed with water, dried and concentrated, and the crude product was separated by preparative chromatography (petroleum ether/ethyl acetate/acetic acid) to give 205mg of a white solid in 68% yield, mp 128-.1H NMR(DMSO-D6)δ ppm 8.15(1H,d,J=8.0Hz,ArH-6),7.83(1H,s,ArH-2),7.74(1H,t,J=8.0Hz,ArH-5),7.50(1H,d,J=8.0Hz,ArH-4),5.94(2H,s,CH2),5.85(2H,s,Py-H),3.76(3H,s,OCH3),2.00(6H,s,Py-CH3×2).
Example 6: 1-carboxymethyl-5- (3- (2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazolium (II-2B) (scheme B)
A10% aqueous NaOH solution (1mL) was added to a solution of II-2a (40mg, 0.13mmol) in methanol (2mL) and reacted at room temperature for 6 hours. Pouring the reaction solution into ice water, adjusting the pH value to acidity by using 10% hydrochloric acid aqueous solution, collecting the solid, washing the solid to neutrality by using water, and drying to obtain the product II-2b34mg with the yield of 87% and the temperature of mp 158-.1H-NMR(DMSO-D6)δ ppm:8.15(1H,d,J=8.0Hz,ArH-6),7.83(1H,s,ArH-2),7.74(1H,t,J=8.0Hz,ArH-5),7.50(1H,d,J=8.0Hz,ArH-4),5.84(2H,s,CH2),5.78(2H,s,Py-H),2.00(6H,s,Py-CH3X 2) mass spectrum (ESI-MS): m/z (%) 296(M-H, 43), 252(M-COOH, 31), 195 (M-H-CH)2COOH-3×N,100).
Example 7: 1-ethoxycarbonylmethyl-5- (3- (3-ethoxycarbonyl-2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole (II-2c) (synthetic route A or B)
Sodium ethoxide (61mg, 0.9mmol) and methyl bromoacetate (0.06mL, 0.6mmol) were added to a solution of II-1d (93mg, 0.3mmol) in ethanol (5mL), and the reaction was refluxed for 4 hours. The solvent was evaporated and the residue dissolved in ethyl acetate, washed with water, dried, concentrated and the crude product was chromatographed (petroleum ether/ethyl acetate) to give II-2c as a white oil 71mg, 60% yield.1H NMR(CDCl3)δ ppm 8.28(1H,d,J=8.0Hz,ArH-6),8.01(1H,s,ArH-2),7.64(1H,t,J=8.0Hz,ArH-5),7.32(1H,d,J=8.0Hz,ArH-4),6.39(1H,s,Py-H),5.47(2H,s,CH2),4.31(4H,m,-OCH 2 CH3×2),2.33(3H,s,Py-CH3),2.02(3H,s,Py-CH3),1.34(6H,m,-CH2 CH 3 ×2).
Example 8: 5- (3- (5- (trifluoromethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazol-e (III-1a) (scheme C)
And (2) cooling a hydrochloride solution (7mL) of 3- (1H-tetrazole-5-yl) aniline (966mg, 6mmol) to 0-5 ℃, and dropwise adding an aqueous solution (4mL) of sodium nitrite (434mg, 6.3mmol) to generate the diazonium salt. An additional aqueous sodium cyanide solution (2mL, 17.2mmol) was added to a mixture of cuprous cyanide (592mg, 6.6mmol) and water (4mL) to form NaCu (CN)3The double salt solution is heated to 60 ℃. The diazonium salt solution was slowly added to the NaCu (CN)3The solution was then stirred for 2 hours at room temperature and cooled to room temperature. The reaction system was slowly poured into cold concentrated hydrochloric acid, extracted with ethyl acetate, dried, and the solvent was removed and separated by preparative chromatography (petroleum ether/ethyl acetate/glacial acetic acid 7:3:0.03) to give 5- (3-cyano) phenyl-1H-tetrazole (C) as a white solid 626mg with a product yield of 61% and mp 145-.
8-Hydroxyquinoline (1mg, 0.0075mmol) was added to a solution of C (513mg, 3mmol) in ethanol (30mL), and then hydroxylamine hydrochloride (446mg, 6.4mmol) and an aqueous solution (5mL each) of sodium carbonate (515mg, 4.9mmol) were added in this order, followed by heating and refluxing for 4 hours. The ethanol is evaporated, the residue is dissolved by adding water (30mL), the pH value is adjusted to acidity by hydrochloric acid aqueous solution, solid is collected, washed to neutrality by water and dried to obtain (Z) -5- (3-amino oxime) phenyl-1H-tetrazole (D), white solid is 465mg, the product yield is 76%, and the temperature is mp194-196 ℃.
Trifluoroacetic anhydride (0.21mL, 1.5mmol) was added to a solution of D (102mg, 0.5mmol) in anhydrous pyridine (3mL) and heated under reflux for 2 h. The reaction was cooled to room temperature and poured into ice water, the pH was adjusted to acidity with aqueous hydrochloric acid, the solid was collected, washed with water to neutrality and dried to give III-1a as a white solid at 121mg, yield 86%, mp 146-.1H-NMR(DMSO-D6) δ ppm: 8.75(1H, s, ArH-2), 8.36(1H, d, J ═ 8.0Hz, ArH-6), 8.30(1H, d, J ═ 8.0Hz, ArH-4), 7.88(1H, t, J ═ 8.0Hz, ArH-5), mass spectrometry (ESI-MS): m/z (%) 281(M-H, 100), 137 (M-tetrazol-phenyl, 23).
Example 9: 5- (3- (5- (chloromethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazol-e (III-1b) (scheme C)
Chloroacetyl chloride (0.1mL, 1.2mmol) was added toD (204mg, 1mmol) in THF (10mL) was heated under reflux for 4 hours. The reaction was cooled to room temperature and poured into ice water, the solid was collected and washed with water to neutrality and separated by preparative chromatography (petroleum ether/ethyl acetate/glacial acetic acid) to give III-1b as a white solid 176mg in 67% yield, mp 160-.1H-NMR(DMSO-D6)δ ppm:8.71(1H,s,ArH-2),8.30(1H,d,J=8.0Hz,ArH-6),8.24(1H,d,J=8.0Hz,ArH-4),7.84(1H,t,J=8.0Hz,ArH-5),5.24(2H,s,CH2) Mass spectrum (ESI-MS): m/z (%) 261(M-H, 100), 263(M-H +2, 28), 117 (M-tetrazolo-phenyl, 35).
Example 10: 5- (3- (5- (hydroxymethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazolium (III-1C) (scheme C)
III-1b (76mg, 0.29mmol) was reacted in 10% aqueous NaOH (2mL) at room temperature for 1.5 h. The reaction solution was poured into ice water, adjusted to acidic pH with 10% aqueous hydrochloric acid, extracted with ethyl acetate, dried, and the solvent was removed and separated by preparative TLC (petroleum ether/ethyl acetate 3:2) to give III-1c as a white solid 39mg in 55% yield, mp 120-.1H-NMR(DMSO-D6)δ ppm:11.60(1H,s,OH),8.47(1H,s,ArH-2),8.22(1H,d,J=8.0Hz,ArH-6),7.96(1H,d,J=8.0Hz,ArH-4),7.75(1H,t,J=8.0Hz,ArH-5),4.44(2H,s,CH2) Mass spectrum (ESI-MS): m/z (%) 243(M-H, 100), 215(M-H-2 XN, 34).
Example 11: 1-ethoxycarbonylmethyl-5- (3- (5- (trifluoromethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazol (III-2a) (scheme C or A)
The preparation method is the same as II-2 c. III-1a (50mg, 0.18mmol) and methyl bromoacetate (0.02mL, 0.22mmol) were reacted to give III-2a as an off-white solid, 35mg, yield 54%, mp 78-81 ℃.1H NMR(CDCl3)δ ppm 8.94(1H,s,ArH-2),8.41(1H,d,J=8.0Hz,ArH-6),8.26(1H,d,J=8.0Hz,ArH-4),7.69(1H,t,J=8.0Hz,ArH-5),5.49(2H,s,CH2),4.31(2H,q,-OCH 2 CH3),1.32(3H,t,-CH2 CH 3 ).
Example 12: 1-Ethyl-5- (3- (5-formyl-furan-2-yl) phenyl) -1H-tetrazolium (IV-1a) (scheme D)
A hydrobromic acid solution (40%, 2mL) of 3- (1H-tetrazol-5-yl) aniline (322mg, 2mmol) was cooled to 0-5 ℃, and an aqueous solution (2mL) of sodium nitrite (166mg, 2.4mmol) was added dropwise to generate the diazonium salt. This diazonium salt solution was slowly added to a solution of cuprous bromide (344mg, 2.4mmol) in hydrobromic acid (40%, 1mL) and reacted overnight at room temperature. The reaction system was slowly poured into ice water, the solid was collected, washed with water to neutrality, and dried to obtain 5- (3-bromo) phenyl-1H-tetrazole as a yellow solid (362 mg, yield 80%). The intermediate (113mg, 0.5mmol) and iodoethane (0.14mL, 1.75mmol) were reacted according to II-2c preparation to give 113mg of 1-ethyl-5- (3-bromophenyl) -1H-tetrazole as an oily substance in 89% yield.
Pd (PPh) under nitrogen protection3)4(22mg, 0.02mmol) was added to a DMF solution (5mL) of 1-ethyl-5- (3-bromophenyl) -1H-tetrazole (113mg, 0.45mmol), followed by addition of an aqueous solution (10mL) of diisopropylethylamine (0.22mL, 1.35mmol) and a DMF solution (5mL) of 5-formyl-2-furanboronic acid (75mg, 0.54mmol), respectively, and reaction at 100 ℃ for 4 hours. The reaction was poured into ice water, extracted with ethyl acetate, dried, and the solvent removed and chromatographed using preparative chromatography (petroleum ether/ethyl acetate 3:1) to give 88mg of a yellow solid, 73% yield, mp77-80 ℃.1H NMR(CDCl3)δ ppm:9.70(1H,s,CHO),8.59(1H,s,ArH-2),8.20(1H,d,J=8.0Hz,ArH-6),7.96(1H,d,J=8.0Hz,ArH-4),7.59(1H,t,J=8.0Hz,ArH-5),7.37&6.6.98(each1H,d,J=4.0Hz,FuranH-4′or-3′),4.74(2H,q,-NCH 2 CH3),1.72(3H,t,-CH2 CH 3 ).
Example 13: 1-Ethyl-5- (3- (5-methylidene- (rhodanin-5-yl) -furan-2-yl) phenyl) -1H-tetrazol (IV-2a) (scheme D)
IV-1a (46mg, 0.17mmol), woundTannin (37mg, 0.28mmol) and anhydrous sodium acetate (42mg, 0.51mmol) were reacted in anhydrous methanol (5mL) at reflux for 13 hours. After removal of the solvent, preparative TLC (petroleum ether/ethyl acetate) was used to obtain IV-2a as a red solid mg, yield%, mp ℃.1H NMR(CDCl3)δ ppm:9.70(1H,s,CHO),8.59(1H,s,ArH-2),8.20(1H,d,J=8.0Hz,ArH-6),7.96(1H,d,J=8.0Hz,ArH-4),7.59(1H,t,J=8.0Hz,ArH-5),7.58(1H,s,C=C-H),7.37 & 6.6.98(each1H,d,J=4.0Hz,FuranH-4′or-3′),4.74(2H,q,-NCH 2 CH3),1.72(3H,t,-CH2 CH 3 ).
Example 14: anti-HIV Activity assay (cell model) with EC50Indicates the activity.
Reference documents to which the assay can be referred are Jiang, s., et al.
50 μ L of compound solutions of different concentrations were mixed with equal volumes of HIV-1in 96 well cell culture platesIIIBVirus strain (100 TCID)50) Mixing, incubating at 37 ℃ for 30 min, and adding 100. mu.L of MT-2 cells (1X 10)5mL, RPIM 1640 medium with 10% serum), mixed well and incubated overnight at 37 ℃. On day 2, 150. mu.L of supernatant was aspirated, an equal volume of fresh medium was replenished, incubation was continued at 37 ℃ for 3 additional days, and Cytopathic (CPE) effects were recorded on day four. Then, 100. mu.L of the culture supernatant was aspirated, and the viral particles were lysed with 5% Triton X-100 to detect p24 antigen by ELISA. Briefly, the ELISA plates were coated with HIVIG (2. mu.g/mL), blocked with 1% nonfat milk, and the viral lysates were added and incubated at 37 ℃ for 60 min. After the plate is fully washed, anti-p 24 monoclonal antibody-183-12H-5C, biotin-labeled goat anti-mouse antibody and avidin-labeled horseradish peroxidase are added in sequence. Then, color was developed with TMB, and the optical density was measured at 450 nm. Calcusyn software was used to calculate half the viral inhibitory concentration (EC) of compounds50)。
TABLE 1 Activity data for inhibition of HIV replication and formation of the target molecule gp41-6 mer
And (3) SI: selection index CC50/EC50(ii) a ND: is under test
TABLE 2 Activity data (cell model) for inhibition of replication of HIV primary strains
Example 15: cytotoxicity assay of Compounds with CC50And (4) showing.
Reference documents to which the assay can be referred are Jiang, s., et al, antimicrob, Agents Chemother.2004, 48, 4349-.
In 96-well cell culture plates, 50. mu.L of different concentrations of compound solutions were mixed with an equal volume of PBS, incubated at 37 ℃ for 30 minutes, and then 100. mu.L of MT-2 cells (1X 10)5mL, RPIM 1640 medium with 10% serum), mixed well and incubated overnight at 37 ℃. After sucking off 150. mu.L of the supernatant on day 2, supplementing an equal volume of fresh medium and further incubating at 37 ℃ for 3 days, 50. mu.L of freshly prepared XTT solution containing PMS (1mg/mL) was added on day four and the optical density at 450nm was measured after 4 h. Calcusyn software was used to calculate half the Cytotoxic Concentration (CC) of the compound50). The results are shown in Table 1.
Example 16: inhibition of gp41 formation of six-membered helix bundle (6-HB), IC for inhibition of Activity50And (4) showing.
References to which assays can be referred are Jiang, S., et al.J.Virol. methods, 1999, 80, 85-96. The results are shown in Table 1.
The existing results show that: the compound of formula I is a non-peptide small molecule inhibitor with a novel framework structure and acting on HIV-1gp 41. They can effectively inhibit the formation of HIV-1gp41 six-membered helix bundle body (6-HB), thereby inhibiting the replication of HIV. The compounds also have inhibitory activity on various types of HIV virus strains clinically isolated, and have wide antiviral spectrum. The compound related by the invention is expected to be developed into a novel anti-HIV drug: a non-peptide small molecule HIV-1 fusion inhibitor taking gp41 as a target.
Claims (8)
1. Tetrazole aryl heterocyclic compounds of formula I:
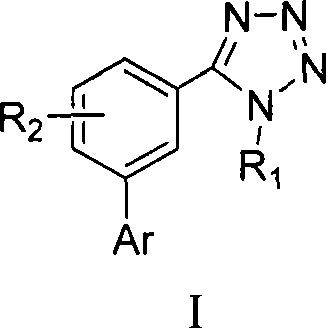
wherein,
R1=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6a hydrocarbyl group;
R2-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Alkyl radical, C1-6Alkoxy, -CF3、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
ar is a five membered heteroaromatic ring containing 1 to 3 heteroatoms selected from N, O, S, selected from:


wherein R is3=CHO、COR’、COOR’、COOH、CF3、CH2R', halogen, C1-6Hydrocarbyl radical, C1-6Alkoxy, -NH2、-OH、-NO2、-CN、-HC=CH-CN、-CH=CH2-C ≡ CH, -C ≡ CR ', -CH ═ CHR ', -CH ═ CHCOR ', or optionally bearing an ester group, a carboxyl group, C ≡ C optionally at available positions in its ring structure1-6Hydrocarbyl, phenyl-substituted heterocyclic groups:

wherein X and Y are each independently selected from C, O, S and NH;
R4=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6hydrocarbyl, phenyl;
R5-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Hydrocarbyl radical, C1-6Alkoxy, -CF3、CHO、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
the five-membered heteroaromatic ring optionally carries an unsaturation selected from the group consisting of aldehyde, keto, ester, carboxyl, cyano, alpha, beta, unsaturated, etc. at an available position on the ringKetones, alkenes, alkynes, C1-6Hydrocarbyl radical, C1-6Alkoxy, halogen, -NH2、-OH、-NO2and-CF3A substituent of (1);
R’=C1-6a hydrocarbyl group; and is
R' is halogen, OH or C1-6An alkoxy group.
2. The compound of claim 1, or a pharmaceutically acceptable salt thereof, having formula II:

wherein,
R1=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6a hydrocarbyl group;
R2-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Alkyl radical, C1-6Alkoxy, -CF3、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R3=CHO、COR’、COOR’、COOH、CF3、CH2r', halogen, C1-6Hydrocarbyl radical, C1-6Alkoxy, -NH2、-OH、-NO2、-CN、-HC=CH-CN、-CH=CH2-C ≡ CH, -C ≡ CR ', -CH ═ CHR', -CH ═ CHCOR ', or optionally bearing an ester group, a carboxyl group, C ≡ CH', or a cyclic structure thereof1-6Hydrocarbyl, phenyl-substituted heterocyclic groups:

wherein X and Y are each independently selected from C, O, S and NH;
R4=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6hydrocarbyl, phenyl;
R5-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Hydrocarbyl radical, C1-6Alkoxy, -CF3、CHO、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R6=H、CH3、CF3halogen or C2-4A hydrocarbyl group;
R’=C1-6a hydrocarbyl group; and is
R' is halogen, OH or C1-6An alkoxy group.
3. The compound of claim 1, or a pharmaceutically acceptable salt thereof, having formula III:

wherein,
R1=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6a hydrocarbyl group;
R2-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Alkyl radical, C1-6Alkoxy, -CF3、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R3=CHO、COR’、COOR’、COOH、CF3、CH2r', halogen, C1-6Hydrocarbyl radical, C1-6Alkoxy, -NH2、-OH、-NO2、-CN、-HC=CH-CN、-CH=CH2-C ≡ CH, -C ≡ CR ', -CH ═ CHR', -CH ═ CHCOR ', or optionally bearing an ester group, a carboxyl group, C ≡ CH', or a cyclic structure thereof1-6Hydrocarbyl, phenyl-substituted heterocyclic groups:
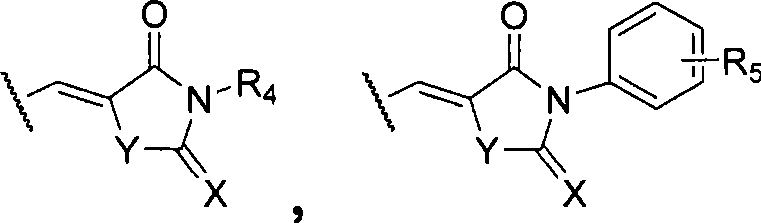
wherein X and Y are each independently selected from C, O, S and NH;
R4=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6hydrocarbyl, phenyl;
R5-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Hydrocarbyl radical, C1-6Alkoxy, -CF3、CHO、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R’=C1-6a hydrocarbyl group; and is
R' is halogen, OH or C1-6An alkoxy group.
4. The compound of claim 1, or a pharmaceutically acceptable salt thereof, having formula IV:
wherein,
R1=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6a hydrocarbyl group;
R2-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Alkyl radical, C1-6Alkoxy, -CF3、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R3=CHO、COR’、COOR’、COOH、CF3、CH2r', halogen, C1-6Hydrocarbyl radical, C1-6Alkoxy, -NH2、-OH、-NO2、-CN、-HC=CH-CN、-CH=CH2、-C≡CH、-C≡CR’、-CH=CHR ', -CH ═ CHCOR', or optionally having an ester group, carboxyl group, C on the ring structure1-6Hydrocarbyl, phenyl-substituted heterocyclic groups:

wherein X and Y are each independently selected from C, O, S and NH;
R4=-H、-CH2COOH、-CH2CH2COOH、-CH=CH-COOH、-CH2COOR’、-CH2CH2COOR’、-CH=CH-COOR’、C1-6hydrocarbyl, phenyl;
R5-H, halogen, -NO2、-NH2、-NHR’、-N(R’)2、-CN、-OH、C1-6Hydrocarbyl radical, C1-6Alkoxy, -CF3、CHO、-COOH、-SO3H、-CONH2-CONHR 'or-COOR';
R’=C1-6a hydrocarbyl group; and is
R' is halogen, OH or C1-6An alkoxy group.
5. The compound of claim 1, or a pharmaceutically acceptable salt thereof, selected from:
5- (3- (2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole;
5- (2-hydroxy-5- (2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole;
5- (2-chloro-5- (2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole;
5- (3- (3-ethoxycarbonyl-2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole;
5- (3- (3-carboxy-2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole;
5- (3- (1H-pyrrole-1-yl) phenyl) -1H-tetrazole (II-1f)
1-carboxymethyl-5- (3- (2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole;
1-carboxymethyl-5- (3- (3-carboxy-2, 5-dimethyl-1H-pyrrol-1-yl) phenyl) -1H-tetrazole;
5- (3- (5- (trifluoromethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazole;
5- (3- (5- (chloromethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazole;
5- (3- (5- (hydroxymethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazole;
1-carboxymethyl-5- (3- (5- (trifluoromethyl) -1, 2, 4-oxadiazol-3-yl) phenyl) -1H-tetrazole;
1-ethyl-5- (3- (5-methine- (rhodanin-5-yl) -furan-2-yl) phenyl) -1H-tetrazole; and
1-carboxymethyl-5- (3- (5-methine- (rhodanin-5-yl) -furan-2-yl) phenyl) -1H-tetrazole.
6. A process for preparing a compound according to claim 1, or a pharmaceutically acceptable salt thereof, which comprises:
route a: general procedure

The cyano group of the substituted benzonitrile compound (A) can react with sodium azide to synthesize a 1H-tetrazole ring to obtain a target compound I-1, and the 1-site nitrogen hydrogen on the tetrazole ring reacts with halohydrocarbon to obtain a compound I-2;
or,
route B: for compounds of formula I wherein Ar is a pyrrole ring:

carrying out Paal-Knorr reaction on substituted 5- (3-amino) phenyl-1H-tetrazole (B) and 2, 5-dimethoxytetrahydrofuran or beta-substituted 1, 4-diketone to obtain an N-arylpyrrole compound (II-1), and reacting with halogenated hydrocarbon to generate a 1-substituted 1H-tetrazole-N-arylpyrrole compound (II-2);
or,
route C: for compounds of formula I wherein Ar is an oxadiazole ring:

substituted 5- (3-cyano) phenyl-1H-tetrazole (C) is used as a raw material, the substituted 5- (3-cyano) phenyl-1H-tetrazole (C) reacts with hydroxylamine hydrochloride to generate an amino oxime intermediate (D), the amino oxime intermediate (D) reacts with an acylation reagent to obtain a 3-aryl-1, 2, 4-oxadiazole compound (III-1), and then a target compound III-2 is obtained through a nitrogen halogenation reaction;
or,
route D: for compounds of formula I wherein Ar is a five-membered heterocyclic (e.g., pyrrole, furan, etc.) aldehyde:
the substituted 5- (3-bromine) phenyl-1H-tetrazole (E) and the aromatic heterocyclic boric acid compound are coupled to generate the target compound IV-1 containing the aromatic heterocyclic through Suzuki reaction. When the aromatic heterocycle contains aldehyde group, a target compound IV-2 can be obtained through condensation and halogenation;
r in the reaction scheme1-R4、R6And X, Y are as previously described for formula I, each U, V, W independently represents a heteroatom selected from N, O, S or is a carbon atom.
7. A pharmaceutical composition comprising at least one compound of formula I as described in any one of claims 1 to 5 or a pharmaceutically acceptable salt thereof, together with one or more pharmaceutically acceptable carriers or excipients.
8. Use of a compound of formula I as described in any one of claims 1-5, or a pharmaceutically acceptable salt thereof, for the manufacture of a medicament for the treatment or prevention of a disease or condition associated with HIV infection.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNA2007103069390A CN101468985A (en) | 2007-12-28 | 2007-12-28 | 5-(3-aromatic heterocyclic substituted phenyl) tetrazole compounds and anti-HIV/AIDS use thereof |
| PCT/CN2008/002105 WO2009094845A1 (en) | 2007-12-28 | 2008-12-29 | 5-(3-heteroaromatic ring substituted phenyl) tetrazole compounds and their uses for anti-hiv/aids |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNA2007103069390A CN101468985A (en) | 2007-12-28 | 2007-12-28 | 5-(3-aromatic heterocyclic substituted phenyl) tetrazole compounds and anti-HIV/AIDS use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101468985A true CN101468985A (en) | 2009-07-01 |
Family
ID=40826820
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2007103069390A Pending CN101468985A (en) | 2007-12-28 | 2007-12-28 | 5-(3-aromatic heterocyclic substituted phenyl) tetrazole compounds and anti-HIV/AIDS use thereof |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN101468985A (en) |
| WO (1) | WO2009094845A1 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010081387A1 (en) * | 2009-01-13 | 2010-07-22 | 中国人民解放军军事医学科学院毒物药物研究所 | N-substituted phenyl-3-methylene heterocyclic aryl-2,5-dimethylpyrrole compounds and anti-hiv/aids uses thereof |
| WO2017076740A1 (en) | 2015-11-04 | 2017-05-11 | Basf Se | Substituted oxadiazoles for combating phytopathogenic fungi |
| US10492494B2 (en) | 2015-11-13 | 2019-12-03 | Basf Se | Substituted oxadiazoles for combating phytopathogenic fungi |
| US10674727B2 (en) | 2015-11-19 | 2020-06-09 | Basf Se | Substituted oxadiazoles for combating phytopathogenic fungi |
| US10687532B2 (en) | 2015-11-13 | 2020-06-23 | Basf Se | Substituted oxadiazoles for combating phytopathogenic fungi |
| US10986839B2 (en) | 2016-04-11 | 2021-04-27 | Basf Se | Substituted oxadiazoles for combating phytopathogenic fungi |
| CN119977956A (en) * | 2024-09-14 | 2025-05-13 | 华东师范大学 | A 3,5-disubstituted-1,2,4-triazole compound and its preparation method and application |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100581199B1 (en) * | 1998-06-19 | 2006-05-17 | 카이론 코포레이션 | Inhibitors of Glycogen Synthase Kinase 3 |
| US7053085B2 (en) * | 2003-03-26 | 2006-05-30 | Merck & Co. Inc. | EP4 receptor agonist, compositions and methods thereof |
| JP2005225819A (en) * | 2004-02-13 | 2005-08-25 | Orient Chem Ind Ltd | Azole compound having pyrrole ring and process for producing the same |
| US7517998B2 (en) * | 2004-06-01 | 2009-04-14 | Boehringer Ingelheim International Gmbh | Non nucleoside reverse transcriptase inhibitors |
-
2007
- 2007-12-28 CN CNA2007103069390A patent/CN101468985A/en active Pending
-
2008
- 2008-12-29 WO PCT/CN2008/002105 patent/WO2009094845A1/en not_active Ceased
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010081387A1 (en) * | 2009-01-13 | 2010-07-22 | 中国人民解放军军事医学科学院毒物药物研究所 | N-substituted phenyl-3-methylene heterocyclic aryl-2,5-dimethylpyrrole compounds and anti-hiv/aids uses thereof |
| WO2017076740A1 (en) | 2015-11-04 | 2017-05-11 | Basf Se | Substituted oxadiazoles for combating phytopathogenic fungi |
| US10492494B2 (en) | 2015-11-13 | 2019-12-03 | Basf Se | Substituted oxadiazoles for combating phytopathogenic fungi |
| US10687532B2 (en) | 2015-11-13 | 2020-06-23 | Basf Se | Substituted oxadiazoles for combating phytopathogenic fungi |
| US10674727B2 (en) | 2015-11-19 | 2020-06-09 | Basf Se | Substituted oxadiazoles for combating phytopathogenic fungi |
| US10986839B2 (en) | 2016-04-11 | 2021-04-27 | Basf Se | Substituted oxadiazoles for combating phytopathogenic fungi |
| CN119977956A (en) * | 2024-09-14 | 2025-05-13 | 华东师范大学 | A 3,5-disubstituted-1,2,4-triazole compound and its preparation method and application |
| CN119977956B (en) * | 2024-09-14 | 2025-09-09 | 华东师范大学 | A 3,5-disubstituted-1,2,4-triazole compound and its preparation method and application |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009094845A1 (en) | 2009-08-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TW492971B (en) | Nitrogen containing heteroaromtics as factor XA inhibitors | |
| US8227500B2 (en) | 5-membered nitrogen containing heterocyclic derivatives and pharmaceutical compositions comprising the same | |
| JP4211394B2 (en) | Propane-1,3-dione derivatives | |
| JP2009532430A (en) | Benzofuran compounds as EP1 receptor antagonists | |
| CN101468985A (en) | 5-(3-aromatic heterocyclic substituted phenyl) tetrazole compounds and anti-HIV/AIDS use thereof | |
| CN102239164A (en) | 2h-chromene compound and derivative thereof | |
| WO2006085685A1 (en) | Pyrazole compound | |
| WO2007055418A1 (en) | Aza-substituted spiro derivative | |
| JPWO2002002533A1 (en) | Propane-1,3-dione derivatives | |
| JP2004502760A (en) | Pyrazole derivative | |
| CZ316995A3 (en) | Pyrrole derivatives | |
| US7951956B2 (en) | Benzoimidazole compound capable of inhibiting prostaglandin D synthetase | |
| WO2005049622A1 (en) | 5-5-membered fused heterocyclic compound and use thereof as hcv polymerase inhibitor | |
| CN112321604A (en) | Macrocyclic JAK2 inhibitors and their applications | |
| CA2718685C (en) | (pyrazolyl carbonyl)imidazolidinone derivatives for the treatment of retroviral diseases | |
| CN106279150B (en) | Thick cyclics of pyridine and its production and use | |
| CN101511354A (en) | Biphenyl substituted spirotetronic acids and their use for the treatment of retroviral diseases | |
| CN104684913B (en) | Three (miscellaneous) arylpyrazole and application thereof | |
| JP2005089298A (en) | Naphthalene compound and its pharmaceutical use | |
| CN101717364A (en) | Paradoximes as HIV reverse transcriptase inhibitor as well as preparation method and purpose thereof | |
| JP3354258B2 (en) | New indoles or their salts | |
| JP2006505625A (en) | Pyrazole amide for treating HIV infection | |
| CN101407476B (en) | M-bis(arene) polysubstituted aniline compound as non-nucleoside HIV reverse transcriptase inhibitor, and preparation and use thereof | |
| CN111285859A (en) | A class of 2,4,5-trisubstituted pyrimidine compounds targeting HIV-1 reverse transcriptase and their preparation method and application | |
| CN101775011B (en) | N-substituted phenyl-3-methylene heterocyclic arene-2,5-dimethylpyrrole compound and application thereof in anti-HIV/AIDS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20090701 |